10 minute read
Reawakening Apoptosis in Cancer Cells: The advent of BH3-mimetic drugs


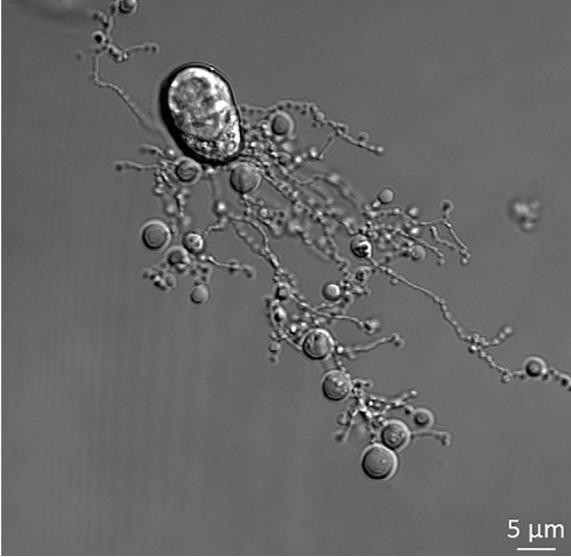
Figure 1. Human THP-1 monocytic cells generating beaded-apoptopodia to regulate the formation of apoptotic bodies. Nowak-Sliwinska P., & Griffioen A. W. (2018). “Apoptosis on the move.” Apoptosis, 23(5-6):251-254.
The uniquely fatal nature of cancer lies in unhindered proliferation, which bypasses the apoptotic machinery within the cells. Apoptosis refers to programmed cell death, a form of ‘cellular suicide’ that is regulated by altering the activity of specific proteins. Apoptosis is an orderly process that is initiated by the permeabilization of the cell’s plasma membrane, which leads to fragmentation of its DNA by activated caspase cascades and ultimately, cell death (Renehan et al., 2001). Without apoptosis, cells can continue to proliferate and grow at uninhibited and potentially threatening rates. There are two widely studied pathways for apoptosis. One is the extrinsic pathway, where an extracellular ligand binds to transmembrane cell-death receptors to stimulate apoptosis. The other is the intrinsic pathway, which involves directly modifying mitochondrial membranes by activating certain conserved signaling pathways within the cell, ultimately inducing caspase activity. An oncogene refers to any gene which can encode a protein that is able to induce cancer within animals. A subsection of oncogenes, termed proto-oncogenes, are derived from mutations in normal cellular genes. The products of proto-oncogenes are directly involved in pathways that control cellular growth; for example, a mutant form of the normal ras proto-oncogene, the rasD oncogene, encodes an oncoprotein which directly promotes uninhibited cellular growth. Since the first discovery of an anti-apoptotic oncogene, BCL-2 (B-cell lymphoma 2), researchers have developed an entirely new class of pharmacological small molecule therapeutics. Cancer cell proliferation is maintained by forced expression of anti-apoptotic BCL-2 proteins, among various other anti-apoptotic proteins, which defend malignant cells from pro-apoptotic pressure. The significance of the equilibrating interactions between antiapoptotic and pro-survival BCL-2 proteins in cancerous cells have given rise to an increasing number of therapeutic agents, such as the dual BCL-2/ BCL-XL inhibitor Navitoclax. The inhibition of anti-apoptotic, pro-survival BCL-2 proteins and the activation of their pro-apoptotic counterparts in early stage tumorigenesis is an area of increasing interest and study. The cell life cycle is governed by the interactions of two broad classes of BCL-2 proteins: pro-apoptotic and pro-survival [Figure 2]. A third class of BCL-2 proteins, the BH3-only proteins, dictate the function of these two BCL-2 proteins types through selective binding, thus regulating the apoptotic commitment of the cell via mitochondrial membrane permeabilization. BH3-only proteins are highly specific, and differential protein–protein binding between pro-survival "Apoptosis refers to programmed cell death, a form of ‘cellular suicide’ that is regulated by altering the activity of specific proteins."
Advertisement
38 | Emory Undergraduate Research Journal
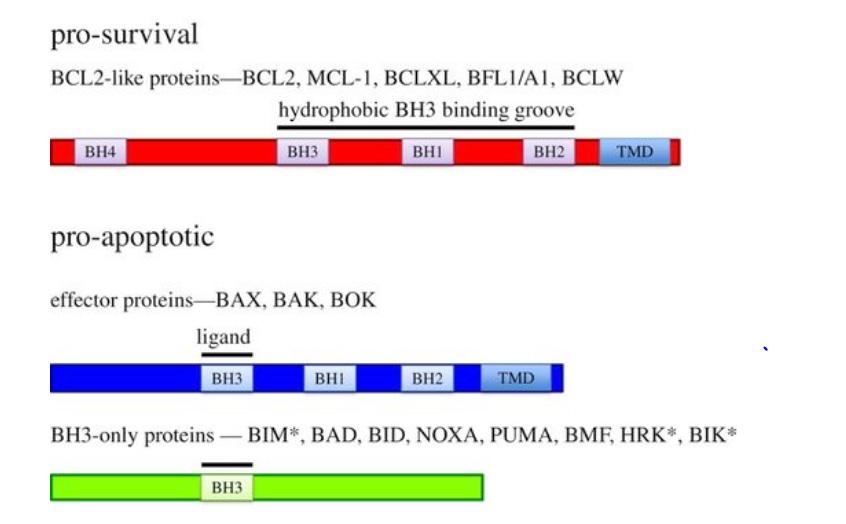
Figure 2. Classes of proteins within the BCL-2 family; these proteins can be classified mainly into pro-survival proteins, proapoptotic proteins, and a further subdivision between the multiBH domain termed as BH3-only proteins, which is similar to the homology of BCL-2 only due to the presence of BH3 effector proteins.
and anti-apoptotic BCL-2 family proteins dictates the ability of pro-apoptotic proteins, particularly BAK and BAX, to induce an apoptotic response through outer membrane permeabilization (Merino et al., 2018). Studies conducted by Liu et al., have demonstrated that apoptosis originates in the mitochondria, with several mitochondrial proteins implicated to directly activate an apoptotic cascade within cells (Liu et al., 1996). These proteins usually occupy the intermembrane space of mitochondria, but in response to apoptotic stimuli are released into the cytosol. There, they promote apoptosis via two complementary and overlapping pathways. The first pathway activates nucleases, or enzymes that cleave phosphodiester bonds between nucleotides of DNA, and caspases, a class of proteases that mediate programmed cell death. The second pathway neutralizes apoptotic inhibitors already present in the cytosol. From these pathways, the BH3-only class of proteins are transcriptionally generated in response to cellular stressors such as oxidative or metabolic stress. They initiate apoptosis either by binding to pro-survival BCL-2 proteins and unshackling BAX/ BAK protein activity, or by directly activating the pro-apoptotic class of proteins and thus, cell death. Unconstraining BAX/BAK leads to the formation of oligomers that form pores to perforate the outer mitochondrial membrane. This induces a cascade of caspase activ
Campbell KJ, & Tait SWG (2018). “Targeting BCL-2 regulated apoptosis in cancer.” Open Biol, 8(5).
Figure 3. Classes of proteins within the BCL-2 family; these proteins can be classified mainly into pro-survival proteins, proapoptotic proteins, and a further subdivision between the multiBH domain termed as BH3-only proteins, which is similar to the homology of BCL-2 only due to the presence of BH3 effector proteins.
Timucin AC, Basaga H, Kutuk O. (2019). “Selective targeting of antiapoptotic BCL-2 proteins in cancer.” Med Res Rev, 39(1):146-175.
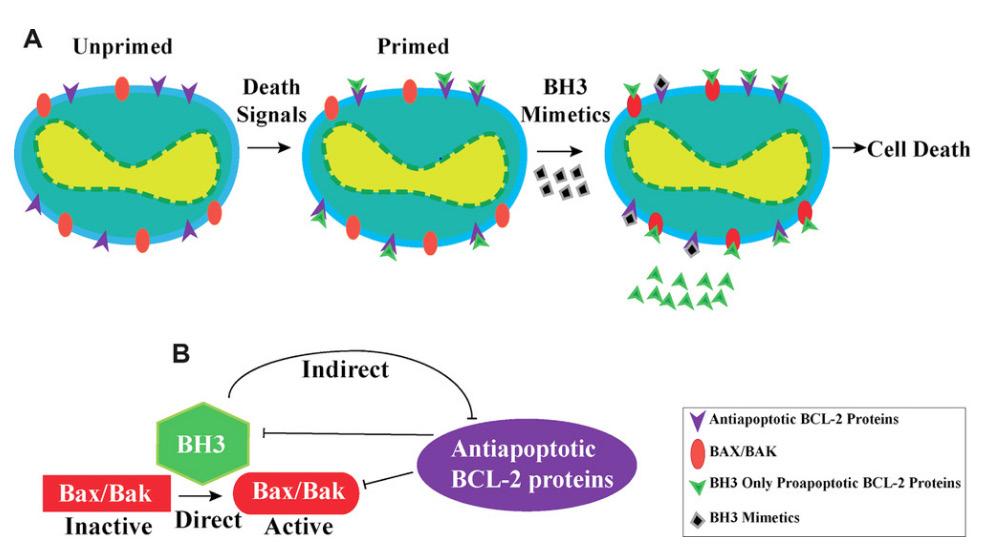
ity by releasing pro-apoptotic proteins into the cytosol, ultimately leading to cell death. The equilibration between the binding of pro-survival and pro-apoptotic members of the BCL-2 family to BAX and BAK is therefore essential in deciding between cell death and survival. The binding system of the BH3-like pro-survival proteins is crucial to initiate efficient cell death. A cell is “primed for death” by death signals, a process wherein its anti-apoptotic BCL-2 protein receptor sites become primarily occupied by pro-apoptotic BCL-2 proteins instead (Certo et al., 2004). As the number of available BH3-binding sites has been altered on the anti-apoptotic BCL-2 protein, the
Volume XVI, Spring 2020 | 39
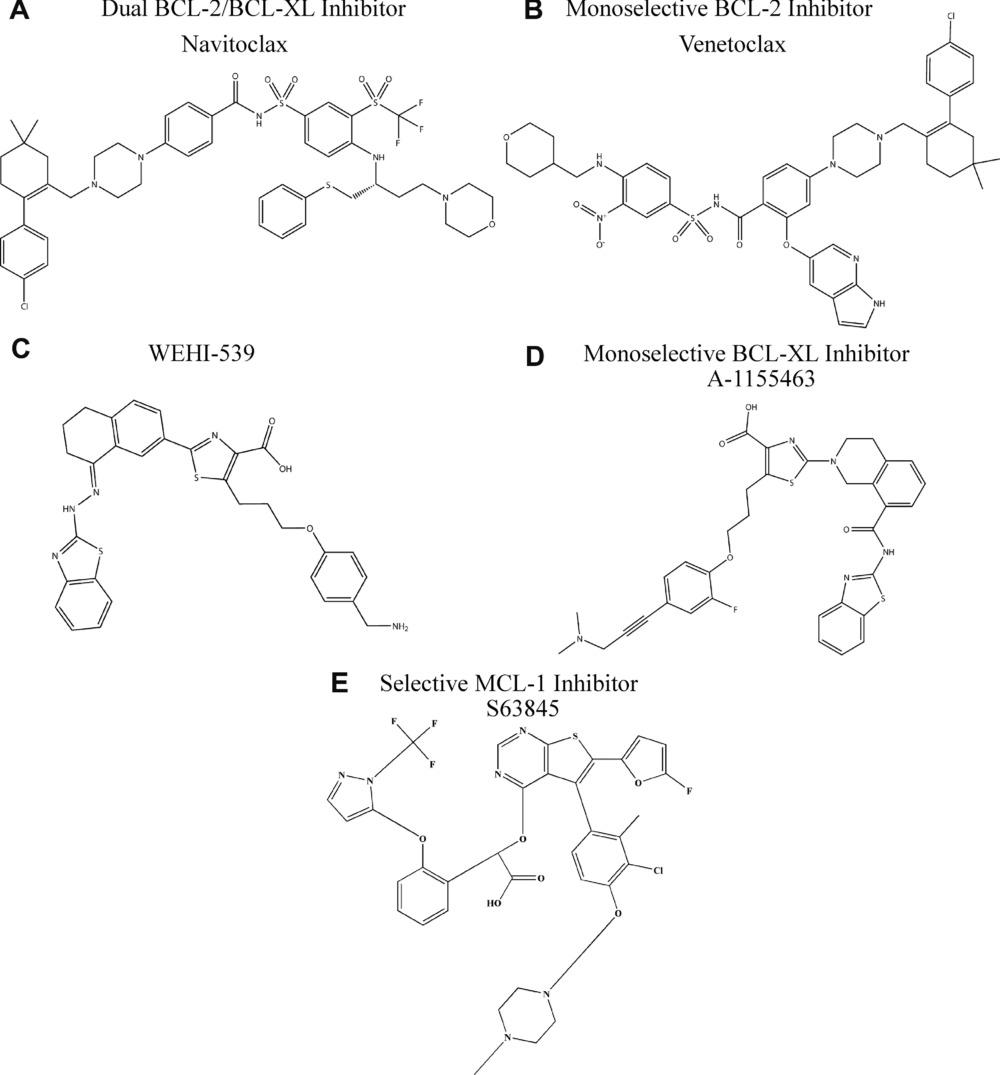
induction of an additional cell death signal through BH3-mimetic drugs or other pro-apoptotic stimuli is more powerful in these primed cells. An alternative approach to mimicking the function of pro-apoptotic BH3-only proteins is direct deletion of pro-survival BCL-2 family proteins. However, the deletion of each pro-survival BCL-2 family protein by genome editing has been shown to elicit drastic physiological effects in mouse models. Mice lacking BCL2 succumb to premature death from polycystic kidney disease, suggesting that BCL-2 is essential for the survival of renal epithelial progenitor cells in embryonic development. Mice lacking BCL-2 also have lowered resting T and B lymphocyte levels, as well as premature reductions of melanocytes causing greying skin. The BCL-XL protein is essential for the development of immature CD4 + and CD8 + T cells as well as B cell progenitors, which are essential in the adaptive immune response (Motoyama et al., 1995). The loss of Bcl2l1, the gene that encodes BCL-XL, exhibits the adverse effect of impairing male fertility and leads to decreased platelet count due to lower platelet viability (Mason et al., 2007). BH3-mimetic drugs that are MCL-1 specific exhibit harmful effects on otherwise healthy tissue and cells. MCL-1, or myeloid cell leukemia 1 is a pro-survival member of the BCL-2 family required for embryonic development, and thus mice lacking MCL-1 die very early in embryogenesis (Rinkenberger et al., 2000). Additionally, regulated deletion of MCL-1 protein shows that the protein plays a functional role in multiple tissues, such as in thymic epithelial survival and function (Jain et al., 2017) and T cell and germinal center B cell activation (Vikstrom et al., 2010). Contemporary pharmaceutical drug development has continually yielded more BH3-mimetic drugs with increased selectivity for specific BCL2 family proteins. This was bolstered by the clinical efficacy of ABT-199 (Venetoclax), the first BCL-2 specific BH3-mimetic drug to gain FDA approval for the treatment of acute myeloid leukemia. Venetoclax seemed to overcome the issue of low platelet count that was associated with AML treatment with Navitoclax, a dual BCL-2/BCL-XL inhibitor. The most remarkable clinical results for Venetoclax were observed when used in tandem with rituximab, a monoclonal antibody. A clinically complete response occurred in 51% of patients with disease-free status lasting for up to 2 years after completion of therapy (Campbell & Tait, 2018). Current BH3-mimetic drugs have demonstrated exceptional efficacy in clinical trials in both selectivity and apoptotic induction, proving to be viable alternatives for carcinomas that are resistant to cytotoxic therapies. Researchers are considering using this type of treatment in patients with solid tumors, in conjunction with conventional therapies. For instance, combination treatment using Navitoclax and Gemcitabine has shown noteworthy success against solid tumors. Furthermore, Navitoclax with Brentuximab has proven effective against Hodgkin’s lymphoma. Another significant instance of combination therapy involves coupling the BH3-mimetic drug ABT-737 with Cisplatin, which has shown to be highly effective against non-small cell lung cancer (Ju et al., 2016). Venetoclax efficacy as a combination drug has also been shown in preclinical models of breast cancer, such as when used in combination with Tamoxifen to treat breast cancer. Another drug approach being investigated directly activates BAX and BAK in order to kill tumor cells (Leshchiner et al., 2013). In terms of direct activation of BAK/BAX, it has been shown that BTSA1, a BAX-activating molecule, has therapeutic effects in acute myeloid leukemia. Figure 4. Chemical structures of clinically significant BH3-mimetic drugs.
Currently, utilizing BH3-mimetics as single therapeutic agents is limited to a small subset of cancer types. Some cancers, such as chronic lymphocytic leukemia, have been theorized to rely predominantly on a single pro-survival BCL-2 family member, making the success for single agent Venetoclax treatment promising; however, other malignancies also appear to rely on BCL-2 proteins for survival and could be targeted. BH3-mimetic drugs may best be used in combination therapy, which increases the potential for clinical efficacy. However, this also increases the potential for toxic complications. Cell pathways derived from leukemia have suggested that there are several resistance mechanisms to BH3-mimetic drugs. These include altered protein binding and reduced expression of the BH3-only protein BIM or the apoptosis effector BAX (Fresquet et al., 2014). Future directions to continue bettering this treatment include identifying predictive biomarkers to selectively identify patients who will most benefit from BH3-mimetic drug treatment, especially using prognostic factors such as tumor type and heterogeneity. Extending BH3-mimetic drug potential to treat solid tumors and improving compatibility with conventional treatments may yield a higher non-recurrence rate in patients, possibly even offering curative treatments for certain types of cancers.
References Campbell KJ, & Tait SWG (2018). “Targeting BCL-2 regulated apoptosis in cancer.” Open Biol, 8(5). Certo M, Del Gaizo Moore V, Nishino M, et al. (2006) “Mitochondria primed by death signals determine cellular addiction to antiapoptotic
BCL-2 family members.” Cancer Cell, 9(5):351-65. Fresquet V, Rieger M, Carolis C, García-Barchino MJ, Martinez-Climent
JA. (2014). “Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma.” Blood, 123(26):4111-9. Jain R, Sheridan JM, Policheni A, et al. (2017). “A critical epithelial survival axis regulated by MCL-1 maintains thymic function in mice.”
Blood, 130(23):2504-2515. Ju W, Zhang M, Wilson KM, Petrus MN, Bamford RN, Zhang X et al. (2016). “Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or Navitoclax in a murine model of human Hodgkin's lymphoma.” Proc Natl Acad Sci USA, 113:1624–1629. Leshchiner ES, Braun CR, Bird GH, Walensky LD. (2013). “Direct activation of full-length proapoptotic BAK.” Proc. Natl Acad. Sci. USA, 110:E986–E995. Liu X., Kim C.N., Yang J., Jemmerson R., Wang X.(1996) “Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c.” Cell, 86:147–157. Mason KD, Carpinelli MR, Fletcher JI, et al. (2007). “Programmed anuclear cell death delimits platelet life span.” Cell, 128(6):1173-86. Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. (2018).
“BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines.”
Cancer Cell, 34(6):879-891. Motoyama N., Wang F., Roth K. A., Sawa H., Nakayama K., Nakayama
K., ... Loh D. Y. (1995). “Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice.” Science, 267(5203):1506- 1510. Renehan A. G., Booth C., & Potten C. S. (2001). “What is apoptosis, and why is it important?” BMJ (Clinical research ed.), 322(7301):1536-8. Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. (2000).
“Mcl-1 deficiency results in peri-implantation embryonic lethality.”
Genes Dev, 14(1):23-7. Vikstrom I, Carotta S, Lüthje K, et al. (2010). “Mcl-1 is essential for germinal center formation and B cell memory.” Science, 330(6007):1095-9.

Anirudh Raghavan ('22C) is a sophomore in the College of Arts and Sciences double majoring in Biology and Chemistry. His article in this issue stems from his past summer experience in India at the Adyar Cancer Institute, a non-profit organization where he volunteered and learned about cancer treatment protocols offered to patients of all socioeconomic statuses. He currently works in the Liang Lab at the Emory School of Medicine, and his current project involves using ligation independent cloning (LIC) to construct Respiratory Syncytial Virus (RSV) co-expression plasmids. He hopes to attend medical school post-graduation. Apart from academics, Anirudh enjoys solving math puzzles, tinkering with audio equipment, and playing table tennis.





