33 minute read
Characterizing the Behavioral and Cellular Effects of the R904S Variant of OPA1 as a Tourette Disorder Probable Risk Gene


Kinza Abbas1† and Cara Nasello2
¹Geisinger Commonwealth School of Medicine, Scranton, PA 18509 ²Rutgers University, Piscataway, NJ 08854 †Doctor of Medicine Program Correspondence: kabbas@som.geisinger.edu
Abstract
Tourette disorder (TD) is a heritable neuropsychiatric and developmental disorder characterized by motor and vocal tics. TD has a complex heterogenous etiology which makes the identification of genes linked to the disorder challenging. However, previous studies have identified optic atrophy 1 (OPA1) as a probable risk gene for Tourette disorder through whole exome sequencing analyses of de novo mutations. The OPA1 protein localizes to the inner membrane of mitochondria in cells throughout the body, where it regulates mitochondrial morphology and function. The Tischfield lab has identified a TD patient with an OPA1 missense mutation, R904S, which we hypothesized would result in abnormal behavioral activity in mutant mice and/or affect the cellular function of mitochondria. We utilized several behavioral paradigms, including motion sequencing, the open field arena test, the marble burying task, and prepulse inhibition. These paradigms test various factors, such as behavioral dynamics, motor activity levels, and sensorimotor gating. To understand the cellular characterization, we observed whole brain expression of the protein, protein expression levels in different brain regions and mitochondria of cortical neurons. Preliminary results of mitochondria imaging show potential mitochondrial aggregates in heterozygous and homozygous mutant cortical neurons, which could indicate an increased preapoptotic state of mutant cells. Although the results of the remaining assays have been inconclusive, there are many variables that complicate the studies. For a better understanding of the variant, mitochondrial function should be tested in the future, such as oxidative phosphorylation activity, cytochrome c levels and reactive oxygen species levels. Cytochrome c levels and apoptosis assays especially should be conducted to better understand the nature of the observed mitochondrial aggregates. Identifying possible behavioral or cellular abnormalities of this variant is an important step in characterizing the mutation, determining its potential role in TD pathology, and developing potential clinical treatments for TD in the future.
Introduction
Tourette disorder Tourette disorder (TD) is a heritable neuropsychiatric and developmental disorder characterized by waxing and waning motor and phonic tics, which are defined by the DSM-V as sudden, rapid, recurrent, semi-voluntary, and nonrhythmic motor movements or vocalizations. The condition is three to four times more likely to occur in males than females and according to the Centers for Disease Control and Prevention, TD diagnosis requires a person to exhibit at least two motor tics, at least one vocal tic and experience persistent symptoms for at least 1 year, with an onset before the age of 18 (1). Therefore, although TD is a tic disorder, not all tic disorders can be classified as TD.
The disorder is often comorbid with other neuropsychiatric disorders — most commonly obsessive-compulsive disorder (OCD) and attention deficit hyperactivity disorder (ADHD). However, autism spectrum disorders (ASD), depression, and anxiety disorders may also be present. This overlap in disorders implies a shared neurological background and a genetic influence. Additionally, family and twin studies have established a strong genetic influence behind TD (2). In studies of premonitory urges, which are often experienced by TD patients, it is reported that these urges were more bothersome than tics, allowed them to better suppress their tics and that their tics were a response to these urges (3). Altogether, these findings demonstrate the complexity of TD etiology, which appears to involve a combination of psychological, neurological, and motor circuitry. This makes it challenging to pinpoint exact pathways or genes contributing to the disease.
Optic atrophy 1 (OPA1) Previous whole-exome sequencing studies have found significant evidence for de novo gene-disrupting variants in TD (4). This study was expanded and optic atrophy 1 (OPA1) was specifically identified as a de novo probable risk TD gene (5). It has been heavily studied for its role in dominant optic atrophy (DOA), which is a neuro-ophthalmic disorder characterized by degeneration of the optic nerve. The OPA1 protein is a dynamin GTPase that mediates inner membrane fusion and enzymatic reactions for membrane remodeling, which in turn regulate mitochondria energetics. A major feature of the inner membrane is the cristae, which are invaginations of the membrane that have important proteins embedded, such as the electron transport chain complexes. OPA1 proteins oligomerize in the cristae to maintain cristae tightness and to function as a regulatory gate. OPA1 proteins also complex between inner membranes of fusing mitochondria to carry out inner membrane fusion (6). Upon apoptosis signaling, OPA1 oligomerization alters cristae structure to allow cytochrome c release (7). The protein’s role in membrane dynamics in turn impacts mitochondrial bioenergetics and general functionality. Mitochondrial dynamics, or the balance between fission and fusion, mediates mitochondrial quality control (MQC). The organelle maintains homeostasis by enforcing the following controls: (a) shifting morphology between filamentous or fragmented according to the energetic demands of the cell, (b) fusing dysfunctional mitochondria to mitigate damage, (c) mitochondria fragmentation, membrane permeability and cytochrome c release signal apoptosis (8).
Although OPA1 is more prominently expressed in the retina and brain, the protein is ubiquitously expressed and well conserved. In fact, OPA1 knockout mice are not viable. The gene encodes 30 coding exons and consists of over 90 kb of DNA on chromosome 3 (9). It is translated in the cytosol after which it is imported into the mitochondria, where it undergoes proteolytic cleavage. The long OPA1-L isoform remains anchored to the inner membrane while the short OPA1-S isoform is released to the inner membrane space. A balance of the two isoforms is required for proper mitochondrial functioning (6).
The R904S variant of OPA1
A TD patient with the OPA1 mutation, R904S, in which there is an arginine conversion to serine at position 904, has previously been identified by the Tischfield laboratory. The amino acid conversion results from a single nucleotide missense mutation and is likely gene disrupting. Arginine is basic, hydrophilic, charged, and large, while serine has a hydroxyl chain group, is neutral, uncharged, and has a small side chain. The gene has several functional regions (9), including a mitochondrial import sequence (MIS) and three dynamin family conserved regions: a GTPase domain, a middle domain, and a GTPase effector domain (GED) which contains a coiled-coil domain (CC2). As previously stated, the gene comprises of 30 coding exons. The R904S mutation is found in exon 27, which is part of the CC2 domain that stretches through exons 27-28. The domain participates in protein-protein interactions; thus, dysfunction of this region may alter the protein’s ability to form complexes (9).
Hypothesis Tourette disorder is not well studied, and due to its complex etiology, it is also difficult to study. With this project, we anticipated on identifying a behavioral or cellular assay for TD with the variant, which would be the first TD model with OPA1. We hypothesized that the R904S variant of the TD risk gene, OPA1, would result in behavioral or cellular abnormalities as compared to wildtype mice/cells. To test this, we employed a mouse model of the mutation generated with CRISPR technology. We utilized motion sequencing, marble burying task, open field arena test, and prepulse inhibition as our behavioral testing paradigms. After behavioral testing, we performed western blots to determine whether there were any significant differences in the level of whole brain OPA1 protein expression between mutant and wildtype mice. We then stained brain slices for OPA1 to determine localization and expression patterns of the mutant protein as compared to the wildtype in various brain regions. Lastly, we stained cortical neurons for mitochondria to determine localization, size and shape of mutant mitochondria. As there are several mitochondrial functions that can be impacted by the OPA1 protein, we began with broad, qualitative cellular tests to determine if there were detectable mitochondrial defects that could potentially pinpoint the mechanism of the mutation. In this way, we could test mitochondrial function according to the mechanism observed.
Behavioral paradigms Motion sequencing Motion sequencing (Moseq) utilizes the Microsoft Xbox 360 Kinect camera, which acquires spatial and temporal data of moving mice using 3D infrared imaging. This allows more detailed characterization of behavior modules and replaces tedious and unreliable human scoring of behavior. Although the system still relies on human perception and intuition, as the initial quantification of behaviors is specified by human observers, this method offers an improved methodology. Using this data, behaviors can be deconstructed into “syllables,” or basic motions, such as a head turn, a body turn, or a pause. The Moseq approach captures pose dynamics of mice — data which are then used to identify behavioral motifs. Computational models can recognize and learn these behavioral modules so comparison to altered structure of behavior after environmental, genetic or neural manipulation can be made (10, 11). Open field The open field chamber is square-shaped and wall-enclosed, separated into an outer and inner zone. Invisible infrared beams are emitted from photocell receptors along the perimeter of the chamber and a computer analyzer can record and analyze beam breaks resulting from subject movement. Numerous motor activity variables can be scored including total ambulatory distance, latency to enter the center and rearing. Latency to enter the center and rearing are considered exploratory behaviors and can be used to measure anxiety (12). Along with anxiety, the open field parameters can also be observed to identify differences in motor activity between subjects and genotypes. As OPA1 is a mitochondrial protein, deficits in the protein function could disrupt mitochondrial function, which could result in increased/decreased motor function of mice.
Marble-burying task The marble-burying test utilizes the natural inclination of mice to dig in natural settings. A greater number of buried marbles indicates greater burrowing and digging activity. Altogether, this test is useful in studying overall mouse activity, and more specifically, obsessive and compulsive behavior (13).
Prepulse inhibition Patients with Tourette disorder exhibit deficits in sensorimotor gating, which is tested by prepulse inhibition (PPI). Sensorimotor gating is a mechanism for filtering information deemed irrelevant to the brain, such as redundant stimuli. This mechanism prevents sensory overload from constant input of unnecessary information. PPI measures sensorimotor gating by measuring startle reflex, which is an involuntary contraction elicited by unexpected stimuli. On average, the subject should have less of a startle response, or greater inhibition, to the primary pulse in the presence of a prepulse stimulus (and increasing inhibition with increasing magnitude of the prepulse stimulus). This would show evidence of functioning sensorimotor gating, which is deficient in TD patients (14). It has also been suggested that premonitory urges can indirectly be assessed by PPI. This is crucial as premonitory urges are a major psychological aspect of TD (15).
Methods
Animals
Our animal protocol was approved by IACUC at Rutgers University, New Brunswick. A total of 224 mice were bred during the span of the project, with 110 males and 114 females.
Fifty-eight mice were wildtypes, 113 were heterozygotes and 38 were homozygous mutants. Approximately 2-month-old C57BL/6 mice were used for the behavioral assays which were run during the morning. The mice were brought into the behavioral annex at least 48 hours before testing to allow environmental acclimation. The tests were run at least 24 hours apart with prepulse inhibition run as the last test, as it induces hearing impairment and increased stress for mice. Mice were allowed to acclimate in the testing room for 15 minutes prior to being tested.
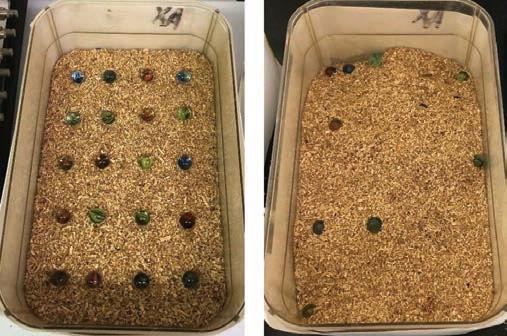
Behavioral testing Motion sequencing (Moseq) The Moseq test was administered by placing mice into circular, opaque black containers with the Microsoft XBox Kinect camera attached above. Mice were filmed for 20 minutes, after which another team obtained and interpreted the footage to extract quantifiable data. Containers were wiped down with bleach, alconox, and 70% ethanol to remove feces, urine, and scents. Marble-burying task The marble-burying task protocol (13) consisted of 20 marbles being uniformly placed in a 5 x 4 pattern on 2 inches of unscented bedding in a 8 in x 13 in housing cage (Figure 1 left). The sides of the cage were covered with paper to prevent distractions from surroundings. Mice were placed in the testing cage for 30-minute sessions (Figure 1 right depicts post-test cage), and the number of marbles buried during the 30-minute session were counted and averaged by three observers. Marbles were considered buried if two-thirds or more of the marble was buried. New bedding was used for each mouse, and marbles were washed with ethanol between different testing sessions to remove feces, urine, and scents. Open field test Mice were placed at the periphery of the open field arena so the data for their interaction with the center of the field would not be skewed. Each mouse was tested for 20-minute sessions. The testing arena was wiped with 70% ethanol between mice to remove feces, urine, and scents.
Figure 1. The left image shows a marble-burying task setup prior to placing the mouse in for testing. Marbles were arranged atop unscented bedding in a 5 x 4 arrangement. The right image depicts a cage after a 30-minute session. The number of buried marbles were then counted by 3 observers and averaged. PPI PPI was used to determine sensorimotor gating deficits using an established protocol involving the SR-LAB system (14). This setup consists of a chamber that administers the auditory stimulus and a stabilimeter placed inside the chamber. The stabilimeter is a plexiglass cylinder in which the mice are held, mounted on a piezoelectric accelerometer that records the movements. A continuous background noise level of 65 dB was maintained to standardize measurements, reduce external noises, and allow comparisons with other literature. The testing session is separated into 4 blocks, each of which presents a different pattern of stimuli. The first and last blocks present stimuli of 120 dB alone for the purpose of calculating habituation throughout the session. Blocks 2 and 3 consist of three different trials presented in a randomized order, including a pulse-alone trial, a prepulse + pulse trial, and a no stimulation (NOSTIM) trial (although the subject’s movements are still recorded during the NOSTIM trial). The pulse-alone trials present a 40 msec stimuli of 120 dB while the prepulse + pulse trials consist of 20 msec prepulses presented before the primary pulse stimulus (the time interval between the two pulses randomly varies between 30 and 500 msec for each trial). The prepulse + pulse trials can also randomly vary in the magnitudes of prepulse used, and in this case, magnitudes of 6, 12, and 16 dB were used (magnitudes of 71, 77, and 81 dB were delivered as the prepulses have to be 6, 12, and 16 dB above the background noise of 65 dB). The percent PPI was calculated for each prepulse intensity using the equation %PPI = 100 × [(pulse-alone) – (prepulse + pulse score)]/pulse-alone score.
Data analysis
GraphPad Prism software was utilized to conduct data analysis. Two-way analysis of variance (ANOVA) SD was used to determine the statistical differences between testing groups.
Cellular testing Restriction digest to confirm genotypes of mice To acquire mouse genotypes, tissue was collected from the ear during ear tagging. DNA from these tissues was extracted by incubating the tissue in the Sigma-Aldrich tissue extraction solution (Sigma-Aldrich). A PCR reaction was then performed to amplify the obtained DNA, followed by a restriction digest reaction with Taq1 restriction enzyme (Thermofisher). This enzyme recognizes and cleaves after an AGC site, allowing for cleavage of the mutant variant which alters an arginine (AGG/ AGA) to a serine (AGC). Western blot to determine OPA1 localization and whole brain levels Mice were first dissected and whole brains were extracted, from which the mitochondria were then isolated using the Abcam Mitochondria Isolation Kit for Tissue (Abcam). A Pierce BCA Protein Assay Kit (Thermofisher) was then utilized to determine protein concentration in both our separated mitochondrial fragment and mitochondrial excluded fragment samples. This protein concentration was needed to calculate and normalize the amount of protein loaded onto a 4–12% Bis-Tris protein gel for each sample. Equal amount of protein loading was crucial to allow comparison between the protein samples.
Samples were then run with gel electrophoresis followed by an electrotransfer of the gel onto polyvinylidene fluoride membrane (PVDF). As three proteins were being identified, OPA1, Hsp60, and GAPDH, the membrane was cut according to the size of each protein. The GAPDH monomer that the antibody recognizes is 36 kDa, Hsp60 is 60 kDa and the two OPA1 peptides are 80–100 kDA. The protein sizes were varying enough to allow us to section the membrane into three to incubate each section with its corresponding antibody. For GAPDH, we used the monoclonal Anti-GAPDH HRP conjugated antibody (Abcam), for Hsp60, the polyclonal antiHsp60 antibody (Abcam) was utilized and for OPA1, we utilized a polyclonal OPA1 antibody (NovusBio). For the secondary antibody, the HRP conjugated monoclonal secondary antibody (Genscript) was utilized for Hsp60 and OPA1. No secondary antibody was required for GAPDH as the primary antibody was already conjugated with HRP. After antibody incubation, the membranes were visualized using the Western Blot Detection Kit (KwikQuant), which uses an enhanced chemiluminescence (ECL) substrate for HRP detection. OPA1 and nuclear staining of whole brain slices To visualize the localization of the R904S variant protein in various brain regions, we obtained brains from mice, sliced them coronally, and stained them with an OPA1 primary antibody (NovusBio). To obtain the brain, we performed a perfusion fixation. The mice were first anesthetized, after which the thoracic cavity was opened to expose the still-beating heart. A saline solution was streamed through the vascular system by inserting a needle into the left ventricle, followed by 4% paraformaldehyde as the fixative solution. Efficient fixation was indicated by a cleared liver color. After fixing, the brains of the mice were extracted and placed in fixation solution for maintenance. The Vibratome was used to slice the brains into coronal sections. The brain slices were then incubated in 0.3% 1X phosphate-buffered saline, 0.1% Tween (PBST) for 10 minutes to permeabilize the slices, followed by a 45-minute incubation in 2% bovine serum albumin (BSA) in 0.1% PBST to block nonspecific antibody binding. Brain slices were then incubated in rabbit polyclonal anti-OPA1 primary antibody (NovusBio) overnight, washed 5 x 5 min in 0.5% BSA in 1X PBS, incubated in goat anti-rabbit 555 fluorescent secondary antibody (Thermofisher) for 1 hour, washed 5 x 5 min in 0.5% BSA in 1X PBS, and again washed 5 x 5 min in 1X PBS. Finally, the slices were incubated with blue Hoechst dye (Invitrogen) for 5 min and washed 3 x 5 min in 1X PBS to stain nuclei. Fluoromount-G mounting medium (Thermofisher) was then used to mount the brain slices onto glass microscope slides for visualization under a fluorescence microscope.
MitoTracker staining in cortical neurons P2 mice were dissected and their brain cortices were extracted. The Pierce Primary Neuron Isolation Kit protocol (Thermofisher) was utilized to enzymatically digest the brain tissue with papain. Cells were counted using a Vi-CELL cell counter and a density of 5 x 105 cells/cm² were plated in a 24well plate with glass coverslips placed in the wells. Neuronal cell culture medium and supplemental medium were applied every 2 to 3 days to reduce non-neuronal cell contamination and promote neuron purity. On approximately day 9, the wells were incubated with MitoTracker Red (Thermofisher) for 30 minutes. The cells were then fixed with a solution of 3% formaldehyde, after which the cells were incubated in Hoechst dye (Invitrogen) for 5 minutes and washed 3 x 5 min with 1X PBS. The glass coverslips were then mounted on glass microscope slides using Fluoromount-G Mounting Medium (Thermofisher) and sealed with clear nail polish. The neurons were then visualized under a fluorescence and confocal microscope.
Results
Mendelian ratio births
As previously stated, TD is 3 to 4 times more likely to occur in males than in females. For this reason, the sex and genotypes of mice were noted as they were born to track whether the mice were born according to Mendelian ratios (Table 1). Aberrant ratios of births of mutant mice would indicate a possible lack of viability or a genetic predisposition. There was no statistically significant difference in the number of mice born between sexes and genotypes.
Behavioral paradigms PPI PPI tests startle response and sensorimotor gating. Subjects startle when presented a loud sound but should startle less if that loud sound is preceded by another sound (a prepulse), which is a concept referred to as sensorimotor gating. The louder the preceding sound is, the lesser the startle response there should be to the main sound. PPI is a TD paradigm as TD patients show a deficiency in sensorimotor gating ability. Thus, we tested PPI as a TD behavioral paradigm. However, there were no significant differences in the percent of prepulse inhibition between the mutant and wildtype mice according to ANOVA analysis (Figure 2). Open field There are several parameters that can be tested with the open field test but only the results for time spent in the vertical (rearing) (Figure 3), total distance traveled (Figure 4), and count of central zone entries (Figure 5) are discussed. The total ambulatory distance covered by the mice can provide insight on the activity levels of mice while rearing behavior and count of central zone entries can be indicative of anxiety-related behavior (wall-hugging behavior or remaining in the periphery suggests higher anxiety levels in mice). Total ambulatory distance is measured in centimeters and tracks the total distance traveled by the mouse. The results for distance traveled and count of central zone entries are also not significantly different with ANOVA analysis, indicating that heterozygous and homozygous mutant mice do not perform differently as compared to wildtype mice in these paradigms. Marble-burying task Mice have a natural inclination to burrow in their environment, which is taken advantage of in the marble burying task. Increased number of buried marbles could indicate increased activity levels, repetitive/obsessive behavior, and/or anxietyrelated behavior. However, the marble-burying task showed no statistically significant differences in the performance of the heterozygous and homozygous mutant mice as compared to the wildtype mice according to ANOVA analysis.
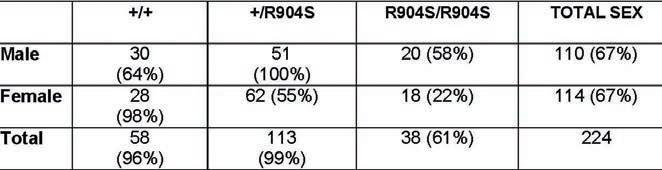
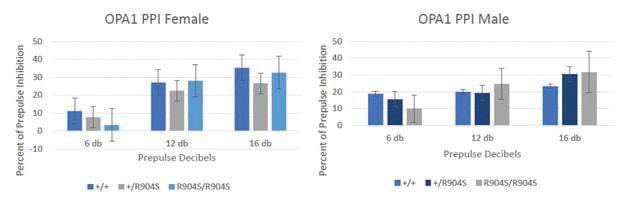
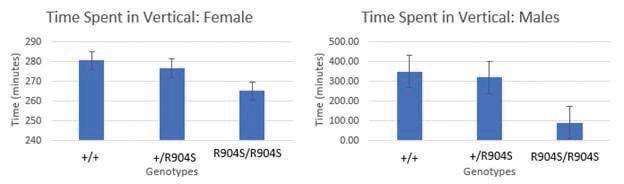
Table 1. Chart displaying number of births for each genotype and sex. Number in parenthesis denotes the percent that births are by chance and following Mendelian ratios. Discrepancy in the total number of births per sex is due to pups that have not been genotyped yet. Average litter size is 7.2.
Figure 2. Graphs depict prepulse inhibition of subjects. Mice should startle less with increasing decibels of the prepulse administered. Thus, there should be a greater percent of prepulse inhibition with increasing decibels of prepulse — as is shown. There was no significant difference in the prepulse inhibition between genotypes for both males and females. This indicates that mutant mice do not show a deficiency in sensorimotor gating (Females: (F[2,81]=.581, p=.561) +/+ n=10, +/R904S n=14, R904S/R904S n=9; Males: +/+ n=14, +/R904S n=15, R904S/R904S n=7).
Figure 3. Graphs depict the amount of time (in minutes) that the subjects spent in vertical, or the amount of time they spent reared up. There was no significant difference in the time spent in the vertical between the genotypes for both males and females (Females: (F[2,41]=.078, p=.925) +/+ n=14, +/R904S n=21, R904S/R904S n=10; Males: (F[2,66]=2.43, p=.096) +/+ n=25, +/R904S n=32, R904S/R904S n=12).
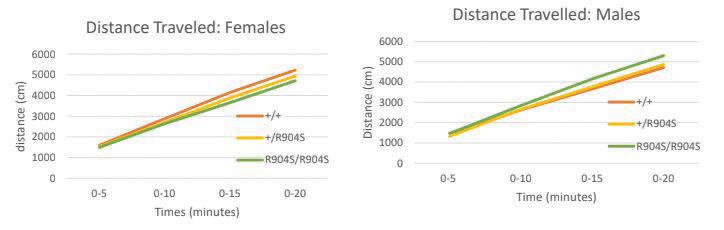
Figure 4. Graphs depict the additive distance traveled by each genotype set for males and females. There is no significant difference in the distance traveled between the genotypes for both males and females (Females: +/+ n=14, +/R904S n=21, R904S/R904S n=10; Males: (F[2,272]=1.836, p=.162) +/+ n=25, +/ R904S n=32, R904S/R904S n=12).
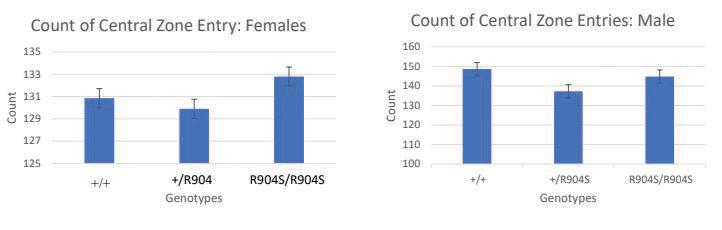
Figure 5. Graphs depict the average number of times subjects of each genotype entered the central zone of the open field area. There was no significant difference in the count of entry into the central entry zone between the genotypes for both males and females (Females: +/+ n=14, +/ R904S n=21, R904S/R904S n=10 (F[2,42]=.67, p=.516); Males: (F[2,49] =.667, p=.518) +/+ n=25, +/R904S n=32, R904S/R904S n=12).
Cellular testing Western blot to determine whole brain OPA1 levels The first cellular test performed was a western blot to determine if there were any differences in whole brain protein levels of OPA1 between genotypes in both males and females (results shown in Figure 6). Mitochondria were separated from the brain lysate to determine whether OPA1 is localizing correctly to the mitochondria alone. Two fragments of OPA1 were observed at approximately 80–100 kDa as OPA1 is proteolytically cleaved to a short and long form in the mitochondria. GAPDH was used as a positive control as it is ubiquitously expressed throughout the brain, thus it was expected to be present in both lysates. The western blot for female mice showed OPA1 solely in the mitochondria and equally expressed in wildtype and homozygous mutant brain lysates. The western blot for males was performed afterwards and includes heterozygous mouse brain lysates and a mitochondrial specific control, Hsp60. Again, OPA1 appeared to be equally expressed in brain lysates of all three genotypes. The western blot for females was subject to human error. First, there appeared to be Hsp60 and OPA1 signals present in the mitochondria exclusion fragment (whole brain lysate without the mitochondria). This could be due to either contamination between the two groups or insufficient separation of mitochondria from the exclusion fragment. There also appeared to be a nonspecific band on the membrane section incubated with Hsp60, which could be due to nonspecific binding, excessive incubation in Hsp60, or insufficient blocking incubation. GAPDH was also not present in every lane as expected, which could be due to excessive washing of that membrane section after incubation. Although there appeared to be light bands of OPA1 in the mitochondria exclusion fragment, we presumed that to be a result of either the contamination or insufficient separation of the mitochondria. However, there appeared to be no difference in the level of OPA1 protein expression between genotypes, which was our primary determination objective in carrying out the western blot. Brain slice staining to determine localization of OPA1 We obtained 10x objective tile images of wildtype and homozygous mutant mice
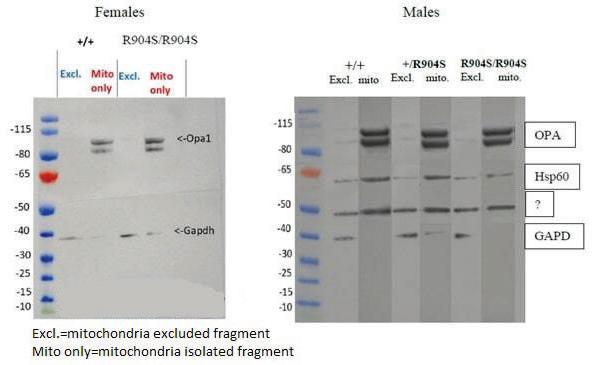
Figure 6: Excl: mitochondria excluded fragment, Mito: isolated mitochondria. This figure depicts the western blot of male and female subjects. Two fragments of OPA1 are seen between 80–100 kDa because OPA1 is proteolytically cleaved to a short and long form. GAPDH served as a positive control, as it is present ubiquitously throughout the brain. Hsp60 served as a positive control for the mitochondrial fragment as it is exclusive to the mitochondria. There are slight Hsp60 and OPA1 bands in the male exclusion fragments, indicating that there was slight contamination of mitochondria in the male exclusion fragments. However, OPA1 clearly appears to be equally strong in the male mitochondrial fragments between the genotypes. There is also a strong nonspecific band appearing in the male Hsp60 fragment of the gel, which can be a result of excessive Hsp60 incubation. Overall, the OPA1 protein bands appear to be equally intense in both wildtype and homozygous mutant of both males and females, indicating that there is no significant difference in whole brain expression between the two genotypes.
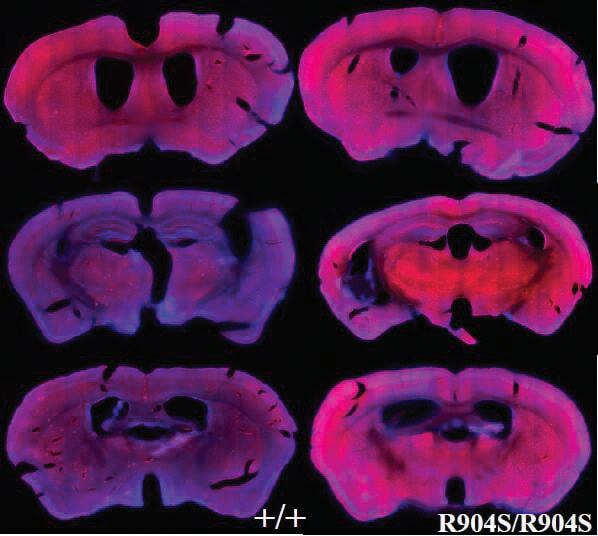
Figure 7. 10x objective tile images of brain slices of wildtype and homozygous mutant mice. Stained for nuclei with Hoechst dye (blue) and OPA1 using an OPA1 primary antibody (NovusBio) and Alexa Fluorescence 555 secondary antibody (Invitrogen).
brain slices for nuclei with Hoechst dye (blue) (Invitrogen), OPA1 using an OPA1 primary antibody (red) (NovusBio) and Alexa Fluorescence 555 secondary antibody (Invitrogen) (Figure 7). Brain slices were stained for OPA1 to determine whether there were brain regions with varying expression in homozygous mutant mice as compared to wildtype mice. We struggled to acquire a strong signal for OPA1. Initially, we utilized Alexa Fluorescent 488 and 657 secondary antibodies (Invitrogen), which were not successful due to low signal output in comparison to the Hoechst stain. This made imaging difficult, thus when we incubated with the 555 Alexa Fluorescent secondary antibody (Invitrogen), we decreased our wash steps to minimize washing away our secondary antibody. This was a preliminary experiment, where our primary goal was to optimize our procedure and imaging process. Therefore, although these results appear to indicate increased OPA1 levels in the homozygous mutant slices, this cannot be supported. There are not enough brain slice comparisons, the brain slices may not be sufficiently aligned to compare between the wildtype and homozygous mutant slices, and the imaging setup may not be optimal. However, this preliminary experiment allowed us to identify the 555 Alexa Fluorescent secondary antibody (Invitrogen) as capable of producing a strong signal that can be visualized with the Hoechst stain.
Cortical neurons stained with MitoTracker to observe mitochondria morphology 63x and 100x objective images utilizing oil on a confocal microscope were taken of cortical neuron nuclei stained blue with Hoechst dye (Invitrogen) and mitochondria stained red with MitoTracker Red (Thermofisher) (Figure 8). The mitochondria appeared to localize correctly throughout the cytoplasm in all genotypes. However, we observed potential mitochondrial aggregates in heterozygous and homozygous mutant neurons that do not seem to be present in wildtype neurons (marked with white arrows). Mitochondrial aggregation may be caused by aberrant OPA1 functioning. A prior study found that siRNA mediated downregulation of OPA1 resulted in an aggregated mitochondria phenotype (16). The R904S mutation could be functioning to downregulate the protein expression by an amount that is not detectable through whole brain western blotting. Previous studies have also found that mitochondrial aggregation could indicate a proapoptotic state as aggregation has been found to be an event that precedes cytochrome c release (17). Thus, the R904S mutant neurons may also be more prone to apoptosis.
Discussion
OPA1 has previously been identified as a probable risk gene for TD (5), which is a heritable, neuropsychiatric and developmental disorder characterized by motor and vocal tics. OPA1 is a protein involved in the fusion and membrane dynamics of the inner mitochondrial membrane. After the Tischfield laboratory identified a TD patient with the R904S mutant of OPA1, we hypothesized that the mutation would result in behavioral and/ or cellular abnormalities as compared to wildtype cells/mice. Essentially, we were searching for a visible phenotype caused by the mutation that could be characterized as a TD model phenotype. This is an important endeavor because TD is not well studied and OPA1’s involvement with the disorder has not been studied previously.
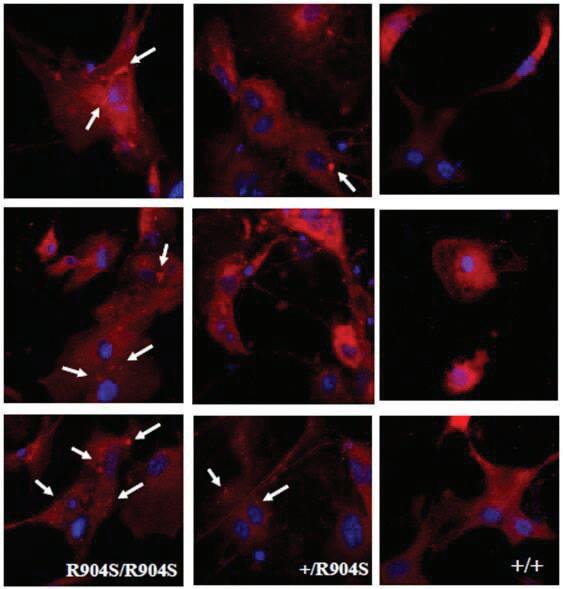
Figure 8. Cortical neurons of homozygous mutant, heterozygous and wildtype mice stained for nuclei with Hoechst dye (blue) and mitochondria with MitoTracker Red. White arrows distinguish possible mitochondrial aggregates that appear to be present in homozygous mutant and heterozygous neurons, while absent in wildtype neurons. Images were taken under 63x and 100x objective utilizing oil on a confocal microscope.
The behavioral paradigms tested were motion sequencing, open field arena test, marble-burying task, and prepulse inhibition. No statistically significant differences have been found in the performance of the mice among these tests. This can be because the mechanism of the variant is not addressed by these assays. Because we cannot know what phenotype the mutation induces, we cannot know which assay will identify it. Although we are aware of the symptoms of TD in humans, we are not certain how tics and other symptoms of TD manifest in mice. Additionally, TD is more than a motor disorder — there are also psychological and neurological aspects which place TD symptoms on a spectrum of severity. Thus, behavioral assays alone cannot encompass the complexity and dynamic nature of the disorder. Cellular testing is crucial because understanding the mechanism of the variant can help to pinpoint how the mutation may be affecting the mouse phenotypically. This in turn can help identify behavioral assays that address the behaviors that may be affected by the mutation. We utilized western blotting to observe whole brain protein expression, which showed no visible differences in expression levels. We were also working to identify certain brain regions with higher or lower OPA1 expression levels to potentially identify regions of interest. A better understanding of the protein’s expression throughout the brain can also provide a better understanding of the potential pathways that the protein may be involved in. Although we obtained preliminary results, they were not sufficient to conclude the nature of OPA1 level in different brain regions, as we did not have an appropriate amount of brain slices for comparison and our staining procedure was not optimized.
Lastly, we visualized mitochondria in cortical neurons and observed potential mitochondrial aggregation in heterozygous and homozygous mutant neurons that was not present in wildtype neurons. Studies have shown that mitochondrial aggregation can be a result of reduced OPA1 expression and that it may suggest a preapoptotic state as aggregation precedes cytochrome c release (16, 17). Thus, it is possible that the R904S mutation either encourages apoptosis or works in downregulating the protein’s expression. As there were no visible decreases in protein expression with the western blot, it is possible that the downregulation is not drastic enough to be qualitatively visible, or the mutation functions in inactivating the protein rather than altering its expression. This project is the first attempt at identifying a behavioral or cellular assay for TD with OPA1. There are several potential explanations for inconclusive results in the assays done thus far. As previously stated, it is possible that the assays done were not relevant to the mutation. For instance, the mutation may be affecting mitochondrial function rather than its appearance and localization. It is also possible that OPA1 is not a gene that contributes to TD, or that the TD patient identified with the R904S mutation has other TD risk genes that are contributing to their disorder. Thus, it is also possible that the TD patient found with the R904S OPA1 mutation may have other genetic mutations that may be contributing to their TD diagnosis. Lastly, the testing thus far has been done on mice between the ages of P2 to approximately 3 months old — thus it is possible that a phenotype manifests in older mice, which is not something that we have investigated yet. So far, we have tested behavior and protein/mitochondrial characteristics, thus additional future directions would be to test mitochondrial function. Due to the observed mitochondrial aggregates, the cristae and its functions specifically should be investigated, such as detailed cristae structure. A previous study has found little to no cristae formation in OPA1-KO mitochondria with transmission EM imaging and loss of cristae tightness in GTPase-defective OPA1 mutant mitochondria (6). Cytochrome c levels and apoptosis potential should also be investigated as mitochondrial aggregation is thought to play a role in apoptosis signaling. Additionally, oxidative phosphorylation and mitochondrial energetics should be studied as there are cases of patient cells with oxidative phosphorylation defects that have no apparent change in mitochondrial morphology (18). Lastly, dOPA1 mutations in Drosophila have been found to cause elevated reactive oxygen species (19). As increased levels of reactive oxygen species indicates stressful conditions in the cell or mitochondria, health of the mitochondria and cell can be tested for using MitoSox and CellRox, which are fluorescent dyes that identify reactive oxygen species in the mitochondria and whole cell respectively. TD is not well studied because of its complex etiology. Understanding the R904S mutation of OPA1 would increase our understanding of Tourette disorder pathology, which may help to develop therapeutic methods to target and ameliorate the debilitating symptoms of the disorder.
Acknowledgments
I would like to acknowledge all members of the Tischfield laboratory for supporting me in my research endeavors. I express my deepest gratitude to my mentors Jay Tischfield, and Cara Nasello for their advice and support throughout the project. I would also like to thank Mikaela Hufnell for her work with maintenance of mouse colonies and her assistance in the animal facility, Sam Iofel and Luis Ramirez for aiding in genotyping mice, Noriko Goldsmith, for her guidance of imaging equipment, Josh Thackray for his technical support, and Yurdiana Garcia for her assistance in perfusion methods.
References
1. Diagnosing Tic Disorders | CDC [Internet]. [cited 2021
May 1]. Available from: https://www.cdc.gov/ncbddd/ tourette/diagnosis.html 2. Georgitsi M, Willsey AJ, Mathews CA, State M, Scharf JM,
Paschou P. The genetic etiology of tourette syndrome:
Large-scale collaborative efforts on the precipice of discovery. Front Neurosci [Internet]. 2016 Aug 3 [cited 2021 May 1];10(AUG):351. Available from: /pmc/articles/
PMC4971013/ 3. Cavanna AE, Black KJ, Hallett M, Voon V. Neurobiology of the premonitory urge in tourette’s syndrome:
Pathophysiology and treatment implications. J
Neuropsychiatry Clin Neurosci [Internet]. 2017 Mar 1 [cited 2021 May 1];29(2):95–104. Available from: /pmc/articles/
PMC5409107/ 4. Willsey AJ, Fernandez T V., Yu D, King RA, Dietrich A, Xing
J, et al. De Novo Coding Variants Are Strongly Associated with Tourette Disorder. Neuron [Internet]. 2017 May 3 [cited 2021 May 1];94(3):486-499.e9. Available from: / pmc/articles/PMC5769876/ 5. Wang S, Mandell JD, Kumar Y, Sun N, Morris MT, Arbelaez
J, et al. De Novo Sequence and Copy Number Variants Are
Strongly Associated with Tourette Disorder and Implicate
Cell Polarity in Pathogenesis. Cell Rep [Internet]. 2018 Sep 25 [cited 2021 May 1];24(13):3441-3454.e12. Available from: /pmc/articles/PMC6475626/ 6. Lee H, Yoon Y. Mitochondrial membrane dynamics— functional positioning of OPA1. Antioxidants [Internet]. 2018 Dec 1 [cited 2021 May 1];7(12). Available from: / pmc/articles/PMC6316456/ 7. Guo R, Gu J, Zong S, Wu M, Yang M. Structure and mechanism of mitochondrial electron transport chain [Internet]. Vol. 41, Biomedical Journal. Elsevier B.V.; 2018 [cited 2021 May 1]. p. 9–20. Available from: /pmc/articles/
PMC6138618/ 8. Alavi MV, Fuhrmann N. Dominant optic atrophy, OPA1, and mitochondrial quality control: Understanding mitochondrial network dynamics [Internet]. Vol. 8,
Molecular Neurodegeneration. BioMed Central; 2013 [cited 2021 May 1]. p. 32. Available from: http:// molecularneurodegeneration.biomedcentral.com/ articles/10.1186/1750-1326-8-32 9. Belenguer P, Pellegrini L. The dynamin GTPase OPA1:
More than mitochondria? Vol. 1833, Biochimica et
Biophysica Acta - Molecular Cell Research. Elsevier; 2013. p. 176–83.
10. Wiltschko AB, Johnson MJ, Iurilli G, Peterson RE, Katon
JM, Pashkovski SL, et al. Mapping Sub-Second Structure in Mouse Behavior. Neuron [Internet]. 2015 [cited 2021
May 1];88(6):1121–35. Available from: /pmc/articles/
PMC4708087/ 11. Datta SR. Understanding Action [Internet]. 2018 [cited 2021 May 1]. Available from: http://datta.hms.harvard.edu/ research/behavioral-analysis/ 12. Seibenhener ML, Wooten MC. Use of the open field maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp [Internet]. 2015 Feb 6 [cited 2021
May 1];(96):52434. Available from: /pmc/articles/
PMC4354627/ 13. Angoa-Pérez M, Kane MJ, Briggs DI, Francescutti DM,
Kuhn DM. Marble burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J Vis
Exp [Internet]. 2013 [cited 2021 May 1];82(82):50978.
Available from: /pmc/articles/PMC4108161/ 14. Geyer MA, Dulawa SC. Assessment of Murine Startle
Reactivity, Prepulse Inhibition, and Habituation. Curr
Protoc Neurosci [Internet]. 2003 Jul 1 [cited 2021
May 1];24(1):8.17.1-8.17.15. Available from: https:// onlinelibrary.wiley.com/doi/10.1002/0471142301. ns0817s24 15. Yael D, Israelashvili M, Bar-Gad I. Animal models of tourette syndrome-from proliferation to standardization.
Front Neurosci [Internet]. 2016 Mar 31 [cited 2021
May 1];10(MAR):132. Available from: /pmc/articles/
PMC4814698/ 16. Kamei S, Chen-Kuo-Chang M, Cazevieille C, Lenaers
G, Olichon A, Bélenguer P, et al. Expression of the
Opa1 mitochondrial protein in retinal ganglion cells: Its downregulation causes aggregation of the mitochondrial network. Investig Ophthalmol Vis Sci [Internet]. 2005 Nov 1 [cited 2021 May 1];46(11):4288–94. Available from: http:// www.ncbi.nlm.nih.gov/Genbank; 17. Haga N, Fujita N, Tsuruo T. Mitochondrial aggregation precedes cytochrome c release from mitochondria during apoptosis. Oncogene [Internet]. 2003 Aug 28 [cited 2021
May 1];22(36):5579–85. Available from: www.nature.com/ onc 18. Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab [Internet]. 2014 Apr 1 [cited 2021 May 1];19(4):630–41. Available from: /pmc/articles/
PMC4018240/ 19. Tang S, Le PK, Tse S, Wallace DC, Huang T. Heterozygous mutation of Opa1 in Drosophila shortens lifespan mediated through increased reactive oxygen species production.
PLoS One [Internet]. 2009 Feb 16 [cited 2021 May 1];4(2).
Available from: /pmc/articles/PMC2637430/








