Free -Range Organic
Edited by Tabbytha Spyrison Ana Risano Langdon J. Martin1
2

O nce, I wandered into an airport bookstore. I rarely wander into stores without an explicit purchase in mind, so I don’t know what drew me into that airport bookstore. Maybe my flight was delayed, or maybe I had arrived even earlier than necessary, or perhaps it just felt like a good time to browse. Nevertheless, I distinctly recall being drawn to the science section, mindlessly scanning the books at eye level. A title caught my eye: Napoleon’s Buttons. Something about that title intrigued me; perhaps it was the juxtaposition of those two words; perhaps the pedant in me wondered if this book had been misfiled. So I pulled it off the shelf and read the subtitle: 17 Molecules That Changed History.¹ As an organic chemist, and therefore a lover of molecules, I opened it and read a few pages. And at that point, it was abundantly clear that I was not leaving the bookstore without purchasing this book, and that whatever I had been planning to do to pass the time on the flight would be supplanted by some reading.
Fast forward to the summer of 2014. I was beginning my third year as chemistry professor at Warren Wilson College. The college was in the midst of adopting new General Education requirements, which included Writing Across the Curriculum (WAC) and therefore increased intentional writing instruction. I wanted to adapt Organic Chemistry I for the new WAC Level-2 designation; I would need to incorporate a writing assignment of 2000+ words with a drafting process. I didn’t think that an extended lab report would be a great way to do this—that sounded tedious to write and mind-numbing to grade—but I struggled to come up with anything better.
Fortunately, my colleague Julie Wilson (Director of the Writing Studio) led a multi-part workshop to help faculty collaborate and think creatively about teaching writing. We wanted to develop assignments that gave students li-
cense to tap the curiosity that brought them to our classes. Sitting in my office one morning, I happened to glance at my (dog-eared) copy of Napoleon’s Buttons. Once again, a chance glance at this title sent me from languor to excitement: What if, for this longer writing assignment, my students created their own chapter in the style of Napoleon’s Buttons? They could pick a molecule or a set of organic compounds as a focus, and dig more deeply they would have agency to explore a topic that had passion for. So, with help from Julie and Brian Conlan (Acquisitions and Learning Technologies Librarian), that became the assignment.
This assignment has changed very little in the years since.2 Students seem to appreciate their control over the course content. Inevitably, everyone has some molecule, or chemical, or material, in which they realize an inherent interest; I’m just giving them a forum to “geek out” on it. As students choose their topics, I emphasize the following direction: This paper should be interesting, accessible, and enjoyable for a broad audience to read. More precisely, this broad audience is meant to include people who are interested in science, but who may have little training in organic chemistry. Their goal is to make complex topics as accessible as possible using creative expositions, stories, and analogies.
I like this assignment as the instructor because each topic is unique: not only does this reduce the tedium of grading, I often learn something! Every semester a few of these chapters really draw me in. The authors of these chapters use an approachable voice to tell a memorable story that teaches me something, all of which reinvigorates my passion for organic chemistry. Starting in Fall 2014, I began to set these gems aside, thinking there might be a way to publish them some day— they really are too good to keep to myself! And, that is what led to this compendium.
¹ Le Couteur, Penny, and Jay Burreson. Napoleon's buttons: 17 molecules that changed history. Penguin, 2004. ² The complete assignment can be viewed here: https://docs.google.com/document/ d/1_SrcYa39E9Q26sd980_oiO_06in2ZjZeYjEpUtFySJ8/edit?usp=sharing
In 2019, Julie Wilson, Ben Feinberg (Prof. of Anthropology) and I wrote and received a grant from the Work Colleges Consortium that included resources to make this publication.
With this momentum, I recruited two student co-editors: Tabby Spyrison (WWC Class of 2022 Biology and Creative Writing Major) and Ana Risano (WWC Class of 2022 Biology Major). Both of them knew much more about editing, style, and aesthetics than I did, and they worked with me to turn the original student pieces into a consistent, readable format.
Around the beginning of 2020, I had a chance conversation with my colleague Jessica White (Prof. of Art) that led to another dimension for this publication. Jessica teaches printmaking, and we realized that this could complement this project. So, Jessica’s Spring 2021 Intro to Printmaking course included an assignment to create a print that would become an editorial illustration for this publication. Students chose a chapter that intrigued them, read the draft, and used the content as inspiration for a piece. (Ana Risano created the image for her own chapter!) There was one student fewer in Jessica’s class than we had chapters, so she also generously contributed a print. These images appear at the beginning of each chapter.
I hope this publication is merely a first edition of many more. There are other existing student papers that I’d like to include, and of course there are many outstanding papers that have simply not yet been written! My students never cease to amaze me with their curiosity and creativity. I hope you enjoy reading these chapters as much as I have enjoyed curating them!
T his publication is divided into four sections organized around key aspects of the chemistry. I’ll begin each section by adding some additional context and explanations to the chemical formalism in these chapters.
The word organic has a handful of definitions and colloquial uses. Herein, the focus is I like the following definition:
Organic molecules contain, first and foremost, one or more carbon atoms. They also, inevitably, contain hydrogen atoms.
While this definition is imperfect, it is concise and it comprises every molecule in this book, so that’s what I’m going with.
Why does carbon have its own subfield of chemistry? Ultimately, this is because there are significantly more known molecules that contain carbon atoms than molecules that lack carbon atoms. This, in turn, is because carbon is the only atom that has both of the following two properties:
1. Carbon can form four stable bonds. Other common elements form fewer bonds. (For example, nitrogen and phosphorus atoms form three bonds; oxygen and sulfur atoms form two bonds; hydrogen, fluorine, and chlorine just one.)
2. Carbon atoms bond strongly and therefore stably to other carbon atoms. They can form linear carbon chains of any length: short chains (e.g. propane—a chain of three carbons surrounded by hydrogens), medium chains (e.g. paraffin wax, a chain of 20–40 carbons), or long chains (e.g. PVC plastic—recyclable plastic #3— chains of billions and billions of carbons). Carbon atoms can also connect to make branched chains (e.g. LDPE plastic recyclable plastic #4), as well as rings and cyclic systems, which I’ll explain more in a little bit.
Molecules do what they do because of what atoms they contain and the way the atoms are put together. Chemical structures are symbolic representations of the molecule that are straightforward to draw. Like any symbolic representation, they provide a lot of information, but you have to know what you’re looking for. Thus, a key challenge of organic chemistry is to learn how to read and interpret these representations of molecules that cannot be seen directly.
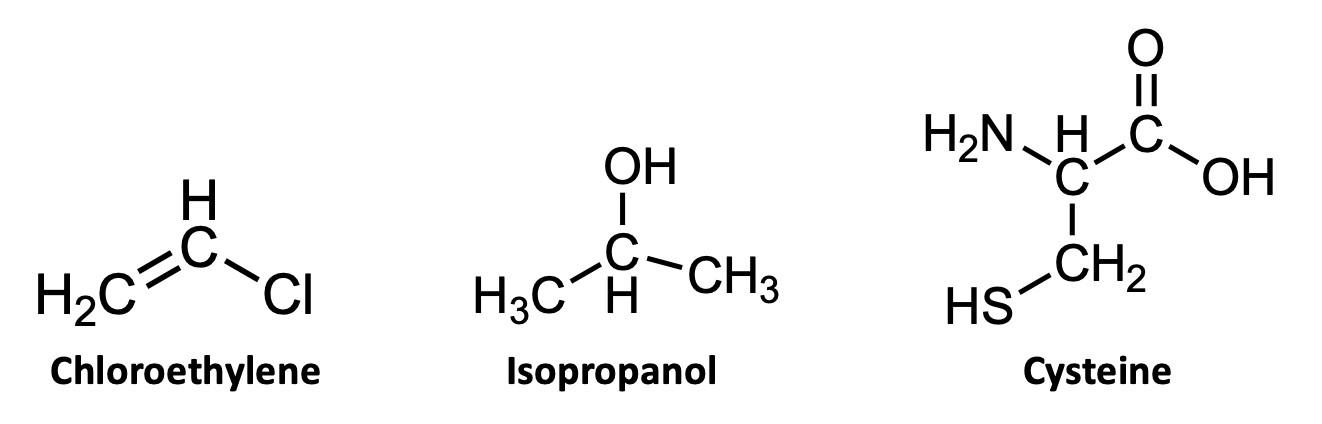
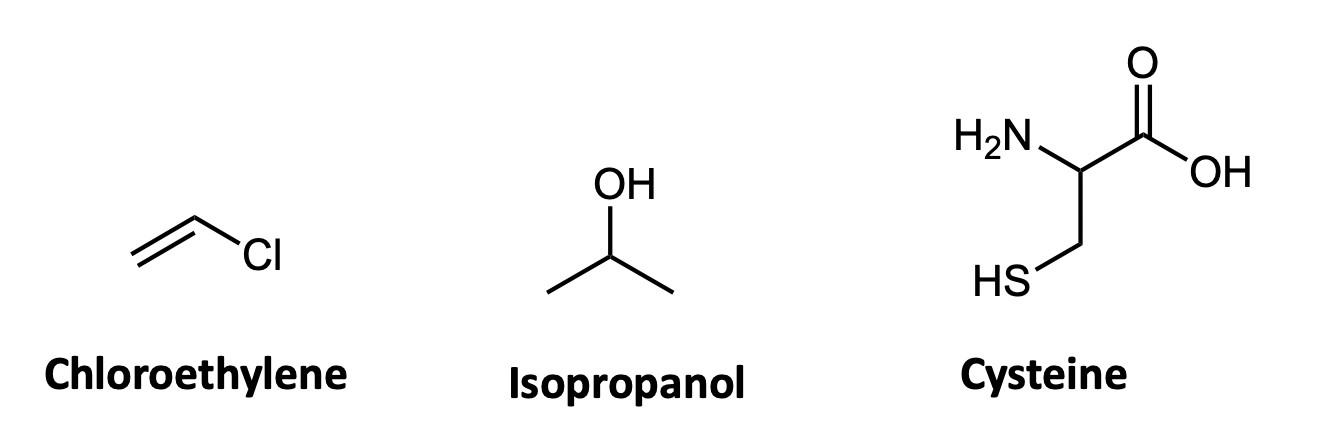
If you’ve ever taken a chemistry course you were almost certainly taught one way of representing chemical structures: using elemental symbols (the ones used on the Periodic Table) to represent the positions of atoms, and lines to represent a pair of bonded electrons that connect the atoms. This system works well for small organic molecules such as chloroethylene (C2H3Cl, the starting material for the synthesis of PVC plastic) or isopropanol (C3H8O, rubbing alcohol); see Figure A1. Even the isopropanol structure, though, with 12 total atoms, is starting to look a little cluttered!
The first student paper, by Grace Girardeau, is a chapter about oxytocin, which is C43H66N12O12S2: 135 atoms! Showing all atoms and bonds is too cluttered in a molecule of this size. Even one building block of oxytocin, amino acid cysteine (Figure A1), is unwieldy. Cysteine has 14 atoms (C3H7NO2S), and although this is just two more than isopropanol, the N, O, and S atoms add additional complexity. So, the best thing to do is to simplify the drawings by relying on some dependable chemical truths.
The first simplification centers on hydrogen atoms. There are a lot of hydrogen atoms in organic compounds—most structures have significantly more hydrogen atoms than carbon atoms. Conveniently, hydrogen atoms always form exactly one bond.
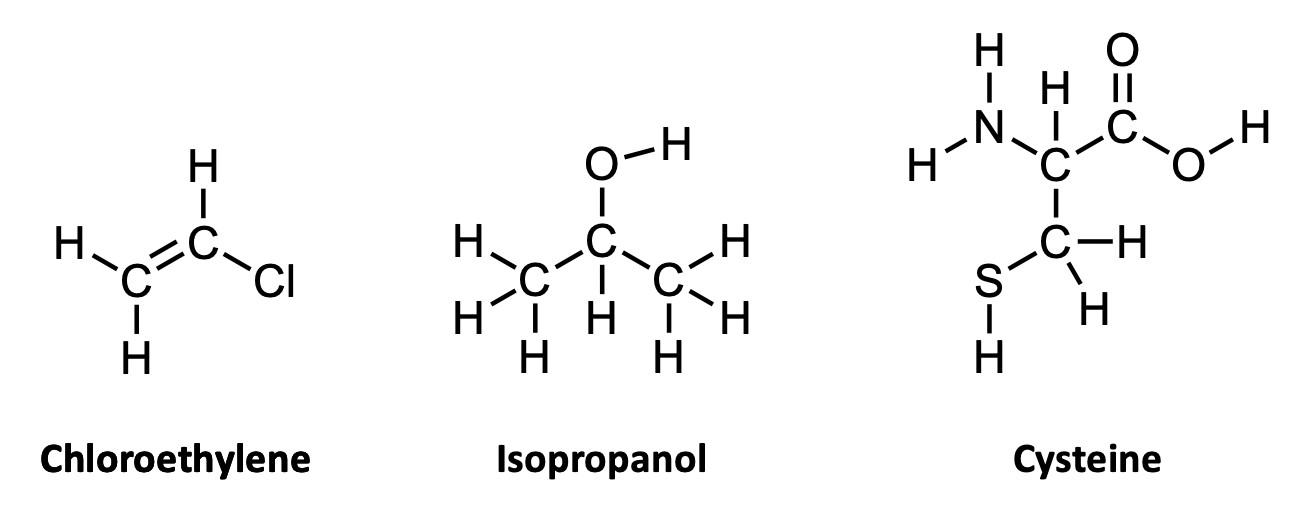
To capitalize on this fact, we use the condensed structure, where bonds connecting to hydrogen atoms are omitted. Instead, the hydrogen [H] atoms are written directly adjacent to the atom to which they are bonded. More than one hydrogen atom is denoted with a subscript as in NH2 or CH3 Figure A2 shows the same three molecules as condensed structures. While the condensed form of chloroethylene has changed little, the larger isopropanol and cysteine structures more easily show key information. For example, notice that cysteine is clearly larger and more complex than isopropanol.
But, more adjustments are often worth making. All organic molecules contain carbon and hydrogen atoms, so often the key chemis-
try comes from the atoms that are neither of these two. Collectively, these atoms are called heteroatoms; common heteroatoms include nitrogen [N], oxygen [O], sulfur [S], and chlorine [Cl]. Because of the outsize importance of heteroatoms to the chemistry of a molecule, and because C’s and H’s are so common, chemists have exploited a few more a few more facts to develop a less-is -more approach to structure drawing:
· Carbon [C] atoms always form exactly four bonds.
· The many carbon–hydrogen bonds in organic molecules are (usually) not very reactive compared to other parts of the molecule.
The line–angle structure relies on these facts. In these structures, we usually do not
write the C for carbon atoms, nor the H’s attached to those carbon atoms.
In a line-angle structure, we still draw all of the bonds that are in condensed structures: all C–C bonds and all C–heteroatom bonds. The H’s attached to heteroatoms are still included, too. To interpret a line–angle structure, note that whenever bonds meet at an angle, there is a carbon atom. And because of the rule that carbon atoms always form exactly four bonds, we can deduce the implicit hydrogen atoms by subtracting the number of bonds shown from 4. Figure A3 shows the same molecules from Figures A 1 & A2 in line-angle form. (This is sometimes called “skeletal form” form, for good reason.)
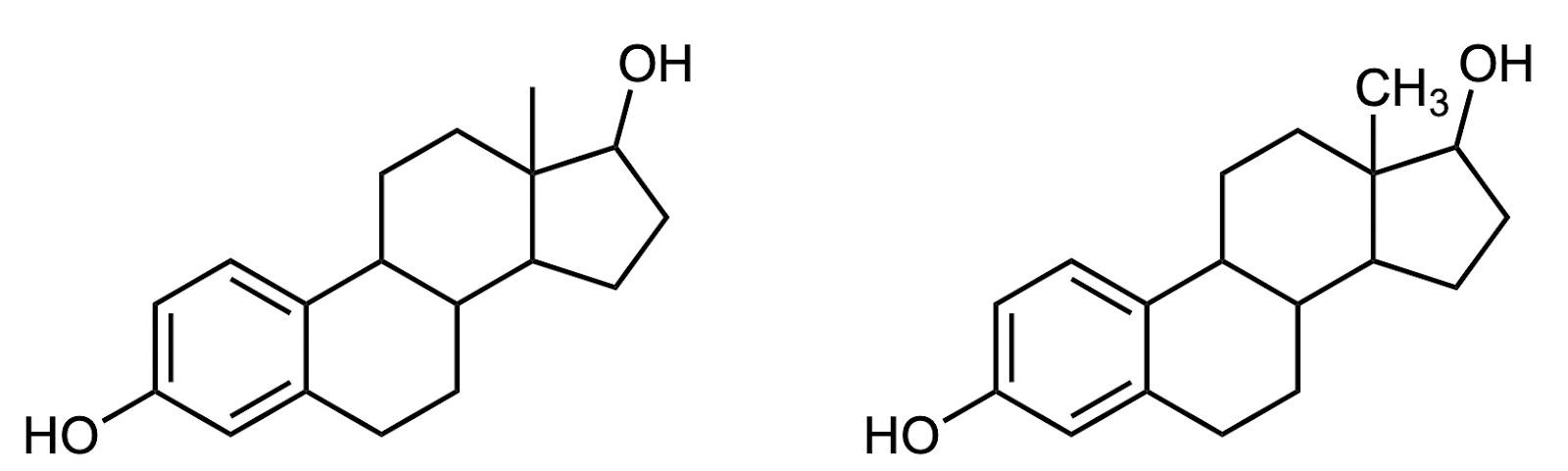
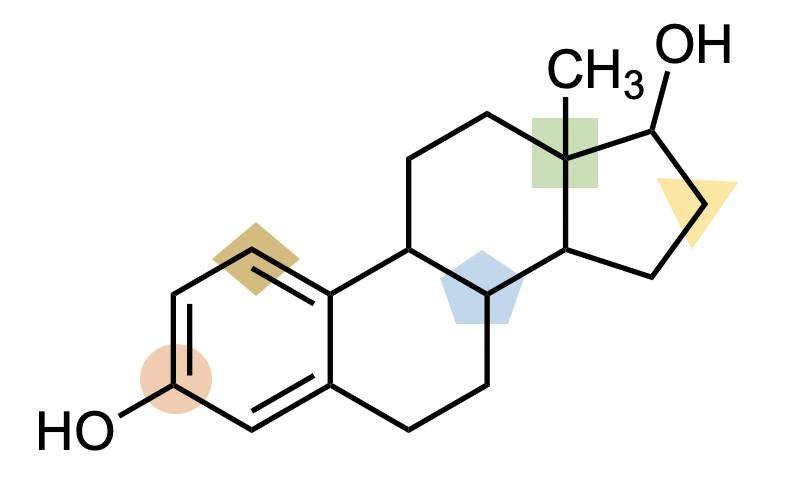
The line–angle formalism is particularly useful for structures with cyclic motifs, which are common. For example, look at the structure line-angle structure of the hormone estrogen (Figure A4). It is quickly apparent that this molecule includes three six-membered rings and one five-membered ring, fused together in a specific way.
When drawing line– angle structures, the one exception, or perhaps compromise, that I like to make is to show terminal carbons explicitly, as –CH3. This can help make the termini of the structure more clear. Estrogen
has one of these –CH3 groups, as shown on the right in Figure A4. I like to think of the terminal –CH3 as the Oxford comma of line–angle structures: not technically necessary, but a way of improving clarity of meaning. That’s the style of chemical structures (and commas!) used herein.
With this introduction to interpreting chemical structures, you are ready to enjoy the first four chapters! After Grace’s chapter on oxytocin are two chapters on psychoactive compounds: Susie Lusardi’s chapter on amphetamines and Taylor Kligerman’s chapter on DMT. This first section concludes with Nicholas Rust’s chapter on florigen.
Of course, being a professor, I can’t help myself from offering a quick end-ofsection review: See if you can practice what you’ve learned about interpreting line-angle structures using this structure of estrogen on the next page (Figure A5) to answer the following questions.
1. How many carbon atoms are in estrogen?
2. How many hydrogen atoms are attached to carbon atoms indicated by the…
A. Green square
B. Yellow triangle
C. Blue pentagon
D. Tan diamond
E. Orange circle
Answers:
1. There are 18 carbon atoms.
2. Hydrogen atoms:
A. Green square: 0. (There are four bonds to other carbons so there’s no room for H)
B. Yellow triangle: 2. (Two bonds to other carbons)
C. Blue pentagon: 1. (Three bonds to other carbons)
D. Tan diamond: 1. (There are three bonds shown to this carbon, including a double-bond)
E. Orange circle: 0. (There are four bonds shown to this carbon, including a double -bond)

O xytocin takes the cake as every quasiscientist’s favorite hormone. After a brief internet search, any curious person might be led to believe that the molecule is responsible for the bond they have with their mother, their inclination to hug their friends, or even human morality. In reality, oxytocin is a member of an intricate network of other hormones and neurotransmitters that moderates every human experience, whether good or bad. Sensational journalism has relished in mystifying this lifegiving chemical, which has come to play a major role in every hospital delivery ward, and in the industry surrounding childbirth.
In 1948, British pharmacologist J. H. Burn developed a method of administering extremely dilute oxytocin intravenously . This procedure is now widespread, and was used to induce approximately 23% and augment 57% of all childbirths in American hospitals in 2010.1 This therapy was a revolutionary medical advancement enabling doctors and families to progress a stalled labor, potentially lowering the risks of infant mortality. However, the overuse and manipulation of this treatment has also become a tool in streamlining healthcare practices and changing attitudes toward women's capability to give birth safely. Researchers are promoting findings that indicate that the administration of exogenous oxytocin during labor can greatly affect maternal health and infant performance.2 Because of this, many healthcare professionals are taking a closer look at uses of oxytocin and promoting a cautious approach to the relatively mysterious chemical.
Sir Henry Dale conducted the first welldocumented studies of the molecule, finding its uterine-contracting effect in 1906 after injecting it into a pregnant cat and observing the birth that ensued.3 He promptly named this peculiar molecule oxytocin, meaning “quick birth” in Greek. Four years later, scientists Ott and Scott unveiled its role in milk secretion in breastfeeding mammals, and began to identify its largely reproductive na-
ture.3 Decades later, in 1952, Pierce isolated the first crystalline derivative of oxytocin, and in 1955 Vincent du Vigneaud was awarded the Nobel Prize for sequencing and synthesizing oxytocin, the first-ever polypeptide hormone to be sequenced and synthesized.3 It wasn’t long before researchers realized that the synthetic form of oxytocin is indistinguishable from natural oxytocin in how it affects the body to bring about labor and lactation.
Oxytocin plays the role of both neurotransmitter and hormone, meaning it may facilitate the transfer of nerve impulses or travel through tissue fluids in order to exact an action. This dual nature makes for a very versatile molecule, and it likely has other mechanisms in the body that are yet to be recognized. Oxytocin is synthesized in the hypothalamus, which is responsible for the autonomic nervous system controlling your breath, heartbeat, and other functions you don’t consciously control. After production, the molecule is transferred to the posterior pituitary gland and released into the bloodstream. Once it reaches the target site (such as the smooth muscles of the uterus or breast), it binds to receptors that advance the function within the tissue.
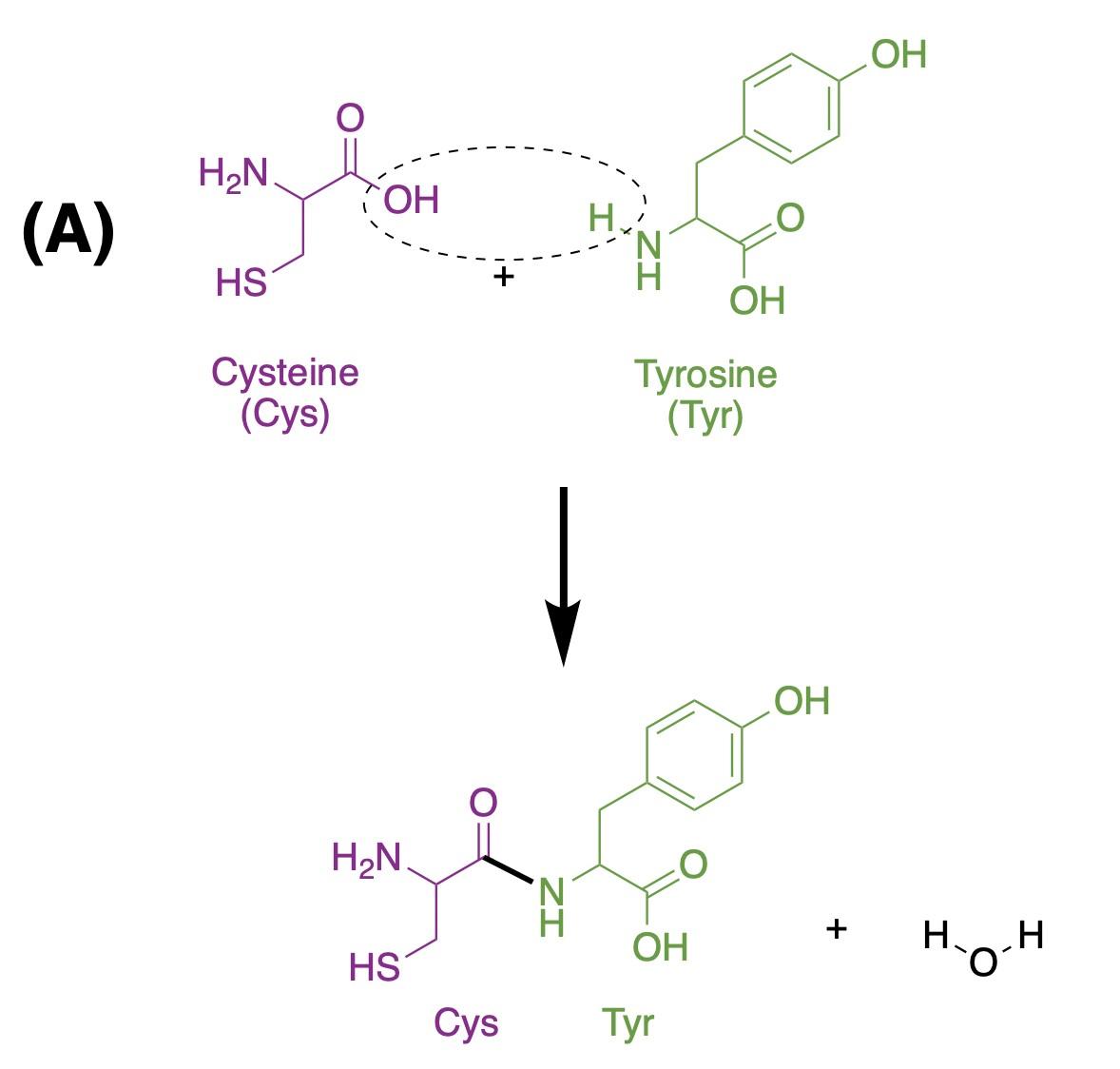
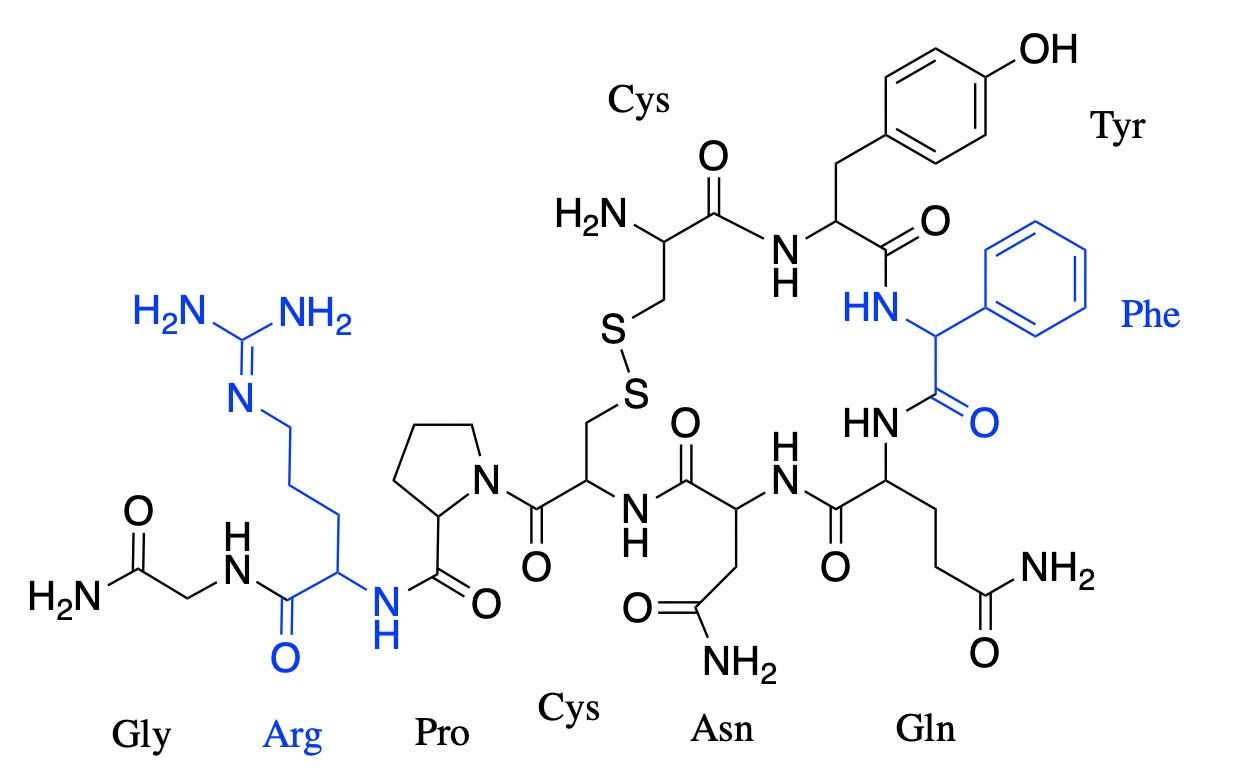
Oxytocin is a simple nonapeptide, meaning it is a peptide composed of nine amino acid residues (Figure 1). Like all peptides and proteins, oxytocin is a specific chain of amino acids. There are 20 distinct amino acids, and these can be assembled in an infinite number of ways to form different shapes and sizes of proteins, ranging from the oxytocin nonapeptide to protein complexes built from thousands of amino acids. When amino acids connect, they form peptide bonds by removing a hydroxyl group (i.e., –OH: an oxygen bonded to a hydrogen) from one amino acid and a hydrogen (H) from the next, releasing a water molecule (H2O) and forming a bond where those groups previously were, also known as dehydration synthesis. Figure 1A shows a representation of this dehydration
synthesis reaction between cysteine and tyrosine, the first two amino acids of oxytocin. Once they are part of a polypeptide, the amino acid building blocks are slightly altered, and are thus referred to as amino acid residues. Figure 1B shows that nine amino acid residues (8 distinct ones) form oxytocin: cysteine, tyrosine, isoleucine, glutamine, asparagine, another cysteine, proline, leucine, glycine; in the figure, each residue is represented by its three-letter abbreviation. Notice, also, that the sulfur atoms of each cysteine residue form a S–S linkage, providing the molecule with a cyclic structure. This feature is critical for binding to the receptor.
mone created in the hypothalamus. Vasopressin regulates water conservation within the body and maintains blood pressure, controlling vital functions, similar to oxytocin. The only structural differences between the two are the third and eighth amino acid residues. The genes for these two hormones are both found on Chromosome 20 in humans; they arose from the duplication of the vasotocin gene. Although both may have similar antidiuretic properties and effects on blood pressure, the two molecules serve very different functions in the body.4
Interestingly, oxytocin bears a striking resemblance to vasopressin, another hor-
Countless online sources hail oxytocin as the “love hormone,” “cuddle chemical,” or whichever nickname seems to do it justice. 5 Although it is imperative that we separate science from pop culture trends and user-based media, oxytocin has aptly earned its fame. For example, greater concentrations of oxytocin appears in the blood plasma of participants who claimed to be falling in love, and in the plasma of both males and females during orgasm.6 Oxytocin even appears across species of mammals. Studies of prairie voles demonstrated the key role that oxytocin plays in forming monogamous partner bonds, and the effect it may have on virgin female sheep developing maternal tendencies toward foreign lambs after an oxytocic spinal treatment.7
It becomes clearer and clearer the role that this molecule may play in social structures, possibly connecting humans and allowing for trusting relationships. Oxytocin is expressed throughout the day in a way that moderates our experiences with others—even when we’re not falling in love. There are countless preliminary studies that explore oxytocin’s role in our levels of trust in strangers, our tendency to gloat, and the significance of our identification with various in -groups based on race, gender, and family.8–10
Today, synthetic oxytocin (often called Pitocin in the healthcare setting) is used primarily in the delivery room: to induce or augment a stalled labor; to prevent postpartum hemorrhage; and to evaluate a fetus’s respiratory capacity. The mechanisms of labor contractions are ruled by positive feedback loops. Oxytocin begins the process by binding to uterine smooth muscle tissue, but contractions also trigger a release of oxytocin, which makes the ensuing contractions more frequent and more intense.11 Practitioners agree that there is a point at which labor carries on too long and intervention is required, so manipulating oxytocin became a revolutionary step in reducing the risk of birth complications and thereby decreasing infant and mother mortality. When executed properly and closely observed by practitioners, supplementing the body’s endogenous oxytocin with exogenous Pitocin has been a highly successful method of achieving a safer and healthier delivery.
Since 1948, little has changed in the ways practitioners administer oxytocin to laboring mothers. They typically use the “continuous” method of administration, “with oxytocin delivered as a continuous infusion with incremental dose rises every 30 minutes until adequate contractions are achieved”.12 The continuous infusion method is preferred because oxytocin has a circulatory half -life of only 3-4 minutes, and is quickly metabolized by the mother.12
For some women, interventions such as oxytocin and cesarean sections may be the best possibility to save their child. For most low-risk mothers, however, intervention is not necessary to labor their children. In the United States, too many healthy women are made to believe that their bodies are not capable of birthing their own children safely. An unnecessary administration of oxytocin, or too high of a dosage, may have adverse effects such as fetal distress, postpartum hemorrhage, and operative delivery.12 Recent trends in rising cesarean and induction rates have led many to reassess the use of oxytocin in the United States and begin to question long-held beliefs about the effectiveness of this drug.
The movie The
showcases the pressure that women face once they’re hospitalized for their labor. Imagine: You and your partner have decided to pursue a drug-free birth in the hospital. After all, you’re a healthy woman and there’s no reason to expect a complication. You’re eight hours into your laboring process, and you’ve become tired of nurses and physicians coming in every 30 minutes and making your worry about how slowly your cervix is dilating. You’re exhausted from the contractions and you’re starting to feel less and less welcome in your hospital bed. There’s already an IV in your arm administering fluids, so it’s all too easy to give in when the doctor says you need Pitocin.
After an hour and a half, your contractions have become overwhelmingly painful, and you’re still not dilating any quicker. The nurses assure you that an epidural is completely safe, and you give in after another painful contraction. Next thing you know, it’s 3:00AM and your epidural is wearing off. It’s time for another epidural and a higher dosage of Pitocin; however, now the nurses are coming in more frequently and checking on your baby’s heart rate and oxygen supply. It seems as if your contractions are proving too stressful for your child, and you have to decide
quickly whether you’ll undergo a cesarean section in order to save your baby. “Of course,” you say. “Do whatever is necessary.”
For many women, the refusal of drugs during their labor process becomes a battle that’s very difficult to fight when they’re scared and in pain. Because exogenous oxytocin is supplementing endogenous oxytocin, the contractions quickly become stronger and more frequent, which is where the epidurals become very helpful. When the mother is no longer feeling pain, the high doses of oxytocin seem harmless, until the uterus becomes hyperstimulated, meaning the contractions are unnaturally forceful and last much longer than they should. When hyperstimulation occurs, the baby’s blood and oxygen supply are compromised, and doctors must quickly perform surgery to save the child.
Depending on the administration of oxytocin, this life-giving drug may endanger the life of the child, and tragedy often strikes when a fetal heart rate is misidentified or contraction patterns are not accurately monitored. American hospitals have historically operated under vague guidelines for oxytocin administration, and the dosage and infusion rate is often at the discretion of individual doctors and nurses.13
Researchers have worked to explore different infusion methods and compare effective outcomes. One is the adoption of pulsatile administration: the mother is infused with oxytocin for 10 seconds every six minutes in order to match the natural oxytocin cycle in the body. One study found the pulsatile method effective for labor induction, lowering the maximum dosage received without adverse effects. It did not lower rates of cesarean following induction, nor did it perform as well for augmentation of labor.12
Various peer-reviewed studies have found widely differing evidence to support or reject the use of oxytocin in all its different forms, which is why some researchers claim
that variation in oxytocic procedures is more of an indicator for fetal stress than actual dosage and treatment methods.13 American hospitals have begun to implement strict checklist-based guidelines for oxytocic interventions based off of European models. The implementation of these guidelines within the Hospital Corporation of America’s system resulted in a 17% decrease of maximum dosage to achieve birth, did not lengthen time of labor or operative outcome, and may have even improved newborn outcome (based on Apgar scores and birthweight). Over the entire hospital system, they saw a decrease from a 23.6% cesarean rate in 2005 to a 21% cesarean rate in 2006 after protocol implementation. The researchers hypothesize that these accomplishments are a result of less uterine hyperstimulation, and “wish to emphasize that the uniform practice pattern achieved with our protocols is probably as important as the actual details of the protocols themselves”.13
Other medical facilities like the Pomona Valley Medical Center are making strides toward avoiding the initiation of Pitocin in the first place. In 2011 they announced the successful elimination of elective inductions before 39 weeks, and noted a 17% decrease in induction post-39 weeks, a 21% decrease in cesarean rates following induction, and decreased admission to the Neonatal Intensive Care Unit.14 By maintaining this restriction on elective inductions, the hope is to continue these downward trends of labor intervention.
There are still many questions surrounding the mechanisms of oxytocin in the human body, and it is clear that either a lack or disregard of sufficient information may be fatal. This research provides evidence that conservative oxytocic treatment may be best for mother and child. The rich history of oxytocin provides a great narrative for those who question the bounds of manipulating natural processes. Practitioners in the medical field should be rightly concerned with learning as much as possible about this mysterious, lifegiving molecule. Oxytocin concentration in
the body, especially for expecting mothers, is a sacred balance that plays a role in dictating the health and prosperity of human life.
Plasma Oxytocin Increases in the Human Sexual Response*. J. Clin. Endocrinol. Metab. 1987, 64 (1), 27–31. https://doi.org/10.1210/ jcem-64-1-27.
(7) Cho, M.; DeVries, A.; Williams, J.; Carter, C. The Effects of Oxytocin and Vasopressin on Partner Preferences in Male and Female Prairie Voles (Microtus Ochrogaster). Behav. Neurosci. 1999, 113, 1071–1079. https:// doi.org/10.1037//0735-7044.113.5.1071.
All figures were created by the author and the editors.
(1) Bell, A. F.; Erickson, E. N.; Carter, C. S. Beyond Labor: The Role of Natural and Synthetic Oxytocin in the Transition to Motherhood. J. Midwifery Womens Health 2014, 59 (1), 35–42. https://doi.org/10.1111/ jmwh.12101.
(2) Oláh, K. S.; Steer, P. J. The Use and Abuse of Oxytocin. Obstet. Gynaecol. 2015, 17 (4), 265–271. https://doi.org/10.1111/tog.12222.
(3) Magon, N.; Kalra, S. The Orgasmic History of Oxytocin: Love, Lust, and Labor. Indian J. Endocrinol. Metab. 2011, 15 (Suppl3), S156–S161. https://doi.org/10.4103/22308210.84851.
(4) Caldwell, H. K. Oxytocin and Vasopressin: Powerful Regulators of Social Behavior. The Neuroscientist 2017, 23 (5), 517–528. https:// doi.org/10.1177/1073858417708284.
(5) Yong, E. The Weak Science Behind the Wrongly Named Moral Molecule https:// www.theatlantic.com/science/ archive/2015/11/the-weak-science-of-thewrongly-named-moral-molecule/415581/ (accessed Nov 20, 2020).
(6) Carmichael, M. S.; Humbert, R.; Dixen, J.; Palmisano, G.; Greenleaf, W.; Davidson, J. M.
(8) Theodoridou, A.; Rowe, A. C.; PentonVoak, I. S.; Rogers, P. J. Oxytocin and Social Perception: Oxytocin Increases Perceived Facial Trustworthiness and Attractiveness. Horm. Behav. 2009, 56 (1), 128–132. https:// doi.org/10.1016/j.yhbeh.2009.03.019.
(9) Shamay-Tsoory, S. G.; Fischer, M.; Dvash, J.; Harari, H.; Perach-Bloom, N.; Levkovitz, Y. Intranasal Administration of Oxytocin Increases Envy and Schadenfreude (Gloating). Biol. Psychiatry 2009, 66 (9), 864–870. https:// doi.org/10.1016/j.biopsych.2009.06.009.
(10) Sheng, F.; Liu, Y.; Zhou, B.; Zhou, W.; Han, S. Oxytocin Modulates the Racial Bias in Neural Responses to Others’ Suffering. Biol. Psychol. 2013, 92 (2), 380–386. https:// doi.org/10.1016/j.biopsycho.2012.11.018.
(11) Nathanielsz, P. W. Comparative Studies on the Initiation of Labor. Eur. J. Obstet. Gynecol. Reprod. Biol. 1998, 78 (2), 127–132. https://doi.org/10.1016/S0301-2115(98)00058X.
(12) Tribe, R. M.; Crawshaw, S. E.; Seed, P.; Shennan, A. H.; Baker, P. N. Pulsatile versus Continuous Administration of Oxytocin for Induction and Augmentation of Labor: Two Randomized Controlled Trials. Am. J. Obstet. Gynecol. 2012, 206 (3), 230.e1-230.e8. https:// doi.org/10.1016/j.ajog.2011.11.001.
(13) Clark, S.; Belfort, M.; Saade, G.; Hankins, G.; Miller, D.; Frye, D.; Meyers, J. Implementation of a Conservative Checklist-Based Pro-
tocol for Oxytocin Administration: Maternal and Newborn Outcomes. Am. J. Obstet. Gynecol. 2007, 197 (5), 480.e1-480.e5. https:// doi.org/10.1016/j.ajog.2007.08.026.
(14) Badertscher, J. “We Just Did It”: Eliminating Elective Inductions Before 39 Weeks. J. Obstet. Gynecol. Neonatal Nurs. 2011, 40, S17–S18. https://doi.org/10.1111/j.15526909.2011.01242_23.x.
Additional Sources Consulted
“Hypothalamus.” You and Your Hormones, Feb. 2018, www.yourhormones.info/glands/ hypothalamus/.
“Oxytocin.” National Center for Biotechnology Information. PubChem Compound Database, U.S. National Library of Medicine, pubchem.ncbi.nlm.nih.gov/ compound/oxytocin#section=ChemicalVendors.
“Oxytocin Molecule.” The Oxytocin Molecule, World of Molecules, www.worldofmolecules.com/emotions/ oxytocin.htm.

C ould you ever imagine giving an elementary school-aged child methamphetamine? But what if I told you that drugs within the same class as methamphetamine are consistently prescribed to millions of children across the United States? Despite common knowledge of methamphetamine—more commonly known as meth, ice, or speed— this powerful drug is part of a family of organic molecules called amphetamines. And surprisingly, amphetamines are among the most popular pharmaceuticals prescribed to children in modern medicine today.1 Amphetamines have had a long history of use in the medical world; however, drugs such as methamphetamine do not have the same medical reputation. This chapter will be an exploration of the legal and illegal uses of amphetamines throughout history and today.
So, how exactly do amphetamines work? Amphetamines are classified as stimulants, meaning that they increase the amount of dopamine and adrenaline in the brain of the recipient. This effect is extremely helpful for those diagnosed with Attention-Deficit Hyperactivity Disorder (ADHD), or other related disorders, and the amount prescribed by a physician typically does not exceed 30 mg per day. This allows the molecule to be effective while remaining relatively non-addictive to those that use it. However, recreational usage of this class of drugs brings an incredible amount of danger to the body. Amphetamines prescription or not have been abused since their initial synthesis, and methamphetamine is only one of the types that are abused.
Methamphetamine is distinctive among amphetamines. All amphetamines are powerful drugs with addictive properties that can lead to dependence or abuse, but methamphetamine is characterized as being particularly addictive, as it is able to stimulate the brain much more quickly.2 This leads to an intense rush—or high—for the individual taking the drug. This feeling is then sought out
repeatedly, resulting in addiction. In recent years, the United States has initiated countless programs to fight back against illegal drug use and abuse. However, the quandary known as the “Opioid Crisis” has cast a large shadow over an equally harmful amphetamine-related crisis that is sweeping the country.
Amphetamine can be first traced to the late 1800s in Germany. Its synthesis is accredited to a Romanian chemist, Lazar Edeleanu, at the University of Berlin in 1887.3 However, this popular drug was not used until the late 1920s, when the American chemist Gordon A. Alles re-synthesized the drug.
Alles is credited with discovering and documenting the psychological effects of amphetamine and other amphetamine derivatives.4 In an attempt to create a new medicine to treat asthma symptoms, Alles triggered a century-long controversy by creating “the first psychoactive prescription drug” from a molecule that had originally been labeled as pharmaceutically useless. On June 3, 1929, he injected himself with 50 milligrams of amphetamine five times the dosage recommended in modern medicine. These are the symptoms that he recorded feeling after the injection: heart palpitations, high blood pressure, feeling “unusually witty,” and racing thoughts.4 After testing on himself, he soon began testing on individuals with asthma. Following a few human trials, he discovered that although the drug causes a sense of euphoria in the individuals taking the drug, it did not effectively treat asthma. Despite this, he proceeded to patent his drug in 1932 and became the inventor of amphetamine sulfate and amphetamine hydrochloride.4 Soon after, Alles reached out to larger partners, as an attempt to turn his compound into a “wonder drug.”
Alles’s drug became a success with the help of his partners. It was put to use by the United States government during World War II. The properties of the drug enabled soldiers to stay alert longer and feel as if they had more energy for longer periods of time. Later on, in the 1960s, it became popular among the typical suburban housewife who needed a little extra “help” getting through the day. It was also around this time that other derivatives of amphetamines became popular in the over-the-counter drug world. The initial rise of methamphetamine usage started at the same time as the beginning of amphetamine use during WWII. German pharmaceutical companies marketed methamphetamine tablets as a nonprescription drug under the brand name Pervitin, for use by soldiers. The drug was effective, as it heightened users’ adrenaline and alertness, perfect for soldiers. This was comparable to the effects that American soldiers felt when taking amphetamines. It wasn’t until the late 1950s that the FDA required a prescription for Benzedrine, which was the major amphetamine-containing drug at the time.
In the 1980s, the United States government acknowledged that the usage of amphetamines and methamphetamine was spiraling out of control. In an attempt to fix the problem, regulations around the sale and use of Ephedrine, Benzedrine, and other amphetamine-containing prescription drugs were tightened. Ephedrine and Benzedrine are both drug precursors that are utilized in the production of crystal meth. However, it wasn’t until the 1990s that use and abuse of methamphetamine exploded in the United States. In the ten years from 1994 to 2004, methamphetamine use rose from just under two percent of the United States adult population to around five percent.5 As of 2017, it has been reported by the National Survey on Drug Use and Health that about three percent of the adult population in the United States were methamphetamine users at the time of the survey.6 This is a shockingly high number of individuals, yet it does not even account for those indi-
viduals who are abusing legal prescription amphetamines. All this information begs the question: How do people become so addicted to a drug that has been prescribed for decades? The answer can be found in the chemistry of the organic molecule’s structures, and how the structures interact with nerve cell proteins.
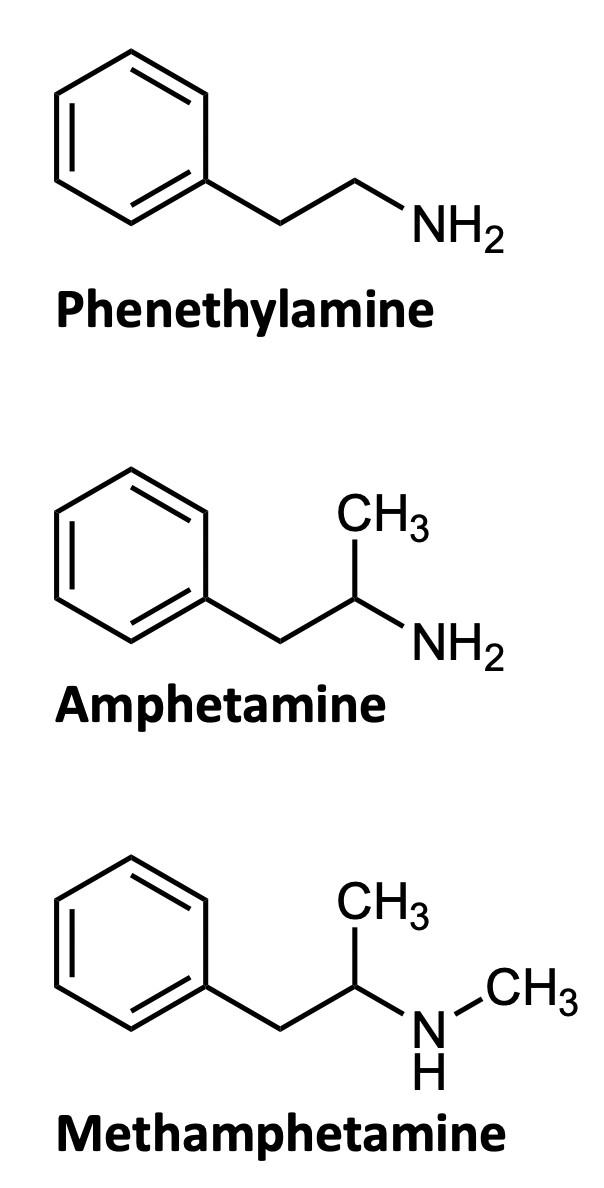
What chemistry causes methamphetamine’s addictive qualities? Methamphetamine is a distinct example of the chemicals that are classified as amphetamines. On the illicit drugs market, the primary representatives of the amphetamines group are amphetamine and methamphetamine. Note that this nomenclature is somewhat confusing: “amphetamines” are a broad class of drugs, and include the specific drug molecules “amphetamine” and “methamphetamine.” These two closely-related synthetic substances are both members of the phenethylamine family (Figure 1).7 The phenethylamine family is a group of organic molecules that are often the base of psychotropic drugs. Simply put, both drugs share many of the same properties and the same central structure (Figure 1). They all have a similar effect on the psychological function of the individual due to these shared properties.
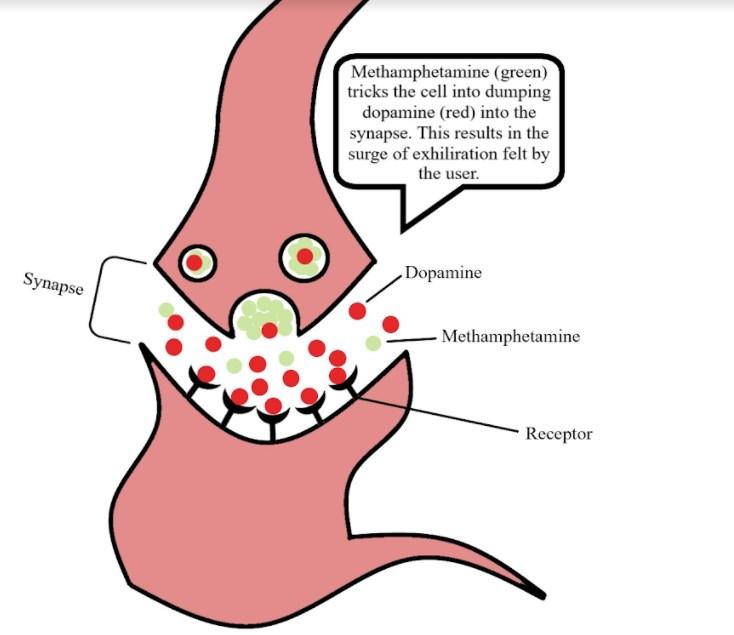
Looking at the three structures in Figure 1, one can tell that the major difference between methamphetamine and amphetamine is the methyl group attached to the end of the nitrogen on the methamphetamine molecule. A methyl group, –CH3, is an extra carbon coming off of a carbon chain, with three hydrogens attached to it. Methamphetamine gets its name from this extra methyl group. This drug in particular is considered an amphetamine anorectic, which means that it is a drug that suppresses the appetite of the individual.8 It is also considered a central nervous system stimulant. This is because the drug works by promoting the release of noradrenaline, dopamine, and serotonin from nerve terminals in the brain, and by preventing their reuptake.8 This process leads to an increased synaptic concentration of dopamine, noradrenaline, and serotonin between neurotransmitters (Figure 2), and causes excess stimulation of postsynaptic receptors. This
chemical process is what leads to the exhilarating psychological high a methamphetamine user experiences. It is this long-lasting high that contributes to the drug’s addictiveness and abuse. 9
Methamphetamine produces a high that lasts anywhere from four to twenty-four hours. This is an eternity when compared to other abused stimulants, like cocaine, which produces a similar high that lasts for a mere fifteen minutes, on average. Due to the length of the high that an individual will experience when using methamphetamine, there are many different stages a user may go through. They are typically as follows:
Stage One: The start of the high, nicknamed “The Rush,” is typically characterized by a surge of dopamine in the brain. A user might expect their heart rate and metabolism to skyrocket as their pupils become dilated. This stage lasts no longer than thirty minutes. 10
Stage Two: Next comes the bulk of the high that a user will experience. After the shock from the large surge of dopamine, the brain begins to adjust, and the user will begin to feel uncharacteristically smarter and intensely focused. This portion of the high can last anywhere from four to sixteen hours. 10
Stage Three: After the high has begun to fade, a third stage, often referred to as “The Binge,” begins. The binge is characterized as a period of days after initial use that a user spends binging on the drug: An individual will continue to use methamphetamine to maintain the high. During this period, a user might not eat or sleep for days. 10
Stage Four: This final stage, called “The Crash,” occurs when the drug no longer provides the user with the desired effect and is often followed by a mental/physical shutdown. This is often manifested as a one- to three-day period of sleep. A user will come out of this stage feeling starved, and mentally, emotionally, and physically exhausted. 10
After learning what the typical methamphetamine user endures when taking this drug, the amount of abuse may seem unreasonable, especially after such an ordeal. Part of the reason for the unusual length of the high is due to the chemistry of methamphetamine. This organic molecule has a half -life of about twelve hours, which is why the actual “high” (Stage Two from the list above) lasts about that long. As a comparison, the average half-life of cocaine another stimulant is only one-and-a-half hours. Due to how long it takes for a user to metabolize methamphetamine, this drug has a much longer period of effectiveness compared to others like it. This is desired by many individuals that abuse drugs as it is seen as a “more bang for your buck” type of deal.
Despite the fact that methamphetamine and amphetamine share so many characteristics, one of these drugs—amphetamine—is more accepted by society than the other. This molecule is found in pharmaceuticals prescribed to millions of children. Amphetamine was synthesized over 100 years ago, and has
transformed from a cure-all drug that was freely available without prescription, into a highly-controlled drug with therapeutic applications restricted to attention deficit hyperactivity disorder (ADHD) and narcolepsy.11
Amphetamines, as a class, are considered socially acceptable in some circumstances, but their risk for abuse is quite high. The 2015 National Survey on Drug Use and Health reported that around 4.8 million people in the United States had abused prescription amphetamine medication that year. This is equivalent to about 1.8 percent of the population over the age of twelve.12 Awareness toward the dangers of amphetamine abuse and addiction are beginning to enter the public eye, as more and more high schools and colleges put forth efforts to inform students of the dangers associated with abusing popular prescription drugs like Adderall. Adderall is one of the most regularly prescribed drugs to children and young adults that contains amphetamines. Unfortunately, despite the efforts of many schools and organizations, rates of abuse for prescription amphetamines are still on the rise.
On a more promising note, the social appeal of methamphetamine seems to be
trending in the opposite direction. Many school-aged children are educated about the harmful, long-term effects of methamphetamine abuse. The fear of these long-term effects such as severe weight loss, extreme paranoia, hallucinations, and dental problems have dissuaded many individuals from even trying the drug. However, in many midwestern and southern states, methamphetamine addiction still prevails in some communities. This drug is highly popular in lower-income, rural areas where youth may not appreciate the dangers before becoming addicted. Despite this, there is still hope for continuing to decrease the use of methamphetamine in the United States. In fact, in 2018 it was reported that less than one percent of twelfth grade students had ever reported using methamphetamine in the United States. 8 Based on these statistics, it is possible that, with continued awareness and support, methamphetamine usage among the younger population will continue to decrease, and the use of the drug will eventually become nonexistent.
https://www.ashleytreatment.org/differencebetween-amphetamine-andmethamphetamine/ (accessed Jan 8, 2021).
(3) Methamphetamine Overview : Origin and History | Methoide https:// methoide.fcm.arizona.edu/infocenter/ index.cfm?stid=164 (accessed Jan 8, 2021).
(4) Hicks, J. Fast Times: The Life, Death, and Rebirth of Amphetamine https:// www.sciencehistory.org/distillations/fasttimes-the-life-death-and-rebirth-ofamphetamine (accessed Jan 8, 2021).
(5) Editors, H. com. History of Meth https:// www.history.com/topics/crime/history-ofmeth (accessed Jan 8, 2021).
(6) Ciulla, A. Meth Addiction and Abuse Statistics. Beach House Rehab Center, 2018.
(7) 2008 Annual report: the state of the drugs problem in Europe | www.emcdda.europa.eu https://www.emcdda.europa.eu/ publications/annual-report/2008_en (accessed Jan 8, 2021).
(8) Abuse, N. I. on D. Methamphetamine DrugFacts https://www.drugabuse.gov/ publications/drugfacts/methamphetamine (accessed Jan 8, 2021).
All figures were created by the author and the editors.
Sources Cited
(1) Iannelli, V. Review the 30 Most Prescribed Drugs in Pediatrics https:// www.verywellhealth.com/the-30-mostprescribed-drugs-in-pediatrics-2633435 (accessed Jan 8, 2021).
(2) Difference Between Amphetamine And Methamphetamine | Ashley Treatment
(9) Drug Use Changes the Brain Over Time https://learn.genetics.utah.edu/content/ addiction/brainchange/ (accessed Jan 8, 2021).
(10) What Does Using Crystal Meth Make You Feel Like? - Drug-Free World https:// www.drugfreeworld.org/drugfacts/ crystalmeth/the-stages-of-the-methexperience.html (accessed Jan 8, 2021).
(11) Heal, D. J.; Smith, S. L.; Gosden, J.; Nutt, D. J. Amphetamine, Past and Present – a Pharmacological and Clinical Perspective. J. Psychopharmacol. Oxf. Engl. 2013, 27 (6), 479–496. https://
(12) Editorial Staff. What’s An Amphetamine? Addiction: Signs, Symptoms, and Treatment
https://americanaddictioncenters.org/ amphetamine (accessed Jan 8, 2021).

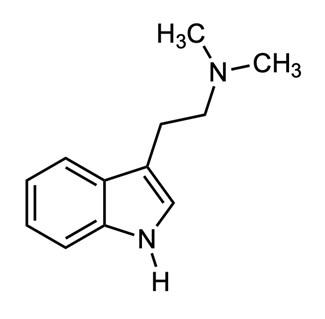
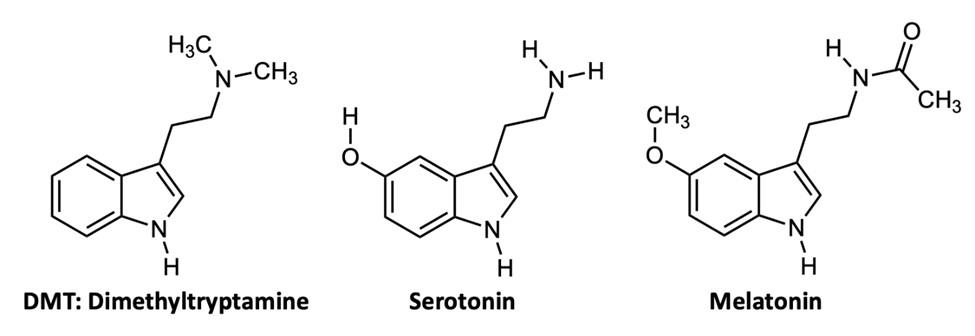
Imagine a molecule so simple, so readily available in nature that all organisms have the ability to synthesize it. This molecule is biologically abundant, but next to nothing is actually known about its purpose in living organisms. This molecule is dimethyltryptamine, which is more commonly known as DMT (Figure 1). This incredible compound's influence on today's global society spans from its ancient cultural uses to its contribution to the 1960s counterculture, and finally to contemporary scientific exploration of its role in human health and consciousness.
cies from ten plant families, as well as in four animal species, three of which are mammals.
DMT is a psychedelic compound that is the structurally simplest member of the tryptamine family of neurotransmitters, which are derived from the amino acid tryptophan . This molecule was first synthesized by Richard Helmuth Fredrick Manske in 1931, and was first discovered as a naturally-occurring chemical by Brazilian chemist, Oswaldo Gonçalves de Lima, in 1945.1 Gonçalves de Lima isolated the alkaloid from Mimosa Tenuiflora, a shrub native to northeast Brazil, with a range that extends to southern areas of Mexico.2 Before the early 2000's, DMT detection was unreliable due to substandard methods which lacked selectivity in measurement. In current research, gas chromatography and mass spectrometry are used to detect and quantify this molecule in animals and plants.3 Since 1945, it has been identified in over fifty plant spe-
Long before this molecule had been introduced to western culture, Amazonian and Andean Amerindian cultures ritually consumed DMT as an active ingredient in ayahuasca (also called yajé, hoasca, caapi, and shori), a drink brewed by a shaman that was used for both religious and healing purposes.4 Most notably used by the Urarina, Quechau, and Aymara people, who are indigenous to northern South America, the ritual consumption is both a religious sacrament and a medicinal purgative. During the religious ritual, the brew affects consciousness for up to six hours with mild physical effects. The purpose of these ceremonies is typically to invoke healing, purification, and spiritual protection. These rites usually fall into the broader practice of ayahuasca shamanism, whose followers believe to have received instruction directly from plant spirits in how to brew ayahuasca. The ayahuasca ritual is conducted by a shaman following strictly prescribed dietary restriction, breath control exercises, singing/ chanting, and an evolution of ceremonial body movements. The resulting psychological experiences are said to be ineffably enlightening and healing. As used for medicinal purposes, ayahuasca can induce intense vomiting and diarrhea that is used to purge the body of intestinal parasites common to the Andean and Amazonian basin region. In addition to this, ayahuasca alkaloids are known for their anthelmintic properties, or their ability to kill common parasites, a wide-spread medical ailment of these regions.5
Consumption of DMT has been observed to produce intense visual hallucinations and euphoria. The molecule has been ingested orally, by inhalation, and by injection. When analyzed in 1957 by the American chemists Anita Paradies and Francis Hochstein, ayahuasca was found to contain both the molecule DMT, and an enzyme inhibitor (Monoamine oxidase inhibitor, or MAOI), found in the Peganum harmala plant.6 Both of
these molecules are necessary for the hallucinogenic effects: Because DMT is rapidly degraded by monoamine oxidase, an enzyme found in the mitochondria of most cells, without the presence of an inhibitory molecule, DMT would be dismantled before psychoactive levels were reached in the body.7,8 To produce hallucinations, the dose of DMT must be sufficient to exceed the enzymatic capabilities of monoamine oxidase, and the inhibition of this enzyme greatly lowers the necessary dose. The amounts of these ingredients vary based on the regional flora, as do each plant's association with local spirits and regional ceremonial value.9
Dr. Timothy Leary, a Harvard psychologist, was perhaps the first to popularize the use of DMT in the western world. Under the Harvard Psilocybin Project, Leary conducted experiments on subjects from 1960 to 1962 alongside Dr. Richard Alpert.10 When Harvard colleagues raised concerns about the viability and safety of the project's research methods, the school's student newspaper published these concerns. The ensuing national attention quickly brought an end to the study, and brought about an official investigation by the Massachusetts Department of Health. In other related experiments, most notably the Marsh Chapel Experiment, concrete evidence of Leary's active participation in the study was well documented. Though both Leary and Alpert were consequently terminated from their positions at the university, they continued as famed advocates for the use of psychedelics, in what became known as the counterculture of the 1960s. The end of the Harvard Psilocybin Project coincided with an increasingly negative portrayal of psychedelics by the media, which was due to a rising public anxiety towards recreational drugs. As a result, DMT was classified as a Schedule I drug under the 1971 UN Convention on Psychotropic Substances.
These events occurred in reaction to a larger social movement of the 1960s and 70s
that was characterized by antiestablishment movements in the Western world. In response to social and political tensions, many of these campaigns—the civil rights movement, protest of the Vietnam war, feminist movements, and the anti-nuclear movement culminated in the collective 1960s counterculture. Popular connotations of this period paint a picture of hippies and free love, protest music, and the expanded use of recreational drugs. DMT was a casually-used substance at the time, forming an enduring association with the movement. In the time since this era, this cultural paradigm shift has been criticized writ large for its futility and irrationality. A continued association with the ‘60s counterculture has led to a similarly poor view of DMT in contemporary society which is often oblivious to a rich history of cultural and medicinal value.
In a 2004 Supreme Court ruling, the Uniao do Vegetal church, a US branch of a Brazilian-based sect, was granted the right to import and consume ayahuasca tea for religious purposes under the Religious Freedom Restoration Act of 1993.10 This specific event exemplifies a larger narrative of the fight for culturally significant practices and substances to be respected in western law and society. Substances used by indigenous groups such as ayahuasca, the coca leaf, and the peyote cactus are listed as Schedule I drugs under the Comprehensive Drug Abuse Prevention and Control Act of 1970. This legislation was signed by President Richard Nixon and was seen as a governmental response both to the counterculture movements and part of the declaration of the War on Drugs.
Our knowledge of DMT has been limited due to the socially and politically reinforced suspension of its scientific study. With a gap of more than 20 years in the research of DMT, there are many things that are just now being learned about the many ways that this incredible molecule may affect the body. When the US government first restricted the
studies, scientists had just begun to explore the short-term effects of DMT on the body. At that time, scientists had begun to find links between levels of DMT in the body and relative rates of schizophrenia and depression.11,12 Though no studies have been able to relate these directly to DMT at a statistically significant level, improved detection methods raise this tantalizing possibility.12 Varying levels of neurotransmitters closely related to DMT, such as serotonin, have historically been linked to feelings of contentment and joy, and thought to be largely responsible for the regulation of sleep cycles, mood, and appetite.13 DMT and serotonin have very similar chemical structures (Figure 2). Due to the similar structures and given that DMT may be produced in the pineal gland (where serotonin is known to be produced), there is building evidence that points to DMT functioning in similar processes in the body.
Due to the cultural pressures following Timothy Leary's fall from grace and the legal classification of DMT, research on this compound ground to a halt for decades until the mid-1990s when primary studies were conducted on DMT's chemistry within the human brain, as well as its association with several mental disorders. Dr. Rick Strassman, a medical doctor, and professor of Psychiatry at the University of New Mexico conducted hundreds of experiments on 60 volunteers from 1990 to 1995. Strassman sought to gather basic information on the chemical's effect on the body through simple measures including heart rate, body temperature, and sensory consciousness.13 Since then, similar studies have allowed scientists to gain a basic knowledge of the effects of DMT on the body. The physical effects of DMT ingestion in humans include slightly elevated blood pressure, body temperature, and heart rate, as well as pupil dilation, and increased
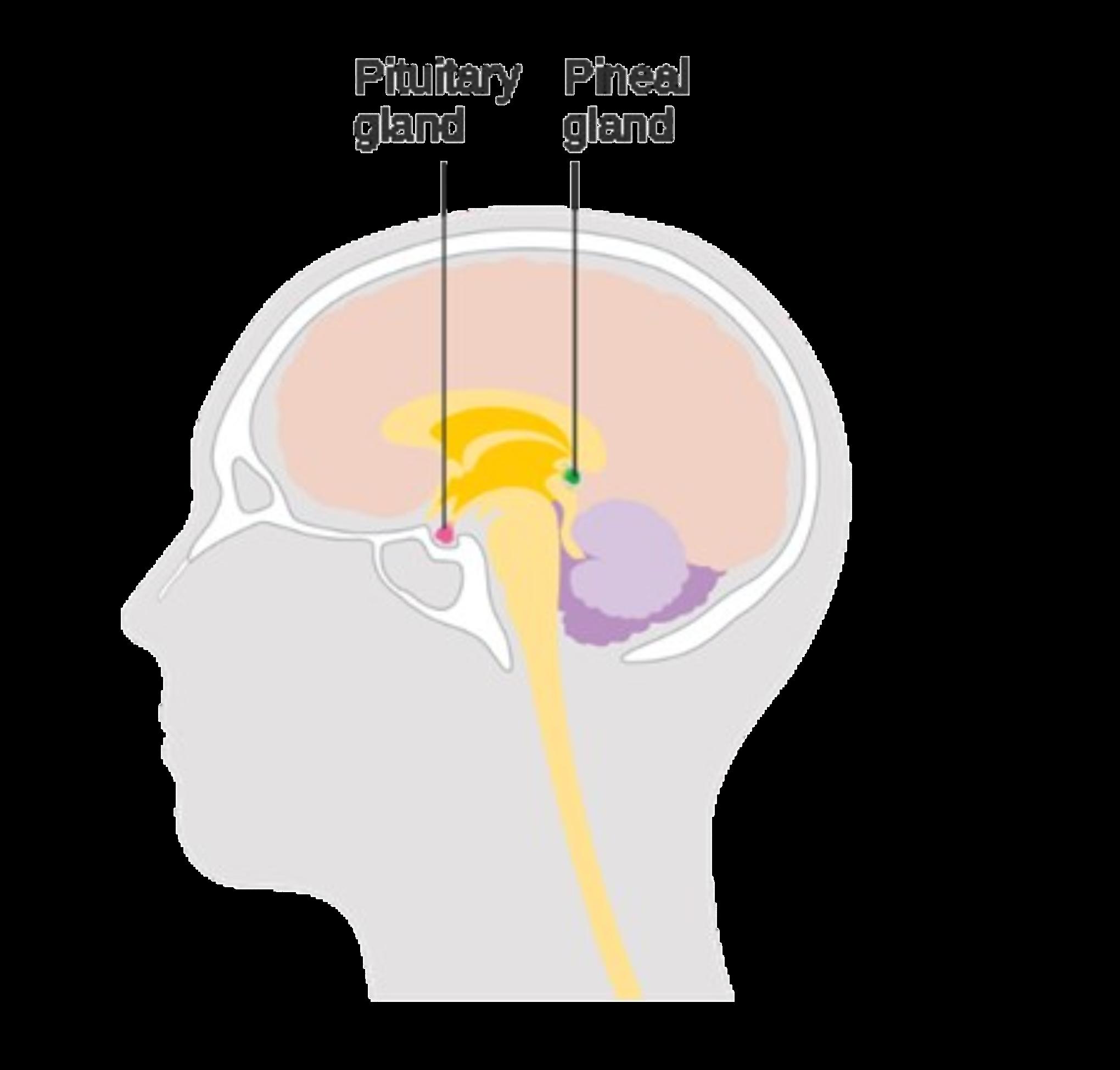
levels of growth hormone, cortisol, prolactin, beta-endorphin, and corticotropin in the blood. The addictive potential and the likelihood of long-term psychological effects were determined to be minimal in a recent psychological study.14 Led by his previous investigations on the pineal gland, Strassman concluded that his experiments supported the notion that the gland and chemical were somehow involved in the reality perception of the human subjects.
The pineal gland (Figure 3) is an endocrine gland in the brain that is responsible for the production of the sleep hormone melatonin. Melatonin is also quite abundant throughout nature, and can be found in plants, animals, fungi, and bacteria.15 DMT is derived by the enzyme amine N-methyltransferase from the amino acid L-tryptophan. The molecule is also structurally analogous to the neurotransmitter melatonin (Figure 2). This, in addition to its hallucinogenic properties, has led some scientists to propose DMT as an important chemical in the process of dreaming during sleep. In another important study, DMT was found in rodent pineal glands in 2013.16 It is hypothesized that the compound may be released by the pineal gland during unique events, such as deep meditation and near-death experiences.
Though human cells possess amine Nmethyltransferase, the enzyme that can produce the molecule, this process has yet to be observed in the human pineal gland.17 Based on a 1961 experiment conducted by an Amer-
ican biochemist, DMT was synthesized from tryptamine by an enzyme extracted from rabbit lung.18 This paper led to the compelling ideas that this molecule could be produced endogenously. The endogenous production of DMT would indicate its importance on a universal level to living organisms. In Strassman's book, The Spirit Molecule, he argues that the endogenous production of DMT points to its elemental significance in the way humans — and perhaps all living organisms perceive and experience consciousness. These experiments have coincided with increasingly welldocumented accounts of near-death experiences and religiously-induced hallucinogenic states that Strassman and others have hypothesized may be the res ult of a massive releas e of DMT from the pineal gland.17
One of the most burdensome consequences of the 1970 Controlled Substances Act was that this molecule was made a cultural taboo. Not only did this have devastating, racially oppressive effects on the indigenous cultures and religions to which it had significance, but it also prevented scientists from studying it with any credibility. Even today, the resurgence of interest in DMTt has been stunted by its association with recreational
drug culture, causing findings to be largely ignored by the medical community at large. For this reason alone, DMT is an important molecule to study further. With primary findings that point to such vast and meaningful ways that DMT may connect with our lives, both medically and psychologically, it is imperative that we continue to explore these theories through a scientific lens.
Figures 1 & 2 were created by the author and the editors.
Figure 3 was created by Cancer Research UK and downloaded from Wikimedia Commons for open-source use.
https://creativecommons.org/licenses/bysa/4.0/
(1) Bigwood, J.; Ott, J. DMT: The Fifteen Minute Trip. Head 1977, 11, 56.
(2) Gaujac, A.; Martinez, S. T.; Gomes, A. A.; de Andrade, S. J.; da Cunha Pinto, A.; David, J. M.; Navickiene, S.; de Andrade, J. B. Application of Analytical Methods for the Structural Characterization and Purity Assessment of N, N-Dimethyltryptamine, a Potent Psychedelic Agent Isolated from Mimosa Tenuiflora Inner Barks. Microchem. J. 2013, 109, 78–83.
(3) Forsström, J. T., J. Kärkkäinen, T. Determination of Potentially Hallucinogenic NDimethylated Indoleamines in Human Urine
by HPLC/ESI-MS-MS. Scand. J. Clin. Lab. Invest. 2001, 61 (7), 547–556.
(4) Rivier, L.; Lindgren, J.-E. “Ayahuasca,” the South American Hallucinogenic Drink: An Ethnobotanical and Chemical Investigation. Econ. Bot. 1972, 26 (2), 101–129.
(5) Andritzky, W. Sociopsychotherapeutic Functions of Ayahuasca Healing in Amazonia. J. Psychoactive Drugs 1989, 21 (1), 77–89. https:// doi.org/10.1080/02791072.1989.10472145.
(6) Callaway, J. C.; Brito, G. S.; Neves, E. S. Phytochemical Analyses of Banisteriopsis Caapi and Psychotria Viridis. J. Psychoactive Drugs 2005, 37 (2), 145–150.
(7) Hochstein, F.; Paradies, A. M. Alkaloids of Banisteria Caapi and Prestonia Amazonicum. J. Am. Chem. Soc. 1957, 79 (21), 5735–5736.
(8) McKenna, D. J.; Towers, G. N.; Abbott, F. Monoamine Oxidase Inhibitors in South American Hallucinogenic Plants: Tryptamine andCarboline Constituents of Ayahuasca. J. Ethnopharmacol. 1984, 10 (2), 195–223.
(9) Descola, P. In the Society of Nature: A Native Ecology in Amazonia; Cambridge University Press, 1996; Vol. 93.
(10) Labate, B. C.; Feeney, K. Ayahuasca and the Process of Regulation in Brazil and Internationally: Implications and Challenges. Int. J. Drug Policy 2012, 23 (2), 154–161.
(11)Hoffer, A.; Osmond, H.; Smythies, J. Schizophrenia: A New Approach. II. Result of a Year’s Research. J. Ment. Sci. 1954, 100 (418), 29–45.
(12) Angrist, B.; Gershon, S.; Sathananthan, G.;
Walker, R. W.; Lopez-Ramos, B.; Mandel, L. R.; Vandenheuvel, W. J. A. Dimethyltryptamine Levels in Blood of Schizophrenic Patients and Control Subjects. Psychopharmacology (Berl.) 1976, 47 (1), 29–32. https://doi.org/10.1007/ BF00428697.
(13) Young, S. N. How to Increase Serotonin in the Human Brain without Drugs. J. Psychiatry Neurosci. JPN 2007, 32 (6), 394.
(14) Gable, R. S. Risk Assessment of Ritual Use of Oral Dimethyltryptamine (DMT) and Harmala Alkaloids. Addiction 2007, 102 (1), 24–34.
(15) Hardeland, R.; Pandi-Perumal, S.; Cardinali, D. P. Melatonin. Int. J. Biochem. Cell Biol. 2006, 38 (3), 313–316.
(16) Barker, S. A.; Borjigin, J.; Lomnicka, I.; Strassman, R. LC/MS/MS Analysis of the Endogenous Dimethyltryptamine Hallucinogens, Their Precursors, and Major Metabolites in Rat Pineal Gland Microdialysate. Biomed. Chromatogr. 2013, 27 (12), 1690–1700. https:// doi.org/10.1002/bmc.2981.
(17) Cozzi, N. V.; Mavlyutov, T.; Thompson, M. A.; Ruoho, A. E. Indolethylamine NMethyltransferase Expression in Primate Nervous Tissue; 2011; Vol. 37.
(18) Thompson, M. A.; Weinshilboum, R. M. Rabbit Lung Indolethylamine NMethyltransferase CDNA AND GENE CLONING AND CHARACTERIZATION. J. Biol. Chem. 1998, 273 (51), 34502–34510.

T hey surround us in our everyday lives. We see them on the sides of busy streets and in the dense forests, both up high and low to the ground. They find their way into every special, sacred moment of our lives: weddings, graduations, anniversaries. These living creatures dominate symbolic beauty, yet their diversity can twist them in every magnificent and sinister way possible. They give us little more than a lovely display, yet we spend over $34 billion dollars on them every single year. These beautiful, odd, even startling organisms are flowers, and we don’t even fully understand how they exist.
The earliest flower that humans have found dates back to the Cretaceous period, or more than 146 million years ago. Flowers existed long before the age of humans and likely even grew among the feet of dinosaurs. Yet, so much about their existence is an enigma. The existence and evolution of flowers hold three main mysteries: How did they evolve and become so common among plants (~80% of plants are angiosperms, or flowering plants); why are humans drawn so powerfully to them; and, how do plants even make such incredible, complex structures such as flowers?
For a market as popular and economically important as flowers, one would think that we would have at least a decent idea about their evolutionary history, but the truth is, we don’t. The question of how plants evolved into angiosperms was raised during the age of the legendary biologist and botanist, Charles Darwin. Darwin’s theory of evolution was viewed as radical, even sacrilegious, among scientists at the time, and so he was obligated to gather as much evidence as he could to support that evolution was more than just some crazy theory. While he collected enough evidence to now be counted among the most well-known scientists, his studies of flowers threw a very large wrench in his hypotheses.
Regarding his thesis, Charles Darwin once wrote “[Evolution] can never take a great and sudden leap, but must advance by short and sure, though slow steps...” While this was consistent with Darwin’s other findings, that was not what he nor other scientists discovered regarding flowering plants. Looking at fossils, it appeared that flowering plants appeared—almost magically—over a very short series of years. Scientists could not find any biological reason for flowering plants to show up so rapidly; neither could they disprove that this was not the way it actually happened.
When scientists discover something that contradicts their work, they begin making theories, and this instance was no different. Several ideas were suggested about how flowers could appear so rapidly, but every idea seemed to be flawed. Some proposed that flowers evolved along with their insect pollinators for the most part. They claimed that the abundance of insects caused enormous amounts of specialization among flowers which led to the huge boom of angiosperms (flowering plants). However, fossil records later revealed that flowering plants existed in abundance and reproduced via wind pollination well before they began such an intimate relationship with pollinators. Others hypothesized that angiosperms simply outcompeted gymnosperms (cone-bearing plants) by having more photosynthetic surface area on their leaves and advanced seed dispersal tactics, but the sheer abundance of the needle-like leaves of gymnosperms and their year-round existence disproved that. Theory after theory was brought up, but every idea fell short. And that hasn’t changed: We are still contemplating these evolutionary relationships.
Regardless of the amount of research put into this “evolutionary puzzle,” the mystery of angiosperm evolution remains unsolved. A recent theory suggested that an increase of veins in the leaves of angiosperms would allow more water and nutrient circula-
tion and therefore more growth. However, the quantity of leaves on a single gymnosperm which would could also lead to a huge amount of surface area and veins, disproving this theory, just like the photosynthetic theory.
The most modern and accepted theory is that there is simply no perfect answer. Flowering plants, like gymnosperms, evolved over time. They began to use living pollinators instead of abiotic methods of pollination (mainly wind) and gradually became modernday plants. While the species diversity—the number of species—of angiosperms is drastically higher than that of gymnosperms, the species richness, or number of individuals per species, of gymnosperms matches and even surpasses angiosperms in some regions. Today, gymnosperms dominate most cold, higher altitude poor-soil regions while angiosperms reign in warmer, tropical climates.
The precise reason for such an abundant, diverse group of flowering plants today is unknown... and the reason humans like them so much is also not generally known! Plants use a variety of different methods to attract their pollinators. Some offer delicious rewards of nectar or pollen. Many release distinctive aromas: these are usually pleasant to humans, with a few notable exceptions like the so-called “corpse flower”. These aromas are important chemical cues that attract insects and birds to their blooms. Some plants, notably the Orchidaceae family, trick insects into pollinating them by using chemical scents and mimicry, offering the insect no reward at all. While some of these methods draw humans to the flower, this does not, in itself, bring about pollination of the flower, a sign that either the flower is poorly evolved, or that these signals are not meant for humans at all.
Humans’ interactions with flowers date back all the way to the Pleistocene Epoch, an era almost 2.6 million years ago. Several ancient human remains were discovered with the remnants of bouquets of flowers of many
varieties and species. Whether for a funeral or an ancient ritual, the allure of flowers has held humans captive since the beginning and that captivity has held tight throughout history.
One interesting aspect of this relationship is the varied symbolism humans gave (and give) to flowers through different cultures. Japanese emperors were given Chrysanthemums to symbolize a long life, while Italians considered this flower a symbol of death. Lilies were depicted in the bible, and were also used in Greek myths and in other Pagan rituals. There have been thousands of poems, paintings, and songs that featured or were centered around flowers. The drastic differentiation among floral symbolism among different cultures proves that the human fascination towards these colorful, little gems is far more intricate than just some chemical attraction: it is a complex, psychological, individualistic one. How did this attraction evolve from picking flowers for a ritual or event to cashing in over $34 billion in their favor every year?
One of the most notable events in the history of human–flower relationships has to do with that of the tulip. During several times in the 16th –18th centuries, the simple tulip caused greed, theft, and murder in several European countries. This series of seemingly insane events, (all over flowers!), began around 1550 when Ogier Ghislain de Busbecq, an ambassador of the Hungarian Hapsburgs, introduced the tulip to Europe. The tulip immediately became a staple across the continent, with Ottoman Turks frenzying over creating the “perfect,” new hybrid and traders in Constantinople buying bulbs for their weight in gold. Historical stories from France claim that a man sold his entire flower mill for a single, rare bulb and a bridegroom accepted a single bulb as his entire dowry. The unexplainable fad over flowers rushed across the continent, but the tulip-craze
Tulipomania as the phenomenon gradually came to be known was worse in the Netherlands than anywhere else.
In 1635, official tulip trades began in the Netherlands and everyone ranging from farmers to botanists to the general populace came from all over the country to buy valuable and rare tulips. At these events, people would sell businesses, homes, and life savings for flowers. When the trade came to an end, sellers would go home rich in coin from their endeavors while buyers would go home poor but rich in tulips.
The tulip trade became an addiction, to the point that people could not wait the several months between the trades, so people opened “colleges” where under-the-table tulip sales were held every few days. Then on February 2, 1637, the time for the official trade came, but people had no money to buy any bulbs, having spent it all at these colleges. Breeders had invested thousands and thousands of coinage into the trade, so when no one was willing to buy, enormous amounts of money were lost and the country nearly went bankrupt.
This event, while ridiculous-sounding, revealed just how important the human -flower relationship was. Leaders and countries alike were willing to invest so heavily in a market of flowers that the failure of the aforementioned trade could literally bankrupt the entire country. That moment in history is what truly codified the relationship between humans and flowers:. They don’t provide food, they don’t provide any sort of physical resource, but people were still willing to invest exorbitant amounts of money into their existence among us. If scientists could just figure out how to make flowers bow to their whims and bloom continuously, they could revolutionize the entire flower industry. But scientists were taking on significantly more than they could imagine with that inquisition.
For over 100 years, scientists have been studying what makes an angiosperm plant actually create flowers and bloom. And for over
100 years, they’ve had found nothing more than theories. That is, until now. Talk of this elusive chemical, (which came to be known as florigen), has always been hypothesized, but it could not be found. Test after test turned up negative, and this near-mythical chemical remained as nothing more than an idea. Yet, in the past few years, scientists believe that they have—maybe—finally discovered what may cause plants to create these beautiful treasures.
The quest for the flowering molecule’s true identity began in Germany in 1865. Julius von Sachs was a historical botanist, whose life goal was to piece together the history of botany and the inner working of plants. During his studies, he realized that transferring the sap of a plant in-flower onto the open-tissue of another plant that was not in-flower would cause the non-flowering plant to bloom. He repeated this test over and over again, even placing the sap of the blooming plant onto a nonblooming plant of another species and genus, and found that the non-blooming plant would almost always begin blooming. This phenomenon gave Sachs the idea that there must be a single molecule that caused plants to flower. Time and time again, he attempted to isolate it, but never succeeded. This molecule was our elusive florigen.
Out of all the “major molecules of biology,” Florigen has arguably had one of the roughest, least satisfying scientific searches of them all. Fast-forward almost 150 years, and the identity of Florigen is still not entirely known. Modern studies on Florigen discovered that Sachs’s experiment with sap did make sense, as Florigen was originally proposed as a single molecule. It was also discovered that semiochemicals (basically plant hormones) were found in extremely small concentrations within the sap of plants as it traveled through the phloem part of the plant’s vascular tissue. Since scientists believed that florigen was one of these small semiochemicals, it meant that scientists could theoretically
find the elusive molecule if they had machines sensitive enough to detect it, yet that still was not the case.
When scientists further studied the molecule that they believed was florigen, they found that the creation of a flower was significantly more complex than the work of a simple little hormone. Various studies turned up with contradictory data as to what kind of molecule florigen was (or if it was even a single molecule). Several studies concluded by saying that florigen was likely a set of molecules that helped with many diverse biological processes. Instead of answering at last what caused plants to flower, these studies only created more and more questions about the seemingly simple process.
A study in 2009 by Shalit et al. concluded that Florigen was a polypeptide a small chain of amino acids that is expressed by a specific sets of genes that led to the evolution of angiosperms in the first place. However, this same study discovered that an overexpression of a specific gene known as the SFT gene (SFT stands for “Single Flower Truss”) led to extremely abundant flowering, but a retardation of growth. They concluded that, since two antithetical factors were controlled by a single gene, Florigen must either have more than one purpose or must, as stated earlier, be more than one molecule.
As the study progressed, they found more and more factors that played a role in the plant flowering. For instance, an overexpression of yet another specific gene sequence could induce flowering regardless of the night/day cycles that the plant was exposed to. After finishing the analyses, this group concluded that, while florigen did help induce flowering, its role is far more complex than they had imagined. They knew that both florigen and local SFT genes, in addition to other proteins and gene sequences, could be used to both induce and terminate flowering in a plant, and that florigen existed in multiple forms.
Yet, florigen’s ability to seemingly transform a vegetative meristem into a reproductive (flowering) one was still a mystery. Florigen seemed to comprise a set of multiple proteins, and possibly even a specific mRNA sequence, which would explain the role of the multiple genes in the flowering process. Clearly, the biochemistry of a flower is an enormous and complex equation!
Scientists have recently come to the conclusion that florigen is not just one molecule. Recent studies agreed with some of the “basic” ideas of some earlier studies, but simplified and organized them significantly more. Today, florigen is accepted as a polypeptide that gains its “magical, flowering powers” when it interacts with another protein and specific gene sequences. Scientists believe that a lengthening of nighttime is what causes this complex reaction to occur which, in turn, induces a plant to create the beautiful structure known as a flower.
What does the identification of florigen mean for society? If humans could harness the power of florigen, the agricultural and botanical industries would be completely revolutionized. The terms “fruit season” or “flower season” would be obsolete. The need for intense and costly heating and lighting strategies to keep plants flowering and producing fruit year -round would be unnecessary. Perhaps gardeners would be able to get any plant to flower regardless of the light it receives, the temperature it grows in, or even the age of the plant. But, as usual, this could lead to other problems.
Attempts to synthesize florigen from scratch have been unsuccessful. Florigen is a polypeptide, and the folding process of polypeptides is often difficult or impossible to do synthetically. Others have focused on extracting florigen from living plant tissue. However, since florigen, which requires another protein and specific DNA/RNA sequences to actually
work, obtaining all of the necessary “puzzle pieces” has proved extremely difficult. Thus, studies and attempts to utilize this incredibly complex molecule continue.
As another day begins, flowers open their sepals and petals to the light. And yet the mystery of flowers remains exactly that: a complex mystery. They captivate the senses of humans and pollinators alike. For humans, the sights and scents of flowers offer rewards that are largely aesthetic. Day by day passes and millions of flowers open and die as time moves forward, a biochemistry that is a subject of ongoing study. Regardless, the flower remains one of the most fascinating, beautiful, and apparently complex sights in the world. Maybe one day we will actually understand them.
(4) Darwin’s Theory Of Evolution https:// www.darwins-theory-of-evolution.com/ (accessed Jan 8, 2021).
(5) Pickup Flowers http:// www.pickupflowers.com/ (accessed Jan 8, 2021).
(6) Hanlon, V. Florigen: The Hidden Hormone https://www.scq.ubc.ca/florigen-the-hiddenhormone/ (accessed Jan 8, 2021).
(7) Kroeze, D. Plant flowering hormone - Florigen | CANNA Gardening USA https:// www.cannagardening.com/florigen (accessed Jan 8, 2021).
(8) Pollan, M. The Botany of Desire: A Plant’sEye View of the World; Random house trade paperbacks, 2002.
(9) Schiestl, F. P.; Johnson, S. D. Flowers and humans – a curious love affair https:// blog.oup.com/2017/02/flowers-humans-loveaffair/ (accessed Jan 8, 2021).
(1) Barras, C. The abominable mystery: How flowers conquered the world https:// www.bbc.com/earth/story/20141017-howflowers-conquered-the-world (accessed Jan 8, 2021).
(2) Britt, R. R.; 2005. Human Affection Altered Evolution of Flowers https:// www.livescience.com/295-human-affectionaltered-evolution-flowers.html (accessed Jan 8, 2021).
(3) Darwin, C.; Bynum, W. F. The Origin of Species by Means of Natural Selection: Or, the Preservation of Favored Races in the Struggle for Life; AL Burt New York, 2009.
(10) Shalit, A.; Rozman, A.; Goldshmidt, A.; Alvarez, J. P.; Bowman, J. L.; Eshed, Y.; Lifschitz, E. The Flowering Hormone Florigen Functions as a General Systemic Regulator of Growth and Termination. Proc. Natl. Acad. Sci. 2009, 106 (20), 8392–8397.
(11) Silady, A. The Economics of Flowers https://smartasset.com/insights/theeconomics-of-flowers (accessed Jan 8, 2021).
(12) Taoka, K.; Ohki, I.; Tsuji, H.; Kojima, C.; Shimamoto, K. Structure and Function of Florigen and the Receptor Complex. Trends Plant Sci. 2013, 18 (5), 287–294.
A ll three of the chapters in Part II deal with pesticides, including aspects of the environmental and social consequences of these molecules. The actions of pesticides, as well as their medium- and long-term impacts, are a result of how each molecule interacts with other natural molecules. A close examination of the structure of each pesticide is often the way to understand it. But, what’s the best way to make such a structural analysis?
The heteroatoms such as nitrogen [N], oxygen [O], and chlorine [Cl] in a line–angle structure tend to stand out, and for good reason: these atoms have outsized importance to the chemistry of a molecule. Heteroatoms feature in structural motifs and functional groups, which are small groups of atoms with specific connectivity and distinctive chemistry. A key example of this is in how molecules interact with water.
You know that water is H2O, and you may have heard that water is “polar.” Polar chemicals like water include some surface area of slight positive charge, and also some surface area of slight negative charge. Since positive and negative charges attract, polar chemicals tend to be attracted to other polar chemi-
cals. (In other words, polar chemicals interact favorably with other polar chemicals.) Nonpolar chemicals tend to avoid polar chemicals, and instead interact more favorably with other nonpolar chemicals. The phrase “like dissolves like” is often taught as a chemical truism: polar solvents (like water) dissolve other polar molecules, whereas nonpolar solvents are needed to dissolve nonpolar molecules.
Acetic acid (C2H4O2) is a great example of a very polar, water-soluble organic molecule. Most vinegars are a solution of about 5% acetic acid in water. Figure A6 includes an illustration of the acetic acid molecule. Vinegar has an equal number of carbon and oxygen atoms, and oxygen atoms give organic molecules at least a little bit of polarity wherever they are located. Even more importantly, one of the oxygen atoms in acetic acid is connected directly to a hydrogen atom. This motif [–OH ] is called the hydroxyl group. Hydroxyl groups are among the most polar, waterfriendly features on molecules. (You can think of a hydroxyl as “H1O”—very similar to H2 O!)
Fats and oils are examples of nonpolar molecules. Vigorously mixing a simple oil–vinegar salad dressing will yield a fleeting mixture that quickly separates into two layers, a result of water and acetic acid (the polar molecules) interacting unfavorably with nonpolar oils. Examine the structure of a molecule that
could be found in coconut oil (Figure A6). As always, start by looking for the heteroatoms: there are some oxygens again. But this time, there is a pretty significant part of the molecule without any heteroatoms: there are three long chains with just carbon and hydrogen atoms. Molecules that are mostly carbons bonded to other carbons and to hydrogen atoms will be very non-polar, and thus very insoluble (not soluble) in water.
But wait what about those six oxygen atoms? Don’t oxygen atoms make parts of a molecule more polar? Yes! So, this molecule is not 100% non-polar; that oxygen-rich part is slightly polar. However, this molecule has no hydroxyl groups, and more importantly, the vast majority of the molecule consists of three long chains of –CH2– groups. Ultimately, most molecules are like this: they have some parts that are more polar than other parts. Heteroatoms such as sulfur (S) and chlorine (Cl) tend to act as nonpolar motifs, whereas motifs with lots of oxygen and nitrogen atoms particularly as –OH and as –NH/NH2 make a molecule more polar.
All this is to say that polarity is a key property to keep in mind as you read these next chapters and look at these chemical structures. Molecules (or parts of molecules) with just carbon and hydrogen will tend to be nonpolar and dissolve in fat so if they are consumed by an animal, they may wind up in that animal’s fat. Fat tends to be long-lasting, so these molecules hang around and accumulate and if the molecule has detrimental effects at higher concentrations, that’s a very bad thing! Also, cell membranes are very nonpolar, which makes them a good barrier to water-soluble molecules. But, molecules that have large nonpolar parts may be more likely to pass into cells more easily...and if we’re talking about a molecule that is not supposed to be there, that’s likewise a bad thing! These concepts are noteworthy in these next chapters: Jenna Joyner’s, on imidacloprid and neonicotinoid pesticides; Caroline Jahn’s, on DDT; and Chloë Moore’s, on Agent Orange.
Bonus Quiz: Suppose I make a very strange salad dressing, starting from my oil/vinegar mixture in Figure A6. Instead of adding garlic and herbs, I add the chemicals shown in Figure A7: a bit of -carotene and a bit of anthocyanin. Where would each of those molecules wind up?
Answer:
· The -carotene, which contains only carbon and hydrogen atoms (it is a “hydrocarbon”) is nonpolar. It is more like the oil than the vinegar, so it would dissolve in the oil layer.
· The anthocyanin, on the other hand, contains five oxygen atoms, including four hydroxyls. This molecule is quite polar and therefore soluble in water. (And, when a molecule has a + or – charge, that also attracts it to polar solvents.)

I n 1874, 24-year-old student and chemist, Othmar Zeidler, in his lab at the University of Vienna, Austria, synthesized what would become one of the most notorious and ultimately devastating insecticides in the world: dichlorodiphenyltrichloroethane, more commonly known as DDT.1 Due to Swiss chemist Paul Müller’s discovery in 1939 that DDT was an effective pesticide, it was being sprayed on croplands throughout the world by the late 1940s.2 Industrial agriculture was in its infancy at the time and DDT was viewed as a “miracle chemical” because of its effectiveness in deterring common crop pests and allowing for increased crop yields at a low cost.2
However, it was not long before the harmful effects of the pesticide on non-target organisms began to emerge. Environmentalists began noting the effects of the pesticide on non-insect animals including birds, mammals, and fish. Studies were showing the link between DDT and eggshell thinning in birds, membrane function in fish, and tumors in mammals such as rats.3 Regulators began vocalizing how the heavy use of the pesticide could lead to a massive collapse of biodiversity, as the underpinning of the food chain was being eradicated. The toxin was not just harming biodiversity, but human health as well. DDT was linked to headaches, nausea, dizziness, obesity, diabetes, and decreased birth weight in humans exposed to relatively small amounts.3 These environmental and health concerns finally led to DDT being banned in the U.S. in 1972, after nearly three decades of use.4
The attributes of DDT that made it so harmful to biodiversity and human health were simple and widespread: DDT was too persistent in the environment and too broadly toxic to non-target organisms. What the world needed was a pesticide that specifically targeted insects, would not persist in the environment, and perhaps most importantly for societal acceptance would not harm human health. This is where the idea for a new type of insecticide, neonicotinoids, came from.
Neonicotinoids, unlike DDT, were designed to naturally degrade and only inhibit biological processes in the insect nervous system. Neonicotinoids have quickly become a worldwide, multi-billion-dollar industry since their inception in the mid-1980s and are now the most prevalent class of pesticides globally.5 The use of neonicotinoids has increased the yield and quality of several indispensable agricultural plants. While they are much less toxic to mammals than other pesticides, they are now believed to be exerting a massive effect on the ecosystem by harming the honeybee, which is one of our most important ecosystem service providers.
For as long as humans have been cultivating plants, they have been looking for ways to manage the pests that inevitably come seeking them. The most common way pests have been managed throughout history and in agriculture today is through the usage of chemical insecticides. Organic molecules derived from insect-resistant plants are some of the most historically prevalent insecticides.1 In ancient China, for example, Dalmatian pyrethrum flowers containing the insecticidal substance pyrethrin were used in the management of mosquitoes, cockroaches, bedbugs, and flies.1 Derivatives from other plants such as elderberry, lavender, garlic, and countless others have been used to exterminate unwanted insects.1,6,7
One of the most notable and effective naturally-occurring insecticides is nicotine, obtained from the tobacco plant. Nicotine is widely known for its properties as an addictive stimulant in smoking and chewing tobacco, but the insecticidal properties of nicotine are familiar to some organic farmers. Nicotine is produced by tobacco plants because it poisons and deters parasitic insects.8 Tobacco leaves steeped in water have even been used as “green” insecticides for small-scale growing operations. Nicotine extracted from the plant has been considered as an alternative to
commercial insecticides on a large scale, but unfortunately it is not as effective as many other pesticides and can be toxic to mammals and non-target organisms.9,10 Research into the method by which nicotine poisons insects has led to the synthesis of neonicotinoids. The term “neonicotinoids” literally translates to “new nicotine-like insecticides” because these molecules were designed to biologically mimic nicotine and result in similar behavioral and lethal effects in invertebrates.11,12
Neonicotinoids are neurotoxins that interact with a specific type of binding site called nicotinic acetylcholine receptors (nAChRs) of neurons required for muscle movement and cognitive functions in insects.13 Neuronal binding sites are normally used by chemical messengers called neurotransmitters, which bind to the site and signal the neuron to transmit information to neighboring cells. However, this system is not foolproof: If a molecule with similar properties is around, it can attach to the binding site as well. When the biologically correct molecule binds to a receptor of a neuron, it causes an electrical impulse to pass down the cell. When this electrical gradient reaches the end of the cell, it can cause the cell to release more neurotransmitters so neighboring cells can fire as well, thereby passing along information.14 A biologically active foreign molecule can disrupt this process easily, and cause significant damage to the organism.
Neuronal receptors and neurotransmitters function like locks and keys, respectively. Using this analogy, a key can only fit into the
lock if it is the right size and has a compatible electrochemical charge.14 In the case of the nAChR “lock,” the neurotransmitter “key” is acetylcholine. Acetylcholine (Figure 1) has a positively charged end consisting of a nitrogen atom connected to four carbon atoms. To activate the receptor, two acetylcholine molecules bind and to form favorable interactions with negative charges on the nAChR receptor.15
This positively charged tip, size, and shape of the molecule is what allows it to bind to the negatively charged binding site of the receptor, and these qualities are mimicked by the chemical properties of nicotine. Nicotine (Figure 2) contains two ring structures, both containing one nitrogen atom. One of these, called a pyrrolidine ring, contains four carbon atoms and one nitrogen in a five-membered ring. The nitrogen in this ring can accept a proton (H+) and become positively charged (Figure 2) just like acetylcholine. This is what allows nicotine to mimic the neurotransmitter and bind to the same enzyme site.15
Both the acetylcholine and nicotine
“keys” can fit into the nAChR “lock” in insects and mammals; however, insect nAChRs possess a quality that mammalian ones do not, and only insect nAChRs will permit neonicotinoids to bind. Unlike acetylcholine and nico-
The neutral form is shown at the left; the protonated, positively-charged form that mimics acetylcholine is shown at right.
tine, neonicotinoids like imidacloprid (Figure 3) do not have strongly positively charged regions. Instead, they contain negatively charged surfaces, consisting of a nitrogen atom bonded to two oxygen atoms (–NO2, called a nitro group).15 This negative charge causes imidacloprid to be attracted by—and bind to a positively charged site that is only present in invertebrate nAChRs.16. This interaction with positively charged amino acids in the insect nAChR can be seen is diagrammed in Figure 3 with imidacloprid, the first neonicotinoid to be synthesized and also the most prevalent.12 Unlike acetylcholine and nicotine, which only interact briefly with the nAChR, the binding of neonicotinoids is irreversible.9 When neonicotinoids bind to an insect nAChR, ion channels of the neuron remain open permanently, forcing the cell to continuously fire until it is unable to reestablish its electrochemical gradient and becomes functionally dead.17 These nAChRs are vital for muscle movement, and when a neonicotinoid forces them into a permanent excitatory response, it can result in apparent intoxication, tremors, paralysis, and death of the insect.18
Because neonicotinoids only bind to receptors in invertebrates, they are considered to be a safer insecticide for use around humans and other mammals when compared to DDT. Unfortunately, neonicotinoids are still
very toxic to other organisms with the same type of nAChR as the target insects. When insecticides are used in the environment, migration to non-target areas is inevitable, and this transfer can have substantial impacts on non-target organisms. Numerous studies have addressed these impacts on a broad range of species. For example, aquatic animals such as crabs, sea urchins, and fish have respectively shown immunosuppression, DNA damage, and developmental abnormalities with exposure to neonicotinoids.19 For terrestrial animals, the vast majority of research has been performed on honeybees; these studies have shown that sublethal amounts of neonicotinoids affect a bee’s ability to forage for food and navigate to its hive.20,21 Other land animals such as birds, amphibians, and reptiles can also be affected.2 The effects of neonicotinoids on all of these animals can also have larger impacts on ecosystems by altering the capacities of these animals to perform their roles in the environment.
Another issue with neonicotinoids is the ease with which they transfer to and persist in the environment, two issues that the toxin was supposed to be designed not to do. Neonico-
The molecular structure of imidacloprid is shown at left.
Shown at right is the binding affinity between a region on imidacloprid with negative polarity and the positively charged amino acids in insect nAChR.
tinoids are systemic insecticides, meaning they are water-soluble and can be taken up by and distributed throughout the targeted plant.22 Water solubility allows them to circulate within the plant, but it also permits the toxins to easily leach into waterways. The process by which this occurs can vary, depending on the method in which the neonicotinoid was applied. In ‘seed dressing,’ for example, seeds of the target crop are coated with a neonicotinoid to allow it to be fully absorbed by the plant as the seed grows, but in actuality, only 2–20% of the active ingredient is actually absorbed.23 The remaining substance binds to soil particles, until they come into contact with an ample amount of water such as during rainfall or irrigation whereby they are released. From there, the insecticide can leach into the groundwater system or travel to larger bodies of water, where it can accumulate and harm aquatic organisms and ecosystems.24 Neonicotinoids can also be released through the decay of treated plants, direct contact with dust resulting from drilling treated seeds, and from topical spray drifting into water bodies.
This process of transfer into watersheds would be inevitable with any water-soluble chemical used for pest treatment, but manufacturers of neonicotinoids gave initial assurance that this process was not cause for concern, as neonicotinoids are designed to degrade in water.25 However, various studies have since demonstrated that this degradation can only occur under certain conditions, most of which are less common in the environment than in certain lab settings.22 Specifically, neonicotinoids degrade through a process called aqueous photolysis, meaning degradation with water and light. The time it takes for half of a concentration of a neonicotinoid to degrade can vary significantly between each type, and neonicotinoids such as imidacloprid can degrade in less than three hours under ideal laboratory conditions.26 Unfortunately, environmental conditions are rarely ideal. Higher pH, increased turbidity, lower temper-
ature, and low amounts of light can slow degradation and increase the persistence of neonicotinoids to an excess of 1,000 days.22,24 Such degradation-inhibiting influences are not unusual; fast-moving streams, deep bodies of water, and even shallow bodies of water with ample tree cover can all accumulate neonicotinoids that will persist in the ecosystem.
After nearly 30 years of use, these negative qualities of neonicotinoids, including the fact that they harm a wide range of nontarget organisms, and considering that they do not readily degrade as intended, are being recognized not only by environmentalists, but by entire countries. The European Union has begun banning neonicotinoids for all outdoor use and the sentiment appears to be growing around the world.27 While the effects of neonicotinoids on animals and the environment in general are different than that of DDT, it is becoming clear that they are harmful enough to garner discontinuation, or at least stricter regulations It seems that a perfect insecticide one which only harms targeted organisms without inducing adverse ecosystem effects— remains yet to be discovered.
All figures were created by the author and the editors.
Sources Cited
(1) Oberemok, V. V.; Laikova, K. V.; Gninenko, Y. I.; Zaitsev, A. S.; Nyadar, P. M.; Adeyemi, T. A. A Short History of Insecticides. J. Plant Prot. Res. 2015, 55 (3), 221–226. https://doi.org/10.1515/jppr-2015-0033.
(2) Scriabine, R. ‘Neonicotinoids: The New DDT?’ Forbidden Knowledge TV, 2014.
(3) Jayaraj, R.; Megha, P.; Sreedev, P. Organochlorine Pesticides, Their Toxic Effects on Living Organisms and Their Fate in the Environment. Interdiscip. Toxicol. 2016, 9 (3–4), 90 –100.
(4) US EPA, O. DDT Ban Takes Effect ddt-bantakes-effect.html (accessed Jan 19, 2021).
(5) AgInfomatics, LLC http:// aginfomatics.com/ (accessed Feb 8, 2021).
(6) Maga, R.; Broussalis, A.; Clemente, S.; Mareggiani, G.; Ferraro, G. 1, 8 Cineol: Responsible for the Insecticide Activity of Lavandula Spica Mill (Lavender). Rev. Latinoam. Quím. 2000, 28 (3), 146–149.
(7) Oparaeke, A.; Dike, M.; Amatobi, C. Insecticide Potential of Extracts of Garlic, Allium Sativum (Linneaus) Bulb and African NutMeg, Monodora Myristica (Gaertn) Dunal Seed for Insect Pest Control on Cowpea.; Entomological Society of Nigeria, 2000; pp 169 –174.
(8) Yamamoto, I. Nicotine to Nicotinoids: 1962 to 1997. In Nicotinoid insecticides and the nicotinic acetylcholine receptor; Springer, 1999; pp 3–27.
(9) Booker, C. J.; Bedmutha, R.; Vogel, T.; Gloor, A.; Xu, R.; Ferrante, L.; Yeung, K. K.-C.; Scott, I. M.; Conn, K. L.; Berruti, F. Experimental Investigations into the Insecticidal, Fungicidal, and Bactericidal Properties of Pyrolysis Bio-Oil from Tobacco Leaves Using a
Fluidized Bed Pilot Plant. Ind. Eng. Chem. Res. 2010, 49 (20), 10074–10079.
(10) Thany, S. H. Neonicotinoid Insecticides. In Insect Nicotinic Acetylcholine Receptors; Springer, 2010; pp 75–83.
(11) Singh, D. Advances in Plant Biopesticides; Springer, 2014.
(12) Kim, U. Neonicotinoid Insecticide. Warn. Getclass Expects Parameter 1 Be Object Array InhomevhostsejournaluserDirhtdocsclassescacheGenericCache Inc Php Line 63 MMG 445 Basic Biotechnol. EJournal 2006, 2 (1), 46–52.
(13) Dupuis, J.; Louis, T.; Gauthier, M.; Raymond, V. Insights from Honeybee (Apis Mellifera) and Fly (Drosophila Melanogaster) Nicotinic Acetylcholine Receptors: From Genes to Behavioral Functions. Neurosci. Biobehav. Rev. 2012, 36 (6), 1553–1564.
(14) Kalat, J. W. Biological Psychology; Nelson Education, 2015.
(15) Matsuda, K.; Kanaoka, S.; Akamatsu, M.; Sattelle, D. B. Diverse Actions and Target-Site Selectivity of Neonicotinoids: Structural Insights. Mol. Pharmacol. 2009, 76 (1), 1–10.
(16) Tomizawa, M.; Lee, D. L.; Casida, J. E. Neonicotinoid Insecticides: Molecular Features Conferring Selectivity for Insect versus Mammalian Nicotinic Receptors. J. Agric. Food Chem. 2000, 48 (12), 6016–6024.
(17) Déglise, P.; Grünewald, B.; Gauthier, M. The Insecticide Imidacloprid Is a Partial Agonist of the Nicotinic Receptor of Honeybee Kenyon Cells. Neurosci. Lett. 2002, 321 (1–2), 13–16. https://doi.org/10.1016/S0304-3940(01) 02400-4.
(18) da Costa, L. M.; Grella, T. C.; Barbosa, R. A.; Malaspina, O.; Nocelli, R. C. F. Determination of Acute Lethal Doses (LD50 and LC50) of
Imidacloprid for the Native Bee Melipona Scutellaris Latreille, 1811 (Hymenoptera: Apidae). Sociobiology 2015, 62 (4), 578–582.
(19) Pisa, L. W.; Amaral-Rogers, V.; Belzunces, L. P.; Bonmatin, J.-M.; Downs, C. A.; Goulson, D.; Kreutzweiser, D. P.; Krupke, C.; Liess, M.; McField, M. Effects of Neonicotinoids and Fipronil on Non-Target Invertebrates. Environ. Sci. Pollut. Res. 2015, 22 (1), 68–102.
(20) Yang, E.; Chuang, Y.; Chen, Y.; Chang, L. Abnormal Foraging Behavior Induced by Sublethal Dosage of Imidacloprid in the Honey Bee (Hymenoptera: Apidae). J. Econ. Entomol. 2008, 101 (6), 1743–1748.
(21) Fischer, J.; Mueller, T.; Spatz, A.-K.; Greggers, U.; Gruenewald, B.; Menzel, R. Neonicotinoids Interfere with Specific Components of Navigation in Honeybees. PloS One 2014, 9 (3), e91364.
(22) Wood, T. J.; Goulson, D. The Environmental Risks of Neonicotinoid Pesticides: A Review of the Evidence Post 2013. Environ. Sci. Pollut. Res. 2017, 24 (21), 17285–17325.
(23) Sur, R.; Stork, A. Uptake, Translocation and Metabolism of Imidacloprid in Plants. Bull. Insectology 2003, 56, 35–40.
(24) Bonmatin, J.-M.; Giorio, C.; Girolami, V.; Goulson, D.; Kreutzweiser, D.; Krupke, C.; Liess, M.; Long, E.; Marzaro, M.; Mitchell, E. A. Environmental Fate and Exposure; Neonicotinoids and Fipronil. Environ. Sci. Pollut. Res. 2015, 22 (1), 35–67.
(25) Bayer Crop Science https:// www.cropscience.bayer.com/ (accessed Feb 8, 2021).
(26) Moza, P.; Hustert, K.; Feicht, E.; Kettrup, A. Photolysis of Imidacloprid in Aqueous Solution. Chemosphere 1998, 36 (3), 497–502.
(27) McDaniel, S. The bees are dying https:// www.baltimoresun.com/opinion/op-ed/bsed-op-0208-bee-health-20190207-story.html (accessed Jan 8, 2021).

I t was September 1943 and the Second World War pressed on across the globe. The Germans invaded Naples, Italy: Ally territory. Before long the city was wrecked. Without sanitation and adequate hygiene, typhuscarrying lice thrived and caused an almost instantaneous epidemic. Civilians were killed by the thousands and Italy seemed close to losing its position as one of the most powerful allies in the war. It was at this time that the United States decided to prioritize aiding Italy in the face of this outbreak. The U.S. government reached out to its various resources. The United States Military Office of Scientific Research and Development was informed of a chemical called dichlorodiphenyltrichloroethane, studied by a man named Paul H. Müller. Dichlorodiphenyltrichloroethane, shortened to DDT, had insecticidal properties including the extermination of lice and the control of the typhus epidemic. So DDT was manufactured in bulk, shipped to Italy, and sprayed on the city of Naples and its people. The effects were astonishing: the death rate plummeted and within a month, typhus was completely eradicated. The miracle insecticide DDT was used soon after to exterminate malaria-carrying mosquitos in other Ally territories, leading to a resurgence of the Allies’ drive. Eventually the Allies were victorious in the war.1
Soon after its success, DDT became a worldwide phenomenon in the pursuit of a means to halt the spread of malaria. In great magnitude and with efficient dispersal, DDT was able to reduce the yearly deaths caused by malaria dramatically.2 In addition, agriculturists discovered the power of the chemical as an insecticide that could greatly increase their yields. In the 1950s American farmers hastily grabbed at an opportunity to eliminate agricultural pests and decided to put their trust in DDT, liberally spraying this chemical on their crops. Unfortunately, they were releasing a compound about which they and the world knew very little, and which would lead to significant consequences. In 1962 Rachel
Carson wrote Silent Spring, a moving treatise concerning the effects of DDT on wildlife.3 In particular, Carson focuses on the effects that DDT has on birds. Though later studies have shown that the chemical itself is not directly lethal to certain species, it has serious effects on the birds’ reproduction processes which directly influence their population sizes. Silent Spring ignited an environmental movement that altered worldviews on insecticides, ecology, and environmental policy.1
The chemical structure of the molecule DDT gives it unique characteristics. This chemical is nearly insoluble in water due to its lack of polarity: the electronegative atoms oxygen and nitrogen, which tend to increase the solubility of organic molecules, are absent from the structure as seen in Figure 1.
DDT is, however, highly soluble in lipids (fats). These two properties have a major influence on the effect of the chemical in the environment. Furthermore, DDT degrades slowly: it has a long lifetime, between two and fifteen years, and so it remains in the environment and in the systems of the organisms for extended periods after the initial application.4
As a lipophilic (fat-soluble) molecule, DDT is able to infiltrate the body of insects. And as an organochlorine (that is, as an organic molecule that contains chlorine atoms), it is able to alter the nervous system of the organism. The molecule binds to the gamma-
aminobutyric acid (GABA) chloride ionophore complex at the picrotoxinin site. This site regulates the chloride gates into the central nervous system; this gate must be able to open and close to function. The presence of DDT effectively blocks this gate, and the flow of chloride to the nerve is obstructed. Eventually, the insect’s movements begin to slow, it seizes, hyperexcites, and then dies.5
DDT is not a naturally-occurring chemical. In 1874 the German chemist Othmar Zeidler was a graduate student doing research at the University of Strassburg. He performed a technique known as the Baeyer Condensation on chlorobenzene and chloral with sulfuric acid additives. This reaction formed dichlorodiphenyltrichloroethane (Figure 1). At the time Zeildler believed this molecule was an ordinary organic compound with few distinguishing properties.6
In the 1930s, a man named Paul H. Müller began to search for an effective organic insecticide with two chloral benzene rings and an additional bond to something such as an oxygen, sulfur, or carbon atom. He was originally looking for a way to exterminate common clothes moths and carpet beetles but soon decided to attempt to find a more general insecticide. In 1939 he resynthesized DDT and discovered its power to kill Calliphora flies, mosquitoes, the Colorado potato beetles, and several other insects. In 1948 he received the Nobel Peace Prize for this work.6
During World War II, American scientists recognized the power of DDT to exterminate disease-carrying insects. The widespread spraying of DDT helped the Allies tremendously, allowing them to regroup and putting them on a path to win the war. Relative peace was restored as the war drew to an end, in part due to DDT’s ability to stifle the dispersal of typhus and malaria. After the
war, the chemical was used across the world in countries that had previously been consumed by these insect-borne diseases, resulting in significant decreases in mortality and benefiting overall public health. DDT was sprayed in households, public places, and even on people themselves. While it was used, DDT is estimated to have saved roughly 50 million lives. It was truly the wonder drug of its day.6
Dichlorodiphenyltrichloroethane was soon introduced to the agricultural world as a crop insecticide. Farmers throughout the world, especially those in the United States, began to shower their fields in the chemical with the hope of eradicating pests. The results were indisputable; crop yields increased everywhere. Over 3.2 billion pounds of DDT were used on approximately 350 different agricultural products such as fruits, cotton, and potatoes. During the height of the DDT craze, the chemical was even used in backyard vegetable gardens and in American households to control clothes moths and carpet beetles.5
As DDT became more prevalent in the environment, especially in aquatic environments, other invertebrates unintentionally ingested the chemical. Due to its lipophilic quality, DDT began to accumulate in the tissues of invertebrates. DDT then traveled up the food chain, accumulating within the bodies of successive predators. Birds including the osprey, pelicans, and eagles began to accumulate relatively high levels of DDT. In high concentrations DDT though not lethal to the individual birds—has serious effects on their cycles of reproduction, and in turn, their population sizes.6,7 While these bird species were still able to produce eggs, the DDT inhibited the deposition of calcium in the eggshells. As a result, the eggs of these
birds could not support the weight of their mother during incubation and would crack, killing the embryo (Figure 2).7

differences in density between the years 1940 (pre-DDT), 1970 (height of DDT use), and 2013 (four decades after the ban). Before DDT was widely used in this area, it was estimated that there were approximately 1,000 pairs of ospreys. Between 1940 and 1970 the population plummeted to approximately 109 pairs. Nests were left empty and sightings became rare. After DDT was banned, the chemical slowly became less prevalent in the environment, especially in shorelines and aquatic environments where ospreys are often found. In 2013 the osprey population was estimated to be greater than 1,200 pairs throughout this area.9 Figure 3 shows an osprey nest on a shore off the bay of Long Island, New York to which osprey families have only returned in the past ten years.
In the 1950s, after the mass use of DDT as an insecticide, populations of the Brown Pelican and the Peregrine Falcon dipped to near extinction in North America. The Golden Eagle and various Osprey followed suit.7,8 This is when ecologists began to investigate the mysterious decrease in bird populations. In 1962, Rachel Carson’s Silent Spring shed light on the possibility that DDT was the main cause of the population decreases. Since then, scientists have investigated that theory and discovered the chemical’s shell -thinning quality.
On December 31, 1972, the United States Environmental Protection Agency banned the use of DDT. Major agricultural operations and other industries were forced to phase out this pesticide. Collectively the United States had dumped 1.35 billion pounds of DDT on its own soil in the years before the ban. Within a year after the ban, all DDT use ceased in the United States.
There have been several studies on the resurgence of certain bird populations in more recent years due to decreased amounts of DDT in the environment. In a study of osprey populations in southern New England and Long Island, researchers found drastic

Today the name of the chemical DDT is recognizable to many. Often it is associated with ideas of harsh chemical insecticides, environmental pollutants, the death of bird species, and an EPA ban. It is far less ordinary to
hear it associated with the concept of a miracle drug or the savior of millions of human lives. DDT is a controversial organic compound with a bad reputation.
Public opinion on the chemical itself, its uses, and its outlaw vary greatly. Many see the publication of Rachel Carson’s Silent Spring as the catalyst for a major environmental movement whose first major accomplishment was the ban of DDT. This organic chemical sparked the interest of many future environmentalists in the United States in the late twentieth century, and cast light on several other agricultural and industrial practices that had potentially harmful ecological consequences. The ban of DDT itself resulted in the recovery of important bird populations and prevented the continued ecological devastation caused by the widespread use of DDT. These, however, are not the most important result of the ban. The ban proved that an environmentally conscious generation could pressure the government and effect real change. This first major accomplishment spurred environmentalists to continue campaigning for environmental justice.
There are some who believe environmentalists overreacted to Silent Spring and blindly demanded the outlaw of a chemical that they did not completely understand. Recognition of DDT’s role in World War II and the large-scale suppression of malaria around the world were swept under the carpet. Recognition of DDT’s role in increasing food production of both large and smallscale American farms has also been overshadowed by its bad reputation. To the untrained eye, DDT is a scar on our history. DDT indisputably had tremendous negative impacts on the environment, but its previous uses and beneficial accomplishments have not been completely lost on today’s scientists who continue to study DDT for the synthesis of new insecticides.
Figure 1 was created by the author and the editors.
Figure 2 was created by Ryan Soma and downloaded from https://www.flickr.com/ photos/ideonexus/4023463558 for opensource use under Creative Commons license: https://creativecommons.org/licenses/bysa/4.0/
Figure 3 was created by the author.
(1) Kinkela, D. DDT and the American Century: Global Health, Environmental Politics, and the Pesticide That Changed the World; Univ of North Carolina Press, 2011.
(2) Prevention, C.-C. for D. C. and. CDC - Malaria - About Malaria - History - Elimination of Malaria in the United States (1947-1951) https://www.cdc.gov/malaria/about/history/ elimination_us.html (accessed Dec 4, 2020).
(3) Carson, R. Silent Spring, 50th anniversary ed.; Penguin Classics: London .a.], 2012.
(4) Sudharshan, S.; Naidu, R.; Mallavarapu, M.; Bolan, N. DDT Remediation in Contaminated Soils: A Review of Recent Studies. Biodegradation 2012, 23 (6), 851–863.
(5) Coats, J. R. Mechanisms of Toxic Action and Structure- Activity Relationships for Organochiorine and Synthetic Pyrethroid Insecticides. 8.
(6) Metcalf, R. L. Century of DDT https:// pubs.acs.org/doi/pdf/10.1021/jf60188a040 (accessed Dec 4, 2020). https:// doi.org/10.1021/jf60188a040.
(7) Ehrilch, P. R.; Dabkin, D. S.; Wheye, D. DDT and Birds https://web.stanford.edu/ group/stanfordbirds/text/uessays/ uDDT_and_Birds.html (accessed Dec 4, 2020).
(8) Ehrlich, P.; Dobkin, D. S.; Wheye, D. Birder’s Handbook; Simon and Schuster, 1988.
(9) Bierregaard, R. O.; David, A. B.; Gibson, L.; Kennedy, R. S.; Poole, A. F.; Scheibel, M. S.; Victoria, J. Post-DDT Recovery of Osprey ( Pandion Haliaetus ) Populations in Southern New England and Long Island, New York, 1970–2013. J. Raptor Res. 2014, 48 (4), 361–374. https://doi.org/10.3356/JRR-OSPR-14-04.1.

I n early morning, American planes flew over Cat Son, releasing a fine, orange mist. The year was 1965, and the Cat Son region was split by the turbulent border between North and South Vietnam. Yet the people of Cat Son who rose from their beds as the strange mist settled over their land were not soldiers, but farmers. Within three days their forests and crops were decimated; foliage withered, fruits disintegrated and the young rice crop which was so relied upon was entirely destroyed. Like in other regions, many farmers fled their homes. Those who stayed were faced with the very real danger of starvation.1
This was one of the first of many uses of Agent Orange by the United States war regime, in its failed attempt to halt communist North Vietnam. Agent Orange was a potent mix of herbicides that led to severe, unintended consequences. It would be many years before the United States government acknowledged the damage done by the application of Agent Orange, not only ecologically, but also in terms of human health.1
The two major components of Agent Orange are organic compounds 2,4-dichlorophenoxyacetic acid and 2,4,5-trichlorophenoxyacetic acid (Figure 1). These are commonly referred to as 2,4-D and 2,4,5-T, and are mixed together in a 1:1 ratio to produce Agent Orange.2 While best known for their use in the Vietnam War, these compounds were actually developed during World War II. Like many other agricultural products of the modern age, 2,4-D and 2,4,5-T were first created as part of the war effort, and only later incorporated into the food system.
The two closely related chemicals, 2,4D and 2,4,5-T, were discovered by four research groups working independently in the early 1940s.3 Testing began shortly thereafter, and the compounds were deemed suitable for wartime crop destruction and for agricultural purposes on the home front.3 In 1945, at war’s end, 2,4-D and 2,4,5-T were put on the agricultural market as effective herbicides.1 This was not uncommon, as many other products of war were transitioned over to agricultural use. Business boomed for companies that could market their wartime products to the enthusiastic farmers willing to transition to new methods of large-scale industrial agriculture. The rise in synthetic pesticides and fertilizers, along with the accompanying rise in industrial farming practices, agricultural productivity, and (arguably) ecological degradation is referred to as the Green Revolution.
Both 2,4-D and 2,4,5-T are what is referred to as hormone herbicides. These compounds mimic auxin, a class of naturallyoccurring phytohormones (plant hormones) that regulate growth.4 Similarities between the structure of 2,4-D and IAA (indole acetic acid), the most common phytohormone within the auxins, can be seen in Figure 2. Plants rely on auxins for various internal signals based on concentration. Thus, the concentration of auxins in one part of a plant may be very different from that in another part, in order to maintain healthy growth patterns. Poor regulation of auxins causes a plant to become overtaxed by futile cycles extraneous metabolic processes related to superfluous and unfavorable
growth. In extreme cases, this leads to stunting or even plant death. While naturallyoccurring auxin molecules are degraded rapidly within a plant’s metabolic processes, synthetic auxins like 2,4-D and 2,4,5-T remain active for far longer. Thus, these molecules overload plant cells, leading to extremely inefficient energy use and rapid death.4
Synthetic auxins are far more harmful to dicots (a diverse class of plants including deciduous trees and most shrub-like understory plants) than they are to monocots (such as grasses).4 Due to the distinction, 2,4-D and 2,4,5-T are referred to as selective herbicides, unlike others that decimate plant life indiscriminately. As such, they are efficient at deforestation, but also at maintaining weed-free monocot crops such as corn, wheat, and the highly-sought-after manicured lawn. Because of this useful specificity, 2,4-D remains as one of the most widely-used agricultural herbicides. On the other hand, 2,4,5-T is now a highly-regulated substance in the United States and around the world, due to the extreme likelihood of it becoming contaminated by a much more dangerous substance during production.5,6
When American soldiers expressed concern about the effects of Agent Orange, they were told they had nothing to fear. Soldiers were often exposed to the powerful herbicidal mixture when clearing away brush surrounding American bases, but more even more regularly when spraying Vietnamese cropland and forests. This tactic was used to diminish guerrilla warfare by eliminating natural cover, and to force poor farmers (who might harbor and feed communist soldiers of North Vietnam) to flee their homes.1 Government agencies and private companies alike assured soldiers, veterans, and other concerned parties that both 2,4-D and 2,4,5-T degraded rapidly within ecosystems and were not harmful to human beings.6 While 2,4-D and 2,4,5-T do indeed have low toxicity, another compound lurked within the Agent Orange mixture—one with a much lower concentration but with severe consequences to human health.
The molecule 2,3,7,8-tetrachlorodibenzo-p-dioxin, commonly referred to simply as “dioxin” or TCDD, (Figure 3) is a highly toxic byproduct of industrial 2,4,5-T synthesis. It was a contaminant of Agent Orange and has had serious, long-term effects on veterans and civilians alike. Unlike 2,4-D and 2,3,5-T, dioxin does not break down quickly. Dioxin is not water-soluble and it binds to soil particles. Though it degrades more quickly when exposed to sunlight, dioxin has a halflife of one hundred years in dimly lit systems such as sediment-filled waterways. Due to its nonpolar structure, dioxin is also fat-soluble
and thus accumulates in animal tissues. It can remain in the human body for a lifetime. Dioxin is also prone to biomagnification, becoming present in higher and higher concentrations as it flows up through a food chain.6 In this manner, the dangerous dioxin can harm far more people than just those who were directly exposed to Agent Orange.
After returning home from the war, Vietnam veterans exposed to Agent Orange began experiencing a host of health problems. High rates of cancer and infertility, along with birth defects in their children were among the many issues.1 This problem was and still is even worse for the people of Vietnam. Upwards of 4.5 million people lived in areas sprayed by Agent Orange, and many more have been exposed through contaminated food sources.7 Intense, direct exposure has caused skin diseases, cancers, and immune deficiencies. Still, the pain experienced by the Vietnamese people does not end there. Birth defects including spina bifida, other neural tube defects (in which the spine and spinal cord do not develop to maturity), and cleft palates are far more likely in the descendants of affected people, along with higher rates of stillbirths.8
The health complications caused by dioxin are rooted in its effects on the transcription factor AHR (aryl hydrocarbon receptor). The role of any transcription factor is to bind to DNA and alter (either increase or decrease) the rate at which a piece of the genetic code is transcribed (rewritten) into RNA. In this way, transcription factors affect how often certain proteins are made by cells. In turn, this affects not only the makeup of the cell but also how rapidly a variety of biological reactions can occur. Transcription factors are thus crucial to proper functioning at the cellular level. AHR is a necessary transcription factor for potentially hundreds of mammalian genes, all of which can thus be mis-regulated by dioxin.9
Within a cell, most AHR molecules are inactive and unneeded at any given moment and are held by other proteins known as cochaperones which prevent them from binding to DNA. Dioxin binds inactive AHR molecules and activates them by separating them from the co-chaperones. Newly active AHR molecules are then free to bind to DNA, and wreak havoc on the cell by drastically altering gene expression.9
Some of dioxin’s potential effects are still debated, for political reasons as much as scientific ones. There has been a great deal of controversy as to which health complications American veterans will be compensated for, and some listed by the VA may not directly be caused by dioxin.10 Despite debate and uncertainty, dioxin appears to have additional carcinogenic and mutagenic effects beyond its interference with AHR, and also to target and disrupt the endocrine system.2,11
With concerns and complaints about health complications on the rise, the U.S. government suspended the usage of 2,4,5-T as an agricultural herbicide in 1970. By the end of the year, military operations in Vietnam also stopped using the contaminated herbicide. Lawsuits from veterans exposed to dioxin began as a trickle, but became a torrent against the U.S. military and private producers alike by the late 1970s and into the 1980s. Due to scientific and political uncertainty, disability compensation for suffering veterans was not secured in full until 2010.1
In the past decade, many millions of dollars have also gone to the American aid effort in Vietnam, though no amount of money can repair the lives destroyed, or eliminate the vast amounts of dioxin still lurking in Vietnamese soils, waterways, plants, animals, and people. The U.S. sprayed an estimated 20 million gallons of toxic herbicides on Vietnamese
land, and it will take many more years before that land is again healthy and safe.12
The environmental impacts of dioxin, 2,4-D and 2,4,5-T have been devastating. With forests destroyed, the huge gaps allowed numerous invasive plant species to flourish. Habitat loss for a variety of large animal species, including tigers, Asian elephants, gaur, water buffalo, wild boar, bears, deer, civets, and leopards was severe, and continues to affect species richness today.12 Vietnamese, American, and other agencies continue to work towards ecological restoration, but such efforts are slow considering the magnitude of contamination.13
The three chemical components of Agent Orange two intentional, one accidental may have started on the same path, but their places in the world today are quite different. The chemical 2,4-D continues to be a common tool of agriculture, and is a perfect representation of the Green Revolution and the largescale, industrial agriculture complex. On the other hand, 2,4,5-T, has been relegated to a few, highly controlled uses, tainted, if not by dioxin, then by its past. Dioxin itself, now feared as the single most toxic compound ever synthesized by humankind, still clings to a land and a people, a deadly reminder of the horrific consequences— intentional or otherwise of modern chemical warfare.
(1) Martini, E. A. Agent Orange: History, Science, and the Politics of Uncertainty; University of Massachusetts Press, 2012.
(2) Mart, M. Pesticides, a Love Story: America’s Enduring Embrace of Dangerous Chemicals; University Press of Kansas, 2015.
(3) Troyer, J. R. In the Beginning: The Multiple Discovery of the First Hormone Herbicides. Weed Sci. 2001, 49 (2), 290–297. https:// doi.org/10.1614/0043-1745(2001)049 [0290:ITBTMD]2.0.CO;2.
(4) Song, Y. Insight into the Mode of Action of 2, 4‐dichlorophenoxyacetic Acid (2,4‐D) as an Herbicide. J. Integr. Plant Biol. 2014, 56 (2), 106 –113.
(5) National Service Center for Environmental Publications (NSCEP). EPA Fact Sheet: Regulatory Status of 2 -4-5T 1978 https:// nepis.epa.gov/Exe/ZyNET.exe/
(6) What Is Dioxin: Chlorine Gone Wild. http://www.agentorangerecord.com/ information/what_is_dioxin/ (accessed Apr 3, 2017)
(7) Health Effects https:// www.aspeninstitute.org/programs/agentorange-in-vietnam-program/health-effects/ (accessed Jan 19, 2021).
(8) Ngo, A. D.; Taylor, R.; Roberts, C. L.; Nguyen, T. V. Association between Agent Orange and Birth Defects: Systematic Review and Meta-Analysis. Int. J. Epidemiol. 2006, 35 (5), 1220 –1230.
All figures were created by the author and the editors.
(9) Tijet, N.; Boutros, P. C.; Moffat, I. D.; Okey, A. B.; Tuomisto, J.; Pohjanvirta, R. Aryl Hydrocarbon Receptor Regulates Distinct DioxinDependent and Dioxin-Independent Gene Batteries. Mol. Pharmacol. 2006, 69 (1), 140–153.
(10) Friedman, S. M.; Dunwoody, S.; Rogers, C. L. Communicating Uncertainty: Media Coverage of New and Controversial Science; Routledge, 1999.
(11) Steenland, K.; Bertazzi, P.; Baccarelli, A.; Kogevinas, M. Dioxin Revisited: Developments since the 1997 IARC Classification of Dioxin as a Human Carcinogen. Environ. Health Perspect. 2004, 112 (13), 1265–1268.
(12) Agent Orange Record. Vietnam https:// agentorangerecord.com/vietnam/ (accessed Jan 19, 2021).
(13) U.S. Agency for International Development. Environmental Remediation | Vietnam https://www.usaid.gov/vietnam/ environmental-remediation (accessed Jan 19, 2021).
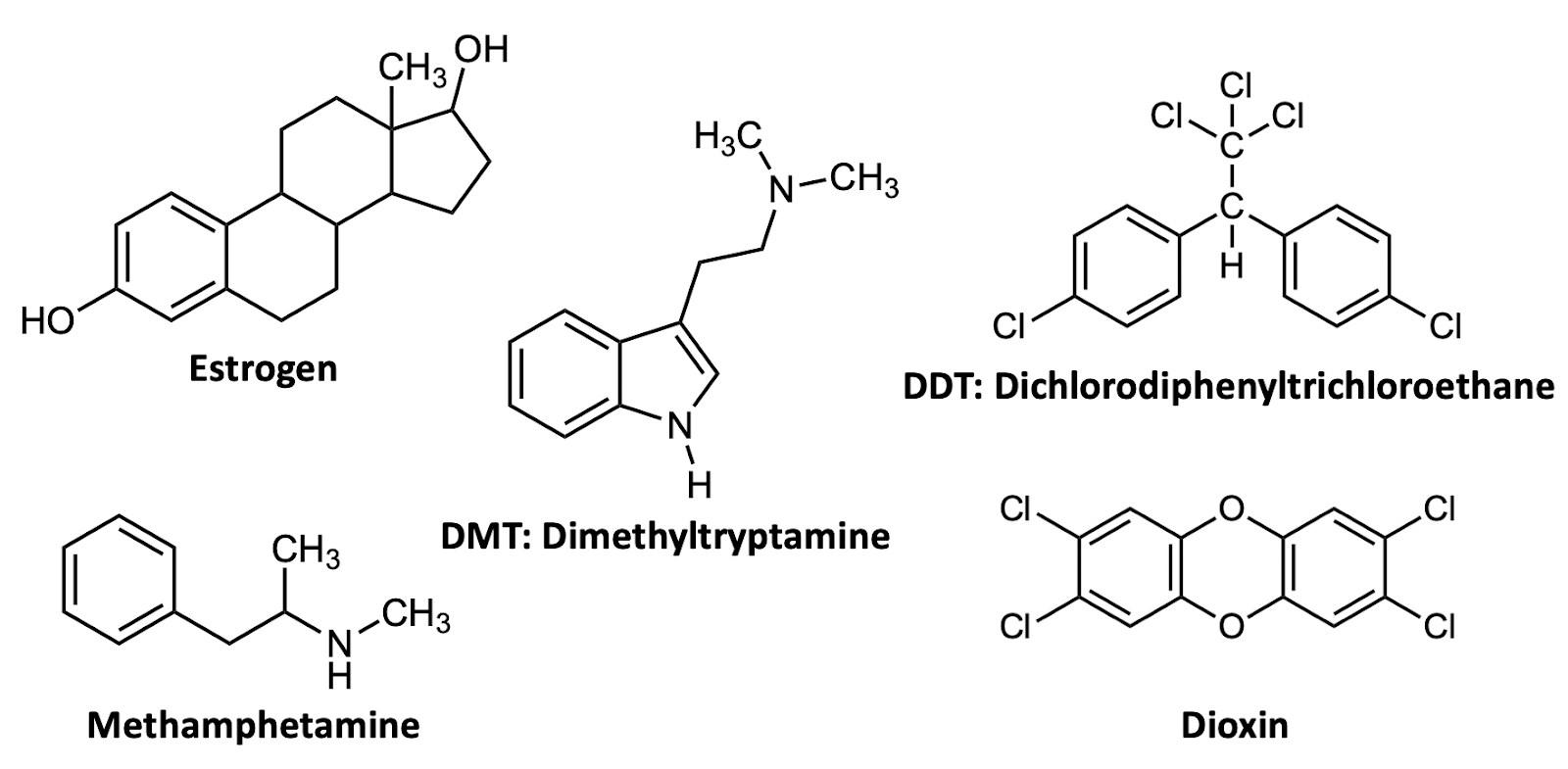
I first introduced the structure of estrogen in the introduction to Part I. There is another feature of this molecule, reprinted in Figure A8, that should catch your eye. In the “southwest” (i.e., lower left) part of the estrogen structure is a special six-membered ring with alternating single- and double-bonded carbon atoms. This motif is called a benzene (or benzene ring) and is extremely common in organic compounds. In fact, more than half of the molecules featured in this publication include it! Four other examples are likewise shown in Figure A8 to emphasize the ubiquity of this motif.
An important concept to define for this section is conjugation. Benzene is an example of a molecule with conjugation (or, said differently, benzene is conjugated). A conjugated molecule has a series of alternating single and double bonds. They may be in a ring, like in benzene, or in a straight line, or in some combination of the two. Some examples of conjugated molecules include -carotene and anthocyanins, shown in Figure A9.
You may have heard of one or both of these molecules and even if not, you’ve
probably eaten both of them recently! Thecarotene molecule, a precursor to vitamin A, is found in a variety of red, orange, and yellow fruits and vegetables, particularly in the carrots, from which the name derives . Anthocyanins (which is actually a class of molecules with similar structures) are found in blue and purple fruits and vegetables, including blueberries and purple cabbage. What’s striking about these molecules is that they are distinctly colorful.
What’s important about these molecules from the perspective of an organic chemist is that they are highly conjugated: there is a long series of alternating single and double bonds. This is no coincidence: highly conjugated molecules are often colorful, and colorful organic molecules are usually ones that have a lot of conjugation.
This is the theme to keep in mind in this next section: four chapters that feature highly conjugated molecules for which color is a key feature: Ana Risano’s, on chlorophyll; Halley McVeigh’s, on carminic acid; Zosha Silverstein-Belden’s, on melanin; and Kait Zinnecker’s, on henna. When you learn about these molecules, make sure to notice their extensive conjugation. (For many of them, their polarity is of great importance, too!)

One of the most vital molecules on Earth looms just beneath our noses. Its functions bring us food, energy, shelter, medicine, and a slew of other things. This molecule is small but mighty, providing a foundation for growth and stability on the planet. It has encompassed life for millions of years, aiding in the development of ecosystems. Can you guess it? Here is a hint:
Recall a time you were outside in a forest, park, or garden. Perhaps you walked down a path bordered by tall, old-growth trees covered in soft moss, or across a field of rough grass scattered with little wildflowers. Have you sat on the damp earth, weeding a bed of tomato plants with young green fruits? Wherever it was, take a moment to think about your place and what it has in common with other green-spaces.
It’s not a trick question: they all contain something green. The trees grow green leaves from their branches and have green moss covering their bark. The ground is covered with green grass. Green’s prevalence in life is extraordinary showing up in unexpected places. The source: molecules of chlorophyll found in plant cells. But what makes chlorophyll green, and why is it so common in nature?
One might recall learning about cells in high school. The mitochondria are the powerhouses of the cell, for example. Organelles — structures within a cell like the mitochondria have critical functions that are specific to the type of cell. There is one organelle specific to plants called the chloroplast. Chloroplasts are the sites of photosynthesis, a process in which plants take in carbon dioxide and liquid water to create sugars, and release oxygen and water vapor as byproducts.1 Chloroplasts are the location of our precious chlorophyll molecule.
Chlorophyll is responsible for absorbing different wavelengths of light. Plants mostly absorb red, blue, and violet wavelengths while reflecting green wavelengths—this is
why they appear green. Our eyes observe the reflected green light, while the plants absorb all other wavelengths and use this energy for photosynthesis.1
Figure 1 illustrates the different wavelengths absorbed by various forms of chlorophyll. Focus for now on Chlorophyll a, the green line in the graph. One of its tallest bands is around 665 nm, indicating the absorption of red visible light. The other tall band is between 400 nm and 465 nm, the wavelength range for violet and blue visible light.2 Between about 500 and 550 nm right at the green light wavelengths—little light is absorbed by any chlorophyll molecules, so this light is reflected for our eyes to see. The red, blue, and purple light taken into the plant is the source of energy that powers photosynthesis. In short, this pigment is essential for plants to gather nutrients and grow, making it more than just a coincidence in color.
Life inhabits the earth in a variety of forms. Plants, animals, bacteria, archaea, and fungi shaped the planet with their living habits, behaviors, and structures; however, they were also shaped by the planet—ecosystems developed and caused organisms to change, or adapt. These adaptations drove the process known as survival of the fittest, often defined as a competition, where only the most welladapted organisms survive within selective habitats.3 This planetary growth increased diversity, resilience, speciation, prosperity...and the curiosity of naturalists and scientists. The study of the natural world focuses on the why and how of everything. Plants, at the forefront, provided a variety of resources, but it took time to learn why they required certain conditions to prosper, how they became abundant, and why they are so necessary.
To understand chlorophyll, it is best to begin with the discovery of photosynthetic properties in plants. The first person to notice
that plants restored an atmospheric substance was Joseph Priestley, who in 1771 found that a sprig of mint could restore the air consumed by candle fire, but he did not incorporate molecules into his explanation of this observation.4 It took another sixty years before other scientists made definitive observations about the reaction of plants to produce the gas that we call oxygen.
In 1817, the source of green in most plants, algae, and cyanobacteria was isolated by Joseph Bienaimé Caventou and Pierre Joseph Pelletier, two chemists who later worked on isolating chemicals used in medicine.5 They named the molecule chlorophyll, meaning pale-green leaf.6 However, little was known about this molecule and what it did.
Twenty years later, Hugo Von Mohl observed chloroplasts as individual green bodies in an examination of plant cells.4 The reasoning behind the green pigment was still un-
known, but the observation led to further studies by Julius Von Sachs, who demonstrated that plants require light to produce starch.4 This was a major discovery, for it led to describing the process of how plants create food. Scientists now understood the importance of chlorophyll in this process.
In 1882, Nathan Prongsheim suggested the role of chlorophyll in light absorption, and experiments displayed a greater production of food with greater absorption of light by chlorophyll. Thus, light and chlorophyll became essentially linked as conditions in what is now called photosynthesis.4
The chemical formula for chlorophyll-a is C55H72O5N4Mg. It turns out, there are actually six known variations of chlorophyll (designated by the suffixes -a through -f). Chlorophyll-a is the most common form, found
in wheat and most plants. Figure 2 shows the structure for Chlorophyll-a.
The chlorophyll molecule includes a chlorin, which is a large ring structure. The chlorin ring is highlighted by the grey box in Figure 2, and is the structure on the left in Figure 3. Chlorins are composed of four smaller, five-membered rings that contain one nitrogen each, and these smaller rings are linked together by one additional carbon. Chlorins are derivatives of porphyrins, which are also commonly found in biological pigments: the most familiar porphyrin is the “heme” group in hemoglobin.7
Figure 3 is a comparison of a chlorin (C20H16N4) and a porphyrin (C20H12N4). The main difference between these two rings is that the chlorin rings have four extra hydrogen
atoms. Two of these hydrogens can be clearly seen attached to nitrogen atoms pointing to the middle of the chlorin. The other two can be inferred from the line-angle structure: the “southeast” fivemembered ring in the chlorin has a carbon–carbon single bond, whereas it is a double-bond in the porphyrin. Thus, in the chlorin this represents –CH2–CH2–, whereas it is –CH=CH– in the porphyrin. Note also that both molecules have a series of alternating single and double bonds that comprise the ring: this feature is key for giving these molecules color.
At the center of the chlorophyll is a magnesium (Mg) atom (Figure 2). Magnesium wasn’t discovered within the chlorophyll structure until 1906 by Richard Willstatter.4 We now know that magnesium is essential for photosynthesis: the magnesium ions are needed to activate a plant enzyme, RuBP carboxylase, that assists in assimilating carbon dioxide.8
Attached to the ring is a long tail of carbons, known as a hydrocarbon tail (Figure 4 shows a close-up of the tail, which can also be seen in an abbreviated form in the gold box in Figure 2). This tail is nonpolar and therefore fat -soluble and water-insoluble.9 The tail acts as an anchor for the chlorophyll, embedding it into the membrane of chambers within the chloroplast called thylakoids.1
Other types of chlorophyll, like chlorophyll-b (Figure 5), differ by small side chains off of the main ring structure. These groups are known as substituents, which are different ar-
rangements of atoms that alter the spectra of light absorbed. The difference between chlorophyll-a and chlorophyll-b is in a single side chain off of the chlorin ring in the “northwest” corner. Compare the blue circles on Figures 5 and 2: Chlorophyll-b contains HC=O (an aldehyde), while chlorophyll-a has CH3 (a methyl group).9 This difference, while subtle, actually changes what light wavelengths are absorbed: compare the green and blue lines on Figure 1. Chlorophyll-b is the second-most common form of chlorophyll in the plant kingdom, usually found in photosynthetic algae and cyanobacteria.2
Scientists wanted to understand the chlorophyll molecule in order to determine whether it can be replicated or adapted. This necessitated a total synthesis: a construction of the molecule by humans in a laboratory. The first such total synthesis of chlorophyll was completed and published by a R.B. Woodward and his team in the early 1960s, and was updated in 1990.10–12 In its entirety, the synthesis involved over forty steps! Ac-
cording to Woodward, chlorophyll is a very sensitive and complicated molecule to work with, making the synthesis a time-consuming process.10
First, Woodward and his team broke the molecule into four building blocks .10 These four pieces (designated I, II, III, and IV in Figure 6) served as the foundation for the synthesis: each of these were engineered starting from available chemicals as the first steps to recreate the molecule. (Note: the Roman numerals used to designate each compound are based on those used in the original publication.11)
Woodward’s team then began combining the pieces, as shown on the next page. In Figure 7, a dipyrromethene (molecule XXXI) was obtained by combining the aldehyde (molecule IV) with the aminoethyl pyrrole (molecule I); water is the other product of this reaction.11 The name “pyrrole” refers to the individual five-membered ring units composed of four carbons and a and a nitrogen that in turn make up the chlorin ring.7 A few minor modifications to this structure (on its “western” half) turned it into molecule XXXIX, shown in Figure 8.
In a separate reaction, they linked molecules II and III to form compound XL. At this point, they essentially had two “halves” of chlorophyll: the “north” half (XL) and the “south” half (XXXI). They connected these two pieces using a motif referred to as a “Schiff Base:” a carbon–nitrogen double-bond. The Schiff Base formation, shown in Figure 8, was brought a out by combining the aldehyde group (on molecule XL) and the amino (–NH2) group (XXXIX), forming the molecule (XLI) that contains all four pyrrole rings (I–IV).10 Note that this reaction also has a water molecule as the byproduct: the two H atoms from the amino and the oxygen from the aldehyde form H2O (shown in blue in Figure 8).
From here, Woodward’s team completed the synthesis in a few more steps. They added the magnesium ion last via a magnesium solution. In the natural creation of chlorophyll within a plant cell, the magnesium ion is likewise added last.10,11
Complex organic molecules require a lot of energy and enzymes to create, so it’s not surprising that the synthesis was difficult. Part of this complexity includes a phenomenon called conjugation alternating single and double bonds that allow the molecule to have color. However, plants do their own synthesis
of this complex molecule regularly, usually without fail. The efficiency of nature is remarkable, including how plants systematically breakdown their chlorophyll before shedding their leaves in order to preserve the magnesium a fairly precious element to plants.
Chlorophyll also has practical applications beyond its natural function of capturing light energy. For instance, it is used as a coloring agent for manufactured food.7 This use of chlorophyll has fluctuated over the years as people become more cautious of consuming artificial colorings, but chlorophyll is unstable in some environments and different pHs, which can affect the color.7 Chlorophyll has also been used medicinally: in some cases, it accelerates wound healing because it stimulates tissue growth.7 An increase in tissue growth corresponds to a decrease in the amount of bacterial growth, acting as an “internal deodorant” for patients—and for this reason it was used during WWII.9 Additionally, ointments with chlorophyll derivatives are known to ease the pain of ulcers and increase the rate of tissue healing.7
As a final note, imagine the world without chlorophyll. Plants would not sequester carbon dioxide from the air and feed themselves with sugars; oxygen would not be produced. The earth’s atmosphere would be completely altered, never having plants to take in the enormous amounts of carbon dioxide produced by living (and non-living) things. In short, the earth would be completely different without chlorophyll. Different forms of life would have to exist because the main sources of nutrients and energy would be altered. Chlorophyll has shaped the way of life for nearly everything we know, including humans.
Figure 1 was created by Serge Helfrich and used under Creative Commons license. https://creativecommons.org/licenses/bysa/4.0/ and https://commons.wikimedia.org/ wiki/User:Helfrich
All other figures were created by the author and by the editors
(1) Alberts, B.; Bray, D.; Hopkin, K.; Johnson, A. D.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Essential Cell Biology; Garland science, 2015.
(2) Chen, M. Chlorophyll Modifications and Their Spectral Extension in Oxygenic Photosynthesis. Annu. Rev. Biochem. 2014, 83, 317 –340.
(3) Bouchard, F. Darwinism without Populations: A More Inclusive Understanding of the “Survival of the Fittest.” Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2011, 42 (1), 106–114.
(4) Gibbs, M. Structure and Function of Chloroplasts. Berlin, Springer-Verlag.; Berlin, Springer-Verlag., 1971.
(5) Haas, L. Pierre Joseph Pelletier (17881842) and Jean Bienaime Caventou (17951887). J. Neurol. Neurosurg. Psychiatry 1994, 57 (11), 1333.
(6) New World Encyclopedia. Chlorophyll https://www.newworldencyclopedia.org/ entry/Chlorophyll (accessed Jan 21, 2021).
(7) Hosikian, A.; Lim, S.; Halim, R.; Danquah, M. K. Chlorophyll Extraction from Microalgae: A Review on the Process Engineering Aspects. Int. J. Chem. Eng. 2010, 2010.
(8) EasyGrow Ltd. Magnesium for Photosynthesis https://www.easy-grow.co.uk/usingmagnesium-to-boost-photosynthesis/ (accessed Jan 21, 2021).
(9) Linus Pauling Institute | Oregon State University. Chlorophyll and Chlorophyllin https:// lpi.oregonstate.edu/mic/dietary-factors/ phytochemicals/chlorophyll-chlorophyllin (accessed Jan 21, 2021).
(10) Woodward, R. B.; Ayer, W.; Beaton, J.; Bickelhaupt, F.; Bonnett, R.; Buchschacher, P.; Closs, G.; Dutler, H.; Hannah, J.; Hauck, F. The Total Synthesis of Chlorophyll. J. Am. Chem. Soc. 1960, 82 (14), 3800–3802.
(11) Woodward, R. B. The Total Synthesis of Chlorophyll. Pure Appl. Chem. 1961, 2 (3–4), 383–404.
(12) Woodward, R. B.; Ayer, W. A.; Beaton, J. M.; Bickelhaupt, F.; Bonnett, R.; Buchschacher, P.; Closs, G. L.; Dutler, H.; Hannah, J.; Hauck, F. P. The Total Synthesis of Chlorophyll a. Tetrahedron 1990, 46 (22), 7599–7659.

C arminic acid has played a prominent role as a vibrant red dye across varying facets of production throughout history. This molecule, found naturally in the world, is used as a colorant for foods, cosmetics, textiles, and pharmaceuticals. Despite its prevalence in everyday life, most people are probably unaware that it originates from tiny insects commonly known as cochineal. It is through the mass production and processing of cochineal that the dye can be isolated. Carminic acid plays a significant role in the history of natural dyes and the story follows the well-known plot of discovery, conquest, and commodification; each component is filled with dramatic rises and downfalls.
The history of cochineal and its derivatives echoes that of other natural dyes such as indigo, logwood, kermes, Tyrian purple, and madder. It follows the story of the New World, acting as a symbol of the elaborate profiteering that occurred. Beginning with the discovery and traditional use by indigenous peoples, followed by the colonization and exploitation of Central and South America, it eventually became entangled in the industrialization and modernization of the developing world. While the use of cochineal has transformed from its original humble beginnings, the pure color of carmine remains dependent on the availability of cochineal insects. Cochineal were used in their native lands for centuries before being commoditized and traded globally. Beginning in the colonial period, the importance and influence of this dye was worldwide and continued in prominence until the discovery of synthetic dyes brought a cheaper alternative.
Much of the recorded history of cochineal is pieced together by art historians and archaeologists. The discovery of art containing carminic acid is used to trace the trajectory of cochineal throughout the world. This detective work has been carried out through various techniques to determine the presence of specific dye colorants. These processes
include thermally assisted hydrolysis, methylation, and surface-enhanced Raman scattering spectroscopy.1 For centuries the carminic acid dye was a secret known only to the indigenous populations of Central and South America. The Aztec, Maya, and Inca civilizations selected for the ideal cochineal organisms that produced the highest vibrancy and quantity of dye product, creating strains that were extremely geographically constrained.2 The cochineal, Dactylopius coccus, are scaled insects that reside on the surface of prickly pear cacti, Opuntia indicamil, and through careful monitoring and selection, they have been bred over centuries to maximize the production of natural dye.3
In Europe, before the colonization of the Americas, red dyes were obtained from molecules other than carminic acid. One such alternative was the root of the madder plant, Rubia tinctorum, which was ground up to extract pigment used to color textiles and artists' paints. The red color from madder, however, did not compare to the deep red saturation of carminic acid.2 Additionally, the royalty of Europe and the Middle East had access to kermes, a scaled insect native to the Mediterranean region, which was used as a rich red dye. Due to the extremely high cost of kermes, its availability was limited to the rich upper class and led to the role of red as a color that symbolized status. Similarly to cochineal, kermes are harvested for kerminic acid; in this case, the molecule is found in the bodies of the larval form.3 While these larvae were found in parasitic galls, on the trunks of oaks across parts of Europe, the highestquality kermes came from Aleppo, Syria.2 Natural dyes were used as colorants in various products, but there were limitations to each, such as in availability, cost, vibrancy, or color fastness.
In the late 15th and 16th centuries as explorers from Spain reached the shores of South America, they encountered more than they had anticipated. Sprawling and devel-

oped civilizations met their eyes, and they were surprised to see the use of new natural resources in ways that they had never before witnessed. One such observation was the abundance of the color red, and not the common madder color they were accustomed to, but a bright red that rivaled kermes. Spaniards immediately began shipping dried cochineal back to their country but continued to believe the color stemmed from plant extracts and not insects. Between 1575 and 1600 exports of cochineal to Spain reached 160 tons annually and shortly after that, 250 tons.2 The only commodities that remained at higher demand were gold and silver.
Over the next 300 years, Spain held a near-monopoly on the cochineal trade, causing adverse effects to the kermes production in the Middle East. This period proved fruitful for acts of piracy on the sea, while the plantations of cochineal in the Spanish New World, called nopalerias, were intensely guarded. In 1686, a French naturalist, C. Plumier, was the first European to realize that the legitimate origin of the dye was the bodies of the dried insects. Shortly thereafter, the Dutch naturalist A. van Leeuwenhoek came to the same understanding.
Despite extensive efforts to cultivate the prickly pear cactus along with stolen cochineal in various climates, there were limited achievements. A French botanist, Thiery de Menonville, was sent to thieve cochineal in order to establish productions elsewhere, however, his successes were restricted to San Domingo.3 Through other such endeavors, both clandestine and not, plantations slowly began to spread in the Caribbean. Around 1820 cochineal was introduced to Guatemala and then to the Canary Islands, where it became one of the biggest exports.
The magnitude of the cochineal enigma which perplexed Europeans for centuries was hidden at the astounding scale of 4–6 mm.2 The cochineal insects are of the order
Hemiptera and superfamily Coccoidea and are minute in size. They parasitize prickly pear cacti hosts by extracting their juices for a food source. Cochineal live in congregations on the surfaces of cacti, or nopals, and resemble a woolly layer. (Figure 1.) This “wool” is due to the combination of a waxy powder and silken threads that coat the female and nymph insects to protect them from sun and rain. The females, which outnumber the males 200 to 1, lack wings and account for much of the persistently sessile specimens. The winged males are able to move to nearby cacti, thus increasing genetic diversity.3
Cochineal insects and their prickly pear hosts are native to Mexico, Peru, Bolivia, and Chile. The carminic acid that can be used as a red colorant can be quantified by the process of high-performance liquid chromatography. Dried cochineal insects contain 17–24% carminic acid, which can be processed in a multistep technique. The highest red dye concentration is found in females that are around 100 days old.3 They are so small that around 70,000 individuals are needed to make one pound of 50% carminic acid. Depending on the pH, carminic acid can create a range of colors, from orange to red to violet. Today, Peru is the largest exporter of cochineal, producing about 200 tons of cochineal dye per year on plantations established to grow cacti under controlled conditions.
This ensures the highest quality of the final product.
Carminic acid has the molecular formula of C22H20O13 and a molar mass of 492.38 g/mol.2 The structural core consists of an anthraquinone structure which is bonded to a glucose sugar group as shown in Figure 2 below. The quinone group is composed of a six-carbon ring double bonded on opposite sides to two oxygen, a structure that exists in nature in many different variations. The glucose sugar is the six-membered ring in the “southwest” corner of the molecule. This structure, with the many polar –OH groups is soluble in solutions of water (at a variety of pH levels), and alcohol, but insoluble in most nonpolar solvents.4
most other insects avoid the carminic acidfilled cochineal, the Laetilia caterpillar of the pyralid moth is not only able to consume the scaled insects, but it can also isolate and concentrate the carminic acid to be used as its own defense weapon.6
In order to sequester the molecule for use as a dye, the cochineal must be processed. Extraction of carminic acid from cochineal has changed significantly over the years. Previous European methods required the use of toxic solvents and metal ions. The many steps include “treatment with organic solvents, alkaline extraction, solid-liquid separation (flocculation and filtration), insoluble lake formation, r ecovery of the precipitate (centrifugation), resolubilisation of carminic acid and concentration.”4 The carminic acid, when combined with aluminum and calcium ions, precipitates as carmine. Newer extraction techniques include pressurized liquid extraction (PLE) and supercritical fluid extraction (SFE); both of these require less time, are more environmentally friendly, and allow for greater selectivity.4
Figure
acid. The anthraquinone core is the six-membered ring with a pair of double-bonded oxygens, fused on either side to benzene rings ; this structure is responsible for the red color.
Carminic acid is found in the blood and muscles of the scaled insect Dactylopius coccus and while the anthropocentric utility of cochineal lies in the natural coloration, the insects have their own use for the compound. Due to its chemical structure containing a quinone, carminic acid holds biological significance for the insects. Other insects use quinones, such as benzoquinones and naphthoquinones, as defense mechanisms. For example, Millipedes and opilionids discharge these chemicals to deter feeding predators. The cochineal defend themselves against predacious ants by using the carminic acid as a chemical weapon.5 Interestingly, while
Carminic acid in all its forms were used extensively in the coloration of products from different departments of consumerism since the time of its commodification. Natural fibers such as wool, cotton, silk, leather, and

fur were dyed with these crushed insects to produce many varying shades of red that were fashionably desirable (Figure 3). Cosmetics products were also dyed with carminic acid when a red was desired. Additionally, inks and paints utilized the addition of this molecule for pigmentation.
Despite carminic acid's desirable effects on fibers and products, the supply of cochineal was not without limit and organic chemists began work to synthesize replacement molecules for production. In 1856, William H. Perkin discovered a synthetic dye which created a mauve color and production of it began at Greenford in England.3,7 Within the next one hundred years, many synthetic dyes were discovered and produced, spanning the spectrum of nearly all desired colors. These dyes were cheaper to produce and therefore natural dyes of botanical and animal origin lost favor.
Today there are phthalocyanines to dye blues and greens, arylides for yellows, and quinacridones that range from orange to violet. Mass production of these synthetic molecules allowed for them to take over the industries of textile dyes, cosmetic colorings, and food additives. Synthetic red aniline dyes, including alizarin crimson, fill the niche of red coloration for various industries. These dyes were formed from petroleum byproducts and proliferated as a cheaper option than natural dyes. The cost of cochineal is around $50–$80 per kg, roughly double the cost of FD&C Red No. 40, which is only $24–$42 per kg.7 While the low production cost and market value made synthetic dyes more popular for some time, a downfall was in their future. From the 1960s to the 1980s environmental activists demanded that synthetic dyes be banned due to carcinogenic factors and toxicological effects, specifically Red Dye No. 2 and Red Dye No. 40. This sentiment spread across the populace and natural dyes resurfaced as a desirable source of color in everyday products.
The current consumer sentiment often desires natural foods and limited synthetic components in manufactured products. In the United States the Food and Drug Administration has restricted the use of synthetic colorants as food additives. The food additive identification code for carmine is E-120, and it can also be identified as Natural Red #4, Carmine, Cochineal Extract, and Carminic acid.8 Carminic acid was renewed as being an alternative to the synthetic dyes due to the health concerns in respect to synthetic food additives. It is one of the most stable of all food, drug, and cosmetic colors and there are no current regulations to limit the amount added to products.
While there seems to be a general consensus that natural additives are preferable to synthetic ones, there continue to be issues with cochineal extract. Recently, there has been outcry towards the use of insect extracts as a colorant, with such headlines as “Don't want to eat bugs? Then don't eat any of these products” and “Bugging Out: Vegetarians Upset With Starbucks' Use of Beetle Extract.” Despite the focus on specific companies and their use of the “crushed bugs,” cochineal extract is found in countless food products in various forms. It can be found in alcoholic drinks, meats, poultry, sausages, desserts, icings, jams, juices, dairy products, and such cosmetics as lipsticks and rouges and in pharmaceuticals as coloration for pills and ointments.3 While carminic acid is considered safer than synthetic red dyes, it can be an allergen for a small percentage of people. It can cause rashes, or in rare cases it can induce anaphylactic shock, which has made it the subject of a fight to add regulations into the labeling of all forms in food and cosmetics.
The complex history of cochineal's carminic acid properties is tied tightly with geopolitical agendas, market incentives, and newly developed environmental concerns. It tells a fascinating tale of the rise and
fall of natural dyes and how popular perception can heavily influence a product. The story about this historically important molecule is ongoing.
Cochineal (Dactylopius Coccus). New Extraction Methods. Food Chem. 2012, 132 (4), 1855–1860.
(5) Eisner, T.; Nowicki, S.; Goetz, M.; Meinwald, J. Red Cochineal Dye (Carminic Acid): Its Role in Nature. Science 1980, 208 (4447), 1039–1042.
(6) Carminic Acid as a Chemical Judas. Science News. 1980, p 357.
Figure 1 was created by Katja Schulz and downloaded from https://www.flickr.com/ photos/treegrow/24239079102/
Figure 2 was created by the author and the editors.
Figure 3 was created by Madelinetosh and downloaded from https://www.flickr.com/ photos/madelinetosh/2202448899/
Figures 1 and 3 are used under Creative Commons license: https://creativecommons.org/licenses/by-nc -nd/2.0/
(7) Venil, C. K.; Zakaria, Z. A.; Ahmad, W. A. Bacterial Pigments and Their Applications. Process Biochem. 2013, 48 (7), 1065–1079.
(8) Greig, J. WHO Food Additives Series 46: Cochineal Extract, Carmine, and Carminic Acid. Food Stand. Agency 2012.
(1) Phipps, E. Cochineal Red: The Art History of a Color; Metropolitan Museum of Art, 2010; Vol. 63.
(2) Dapson, R. The History, Chemistry and Modes of Action of Carmine and Related Dyes. Biotech. Histochem. 2007, 82 (4–5), 173–187.
(3) Baranyovits, F. Cochineal Carmine: An Ancient Dye with a Modern Role. Endeavour 1978, 2 (2), 85–92.
(4) Borges, M.; Tejera, R.; Díaz, L.; Esparza, P.; Ibáñez, E. Natural Dyes Extraction from

I grew up in a household of colors. By this, I certainly do mean vibrant greens of anticipation, blossoming yellows of happiness, complex white castles of cold, striking blue adventures, and overwhelming, unbelievable red love. However, there were colors dividing my home as well. By this, I mean the blue and green eyes of my parents, my brother, and I; and the deep brown eyes of my sister. I mean the chestnut brown hair of my mother, which my brother and I share, and the rich, one-pixel-from-black hair of my sister. (Here my father is exempt due to male-pattern baldness). I mean the fair, tan tone of my parents, my brother, and I; and the deep brown of my sister a color that not even my darkest freckles can match. I find these differences beautiful, while my sister finds them to be a barrier that she cannot cross. She feels a separation from the rest of my family; never will she hear “you look so much like your mother.” Worse, she has already been profiled in a way that my blood-relatives and I have never experienced. In her support groups, she has had classes on how to behave if pulled over by a police officer, so as not to be arrested or shot.
Why is she treated differently from the rest of my family? What differences are there between us? There is but one difference that has driven discrimination and exclusion for the better part of the Modern Era and all of the Cenozoic: skin color. Why does our society discriminate against people based on one characteristic and what is the molecular cause of this characteristic? What are the factual, scientific reasoning for the human reactions to this difference throughout history? Ultimately, differences in skin tones can be attributed to the chemical melanin. How is it that different concentrations of different types of melanin have contributed so strongly to social interactions between different peoples?
Melanin is the chemical in our bodies that is responsible for pigmentation. Pigmentation is the natural coloring of animal and plant tissues. Its presence may be one of the first things that a person notices upon meeting a new acquaintance. The amount of melanin in the body, as well as the type, are responsible for variations in skin color. There are two main types of melanin: eumelanin and pheomelanin. Eumelanin is what most people think of when they think of the chemical melanin, as it is responsible for a rich, dark skin tone; pheomelanin is mainly expressed in red hair and freckles.
While the arrival of most Africans in ancient cultures was due to the importation of enslaved peoples, the original basis for slavery was, surprisingly, not skin color. In ancient Rome, people of African descent could hold the same job, spouse, and amount of land as that of anyone. This equality is also represented in ancient Greek writings, with Homer writing of Eurybates (Odysseus’s herald) in the Odyssey. 1 In fact, ancient Greeks regarded darker -skinned people as god-like due to their resistance to sun damage. What, if not godliness, could give these people such magnificent powers?
While melanin’s precise role in photoprotection is still controversial, it is widely agreed that melanin is the chemical responsible for protection of the skin from highenergy ultraviolet (UV) rays. Shown in Figure 1 are the steps in the formation of both eumelanin and pheomelanin. I will focus here on the formation of eumelanin as that is the chemical attributed to dark skin tones.
Synthesis of both types of melanin start with the same two reactions. First is the conversion of tyrosine (an amino acid), to DOPA through the action of the enzyme tyrosine hydroxylase, as the catalyst of the reaction.2 (This means that tyrosine hydroxylase, an enzyme gets the reaction started.)
After Tyrosine is converted to DOPA, the DOPA is oxidized (meaning that it loses electrons) and becomes DOPA quinone (Figure 1). Here, one of two things can happen, which will determine which kind of melanin is formed. DOPA quinone can react with the amino acid cysteine, taking this molecule towards pheomelanin (the melanin prevalent in redheads and freckles). Alternatively, the pathway may lead towards eumelanin, the type responsible for dark pigmentation (Figure 1).3
So why does eumelanin prevent skin damage? Simply put, free radicals and oxidation can damage the body, and eumelanin is an antioxidant, meaning that it inhibits the oxidation of other molecules.4 Oxidation is the process of one molecule or atom giving one or more electrons to another molecule or atom. A common example of this is the rusting of iron. In the process, free radicals may form; this process happens frequently in our body as well. Free radicals are usually unstable and often overactive. Due to their odd electron (or “free” as in the name), the free
radical will react with whatever it can in order to get rid of this electrical imbalance. Free radicals will react with critical molecules such as proteins and even more notably DNA. When a cell’s DNA is damaged by a free radical the cell must to repair itself, a process that leads to a chance of mutation. Cell mutations then lead to various cancers. When high-energy (ultraviolet) solar radiation hits the skin, it creates free radicals in skin cells. Melanin reduces the cellular damage that can lead to skin cancer by safely disarming the free radicals before they can interact with the DNA.
Why, then, is melanin an antioxidant? A good indicator of an antioxidant is a benzene ring attached to an –OH, called a phenol, and the related phenolic acid structure (Figure 2, next page). Phenol can be identified as a hexagon with alternating single and double bonds (i.e., a benzene) with at least one –OH connected directly to the ring. An example of a phenol is the amino acid tyrosine (Figure 2), the starting material for melanin synthesis (Figure 1).
The synthesis of melanin turns tyrosine into a molecule that resembles phenolic acid, an extremely potent antioxidant. Shown below in Figure 3, phenolic acid looks very structurally similar to the monomer of eumelanin. Phenolic acid is a well-known, powerful antioxidant. The low toxicity of both phenolic acid and eumelanin makes them both very effective in this role. Highlighted on eumelanin is the structural motif that it shares with phenolic acid.
Beyond simply preventing oxidation, eumelanin can also act as a radical scavenger: it can diffuse already-oxidized molecules that are high-energy and therefore poised to do cellular damage. This is where eumelanin is a better protector of the skin than is
pheomelanin. The process of sunlight hitting the skin and entering the system is one that regularly leads to the formation of free radicals, due to the interaction of rays of light and the molecules in skin. Eumelanin is able to scavenge these free radicals, because eumelanin is insoluble, resistant, and very stable.5 Quantitatively, eumelanin has a melting point of 3500 °C: This extremely high number means that it requires a whole lot of energy to separate two molecules of eumelanin in order to make it melt.6 Thus, once eumelanin is in place in the skin, it stays in place. This is the reason that I am much more likely to become sunburnt than my sister who has darker skin and thus more eumelanin.
It is thought that the bulk of melanin’s skin protection is due to melanocytes (which hold on to melanin) forming a physical barrier between the sun’s rays and our cells. These melanocytes can either form a supranuclear cap, which is what eumelanin does, blocking most of the sun, or form “melanin dust,” made by pheomelanin, which leave it much more vulnerable.5
If the presence of melanin means that a person is less susceptible to damage from the sun, why has our culture regarded it as a flaw for so long? The answer to this is wholly unsatisfying. It begins with human advancement: Due to improving technology, countries were able to create a true global market. This meant that demand for goods skyrocketed, and with an increased demand must come an increased workforce. The drawback, however, was that workers require pay. In order to avoid this caveat, European settlers turned to slavery as a form of virtually free labor.
or CO2H, depending on which indole-5,6-quinone precursor was used (refer to Figure 1).
A Dutch ship arrived in Jamestown Virginia in 1619, bringing with it the first 20 African slaves to America.7 European settlers
needed a source of cheap, accessible labor that wasn’t satisfied by indentured servitude; indentured servants work off a debt, and once the debt is repaid the servants are set free. Historically, these indentured servants were a source of cheap labor, until slavery was introduced to the country. In stark contrast, chattel slavery was economically beneficial to white settlers because an enslaved person must work for their entire life and their children become property of the slaveowner.
Slavery grew rapidly when indentured servants began refusing to work on sugar fields in Barbados due to harsh conditions; the colonies needed workers that couldn’t refuse. Interestingly, while most American colonies used African slaves, other countries such as the Netherlands turned to Asia. Also, while slavery was in practice since ancient times, it wasn’t until the Middle Ages that slaves were imported from other countries and distinguished by skin color.
When entering into a practice of dehumanization, one must find a way to justify the practice. The easiest way to do this is to pick a noticeable difference between the enslaved and oneself. This had previously been done with prisoners of war, using “they fought against us” as a justification for maltreatment. The choice to use people with a different skin color than that of the colonists did the same thing, making it easier to separate them from their humanity. European colonists learned to view people with more eumelanin than them as lesser, almost at the level of an animal.
The decision to use the presence of eumelanin in skin was one without reason. It is human nature to view different as bad or lesser, something that society struggles with even today. A cop looks at my sister as more likely to commit a crime than her white siblings. Seeing darker skin tone as “bad” is simply a continuation of our need to justify enslaving human beings. How else can we justify the act that allowed our country to grow into the
global power that it is now? This prejudice has lasted for hundreds of years with absolutely no scientific backing. Humans as a whole are not eager to change; places that relied on slave labor have clung to the belief that slaves are barely human. This is the reason that racism can still be seen on the streets of any city to this day.
The discovery of the melanocyte in the 19th Century, coinciding with the abolition movement, allowed us to gain perspective on the cause of differing skin tones. The first record of a scientist discussing skin pigmentation came from Sangiovanni, who was researching a squid. Some years later, the existence of pigment cells was reported.5 By the mid 19th century scientists agreed on the presence of melanocytes, proving that skin color originates from a minor chemical difference in a human’s makeup.
Once it was realized that humans are almost entirely the same, with only eumelanin and pheomelanin distinguishing skin color, it was harder to argue that it is ethically acceptable to hold people as property. Slavery was finally abolished in the United States in 1865, but racism has lingered. This bias that our ancestors created remains with us today. This bias makes my sister feel unequal, and gives her the idea that she won’t have the same opportunities as me. It is incredible that one molecule was responsible for one of the greatest atrocities of mankind. This molecule is harmless; in fact, it is helpful. It should have been regarded as a strength, yet human interpretation has made it a villain.
All figures were created by the author and the editors.
(1) Black people in history ~ in the Caribbean and the UK http://web.archive.org/ web/20070209204524/http:// www.itzcaribbean.com/ black_people_history.php (accessed Jan 8, 2021).
(2) Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tymi ska, A. Skin Melanocytes: Biology and Development. Adv. Dermatol. Allergol. Dermatol. Alergol. 2013, 30 (1), 30.
(3) Ito, S.; Wakamatsu, K. Chemistry of Mixed Melanogenesis—Pivotal Roles of Dopaquinone. Photochem. Photobiol. 2008, 84 (3), 582 –592.
(4) ScienceDaily. Eumelanin’s secrets: Discovery of melanin structure may lead to better sun protection https:// www.sciencedaily.com/ releases/2014/05/140522115754.htm (accessed Jan 8, 2021).
(5) Solano, F. Melanins: Skin Pigments and Much More Types, Structural Models, Biological Functions, and Formation Routes. New J. Sci. 2014, 2014.
(6) Brenner, M.; Hearing, V. J. The Protective Role of Melanin against UV Damage in Human Skin. Photochem. Photobiol. 2008, 84 (3), 539–549.
(7) Editors, H. com. Slavery in America https://www.history.com/topics/blackhistory/slavery (accessed Jan 8, 2021).
(8) Today in African American History http:// todayinafricanamericanhistory.com/
(9) Editors, H. com. Black History in the United States: A Timeline https:// www.history.com/topics/black-history/black -history-milestones

No wedding is quite as spectacular as the traditional Hindu Indian wedding. These remarkable ceremonies can last up to a week and consist of several different parties. One of the most important celebrations is the Mehndi party, where the bride gets mehndi, or henna, applied onto her hands and feet. All of the women on her guest list attend, often getting a mehndi design themselves. They often will dance and provide entertainment for the bride who may sit for up to 12 hours while multiple mehndi artists apply dye to her skin. This party typically takes place a few days before the actual nuptials in order to allow the henna to stain the skin as much as possible. It is believed that the darker the mehndi on the bride, the better the marriage will be. Henna is also believed to guard against ill fortune, granting the wearer good luck. Henna designs are typically floral or geometric, and are incredibly symbolic. A beautiful red flower isn’t simply a flower; it may represent fertility or hope. Each design is carefully thought out as it is put onto the bride’s skin—and typically her husband’s name is hidden in the design as well.1
Henna dye is obtained from the leaves of Lawsonia inermis, also known as the henna tree, the mignonette tree, and the Egyptian privet. The tree is native to North Africa and Asia and can only grow in very hot and dry climates. The trees grow 12–15 feet high, in heat up to 120 ºF with very dry soil.2 Henna is grown commercially as a dye crop throughout India. L. inermis must be cultivated outside, often propagated by stem-cuttings, and never gets larger than shrub-sized. The hotter the weather, the more pigment present in the leaves. Interestingly, henna grown in hotter areas is typically used for body art; henna grown in the cooler parts of India is often used to make hair dye, and the flowers can be used in perfume making.3,4
Henna has its roots deep in the ancient
world with some of the earliest uses documented 9,000 years ago. In ancient Egypt, the nails of mummies were often painted with henna before they were buried. Cleopatra herself used henna to decorate her body and face. Henna has been widely used in Northern Africa, the Arabian Peninsula (where the name originated from), India, and other parts of Southeastern Asia for thousands of years. Henna was even used as makeup within the Roman Empire.5
In Africa, Moroccans paint the front doors of their houses with henna to bring good fortune to the home, and to ward off evil spirits. Similar to the Indian tradition of mehndi, Moroccans also use henna to decorate the bodies of brides. In North Africa, henna body art typically features geometric shapes and sharp lines.6 One of the reasons henna is so widespread across cultures is because of how accessible it is to all people. Even those living in poverty could adorn themselves with intricate designs.
Besides cosmetic use, henna has also been known to treat skin conditions, aid in health, as well as dye and protect fabric. Henna has natural cooling properties and historically, people who lived in the desert would soak their hands and feet for additional protection. Henna paste can be used as a great sunblock; some farmers apply it to the noses of their animals in order to keep them from getting sunburnt. Henna is also used to treat headaches, stomach pains, burns, open wounds, reduce fever, treat athlete's foot, and prevent hair loss.7 It truly is a wonder drug!
Many of henna’s powers come from a dye, called lawsone, present in its leaves. Lawsone readily binds with the proteins in our skin, hair, and nails resulting in a dark stain.8 In addition to dyeing skin, hair, and nails, henna is widely used in dyeing fabric
such as silk, wool and leather. It can also be used to “moth-proof” fabric since the henna is poisonous or inedible to most insects.9
Body art henna is simple in construction. It consists of powdered henna leaves mixed with sugar, a mildly acidic liquid, and a solvent. The composition of these additional ingredients affect the shade and the shelf-life of the henna.
Henna leaves can be ground into a powder using a mortar and pestle. More commonly, the leaves are extracted industrially using machinery. The powder is milled as finely as possible and sifted carefully.
“Hair-grade” henna, used for dyeing hair, is not sifted carefully and is therefore of lower quality than body art henna. It also often contains other herbs and chemicals in order to condition the hair.
The most important component of the henna paste is the henna powder. Every batch may be different, depending on where the henna came from and what the weather and temperature was like in that specific region. Henna leaves typically contain 1–3% lawsone dye by mass, and henna from hotter and drier climates is preferred by artists, as it has higher concentrations of dye. In addition to the dye, other chemicals present in the leaves can affect the composition of the paste. Leaves with an abundance of polysaccharides long chains of sugars will give the paste a texture that is stringy, similar to paper mâché. Henna made from leaves with a high concentration of calcium oxalate will yield a gritty paste, which can clog bottles and overall be more difficult to work with. Good body art henna should always be made fresh, and henna powder stored in air-tight packaging will keep almost indefinitely.10
The next component of henna paste is a liquid to tune the acidity. Henna paste should be mildly acidic. The leaves of the henna plant contain some acid, but an addi-
tional acidic liquid additive improves the paste. It is common for henna artists to mix henna powder with lemon juice or even coffee or tea. It is important to note that some people’s skin can be sensitive to citrus juices, and caffeine from coffee or tea can penetrate the skin. For these reasons, some artists simply use distilled water, but as this lacks acidity, the shelf life of the henna paste is greatly reduced. In recent times, some artists have used apple juice, which is acidic but does not contain irritating citric acid. However, this is not a perfect solution, and artists must always keep the complete chemical composition of their mixture in mind.10
Sugar is the next component of henna paste: sugar gives flexibility and improves the texture. If apple juice is used, very little extra sugar is needed for the mixture. For other recipes, it is important to add sugar. There are two types of sugars that are important in the formation of henna paste. The first are the simple sugars: monosaccharides and disaccharides, including fructose, glucose, and sucrose, all of which can be added to help the paste stick to the skin and keep the paste from cracking or crumbling when applied. The other added sugars are polysaccharides, which are long chains of sugars, usually in the form of pectin or xanthan gum. This gives the paste a creamy texture to the likeness of egg whites.10
The final component of henna paste is the organic solvent. Some pre-packaged boxed henna dyes contain solvents such as turpentine, which is caustic. Most professional henna artists use safe, monoterpene alcohols (such as those found in tea tree oil) which improves the potency of the paste and, in turn, yields a darker stain. Powdered henna leaves, liquid, sugar, and solvent are all important in the formation of a good henna paste. Without one component, the application and execution can completely fall apart. Like baking, all of the ingredients in the henna mixture must be measured and added in
at just the right time or the release of the dye will not go as planned.10
When liquid is added to the henna powder, the dye molecule lawsone is released. Lawsone (Figure 1) is also known as hennotannic acid and has the chemical formula C10H6O3. Its official chemical name is 2hydroxy-1,4-naphthoquinone. Lawsone belongs to the naphthoquinone family which also includes juglone, the compound produced by black walnut trees, and is used in the dye C.I. Natural Brown 7.12,13 Note the high structural similarity between lawsone and juglone: they differ only by the location of one hydroxyl (–OH) group (Figure 1).
Within the henna tree, the lawsone molecule is not typically present as the structure shown in Figure 1. Instead, a precursor to this molecule 1,2,4-trihydroxynaphthalene (THN) is found attached to a sugar molecule called a glycoside (Figure 2). Two processes can release the dye molecule from the sugar: enzymatic hydrolysis and acidic hydrolysis. Enzymatic hydrolysis occurs when the enzymes in the plant break down the sugars. This is the reason you would have stained fingers if you were to rub a henna leaf between your hands. Acidic hydrolysis is much slower than enzymatic hydrolysis and occurs when the powdered leaves are mixed with an acidic liquid. When this happens, the THN is slowly released (Figure 2).10,14 When the powdered henna leaves mix with a liquid in the presence of oxygen, the molecule is oxidized: it loses two hydrogen atoms and becomes lawsone. (Figure 2) Lawsone is pale yellow in color at this stage, and the molecule is now poised to react with the skin.8,10
Lawsone readily reacts with and binds to keratin, the protein found in our hair, skin, and nails. This is why henna is used to “tattoo” skin, and dye hair and fabric. Henna is often advertised as a non-permanent way to tattoo the skin, but this isn’t entirely true. Lawsone binds to keratin permanently through a process known as the Michael addition reaction (Figure 3).15 What’s happening here is that
one of the carbon-carbon double-bonds on the lawsone is reacting with a sulfur atom (from the amino acid cysteine) found in keratin, resulting in a permanent stain. This reaction causes the lawsone molecule to change color to the well-known deep reds and browns we see in henna tattoos (Figure 3).8,12 Luckily, the epidermis of our skin is not permanent, and it takes about two weeks for the dyed cells to be exfoliated. However, if henna gets on fingernails, or if it’s used to dye hair, it will be there permanently until the nails or hair are grown out and cut.
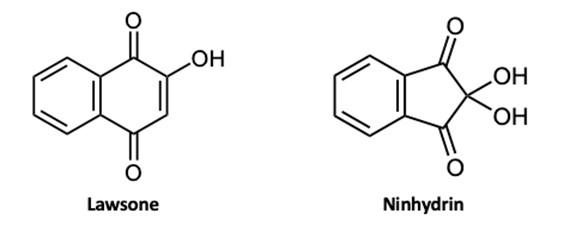
Mehndi artists and all-natural at-home hair dyers aren’t the only ones trying to get their hands on henna. Because of the lawsone molecule’s ability to react with keratin, detectives and forensic scientists are now trying to see what they can do with this compound. Ninhydrin, the primary molecule used today for detecting and visualizing fingerprints, has structural similarity to lawsone (Figure 4). New research has shown that lawsone works as well as if not better than ninhydrin in fingerprint detection.16 This means that in the future, henna could possibly help take down criminals: lawsone can quite literally catch someone red handed!
Despite how cool henna crime-solving sounds, this raises an important point: in the US today, henna is used without much thought towards its cultural significance. It's easy to find henna stands at fair grounds and heavily trafficked tourist spots. At these stands, the history behind henna is often disregarded and tourists can simply pay for fun, nontraditional designs. Today you can find athome henna kits in stores like Target and Michaels; for much of the world henna is simply ornamental.
Additionally, the popularity of henna raises safety concerns: people looking to get henna tattoos need to keep an eye out for black henna. Black henna is not FDAapproved, and has given the whole industry a bad reputation. Black henna is henna that has been treated with the adulterant pphenylenediamine (PPD), which can cause severe allergic reactions resulting in blisters, lesions, and permanent scarring. Some lab studies have even found that this compound can cause cancer in lab mice.11 Although it is dangerous, black henna is thankfully easy to spot because of its dark color.
Some henna traditions continue to stand the test of time. Similar to Indian Mehndi parties, those of Middle Eastern Jewish descent often use henna in pre-wedding celebrations. During the appropriatelynamed Henna Party, the dye is used to stain the palms of the bride and groom. This repre-
sents different things from family to family, but the most common meaning is that the henna represents the unity between bride and groom.17,18 In Morocco, henna is as prevalent as ever, with both Moroccan men and women decorating themselves with henna for religious events and festivals.19
Whether you’re opting to dye your hair the natural way, getting a fun design at a fairground, participating in a friend’s wedding, or trying to solve a crime - lawsone is the molecule responsible! Lawsone has been helping people dye their skin with henna for thousands of years, and it probably isn’t going anywhere soon (especially if Earth’s temperatures continue to rise). The chemistry of henna is a great reminder of the intersection between the most culturally and spiritually significant practices from around the world and science. In this case, we have just a single molecule to thank for helping it leave an indelible mark on the world.
The Geography of Henna http:// www.hennapage.com/henna/encyclopedia/ geography/indiahenna.html (accessed Jan 22, 2021).
(4) Foster, M. The Henna Page - Henna: Germinating Seeds http://www.hennapage.com/ henna/encyclopedia/growing/seeds.html (accessed Mar 1, 2021).
(5) Moazzam, I. A Brief History of Henna. The Express Tribune. August 4, 2014.
(6) Sienna, N. The History of Henna in West Africa. Ayiba Magazine. April 15, 2016.
(7) Pradhan, R.; Dandawate, P.; Vyas, A.; Padhye, S.; Biersack, B.; Schobert, R.; Ahmad, A.; H Sarkar, F. From Body Art to Anticancer Activities: Perspectives on Medicinal Properties of Henna. Curr. Drug Targets 2012, 13 (14), 1777–1798.
(8) Bhuiyan, M. R.; Islam, A.; Ali, A.; Islam, M. Color and Chemical Constitution of Natural Dye Henna (Lawsonia Inermis L) and Its Application in the Coloration of Textiles. J. Clean. Prod. 2017, 167, 14–22.
All figures were created by the author and the editors.
(1) Callaway, N. Bridal Henna Party https:// www.thespruce.com/bridal-henna-party3489631 (accessed Jan 22, 2021).
(2) Henna | plant https:// www.britannica.com/plant/henna (accessed Jan 22, 2021).
(3) Cartwright-Jones, C. The Henna Page -
(9) Nazari, A. Efficient Mothproofing of Wool through Natural Dyeing with Walnut Hull and Henna against Dermestes Maculatus. J. Text. Inst. 2017, 108 (5), 755–765.
(10) Schafer, J. The Chemistry of Henna for Body Art. Henna Muse, 2017.
(11) Nutrition, C. for F. S. and A. Hair Dyes. FDA 2020.
(12) Jordao, A. K.; Vargas, M. D.; Pinto, A. C.; da Silva, F. de C.; Ferreira, V. F. Lawsone in Organic Synthesis. RSC Adv. 2015, 5 (83), 67909–67943.
(13) Feeley, C. Black Walnut: The Killer Tree https://www.extension.iastate.edu/ news/2005/jul/070701.htm (accessed Jan 22,
(14) Chaplin, M. The Use of Enzymes in Starch Hydrolysis. Last Accessed Httpwww Isbu Ac Ukbiologvenzlechglucose Htmlon 2004, 5 (15), 2007.
(15) Little, R. D.; Masjedizadeh, M. R.; Wallquist, O.; Mcloughlin, J. I. The Intramolecular Michael Reaction. Org. React. 2004, 47, 315–552.
(16) Jelly, R.; Lewis, S. W.; Lennard, C.; Lim, K. F.; Almog, J. Lawsone: A Novel Reagent for the Detection of Latent Fingermarks on Paper Surfaces. Chem. Commun. 2008, No. 30, 3513–3515.
(17) Cooper-Benisty, N. Embracing A Jewish Henna Wedding Tradition https:// www.myjewishlearning.com/2015/10/07/ embracing-a-jewish-henna-weddingtradition/ (accessed Jan 22, 2021).
(18) Refael, T. The Charm of Jewish Henna Ceremonies https://jewishjournal.com/ culture/community/277707/the-charm-ofjewish-henna-ceremonies/ (accessed Jan 22, 2021).
(19) Williams, S. A Guide to Henna Traditions in Morocco https://theculturetrip.com/africa/ morocco/articles/a-guide-to-henna-traditions -in-morocco/ (accessed Jan 22, 2021).
A photograph or a sketch uses a twodimensional medium to represent a threedimensional reality. Some information is lost or omitted, but key features are retained—and more importantly, now the format can be saved in a book or displayed on a computer screen! This limitation is much the same in organic chemistry, because most organic structures are three-dimensional arrangements of carbons, hydrogens, and heteroatoms. Yet, drawings of chemical structures, such as in Figures A1–A9, can only show the molecule spread flat. Most of the time this representation is satisfactory, and that has been true for every chapter up to this point. For these last three chapters, though, three-dimensionality plays a particularly important role.
Let’s return to the carbon atom, and its key ability to form four bonds. When those bonds are to four distinct atoms (i.e., no double-bonds), the four bonds spread out as though they were pointing to the four corners of a tetrahedron, which gives rise to the threedimensionality of the molecule. A chemical bond, such as in Figures A1–A9, is shown as a straight line. This implies that the bond is positioned on the plane of the paper or screen — either that, or the three-dimensionality is not germane to the discussion at hand.
When a bond is not actually on the plane of the page and its three-dimensionality is important, we use another type of representation: the wedge and hash bonds. Figure A10 is designed to give you an idea for how this works. In Figure A10, the central, tetrahedral carbon atom is bonded to four atoms : w, x, y, and z. The same molecule is shown three times, in different orientations. (Imagine the central carbon atom as a sphere that you hold with your fingers as you rotate the bonds; you could use something like a marshmallow and four pretzel sticks if you want to build a model!) For each orientation shown, the bonds to “y” and “z”
are in the same plane as the central carbon atom (and everything else on this page). The “w,” however, is coming out towards you, which is what the wedge-shaped bond indicates. In contrast, the “x” is going back away from you—like it’s behind the page—which is what the hash-shaped bond indicates. The molecules in the top row and the bottom row are identical; I use shading in the top row to help visualize the wedge-coming-forward and hashgoing-back, whereas the bottom row shows the style you will see in future images.
The molecular representations you have seen to this point have not included dashes or wedges. Even though almost all molecules have an element of three-dimensionality, it is often better to focus on things like polarity. The next two chapters, Tabby Spyrison’s, on narcan, and James Kitchens’s, on tetrodotoxin, feature big, complex structures with many fused rings that really need hashes and wedges to understand and appreciate the connectivity of the overall structure. And in the case of Brian Wuertz’s chapter about salbutamol, the three dimensionality plays a significant role in the chemistry and efficacy of the molecule. I’ll leave it to the authors to explain why and how!
that it is going behind the diagram and away from you. The top row and the bottom row are identical, but the top row uses shading to further emphasize the three dimensions.

Opioid addiction is regularly portrayed on TV and in movies. Characters may display immediate symptoms a fatigued form is shown collapsing against a wall as their body descends into a debilitating high or be suffering long-term, continued isolation from everything they know and love. It is as if they have fled from their family and friends, and no longer have the ability to carry on with their lives. This is a scary concept, and while many of us imagine it only as the material for an uncontrollable dramatic plot, for some, this is reality.
John Hopkins University defines the symptoms of opioid abuse as everything from “Uncontrollable cravings and drowsiness, to weight loss and frequent flu-like symptoms...” and the list goes on.1 This crisis is just as scary as the television portrays. It permeates every single aspect of an addicted person’s life, preventing day-to-day operations, getting in the way of personal relationships, and simultaneously alienating its victims from the world. Drug abuse is like a black hole, and once an unlucky individual becomes trapped under its influence, it is almost impossible to resist. Sometimes, these individuals will hit rock bottom, and when that occurs, we hear heart-wrenching stories of overdose. No matter how hard we try to protect everyone, there are still individuals who slip through the cracks. This is an ongoing crisis, and this is why a drug such as naloxone, an opiate antagonist, has become so vital these past few years.
The brain is the operation center behind bodily function and control. While the science behind this is very complex, one of the most important aspects of this system is the neuroreceptors. A neuroreceptor is a receptor protein, protruding from a brain cell, that can be activated by specific molecules. Each neuroreceptor controls a different path-
way, and each produces a different effect once activated. In the case of opioid neuroreceptors, an opioid drug such as morphine can bind to these receptors and prevent the regulation or induce the release of molecules such as dopamine, a hormone that can produce the desired “high” at elevated levels. Particularly when taken in lofty amounts, the opioid can negatively impact brain function and can even shut down vital functions.2 This is where opiate antagonists (like naloxone) come it.
“Opiate antagonist” is a classification for chemicals that bind to opioid receptors in the brain, replacing actual opiates. Instead of triggering the regular high the user expects, opiate antagonists instead block the receptors from being activated by any divergent opioid molecules (Figure 1A). The user is unable to feel the usual euphoria because the receptors in the brain that are responsible are blocked, and immune to interaction with the addictive drugs.
These molecules are usually used in emergencies, for overdoses, because an opioid antagonist can reverse the effects of drug use, ejecting the opioid molecules from the brain receptors, and taking their place (Figure 1B). This species of molecules has been used successfully for almost 40 years, and its success has caused an enormous drop in overdose cases.3 In addition, prophylactic use of these molecules is an effective method of rehabilitation for some, by reconditioning an individual to believe that their body no longer responds to the drug. If the receptors are blocked, then no matter how large of a dose of opioids a person may take there will be no stimulation, allowing for effective recovery though this individual will still have to go through the process of withdrawal.
Naloxone is one such opioid antagonist. It is widely available and frequently used in medical emergencies. It is also knowns by brand names, Narcan® or Evzio.
A) Opioids and opioid antagonists both bind to opioid receptors, but only opioids activate the receptor.
B) B) Opioid antagonists bind more strongly to opioid receptors, both kicking out opioids and blocking further binding. Opioid Antagonist Transmitter Interaction
(Ambrosia, Ambrosia Treatment Center)
Naloxone was first recognized as an opiate antagonist in the early 1960s, after it was patented by a Japanese company, Sankyo, in Britain and by Mozes J. Lewenstein and Jack Fishman in the United States. Despite being structurally related to morphine, scientists quickly discovered that Naloxone had no painkilling properties, but worked as an opioid antagonist instead. There was little argument or concern as to its release, as its presence was severely needed to control the ongoing opioid crisis.3 By 1971, the FDA had approved the use of naloxone in the treatment of opioid overdose by intravenous or intramuscular injection. Since then, our need has only increased, and development has grown rapidly. Many scientists have been researching other forms of administration, preferably by a method acces-
sible to the general population; for example, there is now a nasal spray available for use in a crisis. Still, there is much work required as the nasal spray is still not as effective as the usual injection, because the dosage cannot be controlled as accurately. However, if administered properly, Naloxone has been shown to work wonders on patients. For example, a distribution program in Massachusetts showed an 11% decrease in opioid-related deaths. Ever since its discovery, Naloxone has been in the front lines of our society’s war against drugs. Often, it can be obtained without a prescription, so anyone who may need the medication has access to it readily. We can only hope it will make a difference in the days to come.
Naloxone has very few adverse side effects, though patients may often feel the symptoms of withdrawal while on naloxone, causing discomfort. Naloxone can only block receptor function, so the body still has to get used to the absence of hormones. In cases of overdose, naloxone will primarily assist respiratory function. While the individual still requires medical attention after administration, it will ensure the individual has enough bodily control to receive further care. This is perfect for extreme cases, as the molecule can enter without ravaging the body of the individual, and eventually leave without a trace.
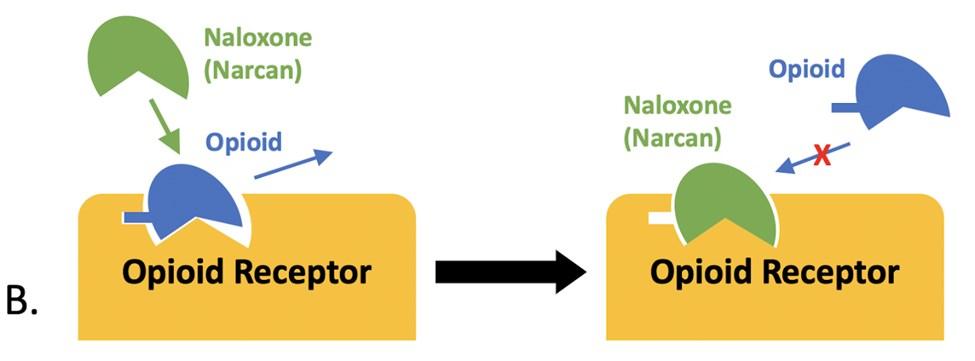
The chemical structure of naloxone (Figure 2) is responsible for its beneficial qualities. This molecule has a chemical formula of C19H21NO4. Its chemical structure features many organic functional groups that contribute to the mechanism of the molecule.
While all this information is very relevant to the identity of the molecule, the most important factor of this structure consists of the polycyclic ring formation at the heart of the molecule. If you compare the opiate alkaloid morphine to naloxone (Figure 2), you can see there is strong similarity between the
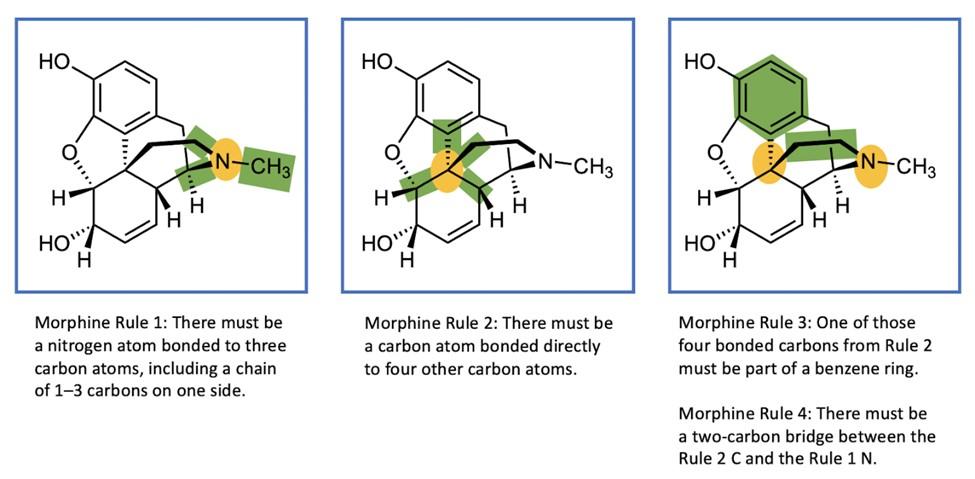
two. This rings true for other opiates as well, such as oxycodone, a synthetic opiate alkaloid.
In all three of these molecules, the same motif appears. These similarities are known as the morphine rule, structural requirements which contribute to the ease of binding to receptors in the brain. As shown in Figure 3, these molecules have: “(1) a ter tiary nitrogen with a small alkyl substituent; (2) a quaternary carbon; (3) a phenyl [benzene] ring or its equivalent attached to the q uaternary carbon; and (4) an ethyl [2-carbon] linker between the quaternary carbon and the tertiary nitrogen”.4 In simpler terms, the molecule must include (1) a nitrogen atom without any bonds to any hydrogens, with a chain of 1–3 carbons on one side. There must be (2) a carbon bonded directly to four other carbon atoms, and one of these four attached must be part of (3) a benzene ring. Finally, there must
be (4) a two -carbon linkage between the nitrogen atom of part (1) and the carbon atom of part (2). Figure 3 depicts this motif in morphine, and if you compare now to the other molecules in Figure 2, you can see that each of them fulfills these requirements, which explains why all these molecules can fit into the brain’s opiate receptors. This structural homology between molecules allows for each to be swapped out with ease.4 They all fit the same mold, so they can take each other’s place.
Despite being closely related to opioid molecules, both structurally and interactively, the most beneficial aspect about naloxone is that, although this molecule fits readily into opiate receptors, it can do so without activating its effect (Figure 1). This is because the molecule is a “competitive inhibitor”: a kind of biologically-active molecule that blocks the function of a receptor or enzyme.5 These molecules typically imitate the structure of the molecule usually employed by the receptor but have small structural changes that ensure the receptor or enzyme is not activated. Going back to the comparison between morphine and naloxone, this is definitely the case. Naloxone is similar enough to bind to the opiate receptors in the brain which morphine usually inhabits, but just different enough to prevent their activation.
The three most relevant opioid receptors in this circumstance are the Mu, Kappa and Delta receptors. All are opiate receptors, and all contribute to the symptoms of an overdose, although each type has its own effect. Mu receptors suppress the coughing reflex and cause congestion. Kappa receptors cause hallucination, while Delta receptors help modulate Mu receptors.5
The primary receptor involved in Naloxone treatment is the Mu receptor: Mu receptors are where the naloxone molecule can
quickly replace any trace of opioids in the system. Since the Mu receptors are involved in respiratory control, the replacement of an opioid with naloxone, as in Figure 1B, enables the person who overdosed continue breathing.5 Once their airway is clear, and the patient is out of respiratory trouble, it becomes easier to care for them at the hospital, and combat the other detrimental effects of the overdose.
Naloxone has proven itself a vital part of the fight against the opioid crisis. Without it, many lives would have been lost to the devastating effects of opiates, as they ravage both the mind and body. This molecule can be part of a second chance: an opportunity for those caught in the web of addiction to pull themselves out. Administration of this molecule gives life to those who are on death’s doorstep, even when all seems hopeless. In a world of sickness and despair, that alone is priceless. This molecule is invaluable, to the sciences and to the world, and it is critical to continue exploring the range of its abilities and the effect it could have upon our society. It truly is our last resort.
All figures were created by the author and the editors.
(1) Butanis, B. Signs of Opioid Abuse https:// www.hopkinsmedicine.org/opioids/signs-ofopioid-abuse.html (accessed Jan 22, 2021).
(2) Tomkins, D. M.; Sellers, E. M. Addiction and the Brain: The Role of Neurotransmitters in the Cause and Treatment of Drug Dependence. Cmaj 2001, 164 (6), 817–821.
(3) Cordant Health Solutions. The History of Naloxone https://cordantsolutions.com/thehistory-of-naloxone/ (accessed Jan 22, 2021).
(4) Chambers, S. A.; DeSousa, J. M.; Huseman, E. D.; Townsend, S. D. The DARK Side of Total Synthesis: Strategies and Tactics in Psychoactive Drug Production. ACS Chem. Neurosci. 2018, 9 (10), 2307–2330. https:// doi.org/10.1021/acschemneuro.7b00528.
(5) Theriot, J.; Azadfard, M. Opioid Antagonists. StatPearls Internet 2019.
National Institute on Drug Abuse. How do medications to treat opioid use disorder work? https://nida.nih.gov/publications/ research-reports/medications-to-treat-opioid -addiction/how -do-medications-to-treatopioid-addiction-work/ (accessed Jan 22, 2021).
National Institute on Drug Abuse. Naloxone for Opioid Overdose: Life-Saving Science. https://nida.nih.gov/publications/naloxoneopioid-overdose-life-saving-science/ (accessed Jan 22, 2021).
National Institute on Drug Abuse. Opioid overdose reversal with naloxone (Narcan, Evzio). https://nida.nih.gov/publications/ drugfacts/naloxone/ (accessed Jan 22, 2021).
I n Brazil in 2014, a large group sat down to share a meal together. The main course was an assortment of fish that had recently been caught. About 20 minutes into the meal, someone began to notice a tingle in their lips that began to grow steadily worse as time passed. Eleven members out of the group soon succumbed to vomiting and a feeling of weakness, a stark contrast to the party-ready attitude they had before the meal began. The affected group was quickly taken to the hospital to be stabilized; three of the individuals were placed in intensive care after struggling to breathe and entering cardiac arrest. Luckily, after spending up to half a week in the hospital recovering from the incident, all of the individuals were released in good health. The culprit: a fisherman had failed to remove a small pufferfish that had been caught with the other edible fish. This resulted in the pufferfish being prepared alongside the other fish, contaminating the batch with tetrodotoxin, a highly lethal chemical found in high concentrations in pufferfish and other select marine animals.
For many individuals, eating pufferfish also called fugu is a once-in-a lifetime experience. Considered a delicacy in Japan, fugu must be meticulously prepared in order to remove tetrodotoxin. Even in low dosages, tetrodotoxin (generally shortened to TTX, Figure 1) can be lethal to humans. The chef must remove all organs in the fish that contain high levels of this toxin, such as the liver and ovaries, and even after this is done the toxin can sometimes be found throughout the fish. Consuming fugu is risky, and it is that thrill that draws people to the experience. According to many individual accounts, the risk actually enhances the taste. Between 40 and 60 people are hospitalized each year due to tetrodotoxin poisoning and about 10% of these individuals ultimately die from consuming improperly prepared fish. At the moment, there is no known antidote for TTX, and much of the treatment at the hospital involves simply attempting to remove or bind the toxin before it
has a chance to interact with the individual’s nervous system. Due to the danger of eating fugu, it is the only dish that the Emperor of Japan is not allowed to eat.
After consumption of an improperly prepared fish, TTX acts rather quickly. TTX binds to the sodium channels of nerve cells, and blocks the channels from opening. This prevents the flow of sodium ions into and out of the cell, which disrupts the passing of electrical potentials through the nervous system, essentially blocking the transmission of movement information. Without the ability to communicate to muscles and organs, the individual can lose function of extremities and become paralyzed. If the dosage is severe enough, respiratory failure is generally the main cause of death. Often, if the poisoning is less severe, the symptoms of TTX poisoning will go away over a week’s time without permanent damage.
Though pufferfish are the figureheads of TTX, they are not the only organisms that contain the toxin: other species that contain detectable amounts of the toxin include horseshoe crabs, starfish, the blue-ringed oc-
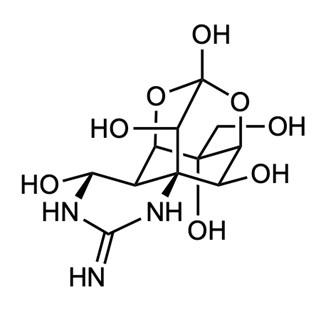
topus, and even certain frogs and salamanders. According to recent publications, the prevailing theory is that TTX is introduced to pufferfish through their diet. Vibrio bacteria produce TTX, and though pufferfish do not eat these bacteria directly, TTX is consumed by other animals in the food chain until it is found in high levels in predatory organisms, a process called biomagnification the same process by which the insecticide DDT accumulated in raptors. Fugu farmers have managed to create toxin-free pufferfish through controlling the diet of farmed pufferfish, though many critics of the process say that this changes the taste of the fish.
Yoshizumi Tahara first isolated TTX in 1909 from pufferfish ovaries. In 1962, R.B. Woodward was the first to definitively determine the structure of TTX (shown in Figure 1), and in 1972, Kishi et al. was the first to develop a synthesis method. The structure of TTX is distinctive, built through the combination of many rings that create a very structurally specific and stable molecule. Where many molecules are able to twist and spin, giving rise to multiple slightly different configurations, TTX is locked into place due to the fused rings. This fact becomes very important when looking into the TTX mechanism of attachment when binding to nerve
cells. TTX is a “key” that fits remarkably well into the “keyhole” found in the sodium channel proteins to which it binds.
To study the effects of TTX on nerve cells, scientists have looked at how TTX binds to gates along squid giant axons. The squid giant axon can be used as a largescale model of the same process that occurs in humans. These cells are enormous, running almost the entire length of the squid’s body. This model system gives scientists a clearer picture of how ions move in and out of the cell in order to transmit the electrical pulse formally known as the “action potential” through the cell.
Through the process of creating and releasing ion gradients and action potentials, a signal can be transmitted through nerve cells, and passed like a wave down through the body until it causes a muscle to contract. To control the passage of ions, there are ion channels along the membrane of the cell which can be opened or closed. When passing the signal through the cell, gates are opened down the cell, allowing for Na+ (sodium ions) to enter into the cell (Figure 2).
Figure 3 shows a diagram of the action potential process. In the beginning, cells have a negative resting potential. When the cell receives the right stimulus, a gate opens (Figure 2, right) and sodium ions rapidly enter the cell. This process, known as depolarization, causes the inside of the cell to switch to a positive electrical potential. In order to repolarize the cell after the action potential has been reached, the sodium ion channels are once again closed (Figure 2, left) while separate potassium ion channels are opened. This allows K+ (potassium ions) to flow out of
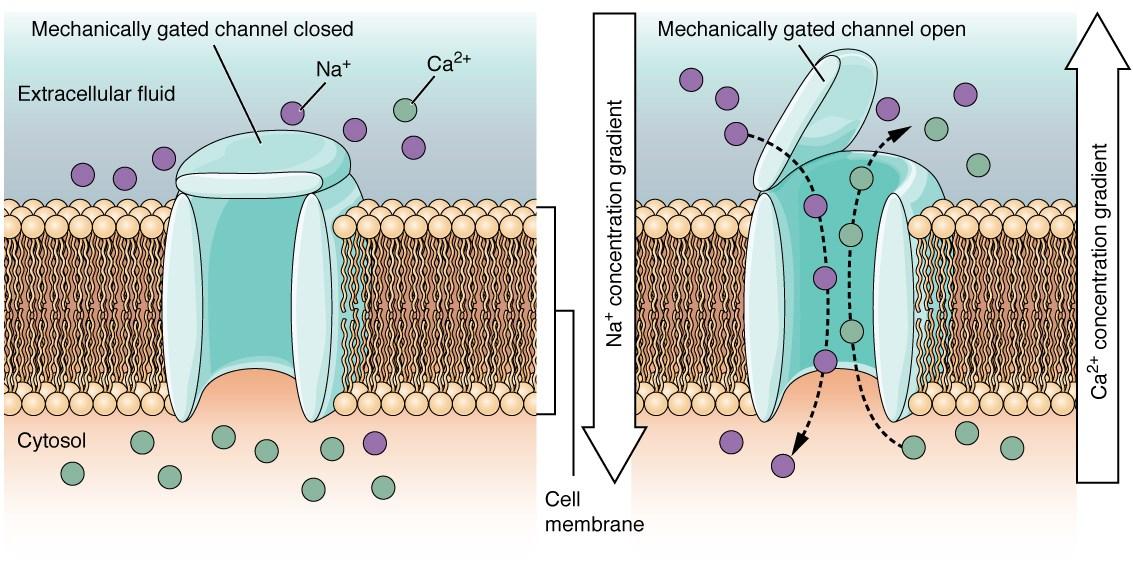
(1) Initially the cell has a negative potential: the “resting potential”.
(2) As a result of a stimulus, depolarization occurs: Na+ ions flow into the cell, giving it a positive electrical potential.
(3) The cell repolarizes. The Na+ ions stop flowing in, and K+ ions begin flowing out, causing the electrical potential to become negative again.
(4) The refractory period. The cell pumps out Na+ ions while pumping in K+ ions, so the original potential and ion gradient is achieved.
the cell and thereby causes the electrical potential in the cell to become negative once again (Figure 3). The cell then enters a refractory period during which sodium ions are pumped back out and potassium ions are pumped back in, until it returns to its resting potential and the original gradient of sodium and potassium ions. This action potential is then passed across the length of the cell, traveling rapidly, but in a stepwise fashion, through the nervous system.
When TTX enters an individual, it binds to the sodium ion channels on the nerve cells, and acts as a plug, blocking the Na+ ions from entering into the cell. Without the influx of sodium ions, the cell cannot reach its action potential. The wave of the signal hits a metaphorical wall, where it can no longer pass along the axon and down the
rest of the nervous system to its target. Symptoms of this include the muscle weakness and loss of motor function. Signal blockage increases as TTX concentration increases; eventually, even the signals to the brain and heart and diaphragm will become blocked, sending the individual into cardiac arrest. For severe poisoning, individuals might need to be put on a ventilator to aid their breathing.
Because there is no known antidote, doctors must simply wait and allow the toxin to be metabolized by the body and removed naturally. But what if doctors could reopen the sodium ion channels and reestablish the action potential? Although, it is still unknown how to properly control the opening and closing of the channels at such exact timings, the answer may lie in another toxin first discovered in the South American rainforests.
For thousands of years, various tribes in South America have coated the tips of blow darts with an excretion from roasted frogs in the family Dendrobatidae, colloquially known as “poison dart frogs.” This particular group of frogs contains the chemical batrachotoxin, known as BTX, in varying quantities (depending on the exact species and individual). The blow dart was a very effective hunting tool for slower, small game. And, by covering the tip of the dart with BTX, hunters were also able to take down larger game. The poison would often prevent their prey from escaping into the bushes before dying where they would be unable to find it.
Similar to tetrodotoxin, batrachotoxin is a neurotoxin that binds to sodium ion channels, but unlike TTX, which closes these channels, BTX floods them all open. Looking again at the graph of electrical potentials (Figure 3), this causes complete depolarization of the cell, and prevents the cell from returning to its resting potential. A constant, severe signal is then passed along the nervous system, contracting all of the associated muscles tightly. It induces the same kind of
paralysis as BTX, just in a different fashion; in both cases, all desired electrical signals are prevented from reaching their destination. Eventually, the toxin begins to interact with crucial nerves and sends the victim into cardiac arrest. This greatly increased the hunting power of the tribes, though the exact mechanism behind BTX’s effectiveness was not known until the late 1960s.
In 1969, the structure of BTX, shown in Figure 4, was first established, and in 1998, BTX was successfully synthesized. Although BTX and TTX are not similar structurally, both contain complex ring structures that are very stable due to the multiple adjacent rings fused together.
When BTX binds to the sodium ion channels, it binds to a different site than TTX. The term to describe this phenomenon is “non-competitive binding,” meaning that both chemicals can attach simultaneously to the same channel. Unfortunately, this does not restore the channel back to normal working order, and so BTX cannot be used as an antidote for TTX poisoning. Nevertheless, their opposite interactions with the sodium ion channels provide a basis for the study of other medical advancements in the field of neuroscience.
In recent medicine, TTX and BTX have been used to study nerve cells in humans. Because of their very specific selection towards the sodium ion channels of the cells, scientists have been able to study the density of these ion channels along cells, as well as determining that there are certain cell channels that are resistant to the effects of TTX.
With these findings another idea arose: because TTX is so targeted to the sodium ion channels on specific nerves, and because we can determine the approximate number of these channels on a given cell, it might be possible to use TTX as a drug to treat chronic pain. In 2012, a paper written by Nieto et al. pulled together a number of studies showing the effects of pain treatment with TTX. The administered doses of TTX are very small, much below the lethal limit, and specifically target a given region of the body. Scientists wished to administer a concentration that would block the pain without preventing the action potential cycle and general mobility of the individual. All of these studies were either performed on animals or within a clinical trial. Many of the papers found that TTX had little to no effect on acute pain signals. But there were other studies that found TTX administration decreased inflammatory pain as well as decreased pain in individuals suffering from neuropathic pain, or debilitating pain associated with malfunctioning nerve signals. The TTX is able to bind to the pain site and prevent the propagation of the action p otentials, which carry the pain signals to the brain. Further studies will have to be performed before a link between TTX and pain mitigation can be made. In addition, the many potential dangers that come from working with TTX makes it a somewhat unlikely candidate for use in regular therapeutic work.
As a result of the arms race between hunters and prey, the pufferfish and poison dart frogs have developed two of the most
lethal defense mechanisms in the world. Though it may seem like an unlikely place to start, toxins and other dangerous chemicals can give an insight into the intricacies of our body. Similar to pulling a computer apart to understand its vital organs and functions, TTX and BTX allow scientists to disrupt and thus fully understand the function of the nervous system. With these deadly toxins, scientists are slowly making medical advancements that have the potential to save and improve lives, rather than end them.
(2) Cataldi, M. Batrachotoxin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S. J., Bylund, D. B., Eds.; Elsevier: New York, 2010; pp 1–9. https://doi.org/10.1016/ B978-008055232-3.63502-0.
(3) Dumbacher, J. P. Batrachotoxin. In Encyclopedia of Toxicology (Third Edition); Wexler, P., Ed.; Academic Press: Oxford, 2014; pp 371–373. https://doi.org/10.1016/ B978-0-12-386454-3.00998-2.
(4) December 18, L. G.; 2014. Close Call! Accidental Pufferfish Diners All Recover https:// www.livescience.com/49181-pufferfishpoison-recovery.html (accessed Jan 8, 2021).
(5) Narahashi, T. Tetrodotoxin. Proc. Jpn. Acad. Ser. B 2008, 84 (5), 147–154.
Figures 1 and 4 were created by the author and the editors.
Figure 2 was created by OpenStax and downloaded from https://commons.wikimedia.org/wiki/ File:1217_Mechanically-gated_Channels02.jpg
Figure 3 was created by Peter Duncan and downloaded from https://commons.wikimedia.org/wiki/ File:Action_potential_schematic.png
Figures 2 and 3 are used under Creative Commons license: https://creativecommons.org/licenses/bysa/4.0/deed.en
(6) Nieto, F. R.; Cobos, E. J.; Tejada, M. Á.; Sánchez-Fernández, C.; González-Cano, R.; Cendán, C. M. Tetrodotoxin (TTX) as a Therapeutic Agent for Pain. Mar. Drugs 2012, 10 (2), 281–305.
(7) Noncompetitive Inhibition https:// www2.chem.wisc.edu/deptfiles/genchem/ netorial/modules/biomolecules/modules/ enzymes/enzyme6.htm
(8) Zhang, X.; Ku a, K.; Dohnal, V.; Dohnalová, L.; Wu, Q.; Wu, C. Military Potential of Biological Toxins. J. Appl. Biomed. 2014, 12 (2), 63–77.
Sources Consulted and Cited
(1) Albuquerque, E.; Brookes, N.; Onur, R.; Warnick, J. Kinetics of Interaction of Batrachotoxin and Tetrodotoxin on Rat Diaphragm Muscle. Mol. Pharmacol. 1976, 12 (1), 82–91.
R unning, jumping, hiking: these are activities we take for granted in the human condition, "granted” being the key word: imagine being unable to hike in the fall and spring or run around with your kids at the park without the constant fear of becoming unable to breathe. Before 1969, this was the bleak reality for many people with moderate to severe asthma.
Asthma is defined as a chronic lung disease, characterized by periodic inflammation of the bronchial tubes or airways. While asthma symptoms vary widely between individuals, the common triggers include allergies, exercise, and infections. These triggers can initiate an inflammatory response and cause airway constriction. Chronic coughing, wheezing, and shortness of breath are common symptoms that asthmatics shoulder on a daily basis. The World Health Organization estimates that approximately 300 million people worldwide have this condition.1 Even though there is still no cure, in 1969 the drug salbutamol revolutionized the way asthma was treated and created the foundation for refining asthma drugs.
The first patients to use salbutamol experienced a liberation that many asthmatics today only experience on a small scale, before their own diagnosis and treatment. The lifestyle of an asthmatic was controlled by their condition because the treatment available was only effective in temporarily relieving the most severe symptoms. Effective long-term management of the disease depended on avoiding triggers such as physical exertion, allergies, and infections. Salbutamol changed the lives of asthmatics because the drug could treat their symptoms more effectively for hours at a time. Salbutamol soon became the model for many drugs currently used in individualized treatment plans.
Doctors and patients have two options when it comes to administration. Salbuta-
mol can be taken in a nebulized form or through an inhaler. Nebulized drugs are inhaled in treatments that typically last about 15 minutes. The drug is stored as a liquid and then is vaporized by an air compressor. The patient inhales the vapor, taking the medicine into their lungs. Alternatively, the inhaler is a less concentrated form of the drug sealed in a compressed chamber. The medicine is aerosolized and inhaled as puffs when needed.
The discovery of salbutamol made treatment options more versatile because this drug can treat symptoms on a regular, everyday basis, not just in an emergency. Albuterol, the market name for salbutamol in the U.S., can even be used as a preventative drug for those who have exercise-induced asthma in the inhaler form of administration. Nebulized salbutamol provided children who could not go to school the ability to attend with only one or two brief interruptions for treatments during the day.
Even as an asthmatic, I struggle to fully comprehend the leap in treatment options that salbutamol created. The incredible leap was twofold. The most obvious jump was the simple fact that salbutamol worked so much better at relieving acute symptoms than the preceding drugs. The other huge difference was that the side effects of the other drugs were so severe that they were not practical for the long-term use or for managing symptoms less severe than emergencies. Of course, salbutamol is no panacea: I dread the prospect of taking nebulized salbutamol because of the side effects, which include elevated blood pressure and heart rate. Nevertheless, these side effects are negligible compared to those from treatments available in 1969: The side effects associated with previous treatments were truly serious. In order to understand the difference in side effects we first have to understand the difference in the ways these chemicals affect the body.
In the first half of the twentieth century, asthma treatment was done with drugs that mimicked the nervous system’s response to acute stress. The response commonly known as “fight or flight” is caused by a spike in the hormone adrenaline. Adrenaline prepares the body to deal with pressing danger. It is one of the main chemicals that cause the sensations we associate with fear, including heightened awareness, increased heart rate, higher blood pressure, and opened airways. Adrenaline (which is sometimes referred to as epinephrine) was injected in a form similar to an EpiPen, to relax the tissue of the lungs and calm an asthma attack. Adrenaline is the body’s natural bronchodilator, meaning that it relaxes the smooth muscle in the lungs and reduces constriction of bronchial tissues to increase airflow.
The side effects of using adrenaline, inhaled or injected, included elevated blood pressure, heart palpitations, tremors, and anxiety that persisted long after the drug’s bronchodilation effects. Usually, people injected with large doses of this drug are sent to the hospital immediately after, to ensure the body doesn’t experience any further acute reactions. The search for a bronchodilator that did not have those adverse side effects initially yielded the drug isoprenaline. Isoprenaline avoided many of the side effects of adrenaline, but still influenced the smooth muscle tissues of the heart and only lasted a short time.
Salbutamol was synthesized as a modification of isoprenaline in 1966.1 Finally there was a drug with fewer side effects that was effective for up to six hours at a time. The function of salbutamol is based on the function of adrenaline, so I will start with a comparison of the structures of adrenaline and salbutamol (Figure 1) to properly contrast their functionality. Even though isoprenaline was used in the synthesis of salbutamol it is more useful to look at the structure of adrenaline because it is the natural chemical made by the body that chemists were trying to mimic and modify. Additionally, understanding the differences between salbutamol and adrenaline is useful for understanding the biological processes responsible for the side effects of salbutamol.
As shown in Figure 1, the two molecules are very similar. These molecules only differ in two substituents. First, compare the green circles: adrenaline has a single methyl (CH3) group attached to the amine (i.e. the nitrogen). That region of the salbutamol molecule is significantly larger than the same region of adrenaline; in this case, there are now four carbon atoms: three methyl groups attached to the carbon next to the amine. Next, look at the gold ovals. One of the alcohol groups (–OH) has an extra carbon between the benzene ring in salbutamol, whereas the alcohol is directly attached to the ring in adrenaline. Everything else is the same, except for a subtle contrast that is hard to infer from these images: chirality.
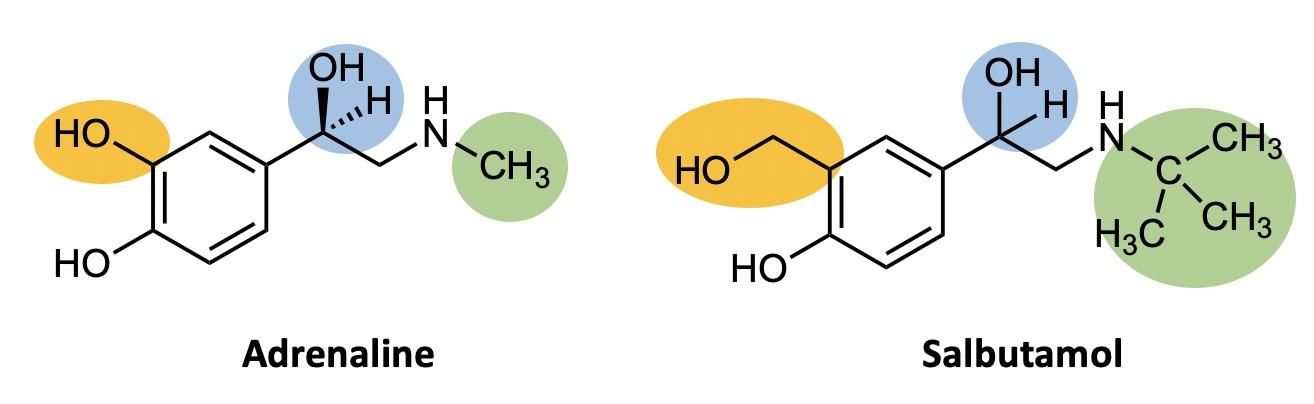
These molecules have a property called chirality, meaning that there are variations of the molecule that are like a right and left hand: they have the same connectivity, but are non-superimposable mirror images. Salbutamol is a mixture of both of the hands, or enantiomers.2 Each “hand,” also called an isomer, is designated as either the (R)isomer or the (S)-isomer, in a manner similar to distinguishing between right and left hands. Drugs with a mixture of isomers are called racemic (ray-SEE-mick) drugs. The blue circle in Figure 1 shows the chirality center that distinguishes between (R)- and (S)-isomers of adrenaline and salbutamol.
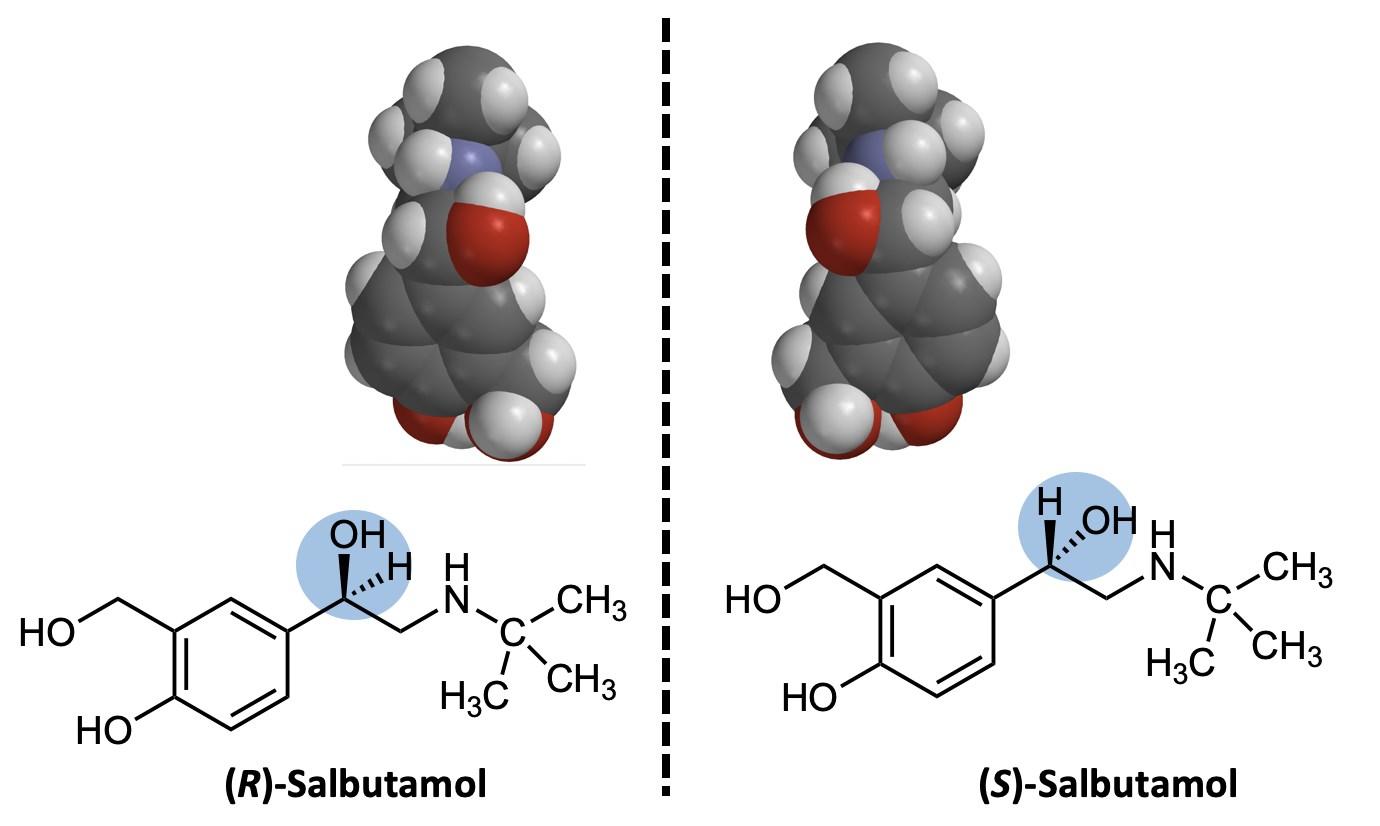
Chirality comes about because carbon atoms form four bonds. Much of the time, the bonds can be thought of as being in the same plane, and this is the default when bonds are shown as straight lines. Now, look at the bond to the –OH (alcohol) group on adrenaline. It is shown with a wedge mark, indicating that the group is really oriented towards us: the bond is coming out from the page (Figure 1). Conversely, the bond to the hydrogen atom is shown with dashes, indicating that it is oriented such that it is going
behind the page. Now look at the structure of salbutamol shown in Figure 2: the –OH and –H group bonds highlighted in blue are both just shown with straight lines. Since the figure represents the mixture of both isomers, the structure does not show a wedge or a dash. It is therefore inferred that the structure represents both isomers, one with the alcohol group coming out of the page (on a wedge) and one with the alcohol group going into the page on a dash.
Refer now to Figure 2. This image shows the structures in wedge and dash form, as well as the space-filling models, more distinctly showing the mirroring images. It may be helpful to think of the central dotted line of the figure as a plane of symmetry: it is like a mirror. Since the body builds specific enantiomers of the chemicals that it needs, the chirality (“handedness”) of a drug is critical in determining how molecules interact with receptors and enzymes. The body only makes adrenaline with the (R) orientation shown in Figure 1. Adrenaline binds to beta-receptors in the smooth muscle of the lungs when stress occurs, causing bronchodilation. Even though they look very similar, the difference in shape of the (R) and (S) isomers is sufficient to determine how well the betareceptors will bind to the molecule. If we think of the two isomers as hands, the receptors can be thought of as a glove. Only the right hand “glove” in the lungs causes bronchodilation when it has a hand in it. Both adrenaline and the (R)isomer of salbutamol fit in that glove, whereas the (S)-
isomer does not fit. It turns out that both enantiomers of salbutamol do not have the same effect on the smooth muscle in the lungs. Only the (R)-isomer binds to the correct beta-receptor and reduces the constriction in the bronchioles to bring open the airway back up.2,3
The side effects of salbutamol are much less severe than those that came from using adrenaline because while the salbutamol molecule fits the proper receptor sites in the lungs, its larger size means that it does not fit as well in receptor sites in the heart.4 Since the salbutamol molecule cannot activate the receptors in the heart as effectively, side effects such as the increased heart rate and blood pressure associated with the fight or flight response caused by adrenaline in the blood system are reduced.
Studies have shown that the side effects can be reduced even more by just using the active (R)-isomer instead of the racemic mixture (of both (R)- and (S)-isomers).2 The (R)-isomer binds to the beta-receptors in the lungs with great affinity; then it is grabbed by an enzyme that metabolizes it quickly. On the other hand, the (S)-salbutamol isomer does not bind well to the receptors in the lungs: instead, it circulates in the bloodstream and binds to some of the adrenoreceptors in heart and nervous tissues, causing those negative side effects. They side effects last for so long after taking the drug because the enzyme that metabolizes the ( R)isomer (and adrenaline) does not fit well around the (S)-isomer. Without enzymemediated degradation, the ( S)-isomer does not metabolize and is only eliminated from the bloodstream when it is filtered out by the kidneys a much slower process. 5 Fortunately, there is now a drug that is composed of just the (R)-isomer, called levalbuterol.
Currently, levalbuterol is very expensive because the process of isolating the ( R)
from the (S) isomer is expensive. The cost of the drug excludes many from having access to the drug with fewer side effects.6 Those who have to buy the drug without health insurance have to pay a much higher cost for the generic levalbuterol than the racemic albuterol or salbutamol. Many health providers and insurers will not cover the levalbuterol because it costs them more.
New ways of isolating the isomers may reduce the price gap, making the better drug accessible to more people. For example, through the process of preferential crystallization, the active (R)-isomer of levalbuterol can be isolated from racemic salbutamol.7 Instead of synthesizing only the (R)-isomer, which takes more steps, the preferential crystallization method takes the abundant racemic drug and forces only the (R)-isomer to crystallize and therefore be isolated from the solution.
The synthesis and isolation processes for airway-specific beta-receptor agonists have been slowly improving since the 1960s, giving rise to creating salbutamol and levalbuterol. Hopefully, the process will continue to be refined and allow more people to have access to the more specific and effective levalbuterol. There is an increased demand every year for effective bronchodilators, including levalbuterol, as the number of people who have asthma worldwide is expected to increase by as much as 100 million in the next ten years.1 Salbutamol is not the final answer for treating exacerbated asthma symptoms, but it is still often the drug of choice for relieving common flare-ups. Despite the side effects it causes, salbutamol has improved the everyday lives of those of us who struggle with asthma. We can only hope that the foundation it provided can be used to make treatment options stronger and more accessible in the future.
All figures were created by the author and the editors.
(1) The American Academy of Allergy, Asthma & Immunology. Asthma Statistics | AAAAI https://www.aaaai.org/about-aaaai/ newsroom/asthma-statistics (accessed Jan 22, 2021).
(2) Ameredes, B. T.; Calhoun, W. J. Levalbuterol versus Albuterol. Curr. Allergy Asthma Rep. 2009, 9 (5), 401–409.
(3) D’Alonzo Jr, G. E. Levalbuterol in the Treatment of Patients with Asthma and Chronic Obstructive Lung Disease. J. Am. Osteopath. Assoc. 2004, 104 (7), 288.
(4) Marasco, C. Salbutamol. Chemical & Engineering News. 2005.
(5) Silverman, R. B.; Holladay, M. W. The Organic Chemistry of Drug Design and Drug Action; Academic press, 2014.
(6) Yuan, J.; Lu, K. Economic Impact of Levalbuterol Versus Albuterol in Low -Income Population. Value Health 2014, 17 (7), A781.
(7) Palacios, S. M.; Palacio, M. A. Enantiomeric Resolution of Albuterol Sulfate by Preferential Crystallization. Tetrahedron Asymmetry 2007, 18 (10), 1170–1175.