All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IPI will be published in Spring 2025. ISSN No.International Pharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
2024 Senglobal Ltd./Volume 17 Issue 1 – Spring – 2025
04 Editor’s Letter
REGULATORY AND MARKETPLACE
06 Without an End-to-end Review of R&D Processes, Efficiency Drives Will Stall
To improve efficiency in pharma R&D, companies must review processes endto-end rather than relying on rigid SOPs. Tobias Hitziger at MAIN5 explains how holistic process transformation requires clarity on outputs, better-defined roles, and targeted automation. Cultural fears and resource concerns hinder change, but streamlined processes enhance compliance, reduce costs, and ease technology integration.
08 We Need to Speak the Same Language: Computer System Validation in the Pharmaceutical Industry from the Supplier's Perspective
Computer system validation is crucial in regulated industries like pharmaceuticals and food production. While customers bear primary responsibility, suppliers must provide compliant, well-integrated systems. Automated solutions simplify validation which reduces complexity and errors. André Schwarz at EyeC GmbH highlights how effective collaboration and standardised documentation, helps to streamline compliance, ensuring efficient validation across multiple locations and regulatory environments.
DRUG DISCOVERY, DEVELOPMENT & DELIVERY
10 Covalent Drug Discovery: Challenges, Advances, and Success Stories
Covalent inhibitors form bonds with target proteins, offering prolonged effects and targeting previously undruggable sites. Historically overlooked due to safety concerns, they are now gaining traction, especially in oncology. Advances in screening and structural biology enhance drug discovery. Dr. Andrew Ratcliffe and Dr. Scott Martin of Domainex explain how successes like sotorasib and omaveloxolone highlight their growing role across therapeutic areas, including neurology and autoimmune diseases.
CLINICAL & MEDICAL RESEARCH
12 Grappling with the Grey Zones in EMA Computerised Systems and Electronic Data Implementation
The EMA’s 2023 guideline on computerised systems in clinical trials mandates stricter data integrity, security, and audit requirements. Expanding beyond clinical data, it challenges sponsors, CROs, and sites to align systems with regulatory expectations. ICON’s Vesta Marciulioniene stresses how ongoing challenges require industry collaboration, clear guidance, and innovative compliance strategies.
MANUFACTURING
14 Innovations in API Manufacturing of Small Molecule Drugs
Innovation is crucial in API manufacturing to address market demands and enhance sustainability. Advances include automation, AI-driven analytics and green chemistry. Dirk-Jan van Zoelen of Ardena describes how the industry applies the 6M methodology to drive efficiency, safety, and regulatory compliance while preparing for future challenges.
18 Combination Filling: The Trend Towards Machine Customisation
Combination filling involves placing multiple active ingredients in a single capsule to enhance efficacy and offer consumer convenience. Used in pharmaceuticals and nutraceuticals, it supports multi-benefit formulations. Bijo Varghese Babu at ACG highlights how advances in technology will further refine precision and adaptability, shaping the future of capsule-based products.
20 Ensuring A Smooth Transition: Tech Transfer Strategies for Monoclonal Antibody Drug Products in Late-phase Clinical and Commercial Launch
The successful tech transfer of a monoclonal antibody (mAb) drug from clinical development to commercial manufacturing is a complex, strategic process. It ensures scalability, regulatory compliance, and supply chain readiness. Shawn Cain of PCI touches on how advanced manufacturing technologies and collaboration accelerate commercialisation, ensuring consistent, high-quality, and safe biologic therapies.
FILL-FINISH
22 Maintain Sterile Pharmaceutical Production with Uninterrupted Power Supply
Pharmaceutical manufacturers rely on uninterrupted power to ensure aseptic production. Power outages risk drug quality and production delays. Kai Schumacher at Vetter explains how despite installation challenges, investing in UPS systems enhances reliability, safeguards drug supply, and ensures consistent pharmaceutical manufacturing amid increasing power disruptions worldwide.
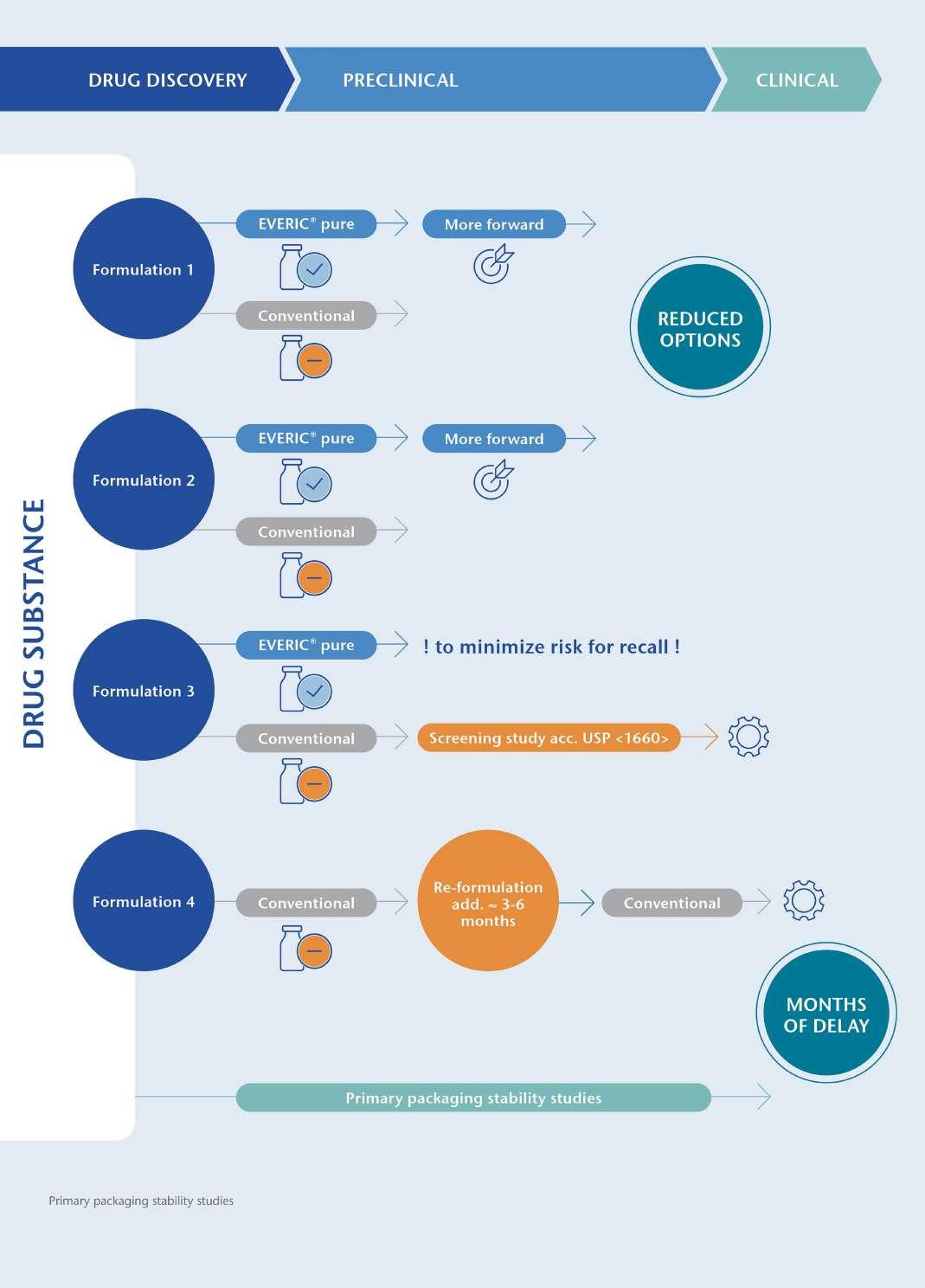
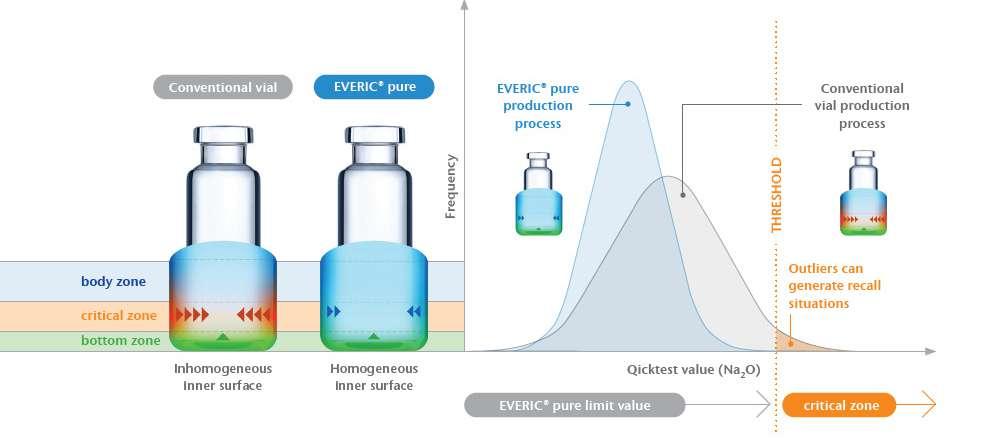
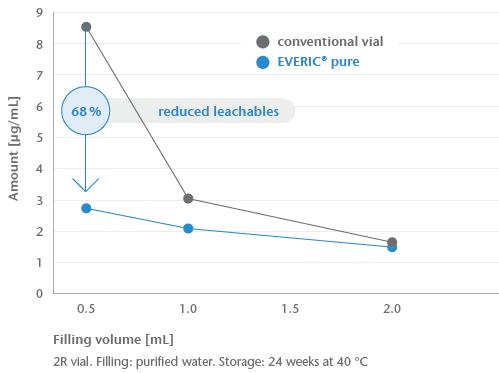
24 EVERIC® pure – The Safe Option Right from the Start: A Drug Containment Platform to Streamline Drug Development
Selecting the wrong vial can jeopardise patient safety and delay drug development. Issues like glass delamination and pH shifts may surface years later, leading to recalls. SCHOTT Pharma’s EVERIC® pure vials prevent these risks with a controlled inner surface, reducing leachables and ensuring drug stability. Diana Löber of SCHOTT Pharma adds how this solution enhances safety, shortens time-to-market, and simplifies adoption.
PACKAGING
30 Labelling and Serialisation to Comply with the EU MDR
The EU MDR enforces stricter regulations on medical device labelling and serialisation to enhance safety and traceability. Manufacturers must adopt UDI codes and suitable printing technologies. Bart Vansteenkiste at Domino Global Life Sciences highlights how future regulations may expand serialisation requirements, benefiting manufacturers in counterfeit prevention and efficiency improvements.
34 Navigating the Ever-changing World of Labelling Compliance
The COVID-19 pandemic exposed flaws in pharmaceutical supply chains, prompting reforms to enhance security, innovation, and affordability. Labelling compliance is crucial for efficiency, safety, and regulatory adherence, yet many firms rely on outdated systems. Kallik’s Gurdip Singh discusses how companies have benefited from digital transformation, which helps to ensure compliance and efficiency amid evolving regulations.
HEALTH OUTCOMES
40 The Impact of Patient Adherence Technology on Global Health Outcomes
AI is transforming medication adherence by integrating with diagnostics, digital health, and biomarker monitoring to personalise treatments. Dr. Hakim Yadi of Closed Loop Medicine discusses how AI-driven insights help tailor medications, address socio-economic disparities, and streamline regulatory processes, ultimately ensuring patients receive the most effective, personalised care.
42 Defying the Odds:
Speeding Patient Access to Life-changing Treatments in Rare Disease
A determined father fought for a gene therapy for his son's rare disease, highlighting the struggle to access rare disease treatments. Veeva Europe’s Chris Moore outlines how, with only 5% of such diseases having approved therapies in the EU, biopharma companies use data, technology, and expert engagement to accelerate market access, improve medical education, and support healthcare professionals in delivering life-changing treatments.
TECHNOLOGY
44 Technology’s Role in Pharmaceutical Manufacturing Excellence Integrating GMP-compliant technologies like LIMS streamlines data management and ensures regulatory compliance. Advanced monitoring and automation improve efficiency, prevent costly failures, and support evolving drug development. Robin Stolzberg of Sapio Sciences stresses that as pharmaceutical manufacturing advances, GMP adherence remains critical for maintaining high-quality, effective treatments.
48 A Five-year Roadmap for Technology-led PV Innovation Technology is revolutionising pharmacovigilance (PV), with AI enhancing data capture, automation, and efficiency. Future priorities include lowering
costs, improving cross-platform integration, and using AI for data interaction rather than just predictions. Martin Holm-Petersen at Qinesca explains how standardised, user-friendly systems and improved data quality will ensure compliance and patient safety in an evolving pharmaceutical landscape.
LOGISTICS & SUPPLY CHAIN MANAGEMENT
52 Six Trends Shaping Patient-centric Pharmaceutical Logistics in 2025
The pharmaceutical cold chain is evolving because of regulations and the need for resilience. Key trends for 2025 focus on direct-to-patient logistics, supply chain diversification, sustainability, and expansion into emerging markets. Mergers and AI-driven logistics will improve efficiency, while blockchain will enhance transparency. Envirotainer’s Delphine Perridy highlights how companies must adopt agile tech solutions for reliable medicine delivery as decentralised trials and small-batch shipments increase.
54 Moving Science Forward in 2025: Emerging trends in the Life Science Logistics Industry
The life sciences logistics industry in 2025 will focus on sustainability, collaboration, and regulatory compliance. Jeff Stone of Biocair discusses how companies must balance reusable packaging with safety, foster partnerships to navigate funding challenges, and stay updated on evolving regulations. Adapting to these trends will ensure efficiency and continued scientific progress.
56 Uncovering the Supply Strategies Shaping More Patient-centric Drug Development
Patient-centric drug development is reshaping clinical trials, prioritising accessibility and efficiency. Decentralised trials, Just-in-Time Manufacturing (JTM), and Direct-to-Patient (DTP) distribution improve patient experience and streamline supply chains. Margaret Radford and David Ergott at Almac Clinical Services add how Managed Access Programmes (MAPs) ensure continued treatment access.
SUBSECTION: NASAL & INHALATION (PART A)
60 Advancing Nasal Protection: Healthcare Innovations Beyond COVID-19 Nasal protection solutions, initially developed for COVID-19 prevention, are evolving to address broader health challenges like seasonal influenza, allergies, and respiratory defence. Innovations include physical barrier and neutralising nasal sprays, which offer protection against pathogens and allergens. Michael Hsu at Birmingham Biotech LTD highlights how ongoing research aims to improve comfort, effectiveness, and non-medicated options.
64 Q&A with Sarah Bunyan: Understanding and Anticipating Regulation in the pMDI Space
As sustainability pressures drive innovation in pressurised Metered Dose Inhalers (pMDIs), the regulatory landscape must adapt to accommodate lowcarbon technology. Regulators, drug developers, and CDMOs are collaborating to transition to environmentally friendly propellants. Bespak’s Sarah Bunyan touches on how the future of pMDI regulations will likely include even lower GWP propellants and better recycling practices.
66 The Role of Ergonomic Studies and Automated Solutions in Nasal Drug Product Development
The high failure rate of orally inhaled and nasal drug products (OINDPs) stems from variability in formulation, device performance, and patient use. Regulatory agencies emphasise human-realistic actuation to improve testing accuracy. Grant Thurston at Proveris Scientific explains how automated systems enhance reproducibility, minimise variability, and streamline regulatory approval, ensuring reliable drug delivery and optimising quality control workflows for better patient outcomes.
APPLICATION NOTE
36 Large-format Pouches in the Medical Device and Pharmaceutical Industry
Christ Packing Systems has developed an automated solution for secondary pouch packaging, helping a U.S. company transition from manual to automated packaging of neurovascular catheters. Jörg Aurbacher of Christ Packing Systems explains how their modular BoxTeq cartoner ensures precise handling, compliance, and efficiency.
PEN CARTRIDGES
Designed for precise integration and functionality with pen systems
Highly accurate dimensional specifications enable precise fit with pen systems and support exact filling volumes
Strong mechanical durability minimizes risk of breakage during fill-finish operationsand end-user handling
High cosmetic quality reduces risk of rejects due to glass defectsand safeguards higher acceptance by end-users
Editor's Letter
As we welcome the spring of 2025, the pharmaceutical industry finds itself at a crucial crossroads – facing both challenges and opportunities that will shape its future. This season of renewal and innovation brings with it a continued focus on advancing drug development, strengthening regulatory frameworks, and ensuring patient access to life-changing treatments.
In this edition, we explore the latest developments across the sector, from the impact of patient adherence technology on global health outcomes, to technology transfer for monoclonal antibody drug products in late-phase clinical and commercial launch. With collaboration between industry leaders, healthcare professionals, and policymakers more critical than ever, we look ahead to how countries around the world can strengthen their positions as global hubs for pharmaceutical innovation.
As always, we remain committed to fostering dialogue and sharing insights that will drive the industry forward. We hope this issue provides valuable perspectives and inspiration for all those dedicated to improving health outcomes through science and innovation.
We open this journal with a write-up from Tobias Hitziger at MAIN5, who highlights the growing pressure on the life sciences industry to improve efficiency across Regulatory Affairs, Quality, and Pharmacovigilance functions. Rising costs, intense global competition, and the growing complexity of pharmaceutical products necessitate a comprehensive transformation of R&D processes. Yet, many organisations remain constrained by traditional Standard Operating Procedures
Editorial Advisory Board
Bakhyt Sarymsakova, Head of Department of International Cooperation, National Research, Center of MCH, Astana, Kazakhstan
Catherine Lund, Vice Chairman, OnQ Consulting
Deborah A. Komlos, Principal STEM Content Analyst, Clarivate
Diana L. Anderson, Ph.D president and CEO of D. Anderson & Company
Franz Buchholzer, Director Regulatory Operations worldwide, PharmaNet development Group
Francis Crawley. Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
(SOPs), limiting the effectiveness of efficiency initiatives. The author emphasises that true progress requires an end-to-end review of processes. By shifting focus to holistic improvements, companies can streamline workflows, enhance compliance, and reduce inefficiencies.
The EMA's guideline on Computerised Systems and Electronic Data in Clinical Trials, released in September 2023, has introduced significant challenges for pharmaceutical and biotech companies. Designed to enhance data integrity, security, and transparency, the framework expands compliance obligations to all trial-related electronic data, impacting sponsors, CROs, and sites worldwide. Despite the deadline passing, many organisations still struggle with implementation complexities, from resource investment to regulatory ambiguity. ICON’s Vesta Marciulioniene delves into the ongoing hurdles of adapting systems to meet these requirements and the broader implications for clinical research, compliance, and the evolving digital landscape in clinical trials.
In the Health Outcomes section, one of my personal favourites is a thought-provoking piece on the unique challenges of rare
diseases, from delayed diagnoses to limited treatment options. Chris Moore of Veeva Europe showcases the inspiring story of a man who defied the odds to develop a gene therapy for his son’s ultra-rare condition. This article also explores the evolving landscape of rare disease treatment, emphasising the need for biopharma agility in launching new therapies. With advanced data insights, connected technology, and strategic medical engagement, companies can accelerate patient access to life-saving treatments. As the industry aims for 1,000 new therapies by 2030, collaboration and innovation remain essential in transforming rare disease care.
We also feature a series on Fill-Finish, which includes an editorial by Kai Schumacher from Vetter. In the highly regulated pharmaceutical industry, maintaining a stable power supply is not just about efficiency – it is essential for ensuring drug safety and production continuity. Any power interruption can disrupt aseptic manufacturing processes, compromise product integrity, and result in significant losses. The author addresses the importance of uninterrupted power and details Vetter’s investment in innovative solutions to safeguard operations. He examines the impact of power fluctuations on pharmaceutical manufacturing and explores technologies and strategies that help mitigate these risks, ensuring a reliable and sustainable production environment.
I hope these discussions spark collaboration, fuel innovation, and drive meaningful progress across the sector. Enjoy this insightful read, and I look forward to connecting with many of you at upcoming exhibitions and events!
Kelly Woods, Editorial & Production Coordinator
Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
Jagdish Unni, Vice President – Beroe Risk and Industry Delivery Lead – Healthcare, Beroe Inc.
Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma
Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
Steve Heath, Head of EMEA – Medidata Solutions, Inc
Patrice Hugo, Chief Scientific Officer, Clearstone Central Laboratories
Heinrich Klech, Professor of Medicine, CEO and Executive Vice President, Vienna School of Clinical Research
Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
Stefan Astrom, Founder and CEO of Astrom Research International HB
Benzalkonium Chloride is a documented broad-spectrum antimicrobial surfactant for use as a preservative and antiseptic active.
It has proven efficacy against both Gram+, Gram- and acid-fast bacteria, fungi and enveloped viruses at very low concentrations. Its properties, combined with our expertise and pharmaceutical grade, make it the ingredient of choice for antimicrobial control in topical formulations.
Learn more at novonordiskpharmatech.com/BKC
the potential benefits will always be compromised.
Inhibitors of Change
So why hasn’t more progress been made, given the scope for holistic process improvement? What is holding back pharma R&D organisations from being bolder and more innovative in their process transformation?
Culturally, some reservations are linked to an instinctive fear of extended transparency, especially during inspections. Where processes are viewed, treated and managed more holistically and continuously across the traditional boundaries or departmental divides in a process management suite, there may be concern that inspectors might extend their curiosity and raise their gaze when reviewing current procedures. This concern is heightened where companies use business process management (BPM suites) to represent their entire process landscape within a single system (with the risk that inspectors can freely “jump” from one process to another). Yet, if processes have been well-defined and are running smoothly, in a closely-tracked way, the ability to see all of this at a glance is a positive development.
More practically, it is the lack of clear process definition (beyond the scope of individual SOPs) that is the greater sticking point. It is only when process owners and process ‘customers’ (those on the receiving end of the output) are agreed on what a good process looks like that this knowledge can be applied effectively to streamline it. This is because, in many cases, there has been a loss of sight of the deliverables of processes, and who and what they are designed to serve – beyond inspectors’ satisfaction that no corners are being cut; that procedure repeatability is ensuring the highest standards of quality, safety and regulatory compliance of a created output. If the output is ultimately wrong, for instance, any gains in ‘throwing it over the wall’ more swiftly will be lost. If the output is a document (e.g. part of a regulatory dossier) which fails to meet the requirements, it will be sent back to the author – creating a costly and timely rework cycle.
Another common fear of investing in real process change is linked to concerns about the likely resources this will consume, both financial and in terms of people’s time – potentially detracting from business
Regulatory & Marketplace
as usual. As long as process reviews and transformation plans are designed for and adapted to the particular needs of the organisation, the justification for the improvements become clear and these barriers to change are soon brought down.
How to go Deeper
True process improvement, especially endto-end transformation across an extended environment, needs to start with clear, ideally in-person communication between the main parties involved – the process owner(s), process participants and the process customer(s). The goal of this should be to identify the most efficient process to create the output as expected by the customer leveraging experiences where repeated issues emerged in the past (e.g. repeated pushback, rework cycles).
Alongside, or to help provide a focus for, discussions about scope for process improvements, process stakeholders can harness intelligent “process mining” tools to identify common bottlenecks and repeat loops.
The greater the scope of the process elaboration, and the broader the range of stakeholders, the greater the timescale needed to evaluate, redesign and optimise the new agreed scenario.
Again, staying focused on the common goals will be important – such as improved and more effortless compliance; greater clarity for all parties about what is needed, when and why; and the scope to alleviate resource pressures through the targeted automation of labour-intensive tasks using appropriate technology (e.g. modern RIM capabilities).
Greater Things will Come from Good Groundwork
Once good, streamlined and aligned processes are in place, teams can pull this
content and create SOPs. Clear definition of process roles (or ‘swim lanes’) meanwhile can help identify the specific activities linked to them. This in turn could be used to inform role-based training, for the individuals fulfilling the processes.
Ultimately, good process elaboration, oriented towards the process customer, enables greater operational efficiency. It also makes it much easier to introduce new technology systems and features, since well-described processes inherently force robust descriptions of user requirements. Certainly, no business ever suffered from having a better understanding of its endto-end processes. By contrast, where a lack of clarity persists – of how process output and any efficiencies gained will flow back into the company and be harnessed by adjacent teams or functions – the scope for transformation (at any level, let alone interdepartment process streamlining) will be significantly compromised.
Tobias Hitziger is a Management Consultant at MAIN5, with over 15 years of experience in the pharmaceutical industry, and heads the firm’s process team. As an expert in business process management within pharma R&D, he specialises in organisational change, lean business transformations, and senior management coaching, with a focus on optimising operations and driving efficiency in global regulatory affairs.
We Need to Speak the Same Language: Computer System Validation in the Pharmaceutical Industry from the Supplier's Perspective
In regulated industries such as pharmaceuticals, cosmetics, and food production, the computer system validation plays a pivotal role in ensuring compliance with the regulations, customers’ and producer's safety, and quality. The responsibility for this process belongs to the company that uses the computerised system in its production process. But as soon as the systems are purchased from a supplier, it is essential that this supplier also knows the industry it is selling into and the requirements of this industry. The system of these suppliers must be integrated seamlessly into the validation framework of the customer. This article examines the topic from the perspective of a supplier to customers in the pharmaceutical, food and medical device industries, with the challenges and solutions for customers and suppliers.
Regulatory Requirements
It always starts with the law – national laws and regulations such as the German Medicinal Products Act (Arzneimittelgesetz, AMG) or the German Drug Manufacturing Regulation (Arzneimittel- und Wirkstoffherstellungsverordnung, AMWHV), to name just two, make direct and indirect reference to the GMP regulations. Therefore, compliance with regulatory standards such as Good Manufacturing Practice (GMP), GAMP5 (Good Automated Manufacturing Practice), EU GMP Annex 11 or in the US sector with 21 CFR Part 11, is mandatory in regulated industries. These regulations outline the requirements for data integrity, traceability and system validation. While the customer bears the main responsibility for the validation of the overall process, the supplier must provide systems that enable this validation process and, ideally, make it as simple as possible for the customer.
What does this mean for the supplier?
First of all, they need to understand the customer's requirements for the system and its validation capability. To do this, it is essential to understand the rules of the regulatory landscape, they must know the relevant laws and ordinances, best
practices and regulations and draw the right conclusions from them: What does all this mean for our product? Where and how does our system fit into this complex process, how does it interact in this system and what are the consequences for the validation?
If software is involved, it must meet the requirements for security and data integrity. The data must be stored securely, the results must be reproducible, and a secure audit trail must record all relevant data. Ultimately, they must develop the product or further develop an existing product, in such a way that it is already designed for the intended use. Without this basic understanding, the delivered system makes the customer’s validation process significantly more complex and time-consuming. The goal for the supplier should be to deliver a system that reduces the complexity of the validation process, saves time and resources, and is perfectly tailored to the needs of the industry.
Simplification Instead of Increased Complexity
Suppliers, as mentioned, play a crucial role in simplifying the validation process for customers. If they can provide a system that is inherently compliant with existing regulations or enables compliance, is well-documented, and includes the necessary features, then it will allow customers to meet regulatory requirements efficiently and with minimal effort.
For example, EyeC offers automated print inspection systems that compare the approved PDF of the artwork against a printed package (folding carton, flexible foil) or a package insert. Errors in the artwork, text, printed 1D and 2D codes, and embossed Braille are detected and visualised. The user can then decide whether the deviations are acceptable or unacceptable. Defective packaging batches can thus be reliably sorted out, with such a sample inspection completed in just a few minutes. It is far superior to any human visual inspection. Even if the four-eyes principle is applied, it can never be completely ruled out that errors will be overlooked. Computer-aided inspections, which analyse pixel by pixel, needs no pause, knows no fatigue and no
overlooking. To help you save time, the system offers unrivaled speed, accuracy and efficiency.
However, one might argue, doesn’t such an automated system increase the complexity of validation? Don’t numerous contingencies need to be considered, especially regarding data handling?
Yes and no – on the one hand, a complex computerised system naturally needs to be understood and integrated into the validation process. For customers, this can be challenging given the multitude of systems involved in their processes. They are often unfamiliar with the systems, have to understand their function and purpose, and need to gather information to know what exactly needs to be done in the validation. More than once, we have received inquiries from validation officers asking, “What exactly did we purchase from you, and what does your system actually do?”
And this is where the aforementioned "no" comes into play – the supplier plays a key role in helping the customer reduce complexity by providing the necessary information, and delivering a system that is easy to understand and to operate.
Role Reversal as a Method
Let’s return to our example of the print inspection system. What can we provide to the customer to help them understand what the system they have purchased does, so they can place it within their own environment and integrate it into the validation framework?
Our first step was to develop a sample URS, where our Validation Support team put themselves in the customer’s shoes and asked: What must such a print inspection system be capable of to meet my requirements for inspecting packaging materials safely and quickly in accordance with GMP regulations? What are my requirements for the system?
Based on these considerations, discussions with customers, and studying laws and regulations, we developed a URS that precisely describes the practical
Covalent Drug Discovery: Challenges, Advances, and Success Stories
Covalent inhibitors possess a functional group that reacts with a nucleophilic residue on a target protein to form a bond that inhibits the protein’s function. Historically, covalent inhibitors have been successfully developed into marketed drugs for a wide range of therapeutic targets, with examples like acetylsalicylic acid (aspirin), β-lactam antibiotics (penicillins), and in particular in the oncogenic area, with the likes of Ibrutinib (for the treatment of lymphoma and chronic lymphocytic leukaemia) and sotorasib (for the treatment of lung cancers with the KRASG12C mutation). Most drugs on the market pre-2000 were only later discovered to have a covalent mechanism of action, one of the most prominent examples being aspirin.1
While representing a significant portion of the available drugs on the market, the pharmaceutical industry has, until relatively recently, been reluctant to deliberately design covalent drugs. This was due to the perception that covalently modifying proteins could pose safety risks, such as autoimmune reactions like idiosyncratic liver toxicity. Designing and tuning the reactivity of the electrophilic warhead (the functional group that forms the covalent bond with the protein), to obtain a suitable selectivity and safety profile, can be challenging. It must balance chemical reactivity with selectivity while maintaining stability and efficacy. Additionally, specialised assays, resistance concerns, and species-specific differences add complexity. Advancements in structural biology, computational modelling, and screening techniques are helping to address these hurdles.
The 2010s marked a significant rise in the development of designed covalent drugs for cancer treatments since the risk of autoimmune reactions was considered less of a concern for oncology. We are now beginning to see covalent drugs being considered for a much wider range of therapeutic areas. An example of a non-oncology covalent compound is omaveloxolone (Skyclarys®), which is derived from a natural product. It
inhibits KEAP1 and is approved in the US for treating Friedreich’s Ataxia.2
Advantages of Covalent Drugs
Covalent drugs exhibit several key features that distinguish them from traditional reversible inhibitors. The formation of the covalent bond with the target protein can result in prolonged pharmacological effects, which can reduce dosing frequency and enhance patient compliance. This extended duration of action is particularly beneficial in cases where the target protein is resynthesised slowly, allowing for sustained therapeutic impact even after the compound has been cleared from circulation. A less frequent drug dosing schedule should help limit the likelihood and severity of drug-drug interactions and adverse events due to off target activities.
Furthermore, covalent drugs are uniquely suited to address cryptic or shallow binding sites that are often inaccessible to reversible inhibitors, making them a valuable tool in targeting proteins previously considered to be undruggable.
Drug Discovery Approaches
There are two essential approaches to discovering covalent drugs. The first one is a ligand first approach and relies on the identification of a reversible inhibitor, to which a covalent warhead can later be attached that interacts with a nucleophilic residue on the target protein. This approach plays very much to the strengths of structural based techniques. The second is an electrophile first approach, which relies on screening a library of fragments containing a reactive warhead, again playing to the strengths of structural based techniques but also to mass spectrometry (MS)-based screening approaches.
Covalent fragment-based drug discovery (FBDD) has become a powerful strategy in preclinical drug discovery. By employing diverse fragment libraries, researchers can screen small, chemically tractable molecules against target proteins to identify initial hits. These hits can then be optimised through iterative cycles of design and testing, ultimately yielding lead compounds with desirable pharmacological properties.
Domainex, a Cambridge based drug discovery CRO, exemplifies this approach through its curated covalent fragment library. Comprising of approximately 700 fragments, this library adheres to fragmentlike properties such as the rule of three. The rule of three refers to fragments having a molecular weight lower than 300Da, no more than three hydrogen bond donors or acceptors, and a calculated logP value of equal to or less than three.3
The inclusion of reversible and irreversible warheads in the fragment library, such as acrylamides and cyanoacrylamides, allows for broad exploration of chemical space, increasing the likelihood of identifying viable leads. Advancements in screening methodologies have played a pivotal role in the resurgence of covalent drug discovery. MS-based covalent screening is used to enable the rapid identification of covalent fragment hits. This approach can provide detailed insights into binding stoichiometry, selectivity, and reactivity, ensuring that only the most promising compounds are progressed.
To improve the quality of the hits further, Domainex established a glutathione (GSH) reactivity real-time kinetic assay to assess the intrinsic reactivity of the covalent compounds. Covalent hits are incubated with excess GSH, then compound disappearance and GSH-adduct formation are measured by LC-MS. Compounds that are deemed to have a half-life of <100 minutes are rejected as containing unfavourable highly reactive warheads which may be more prone to causing off target effects.
Compounds that have the right properties (stoichiometry/GSH half-life/% binding) are selected for binding site identification by LC-MS peptide mapping to confirm covalent binding to the target cysteine. Further characterisation involves determining the second order rate constant Kinact/Ki that measures the covalent binding rate. This can be used to understand the potency of the hits and form part of a screening cascade in the design-maketest cycles for chemistry optimisation during lead identification and lead optimisation.
Regulatory & Marketplace Drug Discovery, Development & Delivery
prioritisation of high-quality leads for covalent preclinical research.
Success Stories and Future Outlook
The success of covalent inhibitors in preclinical development is best illustrated through case studies. The KRASG12C inhibitor sotorasib stands as a landmark achievement, demonstrating the potential of covalent drugs to address highly challenging targets. Similarly, BTK inhibitors like ibrutinib have shown remarkable efficacy in autoimmune disease models, highlighting the versatility of covalent approaches.
Moreover, non-oncology indications are now benefiting from covalent drug discovery. For example, CNS-targeted covalent inhibitors are advancing rapidly in preclinical models, showcasing the potential for these compounds to address unmet needs in neurology and beyond.
While most covalent drugs on the market target cysteine,4 recent years have shown growing interest in developing covalent inhibitors against other amino acids, such as tyrosine, lysine, histidine and threonine. Cysteine is prevalent in only 2.3% of the human proteome, compared to the higher prevalence of lysine and threonine at approximately 5–6%.5 This shift has been accompanied by the emergence of noncysteine targeting covalent warheads, including fluorosulfate and sulfonyl fluoride classes, which effectively target residues like histidine, tyrosine, and lysine. Notably, Revolution Medicine's RMC-98056, a compound targeting the KRASG12D mutation found in pancreatic and colorectal cancers, exemplifies innovative covalent drug design. Employing an aziridine warhead, this compound binds Cyclophilin A, forming a binary complex that subsequently interacts
being beyond the ‘rule of five', RNC-9805 demonstrated encouraging safety signals and preliminary efficacy in Phase 1/1b clinical trials, underscoring the potential of these novel approaches to be developed for a broader range of therapeutic targets and increasing their applicability across disease areas.7
Despite the challenges in design, such as balancing reactivity and selectivity, the advantages of covalent drugs have transitioned from historical successes discovered serendipitously to a deliberate targeted approach in drug discovery and design. Advancements in structural biology, computational modelling and screening techniques are addressing the hurdles enabling their discovery and optimisation. The growing success of covalent drugs in oncology and non-oncology indications, coupled with innovations targeting residues beyond cysteine, highlights their expanding potential across therapeutic areas. Drugs such as sotorasib and omaveloxolone are showing that with strategic design and a refined approach from the pharmaceutical industry, covalent drugs are poised to play an even more significant role in addressing unmet medical needs.
2. Arnold Lee, Omaveloxolone: First Approval, Drugs 2023, 83, 725
3. Jhoti, H., Williams, G., Rees, D. et al. The 'rule of three' for fragment-based drug discovery: where are we now?. Nat Rev Drug Discov 12, 644 (2013). https://doi.org/10.1038/nrd3926-c1
4. Gyorgy M Keseru et al. Covalent fragment approaches targeting non-cysteine residues, Trends in Pharmacological Sciences 2023, 44,
802
Peczka et al. Electrophilic warheads in covalent drug discovery : an overview Expert Opin. Drug Discovery 2022, 17, 413
Discovery of RMC-9805, an oral, RAS(ON) G12Dselective covalent Tri-Complex Inhibitor chromeextension://efaidnbmnnnibpcajpcglclefindmkaj https://www.revmed.com/wp-content/ uploads/2024/04/NDoTH_9805_FINAL.pdf https://www.onclive.com/view/rmc-9805triggers-tumor-regressions-in-kras-g12dmutant-pancreatic-cancer
Dr. Andrew Ratcliffe 'AJ' is a Medicinal Chemist with over 30 years of drug discovery experience covering early hit identification through to candidate nomination. Prior to joining Domainex, AJ has worked at Rhone Poulenc Rorer, Celltech, UCB, Cellzome, Redx Antiinfectives and Novintum Bioscience. He has taken leadership roles cumulating in the nomination of multiple small molecule drug candidates including CDP323 (a VLA4 integrin antagonist) which was progressed to phase II clinical trials. AJ completed his first degree and PhD at the University of Bath and has over 70 publications covering peerreviewed papers and patents.
Dr. Scott Martin has 25 years of experience in bioanalytical/analytical mass spectrometry within drug discovery and development. He has successfully supported projects throughout the drug discovery and development process by driving scientific innovation, matrix managing teams and providing expert opinion in biotransformation and bioanalysis. Before joining Domainex, Scott worked within the Oncology DMPK department at AstraZeneca for 20 years. He has also held positions at Roche, GlaxoWellcome and Zeneca agrochemicals. While at AstraZeneca, Scott successfully completed a part time PhD in drug metabolism at Sheffield Hallam University and has authored/ co-authored 20 papers in peer reviewed journals.
Dr. Andrew Ratcliffe
Dr. Scott Martin
Clinical and Medical Research
Grappling with the Grey Zones in EMA Computerised Systems and Electronic Data Implementation
The integration of computerised systems and electronic data standards continues to play a vital role in the conduct and management of clinical trials as well as in ensuring the clinical trial data reliability and the protection of patient safety and rights from systems and electronic data perspective. The EMA released its guideline on Computerised Systems and Electronic Data in Clinical Trials in September 2023. All biotech or pharmaceutical companies worldwide seeking marketing authorisation of medicinal products in EU are subject to this guideline. As the implementation deadline has come and gone, we explore the ongoing challenges of navigating the boundaries of scope related to compliant implementation – from the high resource investment required to develop and adapt systems across multiple stakeholders to understanding the regulatory layers around impacted data systems.
Bracing for Broad Impacts on Clinical Research
The EMA’s new guideline introduced a future-focused framework for ensuring the integrity, security and traceability of electronic data. Among its key provisions, it reinforces the importance of access controls, user authentication and data encryption to safeguard against unauthorised access and breaches. Additionally, it places greater emphasis on audit trails, ensuring transparency and traceability in data modifications and reinforcing the reliability and authenticity of electronic data in clinical trials.
One of the most significant shifts the guideline introduced is expanding the scope beyond clinical data. Under the new framework, “any trial related data handled in electronic systems for the purposes of conducting and reporting a clinical trial, and relevant for the clinical trial”, as clarified by EMA, is now within its remit. This change has prompted sponsors, sites, CROs and vendors to reevaluate their computerised systems and electronic data management, ensuring
that all relevant systems and system owners align with the new requirements. As organisations reorient around this shift, a critical challenge remains identifying and mitigating potential gaps that now fall under regulatory scrutiny.
Conquering Compliance Challenges
The complexity and broad scope of the EMA’s guideline have presented significant hurdles for the industry, particularly in terms of time and investment. Many underestimated the effort required to adapt or develop their existing systems to meet the future-thinking expectations. The process involves multiple stages: scoping, planning, piloting, testing, validating and implementing; each requiring meticulous execution to ensure compliance.
While sponsor-owned systems tend to be within reasonable control, compliance becomes more of an intricate challenge when accounting for the variability of governmental, regulatory and site-owned systems used in clinical trials. Key concerns include access control discrepancies, functional adequacy, direct access provisions and audit trail consistency – all of which can create compliance gaps.
The EMA took a holistic, forward-thinking approach, which emphasised the need for cross-functional collaboration among sponsors, CROs, investigators and technology vendors. Larger industry players, particularly well-resourced sponsors and CROs, were able to adapt more swiftly. Many of us had a long line of sight on the draft guidelines, allowing for proactive strategies for change management upon final publication. However, for smaller organisations, service providers and investigative sites, the compressed six-month implementation window posed a significant challenge, leaving many grappling to understand the scope of relevant guidelines and struggling to align with the new requirements.
An Issue of Awareness
Compliance requires expertise across process management, system implementation, clinical operations and quality assurance. Without clear guidance, organisations risk misinterpretation or noncompliance.
For global companies, a key challenge has been educating stakeholders outside the EU who may have assumed the guideline did not apply to them. Aligning regional teams, vendors and partners with the new regulatory expectations required significant effort.
Smaller organisations and sites have faced added complexity. While working under different national regulations on computerised systems and electronic data, some still remain unaware or uncertain how the EMA’s guidance fits within their existing compliance frameworks while ensuring they meet other regional and global standards.
Filling in the Cracks
A significant point to consider is the industry expectations of the boundary and overlap between the computerised systems and electronic data owned by sponsors or investigators. The industry must be careful to fill any cracks where data could slip through. These governmental health care electronic data systems fall under the governmental health care system standards of data management in standard health care settings but contain clinical trial data as data source. Rarely are these computerised systems privately owned by the sites or used in isolation for the clinical trial setting.
The EMA guideline did not quite consider how its expectations may extend and overlap, or even clash, with the standards that regulate national healthcare computerised systems and electronic data. The grey zone of overlapping boundaries does not exclude those “digital standards of care” and complicates how the standards of the EMA guideline apply, expecting the same standards in the national healthcare computerised systems and databases.
For privately owned site systems and technology or contracted system providers, the sites themselves should take full accountability for compliance with the EMA guideline. However, in cases when electronic medical records (EMRs) are owned by national or regional authorities, there are ongoing debates around assessing compliance with the guideline and potentially addressing system deficiencies and limitations. As a result, there are paper-
Clinical and Medical Research
based workarounds to avoid inspection issues within a clinical trial.
On the Auditor’s Radar
Auditors quickly recognised the EMA guideline as a comprehensive resource for computerised systems and electronic data in clinical trials. Unlike more targeted guidance, such as the MHRA ‘GXP’ Data Integrity Guidance and Definitions or the EMA guideline on the content, management and archiving of the clinical trial master file, this guideline serves as a single reference for compliance across a range of multiple all-encompassing requirements. Quality Assurance teams have leveraged the guideline extensively for internal and vendor audits, consulting, QMS management and CAPA closure.
With this new ‘auditor’s handbook’ in place, auditors have increased their focus on compliance, prompting sponsors to seek stronger assurances from their vendors. As audit expectations rise, organisations must ensure their systems and processes align with the guideline’s requirements. The question remains: Can all stakeholders confidently demonstrate full compliance?
The Challenges that Remain
As stated, one of the major challenges has been the variability of governmental,
regulatory agency/ethics committee and site-owned systems used in clinical trials. Not all technology systems are within the control of individual sponsors, CROs or sites. For instance, EMR systems are often owned by national or regional institutions and pose unique challenges for sponsors. Another example is the European Medicine Agency (EMA) Clinical Trial Information System (CTIS), which does not have audit trail visibility for end users or reporting tools to the extent the end users may expect those being available in the light of what EMA guideline determines as expected system features.
The unintended consequences of the guideline's application to EMRs, has led to proposals to EMA to address this through defining a "digital standard of care," applicable to technology used in routine medical practice and compliant with national requirements. To date, there is no resolution on whether this approach will be accepted and so stakeholders are left to manage the ambiguity until clarification is provided.
Yesterday’s News, Today’s Challenges
The guideline issued by the EMA on computerised systems and electronic data in clinical trials remains a pertinent subject in 2025. Its complexity, broad scope and the substantial efforts required for
compliance makes it a primary focus for sponsors, investigators, CROs and auditors and it continues to raise questions amid its implementations. As the industry strives to adhere to these standards, continued collaboration with change management experts and commitment to innovative approaches will be essential to the successful implementation of this comprehensive framework across its wide range of applicability.
Ms. Vesta Marciulioniene, Director Regulatory Affairs at ICON, has spent more than 10 years at Pfizer and a decade at Covance & Labcorp. Vesta has extensive clinical research expertise with strong focus in start-up having formerly taken global roles of Head of Start Up Centre of Excellence and Head of Site ID in previous organisations. Vesta has been leading multifunctional teams regionally and globally, aiming to connect start up and regulatory world with clinical project delivery.
Vesta
Marciulioniene
Innovations in API Manufacturing of Small Molecule Drugs
Throughout the years, innovation has been the driving factor of many industries, allowing them to stay viable. This is also the case for the pharmaceutical industry, including API manufacturing. With the cost of new pharmaceuticals steadily rising, innovation is the only way to reduce it and ensure the future availability of new pharmaceutical treatments. Predicting which fine chemicals or APIs will be significant in the following decades is also challenging. Only through innovation is the industry capable of reacting in time to fulfil the market’s needs. This article discusses recent and future innovations in the API manufacturing industry with the potential of reducing manufacturing costs and addressing market demand accurately.
Introduction
Innovation is the practical implementation of ideas that results in introducing new goods or services or improving the offerings of existing goods or services.1 ISO TC 279 in the standard ISO 56000:2020 defines innovation as “a new or changed entity, realising or redistributing value”.2 Others have different definitions, but a common element is the focus on newness, improvement, and the spread of ideas or technologies.
The pharmaceutical industry, and therefore also the manufacturing of APIs, faces many hurdles, including the availability of a skilled workforce, supply chain disruptions, the rising cost of new therapies, and increasing environmental regulations. Innovation is the only way to decrease costs and secure the future availability of new pharmaceutical treatments. Predicting which fine chemicals or APIs will be significant in the following decades is also challenging. Only through innovation is the industry capable of reacting in time to fulfil the market’s needs.
Within the pharmaceutical industry, the 6M methodology has proven a powerful tool for performing root cause analysis or risk assessment. The six factors it is built upon
are manpower, method, machine, material, measurement, and mother nature (the environment).3 Applying the 6M methodology lowers the chance of missing essential root cause analysis or risk assessment items. It helps ensure product quality, safety, and efficacy. This article will use these six factors as a guiding principle when discussing the innovations within API manufacturing of small molecule drugs.
Manpower
The first factor focuses on the human interfaces involved in API manufacturing. Especially in the industrialised part of the world, a highly skilled, trained, and experienced workforce is becoming increasingly costly. Improving welfare and living standards have increased salaries throughout the past decades. Due to the cost, the workforce must be used most efficiently and competently. Employees’ repetitive work and administrative tasks should be minimised as much as possible. One example of such repetitive work is finding the optimum in a chemical conversion regarding selectivity, quality yield, and reaction time, with many process parameters involved and scrutinised. Increasingly, automation is helping with these types of tasks. The group of prof T. Noel recently published an excellent example of such automation, presenting a versatile, allin-one robotic platform for the autonomous optimisation, intensification, and scaling up of photocatalytic reactions in flow.4
Method
The second factor focuses on the methods involved in API manufacturing. When discussing methods, we want to focus on the chemical conversions and different types of chemistry involved in API manufacturing. Catalysis involving rare metals like Pd, Ru, and Pt has been part of the standard toolkit of chemists for many years, helping them develop synthetic routes toward new chemical entities (NCEs). The Suzuki-Miyaura coupling is one of the most versatile ways to create carbon-carbon bonds to produce conjugated systems of alkenes, styrenes, or biaryl compounds.5 During the development of NCEs, the route of synthesis changes over time, typically starting with a “medicinal chemistry” route, which process chemists change into a scalable, safe, and robust
route. The resulting synthesis route is often shorter and, therefore, more cost-efficient. Innovations in chemistry that have been used more frequently by process chemists over the past decade involve photochemistry, electrochemistry, and the application of enzymes. A typical example of photochemistry is the production of Vitamin D3, where 7-dehydrocholesterol is exposed to UVB and UVC light, followed by purification.6 Vitamin D2 (ergocalciferol) is produced similarly using yeast ergosterol as a starting material.6,7 When we look at nature, enzymes are widely used. The human body is an excellent example of enzymes that perform many conversions to produce hormones.23 Of course, in the laboratory and chemical plants, enzymes can also be used to manufacture small molecules. An interesting example is the manufacturing of Sitagliptin24 or Testosterone.25
Machine
The third factor involves the machinery or equipment used in API manufacturing. One of the most challenging aspects of API manufacturing is scalability. Bringing a synthesis route from the lab to the pilot plant on a commercial scale is sometimes hampered by all kinds of physical challenges like dosing times, filtration times, heat transfer, cooling capacity, and the ability to agitate effectively. Also, from a safety point of view, several hurdles must be taken while scaling a process to full-blown commercial manufacturing. Process intensification as continuous manufacturing is being applied increasingly throughout the industry. A recent article on HiGee reactors is a good example of such an application and firm innovation. HiGee are mini reactors that use high gravitational forces, usually by spinning or moving parts, thereby creating high levels of shear. This high shear increases both mass- and heat transfer on a micro level. In this way, process intensification offers a high potential to improve the efficiency of several industrial processes and, thus, reduce the environmental footprint of the chemical industry.8,9,10
Material
The fourth factor focuses on the materials used in API manufacturing or the new types of molecules, concepts, or platforms applied in new therapies. Here, we can mention many
recent innovations and trends. A stronger focus on niche products and orphan drugs is observed within the API industry, with new treatments increasingly discovered and developed for smaller populations. Another trend is the increased potency of APIs. Advancements in drug research, especially in oncology, inflammatory diseases, and antiviral compounds, have driven the development of new therapies with highly potent compounds. The lack of data, particularly concerning the novel pharmacological actions of compounds potentially designated as highly potent active pharmaceutical ingredients (HPAPIs), and the transformation of process intermediates to conform with acceptable occupational exposure levels, poses a significant obstacle. A risk-based approach is considered to overcome this issue in early drug discovery.11,12
Another innovation in the materials focus area is the development of antibody-drug conjugates (ADCs).13 Sometimes, generic APIs like Doxorubicin can be rediscovered by applying them to an ADC.14
The latest innovation areas we want to highlight are so-called PROTACs (proteolysistargeting chimeras) and molecular glues. The earliest-known published description of the concept of chimeric degraders is in a patent filed in 1999 by a biotechnology company, Proteinix, proposing taking over the cellular protein degradation system.15,16
The establishment of the PROTAC strategy was further augmented by the finding of degrader compounds that became known as molecular glues.17,18 Molecular glues are monovalent small molecules (<500 Da) that reshape the surface of an E3 ligase receptor, promoting novel protein−protein interactions (PPIs) and offering many opportunities to engage currently undruggable targets.
Measurement
The fifth factor concerns measurements used in API manufacturing. Analysis is an essential aspect of API manufacturing. Whether during the release of (regulatory) starting materials, in-process analysis, or the release of the final API, the ability to analyse and characterise the materials or conversions is critical. Therefore, strong analytical capabilities are essential in setting up a proper and successful control strategy for filing your investigational new drug (IND) application or investigational medicinal product dossier (IMPD).
Throughout the years, the field of analytics has developed many innovations, and it will continue to do so in the near future, for example, by creating the ability to analyse impurities like nitrosamines or polyand perfluoroalkyl substances (PFAS) at levels that were impossible before. A unique innovation we want to mention here is the application of artificial intelligence (AI) within the analytical area. Of course, nowadays,
no article on innovation can or should be written without mentioning AI. A milestone in AI application in analytical chemistry is its ability to handle heterogeneous and complex data. Traditionally, analysing such data would require extensive expertise and time. However, with AI algorithms, it is possible to extract relevant information quickly and efficiently.19,20
Mother Nature – The Environment
The sixth and final factor focuses on the natural environment in the context of API manufacturing. Besides AI, sustainability is another topic that can not be overlooked. Many API manufacturing innovations are being developed to minimise global warming, decrease of biodiversity, and other critical aspects. We want to tackle two examples in this article: solvent use in solid-phase peptide synthesis (SPPS) and a modular flow platform that streamlines the synthesis of heteroatomCF3 motifs.
SPPS is the preferred technique for synthesising bioactive peptides. However, traditional SPPS generates significant waste and employs hazardous solvents like DMF and DCM. To overcome this challenge, Giovanni Vivenzio et al. developed a novel green solvent mixture by combining anisole with NOP.21 This mixture can swell different resins, and its capability to solubilise all Fmoc-protected amino acids has been
Manufacturing
investigated. The exact mix was also assessed with a green coupling agent, TBEC, in combination with ETT as an additive. Model peptides Aib-enkephalin and Aib-ACP were synthesised, resulting in favourable outcomes in peptide synthesis efficiency, 97.81% and 98.86%, respectively.
The last innovation we want to discuss is preventing the use of PFAS chemicals to introduce the trifluoromethyl group (CF3) on a molecule. The CF3 group is a key functionality in pharmaceutical and agrochemical development, greatly enhancing the efficacy and properties of resulting compounds. However, attaching the CF3 group to heteroatoms such as sulphur, oxygen, and nitrogen poses challenges because of the lack of general synthetic methods and reliance on PFAS chemicals. The method developed uses readily available organic precursors in combination with cesium fluoride as the primary fluorine source, facilitating the rapid generation of N-trifluoromethyl(R) [NCF3(R)], SCF3 (trifluoromethylthio), and OCF3 (trifluoromethoxy) anions on demand without using PFAS chemicals, turning it into a strategy that is far more environmentally friendly.22
Conclusions
Innovation is essential for any industry, and the pharmaceutical industry is no exception. In this article, a few examples have been given to show recent innovations within API manufacturing. We did not intend to create a complete overview, as it is possible to list many more examples of innovation in this field. Nevertheless, the innovations presented here show that the industry keeps evolving and adapting to be ready for future challenges. Applying these innovations can reduce costs, help develop new therapeutic areas, and meet global sustainability targets. Thanks to some of the innovations mentioned in this article, the API manufacturing industry faces a bright future.
REFERENCES
1. Schumpeter, Joseph A., 1883–1950 (1983). The theory of economic development: an inquiry into profits, capital, credit, interest, and the business cycle. Opie, Redvers,, Elliott, John E. New Brunswick, New Jersey
2. ISO 56000:2020, Innovation management. “Fundamentals and vocabulary". ISO. 2020
4. Slattery, A; Wen, Z; Tenblad, P.; Pintossi, D.; Sanjose-Orduna, J.; den Hartog, T. and Noël, T. Automated self-optimization, intensification, and scale-up of photocatalysis in flow. Science, 2024, 383,
5. Miyaura, N.; Yanagi, T.; Suzuki, A. Synth,
Commun. 1981, 11, 513.
6. Holick MF (November 2005)."The vitamin D epidemic and its health consequences".The Journal of Nutrition.135 (11):2739S 2748S
7. Hirsch AL (12 May 2011). "Chapter 6: Industrial Aspects of Vitamin D". In Feldman D, Pike JW, Adam JS (eds.).Vitamin D: Two-Volume Set. Academic Press
8. Hop, C.J.W.; Jansen, R.; Besten, M.; Chaudhuri, A.; Baltussen, M.W.; van der Schaaf, J. The hydrodynamics of a rotor stator spinning disc reactor: Investigations by Large Eddy Simulation. Phys. Fluids 2023, 35, 035105.
9. Kleiner, J.; Münch, B.; Rößler, F.; Fernengel, J.; Habla, F.; Hinrichsen, O. CFD simulation of single-phase heat transfer in arotor-stator spinning disc reactor. Chem. Eng. Process. Process Intensif. 2018, 131, 150–160.
10. Petra Meeuwse and Marit van Lieshout. ChemEngineering 2025, 9, 8
11. F. Pognan, M. Beilmann, H.C.M. Boonen, A. Czich, G. Dear, P. Hewitt, T. Mow, T. Oinonen, A. Roth, T. Steger-Hartmann, J.P. Valentin, F. Van Goethem, R.J. Weaver, P. Newham, The evolving role of investigative toxicology in the pharmaceutical industry, Nat Rev Drug Discov. 22 (2023) 317–335.
12. 14. E.A.G. Blomme, Y. Will, Toxicology Strategies for Drug Discovery: Present and Future, Chem Res Toxicol. 29 (2016) 473–504.
13. A.W.Tolcher Annals of Oncology Volume 27, Issue 12, December 2016, Pages 2168-2172
14. P.A.Trail, D. Willner, S.J. Lasch, et al. Cure of xenografted human carcinomas by BR96doxorubicin immunoconjugates Science, 261 (5118) (1993), pp. 212-215
15. Kenten, J. H.; Roberts, S. F. Controlling protein levels in eukaryotic organisms. US6306663, 1999.
16. Janet M. Sasso, Rumiana Tenchov, DaSheng Wang, Linda S. Johnson, Xinmei Wang, and Qiongqiong Angela Zhou. Biochemistry 2023, 62, 601−623
17. Dong, G.; Ding, Y.; He, S.; Sheng, C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64, 10606−10620.
18. den Besten, W.; Lipford, J. R. Prospecting for molecular glues. Nat. Chem. Biol. 2020, 16, 1157−1158.
19. F. Lussier et al. Deep learning and artificial intelligence methods for Raman and surfaceenhanced Raman scattering TrAC, Trends Anal. Chem.(2020)
20. Rafael Cardoso Rial, Talanta Volume 274, 1 July 2024, 12594
21. Giovanni Vivenzio, Maria Carmina Scala, Giulia
Auriemma, Carla Sardo, Pietro Campiglia and Marina Sala. Green Chemistry Letters and Reviews Volume 17, 2024 -Issue 1
22. Mauro Spennacchio, Miguel Bernús, Jelena Stanić, Daniele Mazzarella, Marco ColellaJames J. Douglas, Omar Boutureira, Timothy Noël. Science 29 Aug 2024 Vol 385, Issue 6712 pp.991996
23. Hiller-Sturmhöfel S, Bartke A. The endocrine system: an overview. Alcohol Health Res World. 1998;22(3):153-64.
24. Christopher K. Savile, Jacob M. Janey, Emily C. Mundorff, Jeffrey C. Moore, Sarena Tam, William R. Jarvis, Jeffrey C. Colbeck, Anke Krebber, Fred J. Fleitz. Science,17 Jun 2010, Vol 329, Issue 5989 pp. 305-30.
25. Zoelen, D-J. V., Heijningen-Ramaekers, M. V., Hoeberichts, W., Maartense, L., Meer, J. Y. V. D., Stock, T., (2023). Development of a secondgeneration Testosterone synthesis route via biocatalysis. J of Pharmaceutical Research, 8(1), 199-20
Dirk-Jan van Zoelen
Dirk-Jan van Zoelen received his Ph.D. in Organic Chemistry from the University of Utrecht in 2008. He started his career at Diosynth Oss, where, over the next 17 years, he fulfilled multiple functions and roles, ranging from Chemical Engineering, Operations, and Supply Chain to R&D. Since December 2023, he has been heading the Drug Substance Business Unit in Ardena Oss (the Netherlands). At Ardena, we are involved in developing over 500 innovative medicines. Within the Drug Substance Unit, we aim to help our customers bring better medicines to patients, faster. We guide you on the best route to the clinic with comprehensive, integrated Development and Manufacturing services, supporting the workflow required to make your Drug Substance available for trials and Market Authorisation.
Fragment hit identi cation against 96 proteins using the Carterra Ultra platform. Maybridge fragment library screening against a kinase panel.
Combination Filling: The Trend Towards Machine Customisation
What is Combination Filling?
Combination filling refers to the process of placing two or more active ingredients within the same capsule. These ingredients can be in the form of powders, pellets, granules, liquids, tablets, micro-tablets, etc. allowing for a wide range of formulation possibilities. The goal is to create a single dose that delivers multiple therapeutic or nutritional benefits, improving patient or consumer convenience and compliance.
Combination filling is being utilised across a broad spectrum of applications in both the pharmaceutical and nutraceutical industries.
In pharmaceuticals, combination capsules are used to treat complex conditions that require multiple therapeutic approaches. For instance, cardiovascular medications often combine antihypertensive agents with cholesterol-lowering drugs to provide a more comprehensive treatment for heart disease.
In the nutraceutical sector, combination capsules are popular for wellness and preventive health. Products that combine antioxidants with vitamins, minerals, and herbal extracts are widely marketed for their holistic health benefits, targeting everything from immune support to cognitive function.
Combination filling is also making inroads into speciality supplements, such as sports nutrition and weight management products. These capsules often combine ingredients such as protein powders, amino acids and agents to support muscle growth, recovery, and fat loss.
Not a New Phenomenon, so Why the Increase in Interest?
Combination filling is gaining popularity due to a number of factors:
• Enhanced Therapeutic Efficacy: Combination filling allows for the simultaneous delivery of complementary or synergistic ingredients, which can enhance the overall therapeutic effect.
For example, combining a pain reliever with an anti-inflammatory agent in a single capsule can provide more comprehensive pain management, reducing the need for multiple medications.
• Improved Patient Compliance:
Simplifying dosage regimens is a critical factor in improving patient compliance, particularly in populations that require multiple medications. By consolidating several active ingredients into one capsule, patients can adhere more easily to their prescribed treatment plans, reducing the likelihood of missed doses.
• Consumer Convenience:
In the nutraceutical market, combination filling is particularly appealing for consumers seeking multi-benefit supplements. For instance, a capsule that combines vitamins, minerals and herbal extracts can provide a comprehensive health supplement in a single dose, eliminating the need to take multiple pills daily.
• Market Differentiation:
In a crowded market, offering combination products can help brands differentiate themselves. Innovative combinations that address specific health needs or conditions can capture consumer interest and loyalty, driving sales growth.
• Customers’ Expectations are Changing – Customisation is Key: When customers are considering the purchase of combination filling encapsulation machines, several key factors influence their decision-making process. These include:
• Flexibility and Customisation: Customers look for machines that can handle/retrofit various types of filling attachments (powders, pellets, liquids, granules, micro-tablets) in the same machine.
• Precision and Accuracy: Accurate dosing of each ingredient within the capsule is critical, particularly
Manufacturing
Emerging Trends
• Digitalisation and Industry 4.0:
• Ease of Operation and Changeover: The ability to quickly and easily changeover between different products or capsule sizes. This reduces downtime and increases overall production efficiency, especially in environments where multiple products are manufactured on the same machine.
• Quality and Consistency: Consistent and uniform filling across all capsules is essential for maintaining product quality and meeting regulatory standards. Machines that ensure minimal variability in capsule fill are preferred.
The integration of digital technologies, such as the Internet of Things (IoT), artificial intelligence (AI) and machine learning, is transforming manufacturing processes. Digitalisation enables realtime monitoring, predictive maintenance and optimisation of machines.
• Focus on Consumer Health and Wellness:
address these health concerns, often combining multiple active ingredients into a single capsule. Encapsulation machines must be adaptable to produce these new and varied formulations efficiently.
• Ageing Populations:
Ageing populations have led to increased demand for pharmaceuticals, particularly in the areas of chronic disease management, oncology and cardiovascular health. This demographic shift has driven growth in the pharmaceutical sector as companies develop products to meet the needs of older adults.
The Future of Combination Filling
The trend toward combination filling in capsules is expected to continue growing as consumer demand for convenient, multibenefit products increases. As technology advances and manufacturers gain more experience with this approach, we can expect to see even more innovative and effective combination products entering the market.
For the pharmaceutical industry, combination filling offers a way to improve patient outcomes and simplify treatment regimens. In the nutraceutical space, it provides an opportunity to create differentiated products that meet the evolving needs of health-conscious consumers.
In conclusion, combination filling is more than just a passing trend, rather it represents a significant shift in how the industry approaches product development. It has the potential to enhance therapeutic efficacy, improve compliance and drive growth in both the pharmaceutical and nutraceutical markets. As the technology and expertise behind combination filling continue to evolve, this approach will likely play an increasingly important role in the future of capsule-based products.
Bijo Varghese Babu is a Sales Manager at ACG with 9 years of experience in the company. He is highly skilled and has successfully managed sales across multiple regions, including the USA, Canada, Europe, and India. in pharmaceuticals. Customers prioritise machines that offer precise control over fill weights and dosing accuracy.
There is an increasing focus on consumer health and wellness, with a growing interest in preventive healthcare, immune support, and mental well-being. Nutraceutical companies are responding by developing new formulations that
Bijo Varghese Babu
The fill-finish stage presents unique challenges, particularly in terms of sterility assurance, container-closure integrity (CCI), and particulate contamination control. Over the years, advancements in technology and process control have significantly improved sterility assurance, product quality, and manufacturing efficiency. Innovations such as single-use systems, coated vials and stoppers, closed barrier technologies, and automation have reshaped sterile drug manufacturing, with isolators and robotics emerging as preferred solutions for contamination-free aseptic processing.
Regulatory expectations for sterile drug product manufacturing have also evolved around the globe, particularly with the latest revisions of EU GMP Annex 1, which emphasises enhanced contamination control strategies and risk-based sterility assurance approaches. Adhering to these guidelines ensures that mAb drug products meet the highest sterility and quality standards.
Analytical Method Transfer and Validation
A critical aspect of tech transfer is the successful transfer and validation of analytical methods used for acceptance of incoming Drug Substance, in-process control, release testing, and stability assessment. Analytical method transfer requires ensuring that critical assays such as high-performance liquid chromatography (HPLC), capillary electrophoresis, mass spectrometry, and bioassays used to measure identity, purity, potency, and impurity levels remain robust and reproducible at the receiving site. This is a key focus of all tech transfers.
To facilitate a smooth method transfer, a comparative testing approach is employed, wherein the sending and receiving sites perform side-by-side testing using the same reference standards and control materials. For complex assays, additional co-validation or revalidation studies may be required to demonstrate assay robustness and accuracy.
Regulatory expectations for analytical method validation are guided by ICH Q2 (R1) and ICH Q6B, which require demonstrating assay specificity, accuracy, precision, linearity, and robustness. Ensuring compliance with these guidelines is essential for regulatory
approval and long-term product quality assurance.
Regulatory Compliance and Post-approval Considerations
Regulatory agencies such as the FDA, EMA, and PMDA require a well-documented tech transfer process to ensure that product quality is maintained at commercial scale. As part of the transfer, the CDMO should work closely with the sponsor company and support regulatory filing updates to reflect process changes, site changes, and new validation data. This includes updates to the Investigational New Drug (IND) application, Biologics License Application (BLA), or Marketing Authorisation Application (MAA). Sponsors should understand the regulatory standing of the receiving site and ensure their Quality systems will meet the regulatory requirements of the regions they plan to apply for authorisation.
Ensuring cGMP compliance at the receiving CDMO commercial facility is critical, as regulatory agencies expect comprehensive process validation, batch release data, and ongoing post-market surveillance to maintain product integrity. Additionally, change control strategies must be in place to address any future process optimisations, changes in excipients, materials or container closures, or site modifications.
Risk Management and Contingency Planning to Secure Supply Chains
The complexity of mAb tech transfer requires proactive risk management to anticipate and address potential challenges. Process failure mode analysis (FMEA) is used to systematically identify high-risk areas that could impact product quality, yield, or regulatory compliance. By developing robust risk mitigation strategies, such as additional process monitoring, operator training programmes, dual sourcing strategies for critical raw materials and redundancy in equipment and facilities, manufacturers can minimise the likelihood of disruptions.
This risk-based approach allows for targeted process adjustments, and together with technical support from subject matter experts ensures that potential challenges are addressed before they impact largescale production. In addition, disaster recovery plans should also be put in place to ensure business continuity, in the event of supply chain disruptions or facilityrelated incidents to secure supply chains and the delivery of life changing therapies to patients.
Manufacturing
Conclusion
The technology transfer of a monoclonal antibody drug product for late-phase clinical and commercial launch is a complex and multifaceted process that requires meticulous planning, robust risk management, and crossfunctional collaboration. Ensuring process scalability, analytical method robustness, regulatory compliance, and supply chain stability is critical for a seamless transition to commercial production.
By leveraging advanced manufacturing technologies, real-time analytical monitoring, and digital process modelling, biopharmaceutical companies and their partnering CDMOs can enhance tech transfer efficiency and accelerate time-to-market for life changing monoclonal antibody therapies. With careful execution, CDMOs can successfully bridge the gap between clinical development and commercial supply, ensuring that patients receive consistent high-quality, safe, and effective biologic treatments.
Shawn Cain
Shawn Cain is PCI’s SVP of Development and Manufacturing and also General Manager of the Bedford, NH Sterile Fill-finish Campus of Excellence. He has over 30 years of experience in combining process engineering and project management to direct the development and manufacture of sterile pharmaceuticals, cell-based biologics, and medical devices. Most recently he was the COO of LSNE, which was acquired by PCI Pharma Services in 2021. Mr. Cain also worked at Organogenesis and was the Director of Operations for another pharmaceutical CDMO, Formatech, Inc. Prior to that, Shawn was Interim President and Chief Executive Officer of Arbios Systems, Inc. Previously, Mr. Cain was employed at Becton Dickinson’s Biologics Business. Mr. Cain was also the Vice President of Operations for Circe Biomedical, Inc., where he led the development of the bioartificial liver technology. Mr. Cain holds six patents, received his M.S. degree in Biological Sciences from the University of Massachusetts and a B.S. in Biological Sciences from Northeastern University.
Maintain Sterile Pharmaceutical Production with Uninterrupted Power Supply
Today’s pharmaceutical manufacturers face a myriad of challenges, including access to constant power supply. Power interruptions, fluctuations and outages are an unfortunate and generally unpredictable occurrence that can have a variety of causes. According to Statista, companies around the world lost approximately 5% of their revenue due to power outages.
Drug product manufacturers require a steady and reliable power grid to support their customers’ pharmaceutical supply chain. Uninterrupted power supply (UPS) is important for aseptic manufacturing processes, which generally use sensitive equipment that operate within an extremely narrow voltage range. Any interruption or loss of power can pose a significant risk to the production processes or even the drug itself, potentially resulting in the entire batch being discarded.
As an expert in electrical engineering at Vetter, I understand the complex nature of power supply in an aseptic production environment. In this article, I will share the impact of power outages on drug manufacturing, how service providers can address power fluctuations and the obstacles they face to maintain constant power supply and protect customers’ drug products.
Frequent Power Outages and the Impact on Aseptic Production
The electrical system can be jeopardised for a number of reasons, resulting in fluctuations in voltage and power levels, network failures and micro cuts. Extreme weather and natural disasters, as well as accidents such as nearby construction, can cause power interruptions. Suspension of the power supply and incidents of voltage spikes and undervoltage can also interrupt the power supply for pharmaceutical facilities.
Even the smallest blip in the electricity flow can affect processes in an aseptic environment. When an interruption occurs, the temporary loss of power can cause equipment to malfunction or even shut
down completely. Any impact can be costly, leading to schedule delays or even quality concerns. Halting production can be difficult for service providers to recover from, as lost time can result in long-term delays and reduced productivity.
Power outages and interruptions can cause failure to production equipment like ventilation systems and cooling generators, which could compromise drug manufacturers’ ability to provide a sterile clean room status. Understanding that regulatory requirements and patient safety are the top priority, as the risks can be detrimental.
Maintaining an uninterrupted and reliable power supply allows for smooth operation and consistent efficiency at pharmaceutical production plants. Pharmaceutical manufacturers require uninterrupted power supply to provide stable and frequent voltage to prevent costly damage to equipment, maintain supply availability and protect the quality and delivery timelines of customer’s drug products. It is essential for pharmaceutical facilities to have a backup plan with additional power resources in place to minimise the impact of power interruptions.
Power Supply Alternatives and Mitigating Risks of Power Loss
There are several technologies and types
of equipment that can support the power supply and provide an extra layer of protection, including ancillary equipment such as smart grid sensors and energy storage systems like batteries. However, two primary technologies can be considered to address the risks of power interruption and failure. Both backup power generators and UPS systems are often used in the pharmaceutical sector as reliable sources of seamless power supply.
Backup power generators, which are more commonly known in the industry, typically use diesel fuel to convert mechanical energy into electrical energy. Separated from the grid, generators can take over the power supply in the event of a power failure. Although generators are instrumental in preventing power outages, they require a short period of time for the equipment to start up and reach full operating levels and therefore cannot provide the uninterrupted power supply that aseptic pharmaceutical production requires.
UPS systems are always on standby and connected to the power grid. Static and dynamic systems are capable of providing continuous and steady power to crucial equipment. Drug product manufacturers value UPS systems for safeguarding their aseptic production. Static UPS systems are usually backup systems with batteries that are charge through the grid. In the case
EVERIC ® pure – The Safe Option Right from the Start: A Drug Containment Platform to Streamline Drug Development
The Wrong Container Choice – A Threat to Patient Safety and Time-To-Market
A common situation is that pharma companies select a certain primary packaging container and in first short term studies, everything is fine… Later, issues could occur during development or after launch – maybe even years after the launch. These issues could be the presence of glass particles due to delamination or a shift in pH because of elevated leachables. From a patient safety perspective, this is disastrous, with a threat of blocked blood vessels, adverse reactions, or unavailability due to market recall respectively drug shortage.
One example is a case from 2010. The drug had been on the market for roughly ten years before suddenly, certain vials showed the presence of particles. A big recall followed, and all containers were retrieved from the market. Glass particles were found in only 0.03 % of the analysed vials. This demonstrates that appearance of glass delamination is driven by production outliers, which makes it very difficult to address.
Pharma companies can also face unpleasant surprises in the early stages of development when specific formulations are not stable in conventional vials. This can
lead to a restriction in packaging options or even product re-formulations that can take many months, resulting in lost time and money.
Controlling the Container’s Inner Surface
As the most relevant contact surface for the drug formulation, a conventional vial container's inner surface may not be chemically stable enough for long term storage through the desired shelf-life. The only chemical durability test pharmacopeia require for Type I glass is for surface hydrolytic resistance, which is insufficient to determine a container's suitability for long term drug storage. The hydrolytic resistance test assesses a glass container's chemical stability by measuring its resistance to releasing soluble ions into water.
The procedure begins with the determination of 90 % of its brimful capacity, its maximum useful storage volume. The containers are repeatedly rinsed to replicate the washing process on bulk filling lines before filling with purified water to 90 % of its brimful volume. After the containers are autoclaved, solutions from the containers are pooled for titration or flame spectrometry measurements for the amount of released alkali. This means that one “very good” and one “bad” container would end up with an average good result.
A Question of Process Control
Achieving a consistently high surface quality is a matter of process control. Theoretically, every vial should have the same inner surface quality if the process is stable. Reality proves different, though: outliers exist that can lead to costly recalls if the criticality is not correctly assessed, and follow-up measures taken. With millions invested in development, drug stability can be put at risk because of the wrong container choice.
The inhomogeneity of the inner surface of these outliers originates from a zone just above the heel of the vial. During the vial production process, the glass tube is heated, which causes evaporates to condense on the nearest cooler region of the vial's interior surface: the aforementioned heel zone. This area is less chemically durable and more susceptible to the processes that lead to delamination.
Figure 1: Particle formation of critical outlier in a conventional vial batch
Figure 2: Filling volumes
The Solution to a Controlled, Homogenous Inner Surface
In combination with a leachable-improved glass tubing (Controlled Hydrolytic Resistance), SCHOTT Pharma has established a patented process that avoids these condensates and leads to a homogenous inner surface in conjunction with a dedicated release test (Quicktest). Imagine having a vial with a bad heel zone – if filled to 90 % of its maximum capacity, the effect is diluted and will normally pass the standard pharmacopeia surface hydrolytic resistance test. It might be a vial with a very bad heel zone but a good remaining quality of the rest of the inner surface – it cannot be identified as a "bad" vial, and the severity will not be detected.
The release test involves mimicking a glass attack by autoclaving and then filling with purified water. In the usual test for hydrolytic resistance, the vial is filled to 90 % of its maximum capacity. For the EVERIC® pure release test, each format is filled so that only the relevant heel zone is covered to determine the effect of that area.
The amount of released sodium is measured and compared with a certified limit value for each format. It is even possible to produce vials that are within the limit for the hydrolytic resistance of pharmacopeia but outside the specified Quicktest limit value. These are the critical outliers, or “bad vials”, that can occur during conventional vial production.
Safeguard the Integrity of Valuable Drugs
A homogeneous inner surface results in
Figure 3: Conventional vial vs. EVERIC® pure
two factors that increase drug stability: no delamination and a lower leachable level.
1. No Delamination
The microscopic images below show that a detachment of flakes took place in a conventional vial filled with a phosphate buffer at pH7 after storage for 24 weeks at 40 °C. In contrast, the EVERIC® pure vial under the same conditions shows no sign of glass attack. This has also been seen with different buffers at different pH levels, but phosphate represents one of the more critical buffer systems.
2. Lower Leachable Level
It has been identified that a lower leachable level is especially relevant for low filling volumes. Since the changed heel zone is the leading cause of the higher leachable level of conventional vials, the impact of an improved, homogenous inner surface is much more pronounced for low-fill volumes.
While the 2 mL (nominal) filling volume shows no significant difference between EVERIC® pure and a conventional vial for leached sodium per ml, a considerable difference can be identified for a filling volume of 0.5 mL. So, for drugs typically filled below the nominal volume, greater attention should be paid to leachables analysis.
Figure 4: Leachables level for different filling volumes
Fill-Finish
EVERIC® pure –
The Right Vial from the Start Choosing EVERIC® pure as your default container solution during drug development offers the most significant reduction of risk, which is vital for patient safety, cost, and time to market. EVERIC® pure delivers reduced leachable levels because of the improved chemical durability of the heel zone. Therefore, there’s an increased likelihood that a formulation remains stable – less risk for the stability of the protein or for the pH shift of a diluent.
Many factors influence stability during drug development, including buffer systems, pH range, post-treatments such as terminal sterilisation, or storage conditions, the interplay of which cannot be foreseen at the beginning of the vial selection process. EVERIC® pure offers a safe platform that provides a broader range of formulation options, potentially avoids additional reformulation cycles if one formulation fails, resulting in a faster time-to-market.
1. Improved patient safety and shorter time-to-market
• Proven drug stability
• Increased formulation options
• No additional screening studies
• Less delays due to reformulation
2. A tried and tested solution
• More than ten years in the market
• More than 100 commercial products
• More than 40 customers
• Only solution with an in-production release test
3. Simple switching
• Available in formats between 2R-50R and in adaptiQ®, a pre-sterilised nest and tub configuration
• No re-registration due to unchanged glass composition
Diana Löber (Global Product Manager Specialty Vials and adaptiQ®) started her career in the medical device industry prior she joined SCHOTT in summer 2018. With now more than 10 years of experience in the area of product management, as global product manager for vials, she is responsible for the product strategy, including the identification of new market opportunities, implementation of lifecycle measures and the development and launch of innovative products.
Email: diana.loeber@schott.com
Diana
Löber
Three Reasons to Choose EVERIC® pure
Labelling and Serialisation to Comply with the EU MDR
The deadline for ensuring compliance with the European Union’s Medical Device Regulations (EU MDR) is looming. These new, stricter regulations are aimed at improving the traceability features and safety management of medical devices for sale within the EU. The new regulations will have significant implications for medical device manufacturers and across production lines. This article discusses the different solutions available for manufacturers seeking compliance with EU MDR, highlights considerations and best practices, and explores what could be next for medical device regulations.
Recap: EU MDR Timeline for Compliance
The EU MDR replaces the EU’s current Medical Device Directive (93/42/EEC) and the Directive on Active Implantable Medical Devices (90/385/EEC). As part of the new regulations, manufacturers of medical devices for sale within the EU must adhere to strict guidelines to ensure their products are safe to use.
The regulations cover all medical devices sold in the EU – including everything from scalpels and needles, pacemakers, and prosthetic limbs to adhesive bandages, gauze dressings, and medical devices with a cosmetic purpose such as coloured contact lenses.
The regulations were originally due to come into force in 2020 but have been faced with a series of administrative delays. In 2023, the European Commission made the most recent extension to the EU MDR transition period, with the publication of Regulation (EU) 2023/607, to address the risk of medical device shortages in the EU market.
In the current timeline, manufacturers of medical devices have until 31st December 2027 or 31st December 2028, depending on the risk class of the device, to ensure compliance with the new regulations.
Labelling Requirements
Under the EU MDR, all medical devices must be assigned a unique device identification
(UDI) code and have their UDI recorded, indexed, and registered in EUDAMED, the Central European Database for Medical Devices.
All medical devices for sale in Europe will need to have a UDI included as part of their packaging, meaning that medical device manufacturers will need to invest in technology to enable the fast and accurate application of traceability coding to products and packaging at the individual item level.
The UDI contains:
• Device identifier – a unique text code for each specific model of a particular device. The information is static, meaning it is the same for all instances of the product model.
• Production identifier – a variable text code comprising one or more variable characteristics, such as the date of manufacture, expiration date, lot number, or, in some cases, a serial number.
In addition, new symbols have been developed to be used with the information supplied by the manufacturer. All symbols included in the ISO 15223-1: 2021 are
internationally recognised symbols and have been harmonised with the EU MDR.
EU MDR UDI labelling rules apply to the lowest packaging format, which protects the product from damage; the rules do not apply to shipping containers or cases containing a multitude or a mix of products.
Failure to comply with these procedures may mean that devices are withdrawn from sale, with device manufacturers no longer able to supply their products to other EU member states.
Serialisation and identification of products down to the individual item is only a requirement for active implantable devices, such as pacemakers, ventilators, and internal glucose monitors. Aggregation is not currently a requirement for serialised products, although it is expected that most manufacturers will look to aggregate product codes for logistics reasons.
Suitable Technologies for EU MDR Compliance
Labelling does not necessarily mean a physical label has to be applied to a product or its packaging; it means “making the required information available on the
Packaging
Medical device manufacturers should speak to their coding and marking providers when deciding which technology to use for UDI compliance. A trusted coding and marking solutions provider will be able to advise on the most appropriate technology to suit specific requirements, as well as carrying out required production line and printing tests to help ensure UDI compliance.
What’s Next for Medical Device Regulations?
The EU MDR legislation currently contains minimal requirements for individual device identification. Serialisation and identification of products down to the individual item level will only be a requirement for active
implantable devices, such as pacemakers, ventilators, and internal glucose monitors.
The future regulatory outlook for medical devices is anything but clear; however, patient safety and counterfeit protection remain a top priority in the pharmaceutical sector more broadly. Future regulations could expand the current requirements for item-level serialisation into more product categories.
A few years ago, when the pharmaceutical industry began preparing for serialisation requirements under the EU FMD and US DSCSA, the total cost of implementing serialisation was at the front of everyone’s minds. Today, the cost of serialisation and the time, effort, and resources needed to upgrade lines have significantly reduced as the knowledge and know-how in this area have grown, making it much more efficient and cheaper to install across a range of markets.
As such, companies whose products do not fall under the category of active implantable devices could still benefit from equipping their production lines with
technology to enable both UDI compliance and serialisation.
The addition of serialisation via 2D codes for medical devices is only a small step beyond what is already required under existing EU MDR compliance – but one that can help reduce the risk of counterfeit products further and bring many untold benefits to end-users and manufacturers.
Domino’s Global Life Sciences Sector Manager Bart Vansteenkiste has a 24year history with the company. He is responsible for developing Domino’s life sciences business and strategy. Over the years, Bart has become a true expert in serialisation and aggregation, regularly attending and presenting at conferences across the world on behalf of Domino.
Email: bart.vansteenkiste@domino-uk.com
Bart Vansteenkiste
Packaging
Navigating the Ever-changing World of Labelling Compliance
First presented by the European Commission way back in 2020, the new pharmaceutical strategy for Europe aimed to make the European pharmaceutical industry, 'fit for the future and competitive'.
Following the transformative impact of the COVID-19 pandemic, and realisation that there were critical flaws hindering the effective production and roll-out of vital vaccines, a reformed proposal was published in April 2023.
The aim? To strengthen security of supply, promote research and innovation, and improve access to affordable medicinal products.
From ensuring better access to treatments and more reliable, cross-border data sharing, to nurturing the creation of both a well-functioning international supply chain and a well performing single market for pharmaceuticals, no corner of the industry has been left untouched.
The Role of Labelling Compliance in Maintaining the Equilibrium
Accurate and effective labelling permeates all of this, and while it's often overlooked or perhaps even dismissed as unimportant or not worth investment, the supply chain would – quite literally – grind to a halt without it, hindering access to treatments and cross-border collaboration.
Let me explain.
In an industry where accuracy, safety, and adherence to regulatory standards is paramount, effective labelling isn't just about clear communication. It's also about maintaining public trust, meeting legal obligations, and safeguarding patient welfare.
Given how quickly global regulatory requirements can change, keeping on top of labelling compliance is becoming an increasing challenge for businesses. Understanding, and navigating, these shifting requirements while also managing a large, diverse product line can feel like an overwhelming task.
Just one error or oversight might lead to a recall, production lines stopping and potentially days of time identifying and resolving the issue, not to mention the potential danger this could pose to patients. The cost can be massive.
Where is the Medical Device and Pharmaceutical Sector Currently?
According to the FDA, labelling and artwork errors are one of the most common reasons for pharmaceutical products to be recalled.
For pharmaceutical manufacturers overseeing the labelling and artwork on hundreds, or even thousands, of products, staying on top of regulatory changes is paramount.
In 2025 alone, we'll see a handful of new regulations come into play that will impact medical devices and pharmaceutical manufacturers and organisations need to be aware. These include the Windsor Framework, and changes to the In Vitro Diagnostic Regulations (IVDR) and EU Medical Device Regulations (MDR).
As of early January, the IVDR and MDR now require manufacturers to notify relevant parties of potential supply interruptions or discontinuations at least six months in advance. While this doesn't directly impact labelling and artwork management, it will require manufacturers to have total visibility of their operation - a job that is made far more difficult for those reliant on disjointed systems and manual processes.
Why Software is Key to Labelling Compliance
Constant regulatory changes and supply chain disruption are, arguably, two of the most common challenges businesses operating in both of these markets face, yet it always surprises me how many firms continue to operate a patchwork of disparate systems with little to no integration with one another. Some use out-dated, legacy systems, while others rely entirely on manual processes.
One lesson learned across all industries during the COVID-19 pandemic was the crucial role digital technologies can play in combating disruption. From improving
interoperability between departments, to boosting overall operational efficiencies and maintaining accuracy, the medical device and pharmaceutical industries must catchup.
For example, a typical manufacturer may have thousands of products to manage at one time, across a number of territories. Software equipped with version control provides much-needed traceability throughout the product's entire life cycle. Pre-approved templates also ensure consistency and accuracy, avoiding the chance of small - yet detrimental - errors creeping into production.
Better Content Management
Deploying an automated labelling and artwork solution is only the first part of the process and some fall victim to implementing a solution that still requires their team to spend hundreds of hours manually inputting artwork data.
This is something we've worked to avoid, as a truly automated, end-to-end, content management process is transformative for any business. No more rifling through files for the latest artwork or liaising with external artwork designers, going back and forth with approvals, and updating them with each change made to the product or the brand's logo and messaging.
Automated workflows also remove repetitive tasks, taking care of artwork generation and automatically applying changes and searching for content. Coupled with version control management and audit trail features, this sort of functionality provides complete visibility over any changes made to your label components.
Accelerating Approval Processes
To ensure total compliance, medical device and pharmaceutical labels are always subject to stringent reviews and approvals.
Take the medical device sector, for example, with a label destined for the EU market. That one label must pass through extensive review and approval processes to ensure medical, marketing and regulatory information – including symbols and
markings – are all accounted for, accurate and consistent.
Then, you've got the challenge of it being translated into more than a dozen languages. Naturally, this is a process that can often result in human error or oversight, leading to a lengthy delay in actually getting the product to the end-user.
Manufacturers will know that time-tomarket is everything. Delays cost money, and can leave retailers without important stock on their shelves.
More significant errors are likely to occur when incorrect symbols or medical information hasn't been properly updated, and then not updated in the other translated versions. Without the appropriate tools to spot the error, your non-compliant products could end up on the shelves with incorrect information on their labels.
The same applies when regulations change. Even the smallest of changes might result in a label needing to be updated. Those shipping products across the EU, UK and U.S. will also know that regulatory requirements often differ, adding yet another layer of complexity to the whole process.
Without a fully integrated workflow, a thorough audit trail and a robust approval process that helps you monitor the performance of your process, and ensure you deliver correct labels each time, you could
be opening yourself, and your customers, up to serious risk.
This Process in Action
Global leader in non-invasive orthopaedics, Ossur, approached us when the new EU MDR regulations were due to come into force.
Its team were stuck using disparate, nonstandardised legacy systems; solutions that meant its IFU labels were stored in separate documents with only a master spreadsheet to chronicle them. While sufficient to create individual IFUs at one time, the requirements to keep costs low and the demands from this latest regulation meant that a more efficient process was needed.
As well as slow approval times on its IFUs, thanks to mismatched data and disparate documents, Ossur's team were also contending with fragmented data and limited visibility across its hundreds of global product lines.
They were also faced with a lack of consistency in tracking and managing information, which led to further challenges when trying to translate each IFU. This called for an overhaul of its legacy system, which saw the introduction of Veraciti.
Implementing the solution meant that Ossur could consolidate all of its IFU phrases into one single system, finally giving them enhanced visibility of all its IFU assets. This streamlining of product information also enabled them to identify key phrases that
could then be reused in multiple product IFUs, while ensuring that each label was accurate and completed in quick time.
Added to this, the changes made from Veraciti's implementation illustrated the scale of the MDR compliance project that was required, giving them a clear vision of the steps needed to be taken to ensure their operations were fully compliant.
As we look ahead to 2025 and beyond, it's clear that organisations need to consider investing in solutions that will not only bring about improved labelling and artwork compliance, but help them to streamline operations and drive efficiencies.
Gurdip Singh is the Chief Executive Officer at Kallik. With decades of experience working in the life sciences and technology space, he enjoys being a hands-on leader, driving organisational transformation and supporting businesses working in heavilyregulated industries. Before joining Kallik, a labelling and artwork software company, Gurdip held senior roles at CSC, a leading life sciences and biotech firm, and DXC Technology, a global leader in IT transformation projects.
Gurdip Singh
Application Note
Challenges in Secondary Packaging
The requirements for secondary packaging are therefore extremely high: the pouches must be securely packed into folding boxes along with various accompanying documents. Afterward, the boxes must be closed and additionally sealed with tamper-evident protection. Different closure techniques, such as tuck-in flaps or hot-melt adhesive closures, are possible. Secure sealing is particularly important for these products, as they are already sterilised in their primary packaging – and in some cases even precoated with active ingredients on the inside of the pouches. An intact and tamper-proof secondary packaging is therefore essential for manufacturers. The folding box’s seal must be designed to allow for one-time opening, with clear and visible evidence of tampering.
Another critical step is the precise application of labels to the folding box, such as U-shaped (i.e. three-sided) labels that ensure readability from all angles. For doctors and medical staff, it is crucial to be able to identify product information quickly and clearly. These labels often contain detailed information such as product type, quantity, weight, and expiration date, and they can cover up to 70% of the packaging surface. In this way, the product is individualised in a legally compliant manner and effectively protected against tampering. In this specific project, a 37 cm (14.5-inch) label was applied to the folding box.
Safety and Precision Through Automation
Additional challenges arise from a diverse product range with numerous variants, which increases complexity and the risk of mix-ups. The packaging process must therefore ensure that the labels are clearly readable and easy to scan, and that the contents – including the product and the accompanying documents – exactly match the information on the folding box. “If a surgeon in the operating room pulls out the wrong catheter from
the pouch, the manufacturer’s credibility would be irreversibly damaged,” summarises Aurbacher.
Especially for manufacturers who still rely on manual packing stations, it is often difficult to imagine that such complexity in secondary packaging can be effectively managed through automation. “However, our customised, precisely coordinated packaging solutions continuously prove that even the most complex requirements can be implemented reliably and efficiently,” explains Aurbacher.
The Modular Cartoner: Key to Project Success
“The neurovascular catheters in this project are manufactured under the most stringent cleanroom conditions and carefully packaged in pouches. But in the secondary packaging process, around 20 employees manually perform the tedious and repetitive task of carefully folding the pouches and placing them, along with accompanying documents, into a flat 30 x 30 cm folding box,” explains Jörg Aurbacher, who has successfully implemented numerous international packaging projects in the medical device and pharmaceutical industry.
“In order to be able to automate such manual packaging processes, we developed our BoxTeq cartoner as a modular base machine. Only through the precisely coordinated interaction of specially designed servo motors, suitable sensory systems, and a highly responsive control system can the movements in the BoxTeq cartoner be synchronised in a way that ensures gentle and secure product handling. Over the decades, all our processes – from design to manufacturing, assembly, and programming
– have been perfectly aligned to meet these requirements.”
Customised Solutions Instead of Standard Machines
Even if the entire packaging line ensures the desired output rate and return on investment, the success of a project ultimately depends on the cartoner. It is the key to transitioning specialised primary packaging into easy-tohandle, automatable secondary packaging. Only when the cartoner is perfectly tailored to the specific requirements will the customer achieve the desired results.
“The transition from primary to secondary packaging is often the most critical stage for our customers. And it is precisely at this point – when the product is placed into the folding carton – that a simple standard machine is no longer sufficient. Only a cartoner specifically designed for the product can deliver the desired outcome,” says Jörg Aurbacher.
Specialised Cartoner: Customised Perfection
“A modular base machine such as our cartoner is designed to adapt perfectly to the respective packaging project, because no two packaging projects are alike,” explains Florian Haertinger, Project Manager at Christ Packing Systems. There are packaging lines in a wide range of performance classes that have to fit into the smallest possible footprint and require a higher level of functional integration. Others require higher output rates or redundant systems for increased reliability. This means the layout of the line can be very different – compact, elongated, or U-shaped. We clarify and document every detail in advance.”
This is why the interdisciplinary development team at Christ Packing Systems assesses the feasibility of the project right from the very beginning and systematically analyses all relevant project factors: product flow, formats, output rates, critical paths, and the adaptation effort on the base cartoning machine. The goal is always to implement the entire line successfully and efficiently.
“Our design department can adapt everything on the modular cartoner – except the packaging itself,” explains Haertinger. “The product packaging is the result of extensive transport and acceptance studies conducted by our customers within their companies and with their end customers. Only with the
Figure 3: Complex secondary packaging – A 37 cm long U-shaped label is to be automatically applied to the folding box.
Figure 4: Suitable sensors are essential for reliable automation.
Application Note
optimal packaging can they obtain approval for their product.”
The combination of modular technology, customised adaptation and precise planning makes the cartoner from Christ Packing Systems the key component of a successful packaging line.
The Cartoner with the Necessary Tact and Sensitivity
As Florian Haertinger and Jörg Aurbacher stand in front of the packaging line developed for their U.S. customer, it quickly becomes clear how much attention to detail went into the design of the cartoner. “On this packaging line, pouches containing neurovascular catheters are packaged into flat folding boxes,” explains Haertinger. “The pouches, which are stacked in transport boxes, are fed through an automatic infeed area with a buffer directly to the heart of the line: the cartoner. Due to its large format, the BoxTeq 1500 forms the basis for the customised cartoner. Its split transport chain and higher number of available stations allows for the integration of additional components such as labellers, printers, and code readers.”
Haertinger demonstrates scanning a barcode from a transport box before placing it into the infeed area. “By scanning, the box is inseparably linked to the batch and reliably documented. Once the batch has fully entered the systems, the packaging process starts automatically – accompanied by seamless documentation.” The central control system records all relevant data, including batch start time and date, the corresponding format,
product information, and batch duration. Even widely varying batch sizes can be reliably processed in this way.
Jörg Aurbacher points to the gripping tool that removes the pouches from the transport box and securely places them onto a bucket in the transport chain. “This is a pouch-specific pick & place tool. Due to the sensitivity of the pouches, we have equipped the top-down gripping tool with high-precision, fast laser distance sensors. These sensors continuously measure the distance during the insertion process to prevent damage. Additionally, the suction force is continuously monitored. If it increases unexpectedly, the picking process is immediately interrupted before any damage to the product can occur. The suction plate on the gripping tool is also designed to disengage to protect against worst-case scenarios, ensuring maximum safety.”
Precision and Protection Through Advanced Sensor Technology
The correct large-format packaging inserts are already in place in the bucket of the transport chain, having been fed by an upstream friction feeder. Haertinger explains: “It was crucial in this process step to ensure that the accompanying documents were the correct ones. We verified this using appropriate sensor technology before feeding them into the system. The properly oriented pouch is then placed on top of these documents and carefully folded at the back end, allowing it to fit into the flat folding box despite its extended length.”
Seamless Coordination: From Transport Chain to Sealed Box
In the insertion station, a servo-driven infeed pusher carefully and precisely guides the bucket’s contents into an already erected folding box. The pre-glued folding boxes, securely held in place by the chain feeder, are picked from a large folding box magazine located above the folding box transport area using vacuum suction. “The magazine is designed in such a way that it can be safely refilled during operation,” emphasises Jörg Aurbacher. “But the real feat is performed by the carton erector: in just fractions of a second, the complex box is erected and positioned perfectly to be filled in synchronisation with the insertion process. This is the result of our extensive motion studies in the CAD system as well as handson testing with our assembly technicians at the real machine.”
Once the bucket’s content is inserted into the folding box, the flap closers securely
close the folding box. As soon as all pouches from a transport box have been processed, the box is automatically discharged, while the chain feeder moves the cartons in perfect rhythm toward the end of the cartoner.
In the cartoner, various sensor systems are used depending on the project – ranging from simple light sensors to complex camerabased pattern recognition. “These systems check serial numbers, print quality, flap closures, barcodes, and labels, ensuring the presence of the correct documents, security labels, and folding boxes,” explains Florian Haertinger. He adds, “Any errors in the packaging process result in immediate rejection. This guarantees maximum process reliability and quality – just as our customers expect.”
"The sensor technology is so precise and reliable that our customers notice here the clearest difference compared to manual packaging – they can truly feel the added security. – Florian Haertinger, Project Manager, Christ Packing Systems
Paving the Way to an Automated Future
Automating complex packaging processes is a crucial step for medical device and pharmaceutical companies to ensure longterm quality, efficiency, and competitiveness. A customised cartoner plays a key role in successfully transitioning from manual packaging to full automation.
At the heart of this transition are maximum safety and efficiency. A cartoner designed precisely to meet customer needs enables gentle product handling while ensuring seamless compliance with the stringent requirements for the transfer from primary to secondary packaging. This allows medical device and pharmaceutical companies to maintain full control over their highly regulated processes – even as production volumes rise and processing speeds increase.
"Every project is a partnership in which we make our customers’ requirements our mission,” emphasises Jörg Aurbacher. “Through our solutions and close collaboration, we help enhance efficiency, optimise processes, and strengthen supply security for patients worldwide. Together, we set automation standards that meet not only today’s challenges but also those of the future.”
Figure 8: Introduction of an internal means of transportation: the pouches stacked in transport boxes are fed to the BoxTeq cartoner.
Application Note
Overview
Situation
A leading U.S. medical device company was searching for a solution to make the secondary packaging of sensitive neurovascular catheters in large-format pouches more efficient and secure. The existing manual packaging process had reached its limits due to increasing production volumes and stringent regulatory requirements.
Challenge
Products developed through decades of medical and pharmaceutical research require scalable, safe, and reliable packaging processes to succeed in the market. This is only achievable with automation solutions specifically designed for what are typically highly sensitive and high-value product.
• Efficiency enhancement: reducing reject rates while increasing processing speed.
• Product protection: ensuring that the sensitive catheters are not damaged during the packaging process.
• Regulatory compliance: implementing serialisation, tamper evidence, and traceability in accordance with strict regulations.
Solution
Christ specialises in automating complex packaging processes for the medical device and pharmaceutical industry. Based on the proven BoxTeq 1500 base machine, the cartoner’s actuators and sensors were specifically adapted for pouch-packaged products. The BoxTeq 1500 cartoner was customised to:
• gently process pouches.
• precisely assign and verify accompanying documents and labels.
• digitally monitor and seamlessly document the entire packaging process.
Result
With the customisable BoxTeq cartoner from
Christ, even supposedly “difficult-to-package” pouches can be safely and reliably placed into large, flat folding boxes. The result is consistently high packaging quality with minimal waste.
Requirements
• 1:1 automation of a highly complex manual packaging process
• Retaining existing materials and processes without revalidation
• Meeting the predefined cycle rate with minimal wastage rates
• Automated folding of pouches without damaging the product
• Precise insertion of the correct user and product information
• Legally compliant documentation of the cartoning process
• Seamless integration into the available production space
Implementation
• Automated infeed system to the cartoner
• Secure pouch handling via sensorassisted suction
• Reliable quality and safety controls through cameras, sensors, and software
• Optimised drive configuration for all product-touching movements to ensure gentle handling
• Integrated downstream processes such as sealing and labelling within the cartoner for optimal space utilisation
Jörg Aurbacher
Jörg Aurbacher joined Christ Packing Systems in 2007 as a design engineer after graduating in mechanical engineering. He became design team lead in 2010, gaining extensive project experience in automating packaging for the pharmaceutical and medical device industries. Working closely with pharmaceutical companies around the world has given him a deep insight into their unique packaging challenges and specific requirements. Since 2024, Jörg has taken over as Managing Director, driving the decisive customer focus and high-tech orientation that Christ Packing Systems represents.
Email: info@christ-ps.com
Figure 5: The Carton erector – Within fractions of a second, the box is folded and precisely positioned for synchronisation with the insertion process.
Figure 6: The BoxTeq cartoner, the centrepiece of the automated packaging line, is modular and customisable.
Health Outcomes
The Impact of Patient Adherence Technology on Global Health Outcomes
How can AI enhance patient adherence to prescribed medications and maximise the therapeutic potential of new and existing drugs?
AI alone won’t solve the challenges of medication adherence – it’s the convergence of AI with advances in point-of-care testing, remote diagnostics, digital health solutions, and passive biomarker monitoring that will truly transform patient care. These technologies allow us to build a complete and real-time picture of a patient’s health. AI and machine learning (ML) can then analyse this data to understand individual behaviours, perceptions, and physiological responses to medications. This enables us to determine not just whether a patient is taking their medication but also why and how –and critically, whether the medication is working as intended.
The most significant breakthrough will come from using AI to personalise medicine at the individual level. Most drugs are developed and tested in controlled clinical trials with highly homogeneous patient groups, which don’t fully represent realworld populations. By integrating AI-driven insights with real-world patient monitoring, we can optimise dosages, minimise side effects, and maximise therapeutic benefits –ensuring each patient gets the most effective treatment tailored to their needs over time.
Can AI Lead to Better Health Outcomes and Improved Efficiency in Healthcare Systems?
Absolutely. Healthcare systems, particularly those like the NHS with a cradle-to-grave patient ID system, hold vast amounts of valuable data at a population level. AI can analyse these datasets to uncover patterns, predict risks, and optimise care delivery which leads to better outcomes and cost efficiencies. However, AI’s impact is only as good as the data it’s trained on.
Many healthcare systems still struggle with fragmented data – where clinical records don’t align with real-world patient experiences, and financial data around
healthcare costs remains siloed. Linking these disparate data sources is crucial. Only when we integrate clinical, economic, and behavioural data will we fully unlock AI’s potential to identify high-impact cost savings and improve system-wide efficiency.
How will AI improve patient engagement and education?
In a world of rapid AI advancements, even experts struggle to keep up with the latest developments – so how can we expect patients to? We have a responsibility to educate the public on their health, chronic disease management, and the steps they can take to lead healthier lives. The COVID-19 pandemic showed us that patients can be empowered with the right knowledge –people learnt to conduct at-home testing, understand their health status, and take informed actions.
AI has the potential to revolutionise patient engagement, but only if we first educate people on its benefits. In a clinical study we published last year, we demonstrated that when patients were given clear, relevant information about their treatment, they engaged more deeply with their care. They were particularly motivated when they understood that their data was directly influencing their treatment decisions. For example, we used AI to help patients personalise their medication dosing, reducing side effects while maintaining blood pressure control. This level of engagement is key to AI’s success in healthcare.
Can AI Potentially Lower Healthcare Costs?
Yes – but not in isolation. Even today, we can make strong health economic arguments for early, high-cost interventions that lead to better long-term outcomes. However, in many healthcare systems, the budget that funds an intervention isn’t the one that benefits from the cost savings down the line which makes implementation challenging.
For AI to truly reduce healthcare costs, we need a comprehensive understanding of patient journeys, integrating both clinical and economic data. A practical starting point is tackling the highest-burden diseases – like hypertension – where AI-driven personali-
sation can drive immediate cost savings and improve patient outcomes. From there, we can scale these approaches across broader healthcare challenges.
Will AI Boost Pharma Revenue and Market Growth?
AI is already being deployed at scale in the pharma and biotech industries, primarily to accelerate drug discovery and development. This is a crucial investment, as it reduces the time and cost required to bring new therapies to market. However, one of the biggest untapped opportunities lies in optimising existing medicines.
We have countless drugs that are already approved and widely used, yet they are rarely optimised for the individual. AI can enhance the tolerability, efficacy, and longterm tracking of these treatments – ensuring patients get the best possible outcomes. Despite this potential, the pharma industry often over-indexes on developing nextgeneration therapies rather than refining the treatments we already have. To change this paradigm, AI should be used to tailor medications to individual patients, not just to the disease, ensuring each patient gets the most effective version of a given drug for them.
Are there any Regulatory & Compliance Benefits?
Regulatory agencies are adapting to the AI revolution, paving the way for more streamlined pathways to evaluate these technologies. In the U.S., for example, the FDA has introduced Prescription Drug Use-Related Software (PDURS), a pioneering regulatory framework that allows software to be paired with a drug to enhance its effectiveness. This includes AI-powered dose optimisation and behaviour-based interventions.
Initiatives like PDURS highlight how AI-driven solutions can create regulatory and compliance advantages for patients, healthcare providers, and the pharma industry alike. By integrating AI with drug therapy, we can not only improve treatment outcomes but also ensure safer, more effective medication use which ultimately benefits everyone involved in the healthcare system.
Health Outcomes
How can AI improve Adherence in Clinical Trials and Real-world Evidence Generation?
AI has the potential to revolutionise adherence by focusing on a deeper understanding of the individual. It’s not just about tracking medication intake – it’s about recognising how a patient’s perceptions, lifestyle, and relationship with their treatment shape their adherence. By analysing these personal factors, AI enables a more personalised approach, ensuring that treatment is tailored to the patient, not just the disease.
How do Socio-economic Factors Influence Adherence, and How can Technology Address Disparities?
Access to high-quality care often depends on socio-economic status. Patients who can afford top-tier physicians receive more personalised adjustments to their therapy, from drug selection to tailored dosing regimens. Technology has the power to democratise this level of care, making it accessible to everyone. AI-driven software can replicate the best clinical practices at scale, ensuring that all patients, regardless of income levels or where they live, receive optimised, data-driven care.
What Ethical Considerations Should be Addressed when Using AI to Monitor Patient Adherence?
Patient data privacy is paramount. AI must be deployed within strict ethical and regulatory frameworks that prioritise patient ownership of their data. Companies like Closed Loop Medicine adhere to rigorous international data protection standards to ensure patient information is used solely for its intended purpose –
enhancing health outcomes – without any risk of misuse.
How can AI-powered Predictive Analytics Help Identify Patients at Risk of Nonadherence?
AI can help predict non-adherence by integrating multiple data sources, from biomarkers of efficacy and side effects to behavioural insights. By reaching a state of hyper-personalisation, we can categorise patients into super-responders, typical responders, and those at high risk of non-adherence. AI’s ability to curate and synthesise fragmented datasets allows for early intervention, ensuring patients receive the support they need to stay on track with their treatment.
What are the Key Barriers to Patient Adherence, and How can Technology Help Overcome them?
A patient’s relationship with their medication, their level of education about their condition, and their overall engagement with treatment are all critical factors in adherence. The most powerful role of technology is to make adherence personal. By showing patients a direct link between taking their medication and tangible health benefits – on a trajectory unique to them – AI can transform adherence from a compliance challenge into an intuitive, personalised health journey.
What is the Future of Patient Adherence Technology?
The future of adherence technology lies in simplicity and passivity. In today’s fastpaced world, patients are bombarded with competing demands, making it difficult to stay engaged with silent conditions like hypertension. The next generation
of technology will integrate high-fidelity wearable monitors and passive tracking systems, seamlessly delivering real-time health insights without requiring active patient input. By leveraging AI to personalise interventions and predict adherence risks, we can help individuals manage their health more effectively and stay healthier for longer. Ultimately, the goal is clear: treat the individual, not just the disease.
Dr. Hakim Yadi is Chief Executive Officer and co-founder of Closed Loop Medicine, a leading TechBio company developing combination prescription drug plus software products, with the aim of redefining precision medicine. The company’s mission is to improve outcomes for patients suffering from long term conditions by creating personalised and integrated treatment pathways, enabled by new closed loop models of care. This approach could level the playing field, providing universal access to the highest quality care, usually only accessible to the few. Before his current role, Hakim was the founding CEO at the Northern Health Science Alliance Ltd, a pan-Northern health partnership that, for the first time, brought together 20 research-based NHS hospitals, the North’s Academic Health Science Networks and universities to collaborate on improving health outcomes across Northern England.
Dr. Hakim Yadi
Health Outcomes
Defying the Odds: Speeding Patient Access to Life-changing Treatments in Rare Disease
To deliver treatments for rare diseases more quickly, companies share how they are adapting their launch strategies and engaging more deeply with physicians and experts.
“The only thing that was impossible was to do nothing.” These are the words of Terry Pirovolakis, CEO of Elpida Therapeutics, and father to Michael who was diagnosed as an infant in 2019 with an ultra-rare neurological condition, SPG50. Despite being told there was no cure, Terry moved mountains over four years to find a breakthrough gene therapy that his son and other affected children all over the world could benefit from.
Michael, Terry, and their family are not alone in facing a long, daunting journey to access life-enhancing therapy for a disease with no cure. In the European Union, less than five percent of rare diseases have at least one approved treatment. It can take up to five years for adults to receive a diagnosis, and half of patients will receive a misdiagnosis first.
By 2030, the goal is to support the development of 1,000 new therapies in Europe. However, rare diseases challenge traditional ways of doing business. Once a medicine is approved, organisations must launch and scale quickly, often across multiple markets. Field medical teams must keep track of complex science at a time when the volume of medical knowledge is doubling every 73 days. They need to identify, engage, and provide education to relevant healthcare professionals (HCPs) and experts even though they’re harder to reach via traditional methods (Veeva Pulse data shows that in-person meetings with physicians have declined in Europe in the last 12 months).
Despite these difficulties, life sciences organisations are improving how their teams organise market access and educate prescribing doctors on new medicines. Companies with comprehensive, accurate healthcare ecosystem data and insights relating to medical experts can prepare
the market pre-launch and hit the ground running to accelerate execution after launch. This helps all customer-facing teams to deliver a consistently high-value scientific exchange in which every interaction builds off the last.
Greater Agility During Launch
Emerging biopharmas play a key role in delivering treatments for affected patients. Of the 192 orphan medicinal products (OMPs) authorised by the European Medicines Agency (EMA) between 2010 and 2022, one in ten successful applicants was designated an ‘SME’. Facing more resource constraints than their competitors (which may have sales organisations multiple times their size), earlystage companies are increasingly seeking the best customer reference data and key opinion leader (KOL) insights to become more nimble when launching new treatments.
Rich information on experts, available in CRM, supported Sweden-headquartered Sobi to launch the first and only medicine for hemophagocytic lymphohistiocytosis (HLH) – a rare, hyperinflammatory condition affecting one in 50,000 resulting in a twomonth life expectancy. In the U.S., MSLs at the company now routinely use real-time intelligence to find the right KOLs, understand their interests, priorities, and activities, and consolidate scientific information and updates. “90% of MSLs found new insights for their next engagements, which is critical for a rare disease company with lean teams,” says Rich Palizzolo, executive director of CX and advanced commercial capabilities at Sobi.
An added complexity is that rare diseases often involve several specialties. MSL teams have to get up to speed quickly on multiple areas before meeting experts – while also being responsible for other (more mainstream) therapeutic areas. ADVANZ PHARMA, which focuses on specialty, hospital, and rare disease medicine, found having scientific resources and activity data in one place helped MSLs use their time efficiently. Head of CRM and Digital Solutions for Global Commercial Excellence, Andy Eeckhout, notes: “Our customer-facing teams need to be agile communicators and effectively switch to a more patient-oriented, in-depth scientific discussion than with generics. Pre-
call planning is crucial for MSLs before and after launch. The more data they can find, including on past interactions, the better.”
Other companies use customer reference and patient data to improve operational agility as they launch and scale their first products across Europe. For example, one late-stage biotech leveraged its data on the healthcare ecosystem to get a head start on launch by identifying market access roles in Spain, Benelux, and the Nordic countries.
One Voice to the Physician
After identifying the right experts, teams can engage them more effectively by ensuring that each HCP interaction builds off the previous one. However, sales, marketing, and medical teams often use disconnected technology. As a result, 65% of HCP engagements are not synchronised.
When these teams are connected in the same system handovers are smoother, and HCPs can find answers quickly or connect with MSLs if needed. ADVANZ PHARMA introduced a pre-launch module in its CRM to help market access, medical, and commercial teams share information compliantly. Eeckhout explains: “Physicians need a direct line to the industry, so they know who to contact when they have questions. Medical and commercial teams need to talk to each other and remain agile across customer conversations.”
ANI Pharmaceuticals, which delivers treatments for certain rare autoimmune and inflammatory conditions, only had 75 days to commercialise following swifter-thanexpected regulatory approval. By using an industry-specific CRM with master data management, it consolidated its view of each HCP to include interactions with medical and sales reps. “Having this information accessible within the CRM system facilitates more thoughtful and helpful conversations with providers, as well as sales teams’ success and high click-through email rates,” explains Bob Acropolis, executive director of operations and analytics at ANI.
The foundation of successful interactions is accurate reference data – on physicians, healthcare organisations (HCOs), or
Health Outcomes
affiliations. If applied across functions as part of a complete life sciences-specific CRM, it helps companies speak with one voice. In most cases, data change requests (DCRs) can now be made (and resolved) in hours, so reps and field medical don’t duplicate attempts to modify account information and instead work from the same database. With greater trust and confidence in reference data, teams save time so they can focus on high-value scientific exchange.
Sharing Medical Content that Engages Europe’s fragmented regulatory landscape and evolving local requirements intensify the pressure on marketing teams: they must provide highly personalised, compliant medical content that field teams can share at scale with scientific experts and physicians. With a single view across the entire content lifecycle, biopharmas can streamline and speed up medical, legal, and regulatory (MLR) reviews.
To deliver highly personalised content across a large rare disease portfolio, marketing teams need clarity on which content to recommend, and when. One global biopharma uses data analytics on its global repository of promotional and medical engagement tools to support content use across 17 areas of focus (and a growing global footprint). As its head of marketing and customer engagement noted, “How we engage with HCPs is critical. We need to know what percentage of our content is being developed and relevant to support different HCPs, whose patients rely on them for their rare disease diagnosis and management.”
As a new generation of digitally-savvy HCPs comes through, companies are considering the most effective tactics to engage them. More scientifically active than their peers and four times more likely to adopt a new treatment, younger HCPs require a different mix of scientific channels and content. They seek medical insights to inform their clinical and commercial decisions, which requires close coordination between medical affairs and field medical. “Gone are the days when medical could just focus on the top-tier scientific thought leaders. The range of stakeholders has broadened, and it’s imperative to expand our engagement strategies beyond traditional experts,” says Angela Smart, director of global medical excellence and operations at ADVANZ PHARMA.
Defying the Odds, then Beating Them Scientific discovery continues to bring hope to patients affected by rare diseases and their families. Life-changing conditions will eventually become chronic illnesses, thanks to the efforts of organisations willing to launch in a high-risk commercial environment. Companies ranging from emerging biotechs to global biopharma are using high-quality customer reference data, deep data on scientific experts, and connected technology to identify, engage, and provide medical education to the most relevant HCPs and KOLs.
Every rare disease patient faces a daunting journey. When Terry Pirovolakis’ son, Michael, was diagnosed, his family was told he was the only child in Canada with SPG50. Life
sciences will do its part to help patients defy overwhelming odds – and eventually beat them.
REFERENCES
1. “The building blocks to make rare disease treatments more common,” European Commission, February 2022
2. “What is Rare Disease,” EURODIS, 2024
3. “Challenges and Opportunities Facing Medical Education,” American Clinical and Climatological Association, 2011
4. “Trends in orphan medicinal products approvals in the European Union between 2010–2022,” Orphanet Journal of Rare Diseases, 2024
As president of Veeva Europe, Chris Moore is responsible for growing the business in the region. A 30+ year veteran of the life sciences industry, Chris started his career at ICI Pharmaceuticals (now AstraZeneca). Chris then joined a start-up called Kinesis, building a team delivering document management solutions for pharmaceutical companies. Through a series of mergers and acquisitions, Kinesis ultimately became PwC; Chris made a partner with PwC in 2001. Chris went on to run both European and US (West Coast) life sciences businesses for IBM before leading the IBM global life sciences consulting Business Analytics and Optimization unit.
Chris Moore
Technology’s Role in Pharmaceutical Manufacturing Excellence
Pharmaceutical manufacturers face increasing compliance demands to ensure the identity, strength, quality, and purity of drug products. As a result, systems used within manufacturing processes must adhere to current Good Manufacturing Practice (GMP) guidelines enforced by regulators like the US Food and Drug Administration (FDA). GMP regulations require drug manufacturers, processors, and packagers to take proactive steps to ensure drugs are produced consistently and that their products meet quality standards for strength, purity, and efficacy.
These core ambitions of the regulations are to prevent errors in the manufacturing process to protect the consumer from purchasing a product that is potentially ineffective or even dangerous. While GMP is applicable to a number of industries, the consequences of failing to follow these guidelines within pharmaceutical manufacturing can have serious consequences by compromising the safety and efficacy of the final product. Failure of firms to comply with GMP regulations can result in severe repercussions, including product recalls, seizures, organisational fines, including consent decrees, and, in some countries, individual jail time.
End-to-end GMP Compliance can Support Drug Development
The success rate of drug candidates that make it through phase I, II, and III clinical trials is just 10%.1 However, industry adoption of new technologies that support the drug discovery workflow is making improvements,2 including software that is used for the collection, management, and storage of data and documentation.
When you consider the time and money involved in drug discovery and the ratio of successes to failures, it is understandable that once a drug is approved, pharmaceutical companies look to maximise revenues and longevity. While pharmaceutical manufacturing is often seen as the final stage in the drug development lifecycle, it is a vital part of a drug's ongoing monetisation,
requiring precision, accuracy, and a hyperfocused approach to quality.
Pharmaceutical companies and CDMOs adhere to GMP regulations as the benchmark to maintain manufacturing excellence. As drug design grows more complex with the development of novel therapies, from personalised and precision medicines to gene therapy and mRNA vaccines, regulatory bodies increase oversight, and shareholders look for improved return on investment, the industry finds itself navigating a landscape where technology is no longer just an asset in the manufacturing process; it is of paramount importance.
Integrating GMP-compliant Technologies to Support Pharmaceutical Manufacturing
The integration of a Laboratory Information Management System (LIMS) into drug discovery and development workflows serves as a useful tool to streamline data management and automate reporting workflows, resulting in time and cost benefits, important considerations for pharmaceutical companies looking to reduce timelines from drug development through to regulatory approval.
To maintain GMP compliance when integrating a LIMS, it is important to consider the advantages of using a LIMS that can support GMP-validated processes and documentation. Configuring a LIMS system to support these processes ensures compliance with stringent regulatory requirements and provides a robust framework for managing the volume and variety of processes involved in pharmaceutical manufacturing.
Key Features of a GMP LIMS
A GMP LIMS must support several key features to ensure regulatory compliance and efficiency in highly regulated industries such as pharmaceutical manufacturing. These include data integrity compliance with 21 CFR Part 11 and EU Annex 11, robust user access controls with role-based permissions, comprehensive data security and traceability, and thorough audit trails that log all user and system actions.
Quality control features such as material and product management, automated review
and approval workflows, environmental monitoring, and stability management are also essential. Ideally, the system should also offer no-code configurability to adapt to changing regulatory requirements and laboratory needs.
Often, GMP LIMS solutions are complex and rigid, leading to high costs and challenges in configuring and adapting the system to specific workflows. Ensuring the system is user-friendly, configurable, and functionally adaptable without requiring extensive coding, customisation, or validation is crucial.
These key features can be further broken down into quality control, environmental and equipment monitoring, and stability testing and monitoring:
Quality Control
GMP or QC LIMS are designed to automate quality control processes, overcoming the issues linked to traditional methods of quality control, which were often time-consuming and prone to human error, such as manual test setup, recording results in spreadsheets, or laborious inventory management.
A QC LIMS, or the QC component of a more comprehensive GMP LIMS, should cover the five main steps in any drug manufacturing process: material and product management, batch creation, sample management, testing and analysis, and finally certificate generation. By closely mirroring the manufacturing process, a GMP-compliant LIMS is able to measure a number of quality control factors, including customisable or predefined acceptance criteria, meaning each batch of pharmaceuticals can be tested against a set of stringent, predefined standards, ensuring consistency and quality across production runs.
An effective GMP LIMS will integrate with lab instruments to allow for direct data capture and eliminate the need for manual data entry. This integration should be based on industry-standard protocols like REST APIs and HL7 to prevent future integration or data issues. A standard-based approach to integration also allows for seamless communication and refined data
mapping, which is essential when trying to build a comprehensive quality picture at a manufacturing facility.
Environmental and Equipment Monitoring
Pharmaceutical manufacturing requires strictly controlled environmental conditions, including the equipment and storage facilities of the production environment. Any deviation from or change to the equipment, facilities, or environment can affect the final product, reducing its efficacy or even making it harmful. In the past, environmental monitoring has been both a manual and reactive process, with manufacturers only becoming aware of an environmental issue after the problem had impacted the process.
With a GMP-compliant LIMS, problems and issues can be spotted before they become critical and affect the manufacturing process or the end product. The functionality required to take this proactive stance includes comprehensive environmental monitoring capabilities such as site, equipment, storage monitoring, and routine testing set-up, along with results measurement, recording, trending, and reporting.
Another critical feature of a GMP-compliant LIMS in a manufacturing environment is the ability to run data visualisation and analysis on the collected data. Having this capability built into the LIMS itself means that manufacturers can run interactive visualisations of the entire manufacturing environment, including trend charts, heat maps, and statistical summaries. This ability to see in real-time specific measurements, locations, or time periods and identify trends and outliers in the data ensure product quality and significantly reduce the possibility of costly batch failures.
Stability Testing and Monitoring
As well as being a GMP requirement, stability testing and monitoring are critical in ensuring the quality and efficacy of pharmaceutical products. Stability testing helps to predict product behaviour under various conditions, ultimately guiding storage, packaging, and shipping decisions to ensure the drug reaches consumers as intended.
The vital nature of this testing means stability studies encompass a wide range of factors, including the product's physical, chemical, microbiological, therapeutic, and toxicological specifications. Managing the stability testing process and the data generated is a complex task, therefore, having a GMP-compliant LIMS can alleviate some of the complexity. For example, a GMP-compliant LIMS can assist the manufacturer in streamlining the testing process, from initiating the study through defining the study parameters, scheduling the tests, and generating the final reports, including information such as trend analysis, degradation pattern identification, and shelflife projections.
Regulatory Compliance
Navigating the complex web of global regulatory requirements is one of the most challenging aspects of pharmaceutical manufacturing, especially as companies expand their operations internationally. Complying with a diverse array of international standards can be simplified with a configurable GMP-compliant LIMS that has the flexibility to accommodate and adapt to the regulatory landscape.
It is vital that whatever LIMS a manufacturer has in place meets regulatory requirements, which includes ensuring
that the LIMS vendor has an established SDLC (Software Development Life Cycle) programme, a Quality Management System (QMS), and that the LIMS is maintained and updated by the vendor in a validated state. It is also important to ensure that the GMP LIMS can keep pace with regulatory changes and international differences.
Adapting to regulatory changes or international differences requires close monitoring of bodies such as the FDA, EMA, or any other regulatory agency. However, being informed is only half the battle, and the LIMS used to manage, monitor, and support regulatory compliance needs to be easily configurable, allowing it to be configured to the changing requirements or international differences without the requirement to invest in extensive coding, customisation, or validation for quick updates.
A second regulatory challenge for anyone involved in drug development or manufacturing is managing the documentation generated and required throughout the entire process. While vital, these processes are seldom prioritised by scientists, researchers, or manufacturers, but in an industry where a single missing document can halt production or delay a product launch, the importance of thorough, accurate documentation cannot be overstated.
A GMP-compliant LIMS should emphasise data reporting and document generation as a core feature, allowing manufacturers to maintain a comprehensive audit trail, tracking and recording data and information across equipment, facilities, storage, raw materials, sample testing, batch release, stability, and quality. This comprehensive level of detail serves a dual function, satisfying regulatory requirements and enabling the identification of any issues that may arise during the manufacturing process.
Looking Ahead
Technology’s role in pharmaceutical manufacturing has been transformative, enabling the cost-effective production of drugs and therapeutics that have helped countless people. As we move into a new era of medical advancement, one that includes more complex drug design, the use of technology in the manufacturing process will become even more central. From automating QA/ QC processes to ensuring global regulatory compliance, GMP LIMS will provide pharmaceutical companies and CRMOs with the ability to operate with the precision, efficiency, and
Technology
transparency required to keep pace with medical advancements.
The journey towards pharmaceutical manufacturing excellence is ongoing, and the adoption of and compliance with GMP means that clinicians and patients can be confident in the quality and efficacy of the
drugs they are prescribing and taking. By adhering to GMP, maintaining flexibility in the face of evolving regulations, and leveraging the tools and capabilities included in a GMP-compliant LIMS, the pharmaceutical industry is well-positioned to maintain a quality-first approach to pharmaceutical manufacturing.
Robin Stolzberg, Global Head of Quality Assurance, Sapio Sciences, brings over 30 years of GxP experience in quality assurance, systems, and engineering. Her experience in the pharmaceutical and biotechnology industries and her strong knowledge of CFR, OECD, ICH, PICs, ISO, CAP/CLIA, and WHO regulations allow her to be a strategic resource to Sapio and its clients. Robin holds a BS degree in Biology from the State University at New Paltz and is a member of the International Society for Pharmaceutical Engineering.
STEAMING SOLUTIONS FOR ALL INDUSTRIES
A Five-year Roadmap for Technology-led PV Innovation
Technology is transforming pharmacovigilance (PV). The promise of artificial intelligence is of course one of the first things that springs to mind, and it is increasingly providing value. More mature technology solutions provide scalable, structured digital platforms to capture adverse event data at source, in multiple languages, across multiple use cases. It is also enabling direct reporting from healthcare professionals and patients with smarter digital ways for people to access, and contribute to, safety data.
The PV Problems of Today and Tomorrow PV consumes around 10–15% of the total allocation of research and development investments in the pharmaceutical industry. COVID-19 raised awareness of the importance of PV but to ensure its continued evolution, and investment, we need to make sure it is fit-for-purpose, not just now but in the future.
In the past we have talked about a direct case-investment metric. More cases that have historically come in – and more complexity – mean we need more funds for PV. However, adverse event reports are no longer slated to increase with a CAGR of approx. 13% as we have been used to, in fact the FDA is reporting a decline from a peak of 2.3 million in 2022 to just over 2 million in 2024.1 This has implications for how we focus our investment and innovations. Instead of planning to deal with increased volumes, we need to focus on improving quality and efficiency and ideally provide savings for the organisation. As an example, we are increasingly capturing adverse event data directly from source, while legacy systems capture this in unstructured formats like emails which make it harder to process and do not ensure quality. We need structured digital platforms which can capture data at source and flow it directly to clients.
Another challenge is the different approaches taken by organisations of different sizes. Big pharma companies have been building complex IT architecture diagrams to try to cater for all their needs. Small companies have been trying to deal
with the complexity with a cornucopia of spreadsheets. About 90% of companies that operate with safety data and really should have a safety database to be compliant, do not have one because of the prohibitive cost. Here, it is somewhat problematic to observe that technology vendors are all trying to invent in the same space – the mid to large pharma segment – and right now this is very much for artificial intelligence (AI) investments. This works if you are a large company with a complex IT infrastructure and want to invest in simplification, but not if you are a spreadsheet company.
Why Don’t We Simplify the Picture by Using AI for Prediction?
Can we simplify the systems architecture? Can we remove some of the legacy support like paper-based and scans? Can we remove parts of the PV ecosystem or the way we operate? Can we get simpler regulatory requirements to operate by? The answer, for the most part, is, no, not easily. We live in a complex reality and innovating in that space is complex.
All the components which make this complex landscape were the solution 20 years ago. Now they are the problem, and we need to apply new technology in smart ways to innovate.
Top Considerations for PV Innovation Over the Next 5 Years
It is important to remember first and foremost that our systems need to deliver compliance but that does not dictate the design. This is a legacy issue where everybody is thinking of the safety database in a specific way. Of course, compliance needs to be there, but we need to break up a bit of that mindset.
Secondly, if you look at the outside world, all of our users and colleagues are used to a completely different world now. They are used to user-friendly IT systems, and are used to using AI in their everyday lives. We are way behind in the PV area. We need to think about how we can generate a better use case for our users. Predicting data points is one thing, but it is not the only thing we need to do. We need to make sure our systems are more applicable and
user-friendly and follow suit with what we are seeing elsewhere.
It is also important to remember that we are all connected in the value chain of PV across the industry. This means that we win or lose together. A larger pharma company has maybe 200 or 300 different partners. They all need to work together and there has not been a lot of investment in trying to solve that piece of the puzzle. Much of the complexity is driven by different partners failing to gel efficiently. We are also connected to the regulators, and they want to see cohesion and effective collaborative working.
Finally, the insights coming from data are key. The results we achieve will derive from the quality of the data.
Strategic Priorities for PV Innovation
There are three strategic priorities for PV from now until 2030 – lowering the cost to operate and standardise, utilising technology enhancements and easing cross-platform data integration.
We need to increase the availability of standardised technology for organisations of all sizes and share best practice processes to ensure all partners can work with efficient, high-quality data. By, for example, making AI and other scraping tools available to all, we can upgrade our quality across the span.
We need to improve user experience and access to information using generative AI (GenAI) and increase automation scope and quality. The focus of this work should be on confidence rather than just accuracy. If you are less confident about a data point you can create a human system to look at those data points which will allow you to utilise this concept.
Getting data to flow, more naturally integrated, is going to be part of the innovation roadmap for the next five years. There are a lot of things happening already in the space that are really interesting to see with more data sources becoming available. However, we also need to focus on quality to ensure there is high quality data for training purposes.
The Most Important Question to Ask
The key question to ask ourselves as we embark on this roadmap is: What questions do I want to ask my PV system? GenAI and chat-based technology is an area that can be used pervasively, for human interaction and supervision. The supervision will remain key. For example, if you are using natural language processing (NLP) for predictions and other technologies for automating, then you will need humans to oversee the control system. Humans must work efficiently with the PV system.
For example, you could ask the system to check for quality issues in the ICSR and emphasise medically important items. If we map out the questions we want to ask, we can have GenAI check its own work and have checks in some of the other models. This is going to be a much more efficient way to work. GenAI is perfectly capable of summarising confidence scores in literature, but it is not necessarily great at data predictions. Instead of focusing on predictions, we should be using GenAI to interact with data and make the voyage to understanding more pleasant.
You could also ask the system to produce a PBRER on Superdrug as an example, for a set period using standard template. GenAI is perfectly capable of pulling data points together and writing nice narratives. What is needed is structured content authoring
in the back. You need some absolute truths that humans have created and then you use AI to pull all that data together.
Another example is asking the system to provide an overview of pending submissions and highlight negative acks. We are focusing too much on using AI to predict things and if we could ask these questions, it would be great because then we can use the data we have already generated, put it into our safety systems and surface them.
The final question is asking the system to schedule a daily overview of reports to a mailbox, sorted by incident seriousness. We can use GenAI to tap into the existing functionalities we have in our safety systems and make them work more efficiently for us.
Conclusion
There are some key steps we need to take on our roadmap to technology-led PV innovation. We need to make sure we expand our PV ecosystem to capture more high-quality data from all partners globally. We need to use the right models, feed them the right data and ask the right questions. We need to use technology not only to make predictions but to help us do our work more efficiently and ensure that the patient voice is heard.
People expect to be able to contribute towards, and access, safety data in smarter, digital ways. Setting out a clear roadmap
for technology-led innovation will help to ensure PV remains a cross-industry priority and adapts to meet the demands of a patient-centric clinical trial landscape.
Martin Holm-Petersen is an executive leader with 20 years of experience in the life sciences Industry. Martin is an experienced leader and strategist, with understanding across the pharma business value chain, and an expert on pharmacovigilance. His current role is Chief Strategy Officer at Qinecsa Solutions, a company that combines best-in-class technology and scientific expertise to connect life science companies with the right safety solutions. In his previous role, he headed the PV Tech Global Industry Pharmacovigilance Technology Network with participation from more than half of the top 100 pharma companies.
Martin HolmPetersen
Logistics & Supply Chain Management
Expanding into Emerging Markets
Despite global tensions and geopolitical conflicts, the pharmaceutical industry is increasingly expanding its reach into underserved markets – and this reach will continue to grow next year. This push is essential for bringing critical medicines to new communities, yet logistics and storage infrastructure in these regions can create additional complications.
Cold chain logistics rely on stable, temperature-controlled environments to protect sensitive medicines. However, outdated infrastructure in emerging markets often means smaller storage spaces, unreliable electrical supplies, and no return logistics for reusable packaging. These factors make it problematic for cold chain providers to maintain the necessary conditions consistently.
To overcome these challenges, market expansion will require specialised packaging and logistics solutions that address local infrastructure limitations. These adaptations will be key to enabling successful market entry and supporting long-term growth.
Rising Mergers, Acquisitions and Strategic Partnerships
We have witnessed an increase in merger and acquisition activity in the pharmaceutical industry in 2024, and we expect to see this continue to rise in 2025. Pharmaceutical companies are also increasing their strategic partnerships with CMOs and CDMOs to accelerate innovation and reduce time-tomarket. By pooling resources and expertise, companies will be able to streamline the R&D process, paving the way for faster production and delivery of high-demand drugs.
The beneficial outcomes will be set to multiply if companies integrate cold chain logistics into these new partnerships. Not only will this support the rapid scale-up of new therapies, but it will also help build a robust supply chain for new markets.
Adapting to Small-batch Shipments
As the demand for smaller, high-value pharmaceutical shipments grow, cold chain logistics will need to become more agile and adaptable to meet these evolving needs. These solutions are essential to ensure highly sensitive medicines, such as personalised treatments, are stored and transported under precise temperaturecontrolled conditions.
Additionally, AI-driven clinical trials are accelerating drug development, leading to faster production cycles that also require efficient, responsive cold chain solutions to handle smaller but high-value, sensitive shipments.
Finally, the growing prevalence of decentralised clinical trials adds to the demand for robust cold chain logistics. These trials, which involve direct delivery of experimental therapies to patients’ homes, rely on precision cold chain networks to maintain the integrity of highly sensitive medicines.
Optimising Cold Chain with Technology
The potential of AI will extend beyond R&D to supply chain management next year. Organisations will begin to use the technology to minimise risk, reduce costs and boost efficiency. Cold chain providers can learn from tech giants like Amazon and Alibaba, who use AI and data-driven insights to improve logistics and access to treatments.
While their focus isn’t exclusively on the cold chain, their innovations may offer valuable ideas for delivering sensitive medicines more efficiently.
Finally, blockchain’s potential to increase supply chain transparency through decentralised, immutable records makes it a key technology to watch. Deloitte’s case studies already demonstrate its practical application in ensuring product integrity, underscoring the importance of blockchain in advancing pharmaceutical logistics.
2024 has seen steady progress in pharmaceutical logistics, with innovations continuing to shape the future. As we move into 2025, the focus will be on smarter packaging, agile supply routes, and solutions that address the growing complexity of global healthcare demands. The year ahead holds significant opportunities for companies willing to adapt, collaborate, and lead the charge in delivering medicines to underserved communities, improving availability and saving lives.
Delphine Perridy
Delphine Perridy is the Chief Commercial Officer at Envirotainer, and responsible for delivering and executing the global commercial strategy. She brings 20+ years of experience acquired in the life sciences industry. She has experience working in EMEA, APAC & Americas, driving high growth expansion plans.
Logistics & Supply Chain Management
for pharma companies to work with their global partners to enable these treatments to progress through clinical trials and onto the market.
Neuroscience, rare diseases and immunology remain important areas of focus in the coming years. New modalities in these growing areas demand different processes and logistics experts must stay ahead of how this will impact their handling requirements to ensure the drug product is protected.
Flexibility and Agility
The impact of recent global events can still be felt throughout the world of supply chain logistics. What is clear now more than ever is the need to be agile and flexible within the rapidly moving industry, where nextgeneration treatments are necessitating new approaches to logistics.
By pioneering cutting-edge innovations and emerging technologies, while at the same time securing valuable collaborations across the industry, companies can keep ahead of the curve.
Deloitte’s 2024 Global Life Sciences Sector Outlook especially highlighted the potential of generative AI to automate tasks, streamline workflows, and optimise processes, allowing for reduced costs, increased efficiency and a personalised patient experience.6 Harnessing new
technologies and combining them with existing processes can help to unlock the potential of the data and information you already possess.
A proactive approach and keeping up with how these tools are being used by other key players in the industry is key to maximising their potential and finding innovative solutions.
Every year, the challenges faced by life science companies evolve, but it remains one of the most innovative and exciting industries. The logistics and supply chain plays a key part in moving science forward and it will remain so through 2025 and beyond.
Jeff Stone has over a decade of experience in various leadership positions across research and development, manufacturing and logistics within the pharmaceutical sector. He began his pharmaceutical career in 2011 working with a leading CDMO, where he gained a diverse range of experience in logistics and clinical trial management across various clinical indications. He went on to work in the specialty logistics market, where he played a pivotal role in building a global Project Management Organisation and infrastructure to support critical cell and gene therapies. This included the creation of a state-of-the-art control centre in North Carolina’s Research Triangle Park. At Biocair, Jeff oversees all aspects of operations and commercial sales in the US. He drives efficiency and effects positive change to enhance the overall client experience.
Jeff Stone
Logistics & Supply Chain Management
A cohort pathway will place full regulatory responsibility on the sponsor. This can increase the burden of operating a MAP, especially in regions like Europe, where each member state has its own legislation relating to how EMA recommendations are implemented.
There are also nuanced supply chain challenges relating to MAPs. Due to the reactive nature of the programme type, demand forecasting is particularly difficult. This can increase the risk of waste or rework if supplies surpass expiry dates while awaiting unsolicited patient requests. A sponsor’s ability to start-up and respond quickly once demand is known is critical to the success of MAP delivery.
To overcome these challenges, the industry is seeking better solutions for MAPs delivery. Solutions like Just in Time Manufacturing (JTM), that helps to deliver a better service to patients and health care professionals (HCPs), drives efficiency gains by enabling more effective management of supplies across MAPs and clinical trials and increases flexibility for packaging and labelling to meet geographical and forecasting complexities. A demand-led, patient-centric production strategy, JTM empowers sponsors to respond quickly to shifting demand and preserve high value product by postponing packaging and labelling until the patient (or site) need arises.
There are many benefits of utilising JTM as part of a MAP. Costs reduce through improved drug conservation and response times can be expedited, allowing for earlier start-ups and improved reaction time to demand changes. The full late-stage customisation of patient kits makes it easier to direct unlicenced medicines to where they are needed both in terms of geography and volume. The added flexibility of JTM means supplies can be used for any country without the need for booklet labels. Furthermore, JTM makes supply pooling possible, allowing sponsors to utilise the same bulk supply
for MAPs and clinical trials concurrently. With JTM, sponsors are also able to adopt patient specific packaging and tailor kits to individual patient need. And as there is less labelled stock in the field, exposure to expiry date events is minimised, along with rework requirements. All these benefits combine to create streamlined and patient-centric MAP operations, while positively engaging key stakeholders, reducing pressure on HCPs and sites and promoting the interests of patients.
Helping Patients at Home with Direct to Patient Distribution
The cost of clinical trials is rising. The average cost of a trail with 250–500 patients is estimated to be $17 million, while the average cost per patient is $41,000, and the average cost to replace patients is $19,533.2
With recruitment costs high and dropout rates tipping 30%, decentralised clinical trials (DCTs) are increasingly being used to ease the patient burden and encourage improved recruitment, retention and overall experience.
Defined by the FDA as ‘A clinical trial that includes decentralised elements where trialrelated activities occur at locations other than traditional clinical trial sites.’, DCTs can encompass many components from electronic consent and remote data gathering to telemedicine, home health nursing and direct-to-patient (DTP) distribution.
The latter refers to a patient-centric by design distribution strategy that empowers patients to receive study drugs at home –either direct from a depot or via a clinical site. The approach provides a workable solution for patients who would otherwise struggle to attend clinic-based appointments. Suitable for temperature-controlled and non-temperature-controlled shipments, DTP distribution is best suited to trials incorporating products with low-risk safety profiles. Trials that include clinic visits where no medical assessment is required for return visits, or medical assessments that can be completed remotely, can also derive considerable value from DTP distribution.
There are a host of benefits that result from operating DCT models that are powered by DTP distribution. Sponsors gain broader access to patient populations, which can positively influence recruitment pace and retention performance. Supply can be optimised, as sponsors can leverage central depot dispensation by a pharmacist and reduce the amount of drug product required to seed sites. Fewer sites and fewer site visits can serve to expedite trial timelines and deliver cost savings.
Most importantly, clinical trials can become more patient focused. Traditionally, the sickest patients often have further to travel, with the average travelling distance for patients enrolled in rare disease studies estimated to be 135 miles.3 DTP distribution reduces patients’ travel burden significantly by allowing them to receive study drugs at home. It can also provide a contingency solution for hybrid trial models for when patients are unable to attend site visits to access medication.
A bigger picture patient benefit is that DCT models powered by DTP distribution enhance the diversity of patients able to participate in clinical trials. This can help to reduce bias and improve health equity, while supporting sponsors to better align their clinical trials with the FDA’s 2022 draft guidance for completing Race and Ethnicity Diversity Plans and the MHRA’s anticipated draft guidance on increasing patient diversity.
To maximise the potential benefit of powering patient-centric DCTs with DTP distribution, there are several study considerations to bear in mind. Regulatory requirements must be appropriately understood, as there are key variations. For instance, in the EU each member state has a different allowance for DTP, with most requiring additional justification for shipping directly from a depot to the patient’s home. Meanwhile, the FDA allows for depot to patient distribution, providing all applicable state laws are followed. Compliance with GDPR and HIPAA, the IRT ordering process, temperature requirements, stakeholder (site, patients, couriers) education, drug returns processes and partner selection are also key considerations.
Ensuring Patient-centric and Successful Operations in the EU
The challenges of operating patient centric clinical trials vary depending on where in the world trial activity is taking place.
Logistics & Supply Chain Management
Success in EU is dependent on developing a robust and effective Qualified Person (QP) release process that delivers full compliance, while safeguarding patient safety and experience.
Within the EU the clinical trial regulatory framework, which has been viewed as challenging by many trial sponsors, has changed with the full implementation of the Clinical Trial Directive EC No. 536/ 2014 on the 31st January 2025. The aim of the Regulation is to harmonise and simplify the processes for assessment and supervision of clinical trials throughout the EU. Prior to the Regulation coming into effect, clinical trial sponsors had to submit separate clinical trial applications to each national competent authorities and ethics committees in each country to gain regulatory approval to execute a clinical trial. The Clinical Trial Regulation framework allows sponsors to submit a single online clinical trial application, and, upon approval, the sponsor can execute multiple trials across EU countries.
Considering these regulatory changes, the importance of Quality and QP resources/ capabilities cannot be understated. In-depth knowledge of cGMP and QP expertise can deliver significant benefit to CT Sponsors, helping them to navigate the new EU requirements and enable timely certification/ release of clinical material - which is essential to maintaining supply continuity to patients at clinical trial sites.
In the EU and UK, the QP plays a crucial role in bringing safe, effective and timely supply of medicinal products to clinical sites. QPs are responsible for ensuring each batch of a medicinal product has been manufactured and tested in compliance with regulatory standards and for ensuring it meets the necessary specifications before it can be released for use by patients.
As a consequence of Brexit, a divergence in the EU and UK clinical trial requirements has the potential to present further challenges for CT sponsors who want to conduct trials in both regions. For Sponsors who utilise an EU QP to support these trials, the batch release process is more complex and costly due to the requirement for an additional UK QP verification, post completion of the EU certification. However, strategically utilising CDMO QP resources situated in Northern Ireland can deliver a compliant, efficient and cost-effective solution to these regulatory challenge for sponsors when considering/ building their clinical study plan.
Through the Northern Ireland Protocol and subsequent Windsor framework, a QP in Northern Ireland can certify and release clinical material to the EU and UK, while maintaining compliance with the regulatory requirements for both regions. This streamlined approach enables a more efficient batch certification process, reduces costs incurred by sponsors and facilitates a more consistent, standardised approach to certification of clinical trial material.
Approached without appropriate expertise or resource, the QP release process can quickly become marred with a lack of oversight that risks non-compliance. In turn, this can create delays that compromise patient access and experience, along with broader clinical trial performance. Contrastingly, by thoroughly understanding requirements and partnering with experts, efficient and effective QP certification processes can offer sponsors significant advantage; helping to avoid compliance chaos, promote timely patient access to study drugs and expedite time to market.
Embracing a Supply with Care Ethos
The mission of delivering the right drug, to the right patient, at the right time and temperature remains the same as ever before yet the challenges standing in the way of achieving this are evolving. While industry perceptions shift to recognise patients as people with individual needs and preferences, and not just clinical trial assets, it is no longer right or effective to retrofit patients into traditional supply chain models. For clinical trials to succeed, they need patients. And for patients to participate, they need clinical trials to be built around them. Designing patient centricity into clinical trial supply chains isn’t just the right thing to do ethically but an opportunity to optimise operations, drive cost-efficiencies, and fast track time to market. By embracing key supply strategies including waste management, MAPs that utilise JTM, DTP distribution, and robust QP release processes patient experience can be enhanced, leading to better performing clinical trials and enhanced opportunity to advance global human health. For in this new era of drug development, it isn’t enough to simply supply, we must Supply with Care.
REFERENCES
1. Citeline Trialtrove, IQVIA Institute, Jan 2023.
2. Brøgger-Mikkelsen M, Ali Z, Zibert JR, Andersen AD, Thomsen SF. Online Patient Recruitment in Clinical Trials: Systematic Review and MetaAnalysis. J Med Internet Res. 2020 Nov 4;22(11): e22179. doi: 10.2196/22179. PMID: 33146627; PMCID: PMC7673977
David Ergott is the Decentralised Clinical Trial Supply Chain Solutions Manager at Almac Clinical Services. As Supply Chain Solutions Manager, David is responsible for enhancing and supporting the Almac to Patient service. This includes ensuring cohesive delivery of new services and products, maintaining positive and collaborative partnerships among supply chain stakeholders, and leading improvement efforts to ensure optimal service quality and client experience. David has 14+ years of supply chain management experience in the healthcare and pharmaceutical industries. He joined Almac in 2019 as a Supply Chain Manager where he successfully managed forecasting, setup, and end-to-end maintenance of clinical trial supply chains before transitioning to the Supply Chain Solutions manager in 2022.
Margaret Radford
Margaret Radford is the Unlicensed Medicine Services Manager at Almac Clinical Services and leads a team of experienced personnel who are responsible for the onboarding and delivery of Managed Access Programmes at all Almac sites globally. Margaret has 20 years of professional experience within Project Management, Business Development, GMP Operations, and People Management. During her time at Almac, Margaret has held various roles including Distribution Project Group Manager and Clinical Supplies Group Manager. She has a demonstrated history of working in the pharmaceutical industry, working at the forefront of strategic project planning, risk management and resource balancing to lead cross functional Teams to achieve global business goals. Margaret graduated from the University of Ulster with a Bachelor’s degree in Business Studies.
Subsection: Nasal and Pulmonary (Part A)
– one sponsor’s emergency-use product faced regulatory deficiencies due to missing Actuation Force specifications, but secured approval upon resubmission after defining and verifying these parameters.3
The Role of Automated Actuation in Nasal Product Development Regulatory agencies such as the FDA and the European Medicines Agency (EMA) mandate rigorous testing methodologies to ensure product consistency and performance. To enhance reproducibility and eliminate human variability, agencies recommend automated actuator systems, which “are expected to decrease variability in drug delivery due to operator factors, thereby increasing the sensitivity for detecting potential differences between products.” 2
Systems like Proveris Vereo Actuators provide precise and consistent control of device actuation during development testing.
Manual actuation, while not prohibited, introduces variability in critical parameters, leading to inconsistencies in product performance data. Figure 2 demonstrates the differences in variability between human actuation and the Vereo NSx actuator when used with the same device.
Although automation is essential, defining correct actuation parameters is equally critical for accurate product expulsion. Variations in stroke length, velocity, acceleration, and applied force can significantly impact CQAs, such as Aerodynamic Particle Size Distribution (APSD), Droplet Size Distribution (DSD), Spray
Regulatory & Marketplace Nasal & Inhalation
Pattern, and Plume Geometry.4,5 Even minor deviations in these parameters can lead to misleading characterisation results, creating misconceptions about product delivery and its effectiveness. Inconsistent drug delivery may affect whether patients receive the proper dose and whether the medication reaches its intended site of action.
For example, comparing Spray Pattern results from a device actuated at two different velocities across increasing formulation viscosities reveals substantial differences. A lower velocity may generate an optimal Spray Pattern for one formulation, while the same device actuated at a higher velocity fails to maintain consistency across all viscosities (Figure 3).
By implementing automated actuation with well-defined parameters, developers can enhance accuracy, reliability, and reproducibility in nasal product testing, ultimately leading to better patient outcomes.
Standardising Actuation Parameters for CQA Testing and Regulatory Success:
1. Establishing a Human-derived Actuation Profile
Human-realistic automated actuation is essential for both novel and generic drug products and is directly referenced in regulatory guidance documents, such as the FDA’s Guidance for Industry: Bioavailability and Bioequivalence Studies for Nasal Aerosols and Nasal Sprays for Local Action.2
A comprehensive human actuation study should serve as the foundation for nasal spray product development. Capturing realworld actuation parameters including stroke length, velocity, acceleration, and hold time, from a representative population informs the development of an operational range of actuation parameters. Once an optimal range of actuation parameters is established, an automated actuation method should be consistently applied across CQA testing to enhance reproducibility and in vivo correlation.
2. Implementation of Human-derived Automated Actuation in Regulatory Testing
Automated actuation based on humanderived parameters ensures consistency and reproducibility across all required performance tests. Automated actuators, such as Vereo Actuators, should be used with analytical tools like the Proveris SprayVIEW® measurement system, cascade impactors, and Malvern Spraytec instruments to improve accuracy and eliminate variability from manual testing.
Regulatory agencies require various spray characterisation and performance tests, all of which benefit from human derived automated actuation. They include:
• Spray Pattern and Plume Geometry (Proveris SprayVIEW) evaluates spray consistency and spatial drug dispersion.
• Spray Velocity and Spray Duration (Proveris SprayVIEW) determines drug dispersion over time.
Figure 3. Spray Pattern results comparison from a device actuated at two different velocities across increasing formulation viscosities.
Nasal & Inhalation
• APSD Testing (Cascade Impactors) measures particle size distribution and deposition behaviour.
• Droplet Size Distribution (DSD) Analysis (Malvern Spraytec System or similar) assesses droplet size distribution in relation to formulation properties.
• Priming and Repriming Studies evaluate spray consistency after storage or multiple actuations.
• Single Actuation Content determines dose uniformity per actuation.
• Actuation Force Testing ensures usability compliance.
3. Scale-up for QC Testing
Automated systems such as the benchtop Vereo Spray, Weigh, Collect (SWC) System and Indizo® Nasal Spray Collection System enable rapid and consistent nasal device testing, optimising both research and
development (R&D) and quality control (QC) workflows.
• Vereo SWC: An automated benchtop nasal workstation for efficient R&D and QC testing.
• Indizo System: Designed for highthroughput QC testing, automating through-life testing for up to 20 devices simultaneously.
• Optimised Laboratory Workflow: These automated systems enhance efficiency by eliminating the need for fume hoods and free analysts from repetitive and tedious tasks.
Ensuring Consistency and Regulatory Compliance Through Automated Actuation As the industry moves toward greater standardisation and efficiency in OINDP development, automation has become essential for ensuring consistent and reliable testing. Manual actuation introduces
variability that can impact dose content uniformity, aerodynamic particle size distribution, and spray characterisation, potentially affecting product performance and regulatory compliance.
By incorporating human-derived automated actuation, manufacturers can better align in vitro testing with real-world patient usage, improving the predictability and reproducibility of test results. Conducting ergonomic studies early allows for patientrelevant actuation parameters to be translated into automated systems, ensuring regulatory adherence and industry best practices from development through submission and batch release. Integrating these solutions throughout the testing process supports the development of safe, effective, and high-quality OINDPs.
REFERENCES
1. Movia, D., & Prina-Mello, A. (2020). Preclinical development of orally inhaled drugs (OIDs)— Are animal models predictive or shall we move towards in vitro non-animal models? Animals, 10(8), 1259. https://doi.org/10.3390/ani10081259
2. U.S. Food and Drug Administration. Bioavailability and Bioequivalence Studies for Nasal Aerosols and Nasal Sprays for Local Action: Guidance for Industry. April 2003
3. Center for Drug Evaluation and Research. (2021, April 23). Review of revised label and labeling (NDA 212045). U.S. Food and Drug Administration (Review)
4. Guo, C., & Doub, W. H. (2006). The influence of actuation parameters on in vitro testing of nasal spray products. Journal of Pharmaceutical Sciences, 95(9), 2029–2040.
5. Doughty, D. V., Hsu, W., & Dalby, R. N. (2014). Automated actuation of nasal spray products: effect of hand-related variability on the in vitro performance of Flonase nasal spray. Drug Development and Industrial Pharmacy, 40(6), 711–718
Grant Thurston is a Product Manager at Proveris Scientific. He is responsible for overseeing the development and enhancement of Proveris product lines, and ensuring the organisation continuously meets and exceeds customer expectations. Grant has a deep understanding of laboratory instrumentation and strong ability to communicate technical concepts effectively. Grant holds Bachelor and master’s degrees in biomedical sciences. He is fluent in English and Spanish.
Automated Analytical Instruments Accelerate Spray Characterization and Inhaled and Nasal Drug Testing
• Human-Realistic Automated Product Actuation
• Spray and Aerosol Characterization
• in vitro Measurements for Potential Clinical Biowaiver
• Formulation-Device Screening
• Reference Product Evaluation
• Efficient CQAs Assessment
Spray Pattern
Plume Geometry
Plume Front Velocity
Spray Duration
Delivered Dose Uniformity
Shot Weight
• Automation of Critical Tasks
Priming/Re-priming
Shot Weight
Pump Delivery
Dose Collection
Dose Weight
Wasting
• CFR Part 11 Compliance
• Incoming Inspection Testing for QC Release
• Scale up Manufacturing for Stability and QC Batch
• Release Testing
• Assess Product Performance Trends
• Review Critical Parameters for Enhancing Workflows
• Streamline Root Cause
• Analysis for OOS/OOT
SOLUTIONS FOR NASAL SPRAYS, pMDIS, AND SOFT MIST INHALERS
Q & A with Sarah Bunyan: Understanding and Anticipating Regulation in the pMDI Space
As sustainability pressures drive innovation in pressurised Metered Dose Inhalers (pMDIs), the regulatory landscape must shift to accommodate new low carbon technology. In a sector that has already undergone one major transition in recent decades, how are regulators, drug developers and contract development and manufacturing organisations (CDMOs) responding? Sarah Bunyan, Head of Regulatory Affairs at Bespak, explores key considerations in this space.
Q: Beginning in the 1990s, chlorofluorocarbon (CFC) propellants were transitioned to hydrofluoroalkanes (HFAs) due to concern over depletion of the ozone layer. How have pMDI regulations around propellants changed since then, and what has prompted these changes?
A: The previous shift from CFCs to HFAs was relatively slow, taking place over a 20-year period from the mid 1990s. pMDIs were already crucial devices in the inhalation space at this time, and it was clear they needed to be updated to avoid the damage to the ozone layer caused by their CFC propellants. Unfortunately, the HFA propellants that were phased in were not without their own environmental impacts. In fact, they still produce emissions today with a higher Global Warming Potential (GWP) than carbon dioxide.1 More than 30 years after the previous transition, environmental awareness has brought this issue into focus and it has become clear that we need more environmentally acceptable options.
There are several factors adding pressure to adopt new propellants. Firstly, whilst pMDIs are only responsible for around 3% of total healthcare-related emissions in the UK,2 they are seen as a relatively quick fix for the industry. As a result, there has been a push towards prescribing propellant-free options such as Dry Powder Inhalers (DPIs) for patients; however, the costs and time implications for managing a switch can be substantial.3
On top of that, much of the impetus for change is driven by the refrigerants industry,
which uses much higher quantities of HFAs than the pharmaceutical industry. Even if the pharmaceutical industry were exempt from quotas or restrictions on HFA use in pMDIs, the cost of HFA propellants would ultimately become prohibitive due to the changing nature of the supply chain.
These drivers have led us to a point where evolving pMDI regulations are now beginning to reflect the inevitable transition to low GWP propellants.
Q: How are different regulatory bodies approaching the transition to low GWP propellants? Are there any streamlined pathways?
A: One major difference is that the EU has been discussing this topic and beginning to take action over the past 3–4 years, whilst the US FDA’s approach has been more reactive, waiting for the industry to start making changes. The EMA has draft guidelines, which are regularly updated. This process has involved dialogue with the industry on what is needed to inform decision-making for clinical trials with low carbon pMDIs. In this way, the EMA has been ahead of the game, whilst up until recently the FDA has been lagging behind. However, towards the end of 2024, the FDA held a 2-day workshop with stakeholders to open up the discussion and map out next steps. This was a significant and welcome step forward, helping to provide a clearer roadmap in the transition from currently used HFAs to low GWP alternatives.
In terms of available pathways, the process will depend on the number of changes needed when updating an existing pMDI to incorporate a new propellant. If the propellant is the only change, another clinical trial may not be needed. However, as the propellant is a complex variable, it is likely to impact key product characteristics from a medicine perspective and manufacturers cannot rule out changes to device components in contact with the new propellant. In these cases, the pathway to approval of the pMDI may be considered a complex type II variation, a line extension, or even a new product from a regulatory perspective.
Q: What steps should pharma and biotech companies developing low carbon pMDIs take to be ready for evolving regulations?
A: Whilst this transition has been initiated by evolving environmental legislation, now that the topic has been brought to the fore, it is a highly collaborative process between regulators and key industry players to pave the path forward. A key step for drug developers will be proactively participating in the conversations that are shaping the regulatory framework. This means engaging with industry forums and workshops – both from the perspective of staying informed, but also in order to input into the decisions made. Membership of key industry groups, such as the International Pharmaceutical Aerosol Consortium on Regulation & Science (IPAC-RS), can be helpful in achieving collaborative input. The industry is stronger when it is united and encourages experts with hands-on experience to work with regulatory authorities on aspects of the new guidelines that they are best placed to understand the impact of. Individual companies commenting and sharing feedback on Q&As and guidance documents is useful to an extent, but bringing together multiple players in the industry in a joint response adds a lot more weight.
Q: What is the role of CDMOs in these regulatory conversations?
A: I personally think CDMOs are incredibly important in both shaping and enacting regulations – they are able to act as supporters for their customers and advocates in the industry due to their significant experience and unique perspectives. And because CDMOs have so many clients, they need to keep their finger on the pulse of every small change. For example, whilst a marketing authorisation holder (MAH) might have two pMDIs in the pipeline, a CDMO could have 20 or 30. This element of scale means that CDMOs can gain experience and insight into technological developments and changing regulations quickly, and can learn from each process iteratively. The key point overall is that this experience allows them to be more proactive for their clients, identifying issues ahead of time and helping to prepare for how the regulations might impact product development and commercialisation processes.
When it comes to regulatory submission, I see CDMOs as the experts in their processes, such as development, manufacturing and testing on incorporating propellants into pMDIs. It can therefore be beneficial to get these CDMO experts to write many of the sections of the submission. Again, because of the experience and numbers of submissions these experts have worked on, it can be far quicker and ultimately more cost-effective to work with a CDMO on a submission.
Q: How do you see pMDI regulations evolving over the next 5–10 years?
A: I anticipate that, ultimately, as technology develops, regulations will evolve to support a transition from low GWP to ‘no GWP’, bringing about a further generation of pMDIs with an even smaller environmental impact. Given that it took 50–60 years to identify the need to move away from CFCs, and 30 years for HFAs, we cannot be sure when further changes will happen, but I am confident that this will not be the last pMDI transition.
A further transition won’t necessarily just be a case of developing new propellants. Of the two current low GWP alternatives, HFO1234ze has a near-zero GWP of ~1, whereas HFA-152a has a GWP of ~138. Based on these metrics, HFO-1234ze is a much stronger candidate under F-gas regulation; however,
it may fall under per-and polyfluoroalkyl substances (PFAS) restrictions.1 This is because it may be a potential precursor to PFAS compounds when it degrades, which can build up into toxic quantities in the environment. There are efforts underway to fully understand this process, which may lead to an exemption for HFO-1234ze from PFAS regulations. So, this is a challenge that the industry is currently facing, and one that may impact the future of zero or near-zero GWP propellants.
In addition to this, I think we will see a much bigger drive towards reducing the overall environmental impact of inhalers including pMDIs, but also DPIs and Soft Mist Inhalers (SMIs). This may manifest in incentives to return used devices for recycling, better recycling practices, and more importance placed on the choice of materials in the device. It will be exciting to see the ongoing relationship between industry and regulators as demand and technology evolve to help reduce the environmental footprint of vital respiratory medicines.
REFERENCES
1. Tewari, SG, Bell, JP, Budgen, N, Platz, S, Gibbs, M, Newham, P, Kimko, H. (2023). Pressurized metered-dose inhalers using next-generation propellant HFO-1234ze(E) deposit negligible
amounts of trifluoracetic acid in the environment. Frontiers in Environmental Science, 11. doi:https://doi.org/10.3389/ fenvs.2023.1297920.
2. Fidler, L, Green, S, Wintemute, K. (2022). Pressurized metered-dose inhalers and their impact on climate change. Canadian Medical Association Journal, 194(12), pp.E460–E460. doi:https://doi.org/10.1503/cmaj.211747.
3. Attar-Zadeh, D, Lewis, H, Orlovic, M. (2021). Health-care Resource Requirements and Potential Financial Consequences of an Environmentally Driven Switch in Respiratory Inhaler Use in England. Journal of Health Economics and Outcomes Research, 8(2). doi:https://doi.org/10.36469/001c.26113.
Sarah Bunyan is a regulatory affairs professional with over 30 years of international experience within the pharmaceutical and biopharmaceutical industry. She has held senior positions in QC, QA and validation, and for over 20 years has held positions of increasing seniority in Respiratory Regulatory Affairs. She is currently Head of Regulatory Affairs and Compliance at Bespak.
The global scientific community continues to advance research on COVID-19 and its long-term effects, commonly referred to as “long COVID.” While the pathogenicity of COVID-19 has weakened due to viral mutations, the importance of infection prevention remains paramount. Repeated infections elevate the risk of long COVID, underscoring the need for sustained vigilance.
Beyond COVID-19, seasonal influenza remains a recurring public health challenge, leading to unpredictable pandemics and posing heightened risks for pregnant women, infants, the elderly, and individuals with chronic illnesses. These vulnerable populations are particularly susceptible to respiratory infections, making preventive measures a continued priority.
Given that nasal passages process thousands of litres of air daily, they act as the first line of defence against airborne pathogens. As a result, their high susceptibility to infections from influenza viruses, coronaviruses, rhinoviruses, and airborne allergens makes effective protective measures essential.1,2
While face masks remain the gold standard for respiratory protection, discomfort and compliance issues have led to growing interest in nasal spray-based protection. Often referred to as “liquid masks,” these sprays do not replace masks but instead provide an additional protective layer.
Two primary types of nasal sprays have been developed:
• Physical Barrier Sprays – These create a protective layer that prevents pathogens from entering the respiratory tract.
• Neutralising Sprays – These contain pharmaceutical ingredients or agents designed to deactivate or disrupt viruses upon contact.
Physical barrier sprays are particularly advantageous due to their broad-spectrum protection. Unlike pharmaceutical interventions that target specific pathogens, “these sprays work by forming a mechanical shield, maintaining the natural integrity of the nasal environment. This makes them an effective safeguard against both emerging viruses and environmental allergens.” said Professor Liam Grover, the Founder-Director of the Healthcare Technologies Institute (HTI) at the University of Birmingham, who is a coinventor of NoriZite Nasal Spray, a safe, nonmedicated physical barrier nasal spray, that was introduced to the market by Birmingham Biotech LTD in 2022 during COVID.
Challenges in Nasal Spray Development
The COVID-19 pandemic led to the rapid development of various nasal protection formulations. However, early research identified challenges in certain formulations that altered the nasal mucosa. Some relied on acidification or the introduction of free radicals, which, despite potential antiviral efficacy, were not widely adopted due to discomfort among users.
Discussions with frontline clinicians and healthcare professionals revealed that “the main discomfort caused by these sprays was the unpleasant sensation” Professor Grover commented. Some sprays were too thin, leading to rapid dripping and inadequate protection, while others were too thick, resulting in a concentrated “jet” effect with minimal coverage. These challenges prompted researchers to refine nasal spray technology, optimising both viscosity and sprayability.
Advances in polymer science and biomaterials research have been instrumental in addressing these issues. By balancing mist distribution and retention on the nasal mucosa, some new formulations have been shown high levels of surface coverage compared to conventional sprays. In laboratory studies,3 these advances have also demonstrated potential in inhibiting SARS-CoV-2 infection, reinforcing their role in respiratory virus prevention.4
The scaling of production, risk assessments, and regulatory approvals has been
essential to ensuring that effective nasal protection solutions are widely accessible. The rapid development of such technologies, while maintaining rigorous scientific validation, has been a key focus in ensuring their success.
As respiratory health remains a public health priority, continued research is focused on expanding access and improving the scientific understanding of nasal protection mechanisms.
Expanding Horizons: Applications Beyond COVID-19
While nasal protection technologies were initially designed for viral infection prevention, their broad-spectrum physical barrier properties have led to increased applications beyond infectious disease control. Emerging uses include:
• Allergy Prevention – Providing a protective barrier against pollen, dust, and airborne pollutants such as PM2.5 particles.
• Travel Comfort – Reducing nasal dryness and irritation during long-haul flights.
• Respiratory Defence – Supporting individuals exposed to high-risk environments, such as healthcare workers and commuters.
To uphold scientific integrity, ongoing research continues to explore the mechanisms of action in these expanded applications. Clinical studies are also assessing the effectiveness of nasal sprays in reducing allergen exposure and improving comfort in varying environments.5,6
Recognising the growing impact of seasonal and environmental allergies, researchers are also developing a nextgeneration anti-allergy nasal spray. This formulation aims to create a physical barrier against allergens, such as pollen, pet dander, and air pollutants, preventing them from triggering allergic reactions.
Key features of next-generation nasal protection solutions include:
Regulatory & Marketplace Nasal & Inhalation
• Barrier Technology: Forms a protective shield over the nasal mucosa to block allergens.
• Long-lasting Protection: Reduces the need for frequent reapplication.
• Non-medicated and Safe: Free from steroids and antihistamines, making it suitable for long-term use.
As interest in non-pharmaceutical approaches to allergy management grows, researchers are conducting clinical trials to assess the effectiveness of nasal sprays in reducing allergen exposure.
The Future of Nasal Protection: A Mainstream Public Health Strategy As nasal protection solutions continue to evolve, regulatory challenges must be
addressed to ensure safe and effective use. Establishing standardised guidelines for approval and commercialisation remains crucial.7,8
Despite regulatory hurdles, demand for effective respiratory protection solutions continues to grow, with nasal sprays emerging as a mainstream component of infection control and allergy prevention.
By integrating nasal protection strategies into global public health efforts, it is possible to significantly reduce the burden of airborne diseases and allergens, leading to a healthier global population.
Final Thoughts: Innovation Through Science and Social Impact
The advancement of nasal protection technologies demonstrates the power of scientific innovation in solving realworld health challenges. Professor Grover shared his three core R&D principles for formulations development.
1. Continuous Learning – Staying informed through extensive reading ensures awareness of emerging knowledge and methodologies.
2. Open-mindedness – Welcoming diverse perspectives fosters objectivity and innovation.
3. Constructive Impatience – Pushing disciplinary boundaries and challenging conventional silos accelerates problemsolving and discovery.
Through continued research, collaboration, and commitment to evidence-based solutions, the field of nasal protection continues to evolve. As scientists, clinicians, and engineers push the boundaries of preventive healthcare, nasal protection strategies will play a critical role in public health efforts worldwide.
Michael Hsu is the founder and CEO of Birmingham Biotech LTD. In partnership with the University of Birmingham, Birmingham Biotech is committed to ensuring that patients in developing countries have access to the latest highquality, affordable healthcare delivered through its rapid global supply chain. The company has developed NoriZite Nasal Spray, a scientifically backed formulation designed to provide a protective barrier in the nasal cavity, helping to trap and block virus particles, thereby reducing the risk of respiratory infections.
Michael Hsu
•
• Biocides
•
IFC
IBC
Page 45
Page 17
Page 70
Page 31
BC
Page 49
Page 51
Page 3
Page 5
Page 29
Page 69
Page 63
Page 68
Page 71
Page 33 & 47
Subscribe today at www.international-pharma.com or email info@senglobalcoms.com
Aptar Group Inc.
Bespak Ltd.
Biopharma Group Limited
Carterra Inc.
Chemspec Europe 2025
Christ Packing Systems GmbH & Co. KG
EyeC GmbH
FUJIFILM Irvine Scientific Inc.
Krautz Temax
Nipro
Novo Nordisk A/S
PCI Pharma Services
Pre-filled Syringes and Injectable Drug Devices (SAE Media Group)
Proveris Scientific
SMI London 2025
The Pharma Days & The Pharma Sustainability Days
Valsteam ADCA
I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry
IPI is also now active on social media. Follow us on: