16 minute read
Activation of Host Kinase Vps34 by Murine Norovirus Non-structural Protein


Scientia Activation of Host Kinase Vps34 by Murine Norovirus Non-structural Protein NS4
Jessica Oros Randall Lab, University of Chicago
Advertisement
Introduction:
Each year, millions of individuals are infected with human norovirus, securing norovirus as the number one worldwide cause
of gastroenteritis and causing about 200,000 deaths each year. Tens of thousands of those deaths occurring in children in developing countries. There are presently no norovirus vaccines or cures, making it a pressing issue to understand the biology of norovirus infection. Unfortunately, human norovirus cannot be easily grown in laboratory cultures. However, murine norovirus (MNV) can serve as a model. MNV is used in the lab to understand the
mechanisms behind infection in murine cell
lines. MNV is a positive-sense RNA virus. Like other viruses within the same class, MNV creates a replication complex upon entry and uncoating inside the cell using host-cell membranes . The purpose of this replication complex seems to be protecting viral genome replication from the hostile environment of the host cell cytoplasm. Luckily for host organisms, these replication complexes can be targetted via an interferongamma (IFNG) mediated antiviral pathway, allowing for lysosomal degradation of the replicating virus. This pathway uses proteins involved in autophagy, a cellular process by which foreign particles or faulty proteins are engulfed and degraded through lysosomal fusion. This recognition by the host cell is done by the Atg12-Atg5-Atg16L1 complex, which is comprised of autophagy-related (Atg) proteins that interact with host membranes to form an
autophagosome . Conjugation of the ubiquitinlike microtubule associated protein light-chain 3 (LC3) with Atg-5 allows interferon-inducible GTPases to localize to LC3 and target the replication complex, inhibiting viral replication . This pathway does not operate via degradative canonical autophagy, but it uses autophagy proteins to prevent MNV from replicating inside the cell . The pathway is called the Targeting by AutophaGy protein (TAG) – dependent antiviral pathway. Prior work in the Randall lab shows that other viruses such as dengue virus (DENV), and encephalomyocarditis virus (EMCV) cannot replicate when the TAG-dependent antiviral pathway is knocked down. If the TAG-dependent antiviral pathway was the only means through which viral replication was controlled, DENV and EMCV would be able to replicate in TAGknockout conditions. Since this is not observed, it indicates that there is some other TAG-
independent antiviral pathway present that is still blocking viral replication. Interestingly, MNV can still replicate in this TAG-deficient model, meaning there is some mechanism by which MNV is evading the immune response. Evasion may be possible at the viral replication stage via the replication complex or through virus-derived factors such as interactions
between viral and host proteins. The aim of this project is to elucidate the mechanisms by which MNV presents a phenotype of immune evasion through interactions with host proteins. One possible player in MNV immune evasion and replication could be the cellular class III phosphatidylinositol-3 kinase (PI3K) vacuolar protein sorting 34 (Vps34), which plays a role in cellular trafficking via initiation of autophagy. Vps34 phosphorylates phosphatidylinositol to produce phosphatidylinositol-3-phosphate (PI3P), which acts as a membrane marker to begin the formation of the autophagosome . In healthy cells, Vps34 is maintained in an inhibitory state by the nutrient-sensing kinase mTOR. Under cellular stress or starvation, mTOR phosphorylates UVRAG, a protein complex involved in endocytosis, which will activate Vps34 to initiate autophagy. The Randall lab has previously shown that knockdown of Vps34 prevents MNV replication, indicating that there may be interactions between the virus and the host lipid kinase. Vps34 activity is tightly regulated by the host cell through acetylation by the acetyltransferase p300, which itself is activated by autoacetylation . Acetylation of Vps34 is inhibitory, and activation by deacetylation contributes to the formation of autophagosomes. Despite Vps34 being used in the host for autophagy, MNV may be utilizing Vps34 to aid the process of viral replication. Thus, we hypothesize that Vps34 may be a point of control by MNV during infection as it may support viral replication in an unknown way. Furthermore, we aim to characterize which viral non-structural protein is responsible for the change in Vps34 activation level. Out of the 6 non-structural proteins of MNV, the most likely candidates are NS4, NS5, and NS6 because of their previously elucidated roles in immune evasion. NS4 has been shown to localize to host
intracellular membranes and thus may play a role in replication complex formation, but its function is largely uncharacterized. The Randall Lab has previously shown that NS5 can produce an immune evasion phenotype upon infection with DEMV and EMCV of BV2 cells transduced
with NS5. Upon entry and uncoating, the viral genome is translated into a polyprotein, and NS6 is the protease responsible for the cleavage and release of individual MNV non-structural
proteins .
In this study, we have shown that infection with MNV results in a decrease in
inhibitory Vps34 acetylation, indicating an increase in activity. Out of the viral nonstructural proteins tested, only non-structural protein 4 (NS4) was able to reproduce the phenotype, indicating that it is responsible for the release of Vps34 from its inhibitory state.
Materials and Methods:
Tissue Culture:
Murine norovirus (MNV) was used as a model to study norovirus activity. Cell culture was done with murine microglial BV2 cells using standard tissue culture protocol, which involves growing the adherent cells in flasks with Gibco Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% each of non-essential amino acids, hydroxyethyl piperazineethanesulfonic acid (HEPES), and PIS. Cells were seeded and used for experiments. In the case of interferon gamma
(IFNG) treatment, 100µL/mL were used for a 24hour period. For infections, MNV strain CW3 was diluted in complete DMEM media and added to cells at 0.05 MOI.
Transductions:
Transductions of viral non-structural
proteins or wild-type Vps34 were done using plasmids previously generated by the Randall lab. Third-generation lentiviral constructs were used, and transfection was prepared for HEK293T cells using CaCl2, HEPES buff ered saline solution, sterilized water, lentivirus packaging plasmids, pseudotyping envelope plasmids, and the previously described plasmids containing V5tagged Vps34 or Flag-tagged MNV non-structural proteins NS4, NS5, or NS6, with the tags being used for immunoprecipitation and immunoblotting. Supernatant was collected and filtered to isolate lentivirus particles 48 hours post transfection, which were added to BV2 cells. Selection for
successfully transduced BV2 cells was done with Thermo Fisher Scientific Hygromycin B at a concentration of 250µg/mL. Gel Electrophoresis and Transfer: After 24 hours post infection (hpi) or post seeding cells transduced with MNV nonstructural proteins, cells were lysed using 2x Laemmli sample buff er. Samples were then heated at 95ºC for 10 minutes before gel electrophoresis was done on an SDS-PAGE to separate samples by size alongside Thermo Fisher Scientific PageRuler 10-250 kDa. Protein samples were transferred from the SDS-PAGE gel to 0.2μm PVDF membranes via semi-dry transfer. Membranes were then blocked in 5% skim milk blocking buff er before immunoblotting with polyclonal antibodies targeting the desired proteins.
Immunoprecipitation: In the case of immunoprecipitation, after 24hpi, cells were treated with cold coimmunoprecipitation lysis buff er was used and comprised 2.5mL 1M Tris at pH 7.5, 1.5mL 5M NaCl, 5mL 10% Triton X-100, proteinase inhibitor cocktail, and 41mL distilled water. Lysed cells were briefl y frozen at -80ºC and thawed to complete lysis. Cell lysates were incubated overnight at 4ºC on a rotator with Cytiva Protein A Sepharose beads and mouse anti-V5 antibody. Precipitated protein was washed using Co-IP wash buff er before 2x Laemmli sample buff er was added, and samples were heated at 95ºC for 10 minutes before an SDS-PAGE and semi-dry transfer was done following the same protocol as whole cell lysates described above.
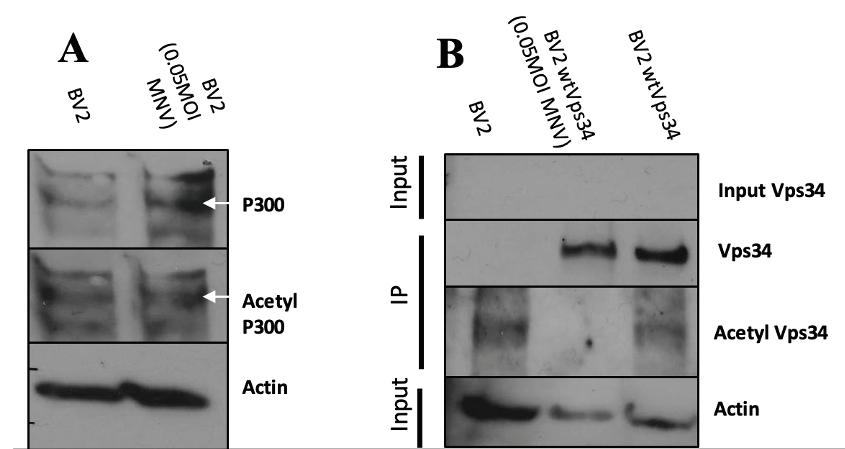
Figure 1: MNV Infection decreases levels of acetylp-300 and acetyl Vps-34 Western blots of control BV2 and control BV2+wtVps34 alongside infected BV2 and infected BV2+wtVps34. Infection was done with 0.05 MOI of MNV for 24 hours. (A) Infected cell lysates are shown in lane 2. Acetylp-300 and p300 bands were fl anked by two unspecific bands on the top and bottom. (B) Immunoprecipitations of cell lysates captured Vps34, with the middle column showing infected lysates. Input Vps34 and actin bands are taken from input samples collected from supernatant of lysates with precipitated
Vps34
Antibodies:
Antibodies for the
immunoprecipitation of Vps34 were Cell Signaling Technology Mouse V5-tag (E9H80). Detection of Vps34 and acetyl-Vps34 via immunoblot was done with Cell Signaling Technology Rabbit V5-tag (D3H80) and Cell Signaling Technology Rabbit Acetylated-Lysine, respectively. Imaging p300 in whole cell lysate was done with Santa Cruz Biotechnology Mouse monoclonal IgG p300 (F-4), while acetyl-p300 was captured with Cell Signaling Technology Rabbit Acetyl-CBP (K1535)/ p300 (K1499). To normalize Western blot bands, actin was detected in each sample using Santa Cruz Biotechnology Mouse monoclonal IgG Betaactin (C4) HRP. Detection of viral non-structural proteins was done with Sigma-Aldrich Rabbit polyclonal Anti-FLAG antibodies. Secondary antibodies used were Thermo Fischer Scientific
goat anti-Rabbit IgG secondary antibody, HRP and Thermo Fischer Scientific goat anti-Mouse IgG secondary antibody, HRP. this phenotype. This indicates that MNV infection results in a decrease of acetylation of the acetyltransferase p300 such that the levels of active p300 are at a lower level. Levels of acetylated Vps34 were measured (Figure 1b) after infection using an immunoprecipitation, as endogenous levels of Vps34 are below detection threshold. When compared to BV2 cells transduced with wild-type Vps34 (BV2+wtVps34), infected cells showed a marked decrease in the levels of acetylated Vps34. Levels of Vps34 were consistent across infected and control cells, meaning the decrease in acetyl-Vps34 is notable. MNV NS4 decreases expression of acetyl-
Vps34 BV2+wtVps34 cells were transduced using lentivirus with NS4, NS5, or NS6, and an immunoprecipitation was run on cell lysates
to precipitate Vps34. Immunoprecipitation products were subject to gel electrophoresis and immunoblotting to detect levels of Vps34 and acetyl-Vps34. Immunoblotting was done to detect the presence of the transduced viral non-structural protein and endogenous actin for normalizing measurements. Additionally, input lanes were included to ensure the eff icacy of the immunoprecipitation. Transduction with NS6 did not replicate the major decrease in acetyl-Vps34 levels (Figure 2a). However, due to problems with either transfer or immunoblotting, repeated instances of noise on the membrane obstructed any detailed view of the acetyl-Vps34 bands. Transductions with NS5 were performed and the level of Vps34 and acetyl-Vps34 were measured without obstruction from noise. NS5 transduction
Results:
MNV Infection decreases levels of acetyl-p300 and acetyl-Vps34 BV2 cells were infected with MNV at a
multiplicity of infection (MOI) of 0.05 for 24 hours, after which cells were lysed and the lysate was run on an SDS-PAGE gel. Western blots using antiacetyl-CBP rabbit polyclonal antibodies and antiCBP mouse polyclonal antibodies were used to detect levels of acetyl-p300 and p300, respectively (Figure 1a). Cells infected with MNV showed a decrease in the fraction of p300 acetylated, as control cells showed a greater increase from p300 to acetyl-p300 while infected cells did not mirror
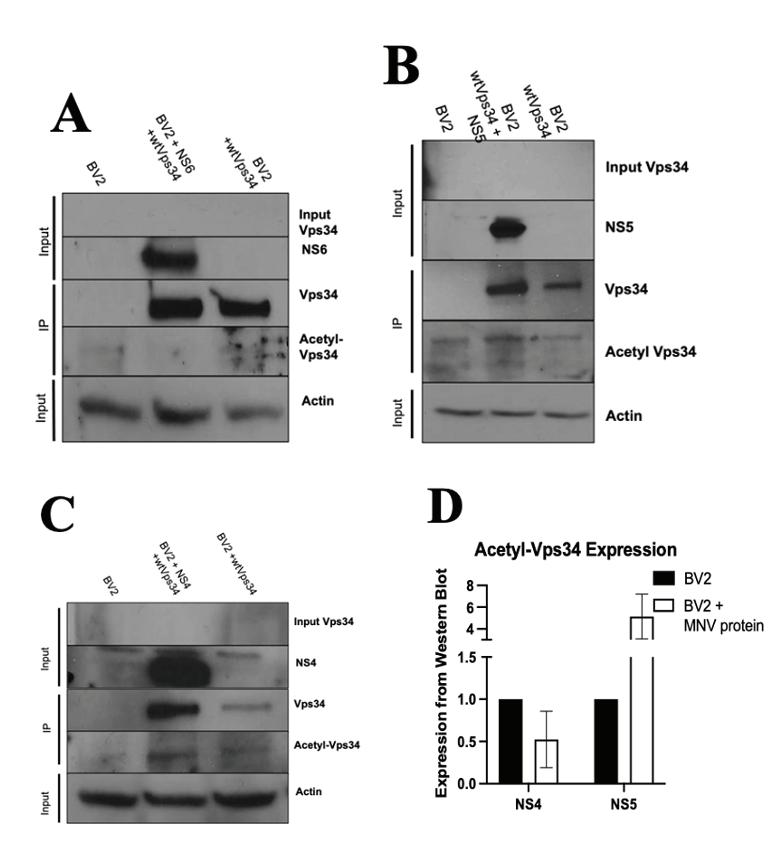
Figure 2: MNVNS5 and NS6 do not produce dramatic decrease in acetyl-Vps34, NS4 decreases acetyl-Vps34 levels Western blots of control BV2 and control BV2-wtVps34 alongside BV2 cells transduced with NS6 (A), NS5 (B), and NS4 (C) are shown with immunoblotting for Vps34 and acetyl-Vps34. Viral proteins were imaged using rabbit anti-Flag antibodies to indicate transduction was successful. Input lanes were included to ensure the eff icacy of immunoprecipitation. Densinometry calculations for expression levels of acetyl-Vps34 were done using data fron NS4-transduced and NS5transduced cells (D).

Figure 3: MNV NS4 decreases acetyl-p300 levels while NS6 does not Western blots of control BV2 cells and
BV2 cells transduced with NS4 (A) and NS6 (B) are shown with immunoblotting for p300 and acetyl-p300. Densinometry calculations for expression level of acetyl-Vps34 were done (C).
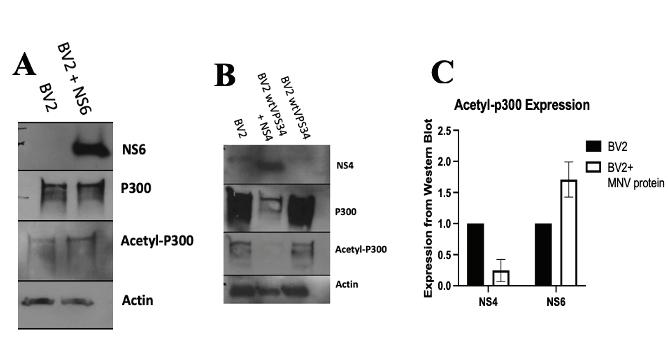
Figure 4: Acetyl-Vps34 levels do not change in NS4 cells treated with IFNG Western blots of control BV2+wtVps34 cells and NS4-transduced cells in response to treatment with IFNG (2uL/mL). Input Vps34 shown for success of immunoprecipitation and NS4 shown for success of transduction. resulted in a minor increase in the levels of Vps34 (Figure 2b), but as compared to the BV2+wtVps34 control, the ratio of acetyl-Vps34 to Vps34 was lower, indicating that NS5 is not responsible for the release of Vps34 from its acetylated inhibitory state. NS4 was transduced into BV2+wtVps34 cells, and a decrease in the level of acetyl-Vps34 was observed (Figure 2c). Because problems with the gel in the case of NS6 resulted in obstruction of acetyl-Vps34 bands, densitometry data was measured and graphed comparing levels of acetylVps34 in NS4 and NS5 transduced cells (Figure 2d). As seen in Figure 2b, an increase in acetyl-Vps34 was present in NS5 transduced cells, but NS4 transduced cells showed a decrease in expression of acetyl-Vps34 when compared to BV2+Vps34 cells without viral protein. This indicates that MNV protein NS4 is responsible for the decrease in Vps34 acetylation observed in infection. MNV NS4 decreases p300 acetylation Cells were transduced with NS4 and NS6, and lysates were run through gel electrophoresis and levels of p300 and acetyl-p300 were detected via immunoblot. NS4 transduction resulted in a notable decrease in not only acetyl-p300 levels, but overall p300 expression as well (Figure 3a). NS6, however, did not show a change in the acetyl-p300 levels as compared to BV2 controls (Figure 3b), indicating that NS6 cannot reproduce the phenotype observed in MNV infection. When compared, it is evident that expression levels of p300 are much lower during NS4 transduction than NS6 transduction (Figure 3c). These results further point to NS4 as being responsible for the phenotype in MNV infection whereby acetylation levels of p300 and Vps34 are decreased. Role of NS4 interaction with Vps34 is not IFNG-dependent To test whether the deacetylation of Vps34 plays a role in viral immune evasion in the presence of interferon gamma (IFNG), cells transduced with NS4 were treated with IFNG at a concentration of 100µL/mL. Immunoprecipitated Vps34 was immunoblotted via Western blot and levels of Vps34 and acetyl-Vps34 were captured (Figure 4). The levels of both Vps34 and acetyl-Vps34 are constant across samples from NS4-transduced BV2 cells incubated with and without
IFNG. This indicates there was no noticeable change in the acetylation state of Vps34 during IFNG treatment. Because IFNG does not alter Vps34 activation in NS4 conditions, NS4 does not appear to play a role in the IFNG antiviral pathway and may instead be involved in viral replication.
Discussion:
The mechanism of MNV replication and immune evasion are not fully characterized, as it is relatively unknown what purposes many non-structural proteins serve upon entry and uncoating. We have shown that activation of the lipid kinase Vps34 is increased during MNV infection through a release of inhibition on Vps34. Furthermore, it was shown that the levels of the active
upstream regulator of Vps34, acetyltransferase p300, decreased during infection. These results suggest that Vps34 does play a role in viral replication and therefore is a point of control in MNV infection. These results were replicated in cells transduced with MNV non-structural protein NS4, while the large decrease in acetylation levels of Vps34 and p300 was not observed in NS5 or NS6 transduced cells. This indicates that, during MNV infection, the phenotype wherein p300 expresses decreased activation and Vps34 expressess increased activation is being brought on by NS4. It is also notable that the interaction between NS4 and Vps34 is not affected by the presence of IFNG. Had there been changes in Vps34 activation in the presence of NS4 and IFNG treatment, it may have led to the belief that Vps34 aids MNV in evading antiviral pathways, as IFNG is responsible for the detection and disruption of the viral replication complex. Instead, it is likely that Vps34 may be involved with aiding viral replication. The Randall lab will perform additional trials of the experiments described in this paper to verify the data and evaluate its statistical significance. It is still unknown exactly what NS4 does in the host cell upon infection. Studies have found it to localize to the Golgi apparatus and endosomesix, leading to a tentative belief that it may be involved in recruiting host membranes for the formation of the viral replication complex. Downstream effects of Vps34 will be studied to further understand how Vps34 creates a pro-viral state within the cell for viral replication. Upon autophagosome initiation, Vps34 functions to produce phosphatidylinositol-3-phosphate (PI3P) at the source of autophagosome membranes . To test whether Vps34 is functioning to form the viral replication complex during infection, immunofluorescence assays are currently underway to see the localization of PI3P in response to MNV infection. Large amounts of colocalization of PI3P with sites of autophagosome membrane sources during infection would indicate that MNV is utilizing the host cell’s autophagy pathway for the construction of the viral replication complex. Additionally, the mechanisms by which Vps34 is aiding viral replication remain unknown. Further work will be done to understand differences in activity of upstream regulators of Vps34 during MNV infection and NS4 transduction, as it is unclear how NS4 is decreasing levels of Vps34 acetylation. It is unlikely that NS4 functions directly as a deacetylase, so the pathway by which Vps34 is deacetylated indirectly during NS4 transduction is yet to be elucidated. Further progress will examine other regulators of Vps34 that install post-translational modifications other than acetylation, such as phosphorylation or ubiquitination.
O’Donnell, T. B., Hyde, J. L., Mintern, J. D., & Mackenzie, J. M. (2016). Mouse Norovirus infection promotes autophagy induction to facilitate replication but prevents final autophagosome maturation. Virology, 492, 130-139. Lopman, B. A. (2015). Global burden of norovirus and prospects for vaccine development. Hyde, J. L., Sosnovtsev, S. V., Green, K. Y., Wobus, C., Virgin, H. W., & Mackenzie, J. M. (2009). Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. Journal of virology, 83(19), 9709-9719. Walczak, M., & Martens, S. (2013). Dissecting the role of the Atg12–Atg5-Atg16 complex during autophagosome formation. Autophagy, 9(3), 424-425. Furlong K, Hwang S. Autophagy and Noroviruses. Viruses. 2019; 11(3):244. https://doi. org/10.3390/v11030244 Biering, S. B., Choi, J., Halstrom, R. A., Brown, H. M., Beatty, W. L., Lee, S., ... & Hwang, S. (2017). Viral replication complexes are targeted by LC3-guided interferon-inducible GTPases. Cell host & microbe, 22(1), 74-85. Biering, S. B., Choi, J., Halstrom, R. A., Brown, H. M., Beatty, W. L., Lee, S., ... & Hwang, S. (2017). Viral replication complexes are targeted by LC3-guided interferon-inducible GTPases. Cell host & microbe, 22(1), 74-85. Hwang, S., Maloney, N. S., Bruinsma, M. W., Goel, G., Duan, E., Zhang, L., ... & Virgin, H. W. (2012). Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell host & microbe, 11(4), 397-409. Kim, J., Kim, Y. C., Fang, C., Russell, R. C., Kim, J. H., Fan, W., ... & Guan, K. L. (2013). Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell, 152(1-2), 290-303. Munson, M. J., Allen, G. F., Toth, R., Campbell, D. G., Lucocq, J. M., & Ganley, I. G. (2015). mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. The EMBO journal, 34(17), 2272–2290. https://doi.org/10.15252/embj.201590992 Su, H., Yang, F., Wang, Q., Shen, Q., Huang, J., Peng, C., ... & Liu, W. (2017). VPS34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy. Molecular cell, 67(6), 907-921. Hyde, J. L., & Mackenzie, J. M. (2010). Subcellular localization of the MNV-1 ORF1 proteins and their potential roles in the formation of the MNV-1 replication complex. Virology, 406(1), 138-148. Herod, M. R., Salim, O., Skilton, R. J., Prince, C. A., Ward, V. K., Lambden, P. R., & Clarke, I. N. (2014). Expression of the murine norovirus (MNV) ORF1 polyprotein is sufficient to induce apoptosis in a virus-free cell model. PLoS One, 9(3), e90679. Boukhalfa, A., Nascimbeni, A.C., Ramel, D. et al. PI3KC2 -dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat Commun 11, 294 (2020). https://doi.org/10.1038/s41467-019-14086-1
Acknowledgements: University of Chicago Randall Lab
Biological Sciences Collegiate Division (BSCD) Summer Fellowship Program


