119 minute read
Hyperactive CREB subpopulations increase during therapy in pediatric B-lineage acute lymphoblastic leukemia


Dino Masic,1 Kayleigh
1Wolfson Childhood Cancer Research Centre, Newcastle University Centre for Cancer, 2Haematology Department, Flow Cytometry Laboratory, Royal Victoria Infirmary, 3Newcastle University Flow Cytometry Core Facility, Newcastle University and 4Innovation, Methodology and Application Research Theme, Newcastle University, Newcastle upon Tyne, UK
Abstract
Correspondence: J. Irving julie.irving@newcastle.ac.uk https://doi.org/10.3324/haematol.2022.281177
Received: June 21, 2022.
Accepted: November 10, 2022.
Early view: November 24, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Persistence of residual disease in acute lymphoblastic leukemia (ALL) during the initial stages of chemotherapy is associated with inferior survival. To better understand clonal evolution and mechanisms of chemoresistance, we used multiparameter mass cytometry, at single-cell resolution, to functionally characterize pediatric B-ALL cells at disease presentation and those persisting during induction therapy. Analysis of ALL cells from presentation samples (n=42) showed that the most abundant phosphosignals were pCREB, pH2AX and pHH3 and we identified JAK-STAT and RAS pathway activation in five of six patients with JAK or RAS genetic aberrations. The clonal composition of ALL was heterogeneous and dynamic during treatment but all viable cell clusters showed pCREB activation. Levels of pCREB in ALL cells were increased or maintained during therapy and high dimensional analysis revealed a subpopulation of ALL cells at presentation that was positive for pCREB/pHH3/pS6 which increased during treatment in some patients, implicating this signaling node in conferring a survival advantage to multi-agent induction therapy. The small molecule CREB inhibitor, 666-15, was shown to reduce CREB transcriptional activity and induce apoptosis in ALL patient-derived xenograft cells of varying cytogenetic subtypes in vitro, both in the presence and absence of stromal support. Together, these data suggest that the cAMP signaling pathway may provide an opportunity for minimal residual disease-directed therapy for many patients at high risk of relapse.
Introduction
Childhood acute lymphoblastic leukemia (ALL) is the most common childhood malignancy and while outcome has improved dramatically over the last 50 years, relapsed ALL remains a major cause of cancer death in children.1,2 There are a number of well-recognized prognostic biomarkers at presentation of ALL including age, peripheral white blood cell count, morphology and key cytogenetic abnormalities.3 However, the most powerful, independent prognostic factor is the response of the leukemia to initial chemotherapy.4 Thus, levels of persisting leukemia cells assessed at 8, 15 or 28 days after the start of induction chemotherapy are highly prognostic. These are monitored initially by morphology, and subsequently by more sensitive methods to evaluate submicroscopic disease, known as minimal residual disease (MRD). Incorporation of residual disease assessment into contemporary trials has enabled risk-directed therapy and has been fundamental in children receiving personalized, optimal therapy.5,6
Genetic analyses of paired ALL samples at presentation and relapse have revealed a number of recurrent pathways implicated in relapse, including RAS, JAK-STAT, cell cycle and B-cell development, as well as genes involved in epigenetic modification.7 Our own data, together with data from others, also implicate cell maturation as a resistance mechanism.8,9 Genomic analyses have revealed extensive clonal diversity and, in most cases, leukemic cells at relapse are related to a major or minor clone of cells found at presentation that have survived therapy and acquired additional mutations to give rise to the relapse.1016 This selection of mutated clones has been noted in the early stages of treatment, when their proportion relative to the total leukemic burden increases during the selective pressure of multi-agent induction therapy.14,17,18 Thus, the genotype, phenotype and therapeutic vulnerabilities of the leukemic clone persisting after induction chemo- therapy may be very different from those at presentation. Drugs targeting MRD and mechanisms of chemoresistance may avert relapse.
MRD is routinely quantified by two different methodologies: molecular analyses of antigen receptor gene rearrangements and flow cytometry of aberrant immunophenotypes.19 Flow MRD relies fundamentally on the characterization of a leukemia-associated immunophenotype (LAIP) at presentation, an antibody combination that discriminates leukemic cells from normal lymphocyte progenitors and can thus be used for ‘on treatment’ samples to discriminate and quantify ALL cells. An advantage of cytometric methods is that, as cell-based assays, they can provide information beyond that of just MRD quantitation, including the mechanism behind the evasion of chemotherapy-induced killing and the presence of therapeutic targets. In this study, we used single-cell, high-dimensional mass cytometry to functionally characterize pediatric B-ALL cells both at presentation and persisting during therapy. We demonstrated the presence of activated cAMP response element-binding protein (CREB) across a broad spectrum of cytogenetic groups at presentation and found minor subpopulations with hyperactive CREB at presentation that appeared to have a selective advantage during induction therapy. We also validated CREB as a therapeutic target in ALL cells using a specific inhibitor of CREB transcriptional function. CREB and its signaling pathway may provide an opportunity for MRD-directed therapy.
Methods
Clinical samples
Bone marrow samples from children with B-lineage ALL were accessed through the Newcastle Haematology Biobank, after appropriate consent (reference numbers 2002/111 and 07/H0906). All patients were registered on the ALL2003 or the UKALL2011 trials which used an induction regimen consisting of three or four drugs, depending on National Cancer Institute risk factors. MRD was assessed using a standardized flow cytometry method that was adapted from four to seven or eight colors.20 The clinical details of the patients included in the study are listed in Online Supplementary Tables S1 and S2 and a CONSORT diagram is shown in Online Supplementary Figure S1
Cell lines and patient-derived xenograft cells
ALL cell lines were obtained from the European Collection of Authenticated Cell Cultures (ECACC), maintained in RPMI-1640 (Sigma-Aldrich, Dorset, UK) supplemented with 10% fetal bovine serum (Gibco, Rugby, UK), and incubated at 37°C in a 5% CO2 atmosphere. Patient-derived xenograft (PDX) cells were originally created by intrafemoral injection of presentation primary bone marrow samples into
NOD SCID γ null mice, as described previously.17 Clinical details of these grafts are also included in Online Supplementary Table S1.
Western blotting
Cells were washed in phosphate-buffered saline and proteins were extracted using PhosphoSafe extraction reagent (Merck, Nottingham, UK) supplemented with protease inhibitors (Roche, Hertfordshire, UK). Western blotting was carried out using a standard methodology with antibodies for pCREB (pS133), CREB, ERK2 (Santa Cruz, Dallas, TX, USA), p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), pSTAT5 (pT694), STAT5 (Cell Signaling Technology, Danvers, MA, USA) and α-tubulin (Sigma-Aldrich, St. Louis, MO, USA) which served as a loading control. Densitometry was carried out using AIDA image analysis software (Raytest, Straubenhardt, Germany).
Pharmacodynamic assays
Externalization of annexin V (Abcam, Cambridge, UK) was assessed by flow cytometry on a FACSCalibur (BD Biosciences, New Jersey, NJ, USA), fitted with a 488 nm laser. The CREB pathway was stimulated by dosing ALL cell lines with 50 mM forskolin and 100 mM IBMX diluted in dimethylsulfoxide for 30 minutes prior to treatment with 666-15. The mRNA levels of CREB gene targets that we had previously identified in ALL cells21 were assessed by quantitative real-time polymerase chain reaction (RQ-PCR) analysis. We included primer probe sets (Invitrogen, Carlsbad, CA, USA) for CXCR4 and MKNK2, with TBP used as a housekeeping control, as described previously.21
Statistical analyses
All statistical analyses were performed using Graphpad Prism. A P value of less than 0.05 was considered statistically significant. Additional methods are provided in the Online Supplementary Material.
Results
Mass cytometry analyses of presentation B-acute lymphoblastic leukemia samples revealed prominent pCREB/ATF1 signaling and identified JAK-STAT and RAS pathway activated leukemia
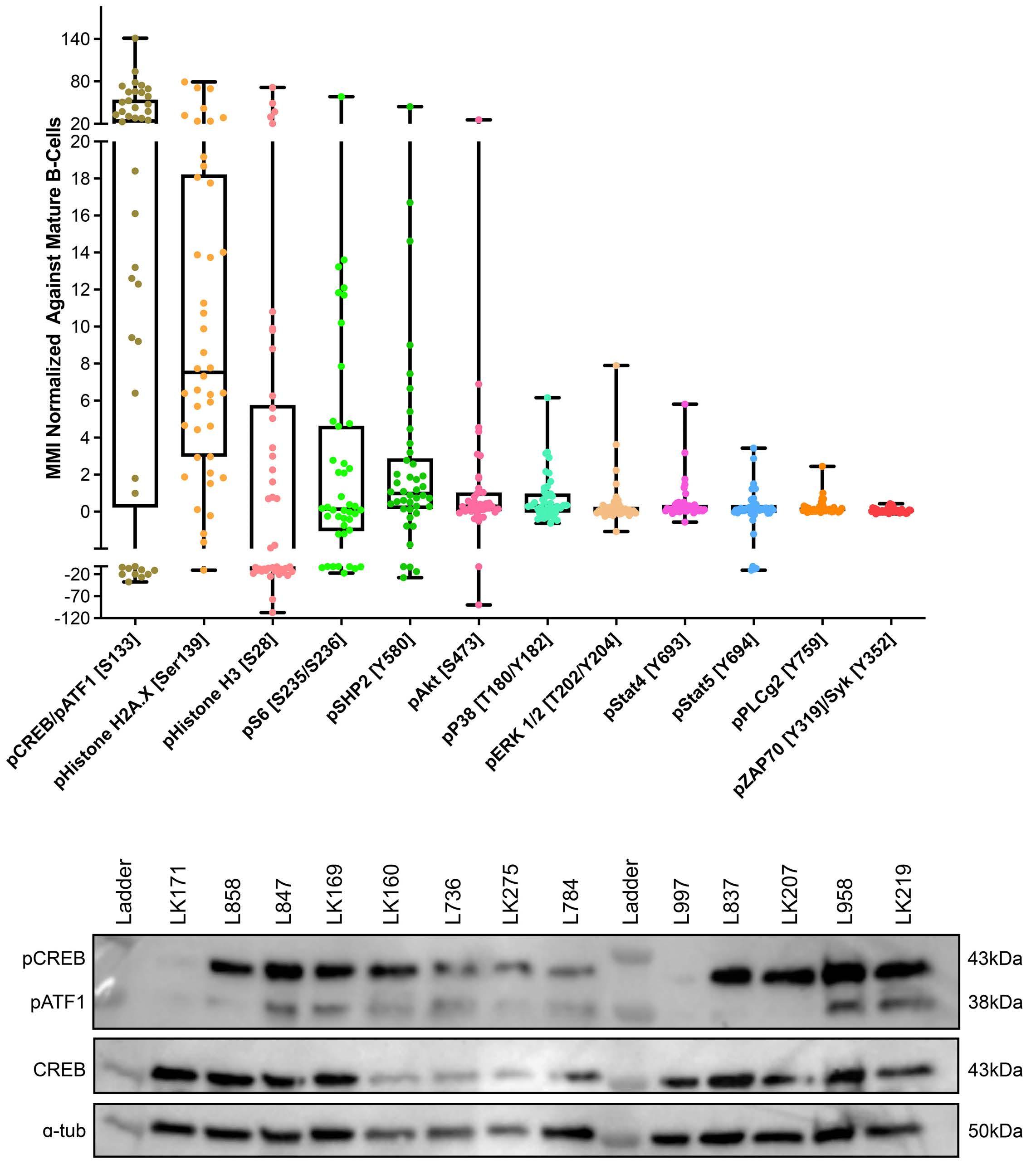
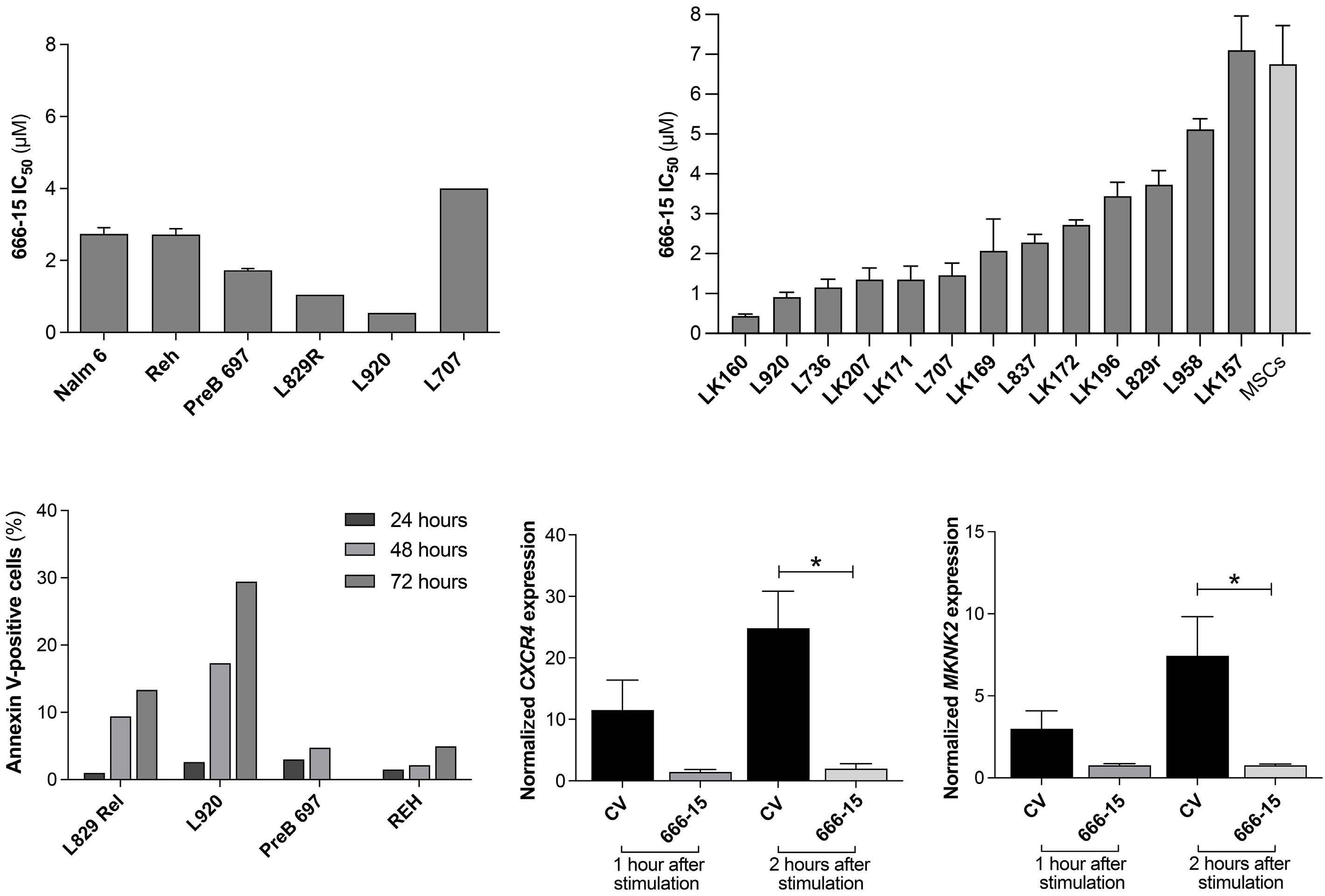
Mass cytometric analyses of B-lineage ALL at presentation (n=42) was performed; live, singlet, non-apoptotic ALL cells were gated by their specific LAIP and mature B cells were identified by the immunophenotype CD34–/CD10–/ CD22+/CD45+. The normalized mean mass intensity (MMI) of all phospho-antibodies in the ALL cells, relative to mature B cells, is shown in Figure 1A. The most prominent phospho-signals were pCREB/pATF1 [S133] (median, 23.95; range, -37.52 to 141.2), pH2AX [S139] (median, 7.53; range, -10.81 to 79.24) which is a marker of double-strand breaks, and pHH3 [Ser28] (median, -2.49; range, -107.1 to 71.5) which is activated in cells undergoing mitosis. For ALL cases at presentation for which there was sufficient stored material, we performed western blot analyses to validate our mass cytometry findings. Western blotting showed high expression of CREB in all ALL cells and confirmed the variable activation of both CREB and its close family member, ATF1 (activating transcription fac- tor 1) (Figure 1B). Western blot analyses for pSTAT5 [Y694] showed that three of seven samples were positive and these had the highest MMI values (Figure 2A, Online Supplementary Figure S2). All three positive samples were Bother ALL, one with a PAX5-JAK2 and another with an IGH-CRLF2 translocation. With regard to pERK, three of seven samples were positive by western blot analyses and two of the positive samples had the highest pERK MMI in the group. These two samples were also B-other ALL, one of which had a known KRAS mutation.
Figure 1. pCREB/pATF1 [S133] is heterogeneously expressed in acute lymphoblastic leukemia at presentation. (A) Box and whisker plot of normalized phospho-signals showing median, upper and lower quartiles and range in acute lymphoblastic leukemia (ALL) samples at presentation normalized to the signals of mature B cells, as detected by mass cytometry. (B) Western blot analysis of ALL lysates for pCREB/ATF1 [S133], CREB and α-tubulin in a representative set of presentation ALL samples that were typical of the cohort.
When the cases of ALL were grouped by cytogenetic risk, levels of pSTAT5 MMI were lower in the good-risk cytogenetic group compared to the intermediate- and poorrisk groups, with means of -2.24 versus 0.48 versus 0.36, respectively (P<0.005) (Figures 2B). There was also a trend for higher pHH3 [Ser28] and pCREB/pATF1 [S133] levels with increasing cytogenetic risk, but the differences did not achieve statistical significance (P=0.067 and P=0.29, respectively) (Figure 2C, D).
Residual disease cells show maturation and increased phospho-signaling in pHH3 and pCREB MMI for antigens were again normalized to mature B cells within each sample and values compared between presentation samples (n=42) and MRD samples (n=15 in total; n=5 at day 8, n=8 at day 28 and n=2 at later time points), identified by sequential gating (Table 1A, Online Supple- mentary Figure S3). MRD levels determined by flow or mass cytometry were highly concordant in this cohort and a pilot cohort (Online Supplementary Figure S4). Compared to presentation ALL cells, MRD showed a significant increase in the expression of the cell surface antigens, CD45 (P<0.01) and CD22 (P<0.05), and a trend to an increase in CD19 (P=0.078), consistent with maturation as previously reported.8 There was also a highly significant increase in the level of pHH3 (P<0.001), from a MMI of3.14 in presentation ALL cells to 62.48 in MRD cells. pCREB levels increased from 26.70 to 69.85 (P<0.01). There were also more modest increases in pp38, pSTAT5 and pZAP70 and a decrease in pSHP2 (P<0.05). In paired presentation and MRD samples (n=10), the same trends in antigen expression were observed but only the increase in pHH3 remained statistically significant (P <0.05) (Table 1B). A decrease of CD10 levels in MRD cells also gained signifi- cance (P<0.05) There was no difference in MMI values in mature B cells in samples taken at presentation or while on treatment, validating their reliability as an internal control (Online Supplementary Table S3).
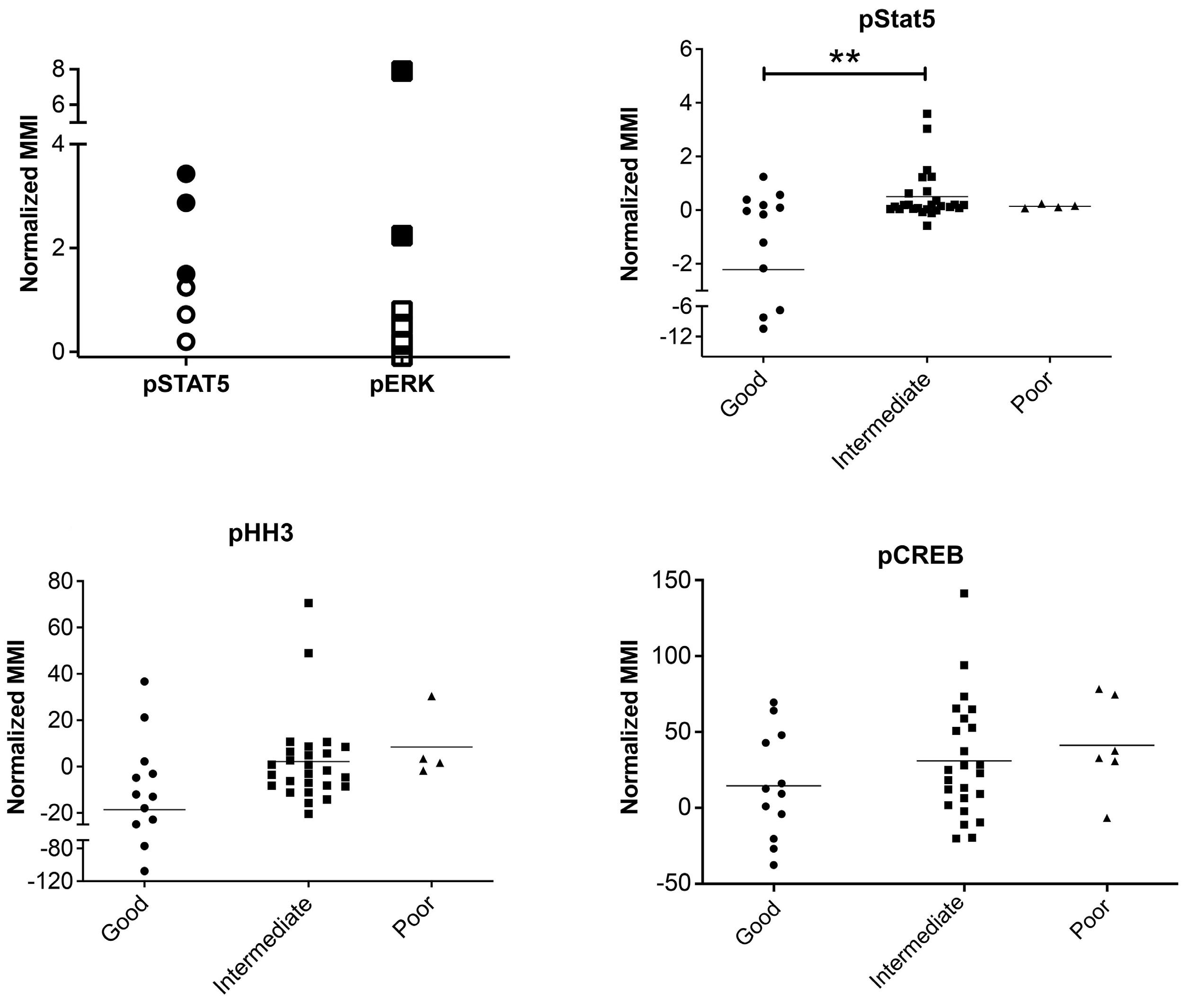
Figure 2. Phospho-signals determined by mass cytometry identify JAK-STAT and RAS pathway activation and a correlation of pSTAT5 activation with cytogenetic risk. (A) Normalized mean mass intensity (MMI) for pSTAT5 and pERK in presentation samples of acute lymphoblastic leukemia (ALL) with pathway activation assessed by both mass cytometry and western blot analysis. Solid black shapes denote ALL samples that are positive by western blot analysis. (B-D) Normalized MMI in good-, intermediate- and poor-risk cytogenetic groups for pSTAT5 (B), pHH3 (C) and pCREB (D). **P<0.01.
The clonal composition of acute lymphoblastic leukemia is dynamic during therapy but all clusters have activated pCREB and high pCREB subpopulations in minimal residual disease are often enriched during induction therapy
To investigate the clonal composition of ALL cells during therapy, we used FlowSOM as part of the R Studio CyTOF workflow package to perform cluster analyses of ALL cells in paired presentation and MRD samples using only phospho-signals and identified 20 unique ALL cell clusters. Most ALL were complex and heterogeneous, showing multiple different clusters at both presentation and in MRD and clonal composition was dynamic during therapy (Figure 3). Despite the varying cytogenetics of the cohort, eight clusters made up more than 94% of the total cluster composition (Figure 4A). All clusters showed expression of pCREB, except clusters 6 and 7 which had very low ex- pression of pHH3 and high levels of pH2AX and were, therefore, likely to be damaged cells that had not yet expressed the apoptotic markers that would have ensured their being gated out from the analyses. One cluster, cluster 10, was significantly more prevalent in MRD cells than in presentation, drug-naïve ALL cells (P<0.05) and was characterized by high levels of pCREB, pHH3 and pS6 (Figure 4B, C). We used the dimension reduction algorithm, Uniform Manifold Approximation and Projection (UMAP) to compare ALL cell clusters at presentation and at MRD. UMAP plots displayed one dominant island comprising the majority of generated clusters and showed an area populated only by presentation ALL at the top left of the plot, housing cluster 11, and two areas at the bottom populated predominantly by MRD (clusters 10, 15 and 16) but most areas having both (both clusters 3, 4 and 14) (Figure 5A, B). Two additional minor islands were generated: one was characterized by pSHP2 expression and comprised clusters 1, 2 and 8, the other expressed pH2AX and comprised clusters 6, 7, 9, and 17 (data not shown). Visualization of pCREB confirmed its ubiquitous ex- pression across the UMAP in both presentation and MRD ALL cells, while pHH3 was more defined and concentrated in the MRD samples (Figure 5C, D). The MRD-enriched areas housed clusters 10, 15 and 16, which are characterized by high pCREB and pHH3. In samples for which there were sufficient MRD cells, individual UMAP showed that in five of seven patients, ALL cells with the highest pCREB levels were enriched during therapy, with pCREB levels often increasing further (Figure 6).
The mean mass intensity (MMI), its range and the statistical significance of differences between values for pre-treatment (presentation) samples of acute lymphoblastic leukemia (ALL) cells and those during treatment (with minimal residual disease, MRD). (A) An unpaired, twotailed, equal variance t test was performed on presentation versus MRD ALL MMI signals. (B) A paired, two-tailed, equal variance t test was performed on presentation versus MRD ALL MMI signals. Statistically significant differences are shown in bold. *P<0.05, **P<0.01 and ***P<0.001.
To investigate the effect of drugs on pCREB activation, we treated PreB 697 cells with the half maximal inhibitory concentration (IC50) of the induction drugs, dexamethasone (67 nM) and vincristine (8.9 nM) on the CREB gene targets, CCRX4 and MKNK2 at various time-points. We documented a modest induction of CCRX4 mRNA levels at the 3-hour time-point after dexamethasone dosing (P<0.01) (Online Supplementary Figure S5).
Figure 3. The clonal composition of acute lymphoblastic leukemia cells is complex and dynamic during therapy. Stacked bar plots show the percentages of clusters within acute lymphoblastic leukemia cells at presentation and at various time-points during treatment.
CREB inhibitors are cytotoxic in acute lymphoblastic leukemia
Since cluster analyses showed pCREB in all clusters of ALL cells, and identi fi ed a signaling node involving pCREB/pHH3/pS6 that is enriched in MRD, we sought to evaluate pCREB as a therapeutic target. We therefore dosed ALL cells with varying concentrations of 666-15, a potent, selective CREB inhibitor, and assessed cell viability relative to that of cells treated with the vehicle control. ALL cell lines (n=3) and PDX cells (n=3) grown in suspension culture were assessed using a metabolic readout and primary (n=10) and PDX cells (n=3), supported on a layer of mesenchymal stromal cells, were analyzed using high throughput imaging microscopy which yielded absolute cell numbers of both ALL blasts and mesenchymal stromal cells. IC50 values for cell lines ranged from 1.7 to 2.7 mM (mean, 2.4 mM), while those for primary/PDX cells ranged from 0.45 to 7 m M (mean, 2.6 mM) (Figure 7A, B). There was no difference in IC50 values of the three PDX samples grown in both suspension and on mesenchymal stromal support (P>0.75). There was no apparent correlation between levels of pCREB as assessed by western blot analysis and IC50 values (n=8, data not shown). Treating ALL cells with IC50 values of 666-15 was associated with induction of apoptosis as determined by phosphatidylserine externalization, with this effect being particularly marked in the PDX cells (P<0.05, paired t test) (Figure 7C). Pharmacodynamic experiments showed significant inhibition of CXCR4 (n=4) (P=0.013) and MKNK2 (n=4) ( P =0.033) gene expression (Figure 7D, E), confirming on- target inhibition of CREB activity at the IC50 values for 666-15.
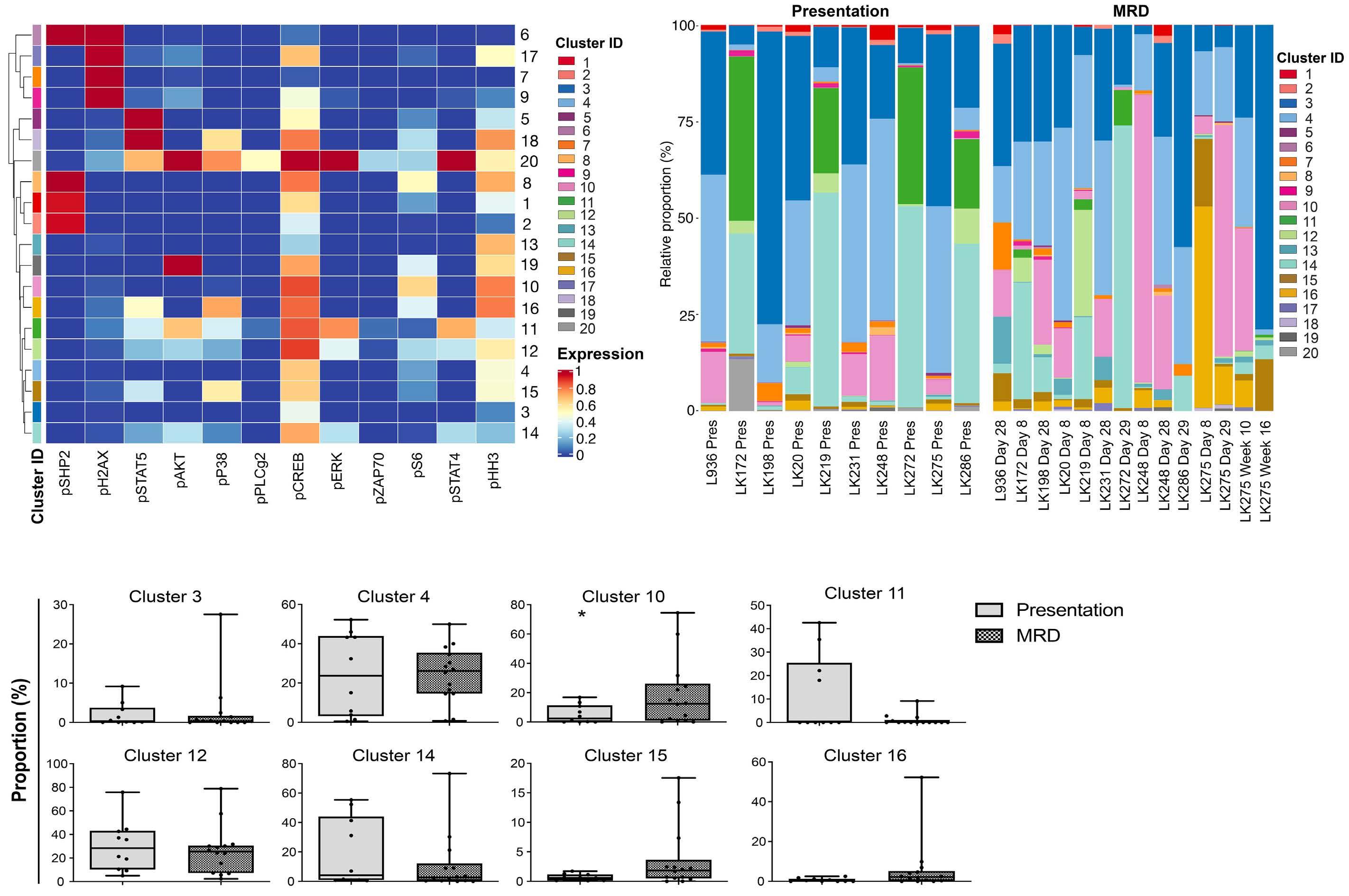
Figure 4. Cluster analysis revealed that phosphorylation of acute lymphoblastic leukemia cells at presentation (drug-naïve) and during treatment (with minimal residual disease) is complex and heterogeneous with all clusters having activated CREB and a cluster characterized by high levels of pCREB, pHH3 and pS6 having an apparent selective advantage during induction therapy. (A) Heatmap showing expression of target markers within generated clusters, with marker expression intensity displayed from low (blue) to high (red). (B) Bar chart showing whole population cluster composition within patients, separated into presentation and minimal residual disease (MRD) samples. (C) Box plots comparing the incidence of the top eight expressed clusters in the cohort of patients, comparing proportions of presentation and MRD samples of acute lymphoblastic leukemia (ALL) cells in the various clusters. *P<0.05.
Discussion
The extensive panel of antibodies used in this mass cytometric analysis adds a new diagnostic, functional dimension for ALL. Normal B cells and ALL cells can be identified prior to and during therapy. Functional parameters such as proliferation, apoptosis, pathway activation and quantification of key antigens were assessed in ALL cells and compared to those of normal B cells within the same sample. Thus in one assay, therapeutically relevant antibody targets, including CD19, CD22 and CD38 and signaling pathways, such as the JAK-STAT and the RAS pathways, can be assessed at the single-cell level. Importantly, this data-rich assay can be performed in samples with high or low leukemic burden. Thus for children with a poor response to induction or reinduction chemotherapy and at high risk of relapse, mass cytometry analysis could evaluate a range of predictive biomarkers to select the optimal targeted therapy directed at residual disease.
In our panel of presentation samples, levels of phosphorylated STAT5 were higher in the intermediate cytogenetic risk group than in the good-risk group, which is likely due to the high incidence of JAK-STAT genetic aberrations reported in Philadelphia chromosome-like and B-other sub- groups of ALL.22 The high pSTAT5 and pERK levels were confirmed by western blotting and were often attributable to genetic aberrations known to activate these pathways and may serve as predictive biomarkers for sensitivity to JAK and MEK inhibitors.17,23 Our data also confirmed the more mature immunophenotype of MRD cells compared to that of cells at presentation,8,9 which suggests that anti-CD22 and anti-CD19 therapies such as inotuzumab and chimeric antigen receptor-modified T cells may be optimal at the end of induction when expression of antigens associated with B-cell maturation may be higher. We also identified a tendency for the levels of phosphorylated histone H3 (pHH3), a typical marker of mitotic cells, to be lower in good-risk cytogenetic ALL than in intermediate- and poor-risk groups. Phosphorylation of histone H3 at either Ser28 or Ser10 is a well-recognized prognostic biomarker in many cancer types but has not been explored in ALL.24 Our data suggest that pHH3 may have a role in prognostic classification in ALL.
Figure 5. Uniform manifold approximation and projection analyses showing enrichment of clusters high in pCREB and pHH3 in minimal residual disease samples. Uniform manifold approximation and projection (UMAP) visualization of all analyzed cells shows variation between presentation and minimal residual disease (MRD) samples. (A) The distribution of presentation and MRD blasts shows a focus of MRD blasts to the south of the figure. (B) UMAP analysis showing the distribution of generated clusters in presentation and MRD blasts, with enrichment of clusters 10, 15, and 16 observed in the MRD blasts. (C) UMAP analysis showing pCREB levels in presentation and MRD blasts, with greater expression observed in MRD-enriched areas. (D) UMAP analysis showing the distribution of pHH3 in presentation and MRD blasts with a focus of pHH3 expression observed in MRD cells.
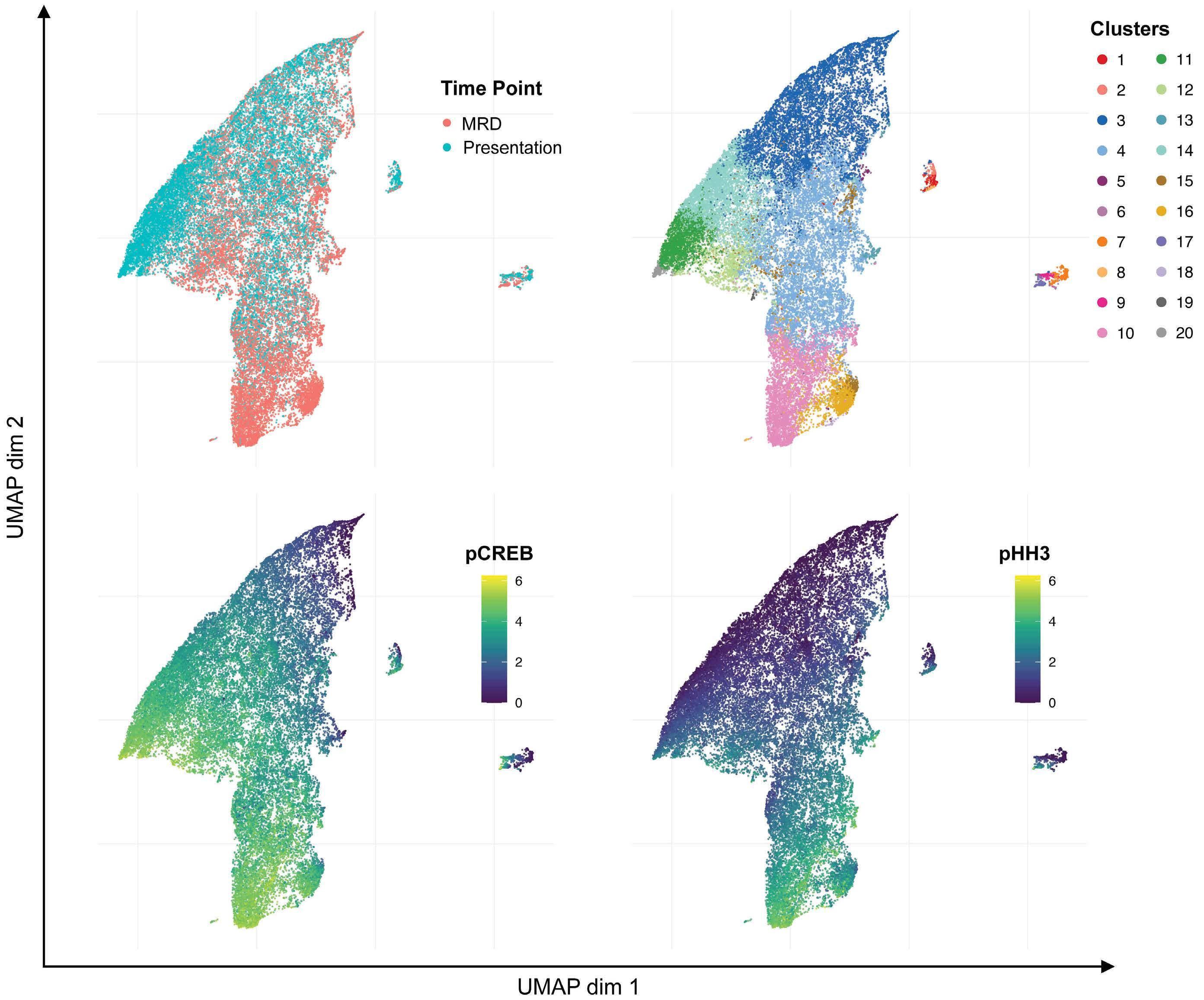
Figure 6. Uniform manifold approximation and projection analysis of individual patients show selection of high pCREB cells during induction therapy. The same uniform manifold approximation and projection (UMAP) as in Figure 5 but with visualization of pCREB levels in presentation and minimal residual disease (MRD) blasts in individual patients.
A major novel finding stemming from our data is the demonstration of high expression of activated CREB as a common feature of ALL, its increase in MRD cells and the demonstration that this increase is commonly due to hyperactive pCREB/pHH3 subpopulations increasing during induction treatment and persisting in MRD. This demonstrates a possible selective advantage of ALL clones that have high pCREB/pHH3/pS6 signaling under chemotherapeutic pressure. Further investigations will be needed to decipher whether this signaling node represents cells in G2M or cells in interphase that have a small fraction of nucleosomes phosphorylated at Ser28 of HH3, related to transcriptional activities.25 Other studies have suggested that MRD cells are in fact dormant.26 Interestingly, our CREB data are supported by a small focused study in
Figure 7. The CREB inhibitor, 666-15, is cytotoxic in acute lymphoblastic leukemia cells. (A) The half maximal inhibitory concentration (IC50) of 666-15 for acute lymphoblastic leukemia (ALL) cell lines and patient-derived xenograft (PDX) ALL cells determined using an Alamar blue assay. The mean and standard error of mean (SEM) of three independent replicate experiments are shown for the cells lines. PDX are technical triplicates. (B) IC50 of 666-15 for primary and PDX ALL cells using mesenchymal stromal cell support. (C) Histogram showing the percentages of annexin V-positive cells after exposure to the specific IC50 of 666-15 for two PDX and two ALL cell lines after 24, 48 and 72 hours of incubation, normalized to that for the vehicle control. (D, E) Histogram of CXCR4 (n=4) (D) and MKNK2 (n=4) (E) gene expression in PreB697 cells after stimulation with forskolin and IBMX and subsequent dosing with the IC50 (1.7 mM) of 666-15 or control vehicle (CV) for 1 and 2 hours. The mean and SEM are shown. *P<0.05.
CRLF2-positive ALL which also showed activation of pCREB at presentation, increased activation in MRD cells and a strong connection between pCREB and pS6.27 Our study is the first to demonstrate this finding in a range of high-risk subtypes of ALL.
CREB and ATF1, along with cAMP response element modulator (CREM), are transcription factors of the basic leucine zipper superfamily which regulate gene expression through the activation of cAMP-dependent or -independent signal transduction. They can homo- or heterodimerize to bind cAMP response elements in target gene promoters. They are phosphorylated and activated by upstream serine-threonine kinases which increase their affinity to a number of transcriptional co-activators, including CREB-binding protein (CBP), p300 and transducers of regulated CREB (TORC). Phosphorylation of CREB at Ser133 is essential for CREBmediated transcription and target gene function in cell proliferation, differentiation and survival. Overexpression and/or overactivation of CREB has been described in many types of cancer and phosphorylation of CREB at Ser133 can be catalyzed by a variety of kinases, including calcium/calmodulin-dependent (Cam) kinases that are activated by calcium fluxes, Akt or p90Rsk which are downstream of ERK, as well as protein kinase A, which is activated by cAMP. Functional analyses suggest that overexpression of CREB contributes to ALL cell proliferation and survival through transcriptional activation of gene targets involved in glycolysis and anti-apoptosis, including Bcl-2, Bcl-xL, Mcl-1 and survivin.28-30 This array of anti-apoptotic CREB targets may explain the observed resistance to multi-agent induction chemotherapy. The importance of CREB in influencing clinical response has also been demonstrated in a study of adult ALL in which high levels of CREB and pCREB were associated with a shorter median overall survival; a similar trend was observed in pediatric disease.30 Furthermore, a recent study identified expanded leukemic cell populations that, if present at the diagnosis of ALL, were associated with relapse.31 One of the features of these expanded populations was high pCREB, again consistent with this pathway contributing to a chemoresistant phenotype.
Given their deregulation in many cancer types, CREB and CREB-specific signaling pathways have been proposed as targets for therapeutic intervention in cancer and inhibitors are being developed and have begun to enter early phase clinical trials.32 Despite the universal role of CREB signaling in cells, pharmacological inhibition ap-
References
1. Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185-3193.
2. Parker C, Waters R, Leighton C, et al. Effect of mitoxantrone on pears well-tolerated, both in preclinical models and according to data emerging from the clinic. This tolerance may be due to cancer cells being differently dependent on CREB activity as compared to their normal counterparts.33 Thus, we preclinically evaluated a novel, specific small molecule CREB inhibitor in pCREB-positive ALL. 666-15 is a potent, selective inhibitor of CREB-mediated gene transcription, with an IC50 of 0.08 mM in cell-free assays. In a xenograft mouse model of breast cancer, it potently inhibited tumor growth, without overt toxicity and blood counts, blood chemistry, and tissue histology from liver, kidney and heart appeared similar to those of controls.34 Our data from cell lines and PDX ALL cells showed that these were sensitive to concentrations of 666-15 that were achievable in mice and that the CREB inhibitor induced robust apoptosis in PDX ALL cells, consistent with a cytotoxic action. Inhibition of CREB transcriptional activity was clearly demonstrated at these cytotoxic concentrations.
In summary, we have established and validated a onestop single-cell assay that can identify antigenic and signaling pathway targets in B-lineage ALL cells at both presentation of the disease and in ‘on treatment’ samples with high MRD. We show a role of hyperactive CREB, associated with an increase in subpopulations found in presentation samples, which appear preferentially selected for during induction therapy, suggesting that this pathway is involved in conferring chemoresistance to multidrug induction therapy. We propose that novel drugs affecting CREB activity or key downstream targets may be promising MRD-directed therapies for the majority of ALL patients at high risk of relapse.
Disclosures
JAEI has been awarded grant funding from Hoffmann LaRoche on an unrelated project. The other authors declare that they have no conflicts of interest to disclose.
Contributions
DM, HLB, KF, SL, GW, MC, DM, and CS performed research. CS, FWvD, HB, DM, JO-G, AF, and JAEI analyzed data. DM and JAEI wrote the manuscript. JAEI gained funding and supervised the study.
Data-sharing statement
Data are available on request.
outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet. 2010;376(9757):2009-2017.
3. Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29(5):551-565.
4. Campana D. Minimal residual disease monitoring in childhood acute lymphoblastic leukemia. Curr Opin Hematol. 2012;19(4):313-318.
5. Vora A, Goulden N, Mitchell C, et al. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standardrisk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2014;15(8):809-818.
6. Vora A, Goulden N, Wade R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14(3):199-209.
7. Irving JA. Towards an understanding of the biology and targeted treatment of paediatric relapsed acute lymphoblastic leukaemia. Br J Haematol. 2016;172(5):655-666.
8. Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112(10):3982-3988.
9. Nicholson L, Evans CA, Matheson E, et al. Quantitative proteomic analysis reveals maturation as a mechanism underlying glucocorticoid resistance in B lineage ALL and resensitization by JNK inhibition. Br J Haematol. 2015;171(4):595-605.
10. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356-361.
11. Davidsson J, Paulsson K, Lindgren D, et al. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia. 2010;24(5):924-931.
12. Dobson SM, Garcia-Prat L, Vanner RJ, et al. Relapse-fated latent diagnosis subclones in acute B lineage leukemia are drug tolerant and possess distinct metabolic programs. Cancer Discov. 2020;10(4):568-587.
13. Kuster L, Grausenburger R, Fuka G, et al. ETV6/RUNX1-positive relapses evolve from an ancestral clone and frequently acquire deletions of genes implicated in glucocorticoid signaling. Blood. 2011;117(9):2658-2667.
14. Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604.
15. van Delft FW, Horsley S, Colman S, et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood. 2011;117(23):6247-6254.
16. Yang JJ, Bhojwani D, Yang W, et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008;112(10):4178-4183.
17. Irving J, Matheson E, Minto L, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124(23):3420-3430.
18. Irving JA, Minto L, Bailey S, Hall AG. Loss of heterozygosity and somatic mutations of the glucocorticoid receptor gene are rarely found at relapse in pediatric acute lymphoblastic leukemia but may occur in a subpopulation early in the disease course. Cancer Res. 2005;65(21):9712-9718.
19. Campana D, Leung W. Clinical significance of minimal residual disease in patients with acute leukaemia undergoing haematopoietic stem cell transplantation. Br J Haematol. 2013;162(2):147-161.
20. Irving J, Jesson J, Virgo P, et al. Establishment and validation of a standard protocol for the detection of minimal residual disease in B lineage childhood acute lymphoblastic leukemia by flow cytometry in a multi-center setting. Haematologica. 2009;94(6):870-874.
21. Dixon ZA, Nicholson L, Zeppetzauer M, et al. CREBBP knockdown enhances RAS/RAF/MEK/ERK signaling in Ras pathway mutated acute lymphoblastic leukemia but does not modulate chemotherapeutic response. Haematologica. 2017;102(4):736-745.
22. Steeghs EMP, Jerchel IS, de Goffau-Nobel W, et al. JAK2 aberrations in childhood B-cell precursor acute lymphoblastic leukemia. Oncotarget. 2017;8(52):89923-89938.
23. Tasian SK, Teachey DT, Li Y, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017;129(2):177-187.
24. Hao Q, Dai C, Deng Y, et al. Pooling analysis on prognostic value of PHH3 expression in cancer patients. Cancer Manag Res. 2018;10:2279-2288.
25. Perez-Cadahia B, Drobic B, Davie JR. H3 phosphorylation: dual role in mitosis and interphase. Biochem Cell Biol. 2009;87(5):695-709.
26. Ebinger S, Ozdemir EZ, Ziegenhain C, et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30(6):849-862.
27. Sarno J, Savino AM, Buracchi C, et al. SRC/ABL inhibition disrupts CRLF2-driven signaling to induce cell death in B-cell acute lymphoblastic leukemia. Oncotarget. 2018;9(33):22872-22885.
28. Pigazzi M, Ricotti E, Germano G, Faggian D, Arico M, Basso G. cAMP response element binding protein (CREB) overexpression CREB has been described as critical for leukemia progression. Haematologica. 2007;92(10):1435-1437.
29. Shabestari RM, Safa M, Alikarami F, Banan M, Kazemi A. CREB knockdown inhibits growth and induces apoptosis in human pre-B acute lymphoblastic leukemia cells through inhibition of prosurvival signals. Biomed Pharmacother. 2017;87:274-279.
30. van der Sligte NE, Kampen KR, ter Elst A, et al. Essential role for cyclic-AMP responsive element binding protein 1 (CREB) in the survival of acute lymphoblastic leukemia. Oncotarget. 2015;6(17):14970-14981.
31. Good Z, Sarno J, Jager A, et al. Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nat Med. 2018;24(4):474-483.
32. Kimura K, Ikoma A, Shibakawa M, et al. Safety, tolerability, and preliminary efficacy of the anti-fibrotic small molecule PRI-724, a CBP/beta-catenin inhibitor, in patients with hepatitis C virusrelated cirrhosis: a single-center, open-label, dose escalation phase 1 trial. EBioMedicine. 2017;23:79-87.
33. Li BX, Gardner R, Xue C, et al. Systemic inhibition of CREB is well-tolerated in vivo. Sci Rep. 2016;6:34513.
34. Xie F, Li BX, Kassenbrock A, et al. Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J Med Chem. 2015;58(12):5075-5087.
Oncogenic TYK2 P760L kinase is effectively targeted by combinatorial TYK2, mTOR and CDK4/6 kinase blockade
Katharina Woess,1 Sabine Macho-Maschler,2 Dorette S. van Ingen Schenau,3 Miriam Butler,3 Caroline Lassnig,1,4 Daniel Valcanover,1 Andrea Poelzl,4 Katrin Meissl,1 Barbara Maurer,5 Tania Brandstoetter,5 Claus Vogl,1 Anna Koren,6 Stefan Kubicek,6 Anna Orlova,1 Richard Moriggl,1 Birgit Strobl,1 Veronika Sexl,5 Frank N. van Leeuwen,3 Roland P. Kuiper3,7 and Mathias Mueller1,4
1Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna, Vienna, Austria; 2Unit of Physiology, Pathophysiology and Experimental Endocrinology, University of Veterinary Medicine Vienna, Vienna, Austria; 3Princess Maxima Center for Pediatric Oncology, Utrecht, the Netherlands; 4University Center Biomodels Austria, University of Veterinary Medicine Vienna, Vienna, Austria; 5Institute of Pharmacology and Toxicology, University of Veterinary Medicine Vienna, Vienna, Austria; 6CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria and 7Department of Genetics, University Medical Center Utrecht, Utrecht, the Netherlands
Abstract
Correspondence: M. Mueller mathias.mueller@vetmeduni.ac.at https://doi.org/10.3324/haematol.2021.279848
Received: September 7, 2021.
Accepted: January 5, 2022.
Early view: January 13, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Tyrosine kinase 2 (TYK2) is a member of the Janus kinase/signal transducer and activator of transcription pathway, which is central in cytokine signaling. Previously, germline TYK2 mutations have been described in two patients developing de novo T-cell acute lymphoblastic leukemias (T-ALL) or precursor B-ALL. The mutations (P760L and G761V) are located within the regulatory pseudokinase domain and lead to constitutive activation of TYK2. We demonstrate the transformation capacity of TYK2 P760L in hematopoietic cell systems including primary bone marrow cells. In vivo engraftment of TYK2 P760L-expressing cell lines led to development of leukemia. A kinase inhibitor screen uncovered that oncogenic TYK2 acts synergistically with the PI3K/AKT/mTOR and CDK4/6 pathways. Accordingly, the TYK2-specific inhibitor deucravacitinib (BMS986165) reduces cell viability of TYK2 P760L-transformed cell models and ex vivo cultured TYK2 P760L-mutated patient-derived xenograft cells most efficiently when combined with mTOR or CDK4/6 inhibitors. Our study thereby pioneers novel treatment options for patients suffering from TYK2-driven acute leukemia.
Introduction
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway links extracellular cytokines with transcriptional regulation and reprogramming. As a core cancer pathway1 it connects to other cancer-driving signaling cascades such as the PI3K/AKT/mTOR or cell cycle progression pathways.2-4 Alterations in JAK/STAT signaling are associated with cancer and immune system disorders.5 Activating JAK1/2/3 mutations are frequent in various hematologic malignancies, whereas the role of the JAK family member tyrosine kinase 2 (TYK2) has only recently emerged.6-8 More evident are TYK2 mutations causative for immune system disorders and inflammatory diseases.9
TYK2 is associated with the cytokine family receptors for type I interferon (including interferon [IFN]-α and -b), interleukin (IL)-12 (including IL-23) and IL-10 (including type III IFN, IL-22 and IL-26). Under physiological conditions TYK2 activity may provoke tyrosine phosphorylation of all
STAT (STAT1-6).10 The sparse reports on aberrant TYK2 activity in cancers include TYK2 locus mutations, fusion proteins and crosstalk to oncogenic pathways.8 The first somatic activating TYK2 mutations have been described in T-cell acute lymphoblastic leukemia (T-ALL) cell lines.11 TYK2 fusion proteins have been found in various hematologic malignancies12-14 and TYK2 cross-talks to the nucleophosmin-anaplastic lymphoma kinase pathway in anaplastic large cell lymphoma.15
Next-generation sequencing is revealing an increasing number of germline gene alterations predisposing to leukemia.16 The germline TYK2 mutations (P760L, G761V) were identified in pediatric patients who developed multiple de novo ALL: the TYK2 P760L carrying patient showed two precursor B-ALLs and the TYK2 G761V patient two T-ALL. Both mutations affect the pseudokinase domain (JAK homology [JH]2 domain) and cause constitutive activation of TYK2 and of downstream STAT1/3/5.17
The first JAK inhibitors developed acted by competing ATP binding at the tyrosine kinase (JH1) domain. The high homology of the kinase domain within the JAK family or other tyrosine kinases limits specificity of such inhibitors.18 The highly specific TYK2 inhibitor deucravacitinib acts differently and stabilizes the negative regulatory pseudokinase JH2 domain.19-21 The efficacy of deucravacitinib is currently evaluated in several advanced clinical trials for the treatment of autoimmune/inflammatory diseases.20,21 Combining inhibitors is beneficial, as combinatorial treatments allow for reduced drug concentrations to avoid side effects and may overcome potential resistances.22 Notably, JAK1/2/3 inhibitors in single or combined drug treatments are already applied for hematological malignancies.23 We here characterized TYK2-activating germline mutations (P760L, G761V) that have been described in childhood leukemia patients with respect to their transformation potential in vitro and in vivo. The TYK2 P760L mutation efficiently transforms hematopoietic cells and provokes cancer upon transplantation in mice. Viability of TYK2transformed cells is efficiently reduced by the highly specific TYK2 inhibitor deucravacitinib. A comprehensive inhibitor-based screen identified pathways co-operating with oncogenic TYK2. We show synergistic action of deucravacitinib with inhibitors of the top hit pathways
PI3K/AKT/mTOR or CDK4/6 in TYK2 P760L-transformed cell lines and in the TYK2 P760L-mutated patient-derived xenograft (PDX) cells. This reveals novel treatment options for acute leukemia in patients harboring gain-of-function (GOF) TYK2.
Methods
Details on plasmids, generation and cultivation of cell lines and primary cells, cell viability assays, flow cytometry, immunoblotting and histochemistry are described in the Online Supplementary Appendix
Ethics statement
Mice were housed under specific pathogen-free conditions according to FELASA guidelines. Animal experiments were approved by the Institutional Ethics and Animal Welfare Committee of the University of Veterinary Medicine Vienna, the Austrian authority according to §§ 26ff. of Animal Experiments Act, Tierversuchsgesetz 2012: TVG 2012 (BMBWF-68.205/0112-WF/V/3b/2016, BMBWF68.205/0174-V/3b/2018) and the Animal Experimental Committee of the Radboud University Medical Center (AVD1030020209324). PDX cells were generated by the Dutch Childhood Oncology Group. Informed consent for the use of spare specimens for research was obtained from study individuals, parents or legal guardians.
Drug screen
CellTiter-Glo Luminescent Cell Viability Assays (Promega, Madison, WI, US) in 384-well plates were performed in an automated high-throughput approach (CeMM Molecular Discovery Platform - Chemical Screening). Six hundred and eighty drugs of kinase inhibitor libraries (L1600, Targetmol, Boston, MA, US and 10505, Cayman Chemicals, Ann Arbor, MI, US) were screened at 10 mM in duplicates on 2x103 Ba/F3 TYK2 P760L 1 cells/well for 3 days. Hits were selected having <30 percentage of control (POC) viability by setting the positive control to 0% (Bortezomib, 10 mM) and the negative control (0.1% dimethyl sulfoxide [DMSO]) to 100%. Two hundred and forty-six drugs from the first round were screened in triplicates with four different 10-fold dilutions on Ba/F3 TYK2 P760L 1 and parental Ba/F3 cells (cultivated with 1 ng/mL IL-3) for 3 days. For hit calling the mean difference of the area under the curve between Ba/F3 TYK2 P760L 1 and parental Ba/F3 cell POC curves was calculated.
In vivo experiments
Male and female age-matched (8-22 weeks) NOD.CgPrkdcscidIl2rgtm1Wjl/Sz (NSG) mice24 were used for xenografts. Bone marrow (BM)-derived patient cells were washed with phosphate-buffered saline (PBS) and digested with DNAse I (130 m g/mL, Roche, Basel, Switzerland) for 10 minutes (min) at room temperature. 5x105 cells were injected intrafemorally (Radboud University Medical Center, Nijmegen, the Netherlands). PDX cells in the blood were assessed bi-weekly and upon detection of human cells weekly. Mice were sacrificed once human cells reached >50% or at the humane endpoint (defined by scoring appearance, behavior, posture and mobility).
Ba/F3 and 32D cell lines were washed with PBS. 1x106 cells were injected intravenously, 5x106 cells subcutaneously. For survival studies mice were sacrificed at the humane endpoint (see above and for subcutaneous injection a tumor volume of 1,500 mm3). Tumor volume (=length*width2/2) was measured with a caliper. Organs were weighed and histologically examined. Blood parameters were measured by VetABC (scil, Viernheim, Germany). Blood was incubated with an ammoniumchloride-potassium lysis buffer for 5 min. Solid tissues were mashed through a 100 mm nylon cell strainer and red blood cell lysis was performed for spleen and liver. All isolated cells were analyzed by flow cytometry or cryopreserved.
Statistical analysis
One-way or two-way ANOVA with Tukey post hoc test and unpaired two-sided t-tests were performed on log or arcsine square root transformed data using GraphPad Prism version 7.0 for Mac (GraphPad Software, San Diego, CA, US). Mean ± standard deviation and statistical significance are shown (*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001).
Results
TYK2 P760L induces colony formation of primary bone marrow cells
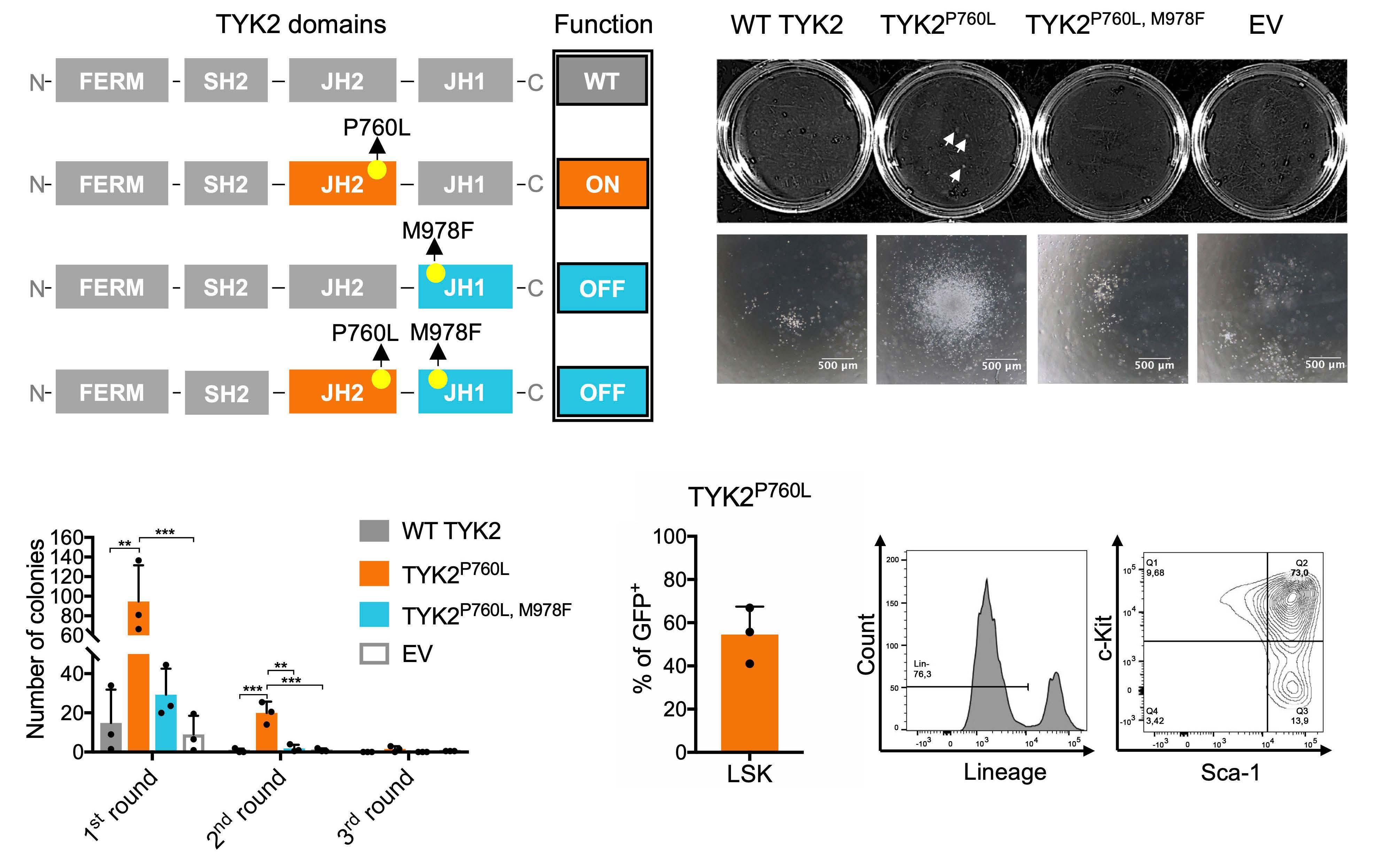
We transduced primary murine BM cells with a retrovirus encoding human TYK2 constructs (Figure 1A) to study the effect of TYK2 P670L on colony formation. In order to test for TYK2-intrinsic kinase activity we introduced M978F as a kinase-inactivation mutation11 in the expression constructs. Co-expression of green fluorescent protein (GFP) allowed for the assessment of the transduction efficiency. On average 15% of cells were retrovirally transduced (Online Supplementary Figure S1) and an equal number of GFP+ cells was seeded into growth factor-free methylcellulose. TYK2 P760L-transduced BM cells formed colonies at largest size (Figure 1B) and highest number compared to cells expressing wild-type (WT) TYK2, TYK2 P760L, M978F or empty GFP vector (EV) (Figure 1C). TYK2 P760L- expressing BM cells were capable of replating for two rounds indicative for an enhanced self-renewal capacity (Figure 1C). The majority of the GFP+ TYK2 P760L-transduced BM cells showed characteristics of hematopoietic stem/progenitor cells (Lin- Sca-1+ c-Kit+)25 (Figure 1D). These data led us to conclude that TYK2 P760L enables colony formation of primary BM cells in growth factor-free conditions.
TYK2 P760L allows factor independence of hematopoietic cell lines and leukemogenesis in vivo
In order to further assess the ability of TYK2 mutants to provide growth factor independence, we used various hematopoietic cell lines including the stem cell factor (SCF)-dependent hematopoietic progenitor cell line HPC7, the IL-3-dependent pro-B cell line Ba/F3 and the BMderived progenitor cell line 32D.
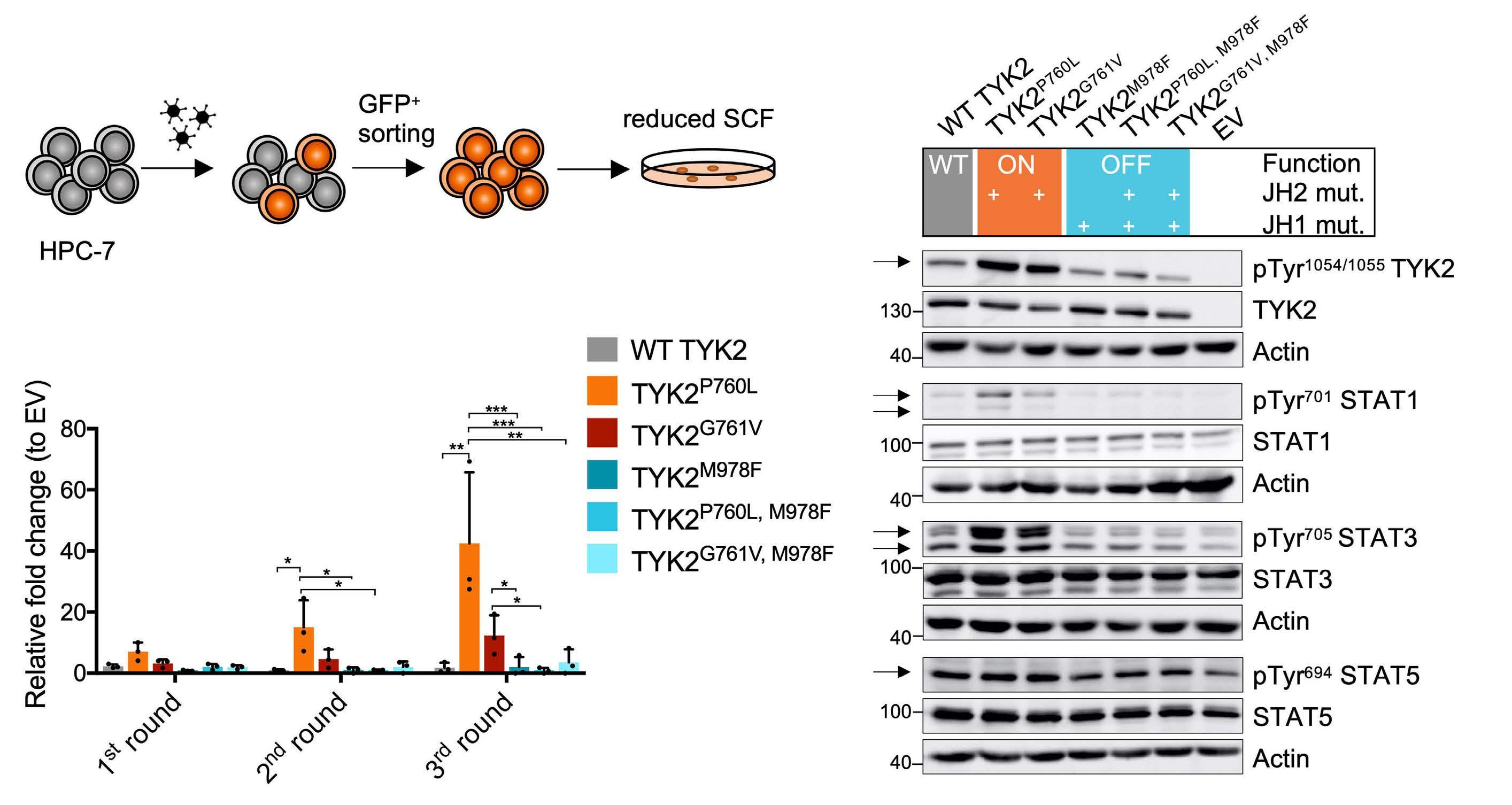
In this experimental setting we employed the TYK2 P760L and the TYK2 G761V mutant, another activating germline TYK2 mutant identified in pediatric ALL. In order to study the activation status of the TYK2-STAT axis, retrovirally transduced and GFP+-sorted HPC-7 cells (Figure 2A) were starved for 6 hours (h) as SCF may activate components of the JAK-STAT signaling pathway.26 As previously shown17 TYK2 P760L and TYK2 G761V enhanced TYK2 phosphorylation and increased phospho-STAT1 and -STAT3 levels. The effects on phospho-STAT5A/B were less consistent. The ectopic overexpression of WT TYK2 also induced activation of STAT1 and STAT3. Introduction of the TYK2 M978F mutation in the expression cassettes showed that the phosphorylation events were a direct consequence of TYK2 kinase activity (Figure 2B). Both TYK2-activating mutants induced the outgrowth of colonies of the immortalized progenitor cell line HPC-7 supplemented with a reduced SCF concentration. Colony numbers raised upon replating to a greater extent in TYK2 P760L cells (Figure 2C; Online Supplementary Figure S2A). This effect did not translate into a growth advantage in suspension culture upon titrated reduction of SCF (Online Supplementary Figure S2B).
Figure 1. TYK2 P760L-transduced primary bone marrow cells and factor-free colony formation. (A) Schematic illustration of combinations of kinase-activating germline and kinase-inactivating mutations: TYK2 consists of a four-point-one, ezrin, radixin, moesin (FERM), an atypical Src-homology 2 (SH2), a JAK homology (JH)2 and a JH1 domain. In wild-type (WT) TYK2 the JH2 pseudokinase domain inhibits the JH1 kinase domain at basal state; the JH1 domain is activated upon cytokine receptor engagement (WT, grey). P760L alters the structure of a conserved motif in the JH2 domain which is predicted to attenuate the kinase inhibitory JH2-JH1 interaction (ON, orange). M978F inactivates the JH1 domain (= OFF, blue). Combination of M978F with P760L abolishes the TYK2 kinase activity. (B) Representative colony pictures of transduced bone marrow (BM) cells, taken with 4-fold magnification objective for single colonies. Scale bars show 500 mm. White arrows indicate visible colonies. (C) Replating experiment of transduced BM cells in factor-free methylcellulose (n=3, duplicates, two-way ANOVA with log transformed data). (D) Percentage of Lin-, Sca-1+, c-Kit+ (LSK) cells among GFP+ TYK2 P760L-transduced BM cells after the first round in methylcellulose (n=3). Cells were gated on single cells, living, GFP+, Lin-, c-Kit+ and Sca-1+. Representative plots are shown. **P<0.01, ***P<0.001.
Next, we electroporated the TYK2 expression cassettes into Ba/F3 and 32D cells (Figure 3A). In both cell lines
TYK2 P760L expression enabled complete IL-3-independent growth while TYK2 G761V was only capable to transform 32D cells (Figure 3B; Online Supplementary Figure S3A). As in HPC-7 cells the TYK2 P760L-transformed cells showed constitutively activated TYK2-STAT1/3 (Online Supplementary Figure S3B). Taken together, both TYK2 germline mutations enhance STAT signaling in a TYK2 kinase-dependent manner and TYK2 P760L has a stronger transforming potential than TYK2 G761V in hematopoietic cell lines. The reasons for this remain to be elucidated. Notably, the activation state level mediated by TYK2 G761V seems to be lower than in TYK2 P760L-expressing cells as indicated by constitutive TYK2 or downstream STAT1 phosphorylation (Figure 2B and Waanders et al.17).
In order to assess the in vivo oncogenic potential, we injected Ba/F3 or 32D TYK2 P760L cells into NSG mice (Figure 3A) and used parental cells as controls. Upon systemic transplantation both cell lines harboring mutated TYK2 induced splenomegaly (Figure 3C; Online Supplementary Figure S3H) and led to high white blood cell counts (Figure 3D). Ba/F3 TYK2 P760L cells additionally caused enlarged livers (Figure 3E; Online Supplementary Figure S3H), thrombocytopenia (Figure 3F) and anemia (Figure 3G). As a consequence, the average survival of mice that had been injected with Ba/F3 TYK2 P760L cells was 14 days and with 32D TYK2 P760L cells 32 days (Figure 3H) whereas mice injected with parental Ba/F3 or 32D cells did not develop leukemia. In line we found that the subcutaneous injection of Ba/F3 or 32D TYK2 P760L cells induced local tumor growth (Figure 3I), while no tumors or signs of disease were detected upon injection of the parental cell lines. Ba/F3 TYK2 P760L cells engraftment was accompanied by hepatosplenomegaly caused by tumor cell infiltration (Online Supplementary Figure S3C to H) confirming the previous notion of oncogene carrying Ba/F3 cells as migratory cells.27
Figure 2. In vitro transformation capacity of TYK2 mutants. (A) Schematic outline of the HPC-7 cell experiment: cells were retrovirally transduced with the different TYK2 constructs (Figure 1A). HPC-7 cells were GFP+-sorted and seeded into methylcellulose containing reduced stem cell factor (SCF). (B) Western blot of transduced and GFP+-sorted HPC-7 cells starved for SCF for 6 hours (h) subjected to (phospho-) TYK2 (human) and STAT1/3/5 (murine) analysis. Wild-type (WT) TYK2 activity can be regulated (grey), JH2 mutations activate TYK2 signaling (ON, orange), JH1 mutation inhibits TYK2 signaling (OFF, blue). Actin was used as loading control. Numbers indicate molecular weight markers in kDa. (C) Replating experiment of transduced and GFP+-sorted HPC-7 cells expressing WT TYK2 (grey), TYK2 P760L (orange), TYK2 G761V (red) or kinase inactive TYK2 (blue) in methylcellulose with reduced SCF (n=3, two-way ANOVA with log transformed data, log colony number of empty vector (EV) transduced cells was subtracted from log number of TYK2-expressing colonies). *P<0.05, **P<0.01, ***P<0.001.
Figure 3. In vitro and in vivo transformation capacity of TYK2 mutants. (A) Schematic outline of the Ba/F3 and 32D cell experiment: cells were electroporated with the different TYK2 constructs (Figure 1A), IL-3 was withdrawn, and outgrowth was monitored. Transformed and parental cells were injected intravenously (i.v.) or subcutaneously (s.c.) into NOD.Cg-PrkdcscidIl2rgtm1Wjl/Sz (NSG) mice. (B) Representative growth curve of Ba/F3 (orange) and 32D cells (red) gaining IL-3 independence by expression of TYK2 P760L (n=3). (C to G) Spleen weight with representative spleen pictures, white blood cell count, liver weight, platelet count and hematocrit (HCT) of i.v. injected mice (Ba/F3 and 32D cells: n=5, Ba/F3 TYK2 P760L cells: n=4 (as humane endpoint was set for 2 mice), 32D TYK2 P760L cells: n=7, unpaired two-tailed t-test with log and arcsine square root [HCT] transformed data, from 2 experiments). (H) Kaplan Meier plot of i.v. injected mice (Ba/F3 and 32D cells [grey]: n=5, Ba/F3 TYK2 P760L cells: n=6, 32D TYK2 P760L cells: n=7, log-rank test: P-value =0.0002 between Ba/F3 TYK2 P760L cells and 32D TYK2 P760L cells, from 2 experiments). (I) Tumor growth of s.c. injected cells into right and left flank (Ba/F3 TYK2 P760L cells: n=5 [individual tumors, one site without tumor formation], 32D TYK2 P760L cells: n=6 [individual tumors]) and individual tumor weight (Ba/F3 TYK2 P760L cells: n=8 [2 sites without tumor formation], 32D TYK2 P760L cells: n=10, from 2 experiments). **P<0.01, ***P<0.001, ****P<0.0001.
Figure 4. Viability of TYK2 P760L-transformed hematopoietic cell lines upon TYK2 inhibition and effect on signaling to STAT. (A and B) Dose response curves and half maximal inhibitory concentration (IC50) values of parental (black) and transformed Ba/F3 and 32D cells (shades of red) treated with deucravacitinib (TYK2inib) for 72 hours (h) supplemented with (dashed line/white filling) and without IL-3 (n≥3, in duplicates or triplicates, not all IC50 could be determined, one-way ANOVA with log transformed data). (C) Analysis of early apoptosis (Annexin V+ and 7-AAD-) and late apoptosis (Annexin V+ and 7-AAD+) of parental and transformed Ba/F3 cells treated with 1 mM deucravacitinib for 24 h, 48 h and 72 h (n=3 in duplicates). (D) Western blot of TYK2 P760L-transformed Ba/F3 and 32D cells and parental cells (with IL-3) subjected to (phospho-) TYK2-STAT1/3 analysis. Cells were treated with 1 mM deucravacitinib (TYK2i) for 6 h. Actin was used as loading control. Numbers indicate molecular weight markers in kDa. ****P<0.0001.
Collectively, these data show that TYK2 P760L-transformed murine cells cause cancer in systemic and local transplantation setups.
Pharmacological TYK2 inhibition abrogates oncogenic TYK2 P760L-driven signaling
We next investigated how TYK2 P760L-transformed cells react to JAK inhibitors (JAKinib). Deucravacitinib (BMS986165) is a highly selective and potent allosteric TYK2inib with excellent pharmacokinetic properties across cell types and species.20,21,28,29 Drug testing was performed on Ba/F3 and 32D TYK2 P760L cells from three outgrowth experiments (1-3; Online Supplementary Figure S3A) and on parental cells by assessing the metabolic activity. All transformed cell lines were highly sensitive to deucravacitinib with a half maximal inhibitory concentration (IC50) ranging from 10 to 90 nM (Figure 4A and B) which is around 100 times lower than IC50 values of control cell lines. Culture of Ba/F3 and 32D TYK2 P760L cells in the presence of IL-3 reversed the effect of TYK2 inhibition, indicating that factor-independent growth was governed by mutated TYK2 (Figure 4A and B). Treatment of Ba/F3 TYK2 P760L cells with deucravacitinib induced apoptosis (Figure 4C; Online Supplementary Figure 4A) and G0 cell cycle arrest (Online Supplementary Figure 4B). The efficacy of deucravacitinib was confirmed by western blotting that showed the decreased phosphorylation state of TYK2 and downstream STAT1/3 (Figure 4D).
In order to demonstrate the specificity of TYK2 inhibition in oncogenic transformed Ba/F3 cells and to exclude potential off-target effects of deucravacitinib, we tested three additional Ba/F3 cell lines driven by known oncogenes, i.e., TEL-JAK2, STAT5B N642H and BCR-ABL1 p210. In these settings IC50 values were comparable to parental Ba/F3 cells cultured with IL-3 (Online Supplementary Figure S5A). These experiments prove that deucravacitinib is selective for TYK2 and specifically interferes with TYK2dependent transformation. Further support for the unique function of TYK2 stems from experiments with the JAK1/3 inhibitor tofacitinib,30 the JAK1/2 inhibitor ruxolitinib31 and the JAK1 inhibitor filgotinib.32 None of these JAKinibwhen employed at specific concentrations - significantly decreased the metabolic activity of TYK2 P760L-transformed cells (except Ba/F3 TYK2 P760L 1 with ruxolitinib) compared to parental Ba/F3 and 32D cells (Online Supplementary Figure S5B to G).
In summary we confirmed that only the TYK2-selective drug deucravacitinib efficiently reduces the cell viability of TYK2 P760L-transformed cells. This underscores the key role of TYK2 in maintaining the transformed state and excludes a major contribution of other JAK family members.
TYK2 P760L signaling co-operates with the PI3K/AKT/mTOR and CDK4/6 pathways
In order to understand whether and how oncogenic TYK2 P760L co-operates with other signaling pathways we designed a drug screen with commercial libraries of 680 approved or investigational kinase inhibitors (Figure 5A).
A first screen in Ba/F3 TYK2 P760L cells identified 246 drugs that reduce metabolically active cells to less than 30% compared to the control. In a second round, we included the Ba/F3 parental cell line as control and counter screen and looked for drugs that specifically block the TYK2 P760L-expressing cells. This narrowed the output down to 34 drugs, of which several target the PI3K/AKT/mTOR (8/34 drugs) and CDK4/6 (3/34 drugs) pathways (Figure 5A; Online Supplementary Table S4). Involvement of these pathways is also supported by increased phosphorylation of AKT in both TYK2 P760L-transformed cell lines and increased levels of CDK6 in transformed 32D cells. No changes in the mTOR target 4EBP1 were observed (Figure 5B).
The compound olverembatinib (GZD824) scored as top hit of the screen (Online Supplementary Table S4). Olverembatinib was initially described as inhibitor of the BCR-ABL1 fusion kinase but also interferes with PI3K/AKT and SRC kinase signaling.33 In order to validate our screening results, we determined dose response curves with inhibitors for the PI3K/AKT/mTOR or CDK4/6 pathway and used everolimus, vistusertib, olverembatinib, LY294002, abemaciclib and palbociclib (Figure 5A). We included the TYK2 P760Ltransformed Ba/F3 and 32D cells, and BCR-ABL1-, TELJAK2- and STAT5B N642H-transformed Ba/F3 cells as controls. TYK2 P760L-transformed Ba/F3 and 32D cells were more sensitive to all tested drugs compared to controls (Figure 5C to E; Online Supplementary Figures S6A to C and S7A to L). mTOR inhibitors everolimus and vistusertib were the most efficacious drugs with highest difference between transformed and parental Ba/F3 cell lines (Figure 5C and Online Supplementary Figure S6A). At the effective concentrations determined in the dose-response curves all tested drugs prevented IL-3-independent growth (Online Supplementary Figure S8A and B).
Cancer therapy regimens rely on drug combinatorial treatments.22 We thus combined the TYK2 inhibitor (deucravacitinib) with all tested co-operating inhibitors and calculated the synergy score by the zero interaction potency (ZIP) method.34,35 For Ba/F3 TYK2 P760L cells 11/18 and for 32D TYK2 P760L cells 16/18 combinations reached a synergy score higher than five indicating additive effects. Synergy scores ≥10 were found in Ba/F3 TYK2 P760L for the combination with PI3K/AKT/mTOR inhibitors (everolimus, LY294002 and olverembatinib), in 32D TYK2 P760L cells for the combination with the PI3K/AKT inhibitor LY294002 and the CDK4/6 inhibitor abemaciclib (Figure 5F; Online Supplementary Figure S8C to H)
Figure 5. Drug screen of TYK2 P760L-expressing cells and cross-talk with other kinase-dependent pathways. (A) Schematic outline of drug screen: Ba/F3 TYK2 P760L cells of the first outgrowth experiment were treated with 680 kinase inhibitors in a high throughput screen. In the second round 246 drugs identified (<30% viability of control) were used for comparison of Ba/F3 TYK2 P760L and parental cells. Among the top hits were mainly drugs targeting the mTOR, PI3K/AKT and CDK4/6 pathway. These targets were validated with the indicated drugs. (B) Western blot of TYK2 P760L-transformed Ba/F3 and 32D cells and parental cells (with IL-3) subjected to (phospho-) AKT, 4EBP1 and CDK6 analysis. Actin was used as loading control. Numbers indicate molecular weight markers in kDa. (C to E) Validation of screen result with dose-response curves and half maximal inhibitory concentration (IC50) values of parental (black dashed line/white filling, with IL-3) and transformed Ba/F3 cells (shades of red) treated for 72 hours (n≥3, in duplicates, not all IC50 could be determined, one-way ANOVA with log transformed data). (F) Synergy scores (n≥3, in duplicates) calculated with the zero interaction potency (ZIP) model. Light grey shows additivity, dark grey synergy. *P<0.05, ***P<0.001, ****P<0.0001.
Taken together, oncogenic TYK2 and co-operating pathways were successfully blocked by combinatorial treatment in murine cellular models.
Combined treatment with TYK2 and mTOR or CDK4/6 inhibitors decreases viability of TYK2 P760L xenograft cells
In order to translate these results in a patient-related setting, we analyzed the combinatorial treatments with deucravacitinib in human PDX cells harboring TYK2 P760L (Online Supplementary Figure S9A) co-cultured with human hTERT mesenchymal stem cells. The patient carrying the TYK2 P760L mutation first developed a leukemia classified as a chromosomal rearranged pre-B-ALL (leukemia 1) and subsequently a hyperdiploid common B-ALL (leukemia 2).17 PDX cells were generated from both leukemias and were co-treated with increasing concentrations of deucravacitinib and inhibitors of the identified co-operating pathways. Adding deucravacitinib to mTOR and CDK4/6 inhibitors resulted in a left-shift of the dose response curves (except for palbociclib in PDX cells leukemia 1) (Figure 6A to D; Online Supplementary Figure S9B to E). For both leukemias the synergy score was higher than 5 for the combination with abemaciclib (Figure 6C and D) and for the leukemia 2 the combination with vistusertib and palbociclib (Online Supplementary Figure S9C and E). The most synergistic area scores, which indicate the peak of the synergy matrix, were ≥5 for both leukemias with all treatments except leukemia 1 with palbociclib and ≥10 for both leukemias with abemaciclib, leukemia 1 with everolimus and leukemia 2 with vistusertib (Figure 6A to D; Online Supplementary Figure S9B to E). Combinations with PI3K/AKT inhibitors (olverembatinib and LY294002) failed to display synergism (data not shown).
These data show that the TYK2 inhibitor improves the efficacy of mTOR and CDK4/6 inhibitors in patient cells.
Discussion
We investigated the TYK2 P760L germline GOF mutation that was found in childhood B-ALL17 with regard to its oncogenic and druggable properties. Using in vitro and in vivo models we demonstrate that constitutively active human TYK2 P760L conferred growth/proliferation advantages on primary and immortalized hematopoietic cells. TYK2 P760L-harboring murine cells led to leukemic disease upon transplantation. A selective TYK2inib, but not JAKinib targeting JAK1-3, was highly efficacious in decreasing the metabolic activity of TYK2 P760L-transformed cells. A kinase inhibitor screen identified pathways co-operating with oncogenic TYK2 and established combinatorial TYK2 inhibition with mTOR or CDK4/6 pathway blockade as therapeutic option to eradicate TYK2-driven leukemia. We established the oncogenic potential of the TYK2 P760L GOF mutation in different cell systems ranging from primary BM cells to hematopoietic cell lines of various differentiation stages as exemplified by enhanced colony formation or growth factor-independent proliferation in vitro and migratory properties in vivo. Thus, the germline TYK2 mutation qualifies for inclusion in the expanding list of cancer driver gene mutations.36 This mutation is predicted to change the structure of the conserved DPG motif of JAK pseudokinases and thereby attenuates the inhibitory function.17 The DPG motif replaces the DFG motif of kinases and is involved in several non-covalent interactions within the JH2 domain.37 DPG mutations of other JAK listed in cancer databases are not further characterized. An activating JAK3 V674A mutation (adjacent to the DPG motif) found in T-ALL patients has been shown to cause leukemia in mice.38 Under the conditions analyzed, constitutively active TYK2 did not suffice to immortalize primary cells or allow for factor-independent growth of all cell lines tested. This is in line with the properties of germline-transmitted leukemia-predisposing mutations that frequently block the differentiation of lymphoid cells but need co-operating factors that enhance indefinite survival for malignant transformation.39 It is also in line with the reported weaker malignant transformation capacity of a given activating mutation of TYK2 compared to other JAK.40,41
As previously shown TYK2 P760L expression leads to constitutive STAT1/3 signaling which is described to shape the oncogenic transcriptome.8,17 We provide strong evidence that these signals stem directly from TYK2 activity as introduction of a kinase-inactivating mutation reverts STAT activation and cellular transformation. In addition, our studies with JAKinib revealed that only the TYK2-specific inhibitor deucravacitinib and not inhibitors with higher sel- ectivity for JAK1-3 showed efficacy in cell viability assays with TYK2 P760L-transformed cells.
D) Cell viability curves of hTERT mesenchymal stem cells co-cultured patient-derived xenograft cells of leukemia 1 and leukemia 2 treated with different concentrations of the indicated drug and different concentrations of deucravacitinib (shades of blue) for 72 hours. A synergy map is shown for each drug combination. Synergy scores and most synergistic area score were calculated with the zero interaction potency (ZIP) model (n=1). Red shows synergism and green antagonism.
Figure 6.
Deucravacitinib belongs to a novel class of JAKinib as it does not block the enzymatic activity in the JH1 domain but rather stabilizes the regulatory pseudokinase domain JH2.19-21 The inhibitor was shown to bind also the JH2 domain of JAK1, albeit with less affinity and selectivity.20,21
Importantly, the biochemical data translate in a weak activity against JAK1-dependent signaling.20,21,28,29 The efficiency of JH1-targeting TYK2 inhibitors in leukemia treatment has been shown for T-ALL xenografts.42 We are the first to prove the successful application of a JH2-targeting drug on a hyperactive TYK2 carrying the mutation in the JH2 domain. This might be important for future development of JAKinib.
Kinases represent one of the largest groups of druggable targets as they drive key signaling pathways and aberrant kinase activity leads or contributes to cancer onset and progression.43 In order to identify co-operation of oncogenic TYK2 with other kinase-driven pathways, we have screened inhibitor libraries using TYK2 P760L-transformed cells and identified the most potent hits in the PI3K/AKT/mTOR and CDK4/6 signaling axes. TYK2-dependent activation of PI3K and a crosstalk between JAK/STAT and mTOR signaling is well established.3,44,45 Aberrant TYK2-PI3K activity has been also reported for prostate cancer46 and T-ALL.11 The PI3K/AKT/mTOR pathway is frequently activated in ALL and specific inhibitors alone or in combination with JAKinib have been successfully applied in Ph-like ALL.47 At the molecular level CDK4/6 is reported to connect cell cycle progression with cell growth via mTOR activation, which may explain the efficacy of combined blocking of CDK4/6 and of PI3K/AKT/mTOR in solid tumors.48 A direct connection between CDK4/6 and TYK2 has not been described. However, STAT3, downstream of TYK2 P760L, is known to activate CDK4/6 during the cell cycle progression.49 CDK6 was shown to interact with STAT3 in tumorigenesis50 and CDK6 hyperactivation has been reported in ALL.51 Importantly, our combinatorial treatment of the TYK2 inhibitor with mTOR or CDK4/6 inhibitors was successfully applied in the TYK2 GOF PDX cells. Ex vivo assays tend to underestimate the effects of cell cycle drugs as even stromal co-cultures do not fully
References
1. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339(6127):1546-1558.
2. Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173(2):321-337.
3. Saleiro D, Platanias LC. Intersection of mTOR and STAT signaling in immunity. Trends Immunol. 2015;36(1):21-29.
4. Steelman LS, Pohnert SC, Shelton JG, et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression support proliferation of ALL xenografts.52 In this context it is important to note that CDK6 also exhibits kinase- and cell cycle-independent functions in tumorigenesis.53,54
In summary, we provide proof of TYK2 acting as an oncogene in hematologic malignancies by firmly establishing the oncogenic potential of TYK2 germline mutations, revealing that TYK2 P760L is involved in leukemia progression. Moreover, our results indicate that combinatorial kinase inhibition in acute leukemia could be a valid strategy to combat hyperactivated TYK2-mutated leukemia.
Disclosures
No conficts of interest to disclose.
Contributions
KW performed and analyzed most experiments; SM-M, BS and MM supervised the project; SM-M, MB, KM, BM, TB and AO provided technical support; AK and SK performed and analyzed the kinase inhibitor screen; SM-M, DSvIS, CL and AP helped with in vivo mouse studies; CV helped with statistical analysis; DV performed some experiments; RM and VS provided crucial material and reagents; SM-M, RM, BS, VS, FNvL, RPK and MM were involved in study design and provided crucial scientific input; KW and MM wrote the manuscript with input from all authors. All authors approved the manuscript.
Acknowledgments
The authors thank Marion Bokor for histological sample preparations and H&E staining, Philipp Jodl for technical support, the mouse facility personnel for their help with in vivo experiments, Thomas Kolbe for provision of some NSG mice and Michael Dworzak for provision of a cell line.
Funding
This work was supported by the Austrian Science Fund (FWF) funded DK W1212 PhD program “Inflammation and Immunity” and the Special Research Program SFB F6101 and F6106 (BS and MM), F6105 (RM) and F6107 (VS).
Data-sharing statement
All novel reagents mentioned are available upon justified request.
and leukemogenesis. Leukemia. 2004;18(2):189-218.
5. O'Shea JJ, Schwartz DM, Villarino AV, et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311-328.
6. Hammaren HM, Virtanen AT, Raivola J, et al. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine. 2019;118:48-63.
7. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32(21):2601-2613.
8. Wöss K, Simonovic N, Strobl B, et al. TYK2: an upstream kinase of STATs in cancer. Cancers (Basel). 2019;11(11):1728.
9. Pellenz FM, Dieter C, Lemos NE, et al. Association of TYK2 polymorphisms with autoimmune diseases: A comprehensive and updated systematic review with meta-analysis. Genet Mol Biol. 2021;44(2):e20200425.
10. Strobl B, Stoiber D, Sexl V, et al. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci (Landmark Ed). 2011;16:3214-3232.
11. Sanda T, Tyner JW, Gutierrez A, et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013;3(5):564-577.
12. Velusamy T, Kiel MJ, Sahasrabuddhe AA, et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood. 2014;124(25):3768-3771.
13. Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27(4):516-532.
14. Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinaseactivating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005-1015.
15. Prutsch N, Gurnhofer E, Suske T, et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia. 2019;33(3):696-709.
16. Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol. 2019;16(4):227-240.
17. Waanders E, Scheijen B, Jongmans MC, et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia. 2017;31(4):821-828.
18. Bryan MC, Rajapaksa NS. Kinase inhibitors for the treatment of immunological disorders: recent advances. J Med Chem. 2018;61(20):9030-9058.
19. Tokarski JS, Zupa-Fernandez A, Tredup JA, et al. Tyrosine kinase 2-mediated signal transduction in T lymphocytes is blocked by pharmacological stabilization of its pseudokinase domain. J Biol Chem. 2015;290(17):11061-11074.
20. Burke JR, Cheng L, Gillooly KM, et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med. 2019;11(502):eaaw1736
21. Wrobleski ST, Moslin R, Lin S, et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J Med Chem. 2019;62(20):8973-8995.
22. Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30(7):679-692.
23. Klein K, Stoiber D, Sexl V, et al. Untwining anti-tumor and immunosuppressive effects of JAK inhibitors-A strategy for hematological malignancies? Cancers (Basel). 2021;13(11):2611.
24. Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174(10):6477-6489.
25. Cheng H, Zheng Z, Cheng T. New paradigms on hematopoietic stem cell differentiation. Protein Cell. 2020;11(1):34-44.
26. Linnekin D, Mou S, Deberry CS, et al. Stem cell factor, the JAKSTAT pathway and signal transduction. Leuk Lymphoma. 1997;27(5-6):439-444.
27. Funakoshi-Tago M, Tago K, Sumi K, et al. The acute lymphoblastic leukemia-associated JAK2 L611S mutant induces tumorigenesis in nude mice. J Biol Chem. 2009;284(19):12680-12690.
28. Chimalakonda A, Burke J, Cheng L, et al. Selectivity profile of the tyrosine kinase 2 inhibitor deucravacitinib compared with janus kinase 1/2/3 inhibitors. Dermatol Ther (Heidelb). 2021;11(5):1763-1776.
29. Catlett IM, Hu Y, Gao L, et al. Molecular and clinical effects of selective TYK2 inhibition with deucravacitinib in psoriasis. J Allergy Clin Immunol. 2021;S0091-6749(21)01690-0.
30. Flanagan ME, Blumenkopf TA, Brissette WH, et al. Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem. 2010;53(24):8468-8484.
31. Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109-3117.
32. Van Rompaey L, Galien R, van der Aar EM, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191(7):3568-3577.
33. Ye W, Jiang Z, Lu X, et al. GZD824 suppresses the growth of human B cell precursor acute lymphoblastic leukemia cells by inhibiting the SRC kinase and PI3K/AKT pathways. Oncotarget. 2017;8(50):87002-87015.
34. Yadav B, Wennerberg K, Aittokallio T, et al. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput Struct Biotechnol J. 2015;13:504-513.
35. Ianevski A, Giri AK, Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020;48(W1):W488-W493.
36. Martinez-Jimenez F, Muinos F, Sentis I, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020;20(10):555-572.
37. Min X, Ungureanu D, Maxwell S, et al. Structural and functional characterization of the JH2 pseudokinase domain of JAK family tyrosine kinase 2 (TYK2). J Biol Chem. 2015;290(45):27261-27270.
38. Degryse S, de Bock CE, Cox L, et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood. 2014;124(20):3092-3100.
39. Tijchon E, Havinga J, van Leeuwen FN, Scheijen B. B-lineage transcription factors and cooperating gene lesions required for leukemia development. Leukemia. 2013;27(3):541-552.
40. Staerk J, Kallin A, Demoulin JB, et al. JAK1 and Tyk2 activation by the homologous polycythemia vera JAK2 V617F mutation: cross-talk with IGF1 receptor. J Biol Chem. 2005;280(51):41893-41899.
41. Shide K, Shimoda K, Kamezaki K, et al. Tyk2 mutation homologous to V617F Jak2 is not found in essential thrombocythaemia, although it induces constitutive signaling and growth factor independence. Leuk Res. 2007;31(8):1077-1084.
42. Akahane K, Li Z, Etchin J, et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br J Haematol. 2017;177(2):271-282.
43. Klaeger S, Heinzlmeir S, Wilhelm M, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367):eaan4368.
44. Rani MR, Leaman DW, Han Y, et al. Catalytically active TYK2 is essential for interferon-beta-mediated phosphorylation of STAT3 and interferon-alpha receptor-1 (IFNAR-1) but not for activation of phosphoinositol 3-kinase. J Biol Chem. 1999;274(45):32507-32511.
45. Kusch A, Tkachuk S, Haller H, et al. Urokinase stimulates human vascular smooth muscle cell migration via a phosphatidylinositol 3-kinase-Tyk2 interaction. J Biol Chem. 2000;275(50):39466-39473.
46. Ide H, Nakagawa T, Terado Y, et al. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem Biophys Res Commun. 2008;369(2):292-296.
47. Tasian SK, Teachey DT, Li Y, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017;129(2):177-187.
48. Romero-Pozuelo J, Figlia G, Kaya O, et al. Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 2020;31(2):107504.
49. Fukada T, Ohtani T, Yoshida Y, et al. STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 1998;17(22):6670-6677.
50. Kollmann K, Heller G, Schneckenleithner C, et al. A kinaseindependent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2013;24(2):167-181.
51. Nebenfuehr S, Kollmann K, Sexl V. The role of CDK6 in cancer. Int J Cancer. 2020;147(11):2988-2995.
52. Frismantas V, Dobay MP, Rinaldi A, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017;129(11):e26-e37.
53. Bellutti F, Tigan AS, Nebenfuehr S, et al. CDK6 antagonizes p53-induced responses during tumorigenesis. Cancer Discov. 2018;8(7):884-897.
54. Uras IZ, Maurer B, Nivarthi H, et al. CDK6 coordinates JAK2 (V617F) mutant MPN via NF-kappaB and apoptotic networks. Blood. 2019;133(15):1677-1690.
Time spent at home among older adults with acute myeloid leukemia receiving azacitidine- or venetoclax-based regimens
Correspondence: D.R. Richardson daniel_richardson@med.unc.edu https://doi.org/10.3324/haematol.2022.280728
Received: January 21, 2022.
Accepted: June 10, 2022.
Early view: July 21, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
Time at home is a critically important outcome to adults with acute myeloid leukemia (AML) when selecting treatment; however, no study to date has adequately described the amount of time older adults spend at home following initiation of chemotherapy. We queried records from a multi-institution health system to identify adults aged ≥60 years newly diagnosed with AML who were treated with azacitidine or venetoclax and evaluated the proportion of days at home (PDH) following diagnosis. Days were considered “at home” if patients were not admitted or seen in the emergency department or oncology/infusion clinic. Assessed covariates included demographics and disease risk. Associations between PDH and baseline characteristics were evaluated via linear regression, adjusted for log length of follow-up. From 2015-2020, 113 older adults were identified. Most received azacitidine plus venetoclax (51.3%) followed by azacitidine monotherapy (38.9%). The mean PDH for all patients was 0.58 (95% confidence interval: 0.54-0.63, median 0.63). PDH increased among survivors over time. PDH did not differ between therapy groups (adjusted mean, azacitidine plus venetoclax: 0.68; azacitidine monotherapy: 0.66; P=0.64) or between disease risk categories (P=0.34). Compared to patients receiving azacitidine monotherapy, patients receiving azacitidine plus venetoclax had longer clinic visits (median minutes: 127.9 vs. 112.9, P<0.001) and infusion visits (median minutes: 194.3 vs. 132.5, P<0.001). The burden of care for older adults with AML treated with “less intense” chemotherapy is high. The addition of venetoclax to azacitidine did not translate into increased time at home. Future prospective studies should evaluate patient-centered outcomes, including time at home, to inform shared decision-making and drug development.
Introduction
The prognosis for older adults diagnosed with acute myeloid leukemia (AML) is poor. Historically, less than 40% of older adults (aged ≥60 years) survived 1 year from the time of diagnosis.1,2 Recently, however, treatment decisionmaking paradigms have shifted with the addition of venetoclax to azacitidine, a prior standard therapy. This combination has been associated with superior remission rates and overall survival compared to azacitidine alone.3 Prior research has demonstrated that patients with AML prefer treatments that allow increased time at home.4 In a national survey of patients with AML, most were willing to accept a reduction in remission rates in exchange for an increase in the amount of time spent at home, al- though preferences for treatment outcomes varied.5 An accurate understanding of the amount of time patients can anticipate spending at home is therefore critical to inform patient-centered treatment decisions for older adults with AML.
Descriptions of patients’ time at home and treatment burden in the setting of advanced solid tumors have recently been published.6,7 However, no study has adequately described the amount of time older patients spend at home while receiving therapy for AML. To address this knowledge gap, we aimed to quantify the amount of time older patients with AML spend at home and the amount they spend engaged in AML-related care with initiation of first-line azacitidine- or venetoclax-containing regimens.
Methods
Setting and patients
We queried records from University of North Carolina (UNC) Health to identify adults aged ≥60 years diagnosed with AML from 2015-2020. Those receiving first-line azacitidine and/or venetoclax were included. Records from 12 other health systems were available via electronic health data exchanges. Individuals receiving oncology care not reflected in available records were excluded. The UNC institutional review board approved the protocol.
Demographic and clinical variables
Demographic variables included age, race, sex, marital status, employment, and area-level measures (rurality8 and median household income9). Driving times/distances to clinical encounters from patients’ addresses were calculated via the Google Maps Application Program Interface (Google, LLC, Mountain View, CA, USA).10 Clinical variables included disease risk according to European LeukemiaNet (ELN) 2017 criteria11 and dates of diagnosis, death/last follow-up, and all clinic/emergency department/inpatient encounters.
Outcomes
The primary outcome was proportion of days at home (PDH),6,7,12 defined as the number of days subjects were not engaged in cancer-related care divided by total follow-up days. Individuals were deemed “engaged in care” if hospitalized (for any cause), seen in an emergency department (for any cause), or seen in an oncology/infusion clinic. Outpatient visits in non-oncology settings were not counted as engaged in cancer-related care. Counting began at diagnosis, with censoring at the end of 2020. Time commitment was also quantified via visit durations, calculated from check-in/check-out timestamps. Only appointments at UNC Health were included, as timestamps were not available for other health systems. Overall survival was calculated via the Kaplan-Meier method. Treatment response was assessed, with individuals achieving a best response of complete remission, complete remission with incomplete hematologic recovery, or a morphological leukemia-free state being considered to have achieved remission.11
Analysis
Descriptive statistics are provided as medians and interquartile ranges (IQR) or frequencies and percentages. Associations between patients’ characteristics and treatment regimens were assessed via modified Poisson models and characterized using relative risks. Cox proportional hazards models were used to examine associations between overall survival and demographic, disease, and treatment variables. Remission was used as a time-varying covariate in a Cox proportional hazards model to assess association between remission and survival. Competing risks analysis was used to analyze the association between baseline characteristics and cumulative incidence of remission while adjusting for deaths. Paired t-tests were used to evaluate the impact of remission on pre- and post-remission time at home among those who entered remission. Median visit durations were stratified by treatment group with hypothesis testing via Wilcoxon rank-sum.
PDH was evaluated in terms of total PDH over the entire study period and on a monthly (30 days) basis. For monthly analyses, only patients surviving the entirety of a particular month were included in that month’s total. PDH was described via summary statistics for the overall cohort and stratified by categorical variables. Associations between PDH and baseline characteristics were evaluated via linear regression models. Regression models were adjusted for the log of duration of follow-up, and adjusted means are presented. Adjusted mean PDH values presented in this analysis were evaluated at the average follow-up time of 8.6 months. A multivariable linear regression model with distance from UNC Medical Center, age, ELN risk, and adjustment for duration of follow-up was performed to further characterize these associations.
Results
Demographics
We identified 372 individuals aged ≥60 years with a new diagnosis of AML treated within the UNC Health system from 2015 to 2020. Of these, 137 received azacitidine and/or venetoclax as first-line therapy. Twenty-four individuals were excluded because of incomplete records, yielding a final cohort of 113 patients.
The median age of these 113 individuals was 73 years (range, 61-95 years), with the majority (51.3%) of patients being between 70 and 79 years old (Table 1). The majority of the cohort was white (80.4%), and 56.6% were male. Most were retired or otherwise not currently employed (89.6%). Most were living with a spouse or long-term partner (66.4%) and had health insurance (89.4%). The median distance from home address to the UNC Medical Center was 42.3 miles (IQR, 27-93 miles).
Disease and treatment
ELN risk category was favorable for 8.1%, intermediate for 30.6%, and adverse for 61.3% of the cohort (Table 1). Patients received azacitidine plus venetoclax combination therapy (51.3%), azacitidine monotherapy (38.9%), and other venetoclax-containing regimens (9.7%) as first-line treatment. Baseline demographic covariates and ELN risk were similar across therapy groups (P for all associations >0.05) except that there were more individuals aged 80-
89 years in the azacitidine plus venetoclax group compared to the azacitidine monotherapy group (27.6% vs. 9.1%, respectively; P=0.036). Treatment was initiated in an inpatient setting for 10.9% of individuals in the azacitidine plus venetoclax group and for 0% of the azacitidine monotherapy group.
AML: acute myeloid leukemia; AZA: azacitidine; VEN: venetoclax; ELN: European LeukemiaNet. Missing data not available in records: race (n=1 subject), employment status (n=17), marital status (n=3), rurality (n=2), household income (n=3), ELN risk (n=2). *Comparison between categorical variables assessed via the c2 test. Statistically significant result shown in bold.
Response to treatment and survival
Among the full cohort, 34.5% of patients achieved remission (complete remission, complete remission with incomplete hematologic recovery, or a morphological leukemia-free state). The 6-month competing risk-adjusted event rate of remission was 0.31. Higher remission rates were observed among those receiving azacitidine plus venetoclax (6-month competing risk-adjusted event rate 0.50, unadjusted 58.9%) than among those receiving azacitidine alone (6-month competing risk-adjusted event rate 0.10, unadjusted 9.5%) (hazard ratio [HR]=7.50, 95% confidence interval [95% CI]: 2.76-20.40, P<0.001) (Online Supplementary Table S1). Increased distance from the UNC Medical Center was associated with lower remission rates in these competing risk-adjusted analyses (HR=0.93 for every 10 additional miles away, 95% CI: 0.870.997, P=0.042). No other factors, including demographic variables and ELN risk category, were associated with having achieved remission (P for all associations >0.05). The median overall survival for the full cohort was 7.7 months (95% CI: 3.8-10.9) and was similar across therapy groups (Online Supplementary Table S2). There were also no significant associations observed between overall survival and ELN risk, race, sex, or other demographic variables (P for all associations >0.05). When evaluated as a time-varying covariate, having achieved remission was associated with superior survival, with a hazard ratio for death of 0.56 (P=0.043), indicating achieving remission was associated with longer survival.
Proportion of days at home
Over the full follow-up period, the mean PDH was 0.58 (95% CI: 0.54-0.63) with a median of 0.63. PDH was positively associated with length of follow-up, with a statistically significant association between PDH and log length of follow-up time (regression coefficient 0.15, 95% CI: 0.120.17, P<0.0001). After adjusting for log follow-up time, the adjusted mean PDH was 0.67 (95% CI: 0.63-0.71). PDH was similar among those with adverse-risk (adjusted mean 0.65), intermediate-risk (0.69, P=0.34), and favorable-risk AML (0.64, P=0.81). PDH did not differ significantly among therapy groups: azacitidine plus venetoclax (adjusted mean 0.68), azacitidine alone (0.66, P=0.64 when compared to azacitidine plus venetoclax), and other venetoclax-containing regimens (0.65, P=0.65). There was also no significant difference in the proportion of days spent as inpatients or in outpatient clinics between patients in the azacitidine and azacitidine plus venetoclax groups (Online Supplementary Table S3).
Patients who had longer driving distances to UNC spent more time engaged in oncologic care, and thus less time at home (regression coefficient for PDH -0.0008 per mile, 95% CI: -0.0013 to -0.0002, P=0.011). Patients who lived farther than 50 miles from UNC spent approximately 7% more days (amounting to 2.1 more days per month) engaged in oncology care than those who lived closer (adjusted mean PDH 0.63 vs. 0.70 for those <50 miles, P= 0.04). Patients who lived farther from UNC spent more days in hospital (26% of all follow-up days spent as inpatients for those living ≥50 miles vs. 20% for those living <50 miles from UNC). After adjusting for age, ELN risk, and median household income, the association between increased distance from the medical center and decreased PDH remained but was no longer statistically significant (P=0.08). There was no association seen between distance from UNC and chemotherapy regimen received. Other than driving distance, no individual covariate (including patient’s age) had a significant association with PDH when adjusted for length of follow-up (all P>0.05).
When evaluated on a month-by-month basis, the PDH rose over time among survivors (Table 2). For example, among patients who survived the full month, the mean PDH was 0.51 (95% CI: 0.47-54, n=105) in month 1 following diagnosis, 0.64 (95% CI: 0.58-0.70, n=75) in month 3, and 0.77 (95% CI: 0.72-0.82, n=57) in month 6. This pattern is demonstrated in Figures 1 and 2, which summarize person-days at home versus person-days engaged in care for 12 months following diagnosis, including contributions of person-days from those who did not survive the full month. Early in follow-up, most care days consisted of inpatient hospitalization (28% of all person-days in the first month), with a decline in hospitalization burden over the ensuing 12 months among survivors (4% of all person-days in month 12). The proportion of days seen in a clinic remained relatively constant over the year following diagnosis (in a range of 16-21% of all person-days). Patterns in monthly PDH were similar between those receiving azacitidine monotherapy and those receiving azacitidine plus venetoclax (Figure 3, Table 2). There was no significant difference in types of visits between those receiving azacitidine monotherapy and those receiving azacitidine plus venetoclax. The rate of early hospitalizations (days hospitalized during the first 30 days following diagnosis) was also similar between recipients of azacitidine monotherapy and azacitidine plus venetoclax, despite the higher percentage of patients within the azacitidine plus venetoclax group initiating therapy while admitted. Among the 38 patients who entered remission, more days were spent at home following remission than prior to remission. For this group, 53% (95% CI: 46-60%) of days prior to remission were spent at home compared to 72% (95% CI: 66-78%, P<0.0001) of days after remission.
Time spent in a clinic
For visits occurring within the UNC Health system, the median duration of oncology provider visits was 123.3 minutes (IQR, 80.1-185.2 minutes) (Table 3). The median duration of an infusion encounter was 169.3 minutes (IQR,
Table 2. Proportion of person-days spent at home by month from diagnosis among older adults with acute myeloid leukemia, stratified by first-line therapy.
AZA: azacitidine, VEN: venetoclax, PDH: proportion of days at home, 95% CI: 95% confidence interval. Number of subjects (N) reflects number of individuals in each group surviving at the end of the respective month. PDH for each month was calculated as the proportion of persondays not engaged in oncologic care among these survivors.
93.8-307.7) and the median duration of a laboratory encounter was 43.3 minutes (IQR, 25.7-83.3). Receipt of azacitidine plus venetoclax was associated with longer clinic visits (median 127.9 vs. 112.9 minutes, P<0.001) and infusion visits (median 194.3 vs. 132.5 minutes, P<0.001) compared to receipt of azacitidine monotherapy. Patients spent a median of 66 minutes (IQR, 30-110 minutes) in the two-way drive time to/from clinic for each day with an appointment. The median drive time was shorter among those receiving azacitidine plus venetoclax monotherapy than among those receiving azacitidine monotherapy (median 50 vs. 66 minutes, P<0.0001).
Discussion
The recent development of multiple novel effective chemotherapeutic agents has expanded treatment options for older adults with AML. Increasingly, patients are empowered to participate in a shared decision-making process that considers their values and preferences with respect to the anticipated outcomes of treatments. Time spent at home represents a critical but under-reported outcome of cancer therapies.7,13,14 We have previously shown that patients with AML are willing to sacrifice remission rates to achieve time at home.5 Currently, clinical trials in AML do not routinely capture, nor report, the time at home experienced by patients following chemotherapy. Reliably capturing this outcome in routine care and clinical trials will enable rational, data-driven shared decisionmaking regarding chemotherapy. Here, we demonstrate the feasibility of quantifying the time at home older patients achieve following first-line therapy and report, for the first time, a comparison between current treatments. Older AML patients receiving azacitidine, azacitidine plus venetoclax, or other venetoclax-containing regimens spent an average of 42% of their days engaged in oncology care. This time commitment was observed for AML patients despite patients receiving “less intensive” chemotherapy compared to intensive induction chemotherapy that often necessitates a lengthy period as an inpatient. Even among survivors 1 year from diagnosis, 24% of days were devoted to their cancer treatment, reflecting the need for ongoing close monitoring and potential transfusion support among those with AML. These data are similar to those from a smaller group of patients receiving hypomethylating agents in an Australian study published prior to the approval of venetoclax.12
The time commitment faced by patients with AML is striking when compared to that by patients with advanced solid cancers. Rocque and colleagues reported that women with newly diagnosed metastatic breast cancer spent 7 to 10% of days in the 3 months following diagnosis engaged in oncology care.6 Among adults with newly di- agnosed metastatic pancreatic cancer, who face a similarly poor prognosis as that of older adults with AML, Bange and colleagues described a cohort median of 10% of days devoted to cancer care following diagnosis.7 Older patients with AML spend over four times as many days engaged in oncology care as these patients. Time at home for patients with other hematologic malignancies has not been routinely reported outside of the context of end-oflife or hospice care.15
Figure 1. Proportion of person-days spent at home and engaged in cancer care among older adults with acute myeloid leukemia treated with azacitidine and/or venetoclax. The figure displays the person-days either at home (not engaged in care) or seen in each venue (clinic, emergency department, or hospital) among survivors. The proportion of days spent at home rose in the year following diagnosis, with a decline in the proportion of care-days consisting of inpatient hospitalization over the same period Values of the proportion of days at home in this figure differ slightly from monthly proportion values (PDH) in Table 2, as the PDH values in Table 2 were calculated only among survivors for a full month, whereas this figure reflects person-day contributions from individuals who did not survive the entirety of the respective month. ED: emergency department.
In a phase III trial of adults deemed ineligible for intensive chemotherapy, the addition of venetoclax to azacitidine increased remission rates and improved median overall survival from 9.6 to 14.7 months.16 In the current study, patients receiving azacitidine plus venetoclax had superior remission rates; however, this did not translate into a greater proportion of time at home for this group in aggregate. Patients receiving venetoclax also had longer clinic visits and longer visits for infusion, potentially related to a greater need for transfusion support. These data suggest that the clear benefit seen from the addition of venetoclax in clinical trials may not result in a substantial improvement in time spent at home for patients. This finding was surprising as we anticipated that remission would translate into prolonged survival, fewer clinic visits, fewer visits to the infusion center, and fewer admissions. We therefore performed a post hoc analysis looking at time at home before and after remission. Among the minority of individuals who entered remission, more days were spent at home after remission than prior to achieving remission. However, in aggregate, remission status did not correlate with the overall PDH throughout follow-up. Assessment of this relationship is complicated, as multiple factors may contribute to increasing time at home further from diagnosis, including achieving remission, better management of an individual’s symptoms further into treatment, and early deaths of the sickest individuals who may account for the greatest time engaged in care. Survival in our azacitidine plus venetoclax cohort was also shorter than that described in some other realworld-data analyses17,18 which likely resulted in decreased time at home achieved by these patients. However, similar survival results have been described in other retrospective cohorts.19 Furthermore, although we examined multiple variables when comparing therapies, confounding by other factors may be present. In particular, we did not quantify comorbidity or frailty other than by age alone. Adjusting for these factors may have altered our findings, especially for patients between the age of 80 and 89 years, who were over-represented in the azacitidine plus venetoclax cohort. Additionally, developing expertise in optimizing delivery of a complicated treatment regimen such as azacitidine plus venetoclax requires time and experience. As oncologists and cancer centers become more familiar with delivering venetoclax-based regimens, time at home may improve.
No significant differences were noted in the PDH following AML diagnosis based on age, race, sex, or disease risk according to ELN criteria in our cohort. We did observe a significantly lower PDH among those living farther from our primary referral hospital and comprehensive cancer center. This difference is partially attributable to more inpatient days experienced by patients living farther from the cancer center, a care-delivery pattern that has been described in the setting of other malignancies.10 The as- sociation between increased distance from the medical center and decreased PDH was no longer significant when adjusted for age, ELN risk, and median household income, possibly related to a degree of collinearity between driving distance and patients’ age.
This study has several additional limitations. Although records were captured from several health systems, this study includes only data from patients who were seen at least once at a single academic referral center. Consequently, the results may not be generalizable to adults treated entirely in community settings in which the burden of care may be different. Furthermore, the need for complete follow-up records to evaluate PDH may have biased our sample toward individuals with shorter survival and less time at home, as older adults with more stable disease may have experienced greater co-management with a local oncologist outside of the captured records. Simi- on the relative value patients with AML place on the specific health states in this study (e.g., clinic visits, infusion visits, hospitalization). We were, therefore, unable to adjust the reported time at home for perceived quality of this time to patients. Studies identifying the relative value of these health states would be informative to allow for quality-adjustment of the reported findings. This study represents an important step in understanding the experience of older adults with AML. A patient’s time is a finite resource that is increasingly consumed by complex cancer care. When compared to those with other advanced cancers, older adults with AML face a markedly increased burden on their time due to oncology care. The recent therapeutic paradigm shift in AML is an opportunity to leverage these remarkable advancements into meaningful improvements in patients’ lives. Future prospective studies should evaluate time at home as an endpoint as part of a broader strategy to incorporate patient-centered outcomes into drug development and shared decisionmaking strategies. Additionally, interventions directed at increasing time at home, such as utilization of virtual visits/telehealth or clustering of care, are critically needed to maximize patients’ time at home. Early studies of increased utilization of telehealth in the context of the COVID-19 pandemic suggest patients often find this caredelivery approach satisfactory.14 More intensive outpatient care models that support patients’ time at home have also been described,20 including all-outpatient “intensive” in- duction strategies for AML.21 Whenever possible, such approaches to increase patients’ time at home should be implemented as overall survival remains tragically short for older patients with AML.
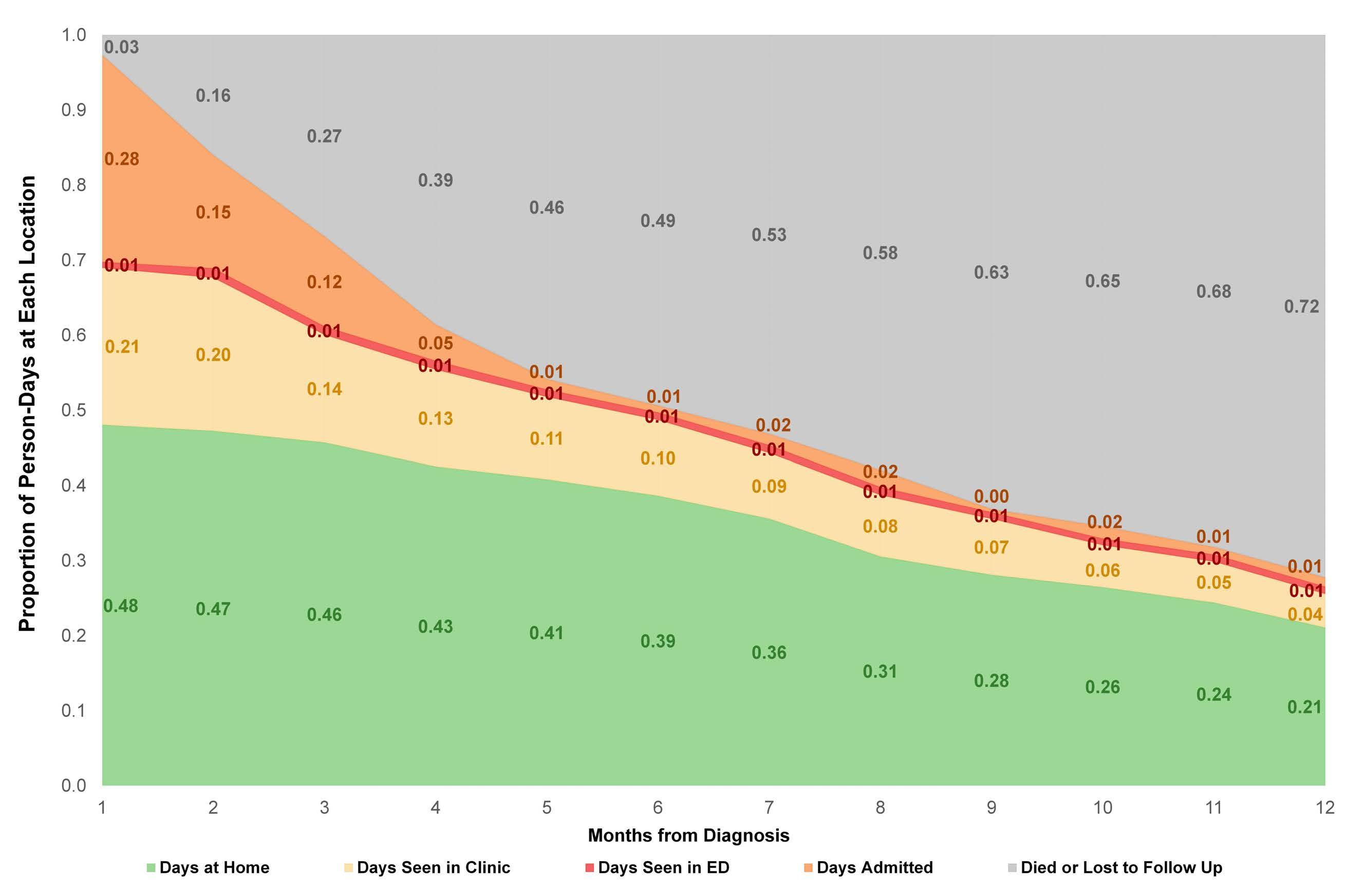
Figure 3. Proportion of person-days spent at home among older adults with acute myeloid leukemia treated with azacitidine with or without venetoclax in the year following diagnosis. Among survivors in each group, the proportion of days spent at home (and not engaged in oncologic care) rose over time, with generally similar proportions observed in each treatment group at each time point. Adjacent values for the two treatment groups are contemporaneous and have been offset for legibility. Error bars reflect 95% confidence intervals.
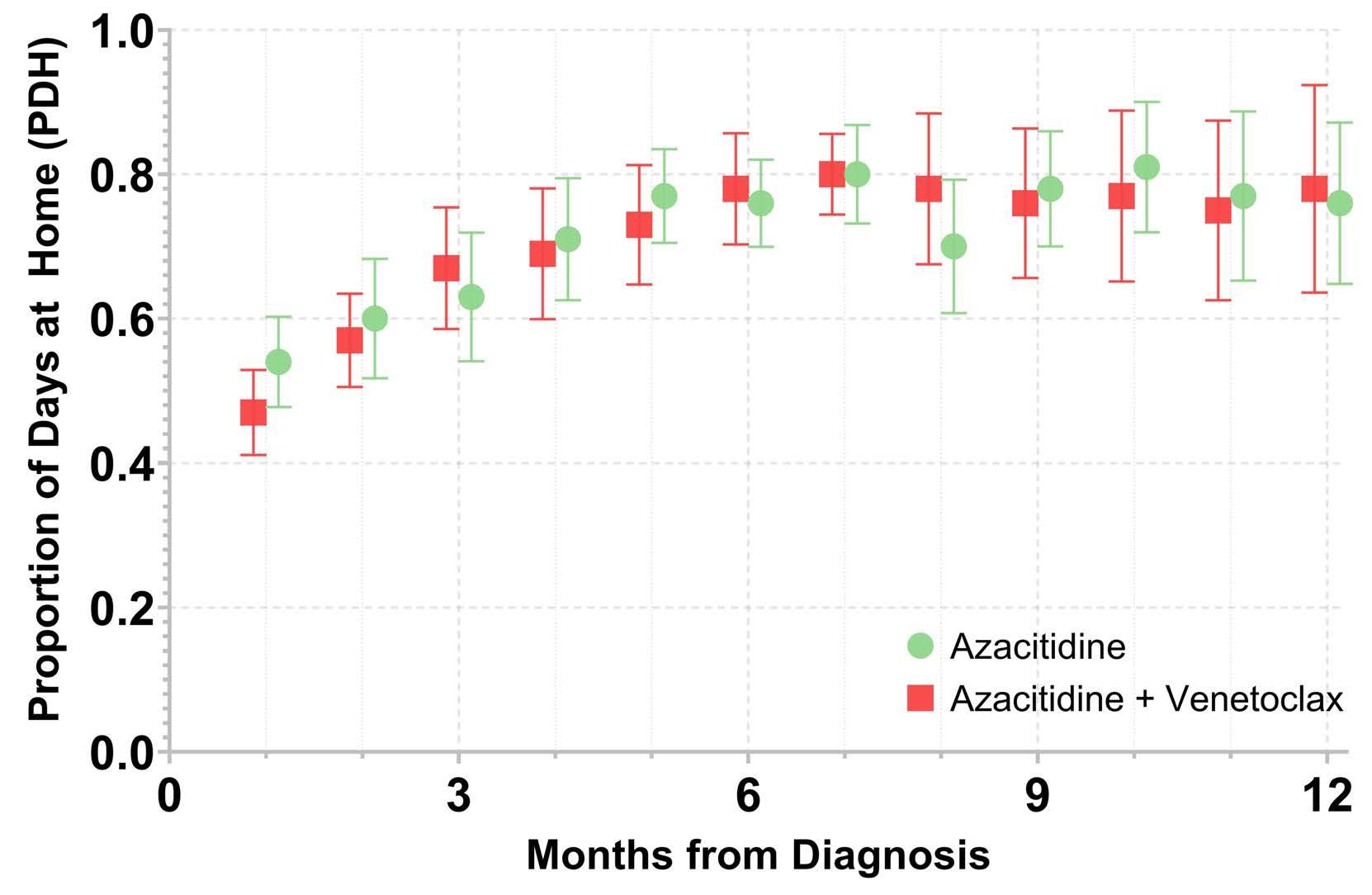
AZA: azacitidine; VEN: venetoclax. Median duration based on data from the University of North Carolina (UNC) Health system only. Numbers for total visits include data from UNC and other health systems (for provider visits and infusion visits) or UNC Health system only (for laboratory visits). *Oncology provider visits include encounters with physicians, nurse practitioners, physician assistants, or clinical pharmacist practitioners. †Drive times are calculated to the nearest minute with typical traffic and reflect time to/from the location of the clinical encounter, starting from the patients’ home addresses. One patient from the azacitdine group was excluded from the drive-time calculations because of a documented home address outside of North Carolina/bordering states. ‡P values reflect comparisons of duration of visits or drive time between therapy groups via the Wilcoxon rank-sum test.
Disclosures
None of the authors has a relevant conflict of interest. CEJ reports research funding (Conquer Cancer) outside the submitted work. ALB reports research funding (Carevive, Jazz Pharmaceuticals) and honoraria (Servier Pharmaceuticals) outside the submitted work. LAC reports research funding (Alexion Pharmaceuticals , Machaon Diagnostics) outside the submitted work. MCF reports research funding (Bellicum Pharmaceuticals , Macrogenics, Rafael Pharmaceuticals) and consulting (Macrogenics, Daiichi Sankyo, Agios) outside the submitted work. DRR reports research funding (Conquer Cancer, Palliative Care Research Cooperative Group) outside the submitted work.
Contributions
CEJ and DRR conceptualized the study and identified the study cohort; CEJ and KEB extracted data from charts; CEJ,
References
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136-1152.
2. Acute myeloid leukemia: SEER stat fact sheets: 2019. Surveillance Epidemiology and End Results (SEER). Vol. 2019: National Cancer Institute. 2019.
3. DiNardo CD, Wei AH. How I treat acute myeloid leukemia in the era of new drugs. Blood. 2020;135(2):85-96.
4. Richardson DR, Oakes AH, Crossnohere NL, et al. Prioritizing the worries of AML patients: quantifying patient experience using best-worst scaling. Psychooncology. 2021;30(7):1104-1111.
5. Richardson DR, Crossnohere NL, Seo J, et al. Age at diagnosis and patient preferences for treatment outcomes in AML: a discrete choice experiment to explore meaningful benefits. Cancer Epidemiol Biomarkers Prev. 2020;29(5):942-948.
6. Rocque GB, Williams CP, Ingram SA, et al. Health care‐related time costs in patients with metastatic breast cancer. Cancer Med. 2020;9(22):8423-8431.
7. Bange EM, Doucette A, Gabriel PE, et al. Opportunity costs of receiving palliative chemotherapy for metastatic pancreatic ductal adenocarcinoma. JCO Oncol Pract. 2020;16(8):e678-e687.
8. United States Census Bureau. 2010 Census Urban and Rural Classification and Urban Area Criteria. Vol. 2021: United States Census Bureau. 2019.
9. United States Census Bureau. American Community Survey (ACS). Vol. 2021: United States Census Bureau. 2020.
10. Rocque GB, Williams CP, Miller HD, et al. Impact of travel time on health care costs and resource use by phase of care for older patients with cancer. J Clin Oncol. 2019;37(22):1935-1945.
11. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
12. Sharplin K, Wee LYA, Singhal D, et al. Outcomes and health care utilization of older patients with acute myeloid leukemia. J
HMH, AMD, and DRR designed the data analysis; HMH and AMD performed the data analysis; CEJ, KEB, ALB, LAC, MCF, and DRR contributed to interpretation of the results; CEJ drafted the manuscript. All authors reviewed the final manuscript.
Funding
The project described was supported by the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) through Grant Award Number UL1TR002489 and by a National Research Service Award Post-Doctoral Traineeship from the Agency for Healthcare Research and Quality sponsored by The Cecil G. Sheps Center for Health Services Research at The University of North Carolina at Chapel Hill through Grant Award Number T32-HS000032. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Data-sharing statement
Data can be requested via e-mail to the corresponding author.
Geriatr Oncol. 2021;12(2):243-249.
13. Fundytus A, Prasad V, Booth CM. Has the current oncology value paradigm forgotten patients’ time? JAMA Oncol. 2021;7(12):1757-1758.
14. Banerjee R, George M, Gupta A. Maximizing home time for persons with cancer. JCO Oncol Pract. 2021;17(9):513-516.
15. Cheung MC, Croxford R, Earle CC, Singh S. Days spent at home in the last 6 months of life: a quality indicator of end of life care in patients with hematologic malignancies. Leuk Lymphoma. 2020;61(1):146-155.
16. Dinardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629.
17. Garcia JS, Wolach O, Vachhani P, et al. Comparative effectiveness of venetoclax combinations vs other therapies among patients with newly diagnosed acute myeloid leukemia: results from the AML real world evidence (ARC) initiative. Blood. 2021;138(Suppl 1):2328.
18. Bouligny IM, Maher KR. Outcomes of induction with venetoclax in combination with decitabine, azacitidine, or low-dose cytarabine for treatment of AML: a real-world retrospective analysis. Blood. 2021;138(Suppl 1):2335.
19. Vachhani P, Abbas JA, Flahavan EM, et al. Real world treatment patterns and outcomes of venetoclax (Ven) and hypomethylating agents (HMA) in patients with newly diagnosed acute myeloid leukemia (AML) in the United States. Blood. 2021;138(Suppl 1):2290.
20. Handley NR, Bekelman JE. The oncology hospital at home. J Clin Oncol. 2019;37(6):448-452.
21. Mabrey FL, Gardner KM, Shannon Dorcy K, et al. Outpatient intensive induction chemotherapy for acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood Adv. 2020;4(4):611-616.
Characterization of therapy-related acute myeloid leukemia: increasing incidence and prognostic implications
Christer
11Department of Medicine, Huddinge, Division of Hematology, Karolinska Institutet, Stockholm; 2Department of Medical Sciences, Uppsala University, Uppsala; 3Department of Hematology, Sahlgrenska University Hospital, Gothenburg; 4Department of Hematology, Skåne University Hospital, Lund, and Department of Hematology, Stem Cell Center, Department of Laboratory Medicine, Lund University, Lund; 5Department of Hematology, Linköping University Hospital, Linköping; 6Department of Hematology, Norrland University Hospital, Umeå and 7Department of Hematology, Sundsvall Hospital, Sundsvall, Sweden
Abstract
Correspondence: C. Nilsson christer.nilsson@ki.se https://doi.org/10.3324/haematol.2022.281233
Received: April 18, 2022.
Accepted: July 21, 2022.
Early view: August 25, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Studies of therapy-related AML (t-AML) are usually performed in selected cohorts and reliable incidence rates are lacking. In this study, we characterized, defined the incidence over time and studied prognostic implications in all t-AML patients diagnosed in Sweden between 1997 and 2015. Data were retrieved from nationwide population-based registries. In total, 6,779 AML patients were included in the study, of whom 686 (10%) had t-AML. The median age for t-AML was 71 years and 392 (57%) patients were females. During the study period, the incidence of t-AML almost doubled with a yearly increase in t-AML of 4.5% (95% confidence interval: 2.8%-6.2%), which contributed significantly to the general increase in AML incidence over the study period. t-AML solidly constituted over 10% of all AML cases during the later period of the study. Primary diagnoses with the largest increase in incidence and decrease in mortality rate during the study period (i.e., breast and prostate cancer) contributed significantly to the increased incidence of t-AML. In multivariable analysis, t-AML was associated with poorer outcome in cytogenetically intermediate- and adverse-risk cases but t-AML had no significant impact on outcome in favorable-risk AML, including core binding leukemias, acute promyelocytic leukemia and AML with mutated NPM1 without FLT3-ITD. We conclude that there is a strong increase in incidence in t-AML over time and that t-AML constitutes a successively larger proportion of the AML cases. Furthermore, we conclude that t-AML confers a poor prognosis in cytogenetically intermediate- and adverse-risk, but not in favorable-risk AML.
Introduction
Therapy-related AML (t-AML) is a feared complication of treatment with chemotherapy and/or radiation. The World Health Organization (WHO) defines the disease entity therapy-related myeloid neoplasms as myeloid neoplasms secondary to cytotoxic treatment, in which t-AML is categorized along with therapy-related myelodysplastic syndrome (MDS) and therapy-related myeloproliferative neoplasms (MPN).1 Between 5-10% of all AML cases have been reported to be therapy-related,2,3 and treatment for a solid cancer increases the risk of AML up to 10-fold with chemotherapy4 and 2.5-fold after treatment with radiotherapy.5 While solid and hematologic tumors dominate as the primary disease, t-AML may also occur after treatment of non-malignant diseases, especially inflammatory dis- orders.3,6-8 The mechanism behind the increased risk of AML after chemo- and/or radiation therapy are not fully understood, but it is currently regarded as a combination of mutagenic effects of chemotherapy and/or radiation and a selective pressure by this treatment, favoring existing premalignant clones in the blood and bone marrow.9-13 This may lead to clonal expansion of the premalignant clone as well as clonal evolution with accumulation of additional leukemogenic genetic aberrations that evolve to t-AML. t-AML is associated with a worse prognosis compared to that of de novo AML, and the disease presents with a higher rate of adverse genetic aberrations compared to de novo AML.3 Alkylating agents and radiation therapy have typically been associated with aberrations in chromosomes 5 and 7, while treatment with topoisomerase II inhibitors has been linked to rearrangements of chromosome 11q23, involving the KMT2A gene.14 However, as chemotherapy treatment usually consists of a combination of agents, this subdivision of t-AML is difficult to identify clinically, and it is no longer part of the WHO classification.15 Complex and monosomal karyotypes as well as TP53 mutations are also overrepresented in t-AML.7,16,17 The spectrum of tumors that precedes t-AML depends on incidence rates, type of treatment and the likelihood of long-term survival for the particular cancer type.4,18 Thus, tumors treated with high intensity chemotherapy followed by high cure rates, are overrepresented as pre-t-AML diagnoses.3,19 Consequently, malignancies that are less frequently treated with intensive chemotherapy, such as prostate cancer, or malignancies with short survival are generally underrepresented as primary diseases in tAML.3,20
Most studies of t-AML are either small or performed in selected cohorts and within clinical trials. Larger populationbased studies in which t-AML is defined from a real-life perspective, and from which true incidence rates can be calculated, are exceedingly rare. Here, we use populationbased quality registries in Sweden to investigate changes in incidence and survival in t-AML over time, as well as to characterize t-AML in a large real-life AML cohort. Furthermore, we study prognostic factors for t-AML patients, as well as t-AML in itself as a prognostic factor within genetic subtypes of AML.
Methods
Data collection
Data were collected from three nationwide registries: the Swedish AML Registry (SAMLR), the Swedish Cancer Registry (SCR) and the Swedish Rheumatology Quality Register (SRQ). SAMLR has been validated against the SCR defining the coverage of SAMLR to 98% of all AML patients nationwide.21 Each registry, its data and the methods to collect data are described in more detail in the Online Supplementary Methods. All registries use national unique personal identification numbers enabling identification of individuals across the registries.
Patients
The study included all AML patients ≥8 years reported to SAMLR between January 1, 1997 and December 31, 2015.
Two t-AML patients with a prior AML diagnosis reported as the antecedent disease were excluded. The median follow-up time was 101 months during which 4,406 deaths were registered; 10 patients were lost to follow-up and 1,076 patients were still alive at the end of follow-up. Intensively treated patients received induction and consolidation therapy according to Swedish guidelines,22 with a chemotherapy intensity comparable to that of the classical
“3 + 7” AML induction. The ethical review board in Gothenburg approved the study (Dnr 781/13).
Definitions t-AML was defined as AML with a prior diagnosis of a malignant or non-malignant disease treated with chemotherapy and/or radiation therapy. All chemotherapy regimens were considered, including methotrexate and cyclophosphamide for autoimmune diseases, but excluding immunosuppressive treatment. Patients treated with chemotherapy for MDS or MPN and progressing to AML were not defined as therapy-related cases, whereas patients developing MDS or MPN during the period between the treatment for the primary disease and the diagnosis of AML were considered to have t-AML. The 2010 European LeukemiaNet (ELN) criteria23 were used to stratify patients into cytogenetic risk groups. For risk classification of patients with available data on NPM1 and FLT3-ITD mutational status (starting from 2007) the ELN2010 criteria for NPM1 and FLT3-ITD were used. Complete remission was defined as <5% blasts in the bone marrow and recovery of peripheral blood counts.
Statistical analyses
Between-group comparisons of categorical and continuous variables were performed using the Pearson c2 test and the Mann-Whitney U test, respectively. The unequal variances t test was used to compare mean incidence rates between time periods. Overall survival was estimated using the Kaplan-Meier method and compared with the log-rank test or univariable Cox regression. Multivariable Cox regression was used for survival analyses with covariates specified where used. The proportional hazards assumption was tested for each covariate by graphical inspection of the scaled Schoenfeld residuals. Two-tailed tests with a 0.05 level of significance were used throughout the analyses. No imputation of missing data was performed. All data preparation and statistical analyses were conducted in R, version 3.6.0.24 Additional information about the statistical methods can be found in the Online Supplementary Material.
Results
Patients’ characteristics
A total of 5,492 patients were diagnosed with either t-AML (n=686) or de novo AML (n=4,806) in Sweden between 1997 and 2015, with an additional 1,287 patients being diagnosed with AML with an antecedent hematologic disease (AHD-AML). The aim of this study was to characterize t-AML patients specifically, which is why details on AHDAML patients are not further described. Of all AML cases in the study cohort, the proportion of t-AML was 10%, de novo AML 71% and AHD-AML 19%.
The clinical characteristics of patients with t-AML and de novo AML are shown in Table 1. Age at diagnosis was similar between t-AML and de novo AML (median 71 vs. 70 years, respectively) while t-AML had a female predominance (57%, compared to 49% in de novo AML). Performance status was similar with the majority of patients having Eastern Cooperative Oncology Group grade 0 or 1 (61% vs 63%, t-AML and de novo AML, respectively). Adverse cytogenetic risk was more common (46% vs. 28%, P<0.001) in t-AML, while favorable risk was somewhat less common
(12% vs. 16%, P=0.030). The proportion of APL was similar in t-AML compared to de novo AML (4% vs. 5%). Minor differences were observed in standard clinical parameters, with patients with t-AML having lower median bone marrow blast percentages (46% vs. 50%), lower white blood cell counts (6.1 vs. 8.7x109/L) and lower platelet counts (50 vs. 65 x109/L) compared to those with de novo AML. Inflammatory diseases and unspecified chronic diseases were more prevalent in t-AML than in de novo AML (13% vs. 3% and 23% vs. 9%, respectively), while the prevalence
Note: Data on FLT3-ITD, NPM1, CEBPA, comorbidities and blood chemistry are only reported since 2007. AML: acute myeloid leukemia; t-AML: therapy-related AML; ECOG: Eastern Cooperative Oncology Group; APL: acute promyelocytic leukemia; CR: complete remission; allogeneic stem cell transplantation; CR1: first of other comorbidities was similar (Online Supplementary Table S1).
Patients with t-AML were less likely to receive intensive induction treatment (60% vs. 71%, P<0.001) and intensively treated patients were less likely to reach complete remission (58% vs. 75%; P<0.001) compared to those with de novo AML. Allogeneic hematopoietic cell transplantation (HCT), regardless of disease state, was performed in 9% of the patients with t-AML compared to 16% of those with de novo AML (P<0.001). Corresponding rates of HCT in first remission were 7% in t-AML compared to 12% in de novo AML (P=0.002). There was no increase or decrease of transplantation rates over time (data not shown).
The incidence of therapy-related acute myeloid leukemia is increasing over time
The age-standardized incidence rate of t-AML increased during the study period from a mean of 0.39 cases per 100,000 adult inhabitants between 1997 and 2006 to 0.63 between 2007 and 2015 (P=0.004) (Figure 1A). During the last 2 years of the study period (2014-2015), the average age-adjusted incidence rate of t-AML was 0.95. There was also a slight increase in the incidence of de novo AML from 3.42 to 3.66 between the two study periods (P=0.02). For all AML (including de novo AML, t-AML and AHD-AML), the incidence rate increased from 4.69 (1997-2006) to 5.32
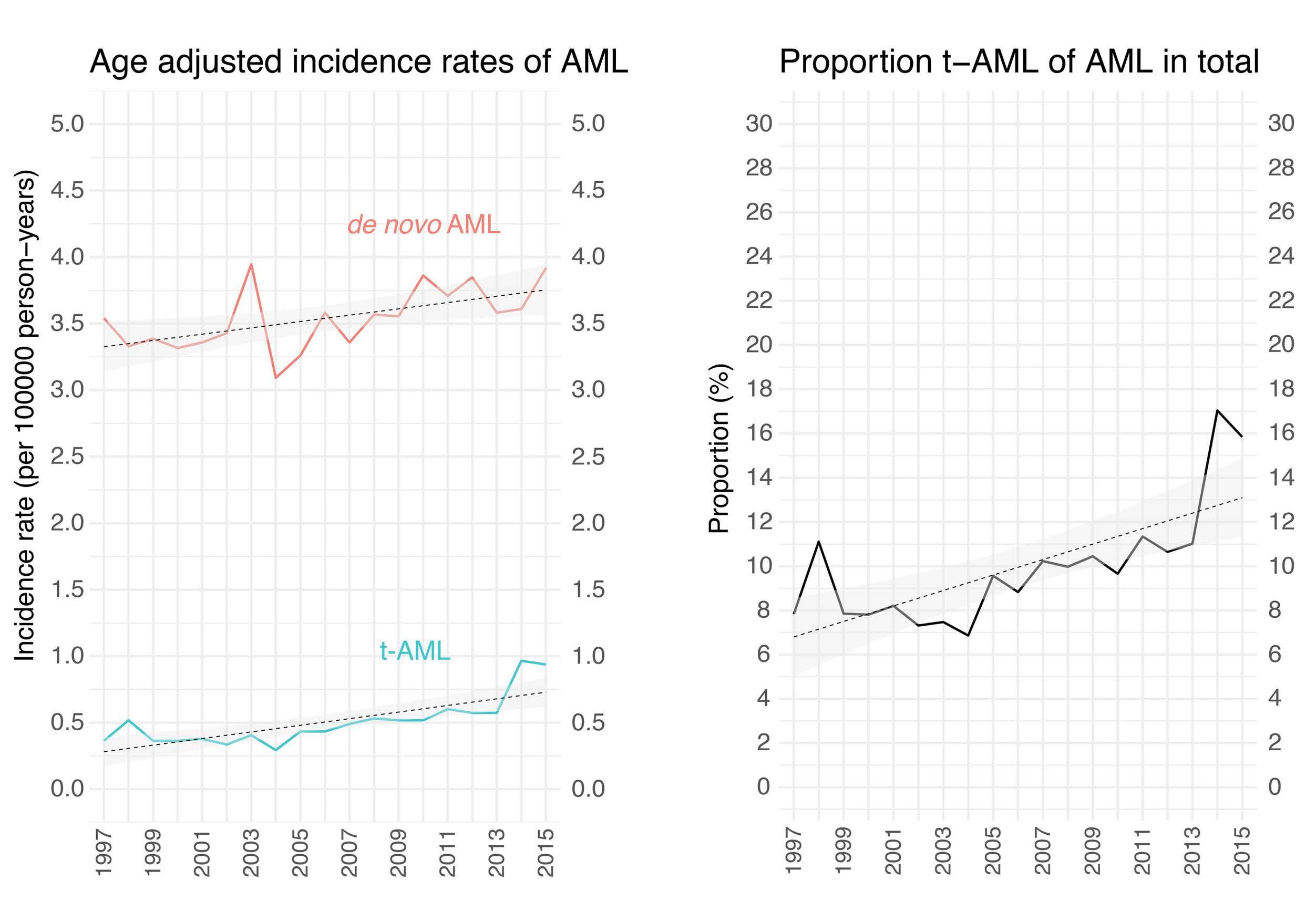
(2007-2015) (P=0.0006). The estimated annual percentage change was 4.5 % in t-AML (95% confidence interval [95% CI]: 2.8%-6.2%) compared to 0.7% in de novo AML (95% CI: 0.2%-1.2%). The relative increase in t-AML was substantial and contributed to a large proportion of the increased incidence of AML as a whole. As a result, the proportion of tAML of all AML cases increased from 8.3% in 1997-2006 to 11.8% in 2007-2015 (P=0.004, t test) (Figure 1B). For the last 2 years of the study period (2014-2015), the average proportion of t-AML was 16%.
To define how different primary tumors contributed to the increase in t-AML incidence, we compared the average number of t-AML cases per year per primary tumor disease between 1997-2002 and 2009-2015. Non-malignant diseases, breast cancer and prostate cancer contributed most to the rise in t-AML incidence with an absolute increase of 8.2, 5.6 and 5.2 cases per year per 10 million inhabitants, representing 40%, 27% and 25% of the increase in t-AML, respectively. A slightly increased contribution was also seen for lymphoma and gastrointestinal cancers. In contrast, gynecological cancers and multiple myeloma displayed a relatively stable or decreased contribution to tAML over time (Online Supplementary Table S2 and Online Supplementary Figure S1). To understand the contribution of the different primary diagnoses, we used official data from the Swedish Cancer Registry to compare the change in t-AML incidence to the change in incidence and mortality rate for the malignant diagnoses during the same period. As shown in Online Supplementary Figure S2A-L, the primary diagnoses that contributed most to the increase in t-AML, i.e. breast and prostate cancer, also displayed the largest increase in incidence and most prominent decrease in mortality rates. For gynecological cancers, incidence and mortality rates remained relatively unchanged for cervix and uterine corpus cancer while they decreased for ovarian carcinoma. Lymphoma displayed a small increase in incidence and a slightly improved survival. Thus, the increase in t-AML incidence seems to be at least partly driven by increased incidence and improved survival of some major tumors.
Characterization of diagnoses prior to therapy-related acute myeloid leukemia
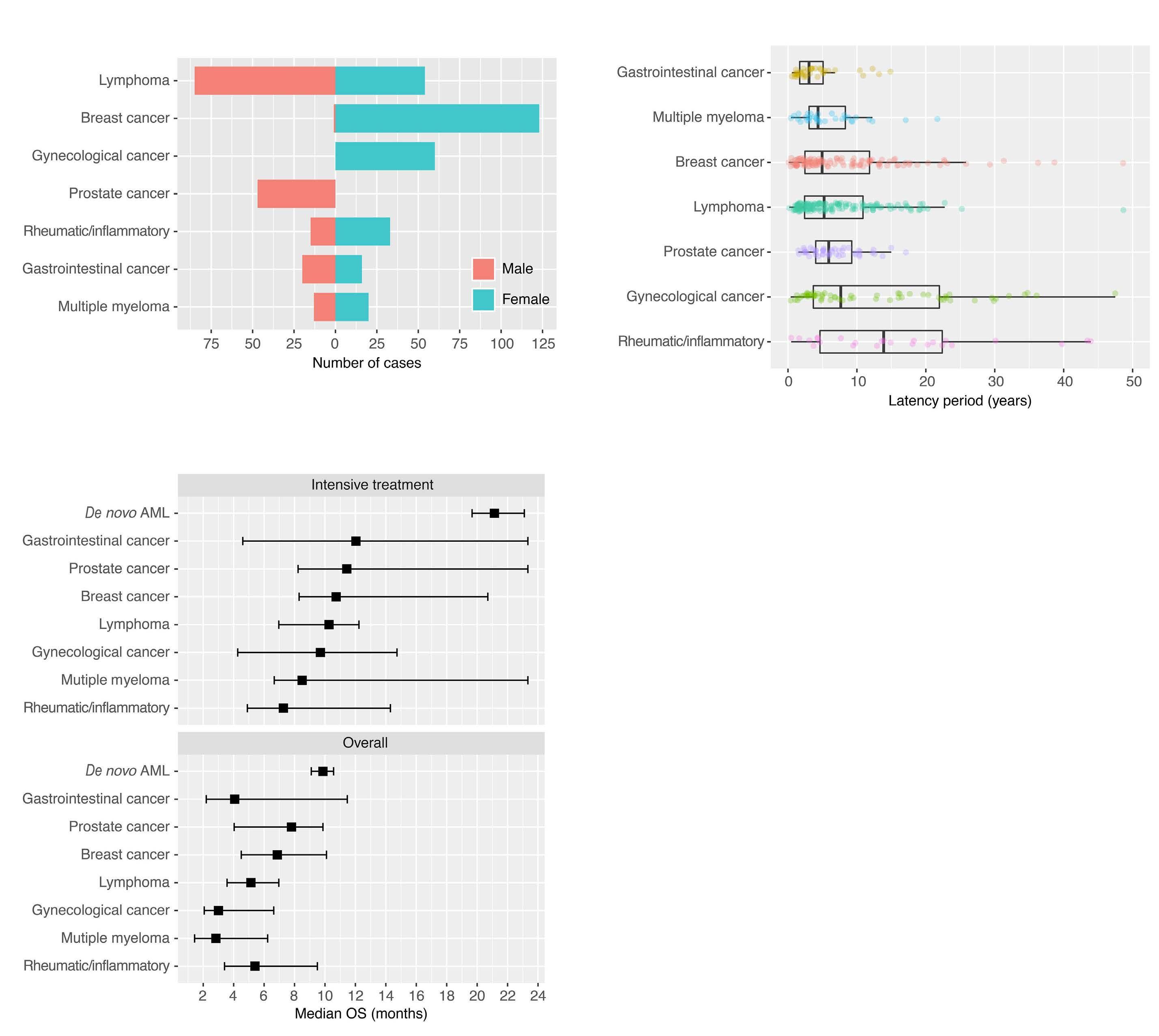
Of primary disorders, for which chemotherapy and/or radiation was given, 55% were non-hematologic solid cancers, 25% hematologic cancers and 18% non-malignant diseases. Figure 2A shows the number of cases and the gender distribution for each primary diagnosis and more details are provided in Online Supplementary Table S3. The most common primary diagnoses were lymphoma (n=139), followed by breast cancer (n=124), rheumatic and inflammatory disease (n=122), gynecological malignancies (n=60), prostate cancer (n=47), gastrointestinal cancer (n=36), and myeloma (n=33). Treatment for the primary disease was chemotherapy alone in 49%, radiation therapy alone in 25%, and chemotherapy and radiation therapy combined in 26% of the patients (Online Supplementary Table S3).
Figure 2. Primary diagnosis, gender, latency times and survival in therapy-related acute myeloid leukemia. (A) Number of cases and gender based on primary diagnoses. (B) Latencies between diagnosis of the primary disease and acute myeloid leukemia related to different primary diagnoses. (C) Median overall survival in intensively treated patients and overall in therapy-related acute myeloid leukemia grouped by primary disease. AML: acute myeloid leukemia; t-AML: therapy-related acute myeloid leukemia;
Nineteen percent (n=133) of t-AML patients had more than one tumor diagnosis before the onset of t-AML, either occurring before, or more often after the malignancy for which the first chemotherapy and/or radiation therapy was given. Figure 3 displays in detail the complexity of sequential relations between the different diagnoses. Seventeen percent (n=118) of the patients had a diagnosis of MDS between the primary disease and t-AML.
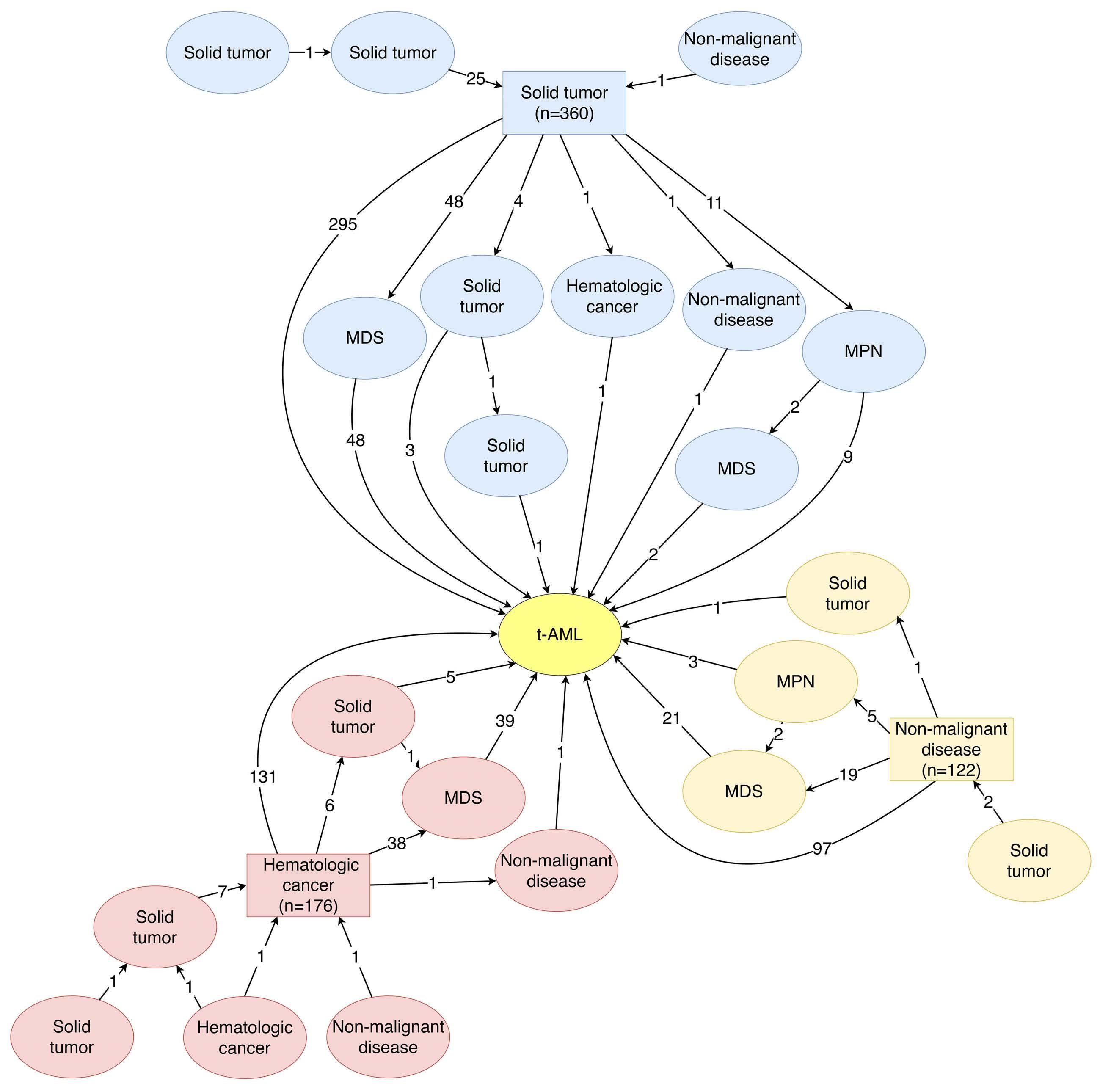
Latency periods
The latency period after a primary hematologic malignancy or a solid cancer to the diagnosis of t-AML was similar (median 57 vs. 61 months, respectively; P=0.287) while the latency period after diagnosis of a primary non-malignant disease (i.e., inflammatory or rheumatic disease) was longer (173 months; P<0.001), but also more variable (Online Supplementary Figure S3A). Figure 2B displays latency periods related to type of primary disease. The latency also varied with the type of primary treatment with similar la- tency after chemotherapy alone and combination therapy (chemotherapy + radiation) (median 54 vs. 55 months, respectively; P=0.848), but longer after radiation alone (median 97 months; P<0.001) (Online Supplementary Figure S3B). Both a primary non-malignant disease and radiation alone were associated with a longer latency period in multivariable analysis adjusting for age at primary diagnosis, type of primary treatment, type of primary disease and cytogenetic risk at diagnosis (Online Supplementary Table S4). The median latency periods of patients with -5/-7, 11q23/MLL and t(15;17) were 101 months (95% CI: 74-126 months), 41 months (95% CI: 29-75 months) and 84 months (95% CI: 41-190 months), respectively, compared to 62 months in t-AML overall (95% CI: 56-71 months). There was no association between the latency period and favorable, intermediate or adverse risk at large.
Figure 3. Paths to therapy-related acute myeloid leukemia. The figure shows the sequence of diagnoses of all malignant and/or hematologic diseases preceding the diagnosis of therapy-related acute myeloid leukemia (t-AML). Squares denote the primary disease for which cytotoxic treatment was given and ovals denote additional diseases, either prior to the primary disease or between the primary disease and t-AML. Numbers between squares and ovals represent number of cases. Twenty-eight cases for which the order of the disease could not be determined were excluded from the figure. MDS: myelodysplastic syndromes; MPN: myeloproliferative neoplasms.
Improved survival for therapy-related acute myeloid leukemia patients over time
The estimated 5-year survival of patients with t-AML was 10% overall, 17% in intensively treated patients (n=385), and 48% in patients who underwent HCT (n=68). As a comparison, the 5-year survival rates in patients with de novo AML were 23% overall, 34% in intensively treated patients, and 57% in transplanted patients. Similarly, the median overall survival was 5.0 months for all t-AML, 9.5 months in intensively treated patients, and 48 months in patients who underwent HCT (Online Supplementary Figure S4A-C). Corresponding median overall survival times patients with de novo AML were 9.7 months overall, 20.7 months in intensively treated patients, and 113 months in transplanted patients. Overall survival among patients transplanted in first complete remission was worse in t-AML patients than in de novo AML patients in both univariable analysis (hazard ratio [HR]=1.56, 95% CI: 1.06-2.29) and after adjusting for age and cytogenetic risk (HR=1.50, 95% CI: 1.02-2.22). Outcome of patients with t-AML improved over time with a 5-year overall survival in intensively treated patients of 11%, 16% and 23% for 1997-2002, 2003-2008 and 20092015, respectively (Online Supplementary Figure S5A, B). Patients with an antecedent diagnosis of MDS had a worse outcome in univariable analysis compared to patients without prior MDS (HR=1.52, 95% CI: 1.09-2.12, P=0.012) but when adjusting for age and cytogenetic risk, no significant survival difference was observed (Online Supplementary Table S5). Patients with t-AML did worse compared to those with de novo AML regardless of primary disease (Figure 2C, details in Online Supplementary Table S6) and type of treatment for the primary disease (chemotherapy, radiotherapy or chemo-radiotherapy; Online Supplementary Figure S6A, B). No correlation between the type of cytotoxic treatment and cytogenetic risk was found (distribution of risk in patients treated with radiotherapy only vs. patients treated with chemotherapy or combination therapy,
P=0.06). The cumulative incidence of death and relapse was higher in patients older than 60 years, although the relapse difference was not statistically significant (Online Supplementary Figure S7).
Therapy-related acute myeloid leukemia had a strong negative impact on survival in cytogenetically intermediate- and adverse- but not favorable-risk acute myeloid leukemia
How t-AML contributes to prognosis within different AML risk groups remains unclear. Therefore, we analyzed the outcomes of patients with t-AML compared to those with de novo AML in each cytogenetic risk group. In crude analyses, t-AML did not have a statistically significant impact on outcome in favorable-risk AML, regardless of whether acute promyelocytic leukemia (APL) was included in the favorable-risk group or not. In contrast, there was a significant and strong negative impact of t-AML in intermediate- and adverse-risk AML (Online Supplementary Figure S8A-E). After adjusting for age and performance status, survival was still similar in favorable-risk t-AML (with or without APL) and in APL alone compared to de novo AML: HR=0.99 (P=0.95); HR=1.11 (P=0.73), and HR=0.89 (P=0.78), respectively. In contrast, after the same adjustments in the intermediate- and adverse-risk groups, t-AML had a strong negative impact on overall survival: HR=1.53 (P<0001) and HR=1.59 (P<0.001), respectively (Table 2).
Therapy-related acute myeloid leukemia does not confer poor prognosis in acute myeloid leukemia with mutated NPM1 and absence of FLT3-ITD
We further analyzed the impact of NPM1 mutations and FLT3-ITD (data available from 2007). There were 112 t-AML patients for whom information on NPM1 status was available and 119 for whom FLT3-ITD data were available; among de novo AML patients, the corresponding numbers were 1,041 and 1,124 patients, respectively. In t-AML patients, FLT3-ITD alone did not add prognostic information (Figure 4A), while t-AML patients with NPM1mut had significantly better overall survival compared to NPM1wt t-AML patients (Figure 4B). t-AML patients with NPM1mut in combination with FLT3wt had better survival compared to t-AML patients with any other combination of NPM1 and FLT3 status (Figure 4C). t-AML patients with NPM1mut/FLT3wt had a similar overall survival compared to that of NPM1mut/FLT3wt de novo AML patients (HR=0.82, 95% CI: 0.40-1.68, P=0.584 for t-AML vs. de novo AML) (Figure 4D). The comparison showed similar results when adjusting for age, performance status and cytogenetic risk (t-AML vs. de novo AML: HR=0.75, 95% CI: 0.36-1.58, P=0.454).
The role of t-AML in relation to the ELN2017 classification including a more comprehensive mutational characterization was outside the scope of the study as the study was based on retrospective registry data from a period before comprehensive mutational analyses was performed routinely. Nevertheless, full mutational characterization was available for 58 of the patients and these data are provided in Online Supplementary Figure S9.
Discussion
In this study, we identified several features of t-AML that point to important current trends regarding t-AML and that provide real-world information on the role of t-AML in the prognostic outcome of AML. Firstly, we identified a significant increase in the incidence of t-AML during the last two to three decades. This increase is substantial and approaches a doubling during the study period. Consequently, it is much more likely that a newly diagnosed AML is a t-AML today compared to 20 to 30 years ago. This increase in t-AML was seen for primary tumors that have shown an increased incidence and decreased mortality during the same period, such as breast and prostate cancers. This is in part due to improvement of cancer therapy, leading to better long-term survival after mutagenic cancer treatments.25,26 With continuous improvements in survival of patients with malignant diseases, the number of individuals at risk of t-AML will continue to rise. However, the use of less mutagenic non-chemotherapeutic agents in cancer treatment may in part counteract this development.
Over the whole study period, the incidence of t-AML in our study was 0.51 cases per 100,000 inhabitants, which is substantially higher than that found in a recent American study using data from the Surveillance, Epidemiology, and End Results (SEER) registry in which the incidence rate of therapy-related myeloid neoplasms was 0.13 cases per 100,000 during 2001–2014.19 The reason for the discrepancy is unclear. Although it may be related to differences concerning treatment and/or survival of malignancies in general, it more likely reflects a difference in reporting and coverage of the registries as well as their sensitivity and specificity for recording previous treatment with chemotherapy and radiation. The advantage of the Swedish reporting system is the 98% coverage of the Swedish AML Registry, the compulsory reporting of cancer diagnoses to the Swedish Cancer Registry and the possibility of crosslinking the registries for each individual using personal identification numbers.
Regarding the basic characterization of the t-AML patients in this study, it is in line with most previous studies2,7,8 while we here can provide data from the largest population-based cohort of AML so far. The good performance status of t-AML patients is somewhat surprising, however, it is confirmed by several other studies.27,28 The good performance status contrasts with the fact that t-AML patients were less likely to receive intensive chemotherapy and allogeneic transplantation. This could be due to fact that other factors rather than performance status can affect the treating physician’s assessment of the patient, such as sequelae from previous exposure to chemotherapy or radiation as well as the presence of a still existing primary tumor. Regarding the latency periods, they were associated with factors such as type of treatment (chemotherapy or radiation) and type of primary disease (malignant vs. non-malignant). In line with previous studies, patients with 11q23/MLL-rearranged AML had shorter latency periods, while those with -5/-7 had longer latency periods.7,29
Table 2. Multivariable Cox regression analysis of overall survival in therapy-related acute myeloid leukemia compared to de novo AML in intensively treated patients according to cytogenetic risk.
AML: acute myeloid leukemia; t-AML: therapy-related AML; APL: acute promyelocytic leukemia; OS: overall survival; HR: hazard ratio; CI: confidence interval; ECOG: Eastern Cooperative Oncology Group; PS: performance status.
Secondly, we were able to examine and describe the role of t-AML in the prognosis within AML risk groups. It is well established that karyotype is an independent prognostic factor among intensively treated patients with t-AML and that poor-risk karyotypes are overrepresented in tAML.7,29,30 However, data on the impact of t-AML within each cytogenetic risk group are conflicting. Our data showed a significantly worse survival in t-AML patients within the intermediate and adverse AML risk groups. In contrast, t-AML patients with favorable-risk AML had the same survival as that of patients with de novo AML, also when correcting for other prognostic factors. Schoch et al. previously reported data contrasting with ours, with no survival differences in intermediate- and adverse-risk patients but a significant difference in the favorable-risk group.29 However, their study was considerably smaller with 93 t-AML patients in total, making the numbers of patients in each cytogenetic risk group very small. Aldoss and Pullarkat have reviewed whether the outcome of patients with favorable-risk t-AML is comparable to that of those with favorable risk de novo AML.31 The concluded that a therapy-related disease does not affect survival in APL but that it negatively affects outcome in core-binding factor AML, although not to a degree that should change transplantation indications in therapy-related core-binding factor leukemia. Our data support the approach that indications for allogeneic transplantation should remain the same in t-AML as de novo AML in patients with favorable-risk cytogenetics.
Mutational screening of NPM1 and FLT3-ITD is part of clinical routine as well as ELN recommendations for prognostic classification32 and for the first time, we could study the role of t-AML in these mutational subcategories of AML. In our study, the presence of FLT3-ITD did not have a negative impact on survival in t-AML patients, while patients with NPM1mut had a significantly better survival compared to those with NPM1wt t-AML. The lack of impact of FLT3-ITD on survival may be due to the different cytogenetic pattern in t-AML, in which the prognostic role of FLT3-ITD can be dependent on the concomitant cytogenetic aberrations.33 Also, all FLT3-ITD mutations, regardless of allelic ratio were included, which may impaired the prognostic power of our analysis. As in de novo AML, the survival of patients with NPM1mut/FLT3wt was better than that of patients with any other combination of these mutations.34,35 Importantly, survival of t-AML patients with NPM1mut/FLT3wt, defined as favorable risk in ELN, did not differ significantly from that of NPM1mut/FLT3wt de novo patients in a multivariable analysis. This suggests that, similarly to cytogenetically favorablerisk AML, t-AML patients with NPM1mut/FLT3wt could be approached with the same treatment strategy as used for de novo NPM1mut/FLT3wt AML patients.
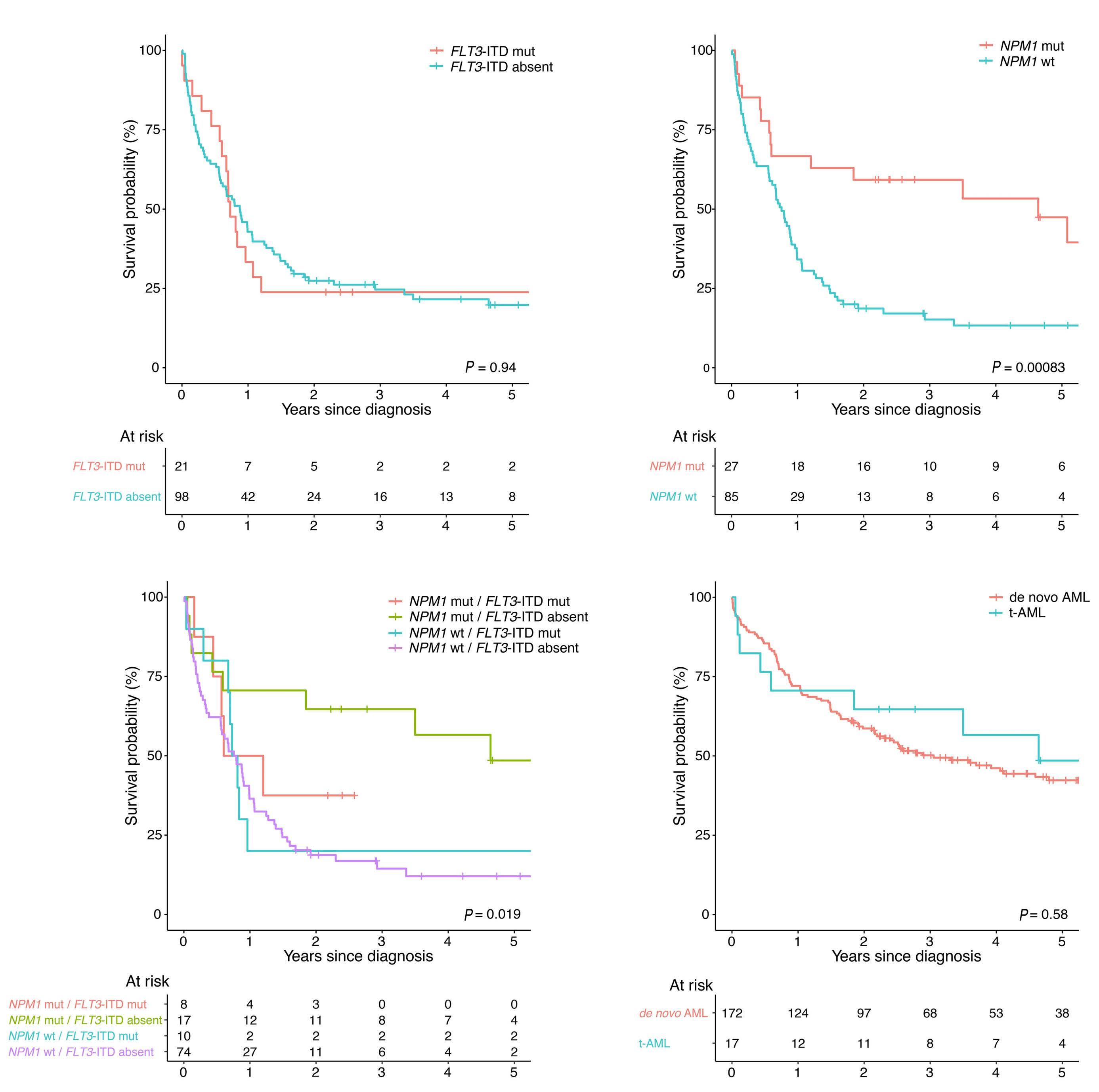
The reason for the worse outcome of intermediate- and poor-risk patients with t-AML, compared to their de novo counterparts, also when adjusting for other factors such as cytogenetics, age and performance status, is likely multifactorial. A higher frequency of unfavorable mutations and/or an enrichment of mutations originating from clonal hematopoiesis in patients with t-AML may contribute.9,13,36
As data in this study emerge from the clinical routine, which did not include mutational screening until very late during the study period, mutational data are lacking in this study. Therefore, there is a need for future studies that elucidate the role of t-AML in the setting of full mutation information. Standard induction treatment for AML remained mainly unchanged during the study period. More recently, new therapeutic options have become available, and these could potentially change the course of the disease in the future. For instance, prolonged survival in AML, including t-AML, has been seen among patients treated with CPX351.37 A broader use of hypomethylating agents alone or in combination with antibodies or novel inhibitors targeting FLT3, IDH1/2 and BCL2 also has the potential to im-
References
1. Swerdlow S, Campo E, Harris N, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). Lyon: IARC, 2017.
2. Granfeldt Ostgard LS, Medeiros BC, Sengelov H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national populationbased cohort study. J Clin Oncol. 2015;33(31):3641-3649.
3. Hulegardh E, Nilsson C, Lazarevic V, et al. Characterization and prognostic features of secondary acute myeloid leukemia in a prove outcome in certain subtypes of t-AML.
In summary, this large population-based study reveals a significant increase in the incidence of t-AML over time, which points to the reality of an increasing likelihood of encounter patients with this malignancy in the clinical practice. Thus, we need more knowledge of, as well as better treatment approaches for, these patients. Given the poor survival in t-AML it is especially important to develop non-chemotherapeutic treatment approaches to malignant and inflammatory diseases, which should result in a decreased risk of t-AML. Our data support an approach in which the indication for HCT should be the same in patients with favorable-risk cytogenetics or NPM1mut/FLT3wt with respect to previous chemotherapy or radiation treatment. On the contrary, intermediate- and adverse-risk patients with t-AML have even poorer survival compared to their de novo counterparts and novel treatment approaches for these patients are highly warranted.
Disclosures
No conflicts of interest to disclose.
Contributions
CN and SL designed the study and gathered, assembled and classified all data. CN and SL performed all analyses and interpreted the data with contributions from FL, EH, HG and LM. CN and SL wrote the manuscript. All other authors contributed to the Swedish AML registry. All authors critically reviewed and approved the manuscript.
Funding
The study was supported by the Swedish Cancer Foundation, Swedish Research Council and Stockholm County Council.
Data-sharing statement
The datasets generated during and/or analyzed during the current study are not publicly available due to privacy concerns and limitations from the ethical review board but are available from the corresponding author upon reasonable request.
population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol. 2015;90(3):208-214.
4. Morton LM, Dores GM, Schonfeld SJ, et al. Association of chemotherapy for solid tumors with development of therapyrelated myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019;5(3):318-325.
5. Teepen JC, Curtis RE, Dores GM, et al. Risk of subsequent myeloid neoplasms after radiotherapy treatment for a solid cancer among adults in the United States, 2000-2014.
Leukemia. 2018;32(12):2580-2589.
6. Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102(1):43-52.
7. Kayser S, Dohner K, Krauter J, et al. The impact of therapyrelated acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. 2011;117(7):2137-2145.
8. Fianchi L, Pagano L, Piciocchi A, et al. Characteristics and outcome of therapy-related myeloid neoplasms: report from the Italian network on secondary leukemias. Am J Hematol. 2015;90(5):E80-85.
9. Bolton KL, Ptashkin RN, Gao T, et al. Oncologic therapy shapes the fitness landscape of clonal hematopoiesis. bioRxiv. 2019 Nov 20. doi: org/10.1101/848739 [preprint; not peer reviewed].
10. Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18(1):112-121.
11. Joannides M, Grimwade D. Molecular biology of therapy-related leukaemias. Clin Transl Oncol. 2010;12(1):8-14.
12. Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 2017;18(1):100-111.
13. Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552-555.
14. Andersen MK, Johansson B, Larsen SO, Pedersen-Bjergaard J. Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica. 1998;83(6):483-488.
15. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
16. Ok CY, Patel KP, Garcia-Manero G, et al. Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res. 2015;39(3):348-354.
17. Shih AH, Chung SS, Dolezal EK, et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica. 2013;98(6):908-912.
18. Morton LM, Dores GM, Tucker MA, et al. Evolving risk of therapyrelated acute myeloid leukemia following cancer chemotherapy among adults in the United States, 1975-2008. Blood. 2013;121(15):2996-3004.
19. Guru Murthy GS, Hamadani M, Dhakal B, Hari P, Atallah E. Incidence and survival of therapy related myeloid neoplasm in United States. Leuk Res. 2018;71:95-99.
20. Howlader N, Noone A, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2016. 2019 Available from: https://seer.cancer.gov/csr/1975_2016/
21. Juliusson G, Lazarevic V, Horstedt AS, Hagberg O, Hoglund M, Swedish Acute Leukemia Registry Group. Acute myeloid leukemia in the real world: why population-based registries are needed. Blood. 2012;119(17):3890-3899.
22. The Swedish AML group. Swedish national AML care program for adult patients. Akut myeloisk leukemi (AML) - Nationellt vårdprogram 2019.
23. Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453-474.
24. R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, 2019.
25. Arnold M, Rutherford MJ, Bardot A, et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995-2014 (ICBP SURVMARK-2): a population-based study. Lancet Oncol. 2019;20(11):1493-1505.
26. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975-2014, featuring survival. J Natl Cancer Inst. 2017;109(9):djx030.
27. Nagel G, Weber D, Fromm E, et al. Epidemiological, genetic, and clinical characterization by age of newly diagnosed acute myeloid leukemia based on an academic population-based registry study (AMLSG BiO). Ann Hematol. 2017;96(12):1993-2003.
28. Østgård LS, Nørgaard JM, Sengeløv H, et al. Comorbidity and performance status in acute myeloid leukemia patients: a nation-wide population-based cohort study. Leukemia. 2015;29(3):548-555.
29. Schoch C, Kern W, Schnittger S, Hiddemann W, Haferlach T. Karyotype is an independent prognostic parameter in therapyrelated acute myeloid leukemia (t-AML): an analysis of 93 patients with t-AML in comparison to 1091 patients with de novo AML. Leukemia. 2004;18(1):120-125.
30. Kern W, Haferlach T, Schnittger S, Hiddemann W, Schoch C. Prognosis in therapy-related acute myeloid leukemia and impact of karyotype. J Clin Oncol. 2004;22(12):2510-2511.
31. Aldoss I, Pullarkat V. Therapy-related acute myeloid leukemia with favorable cytogenetics: still favorable? Leuk Res. 2012;36(12):1547-1551.
32. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
33. Tao S, Wang C, Chen Y, et al. Prognosis and outcome of patients with acute myeloid leukemia based on FLT3-ITD mutation with or without additional abnormal cytogenetics. Oncol Lett. 2019;18(6):6766-6774.
34. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752-1759.
35. Thiede C, Koch S, Creutzig E, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107(10):4011-4020.
36. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367-1376.
37. Kolitz JE, Strickland SA, Cortes JE, et al. Consolidation outcomes in CPX-351 versus cytarabine/daunorubicin-treated older patients with high-risk/secondary acute myeloid leukemia. Leuk Lymphoma. 2020;61(3):631-640.
Cell Transplantation
Correspondence: M. Hudspeth hudspeth@musc.edu
Received: May 30, 2022.
1Medical University of South Carolina Children’s Hospital/Hollings Cancer Center, Charleston, SC, USA; 2Sarah Cannon Transplant and Cellular Therapy Program, Mountain View Hospital, Las Vegas, NV, USA; 3University Hospital for Internal Medicine V, Hematology & Oncology, Medical University, Innsbruck, Austria; 4Hospital Universitario Virgen del Rocío, Instituto de Biomedicina de Sevilla (IBIS/CISC), Universidad de Sevilla, Sevilla, Spain; 5Medizinische Klinik und Poliklinik 1, Universitätsklinikum Carl Gustav Carus an der TU Dresden, Dresden, Germany; 6University of North Carolina at Chapel Hill, Chapel Hill, NC, USA; 7Jazz Pharmaceuticals, Palo Alto, CA, USA; and 8Jazz Pharmaceuticals, Philadelphia, PA, USA and 9CHU de Lille, INSERM U1286, Infinite, Lille, France
Abstract
Accepted: December 2, 2022.
Early view: December 15, 2022.
https://doi.org/10.3324/haematol.2022.281471
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Acute graft-versus-host disease (aGvHD) is a life-threatening complication typically occurring within 100 days after allogeneic hematopoietic cell transplantation (allo-HCT). This hypothesis-generating, phase II, prospective, open-label, randomized study (clinicaltrials gov. Identifier: NCT03339297) compared defibrotide added to standard-of-care (SOC) GvHD prophylaxis (defibrotide prophylaxis arm) versus SOC alone (SOC arm) to prevent aGvHD post-transplant. This study estimated incidences of aGvHD and was not statistically powered to assess differences among treatment arms. Patients were randomized 1:1 to defibrotide prophylaxis arm (n=79; median age 57 years; range, 2-69 years) or SOC arm (n=73; median age 56 years; range, 2-72 years). Patient demographics in the two arms were similar except for conditioning regimen type (myeloablative: defibrotide, 76% vs. SOC, 61%) and stem cell source for allo-HCT (bone marrow: defibrotide, 34% vs. SOC, 26%). In the intent-to-treat primary endpoint analysis, the cumulative incidence of grade B-D aGvHD at day 100 post-transplant was 38.4% in the defibrotide prophylaxis arm versus 47.1% in the SOC arm (difference: –8.8%, 90% confidence interval [CI]: –22.5 to 4.9). The difference noted at day 100 became more pronounced in a subgroup analysis of patients who received antithymocyte globulin (defibrotide: 30.4%, SOC: 47.6%; difference: –17.2%; 90% CI: –41.8 to 7.5). Overall survival rates at day 180 post-transplant were similar between arms, as were the rates of serious treatment-emergent adverse events (defibrotide: 42%, SOC: 44%). While the observed differences in endpoints between the two arms were not substantial, these results suggest defibrotide prophylaxis may add a benefit to currently available SOC to prevent aGvHD following allo-HCT without adding significant toxicities.
Introduction
Graft-versus-host disease (GvHD), the most important life-threatening complication after allogeneic hematopoietic cell transplantation (allo-HCT), occurs when donor T cells are activated in response to major or minor histocompatibility mismatch or gene polymorphisms from the recipient, causing a cytotoxic effect in healthy tissues and organs.1,2 Acute GvHD (aGvHD) typically occurs within the first 100 days after allo-HCT.3,4 The pathophysiology of aGvHD broadly follows a three-stage process whereby tissue damage from the conditioning regimen activates the host antigen-presenting cells, which in turn activate donor
T cells to initiate GvHD. Subsequently, T-cell–induced cellular and inflammatory factors cause damage to organs,5 namely the skin, gastrointestinal tract, and liver.6 Patients who develop aGvHD exhibit a greater degree of endothelial damage and dysfunction compared to patients without this complication,7,8 as well as elevated biomarkers associated with endothelial cell damage (endothelial microparticles, E-selectin, intercellular adhesion molecule–1 [ICAM-1], and von Willebrand factor).9-12 Furthermore, factors in serum from patients with aGvHD have been shown to promote endothelial cell activation.7
Prophylactic regimens used to prevent aGvHD usually include a calcineurin inhibitor (e.g., cyclosporine A [CSA], ta- crolimus) and methotrexate or mycophenolate mofetil.2,14 Despite the use of these immunosuppressive regimens, approximately 39% to 59% of patients receiving allo-HCT develop grade B-D aGvHD.15 Antithymocyte globulin (ATG) has been shown to lower the incidence of aGvHD after allo-HCT from an unrelated or sibling donor.16-18 Furthermore, cyclophosphamide taken post-HCT (PTCy) is widely used in both haploidentical and matched unrelated donor transplants.19,20 Although PTCy is on the path to becoming the new standard-of-care (SOC) for post-HCT aGvHD prophylaxis, it has been associated with graft dysfunction and infection.21 The mechanism of action of ATG and presumably of PTCy in the prevention of aGvHD involves T-cell depletion in blood and peripheral lymphatic tissues.20,22-24 Additionally, the selective T-cell co-stimulation modulator abatacept was recently approved by the US Food and Drug Administration for aGvHD prophylaxis when in combination with a calcineurin inhibitor and methotrexate.25 However, as abatacept is also a cytotoxic Tcell immunoglobulin, its primary mechanism of action involves immune suppression.25 The effectiveness of current aGvHD prophylactic regimens remains unsatisfactory, resulting in a need for safe and more effective therapies for the prevention of aGvHD.26
Defibrotide is a polydisperse mixture of predominantly single-stranded polydeoxyribonucleotide sodium salts.27 It reduces endothelial cell activation and enhances protection and stabilization of endothelial cells through antiinflammatory and anti-adhesive mechanisms and has been shown to protect the endothelium from toxic, inflammatory, and ischemic damage.27-29 In vitro evidence suggests that defibrotide protects endothelial cells, restores the thrombotic-fibrinolytic balance, and has immunosuppressive effects by inducing synthesis of prostaglandins that inhibit T-cell proliferation.30,31 Defibrotide has also been shown to suppress heparanase gene expression, and high levels of heparanase have been postulated as a risk factor for aGvHD development.32,33 Defibrotide has been approved for the treatment of hepatic veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS),34,35 a disease characterized by endothelial cell dysfunction.
The current SOC for prophylaxis of aGvHD works to suppress the immune system through either inhibition or depletion of T-cell lymphocytes or induction of tolerance to overcome the immune response from donor T-cell recognition that induces aGvHD; however, this can attenuate the beneficial graft-versus-tumor effect and increase the risk of opportunistic infection and disease relapse.2,36,37 Due to high morbidity and mortality associated with GvHD and the limitations of current therapies, prevention of aGvHD remains an area with significant unmet need. New treatment strategies for aGvHD prophylaxis are needed to improve clinical outcomes. Defibrotide is postulated to reduce the incidence of aGvHD without an increase in opportunistic infections and relapse by (i) protecting endothelial cells in GvHD target organs from donor alloreactive T-cell infiltration and damage, (ii) not directly depleting T cells involved in the graft-versus-tumor effect, and (iii) ameliorating the inflammatory response and tissue damage associated with this immunopathological disease, reducing its incidence and severity.26
Several clinical studies have examined whether defibrotide can reduce the incidence of aGvHD.39-42 We designed the present hypothesis-generating, phase II, prospective, open-label, randomized study (clinicaltrials gov. Identifier: NCT03339297) to evaluate defibrotide added to SOC GvHD prophylaxis compared with SOC GvHD prophylaxis alone for the prevention of aGvHD following allo-HCT.
Methods
Study design and patients
Overall, 150 patients were planned for enrollment. Patients were stratified by age at screening (<17 years vs. ≥17 years), geographic region (North America vs. Europe), and use of ATG and were randomized 1:1 to defibrotide prophylaxis plus SOC (defibrotide prophylaxis arm) or SOC alone (SOC arm; Online Supplementary Figure S1). ATG type and dose were per the site SOC and varied by region. ATG use was limited to 30% of patients. Eligible patients (age ≥1 year) had to have a diagnosis of acute leukemia (in morphologic complete remission) or myelodysplastic syndrome and, after myeloablative or reduced-intensity conditioning, were scheduled for CD3+ T-cell replete peripheral blood stem cell or non-manipulated bone marrow graft transplantation from a human leukocyte antigen-matched or single-allele mismatched, unrelated donor. Key exclusion criteria were prior autologous or alloHCT and clinically significant acute bleeding within 24 hours before study treatment initiation.
Institutional Review Boards at participating centers approved the study, which was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference on Harmonization. All patients, parents, or legal guardians provided written informed consent.
Treatment
Patients in the defibrotide prophylaxis arm received defibrotide 25 mg/kg/day administered as 2-hour intravenous infusions of 6.25 mg/kg/dose every 6 hours prior to the start of conditioning therapy (1-4 doses) and continued for ≥21 days (ending no later than day 30 post-transplant). SOC GvHD immunoprophylaxis consisted of methotrexate or mycophenolate mofetil plus CSA or tacrolimus with or without ATG starting on the day of the conditioning regimen. During the study, patients who developed VOD/SOS in either treatment arm could be treated with defibrotide. Per protocol, the use of medications that increased the risk of bleeding was closely monitored, and patients on defibrotide would be discontinued from defibrotide when bleeding developed or when taking medications that increased the risk of bleeding during the study. Thromboprophylaxis with heparin was allowed throughout the study for patients in both arms (maximum of 100 U/kg/day). Patients were followed for up to 180 days after transplantation.
Endpoints and assessments
The primary endpoint was cumulative incidence of grade B-D aGvHD by day 100 post-transplant. Key secondary endpoints included cumulative incidence of grade B-D aGvHD by day 180 post-transplant, grade C-D aGvHD by days 100 and 180 post-transplant, and safety. Grading of aGvHD for assessment of the primary and applicable secondary efficacy endpoints was based on the International Bone Marrow Transplant Registry Severity Index.43 An exploratory endpoint was overall survival (OS) by day 180 post-transplant. Safety assessments included monitoring treatment-emergent adverse events, serious treatmentemergent adverse events, and treatment-related treatment-emergent adverse events. Investigators classi fied adverse events using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.
Statistical analysis
This study was designed to obtain estimates of the treatment difference of the cumulative incidence rates of aGvHD between the two treatment arms to compare the efficacy of defibrotide added to SOC (defibrotide prophylaxis arm) versus SOC alone (SOC arm). The study was for hypothesis generating and was not powered to detect minimal clinically meaningful differences between treatment arms at a significant level of 5%. A sample size of 75 patients per arm was estimated to provide a 90% confidence interval (CI): –0.28 to –0.03 for the treatment difference of the primary endpoint, assuming a cumulative incidence of 28.6% for the defibrotide prophylaxis arm and 44% for the SOC arm.41
Data were summarized by treatment arms using descriptive statistics for continuous variables, and numbers and percentages of patients for categorical variables. Computations were performed using SAS 9.4 (SAS Institute Inc., Cary, NC, USA). The primary analysis was performed on the intent-to-treat (ITT) population of all randomized patients. The safety population included all patients randomized to the defibrotide prophylaxis arm who received ≥1 dose of defibrotide and all patients randomized to the SOC arm. Analysis of the primary endpoint is detailed in the Online Supplementary Appendix.
Results
Patients
From February 21, 2018 to May 12, 2020, 152 patients participated in this study at 43 sites across 11 countries. The ITT population comprised 79 patients randomized to the defibrotide prophylaxis arm and 73 patients randomized to the SOC arm (Online Supplementary Figure S2). Five patients randomized to defibrotide prophylaxis did not undergo alloHCT and did not receive defibrotide; three patients randomized to the SOC did not undergo allo-HCT. The safety population included 74 patients in the defibrotide prophylaxis arm and 70 patients in the SOC arm. Fifty-six patients (71%) in the defibrotide prophylaxis arm and 59 patients (81%) in the SOC arm completed the study (Online Supplementary Figure S2).
The two treatment arms had similar baseline demographic characteristics (Table 1); however, the proportion of patients who received myeloablative conditioning prior to allo-HCT was higher in the defibrotide prophylaxis arm than in the SOC arm (56/74 patients [76%] vs. 43/70 patients [61%], respectively). Similarly, patients in the defibrotide prophylaxis arm more frequently received bone marrow as the source of stem cells compared to patients in the SOC arm (25/74 [34%] vs. 18/70 [26%], respectively). Among patients stratified to ATG use, baseline characteristics between the defibrotide prophylaxis (n=24) and SOC (n=21) arms followed a similar pattern as the total population (Table 2).
Treatment
The same percentage of patients (74%) in both the defibrotide prophylaxis and SOC arms received GvHD prophylaxis with methotrexate-based regimens (Online Supplementary Table S1), and 30% of patients received ATG in both study arms, per protocol. The percentage of patients who received CSA (47% vs. 47%) or tacrolimus (51% vs. 53%) was similar in the defibrotide prophylaxis and SOC arms, respectively. Median duration of exposure to defibrotide and duration of defibrotide treatment were both 25 days (range, 11-40 days) among the 74 patients who received defibrotide prophylaxis. A total of four (5%) patients in the defibrotide prophylaxis arm discontinued defibrotide treatment; three (4%) were due to adverse events and one (1%) was due to patient withdrawal. One patient (1%) in the defibrotide prophylaxis arm and four patients (6%) in the SOC arm received defibrotide for the treatment of VOD/SOS.
Cumulative incidence of acute graft-versus-host disease
Per the primary endpoint ITT analysis, where death without experiencing grade B-D aGvHD was treated as a competing risk, the cumulative incidence of grade B-D aGvHD by day 100 post-transplant in the defibrotide prophylaxis arm versus the SOC arm was 38.4% versus 47.1%, respectively (difference: –8.8%; 90% CI: –22.5 to 4.9; Figure 1A). In a planned aPercentages may not total 100 due to rounding. bIn either treatment arm. cIncludes MDS with single-lineage dysplasia, ringed sideroblasts, multilineage dysplasia, or isolated del5q and unclassifiable MDS. dIncludes AML with recurrent genetic abnormality, myelodysplasia-related changes, or not otherwise specified AML. eFive patients in the defibrotide prophylaxis arm and 3 patients in the SOC arm did not receive HCT. AML: acute myeloid leukemia; HCT: hematopoietic cell transplantation; HLA: human leukocyte antigen; MDS: myelodysplastic syndrome; SOC: standard-of-care. sensitivity analysis for the primary endpoint using disease relapse as a competing risk in addition to death, the cumulative incidence of grade B-D aGvHD by day 100 post-transplant in the defibrotide prophylaxis arm compared to the SOC arm was 37.0% versus 45.7%, respectively (difference: –8.7; 90% CI: –22.4 to 4.9; Figure 1B). Both treatment arms had similar cumulative incidences of grade B-D aGvHD by day 180 post-transplant (49.0% vs. 50.2%, respectively).
Table 1. Baseline demographic and clinical characteristics (intent-to-treat population).
In patients who received ATG, the cumulative incidence of grade B-D aGvHD by day 100 was consistent with the ITT population; however, the incidence was numerically lower in the defibrotide prophylaxis arm compared with the SOC arm. The cumulative incidence of grade B-D aGvHD by day 100 post-transplant in the defibrotide prophylaxis arm compared to the SOC arm was 30.4% versus 47.6%, respectively (difference: –17.2%; 90% CI: –41.8 to 7.5; Figure 2A). In patients who did not receive ATG, the cumulative incidence rates of grade B-D aGvHD by day 100 were similar between the defibrotide prophylaxis and SOC arms (42.0% vs. 46.9%, respectively; difference: –4.9%; 90% CI: –21.6 to 11.7; Figure 2B). Results of the cumulative incidence of grade C-D aGvHD in ATG subgroups (Figure 3; Table 3) followed a similar pattern as the grade B-D aGvHD ATG subgroup analysis but with a more pronounced lowering of the cumulative incidence of grade C-D aGvHD by day 100 post-transplant with defibrotide prophylaxis versus SOC (4.3% vs. 28.9%, respectively; difference: –24.5%; 90% CI: –42.9 to –6.2). As shown in Table 2, in the ATG use subgroup, there was a slightly higher proportion of mismatched donors in the SOC arm (24% [5/21]) vs. the defibrotide prophylaxis arm (13% [3/24]); these proportions are somewhat higher than those seen in the non-ATG groups (DP: 7%; SOC 6%).
As in the subgroup of patients who received ATG, decreases in the cumulative incidence of grade B-D aGvHD at day 100 post-transplant were noted with defibrotide prophylaxis (n=56) compared with SOC alone (n=43) in patients who received myeloablative conditioning (41.8% vs. 55.8%, respectively; difference: –14.0%; 90% CI: –30.8 to 2.9; Table 3). In patients who did not receive myeloablative conditioning, the cumulative incidence of grade B-D aGvHD by day 100 was 27.8% in the defibrotide prophylaxis arm (n=18) versus 33.3% in the SOC arm (n=27; aTwo patients in the defibrotide prophylaxis arm were randomized and stratified as receiving ATG per the interactive response technology but were verified as not receiving ATG. bPercentages may not total 100 due to rounding. cIn any treatment subgroup. dIncludes MDS with single-lineage dysplasia, ringed sideroblasts, multilineage dysplasia, or isolated del5q and unclassifiable MDS. eIncludes AML with recurrent genetic abnormality, myelodysplasia-related changes, or not otherwise specified AML. fAmong patients stratified to ATG use, 1 patient in the defibrotide prophylaxis arm did not receive HCT. Among patients stratified to no ATG use, 4 patients in the defibrotide prophylaxis arm and 3 patients in the SOC arm did not receive HCT. AML: acute myeloid leukemia; ATG: antithymocyte globulin; HCT: hematopoietic cell transplantation; HLA: human leukocyte antigen; MDS: myelodysplastic syndrome; SOC: standard-of-care.
18.0; Table 3). Similarly, in patients who received bone marrow as the source of stem cells, decreases in the cumulative incidence of grade B-D and grade C-D aGvHD at day 100 post-transplant were noted with defibrotide prophylaxis (n=25) compared with SOC alone (n=18; Table 3).
Overall survival
OS rates were similar in the defibrotide prophylaxis and SOC arms by day 180 post-transplant (86.0% vs. 86.9%, respectively; Figure 4).
Safety
All patients experienced ≥1 treatment-emergent adverse event (Table 4). Fewer patients in the defibrotide prophylaxis arm experienced bleeding events compared to patients in the SOC arm (34% vs. 41%, respectively). Nausea (78% vs 70%), diarrhea (65% vs. 76%), stomatitis (57% vs. 51%), and vomiting (53% vs. 54%) were the most common treatmentemergent adverse events reported in patients in both the defibrotide prophylaxis and SOC arms, respectively. In the defibrotide prophylaxis arm, 12 patients (16%) had treatment-related adverse events (Online Supplementary Table S2). No patients discontinued defibrotide due to treatmentrelated treatment-emergent adverse events.
Serious treatment-emergent adverse events were reported in 42% of patients in the defibrotide prophylaxis arm and 44% of patients in the SOC arm; none of the serious events were deemed to be related to defibrotide. Five patients in the defibrotide prophylaxis arm and three patients in the SOC arm had treatment-emergent adverse events leading to death. The events leading to death were aGvHD (n=2), bacterial sepsis (n=1), respiratory failure (n=1), and VOD/SOS (n=1) in the defibrotide prophylaxis arm and multiple organ failure (n=1), lung disorder (n=1), and shock (n=1) in the SOC arm. None of these events were related to defibrotide.
Discussion
This hypothesis-generating, phase II, prospective, openlabel, randomized study is the first multi-national prevention study to use a novel approach directed towards endothelial injury. Here, defibrotide plus SOC GvHD pro- phylaxis versus SOC alone was evaluated for the prevention of aGvHD after allo-HCT in 152 patients with acute leukemia or myelodysplastic syndrome. Patient baseline demographic and clinical characteristics were mostly similar between the two arms of the study and were consistent with those of a population at risk for aGvHD. There were differences between treatment arms in the type of conditioning regimen (myeloablative: defibrotide, 76% vs SOC, 61%) and source of stem cells (bone marrow: defibrotide, 34% vs. SOC, 26%). While the data from this study many only suggest a modest treatment effect of defibrotide, the findings add to the relatively small body of literature of randomized assessments for GvHD prevention.
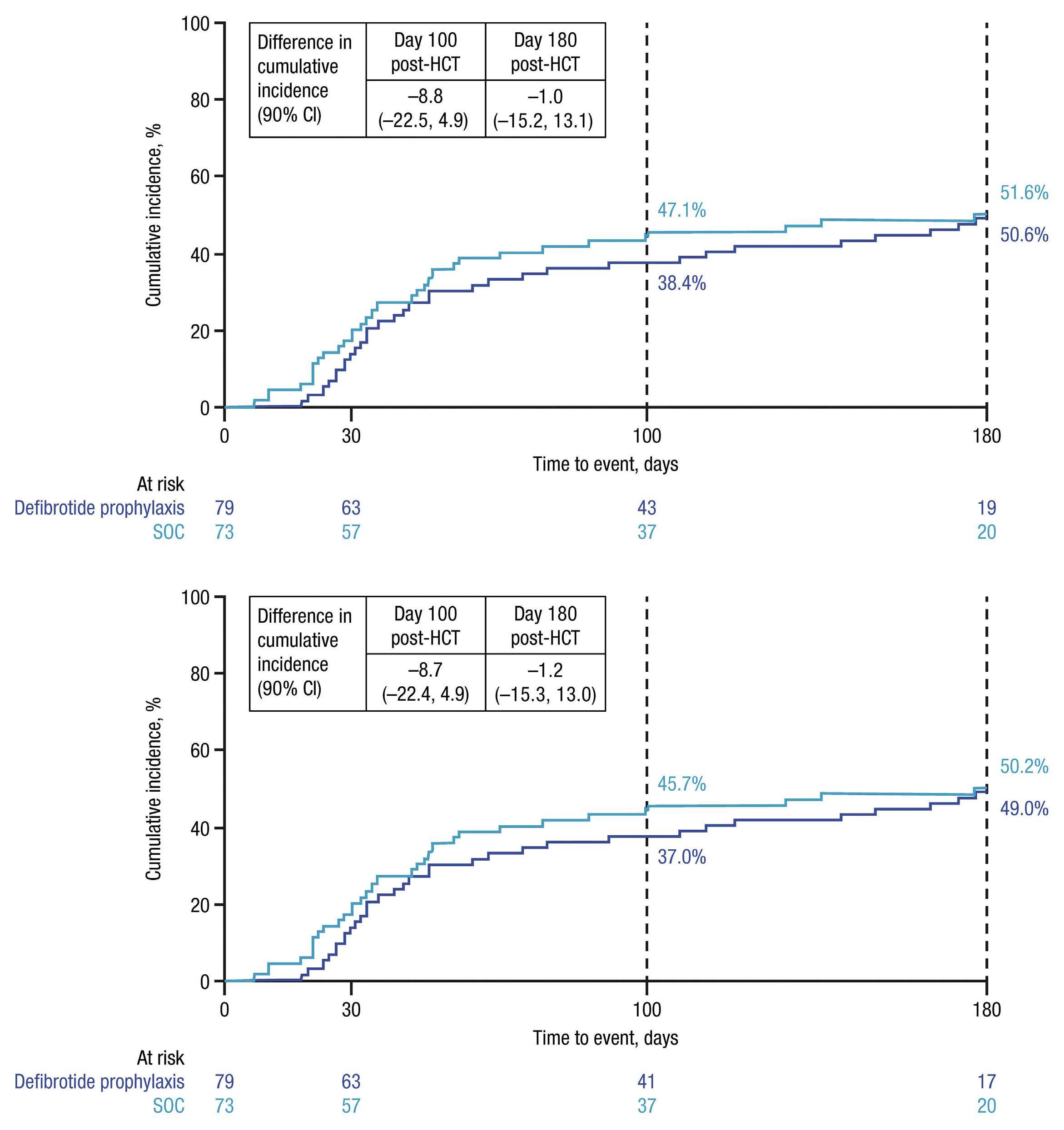
ITT analysis of the primary endpoint revealed that the cumulative incidence of grade B-D aGvHD by day 100 posttransplant was numerically lower in the defibrotide prophylaxis arm (38.4%) compared with the SOC arm (47.1%). By day 180 post-transplant, patients in the two treatment arms had similar cumulative incidences of grade B-D aGvHD. Similar results were reported in a study by Corbacioglu et al.41,42 in which patients who received defibrotide prophylaxis for VOD/SOS had a lower incidence of aGvHD by day 100 post-transplant versus control (no defibrotide; 47% vs. 65%, respectively; P=0.0046), and the inci- dence of chronic GvHD by day 180 did not differ between study arms (defibrotide prophylaxis, 9%; control, 10%; P=0.8022). The absence of a noted effect of defibrotide on the incidence of chronic GvHD by day 180 may be explained by the different pathophysiologies of acute and chronic forms of this disease. Another study by Strouse et al.44 found notable differences in the cumulative incidence of grade B-D acute GvHD at day 100 post-HCT in patients who received defibrotide versus those who did not (23.1% vs. 37.7%; difference, –14.6; 95% CI: –33.1 to 3.9]). Furthermore, a study by use of defibrotide prior to transplantation and concurrently with the conditioning regimen may decrease the incidence of aGvHD, and a separate study by Chalandon et al.40 indicated that defibrotide prophylaxis significantly reduced 1-year cumulative incidence of aGvHD. Interestingly, a retrospective study by Tilmont et al.45 showed no protective effect of defibrotide on the development or severity of aGvHD versus control (no defibrotide) in patients undergoing allo-HCT. However, in contrast to the current study, this was a retrospective observational study that included a small number of patients who received defibrotide, the majority of whom received defibrotide for the treatment of VOD/SOS, and only a small number who received defibrotide as prophylaxis. Furthermore, more patients in the defibrotide group had progressive disease, which may have contributed to poorer outcomes.
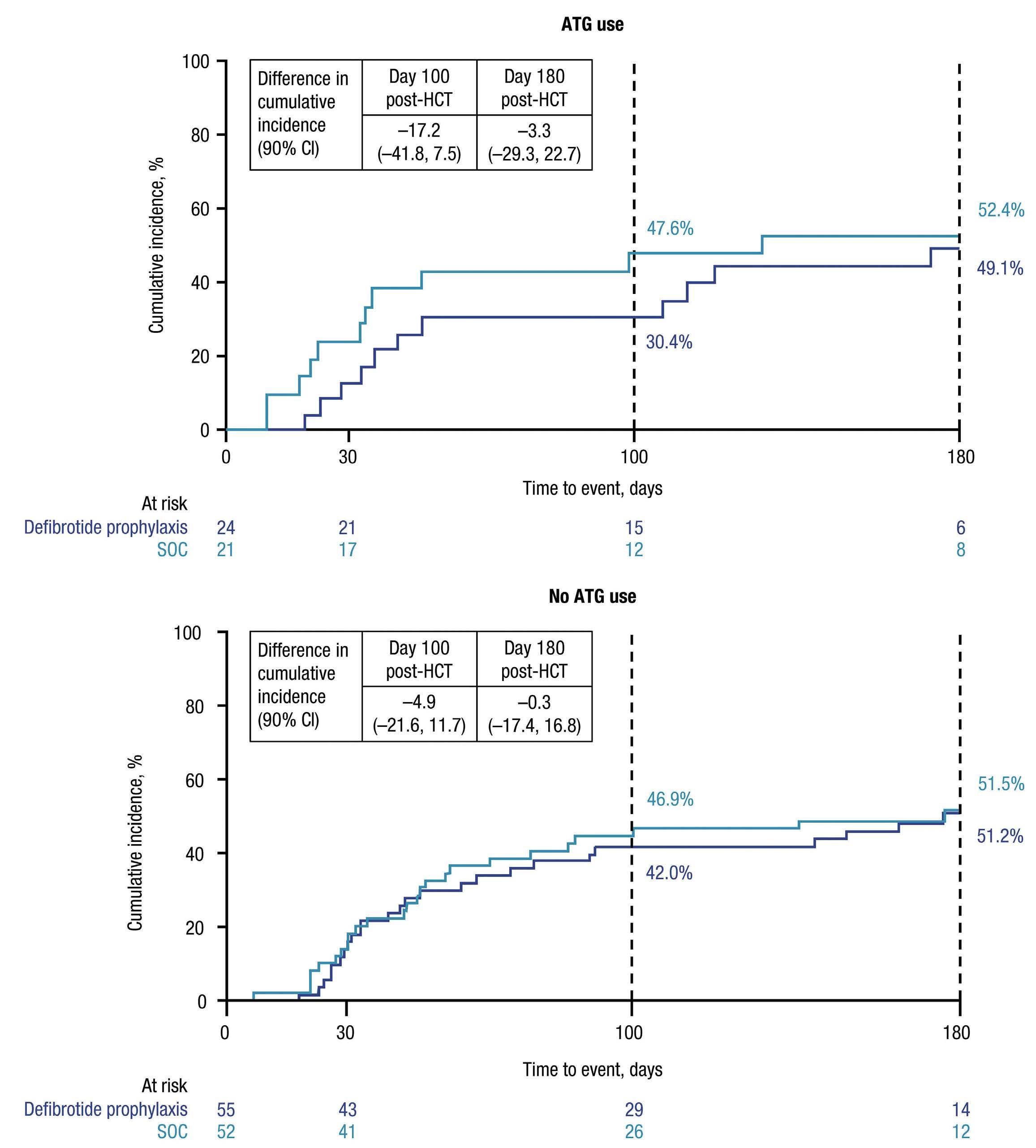
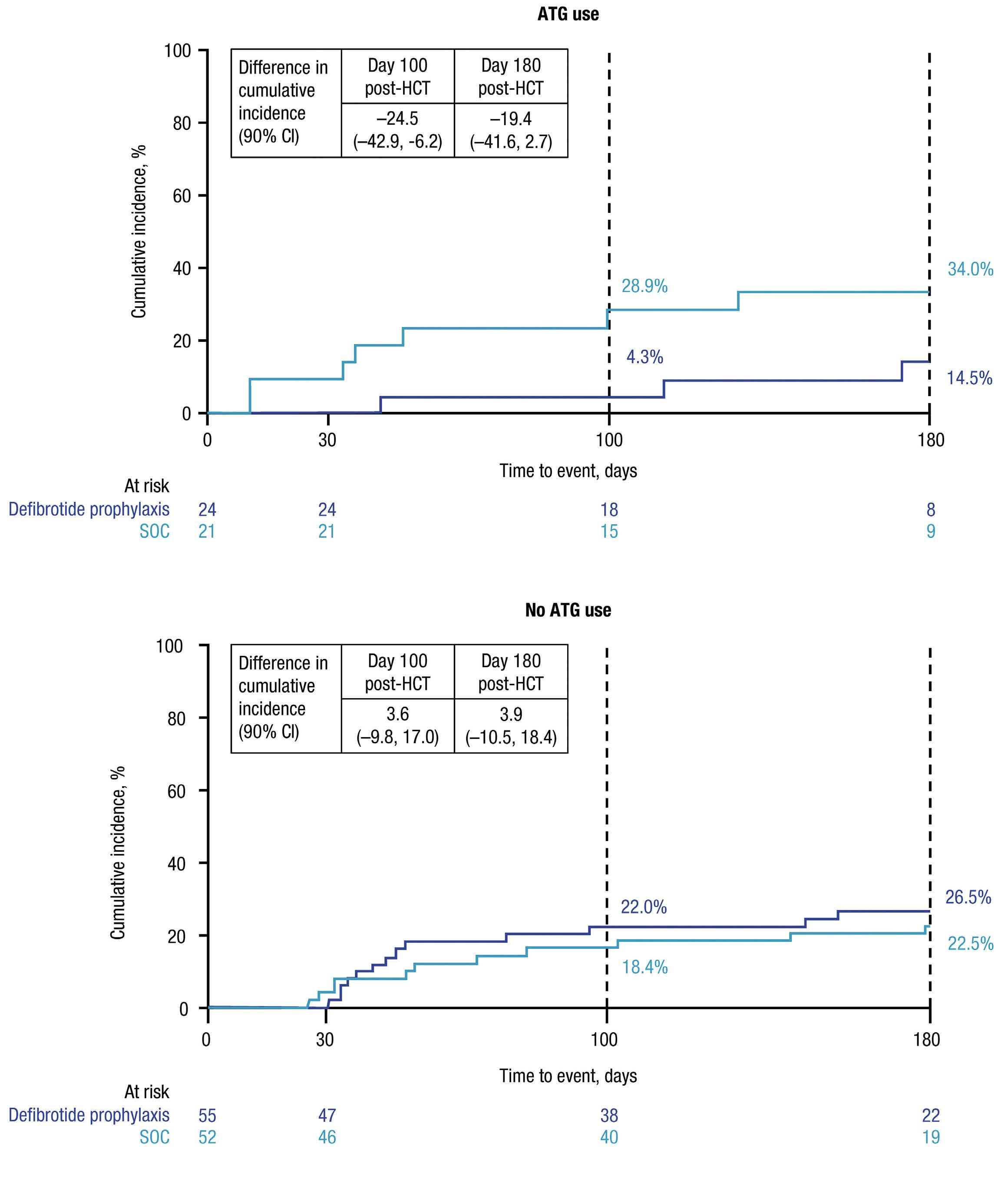
In our study, the numerical difference noted by day 100 post-transplant in the ITT population became more pronounced in subgroup analyses of patients stratified by ATG use. T-cell depletion with ATG in addition to standard
GvHD prophylaxis has been shown to significantly reduce the occurrence and severity of GvHD in patients undergoing allo-HCT; ATG is also associated with impaired immune reconstitution and increased risk of infections.46,47 In patients receiving ATG in the current study, the cumulative incidence of the more severe grade C-D aGvHD by day 100 was lower with defibrotide prophylaxis compared to SOC alone; this difference was maintained through day 180 post-transplant. There was a slightly higher proportion of mismatched donors with grade C-D aGvHD in the SOC arm (24%) versus the defibrotide prophylaxis arm (13%). Although the small patient numbers in each group precludes the ability to draw solid conclusions, this could have led to the somewhat higher incidence of grade C-D aGvHD in the ATG SOC group. These results are consistent with those of Corbacioglu et al., 41 in which prophylaxis with defibrotide significantly reduced the occurrence and severity of aGvHD versus control; adjusting for ATG as a covariate confirmed the significant effects of defibrotide (adjusted risk difference for aGvHD grade B-D: –0.1470; 95% CI: –0.2618 to –0.0322; P=0.0121).42 Furthermore, the authors noted that defibrotide did not seem to interfere with the graft- versus-leukemia effect.42 In the current study, the two treatment arms had similar OS by day 180 post-transplant.
The effect of defibrotide on the cumulative incidence of aGvHD also appeared more pronounced in patients who had received bone marrow as the source of the stem cells. How- ever, other studies have shown no difference in the incidence of aGvHD with these sources of progenitor cells.48,49 Similarly, in patients who received myeloablative conditioning, the cumulative incidence of aGvHD was also lower in the defibrotide prophylaxis arm versus the SOC arm. HCT recipients are exposed to insults that can stem from stressors such as conditioning regimen, engraftment, and infections that can cause endothelial cell activation and direct endothelial damage.50 Endothelial cells may be an important target for prophylaxis and therapeutic intervention for complications like GvHD, especially due to the role of endothelium in the pathophysiology of the condition. Despite the potential higher risk of developing aGvHD associated with myeloablative conditioning,15 defibrotide prophylaxis showed some benefit, which we hypothesize could be due to the known protective effect of defibrotide on the acute endothelial damage inflicted on these patients during conditioning.51 We speculate that the more intense the conditioning, the higher the endothelial damage. Preclinical studies suggest that defibrotide downregulates expression of key endothelial adhesion molecules (e.g., E-selectin, vascular cell adhesion molecule–1) involved in trafficking alloreactive immune cells to aGvHD target tissues.26 In a mouse model of allo-HCT, defibrotide prophylaxis prevented T cell and neutrophil tissue infiltration and aGvHD-associated tissue damage, resulting in reduced incidence of aGvHD and significantly improved survival versus untreated controls.26 Additionally, defibrotide may be acting synergis- aGvHD: acute graft-versus-host disease; SOC: standard-of-care. aIncidence was based on the number of patients, not the number of events. Percentages were calculated using the number of patients in each arm from the safety population as the denominator. bCoding was based on MedDRA version 21.1. cIn either treatment arm. tically with ATG’s polyclonal nature to produce a better response in patients who had received ATG. In support of a synergistic effect of defibrotide with other immunosuppressive agents, results from a preliminary study suggest that defibrotide prophylaxis combined with ATG, post-transplant cyclophosphamide, and CSA may be an effective strategy for preventing aGvHD.52 Furthermore, the role of cell subsets other than T cells, such as endothelial cells, in the pathophysiology of GvHD might be more pronounced in the absence of T lymphocytes, as it occurs with natural killer cell and killer Ig–like receptor disparities in the haploidentical transplant setting.53
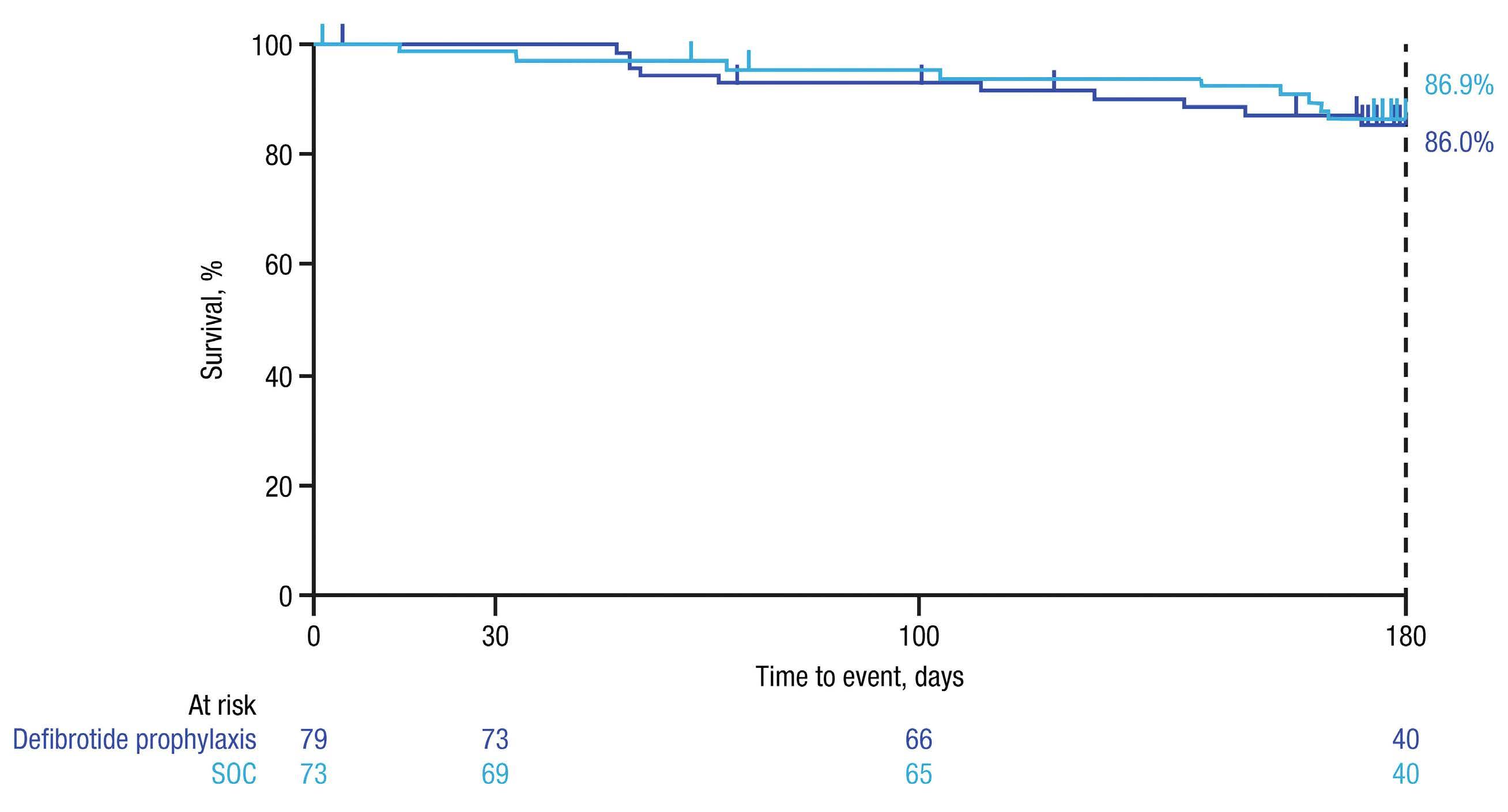
The potential benefits of defibrotide in lowering the incidence of aGvHD, most commonly occurring in the first 100 days following HCT, may reflect defibrotide’s mechanism of action, especially its anti-inflammatory and endothelial protective properties, along with the suppression of heparanase gene expression. Additional studies are needed to further evaluate the effect of defibrotide in preventing aGvHD. Safety results in this study were consistent with the safety profile of defibrotide reported in other randomized studies.41,54 Importantly, there was no increased incidence of bleeding events with defibrotide prophylaxis compared to SOC, and there were no defibrotide-related serious treatment-emergent adverse events or deaths.
The main limitation of this study is the small sample size, which offered a low statistical power to detect differences between arms, particularly for the ATG and myeloablative conditioning subgroups. The study was intended to be hypothesis generating, with a goal of providing estimates of the cumulative incidence of aGvHD for defibrotide prophylaxis compared with SOC. The greater reductions in the incidence and severity of aGvHD with defibrotide prophylaxis versus SOC reported by Corbacioglu et al.41 in the phase III VOD/SOS prevention study may have been the result of a larger number of patients. Furthermore, the study by Corbacioglu et al.41 was performed in pediatric patients while this study included only a few pediatric patients. Another limitation of our study is the potential variability in SOC among patients, given that it was primarily defined by institutional guidelines that may vary among sites and regions. ATG type and dose were not specified in the protocol to be collected in the trial but rather were administered per the site SOC. In addition, although this study included many global HCT centers to include as many diverse populations as possible, the trial ended up with limited enrollment of minority populations and pediatric patients by nature. In order to extend the relevance of our findings to a broader patient population, future studies should have more stringent management of enrollment to ensure patient diversity. While not conclusive, the results of our study suggest that there may be a benefit to addition of defibrotide prophylaxis to SOC for the prevention of aGvHD after allo-HCT; however, further work is needed in the context of recently adopted therapeutic approaches. Future studies are needed to determine which subgroups of patients might derive the most clinical benefit from defibrotide prophylaxis added to standard GvHD prophylaxis.
Disclosures
MH has participated on a Data Safety Monitoring Board or Advisory Board for Jazz Pharmaceuticals , Mesoblast, and Novartis. SM has received payment for speaker bureaus from Incyte Pharmaceuticals. JAP-S has received the following from Novartis, Jazz Pharmaceuticals, Janssen, Gilead, BMS, Amgen, Takeda, MSD, Alexion, and Abvvie: grants or contracts; payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events; payment for expert testimony; and support for attending meetings and/or travel. FS has served on advisory boards and received honoraria from Jazz Pharmaceuticals, and has received support for attending meetings and/or travel from Jazz Pharmaceuticals . MR is a member of a Data Safety Monitoring Committee for Gamida Cell, is chair of the IT committee for the American Society of Transplantation and Cellular Therapy, is an employee of IQVIA Biotech, and her institution has receiv ed research funding and grants/contracts from Atara Biotherapeutics and Jazz Pharmaceuticals. WW, PZ, and SA are employees of Jazz Pharmaceuticals and hold stock and/or stock options in Jazz Pharmaceuticals. IY-A has received grants or contracts and consulting fees from Jazz Pharmaceuticals. DN has no other conflicts of interest to disclose. All authors received medical writing and editorial assistance for the development of this manuscript that was funded by Jazz Pharmaceuticals.
Contributions
MH, SM, MR, WW, SA, and IY-A performed the research, participated in the acquistion, analysis, or interpretation of the data, and critically revised the manuscript; DN, JAP-S, FS, and PZ participated in the acquisition, analysis, or interpretation of the data and critically revised the manuscript.
Acknowledgments
The authors would like to thank all the study participants and their families and caregiv ers. Medical writing and editorial assistance were provided by Monica Nicosia, PhD, and Heather Nyce, PhD, CMPP™, of Lumanity Scientific Inc ., and were financially supported by Jazz Pharmaceuticals.
Funding
The study was funded by Jazz Pharmaceuticals , Inc. (Palo Alto, CA, USA).
Data-sharing statement
All relevant data are provided within the manuscript and supporting files.
References
1. Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007;7(5):340-352.
2. Zeiser R, Blazar BR. Acute graft-versus-host disease - biologic process, prevention, and therapy. N Engl J Med. 2017;377(22):2167-2179.
3. Nassereddine S, Rafei H, Elbahesh E, Tabbara I. Acute graft versus host disease: a comprehensive review. Anticancer Res. 2017;37(4):1547-1555.
4. Ferrara JLM, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550-1561.
5. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT-related major complications. Front Immunol. 2017;8:79.
6. Harris AC, Ferrara JL, Levine JE. Advances in predicting acute GVHD. Br J Haematol. 2013;160(3):288-302.
7. Mir E, Palomo M, Rovira M, et al. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant. 2017;52(9):1317-1325.
8. Cordes S, Mokhtari Z, Bartosova M, et al. Endothelial damage and dysfunction in acute graft-versus-host disease. Haematologica. 2021;106(8):2147-2160.
9. Palomo M, Diaz-Ricart M, Carbo C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transplant. 2010;16(7):985-993.
10. Pihusch V, Rank A, Steber R, et al. Endothelial cell-derived microparticles in allogeneic hematopoietic stem cell recipients. Transplantation. 2006;81(10):1405-1409.
11. Matsuda Y, Hara J, Osugi Y, et al. Serum levels of soluble adhesion molecules in stem cell transplantation-related complications. Bone Marrow Transplant. 2001;27(9):977-982.
12. Biedermann BC, Tsakiris DA, Gregor M, Pober JS, Gratwohl A. Combining altered levels of effector transcripts in circulating T cells with a marker of endothelial injury is specific for active graft-versus-host disease. Bone Marrow Transplant. 2003;32(11):1077-1084.
13. Dietrich S, Falk CS, Benner A, et al. Endothelial vulnerability and endothelial damage are associated with risk of graft-versushost disease and response to steroid treatment. Biol Blood Marrow Transplant. 2013;19(1):22-27.
14. Gooptu M, Antin JH. GVHD prophylaxis 2020. Front Immunol. 2021;12:605726.
15. Jagasia M, Arora M, Flowers ME, et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119(1):296-307.
16. Finke J, Bethge WA, Schmoor C, et al. Standard graft-versushost disease prophylaxis with or without anti-T-cell globulin in haematopoietic cell transplantation from matched unrelated donors: a randomised, open-label, multicentre phase 3 trial. Lancet Oncol. 2009;10(9):855-864.
17. Brissot E, Labopin M, Moiseev I, et al. Post-transplant cyclophosphamide versus antithymocyte globulin in patients with acute myeloid leukemia in first complete remission undergoing allogeneic stem cell transplantation from 10/10 HLAmatched unrelated donors. J Hematol Oncol. 2020;13(1):87.
18. Bonifazi F, Rubio MT, Bacigalupo A, et al. Rabbit ATG/ATLG in preventing graft-versus-host disease after allogeneic stem cell transplantation: consensus-based recommendations by an international expert panel. Bone Marrow Transplant. 2020;55(6):1093-1102.
19. Ciurea SO, Zhang MJ, Bacigalupo AA, et al. Haploidentical transplant with posttransplant cyclophosphamide vs matched unrelated donor transplant for acute myeloid leukemia. Blood. 2015;126(8):1033-1040.
20. Bolaños-Meade J, Reshef R, Fraser R, et al. Three prophylaxis regimens (tacrolimus, mycophenolate mofetil, and cyclophosphamide; tacrolimus, methotrexate, and bortezomib; or tacrolimus, methotrexate, and maraviroc) versus tacrolimus and methotrexate for prevention of graft-versus-host disease with haemopoietic cell transplantation with reduced-intensity conditioning: a randomised phase 2 trial with a non-randomised contemporaneous control group (BMT CTN 1203). Lancet Haematol. 2019;6(3):e132-e143.
21. Irene GC, Albert E, Anna BV, et al. Patterns of infection and infectious-related mortality in patients receiving posttransplant high dose cyclophosphamide as graft-versus-host-disease prophylaxis: impact of HLA donor matching. Bone Marrow Transplant. 2021;56(4):818-827.
22. Martin PJ, Hansen JA, Buckner CD, et al. Effects of in vitro depletion of T cells in HLA-identical allogeneic marrow grafts. Blood. 1985;66(3):664-672.
23. Choi SW, Reddy P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat Rev Clin Oncol. 2014;11(9):536-547.
24. Mohty M. Mechanisms of action of antithymocyte globulin: Tcell depletion and beyond. Leukemia. 2007;21(7):1387-1394.
25. Orencia® (abatacept) [prescribing information]. Princeton, NJ: Bristol-Myers Squibb Company; 2021. https://packageinserts.bms.com/pi/pi_orencia.pdf. Accessed 1 December 2022.
26. Garcia-Bernal D, Palomo M, Martinez CM, et al. Defibrotide inhibits donor leucocyte-endothelial interactions and protects against acute graft-versus-host disease. J Cell Mol Med. 2020;24(14):8031-8044.
27. Richardson PG, Palomo M, Kernan NA, Hildebrandt GC, Chao N, Carreras E. The importance of endothelial protection: the emerging role of defibrotide in reversing endothelial injury and its sequelae. Bone Marrow Transplant. 2021;56(12):2889-2896.
28. Bonifazi F, Barbato F, Ravaioli F, et al. Diagnosis and treatment of VOD/SOS after allogeneic hematopoietic stem cell transplantation. Front Immunol. 2020;11:489.
29. Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood. 2016;127(13):1719-1727.
30. Richardson PG, Corbacioglu S, Ho VT, et al. Drug safety evaluation of defibrotide. Expert Opin Drug Saf. 2013;12(1):123-136.
31. Ferraresso M, Rigotti P, Stepkowski SM, Chou TC, Kahan BD. Immunosuppressive effects of defibrotide. Transplantation. 1993;56(4):928-933.
32. Ostrovsky O, Shimoni A, Rand A, Vlodavsky I, Nagler A. Genetic variations in the heparanase gene (HPSE) associate with increased risk of GVHD following allogeneic stem cell transplantation: effect of discrepancy between recipients and donors. Blood. 2010;115(11):2319-2328.
33. Mitsiades CS, Rouleau C, Echart C, et al. Preclinical studies in support of defibrotide for the treatment of multiple myeloma and other neoplasias. Clin Cancer Res. 2009;15(4):1210-1221.
34. Defitelio® (defibrotide sodium) [prescribing information]. Palo Alto, CA: Jazz Pharmaceuticals, Inc.; 2016. https://pp.jazzpharma.com/pi/defitelio.en.USPI.pdf. Accessed 1
December 2022.
35. Defitelio® (defibrotide sodium) [summary of product characteristics]. Villa Guardia, Italy: Gentium SpA; 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/defitelio Accessed 1 December 2022.
36. Naserian S, Leclerc M, Shamdani S, Uzan G. Current preventions and treatments of aGVHD: from pharmacological prophylaxis to innovative therapies. Front Immunol. 2020;11:607030.
37. Jiang H, Fu D, Bidgoli A, Paczesny S. T cell subsets in graft versus host disease and graft versus tumor. Front Immunol. 2021;12:761448.
38. Martinez-Sanchez J, Hamelmann H, Palomo M, et al. Acute graft-vs.-host disease-associated endothelial activation in vitro is prevented by defibrotide. Front Immunol. 2019;10:2339.
39. Tekgunduz E, Kaya AH, Bozdag SC, et al. Does defibrotide prophylaxis decrease the risk of acute graft versus host disease following allogeneic hematopoietic cell transplantation? Transfus Apher Sci. 2016;54(1):30-34.
40. Chalandon Y, Simonetta F, Dantin C, et al. Efficient prophylaxis with defibrotide for sinusoidal obstruction syndrome (SOS) after allogeneic hematopoietic ttem cell transplantation (HSCT). Blood. 2016;128(22):2204-2204.
41. Corbacioglu S, Cesaro S, Faraci M, et al. Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet. 2012;379(9823):1301-1309.
42. Corbacioglu S, Cesaro S, Faraci M, et al. Impact of prophylaxis with defibrotide on the occurrence of acute GvHD in allogeneic HSCT. Blood. 2013;122(21):4591-4591.
43. Rowlings PA, Przepiorka D, Klein JP, et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol. 1997;97(4):855-864.
44. Strouse C, Richardson P, Prentice G, et al. Defibrotide for treatment of severe veno-occlusive disease in pediatrics and adults: an exploratory analysis using data from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. 2016;22(7):1306-1312.
45. Tilmont R, Yakoub-Agha I, Ramdane N, et al. Impact of defibrotide in the prevention of acute graft-versus-host disease following allogeneic hematopoietic cell transplantation. Ann Pharmacother. 2022;56(9):1007-1015.
46. Yang X, Li D, Xie Y. Anti-thymocyte globulin prophylaxis in patients with hematological malignancies undergoing allogeneic hematopoietic stem cell transplantation: an updated metaanalysis. Front Oncol. 2021;11:717678.
47. Kekre N, Antin JH. ATG in allogeneic stem cell transplantation: standard of care in 2017? Counterpoint. Blood Adv. 2017;1(9):573-576.
48. Stem Cell Trialists' Collaborative Group. Allogeneic peripheral blood stem-cell compared with bone marrow transplantation in the management of hematologic malignancies: an individual patient data meta-analysis of nine randomized trials. J Clin Oncol. 2005;23(22):5074-5087.
49. Anasetti C, Logan BR, Lee SJ, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med. 2012;367(16):1487-1496.
50. Hildebrandt GC, Chao N. Endothelial cell function and endothelial-related disorders following haematopoietic cell transplantation. Br J Haematol. 2020;190(4):508-519.
51. Luft T, Dreger P, Radujkovic A. Endothelial cell dysfunction: a key determinant for the outcome of allogeneic stem cell transplantation. Bone Marrow Transplant. 2021;56(10):2326-2335.
52. Akpinar S, Kayikci O, tekgunduz E. Defibrotide combined with triple therapy including posttransplant cyclophosphamide, low dose rabbit anti-t-lymphocyte globulin and cyclosporine is effective in prevention of graft versus host disease after allogeneic peripheral blood stem cell transplantation for hematologic malignancies. Transfus Apher Sci. 2022;61(1):103367.
53. Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097-2100.
54. Richardson PG, Soiffer RJ, Antin JH, et al. Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol Blood Marrow Transplant. 2010;16(7):1005-1017.







