
56 minute read
An objective assessment in newly diagnosed multiple myeloma to avoid treatment complications and strengthen therapy adherence


Correspondence: M. Engelhardt monika.engelhardt@uniklinik-freiburg.de
Received: May 30, 2022.
1Department of Medicine I Hematology and Oncology; 2Comprehensive Cancer Center Freiburg (CCCF), and 3Clinical Trials Unit, Medical Center - University of Freiburg, Faculty of Medicine, Freiburg, Germany
Abstract
Accepted: October 27, 2022.
Early view: November 3, 2022.
https://doi.org/10.3324/haematol.2022.281489
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
In heterogeneous multiple myeloma (MM) patients treatment decisions are challenging. The hypothesis was that adaptation of treatment intensity (dose reduction [DR] vs. none) according to an objective risk score (revised-myeloma comorbidity index [R-MCI]) rather than physician judgement alone may improve therapy efficacy and avoid toxicities. We performed this study in 250 consecutive MM patients who underwent a prospective fitness assessment at our center, after having received induction protocols based on physicians’ judgement. DR, serious adverse events (SAE), response, progression-free survival (PFS) and overall survival (OS) were compared in fitness (fit, intermediate-fit, frail), age (<60, ≥70 years [y]) and therapy intensity subgroups at baseline and follow-up. Fit and <60 y patients were mostly treated with full intensity, whereas frail and ≥70 y patients usually received DR. Hematological and non-hematological SAE were more frequently seen in frail versus ≥70 y patients. Dose adaptations were mainly necessary in frail patients. OS and PFS were similar in fit and intermediate-fit but significantly worse in frail patients (P=0.0245/P<0.0001), whereas in age-based subgroups, OS and PFS differences did not reach significance (P=0.1362/P=0.0569). Non-hematological SAE were another negative predictor for impaired OS and PFS (P=0.0054/P=0.0021). In the follow-up performed at a median of 11 months after the first fitness assessment, the R-MCI improved or remained stable in 90% versus deteriorated in only 10% of patients. In conclusion, separation by R-MCI/frailty-defined subgroups was superior to age-based subgroups and can be used to improve tailored treatment. Fitter patients benefit from intensive therapies, whereas frail patients bear a need for initial DR.
Introduction
Multiple myeloma (MM) is a hematological disease, which typically affects elderly patients.1 In the past decade, treatment options have substantially evolved and with inclusion of proteasome inhibitors (PI), immunomodulatory drugs (IMiDs) and antibodies (Ab)/immunotherapies into induction and relapse protocols, response rates, progression-free survival (PFS) and overall survival (OS) have improved impressively.2 Standard treatment in newly diagnosed MM (NDMM) includes triplets or quadruplets, plus - if patients are deemed fit enough - autologous stem cell transplantation (ASCT), followed by maintenance therapy.3,4 Even though patient assessment, via age,
Karnofsky Performance Status (KPS), comorbidities, patient history and examination, is performed, MM patients’ actual constitution and fitness can be over- or underestimated.5-8 Additionally, inclusion in clinical trials is especially rare in elderly patients over the age of 70 years.5,6 This suggests that more objective tools may assist physicians to find most suitable treatment regimens and to adapt dose intensity for elderly and/or MM-stricken patients.
In line, the long continuing COVID-19 pandemic imposes the need to prevent any unnecessary serious adverse events (SAE), hospitalization, time-consuming dose adjustments and therapy cessations.9-13 However, therapy decisions are often made without an objective fitness as- sessment.7,14,15 In recent years, MM-specific risk scores (e.g., International Myeloma Working Group (IMWG)-frailty score, revised-myeloma comorbidity index (R-MCI), Mayorisk score, UK Myeloma Research Alliance Risk Profile) were developed to assist in this unsolved matter.16-19
The hypothesis of this study was that adaptation of therapy intensity according to an objective risk score (via RMCI; Online Supplementary Figure S1), rather than via physician judgement alone, may improve therapy efficacy and avoid therapy toxicities and discontinuation. More studies are now testing the feasibility of MM-specific riskscores for treatment assistance in MM patients, albeit additional studies should further be performed.14,20-22 We used the R-MCI, because this constitutes a repeatedly validated MM-specific risk tool, used routinely for MM patients at our institution, that has been integrated into our electronic tumor board (TB) online system.17,23 The R-MCI contains of five weighted risk factors, namely renal and lung function, KPS, frailty and age, plus allows to include cytogenetics.17,23 The R-MCI web tool allows the immediate calculation of the weighted R-MCI, which can be likewise performed by physicians or research assistants (www.myelomacomorbidityindex.org). We here investigated the applicability of the R-MCI for future therapy decision support by performing an analysis of patients receiving first-line treatment. Main aspects of this study were to track patients’ induction treatment, comparing RMCI- versus age subgroups in terms of therapy adaptations, SAE, response, OS and PFS. Since patients’ constitution and disease burden may change during treatment, we also re-evaluated constitution, fitness and R-MCI changes in a follow-up analysis (Online Supplementary Figure S2).
Methods
Data sources and study design
We performed this exploratory study in 250 consecutive MM patients, who received induction treatment ideally continuing until intolerance or progression at our Comprehensive Cancer Center Freiburg (CCCF) and catchment area of the Black Forest from 2000 to 2018. All patients were fully documented at our CCCF. The cohort with prospectively assessed R-MCI was pooled from two prior conducted analyses,8,17 retrieving patient- and therapy-relevant data through our electronic documentation system ‘Medoc’. Of the initial 359 patients, 109 had to be excluded either due to ongoing induction at the time of assessment (n=50) or incomplete data (n=59; Online Supplementary Figure S2 ). Patients’ characteristics included the International Staging System (ISS), R-MCI and IMWG-frailty scores at baseline. Via R-MCI and IMWGfrailty scores, all patients were grouped as fit, intermedi- ate-fit or frail (Table 1; Online Supplementary Figure S1). Decisions of induction regimen, treatment intensity and dose reductions (DR) were based on physicians’ choice. Comparisons were performed for frailty- (R-MCI-fit, -intermediate-fit, vs. -frail), age- (<60, 60-69 vs. ≥70 years) and therapy-intensity (with vs . without initial DR) subgroups.
The study was performed according to the guidelines of the Declaration of Helsinki and Good Clinical Practice. All patients gave their written informed consent for institutionally initiated research studies and analyses of clinical outcome studies conforming to the institutional review board guidelines. The trial protocol was approved by the ethics committee of the University of Freiburg (EV 81/10).
Induction, dose reductions, serious adverse events, response and follow-up analysis
For each induction regimen, one lead agent was determined. Albeit any DR of any drug in combination first-line treatment could have been counted as a dose modification, this would not have allowed a less complex intensity calculation. Thus, any decrease in the lead agents’ standard dose intensity or change from triplets to doublets was defined as DR. Lead agents were defined, with regard to the severity of adverse events (AE) in the following order of priority: alkylating agents, subsequently IMiDs and PI/Ab, last anthracyclines or glucocorticoids. The standard dose was consistent with NCCN/EMN-guidelines and CCCF-chemotherapy manual.24
We used the Common Terminology Criteria (CTC) for AE version 5.0 to assess grade 3-4 hematological SAE (anemia, leukocytopenia, thrombocytopenia) and non-hematological SAE grades 3-5 (infections, renal, pulmonary, cardiac impairment).
Quality of response was assessed via IMWG-remission criteria until the end of induction.25,26
Six to 24 months after the first fitness assessment, we analyzed, if changes in remission status and patients’ constitution via a follow-up R-MCI analysis had occurred.
Statistical analysis
OS was defined as the time from start of induction to death from any cause and PFS as the time from start of induction to cancer recurrence or death from any cause. Data for patients alive at the time of the analysis were censored at the last follow-up. Probabilities of PFS and OS were estimated using Kaplan-Meier method and compared with log-rank tests. In order to avoid an immortal time bias, plots of OS and PFS of patients showing no or at least one SAE, included only those who survived at least 6 months after the start of induction treatment. Chi-squareand Mantel-Haenszel tests were utilized as appropriate in the comparisons of therapy protocols, DR, SAE, response rates in R-MCI and age groups. A P value of <0.05 was con- sidered as statistically significant. Data were analyzed with SAS 9.2 (SAS Institute, Inc, Cary, North Carolina).
Results
Patient characteristics’ and induction regimen
Among the 250 analyzed patients the median age at baseline was 62 years and 61% were males. For a cohort, where first-line treatment was mainly applied at a tertiary and referral center, patient characteristics were typical (Table 1), likewise myeloma subtypes, ISS, median bone marrow infiltration (45%) underlying AL-amyloidosis rate (6%), and cytogenetics. ISS stage 2 and 3 were most common with 66% (Table 1).
Median IMWG-frailty score and R-MCI were 1 and 4, respectively, in line with prior test and validation analyses.15,17,23,27,28 Impaired constitution via KPS ≤70% was present in 42% and moderate or severe frailty was reported in 36%.8,17,23 Renal impairment with eGFR <60 mL/min/1.73m2 was present in 40% and moderate or se- vere lung dysfunction as defined via R-MCI website in 12%, in line with previous analyses.8,17,23,29,30
VCD, VRd/RAD or other induction (Online Supplementary Figure S3) was predominantly performed with VCD and whenever possible in German study group (DSMM)-protocols (DSMM XI, XII, XIV).31,32 Induction was often initiated at the CCCF, but also at regional hospitals and private practices. Stem cell transplantation (SCT) was performed in 72% (Table 1).
Comparisons of patient- and therapy-relevant parameters in entire cohort, and in revised myeloma comorbidity index, age and dose intensity subgroups Patient and therapy parameters are summarized in Table 2 in entire cohort and in fit versus frail, younger versus older and full-dosed (no DR) and dose-reduced (initial DR) subgroups. No initial DR versus DR were performed in 59% and 41%, respectively, reflecting the complexity to treat an even fairly young MM cohort, and that initial DR was frequently performed. The number of hematological and non-hematological SAE were frequent with 157 and 123,
VCD/VRD/RAD/other*4
Place
Performed SCT/non-SCT
176/35/39 (70/14/16)
198/52 (79/21)
179/71 (72/28) which accounted for SAE per patient in 0.63 and 0.49, respectively. The median induction duration was 62 days. Best IMWG response (≥ partial response [PR]) after induction was observed in 75% in line with prior data (Table 2).31 Results of R-MCI fit and younger (<60 years) patients were similar and merely differed in used protocols and SAE (Table 2). Intermediate-fit and frail patients (combined: 71%) showed increased median ages of 66 and 74 years, respectively. VRd/RAD first-line treatment in fit, intermediate-fit and frail patients were performed in 37%, 15% and 6%, thus decreased with frailty, whereas DR substantially increased (19%, 45% and 72%, respectively; Table 2). In line, SAE per patient increased from 0.23 in fit, to 0.72 in intermediate-fit and 1.1 in frail patients for hematological SAE, and 0.23, 0.48 and 1.13 for non-hematological SAE. Median induction duration in intermediate-fit patients was similar to fit patients, basically because SCT in intermediate-fit patients remained considerable with 72%. As expected, in frail patients longer induction (6-9 cycles plus maintenance) was performed and less SCT (Table 2). Divided into fit, intermediate-fit and frail patients, the response rates were 73%, 77% and 69%, respectively, thus higher in both former than latter subgroup (Table 2).
IMWG: International Myeloma Working Group; interm.: intermediate-fit; R-MCI: revised myeloma comorbidity score; KPS: Karnofsky performance status; eGFR: estimated glomerular filtration rate; VCD: bortezomib, cyclophosphamide, dexamethasone; VRd: bortezomib, lenalidomide, dexamethasone; RAD: lenalidomide, adriamycin, dexamethasone; CCCF: Comprehensive Cancer Center Freiburg; SCT: stem cell transplant. *1Unfavorable: del17p13, t(4;14), t(14;16), t(14;20), chromosome 1 abnormalities, c-myc, del(13q14), hypodiploidy. *2Frailty8,17,23: Karnofsky Index ≤70%; Time Up/Go >10 seconds; IADL ≤4 points; subjective fitness grade E or F. *3Lung dysfunction: mild: FEV1 <80%; moderate: FEV1 ≤60% or diffusion capacity ≤61%, severe: FEV1 <50%; mild: 0/1 parameter is correct; moderate: 2 parameters are correct; severe: >2 parameters are correct. *4Others: proteasome inhibitors, antibodies, anthracyclines, glucocorticoids. *5Others: regional hospitals/private practices.
According to age subgroups, DR increased in <60, 60-69 and ≥70-year-old patients from 21% to 43% and 66%, respectively. SAE per patients increased less and seemed less predictable than in R-MCI subgroups (Table 2). Transplant-eligible patients aged 60-69 years did hardly show any differences in non-hematological SAE compared to patients ≥70 years (0.58/patient vs. 0.62/patient). SCT frequencies were typical in young in 93%, 81% in 60-69 and 32% in ≥70-year-old patients. Transplant-ineligible patients (≥70 years) were at treatment initiation in 29 of 73 (40%) >75 years old. Best responses (≥PR) in age cohorts ranged from 66-79% (Table 2).
In dose intensity subgroups, for patients without versus with DR, median age differences and R-MCI-differences were notable with 58 versus 69 years and 4 versus 5, respectively. This was in line with lesser performed SCT in the latter group (Table 2). Whereas hematological SAE/patient were similar in patients without versus with DR, these almost doubled for non-hematological SAE (0.36 versus 0.69/patient, respectively). Median induction duration was similar in both groups, while as expected, response rates (≥PR) were increased in patients without DR (Table 2).
DR: dose reduction; R-MCI: revised-myeloma comorbidity index; VCD: bortezomib, cyclophosphamide, dexamethasone; IMiDs: immunomodulatory drug; RAD: lenalidomide, adriamycin, dexamethasone; VRd: bortezomib, lenalidomide, dexamethasone; SAE: serious adverse event; SCT: stem cell transplant; CR: complete remission; vgPR: very good partial remission; PR: partial remission; SD: stable disease; PD: progressive disease. *1Lead agent prioritization was based on expected severity of adverse events; lead agents were rated: alkylating agents (mainly VCD) first, subsequently IMiDs (mainly RAD/VRd) and PI/Ab, and last anthracyclines or glucocorticoids; i.e., in VCD: cyclophosphamide was the lead agent. *2Others: proteasome inhibitors, antibodies, anthracyclines, glucocorticoids.
Serious adverse events: subgroup distribution for hematological and non-hematological serious adverse events
Distinct differences in types of hematological and nonhematological SAE are shown in Figure 1A and B, being more prevalent in frail than intermediate-fit or fit patients.
Anemia, leukocytopenia and thrombocytopenia SAE (CTC grade 3-4) occurred in 76, 56 and 25 patients, respectively. Leukocytopenia was equally prevalent in frail and intermediate-fit patients (44% and 45%, respectively), while anemia and thrombocytopenia were predominantly observed in frail patients (59% and 68%, respectively; Figure 1A). Hematological SAE appeared in patients treated with alkylating agents (VCD) in 47% and with IMiD-based protocols (VRd/RAD) in 20% (‘others’ were too few for meaningful conclusions).
Figure 1. Serious adverse events in revised-myeloma comorbidity index subgroups. (A) Hematological serious adverse events (SAE) common toxicity criteria (CTC) 3-4: distribution in entire cohort and revised-myeloma comorbidity index (R-MCI) subgroups in percentage. Leukocytopenia: P=0.0008; thrombocytopenia: P=0.0007; anemia: P<0.0001. (B) Non-hematological SAE CTC grades 3-5 in R-MCI subgroups per patient. pt.: patient. Intermed: intermediate.
Of 123 registered non-hematological SAE, 69 were attributed to infectious, 23 to renal, 18 to pulmonary and 13 to cardiac causes (Figure 1B). Comparison of different R-MCI subgroups with infectious, renal, pulmonary and cardiac SAE showed significant increases in frail as compared to fit or intermediate-fit patients, reaching significance in all except pulmonary SAE (Figure 1B).
Therapy adaptations in frailty and age subgroups
Our assessment of SAE in patients without or with initial DR is depicted in the Online Supplementary Table S1A and B. The occurrence of hematological SAE without or with DR did not show distinct differences in frailty subgroups. Therapy adaptations (DR, therapy pauses or discontinuation) after hematological SAE occurred in intermediatefit or frail patients only, both without and with performed initial DR (Online Supplementary Table S1A).
Non-hematological SAE occurred in R-MCI subgroups likewise more often in patients without DR than if performed, whereas this was less strikingly found for age subgroups (Online Supplementary Table S1B). Therapy adjustments or therapy discontinuations after non-hematological SAE increased both with frailty and age, and were more prevalent with full doses than if DR had been performed (Online Supplementary Table S1B).
Therapy discontinuation occurred in only eight of 250 (3%) patients (Online Supplementary Table S2), showing mostly advanced age and impaired R-MCI scores. Initial dose reductions had been performed in seven of eight patients. Patient constitution complications, myelomaand/or therapy-induced complications occurred in already the 1st (n=3) to 4th (n=3) induction cycle (range, 14). Retrospectively assessed therapy intensity by us (MH, ME) suggested in seven of eight patients, that these were overdosed. Thus, if on top of clinical judgement, R-MCItailored therapy had been performed - SAE and therapy cessation might have been avoided in seven of eight patients.
Serious adverse events leading to early death (<60 days after induction) or concomitant serious adverse events
SAE during induction which led to death were seen in three patients (3/250 i.e., in 1.2%). These showed cardiac complications, two of three associated with underlying AL-amyloidosis. Sixteen patients with concomitant AL-amyloidosis contributed to 28% of cardiac (n=13) and renal (n=23) SAE (CTC 3-5) of the entire cohort, thus showed a 5.6 higher risk for these complications. As expected, patients with CTC grade 3-4 leukocytopenia were more likely to acquire severe infections (Online Supplementary Table S3).
Overall survival and progression-free survival of entire cohort and in various subgroups
The median follow-up from start of induction was 65 months (range, 1-246). Detailed response in fit, intermediate-fit and frail patients are summarized in the Online Supplementary Table S4. The Kaplan-Meier curves for OS and PFS analyses are displayed in Figures 2 and 3. The estimated 3-year OS and PFS in the entire cohort were 85% (95% confidence interval [CI]: 79-89) and 53% (95% CI: 4760), respectively (Figure 2A and B). The three R-MCI subgroups in Figure 2C and D showed similar results for both fit and intermediate-fit patients. The 3-year OS in R-MCI fit, intermediate-fit and frail was 89% (95% CI: 79-94), 85% (95% CI: 77-90) and 70% (95% CI: 47-85), respectively (P=0.0688; Figure 2C). The 3-year PFS was 60% (95% CI: 47-70), 55% (95% CI: 47-63) and 21% (95% CI: 6-41), respectively (P<0.0001; Figure 2D).
Since results of fit and intermediate-fit patients were comparable and very different to frail patients, these were combined as displayed in Figure 2E and F. Of note, 3-year OS and PFS in fit/intermediate versus frail patients showed highly significant differences of 86% (95% CI: 81-90) versus 70% (95% CI: 47-85) and 57% (95% CI: 50-63) versus 21% (95% CI: 6-41), respectively (P=0.0245/P<0.0001).
Age subgroups revealed differences with best OS/PFS for <60-year-old patients, whereas 60-69 and ≥70-year subgroups revealed similar Kaplan Meier curves. Notably, 3year OS/PFS results via age displayed lesser and insignificant group distinctions (P=0.1362/P=0.0569; Figure 2G and H).
Serious adverse events and dose reduction impact on overall survival and progression-free survival
Any SAE led to impaired OS and PFS (Figure 3A and B), both for hematological (Figure 3C and D) and non-hematological SAE (Figure 3E and F), with more striking OS and PFS differences for the latter SAE. In line, DR versus no DR led to both OS and PFS differences in favor of patients with no DR, reflecting standard doses to be easier applied in fitter and younger patients and therefore accounting for these differences (Figure 3G and H).
Revised myeloma comorbidity index follow-up analysis
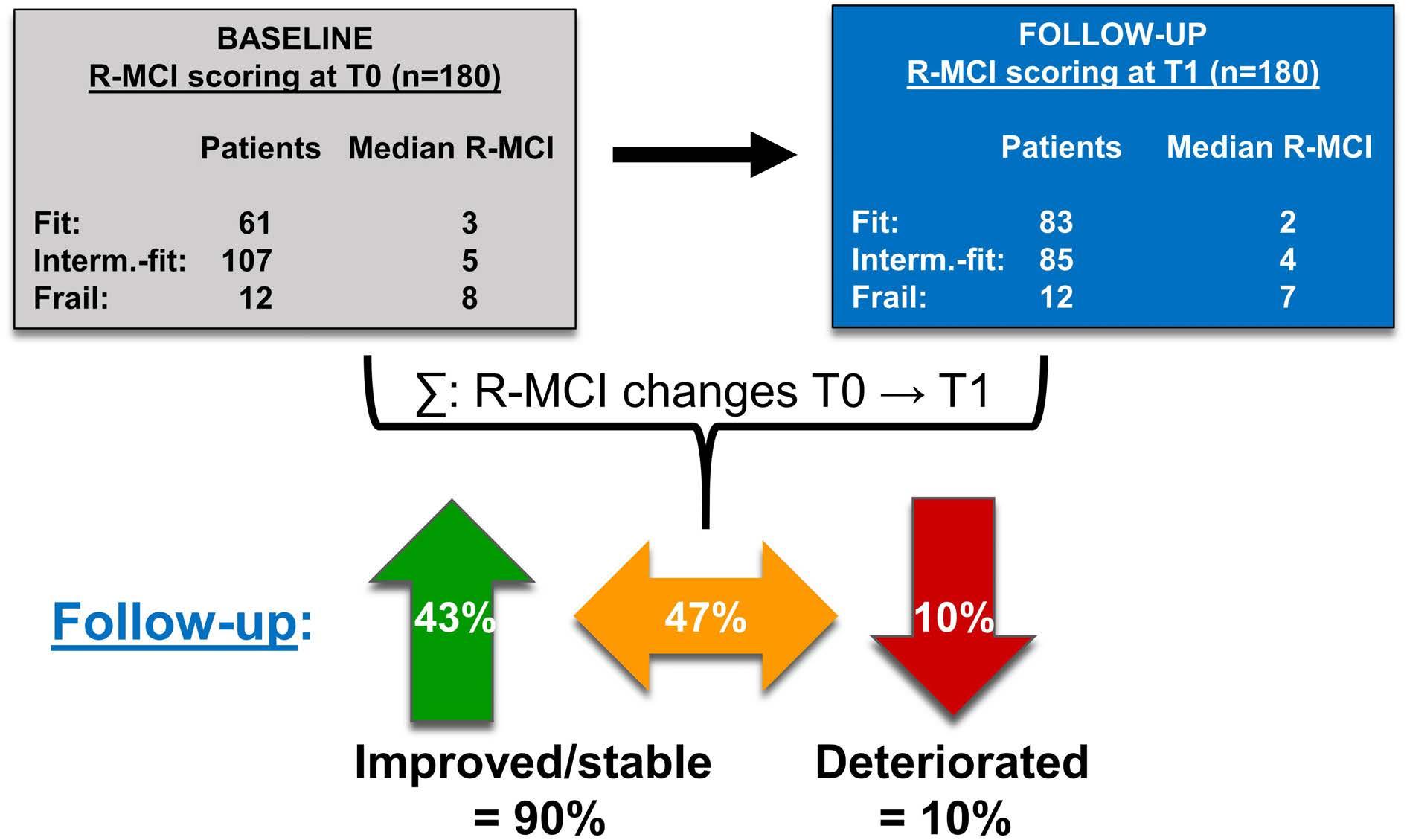
The follow-up analysis (T0 → T1) was possible in 180 patients (72%; Figure 4; Online Supplementary Figure S2). The median follow-up was 11 months (range, 6-24), in line with our previous study.8 Median R-MCI scores at T0 and T1 were both 4, reflecting intermediate-fit patients. Assessing also mean T0 versus T1 differences, the R-MCI improved from 4.3 to 3.7, respectively and accounted for a mean improvement of 0.6 points over an 11 month period (P<0.0001). Among all follow-up patients, 77 (43%) achieved a better, 84 (47%) a stable and only 19 (10%) a worse R-MCI (Figure 4). Maximum R-MCI changes were improvements by four points (R-MCI 6 → 2) and deteriorations by three points (R-MCI 4 → 7), impressively illustrating that R-MCI changes within a ~1 year follow-up can be more drastic than age shifts within this period.
Figure 3. Kaplan-Meier plots for patients without or with serious adverse events and patients without or with initial dose reduction. (A) Overall survival (OS) and (B) progression-free survival (PFS) in patients without or with any serious adverse events (SAE). (C) OS and (D) PFS in patients without or with any hematological SAE. (E) OS and (F) PFS in patients with or without any non-hematological SAE. (G) OS and (H) PFS in patients with or without DR. R-MCI: revised-myeloma comorbidity index; SAE: serious adverse events dose reduction; DR: dose reduction.
Discussion
One crucial aspect for MM patients and physicians is the individual selection of the initial (and subsequent) treatment and its intensity.7,33 Our message is that we apply evidence generated from clinical trials that rarely include old or frail patients to treat such patients without knowledge of the need for modifications of the drugs used, the dosing or schedule. Although our study population received firstline MM treatment with dose modifications according to best clinical judgment and a prospective frailty assessment was performed, the frailty assessment was not used for decision making. Thus, clinicians were blinded to this information. This allowed to assign patients into those with versus without DR as compared to comorbidity and age subgroups (Table 2). Hence, we investigated physicians’ therapy decisions in a NDMM population during induction by analyzing the course of therapy and patient outcome. Our data demonstrates that the use of the R-MCI can assist to anticipate the likelihood of SAE. Those predictions can subsequently be used to optimize induction dose intensity (Table 2; Figure 1A and B) and identify patients at risk of treatment discontinuation with the associated risk of a more unfavorable outcome (Figure 2 and 3). We found compelling differences between R-MCI subgroups regarding protocols and doses prescribed. Overall, alkylating agents were the most used leading agent, in line with VCD being a commonly applied DSMM/GMMG and EMN-study regimen.31,34 With a median start of induction in 2016, most frail patients in our cohort were treated in compliance with guidelines for dose-adjusted VCD or Vd as first-line treatment.34 The EHA-ESMO guideline from 2021 propagates nowadays to add a CD38-antibody (e.g., Dara-VMP, DaraRd) in NDMM patients ineligible for ASCT, reiterating that R-MCI- or other risk-tool-adapted triplets and quadruplets may profit from our approach described here to avoid SAE and therapy cessations.33
We were also able to show that patients at risk were mostly correctly identified and reasonable dose adaptations were made, but potential improvements remain. Moreover, we determined that fit patients had few initial DR (19%) and a low incidence of hematological or nonhematological SAE (both 0.14 per patient). In contrary, fulldosed frail patients (28%) were nine times more likely to suffer from hematological SAE and had a four times higher rate of non-hematological SAE (Online Supplementary Table S1A and B). The conjecture that some frail patients were possibly overtreated and dose-reduced fit patients undertreated is therefore plausible and has been described previously.6,17,23,34 Besides, the 3-year OS was superior in frail patients with initial DR versus in frail patients without initial DR (73% vs. 63%). Although numbers of this subgroup comparison were limited, other studies confirm this observation.11,22,35,36 Moreover, we observed that two of three events of death were associated with subclinically underlying AL-amyloidosis. These findings confirmed the need to critically evaluate induction protocols and protocol doses to avoid therapy complications and to reliably detect AL-amyloidosis.37,38 Indeed, the occurrence of any SAE was associated with a worse outcome (Figure 3A), validating prior studies.11,36,37
Since various treatment pathways continue to suggest age cut-offs (i.e., >60 or >70-years), we divided our cohort into patients aged <60, 60-69 and ≥70 years and compared them to R-MCI and therapy intensity (DR vs. no DR performed) subgroups (Table 2). In line with our and other prior analyses, R-MCI subgroups differed much more than age or dose intensity subgroups, supporting the paradigm of an objective, functional GA and risk score.5,8,39-42 Notably, ≥70 and 60-69-year-old (transplant-eligible) patients showed similar results regarding SAE, OS and PFS and thereby differed from <60-year-old patients, whereas for fitness groups, fit and intermediate-fit patients were comparable and very distinct from frail patients. Since patients ≥70 years are often excluded from clinical trials and more intensive therapies, we assessed this age group more thoroughly.6,34,42,43 Of interest, 32% of our ≥70-year-old patients received a SCT after physicians’ appraisal (Table 2). Nevertheless, 56% (n=28) of patients ≥70 years, who did not receive a SCT, were classified as either intermediatefit or fit. Thus, those patients could have received intensive treatment. Larocca et al. elegantly demonstrated the benefit of a therapy-decision approach (dose-adjusted Rd-R vs continuous Rd) via IMWG-frailty index in intermediate-fit patients in a prospective study.20 An ongoing, equally important Medical Research Councils study randomizes unaltered to adjusted treatment according to fitness results (FiTNEss study, clinicaltrails gov. Identifier: NCT03720041, PI: G.Cook), both supporting our findings. Concerning patients’ outcome, we observed a substantial OS and PFS advantage in fit and intermediate-fit versus frail patients (Figure 2A and B). In line, Facon et al. propagated a simpler approach of the IMWG-frailty score, dividing patients likewise into non-frail versus frail patients, which seems straightforward for clinical routine and clinical trials.21 An understandable request is that these riskassessments should not be time-consuming and prospectively performed.7 Both, the R-MCI and IMWGfrailty index offer online tools for their automatic calculation.
Figure 4. Changes in revised-myeloma comorbidity index scoring from start of induction (T0) to 6-24 months after first revised-myeloma comorbidity index assessment (T1) in 180 follow-up patients. R-MCI: revised-myeloma comorbidity index; Interm.-fit: intermediate-fit.
Strengths of our analysis were the meticulous examination of a NDMM patient cohort, of performed treatment, doses, SAE, PFS and OS in R-MCI, age and therapy intensity subgroups. Moreover, 72% of the initially assessed patients could be included in our follow-up analysis and their constitution and fitness, as measured via R-MCI, revealed improvement or stabilization in 90% and deterioration in only 10%. We have previously described in an even more detailed functional analysis using 12 different comorbidity scores and functional tests: the assessments more frequently and significantly changed in younger patients (<70 years) and those with good response (≥PR), suggesting a better functional reconstitution in younger and responsive than in older and less responsive myeloma patients.8 These 10% of patients, whose R-MCI deteriorated in our follow-up analysis, should be reliably identified, because fittingly chosen therapies are relevant to perform and will ideally improve patients’ quality of life (QoL).8 This was also reflected in frequencies of nonhematological SAE, which were much lower in patients with improved or stable R-MCI (both 0.39/per patient) than in those with deteriorated R-MCI (0.53/per patient). No difference in infections was observed, most likely, due to the use of detailed chemotherapy treatment plans, including strict antibiotic prophylaxis schedules therein.24 Although OS differences between these subgroups were hampered by limited patient numbers with deteriorated R-MCI, our follow-up analysis confirmed that MM treatment may indeed improve patients’ constitution. Renal impairment and/or frailty (including KPS) may recover under MM therapies, whereas in those not improving and deteriorating with QoL domains, therapeutic adjustments are important to consider.8 Additionally, our median observation period of 5.4 years was substantial, therefore our Kaplan-Meier results in all patients, in R-MCI, age, SAE-experiencing and dose-reduced patients were robust and mature. So far, there are few studies on prospective MM-specific risk tools for therapy-decision-support for NDMM and even lesser for relapsed/refractory (RR)MM patients, albeit these data and currently ongoing GIMEMA, MRC, HOVON and other studies support these endeavors.44-46 Lastly, these data impressively confirm that the R-MCI seems superior to age-based treatment pathways. Limitations of our study were the single institution approach, yet due to strict inclusion criteria regarding patients’ and therapy data, all patients included provided infinitely detailed information. Another criticism could be the heterogeneity in patients (age range, 27-92 years), with a considerable number of patients <70 years (71%), as is typical for tertiary centers in Germany (vs. more centralized institutions and countries). Since our university and catchment area-treated patient population was relatively young and the majority received ASCT, we refrained from non-ASCT versus ASCT-based subgroup analyses, but considered all patients as one group. Besides, one could criticize the use of other than VCD-induction protocols in rare subgroups. Underlying AL-amyloidosis could also been argued to be possibly excluded, which we decided against, because all patients were initially diagnosed with MM only, but determined with AL amyloidosis by us, thus initially remaining undetected.38 Lastly, we did not analyze the event-free survival as shown in prior studies,20 as we focused on OS/PFS. The former will be part of another upcoming study at our institution.
In conclusion, our results demonstrate the higher frequency of SAE, higher discontinuation rates and early mortality in frail patients, supporting MM patients' need for individualized induction and relapse protocols.17,23,46,47 Full-dose intensity for fit and reduced doses for frail patients appears pertinent, whereas intermediate-fi t patients need continuing consideration. The precise fitness assessment in MM, similar to other hematological malignancies, seems relevant to achieve favorable treatment results, less DR, SAE, as few unscheduled re-hospitaliza- tions and preserved QoL.7,12,42,48,49 The latter demonstrated itself to be possible even after intensive regimens, after allogeneic transplantation or quadruplet RRMM treatment.8,17,23,27,28 The implementation of functional assessments in myeloma TB may also support physicians in treatment decisions, since this adds an objective assessment of patients’ individual constitution and possible treatment endurance. Future studies are needed to evaluate the benefits of a functionally adapted treatment approach versus ´treatment as usual´. Prospective studies using the R-MCI in TB for therapeutic decision support are in process at our CCCF.
Disclosures
No conflicts of interest to disclose.
Contributions
MH, ME, and all other authors performed the analysis. MH,
References
1. Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, riskstratification and management. Am J Hematol. 2020;95(5):548-567.
2. Kumar SK, Rajkumar V, Kyle RA, et al. Multiple myeloma. Nat Rev Dis Primer. 2017;3:17046.
3. Mateos M-V, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528.
4. Dimopoulos MA, Jakubowiak AJ, McCarthy PL, et al. Developments in continuous therapy and maintenance treatment approaches for patients with newly diagnosed multiple myeloma. Blood Cancer J. 2020;10(2):17.
5. Soto-Perez-de-Celis E, Li D, Yuan Y, Lau YM, Hurria A. Functional versus chronological age: geriatric assessments to guide decision making in older patients with cancer. Lancet Oncol. 2018;19(6):e305-e316.
6. Fakhri B, Fiala MA, Tuchman SA, Wildes TM. Undertreatment of older patients with newly diagnosed multiple myeloma in the era of novel therapies. Clin Lymphoma Myeloma Leuk. 2018;18(3):219-224.
7. Goede V, Neuendorff NR, Schulz R-J, Hormigo A-I, MartinezPeromingo FJ, Cordoba R. Frailty assessment in the care of older people with haematological malignancies. Lancet Healthy Longev. 2021;2(11):e736-e745.
8. Scheubeck S, Ihorst G, Schoeller K, et al. Comparison of the prognostic significance of 5 comorbidity scores and 12 functional tests in a prospective multiple myeloma patient cohort. Cancer. 2021;127(18):3422-3436.
9. Voorhees PM, Kaufman JL, Laubach J, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplanteligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136(8):936-945.
10. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115.
11. Bringhen S, Mateos MV, Zweegman S, et al. Age and organ damage correlate with poor survival in myeloma patients: meta-analysis of 1435 individual patient data from 4
GI, HR and ME analyzed results. MH, GI and ME prepared tables and figures. ME, RW, GI designed the research and MH and ME wrote the paper. All authors approved and carefully revised the paper.
Acknowledgments
The authors thank DSMM, GMMG, EMN and IMWG experts for their support and prior recommendations on this study. We thank all MM patients who participated in this study.
Funding
This work was supported by the Deutsche Krebshilfe (grants 1095969 and 111424 to ME and RW).
Data-sharing statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
randomized trials. Haematologica. 2013;98(6):980-987.
12. Klepin HD, Sun C-L, Smith DD, et al. Predictors of unplanned hospitalizations among older adults receiving cancer chemotherapy. JCO Oncol Pract. 2021;17(6):e740-e752.
13. Terpos E, Engelhardt M, Cook G, et al. Management of patients with multiple myeloma in the era of COVID-19 pandemic: a consensus paper from the European Myeloma Network (EMN). Leukemia. 2020;34(8):2000-2011.
14. Isaacs A, Fiala M, Tuchman S, Wildes TM. A comparison of three different approaches to defining frailty in older patients with multiple myeloma. J Geriatr Oncol. 2020;11(2):311-315.
15. Engelhardt M, Ihorst G, Duque-Afonso J, et al. Structured assessment of frailty in multiple myeloma as a paradigm of individualized treatment algorithms in cancer patients at advanced age. Haematologica. 2020;105(5):1183-1188.
16. Palumbo A, Bringhen S, Mateos M-V, et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: an International Myeloma Working Group report. Blood. 2015;125(13):2068-2074.
17. Engelhardt M, Domm A-S, Dold SM, et al. A concise revised Myeloma Comorbidity Index as a valid prognostic instrument in a large cohort of 801 multiple myeloma patients. Haematologica. 2017;102(5):910-921.
18. Milani P, Vincent Rajkumar S, Merlini G, et al. N-terminal fragment of the type-B natriuretic peptide (NT-proBNP) contributes to a simple new frailty score in patients with newly diagnosed multiple myeloma. Am J Hematol. 2016;91(11):1129-1134.
19. Cook G, Royle K-L, Pawlyn C, et al. A clinical prediction model for outcome and therapy delivery in transplant-ineligible patients with myeloma (UK Myeloma Research Alliance Risk Profile): a development and validation study. Lancet Haematol. 2019;6(3):e154-e166.
20. Larocca A, Bonello F, Gaidano G, et al. Dose/schedule-adjusted Rd-R vs continuous Rd for elderly, intermediate-fit patients with newly diagnosed multiple myeloma. Blood. 2021;137(22):3027-3036.
21. Facon T, Dimopoulos MA, Meuleman N, et al. A simplified frailty scale predicts outcomes in transplant-ineligible patients with newly diagnosed multiple myeloma treated in the FIRST (MM020) trial. Leukemia. 2020;34(1):224-233.
22. Brioli A, Manz K, Pfirrmann M, et al. Frailty impairs the feasibility of induction therapy but not of maintenance therapy in elderly myeloma patients: final results of the German Maintenance Study (GERMAIN). J Cancer Res Clin Oncol. 2020;146(3):749-759.
23. Engelhardt M, Dold SM, Ihorst G, et al. Geriatric assessment in multiple myeloma patients: validation of the International Myeloma Working Group (IMWG) score and comparison with other common comorbidity scores. Haematologica. 2016;101(9):1110-1119.
24. Engelhardt M, Mertelsmann R, Duyster J. Das Blaue Buch Chemotherapie-Manual Hämatologie und Onkologie. 7. Auflage. Springer Verlag. 2020.
25. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346.
26. Durie BGM, Harousseau J-L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20(9):1467-1473.
27. Greil C, Engelhardt M, Ihorst G, et al. Allogeneic transplantation of multiple myeloma patients may allow longterm survival in carefully selected patients with acceptable toxicity and preserved quality of life. Haematologica. 2019;104(2):370-379.
28. Waldschmidt JM, Keller A, Ihorst G, et al. Safety and efficacy of vorinostat, bortezomib, doxorubicin and dexamethasone in a phase I/II study for relapsed or refractory multiple myeloma (VERUMM study: vorinostat in elderly, relapsed and unfit multiple myeloma). Haematologica. 2018;103(10):e473-e479.
29. Dold SM, Möller M-D, Ihorst G, et al. Validation of the revised myeloma comorbidity index and other comorbidity scores in a multicenter German study group multiple myeloma trial. Haematologica. 2021;106(3):875-880.
30. Möller M-D, Ihorst G, Pahl A, et al. Physical activity is associated with less comorbidity, better treatment tolerance and improved response in patients with multiple myeloma undergoing stem cell transplantation. J Geriatr Oncol. 2021;12(4):521-530.
31. Einsele H, Engelhardt M, Tapprich C, et al. Phase II study of bortezomib, cyclophosphamide and dexamethasone as induction therapy in multiple myeloma: DSMM XI trial. Br J Haematol. 2017;179(4):586-597.
32. Bachmann F, Schreder M, Engelhardt M, et al. Kinetics of renal function during induction in newly diagnosed multiple myeloma: results of two prospective studies by the German Myeloma Study Group DSMM. Cancers. 2021;13(6):1322.
33. Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(3):309-322.
34. Tuchman SA, Moore JO, DeCastro CD, et al. Phase II study of dose-attenuated bortezomib, cyclophosphamide and dexamethasone (“VCD-Lite”) in very old or otherwise toxicityvulnerable adults with newly diagnosed multiple myeloma. J
Geriatr Oncol. 2017;8(3):165-169.
35. Mai EK, Miah K, Bertsch U, et al. Bortezomib-based induction, highdose melphalan and lenalidomide maintenance in myeloma up to 70 years of age. Leukemia. 2021;35(3):809-822.
36. Ludwig H, Delforge M, Facon T, et al. Prevention and management of adverse events of novel agents in multiple myeloma: a consensus of the European Myeloma Network. Leukemia. 2018;32(7):1542-1560.
37. Bringhen S, Milan A, Ferri C, et al. Cardiovascular adverse events in modern myeloma therapy - incidence and risks. A review from the European Myeloma Network (EMN) and Italian Society of Arterial Hypertension (SIIA). Haematologica. 2018;103(9):1422-1432.
38. Ihne S, Morbach C, Sommer C, Geier A, Knop S, Störk S. Amyloidosis - the diagnosis and treatment of an underdiagnosed disease. Dtsch Arzteblatt Int. 2020;117(10):159-166.
39. Möller M-D, Gengenbach L, Graziani G, Greil C, Wäsch R, Engelhardt M. Geriatric assessments and frailty scores in multiple myeloma patients: a needed tool for individualized treatment? Curr Opin Oncol. 2021;33(6):648-657.
40. Antoine-Pepeljugoski C, Braunstein MJ. Management of newly diagnosed elderly multiple myeloma patients. Curr Oncol Rep. 2019;21(7):64.
41. Cordoba R, Eyre TA, Klepin HD, Wildes TM, Goede V. A comprehensive approach to therapy of haematological malignancies in older patients. Lancet Haematol. 2021;8(11):e840-e852.
42. Rosko AE, Huang Y, Benson DM, et al. Use of a comprehensive frailty assessment to predict morbidity in patients with multiple myeloma undergoing transplant. J Geriatr Oncol. 2019;10(3):479.485.
43. Soekojo CY, Kumar SK. Stem-cell transplantation in multiple myeloma: how far have we come? Ther Adv Hematol. 2019;10:2040620719888111.
44. D’Agostino M, De Paoli L, Conticello C, et al. Continuous therapy in standard- and high-risk newly-diagnosed multiple myeloma: a pooled analysis of 2 phase III trials. Crit Rev Oncol Hematol. 2018;132:9-16.
45. Stege CAM, Nasserinejad K, Klein SK, et al. Improving the identification of frail elderly newly diagnosed multiple myeloma patients. Leukemia. 2021;35(9):2715-2719.
46. Cook G, Larocca A, Facon T, Zweegman S, Engelhardt M. Defining the vulnerable patient with myeloma-a frailty position paper of the European Myeloma Network. Leukemia. 2020;34(9):2285-2294.
47. Larocca A, Dold SM, Zweegman S, et al. Patient-centered practice in elderly myeloma patients: an overview and consensus from the European Myeloma Network (EMN). Leukemia. 2018;32(8):1697-1712.
48. Stauder R, Eichhorst B, Hamaker ME, et al. Management of chronic lymphocytic leukemia (CLL) in the elderly: a position paper from an international Society of Geriatric Oncology (SIOG) Task Force. Ann Oncol. 2017;28(2):218-227.
49. Abel GA, Klepin HD. Frailty and the management of hematologic malignancies. Blood. 2018;131(5):515-524.
Immune-mediated thrombotic thrombocytopenic purpura plasma induces calcium- and IgG-dependent endothelial activation: correlations with disease severity
Edwige Tellier,1,2 Agnès Widemann,1 Raphaël Cauchois,2,3 Julien Faccini,1,2 Marie Lagarde,1,2 Marion Brun,3 Philippe Robert,4 Stéphane Robert,1 Richard Bachelier,1 Pascale Poullin,2,5 Elien Roose,6 Karen Vanhoorelbeke,6 Paul Coppo,2,7 Françoise Dignat-George1,8 and Gilles Kaplanski2,3 on behalf of the ENDO-13 study group
1Aix-Marseille Université, INSERM, INRAE, C2VN, Marseille, France; 2French Reference Center for Thrombotic Microangiopathies, Hôpital Saint-Antoine, Paris, France; 3Aix-Marseille Université, APHM, INSERM, INRAE, C2VN, CHU Conception, Service de Médecine Interne et Immunologie Clinique, Marseille, France; 4Aix-Marseille Université, APHM, CNRS, INSERM, CHU Conception, Laboratoire Immunologie, Marseille, France; 5APHM, Service d’Hémaphérèse, CHU Conception, Marseille, France; 6Laboratory for Thrombosis Research, IRF Life Sciences, KU Leuven Campus Kulak Kortrijk, Kortrijk, Belgium; 7Service d’Hématologie, Hôpital Saint-Antoine, APHP.6-SU, INSERM, UMRS 1138, Centre de Recherche des Cordeliers, Paris, France and 8Aix-Marseille Université, APHM, INSERM, INRAE, C2VN, CHU Conception, Laboratoire d’Hématologie, Marseille, France
Abstract
Correspondence: E. Tellier edwige.tellier@univ-amu.fr https://doi.org/10.3324/haematol.2022.280651
Received: January 12, 2022.
Accepted: November 21, 2022.
Early view: December 1, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is characterized by a severe ADAMTS13 deficiency due to the presence of anti-ADAMTS13 auto-antibodies, with subsequent accumulation of circulating ultra-large von Willebrand factor (VWF) multimers. The role of endothelial cell activation as a trigger of the disease has been suggested in animal models but remains to be demonstrated in humans. We prospectively obtained plasma from the first plasma exchange of 25 patients during iTTP acute phase. iTTP but not control plasma, induced a rapid VWF release and P-selectin exposure on the surface of dermal human micro-vascular endothelial cell (HMVEC-d), associated with angiopoietin-2 and endothelin-1 secretion, consistent with Weibel-Palade bodies exocytosis. Calcium (Ca2+) blockade significantly decreased VWF release, whereas iTTP plasma induced a rapid and sustained Ca2+ flux in HMVEC-d which correlated in retrospect, with disease severity and survival in 62 iTTP patients. F(ab)’2 fragments purified from the immunoglobulin G fraction of iTTP plasma mainly induced endothelial cell activation with additional minor roles for circulating free heme and nucleosomes, but not for complement. Furthermore, two anti-ADAMTS13 monoclonal antibodies purified from iTTP patients’ B cells, but not serum from hereditary TTP, induced endothelial Ca2+ flux associated with Weibel-Palade bodies exocytosis in vitro, whereas inhibition of endothelial ADAMTS13 expression using small intering RNA, significantly decreased the stimulating effects of iTTP immunoglobulin G. In conclusion, Ca2+-mediated endothelial cell activation constitutes a “second hit” of iTTP, is correlated with the severity of the disease and may constitute a possible therapeutic target.
Introduction
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is a rare and life-threatening thrombotic microangiopathy (TMA) characterized by a severe thrombocytopenia (<30.109/L) and a mechanical hemolytic anemia. Consequently, ischemic events of variable severity occur, mainly affecting the brain, the heart, and the mesenteric tract. The diagnosis of iTTP relies on the demonstration of ADAMTS13 protease functional deficiency (<10%), due to the presence of anti-ADAMTS13 auto-antibodies.1–3 ADAMTS13 is responsible for the cleavage of ultra-large von Willebrand factor (UL-VWF) into smaller and less thrombotic multimers.4,5 Deficiency of ADAMTS13 activity leads to the accumulation of highly prothrombotic UL-VWF in the patient plasma inducing the formation of multiple platelet-rich thrombi into the microcirculation, consumptive thrombocytopenia, mechanical hemolysis and clinical symptoms.1–3 Despite treatments based on therapeutic plasma exchange (PEX) and immunosuppressive drugs, the mortality rate remains as high as 5-10%.
Animal models demonstrate that in addition to ADAMTS13 deficiency, endothelial UL-VWF exocytosis is necessary to reproduce the disease, suggesting that endothelial cells (EC) may participate in a “second hit” of the disease.6 ULVWF is the main constituent of endothelial Weibel-Palade bodies (WPB), from which it can rapidly be released upon EC activation.6 In order to obtain a TTP-like disease, ADAMTS13 knockout (KO) mice need to be crossed with mice expressing high intracellular concentrations of VWF, then to be injected with shigatoxin to induce WPB degranulation leading to UL-VWF release, showing that inactivation of the adamts13 gene is not sufficient to induce TTP-like manifestations.7,8,9 In agreement, injections of large concentrations of recombinant VWF can induce TTP in ADAMTS13 KO mice. Moreover, vwf gene deletion results in complete protection in the shigatoxin-induced TTP murine model, demonstrating the absolute requirement of VWF to develop TTP.10,11 Another TTP model consisting in injection of murine anti-ADAMTS13 inhibitory monoclonal antibodies (mAb) into wild-type mice, led to plasma ADAMTS13 deficiency and UL-VWF accumulation without TTP-like symptoms.12 The additional injection of recombinant VWF in this model induces an iTTP-like disease, further demonstrating the essential role of large concentrations of circulating VWF. In a primate model of TTP, injection of a murine anti-human ADAMTS13 inhibitory antibody induced TTP, as demonstrated by the appearance of severe thrombocytopenia, hemolytic anemia, elevated LDH, schistocytes and the occurrence of microthrombi in kidney, heart, brain and spleen.13 However, the primates did not develop end-stage disease, suggesting once again that inhibition of ADAMTS13 alone, may not be sufficient to reproduce a full-spectrum human TTP.
Thus, these experimental models suggest that induction of TTP in animals is a “two-hit” process requiring first, ADAMTS13 protease inactivation and second, an increased VWF release by activated EC. Similar mechanisms however, remain to be demonstrated in humans. In this context, we asked whether iTTP-patient plasma was able to induce WPB exocytosis and tried to identify possible endothelial activators in iTTP-patient plasma. Using plasma prospectively collected from patients during the acute phase of iTTP, we observed induction of UL-VWF release from EC via WPB exocytosis in a Ca2+-dependent pathway. We identified IgG from iTTP patient plasma as the main inducer of endothelial activation and observed that Ca2+-dependent endothelial activation intensity correlated with disease severity.
Methods
Patient characteristics
We conducted a prospective study between 2008 and 2011 consisting in a National Clinical Research Project (#2007/23) approved by the Ethical Committee of the “Assistance Publique-Hôpitaux de Marseille”. Informed consent was provided according to the Declaration of Helsinki. Detailed information about the 62 patients can be found in the Online Supplementary Appendix and Table 1.
Immunofluorescence studies
Confluent human microvascular dermal EC (HMVEC-d) were grown in EC growth basal medium-2 (EBM2) containing 1% fetal bovine serum for 16 hours (h) and activated for 1 hour (h) with either control or iTTP plasma (1/100 in EBM2), washed twice in phosphate buffer saline (PBS), fixed in 1% paraformaldehyde for 10 minutes (min) and labeled with anti-VWF, anti-P-selectin antibodies or with rabbit non-immune serum like described in supplementary methods.
In order to visualize ADAMTS13, HMVEC-d were activated for 20 min with control or iTTP immunoglobulin G (IgG) (30 m g/mL), washed in PBS and labeled as described in the Online Supplementary Appendix. In order to exclude the influence of possible endotoxin contamination of biological samples, all experiments were performed in the presence of 10 m g/mL polymyxin B (Sigma-Aldrich, Saint Louis, USA).
iTPP: immune-mediated thrombotic thrombocytopenic purpura; IQR: interquartile range; LDH: lactate dehydrogenase.
Soluble von Willebrand factor, endothelin-1 and angiopoietin-2 measurements
Specific enzyme-linked immunosorbent assays (ELISA) were performed to determine the concentrations of soluble VWF (American Diagnostica, Stanford, USA), endothelin-1 or angiopoietin-2 (R&D system, Minneapolis, USA), according to the manufacturer’s instructions.
Intracellular Ca2+ flux
HMVEC-d were incubated for 1 h with 5 mM fluo-4 AM fluorescent dye (ThermoFisher Scientific, USA) in PBS containing 1% bovine serum albumin (BSA), washed and incubated in PBS 1% BSA for 30 min at 37°C. Cells were stimulated with A23187 (10 µM in EBM2), thrombin (4 IU/mL or 0.5 mM), control or iTTP plasma samples (1/100 in EBM2) for 20 to 1,800 seconds, or with IgG (30 mg/mL in EBM2). Fluorescence analysis used a Cytofluor®, fluorescence multi-well plate reader (PerSeptive-Biosystems, Framingham, USA). Total fluorescence intensity was expressed in arbitrary unit (AU). Intracellular Ca2+ flux was also measured by microscopy (see the Online Supplementary Appendix).
Preparation of IgG-depleted plasma and purification of the IgG fraction
After washing with binding buffer (PBS containing 100 mM Na2HPO4 at pH 7.4), 100 mL of protein A magnetic sepharose beads (GEHealthcare) were incubated for 30 min at 4°C with 300 mL of 1:5 diluted plasma from control or iTTP patients. Beads were retrieved using a magnetic bench and plasma was used for Ca2+ flux induction and IF studies. IgG were eluted using 100 mL of elution buffer (100 mM glycine at pH 12.8) and 5 mL of 1 M TRIS hydrochloride at pH 9.
Human anti-ADAMTS13 monoclonal antibodies
The two anti-human ADAMTS13 mAb TTP73-1 and ELH2-1 were isolated and cloned from B cells of two different iTTP-patients. As previously reported, these two antibodies recognize cryptic epitopes in ADAMTS1314 and were used at 30 mg/mL in experiments.
ADAMTS13 gene silencing
Downregulation of ADAMTS13 mRNA level was achieved using small interfering RNA (siRNA) directed against ADAMTS13 (Ambion, ThermoFisher-Scientific, USA) and Jet Prime kit (Polyplus-transfection, USA) according to the manufacturer’s instructions. As the decrease in ADAMTS13 mRNA level was achieved 24 h after transfection by quantitative real-time polymerase chain reaction (RT-qPCR) and remained stable for 96 h, Ca2+ flux experiments were performed 48 h after transfection.
Statistical analysis
Graphpad Prism v.9.2.0 software (GraphPad-Software Inc.,
San Diego, USA) was used for statistical analysis. Further details of the methods are available in the Online Supplementary Appendix.
Results
Plasmas from immune-mediated thrombotic thrombocytopenic purpura patients induce von Willebrand factor string formation on the surface of HMVEC-d via Weibel-Palade bodies exocytosis
First, we examined VWF string formation on HMVEC-d by confocal microscopy using anti-VWF labeling on permeabilized HMVEC-d. After incubation with control plasma samples, anti-VWF labeling showed punctuated fluorescent spots, whereas incubation with iTTP plasma samples induced the appearance of typical UL-VWF strings (Figure 1A). Quantification of the VWF string fluorescence was performed after 30 min of incubation and demonstrated higher VWF fluorescence intensity (FI) when HMVEC-d were stimulated with iTTP than with control plasma samples (FI=6.77 AU/cellx104; interquartile range [IQR], 4.8-8.79 vs. FI=0.53 AU/cellx104; IQR, 0.36-0.87, respectively; P<0.0001; Figure 1B). Kinetic studies showed that VWF strings on the endothelial surface in response to iTTP plasma was observed after 5 min incubation and increased for up to 60 min (Figure 1C) (FI=6.75 AU/cellx104; IQR, 4.81-7.38; FI=8.62 AU/cellx104, IQR, 7.03-14.82; FI=14.5 AU/cellx104; IQR, 10.1-21.68 vs. FI=0.76 AU/cellx104, IQR, 0.712.99; P=0.0159; P=0.0079; P=0.0079 respectively at 5, 15 and 60 min vs. 0 min; Figure 1D)
Similar data were obtained in various conditions such as dynamic using a flow chamber device (Online Supplementary Figure S1A), macrovascular HUVEC (Online Supplementary Figure S1B), or when using either plasma obtained on citrated tubes or serum, instead of plasma from PEX in order to eliminate a possible Ca2+ overload due to the PEX process (Online Supplementary Figure S1C). In order to exclude possible passive absorption of soluble VWF contained in iTTP plasma on HMVEC-d surface, we used a VWF-affinity column to completely eliminate soluble VWF contained in plasma. Using ELISA, we measured soluble VWF in the supernatant of HMVEC-d stimulated with either VWF-free iTTP or VWF-free control plasma samples for 1 h and observed significantly higher concentrations of endothelial soluble VWF in the former condition (2.13 ng/mL, IQR, 1.76-2.50 vs. 1.05 ng/mL, IQR, 0.98-1.61; P=0.0159; Online Supplementary Figure S1D).
In order to determine whether VWF string formation on HMVEC-d was effectively due to WPB exocytosis, we measured the concentrations of endothelin-1 and of angiopoietin-2, two molecules stored in WPB. Significantly higher concentrations of endothelin-1 (19.02 pg/mL; IQR, 16.88-20.21 vs. 11.55 pg/mL; IQR, 5.79-17.9, respectively;
Figure 1. Induction of Weibel-Palade bodies exocytosis on endothelial cells after stimulation with immune-mediated thrombotic thrombocytopenic purpura plasma. (A) Confocal microscopy analysis of permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubated with either control plasma (A) or immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples (B) using rabbit anti-von Willebrand Factor (VWF) antibodies and Alexa Fluor 594-conjugated donkey anti-rabbit immunoglobulin G (IgG) (original magnification X63, scale bar=10 mm). (B) Quantification of VWF fluorescence intensity (FI) after HMVEC-d stimulation with either control plasma (Control: n=9) or iTTP plasma samples during the acute phase (iTTP: n=14, ***P<0.001). (C) Kinetics of VWF HMVEC-d exocytosis visualized by fluorescence microscopy after incubation with iTTP plasma samples (original magnification X40, scale bar=10 mm). (D) Quantification of VWF FI over time on HMVEC-d stimulated with iTTP plasma samples (iTTP: n=5, *P<0.05, **P<0.01). (E) Endothelin-1 and (F) angiopoietin-2 concentrations in HMVEC-d supernatants stimulated with either control plasma (n=6) or iTTP plasma samples for 1h (n=25, *P<0.05). (G) P-selectin expression detected by confocal microscopy analysis using anti-rabbit P-selectin antibodies and Alexa Fluor 488-conjugated donkey anti-rabbit IgG (green) in HMVEC-d incubated with control (A) or iTTP plasma samples (B) (original magnification X63, scale bar=10 mm). (H) Percentage of P-selectin FI per cell on HMVEC-d stimulated with control plasma (n=10) (representing 100%) or iTTP plasma samples (n=23, ***P<0.001).
P=0.0102; Figure 1E) and angiopoietin-2 (978 pg/mL; IQR, 619.5-1,200 vs. 614.5 pg/mL; IQR, 445.8-868.3, respectively; P=0.0355; Figure 1F), were present in the supernatants of HMVEC-d when cultured with iTTP compared to control plasma samples. In addition, membrane P-selectin expression on HMVEC-d was observed after stimulation for 10 min with iTTP, but not with control plasma samples (Figure 1G), associated with a 3-time-increased P-selectin FI (FI=290.3 AU/cellx102; IQR, 188.2-596.6 vs. FI=81.23 AU/cellx102; IQR: 61.55-114.3; P<0.0001; Figure 1H). The same data concerning endothelin-1 secretion were observed when HUVEC were used instead of HMVEC-d (40.58 pg/mL; IQR, 25.66-43.52 vs. 19.28 pg/mL; IQR, 16.45-21.56; P=0.0070; Online Supplementary Figure S1E). The endothelin-1 secretion was equivalent when citrated plasma or serum of iTTP-patients were using instead of plasma from PEX (24.65 pg/mL; IQR, 20.39-28.41 and 25.33 pg/mL; IQR, 20.84-32.92 vs. 24.10 pg/mL; IQR, 19.9-24.9; Online Supplementary Figure S1F). Altogether, these data are consistent with the fact that iTTP plasmas obtained after PEX were able to induce WPB exocytosis.
Weibel-Palade bodies exocytosis induced by immune-mediated thrombotic thrombocytopenic purpura plasma is Ca2+-mediated
Two pathways of WPB exocytosis are known: a Ca2+-dependent pathway and a cAMP-dependent pathway. In order to determine whether the Ca2+ signaling cascade was involved in WPB exocytosis induced by iTTP plasma, we used the pharmacological inhibitor MAPTAM. Compared with iTTP plasma samples alone, we observed a significant decrease of VWF FI in the presence of MAPTAM (FI=1.09 AU/Cellx104; IQR, 0.78-1.79 vs. FI= 7.59 AU/Cellx104; IQR, 7.52-8.85; P=0.0079; Figure 2A, B). iTTP plasma-induced endothelin-1 secretion was also reduced by 92% in the presence of MAPTAM (82.1%; IQR, 73-134.8 vs. 8.5%; IQR, 6.9-9.6; P<0.0001; Figure 2C).
We then measured the ability of iTTP plasma to induce Ca 2+ -flux in EC, using a single-wavelength Ca 2+ fluorescent dye. After a 20-second incubation, iTTP plasma samples induced a significantly higher FI level than control plasma samples in HMVEC-d (94 AU; IQR, 78-125.5 vs. 45 AU; IQR, 35.5-76 respectively; P<0.0001; Figure 2D) or in HUVEC (68 AU; IQR, 55.38-114.9 vs . 41.75 AU; IQR, 37.75-54; P =0.0063; Online Supplementary Figure S1G). We also calculated the AUC for Ca2+ flux after 30 min of stimulation and observed that iTTP plasma induced significantly higher Ca 2+ mobilization than control plasma samples (1,011 AU; IQR, 472-1,334 vs. 242 AU, IQR, 43.5388.5; P=0.0002; Figure 2E). The endothelial Ca2+ mobilization was equivalent when using citrated plasma or serum instead of PEX plasma (74.5 AU; IQR, 59.5-94.75 and 66 AU; IQR, 41.75-85 vs. 70 AU; IQR, 48.75-87.5; Online Supplementary Figure S1H).
Immune-mediated thrombotic thrombocytopenic purpura plasma-induced endothelial Ca2+ flux correlates with immune-mediated thrombotic thrombocytopenic purpura severity
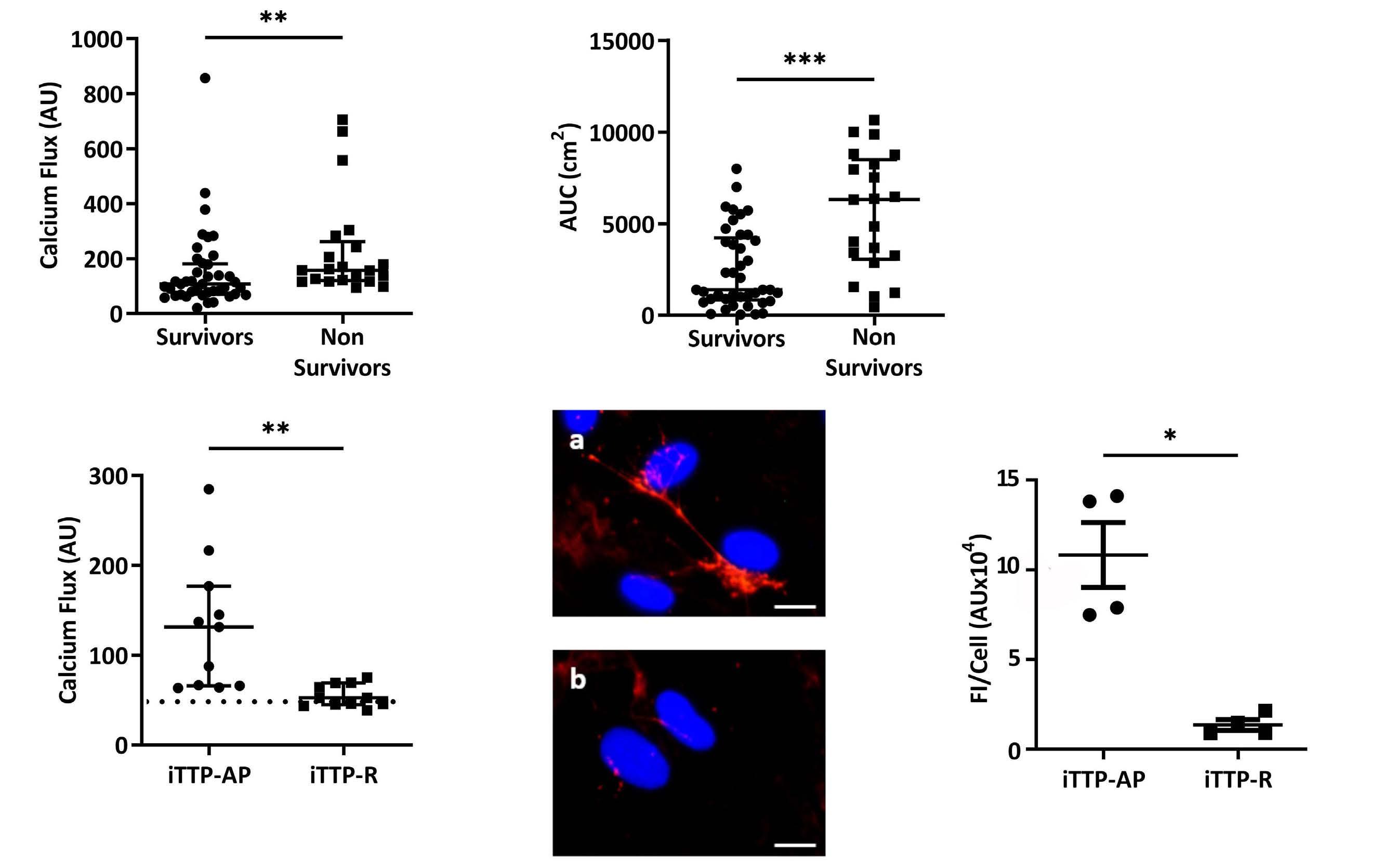
In order to establish whether iTTP-induced Ca2+ flux correlated with disease severity, we studied sera from 62 iTTP patients (25 from Marseille added with 37 from Paris) collected during the acute phase, and classified in two different groups depending on survival (Table 1). At 20 seconds, Ca2+ flux appeared significantly higher in cells treated with plasma from non-survivor patients compared with those of survivors (157 AU; IQR, 119.7-262.3 vs. 107 AU; IQR, 70-181, respectively; P=0.0031; Figure 3A). Similarly, after 30 min, area under the curve (AUC) from non-survivors was much higher than those in survivors (6,316 cm2; IQR, 3,066-8,503 vs. 1391 cm2; IQR: 833.5-4,235, respectively; P=0.0002; Figure 3B). In addition, plasma from iTTP patients in remission induced significantly less Ca2+ flux than plasma from the same patients during the acute phase (52.5 AU; IQR: 45-69 vs. 131.5 AU; IQR, 66-177; P=0.0013; Figure 3C) and levels of Ca2+ flux were not different from those induced by control plasma. In agreement, plasma obtained from patients in complete remission did not induce VWF release by HMVEC-d (Figure 3D) which was confirmed by fluorescence quantification (1.19 AUx104; IQR, 0.89-1.99) during remission versus 10.85 AUx104; IQR, 7.59-14.04 during the acute phase; P=0.0286; Figure 3E).
The purified IgG fraction from immune-mediated thrombotic thrombocytopenic purpura plasma induced Ca2+-dependent Weibel-Palade bodies exocytosis
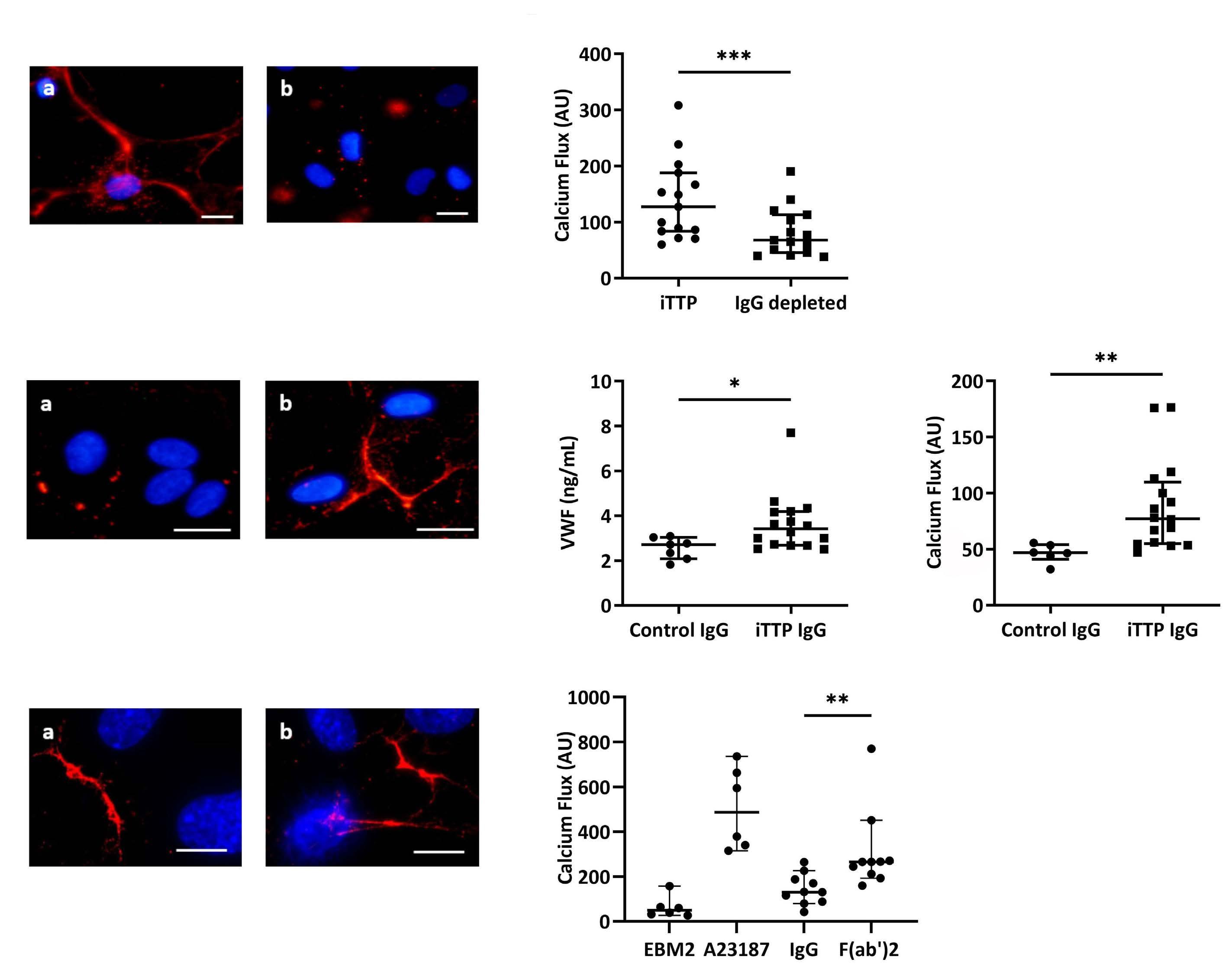
In order to identify the soluble mediators contained in iTTP plasma involved in WPB exocytosis, we studied several candidates. As iTTP is an autoimmune disease, we first investigated the role of circulating IgG. After depletion of the IgG fraction from iTTP plasma, we observed a significant decrease of VWF FI release by HMVEC-d (Figure 4A). In agreement, the Ca2+ flux was reduced by 47% in HMVEC-d cells treated with IgG-depleted plasma compared with complete plasma (127.5 AU; IQR, 83.5-188 vs. 68 AU; IQR, 45.5-113, respectively; P=0.0003; Figure 4B). Conversely, we tested the ability of the purified IgG fraction from iTTP or control plasma to activate HMVEC-d. VWF exocytosis was observed in HMVEC-d cultured with iTTP IgG, but not with control IgG (Figure 4C). Using ELISA, we measured soluble VWF released in the supernatants of HMVEC-d and observed significantly increased VWF concentrations in HMVEC-d stimulated by iTTP IgG compared with control IgG (3.42 ng/mL; IQR, 2.68-4.18 vs. 2.72 ng/mL; IQR, 2.083.03, respectively; P=0.0273; Figure 4D). In agreement, we observed a significantly higher Ca2+ flux in HMVEC-d stimulated with iTTP IgG than with control IgG fractions (77.25 AU; IQR, 54.88-109.8 vs. 46.75 AU; IQR, 41-54, respectively;
Figure 2. Weibel-Palade bodies exocytosis induced by immune-mediated thrombotic thrombocytopenic purpura plasma is Ca2+mediated. (A) Von Willebrand Factor (VWF) secretion detected by fluorescence microscopy, using rabbit anti-VWF antibodies and Alexa Fluor 594-conjugated donkey anti-rabbit immunoglobulin G (IgG) after incubation of permeabilized human microvascular endothelial cells from derm (HMVEC-d) with immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples in the absence (A, B) or in the presence of the Ca2+ chelator MAPTAM (C, D) (original magnification X40, scale bar=30 mm). (B) VWF fluorescence intensity (FI) quantification by cell comparing HMVEC-d stimulated with iTTP plasma samples in absence or presence of MAPTAM (iTTP: n=5, **P<0.01). (C) Endothelin-1 concentration measured in the supernatants of HMVEC-d incubated with iTTP plasma samples (n=10) in the absence (representing 100%) or in the presence of MAPTAM (***P<0.001). (D) Fluorescence intensity (AU) of the Ca2+ flux in HMVEC-d incubated for 20 seconds with either control plasma samples (n=13), iTTP plasma samples (n=25, ***P<0.001) or A23187 Ca2+ ionophore (n=3). (E) Area under the curve (AUC) (cm2) from Ca2+ flux curves after 30 minutes of HMVEC-d incubation with either control plasma (n=13), iTTP plasma samples (n=25, ***P<0.001) or A23187 Ca2+ ionophore (n=3).
P=0.0011; Figure 4E). These results were confirmed by directly measuring Ca2+ flux in living cells stimulated with either iTTP or control IgG, using microscopy (Online Supplementary Figure S2A, B). In order to determine which fraction of the immunoglobulin was involved in EC activation, we purified the F(ab’)2 fragments from the IgG fraction, and observed that F(ab’)2 fragments induced both VWF string formation (Figure 4F) and a twice higher endothelial Ca2+ flux compared to the complete iTTP IgG fraction (130.8; IQR, 86.11-197.2 vs. 265.3; IQR, 206.6-315.3; P=0.002; Figure 4G).
Immune-mediated thrombotic thrombocytopenic purpura IgG induced Weibel-Palade bodies degranulation partly via ADAMTS13 recognition on endothelial cells
In order to identify the endothelial antigen target of iTTPIgG, we concentrated on ADAMTS13, the main protein involved in iTTP pathogenesis. Since it was impossible to create an ADAMTS13 affinity column in order to eliminate anti-ADAMTS13 auto-antibodies contained in iTTP-purified IgG, we used different procedures. First, we compared the ability of hereditary TTP (hTTP) serum samples which do not contain anti-ADAMTS13 auto-antibodies to induce Weibel-Palade bodies degranulation, with iTTP serum samples. Compared to that induced by iTTP serum samples, we observed that VWF release induced by hTTP serum was 65% lower (6.61 ng/mL; IQR, 4.45-12.84 vs. 17.27 ng/mL; IQR, 11.09-24.88; P=0.0317; Figure 5A) and that the Ca2+ flux induced by hTTP serum samples appeared significantly reduced (113.5 AU; IQR, 112.7-118.4 vs. 126.7 AU; IQR, 123.3-131.7; P=0.0079; Figure 5B). After we verified that extracellular expression of endothelial ADAMTS13 was not modified when EC were stimulated with IgG from iTTPpatients or control plasma samples (Online Supplementary Figure S3A) (90.4%; IQR, 62.78-116.2 vs. 116.3 %; IQR, 73.37144.7; P=0.3357; Online Supplementary Figure S3B), we inhibited endothelial ADAMTS13 expression using specific siRNA (70% of inhibition, 1; IQR, 1-1 vs. 0.29; IQR, 0.16-0.41;
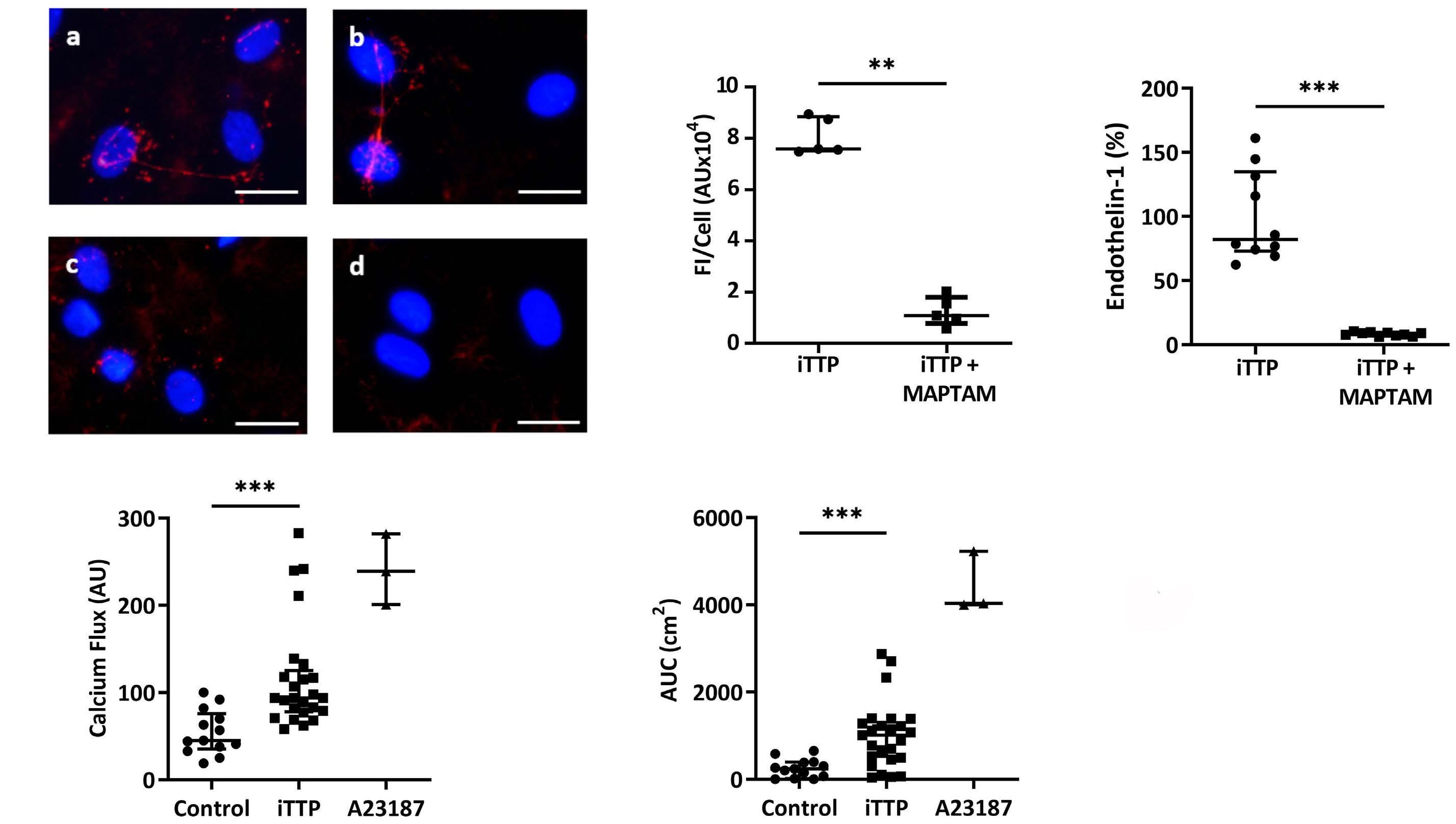
Figure 3. In vitro endothelial Ca2+ flux intensity induced by immune-mediated thrombotic thrombocytopenic purpura plasma is associated with clinical severity and prognosis. (A) Fluorescence intensity (AU) of the Ca2+ flux measured at 20 seconds in permeabilized human microvascular endothelial cells from derm (HMVEC-d) stimulated with immune-mediated thrombotic thrombocytopenic purpura (iTTP) plasma samples of survivors (n=41) or non-survivors patients (n=21, **P<0.01). (B) Area under the curve (AUC) from Ca2+ flux curves after 30 minutes of HMVEC-d stimulated with iTTP plasma samples of survivors (n=41) or non-survivors (n=21, ***P<0.001). (C) Fluorescence intensity (AU) of the Ca2+ flux in HMVEC-d incubated for 20 seconds with plasma samples from iTTP patients in acute phase (iTTP-AP) or from same patients in remission (iTTP-R) (n=11, **P<0.01). Horizontal dotted line represents median Ca2+ mobilization induced in HMVEC-d by control plasma. (D) Von Willebrand Factor (VWF) secretion and tethering detected by fluorescence microscopy using rabbit anti-VWF antibodies and Alexa Fluor 594conjugated donkey anti-rabbit immunoglobulin (red) after 1-hour-incubation of HMVEC-d with iTTP plasma samples collected in acute phase (A) or during remission (B). Cell nuclei are labeled in blue using DAPI (original magnification X40, scale bar=10 mm). (E) VWF fluorescence quantification after the HMVEC-d stimulation with iTTP plasma from patients in acute phase (iTTP-AP: n=4), or in remission (iTTP-R: n=4, *P<0.05).
P=0.0313; Figure 5C,) and observed that inhibition of ADAMTS13 expression was associated with a significant reduction of the iTTP IgG-induced Ca2+ flux (33.44 AU; IQR, 25.88-35.22 vs. 47.15 AU; IQR, 31.42-73.9; P=0.0084; Figure 5D). We further tested the ability of two anti-ADAMTS13 antibodies, TTP73-1 or ELH2-1, previously isolated and cloned from the B cells of two iTTP-patients,14 to induce WPB exocytosis in HMVEC-d. Both TTP73-1 and ELH2-1 weakly induced VWF tethering on HMVEC-d membranes (Figure 5E). When cultured in the presence of TTP73-1 but not of or ELH2-1, higher soluble VWF concentrations were measured in HMVEC-d supernatants compared to stimulation with control IgG (3.7 ng/mL; IQR, 3.3-4.3 for TTP731 and 1.7 ng/mL; IQR, 1.6-2 for ELH2-1 vs. 0.8 ng/mL; IQR, 0.7-0.99; P=0.0357 and P=0.0571 respectively; Figure 5F), as well as higher endothelin-1 secretion (14.1 pg/mL; IQR, 11.8-15.4 for TTP73-1 and 11.5 pg/mL; IQR, 10.8-14.4 for
ELH2-1 vs. 6.4 pg/mL; IQR, 4.2-11.5; P=0.0317 and P=0.0952 respectively; Figure 5G). In addition, an increased but nonsignificant Ca2+ flux was also induced by TTP73-1 and ELH2-1, compared to control IgG (15.01 AU, IQR, 11.55-19.31 and 14.91 AU; IQR, 6.26-18.39, respectively, vs. 3.2 AU, IQR, 0.59-5.83; P=0.10; Figure 5H).
Free heme and nucleosomes, but not complement, play minor roles in immune-mediated thrombotic thrombocytopenic purpura plasma-induced
Weibel-Palade bodies exocytosis
The concentrations of free heme were significantly increased in iTTP compared to control plasma samples (11.2 mM; IQR, 6.98-20.35 vs. 9 mM; IQR, 8.2-9.6; P=0.0369; Figure 6A), but returned to control levels after remission (0.63 mM; IQR, 0.44-1.03 vs. 11.2 mM; IQR, 6.98-20.35; P<0.0001; Figure 6A). Free heme concentrations however, were equivalent between survivor and non-survivor patients (10.96 m M; IQR, 8.2-14.07 vs. 12.96 m M; IQR, 2.55-43.56; P=0.6368; Online Supplementary Figure S4A). Inhibition of heme by addition of large concentrations of hemopexin, weakly reduced iTTP plasma-induced VWF exocytosis (Figure 6B), endothelin-1 release (69.86%; IQR, 27.59-136.1 vs. 79.86%; IQR, 54.79-162.6; P<0.0494; Figure 6C) and iTTPinduced Ca2+ flux (30% inhibition, 133.3 AU; IQR, 89.25- iTTP plasma also contained significant enrichment in nucleosomes concentrations compared to control plasma samples (6.34-fold increase; IQR, 3.37-12.14 vs. 1.15-fold increase; IQR; 0.12-1.78; P<0.0001; Figure 6E) which was no longer observed after remission (0.81-fold increase; IQR, 0.53-1.52 vs. 6.34-fold increase; IQR, 3.37-12.14 in acute phase; P<0.0001; Figure 6E), in accordance with previous studies.15 Nucleosomes enrichment however, was not different between survivors and non-survivors (3.57-fold increase; IQR, 1.76-5.24 vs. 3.51-fold increase; IQR, 1.24-7.49; P=0.9295; Online Supplementary Figure S4B). Nucleosomes signalization is dependent of the phosphatidylserine activation and can be inhibited by Annexin V. Addition of Annexin V to iTTP plasma weakly but significantly reduced VWF exocytosis (Figure 6F) and endothelin-1 release by HMVEC-d (10% inhibition, 72.62%; IQR, 64.16-126.1 vs 80.11%; IQR; 67.52-149.6; P=0.0052; Figure 6G), as well as Ca2+ flux induced by iTTP plasma (13%-reduction, 216 AU; IQR, 102.8-262.8 vs. 246.5 AU; IQR, 128-304.5; P=0.0011; Figure 6H).
Figure 4. Immunoglobulin G contained in immune-mediated thrombotic thrombocytopenic purpura plasma are mainly involved in permeabilized human microvascular endothelial cells from derm activation. (A) Von Willebrand Factor (VWF) secretion detected by fluorescence microscopy after permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubation with complete immune-mediated thrombotic thrombocytopenic purpura (iTTP) (A) or immunoglobulin (IgG)-depleted (B) plasma samples (original magnification X20, scale bar=30 mm). (B) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20-second incubation with complete iTTP or IgG-depleted plasma samples (n=15, ***P<0.001). (C) VWF secretion detected by fluorescence microscopy after HMVEC-d incubation with IgG purified from control (A) or iTTP plasma samples (b) (original magnification X40, scale bar=30 mm). (D) VWF quantification by enzyme-linked immunosorbant assay in the supernatants of cells treated with IgG purified from control (n=7) or iTTP plasma samples (n=16, *P<0.05). (E) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d after 20 seconds incubation with IgG from control (n=6) or iTTP plasma samples (n=16, **P<0.01). (F) VWF secretion detected by fluorescence microscopy after HMVEC-d incubation with IgG (a) from iTTP plasma samples or F(ab)’2 fragments (b) purified from these IgG (original magnification X63, scale bar=20 mm). (G) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d after a 20-second of incubation with medium alone (EBM2), A23187 calcium (Ca2+) ionophore, IgG (IgG) from iTTP plasma samples, or F(ab)’2 fragments purified from these IgG (n=10, **P<0.01).
242.8 vs. 107.5 AU; IQR, 61.88-208.3; P=0.0063; Figure 6D).
Figure 5. ADAMTS13 is involved in immune-mediated thrombotic thrombocytopenic purpura IgG-induced Weibel-Palade bodies degranulation. (A) Von Willebrand Factor (VWF) quantification by enzyme-linked immunosorbant assay in the supernatants of cells treated with immune-mediated thrombotic thrombocytopenic purpura (iTTP) serum (n=5) or hereditary TTP serum samples (hTTP, n=5, *P<0.05). (B) Fluorescence intensity (AU) of Ca2+ flux induction in permeabilized human microvascular endothelial cells from derm (HMVEC-d) after a 20-second incubation with serum from iTTP patients (n=5) or from hTTP patients (n=5, **P<0.01). (C) ADAMTS-13 mRNA relative quantification in HMVEC-d cultured in the presence of control small interfering RNA (siRNA) (Ct si) or siRNA targeting ADAMTS13 (ADAMTS13 Si) (n=6, *P<0.05). (D) Fluorescence intensity (AU) of Ca2+ flux induction in HMVEC-d transfected with control or anti-ADAMTS13 siRNA, then stimulated for 20 seconds with IgG from iTTP plasma samples (n=15, **P<0.01). (E) VWF expression detected by fluorescence microscopy in HMVEC-d incubated with IgG from control patient samples (a) TTP73-1 monoclonal antibody (mAb) (B) or ELH2-1 mAb (C) (original magnification X40, scale bar=30 mm). (F) VWF or (G) endothelin-1 concentrations in supernatants of HMVEC-d incubated with IgG from control patient samples (Control) (n=3), TTP73-1 or ELH2-1 mAb (n=5, *P<0.05). (H) Fluorescence intensity (AU) of Ca2+ flux measured at 20 seconds in HMVEC-d stimulated in a similar fashion as in (G) (n=3, ns: not significant).
Figure 6. Heme and nucleosomes participate of the immune-mediated thrombotic thrombocytopenic purpura patient’s plasma induced Ca2+-mediated endothelial activation. (A) Free heme concentration (mM) measured in control (n=13), immune-mediated thrombotic thrombocytopenic purpura (iTTP) patients in acute phase (iTTP-AP, n=62) or iTTP patients in remission (iTTP-R, n=14) plasma samples (*P<0.05, ***P<0.001). (B) Von Willebrand Factor (VWF) expression detected by fluorescence microscopy after permeabilized human microvascular endothelial cells from derm (HMVEC-d) incubation with heme or iTTP plasma samples in the absence (A, B) or in the presence (C, D) of hemopexin (+Hx) (original magnification X40, scale bar=30 mm). (C) Endothelin-1 concentrations in the supernatants of HMVEC-d incubated with iTTP plasma samples in the absence (representing 100%) or in the presence of hemopexin (n=14, *P<0.05). (D) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20second incubation with iTTP plasma samples alone or after preincubation with hemopexin (n=18, **P<0.01). (E) Total nucleosomes in control (n=13), iTTP patients in acute phase (iTTP-AP, n=62) or iTTP patients in remission (iTTP-R, n=14) plasma samples (***P<0.001). (F) VWF secretion detected by fluorescence microscopy after incubation of HMVEC-d with iTTP plasma samples in the absence (A) or in the presence of Annexin V (B) (original magnification X40, scale bar=30 mm). (G) Endothelin-1 concentrations in the supernatants of HMVEC-d incubated with iTTP plasma samples in the absence (representing 100%) or in the presence of Annexin V (n=16, **P<0.01). (H) Fluorescence intensity (AU) of the Ca2+ flux induction in HMVEC-d after a 20second incubation with either iTTP plasma samples alone, or in the presence of Annexin V (n=17, **P<0.01).
In order to investigate the role of complement on iTTP plasma-induced endothelial activation, we performed the same experiments with heated plasma or in the presence of eculizumab. None of these treatments significantly influenced endothelial VWF (Online Supplementary Figure S5A) or endothelin-1 secretion after 1 hour of stimulation (29.45 pg/mL; IQR, 23.61-41.76 and 20.85 pg/mL; IQR, 16.0127.10 vs. 23.27 pg/mL; IQR, 19.29-30.39, not significant; Online Supplementary Figure S5B), as well as endothelial Ca2+ flux intensity after 20 seconds (111.8 AU; IQR, 74.85-148.8 and 100.2 AU; IQR, 79.43-287.4 vs. 99.41 AU; IQR, 72.49210.7, not significant; Online Supplementary Figure S5C).
Since iTTP plasma is likely to contain thrombin, we reproduced the same experiments in the presence of hirudin and observed no significant modification (187.5 AU; IQR, 82.75-240 vs. 180 AU; IQR, 89.75-262.5, not significant; Online Supplementary Figure S5D). We also used the general serine protease inhibitor PPACK on the iTTP plasma induced VWF release and Ca2+ flux increase and observed no significant difference (6.298 ng/mL; IQR, 5.859-6.924 vs 6.345 ng/mL; IQR, 6.095-8.615, not significant; Online Supplementary Figure S5E, and 149.3 AU; IQR, 134.2-154.8 vs 144.4 AU; IQR, 138.5-155.9, not significant; Online Supplementary Figure S5F, respectively).
Discussion
In the present study, we observed for the first time that plasma or serum from iTTP patients in acute phase, but not in remission, were able to induce VWF secretion via WPB exocytosis by a mechanism involving a Ca2+ flux. We ruled out a possible passive adhesion of soluble VWF contained in iTTP plasma on the EC surface, since VWF tethering remained present on the EC membrane even after complete depletion of VWF contained in iTTP plasma and concluded that VWF secretion was an active mechanism likely due to WPB exocytosis. WPB exocytosis was indeed confirmed by the fact that iTTP plasma not only induced VWF and P-selectin membrane exposure, but also endothelin-1 and angiopoietin-2 secretion by EC, all com- ponents of the WPB. WPB exocytosis is known to involve two different signaling pathways. One is rapid and Ca2+mediated, whereas the other is linked to adenylate cyclase activation.6 Both the WPB exocytosis and the rapid and sustained intracellular Ca2+ flux induced by iTTP plasma were inhibited by the Ca2+ chelator MAPTAM, demonstrating a Ca2+-mediated signaling pathway. The possible Ca2+ overload of iTTP plasma due to the PEX process was excluded by the observation that the serum of the iTTP-patients had similar effects as plasma and that plasma of non-TTP patients treated with PEX, was not able to activate EC.
Our objective was then to identify the main endothelial activators contained in iTTP plasma/serum and we showed that the IgG fraction purified from iTTP plasma was able to induce a Ca2+ flux and subsequent WPB exocytosis associated with VWF secretion, whereas IgG depletion resulted in an almost 60% reduction of EC activation by iTTP plasma. Noteworthy, was the fact that IgG stimulating effects were reproduced by the F(ab’)2 fractions and were thus linked to a putative antigen recognition on EC. Since iTTP is known to be caused by acquired antibodies directed against ADAMTS13, we investigated the role of these antibodies in EC activation. We did not succeed in directly purifying anti-ADAMTS13 Ab from the iTTP IgG fraction, thus we used different ways to determine whether ADAMTS13 played a role in endothelial activation by purified IgG. First, we compared the ability of serum from iTTP with those of hTTP to induce endothelial activation. Hereditary TTP are due to ADAMTS13 genetic deficiency and do not contain anti-ADAMTS13 auto antibodies. We observed a significant reduction (almost 60%) of endothelial activation using hTTP serum samples. Second, we observed that efficient inhibition of ADAMTS13 expression in HMVEC-d using siRNA was associated with a significant reduction of EC degranulation induced by iTTP IgG fraction. Third, we used two previously described human antiADAMTS13 mAb, cloned from iTTP isolated B cells,14 and observed that both of them weakly induced Ca2+ flux in EC as well as VWF and endothelin-1 secretion, confirming that anti-ADAMTS13 IgG were at least in part, responsible for iTTP-induced Ca2+-mediated EC activation. In most of the patients, anti-ADAMTS13 antibodies behave as inhibitors of the ADAMTS13 protease, but in 10-20% of iTTP-patients, these antibodies demonstrate other mechanisms of interaction with ADAMTS13, such as increased clearance,16,17 conformational changes,14,18 or increased metalloprotease activity19 without modifying ADAMTS13 quantification in ELISA.20,21 Our data suggest that in addition to these various interactions, anti-ADAMTS13 antibodies may also induce endothelial activation and VWF exocytosis, the well-known second hit of the disease.8 This observation may appear contradictory with animal models suggesting that antiADAMTS13 antibodies do not induce VWF release. Indeed, injection of an inhibiting anti-human ADAMTS13 mAb into baboons did not increase circulating VWF concentrations, but induced histological lesions of TTP, therefore suggesting that at least local VWF occured.13 Also in humans the sole presence of anti-ADAMT13 auto antibodies seems insufficient to induce the disease, which appears frequently related to precipitating events such as pregnancy, surgery or infections.22 These apparently contradictory results may have several explanations. First that our observations in vitro may not have an important significance in vivo. However, despite some heterogeneity, our results show a significant association of Ca2+-induced endothelial activation in vitro with TTP prognosis and survival. Second, that not all anti-ADAMTS13 auto-antibodies have the capacity to induce WPB degranulation or that iTTP IgG may recognize additional endothelial targets to induce this effect. This is consistent with the fact that in our experiments, inhibition of ADAMTS13 expression did not completely suppress the effects of iTTP IgG and that anti-ADAMTS13 mAb weakly activated EC in vitro. Other anti-EC antibodies, as suggested by previous reports in iTTP and other autoimmune diseases, may participate in endothelial activation.23 Moreover, in support of the role of anti-ADAMTS13 in endothelial VWF release, it may be noticeable that during hTTP, even very low circulating concentrations of ADAMTS13 may not be associated with disease bouts, and that serum of hTTP which do not contain anti-ADAMTS13 auto-antibodies did not induce a strong endothelial activation in our experiments.
We also observed high free heme concentrations in iTTP plasma, in the order of magnitude of those observed in sickle cell disease and HUS patients.24,25 Heme-induced VWF exocytosis and participated in iTTP-induced Ca2+-mediated EC activation since VWF exocytosis was decreased by 30% by hemopexin. Free heme binds to TLR4 on EC and has been shown to induce WPB exocytosis.26,27 Interestingly, TLR4, also the receptor for LPS, may mediate Ca2+ flux and endothelial permeability, as shown in lung EC.28 Moreover, hemoglobin resulting from intravascular hemolysis has been shown to inactivate ADAMTS13.29 Furthermore, in accordance with previous reports showing that histones could induced WPB exocytosis,30 we observed elevated concentrations of nucleosomes in iTTP plasma samples which weakly participated in iTTP-induced Ca2+-mediated EC. DNA/histone complexes known to contribute to thrombosis have been reported elevated in the plasma of TMA patients15 and have been shown to induce TTP in a zebrafish model, underscoring their potential importance in iTTP pathogenesis.31 Despite previous evidence showing the amplifying role of complement in endothelial activation,32 our experiments did not show a role for complement in this very initial endothelial activation during iTTP. To date, except for markers of tissue ischemic injury, no real prognostic factor has been reported in iTTP, although anti-ADAMTS13 antibodies of various isotypes, including IgA, may be associated with more severe forms.33,34 In our study, the amount of in vitro endothelial Ca2+ flux induced by 62 iTTP plasma or serum samples appeared to be associated with disease severity and death, whereas neither free heme, nor nucleosome concentrations correlated with iTTP severity. This result may indicate that several plasmatic components including anti-ADAMTS13 IgG, free heme, nucleosomes and possibly others converge to induce Ca2+-mediated EC activation. We have previously observed in a prospective study that circulating EC counts correlated with disease severity.35 Thus, iTTP early prognosis and death may be related to the intensity of endothelial activation/injury. Moreover, the identification of Ca2+ as the main cellular messenger of iTTP-induced WPB degranulation may possibly help to design a new therapeutic strategy targeting endothelial activation.
Disclosures
PC is a consultant for Sanofi, Alexion, Takeda and Roche. KV is a member of the scientific advisory boards of Ablynx-Sanofi and Shire-Takeda. PP is a member of the scientific advisory boards of Ablynx-Sanofi. All other authors have no conflicts of interest to disclose.
Contributions
ET, AW, MB, JF, RC and ML conducted the experiments and interpreted the data; MB, PP, PC, ENDO-13 study group members, ER, KV and GK contributed to sample collection and analysis of clinical data; ET, SR and JF contributed to image capture; PR and ET contributed to flow chamber experiments and fluorescence microscopy analysis; ER and KV provided TTP73 and ELH2-1 for the study and critically reviewed the manuscript; FDG handled funding and made a critical revision of the manuscript; RB critically reviewed the manuscript; GK took care of patients, handled funding and supervision of the project, helped with the interpretation of the experiments, and wrote the manuscript together with ET.
Acknowledgments
Members of the french ENDO-13 study group include D. Bourdessoule, CHU de Limoges; K. Clabault, CHU de Rouen; C. Daubin, CHU de Caen and G. Choukroun, CHU Amiens, France.
Funding
This study was supported by research funding from the French ministry of Health (Projet National de Recherche Clinique 2007, #2007/23) to GK.
Data-sharing statement
The original data and protocols are available to others investigators without unreasonable restrictions.
References
1. Kremer Hovinga JA, Coppo P, Lämmle B, Moake JL, Miyata T, Vanhoorelbeke K. Thrombotic thrombocytopenic purpura. Nat Rev Div Primers. 2017;3:17020.
2. Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836-2846.
3. Scully M, Cataland S, Coppo P, et al. Consensus on the standardization of terminology in thrombotic thrombocytopenic purpura and related thrombotic microangiopathies. J Thromb Haemost. 2017;15(2):312-322.
4. Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585-1594.
5. Furlan M, Robles R, Galbusera M, et al. von Willebrand factorcleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med. 1998;339(22):1578-1584.
6. Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26(5):1002-1007.
7. Banno F, Kokame K, Okuda T, et al. Complete deficiency in ADAMTS13 is prothrombotic, but it alone is not sufficient to cause thrombotic thrombocytopenic purpura. Blood. 2006;107(8):3161-3166.
8. Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115(10):2752-2761.
9. Nolasco LH, Turner NA, Bernardo A, et al. Hemolytic uremic syndrome-associated Shiga toxins promote endothelial-cell secretion and impair ADAMTS13 cleavage of unusually large von Willebrand factor multimers. Blood. 2005;106(13):4199-4209.
10. Schiviz A, Wuersch K, Piskernik C, et al. A new mouse model mimicking thrombotic thrombocytopenic purpura: correction of symptoms by recombinant human ADAMTS13. Blood. 2012;119(25):6128-6135.
11. Chauhan AK, Walsh MT, Zhu G, Ginsburg D, Wagner DD, Motto DG. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood 2008;111(7):3452-3457.
12. Deforche L, Tersteeg C, Roose E, et al. Generation of antimurine ADAMTS13 antibodies and their application in a mouse model for acquired thrombotic thrombocytopenic purpura. PLoS One. 2016;11(8):e0160388.
13. Feys HB, Roodt J, Vandeputte N, et al. Thrombotic thrombocytopenic purpura directly linked with ADAMTS13 inhibition in the baboon (Papio ursinus). Blood. 2010;116(12):2005-2010.
14. Roose E, Vidarsson G, Kangro K, et al. Anti-ADAMTS13 Autoantibodies against cryptic epitopes in immune-mediated thrombotic thrombocytopenic purpura. Thromb Haemost. 2018;118(10):1729-1742.
15. Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lämmle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120(6):1157-1164.
16. Shelat SG, Smith P, Ai J, Zheng XL. Inhibitory autoantibodies against ADAMTS-13 in patients with thrombotic thrombocytopenic purpura bind ADAMTS-13 protease and may accelerate its clearance in vivo. J Thromb Haemost. 2006;4(8):1707-1717.
17. Thomas MR, de Groot R, Scully MA, Crawley JTB. Pathogenicity of anti-ADAMTS13 autoantibodies in acquired thrombotic thrombocytopenic purpura. EBioMedicine. 2015;2(8):942-952.
18. Roose E, Schelpe A-S, Tellier E, et al. Open ADAMTS13, induced by antibodies, is a biomarker for subclinical immune-mediated thrombotic thrombocytopenic purpura. Blood. 2020;136(3):353-361.
19. Schelpe A-S, Petri A, Roose E, et al. Antibodies that conformationally activate ADAMTS13 allosterically enhance metalloprotease domain function. Blood Adv. 2020;4(6):1072-1080.
20. Dekimpe C, Roose E, Tersteeg C, et al. Anti-ADAMTS13 autoantibodies in immune-mediated thrombotic thrombocytopenic purpura do not hamper ELISA-based quantification of ADAMTS13 antigen. J Thromb Haemost. 2020;18(4):985-990.
21. Dekimpe C, Roose E, Kangro K, et al. Determination of antiADAMTS-13 autoantibody titers in ELISA: influence of ADAMTS-13 presentation and autoantibody detection. J Thromb Haemost. 2021;19(9):2248-2255.
22. Morgand M, Buffet M, Busson M, et al. High prevalence of infectious events in thrombotic thrombocytopenic purpura and genetic relationship with toll-like receptor 9 polymorphisms: experience of the French Thrombotic Microangiopathies Reference Center. Transfusion. 2014;54(2):389-397.
23. Praprotnik S, Blank M, Levy Y, et al. Anti-endothelial cell antibodies from patients with thrombotic thrombocytopenic purpura specifically activate small vessel endothelial cells. Int Immunol. 2001;13(2):203-210.
24. Frimat M, Tabarin F, Dimitrov JD, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122(2):282-292.
25. Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383-1389.
26. Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vasoocclusion in murine sickle cell disease. Blood. 2014;123(3):377-390.
27. Figueiredo RT, Fernandez PL, Mourao-Sa DS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282(28):20221-20229.
28. Tauseef M, Knezevic N, Chava KR, et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxininduced lung vascular permeability and inflammation. J Exp Med. 2012;209(11):1953-1968.
29. Studt J-D, Kremer Hovinga JA, Antoine G, et al. Fatal congenital thrombotic thrombocytopenic purpura with apparent ADAMTS13 inhibitor: in vitro inhibition of ADAMTS13 activity by hemoglobin. Blood. 2005;105(2):542-544.
30. Michels A, Albánez S, Mewburn J, et al. Histones link inflammation and thrombosis through the induction of WeibelPalade body exocytosis. J Thromb Haemost. 2016;14(11):2274-2286.
31. Zheng L, Abdelgawwad MS, Zhang D, et al. Histone-induced thrombotic thrombocytopenic purpura in adamts13-/- zebrafish depends on von Willebrand factor. Haematologica. 2020;105(4):1107-1119.
32. Ruiz-Torres MP, Casiraghi F, Galbusera M, et al. Complement activation: the missing link between ADAMTS-13 deficiency and microvascular thrombosis of thrombotic microangiopathies. Thromb Haemost. 2005;93(3):443-452.
33. Ferrari S, Scheiflinger F, Rieger M, et al. Prognostic value of anti-ADAMTS 13 antibody features (Ig isotype, titer, and inhibitory effect) in a cohort of 35 adult French patients undergoing a first episode of thrombotic microangiopathy with undetectable ADAMTS 13 activity. Blood. 2007;109(7):2815-2822.
34. Ferrari S, Mudde GC, Rieger M, Veyradier A, Kremer Hovinga JA, Scheiflinger F. IgG subclass distribution of anti-ADAMTS13 antibodies in patients with acquired thrombotic thrombocytopenic purpura. J Thromb Haemost. 2009;7(10):1703-1710.
35. Widemann A, Pasero C, Arnaud L, et al. Circulating endothelial cells and progenitors as prognostic factors during autoimmune thrombotic thrombocytopenic purpura: results of a prospective multicenter French study. J Thromb Haemost. 2014;12(10):1601-1609.







