15 minute read
Mutant p53 and its Isoforms in the Onset & Treatment of Breast Cancer Brooklyn Schroeder '22


Mutant p53 and its Isoforms in the Onset & Treatment of Breast Cancer
BY BROOKLYN SCHROEDER ’22
Advertisement
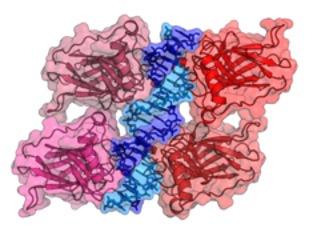
Cover Image: P53 is a tetramer tumor suppressor protein. The above depicts the crystal structure of the p53 binding domains which are attached to the respective DNA binding site. Image Source: Wikimedia Commons Introduction
The molecular mysteries of tumor onset and progression and their role in the development of a variety of cancers still perplex the modern world of science. Breast cancer, the most frequently diagnosed cancer in women in the United States, is no exception to this enigma. The manner in which a breast tumor develops is unique to a case-by-case basis. With this being said, regulation of cellular pathways, and especially their tumor suppressing properties, are critical in preventing this onset. The p53 protein, a tumor suppressor protein, is an essential transcription factor encoded by the TP53 gene and is essential in the regulation of a variety of cellular processes associated with apoptosis (a form of programmed cell death), cellular proliferation, cellular senescence (the ceasing of cell division), and also plays a critical role in DNA replication and repair activity. Most notably, in consideration of the development of cancerous tumors, especially in light of breast cancer, mutations in the p53 protein are characteristic of a considerable fraction of breast cancer cases (Ziyaie et al., 2000). The p53 protein is a vitally important point of molecular focus in recognizing the onset of tumor development and can therefore be a biological marker for breast cancer treatments.
p53 Structure & Function
The molecular structure of the p53 protein gives it characteristic function in the regulation of cell cycle arrest, DNA repair, and apoptotic pathways. By dissecting the structure of the p53 protein and its various domains, one can further recognize the extent to which this protein becomes nonfunctional by structural mutation. The p53 protein contains 393 amino acids, which are divided into various domains: the functional domain, which consists of a specific region called the transactivation region, a sequence-specific DNA binding region, oligomerization region, and a nuclear localization sequence (Harris, 1996). The other two key domains are categorized as evolutionary conserved domains deemed to be hotspots of mutational events (Harris, 1996).
As previously mentioned, the p53 protein is an essential component within cell cycle checkpoints and apoptotic pathways. Depending on the type of signaling, p53 can respond in various ways. The p53 protein plays an essential role in controlling the transition from G1/S to G2/M phases in the cell cycle in addition to its role in DNA repair, cellular senescence, apoptosis, autophagy and mitotic catastrophe (Varna et al., 2011). However, to focus upon the implications of such processes on oncogenic activity, one must identify the importance of p53 activation in inducing cell cycle arrest and the process by which damaged DNA is repaired. One critical target of p53 is p21, which binds to cyclin E/ Cdk2 and cyclin D/Cdk4 complexes to cause G1 arrest in the cell cycle (Chen, 2016). As a result of inhibited Cdk2 and Cdk4, pRb is unable to be phosphorylated, and therefore binds to E2F1, which silences its downstream targets that are essential for the process of DNA replication and progression through the cell cycle (Chen, 2016). In the case of DNA damage, a variety of protein kinases, whether that be Cdc2, protein kinase C, casein kinase II, etc., become activated and can then phosphorylate the p53 protein (Maclaine & Hupp, 2009). The phosphorylation of p53 allows it to act as an active, stable transcription factor capable of binding upstream regulatory regions of various genes and promoting their transcription, while non-phosphorylated p53 would be targeted for degradation by its interaction with the protein MDM2 (Maclaine & Hupp, 2009). In summary, it becomes evident that as long as there is damage present, a cascade of events beginning with the phosphorylation of p53 and the subsequent translation of p21 will ensure the cell cycle does not proceed into the S phase until any detectable damage is repaired (in which case, this described signaling pathway would not be active). In relevance to carcinogenesis, if there was a case in which p53 is mutated, the cell cycle would then proceed into S phase with the DNA damage present. This would result in proliferation of this mutation, which not only amplifies this damage but can also lead to a variety of new mutations.
In the context of generalized mutations, there is no case in which one mutation accurately depicts all that occur. Rather, within the p53 protein there are a variety of mutations that can occur, and the extent to which they contribute to carcinogenesis can either be attributed to loss of function (LOF) or gain of function (GOF) characteristics. LOF mutations include mutations "it becomes evident that as long as there is damage present, a cascade of events beginning with the phosphorylation of p53 and the subsequent translation of p21 will ensure the cell cycle does not proceed into the S phase until any detectable damage is repaired"
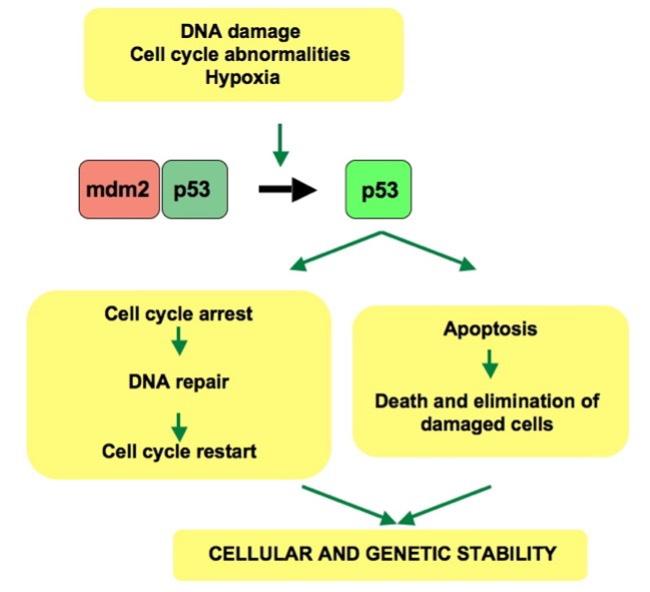
Figure 1: The activated p53 protein plays a critical role in establishing the outcome of the cell cycle arrest and apoptosis processes. This activated p53 protein depends heavily on the relationship between p53 and mdm2.
Image Source: Wikimedia Commons
that diminish p53’s ability to bind DNA sequence motifs, which are key in directing transcription of genes regulated by p53 (Baugh et al., 2018). The idea that mutant p53 proteins could actually acquire GOF capabilities was not discovered until much after those LOF cases. A study conducted by Dittmer et al. in 1993, om which cDNA was added to a TP53 mutant, revealed this acquisition of oncogenic activity in mice, such as more rapid cell proliferation, loss of contact inhibition activities, larger tumorigenic potential, and larger tumor outcomes (Baugh et al., 2018).
DNA sequencing has revealed 74% of p53 mutations in cancers to be missense mutations which most notably occur within the protein’s centralized DNA binding domain (amongst amino acids 102-292), in which more of 30% of said mutations occur in the aforementioned “hotspot” regions of the p53 protein (Goh et al., 2011). Nearly 10% of p53 mutations are LOF, and result in no protein being translated at all (Baugh et al., 2018).
As previously stated, there are a wide array of outcomes that can occur as a result of the p53 mutant, and the majority of such possibilities are strategically outlined in the study conducted by Muller & Vousden, 2013, in which four overarching models are: 1) alterations in the DNA binding abilities of p53 2) changes in the interaction of mutant p53 with other proteins 3) changes in the interaction of mutant p53 with other transcription factors and 4) proteins not related to the general regulation of gene expression (Muller & Vousden, 2013).
It is universally acknowledged that carcinogens, aside from being genetically predisposed, bear momentous implications upon the potential of carcinogenesis and oncogenic activity in humans. Carcinogens, which are agents capable of causing cancer in human tissues, hold a notable relationship with mutations in the p53 protein. To illustrate this relationship, let us consider the most commonly recognized carcinogens -- aflatoxin B1, tobacco smoke, and UV radiation associated with sun exposure. In the case of aflatoxin B1, multiple studies have shown evidence for aflatoxin B1 to cause a G:C to T:A transversion within codon 249 of the p53 gene (Rivlin et al., 2011). Similarly, tobacco smokers are frequently found to have an abundance of G to T transversions, while UV radiation exposure induced C to T and CC to TT transitions within nearly 50% of cases (Rivlin et al., 2011). Needless to say, there is indeed an evident correlation between carcinogens and p53 protein mutations. The TP53 mutations, specifically missense mutations, are the most common mutation in human cancers (Baugh et al., 2018). This review will take a concentrated view looking at the frequency and implications of these previously discussed p53 mutations in breast cancer (Bellazzo et al., 2018). Interestingly, although much discussion has been given to missense mutations in light of p53 due to their frequency in nature, germ-line mutations in p53 occur most frequently in humans with Li-Fraumeni cancer susceptibility syndrome, which actually bestows a higher risk of developing breast cancer (Gasco et al., 2002).
The prevalence of p53 mutations in breast cancer, at 20%, is not necessarily considerable (Gasco et al., 2002), but these mutations have significant ties to particular germ-line meaning they have the capability to be passed on to progeny. Numerous recent studies over the course of the last decade have recognized increasing rates of p53 mutations in cancers specific to carriers of germ-line BRCA1 & BRCA2 mutations – genes that are of utmost focus in breast cancer discussions. In fact, modern biological study has deemed BRCA1 as “the breast cancer gene” (Gasco et al., 2002).
We have seen the general consequences of p53 alteration in cancer, and these observations remain consistent with breast cancer as well; specifically, loss of p53 wildtype activity has been posited as a primary element in breast cancer onset (Ziyaie et al., 2000). Most, notably though, what differentiates p53 mutations in breast cancers, versus those of other human cancers, is their ability to radically alter the severity and outcomes of breast cancer patients. Gasco et al. revealed that those p53 mutations acting directly upon amino acids essential for DNA binding, were most prevalent in the most aggressive cases of cancer (2002). Missense mutations and other null mutations were more closely associated with an undetermined phenotype (Gasco et al., 2002). In essence, though, the importance of a clear-cut focus upon p53 mutations and their ubiquity in human breast cancers is the general association of such mutations with worse survival rates amongst these individuals.
p53 Isoforms in Breast Cancer
The modern field of breast cancer oncology has paid a great deal of attention toward expression of p53 isoforms proteins that have comparable functionality but differ in their amino acid sequencing) , and the varying extent to which these isoforms contribute to the regulation of
normal-functioning cells and cells with oncogenic activity in response to stressors (Kim & An, 2016). The TP53 gene undergoes translation to encode 12 distinguished p53 isoforms. These isoforms are produced through varying means: alternative promoter usage, alternative splicing and alternative initiation of translation (Avery-Kiejda et al., 2014). Milićević et a.l (2014) concluded that ΔNp53 (47 kDa) and Δ133p53�� (35 kDa) (which are dominant-negative repressors of p53 function) were the most frequently occurring isoforms in invasive breast carcinoma cells, and Avery-Kiejda et al. (2014) found Δ40p53 to be the most expressed p53 isoform within breast cancer. Indeed, Milićević et al. used thirty random breast tumors for analysis of p53 isoforms through use of a classic Western blot analysis, which revealed that none of the tumors had the same p53 isoform expression pattern (2014). These results can contribute to the general conclusion of a multitude of studies that the implications of p53 upon molecular processes in breast cancer is still not completely known. However, with data supporting particular isoforms to be highly expressed compared to others, this provides a starting point for further focus within p53 mutant studies and target treatment in breast cancers in the future.
Targeting p53 for Treatment of Breast Cancer
Since the p53 mutation has been consistently identified across numerous forms of human cancers, it is a reasonable area of focus in terms of biomarkers for cancer treatments. The unique fact about p53, and its complex nature, is that it had been considered “undruggable” until recent years. Since then, there have been a couple notable compounds that are believed to have a potential impact on mutant p53 -- PRIMA-1 and APR-246 (a methylated derivative and structural analogue of PRIMA-1) (Duffy et al., 2017). Both compounds reactivate mutant p53, allowing it to regain its wild-type characteristics. Aside from the general acquisition of wildtype properties, other approaches for restoring function in p53 entail depletion of the mutation p53 altogether, blocking negative regulator proteins MDM2 and MDM4, and treatments that promote close assessment of premature termination codes (Duffy et al., 2017).
The revolutionary characteristic of PRIMA-1 in its ability to restore the wildtype function of p53 is restoration of sequence-specific DNA binding and ability to induce the apoptotic pathway (Duffy et al., 2018). The overarching outcome to focus on with PRIMA-1 is the biological evidence supporting its ability to inhibit p53 mutant breast cancer cells. Similarly, APR-246 is also characteristic of inhibiting excessive cell proliferation and promoting the occurrence of the apoptotic pathway (Duffy et al., 2014). Given the recent discovery that p53 is not in fact undruggable, there are minimal options in terms of chemicals to specifically target p53 mutants (Fedorova et al., 2020). PRIMA-1 and APR-246 are considered as the first mutant p53 reactivating compounds that progressed to clinical trials (Duffy et al., 2017). On a broader scale of common cancer treatments, it has been consistently concluded that p53 mutations are frequently associated with poor responses to the realms of chemotherapy, hormonal therapy and radiotherapy (Varna et al., 2011).
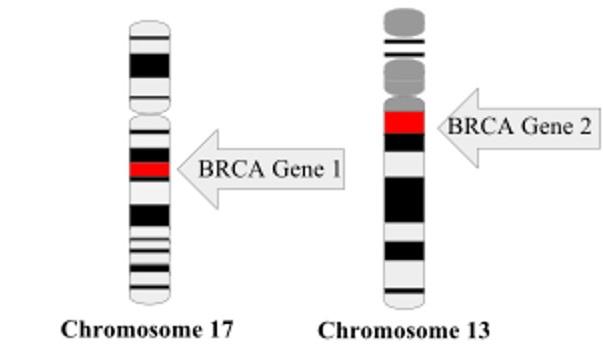
Figure 2: There are increasing cases of p53 mutations within carriers of germline BRCA 1 and BRCA 2 mutations. BRCA 1 has most recently been recognized to be the “breast cancer gene”. Image Source: Wikimedia Commons
While on a surface level it seems unfathomable that there are infinite layers in terms of targeting p53 as a biomarker for cancer therapies, there is immeasurable complexity associated with this protein and its mutant form(s). In the case that a given chemical is able to target mutated p53, it shall still be considered that such findings may be confined to those bearing very specific molecular subtypes of breast cancer or to those receiving this chemical in addition to a specific form of cancer therapy (Duffy et al., 2017). With this being said, as has been previously determined, individuals may have their p53 mutation present on a different region of the TP53 gene, which then makes it that much more specified in discerning a targeting therapy. In addition to this, the isoforms of mutated p53 add another level of specificity to breast cancer patients. In a study conducted by Marcel et al., their findings suggest that in the presence of p53γ, but not p53β in breast cancer patients, there are comparable survival rates overall compared to those with wildtype p53 (2011). However, in another study, results show that expression of p53β but not the p53γ isoform was discovered to be protective in patients with p53 mutant tumors (Avery-Kiejda et al., 2014).
Conclusion
Although recognized as the most prevalent mutation in human cancers, there is evidently still a great deal of complexity associated with the mutated p53 protein. Especially in light of breast cancer, there are a multitude of factors that go alongside these aforementioned p53 mutations as they can occur at varying locations along the TP53 gene. Not only this, but the twelve isoforms of the p53 protein make the approach to potential treatments and therapies that much more difficult given their respective specificities. However, there is a promising future in terms of targeting p53 as a biomarker in breast cancer treatments given the recent rise of compounds to reactivate p53 mutant protein to regain its wildtype properties.
References
Avery-Kiejda, K. A., Morten, B., Wong-Brown, M. W., Mathe, A., & Scott, R. J. (2014). The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis, 35(3), 586–596. https://doi. org/10.1093/carcin/bgt411
Baugh, E. H., Ke, H., Levine, A. J., Bonneau, R. A., & Chan, C. S. (2018). Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death & Differentiation, 25(1), 154–160. https://doi.org/10.1038/cdd.2017.180
Bellazzo, A., Sicari, D., Valentino, E., Sal, G. D., & Collavin, L. (2018). Complexes formed by mutant p53 and their roles in breast cancer. Breast Cancer: Targets and Therapy, 10, 101–112. https://doi.org/10.2147/BCTT.S145826 Chen, J. (2016). The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harbor Perspectives in Medicine, 6(3). https://doi.org/10.1101/ cshperspect.a026104
Duffy, M. J., Synnott, N. C., & Crown, J. (2018). Mutant p53 in breast cancer: Potential as a therapeutic target and biomarker. Breast Cancer Research and Treatment, 170(2), 213–219. https://doi.org/10.1007/s10549-018-4753-7
Duffy, M. J., Synnott, N. C., McGowan, P. M., Crown, J., O’Connor, D., & Gallagher, W. M. (2017). P53 as a target for the treatment of cancer. Cancer Treatment Reviews, 40(10), 1153–1160. https://doi.org/10.1016/j.ctrv.2014.10.004
Fedorova, O., Daks, A., Shuvalov, O., Kizenko, A., Petukhov, A., Gnennaya, Y., & Barlev, N. (2020). Attenuation of p53 mutant as an approach for treatment of Her2-positive cancer. Cell Death Discovery, 6(1), 1–8. https://doi.org/10.1038/s41420-020-003374
Gasco, M., Shami, S., & Crook, T. (2002). The p53 pathway in breast cancer. Breast Cancer Research: BCR, 4(2), 70–76. https://doi.org/10.1186/bcr426
Goh, A. M., Coffill, C. R., & Lane, D. P. (2011). The role of mutant p53 in human cancer. The Journal of Pathology, 223(2), 116–126. https://doi.org/10.1002/path.2784
Harris, C. C. (1996). Structure and Function of the p53 Tumor Suppressor Gene: Clues for Rational Cancer Therapeutic Strategies. JNCI: Journal of the National Cancer Institute, 88(20), 1442–1455. https://doi.org/10.1093/jnci/88.20.1442
Kim, S., & An, S. S. A. (2016). Role of p53 isoforms and aggregations in cancer. Medicine, 95(26), e3993. https://doi. org/10.1097/MD.0000000000003993
Maclaine, N. J., & Hupp, T. R. (2009). The regulation of p53 by phosphorylation: A model for how distinct signals integrate into the p53 pathway. Aging, 1(5), 490–502.
Marcel V, Dichtel-Danjoy ML, Sagne C, Hafsi H, Ma D, Ortiz- Cuaran S, Olivier M, Hall J, Mollereau B, Hainaut P, Bourdon JC (2011). Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ 18:1815–1824
Milićević, Z., Bajić, V., Živković, L., Kasapović, J., Andjelković, U., & Spremo-Potparević, B. (2014). Identification of p53 and Its Isoforms in Human Breast Carcinoma Cells. The Scientific World Journal, 2014, e618698. https://doi. org/10.1155/2014/618698
Muller, P. A. J., & Vousden, K. H. (2013). P53 mutations in cancer. Nature Cell Biology, 15(1), 2–8. https://doi.org/10.1038/ ncb2641
Rivlin, N., Brosh, R., Oren, M., & Rotter, V. (2011). Mutations in the p53 Tumor Suppressor Gene. Genes & Cancer, 2(4), 466–474. https://doi.org/10.1177/1947601911408889
Varna, M., Bousquet, G., Plassa, L.-F., Bertheau, P., & Janin, A. (2011). TP53 Status and Response to Treatment in Breast Cancers. Journal of Biomedicine and Biotechnology, 2011, e284584. https://doi.org/10.1155/2011/284584
Ziyaie, D., Hupp, T. R., & Thompson, A. M. (2000). P53 and breast cancer. The Breast, 9(5), 239–246. https://doi.org/10.1054/ brst.2000.0199






