17 minute read
Don’t Be Afraid of Contamination Testing


Bridget Gegorski, PharmD, was convinced her hospital system had done an outstanding job preparing for the implementation of USP General Chapter <800>.
“Particularly for [NIOSH] Table 1 drugs and chemotherapy, we had implemented a number of processes that significantly cut down on hazardous drug contamination,” Dr. Gegorski, the medication safety officer at University Hospitals Health System, in Cleveland, told Pharmacy Practice News. These steps included both administrative controls, such as staff training, limiting access to areas containing hazardous drugs (HDs) and good housekeeping practices, as well as engineering controls such as biological safety cabinets, proper ventilation and negative pressure rooms.
But surface contamination testing told her those steps had not been sufficient. “When we conducted surface contamination testing, we were still getting at least low positives in the environment, in places where we wouldn’t expect it—areas where we know the operators are doing things correctly and probably are even more cautious than I am about processes, so we didn’t expect to find contamination there,” said Dr. Gegorski, who spoke with Pharmacy Practice News in advance of a presentation on challenges in surface contamination testing at the 2021 virtual annual meeting of the National Home Infusion Association (NHIA).
Studies have documented the importance of wipe sampling programs for identifying the sources of surface contamination in facilities that manage HDs. In 2019, researchers from the University of North Carolina retrospectively analyzed 5,842 individual surface wipe samples from 338 pharmacies over six years. Depending on the location and surface tested, 3.94% to 25.96% of samples had high levels of HD contamination, and the researchers concluded the highest levels of contamination were in preparation areas. Repeated wipe sampling lowered overall HD contamination: 45.24% of samples detected HDs with the first wipe compared with 31.64% for subsequent wipes (Am J Health Syst Pharm 2019;76[9]:591-598).
After the first round of surface wipe sampling in December 2018 detected contamination in several areas, University Hospitals engaged in all the recommended steps that should follow a finding of HD contamination, including: • cleaning the location with a deactivation/decontamination agent; • evaluating proper use of closed system drug-transfer devices; • observing work practice controls and the use of personal protective equipment (PPE); • revisiting PPE doffing procedures; • evaluating cleaning procedures and the cleaning and decontamination agents used; and • assessing the appropriateness of cleaning frequency.
“We did all that, and we were still finding positives, so we were trying to figure out where we were having particles escape,” Dr. Gegorski said. “Finally, we realized that HD particles found on the outside of vials from our wholesaler could be a significant source of contamination.”
Several recent studies demonstrated that shipments of HDs can come into hospital pharmacies with existing surface contamination. For example, a 2020 literature review of 24 articles from 11 countries concluded that the majority of antineoplastic vials have surface contamination when they arrive (Eur J Hosp Pharm 2020;27[5]:313-314). So University Hospitals added a new step to its HD mitigation processes. “While USP <800> does not require this, our wholesaler has consented to labeling totes that contain HDs,” Dr. Gegorski said. “Those totes are immediately taken to a receiving area within the HD cleanroom where we store all oncology products. We don’t open the tote until we are in that area and then we wipe down all the container surfaces.”
Pharmacy leadership may fear implementing a surface wipe contamination testing plan because they don’t want to find positives. “USP <800> says that you should do this testing, not that you must do it,” she noted. “But if you start testing and you find positives—and you’re almost certainly going to find positives—then ‘should’ becomes ‘must.’ You have to mitigate, document your action and then retest. People may not be too keen on that, but fortunately at our institution, our leaders supported our implementation of the process. Having leadership support is key.”
Staff education is vital to a good sampling program, Dr. Gegorski said. “After our first round of sampling in December 2018, we realized that there was a lot of confusion and misinformation. We created education to explain the benefits of testing for HD contamination, the controls that are currently in place to prevent occupational exposure,

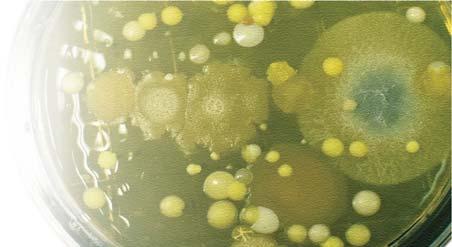
The number of colony-forming units on this cleanroom sample exceeded the action level and mold was recovered. These results prompted an investigation into the source of the contamination and remediation efforts to prevent recurrence.
see CONTAMINATION TESTING, page 12
continued from page 10
how to reduce HD contamination in everyday practices, and next steps to take if a positive result is found.”
A town hall, held in early 2019, offered an open forum to staff to ask questions related to HD surface sampling.
University Hospitals’ second round of HD sampling took place in February 2019. “We got better results at some of our sites that had been struggling during that round, and we realized that a lot of the December bad samples had come from older compounding facilities,” Dr. Gegorski said. “We conducted a refresh on our compounding areas and now just about every one of our sites has a sterile compounding suite that is, if not brand new, at least refurbished, including deep cleaning of older hoods that was able to cut contamination back.”
Her key message is don’t be afraid of surface contamination testing. “People are afraid of what they’ll find if they start testing, so they don’t do it,” she said. “That’s misguided. I actually think it should be required, not optional. This is an essential thing we need to do to protect our employees, to identify where we’re having problems and do focused improvement.”
Microbial Sampling
Sampling in the cleanroom isn’t just for HD contamination. The 2019 version of USP Chapter <797> lays out requirements for air and surface sampling programs for viable microbial contamination (bioburden) in sterile compounding facilities. Although it is more specific about certain elements of the program, such as air sample volumes, having a diagram of sampling locations and specifying collection procedures for those locations, “the requirements in the chapter still don’t go into enough detail,” said Abby Roth, the senior director of business operations for CriticalPoint. “It tells you what you need to do, but not how to clearly do it. However, in 2020, the Controlled Environmental Testing Association (CETA) updated its testing guide, ‘CAG-009 Viable Environmental Monitoring for Sterile Compounding Facilities,’ and it provides best practices recommendations to close the gaps in the chapters. It’s a great reference to help you ensure you have a good sampling program.” (The document is available to CETA members; membership costs $245 annually.)
The testing guide provides a list of all the documentation you should have after conducting a sampling session, she said. “That’s really where pharmacies tend to not have all the pieces to put the whole story together,” Ms. Roth told Pharmacy Practice News. For example: “Have you captured the purpose of the sampling session? Is it routine monthly sampling or is it investigational due to environmental excursions? If you do an investigational sampling and that’s not marked on the report, surveyors will ask what this is about,” she said. “It also addresses things such as controls for your media, how to ship the sampling plates to a lab, and how to interpret reports that you receive.”
Another key element of a good microbial sampling program that many institutions neglect, Ms. Roth said, is the control plate. “People often fail to understand what the purpose of control plates would be and why they should use them. They tend to use ‘negative controls,’ sending unused media to the lab to be incubated with the samples, hoping nothing grows. But you never know what happens as your packages go through shipping. If you send an additional ‘positive sample’ to the lab, which is an unused plate the lab will purposely inoculate with organisms, you have a mechanism to know that the media is still good.”
Depending on what your viable sampling report yields, Ms. Roth advised doing a methodical investigation and only addressing one variable at a time to identify the root cause if environmental excursions exceed the action levels described in the chapter. “You don’t want to attack every possible source of the contamination at once because then you won’t be able to pinpoint where the actual source of contamination is coming from.”
In a presentation at the NHIA meeting, Ms. Roth advised pharmacies to have a standard operating procedure for postsampling investigations. “Go in with an open mind as to the source of the contamination. Let the identification of the recovered microorganisms point to where you need to spend your time investigating,” she said. “You can start with lowhanging fruit, such as garbing, and then move on to something like material handling. Make those adjustments and then resample and see what comes back. If you continue to have persistent contamination, then you’re looking at other things like environmental concerns. Is there a facility issue? Is something not sealed properly and you’re getting ambient outside air into the cleanroom?”
The type of organism identified in your sterile compounding environment will be key and will provide clues to identifying the point sources of air and surface microbial contamination, although some identified microorganisms may provide more specific direction than others. “We tend not to see a lot of gram-negatives in the cleanroom suite, so if you see something like Pseudomonas, which tends to breed in damp areas, there’s a good chance you have a problem with your sink,” Ms. Roth said. “On the other hand, something like Bacillus is an ‘everywhere organism,’ so ubiquitous that it is probably going to take multiple attempts to figure out where it is coming from.” —Gina Shaw

Documenting the sampling session is essential. This information can yield clues in the event of a microbial excursion or hazardous drug contamination.
The sources reported no relevant fi nancial disclosures.
NOW AVAILABLE The First and Only Antiemetic Approved for Rescue Treatment of PONV Despite Prophylaxis



• Barhemsys is a selective dopamine-2 and dopamine-3 receptor antagonist, offering a pharmacological option with demonstrated 1 • in patients who failed prophylaxis: 42% vs 29% achieved the primary endpoint of complete response; P=0.0031,2,* • In patients who failed prophylaxis, 70% (160/230) of patients met the criteria for complete response at 2 hours after receiving a 10 mg dose of Barhemsys compared with 49% (116/235) of placebo-treated patients (secondary endpoint)2,* • The most common adverse reaction reported for Barhemsys 10 mg (6% vs 4%)1,†
Scan for additional information or visit Barhemsys.com


Packaging and vial shown are not actual size.
Indications
Barhemsys is a selective dopamine-2 (D2) and dopamine-3 (D3) receptor antagonist indicated in adults for: • •

Select Important Safety Information
Please see Brief Summary of Prescribing Information for Barhemsys on next page.


† 1. 2. Anesthesiology. 2019;130:203-212.

Delivers when it matters most ™
See package insert for full Prescribing Information
Indications: Barhemsys is a selective dopamine-2 (D
receptor antagonist indicated in adults for: 2) and dopamine-3 (D3) • prevention of postoperative nausea and vomiting (PONV), either alone or in combination with an antiemetic of a different class • treatment of PONV in patients who have received antiemetic prophylaxis with an agent of a different class or have not received prophylaxis Dosage & Administration: The recommended adult dosage of Barhemsys: • Prevention of PONV, either alone or in combination with another antiemetic: 5 mg as a single intravenous dose infused over 1 to 2 minutes at the time of induction of anesthesia. • Treatment of PONV: 10 mg as a single intravenous dose infused over 1 to 2 minutes in the event of nausea and/or vomiting after a surgical procedure. Protect from light. Barhemsys is subject to photodegradation. Administer Barhemsys within 12 hours of removal of the vial from the protective carton. See full prescribing information for preparation and administration instructions. Dosage Forms and Strength: Injection: 5 mg/2 mL (2.5 mg/mL) or 10 mg/4 mL (2.5 mg/mL) as a clear, colorless sterile solution in a single-dose vial. Contraindication: Barhemsys is contraindicated in patients with known hypersensitivity to amisulpride. QT Prolongation: Barhemsys causes dose- and concentration-dependent prolongation of the QT interval. The recommended dosage is 5 mg or 10 mg as a single intravenous (IV) dose infused over 1 to 2 minutes. Avoid Barhemsys in patients with congenital long QT syndrome and in patients taking droperidol. Electrocardiogram (ECG) monitoring is recommended in patients with pre-existing arrhythmias/cardiac conduction disorders, electrolyte abnormalities (e.g., hypokalemia or hypomagnesemia), congestive heart failure, and in patients taking other medicinal products (e.g., ondansetron) or with other medical conditions known to prolong the QT interval. Adverse Reactions: Common adverse reactions reported in ≥ 2% of adult patients who received Barhemsys 5 mg (N=748) and at a higher rate than placebo (N=741) in clinical trials for the prevention of PONV were: chills (4% vs. 3%), hypokalemia (4% vs. 2%), procedural hypotension (3% vs. 2%), and abdominal distention (2% vs. 1%). Serum prolactin concentrations were measured in one prophylaxis study where 5% (9/176) of Barhemsys-treated patients had increased blood prolactin reported as an adverse reaction compared with 1% (1/166) of placebo-treated patients. The most common adverse reaction, reported in ≥ 2% of adult patients who received Barhemsys 10 mg (N=418) and at a higher rate than placebo (N=416), in clinical trials for the treatment of PONV was infusion site pain (6% vs. 4%).
Drug Interactions:
• Barhemsys causes dose- and concentration-dependent QT prolongation. To avoid potential additive effects, avoid use of Barhemsys in patients taking droperidol. • ECG monitoring is recommended in patients taking other drugs known to prolong the QT interval (e.g., ondansetron). • Reciprocal antagonism of effects occurs between dopamine agonists (e.g., levodopa) and Barhemsys. Avoid using levodopa with Barhemsys. Postmarketing Experience: The following adverse reactions have been identified during postapproval chronic oral use of amisulpride outside of the United States (Barhemsys is not approved for oral dosing or chronic use). Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure: Blood and Lymphatic System Disorders: agranulocytosis; Cardiac Disorders: bradycardia, torsades de pointes, ventricular tachycardia, prolonged QT by electrocardiogram; General Disorders: neuroleptic malignant syndrome; Immune System Disorders: angioedema, hypersensitivity, urticaria; Hepatic Disorders: increased hepatic enzymes; Nervous System Disorders: agitation, anxiety, dystonia, extrapyramidal disorder, seizure; Psychiatric Disorders: confusional state, insomnia, somnolence; Vascular Disorders: hypotension. Use in Specific Populations: Pregnancy—Risk Summary: Available data with amisulpride use in pregnant women are insufficient to establish a drug associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, there were no adverse developmental effects observed with oral administration of amisulpride in rats and rabbits during the period of organogenesis at exposures about 43 and 645 times, respectively, the exposure delivered by the highest recommended human dose (see Data). The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. Data—Animal Data: Reproduction studies of amisulpride were conducted in pregnant rats administered oral doses up to 160 mg/ kg/day (43 times the exposure based on area under the curve (AUC) at the highest recommended dose of 10 mg) throughout the period of organogenesis. No adverse embryo-fetal developmental effects were observed at any dose level. Maternal animals exhibited a dose-related decrease in overall mean body weight gain. In rabbits administered amisulpride throughout the period of organogenesis, oral doses up to 210 mg/kg/day (645 times the exposure based on AUC at the highest recommended dose of 10 mg) had no adverse developmental effects on the fetus. Maternal animals exhibited reduced mean body weight gain at doses of 100 and 210 mg/kg/day and reduced food intake was observed at 210 mg/kg/day. The pre- and post-natal developmental effects of amisulpride were assessed in rats administered oral doses of 60, 100 or 160 mg/kg/day during the periods of organogenesis and lactation. At 160 mg/kg/day (43 times the exposure based on AUC at the highest recommended dose of 10 mg), maternal animals exhibited a reduction in mean body weight gain and decrease in food intake during lactation. Amisulpride had no effect on maternal pregnancy parameters, litter survival or pup growth, development or maturation at any dose tested. Lactation—Risk Summary: Based on case reports in published literature, amisulpride is present in human milk at concentrations that are 11- to 20-fold higher than human plasma in patients taking multiple oral doses of amisulpride (200 to 400 mg/day). The estimated infant daily dose ranged from 5% to 11% of the maternal dose. There are ways to minimize drug exposure to a breastfed infant (see Clinical Considerations). There are no reports of adverse effects on the breastfed child and no information on the effects of amisulpride on milk production. The pharmacological action of amisulpride, a dopamine-2 (D2) receptor antagonist, may result in an increase in serum prolactin levels, which may lead to a reversible increase in maternal milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Barhemsys and any potential adverse effects on the breastfed child from Barhemsys or from the underlying maternal condition. Clinical Considerations: A lactating woman may consider interrupting breastfeeding and pumping and discarding breast milk for 48 hours after Barhemsys administration to minimize drug exposure to a breastfed infant. Females and Males of Reproductive Potential—Infertility: In animal fertility studies, administration of repeated doses of amisulpride over a 10-day period to female rats resulted in infertility that was reversible. Pediatric Use—Safety and effectiveness in pediatric patients have not been established. Geriatric Use—No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Renal Impairment—Avoid Barhemsys in patients with severe renal impairment (estimated glomerular filtration rate [eGFR] < 30 mL/min/1.73 m2). The pharmacokinetics of amisulpride in patients with severe renal impairment have not been adequately studied in clinical trials. Amisulpride is known to be substantially excreted by the kidneys, and patients with severe renal impairment may have increased systemic exposure and an increased risk of adverse reactions. No dosage adjustment is necessary in patients with mild to moderate renal impairment (eGFR ≥ 30 mL/min/1.73 m2). Overdosage: Doses of oral amisulpride (Barhemsys is not approved for oral dosing) above 1200 mg/day have been associated with adverse reactions related to dopamine-2 (D2) antagonism, in particular: • cardiovascular adverse reactions (e.g., prolongation of the QT interval, torsades de pointes, bradycardia and hypotension). • neuropsychiatric adverse reactions (e.g., sedation, coma, seizures, and dystonic and extrapyramidal reactions). There is no specific antidote for amisulpride overdose. Management includes cardiac monitoring and treatment of severe extrapyramidal symptoms. Since amisulpride is weakly dialyzed, hemodialysis should not be used to eliminate the drug. How Supplied/Storage and Handling: Barhemsys is supplied as follows: • NDC 71390-125-20: Package of 10 cartons. Each carton (NDC 71390-125-21) contains one single-dose vial of Barhemsys (amisulpride) injection, 5 mg in 2 mL (2.5 mg/mL). • NDC 71390-125-50: Package of 10 cartons. Each carton (NDC 71390-125-51) contains one single-dose vial of Barhemsys (amisulpride) injection, 10 mg in 4 mL (2.5 mg/mL). Store vials at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature]. Patient Counseling Information: QT Prolongation—Instruct patients to contact their healthcare provider immediately if they perceive a change in their heart rate, if they feel lightheaded, or if they have a syncopal episode. Drug Interactions—Advise patients to report to their healthcare provider if they are taking drugs which prolong the QT interval. Lactation—Women may consider reducing infant exposure through pumping and discarding breastmilk for 48 hours after Barhemsys administration.
BAR HCP BS 10/2020






