
Robert Mulroy
Senior Thesis | 2024

Robert Mulroy
Dr. Krivacek
April 10, 2024
Characterizing Recruitment of CBP Binding Domains
Abstract The CREB-binding protein (CBP) is a cofactor (COF) responsible for the regulation of over 16,000 different genes in humans. CBP regulates gene expression by binding to transcription factors (TFs) to form TF-COF complexes. CBP binds to TFs at specific areas of the protein called binding domains. The recruitment of CBP by TFs is complex and not well understood. This research focused on 5 binding domains: CH1, CH3, IBID, NRID, and KIX. These domains were cloned via Gibson Assembly into a lentivirus plasmid to prepare them for use in a proteinbinding microarray (PBM). The PBM involves pairing the binding
domains with thousands of transcription factors and quantifying which TFs recruit which domains most effectively.
Background
Cells in the human body vary widely in form and function. Yet, from your blood cells to the lining of your stomach to the neurons in your brain, all of these cells contain exactly the same genetic information. So how does the same set of genes create such distinct cell types? The answer is gene regulation. There are many ways in which a cell can control which genes are expressed and which are not. Transcription factors (TFs) are proteins that bind to the promoter regions of genes to regulate gene expression. TFs can either promote or inhibit gene expression. However, genes are not simply regulated by one TF. Cofactors (COFs) are proteins that bind to the TFs to form TF-COF complexes, as shown in Figure 1. These cofactors can then bind to other segments of DNA, altering
the 3D structure of the DNA. This research was focused on one particular cofactor called the CREB Binding Protein (CBP).
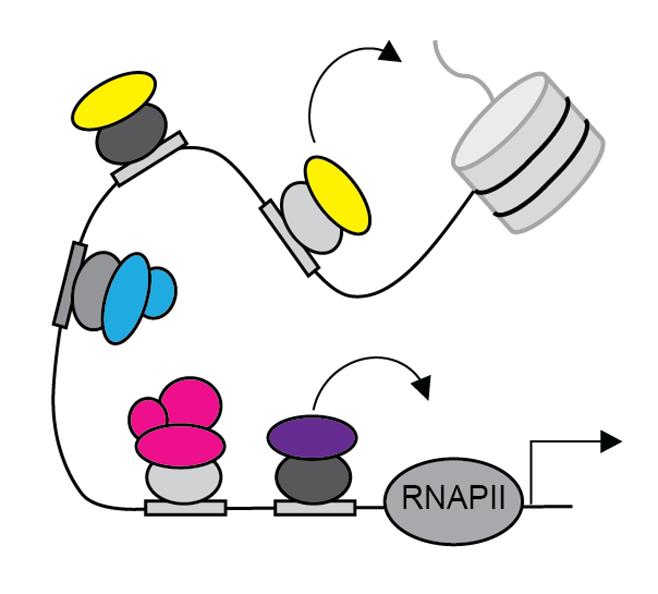
Figure 1. Cofactors, shown by the colored ovals, bind to transcription factors, shown by the gray ovals. These TF-COF complexes can then interact with other parts of the transcription machinery to regulate gene expression. The figure shows cofactors interacting with RNA polymerase II and a histone tail.
CBP is a cofactor that plays a vital role in the regulation of over 16,000 different genes in humans (Ramos et al., 2010). CBP operates via histone acetylation, which involves adding acetyl groups to the lysine residues of histone tails. These tails are positively charged, which generates an attractive force between them and the negatively charged DNA helix. This keeps the DNA tightly wrapped around the histones, making it difficult for RNA polymerase to access and transcribe the gene. When negatively charged acetyl groups are added to a histone tail, this attractive force decreases and eventually becomes repellent. This means the DNA is less tightly wrapped around the histones, allowing easier access for the RNA polymerase to begin gene expression (Loredana et al., 2006). Through this process, CBP is able to upregulate the expression of certain genes.
CBP is particularly important because of its role in the immune system. CBP interacts as a coactivator with a family of TFs called E2F that play a crucial role in the cell cycle. CBP also
interacts with the anaphase promoting complex/cyclosome (APC/C) which kicks off the transition from metaphase and anaphase during mitosis. Without CBP present, APC/C cannot perform its usual functions and in some cases, mitosis completely stops. Through these interactions as well as its primary histone acetylation function, CBP regulates several phases of the cell cycle. This makes CBP a proto-oncogene, a gene that has the potential to cause cancer. Proto-oncogenes usually regulate the cell cycle or cell growth, which means that if they have a mutation, they can cause cancer. CBP has been linked to every stage of tumor development. Overexpression of CBP is linked to many types of cancer, including lung cancer, head and neck cancer, acute leukemia, colorectal cancer, breast cancer, and prostate cancer. However, the exact mechanism with which CBP causes cancer is not fully understood. CBP is linked to many other conditions, including diabetes, Alzheimer's, schizophrenia, depression, and
Huntington’s disease, among others (Lee & Young, 2013; Kimura et al., 2022).
How and where does CBP bind? CBP has five different binding domains, the regions that actually interact with the transcription factors. These domains are CH1 (cysteine-histidinerich domain 1), CH3 (cysteine-histidine-rich domain 3), KIX (kinase inducible domain interacting domain), NRID (nuclear receptor interaction domain), and IBiD (interferon response factor binding domain) (Akinsiku et al., 2021). These five domains are highlighted in Figure 2, which shows the organization of CBP’s most important regions.
are the binding domains, the regions transcription factors

Unfortunately, the interactions between these domains and the TFs are very complex, as multiple different domains can be recruited by different TFs at the same time. CBP has a very flexible 3D structure which allows the domains to shift around relative to each other. This means there is no exclusivity; the same domain can bind to different TFs and the same TF can recruit different binding domains (Dyson and Wright, 2016).
Methods
The final goal of this project was to use a protein-binding microarray (PBM) to quantify the binding events between the five CBP binding domains and various TFs. However, the PBM requires a significant amount of preparation. First, PCR was used to create many copies of the CBP binding domain plasmids. Then, the recipient plasmids with 3x HA tags and 3x NLS tags were digested before the CBP domains were cloned into them using a Gibson assembly. Next, the domains were cloned with the tags into a lentivirus plasmid. Finally, the lentivirus cell nuclei were extracted and applied to a microarray. There are many additional steps along the way to confirm that reactions worked properly and to purify reaction products.
First, the 5 binding domains were amplified via PCR A separate 0.2 mL PCR tube was used for each of the five CBP binding domains and a sixth tube for a negative control. For each
domain, 1 μL of plasmid was mixed with 12.5 μL of Master Mix, 2.5 μL of forward primer, 2.5 μL of reverse primer, and 6.5 μL of water. The negative control tube contained the same volume of both primers and Master Mix but had an extra 1 μL of water instead of the plasmid.
Gel electrophoresis was used to verify the lengths of the PCR products, which have known lengths. First, the gel was made by pouring an agarose solution into a mold and letting it set. The solution was 1% agarose and 99% TAE buffer, as well as a few μL of gel dye. Next, the PCR products were prepared by combining them with DNA dye and water to dilute them. Finally, everything was added to the gel. Eight lanes were used: One for a 1000 bp ladder, one for a 100 bp ladder, one for a negative control, and five for the five CBP binding domains. Then, 85 volts were run through the gel for about 30 minutes, at which point the bands were clearly separated. The expected lengths for the CBP domains are 221 bp for IBiD, 344 bp for CH1, and 311 bp for NRID, KIX, and CH3.
The 100 bp ladder used had bands at 200, 300, and 400 base pairs, so the samples were compared to the bands to make sure they were the correct size.
Next, the plasmid was digested with restriction enzymes to linearize it. The miniTurbo plasmid with 3xHA and 3xNLS tags was added to a solution with EcoRL and NheI restriction enzymes, as well as a buffer. The reaction was placed in a 37° C water bath for two hours. It was then taken to 65 C to stop the restriction enzymes. After the digestion, the linearized vector and miniTurbo were still in the same solution together and needed to be separated.
The vector is much larger than the miniTurbo fragment, so gel electrophoresis was used to separate the two. When the gel was finished and there were two distinct bands, the larger one (the vector) was cut out using a razor blade. The slice of gel was placed in a small tube and weighed. A binding buffer was added at a ratio of 1 μL of buffer per 1 mg of gel. The gel mixture was incubated at 50° C for 10 minutes to dissolve the gel. The solution was moved
to a GeneJet purification column, which was then centrifuged for 60 seconds at 13,000 rpm. Next, 700 μL of a wash buffer was added and the column was centrifuged for another 90 seconds. The column was centrifuged for an additional 90 seconds to remove any leftover wash buffer. Finally, the column was transferred to a clean collection and tube 20 μL of water was added to elute DNA from the membrane. The tube was incubated for 5 minutes at 50°
C. The digested vector was then ready for the Gibson Assembly.
To perform the Gibson Assembly, the PCR product was added to 50 ng of the digested vector and 5 μL of a master mix.
5.96 ng of PCR product was used for IBiD, 9.27 ng was used for CH1, and 8.38 ng was used for CH3, NRID, and KIX. The solution was incubated in a thermal cycler at 50 C for one hour.
The next step was the transformation, which is used to put the finished vectors inside bacteria that can then be cultured to make many copies of the vector. To do this, 1 μL of ligase reaction was added to 10 μL of bacteria. The bacteria used was NEB® 5-
alpha chemically competent E. coli. The ligation product and bacteria solution was left on ice to incubate for 30 minutes. The bacteria were then placed in a 42° C water bath for exactly 30 seconds. After 30 seconds, the bacteria were quickly removed and placed on ice for 5 minutes. Next, 200 mL of S.O.C. media was added to the bacteria. The bacteria with S.O.C. media was placed in a shaker at 37° C for 1 hour to recover. Finally, the bacteria were plated. Each of the 5 samples was plated twice, one plate with 20 μL of bacteria and one with 180 μL. Depending on how efficient the Gibson and transformation were, the 20 μL plate may not be enough to grow or the 180 μL plate may overgrow, so having both gives a better chance of getting a usable plate. The plates were placed in a 37° C incubator for 19 hours. Bacterial colonies were picked from the plate to be grown in liquid cultures with carbenicillin. Figure 3 shows a summary of the Gibson Assembly process where the CBP domains are amplified via PCR
and the miniTurbo plasmid is digested before the domains are inserted into the plasmid.
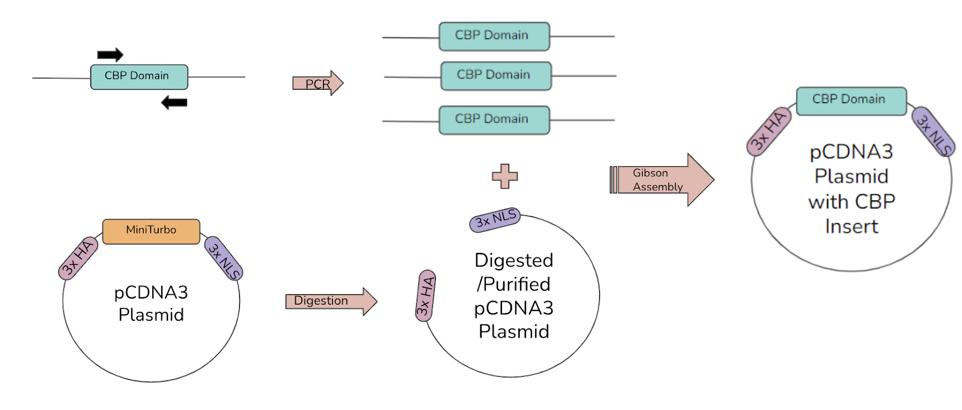
Figure 3. The first Gibson Assembly process. The five CBP domains are amplified by PCR, with the black arrow representing the forward and reverse primers. Then, the plasmid is digested to remove the miniTurbo, creating space for the insert. Finally, the linearized plasmid and CBP domain inserts are ligated via Gibson Assembly. The result is a plasmid with the CBP domain next to the 3xHA and 3xNLS tags.
The entire Gibson process was then repeated to get the CBP binding domains into a lentivirus vector. First, the plasmids had to be removed from the bacteria using a miniprep. The inserts, including the 3xHA and 3xNLS tags, were excised from the plasmid and copied via PCR. The plasmid this time is a lentivirus
which can later be used to infect mammalian cells. The plasmid was digested and the insert was cloned into it in the same way as before.
The miniprep was used to extract the plasmid DNA back from the bacteria in order to create more copies of the plasmid.
First, 5 mL of bacteria was pelleted in a centrifuge at 4000 rpm for 5 minutes. The pelleted bacteria were then resuspended in 250 μL of Buffer P1 from the QIAprep Spin Miniprep Kit. Then, 250 μL of Buffer P2 was added and the solution was mixed. Then, 350 μL of Buffer N3 was added and mixed. The solution was centrifuged for 10 minutes at 13,000 rpm (and all subsequent centrifugations were also carried out at 13,000 rpm). 800 μL of supernatant was pipetted into a QIAprep 2.0 spin column, avoiding the pellet. The solution was centrifuged for 60 seconds, pulling it through the membrane while the DNA got stuck. The flow-through was discarded. Next, 750 μL of Buffer PE was added, the spin column was centrifuged for 90 seconds, and the flow-through was
discarded. The column was centrifuged for another 90 seconds to remove any leftover wash buffer. The column was then left to sit for 3 minutes to allow any ethanol from the wash buffer that may be in the membrane to evaporate. Finally, the spin column with the membrane was transferred to a clean centrifuge tube to collect the DNA. 50 μL of water was added to the spin column, which was then centrifuged for 60 seconds (QIAprep Spin Miniprep Kit). The miniprep products were then amplified via PCR and a lentiviral plasmid was digested. Another Gibson Assembly was performed to insert the PCR product into the lentivirus, as shown in Figure 4.
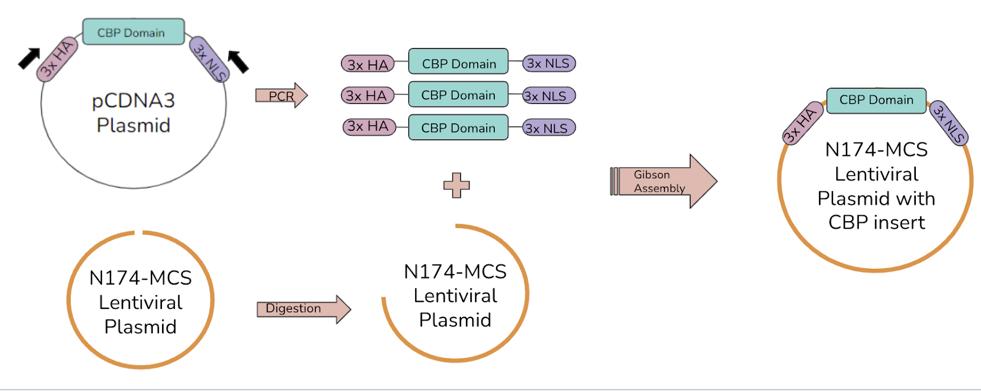
Figure 4. The second Gibson Assembly process. The five CBP domains, now with the 3xHA and 3xNLS tags, are amplified by PCR. The lentivirus plasmid is digested. The CBP domain inserts and linearized lentivirus plasmid are ligated to create a lentivirus that can be used to infect cells, which will then express the CBP domains.
Results
Gel electrophoresis was used to verify the results of the PCR reactions and plasmid digestions. Gel electrophoresis separates strands of DNA by their length. A DNA ladder that contains strands of known lengths is used as a comparison to experimental results.
Figure 5 shows the gel after the first PCR amplification of the CBP domains. The five domains have known lengths, so the lanes on the gel can be compared to the DNA ladder to confirm that the PCR products are the correct size. The far left lane contains the ladder. The next five lanes moving right contain negative controls for the five domains. The five rightmost lanes contain the actual PCR products. While all five domains are the correct size in this gel, note that there is a band in the CH3 negative control lane.
Also note in Figure 5 that the CH3 negative control lane has a band while the other 4 negative controls do not. The negative control lanes do not contain the plasmid. They only have the primers, master mix, and water, so there shouldn’t be any DNA that shows up. There were a lot of issues with CH3 in particular. Significant troubleshooting was required to test any possible sources of contamination.

Figure 5. Agarose gel of PCR-amplified inserts. The far left is a 100 base pair DNA ladder, with the lowest band at 100 bp and the highest at 1517 bp. The next 5 lanes are negative controls. The one labeled “-” that shows a band is the CH3 negative control. Lane 1 is IBiD (221 bp), 2 is NRID (311 bp), 3 is CH1 (344 bp), 4 is KIX (311 bp), and 5 is CH3 (311 bp).
Figure 6 shows the results of the first digestion of the plasmid. All six lanes contain different samples of the same vector. All six samples of the linearized vector matched the expected length, and the miniTurbo that was removed from the plasmid is clearly separated. Two different DNA ladders were used because
the two products are very different in size. The 5567 bp vector can be compared to the 1k bp ladder (in the far left lane) and the miniTurbo can be compared to the 100 bp ladder (in the second lane from the left).

Figure 6. Agarose gel of digested and purified vector. The far left lane has a 1k bp DNA ladder. The lowest band is 500 bp and the highest is 10,000. The vector is 5567 bp and the miniTurbo fragments are 771 bp.
The products of the first Gibson Assembly are shown in Figure 7. The leftmost lane contains the DNA ladder. All five of the ligated vectors in lanes 1-5 were the correct length.
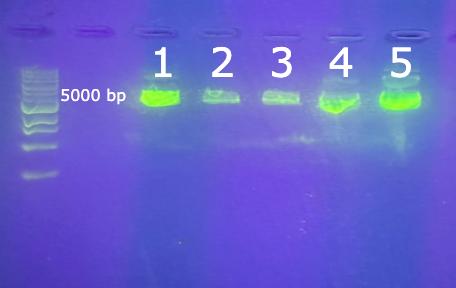
Figure 7. Agarose gel of the completed Gibson assembly. The leftmost lane has a 1k bp DNA ladder. The lane labeled 1 is IBiD (5738 bp), 2 is NRID (5828 bp), 3 is CH1 (5861 bp), 4 is KIX (5828 bp), and 5 is CH3 (5828 bp).
Discussion
Polymerase chain reaction (PCR) is a process used to create millions of copies of a certain segment of DNA. PCR utilizes primers to show the DNA polymerase where to start synthesizing the DNA. Primers are short sequences of DNA, usually 18 to 25 nucleotides long. These primers are specifically chosen to be complementary to the start of the DNA sequence being cloned. For
this experiment, the sequence was one of the CBP binding domains. Two different primers are used, the forward and reverse. This is because the DNA will split into two complementary strands. In order to clone both of them, the forward primer starts at the beginning of a strand and the reverse primer starts at the end of the complement. DNA polymerase can only synthesize them in the 3’ to 5’ direction, so starting at the beginning or end depending on the strand is crucial.
To perform the PCR, the binding domain plasmid, as well as forward and reverse primers are added to a master mix. The master mix contains the DNA polymerase, dNTPS (which are the nucleotides that DNA polymerase uses), and a buffer (PCR Master Mix). These components are combined in a tube and placed in a thermal cycler which can precisely control the temperature of the mixture. The PCR takes place in three steps which are repeated until the desired quantity of DNA copies is reached. These different steps are all initiated by the temperature of the thermal
cycler. First, the DNA is denatured at 98° C. The high heat causes the two complementary strands of DNA to separate from one another. Second is annealing at 69° C, in which the primers attach to the now separated template DNA strands. The final step is extension which occurs at 72° C. During extension, the DNA polymerase constructs the new copy of the template strand using the dNTPS. The two complementary template strands have now become four, meaning the quantity of the target DNA sequence doubles with each cycle (Wages Jr., 2005). This means that the quantity of copies grows very rapidly after only a few iterations, as seen in Figure 3. These three steps take about two minutes in total, so the thermal cycler was programmed to complete 35 cycles, giving millions of copies of each binding domain.
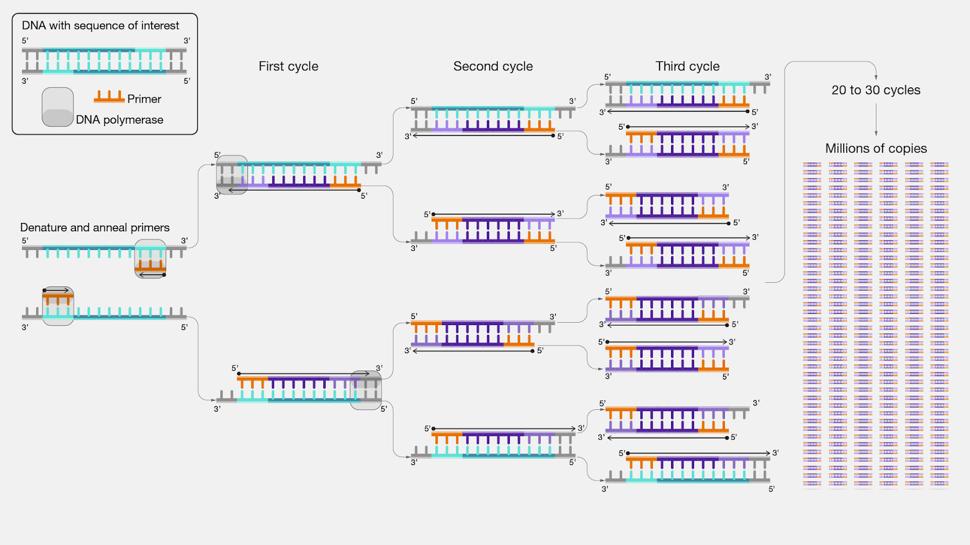
Figure 8. Polymerase chain reaction (PCR) allows for the rapid duplication of a certain sequence of DNA. The number of copies doubles with each cycle of the process performed, and this exponential growth means that millions of copies can be produced in about 30 cycles.
Along with PCR amplifying the five CBP binding domains, a negative control was also used that contained water instead of the CBP domain. While control experiments are always important, they are especially important for a PCR. Because of the exponential growth, any small error or contamination at the start will be massively amplified. The negative control can confirm that
the actual CBP plasmid is being copied and not a random sequence of DNA that accidentally got into the solution.
Gel electrophoresis was also used to check the results of the PCR. Gel electrophoresis uses the fact that DNA is negatively charged to separate DNA strands of different sizes. A gel is added to the DNA samples beforehand. The dye stains the DNA during electrophoresis, making it visible under UV light afterward. The DNA is then placed at one end of an agarose gel submerged in a buffer solution. An electrical current is run through the gel, pulling the negatively charged DNA towards the positive end. Smaller DNA strands move more quickly while larger strands move more slowly. The electrical current is stopped after a set amount of time, leaving the DNA separated based on size. A DNA ladder is used to judge the actual length of the strands. The ladder contains various DNA strands of known lengths, so the strands being tested can be compared to it. The strands in the ladder will be pulled through the gel in the same way as the other DNA, so looking at the band of
the ladder closest to the test sample can confirm whether it is the right length or not.
The central technique for this project was the Gibson Assembly, a method for assembling two different strands of DNA. The first step is to digest the plasmid to linearize it. Restriction enzymes are used to remove the miniTurbo, leaving the 3xHa and 3xNLS tags. The digested vector is then thoroughly purified to remove any impurities. The vector and the insert both have a matching sequence of 20 - 30 base pairs on either end. When the restriction enzymes cut the DNA, they leave sticky ends, which means that they cut the two complementary DNA strands a few base pairs apart, so that one of the strands overhangs the other.
This allows for an insert that has complementary sticky ends to be put into that gap in the vector, reconnecting the two ends, as seen in Figure 4.
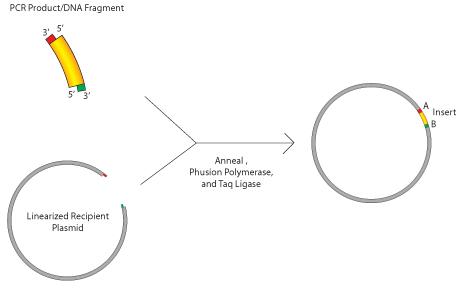
Figure 9. Gibson Assembly uses complementary sticky ends on the recipient vector and the insert to combine the two. Both the vector and insert had the green and red sequences which become the sticky ends after being cut.
For the first Gibson Assembly, EcoRL and NheI enzymes were used. They both have specific nucleotide sequences that they look for and cut. These two particular enzymes are used to remove the miniTurbo from the plasmid while leaving sticky ends for the Gibson Assembly. Then, the master mix puts the insert and vector together. The master mix contains three different enzymes that all work together to anneal the DNA. First, an exonuclease cuts back on the 5’ end of the overlapping region shared by the vector and
insert, creating an overhang. These complementary overhangs allow the vector and insert to anneal. Then DNA polymerase fills in any gaps in the newly formed vector before DNA ligase fixes any knicks in the DNA sequence (Lehman, 1974).
For the transformation, NEB® 5-alpha chemically competent E. coli was used. “Chemically competent” means that the bacteria have been treated to better accept foreign DNA. They are treated with calcium chloride which helps attach foreign DNA to the cell membrane (Chemically Competent Cells). Then, placing the bacteria in a hot water bath heat-shocks the bacteria, expanding the pores in their membranes and allowing the plasmids to enter. Finally, the bacteria is plated in S.O.C. media. S.O.C. media (Super Optimal broth with Catabolite suppression) is a solution that provides ideal conditions for E. Coli growth by providing the zbacteria with glucose. Extra precautions have to be taken while working with the S.O.C. media to make sure that everything stays
sterile, as even a tiny bit of bacteria could quickly grow and contaminate the entire plate or bottle (S.O.C. Medium).
The entire Gibson process now has to be repeated to insert the CBP bindings domains into a lentivirus plasmid. The reason for inserting the CBP domains into the other plasmid first instead of just putting them straight into the lentivirus is to add the 3xHA and 3xNLS tags. Once the lentivirus has infected a cell, these tags will cause the CBP domain to be expressed. The HA (human influenza hemagglutinin) tags bind the virus to the cell. The NLS (nuclear localization sequence) tags mark the insert to be brought into the cell nucleus (Giraud et al., 2014).
Before the second PCR, the CBP domains have to be removed from the bacteria via miniprep. First, the centrifuge causes the heavier bacteria cells to separate from the growth media and pellet at the bottom of the tube. Then, buffers P1 and P2 lyse the E. coli cells, meaning they break down the cell membranes. The solution is then centrifuged again to pellet all of the broken-
down cell membranes at the bottom of the tube, leaving the cell insides in the rest of the solution. The supernatant is then transferred to a QIAprep 2.0 spin column. The spin column is a centrifuge tube with a silica membrane in the middle that can bind and release DNA depending on the amount of chaotropic salt present. Buffer PE is added to the spin column and centrifuged and then water is added to the column and centrifuged. Buffer PE is the wash buffer that removes any extra contaminants caught in the silica membrane with the DNA. The water dilutes the concentration of chaotropic salts, eluting the DNA from the membrane. The next step for this project would be to infect mammalian cells with the lentivirus. Then, the cell nuclei can be extracted and used for the PBM to quantify thousands of binding events between the five CBP domains and various transcription factors.
References
Akinsiku OE, Soremekun OS, Soliman MES. (2021). Update and
Potential Opportunities in CBP [Cyclic Adenosine Monophosphate (cAMP) Response Element-Binding
Protein (CREB)-Binding Protein] Research Using Computational Techniques. Protein J. 40(1):19-27. doi: 10.1007/s10930-020-09951-8. Update and Potential
Opportunities in CBP [Cyclic Adenosine Monophosphate (cAMP) Response Element-Binding Protein (CREB)-
Binding Protein] Research Using Computational Techniques - PMC (nih.gov).
Berger MF, Bulyk ML. (2006). Protein binding microarrays (PBMs) for rapid, high-throughput characterization of the sequence specificities of DNA binding proteins. Methods Mol Biol. 338:245-60. doi: 10.1385/1-59745-097-9:245.
Protein Binding Microarrays (PBMs) for the Rapid, High-
Throughput Characterization of the Sequence Specificities of DNA Binding Proteins - PMC (nih.gov).
Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, Ting AY. (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 36(9):880-887. doi: 10.1038/nbt.4201. Efficient proximity labeling in living cells and organisms with TurboID - PubMed (nih.gov).
Bray D, Hook H, Zhao R, Keenan JL, Penvose A, Osayame Y, Mohaghegh N, Chen X, Parameswaran S, Kottyan LC, Weirauch MT, Siggers T. (2022). CASCADE: highthroughput characterization of regulatory complex binding altered by non-coding variants. Cell Genom. 2(2):100098. doi: 10.1016/j.xgen.2022.100098. PMID: 35252945; PMCID: PMC8896503.CASCADE: high-throughput characterization of regulatory complex binding altered by non-coding variants - PMC (nih.gov).
Chemically Competent Cells. Thermo Fisher Scientific.
Chemically Competent Cells | Thermo Fisher ScientificUS.
Chen Q, Yang B, Liu X, Zhang XD, Zhang L, Liu T. (2022).
Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics.12(11):4935-4948. doi: 10.7150/thno.73223.
Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agentsPMC (nih.gov).
Dyson HJ, Wright PE. (2016). Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J Biol Chem. 291(13):6714-22. doi: 10.1074/jbc.R115.692020. Role of Intrinsic Protein
Disorder in the Function and Interactions of the
Transcriptional Coactivators CREB-binding Protein (CBP) and p300 - PMC (nih.gov).
Fauquier, L., Azzag, K., Parra, M.A.M. et al. (2018).CBP and P300 regulate distinct gene networks required for human primary myoblast differentiation and muscle integrity. Sci Rep 8, 12629. CBP and P300 regulate distinct gene networks required for human primary myoblast differentiation and muscle integrity | Scientific Reports (nature.com).
Gibson D, Young L, Chuang RY, Venter J, Hutchinson III C, Smith H. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. doi: https://doi.org/10.1038/nmeth.1318. Enzymatic assembly of DNA molecules up to several hundred kilobases | Nature Methods.
Giraud G, Stadhouders R, Conidi A, Dekkers DH, Huylebroeck D, Demmers JA, Soler E, Grosveld FG. (2014). NLS-tagging:
an alternative strategy to tag nuclear proteins. Nucleic Acids Res. 42(21):e163. doi: 10.1093/nar/gku869. NLStagging: an alternative strategy to tag nuclear proteinsPMC (nih.gov).
Kimura M, Nishikawa K, Osawa Y, Imamura J, Yamaji K, Harada K, Yatsuhashi H, Murata K, Miura K, Tanaka A, Kanto T, Kohara M, Kamisawa T, Kimura K. (2022). Inhibition of CBP/β-catenin signaling ameliorated fibrosis in cholestatic liver disease. Hepatol Commun. 6(10):2732-2747. doi: 10.1002/hep4.2043. Inhibition of CBP/β-catenin signaling ameliorated fibrosis in cholestatic liver disease - PubMed (nih.gov).
Lee T, Young R. (2013). Transcriptional Regulation and Its Misregulation in Disease. Cell, 152(6). doi: https://doi.org/10.1016/j.cell.2013.02.014. Transcriptional Regulation and Its Misregulation in Disease: Cell.
Lehman IR. (1974) DNA ligase: structure, mechanism, and function. Science. 186(4166):790-7. doi: 10.1126/science.186.4166.790. DNA ligase: structure, mechanism, and function - PubMed (nih.gov).
Loredana Verdone, Eleonora Agricola, Micaela Caserta, Ernesto Di Mauro. (2006). Histone acetylation in gene regulation. Briefings in Functional Genomics, 5(3), 209–221, doi:https://doi-org.ezproxy.bu.edu/10.1093/bfgp/ell028.
Histone acetylation in gene regulation | Briefings in Functional Genomics | Oxford Academic (bu.edu).
Ngan V, Richard G. (2001). CREB-binding Protein and p300 in Transcriptional Regulation. Journal of Biological Chemistry, 276(17), 13505-13508. doi: https://doi.org/10.1074/jbc.R000025200. CREB-binding Protein and p300 in Transcriptional RegulationScienceDirect (bu.edu).
PCR Master Mix. Sigma-Aldrich. PCR Master Mix (sigmaaldrich.com).
QIAprep Spin Miniprep Kit. QIAGEN. QIAprep Spin Miniprep Kit (qiagen.com).
Ramos YF, Hestand MS, Verlaan M, Krabbendam E, Ariyurek Y, van Galen M, van Dam H, van Ommen GJ, den Dunnen JT, Zantema A, 't Hoen PA. (2010). Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res. 38(16):5396-408. doi: 10.1093/nar/gkq184. Epub 2010 Apr 30. PMID: 20435671; PMCID: PMC2938195. Genome-wide assessment of differential roles for p300 and CBP in transcription regulation - PMC (nih.gov).
S.O.C. Medium. Thermo Fisher Scientific. S.O.C. Medium (thermofisher.com).
Wages Jr. JM. (2005). POLYMERASE CHAIN REACTION. Encyclopedia of Analytical Science, 2, 243-250. doi:
https://doi.org/10.1016/B0-12-369397-7/00475-1.
CHAIN REACTION - ScienceDirect (bu.edu).