10 minute read
Phenylketonuria


PHENYLKETONURIA: A LIFE-LONG JOURNEY
Phenylketonuria (PKU) is a rare inherited error of metabolism caused by a deficiency in the enzyme phenylalanine hydroxylase (PAH).1 Loss of this enzyme results in mental retardation and organ damage. A low-protein diet for life is the mainstay of treatment and is especially important in maternal PKU, which can severely compromise pregnancy.
Advertisement
Dr Shazia Faisal ANutr
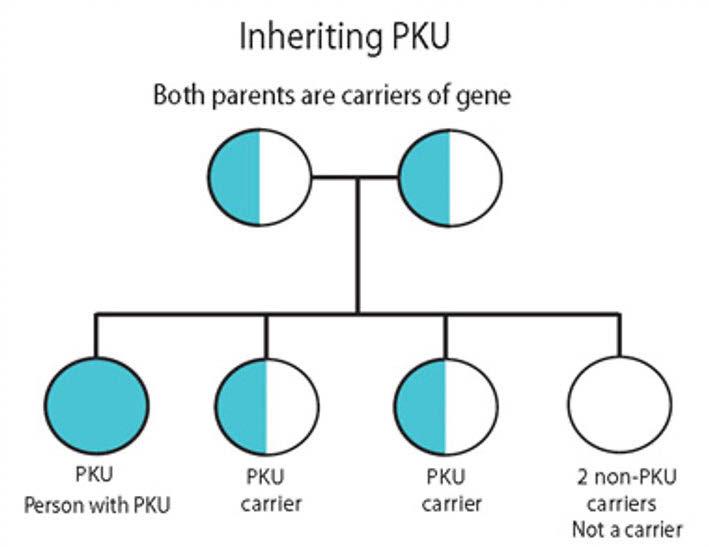
PKU affects around 1 in 10,000 babies born in the UK every year, with males and females equally affected. Globally, PKU varies amongst ethnic groups, races and geographic regions.3 It is an autosomal recessive disorder, meaning an individual needs two copies of the mutant gene that causes PKU.5 The human gene for PAH is located at chromosome 12q23.2, and more than 600 mutations of the PAH gene have been reported by scientists.5,6,7 If both parents are carriers of a mutant gene, then there is a one-in-four chance of the child being affected by PKU.
PAH is responsible for the conversion of phenylalanine (Phe) into tyrosine and other by-products (see Figure 1). Its deficiency, if untreated, results in the accumulation of Phe in the body, which can cause irreversible brain damage.1,2
PKU was first described by Norwegian physician Dr Asbjørn Følling in 1934.1,2,3 He found higher levels of phenylpyruvic acid, an intermediate product of Phe metabolism, in the urine of intellectually impaired individuals and thus named the disease. In 1969, the universal screening programme for the early detection of PKU was introduced in several developed countries including the UK, Europe and the USA.8 It is a simple heel prick test performed in the first 24-72 hours after birth. Dried blood spots are the best sample for the simultaneous measurement of Phe, pterins and dihydropterin reductase activity from a single specimen.4,5
LEVELS AND SYMPTOMS There is no specific classification of PKU. Nevertheless, on the basis of PAH activity levels and blood Phe levels on diagnosis or when untreated, it is divided into three types: • Classic PKU is when PAH is completely missing and Phe levels are 1200µmol/L or above.9,10 • Moderate PKU is caused by residual enzyme activity (Phe levels are 900-1200µmol/L). • Mild PKU is when Phe concentration is 600-900µmol/L.
Shazia is a Medical Doctor with a special interest in metabolic and lifestyle medicine. She is a Community Nutritionist and a Nutritional Blogger.
REFERENCES Please visit: nhdmag.com/ references.html
Figure 1: Phenylalanine metabolism inside the human body
Dietary protein
Phenylpyruvic acid Phenylalanine Tyrosine Phenylketonuria
KETOGENICS
600K
people in the UK diagnosed with epilepsy and receiving anti-epileptic drug (AED) treatment; that’s 1 in every 103 people1
36%
Epilepsy patients have inadequate control of seizures with AEDs2
Drug Resistant Epilepsy
Failure of two or more appropriately chosen AEDs to achieve seizure freedom2
l
Increased risks
• Injury • SUDEP • Hospital visits • Depression and anxiety • Developmental issues4
AED side e ects
commonly associated with drowsiness, blurred vision, dizziness, nausea and vomiting3
Ketogenic Diet Therapies 50%
chance of reducing seizures6
More palatable than ever before
This information is intended for healthcare professionals only. The Ketocal range are Foods for Special Medical Purposes for the dietary management of drug resistant epilepsy or other conditions where the ketogenic diet is indicated and must be used under medical supervision.
Candidates for Ketogenic Diet
Therapies5
• Angelman syndrome • Complex 1 mitochondrial disorders • Dravet syndrome • Epilepsy with myoclonic-atonic seizures (Doose syndrome) • Glucose transporter protein 1 (Glut-1) deficiency syndrome • Febrile infection-related epilepsy syndrome (FIRES) • Formula-fed (solely) children or infants • Infantile spasms • Ohtahara syndrome • Pyruvate dehydrogenase deficiency (PDHD) • Super-refractory status epilepticus • Tuberous sclerosis complex • The failure of 2 or more AEDs
References
1. Joint Epilepsy Council (2011) ‘Epilepsy Prevalence, Incidence and Other Statistics’,
Available at: www.jointepilepsycouncil.org.uk (Accessed: Sept 2020) 2. Kwan, P., Arzimanoglou, A. and Berg, A (2010) ‘Definition of Drug-resistant Epilepsy:
Consensus Proposal by the Ad Hoc Task Force of the ILAE Commission on Therapeutic
Strategies’, Epilepsia, 51(6), pp. 1069–1077 3. Epilepsy Foundation (2018) ‘Risks with Epilepsy’, Available at: www.epilepsysociety.org.uk/risks-epilepsy#.W6DyWehKjIU (Accessed: Sept 2020) 4. Epilepsy Society (2018) ‘Risks with Epilepsy’, Available at: www.epilepsysociety.org.uk/risks-epilepsy#.W6DyWehKjIU (Accessed: Sept 2020) 5. Kossoff, E. et al., 2018 ‘Optimal Clinical Management of Children Receiving Dietary
Therapies for Epilepsy: Updated Recommendations of the International Ketogenic Diet
Study Group’. Epilepsia Open: 1–18 6. Martin K et al. Cochrane Database of Systematic Reviews 2016:CD001903.pub3
DRUG RESISTANT EPILEPSY Medication is not always the answer.
Various conditions, such as pterin defects, liver disease and high protein intake, also cause high Phe levels, so must be differentiated in the neonatal period.9,10
Clinical manifestations of PKU depend on the blood levels of Phe and time of diagnosis. Symptoms of untreated PKU include growth failure, intellectual disabilities, seizures, mood disorders and eczema.5,6,7 Early detection and treatment in newborns may reduce the severity of symptoms of PKU, but some studies report that psychomotor abnormalities still present in some early treated PKU patients who have poor metabolic control.10,11 Nevertheless, immediate dietary intervention before 10-14 days of age is essential for the prognosis so that patients can achieve almost normal clinical outcomes, as long as blood Phe concentrations are maintained at the correct level.
PROGRESSION OF THE DISEASE The exact pathogenesis of PKU remains unclear. How higher levels of Phe cause brain damage and impair cognitive function in patients is still questionable. However, there are some hypotheses that suggest different factors, such as decreased production of neurotransmitters, white matter abnormalities, imbalances of large neutral amino acids (LNAA) and oxidative stress.13-15 PAH deficiency blocks the conversion of Phe into tyrosine, which is a precursor of neurotransmitters such as dopamine, adrenaline and noradrenaline. Low levels of these important brain chemicals result in abnormal brain development, intellectual disability and psychological disorders.
Some studies have shown MRI scans of PKU patients to reveal abnormalities in white matter.14,15 White matter is mainly composed of neurons that process and transmit information. Hypothetically, higher levels of Phe damage the myelin sheath of neurons (hypomyelination) and slow down the process and transport of information, as evidenced by poor executive function performance in PKU patients.12-15 The blood-brain barrier is a special system, which controls the influx of nutrients and toxins to the brain cells.16 LNAA (Phe, tyrosine, tryptophan, leucine and isoleucine) are transported to the brain via a carrier system, which has a high affinity to Phe. When blood Phe levels are high, this transport system becomes saturated and the migration of other LNAA is inhibited.16,17 The protein substitutes taken by patients with PKU as a mainstay of their dietary regimen, all contain LNAA.
DIETARY MANAGEMENT: DIET FOR LIFE The goal of dietary management for those with PKU is to maintain plasma Phe concentration to support optimal growth, development and mental functioning, while providing a nutritionally complete diet. In order to do this, PKU management is by way of a low-protein diet virtually devoid of Phe and intake of protein substitutes available as phenylalanine-free aminoacid mixtures (AAM), glycomacropeptide-based protein substitutes (GMP) and LNAA. The PKU diet is well established, safe and effective, and, consequently, remains the cornerstone of PKU management.
Diet for life is now encouraged, as the prefrontal cortex development continues through adolescence and is not complete until adulthood, and so the toxicity of Phe is, therefore, present well into adult life. This was corroborated in the 2017 European Guidelines for Diagnosis and Management of PKU. 26 It appears that adult patients who stop the PKU diet seem to experience poor executive functioning, information processing and mood, including anxiety, depression and low self-esteem.18,32-35 Reports of neurological problems have also been noted, even with good metabolic control and continued Phe restriction.36,37 Research shows that these complications can be reversed in patients who return to a Phe-restricted diet.20,38
In most people with PKU, the natural protein or dietary Phe is restricted to 25% or less of a regular intake to maintain blood phenylalanine concentrations within European PKU guidelines target ranges.26 This usually requires avoidance/ restriction of all high-protein foods. There are many regular foods which are naturally very low in protein that can be eaten without measurement.
Although all fruit and vegetables containing phenylalanine ≤75mg/100g contribute a small amount of daily Phe, it’s generally not enough to affect blood control, and are given without restriction. Certain foods such as potatoes and beetroot and parsnip crisps are concentrated in Phe and can be calculated/measured in the daily Phe allowance.
The NSPKU website gives plenty of expert advice on low-protein food and food exchanges/ measures: www.nspku.org/dietary-information.
ISSUES WITH RESTRICTING THE LIFELONG DIET A low- or restricted-Phe diet for life is very complex and restrictive. Low adherence often leads to the total discontinuation of the diet.22,23
Whilst certain foods like low-Phe flour, pasta and cookies have increased options for PKU patients, the diet is challenging in respect of meal preparation, limited food choices and palatability of protein supplements.21,22 It is reported by one study that around 45% of the UK PKU adult
population are not following their low-protein diet.19 However, studies have shown that high blood Phe is inversely related with IQ levels in adults who stop their diet in their childhood or early adulthood and also causes learning disabilities, psychomotor abnormalities and even neurological complications in people with PKU.18,19
NUTRITIONAL DEFICIENCIES A Phe-restricted diet may cause nutrient deficiencies in zinc, iron, vitamin A and vitamin D, for example.22 However, most of the modern protein substitutes, which are part of the PKU dietary regimen, supply these micronutrients and, if taken in the correct dosage, keep micronutrient deficiencies minimal. Low bone mass density (BMD) is associated with osteopenia and fractures, as reported by some studies. The exact cause of low BMD in patients with PKU is unclear; however, it may be linked to low intake of calcium, phosphorus and vitamin D.26
The lifelong restriction of protein intake for PKU patients may be accompanied by insufficient dietary intake of long-chain polyunsaturated fatty acids (LCPUFA) like arachidonic acid (AA) and docosahexaenoic acid (DHA). DHA is essential for normal brain development and visual development.22-24 In children with PKU, low dietary DHA results in low blood and tissue DHA levels and causes adverse consequences for the central nervous system, such as learning difficulties, behavioural problems and visual problems. Currently, many protein substitutes for PKU patients are formulated with DHA.25
MATERNAL PKU Maternal PKU is considered high risk, as metabolic control is required throughout pregnancy. It is recommended by the European PKU guidelines, that women should maintain Phe levels in the range of 120-360µmol/L before conception and during pregnancy, by following the low-protein diet.25,26 During pregnancy, Phe crosses the placenta by active transport, resulting in a 70% to 80% increase in foetal concentration of Phe compared with maternal concentration.27,28 Tight metabolic control in early pregnancy is critical for the development of the foetus, and higher Phe levels are teratogenic, causing complications like microcephaly, congenital heart disease and developmental delay.28,29
Nausea and vomiting are common among all pregnancies, but, in women with PKU, these symptoms are associated with poor metabolic control. Decreased appetite, low caloric food and poor adherence to protein supplements, all increase the blood Phe levels during pregnancy.29 Women who are unable to maintain their metabolic control can be prescribed sapropterin (Kuvan) in the UK (see below).26,28
Breastfeeding should always be encouraged as breast milk is low in Phe (46mg/100ml).26 Newborns with inherited PKU can take their mother’s breast milk along with Phe-free infant formula under strict monitoring. Many women with PKU stop their low-protein diet following the birth of their baby.29 They may not perceive a need for continued treatment, or they may consider the diet too challenging whilst caring for their infant.30,31
According to the European PKU guidelines, all women should receive regular nutritional support post-pregnancy.26 Women also need to be aware that the postpartum period is high risk for unintended pregnancy, so family planning and contraceptive advice remain important.26,30
OTHER TREATMENTS Sapropterin dihydrochloride is a synthetic formulation of tetrahydrobiopterin (BH4), a naturally occurring co-factor for the enzyme PAH. Sapropterin is licensed in the UK for the treatment of mild to moderate PKU in adults and children of all ages who have been shown to be responsive to it. It may increase daily Phe tolerance two- to threefold improving metabolic control.
CONCLUSION PKU is an inherited genetic disorder, which is screened for at birth with the newborn blood spot test. Once diagnosed, treatment can begin straight away to reduce the risk of serious complications. A strict low-protein diet for life allows individuals with PKU to live long and healthy lives. Due to the restrictive nature of the diet, further research should be focused on the pathophysiology of this disorder, aiming for better treatment options.






