
Investigating the Effects of Sodium-Glucose Co-Transporter 2 Inhibitors on Rat Cardiomyocytes
Suzanna Luptakova
Senior Thesis | 2024

Title: Investigating the Effects of Sodium-Glucose Co-Transporter 2 Inhibitors on Rat Cardiomyocytes
Author: Suzanna Luptakova
Abstract:
Sodium-glucose co-transporter 2 (SGLT2) inhibitors, such as Ertugliflozin (ERTU), are recognized for their glucose-lowering effects in Type-2 diabetes by causing the kidneys to release glucose through urine. These inhibitors have been found to improve cardiovascular outcomes which have never been demonstrated with other diabetes drugs and emerging evidence suggests a novel role in improving cardiovascular outcomes. However, the direct impact on cardiac cells remains largely unexplored. Our research, conducted under Dr. David Pimentel at Boston Medical Center, delves into the effects of ERTU on cardiomyocytes amidst simulated heart failure conditions.
Our data show that when ERTU is given to an animal overexpressing cardiac specific Gαq protein (Gq), which is responsible for the initiation of stress response signaling cascade, allows for cellular growth and progression to heart failure via mitochondrial energetic changes, there is attenuation of these responses. To determine if SGLT2 inhibition could directly affect the cardiomyocyte and thus the heart directly, we used isolated rat cardiac myocytes to measure the energetics of individual cells that overexpressed Gq in the presence and absence of ERTU. Utilizing Perceval which is a fluorescent biosensor that allows us to perform real-time measures of the ratio of ATP to ADP intracellularly, we found that an SGLT2 inhibitor increased the ratio of ATP to ADP production in isolated cardiac myocytes. However, unlike the animal model, we found no change in this ratio with go overexpression. To further define if this event was due to calcium changes, we measured mitochondrial calcium levels using a
mitochondrial-directed Cameleon biosensor that can measure nanomolar calcium concentrations. We found that there were trends to higher mitochondrial calcium levels with ERTU but there were no significant changes. We then inhibited the entry of calcium into the mitochondria using an inhibitor of the mitochondrial uniporter (MCU) and found that this did not change the ATP to ADP ratio suggesting that ERTU may directly affect the mitochondria to lead to increased ATP:ADP concentrations independent of calcium.
Introduction
Heart failure is a clinical syndrome that leads to symptoms or signs caused by a situation where the myocardium can no longer provide enough oxygen for the demand of the tissues. There are numerous conditions such as myocardial infarction, also known as a heart attack, that weaken the myocardium - the muscular tissue of the heart - reduce its relaxation or increase oxygen demand by the body tissue beyond the capability of the heart to deliver. Clinically two separate entities exist, heart failure with reduced systolic function and heart failure with preserved function. Each of these clinical syndromes has medications that are beneficial based on the presumed pathogenesis. For example, individuals with heart failure with reduced systolic function are felt to have an increase in the renin-angiotensin system, a hormone system that regulates blood pressure through the kidney,1 and noradrenergic system, the system controlling norepinephrine, a neurotransmitter that regulates cellular metabolism,2 which have led to medications targeting these pathways such as angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and beta-blockers to be beneficial for patient survival. Each of these drugs works in inhibiting distinct pathways, thus reducing fluid buildup and improving myocardial performance.3 After a myocardial infarction, the released neurohormones like angiotensin stimulate the Gq
receptor leading to another disease. The angiotensin inhibitors block the formation of angiotensin or binding to its receptor which would then activate a G-coupled protein, Gαq. It is responsible for the initiation of the stress response signaling cascade, allowing for cellular growth and progression to heart failure via mitochondrial energetic changes. To understand heart failure better, researchers have developed animal models that overexpress Gq, simulating the heart conditions observed in heart failure.
One of the most frequent causes of heart failure is myocardial infarction.4 Myocardial infarction is highly associated with Type-2 diabetes, which is a chronic condition that affects the way the body processes glucose. It is a global health concern due to its rapidly increasing prevalence, often linked to lifestyle factors like obesity and sedentary behavior.5 Persistently elevated glucose levels can damage blood vessels and nerves. This damage can lead to atherosclerosis due to the buildup of plaque in the arteries, which reduces the flow of oxygen-rich blood to the heart and can lead to conditions like coronary artery disease and myocardial infarction.6
There are many different types of treatments for Type-2 diabetes such as metformin, sulfonylureas, and insulin. Metformin primarily lowers blood glucose levels by decreasing the amount of glucose produced by the liver. It also helps in making muscle tissue more sensitive to insulin so that glucose can be utilized for energy. It also improves insulin sensitivity, which helps in better glucose management.7 Sulfonylureas lower blood glucose by stimulating the secretion of insulin in the body, thereby increasing insulin levels in the blood.7 Insulin directly supplies insulin to control and prevent diabetes complications. It works similarly to the natural hormone insulin that the body usually produces, which comes from an organ in the stomach area called the pancreas.8 Unfortunately, despite the ability of these medications to control blood glucose levels none of these treatments have been able to improve cardiovascular
morbidity or mortality. Thus, there is a need for more effective and safer treatments.
Approximately 10 years ago, a novel class of diabetes medications, sodium-glucose co-transporter 2 (SGLT2) inhibitors were approved by the FDA. These drugs are a class of drugs that reduce blood glucose by causing glycosuria or that allow the kidneys to remove sugar from the body through urine. They block the reabsorption of glucose in the kidneys, leading to glucose excretion.9 They are used to control blood glucose levels in Type-2 diabetes. Unlike all other diabetic medications, SGLT2 inhibitors (SGLT2is) have been shown to improve cardiovascular outcomes, a benefit not observed with many other diabetes drugs. SGLT2is significantly reduced major adverse cardiovascular events, all-cause mortality, hospitalization for heart failure, myocardial infarction, stroke, cardiovascular mortality, and heart failure.10 They also resulted in cardiovascular benefits in patients with Type-2 diabetes. While these drugs exhibit cardiovascular benefits, the exact mechanisms by which they impact the heart remain unknown. Building on the understanding that SGLT2is, such as Ertugliflozin (ERTU), have a systemic impact on cardiovascular outcomes, this research aims to delve deeper into the direct effects these inhibitors may have on cardiac tissue, particularly on energetics. 11 This could redefine the therapeutic use of these drugs, extending beyond their current role in glucose excretion and potentially leading to new interventions that address the cardiac complications often associated with Type-2 diabetes.
Dr. Chambers found that oxidative phosphorylation changes with ERTU through a gene array. The ERTU treatment increased oxidative phosphorylation in both the Wild Type and Gq treatment. ERTU also increased fatty acid metabolism in both wild-type and Gq treatment. This means that the cells run through the electron transport chain (ETC) rather than glycolysis. In other words, it has become more of a fatty acid consumer to create ATP rather than a
glucose consumer. The heart usually consumes mostly fatty acids to create energy (about 70% fatty acids, 30% sugars), but the overexpression of the G-coupled protein causes it to become more of a glucose eater, similar to how during heart failure the heart becomes more of a sugar eater (50-60% fatty acids). The information that Dr. Chambers found shows that ERTU reverses the change that Gq causes. The goal of this study is to find out why this happens.
Methods
Chemicals were purchased from Sigma-Aldrich unless otherwise stated. The animal protocol was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine.
Experimental Animals
Wild type and cardiac-specific Gαq-overexpressing mice (6) were randomized to chow formulated (Research Diets, New Brunswick, NJ) with or without ertugliflozin (Merck; 0.5 mg/g of chow) which was begun at 4 weeks of age and continued for 16 weeks. Body weight and food consumption were measured weekly. Fasting blood glucose was measured in whole blood collected from the tail vein following an overnight fast using a ContourNext glucometer as per the manufacturer’s instructions. The protocol was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine.
Echocardiography
Tissue doppler echocardiography was measured with a VisualSonics Vevo 2100 high-resolution imaging system equipped with a 22-5-MHz MS550D transducer, as previously described33.
Mice were anesthetized via isoflurane, and the apical view fourchamber of the heart was used to record myocardial tissue movement at the mitral annulus. Myocardial peak early diastolic velocity (Em) was measured via this method over 5 consecutive cardiac cycles, averaged, and analyzed via Vevo 2100 Analytic software.
RNA isolation and sequencing
RNA was isolated from the frozen left ventricle (LV), sequenced, and analyzed as previously described. In brief, LV from 4 - 5 hearts per treatment group was homogenized, and mRNA was extracted with the RNeasy Universal Mini kit (Qiagen) according to the manufacturer’s instructions. The Boston University Microarray and Sequencing Resource Core Facility performed RNA quantity and quality measurements, library preparation, and RNA sequencing. Sequencing libraries were generated using the NEBNext Ultra II RNA Kit with poly(A) selection and run on an Illumina NextSeq 500 platform with 75-bp paired-end reads. FASTQ files were aligned to mouse genome build mm10 using STAR (Dobin et al., 2013) (version 2.6.0c). EnsemblGene-level counts for non-mitochondrial genes were generated using featureCounts (Subread package, version 1.6.2) and Ensembl annotation build 100 (uniquely aligned proper pairs, same strand). Separately, SAMtools (version 1.9) was used to count reads aligning in proper pairs at least once to either strand of the mitochondrial chromosome (chrM) or to the sense or antisense strands of Ensembl loci of gene biotype “rRNA” or of nonmitochondrial RepeatMasker loci of class “RNA” (as defined in the RepeatMasker track retrieved from the UCSC Table Browser). FASTQ quality was assessed using FastQC (version 0.11.7), and alignment quality was assessed using RSeQC (version 3.0.0). Variance-stabilizing transformation (VST) was accomplished using the varianceStabilizingTransformation
function in the DESeq2 R package (version 1.23.10) (Love et al., 2014). Principal Component Analysis (PCA) was performed using the prcomp R function with variance stabilizing transformed (VST) expression values that were z-normalized (set to a mean of zero and a standard deviation of one) across all samples within each gene. Differential expression was assessed using the Wald test implemented in the DESeq2 R package. Correction for multiple hypothesis testing was accomplished using the Benjamini-Hochberg false discovery rate (FDR). Human homologs of mouse genes were identified using HomoloGene (version 68). All analyses were performed using the R environment for statistical computing (version 3.6.0).
Gene set enrichment analysis
Gene set enrichment analysis (GSEA; version 2.2.1) (22) was used to identify biological terms, pathways and processes that are coordinately up- or down-regulated within each pairwise comparison, as we have described (18). In brief, the Entrez Gene identifiers of the human homologs of all genes in the Ensembl Gene annotation were ranked by the Wald statistic computed for each effect in the two-factor model and each pairwise comparison. Ensembl Genes matching multiple mouse Entrez Gene identifiers, and mouse genes with multiple human homologs (or vice versa), were excluded prior to ranking, so that the ranked list represents only those human Entrez Gene IDs that match exactly one mouse Ensembl Gene. Each ranked list was then used to perform preranked GSEA analyses (default parameters with random seed 1234) using the Entrez Gene versions of the Hallmark, Biocarta, KEGG, Reactome, PID, Gene Ontology (GO), and transcription factor and microRNA motif gene sets obtained from the Molecular Signatures Database (MSigDB), version 7.1 (22).
Gene set size was determined by the number of genes in the gene set that overlap with genes in the ranked list. Enrichment scores, a measure of skewness of each gene set corresponding to a trend for upregulated or downregulated genes with respect to the reference group, were normalized to the mean enrichment score measured across 1000 random distributions of the members of the gene set across the ranked list. Leading-edge heatmaps were generated from each ranked list. The colors of each matrix were assigned as z-scores normalized to a mean of 0 and a standard deviation of 1 across all samples in each row. The z-scores were trimmed to a range of -2 (blue) to 2 (red).
Adult Rat Ventricular Myocyte Isolation
Adult Rat Ventricular Myocytes (ARVMs) were isolated from 150-200g male Sprague Dawley rats, as previously described.12 In brief, rats were anesthetized with isoflurane in a bell jar until nonreactive to a toe pinch, the heart was rapidly excised and cannulated by the aorta onto a retrograde perfusion system (Radnoti MasterFlex), and perfused with 37°C Krebs Henseleit (KH) buffer containing (in mM): 118 NaCl, 4.75 KCl, 1.2 MgSO4, 1.2 KH2PO4, and 25 NaHCO3, pH 7.4, for the initial 2 minutes of perfusion. The perfusate was then switched to KH containing collagenase type II (Worthington) and hyaluronidase, which was allowed to recirculate for 16 minutes. The heart was removed from the cannula, ventricles minced, and incubated in KH containing collagenase, hyaluronidase, DNAse, trypsin, and 1 mM CaCl2 in a shaking water bath for 20 minutes. The ventricular slurry was pipetted up and down to further dissociate cells, then passed through a 100 μm filter. Cells were centrifuged at 500 rpm for 3 minutes, supernatant was removed, and the ventricular myocyte pellet was resuspended in a wash buffer comprised of equal parts KH and DMEM (12320-032, Life Technologies). Cells were allowed to settle by gravity for 10
minutes, supernatant was removed, and resuspended in the wash buffer. This step was repeated, and upon final resuspension, cells were layered atop a 5% Bovine Serum Albumin (BSA) in DMEM gradient and allowed to settle for 11 minutes. The resulting pellet comprised primarily viable cardiomyocytes, which were resuspended and plated onto laminin-coated dishes. After 30-60 minutes, the media was changed by a gentle aspiration to remove unattached and non-viable cells, and experiment-specific media and treatments were added to the plates (described in next section).
In Vitro imaging.
The cells were transfected for 48 hours with an adenovirus encoding Perceval-HR, the ATP:ADP fluorescent probe,13 empty vector, and GαqQ209L (empty vector and Gαq Q209L were kind gifts from Dr. Joan Heller Brown and Dr. Ulrike Mende) were applied.14 The Perceval HR adenovirus was constructed as previously described.15 RoGFP was used to measure oxidative stress and a virus was created as previously described.16 Ertugliflozin 200ng/ml (Ertugliflozin (Steglatro): A New Option for SGLT2 Inhibition - PMC (nih.gov)) was given to cells for 24 hours prior to the start of the experiment and media was changed 6 hours prior to the start of the experiment with replenishment of the ertugliflozin. Cells were imaged at 37° using an inverted Olympus Spinning Disk confocal microscope in wide field mode as previously described. All optical filters were purchased from Chroma: excitation 390/70 nm, GFP 470/510 nm, and emission EGFP(FITC/Cy2). Cells were excited at 390 and 480 nm and emission was collected with a rapid emission filter changer. The cells were imaged after being in 37oC PBS with Hepes 25 mM for 10 minutes. Ratiometric images were calculated after background subtraction by dividing GFP-excited images over 390 excited images. Images were analyzed using the
NIS Elements software (Nikon). Only rod-shaped cells were used for this analysis.
Statistics
Data shown as mean ± SEM. Statistics were performed using unpaired t-test or ANOVA with bonferroni post-hoc tests, where appropriate, calculated using Graphpad Prism 6. For all analyses, a p-value of < 0.05 was considered to be statistically significant.
Results
SGLT2 inhibitors attenuate cardiac heart failure.
Several features of adverse cardiac remodeling that suggest worsened heart failure include left ventricular dilation in diastole and systole, thinning wall sizes, and reduction of LV function. In vivo, overexpression of Gq leads to increases in left ventricular enddiastolic dimensions along with left ventricular larger LV systolic volumes. ERTU can lead to attenuation of these parameters after 8 weeks of therapy and is maintained (Figure 1A and 1B). On the other hand, left ventricular wall thinning is noted to reverse with ERTU by week 4 with improvements in LV function occurring simultaneously (Fig 1C and 1D).
SGLT2 inhibitors lead to transcriptional changes favoring oxidative phosphorylation/ fatty acid metabolism.
Cardiac tissue favors fatty acid metabolism for energetic usage and with heart failure, there is a substrate switch to glycolysis.17 Gq overexpression changes transcriptional regulation to less oxidative phosphorylation and fatty acid utilization. ERTU, on the other hand, increases both (Fig 2A and 2B). Furthermore, ERTU
mitigates the Gq decrease in oxidative phosphorylation and fatty acid utilization.
SGLT2 inhibitors act directly on cardiac myocytes altering energetics.
When treated with ERTU, isolated cardiomyocytes demonstrate an increase in ATP/ADP ratio using PercevalHR Fig 3 (n= 6, p<0.05). Gq has no significant effect on the cells energetics.
SGLT2 inhibitors effect on energetics is independent of mitochondrial calcium and oxidative stress.
Mitochondrial calcium has been reported to increase with SGLT2 inhibitors. ERTU trended with a similar increase that was not significant (figure 4A n= 6, p>0.1) with no change with Gq overexpression. In addition, the inhibition of mitochondrial calcium entry with a dominant negative form of mitochondrial calcium uniporter (DN-MCU) showed no change in ERTU-induced increases in ATP/ADP ratio (n =, p< 0.05 for control versus DNMCU ERTU). Lastly, ERTU showed no changes in oxidative stress measure using ro-GFP (n=, p>0.30).
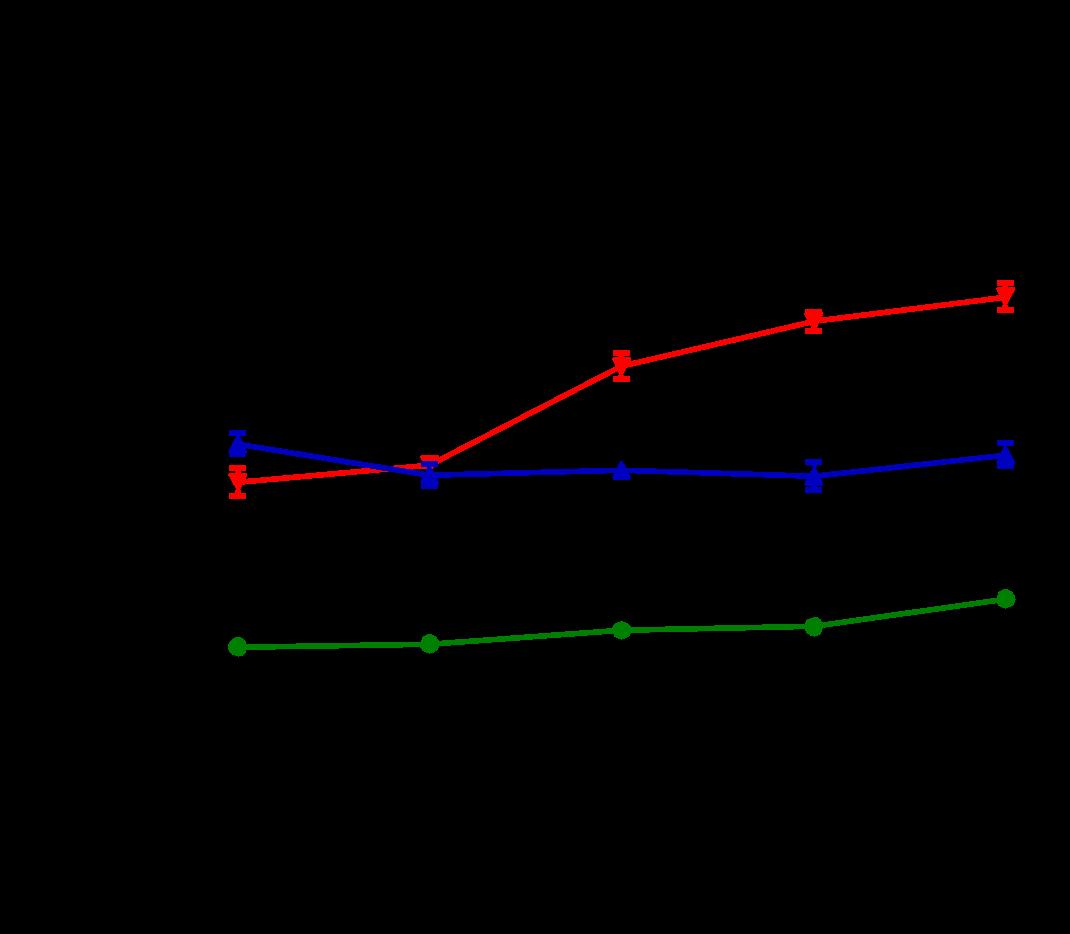
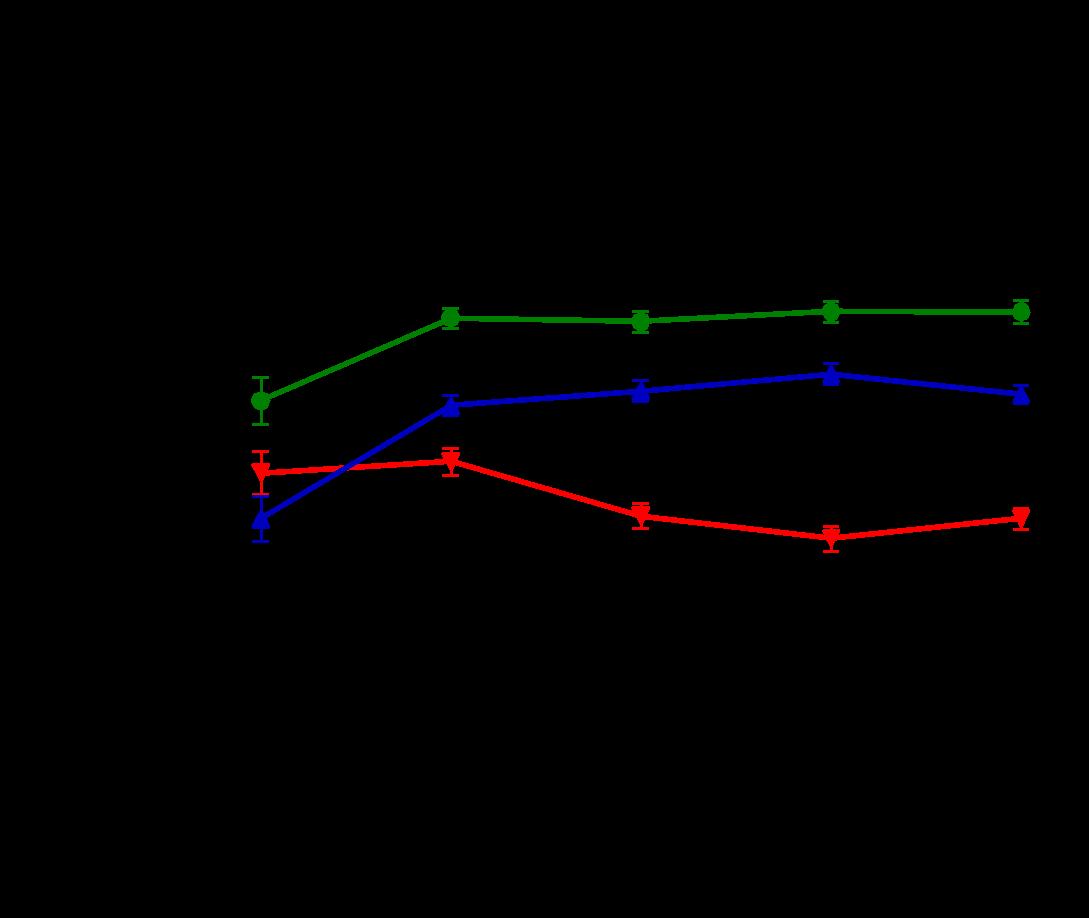
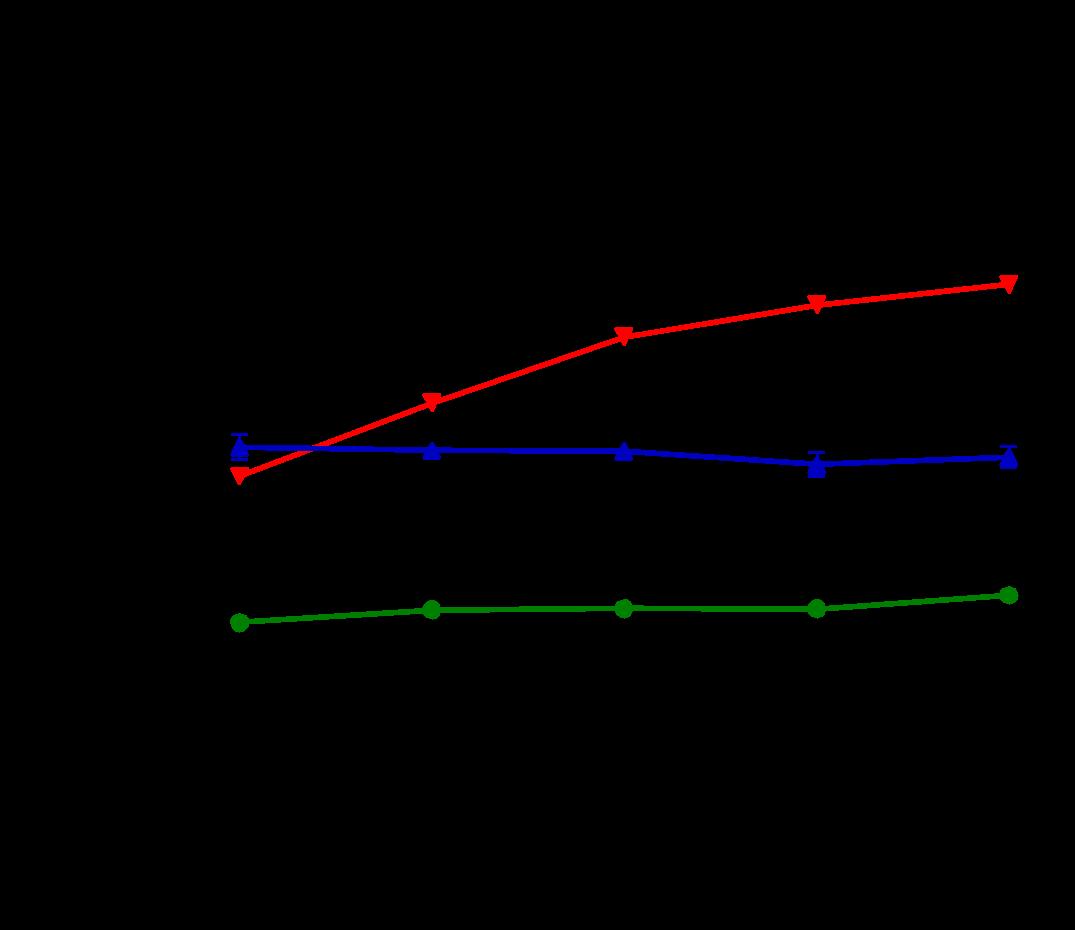
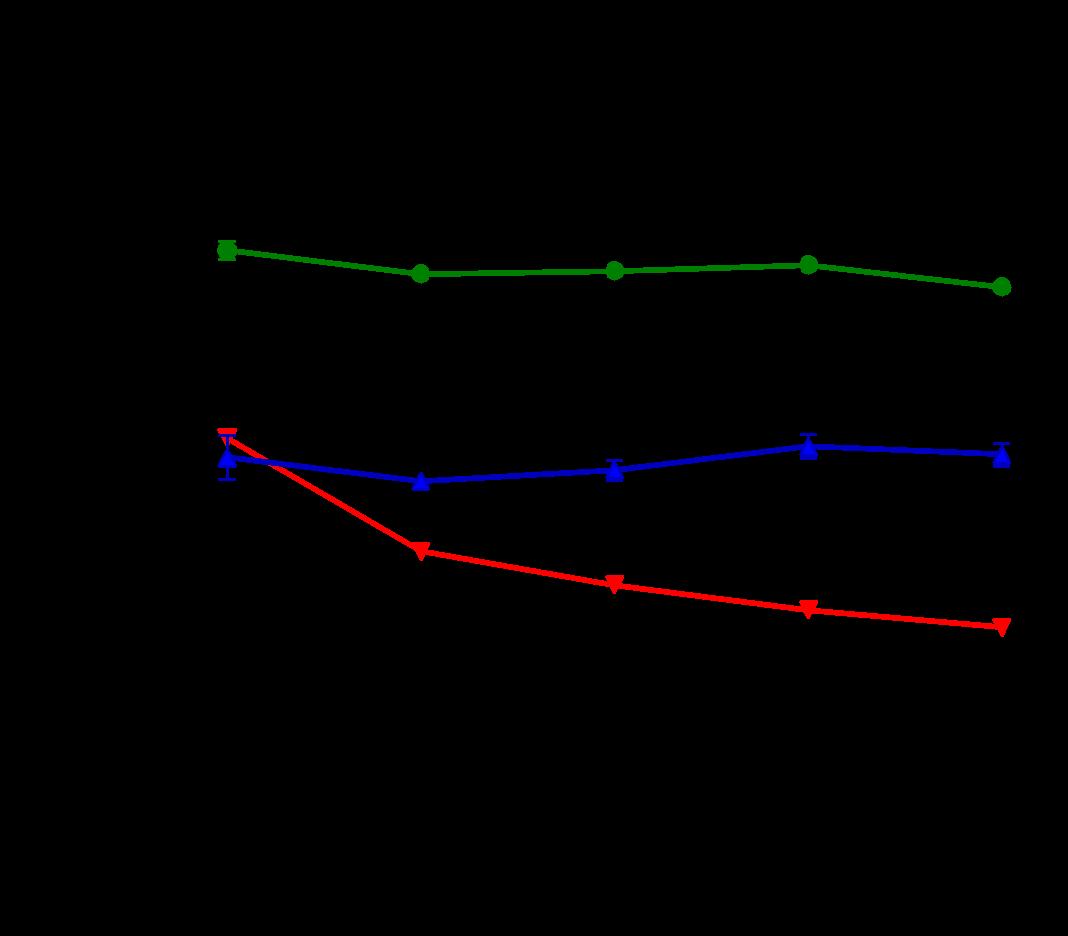
Figure 1. ERTU prevents left ventricular (LV) dilation, wall thinning, and systolic failure in α mice. Beginning at 4 weeks of age, wild type (WT) and cardiac-specific Gαq-overexpressing mice were randomized to chow formulated with ertugliflozin (ERTU; Merck; 0.5 mg/g of chow) or without ERTU (CD). Over the subsequent 16 weeks, Gαq over-expressing mice exhibited progressive LV dilation (Panels A and B) with LV wall thinning (Panel C) and a decrease in LV fractional shortening (Panel D) as assessed by echocardiography. Treatment with ERTU prevented LV dilation, wall thinning, and progressive systolic dysfunction. Data presented as mean ± SEM; n=10-16/group); * = p< 0.001 Gαq vs. Gαq ERTU. Black = WT CD, green = WT ERTU, red = Gαq CD, blue = Gαq ERTU.
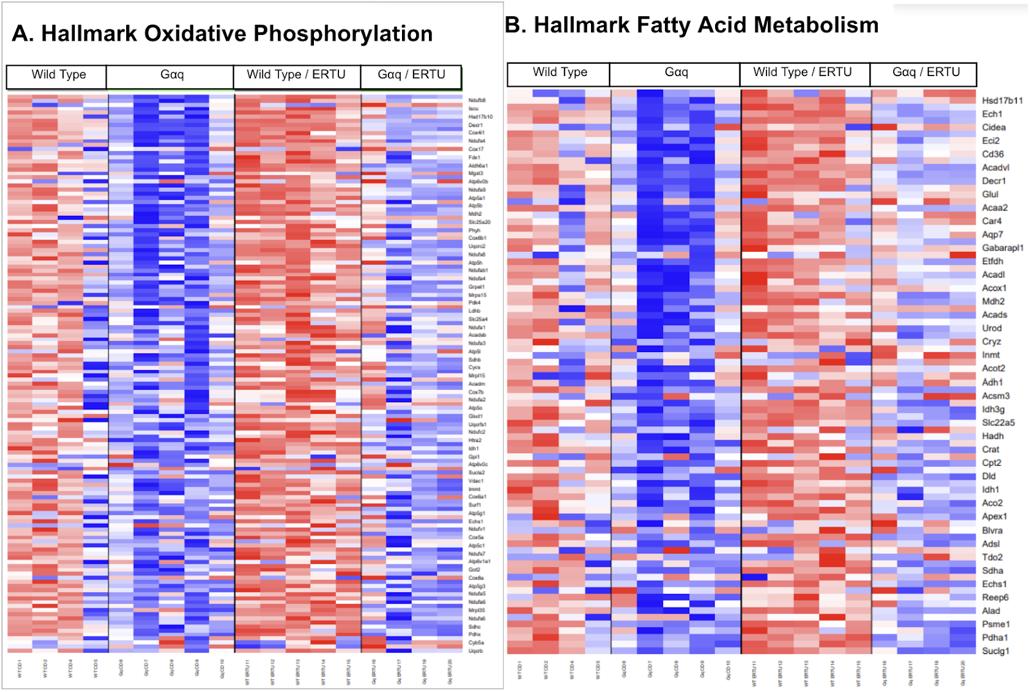
Figure 2. ERTU treatment up-regulates myocardial genes for oxidative phosphorylation and fatty acid metabolism. After 16 weeks of treatment with ERTU, 19 of the 20 most enriched GO gene sets were for mitochondrial structure and function (see also Table 1 for details). Hallmark GSEA showed that ERTU treatment markedly enriched gene sets for oxidative phosphorylation (OXPHOS; Panel A; NES 3.00, p < 0.0001, q < 0.0001) and fatty acid metabolism (FAO; Panel B; NES 2.61, p < 0.0001, q < 0.0001). The scale bar represents the z-scores of the expression values (i.e., the number of standard deviations from the mean of each row), with the maximum and minimum z-scores set to +2 (red) and -2 (blue), respectively (n=4-5/group).

Figure 3. ATP:ADP ratio is higher with ERTU in Gq-infected cells and control cells. Perceval is a construct created by taking a piece of a bacterial protein GLNK1 which is an energy sensor of ATP/ADP. The construct is created so it is attached to a circularly permutated fluorescence protein and when ATP and ADP bind the fusion protein change’s structure and leads to a differential color expression for each of the ATP and ADP. We use pseudo color to determine its range from blue to red. Gq QL is a mutation of Gq which leads to an active form of the protein by altering the active site of the protein. This allows the protein to constantly signal. After the addition of ERTU for 36 hours, there is in a change in energetics. ERTU leads to higher ATP/ADP ratio versus control cells (* p<0.05) and active Gq/ERTU cells also have higher energetics than its control. There was no difference between Active GQ cells versus control Cells. The more red indicates more ATP/ADP ratio trending to blue means less ATP:ADP ratio.

Figure 4. Oxidative stress does not change with ERTU. Ro GFP is a circularly permutated GFP that has been mutated so it is sensitive to changes in cellular oxidation state and similar to Perceval the conformational change can demonstrate the change from oxidation: reduction. This data shows that an SGLT2i does not change oxidative stress in a cardiac myocyte. The two presumed mechanisms of changes in ATP levels from ERTU has been thought to be a reduction in oxidative stress and an increase in mitochondrial calcium. We now show that neither is changed with an SGLT2i.

Figure 5. Inhibiting mitochondrial calcium does not change ERTU-induced energetic changes. Using a clone that inhibits calcium entry into the mitochondria, the Dominant negative mitochondrial calcium uniporter (DN MCU) does not change energetics. Lac-Z is a control protein (beta-galactosidase) that is expressed in cells while DNMCU is the mitochondrial uniporter that is rendered inactive by a point mutation in the protein, so the active site no longer works.
Discussion
SGLT2 inhibitors have been shown to modify several cardiac illnesses like myocardial infarction and severe hypertension. We now extend this finding to a model of progressive heart failure. We show several findings 1) SGLT2is can inhibit cardiac hypertrophy and the progression to heart failure 2) genetic changes occur that reprogram the heart back to utilization of oxidative phosphorylation/ fatty acids and 3) the drug seems to have a direct effect on the myocytes’ energy utilization which is not modulated by oxidative stress or mitochondrial calcium.
Our study presents new insights into the direct impact of ERTU on cardiomyocytes. The central finding from our experiments, as highlighted in Figure 1, is the significant alteration of ATP/ADP ratios in the presence of ERTU, independent of Gq overexpression. This suggests a mitochondrial modulatory role for ERTU, distinct from its established glucose-lowering effect in Type-2 diabetes. The results show that ERTU prevents LV dilation, wall thinning, and systolic failure.
ERTU affects myocardial remodeling in heart failure.
SGLT2 inhibitors have been shown to reverse adverse cardiac remodeling- left ventricular dilation, wall thinning, and reduction in systolic function.18 Gq is known to work via a distinct signaling pathway with activation of the mitochondrial dysfunction leading to LV dilation.19 ERTU can attenuate this adverse remodeling but not return it to control levels. Transcriptional analysis showed very little change in genes that would cause dilation like apoptosis, or wall thinning like hypertrophic or atrophic genes (data not shown). There were very structural changes that were present in transcriptional analysis (data not shown). RNAseq did, however, show marked changes in mitochondrial ATP production with the prevention of a metabolic switch to glycolysis. SGLT2is
has been shown to lead to substrate preference in other organs and can change the substrate utilization in the myocardium.20,21,22 Given the known changes in mitochondrial function that lead to LV dilation in the Gq model, it is reasonable to infer that this is a significant factor in the phenotypic changes seen. At the very least, the glycolytic switch was changed as noted by transcriptomics.
ERTU and Mitochondrial Energetics
In this study, the ATP to ADP ratio changed notably in cardiomyocytes treated with ERTU, a finding that stands out due to its statistical significance (p=0.5). The ratio is an indicator of cellular energy status, and its alteration suggests that ERTU directly affects mitochondrial function and directly affects the cardiac myocyte at physiologic levels (circulating levels in humans is ~50ng-300ng/ml.13 There is controversy about whether SGLT2 inhibitors can affect other organs and what the off-target may be in those tissues. Although the myocardium has sparse SGLT1 receptors it has minimal SGLT2 receptors.23 It has been speculated that it may affect the sodium hydrogen exchanger.24 It is likely the drug has other off-target effects as well. The change in myocardial energetics is consistent with other groups’ findings. The change in mitochondrial energetics, indicated by this change in ATP/ADP ratio, could be a pivotal factor in the cardioprotective effects observed with SGLT2 inhibitors. Several animal models (ACC knock-out mouse/ATPIF1 mouse) show that improving ATP production can prevent heart failure and left ventricular dilation. This direct influence on cellular energetics, as opposed to mere systemic glucose lowering, opens new pathways for understanding and potentially enhancing the therapeutic effects of drugs like ERTU in heart failure management.
Interestingly, our results showed no significant change in ATP/ADP ratios with Gq overexpression alone. This observation is
intriguing, considering Gq’s role in initiating stress response signaling and its association with heart failure progression. The fact that Gq overexpression did not alter ATP/ADP ratios in the same way as ERTU treatment suggests that the effects of ERTU are not mediated through the typical Gq signaling pathways. This could imply a more direct mitochondrial action of ERTU, independent of surface receptor-mediated pathways. This finding shows the complexity of signaling cascades and the need for further research in the context of heart failure.
The Role of Calcium and Oxidative Stress in Cardiac Energetics
Mitochondrial ATP production is felt to be modulated by oxidative stress as well as calcium. Several calcium-sensitive mitochondrial proteins can help increase ATP production. Although ERTU shows a trend toward an increase in mitochondrial calcium, inhibition of calcium entry shows no change in ATP. Furthermore, there is no change in oxidative stress. Since these two factors are typically closely linked to mitochondrial function and cardiac energetics, it is intriguing that there is an absence of significant changes in calcium dynamics and oxidative stress. Based on the data, it is entirely feasible that the increase in the ATP concentration is leading to increased calcium. The independence from these traditional pathways suggests that ERTU may be operating through a different mechanism. This could potentially involve direct modulation of mitochondrial enzymes or transporters, or alterations in substrate availability or utilization within the mitochondria.
Comparing Our Findings with Other Studies
Our findings diverge from existing literature that heavily implicates calcium in mitochondrial and cardiac function. Other research groups have highlighted the pivotal role of calcium in
mitochondrial energetics and cardiac function, often correlating changes in calcium levels with significant alterations in cardiac health.25 Our study adds a different perspective to this discourse, suggesting that while calcium is important, other mechanisms, possibly independent of calcium flux, can also significantly influence cardiac energetics. This suggests a need to re-evaluate the understanding of cardiac energetics, particularly in the context of heart failure and the therapeutic action of SGLT2 inhibitors. It points to the possibility that these drugs may exert their cardioprotective effects through mechanisms that are not yet fully understood or explored.
Challenges with Measuring ATP:ADP
The problem with only measuring the ATP to ADP ratio is that the ratio could change with both the creation and degradation of either. The ratio can increase with the phosphorylation of ADP, thus creating more ATP and lowering the amount of ADP. This is the first thing that comes to mind when seeing the increased ratio, it can also increase with the degradation of ADP or changes in nucleotide transport across mitochondrial membranes. To find the reason for the increase in the ATP to ADP ratio, further research should inhibit the electron transport chain (ETC). If even with the inhibition the ratio increases, then the ADP is most likely being degraded as the ETC creates most of the ATP. If with the inhibition the ATP to ADP ratio decreases, then ATP is most likely being produced.
Implications for Future Research and Therapeutic Strategies
Our study opens new avenues for research into the direct cardiac effects of SGLT2 inhibitors. The potential of ERTU to modulate mitochondrial energetics independently of traditional pathways like calcium signaling and oxidative stress could redefine
its therapeutic role. Future research should focus on finding the precise mechanisms through which ERTU influences cardiomyocyte energetics and exploring its potential in mitigating heart failure progression. Additionally, investigating the role of ERTU in different models of heart failure, and under various physiological and pathological conditions, could provide more comprehensive insights into its therapeutic potential. Furthermore, expanding the research to include human cardiomyocytes, in addition to animal models, could enhance the relevance and applicability of the findings. These insights could pave the way for developing new therapeutic strategies that target mitochondrial function directly, offering hope for more effective management of heart failure, particularly in patients with Type-2 diabetes where they would be able to treat two things at once.
References
1. Maron BA, Leopold JA. The Role of the Renin‐Angiotensin‐Aldosterone System in the Pathobiology of Pulmonary Arterial Hypertension (2013 Grover Conference Series). Pulm Circ. 2014;4(2):200-210. doi:10.1086/675984
2. O’Donnell J, Zeppenfeld D, McConnell E, Pena S, Nedergaard M. Norepinephrine: A Neuromodulator That Boosts the Function of Multiple Cell Types to Optimize CNS Performance. Neurochem Res. 2012;37(11):2496-2512. doi:10.1007/s11064-012-0818-x
3. Olokoba AB, Obateru OA, Olokoba LB. Type 2 Diabetes Mellitus: A Review of Current Trends. Oman Med J. 2012;27(4):269-273. doi:10.5001/omj.2012.68
4. Jenča D, Melenovský V, Stehlik J, et al. Heart failure after myocardial infarction: incidence and predictors. ESC Heart Fail. 2021;8(1):222-237. doi:10.1002/ehf2.13144
5. Marín-Peñalver JJ, Martín-Timón I, Sevillano-Collantes C, Cañizo-Gómez FJ del. Update on the treatment of type 2 diabetes mellitus. World J Diabetes. 2016;7(17):354. doi:10.4239/wjd.v7.i17.354
6. Ebong IA, Goff DC, Rodriguez CJ, Chen H, Bertoni AG. Mechanisms of heart failure in obesity. Obes Res Clin Pract. 2014;8(6):e540-e548. doi:10.1016/j.orcp.2013.12.005
7. Madsen KS, Kähler P, Kähler LKA, et al. Metformin and second- or third-generation sulphonylurea combination therapy for adults with type 2 diabetes mellitus. Cochrane Database of Systematic Reviews. Published online April 18, 2019. doi:10.1002/14651858.CD012368.pub2
8. Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev. 2018;98(4):2133-2223. doi:10.1152/physrev.00063.2017
9. Croteau D, Baka T, Young S, et al. SGLT2 inhibitor ertugliflozin decreases elevated intracellular sodium, and improves energetics and contractile function in diabetic cardiomyopathy. Biomedicine & Pharmacotherapy. 2023;160:114310. doi:10.1016/j.biopha.2023.114310
10. Wong CKH, Lau KTK, Tang EHM, et al. Cardiovascular benefits of SGLT2 inhibitors in type 2 diabetes, interaction with metformin and role of erythrocytosis: a self-controlled case series study. Cardiovasc Diabetol. 2022;21(1).
doi:10.1186/s12933-022-01520-w
11. Sharma R, Razda K, Kuhad A, Kuhad A. Ertugliflozin for the treatment of type 2 diabetes. Drugs of Today. 2019;55(3):167. doi:10.1358/dot.2019.55.3.2904972
12. Goodman JB, Qin F, Morgan RJ, et al. Redox-Resistant SERCA [Sarco(endo)plasmic Reticulum Calcium ATPase] Attenuates Oxidant-Stimulated Mitochondrial Calcium and Apoptosis in Cardiac Myocytes and Pressure Overload–Induced Myocardial Failure in Mice. Circulation. 2020;142(25):2459-2469.
doi:10.1161/CIRCULATIONAHA.120.048183
13. Tantama M, Martínez-François JR, Mongeon R, Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 2013;4(1):2550. doi:10.1038/ncomms3550
14. Hao J, Michalek C, Zhang W, Zhu M, Xu X, Mende U. Regulation of cardiomyocyte signaling by RGS proteins: Differential selectivity towards G proteins and susceptibility to regulation. J Mol Cell Cardiol. 2006;41(1):51-61.
doi:10.1016/j.yjmcc.2006.04.003
15. Sverdlov AL, Elezaby A, Qin F, et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet‐Induced Metabolic Heart
Disease. J Am Heart Assoc. 2016;5(1). doi:10.1161/JAHA.115.002555
16. Waypa GB, Marks JD, Guzy R, et al. Hypoxia Triggers Subcellular Compartmental Redox Signaling in Vascular Smooth Muscle Cells. Circ Res. 2010;106(3):526-535. doi:10.1161/CIRCRESAHA.109.206334
17. Yamamoto, T., & Sano, M. (2022). Deranged Myocardial Fatty Acid Metabolism in Heart Failure. International Journal of Molecular Sciences, 23(2), 996.
https://doi.org/10.3390/ijms23020996
18. Carluccio, E., Biagioli, P., Reboldi, G., Mengoni, A., Lauciello, R., Zuchi, C., D’Addario, S., Bardelli, G., & Ambrosio, G. (2023). Left ventricular remodeling response to SGLT2 inhibitors in heart failure: an updated meta-analysis of randomized controlled studies. Cardiovascular Diabetology, 22(1), 235. https://doi.org/10.1186/s12933-023-01970-w
19. Wang, J., Gareri, C., & Rockman, H. A. (2018). G-Protein–Coupled Receptors in Heart Disease. Circulation Research, 123(6), 716–735.
https://doi.org/10.1161/CIRCRESAHA.118.311403
20. Dutka, M., Bobiński, R., Ulman-Włodarz, I., Hajduga, M., Bujok, J., Pająk, C., & Ćwiertnia, M. (2021). Sodium glucose cotransporter 2 inhibitors: mechanisms of action in heart failure. Heart Failure Reviews, 26(3), 603–622. https://doi.org/10.1007/s10741-020-10041-1
21. Yaribeygi, H., Maleki, M., Jamialahmadi, T., Moallem, S. A., & Sahebkar, A. (2023). Hepatic benefits of sodium-glucose cotransporter 2 inhibitors in liver disorders. EXCLI Journal, 22, 403–414. https://doi.org/10.17179/excli2023-6022
22. Croteau, D., Luptak, I., Chambers, J. M., Hobai, I., Panagia, M., Pimentel, D. R., Siwik, D. A., Qin, F., & Colucci, W. S. (2021). Effects of Sodium‐Glucose Linked Transporter 2 Inhibition With Ertugliflozin on Mitochondrial Function,
Energetics, and Metabolic Gene Expression in the Presence and Absence of Diabetes Mellitus in Mice. Journal of the American Heart Association, 10(13).
https://doi.org/10.1161/JAHA.120.019995
23. Zhao, M., Li, N., & Zhou, H. (2023). SGLT1: A Potential Drug Target for Cardiovascular Disease. Drug Design, Development and Therapy, Volume 17, 2011–2023.
https://doi.org/10.2147/DDDT.S418321
24. Baartscheer, A., Schumacher, C. A., Wüst, R. C. I., Fiolet, J. W. T., Stienen, G. J. M., Coronel, R., & Zuurbier, C. J. (2017). Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia, 60(3), 568–573. https://doi.org/10.1007/s00125016-4134-x
25. Holmström KM, Pan X, Liu JC, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol. 2015;85:178-182. doi:10.1016/j.yjmcc.2015.05.022