
13 minute read
THE POWER WITHIN


FIGHTING CANCER WITH ADOPTIVE CELL THERAPY
By XIYA WU SWATHI BALAJI
Advertisement
In 2012, a clinical trial made headlines throughout the nation as 7-year-old Emily Whitehead became the first pediatric patient to undergo T-cell therapy. For Emily, CAR T-cell therapy was a last resort, after chemotherapy had twice resulted in relapse. The weeks following the reintroduction of the engineered T-cells into her veins were difficult. She developed a high-grade fever and her blood pressure dropped dangerously in what we now know are the effects of an inflammatory response known as cytokine release syndrome (CRS) 3 . Ultimately, the treatment was a success, paving the way for T-cell therapy to be recognized as one of the most promising fields in the future of cancer treatment.
In the last decade, adoptive cell therapy (ACT) has taken the world of cancer treatment research by storm. A type of ACT, CAR T-cell therapy has made remarkable strides from its use in small clinical trials
Emily Whitehead and her mother Kari Whitehead. Taken by Jeff Swensen for the New York Times 4 .
to its use in the United States as the first type of gene-based therapy approved by the Food and Drug Administration (FDA) 1 . At the core of ACT is the idea of harnessing the patient’s own immune system to fight against cancer. For the standard cold or flu, the immune cells in our bodies are usually able to mount a highly effective response, and the accompanying symptoms are only temporarily uncomfortable. More impressively, the immune system has memory of the diseases it has encountered before. If the same virus comes knocking again, the secondary response from the immune system is both faster and stronger because the accumulated memory cells "remember" the antigens in question. Recognition of outsiders is crucial to the immune system's function. To prevent our immune system from turning on the body, immune cells must be able to distinguish "self " from "non-self." Failure to do so is what results in autoimmune diseases, where the immune system launches an immune response against its own antigens.
Cancer, however, hinges on the opposite problem. Because cancer cells are the descendants of our normal cells, they can evade the immune system by disguising themselves as "self " despite their existence being harmful to the body. They are wolves in sheep's clothing. T-cell therapy, then, involves the shearing away of this disguise. Researchers generate CAR T-cells by genetically engineering the patient’s T-cells to produce chimeric antigen receptors (CARs) on the cell surfaces 1 . Like the Greek Chimera that existed as a hybrid of animal forms, these receptors are a fusion of different components decided by the researcher. The modified cells are then reintroduced into the patient and allowed to proliferate, truly earning them
T-cell activation or immune cell lysis as the result of T-cell therapy induces release of various cytokines. These cytokines activate other immune cells, leading to greater cytokine release and a positive feedback loop that results in a "cytokine storm." Graphic by Shimabukuro-Vornhagen et al (2018) 12 .
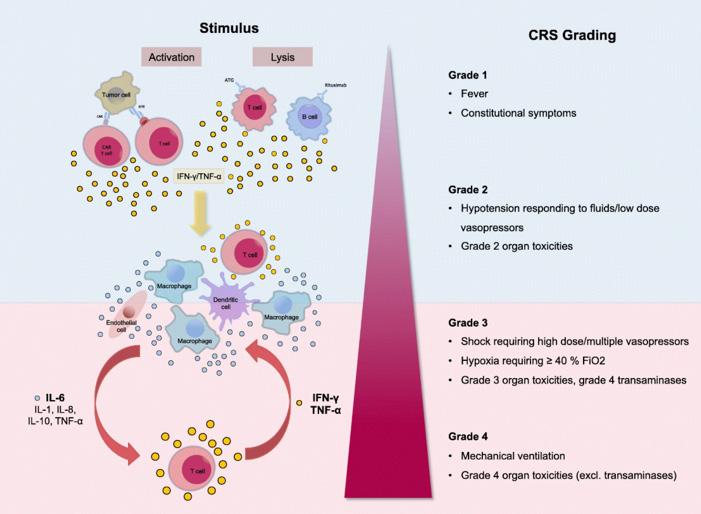
the moniker of a "living drug." The presence of CARs gives the T-cells new vision, allowing them to recognize common antigens present on cancer cells. The cancer cell's disguise is stripped away, and the T-cells now see the wolf for what it is 2 . Nevertheless, with every new treatment comes new side effects and downsides that need to be overcome with more research. Around the same time that Emily was declared to be in remission, another child, Poppy*, was treated with the new therapy. Poppy improved similarly for a period of time before her cancer relapsed and no longer responded to the same treatment. The cause? Her leukemia cells had returned with new antigens, made once again invisible to the CAR T-cells engineered to look for different markers 4 . Finding solutions to this "antigen escape," along with mitigating the effects of CRS that many patients who undergo T-cell therapy experience, are one of the primary goals of new research in this field. In recent years, T-cell therapy has evolved beyond the comparatively simple constructions of before, now including new lines such as bispecific T-cell engagers (BiTEs), and engager T-cells, and dual CAR T-cells.
T Mu h f a G Th g: Cyt k R l a Sy r m As described in Emily's case, a common side effect of T-cell therapy is cytokine release syndrome (CRS). As the name suggests, CRS involves the release of cytokines in the body. Under normal conditions, these cytokines are critical for effective functioning of the immune system. They are responsible for modulating
the activation, proliferation and differentiation of various cell types and regulating antibody production. When functioning as expected, cytokines can control the intensity and length of the immune response 5 . However, CRS occurs when immune cells release quantities of inflammatory cytokines many times, sometimes thousands of times higher than expected. This is when problems arise. The various cytokines flood the body and can even prompt other immune cells to produce additional cytokines in what is known as a positive feedback loop, a vicious cycle of immune cell activation and cytokine release that culminates in a "cytokine storm." Clinically, CRS presents as a range of symptoms, including anything from mild, flu-like symptoms to severe symptoms such as hypotension, high fever and eventually multiple organ failure. Children in particular are at a higher risk of developing CRS, hypothesized to be the result of their more immature immune systems 6 . CRS can be deadly. In Emily’s case, were it not for immediate and fortunate interventions taken to treat the syndrome, the happy story may very well have taken a different turn. At the same time, Emily didn’t have very many good options left; CAR T-cell therapy had been the last resort. However, the problem with CAR T-cells is that it is administered in a single dose and it is hard to predict how much the cells expand once inside the body 6 . There would be no way to stop the treatment midway if, for example, the effects of CRS became too severe. It's all or nothing.
Thus, researchers turned their attention to ways to address this problem. How do we make T-cell therapy safer for patients? In one clever treatment alternative, CAR T-cells are discarded in favor of smaller, injectable proteins called bispecific T-cell engagers (BiTEs). These BiTEs consist of two single-chain variable fragments connected by a short linker, and their name, much like many other inventions in this field, is highly suggestive of their function. Since BiTEs are proteins, not cells themselves, they work by directing T-cells already present within the patient's body to kill tumor cells. These resident T-cells are normally, as discussed, oblivious to the tumor threat. BiTEs force the T-cells to pay attention by binding to both tumor cells and T-cells simultaneously. One of the fragments in its structure is specific to a tumor antigen and the other specific to a cell-surface molecule on the T-cells. Once both cell types have been lassoed, BiTEs mediate T-cell responses and the killing of the tumor cell 7 . Unlike CAR T-cells, BiTEs can be given in multiple doses, so treatment can be interrupted if necessary, such as if a patient experiences severe CRS. Despite this safety benefit, BiTEs unfortunately have a very short half-life and are unable to proliferate in the body. Since one of the biggest draws of ACT is that they involve living drugs, BiTEs by themselves suffer a comparative disadvantage.
Another common safety feature now engineered into T-cells used in immunotherapy is the inclusion of a "safety switch," usually in the form of a suicide gene that responds to the addition of some drug. For example, for T-cells engineered to have a CD20 suicide gene, addition of the anti-CD20 antibody rituximab resulted in significant T-cell death 8 . With this safety feature, even though CAR T-cell therapy functions along the lines of "all-or-nothing," there is at least the option to pull the plug on the treatment if circumstances, such as uncontrollably severe CRS, necessitate such action.
What G Up: CAR T- ll Exhau t Though the selling point of CAR T-cell therapy is that it involves the use of living T-cells themselves, this fact is also a potential weakness. T-cells, like all cells in the body provided they're functioning correctly, cannot proliferate forever, nor are their existence static. The types of T-cells in a T-cell pool changes over time. With increasing age, there is a shift towards more effector and memory T-cells as opposed to undifferentiated naïve T-cells. Naïve T-cells are essentially "younger" T-cells. Compared to
their differentiated, older counterparts, naïve T-cells have improved proliferation and cytotoxicity, the ability to kill tumor cells. Before being injected into the patient’s body, T-cells need to first be extracted and engineered into CAR T-cells. The process takes days, so when the cells are reintroduced into the patient, they are older and thus more susceptible to exhaustion. Though the mechanisms of CAR T-cell exhaustion are not completely understood, researchers have found that exhausted CAR T-cells have poor expansion and cytotoxicity, which makes the therapy less likely to work.
In other words, time is of the essence. Methods have been used to improve T-cell expansion and persistence modifying the structure of the receptor. Another option is to rely on more than just the engineered cells. Patients often have T-cells present within the body, known as resident or bystander T-cells, that are unmodified but have potential to be recruited by engineered cells to help attack the tumor burden. These types of engineered T-cells, capable of recruiting resident T-cells, are called engager T-cells.
Engager T-cells can be thought of as a balance between BiTEs and CAR T-cells. BiTEs can redirect resident T-cells to tumor cells, but they are incapable of self-amplifying and have a relatively short half-life. Meanwhile, CAR T-cells have a much longer half
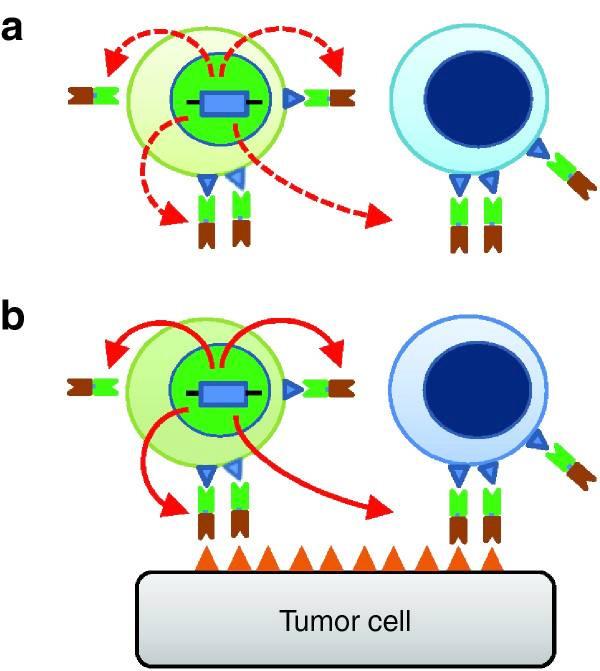
life but are not able to take advantage of the resident T-cells within the body. Engager T-cells possess the strengths of both of these alternatives. They are engineered T-cells, so they can proliferate in the body just like CAR T-cells, but engager T-cells are also capable of secreting engager molecules, which function just like BiTEs in recruiting resident T-cells. In other words, engager T-cells have both direct cytotoxic effects on tumor cells as well as indirect effects through the secretion of these engager molecules 11 . In these ways, T-cell therapy is paving promising paths to overcome and circumvent T-cell exhaustion.
Cat h M If Y u Ca : A t g E ap Poppy hadn’t been as lucky as Emily. Her cancer initially responded well to CD19 CAR T-cell therapy, but it relapsed, returning with a different uniform. The same CD19 CAR T-cells that worked so well were now useless because the relapsed cancer no longer expressed CD19 antigens. The antigen had escaped. The reasoning behind this occurrence is simple despite its devastating implications. Even if 99% of tumor cells express the CD19 antigen and are successfully targeted and killed by the CD19 CAR T-cells, the 1% that survive would be CD19 negative. The surviving tumor cells are capable, then, of proliferating and forming a new growth that is now 100% CD19 negative, effectively "immune" to the old treatment.
This is an all too common problem with T-cell therapy, one that's provoked many different suggestions on how to improve the treatment. One such suggestion involves the production of dual CAR T-cells. These T-cells, once again as the name suggests, would be engineered to target two different tumor antigens at once. For example, one Stanford team has engineered a CAR T-cell that would simultaneously target both CD19 and CD22 9 . The thought is that even if some percentage of tumor cells are CD19 negative, they can still be identified by this dual CAR T-cell if they express CD22.
Engager T-cells in action. Engager T-cells (green) secrete engager molecules, which consist of two fragments. One fragment (neon green) binds to antigens on both engager T-cells and the patient’s resident T-cells (blue). The other fragment binds to antigens on the tumor cell. In this way, engager T-cells can "teach" resident T-cells to recognize and kill tumor cells. Graphic by Iwahori et al (2015) 11 .
30 || pulse Though the potential of these T-cells is great, there are still many variables that need to be considered when engineering something like dual CAR T-cells. Though CD19 has been a tried and true tumor antigen to target, choosing a second antigen requires careful scrutiny. First, is this antigen highly expressed on tumor cells? More importantly, is this antigen not expressed on other tissue cells in the body or other T-cells? To choose an antigen that may be highly expressed on tumor cells but is also highly expressed elsewhere may lead to a phenomenon known as fratricide, where the engineered T-cells may end up killing fellow T-cells or even attacking healthy cells in the body 10 . Furthermore, dual CAR T-cells are, simply put, difficult to engineer. For some patients, even regular CAR T-cell therapy is not an option. Their T-cells do not respond to attempts to engineer them with necessary receptors, either because the cells are too fragile or exhausted from chemotherapy treatment or due to other unavoidable causes.
Nevertheless, advancements in T-cell therapy continue to persist as one of the brightest hopes for the future of cancer treatment. Evolving from CAR T-cells to now a whole host of different adoptive cell therapies, this field of research has only scratched the surface of its potential.
*Note: The real name of this patient has not been released to the public. She is referred to as "Poppy" in this article.
1. "CAR T Cells: Engineering Immune Cells to Treat Cancer." National Cancer Institute. Accessed February 23, 2020. https://www.cancer.gov/ about-cancer/treatment/research/car-t-cells. 2. Ruella, Marco, and Marcela V Maus. "Catch Me If You Can: Leukemia Escape after CD19-Directed T Cell Immunotherapies." Computational and structural biotechnology journal. Research Network of Computational and Structural Biotechnology, September 28, 2016. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5061074/. 3. "Relapsed Leukemia: Emily's Story." The Children's Hospital of Philadelphia, January 28, 2014. https://www.chop.edu/stories/relapsedleukemia-emilys-story. 4. Grady, Denise. "In Girl's Last Hope, Altered Immune Cells Beat Leukemia." The New York Times. The New York Times, December 9, 2012. https://www.nytimes.com/2012/12/10/health/a-breakthroughagainst-leukemia-using-altered-t-cells.html. 5. Cohen, Marion C, and Stanley Cohen. "Cytokine Function: A Study in Biologic Diversity. Retrieved from Cytokine Function A Study in Biologic Diversity." American Journal of Clinical Pathology, May 1996. https://academic.oup.com/ajcp/article-pdf/105/5/589/24878191/ ajcpath105-0589.pdf. 6. Shimabukuro-Vornhagen, Alexander, Philipp Gödel, Marion Subklewe, Hans Joachim Stemmler, Hans Anton Schlößer, Max Schlaak, Matthias Kochanek, Boris Böll, and Michael S von Bergwelt-Baildon. "Cytokine Release Syndrome." Journal for Immunotherapy of Cancer. BioMed Central, June 15, 2018. https://www.ncbi.nlm.nih.gov/pmc/articles/ PMC6003181/. 7. Slaney, Clare Y, Pin Wang, Phillip K Darcy, and Michael H Kershaw. "CARs versus BiTEs: A Comparison between T Cell–Redirection Strategies for Cancer Treatment." Cancer Discovery. American Association for Cancer Research, July 16, 2018. https://cancerdiscovery. aacrjournals.org/content/candisc/8/8/924.full.pdf. 8. Griffioen, Marieke, Esther H M van Egmond, Michel G D Kester, et al. "Retroviral Transfer of Human CD20 as a Suicide Gene for Adoptive T-Cell Therapy." Haematologica. Ferrata Storti Foundation, September 2009. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2738728/. 9. "Two Is Better Than One: Arming the T-Cells with Dual CancerTargeting Antibodies." Stanford Medicine BMT, n.d. http://med.stanford. edu/bmt/news/news-archive/two-is-better-than-one.html. 10. Breman, Eytan, Benjamin Demoulin, Sophie Agaugué, et al. "Overcoming Target Driven Fratricide for T Cell Therapy." Frontiers in immunology. Frontiers Media S.A., December 12, 2018. https://www. ncbi.nlm.nih.gov/pmc/articles/PMC6299907/. 11. Iwahori, Kota, Sunitha Kakarla, Mireya P Velasquez, et al. "Engager T Cells: a New Class of Antigen-Specific T Cells That Redirect Bystander T Cells." Molecular therapy: The Journal of the American Society of Gene Therapy. Nature Publishing Group, January 23, 2015. https://www.ncbi. nlm.nih.gov/pmc/articles/PMC4426792/. 12. Shimabukuro-Vornhagen, Alexander, Philipp Gödel, et al. "Cytokine Release Syndrome." Journal for Immunotherapy of cancer. BioMed Central, June 15, 2018. https://www.ncbi.nlm.nih.gov/pmc/articles/ PMC6003181/.






