Combination Advanced Therapy in the Treatment of Infl ammatory Bowel Disease
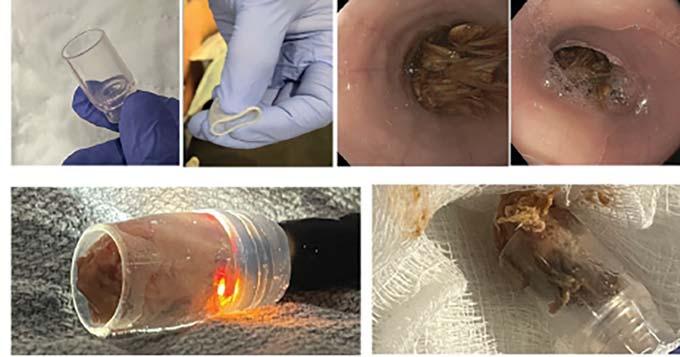
Recurrent Intentional Foreign Body Ingestion: Technical and Nontechnical Challenges
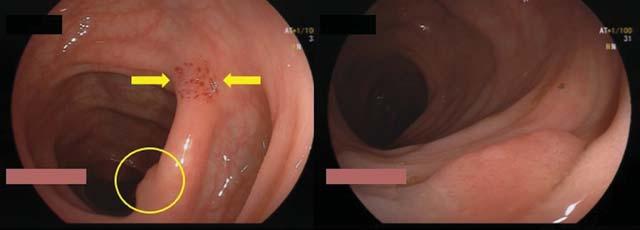
Diagnosis of Achalasia
SPOTLIGHT SECTIONS
IBD: Pouchitis Guidelines and #MondayNightIBD on Obesity Management
Endoscopy: Reprocessing Strategies, EndoHacks
Hepatology Guidelines: Acute Liver Failure And Alcohol-Associated Liver Disease
Guideline on EET for Barrett’s Esophagus
Microbiome Zone: Microbial Imbalances And Comparing FMT Therapies
For your adult patients with moderately to severely active ulcerative colitis (UC) who had inadequate response to their current treatment1
MAKE THE URGENT CHANGE WITH OMVOH
Omvoh demonstrated sustained clinical remission and reduced bowel urgency at Week 521
Nearly 2 in 3 patients taking Omvoh achieved clinical response at Week 121 65% of patients (n=517/795) taking Omvoh achieved clinical response* after 12 weeks of induction dosing vs 43% (n=115/267) with placebo (secondary endpoint), and nearly 1 in 4 (24%, n=191/795) achieved clinical remissiona vs 15% (n=40/267) with placebo (primary endpoint).1
aClinical remission based on mMS is defined as: SF=0 or 1, RB=0, and centrally read ES=0 or 1 (excluding friability).1
*Clinical response is defined as a decrease in the mMS of ≥2 points with ≥30% decrease from baseline, and either a decrease of ≥1 point in RB from baseline or RB=0 or 1.1
INDICATION
Omvoh™ is an interleukin-23 antagonist indicated for the treatment of moderately to severely active ulcerative colitis in adults.1
SELECT IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS: Omvoh is contraindicated in patients with a history of serious hypersensitivity reaction to mirikizumab-mrkz or any of the excipients.
Please see Important Safety Information below.
UC-1 AND UC-2 TRIAL DESIGN
Omvoh was studied in two Phase 3, randomized, doubleblind, placebo-controlled clinical trials of adult patients with moderately to severely active UC. Patients (N=1279) were randomized 3:1 to receive Omvoh 300 mg IV infusion or placebo every 4 weeks (Q4W) for 12 weeks (Week 0, 4, and 8) in the induction study (UC-1). Patients who achieved clinical response with Omvoh at Week 12 in UC-1 (N=581) were re-randomized 2:1 to receive Omvoh 200 mg SC injection or placebo Q4W for 40 weeks in the maintenance study (UC-2) (52 weeks of continuous therapy). The primary endpoint was the proportion of patients in clinical remission at Week 12 in UC-1 and Week 40 in UC-2.1
At baseline of UC-1, all patients had inadequate response, loss of response, or intolerance to at least one corticosteroid, immunomodulator, biologic treatment (TNF blocker, vedolizumab), or tofacitinib. In UC-2, patients who were on concomitant UC therapies during UC-1 were required to continue on stable doses of oral aminosalicylates and immunomodulator agents. Corticosteroid tapering was required for patients who were receiving oral corticosteroids at baseline and achieved clinical response in UC-1.1
Patients with an mMS of 5 to 9 at baseline of UC-1 were included for efficacy analyses. Patients had a median mMS of 7, and 58% had severely active disease (mMS 7 to 9). Patients’ baseline therapies included 41% of patients receiving oral corticosteroids, 24% receiving immunomodulators, and 75% receiving aminosalicylates. Patients’ prior treatment experiences include: 57% were biologic- and JAKi-naive, 41% had failed at least one biologic, 3% had failed a JAKi, and 2% had previously received but had not failed a biologic or JAKi.1
Bowel urgency was assessed using an Urgency Numeric Rating Scale (UNRS) ranging from 0 (no urgency) to 10 (worst possible urgency) during UC-1 and as a secondary endpoint in UC-2. Bowel urgency improvement was evaluated as the proportion of patients with a baseline UNRS weekly average score of ≥3 achieving a weekly average score of 0 to 1 at Week 12 in UC-1 and Week 40 in UC-2.1,2,4
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS - Omvoh is contraindicated in patients with a history of serious hypersensitivity reaction to mirikizumab-mrkz or any of the excipients.
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis during intravenous infusion, have been reported with Omvoh administration. Infusion-related hypersensitivity reactions, including mucocutaneous erythema and pruritus, were reported during induction. If a severe hypersensitivity reaction occurs, discontinue Omvoh immediately and initiate appropriate treatment.
Infections
Omvoh may increase the risk of infection. Do not initiate treatment with Omvoh in patients with a clinically important active infection until the infection resolves or is adequately treated. In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing Omvoh. Instruct patients to seek medical advice if signs or symptoms of clinically important acute or chronic infection occur. If a serious infection develops or an infection is
not responding to standard therapy, monitor the patient closely and do not administer Omvoh until the infection resolves.
Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Omvoh. Do not administer Omvoh to patients with active TB infection. Initiate treatment of latent TB prior to administering Omvoh. Consider anti-TB therapy prior to initiation of Omvoh in patients with a history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after Omvoh treatment. In clinical trials, subjects were excluded if they had evidence of active TB, a history of active TB, or were diagnosed with latent TB at screening.
Hepatotoxicity
Drug-induced liver injury in conjunction with pruritus was reported in a clinical trial patient following a longer than recommended induction regimen. Omvoh was discontinued. Liver test abnormalities eventually returned to baseline. Evaluate liver enzymes and bilirubin at baseline and for at least 24 weeks of treatment. Monitor thereafter according to routine patient management. Consider other treatment options in patients with evidence of liver cirrhosis. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. Interrupt treatment if druginduced liver injury is suspected, until this diagnosis is excluded. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of hepatic dysfunction.
Immunizations
according to current immunization guidelines. No data are available on the response to live or non-live vaccines in patients treated with Omvoh.
ADVERSE REACTIONS
Most common adverse reactions (≥2%) associated with Omvoh treatment are upper respiratory tract infections and arthralgia during induction, and upper respiratory tract infections, injection site reactions, arthralgia, rash, headache, and herpes viral infection during maintenance.
MR HCP ISI UC APP
See Brief Summary of Prescribing Information on subsequent pages. See Instructions for Use included with the device.
References: 1. Omvoh (mirikizumab-mrkz). Prescribing Information. Lilly USA, LLC.
2. D'Haens G, Dubinsky M, Kobayashi T, et al. Mirikizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2023;388(26):2444-2455. doi:10.1056/NEJMoa2207940 3. Data on File. DOF-MR-US-0018. Lilly USA, LLC.
4. Dubinsky MC, Irving PM, Panaccione R, et al. Incorporating patient experience into drug development for ulcerative colitis: development of the Urgency Numeric Rating Scale, a patient-reported outcome measure to assess bowel urgency in adults. J Patient Rep Outcomes. 2022;6(1):31. doi:10.1186/s41687-022-00439-w
Avoid use of live vaccines in patients treated with Omvoh. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating therapy, complete all age-appropriate vaccinations
OmvohTM (mirikizumab-mrkz) injection, for intravenous or subcutaneous use
Brief Summary: Consult the package insert for complete prescribing information.
INDICATIONS AND USAGE
Omvoh is an interleukin-23 antagonist indicated for the treatment of moderately to severely active ulcerative colitis in adults.
CONTRAINDICATIONS
Omvoh is contraindicated in patients with a history of serious hypersensitivity reaction to mirikizumab-mrkz or any of the excipients [see Warnings and Precautions]
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis during intravenous infusion, have been reported with Omvoh administration. Infusion-related hypersensitivity reactions, including mucocutaneous erythema and pruritis, were reported during induction [see Adverse Reactions]. If a severe hypersensitivity reaction occurs, discontinue Omvoh immediately and initiate appropriate treatment.
Infections
Omvoh may increase the risk of infection [see Adverse Reactions].
Do not initiate treatment with Omvoh in patients with a clinically important active infection until the infection resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing Omvoh. Instruct patients to seek medical advice if signs or symptoms of clinically important acute or chronic infection occur. If a serious infection develops or an infection is not responding to standard therapy, monitor the patient closely and do not administer Omvoh until the infection resolves.
Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with Omvoh.
Do not administer Omvoh to patients with active TB infection. Initiate treatment of latent TB prior to administering Omvoh. Consider anti-TB therapy prior to initiation of Omvoh in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after Omvoh treatment.
In clinical trials, subjects were excluded if they had evidence of active TB, a past history of active TB, or were diagnosed with latent TB at screening.
Hepatotoxicity
A case of drug-induced liver injury (alanine aminotransferase [ALT] 18x the upper limit of normal (ULN), aspartate aminotransferase [AST] 10x ULN, and total bilirubin 2.4x ULN) in conjunction with pruritus was reported in a clinical trial subject following a longer than recommended induction regimen. Omvoh was discontinued. Liver test abnormalities eventually returned to baseline.
Evaluate liver enzymes and bilirubin at baseline and for at least 24 weeks of treatment. Monitor thereafter according to routine patient management.
Consider other treatment options in patients with evidence of liver cirrhosis. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. Interrupt treatment if drug-induced liver injury is suspected, until this diagnosis is excluded. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of hepatic dysfunction.
Immunizations
Avoid use of live vaccines in patients treated with Omvoh. Prior to initiating therapy with Omvoh, complete all age-appropriate vaccinations according to current immunization guidelines. No data are available on the response to live or non-live vaccines in patients treated with Omvoh.
ADVERSE REACTIONS
The following topics are also discussed in detail in the Warnings and Precautions section: Hypersensitivity Reactions Infections
Omvoh™ (mirikizumab-mrkz) injection, for intravenous or subcutaneous use
Tuberculosis
Hepatotoxicity
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Omvoh was studied up to 12 weeks in subjects with moderately to severely active ulcerative colitis in a randomized, double-blind, placebo-controlled induction study (UC-1). In subjects who responded to induction therapy in UC-1, long-term safety up to 52 weeks was evaluated in a randomized, double-blind, placebo-controlled maintenance study (UC-2) and a long-term extension study [see Clinical Studies].
In the induction study (UC-1), 1279 subjects were enrolled of whom 958 received Omvoh 300 mg administered as an intravenous infusion at Weeks 0, 4, and 8. In the maintenance study (UC-2), 581 subjects were enrolled of whom 389 received Omvoh 200 mg administered as a subcutaneous injection every 4 weeks.
Table 1 summarizes the adverse reactions reported in at least 2% of subjects and at a higher frequency than placebo during UC-1.
Table 1: Adverse Reactionsa in Subjects with Ulcerative Colitis through Week 12 in a Placebo-Controlled Induction Study (UC-1)
a Reported in at least 2% of subjects and at a higher frequency than placebo.
b Omvoh 300 mg as an intravenous infusion at Weeks 0, 4, and 8.
c Upper respiratory tract infections includes related terms (e.g., COVID-19, nasopharyngitis, pharyngitis, rhinitis, sinusitis, and upper respiratory tract infection).
In the induction study (UC-1), infusion-related hypersensitivity reactions were reported by 4 (0.4%) subjects treated with Omvoh and 1 (0.3%) subject treated with placebo.
Table 2 summarizes the adverse reactions reported in at least 2% of subjects and at a higher frequency than placebo during the 40-week controlled period of UC-2.
Table 2: Adverse Reactionsa in Subjects with Ulcerative Colitis through Week 40 In a Placebo-Controlled Maintenance Study (UC-2)
Adverse Reactions OMVOH
200-mg Subcutaneous
Upper respiratory tract infectionsc
Injection site reactionsd
(9%)8 (4%)
Arthralgia26 (7%)8 (4%)
Rashe 16 (4%)2 (1%)
Headache16 (4%)2 (1%)
Herpes viral infectionf 9 (2%)1 (1%)
a Reported in at least 2% of subjects and at a higher frequency than placebo
b Omvoh 200 mg as a subcutaneous injection at Week 12 and every 4 weeks thereafter for up to an additional 40 weeks.
c Upper respiratory tract infections includes related terms (e.g., COVID-19, nasopharyngitis, pharyngitis, rhinitis, sinusitis, and upper respiratory tract infection).
d Injection site reactions includes related terms (e.g., erythema, hypersensitivity, pain, reaction, and urticaria at the injection site).
e Rash is composed of several similar terms.
f Herpes viral infection includes related terms (e.g., herpes zoster, herpes simplex, and oral herpes.)
Omvoh™ (mirikizumab-mrkz) injection, for intravenous or subcutaneous use
Infections
In UC-1 through Week 12, infections were reported by 145 (15%) subjects treated with Omvoh 300 mg and 45 (14%) subjects treated with placebo. Serious infections were reported by less than 1% in both groups. Serious infections in the Omvoh group included intestinal sepsis, listeria sepsis, and pneumonia.
In the maintenance study (UC-2) through Week 40 (a total of 52 weeks of treatment), infections were reported by 93 (24%) subjects treated with Omvoh 200 mg and 44 (23%) subjects treated with placebo. A case of COVID-19 pneumonia was reported as a serious infection in the Omvoh group.
Hepatic Enzyme Elevations
In UC-1 through Week 12, alanine aminotransferase (ALT) ≥5X ULN was reported by 1 (0.1%) subject treated with Omvoh 300 mg and 1 (0.3%) subject treated with placebo. Aspartate aminotransferase (AST) ≥5X ULN was reported by 2 (0.2%) subjects treated with Omvoh 300 mg and no subject treated with placebo. These elevations have been noted with and without concomitant elevations in total bilirubin.
In the maintenance study (UC-2) through Week 40 (a total of 52 weeks of treatment), 3 (0.8%) subjects treated with Omvoh 200 mg reported ALT ≥5X ULN and 3 (0.8%) subjects reported AST ≥5X ULN; with or without concomitant elevations in total bilirubin. No subjects treated with placebo experienced similar elevations [see Warnings and Precautions]
USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Omvoh during pregnancy. Pregnant women exposed to Omvoh and healthcare providers are encouraged to call Eli Lilly and Company at 1-800-Lilly-Rx (1-800-545-5979).
Risk Summary
Available data from case reports of mirikizumab-mrkz use in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Although there are no data on mirikizumabmrkz, monoclonal antibodies can be actively transported across the placenta, and mirikizumab-mrkz may cause immunosuppression in the in utero-exposed infant. An enhanced pre- and post-natal development study conducted in pregnant monkeys at a dose 69 times the maximum recommended human dose (MRHD) revealed no adverse developmental effects to the developing fetus, or harm to infant monkeys from birth through 6 months of age. There are risks of adverse pregnancy outcomes associated with increased disease activity in women with inflammatory bowel disease (see Clinical Considerations)
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defects, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and Embryo/Fetal Risk
Published data suggest that the risk of adverse pregnancy outcomes in women with inflammatory bowel disease (IBD) is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Fetal/Neonatal Adverse Reactions
Transport of endogenous IgG antibodies across the placenta increases as pregnancy progresses, and peaks during the third trimester. Because mirikizumab-mrkz may interfere with immune response to infections, risks and benefits should be considered prior to administering live vaccines to infants exposed to Omvoh in utero. There are no data regarding infant serum levels of mirikizumab-mrkz at birth and the duration of persistence of mirikizumab-mrkz in infant serum after birth. Although a specific timeframe to delay live virus immunizations in infants exposed in utero is unknown, a minimum of 2 months after birth should be considered because of the half-life of the product.
Omvoh™ (mirikizumab-mrkz) injection, for intravenous or subcutaneous use
Data
Animal Data
An enhanced pre- and postnatal development study was conducted in cynomolgus monkeys administered mirikizumab-mrkz by intravenous injection during organogenesis to parturition at a dose of 300 mg/kg twice weekly (69 times the MRHD based on exposure comparisons). Mirikizumab-mrkz crossed the placenta in monkeys. No maternal toxicity was noted in this study. No mirikizumab-mrkz-related effects on morphological, functional, or immunological development were observed in infant monkeys from birth through 6 months of age. However, incidences of embryo/fetal loss were higher in the treated groups compared to control (6.7% [1 of 15] in controls vs 26.7% [4 of 15] at 300 mg/kg (69 times the MRHD, based on exposure comparisons) but were within the range of historical control data. Following delivery, most adult female cynomolgus monkeys and all infants from the mirikizumab-mrkz-treated group had measurable serum concentrations up to 28 days postpartum. In the infant monkeys, mean serum concentrations were approximately 4.8 times the respective mean maternal concentrations.
Lactation
Risk Summary
There are no data on the presence of mirikizumab-mrkz in human milk, the effects on the breastfed infant, or the effects on milk production. Endogenous maternal IgG and monoclonal antibodies are transferred in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to mirikizumab-mrkz are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Omvoh and any potential adverse effects on the breastfed infant from Omvoh or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of Omvoh have not been established in pediatric patients.
Geriatric Use
Of the 795 Omvoh-treated subjects in the two clinical trials, 64 subjects (8%) were 65 years of age and older, while 10 subjects (1%) were 75 years of age and older. These clinical studies did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger adult subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger subjects. No clinically meaningful differences in the pharmacokinetics of mirikizumab-mrkz were observed in subjects 65 years of age and older compared to younger adult subjects [see Clinical Pharmacology]
DOSING
Recommended Dosage
Induction Dosage
The recommended induction dosage of Omvoh is 300 mg administered by intravenous infusion over at least 30 minutes at Week 0, Week 4, and Week 8 [see Dosage and Administration]
Maintenance Dosage
The recommended maintenance dosage of Omvoh is 200 mg administered by subcutaneous injection (given as two consecutive injections of 100 mg each) at Week 12, and every 4 weeks thereafter [see Dosage and Administration]
PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Hypersensitivity Reactions
Advise patients to discontinue Omvoh and seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions].
Infections
Advise patients that Omvoh may lower the ability of their immune system to fight infections and to contact their healthcare provider immediately if they develop any symptoms of infection [see Warnings and Precautions]
Tuberculosis
Advise patients to contact their healthcare provider if they experience symptoms suggestive of TB (e.g., unexplained fever, cough, or difficulty breathing) [see Warnings and Precautions]
Omvoh™ (mirikizumab-mrkz) injection, for intravenous or subcutaneous use
Hepatotoxicity
Inform patients that Omvoh may cause liver injury. Advise patients to seek immediate medical attention if they experience symptoms suggestive of liver dysfunction (e.g., unexplained rash, nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine) [see Warnings and Precautions].
Immunizations
Advise patients that vaccination with live vaccines is not recommended during Omvoh treatment and immediately prior to or after Omvoh treatment. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Instruct patients to inform their healthcare provider that they are taking Omvoh prior to receiving a vaccination [see Warnings and Precautions]
Pregnancy
Advise patients who are exposed to Omvoh during pregnancy to contact Eli Lilly and Company [see Use in Specific Populations]
Administration
Instruct patients in preparation and administration of Omvoh, including choosing anatomical sites for subcutaneous administration, and proper subcutaneous injection technique. Instruct patients in the technique of pen disposal [see Instructions for Use]
Instruct patients or caregivers to administer two 100-mg prefilled pens to achieve the full 200-mg dose of Omvoh.
Additional information can be found at www.Omvoh.com
See Instructions for Use accompanying the product device.
MR HCP BS UC APP
PP-MR-US-0147
Omvoh™ (mirikizumab-mrkz) injection, for intravenous or subcutaneous use
MR HCP BS UC APP
EDITORIAL ADVISORY BOARD
ANDREW ALBERT, MD, MPH Chicago, Illinois
MANOOP S. BHUTANI, MD, FASGE Houston, Texas
BROOKS D. CASH, MD, AGAF, FACG, FACP, FASGE Houston, Texas
ALINE CHARABATY, MD, AGAF Washington, D.C.
AUSTIN CHIANG, MD, MPH Philadelphia, Pennsylvania
ALAN F. CUTLER, MD, FACG, FACP, AGAG Farmington Hills, Michigan
SARAH ENSLIN, PA-C Rochester, New York
RONNIE FASS, MD, MACG Cleveland, Ohio
HARISH K. GAGNEJA, MD, FACG, FASGE, AGAF Austin, Texas
FRANK G. GRESS, MD, MACG, FASGE New York, New York
SETH GROSS, MD, FASGE New York, New York
VIVEK KAUL, MD, FACG, FASGE, AGAF Rochester, New York
GARY R. LICHTENSTEIN, MD, FACG Philadelphia, Pennsylvania
JENIFER R. LIGHTDALE, MD, MPH Worcester, Massachusetts
DANA J. LUKIN, MD, PHD, FACG New York, New York
PETER R. MCNALLY, DO Fort Carson, Colorado
KLAUS MERGENER, MD, PHD, MBA, MASGE Tacoma, Washington
SATISH RAO, MD, PHD Augusta, Georgia
JOEL E. RICHTER, MD, MACG Tampa, Florida
DAVID ROBBINS, MD New York, New York
ELLEN J. SCHERL, MD New York, New York
PRATEEK SHARMA, MD, FACG, FACP, FASGE Kansas City, Kansas
ASHWANI K. SINGAL, MD, MS, FACG, FAASLD, AGAF Louisville, Kentucky
Vice President, Medical Education jmalichio@mcmahonmed.com
ART AND PRODUCTION
MICHELE MCMAHON VELLE
Creative Director
JEANETTE MOONEY
Senior Art Director
JAMES O’NEILL
Senior Systems Manager
RON REDFERN
Production Manager
ROB SINCLAIR
Circulation Manager
MCMAHON PUBLISHING
VAN VELLE President, Partner
MATTHEW MCMAHON General Manager, Partner
LAUREN SMITH
MICHAEL P. MCMAHON
MICHELE MCMAHON VELLE Partners
RAY AND ROSANNE MCMAHON Co-founders
DISCLAIMER—The reviews in this issue are designed to be a summary of information, and they represent the opinions of the authors. Although detailed, the reviews are not exhaustive. Readers are strongly urged to consult any relevant primary literature, the complete prescribing information available in the package insert of each drug, and the appropriate clinical protocols. No liability will be assumed for the use of these reviews, and the absence of typographical errors is not guaranteed.
Using the Soft, Mega, Distal Transparent Cap to Remove Food Bolus
45 EndoHacks
Suction Mark Technique for Identifying And Resecting Flat Colon Polyps
47 Between the Guidelines New ACG Guideline on Acute Liver Failure
48 Between the Guidelines ACG Issues Updated Guidance on Alcohol-Associated Liver Disease
51 Between the Guidelines AGA Issues Updated Guideline on Endoscopic Eradication Therapy for BE
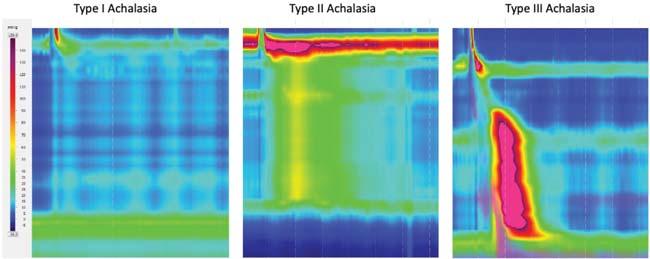
55 Diagnosis of Achalasia
Ofer Z. Fass, MD
Ronnie Fass, MD, MACG
Microbiome Zone New Data Bring Answers to Microbial Imbalance
Microbiome Zone How Have FDA Approvals Changed the Landscape of Microbiome Therapeutics?
Combination Advanced Therapy in the Treatment of Inflammatory Bowel Disease
MICHAEL J. MINTZ, MD
DANA J. LUKIN, MD, PHD, AGAF, FACG
Jill Roberts Center for Inflammatory Bowel Disease
Weill Cornell Medicine
New York, New York
The development of effective biologics and small molecule targeted therapies has revolutionized the treatment of inflammatory bowel disease. Physicians and patients have an increasing number of treatment options, making therapy choice more nuanced and personalized.
In choosing a therapy, one must consider disease phenotype and behavior, presence of extraintestinal manifestations (EIMs), response to prior therapies, safety, patient preference, and preferred route of administration.1 However, the ceiling in achieving clinical and endoscopic remission with a single biologic or small molecule therapy is approximately 50%.2,3 In addition, many patients experience a loss of response to a therapy that had previously been effective. Recent guidelines suggest that both clinical remission and complete endoscopic healing are essential treatment targets to prevent future complications of disease, including malignancy, need for surgery, and penetrating complications.4 In pursuing these treatment targets, patients often are cycled through multiple lines of therapy and eventually run out of effective treatment options.In addition, although a certain therapy may optimally control a patient’s luminal disease, it may not be effective in treating EIMs, perianal disease, or concurrent rheumatologic disease.5
To address these unmet needs, researchers have been exploring the potential role of combination therapy in IBD. The goal of combination therapy is to use
multiple synergistic therapeutic mechanisms to achieve improved disease outcomes. 6 High-quality data are starting to emerge that suggest that combination therapy may be effective for refractory disease. However, several challenges remain, including a lack of long-term efficacy data, unknown infectious and/or neoplastic risks,7 and insurance coverage limitations. This article reviews the growing efficacy and safety data on combination therapy and strategies for its implementation.
Clinical Efficacy Data
A growing number of studies demonstrate efficacy of both traditional combination therapy (TCT) and advanced combination therapy (ACT).8 TCT consists of an anti–tumor necrosis factor (TNF) agent combined with an immunomodulator such as a thiopurine or methotrexate. ACT refers to the concurrent use of multiple biologics and/ or small molecule inhibitors.Data are available from several clinical trials (Table 1), 9-13 retrospective cohort studies (Table 2),14-20 and systematic reviews with meta-analyses looking at both TCT and ACT in IBD.
Randomized Clinical Trials in TCT
The landmark SONIC and UC-SUCCESS trials demonstrated clear efficacy data of TCT in IBD. 9,10 SONIC was a multicenter randomized controlled trial that evaluated the combination of infliximab and weight-based azathioprine compared with infliximab or azathioprine monotherapy in patients with moderate to severe Crohn’s disease (CD). Combination therapy resulted in improved clinical outcomes over either monotherapy group. At week 26, 56.8% of the patients who received combination therapy were in corticosteroid-free clinical remission, compared with 44.4% in the infliximab monotherapy group and 30.0% in the azathioprine monotherapy group.
SONIC further showed that combination therapy resulted in pharmacologic optimization of infliximab, boosting drug levels and decreasing immunogenicity.9 This trial clearly demonstrated the benefits and rationale for combination therapy, as it strongly supported the notion that combining 2 therapies with unique mechanisms of action could lead to improved clinical outcomes and pharmacologic synergy.
A similar result was found in the UC-SUCCESS trial.10 Similar to SONIC, this randomized controlled trial examined infliximab and azathioprine alone or in combination in patients with moderate to severe ulcerative colitis (UC). Combination therapy resulted in higher rates of corticosteroid-free remission at week 16 of treatment (combination, 39.7%; infliximab monotherapy, 22.1%; azathioprine monotherapy, 23.7%) and higher rates of mucosal healing (combination, 62.8%; infliximab monotherapy, 54.6%; azathioprine monotherapy, 36.8%).
Although combination treatment with an anti-TNF agent plus an immunomodulator has strong efficacy data,9,10 data supporting immunomodulator use with non–anti-TNF therapies are limited. A large retrospective study of patients who received either vedolizumab (Entyvio, Takeda) or ustekinumab (Stelara, Janssen) did not demonstrate improved clinical or endoscopic outcomes when combined with thiopurines or methotrexate at 1 year, 21 which may be at least partly attributable to the less clear dose–response relationship seen with these agents, their lower immunogenicity, and a less prominent impact of thiopurines on boosting serum trough levels.
Clinical Trials in ACT
The first clinical trial to examine advanced combination therapy in IBD was published by Sands et al in 2007.11 This study randomized patients with active CD on infliximab to receive 3 additional infusions of natalizumab (Tysabri, Biogen) or placebo every 4 weeks. The patients who received the natalizumab infusions experienced a decrease in their mean Crohn’s Disease Activity Index score, while patients who received placebo did not see a decrease (natalizumab, –37.7; placebo, 3.5). However, this difference did not reach statistical significance. There were no increased adverse events (AEs) in the combination group at the 32-week follow-up period.
More recently, the EXPLORER and VEGA trials looked at the efficacy and safety of ACT.12,13
The EXPLORER trial was a single-arm, phase 4 study that assessed triple combination therapy with vedolizumab, adalimumab, and methotrexate in patients with moderateto high-risk CD.12 The EXPLORER investigators found that combination therapy achieved high rates of clinical remission (61.8% and 54.5% at weeks 10 and 26, respectively). Of note, there was no control arm in this study, so data are not available regarding vedolizumab or adalimumab monotherapy within the study population.
In comparison, the registration trials for these agents noted a week 6 clinical remission rate of 14.5% for vedolizumab (GEMINI-2) 22 and week 4 clinical remission rate of 36% for adalimumab (CLASSIC-1). 23 Among treatment responders, vedolizumab every 8 weeks was associated with maintenance of remission in 39.0% (week 52) 22 and adalimumab in 36% (week 56) (CHARM).24 Although these studies were performed among unique populations and had different designs, the therapeutic efficacy observed in EXPLORER clearly suggests a benefit for combination therapy.
VEGA was a randomized controlled, phase 2a, proofof-concept trial of a combination of the interleukin (IL)-23 agent guselkumab (Tremfya, Janssen) and the anti-TNF agent golimumab (Symponi Aria, Janssen), which is not approved to treat IBD, versus the 2 agents as monotherapy in patients with moderately to severely active UC.13
At week 12 of the study, 83% of the patients in the combination group had a clinical response, compared with 75% and 61% in the guselkumab and golimumab monotherapy groups, respectively. Although numerically higher, the clinical response rate between the combination therapy group and guselkumab monotherapy group did not meet statistical significance. However, 37% of the patients in the combination group achieved clinical remission, compared with 22% and 21% of the golimumab and guselkumab monotherapy groups, respectively, which reached statistical significance. Furthermore, the combination group achieved higher rates of endoscopic improvement (49%), compared with the golimumab monotherapy group (25%) and the guselkumab monotherapy group (30%).
Positive results from VEGA have led to 2 large randomized controlled trials of combination golimumab and guselkumab in CD (DUET-CD; ClinicalTrials.gov Identifier: NCT05242471) and UC (DUET-UC; NCT05242484). These trials, which are ongoing, will report both induction and maintenance outcomes. In addition, prospective combination phase 4 therapeutic trials with vedolizumab are in progress in both CD (EXPLORER-2; vedolizumab + adalimumab or ustekinumab; NCT0604574) and UC (ExiGem; vedolizumab + tofacitinib in TNF antagonist–exposed patients; NCT06095128). These trials may provide the first glimpse of high-quality efficacy and safety data supporting the use of ACT.
Retrospective Cohort Studies
The majority of ACT data come from published case series and retrospective cohort studies. More than 30 published studies report effectiveness and safety data for dual biologic or small molecule therapy.25 The majority of these studies
have less than 100 participants and include multiple combinations of biologics and/or small molecule therapies. A summary of several larger cohorts is detailed in Table 2.14-20
Two systematic reviews and meta-analyses have been published that have helped consolidate some of these data, both reporting positive clinical effectiveness of various ACT regimens.25,26 Ahmed et al included 279 patients, the majority of whom received combination therapy for medically refractory luminal disease. 25 The most common regimens were anti-TNF and anti-integrin (48%) and ustekinumab and anti-integrin (19%). Another common combination was a small molecule inhibitor with either vedolizumab or ustekinumab. There were notably fewer studies looking at a small molecule inhibitor combined with an anti-TNF agent, likely due to safety concerns.
Ahmed et al reported an ACT pooled clinical remission rate of 59% and endoscopic remission rate of 34%. They did not report effectiveness data for specific combinations.
Alayo et al published a systematic review including 13 studies with a total of 266 patients.26 This review included effectiveness rates of specific combinations as opposed to a pooled analysis. They reported the following clinical response and remission rates: 77.9% and 55.1% for vedolizumab plus anti-TNF, 83.9% and 47% for vedolizumab plus ustekinumab, and 59.9% and 47.8% for vedolizumab plus tofacitinib.
Fewer data exist regarding more recently approved therapies being used in combination. This includes upadacitinib (Rinvoq, AbbVie), risankizumab (Skyrizi, AbbVie), ozanimod (Zeposia, Bristol Myers Squibb), etrasimod (Velsipity, Pfizer), and mirikizumab (Omvoh, Lilly). Given greater selectivity in mechanism of action and/or optimization of the induction dosing regimens used for these agents, it is feasible that these combinations may demonstrate further improvements in therapeutic efficacy.
Although much of the real-world data are encouraging, there are challenges in interpreting many of these studies.
SONIC9 RCTModerate to severe bio-naive CD
UC-SUCCESS10 RCTModerate to severe bio-naive UC
Sands et al, 200711 RCTActive CD despite infliximab
EXPLORER12 Phase 4, single-arm, open-label
VEGA13 Phase 2a, RCT
EXPLORER-2 (NCT06045754)
EXIGEM (NCT06095128)
DUET-UC (NCT05242484)/ DUET-CD (NCT05242471)
Phase 4, open-label
Phase 4, open-label
Phase 2b, RCT
Bio-naive newly diagnosed moderate- to high-risk CD
There is notable heterogeneity of patient characteristics, study design, and outcomes among the cohort studies. In addition, because there are numerous potential combinations of therapy, there are generally a small number of patients receiving a specific combination, making overall effectiveness data difficult to interpret.
Safety Data
The short- and long-term safety implications of combination therapy must be elucidated before such regimens may be considered for more widespread use. Most studies to date have reported no concerning increase in short-term infectious complications, albeit most data are subject to the biases of retrospective analyses. However, less is known about potential long-term infectious and/or neoplastic complications attributable to combination therapy.
With respect to TCT, neither the SONIC trial nor the
UC-SUC CESS trial reported an increase in adverse events for the combination group during the study period.9,10 As a result, it was generally accepted that the risk–benefit profile was favorable. However, reports of hepatosplenic T-cell lymphoma, specifically in young men receiving infliximab and azathioprine, made many providers wary of prescribing this combination.27 While rare, revelation of this potentially fatal consequence was enough to change practice patterns. Experience using combination therapy in rheumatology has raised additional safety concerns. A meta-analysis of combination therapy in rheumatoid arthritis showed a significant increase in serious AEs (SAEs), including serious infections, with combination biologic therapy. 28 Whether the experience in rheumatology can be extrapolated to combination therapy use in IBD is unclear.
More recent data from the retrospective cohort studies suggest short-term SAEs of ACT are similar to those
Glassner et al, 202014
Ustekinumab + vedolizumab (n=25)
Vedolizumab + tofacitinib (n=8)
Infliximab + tofacitinib (n=4)
Goessens et al, 202115 Anti-TNF + vedolizumab (n=41)
Ustekinumab + vedolizumab (n=21)
Tofacitinib + vedolizumab (n=13)
McShane et al, 202316
Ustekinumab + vedolizumab (n=32)
Adalimumab + vedolizumab (n=16)
Vedolizumab + tofacitinib (n=12)
Alayo et al, 202117
Yang et al, 202018
Lee et al, 202219
Llano et al, 202120
Vedolizumab + tofacitinib (n=24)
Infliximab + tofacitinib (n=6)
Ustekinumab + tofacitinib (n=5)
Ustekinumab + vedolizumab (n=8)
Infliximab + vedolizumab (n=6)
Adalimumab + ustekinumab (n=2)
Tofacitinib + ustekinumab (n=11)
tofacitinib + vedolizumab (n=7)
tofacitinib + certolizumab (n=1)
Vedolizumab + tofacitinib (n=9)
Vedolizumab + ustekinumab (n=3)
Vedolizumab + adalimumab (n=2)
Improved rates of pooled clinical and endoscopic remission after treatment vs baseline (50% vs 14%) and endoscopic remission (34% vs 6%)
Decrease in IBD disease activity in 70% of patients; improvement in EIMs in 81% of patients
76.5% remained on combination therapy at median 40.7-wk follow-up period, which the authors concluded as representing clinical efficacy
50% clinical response rate at week 8; 90% clinical response rate and 70% clinical remission rate at week 26
Table 2. Selected Retrospective Cohort Studies Using ACT in IBD
seen with monotherapy. The aforementioned systematic reviews and meta-analyses demonstrated relatively low SAEs and infection rates. 25,26 Ahmed et al reported a pooled combination therapy SAE rate of 6.5% over a median follow-up period of 32 weeks. 25 Alayo et al subsequently reported an SAE rate range of 0% (anti-TNF and ustekinumab) to 12.3% (vedolizumab and ustekinumab). 26 Both meta-analyses did not identify any new infectious or neoplastic safety signals. VEGA did not suggest an increased AE rate in the combination therapy arm.13
As for TCT, we do not yet know the long-term infection and malignancy risk of ACT. Although there are reassuring data to suggest biologic monotherapy does not increase the incidence of solid-organ tumors in high-risk patients with previous cancers, 29,30 we do not yet know the potential malignancy risk resulting from dual immune manipulation. Long-term safety data of ACT are unlikely to be available in the near future.
Strategies for Use
There is little guidance on how to implement ACT in clinical practice. As high-quality clinical trial data and FDAapproved regimens are lacking, none of the major GI societies have drafted guidelines on when it is appropriate to consider ACT. Currently, ACT is mostly being used at highvolume IBD centers by clinical experts. As such, patients who may be candidates for ACT should be referred to an IBD center.
ACT requires a patient with a very specific clinical indication and favorable risk profile (Figure). The most common indication for ACT is medically refractory disease in a patient who has prior exposure to multiple advanced monotherapies. 25 This includes primary nonresponse or attenuation to FDA-approved regimens, an incomplete response to optimized monotherapies, or the responsiveness to one aspect of disease but not others (eg, healing
Indications
• Medically refractory luminal disease
• Uncontrolled perianal disease
• Active EIMs
• Concomitant immune-mediated disease
Patient characteristics
• Limited comorbidities
• Limited history of infections
• No active malignancy
• No good surgical alternatives/averse to surgery
• Not interested/does not qualify for clinical trial enrollment
of luminal inflammation but with persistent perianal fistula). When discussing ACT with a patient, surgical management or enrollment in a clinical trial also should be considered because these often are viable alternative options.
Outside of medically refractory disease, ACT also may be used to treat EIMs or concomitant immune-mediated disease. In this scenario, careful collaboration of the patient’s care team, including the primary care physician, colorectal surgeon, and subspecialists such as rheumatologists, dermatologists, and ophthalmologists, is essential. In addition, clinicians should carefully examine the patient’s phenotype and ensure that prior therapeutic regimens have been optimized to confirm whether prior options have truly been exhausted and not deemed unsuccessful because they did not improve a noninflammatory condition (fibrostenotic CD, bile salt–associated diarrhea, or superimposed irritable bowel syndrome).
When considering ACT, a patient’s risk profile also must be assessed. There are presumed additive risks when multiple immunomodulatory therapies are used in combination, but the potential degree and details of this are unknown. The ideal candidate for ACT should have minimal comorbidities and be in otherwise good physical health. Caution should be used in patients with a history of cancer or previous serious infections, as well as contraindications to each of the therapeutic agents being considered.
There are also limited data to suggest which advanced therapies a provider should choose to use in combination. As mentioned previously, the most common ACT regimens studied in cohorts are vedolizumab and an anti-TNF agent, ustekinumab and anti-integrins, and an anti-TNF agent and ustekinumab.25 The rational selection of agents for a combination regimen often focuses on avoidance of 2 systemically immunosuppressant medications (eg, an anti-TNF and a JAK [Janus kinase] inhibitor) to reduce the likelihood
Ideal therapy choices
• Novel to the patient
• Not novel but previously effective and/or low immunogenicity risk
• Good long-term safety data
• Addresses any EIMs or concomitant immune-mediated disease
Monitor and adjust as needed
• Follow clinical, endoscopic, and biochemical response
• Watch closely for infections or other AEs
• Consider de-escalating to monotherapy if remission is achieved
• Discontinue if not clinically effective or SAEs emerge
Figure. Strategies and considerations for advanced combination therapy use in IBD. AEs, adverse events; EIMs, extraintestinal manifestations; IBD, inflammatory bowel disease; SAEs, serious AEs.
of SAEs. A personalized care approach should be implemented when making this decision, with consideration of a patient’s medication exposure/response history, comorbidities, and presence of EIMs. Ideally, a patient would receive 2 therapies with mechanisms to which they are naive. Alternatively, a patient could receive therapies to which they previously had a positive clinical response. It is unclear whether previously ineffective therapies can be “recycled” and effective when used a second time in combination. When choosing a combination regimen, it is also important to select therapies that address each aspect of the patient’s disease phenotype and EIMs. For example, an anti-TNF agent should be considered in fistulizing disease, 31 ustekinumab or risankizumab can be considered in patients with concomitant psoriasis,32 and JAK inhibitors can be considered in patients with inflammatory joint conditions.33 Vedolizumab, in contrast, is less likely to effectively treat EIMs because it is considered more gut selective.22
Safety also must be considered in selecting which agents to combine. It is important to prioritize using agents with clear long-term safety data, such as vedolizumab34 or ustekinumab.35
Significant questions also remain about the acceptable duration of combination therapy. Many providers will choose to continue both therapies in combination indefinitely as long as there is sustained clinical effectiveness and a lack of significant AEs. However, as there is a paucity of long-term safety data, many providers will want to use combination therapy as a short-term treatment strategy or bridge to long-term monotherapy. One strategy is to use combination therapy during induction, and once a
References
1. Chang S, et al. Am J Gastroenterol. 2024;119(1):55-80.
2. Stalgis C, et al. Gastroenterology. 2021;161(2):394-399.
3. Peyrin-Biroulet L, et al. Aliment Pharmacol Ther. 2011;33(8):870-879.
4. Turner D, et al. Gastroenterology. 2021;160(5):1570-1583.
5. Balderramo D. World J Gastroenterol. 2022;28(47):6743-6751.
6. Sultan KS, et al. World J Gastrointest Pharmacol Ther 2017;8(2):103-113.
7. Hirten RP, et al. Clin Gastroenterol Hepatol. 2018;16(9):1374-1384.
8. Solitano V, et al. Gastroenterol Hepatol (N Y). 2023;19(5):251-263.
9. Colombel JF, et al. N Engl J Med. 2010;362(15):1383-1395.
10. Panaccione R, et al. Gastroenterology. 2014;146(2):392-400.
11. Sands BE, et al. Inflamm Bowel Dis. 2007;13(1):2-11.
12. Colombel JF, et al. Clin Gastroenterol Hepatol 2024;22(7):1487-1496.
13. Feagan BG, et al. Lancet Gastroenterol Hepatol. 2023;8(4):307-320.
14. Glassner K, et al. J Dig Dis. 2020;21(5):264-271.
15. Goessens L, et al. United European Gastroenterol J 2021;9(10):1136-1147.
16. McShane C, et al. J Crohns Colitis. 2023;17(suppl 1):i825-i826.
17. Alayo QA, et al. Inflamm Bowel Dis. 2021;27(10):1698-1702.
18. Yang E, et al. Aliment Pharmacol Ther. 2020;51(11):1031-1038.
19. Lee SD, et al. Inflamm Bowel Dis. 2022;28(2):309-313.
patient has achieved clinical remission, to withdraw one medication and use the other as monotherapy. The SPARE trial demonstrated that there was no increased relapse rate in patients previously maintained on TCT after the immunomodulator was withdrawn.36 In addition, one could consider using a rapidly effective agent, such as a JAK inhibitor, 37 while co-inducing a second therapy with a longer onset of action, such as vedolizumab or ustekinumab. One must, however, also consider the potential for immunogenicity or loss of responsiveness to an effective therapy if the above strategies are used.
Conclusion
Despite major advances in IBD therapeutics over the past several decades, additional treatment options are still needed. For subsets of patients with medically refractory and/or severe disease as well as those with EIMs or multiple immune-mediated inflammatory diseases, ACT has emerged as a potential strategy, based on the principles demonstrated with azathioprine and infliximab in the SONIC and UC-SUCCESS trials. 9,10 Although this remains an active area of investigation, promising data are starting to emerge to support advantages of ACT over optimized monotherapy with an acceptable short-term risk profile. Careful patient selection by an experienced IBD clinician is paramount. Current challenges include a lack of long-term outcome data, FDA-approved combination regimens, and GI society guidance, as well as difficulty in obtaining insurance authorization. Data from ongoing randomized controlled trials will help to inform the safety and efficacy of combination therapy approaches in IBD.
20. Llano EM, et al. Crohns Colitis 360. 2021;3(3):otab030.
21. Hu A, et al. Clin Gastroenterol Hepatol. 2021;19(7):1366-1376.
22. Sandborn WJ, et al. N Engl J Med. 2013;369(8):711-721.
23. Hanauer SB, et al. Gastroenterology. 2006;130(2):323-333.
24. Colombel JF, et al. Gastroenterology. 2007;132(1):52-65.
25. Ahmed W, et al. Clin Gastroenterol Hepatol. 2022;20(3):e361-e379.
26. Alayo QA, et al. Crohns Colitis 360. 2022;4(1):otac002.
27. Mackey AC, et al. J Pediatr Gastroenterol Nutr. 2007;44(2):265-267.
28. Boleto G, et al. Semin Arthritis Rheum. 2019;49(1):35-42.
29. Vedamurthy A, et al. Clin Gastroenterol Hepatol. 2022;20(1):88-95.
30. Axelrad J, et al. Clin Gastroenterol Hepatol. 2016;14(1):58-64.
31. Sands BE, et al. N Engl J Med. 2004;350(9):876-885.
32. Papp KA, et al. N Engl J Med. 2017;376(16):1551-1560.
33. Yamaoka K, et al. Immunol Med. 2023;46(3):143-152.
34. Schreiber S, et al. J Gastroenterol. 2018;53(9):1048-1064.
35. Sandborn WJ, et al. Clin Gastroenterol Hepatol. 2022;20(3):578-590.
36. Louis E, et al. Lancet Gastroenterol Hepatol. 2023;8(3):215-227.
37. Singh A, et al. Am J Gastroenterol. 2024;119(7):1365-1372.
Dr. Lukin reported financial relationships with AbbVie, Boehringer Ingelheim, Bristol Myers Squibb, Fresenius Kabi, Janssen, Lilly, Pfizer and Takeda. Dr. Mintz reported no relevant financial disclosures.
Between the Guidelines AGA Issues Guideline on Management of Pouchitis
The American Gastroenterological Association recently published a guideline on the management of pouchitis and inflammatory pouch disorders (Gastroenterology 2024;166[1]:59-85). GEN’s Sarah Tilyou spoke with lead author Edward Barnes, MD, MPH, an IBD specialist and assistant professor of medicine at the University of North Carolina at Chapel Hill, about the new guideline and its implications for practice.
GEN: What prompted the guideline?
Dr. Barnes: This is the first society-sponsored guideline related to pouchitis and inflammatory conditions of the pouch. There’s been a push in the last five or 10 years to standardize our approach and delve into the evidence on treating patients after they undergo an ileal pouch−anal anastomosis (IPAA) surgery and have these inflammatory conditions in the pouch. There have been several different consensus statements and different research groups that have led this push to standardize, but now we have come together as a group to develop consensus definitions on what these inflammatory conditions are, what the state of the evidence is and what the future directions are.
GEN: What’s new in the guideline that clinicians need to know?
Dr. Barnes: There are a couple of important points that are new and that codify what we have known for a long time. I separate those into two areas.
One is that we were very deliberate in how we tried to define these inflammatory conditions of the pouch. There is some new terminology, for example, the idea of intermittent pouchitis, which is really the most common complication after IPAA. In the literature, for a long time, this has been referred to as acute pouchitis. But it has been difficult to determine what acute pouchitis is and when that patient starts to transition to chronic pouchitis. Patients often have intermittent episodes of pouchitis, in which they have a defined increased activity that’s presumably related to inflammation in their pouch, but when we treat them with antibiotics or some other therapy, they undergo long periods of normal pouch function without additional therapy. We chose the term intermittent pouchitis for this, moving away from the less accurate term acute pouchitis.
The second thing we tried to do is better define chronic pouchitis. We wanted to get away from numeric definitions of more than four episodes per year, which has been the traditional definition in the literature. Some patients who are treated with antibiotics have recurrence of symptoms once the antibiotics are stopped. The condition is not intermittent anymore in these patients. They really need antibiotics all the time and would be considered to have chronic antibiotic-dependent pouchitis. In some patients— those with chronic antibiotic-refractory pouchitis—antibiotics stop working, and we start thinking about using immunosuppressive therapies and advanced therapies such as biologics and small molecules.
Those definitions, plus the definition of Crohn’s-like disease of the pouch, a term that has been used over the past several years, are some things that were new.
Another new idea we discussed was when the appropriate time is to introduce advanced therapies for patients with chronic pouchitis. That was informed by some really interesting discussions the guideline panel had, as well as
by the landmark EARNEST trial published last year, which evaluated vedolizumab [Entyvio, Takeda] in the treatment of chronic pouchitis [N Engl J Med 2023;388(13):1191-1200].
In the trial, investigators treated patients with antibiotics at the time of enrollment, then gave them vedolizumab or placebo. Vedolizumab was associated with improved remission rates in these patients with chronic pouchitis after IPAA. We are trying to get at the idea of whether we should be treating patients with chronic pouchitis earlier with advanced therapies, whether there is a potential advantage of that if a patient doesn’t want to take chronic antibiotics, with shared decision making between the treating provider and the patient.
GEN: Is there pushback from payors on use of biologics for patients with pouchitis?
Dr. Barnes: One thing that we are always cognizant of is how do we get patients appropriate access to medicines when they need them, and in the United States, insurance companies are a big piece of that puzzle. Although patients no longer technically meet a definition of ulcerative colitis after they have surgery and an IPAA because they don’t have a colon anymore, they still have the underlying IBD phenotype. I think that’s important for us as treating providers to remember, and it’s also important for the insurance companies to remember. That underlying phenotype that made that patient have such bad ulcerative colitis that they didn’t respond to available treatments and required surgery is still there. Some of these patients develop these chronic inflammatory conditions after surgery and will need reintroduction of these advanced therapies. We tried to be cognizant of that when we talked about what therapies potentially could be useful in that scenario.
Although no advanced therapies are specifically approved for pouchitis, current evidence indicates that anything that’s been approved for ulcerative colitis or Crohn’s disease conceivably could be used in that situation. We don’t have large randomized controlled trials or head-to-head comparative effectiveness studies outside of the EARNEST study and one small study of adalimumab. So we’re really trying to make these decisions on a oneon-one basis, considering a patient’s individual characteristics. Sometimes we do have pushback from insurance companies, but in the guidelines we aimed to help treating providers avoid some of this pushback in the future, so we can get these effective therapies to patients earlier.
GEN: How might the guideline change practice?
Dr. Barnes: I think a couple of different ways. One is that for a long time, there have been gray areas in how we treat pouchitis and inflammatory conditions of the pouch to some degree, and if you weren’t practicing in major
Pouchitis continued, page 61
For adults with moderately to severely active Crohn’s disease1
SKYRIZI PROVIDES THE OPPORTUNITY FOR ENDOSCOPIC AND SYMPTOM CONTROL. FOR YOUR PATIENTS, THAT’S EVERYTHING.
ENDOSCOPIC AND SYMPTOM
CO-PRIMARY ENDPOINT: Endoscopic Response (SES-CD)
CO-PRIMARY ENDPOINT: Clinical Remission (CDAI)
SECONDARY ENDPOINT:
Endoscopic Remission
SKYRIZI 40%, PLACEBO 12%
SKYRIZI 45%, PLACEBO 25%
SKYRIZI 24%, PLACEBO 9%
SKYRIZI 29%, PLACEBO 11%
SKYRIZI 42%, PLACEBO 20%
SKYRIZI 19%, PLACEBO 4%
SKYRIZI 600 mg IV n=336, PLACEBO n=175 p<0.001
SKYRIZI 360 mg 48%, SKYRIZI 180 mg 50%, PBO (Induction Responders) 22% p<0.05; all p-values are SKYRIZI treatment arms vs placebo.
SKYRIZI 360 mg 57%, SKYRIZI 180 mg 61%, PBO (Induction Responders) 46% p<0.05; all p-values are SKYRIZI treatment arms vs placebo.
This endpoint was not statistically significant under the prespecified multiple testing procedure.
SKYRIZI 600 mg IV n=191, PLACEBO n=187 p<0.001
Placebo (Induction Responders): Patients who achieved CDAI clinical response (CR-100) to SKYRIZI induction therapy and were randomized to receive placebo in the maintenance study.
Clinical Remission: Defined as a CDAI score <150 points.1
Endoscopic Remission: SES-CD ≤4 and at least a 2-point reduction vs baseline and no subscore >1 in any individual variable, as scored by a central reviewer.
Endoscopic Response: Defined as a decrease in SES-CD >50% from baseline, or a decrease of at least 2 points for subjects with a baseline score of 4 and isolated ileal disease, based on central reading. The sections evaluated on endoscopy are the rectum, sigmoid and left colon, transverse colon, right colon and ileum (per SES-CD assessment).
PIVOTAL TRIAL STUDY DESIGNS
SKYRIZI is indicated for the treatment of moderately to severely active Crohn’s disease in adults.
IMPORTANT SAFETY INFORMATION1
Hypersensitivity Reactions
SKYRIZI ® (risankizumab-rzaa) is contraindicated in patients with a history of serious hypersensitivity reaction to risankizumab-rzaa or any of the excipients. Serious hypersensitivity reactions, including anaphylaxis, have been reported with the use of SKYRIZI. If a serious hypersensitivity reaction occurs, discontinue SKYRIZI and initiate appropriate therapy immediately.
ADVANCE (N=850) and MOTIVATE (N=569) Induction studies were 12-week, randomized, double-blind, placebo-controlled studies that evaluated the efficacy and safety of SKYRIZI in patients with moderately to severely active Crohn’s disease who demonstrated prior treatment failure to conventional and/or biologic treatment. 2 Patients received an IV infusion of SKYRIZI 600 mg, 1200 mg, or placebo at Weeks 0, 4, and 8.1
FORTIFY (N=382) Maintenance study was a 52-week study that evaluated the efficacy and safety of SKYRIZI in patients who achieved clinical response (decrease in CDAI ≥100) from SKYRIZI induction in the ADVANCE and MOTIVATE studies. Patients were randomized to SKYRIZI 180 mg SC, SKYRIZI 360 mg SC, or placebo at Week 12 and every 8 weeks thereafter.1
INDICATION AND IMPORTANT SAFETY INFORMATION FOR SKYRIZI1 INDICATION1
Tuberculosis (TB)
Infection
SKYRIZI may increase the risk of infection. Do not initiate treatment with SKYRIZI in patients with a clinically important active infection until it resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefi ts prior to prescribing SKYRIZI. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If a patient develops such an infection or is not responding to standard therapy, closely monitor and discontinue SKYRIZI until the infection resolves.
Prior to initiating treatment with SKYRIZI, evaluate for TB infection and consider treatment in patients with latent or active TB for whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after SKYRIZI treatment. Do not administer SKYRIZI to patients with active TB.
Hepatotoxicity in Treatment of Crohn’s Disease
Drug-induced liver injury was reported in a patient with Crohn’s disease who was hospitalized for a rash during induction dosing of SKYRIZI. For the treatment of Crohn’s disease, evaluate liver enzymes and bilirubin at baseline and during induction (12 weeks); monitor thereafter according to routine patient management. Consider an alternate treatment for patients with evidence of liver cirrhosis. Interrupt treatment if druginduced liver injury is suspected, until this diagnosis is excluded. Instruct your patient to seek immediate medical attention if they experience symptoms suggestive of hepatic dysfunction.
Administration of Vaccines
Avoid use of live vaccines in patients treated with SKYRIZI. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating SKYRIZI, complete all age appropriate vaccinations according to current immunization guidelines.
STUDY DESIGN
SEQUENCE was a Phase 3, multicenter, randomized, open-label, effi cacy assessment-blinded ‡ study of SKYRIZI (n=255) compared to STELARA ® (ustekinumab) § (n=265) for the treatment of adult patients with moderate to severe Crohn’s disease who have failed anti-TNF therapy. Eligible patients were randomized (1:1) to receive either SKYRIZI (600 mg IV to 360 mg SC) or STELARA ® (ustekinumab) (weight-based|| IV to 90 mg SC). After induction dosing was completed, patients remained on their respective therapy throughout the duration of the maintenance period (treat-through study design). Dosing for both treatment arms was aligned to the US Prescribing Information with no dose escalation allowed throughout the trial.
POWERFUL SUPERIORITY DATA3,4
Endoscopic Remission at Week 48 (Superiority Endpoint, NRI-MI)
Patients at baseline had an average disease duration of ~9 years and average SES-CD score of 14
Clinical Remission at Week 24 (Non-inferiority Endpoint, NRI-MI)
‡ The investigator and site personnel were blinded to the results of the clinical outcomes (CDAI) for the duration of the study, and endoscopies were centrally read with assessors blinded to study drug.
§ Active Comparator: 31 patients received US-approved ustekinumab. All other patients received European Union-approved ustekinumab. The comparability between US- and non-US-approved ustekinumab has not been established.
Superiority Endpoint: This primary endpoint was evaluated based on a 0.05, 2-sided significance level.
Endoscopic Remission: SES-CD ≤4 and at least a 2-point reduction vs baseline and no subscore >1 in any individual variable, as scored by a central reviewer.
Non-inferiority Endpoint: This primary endpoint was measured in ~50% of total population. This measure was based on a non-inferiority margin of 10% at the 0.05, 2-sided significance level, where a margin of 10% was selected based on physicians’ perspective on the clinical meaningfulness of infl ammatory bowel disease trial results: an International Organization for the Study of Inflammatory Bowel Disease (IOIBD) survey.5
Adverse Reactions
Most common (>3%) adverse reactions associated with SKYRIZI in Crohn’s disease are upper respiratory infections, headache, and arthralgia in induction and arthralgia, abdominal pain, injection site reactions, anemia, pyrexia, back pain, arthropathy, and urinary tract infection in maintenance.
Lipid Elevations: Increases from baseline and increases relative to placebo were observed at Week 4 and remained stable to Week 12 in patients treated with SKYRIZI in Crohn’s disease.
Dosage Forms and Strengths: SKYRIZI is available in a 600 mg/10 mL single-dose vial for intravenous infusion and a 180 mg/1.2 mL or 360 mg/2.4 mL single-dose prefi lled cartridge with on-body injector.
Please see the brief summary of the full Prescribing Information on the following pages. DEMONSTRATED SYMPTOM RELIEF DATA3,4
Clinical Remission: Defined as a CDAI score <150 points.1
NRI-MI: Non-responder imputation for missing data with the exception that if the reason for missing data is due to COVID-19 infection or logistical restriction due to pandemic or geopolitical confl ict, the patient’s assessment will be imputed using multiple imputation.
* The mixed population includes patients who had inadequate response, loss of response, or intolerance to one or more biologics (biologic failure), as well as patients who had never demonstrated inadequate response, loss of response, or intolerance to a biologic (bio-naïve; includes 13% in ADVANCE and 8% in FORTIFY who were bio-exposed).
INTERESTED IN SEEING ADDITIONAL RESULTS?
WWW.SKYRIZIHCP.COM
† Prior biologic failure includes inadequate response, loss of response, or intolerance to one or more biologics.
|| Baseline STELARA® (ustekinumab) IV dose is weight-based: ≤55 kg: 260 mg dose, >55 kg to 85 kg: 390 mg dose, or >85 kg: 520 mg dose.
STELARA ® is a registered trademark of Johnson & Johnson. See US Prescribing Information for further information.
References: 1. SKYRIZI [package insert]. North Chicago, IL: AbbVie Inc. 2. D’Haens G, Panaccione R, Baert F, et al. Risankizumab as induction therapy for Crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet. 2022; 399(10340):2015-2030. 3. Peyrin-Biroulet L, Chapman JC, Colombel J-F, et al. Risankizumab versus ustekinumab for patients with moderate to severe Crohn’s disease: results from the Phase 3b SEQUENCE study. Presented at United European Gastroenterology Week (UEGW 2023), October 14 – 17, 2023, Copenhagen, Denmark. 4. Data on File, AbbVie Inc, ABVRRTI76928. 5. Olivera P, Sandborn WJ, Panés J, et al. Physicians’ perspective on the clinical meaningfulness of infl ammatory bowel disease trial results: an International Organization for the Study of Infl ammatory Bowel Disease (IOIBD) survey. Aliment Pharmacol Ther. 2018;47(6):773-783.
SKYRIZI® (sky-RIZZ-ee) (risankizumab-rzaa) injection, for subcutaneous or intravenous use
90 mg/mL single-dose prefilled syringe
150 mg/mL single-dose pen and prefilled syringe
600 mg/10 mL single-dose vial for intravenous infusion
180 mg/1.2 mL single-dose prefilled cartridge with on-body injector
360 mg/2.4 mL single-dose prefilled cartridge with on-body injector
INDICATIONS AND USAGE
Plaque Psoriasis
SKYRIZI® is indicated for the treatment of moderate-to-severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy.
Psoriatic Arthritis
SKYRIZI is indicated for the treatment of active psoriatic arthritis in adults.
Crohn’s Disease
SKYRIZI is indicated for the treatment of moderately to severely active Crohn’s disease in adults.
CONTRAINDICATIONS
SKYRIZI is contraindicated in patients with a history of serious hypersensitivity reaction to risankizumab-rzaa or any of the excipients [see Warnings and Precautions]
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been reported with use of SKYRIZI. If a serious hypersensitivity reaction occurs, discontinue SKYRIZI and initiate appropriate therapy immediately [see Adverse Reactions]
Infections
SKYRIZI may increase the risk of infections [see Adverse Reactions] Treatment with SKYRIZI should not be initiated in patients with any clinically important active infection until the infection resolves or is adequately treated.
In patients with a chronic infection or a history of recurrent infection, consider the risks and benefits prior to prescribing SKYRIZI. Instruct patients to seek medical advice if signs or symptoms of clinically important infection occur. If a patient develops such an infection or is not responding to standard therapy, monitor the patient closely and do not administer SKYRIZI until the infection resolves.
Tuberculosis
Evaluate patients for tuberculosis (TB) infection prior to initiating treatment with SKYRIZI. Across the Phase 3 psoriasis clinical studies, of the 72 subjects with latent TB who were concurrently treated with SKYRIZI and appropriate TB prophylaxis during the studies, none developed active TB during the mean follow-up of 61 weeks on SKYRIZI. Two subjects taking isoniazid for treatment of latent TB discontinued treatment due to liver injury. Of the 31 subjects from the PsO-3 study with latent TB who did not receive prophylaxis during the study, none developed active TB during the mean follow-up of 55 weeks on SKYRIZI. Consider anti-TB therapy prior to initiating SKYRIZI in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed. Monitor patients for signs and symptoms of active TB during and after SKYRIZI treatment. Do not administer SKYRIZI to patients with active TB.
Hepatotoxicity in Treatment of Crohn’s Disease
A serious adverse reaction of drug-induced liver injury in conjunction with a rash that required hospitalization was reported in a patient with Crohn’s disease (ALT 54x ULN, AST 30x ULN, and total bilirubin 2.2x ULN) following two 600 mg intravenous doses of SKYRIZI. The liver test abnormalities resolved following administration of steroids. SKYRIZI was subsequently discontinued.
For the treatment of Crohn’s disease, evaluate liver enzymes and bilirubin at baseline, and during induction at least up to 12 weeks of treatment. Monitor thereafter according to routine patient management.
Consider other treatment options in patients with evidence of liver cirrhosis. Prompt investigation of the cause of liver enzyme elevation is recommended to identify potential cases of drug-induced liver injury. Interrupt treatment if drug-induced liver injury is suspected, until this diagnosis is excluded. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of hepatic dysfunction.
Administration of Vaccines
Avoid use of live vaccines in patients treated with SKYRIZI. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Prior to initiating therapy with SKYRIZI, complete all age-appropriate vaccinations according to current immunization guidelines. No data are available on the response to live or inactive vaccines.
ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of labeling:
•Hypersensitivity Reactions [see Warnings and Precautions]
•Infections [see Warnings and Precautions]
•Tuberculosis [see Warnings and Precautions]
•Hepatotoxicity in Treatment of Crohn’s Disease [see Warnings and Precautions]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse drug reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Plaque Psoriasis
A total of 2234 subjects were treated with SKYRIZI in clinical development trials in plaque psoriasis. Of these, 1208 subjects with psoriasis were exposed to SKYRIZI for at least one year.
Data from placebo- and active-controlled trials were pooled to evaluate the safety of SKYRIZI for up to 16 weeks. In total, 1306 subjects were evaluated in the SKYRIZI 150 mg group.
Table 1 summarizes the adverse drug reactions that occurred at a rate of at least 1% and at a higher rate in the SKYRIZI group than the placebo group during the 16-week controlled period of pooled clinical trials.
Table 1. Adverse Drug Reactions Occurring in ≥ 1% of Subjects on SKYRIZI through Week 16
Adverse Drug Reactions SKYRIZI N = 1306 n (%) Placebo N = 300 n (%)
Upper respiratory infectionsa 170 (13.0)29
Injection site reactionsd
(1.5)3 (1.0) Tinea infectionse 15 (1.1)1 (0.3)
a Includes: respiratory tract infection (viral, bacterial or unspecified), sinusitis (including acute), rhinitis, nasopharyngitis, pharyngitis (including viral), tonsillitis
b Includes: headache, tension headache, sinus headache, cervicogenic headache
c Includes: fatigue, asthenia
d Includes: injection site bruising, erythema, extravasation, hematoma, hemorrhage, infection, inflammation, irritation, pain, pruritus, reaction, swelling, warmth
e Includes: tinea pedis, tinea cruris, body tinea, tinea versicolor, tinea manuum, tinea infection, onychomycosis
Adverse drug reactions that occurred in < 1% but > 0.1% of subjects in the SKYRIZI group and at a higher rate than in the placebo group through Week 16 were folliculitis and urticaria.
Specific Adverse Drug Reactions Infections
In the first 16 weeks, infections occurred in 22.1% of the SKYRIZI group (90.8 events per 100 subject-years) compared with 14.7% of the placebo group (56.5 events per 100 subject-years) and did not lead to discontinuation of SKYRIZI. The rates of serious infections for the SKYRIZI group and the placebo group were ≤0.4%. Serious infections in the SKYRIZI group included cellulitis, osteomyelitis, sepsis, and herpes zoster. In Studies PsO-1 and PsO-2, through Week 52, the rate of infections (73.9 events per 100 subject-years) was similar to the rate observed during the first 16 weeks of treatment.
Safety Through Week 52
Through Week 52, no new adverse reactions were identified, and the rates of the adverse reactions were similar to those observed during the first 16 weeks of treatment. During this period, serious infections that led to study discontinuation included pneumonia.
Psoriatic Arthritis
The overall safety profile observed in subjects with psoriatic arthritis treated with SKYRIZI is generally consistent with the safety profile in subjects with plaque psoriasis. Additionally, in the Phase 3 placebo-controlled trials the incidence of hepatic events was higher in the SKYRIZI group (5.4%, 16.7 events per 100 patient years) compared to the placebo group (3.9%, 12.6 events per 100 patient years). Of these, the most common events that were reported more frequently in both the placebo group and the SKYRIZI group were ALT increased (placebo: n=12 (1.7%); SKYRIZI: n=16 (2.3%)), AST increased (placebo: n=9 (1.3%); SKYRIZI: n=13 (1.8%)), and GGT increased (placebo: n=5 (0.7%); SKYRIZI: n=8 (1.1%)). There were no serious hepatic events reported. The incidence of hypersensitivity reactions was higher in the SKYRIZI group (n=16, 2.3%) compared to the placebo group (n=9, 1.3%). In the Phase 3 placebo-controlled trials, hypersensitivity reactions reported at a higher rate in the SKYRIZI group included rash (placebo: n=4 (0.6%); SKYRIZI: n=5 (0.7%), allergic rhinitis (placebo: n=1 (0.1%); SKYRIZI: n=2 (0.3%), and facial swelling (placebo: n=0 (0.0%); SKYRIZI n=1 (0.1%). One case of anaphylaxis was reported in a subject who received SKYRIZI in the Phase 2 clinical trial.
Crohn’s Disease
SKYRIZI was studied up to 12 weeks in subjects with moderately to severely active Crohn’s disease in two randomized, double-blind, placebo-controlled induction studies (CD-1, CD-2) and a randomized, double-blind, placebocontrolled, dose-finding study (CD-4; NCT02031276). Long-term safety up to 52 weeks was evaluated in subjects who responded to induction therapy in a randomized, double-blind, placebo-controlled maintenance study (CD-3). In the two induction studies (CD-1, CD-2) and the dose finding study (CD-4), 620 subjects received the SKYRIZI intravenous induction regimen at Weeks 0, 4 and 8. In the maintenance study (CD-3), 297 subjects who achieved clinical response, defined as a reduction in CDAI of at least 100 points from baseline after 12 weeks of induction treatment with intravenous SKYRIZI in studies CD-1 and CD-2, received a maintenance regimen of SKYRIZI either 180 mg or 360 mg subcutaneously at Week 12 and every 8 weeks thereafter for up to an additional 52 weeks.
Adverse reactions reported in > 3% of subjects in induction studies and at a higher rate than placebo are shown in Table 2.
PROFESSIONAL BRIEF SUMMARY CONSULT PACKAGE INSERT FOR FULL PRESCRIBING INFORMATION
Table 2. Adverse Drug Reactions Reported in > 3% of Subjects with Crohn’s Disease Treated with SKYRIZI in Placebo-Controlled 12-Week Induction Studies
Adverse
a SKYRIZI 600 mg as an intravenous infusion at Week 0, Week 4, and Week 8.
Adverse reactions reported in >3% of subjects in the maintenance study and at a higher rate than placebo are shown in Table 3.
Table 3. Adverse Reactions Reported in >3% of Subjects with Crohn’s Disease Treated with SKYRIZIa in PlaceboControlled 52-Week Maintenance Study (CD-3)
Adverse Drug Reactions
tract infection1 (0.6)5 (3.5)4 (2.8) a SKYRIZI 180 mg or 360 mg at Week 12 and every 8 weeks thereafter for up to an additional 52 weeks
b Includes: abdominal pain, abdominal pain upper, abdominal pain lower c Includes: injection site rash, injection site erythema, injection site swelling, injection site urticaria, injection site warmth, injection site pain, injection site hypersensitivity, injection site reaction
d Some subjects had multiple occurrences of injection site reactions. In this table, injection site reactions are counted only once per subject for the rate calculations.
Specific Adverse Drug Reactions
Infections
In the maintenance study (CD-3) through Week 52, the rate of infections was 32.3% (50.2 events per 100 subject-years) in subjects who received SKYRIZI 180 mg and 36.6% (60.8 events per 100 subject-years) in subjects who received SKYRIZI 360 mg compared to 36.4% (60.3 events per 100 subject-years) in subjects who received placebo after SKYRIZI induction. The rate of serious infections was 2.6% (2.7 events per 100 subject-years) in subjects who received SKYRIZI 180 mg and 5.6% (7.4 events per 100 subject-years) in subjects who received SKYRIZI 360 mg compared to 2.1% (2.4 events per 100 subject-years) in subjects who received placebo after SKYRIZI induction.
Lipid Elevations
Elevations in lipid parameters (total cholesterol and low-density lipoprotein cholesterol [LDL-C]) were first assessed at 4 weeks following initiation of SKYRIZI in the induction trials (CD-1, CD-2). Increases from baseline and increases relative to placebo were observed at Week 4 and remained stable to Week 12. Following SKYRIZI induction, mean total cholesterol increased by 9.4 mg/dL from baseline to a mean absolute value of 175.1 mg/dL at Week 12. Similarly, mean LDL-C increased by 6.6 mg/dL from baseline to a mean absolute value of 92.6 mg/dL at Week 12. Mean LDL-C increased by 3.1 mg/dL from baseline to a mean absolute value of 99.0 mg/dL at Week 52 with SKYRIZI 180 mg maintenance treatment and by 2.3 mg/dL from baseline to a mean absolute value of 102.2 mg/dL at Week 52 with SKYRIZI 360 mg maintenance treatment.
Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products, including other risankizumab products, may be misleading.
Plaque Psoriasis
By Week 52, approximately 24% (263/1079) of subjects treated with SKYRIZI at the recommended dose developed antibodies to risankizumabrzaa. Of the subjects who developed antibodies to risankizumab-rzaa, approximately 57% (14% of all subjects treated with SKYRIZI) had antibodies that were classified as neutralizing. Higher antibody titers in approximately 1% of subjects treated with SKYRIZI were associated with lower risankizumab-rzaa concentrations and reduced clinical response.
Psoriatic Arthritis
By Week 28, approximately 12.1% (79/652) of subjects treated with SKYRIZI at the recommended dose developed antibodies to risankizumab-rzaa.
None of the subjects who developed antibodies to risankizumab-rzaa had antibodies that were classified as neutralizing. Antibodies to risankizumabrzaa were not associated with changes in clinical response for psoriatic arthritis. A higher proportion of subjects with anti-drug antibodies experienced hypersensitivity reactions (6.3% (5/79)) and injection site reactions (2.5% (2/79)) compared to subjects without anti-drug antibodies (3.8% (22/574) with hypersensitivity reactions and 0.7% (4/574) with injection site reactions). None of these hypersensitivity and injection site reactions led to discontinuation of risankizumab-rzaa.
Crohn’s Disease
By Week 64, antibodies to risankizumab-rzaa developed in approximately 3.4% (2/58) of subjects treated with SKYRIZI induction followed by 360 mg maintenance regimen. No subjects (0/57) treated with SKYRIZI induction followed by 180 mg maintenance regimen developed antibodies to risankizumab-rzaa. None of the subjects who developed antibodies to risankizumab-rzaa had antibodies that were classified as neutralizing.
Postmarketing Experience
The following adverse reactions have been reported during post-approval of SKYRIZI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to SKYRIZI exposure:
• Skin and subcutaneous tissue disorders: eczema and rash USE IN SPECIFIC POPULATIONS
Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors outcomes in women who become pregnant while treated with SKYRIZI. Patients should be encouraged to enroll by calling 1-877-302-2161 or visiting http://glowpregnancyregistry.com.
Risk Summary
Available pharmacovigilance and clinical trial data with risankizumab use in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Although there are no data on risankizumab-rzaa, monoclonal antibodies can be actively transported across the placenta, and SKYRIZI may cause immunosuppression in the in utero-exposed infant There are adverse pregnancy outcomes in women with inflammatory bowel disease (see Clinical Considerations)
In an enhanced pre- and post-natal developmental toxicity study, pregnant cynomolgus monkeys were administered subcutaneous doses of 5 or 50 mg/kg risankizumab-rzaa once weekly during the period of organogenesis up to parturition. Increased fetal/infant loss was noted in pregnant monkeys at the 50 mg/kg dose (see Data). The 50 mg/kg dose in pregnant monkeys resulted in approximately 10 times the exposure (AUC) in humans administered the 600 mg induction regimen and 39 times the exposure (AUC) to the 360 mg maintenance doses, respectively. No risankizumab-rzaa-related effects on functional or immunological development were observed in infant monkeys from birth through 6 months of age. The clinical significance of these findings for humans is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and embryo/fetal risk
Published data suggest that the risk of adverse pregnancy outcomes in women with inflammatory bowel disease is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Fetal/Neonatal adverse reactions
Transport of endogenous IgG antibodies across the placenta increases as pregnancy progresses, and peaks during the third trimester. Because risankizumab may interfere with immune response to infections, risks and benefits should be considered prior to administering live vaccines to infants exposed to SKYRIZI in utero. There are insufficient data regarding infant serum levels of risankizumab at birth and the duration of persistence of risankizumab in infant serum after birth. Although a specific timeframe to delay live virus immunizations in infants exposed in utero is unknown, a minimum of 5 months after birth should be considered because of the half-life of the product.
Data
Animal Data
An enhanced pre- and post-natal developmental toxicity study was conducted in cynomolgus monkeys. Pregnant cynomolgus monkeys were administered weekly subcutaneous doses of risankizumab-rzaa of 5 or 50 mg/kg from gestation day 20 to parturition, and the cynomolgus monkeys (mother and infants) were monitored for 6 months after delivery. No maternal toxicity was noted in this study. There were no treatmentrelated effects on growth and development, malformations, developmental immunotoxicology, or neurobehavioral development. However, a dosedependent increase in fetal/infant loss was noted in the risankizumabrzaa-treated groups (32% and 43% in the 5 mg/kg and 50 mg/kg groups, respectively) compared with the vehicle control group (19%). The increased fetal/infant loss in the 50 mg/kg group was considered to be related to risankizumab-rzaa treatment. The no observed adverse effect level (NOAEL) for maternal toxicity was identified as 50 mg/kg and the NOAEL for developmental toxicity was identified as 5 mg/kg. On an exposure (AUC) basis, the 5 mg/kg dose in pregnant monkeys resulted in approximately 1.24 times the exposure in humans administered the 600 mg induction regimen and 5 times the exposure in humans administered the 360 mg maintenance doses, respectively. In the infants, mean serum concentrations increased in a dose-dependent manner and were approximately 17%-86% of the respective maternal concentrations. Following delivery, most adult female cynomolgus monkeys and all infants from the risankizumab-rzaatreated groups had measurable serum concentrations of risankizumab-rzaa up to 91 days postpartum. Serum concentrations were below detectable levels at 180 days postpartum.
Lactation
Risk Summary
There are no data on the presence of risankizumab-rzaa in human milk, the effects on the breastfed infant, or the effects on milk production. Endogenous maternal IgG and monoclonal antibodies are transferred in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to risankizumab-rzaa are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SKYRIZI and any potential adverse effects on the breastfed infant from SKYRIZI or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of SKYRIZI have not been established in pediatric patients.
Geriatric Use
Of the 2234 subjects with plaque psoriasis exposed to SKYRIZI, 243 subjects were 65 years or older and 24 subjects were 75 years or older. No overall differences in SKYRIZI exposure, safety, or effectiveness were observed between older and younger subjects who received SKYRIZI. However, the number of subjects aged 65 years and older was not sufficient to determine whether they respond differently from younger subjects.
Clinical studies of SKYRIZI for the treatment of Crohn’s disease did not include sufficient numbers of subjects 65 years of age and older to determine whether they respond differently from younger adult subjects. No clinically meaningful differences in the pharmacokinetics of risankizumab-rzaa were observed in geriatric subjects compared to younger adult subjects with Crohn’s disease.
PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Hypersensitivity Reactions
Advise patients to discontinue SKYRIZI and seek immediate medical attention if they experience any symptoms of serious hypersensitivity reactions [see Warnings and Precautions].
Infections
Inform patients that SKYRIZI may lower the ability of their immune system to fight infections. Instruct patients of the importance of communicating any history of infections to the healthcare provider and contacting their healthcare provider if they develop any symptoms of an infection [see Warnings and Precautions]
Hepatotoxicity in Treatment of Crohn’s Disease
Inform patients that SKYRIZI may cause liver injury, especially during the initial 12 weeks of treatment. Instruct patients to seek immediate medical attention if they experience symptoms suggestive of liver dysfunction. (e.g., unexplained rash, nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine) [see Warnings and Precautions].
Administration of Vaccines
Advise patients that vaccination with live vaccines is not recommended during SKYRIZI treatment and immediately prior to or after SKYRIZI treatment. Medications that interact with the immune system may increase the risk of infection following administration of live vaccines. Instruct patients to inform the healthcare practitioner that they are taking SKYRIZI prior to a potential vaccination [see Warnings and Precautions]
Administration Instruction
Instruct patients or caregivers to perform the first self-injected dose under the supervision and guidance of a qualified healthcare professional for training in preparation and administration of SKYRIZI, including choosing anatomical sites for administration, and proper subcutaneous injection technique.
If using SKYRIZI 90 mg/mL, instruct patients or caregivers to administer two 90 mg single-dose syringes to achieve the full 180 mg maintenance dose or four 90 mg single-dose syringes to achieve the full 360 mg maintenance dose of SKYRIZI for Crohn’s disease.
Instruct patients or caregivers in the technique of pen or syringe disposal.
Pregnancy
Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to SKYRIZI during pregnancy [see Use in Specific Populations]
A #MondayNightIBD Conversation Treating Patients With Inflammatory Bowel Disease And Obesity
Patricia Kaazan, MD, AMCc, FRACP
Gastroenterologist IBDSA
The University of Adelaide
Adelaide, Australia
@KaazanPatricia
Poonam Beniwal-Patel, MD
Associate Professor of Medicine
Division of Gastroenterology & Hepatology
Medical College of Wisconsin
Milwaukee, Wisconsin
@BeniwalPatelMD
Mehak Bassi, MD
Gastroenterology Fellow
Saint Peter’s Universtiy Hospital/Rutgers University
New Brunswick, New Jersey
@BassiMehak
Ifrah Fatima, MD
PGY-3 Internal Medicine Resident
University of Missouri–Kansas City
Kansas City, Missouri
@ifrahfatima
AAline Charabaty, MD, AGAF, FACG
Associate Professor of Clinical Medicine
The Johns Hopkins University School of Medicine
Baltimore, Maryland
Clinical Director, IBD Center
Johns Hopkins–Sibley Memorial Hospital
Washington, DC
@DCharabaty
n estimated 41% of the US population is obese (defined as a body mass index ≥ 30 kg/m 2), including 22% of individuals aged 12 to 19 years,1,2 while nearly 1 of 100 Americans has a diagnosis of inflammatory bowel disease. 3 An estimated 15% to 40% of individuals with IBD are obese and 20% to 40% are overweight (BMI, 25-30 kg/m 2). 4
Obesity, particularly visceral obesity, which itself is a chronic inflammatory state in white adipose tissue, may be a risk factor for developing IBD, and obese patients are at a higher risk for IBD-associated complications compared with nonobese IBD patients. Each increase of 5 kg/m 2 in BMI is associated with a 16% increased risk of developing Crohn’s disease. 5 Obese IBD patients are more likely to experience flares, complications from IBD surgeries, and IBD-related hospitalizations, as well as lower overall quality of life. 4
Although earlier studies evaluated obesity using BMI, more recently investigators are using body composition, which looks at fat and lean mass alterations, as a more accurate and specific measure of obesity. A prospective study of 141 patients with IBD and 51 controls found that patients with higher intraabdominal visceral adipose tissue (IA-VAT) were less likely than those with lower IA-VAT percentages to achieve corticosteroid-free deep remission (P<0.001) and/or endoscopic remission (P=0.02).6
Tailoring IBD therapy in patients with obesity requires an understanding of how obesity affects IBD disease course and prognosis, efficacy of drug therapy, and surgical outcomes. Comprehensive IBD care of this population should include interventions for weight loss.
In this article, we summarize 2 #MondayNightIBD case conversations that focused on managing patients with IBD and obesity, providing information to guide clinicians in the treatment of these patients. We explore factors to consider in treatment decisions in this population based on the available data and expert opinions, reviewing the efficacy and safety data on anti-obesity medications and endoscopic and surgical procedures in patients with IBD.
Take-Home Messages for the Management of IBD With Obesity
Some conclusions can be drawn from the available data and discussion on #MondayNightIBD to help guide clinicians in their treatment decisions for patients with IBD and obesity:
• Up to 40% of patients in the United States with IBD are obese.
• Obesity may increase the risk for developing Crohn’s disease and for poor IBD outcomes (eg, increased risk for flares, need for steroids, and need for hospitalization).
• Obesity is associated with an increased risk for medical treatment failure, whether patients are treated with weight-based therapies (thiopurines, IV infliximab) or “fixed-dose subcutaneous antiTNFs or non–anti-TNF therapies. There are limited data on the effects of obesity on the efficacy of ustekinumab (Stelara, Janssen), the anti–IL-23 p19 antibodies mirikizumab (Omvoh, Lilly) and risankizumab (Skyrizi, AbbVie), and JAKi.
• IV infliximab may confer an advantage over subcutaneous anti-TNF medications, with data showing an increased risk for accelerated time to loss of response with subcutaneous adalimumab compared with IV infliximab in obese patients.
• Obesity does not seem to affect the pharmacodynamics and pharmacokinetics of
Clinical Case Scenario 1
The first case described a patient with pan-ulcerative colitis (UC) and a BMI of 54 kg/m2, with secondary loss of response to infliximab due to antidrug antibodies and no response to vedolizumab (Entyvio, Takeda). Our IBD poll asked gastroenterologists and other healthcare professionals how they would advise the patient in this case (Figure 1).
Obesity and IBD Drug Efficacy
Clinicians responding to our poll on treatment decisions in the first case cited data showing that obesity leads to accelerated drug clearance and increased central volume of distribution, leading to reduced drug concentration (and subsequently decreased efficacy), particularly with anti–tumor necrosis factor (anti-TNF) drugs.7 A retrospective study of 124 bio-naive patients found that patients with Crohn’s disease and obesity and those with UC who are overweight had an earlier time to loss of response to infliximab.8 Another study found that BMI was independently associated with an increased risk for treatment failure in 160 bio-naive patients with UC, whether they were treated with weight-based IV infliximab, fixed-dose subcutaneous anti-TNFs, or IV vedolizumab. Each 1-kg/m2 increase
JAKi, but the potential risk for cardiovascular events in this patient population needs to be carefully assessed when considering JAKi therapies.
• Anti-TNF agents and non–antiTNF therapies are associated with a moderate weight gain in patients with IBD, but a small percentage of patients can experience significant weight gain with IV infliximab.
• Obesity increases the risk for IBD-related surgery complications, including surgical site infections, impaired wound healing, impaired ostomy function, and thromboembolic events. In addition, obesity causes technical difficulties during surgery, particularly during creation of an ostomy and IPAA.
• Anti-obesity medications and procedures have been underused in patients with IBD. The limited data available from small case series suggest that GLP-1 agonist anti-obesity medications and bariatric procedures (endoscopic or surgical) are effective and safe in patients with IBD. In addition, the weight loss following these interventions has been associated with improved IBD outcomes, suggesting that weight loss has a positive effect on gut inflammation.
in BMI was associated with a 4% to 8% higher risk for adverse outcomes, including treatment failure, IBD-related surgery, and/ or hospitalization.9 A pooled meta-analysis of randomized controlled trials and observational cohort studies that investigated the outcome of anti-TNF therapies, with stratification according to BMI, showed that obesity increased the odds of treatment failure of both dose-fixed (eg, subcutaneous adalimumab) and weight-based anti-TNF agents (eg, IV infliximab) in patients with UC but not in those with Crohn’s disease.10 A study of 181 bionaive patients had similar findings, particularly for those taking adalimumab. Although 68% of patients in the study had loss of response to either infliximab or adalimumab, in those with a BMI of at least 30 kg/m2, up to 83% had loss of response to adalimumab and 61% to infliximab. While there was no difference in time to loss of response to infliximab based on BMI (P=0.177), there was an increased accelerated time to loss of response for adalimumab-treated patients with a BMI of at least 30 kg/m2 compared with less than 30 kg/m2 (P=0.026).11
In patients who have developed anti-infliximab antibodies, such as the patient in this case, it is recommended that if adalimumab is to be started, it should be used in
Colectomy
Figure 1. Clinician poll on case No. 1.
A patient with pan-ulcerative colitis and a BMI of 54 kg/m2, with secondary loss of response to infliximab due to antidrug antibodies and no response to vedolizumab (Entyvio, Takeda). What is the next best option for this patient?
BMI, body mass index; JAKi, Janus kinase inhibitor; IL, interleukin; TNF, tumor necrosis factor.
Source: @MondayNightIBD
combination with thiopurines to prevent antibodies to the second anti-TNF. Although thiopurines doses are weightbased, increasing BMI results in lower 6-thioguanine nucleotide (6-TGN) and higher 6-methylmercaptopurine nucleotide levels. One study found that every 5-kg/m2 increase in BMI was related to an 8% decrease in 6-TGN, and patients with obesity were found to be more likely to have subtherapeutic levels of 6-TGN.12 Another retrospective review found that patients with a BMI higher than 25 kg/m2 had reduced response to azathioprine compared with those with a BMI below that.13
In the post hoc analysis of the IM-UNITI trial, BMI influenced drug levels of the anti-interleukin-12/23 p40 antibody ustekinumab (Stelara, Janssen) but did not affect clinical response.14 Another post hoc analysis looked at patients with UC treated with placebo versus tofacitinib and stratified patients by BMI (<25, 25 to <30, and ≥30 kg/m2).15 The study did not find that BMI was a significant predictor of remission,
Clinical Case Scenario 2
The second case described a patient with Crohn’s disease in remission on IV infliximab and thiopurines who had a BMI of 40 kg/m2 and chose to discontinue infliximab due to drugassociated weight gain. She remained in remission on thiopurine monotherapy for 6 years but subsequently developed a sigmoid stricture and an associated abdominal abscess, requiring surgical resection of the affected segment and a colostomy. Our IBD poll asked gastroenterologists and other healthcare professionals how they would advise the patient (Figure 2).
IBD Medications and Weight Gain
Clinicians reported instances of weight gain in patients receiving biologics, noting that the weight gain was modest most of the time and did not warrant discontinuing therapy. Indeed, in a retrospective study, the mean change in weight
although serious infections were numerically greater in the higher BMI subgroup (3.2%) versus other subgroups (0.4%). In addition, the risks and benefits of Janus kinase inhibitor (JAKi) therapy should be assessed in the context of a potential increased risk for major adverse cardiac events, given a patient’s age and the presence of cardiovascular risk factors of obesity and hyperlipidemia. If a JAKi therapy is initiated, lipid levels should be monitored, and anti-lipid drugs may need to be adjusted.16
Finally, the consideration of colectomy sparked extensive debate among the participants, primarily due to the association of high BMI with an increased risk for surgical complications, compromised ostomy function, and the challenges involved in performing an ostomy or an ileal pouch–anal anastomosis (IPAA) in patients with central obesity. 4 However, most clinicians agreed that surgery should be explored and discussed with a patient with active UC after the loss or lack of response to 2 effective therapies.
on infliximab was 3.3 kg (±6.5 kg), or 6.6 lb (±13 lb).17 Weight gain is thought to be due to the control of gut inflammation and symptoms, decreased intestinal permeability, improved dietary intake, and increased absorption of nutrients.18 A retrospective study of 294 patients noted more weight gain in patients treated with infliximab compared with those receiving adalimumab or vedolizumab. Although most patients had modest weight gain, up to 5% of the cohort patients, most on infliximab, had marked weight gain (up to 24.3 kg, or 53.46 lb).19 Some clinicians confirmed they noted isolated cases in their practice of excessive weight gain on infliximab out of proportion with what is expected with gut healing. In a retrospective study, infliximab-associated weight gain was more significant in patients with Crohn’s disease (4.97 kg, or 11 lb) than in those with rheumatoid arthritis (1.1 kg, or 2.42 lb),
Figure 2. Clinician poll on case No. 2.
A patient with Crohn’s disease in remission on IV infliximab and thiopurines who had a BMI of 40 kg/m2 and chose to discontinue infliximab due to drug-associated weight gain. She remained in remission on thiopurine monotherapy for 6 years but subsequently developed a sigmoid stricture and an associated abdominal abscess, requiring surgical resection of the affected segment and a colostomy. What is the next best option for this patient?
BMI, body mass index; GLP-1, glucagon-like peptide-1; JAKi, Janus kinase inhibitor; TNFi, tumor necrosis factor inhibitor.
Source: @MondayNightIBD
Figure 3. Patient poll No. 1.
During the course of your disease, did your IBD symptoms or treatment (medical or surgical) affect your body image?
Source: @MondayNightIBD
suggesting a potential direct effect of infliximab on weight gain in patients with IBD that is not related solely to the control of gut inflammation.20
It is unclear whether switching from IV to subcutaneous infliximab will reduce the risk for weight gain in this patient. Given the history of Crohn’s disease complications and the fact that the patient has been maintained on thiopurines after stopping infliximab (hence preventing the development of anti-infliximab antibodies), infliximab remains an appropriate option for this patient.
The role of glucagon-like peptide-1 (GLP-1) agonists is crucial in facilitating weight loss while reintroducing infliximab.
Non–anti-TNF options include ustekinumab, the anti–IL-23 p19 antibody risankizumab (Skyirzi, AbbVie), and the JAKi upadacitinib (Rinvoq, AbbVie), which are all effective options in patients with moderate to severe Crohn’s disease, including those who were previously exposed to biologics.
Finally, clinicians who chose to resume bowel continuity and monitor for disease recurrence before changing therapy argued that the patient remained in clinical remission for 6 years on thiopurines alone before developing complications. However, all agreed that clinical monitoring alone is not sufficient to diagnose Crohn’s disease recurrence, and
Recurrent Intentional Foreign Body Ingestion
Technical Challenges, Tips and Tricks
CLIVE J. MIRANDA, DO, MSC
Department of Gastroenterology, Hepatology, and Nutrition
University at Buffalo
Buffalo, New York
Division of Gastroenterology
Catholic Health Initiatives Health
Creighton University–Bergan Mercy Medical Center Omaha, Nebraska
Adult recurrent intentional foreign body ingestion (RIFBI), a phenomenon characterized by the deliberate and repetitive ingestion of nonnutritive substances or objects, constitutes a distinctive challenge at the intersection of psychology and healthcare economics. The problem, which affects hospital resources and staff morale, is not unique to the United States, with healthcare centers globally affected by the problem.
STEVEN J. BOLLIPO, MBBS, FRACP
Department of Gastroenterology and Endoscopy
John Hunter Hospital
Newcastle, New South Wales, Australia
School of Medicine and Public Health
University of Newcastle Callaghan, New South Wales, Australia
A fascinating variety of ingested foreign bodies created using an AI art generator. Courtesy of Steven J. Bollipo, MBBS, FRACP.
There is a paucity of literature on the magnitude of impact of RIFBI and little guidance on optimal management of patients who engage in this practice from a systems perspective. This review aims to outline the reasons for adult RIFBI and the extent of its burden on healthcare systems globally and proposes guidance for innovative, cost-effective, and safe management of this condition.
To Scope or Not to Scope
The majority of foreign objects (»80%) will pass spontaneously, with approximately 20% requiring endoscopic intervention and less than 1% necessitating surgery.1-3 Sharp and pointed objects pose more of a retrieval urgency before they pass through the stomach because intestinal perforation otherwise will occur in 15% to 35% of cases.4 In addition to this complication, other reported complications of sharp object ingestion include aortoesophageal or tracheoesophageal fistula and cardiac tamponade.5,6
Guidelines for endoscopic management of foreign body ingestion are determined by the tangible characteristics of the object, where the object is located in the gastrointestinal tract, and how long the object has been there. Multiple societies, including the American Society for Gastrointestinal Endoscopy and the European Society of Gastrointestinal Endoscopy, have published algorithms to guide gastroenterologists on foreign body management, breaking down what constitutes an emergent, urgent, and nonurgent endoscopy (Table).4,7
The Standard Toolbox
More often than not, conventional tools and techniques are all that is needed for safe and effective retrieval of ingested foreign bodies. This section discusses standard retrieval and protective devices in the market that encompass the majority of RIFBI cases.
The forward-viewing flexible endoscope is the first-line
Table. Guidelines on Timing of Endoscopic Intervention
For Foreign Body Ingestion From the ASGE and ESGE
Emergent endoscopy
ASGE guidelines (<24 h)
• Esophageal obstruction (including food bolus)
• Disk batteries in esophagus
• Sharp-pointed objects in esophagus
Urgent endoscopy
ASGE guidelines (variable, usually <48 h)
• Blunt objects in esophagus
• Esophageal food bolus without complete obstruction
• Sharp-pointed objects in stomach/duodenum
• Objects >6 cm at or above proximal duodenum
• All magnets within endoscopic reach
ESGE guidelines (within 2 or <6 h)
• Esophageal obstruction (including food bolus)
• Batteries of any type in esophagus
• Sharp-pointed objects in esophagus
Nonurgent endoscopy
ASGE guidelines (variable, usually <72 h)
• Coins in esophagus after failed watchful waiting for 12-24 h in asymptomatic patient
• Objects in stomach with diameter >2.5 cm
• Batteries in stomach after failed watchful waiting up to 48 h in asymptomatic patient
ESGE guidelines (within 24 h)
• Blunt and small objects <2-2.5 cm diameter in esophagus
• Blunt and medium objects >2-2.5 cm diameter in esophagus
• Esophageal food bolus without symptoms or complete obstruction
• Sharp-pointed objects in stomach/small bowel
• Objects >5-6 cm in esophagus/small bowel
• Magnets in stomach/small bowel
• Batteries in stomach/small bowel
ESGE guidelines (within 72 h)
• Blunt and small objects <2-2.5 cm diameter in stomach/small bowel
• Blunt and medium objects >2-2.5 cm diameter in stomach/small bowel
ASGE, American Society for Gastrointestinal Endoscopy; ESGE, European Society of Gastrointestinal Endoscopy.
choice for objects in the stomach and duodenum, with success rates greater than 95% and complication rates under 5%.8-10 Additional devices to facilitate retrieval come in all shapes and sizes. Overtubes, hoods, and clear caps are employed for mucosal protection upon object retrieval, whereas snare nets, graspers, and forceps are used for object capture. For example, overtubes provide protection to the airway, esophagus, and gastroesophageal junction from laceration and facilitate passage of multiple objects or piecemeal food boluses during endoscopic removal.11,12 Retrieval snare nets or stone retrieval baskets facilitate successful removal of button cell or small disk batteries that avoids necrosis due to release of the batteries’ metallic salts or alkaline fluids.13
Devices for Protection
Overtubes
Overtubes are the most standard protective devices
used in the removal of sharp foreign bodies to avoid tracheal aspiration and injury to the esophagus upon retrieval. Constructed from semirigid plastic with a proximal closure and tapered soft distal end, overtubes can overcome difficult anatomic strictures and limited insertion depths, thereby facilitating repeated insertion and withdrawal (Figure 1).14 Overtubes come in different lengths and diameters; and, depending on the foreign body in question, a larger diameter overtube may be needed to accommodate the object safely in its lumen. Therefore, it is remarkably helpful to have actual dimensions of the foreign body before removal to choose the most appropriate overtube for the procedure. These measurements can be based on preprocedural radiographs; although inaccuracies can occur due to the 2D nature of the images, they can provide at least a rough idea of the object’s measurements. Preprocedural radiographs provide more accurate measurements of objects lodged in the esophagus because the axis of the object often is
Figure 3. Olympus distal attachment clear caps can be highly effective in protecting tissue while optimizing visualization.
2. Cantel foreign body retrieval hood (40-mm distal end diameter, 8.3-mm proximal end diameter).
4. Diagmed Healthcare Roth Net with 2.5-mm catheter diameter, 230-cm length, and 3x6-cm net size.
Figure 1. STERIS 50-cm Guardus gastric overtube can facilitate foreign body retrieval from the stomach.
Figure
Figure
parallel to the inserted endoscope. Nonetheless, at least 2 different radiographic views are recommended to account for variable object widths and heights. It is much more useful to obtain object measurements outside the body if there are objects at hand similar to the one that was ingested. For example, if the patient ingested standard safety pins, the physician should obtain measurements of commercially available standard safety pins, rather than try to ascertain this from radiographs of the patient.
One issue with an overtube is that it is flexible, not rigid. Therefore, a long, solid foreign body (eg, a pencil) can get stuck behind the cricoid cartilage at the cricopharyngeus muscle, a region that entraps the majority of swallowed foreign bodies.15 If this occurs peri-procedure, it is important to straighten the neck and possibly even have the patient in the supine position and extend their neck, similar to the position used for a rigid esophagoscopy. This manipulative technique allows straightening of the overtube to facilitate pulling of the
foreign body into the top half of the mouth if it enters the oropharynx. The gastroenterologist can start the procedure with the patient in the left lateral position. Once the foreign body is pulled into the proximal esophagus, they potentially can extend the patient’s neck while the patient is in the left lateral position or consider placing the patient supine to do this, although the latter might be difficult to do ergonomically.
Retrieval Hoods
A foreign body retrieval hood is used to remove safety pins and small blades. Designed to protect the cardiac sphincter, esophagus, and pharynx during retrieval, the hood flips over on itself upon traversing past the lower esophageal sphincter, thereby preventing the foreign object encased within it from causing injury to the surrounding tissues (Figure 2).
However, the hood has its limitations. Sometimes, for atypical objects, the hood length is not as long as the foreign
Figure 6. Micro-Tech UK biopsy forceps with 9-mm distal end.
Figure 5. Diagmed Healthcare Raptor forceps with 10-cm jaw opening, 6.6-mm jaw length, 230-cm length, and 2.4-mm sheath diameter.
Figure 8. Karl Storz oval polypectomy snare with 15-mm diameter and 210-cm working length.
Figure 7. Olympus Coagrasper Hemostatic Forceps.
body itself. When this happens, the lower part of the foreign body is not covered by the hood. This will result in mucosal damage upon extraction back up the alimentary canal.
Clear Caps
The simple, standard distal clear cap is an invaluable protective device for object retrieval. For simple, small objects, this often is all that is needed. The distal clear cap often is used in endoscopic mucosal resections and has been revolutionary in the field of interventional endoscopy. It comes in many shapes and sizes and neatly fits onto the end of the endoscope (Figure 3). When possible, this should be the go-to device for retrieval because it is inexpensive, easy-touse, and widely available.
Devices for Object Capture
The Roth Net (Diagmed Healthcare) employs a flexible, reinforced net that can rotate 360 degrees, offering full visibility upon object retrieval and easily capturing objects in retroflexed and tortuous positions along the gastrointestinal tract (Figure 4).
Figure 9. Duomed brained polypectomy snare made with 0.36-mm braided stiff wire.
Figure 10. The Xcavator (Ovesco) over-thescope grasper with allowance for a free working channel for additional instrumentation for tissue/foreign body removal.
Figure 11. DeRoyal endoscopic specimen retrieval pouch to facilitate foreign body retrieval.
Figure 12 This pediatric anesthesia circuit bag (A) was cut at the lower half to create a hood. The narrow tip of the bag is placed over the tip of the scope, (B) and the scope is introduced into the stomach in this position. After the blade has been grabbed with a snare or forceps, the scope is withdrawn. While being withdrawn through the lower esophageal sphincter, the hood flips over and covers the blade (C), thereby protecting the esophagus from injury. As can be seen (D), the blades usually are smaller than a commercially available hood.
Courtesy of Steven J. Bollipo, MBBS, FRACP.
However, because the Roth Net is not that large, it is more appropriate for the capture of smaller blunt objects and can have trouble capturing large or long objects. The device would have difficulty capturing an AA battery, for example. In addition, small sharp objects, such as pins and clips, are not suitable for capture within a Roth Net because sharp edges can poke through the net mesh and injure the mucosa upon extraction.
Another method of retrieval is with the Raptor (Diagmed) forceps (Figure 5). Combining alligator and rat-tooth capabilities, raptor forceps have serrated edges but are not sharp, so they are ideal for picking up foreign objects lodged in the esophagus and stomach.
Biopsy forceps (Figure 6) also are useful for grasping foreign objects. However, these are sharp, so the risk for mucosal injury is significantly higher than that associated with raptor forceps.
A Coagrasper Hemostatic Forceps (Olympus) can be used for foreign body retrieval as well due to its blunt
nature and anti-slip serrated teeth. Make sure that no heat is used when extracting objects, as you do not want to inadvertently burn healthy mucosa (Figure 7).
A polypectomy snare is another option for extraction (Figure 8). In fact, for most foreign bodies, this is an excellent retrieval option and the most secure means to endoscopically hold an object. Because of their extensive training, gastroenterologists are well versed in polypectomy snares and how to maneuver them. Snares are particularly useful for round, cylindrical objects (ie, batteries). However, sometimes foreign bodies slip through the snare. If there is a risk for this, a braided snare might have better grip (Figure 9).
A number of ergonomic tips can be employed when using a snare. As soon as you close the snare around an object (eg, a pen), it may flip perpendicularly. You would then have a foreign body positioned at a right angle to the tip of the scope, making it unable to traverse the gastroesophageal junction. If the object is sharp, you risk injuring exposed mucosa repeatedly. It is, therefore, important to
or other
objects
be placed inside the glove tip using the forceps. The open end of the glove tip is closed using the
and the whole setup is retrieved together with the scope. A snare can be used instead of forceps.
Figure 13. The glove tip is taken into the stomach using a biopsy forceps or a rat-tooth forceps. Blades
small, sharp
can
forceps,
Courtesy Steven J. Bollipo, MBBS, FRACP.
Figure 14. Karl Storz modified Magill forceps to facilitate foreign body extraction if the object in question is lodged at, but was able to partially traverse, the oropharynx. The assistance of an anesthesiologist is required.
not close the snare too tightly, thereby avoiding inadvertent sideways positioning of the foreign body. The axis of the foreign body should align with the longitudinal axis of the endoscope. A general guiding principle is to gently grasp a long foreign body with a snare at the object’s proximal or distal 10%—not in the middle of the object. Once the object is in the esophagus, you can tighten the snare’s grip, as the esophageal walls prevent accidental rotation of the object while it is in the snare’s grasp.
The over-the-scope grasper Xcavator (Ovesco) is a newer cap that attaches to the distal end of a gastroscope, allowing for grasping function during endoscopy for foreign body extraction. In addition, it allows for additional endoscopic instruments (ie, guidewires, suction flushing pumps, through-the-scope instruments) to be passed through the working channel of the endoscope concurrently (Figure 10).
Thinking Outside the Box
Commercially available protective and extractive devices often come in standard shapes and sizes and cannot accommodate all foreign bodies. Endoscopists, therefore, need to think outside the standard toolbox and should be comfortable employing innovative techniques and instrumentation when needed.
Endoscopic retrieval bags and surgical laparoscopic retrieval bags (eg, for cholecystectomies and appendectomies) are another protective option for sharp or atypical foreign bodies (Figure 11). The bag can be dropped in the stomach into which the object can be placed. The bag can then be closed with a snare and the whole setup can be pulled out, protecting the surrounding tissues along the way.
It is important to prevent esophageal injury when retrieving sharp objects. However, some objects may be too wide or long to traverse an overtube, and the commercially available foreign body hood may be too short to adequately cover longer sharp foreign bodies, such as blades.
Such a scenario requires creative methods to construct a hood using an anesthesia bag (Figure 12) or using the wrist part of a glove to create a skirt on the tip of the scope that can be secured by tape or thread. This also is useful if a commercially available hood is not available in the endoscopy suite (Figure 13).
Longer objects such as pens and spoons can be retrieved with a polypectomy snare, and bezoars can be tackled with stone baskets or the lithotripter for fragmentation before removal.8
Other unique extraction methods have been published in the literature. Two reports describe employment of incisions in the distal end of an overtube to create an oval flap to accommodate razor blades, showing the overtube’s unique attribute of accommodating sharp-edged objects much larger than its diameter after some innovative modification.16,17 In the case that a sharp object is too large to fit into the overtube, use of video laryngoscopy adjacent to the flexible endoscope can facilitate safe extraction.18 Double-balloon enteroscopy also can be used for successful retrieval and was reported for extraction of a screw nail in the jejunum.19 More recently, innovative removal of a large 20-cm kitchen knife was performed at our institution; the knife became impacted within the overtube at the oropharyngeal angulation and required use of McGill forceps for final removal.20
Finally, long foreign bodies occasionally can become stuck in the proximal region of an overtube at the cricopharynx. If this happens, one may have to grab the tip of the foreign body with Magill forceps via the help of anesthesia (Figure 14). However, a Magill forceps cannot go through the overtube itself. Therefore, if one runs into this challenge, the overtube can be cut at the mouth, exposing a larger lumen to insert the Magill forceps through to pull the foreign body out from the proximal oropharynx. Occasionally, however, the foreign body can get stuck before exiting the cricopharynx. If this happens, the assistance of an otolaryngologist should be sought. Remember that the object only will be visible in the throat if it lies in the oropharynx. It is good form at the beginning of the procedure, based on the size of the ingested object and limitations of endoscopy suite equipment, to have the otolaryngology team on standby just in case.
Knowing When to Call the Surgeon
As exciting as it is to sometimes think outside the box, one should not mitigate the value of surgical intervention. In almost all cases in which a foreign body is ingested, it can be endoscopically removed—with some creativity, at times— so that unnecessary surgery can be avoided. However, it is important to strike a fine balance. If endoscopists lose
References
1. Webb WA. Management of foreign bodies of the upper gastrointestinal tract: update. Gastrointest Endosc. 1995;41(1):39-51.
2. Smith MT, Wong RK. Foreign bodies. Gastrointest Endosc Clin N Am. 2007;17(2):361-382.
3. Schwartz GF, Polsky HS. Ingested foreign bodies of the gastrointestinal tract. Am Surg. 1976;42(4):236-238.
4. ASGE Standards of Practice Committee, Ikenberry SO, Jue TL, et al. Management of ingested foreign bodies and food impactions. Gastrointest Endosc. 2011;73(6):1085-1091.
5. Sica GS, Djapardy V, Westaby S, et al. Diagnosis and management of aortoesophageal fistula caused by a foreign body. Ann Thorac Surg. 2004;77(6):2217-2218.
6. Vesna D, Tatjana A, Slobodan S, et al. Cardiac tamponade caused by migration of a swallowed sewing needle. Forensic Sci Int 2004;139(2-3):237-239.
7. Birk M, Bauerfeind P, Deprez PH, et al. Removal of foreign bodies in the upper gastrointestinal tract in adults: European Society of Gastrointestinal Endoscopy (ESGE) Clinical Guideline. Endoscopy 2016;48(5):489-496.
8. Conway WC, Sugawa C, Ono H, et al. Upper GI foreign body: an adult urban emergency hospital experience. Surg Endosc 2007;21(3):455-460.
9. Ginsberg GG. Management of ingested foreign objects and food bolus impactions. Gastrointest Endosc. 1995;41(1):33-38.
10. Webb WA. Management of foreign bodies of the upper gastrointestinal tract: update. Gastrointest Endosc. 1995;41(1):39-51.
11. Spurling TJ, Zaloga GP, Richter JE. Fiberendoscopic removal of a gastric foreign body with overtube technique. Gastrointest Endosc 1983;29(3):226-227.
perspective and see retrieval as a personal challenge, this may end up causing injury when a gastrotomy by surgeons could have solved the issue. Endoscopic innovation for RIFBI is exciting and constantly evolving, but remember the motto: “Don’t give up too easily, but also know when to give up.”
Summary and What’s Next
RIFBI is a globally debilitating problem, affecting hospital resources, staff morale, and healthcare finances despite initiatives aimed to combat it. Although some cases can be managed with watchful waiting, endoscopic removal is required in certain situations, such as sharp esophageal foreign bodies, longer objects, or dangerous items such as batteries or magnets in the stomach. Removal of such objects sometimes can be technically challenging, and endoscopists should make themselves familiar with the various devices for retrieval—such as snares, forceps, overtubes, and protective hoods—in the standard toolbox. Endoscopists also need to be creative and think outside the box when these standard tools fail. However, endoscopic removal techniques have their limitations. Endoscopists should realize this proactively and be prepared to request assistance from their otolaryngology, anesthesiology, or general surgery colleagues when appropriate to ensure that patient safety takes precedence.
12. ASGE Technology Committee, Tierney WM, Adler DG, et al. Overtube use in gastrointestinal endoscopy. Gastrointest Endosc 2009;70(5):828-834.
13. Faigel DO, Stotland BR, Kochman ML, et al. Device choice and experience level in endoscopic foreign object retrieval: an in vivo study. Gastrointest Endosc. 1997;45(6):490-492.
14. Kim SH. Usefulness of an overtube device in gastrointestinal endoscopy. Clin Endosc. 2019;52(3):203-204.
15. Long B, Koyfman A, Gottlieb M. Esophageal foreign bodies and obstruction in the emergency department setting: an evidencebased review. J Emerg Med. 2019;56(5):499-511.
16. Alhaji M, Atreja A, Upchurch BR. Razor blade removal from the stomach utilizing a novel modification of the overtube. Endoscopy 2009;41(suppl 2):E166.
17. Inayat F, Zafar F, Lodhi HT, et al. Endoscopic removal of large sharp-edged foreign bodies in the gastrointestinal tract using an innovative modification of the overtube. Cureus. 2018;10(9):e3264.
18. Hiller KN, Hagberg CA. Use of a video laryngoscope to facilitate removal of a long, sharp-pointed blade from the esophagus. J Clin Anesth. 2016;32:4-6.
19. Kim DJ, Sim MK, Lee SW, et al. Successful removal of a screw nail in the jejunum using double-balloon enteroscopy. Clin Endosc 2015;48(5):444-446.
20. Zala A, Bollipo SJ, Pockney P, et al. Endoscopic removal of a large kitchen knife. Gastrointest Endosc. 2017;85(5 suppl):AB126. Abstract 1038.
Drs. Bollipo and Miranda reported no relevant financial disclosures.
Recurrent Intentional Foreign Body Ingestion Nontechnical Challenges
CLIVE J. MIRANDA, DO, MSC
Department of Gastroenterology, Hepatology, and Nutrition University at Buffalo Buffalo, New York
Division of Gastroenterology
Catholic Health Initiatives Health
Creighton University–Bergan Mercy Medical Center Omaha, Nebraska
STEVEN J. BOLLIPO, MBBS, FRACP
Department of Gastroenterology and Endoscopy
John Hunter Hospital Newcastle, New South Wales, Australia
School of Medicine and Public Health University of Newcastle Callaghan, New South Wales, Australia
Disclaimer: The following case is not real, and any resemblance to an actual person or event is purely coincidental.
It is 11 PM on a Saturday. You, the endoscopist, are on call for the weekend. Covering 3 hospitals has not been easy. You have a full procedure list and the phone is ringing yet again.
“Hey, it’s Tom in the ER. Jane is back and she has swallowed a nail clipper this time.”
Jane, 21 years old, resides in a psychiatric facility and is well known to the gastroenterology service. This is the 16th time this year she has presented for foreign body ingestion and the 7th time when you have been on call. In fact, just last week, you removed a toothbrush from her duodenum.
You review her ER course. A plain x-ray was done, and it shows a nail clipper in her stomach. It measures 5 cm long and has sharp edges on one side where the clippers are. The patient is asymptomatic.
The ER is requesting an endoscopy. What would you do?
• Observe overnight in the ER and repeat an x-ray in the morning.
• Immediate gastroscopy and return her to her mental health facility afterward.
• Admit to the hospital and repeat x-ray in the morning and do an endoscopy if the object doesn’t pass.
• Send her back to her mental health facility now.
A surreal scene of an endoscopy unit, generated by AI, showing the seemingly endless and global problem of RIFBI and the debilitating impact on hospital resources and staff morale.
Courtesy of Clive J. Miranda, DO, MSc.
This challenging case sets the scene for our review article.
Recurrent intentional foreign body ingestion (RIFBI) is a global challenge on both the health-system level and the individual level for clinicians and ancillary staff, with proposed mechanisms to tackle the issue falling remarkably short in efficacy and sustainability.
With a notable negative impact on hospital finances and staff morale on top of concerns about patient safety, psychosocial management, and ethical principles, the prospects for managing RIFBI safely and cost-effectively in the near future appear bleak.
In this review, we focus on the nontechnical discussions regarding RIFBI, including epidemiology, biopsychosocial aspects, the impact on health systems including costs and staff burnout, medicolegal and ethical dilemmas, and a proposal for algorithmic approaches for multidisciplinary management of this condition.
Factors in Ingestion: What and Why
Foreign body ingestion (FBI) can be accidental or intentional. While accidental ingestion can happen to anyone, intentional ingestion often is seen in people who have underlying psychiatric illnesses, as well as in incarcerated individuals. The condition also occasionally occurs in people without chronic mental illness who are faced with a situational crisis or are under the influence of drugs or alcohol. These are often one-time episodes. However, RIFBI is a distinct entity, and in this section, we elaborate on the underlying reasons it is done and the various types of nonnutritious objects commonly ingested.
RIFBI falls under one of many forms of self-injurious behaviors, the motivations for which include pain relief via endogenous opioid system induction, communication of internal distress, frank anger and acting out, and environmental manipulation with identifiable secondary gain (attempts at self-injury, escape, and suicide).1,2 Quite often, however, the underlying motivation for ingestion remains unclear. Objects range across the spectrum and can be metallic (batteries, nails), sharp (needles, blades), or large (toothbrushes, pens).3
Factitious disorder and Munchausen syndrome are not uncommon diagnoses attributed to recurrent swallowers, and studies on the coexistence of personality disorders in repeat swallowers have been around since the 1950s.4
A study by Palta et al estimated that a concomitant psychiatric diagnosis is present in roughly 80% of intentional FBI patients, and another study by Grimes et al noted that patients with RIFBI are 2.5 times more likely than those with single ingestion events to have a psychiatric diagnosis.5,6
The magnitude of the problem is particularly high in the prison population (much of which, not surprisingly, also suffers from psychiatric comorbidities but their intention may be secondary gain from hospitalization rather than due to their underlying psychiatric illness). In the United States, these populations tend to be cared for in safety net hospitals, making select hospitals hot spots for encountering RIFBI. Of note, prisoners also tend to be more daring with repeat ingestions, with ingested objects becoming larger, more dangerous, and more toxic upon subsequent episodes.7
Extent of the Problem and Impact On Healthcare Utilization
The incidence of FBI is increasing, nearly doubling from 3 per 100,000 people in 2000 to 5.3 per 100,000 in 2017,8 with an estimated 1,500 deaths attributed to intentional FBI annually in the United States.9
The financial burden imposed by RIFBI on health systems globally is substantial. The overall annual costs of RIFBI in 44 of 50 US states has been estimated to exceed $6 billion.10 A 2010 study of 305 intentional FBI cases at Rhode Island Hospital demonstrated an average hospital cost of $6,616.63 per presentation, with hospital costs over an 8-year period totaling more than $2 million.1 In Ontario, Canada, a 2009 study of health systems showed that the average cost per hospital visit for intentional FBI, including emergency department (ED) care, inpatient stays, ancillary charges, and procedural/operative intervention was approximately $2,305 (Canadian dollars).11 A study at University Hospital Southampton, in England, showed that fiscal expenses for intentional FBI during a 5-year period were roughly £1,500 per patient per episode, or currently about $1,887 in US dollars.3
Costs for prisoners have been shown to be higher than for other patients. Median charges for RIFBI in prisoners ranged between $4,683 and $7,698 for both ED evaluations and subsequent admissions in a study of prisoners in Ohio.12 For those requiring hospital admission, median charges per admission exceeded $14,000 and cumulative charges for RIFBI studied in the same cohort neared $50,000.12
These extraordinary hospital costs and resource depletions understandably can contribute to significant provider and staff burnout. Urgent, middle-of-the-night endoscopies coupled with continued depreciation of resources harm provider morale and, thereby, risk a reduction in quality of patient care and bedside manner.
Back to Jane: There are merits in nonoperative management in this clinical vignette.
Given that the nail clipper with a sharp edge is in the stomach, most people would have removed it if this were the patient’s first presentation. Given that this is a recurrent presentation with intentional ingestion, however, you veer toward nonoperative management so as to not
reinforce her behavior of repeat ingestion.
While nonoperative management is useful, it is important for the endoscopist to be certain that the decision to not intervene is in the best interest of the patient rather than out of frustration and burnout from having already done multiple procedures for the same problem.
Issues With Preventive Measures
The correlation between advancing psychiatric illness and RIFBI is an impetus for the implementation of effective prevention systems to curtail this problem and early and aggressive psychiatric intervention to help reduce the RIFBI burden. Another impetus for prevention is the financial burden that RIFBI places on health systems because with increasing complexity of care and frequency of surgical intervention with recurrent presentations, the cost of care also inordinately increases.12
It is important to note that RIFBI is resistant to medical treatment.2 When RIFBI is caused by a mood or psychotic disorder, pharmacologic interventions such as antipsychotic agents, antidepressants, or mood stabilizers are the treatments of choice. However, because RIFBI often is volitional and malingering in nature, such pharmacotherapy has limited effectiveness.1 In addition, RIFBI usually is not an acute issue or related to suicidal tendencies, so overnight admission to inpatient psychiatric care generally is not warranted. Behavioral therapies have been recommended but have not been studied adequately in RIFBI situations. Nonetheless, psychiatric consultation is imperative, and one-to-one supervision almost always is required.
The issue of implementing strong preventive measures does pose ethical risks. Incapacitation is the one assured means of preventing RIFBI, but providers must remain aware of balancing individual rights and freedoms of the patient against saving healthcare costs. There is a temptation for clinicians to confine repeat swallowers in 24-hour psychiatric facilities, but there are several issues with this. As mentioned, the desire for containment may stem from provider frustration rather than genuine efforts at medical care, with inpatient psychiatric admission being a proxy for unethical “imprisonment.” In addition, psychiatric admission often is futile in recalcitrant patients because recurrent swallowers will find a way to continue this habit (Figure 1). Lastly, facility containment is contingent on facility acceptance and, quite often, the psychiatry team may not see RIFBI as a reason to admit the patient.
Jane swallowed a toothbrush last week, a nail clipper today, and who knows what she could swallow tomorrow. For years, she has been seen in the endoscopy suite for multiple (often dangerous) ingested foreign bodies. She currently lives in an inpatient psychiatric facility, but this does not stop her from repeatedly swallowing objects.
You ask yourself: “Is my decision to pursue nonoperative care coming from a place of spite, retaliation, and frustration, or out of genuine care for Jane?”
If the former is true, there is potentially the risk you could be making the suboptimal decision to inappropriately lean toward nonoperative management when you otherwise would have not done so if this were Jane’s first presentation.
Ethical Dilemmas
Endoscopic retrieval of foreign bodies is an obvious option in most cases of esophageal and stomach FBI. However, with increasing realization that operative management indirectly is reinforcing the pattern of ingestion and hospitalization, it is reasonable to raise the bar for intervention and pursue nonoperative management, but this can result in inappropriate withholding of essential treatment, thus leading to ethical and medicolegal dilemmas.
RIFBI patients exhibit affective instability, volatility, impulsiveness, limited interpersonal skills, and little to no consideration for care plan adherence, making them one of the most difficult populations to manage. Provider anger and frustration are not uncommon because providers can feel that the patient is not only rejecting help but is actively participating in self-sabotage.13 In addition, the inordinate recurrences of ingestion in these psychiatric patients instill a sense of futility, and providers may indeed question whether it makes sense to continue to provide treatment.13
Often, when these patients appear in the hospital, they first interact with the on-call team. These teams may have limited familiarity with the patient, and their compassion can be tested by the intentionality and self-injurious nature of RIFBI.14
It is worth briefly exploring the nature of provider burnout when managing RIFBI patients. The evident drain on time and hospital resources is a tangible measure of burnout, but there is also a more nuanced psychological reason. Often, the secondary gain intended by these patients prompts clinicians to wonder whether nonoperative management might, perhaps, be a form of behavior modification. If this secondary gain is not achieved, would the rate of RIFBI diminish over time? The ethical considerations in this dilemma are complex; however, the gastroenterologist’s morals strongly urge him or her to act in the best interest of the patient. This often means providing endoscopic intervention, knowing full well that it is both therapeutic yet enabling to the patient. The continued cycle with multiple patients can lead clinicians to become jaded, which risks them intervening with subconscious resentment, the manifestations of which can
range from poor bedside manner to careless procedural skills in the endoscopy suite. Thus, it is evident that management of RIFBI patients clearly results in strong moral afflictions and resulting burnout for gastroenterologists, regardless of what intervention is provided—endoscopic or nonoperative.
While frustration with recurrent foreign body swallowers is understandable, when dealing with these patients, clinicians must adhere to the ethical principles of non-maleficence and beneficence that they have vowed to uphold throughout their medical training, especially when endoscopic management is being withheld. Clinicians have an obligation to protect patient safety (even if the patients would not do so on their own) and to balance treatment benefits with its associated risks.14 Countertransference is highly likely, and physicians should ensure that the care decisions they make are not related to this or to their frustrations toward the patient.
There is an ethical obligation to provide whatever care
Despite 24/7 observation, objects provided to these patients for hygiene, entertainment, or basic living can very easily be ingested by those who are determined to do so (as shown by the toothbrush, plastic cutlery, and colored pencils).
Courtesy of Clive J. Miranda, DO, MSc.
Figure 1. A realistic scene of an incapacitated psychiatric patient generated by AI.
RIFBI patient identified Psychiatry consultation
Ingestion event
Multidisciplinary conference involving facility personnel, along with ED, radiology, gastroenterology, anesthesia
Facility-provided instructions for future ingestion events
Management protocol created incorporating facility-provided instructions
Living environment assessment and modification
Periodic revision of protocol as needed if ingestion events recur
Referral to ED if red-flag symptoms are present
Facility to contact gastroenterology service with history, obtain x-ray/CT imaging if recommended
From history and imaging studies, gastroenterology team determines management based on existing guidelines
Endoscopy not required
• Patient stays at facility and is monitored
Endoscopy required
• Patient is transported directly for endoscopy, recovers, and returns immediately to facility
• ED evaluation and hospital admission are circumvented unless there are extenuating circumstances
Figure 2. Identification of RIFBI and creation of a tailored management protocol in a patient presenting with foreign body ingestion from a psychiatric facility (pink); example management protocol (gray).
ED, emergency department; RIFBI, recurrent intentional foreign body ingestion.
is necessary if it will benefit the patient, and “bedside rationing” is not justifiable. Indeed, patients have a right to treatment. The principle of autonomy can apply when patients understand the risks and consequences of RIFBI treatment, but this principle may not always apply to psychiatric patients because they often lack decision-making capacity due to the absence of 1 or more factors of selfdetermination (cognitive ability, rationality, self-identity, and the ability to reason hypothetically).15 Endoscopists need to understand that they have a “duty of care” and to provide such care where it is warranted. Nonoperative management, while useful in certain circumstances, should not transform what should be a “duty of care” into a “denial of care.”
From a medicolegal perspective, despite the drain on resources and personnel, it would not be justified to withhold endoscopic intervention from patients presenting for intentional FBI who would definitely benefit from it. The 1986 Emergency Medical Treatment and Labor Act (EMATLA) dictates that anyone presenting to an ED be
stabilized and treated accordingly.16 Endoscopists must aim to treat each ingestion episode on its own merits (based on type/location of object and feasibility of retrieving it vs watchfully waiting) rather than risk one’s judgment to be clouded by previous RIFBI episodes of the patient and the endoscopist’s own emotional liability. This is especially important if a decision to pursue nonoperative management leads to a perforation with surgical intervention or patient decompensation, where the decision to not endoscopically retrieve the object initially would then be brought into question. Meetings with the medicolegal department and ethics teams in institutions should be commonplace to allow for the crafting of strategic management frameworks for RIFBI patients and to determine whether and when it would be appropriate for invasive procedures to be provided versus withheld.
Ideally, constant supervision, multidisciplinary care, and restricted access to almost all objects are what is necessary to prevent RIFBI. Low Kapalu provided an insightful commentary in 2020, stressing that, when providing care,
physicians should ask themselves several questions to employ the best possible ethical care for these patients: Is this being done in the name of beneficence? Is the treatment of last resort? Is the decision nonretaliatory? Is the decision therapeutic or preventive? 14 Consulting with ethics team members and colleagues can help endoscopists sift through these nuanced points and facilitate a process of self-reflection when they are managing these difficult patients.
Jane undergoes nonoperative management for her ingested nail clipper. You are wary that you could be medically and legally liable if harm was done.
You ask yourself: “Am I benefiting the patient by not endoscopically intervening and, therefore, preventing secondary gain, or am I frustrated with her and actually being subconsciously punitive?”
Fortunately, Jane excretes the nail clipper after 2 days of watchful waiting and you breathe a sigh of relief. She is a difficult patient, but your medical judgment in this episode was correct and sound.
Multidisciplinary Approaches And Natural History of Swallowers
Due to nuanced complexities surrounding RIFBI in adult psychiatric patients, a one-size-fits-all approach to management is virtually impossible. Multiple initiatives have been tried and tested and have yielded little ground in promoting cost-effective care and provider well-being. A multidisciplinary approach is imperative, with algorithms tailored to each patient’s circumstances. All parties (including stakeholders and hospital administration) should be aligned with the aim of reducing unnecessary tests and procedures, avoidable hospital admissions, and length of stay for each episode.
There is a reflex to promote defensive medicine, particularly in the United States, as one’s clinical practice goes hand in hand with consideration of the legal ramifications of one’s actions. Annual medical malpractice−related costs account for 2% to 3% of healthcare spending, totaling just under $60 billion, and this sum does not include costs incurred by defensive medicine.17-19 From the top down, emphasis should be placed on evidence-based clinical practice, even if it means a reduction in testing and
References
1. Huang BL, et al. Clin Gastroenterol Hepatol. 2010;8(11):941-946.
2. Villalba R, et al. Semin Clin Neuropsychiatry. 2000;5(4):215-226.
3. Yadollahi S, et al. Frontline Gastroenterol. 2021;13(2):98-103.
4. Carp L. Arch Surg (1920). 1950;60(6):1055-1075.
5. Palta R, et al. Gastrointest Endosc. 2009;69(3 pt 1):426-433.
6. Grimes IC, et al. Can J Gastroenterol. 2013;27(1):e1-4.
7. Dalal PP, et al. J Surg Res. 2013;184(1):145-149.
8. Hsieh A, et al. Gastrointest Endosc. 2020;91(2):350-357.
9. Barros JL, et al. World J Surg. 1991;15(6):783-788.
10. Poynter BA, et al. Gen Hosp Psychiatry. 2011;33(5):518-524.
11. The Pew Charitable Trusts. Managing prison health care spending. Published May 15, 2014. Accessed May 13, 2024. pewtrusts.org/en/research-and-analysis/reports/2014/05/15/ managing-prison-health-care-spending
intervention, to provide safe and cost-effective care to these patients.
The clinical course for these patients is highly variable. With endoscopic intervention and appropriate psychosocial therapy, many patients recover and never have a RIFBI episode again. Other patients, however, respond minimally to treatment and, as mentioned earlier, use RIFBI as a form of tangible or abstract secondary gain. The pleasure derived from opioid administration during object retrieval or from the attention and sympathy as a result of being hospitalized, for example, are not easy for patients to give up. RIFBI risks further complications (including perforation, operative intervention, and superimposed infection) and results in increased morbidity and mortality.
There needs to be an individualized algorithm for intervention to manage patients, particularly those who present from psychiatric facilities. This should be done in consultation with hospital management, hospital ethics teams, and legal departments, and should be tailored to each patient type—taking into account their unique circumstances. Such factors are the presence of factitious disorder/malingering, whether the patient is a recurrent swallower or a first timer, the type of object ingested, the presence or absence of psychiatric disease, and experiences during the patient’s previous hospitalization. An algorithm highlighting a safe and cost-effective multidisciplinary approach to manage RIFBI in patients presenting from psychiatric facilities was presented at Digestive Disease Week 2023 (Figure 2).20
Conclusion
RIFBI is a difficult problem due to vast differences in ingested objects as well as the backgrounds, psychosocial demographics, and determinants of health of patients with this disorder. Several innovative endoscopic techniques are available for removal of foreign objects, but because the sizes and shapes of objects and the nature of their ingestion are so varied, there are no definitive guidelines for removal. Although these experiences often are shared anecdotally and/or on social media, they are not prevalent in the medical literature. We hope this review promotes a more cost-effective, evidence-based, and multidisciplinary approach across all parties in healthcare pyramids to tackle this global problem.
12. Otey JA, et al. OPUS 12 Scientist. 2014;8(1):6-8.
13. Lytle S, et al. J Clin Ethics. 2013;24(2):91-97.
14. Low Kapalu C, et al. Pediatrics. 2020;145(1):e20191515.
15. Larcher V. BMJ. 2005;330(7487):353-356.
16. American College of Emergency Physicians. Understanding EMTALA. Accessed May 13, 2024. acep.org/life-as-a-physician/ ethics--legal/emtala/emtala-fact-sheet
17. Albolino S, et al. Curr Pharm Biotechnol. 2019;20(8):615-624.
18. Meng X, et al. J Biomed Inform. 2019;93:103169.
19. Fogel AL, et al. JAMA. 2019;321(13):1309-1310.
20. Miranda CJ, et al. Gastroenterology. 2023:164:S-546. Abstract Su1148.
Drs. Bollipo and Miranda reported no relevant financial disclosures.
How to Fix the 2 Achilles’ Heels Of Reprocessing
Despite progress in cleaning techniques and guidelines, flexible endoscope reprocessing remains inconsistent, putting patient safety at risk and bolstering the argument for single-use scopes. Addressing two “Achilles’ heels” could drastically reduce discrepancies and ensure reprocessed scopes are safe, said Michelle Alfa, PhD, MSc, FCCM, a certified clinical microbiologist (retired) and the CEO of AlfaMed Consulting, in Winnipeg, Manitoba. Solutions do exist, she said. The challenge is getting sites to implement them.
“Every time you add to the cost, it has an impact on the bottom line and, therefore, there’s a lot of hesitancy when it comes to trying to proactively comply with these types of issues,” Dr. Alfa said. But it’s ultimately worth that cost, she explained to Gastroenterology & Endoscopy News
Inconsistencies in Manual Reprocessing Neglecting IFU
The first hurdle is inconsistencies in manual reprocessing steps. The fact that this part of the process is manually controlled leaves ample room for error, she said, especially when there is a lack of adherence to manufacturers’ instructions for use (IFU).
“In some sites, it takes them less than five minutes to manually clean the scope,” Dr. Alfa said, despite the correct process taking 15 to 20 minutes. “They don’t even know they’re not getting it clean.”
A Brazilian study published last year investigated eight gastroscopes and five colonoscopes used at sites that perform between 140 and 530 scope-related examinations per month (Gastroenterol Nurs 2023;46[6]:455-464). After reprocessing, 46% still harbored debris.
for reprocessing, or through scope manufacturers, which sometimes offer on-site assessments, Mr. Czarnowski said at the very least, monitoring evaluations should be done by a clinical educator or manager—“someone who is knowledgeable but doesn’t do the procedure every day.”
An initial competency assessment will reveal how a technician was trained. “If I look at how a new tech cleans a scope, then I know how a new tech was taught to clean the scope,” Mr. Czarnowski said. If there are errors, “then I can go back to the whole team to address everyone on the team to ensure everyone is on the same page.”
The Drying Dilemma
Scope storage and drying is the second major hurdle in endoscopy reprocessing, but it’s one that may not be solved with closer adherence to IFU.
‘Break down that IFU into a chart, a graph or something that is very usable for the people reprocessing the scopes.’
Casey Czarnowski, BA, CRCST, CSPDT, CIS, CER, a perioperative consultant in Rochester, Minn., who specializes in sterile processing, said facilities should review each device’s IFU and post clear, easy-to-follow instructions in the rooms in which cleaning takes place. “[The IFUs] will go down to the minute for how long each task is going to take, whether it’s brushing or flushing. Break down that IFU into a chart, a graph or something that is very usable for the people reprocessing the scopes,” Mr. Czarnowski said.
“The manufacturer says you must store the scope totally dry, but they don’t tell the site how to do that or how to measure the adequacy of drying,” said Dr. Alfa, adding that finishing cleaning a scope in a reprocessor that has an alcohol rinse and drying cycle is not enough.
Instead, the best option is to invest in an active drying cabinet that pushes medical-grade air through the scopes, Mr. Czarnowski said.
“If the choice is made to not do that, ensure that everyone at the very least is hanging scopes to dry in a cabinet made for flexible scopes, and not storing them coiled laying on their side,” he said.
Some observers worry that without the threat of legal ramifications there won’t be enough incentive for hospitals to make costly investments in scope reprocessing equipment and training.
Two important ways to handle inconsistencies—monitoring regularly and correcting any errors—can work in tandem.
“We know there are lots of sites that are taking shortcuts because there’s no monitoring,” Dr. Alfa said, noting that monitoring by using testing strips to test for moisture and debris should be a step in reprocessing.
Mr. Czarnowski said people, not just equipment, should be monitored. If a facility does not opt for third-party accreditation through an organization such as The Joint Commission, which will conduct competency evaluations
“In my mind, a lot of these things will only be fixed by very strict regulation. If you don’t [follow the regulations], you will be in trouble,” said Mohammad Yassin, MD, PhD, an associate professor of infectious diseases and microbiology at the University of Pittsburgh School of Public Health.
Dr. Alfa pointed to the Association for the Advancement of Medical Instrumentation’s new scope reprocessing guidelines as an example of industry leaders outlining what healthcare facilities should be doing.
“We’re getting some practical advice on how to do this,” she said. “We need to start integrating it.”
—Kaitlin Sullivan
The sources reported no relevant financial disclosures.
Thinking Outside the Traditional Endoscopy Box
Using the Soft, Mega, Distal Transparent Cap to Remove Food Bolus
Section Editors
Klaus Mergener, MD, PhD, MBA, MASGE
Affiliate Professor of Medicine
University of Washington School of Medicine Seattle, Washington
Klaus Mönkemüller, MD, PhD, FASGE, FJGES
Professor of Medicine
Virginia Tech Carilion School of Medicine Roanoke, Virginia
Universidad de La República Montevideo, Uruguay
Contributors
Reid Wasserman, DO
Carilion Roanoke Memorial Hospital
Virginia Tech Carilion School of Medicine Roanoke, Virginia
Maren Haslach-Häfner, BA Berufsfachschule für Pflege Helios Bildungszentrum Kronach, Germany
Klaus Mönkemüller, MD, PhD, FASGE, FJGES
Professor of Medicine
Virginia Tech Carilion School of Medicine Roanoke, Virginia
Universidad de La República Montevideo, Uruguay
There are myriad instruments to remove impacted food
boluses, including nets, snares, forceps, tripod prongs, overtubes, and various distal scope attachment caps (soft, short, stiff, capuchon).
In our experience, using a distal transparent cap is the easiest, fastest, and safest method to remove impacted meat from the esophagus.
In this installment of EndoHacks, we report on the novel use of the soft, elastic cap (remOVE FBR Set, Ovesco), which originally was developed to remove metal pieces, specifically clip fragments of the overthe-scope clip (OTSC System, Ovesco). We have found this cap to be very useful to remove impacted meat from the esophagus (Figure). Because this cap is 3 times larger than traditional “hard” caps (ie, those used for banding of esophageal varices) and has large capacity volume, we refer to it as the “mega cap.”
The cap is larger than any existing transparent cap and allows for suctioning of most of the impacted meat inside of it. Other advantages of this cap are its pliability and elasticity, which allow for smooth introduction into the esophagus.
The cap is large and flexible. This flexibility allows for easy insertion through the oropharynx and upper esophageal sphincter. In this case the food bolus was completely obstructing the esophageal lumen. The cap could be advanced toward the food bolus to allow suctioning and complete extraction of the bolus.
Images from EndoCollab. See endocollab.com for more information, including videos, quick tips, and lectures on these and many other practical endoscopy tricks and techniques.
We recommend the following steps to remove impacted meat with the “mega cap”:
Slowly place the distal part of the cap onto the meat.
While gently pushing, gently torque the scope to the right and left to allow the proximal part of the impaction to enter the cap.
Apply constant suction and continue the maneuver in step 2.
When the impaction occludes the visual field, this indicates that it is inside the cap.
Gently pull the scope out, while continuously torquing right and left. The torquing maneuver allows for the meat to detach itself from the impacted site. Also, by torquing the scope, there is less of a chance that the caught piece of meat gets hung up on any of the esophageal contractions or anatomic stenotic areas.
Since this cap can hold larger volumes of meat, there is a higher probability of removing the entire food bolus at once. In addition, because the cap holds most of the food bolus, there is less chance that the meat will detach and fall into the oropharynx while you are removing it. Therefore, this cap appears superior to existing ones.
In summary, this novel cap is a new addition to the armamentarium for handling food bolus impactions, as it is easy to handle, has a large volume, catches larger pieces of meat and saves time during food bolus removal, and is ideal for removal of other foreign bodies, such as sharp or pointed objects, from inside the gastrointestinal tract. A potential disadvantage is its higher cost, since the cap currently is not available separately but comes in a set with a grasping forceps.
The researchers reported no relevant financial disclosures. Dr. Mergener is a member of the Gastoenterology & Endoscopy News editorial board.
Figure. Use of the remOVE FBR Set (Ovesco) to remove impacted meat from the esophagus.
Thinking Outside the Traditional Endoscopy Box
Suction Mark Technique for Identifying and Resecting Flat Colon Polyps
Section Editors
Klaus Mergener, MD, PhD, MBA, MASGE
Affiliate Professor of Medicine
University of Washington School of Medicine
Seattle, Washington
Klaus Mönkemüller, MD, PhD, FASGE, FJGES
Professor of Medicine
Virginia Tech Carilion School of Medicine
Roanoke, Virginia
Universidad de La República Montevideo, Uruguay
Contributors
Reid Wasserman, DO
Carilion Roanoke Memorial Hospital
Virginia Tech Carilion School of Medicine
Roanoke, Virginia
Klaus Mönkemüller, MD, PhD, FASGE, FJGES
Professor of Medicine
Virginia Tech Carilion School of Medicine
Roanoke, Virginia
Universidad de La República Montevideo, Uruguay
Flatlesions are found commonly during colonoscopy. However, and not infrequently, these lesions “disappear” from the endosopist’s and assistant’s eyes, especially when the colon is not completely clean, the colon motility increases, or the scope moves away from the lesion.
A useful trick is to focus on such a lesion, push the scope toward it, and suction the mucosa right next to it. This creates a red submucosal suction mark (Figure 1, yellow arrows), which then becomes pivotal to re-identify or relocate the polyp (Figure 1, yellow circle).
We also have developed the suction, reshape, and resect technique, which has the advantage of relocating the polyp and improving its resection (Figure 2).
Here are the key steps for this technique: application, reshape, and resection.
Application: Once the flat lesion is seen (Figure 2A and B), place the scope directly on it and suction it into the working channel (Figure 2C).
Revisualization and reshape: The suctioning creates a suction mark and “reshapes” the polyp into a “sessile” lesion (Figure 2D). While applying suction, insert the snare into the working channel. What began as a flat lesion now morphed into a pseudo-polyp or “sessile” lesion that also is marked (Figure 2D).
Execution (resection): The advancement of the snare will displace the air of the dead space of
the working channel and likely “detach” it. Detachment of the suctioned polyp also occurs when the suction is discontinued. If for some reason we lose scope position, the polyp is marked already and easy to find. If scope position was not lost, we proceed to immediate resection (cold or hot) (Figure 2E and F).
In summary, these suction strategies in colonoscopy aid in
the effective treatment of lesions and streamline the process. The time wasted in re-identifyng a “lost” flat lesion can be substantial. The transformation of a flat lesion into a pseudopolyp using suction provides an enhanced visual and practical advantage, enabling a more precise resection. Such techniques are essential tools in the repertoire of gastroenterology practices.
Images from EndoCollab. Go to endocollab.com for more information, including videos, quick tips and lectures on these and many other practical endoscopy tricks. Dr. Mergener is a member of the Gastroenterology & Endoscopy News editorial board.
Figure 1. Use the suction mark technique to create a temporary marking by applying suction (arrows) to the mucosa close to the flat polyp (circle).
Figure 2. With the suction, reshape, and resect technique, you can relocate the polyp and improve its resection.
Between the Guidelines New ACG Guideline on Acute Liver Failure
The American College of Gastroenterology recently released a guideline on acute liver failure ( Am J Gastroenterol 2023;118[7]:1128-1153). GEN ’s
Sarah Tilyou spoke with guideline senior author Lafaine Grant, MD, an associate professor of internal medicine in the Division of Digestive and Liver Diseases at UT Southwestern Medical Center, in Dallas, about the impetus for the guideline and what it means to GI practice.
GEN: What prompted the guideline?
Dr. Grant: It’s well recognized that acute liver failure is a devastating disease. But it only occurs in approximately 2,000 patients in the United States annually, and so it’s a rare disease, with any single center seeing only a handful of cases annually. So, there was a need for readily available, evidence-based guidance on how to care for these patients with acute liver failure.
GEN: What’s new in the guideline that clinicians need to know?
Dr. Grant: One of the things we discussed a lot was a new clinical syndrome that’s been described in recent years: acute-on-chronic liver failure. Patients with acute-on-chronic liver failure can be very sick, like patients with acute liver failure. But it’s really crucial to distinguish between acute liver failure and acute-on-chronic liver failure because they are two clinical entities that are treated very differently. In a patient with acute liver failure, time is of the essence. So, in the beginning of the guideline, we have a section describing what acute liver failure is not, and we made sure to emphasize the differences between acute-on-chronic liver failure—which is typically seen in patients with cirrhosis—and acute liver failure, which except for a few special cases, typically is seen in patients without
preexisting liver disease. We really wanted to distinguish between those two clinical entities because their treatments are vastly different.
One of the major causes of acute liver failure is a special case of druginduced liver failure, acetaminophen toxicity. Liver failure due to acetaminophen toxicity behaves differently from liver failure associated with other drugs, and we discussed this in detail.
However, the guideline also discusses other etiologies that occur less commonly. We felt it was important to have guidelines for what to do when you see those rare cases, such as mushroom toxicity or Budd-Chiari syndrome, for example, that you certainly won’t see every day or even every month, especially depending on where you live in the country.
GEN: How might the new guideline change
practice?
Dr. Grant: One of the things we considered was how we would present the new guidelines. We really wanted it to be easy to use in practice because this is guidance for clinicians at the bedside when they first evaluate the patient. Recognizing that evaluation and triaging need to be timely, we had a system-specific approach to the recommendations. Therefore, the clinician at the bedside can go directly to that organ system that they may have a particular question about and find the relevant information. For example, if they have a question about how to manage altered mentation or how to manage coagulopathy,
we have addressed each system separately, so you can go quickly through each section and get clarification. Then, each section is followed by key concepts and recommendations. So, even within the sections, if you just want to know, for example, should I correct coagulopathy, you can quickly go to the key concepts and see what’s recommended by expert opinion or evidence to help you with that. Ease of use of the guidelines was critically important to the guideline panel. So, there are sections with detailed descriptions, but also key concepts to take away and places to find immediate answers for specific questions.
GEN: What were some of the hottest points of debate among the guidelines panelists and how did you resolve them?
Dr. Grant: Acute liver failure has multiple etiologies, so one challenge was to present all the necessary information and keep it concise and practical. That was a recurring theme, keeping in mind our audience, the clinician who is taking care of these patients. To be able to keep it concise, we always had to corral the tendency to want to give more information. When you are in a group effort, you really have to agree on what to eliminate because there is a lot of information out there, and we want to be the funnel through which to give the best evidence-based information that’s concise and practical to use.
Acute Liver Failure continued, page 48
Lafaine Grant, MD
Between the Guidelines
ACG Issues Updated Guidance On Alcohol-Associated Liver Disease
The American College of Gastroenterology recently published a guideline on alcohol-associated liver disease (Am J Gastroenterol 2024;119[1]:3054). GEN ’s Sarah Tilyou spoke with lead author Loretta Jophlin, MD, PhD, an
assistant professor of medicine and the medical director of the liver transplantation program at the UofL School of Medicine, in Louisville, Ky., about the guideline and its implications for GI practice.
GEN: What prompted the guideline?
Dr. Jophlin: It was time for an update. The last guideline was published in 2018, and we’ve been through a lot since then. We’ve seen some startling trends in alcohol-associated liver disease (ALD), with rising prevalence in young women, younger people in general and certain minority groups. In addition, more centers are offering liver transplantation for alcohol-associated liver disease, particularly alcohol-associated hepatitis. Since publication of the ACCELERATE-AH [American Consortium of Early Liver Transplantation for Alcoholic Hepatitis] trial data and the Dallas Consensus Conference on Liver Transplantation
Acute Liver Failure
continued from page 47
GEN: What are the biggest remaining gaps in evidence?
Dr. Grant: Two things come to mind.
One is a subject that everyone in medicine is still learning more about— COVID-19. That’s an evolving area about which there’s not definitive information because we’re learning as we go. For example, the timing of liver transplant in a patient with COVID-19: We know that liver transplant can be lifesaving for patients with acute liver failure, but what happens when that patient also is infected with SARS-CoV-2? Right now, there’s some general guidance as to the timing of liver transplant. But I think
for Alcohol Associated Hepatitis, which support the selective use of LT for patients with severe AH—many programs have written their own protocols to offer transplant to selected individuals.
GEN: What’s new in the guideline that clinicians need to know?
Dr. Jophlin: There are several new key recommendations and key statements.
The first ties into our recognition of metabolic-associated steatohepatitis (MASH) or liver disease (MASLD), and the clinical entity of metALD, the overlap of ALD and metabolic
that is a moving target as we gain more experience with taking care of patients with COVID-19.
Another longstanding gap relates to the idea of artificial liver support. There are two systems available for investigational purposes and for compassionate use, but there’s still no FDA-approved liver support device that we can readily use as a lifesaving bridge to transplant or stabilization until liver recovery. That’s another big gap in our scientific knowledge that is a continuing work in progress.
GEN: What’s the take-home message for our readers?
Dr. Grant: One of the most important things is recognizing acute liver failure, and getting in touch with a transplant center very early on in the process. These patients are complex, and they require interprofessional cooperation and collaborative care. I would say to my gastroenterology colleagues: Do not hesitate to contact your closest transplant center. It’s better to err on the side of sending the patient and the patient recovering than the other way around. So, my No. 1 message would be get in touch early with a transplant center if you have any concerns about a patient with acute liver failure. Sometimes, the referral centers can help with the diagnosis, and they certainly can help with the transition if a patient needs to be transferred. ■
The full interview was edited for clarity and brevity.
Loretta Jophlin, MD, PhD
liver disease. One important recommendation is that individuals who have risk factors for MASH or MASLD should not consume alcohol.
We also have made the recommendation, based on a growing body of literature, that individuals who have a history of bariatric surgery or bariatric interventions should not partake in any alcohol, given their risk for accelerated liver disease.
We’ve also presented the concept of early ALD. This is the stage of disease where there may be inflammation and some scar tissue from drinking, but cirrhosis has not yet become established. We recommended the use of noninvasive testing with liver elastography and FIB-4 to estimate fibrosis in those with risk factors for ALD or early ALD, with the hope of changing the trajectory of people who are on their way to developing cirrhosis.
We’ve expanded our discussion on alcohol regulation and policy, to give the general practitioner information on global efforts aimed at primary prevention.
GEN: How might the guidelines change practice?
Dr. Jophlin: We included a comprehensive section on alcohol use disorder (AUD) including guidance for screening, diagnosis and treatment. We hope every practitioner at every healthcare encounter is empowered to screen patients. The AUDIT-C [Alcohol Use Disorders Identification Test−Consumption] is quick and performs well for predicting high-risk alcohol use.
For a positive screen, providers can refer to subspeciality care for AUD treatment, but we also recognize that many communities and healthcare systems lack robust pipelines for such referrals and that patients can get lost to follow up. With the growing body of literature supporting the safety of medication-assisted treatment, we make recommendations for providers to prescribe selected medications to support their patient in maintaining sobriety. For example, baclofen— a safe medication—can be prescribed off-label, or the FDAapproved drugs naltrexone and acamprosate can be used for AUD in individuals with careful monitoring. A practitioner must understand the patient’s comorbidities to choose the right medication, but if the practice is incorporated into primary care, it could prevent the progression from early ALD to cirrhosis and decompensated cirrhosis. That’s truly what we want to prevent.
GEN: What
were some of the hottest points of debate among the guideline panel, and how did you resolve them?
Dr. Jophlin: One of the hottest areas of debate was that we are all frustrated with the lack of treatment options. We have no FDA-approved medication for alcohol-associated hepatitis, so we continue to scrutinize the literature and hope for a clinical trial that can be validated beyond the pilot phase. Every center that takes care of liver patients has its own culture and approach to therapies. For example, I
don’t think my center will step away from using pentoxifylline even though it’s not recommended in the guideline because it does have a signal in hepatorenal syndrome (HRS). Until we get terlipressin (Terlivaz, Mallinckrodt) on our formulary, we’re going to do everything we can to try to prevent HRS. In the guideline, we had to stick with the literature and not let anything sneak in that wasn’t supported by evidence, so we did have some debates on what to include and exclude.
We also recognized that there are at least two phenotypes of people who present meeting the NIAAA [National Institute on Alcohol Abuse and Alcoholism] criteria for alcohol-associated hepatitis: the person who has not been drinking for decades, who lacks longstanding cirrhosis in the background versus someone with established ALD cirrhosis with a superimposed steatohepatitis. The former may represent true alcohol-associated hepatitis and the latter may be acute-on-chronic liver failure. These two phenotypes may be diminishing the signal in clinical trials. We struggled to integrate this into our discussion on alcoholassociated hepatitis. At the end of the day, we recognize that it’s still a work in progress.
GEN: What are the remaining gaps in evidence?
Dr. Jophlin: We don’t have the best tools for predicting return to drinking post-transplant. Right now, we use the SALT [Sustained Alcohol Use Post−Liver Transplant score]; we use HALT [Harmful Alcohol Use Post−Liver Transplant score]; some centers use SIPAT [Stanford Integrated Psychosocial Assessment for Transplantation (pre-transplant screening)]. I have this conversation with our social workers and addiction professionals every day. Every time something new is published, I think, ‘ Should we integrate this into our center protocol?’ but ultimately decide to stay the course for now. Centers need to eventually share and consolidate their experiences. I believe that is how we’re going to determine the ideal tool. At some point, OPTN [Organ Procurement and Transplantation Network] should get involved, whether that’s by regulating all of us or coming out with their own statements that give us some guidance to follow so that we’re all practicing in a similar manner. I think that’s going to be the fairest for the transplant landscape.
GEN: Do you have any take-home messages for our readers?
Dr. Jophlin: The take-home message is that the guideline should be seen as a living tool. There will be a new guideline in five years, but right now, I feel confident that with these key statements and recommendations, any provider caring for patients with ALD can provide good care. At its end stage, ALD is challenging to care for and the impacts on the individual, family and community are devastating. Early-stage ALD is an intervenable timepoint where providers can make a huge impact. ■
The full interview was edited for clarity and brevity.
It’s time to think BEyond dysplasia
TissueCypher goes beyond the watch-and-wait approach to non-dysplastic BE. This extensively validated, precision medicine test analyzes 9 protein biomarkers in the context of 7 tissue structures to objectively predict a patient’s 5-year risk of progression to esophageal cancer. Non-dysplastic patients scoring high-risk on TissueCypher progress at a rate similar to expert-confirmed low-grade dysplasia.
TissueCypher predicts progression of Barrett’s esophagus to high-grade dysplasia or esophageal adenocarcinoma
Provides individualized risk stratification independent of histological classification (or morphology) and other clinical risk factors
Transforms patient management by enabling upstaging or downstaging based on individual patient risk
Between the Guidelines AGA Issues Updated Guideline On Endoscopic Eradication Therapy for BE
The American Gastroenterological Association recently published a clinical practice guideline on endoscopic eradication therapy for Barrett’s esophagus and related neoplasia (Gastroenterology 2024;166[6]:10201055). GEN ’s Sarah Tilyou spoke with Joel H. Rubenstein, MD, a research scientist at the Veterans Affairs Center for Clinical Management Research and professor of gastroenterology and the director of the Barrett’s Esophagus Program at the University of Michigan, in Ann Arbor, about the guideline and its implications for GI practice.
GEN: What prompted the guideline update?
Dr. Rubenstein: It’s basically a fresh new guideline, because the last full AGA guideline on Barrett’s was published in 2011. There have been a lot of changes over the last 13 years in terms of what’s available for treating Barrett’s esophagus and knowledge about which patients should be treated and how, so that’s why we wanted to address this now.
GEN: What’s in the updated guideline that clinicians need to know?
Dr. Rubenstein: For the most part, our main recommendations reinforce the prior clinical practice updates and guidance from other societies, but there are differences in
the nuances of how we address the management of lowgrade dysplasia (LGD). In addition, our recommendation regarding which form of endoscopic resection to use differs slightly from some prior guidance. We also made several new implementation statements, which are based more on expert opinion than high-quality evidence since the evidence was lacking in those areas.
For example, we have implementation statements related to counseling patients with Barrett’s on tobacco cessation and weight loss if they are overweight, optimizing acid reflux control before endoscopic eradication therapy (EET), and discussions with patients about the risks and benefits of EET, which cover the need for adherence with reflux control and continued surveillance after completion of therapy and emphasize consideration of patient preferences. In addition, we provided some specific guidance on what the endoscopic
and histologic end point should be when performing EET. We also gave some updates on surveillance intervals following completion of EET.
GEN: How might the guideline change practice?
Dr. Rubenstein: I think that’s a great question. I hope the most important takeaway is in the management of LGD, which is very frequently overdiagnosed.
In high-grade dysplasia (HGD), there’s a risk for prevalent missed cancer of roughly 30%. Even in the absence of prevalent cancer, the incidence of new cancer is about 6% per year. In randomized controlled trials (RCTs), EET decreased the risk for progression to cancer by 60%. So, we’re moderately certain of the benefits of EET in HGD.
In LGD, there’s about a 10% risk for missed prevalent cancer. So, all cases of dysplasia should be reviewed by expert pathologists and referred to endoscopists who are experts in EET. But the risk for incident cancer is much lower than in those with HGD (around 1%-2% per year). So, the maximum possible benefit is a lot smaller than in HGD. Also, it’s not clear from the RCTs that EET is any better than surveillance for preventing cancer in those with LGD. In addition, in the available RCT data, almost all patients with LGD undergoing surveillance who develop cancer still can be treated with EET.
So, the benefits for patients with LGD are smaller than in the setting of HGD, and, therefore, any risks with EET get relatively magnified. Those risks include bleeding and perforation, which are uncommon but real, and stricture formation, which is not uncommon but usually easy for us to treat. Chest pain following EET is nearly universal, and EET necessitates multiple procedures with transportation with a chaperone. And there’s still a need for continued surveillance after we complete EET.
So, we felt strongly that endoscopists need to discuss these trade-offs with patients in a clinic setting, with adequate time to gauge the patient’s preferences and help them decide what’s best for them and their situation. Shared decision making is important in all areas of medicine, but its particularly important when the risks and benefits are nearly equal, as is the case with LGD.
GEN: What are some of the hottest points of debate among the guideline panel, and how did you resolve them?
Dr. Rubenstein: I think among Barrett’s endoscopists the current hottest topic is the role of endoscopic submucosal dissection (ESD) versus endoscopic mucosal resection (EMR). While EMR is performed piecemeal—the largest pieces about 15 to 20 mm—ESD aims to remove lesions en bloc with negative lateral margins and also might be able to get deeper into the submucosa to obtain negative deep margins.
Although studies have shown ESD to be more likely to result in negative margins than EMR, there was no difference in the control of cancer for ESD compared with EMR, and there was greater risk for strictures with ESD. This was
primarily based on observational studies rather than RCTs, and there may have been differences in which patients were selected for ESD in those observational studies. It could be that an RCT might be able to show that ESD is better, but we just don’t have data showing that currently.
There are technical reasons why some lesions might benefit from ESD. If the lesion is very bulky, it might not fit into the cap device for performing EMR. Or if the lesion is highly suspicious of a T1b lesion invading the submucosa, then ESD may be more likely to achieve a negative deep margin and get underneath that. But our guideline differs from some other earlier guidelines, which had set a size limit above which ESD should be favored over EMR. That was based on the thought that we should be resecting the lesion in one piece. Although we found a striking effect in favor of ESD over EMR for achieving negative margins, it didn’t translate into cancer control, suggesting that the negative lateral margins seen with ESD are not all that relevant when you perform an EMR with good quality, having overlapping, piecemeal contiguous pieces.
GEN: What are the remaining gaps in evidence?
Dr. Rubenstein: A lot of research is trying to identify ways of selecting patients with LGD who would most benefit from EET. Similarly, there might be ways of identifying patients with nondysplastic Barrett’s who might benefit from EET.
As we discussed, we need more research on comparing ESD and EMR, particularly in those high-risk lesions to tell whether ESD is of benefit and worth perhaps the increased risk. Hopefully, we’ll get better research to understand how to better control pain after EET because, as I mentioned, 95% of patients undergoing radiofrequency ablation have pain for two to four weeks after the procedure. We also need better research to understand how to prevent strictures after EET. And then finally, I would say that we don’t really have a lot of data guiding us on how frequently we should be doing surveillance after completion of EET, in particular on when we can stop doing surveillance considering a patient’s age and comorbidities.
GEN: Are there any take-home messages for the readers?
Dr. Rubenstein: The main takeaway is that patients with Barrett’s dysplasia of any degree should be referred to experts who see a lot of Barrett’s dysplasia to manage, even those with LGD or indefinite for dysplasia. We not infrequently find cancers when they’re initially diagnosed with LGD or indefinite for dysplasia. That being said, once we exclude that, the risk for progression to cancer is limited. But it’s certainly not zero, and it’s not clear whether we should be treating those patients or not, so we really need to take the time to discuss that with those patients. ■
Dr. Rubenstein reported a financial relationship with Lucid Diagnostics. The full interview was edited for clarity and brevity
•
•
Diagnosis of Achalasia
AOFER Z. FASS, MD
Division of Gastroenterology and Hepatology
Stanford University School of Medicine
Redwood City, California
RONNIE FASS, MD, MACG
Esophageal and Swallowing Center
Division of Gastroenterology and Hepatology
MetroHealth Medical System
Case Western Reserve University Cleveland, Ohio
chalasia is a rare esophageal disorder characterized by impaired relaxation of the lower esophageal sphincter (LES) and either absent or spastic contractions of the esophageal body.1 The incidence of achalasia is estimated to range from approximately 0.8 to 2.2 cases per 100,000 individuals, with a prevalence of 7.0 to 15.3 cases per 100,000 people.2-4 The economic burden of achalasia is significant, with costs surpassing $400 million annually in the United States alone.5
The underlying cause of achalasia is believed to be inflammatory degeneration of inhibitory neurons within the esophageal wall, which results in abnormal peristalsis and incomplete relaxation of the LES.6 While the exact trigger of inflammation for primary achalasia remains unknown, genetic studies suggest an autoimmune origin associated with changes to HLA-DQ alleles.7,8
Secondary achalasia, on the other hand, arises from other conditions that lead to esophageal motor abnormalities similar to those observed in primary achalasia. These conditions
include malignancies (“pseudoachalasia”), Chagas disease, and others.9,10 This review focuses on primary achalasia.
The most prominent symptoms of achalasia include a progressive difficulty swallowing solids and liquids that often is accompanied by weight loss.11 Patients frequently experience regurgitation of undigested food and saliva, which can potentially lead to complications such as bronchitis or recurrent aspiration pneumonia. Diagnosing achalasia can be challenging since dysphagia has a broad range of potential causes, necessitating various
Table 1. Eckardt Score
examinations and supportive tests. Achalasia is not uncommonly encountered in gastroenterology clinics, emphasizing the importance of maintaining a high degree of clinical suspicion for this condition.
Overview of Diagnosis
Achalasia is a condition that progresses slowly over the course of several years, and its symptoms can be subtle. In a prospective study involving 87 individuals with achalasia, the average duration of symptoms before diagnosis was 4.7 years.12 This highlights the need for healthcare practitioners to maintain a high level of suspicion when evaluating patients who exhibit symptoms that may be suggestive of achalasia.
The evaluation of achalasia begins with patients presenting with suspicious symptoms. The most common symptom reported by patients is dysphagia (94%) for liquids and solids.13 Other frequently reported symptoms include regurgitation of undigested food and saliva (76%), particularly when in a supine position, heartburn (52%), chest pain (41%), and weight loss (35%).13,14 The regurgitation may not respond to proton pump inhibitor treatment.11 In some cases, regurgitation may lead to complications such as bronchitis or recurrent aspiration pneumonia.1,11 The differential diagnosis for achalasia includes gastroesophageal reflux disease (GERD), eosinophilic esophagitis (EoE), stricture, other esophageal motor disorders, and malignancy.
The Eckardt score is a standardized tool that was initially developed to measure treatment response based on 4 principal symptoms of achalasia: dysphagia, regurgitation, chest pain, and weight loss (Table 1).15 It uses a 4-point scale (0-3) for each symptom, with a maximum score of 12. A score of no more than 3 indicates an adequate treatment outcome. Although the Eckardt score has not been validated as a patient-reported outcome, it serves as a valuable tool for assessing the severity of symptoms upon the initial diagnosis of achalasia.16,17
Endoscopy
The initial diagnostic step in evaluating achalasia should be an upper endoscopy. The primary purpose of endoscopy is to identify alternative causes of esophageal symptoms, such as mechanical obstructions or pseudoachalasia, which require prompt evaluation and can mimic achalasia during esophageal manometry.18-20 However, endoscopy also can provide clues that increase suspicion for achalasia and warrant further testing. Although the overall diagnostic yield of endoscopy for achalasia is low,11 certain endoscopic findings may raise suspicion for achalasia and prompt referral for manometric testing.
During endoscopy, the following features may suggest achalasia (Table 2):
• dilated or tortuous esophageal lumen with a tight, but not strictured, esophagogastric junction (EGJ);
• retained food or saliva, along with signs of stasis esophagitis;
• rosette-like esophageal folds in the lower esophagus that appear after deep inspiration (known as the esophageal rosette sign)21; and
• the “champagne glass sign,” which is characterized by the distal end of the contracted LES being located
proximal to the squamocolumnar junction, while the squamocolumnar junction itself appears dilated in the retroflexed view.22
Although biopsies obtained during upper endoscopy may be used to rule out EoE, they do not have diagnostic value for achalasia. In cases of achalasia, mucosal eosinophils may be elevated due to primary autoimmune inflammation or secondary to stasis inflammation.11 Differentiating between EoE and achalasia based solely on these findings can be challenging. However, the presence of dysphagia to liquids and solids, along with classic manometric findings, can support a diagnosis of achalasia.
Barium Esophagram
Contrast studies using a barium esophagram play an important role in supporting the diagnosis of achalasia and providing prognostic information. This test is also valuable for assessing the response to therapeutic interventions.
The classic finding on a barium esophagram in achalasia is a dilated esophagus with a tapered EGJ that resembles a “bird’s beak” appearance. Other possible findings include aperistalsis (lack of coordinated contractions), delayed barium emptying, and in late-stage achalasia, a tortuous or severely dilated esophagus (sigmoid esophagus).
Timed Barium Esophagram
To increase the diagnostic yield, a timed barium esophagram (TBE) with a 13-mm barium tablet can be performed. Including a 13-mm barium tablet improves the diagnostic yield for untreated achalasia from 79.5% to 100%, and is recommended by the American College of Gastroenterology guidelines.23,24 To perform a TBE, the patient typically is instructed to consume 200 mL of liquid barium sulfate rapidly. Upright x-ray images are then taken at different time intervals—1 minute, 2 minutes, and 5 minutes—to measure the height of the barium column within the esophagus.25 Abnormal findings on the barium esophagram that support a diagnosis of achalasia
Table 2. Endoscopic Findings
Suggestive of Achalasia
Endoscopic finding
Dilated esophageal lumen with a tight (not strictured) EGJ
Retained food or saliva with stasis esophagitis
Esophageal rosette sign: rosette-like folds in the distal esophagus that appear after deep inspiration
Champagne glass sign: The distal end of contracted LES is located proximal to the squamocolumnar junction, while the squamocolumnar junction itself appears dilated in the retroflexed view
include a barium height more than 5 cm at 1 minute (sensitivity 86%, specificity 71%) and a height greater than 2 cm at 5 minutes (sensitivity 80%, specificity 86%).24
In cases where high-resolution esophageal manometry (HRM) studies yield inconclusive results, a TBE is recommended to distinguish between EGJ outlet obstruction (EGJOO) and achalasia. 25,26 The TBE is relatively easy to perform and provides reproducible results, making it a valuable tool in such cases.
Manometry
Achalasia is diagnosed based on an elevated median integrated relaxation pressure (IRP) and the absence of peristalsis during 100% of swallows.26 In other words, all swallows either occur prematurely or fail to occur. Achalasia is further divided into 3 subtypes based on manometric features ( Figure 1). These subtypes reflect differences in disease chronicity, potential pathophysiology, response to treatment, and overall prognosis.
Type I (classic) achalasia represents an advanced form of the disease characterized by progressive loss of neuronal cell function in the distal esophagus and LES that leads to progressive dilation of the esophageal body.27 During HRM, type I achalasia is distinguished by absent contractility and an increase in IRP.26
In contrast, type II achalasia is considered an earlier form of the disease and the most common subtype identified during HRM. It is characterized by a panesophageal pressure wave of at least 30 mm Hg. 26 This pressure wave is generated by non-lumen obliterating esophageal contractions, similar to what occurs when a water balloon is squeezed.27
Type III (spastic) achalasia is the least common subtype and may involve a different pathophysiology compared with types I and II, with less destruction of neurons from the myenteric plexus within the distal esophagus.28,29 It is characterized by HRM findings that include at least 20% premature and spastic swallows, defined by a distal latency of less than 4.5 seconds and a distal contractile integral of at least 450 mm Hg·s·cm.26 The remaining swallows that are not premature must either fail or fail with panesophageal pressurization.
In certain cases, the diagnosis of achalasia type I or II may be inconclusive. This can occur if a patient with HRM patterns consistent with achalasia type I or II develops noticeable peristalsis when transitioning between the supine and upright positions. 26 In addition, achalasia patients may demonstrate 100% absent contractility or panesophageal pressurization but with borderline or normal IRP. This may occur due to a tight LES, diverticulum, or tortuous esophagus that results in an inability to pass the probe into the stomach.
To confirm the diagnosis of achalasia in cases of inconclusive findings and dysphagia symptoms, the Chicago Classification version 4.0 recommends supportive testing with either TBE or a functional lumen imaging probe (FLIP). In untreated patients, a barium height more than 2 cm at 5 minutes has a sensitivity of 80% and specificity of 86% for differentiating achalasia from other disorders.24 In a study evaluating FLIP, 13 patients with typical achalasia symptoms, absent peristalsis on HRM, and an IRP at the upper limit of normal showed lower EGJ distensibility than 15 healthy controls.30 In addition, these patients responded favorably to achalasia-specific treatments, showing improvements in
1. Achalasia subtypes defined by high-resolution esophageal manometry. All achalasia subtypes must have an elevated median integrated relaxation pressure and absence of peristalsis during 100% of swallows. Type I achalasia is distinguished by a distal contractile integral of <100 mm Hg·s·cm and the absence of panesophageal pressurization. Type II achalasia is characterized by a panesophageal pressure wave ≥30 mm Hg. Type III achalasia is represented by ≥20% premature and spastic swallows, defined by a distal latency of <4.5 s and a distal contractile integral ≥450 mm Hg·s·cm.26
Figure
Figure from Ronnie Fass, MD.
both reported Eckardt score and EGJ distensibility.
HRM testing also can play a role in differentiating opioidinduced esophageal motility disorders from type III achalasia. Chronic opioid use has been shown to cause elevated IRP and peristaltic abnormalities resembling those seen in patients with EGJOO and type III achalasia.31 Ideally, HRM should be performed when patients are not taking opioids, but discontinuing opioid medications may not always be feasible for patients. In such cases, pharmacologic provocation with amyl nitrite and cholecystokinin has been shown to differentiate between opioid-induced esophageal motility disorder and type III achalasia.32 Opioid-exposed patients demonstrate an attenuated rebound contraction of the LES during amyl nitrite recovery and esophageal contraction during the first phase of cholecystokinin response.
The FLIP Assessment
Various studies have established the utility of FLIP in identifying achalasia (Figure 2). In one study involving 40 patients referred for endoscopy and HRM, all 9 patients with achalasia showed abnormal FLIP metrics, characterized by reduced EGJ opening (EGJ distensibility index <2.8 mm2/mm Hg) and an abnormal contractile response to distension.33 Another study of 145 patients with nonobstructive dysphagia found that among the 70 patients with confirmed achalasia on HRM, all exhibited abnormal FLIP measurements of reduced EGJ opening and abnormal contractility. However, the contractility patterns were more heterogeneous among patients with type II achalasia.34
Furthermore, a large retrospective study involving 240 achalasia patients demonstrated that a low EGJ distensibility index is valuable for distinguishing patients from healthy controls. In addition, specific components of the
Figure 2. Characteristics of achalasia defined by FLIP.
A FLIP study in a patient with achalasia. Balloon volume at 50 mL demonstrates a distensibility index of 0.52, diameter of 4.7 mm, pressure of 33.1 mm Hg, and absent contractility.33 FLIP, functional lumen imaging probe.
Figure from Ronnie Fass, MD.
distensibility index, such as a maximum EGJ diameter less than 16 mm, can increase the sensitivity of FLIP in diagnosing achalasia even when the EGJ distensibility index appears normal.35
FLIP also has the potential to identify patients with achalasia-like symptoms but an inconclusive diagnosis, who may benefit from achalasia-directed treatment. Studies have shown that patients with achalasia symptoms and absent peristalsis on HRM but IRP values at the upper limit of normal have a lower EGJ distensibility index compared with healthy controls and respond favorably to achalasia treatment. 30 These results have been supported by other studies.36 Similarly, a retrospective study of 89 patients undergoing peroral endoscopic myotomy (POEM) identified 24 individuals with achalasia-like symptoms, failed peristalsis, and normal IRP values. These patients had a higher EGJ distensibility index (mean value, 2.75 mm2/mm Hg) but responded well to POEM, with a reduction in Eckhardt score.37
FLIP serves as a complementary diagnostic tool to HRM and can be used as confirmatory testing when there is ambiguity in diagnosing achalasia. However, FLIP has not yet been shown to accurately differentiate between the subtypes of achalasia.
Conclusion
Achalasia is a complex esophageal motility disorder that has a significant impact on patients’ well-being and healthcare resources. Its etiology involves the inflammatory degeneration of inhibitory neurons within the esophageal wall, contributing to its subtle and progressive nature. While endoscopy is crucial for excluding secondary causes, HRM stands out as the definitive diagnostic tool for primary achalasia. In instances of diagnostic ambiguity, TBE and FLIP assume pivotal roles, not only in clarifying diagnoses but also in evaluating treatment responses.
The diagnostic process hinges on maintaining a high clinical suspicion, particularly when patients present with hallmark symptoms such as dysphagia, regurgitation, heartburn, chest pain, and weight loss. This recognition is paramount for timely intervention and improved patient outcomes. The comprehensive understanding of achalasia’s diagnostic nuances, coupled with the strategic use of advanced techniques like HRM, TBE, and FLIP, equips clinicians to navigate the complexities of this condition and offer optimal care to those affected.
References
1. Savarino E, et al. Nat Rev Dis Primers. 2022;8(1):28.
2. Sato H, et al. J Gastroenterol. 2019;54(7):621-627.
3. Sadowski DC, et al. Neurogastroenterol Motil 2010;22(9):e256-e261.
4. van Hoeij FB, et al. Neurogastroenterol Motil. 2018;30(2). doi:10.1111/nmo.13195
5. Gaber CE, et al. Clin Gastroenterol Hepatol. 2022;20(2):342-352.
6. Behar J, et al. Gastroenterology. 1993;105(1):111-118.
7. Wong RK, et al. Dig Dis Sci. 1989;34(3):349-352.
8. Verne GN, et al. Gastroenterology. 1999;117(1):26-31.
9. de Oliveira RB, et al. Am J Gastroenterol. 1995;90(7):1119-1124.
10. Costigan DJ, et al. Dig Dis Sci. 1983;28(8):763-765.
11. Vaezi MF, et al. Am J Gastroenterol. 2020;115(9):1393-1411.
12. Eckardt VF, et al. Dig Dis Sci. 1997;42(3):580-585.
13. Fisichella PM, et al. World J Surg. 2008;32(9):1974-1979.
14. Eckardt VF, et al. Gastroenterology. 1999;116(6):1300-1304.
15. Eckardt VF, et al. Gastroenterology. 1992;103(6):1732-1738.
16. Urbach DR, et al. Am J Gastroenterol. 2005;100(8):1668-1676.
17. Patel DA, et al. Dis Esophagus. 2017;30(5):1-23.
18. Tucker HJ, et al. Ann Intern Med. 1978;89(3):315-318.
19. Kahrilas PJ, et al. Am J Med. 1987;82(3):439-446.
20. Dodds WJ, et al. AJR Am J Roentgenol. 1986;146(1):21-23.
21. Iwakiri K, et al. J Gastroenterol. 2010;45(4):422-425.
22. Gomi K, et al. Dig Endosc. 2016;28(6):645-649.
23. Gyawali CP, et al. Am J Gastroenterol. 2020;115(9):1412-1428.
24. Blonski W, et al. Am J Gastroenterol. 2018;113(2):196-203.
25. Kostic S, et al. Dis Esophagus. 2005;18(2):96-103.
26. Yadlapati R, et al. Neurogastroenterol Motil. 2021;33(1):e14058.
27. Pandolfino JE, et al. JAMA. 2015;313(18):1841-1852.
#MondayNightIBD
continued from page 25
colonoscopy should be performed regularly and a change in therapy initiated when there is endoscopic recurrence of the disease.
Patient Experience
During our patient-focused discussions, we delved into the profound effects of IBD symptoms and medication side effects (eg, weight fluctuations, skin manifestations, perianal
28. Tøttrup A, et al. A possible pathogenic role. Dig Dis Sci 1989;34(12):1894-1899.
29. Rieder E, et al. Ann N Y Acad Sci. 2020;1482(1):85-94.
30. Ponds FA, et al. Neurogastroenterol Motil. 2017;29(1):e12908.
31. Babaei A, et al. Neurogastroenterol Motil. 2019;31(7):e13601.
32. Babaei A, et al. Clin Gastroenterol Hepatol. 2020;18(4):813-821.
33. Carlson DA, et al. Gastrointest Endosc. 2019;90(6):915-923.
34. Carlson DA, et al. Am J Gastroenterol. 2016;111(12):1726-1735.
35. Rooney KP, et al. Clin Gastroenterol Hepatol. 2021;19(5):1058-1060.
36. Triggs JR, et al. Clin Gastroenterol Hepatol. 2020;18(10):2209-2217.
37. Kim E, et al. J Neurogastroenterol Motil. 2020;26(2):274-280.
Ofer Fass reported no relevant financial disclosures. Ronnie Fass reported financial relationships with Diversatek, Laborie and Medtronic. He is a member of the Gastroenterology & Endoscopy News editorial board.
disease, and fatigue) on body image perception. Up to 74% of patients conveyed that these factors significantly influenced their negative body image (Figure 3, see page 25). A substantial percentage (59%) revealed that these challenges prompted them to avoid social situations and activities (Figure 4). In particular, surgeries, encompassing procedures, such as J-pouch and ostomies, posed a personal uphill challenge. Although overcoming these challenges instilled a sense of
4. Patient poll No. 2.
If IBD has negatively affected your body image or self-confidence, have you avoided social situations or activities because of it?
Source: @MondayNightIBD
Figure
pride among patients, they acknowledged that, at different points, these experiences took a toll on their mental health. They noted that patient support groups and safe spaces to share personal stories were not only inspiring but also fostered a sense of community that empowered individuals navigating the complexities of the IBD journey.
Anti-Obesity Therapy in IBD
Medical Management
Incretins such as glucose-dependent insulinotropic polypeptide and GLP-1 agonists are nutrient-induced gut hormones whose main physiologic role is to increase insulin secretion and delay gastric emptying. The GLP-1 agonists exenatide (Byetta/Bydureon, AstraZeneca), liraglutide (Saxenda/Victoza, Novo Nordisk), and semaglutide (Ozempic/ Wegovy, Novo Nordisk) are approved to treat type 2 diabetes and are effective anti-obesity medications.21
The use of anti-obesity pharmacotherapy remains limited in patients with IBD, despite the increased prevalence of obesity in this population throughout the last decade. In a retrospective analysis of population-level data using a commercial database from 2010 to 2019, only 2.8% of eligible adults with IBD and obesity were prescribed anti-obesity pharmacotherapy.22 There are limited data on the use of GLP-1 medications for obesity in patients with IBD, but this class of drugs appears to be safe in IBD and could have potential anti-inflammatory effects and a positive impact on the course of IBD. In a nationwide Danish registry study, among 3,751 patients with IBD and type 2 diabetes, those who received GLP-1 therapies had a lower risk for IBDrelated adverse clinical events (eg, the need for steroids, biologics, and hospitalization) compared with those treated with other antidiabetic medications.23
References
1. CDC. Adult obesity facts. May 17, 2022. Accessed April 4, 2024. cdc.gov/obesity/data/adult.html
2. CDC. Childhood obesity facts. May 17, 2022. Accessed April 4, 2024. cdc.gov/obesity/data/childhood.html
3. Lewis JD, et al. Gastroenterology. 2023;165(5):1197-1205.
4. Singh S, et al. Nat Rev Gastroenterol Hepatol. 2017;14(2):110-121.
5. Kim JH, et al. World J Gastroenterol. 2023;29(12):1779-1794.
6. Yarur AJ, et al. Gastroenterology. 2023;165(4):963-975.
7. Bassi M, et al. BioDrugs. 2022;36(2):197-203.
8. Harper JW, et al. Inflamm Bowel Dis. 2013;19(10):2118-2124.
9. Kurnool S, et al. Aliment Pharmacol Ther. 2018;47(11):1472-1479.
10. Dai ZH, et al. Ann Pharmacother. 2020;54(8):729-741.
11. Chuck W, et al. Eur J Gastroenterol Hepatol. 2022;34(6):622-629.
12. Poon SS, et al. J Crohns Colitis. 2015;9(8):640-646.
13. Holtmann MH, et al. Dig Dis Sci. 2010;55(4):1066-1078.
14. Wong ECL, et al. Inflamm Bowel Dis. 2021;27(6):848-854.
15. Farraye FA, et al. Aliment Pharmacol Ther. 2021;54(4):429-440.
16. Sands BE, et al. Inflamm Bowel Dis. 2021;27(6):797-808.
17. Kaazan P, et al. Intern Med J. 2020;50(9):1134-1138.
Endoscopic and Surgical Bariatric Procedures
There are limited data on the outcomes of bariatric procedures in patients with IBD and obesity, and there has been a concern that bariatric surgery may lead to fistulizing complications in patients with Crohn’s disease or exacerbation of IBD. However, a recent review of published case series totaling 101 patients with IBD (61 with Crohn’s disease) highlighted that bariatric surgery is effective and safe in this population.24 Although 10 patients experienced a flare postoperatively, 7 were able to discontinue immunosuppressive therapies. In a small case series, patients with IBD who achieved weight loss after bariatric surgery had fewer IBD-related complications, including steroid use and IBD-related surgery, compared with matched controls during a median follow-up of 7 years.25 In a prospective study of 10 patients (8 with Crohn’s disease), an excess weight loss of 71% after bariatric surgery was reported, along with cessation of mesalamine therapy in some patients during a 2-year follow-up period.26 Endoscopic bariatric therapies (3 intragastric balloon procedures and 4 endoscopic sleeve gastroplasty) performed in a single center in 7 patients with IBD (6 with Crohn’s disease) were found to be safe and effective in patients with IBD and obesity.27
Conclusion
Obesity is associated with an increased risk for IBD and poor treatment response and clinical outcomes. Conversely, weight loss could have potential benefits on the course of IBD. Obesity management should be approached with a multidisciplinary team, including a gastroenterologist, psychologist, registered dietitian, and weight-loss specialists (medical specialist, bariatric endoscopist, and surgeon). Small case series suggest that GLP-1 agonist anti-obesity therapies as well as bariatric procedures are safe and effective in patients with IBD.
18. Winter RW, et al. Am J Gastroenterol. 2022;117(5):777-784.
19. Kaazan P, et al. Dig Dis Sci. 2022;67(12):5628-5636.
20. Brown C, et al. Presented at: American College of Gastroenterology 2007 Annual Scientific Meeting; October 12-17, 2007; Philadelphia, PA. Abstract P270.
21. Wang JY, et al. Front Endocrinol (Lausanne). 2023;14:1085799.
22. Elangovan A, et al. Obes Surg. 2021;31(3):1105-1112.
23. Villumsen M, et al. EClinicalMedicine. 2021;37:100979.
24. Hudson JL, et al. Inflamm Intest Dis. 2019;3(4):173-179.
25. Braga Neto MB, et al. Inflamm Bowel Dis. 2020;26(7):1089-1097.
26. Keidar A, et al. Surg Obes Relat Dis. 2015;11(1):132-136.
27. Johnson AM, et al. Obes Surg. 2023;33(2):676-681.
Dr. Charabaty, the founder of @MondayNightIBD, is a member of the Gastroenterology & Endoscopy News editorial board. She reported financial relationships with AbbVie, Lilly, Janssen, Pfizer, and Takeda. Drs. Bassi, Beniwal-Patel, Fatima, and Kaazan reported no relevant financial disclosures.
Pouchitis
continued from page 17
academic IBD centers, you didn’t see a lot of patients with pouchitis and inflammatory conditions of the pouch. When I would talk to treating providers, they would have a lot of questions about pouchitis and did not have a sense of comfort with how they were taking care of patients in this scenario. The biggest thing we are trying to do is minimize these gray areas where people have all these questions and help clinicians guide their management approach from the beginning according to the available evidence.
GEN: What were some of the hottest points of debate among the guideline panel, and how did you resolve them?
Dr. Barnes: One topic about which there was a little bit of a debate, and that has generated a lot of questions from patients for years, relates to the role of probiotic therapy in both the prevention and the treatment of pouch-related disorders. This has been covered to some degree by prior AGA guidelines, although they were focused on probiotics and probiotic therapy, not on pouchitis.
One reason this has generated a lot of discussion is that the evidence for the use of probiotics, particularly in the area of primary prevention, and even in secondary prevention, is relatively robust. There are some randomized controlled trials, and they showed a pretty big delta when you compare those probiotic therapies versus placebo. What is sort of lacking there is the translation into the real world. Even in some of the real-world studies where they’ve used the same formulations or the same probiotics, patients haven’t had the same effect. That generated a lot of discussion among the guideline panelists regarding weighing real-world evidence versus what we know from a few small randomized controlled trials.
The second thing that also generated a lot of discussion was the role of endoscopic evaluation, or specifically a pouchoscopy, among patients with a suspected inflammatory condition in the pouch. For a long time, people would say if you suspect that a patient has pouchitis, you need to do a pouchoscopy to confirm that before you treat them with antibiotics. I don’t disagree that it would be nice to confirm in every patient that their symptoms are being driven by active inflammation of the pouch. But we know a couple of things. One is that clinical symptoms don’t always track with the degree of inflammation that you see on pouchoscopy. And we also know that, logistically, in terms of practice patterns in the United States across most centers, if a patient calls you and says, “I have increased frequency” or “I have abdominal pain” or “I’m having unusual nighttime stools that are waking me up,” it’s difficult to say, “Come in and we’ll do a pouchoscopy tomorrow morning.” That’s just not how most practices function. But we can write a prescription for antibiotics, and in most cases, their symptoms are going to get better within 24 to 48 hours.
So, as a guideline panel, we said endoscopy is not
necessary for every first episode of pouchitis. But if a patient is having recurring symptoms, or you have a reason to suspect that something else is going on, certainly you should be following that up with a pouchoscopy to make sure that you understand exactly what’s driving that patient’s symptoms.
GEN: What are the biggest gaps in evidence remaining in
this area?
Dr. Barnes: I think one big gap is to determine who’s at risk for pouchitis and the role of prevention. There is an opportunity for earlier intervention that is fueled in some ways by some of what I mentioned before about probiotics. We made a recommendation that secondary prevention could be considered in patients who are interested in taking probiotics. The other reason I mention prevention is that there’s a suggestion that the incidence rate of pouchitis may be increasing. We still don’t know who is at the highest risk for pouchitis outside of those with some classic predictors, such as primary sclerosing cholangitis. Better defining the high-risk groups, by either clinical or translational approaches—that’s a big gap that needs to be filled.
A second big gap is that we still need better study design and a better evidence base when we think about determining how to best treat patients after they develop these inflammatory conditions of the pouch. I mentioned the landmark EARNEST study, which is a randomized controlled trial. I don’t think every single study for every potential therapy to treat patients with a pouch needs to be a randomized controlled trial. We can consider comparative effectiveness studies, pragmatic clinical trials, and don’t necessarily need a placebo control in every trial. We probably need to think a little bit outside the box in terms of our study design. Another approach is large prospective registries. I’m a little bit biased because that’s the kind of research that we’ve been doing at UNC. With these multicenter registries, we probably will be able to more quickly answer some of these questions.
GEN: Is there any other take-home message that you want to end with to wrap up?
Dr. Barnes: It was very exciting to be a part of the first set of society guidelines and going through the evidence base supporting management of patients with pouchitis. But perhaps just as exciting was the opportunity to really codify and try to standardize and spell out an approach to managing these patients. This is not the one end-all, be-all; we’re going to continue to reevaluate the new evidence and incorporate that into future guidelines, but I hope that treating clinicians find this first iteration valuable to fill in some of the gaps. ■
The full interview was edited for clarity and brevity.
Microbiome Zone New Data Bring Answers To Microbial Imbalance
Research has shown that imbalances in the gut microbiome can have negative health consequences, but the jury is still out on how to best test for, identify and treat problems of the microbiome. However, new data are adding to the field.
For this edition of Microbiome Zone, Elena Ivanina, DO, MPH, an integrative gastroenterologist and the founder of the Center for Integrative Gut Health, in New York City, interviewed Mark Pimentel, MD, the executive director of the Medically Associated Science and Technology program at Cedars-Sinai, in Los Angeles, on research he and his team presented at DDW 2024. The topics span imbalances of the microbiome, including small intestinal bacterial overgrowth (SIBO), intestinal sulfide overproduction (ISO), intestinal methanogen overgrowth (IMO) and more.
Dr. Ivanina: You have upward of 12 to 14 posters that you’re presenting. What is the most exciting research that you are presenting at this conference?
Dr. Pimentel: I think one of the questions in regard to breath testing and bacterial overgrowth or the gut microbiome has
been whether we can equate the breath test to symptoms and the microbiome at the same time. And for the first time, we show that if you have hydrogen sulfide on the three-gas breath test, you have hydrogen sulfide producers in the small bowel, and it’s proportional. We also show the same for methane. If you have methane in the breath test result, you have methanogens proportionally higher in the small intestine. So—it seems really weird—but for the first time, breath testing is validated using this three-gas breath test, because we’ve never been able to do that or to validate it in that way.
Dr. Ivanina: Incredible. This is looking at at-home breath testing, correct?
Dr. Pimentel: Yes, that’s the other thing. We have another abstract later where the at-home breath test predicts methane … and that predicted constipation in a nationwide study.
So, at-home breath testing works. It’s pretty cool.
Dr. Ivanina: That’s great. Another interesting concept that I think is being introduced here at DDW is the concept of ISO, or intestinal sulfide overgrowth. Can you talk to us about the story behind ISO?
Dr. Pimentel: I like to make things complicated, but simple. ISO is intestinal sulfide overproduction. It confuses people because overgrowth implies it’s one bug overgrowing, and we see that with SIBO and with IMO, it’s Methanobrevibacter smithii. But with sulfide production, it’s complicated because there are so many bugs that can produce hydrogen sulfide. We’re presenting on ISO, which is caused by a group of bugs, but Proteus mirabilis is the champion of all of them. But it’s not just that one, which again complicates the terminology.
Dr. Ivanina: Interesting. It’s going to keep us all on our toes. You’ve also written some interesting information about hydrogen sulfide (H2S) and even its effect on the mitochondria. Can you say a little bit about that?
Dr. Pimentel: So H2S does a number of things, and it’s been known for a while. Even in the 1990s, research showed it was associated with ulcerative colitis. It’s inflammatory, but it damages your ability to make energy in cells in the lining of the gut, and that can affect motility. It can affect pain signaling. Now, in some of the studies we’re presenting here, we’ve shown that pain is augmented because of the pro-inflammatory nature of H2S. You die if you have a room with too much H 2S in it. That’s the reason behind the canary in the coal mine. It is very toxic, so your body has all sorts of enzymes to try to break it down to prevent toxicity.
Dr. Ivanina: Yes, prior studies have shown a connection with colorectal cancer and ulcerative colitis, but for some reason, it never stuck as something that you really need to start thinking about, right?
Dr. Pimentel: What’s interesting is in the 1990s, people were getting close to this, they were really getting to this point. And then all of a sudden anti−tumor necrosis factor biologics jumped in the arena, and everybody jumped to them, and then that research kind of faded and disappeared. So we’ll see. The IBD people are already starting to reexamine H2S again. There are a couple of abstracts by others on H 2S. Maybe there will be a reinvigoration of this understanding of this group of bugs and inflammatory disease.
Dr. Ivanina: I saw another abstract looking at SIBO-SIFO [small intestinal fungal overgrowth] aspiration in Crohn’s disease, and there was such a high prevalence. These microbiome dysbioses are really connected with IBD. Something else I wanted to bring up is what I believe is the first publication on the elemental formula called mBIOTA (mBIOTA Labs). I think this is probably the first time I saw that there was 100% compliance with an elemental diet. Can you speak about mBIOTA? Why is it different? And how do you use it in your practice for SIBO?
Dr. Pimentel: Well, if you go back to 2003, we published the first paper on an elemental diet for SIBO because it worked when all other antibiotics failed. The problem is it tastes so bad. One thousand people did it in my practice over the years, but it was for the champions of my practice because they really were able to sustain two weeks of this very foul stuff. So, it wasn’t until just the last couple of years that we’ve been assisting this company to really get it to taste good. And they did, and it works amazingly. Compliance is the most important thing for antibiotics for SIBO, and for diet for SIBO, so if you don’t take it, you’re not going to succeed. And it works really, really well.
Dr. Ivanina: Amazing, and just one other question about that. Patients went from mBIOTA to a regular diet, and they maintained some of this efficacy?
Dr. Pimentel: So, we don’t know all the follow-up yet because it wasn’t part of the initial protocol, but we’re going to follow them over time and then maybe do a retrospective study to see how they did. But remarkably, the stools got better. Everything was much, much better. Elemental works.
Dr. Ivanina: Very promising. You have another really fascinating study about N-acetylcysteine (NAC) and its use as a mucolytic with rifaximin (Xifaxan, Salix) for SIBO. Can you talk about that study?
Dr. Pimentel: We always knew rifaximin worked for SIBO and irritable bowel syndrome. It’s approved for IBS on the basis that IBS is a microbiome condition. What we didn’t know then, that we know now, is that a lot of the microbes that are part of SIBO—Escherichia coli and Klebsiella—live in the thick mucus layer that rifaximin can’t penetrate. So, we quickly learned that if we’re able to create a formulation to dissolve the mucus, rifaximin works better. In this animal study, it worked better than rifaximin alone. We also already have a human study, which we haven’t published yet. Another study was presented at DDW on rifamycin MMX (Aemcolo, Redhill) from Europe. So maybe we’re going to move past rifaximin sometime very soon, because rifaximin works, but there are better things coming.
Dr. Ivanina: Fascinating, and I’m assuming we’re not ready for prime time yet, using NAC with rifaximin. We’ll wait for your other published work.
Dr. Pimentel: We’re going to have to wait for the clinical trial.
Dr. Ivanina: OK, coming soon. Any other updates you want to share, anything else that you’re excited about?
Dr. Pimentel: I’m just really excited. This DDW has been very good. I think microbiome research is really taking off at the meeting in different conditions, and I think we’re getting better at understanding the microbiome. ■
This interview was edited for clarity and brevity.
Microbiome Zone
How Have FDA Approvals Changed the Landscape of Microbiome
Therapeutics?
WASHINGTON—Within the past 18 months, the FDA has approved two fecal microbiota products.
“This has been a tremendous year in this field. A lot is changing with regard to the treatment landscape,” said Jessica Allegretti, MD, MPH, the medical director of the Crohn’s and Colitis Center at Brigham and Women’s Hospital and an associate professor of medicine at Harvard Medical School, in Boston.
At DDW 2024, Dr. Allegretti, who also leads her hospital’s Fecal Microbiota Transplantation Program, described these new treatments for recurrent Clostridioides difficile and how she incorporates them into her practice.
The FDA approvals of fecal microbiota, live-jslm (Rebyota, Ferring) and fecal microbiota spores, live-brpk capsules (Vowst, Seres) have changed the field, she explained. Both are indicated for preventing a recurrence of C. difficile after standard antibiotic treatment. “This expanded the options for patients, … although workflow, patient assistance programs and payor coverage remain challenging,” she said.
Comparing and Contrasting
Rebyota is a broad-consortium microbiota-based therapy given as a single rectal installation by enema or colonoscopy in the provider’s office or hospital. In the PUNCH CD3 phase 3 trial, the estimated cure rate was 70.6% with Rebyota versus 57.5% with placebo ( Drugs 2022;82[15]:1527-1538). The product is kept frozen until needed and can be refrigerated for up to five days.
Vowst is a narrow-consortium therapy composed of live purified Firmicutes spores. In the ECOSPOR III study, recurrences were observed in 12% of the Vowst arm compared with 40% of the placebo arm (N Engl J Med 2022;386[3]:220-229). Patients take four capsules once daily for three consecutive days after completing antibiotics two to four days before. Patients should drink 10 ounces of magnesium citrate the day before—at least eight hours before—the first dose of the product.
Although approved in recurrent cases, there are some high-risk patients who may benefit with earlier
treatment, Dr. Allegretti said, “such as in patients who are elderly, immunocompromised or have inflammatory bowel disease, [or are] residents of skilled nursing facilities. The flexibility of the label gives us the ability to use these earlier in the treatment paradigm.”
FMT Still a Good Option
Non–FDA-approved FMT remains an option, she said. “I think there is some ongoing confusion about access to FMT and how to do it, but it’s very simple to order,” she said.
Although many centers halted manufacturing during the COVID-19 pandemic, OpenBiome is still shipping products. It distributes investigational FMT preparations manufactured by the University of Minnesota under an Investigational New Drug (IND) application, Dr. Allegretti noted. Registered physicians may request investigational FMT preparations for the treatment of recurrent C. difficile infection that has not responded to standard therapies. The IND requirement responsibility, as of now, remains with the stool bank and not the recipient site. There are two preparations: one for the prevention of recurrent infection and a higher dose preparation for the treatment of fulminant C. difficile.
However, there are concerns about non-approved FMT, Dr. Allegretti said, including costs of about $1,700 (which is not covered by payors), storage requirements and safety concerns (infections).
How to Choose the Appropriate Product
Dr. Allegretti looks at several factors to guide her choice among the two FDA-approved products and the non–FDA-approved version of FMT:
• Upper versus lower administration: ability to swallow capsules, contraindication to rectal administration or colonoscopy, need for a mucosal assessment.
• Location of patient: distance from center (Vowst is shipped to the patient’s home); inpatient versus outpatient.
• Speed: amount of time available to wait for treatment; insurance approval can take weeks, but non–FDA-approved FMT is readily available. Patient assistance programs are emerging but require that patients apply.
• Approval status: importance of FDA approval to the patient.
“Despite all this, FMT has a well-established role for the prevention of recurrent C. difficile.… I think there may always be a need for the non–FDA-approved version of FMT for a subset of patients.”
On the Horizon
The first non–donor-derived product, VE303, is a rationally defined live bacterial consortia that contains eight types of clonal human commensal bacteria, Dr. Allegretti noted. Administration is oral and no bowel prep is required. In a phase 2 trial, VE303 was very effective in resolving C. difficile (JAMA 2023;329[16]:1356-1366). A phase 3 trial is underway.
More than a dozen other products are in early-phase testing for a variety of conditions beyond C. difficile . “Not all are going to be hits,” she said, “but there’s a lot of interest.”
—Caroline Helwick Dr. Allegretti reported financial relationships with Ferring, Merck and Seres.
BARS® Defined lumen reduction.
Endoscopic anastomosis reduction, e.g. after gastric bypass
BARS® is based on the well-established OTSC® System which already demonstrated safety and effectiveness in anastomosis treatment and lumen reduction after previous gastric bypass.1
BARS® enables the simultaneous use of three different instruments with a conventional singlechannel endoscope. Tissue is retracted into the cap by two anchors, allowing for tissue mobilization on both sides of the enlarged anastomosis. By applying the clip, the enlarged anastomosis is reduced while the remaining lumen is defined by a space keeper balloon.
1 Heylen AM, Jacobs A, Lybeer M, Prosst RL. The OTSC® -Clip in Revisional Endoscopy Against Weight Gain After Bariatric Gastric Bypass Surgery. Obesity Surgery. 2011, 21(10):1629-33; 10.1007/s11695-010-0253-5.