4 minute read
Chronic Thromboembolic Pulmonary Hypertension


By Daniel T. Layish, MD
There are several categories of pulmonary hypertension. WHO Group I includes patients with idiopathic pulmonary hypertension, familial pulmonary hypertension, drug and toxin related (fen-phen) portopulmonary hypertension, HIV related pulmonary hypertension and pulmonary arterial hypertension associated with connective tissue disorders (such as scleroderma). WHO Group II pulmonary hypertension is often referred to as pulmonary venous hypertension. This includes patients with left ventricular systolic or diastolic dysfunction or valvular heart disease. Essentially, the WHO Group II category includes patients who have an elevated pulmonary capillary wedge pressure and/ or elevated left ventricular end diastolic pressure. WHO Group III pulmonary hypertension consists of patients with COPD, interstitial lung disease, or other conditions in which hypoxia causes vasoconstriction. The remainder of this article will focus on WHO Group IV pulmonary hypertension (chronic thromboembolic pulmonary hypertension or CTEPH). Although WHO Group IV patients are relatively rare, it is crucial to identify them because this is the only type of pulmonary hypertension which is potentially surgically curable.
Advertisement
After acute pulmonary embolism, most patients will recover and have normal pulmonary hemodynamics, gas exchange, and exercise tolerance. It is believed that 1-4% of patients with acute pulmonary embolism will go on to develop CTEPH within two years. It is not clear why some patients with acute pulmonary embolism develop CTEPH. Risk factors include hypercoagulable states, myeloproliferative syndromes, splenectomy and chronic indwelling central venous catheters. Patients with CTEPH present with dyspnea, which can have a gradual onset. Many patients with CTEPH will not have a known previous diagnosis of acute pulmonary embolism. As with other patients with pulmonary hypertension, patients with CTEPH may not show findings on physical exam until pulmonary hypertension is in the late stages. Findings include a right ventricular lift, jugular venous distention, fixed splitting of the second heart sound, hepatomegaly, ascites, and peripheral edema. Patients with CTEPH may have “flow murmurs” heard over the lung fields because of turbulent flow through partially obstructed or recanalized pulmonary arteries. These tend to be accentuated during inspiration.
Acute pulmonary embolism is the trigger for CTEPH. In some patients this triggers a small vessel vasculopathy (for unclear reasons) that contributes to the extent of pulmonary hypertension. This may explain why up to 35 percent of patients who undergo succesful pulmonary thromoendarterectomy can have some degree of postoperative pulmonary hypertension.
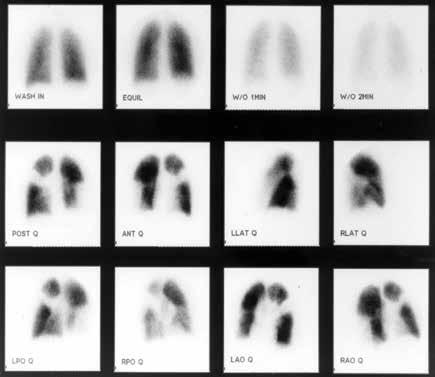
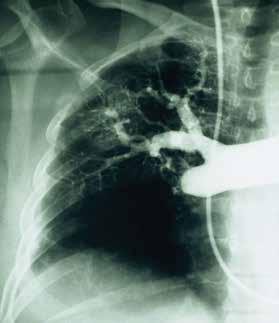
Although VQ scanning has become less commonly used for diagnosis of acute pulmonary embolism this remains the initial imaging study of choice in patients with pulmonary hyperten- sion to separate “small vessel” variants (Idiopathic pulmonary arterial hypertension) from “large vessel” disease (CTEPH) A normal VQ scan essentially excludes the diagnosis of CTEPH. A scan with one or more mismatched segmental defects is suggestive of the diagnosis. However, it is important to note that VQ scan can often understate the extent of central pulmonary vascular obstruction. Once the VQ scan is found to be abnormal then further testing should be undertaken (such as CT angiogram and/or pulmonary angiography). The angiographic findings in CTEPH are distinct from those of acute pulmonary embolism. They can include pouch defects and pulmonary artery webs. Patients with severe pulmonary hypertension have been found to tolerate performance of angiography as well as VQ scan without significant complication rate.
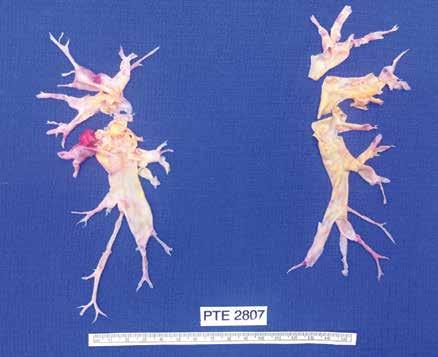
The surgery for CTEPH is quite different from surgical intervention for an acute pulmonary embolism. Surgery for CTEPH is called a pulmonary thromboendarterectomy (PTE), which requires median sternotomy and cardiopulmonary bypass. It requires an often tedious intimal dissection of fibrotic recannalized thrombus from the native pulmonary arterial wall. IVC filter placement is usually recommended before pulmonary thromboendarterectomy. These patients can have a complicated postoperative course and this type of surgery is only done at a few specialized centers in the country. The center which is best known for this type of surgery is the University of California (San Diego). Patients who have undergone PTE are typically maintained on lifelong anticoagulation. To be a candidate for this surgery, a patients must have central, surgically accesible chronic thromboemboli. A significant postoperative complication is pulmonary artery steal, which refers to redistribution of pulmonary arterial blood flow from well-perfused segments into the newly opened segments resulting in ventilation perfusion mismatch and hypoxia. This redistribution of flow resolves over time. Approximately, 30% of PTE patients can develop reperfusion pulmonary edema. The perioperative mortality of pulmonary thromboendarterectomy can be in the range of 2-3% in experienced centers. Outcome is clearly better in high voluime centers (more than fifty PTE surgeries/year). Approximately 5000 thromboendarterectomy procedures have been performed worldwide, 3000 at UCSD alone.
Surgery for CTEPH is clearly the best therapeutic option. However, there are some patients with CTEPH who are inoperable or who have persistent or recurrent pulmonary hypertension after undergoing pulmonary thromboendarterectomy. There is now a medical therapy available for these patients. Riociguat (Adempas) was approved by the FDA in October 2013. It is a member of a new class of compounds-soluble guanylate cyclase stimulators. In the multicenter study by Ghofrani et al that was published in the New England Journal of Medicine in July 2013,
261 patients were randomized prospectively to receive riociguat versus placebo. Riociguat was shown to significantly improve exercise capacity and pulmonary vascular resistance. Side effects include systemic hypotension. Prior smaller studies have also shown some benefits to medical therapy in CTEPH (inoperable or with post-operative PH) with oral agents such as bosentan and sildanefil, inhaled iloprost and subcutaneous treprostinil. Medical therapy has also been used as a “bridge” before PTE.
Although relatively rare, CTEPH is an important cause of PH since it is potentially curable with pulmonary endarterectomy. This surgery should only be performed in very experienced, specialized centers. PTE surgery should always be the treatment of choice for CTEPH. However, medical therapy can have a role as a bridge to PTE,in patients who are not surgical candidates or in those who have persistent pulmonary hypertension despite undergoing PTE.
I would like to express my gratitude to Dr. Peter Fedullo (University California San Diego) for his review of this manuscript and providing the photographs.
Daniel Layish, MD, graduated magna cum laude from Boston University Medical School in 1990. He then completed an Internal Medicine Residency at Barnes Hospital (Washington University) in St.Louis, Missouri and a Pulmonary/Critical Care/ Sleep Medicine Fellowship at Duke University in Durham, North Carolina. Since 1997, he has been a member of the Central Florida Pulmonary Group in Orlando. He serves as Co-director of the Adult Cystic Fibrosis Program in Orlando. He may be contacted at 407-841-1100 or by visiting www.cfpulmonary.com.