Journal of the Ferrata Storti Foundation


VOL. 109 JUNE 2024 haematologica.org ISSN 0390 - 6078

Journal of the Ferrata Storti Foundation

Much cited Journal

Impact Factor 2022: 10.1
CiteScore 2022: 13.3
Fast review process

Submission ® 1st decision (submit to peer review or quick rejection): 3 days
Submission ® 2nd decision for peer-reviewed papers (accept, reject or make changes): 24 days
Low publication cost
The publisher is a non-profit Foundation that keeps the cost for authors as low as possible
Journal of the Ferrata - Storti Foundation
Editor-in-Chief
Jacob M. Rowe (Jerusalem)
Deputy Editors
Carlo Balduini (Pavia), Jerry Radich (Seattle)
Associate Editors
Michael Deininger (Milwaukee), Shai Izraeli (Tel Aviv), Pier Mannuccio Mannucci (Milan), Jessica Okosun (London), Pavan Reddy (Ann Arbor), David C. Rees (London), Paul G. Richardson (Boston), Francesco Rodeghiero (Vicenza), Gilles Salles (New York), Kerry Savage (Vancouver), Aaron Schimmer (Toronto), Richard F. Schlenk (Heidelberg)
Statistical Consultant
Catherine Klersy (Pavia)
AI Consultant
Jean Louis Raisaro (Lausanne)
Editorial Board
Walter Ageno (Varese), Sarit Assouline (Montreal), Andrea Bacigalupo (Roma), Taman Bakchoul (Tübingen), Pablo Bartolucci (Créteil), Katherine Borden (Montreal), Marco Cattaneo (Milan), Corey Cutler (Boston), Kate Cwynarski (London), Ahmet Dogan (New York), Mary Eapen (Milwaukee), Francesca Gay (Torino), Ajay Gopal (Seattle), Alex Herrera (Duarte), Martin Kaiser (London), Marina Konopleva (Houston), Nicolaus Kröger (Hamburg), Austin Kulasekararaj (London), Shaji Kumar (Rochester), Ann LaCasce (Boston), Matthew J. Mauer (Rochester) Neha Mehta-Shah (St. Louis), Moshe Mittelman (Tel Aviv), Alison Moskowitz (New York), Yishai Ofran (Haifa), Farhad Ravandi (Houston), John W. Semple (Lund), Liran Shlush (Toronto), Sarah K. Tasian (Philadelphia), Pieter van Vlieberghe (Ghent), Ofir Wolach (Haifa), Loic Ysebaert (Toulouse)
Managing Director
Antonio Majocchi (Pavia)
Editorial Office
Lorella Ripari (Office & Peer Review Manager), Simona Giri (Production & Marketing Manager), Paola Cariati (Graphic Designer), Giulia Carlini (Graphic Designer), Debora Moscatelli (Graphic Designer), Igor Poletti (Graphic Designer), Diana Serena Ravera (Peer Review), Laura Sterza (Account Administrator), Andrew Sturgeon (Peer Review)
Assistant Editors
Luca Arcaini (Scientific Consultant), Luk Cox (Graphic Artist), Britta Dost (English Editor), Anne Freckleton (English Editor), Rosangela Invernizzi (Scientific Consultant), Marianna Rossi (Scientific Consultant), Massimo Senna (Information Technology), Rachel Stenner (English Editor)
Haematologica (print edition, pISSN 0390-6078, eISSN 1592-8721) publishes peer-reviewed papers on all areas of experimental and clinical hematology. The journal is owned by a non-profit organization, the Ferrata Storti Foundation, and serves the scientific community following the recommendations of the World Association of Medical Editors (www. wame.org) and the International Committee of Medical Journal Editors (www.icmje.org).
Haematologica publishes Editorials, Original articles, Review articles, Perspective articles, Editorials, Guideline articles, Letters to the Editor, Case reports & Case series and Comments. Manuscripts should be prepared according to our guidelines (www.haematologica.org/information-for-authors), and the Uniform Requirements for Manuscripts Submitted to Biomedical Journals, prepared by the International Committee of Medical Journal Editors (www.icmje.org).
Manuscripts should be submitted online at http://www.haematologica.org/.
Conflict of interests. According to the International Committee of Medical Journal Editors (http://www.icmje. org/#conflicts), “Public trust in the peer review process and the credibility of published articles depend in part on how well conflict of interest is handled during writing, peer review, and editorial decision making”. The ad hoc journal’s policy is reported in detail at www.haematologica.org/content/policies.
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articles to the Ferrata Storti Foundation. No formal permission will be required to reproduce parts (tables or illustrations) of published papers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with commercial intent will require written permission and payment of royalties.
Subscription. Detailed information about subscriptions is available at www.haematologica.org. Haematologica is an open access journal and access to the online journal is free. For subscriptions to the printed issue of the journal, please contact: Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, E-mail: info@haematologica.org).
Rates of the printed edition for the year 2022 are as following:
Institutional: Euro 700
Personal: Euro 170
Advertisements. Contact the Advertising Manager, Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, e-mail: marketing@haematologica.org).
Disclaimer. Whilst every effort is made by the publishers and the editorial board to see that no inaccurate or misleading data, opinion or statement appears in this journal, they wish to make it clear that the data and opinions appearing in the articles or advertisements herein are the responsibility of the contributor or advisor concerned. Accordingly, the publisher, the editorial board and their respective employees, officers and agents accept no liability whatsoever for the consequences of any inaccurate or misleading data, opinion or statement. Whilst all due care is taken to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this journal, should only be followed in conjunction with the drug manufacturer’s own published literature.
Direttore responsabile: Prof. Carlo Balduini; Autorizzazione del Tribunale di Pavia n. 63 del 5 marzo 1955. Printing: Press Up, zona Via Cassia Km 36, 300 Zona Ind.le Settevene - 01036 Nepi (VT)
Associated with USPI, Unione Stampa Periodica Italiana. Premiato per l’alto valore culturale dal Ministero dei Beni Culturali ed Ambientali
Image taken from the Introduction to the Review Series by Locatelli in this issue.
1629 Genetics as predictive marker for consolidation therapy with high-dose cytarabine in acute myeloid leukemia
R.F. Schlenk
https://doi.org/10.3324/haematol.2024.285624
1631 Chimeric antigen receptor therapy for T-cell acute lymphoblastic leukemia: finally catching up with B-cell leukemia?
O. Beyar-Katz and J.M. Rowe
https://doi.org/10.3324/haematol.2024.284982
1634 CApSiZing T-cell acute lymphoblastic leukemia
K. Mandleywala and D. Herranz
https://doi.org/10.3324/haematol.2023.284714
1637 Anatomy of a crime: how IL7R and NRAS join forces to drive T-cell acute lymphoblastic leukemia
J.T. Barata
https://doi.org/10.3324/haematol.2023.284660
1640 SEQ-ing the genetic constellation of acute lymphoblastic leukemia
Z. Li and A.E.J. Yeoh
https://doi.org/10.3324/haematol.2023.284456
1643 Charting a path through resistance: histone deacetylase inhibitors for TP53-mutated B-cell acute lymphoblastic leukemia
E. Kugler
https://doi.org/10.3324/haematol.2023.284796
1646 Be aware of the X: BCOR mutations in myeloid neoplasms
G. Ramil et al.
https://doi.org/10.3324/haematol.2023.284748
1648 Anticoagulation and thrombocytopenia in cancer: what more can we learn from existing randomized controlled trials
A. Falanga and C. Giaccherini
https://doi.org/10.3324/haematol.2023.284291
1651 Adenovirus-associated thrombosis and thrombocytopenia: an emerging anti-PF4 disorder
A.B. Song and H. Al-Samkari
https://doi.org/10.3324/haematol.2023.284460
1653 Introduction. Immunotherapy for childhood malignancies: the future is now
F. Locatelli
https://doi.org/10.3324/haematol.2023.284553
1656 Chimeric antigen receptor T-cell therapy in childhood acute myeloid leukemia: how far are we from a clinical application?
S. Naik et al
https://doi.org/10.3324/haematol.2023.283817
1668 Bispecific T-cell engagers in childhood B-acute lymphoblastic leukemia
K.U. Lyons and L. Gore.
https://doi.org/10.3324/haematol.2023.283818
1677 Chimeric antigen receptor T-cell therapy for T-cell acute lymphoblastic leukemia
B.L.Z. Oh et al.
https://doi.org/10.3324/haematol.2023.283848
1689 Allogeneic chimeric antigen receptor T cells for children with relapsed/refractory B-cell precursor acute lymphoblastic leukemia
F. Locatelli et al.
https://doi.org/10.3324/haematol.2023.284604
1700 Naked antibodies and antibody-drug conjugates: targeted therapy for childhood acute lymphoblastic leukemia
E. Brivio et al.
https://doi.org/10.3324/haematol.2023.283815
Acute Lymphoblastic Leukemia
1713 CASZ1 upregulates PI3K-AKT-mTOR signaling and promotes T-cell acute lymphoblastic leukemia
B.A. Cardoso et al.
https://doi.org/10.3324/haematol.2023.282854
Acute Lymphoblastic Leukemia
1726 Mechanism of co-operation of mutant IL-7Ra and mutant NRAS in acute lymphoblastic leukemia: role of MYC
H. Winer et al.
https://doi.org/10.3324/haematol.2023.283559
Acute Lymphoblastic Leukemia
1741 MD-ALL: an integrative platform for molecular diagnosis of B-acute lymphoblastic leukemia
Z. Hu et al.
https://doi.org/10.3324/haematol.2023.283706
Acute Lymphoblastic Leukemia
1755 Histone deacetylase inhibition sensitizes p53-deficient B-cell precursor acute lymphoblastic leukemia to chemotherapy
W.P.J. Cox et al.
https://doi.org/10.3324/haematol.2023.284101
Acute Myeloid Leukemia
1766 Multi-gene measurable residual disease assessed by digital polymerase chain reaction has clinical and biological utility in acute myeloid leukemia patients receiving venetoclax/azacitidine
A.C. Winters et al.
https://doi.org/10.3324/haematol.2023.283790
Acute Myeloid Leukemia
1779 Genetic landscape and clinical outcomes of patients with BCOR mutated myeloid neoplasms
A. Baranwal et al.
https://doi.org/10.3324/haematol.2023.284185
Blood Transfusion
1792 Granulocyte transfusions in severe aplastic anemia
R.V. Rajput et al.
https://doi.org/10.3324/haematol.2023.283826
Hematopoiesis
1800 PELI2 regulates early B-cell progenitor differentiation and related leukemia via the IL-7R expression
Y. Xu et al.
https://doi.org/10.3324/haematol.2023.284041
Neoplasms of Lymphoid Tissue
1815 Massive parallel sequencing unveils homologous recombination deficiency in follicular dendritic cell sarcoma
L. Lorenzi et al.
https://doi.org/10.3324/haematol.2023.283669
Myeloid Neoplasms
1825 STAT5B mutations in myeloid neoplasms differ by disease subtypes but characterize a subset of chronic myeloid neoplasms with eosinophilia and/or basophilia
C. Yin et al.
https://doi.org/10.3324/haematol.2023.284311
Hemostasis
1836 Differences in venous clot structures between hemophilic mice treated with emicizumab versus factor VIII or factor VIIIFc
T. Sefiane et al.
https://doi.org/10.3324/haematol.2023.284142
Coagulation & its Disorders
1849 Impact of mild thrombocytopenia on bleeding and recurrent thrombosis in cancer
R. Patell et al.
https://doi.org/10.3324/haematol.2023.284192
Non-Hodgkin Lymphoma
1857 Long-term analysis of the RiBVD phase II trial reveals the unfavorable impact of TP53 mutations and hypoalbuminemia in older adults with mantle cell lymphoma; for the LYSA group
S. Carras et al.
https://doi.org/10.3324/haematol.2023.283724
Chronic Lymphocytic Leukemia
1866 Pirtobrutinib versus venetoclax in covalent Bruton tyrosine kinase inhibitor-pretreated chronic lymphocytic leukemia: a matching-adjusted indirect comparison
O. Al-Sawaf et al.
https://doi.org/10.3324/haematol.2023.284150
Plasma Cell Disorders
1874 t(11;14) status is stable between diagnosis and relapse, and concordant between detection methodologies based on fluorescence in situ hybridization and next-generation sequencing in patients with multiple myeloma
H. Avet-Loiseau et al.
https://doi.org/10.3324/haematol.2023.284072
Plasma Cell Disorders
1882 Real-world multiple myeloma risk factors and outcomes by non-Hispanic Black/African American and non-Hispanic White race/ethnicity in the United States
T. Buck et al.
https://doi.org/10.3324/haematol.2023.282788
Plasma Cell Disorders
1893 Epigenetic dysregulation of eukaryotic initiation factor 3 subunit E (eIF3E) by lysine methyltransferase
REIIBP confers a pro-inflammatory phenotype in t(4;14) myeloma
P.S.Y. Chong et al.
https://doi.org/10.3324/haematol.2023.283467
Plasma Cell Disorders
1909 Recovery of uninvolved heavy/light chain pair immunoparesis in newly diagnosed transplant-eligible myeloma patients complements the prognostic value of minimal residual disease detection
S. Lakhwani et al.
https://doi.org/10.3324/haematol.2023.284154
Red Cell Biology & its Disorders
1918 Epeleuton, a novel synthetic ω-3 fatty acid, reduces hypoxia/reperfusion stress in a mouse model of sickle cell disease
A. Mattè et al.
https://doi.org/10.3324/haematol.2023.284028
1933 An extensive database analysis demonstrates significant increase in platelet quantity in unselected hospitalized patients following treatment with oseltamivir
C. Muthiah et al.
https://doi.org/10.3324/haematol.2023.283731
1936 A retrospective analysis of gene fusions and treatment outcomes in pediatric acute megakaryoblastic leukemia without Down syndrome
K. Suzuki et al.
https://doi.org/10.3324/haematol.2023.283760
1941 Hepatosplenic T-cell lymphoma displays an original oyster-shell cytological pattern and a genomic profile distinct from that of γδ T-cell large granular lymphocytic leukemia
A. Desmares et al.
https://doi.org/10.3324/haematol.2023.283856
1947 Safety and efficacy of anakinra in hemophagocytic lymphohistiocytosis associated with acute leukemia
H. Al-Yousuf et al.
https://doi.org/10.3324/haematol.2023.283879
1951 Postnatal origin of the chromosomal gains in older patients with high hyperdiploid acute lymphoblastic leukemia
M. Yang et al.
https://doi.org/10.3324/haematol.2023.284128
1956 Update and European consensus on a patient-centered core outcome set for multiple myeloma in clinical practice and research
S. Oerlemans et al.
https://doi.org/10.3324/haematol.2023.284282
1960 Longitudinal dynamics and clinically available predictors of poor response to COVID-19 vaccination in multiple myeloma
G. Agarwal et al.
https://doi.org/10.3324/haematol.2023.284286
1966 TENT5C/FAM46C modulation in vivo reveals a trade-off between antibody secretion and tumor growth in multiple myeloma
M. Resnati et al.
https://doi.org/10.3324/haematol.2023.284299
1973 Randomized phase III GnG study on two schedules of gemtuzumab ozogamicin as adjunct to intensive induction therapy and double-blinded intensive post-remission therapy with or without glasdegib in patients with newly diagnosed acute myeloid leukemia
S. Jaramillo et al.
https://doi.org/10.3324/haematol.2023.284346
1977 A randomized, double-blind study of zinpentraxin alfa in patients with myelofibrosis who were previously treated with or ineligible for ruxolitinib: stage 2 of a phase II trial
S. Verstovsek et al.
https://doi.org/10.3324/haematol.2023.284410
1984 Targeting CCR4 with mogamulizumab in refractory CD3-CD4+ lymphocytic-variant hypereosinophilic syndrome
E. Ledoult et al.
https://doi.org/10.3324/haematol.2023.284429
1989 Role of red cell mass evaluation in myeloproliferative neoplasms with splanchnic vein thrombosis and normal hemoglobin value: a study of the France Intergroupe des Syndromes myeloprolifératifs
J. Galtier et al.
https://doi.org/10.3324/haematol.2023.284488
1994 A phase II randomized, placebo-controlled, multicenter trial to evaluate the efficacy of cytomegalovirus PepVax vaccine in preventing cytomegalovirus reactivation and disease after allogeneic hematopoietic stem cell transplant
R. Nakamura et al.
https://doi.org/10.3324/haematol.2023.284544
2000 The outcome of allogeneic hematopoietic stem cell transplantation among elderly patients with severe aplastic anemia and a predictive model from the Chinese Blood and Marrow Transplant Registry group
Z. Xu et al.
https://doi.org/10.3324/haematol.2023.284581
2005 Lenalidomide, rituximab, and methotrexate are effective in newly diagnosed primary central nervous system lymphoma
X. Yuan et al.
https://doi.org/10.3324/haematol.2023.284834
2010 Cerebral venous sinus thrombosis and thrombocytopenia due to heparin-independent anti-PF4 antibodies after adenovirus infection
G. Uzun et al.
https://doi.org/10.3324/haematol.2023.284127
2016 The aggravating fury rituximab obliterated
H. Mahadevia et al.
https://doi.org/10.3324/haematol.2023.284309
2019 Treatment of immune-mediated thrombotic thrombocytopenic purpura without plasma exchange
M. Capecchi et al.
https://doi.org/10.3324/haematol.2023.284438
Richard F. Schlenk
Department of Internal Medicine V, Heidelberg University Hospital; NCT-Trial Center, National Center of Tumor Diseases, Heidelberg University Hospital and German Cancer Research Center, Heidelberg, Germany
E-mail: richard.schlenk@nct-heidelberg.de
https://doi.org/10.3324/haematol.2024.285624
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
TITLE
AUTHORS
JOURNAL

Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype.
Bloomfield CD, Lawrence D, Byrd JC, et al.
Cancer Research 1998;58(18):4173-4179. PMID: 9751631.
High-dose cytarabine was established as the major component of consolidation chemotherapy in acute myeloid leukemia (AML) by the randomized study performed by the prestigious US Cancer and Leukemia Study Group B. The trial recruited between 1985 and 1990 and was published by Robert J. Mayer and colleagues in 1994 in the New England Journal of Medicine. 1 This study established the concept of a dose-response effect for cytarabine (100 mg vs. 400 mg vs. 3 g) in younger patients with AML in first complete remission. However, although disease-free
Figure 1. Cytogenetics is not only a prognostic factor independent of treatment, but also a predictive marker. CR: complete remission; CBF: core-binding factor; AML: acute myeloid leukemia. Figure adapted from Figures 1 and 4 in the paper by Bloomfield et al.2
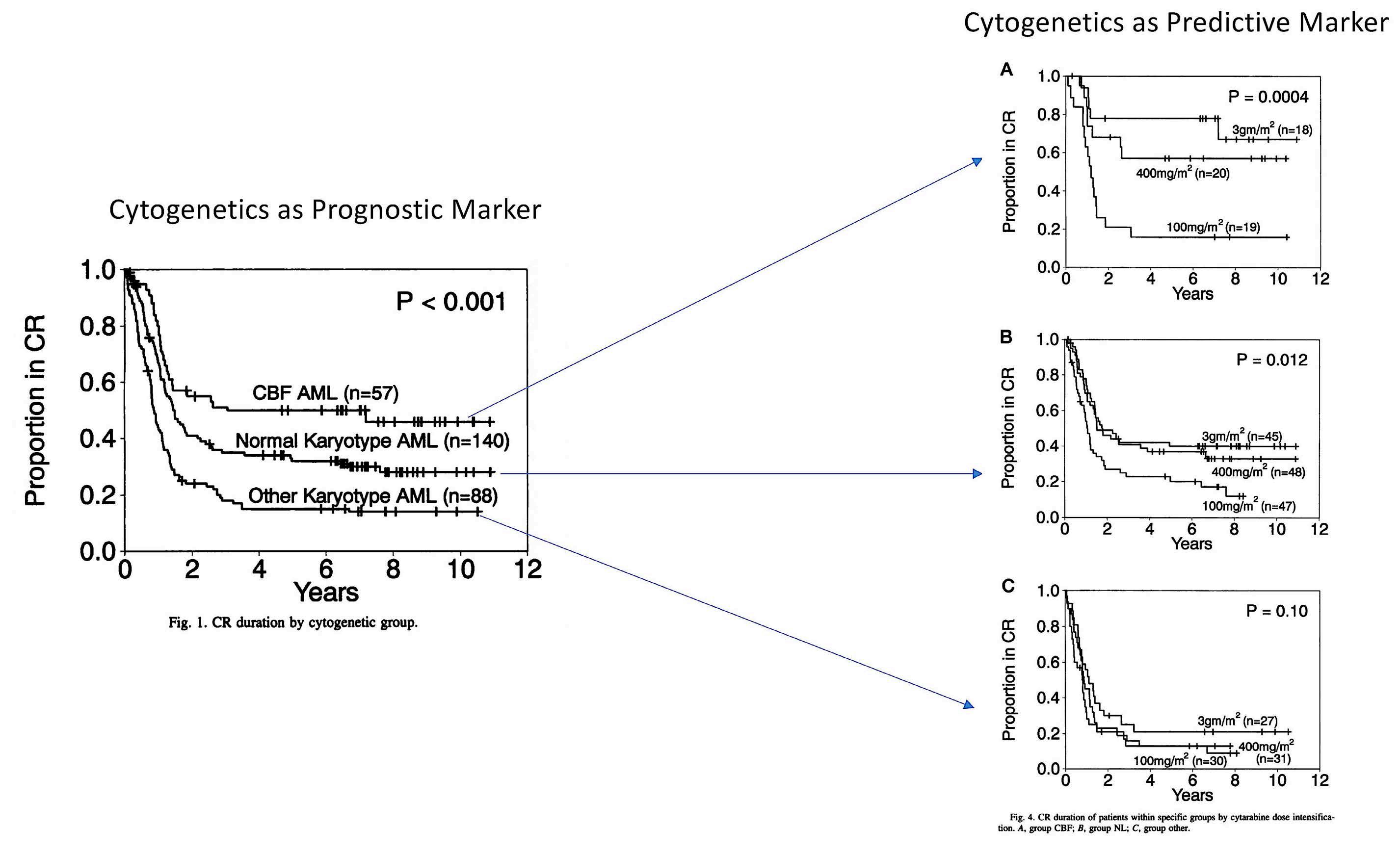
survival after 4 years was improved from 24% in the 100 mg group, to 29% in the 400 mg group and to 44% in the 3 g group not all patients benefited equally from dose-intensification.
At this point, the companion genetic diagnostics study including patients with adequate, pretreatment, centrally reviewed cytogenetics came into focus.2 It was already clear that cytogenetics was one of the major prognostic markers, identifying better outcome in AML patients with so-called core binding factor (CBF) abnormalities [t(8;21)(q22;q22) and inv(16)(p13q22) or t(16;16)(p13;q22)] and those exhibiting a normal karyotype.3 However, for the first time it was possible to show that cytogenetics is not only a prognostic factor independent of treatment, but also a predictive marker indicating better efficacy of high-dose cytarabine as
1. Mayer RJ, Davis RB, Schiffer CA, et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. Cancer and Leukemia Group B. N Engl J Med. 1994;331(14):896-903.
2. Bloomfield CD, Lawrence D, Byrd JC, et al. Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58(18):4173-4179.
3. Mrózek K, Heinonen K, de la Chapelle A, Bloomfield CD. Clinical significance of cytogenetics in acute myeloid leukemia. Semin
consolidation therapy in distinct genetically defined subgroups (Figure 1).
From different perspectives the study published in 1998 by Clara D. Bloomfield was a pivotal study: (i) it set the standard for consolidation therapy in AML patients with CBF abnormalities;4,5 (ii) it demonstrated impressively how companion diagnostics can guide results from randomized clinical trials; and (iii) it paved the way for the design of modern clinical trials - particularly with respect to biobanking, long-term follow-up and patient-reported outcomes - to allow maximal gain of knowledge through a multidimensional approach.
No conflicts of interest to disclose.
Oncol. 1997;24(1):17-31.
4 National Comprehensive Cancer Network. Acute myeloid leukemia (version 2.2024). https://www.nccn.org/professionals/ physician_gls/pdf/aml.pdf. Accessed April 12, 2024.
5. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345-1377.
1Department of Hematology and Bone Marrow Transplantation, Rambam Health Care Campus, Haifa; 2The Ruth and Bruce Rappaport Faculty of Medicine, Technion, Israel Institute of Technology, Haifa and 3Shaare Zedek Medical Center, Jerusalem, Israel
Correspondence: O. Beyar Katz o_katz@rmc.gov.il
Received: February 6, 2024. Accepted: February 22, 2024.
https://doi.org/10.3324/haematol.2024.284982
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
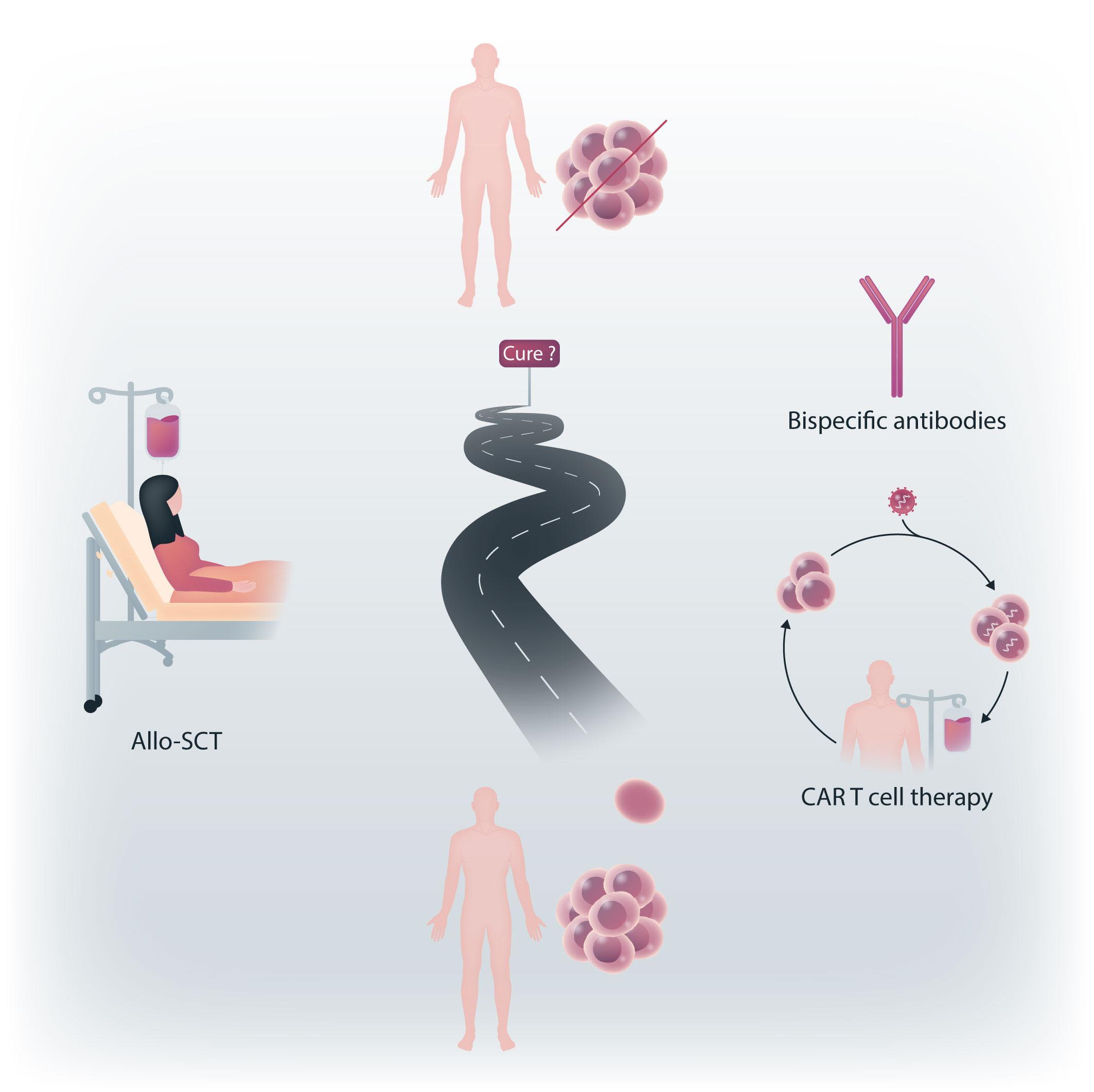
Chimeric antigen receptor (CAR) T-cell therapy is a treatment in which an artificial gene causes expression of a CAR within T cells. There are currently six CAR T-cell products approved by the Food and Drug Administration in the United States and by the European Union, targeting either CD19 or B-cell maturation antigen (BCMA) for several hematologic indications, all involving B-cell malignancies. These therapies have reshaped clinical practice leading to deep and sustained responses in patients diagnosed with these malignancies.
Until recently, the major advances in the therapy of acute lymphoblastic leukemia (ALL) have been confined, almost exclusively, to the B-lineage variant. Experience from more than a decade of therapy with rituximab taught us, rather surprisingly, that prolonged B-cell aplasia and hypogammaglobulinemia can be tolerated. This was a crucial forerunner to the development of bispecific antibodies targeting CD19 (as in blinatumumab) or antibody-drug conjugates targeting CD22 (as in inotuzumab ozogamicin) and more recently CAR T-cell therapy targeting CD19 or CD22 in leukemia and lymphoma. These were dramatic scientific and clinical developments that altered the landscape and standard of care for patients with B-cell leukemia and lymphoma. In contrast, T-cell leukemia and lymphoma, with a grim prognosis, appeared to be left behind. How could one target a T-cell antigen with a CAR T cell without committing ‘fratricide’? And, recalling the difficulty in developing CAR T cells for acute myeloid leukemia due to the inevitable neutropenia, how would one tolerate CAR T cells for T-ALL with the predicted lymphopenia?
In this issue of Haematologica, Oh et al.1 beautifully review the development and potential of applying CAR T-cell therapy to T-cell ALL with a focus on CD7 as an ideal target. The choice of the target antigen is extremely important to improve both the efficacy and safety of CAR T-cell ther-
apy. CD7 is the most widely explored and presented as a selected target due to its consistent expression in the majority of patients with T-ALL and, particularly, in refractory subtypes such as early T-cell precursor ALL. Thus, most CAR are designed to target CD7, with additional studies targeting mainly CD5 and CD38.
There are several inherent challenges associated with redirecting one T cell towards another T cell (Figure 1). The first challenge is based on the fact that rarely is there a tumor-specific antigen to target, so one uses a tumor-associated antigen, present on normal cells as well as malignant cells. Furthermore, in the context of T-cell malignancies, effector cells and target cells express the same antigens. Thus, CD7 targeting by CAR T cells will result in cytotoxic killing, a phenomenon widely known as “fratricide”. To overcome this challenge some groups are exploring ways to reduce or eliminate the target expression on T cells, either by blocking the protein expression on the surface, selecting for a T-cell population not expressing the target antigen, or by genetic editing of the CAR T cells. Although reduction of target antigen expression seems rational, some groups have shown impressive clinical responses while avoiding such manipulations in T-cell leukemia and lymphoma.2,3 This could be explained by a reported detection of some cells transduced with a CAR that would result in masking or intracellular sequestration of CD7 expression, leading to resistance to fratricide.3 On this basis, one could hypothesize that with some tumor-associated antigens, the fratricide would be minimal and not harmful for the final CAR T-cell product.
The second challenge is the risk of leukemic contamination of the final product. In 2018, a group from the University of Pennsylvania reported a dreaded complication following CAR T-cell treatment, defined as CAR-transduced B-cell leukemia (CARB) cells.4 In that report, a patient with B-ALL
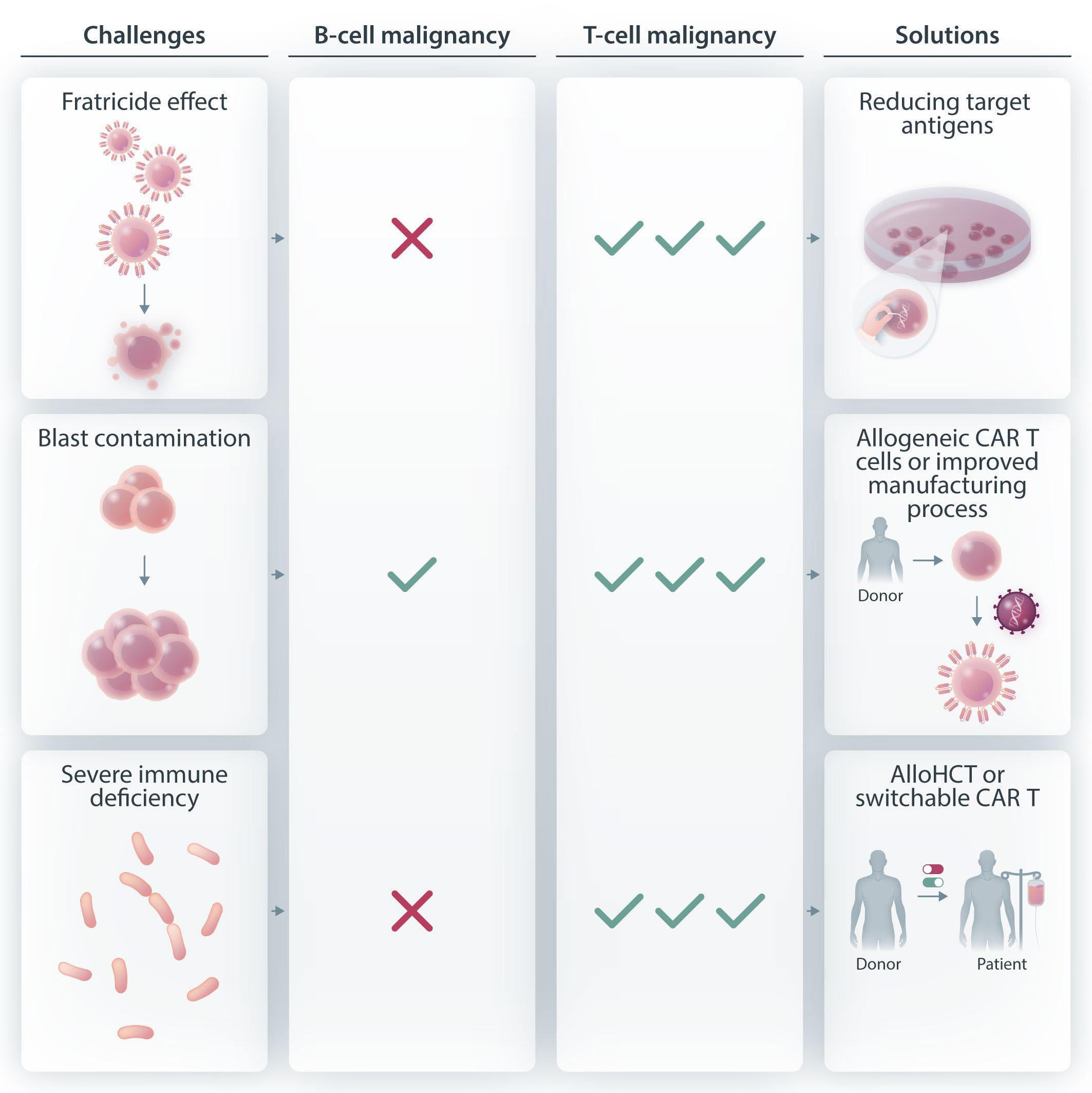
Figure 1. Unique challenges of developing autologous chimeric antigen receptor T cells for T-cell malignancies. There are several unique challenges presented by virtue of targeting T-cell malignancies using chimeric antigen receptor (CAR) T cells. Fratricide is described in the setting of T-cell malignancies but not in B-cell malignancies. This can be addressed by reducing or eliminating the target antigen from the surface of the T cells. Blast contamination can complicate any CAR T-cell therapy but is more prominent in the setting of T-cell acute lymphoblastic leukemia. This hurdle can be overcome by using allogeneic CAR T cells or by improving manufacturing techniques so that blast cells are certainly excluded. The risk of infections is greater with prolonged T-cell aplasia than with B-cell aplasia. This toxicity is reasonably well tolerated during CAR T-cell therapy targeting B-cell malignancies, with immunoglobulin supplementation for patients experiencing recurrent infections. However, there is no available therapy for prolonged T-cell aplasia. Potential ways to overcome this toxicity is by consolidative allogeneic hematopoietic cell transplantation or a switchable CAR T-cell platform. AlloHCT: allogeneic hematopoietic cell transplantation.
experienced an aggressive leukemia relapse 252 days following CAR T-cell administration with transduction of a single blast cell. This led to the generation of CAR19-expressing B-ALL cells that masked the CD19 antigen and created a CAR T-cell-resistant leukemia. This case emphasizes the need to improve manufacturing methods so as to exclude any possibility of product contamination by blast cells. Since this is particularly difficult to ensure in T-ALL, careful analysis of the final product is required. A potential approach to address this challenge is the utilization of allogeneic CAR T cells, with which there are, obviously, no concerns about blast contamination.
The third challenge involves the long-term and durable T-cell depletion associated with CAR T cells targeting the T-cell lineage (on-target, off-tumor). In B-cell malignancies, the depletion of B cells and hypogammaglobulinemia are manageable in most patients who do not experience serious infections or require immunoglobulin supplementation. Conversely, based on the evidence obtained in the setting of T-cell-depleted allogeneic transplants, reduced anti-microbial responses are expected and severe life-threatening infections are the rule. Moreover, T-cell aplasia is commonly accompanied by immune effector-cell-associated hematotoxicity, such as pancytopenia, developing after CAR T-cell
administration, further increasing the risk of infection. In order to circumvent this problem, subsequent allogeneic transplantation or “switching off” the CAR T-cell product upon malignant cell killing must be offered to ensure T-cell reconstitution. Nevertheless, in a recent study in which CAR T cells targeting CD5 were administered to nine patients with mature T-cell lymphoma,2 two patients declined to proceed to allogeneic hematopoietic cell transplantation and, surprisingly, this was not associated with prolonged T-cell aplasia or severe infectious complications. Although hard to draw conclusions from two patients, this raises the issue of whether allogeneic transplantation is always critical for reducing prolonged T-cell aplasia. This is further emphasized in a report of manageable T-cell aplasia in 12 patients treated with CD7-CAR T cells not proceeding to allogeneic transplantation.5 Irrespectively, the aggressive nature of the underlying disease may in and of itself mandate using CAR T cells as a bridge to transplantation. This is uncertain territory also in the setting of B-ALL, for which many more data are available. In other reports assessing CAR T cells for the treatment of T-ALL/T-lymphoblastic lymphoma, initial responses appear very promising, with an 85-95% complete response rate by day 28, but the durability of the effect is unknown since many of these patients underwent consol-
1. Oh BLZ, Vinanica N, Wong DMH, Campana D. Chimeric antigen receptor-directed T-cell therapy for T-cell acute lymphoblastic leukemia. Haematologica. 2024;109(6):1677-1688.
2. Hill LC, Rouce RH, Wu M, et al. Anti-tumor efficacy and safety of unedited autologous CD5.CAR T cells in relapsed/refractory mature T-cell lymphomas. Blood. 2024;143(13):1231-1241
3. Lu P, Liu Y, Yang J, et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human
idative allogeneic hematopoietic cell transplantation.3,5 Currently, there are 16 CAR T-cell trials for T-cell malignancies listed in the ClinicalTrial.gov website, with eight of these trials actively recruiting patients. Eleven trials are in China, three in the USA and two in Europe. Most of these trials target CD7 (n=11), whereas other less common targets include CD5, CD1a, TRBC1, and OC-1.
Clearly, the field of CAR T-cell therapy for T-cell malignancies is evolving rapidly. While very significant obstacles persist, and we are still far from adopting such as standard of care, there is at last excitement and hope that we are getting closer to overcoming what hitherto appeared to be insurmountable.
No conflicts of interest to disclose.
Both authors contributed equally.
The authors wish to acknowledge with thanks the assistance of Sonia Kamenetsky in the preparation of this manuscript.
phase 1 clinical trial. Blood. 2022;140(4):321-334.
4 Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499-1503.
5. Pan J, Tan Y, Wang G, et al. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: first-in-human, phase I trial. J Clin Oncol. 2021;39(30):3340-3351.
1Rutgers Cancer Institute of New Jersey, Rutgers University, New Brunswick, NJ; 2Department of Pharmacology, Robert Wood Johnson Medical School, Rutgers University, Piscataway, NJ and 3Department of Pediatrics, Robert Wood Johnson Medical School, Rutgers University, New Brunswick, NJ, USA
In this issue of Hematologica, Cardoso et al. identify CASZ1 as a novel player in T-cell acute lymphoblastic leukemia (T-ALL), an aggressive hematological malignancy with a high risk of relapse and associated long-term complications.1 Thus, discovering new factors involved in the development of leukemia will enhance our basic understanding of the pathophysiology of this disease and may lead to the identification of novel therapeutic targets for T-ALL treatment. In this context, it is noteworthy that CASZ1 is particularly enriched in patients expressing elevated levels of TAL1, a well described major oncogene in T-ALL.2
CASZ1, the mammalian homolog of the Drosophila zinc finger transcription factor Castor, is known for its critical role in vascular and neural development.3,4 CASZ1 consists of two alternatively spliced isoforms (CASZ1a and CASZ1b) which, however, seem to play similar roles. Interestingly, embryonic deletion of Casz1 leads to abnormal heart development and lethality in mice.5 Moreover, the potential role of CASZ1 in cancer is complex. Caren et al. first showed that the loss of several genes in the chromosome region 1p36, particularly CASZ1 and PIK3CD, is associated with the development of neuroblastoma.6 This initial discovery sparked a cascade of investigations into the diverse roles of CASZ1 across various cancer types and physiological processes. Beyond its tumor suppressor role in neuroblastoma, where CASZ1 low expression also significantly correlates with poor clinical outcomes,7 it has been shown that overexpression of CASZ1 is associated with metastasis in ovarian cancer,8 highlighting its potential tissue-specific role in cancer development. In the present study, the authors start dissecting the role of CASZ1 in T-ALL by examining its interplay with key oncogenes and T-ALL-specific mutations. Taking advantage of the BloodSpot database, they found that the CASZ1b isoform (which shows higher evolutionarily conservation than CASZ1a), was significantly upregulated in T-ALL cell lines and patient samples. Interestingly, CASZ1b upregulation was especially marked in cases with high TAL1 expression, suggesting that TAL1 might regulate CASZ1b. Indeed, TAL1 overexpression or knockdown in different human T-ALL cell lines led to
Correspondence: D. Herranz dh710@cinj.rutgers.edu
Received: December 22, 2023. Accepted: January 9, 2024. Early view: January 18, 2024.
https://doi.org/10.3324/haematol.2023.284714
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
upregulation or downregulation of CASZ1b, respectively. Moreover, the authors found that TAL1 directly binds to the CASZ1b promoter, further reinforcing the positive correlation between TAL1 and CASZ1 in T-ALL. Still, CASZ1 was generally overexpressed in T-ALL compared to normal T cells, suggesting that additional mechanisms might be involved in the regulation of CASZ1 in TAL1-negative T-ALL cases and, more broadly, supporting a relevant role for CASZ1 in T-ALL overall. Next, the authors demonstrate that CASZ1 overexpression is sufficient to confer interleukin (IL)-3-independent growth in the otherwise IL-3-dependent Ba/F3 murine pro-B cell line, suggesting a pro-oncogenic role for CASZ1. In order to dissect the underlying mechanism, the authors performed gene expression profiling analyses in this setting and found that CASZ1 correlated with overexpression of the PI3K-AKTmTOR signaling axis, which is well known to play a critical role in T-ALL.9 Notably, pharmacological inhibition of the PI3K/mTOR pathway rescued the oncogenic effects driven by CASZ1 in Ba/F3 cells, both in vitro and in vivo. Similarly, CASZ1 also positively correlates with the PI3K-AKT pathway in T-ALL cells, underscoring the central role of the PI3K/AKT/ mTOR pathway downstream of CASZ1. Still, how might CASZ1 contribute to regulating the PI3K-AKT pathway remains a key lingering question.
Building upon these findings, the authors next used a zebrafish model of NOTCH1-induced T-ALL to demonstrate that CASZ1 not only accelerated thymic hyperplasia but also actively promoted the development of NOTCH1-induced leukemia in vivo. Next, the authors performed a variety of experiments in human T-ALL cell lines in vitro in order to investigate the functional relevance of CASZ1. Under normal conditions, overexpressing CASZ1 had no impact on the viability or proliferation of human T-ALL cells. However, under stress conditions such as serum starvation, CASZ1 overexpression displayed a prosurvival role. Moreover, CASZ1 also conferred resistance to a variety of chemotherapeutic drugs commonly used in T-ALL treatment, such as daunorubicin, dexamethasone or L-asparaginase, suggesting a broader protective role from different types of cellular stress. Finally,
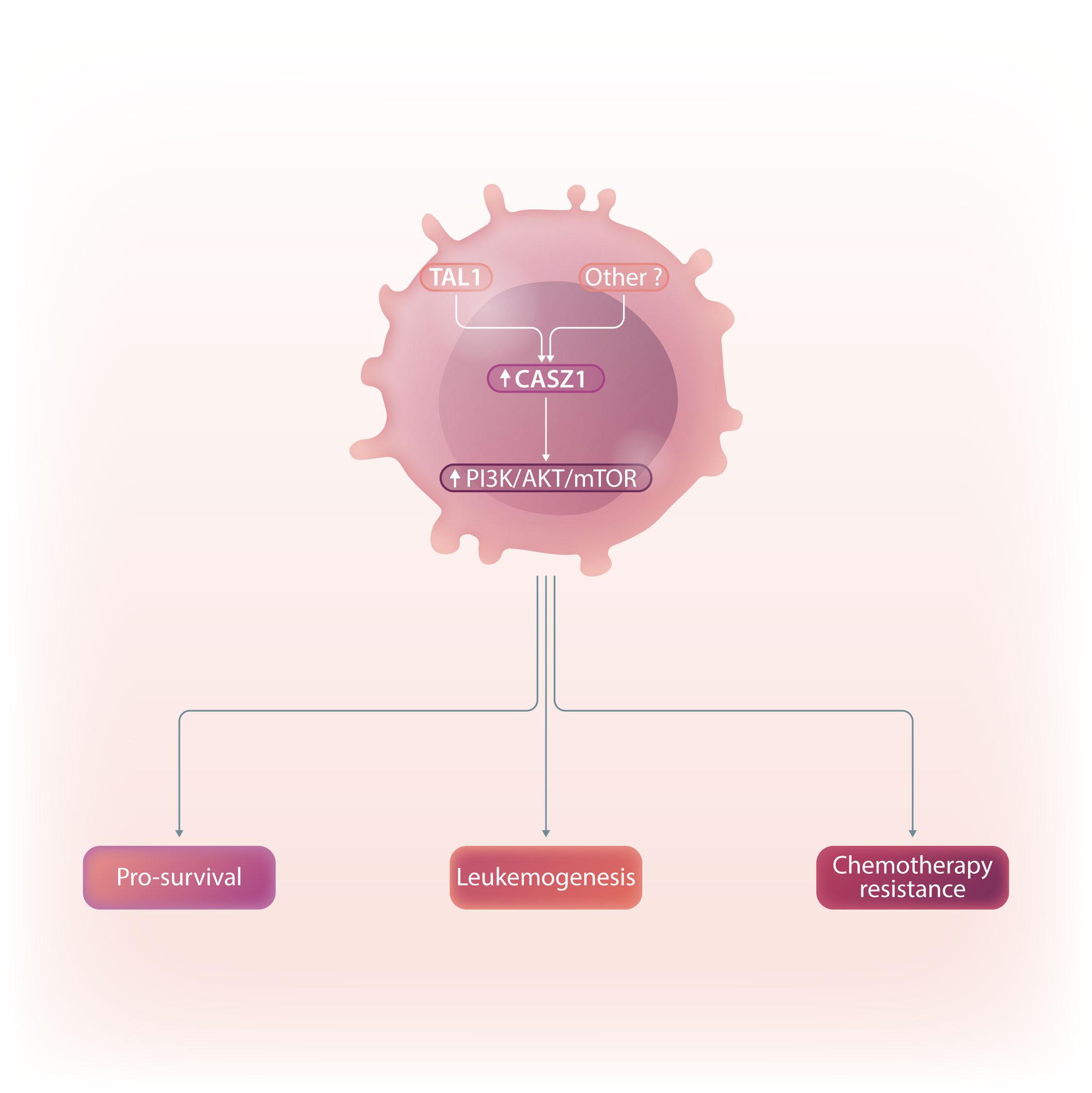
although CASZ1 levels did not stand out as an independent prognostic factor in newly diagnosed cases of T-ALL, high levels of CASZ1 were associated with poorer prognosis in patients with relapsed T-ALL. Overall, this report uncovers a previously unknown oncogenic role for CASZ1 in T-ALL, which might be of particular relevance in the response to common anti-leukemic drug treatments and in the progression of (heavily pretreated) relapsed T-ALL cases. Thus, further studies are warranted to investigate the potential role of CASZ1 as a novel therapeutic target in T-ALL treatment.
1. Cordo V, van der Zwet JCG, Cante-Barrett K, Pieters R, Meijerink JPP. T-cell acute lymphoblastic leukemia: a roadmap to targeted therapies. Blood Cancer Discov. 2021;2(1):19-31.
Disclosures
No conflicts of interest to disclose.
Contributions
KM and DH contributed equally.
Funding
Work in the laboratory of DH is supported by The Leukemia & Lymphoma Society (Scholar Award 1386-23). KM is supported by a Fellowship from the New Jersey Commission on Cancer Research (COCR24PRF011).
2. Cardoso BA, Duque M, Girio A, et al. CASZ1 upregulates PI3KAKT-mTOR signaling and promotes T-cell acute lymphoblastic leukemia. Haematologica. 2024;109(6):1713-1725.
3. Charpentier MS, Christine KS, Amin NM, et al. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev Cell. 2013;25(2):132-143.
4 Mattar P, Jolicoeur C, Dang T, Shah S, Clark BS, Cayouette M. A Casz1-NuRD complex regulates temporal identity transitions in neural progenitors. Sci Rep. 2021;11(1):3858.
5. Liu Z, Li W, Ma X, et al. Essential role of the zinc finger transcription factor Casz1 for mammalian cardiac morphogenesis and development. J Biol Chem. 2014;289(43):29801-29816.
6. Caren H, Fransson S, Ejeskar K, Kogner P, Martinsson T. Genetic and epigenetic changes in the common 1p36 deletion in neuroblastoma tumours. Br J Cancer. 2007;97(10):1416-1424.
7 Liu Z, Yang X, Li Z, et al. CASZ1, a candidate tumor-suppressor
gene, suppresses neuroblastoma tumor growth through reprogramming gene expression. Cell Death Differ. 2011;18(7):1174-1183.
8. Wu YY, Chang CL, Chuang YJ, et al. CASZ1 is a novel promoter of metastasis in ovarian cancer. Am J Cancer Res. 2016;6(6):1253-1270.
9 Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti-NOTCH1 therapy in T-cell acute lymphoblastic leukemias and lymphomas. Clin Cancer Res. 2008;14(17):5314-5317.
Instituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina, Universidade de Lisboa, Lisboa, Portugal
Correspondence: J.T. Barata joao_barata@medicina.ulisboa.pt
Received: January 9, 2024.
Accepted: January 30, 2024. Early view: February 8, 2024.
https://doi.org/10.3324/haematol.2023.284660 ©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Winer et al.1 demonstrate that MYC is crucial to the oncogenic cooperation between IL7R and NRAS driving T-cell acute lymphoblastic leukemia (T-ALL) development.2 They further show that the kinase PLK-1 may contribute to MYC protein stability and that MYC-modulating drugs can be of therapeutic value against T-ALL driven by activating mutations in both IL7R and NRAS. Why the relevance of diving deeper into the mechanisms underlying this collaboration? Roughly 10% of T-ALL patients display IL7R gain-of-function mutations, and a much larger fraction (some 50-80% of the cases) express IL7R and may benefit from IL7 produced in the leukemia milieu.3-7 IL7R-mediated signaling (because of IL7, high IL7R levels or mutational activation of the receptor or downstream effectors) can promote T-ALL establishment and maintenance, and resistance to glucocorticoids.5 RAS activating mutations in general occur in around 2% of the cases, and NRAS alterations are infrequent. However, full appreciation of the importance of RAS signaling in T-ALL must consider other lesions, including inactivating mutations in the RAS-MEK-ERK pathway negative regulator NF1 or RasGRP1 overexpression. Interestingly, MEK-ERK pathway can be activated also by IL7/IL7R in T-ALL, contributing to IL7R-mediated resistance to glucocorticoids.8
To understand why IL7R or NRAS alone cannot drive T-ALL in a transplant mouse model, whereas their combination is clearly leukemogenic,2 Winer et al. have now analyzed the transcriptome of immature mouse thymocytes transduced with mutant IL7R (a particular type 1a IL7R activating mutation, hereafter referred to as mutIL7R),3,5 mutant NRAS (coding for NRAS G13D, which leads to NRAS activation; hereafter referred to as mutNRAS) or their combination. They found no evidence of Myc activation (as measured by the upregulation of Myc target genes) by mutIL7R alone, whereas mutNRAS activated Myc and, importantly, this effect was augmented by the combination of mutIL7R and mutNRAS. In agreement, Myc itself was up-regulated
by mutNRAS and mutNRAS+mutIL7R, but not by mutIL7R alone. The fact that mutIL7R was unable to activate Myc is intriguing, as MYC is a downstream target of IL7R-mediated signaling in human thymocytes, and in zebrafish models of T-ALL.9 The reasons for these discrepancies are unclear. They may relate to the stage of disease development at which the analyses were conducted: D1 cells, being p53null, are one step closer to transformation than healthy thymocytes, and the analyses in zebrafish and human T-ALL focused on fully transformed cells.
These considerations apart, the authors provide convincing evidence linking RAS signaling and MYC. They show that MYC overexpression phenocopied mutant NRAS in its ability to collaborate with mutIL7R to drive T-ALL. Both mutIL7R+mutNRAS and mutIL7R+MYC led to CD4+CD8+ T-ALL, with relatively similar expression of TCR Va and predominance of aβ over γδ T-cells. Whether this resemblance extends to the transcriptional profile was not addressed. Nonetheless, these findings align with previous studies showing that mutIL7R and MYC collaborate to drive T-ALL in zebrafish.9 The relevance of MYC in mutIL7R+mutNRAS-driven leukemias was further exposed by experiments showing that Myc deletion decreased leukemia burden in vivo and that Myc silencing decreased the fitness of D1 cells and primary thymocytes transduced with the combination of the two oncogenes.
Winer et al. further combined RNA-sequencing and mass-spectrometry to show that Bcl-2 transcript and protein were up-regulated.1 Contrary to Myc, Bcl-2 upregulation was due to mutIL7R and not mutNRAS, and the combination of the two oncogenes did not potentiate mutIL7R effects. That IL7-IL7R-mediated signaling up-regulates Bcl-2 is well known, although (in contrast to the illustration in Figure 8 of the paper) this is unlikely to be mediated by STAT5 in T-ALL.8,10
Contrary to Bcl-2, Myc was up-regulated exclusively at the protein level, and the authors propose that NRAS collabo-
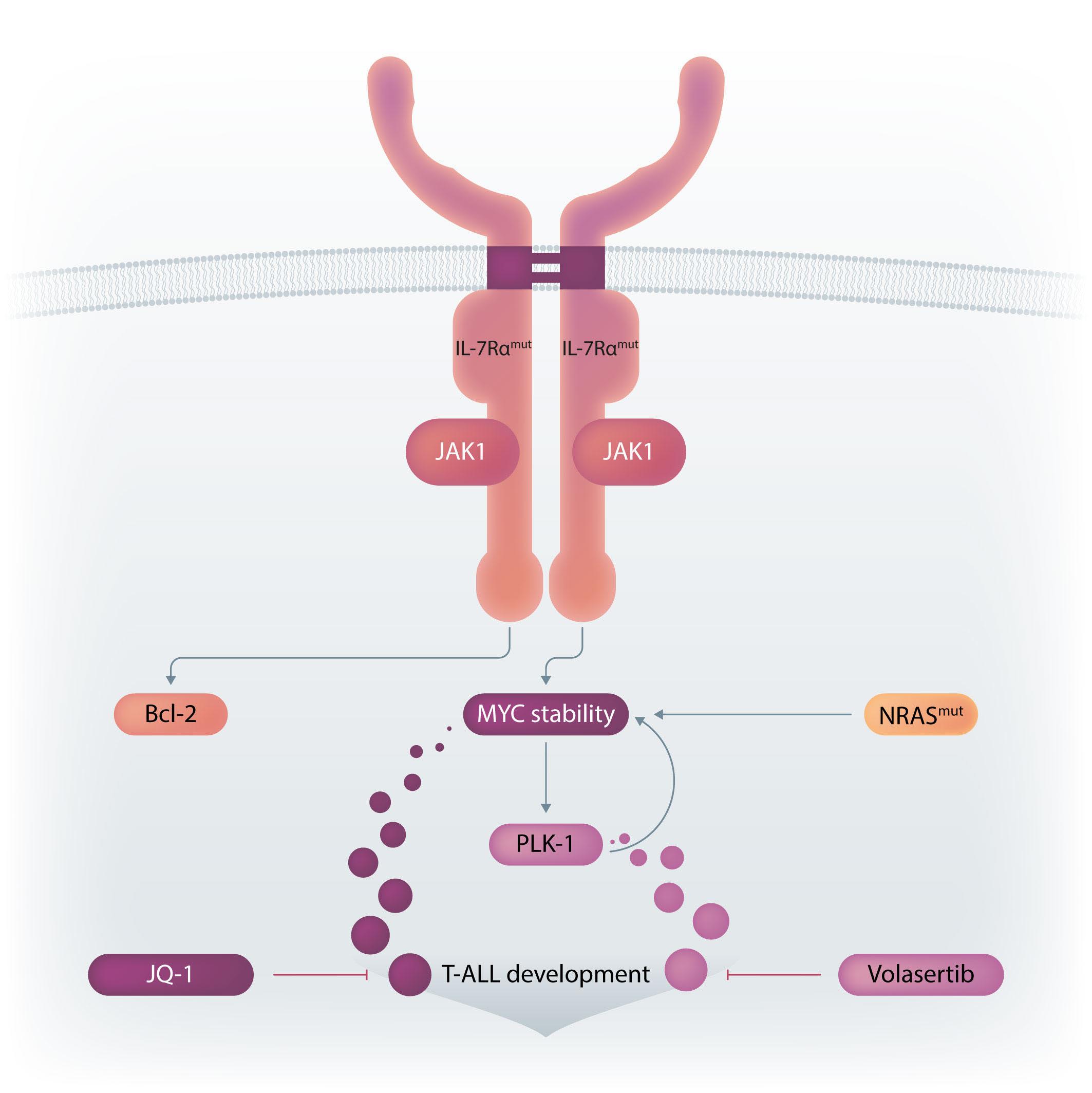
Figure 1. Winer et al. demonstrate that gain-of-function (i.e., activating) mutations in IL7R and NRAS cooperate to drive T-cell acute lymphoblastic leukemia (T-ALL) in mice mainly due to the ability to activate MYC, in particular via NRAS-dependent upregulation of MYC protein levels. NRAS likely promotes MYC protein stabilization by at least two mechanisms: directly, by phosphorylation of MYC, and indirectly, via transcriptional activation of PLK-1, whose increased expression prevents MYC proteasomal degradation. Bcl-2 is also up-regulated (essentially due to IL7R-mediated signaling), although its exact role in the cooperative oncogenic effects of IL7R and NRAS is unclear. The use of a chemical inhibitor of PLK-1 (volasertib) or of a drug that down-regulates MYC (and IL7R) transcription (JQ-1) diminishes leukemia burden in vivo. Pre-clinical studies evaluating the value of these drugs against human T-ALL with IL7R and RAS signaling pathway mutations are warranted. Final version by somersault18:24.
rated with IL7R essentially by stabilizing Myc. How activated NRAS, and its combination with mutant IL-7R, promoted Myc stabilization may involve two mechanisms. RAS-MEKERK signaling is known to phosphorylate Myc at Serine 62 (S62), and this phosphorylation contributes to Myc stability. Indeed, Myc S62 was up-regulated by mutIL7R and even more so by mutNRAS, although not further increased by the combination. On the other hand, AKT-dependent Myc Threonine 58 (T58) phosphorylation was up-regulated by each oncogene alone and by their combination. This should mark Myc for degradation, which obviously was not the case. So, there should be a mechanism counterbalancing the effects of Myc T58-phosphorylation. Winer et al. no-
ticed that PLK-1 (which contributes to MYC stabilization by preventing its proteasomal degradation) was up-regulated by NRAS and its combination with IL7R. Thus, they used volasertib, a PLK-1 inhibitor, to test the impact on Myc expression. Volasertib not only down-regulated Myc protein but also, surprisingly, Myc transcript levels, suggesting that PLK-1 may up-regulate Myc via different mechanisms. In the absence of PLK-1 genetic manipulation, and given that pharmacological inhibitors often have off-target effects, it may be that volasertib impacted Myc expression, particularly at the transcript level, by PLK-1-independent mechanisms. Nonetheless, the authors provide good evidence that PLK1 and MYC are likely involved in a positive-feedback loop
that partakes in leukemia development promoted by the combination of mutIL7R and mutNRAS Evidently, the question that remained was whether these findings have translational potential. Using their transplant mouse model of mutIL7R+mutNRAS T-ALL, Winer et al. show that volasertib diminished Myc levels and leukemia burden in vivo. There was no benefit in combining volasertib with the Bcl-2 inhibitor venetoclax. This is surprising, given the importance of Bcl-2 for IL-7R-mediated viability in T-ALL, and how MYC (which is down-regulated by volasertib in vivo) and BCL2 synergize to promote cancer development. The authors also tested JQ-1, a BET bromodomain BRD4 inhibitor, which down-regulates MYC and IL7R transcription. JQ-1 demonstrated similar in vivo effects to volasertib, yet another demonstration of the importance of MYC in these leukemias.
Overall, the studies by Winer et al. not only allow a better understanding of how oncogenic IL7R cooperates with RAS
1. Winer H, Li W, Rodrigues G, et al. Mechanism of co-operation of mutant IL-7Ralpha and mutant NRAS in acute lymphoblastic leukemia: role of MYC. Haematologica. 2024;109(6):1726-1740.
2. Cramer SD, Hixon JA, Andrews C, et al. Mutant IL-7Ralpha and mutant NRas are sufficient to induce murine T cell acute lymphoblastic leukemia. Leukemia. 2018;32(8):1795-1882.
3. Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain-offunction mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43(10):932-939.
4 Shochat C, Tal N, Bandapalli OR, et al. Gain-of-function mutations in interleukin-7 receptor-{alpha} (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208(5):901-908.
5. Barata JT, Durum SK, Seddon B. Flip the coin: IL-7 and IL-7R in health and disease. Nat Immunol. 2019;20(12):1584-1593.
6. Silva A, Almeida ARM, Cachucho A, et al. Overexpression of
signaling in driving T-ALL (Figure 1), but also pave the way for preclinical studies testing the value of PLK-1 and/or MYC inhibitors in human T-ALL patient samples, and patient-derived xenograft models, with IL7R and RAS pathway mutations.
Disclosures
No conflicts of interest to disclose.
Funding
The work in João T. Barata’s lab was supported by grants from the following funding agencies: European Research Council (ERC-PoC-101069429), la Caixa Foundation (HR2100761) and Fundação para a Ciência e a Tecnologia (PTDC/ MEC-ONC/4606/2021).
The original cartoon for Figure 1 was created by Marta Fernandes.
wild-type IL-7Ralpha promotes T-cell acute lymphoblastic leukemia/lymphoma. Blood. 2021;138(12):1040-1052.
7. Courtois L, Cabannes-Hamy A, Kim R, et al. IL-7 receptor expression is frequent in T-cell acute lymphoblastic leukemia and predicts sensitivity to JAK inhibition. Blood. 2023;142(2):158-171.
8. Van der Zwet JCG, Cordo V, Buijs-Gladdines J, et al. STAT5 does not drive steroid resistance in T-cell acute lymphoblastic leukemia despite the activation of BCL2 and BCLXL following glucocorticoid treatment. Haematologica. 2023;108(3):732-746.
9 Oliveira ML, Veloso A, Garcia EG, et al. Mutant IL7R collaborates with MYC to induce T-cell acute lymphoblastic leukemia. Leukemia. 2022;36(6):1533-1540.
10 Ribeiro D, Melao A, van Boxtel R, et al. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018;2(17):2199-2213.
1Department of Pharmacy and Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN, USA; 2Department of Pediatrics, Yong Loo Lin School of Medicine, National University of Singapore, Singapore and 3VIVA-University Children’s Cancer Center, Khoo Teck Puat-National University Children’s Medical Institute, National University Health System, Singapore
Correspondence: A.E.J. Yeoh allen.yeoh@nus.edu.sg
Received: November 20, 2023. Accepted: December 12, 2023. Early view: December 21, 2023. https://doi.org/10.3324/haematol.2023.284456 ©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Acute lymphoblastic leukemia (ALL) is a heterogeneous cancer driven by a constellation of diverse recurrent genetic aberrations. The ease to sample bone marrow allows easy access to the cancer cells and enables deep exploration of the genetics that drive ALL. Naturally, with every new genetic tool, the genetic constellation of ALL is often the first frontier to be explored. These deep explorations result in a detailed map of the genetic constellation of ALL (Figure 1) which is the basis of World Health Organization classification of tumors of hematopoietic and lymphoid tissues. From the 1960s, when karyotyping and chromosomal banding were established, investigators embarked on this 60-year journey of discovery. This discovery started with abnormal whole chromosome copy numbers termed aneuploidy. Excess chromosomes >50, also known as hyperdiploidy, was the most common driver (Figure 1). Translocations, where bits of chromosomes were aberrantly fused, led to the discovery of Philadelphia (Ph) chromosome t(9;22)/ BCR::ABL1 and t(1;19)/TCF3::PBX1. Translocations which do not change the banding patterns like t(12;21)/ETV6::RUNX1 took a while longer before yielding to discovery. Paralleling this discovery is better treatment. With better treatment, investigators found that these genetic drivers are prognostic i.e., they predict the risk of relapse. This prognostic value of genetic subtypes gave birth to genetic risk stratification and eventually genetically driven treatment like addition of imatinib and dasatinib for Ph ALL. However, the difficulty to karyotype lymphoblasts and the need for many different diagnostic platforms, like multiple fluorescence in situ hybridization (FISH) probes, limited widespread use of genetic stratification.
In the 2000s, gene arrays enticingly promised a single platform to interrogate genetic drivers of ALL. Gene expression microarrays, which measure the expression levels of tens of thousands genes at the same time, allowed the discovery of the “novel” subtype1 (later found to be the DUX4 subtype) and the Ph-like subtype.2 Using single
nucleotide polymorphism (SNP) arrays which simultaneously genotype hundreds of thousands of SNP, deletion of a segment in pseudoautosomal region 1 (PAR1) next to CRLF2 was identified.3
In late 2010s, transcriptomic sequencing (RNA-seq) promised another revolution. With RNA-seq, we can study both genetic expression profiles and the sequences of mRNA. Together, RNA-seq allowed us to identify gene rearrangements, karyotype, gene expression patterns as well as sequence mutations. Using RNA-seq of leukemic blasts, Gu et al. elegantly showed that >90% of ALL patients can be assigned to a specific genetic subtype.4 We and others have tried to implement RNA-seq in clinical practice.5,6 With standardization of RNA-seq library preparations and affordable sequencing services, perhaps the most significant obstacle remaining was bioinformatics analysis. In this issue of Haematologica, Hu et al 7 shared the Molecular Diagnosis of ALL (MD-ALL), an integrated analysis software for ALL subtype classification using RNA-seq. Using published RNA-seq data, they carefully selected the feature genes responsible for each subtype distinction, constructed machine learning models to perform gene expression analysis, and combined gene expression and genomic alterations to classify ALL subtypes. MD-ALL advanced the bioinformatics analysis for RNA-seq-based ALL classification by addressing three key areas:
i) a reliable reference dataset. Hu et al. assembled an RNA-seq dataset with 2,955 ALL cases around the world, representing more than 20 subtypes from both children and adult patients.
ii) standardization of gene expression analysis. With different analysis methods or features used, gene expression defined subtypes can be variable. For example, the BCR::ABL1like subtype defined by European researchers have minor variations compared to the Ph-like subtype defined by St. Jude investigators.2,3 Hu et al. tested the different feature selection methods and streamlined gene expression analysis
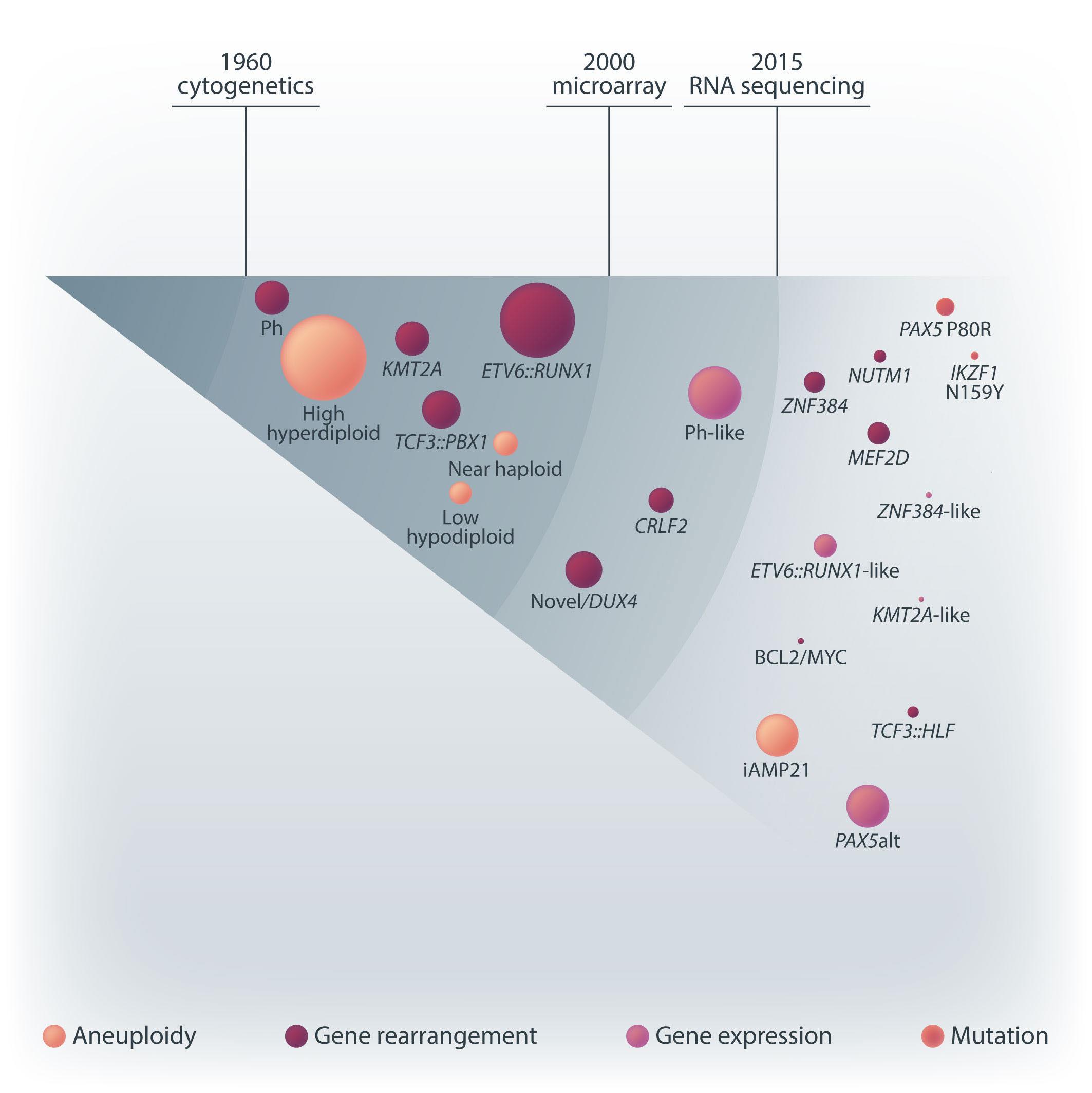
Figure 1. A brief history of acute lymphoblastic leukemia subtype classification. This figure summarizes the main technologies available and the subtypes discovered during different time periods. Sizes of the circles indicate approximate relative frequencies of acute lymphoblastic leukemia subtypes in children.
using multiple machine learning methods. This enhanced reproducibility and robustness for clinical use. iii) integration of multiple types of information into a final call. Though majority of cases can be uniquely assigned to a subtype, multiple genetic events may appear together. For example, high hyperdiploidy can occur with BCR::ABL1 fusion, and low hypodiploidy with TP53 mutations. A decision-making workflow is implemented in MD-ALL. The recent International Consensus Classification of acute lymphoblastic leukemia/lymphoma included nearly 30 subtypes. Efforts like MD-ALL are important for clinical use of the newly discovered subtypes, particularly in resource-constrained settings.
ALL subtypes have distinct sensitivity patterns to commonly used chemotherapy agents, 8 targeted therapy, and even to immune therapy.9 How to integrate these subtypes into risk stratification or treatment protocols need further
investigations. For example, the DUX4 subtype, despite poorer end of induction minimal residual disease (hence treated with intense treatment), have excellent outcomes. Yet, de-intensification for this favorable subtype needs to be done cautiously. On the other hand, intensifying therapy or use of novel treatment for newly discovered unfavorable subtypes, such as TCF3::HLF and MEF2D , is necessary. In additional, targeted or immune therapy could be used for certain subtypes, e.g., ABL1 inhibitors and blinatumomab for Ph ALL creating a chemotherapy free regimen is exciting.10
We are on the cusp of a brave new world of ALL: better understanding of the biological basis of each genetic subtype and better ways to treat them. With better and more ways to treat ALL, exploration of the genetic constellation of ALL is no longer an academic exercise, it transforms care.
Disclosures
No conflicts of interest to disclose.
1. Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1(2):133-143.
2. Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125-134.
3. Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndromeassociated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243-1246.
4. Gu Z, Churchman ML, Roberts KG, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296-307.
5. Chin WHN, Li Z, Jiang N, et al. Practical considerations for using RNA sequencing in management of B-lymphoblastic leukemia: Malaysia-Singapore Acute Lymphoblastic Leukemia 2020
Contributions
Both authors wrote, reviewed and approved the manuscript.
Implementation Strategy. J Mol Diagn. 2021;23(10):1359-1372.
6. Yu CH, Wu G, Chang CC, et al. Sequential approach to improve the molecular classification of childhood acute lymphoblastic leukemia. J Mol Diagn. 2022;24(11):1195-1206.
7 Hu Z, Jia Z, Liu J, et al. MD-ALL: an integrative platform for molecular diagnosis of B-cell acute lymphoblastic leukemia. Haematologica. 2024;109(6):1741-1754.
8. Lee SHR, Yang W, Gocho Y, et al. Pharmacotypes across the genomic landscape of pediatric acute lymphoblastic leukemia and impact on treatment response. Nat Med. 2023;29(1):170-179.
9 Li Y, Moriyama T, Yoshimura, et al. PAX5 epigenetically orchestrates CD58 transcription and modulates blinatumomab response in acute lymphoblastic leukemia. Sci Adv. 2022;8(50):eadd640.
10. Foà R, Bassan R, Vitale A, et al. Dasatinib–blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med. 2020;383(17):1613-1623.
Department of Leukemia, UT MD Anderson Cancer Center, Houston TX, USA and Rabin Medical Center and Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
Correspondence: E. Kugler ekugler@mdanderson.org
Received: January 26, 2024. Accepted: February 7, 2024. Early view: February 15, 2024.
https://doi.org/10.3324/haematol.2023.284796
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
In this issue of Haematologica, Cox et al. describe the therapeutic vulnerabilities of relapsed acute lymphoblastic leukemia (ALL) with TP53 aberration, a disease subset characterized by very poor prognosis. Utilizing a pediatric cancer drug library and high-throughput screening, the authors aimed to map the drug sensitivity profile of TP53-mutated B-ALL. They discovered that when combined with cytarabine, the class I histone deacetylase (HDAC) inhibitor romidepsin effectively restored chemotherapy sensitivity in TP53-deficient B-ALL.1
TP53 is the gene most commonly mutated in cancer.2 It frequently displays missense mutations that lead to the production of a p53 protein with impaired function, which fails to bind DNA and activate target genes. Mutant p53 can also have a dominant-negative effect on wild-type p53 and harbor gain-of-function activities by interacting with other transcription factors to drive oncogenic gene transcription.3,4 In the context of ALL, the incidence of TP53 aberrations at diagnosis differs significantly between pediatric and adult patients. While these aberrations are relatively rare in children, with an occurrence rate of about 2-3%, they are more frequent in adults, affecting 6-19% of all cases.5,6 These aberrations are especially prevalent in certain subtypes of ALL, particularly low hypodiploid and near triploid ALL, and are more frequently observed during relapse.6 In essence, TP53 mutations contribute significantly to the development of treatment-resistant clones in ALL, leading to early relapses and poorer survival outcomes. TP53 has long been considered undruggable. Since most small molecule drugs inhibit excessive protein activity, reactivating mutant proteins to restore their tumor suppressive properties can be challenging. Several strategies have been developed to target mutant TP53 in hematologic malignancies, with the aim of restoring a certain level of wild-type activity (PRIMA-1, APR-246, APR-538, arsenic tri-
oxide) or exploiting vulnerabilities caused by mutant TP53 (immune checkpoint inhibition).7 While promising in early phase studies, these strategies have not yet demonstrated efficacy in phase III trials, highlighting an urgent unmet need for novel therapeutic approaches.
Cox et al. sought to identify therapeutic vulnerabilities in TP53-deficient B-ALL. To this end, they employed CRISPR/ Cas9 to create isogenic pairs of TP53 wild-type (WT) and knockout (KO) from the relapsed B-cell precursor ALL cell lines, Nalm6 and RCH-ACV. This was followed by a high-throughput screening of 198 compounds used in standard care. The authors did not identify any compound that selectively targets TP53KO cells; however, romidepsin, a class I HDAC inhibitor, emerged as a potent suppressor of proliferation of both TP53KO and TP53WT cells.
In a series of in vitro experiments on TP53KO and TP53WT Nalm6 and RCH-ACV cells, the authors illustrated that romidepsin, when used in conjunction with cytarabine, elicited a synergistic effect. The synergy was consistently observed with other standard B-ALL chemotherapeutic agents, and it remained significant regardless of the TP53 status of the cells. Further validation of these findings in an expanded panel of B-ALL cell lines that included both TP53 wild-type and mutant forms strengthened the reliability of the results.
To elucidate the mechanism underlying the synergistic effect observed, the authors performed RNA sequencing on TP53KO and TP53WT Nalm6 and RCH-ACV cells. Notably, while sets of genes associated with apoptosis were enriched in samples treated with romidepsin, there was no observed upregulation of TP53 target genes, indicating that romidepsin may induce apoptosis through TP53-independent pathways.
Additionally, and perhaps counterintuitively, it was found that gene repression rather than de-repression was the
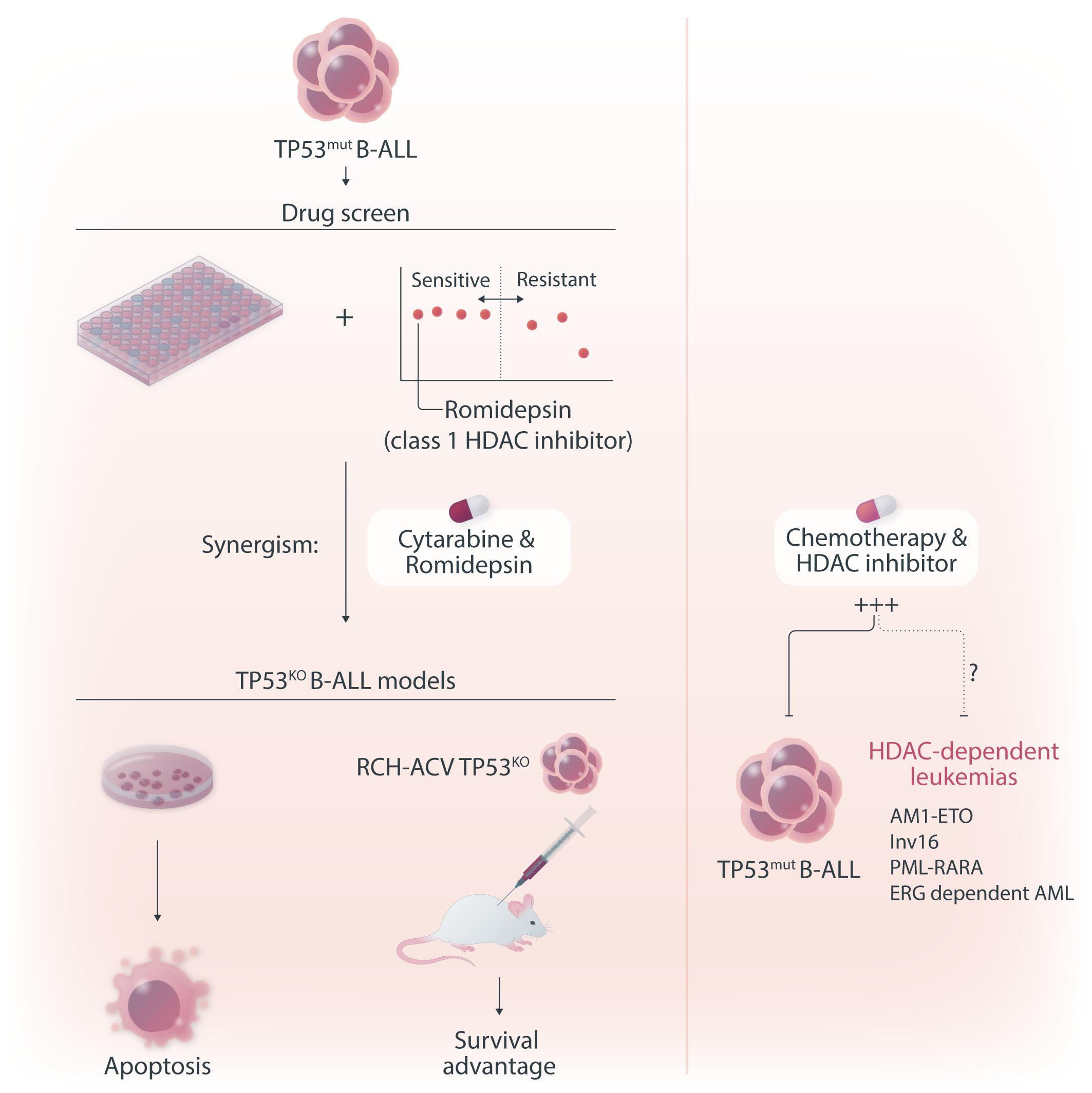
Figure 1. The role of histone deacetylase inhibition in the treatment of acute leukemia. In this issue of Haematologica, Cox et al studied the therapeutic vulnerabilities of relapsed acute lymphoblastic leukemia (ALL) with TP53 aberrations.1 The high-throughput drug screening they conducted demonstrated that combining the class I histone deacetylase (HDAC) inhibitor romidepsin with cytarabine is synergistic and can overcome the inherent chemoresistance of this high-risk subtype of ALL. Gene expression analysis suggested that romidepsin induces a distinct anti-leukemic effect independent of the TP53 pathway. The diagram on the right-hand side illustrates additional types of acute leukemia that are modulated by HDAC, highlighting the necessity for more research to determine which classes of HDAC inhibitors could be effective in treating various forms of acute leukemia.
predominant phenomenon in samples treated with romidepsin compared to untreated samples. These genes were associated with ribosome biogenesis and proteasome assembly pathways; however, the effect of romidepsin was not recapitulated upon treatment of B-ALL cell lines with proteasome inhibitors and cytarabine, suggesting an additional, distinct anti-leukemic effect induced by HDAC inhibition.
Finally, the study convincingly demonstrated that the findings extend to in vivo models. Immunodeficient mice transplanted with luciferase-expressing RCH-ACV p53KO cells had a reduced leukemia burden and survived longer when treated with the romidepsin-cytarabine combination than with cytarabine alone.
HDAC play a pivotal role in multilineage development and
hematopoietic stem cell fate.8 Simultaneous knockdown of HDAC1 and HDAC2 leads to early myeloid differentiation and loss of hematopoietic stem cells.9 Furthermore, the loss of HDAC1 and HDAC2 is linked to a marked impediment in pre-B-cell development, manifested by G1 arrest and apoptosis, underscoring the importance of HDAC in the early stages of B-cell development and terminal maturation.8 The development of HDAC inhibitors for therapy of hematopoietic malignancies originated from the observation that several compounds that induced differentiation of leukemic cell lines were inhibitors of HDAC.
HDAC have been implicated in the pathogenesis of certain subtypes of acute myeloid leukemia with distinct chromosomal translocations, such as AML1-ETO, Inv16, PML-RARA, and those involving high ERG expression and
dependency.10,11 The chimeric proteins recruit HDAC and co-repressor complexes to repress genes crucial for myeloid differentiation.11 Similarly, in the context of ALL, HDAC inhibitors suppress the MLL-AF4 fusion protein and other proto-oncogenes, triggering apoptosis in leukemic cells with KMT2A rearrangements.12 While the antileukemic mechanisms of HDAC inhibitors are not fully elucidated, they extend beyond histone deacetylation and include a wide array of biological effects on cancer cells. These effects encompass cell cycle arrest, metabolic reprogramming, autophagic cell death induction, generation of reactive oxygen species, and impairment of the DNA damage response.13
One strength of the study by Cox et al. is that it demonstrated that HDAC inhibitors elicit apoptosis and an anti-leukemic response which overcomes the intrinsic chemoresistance characteristic of TP53-mutated B-ALL. Previous observations support these findings, showing that HDAC
1. Cox WPJ, Evander N, van Ingen Schenau DS, et al. Histone deacetylase inhibition sensitizes p53-deficient B-cell precursor acute lymphoblastic leukemia to chemotherapy. Haematologica. 2024;109(6):1755-1765.
2. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333-339.
3. Boettcher S, Miller PG, Sharma R, et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science. 2019;365(6453):599-604.
4 Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ. 2018;25(1):161-168.
5. Chitadze G, Stengel A, John-Klaua C, et al. Somatic TP53 mutations are preleukemic events in acute lymphoblastic leukemia. Blood. 2023;141(13):1640-1644.
6. Hof J, Krentz S, van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185-3193.
7 Hassin O, Oren M. Drugging p53 in cancer: one protein, many targets. Nat Rev Drug Discov. 2023;22(2):127-144.
8. Wang P, Wang Z, Liu J. Role of HDACs in normal and malignant
inhibitors induce apoptosis in cancer cells via both TP53-dependent and independent pathways.14 Whether this effect is specific to an HDAC class, or a cell-intrinsic characteristic remains unknown.
In summary, the data published by Cox et al. provide a novel perspective on an established class of drugs. It is important to recognize that despite the convincing anti-cancer potential, the results of several advanced phase clinical trials of HDAC inhibitors in leukemia were disappointing. Notably, these trials predominantly utilized pan-HDAC inhibitors, indicating that future research should focus on determining which specific HDAC inhibitor classes may be effective in treating particular forms of acute leukemia (Figure 1). The work by Cox et al. underscores the value of this research direction.
No conflicts of interest to disclose.
hematopoiesis. Mol Cancer. 2020;19(1):5.
9 Heideman MR, Lancini C, Proost N, Yanover E, Jacobs H, Dannenberg JH. Sin3a-associated Hdac1 and Hdac2 are essential for hematopoietic stem cell homeostasis and contribute differentially to hematopoiesis. Haematologica. 2014;99(8):1292-1303.
10. Kugler E, Madiwale S, Yong D, et al. The NCOR-HDAC3 corepressive complex modulates the leukemogenic potential of the transcription factor ERG. Nat Commun. 2023;14(1):5871.
11. Zhang J, Gao X, Yu L. Roles of histone deacetylases in acute myeloid leukemia with fusion proteins. Front Oncol. 2021;11:741746.
12. Cruickshank MN, Ford J, Cheung LC, et al. Systematic chemical and molecular profiling of MLL-rearranged infant acute lymphoblastic leukemia reveals efficacy of romidepsin. Leukemia. 2017;31(1):40-50.
13. Li G, Tian Y, Zhu WG. The roles of histone deacetylases and their inhibitors in cancer therapy. Front Cell Dev Biol. 2020;8:576946.
14 Sonnemann J, Marx C, Becker S, et al. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br J Cancer. 2014;110(3):656-667.
Department of Hematology, Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona and IIB-Sant Pau, Institut Josep Carreras (IJC), Barcelona, Spain
In this issue of Haematologica, Baranwal et al. 1 analyze the clinical and biological features of myeloid neoplasms carrying mutations in the BCL6 corepressor (BCOR) gene. The introduction of next-generation sequencing (NGS) to the diagnosis of myeloid neoplasms such as acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) has shed light on the molecular pathogenesis of these disorders, especially for those cases without recurrent chromosomal alterations. Over the past decade, numerous somatic single-nucleotide variants (SNV) have been recognized as driver mutations and prognostic biomarkers in myeloid neoplasms.2 This growing evidence has been incorporated into AML and MDS classifications and predictive scores, including new molecularly-defined diagnostic categories and several mutations in risk stratification models.
The recent WHO and International Consensus classifications3,4 have proposed a new group called “AML with myelodysplasia-related gene mutations,” defined by somatic mutations in a set of genes, including BCOR. This category has also been incorporated into the European LeukemiaNet (ELN) 2022 AML classification5 within the adverse risk group. Along these lines, the Molecular International Prognostic Scoring System (IPSS-M) risk score for MDS has considered BCOR, among other genes, a predictor of poor prognosis for MDS patients.6
BCL6 corepressor (BCOR) is a gene located in chromosome X encoding for a transcriptional repressor that participates in one form of the Polycomb repressive complex 1. This multi-protein complex regulates gene expression through histone modification, and its function is crucial for hematopoiesis and lymphoid differentiation.7 BCOR gene alterations have been recurrently found in various human cancers, supporting its role as a tumor suppressor gene.8 In hematologic malignancies, somatic mutations in BCOR have been reported in myeloid and lymphoid neoplasms,9,10 and other non-malignant disorders like aplastic anemia.11
Baranwal et al. 1 sought to characterize BCOR-mutated (mBCOR) myeloid neoplasms. To investigate their incidence, they screened for BCOR mutations through NGS in a consecutive
Correspondence: J. F. Nomdedéu jnomdedeu@santpau.cat
Received: January 17, 2024. Accepted: January 26, 2024. Early view: February 8, 2024.
https://doi.org/10.3324/haematol.2023.284748
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
cohort of 6,887 adults treated at the Mayo Clinic from 2015 to 2017. They detected 138 (2%) patients carrying any SNV in BCOR. The authors describe the clinical features and outcomes of mBCOR patients compared to a wild-type cohort, and report that BCOR mutations are enriched in high-risk MDS and AML with an increasing incidence with age.
Interestingly, mBCOR MDS and AML display a distinct genetic signature, with a solid association to RUNX1 and U2AF1 mutations and mutual exclusion to other common mutations such as NPM1 or TP53. Regarding cytogenetics, most patients had a normal karyotype, with a minority having complex karyotypes or other high-risk abnormalities. Mutations in BCOR were distributed along the coding sequence with no particular hotspot and were mostly frameshift.
The prognosis of mBCOR AML and MDS in this study was poor, with a median overall survival (OS) of 15 months, irrespective of blast count or initial diagnosis. While only 52.8% of AML patients in this study were initially assigned to the ELN 2017 adverse risk category, the results of the AML cohort are comparable to the ELN adverse group.5 These data validate the inclusion of mBCOR AML into the adverse risk category of the ELN 2022 classification. Patients with complex karyotypes had the worst survival rates, while other co-occurring mutations made no substantial negative impact.
The authors demonstrate the beneficial effect of allogeneic stem cell transplant (alloSCT) in this group of myeloid neoplasms, suggesting that all mBCOR AML/MDS patients should be evaluated for upfront alloSCT when possible, given that the OS rate for alloSCT recipients was 61.1% at three years. Factors that worsened post-transplant outcomes in this cohort were RUNX1 mutations and the presence of complex karyotypes.
The results published in this paper support the view that BCOR mutations identify a high-risk subgroup of myeloid neoplasms with a unique genetic signature and unfavorable prognosis that can be partially modified with alloSCT. Further research is needed to elucidate the impact of BCOR mutations on the survival of AML/MDS patients treated with venetoclax-based regimens or other target therapies.
Disclosures
No conflicts of interest to disclose.
1. Baranwal A, Gurney M, Basmaci R, Katamesh B, He R, Viswanatha DS, et al. Genetic landscape and clinical outcomes of patients with BCOR mutated myeloid neoplasms. Haematologica. 2024;109(6):1779-1791.
2. Cancer Genome Altas Research Network; Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074.
3. Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200-1228.
4 Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703-1719.
5. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood.
Contributions
All authors discussed the results of the paper and wrote the editorial.
2022;140(12):1345-1377.
6. Bernard E, Tuechler H, Greenberg PL, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022;1(7):EVIDoa2200008.
7. Kelly MJ, So J, Rogers AJ, et al. Bcor loss perturbs myeloid differentiation and promotes leukaemogenesis. Nat Commun. 2019;10(1):1347.
8. Astolfi A, Fiore M, Melchionda F, Indio V, Bertuccio SN, Pession A. BCOR involvement in cancer. Epigenomics. 2019;11(7):835-855.
9 Sportoletti P, Sorcini D, Falini B. BCOR gene alterations in hematologic diseases. Blood. 2021;138(24):2455-2468.
10 Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2023;122(8):3169-1277.
11. Yoshizato T, Dumitriu B, Hosokawa K, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373(1):35-47.
1Department of Medicine and Surgery, University of Milan Bicocca, Milan and 2Division of Immunohematology and Transfusion Medicine, Hospital Papa Giovanni XXIII, Bergamo, Italy
Correspondence: A. Falanga annafalanga@yahoo.com
Received: November 30, 2023. Accepted: December 7, 2023. Early view: December 14, 2023.
https://doi.org/10.3324/haematol.2023.284291 ©2024 Ferrata Storti Foundation Published under a CC BY-NC license
In this issue of Haematologica, Patell and colleagues,1 present a post hoc analysis of the Hokusai VTE Cancer study, a randomized phase III trial comparing edoxaban with dalteparin for treatment of acute venous thromboembolism (VTE) in patients with cancer.2 The aim was to evaluate the outcomes of major bleeding, clinically relevant non-major bleeding (CRNMB), recurrent VTE, and survival, in cancer patients with thrombocytopenia (TP) (i.e., platelet count <100x106/L at one or more specified time points during the trial) who were undergoing anticoagulation for acute VTE. The results show that patients with TP experienced significantly higher major bleeding (9.0% vs. 4.0%, sub-distribution hazard ratio [SHR]=2.4, 95% confidence interval [CI]: 1.19-5.06) and CRNMB (17.9% vs. 9.6%, SHR=2.0, 95% CI: 1.21-3.32) than patients without TP. In addition, TP did not reduce recurrent VTE (9.8% vs. 7.4%, SHR=1.3, 95% CI: 0.7-2.6).
In a group of patients with TP and gastrointestinal (GI) cancer the rate of major bleeding was higher with edoxaban compared to dalteparin (16.8% vs. 0%), whereas in patients with TP and hematologic malignancies this rate was higher with dalteparin compared to edoxaban (19.0% vs. 0%).
TP exposes patients to bleeding complications and represents a relevant limiting factor for use of antithrombotic medications, which are often required in malignant disease due to the increased risk of both venous and arterial thrombosis.3 Notably, the presence of TP is not protective of VTE.4 In cancer patients, TP is rather frequent, as a result of the bone marrow primary or secondary involvement by malignant disease or as a consequence of anti-cancer treatments. A thoughtful balance between the severity of TP and the need for anticoagulation must be accomplished when TP occurs in patients with an acute VTE event or in those who are already on chronic anticoagulation for one known indication (i.e., prevention of stroke in atrial fibrillation, or recurrent VTE). In those scenarios, both the thrombotic and
bleeding risks of the individual patient should be carefully considered (Figure 1). The perception of a prevailing bleeding risk supports the decision by physician to hold or reduce the dose of antithrombotic drugs. Differently, the perception of a high thrombotic risk drives the decision towards continuing antithrombotic therapy at a full or reduced dose with or without supportive platelet transfusions.5 As reviewed recently,3 the results of a number of randomized controlled trials (i.e., HOKUSAI VTE, SELECT-D, ADAM-VTE, CARAVAGGIO, CANVAS, and CASTA-DIVA studies) have consolidated the recommendation by international guidelines6,7 for the use of anti-Xa direct oral anticoagulants (DOAC) as a first line option for treatment of cancer-associated VTE. However, there are still areas of uncertainties, particularly for the use of these drugs in the presence of concomitant TP. Available evidence suggests that in mild to moderate TP (i.e., 50-100x106/L platelet) full-dose anticoagulation in patients with cancer-associated VTE is generally safe,8 however, according to recent European Hematology Association guidelines,9 when TP is not stable and is expected to drop <50x106/L in the next days to weeks, low molecular weight heparin (LMWH) should be preferred over DOAC and VKA. Furthermore, due to lack of data, these guidelines recommend not to use DOAC in conditions of severe TP (i.e., <50x106/L). Given the increasingly widespread use of DOAC for treatment of VTE in patients with cancer, the need to manage this type of anticoagulation in TP cancer patients may occur with growing frequency, and it is therefore important to determine the behaviour of DOAC and possible dose adjustments in this specific setting. As the current guidelines on TP and anticoagulation in cancer patients are mainly based on consensus guides and expert opinions, with their work, Patell and colleagues give a great impulse, that should be followed by others, to improve evidence-based decisions by clinicians. Indeed, up
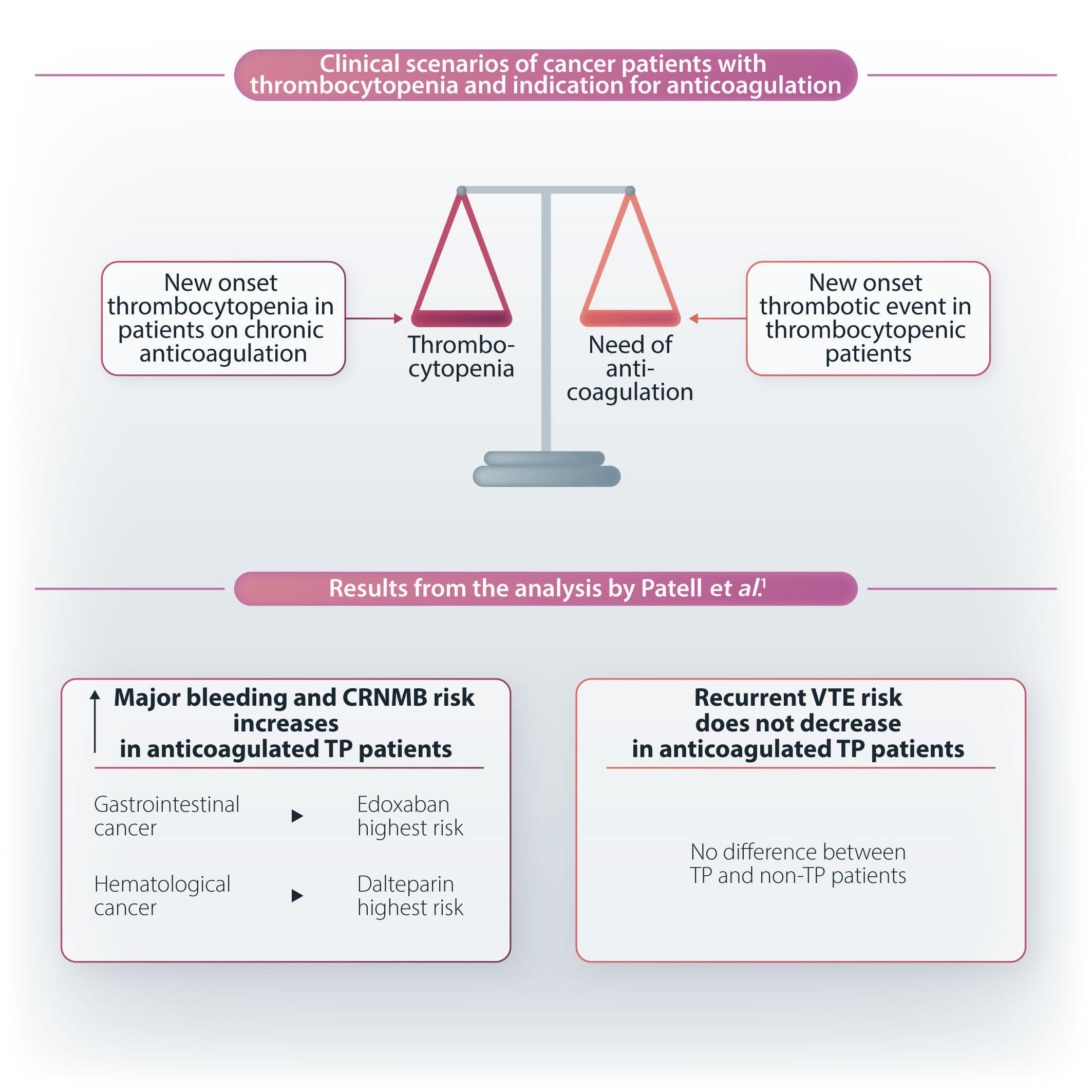
Figure 1. Anticoagulation plus thrombocytopenia in cancer patients: evidence from the HOKUSAI VTE post hoc analysis. CRNMB: clinically relevant non-major bleeding; TP: thrombocytopenia; VTE: venous thromboembolism.
to now there are no ad hoc studies or randomized clinical trials to test strategies of anticoagulation in cancer patients with TP, to the opposite thrombocytopenic cancer patients are often excluded from enrolment in trials testing efficacy and safety of anticoagulant drugs. Therefore using available data collected from prospective registries, as done by the investigators of the TROVE study,10 or using post hoc analysis of existing phase III randomized clinical trials investigating DOAC for cancer-associated VTE treatment, as done by this post hoc analysis, are currently the best possible approaches to take a step forward. Although these studies have limitations in that they have enrolled a small percentage of patients with TP and have excluded
1. Patell R, Hsu C, Shi M, et al. Impact of mild thrombocytopenia on bleeding and recurrent thrombosis in cancer. Haematologica. 2024;19(6):1849-1856.
severe TP forms, they still provide interesting information on mild-moderate TP. These data will help to lay the foundation for future clinical studies that, due to consistent sample size and high quality, will be able to dictate specific strategies for the management of TP in patients with cancer-associated VTE, who are receiving all different types of anticoagulant drugs, including DOAC.
Disclosures
No conflicts of interest to disclose.
Contributions
Both authors contributed equally.
2. Raskob GE, van Es N, Verhamme P, et al. Edoxaban for the treatment of cancer-associated venous thromboembolism. N Engl J Med. 2018;378(7):615-624.
3. Falanga A, Marchetti M. Cancer-associated thrombosis: enhanced awareness and pathophysiologic complexity. J Thromb Haemost. 2023;21(6):1397-1408.
4 Leader A, ten Cate H, Spectre G, et al. Antithrombotic medication in cancer-associated thrombocytopenia: current evidence and knowledge gaps. Crit Rev Oncol Hematol. 2018;132:76-88.
5. Leader A, Ten Cate V, Ten Cate-Hoek AJ, et al. Anticoagulation in thrombocytopenic patients with hematological malignancy: a multinational clinical vignette-based experiment. Eur J Intern Med. 2020;77:86-96.
6. Lyman GH, Carrier M, Ay C, et al. American Society of Hematology 2021 guidelines for management of venous thromboembolism: prevention and treatment in patients with cancer. Blood Adv. 2021;5(4):927-974.
7 Falanga A, Ay C, Di Nisio M, et al. Venous thromboembolism in cancer patients: ESMO clinical practice guideline. Ann Oncol. 2023;34(5):452-467.
8. Samuelson Bannow BT, Lee A, Khorana AA, et al. Management of cancer-associated thrombosis in patients with thrombocytopenia: guidance from the SSC of the ISTH. J Thromb Haemost. 2018;16(6):1246-1249.
9 Falanga A, Leader A, Ambaglio C, et al. EHA guidelines on management of antithrombotic treatments in thrombocytopenic patients with cancer. Hemasphere. 2022;6(8):e750.
10 Carney BJ, Wang TF, Ren S, et al. Anticoagulation in cancerassociated thromboembolism with thrombocytopenia: a prospective, multicenter cohort study. Blood Adv. 2021;5(24):5546-5553.
Division of Hematology Oncology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
Correspondence: H. Al-Samkari hal-samkari@mgh.harvard.edu
Received: November 14, 2023.
Accepted: January 26, 2024. Early view: February 8, 2024.
https://doi.org/10.3324/haematol.2023.284460 ©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Uzun and colleagues illustrate a rare case of adenovirus infection associated with thrombocytopenia, cerebral venous sinus thrombosis, and heparin-independent anti-platelet factor 4 (PF4) antibodies.1 In classic heparin-induced thrombocytopenia (HIT), negatively charged heparin molecules charge, neutralize and facilitate close approximation of positively charged PF4 tetramers while causing neo-antigen sites to form on two ends of the PF4 tetramer. Heparin-dependent anti-PF4/heparin complex antibodies subsequently bind to these sites on platelet membranes and elicit FcγIIa receptor-mediated platelet activation, thrombocytopenia, and thrombosis. In contrast, vaccine-induced immune thrombocytopenia and thrombosis (VITT) involves heparin-independent antibodies targeting the heparin-binding domain of PF4, distinct from those in classic HIT.2
VITT was first characterized in 2021 during the COVID-19 pandemic in association with the two adenovirus vector vaccines ChAdOx1 nCoV-19 and Ad26.COV2 and has subsequently garnered substantial global health interest.3 VITT is a particularly severe anti-PF4 disorder with most patients experiencing symptomatic thrombotic complications, often in otherwise unusual sites such as splanchnic vein thrombosis and cerebral venous sinus thrombosis (CVST). Despite occurring in only 1-2 per 100,000 people in the general population annually and being a rare occurrence with HIT, CVST is the most common thrombotic complication of VITT, occurring in 25-60% of such patients.2,4 The patient presented by Uzun and colleagues had not received an adenovirus vector vaccine but instead had an adenovirus infection subsequently leading to severe thrombocytopenia, CVST, and an antibody profile analogous to that of VITT. PF4/heparin enzyme immunoassay (EIA) showed a strong response which is consistent with the high sensitivity of IEA assays for both HIT and VITT antibodies. A major diagnostic branchpoint here was that a heparin-induced platelet activation (HIPA) assay with
low heparin concentration was negative, making classic HIT unlikely, whereas a modified assay with addition of exogenous PF4 was positive (PF4-enhanced HIPA or PIPA). These results are consistent with a VITT-like profile. Two additional patients have recently been described with adenovirus infection, thrombotic events (one with CVST), and VITT-like antibody profiles.5
Improved recognition and diagnosis of this clinical entity is important due to the different treatment paradigms for VITT (and VITT-like disorders) versus classic HIT. Guidelines for treatment of VITT recommend the use of intravenous immunoglobulin (IVIG),6 and mechanistically IVIG has been shown to inhibit FcγIIa-mediated platelet activation and subsequent thrombocytopenia and thrombosis. Uzun and colleagues’ patient with adenovirus infection demonstrated significant improvement in platelet count and clinical stabilization with high-dose IVIG. Subsequently, ex vivo analysis of the patient’s serum demonstrated the ability to induce procoagulant platelet formation along with abrogation by IVIG. This case, along with the others that have been published, suggests that adenovirus-associated thrombosis and thrombocytopenia syndrome with a consistent VITTlike antibody profile could be managed similarly to VITT. This may be particularly relevant in cases of severe and unusual thrombosis like CVST requiring urgent treatment. Although typically contraindicated in HIT, heparin was used for anticoagulation in this patient due to initial concern for ITP rather than HIT. The patient did well and did not experience rebound thrombocytopenia after heparin exposure. This observation is consistent with existing data suggesting that heparin is safe and effective for anticoagulation in VITT. Additionally, heparin has been shown to inhibit binding of VITT anti-PF4 antibodies to their culprit heparin-binding site.7 Given that both VITT and adenovirus-associated thrombosis and thrombocytopenia are newly recognized clinical entities, clinicians may appropriately have reservations about usage of heparin given that these are PF4 dis-
orders. This case is illustrative of the importance of better understanding the nuances of the pathophysiology unique to each PF4 disorder to facilitate optimal management of these life-threatening disorders. What implications do these VITT-like manifestations of adenovirus infection and the ChAdOx1 nCoV-19 and Ad26. COV2 vaccines have on the future safety analyses and development of adenovirus-based vaccines such as those being developed for influenza, Ebola, Zika, malaria, and others? One recent meta-analysis of clinical trial of adenovirus vector-based vaccines did not observe a class-wide effect towards either thrombocytopenia or coagulopathy/ thrombotic events in the general populations or in the pregnant population, although more prospective data is needed.8 Further data characterizing any potential association between other viral infections/vectors with a thrombosis and thrombocytopenia syndrome would be helpful, although the fact that VITT itself is a recently discovered phenomenon, along with understanding of its PF4 antibody profiles, limits the ability of retrospective research to potentially associate this pathophysiology as a complication of other viral infections and better understand its actual incidence in adenoviral infections. It is interesting to note that a VITT-like syndrome was recently described in a patient after a human papilloma virus vaccine.9 And although cases of VITT have been reported with the mRNA-based COVID-19 vaccines, these have been much rarer on an epidemiological scale.
Recent data from Warkentin and colleagues shows that
1. Uzun G, Zlamal J, Althaus K, et al. Cerebral venous sinus thrombosis and thrombocytopenia due to heparin-independent anti-PF4 antibodies after adenovirus infection. Haematologica. 2024;109(6);2010-2015.
2. Warkentin TE. Platelet-activating anti-PF4 disorders: an overview. Semin Hematol. 2022;59(2):59-71.
3. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med. 2021;384(22):2092-2101.
4. Palaiodimou L, Stefanou MI, Katsanos AH, et al. Cerebral venous sinus thrombosis and thrombotic events after vectorbased COVID-19 vaccines: a systematic review and metaanalysis. Neurology. 2021;97(21):e2136-e2147.
5. Warkentin TE, Baskin-Miller J, Raybould AL, et al. Adenovirusassociated thrombocytopenia, thrombosis, and VITT-like
sera obtained from selected patients prior to the COVID-19 pandemic demonstrated VITT-like characteristics.10 These patients were identified by history of thrombocytopenia and/or thrombosis with strong reactivity in anti-PF4/heparin IgG EIA but negative HIPA and a significant percentage subsequently tested positive by newer rapid anti-PF4 assays. The authors convey that this VITT-like antibody signal temporally could not have been associated with COVID-19 or COVID-19-directed adenovirus vector-based vaccines. The greater understanding of this clearly rare but severe complication of adenoviral infection (and possibly other viral infections) contributed by the case published by Uzun and colleagues is valuable, and reminds us that we must consider the possibility of a rare anti-PF4 disorder in patients with apparent thrombosis and thrombocytopenia syndrome but without antecedent heparin exposure.
HA-S discloses research funding to institution (Agios, Sobi, Novartis, Vaderis and Amgen) and consultancy (Agios, Sobi, Moderna, Novartis, Rigel, argenx, Forma and Pharmacosmos). ABS has no conflicts of interest to disclose.
Contributions
ABS and HA-S wrote and edited this editorial.
Funding
HA-S is funded by the National Heart, Lung, and Blood Institute (1K23HL159313).
antibodies. N Engl J Med. 2023;389(6):574-577.
6. Warkentin TE, Cuker A. COVID-19: vaccine-induced immune thrombotic thrombocytopenia (VITT). UpToDate. 2023 Dec 7.
7. Huynh A, Kelton JG, Arnold DM, Daka M, Nazy I. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature. 2021;596(7873):565-569.
8. Pischel L, Patel KM, Goshua G, Omer SB. Adenovirus-based vaccines and thrombosis in pregnancy: a systematic review and meta-analysis. Clin Infect Dis. 2022;75(7):1179-1186.
9. Kanack AJ, Laegreid IJ, Johansen S, Reikvam H, Ahlen MT, Padmanabhan A. Human papilloma virus vaccine and VITT antibody induction. Am J Hematol. 2022;97(10):E363-E364.
10. Schonborn L, Esteban O, Wesche J, et al. Anti-PF4 immunothrombosis without proximate heparin or adenovirus vector vaccine exposure. Blood. 2023;142(26):2305-2314.
Correspondence: F. Locatelli franco.locatelli@opbg.net
Received: January 15, 2024. Accepted: January 22, 2024.
https://doi.org/10.3324/haematol.2023.284553
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
The improvements in survival probability of children with hematologic malignancies observed in the last three decades represent one of the most remarkable successes of modern medicine. Nowadays, almost 90% of children with acute lymphoblastic leukemia (ALL) and more than 70% of those with acute myeloid leukemia (AML) are cured with multiagent chemotherapy protocols, together with, in some selected subsets of patients, allogeneic hematopoietic stem cell transplantation.1,2 However, despite these impressive results, there is still a proportion of patients with acute leukemia for whom innovative approaches are desperately needed to rescue them from their relapsed/ refractory malignancies. In addition, there is growing evidence that the optimization of conventional treatments has reached its limit and we cannot further increase the intensity of chemo/radiotherapy in children with either ALL or AML, also considering that a relevant proportion of patients experience both acute and long-term severe toxicities, including life-threatening infections, pancreatitis, osteonecrosis, cardiomyopathy, neurocognitive defects, loss of fertility and endocrinopathies.3-6
Immunotherapy has emerged in the last decade as one of the most promising approaches for treating patients with relapsed/refractory hematologic malignancies and for sparing severe toxicities related to intense chemo/radiation therapy. The new immunotherapy agents that have been developed include antibodies targeting checkpoint inhibitors, naked antibodies directed against antigens expressed on tumor cells, antibody-drug conjugates,7 bispecific T-cell engagers8,9 and T cells re-directed against the tumor elements through the use of chimeric antigen receptors (CAR).10
In this issue of Haematologica, five different contributions prepared by experts in the field analyze the current use and the future prospective applications of different types of immunotherapies in pediatric patients with either ALL or AML (Figure 1).11-15
Some of these immunotherapies, such as blinatumomab (the prototype of bispecific T-cell engagers) and CD19-tar-
geting CAR T cells (tisagenlecleucel) have already been approved by the Food and Drug Administration in the USA and the European Medicines Agency in the European Union for clinical use in children with relapsed/refractory B-ALL, while others are under investigation and/or early clinical development (e.g., inotuzumab, CAR T cells for T-ALL or for AML). The studies so far conducted have clearly documented that these therapies are characterized by selective mechanisms of action, thus potentially sparing the acute and chronic toxicities associated with intensive, multiagent chemotherapy courses, which may have a particularly detrimental effect in subjects with a long expectancy of life. However, novel treatment-related toxicities have also emerged, such as the cytokine release syndrome and immune cell-associated neurotoxicity, which were unknown before the development and implementation of these treatments. Efforts aimed at properly managing these specific complications with targeted treatments, without jeopardizing the therapeutic effects of immune treatments are key to optimizing the benefit-to-risk ratio for the patients. In addition, we are still in the learning curve for already approved therapies with regard to the role played by factors influencing clinical efficacy (e.g., leukemia burden, T-cell function, downregulation or loss of the target antigen, manufacturing processes, cell doses, extramedullary leukemia relapse).16-19 Future studies will have to elucidate the best place in which these novel approaches should be inserted in the patient’s therapeutic journey and how these therapies influence each other.20 We also still have to further elaborate and refine strategies to optimize the benefit deriving from the application of these therapies, including, for example, administration of blinatumomab subcutaneously instead of by continuous intravenous infusion or the use of humanized CAR constructs potentially able to lead to longer persistence and greater efficacy.
What is certain is that, in the forthcoming years, the therapeutic scenario of childhood ALL and, it is to be hoped,
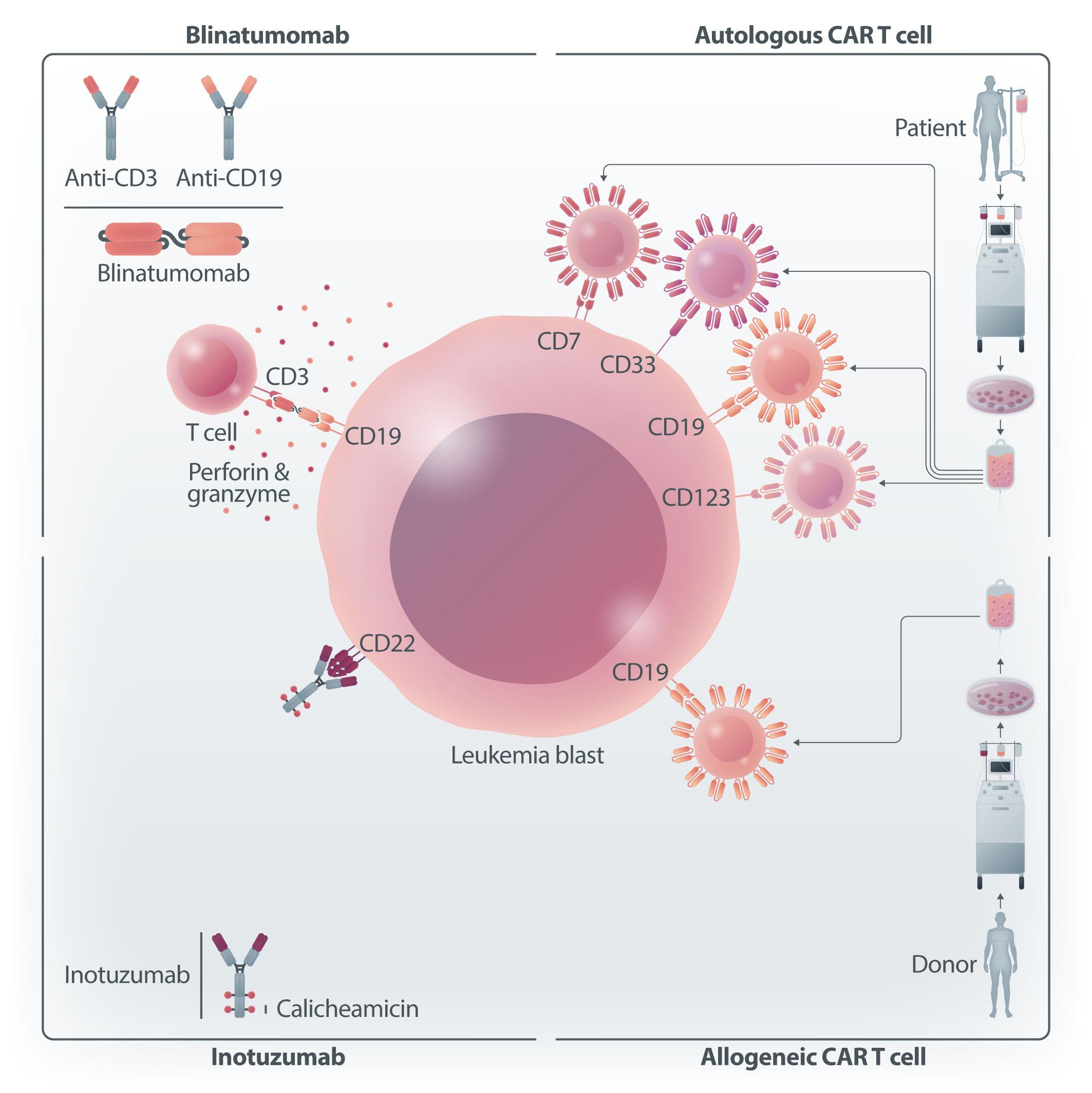
Figure 1. Schematic representation of different immunotherapy approaches for the treatment of childhood hematologic malignancies. Bispecific, CD3-CD19, T-cell engager: blinatumomab. Antibody-drug conjugate targeting CD22 and carrying calicheamicin: inotuzumab. Autologous chimeric antigen receptor (CAR) T cells directed towards CD19, CD7, CD33 or CD123. Allogeneic CAR T cells directed towards CD19. Figure created with BioRender.
AML will be marked by the introduction of several immunotherapy approaches not only in patients with relapsed/ refractory disease, but also in the treatment strategy of newly diagnosed patients, as recently demonstrated in infants with B-ALL.21 The five contributions published in this issue of Haematologica will offer readers the most
1. Pieters R, Mullighan CG, Hunger SP. Advancing diagnostics and therapy to reach universal cure in childhood ALL. J Clin Oncol. 2023;41(36):5579-5591.
2. Zwaan CM, Kolb EA, Reinhardt D, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol. 2015;33(27):2949-2962.
recent and up-to-date information and perspective of use of these therapies which hold the promise to represent a cornerstone in the field of pediatric hematology.
Disclosures
No conflicts of interest to disclose.
3. Schmiegelow K, Attarbaschi A, Barzilai S, et al. Consensus definitions of 14 severe acute toxic effects for childhood lymphoblastic leukaemia treatment: a Delphi consensus. Lancet Oncol. 2016;17(6):e231-e239.
4 Andrés-Jensen L, Attarbaschi A, Bardi E, et al. Severe toxicity free survival: physician-derived definitions of unacceptable
long-term toxicities following acute lymphocytic leukaemia. Lancet Haematol. 2021;8(7):e513-e523.
5. Dixon SB, Liu Q, Chow EJ, et al. Specific causes of excess late mortality and association with modifiable risk factors among survivors of childhood cancer: a report from the Childhood Cancer Survivor Study cohort. Lancet. 2023;401(10386):1447-1457.
6. Gibson TM, Mostoufi-Moab S, Stratton KL, et al. Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970-99: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2018;19(12):1590-1601.
7 Pennesi E, Michels N, Brivio E, et al. Inotuzumab ozogamicin as single agent in pediatric patients with relapsed and refractory acute lymphoblastic leukemia: results from a phase II trial. Leukemia. 2022;36(6):1516-1524.
8. von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/ refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
9. Locatelli F, Zugmaier G, Rizzari C, et al. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia. A randomized clinical trial. JAMA. 2021;325(9):843-854.
10 Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517.
11. Brivio E, Bautista Sirvent FJ, Zwaan CM. Naked antibodies and antibody-drug conjugates: targeted therapy for childhood acute lymphoblastic leukemia. Haematologica. 2024;109(6):1700-1712.
12. Naik S, Velasquez MP, Gottschalk S. Chimeric antigen receptor T-cell therapy in childhood acute myeloid leukemia: how far are we from a clinical application? Haematologica. 2024; 109(6):1656-1667.
13. Lyons KU, Gore L. Bispecific T-cell engagers in childhood B-acute lymphoblastic leukemia. Haematologica 2024;109(6):1668-1676.
14 Locatelli F, Del Bufalo F, Quintarelli C. Allogeneic chimeric antigen receptor T cells for children with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Haematologica. 2024;109(6):1689-1699.
15. Oh BLZ, Vinanica N, Wong DMH, Campana D. Chimeric antigen receptor T-cell therapy for T-cell acute lymphoblastic leukemia. Haematologica. 2024;109(6):1677-1688.
16. Queudeville M, Stein AS, Locatelli F, et al. Low leukemia burden improves blinatumomab efficacy in patients with relapsed/ refractory B-cell acute lymphoblastic leukemia. Cancer. 2023;129(9):1384-1393.
17. Schultz LM, Baggott C, Prabhu S, et al. Disease burden affects outcomes in pediatric and young adult B-cell lymphoblastic leukemia after commercial tisagenlecleucel: a pediatric realworld chimeric antigen receptor consortium report. J Clin Oncol. 2022;40(9):945-955.
18. Sotillo E, Barrett D, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282-1295.
19. Jacoby E, Ghorashian S, Vormoor B, et al. CD19 CAR T-cells for pediatric relapsed acute lymphoblastic leukemia with active CNS involvement: a retrospective international study. Leukemia. 2022;36(6):1525-1532.
20 Myers RM, Taraseviciute A, Steinberg SM, et al. Blinatumomab nonresponse and high-disease burden are associated with inferior outcomes after CD19-CAR for B-ALL. J Clin Oncol. 2022;40(9):932-944.
21. van der Sluis IM, de Lorenzo P, Kotecha RS, et al. Blinatumomab added to chemotherapy in infant lymphoblastic leukemia. N Engl J Med. 2023;388(17):1572-1581.
Department of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN, USA
Correspondence: S. Gottschalk stephen.gottschalk@stjude.org
Received: November 7, 2023. Accepted: February 28, 2024.
https://doi.org/10.3324/haematol.2023.283817
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Abstract
Recurrent and/or refractory (R/R) pediatric acute myeloid leukemia (AML) remains a recalcitrant disease with poor outcomes. Cell therapy with genetically modified immune effector cells holds the promise to improve outcomes for R/R AML since it relies on cytotoxic mechanisms that are distinct from chemotherapeutic agents. While T cells expressing chimeric antigen receptors (CAR T cells) showed significant anti-AML activity in preclinical models, early phase clinical studies have demonstrated limited activity, irrespective of the targeted AML antigen. Lack of efficacy is most likely multifactorial, including: (i) a limited array of AML-specific targets and target antigen heterogeneity; (ii) the aggressive nature of R/R AML and heavy pretreatment of patients; (iii) T-cell product manufacturing, and (iv) limited expansion and persistence of the CAR T cells, which is in part driven by the immunosuppressive AML microenvironment. Here we review the results of early phase clinical studies with AML-specific CAR T cells, and avenues investigators are exploring to improve their effector function.
The overall survival of pediatric patients with newly diagnosed acute myeloid leukemia. (AML) has improved over the last two decades with rates reported to be close to 70%.1-4 However, the event-free survival of these patients has plateaued at about 55% despite the use of intensive therapies.1-3 Furthermore, the outcome of children with relapsed and/or refractory (R/R) disease remains poor. Recent data from the Berlin Frankfurt Münster (BFM) group and Children’s Oncology Group (COG) showed that the overall survival rates of pediatric patients with first relapse of AML were 42% and 35%, respectively.4 For patients with second relapse of AML, the overall survival at 5 years remains dismal (15 ± 4%) based on BFM and COG data.5 Similar outcomes were reported in a recent analysis by a Nordic-Dutch-Belgian-Spain-Hong-Kong-Israel-Portugal (NOPHO-DB-SHIP) consortium.6 This high rate of treatment failure has been attributed in part
to the existence of chemotherapy-resistant leukemic stem cells, a minor fraction of leukemic cells that are capable of maintaining and re-initiating the disease.7,8 While allogeneic hematopoietic stem cell transplant (HSCT) at present offers the highest probability of long-term sustained remission,3 its success relies on several factors with remission status prior to HSCT being one of the most important prognostic factors.9 However, it is challenging to re-induce remission in these patients with salvage chemotherapy due to toxicities and limited efficacy of standard or investigational therapies. Many patients are therefore unable to proceed to allogeneic HSCT.
Adoptive transfer of chimeric antigen receptor (CAR) T cells has emerged as a promising treatment option for pediatric patients with CD19-positive B-cell acute lymphoblastic leukemia.10,11 CD19-CAR T cells have shown potent anti-leukemia activity in the R/R setting, leading to their approval by the Food and Drug Administration in 2017.10-14 In contrast, the clinical experience with AML-specific CAR T cells is
limited with information on several studies only reported in abstract form. Overall, the clinical activity of AML-specific CAR T cells, irrespective of the targeted antigen, has been underwhelming and has shown variable complete response (CR) rates.15-33 Importantly, early phase clinical and preclinical studies have highlighted several roadblocks (Figure 1), including: (i) a limited array of AML-specific targets and target antigen heterogeneity; (ii) the aggressive nature of R/R AML and heavy pretreatment of patients; (iii) T-cell product manufacturing; and (iv) limited expansion and persistence of the CAR T cells, which is in part driven by the immunosuppressive AML microenvironment.18,34-39 In this review we summarize published results of early phase clinical studies with AML-specific CAR T cells from centers around the globe, including the USA, Europe, Australia and China, presenting pediatric as well as adult studies. In the second part of the review, we then outline potential ways to overcome current roadblocks to effective CAR T-cell therapies for AML. This review is
not meant to be comprehensive, and we refer the reader to other excellent reviews on CAR immune cell therapies for AML that have been published recently.36,40-42 Likewise, due to space limitations, we will not review combinatorial approaches in which CAR T cells are combined with other treatment modalities.
An ideal antigen for CAR T-cell therapy is a cell surface antigen that is highly expressed in all malignant cells but not expressed in healthy tissues. As for many other malignancies, such an ideal target remains elusive for AML. To date, CD33, CD123, and CLL1 (CLEC12A) have been the most frequently targeted AML antigens.43-49 While these are overexpressed on the cell surface of AML blasts, they are also expressed on normal hematopoietic cells, raising

Figure 1. Strategies to overcome acute myeloid leukemia-specific chimeric antigen receptor T-cell therapy roadblocks. Roadblocks to acute myeloid leukemia (AML)-specific chimeric antigen receptor (CAR) T-cell therapy include: (i) a limited array of AML-specific targets and target antigen heterogeneity (the antigen dilemma); (ii) the aggressive nature of relapsed/refractory AML and the fact that patients are heavily pretreated (fragile) (clinical challenges); (iii) T-cell product manufacturing (cell production); and (iv) limited expansion and persistence (limited fitness) of the CAR T cells. Potential strategies to overcome each roadblock are highlighted. For additional details see the text. HSC: hematopoietic stem cells.
concerns about myelotoxicity, with CD33 and CD123 being pan-myeloid antigens that are also expressed on hematopoietic stem cells and CLL1 being expressed on mature myeloid cells, including mature granulocytes. Nevertheless, the safety of targeting CD33 and CD123 has been shown in the pediatric clinical setting with the use of gemtuzumab, a CD33-specific monoclonal antibody drug conjugate,50,51 leading to its approval by the Food and Drug Administration, and flotetuzumab, a bispecific CD123xCD3 antibody. 52 Early phase clinical trials of CAR T cells targeting CD123, CD33 and CLL1, as described in the next section, have used either safety switches or adopted a bridge-to-transplant strategy to overcome this issue. Targets that are expressed on the cell surface of AML blasts and not on normal myeloid cells are also being explored, including NKG2D, CD70, and GRP78.29,53-55 However, these are also associated with potential on-target/off-cancer toxicity, including being upregulated in normal cells under conditions of cellular stress (NKG2D, GRP78) or in subsets of activated immune cells (CD70). Other AML targets that have been or are being actively explored include CD38, CD44v6, CD117 (c-KIT) CD276 (B7-H3), CD327 (Siglec-6), FLT3, FRβ, GRP78, LILRB3, LILRB4, and TIM3.56-65 While these promise to have limited myelotoxicity, this aspect needs to be carefully evaluated in preclinical studies. For example, CD38-CAR T cells had potent anti-AML activity and prolonged survival in xenograft models, but limited hematopoietic stem cell toxicity was observed, consistent with the pattern of CD38 expression.56 Finally, a recent single-cell transcriptomic study of AML blasts discovered and validated CD86 and CSF-1R as AMLCAR T-cell therapy targets.35
In the following section we summarize results of clinical studies (Table 1) which have been either published or presented in abstract form. Data from the USA, UK, Europe, and China are included.
Lewis-Y-chimeric antigen receptor T cells
Lewis-Y (LeY) is an aberrantly glycosylated carbohydrate antigen that is overexpressed on AML blasts but has limited expression on normal tissues. While to our knowledge, LeY is no longer being pursued as an AML target, LeY-CAR T cells were the first CAR T-cell product that was evaluated in a phase I clinical study for AML (NCT03851146).15 Four adult patients with AML received LeY.CD28.ζ-CAR T cells. No significant toxicities were reported, but only transient responses or stable disease were observed. However, this study provided valuable information since CAR T cells were labeled with indium enabling tracking by single-photon
emission computed tomography, which demonstrated CAR T-cell trafficking to bone marrow and at extramedullary disease sites, highlighting the potential benefit of using CAR T-cell therapy for AML.
CD123-chimeric antigen receptor T cells
Autologous CD123-chimeric antigen receptor T cells
Investigators evaluated T cells that expressed a CD123. CD28.ζ-CAR and a truncated EGFR safety switch which were infused after lymphodepletion induced by fludarabine/cyclophosphamide (NCT02159495). From this study, the outcome of six patients with AML, who were infused with CD123-CAR T cells at either dose level 1 (DL1: 5x107 CAR T cells) or dose level 2 (DL2: 2x108 CAR T cells), have been reported.16 Two patients achieved a CR and proceeded to allogeneic HSCT. Blood counts recovered in all patients following initial pancytopenia. Four patients showed either grade 1 or 2 cytokine release syndrome (CRS) but no dose-limiting toxicities were observed.
T cells that express CD123.CD28.ζ-CAR and a CD20 safety switch are actively being evaluated in a phase I clinical trial (NCT04318678) and preliminary results have been presented.17 Following lymphodepletion with fludarabine/ cyclophosphamide, two patients were infused at DL1 (3x105 CAR T cells/kg), and four patients at DL2 (3x105 CAR T cells/ kg), with one patient being infused off protocol. Isolated fevers occurred after infusion but resolved within 24 hours, and no dose-limiting toxicities were observed. At DL2, one patient with extramedullary disease achieved a CR that lasted for 2 months; she then received a second infusion and again achieved a short-lived CR. One additional patient was infused at DL2 off protocol and achieved a morphological CR but with detectable low-level minimal residual disease (MRD).
To overcome the potential concern regarding prolonged myelosuppression and vascular toxicity, investigators have evaluated T cells that were transfected with messenger RNA encoding a CD123.41BB.ζ-CAR, which results in transient CAR expression in adoptively transferred T cells (NCT02623582). Five patients received one to three serial infusions of CD123-CAR T cells following lymphodepleting chemotherapy. All five patients had grade ≥2 CRS, but the study was terminated as no patient had a clinical response. However, the lack of significant vascular toxicity prompted the development of a follow-up study utilizing a lentiviral vector to generate CD123-CAR T cells (NCT03766126).18,19 Another unique approach to overcome the risk of myelosuppression has been the use of switchable CAR. The universal CAR platform (UniCAR) is a two-component, second-generation CD28.ζ-based CAR T-cell platform in which an adapter molecule, targeting module (TM), confers specificity against the cancer antigen of choice.66 The targeting module has a short half-life of less than 30 minutes, enabling a rapid switch-off. Ten patients have been treated on a phase I trial with UniCAR-T cells directed at
Table
NCT04230265
1
1 MRD–
grade 1 to 2 CRS (N=12), grade 4 CRS (N=2), grade 5 CRS (N=1), grade 3 ICANS (N=1)
(N=4)
*Actual enrollment according to the clinicaltrials.gov webpage; unclear how many patients were infused. **The only product that was generated by mRNA transfection; all other utilized viral vectors. ***Seven out of eight patients received fludarabine/cyclophosphamide; #One patient received a chimeric antigen receptor T-cell product from a matched sibling donor. ID: identity; LD: lymphodepletion; Peds: pediatric population; N: number; Ref: reference; auto: autologous; CAR: chimeric antigen receptor; Flu: fludarabine; SPECT: single photon emission computed tomography scanning; Cy: cyclophosphamide; N/A: data not available; CR: complete response; CRS: cytokine release syndrome; EMD: extramedullary disease; MRD: measurable residual disease; CRi: complete response with incomplete hematologic recovery; allo: allogeneic; PR: partial response; KO: knock out; TRAC: T-cell receptor a constant; SD: stable disease; MLFS: morphological leukemia-free state; ICANS: immune effector cell-associated neurotoxicity syndrome; CRES: T cell-related encephalopathy syndrome; NR: no response; iC9: inducible caspase 9; AML: acute myeloid leukemia; MDS: myelodysplastic syndrome; costim?: co-stimulatory domain not disclosed.
the CD123 antigen (NCT04230265).20,21 Following lymphodepletion, patients received continuous intravenous infusion of a CD123 targeting module (TM123; 0.5 to 1 mg/day) from days 0 to 24. CD123-directed UniCAR-T-cell therapy was safe and tolerable with limited toxicities. Encouraging anti-AML activity was observed, including one CR, two CR with incomplete hematologic recovery (CRi), and four partial responses (PR).
Allogeneic CD123-chimeric antigen receptor T cells
To overcome the concern about T-cell fitness and in an effort to enhance persistence, allogeneic CD123-CAR T cells have been evaluated, including an off-the-shelf, ‘universal’ CD123-CAR T-cell product, which is generated by transduction with a lentiviral vector encoding a CD123.41BB.ζ-CAR and gene edited with transcription activator-like effector nuclease to disrupt the T-cell receptor a constant gene locus to minimize risk of graft-versus-host disease and the CD52 locus to enable the use of alemtuzumab (anti-CD52) as part of the lymphodepletion regimen (NCT04106076).22
Sixteen patients received off-the-shelf CD123-CAR T-cell infusions with half undergoing lymphodepletion with fludarabine/cyclophosphamide and the other half receiving fludarabine/cyclophosphamide plus alemtuzumab. Four patients experienced dose-limiting toxicities including grade 4 CRS (n=2), grade 5 CRS (n=1), and grade 3 immune effector cell-associated neurotoxicity syndrome (ICANS) (n=1) irrespective of the lymphodepletion used. Low-grade (<3) CRS occurred in 12 patients. Adding alemtuzumab to fludarabine/cyclophosphamide was associated with improved lymphodepletion and significantly higher CAR T-cell expansion and persistence. Four patients had a response, including two cases of stable disease, one morphological leukemia-free state, and one durable MRD-negative CR. In addition to this study, a case report of a single patient given donor-derived CD123-CAR T cells as part of a reduced intensity conditioning regimen prior to HSCT has been published.67
Investigators evaluated the feasibility and safety of autologous T cells modified to express a CD33.41BB.ζ-CAR and truncated human epidermal growth factor receptor (HER1t) (NCT03126864).23 Ten adults with R/R AML were enrolled; T cells were collected by apheresis from eight, although only four T-cell products were successfully manufactured, and three patients were infused at the lowest dose level of 0.3x106 cells/kg. There were no dose-limiting toxicities; two patients had CRS (grades 2 and 3) and one had ICANS (grade 2). Although CD33-CAR T cells were detected in blood samples from patients following infusion, with associated symptoms and increased cytokine levels, no anti-leukemic responses were seen. One case report on the use of CD33.41BB.ζ-CAR T cells for R/R AML described transient clinical activity but also pancytopenia.24 Finally, there is
an ongoing multicenter study, being conducted through the Pediatric Transplantation and Cellular Therapy Consortium, evaluating the safety and efficacy of autologous CD33.CD28.ζ-CAR T cells in pediatric patients with R/R AML (NCT03971799). Interim results from this study were presented at the American Society of Hematology annual meeting in 2023. Nineteen of the 24 patients enrolled were infused with CD33 CAR T cells. CRS was early in onset and was reported in 13 (68%) patients, being high grade (≥3) in four (21%) of these patients. Complete remission was seen at DL4, with two patients achieving MRD-negative CR. Dose escalation is currently underway in the phase II portion of the study.68
CLL1.41BB.ζ-CAR T-cell products have been evaluated in a phase I clinical trial in eight pediatric patients with AML.25 The children were given 0.35-1x106 CAR T cells/kg as a single dose after fludarabine/cyclophosphamide lymphodepleting chemotherapy. All developed grade 1-2 CRS and pancytopenia following the infusion. Five patients achieved a morphological leukemia-free state, being MRD-negative in four cases and MRD-positive in one case, one patient had an MRD-positive CRi, one patient had a PR, and one patient has stable disease. In a second report, four pediatric patients received T cells expressing CLL1.CD28.CD27.ζ-CAR and an inducible caspase 9 suicide gene after lymphodepleting chemotherapy. Infusions were well tolerated with only low-grade CRS (≤2) and/or ICANS (≤2). Three patients achieved an MRD-negative CR.26 The investigators subsequently compared autologous T cells expressing CLL1.CD28. CD27.ζ-CAR and the inducible caspase 9 suicide gene (n=4) or CLL1.41BB.ζ-CAR (n=3) in seven children with R/R AML following lymphodepleting chemotherapy.27 The CAR T-cell infusions were well tolerated and only grade ≤2 CRS and one grade 2 ICANS were observed. Five patients achieved CR (3 negative and 2 positive for MRD) with a 1-year overall survival rate of 57.1%. Due to the small number of patients, no significant differences were noted between the two CAR T-cell products.
Finally, CLL1.41BB.ζ-CAR T cells have been evaluated in ten adults with R/R AML.28 Patients received fludarabine/cyclophosphamide lymphodepleting chemotherapy followed by an infusion of a single dose of 1-2 x106 CAR T cells/kg. All patients developed CRS, including grade 2 (n=4) and grade 3 (n=3) cases, and one patient developed T cell-related encephalopathy syndrome. Severe pancytopenia occurred in all patients, and two patients died of severe infection due to chronic agranulocytosis. The CR/CRi rate was 70% and, at a median follow-up time of 173 days, six patients were alive.
NKG2D ligand-chimeric antigen receptor T cells
NKG2D ligand (NKG2D-L)-positive tumor cells can be targeted with human leukocyte antigen (HLA) killer immu-
noglobulin-like receptor mismatched natural killer (NK) cells.69 Another approach relies on expressing a NKG2D-LCAR which consists of NKG2D directly linked to the CD3ζ chain.70 NKG2D-L-CAR T cells were infused without prior lymphodepletion to alleviate concerns that chemotherapy might induce NKG2D-L expression on healthy tissues.29 Seven patients with NKG2D-L-positive R/R AML received NKG2D-L-CAR T cells in four escalating dose levels (1x106 to 3x107 total viable T cells). No dose-limiting toxicities were reported, but no objective responses were noted either. It is likely that the low cell dose without antecedent chemotherapy played a role in limited expansion and persistence of CAR T cells, resulting in limited anti-AML activity.
Building on these initial results and preclinical data supporting the use of multiple NKG2D-L-CAR T-cell infusions, a follow-up study was conducted (NCT03018405).30,31 This multicenter study enrolled 25 patients of whom 16 were infused at three dose levels, DL1: 3x108, DL2: 1x109, and DL3: 3x109 T cells per infusion. Patients received three infusions at 2-week intervals followed by potential consolidation treatment consisting of three additional infusions. Of the infused patients, one had myelodysplastic syndrome, three had multiple myeloma, and 12 had R/R AML. Five patients across all dose levels had grade 3 or 4 CRS, and one dose-limiting toxicity was observed in a patient on DL3. Three of 12 evaluable patients with R/R AML (n=11) or myelodysplastic syndrome (n=1) had CR/CRi. Among responders, two patients with R/R AML proceeded to allogeneic HSCT with durable ongoing remissions (at 5 and 61 months).
Multi-antigen-specific chimeric antigen receptor T cells
In addition to monospecific CAR T-cell products, multi-antigen-specific CAR T cells are starting to be evaluated in early phase clinical studies.32,33 Nine patients received escalating doses (1-3x106/kg) of CAR T cells expressing CLL-1- and CD33CAR as single or split doses following lymphodepletion with fludarabine/cyclophosphamide. The T-cell products were autologous in eight cases and from an HLA-matched sibling donor in one case. Toxicities included grade 4 pancytopenia (n=9), grade 1-3 CRS (n=8), and grade 1-3 neurotoxicity (n=4). Seven of the nine patients achieved a CR/CRi (MRD negative by flow cytometry), while two had no response (the AML blasts of one of the non-responders expressed only CD33).
Six of the seven patients who achieved MRD-negative CR proceeded to a subsequent HSCT.
Based on the available clinical experience with CAR T cells for AML, there is a long road ahead before these cells can
be applied in the routine clinical setting akin to CD19-CAR T cells. The timeline until CAR T-cell therapy for AML achieves similar effectiveness as CD19-CAR T cells for B-cell acute lymphoblastic leukemia remains undefined. The current status of the field of AML-CAR immune cell therapy is reminiscent of the early days of HSCT in the 1960s, when outcomes were poor and many experts believed that this approach would never become part of our treatment armamentarium for patients with hematologic malignancies.71 Current preclinical and clinical studies have identified formidable roadblocks that center around: (i) which antigen(s) to target and heterogenous antigen expression (the antigen dilemma); (ii) limited T-cell effector function through intrinsic and extrinsic mechanisms; and (iii) the ideal starting cell source for generating AML-specific T-cell products. In the following we summarize current efforts to overcome these roadblocks,, which are also illustrated in Figure 1.
A major obstacle to AML-specific CAR T-cell therapies is the identification of target antigens that are expressed on leukemic blasts but are absent or expressed at low levels on normal myeloid cells and/or other healthy tissues.34,35,72,73 Moreover, AML blasts and leukemic stem cells are highly heterogeneous cell populations and more than one antigen may need to be targeted to increase antitumor activity.7 While target antigen discovery is ongoing, we believe that targeting a single antigen is highly unlikely to achieve outstanding cure rates, i.e., we will not suddenly discover a CD19-equivalent pan-AML target that is consistently expressed with an acceptable on-target/off-cancer toxicity profile. We will likely have to develop CAR T-cell products directed at certain AML subsets. The majority of CAR-targeting approaches have relied on targeting cell surface molecules. However several studies have highlighted that CAR with an antigen recognition domain which recognize a peptide in the context of an HLA molecule can target intracellular proteins in cancer cells, including AML.74-76
While this approach increases the repertoire of targetable antigens it is HLA-restricted and therefore raises feasibility concerns for small populations unless we revolutionize cell production to enable the generation of patient-specific genetically modified T-cell products in a cost-effective and timely manner.
In addition to antigen discovery, investigators have focused on developing dual targeting strategies for AML.34,72,77,78 This approach has the potential not only to mitigate the risk of antigen loss variants, but also specificity since CAR immune cells will only be fully activated in the presence of both antigens. For example, a multiomic approach systematically analyzed potential AML target antigen combinations,34 and based on these data an AML-specific T-cell product was developed that expresses an ADGRE2-CAR and a CLEC12A-chimeric co-stimulatory receptor (CCR).78 Other CAR/CCR combinations that are being explored include
CD123-CAR/CD33-CCR and CLEC12A-CAR/TIM3-CCR.79,80
Other studies have suggested that targeting either CD33 and TIM3 or CLL1 and TIM3 has the potential to overcome heterogenous antigen expression of AML blasts.72 Finally, dual targeting of CD123 and GRP78 is being explored as an approach to prevent immune escape.81 In this regard, utilizing NK cells as immune cells to express CAR may be beneficial due to their inherent ability to target NKG2D-L-positive tumor cells (see the section on NKG2D-L CAR T cells). Other efforts are focused on limiting on-target/ off-cancer toxicities, including myelotoxicity, by deleting AML target antigens, including CD33, from hematopoietic stem cells or by using base editing to delete a specific epitope of an AML target in the hematopoietic stem cells (e.g. CD45).82,83 Finally, investigators have focused on fine-tuning antigen recognition and logical gating approaches to drive the specificity of CAR T cells.84-88
Roadblock #2: limited effector function through T-cell intrinsic and extrinsic mechanisms
Intrinsic and extrinsic mechanisms contribute to the limited effector function of CAR T cells for cancers including AML. The immunosuppressive AML microenvironment presents a key extrinsic mechanism, which has been recently reviewed, and we defer the interested reader to these publications.37-39 Clearly, the microenvironment needs to be studied in pediatric as well as adult AML since these are distinct disease entities.89 Several strategies have been evaluated to enhance the effector function of CAR T cells in preclinical models focusing on genetic engineering.90,91 While many of these have not been directly evaluated in AML models, they could potentially also enhance the anti-AML activity of CAR T cells. Conceptually, additional genetic modifications of CAR T cells can be divided into two approaches. One approach is focused on identifying and deleting negative regulators of immune cell function, and the other approach relies on transgenic expression of molecules to enhance their function. The former strategy includes targeting molecules that play a role in (i) limiting T cell activation,92,93 and (ii) regulating T-cell fate through epigenetic mechanisms.94-98 The latter approach includes expressing (i) transcription factors,99 (ii) secretory
or membrane-bound cytokines,100-104 (iii) chimeric cytokine receptors that are either constitutively active or ligand-activated,105-107 (iv) chimeric switch receptors that convert a T-cell inhibitory signal into an activating signal,108,109 or (v) co-stimulatory receptors.110,111 Specific examples for each approach are listed in Table 2. Some of these approaches are actively being explored for CAR immune cell therapies for solid tumors, and it is unclear which approach will have the desired outcome of achieving long-term CR. It is likely that several strategies will have to be combined to generate effective AML-specific CAR T cells, for example improving T-cell activation and manipulating epigenetic pathways. In addition, other factors need to be considered. For example, pro-inflammatory cytokines have been reported to promote leukemogenesis, which would make the transgenic expression of cytokines in AML-specific immune cells less desirable.112
The optimal CAR design, CAR immune cell production, as well as the donor source of AML-specific CAR immune cells remain unknown. The original CAR design celebrates its 30th anniversary this year,113,114 and is still being used by the majority of synthetic T-cell receptors that are currently being evaluated in clinical studies, including those on AML. It has a cytoplasmic signaling domain that consists of a signaling domain derived from one or two co-stimulatory molecules and an activation domain derived from the CD3ζ chain.115 Several preclinical studies have highlighted that adding a signaling domain from cytokine receptors or replacing CD3ζ with ZAP70 improves CAR functionality.85,116 Likewise, adding a domain to enhance recruitment of intracellular signaling molecules has been shown to improve the effector function of CAR T and NK cells.117 Novel synthetic T-cell receptors have also been developed, including T-cell antigen couplers (TAC),118 T-cell receptor fusion constructs (TRuC),119 HLA-independent T-cell receptors (HIT),120 and synthetic T-cell receptor and antigen receptors (STAR),121 which endow T cells with enhanced effector function in comparison to CAR T cells. Finally, modifying T cells with a bispecific T-cell engager is another approach to generate
T cells that recognize AML blasts.122 While it will be impossible to compare all these different approaches in clinical studies, revisiting which genetic engineering approach is optimal to generate tumor-specific immune cells for AML in preclinical models seems advisable, given their limited clinical activity thus far.
Currently, most investigators use the same CAR T-cell-product manufacturing approach irrespective of the targeted disease. At least for autologous products it is likely that cytokine requirements might differ to generate CAR T cells with optimal antitumor activity, since for example T cells from patients with AML or acute lymphoblastic leukemia have been exposed to very different classes of chemotherapeutic agents, and the leukemia microenvironments differ. To our knowledge, there are no published reports of studies investigating the effects of different cytokine cocktails or other culture media supplements on the anti-AML activity of autologous CAR T cells. Integrative bulk- and single-cell sequencing approaches have been used to characterize CD19-CAR T-cell products in detail and track infused T cells at a clonal level.123,124 We are hopeful that insights gained from these types of studies conducted on autologous AML-specific CAR T-cell products will direct the development of a manufacturing process to optimize their effector function in the future. What is the ideal donor source of immune cells to develop AML-specific immune cell therapies? This of course depends on the type of immune cells. For example, while autologous CAR T cells can be generated from AML patients, this might be less than ideal since the effector function of autologous T cells is compromised by previous exposure to chemotherapeutic agents.125,126 Likewise, it presents a logistical challenge since patients must stop their AML-targeted therapies for 1 to 2 weeks (or even longer) before T cells can be collected for manufacturing. These challenges could be overcome by using allogeneic donors. These include the previous HSCT donor for patients who relapse after a transplant or off-the-shelf CAR T-cell products from healthy donors. While generating CAR T-cell products from the previous HSCT donor requires no additional genetic modifications, off-the-shelf CAR T-cell products require additional modifications to prevent graft-
1. Hoffman AE, Schoonmade LJ, Kaspers GJ. Pediatric relapsed acute myeloid leukemia: a systematic review. Expert Rev Anticancer Ther. 2021;21(1):45-52.
2. Miller KD, Fidler-Benaoudia M, Keegan TH, et al. Cancer statistics for adolescents and young adults, 2020. CA Cancer J Clin. 2020;70(6):443-459.
3. Rasche M, Zimmermann M, Borschel L, et al. Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia. 2018;32(10):2167-2177.
4 Rasche M, Zimmermann M, Steidel E, et al. Survival following
versus-host disease and/or allorejection.127,128 In addition, allogeneic NK cells, including cytokine-induced memory NK cells and CAR NK cells, are actively being explored as a source of immune effector cells for the immunotherapy of AML.76,129-131
We believe that developing safe and effective CAR T-cell therapy for AML is not a pipe dream. However, there are formidable challenges that have to be addressed, including targeting multiple antigens to prevent immune escape and on-target/off-cancer toxicities, and to overcome the immunosuppressive AML microenvironment. Likewise, T-cell production as well as evaluating these therapies initially in heavily pretreated patients poses significant challenges. Clearly a concerted team effort will be required in which basic/translational science and clinical investigators from multiple centers work together to achieve this goal. We hope that this review will stimulate discussions on how to reach the summit of effective cell therapies for AML.
Disclosures
SG and MPV are co-inventors of products with patents or patent applications in the fields of T-cell and gene therapy for cancer. SG is a member of the Scientific Advisory Board of Be Biopharma and CARGO, and the Data and Safety Monitoring Board (DSMB) of Immatics and has received honoraria from TESSA Therapeutics within the last year.
Contributions
SN, MPV, and SG co-wrote the manuscript.
Funding
MPV is supported by a St. Baldrick’s scholar grant. The research efforts of SN, MPV, and SG focused on hematologic malignancies are supported by grants from the V Foundation, the Leukemia Lymphoma Society, Cookies for Kids’ Cancer, Assisi Foundation of Memphis and American Lebanese Syrian Associated Charities (ALSAC).
relapse in children with acute myeloid leukemia: a report from AML-BFM and COG. Cancers (Basel). 2021;13(10):2336.
5. Rasche M, Steidel E, Zimmermann M, et al. Second relapse of pediatric patients with acute myeloid leukemia: a report on current treatment strategies and outcome of the AML-BFM study group. Cancers (Basel). 2021;13(4):789.
6. White T, Kaspers G, Abrahamsson J, et al. Clinical outcomes of second relapsed and refractory first relapsed paediatric AML: a retrospective study within the NOPHO-DB SHIP consortium. Br J Haematol. 2022;197(6):755-765.
7 Thomas D, Majeti R. Biology and relevance of human acute
myeloid leukemia stem cells. Blood. 2017;129(12):1577-1585.
8. Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bonemarrow endosteal region. Nat Biotechnol. 2007;25(11):1315-1321.
9 Jentzsch M, Grimm J, Bill M, et al. Prognostic relevance of remission and measurable residual disease status in AML patients prior to reduced intensity or non-myeloablative allogeneic stem cell transplantation. Blood Cancer J. 2021;11(4):80.
10 Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439-448.
11. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517.
12. Fabrizio VA, Phillips CL, Lane A, et al. Tisagenlecleucel outcomes in relapsed/refractory extramedullary ALL: a Pediatric Real World CAR Consortium report. Blood Adv. 2022;6(2):600-610.
13. Ghorashian S, Jacoby E, De Moerloose B, et al. Tisagenlecleucel therapy for relapsed or refractory B-cell acute lymphoblastic leukaemia in infants and children younger than 3 years of age at screening: an international, multicentre, retrospective cohort study. Lancet Haematol. 2022;9(10):e766-e775.
14. Moskop A, Pommert L, Baggott C, et al. Real-world use of tisagenlecleucel in infant acute lymphoblastic leukemia. Blood Adv. 2022;6(14):4251-4255.
15. Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122-2129.
16. Budde L, Song JY, Kim Y, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: a first-inhuman clinical trial. Blood. 2017;130(Supplement 1):811.
17 Naik S, Madden RM, Lipsitt A, et al. Safety and anti-leukemic activity of CD123-CAR T cells in pediatric patients with AML: preliminary results from a phase 1 trial. Blood. 2022;140(Supplement 1):4584-4585.
18. Mardiana S, Gill S. CAR T cells for acute myeloid leukemia: state of the art and future directions. Front Oncol. 2020;10:697.
19. Cummins KD, Gill S. Will CAR T cell therapy have a role in AML? Promises and pitfalls. Semin Hematol. 2019;56(2):155-163.
20 Wermke M, Kraus S, Ehninger A, et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-TCD123 in relapsed/refractory AML. Blood. 2021;137(22):3145-3148.
21. Ehninger G, Kraus S, Sala E, et al. Phase 1 dose escalation study of the rapidly switchable universal CAR-T therapy Unicar-TCD123 in relapsed/refractory AML. Blood. 2022;140(Suppl 1):2367-2368.
22. Sallman DA, DeAngelo DJ, Pemmaraju N, et al. Ameli-01: a phase I trial of UCART123v1.2, an anti-CD123 allogeneic CAR-T cell product, in adult patients with relapsed or refractory (R/R) CD123+ acute myeloid leukemia (AML). Blood. 2022;140(Suppl 1):2371-2373.
23. Tambaro FP, Singh H, Jones E, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. 2021;35(11):3282-3286.
24. Wang QS, Wang Y, Lv HY, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23(1):184-191.
25. Zhang H, Bu C, Peng Z, et al. Characteristics of anti-CLL1 based CAR-T therapy for children with relapsed or refractory acute myeloid leukemia: the multi-center efficacy and safety interim analysis. Leukemia. 2022;36(11):2596-2604.
26. Zhang H, Wang P, Li Z, et al. Anti-CLL1 chimeric antigen receptor T-cell therapy in children with relapsed/refractory acute myeloid leukemia. Clin Cancer Res. 2021;27(13):3549-3555.
27. Pei K, Xu H, Wang P, et al. Anti-CLL1-based CAR T-cells with 4-1-BB or CD28/CD27 stimulatory domains in treating childhood refractory/relapsed acute myeloid leukemia. Cancer Med. 2023;12(8):9655-9661.
28. Jin X, Zhang M, Sun R, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol Oncol. 2022;15(1):88.
29. Baumeister SH, Murad J, Werner L, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. 2019;7(1):100-112.
30 Sallman DA, Kerre T, Havelange V, et al. CYAD-01, an autologous NKG2D-based CAR T-cell therapy, in relapsed or refractory acute myeloid leukaemia and myelodysplastic syndromes or multiple myeloma (THINK): haematological cohorts of the dose escalation segment of a phase 1 trial. Lancet Haematol. 2023;10(3):e191-e202.
31. Sallman DA, Brayer J, Sagatys EM, et al. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/ refractory acute myeloid leukemia patient. Haematologica. 2018;103(9):e424-e426.
32. Liu F, Hongyu Z, Sun L, et al. First-in-human CLL1-CD33 compound CAR (CCAR) T cell therapy in relapsed and refractory acute myeloid leukemia. EHA Library. 2020;294969:S149.
33. Liu F, Cao Y, Pinz K, et al. First-in-human CLL1-CD33 compound CAR T cell therapy induces complete remission in patients with refractory acute myeloid leukemia: update on phase 1 clinical trial. Blood. 2018;132(Supplement 1):901.
34 Perna F, Berman SH, Soni RK, et al. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell. 2017;32(4):506-519.
35. Gottschlich A, Thomas M, Grunmeier R, et al. Single-cell transcriptomic atlas-guided development of CAR-T cells for the treatment of acute myeloid leukemia. Nat Biotechnol. 2023;41(11):1618-1632.
36. Daver N, Alotaibi AS, Bucklein V, Subklewe M. T-cell-based immunotherapy of acute myeloid leukemia: current concepts and future developments. Leukemia. 2021;35(7):1843-1863.
37. Epperly R, Gottschalk S, Velasquez MP. A bump in the road: how the hostile AML microenvironment affects CAR T cell therapy. Front Oncol. 2020;10:262.
38. Tettamanti S, Pievani A, Biondi A, Dotti G, Serafini M. Catch me if you can: how AML and its niche escape immunotherapy. Leukemia. 2022;36(1):13-22.
39. Vadakekolathu J, Rutella S. Escape from T-cell targeting immunotherapies in acute myeloid leukemia. Blood. 2023 Jul 19. doi: 10.1182/blood.2023019961. [Epub ahead of print]
40. Atilla E, Benabdellah K. The black hole: CAR T cell therapy in AML. Cancers (Basel). 2023;15(10):2713.
41. Vishwasrao P, Li G, Boucher JC, Smith DL, Hui SK. Emerging CAR T cell strategies for the treatment of AML. Cancers (Basel). 2022;14(5):1241.
42. Gurney M, O’Dwyer M. Realizing innate potential: CAR-NK cell therapies for acute myeloid leukemia. Cancers (Basel).
2021;13(7):1568.
43. O’Hear C, Heiber JF, Schubert I, Fey G, Geiger TL. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica. 2015;100(3):336-344.
44 Tettamanti S, Marin V, Pizzitola I, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389-401.
45. Riberdy JM, Zhou S, Zheng F, et al. The art and science of selecting a CD123-specific chimeric antigen receptor for clinical testing. Mol Ther Methods Clin Dev. 2020;18:571-581.
46. Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123(15):2343-2354.
47. Mardiros A, Dos SC, McDonald T, et al. T cells expressing CD123specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138-3148.
48. Tashiro H, Sauer T, Shum T, et al. Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to C-type lectin-like molecule 1. Mol Ther. 2017;25(9):2202-2213.
49 Wang J, Chen S, Xiao W, et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J Hematol Oncol. 2018;11(1):7.
50 Niktoreh N, Lerius B, Zimmermann M, et al. Gemtuzumab ozogamicin in children with relapsed or refractory acute myeloid leukemia: a report by Berlin-Frankfurt-Münster study group. Haematologica. 2019;104(1):120-127.
51. Pollard JA, Guest E, Alonzo TA, et al. Gemtuzumab ozogamicin improves event-free survival and reduces relapse in pediatric KMT2A-rearranged AML: results from the phase III Children’s Oncology Group trial AAML0531. J Clin Oncol. 2021;39(28):3149-3160.
52. Lamble AJ, Liu X, Minard C, et al. Safety and activity of flotetuzumab in pediatric and young adult patients with relapsed/refractory acute myeloid leukemia: results from the COG PEPN1812 phase 1 trial. Blood. 2022;140(Suppl 1):6209-6210.
53. Sauer T, Parikh K, Sharma S, et al. CD70-specific CAR T-cells have potent activity against acute myeloid leukemia (AML) without HSC toxicity. Blood. 2021;138(4):318-330.
54. Leick MB, Silva H, Scarfo I, et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell. 2022;40(5):494-508.
55. Hebbar N, Epperly R, Vaidya A, et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat Commun. 2022;13(1):587.
56. Glisovic-Aplenc T, Diorio C, Chukinas JA, et al. CD38 as a panhematologic target for chimeric antigen receptor T cells. Blood Adv. 2023;7(16):4418-4430.
57 Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461-3472.
58. Myburgh R, Kiefer JD, Russkamp NF, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117expressing hematopoietic cells. Leukemia. 2020;34(10):2688-2703.
59 Lichtman EI, Du H, Shou P, et al. Preclinical evaluation of B7-
H3-specific chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Clin Cancer Res. 2021;27(11):3141-3153.
60 Jetani H, Navarro-Bailon A, Maucher M, et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. Blood. 2021;138(19):1830-1842.
61. Niswander LM, Graff ZT, Chien CD, et al. Potent preclinical activity of FLT3-directed chimeric antigen receptor T-cell immunotherapy against FLT3-mutant acute myeloid leukemia and KMT2A-rearranged acute lymphoblastic leukemia. Haematologica. 2023;108(2):457-471.
62. Lynn RC, Poussin M, Kalota A, et al. Targeting of folate receptor beta on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125(22):3466-3476.
63. Mai S, Hodges A, Chen HM, et al. LILRB3 modulates acute myeloid leukemia progression and acts as an effective target for CAR T-cell therapy. Cancer Res. 2023;83(24):4047-4062.
64 John S, Chen H, Deng M, et al. A novel anti-LILRB4 CAR-T cell for the treatment of monocytic AML. Mol Ther. 2018;26(10):2487-2495.
65. Lee WS, Ye Z, Cheung AMS, et al. Effective killing of acute myeloid leukemia by TIM-3 targeted chimeric antigen receptor T Cells. Mol Cancer Ther. 2021;20(9):1702-1712.
66. Loff S, Dietrich J, Meyer JE, et al. Rapidly switchable universal CAR-T cells for treatment of CD123-positive leukemia. Mol Ther Oncolytics. 2020;17:408-420.
67. Yao S, Jianlin C, Yarong L, et al. Donor-derived CD123-targeted CAR T cell serves as a RIC regimen for haploidentical transplantation in a patient with FUS-ERG+ AML. Front Oncol. 2019;9:1358.
68. Shah NN, Tasian SK, Kohler ME, et al. CD33 CAR T-cells (CD33CART) for children and young adults with relapsed/ refractory AML: dose-escalation results from a phase I/II multicenter trial. Blood. 2023;142(Suppl 1):771.
69 Davis CT, Rizzieri D. Immunotherapeutic applications of NK cells. Pharmaceuticals (Basel). 2015;8(2):250-256.
70. Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106(5):1544-1551.
71. Little MT, Storb R. History of haematopoietic stem-cell transplantation. Nat Rev Cancer. 2002;2(3):231-238.
72. Haubner S, Perna F, Kohnke T, et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia. 2019;33(1):64-74.
73. Willier S, Rothamel P, Hastreiter M, et al. CLEC12A and CD33 coexpression as a preferential target for pediatric AML combinatorial immunotherapy. Blood. 2021;137(8):1037-1049.
74. Rafiq S, Purdon TJ, Daniyan AF, et al. Optimized T-cell receptormimic chimeric antigen receptor T cells directed toward the intracellular Wilms tumor 1 antigen. Leukemia. 2017;31(8):1788-1797.
75. Kirkey DC, Loeb AM, Castro S, et al. Therapeutic targeting of PRAME with mTCRCAR T cells in acute myeloid leukemia. Blood Adv. 2023;7(7):1178-1189.
76. Dong H, Ham JD, Hu G, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc Natl Acad Sci U S A. 2022;119(25):e2122379119.
77 Nixdorf D, Sponheimer M, Berghammer D, et al. Adapter CAR T cells to counteract T-cell exhaustion and enable flexible targeting in AML. Leukemia. 2023;37(6):1298-1310.
78. Haubner S, Mansilla-Soto J, Nataraj S, et al. Cooperative CAR
targeting to selectively eliminate AML and minimize escape. Cancer Cell. 2023:41(11):1871-1891.
79. Perriello VM, Rotiroti MC, Pisani I, et al. IL-3-zetakine combined with a CD33 costimulatory receptor as a dual CAR approach for safer and selective targeting of AML. Blood Adv. 2023;7(12):2855-2871.
80 Van Der Schans JJ, Vishwasrao P, Van Gils N, et al. Dual splitsignaling TIM3+CLEC12a targeting CAR T-cells with optimized signaling as a safe potential therapy for acute myeloid leukemia. Blood. 2023;142(Suppl 1):883.
81. Zoine JT, Immadisetty K, Ibanez-Vega J, et al. Peptide-scFv antigen recognition domains effectively confer CAR T cell multiantigen specificity. Cell Rep Med. 2024;5(2):101422.
82. Borot F, Wang H, Ma Y, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc Natl Acad Sci U S A. 2019;116(24):11978-11987.
83. Wellhausen N, O’Connell RP, Lesch S, et al. Epitope base editing CD45 in hematopoietic cells enables universal blood cancer immune therapy. Sci Transl Med. 2023;15(714):eadi1145.
84 Hamieh M, Mansilla-Soto J, Riviere I, Sadelain M. Programming CAR T cell tumor recognition: tuned antigen sensing and logic gating. Cancer Discov. 2023;13(4):829-843.
85. Tousley AM, Rotiroti MC, Labanieh L, et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. 2023;615(7952):507-516.
86. Choe JH, Watchmaker PB, Simic MS, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591):eabe7378.
87. Hyrenius-Wittsten A, Su Y, Park M, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med. 2021;13(591):eabd8836.
88. Richards RM, Zhao F, Freitas KA, et al. NOT-gated CD93 CAR T cells effectively target AML with minimized endothelial crossreactivity. Blood Cancer Discov. 2021;2(6):648-665.
89 Bolouri H, Farrar JE, Triche T Jr, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24(1):103-112.
90. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T cell therapy for solid tumors: bright future or dark reality? Mol Ther. 2020;28(11):2320-2339.
91. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. 2023;614(7949):635-648.
92. Carnevale J, Shifrut E, Kale N, et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature. 2022;609(7925):174-182.
93. Wei J, Long L, Zheng W, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019;576(7787):471-476.
94. Prinzing B, Zebley CC, Petersen CT, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. 2021;13(620):eabh0272.
95. Jain N, Zhao Z, Feucht J, et al. TET2 guards against unchecked BATF3-induced CAR T cell expansion. Nature. 2023;615(7951):315-322.
96. Talleur AC, Qudeimat A, Metais JY, et al. Preferential expansion of CD8+ CD19-CAR T cells postinfusion and the role of disease burden on outcome in pediatric B-ALL. Blood Adv. 2022;6(21):5737-5749.
97. Weber EW, Parker KR, Sotillo E, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. 2021;372(6537):eaba1786.
98. Fraietta JA, Nobles CL, Sammons MA, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558(7709):307-312.
99 Lynn RC, Weber EW, Sotillo E, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576(7786):293-300.
100 Krenciute G, Prinzing BL, Yi Z, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;5(7):571-581.
101. Avanzi MP, Yeku O, Li X, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018; 23(7):2130-2141.
102. Pegram HJ, Lee JC, Hayman EG, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133-4141.
103. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145-1154.
104 Hurton LV, Singh H, Najjar AM, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. 2016;113(48):E7788-E7797.
105. Sockolosky JT, Trotta E, Parisi G, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018;359(6379):1037-1042.
106. Bell M, Gottschalk S. Engineered cytokine signaling to improve CAR T cell effector function. Front Immunol. 2021;12:684642.
107. Shum T, Omer B, Tashiro H, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7(11):1238-1247.
108. Leen AM, Sukumaran S, Watanabe N, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther. 2014; 22(6):1211-1220.
109 Wilkie S, Burbridge SE, Chiapero-Stanke L, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285(33):25538-25544.
110 Omer B, Cardenas MG, Pfeiffer T, et al. A costimulatory CAR improves TCR-based cancer immunotherapy. Cancer Imunol Res. 2022;10(4):512-524.
111. Katsarou A, Sjostrand M, Naik J, et al. Combining a CAR and a chimeric costimulatory receptor enhances T cell sensitivity to low antigen density and promotes persistence. Sci Transl Med. 2021;13(623):eabh1962.
112. Binder S, Luciano M, Horejs-Hoeck J. The cytokine network in acute myeloid leukemia (AML): a focus on pro- and antiinflammatory mediators. Cytokine Growth Factor Rev. 2018;43:8-15.
113. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720-724.
114 Brocker T, Peter A, Traunecker A, Karjalainen K. New simplified molecular design for functional T cell receptor. Eur J Immunol. 1993;23(7):1435-1439.
115. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64-73.
116. Kagoya Y, Tanaka S, Guo T, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352-359.
117 Chockley PJ, Ibanez-Vega J, Krenciute G, Talbot LJ, Gottschalk S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat Biotechnol. 2023;41(10):1434-1445.
118. Helsen CW, Hammill JA, Lau VWC, et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat Commun. 2018;9(1):3049.
119 Baeuerle PA, Ding J, Patel E, et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat Commun. 2019;10(1):2087.
120 Mansilla-Soto J, Eyquem J, Haubner S, et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat Med. 2022;28(2):345-352.
121. Liu Y, Liu G, Wang J, et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci Transl Med. 2021;13(586):eabb5191.
122. Bonifant CL, Szoor A, Torres D, et al. CD123-engager T cells as a novel immunotherapeutic for acute myeloid leukemia. Mol Ther. 2016;24(9):1615-1626.
123. Wilson TL, Kim H, Chou CH, et al. Common trajectories of highly effective CD19-specific CAR T cells identified by endogenous T-cell receptor lineages. Cancer Discov. 2022;12(9):2098-2119.
124. Chen GM, Chen C, Das RK, et al. Integrative bulk and single-cell profiling of premanufacture T-cell populations reveals factors
mediating long-term persistence of CAR T-cell therapy. Cancer Discov. 2021;11(9):2186-2199.
125. Das RK, Vernau L, Grupp SA, Barrett DM. Naive T-cell deficits at diagnosis and after chemotherapy impair cell therapy potential in pediatric cancers. Cancer Discov. 2019;9(4):492-499.
126. Das RK, O’Connor RS, Grupp SA, Barrett DM. Lingering effects of chemotherapy on mature T cells impair proliferation. Blood Adv. 2020;4(19):4653-4664.
127. Caldwell KJ, Gottschalk S, Talleur AC. Allogeneic CAR cell therapy-more than a pipe dream. Front Immunol. 2020;11:618427.
128. Aparicio C, Acebal C, Gonzalez-Vallinas M. Current approaches to develop “off-the-shelf” chimeric antigen receptor (CAR)-T cells for cancer treatment: a systematic review. Exp Hematol Oncol. 2023;12(1):73.
129. Bednarski JJ, Zimmerman C, Berrien-Elliott MM, et al. Donor memory-like NK cells persist and induce remissions in pediatric patients with relapsed AML after transplant. Blood. 2022;139(11):1670-1683.
130 Berrien-Elliott MM, Foltz JA, Russler-Germain DA, et al. Hematopoietic cell transplantation donor-derived memory-like NK cells functionally persist after transfer into patients with leukemia. Sci Transl Med. 2022;14(633):eabm1375.
131. Daher M, Melo Garcia L, Li Y, Rezvani K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin Transl Immunology. 2021;10(4):e1274.
1Children’s Hospital Colorado; 2University of Colorado School of Medicine and 3University of Colorado Cancer Center, Aurora, CO, USA
Correspondence: L. Gore
Lia.Gore@cuanschutz.edu
Received: November 2, 2023. Accepted: January 26, 2024.
https://doi.org/10.3324/haematol.2023.283818
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Abstract
Immunotherapy has revolutionized treatment for a wide variety of cancers yet its use has been relatively limited in childhood malignancies. With the introduction of bispecific T-cell engagers (BiTE®) and chimeric antigen T-cell receptor technologies, previously refractory patients have attained remission, including molecularly negative states of disease, thus providing the possibility of long-term cure. Blinatumomab is a widely available CD3-CD19 BiTE that has dramatically changed the landscape of therapy for some children with precursor-B acute lymphoblastic leukemias (B-ALL) and lymphoblastic lymphomas. Challenges remain with using BiTE in a broader population although the appeal of now-confirmed reduced toxicity and deeper molecular remissions suggests that this approach will be an essential part of future treatment of childhood B-ALL. Herein, we review some of the pertinent literature covering clinical trials with blinatumomab and address future approaches and combination trials including BiTE.
Bispecific T-cell engager (BiTE®) immunotherapy has revolutionized the treatment of B-lineage malignancies in children. These agents offer tumor antigen-directed T-cell-mediated killing with favorable toxicity profiles in clinical practice.1 Blinatumomab, a CD19/CD3 BiTE, has shown remarkable efficacy in reducing subsequent relapse and improving overall survival in patients with relapsed and refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL) compared with conventional multi-agent chemotherapeutic regimens and is the only BiTE with regulatory approval for use in children.2-4 As such, blinatumomab received accelerated approval initially by the United States Food and Drug Administration (US FDA) for the treatment of R/R and minimal residual disease (MRD)-positive B-ALL in adults and children and has subsequently received regulatory approval in more than 50 countries worldwide. Despite the successes of blinatumomab, response is variable, and resistance has been reported. This highlights the need for additional BiTE therapies as well as strategies to overcome the likely class-specific limitations of these agents, such as improved efficacy in extramedullary disease and target-
ing resistance mechanisms including antigen loss, lineage switch, and microenvironmental inhibition.1,2,5-7
BiTE employ a targeted, immune-directed strategy through the simultaneous binding of tumor-associated antigens and a component of the patient’s own T-cell receptor, most commonly a CD3ε within the T-cell receptor complex.8 BiTE traffic T cells directly to the designated antigen on the tumor cell of interest, inducing T-cell activation and targeted cell lysis in the absence of co-stimulatory signaling. Vital to their function, BiTE are small molecules devoid of the antibody constant region typical of monoclonal antibodies. Instead, they consist of two single-chain variable fragments (scFv). This allows the formation of tight immune synapses, facilitating perforin and granzyme-mediated target cell lysis.1,8 Blinatumomab transiently links CD19 on B cells and many B-cell malignancies to CD3 on a patient’s endogenous cytotoxic T cells. CD19 represents an ideal target for B-cell malignancies as it is expressed in nearly all stages of B-cell development. Additionally, in most patients, CD19 is ex-
pressed on nearly 100% of B-lymphoid malignant cells.9 In the presence of CD19, blinatumomab can independently activate resting T cells, leading to upregulation of activation markers, release of inflammatory cytokines, and induction of robust T-cell proliferation in vitro 10 Like other BiTE, blinatumomab causes T-cell activation independently, and these activated T cells have the potential to eliminate multiple target cells without additional stimulation by the drug.11 In preclinical models, repeated target lysis of CD19/CD3 double-positive T cells is presumed to represent transfer of CD19 from B cells to T cells when synapses form, triggering subsequent cytolysis.11 This capacity of blinatumomab, in conjunction with its small size of 54.1 kDa and transient direct action, likely contributes to the improved efficacy and favorable toxicity profile observed with this BiTE compared with other antibody-targeted approaches.
Despite decades of advances in outcomes of children with B-ALL, 10-15% of patients will relapse.12 The 5-year overall survival of these patients in the era of intense, conventional multi-agent chemotherapy is between 35% and 50%.13,14 Blinatumomab has demonstrated efficacy in improving the event-free survival and overall survival in these patients, with a significant reduction in severe adverse events.1-4,8 Following the early successes of blinatumomab in adults with relapsed, refractory, and MRD-positive B-ALL, a phase I/II study examining blinatumomab in children with R/R B-ALL was conducted.15-17 This study established the recommended dose of blinatumomab of 5 µg/m2 with escalation to 15 µg/m2 after 1 week of therapy to modulate cytokine release syndrome, observed primarily in patients with high disease burden in the bone marrow. With 39% of patients achieving complete remission with monotherapy, blinatumomab exhibited previously unobserved single-agent antileukemic activity, including MRD-negative responses in patients with multiply R/R B-ALL, with a tolerable toxicity profile.3 These findings launched a series of pediatric trials and led to the accelerated approval of blinatumomab for R/R B-ALL in children.
In children and young adults with intermediate and high-risk B-ALL in first relapse, the Children’s Oncology Group (COG) study AALL1331 demonstrated a 2-year event-free survival of 54% with blinatumomab treatment after reinduction compared to 39% in those treated with chemotherapy.4 While the improvement in event-free survival was not statistically significant, this study was terminated early because of the substantial toxicity seen in the chemotherapy arm and MRD advantages for blinatumomab. The international, expanded-access RIALTO trial found that 59% of children with multiply relapsed or refractory disease achieved a complete remission within two cycles of blinatumomab and 65% of patients proceeded to allogeneic transplant with a trend
toward improved overall survival in this cohort through the follow-up period of 18 months.18 Substitution of one cycle of blinatumomab for a third consolidative chemotherapy cycle in pediatric patients with high-risk, first-relapse B-ALL significantly improved event-free survival (69% vs. 43%) in an important study run in parallel in Europe,19 with nearly identical findings observed in AALL1331.4,9 This effect was seen across subgroups and was independent of MRD following two cycles of consolidation therapy.4,19
AALL1331 also studied blinatumomab for patients with low-risk B-ALL in first relapse, randomizing patients to receive either blinatumomab or chemotherapy as block 3 of consolidation. Patients in the blinatumomab arm had the drug intercalated into two continuation blocks. This significantly improved disease-free survival and overall survival in the two-thirds of patients with bone marrow relapse with or without extramedullary relapse for patients with low-risk disease. Patients with isolated extramedullary relapse fared poorly in both arms, particularly those with isolated central nervous system (CNS) relapse, and it is known that blinatumomab, like other BiTE, does not cross the blood-brain barrier with any consistency. This study highlighted the inefficacy of blinatumomab in treating CNS leukemia, which can be mitigated by CNS-directed therapy delivered concurrently with blinatumomab cycles, typically on days 1 or 1 and 15 of each 28-day cycle of blinatumomab depending on individual patients’ factors, and including single or multi-agent intrathecal agents. AALL1331 was the first trial to suggest that extramedullary disease may be more refractory to blinatumomab, a finding that was not appreciated in the intermediate and high-risk arm of AALL1331. It is important to note that the intensity of CNS-directed therapy was reduced compared to that of previous studies of relapsed B-ALL, an important consideration for future trials 2 and when delivering standard-of-care therapy. Currently, the International Frankfurt Berlin Münster (IBFM) group, the COG, and individual institutions are assessing the utility of blinatumomab in improving disease-free survival as an addition to up-front therapy in newly diagnosed standard-risk B-ALL in combination with conventional chemotherapy and, in some cases, other immunotherapeutic agents. In the Cancer Research United Kingdom (CRUK)-sponsored AllTogether-1 study conducted in the UK and European Union, approximately 8,000 children, adolescents, and young adults up to the age of 29 years will be enrolled in a series of risk-adapted arms. Importantly, AllTogether-1 will treat patients with Down syndrome with blinatumomab in the intermediate-risk group. This population of patients may benefit from receiving less cytotoxic chemotherapy and the concerted effort to identify both toxicity and response for patients with Down syndrome will contribute to a wider understanding in this context.
Blinatumomab recently demonstrated remarkable efficacy
Table 1. Summary of selected studies with blinatumomab in pediatric patients with B-lineage malignancies.
Study and publication date
2016
von Stackelberg A et al.3
2020
Locatelli F et al.18
Phase and aim
Phase I/II. To determine the safety and pharmacokinetics of blinatumomab in children with R/R B-ALL.
Phase II: RIALTO. An open-label, single-arm, expanded access trial to determine the safety and efficacy of blinatumomab in pediatric patients with R/R BCPALL.
Results Additional notes
Safety and dosing established: 5 µg/ m2 for 7 days, followed by escalation to 15 µg/m2 in two 28-day cycles. CR rate of 39% (95% CI: 27-51%).
Tolerable safety profile. Of patients with ≥5% blasts, 59.2% had CR and 79.3% of those became MRD negative. Of patients with ≤5% blasts, 91.7% had an MRD-negative response.
MRD-negative remissions noted across dose levels. All patients enrolled had highly refractory, multiply relapsed B-ALL.
2021
Brown PA et al.4
Study interpretation limited by early termination recommended by the safety monitoring committee due to toxicity and MRD advantages of blinatumomab, however possibly underpowering the primary endpoint. 2021
F et al.19
2023
Hogan LE et al.2
2023
Van der Sluis IM et al.20
Phase III: COG AALL1331. To compare the survival of patients with high- and intermediate-risk first relapse of B-ALL treated with chemotherapy or chemotherapy plus blinatumomab.
Phase III. To evaluate EFS of children with high-risk B-ALL in first relapse following blinatumomab or chemotherapy as a third consolidative course prior to HSCT.
Phase III: COG AALL1331. To compare the survival of patients with low-risk first relapse of B-ALL treated with chemotherapy or chemotherapy plus blinatumomab.
Phase II. To assess the safety and efficacy of blinatumomab added to Interfant-06 in infants with KMT2A-rearranged ALL.
2-year DFS of 54.4% for the blinatumomab arm versus 39% for the chemotherapy arm. One-sided P=0.03, non-significant. 2-year OS of 71.3% for the blinatumomab arm versus 58.4% for the chemotherapy arm (1-sided P=0.02), significant.
EFS in the blinatumomab arm was 69% versus 43% in the chemotherapy arm with a median follow-up of 22.4 months. OS was 85.2% in the blinatumomab arm versus 70.4% in the chemotherapy arm.
4-year DFS of 61.2% ± 5% and OS of 90.4% ± 3% in blinatumomab arm versus 49.5 ± 5.2% DFS and 79.6 ± 4.3% OS in chemotherapy arm (P=0.89/P=0.11), non-significant.
Incidence of adverse events in the blinatumomab versus chemotherapy arms was 31% versus 57%.
For BM ± EM relapses, 4-year DFS/OS were 72.7 ± 5.8%/97.1% ± 2.1% for blinatumomab versus 53.7% ± 6.7%/84.8% ± 4.8% for chemotherapy (P=0.015/ P=0.020). Poor outcomes for patients with CNS disease, although traditional CNS-directed therapy was reduced.
No toxic effects meeting the definition of the primary end point occurred.
2-year DFS of 81.6% and OS of 93.3% versus 49.4% and 65.8% in Interfant-06.
R/R: relapsed/refractory; ALL: acute lymphoblastic leukemia; CR: complete response; 95% CI: 95% confidence interval; MRD: minimal residual disease; BCP: B-cell precursor; DFS: disease-free survival; OS: overall survival; EFS: event-free survival; HSCT: hematopoietic stem cell transplantation; BM: bone marrow; EM: extramedullary; CNS: central nervous system.
when added to the Interfant-06 regimen for KMT2A-rearranged ALL in infants. Thirty infants were enrolled, each of whom received a single post-induction cycle of blinatumomab. The 2-year disease-free survival rate among these patients was 81.6% compared to 49.4% in the Interfant-06 trial, with corresponding overall survival rates of 93.3% and 65.8%, respectively. Additionally, 30% of patients experienced serious adverse events, and no fatal adverse events were reported.20 The purposes and findings of selected studies of blinatumomab in children are summarized in Table 1. Intensification of chemotherapy has failed to significantly improve outcomes for these patients for nearly 40 years, and so the dramatic improvement with blinatumomab highlights a promising strategy for patients with infant ALL. The Interfant-21 study will prospectively seek to expand these
very promising observations in the newest multinational study (NCT05327894) of infants under the age of 1 year with KMT2A-rearranged and multilineage/mixed phenotype acute leukemia.
Indeed, little is known about the efficacy of blinatumomab in patients with mixed-phenotype acute leukemia, although as of the date of writing this review, three adult patients have been reported to respond at least transiently to blinatumomab; however, no prospective or randomized studies of mixed-phenotype acute leukemia treated with blinatumomab have been conducted to determine the true potential efficacy of the BiTE in this setting. The Interfant-21 study noted above, sponsored by the Princes Maxima Center in the Netherlands, will contribute important information regarding this very rare population of patients.
Randomized trials of blinatumomab in pediatric B-ALL thus far have demonstrated a decrease in grade ≥3 adverse events among patients receiving blinatumomab. AALL1331 found a significant difference in febrile neutropenia (3% vs. 48%), infections (5% vs. 51%), sepsis (0% vs. 11%), and anemia (13% vs. 58%) among low-risk patients receiving blinatumomab for block 3 compared to those receiving conventional chemotherapy,2 Similar advantages favoring blinatumomab were observed in the intermediate- and high-risk groups.4 These data are of particular importance for patients with Down syndrome, who are far more vulnerable to serious infectious complications and non-relapse morbidity and mortality.21 As such, the frontline trial for standard-risk patients in COG was designed to substitute blinatumomab for some intensive elements of therapy in high-risk Down syndrome patients as well. While blinatumomab exhibits a favorable overall toxicity profile, it still presents distinct and potentially significant adverse effects. Cytokine release syndrome (CRS), an on-target phenomenon, is seen in 4-22% of pediatric patients, with grade ≥3 syndrome occurring in 0-3% of patients.2,4,18,19 The median time to onset of CRS is 24-48 hours following blinatumomab initiation, and the syndrome is correlated with a variety of cytokines, most notably interferon- γ and interleukin-6, and less so with tumor necrosis factors.22 Tumor burden is correlated with the risk and severity of CRS, and reduction of disease burden prior to blinatumomab has demonstrated efficacy in reducing this risk.22 Established as the standard of care, dose-escalation can mitigate the effects of CRS.3 Additionally, patients with a high disease burden can be premedicated with dexamethasone in conjunction with slower dose-escalation.22 In a 2022 meta-analysis examining blinatumomab safety in pediatric patients, no difference in the frequency of CRS was found between patients in the blinatumomab and chemotherapy arms. The AALL1331 trial did not provide details of CRS events in the chemotherapy arm.23 Tocilizumab, an interleukin-6 receptor antagonist, is approved for use in CAR T-cell mediated CRS, as interleukin-6 is the predominant driver in this process. Treatment with tocilizumab has not been shown to reduce T-cell efficacy when used in this context24 and has been given to adults and children with CRS caused by blinatumomab, although published evidence for its use in this setting is somewhat limited.25 Immune effector cell-associated neurotoxicity syndrome (ICANS) is an adverse effect associated with targeted immunotherapies. Manifestations include tremor, dizziness, confusion, encephalopathy, and, less commonly, seizure. Blinatumomab carries a higher risk of encephalopathy than does more conventional chemotherapy, with comparable risks of seizure.23 The incidence of neurotoxicity is 3.724% in pediatric patients, with grade ≥3 adverse events in
2-3.6%.2,4,18,19 The incidence is as high as 52% in adults, with 17% suffering grade ≥3 adverse events. Neurological abnormalities are typically present within the first 2 weeks of treatment with a median onset at 9 days in adults.26 Given the short half-life of blinatumomab, temporary cessation of the drug with or without steroids is often sufficient to manage neurological toxicity, and most patients can be successfully retreated with the same or a reduced dose of blinatumomab to complete the recommended treatment course. Blinatumomab should be stopped at the onset of grade ≥3 adverse events, and dexamethasone can be added for patients with severe adverse events. Premedication with dexamethasone may be considered for subsequent doses, and secondary seizure prophylaxis at re-initiation of therapy should be started for patients with severe neurological adverse events. The choice of anti-seizure medication is left to the discretion of the treating physician, and is made easier by the lack of enzyme-inducing agent contraindications with BiTE therapies overall. It is recommended that blinatumomab be permanently discontinued following any seizure, grade 4 adverse event, or a delay in neurological recovery beyond 7 days.27
The efficacy of blinatumomab is variable, and while certain factors may aid in predicting response, understanding of the complex interplay between leukemia-intrinsic and environmental influences in the context of this therapy remains limited. Conventional indicators used to predict response to chemotherapy, such as age, duration of prior complete remission, lack of sensitivity to prior chemotherapy, and post-transplant relapse, do not appear to affect response to blinatumomab. Duration of therapy with blinatumomab is also individualized for patients, although common regimens include two or four 28-day cycles intercalated into a multi-agent chemotherapy regimen. The observation that conventional predictors of response do not match blinatumomab response aligns with the fact that blinatumomab bypasses many of the mechanisms associated with chemotherapy resistance.28 Lower tumor burden has been associated with higher rates of complete remission across multiple studies,3,18,19,28 influencing relapse-free and overall survival.29 To identify additional factors affecting resistance or sensitivity to blinatumomab, pre-treatment samples from 44 adult B-ALL patients were analyzed using bulk tumor and single-cell sequencing: 55% of these patients achieved a complete remission, with a more favorable response observed in CRLF2-rearranged Philadelphia chromosome-like ALL (75%).30 Other leukemogenic genetic aberrations did not predict response, though sample size
in most subgroups was small. Tumor cell immune response genes were enriched among those who achieved complete remission suggesting that blinatumomab-induced T-cell activation may be influenced by leukemia-intrinsic factors. There is growing evidence to suggest that endogenous T-cell function and T-cell subsets influence responses to blinatumomab and other immunotherapies. Enrichment of regulatory T cells in the peripheral blood predicted response to blinatumomab in vitro, influenced by interleukin-10-mediated T-cell suppression.31 T-cell exhaustion markers, PD-1 and TIM3, were increased in samples from pediatric patients with poor response to blinatumomab, highlighting the role of the tumor microenvironment and immune escape/T-cell exhaustion in influencing BiTE efficacy.5 Preclinical studies exploiting immune checkpoint inhibition in conjunction with blinatumomab have shown improved T-cell-mediated tumor killing and T-cell proliferation.32 and in some cases, have reversed the T-cell exhaustion phenotype. These findings have prompted clinical trials combining these two targets. Phase 1 data on blinatumomab plus nivolumab, a PD-1 inhibitor, in adults demonstrated tolerable safety.33 This same approach is being studied in COG study AALL1821 in pediatric patients in first relapse of B-ALL.
Low-risk patients with isolated extramedullary relapse had poor outcomes on AALL1331, particularly those with isolated CNS disease, which again is predictable based on blinatumomab’s known poor penetrance of the CNS when delivered into the central circulation. Patients with isolated testicular disease did well with or without blinatumomab, suggesting that one limitation of blinatumomab in patients with B-ALL may be restricted to efficacy within the CNS.2 It is unlikely, however, that trafficking to extramedullary sites is the sole limitation in extramedullary disease. Among adult responders to blinatumomab who experience subsequent relapse, 40% will do so at extramedullary sites, including sites outside of the CNS. While trafficking of BiTE may be a challenge in these cases, tumor microenvironmental factors may also restrict T-cell recruitment or efficacy in extramedullary sites. In support of this principle, B-ALL appears to be more sensitive than non-Hodgkin lymphoma (NHL) to blinatumomab at low doses, with higher-dose blinatumomab (60 µg/m2/day) for NHL bridging this gap in response.34 Studies are needed to examine the role of higher-dose blinatumomab in patients with extramedullary ALL.
CD19 loss or reduction in antigen density are described consequences CD19-targeted immunotherapies. In the largest retrospective real-world analysis of adult patients receiving blinatumomab, 9.8% of patients relapsed with CD19-negative disease, representing 34.2% of relapsed cases,35 which is a higher percentage than what has been observed in clinical trials. In a meta-analysis reviewing 27 blinatumomab studies, 4.5% (median) of patients relapsed with CD19-negative disease, and 22.5% (median) of relapse
cases overall. Findings in adults and children were comparable.6 Regarding patients undergoing sequential therapy with blinatumomab and CAR-T cells, prior blinatumomab exposure has been associated with higher remission failure, subsequent remission loss, and antigen loss.36 In the largest report to date, an analysis of 420 B-ALL patients, nonresponse to blinatumomab was independently associated with inferior event-free survival following CAR-T-cell therapy. Additionally, patients who received blinatumomab were more likely to exhibit CD19-dim or partial expression prior to CAR-T-cell treatment.37 Notably, outcomes were comparable in blinatumomab-naïve patients and in blinatumomab responders. Therefore, while CD19 antigen loss is a mechanism by which leukemia can evade CD19-targeted immunotherapy, it is not the sole driver of treatment failure among these patients.
Several mechanisms for CD19 antigen loss have been described. In an analysis of patients’ samples with antigen loss following blinatumomab treatment, CD19 mutations universally involved the extracellular domain of the protein and included frameshift and nonsense mutations, splicesite variants, and in-frame deletions. These mutations were not present prior to exposure to blinatumomab, which was confirmed using targeted deep sequencing. One sample harbored a mutation in CD81, a chaperone protein that partners with CD19 to stabilize its surface expression. Thus, abnormalities in CD81 may contribute to a decrease or loss of CD19 expression in the absence of CD19 aberrations. Patients with hypodiploid ALL and chromosome 16 loss are likely more vulnerable to single allelic abnormalities in CD19, because its locus is on chromosome 16. This pattern was described in two of the evaluated samples. However, unlike observations in the setting of CAR-T-cell-mediated antigen loss, no samples had copy-neutral loss of heterozygosity at this locus.30 In a retrospective analysis of blinatumomab-treated patients, patients with CD19-negative relapse maintained an identical cytogenetic profile to that at diagnosis.28
Lineage switch is a rare complication of CD19-directed therapies, including both blinatumomab and CAR T cells. Lineage switch to acute myeloid leukemia is most common following blinatumomab, although mixed-phenotype acute leukemia and unclassifiable leukemias have also been reported.6 In a retrospective review of 161 relapsed B-ALL cases, nine patients experienced lineage switch.37 KMT2A rearrangements are the most commonly described cytogenetic abnormalities in cases of lineage switch, although others including TCF3-ZNF384 fusion, FLT3-ITD and PAX5 polymorphisms have been described.6 Lineage switch is a rare phenomenon, and its mechanisms are not well understood. Unsurprisingly, standardized treatment is lacking in these cases, and outcomes are generally very poor and also inconsistently reported. The prospective studies including patients with mixed-phenotype acute leukemia will be highly informative in this regard.
The first human pharmacokinetic data for blinatumomab emerged from three phase I studies examining its safety in the context of R/R NHL. Patients in these trials received blinatumomab as intravenous (IV) infusions over 2-4 hours at doses ranging from 0.75-13 µg/m2, two to three times weekly.1,8 Grade 3 cytokine release syndrome, neurological toxicity and infections were experienced in the absence of objective clinical responses, leading to the early termination of these studies. Subsequent pharmacokinetic data determined the short serum half-life of blinatumomab in humans of 1.25-3 hours. Consequently, continuous IV infusion of blinatumomab was examined in a subsequent phase I study, with doses in the range of 0.5-90 µg/m2/ day over a 4- or 8-week period in NHL patients. Sustained persistence of blinatumomab was confirmed, allowing for predictable serum levels of the drug and dose linearity. Additionally, prolonged T-cell activation and reduced toxicity were demonstrated with the continuous infusion approach, with CD19-positive B-cell depletion at doses as low as 5-15 µg/m2/day.38
Continuous IV infusion of blinatumomab has been widely accepted as the standard of care due to the drug’s short half-life and demonstrated efficacy. However, this can be a major challenge for patients receiving this agent, particularly in pediatrics, in which access to medical centers or pharmacy services able to dispense the product may be limited, and disproportionally so for those patients/families with more limited resources and transportation access. Alternative means of more convenient and patient-friendly delivery, such as subcutaneous or extended half-life IV infusions, could be considered an unmet need for patients receiving blinatumomab. AMG562 is a half-life extended BiTE administered IV, engaging CD19-expressing cells and CD3 on T cells. In a phase I study in patients with R/R NHL, seven of nine patients treated with AMG562 had disease progression; serum pharmacokinetic data were unobtainable because of immeasurably low serum concentrations at most post-dose timepoints.39 The study was, therefore, terminated early, and there are no other half-life extended BiTE in clinical trials to date.
Early clinical trials of a subcutaneous form of blinatumomab for adults with R/R B-ALL are starting to return results. A phase Ib dose-escalation study assessed responses following daily to thrice weekly administration of subcutaneous blinatumomab in this population. Among the highdose cohort, average concentrations at steady state were consistent with those of the approved IV blinatumomab regimen after a 26-day period. Overall, 64.3% of patients achieved an MRD-negative (MRD <10-4) response, with 80% achieving these results in the high-dose cohort. Additionally, no dose-limiting toxicities were reported, although 85% of patients overall experienced a grade ≥3 adverse
event.40 While promising, additional data and longer duration of follow-up are necessary to determine efficacy and a more extensive comparison to the continuous IV infusion formulation in pediatric patients will be important. Only if non-inferior outcomes and similarly acceptable or better side effect profiles are observed in pediatric patients would this alternative be widely considered for use.
In addition to demonstrating remarkable efficacy in pediatric B-ALL, blinatumomab has a favorable toxicity profile compared with chemotherapy. While this agent is associated with unique acute toxicities, they are rarely severe and usually manageable and reversible. Given the relatively recent approval of blinatumomab in pediatrics, late/longterm effects of this drug are not known. Conversely, late effects of conventional chemotherapy commonly used in ALL treatment have been extensively evaluated and are well-described. For example, osteopenia and osteonecrosis are well-described complications of glucocorticoid use. The incidence of osteonecrosis varies from 10% to upwards of 44.6% in adolescents treated for ALL,41 and not infrequently necessitates surgical intervention or joint replacement, often at a very young age relative to the expected lifecycle of prosthetic joints.
Additionally, cardiac abnormalities are common and often progressive following anthracycline therapy among survivors of pediatric ALL, leading to increased cardiomyopathy and mortality.42 Delayed intensification phases of therapy not only employ both steroids and anthracyclines, but remain a high-risk period for febrile neutropenia, infection, sepsis, the possibility of prolonged hospitalization, and occasionally fatal treatment-related toxicity. Each of these complications was reduced significantly when blinatumomab substituted chemotherapy in global clinical trials.4,19 Therefore, the potential to substitute daunorubicin and glucocorticoids with blinatumomab or employ dose reductions in conjunction with blinatumomab to improve toxicities without compromising efficacy can be considered a next landmark to evaluate in pediatric ALL therapy overall and is already being done in adult treatment protocols for ALL.
The high costs of blinatumomab and the associated administration and preparation costs are important considerations, although a reduction in the above-described acute and late effects of chemotherapy would likely improve the overall financial burden of ALL therapy significantly. Additionally, the potential for generic formulations to become available when the drug patent expires would further reduce potential costs. At the time of this publication, there were 19 active studies listed in ClinicalTrials.gov for pediatric patients using blinatumomab (classic.clinicaltrials.gov/ct2/results?cond=leukemia&term=blinatumomab&cntry=&state=&city=&dist=&Search=Search&recrs=a&recrs=d&recrs=f&age=0; Accessed
16 December 2023), indicating a strong desire among the pediatric oncology community, and beyond, to explore a variety of combinations and strategies incorporating BiTE therapy. As more BiTE with targets of interest are developed, the portfolio of trials should expand appropriately. As noted above, further development of subcutaneous blinatumomab is proceeding in adult patients and there is strong enthusiasm in the pediatric community to advance this agent to children as soon as appropriate adult data are confirmed. A subcutaneous formulation, if proven equally efficacious, would improve convenience and quality of life for patients and families during treatment, and would bring the potential for greater health equity to patients who are currently limited by travel and frequency of clinic visits or home care support necessitated by the continuous IV formulation. Currently, patients with fewer financial, transportation, and health care access resources to travel to treatment centers are limited in their ability to receive the continuous infusion formulations and have less access to what is now considered a new standard of care for many pediatric and adult patients with B-ALL and B-lymphoblastic lymphoma. Removing the current barriers inherent in the continuous infusion formulation could enhance patients’ therapy experience substantially as long as efficacy is not compromised.
At present, there is no specific guidance from regulatory authorities on what data are or would be needed to demonstrate that subcutaneous blinatumomab has both bioequivalence and therapeutic equivalence/non-inferiority and in general, the benchmarking for how best to transition between formulations can vary widely among different drug products. The pharmacokinetics of the continuous infusion formulation of blinatumomab are virtually identical in children and adults.2,3,15,17,22 Once safety and toxicity have been established in adult patients, it may be possible to consider study designs that would allow pediatric patients to enroll on subcutaneous blinatumomab trials, including staggering pediatric patients to enroll one dose level behind adults as long as non-inferiority pharmacoequivalence can be proven. Similarly, enrolling patients who are post-pubertal but under the age of 18 onto adult clinical trials studying subcutaneous blinatumomab before enrolling younger patients would allow for careful consideration of the potential for unique considerations in pediatrics while shortening the duration of clinical development with the goal of making subcutaneous blinatumomab available to children more quickly. Study endpoints on these trials could include pharmacokinetic data, short-term efficacy including marrow and MRD responses at defined time periods with a longer follow-up, as has been done already with the continuous infusion formulation.
While blinatumomab is moving into frontline therapy for pediatric B-ALL, evidence for its efficacy in pediatric lymphomas is more limited. Blinatumomab has produced responses in diffuse large B-cell lymphoma, follicular lymphoma, man-
tle cell lymphoma and more recently, relapsed/refractory Burkitt lymphoma among adults.8,43 Phase I and II studies have shown promise in pediatric NHL,34 though advanced phase trials of blinatumomab in children are warranted to potentially limit toxicity and improve outcomes in the upfront and relapsed settings.
Novel BiTE are under increasing investigation in liquid malignancies such as acute myeloid leukemia, multiple myeloma, and B-cell lymphomas. Odronextamab is a novel human CD20-CD3 bispecific antibody that has produced high response rates and durable remissions in R/R B-cell lymphomas in adults.44 The phase I/II study NCT05991388 will evaluate the drug in children and adolescents with R/R B-cell lymphomas, although access to this drug is currently limited to compassionate use. A first-in-human trial of the CD33-CD3 BiTE AMG 330 demonstrated tolerability and anti-leukemic activity in patients with R/R acute myeloid leukemia, although further dose escalation evaluations are needed.45 If efficacious, this compound could be considered for pediatric patients with ALL, multi-lineage acute leukemias, acute myeloid leukemia, or ALL cases with CD33 expression.
As discussed, coupling these agents with immune-checkpoint inhibition may overcome resistance to BiTE therapy. Further exploiting this mechanism are newly developed bifunctional checkpoint-inhibitory T-cell engagers (CiTE). These agents consist of a BiTE core with the addition of an anti-PD-1 or anti-PD-L1 to enhance efficacy and potentially limit off-target toxicity. This strategy has been demonstrated in a preclinical model targeting acute myeloid leukemia,46 and additional strategies targeting exhaustion markers CTLA4 and TIM3 are being explored.47
Consideration of the role of blinatumomab in antibody-drug conjugate combinations and/or with CAR-T-cell therapy is beyond the scope of this review and is addressed elsewhere. There is also little known about the optimal duration of treatment with blinatumomab, including how many cycles are necessary or ideal, and whether or not blinatumomab should be used in phases of therapy other than those tested already. For example, blinatumomab in induction therapy such as in pre-phase48 or induction cycles has not been studied sufficiently to date.
Consolidation therapy after stem cell transplant for patients with high MRD early after transplant or with early loss of B-cell aplasia after CAR-T-cell therapy has been suggested as an opportunity for blinatumomab use that has yet to be formally studied in children with B-ALL or B-lymphoblastic lymphoma. In summary, blinatumomab has transformed the care of children, adolescents, and adults with B-ALL and B-NHL, and is known to be both effective and no more toxic in patients with Down syndrome, bringing previously unobserved single-agent activity with negative MRD in refractory cases, advantages for toxicity over conventional multi-agent chemotherapy, and the ability for patients with refractory disease to proceed to more definitive therapies
including stem cell transplant at a higher rate, thus optimizing their chance for cure. In time, it is expected that these and many other important questions about how best to incorporate BiTE therapy for childhood ALL will be answered. New combinations of therapies, investigation in the front line, use in special populations including infants, and more patient-friendly formulations all lie ahead in the continuously rising trajectory of blinatumomab use, always with the goal of improving outcomes and quality of life for all.
1. Goebeler ME, Bargou RC. T cell-engaging therapies - BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418-434.
2. Hogan LE, Brown PA, Ji L, et al. Children’s Oncology Group AALL1331: phase III trial of blinatumomab in children, adolescents, and young adults with low-risk B-cell ALL in first relapse. J Clin Oncol. 2023;41(25):4118-4129.
3. von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/ refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
4 Brown PA, Ji L, Xu X, et al. Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on diseasefree survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 2021;325(9):833-842.
5. Feucht J, Kayser S, Gorodezki D, et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget. 2016;7(47):76902-76919.
6. Locatelli F, Shah B, Thomas T, et al. Incidence of CD19-negative relapse after CD19-targeted immunotherapy in R/R BCP acute lymphoblastic leukemia: a review. Leuk Lymphoma. 2023;64(10):1515-1633.
7 Tian Z, Liu M, Zhang Y, et al. Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J Hematol Oncol. 2021;14(1):75.
8. Nagorsen D, Kufer P, Baeuerle PA, et al. Blinatumomab: a historical perspective. Pharmacol Ther. 2012;136(3):334-342.
9 Raponi S, De Propris MS, Intoppa S, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52(6):1098-1107.
10 Brandl C, Haas C, d’Argouges S, et al. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific singlechain antibody construct. Cancer Immunol Immunother. 2007;56(10):1551-1563.
11. Hoffmann P, Hofmeister R, Brischwein K, et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3bispecific single-chain antibody construct. Int J Cancer. 2005;115(1):98-104.
12. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541-1552.
LG serves as an unpaid advisor to Amgen, OnKure, and Pfizer regarding the use of oncology products in childhood cancers. LG receives salary support from U10CA180886, UM1CA228823, P30CA046934, and the Ergen Family Chair at Children’s Hospital Colorado.
Contributions
KUL and LG conceived, researched, wrote, revised and edited the manuscript, and approved the initial and final submissions.
13. Ko RH, Ji L, Barnette P, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol. 2010;28(4):648-654.
14 Freyer DR, Devidas M, La M, et al. Postrelapse survival in childhood acute lymphoblastic leukemia is independent of initial treatment intensity: a report from the Children’s Oncology Group. Blood. 2011;117(11):3010-3015.
15. Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57-66.
16. Gökbuget N, Zugmaier G, Klinger M, et al. Long-term relapsefree survival in a phase 2 study of blinatumomab for the treatment of patients with minimal residual disease in B-lineage acute lymphoblastic leukemia. Haematologica. 2017;102(4):e132-e135.
17 Gökbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
18. Locatelli F, Zugmaier G, Mergen N, et al. Blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia: results of the RIALTO trial, an expanded access study. Blood Cancer J. 2020;10(7):77.
19 Locatelli F, Zugmaier G, Rizzari C, et al. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 2021;325(9):843-854.
20. van der Sluis IM, de Lorenzo P, Kotecha RS, et al. Blinatumomab added to chemotherapy in infant lymphoblastic leukemia. N Engl J Med. 2023;388(17):1572-1581.
21. Lange B. The management of neoplastic disorders of haematopoiesis in children with Down’s syndrome. Br J Haematol. 2000;110(3):512-524.
22. Topp MS, Gökbuget N, Zugmaier G, et al. Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol. 2014;32(36):4134-4140.
23. Marrapodi MM, Mascolo A, di Mauro G, et al. The safety of blinatumomab in pediatric patients with acute lymphoblastic leukemia: a systematic review and meta-analysis. Front Pediatr. 2022;10:929122.
24. Gardner RA, Ceppi F, Rivers J, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation
of antileukemic efficacy. Blood. 2019;134(24):2149-2158.
25. Choudhry J, Parson, M., Wright, J. A retrospective review of tocilizumab for the management of blinatumomab (a bispecific T cell engager)-induced cytokine release syndrome (CRS). Blood. 2018;132(Suppl 1):5211.
26. Stein AS, Schiller G, Benjamin R, et al. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: management and mitigating factors. Ann Hematol. 2019;98(1):159-167.
27. Bhojwani D, Bansal R, Wayne AS. Managing therapy-associated neurotoxicity in children with ALL. Hematology Am Soc Hematol Educ Program. 2021;2021(1):376-383.
28. Aldoss I, Song J, Stiller T, et al. Correlates of resistance and relapse during blinatumomab therapy for relapsed/refractory acute lymphoblastic leukemia. Am J Hematol. 2017;92(9):858-865.
29 Cabannes-Hamy A, Brissot E, Leguay T, et al. High tumor burden before blinatumomab has a negative impact on the outcome of adult patients with B-cell precursor acute lymphoblastic leukemia. A real-world study by the GRAALL. Haematologica. 2022;107(9):2072-2080.
30. Zhao Y, Aldoss I, Qu C, et al. Tumor-intrinsic and -extrinsic determinants of response to blinatumomab in adults with B-ALL. Blood. 2021;137(4):471-484.
31. Duell J, Dittrich M, Bedke T, et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia. 2017;31(10):2181-2190.
32. Wunderlich M, Manning N, Sexton C, et al. PD-1 inhibition enhances blinatumomab response in a UCB/PDX model of relapsed pediatric B-cell acute lymphoblastic leukemia. Front Oncol. 2021;11:642466.
33. Webster J, Luskin MR, Prince GT, et al. Blinatumomab in combination with immune checkpoint inhibitors of PD-1 and CTLA-4 in adult patients with relapsed/refractory (R/R) CD19 positive B-cell acute lymphoblastic leukemia (ALL): preliminary results of a phase I study. Blood. 2018;132(Suppl 1):557.
34 Goebeler ME, Knop S, Viardot A, et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J Clin Oncol. 2016;34(10):1104-1011.
35. Aldoss I, Otoukesh S, Zhang J, et al. Extramedullary disease relapse and progression after blinatumomab therapy for treatment of acute lymphoblastic leukemia. Cancer. 2022;128(3):529-535.
36. Pillai V, Muralidharan K, Meng W, et al. CAR T-cell therapy is effective for CD19-dim B-lymphoblastic leukemia but is impacted by prior blinatumomab therapy. Blood Adv.
2019;3(22):3539-3549.
37. Myers RM, Taraseviciute A, Steinberg SM, et al. Blinatumomab nonresponse and high-disease burden are associated with inferior outcomes after CD19-CAR for B-ALL. J Clin Oncol. 2022;40(9):932-944.
38. Zhu M, Wu B, Brandl C, et al. Blinatumomab, a bispecific T-cell engager (BiTE(®)) for CD-19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin Pharmacokinet. 2016;55(10):1271-1288.
39 Mead MD, Popplewell LL, Subklewe M, et al. Phase I study of the CD19/CD3 half-life extended BiTE molecule AMG 562 in relapsed/refractory diffuse large B cell lymphoma, mantle cell lymphoma and follicular lymphoma. Hematol Oncol. 2021;39(S2):2881.
40 Sánchez PM, Zugmaier G, Gordon P, et al. Safety and pharmacokinetics of subcutaneous blinatumomab (SC blinatumomab) for the treatment of adults with relapsed or refractory B cell precursor acute lymphoblastic leukemia (R/R B-ALL); results from a phase 1b study. Blood. 2022;140(Suppl 1):6122-6124.
41. Rao SS, El Abiad JM, Puvanesarajah V, et al. Osteonecrosis in pediatric cancer survivors: epidemiology, risk factors, and treatment. Surg Oncol. 2019;28:214-221.
42. Lipshultz SE, Lipsitz SR, Sallan SE, et al. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23(12):2629-2636.
43. Bohler J, Bacher U, Banz Y, et al. Blinatumomab in relapsed/ refractory Burkitt lymphoma. Cancers (Basel). 2022;15(1):44.
44 Bannerji R, Allan JN., Arnason JE, et al. Odronextamab (REGN1979), a human CD20 x CD3 bispecific antibody, induces durable, complete responses in patients with highly refractory B-cell non-Hodgkin lymphoma, including patients refractory to CAR T therapy. Blood. 2020;136(Suppl 1):42-43.
45. Ravandi F, Stein AS, Kantarjian HM, et al. A phase 1 first-inhuman study of AMG 330, an anti-CD33 bispecific T-cell engager (BiTE®) antibody construct, in relapsed/refractory acute myeloid leukemia (R/R AML). Blood. 2018;132(Suppl 1):25.
46. Herrmann M, Krupka C, Deiser K, et al. Development of a bifunctional checkpoint inhibitory T cell engager (CiTE) to reverse adaptive immune escape in AML. Blood. 2018;132(Suppl 1):4069.
47. Böhme M, Kayser S. Immune-based therapeutic strategies for acute myeloid leukemia. Cancers (Basel). 2021;14(1):105.
48. Rijneveld A, Gradowska P, Bellido M, et al. Blinatumomab added to prephase and consolidation therapy in newly diagnosed precursor B-ALL in adults. A phase II HOVON trial. Hemasphere. 2022;6(S3):266-267.
1Viva-University Children’s Cancer Center, Khoo Teck Puat-National University Children’s Medical Institute, National University Hospital, National University Health System; 2Department of Pediatrics, Yong Loo Lin School of Medicine, National University of Singapore and 3Cancer Science Institute, Yong Loo Lin School of Medicine, National University of Singapore, Singapore
Correspondence: B.L.Z. Oh paeolzb@nus.edu.sg
Received: October 4, 2023. Accepted: January 11, 2024.
https://doi.org/10.3324/haematol.2023.283848
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Chimeric antigen receptor (CAR) T-cell therapy is a new and effective treatment for patients with hematologic malignancies. Clinical responses to CAR T cells in leukemia, lymphoma, and multiple myeloma have provided strong evidence of the antitumor activity of these cells. In patients with refractory or relapsed B-cell acute lymphoblastic leukemia (ALL), the infusion of autologous anti-CD19 CAR T cells is rapidly gaining standard-of-care status and might eventually be incorporated into frontline treatment. In T-ALL, however, leukemic cells generally lack surface molecules recognized by established CAR, such as CD19 and CD22. Such deficiency is particularly important, as outcome is dismal for patients with T-ALL that is refractory to standard chemotherapy and/or hematopoietic stem cell transplant. Recently, CAR T-cell technologies directed against T-cell malignancies have been developed and are beginning to be tested clinically. The main technical obstacles stem from the fact that malignant and normal T cells share most surface antigens. Therefore, CAR T cells directed against T-ALL targets might be susceptible to self-elimination during manufacturing and/or have suboptimal activity after infusion. Moreover, removing leukemic cells that might be present in the cell source used for CAR T-cell manufacturing might be problematic. Finally, reconstitution of T cells and natural killer cells after CAR T-cell infusion might be impaired. In this article, we discuss potential targets for CAR T-cell therapy of T-ALL with an emphasis on CD7, and review CAR configurations as well as early clinical results.
T-cell acute lymphoblastic leukemia (T-ALL) accounts for about 10-15% of childhood ALL and 20-25% of adult ALL.1,2 Historically, T-ALL had a poorer overall outcome when compared with B-lineage ALL, and patients with T-ALL were often regarded as having high-risk ALL.1,3 Among patients with T-ALL, those with early T-cell progenitor (ETP)-ALL have a particularly poor response to remission induction chemotherapy and often require intensive chemotherapy and/or hematopoietic stem cell transplant (HSCT).4-9 For patients with T-ALL who are refractory to chemotherapy or relapse, new and effective therapies are urgently needed.10-12 In this context, the success of chimeric antigen receptor (CAR) T-cell therapy in B-lineage ALL provides a strong impetus to develop similar technologies applicable to T-ALL. CAR can direct T lymphocytes against antigens expressed by tumor cells. Antigen binding transduces signals which
induce T-cell activation, resulting in both tumor cell killing and T-cell proliferation.13 Consequently, CAR T cells can expand and persist after infusion, reducing tumor load and eliminating minimal residual disease (MRD). The antitumor potential of this approach has been amply demonstrated through the clinical use of CAR T cells in B-cell malignancies with CAR targeting CD19 or CD22.14-26 Both target antigens are detectable by flow cytometry on the surface membrane of the vast majority of leukemic cells in most cases of B-lineage ALL and other B-cell malignancies.27-31 CD19 and CD22 are normally expressed only by B lymphoid cells.32 Although the infusion of the corresponding CAR T cells induces B-cell lymphopenia and agammaglobulinemia, these are clinically manageable.33 Moreover, CD19 and CD22 are not expressed by T cells and their targeting does not affect CAR T-cell manufacturing.
The surface marker profile of T-ALL and of its lymphomatous counterpart, T-cell lymphoblastic lymphoma (T-LL),34
is considerably different from that of B-cell leukemias and lymphomas. Proven CAR T-cell targets, such as CD19 and CD22, are generally absent and new targets must be identified. Moreover, T-ALL and T-LL typically share surface antigens with peripheral blood T lymphocytes.4,27,34 This poses three major potential problems. First, the manufacturing of viable CAR T cells might be complicated by CAR-mediated killing within the CAR T-cell population, the so-called “fratricide” effect.35,36 Secondly, T lymphocytes are difficult to separate from T-ALL cells should the latter contaminate the cell source for CAR T-cell manufacturing. Finally, the degree of T-cell ablation resulting from the infusion of CAR T cells directed against T-cell antigens and its clinical consequences are unclear. One of the important factors for the success of autologous anti-CD19 CAR T-cell therapy is the expansion and prolonged persistence of CAR T cells,14,17,18 but infusion of autologous CAR T cells directed against T-cell antigens might require HSCT soon after to correct T-cell aplasia.
We here review potential targets for CAR T-cell therapy of T-ALL with an emphasis on CD7, which has been the focus of most clinical studies reported to date. We discuss CAR design and testing, approaches to avert fratricide and lessons learned from the early clinical experience.
CD7 expression
CD7 is a 40 kDa type I transmembrane glycoprotein belonging to the immunoglobulin superfamily and is one of the earliest surface markers in the developmental pathway of human T cells.37-39 CD7 is widely used as a key marker for the diagnosis of T-ALL because of its high and often uniform expression in leukemic blasts.40-43 Importantly, CD7 is also expressed in ETP-ALL; by definition, this T-ALL subtype lacks some markers often expressed in non-ETP T-ALL, such as CD5 and CD1a.4 We studied leukemic cells from diagnostic bone marrow samples of 49 T-ALL patients (14 with ETP-ALL) and found that the median percent CD7 expression exceeded >99% in 46 cases.36 In addition to high levels of expression, an effective CAR T-cell target must be present in chemoresistant leukemic clones which drive relapse. We studied 14 samples collected at relapse and found that CD7 expression in these samples was as high as that in samples studied at diagnosis.36 We also determined whether chemotherapy would alter CD7 expression by measuring it in leukemic cells from bone marrow samples examined for MRD during chemotherapy. In 54 samples (from 21 patients), >99% of leukemic cells expressed CD7.36 Sequential samples were available from 18 patients and the results clearly showed high stability of
CD7 expression.36 Taken together, these results provided a strong rationale for targeting CD7 with CAR T cells in T-ALL. CD7 is also expressed by most normal T cells in peripheral blood, and CD7 mRNA expression in T cells rapidly increases after activation.44,45 CD7 function in human T cells is unclear, and CD7-deficient mice showed normal lymphocyte populations in primary and secondary lymphoid tissues without significant immunodeficiency.46,47 Nevertheless, it was noted that mice had reduced in vitro antigen-specific interferon-γ production and CD8 cytotoxic capacity, and in vivo resistance to lipopolysaccharide-induced shock syndromes.48 Human peripheral blood T cells contain a small CD7-negative fraction typically accounting for <10% of T cells,49 which progressively increases with age.50 This CD7-negative T-cell population contains a higher proportion of CD4-positive cells than that in the CD7-positive cell population and a relatively higher proportion of cells with a memory phenotype.50,51
Among normal tissues, CD7 expression is generally limited to the T-cell lineage.36,40,42-44 An anti-CD7-ricin A chain immunotoxin did not seem to affect other cells when administered to patients with T-cell lymphoma; the vascular leak syndrome observed in some patients appeared to be unrelated to the CD7 specificity, as there was no anti-CD7 reactivity with endothelial cells.52
Anti-CD7 chimeric antigen receptor design and expression
CD7 holds potential as a CAR T-cell target because of its high expression in T-ALL/T-LL cells and its lack of expression in non-hematopoietic cells. We designed an anti-CD7 CAR composed of the single-chain variable fragment (scFv) of the anti-CD7 antibody TH-69 joined to the signaling domains of 4-1BB (CD137) and CD3ζ via the hinge and transmembrane domain of CD8a. In addition to the scFv derived from the antibody TH-69,53 other scFv derived from the 3A1 antibody and other 3A1 hybridomas44 have also been studied (Table 1). TH-69 has been reported to have the highest antigen binding affinity.54,55
Because an scFv construct does not always recapitulate the binding profile of the original antibody, it is important to test its specificity and binding capacity. To this end, we generated a TH-69 scFv in soluble form and ensured that it could only label cell lines known to express CD7.36 Further assurance was provided by experiments in which preincubation with the scFv inhibited staining of CD7-positive cells with an anti-CD7 monoclonal antibody.36 Finally, we used two differently labeled CD7-positive cell lines and found that the TH-69 scFv induced significant and specific cell aggregation.36 This extensive set of experiments provided solid data regarding the capacity of the TH-69-derived scFv to bind CD7 strongly and specifically.
After incorporating the TH-69 scFv into the CAR construct, we used a retroviral vector to express the anti-CD7-41BBCD3ζ CAR in the Jurkat T-cell line. CAR expression was high
Table 1. Targets and receptor design for chimeric antigen receptor T-cell therapy in T-cell acute lymphoblastic leukemia/T-cell lymphoblastic lymphoma.
TH-69
CD8a CD8a 4-1BB CD3ζ Png et al.36
VHH6a IgG1 CD8a ICOS and 4-1BB CD3ζ Zhang et al.65
3A1e IgG1 CD28 CD28 CD3ζ Gomes-Silva et al.59
TH-69 CD8a CD28 CD28 and 4-1BB CD3ζ Cooper et al.60
CD7 >99
3A1e CD8a CD8a 4-1BB CD3ζ Georgiadis et al.61
3A1e CD8a CD8a CD28 CD3ζ Diorio et al.70
TH-69 CD8a CD28 4-1BB CD3ζ Lu et al.80
3A1e CD8a CD8a CD28 CD3ζ Watanabe et al.77
3A1e IgG1 CD28 CD28 CD3ζ Freiwan et al.81
CD5 ~80 H65 IgG1 CD28 CD28 CD3ζ Mamonkin et al.35 H65, FHVH1b, FHVH3b
IgG1 CD28 4-1BB
ζ Mamonkin et al.86
CD1a ~40 NA1, 34.HLK CD8a CD8a 4-1BB CD3ζ Sanchez-Martinez et al.93
TRBC1 ~30 JOVI-1 CD28 CD28 CD28 and OX40 CD3ζ Maciocia et al.95
CD38 >80 THB-7 CD8a CD8a 4-1BB CD3ζ Mihara et al.104 028, 056, 026, daratumumab CD8a CD8a 4-1BB CD3ζ Drent et al.107 Nb-1G3c CD8a CD8a 4-1BB CD3ζ An et al.129 MOR202, AB79, MOR3080 CD8a CD8a 4-1BB CD3ζ Glisovic-Aplenc et al.108
aDerived from llama immunized with CD7-positive Jurkat cells. bDerived from fully human heavy-chain-only phage display antibody library. cDerived from camel immunized with recombinant CD38.
and its ligation induced signal transduction, as revealed by increases in the expression of T-cell activation markers, such as CD69 and CD25.36
We selected 4-1BB (CD137) as a co-stimulatory molecule in our second-generation CAR.36 4-1BB is the co-stimulatory molecule in the CAR for B-lineage ALL originally developed in our laboratory,56 and is the one incorporated in the first CAR T-cell product approved for the treatment of B-lineage ALL (tisagenlecleucel).26 The addition of 4-1BB to CAR markedly increases their capacity to induce and sustain T-cell expansion and antitumor activity.56-58 As summarized in Table 1, most reported CAR directed against CD7 or other T-cell antigens have either 4-1BB or CD28 as co-stimulatory molecules, while CD3ζ is universally used to deliver the primary activation stimulus.
The problem of chimeric antigen receptor T-cell fratricide and its solutions
Given the high expression of CD7 in peripheral blood T cells, it was not surprising that expression of anti-CD741BB-CD3ζ CAR dramatically reduced cell viability, regardless of whether the CAR was permanently expressed by retroviral transduction or only transiently expressed by mRNA electroporation; in 17 experiments, CAR expression
reduced viable cell recovery to about one-third 24 hours after CAR introduction and cell viability decreased with time.36 The occurrence of CAR T cells that killed other CAR T cells (i.e., fratricide) was corroborated by the appearance of exocytosis of lytic granules in the CAR T cells. Similar findings were also reported by other investigators studying different anti-CD7 CAR constructs, all of whom noted poor viability and limited in vitro expansion of CAR T cells.59-62 These results collectively indicate that downregulation of CD7 in CAR T cells is a critical step for robust manufacturing of anti-CD7 CAR T cells.
Several approaches have been developed to mitigate the issue of fratricide by generating CD7-negative anti-CD7 CAR T cells, including post-translational interference of CD7 expression, gene editing, and the use of naturally occurring CD7-negative T cells.
To interfere with the expression of CD7 on the surface membrane of T cells we used the protein expression blocker (PEBL) technology previously developed in our laboratory.63,64 These blockers consist of a scFv (anti-CD7 in this case) linked to amino acid sequences containing endoplasmic reticulum/Golgi retention domains. We observed that transduction of anti-CD7 PEBL resulted in almost immediate suppression of CD7 expression while CD7 mRNA expression Target
was retained.36 A similar approach using a construct that contains a heavy-chain-only CD7 binding domain instead of a scFv was subsequently reported.65 Expression of anti-CD7 PEBL and downregulation of CD7 do not appear to affect the expression of other surface antigens.36 Moreover, T-cell survival in culture was not altered; when the T cells rendered CD7-negative by PEBL were transduced with an anti-CD19 CAR, they were functionally indistinguishable from CD7-positive anti-CD19 CAR T cells.36 In sum, PEBL expression and the formation of PEBL-CD7 complexes in the endoplasmic reticulum did not have discernible effects on T-cell function. Lack of CD7 did not produce any detectable functional impairment. When the PEBL-transduced CD7-negative T cells were transduced with the anti-CD7 CAR, post-transduction viability improved markedly, demonstrating that downregulation of CD7 prevented fratricide.36 In several experiments in vitro and in xenograft models of leukemia, anti-CD7 PEBL CAR T cells were remarkably effective anti-leukemic agents.36
During the testing of anti-CD7 PEBL CAR T cells we compared their potency to that of T cells expressing the antiCD19-41BB-CD3ζ CAR, using T-ALL cell lines transduced with CD19. Cytotoxicity and long-term proliferative capacity were similar, indicating that the anti-CD7 CAR T cells would be expected to produce an antitumor effect similar to that of the established anti-CD19 CAR T-cell product.36 We also compared their antitumor capacity to that of residual T cells recovered after CAR expression without prior CD7 downregulation. In 45 experiments, the former were consistently more potent, and their superiority extended to exocytosis of lytic granules, cytokine production, and proliferative capacity in co-cultures with CD7-positive target cells.36 These results indicate that CD7 downregulation is not only essential to avoid fratricide but also benefits CAR T-cell function. We speculate that apparently CD7-negative cells may express CD7 during CAR signaling and activation resulting in engagement of the CAR by self-CD7, which would create a decoy that diverts the CAR from antigen expressed by target cells.
A unique aspect of the PEBL technology as applied to CAR T-cell therapy manufacturing is that it fits well with current Good Manufacturing Practice (GMP) protocols. For example, PEBL and CAR genes can be included in a single bicistronic vector allowing CAR T-cell production with minimal fratricide using a work schedule essentially identical to that used for anti-CD19 CAR T-cell manufacturing.36 Therefore, it can be easily implemented by any GMP facility familiar with CAR T-cell manufacturing. Because both CAR and PEBL genes are within a single viral vector, there should not be an increased risk of integration in potentially oncogenic sites as compared to expression of a CAR gene alone. The CAR-PEBL technology has been translated into a process that meets current GMP regulations and is, at present, being studied in an ongoing clinical trial (ClinicalTrials.gov Identifier: NCT05043571).
Gene editing technologies have also been used to eliminate CD7 expression for anti-CD7 CAR T-cell manufacturing. Cytidine deamination guided by clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 has been successfully utilized to delete CD7 and prevent CAR-mediated fratricide.59,60,62 While CRISPR-Cas9 is effective in knocking out targets, the formation of double-stranded DNA breaks might result in genetic aberrations, especially in multiplexed editing scenarios.61,66,67 Base-editors rely on CRISPR-Cas9 technology, wherein a defective Cas9 that cannot cause double-stranded breaks is linked to a cytosine or adenine deaminase, and uracil glycosylase inhibitor.61,68,69 When base-editors are guided to the target DNA sequence by guide RNA, the deaminase converts cytosine to uracil or adenine to inosine. The resulting mismatch is then repaired by cellular machinery, resulting in CG to AT, or AT to CG mutations.68 Base-editors have been used for multiplexed editing of anti-CD7 CAR T cells, primarily to prevent fratricide and, in the setting of allogeneic CAR T cells, to knockout other surface proteins such as T-cell receptor (TCR) (to prevent graft-versus-host disease [GvHD]) and CD52 (to protect CAR T cells from alemtuzumab administered to suppress rejection).70,71
While gene editing completely eliminates the expression of a specific target protein, off-target mutations may potentially lead to carcinogenicity or cytotoxicity in normal cells.66,67 Additionally, genome-wide, non-specific editing may also arise from multiplexed editing.68,69 Although unbiased genome-wide screening can be used to detect potential off-target mutations, such technologies are limited by the 0.1% detection limit of next-generation sequencing.72,73 Next-generation base-editors might have a reduced non-specificity but this may be accompanied by a lower on-target efficiency and higher sequence-specific requirements.74-76
Other investigators have proposed alternative approaches to anti-CD7 CAR T-cell manufacturing that do not include downregulation of CD7. Thus, Watanabe et al.77 used ibrutinib and dasatinib which inhibit tyrosine kinases, such as ITK and LCK, involved in CAR signaling,78,79 to allow survival and expansion of anti-CD7 CAR T cells during manufacturing. The cytotoxic, proliferative, and cytokine secreting functions of the resulting CAR T cells were restored following removal of dasatinib. While this method allows production of sufficient functional CAR T cells ex vivo, it is unclear whether fratricide after dasatinib wash-out will result in lower persistence of these cells in vivo, possibly reducing longevity of the therapy.
The selective transduction of naturally occurring CD7-negative T cells through magnetic bead separation with CD7 depletion followed by CD3 enrichment has been described as an approach to bypass fratricide.80,81 Through this approach, CD7-negative T cells were engineered to express a CD7-targeting CAR that was shown to trigger cytotoxic activity in vitro with good cell viability and expansion. How-
ever, the major foreseeable challenge to clinical application of this approach is the potentially low cell numbers, particularly in heavily pre-treated patients from whom it is often difficult to obtain sufficient numbers of T cells regardless of their phenotype. Moreover, as discussed above, anti-CD7 CAR T cells with downregulated CD7 were more potent than those derived from T cells surviving fratricide during manufacturing.36
CD5
CD5 is a 67 kDa cell surface glycoprotein expressed in approximately 80% of T-ALL.4,27,82 In comparison to CD7, its expression is generally lower, and dim or absent in ETPALL.4,9,83 CD5 is also expressed by peripheral blood T cells but Mamonkin et al. was able to develop anti-CD5 CAR T cells without prior CD5 downregulation,35 perhaps because of the relatively weak expression of CD5 in some T-cell subsets. Subsequent studies relied on CD5 knockout by CRISPR-Cas9 prior to transduction of the CD5 CAR, which led to higher CAR expression and reduced tonic activation, as compared to unedited CAR T cells.84,85 In a separate approach using a system wherein CAR expression is controlled by doxycycline, anti-CD5 CAR T cells were cultured with doxycycline and T-cell expansion improved due to reduced fratricide. CAR expression was then restored after washout and allowed the elimination of CD5-positive targets in a mouse model.86
CD1a is related to the major histocompatibility complex and is expressed during T-cell development in the thymus.87,88 It is also expressed in approximately 40% of T-ALL cases and is a defining feature of the cortical T-ALL subtype.27,89-92
Absence of CD1a expression is one of the defining features of ETP-ALL.4 CD1a is not expressed by peripheral blood T cells and Sanchez-Martinez et al.93 showed that fratricide was not an issue when manufacturing anti-CD1a CAR T cells, which had specific cytotoxicity against CD1a-positive T-ALL cell lines and primary leukemic cells. Obviously, the main restriction of using CD1a as a target is the limited number of T-ALL cases that express CD1a.
TCR β-chains can be encoded by either the TRBC1 or the TRBC2 gene, and their expression is mutually exclusive.94,95 Thus, targeting TRBC1 would eliminate TRBC1-expressing normal T cells but spare a substantial number of normal T cells expressing TRBC2. Maciocia et al. 95 generated anti-TRBC1 CAR T cells and showed that these cells had the capacity for specific killing of TRBC1-positive cell lines and
primary cells in vitro and in mouse models of leukemia. In the context of T-ALL, the application of this elegant approach is limited by the fact that CD3/TCRaβ expression is found in a minority of cases and expression is often dim.27,90,96
CD38 is a 45 kDa type II, transmembrane glycoprotein expressed on activated T cells, terminally differentiated B cells, monocytes and natural killer (NK) cells,97 and is reportedly not expressed by multipotent hematopoietic stem cells.98-100 CD38 is also expressed in hematologic malignancies, including ALL, acute myeloid leukemia, lymphoma and multiple myeloma. CD38 is expressed in most cases of T-ALL,101 suggesting that it could be possible to target T-ALL with the anti-CD38 antibody daratumumab.102,103 Anti-CD38 CAR T cells were originally developed to target B-cell non-Hodgkin lymphoma and multiple myeloma.104-107 CD38 is also expressed in normal activated T cells; cell recovery after CAR expression was reported to be low due to cell fratricide and was improved by the addition of a blocking anti-CD38 antibody during T-cell manufacturing.104 More recently, the anti-CD38 CAR approach was extended to preclinical models of T-ALL.108 In these studies, fratricide did not appear to be an issue.
Table 2 summarizes data from clinical studies of CAR T cells in T-ALL/T-LL which have used either autologous or allogeneic cells as a source for CAR T-cell manufacturing. The studies reported so far have targeted either CD7 or CD5.
Autologous cell products
Zhang et al.65 used autologous anti-CD7 CAR T cells with nanobody-derived fratricide resistance to treat seven patients with T-ALL. Despite significant disease burden prior to treatment, six of these patients achieved complete responses with tolerable toxicities, although four relapsed 3 months later.65 Information regarding the quality of the manufactured products, in terms of transduction efficiency and leukemic cell contamination, was not extensive in the report, and it is not clear whether these factors could have contributed to the relapses. Lu et al.80 used autologous anti-CD7 CAR T cells without any additional modifications to reduce fratricide to treat 20 patients with T-ALL/T-LL; 16 had bone marrow involvement prior to CAR T-cell infusion and 15 of these achieved a complete response 1 month later. Twelve of these 15 patients received a HSCT within 100 days of infusion.80 Autologous cells have also been tested in the context of CD5-specific CAR. Of four reported
Table 2. Data from clinical studies of chimeric antigen receptor T cells in T-cell acute lymphoblastic leukemia/T-cell lymphoblastic lymphoma. Cell
Post-translational interference of CD7
Allogeneic from HLA-matched or haploidentical donors CD7
Post-translational interference of CD7 expression
Allogeneic not from HSCT donors CD7
Deletion of CD7 by base-editing
Deletion of TRBC, CD52 3 2
Flu 90 mg/m2
Cy 750 mg/m2 or 90 mg/kg (b~3,150 mg/m2) for new donor-derived CAR T cells
Flu 150 mg/m2
Cy 120 mg/kg (b~4,200 mg/m2)
Alemtuzumab: 1 mg/kg Chiesa
Cy 1,200-3,600 mg/m2
Deletion of TRAC 2 2 Flu 180-222 mg/m2
Deletion of CD7 by CRISPR-Cas9
Deletion of TRAC, RFX5; expression of E-cadherinCD28, IL2RG
of
Prednisone 240 mg/m2 + melphalan 74 mg/m2 or methylprednisolone 360 mg/m2
Flu 150 mg/m2
Cy 1,500 mg/m2
Etoposide 500 mg
mg/m2
mg/m2
aIn some but not all patients with T-cell acute lymphoblastic leukemia, complete remission was confirmed by flow cytometry and/or polymerase chain reaction. In some patients with T-cell lymphoblastic lymphoma, the definition of complete remission was based on imaging only. bBased on a 70 kg adult with an estimated body surface area of 2 m2. Pts: patients; CR: complete remission; Flu: fludarabine; Cy: cyclophosphamide; CAR: chimeric antigen receptor; HSCT: hematopoietic stem cell transplant; TRBC: T cell receptor beta constant gene; TRAC: T-cell receptor alpha constant gene; RFX5: regulatory factor X 5 gene; IL2RG: interleukin 2 receptor subunit gamma gene.
patients with T-ALL, only one had an initial response but could not proceed with the planned HSCT and relapsed with CD5-positive disease.109
None of the clinical studies of autologous CAR T cells in T-ALL/T-LL reported to date has indicated the occurrence of relapse driven by reinfusion of leukemic cells that survived CAR T-cell manufacturing. This is, however, a potential risk of autologous CAR T-cell infusions. If circulating leukemic cells survive the culture period required for CAR T-cell manufacturing, they may be genetically modified and express the CAR, which may mask the target antigen and cause escape from CAR T-cell cytotoxicity, as has been reported with anti-CD19
CAR T cells.110 This occurrence is likely to be rare because ALL cells require mesenchymal cell support to survive in vitro;111,112 they are unlikely to persist and even less likely to be amenable to viral vector transduction under the culture conditions used to manufacture CAR T cells. Nevertheless, levels of MRD in peripheral blood in T-ALL are generally higher than in B-lineage ALL,113,114 increasing the risk of leukemic cell contamination in the starting material. Moreover, enrichment of normal T cells during manufacturing using antibodies is often impossible in T-ALL, with the exception of a minority of cases in which leukemic cells lack surface expression of CD3 or CD4 and CD8.80 It is therefore important to reduce
levels of peripheral blood MRD as much as possible prior to leukapheresis, and test for the presence of residual T-ALL cells at the end of the cultures, including their expression of CAR and targeted antigen.
Allogeneic chimeric antigen receptor T cells derived from HLA-matched hematopoietic stem cell donors In patients with refractory T-ALL, it may be challenging to obtain an adequate number of peripheral blood mononuclear cells for CAR T-cell manufacturing. Such patients often have high numbers of circulating leukemic cells and chemotherapy to reduce the tumor burden also reduces lymphocyte counts. Thus, in patients who relapse after HSCT, the use of HLA-matched donors as a source of peripheral blood is an attractive option. As a caveat, this approach has the potential to trigger GvHD as CAR T cells are strongly stimulated during manufacturing and the numbers of T cells infused (including those not expressing the CAR) might exceed the threshold regarded as safe during HSCT and for donor lymphocyte infusions.115,116 Pharmacological interventions to control GvHD are generally targeted at T cells and would most likely nullify any benefits derived from CAR T-cell therapy. Ultimately, decisions to use matched-donor cells should be weighed against individual risks that are both specific to the type of product infused and the patient’s clinical status and previous treatment. Pan et al.117 obtained peripheral blood from HLA-matched or haploidentical HSCT donors to manufacture anti-CD7 CAR T cells. CD7 expression was downregulated using an approach seemingly identical to the PEBL strategy described earlier.36 The study included patients who had already undergone HSCT who received cells from their previous donors as well as patients who had not undergone HSCT who received cells from prospective matched donors. The latter group of patients were given higher doses of lymphodepleting cyclophosphamide (30 mg/kg/day vs. 250 mg/m2/ day). Among 20 patients treated, 17 achieved MRD-negative complete remission; all seven patients who had received cells from new donors went on to receive HSCT after CAR T-cell infusion. Fifteen of the 17 initial responders remained in remission, with a median follow-up of 6.3 months. Of note, all patients who had received cells manufactured from previous donors had complete chimerism. Although early-onset GvHD was reported in 60% of patients, most cases were grade 1 involving the skin. The follow-up of this study extended to 2 years was reported more recently.118 Six patients relapsed within 4 to 11 months after CAR T-cell infusion, and four of these lacked CD7 expression. A follow-up paper further reported on the initial cohort of patients who received HSCT consolidation with the addition of another six patients.119
Matched-donor cells were also tested in the clinical trial by Lu et al.80 who used anti-CD7 CAR transduced cells that had survived fratricide during manufacturing despite the lack of CD7 downregulation. The two patients treated
both had complete responses (1 with mild skin GvHD) and subsequently received HSCT.
Some inroads have been made in the generation and clinical testing of allogeneic CAR T cells not derived from HSCT donors. A recent paper reported interim results of a phase I study in which base-editing was used to remove CD7 (to avoid fratricide after anti-CD7 CAR expression), the β chain of the aβ TCR (to avoid GvHD) as well as CD52 (to allow the possible use of alemtuzumab to delay rejection).71 Of the three patients who received cells derived from various healthy donors, one achieved a MRD-negative status within 1 month. She went on to receive HSCT on day 49, ahead of reduced toxicity conditioning and antithymocyte globulin to remove allogeneic CAR T cells and prevent GvHD. The patient was in remission at the time of the report, 9 months later. Another patient remained MRD-positive, as determined by polymerase chain reaction analysis, and died of a fungal infection. The third patient achieved MRD negativity and had HSCT with no further follow-up at the time of the report.
Li et al.120 reported the use of “off-the-shelf” anti-CD7 CAR T cells. They used CRISPR-editing of CD7 to avoid fratricide and of TRAC to abrogate TCR expression. Of the two treated patients, one remained in remission for a year after the transplant and one relapsed 48 days later.
Hu et al.121 introduced a multitude of other gene modifications, including deletion of CD7, TCR, and HLA class II, expression of a NK inhibiting receptor (e-cadherin + CD28), and expression of the interleukin 2-receptor γ chain, an approach that seems likely to considerably increase the risk of non-specific genetic alterations. Remission was achieved in five of eight evaluable patients with T-ALL/T-LL but only two remained in remission, one after a transplant. Overall, the three studies provide evidence that allogeneic anti-CD7 CAR T cells can produce tumor responses and that GvHD risk can be markedly reduced by removing TCR expression. In the studies by Li et al.120 and Hu et al. 121 the number of cells infused was 1x107 cells/kg or greater (a dose 10 times higher than that in the study by Chiesa et al.71) but no GvHD was reported. The usefulness of targeting other genes beyond CD7 and TCR remains unclear in the context of using CAR T-cell therapy as a bridge to transplant.
Regardless of the autologous or allogeneic origin of CAR T cells, their infusion is commonly preceded by lymphodepleting chemotherapy which creates an environment that favors engraftment of CAR T cells.122 The intensity of lymphodepletion can affect the degree of expansion of autologous CAR T cells after infusion.17 In the case of allogeneic CAR T cells,
lymphodepletion also temporarily suppresses rejection. Table 2 summarizes the lymphodepleting regimens that have been used prior to CAR T-cell infusion in patients with T-ALL/T-LL. Given the small numbers of patients, it is unclear whether the differences in lymphodepletion regimens have had any impact on clinical responses.
Pancytopenia was one of the most common toxicities reported after CD7-directed CAR T-cell therapy, and it was associated with viral reactivation and fungal infections.71,117 It is unclear whether specific targeting of CD7 affects the severity and duration of pancytopenia. However, patients with T-ALL who received CAR T-cell therapy in the reported studies had usually been given multiple lines of intensive chemotherapy and many had undergone HSCT, as well as further bridging chemotherapy and lymphodepletion prior to receiving the CAR T cells. Pancytopenia was not, therefore, unexpected. The types of infections reported following CAR T-cell therapy were not distinctly different from those observed in patients with prolonged pancytopenia. Reported infections after CAR T-cell infusion in patients with T-ALL appeared to be primarily associated with pancytopenia and not due to CAR T-cell-driven immune cell deficiencies. The toxicity profile typically associated with CAR T cells, including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS),33 does not appear different from that when CAR T cells target CD7. No dose-limiting toxicities were reported in the reviewed clinical studies; manageable grade 1-2 CRS was the most common adverse effect reported, while grade 3-4 CRS occurred rarely.80,117 The severity of reported cases of ICANS was also mild; the cases resolved spontaneously in most studies and were associated with a high burden of disease.71,80,117 CD7 expression in T-ALL cells is typically higher than in normal T cells36 and some normal T cells may have very dim or undetectable CD7 expression.49-51 Whether such T cells can repopulate the T-cell compartment after anti-CD7 CAR T-cell therapy is unclear. As mentioned earlier, CD7-deficient mice did not have significant lymphopenia or immunodeficiency,46,47 but the specific deficiency of CD7 has not been described in humans. In the study by Pan et al.117 with donor-derived CAR T cells, there was recovery of T and NK cells that lacked CD7 expression after initial depletion of CD7-positive immune cells by the CAR T cells. The T cells were also reactive to stimulation with fungal and viral peptides. Cell profiling by single-cell RNA-sequencing-based profiling indicated a higher expression of T-cell functional pathways as compared to T cells prior to CAR T-cell therapy.123 Further studies are required to determine the long-term impact of the loss of CD7 expression on T cells and NK cells.
Effective CAR T-cell technologies targeting antigens expressed by T-ALL and T-LL have been developed and are
being translated into clinical-grade products. The collective data reported so far show that their infusion produces marked antitumor responses with an acceptable safety profile. Although some investigators rely on a minority of T cells that escape killing by the CAR T cells during manufacturing, others prefer to downregulate the antigen targeted by the CAR in the CAR T cells to avert fratricide, as well as maximize the potency and T-cell repertoire of the infused T cells.
Because of its high expression in T-ALL, including ETP-ALL, CD7 is an excellent target for CAR T-cell therapy of T-ALL and T-LL. Other targets, however, could be important in cases in which not all leukemic cells are CD7-positive; such cells may drive CD7-negative relapse after anti-CD7 CAR T-cell infusion.65,80,117 Should CD5, CD1a, TRBC1, CD38 and other targets be expressed in the CD7-negative cells, they could provide additional opportunities for treatment, and might also be useful in treating patients with more mature T-cell malignancies, which often lack CD7.124 Conceivably, levels of target antigen expression in leukemic cells influence the cells’ susceptibility to CAR T-cell killing, and leukemic cells with undetectable target antigen by flow cytometry are likely to escape cytotoxicity. If these cells have clonogenic potential, they can be the source of relapse after CAR T-cell infusion. Nevertheless, data are still lacking to determine specific thresholds to use as inclusion criteria.
The use of autologous cells remains the most widely used form of CAR T-cell therapy for ALL. The development of methods that allow manufacturing of autologous CAR T cells on site, i.e., at the point-of care and not in a remote facility, should further widen their accessibility and timely infusion.125,126 Cells derived from healthy donors should have more predictable expansion and better capacity for persistence than those derived from patients who received multiple courses of chemotherapy; culture conditions that promote expansion of cells with superior longevity and antitumor capacity could further enhance the quality of allogeneic products.13,127,128 The immediate availability of allogeneic off-the-shelf products would make them clinically valuable, particularly for patients with rapidly progressing disease, when it might be difficult to collect a sufficient number of effective T cells and the risk of transducing and/or genetically modifying contaminating leukemic cells is high. While allogeneic cells have the potential to cause GvHD, this can be effectively prevented by abrogating TCR expression in CAR T cells.13,127,128 Without any further modification, however, allogeneic CAR T cells are likely to be quickly rejected and have insufficient time to eradicate leukemia. Therefore, at present, their use is limited to being a last-ditch effort to reduce MRD before HSCT. By contrast, autologous CAR T cells (or those derived from HSCT donors) have the potential to persist for years after infusion and, hence, exert a profound and durable anti-leukemic effect. It remains to be seen whether allogeneic CAR T cells can
be used only as a bridge to HSCT (or to autologous CAR T cells) or can represent a stand-alone treatment. In sum, CAR T-cell therapy is an emerging new treatment option for patients with T-ALL/T-LL. Much work still needs to be done but the tremendous knowledge already gained with CAR T-cell therapy in B-cell malignancies will serve as a guide and hopefully accelerate progress in this area, which holds promise for cure in patients with an otherwise lethal disease.
Disclosures
BLZO has no conflicts of interest to disclose. NV, DMHW, and DC receive royalties for patents related to the development of CAR
1. Teachey DT, Pui CH. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019;20(3):e142-e154.
2. Marks DI, Paietta EM, Moorman AV, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. 2009;114(25):5136-5145.
3. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166-178.
4 Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147-156.
5. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157-163.
6. Inukai T, Kiyokawa N, Campana D, et al. Clinical significance of early T-cell precursor acute lymphoblastic leukaemia: results of the Tokyo Children’s Cancer Study Group study L99-15. Br J Haematol. 2012;156(3):358-365.
7. Neumann M, Heesch S, Gokbuget N, et al. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012;2(1):e55.
8. Jain N, Lamb AV, O’Brien S, et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: a high-risk subtype. Blood. 2016;127(15):1863-1869.
9 Wood B, Devidas M, Summers RJ, et al. Prognostic significance of ETP phenotype and minimal residual disease in T-ALL: a Children’s Oncology Group study. Blood. 2023;142(24):2069-2078.
10 Campana D, Pui CH. Minimal residual disease-guided therapy in childhood acute lymphoblastic leukemia. Blood. 2017;129(14):1913-1918.
11. Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood. 2017;129(9):1134-1142.
12. Eckert C, Parker C, Moorman AV, et al. Risk factors and outcomes in children with high-risk B-cell precursor and T-cell relapsed acute lymphoblastic leukaemia: combined analysis of ALLR3 and ALL-REZ BFM 2002 clinical trials. Eur J Cancer. 2021;151:175-189.
13. Labanieh L, Mackall CL. CAR immune cells: design principles,
T cells for T-cell malignancies. DC also receives patent royalties from Juno Therapeutics; is scientific founder, consultant and stockholder of Nkarta Therapeutics and Medisix Therapeutics; and is a consultant for Locus Cell and Jeito Capital.
Contributions
BLZO led the writing of the article with input from the other authors.
We thank Yi Tian Png, Takahiro Kamiya, Elaine Coustan-Smith, Noriko Shimasaki and Allen Yeoh for their contributions to the development of anti-CD7 PEBL CAR T-cell therapy.
resistance and the next generation. Nature. 2023;614(7949):635-648.
14 Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Eng J Med. 2011;365(8):725-733.
15. Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptormodified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509-1518.
16. Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra138.
17 Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123-2138.
18. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439-448.
19 Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19targeted CAR immunotherapy. Nat Med. 2018;24(1):20-28.
20 Wayne AS, Huynh V, Hijiya N, et al. Three-year results from phase I of ZUMA-4: KTE-X19 in pediatric relapsed/refractory acute lymphoblastic leukemia. Haematologica. 2023;108(3):747-760.
21. Laetsch TW, Maude SL, Rives S, et al. Three-year update of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia in the ELIANA trial. J Clin Oncol. 2023;41(9):1664-1669.
22. Shah NN, Lee DW, Yates B, et al. Long-term follow-up of CD19CAR T-cell therapy in cchildren and young adults with B-ALL. J Clin Oncol. 2021;39(15):1650-1659.
23. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531-2544.
24. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45-56.
25. Westin JR, Oluwole OO, Kersten MJ, et al. Survival with axicabtagene ciloleucel in large B-cell lymphoma. N Engl J Med. 2023;389(2):148-157.
26. Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nature Rev Clin Oncol.
2023;20(6):359-371.
27. Campana D, Behm FG. Immunophenotyping of leukemia. J Immunol Methods. 2000; 243(1-2):59-75.
28. Jasper GA, Arun I, Venzon D, et al. Variables affecting the quantitation of CD22 in neoplastic B cells. Cytometry B Clin Cytom. 2011;80(2):83-90.
29 van Dongen JJ, Lhermitte L, Bottcher S, et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia. 2012;26(9):1908-1975.
30 Haso W, Lee DW, Shah NN, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121(7):1165-1174.
31. Myers RM, Taraseviciute A, Steinberg SM, et al. Blinatumomab nonresponse and high-disease burden are associated with inferior outcomes after CD19-CAR for B-ALL. J Clin Oncol. 2022;40(9):932-944.
32. Campana D, Janossy G, Bofill M, et al. Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. J Immunol. 1985;134(3):1524-1530.
33. Mahadeo KM, Khazal SJ, Abdel-Azim H, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nature Rev Clinl Oncol. 2019;16(1):45-63.
34. Coustan-Smith E, Sandlund JT, Perkins SL, et al. Minimal disseminated disease in childhood T-cell lymphoblastic lymphoma: a report from the Children’s Oncology Group. J Clin Oncol. 2009; 27(21):3533-2539.
35. Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-celldirected chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126(8):983-992.
36. Png YT, Vinanica N, Kamiya T, et al. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017;1(25):2348-2360.
37. Lobach DF, Hensley LL, Ho W, Haynes BF. Human T cell antigen expression during the early stages of fetal thymic maturation. J Immunol. 1985;135(3):1752-1759.
38. Campana D, van Dongen JJ, Mehta A, et al. Stages of T-cell receptor protein expression in T-cell acute lymphoblastic leukemia. Blood. 1991;77(7):1546-1554.
39. Hao QL, George AA, Zhu J, et al. Human intrathymic lineage commitment is marked by differential CD7 expression: identification of CD7- lympho-myeloid thymic progenitors. Blood. 2008;111(3):1318-1326.
40 Vodinelich L, Tax W, Bai Y, et al. A monoclonal antibody (WT1) for detecting leukemias of T-cell precursors (T-ALL). Blood. 1983;62(5):1108-1113.
41. Link M, Warnke R, Finlay J, et al. A single monoclonal antibody identifies T-cell lineage of childhood lymphoid malignancies. Blood. 1983;62(4):722-728.
42. Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1(2):133-143.
43. Janossy G, Coustan-Smith E, Campana D. The reliability of cytoplasmic CD3 and CD22 antigen expression in the immunodiagnosis of acute leukemia: a study of 500 cases. Leukemia. 1989;3(3):170-181.
44 Haynes BF, Eisenbarth GS, Fauci AS. Human lymphocyte antigens: production of a monoclonal antibody that defines functional thymus-derived lymphocyte subsets. Proc Natl Acad Sci U S A. 1979;76(11):5829-5833.
45. Sempowski GD, Lee DM, Kaufman RE, Haynes BF. Structure and function of the CD7 molecule. Critical Rev Immunol. 1999;19(4):331-348.
46. Bonilla FA, Kokron CM, Swinton P, Geha RS. Targeted gene disruption of murine CD7. Int Immunol. 1997;9(12):1875-1883.
47. Lee DM, Staats HF, Sundy JS, et al. Immunologic characterization of CD7-deficient mice. J Immunol. 1998;160(12):5749-5756.
48. Sempowski GD, Lee DM, Scearce RM, Patel DD, Haynes BF. Resistance of CD7-deficient mice to lipopolysaccharide-induced shock syndromes. J Exp Med. 1999;189(6):1011-1016.
49 Reinhold U, Abken H, Kukel S, et al. CD7- T cells represent a subset of normal human blood lymphocytes. J Immunol. 1993;150(5):2081-2089.
50 Kukel S, Reinhold U, Oltermann I, Kreysel HW. Progressive increase of CD7- T cells in human blood lymphocytes with ageing. Clin Exp Immunol. 1994;98(1):163-168.
51. Aandahl EM, Sandberg JK, Beckerman KP, et al. CD7 is a differentiation marker that identifies multiple CD8 T cell effector subsets. J Immunol. 2003;170(5):2349-2355.
52. Frankel AE, Laver JH, Willingham MC, et al. Therapy of patients with T-cell lymphomas and leukemias using an anti-CD7 monoclonal antibody-ricin A chain immunotoxin. Leuk Lymphoma. 1997;26(3-4):287-298.
53. Baum W, Steininger H, Bair HJ, et al. Therapy with CD7 monoclonal antibody TH-69 is highly effective for xenografted human T-cell ALL. Br J Haematol. 1996;95(2):327-338.
54. Pauza ME, Doumbia SO, Pennell CA. Construction and characterization of human CD7-specific single-chain Fv immunotoxins. J Immunol. 1997;158(7):3259-3269.
55. Pennell CA, Pauza ME. CD7-specific single chain Fv immunotoxins. Design and expression. Methods Mol Biol. 2001;166:17-29.
56. Imai C, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676-684.
57 Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453-1464.
58. Campana D, Schwarz H, Imai C. 4-1BB chimeric antigen receptors. Cancer J. 2014;20(2):134-140.
59 Gomes-Silva D, Srinivasan M, Sharma S, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130(3):285-296.
60 Cooper ML, Choi J, Staser K, et al. An “off-the-shelf” fratricideresistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32(9):1970-1983.
61. Georgiadis C, Rasaiyaah J, Gkazi SA, et al. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia. 2021;35(12):3466-3481.
62. Jiang J, Chen J, Liao C, et al. Inserting EF1alpha-driven CD7specific CAR at CD7 locus reduces fratricide and enhances tumor rejection. Leukemia. 2023;37(8):1660-1670.
63. Kamiya T, Wong D, Png YT, Campana D. A novel method to generate T-cell receptor-deficient chimeric antigen receptor T cells. Blood Adv. 2018;2(5):517-528.
64 Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest. 2019; 12;130.
65. Zhang M, Chen D, Fu X, et al. Autologous nanbody-derived fratricide-resistant CD7-CAR T-cell therapy for patients with
relapsed and refractory T-cell acute lymphoblastic leukemia/ lymphoma. Clin Cancer Res. 2022;28(13):2830-2843.
66. Cameron P, Fuller CK, Donohoue PD, et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods. 2017;14(6):600-606.
67. Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481).
68. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without doublestranded DNA cleavage. Nature. 2016;533(7603):420-424.
69 Doman JL, Raguram A, Newby GA, Liu DR. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat Biotechnol. 2020;38(5):620-628.
70. Diorio C, Murray R, Naniong M, et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140(6):619-629.
71. Chiesa R, Georgiadis C, Syed F, et al. Base-edited CAR7 T cells for relapsed T-cell acute lymphoblastic leukemia. N Engl J Med. 2023;389(10):899-910.
72. Kim D, Bae S, Park J, et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12(3):237-243.
73. Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017;14(6):607-614.
74. Yu Y, Leete TC, Born DA, et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat Commun. 2020;11(1):2052.
75. Grunewald J, Zhou R, Lareau CA, et al. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat Biotechnol. 2020;38(7):861-864.
76. Lam DK, Feliciano PR, Arif A, et al. Improved cytosine base editors generated from TadA variants. Nat Biotechnol. 2023;41(5):686-697.
77 Watanabe N, Mo F, Zheng R, et al. Feasibility and preclinical efficacy of CD7-unedited CD7 CAR T cells for T cell malignancies. Mol Ther. 2023;31(1):24-34.
78. Mestermann K, Giavridis T, Weber J, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Trnasl Med. 2019;11(499):eaau5907.
79 Weber EW, Lynn RC, Sotillo E, et al. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019;3(5):711-717.
80. Lu P, Liu Y, Yang J, et al. Naturally selected CD7 CAR-T therapy without genetic manipulations for T-ALL/LBL: first-in-human phase 1 clinical trial. Blood. 2022;140(4):321-334.
81. Freiwan A, Zoine JT, Crawford JC, et al. Engineering naturally occurring CD7- T cells for the immunotherapy of hematological malignancies. Blood. 2022;140(25):2684-2696.
82. Jones NH, Clabby ML, Dialynas DP, et al. Isolation of complementary DNA clones encoding the human lymphocyte glycoprotein T1/Leu-1. Nature. 1986;323(6086):346-349.
83. Tembhare PR, Chatterjee G, Khanka T, et al. Eleven-marker 10-color flow cytometric assessment of measurable residual disease for T-cell acute lymphoblastic leukemia using an approach of exclusion. Cytometry B Clin Cytom. 2021;100(4):421-433.
84 Raikar SS, Fleischer LC, Moot R, et al. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology. 2018;7(3):e1407898.
85. Dai Z, Mu W, Zhao Y, et al. The rational development of CD5targeting biepitopic CARs with fully human heavy-chain-only antigen recognition domains. Mol Ther. 2021;29(9):2707-2722.
86. Mamonkin M, Mukherjee M, Srinivasan M, et al. Reversible transgene expression reduces fratricide and permits 4-1BB costimulation of CAR T cells directed to T-cell malignancies. Cancer Immunol Res. 2018;6(1):47-58.
87. van de Rijn M, Lerch PG, Knowles RW, Terhorst C. The thymic differentiation markers T6 and M241 are two unusual MHC class I antigens. J Immunol. 1983;131(2):851-855.
88. Martin LH, Calabi F, Lefebvre FA, Bilsland CA, Milstein C. Structure and expression of the human thymocyte antigens CD1a, CD1b, and CD1c. Proc Natl Acad Sci U S A. 1987;84(24):9189-9193.
89 Amiot M, Dastot H, Schmid M, Bernard A, Boumsell L. Analysis of CD1 molecules on thymus cells and leukemic T lymphoblasts identifies discrete phenotypes and reveals that CD1 intermolecular complexes are observed only on normal cells. Blood. 1987;70(3):676-685.
90 Pui CH, Behm FG, Singh B, et al. Heterogeneity of presenting features and their relation to treatment outcome in 120 children with T-cell acute lymphoblastic leukemia. Blood. 1990;75(1):174-179.
91. Niehues T, Kapaun P, Harms DO, et al. A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. Leukemia. 1999;13(4):614-617.
92. van Grotel M, Meijerink JP, Van Wering ER, et al. Prognostic significance of molecular-cytogenetic abnormalities in pediatric T-ALL is not explained by immunophenotypic differences. Leukemia. 2008;22(1):124-131.
93. Sanchez-Martinez D, Baroni ML, Gutierrez-Aguera F, et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood. 2019;133(21):2291-2304.
94 Sims JE, Tunnacliffe A, Smith WJ, Rabbitts TH. Complexity of human T-cell antigen receptor beta-chain constant- and variable-region genes. Nature. 1984;312(5994):541-545.
95. Maciocia PM, Wawrzyniecka PA, Philip B, et al. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat Med. 2017; 23(12):1416-1423.
96. Campana D, Thompson JS, Amlot P, Brown S, Janossy G. The cytoplasmic expression of CD3 antigens in normal and malignant cells of the T lymphoid lineage. J Immunol. 1987;138(2):648-655.
97. Reinherz EL, Kung PC, Goldstein G, Levey RH, Schlossman SF. Discrete stages of human intrathymic differentiation: analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc Natl Acad Sci U S A. 1980;77(3):1588-1592.
98. Huang S, Terstappen LWMM. Lymphoid and myeloid differentiation of single human CD34+, HLA-Dr+, CD38hematopoietic stem cells. Blood. 1994;83(6):1515-1526.
99. Bhatia M, Wang JCY, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci U S A. 1997;94(10):5320-5325.
100 Taussig DC, Vargaftig J, Miraki-Moud F, et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood. 2010;115(10):1976-1984.
101. Tembhare PR, Sriram H, Khanka T, et al. Flow cytometric evaluation of CD38 expression levels in the newly diagnosed
T-cell acute lymphoblastic leukemia and the effect of chemotherapy on its expression in measurable residual disease, refractory disease and relapsed disease: an implication for antiCD38 immunotherapy. J Immunother Cancer. 2020;8(1):e000630.
102. Ofran Y, Ringelstein-Harlev S, Slouzkey I, et al. Daratumumab for eradication of minimal residual disease in high-risk advanced relapse of T-cell/CD19/CD22-negative acute lymphoblastic leukemia. Leukemia. 2020;34(1):293-295.
103. Bride KL, Vincent TL, Im SY, et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood. 2018;131(9):995-999.
104 Mihara K, Yanagihara K, Takigahira M, et al. Activated T-cellmediated immunotherapy with a chimeric receptor against CD38 in B-cell non-Hodgkin lymphoma. J Immunother. 2009;32(7):737-743.
105. Mihara K, Yanagihara K, Takigahira M, et al. Synergistic and persistent effect of T-cell immunotherapy with anti-CD19 or anti-CD38 chimeric receptor in conjunction with rituximab on B-cell non-Hodgkin lymphoma. Br J Haematol. 2010;151(1):37-46.
106. Mihara K, Bhattacharyya J, Kitanaka A, et al. T-cell immunotherapy with a chimeric receptor against CD38 is effective in eliminating myeloma cells. Leukemia. 2012;26(2):365-367.
107. Drent E, Groen RW, Noort WA, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica. 2016;101(5):616-625.
108. Glisovic-Aplenc T, Diorio C, Chukinas JA, et al. CD38 as a panhematologic target for chimeric antigen receptor T cells. Blood Adv. 2023;7(16):4418-4430.
109. Mamonkin M, Brenner MK, Heslop H, et al. Safety and antitumor activity of CD5 CAR T-cells in patients with relapsed/ refractory T-cell malignancies. Blood. 2019;134;(Supplement_1):199.
110 Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499-1503.
111. Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, Campana D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood. 1992;79(9):2370-2377.
112. Winter SS, Sweatman JJ, Lawrence MB, et al. Enhanced T-lineage acute lymphoblastic leukaemia cell survival on bone marrow stroma requires involvement of LFA-1 and ICAM-1. Br J Haematol. 2001;115(4):862-871.
113. Coustan-Smith E, Sancho J, Hancock ML, et al. Use of peripheral blood instead of bone marrow to monitor residual disease in children with acute lymphoblastic leukemia. Blood. 2002;100(7):2399-2402.
114. van der Velden V, Jacobs DC, Wijkhuijs AJ, et al. Minimal residual disease levels in bone marrow and peripheral blood are comparable in children with T cell acute lymphoblastic leukemia, but not in precursor-B-ALL. Leukemia. 2002;16(8):1432-1436.
115. Muller S, Schulz A, Reiss U, et al. Definition of a critical T cell threshold for prevention of GVHD after HLA non-identical PBPC
transplantation in children. Bone Marrow Transplant. 1999;24(6):575-581.
116. Dholaria B, Savani BN, Labopin M, et al. Clinical applications of donor lymphocyte infusion from an HLA-haploidentical donor: consensus recommendations from the Acute Leukemia Working Party of the EBMT. Haematologica. 2020;105(1):47-58.
117 Pan J, Tan Y, Wang G, et al. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: firstin-human, phase I trial. J Clin Oncol. 2021;39(30):3340-3351.
118. Tan Y, Shan L, Zhao L, et al. Long-term follow-up of donorderived CD7 CAR T-cell therapy in patients with T-cell acute lymphoblastic leukemia. J Hematol Oncol. 2023;16(1):34.
119 Li Z, An N, Yang K, et al. Donor CD7 chimeric antigen receptor T cell bridging to allogeneic hematopoietic stem cell transplantation for T cell hematologic malignancy. Transplant Cell Ther. 2023;29(3):167-173.
120 Li S, Wang X, Yuan Z, et al. Eradication of T-ALL cells by CD7targeted universal CAR-T cells and initial test of ruxolitinibbased CRS management. Clin Cancer Res. 2021;27(5):1242-1246.
121. Hu Y, Zhou Y, Zhang M, et al. Genetically modified CD7-targeting allogeneic CAR-T cell therapy with enhanced efficacy for relapsed/refractory CD7-positive hematological malignancies: a phase I clinical study. Cell Res. 2022;32(11):995-1007.
122. Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6(5):383-393.
123. Chen W, Shi H, Liu Z, et al. Single-cell transcriptomics reveals immune reconstitution in patients with R/R T-ALL/LBL treated with donor-derived CD7 CAR-T therapy. Clin Cancer Res. 2023;29(8):1484-1495.
124. Vega F, Medeiros LJ. A suggested immunohistochemical algorithm for the classification of T-cell lymphomas involving lymph nodes. Human Pathol. 2020;102:104-116.
125. Shah NN, Johnson BD, Schneider D, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med. 2020;26(10):1569-1575.
126. Del Bufalo F, Becilli M, Rosignoli C, et al. Allogeneic, donorderived, second-generation, CD19-directed CAR-T cells for the treatment of pediatric relapsed/refractory BCP-ALL. Blood. 2023;142(2):146-157.
127. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423-431.
128. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nature Rev Clin Oncol. 2020;17(3):147-167.
129. An N, Hou YN, Zhang QX, et al. Anti-multiple myeloma activity of nanobody-based anti-CD38 chimeric antigen receptor T cells. Mol Pharm. 2018;15(10):4577-4588.
130. Hill LC, Rouce RH, Smith TS, et al. Safety and anti-tumor activity of CD5 CAR T-cells in patients with relapsed/refractory T-cell malignancies. Blood. 2019;134;(Supplement_1):199.
131. Ghobadi A, Aldoss I, Maude SL, et al. Phase 1/2 dose-escalation study of anti-CD7 allogenic CAR-T cell in relapsed or refractory T-cell acute lymphoblastic leukemia/lymphoblastic lymphoma. HemaSphere. 2023;7(10):e957.
1Department of Hematology/Oncology, Cell and Gene Therapy – IRCCS, Bambino Gesù Children’s Hospital, Rome; 2Catholic University of the Sacred Heart, Department of Life Sciences and Public Health, Rome and 3Department of Clinical Medicine and Surgery, University of Naples Federico II, Naples, Italy
Correspondence: F. Locatelli franco.locatelli@opbg.net
Received: November 14, 2023. Accepted: February 1, 2024. https://doi.org/10.3324/haematol.2023.284604
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Chimeric antigen receptor (CAR) T-cell therapy has emerged as a breakthrough cancer therapy over the past decade. Remarkable outcomes in B-cell lymphoproliferative disorders and multiple myeloma have been reported in both pivotal trials and real-word studies. Traditionally, the use of a patient’s own (autologous) T cells to manufacture CAR products has been the standard practice. Nevertheless, this approach has some drawbacks, including manufacturing delays, dependence on the functional fitness of the patient’s T cells, which can be compromised by both the disease and prior therapies, and contamination of the product with blasts. A promising alternative is offered by the development of allogeneic CAR-cell products. This approach has the potential to yield more efficient drug products and enables the use of effector cells with negligible alloreactive potential and a significant CAR-independent antitumor activity through their innate receptors (i.e., natural killer cells, γδ T cells and cytokine induced killer cells). In addition, recent advances in genome editing tools offer the potential to overcome the primary challenges associated with allogeneic CAR T-cell products, namely graft-versus-host disease and host allo-rejection, generating universal, off-the-shelf products. In this review, we summarize the current pre-clinical and clinical approaches based on allogeneic CAR T cells, as well as on alternative effector cells, which represent exciting opportunities for multivalent approaches and optimized antitumor activity.
Autologous, patient-derived T cells engineered to express a chimeric antigen receptor (CAR) represent a breakthrough treatment that has revolutionized the management of patients with B-cell malignancies.1-5 In 2018, the results of the ELIANA study, a global trial testing the use of tisagenlecleucel in pediatric patients with relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL), showed that a substantial proportion of these patients in whom many lines of previous therapies had failed, including allogeneic hematopoietic stem cell transplantation (HSCT), can be rescued with the approach, leading to the first ever approval of a CAR T-cell product by the American and European Regulatory Agencies, the Food and Drug Administration and European Medicines Agency, respectively.1 Recently, the 3-year update of the ELIANA study was published
and confirmed an overall remission rate of 82%, with a 3-year event-free survival of 44%.2 After the approval of tisagenlecleucel, several real-word studies confirmed the extraordinary ability of these autologous, CD19-directed, second-generation CAR T cells to induce high rates of minimal residual disease (MRD)-negative complete remission (CR) in children with relapsed/refractory B-ALL and provided further evidence on their therapeutic potential. In particular, several studies documented that tisagenlecleucel was able to induce CR in 85-87% of the patients, with the remission being MRD-negative in most of the cases, and 1-year and 2-year event-free survival rates of 50-52.4% and 45.3%, respectively.3-5 A high response rate can also be achieved in patients relapsing after receiving allogeneic HSCT, with the time to relapse after the transplant being a strong predictor of outcome.4 Importantly, CAR T cells are able to induce similar results across cytogenetic cat-
egories and are associated with promising outcomes also in infants, with an unclear impact on the risk of myeloid switch.6-8 In addition, some preliminary evidence of efficacy of CD19-CAR T cells in patients with central nervous system involvement has been reported, especially if disease control is achieved before infusion of the cell product.9-12 Despite the success of CD19-specific CAR T-cell therapy, several limitations of the approach have emerged and will be discussed in this review, together with the possible solutions offered by the use of allogeneic CAR T cells.
The manufacture of autologous CAR T cells involves a complex productive procedure that starts with the collection of
the patient’s peripheral blood mononuclear cells to obtain viable T cells. The quality of the leukapheresis product is key in ensuring the success of the entire manufacturing process (Figure 1). Thus, collecting a sufficient number of T lymphocytes with preserved cytotoxic functions is a crucial prerequisite in autologous CAR T-cell therapy. The timing of this procedure may present a considerable challenge when dealing with heavily pre-treated patients, who are experiencing rapid disease progression and have been exposed to agents impairing T-cell function. As previously reported, the collection of peripheral blood mononuclear cells in pediatric patients poses some peculiar technical challenges, especially in, but not limited to, infants, including: the difficulty of obtaining an adequate venous access; a relatively large extracorporeal volume of the cell separator device, occasionally requiring priming with packed red blood cells; and metabolic complications due
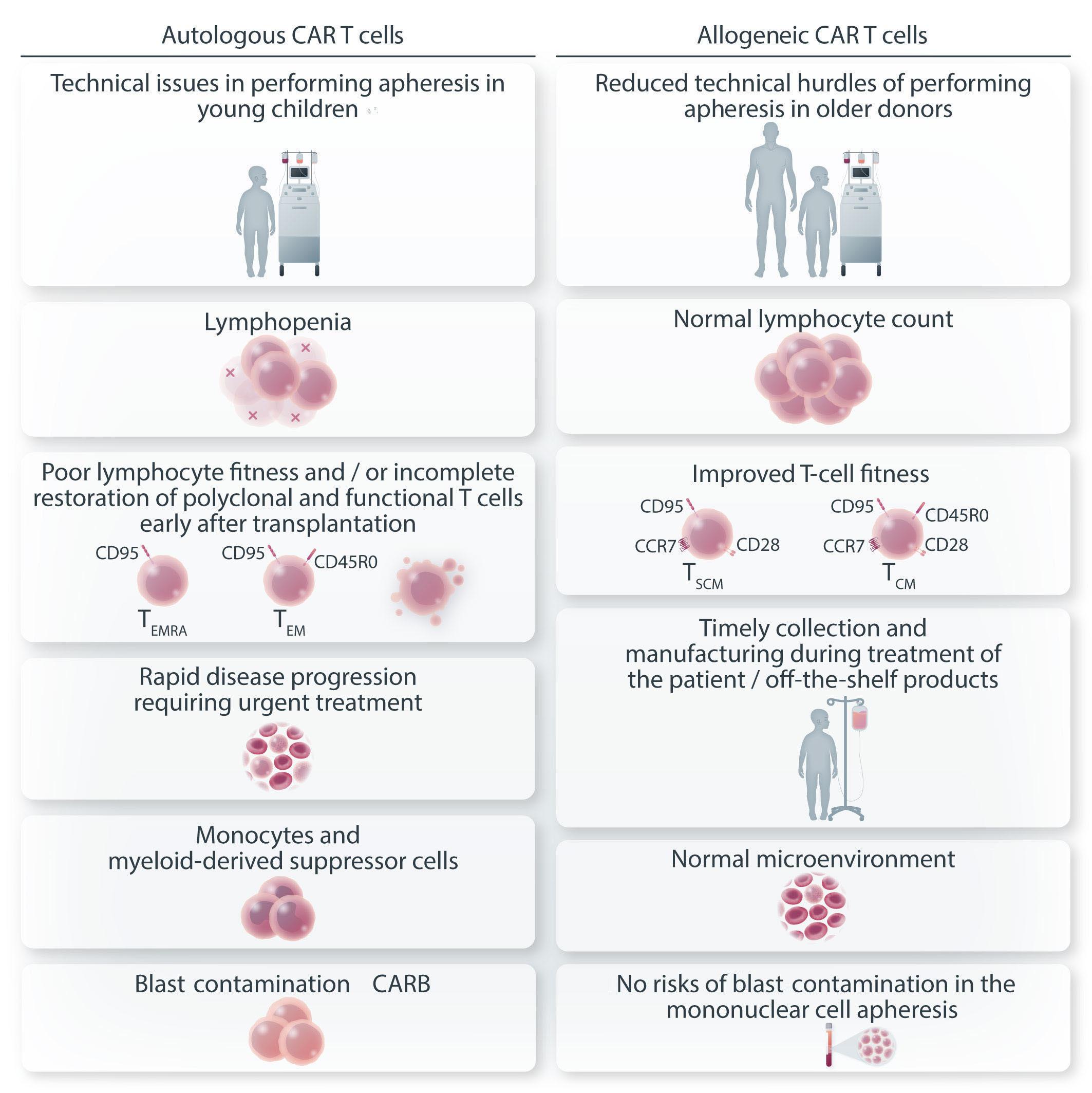
to citrate toxicity.13 Although reports on the use of CAR T-cell therapies in the real-world setting have shown that most centers have gained extensive experience in the performance of leukapheresis, demonstrating the ability to obtain peripheral blood mononuclear cells from both very young/infant patients7,8,14 and heavily pre-treated adults (all patients receiving CAR T cells as third or further lines of treatment), some patients are unfortunately not eligible for this step, due to marked lymphopenia. Data on the actual percentage of those patients for whom a sufficient number of T cells cannot be harvested are limited and these patients are often excluded from studies on CAR T cells.15 The use of autologous lymphocytes also has another relevant clinical limitation: patients with rapidly progressing disease, a scenario frequently associated with relapsed/ refractory B-ALL, require urgent treatment and may not be able to undergo the collection and manufacturing; alternatively, these procedures can ultimately be performed, but not in a timely manner before patients experience massive progression or disease-related complications, preventing the opportunity to receive CAR T cells. These important considerations are reflected in the difference in the overall CR rates in the intention-to-treat analysis of the ELIANA study, which dropped from 81% to 66%, with one patient not enrolled for apheresis-related issues and seven additional patients who died after enrollment, during the manufacturing period.1
Beyond lymphocyte count, other cellular components in the apheresis product can affect CAR T-cell production, including both circulating blasts and non-malignant cells, such as myeloid cells (Figure 1). Cells of myeloid origin with immunosuppressive properties have been found in patients with cancer and have been associated with manufacturing failures related to their inhibitory effects on T-cell proliferation.16,17 Contamination of the apheresis product with blasts represents another relevant limitation to the use of autologous products, since blasts can be inadvertently transduced and aberrantly express the CD19-CAR on their surface (generating the so-called CARB). Although rare, such an event has been described and is associated with binding in cis of the CD19 epitope leading to its masking from recognition by CD19-CAR T cells, thereby conferring an intrinsic mechanism of resistance to the therapy.18 The quality of the lymphocytes used as starting material for CAR T-cell manufacturing is extremely important for the quality of the final product and, therefore, for its subsequent clinical activity, especially in the long-term. In patients with cancer, both transcriptional and functional heterogeneities have been identified in T cells, leading to the classification of these cells into distinct categories, including pre-dysfunctional, early dysfunctional, and late dysfunctional T cells.19 These T-cell categories are characterized by elevated expression of inhibitory receptors on the cell surface, including programmed cell death-1 (PD1), lymphocyte activation gene 3 protein (LAG3), T-cell immu-
noglobulin mucin receptor 3 (TIM3; encoded by HAVCR2), 2B4, CD200, and cytotoxic T-lymphocyte antigen 4 (CTLA4). Additionally, these cells exhibit a reduced capacity to perform classical CD8+ T-cell effector functions, such as the ability to produce cytokines including tumor necrosis factor, interleukin (IL)-2, and interferon-γ. This reduction in functionality is observed directly ex vivo or after stimulation with cognate antigens or low doses of anti-CD3 antibodies. It is intuitive that the clinical efficacy of CAR T cells generated by cancer patients may be limited by the high frequencies of these dysfunctional T cells present in both the starting material and the final CAR T-cell product. The overall fitness of patient-derived T cells is influenced by several impairing factors, including age, disease burden, and prior cancer treatments and is reasonably a constraining factor for CAR T-cell therapy. By contrast, it has been consistently demonstrated that individuals whose leukapheresis products contain a higher proportion of proliferative, less differentiated T-cell subsets tend to achieve more favorable outcomes. Indeed, the use of T cells with central memory or stem cell-like memory phenotypes for manufacturing CAR T cells has been associated with greater expansion in culture and final products characterized by superior antitumor activity and persistence than products obtained from bulk T cells.20-22 Considering that approximately 50% of patients treated with CAR T cells experience a relapse of disease, in several cases characterized by loss of persistence of the engineered T cells and CD19+ blasts, identifying potential approaches able to improve the persistence of the engineered cells remains crucial. In addition to the abovementioned limitations, a recent German real-world experience highlighted another important obstacle to the efficacy of autologous CAR T cells. Interestingly, the German study showed that in patients relapsing after allogeneic HSCT, the use of tisagenlecleucel within 6 months after transplantation was associated with significantly worse long-term clinical outcomes and shorter duration of B-cell aplasia than those of patients relapsing >6 months after the transplant.4 These findings suggest a lower persistence of CAR T cells generated early after allogeneic HSCT and the authors speculated that this may be associated with an incomplete restoration of polyclonal and functional T cells early after transplantation.4 The use of mononuclear cells obtained from healthy donors, i.e. allogeneic CAR T cells, could be a way of tackling all these issues (Figure 1). Allogeneic CAR T cells have many potential advantages, including: greater fitness, since the T cells have not been exposed to either chemotherapy, or to the immune-suppressive effects of myeloid cells and blasts; they can be obtained from all donors, overcoming the potential issue of lymphopenia, in a timely manner without the need of a wash-out windows from the therapies that would require an halt in the treatment of a patient; and they do not run the risk of being contaminated by blasts. The creation of universal, off-the-shelf products
also has the advantage of being able to build a bank of drug products that can be available anytime upon clinical need, also reducing manufacturing costs by scaling up a single process.
Several allogeneic approaches, tested in the preclinical and clinical settings, are discussed in this review.
The clinical use of allogeneic CAR T cells has been limited by the potential occurrence of graft-versus-host disease (GvHD) leading, on the one hand, to the underdevelopment of this strategy and prompting, on the other hand, the identification of strategies to develop universal CAR T cells, engineered to abrogate the alloreactivity. The results of the clinical studies testing allogeneic, donor-derived CAR T cells are reported in Table 1.
The first experience on the use of allogeneic, donor-derived CAR T cells was reported in 2013 by Cruz et al.23 who gave donor-derived, virus-specific cytotoxic T cells genetically modified to express a second-generation (incorporating CD28 as a costimulatory domain) CD19-specific CAR (CD19. CAR-VST) to patients with B-cell malignancies who either had disease relapse or were at high risk of disease relapse after allogeneic HSCT. Six adults and two children (9 and 12 years of age) with either chronic lymphoblastic leukemia or B-ALL were treated with CD19.CAR-VST, generated from either an unrelated donor or an HLA-matched sibling donor. An objective response was observed in two of six patients, lasting for 3 months and 8 weeks. Importantly, none of the patients developed GvHD.23 In the same year, Kochenderfer et al.24 published their preliminary experience on ten adults affected by chronic lymphoblastic leukemia, diffuse large B-cell lymphoma or mantle cell lymphoma persisting after allogeneic HSCT and at least one donor lymphocyte infusion. CAR T cells were generated from either an unrelated donor, 10/10 HLA-matched, or matched sibling donor, using a second-generation (CD28), CD19-directed construct, and infused at doses of 0.4-7.8x106 CAR+ T cells/ kg recipient body weight, without prior lymphodepletion. Three patients had regression of their tumors and, importantly, none of the ten patients developed GvHD.24 In 2016, the same group reported updated results on a cohort of 20 adult patients with B-cell malignancies, including five patients with B-ALL.25 Interestingly, but not surprisingly, patients with B-ALL had the best response to treatment, with four out of the five (80%) obtaining an MRD-negative CR after infusion; overall, eight of the 20 patients obtained either CR or partial remission. Also in the updated cohort, no new onset GvHD was observed in any of the patients.
In 2015, Dai et al.26 reported on an adolescent and a young adult (15 and 23 years of age) treated with allogeneic, donor-derived, second-generation (4.1bb) CD19-CAR T cells at the doses of 4.5 and 4.2x106 CAR+ T cells/kg of recipient body weight, respectively, for B-ALL. No details on preparation with lymphodepletion or on the characteristics of the donors were reported. Neither patient had a sustained response and both developed grade 2-3 acute GvHD, requiring short-term use of corticosteroids and/or cyclosporin A. Chen et al.27 treated five adults and one child who had relapsed after allogeneic HSCT with allogeneic, third-generation (CD28-CD27) CAR T cells derived from haploidentical donors and infused after administration of lymphodepleting chemotherapy, mostly fludarabine and cyclophosphamide-based. The characteristics of the CAR construct employed in the study were not reported. MRD-negative CR was obtained by 83% of the patients and 50% of the patients developed grade 2-3 GvHD after infusion, which is similar to the rate previously reported by the same group with the use of donor lymphocyte infusion from haploidentical donors.28 All patients had a short duration of remission with relapses occurring 2-7 months after response.27
Using a different transduction system, namely the Sleeping Beauty transposon/transposase system, Kebriaei et al. 29 treated 19 adults with either ALL (n=17) or diffuse large B-cell lymphoma (n=2) with second-generation (CD28) CAR T cells generated by the lymphocytes of their matched sibling donors (n=11) or haploidentical donors (n=8). CAR T cells were infused at doses ranging from 106/m2 to 108/ m2, without prior lymphodepletion, at a median of 64 days after allogeneic HSCT. The 1-year progression-free and overall survival rates were 53% and 63%, respectively. Three patients developed GvHD, so the rate of this complication was comparable to that expected after allogeneic HSCT and lower than that observed after donor lymphocyte infusion.29 Very limited experience has been reported to date in the pediatric population. In 2021, Zhang et al.30 described their experience using second-generation (either CD28 or 4.1bb) CD19-CAR T cells in 43 subjects, aged from 4 up to 60 years old, with B-ALL relapsing after allogeneic HSCT: 79% had a morphological CR and there were only two cases of acute GvHD, both grade ≤2. Unfortunately, no specific details on the outcome of the children were provided. Recently, we reported our experience on the use of donor-derived, second-generation (4.1bb) CD19-CAR T cells in 13 children and young adults with B-ALL that had relapsed after allogeneic HSCT or was extremely refractory.31 Doses ranged between 1×106 and 3×106 CAR+ T cells/kg and all patients underwent fludarabine/cyclophosphamide-based lymphodepletion before infusion. Only one patient developed acute GvHD, namely a young adult who was treated early after allogeneic HSCT and who had previously developed grade 2 acute GvHD.31 Together with a mild toxicity profile, we obtained 100% MRD-negative CR in the bone marrow
Table 1. Studies describing clinical experience with the use of allogeneic, donor-derived, CD19-chimeric antigen receptor T cells.
Adults and children CLL B-ALL 8 (6 adults and 2 children) 2nd gen, CD28 Retrovirus No
5.8x1071.13x108 (5 pts multiple infusions) URD, MSD Yes (2 pts only) No
Adults CLL B-NHL 10 2nd gen, CD28 Retrovirus No 0.4-7.8x106/kg URD, MSD Yes No
Adults B-NHL B-ALL 2 2nd gen, 4.1bb Lentivirus Not reported
4.5x106/kg 4.2x106/kg Not reported Yes
Adults B-ALL B-NHL CLL 20 2nd gen, CD28 Retrovirus No 0.4-7.8x106/kg URD 10/10 or 9/10; MSD Yes
Adults B-ALL (>5% blasts) 6 3rd gen (CD28/ CD27) Lentivirus Flu-Cy
G2-3 GvHD in both 1 CR in BM and PR in EM 1 PR (2) 26
GvHD in 2 pts (preexisting)
0.38-4.1x108/ kg (4 pts: 2 infusions) Haplo Yes 50% GvHD (G2-3) 4 CR (all relapsed subsequently) 1 not evaluable 1 NR 27
Adults B-NHL B-ALL 19 2nd gen, CD28 Sleeping Beauty No 106/m2-108/m2 MSD, haplo No
Adults and children B-ALL 43 2nd gen, CD28 or 4.1bb Lentivirus Mainly Flu-CY 0.4-12×106/kg MSD, haplo Yes
Children and young adults B-ALL 13 2nd gen, 4.1bb Retrovirus or lentivirus Flu-Cy 1-3x106/kg
G1-4 GvHD in 3 pts 9 CCR 1 CR 2 AWD 2 DIR 5 DOD 29
G1-2 GvHD in 2 pts 2 died of toxicity 34 morph. CR 30
MSD, URD, haplo Yes G3 GvHD in 1 pt 12 CR in BM and EM 1 CR in the BM and PR in EM 31
Pts: patients; CAR: chimeric antigen receptor; LD: lymphodepletion; DLI: donor lymphocyte infusion; GvHD: graft-versus-host disease; Ref: references; CLL: chronic lymphocytic leukemia; B-ALL: B-cell acute lymphoblastic leukemia; B-NHL: B-cell non-Hodgkin lymphoma; gen: generation; URD: unrelated donor; MSD: matched sibling donor; CR: complete remission; PR: partial response; SD: stable disease; PD: progressive disease; BM: bone marrow; EM: extramedullary; G: grade; MRD: measurable residual disease; Flu: fludarabine; Cy: cyclophosphamide; NR: non-response; haplo: haploidentical donor; CCR: continuous complete remission; AWD: alive with disease; DIR: dead in remission; DOD: dead of disease; morph.: morphological.
and CR in five of the six cases of extramedullary diseases which, with a median follow-up of 12 months, was maintained in 62% of the patients.31 Overall, all these studies, although extremely heterogeneous in terms of patient population, diseases, CAR constructs and lymphodepletion, highlight the feasibility of the use of allogeneic, donor-derived CAR T cells, with a low incidence of GvHD. Together with the encouraging data on the antileukemic activity of allogeneic products, these reports suggest that the approach deserves further investigation. Interestingly, none of the group experienced rejection of the allogeneic, donor-derived CAR T cells. It has been extensively shown, in the context of organ and umbilical cord blood transplantation, that matching of HLA-A, HLA-B and HLA-DR alleles is sufficient to significantly reduce the
incidence of allograft rejection.32,33 The use of donor-derived T cells for the generation of allogeneic CAR T cells overcomes the issue of rejection thanks to the HLA-compatibility between donor and recipient of the cells, in the case of unrelated and matched sibling donors, and to the immune-reconstitution of the patient with the immune system of the donor before generation and infusion of allogeneic CAR T cells. This aspect also contributes to the long-term persistence of these cells. The role of the possible occurrence of donor-specific antibodies will have to be further investigated in this setting, since it may impair the persistence of allogeneic, donor-derived CAR T cells. In order to develop a universal, off-the-shelf CAR T-cell product for the timely treatment of patients, readily available when clinically needed, some groups have focused
on different gene-editing approaches aimed at disrupting the endogenous T-cell receptor (TCR) locus, to abrogate the potential alloreactivity of allogeneic T cells. Moreover, to overcome the second obstacle associated with the potential rejection of the product by the patient, these approaches have simultaneously disrupted the CD52 locus in the donor cells in order to induce resistance to the CD52-directed monoclonal antibody alemtuzumab, which can then be introduced in the lymphodepleting regimen of the patients to ablate the endogenous T cells before infusing allogeneic CAR T cells.34 The clinical studies testing these approaches are summarized in Table 2. The clinical proof of concept of the potential of such an approach (named UCART19), using transcription activator-like effector nuclease (TALEN) technology for gene editing of the TCR a chain (to abrogate alloreactivity) and CD52 (to induce resistance to alemtuzumab, used in the lymphodepleting chemotherapy) gene loci, was provided by two infants.35 Both patients achieved MRD-negative CR within 28 days after infusion, enabling consolidation with allogeneic HSCT. Subsequently, two phase I studies evaluated the feasibility, safety and antileukemia activity of UCART19 in adults (CALM trial, n=14) and children (PALL trial, n=7).36 All patients received lymphodepletion with fludarabine and cyclophosphamide with (n=17) or without (n=4) alemtuzumab; then, children received 1.1-2.3×10⁶ UCART19 cells/kg and adults received doses of 6×10⁶, 6-8×10⁷ or 1.8-2.4×10⁸ UCART19 cells in a dose-escalation study. Overall, 67% of patients achieved CR after infusion and alemtuzumab proved essential for allowing UCART19 expansion and anti-leukemia activity. Subsequently, 71% of the patients proceeded to allogeneic HSCT. At data cut-off, only two children and three adults were alive and in CR.36
Among the seven children treated, six (86%) obtained CR and five subsequently underwent allogeneic HSCT. UCART19 expanded with kinetics comparable to that of autologous CAR T-cell products, but had low persistence, generally up to 28 days. With regard to toxicity, one child and one adult developed grade 1, skin acute GvHD and the toxicity profile was comparable to that reported with autologous CAR T cells.36 More recently, Ottaviano et al.37 described the results obtained with the use of TT52CAR19 T cells, universal, CD19 CAR T cells generated by disrupting the TCR a chain (TRAC) and CD52 loci with next-generation CRISPR-Cas9 editing, for the treatment of six children with relapsed/refractory B-ALL in a phase I clinical trial. Patients underwent lymphodepletion with fludarabine, cyclophosphamide and alemtuzumab, followed by a single infusion of 0.8-2.0x106 TT52CAR19 T cells/kg. TT52CAR19 T cells expanded after 7-14 days in four of the six children and induced clearance of the leukemia, enabling consolidation with allogeneic HSCT; in two children, no CAR T-cell expansion was observed and both patients experienced progression of their disease. One patient developed skin GvHD.37 In 2021, Hu et al. 38 reported on the first six patients treated with CTA101, a universal, second-generation (4.1bb) CD19/CD22 tanCAR, obtained by disrupting TCR a chain (TRAC) and CD52 loci with CRISPR-Cas9 editing. Patients underwent fludarabine-cyclophosphamide-alemtuzumab lymphodepletion followed by the infusion of either 1x106 or 3x106 CTA101 cells/kg. No case of GvHD was reported and five of the six (83%) patients obtained MRD-negative CR, which was sustained in three of the five patients with a median follow-up of 4.8 months (range, 2-8). The median duration of CTA101 persistence, measured by quantitative polymerase chain reaction, was 42 days (range, 21-114).38
Table 2. Studies describing the clinical experience with the use of universal, gene-edited CD19-chimeric antigen receptor T cells.
2
14 and 7
Children
Adults
(N=4)
alemtuzumab (N=17)
Ped: 1.1-2.3 × 10⁶ cells/kg
Adults: 6×10⁶, 6-8×10⁷ or 1.8-2.4×10⁸
GvHD (1 child and 1 adult) 14 CR (71%
6
6 CTA101 (CD19/CD22 dual targeting)
Pts: patients; CAR: chimeric antigen receptor; LD: lymphodepletion; GvHD: graft-versus-host disease; Ref: references; B-ALL: B-cell acute lymphoblastic leukemia; TALEN: transcription activator-like effector nucleases; Flu: fludarabine; Cy: cyclophosphamide; G: grade; MRD: measurable residual disease; CR: complete remission; CRISPR: clustered regularly interspaced short palindromic repeats; Ped: pediatrics; NR: non-response.
Interestingly, viral and/or fungal infections were reported in all the studies using universal CAR T cells, suggesting that the prolonged lymphodepletion induced by alemtuzumab may expose patients to a higher risk of infections. However, as mentioned, the use of alemtuzumab was shown to be crucial for allowing the expansion of universal CAR T cells.36 Moreover, the curtailed persistence of these cells, linked to both rejection by the host and the complex and prolonged T-cell manipulation during manufacturing that impairs the function of T cells, remains a limitation of the approach.
In addition to conventional effector cells, allogeneic CAR cells can be produced using alternative cell sources, which encompass stem cell and progenitor cell populations, such as induced pluripotent stem cells (iPSC) and precursor T cells. iPSC, with their nearly limitless proliferative potential, can be directed to differentiate into various cell types, including T and NK cells. This unique feature makes iPSC an excellent renewable source for potentially standardized cell-based immunotherapies. Moreover, iPSC can be genetically modified to potentiate the characteristics of the resulting immune cells. The possibility of generating CAR T cells from iPSC was confirmed by the demonstration of the successful transduction of iPSC derived from peripheral blood lymphocytes with a lentiviral vector encoding for a second-generation CD19-directed CAR.39 In detail, after hematopoietic differentiation, the authors employed the T-lymphoid commitment co-culture protocol to generate anti-CD19-CAR-T-iPSC-T cells. These iPSC-derived CAR T cells were directly compared with TCR-aβ and TCR-γδ peripheral blood lymphocytes from the same donor, transduced with the same CAR. The study demonstrated that iPSC-derived CAR T cells exhibited anti-cancer activity comparable to that of CAR TCR-γδ cells in an immunodeficient mouse xenograft tumor model.39 T-cell-derived iPSC hold promise as a source for producing allogeneic ‘off-the-shelf’ CAR T cells, but their in vitro differentiation often leads to effector cells with less-than-ideal characteristics. Several approaches have been developed recently to overcome this limitation. It has been demonstrated that inducing premature expression of the TCR or a continuously active CAR in T-cell-derived iPSC promotes the development of an innate-like phenotype. This issue can be circumvented by deactivating the TCR and relying on the CAR to drive differentiation instead. By delaying CAR expression and fine-tuning its signaling strength in T-cell-derived iPSC, human TCR- CD8aβ+ CAR T cells with a similar activity
compared to standard CD8aβ+ CAR T cells derived from peripheral blood can be successfully generated. Indeed, these iPSC-derived CAR T cells demonstrated the ability to effectively control tumors when administered systemically in a mouse model of leukemia, without triggering GvHD.40 Moreover, an approach to optimize the proliferation and persistency of allogeneic CAR T cells derived from iPSC has been recently published and is based on the genetic knockout of diacylglycerol kinase, which inhibits antigen-receptor signaling, and the transduction of cells with genes encoding for membrane-bound IL-15 and its receptor subunit IL-15Ra. 41 Another great innovation in the field of iPSC-derived CAR T cells is represented by artificial thymic organoids, used to facilitate the specific differentiation of CAR-transduced human iPSC into CAR T cells.42 This innovative approach has enabled the production of human CD19-CAR T cells closely resembling conventional CAR T cells. Remarkably, these engineered CAR-T cells have proven to be highly effective in controlling human CD19+ leukemia in animal models.42 Both approaches, leveraging CAR-driven maturation in T-derived iPSC, could pave the way for the large-scale production of potent allogeneic aβ+ CAR T cells.
Another strategy for developing allogeneic CAR-cell products involves bypassing the use of aβ T cells and genetically modifying different cell types to express a CAR. The ideal cell suitable for CAR adoptive therapy should be: endowed with inherent cytotoxic abilities that can be redirected towards a cancer cell through the expression of a cell surface receptor; amenable to collection from accessible sources such as peripheral blood or the umbilical cord; and relatively straightforward to transduce and expand. NK cells, originally recognized for their tumor-killing capabilities, play a central role in natural tumor immunosurveillance mechanisms and meet all the above-mentioned criteria. Several different sources of NK cells have been considered for clinical development of allogeneic CAR NK cells, including peripheral blood, umbilical cord blood, iPSC or immortalized cell lines. Umbilical cord blood-derived NK cells genetically modified using an inducible caspase 9/ CAR.19/IL-15 vector have demonstrated promising efficacy in treating relapsed or refractory CD19+ cancers (non-Hodgkin lymphoma or chronic lymphoblastic leukemia).43 CAR-NK cells were administered in a single infusion at one of three doses (1×105, 1×106, or 1×107 CAR-NK cells/kg) after fludarabine/cyclophosphamide-based lymphodepleting chemotherapy and obtained a 73% response rate, with seven of eight responders showing CR.43 Although this study paved
the way for the clinical use of allogeneic CAR-NK cells, it also showed the difficulties in generating an off-the-shelf product, as well as underscoring a limited persistence of the engineered NK cells. With regard to the first issue, several pre-clinical models are under development to overcome this limitation, including the large expansion of allogeneic CAR.CD19 NK cells starting from either peripheral blood mononuclear cells or leukapheresis, from which a larger number of NK cells can be derived, or from iPSC.44 The allogeneic iPSC approach was exploited to differentiate NK cells, subsequently engineered with CAR characterized by standard constructs or by the NK co-stimulatory machinery represented by the transmembrane domain of NKG2D and the co-stimulatory domain of 2B4.45-47 These preclinical models have shown that iPSC-derived CAR-NK cells hold the potential to serve as an effective off-theshelf product. In the search of a strategy to improve the long-term persistence of allogeneic CAR NK cells, memory-like NK cells (MLNK) have been explored. MLNK cells, induced by the presence of IL-12, IL-15, and IL-18, display enhanced and prolonged responses upon tumor re-stimulation, making them an appealing choice for adoptive cellular immunotherapy. In a recent study, researchers demonstrated efficient and stable gene delivery of a CD19-CAR construct to MLNK cells using retroviral vectors, achieving a transduction efficiency comparable to that seen in conventional NK cells.48 When compared to conventional CAR NK cells, CAR-MLNK cells exhibited significantly increased production of interferon-γ and degranulation in response to CD19+ target cells, resulting in enhanced cytotoxic activity against CD19+ leukemia and lymphoma cells. Moreover, the memory-like properties induced by IL-12, IL-15, and IL-18 led to prolonged persistence of effector cells in the CD19+ tumor-bearing mice.48
NK T cells (NKT cells) represent another source of allogeneic effector cells that could be used in alternative to conventional T cells. They are a subset of T lymphocytes that exhibit NK-cell surface markers. Within NKT cells, ‘invariant NKT cells’ (iNKT cells) are characterized by a TCR that recognizes particular lipid antigens presented by CD1d, a non-polymorphic HLA class I-like molecule found on various cells, including B cells, antigen-presenting cells, and select epithelial tissues. In preclinical models, iNKT cells engineered with a CD19-targeted CAR displayed potent anti-lymphoma properties by simultaneously targeting CD19 and CD1d molecules expressed on lymphoma cells.49 Moreover, it has been shown that allogeneic CAR iNKT cells, in addition to their established direct cytotoxic activity, trigger host CD8 T-cell antitumor responses. This activation has a robust and enduring antitumor effect, lasting even beyond the persistence of the allogeneic cells.50
Cytokine-induced killer (CIK) cells constitute a heterogeneous cell population that includes NK, NKT, and T cells. They are generated through the culture of peripheral blood mononuclear cells in the presence of interferon-γ, anti-CD3
antibody, and IL-2 for 2-3 weeks. CIK cells possess a unique combination of phenotypic and functional characteristics combining the advantages of both T and NK cells. This makes them capable of recognizing and eliminating tumor targets through mechanisms that involve both MHC-dependent and MHC-independent pathways. Previous clinical data have confirmed the safety of donor-derived CIK-cell therapy, without occurrence of GvHD and with demonstration of CR in eight of 13 patients 28 days after infusion.51 The concept of enhancing the efficacy and reducing the GvHD potential of allogeneic CAR T-cell therapy has also been explored by the use of γδ T cells. The first description of CAR-modified γδ T cells was by Rischer et al 52 who demonstrated specific in vitro tumor cell lysis using zoledronate-expanded Vγ9Vδ2 T cells equipped with CD19- or GD2-directed CAR. Subsequent studies confirmed these findings, showing the potential of γδ T cells bearing CAR against a variety of targets.53,54 Notably, CAR-modified Vγ9Vδ2 T cells retained their ability to cross-present tumor antigens to aβ T cells in vitro, suggesting the possibility of prolonging anti-tumor efficacy. Furthermore, γδ T cells with a CD19CAR, unlike conventional CD19-aβ CAR T cells, exhibited reactivity against both CD19+ and CD19– cells in vitro and in vivo. This effect was augmented by zoledronate, indicating that CD19-directed γδ CAR T cells might be effective against leukemic cells even after antigen loss, maintaining their specificity against phospho-antigens through their TCR.55,56 More recently, it has been demonstrated that γδ CAR T cells can also be derived from iPSC. Wallet et al. introduced iPSC-derived γδ CAR T cells (γδ CAR-iT)57 and demonstrated that, in the presence of IL-15, γδ CAR-iT cells exhibited sustained in vitro tumor cell killing, while producing significantly less interferon-γ and other inflammatory cytokines compared to conventional aβ CAR-T cells. This suggests a potentially lower risk of cytokine release syndrome. Moreover, a single dose of γδ CAR T cells resulted in potent inhibition of tumor growth in a xenograft mouse model.55
Allogeneic CAR platforms are gaining increasing attention thanks to their capability to overcome most of the main limitations of autologous CAR T cells. Three major strategies have made their way to the clinics showing promising results: the use of donor-derived allogeneic T cells, the use of off-the-shelf, gene-edited universal T cells and the use of CAR-NK/CIK cells. Donor-derived CAR T cells are associated with no rejection by the patient, promising long-term persistence and encouraging antitumor efficacy, but are limited by the availability of a cell donor. The experiences on the use of T cells derived from haploidentical donors suggest comparable results, in terms of risks of either GvHD or rejection, to those from unrelated donors and matched sibling donors, significantly enlarging the
range of patients who could potentially benefit from such an approach. Moreover, our experience showed that in the case of a matched sibling donor, donor-derived CAR T cells can potentially be used also in patients with highly refractory diseases who did not receive a prior allogeneic HSCT.31 Universal, off-the-shelf CAR T cells represent a fascinating approach, potentially readily available for every patient upon clinical need, and have shown an acceptable toxicity profile and promising efficacy. Nevertheless, their limited long-term persistence indicates that this approach needs further optimization. Preclinical translational data and shortened, optimized manufacturing strategies will be needed to determine the most efficient approach to avoid rejection of allogeneic CAR T cells and prolong their persistence. Moreover, the requirement for lymphodepletion that ensures prolonged ablation of the patient’s T cells increases the risk of infections, limiting the approach to well-selected patients. CAR-NK/CIK cells have shown antitumor activity with negligible toxicity in the first clinical
1. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439-448.
2. Laetsch TW, Maude SL, Rives S, et al. Three-year update of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia in the ELIANA trial. J Clin Oncol. 2023;41(9):1664-1669.
3. Pasquini MC, Hu ZH, Curran K, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020;4(21):5414-5424.
4 Bader P, Rossig C, Hutter M, et al. CD19 CAR T cells are an effective therapy for posttransplant relapse in patients with B-lineage ALL: real-world data from Germany. Blood Adv. 2023;7(11):2436-2448.
5. Schultz LM, Baggott C, Prabhu S, et al. Disease burden affects outcomes in pediatric and young adult B-cell lymphoblastic leukemia after commercial tisagenlecleucel: a Pediatric RealWorld Chimeric Antigen Receptor Consortium report. J Clin Oncol. 2022;40(9):945-955.
6. Leahy AB, Devine KJ, Li Y, et al. Impact of high-risk cytogenetics on outcomes for children and young adults receiving CD19directed CAR T-cell therapy. Blood. 2022;139(14):2173-2185.
7 Moskop A, Pommert L, Baggott C, et al. Real-world use of tisagenlecleucel in infant acute lymphoblastic leukemia. Blood Adv. 2022;6(14):4251-4255.
8. Ghorashian S, Jacoby E, De Moerloose B, et al. Tisagenlecleucel therapy for relapsed or refractory B-cell acute lymphoblastic leukaemia in infants and children younger than 3 years of age at screening: an international, multicentre, retrospective cohort study. Lancet Haematol. 2022;9(10):e766-e775.
9. Fabrizio VA, Phillips CL, Lane A, et al. Tisagenlecleucel outcomes in relapsed/refractory extramedullary ALL: a Pediatric Real World CAR Consortium report. Blood Adv. 2022;6(2):600-610.
10 Leahy AB, Newman H, Li Y, et al. CD19-targeted chimeric antigen receptor T-cell therapy for CNS relapsed or refractory
trials, paving the way for their development also for other clinical indications. Multiple infusions will probably be required to ensure long-term control of the disease, considering the limited persistence of these cells. Preclinical studies have shown promising results also with alternative cell platforms and the clinical testing of these strategies will provide crucial information on their potential. Important restrictions to the use of these approaches are limited frequency and yield and optimal strategies for expansion or differentiation systems are under investigation.
Disclosures
No conflicts of interest to disclose.
FdB, CQ and FL contributed equally to this manuscript, reviewing the available literature, supervising the preparation of the paper and writing the manuscript. The final version was approved by all the authors.
acute lymphocytic leukaemia: a post-hoc analysis of pooled data from five clinical trials. Lancet Haematol. 2021;8(10):e711-e722.
11. Jacoby E, Ghorashian S, Vormoor B, et al. CD19 CAR T-cells for pediatric relapsed acute lymphoblastic leukemia with active CNS involvement: a retrospective international study. Leukemia. 2022;36(6):1525-1532.
12. Qi Y, Zhao M, Hu Y, et al. Efficacy and safety of CD19-specific CAR T cell-based therapy in B-cell acute lymphoblastic leukemia patients with CNSL. Blood. 2022;139(23):3376-3386.
13. Hutt D, Bielorai B, Baturov B, et al. Feasibility of leukapheresis for CAR T-cell production in heavily pre-treated pediatric patients. Transfus Apher Sci. 2020;59(4):102769.
14 Shalabi H, Shah NN. CD19 CAR T cells for infants and young children. Lancet Haematol. 2022;9(10):e712-e714.
15. Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372-385.
16. Stroncek DF, Ren J, Lee DW, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy. 2016;18(7):893-901.
17 De Veirman K, Van Valckenborgh E, Lahmar Q, et al. Myeloidderived suppressor cells as therapeutic target in hematological malignancies. Front Oncol. 2014;4:349.
18. Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499-1503.
19. van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. 2020;20(4):218-232.
20. Singh N, Perazzelli J, Grupp SA, et al. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. 2016;8(320):320ra3.
21. Daudt L, Maccario R, Locatelli F, et al. Interleukin-15 favors the expansion of central memory CD8+ T cells in ex vivo generated, antileukemia human cytotoxic T lymphocyte lines. J
Immunother. 2008;31(4):385-393.
22. Arcangeli S, Bove C, Mezzanotte C, et al. CAR T cell manufacturing from naive/stem memory T lymphocytes enhances antitumor responses while curtailing cytokine release syndrome. J Clin Invest. 2022;132(12):e150807.
23. Cruz CR, Micklethwaite KP, Savoldo B, et al. Infusion of donorderived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122(17):2965-2973.
24. Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122(25):4129-4139.
25. Brudno JN, Somerville RP, Shi V, et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol. 2016;34(10):1112-1121.
26. Dai H, Zhang W, Li X, et al. Tolerance and efficacy of autologous or donor-derived T cells expressing CD19 chimeric antigen receptors in adult B-ALL with extramedullary leukemia. Oncoimmunology. 2015;4(11):e1027469.
27. Chen Y, Cheng Y, Suo P, et al. Donor-derived CD19-targeted T cell infusion induces minimal residual disease-negative remission in relapsed B-cell acute lymphoblastic leukaemia with no response to donor lymphocyte infusions after haploidentical haematopoietic stem cell transplantation. Br J Haematol. 2017;179(4):598-605.
28. Yan CH, Liu DH, Xu LP, et al. Modified donor lymphocyte infusion-associated acute graft-versus-host disease after haploidentical T-cell-replete hematopoietic stem cell transplantation: incidence and risk factors. Clin Transplant. 2012;26(6):868-876.
29 Kebriaei P, Singh H, Huls MH, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest. 2016;126(9):3363-3376.
30 Zhang C, Wang XQ, Zhang RL, et al. Donor-derived CD19 CAR-T cell therapy of relapse of CD19-positive B-ALL post allotransplant. Leukemia. 2021;35(6):1563-1570.
31. Del Bufalo F, Becilli M, Rosignoli C, et al. Allogeneic, donorderived, second-generation, CD19-directed CAR-T cells for the treatment of pediatric relapsed/refractory BCP-ALL. Blood. 2023;142(2):146-157.
32. Opelz G, Dohler B. Effect of human leukocyte antigen compatibility on kidney graft survival: comparative analysis of two decades. Transplantation. 2007;84(2):137-143.
33. Kurtzberg J, Prasad VK, Carter SL, et al. Results of the Cord Blood Transplantation Study (COBLT): clinical outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with hematologic malignancies. Blood. 2008;112(10):4318-4327.
34 Depil S, Duchateau P, Grupp SA, et al. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185-199.
35. Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9(374):eaaj2013.
36. Benjamin R, Graham C, Yallop D, et al. Genome-edited, donorderived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet. 2020;396(10266):1885-1894.
37. Ottaviano G, Georgiadis C, Gkazi SA, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. 2022;14(668):eabq3010.
38. Hu Y, Zhou Y, Zhang M, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/ refractory B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2021;27(10):2764-2772.
39. Themeli M, Kloss CC, Ciriello G, et al. Generation of tumortargeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31(10):928-933.
40 van der Stegen SJC, Lindenbergh PL, Petrovic RM, et al. Generation of T-cell-receptor-negative CD8aβ-positive CAR T cells from T-cell-derived induced pluripotent stem cells. Nat Biomed Eng. 2022;6(11):1284-1297.
41. Ueda T, Shiina S, Iriguchi S, et al. Optimization of the proliferation and persistency of CAR T cells derived from human induced pluripotent stem cells. Nat Biomed Eng. 2023;7(1):24-37.
42. Wang Z, McWilliams-Koeppen HP, Reza H, et al. 3D-organoid culture supports differentiation of human CAR(+) iPSCs into highly functional CAR T cells. Cell Stem Cell. 2022;29(4):515-527.e8.
43. Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545-553.
44 Quintarelli C, Sivori S, Caruso S, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia. 2020;34(4):1102-1115.
45. Ueda T, Kumagai A, Iriguchi S, et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptorexpressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111(5):1478-1490.
46. Tang SY, Zha S, Du Z, et al. Targeted integration of EpCAMspecific CAR in human induced pluripotent stem cells and their differentiation into NK cells. Stem Cell Res Ther. 2021;12(1):580.
47. Li Y, Hermanson DL, Moriarity BS, et al. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181-192.e5.
48. He B, Mai Q, Pang Y, et al. Cytokines induced memory-like NK cells engineered to express CD19 CAR exhibit enhanced responses against B cell malignancies. Front Immunol. 2023;14:1130442.
49. Rotolo A, Caputo VS, Holubova M, et al. Enhanced antilymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell. 2018;34(4):596-610.e11.
50. Simonetta F, Lohmeyer JK, Hirai T, et al. Allogeneic CAR invariant natural killer T cells exert potent antitumor effects through host CD8 T-cell cross-priming. Clin Cancer Res. 2021;27(21):6054-6064.
51. Magnani CF, Gaipa G, Lussana F, et al. Sleeping Beautyengineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Invest. 2020;130(11):6021-6033.
52. Rischer M, Pscherer S, Duwe S, et al. Human gammadelta T cells as mediators of chimaeric-receptor redirected antitumour immunity. Br J Haematol. 2004;126(4):583-592.
53. Deniger DC, Switzer K, Mi T, et al. Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol Ther.
2013;21(3):638-647.
54 Polito VA, Cristantielli R, Weber G, et al. Universal ready-to-use immunotherapeutic approach for the treatment of cancer: expanded and activated polyclonal γδ memory T cells. Front Immunol. 2019;10:2717.
55. Capsomidis A, Benthall G, Van Acker HH, et al. Chimeric antigen receptor-engineered human gamma delta T cells: enhanced cytotoxicity with retention of cross presentation. Mol Ther.
2018;26(2):354-365.
56. Rozenbaum M, Meir A, Aharony Y, et al. Gamma-delta CAR-T cells show CAR-directed and independent activity against leukemia. Front Immunol. 2020;11:1347.
57 Wallet MA, Nishimura T, Del Casale C, et al. Induced pluripotent stem cell-derived gamma delta CAR-T cells for cancer immunotherapy. Blood. 2021,138(Suppl 1):2771.
1Princess Máxima Center for Pediatric Oncology, Utrecht and 2Pediatric Oncology, Erasmus MC-Sophia Children’s Hospital, Rotterdam, the Netherlands
Correspondence: C.M. Zwaan
c.m.zwaan@prinsesmaximacentrum.nl
Received: November 8, 2023.
Accepted: March 15, 2024.
https://doi.org/10.3324/haematol.2023.283815
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
The treatment of childhood acute lymphoblastic leukemia (ALL) has reached overall survival rates exceeding 90%. The present and future challenges are to cure the remainder of patients still dying from disease, and to reduce morbidity and mortality in those who can be cured with standard-of-care chemotherapy by replacing toxic chemotherapy elements while retaining cure rates. With the novel therapeutic options introduced in the last years, including immunotherapies and targeted antibodies, the treatment of ALL is undergoing major changes. For B-cell precursor ALL, blinatumomab, an anti-CD19 bispecific antibody, has established its role in the consolidation treatment for both high- and standard-risk first relapse of ALL, in the presence of bone marrow involvement, and may also have an impact on the outcome of high-risk subsets such as infant ALL and Philadelphia chromosome-positive ALL. Inotuzumab ozogamicin, an anti-CD22 drug conjugated antibody, has demonstrated high efficacy in inducing complete remission in relapsed ALL, even in the presence of high tumor burden, but randomized phase III trials are still ongoing. For T-ALL the role of CD38-directed treatment, such as daratumumab, is gaining interest, but randomized data are needed to assess its specific benefit. These antibodies are currently being tested in patients with newly diagnosed ALL and may lead to major changes in the present paradigm of treatment of pediatric ALL. Unlike the past, lessons may be learned from innovations in adult ALL, in which more drastic changes are piloted that may need to be translated to pediatrics.
Significant improvements in the outcome of children with acute lymphoblastic leukemia (ALL) have been made in the past decades, with current rates of overall survival now exceeding 90%.1 These results have been reached with the use of very intensive chemotherapeutic regimens, which are associated with acute toxicity as well as long-term side effects.2 New therapeutic approaches are needed not only to cure the patients who currently still relapse and die from disease, but also to replace toxic therapy elements to mitigate treatment intensity and side-effects in those who can be cured with current chemotherapy, while maintaining cure rates.
With the novel therapeutic options introduced in the last years, including immunotherapies and targeted antibodies, ALL treatment is undergoing major changes. Treatment options for patients with B-cell precursor ALL (BCP-ALL)
have increased significantly with the marketing authorization of blinatumomab and chimeric antigen receptor T cells (CAR T cells), changing the landscape for relapsed BCP-ALL.3,4 For T-cell ALL, despite current high survival rates in first complete remission, first-relapse salvage rates remain dismal and there is no unified approach to relapse.5 Nevertheless new approaches for relapsed T-ALL are also becoming available.6,7 Among the novel therapeutic options, in this review we focus on the role of targeted antibodies, which have proven to be particularly effective for specific groups of pediatric ALL.
Targeted antibodies are designed with different mechanisms of action (Figure 1). Monoclonal antibodies, after binding a surface antigen, induce lysis through different cytotoxic mechanisms, including complement-dependent, cell-mediated and antibody-dependent cellular phagocytosis. Bispecific antibodies bind two distinct antigens simultaneously, linking antigens on target cells to immune effector cells (i.e.,
T cells, natural killer cells, or macrophages). Antibody-drug conjugates combine the targeting capabilities of monoclonal antibodies with cancer-killing abilities of cytotoxic drugs: once the monoclonal antibody-drug conjugates to the tumor antigen, the antigen-antibody complex is internalized and the cytotoxic agent is delivered inside the targeted tumor cells, resulting in a significantly improved therapeutic index and less toxicity to the normal cell compartment.8 The characterization of suitable target antigens is essential to the further development of new targeted antibodies. An ideal antigen should be exclusively and highly expressed on malignant cells, to minimize on-target/off-tumor side effects and maximize anticancer activity; it should be highly expressed in the majority of patients with a disease or even by different cancers types and the ability of rapid internalization after binding an antibody is another desirable characteristic especially for antibody-drug conjugates.9 Multiple surface antigens have been identified as (potential) treatment targets in ALL. CD19 is expressed on the vast majority of B-cell malignancies, including 80% of ALL. CD22 is uniquely expressed in B-lymphocytes and in virtually all BCP-ALL.10 CD20 is highly expressed in mature B-lineage cells and in a variable degree (30-50%) of
BCP-ALL blasts.11 CD123 represents another potential target, expressed in different genetic subtypes of BCP-ALL patients, while it is absent in T-ALL.12 CD38 is expressed in pediatric ALL, including T-ALL.13 Other potential targets for T-ALL are being explored, mainly in the development of CAR T-cell therapies: CD5, CD7 which is expressed on T lymphoblasts but also on effector T cells, and CD1a which is a target for cortical T-ALL.14
In this review we aim to present the available antibodies for the treatment of childhood ALL, focusing on the evidence generated so far and the future perspective of the use of antibodies in the treatment of childhood ALL.
The website https://clinicaltrials.gov was scrutinized using the advanced search mode to identify clinical trials exploring relevant monoclonal antibodies addressed to patients with ALL. Thirteen searches were conducted. For all searches, the pre-defined term “acute lymphoblastic leukemia” was used in the “condition or disease” box. In addition to this term, each search contained one of the
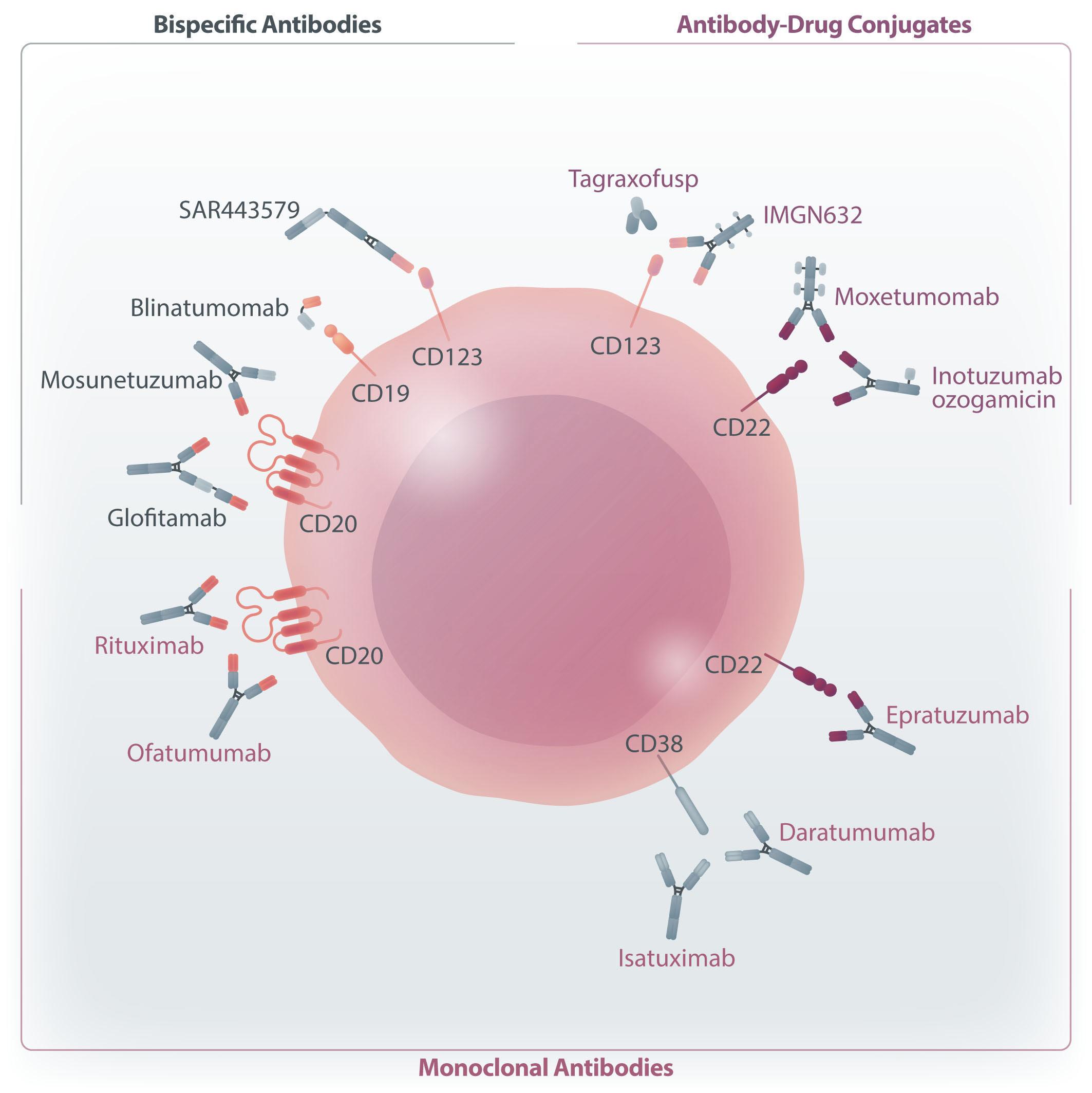
following pre-defined terms to identify relevant trials for the drugs covered in this review: “inotuzumab ozogamicin”, “blinatumomab”, “isatuximab”, “daratumumab”, “moxetumomab”, “epratuzumab”, “ofatumumab”, “rituximab”, “mosunetuzumab”, “glofitamab”, “tagraxofusp”, “SAR443579”, “IMGN632”. Eligibility criteria were defined a priori: only interventional clinical trials in children were searched in the period from January 1, 2000 to August 1, 2023 and studies with unknown status were excluded. The search was performed on August 28, 2023. In the results, we discuss the trials performed in children with ALL for which results have already been published (Table 1) or that are currently ongoing (Table 2).
The search strategy at clinicaltrials.gov yielded 119 studies (Online Supplementary Figure S1, Online Supplementary Table S1). Of those, 84 fulfilled the eligibility criteria. Two studies were identified through cross-references.15,16
Anti-CD19: blinatumomab
Blinatumomab is a bispecific antibody that directs CD3-positive effector T cells to CD19-positive target cells. Blinatumomab is currently approved by the Food and Drug Administration as monotherapy for the treatment of children with relapsed or refractory (R/R) BCP-ALL, or in first or second complete remission with persisting positive minimal residual disease (MRD). In Europe, blinatumomab is indicated for patients above the age of 1 year, with second or beyond R/R BCP-ALL, or with high-risk first relapse BCP-ALL as part of consolidation therapy.
A phase I/II study of blinatumomab as a single agent in children with R/R B-BCP and bone marrow blasts ≥25% identified the stepwise dosage of 5/15 µg/m2/day for further evaluation. The overall response rate at the recommended phase II dose was 39%, with 52% of those patients being MRD-negative.3 This single-agent trial identified neurological toxicity (experienced by 24% of patients) and cytokine release syndrome (11% of patients) as blinatumomab-specific toxicities, the latter as a result of high tumor burden.3 The RIALTO (NCT02187354) trial tested blinatumomab in the same R/R population, also allowing patients with persisting or re-emerging MRD ≥0.1% to be included, and documented a similar rate of MRD-negative complete remissions and low incidences of grade 3/4 cytokine release syndrome and neurological toxicity (1.8% and 3.6%, respectively). This trial clearly showed the need for consolidation with hematopoietic stem cell transplantation (HSCT) after blinatumomab treatment reinduction (1-year overall survival with vs. without allogeneic HSCT after blinatumomab: 87% vs. 29%, respectively).17,18 After these initial experiences with blinatumomab in multiply R/R ALL, two randomized trials have highlighted the superiority (compared to chemothera-
py) of one or two courses of blinatumomab monotherapy as post-reinduction consolidation treatment for first relapse ALL. This was due both to enhanced antileukemic activity as well as to lower toxicity than that with chemotherapy in the standard arm, especially inducing less hematologic toxicity and fewer infections. In the Children’s Oncology Group (COG) phase III AALL1331 trial (NCT02101853) in intermediate- and high-risk first relapse of BCP-ALL, two cycles of blinatumomab as post-reinduction consolidation treatment produced a higher disease-free survival rate at 2 years compared to conventional chemotherapy (54.4% vs. 39.0%, respectively).19 The NCT02393859 randomized trial demonstrated superior event-free survival for children with high-risk first relapse of BCP-ALL treated with blinatumomab as compared to those given a third block of consolidation chemotherapy before HSCT (2-year eventfree survival: 66.2% vs. 27.1%, respectively).20 More recently, blinatumomab has also been studied in standard-risk first relapse ALL in a randomized phase III trial in which blinatumomab was intercalated to continuation chemotherapy before maintenance, and tested against standard chemotherapy treatment. The 4-year disease-free and overall survival rates were superior for the blinatumomab group in the patients with bone marrow relapse (with or without extramedullary involvement), being 72.7% and 97.1% in the group treated with blinatumomab versus 53.7% and 84.8% in the group given chemotherapy. In contrast, for the patients with isolated extramedullary relapses, similar outcomes and poor disease-free survival were observed in both arms, with a 4-year disease-free survival rate of 36.6% for blinatumomab versus 38.8% for chemotherapy.21 Based on these results, blinatumomab has become a new standard of care for post-induction consolidation therapy in high-risk first relapse ALL, and low-risk first relapse ALL in the case of bone marrow involvement. By contrast, unsatisfactory results were obtained in patients with isolated extramedullary relapse, possibly explained by the fact that blinatumomab does not cross the blood-brain barrier. Overall, as in adults, blinatumomab seems to be more effective in circumstances of lower disease burden.22 Importantly, in a study by Gokbuget et al., although the MRD clearance rate was high also in patients with multiply relapsed ALL, these patients had substantially inferior relapse-free survival and overall survival compared with those treated in first remission.22 This shows that the impact of clearance of MRD on survival is greatest when achieved early in the disease history.
Infants with ALL, carrying the KMT2A rearrangement in 80% of cases, have a poor prognosis, with a 4-year eventfree survival rate of 47% and a survival rate after relapse of approximately 20%.23,24 In this context of high medical need, a block of blinatumomab monotherapy was studied in newly diagnosed patients, as an addition to front-line chemotherapy. In a pilot study, one post-induction course of blinatumomab was added to the Interfant-06 back-
COG AALL1331
Blina Infant EudraCT2016-004674-17
(added postinduction to Interfant-06 chemotherapy)
Continued on following page.
Study Agent Disease status
Epratuzumab
COG Pilot Phase I
COG ADVL04P2 Phase II
Epratuzumab + chemotherapy BCP-ALL ≥1st relapse 15 60 77 - - 52
Epratuzumab + chemotherapy HR BCP-ALL 1st relapse
54/60 (weekly/ bi-weekly epratuzumab)
65/66 (weekly/ bi-weekly epratuzumab)
31/39 (weekly/ bi-weekly epratuzumab)
2-yr: 34.2%/49.3% (weekly/ bi-weekly epratuzumab)
2-yr: 25.9%/ 39.9% (weekly/ bi-weekly epratuzumab) 53
*When disease-free survival or relapse-free survival is used, this is specified in the table. N: number; CR: complete remission; MRD: minimal residual disease; OS: overall survival; EFS: event-free survival; Ref: references; R/R: relapsed/refractory; BCP-ALL: B-cell precursor acute lymphoblastic leukemia; HSCT: hematopoietic stem cell transplantation; RFS: relapse-free survival; HR: high risk; yr: year; COG: Children’s Oncology Group; IR: intermediate risk; SR: standard risk; DFS: disease-free survival; VXLD: vincristine, prednisone, pegylated asparaginase, and doxorubicin; InO: inotuzumab ozogamicin.
bone for infants with KMT2A-rearranged ALL. This addition provided a clear benefit to this group of patients, significantly increasing the rate of MRD negativity, with a 2-year disease-free survival of 81.6% in the study, as compared with 49.4% in the historical Interfant-06 trial, with corresponding overall survival values of 93.3% and 65.8%.25 Despite the fact that these are non-randomized data, the effect size suggests that this should be a new standard of care also in infants with KMT2A-rearranged ALL, which will be confirmed prospectively in the Interfant-21 trial (NCT05327894). These excellent results and the promising findings with the menin-inhibitor revumenib may alter the poor prognosis of these patients in the near future.26 Currently, research is focused on moving blinatumomab to upfront treatment also in other groups of patients. The COG AALL1731 trial (NCT03914625), and the Berlin-Frankfurt-Münster (BFM)/Associazione Italiana Emato-Oncologia Pediatrica (AIEOP) (NCT03643276) trial include randomized questions of blinatumomab during consolidation. The ALLTogether1 trial (NCT03911128) evaluates blinatumomab as consolidation treatment in patients with Down syndrome, who have poor tolerance to chemotherapy. A warning about an increased risk of seizures during blinatumomab infusion in patients with Down syndrome >10 years old has been raised.27 In clinical practice, blinatumomab has also been given to patients intolerant of chemotherapy during front-line treatment.28 A retrospective study on 105 patients showed that the 2-year outcome of patients treated with blinatumomab as replacement for post-remission intensive chemotherapy was comparable to that of the chemotherapy-treated control group, with only one grade 3-4 adverse event occurring in the blinatumomab group.29 In adults, the ECOG ACRIN E1910 randomized phase III trial showed that patients with newly diagnosed ALL who become MRD-negative (<0.01%) after induction chemotherapy have better survival when they are given four cycles of blinatumomab during consolidation chemotherapy than when they are given only chemotherapy. In contrast, a benefit could not be confirmed in patients who received only one or two cycles of blinatumomab.30,31
Novel strategies to overcome mechanisms of blinatumomab resistance are under investigation, including the combination with immune checkpoint inhibitors, such as nivolumab (COG trial AALL1821, NCT04546399).32 Preclinical studies have shown increased PD-L1 expression on leukemic blasts in patients who are refractory to or relapse after blinatumomab treatment. Furthermore, in vitro studies suggest that the addition of PD-1 blockade to blinatumomab and ALL blasts leads to increased T-cell proliferation and enhanced blinatumomab-mediated cytotoxicity.33
Another area of interest is related to the use of this drug after HSCT to prevent relapse.34 Blinatumomab seems a very good candidate for this use, considering its good tolerability, with low risk of infections and low rate of other toxicities such as liver toxicity, which prevents the use of many other drugs after a transplant. An MD Anderson trial, including pediatric patients, tested blinatumomab as a prophylactic treatment in patients at high risk of relapse after HSCT. The treatment was well tolerated, without requiring interruptions secondary to cytopenia. In this small cohort, the comparison to a matched control group did not demonstrate a clear clinical benefit, similar to what was observed in adults; nevertheless, it was highlighted that response to blinatumomab is dependent on the immune reconstitution following HSCT.35 Other trials are using a pre-emptive approach, rather than prophylactic, treating patients with evidence of MRD positivity after HSCT (St. Jude trial NCT02790515). The FORUM consortium in Europe has designed a study with a dual approach, administering blinatumomab as a prophylactic treatment to patients undergoing HSCT with positive MRD and as a pre-emptive treatment for those becoming MRD-positive after the transplant (NCT04785547).
There are some data, mainly generated in adults, showing that prolonged blinatumomab administration may result in cure, and hence further studies also need to be performed to study the right schedule and duration of blinatumomab therapy and to determine its real potential to eradicate the disease without subsequent consolidation with HSCT.22 Potential long-term side-effects will have to be considered
Table 2. Targeted antibodies in ongoing clinical trials in pediatric acute lymphoblastic leukemia.
Compound
Blinatumomab Front-line ALL III
≥1 year ≤31 years COG ALL1731 NCT03914625
Blinatumomab Front-line ALL III ≤1 year Interfant-21 NCT05327894
Blinatumomab Front-line ALL III <18 years AIEOP-BFM ALL 2017 NCT03643276
Blinatumomab Front-line ALL III ≤45 years ALL together NCT03911128
Blinatumomab Post-HSCT II ≥6 months ≤21 years FORUM NCT04785547
Blinatumomab Post-HSCT (prophylactic) II ≤21 years St. Jude NCT02790515, NCT03849651
Blinatumomab + nivolumab 1st relapse ALL III ≥1 year ≤31 years COG AALL1821 NCT04546399
Daratumumab T-ALL post-HSCT TBI-based conditioning I ≤39 years NCT04972942
Tagraxofusp R/R ALL I/II ≥1 year ≤21 years NCT05476770 SAR443579 R/R ALL I/II ≥1 year NCT05086315
InO 1st relapse VHR ALL II ≥1 year ≤18 years ITCC-059 EudraCT 2016-000227-71
InO 1st relapse HR ALL II with randomization
InO
InO Front-line ALL III
InO Post-HSCT (prophylactic) I/II
InO Post-HSCT (MRD positivity) II
NCT05748171
NCT03959085
years ALL Together NCT04307576
years NCT03104491
years NCT05940961
InO R/R MRD positive II ≤21 years St. Jude NCT03913559 InO + blinatumomab+ rituximab R/R ALL II
NCT05645718
InO + blinatumomab+ hyper-CVAD + rituximab or ofatumumab Front-line ALL II
Rituximab or ofatumumab + CEC + liposomal vincristine + bortezomib R/R ALL II
ALL: acute lymphoblastic leukemia; COG: Children’s Oncology Group; AIEOP: Associazione Italiana Emato-Oncologia Pediatrica; BFM: Berlin-Frankfurt-Münster; HSCT: hematopoietic stem cell transplantation; TBI: total body irradiation; R/R: relapsed/refractory; InO: inotuzumab ozogamicin; VHR: very high risk, HR: high risk; MRD: minimal residual disease; CVAD: cyclophosphamide, vincristine, doxorubicin, dexamethasone; CEC: clofarabine, etoposide, cyclophosphamide.
in such trials as well. Indeed, decreased serum concentrations of immunoglobulins have been reported during and after treatment with blinatumomab (similarly to the changes after rituximab). Therefore, periodic monitoring of immunoglobulin levels may be considered in patients who are receiving or have received blinatumomab (as well as other antibodies against B cells) and IgG substitution treatment could be considered in patients experiencing severe infections.36
Anti-CD22
Inotuzumab ozogamicin
Inotuzumab ozogamicin (InO) is an anti-CD22 drug-conjugated antibody linked to calicheamicin, a potent cytotoxic agent, which causes cell death by inducing double-stranded DNA breaks.37 In vitro studies demonstrated high efficacy of InO against BCP-ALL, related to the intrinsic high sensitivity of ALL blasts to calicheamicin, but also to the spe-
cific CD22 ligand-induced internalizing properties, which make repetitive loops of CD22 saturation, internalization and renewed CD22 expression not necessary to achieve intracellular threshold levels of calicheamicin sufficient for apoptosis.38 Clinical data confirmed the high efficacy of InO in ALL, while failing to provide evidence of efficacy in non-Hodgkin lymphomas. The INO-VATE adult phase III study (NCT01564784), showed a complete remission rate of 80.7% in the InO arm as compared to 29.4% in the standard intensive chemotherapy control arm, and led to the approval of InO for adults with R/R CD22-positive BCP-ALL in 2017 by the Food and Drug Administration and European Medicines Agency.39 The long-term follow-up also showed a benefit in survival with InO (2-year OS rates of 22.8% and 10.0%, respectively).
The first data on pediatric R/R BCP-ALL were collected from a compassionate use program, showing that InO as a single agent resulted in complete remissions in 67% of patients.40 A phase I study in children with R/R BCP ALL
within the European Innovative Therapies for Children with Cancer Consortium (ITCC; the ITCC-059 trial) established that the recommended phase II dose of InO was 1.8 mg/ m2 fractionated in three dosages with an initial loading dose, as in adults, resulting in a high overall response rate of 80%. The phase II part of the study confirmed the very high response rate in the setting of multiple relapses and in patients relapsing after HSCT.15,16 The COG phase II study, AALL1621, in the same subgroup of children and young adults with multiply R/R BCP-ALL, demonstrated an overall response rate of 58.6%.41 It should be noted that none of these trials included children <1 year of age and experience with InO in infants is limited. Infant ALL, in the presence of a KMT2A rearrangement, is characterized by intrinsic resistance to chemotherapy, and by possibly lower CD22 expression, associated with immature leukemias arrested in the pro-B-cell stage.10 A retrospective study showed a complete remission rate of 47% in R/R infant ALL (with a large majority of patients having a KMT2A rearrengement).42
A particular concern with the use of InO is sinusoidal obstruction syndrome, likely due to the calicheamicin component. Warnings were raised by studies in adults, showing a high incidence in the transplant population (20% of transplanted patients following InO treatment).43 The higher incidence of post-HSCT sinusoidal obstruction syndrome was confirmed in pediatric studies (26.1% in the ITCC-059 trial, 28.6% in the AALL1621 study).16,41 In adult trials, conditioning regimens containing dual alkylators, high bilirubin levels and the number of treatment cycles received were identified as risk factors.43 In the pediatric population, risk factors for developing post-HSCT sinusoidal obstruction syndrome are not yet supported by statistical evidence due to the limited sample size studied. The ITCC-059 trial identified a shorter time interval between the last InO dose and HSCT as the only significant risk factor.15 In adults, InO has already been studied in different combination strategies.44,45 In the pediatric ITCC-059 trial, the combination of InO with a modified R3 reinduction regimen (5-day blocks of 20 mg/m2 dexamethasone, weekly vincristine) showed comparable results to those in the single-agent part of the study, not providing evidence of further improvement with the addition of chemotherapy. Furthermore, liver toxicity was a limiting factor, requiring dexamethasone dose reduction (to 10 mg/m2) and prohibiting the addition of asparaginase to the combination regimen. Nevertheless, heavily pretreated patients were included in this trial (after multiple relapses and/or after HSCT) and this might have had an impact on the added value of chemotherapy as well as the liver toxicity.46 In the front-line setting, InO is also being studied in combination with chemotherapy, using a sequential model. The COG AALL1732 trial is testing InO in a phase III, randomized trial in newly diagnosed, high-risk, CD22-positive BCP-ALL (NCT03959085). Patients are randomized to chemotherapy or chemotherapy plus two cycles of InO after consolida-
tion. After a first safety analysis showed increased rates of delayed methotrexate clearance following, and more sepsis events during, delayed intensifications in the arm including InO, the dose of this drug was reduced to 1.2 mg/ m2/cycle.47 A later safety analysis raised concerns about the occurrence of sinusoidal obstruction syndrome during treatment, especially during thioguanine administration after InO, and so additional changes to the study are planned.48 In the ALLTogether group, InO is being studied during consolidation for newly diagnosed ALL patients with persistent, high MRD (NCT04307576). In the front-line setting for adult ALL, the MD Anderson center is testing a different approach, with sequential use of hyperfractionated chemotherapy (hyper-CVAD) and lower-dose fractioned InO followed by blinatumomab (with or without CD20 targeted therapy with rituximab or ofatumumab), to shorten the duration of intensive chemotherapy, while improving safety and efficacy (NCT02877303, enrolling patients from 14 years of age).49 Very high response rates are reported with such regimens, including in elderly ALL patients who cannot tolerate intensive chemotherapy.
Currently, the ITCC-059 trial is including a cohort of patients with very high-risk ALL in first relapse, defined as very early relapse within 18 months after the initial diagnosis and/or the presence of high-risk genetic features, who receive re-induction with single agent InO followed by consolidation with HSCT or CAR T cells once in complete remission. In a company-sponsored, randomized trial (NCT05748171) in patients with high-risk ALL in first relapse, defined in this case as relapse occurring within 18 to 30 months of the original diagnosis, and lacking any identified very high-risk genetic abnormalities, re-induction with InO monotherapy is being tested against ALL R3 block treatment (dexamethasone, vincristine, mitoxantrone, PEG-asparaginase). The plan is to further test InO against regular reinduction therapy also in standard-risk BCP-ALL in first relapse within the European IntReALL group. An ongoing trial at St. Jude Children’s Hospital is testing InO in R/R patients with persisting MRD positivity (NCT03913559) rather than as re-induction treatment. As with blinatumomab, a few trials including adolescents are testing the use of InO after HSCT, as prophylactic or pre-emptive treatment (NCT03104491, NCT05940961).
Mechanisms of resistance to InO still need to be fully understood. The ITCC-059 trial reported intrinsic resistance to calicheamicin, measured ex-vivo with a 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT)based assay, broadly used to measure in vitro cytotoxic effects of drugs on cell lines, as the only factor related to the response.15 Others reported low CD22 expression combined with high BCL-2 expression as a predictive factor for response to InO.50 Potential synergistic mechanisms with BCL-2 inhibitors, such as venetoclax, as suggested by murine models, are being explored in adults (NCT05016947).51
Epratuzumab
Epratuzumab is a monoclonal antibody that binds CD22. A COG pilot study showed that epratuzumab in combination with standard reinduction chemotherapy was tolerable in children with first or later relapsed CD22-positive ALL, with a high rate of complete molecular remissions (47%).52 Therefore, a phase II trial in early first relapse ALL was performed, but did not show improved rates of second complete remission compared to those in historical controls.53 The European group InReALL tested epratuzumab in standard-risk ALL in first relapse in a randomized fashion against standard chemotherapy. Although the results of this trial have not yet been published, the randomization was prematurely suspended because of a stop in production of the drug.
Moxetumomab
Moxetumomab pasudotox, an anti-CD22 immunotoxin, failed to demonstrate significant activity in a phase II pediatric study,54 preventing further investigation in children with ALL and the drug has been withdrawn from the US and European markets.55
Anti-CD20
Rituximab
Rituximab, a monoclonal antibody that binds to CD20, is currently the standard of care in addition to chemotherapy in mature B-cell non-Hodgkin lymphoma, and also as a single agent in post-transplant lymphoproliferative disease, especially in the case of Epstein-Barr virus-positive disease.56 Its role in ALL is less well established, with only 30 to 50% of BCP-ALL blasts expressing CD20, including pediatric ALL.11 CD20 expression has an adverse prognostic significance in adult ALL, while its impact in pediatric ALL is controversial.57
A French trial in adults, in which rituximab was added to the ALL chemotherapy protocol, demonstrated an improved outcome for younger adults with CD20-positive, Philadelphia chromosome-negative ALL, and hence rituximab is now used in many adult treatment protocols.58 The UKALL14 trial did not confirm the benefit of additional rituximab, but only four doses of rituximab were administered compared to 16–18 doses in the GRAAL trial.59
Novel anti-CD20 antibodies are being explored in the treatment of ALL, mainly for adult patients. These include ofatumumab, which targets a juxtamembrane, small-loop, extracellular epitope of CD20 and shows more potent in vitro complement-dependent cytotoxicity than rituximab. Research at MD Anderson showed that hyper-CVAD plus ofatumumab was associated with better outcomes than hyper-CVAD plus rituximab in newly diagnosed BCP-ALL. Although the trial was designed to include pediatric patients, only adults were enrolled.60 Currently a study is open in MD Anderson for inclusion of patients from 14 years of age with R/R ALL (and Burkitt leukemia/lymphoma), testing CEC (cyclophosphamide, etoposide, clofarabine) and liposomal vincristine plus bortezomib and ofatumumab or rituximab (NCT03136146).
Another study at MD Anderson is testing anti-CD20 antibodies in the Pedi-cRIB regimen (NCT05645718), which combines low-intensity chemotherapy with blinatumomab, InO and rituximab in a condensed regimen.
Other bispecific T-cell engagers such as glofitamab and mosunetuzumab, may be of interest for future studies in mature B-cell ALL or in non-Hodgkin lymphoma, as well as in CD20-positive BCP-ALL.61 No studies open for pediatric patients were found in our search regarding these bispecific antibodies; nevertheless, they could gain a role for patients without other targets available (CD19, CD22), after multiple lines of therapy.
CD123 is widely expressed on a variety of hematologic malignancies, including some subtypes of BCP-ALL.12 Multiple compounds targeting CD123 are currently in development. Tagraxofusp is a protein-drug conjugate consisting of human interleukin-3 fused to a truncated diphtheria toxin, approved as a single agent for the treatment of patients (from the age of 2 years in the USA) with blastic plasmacytoid dendritic cell neoplasm.62 A Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) consortium phase I/II trial of tagraxofusp, as a single agent and in combination with chemotherapy, is enrolling pediatric patients with R/R hematologic malignancies, including ALL (NCT05476770). IMGN632 is another humanized anti-CD123 antibody linked to a novel DNA-alkylating payload of the indolinobenzodiazepine pseudodimer (IGN) class, approved by the Food and Drug Administration for the treatment of blastic plasmacytoid dendritic cell neoplasm.63 Nevertheless, its development for pediatric leukemias has been discontinued (COG trial NCT05320380 withdrawn). SAR443579 is a natural killer-cell engager targeting CD123 that is currently being tested in a phase I/II trial open to children and adults with different hematologic malignancies, including ALL (NCT05086315).
Therapeutic CD38-targeting monoclonal antibodies (daratumumab, isatuximab; both G2 class antibodies), approved to treat adults with multiple myeloma, have been explored in pediatric ALL. Although CD38 expression in pediatric ALL might be lower than in multiple myeloma, promising preclinical data and the robust CD38 surface expression at diagnosis and relapse, especially in T-ALL, have led to the development of clinical trials.13,64
Daratumumab
Daratumumab is a monoclonal antibody that induces lysis through cytotoxic mechanisms (complement-dependent, cell-mediated and antibody-dependent cellular phagocytosis). It is approved as monotherapy and in combination with standard-of-care regimens for newly diagnosed and R/R multiple myeloma and in combination with standard-of-care regimens for systemic light chain amyloidosis. Daratumomab
has been evaluated in pediatric patients with R/R T-ALL and lymphoblastic lymphomas in a non-randomized phase I/II trial in combination with standard four-drug re-induction (vincristine, prednisone, PEG-asparaginase, and doxorubicin). Similar to other monoclonal antibodies, infusion reactions have been reported as the most common adverse event, for daratumomab as well as for isatuximab. Among T-ALL patients, 41.7% (n=10) achieved complete remission at the end of cycle 1, reaching the prespecified response rate.6 However, these results are in the same range as, for example, the response rate of 33% (4/12) obtained in R/R T-ALL in the NECTAR trial (testing cyclophosphamide, etoposide, and nelarabine),65 and hence a randomized trial is needed to establish the potential added benefit of treatment with daratumomab. A phase I trial open in the US is testing daratumumab monotherapy following a total body irradiation-based conditioning regimen and allogeneic HSCT in T-ALL (in patients aged 0-39 years) as prophylactic treatment to prevent post-transplant relapse (NCT04972942). Some reports in adults have described activity of daratumumab as a single agent in patients with T-ALL and molecular or morphological relapse.66,67 There are also some reports of preclinical development of an antibody-drug conjugate targeting CD38, which could be of potential relevance for T-ALL in the future.68
A specific diagnostic challenge related to treatment with daratumomab is the long persistence of this antibody on the cell surface. Due to the fact that all diagnostic CD38 antibodies bind to epitopes overlapping with the daratumumab binding site, this may interfere with CD38 detection by flow cytometry for a long period (up to several months, as reported in multiple myeloma), especially in the context of MRD.69 Moreover CD38 is expressed at low levels on red blood cells and daratumumab may mask the detection of antibodies in a patient’s serum, interfering with the compatibility tests that are part of a routine pre-transfusion work-up.70
Isatuximab
Isatuximab is an immunoglobulin G1 class monoclonal antibody, also binding CD38, which exerts its activity by the same mechanisms as daratumumab.71 The ISAKIDS study (NCT03860844) tested isatuximab in combination with chemotherapy for pediatric BCP/T-ALL and acute myeloid leukemia. A complete remission with or without complete hematologic recovery was observed in 13/25 (52.0%) patients in the B-ALL cohort, 5/11 (45.5%) in the T-ALL cohort, and 14/23 (60.9%) in the cohort with acute myeloid leukemia. These remission rates in individual cohorts did not meet the prespecified criteria to proceed to stage 2 of the ISAKIDS trial, which was therefore terminated.72
Targeted antibodies have already gained an established
role in the treatment of pediatric ALL, mainly in the setting of relapsed BCP-ALL. Following the results of randomized trials, blinatumomab has established its role in consolidation treatment for both high- and standard-risk ALL with low tumor burden in first relapse, including cases with bone marrow involvement, while failing in the setting of standard-risk extramedullary relapse.19-22 InO has demonstrated high efficacy in inducing MRD-negative complete remissions in relapsed ALL, even in the presence of high tumor burden, but randomized phase III trials are still ongoing.15,16,41 In T-ALL, the role of daratumomab is gaining interest, but randomized data are still needed to really prove its benefit. CD20-directed therapy may be of use for patients who express this antigen, but has never been extensively investigated in children.
Novel targets are being explored to further expand the role of targeted therapy for ALL: CD47 is expressed on pediatric T-ALL blasts and blocking CD47 allows macrophages to phagocytose T-ALL blasts. Co-targeting CD47 and CD38 may have a synergistic effect, based on the fact that CD38 also plays a role in the regulation of phagocytosis.73 CD79 is a B-cell-specific antigen expressed across the whole B-cell development. Antibody-drug conjugates targeting CD79 are in development for the treatment of lymphomas and could be potentially effective in BCP-ALL.74 CD127 expression was found to high in T-ALL; the OSE-127 antibody, currently being evaluated in phase II trials in inflammatory and autoimmune diseases, is potentially attractive for the treatment of T-ALL.75
With multiple options of novel therapies (blinatumomab, InO, CAR T cells) available for R/R BCP-ALL, the best combination or sequence of treatment for a relapse or refractory state remains one of the most important unanswered questions at present. Some trials are already testing a combination of multiple antibodies, such as the multiagent regimen under investigation at MD Anderson, which combines low-intensity chemotherapy with blinatumomab, InO and rituximab in a condensed regimen (Pedi-cRIB regimen, NCT05645718).76
InO and/or blinatumomab are not considered stand-alone therapies and consolidation with an allogeneic HSCT or CAR T cells is recommended in the pediatric setting. Data regarding the safety profile of allogeneic HSCT after InO and blinatumomab are well known, with the important highlight of the risk of sinusoidal obstruction syndrome after HSCT following InO. Whether or not blinatumomab and InO are suitable bridging therapies before the administration of CAR T cells is the focus of more recent studies. Prior therapy with blinatumomab, directed to CD19 as the available CAR T-cell products, has been associated with higher rates of CAR T-cell failure.51 Nevertheless, it was found that only patients with no response to blinatumomab had an inferior event-free survival after subsequent CAR T-cell therapy.52 A possible effect of InO on CAR T-cell efficacy may be mediated by the reduction of CD19-positive
B cells, both leukemic and native, following InO treatment. It was previously reported that a bone marrow CD19-positive antigen load of less than 15% can be correlated with suboptimal expansion and persistence of CAR T cells.53 However, a retrospective study showed similar response rates, overall survival and event-free survival for patients treated with InO prior to CAR T-cell therapy compared to published data regarding patients treated with CAR T cells without prior exposure to InO.54
The poorer outcome in the adult setting enables pilot trials of different strategies in larger populations with a dismal prognosis. An example is the combination of steroids, blinatumomab and tyrosine kinase inhibitors for induction in Philadelphia chromosome-positive ALL in adults, which opens the door to a possible ‘chemo-free’ reinduction.77,78 Innovations in the front-line setting for pediatric ALL have to compete with an overall survival rate of about 90% currently reached with the available, standard-of-care, poly-chemotherapy regimens.79 InO and blinatumomab are now being introduced in front-line treatment for children with newly diagnosed ALL, as single agents given in treatment cycles during consolidation therapy (COG ALL1731, COG AALL1732, ALLTogether, AIEOP-BFM 2017). Although the introduction of novel agents in the front-line setting might be challenging because of the high overall survival rate reached with standard chemotherapy, the need to replace toxic elements is clear. Immunotherapy, antibodies and other targeted therapies (such as menin-inhibitors) will allow reductions in toxic deaths and morbidity rates from front-line treatment and improve the quality of life of survivors. Whether the current risk stratification of patients, based on historical survival data after chemotherapy treatment, would be the most appropriate one also for patients treated with reg-
1. Pieters R, Mullighan CG, Hunger SP. Advancing diagnostics and therapy to reach universal cure in childhood ALL. J Clin Oncol. 2023;41(36):5579-5591.
2. Sun W, Orgel E, Malvar J, et al. Treatment-related adverse events associated with a modified UK ALLR3 induction chemotherapy backbone for childhood relapsed/refractory acute lymphoblastic leukemia. Pediatr Blood Cancer. 2016;63(11):1943-1948.
3. von Stackelberg A, Locatelli F, Zugmaier G, et al. Phase I/phase II study of blinatumomab in pediatric patients with relapsed/ refractory acute lymphoblastic leukemia. J Clin Oncol. 2016;34(36):4381-4389.
4 Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439-448.
5. Reismüller B, Attarbaschi A, Peters C, et al. Long-term outcome of initially homogenously treated and relapsed childhood acute lymphoblastic leukaemia in Austria – a population-based report of the Austrian Berlin-Frankfurt-Münster (BFM) study group. Br J Haematol. 2009;144(4):559-570.
imens including novel agents will need to be verified. For example, InO is very effective in reinduction but a low level of MRD after InO monotherapy may not have the same implications as after four-drug induction. In addition, high-risk cytogenetics seem not to influence the outcome of newly diagnosed (adult) ALL patients treated with blinatumomab and/or InO.80 Prognostic factors for response to antibody treatment are still largely unknown.15,81 Lastly, the very competitive arena and the commercial interests of manufacturers are critical in the process of selecting and prioritizing new agents for pediatric indications to avoid gaps and interruptions in the development of pediatric studies, highlighting the importance of collaborative efforts among stakeholders in order that childhood cancer drug development can prosper.82
CMZ has served as a consultant/advisory board member for Incyte, Takeda, Johnson & Johnson, Sanofi, Syndax, Agios, Bristol Meyers Squibb, Agios, Roche, Nektar Therapeutics, Kura Oncology Inc., Novartis, Pfizer, AbbVie, Daiichi Sankyo, Servier, and AstraZeneca; has sat on a steering committee for the Children’s Oncology Group; has received study funds from Sanofi, Pfizer, and Novartis; and has received financial support for academic conferences and symposia from Allucent, Gilead, Kura Oncology, Novartis Pharma BV, Syndax, and Roche. EB and FB have no conflicts of interest to disclose.
All authors conceived the review. EB and FB performed the literature search and revision. EB wrote the original draft. All authors were involved in writing, reviewing and editing the final manuscript.
6. Hogan LE, Bhatla T, Teachey DT, et al. Efficacy and safety of daratumumab (DARA) in pediatric and young adult patients (pts) with relapsed/refractory T-cell acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LL): results from the phase 2 DELPHINUS study. J Clin Oncol. 2022;40(16_suppl):10001.
7 Patel J, Gao X, Wang H. An update on clinical trials and potential therapeutic strategies in T-cell acute lymphoblastic leukemia. Int J Mol Sci. 2023;24(8):7201.
8. Attarwala H. Role of antibodies in cancer targeting. J Nat Sci Biol Med. 2010;1(1):53-56.
9. Bauer J, Nelde A, Bilich T, Walz JS. Antigen targets for the development of immunotherapies in leukemia. Int J Mol Sci. 2019;20(6):1397.
10. Shah NN, Stevenson MS, Yuan CM, et al. Characterization of CD22 expression in acute lymphoblastic leukemia. Pediatr Blood Cancer. 2015;62(6):964-969.
11. Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed
immunotherapy. Blood. 2008;112(10):3982-3988.
12. Bras AE, de Haas V, van Stigt A, et al. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytometry B Clin Cytom. 2019;96(2):134-142.
13. Bras AE, Beishuizen A, Langerak AW, et al. CD38 expression in paediatric leukaemia and lymphoma: implications for antibody targeted therapy. Br J Haematol. 2018;180(2):292-296.
14. Cordo’ V, van der Zwet JCG, Canté-Barrett K, Pieters R, Meijerink JPP. T-cell acute lymphoblastic leukemia: a roadmap to targeted therapies. Blood Cancer Discov. 2021;2(1):19-31.
15. Pennesi E, Michels N, Brivio E, et al. Inotuzumab ozogamicin as single agent in pediatric patients with relapsed and refractory acute lymphoblastic leukemia: results from a phase II trial. Leukemia. 2022;36(6):1516-1524.
16. Brivio E, Locatelli F, Lopez-Yurda M, et al. A phase I study of inotuzumab ozogamicin in pediatric relapsed/refractory acute lymphoblastic leukemia (ITCC-059 study). Blood. 2021;137(12):1582-1590.
17 Locatelli F, Zugmaier G, Mergen N, et al. Blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia: results of the RIALTO trial, an expanded access study. Blood Cancer J. 2020;10(7):77.
18. Locatelli F, Zugmaier G, Mergen N, et al. Blinatumomab in pediatric relapsed/refractory B-cell acute lymphoblastic leukemia: RIALTO expanded access study final analysis. Blood Adv. 2022;6(3):1004-1014.
19. Brown PA, Ji L, Xu X, et al. Effect of postreinduction therapy consolidation with blinatumomab vs chemotherapy on diseasefree survival in children, adolescents, and young adults with first relapse of B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 2021;325(9):833-842.
20 Locatelli F, Zugmaier G, Rizzari C, et al. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia: a randomized clinical trial. JAMA. 2021;325(9):843-854.
21. Hogan LE, Brown PA, Ji L, et al. Children’s Oncology Group AALL1331: phase III trial of blinatumomab in children, adolescents, and young adults with low-risk B-cell ALL in first relapse. J Clin Oncol. 2023;41(25):4118-4129.
22. Gökbuget N, Dombret H, Bonifacio M, et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood. 2018;131(14):1522-1531.
23. Pieters R, De Lorenzo P, Ancliffe P, et al. Outcome of infants younger than 1 year with acute lymphoblastic leukemia treated with the Interfant-06 protocol: results from an international phase III randomized study. J Clin Oncol. 2019;37(25):2246-2256.
24. Tomizawa D, Koh K, Hirayama M, et al. Outcome of recurrent or refractory acute lymphoblastic leukemia in infants with MLL gene rearrangements: a report from the Japan Infant Leukemia Study Group. Pediatr Blood Cancer. 2009;52(7):808-813.
25. van der Sluis IM, de Lorenzo P, Kotecha RS, et al. Blinatumomab added to chemotherapy in infant lymphoblastic leukemia. N Engl J Med. 2023;388(17):1572-1581.
26. Issa GC, Aldoss I, DiPersio J, et al. The menin inhibitor revumenib in KMT2A-rearranged or NPM1-mutant leukaemia. Nature. 2023;615(7954):920-924.
27. Li AM, Rabin KR, Kairalla J, et al. Blinatumomab associated seizure risk in patients with Down syndrome and B-lymphoblastic leukemia: an interim report from Children’s Oncology Group (COG) study AALL1731. Blood.
2021;138(Suppl 1):2304.
28. Gupta S, Casey J, Lasky J. Case report: blinatumomab as upfront consolidation and maintenance therapy in a pediatric patient with high-risk B-cell acute lymphoblastic leukemia. Front Oncol. 2023;13:1246924.
29. Hodder A, Mishra AK, Enshaei A, et al. Blinatumomab for firstline treatment of children and young persons with B-ALL. J Clin Oncol. 2024;42(8):907-914.
30. Litzow MR, Sun Z, Paietta E, et al. Consolidation therapy with blinatumomab improves overall survival in newly diagnosed adult patients with B-lineage acute lymphoblastic leukemia in measurable residual disease negative remission: results from the ECOG-ACRIN E1910 randomized phase III National Cooperative Clinical Trials Network trial. Blood. 2022;140(Suppl 2):LBA-1.
31. Luger SM, Sun Z, Mattison RJ, et al. Assessment of outcomes of consolidation therapy by number of cycles of blinatumomab received in newly diagnosed measurable residual disease negative patients with B-lineage acute lymphoblastic leukemia: in the ECOG-ACRIN E1910 randomized phase III National Clinical Trials Network trial. Blood. 2023;142(Suppl 1):2877.
32. Webster J, Luskin MR, Prince GT, et al. Blinatumomab in combination with immune checkpoint inhibitors of PD-1 and CTLA-4 in adult patients with relapsed/refractory (R/R) CD19 positive B-cell acute lymphoblastic leukemia (ALL): preliminary results of a phase I study. Blood. 2018;132(Suppl 1):557.
33. Feucht J, Kayser S, Gorodezki D, et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget. 2016;7(47):76902-76919.
34 Ilan U, Brivio E, Algeri M, et al. The development of new agents for post-hematopoietic stem cell transplantation noninfectious complications in children. J Clin Med. 2023;12(6):2149.
35. Gaballa MR, Banerjee P, Milton DR, et al. Blinatumomab maintenance after allogeneic hematopoietic cell transplantation for B-lineage acute lymphoblastic leukemia. Blood. 2022;139(12):1908-1919.
36. Zugmaier G, Topp MS, Alekar S, et al. Long-term follow-up of serum immunoglobulin levels in blinatumomab-treated patients with minimal residual disease-positive B-precursor acute lymphoblastic leukemia. Blood Cancer J. 2014;4(9):244.
37. DiJoseph JF, Armellino DC, Boghaert ER, et al. Antibodytargeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood. 2004;103(5):1807-1814.
38. de Vries JF, Zwaan CM, De Bie M, et al. The novel calicheamicinconjugated CD22 antibody inotuzumab ozogamicin (CMC-544) effectively kills primary pediatric acute lymphoblastic leukemia cells. Leukemia. 2012;26(2):255-264.
39 Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
40 Bhojwani D, Sposto R, Shah NN, et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia. 2019;33(4):884-892.
41. O’Brien MM, Ji L, Shah NN, et al. Phase II trial of inotuzumab ozogamicin in children and adolescents with relapsed or refractory B-cell acute lymphoblastic leukemia: Children’s Oncology Group protocol AALL1621. J Clin Oncol. 2022;40(9):956-967.
42. Brivio E, Chantrain CF, Gruber TA, et al. Inotuzumab ozogamicin in infants and young children with relapsed or refractory acute lymphoblastic leukaemia: a case series. Br J Haematol. 2021;193(6):1172-1177.
43. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer. 2019;125(14):2474-2487.
44 Jabbour E, Ravandi F, Kebriaei P, et al. Salvage chemoimmunotherapy with inotuzumab ozogamicin combined with mini-hyper-CVD for patients with relapsed or refractory Philadelphia chromosome-negative acute lymphoblastic leukemia: a phase 2 clinical trial. JAMA Oncol. 2018;4(2):230-234.
45. Short N, Jabbour E, Ravandi F, et al. The addition of inotuzumab ozogamicin to hyper-CVAD plus blinatumomab further improves outcomes in patients with newly diagnosed B-cell acute lymphoblastic leukemia: updated results from a phase II study. Blood. 2022;140(Suppl 1):8966-8968.
46. Pennesi E, Brivio E, Ammerlaan ACJ, et al. Inotuzumab ozogamicin (InO) combined with UKALL-R3 modified chemotherapy in pediatric patients with B-cell precursor CD22+ acute lymphoblastic leukemia (BCP-ALL) - results from the ITCC-059 phase 1B trial. Blood. 2022;140(Suppl 1):8976-8978.
47. McNeer JL, O’Brien MM, Rheingold SR, et al. A phase 3 randomized trial of inotuzumab ozogamicin for newly diagnosed high-risk B-ALL: safety phase results from Children’s Oncology Group protocol AALL1732. Blood. 2021;138(Suppl 1):3398.
48. O’Brien MM, McNeer JL, Rheingold SR, et al. A phase 3 trial of inotuzumab ozogamicin for high-risk B-ALL: second safety phase results from Children’s Oncology Group AALL1732. J Clin Oncol. 2023;41(16_suppl):10016.
49 Jabbour E, Short NJ, Jain N, et al. The evolution of acute lymphoblastic leukemia research and therapy at MD Anderson over four decades. J Hematol Oncol. 2023;16(1):22.
50 Wintering A, Ishiyama K, Tamaki S, et al. CD22low/Bcl-2high expression identifies poor response to inotuzumab ozogamicin in relapsed/refractory acute lymphoblastic leukemia. Blood Adv. 2023;7(2):251-255.
51. Kirchhoff H, Karsli U, Schoenherr C, et al. Venetoclax and dexamethasone synergize with inotuzumab ozogamicin–induced DNA damage signaling in B-lineage ALL. Blood. 2021;137(19):2657-2661.
52. Raetz EA, Cairo MS, Borowitz MJ, et al. Chemoimmunotherapy reinduction with epratuzumab in children with acute lymphoblastic leukemia in marrow relapse: a Children’s Oncology Group pilot study. J Clin Oncol. 2008;26(22):3756-3762.
53. Raetz EA, Cairo MS, Borowitz MJ, et al. Re-induction chemoimmunotherapy with epratuzumab in relapsed acute lymphoblastic leukemia (ALL): phase II results from Children’s Oncology Group (COG) study ADVL04P2. Pediatr Blood Cancer. 2015;62(7):1171-1175.
54 Wayne AS, Shah NN, Bhojwani D, et al. Phase 1 study of the anti-CD22 immunotoxin moxetumomab pasudotox for childhood acute lymphoblastic leukemia. Blood. 2017;130(14):1620-1627.
55. Shah NN, Bhojwani D, August K, et al. Results from an international phase 2 study of the anti-CD22 immunotoxin moxetumomab pasudotox in relapsed or refractory childhood B-lineage acute lymphoblastic leukemia. Pediatr Blood Cancer.
2020;67(5):e28112.
56. Minard-Colin V, Aupérin A, Pillon M, et al. Rituximab for highrisk, mature B-cell non-Hodgkin’s lymphoma in children. N Engl J Med. 2020;382(23):2207-2219.
57 Jeha S, Behm F, Pei D, et al. Prognostic significance of CD20 expression in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2006;108(10):3302-3304.
58. Maury S, Chevret S, Thomas X, et al. Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med. 2016;375(11):1044-1153.
59 Marks DI, Kirkwood AA, Rowntree CJ, et al. Addition of four doses of rituximab to standard induction chemotherapy in adult patients with precursor B-cell acute lymphoblastic leukaemia (UKALL14): a phase 3, multicentre, randomised controlled trial. Lancet Haematol. 2022;9(4):e262-e275.
60. Sasaki K, Kantarjian HM, Morita K, et al. Hyper-CVAD plus ofatumumab versus hyper-CVAD plus rituximab as frontline therapy in adults with Philadelphia chromosome–negative acute lymphoblastic leukemia: a propensity score analysis. Cancer. 2021;127(18):3381-3389.
61. Lussana F, Gritti G, Rambaldi A. Immunotherapy of acute lymphoblastic leukemia and lymphoma with T cell–redirected bispecific antibodies. J Clin Oncol. 2021;39(5):444-455.
62. Pemmaraju N, Lane AA, Sweet KL, et al. Tagraxofusp in blastic plasmacytoid dendritic-cell neoplasm. N Engl J Med. 2019;380(17):1628-1637.
63. Kovtun Y, Jones GE, Adams S, et al. A CD123-targeting antibodydrug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018;2(8):848-858.
64. Bride KL, Vincent TL, Im S-Y, et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood. 2018;131(9):995-999.
65. Whitlock JA, Malvar J, Dalla-Pozza L, et al. Nelarabine, etoposide, and cyclophosphamide in relapsed pediatric T-acute lymphoblastic leukemia and T-lymphoblastic lymphoma (study T2008-002 NECTAR). Pediatr Blood Cancer. 2022;69(11):e29901.
66. Prejzner W, Piekoś O, Bełdzińska K, et al. The role of daratumumab in relapsed/refractory CD38 positive acute leukemias-case report on three cases with a literature review. Front Oncol. 2023;13:1228481.
67. Cerrano M, Bonifacio M, Olivi M, et al. Daratumumab with or without chemotherapy in relapsed and refractory acute lymphoblastic leukemia. A retrospective observational Campus ALL study. Haematologica. 2022;107(4):996-999.
68. Li L, Tong W, Lau M, et al. Preclinical development of an antiCD38 antibody-drug conjugate for treatment of hematological malignancies. Blood. 2019;134(Suppl_1):5621.
69 Oberle A, Brandt A, Alawi M, et al. Long-term CD38 saturation by daratumumab interferes with diagnostic myeloma cell detection. Haematologica. 2017;102(9):e368-e370.
70. Lancman G, Arinsburg S, Jhang J, et al. Blood transfusion management for patients treated with anti-CD38 monoclonal antibodies. Front Immunol. 2018;9:2616.
71. Deckert J, Wetzel MC, Bartle LM, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20(17):4574-4583.
72. Baruchel A, Bertrand Y, Nysom K, et al. Isatuximab plus chemotherapy for pediatric relapsed/refractory acute lymphoblastic leukemia or acute myeloid leukemia (ISAKIDS):
interim efficacy analysis. Hemasphere. 2023;7(S3):e121813.
73. Müller K, Vogiatzi F, Winterberg D, et al. Combining daratumumab with CD47 blockade prolongs survival in preclinical models of pediatric T-ALL. Blood. 2022;140(1):45-57.
74. Ferreri AJM. Targeted therapies make room, anti-CD79b agents are coming. Lancet Oncol. 2019;20(7):898-900.
75. Baccelli I, Lenk L, Laqua A, et al. CD127 is expressed by acute lymphoblastic leukemias and is efficiently targeted by the IL7R-antagonist OSE-127 through macrophage-mediated antibody dependent phagocytosis. Cancer Res. 2023;83(7_Supplement):2957.
76. McCall D, Jabbour E, Roth M, Nunez C, Cuglievan B. Mini-hyper CVD + CRIB (condensed rituximab, inotuzumab ozogamicin, and blinatumomab) for refractory pediatric B-acute lymphoblastic leukemia. Pediatr Blood Cancer. 2023;70(1):e29939.
77. Foà R, Bassan R, Vitale A, et al. Dasatinib-blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med. 2020;383(17):1613-1623.
78. Jabbour E, Short NJ, Jain N, et al. Ponatinib and blinatumomab for Philadelphia chromosome-positive acute lymphoblastic leukaemia: a US, single-centre, single-arm, phase 2 trial. Lancet Haematol. 2023;10(1):e24-e34.
79. Pui CH, Yang JJ, Hunger SP, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol.
2015;33(27):2938-2948.
80 Senapati J, Jabbour E, Short NJ, et al. Impact of high-risk cytogenetics (HR-CTG) on the outcome of newly diagnosed adult patients with Philadelphia negative B-cell acute lymphoblastic leukemia (B-ALL) treated with frontline blinatumomab (Blina) and/or inotuzumab ozogamicin (Ino) containing HyperCVAD (HCVAD) therapy. Blood. 2023;142(Suppl 1):1500.
81. Schultz L, Gardner R. Mechanisms of and approaches to overcoming resistance to immunotherapy. Hematology Am Soc Hematol Educ Program. 2019;2019(1):226-232.
82. Pearson ADJ, Weiner SL, Adamson PC, et al. ACCELERATE - Five years accelerating cancer drug development for children and adolescents. Eur J Cancer. 2022;166:145-164.
83. Baruchel A, Abrahamsson J, Bertrand Y, et al. Isatuximab in combination with chemotherapy in pediatric patients with relapsed/refractory acute lymphoblastic leukemia or acute myeloid leukemia (ISAKIDS): interim analysis. Blood. 2021;138(Suppl 1):516.
84 Nakayama H, Ogawa C, Sekimizu M, et al. A phase I study of inotuzumab ozogamicin as a single agent in pediatric patients in Japan with relapsed/refractory CD22-positive acute lymphoblastic leukemia (INO-Ped-ALL-1). Int J Hematol. 2022;116(4):612-621.
Bruno A. Cardoso,1* Mafalda Duque,1* Ana Gírio,1 Rita Fragoso,1 Mariana L. Oliveira,1 James R. Allen,2 Leila R. Martins,1 Nádia C. Correia,1 André Bortolini Silveira,3 Alexandra Veloso,2 Shunsuke Kimura,4 Lisa Demoen,5 Filip Matthijssens,5 Sima Jeha,6,7 Cheng Cheng,8 Ching-Hon Pui,6,7,9 Ana R. Grosso,10,11 João L. Neto,1 Sérgio F. de Almeida,1 Pieter Van Vlieberghe,5 Charles G. Mullighan,4 J. Andres Yunes,3 David M. Langenau,2 Françoise Pflumio12,13 and João T. Barata1
1Instituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina, Universidade de Lisboa, Lisbon, Portugal; 2MGH Pathology and Harvard Medical School, Charlestown, MA, USA; 3Laboratório de Biologia Molecular, Centro Infantil Boldrini, Campinas, São Paulo, Brazil; 4Department of Pathology, Center of Excellence for Leukemia Studies, and Hematological Malignancies Program, St. Jude Children’s Research Hospital, Memphis, TN, USA; 5Department of Biomolecular Medicine, Ghent University, and Cancer Research Institute Ghent (CRIG), Ghent, Belgium; 6Department of Oncology, St. Jude Children’s Research Hospital and the University of Tennessee Health Science Center, Memphis, TN, USA; 7Department of Global Pediatric Medicine, St. Jude Children’s Research Hospital and the University of Tennessee Health Science Center, Memphis, TN, USA; 8Department of Biostatistics, St. Jude Children’s Research Hospital and the University of Tennessee Health Science Center, Memphis, TN, USA; 9Department of Pathology, St. Jude Children’s Research Hospital and the University of Tennessee Health Science Center, Memphis, TN, USA; 10Associate Laboratory i4HB - Institute for Health and Bioeconomy, NOVA School of Science and Technology, Universidade NOVA de Lisboa, Caparica, Portugal; 11UCIBIO - Applied Molecular Biosciences Unit, Department of Life Sciences, NOVA School of Science and Technology, Universidade NOVA de Lisboa, Caparica, Portugal; 12Université Paris-Saclay, INSERM, iRCM/IBFJ CEA, UMR Stabilité Génétique Cellules Souches et Radiations, Fontenayaux-Roses, France and 13OPALE Carnot Institute, the Organization for Partnerships in Leukemia, Saint-Louis Hospital, Paris, France
*BAC and MD contributed equally as first authors.
Abstract
Correspondence: J.T. Barata joao_barata@medicina.ulisboa.pt
Received: February 10, 2023.
Accepted: November 27, 2023. Early view: December 7, 2023.
https://doi.org/10.3324/haematol.2023.282854
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
CASZ1 is a conserved transcription factor involved in neural development, blood vessel assembly and heart morphogenesis. CASZ1 has been implicated in cancer, either suppressing or promoting tumor development depending on the tissue. However, the impact of CASZ1 on hematological tumors remains unknown. Here, we show that the T-cell oncogenic transcription factor TAL1 is a direct positive regulator of CASZ1, that T-cell acute lymphoblastic leukemia (T-ALL) samples at diagnosis overexpress CASZ1b isoform, and that CASZ1b expression in patient samples correlates with PI3K-AKT-mTOR signaling pathway activation. In agreement, overexpression of CASZ1b in both Ba/F3 and T-ALL cells leads to the activation of PI3K signaling pathway, which is required for CASZ1b-mediated transformation of Ba/F3 cells in vitro and malignant expansion in vivo. We further demonstrate that CASZ1b cooperates with activated NOTCH1 to promote T-ALL development in zebrafish, and that CASZ1b protects human T-ALL cells from serum deprivation and treatment with chemotherapeutic drugs. Taken together, our studies indicate that CASZ1b is a TAL1-regulated gene that promotes T-ALL development and resistance to chemotherapy.
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematological malignancy that results from transfor-
mation and clonal expansion of developmentally-arrested T-cell progenitors.1,2 Conventional risk-adjusted multi-agent chemotherapy allows for high 5-year event-free survival
rates in children. However, a significant number of patients still relapse or do not respond to therapy (features that are striking in adults), and the intensive-treatment regimens are often associated with severe complications. Consequently, there have been considerable efforts to better understand the cell-intrinsic lesions and microenvironmental underpinnings of the disease, resulting in the identification of numerous key genetic,3-7 epigenetic or posttranscriptional,8-12 and cell-extrinsic13-15 alterations involved in the development and resistance to treatment of T-ALL. Amongst these, several have revealed potential to translate into novel, hopefully less toxic and more efficient therapies,2,16-19 which may contribute to circumventing resistance to chemotherapy. Thus, identification of new molecular regulators of T-ALL should contribute to a better understanding of the disease and, consequently, to the improvement of therapeutic approaches.
CASZ1 is a highly conserved20 zinc finger transcription factor essential for blood vessel assembly,21 cardiomyocyte differentiation and proliferation22,23 and heart morphogenesis.24,25 CASZ1 is also critical during neural development. 26,27 In accordance, loss of CASZ1 expression associates with high risk and poor prognosis in neuroblastoma.28,29 In agreement with a tumor suppressor role for CASZ1, restoration of CASZ1 expression in neuroblastoma cell lines resulted in cell differentiation, increased adhesion and reduced migration, and inhibition of cell growth in vitro and in vivo 28,29 CASZ1 displays an anti-oncogenic role also in hepatocellular carcinoma30 and embryonal rhabdomyosarcoma.31 Nevertheless, the involvement of CASZ1 in cancer may vary depending on the tissue. For instance, CASZ1 is upregulated in epithelial ovarian cancer cells and promotes epithelial-mesenchymal transition, cell migration, invasion and metastasis.32
There are two known CASZ1 isoforms in humans: CASZ1a (also known as CASZ11 or CASZ1 transcript variant 1), which has 21 exons, is roughly 8 Kb-long and encodes for a nuclear protein containing 11 zinc fingers; and CASZ1b (CASZ5 or CASZ1 transcript variant 2), which has 16 exons, is 4.4 kb-long and encodes for a mainly nuclear but also cytoplasmic protein with five zinc fingers. The sequence of CASZ1b is identical to that of the longer CASZ1 isoform until the end of exon 16, where a 5’ splice donor located 1 bp before the stop codon of CASZ1b can be used with a 3’ splice acceptor in exon 17 of CASZ1a to produce the latter by alternative splicing.20 Interestingly, CASZ1a and CASZ1b are often co-expressed (although not always regulated in a similar fashion) and appear to exert similar functions in particular tissues.29 For example, both isoforms were shown to have anti-tumoral activity in neuroblastoma,28,29 while exerting protumoral effects in ovarian cancer.32 In the current study, we aimed to define the role of CASZ1 in T-ALL. We demonstrate that CASZ1b, which is upregulated in T-ALL cells in part via transcriptional activity of TAL1, has an oncogenic role, transforming Ba/F3 cells and cooperating with NOTCH1 in promoting T-ALL in zebrafish. In human T-ALL
cells, CASZ1b expression correlates with poor prognosis and activation of the PI3K-AKT-mTOR signaling pathway. Consistent with these observations, CASZ1b overexpression leads to PI3K-AKT-mTOR signaling activation, which is required for T-ALL cell viability. Overall, our findings establish CASZ1 as a putative novel T-cell oncogene and suggest that PI3K targeting drugs may offer clinical benefit for relapsed T-ALL cases exhibiting high CASZ1 levels.
Primary T-cell acute lymphoblastic leukemia samples, cell lines and culture conditions
Primary T-ALL cells collected from pediatric patients at diagnosis were isolated as described.14 Informed consent was obtained in accordance with the Declaration of Helsinki after institutional ethical review board approval (authorization #13-105-1 obtained from the Institutional Review Board [IRB00003888] of the French Institute of Medical Research and Health). Upon isolation, primary samples were cultured in RPMI 1640 medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS, Biowest), hereafter named RPMI-10. The human T-ALL cell lines CEM, DND-41, HPB-ALL, Jurkat, Loucy, MOLT4 and P12 were obtained commercially and cultured in RPMI-10. The IL-3-dependent murine pro-B cell line Ba/F3 was maintained in RPMI-10 with IL-3. Mouse IL-3 was produced by WEHI3B cells, whose supernatant was used to maintain Ba/F3 cells. The human embryonic fibroblast 293T cell line was maintained in DMEM medium (Life Technologies) supplemented with 10% FBS (DMEM-10). T-ALL primary cells, cell lines and Ba/F3 cells were cultured at 37°C with a 5% CO2 atmosphere in RPMI-10 alone (with the appropriate vehicle), RPMI (when indicated) or in RPMI-10 plus the following pharmacological agents: 4-hydroxy-tamoxifen (4OHT; Sigma); NVP-BEZ235 (Selleckchem); LY294002 (Cayman Chemical), daunorubicin (Selleckchem); dexamethasone (Sigma) and L-asparaginase (Sigma). At defined time points, cells were harvested and processed as indicated below for assessment of cell viability, RNA extraction and immunobloting.
Determination of cell viability was performed by flow cytometry analysis of forward scatter versus side scatter (FSC vs. SSC) distribution using a LSRFortessa cell analyzer (BD Biosciences). We have confirmed previously that this strategy measures lymphocyte viability as accurately as when using annexin V and propidium iodide staining.8 Cell size, as a measure of cell activation, was assessed by FSC versus SSC gated on the live cell population. The samples were acquired using a LSRFortessa cell analyzer with a high-throughput-sampler (HTS) with a fixed volume of sample to be analyzed, which allowed us to extrapolate the number of cells that were analyzed and thus calculate proliferation.
Immunobloting
Cell lysates were prepared as described16 and equal amounts of protein were analyzed by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred onto nitrocellulose membranes, and immunoblotted with antibodies against p-mTOR (S2448), p-AKT (S473), AKT, p-S6 (S235/236) and S6 (all from Cell Signaling Technology), CASZ1 (Rockland), Lamin B (Santa Cruz Biotechnology), TAL1 (EMD Millipore), and p27Kip1 (BD Transduction Labs). Immunodetection was performed by incubation with horseradish peroxidase-conjugated appropriate secondary antibodies and developed by chemiluminescence. Where indicated, densitometry analysis was performed using Adobe Photoshop CS3 software (version 10.0, Adobe Systems Incorporated, San Jose, CA, USA). Each band was analyzed with a constant frame and normalized to the respective loading control.
The GraphPad Prism software was used for statistical analysis. Differences between mean values were calculated using two-tailed Student’s t test and one-way analysis of variance, as appropriate. Correlation in gene expression levels was determined using the Pearson correlation coefficient. Differences in survival curves were analyzed using the log-rank (Mantel-Cox) test. Differences were considered significant at P<0.05.
Information on plasmid generation, electroporation, viral transduction, mouse and zebrafish in vivo models, quantitative PCR (including primer sequences: Online Supplementary Table S1), chromatin immunoprecipitation, RNA sequencing and bioinformatics analyses are available in the Online Supplementary Appendix
CASZ1b is upregulated in T-cell acute lymphoblastic leukemia, especially in TAL1-positive cases
We first analyzed the pattern of Casz1 expression during normal mouse hematopoiesis using the BloodSpot database.33 Casz1 was expressed at low levels in long-term hematopoietic stem cells (HSC) and upregulated in short-term HSC (Online Supplementary Figure S1). Casz1 expression appeared to be highest in common lymphoid progenitors and it was downregulated upon commitment to either T or B lymphoid lineage, with both B- and T-cell precursors displaying low Casz1 levels (Online Supplementary Figure S1).
Because T-ALL arises from clonal expansion of T-cell precursors arrested at different stages of differentiation, we next used a publicly available dataset to assess CASZ1 expression in leukemia samples collected from T-ALL patients at presentation, as compared to normal thymocyte subsets representative of the main T-cell developmental stages.34 We found that, in contrast to CASZ1a (Figure 1A), the short isoform of CASZ1 (CASZ1b) was significantly up-
regulated in T-ALL cells (Figure 1B). These observations, together with the fact that CASZ1b is the most conserved of the two CASZ1 isoforms,20 drove us to focus on CASZ1b (hereafter referred to simply as CASZ1) for the remainder of our studies.
Although CASZ1 levels were generally increased in the T-ALL patients (Figure 1A, B) and T-ALL cell lines (Online Supplementary Figure S2) as compared to normal T-cell precursors, we noticed that TAL1 high cases expressed higher levels of CASZ1 than TAL1 low samples in three different T-ALL patient cohorts (Figure 1C; Online Supplementary Figure S3).35,36
TAL1 upregulates CASZ1 in T-cell acute lymphoblastic leukemia cells
The association between TAL1 and CASZ1 expression in T-ALL patient samples prompted us to evaluate whether TAL1 might transcriptionally regulate CASZ1. Forced expression of TAL1 in two TAL1-negative T-ALL cell lines, P12 (data not shown) and MOLT4 (Online Supplementary Figure S4A), led to the upregulation of CASZ1 expression (Figure 2A), whereas TAL1 inactivation in TAL1-positive Jurkat and CEM cells, using CRISPR/cas9 technology (Figure 2B; Online Supplementary Figure S4B), downregulated CASZ1 (Figure 2B). Importantly, TAL1 silencing in two primary T-ALL patient samples also resulted in CASZ1 downregulation (Figure 2C), further reinforcing the positive link between TAL1 and CASZ1 in T-ALL.
CASZ1 transforms Ba/F3 cells through the activation of PI3K-AKT-mTOR signaling pathway
Given that CASZ1 is overexpressed in T-ALL cells, including downstream of the major T-cell oncogene TAL1, we postulated that CASZ1 should have an oncogenic role in lymphoid cells. To test this possibility, we ectopically expressed CASZ1 in a stable manner in IL-3-dependent Ba/ F3 cells7 (Online Supplementary Figure S5A). CASZ1 rescued viability, cell size and proliferation of Ba/F3 cells under IL3 deprivation (Online Supplementary Figure S5B, C), which allowed Ba/F3 cells expressing CASZ1 in the absence of IL-3 to maintain their viability (Figure 3A) and proliferate (Figure 3B) throughout time to similar levels as mock-transduced control cells cultured with the growth factor. These findings indicate that CASZ1 has the capacity to transform Ba/F3 cells, rendering them growth factor-independent. To identify potential mechanisms by which CASZ1 exerted its oncogenic effects, we analyzed the transcriptional program engaged by CASZ1 in Ba/F3 cells. RNA-sequencing analysis showed that CASZ1 affected the expression of 1,207 genes (Online Supplementary Figure S6; Online Supplementary Table S2), and subsequent KEGG pathway overrepresentation analysis indicated that PI3K-AKT signaling was highly enriched in CASZ1 overexpressing cells (Figure 3C). In agreement, immunoblot analysis showed that CASZ1 overexpression in Ba/F3 cells (Online Supple-
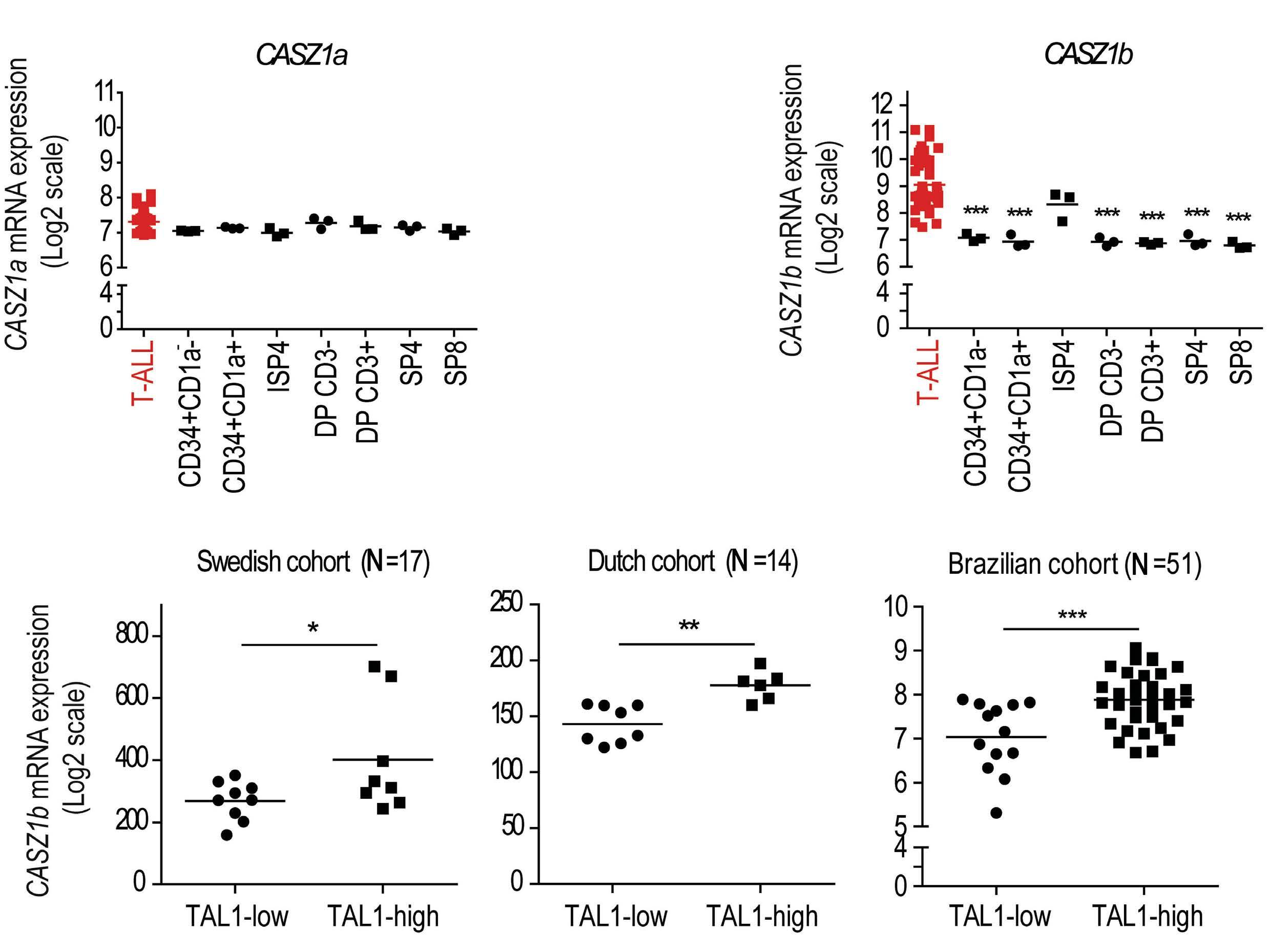
Figure 1. CASZ1b is overexpressed in T-cell acute lymphoblastic leukemia. (A, B) CASZ1a (A) and CASZ1b (B) transcript levels in T-cell acute lymphoblastic leukemia (T-ALL) patients versus normal thymocyte subpopulations. Gene expression data set GSE33469-33470. ISP4 - immature single positive CD4; DPCD3- - CD4 CD8 double positive, without CD3 expression; DPCD3+CD4 CD8 double positive, with CD3 expression; SP4 - single positive CD4; SP8 - single positive CD8. (C) CASZ1b transcript levels in 3 different T-ALL patients cohorts (Swedish - GSE41621, Dutch - GSE18497 and Brazilian - GSE51001 and GSE66638) analyzed according to the expression levels of the TAL1 transcript (TAL1-high vs. TAL1-low). *P<0.05, **P<0.01; ***P<0.001, student’s t test.
mentary Figure S7A) upregulated the phosphorylation levels of members of the PI3K-AKT-mTOR signaling pathway, such as AKT, mTOR and S6 (Figure 3D). In addition, CASZ1 promoted the downregulation of p27Kip1, a readout of cell cycle progression consistent with PI3K-AKT activation.37 Notably, CASZ1 upregulated Pik3cd (the gene that encodes PI3Kδ) transcript levels (Online Supplementary Figure S6B) and PI3Kδ protein expression (Online Supplementary Figure S8A), suggesting that CASZ1 may promote PI3K-AKT-mTOR pathway activation, at least in part, via upregulation of PI3KCD/PI3Kδ.
In order to test the involvement of PI3K-AKT-mTOR signaling in CASZ1-mediated transformation, we treated CASZ1-expressing Ba/F3 cells with two distinct specific small molecule PI3K/mTOR inhibitors. As expected, NVP-BEZ235 (dactolisib)38 and LY294002 dampened PI3K-AKT-mTOR signaling (Figure 3E; Online Supplementary Figure S9A), and abrogated the effects of CASZ1 on viability (Figure 3F; Online Supplementary Figure S9B) and proliferation (Fig-
ure 3G; Online Supplementary Figure S9C) of Ba/F3 cells. These results demonstrate the importance of maintaining the integrity of the PI3K-AKT-mTOR signaling cascade for CASZ1-mediated transformation.
CASZ1 expression activates the PI3K-AKT-mTOR signaling pathway in T-cell acute lymphoblastic leukemia
We next sought to evaluate whether, similar to Ba/F3 cells, CASZ1-mediated effects in T-ALL cells involved upregulation of PI3K-AKT-mTOR signaling. We compared expression levels of all the transcripts to those of CASZ1 in the T-ALL GSE18497 dataset36 and calculated their Pearson correlation coefficient. Genes whose correlation coefficient was ≥0.7 (indicating a strong positive correlation with CASZ1) were subsequently employed to perform KEGG pathway overrepresentation analysis. Our analysis shows that genes whose expression correlates with that of CASZ1 were enriched in different KEGG pathways, including PI3K-AKT signaling
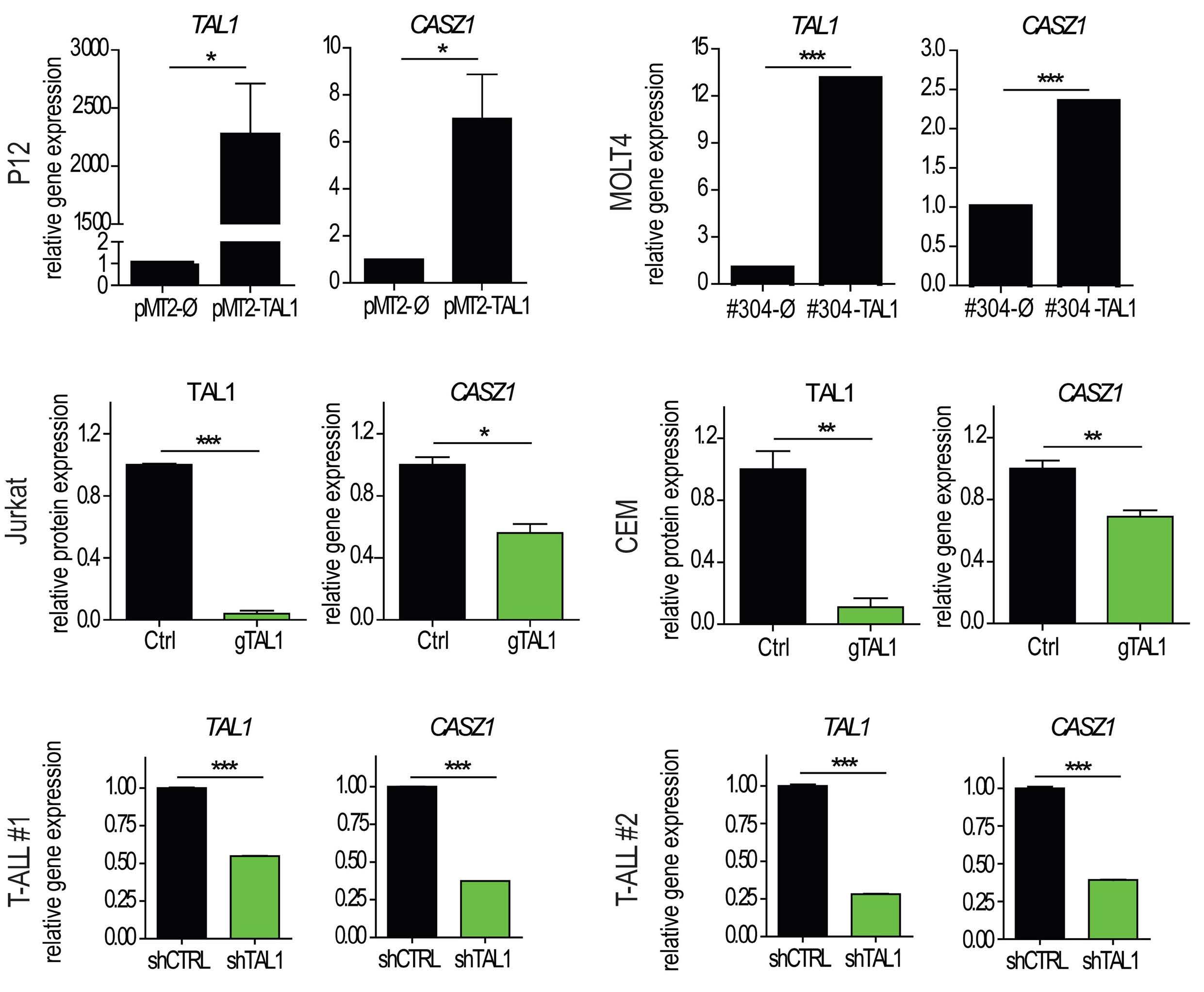
Figure 2. CASZ1b is regulated by TAL1 in T-cell acute lymphoblastic leukemia cells. (A) P12 and MOLT4 cells ectopically expressing the TAL1 protein were evaluated for TAL1 and CASZ1 expression by quantitative polymerase chain reaction (qPCR). Values were normalized to the respective control condition (empty vector). (B) TAL1 expression was inactivated in Jurkat and CEM cells by CRISPR/cas9. Three control clones (Ctrl) and 3 clones with inactivated TAL1 (gTAL1) from each of the cell lines were analyzed for TAL1 protein expression and CASZ1 mRNA levels. Values were normalized to the average of the control clones. (C) Two primary T-cell acute lymphoblastic leukemia (T-ALL) patient samples collected at diagnosis were transduced with short hairpin RNA (shRNA) against TAL1. Cells were expanded for 72 hours, and TAL1 (left) and CASZ1 (right) transcript levels were detected by qPCR. Values were normalized to the control condition (shCTRL). Values represent the mean ± standard deviation of experimental triplicates of a representative experiment (N=3). *P<0.05, **P<0.01; ***P<0.001, student’s t test.
pathway (Figure 4A). Immunoblot analysis confirmed that T-ALL cell lines overexpressing CASZ1 (Online Supplementary Figure S7B) displayed higher levels of PI3K-AKT-mTOR signaling activation than mock-transduced controls (Figure 4B). In agreement with our transcriptomics data indicating elevated levels of Pik3cd (the gene that encodes PI3Kδ) in CASZ1-overexpressing Ba/F3 cells (Online Supplementary Figure S6B), we found that CASZ1 upregulates PI3Kδ protein levels in DND-41 cells (Online Supplementary Figure S8B). Interestingly, PI3Kβ was the isoform that was upregulated by CASZ1 in MOLT4 cells (Online Supplementary Figure S8C).
CASZ1 promotes tumorigenesis in vivo
Next, we evaluated whether CASZ1 was able to promote
tumorigenesis in vivo. We subcutaneously transplanted empty vector- and CASZ1-transduced Ba/F3 cells into opposite flanks of each recipient NOD/SCID mouse (Figure 5A). Whereas none of the control transplants originated any detectable tumors, ten of ten CASZ1-expressing transplants originated large tumor masses within 20 days (Figure 5B). Then, to ascertain whether PI3K-AKT-mTOR signaling pathway is essential for the maintenance of CASZ1-triggered tumors in vivo, we let tumors from subcutaneously transplanted Ba/F3 cells overexpressing CASZ1 grow until 500 mm3, at which point the mice were randomly divided into two groups that either received vehicle or NVP-BEZ235/ dactolisib (Figure 5C). Treatment with NVP-BEZ235/dactolisib clearly delayed tumor growth (Figure 5D) and the effect
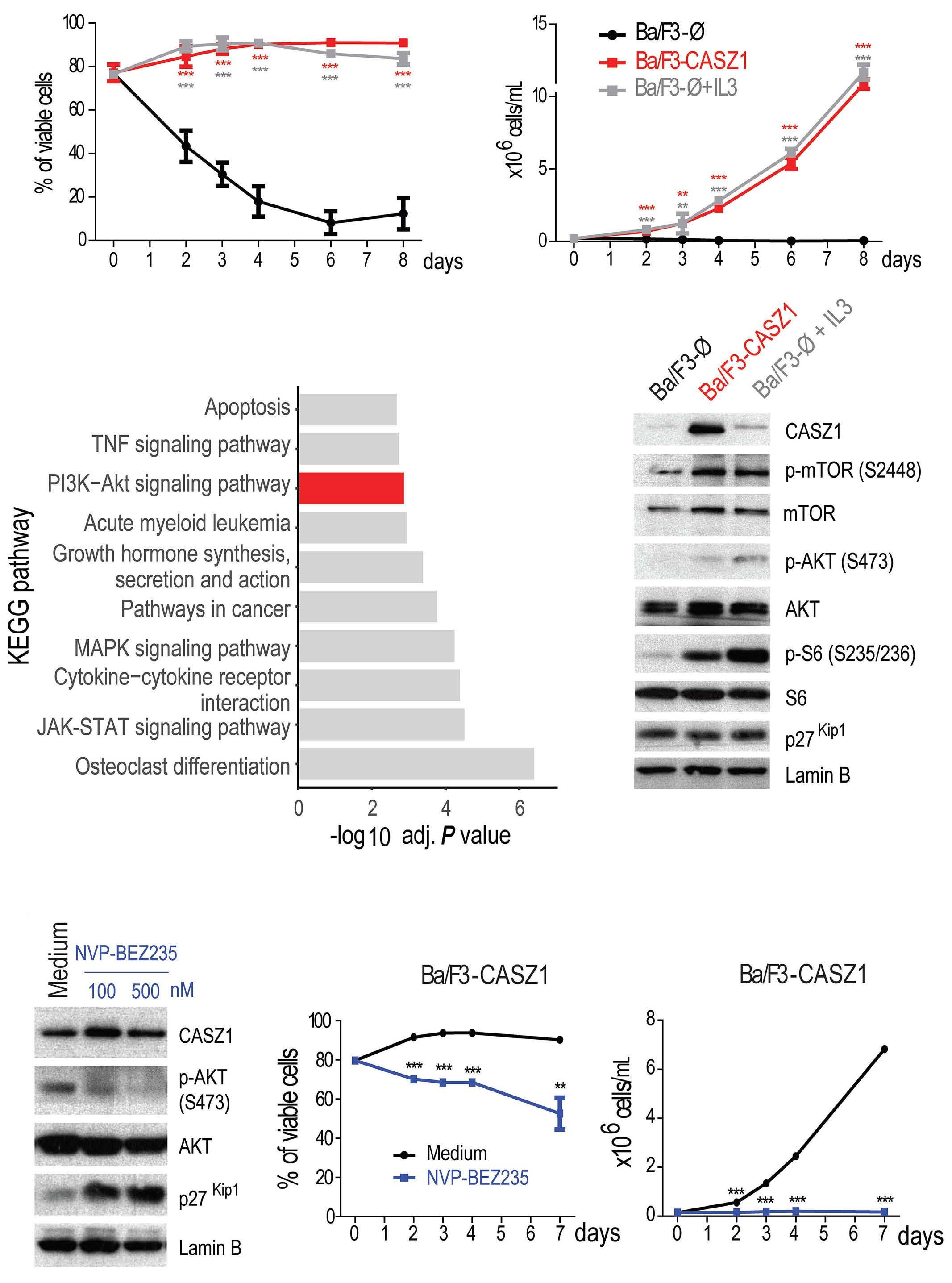
Figure 3. CASZ1 overexpression transforms Ba/F3 cells by activating the PI3K-AKT-mTOR signaling pathway. (A-B) Ba/F3 cells stably transduced with empty vector (Ba/F3-Ø) or CASZ1 (Ba/F3-CASZ1) were cultured in medium without growth factors. As a positive control, Ba/F3-Ø cells were also cultured in the presence of IL-3 (Ba/F3-Ø + IL-3). Viability (A) and proliferation (B) were determined at the indicated time points. Values represent the mean ± standard deviation of at least 3 independent experiments. **P<0.01; ***P<0.001 (C) Top 10 most significantly enriched KEGG pathways (adj. P<0.05) from upregulated genes (adj. P<0.05, log2 fold change>1) in the RNA-sequencing analysis of Ba/F3-CASZ1 versus Ba/F3-Ø cells. (D) Ba/F3-Ø and Ba/F3-CASZ1 cells were cultured in the indicated conditions for 24 hours. PI3K signaling activation was evaluated by immunoblot detection of the phosphorylation levels of AKT and S6. Total levels of CASZ1 and p27Kip1 were also analyzed. Lamin B was used as loading control. Data are representative of 2 independent experiments. (E) Ba/F3-CASZ1 cells were cultured for 24 hours in the presence of 100 or 500 nM of NVP-BEZ235 or vehicle (medium). PI3K signaling activation was evaluated by immunoblot detection of the phosphorylation levels of AKT. Total levels of CASZ1 and p27Kip1 were also analyzed. Lamin B was used as loading control. Data are representative of 2 independent experiments. (F, G) Ba/F3-CASZ1 cells were cultured in the presence or absence of 500 nM of NVP-BEZ235. Viability (F) and proliferation (G) were determined at the indicated time points. Values represent the mean ± standard deviation of experimental triplicates of a representative experiment (N=3). **P<0.01; ***P<0.001, one-way analysis of variance.
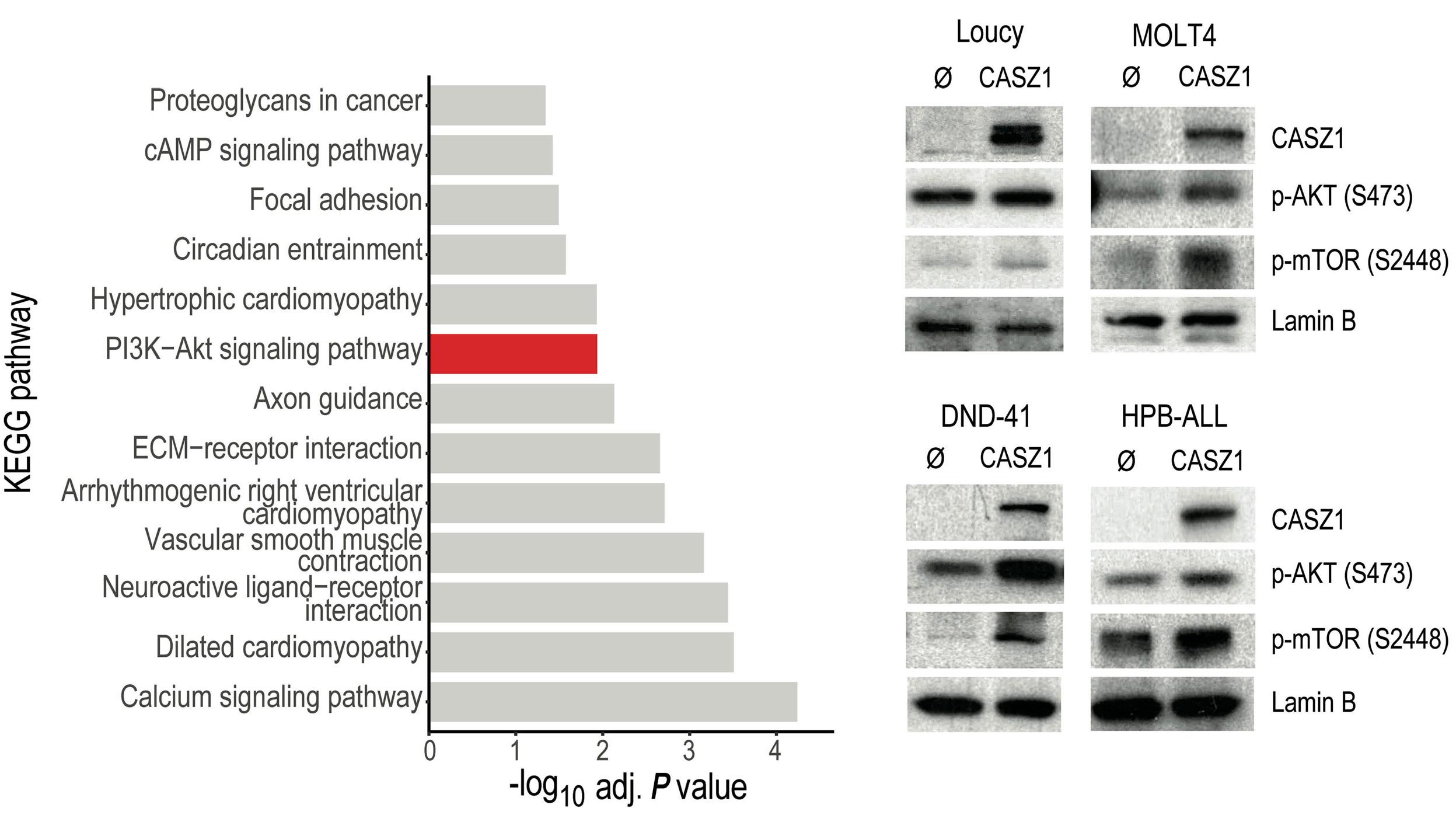
Figure 4. CASZ1 overexpression activates PI3K-AKT-mTOR signaling in T-cell acute lymphoblastic leukemia. (A) KEGG pathway analysis of the genes whose expression levels display a correlation coefficient of at least 0.7 with CASZ1 expression in a cohort of 14 T-cell acute lymphoblastic leukemia (T-ALL) patient samples (GEO database, accession number GSE18497). (B) T-ALL cell lines without (Ø) or with CASZ1 overexpression were assessed for PI3K signaling activation by immunoblot detection of the phosphorylation levels of AKT and mTOR. Total CASZ1 levels were also examined. Lamin B was used as loading control. Data are representative of 2 independent experiments.
of the PI3K-AKT-mTOR pathway inhibitor was specific, as shown by the downregulation of S6 phosphorylation levels (Figure 5E, F). Taken together, these results clearly indicate that CASZ1 promotes in vivo tumorigenesis by activating PI3K-AKT-mTOR signaling.
To determine whether CASZ1 promotes tumorigenesis in the context of T-ALL, we next tested whether CASZ1 accelerates NOTCH-induced T-cell leukemia in zebrafish.39,40 We generated mosaic zebrafish lines expressing intracellular NOTCH1 (ICN1) and/or CASZ1. Although with long latency, we found that CASZ1 accelerated the development of thymic hyperplasia with subsequent invasion of adjacent tissues (Figure 6A) leading to leukemia/lymphoma development in six of 20 animals expressing CASZ1 and ICN1 (starting at day 77, median survival: 259 days), as opposed to only one of 41 animals expressing ICN1 alone that developed leukemia by day 190 (median survival not reached during the duration of the experiment; Figure 6B). The lymphoid neoplastic cells from CASZ1+ICN1 zebrafish displayed a typical blast morphology, which contrasted to the cells collected from the vast majority of the ICN1 animals (Figure 6C). As expected, leukemia/lymphoma cells were of T-cell origin, as determined by the expression of CD4, CD8 and LCK, and the absence of B-cell markers such as PAX5, IGM or CD79a (Figure 6D). Finally, malignant cells in CASZ1+ICN1 zebrafish displayed decreased apoptosis
(Figure 6E, F) compared to control cells in ICN1 zebrafish. Altogether, our results indicate that CASZ1 can have a tumorigenic effect in lymphoid cells and cooperate with ICN1 to promote T-cell leukemia in vivo.
CASZ1 protects human T-cell acute lymphoblastic leukemia cells from death triggered by serum starvation and chemotherapy
To investigate the biological impact of CASZ1 in human T-ALL, we tried to silence CASZ1 in T-ALL cell lines. However, we were able to do so only mildly in Jurkat cells, after transfection with small interfering RNA (siRNA) for CASZ1. Although CASZ1 knock down was only around 25-30% (Online Supplementary Figure S10A ), there was a tendency for decreased viability in cells with lower CASZ1 levels (Online Supplementary Figure S10B) and a statistically significant negative impact on proliferation (Online Supplementary Figure S10C ). We then ectopically expressed CASZ1 in several T-ALL cell lines using lentiviral transduction ( Online Supplementary Figure S11A ). Overexpression of CASZ1 had no impact on T-ALL cell viability (Online Supplementary Figure S11B) or proliferation (Online Supplementary Figure S11C ). Basal viability and growth rate of T-ALL cell lines in regular culture conditions are high. This may mask positive effects of some oncogenes. For instance, in vitro prosurvival effects of TAL1 in T-ALL
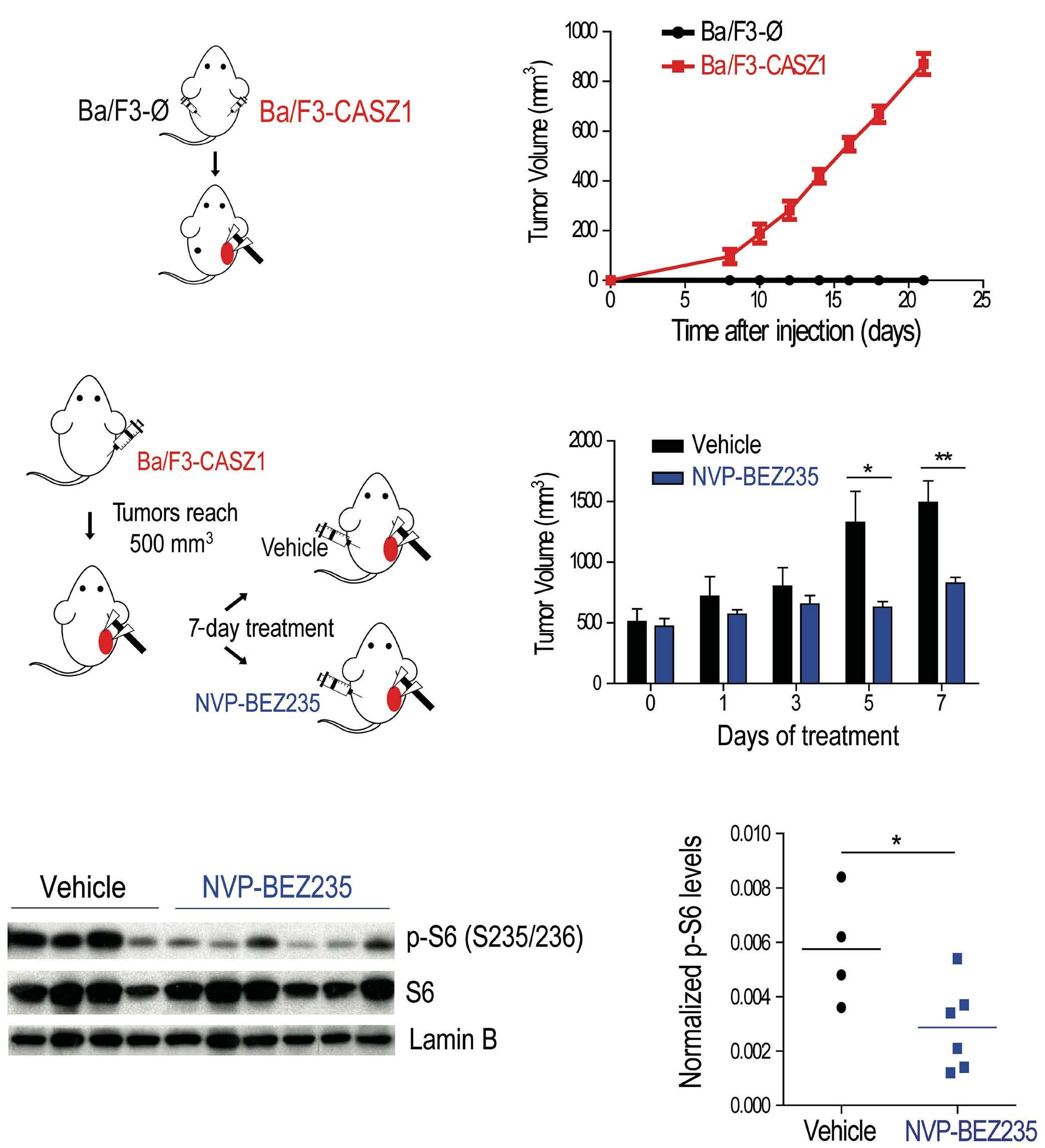
Figure 5. CASZ1 overexpression in Ba/F3 cells drives in vivo tumorigenesis by activating the PI3K-AKT-mTOR signaling pathway. (A, B) Eight-week old NOD/SCID mice (N=10) were injected subcutaneously with 107 Ba/F3-Ø or Ba/F3-CASZ1 cells and tumor growth was measured as described in the methods. (A) Schematic representation of the experimental layout. (B) Longitudinal analysis of tumor growth. (C-F) Eight-week-old NSG mice were injected subcutaneously with 107 Ba/F3-CASZ1 cells. Once the tumors reached 500 mm3, mice were randomized to receive either NVP-BEZ235 (N=6) or vehicle (N=5) for 7 consecutive days. (C) Schematic representation of the experimental layout. (D) Longitudinal analysis of tumor growth upon treatment initiation. * P<0.05; **P<0.01, one-way analysis of variance. (E) Vehicle- or NVP-BEZ235-treated tumors were collected and phosphorylation and total levels of S6 assessed by immunoblot analysis. Lamin B was used as loading control. (F) S6 phosphorylation levels in (E) were measured by densitometry analysis, and then normalized to total S6 and Lamin B levels. *P<0.05; **P<0.01, student’s t test.
cells were exposed only upon demonstration that TAL1 protected leukemia cells from stress-induced apoptosis. 41
Similar to TAL1, CASZ1 overexpression protected T-ALL cells from serum starvation-induced apoptosis (Figure 7A). Moreover, CASZ1 conferred resistance to treatment with daunorubicin, dexamethasone and L-asparaginase (Figure 7B), chemotherapeutic drugs currently used to treat patients with T-ALL.
Based on our findings that CASZ1 overexpression could rescue T-ALL cell viability under stress conditions and
contribute to in vitro resistance to chemotherapeutic drugs commonly used in the treatment of T-ALL patients, we hypothesized that high expression of CASZ1 might correlate with unfavorable outcome. However, we did not observe any significant association between CASZ1 expression levels and relapse-free or overall survival in children with newly diagnosed T-ALL enrolled in the St. Jude Children’s Research Hospital Total Therapy 15 and 16 studies (Online Supplementary Figure S12A, B), even when specifically examining relapse cases (Online Supplementary Figure S12C).
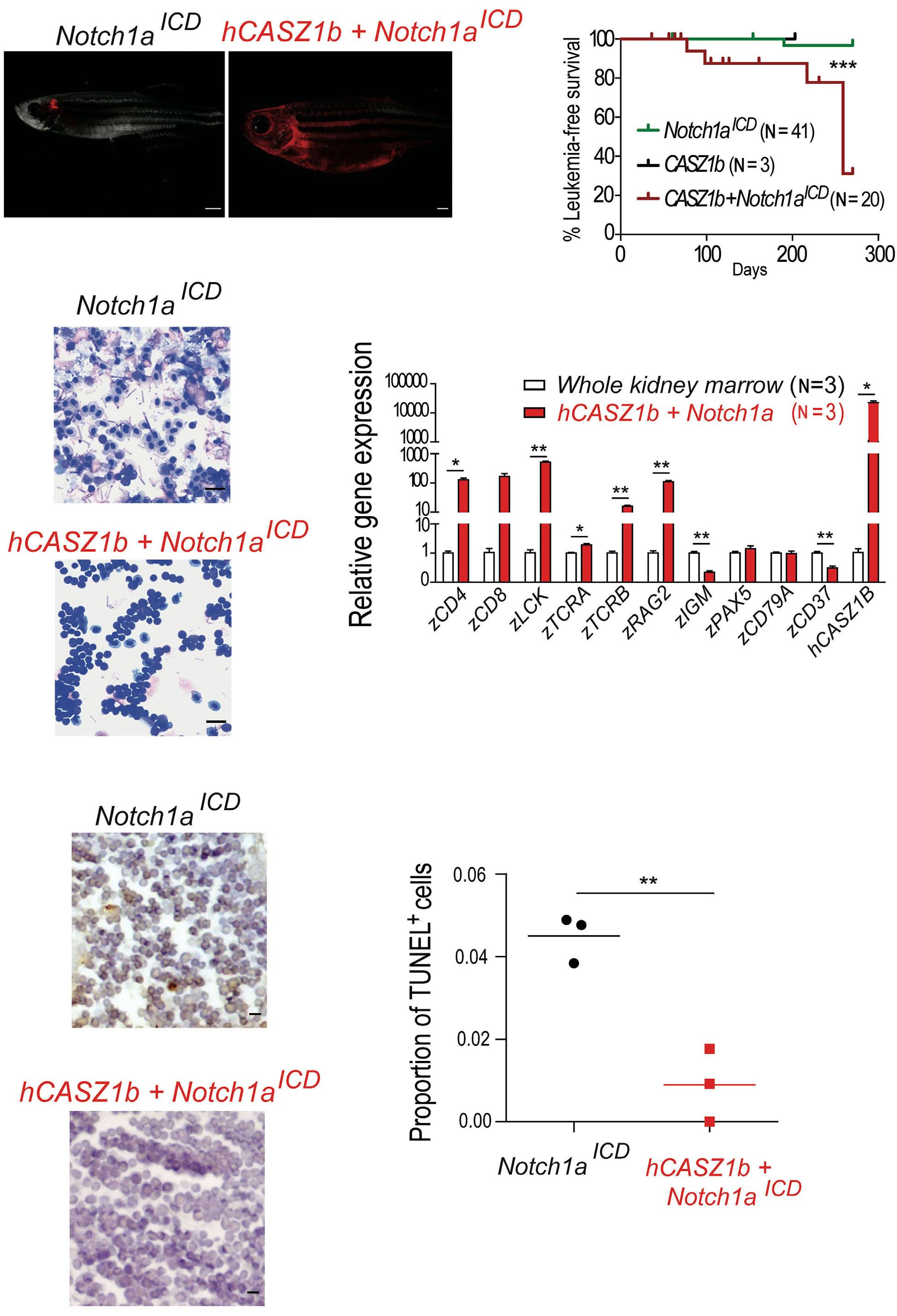
Nevertheless, in another cohort comprising exclusively of relapse T-ALL patients (GSE18497), for which gene expression and clinical parameters are publicly available,36 high CASZ1 levels associated with particularly poor prognosis, as the CASZ1-high group displayed faster progression to disease relapse and decreased overall survival (Online Supplementary Figure S12D-F). Interestingly, in agreement with our findings that TAL1 regulates CASZ1 in T-ALL, TAL1 was also associated with poor prognosis in the GSE18497 dataset (Online Supplementary Figure S12G-I). Overall,
Figure 6. CASZ1 cooperates with NOTCH1 to drive T-cell leukemogenesis in transgenic zebrafish. Tu/AB strain embryos were injected at one cell stage with rag2:notch1aICD, rag2:hCASZ1b or a mixture of both. (A) Images representative of mosaic transgenic zebrafish at 77 days post injection. (B) Kaplan-Meier analysis of leukemic fish comparing Notch1aICD (N=15), hCASZ1b (N=3, sacrificed at 203 days without evidence of disease) and hCASZ1b + Notch1aICD (N=20). ***P<0.001, log-rank (Mantel-Cox) test. (C) May-Grünwald and Wright-Giemsa stained cytospins showing lymphoblast morphology; 400x magnification. Images are representative of at least 3 independent fish analyzed of each genotype up to 100 days post injection. Scale bar equals 10 µm. (D) Transcript levels of the indicated genes were determined by quantitative polymerase chain reaction (qPCR) and normalized to β-actin. The values were further normalized to the control condition (whole kidney marrow) and represent the mean ± standard deviation of at least 2 independent replicates. (E) Representative hematoxylin- and eosin-stained histological sections juxtaposed to immunohistochemistry for TUNEL. Scale bar equals 10 μm. (F) Values are the quantification of the immunohistochemistry data represented in (E). * P<0.05; **P<0.01, student’s t test.
these analyses suggest that CASZ1 is not an independent prognostic factor in T-ALL, although it may associate with poor prognosis in relapse T-ALL depending on the specific treatment protocol.
CASZ1 is a conserved zinc finger transcription factor that is essential for heart and neural development.20,22-25,28,29 Not-
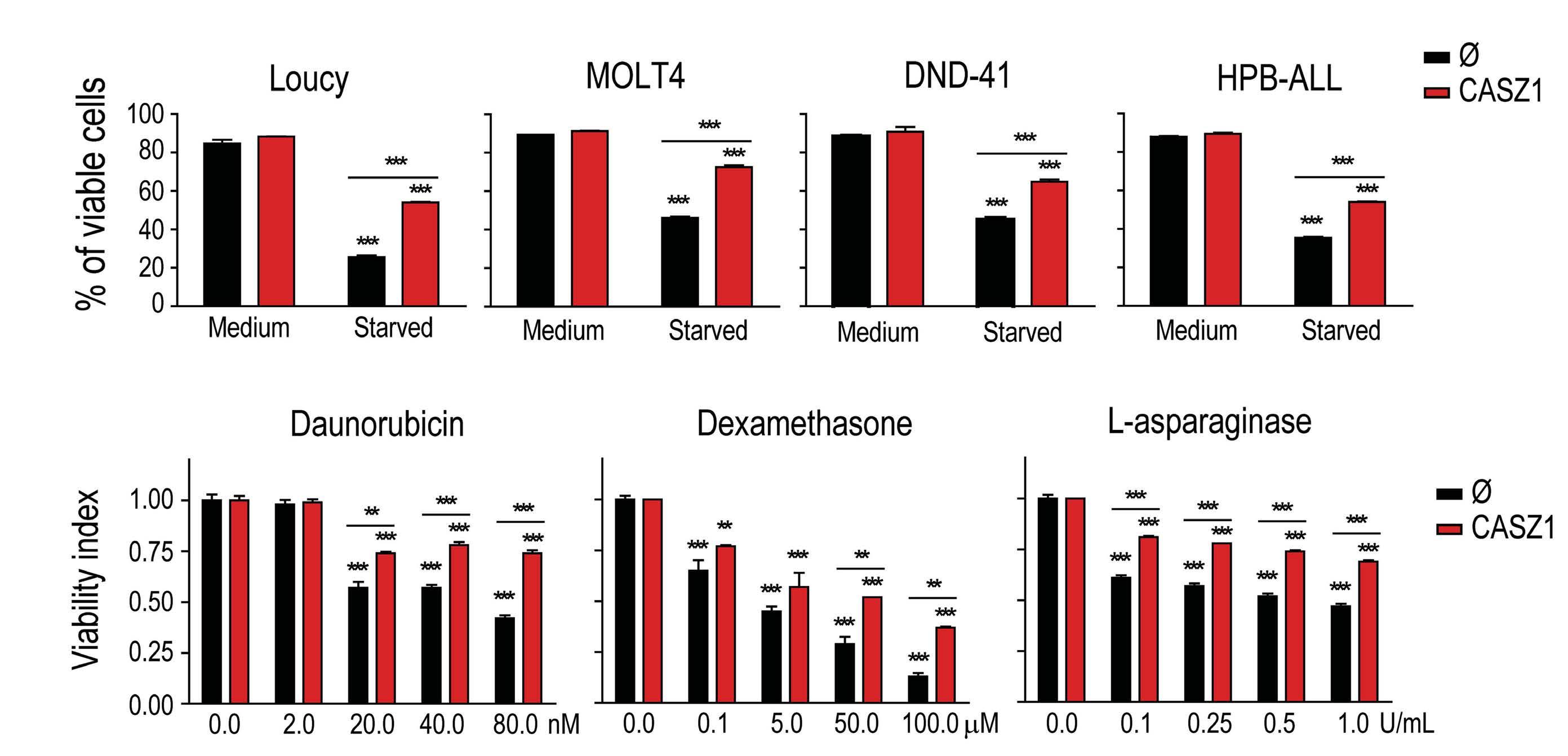
Figure 7. CASZ1 overexpression promotes T-cell acute lymphoblastic leukemia resistance to chemotherapy. (A) T-cell acute lymphoblastic leukemia (T-ALL) cells without (Ø) or with CASZ1 overexpression were cultured in regular culture conditions (medium) or under serum starvation (starved) for 48 hours [h] (Loucy), 72 h (MOLT4) or 144 h (DND-41 and HPB-ALL). (B) Loucy cells were cultured with the indicated concentrations of daunorubicin (72h), dexamethasone (72 h) and L-asparaginase (24 h). (A, B) Viability was determined, and the indicated values represent the mean ± standard deviation of at least 3 independent experiments *P<0.05; **P<0.01; ***P<0.001, student’s t test.
withstanding its pivotal role in embryogenesis, CASZ1 has been also implicated in cancer, either by acting as tumor suppressor in neuroblastoma,28,29 embryonal rhabdomyosarcoma31 and hepatocellular carcinoma30 or by promoting tumor metastasis in an ovarian cancer model.32 Here, we demonstrate that CASZ1 is overexpressed and acts in an oncogenic manner in T-ALL.
The transcription factor TAL1 is a major oncogene in the context of T-ALL,42 being overexpressed in a large portion of the patients and defining one of the subgroups of the disease.43 TAL1 transgenic mice develop aggressive T-cell leukemia albeit with long latency.44,45 Previous studies unveiled the transcriptional program associated with TAL1 in T-ALL.46-48 Here, we demonstrated that TAL1 can regulate CASZ1. However, analysis of publicly-available ChIP-seq data revealed that TAL1 binding to the CASZ1 locus in T-ALL cells is limited, with small and non-conserved peaks (Online Supplementary Figure S13), suggesting that TAL1 may have, at best, a mild direct regulatory effect on CASZ1. In addition, the negative impact of TAL1 genetic inactivation on CASZ1 expression varied in degree amongst the T-ALL cell lines and primary leukemia cells we analyzed. This, together with the fact that T-ALL samples in general (including TAL1-negative cases) displayed upregulation of CASZ1 as compared to normal thymocytes, suggests that additional factors should contribute to CASZ1 upregulation in T-ALL. Preliminary analyses of publicly-available data and treatment of T-ALL cell lines with small molecule inhibi-
tors did not find evidence for NOTCH1, MYC or EZH2 being consistent upstream regulators of CASZ1 (data not shown). Thus, the identification of CASZ1 upstream regulators in T-ALL, including transcription factors directly activating CASZ1, warrants further investigation.
The role of CASZ1 in hematopoietic differentiation remains unaddressed. However, our analyses of a mouse dataset from the BloodSpot database33 allowed us to trace CASZ1 expression throughout T-cell ontogeny. CASZ1 expression is low in developing and terminally differentiated T cells, paralleling that of TAL1 expression.49 Compatible with an oncogenic role for CASZ1 overexpression, CASZ1 levels are normally low in healthy T-cell progenitors, which are the targets for malignant transformation in T-ALL.1 In agreement, we showed that CASZ1 is able to cooperate with ICN1 to drive T-cell leukemia in zebrafish, by promoting the survival of T-ALL blasts (effects that are recapitulated upon CASZ1-mediated transformation of Ba/F3 cells). Interestingly, in established human leukemia, the prosurvival role of CASZ1 was exposed (reminiscent of what happens with TAL141) when T-ALL cells were cultured under stress conditions, either via serum starvation or treatment with chemotherapeutic drugs. This suggests that CASZ1 may protect, to some extent, against chemotherapy. In agreement with this assumption, we found that, among patients with relapsed T-ALL, high CASZ1 expression can associate with accelerated time to relapse and decreased overall survival. The PI3K-AKT-mTOR signaling pathway regulates multiple
cellular processes, including those that are associated with the tumorigenic process, such as viability, proliferation, migration and metabolism. The pivotal contribution of this signaling pathway to the malignant process in T-ALL has been established.5,8,50 In the present study, we demonstrated that CASZ1 expression activates PI3K-AKTmTOR signaling in both Ba/F3 cells and T-ALL cells, which is required for CASZ1-mediated cell transformation. These results demonstrate that one of the major molecular events elicited by CASZ1 expression in T-ALL is the activation of the PI3K-AKT-mTOR pathway. The mechanisms by which CASZ1 activates PI3K signaling in lymphoid cells warrant investigation. Our bioinformatics analyses of RNA-sequencing data also indicated that CASZ1 can activate JAK-STAT5 and MAPK pathways (Figure 5C). We confirmed these findings at the protein level (Online Supplementary Figures S14 and S15). Whereas we did not test the functional impact of JAK-STAT signaling downstream from CASZ1, we found that MAPK is required for in vitro transformation of Ba/F3 cells but dispensable for CASZ1-mediated expansion of Ba/ F3 cells in vivo (Online Supplementary Figure S16). Overall, our studies demonstrate that CASZ1 is upregulated in T-ALL (at least in part via TAL1 activity), protecting T-ALL cells from stress induced apoptosis and inducing the activation of the PI3K-AKT-mTOR signaling pathway, which is required for transformation in vitro and tumor growth in vivo. Our findings identify CASZ1 as a putative novel oncogene in T-ALL.
Disclosures
No conflicts of interest to disclose.
Contributions
BAC designed and performed experiments, analyzed and interpreted data, and drafted the manuscript. MD, AG, RF, MLO, JRA, LRM, NCC, AV, LD, FM and ABS designed, performed and interpreted experimental data. ARG supervised and performed bioinformatics analysis together with JLN. SK,
1. Girardi T, Vicente C, Cools J, De Keersmaecker K. The genetics and molecular biology of T-ALL. Blood. 2017;129(9):1113-1123.
2. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16(8):494-507.
3. Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269-271.
4 Clappier E, Cuccuini W, Kalota A, et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood. 2007;110(4):1251-1261.
SJ, CC, C-HP and CGM were involved in the collection and analysis of data on gene expression versus clinical outcome (St. Jude’s cohort). SFdA, PVV, JAY, DML and FP contributed to the design of the study with additional experimental suggestions. JTB designed the project structure, interpreted data, coordinated the studies and wrote the manuscript. All authors critically read and contributed to the final version of the manuscript.
We thank Dr Thiele for kindly providing the CASZ1 plasmids and Francisco Alexandrino, Hannah Taylor, Ana Sofia Moreira, Danyl Shatalov and Veronika Waas for their technical support. We also thank the mouse, zebrafish and flow cytometry core facilities of Instituto de Medicina Molecular João Lobo Antunes for their technical support.
This work was supported by the ERC-CoG-648455 and ERCPOC-101069429 grants from the European Research Council, under the European Union’s Horizon 2020 research and innovation program; PTDC/SAU-OBD/69974 grant from Fundação para a Ciência e a Tecnologia (FCT), Portugal; and a grant from Children with Leukemia (now Children with Cancer) Charity, UK (to JTB). The work was also supported by EXPL/ MEC-HEM/0571/2021 grant from FCT (to BAC), and grants R01CA211734 and MGH Scholars Award (to DML); and US NCI CA21765 grant to the Cancer center of St. Jude Children’s Research Hospital. ARG and RF were the recipients of FCT Investigator Grants (CEECIND/02699/2017 and CEECIND/03459/2018, respectively). JRA is the recipient of a Howard Hughes Medical Institute Hanna H. Gray Fellowship. ABS and JAY (301596/20174) received a fellowship from the Brazilian National Counsel of Technological and Scientific Development (CNPq).
Data-sharing statement
For original data that are not publicly deposited, please contact the corresponding author.
5. Gutierrez A, Sanda T, Grebliunaite R, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009;114(3):647-650.
6. Homminga I, Pieters R, Langerak AW, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19(4):484-497.
7 Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain-offunction mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43(10):932-939.
8. Silva A, Yunes JA, Cardoso BA, et al. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest. 2008;118(11):3762-3674.
9 Mansour MR, Sanda T, Lawton LN, et al. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J Exp Med. 2013;210(8):1545-1557.
10 Durinck K, Wallaert A, Van de Walle I, et al. The Notch driven long non-coding RNA repertoire in T-cell acute lymphoblastic leukemia. Haematologica. 2014;99(12):1808-1816.
11. Correia NC, Fragoso R, Carvalho T, Enguita FJ, Barata JT. MiR146b negatively regulates migration and delays progression of T-cell acute lymphoblastic leukemia. Sci Rep. 2016;6:31894.
12. Zhou Y, Han C, Wang E, et al. Posttranslational regulation of the exon skipping machinery controls aberrant splicing in leukemia. Cancer Discov. 2020;10(9):1388-1409.
13. Silva A, Laranjeira AB, Martins LR, et al. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. 2011;71(14):4780-4789.
14 Uzan B, Poglio S, Gerby B, et al. Interleukin-18 produced by bone marrow-derived stromal cells supports T-cell acute leukaemia progression. EMBO Mol Med. 2014;6(6):821-834.
15. Pitt LA, Tikhonova AN, Hu H, et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell. 2015;27(6):755-768.
16. Cardoso BA, de Almeida SF, Laranjeira AB, et al. TAL1/SCL is downregulated upon histone deacetylase inhibition in T-cell acute lymphoblastic leukemia cells. Leukemia. 2011;25(10):1578-1586.
17 Peirs S, Matthijssens F, Goossens S, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2014;124(25):3738-3747.
18. Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759-1767.
19 De Smedt R, Morscio J, Reunes L, et al. Targeting cytokine- and therapy-induced PIM1 activation in preclinical models of T-cell acute lymphoblastic leukemia and lymphoma. Blood. 2020;135(19):1685-1695.
20 Liu Z, Yang X, Tan F, Cullion K, Thiele CJ. Molecular cloning and characterization of human Castor, a novel human gene upregulated during cell differentiation. Biochem Biophys Res Commun. 2006;344(3):834-844.
21. Charpentier MS, Christine KS, Amin NM, et al. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev Cell. 2013;25(2):132-143.
22. Christine KS, Conlon FL. Vertebrate CASTOR is required for differentiation of cardiac precursor cells at the ventral midline. Dev Cell. 2008;14(4):616-623.
23. Dorr KM, Amin NM, Kuchenbrod LM, et al. Casz1 is required for cardiomyocyte G1-to-S phase progression during mammalian cardiac development. Development. 2015;142(11):2037-2047.
24. Huang RT, Xue S, Wang J, et al. CASZ1 loss-of-function mutation associated with congenital heart disease. Gene. 2016;595(1):62-68.
25. Sojka S, Amin NM, Gibbs D, Christine KS, Charpentier MS, Conlon FL. Congenital heart disease protein 5 associates with CASZ1 to maintain myocardial tissue integrity. Development. 2014;141(15):3040-3049.
26. Mellerick DM, Kassis JA, Zhang SD, Odenwald WF. castor encodes a novel zinc finger protein required for the development of a subset of CNS neurons in Drosophila. Neuron. 1992;9(5):789-803.
27. Isshiki T, Pearson B, Holbrook S, Doe CQ. Drosophila neuroblasts
sequentially express transcription factors which specify the temporal identity of their neuronal progeny. Cell. 2001;106(4):511-521.
28. Liu Z, Yang X, Li Z, et al. CASZ1, a candidate tumor-suppressor gene, suppresses neuroblastoma tumor growth through reprogramming gene expression. Cell Death Differ. 2011;18(7):1174-1183.
29 Liu Z, Naranjo A, Thiele CJ. CASZ1b, the short isoform of CASZ1 gene, coexpresses with CASZ1a during neurogenesis and suppresses neuroblastoma cell growth. PLoS One. 2011;6(4):e18557.
30 Wang JL, Yang MY, Xiao S, Sun B, Li YM, Yang LY. Downregulation of castor zinc finger 1 predicts poor prognosis and facilitates hepatocellular carcinoma progression via MAPK/ERK signaling. J Exp Clin Cancer Res. 2018;37(1):45.
31. Liu Z, Zhang X, Lei H, et al. CASZ1 induces skeletal muscle and rhabdomyosarcoma differentiation through a feed-forward loop with MYOD and MYOG. Nat Commun. 2020;11(1):911.
32. Wu YY, Chang CL, Chuang YJ, et al. CASZ1 is a novel promoter of metastasis in ovarian cancer. Am J Cancer Res. 2016;6(6):1253-1270.
33. Bagger FO, Sasivarevic D, Sohi SH, et al. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2016;44(D1):D917-D924.
34 Van Vlierberghe P, Ambesi-Impiombato A, Perez-Garcia A, et al. ETV6 mutations in early immature human T cell leukemias. J Exp Med. 2011;208(13):2571-2579.
35. Borssen M, Palmqvist L, Karrman K, et al. Promoter DNA methylation pattern identifies prognostic subgroups in childhood T-cell acute lymphoblastic leukemia. PLoS One. 2013;8(6):e65373.
36. Staal FJ, de Ridder D, Szczepanski T, et al. Genome-wide expression analysis of paired diagnosis-relapse samples in ALL indicates involvement of pathways related to DNA replication, cell cycle and DNA repair, independent of immune phenotype. Leukemia. 2010;24(3):491-499.
37. Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med. 2004;200(5):659-669.
38. Seront E, Rottey S, Filleul B, et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma. BJU Int. 2016;118(3):408-415.
39 Blackburn JS, Liu S, Raiser DM, et al. Notch signaling expands a pre-malignant pool of T-cell acute lymphoblastic leukemia clones without affecting leukemia-propagating cell frequency. Leukemia. 2012;26(9):2069-2078.
40 Chen J, Jette C, Kanki JP, Aster JC, Look AT, Griffin JD. NOTCH1induced T-cell leukemia in transgenic zebrafish. Leukemia. 2007;21(3):462-471.
41. Bernard M, Delabesse E, Novault S, Hermine O, Macintyre EA. Antiapoptotic effect of ectopic TAL1/SCL expression in a human leukemic T-cell line. Cancer Res. 1998;58(12):2680-2687.
42. Correia NC, Arcangeli ML, Pflumio F, Barata JT. Stem cell leukemia: how a TALented actor can go awry on the hematopoietic stage. Leukemia. 2016;30(10):1968-1978.
43. Ferrando AA, Neuberg DS, Staunton J, et al. Gene expression signatures define novel oncogenic pathways in T cell acute
lymphoblastic leukemia. Cancer Cell. 2002;1(1):75-87.
44 O’Neil J, Shank J, Cusson N, Murre C, Kelliher M. TAL1/SCL induces leukemia by inhibiting the transcriptional activity of E47/HEB. Cancer Cell. 2004;5(6):587-596.
45. Kelliher MA, Seldin DC, Leder P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. Embo J. 1996;15(19):5160-5166.
46. Sanda T, Lawton LN, Barrasa MI, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22(2):209-221.
47. Palomero T, Odom DT, O’Neil J, et al. Transcriptional regulatory networks downstream of TAL1/SCL in T-cell acute
lymphoblastic leukemia. Blood. 2006;108(3):986-992.
48. Kusy S, Gerby B, Goardon N, et al. NKX3.1 is a direct TAL1 target gene that mediates proliferation of TAL1-expressing human T cell acute lymphoblastic leukemia. J Exp Med. 2010;207(10):2141-2156.
49. Herblot S, Steff AM, Hugo P, Aplan PD, Hoang T. SCL and LMO1 alter thymocyte differentiation: inhibition of E2A-HEB function and pre-T alpha chain expression. Nat Immunol. 2000;1(2):138-144.
50 Palomero T, Sulis ML, Cortina M, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13(10):1203-1210.
1Cytokines and Immunity Section, Cancer Innovation Laboratory (CIL) and 2CCR Collaborative Bioinformatics Resource (CCBR), National Cancer Institute (NCI), National Institutes of Health (NIH), Frederick, MD, USA
*HW and WL contributed equally as first authors.
Correspondence: S. Durum durums@mail.nih.gov
Received: May 25, 2023. Accepted: November 22, 2023. Early view: November 30, 2023. https://doi.org/10.3324/haematol.2023.283559 ©2024 NIH (National Institutes of Health)
Abstract
Acute lymphoblastic leukemia (ALL) is an aggressive leukemia which can be derived from either T-cell or B-cell precursors. With current treatments, the survival rate is high, but the treatments are highly toxic with severe side effects. Individual mutations in IL7Ra and RAS pathways have been previously shown to be prevalent in ALL, and especially in relapsed patients. The relationship of IL-7Ra and RAS was investigated by transducing immature mouse thymocytes with the combination of these mutants. The resultant ALL cells were analyzed to identify the regulators and the oncoproteins that are up-regulated or down-regulated by the combination of IL7Ra with NRAS. Leukemia cells showed a significant increase in IL7Ra-mediated BCL2 expression, and an increase in MYC protein levels was mainly induced by NRAS signaling. MYC was both necessary and sufficient to replace mutant NRAS, and drugs targeting the MYC pathway showed a therapeutic benefit in IL-7Ra/NRAS T-ALL. We suggest that MYC protein stability can be regulated by PLK-1 kinase, which was increased mainly by the NRAS signal. These studies identify novel pathways of oncogenesis and new targets for intervention that could lead to better therapeutic development.
Acute lymphoblastic leukemia (ALL) is the most common cancer in children. Current treatments have 5-year overall survival (OS) rates of up to 90%; however, the treatments are highly toxic with severe side effects.1 In addition, relapse is relatively common in T-ALL and is exceptionally difficult to salvage with hematopoietic stem cell transplantation, which is essentially the only treatment.2,3 Thus, a better understanding of T-ALL etiology and subtypes is required to develop more targeted therapies. IL7Ra, one of the two IL7R subunits, is essential for T-cell development and survival, and mutations in the receptor itself or components of its signal pathways can lead to ALL.4-6 Somatic gain of function mutations in IL7R a usually include insertion of cysteine into the receptor transmembrane region. This leads to disulfide bonds between two IL7Ra subunits causing constitutive signaling independent of IL-7.7,8 Recent studies showed that mutations in IL7Ra alone are insufficient to cause T-ALL in mice and other mutations that co-operate with IL7Ra are needed.4,9,10 A
recent study in zebrafish11 did observe that mutant IL-7R transgenes were sufficient to induce T-ALL. However, the long latency of 20 weeks and oligoclonality led the authors to propose that additional events were required. Their results suggested one such event was MYC upregulation. RAS mutations in both B and T lineage-ALL occur in approximately 15% of patients, with NRAS being the most dominant.12 In early T-cell precursor ALL (ETP), mutations in the RAS signaling pathway are found in approximately 67% of cases, with 19% of them in the NRAS gene.12 Moreover, other studies showed that RAS pathway mutations account for 30-50% of cases of relapse in ALL.13 Therefore, we tested the effect of combining IL7Ra with NRAS. We had previously showed that mutant human NRAS (G13D) in combination with a somatic gain of function mutation in human IL-7Ra (c.731_732insTTGTCCCAC) caused severe T-ALL in mice compared to expression of each oncogene alone or empty vector.1 In this study, we focused on the signaling pathways and factors that are activated or inhibited when mutant NRAS and IL-7Ra are transduced into murine T-cell progenitors, causing T-ALL.
It was first shown in the 1990s that BCL2 can co-operate with MYC, which together induce transformation in vitro 14 Since then, many studies have showed the involvement of these two oncoproteins in different cancer types, both in humans and in murine models.15,16 As mentioned above, a recent study showed that mutant IL7Ra collaborated with MYC to induce T-ALL in transgenic zebrafish; however, the mechanism that is responsible for increased MYC is unclear.11 Here, using a murine model, we shed light on the mechanism that regulates MYC in ALL, on the collaboration of MYC and BCL2, and the relative roles of mutated NRAS and IL-7Ra.
Cell culture and transduction
Thymuses were harvested from 3-6 week-old C57Bl/6J female mice, depleted for CD4+ CD8+ cells, processed and transduced as previously described.1 cDNA were subcloned into a pMIG or IRES-mCherry (Addgene) retroviral vector by PCR cloning. The hIL-7Ra mutant sequence was with in-frame insertion of c.731_732insTTGTCCCAC, based on subject P2.7 Mutant human NRAS contained a point mutation (G13D). Human MYC was from pcDNA3-MYC (Addgene). Phoenix cells were transfected with pCL-Eco (Addgene) helper plasmid and the retroviral vector pMIG carrying either mutant hIL7Ra, mutant NRAS, or the combination of both mutIL7Ra-T2A-mutNRAS or IRES-mCherry containing MYC or Cre. Transfection was performed using lipofectamine and OPTI-MEM I reduced serum medium (Invitrogen). After transduction, thymocytes were maintained on OP9-DL4 cells prior to injection or sequencing.
Animal experiments
Transduced thymocytes were injected into sub-lethally irradiated (3 Gy), 6-15 week-old Rag1-/- (B6.129S7-Rag1tm1Mom/J) mice via tail vein (5x105 cells / 200 µL/ mouse). For studies combining mutNRAS or MYC, the experimental groups and the numbers of animals in each group were: mutIL-7RaT2A-mutNRAS (N=7), mutIL7Ra (N=7), MYC (N=7), MYC+ mutIL-7Ra (N=7). MYC floxed mice were kindly provided by the Susan Mackem lab (NCI). C57 thymocytes or MYC-floxed thymocytes were transduced with Cre (mCherry) and mutIL-7Ra-T2A-mutNRAS (GFP) (N=3 for each group). Twenty-four hours after transduction, thymocytes were sorted on BD FACSAria II for mCherry+/ GFP+ thymocytes and immediately injected intravenously into RAG1-/- mice, which were monitored for up to 35 days. Peripheral blood was collected weekly (submandibular) for leukemic burden assessment, and mice showing signs of morbidity were euthanized and harvested for spleen and blood.
For drug studies, mutIL-7Ra-T2A-mutNRAS transduced thymocytes were injected into RAG1-/- mice as described
above. Twenty-four hours after injection, mice were treated with: JQ-1 (Selleckchem) administered intraperitoneally (i.p.) at 25 mg/kg (N=7), daily for 21 days, for controls (no treatment) 2.5% DMSO + 30% PEG300 + 5% TWEEN-80 + dH2O was injected into mice (N=7). Volasertib (BI6727Selleckchem) was administered at 10 mg/kg (N=6), i.p. daily for 21 days, for controls (no treatment) mice were injected with 2% DMSO in PBS (N=6). For the combination of volasertib and venetoclax (ABT-199-; DC chemicals), mice were treated with either control (no treatment) with one injection with 2% DMSO in PBS followed three hours (hr) later with an additional injection of 5% DMSO + 50% PEG300 + 5% Tween-80 + dH2O, or with volasertib alone (10 mg/kg), venetoclax alone (15 mg/kg) or the combination of both drugs in separate injections. All drugs were injected i.p. five days per week.
All animal experimental procedures were conducted according to the US NIH guidelines for the Care and Use of Laboratory Animals, and were approved by the NCI Animal Care and Use Committee.
Statistical analysis was performed using GraphPad Prism 7. Differences were calculated with unpaired Student t test or one-way ANOVA for multiple comparisons. P<0.05 was considered statistically significant. Error bars are Standard Deviation.
A detailed description of other methods used is provided in the Online Supplementary Appendix
Identifying the molecular pathways that lead to T-ALL caused by mutIL7Ra and mutNRAS
Previously our lab showed that an IL7Ra gain of function mutation is not sufficient to cause T-ALL.1 By combining different known mutations that are common in ALL, Cramer et al. found that combination of mutant human IL7Ra (P7=c.731_732insTTGTCCCAC) with mutant human NRAS (G13D) can lead to severe T-ALL in mice.1 In addition to these two specific mutations, we also tested combination of mutIL7R (P7) with mutKRAS (G12D or G12V) and mutIL7R (P1= c.726_727insAACCCATGC) with mutNRAS(G13D), and found that all gave leukemia in mice, indicated by an increase in GFP percentage in the spleen, marking leukemic cells (Online Supplementary Figure S1A). We found that combining mutIL7R(P7) and mutNRAS (G13D) gave the strongest leukemia, making it easier to study and identify the molecular and signaling pathways, and explain why combining these two oncogenes leads to ALL. To answer this question, RNA sequencing and mass spectrometry were performed on double negative primary immature mouse thymocytes, transduced with either: empty vector, mutant human IL7Ra, mutant human NRAS, or both oncogenes combined
on a single bicistronic vector (Figure 1A). RNA sequencing and differential expression of genes (DEG) analysis highlighted the differences between the four groups (Online Supplementary Figure S1B). Gene set enrichment analysis (GSEA) revealed significant changes in gene sets expressed in thymocytes that contain both mutations compared to each mutation alone or empty vector. The top six significant HALLMARK pathways as determined by GSEA are shown in enrichment plots comparing: both mutations to empty vector (Figure 1B), both mutations to mutant IL7R alone (Online Supplementary Figure S1C), and both mutations to mutant NRAS alone (Online Supplementary Figure S1D).
One of the pathways that is predicted to be activated in all three comparisons in the presence of both mutations was the HALLMARK_MYC_TARGETS_V2 pathway (Figure 1B, Online Supplementary Figure S1C, D). On the other hand, one of the pathways that was predicted to be significantly deactivated in the presence of both mutations was in the HALLMARK_APOPTOSIS pathway (Figure 1B). However, as most apoptotic-related genes are regulated in protein level, we also confirmed our results by western blot (Figure 2A).
Heatmap of the HALLMARK_MYC_TARGETS_V2 GSEA leading edge genes reveals an increase in the MYC positive regulator, PLK1, but only in the presence of both mutations (Figure 3). Finally, combining RNA sequencing with mass spectrometry results using a Venn diagram showed an increase in BCL2 gene and protein levels that are expressed in thymocytes containing both mutations, compared to empty vector (Online Supplementary Figure S1E).
Expression of mutIL7R and mutNRAS induces increased MYC and BCL2 protein and mRNA levels
Next, we validated the bioinformatic analysis by immunoblotting in both primary thymocytes and similarly transduced D1 thymocyte cell line. In primary thymocytes, MYC protein was significantly increased in the oncogene combination compared to mutIL7R alone or empty vector, and overexposure suggested that the MYC increase is mostly due to mutant NRAS (Figure 2A). The BCL2 protein level increased in mutIL7Ra alone and in thymocytes expressing both mutIL7Ra and mutNRAS. The pro-apoptotic protein BAK1 significantly decreased in the presence of both mutations (Figure 2A). The D1 cell line is IL-7-dependent for survival and growth in vitro. In the presence of IL-7, high levels of MYC protein were observed, and the levels declined dramatically after IL-7 withdrawal (Online Supplementary Figure S2A). In this cell line after IL-7 removal, either oncogene or the combination, maintained MYC levels, albeit not up to the same level as in the presence of IL-7. Moreover, we observed an increase in PIM-1, a known oncoprotein which is regulated by the IL7R pathway.17 Both the short and the long forms of PIM-1 were significantly higher in the presence of both mutations compared to each mutation alone or empty vector (Online Supplementary Figure S2A). In addition, the pro-apoptotic protein BAD phosphorylation
(inhibitory form) was increased mainly with both mutations, consistent with an anti-apoptotic fate when both IL7Ra and NRAS mutations are present (Online Supplementary Figure S2A). To determine whether MYC and BCL2 were controlled at the transcription, transitional or protein level, we first tested mRNA levels of MYC, BCL2, and BAK1. MYC and BAK1 mRNA levels did not show any major changes. On the other hand, BCL2 mRNA increased 5- and 3-fold with mutIL7Ra alone and in the presence of both oncogenes, respectively (Figure 2B). This supports our bioinformatic analysis in addition to other studies that showed BCL2 transcription is up-regulated by the IL7R pathway.18-20 To further investigate the increase in MYC protein, we used MK-2206, an AKT inhibitor, which is also known to inhibit MYC translation.21,22 We did not find any significant change in MYC protein levels in either primary thymocytes or D1 cell line transduced with mutIL7R and mutNRAS (Online Supplementary Figure S2B). This suggests that, in this scenario, MYC increased by increasing its stability, a wellknown MYC regulatory mechanism.
We conclude that in the presence of both mutant IL7Ra and NRAS, MYC stability is regulated at the protein level and BCL2 is increased at the transcriptional level. Since the combination of MYC and BCL2 is known to be highly oncogenic in many cancer types, including different types of leukemia and lymphoma,14,16,23 we suggest this combination is also responsible for the severity of T-ALL caused by mutations in IL7Ra and NRAS.
MYC overexpression can replace mutant NRAS in combination with mutant IL7Ra to create T-ALL in mice
To evaluate whether the mutant NRAS contribution to leukemogenesis is explained by stabilization of MYC, we tested whether overexpression of MYC was sufficient to replace mutant NRAS. Double negative thymocytes were transduced with mutant IL-7Ra/GFP together with MYC/ mCherry, injected into RAG1-/- mice and observed for leukemia progression (Online Supplementary Figure S3A). Disease appeared approximately 16 days after injection only in the group receiving both mutIL7Ra and MYC over-expressing thymocytes, displaying increased liver and spleen size (Figure 4A, Online Supplementary Figure S3B) and increased white blood count (WBC) in both blood and spleen, all indicators for leukemia (Figure 4B). To characterize the leukemia, we first evaluated the expression of GFP and mCherry thymocytes in the blood and observed a significant increase in cells that contained both mutIL7Ra and MYC compared to controls (Online Supplementary Figure S3C). Interestingly, cells positive for both mutations were mostly Thy 1.2, CD4, and CD8 positive, marking intermediate stage thymocytes (Figure 4C). Finally, we observed that the mutIL7Ra/MYC leukemia was transferrable by injection into recipient mice, generating a leukemia of similar aggressiveness and phenotype (data not shown). Overall, we conclude that the combination of mutIL7Ra and MYC
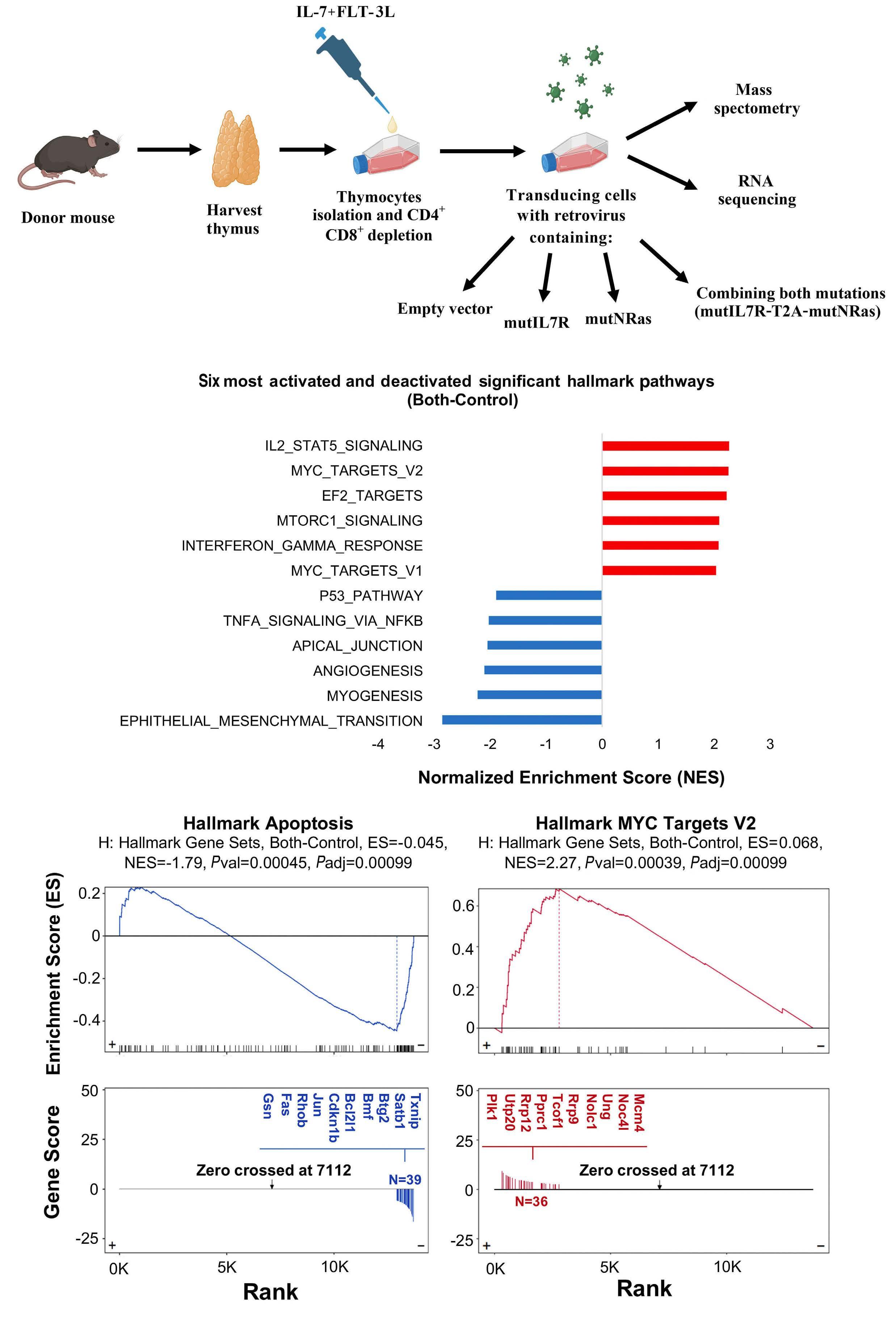
Continued on following page.
Figure 1. Identifying the molecular pathway that leads to acute lymphoblastic leukemia caused by mutIL7R and mutNRAS. (A) Schematic diagram describing the experimental design for conducting mass spectrometry and RNA sequencing. (B) (Top) Bar chart showing the top 6 GSEA Hallmark pathways in mutIL7R + mutNRAS relative to control empty vector. The x-axis shows normalized enrichment scores, with activated pathways given red bars and deactivated pathways in blue. (Bottom) GSEA enrichment plots for both the deactivated HALLMARK_APOPTOSIS pathway and the activated HALLMARK_MYC_TARGETS_V2 pathway. Both P values (Pval) and adjusted P values (Padj) are shown.

Figure 2. Expression of mutIL7R and mutNRAS lead to changes in MYC protein level and BCL-2 protein and mRNA level. (A) Primary thymocytes transduced with: empty vector, mutIL7R, mutNRAS or combination of both mutations (Both), 72 hours from transduction cells were lysed and analyzed by western blot. (This was repeated with at least 3 biological experiments). (B) RT-PCR analysis of mRNA from primary thymocytes transduced with the different mutations or empty vector tested for MYC, BCL2 and BAK1. Each point represents biological repeat, and each biological repeat was done in triplicates (N=3).
can lead to ALL, and that the leukemogenic contribution of mutant NRAS can be explained by its stabilization of MYC.
Acute lymphoblastic leukemia from mutant IL7R and NRAS has a similar phenotype to mutant IL7R and MYC
The two ALL combinations were compared to assess whether MYC plus mutIL-7R generated a phenotype similar to mutNRAS plus mutIL-7R. Double negative primary thymocytes were transduced with either mutIL7R a plus mutNRAS (encoded on a single plasmid together with GFP) ( Online Supplementary Figure S4A) or with mutIL7R a and over-expressed MYC (encoded on separate plasmids with GFP or mCherry expression, respectively) ( Online Supplementary Figure S4B ). Following transduction, thymocytes were injected into RAG1 -/- mice and monitored for leukemia progression by both flow cytometry and disease symptoms for approximately two weeks. In both combinations, the percentage of leukemic cells in the spleen, was around 90% ( Online Supplementary Figure S4 ), consistent with a similar growth rate. Leukemic cells in both models also resembled the CD4 + CD8 + developmental stage (Figure 5A). TCRV a expression in the
combinations were also similar between the models (Figure 5B) and the TCR a β lineage predominated over TCR γδ lineage in both models (Figure 5C). We conclude that MYC plus mutIL-7R a generated an ALL that was very similar to mutNRAS plus mutIL-7R a . This is consistent with the mutNRAS contribution being mediated via increased MYC stability.
Targeting MYC reduced acute lymphoblastic leukemia progression caused by mutIL7Ra and mutNRAS
To further assess the involvement of MYC in our ALL model, we used several approaches to determine if MYC was required. Volasertib (BI6727), a dihydropterinone that targets PLK1, has shown efficacy in several cancer clinical trials, and was previously shown to destabilize MYC protein by increasing MYC proteasomal degradation.15,21,24-27 Double negative thymocytes expressing both mutIL7Ra and mutNRAS were injected into RAG1-/- treated with 10 mg/kg volasertib (BI6727) for 21 days, starting 24 hr after injection, or injected with buffer alone as control (no treatment) (Online Supplementary Figure S5A). At the end of treatment, GFP+ leukemia cells were significantly decreased in mice treated with
volasertib, in both blood and spleen, compared to controls (Figure 6A). In addition, non-treated mice had significantly higher WBC in both blood and spleen, significant weight loss, and increased spleen weight compared to volasertib-treated mice (Figure 6B). MYC protein levels were significantly reduced in the presence of volasertib compared to control
mice (Online Supplementary Figure S5B). Our model predicts dependency on combined overexpression of MYC and BCL2 (see Figure 2). Therefore, we next treated leukemic RAG1-/mice with volasertib and venetoclax (a BCL2 inhibitor) separately and combined (Online Supplementary Figure S5C), possibly expecting an additive effect. The GFP percentage of
Figure 3. Heatmap of all GSEA leading edge genes from the HALLMARK_MYC_TARGETS_V2 gene. The heat is from row-centered and normalized expression z-scores, colored annotation bars along the top indicate treatment groups, and phylograms on the top and left side are the result of unsupervised clustering of samples and genes, respectively.
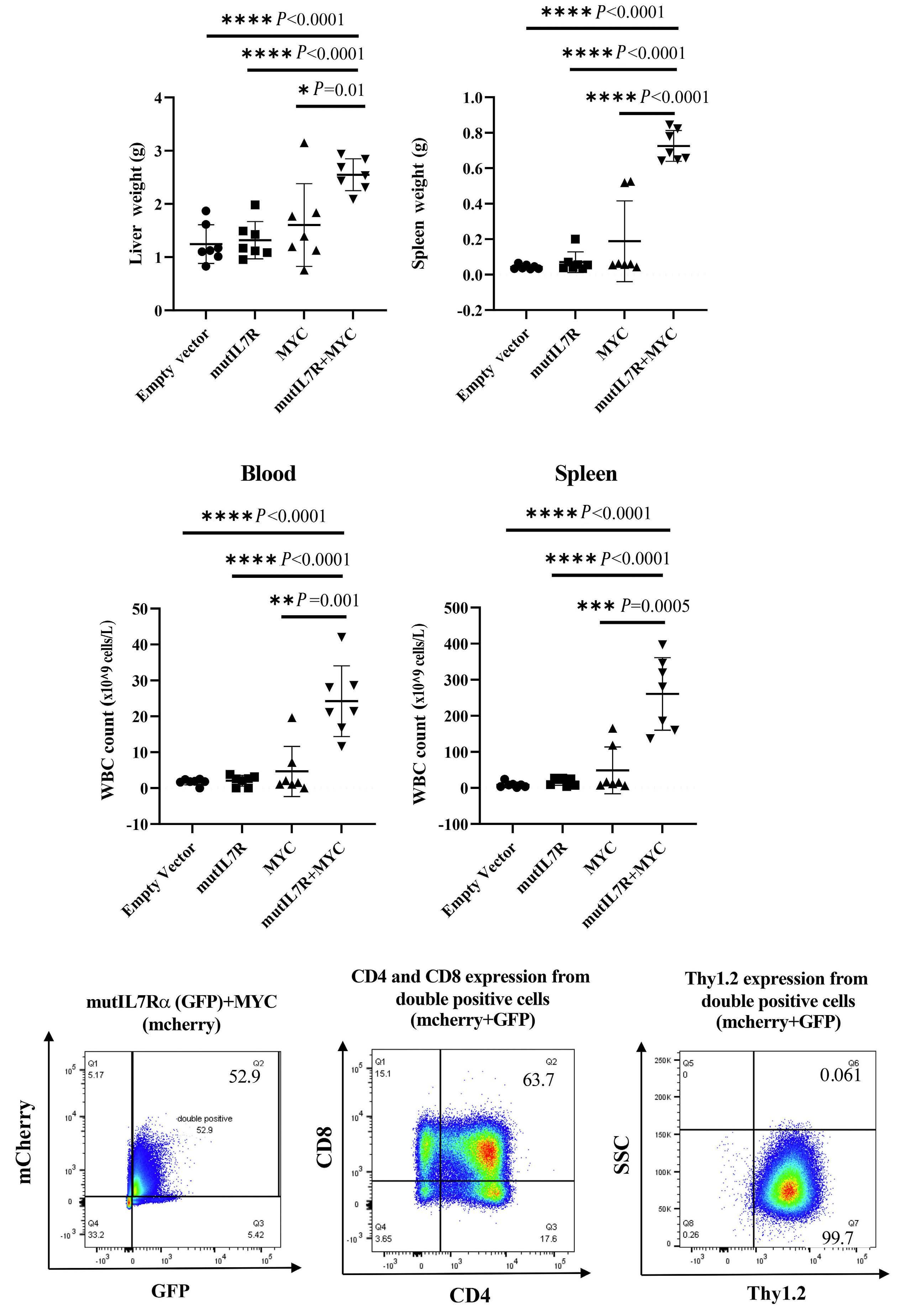
Figure 4. MYC replaces mutant NRAS in combination with mutant IL7Ra to create acute lymphoblastic leukemia in mice. RAG1-/- mice were injected with thymocytes transduced with either: empty vector, mutIL7R, MYC or combination of both IL7R and MYC. After 16 days from injection, mice (N=7) were harvested for liver, spleen, and blood, and analyzed as follows: (A) liver and spleen weights, (B) white blood cell (WBC) counts from both blood and spleen, (C) flow cytometry analysis from blood detecting, left to right: GFP and mCherry which represent the expression of mutIL7R and MYC respectively (double positive for cells express both), CD4 versus CD8 expression and Thy1.2 expression, that represent thymocytes marker, gated on double positive thymocytes.
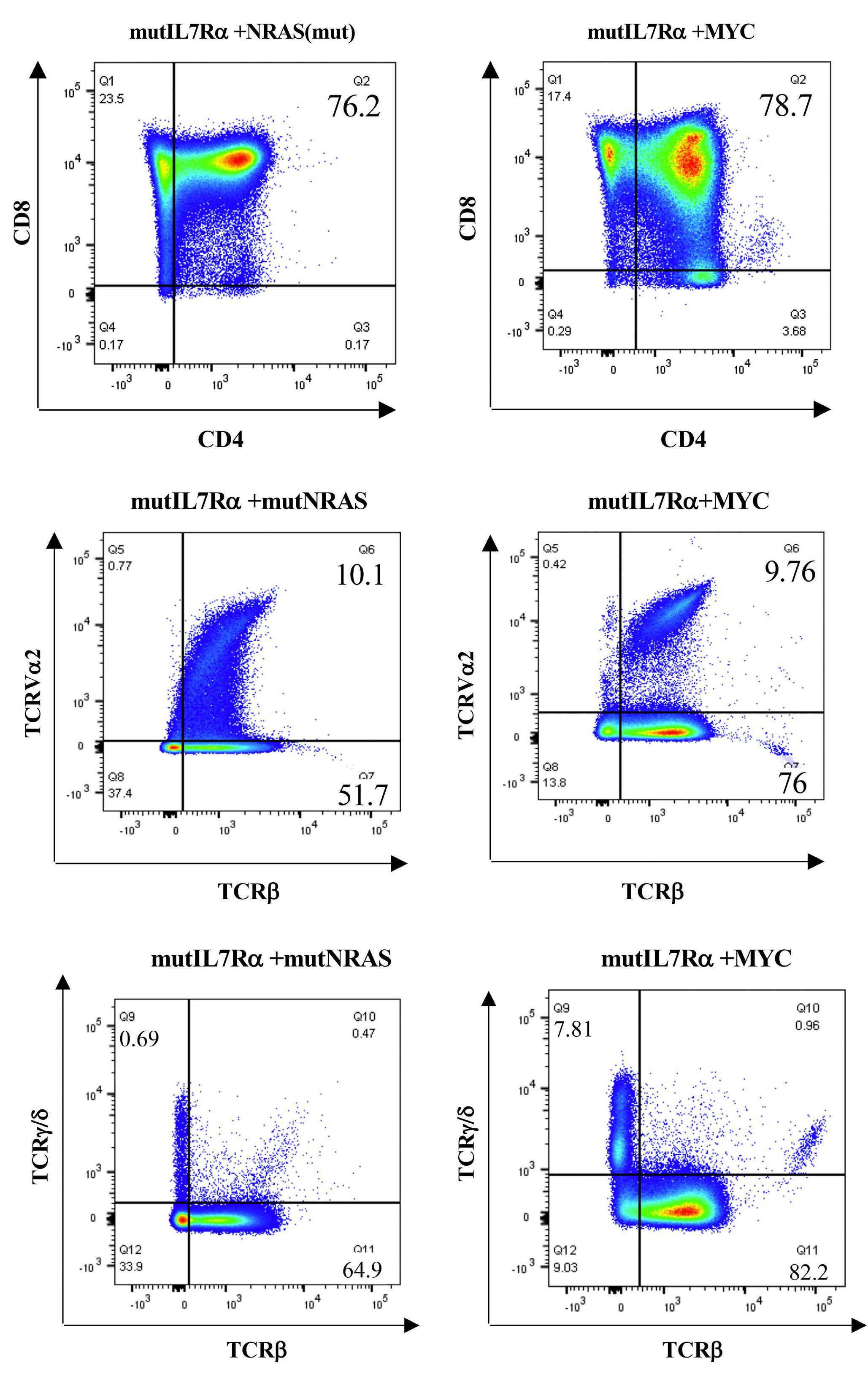
Figure 5. Acute lymphoblastic leukemia from mutant IL7Ra and mutNRAS has similar phenotype to mutant IL7Ra and over-expressed MYC. RAG1-/- mice were injected with thymocytes transduced with either mutIL7R and mutNRAS or with mutIL7R and MYC. After 16-21 days from injection, spleen was harvested from mice (N=7) and analyzed by flow cytometry as follows: all markers’ expression is from either GFP or GFP and mCherry (double positive) gated thymocytes. (A) CD4 versus CD8 (B) TCRVa2 versus TCRβ (C) TCRβ versus TCRγ/δ
Figure 6. Targeting MYC reduced acute lymphoblastic leukemia progression caused by mutIL7Ra and mutNRAS. RAG1-/- mice injected with thymocytes transduced with mutIL7R and mutNRAS, were treated with volasertib. (A) Percentage of GFP leukemic cell expression in blood and spleen in either Control-no treatment (N=6) or treated with 10 mg/kg volasertib (N=6). (B) White blood cell (WBC) count in the spleen and blood, spleen weight and percentage of loss or gain body weights in mice treated with or without 10 mg/kg volasertib. (C) RAG1-/- were injected with mutIL7R and mutNRAS thymocytes, Twenty-four hours after mice were treated with either buffers alone (no treatment), volasertib (10 mg/kg), venetoclax (15 mg/kg) or with both volasertib and venetoclax. (N=5 in each group), mice were euthanized when they showed morbid symptoms and analyzed for GFP leukemic cells in the blood by flow cytometry: representative flow image (top) and quantification of GFP leukemic cells percentage in each group (bottom).
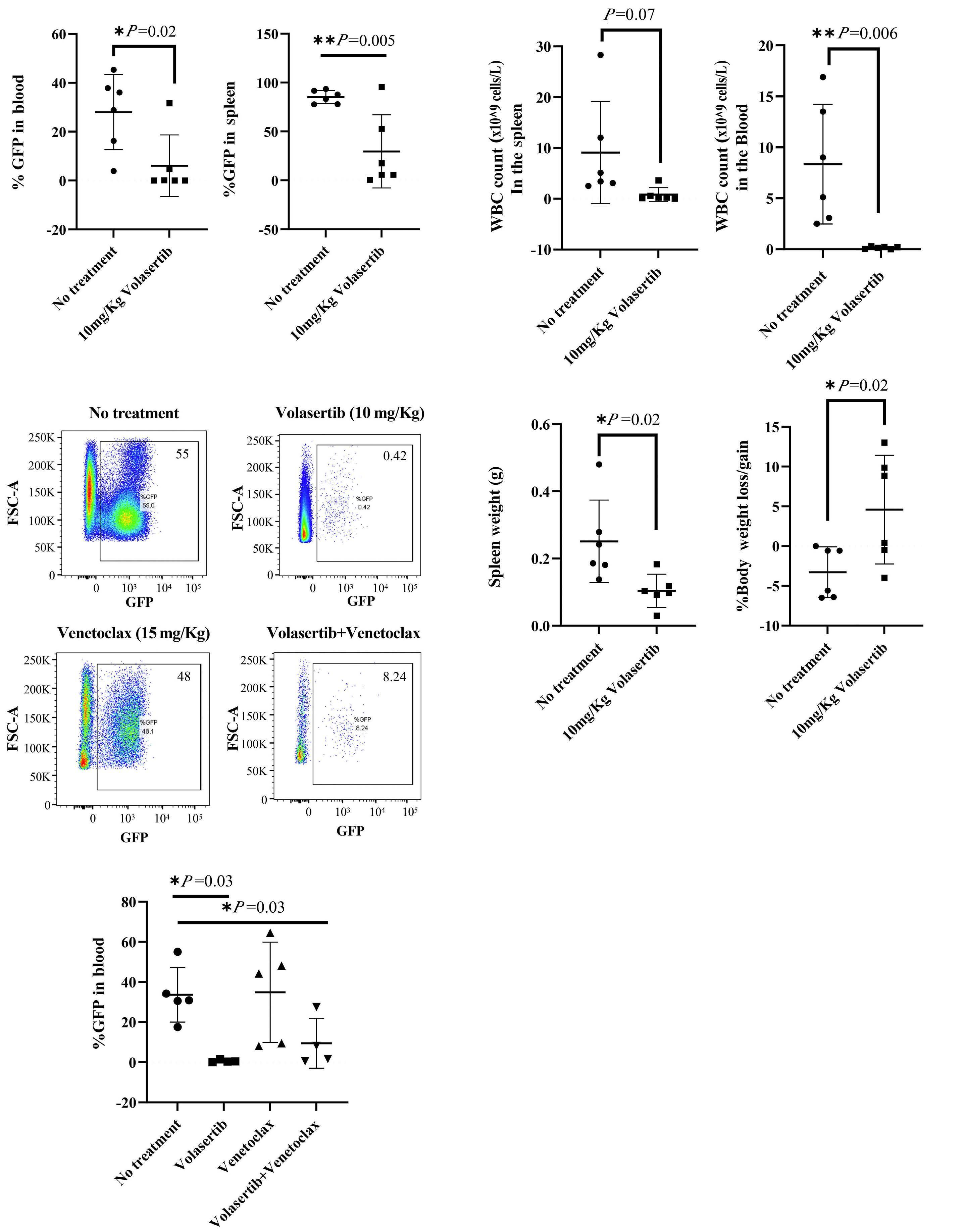
leukemic cells in the blood was significantly reduced with volasertib alone and in combination with ventoclax (Figure 6C). Therefore, volasertib alone in this regimen was quite efficient in slowing leukemia progression and there was no added benefit of venetoclax. This result suggests that volasertib is more efficient in treating ALL then venecolax and it should be considered as single drug for ALL with high MYC expression or with RAS mutations. We conclude that inhibition of PLK1 lowers MYC in leukemia cells and is associated with reducing progression of ALL driven by mutIL-7Ra plus mutNRAS. Another drug that targets MYC is JQ-1, a selective small molecule, BET bromodomain BRD4 inhibitor. BET bromodomain proteins act as regulatory factors for MYC and inhibition by JQ-1 was found to down-regulate MYC transcription followed by downregulation of its target genes in multiple mouse cancer models including ALL.21,28-30 Leukemic RAG1-/- mice were treated with either control-buffer alone or with 25 mg/kg JQ-1 (Online Supplementary Figure S6A). Untreated mice showed significantly increased WBC in the blood and spleen, in addition to enlarged spleen and liver compared to mice treated with JQ-1 (Online Supplementary Figure S6B). Moreover, mice that were treated with JQ-1 showed significantly reduced GFP positive cells in both spleen and blood (Online Supplementary Figure S6C, D), a therapeutic effect similar to the other MYC inhibitor, volasertib (Figure 6).
In another approach to implicate MYC in this ALL model, the Flox-Cre system was used to delete MYC in leukemia cells. Thymocytes were harvested from either MYC floxed mice or from normal C57BL/6J mice, depleted for CD4+ CD8+ and transduced with mutIL7Ra-T2A- mutNRAS/GFP and with Cre/mCherry (Online Supplementary Figure S7A). Following transduction, thymocytes were sorted for both GFP and mCherry to ensure cells contained both the oncogenes together with the Cre plasmids. Sorted cells were injected to RAG1-/- mice monitored for disease symptoms and WBC fluorescence (Online Supplementary Figure S7A). To confirm MYC deletion with Flox-Cre, we transduced either Cre or empty vector into thymocytes from MYC floxed mice and observed by immunoblotting a reduction in MYC levels in the presence of Cre (Online Supplementary Figure S7B).
Mice that were injected with normal C57BL/6J thymocytes containing Cre+ mutIL7Ra and mutNRAS, showed the most severe leukemia symptoms 21 days after injection. On the other hand, mice injected with thymocytes bearing MYC floxed and Cre+ mutIL7Ra and mutNRAS showed morbid symptoms (of an unknown basis) 35 days after injection. However, these symptoms were not a result of leukemia, which was not detected. Mice injected with normal C57BL/6J thymocytes had increased WBC in the blood and spleen (Online Supplementary Figure S7C) and enlarged spleens (Online Supplementary Figure S7D) compared to mice injected with thymocytes that have MYC floxed and Cre. Moreover, flow cytometry analysis showed that at least 8% of splenocytes containing Cre and mutIL7Ra + mutNRAS, but not floxed MYC, transformed to
leukemic cells, as seen by the GFP and mCherry in the splenocytes (Online Supplementary Figure S7E).
To further confirm the requirement for MYC in our ALL model, shRNA against MYC was introduced into both primary thymocytes and the D1 thymic cell line. Two of the MYC shRNA sequences were able to reduce MYC protein levels compared to control shRNA in the D1 cell line (Online Supplementary Figure S8A). To monitor D1 cell survival, we transduced cells expressing mutIL7Ra+ mutNRAS + GFP with shRNA scrambled/MYC expressed together with mCherry (Online Supplementary Figure S8B). Approximately 50% of D1 cells expressed both GFP and mCherry (Online Supplementary Figure S8B). Next, we followed cells expressing the double positive fluorophores and observed a reduction in the percentage of cells with MYC shRNA1891 and 2105, but not with scrambled shRNA, which showed no change in survival (Online Supplementary Figure S8C). In addition to shRNA experiments in the D1 cell line, we also tested the effect of MYC shRNA in primary thymocytes transduced with mutIL7Ra plus mutNRAS. The percentage of double positive thymocytes, were significantly reduced over time in the presence of MYC shRNA compared to scrambled shRNA (Online Supplementary Figure S8D).
PLK-1 involvement in MYC stability in acute lymphoblastic leukemia induced by mutIL7Ra and mutNRAS
MYC stability can be regulated in different stages and by several different regulators. Many past studies revealed a relationship between the RAS family and MYC protein stability.31-33 One of the mechanisms identified by Sears et al.33 is that RAS can regulate MYC stability by activating the Raf/ MEK/ERK signal: ERK can then phosphorylate MYC at Ser62, which stabilizes MYC (as well as activating its transcriptional activity). A second MYC stabilization mechanism downstream of RAS can occur via activating the PI3K-AKT pathway, which can phosphorylate GSK-3 and down-regulate its activity. This would prevent MYC phosphorylation on Thr58 which would otherwise mark MYC for ubiquitination and proteasomal degradation.33 We, therefore, tested both pathways in the D1 thymic cell line transduced with mutIL7Ra and mutNRAS both separately and combined, and observed an increase in p-ERK as well as at p-AKT and p-GSK-3 protein levels (Online Supplementary Figure S9A, B). Although, p-AKT was increased in the presence of the mutations (separate and combined), in the absence of IL-7, p-AKT was more stable only in the presence of both mutations (Online Supplementary Figure S9B). Moreover, GSK-3a/β phosphorylation was most dramatically elevated in the presence of both IL7Ra and NRAS combined, which parallels the increase in AKT activity (Online Supplementary Figure S9B). To further confirm the involvement of NRAS in MYC stabilization in our model, we also tested MYC phosphorylation in Ser62, which is crucial for MYC stabilization and known to be induced by ERK.33 Primary thymocytes transduced with both oncogenes
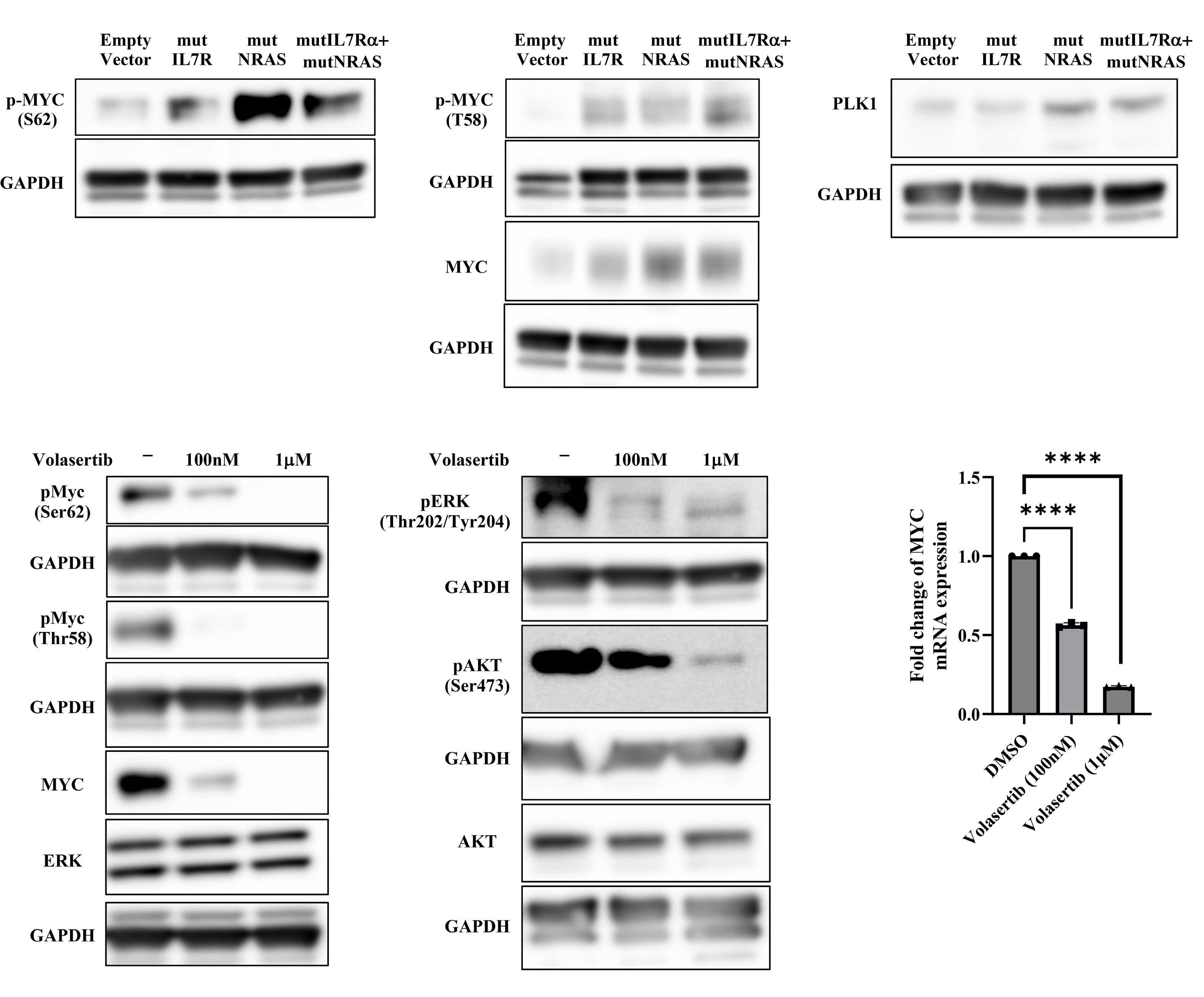
Figure 7. Suggested mechanism for MYC regulation in acute lymphoblastic leukemia, caused by mutant IL7R and NRAS. Primary thymocytes were transduced with either: empty vector, mutant IL7R, mutant NRAS or both mutIL7R and mutNRAS, after 72 hours (hrs) cells were lysed and analyzed by western blot for: (A) p-MYC (Ser 62), (B) p-MYC (Thr58), MYC and (C) PLK1. (D and E) Primary thymocytes transduced with both mutIL7R and mutNRAS treated with DMSO, 100 nM or 1µM volasertib for 24 hrs were analyzed as follows: (D) immunoblot analysis testing MYC, ERK and AKT phosphorylation and total protein (presentative analysis from 3 biological repeats). (E) RT-PCR analysis testing MYC mRNA expression. Each point represents biological repeat, and each biological repeat was done in triplicate (N=3). **** P<0.0001.
combined or NRAS alone showed increased p-MYC (Ser62) (Figure 7A), similar results were also observed in the thymic D1 cell line, although only with both oncogenes combined and in the absence of IL-7 (Online Supplementary Figure S9C). Interestingly, we also observed an increase in MYC phosphorylation in Thr58 which can induce MYC degradation (Figure 7B). Therefore, an additional mechanism may be operating which inhibits MYC degradation. In the last few years, PLK1 has been shown to regulate MYC stability in various cancer types.15,34,35 PLK1 was significantly increased in the presence
of mutant NRAS alone and in both mutations combined, which implies PLK1 regulation by the RAS signaling, which is responsible for MYC stabilization (Figure 7C). This also validates the bioinformatic analysis (Figures 1B, 3), which showed increased MYC pathways, including upregulation of PLK1. To further investigate the involvement of PLK-1 in NRAS signaling, we treated primary thymocytes transduced with mutIL7R+mutNRAS with different concentration of volasertib in vitro and found that, in addition to a significant reduction in MYC protein level (Figure 7D), there was also a significant
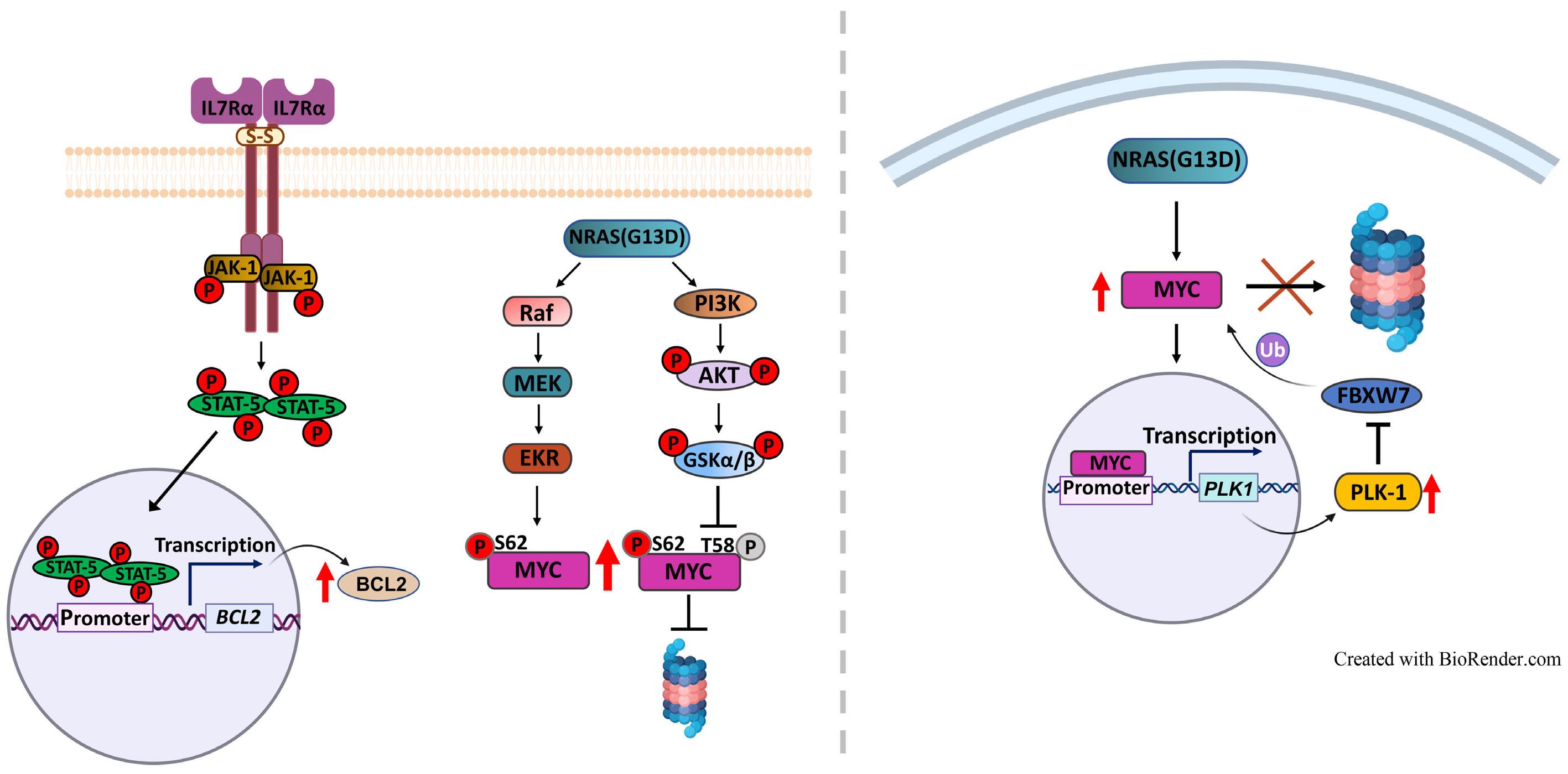
Figure 8. Schematic model of acute lymphoblastic leukemia induction by mutant human IL7R and NRAS. (A) mutIL7R induced expression of BCL2 by JAK/STAT signal. p-JAK-1 phosphorylates STAT-5, which activated its transcription activity. This leads to increased levels of BCL2 mRNA and protein. MutNRAS contributes to MYC stabilization through Raf/ERK and PI3K/AKT activation. The Raf/ERK pathway increases the stable form of MYC by phosphorylation of (Ser62). In addition, the PI3K/AKT signal contributes indirectly to MYC stability by increasing p-AKT and inhibiting GSK-3 activity, which results in inhibition of MYC degradation. (B) PLK-1 that is positively regulated by MYC activity can contribute to MYC stabilization through inhibition of FBXW-7, which leads to inhibition of MYC proteasomal degradation and creates a positive feedback loop.
decrease in MYC transcripts compared to control, non-treated cells (Figure 7E). Interestingly, we also observed a reduction in phosphorylated ERK and AKT in addition to a significant reduction in MYC phosphorylated form in Ser62 and Thr58, which can suggest that PLK-1 may regulate more up-stream factors in the RAS pathway, which eventually effect both MYC protein stability and its transcripts (Figure 7D). To confirm that the mutant oncogenes are over-expressed in transduced thymocytes, we also examined levels of NRAS and hIL7Ra showing high levels in the groups expressing hIL7Ra or NRAS alone compared to both mutations combined (Online Supplementary Figure S9D; see also Figure 2A). The combined group showed less of each oncogene than the single oncogene, presumably because the two were encoded on the same construct. We combined them on the same plasmid to assure that every transduced thymocyte received both oncogenes.
Mutations in the IL7Ra gene were shown in several studies to cause ALL;7,8,36 however, mutations in IL7Ra alone is not sufficient to lead to T-ALL and other mutations are necessary.1 Our lab showed that combining mutant IL7Ra
with mutant NRAS can cause severe ALL in mice, but the mechanism of this oncogene co-operation was not determined.1 In B-ALL, nearly half of IL-7R mutants were reported to also harbor mutations in the Ras pathway.6 In T-ALL, approximately 2% of IL-7R mutants were reported to have mutations in K/N RAS.37 However, mutations in regulators that activate RAS may be a more important mechanism in T-ALL. Approximately 7% of IL-7R mutants were reported with inactivating mutations in the exchange factor NF1 which would result in RAS activation. In other studies, the RAS loading factor RasGRP1 was over-expressed in a majority of T-ALL (although the association with mut IL-7R has not been reported) and can induce T-ALL in mice.38 Finally, WT IL-7R is expressed on approximately 70% of T-ALL, is activated by IL-7 in vivo, and could co-operate with the RAS pathway. Approximately 17% of WT IL-7R T-ALL harbor K/N RAS mutations.37 Our study shows that co-operation between the IL-7R and RAS pathways is powerfully leukemogenic.
Here we show that in the presence of both mutant IL7Ra and NRAS there is an increase in both BCL2 and MYC protein levels, which together have long been known to be a powerful pro-oncogenic combination14 (Figure 8). BCL2 has long been identified as a major IL-7-induced anti-apoptotic mediator,36 and this was confirmed in our
model at the mRNA and protein levels (Figures 2, 8A).
We considered that increasing MYC could explain part of the mechanism of oncogene co-operation, as MYC target genes were over-expressed when both mutant IL7Ra and NRAS were present. We observed that while MYC protein increased with NRAS (G13D) alone, a much greater increase occurred when combining mutant NRAS with mutant IL7Ra. It has previously been shown that RAS protein can stabilize MYC by increasing its phosphorylation at Ser62 through PI3K/AKT and ERK activation.33 We showed that MYC increase at the level of protein stability, whereas the BCL2 increase is largely at the transcriptional level19,36,39 (Figures 2, 8A) An additional pro-survival effect that we observed in both primary thymocytes and a thymocyte cell line was a decrease in pro-apoptotic signals regulated by proteins such as BAK1 and BAD. The BAK1 decrease was induced by NRAS (G13D) mutant alone, which has not previously been reported. On the other hand, BAD phosphorylation (which leads to its sequestration) seemed much more stable in the presence of both IL7Ra and NRAS mutants compared to each one expressed alone.
Another potential contributor to leukemogenesis is the increase in pro-oncogene PIM-1, which is known to be induced by the JAK-STAT pathway and was shown to be activated in T-ALL40 in the presence of mutations in the IL7Ra itself or in its signaling pathway.17 In addition, it has previously been shown that PIM-1 can enhance MYC transcriptional activity by increasing phosphorylation on Ser62 and decreasing degradation by inhibiting its phosphorylation on Thr58.41 Thus PIM-1 could also contribute to the oncogenic mechanisms in ALL. To test the hypothesis that NRAS acts by regulating MYC stability, we replaced NRAS (G13D) expression with MYC overexpression together with mutant IL7Ra which induced leukemia with the same phenotype. This result is consistent with the recent study of Oliveira et al. in zebra fish showing T-ALL transformation by combining mutant IL7Ra with MYC overexpression.11
One goal of this study was to identify new targets for ALL therapy. We used several different methods to inhibit MYC levels in vivo and in vitro, and followed ALL progression. Since, so far, no drug has been found that can inhibit MYC directly, we used two clinically tested drugs that inhibit MYC indirectly: volasertib and JQ-1. Both drugs significantly reduced the progression of ALL in mice caused by the combination of mutant IL7Ra with mutant NRAS. Deletion of MYC with cre-flox also blocked ALL in vivo. In addition to our in vivo model, we also used in vitro systems, inhibiting MYC with shRNA in both primary thymocytes and a thymocyte cell line, and observed a significant reduction in cell proliferation. Altogether, we suggest that increased MYC stability is a key mechanism in this ALL model.
Finally, to understand the mechanism that could be responsible for MYC stability we tested one pathway that could be involved. We saw, in both enrichment analysis and at the protein level, that the serine/threonine kinase PLK-1
was significantly increased when both IL7Ra and NRAS mutants were expressed. Most importantly, NRAS (G13D) is that which is responsible for this increase (Figure 7C), which demonstrates that NRAS could be one of the PLK-1 regulators. PLK-1 has previously been shown to be a MYC positive regulator by inhibiting the FBWX-7 E3 ligase which targets MYC for degradation.15,42 Since PLK-1 is also one of the MYC target genes, increases in either PLK-1 or MYC can induce a positive feedback loop increasing both proteins (Figure 8B). There are two major pathways regulated by RAS that are known to stabilize MYC: 1) the Raf/ERK pathway, which phosphorylates MYC on Ser62 that is required for its stabilization33 (Figure 8A); 2) the PI3K/AKT pathway which indirectly increases the stabilization of phosphorylated MYC in Ser62 by inhibiting GSKa/β activation; the latter phosphorylates MYC on Thr58, targeting it for degradation22,33 (Figure 8A). Our data show that both p-MYC on Thr58 and Ser62 are accumulated in ALL primary cells, which suggests that GSK-3a/β retains some activity; nevertheless, MYC is not sent for proteasomal degradation. The latter supports our hypothesis that PLK-1 is involved in MYC stabilization, possibly through inhibiting the E3 ligase, FBWX-7 (Figure 8B). Overall, our study shows that combination of a gain-offunction mutation in human IL7Ra together with overexpression of MYC, generates severe ALL in mice. We suggest that NRAS (G13D) mutation acts via increasing MYC stability, and, therefore, offers MYC as an attractive target in ALL, which frequently involves the IL-7R pathway together with RAS mutations.
No conflicts of interest to disclose.
Contributions
HW and WL designed and performed experiments, and wrote the paper. GR contributed experiments and discussion. TG performed animal studies. TJM analyzed data. JH contributed experiments. SKD directed the study.
We are grateful to Megan Karwan and Jeff Carell from the Flow Cytometric Unit. We thank Susan Mackem for providing MYC floxed mice. We also thank Elijah Edmondson and his team at the IHC unit NCI/NIH, Maggie Cam from the CCBR unit NCI/NIH, and Lisa M. Miller Jenkins from the Laboratory of Biochemistry and Molecular Biology NCI/NIH. All illustrations were created using BioRender software.
This research was supported by the intramural program of the National Cancer Institute, National Institutes of Health.
All data for this work are available either through the detailed Online Supplementary Appendix or upon request.
1. Cramer SD, Hixon JA, Andrews C, et al. Mutant IL-7Ra and mutant NRas are sufficient to induce murine T cell acute lymphoblastic leukemia. Leukemia. 2018;32(8):1795-1882.
2. McMahon CM, Luger SM. Relapsed T cell ALL: current approaches and new directions. Curr Hematol Malig Rep. 2019;14(2):83-93.
3. Lato MW, Przysucha A, Grosman S, Zawitkowska J, Lejman M. The new therapeutic strategies in pediatric T-cell acute lymphoblastic leukemia. Int J Mol Sci. 2021;22(9):4502.
4 Rodrigues GOL, Cramer SD, Winer HY, et al. Mutations that collaborate with IL-7Ra signaling pathways to drive ALL. Adv Biol Regul. 2021;80:100788.
5. Oliveira ML, Akkapeddi P, Ribeiro D, Melao A, Barata JT. IL-7Rmediated signaling in T-cell acute lymphoblastic leukemia: an update. Adv Biol Regul. 2019;71:88-96.
6. Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinaseactivating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005-1015.
7 Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain-offunction mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43(10):932-939.
8. Shochat C, Tal N, Bandapalli OR, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. 2011;208(5):901-908.
9 Yokoyama K, Yokoyama N, Izawa K, et al. In vivo leukemogenic potential of an interleukin 7 receptor alpha chain mutant in hematopoietic stem and progenitor cells. Blood. 2013;122(26):4259-4263.
10. Tremblay CS, Curtis DJ. The clonal evolution of leukemic stem cells in T-cell acute lymphoblastic leukemia. Curr Opin Hematol. 2014;21(4):320-325.
11. Oliveira ML, Veloso A, Garcia EG, et al. Mutant IL7R collaborates with MYC to induce T-cell acute lymphoblastic leukemia. Leukemia. 2022;36(6):1533-1540.
12. Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120(17):3397-3406.
13. Tasian SK, Loh ML, Hunger SP. Childhood acute lymphoblastic leukemia: integrating genomics into therapy. Cancer. 2015;121(20):3577-3590.
14 Fanidi A, Harrington EA, Evan GI. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature. 1992;359(6395):554-556.
15. Xiao D, Yue M, Su H, et al. Polo-like kinase-1 regulates Myc stabilization and activates a feedforward circuit promoting tumor cell survival. Mol Cell. 2016;64(3):493-506.
16. Wang J, Zhou M, Xu JY, Chen B, Ouyang J. Combination of BCL-2 and MYC protein expression improves high-risk stratification in diffuse large B-cell lymphoma. Onco Targets Ther. 2015;8:2645-2650.
17. De Smedt R, Morscio J, Reunes L, et al. Targeting cytokine- and therapy-induced PIM1 activation in preclinical models of T-cell acute lymphoblastic leukemia and lymphoma. Blood. 2020;135(19):1685-1695.
18. Senkevitch E, Li W, Hixon JA, et al. Inhibiting Janus Kinase 1 and BCL-2 to treat T cell acute lymphoblastic leukemia with IL7Ralpha mutations. Oncotarget. 2018;9(32):22605-22617.
19 Silva A, Laranjeira AB, Martins LR, et al. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias.
Cancer Res. 2011;71(14):4780-4789.
20. Hofmeister R, Khaled AR, Benbernou N, et al. Interleukin-7: physiological roles and mechanisms of action. Cytokine Growth Factor Rev. 1999;10(1):41-60.
21. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5.
22. Blackburn JS, Liu S, Wilder JL, et al. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell. 2014;25(3):366-378.
23. Fairlie WD, Lee EF. Co-operativity between MYC and BCL-2 pro-survival proteins in cancer. Int J Mol Sci. 2021;22(6):2841.
24. Rudolph D, Steegmaier M, Hoffmann M, et al. BI 6727, a Pololike kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15(9):3094-3102.
25. Gorlick R, Kolb EA, Keir ST, et al. Initial testing (stage 1) of the Polo-like kinase inhibitor volasertib (BI 6727), by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2014;61(1):158-164.
26. Abbou S, Lanvers-Kaminsky C, Daudigeos-Dubus E, et al. Polo-like kinase inhibitor volasertib exhibits antitumor activity and synergy with vincristine in pediatric malignancies. Anticancer Res. 2016;36(2):599-609.
27. Li J, Ohmura S, Marchetto A, et al. Therapeutic targeting of the PLK1-PRC1-axis triggers cell death in genomically silent childhood cancer. Nat Commun. 2021;12(1):5356.
28. Roderick JE, Tesell J, Shultz LD, et al. c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood. 2014;123(7):1040-1050.
29 Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904-917.
30 Vanden Bempt M, Demeyer S, Broux M, et al. Cooperative enhancer activation by TLX1 and STAT5 drives development of NUP214-ABL1/TLX1-positive T cell acute lymphoblastic leukemia. Cancer Cell. 2018;34(2):271-285.e7.
31. Mahauad-Fernandez WD, Felsher DW. The Myc and Ras partnership in cancer: indistinguishable alliance or contextual relationship? Cancer Res. 2020;80(18):3799-3802.
32. Kerkhoff E, Houben R, Loffler S, et al. Regulation of c-myc expression by Ras/Raf signalling. Oncogene. 1998;16(2):211-216.
33. Sears R, Nuckolls F, Haura E, et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14(19):2501-2514.
34. Ren Y, Bi C, Zhao X, et al. PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas. J Clin Invest. 2018;128(12):5517-5530.
35. Murga-Zamalloa C, Polk A, Hanel W, et al. Polo-like-kinase 1 (PLK-1) and c-myc inhibition with the dual kinasebromodomain inhibitor volasertib in aggressive lymphomas. Oncotarget. 2017;8(70):114474-114480.
36. Winer H, Rodrigues GOL, Hixon JA, et al. IL-7: comprehensive review. Cytokine. 2022;160:156049.
37. Kim R, Boissel N, Touzart A, et al. Adult T-cell acute lymphoblastic leukemias with IL7R pathway mutations are slow-responders who do not benefit from allogeneic stem-cell transplantation. Leukemia. 2020;34(7):1730-1740.
38. Klinger MB, Guilbault B, Goulding RE, Kay RJ. Deregulated expression of RasGRP1 initiates thymic lymphomagenesis
independently of T-cell receptors. Oncogene. 2005;24(16):2695-2704.
39. Jiang Q, Li WQ, Hofmeister RR, et al. Distinct regions of the interleukin-7 receptor regulate different Bcl2 family members. Mol Cell Biol. 2004;24(14):6501-6513.
40. La Starza R, Messina M, Gianfelici V, et al. High PIM1 expression is a biomarker of T-cell acute lymphoblastic leukemia with JAK/ STAT activation or t(6;7)(p21;q34)/TRB@-PIM1 rearrangement.
Leukemia. 2018;32(8):1807-1810.
41. Zhang Y, Wang Z, Li X, Magnuson NS. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene. 2008;27(35):4809-4819.
42. Wang D, Pierce A, Veo B, et al. A Regulatory loop of FBXW7MYC-PLK1 controls tumorigenesis of MYC-driven medulloblastoma. Cancers (Basel). 2021;13(3):387.
1Department of Computational and Quantitative Medicine, Beckman Research Institute of City of Hope; 2Department of Systems Biology, Beckman Research Institute of City of Hope; 3Department of Hematology and Hematopoietic Cell Transplantation; 4Irell and Manella Graduate School of Biological Sciences of City of Hope and 5Research Informatics, City of Hope National Medical Center, Duarte, CA, USA
Correspondence: Z. Gu zgu@coh.org
Received: June 7, 2023.
Accepted: November 6, 2023. Early view: November 16, 2023.
https://doi.org/10.3324/haematol.2023.283706
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
B-acute lymphoblastic leukemia (B-ALL) consists of dozens of subtypes defined by distinct gene expression profiles (GEP) and various genetic lesions. With the application of transcriptome sequencing (RNA sequencing [RNA-seq]), multiple novel subtypes have been identified, which lead to an advanced B-ALL classification and risk-stratification system. However, the complexity of analyzing RNA-seq data for B-ALL classification hinders the implementation of the new B-ALL taxonomy. Here, we introduce Molecular Diagnosis of Acute Lymphoblastic Leukemia (MD-ALL), an integrative platform featuring sensitive and accurate B-ALL classification based on GEP and sentinel genetic alterations from RNA-seq data. In this study, we systematically analyzed 2,955 B-ALL RNA-seq samples and generated a reference dataset representing all the reported B-ALL subtypes. Using multiple machine learning algorithms, we identified the feature genes and then established highly sensitive and accurate models for B-ALL classification using either bulk or single-cell RNA-seq data. Importantly, this platform integrates multiple aspects of key genetic lesions acquired from RNA-seq data, which include sequence mutations, large-scale copy number variations, and gene rearrangements, to perform comprehensive and definitive B-ALL classification. Through validation in a hold-out cohort of 974 samples, our models demonstrated superior performance for B-ALL classification compared with alternative tools. Moreover, to ensure accessibility and user-friendly navigation even for users with limited or no programming background, we developed an interactive graphical user interface for this MD-ALL platform, using the R Shiny package. In summary, MD-ALL is a user-friendly B-ALL classification platform designed to enable integrative, accurate, and comprehensive B-ALL subtype classification. MD-ALL is available from https://github.com/gu-lab20/MD-ALL.
B-acute lymphoblastic leukemia (B-ALL) is a highly heterogeneous disease, which consists of dozens of subtypes with distinct gene expression profiles (GEP) and constellations of genetic alterations.1 Through the application of transcriptome sequencing (RNA sequencing [RNA-seq]), multiple novel B-ALL subtypes have been identified harboring recurrent genetic lesions and distinct GEP. 2-4 The current World Health Classification (5 th edition) of Hematolymphoid Tumors (WHO-HAEM5), 5 along with the International Consensus Classification of Myeloid Neoplasms and Acute Leukemia (ICC),6 recognize a total of 11 and 26 molecular subtypes of B-ALL, respectively. Currently, clinical diagnosis and classification of B-ALL rely on a range of assays such as flow cytometry,
fluorescence in situ hybridization (FISH), cytogenetic karyotyping, and panel-based sequencing assays.7,8 The data generation and analysis using these platforms are time-consuming, expensive, and error-prone. Furthermore, they are inadequate to identify specific subtypes defined by cryptic genetic lesions (e.g., DUX4 and MEF2D rearrangements) or the ones primarily defined by GEP (e.g., Ph-like and ETV6::RUNX1-like).
With rapid progress in discovering novel B-ALL subtypes, updating clinical test assays accordingly has become a challenging task. Alternatively, the application of RNAseq for clinical diagnosis of B-ALL subtypes has been investigated by multiple institutions and led to encouraging outcomes. 9,10 With its easy-to-follow protocol and multiple layers of information, RNA-seq is poised to revolutionize the classification of B-ALL in both research
and clinical settings. However, bioinformatics analysis of RNA-seq data to extract both the sentinel genetic lesions and the GEP signatures for classification is still challenging. Although a few bioinformatics tools have been developed for this purpose,11-13 they solely rely on GEP for B-ALL subtyping. Here, we present Molecular Diagnosis of Acute Lymphoblastic Leukemia (MD-ALL), a user-friendly bioinformatics platform that integrates genetic and transcriptomic features from RNA-seq to provide integrative, accurate, and comprehensive B-ALL subtype classification.
Methods
RNA-sequencing datasets
Raw RNA-seq data of 3,005 B-ALL samples were collected from multiple published studies.1,4,14-22 After removing potential duplicates (inferred by KING23) and samples with low coverage, 2,955 samples were kept as the primary cohort for this study ( Online Supplementary Table S1 ).
RNA-sequencing data analysis
The sequencing reads were aligned to the human genome reference (GRCh38) using STAR. 24 Then Picard (see URL) was used to mark PCR duplicates.
Gene expression
Read counts were calculated by HTSeq 25 and FeatureCount, 26 and then normalized by DESeq2. 27 The ComBat function in the sva R package 28 was used to correct potential batch effects introduced by different library preparation approaches (mRNA vs. total RNA and stranded vs. unstranded) and variable sequencing lengths ( Online Supplementary Figure S1 ). t-distributed stochastic neighbor embedding (tSNE) and uniform manifold approximation and projection (UMAP) were used for dimensionality reduction visualization.
Mutations
Single nucleotide variants (SNV) and insertions/deletions (Indel) were called by following the best practice pipeline from GATK (see Online Supplementary Methods ). 29
Deconvolution of bulk gene expression profiles
Single-cell RNA-seq (scRNA-seq) data from the 1-Million Immune Cells Project (see Online Supplementary Methods ) were reanalyzed to establish a GEP reference representing 20 primary blood cell types. Then, CIBERSORTx 30 was used to deconvolute the bulk GEP of B-ALL to estimate their leukemic cell ratios and granular B-cell composition.
Fusion calling CICERO 31 and FusionCatcher 32 were used as they can
identify gene rearrangements involving highly repetitive regions such as the IgH locus.
Copy number variation calling
With read counts and SNV called from RNA-seq, the RNAseqCNV package 33 was used to detect chromosomal-level copy number variation (CNV).
Ancestry inference
The samples’ ancestral background was estimated using iAdmix, 34 with the genotype of SNP from the 1K-Genome Project used as the reference. 35,36
Gene expression profile reference of B-acute lymphoblastic leukemia subtype
Through analyzing the RNA-seq data of the 2,955 B-ALL samples, 26 subtypes were identified, with 19 having distinct GEP features. In order to construct a GEP reference for B-ALL classification, PhenoGraph clustering 37 and k-nearest neighbor analysis of two-dimensional UMAP were performed to identify the representative samples of each subtype.
Feature gene selection
Since the reference cohort is not evenly distributed across B-ALL subtypes, SMOTE algorithm 38 was used to subsample or artificially construct additional samples, which resulted in cohorts with the same sample size per subtype. Then Boruta 39 was used to identify the genes confirmed as contributing features for distinguishing different subtypes.
Gene expression profile-based B-acute lymphoblastic leukemia classification
Two GEP-based B-ALL prediction models were constructed: i) support vector machine (SVM) classification; among multiple tested machine learning algorithms, SVM performed the best; ii) PhenoGraph clustering; 37 PhenoGraph is a clustering algorithm originally developed to identify and partition cells into subpopulations.
Integration of genetic lesions and gene expression profile features
GEP-based subtype prediction and key genetic lesions identified from RNA-seq were integrated for definitive B-ALL classification. A detailed description of integrating GEP-based prediction and sentinel genetic lesions for B-ALL classification is summarized in Table 1.
Single-cell RNA-sequencing analysis and B-acute lymphoblastic leukemia classification
scRNA-seq reads were mapped to the GRCh38 reference. After quality control, the Seurat package 40 was used for gene expression normalization and variable gene selection. With the GEP reference of blood cell types and
Genetic alteration GEP subtype GEP feature
BCL2, MYC or BCL6 rearrangement
CDX2 overexpression & UBTF::ATXN7L3 fusion
BCL2/MYC
CDX2/UBTF
CRLF2 rearrangement Not Ph/Ph-like
DUX4 rearrangement
ETV6::RUNX1 fusion
No
ETV6::RUNX1 fusion
HLF rearrangement
DUX4
ETV6::RUNX1
ETV6::RUNX1
HLF
Chromosome number ≥51 Hyperdiploid
iAMP21 iAMP21
IKZF1 N159Y mutation IKZF1 N159Y
Distinct
Highly distinct
Non-distinct
Highly distinct
Highly distinct
Highly distinct
Distinct
Distinct
Subtype Note
BCL2/MYC The rearrangements can involve genes adjacent to MYC
CDX2/UBTF CDX2 overexpression
CRLF2(non-Ph-like) Less recognized subtype
DUX4
ETV6::RUNX1 -
ETV6::RUNX1-like
DUX4 gene family overexpression
Commonly seen with ETV6 or IKZF1 rearrangements
HLF HLF overexpression
Hyperdiploid -
Less distinct iAMP21 iAMP21 can be identified by RNAseqCNV
Highly distinct IKZF1 N159YKMT2A rearrangement KMT2A
No KMT2A rearrangement KMT2A
Chromosome number 47-50
Distinct KMT2A -
Distinct KMT2A-like
Hyperdiploid
Chromosome number 31-39 Low hypodiploid
MEF2D rearrangement MEF2D
Chromosome number 24-30
Hyperdiploid
PAX5 P80R mutation
PAX5::ETV6
PAX5 P80R
Minor subtype; reported with AFF1 fusion
Distinct Low hyperdiploid Less recognized subtype
Distinct Low hypodiploid
Highly distinct
MEF2D
Non-distinct Near haploid
Highly distinct
PAX5::ETV6 Distinct
PAX5 P80R
Commonly seen with TP53 mutations
Commonly seen with chromothripsis around MEF2D
Less frequently with GEP of Low hypodiploid
NUTM1 rearrangement NUTM1 Less distinct NUTM1 NUTM1 overexpression
Abnormal MEGF10 isoform overexpression
PAX5::ETV6 Originally reported as PAX5alt
PAX5 alteration PAX5alt Distinct PAX5alt
BCR::ABL1 fusion
Non-Ph kinase-activating alteration*
TCF3::PBX1 fusion
ZNF384 rearrangement
Ph/Ph-like Distinct Ph
Ph/Ph-like
TCF3::PBX1
ZNF384
No ZNF384 rearrangement ZNF384
Distinct
Highly distinct
Highly distinct
Highly distinct
Ph-like
TCF3::PBX1
ZNF384
ZEB2/CEBP Minor subtype Table 1. Integrative
Featured with PAX5 fusion, mutation, or iAmp, but not deletion
At least two GEP subclusters observed within Ph group
Commonly seen with kinase activating fusions
Rare fusions with EWSR1 have been reported
Also observed in mixed phenotype acute leukemia
ZNF384-like Minor subtype; reported with ZNF362 fusion
ZEB2 H1038R mutation and/or CEBP fusion
ZEB2/CEBP Distinct
If genetic lesions do not agree with gene expression profile (GEP)-based prediction, genetic lesions determine the primary subtypes, while GEP guide the decision on the secondary subtypes. *Gene rearrangements involving ABL1, ABL2, CSF1R, PDGFRA, PDGFRB, LYN, CRLF2, JAK2, EPOR, TSLP, TYK2, IL2RB, NTRK3, PTK2B, FGFR1, FLT3, DGKH, BLNK, and CBL. MD-ALL: Molecular Diagnosis of Acute Lymphoblastic Leukemia; iAmp: intragenic amplification.
B-ALL subtypes described above, SingleR41 was used to annotate cell types and B-ALL subtypes for each cell.
Characteristics of the RNA-sequencing cohort
In total, 2,955 B-ALL samples with high-quality RNA-seq data were included in this study (Online Supplementary Table S1). This cohort comprises 67.8% pediatric and 28.4% adult cases from different racial/ethnic backgrounds, with a relative higher proportion of male patients (56.1%) (Online Supplementary Figure S2). Through manual curation of the genetic lesions, 3,304 gene rearrangements, 2,979 sequence mutations, and 95 FLT3 internal tandem duplications (ITD) were identified (Online Supplementary Tables S2-4). Subsequently, sentinel gene fusions and mutations were compiled to facilitate B-ALL classification (Online Supplementary Tables S5 and S6). Through integration of genetic lesions and GEP-based predictions, the cohort was classified into 26 molecular subtypes (Figure 1A). In summary, this well-curated large cohort encompasses all the reported B-ALL subtypes across different age groups, sex, and racial/ethnical backgrounds, making it an excellent resource for constructing and evaluating B-ALL subtype prediction models, as well as advancing our understanding of the genetic and transcriptomic features of each B-ALL subtype.
High accuracy of gene expression profile-based B-acute lymphoblastic leukemia classification by MD-ALL In order to generate a GEP reference for subtype prediction, 1,821 samples confirmed by sentinel genetic lesions and stable GEP clusters were selected as the training cohort, representing 19 B-ALL subtypes with distinct GEP (Figure 1B). Using this GEP reference cohort, 1,058 feature genes were consistently confirmed by the Boruta algorithm in eight SMOTE-resampled cohorts (Online Supplementary Table S7). Due to the substantial batch effect between mRNA-seq and total RNA-seq library preparation approaches (Online Supplementary Figure S1), only the protein-coding genes were considered for feature selection to accommodate both library types. Each feature gene was assigned an importance score by Boruta, which was used to rank their significance for distinguishing different subtypes. Based on the reference cohort and selected feature genes, MDALL employs SVM and PhenoGraph algorithms to predict the subtypes of the test samples. Considering that the user-provided test RNA-seq data may use different library preparation strategies and the sample size may not be sufficient for reliable batch effect correction, our prediction models were evaluated using the test samples’ GEP data without batch effect correction.
For the training cohort, 100% accuracy was achieved by both SVM and PhenoGraph algorithms as expected (Figure 2A). For the test cohort, subtypes with non-distinct GEP, such as Near haploid, and less recognized subtypes, such
Figure 1. Gene expression profiles of B-acute lymphoblastic leukemia subtypes. The t-distributed stochastic neighbor embedding (tSNE) plots display the gene expression profiles (GEP) distribution using 1,058 signature coding genes identified from reference B-acute lymphoblastic leukemia (B-ALL) subtypes (see Methods). GEP are derived from bulk RNA-sequencing data, with each dot representing an individual sample. A perplexity parameter of 10 was used in tSNE analysis to better visualize the minor subtypes. B-ALL subtypes are color-coded and annotated, while less recognized ones such as CRLF2 (non-Ph-like), Low hyperdiploid, ZNF384-like, KMT2A-like, and unclassified are shown in grey. (A) tSNE plot of 2,955 B-ALL samples, which represents the total cohort of this study. (B) tSNE plot of reference samples (N=1,821) from 19 B-ALL subtypes with distinct GEP. For GEP-based classification, Ph and Ph-like are combined as one Ph/Ph-like group.
as Low hyperdiploid and CRLF2 (non-Ph-like), as well as unclassified cases were excluded. In order to evaluate the performance across different tools, phenocopy subtypes, including Ph-like, ETV6::RUNX1-like, KMT2A-like, and ZNF384-like, were merged with their canonical counterparts to accommodate the different strategies used by different tools for identifying them. Moreover, PAX5alt and Ph-like subtypes are primarily defined by GEP, but their GEP features are less distinct compared with others. In order to avoid potential bias of evaluating different tools for these two subtypes, only the PAX5alt and Ph-like cases confirmed by sentinel genetic lesions (i.e., PAX5 mutation, fusion, or intragenic amplification in PAX5alt, and rearrangements involving kinase activating genes in Ph-like; see Table 1) were kept in the test cohort. Although this study enrolled a large number of samples, seven minor subtypes have fewer than 30 qualified samples, which include BCL2/MYC (N=29), PAX5::ETV6 (N=23), ZEB2/CEBP (N=19), NUTM1 (N=18), IKZF1 N159Y (N=14), HLF (N=11), and CDX2/UBTF (N=9). Following the training versus testing sample size ratio of 2:1 set for the major subtypes, fewer than ten samples would be left for testing. Therefore, a leave-one-out validation was used to evaluate the prediction models for these minor subtypes, eventually resulting in a test cohort of 974 samples (Online Supplementary Table S8).
Through GEP-based prediction, SVM and PhenoGraph successfully classified 971 and 972 samples into distinct subtypes, respectively, with high overall accuracy achieved in both models (SVM: 96.1%, N=936; PhenoGraph: 92.7%, N=903). Despite the high accuracy of both models, SVM surpassed PhenoGraph in discerning multiple subtypes such as intrachromosomal amplification of chromosome 21 (iAMP21) and Ph/Ph-like, whereas PhenoGraph demonstrated superior performance over SVM in identifying the ETV6::RUNX1/-like subtype (Figure 2B, C). In summary, the GEP-based models in MD-ALL can achieve high classification rate as well as high accuracy for B-ALL classification.
MD-ALL classification is superior compared with alternative tools
Currently, there are three alternative tools providing the functionality of B-ALL classification, which are ALLSpice,11 ALLSorts,12 and ALLCatchR.13 The subtype prediction by these tools is solely based on GEP; therefore, the comparison with them is restricted to the GEP prediction results of MD-ALL. Additionally, it should be noted that the holdout test cohort of this study partially overlaps with the training cohort of the other tools, since the majority of B-ALL RNA-seq data used in MD-ALL and these alternative tools are from our previous study, which comprises 1,988 B-ALL samples.14 This overlap may lead to overestimated accuracy of the alternative tools. Additionally, the PAX5::ETV6 fusion, originally reported as one of the sentinel
alterations of PAX5alt subtype,14 is still considered as PAX5alt by other tools. Therefore, the PAX5::ETV6 cases were annotated as PAX5alt when comparing the performance of different models.
In the same test cohort of 974 samples, a much higher number of samples remained unclassified by ALLCatchR (N=36), ALLSorts (N=142), and ALLSpice (N=327) when compared to MD-ALL. The overall accuracies were 91.3% (889/974), 81.2% (791/974), and 58.8% (573/974) for each method, respectively, which were significantly lower than those achieved by both models in MD-ALL. When considering only the samples with assigned subtypes, the accuracies of ALLCatchR, ALLSorts, and ALLSpice were 94.8% (889/938), 95.1% (791/832), and 88.6% (573/647), respectively (Figure 2B). Therefore, the MD-ALL SVM prediction surpassed all other models in terms of classification rate and accuracy. For the MD-ALL PhenoGraph model, when evaluating solely the samples classified by other tools, the accuracies reached 93.7% (879/938 ALLCatchR-classified), 94.8% (789/832 ALLSorts-classified), and 97.1% (628/647 ALLSpice-classified), indicating that PhenoGraph is also a highly reliable prediction model for B-ALL subtyping (Online Supplementary Table S8). Among the prediction models, ALLSpice had the lowest number of correctly classified samples (N=573). Moreover, key B-ALL subtypes, such as Ph-like and ZEB2/CEBP, are not included in ALLSpice, significantly limiting its potential for clinical use. Therefore, ALLSpice will be excluded from further comparisons.
In terms of specificity, MD-ALL (SVM and PhenoGraph), ALLCatchR and ALLSorts demonstrated excellent performance for most subtypes. However, differences were observed in certain subtypes: MD-ALL algorithms outperformed ALLCatchR and ALLSorts in Ph/Ph-like subtype, while ALLCatchR and ALLSorts excelled in Hyperdiploid subtype (Figure 2C). As for sensitivity, ALLSorts consistently underperformed compared with MD-ALL and ALLCatchR in most subtypes, particularly those with less distinct GEP clusters, such as iAMP21 (35.6%), Low hypodiploid (50.0%), PAX5alt (72.4%), and Hyperdiploid (70.6%). Of note, ALLCatchR performed very well in the test cohort; especially in the Ph/Ph-like group, ALLCatchR surpassed both MDALL algorithms in sensitivity (97.2%) at the expense of reduced specificity (95.9%) compared to MD-ALL. As both MD-ALL SVM and ALLCatchR use the SVM algorithm, the high sensitivity levels achieved by these two models are anticipated. However, MD-ALL SVM surpassed ALLCatchR in terms of sensitivity for multiple major subtypes, such as iAMP21 (81.4% vs. 59.3%), PAX5alt (99.0% vs. 79.0%), Hyperdiploid (94.5% vs. 89.9%), ETV6::RUNX1/-like (96.0% vs. 91.3%), ZNF384 (100% vs. 97.1%), and Low hypodiploid (100% vs. 97.5%; Figure 2C)
In conclusion, the GEP-based models in MD-ALL demonstrate superior performance over alternative tools in B-ALL classification, even for the challenging subtypes.
Integrative RNA-sequencing analyses provide reliable and definitive B-acute lymphoblastic leukemia classification
Although GEP alone can provide highly accurate B-ALL classifications, sentinel genetic lesions may take precedence when GEP results are ambiguous or conflict with the genetic lesions. Additionally, genetic lesions found in
the same samples may also lead to different subtypes. For example, among the 202 Ph-positive cases in this study, 22 (10.9%) carry more than 50 chromosomes, which fit the definition of Hyperdiploid subtype. Considering the associated prognosis and potential benefit of using tyrosine kinase inhibitors, Ph subtype overrides Hyperdiploid when both sentinel genetic lesions are identified, even though
Continued on following page.
Figure 2. High accuracy of B-acute lymphoblastic leukemia subtyping with MD-ALL. (A) A heatmap showing the study cohort (N=2,955) highlights B-ALL subtypes and metadata. Each column represents a sample. Two gene expression profile (GEP)-based subtype prediction models, support vector machine (SVM) and PhenoGraph, were established within Molecular Diagnosis of Acute Lymphoblastic Leukemia (MD-ALL). *Phenocopy subtypes are identified by their similar GEP to their corresponding canonical subtypes and are thus annotated with the same colors. For the training/testing annotation, leave-one-out validation was used to evaluate the prediction for minor subtypes, which made samples in these subtypes as both training and testing data. Sex information was inferred using the package (see Methods), while race/ethnicity information was determined by the iAdmix package (see Methods). (B) A confusion matrix compares subtype predictions made by MD-ALL and alternative tools. The ground-truth subtypes of the 974-sample test cohort are displayed on the left side of each matrix, while prediction results from different models are shown at the bottom. The phenocopy subtypes and their corresponding canonical subtypes are merged for evaluation. MD-ALL, comprising SVM and PhenoGraph models, is compared with ALLCatchR, ALLSorts, and ALLSpice, with ALLSpice showing the largest number of unclassified samples. (C) Sensitivity and specificity of GEP-based B-ALL classification. The same test cohort (N=974) described above was used to evaluate all different models. The ZEB2/CEBP and CDX2 (CDX2/UBTF) subtypes are not available in the ALLSorts model. Detailed sensitivity and specificity values are labeled for conditions where they are not 100%. The evaluated sample sizes per subtype are annotated in parentheses.
strong Hyperdiploid GEP was observed in eight cases (Online Supplementary Table S1). By integrating multiple aspects of information, a more well-rounded subtyping result can be achieved. For example, 43 Near haploid cases were identified based on the total chromosome number (≤30). These cases were predicted as Hyperdiploid (N=40) or Low hypodiploid (N=3) by our GEP models. Of the three cases with Low hypodiploid GEP, they all carry 28 chromosomes, which are on the boundary of defining Near haploid and Low hypodiploid subtypes. Furthermore, they all carry TP53 hotspot mutations with high mutant allele frequency (>90%), which resembles the features of Low hypodiploid.42 Therefore, they should be categorized as Low hypodiploid subtype. This scenario highlights the importance of integrating GEP predictions with signature genetic lesions to accurately determine the subtypes (Online Supplementary Table S1).
In MD-ALL, users can provide raw translocations and sequence mutations for integrative B-ALL classification. Upon re-analysis of 2,955 RNA-seq samples, 96 sentinel gene rearrangements and 587 mutations were identified (Online Supplementary Tables S5 and S6). By integrating GEP and mutation information, MD-ALL calls RNAseqCNV to identify aneuploid subtypes, such as Hyperdiploid, Low hypodiploid, Near haploid, and even iAMP21. Our previous work on RNAseqCNV33 demonstrated 100% accuracy in determining aneuploid subtypes, though iAMP21 detection was not mentioned. In this study, we observed high ac-
curacy (35/36) of detecting iAMP21 in B-ALL samples with confirmed iAMP21 status (by SNP array), further broadening the utility of RNA-seq for defining B-ALL subtypes (Online Supplementary Figure S3; Online Supplementary Table S9). In addition, MD-ALL provides visualization of subtyping results for test sample in SVM and PhenoGraph models using different numbers of genes (Figure 3A). This visualization aids in assessing the stability of the subtyping results. Furthermore, a UMAP plot of the test sample mapped to the reference cohort using all the feature genes (N=1,058) offers an insightful overview of the sample’s relationship to the reference (Figure 3B). As certain gene rearrangements are strongly associated with specific gene expressions, such as CRLF2 overexpression commonly seen in CRLF2-rearranged cases, MD-ALL can display a gene’s expression across all B-ALL subtypes to verify the reliability of specific fusions or subtypes (Figure 3C). The JAK2 p.R683 hotspot mutations, known for their high concurrence in CRLF2-rearranged cases,43 further confirm the reliability of the IGH::CRLF2 fusion. MD-ALL then compiles all input information to assist the final subtype classification. For instance, a sample with an IGH::CRLF2 fusion and GEP-based Ph/Ph-like prediction, but lacking BCR::ABL1 fusion, can be definitively classified as Ph-like (Figure 3D). In order to facilitate definitive B-ALL classification for all subtypes, MD-ALL incorporates a knowledge-based subtyping guideline that integrates both genetic lesions and GEP features (Table 1).
With the technical, biological, and clinical considerations
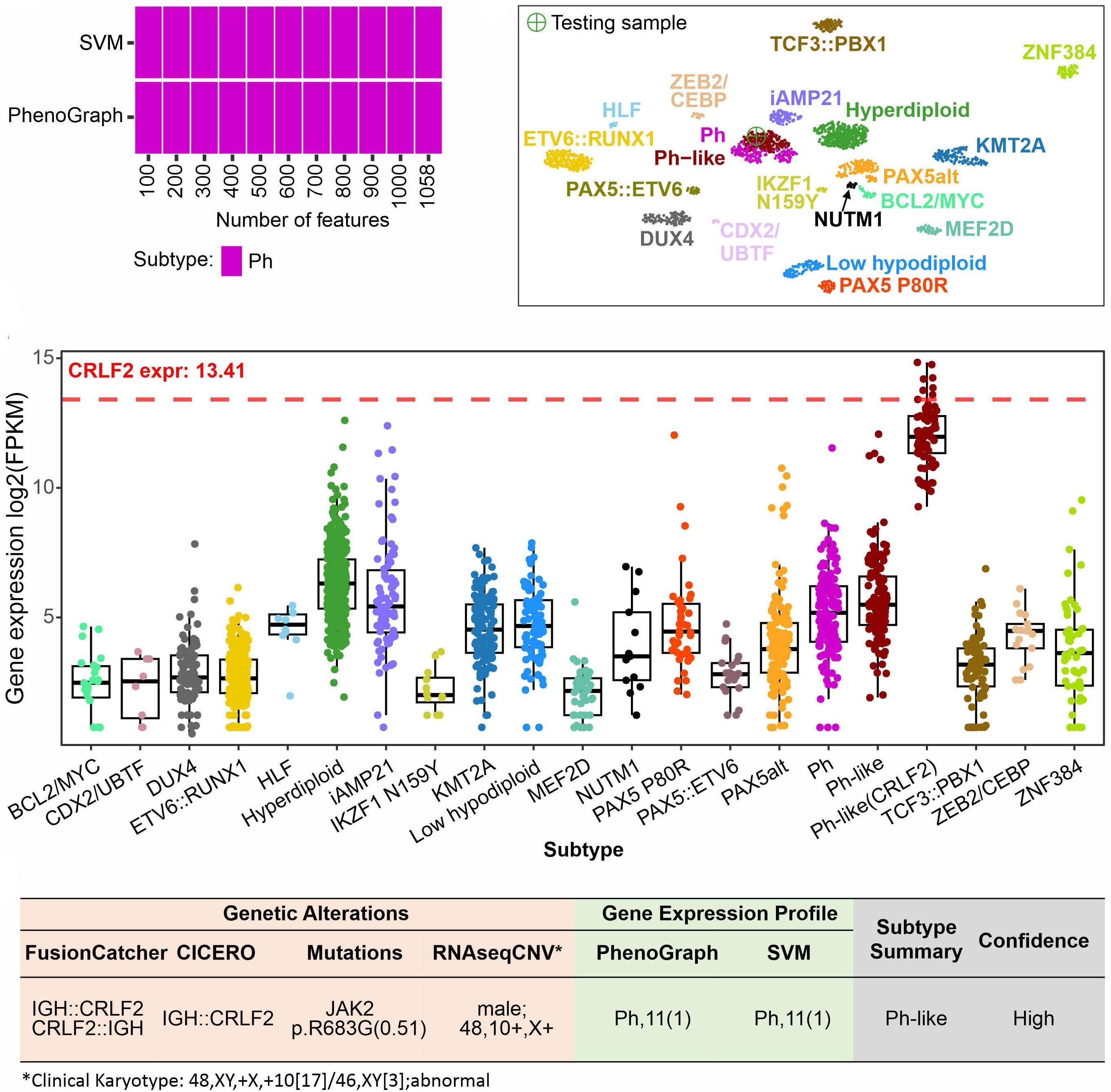
Figure 3. Integrative summary of B-acute lymphoblastic leukemia classification by MD-ALL. (A) Gene expression profile (GEP) -based subtype prediction by support vector machine (SVM) and PhenoGraph models. Different numbers of feature genes are used in the prediction models to evaluate classification robustness. The test sample was consistently predicted as the Ph subtype. (B) The test sample is mapped to a predefined uniform manifold approximation and projection (UMAP) space for visualizing GEP-based classification. The UMAP uses 1,058 features genes. The test sample clusters with the Ph/Ph-like group, which agrees with the SVM and PhenoGraph prediction. (C) Expression of a specific gene across different B-acute lymphoblastic leukemia (B-ALL) subtypes. Ph-like (CRLF2) is shown as a separate group here for confirming CRLF2 rearrangements. Users can specify a gene to examine its expression for validating genetic lesions (e.g., overexpression of CRLF2 in CRLF2-rearranged cases) or potential subtypes. (D) Summary of Molecular Diagnosis of Acute Lymphoblastic Leukemia (MD-ALL) to assist B-ALL classification. The genetic lesions, which include fusions, mutations, large-scale copy number variation (CNV), are integrated with GEP-based prediction by PhenoGraph and SVM to assist the classification of the test sample’s B-ALL subtype.
applied in the MD-ALL platform, we developed a decision-tree-based pipeline to integrate multiple aspects of information acquired from RNA-seq to accurately determine B-ALL subtypes and the associated confidence score (Figure 4). Basically, a step-by-step process is taken for each sample to determine the subtype based on the GEP and signature genetic lesions, and then assigns the confidence score.
Of the total cohort comprising 2,955 samples, 2,689 (91.0%)
were classified with high confidence. Among these, 2,682 (99.7%) were consistent with the manually curated subtypes (Online Supplementary Table S10). In the seven samples with discrepancies:
• Two curated B-other cases without detectable iAMP21 alteration were predicted as iAMP21, based on GEP and chr21 gain.
• In contrast, two curated iAMP21 cases were defined as Hyperdiploid (by GEP and 52 chromosomes) and PAX5alt
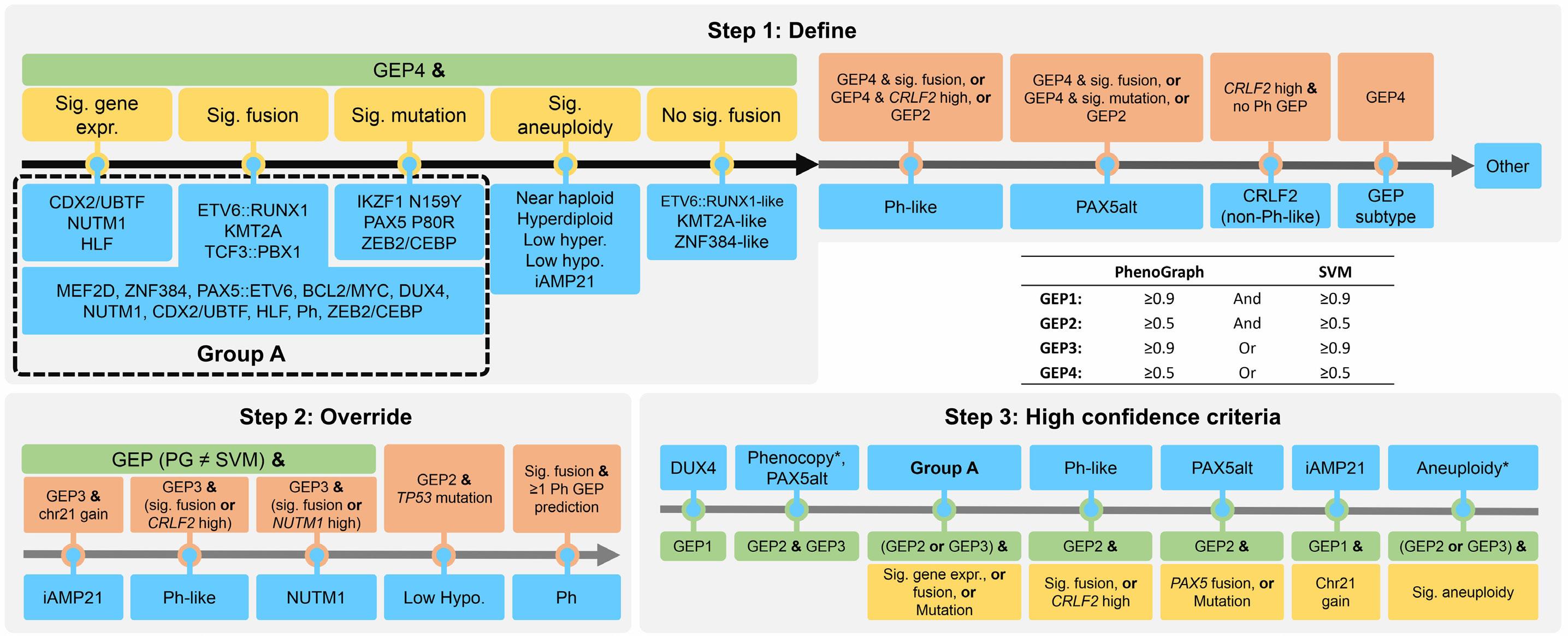
Figure 4. Integrative B-acute lymphoblastic leukemia classification pipeline. The integrative B-ALL classification pipeline implemented in Molecular Diagnosis of Acute Lymphoblastic Leukemia (MD-ALL) consists of 3 steps: Step 1. Define. In this step, MD-ALL integrates gene expression profiles (GEP), signature (sig.) gene expression (expr.), fusions, mutations, and aneuploidies to define different B-ALL subtypes. The sequence in which subtypes are defined in this step is carefully orchestrated, primarily following the order from the most distinct subtypes, such as the ones in Group A, to less distinct ones, such as the aneuploid and phenocopy subtypes. Step 2. Override. Due to the potential overlap of some subtypes, 5 additional rules were implemented to override the subtypes defined in Step 1, which include, Ph-like, NUTM1, Low hypodiploid, and Ph. Step 3. Define high confidence score. With the subtypes defined in Step 1 and 2, a high confidence score will be assigned if they meet specific criteria, which are developed based on the GEP prediction scores and signature genetic alterations. For the less recognized subtypes such as Low hyperdiploid and CRLF2 (non-Ph-like), a low confidence is assigned. Hyper.: Hyperdiploid; hypo.: hypodiploid; GEP1 to GEP4 are defined based on the GEP prediction by PhenoGraph (PG) and SVM shown in a table. In Step 3, Phenocopy* subtypes include Ph-like, ETV6::RUNX1-like, KMT2A-like, and ZNF384-like, and Aneuploidy* subtypes include Near haploid, Hyperdiploid, and Low hypodiploid.
(by GEP) by MD-ALL, respectively.
• One curated KMT2A-like case was predicted as KMT2A subtype, based on GEP and a KMT2A::BIRC3 rearrangement. Due to the low confidence in the KMT2A rearrangement, which was supported by only four reads, the sample was eventually classified as KMT2A-like after manual curation.
• One curated Ph case was classified as Low hypodiploid based on GEP and a TP53 mutation. However, this classification was overridden and labeled as Ph subtype due to the detected BCR::ABL1 fusion.
• One curated ETV6::RUNX1 case with a predicted Phlike subtype, because of a strong Ph GEP signature, was eventually classified as ETV6::RUNX1 subtype based on an ETV6::RUNX1 fusion.
Of the samples with low confidence scores (N=266), 53.0% (N=141) are concordant with the manually curated results.
Approximately half of the 266 samples (N=130) are classified as aneuploid or iAMP21 subtypes, which can be easily confirmed by manually checking the RNAseqCNV or available karyotype information. In the remaining 136 samples, the subtypes can be distinguished by checking the GEP-based predictions and the signature genetic alterations provided by MD-ALL.
In summary, MD-ALL integrates multiple aspects of information derived from RNA-seq data to provide highly
accurate and definitive B-ALL classification.
Using high-quality scRNA-seq data, we compiled a GEP reference consisting of over 10K cells that represent 20 major blood cell types (see Methods; Figure 5A). Subsequently, we used the single-cell GEP reference to deconvolute the bulk RNA-seq GEP of different B-ALL subtypes (Online Supplementary Table S11). Our analysis revealed that the PAX5 P80R and KMT2A subtypes carry a strong Pro B1 (pre-pro B stage) signature, indicating that they are at the very early stage of B-cell development. By contrast, the BCL2/MYC subtype exhibits a strong enrichment of pre B2 and even immature B-cell signatures (Figure 5B). This suggests that the leukemic B cells are more mature, which is consistent with the observation that BCL2 and MYC rearrangements are more commonly seen in B-cell lymphomas,44 a malignancy transformed from more mature B lymphocytes. These conclusions agree with clinically reported immunophenotypic features of B-ALL subtypes18 as well as other digital deconvolution reports.45
In order to validate the digital deconvolution results, we compared the clinically reported B-cell blast ratio from 70 B-ALL samples and their inferred B-cell ratio by CIBER-
SORTx, and a high correlation was observed (correlation=0.85; 95% confidence interval [CI]: 0.76-0.9; Figure 5C; Online Supplementary Table S12). Therefore, digital deconvolution can be used to assess the potential normal cell contamination in bulk samples. In addition, we observed that samples without classified subtypes were enriched with low B-cell ratio (35.9% of 64 samples have <50% B-cell ratio) compared to those with defined subtypes (3.1% of 2,718 samples have <50% B-cell ratio). This finding indicates that contamination of normal cells can interfere with classification of B-ALL subtypes.
High sensitivity B-acute lymphoblastic leukemia subtyping at a single-cell level
In bulk RNA-seq, it is critical to obtain pure leukemic cells prior to RNA-seq assay to ensure that the GEP represents the disease. However, in clinical settings, patient samples often contain a low proportion of leukemic cells. As a result, B-cell blasts require proper enrichment prior to analysis. Even with B-cell enrichment, samples may still be contaminated by normal B-cell blasts, or contain an inadequate number of enriched cells for bulk RNA-seq. In order to address these challenges, we explored the potential of using single-cell GEP to identify B-cell blasts (proto pre-B cells) using the GEP reference representing major blood cell types (Figure 5A). After identifying the blast cells,
we annotated them to different B-ALL subtypes using the GEP reference compiled from bulk RNA-seq (Figure 1B). By using public scRNA-seq datasets,46,47 we can reliably (>50% of the B-cell blasts are correctly predicted) identify multiple B-ALL subtypes, such as KMT2A, ETV6::RUNX1/-like, Hyperdiploid, Ph, DUX4, MEF2D, PAX5alt, TCF3::PBX1, and ZNF384 (Figure 6; Online Supplementary Figure S4), even in samples with blast percentages below 20% (Figure 6B). Furthermore, a cluster of B cells was observed with a mixture of different B-ALL subtypes in the KTM2A case (Figure 6A), indicating that they are normal B-cell blasts. In summary, our study highlights the potential of single-cell analysis in the sensitive and accurate detection of leukemic cells and their B-ALL subtypes. With the advent of more cost-effective scRNA-seq platforms and the continual decrease in sequencing costs, single-cell analysis is expected to revolutionize clinical diagnosis of granular disease subtypes.
MD-ALL: an integrative platform for B-acute lymphoblastic leukemia classification MD-ALL integrates both GEP and signature genetic lesions to provide a one-stop solution for B-ALL classification. This is especially important to distinguish the canonical subtypes (e.g., Ph and ETV6::RUNX1) from their phenocopy counterparts (e.g., Ph-like, and ETV6::RUNX1-like, respectively). In
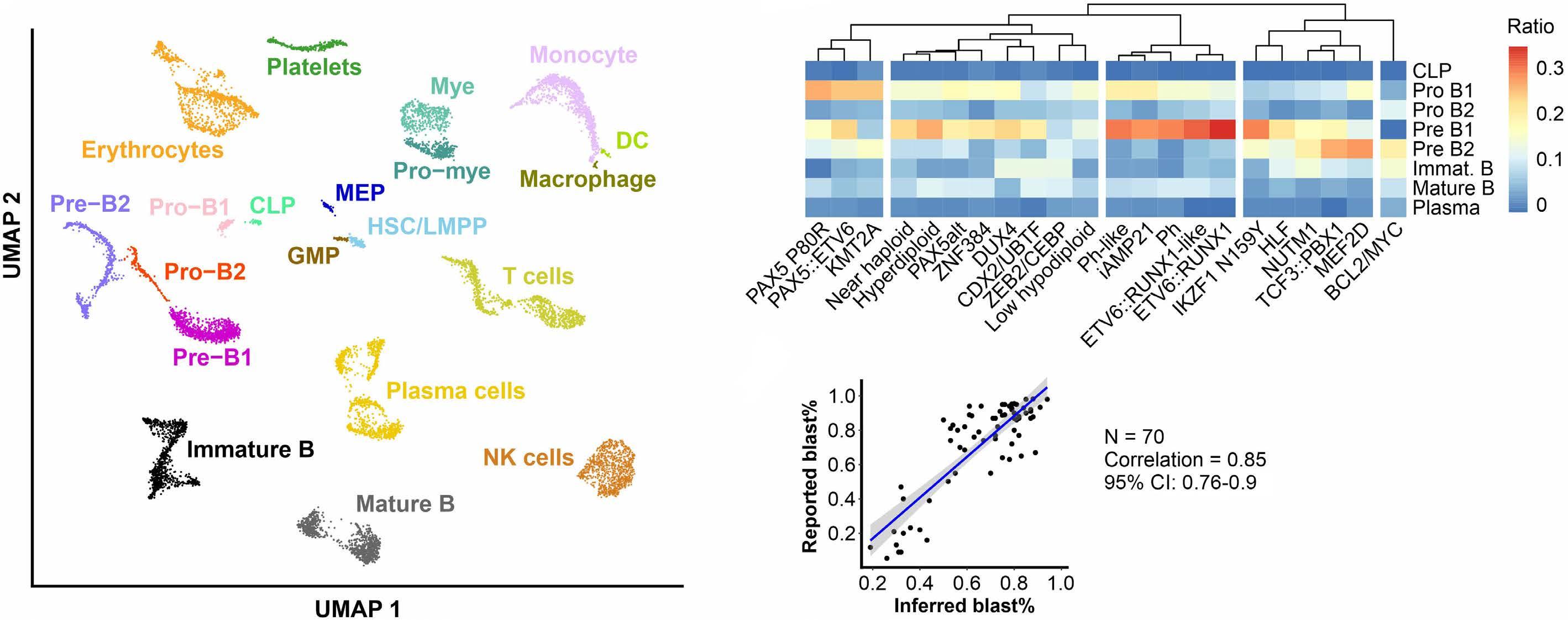
Figure 5. Deconvolution of bulk gene expression profile of B-acute lymphoblastic leukemia subtypes. (A) Uniform manifold approximation and projection (UMAP) of single-cell gene expression reference of the primary blood cell types. Over 10K cells representing 20 primary blood cell types were selected from the 1-Million Immune Cells project (see Online Supplementary Methods). (B) Cells are classified into granular B cell differentiation stages, including common lymphoid progenitor (CLP), pro-B1 (early pro-B), pro-B2 (late pro-B), pre-B1 (large pre-B), and pre-B2 (small pre-B). HSC: hematopoietic stem cell; LMPP: lymphoid-primed multipotential progenitor; DC: dendritic cell; Mye: myelocytes; Pro-mye: promyelocytes; GMP: granulocyte-monocyte progenitor; MEP: megakaryocyte-erythrocyte progenitor; NK cell: natural killer cell. (B) Heatmap of different B-acute lymphoblastic leukemia (B-ALL) subtypes and their inferred B-cell differentiation stages. For each subtype, the median value of each B-cell stage is calculated and presented in the heatmap. The Euclidean distance and Ward’s minimum variance clustering method were used to generate the clusters. (C) Correlation of digitally inferred and clinically reported blast percentage (blast%). The inferred blast% is estimated by combining B-lineage cells from pro B1 to mature B stages (see Methods). Seventy samples from a cohort provided by the ALLSorts package were used in this analysis.
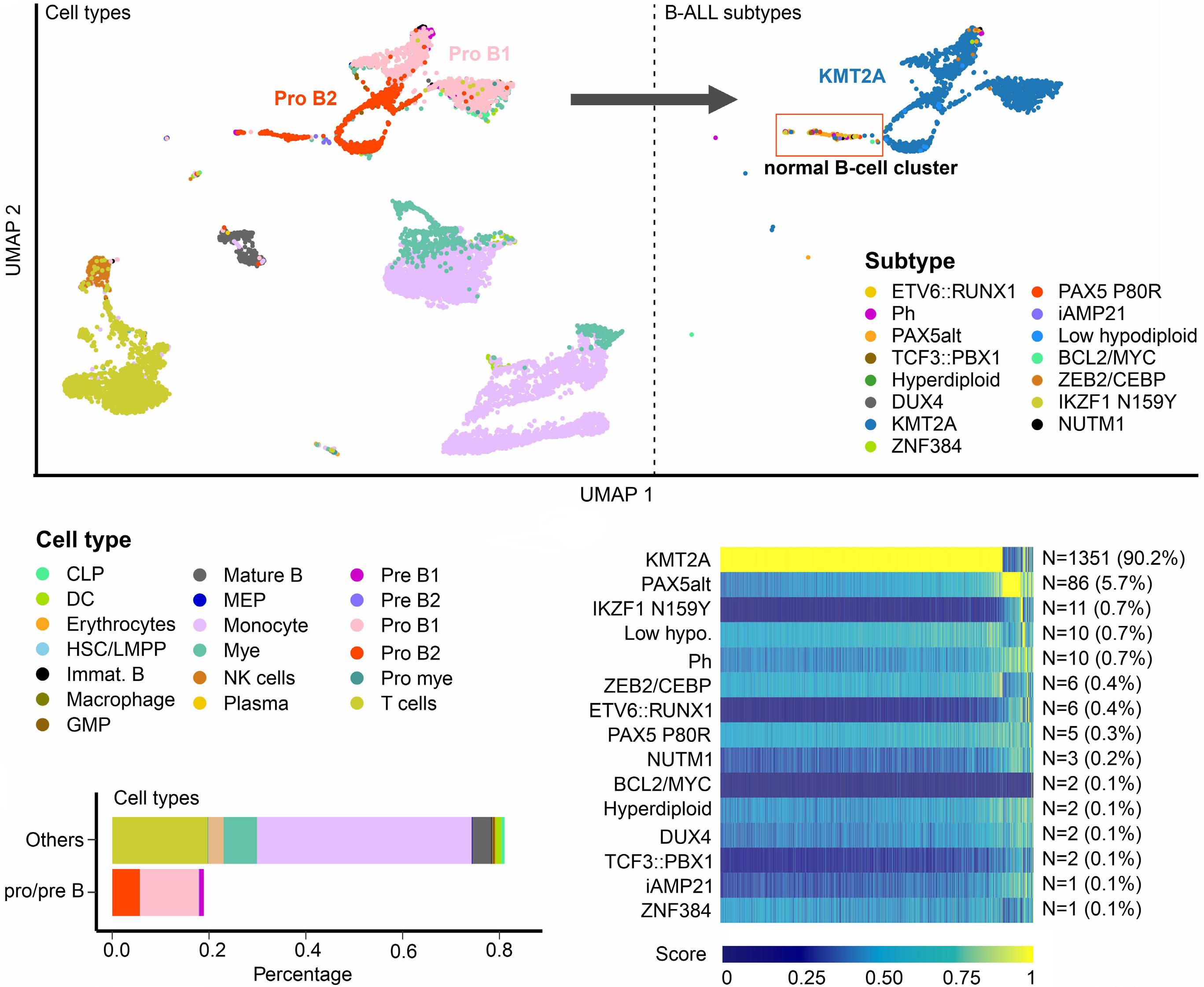
Figure 6. B-acute lymphoblastic leukemia subtype classification at a single-cell level. (A). Single-cell RNA sequencing (scRNAseq) of a B-acute lymphoblastic leukemia (B-ALL) sample at diagnosis shown in a UMAP plot. The abnormally enriched B-cell blasts (pro- to pre-B cells) represent the leukemic cells. With the gene expression profile (GEP) reference of the B-ALL subtypes, the majority of the B-cell blasts are reliably predicted as KMT2A subtype, which is consistent with the reported subtype. A small cluster (highlighted in a red rectangle) observed with a mixture of different B-ALL subtypes indicates that they are normal B-cell blasts. (B) A bar graph shows the distribution of different cell types. Less than 20% of the test sample are B-cell blasts, which could be challenging to be accurately identified as KMT2A subtype based on bulk GEP prediction. (C) Heatmap of subtype prediction score shows that over 90% of the B-cell blasts exhibit highly reliable KMT2A GEP signature. Low hypo.: Low hypodiploid; CLP: common lymphoid progenitor; HSC: hematopoietic stem cell; LMPP: lymphoid-primed multipotential progenitor; DC: dendritic cell; Mye: myelocytes; Pro-mye: promyelocytes; GMP: granulocyte-monocyte progenitor; MEP: megakaryocyte-erythrocyte progenitor; NK cell: natural killer cell.
addition, an interactive graphical interface was provided within MD-ALL, making the tool accessible to users with limited or no computational background. The minimum required input is the raw read count from RNA-seq data. The test samples will be normalized against an internal reference cohort, which consists of 234 samples representing all reported subtypes (Online Supplementary Table S13). This reference cohort was sequenced using various library preparation kits, sequencing lengths, and strandness. Therefore, normalization against this reference helps
minimize potential batch effects. Users may also provide raw output of gene rearrangements and mutations to MDALL to perform automatic filtering and genetic alteration identification based on the signature lesions identified in the large B-ALL cohort. Subsequently, MD-ALL will integrate the information of genetic alterations and GEP for robust B-ALL classification (Figure 7A). Furthermore, MDALL also provides the functionality for single-cell B-ALL classification, requiring only the raw read count output from standard scRNA-seq analysis (Figure 7B).
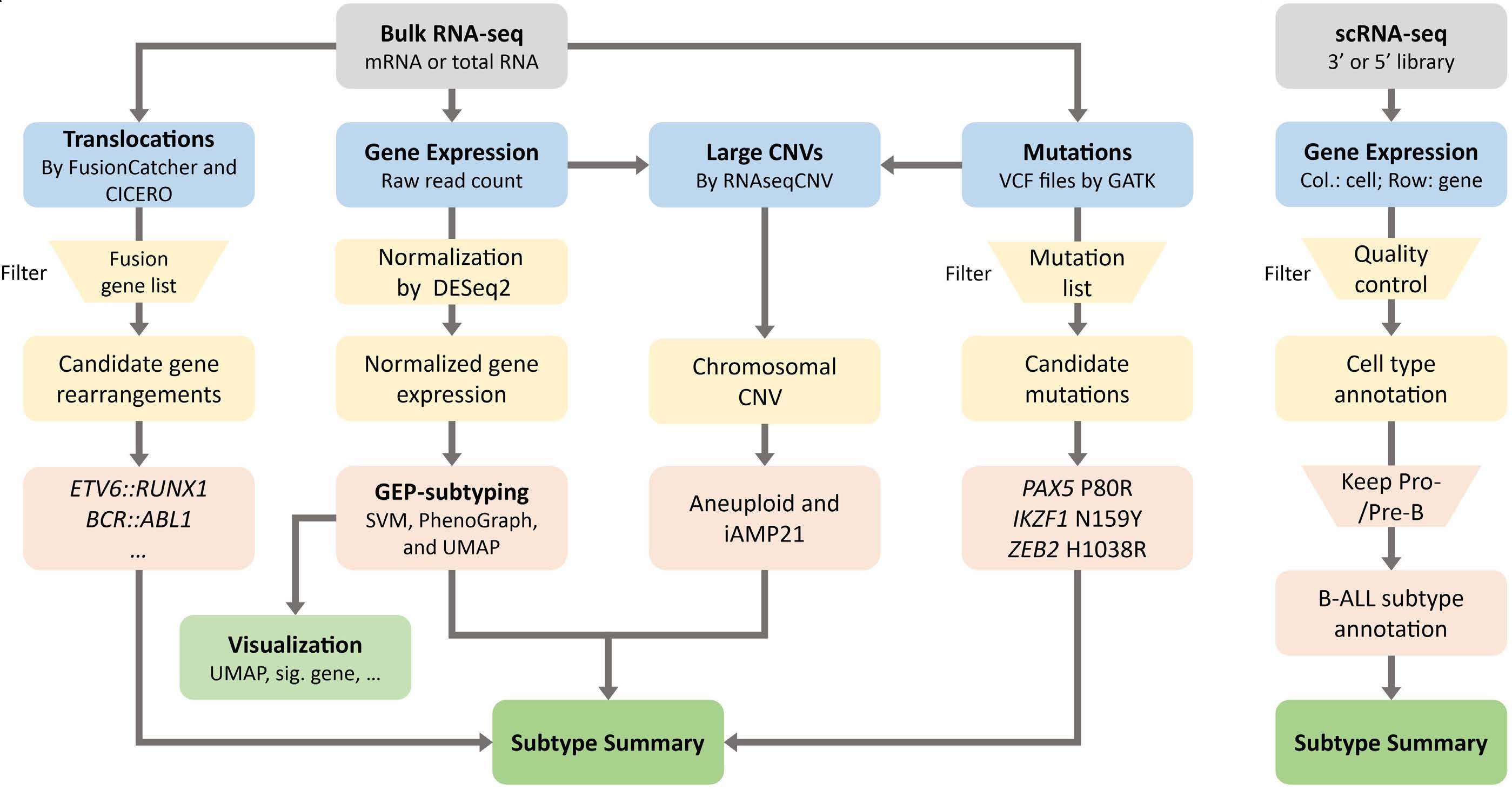
Figure 7. Summary of integrative B-acute lymphoblastic leukemia classification by MD-ALL. Molecular Diagnosis of Acute Lymphoblastic Leukemia (MD-ALL) accepts both bulk and single-cell (sc) RNA-sequencing (RNA-seq) data for B-acute lymphoblastic leukemia (B-ALL) classification. (A) Bulk analysis is the main function of MD-ALL, which accepts 3 types of standard output from bulk RNA-seq data: translocations (optional; raw output from FusionCatcher and/or CICERO), gene expression read count (required; called by HTSeq or FeatureCount), and sequence mutations (optional; Variant Call Format [VCF] files called by GATK). Based on the input data, four aspects of information will be identified: i) the input translocations are compared with an internal reference to identify signature fusion genes; ii) the gene expression data normalized from raw read count are analyzed by support vector machine (SVM) and PhenoGraph to predict the subtype and shown in a uniform manifold approximation and projection (UMAP) plot; iii) the variants in the provided VCF files are annotated to identify the signature gene mutations; and iv) the gene expression and mutation information are integrated by the RNAseqCNV package (see Methods) to identify chromosomal CNV, which will assist the identification of aneuploid and intrachromosomal amplification of chromosome 21 (iAMP21) subtypes. Then, a comprehensive subtype summary from the 4 aspects of information will be integrated to determine the subtypes of the test samples. (B) For scRNA-seq-based B-ALL classification, the input data is a count matrix with genes in rows and cells in columns. This read count matrix can be generated from either 3’ or 5’ scRNA-seq libraries using standard analysis pipelines. A basic quality control analysis is then performed to remove cells or genes with low sequencing coverage (see Online Supplementary Appendix). With the cell type gene expression profile reference, each test cell is annotated and only the B-lineage blast cells, which are pro- and pre-B cells, are retained for subsequent B-ALL subtyping, with results summarized in the report.
Thus, with minimal bioinformatics assistance to generate the raw information of GEP and genetic lesions, users can manage the subsequent analysis using MD-ALL to achieve integrative B-ALL classification.
In this study, we present the first RNA-seq analysis platform capable of integrating both genetic lesions and GEP features for B-ALL classification. For more than 90% of the study cohort, the integrative analysis led to highly accurate B-ALL classification based on multiple layers of information. Additionally, the platform supplies detailed information for users to review and adjust the results as necessary.
This study is based on one of the largest B-ALL RNA-seq cohorts to establish a GEP reference representing all reported B-ALL subtypes, achieving high accuracy and sensitivity compared with alternative tools. By integrating genetic lesions, which other tools lack, subtypes can be determined more accurately, making this approach more feasible for future translational application in clinical settings. Using the GEP reference compiled from bulk RNA-seq, we also explored the B-cell differentiation stages of different B-ALL subtypes. Our observations confirmed that certain B-ALL subtypes are blocked at early B-cell progenitor stages, while others progress to more mature stages. Moreover, some subtypes have been observed to have overlapping GEP features, such as iAMP21, PAX5alt, and Ph/Ph-like. Incorporating distinct B-cell differentiation patterns of different subtypes might be beneficial for better separation
of these subtypes.
As genomic analysis advances towards single-cell resolution, we have demonstrated the feasibility of using GEP reference derived from bulk RNA-seq for accurate single-cell B-ALL classification in multiple subtypes. Currently, generating comparable samples size of single-cell data remains challenging due to technological and cost limitations. Moreover, scRNA-seq is unable to provide as comprehensive transcript abundance as bulk RNA-seq, and different scRNA-seq library preparation kits have been reported with larger batch effects compared with bulk RNA-seq. As a result, bulk RNA-seq remains the optimal platform for generating bona fide GEP signatures for each B-ALL subtype.
The classification of B-ALL subtypes using RNA-seq is revolutionizing clinical practice. Moreover, genomic data such as whole-genome sequencing can provide a more comprehensive understanding of genetic alterations. These results can further confirm the subtypes identified by RNA-seq. Importantly, genetic alterations can further differentiate patients within the same subtypes into more granular prognosis subgroups, making them critical complementary assays for B-ALL classification.48,49
In conclusion, we introduce MD-ALL, a highly reliable and accurate bioinformatics platform that serves the research and clinical fields for integrative B-ALL classification based on bulk or single-cell RNA-seq.
1. Brady SW, Roberts KG, Gu Z, et al. The genomic landscape of pediatric acute lymphoblastic leukemia. Nat Genet. 2022;54(9):1376-1389.
2. Zhang J, McCastlain K, Yoshihara H, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48(12):1481-1489.
3. Gocho Y, Kiyokawa N, Ichikawa H, et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2015;29(12):2445-2448.
4 Gu Z, Churchman M, Roberts K, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331.
5. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
6. Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200-1228.
7. Hiemenz MC, Oberley MJ, Doan A, et al. A multimodal genomics approach to diagnostic evaluation of pediatric hematologic malignancies. Cancer Genet. 2021;254-255:25-33.
8. Pui CH, Roberts KG, Yang JJ, Mullighan CG. Philadelphia chromosome-like acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. 2017;17(8):464-470.
9. Tran TH, Langlois S, Meloche C, et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: a report from the
No conflicts of interest to disclose.
ZH and ZG conceived and designed the study, analyzed the genomics data, developed classification models, and wrote the manuscript. ZJ, JL and HH assisted the data analysis. AM provided HPC support for the data analysis.
The authors would like to acknowledge the COH Center for Informatics, and the utilization of the POSEIDON platforms for data exploration, visualization, and analysis.
Funding
This work was supported by the Research Start-Up Budget from the Beckman Research Institute of the City of Hope, the Leukemia and Lymphoma Society Special Fellow Award, NIH/NCI Pathway to Independence Award R00 CA241297, Andrew McDonough B+ Childhood Cancer Research Grant, Leukemia Research Foundation New Investigator Grant, and The V Foundation for Cancer Research V Scholar Award to ZG.
Data-sharing statement
MD-ALL code, relevant datasets, and detailed tutorial are freely available from https://github.com/gu-lab20/MD-ALL.
DFCI ALL Consortium Protocol 16-001. Blood Adv. 2022;6(4):1329-1341.
10 Walter W, Shahswar R, Stengel A, et al. Clinical application of whole transcriptome sequencing for the classification of patients with acute lymphoblastic leukemia. BMC Cancer. 2021;21(1):886.
11. Makinen VP, Rehn J, Breen J, Yeung D, White DL. Multi-cohort transcriptomic subtyping of B-cell acute lymphoblastic leukemia. Int J Mol Sci. 2022;23(9):4574.
12. Schmidt BM, Brown LM, Ryland G, et al. ALLSorts: a RNA-Seq subtype classifier for B-cell acute lymphoblastic leukemia. Blood Adv. 2022;6(14):4093-4097.
13. Beder T, Hansen B-T, Hartmann AM, et al. The gene expression classifier ALLCatchR identifies B-precursor ALL subtypes and underlying developmental trajectories across age. Hemasphere. 2023;7(9):e939.
14 Gu Z, Churchman ML, Roberts KG, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296-307.
15. Waanders E, Gu Z, Dobson SM, et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020;1(1):96-111.
16. Montefiori LE, Bendig S, Gu Z, et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage-ambiguous stem cell leukemia. Cancer Discov. 2021;11(11):2846-2867.
17 Kimura S, Montefiori L, Iacobucci I, et al. Enhancer retargeting of CDX2 and UBTF::ATXN7L3 define a subtype of high-risk B-progenitor acute lymphoblastic leukemia. Blood.
2022;139(24):3519-3531.
18. Paietta E, Roberts KG, Wang V, et al. Molecular classification improves risk assessment in adult BCR-ABL1-negative B-ALL. Blood. 2021;138(11):948-958.
19 Jeha S, Choi J, Roberts KG, et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease–directed therapy. Blood Cancer Discov. 2021;2(4):326-337.
20. Li Z, Lee SHR, Chin WHN, et al. Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv. 2021;5(23):5226-5238.
21. Li Z, Jiang N, Lim EH, et al. Identifying IGH disease clones for MRD monitoring in childhood B-cell acute lymphoblastic leukemia using RNA-Seq. Leukemia. 2020;34(9):2418-2429.
22. Qian M, Zhang H, Kham SK-Y, et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities ofEP300andCREBBP. Genome Res. 2017;27(2):185-195.
23. Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867-2873.
24. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21.
25. Anders S, Pyl PT, Huber W. HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166-169.
26. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923-930.
27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
28. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882-883.
29. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303.
30. Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773-782.
31. Tian L, Li Y, Edmonson MN, et al. CICERO: a versatile method for detecting complex and diverse driver fusions using cancer RNA sequencing data. Genome Biol. 2020;21(1):126.
32. Nicorici D, Şatalan M, Edgren H, et al. FusionCatcher – a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv. 2014;011650. doi: https://doi.org/10.1101/011650 [preprint, not peer-reviewed].
33. Barinka J, Hu Z, Wang L, et al. RNAseqCNV: analysis of largescale copy number variations from RNA-seq data. Leukemia. 2022;36(6):1492-1498.
34. Bansal V, Libiger O. Fast individual ancestry inference from DNA
sequence data leveraging allele frequencies for multiple populations. BMC Bioinformatics. 2015;16:4.
35. Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68-74.
36. Lee SHR, Antillon-Klussmann F, Pei D, et al. Association of genetic ancestry with the molecular subtypes and prognosis of childhood acute lymphoblastic leukemia. JAMA Oncol. 2022;8(3):354-363.
37. Levine JH, Simonds EF, Bendall SC, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162(1):184-197.
38. Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE: synthetic minority over-sampling technique. J Artif Intell Res. 2002;16:321-357.
39 Kursa MB, Rudnicki WR. Feature selection with the Boruta Package. J Stat Softw. 2010;36(11):1-13.
40. Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33(5):495-502.
41. Aran D, Looney AP, Liu L, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163-172.
42. Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242-252.
43. Harvey RC, Mullighan CG, Chen IM, et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood. 2010;115(26):5312-5321.
44 Rosenthal A, Younes A. High grade B-cell lymphoma with rearrangements of MYC and BCL2 and/or BCL6: double hit and triple hit lymphomas and double expressing lymphoma. Blood Rev. 2017;31(2):37-42.
45. Khabirova E, Jardine L, Coorens THH, et al. Single-cell transcriptomics reveals a distinct developmental state of KMT2A-rearranged infant B-cell acute lymphoblastic leukemia. Nat Med. 2022;28(4):743-751.
46. Witkowski MT, Dolgalev I, Evensen NA, et al. Extensive remodeling of the immune microenvironment in B cell acute lymphoblastic leukemia. Cancer Cell. 2020;37(6):867-882.
47. Caron M, St-Onge P, Sontag T, et al. Single-cell analysis of childhood leukemia reveals a link between developmental states and ribosomal protein expression as a source of intraindividual heterogeneity. Sci Rep. 2020;10(1):8079.
48. Ryan SL, Peden JF, Kingsbury Z, et al. Whole genome sequencing provides comprehensive genetic testing in childhood B-cell acute lymphoblastic leukaemia. Leukemia. 2023;37(3):518-528.
49 Leongamornlert D, Gutierrez-Abril J, Lee SW, et al. Diagnostic utility of whole genome sequencing in adults with B-other acute lymphoblastic leukemia. Blood Adv. 2023;7(15):3862-3873.
Willem P.J. Cox,1 Nils Evander,1 Dorette S. van Ingen Schenau,1 Gawin R. Stoll,1 Nadia Anderson,1 Lieke de Groot,1 Kari J.T. Grünewald,1 Rico Hagelaar,1,2 Miriam Butler,1 Roland P. Kuiper,1,3 Laurens T. van der Meer1# and Frank N. van Leeuwen1#
1Princess Máxima Center for Pediatric Oncology and 2Oncode Institute and 3Department of Genetics, Utrecht University Medical Center, Utrecht University, Utrecht, the Netherlands
#LTvdM and FNvL contributed equally as senior authors.
Correspondence: F.N. van Leeuwen
f.n.vanleeuwen@prinsesmaximacentrum.nl
Received: August 17, 2023.
Accepted: December 13, 2023. Early view: December 21, 2023.
https://doi.org/10.3324/haematol.2023.284101
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
In pediatric acute lymphoblastic leukemia (ALL), mutations/deletions affecting the TP53 gene are rare at diagnosis. However, at relapse about 12% of patients show TP53 aberrations, which are predictive of a very poor outcome. Since p53-mediated apoptosis is an endpoint for many cytotoxic drugs, loss of p53 function frequently leads to therapy failure. In this study we show that CRISPR/Cas9-induced loss of TP53 drives resistance to a large majority of drugs used to treat relapsed ALL, including novel agents such as inotuzumab ozogamicin. Using a high-throughput drug screen, we identified the histone deacetylase inhibitor romidepsin as a potent sensitizer of drug responsiveness, improving sensitivity to all chemotherapies tested. In addition, romidepsin improved the response to cytarabine in TP53-deleted ALL cells in vivo. Together, these results indicate that the histone deacetylase inhibitor romidepsin can improve the efficacy of salvage therapies for relapsed TP53-mutated leukemia. Since romidepsin has been approved for clinical use in some adult malignancies, these findings may be rapidly translated to clinical practice.
With an overall survival rate that exceeds 90%, pediatric acute lymphoblastic leukemia (ALL) has one of the best outcomes among all pediatric cancers.1 However, relapsed ALL remains a significant clinical problem. Aberrations affecting the TP53 gene, although not very common at diagnosis with an incidence of less than 3%, are associated with a dismal prognosis in relapsed ALL in both children and adults in whom the incidences are 12% and 35%, respectively.2-4 Consequently, relapsed TP53-deleted ALL is now classified as ‘very high risk’ in children. Most often the function of both TP53 alleles is affected, either through direct perturbation of the allele or through dominant-negative effects of one mutated allele over the remaining allele. The encoded p53 protein acts as a transcription factor that coordinates responses to a variety of cellular stressors by controlling the expression of genes involved in cell cycle regulation, apoptosis, and metabolism.5 Importantly, p53-mediated apoptosis is an endpoint for many anti-cancer agents. Loss of p53 functions consequently
induces failure not only of classical chemotherapeutics but also of many newly introduced treatments. Hence, there is a continued need for the identification of drugs or drug combinations that effectively eradicate TP53-mutated leukemic cells. In this study, we modeled TP53 deletions in B-cell precursor (BCP)-ALL cell lines and exposed these models to the armamentarium used in the treatment of relapsed ALL. Moreover, we performed a high-throughput drug screen to identify targeted agents that could be used to restore response to current (chemo)therapies.
Information on model generation, reagents, primers, plasmids, and antibodies can be found in the Online Supplementary Material.
Cell viability assays
Cells were seeded at 0.5x106 cells/mL in 96-well plates and
cultured in the presence of test compounds for 72 hours. Viability was assessed by measuring membrane integrity or metabolic activity. Membrane integrity was measured by flow cytometry using amine staining (LIVE/DEAD™ Fixable Far-Red Dead Cell Stain Kit, L34974; Thermo Fisher Scientific) and metabolic activity was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 475989; Sigma-Aldrich) conversion as the cell readout, both according to the manufacturers’ instructions. Ex vivo co-culture was performed as described previously.6 For drug screening, 0.4x106 cells/mL were seeded in 384-well plates using a multidrop combi reagent dispenser (Thermo Scientific). In a fully automated system, drugs, dissolved in dimethylsulfoxide or water, were added shortly after seeding of the cells with a Biomek i7 liquid handler at the high-throughput screening facility of Princess Máxima Center. An Echo550 dispenser was used for direct drug transfers. MTT conversion was used as the cell readout according to the manufacturer’s protocol and area under the curve (AUC) values were calculated. For further downstream analysis, AUC values of p53-knockout (p53KO) cells were normalized for the wild-type (p53WT) cells per drug within each cell line.
Proteins were extracted from cells with Laemmli buffer and boiled. Lysates from equal numbers of cells were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis and subsequently transferred to a polyvinylidene fluoride membrane. Blocking and staining conditions are described in Online Supplementary Table S1. Proteins were visualized with the Odyssey® CLx and accompanying Image Studio software (LI-COR Biotechnology).
RNA sequencing
Cells were treated for 16 hours with 2 nM romidepsin, 200 nM cytarabine, or a combination thereof (11 conditions in triplicate, 4 in duplicate, 1 in quadruplicate). mRNA was purified from cell cultures using a NucleoSpin RNA isolation kit (740955, Machery-Nagel). RNA sequencing and data processing on samples were conducted by NovoGene (Cambridge, UK). Gene set enrichment analysis (GSEA) was performed on all genes with sum of fragments per kilobase million (FPKM) values greater than three per cell line for published gene sets in the Molecular Signatures Database (MSigDB).7-9 Differential gene expressions were filtered by P value (excluded P values <0.05) and fold changes (excluded log2 values between -0.5 and 0.5) prior to further analysis.
Animal experiments were approved by the Animal Experimental Committee of Radboud University (RU-DEC-2019-0036). Luciferase-positive RCH-ACV p53KO cells were intrafemorally engrafted in female NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl Tg (CMV-IL3,CSF2,KITLG)1Eav/J (NRG-SGM3, Jackson
Laboratory) mice. Starting 3 days after engraftment, mice were randomly assigned to groups of seven mice treated with either 17.5 mg/kg cytarabine (in double distilled H2O; 5 days on, 2 days off), 1.5 mg/kg romidepsin (2% dimethylsulfoxide, 30% PEG 300, 5% Tween 80, 62% double distilled H2O; every 1st and 4th day of the week), and/or placebo(s) for 21 days or until an endpoint was reached. Tumor load was monitored twice weekly via bioluminescence after intravenous injection of luciferin (D-luciferin, #ab143655, Abcam) and subsequent measurement of released photons using the IVIS Spectrum In Vivo Imaging System (Xenogen, now Perkin-Elmer).
Loss of p53 functions confers resistance to most drugs used to treat B-cell precursor acute lymphocytic leukemia
Aberrations that affect the TP53 gene are enriched in relapsed ALL and predict therapy failure, although how TP53 affects responses to specific drugs used for relapsed ALL has not been explored. Therefore, we generated isogenic TP53 wild-type/knockout pairs from the p53WT BCP-ALL cell lines Nalm6 and RCH-ACV (Figure 1A). For initial validation, cells were treated with either the MDM2 inhibitor nutlin-3 (which prevents proteasomal degradation of p53 by inhibiting its ubiquitination) or the anthracycline daunorubicin. Both drugs induced elevated levels of p53, as well as the two key p53 effector proteins cyclin-dependent kinase inhibitor 1 (p21) and p53 upregulated modulator of apoptosis (PUMA), in p53-proficient cells (Figure 1A, B). In addition, apoptosis was induced in response to exposure to either drug, as determined by poly-ADP ribose polymerase (PARP) cleavage. In contrast, these effects were not observed in the p53KO cells. Consistently, p53KO models were significantly more resistant to drug-induced apoptosis than were their wild-type counterparts (Figure 1C, D). Having validated these models, we tested differential sensitivity to every drug currently used in the treatment of BCP-ALL. Both p53KO cell lines showed increased resistance, relative to their p53WT counterparts, to every drug tested (Figure 1E) and exhibited a clear survival advantage after drug treatment when combined in one culture (Online Supplementary Figure S1A). These experiments indicate that ALL cells lacking TP53 are much less responsive to standardof-care drugs used to treat BCP-ALL.
Romidepsin sensitizes B-cell precursor acute lymphocytic leukemia cells to therapy
To identify therapies that can potentially restore treatment response in TP53-deficient leukemia, we performed a high-throughput drug screen to determine the sensitivity of the p53WT and p53KO cells to a custom pediatric cancer drug library, comprising 198 standard-of-care drugs as
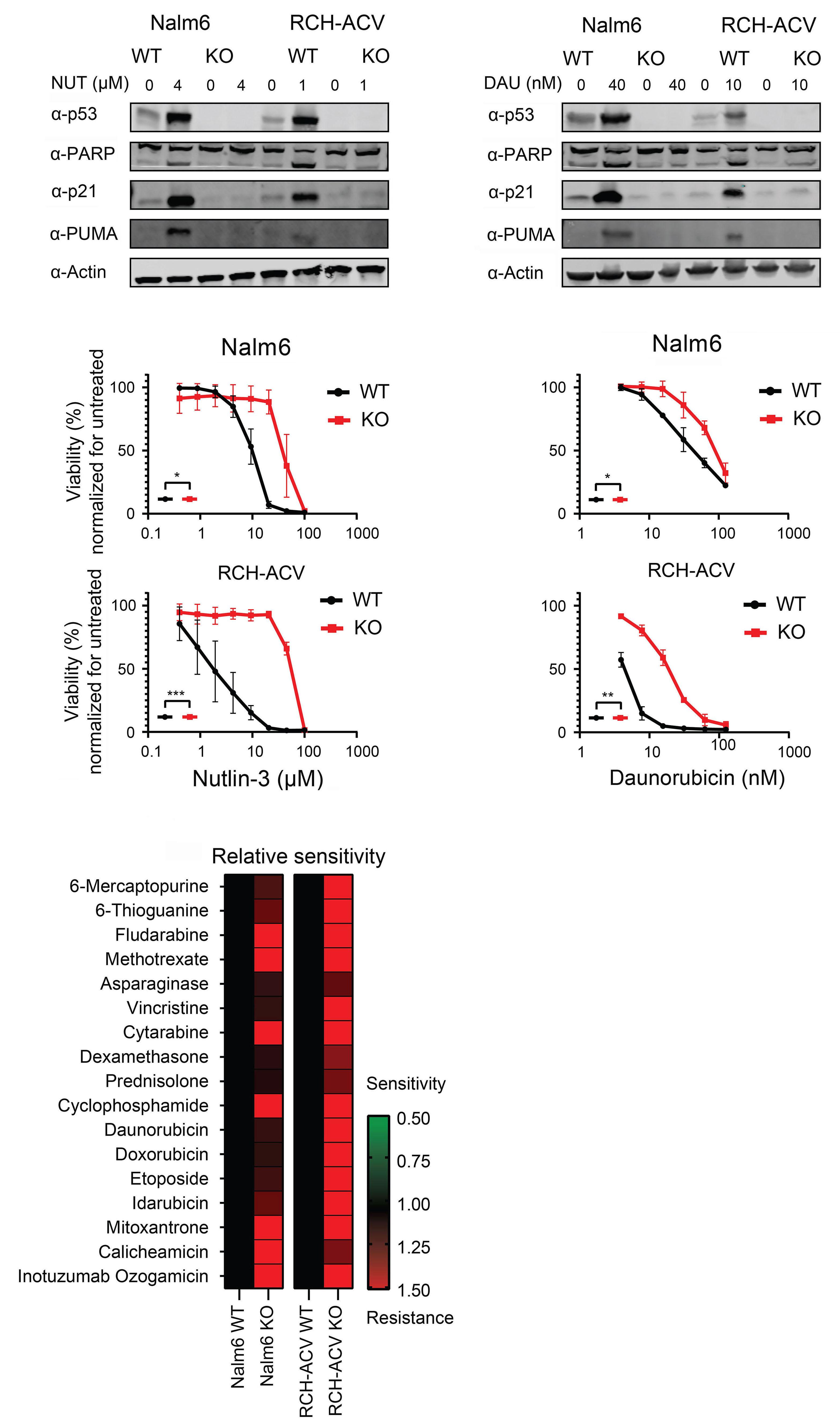
Figure 1. Loss of p53 protects B-cell precursor acute lymphocytic leukemia from chemotherapy treatment. (A, B) Western blot showing p53, PARP, p21 and PUMA expression in p53WT and p53KO cells treated with Nutlin-3 (A) or daunorubicin (B), with actin as the loading control. A representative example of three independent experiments is shown. (C, D) Dose-response curves showing viability of Nalm6 and RCH-ACV p53WT and p53KO cells treated with the indicated concentrations of Nutlin-3 (C) or daunorubicin (D) for 72 hours. Cell viability was determined by flow cytometric quantification of cells positive for amine-reactive dyes. Each data point represents the mean (± standard deviation) of two independent experiments. The area under the curve (AUC) was determined and differences were tested for statistical significance using an unpaired two-tailed t test. (E) Heatmap of results showing relative viability of p53KO Nalm6 and RCH-ACV cells as AUC ratios of dose-response curves, calculated by dividing the AUC value of each p53KO cell by the p53WT control for two independent experiments. Flow cytometric quantification of cells positive for amine-reactive dyes was used as a measure of viability after 72 hours of treatment with a range of concentrations of the indicated drugs. *P<0.05, **P<0.01, ***P<0.001. WT: wild-type; KO: knockout; NUT: Nutlin-3; DAU: daunorubicin; PARP: poly (ADP-ribose) polymerase; PUMA: p53 upregulated modulator of apoptosis.
well as targeted compounds in early-phase clinical trials. Consistent with our earlier observations, p53KO cells were resistant to MDM2 inhibitors such as idasanutlin, milademetan, and siremadlin as well as to nearly all included standard-of-care drugs (Figure 2A, extended results in Online Supplementary Figure S1B). Although we did not find any drug that selectively killed p53KO cells, we did identify compounds that were effective against both wild-type and p53KO cells. One of these compounds, the histone deacetylase (HDAC) 1/2 inhibitor romidepsin, effectively increased histone 3 acetylation and induced apoptosis in the low nanomolar range (Online Supplementary Figure S2A-C). While HDAC inhibitors frequently have little clinical activity as monotherapy, these compounds show promise when they are used to enhance conventional chemotherapy.10 We therefore tested whether romidepsin was able to potentiate the efficacy of cytarabine, an important drug in the current treatment protocols for relapsed/refractory ALL. We observed that romidepsin was able to fully restore therapy response in TP53-deficient ALL cells treated with cytarabine and enhanced response in TP53-proficient cells (Figure 2B, C). The observed synergy between cytarabine and romidepsin was recapitulated in all tested BCP-ALL cell lines, irrespective of their p53 status (Online Supplementary Figure S2D). Similarly, a combination of cytarabine with HDAC inhibitors tucidinostat and entinostat (inhibiting HDAC 1, 2, 3, and 10, and HDAC 1 and 3, respectively) produced comparable synergies (Online Supplementary Figure S2E), showing that this effect is not restricted to the specific chemical properties of romidepsin. However, as its effective concentration is up to 250 times lower than that of its tested peers, romidepsin was chosen to test the effects of HDAC inhibition on other drugs used to treat ALL. To confirm observations in cell lines, we tested the efficacy of combining romidepsin and cytarabine in 19 patient-derived xenografts (PDX), of which three p53 wildtype, six p53-aberrant (mutant and/or deleted), and ten with an undetermined p53 status (Figure 2D, viability curves in Online Supplementary Figure S3, p53 status in Online Supplementary Table S2). Regardless of p53 status, 17 PDX showed clear reductions in AUC values of viability when treated with both romidepsin and cytarabine compared to cytarabine alone. Moreover, the combination of romidepsin and cytarabine proved to be synergistic in ten of these 17 PDX. To expand the potential of romidepsin in combination with other drugs, we first combined it with inotuzumab ozogamicin, a recently introduced antibody-drug conjugate that is currently being used as a last-resort drug for high-risk relapsed BCP-ALL and confirmed the synergistic potential of this combination (Figure 2C, E). Finally, we combined romidepsin with all other therapy drugs and observed that romidepsin enhanced response to each of the tested drugs (Figure 3, full synergy landscapes in Online Supplementary Figure S4) sensitizing both p53KO and p53WT cells. Together, these results indicate that romidepsin
effectively improves the therapeutic efficacy of commonly used drugs regardless of p53 status, both in BCP-ALL cell lines and in patient-derived cells.
Romidepsin increases cytarabine-induced apoptosis by downregulation of components of the proteasome and ribosome biogenesis
Since histone acetylation induces global changes in gene expression, HDAC inhibition may act on many different pathways to affect response to therapy. To obtain further insights into these mechanisms, we performed bulk RNA sequencing on p53-deficient and p53-proficient cells treated with romidepsin, cytarabine, or their combination. We hypothesized that p53-deficient cells would show a reduced ability to induce p53 target genes and genes involved in the execution of apoptosis. Using GSEA with the MSigDB hallmark gene set collection7 (comprising 50 gene sets, including DNA damage and repair, apoptosis, glycolysis and p53 pathway gene sets) on the comparison of cytarabine-treated wild-type cells versus knockout cells, we confirmed enrichment of both p53 target genes and apoptosis-related genes in p53-proficient cells compared to p53-deficient cells (Online Supplementary Figure S5A). We confirmed by quantitative real-time polymerase chain reaction (qRT-PCR) analysis that induction of the p53 target genes CDKN1A (protein: p21), BBC3 (PUMA), and GADD45A (GADD45a) was compromised in p53-deficient cells (Online Supplementary Figure S5B), while differences in apoptosis induction were already established (Figure 2C). PMAIP1 (NOXA) induction was not affected by p53 loss, consistent with the notion that this gene, apart from being a p53 target, can mediate apoptosis in a p53-independent manner in hematologic malignancies.11
To test whether romidepsin may act to (re)activate p53 target genes and thereby induce apoptosis, we performed GSEA, again using the MSigDB hallmark gene set collection which includes gene sets that cover p53 pathway or apoptosis, comparing both romidepsin-only versus control treatment and combination versus cytarabine-only treatment (normalized enrichment scores, P values and false discovery rate q values of HALLMARK_APOPTOSIS and HALLMARK_P53_PATHWAY are shown in Online Supplementary Table S3). We found significantly enhanced apoptosis in three out of four models for both comparisons (non-significant enrichment of RCH-ACV wild-type for romidepsin versus control, normalized enrichment score 1.23, P=0.133; and Nalm6 wild-type near significant in combination versus cytarabine, normalized enrichment score 1.31, P=0.06). However, we did not observe a consistently significant enrichment of the p53 pathway across our models when comparing either romidepsin-only to control treatment, or combination treatment to cytarabine-only (Online Supplementary Table S3). Thus, while romidepsin does induce apoptosis, this does not appear to require p53. To identify other mechanisms of action of romidepsin,
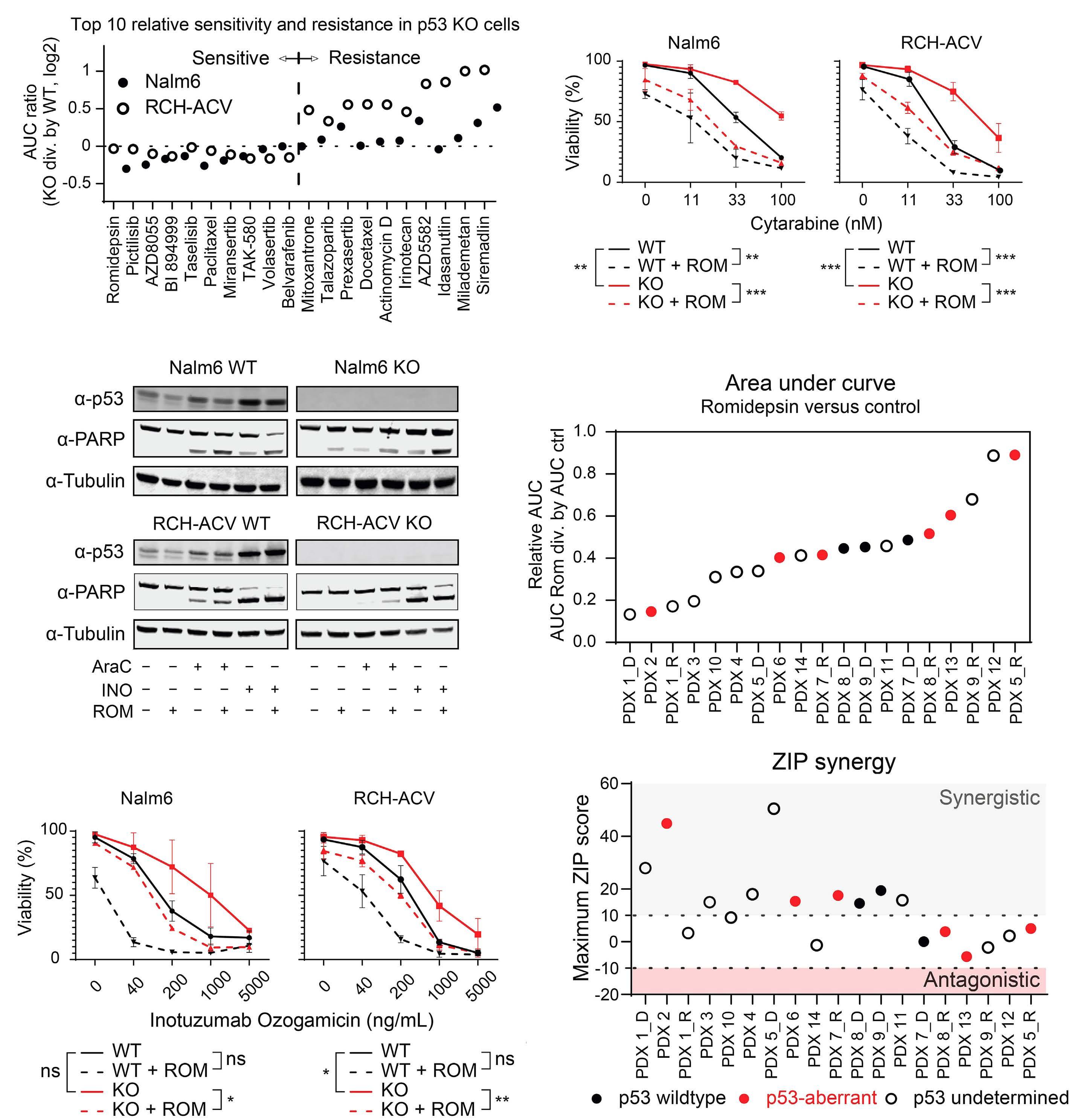
Figure 2. Romidepsin sensitizes p53-deficient and p53-proficient cells to cytarabine and inotuzumab. (A) Results of the drug screen showing the relative viability of p53KO Nalm6 and RCH-ACV cells as area under the curve (AUC) ratios, calculated by dividing the AUC value of each p53KO cell by that of the p53WT control. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) conversion was used as a measure of viability after treatment with a range of concentrations of the indicated drug for 72 hours (n=1). (B) Dose-response curves showing viability of Nalm6 and RCH-ACV p53WT and p53KO cells treated with the indicated concentrations of cytarabine in the presence or absence of romidepsin (2.25 nM) for 72 hours as determined by flow cytometric quantification of cells positive for amine-reactive dyes. Each data point represents the mean (± standard deviation [SD]) of two independent experiments. The AUC was determined and differences were tested for statistical significance using analysis of variance (ANOVA) followed by Tukey multiple comparisons tests. (C) Western blot showing p53 and PARP expression in p53WT and
Continued on following page.
p53KO cells treated with romidepsin (2 nM), cytarabine (30 nM), and/or inotuzumab ozogamicin (Nalm6, 200 ng/mL; RCH-ACV, 100 ng/mL) for 24 hours, with tubulin as the loading control. A representative example of three independent experiments is shown. (D) Fourteen patient-derived xenograft (PDX) samples from subjects with B-cell precursor acute lymphoblastic leukemia were seeded on hTERT-immortalized mesenchymal stromal cells and treated with the indicated concentrations of romidepsin and/or cytarabine for 72 hours. Flow cytometric quantification of cells positive for amine-reactive dyes was used as a measure of viability and as an input for AUC and zero interaction potency (ZIP) synergy calculations. AUC ratios were calculated by dividing the AUC value of the dose-response curve of the combination of romidepsin (2 or 2.25 nM, depending on the PDX) and a range of cytarabine concentrations by that of the cytarabine-only curve. Maximum ZIP synergy scores were computed from the viability response after romidepsin (at a concentration ranging from 0.5-8 nM) and/or cytarabine treatment (at a concentration ranging from 0.1 nM-100 µM ). Diagnosis-relapse pairs of samples from the same patient are indicated by suffixes _D and _R. Data shown are n=1 for each PDX. (E) Dose-response curves showing viability of Nalm6 and RCH-ACV p53WT and p53KO cells treated with the indicated concentrations of inotuzumab ozogamicin in the presence or absence of romidepsin (2.25 nM) for 72 hours as determined by flow cytometric quantification of cells positive for amine-reactive dyes. Each data point represents the mean (± SD) of two independent experiments. The AUC was determined and differences were tested for statistical significance using ANOVA followed by Tukey multiple comparisons tests. ns: not statistically significant, *P<0.05, **P<0.01, ***P<0.001. KO: knockout; WT: wild-type; ROM: romedepsin; PARP: poly (ADP-ribose) polymerase; AraC: cytarabine; INO; inotuzumab ozogamicin; PDX: patient-derived xenograft; ZIP: zero interaction potency.
we performed differential gene expression calculations for every possible treatment combination per cell model, filtered for significance (P<0.05) and fold change (log2 fold change smaller than -0.5 or greater than 0.5), calculated z-scores per filtered gene for both genotypes per cell line and generated heatmaps (Nalm6 p53KO shown in Figure 4A, remaining models in Online Supplementary Figure S6A). These heatmaps showed clusters of genes that are either upregulated or downregulated in both romidepsin-only versus control and combination versus cytarabine-only comparisons (as indicated in Figure 4A and Online Supplementary Figure S6A). Only a small number of genes (21 genes) was consistently activated in all four cell models (Figure 4B), and pathway analysis of these genes did not lead to significant pathway correlation. In contrast, gene suppression as a result of romidepsin treatment was a more general effect, with 237 downregulated genes shared by all models (Figure 4B), and subsequent Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed significant enrichment for genes involved in ribosome biogenesis and proteasome assembly (Figure 4C). We again validated this downregulation with qRT-PCR, assessing the mRNA expression of four genes per pathway included in the downregulated clusters (Online Supplementary Figure S6B, C). To assess whether the downregulation of these pathways renders cells more sensitive to chemotherapy, we combined inhibitors of the proteasome (carfilzomib and bortezomib) or ribosome biogenesis (actinomycin D) with cytarabine (Figure 5). Indeed, both proteasome inhibition and ribosome biogenesis perturbation showed synergy with cytarabine, albeit with lower synergy values compared to those with romidepsin, suggesting that neither one of these effects fully recapitulates the effects of romidepsin in enhancing therapy-induced apoptosis (Online Supplementary Figure S6D). We conclude from these results that romidepsin treatment leads to the downregulation of components of ribosome biogenesis and the proteasome and that this may, at least in part, contribute to an increased sensitivity to cytarabine.
Romidepsin improves the response to cytarabine in vivo
We next tested whether the combination of cytarabine and romidepsin would be effective in vivo. In order to do this, immunocompromised mice were transplanted with luciferase-expressing RCH-ACV p53KO cells. Three days after trans-
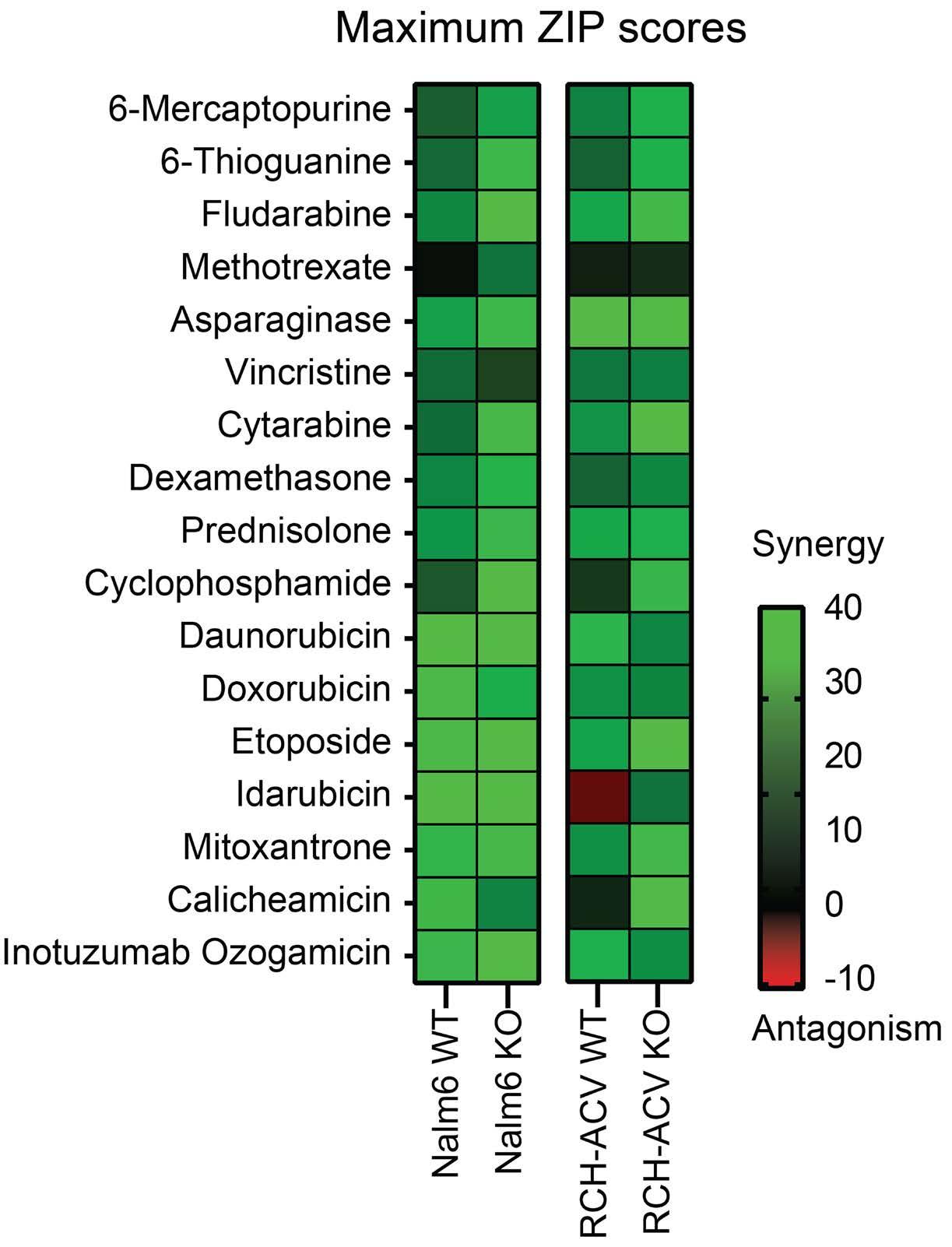
Figure 3. Romidepsin sensitizes p53-deficient and p53-proficient cells to therapy for B-cell precursor acute lymphocytic leukemia therapy in vitro. Heatmap of maximum zero interaction potency synergy scores for two independent experiments. Nalm6 and RCH-ACV p53KO cells were exposed to five concentrations of romidepsin (0-3.375 nM) and five concentrations of the drug listed per row for 72 hours. Flow cytometric quantification of cells positive for amine-reactive dyes was used as a measure of viability and as input for synergy calculations. ZIP: zero interaction potency; WT: wild-type; KO: knockout.
plantation, the mice were treated with vehicle, cytarabine, romidepsin, or a combination of both drugs (Figure 6A). This treatment regimen was well tolerated as evidenced by a constant body weight during treatment (Online Supplementary Figure S7). Leukemia burden was assessed during and after treatment by measurement of photon flux following lucifer-
in injection. Mice treated with romidepsin as a single agent did not benefit from treatment, whereas a short course of treatment with cytarabine was sufficient to delay leukemia development (Figure 6B, C). Importantly, in mice treated with the combination regimen, leukemia development was significantly delayed compared to that in the animals treated
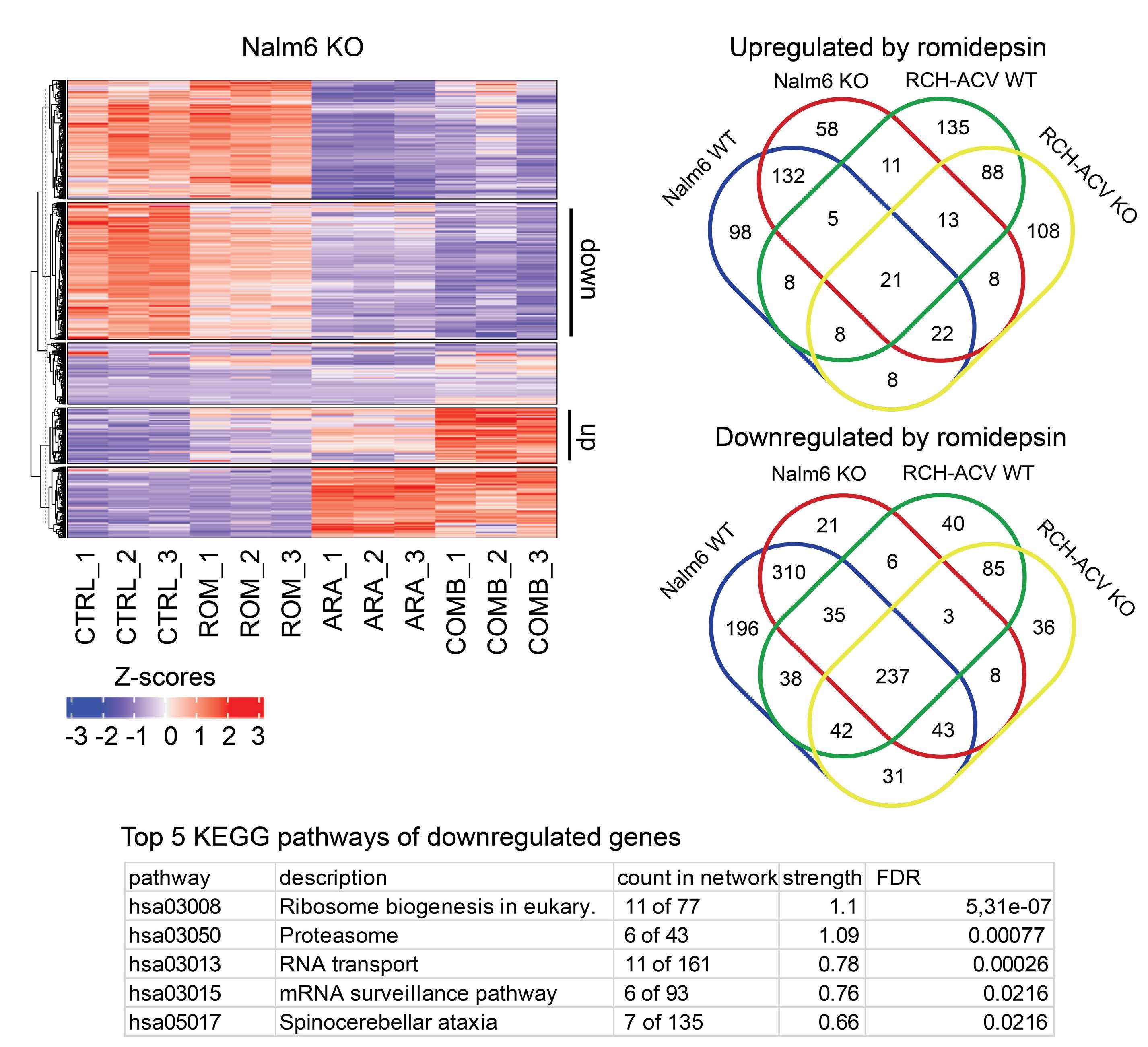
Figure 4. RNA sequencing reveals transcriptional effects of cytarabine and romidepsin treatment. (A) Heat map showing gene-expression levels from RNA-sequencing analysis in Nalm6 p53KO cells treated with cytarabine and/or romidepsin. Calculations of differentially expressed genes were performed for every possible treatment combination within a cell line and filtered for genes with a P value of less than 0.05 and a log2 cutoff of smaller than -0.5 or greater than 0.5. Gene lists were then combined for both genotypes per cell line, Z scores were computed and heatmaps were generated for each model in RStudio using unsupervised clustering. Clusters of up- (up) and downregulated (down) genes in both the comparisons of romidepsin versus control treatment and combination versus cytarabine treatment are indicated. (B) Venn diagram of overlapping differentially expressed gene lists either consistently upregulated (top) or downregulated (bottom) upon romidepsin treatment in the four tested models. (C) The top five Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways associated with the shared genes are depicted in the bottom panel of (B) according to the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) algorithm. KO: knockout; CTRL: control; ROM: romidepsin; ARA: cytarabine; COMB: combination therapy; WT: wild-type; FDR: false discovery rate.
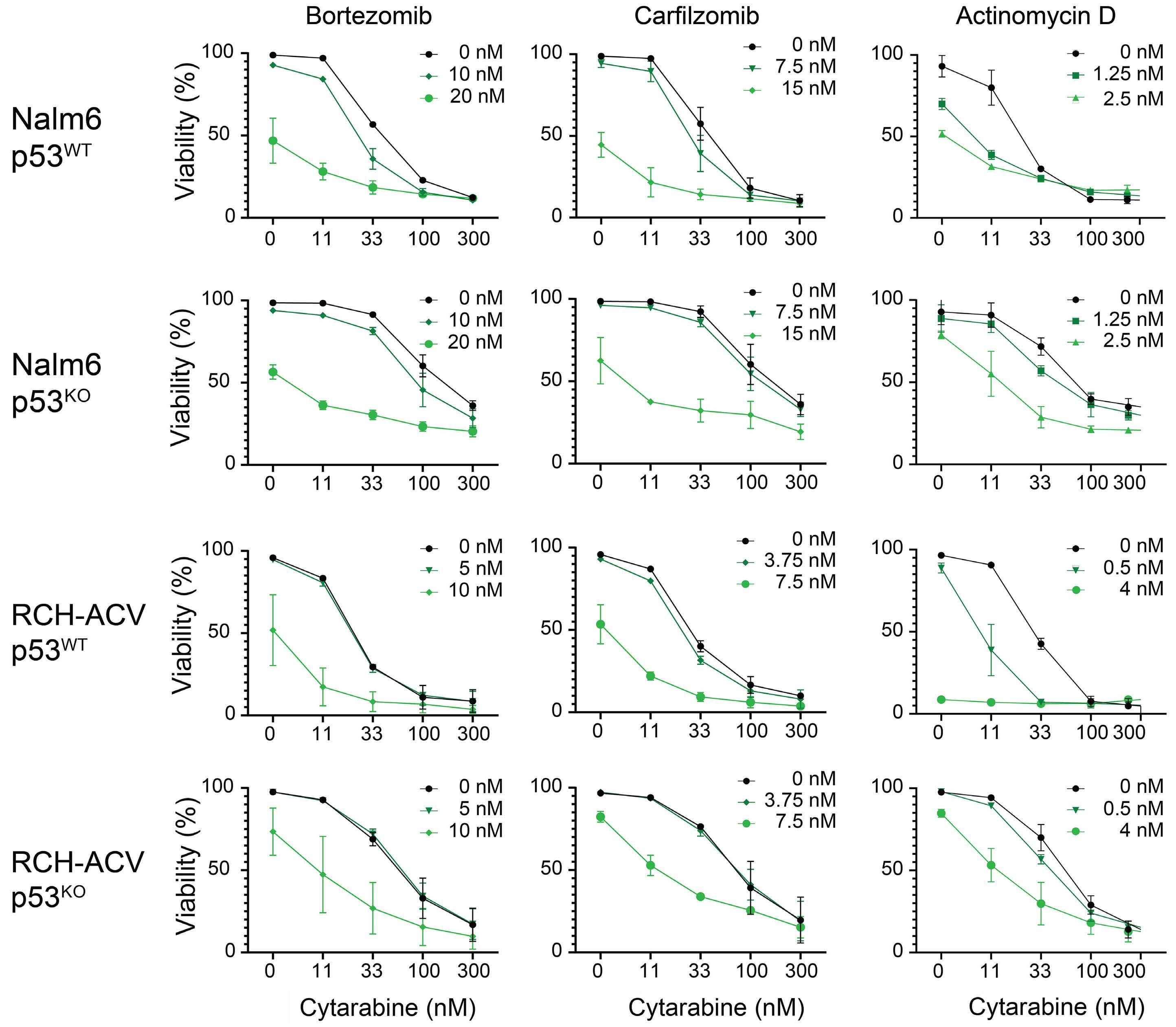
Figure 5. Inhibition of ribosome biogenesis or the proteasome sensitizes B-cell precursor acute lymphocytic leukemia cells to cytarabine in vitro. Dose-response curves showing the viability of Nalm6 and RCH-ACV p53WT and p53KO cells treated with the indicated concentrations of cytarabine in the presence or absence of the proteasome inhibitors bortezomib and carfilzomib, and the ribosome biogenesis inhibitor actinomycin D for 72 hours as determined by flow cytometric quantification of cells positive for amine-reactive dyes. Each data point represents the mean (± standard deviation) of two independent experiments. WT: wild-type; KO: knockout.
with cytarabine only, showing that romidepsin synergizes with cytarabine in vivo without causing any overt signs of toxicity. We conclude from these results that romidepsin may potentially be used to enhance response to therapy in relapsed/refractory TP53-deficient leukemias.
The treatment of relapsed BCP-ALL involves high-dose chemotherapeutics that may be followed by hematopoi-
etic stem cell transplantation. Ongoing clinical trials are studying the potential benefit of various immunotherapies, including antibody-drug conjugates, T-cell engagers, and chimeric antigen receptor (CAR) T-cell therapy. Loss of p53 functions predicts a dismal outcome in relapsed BCP-ALL that is treated with conventional protocols and initial observations indicate a poor response to CAR T-cell therapy.12 Similarly, the efficacy of inotuzumab ozogamicin, an antibody-drug conjugate that is highly effective for the treatment of relapsed B-cell ALL, is impaired in TP53-mutated leukemia as reported by Tirrò et al.13 and shown in
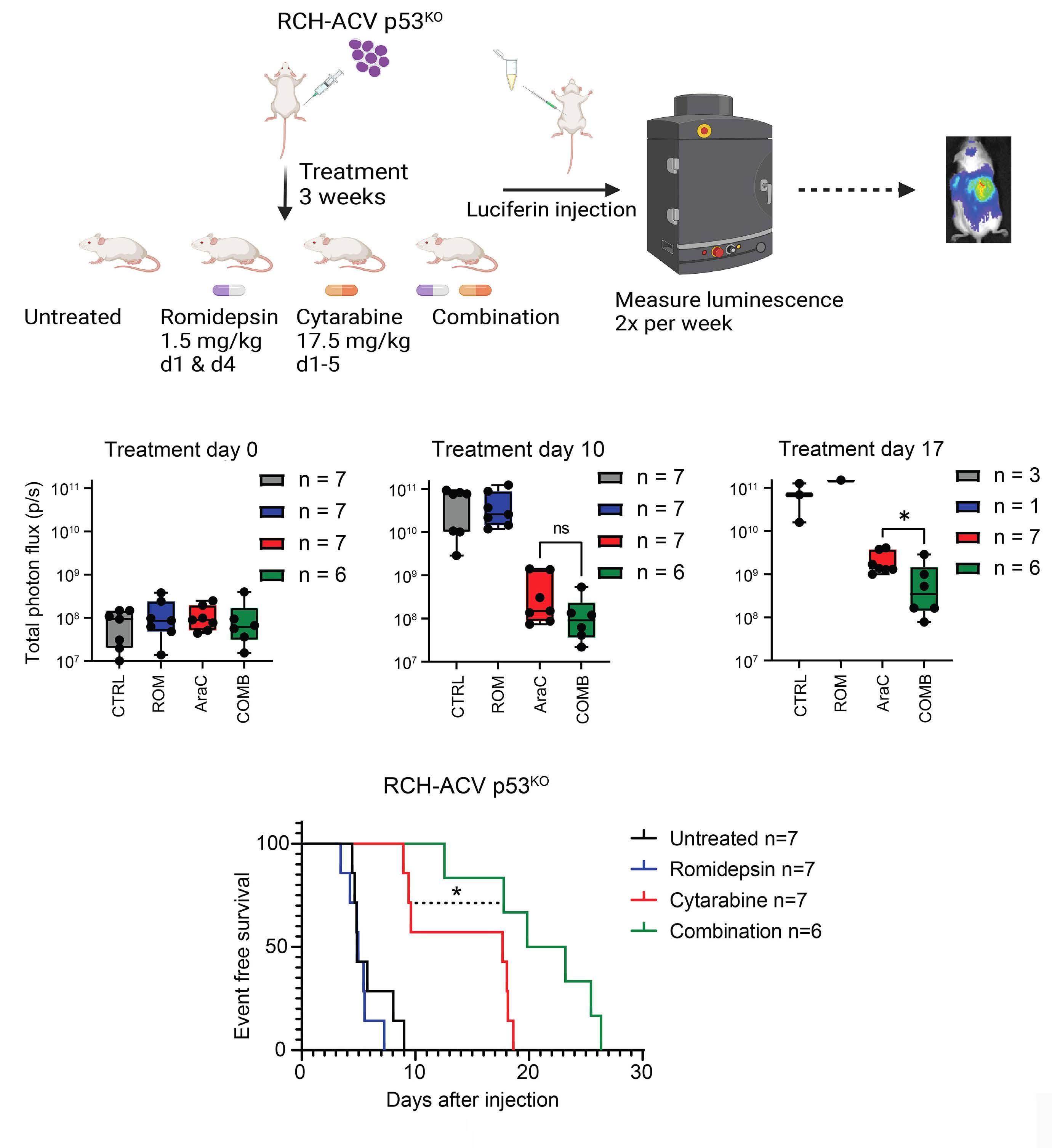
Figure 6. Romidepsin sensitizes p53-deficient cells to cytarabine in vivo. (A) Schematic representation of the setup of the in vivo experiment. Luciferase-expressing RCH-ACV p53KO cells were injected into NRG-SM3 mice and treated for 3 weeks with cytarabine, romidepsin, or their combination. Leukemia development was followed by measuring luciferase-induced photon flux. Image created with BioRender.com. (B) Total photon flux is visualized for the start of treatment and days 10 and 17 of treatment. Differences were tested for statistical significance using a Mann-Whitney test comparing the cytarabine and combination groups as data in these groups were not normally distributed. (C) Kaplan-Meier plot of NRG-SM3 mice transplanted with luciferase-expressing RCH-ACV p53KO cells under treatment with cytarabine, romidepsin or their combination. An event was defined as a tumor load reaching a total photon flux of 5x108 photons per second. Differences were tested using a log-rank test comparing the cytarabine and combination groups. ns: not significant; *P<0.05. KO: knockout; CTRL: control; ROM: romidepsin; AraC: cytarabine; COMB: combination therapy; WT: wild-type.
Figure 2C, E. Hence, despite the advances that are being made for other subtypes of relapsed BCP-ALL, there is an urgent clinical need for the development of novel therapeutic strategies that effectively eradicate TP53-mutated leukemia.
In the past few decades, drugs that modulate the epigenome, including hypomethylating agents and HDAC inhibitors have shown promise in anti-cancer treatment.14,15 In hematologic malignancies, in particular, oncogenic driver mutations are commonly found in genes involved in epigenetic modifications, suggesting that these tumors may be sensitive to drugs targeting epigenetic regulators. While the clinical efficacy of these drugs as monotherapy is limited, the combination of a hypomethylating agent and a Bcl-2 inhibitor was shown to be highly effective in the treatment of patients with acute myeloid leukemia who are unfit for curative chemotherapy-based protocols.16,17 Altogether, our results indicate that loss of p53 function renders BCP-ALL cells resistant to most currently used (chemo)therapies, but that HDAC inhibition by romidepsin can resensitize these cells to therapy. In contrast to earlier findings by Yan et al.18 in acute myeloid leukemia, we did not observe specific transcriptional (re)activation of the p53 pathway upon romidepsin treatment in p53-deficient cells, which may be explained by the different cellular context that was tested. The transcriptome data and validations presented here instead suggest that inhibition of ribosome biogenesis and the proteasome, both associated with apoptosis given the appropriate context, may be involved in the increased apoptotic response observed when cytarabine treatment is combined with romidepsin. Potential mechanisms through which class I histone deacetylases such as HDAC1 and HDAC2, the main targets of romidepsin, can affect gene expression may include regulating histone acetylation and thereby chromatin accessibility,19 as well as allowing the formation of a complex with corepressors such as NCoR or SMRT which may lead to gene repression.20-22 The latter has been implicated in differentiation of acute myeloid leukemia, most notably in the context of the fusion genes PML-RAR and AML1-ETO 23 Of note, both Nalm6 (IGH-DUX4) and RCH-ACV (TCF3-PBX1) carry gene fusions. However, it remains to be seen to what extent such mechanisms may contribute to the observed synergy involving HDAC inhibition in BCP-ALL.
Consistent with our findings that romidepsin improves response to therapy regardless of p53 status, romidepsin was previously found to improve response to therapy in KMT2A-rearranged leukemia.24,25 It appears that romidepsin is effective in both p53-deficient as well as some p53-proficient subsets of ALL and may therefore represent a valuable addition to the current treatment protocols for (relapsed) BCP-ALL. As the effects on the proteasome seem to contribute to the observed synergy of romidepsin, it is
important to note that the proteasome inhibitor bortezomib has previously been added to a chemotherapy backbone in relapsed pediatric B-ALL and T-ALL patients, demonstrating manageable toxicity and clinical efficacy.26-29 However, our results show that proteasome inhibition alone does not fully recapitulate the synergy observed with romidepsin, and therefore romidepsin may be even more effective in improving therapy response on a (chemo)therapy backbone. Clinically, romidepsin was previously tested as a single agent in both adults and children, showing manageable toxicities.30,31 More recently, romidepsin was combined with doxorubicin as well as the immunomodulatory agent lenalidomide in phase I or II trials, showing clinical efficacy and feasibility in the treatment of hematologic malignancies.32,33 Together, these results highlight a potential for romidepsin combination regimens in hematologic malignancies. Further (pre)clinical studies should reveal whether romidepsin may be used to improve current or experimental salvage therapies for relapsed TP53-deficient ALL.
No conflicts of interest to disclose.
Contributions
FNvL, LTvdM, MB, and WPJC conceptualized the project. FNvL and LTvdM supervised the work. WPJC performed the experiments with assistance from NE, DSvIS, GRS, NA, LdG, and MB. KJTG, RH, and RPK contributed to analyzing the data. RPK assisted with genotyping samples. DSvIS generated the xenografts. WPJC, LTvdM, and FNvL drafted the manuscript which was reviewed, and approved by all the authors.
The authors thank members of the flow cytometry facility, the high throughput screening facility (https://research. prinsesmaximacentrum.nl/en/core-facilities/high-throughput-screening) and the Kuiper research group in the Princess Máxima Center, and the PRIME department of RadboudUMC animal facility for valuable technical support. In addition, the authors thank MRC Holland for providing digital MLPA data and Vaskar Saha for providing the pLNT-Sffv-luciferase plasmid. pSpCas9(BB)-2A-GFP was a gift from Feng Zhang (Addgene plasmid # 48138; RRID: Addgene_48138).
Funding
This work was supported by the Princess Máxima Center for Pediatric Oncology.
Data-sharing statement
The data generated in this study are available within the article and its supplementary data files. Transcriptome data generated in the study are publicly available in Gene Expression Omnibus (GEO) (GSE234091).
1. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105(11):2524-2539.
2. Donehower LA, Soussi T, Korkut A, et al. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. Cell Rep. 2019;28(5):1370-1384.
3. Hof J, Krentz S, Van Schewick C, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol. 2011;29(23):3185-3193.
4 Kanagal-Shamanna R, Kantarjian HM, Khoury JD, et al. Distinct prognostic effects of TP53 mutations in newly diagnosed versus relapsed/refractory (R-R) patients (pts) with B-acute lymphoblastic leukemia (ALL) treated with mini-Hcvdinotuzumab ozogamicin with or without blinatumomab regimens. Blood. 2020;136(Suppl 1):41-43.
5. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170(6):1062-1078.
6. Frismantas V, Dobay MP, Rinaldi A, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017;129(11):26-37.
7. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417-425.
8. Mootha VK, Lindgren CM, Eriksson K-F, et al. PGC-1a-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267-273.
9 Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-15550.
10 Hontecillas-Prieto L, Flores-Campos R, Silver A, de Álava E, Hajji N, García-Domínguez DJ. Synergistic enhancement of cancer therapy using HDAC inhibitors: opportunity for clinical trials. Front Genet. 2020;11:578011.
11. Fidyt K, Pastorczak A, Cyran J, et al. Potent, p53-independent induction of NOXA sensitizes MLL-rearranged B-cell acute lymphoblastic leukemia cells to venetoclax. Oncogene. 2022;41(11):1600-1609.
12. Zhang X, Lu X-A, Yang J, et al. Efficacy and safety of anti-CD19 CAR T-cell therapy in 110 patients with B-cell acute lymphoblastic leukemia with high-risk features. Blood Adv. 2020;4(10):2325-2338.
13. Tirrò E, Massimino M, Romano C, et al. Chk1 inhibition restores inotuzumab ozogamicin citotoxicity in CD22-positive cells expressing mutant p53. Front Oncol. 2019;9:57.
14 Bennett RL, Licht JD. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol. 2018;58(1):187-207.
15. Ho TCS, Chan AHY, Ganesan A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J Med Chem. 2020;63(21):12460-12484.
16. San José-Enériz E, Gimenez-Camino N, Agirre X, Prosper F. HDAC Inhibitors in acute myeloid leukemia. Cancers (Basel). 2019;11(11):1794.
17 DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629.
18. Yan B, Chen Q, Xu J, Li W, Xu B, Qiu Y. Low-frequency TP53
hotspot mutation contributes to chemoresistance through clonal expansion in acute myeloid leukemia. Leukemia. 2020;34(7):1816-1827.
19 Gallinari P, Marco SD, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17(3):195-211.
20. Heinzel T, Lavinsky RM, Mullen T-M, et al. A complex containing N-CoR, mSln3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387(6628):43-48.
21. Wen Y-D, Perissi V, Staszewski LM, et al. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc Natl Acad Sci U S A. 2000;97(13):7202-7207.
22. Alland L, Muhle R, Hou H Jr, et al. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387(6628):49-55.
23. Minucci S, Nervi C, Lo Coco F, Pelicci PG. Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene. 2001;20(24):3110-3115.
24. Cruickshank MN, Ford J, Cheung LC, et al. Systematic chemical and molecular profiling of MLL-rearranged infant acute lymphoblastic leukemia reveals efficacy of romidepsin. Leukemia. 2017;31(1):40-50.
25. Cheung LC, Cruickshank MN, Hughes AM, et al. Romidepsin enhances the efficacy of cytarabine in vivo, revealing histone deacetylase inhibition as a promising therapeutic strategy for KMT2A-rearranged infant acute lymphoblastic leukemia. Haematologica. 2019;104(7):300-303.
26. Horton TM, Whitlock JA, Lu X, et al. Bortezomib reinduction chemotherapy in high-risk ALL in first relapse: a report from the Children’s Oncology Group. Br J Haematol. 2019;186(2):274-285.
27. Wang R, Wang W, Liu X, et al. Treatment for a B-cell acute lymphoblastic leukemia patient carrying a rare TP53 c.C275T mutation: a case report. Front Oncol. 2022;12:1018250.
28. Miyagawa N, Goto H, Ogawa A, et al. Phase 2 study of combination chemotherapy with bortezomib in children with relapsed and refractory acute lymphoblastic leukemia. Int J Hematol. 2023;118(2):267-276.
29. Teachey DT, Devidas M, Wood BL, et al. Children’s Oncology Group trial AALL1231: a phase III clinical trial testing bortezomib in newly diagnosed T-cell acute lymphoblastic leukemia and lymphoma. J Clin Oncol. 2022;40(19):2106-2118.
30 Fouladi M, Furman WL, Chin T, et al. Phase I study of depsipeptide in pediatric patients with refractory solid tumors: a Children’s Oncology Group report. J Clin Oncol. 2006;24(22):3678-3685.
31. Whittaker SJ, Demierre MF, Kim EJ, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol. 2010;28(29):4485-4491.
32. Ruan J, Zain J, Palmer B, et al. Multicenter phase 2 study of romidepsin plus lenalidomide for previously untreated peripheral T-cell lymphoma. Blood Adv. 2023;7(19):5771-5779.
33. Vu K, Wu CH, Yang CY, et al. Romidepsin plus liposomal doxorubicin is safe and effective in patients with relapsed or refractory T-cell lymphoma: results of a phase I doseescalation study. Clin Cancer Res. 2020;26(5):1000-1008.
Amanda C. Winters,1 Mohd Minhajuddin,2 Brett M. Stevens,2 Ajay Major,2 Grace Bosma,3 Diana Abbott,3 Nicholas Miltgen,4 Ji Yuan,4 Amy L. Treece,5 Bradford J. Siegele,6 Mark D. Ewalt,7 Jonathan A. Gutman,2 Craig T. Jordan2 and Daniel A. Pollyea2
1Center for Cancer and Blood Disorders, Department of Pediatrics, University of Colorado, Aurora, CO; 2Division of Hematology, Department of Medicine, University of Colorado, Aurora, CO; 3Department of Biostatistics and Informatics, University of Colorado, Aurora, CO; 4Molecular Diagnostics, Children’s Hospital Colorado, Aurora, CO; 5Pediatric Pathology, Children’s of Alabama, Birmingham, AL; 6Department of Pathology, University of Colorado, Aurora, CO and 7Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY, USA
Abstract
Correspondence: A.C. Winters amanda.winters@childrenscolorado.org
Received: June 19, 2023.
Accepted: December 7, 2023. Early view: December 14, 2023.
https://doi.org/10.3324/haematol.2023.283790
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Venetoclax with azacitidine (ven/aza) is a lower-intensity therapeutic regimen that has been shown to improve outcomes in elderly patients with acute myeloid leukemia (AML). Measurable residual disease (MRD) using flow cytometry is a valuable tool for the prediction of relapse in AML using conventional therapies and ven/aza; however, the prognostic value for broadscale molecular MRD after ven/aza treatment is less clear. We aimed to determine the utility of retrospective assessment using multi-gene molecular MRD by droplet digital polymerase chain reaction (ddPCR). We found this approach correlates with outcomes in a cohort of patients receiving frontline ven/aza for AML. The predictive value of ddPCR MRD persisted when NPM1 mutations were removed from analysis, as well as after adjustment for the impact of stem cell transplant on outcomes. Late achievement of MRD negativity, including after SCT, was still associated with superior outcomes compared to persistently detectable MRD. We further explored the impact of ven/aza on the burden of different classes of mutations, and identified the persistence of splicing factor mutations, commonly associated with MDS, as a consistent finding after ven/aza treatment. These data add to our understanding of the effects of ven/aza on AML disease biology and provide details on molecular depth of remission that can guide prospective trials in the future.
Measurable residual disease (MRD) is a sensitive tool in the post-treatment setting to predict relapse in acute myeloid leukemia (AML).1,2 The majority of clinical MRD studies in AML have been performed via multi-channel flow cytometry (MCF).1,3 Despite its utility as a prognostic marker, 20-30% of MCF MRD-negative patients relapse.1 Targeted molecular MRD has also proven to be a powerful tool for predicting outcomes in AML patients,3-6 however, no molecular MRD platform is widely established for clinical use in AML. This is in part because of a lack of standardized methods and thresholds to monitor molecular MRD, 3 and uncertainty around which persistent mutations have immediate prognostic value.7,8 Approximately 20% of adults with AML have recurrent chro-
mosomal translocations and 27% have NPM1 mutations,9,10 limiting the utility of standard quantitative polymerase chain recation (qPCR) whose use is largely restricted to these mutation subtypes. However, over 95% of patients have single nucleotide variants or small insertions/deletions9,10 which can be captured by droplet digital PCR (ddPCR) or next-generation sequencing (NGS). NGS platforms are in general quite expensive and historically have had low sensitivity (2-3% limit of detection) prohibiting their use as MRD tools, although more sensitive platforms are being validated.6,11 ddPCR has been identified as a highly sensitive and precise modality for mutation monitoring in AML and other malignancies, with limits of detection reported to be 10-4 to 10-5,12 but its primary limitation has been assay specificity to individual mutations or hotspots.13,14 Accordingly, to date the range of mutations evaluated by
ddPCR has been limited.
The introduction of venetoclax with hypomethylating agents (HMA) or low-dose cytarabine for the treatment of elderly patients with AML has led to enhanced remission rates and improved survival in a poor-risk patient population.15-17 While promising, venetoclax combinations do not have universal efficacy,18-20 and most patients will ultimately relapse in the absence of consolidative stem cell transplant (SCT).21 Given the high likelihood of relapse in these patients,18 and poor prognosis when it occurs,18,22 it is important to identify early warning signs so that, where possible, these patients might be offered other treatment options to prevent relapse. While there are now four retrospective studies confirming the prognostic value of MCF MRD in patients receiving venetoclax-based therapies,23-26 robust data on the relevance of molecular MRD for venetoclax-based therapies are lacking. To date, only NPM1-based molecular MRD has been evaluated in this context.27,28 Our laboratory has previously provided preliminary ddPCR data demonstrating molecular depth of remission in cohorts of patients receiving venetoclax/azacitidine (ven/aza),29-31 but has not published long-term outcome data on all ven/aza patients at our center based on ddPCR MRD status.
Here we demonstrate the feasibility of retrospectively detecting MRD with a broad panel of ddPCR assays for mutations identified by diagnostic NGS in adult AML patients. We confirm the association of ddPCR MRD status with outcomes in patients receiving ven/aza, including a subset of patients proceeding to SCT. Our findings also illuminate the relative responsiveness of different AML subclones to ven/aza selective pressure. To our knowledge, this is the first report of molecular MRD beyond NPM1 mutations in the context of venetoclax-based therapy.
Patient selection
For this retrospective analysis, all patients ≥18 years of age with a diagnosis of AML treated at the University of Colorado were considered. Inclusion/exclusion criteria are summarized in Figure 1 and detailed in the Online Supplementary Appendix. A total of 64 patients were evaluated for MRD by ddPCR. All patients signed Colorado Multiple Institution Review Board (COMIRB)-approved consent for collection of tissue used in this analysis, and an additional IRB approval allowed the retrospective demographic and outcome data analysis.
DNA extraction
Genomic DNA was extracted from whole bone marrow aspirates in the Children’s Hospital Colorado Molecular Diagnostics Core using the Qiagen QIAsymphony DSP DNA kits, as per institutional standards. Concentration and quality of DNA were evaluated via Qubit spectrophotom-
eter. DNA was stored at -20°C.
Droplet digital polymerase chain reaction measurable residual disease monitoring
Based on diagnostic NGS results, a total of 50 AML-associated mutations were evaluated in this patient cohort (Online Supplementary Table S1). Mutations in DNMT3A, TET2, and ASXL1 (“DTA mutations”) were excluded from evaluation given the previous literature showing lack of correlation of these clonal hematopoiesis mutations with relapse outcomes.8 In general, large insertions/deletions (such as FLT3 ITD) were not amenable to ddPCR assay design and were excluded from evaluation. Mutation-specific primer/probe ddPCR assays were purchased commercially from BioRad or were custom designed. All assays were validated to a limit of detection of 0.02-0.15% variant allelic frequency (VAF). ddPCR was performed with 150 nanograms gDNA input on a BioRad QX200 Droplet Digital PCR instrument and data were analyzed via the BioRad QuantaSoft Analysis Pro v1.0.596 software. Additional details about assay design and validation are described in the Online Supplementary Appendix.
Retrospective chart review
Demographic information, diagnostic mutational data, dates of bone marrow evaluations and concurrent disease status, and outcome data were extracted from the electronic medical record.
Histologic assessment of dysplasia
Archived bone marrow slides from remission time points corresponding to ddPCR MRD evaluations were reviewed by two hematopathologists (ME and BJS) and the degree of dysplasia and involved lineages were scored according to clinical guidelines.32
Statistical analyses
Relapse-free survival (RFS) and overall survival (OS) were defined from the date of diagnosis to the respective endpoint. If no events occurred, individuals were censored at date of last follow-up. Median survival times were created using Kaplan-Meier product-limit estimates. Kaplan-Meier curves and Mantel-Cox log-rank tests were used to compare survival times. Summary statistics for variables of interest are presented alongside P values for the corresponding non-parametric tests based on variable class: Kruskal-Wallis for continuous variables, χ2 test for categorical variables with expected call counts greater than or equal to 5, and Fisher exact tests for categorical variables where any expected cell counts do not meet this requirement. Two multivariate Cox proportional hazard models were explored with transplant, age at diagnosis, ddPCR MRD status, and type of mutation as covariates of interest: the first using OS outcome definition and the second using RFS outcome definition.
Figure 1. Cohort selection and mutational details. (A) Algorithm for patient cohort selection. Patients with acute myeloid leukemia (AML) newly diagnosed and induced with venetoclax/azacitidine (ven/aza) at the University of Colorado between 2015 and 2020 were evaluated for the present study. Patients who were primary refractory to therapy, those without diagnostic mutations amenable to droplet digital polymerase chain reaction (ddPCR), or those for whom no remission bone marrow samples were available were excluded from analysis. A final cohort of 64 patients was selected for ddPCR measurable residual disease (MRD) analysis. (B) Co-mutation table for individual patients. Numbers listed across the top of the table are research identification numbers for individual patients. Rows list specific acute myeloid leukemia (AML)-associated genes and squares are colored if a patient has a given mutation. Mutations in green cleared with ven/ aza treatment (pre-transplant); mutations in red did not clear with ven/aza alone. Mutations in gray (including cytogenetic abnormalities [“Abn Cyto”]) were not evaluated by ddPCR. Overall MRD status (factoring in stem cell transplant [SCT]) is shown at the bottom, with MRD-negative patients on the left of the plot and MRD-positive patients on the right. Relapse status is also shown at the bottom of the plot. Note: all FLT3 mutations monitored via ddPCR were tyrosine kinase domain mutations (TKD) and those excluded from monitoring were FLT3 internal tandem duplications.
Demographics and disease biology
Figure 1A describes inclusion/exclusion criteria for the patients in our ven/aza cohort. A date range of January 1, 2015 through December 31, 2020 was chosen for this study; prior to 2015 ven/aza was not used at our site, while patients diagnosed subsequent to 2020 had insufficient follow-up for outcome measurements. A total of 145 patients received ven/aza as frontline therapy. Of those, 39 individuals (27%) had primary refractory disease and therefore were not eligible for MRD evaluation. Another 31 (21%) had no remission bone marrow samples available retrospectively.
Finally, 11 (8%) had no amenable mutations for ddPCR MRD monitoring. The final 64 patients (44%) were included in this analysis. These 64 individuals had a median of three (range, 1-7) mutations identified by targeted NGS at diagnosis (VAF 8-80%). The frequency of mutations in our patient cohort closely followed that described in larger cohorts of adult AML patients9,10 (Online Supplementary Figure S1). Figure 1B shows a plot of mutation co-occurrence in individual patients. After eliminating DTA mutations (DNMT3A, TET2, and ASXL1), which are associated with clonal hematopoiesis,8 patients had a median of two (range, 1-6) mutations, of which at least one (and up to 5) mutation was monitored by ddPCR. A median of five (range, 1-14) post-remission bone
Subtype, N
AML, N
1Wilcoxon rank sum test, Pearson’s χ2 test, Fisher’s exact test; MRD: measurable residual disease; IQR: interquartile range; ELN: European LeukemiaNet; SWOG: Southwest Oncology Group; FAB: French-American-British morphology subtype; AML: acute myeloid leukemia; allo: allogeneic.
marrow time points were evaluated per patient. Patients were classified as ddPCR MRD-negative if they had undetectable VAF of all monitored mutations at any single remission time point (termed time of best response or TBR), including after SCT (Online Supplementary Table S2). Otherwise, they were classified as ddPCR MRD-positive. Twenty-nine patients (45%) achieved MRD negativity by ddPCR; 35 patients (55%) remained MRD-positive. Demographics of the patient cohort by ddPCR MRD status are shown in Table 1. The only significant factors associated with ddPCR MRD status were age, sex, and transplant status. There were no differences in genetic risk classification, including incidence of TP53 mutations, initial marrow disease burden, French-American-British (FAB) AML subtype, or proportion of patients with therapy-related AML or AML secondary to MDS between MRD-positive and MRD-negative cohorts.
Molecular measurable residual disease by droplet digital polymerase chain reaction correlates with outcomes after venetoclax/azacitidine therapy
Stratification of the frontline ven/aza cohort into MRD-negative and MRD-positive groups by ddPCR, as above, demonstrated an association with notable differences in outcomes (Figure 2A-C). Median follow-up for the entire cohort was 21 months (95% confidence interval [CI]: 17.3-43.2). Twoyear RFS and OS were 73% versus 20% (P<0.0001) and 73% versus 26% (P=0.00015) in the ddPCR MRD-negative versus MRD-positive groups, respectively. Two-year cumulative incidence of relapse (CIR) was 80% in the ddPCR MRD-positive group versus 28% in the ddPCR MRD-negative group (P<0.0001). No patient in either cohort relapsed beyond 5 years. Of note, three of the 29 MRD-negative patients had recurrence of detectable mutation(s) by ddPCR - all three patients relapsed. Online Supplementary Figure S2
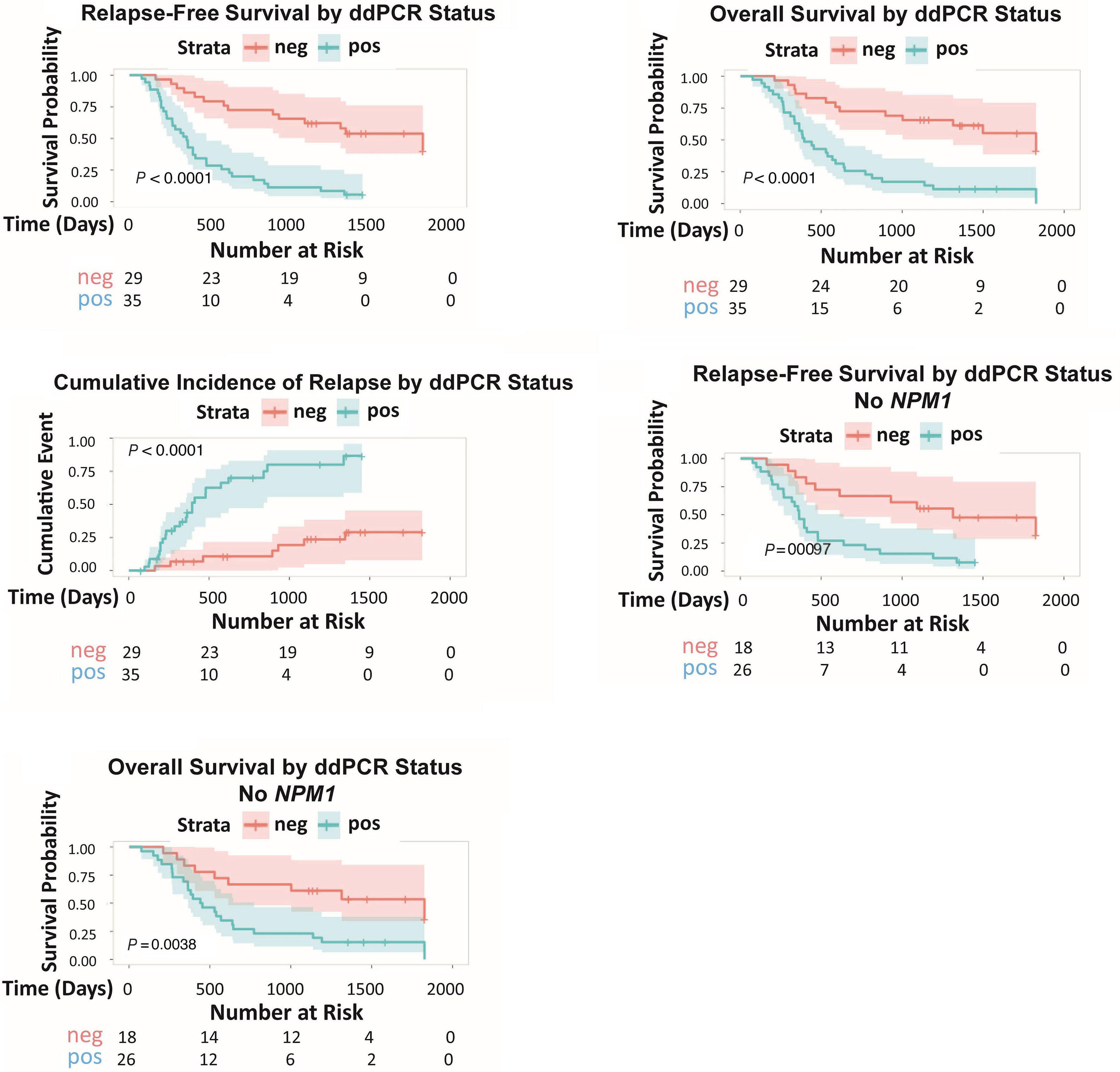
Figure 2. Droplet digital polymerase chain reaction measurable residual disease correlates with survival. (A) Relapse-free survival (RFS), (B) overall survival (OS), and (C) cumulative incidence of relapse (CIR) for the entire 64-patient cohort, stratified by droplet digital polymerase chain reaction (ddPCR) measurable residual disease (MRD) status, are shown with P values. Given that NPM1 status is a well established molecular MRD marker, we assessed the impact of ddPCR MRD in the absence of this gene. (D) RFS and (E) OS for the subset of patients (N=44) without NPM1 mutations are still significantly different depending on ddPCR MRD status. Pos: positive; neg: negative.
shows re-stratification of these individuals into the ddPCR MRD-positive group, which did not change the predictive value of the modality.
NPM1 is the only established gene mutation for clinically actionable MRD evaluation in adult AML,3,5 whereas there is controversy about the prognostic value of other genes.7 Twenty of our 64 patients had an NPM1 mutation that was monitored by ddPCR. When we removed those patients from the analysis (and thereby the effect of NPM1-based MRD on prognosis), we still observed significant differences in RFS and OS in ddPCR negative and positive cohorts (Figure 2D, E). These data confirm for the first time that multi-gene molecular MRD is a valuable tool for risk stratification in patients with AML receiving ven/aza.
Time to relapse depends on the nature of persisting mutations
One of the challenges of molecular MRD is that ramifications for disease recurrence seem to differ based on the mutation in question, likely related to associations with disease ontogeny.7,33 We evaluated the impact on survival and time to relapse for persistence of different classes of mutations: (i) NPM1 mutation, (ii) “late” mutations (FLT3 tyrosine kinase domain/TKD, NRAS, PTPN11), (iii) IDH1 or IDH2 mutations, (iiii) splicing factor mutations (SRSF2, SF3B1, U2AF1), or (iv) multiple. In the latter category we particularly noted persistence of co-occurring IDH1/2 and
splicing factor mutations in nine patients. For this specific analysis, we considered mutations “persistent” if they were detectable throughout all pre-SCT assessments, even if they cleared post-SCT. Patients with persistence of “late” mutations including NPM1 had a significantly worse prognosis than other groups with persistent mutations (Figure 3A). Patients with persistent IDH1/2 mutations, persistent splicing factor mutations, or multiple persistent mutations fared better, with no significant difference in RFS between these groups. We hypothesized that, in addition to any inherent differences in disease biology associated with these different mutations, time to relapse played a major role since this likely impacted whether a patient could be successfully salvaged by SCT or other therapies. We divided patients who relapsed (N=31) into those with no persistent mutations by ddPCR (“negative”), the mutation categories listed above, persistent TP53 mutation, or a basket category of “other.” We combined NPM1 and other “late” mutations for statistical analysis given small sample sizes. Consistent with findings from previous mutational analyses, patients with persistence of NPM1 or RAS pathway mutations had the shortest time to relapse (Figure 3B), which was significantly shorter than both those with no persistent mutations (P=0.03) and those with persistent splicing factor mutations (P=0.03). There was no significant difference in time to relapse when any of the other mutation groups were compared against those without persistent mutations (splicing factor vs. negative comparison shown). Of note,
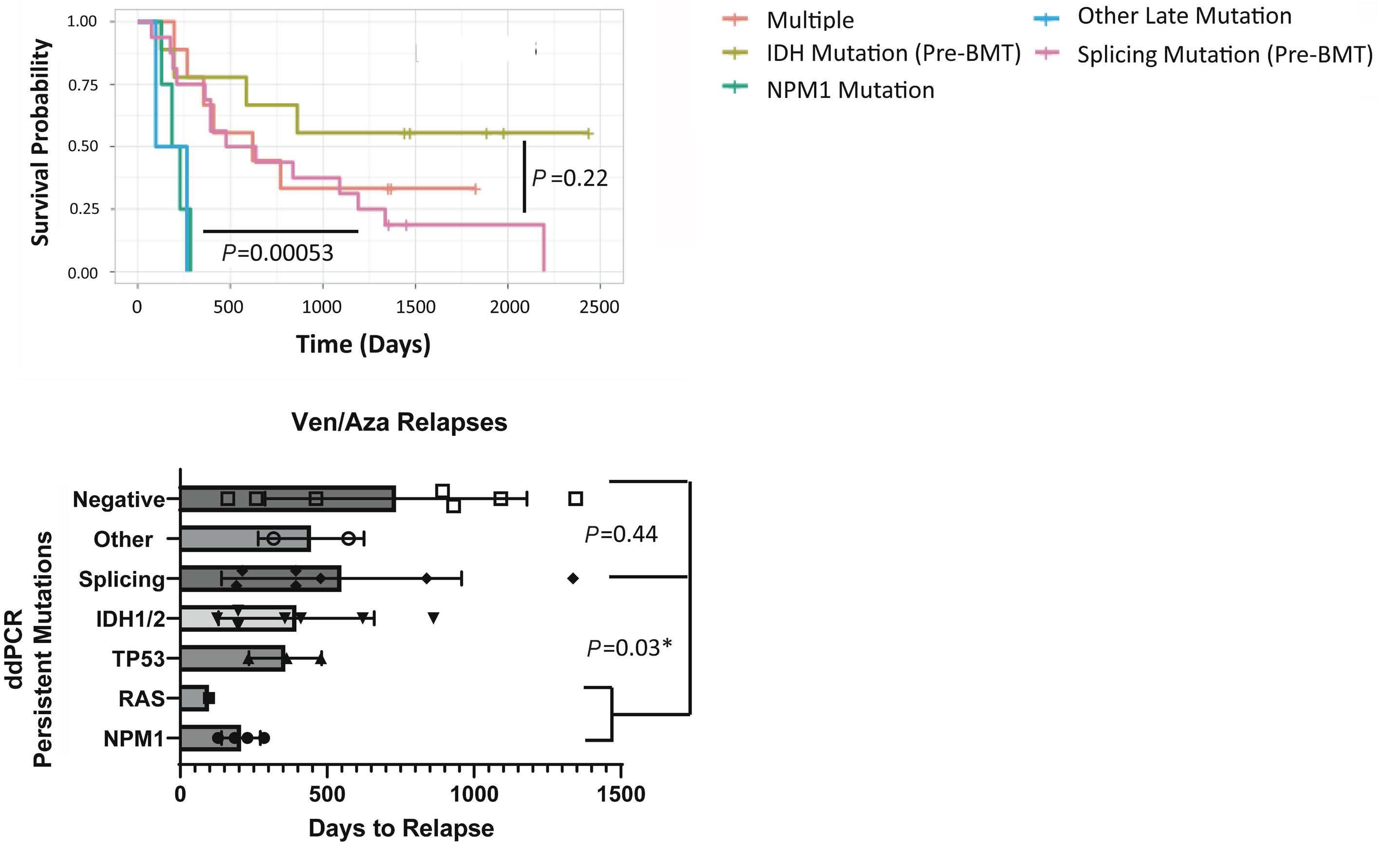
Figure 3. Various classes of genes have differential impact on the timing of relapse. (A) Relapse-free survival (RFS) was calculated for patients with persistent mutations pre-stem cell transplant (SCT) or in the absence of SCT, by category. NPM1 and “late” mutations (signaling pathway mutations) when persistent portended the worst survival, whereas persistent IDH1, IDH2, or splicing factor mutations (or a combination thereof) were indistinguishable and had relatively improved RFS compared to NPM1 and “late” mutations. (B) For patients who relapsed, time to relapse was evaluated based on whether the patient had no persisting mutations (“negative”) versus persistent mutations of various classes. Once again, NPM1 and RAS pathway mutations when persistent were associated with rapid onset of relapse compared to MRD-negative patients (P=0.03) and compared to patients with persistent splicing factor mutations (P=0.03). Ven/aza: venetoclax/azacitidine.
we did not see any differences in rate of clearance of IDH1 or IDH2 mutations based on whether they were clonal (VAF ≥40% at diagnosis) versus subclonal (VAF <40% at diagnosis), as shown in Online Supplementary Table S3.
Persistence of splicing factor mutations suggests an “myodisplatic syndrome reset” phenomenon
Splicing factor mutations such as SRSF2, SF3B1, and U2AF1 have been shown to be associated with MDS and MDS-related (“secondary”) AML.33,34 These mutations are included in the new World Health Organization and International Consensus Classification algorithms as defining AML with MDS-related gene mutations.35,36 Previous studies have demonstrated persistence of these mutations in the con-
text of cytotoxic induction chemotherapy and epigenetic modifiers.33,37 While there was variability in the clearance of other classes of mutations with ven/aza, splicing factor mutations uniformly persisted after ven/aza therapy with one exception (a patient with U2AF1 mutation); three other patients cleared their splicing factor mutations only after SCT (Figure 4A). As can be seen in the figure, and summarized in detail in Online Supplementary Table S4, approximately half of patients with persistent splicing factor mutations relapsed unless they proceeded to consolidative SCT with disappearance of their mutation. The patients who did not receive SCT yet did not relapse had splicing factor mutation VAF between 0.5% and 50% at best response. While ven/aza is effective at clearing leukemia cells, including
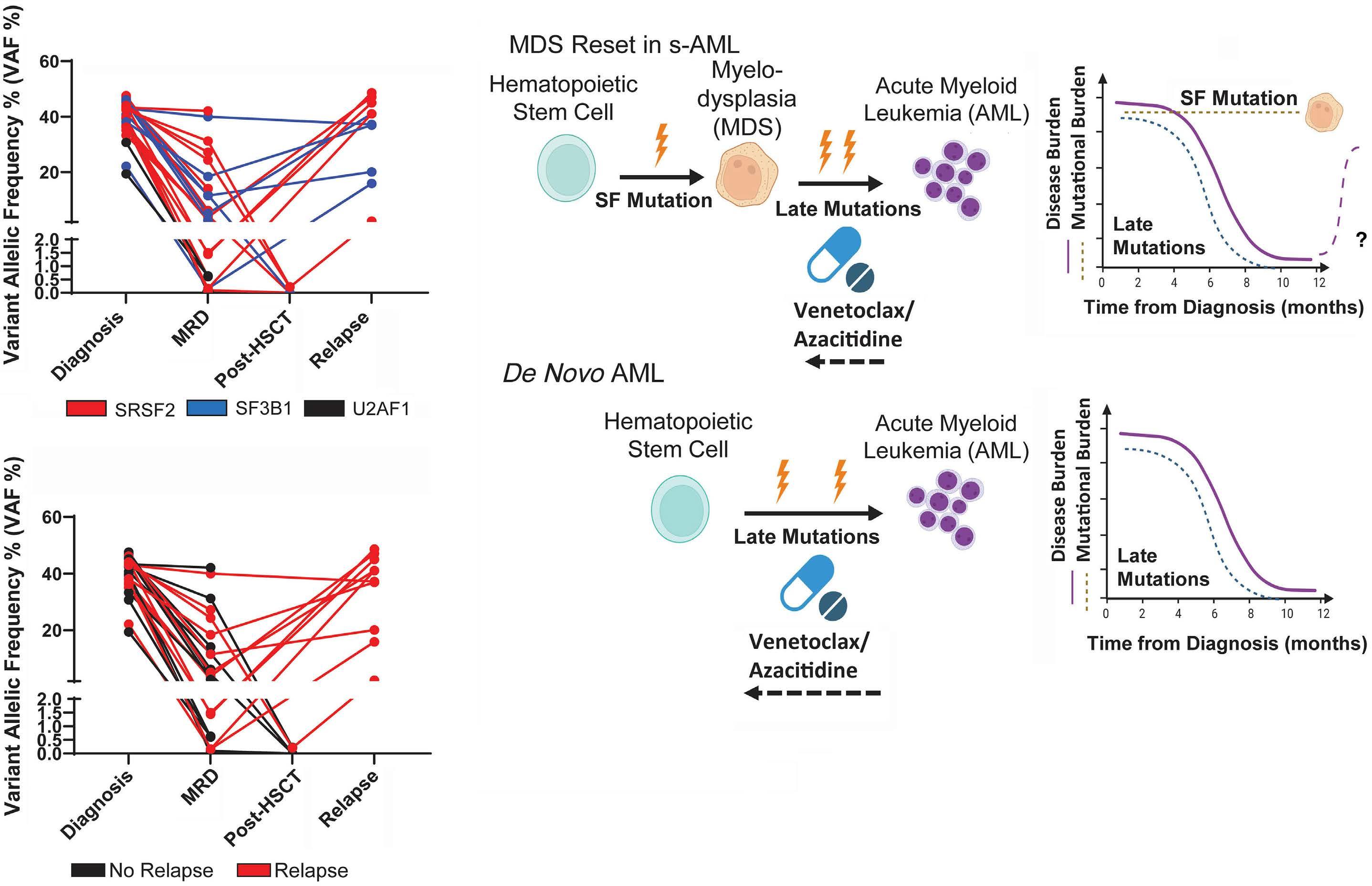
Figure 4. Splicing factor mutations persist in the absence of stem cell transplant, suggestive of persistence of an myelodysplastic syndrome clone. (A) The top graph shows individual patient time series by mutation, with variant allelic frequency (VAF) on the y-axis and time point during therapy on the x-axis. Measurable residual disease (MRD) signifies the lowest VAF achieved pre-transplant (if applicable). The bottom graph shows the same data but colored for outcome, with patients who relapsed in red and those who did not relapse in black. Save those who lost their splicing factor mutation after transplant, about half of these patients relapsed. (B) Schematic diagram of the “MDS reset” phenomenon (created with BioRender.com). Patients with myelodysplastic syndrome (MDS) preceding their acute myeloid leukemia (AML), as evidenced by dysplasia or splicing factor mutations (or both), tend to respond to venetoclax/azacitidine (ven/aza) with disappearance of their later mutations but persistence of MDS mutations. This reversion back to the pre-leukemic clone may ultimately lead to recurrence of AML at longer follow-up. Conversely, patients with de novo AML who have only later mutations and no dysplasia tend to lose all AML-associated mutations upon treatment with ven/aza and were more likely to be categorized as “MRD-negative” by droplet digital polymerase chain reaction. HSCT: hematopoietic stem cell transplant.
leukemia stem cells (LSC),29 we hypothesized that the regimen may not effectively eradicate pre-leukemic stem cells and therefore essentially “resets” a patient’s bone marrow to an MDS-like phenotype post-AML remission. We accessed bone marrow biopsy slides from these patients at all available MRD time points and sought to correlate dysplasia in the marrow (evaluated independently by 2 hematopathologists, MDE and BJS) with VAF of persistent splicing factor mutations. In total, there was available histology on 37 patients from our MRD cohort, 22 of whom had no splicing factor mutation and 15 of whom had a splicing factor mutation. Some were characterized clinically as having secondary AML (s-AML). Seventeen patients (6 with clinically defined s-AML) had no splicing factor mutations at diagnosis, nor did they have dysplasia meeting criteria for MDS on their remission marrows (Online Supplementary Table S5). Seven patients (1 with clinically defined s-AML), had a splicing factor present but no dysplasia on any follow-up marrows, despite persistence of these mutations in six of the patients. Five patients (2 with clinically defined s-AML) without splicing factor mutations had dysplasia - two with TP53 mutations, one with DNMT3A mutation, and two with IDH1 mutations - and in all cases the degree of dysplasia correlated with VAF of their primary mutation. Finally, eight patients had splicing factor mutations and dysplasia (4 with clinically defined s-AML). Two patients had poor-quality samples limiting correlation of histology with ddPCR; the other six patients had rising VAF of their splicing factor mutation
that immediately preceded or coincided with re-emergence or increased prominence of dysplastic features. Thus, the incidence of dysplasia was enriched in but not exclusive to patients with splicing factor mutations in our cohort. In all cases of dysplasia with sufficient quality of remission samples, persistence or re-emergence of dysplasia correlated strongly with the VAF of splicing factor or other founder mutations (TP53, IDH1), suggestive of an MDS reset phenomenon (Figure 4B). There were no differences observed between groups with respect to initial bone marrow blast percentage, percent identified clinically as s-AML, proportion of patients with abnormal cytogenetics, or proportion of patients who relapsed (Online Supplementary Table S6).
Droplet digital polymerase chain reaction measurable residual disease does not correlate with muti-channel flow cytometry measurable residual disease in our patient cohort
Since MCF MRD is the current clinical standard for AML, we evaluated the agreement between MCF MRD and ddPCR MRD, considering only MCF MRD performed at a reference laboratory with a validated assay. Forty-one patients had such a test performed. Due to small sample size, likely time period bias (i.e., patients diagnosed more recently having higher likelihood of MCF MRD obtained), and sampling error (MCF MRD at fewer time points than ddPCR MRD), MCF MRD status did not correlate with outcomes (Figure 5), contrary
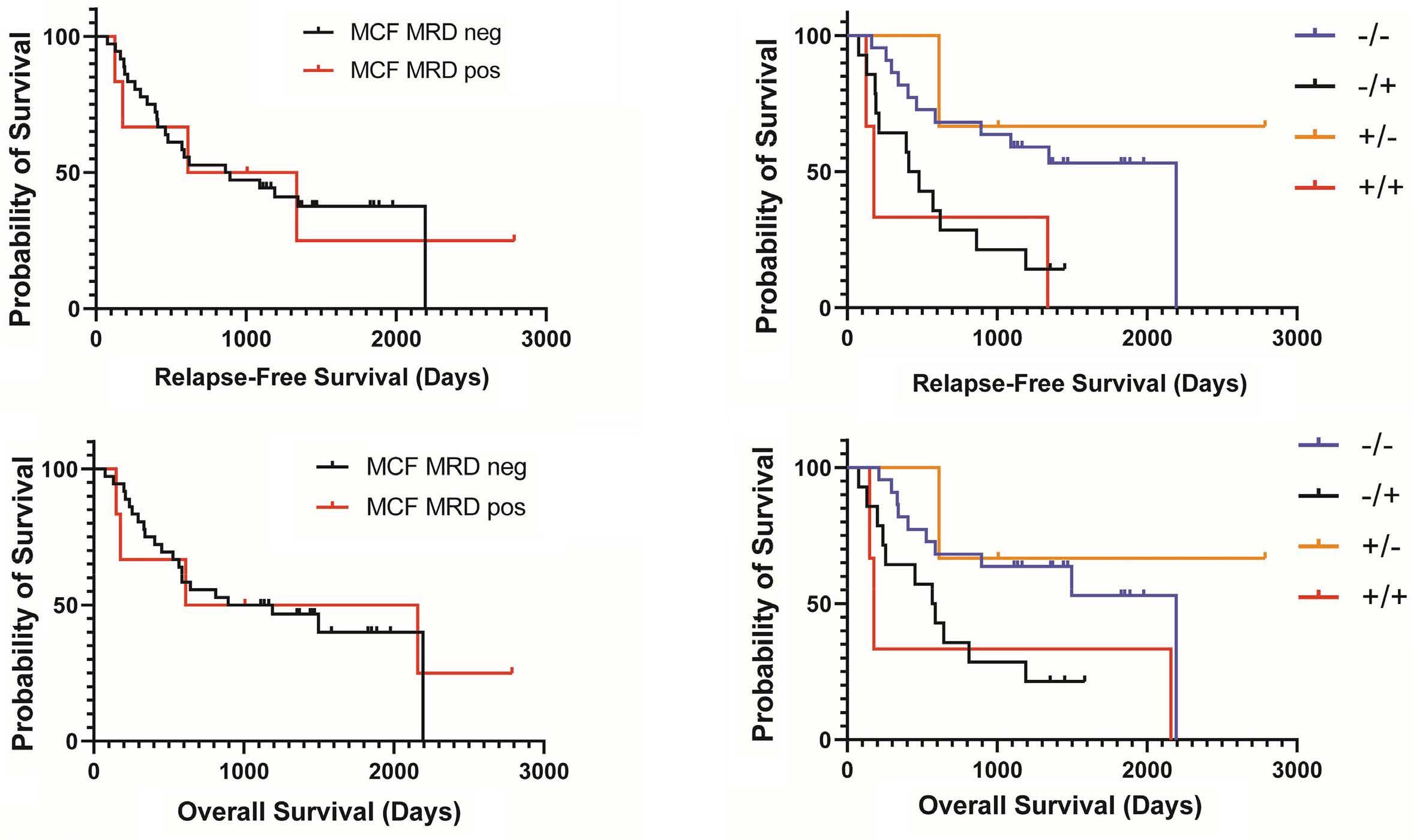
Figure 5. Multi-channel flow cytometry measurable residual disease was not prognostic of outcome in our patient cohort. (Left) Relapse-free survival (RFS) and overall survival (OS) by MCF measurable residual disease (MRD) status alone (N=41 patients). (Right) RFS and OS stratified by both multi-channel flow cytometry (MCF) MRD and droplet digital polymerase chain reaction (ddPCR) MRD in our patient cohort, where both were available (N=41 patients). Survival correlates better with ddPCR MRD. Blue lines = MCF MRD-negative (neg), ddPCR MRD-negative; black lines = MCF MRD-negative, ddPCR MRD-positive (pos); orange lines = MCF MRD-positive, ddPCR MRD-negative; red lines = MCF MRD-positive, ddPCR MRD-positive.
to what has been consistently shown in the literature1,2 and to what we show for ddPCR MRD. While analysis of 41 patients is not sufficient to rigorously evaluate MCF versus ddPCR, we note that only three of 17 patients scored as MRD-positive by ddPCR were also positive by MCF; whereas 22 of 24 individuals scored as MRD-negative by ddPCR were also negative by MCF. The two MRD modalities were in agreement only 59.5% of the time due to most of the ddPCR MRD-positive patients being classified as MRD-negative by MCF. Nevertheless, within this 41-patient subset ddPCR MRD
still stratified patients with differing RFS and OS (Figure 5). These findings suggest that ddPCR has considerably greater predictive power for relapse than MCF.
Correlation of digital droplet polymerase chain reaction measurable residual disease with survival persists in the absence of stem cell transplantation
We have previously published that patients receiving ven/ aza and then proceeding to SCT have better outcomes than patients who receive ven/aza in the absence of consolidative
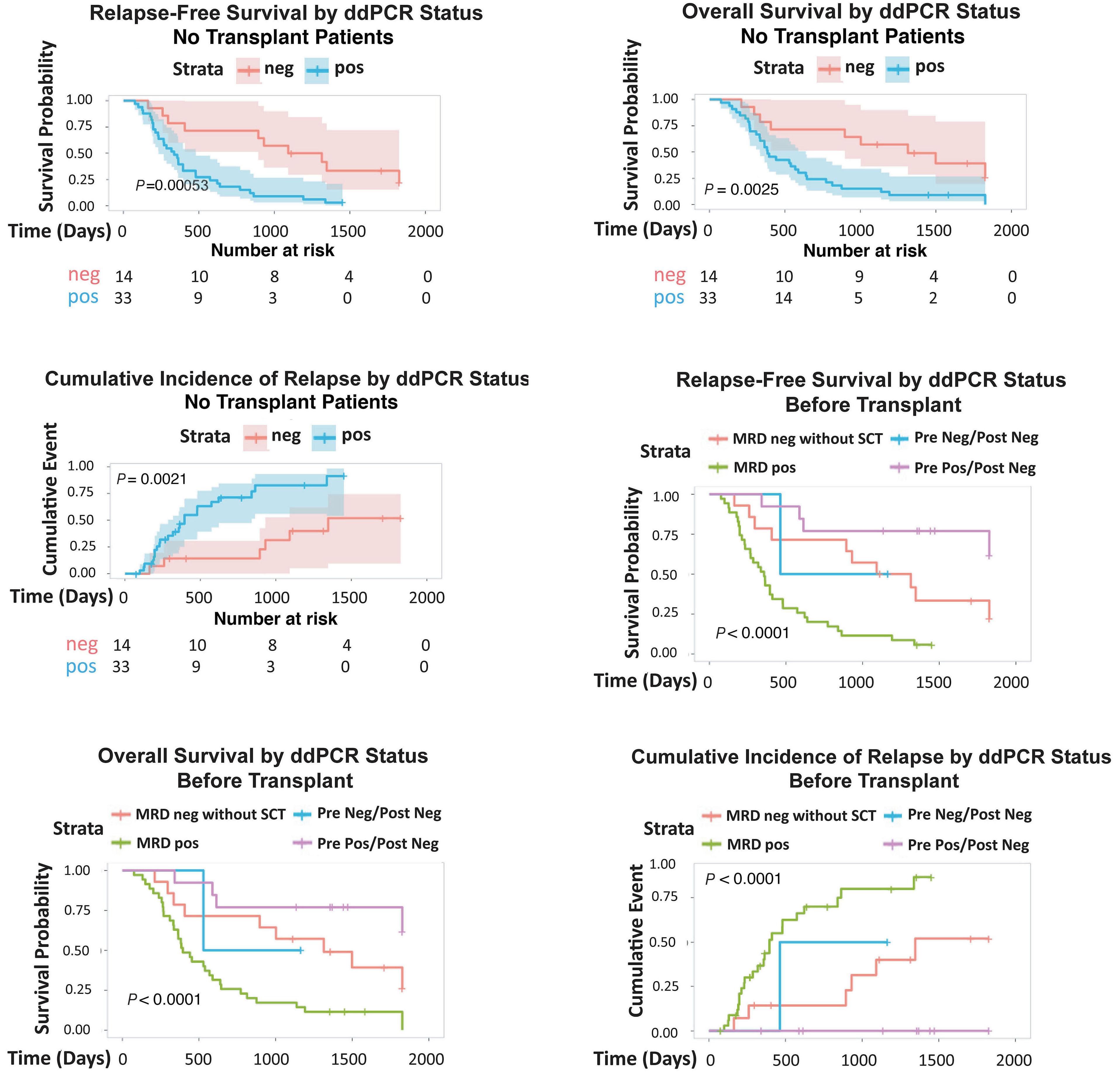
Figure 6. Droplet digital polymerase chain reaction measurable residual disease is a valuable prognostic tool after venetoclax/ azacitidine with or without stem cell transplant. (A) Relapse-free survival (RFS), (B) overall survival (OS), and (C) cumulative incidence of relapse (CIR) of all venetoclax/azacitidine (ven/aza) patients who did not receive stem cell transplantation (SCT), stratified by droplet digital polymerase chain reaction (ddPCR) measurable residual disease (MRD) status. When the total cohort (N=64) was reclassified according to ddPCR MRD status pre-SCT, the presence of SCT did further improve outcomes relative to no SCT in the MRD-negative (neg) group, but MRD-positive (pos) patients still did the worst. (D) RFS, (E) OS, and (F) CIR.
SCT.21 In the present cohort it was notable that the vast majority of individuals in the ddPCR MRD-negative group received SCT, many of them becoming MRD-negative only after SCT. Therefore we evaluated whether the predictive value of ddPCR MRD after ven/aza was only valid because of the role of SCT. We censored patients who received SCT and evaluated only patients receiving ven/aza alone for AML therapy (N=47). For this subset, ddPCR MRD status was still associated with differential RFS, CIR, and OS (Figure 6A-C), although this significance looked to be related to delays in relapse and death rather than total cure as the ddPCR MRD-negative patients without SCT still ultimately relapsed at high rates. Next, we considered the full cohort (N=64) and divided the ddPCR MRD negative group into those negative prior to SCT versus those only negative after SCT (Figure 6D-F). There was a differential outcome for these groups. Patients who were ddPCR MRD-positive, including the two patients whose MRD persisted despite SCT, did worst. Patients who were ddPCR MRD-negative prior to transplant (N=2) or who did not receive SCT (N=14) had intermediate outcomes. Patients who became MRD-negative after SCT (N=13) had the best outcome, with CIR of 0%. These data support the hypothesis that, while SCT undoubtedly has benefit in this patient population, ddPCR MRD status is still an important variable to consider in clinical decision-making after ven/aza treatment.
Multivariate analysis confirms molecular measurable residual disease status as the sole predictor of outcome We performed multivariate analysis using a Cox PH regression model to evaluate factors contributing to RFS and OS in this cohort. We considered as covariates age, SCT, ddPCR MRD status, and persistent mutation class. Results for both RFS and OS outcomes were similar: only ddPCR MRD-positive status in general and persistent NPM1 mutation in particular were predictive of inferior outcomes with hazard ratios between 4 and 5, though wide confidence intervals indicate these measures are inflated (Table 2). These results confirm previous literature27 demonstrating the value of monitoring NPM1 mutational burden in patients with NPM1-mutant AML receiving ven/aza.
This is the first study to demonstrate the utility of multigene ddPCR-based MRD for predicting outcomes in patients with AML receiving ven/aza. We show that ddPCR MRD status correlated with RFS, OS, and CIR, both including and excluding patients with NPM1 mutations. We show that time to relapse is shortest in patients with NPM1 or “late” signaling pathway mutations, but many patients with persistence of mutations such as IDH1, IDH2, and splicing factors also relapse in the absence of SCT, albeit with longer remis-
sion duration. We confirm the existence of an “MDS reset” phenomenon with ven/aza therapy, whereby persistence of MDS-associated mutations precedes or coincides with re-emergence of dysplasia. It remains to be seen whether “MDS reset” patients will have higher incidences of relapse with longer follow-up. However, this raises the question of whether venetoclax and HMA, currently in clinical trials for MDS, are capable of curing patients. Finally, we confirm the value of SCT as consolidative therapy for ven/aza, and demonstrate that MRD status can add value for SCT timing considerations. In contrast to MRD after cytotoxic chemotherapy, which is most prognostic post-induction,3 we find that ddPCR MRD negativity at TBR, whether occurring early or late in therapy and whether pre-SCT or post-SCT, correlates with lowest relapse rates. These findings are in agreement with those published from the VIALE-A cohort,
Table 2. Multi-variate analysis of factors impacting relapse-free survival and overall survival.
mutation (pre-SCT)
or IDH2 mutation
mutation
late mutation
factor mutation
ddPCR MRD
Negative
Persistent mutation (pre-SCT)
None
Multiple IDH1 or IDH2 mutation
NPM1 mutation
Other late mutation
Splicing factor mutation
(0.15-1.51)
(0.08-1.28) 4.81 (1.27-18.2) 3.02 (0.54-17.0)
(0.18-1.01)
HR: hazard ratio; CI: confidence interval; ddPCR: droplet digital polymerase chain reaction; MRD: measurable residual disease; SCT: stem cell transplant.
where MCF MRD negativity after multiple cycles of ven/aza showed no decrement in survival compared to MRD negativity after one cycle.23 Conversely, patients receiving SCT did best when they proceeded to SCT with MRD, suggesting that low-level persistence of mutations post-ven/aza can be an impetus for early SCT in eligible patients. ddPCR has been described as highly sensitive and relatively cost-effective yet limited in clinical utility due to its restriction to one mutation per assay. Indeed, previous reports utilizing ddPCR in the context of AML have been restricted to a handful of frequently occurring mutations.14,38 We utilized 50 unique ddPCR assays for the current ven/aza cohort, which were a mixture of 27 commercially available assays and 23 custom designed constructs. Since the inception of our project, availability of commercial assays has expanded; today only eight of our custom assays would not be available for purchase from an established vendor. Based on our retrospective experience, rapid design and ordering of custom assays can typically occur in 1-2 days and delivery of both custom and commercial assays in 2 weeks. Given the 2–3-week turnaround for diagnostic NGS, a provider could have a relevant ddPCR assay around the time of completion of cycle 1 of ven/aza, allowing for early MRD assessment. We acknowledge differences in cost and person-hours between laboratory-grade assay validation versus CLIA certification of an assay for prospective clinical use. However, newer iterations of ddPCR such as the development of dropoff assays for hotspot mutations39,40 could further simplify the workflow and make development of ddPCR for clinical MRD more appealing. It is worth noting that only 8% of the attrition in our ven/aza patient cohort was due to inability to design an assay for patient-relevant mutations, suggesting that >90% of patients would be eligible for ddPCR MRD monitoring. In addition, while targeted gene panels for NGS will likely become the standard of care for molecular MRD in much of the United States and Europe over the next decade, under-resourced countries might preferentially benefit from the relatively inexpensive (~US $5/sample) and less labor- and time-intensive ddPCR workflow based on more focused diagnostic mutational assessment. A limitation of the present study was our relatively small cohort size relative to other historic MRD analyses in the literature, limiting our capacity to perform subgroup analyses. However, we note that the number of patients in our cohort was comparable to existing MRD evaluations of ven/aza patients,24,26,28 with the exception of the VIALE-A cohort.23 Ours is also the most comprehensive molecular MRD analysis of ven/aza patients to date.27,28 Although our data suggest that the receipt of SCT did not fully explain the beneficial effects of ddPCR MRD-negative status in our cohort, given our small sample sizes we cannot fully rule out that disease biology or patient factors such as age do not impact our results. Therefore, to enable more in-depth subset analysis we are continuing these studies in ven/aza patients. Finally, although ddPCR MRD strongly correlated with out-
comes in our cohort, we were not able to monitor every mutation in every patient, particularly FLT3 ITD and other large insertions/deletions. The addition of a FLT3 ITD assay to our panel, which was not logistically possible at the time of our analysis and was a limitation of our MRD coverage, would further add to the power of this modality and is an active area of development. This could have impacted our results and led to misclassification of patients as MRD-negative who actually had residual disease. An ultra-sensitive NGS platform such as those recently described6,8,41,42 is another alternative for molecular MRD monitoring, although currently these platforms are more labor-intensive and more costly than ddPCR. Consortium efforts to standardize molecular MRD for future studies are underway and will be essential to the establishment of this modality for clinical use. In summary, multi-gene molecular MRD utilizing ddPCR is feasible and correlates with outcomes after ven/aza therapy. While persistence of AML-associated mutations such as NPM1 portend more imminent relapse, consideration of other mutations such as IDH1, IDH2, and potentially splicing factor mutations may also provide a comprehensive assessment of disease status and contribute to clinical decision-making.
DAP has received research funding and served as a consultant to Abbvie. The other authors have no conflicts of interest to disclose.
ACW conceptualized the project, designed the assays, performed ddPCR experiments, collected clinical data from the electronic medical record, and analyzed data. MM and AM performed ddPCR experiments and data analysis. BMS, JAG, DAP, and CTJ contributed to the design of the project and provided feedback on the work. JAG and DAP consented patients for biobanking and clinical data acquisition. NM, JY, and ALT performed diagnostic and relapse NGS and processed bone marrow samples for DNA isolation. MDE and BJS reviewed bone marrow slides for dysplasia to correlate with persistence of splicing factor mutations. All authors contributed to the writing of the manuscript.
The authors would like to acknowledge the patients who contributed to this research and the outstanding team at the Blood Disorders Center at the University of Colorado.
ACW is funded by Career Enhancement Program funds from the University of Colorado Department of Medicine, by Swim Across America, by the Morgan Adams Foundation, and by NIH 1K08CA279762-01. CTJ is supported by the Nancy Carroll Allen Chair in Hematology Research, a Leukemia and Lymphoma Society SCOR grant (7020-19), NIH R35CA242376,
and Veterans Administration merit award BX004768-01. DAP is supported by the Robert H. Allen MD Chair in Hematology and the Leukemia and Lymphoma Society Scholar in Clinical Research.
1. Short NJ, Zhou S, Fu C, et al. Association of measurable residual disease with survival outcomes in patients with acute myeloid leukemia: a systematic review and meta-analysis. JAMA Oncol. 2020;6(12):1890-1899.
2. Buckley SA, Wood BL, Othus M, et al. Minimal residual disease prior to allogeneic hematopoietic cell transplantation in acute myeloid leukemia: a meta-analysis. Haematologica. 2017;102(5):865-873.
3. Heuser M, Freeman SD, Ossenkoppele GJ, et al. 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138(26):2753-2767.
4 Klco JM, Miller CA, Griffith M, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA. 2015;314(8):811-822.
5. Ivey A, Hills RK, Simpson MA, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374(5):422-433.
6. Dillon LW, Higgins J, Nasif H, et al. Quantification of measurable residual disease using duplex sequencing in adults with acute myeloid leukemia. medRxiv. 2023 Mar 27. doi: 10.1101/2023.03.26.23287367 [preprint, not peer-reviewed].
7 Hasserjian RP, Steensma DP, Graubert TA, Ebert BL. Clonal hematopoiesis and measurable residual disease assessment in acute myeloid leukemia. Blood. 2020;135(20):1729-1738.
8. Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189-1199.
9 Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221.
10 Cancer Genome Atlas Research N, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074.
11. Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet. 2018;19(5):269-285.
12. Maier J, Lange T, Cross M, Wildenberger K, Niederwieser D, Franke GN. Optimized digital droplet PCR for BCR-ABL. J Mol Diagn. 2019;21(1):27-37.
13. Bacher U, Dicker F, Haferlach C, et al. Quantification of rare NPM1 mutation subtypes by digital PCR. Br J Haematol. 2014;167(5):710-714.
14 Brambati C, Galbiati S, Xue E, et al. Droplet digital polymerase chain reaction for DNMT3A and IDH1/2 mutations to improve early detection of acute myeloid leukemia relapse after allogeneic hematopoietic stem cell transplantation. Haematologica. 2016;101(4):e157-161.
15. Wei AH, Strickland SA, Jr., Hou JZ, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol. 2019;37(15):1277-1284.
16. Pollyea DA, Pratz K, Letai A, et al. Venetoclax with azacitidine or
Data-sharing statement
Original data, primer/probe sequences, and protocols are available upon request by contacting the corresponding author.
decitabine in patients with newly diagnosed acute myeloid leukemia: Long term follow-up from a phase 1b study. Am J Hematol. 2021;96(2):208-217.
17 DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629.
18. DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791-803.
19 Pei S, Pollyea DA, Gustafson A, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 2020;10(4):536-551.
20 Stevens BM, Jones CL, Pollyea DA, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer. 2020;1(12):1176-1187.
21. Pollyea DA, Winters A, McMahon C, et al. Venetoclax and azacitidine followed by allogeneic transplant results in excellent outcomes and may improve outcomes versus maintenance therapy among newly diagnosed AML patients older than 60. Bone Marrow Transplant. 2022;57(2):160-166.
22. Chua CC, Hammond D, Kent A, et al. Treatment-free remission after ceasing venetoclax-based therapy in patients with acute myeloid leukemia. Blood Adv. 2022;6(13):3879-3883.
23. Pratz KW, Jonas BA, Pullarkat V, et al. Measurable residual disease response and prognosis in treatment-naive acute myeloid leukemia with venetoclax and azacitidine. J Clin Oncol. 2022;40(8):855-865.
24. Maiti A, DiNardo CD, Wang SA, et al. Prognostic value of measurable residual disease after venetoclax and decitabine in acute myeloid leukemia. Blood Adv. 2021;5(7):1876-1883.
25. Bazinet A, Kadia TM, Short NJ, et al. Undetectable measurable residual disease is associated with improved outcomes in AML irrespective of treatment intensity. Blood Adv. 2023;7(13):3284-3296.
26. Ong SY, Tan Si Yun M, Abdul Halim NA, et al. Real-world experience of measurable residual disease response and prognosis in acute myeloid leukemia treated with venetoclax and azacitidine. Cancers (Basel). 2022;14(15):3576.
27. Tiong IS, Dillon R, Ivey A, et al. Venetoclax induces rapid elimination of NPM1 mutant measurable residual disease in combination with low-intensity chemotherapy in acute myeloid leukaemia. Br J Haematol. 2021;192(6):1026-1030.
28. Othman J, Tiong IS, O’Nions J, et al. Molecular MRD is strongly prognostic in patients with NPM1-mutated AML receiving venetoclax-based non-intensive therapy. Blood. 2024;143(4):336-341
29 Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859-1866.
30. Winters AC, Gutman JA, Purev E, et al. Real-world experience of venetoclax with azacitidine for untreated patients with acute
myeloid leukemia. Blood Adv. 2019;3(20):2911-2919.
31. Gutman JA, Winters A, Kent A, et al. Higher-dose venetoclax with measurable residual disease-guided azacitidine discontinuation in newly diagnosed acute myeloid leukemia. Haematologica. 2023;108(10):2616-2625.
32. Valent P, Orazi A, Steensma DP, et al. Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget. 2017;8(43):73483-73500.
33. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367-1376.
34 McCarter JGW, Nemirovsky D, Famulare CA, et al. Interaction between myelodysplasia-related gene mutations and ontogeny in acute myeloid leukemia. Blood Adv. 2023;7(17):5000-5013.
35. Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200-1228.
36. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703-1719.
37. Uy GL, Duncavage EJ, Chang GS, et al. Dynamic changes in the
clonal structure of MDS and AML in response to epigenetic therapy. Leukemia. 2017;31(4):872-881.
38. Mencia-Trinchant N, Hu Y, Alas MA, et al. Minimal residual disease monitoring of acute myeloid leukemia by massively multiplex digital PCR in patients with NPM1 mutations. J Mol Diagn. 2017;19(4):537-548.
39 Grassi S, Guerrini F, Ciabatti E, et al. Digital droplet PCR is a specific and sensitive tool for detecting IDH2 mutations in acute myeloid leuKemia patients. Cancers (Basel). 2020;12(7):1738.
40 Rausch C, Rothenberg-Thurley M, Buerger SA, et al. Double prop-off droplet digital PCR: a novel, versatile tool for mutation screening and residual disease monitoring in acute myeloid leukemia using cellular or cell-free DNA. J Mol Diagn. 2021;23(8):975-985.
41. Hourigan CS, Dillon LW, Gui G, et al. Impact of conditioning intensity of allogeneic transplantation for acute myeloid leukemia with genomic evidence of residual disease. J Clin Oncol. 2020;38(12):1273-1283.
42. Bae JH, Liu R, Roberts E, et al. Single duplex DNA sequencing with CODEC detects mutations with high sensitivity. Nat Genet. 2023;55(5):871-879.
Anmol Baranwal,1,2 Mark Gurney,1 Rami Basmaci,1 Bahga Katamesh,1 Rong He,3 David S. Viswanatha,3 Patricia Greipp,3 James Foran,4 Talha Badar,4 Hemant Murthy,4 Cecilia Arana Yi,5 Jeanne Palmer,5 Abhishek A. Mangaonkar,1 Mrinal M. Patnaik,1 Mark R. Litzow,1 William J. Hogan,1 Kebede Begna,1 Naseema Gangat,1 Ayalew Tefferi,1 Aref Al-Kali,1 Mithun V. Shah1 and Hassan B. Alkhateeb1
1Division of Hematology, Department of Medicine, Mayo Clinic, Rochester, MN; 2Cancer Centers of Southwest Oklahoma, Lawton, OK; 3Division of Hematopathology, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN; 4Division of HematologyOncology, Blood and Marrow Transplantation Program, Department of Medicine, Mayo Clinic, Jacksonville, FL and 5Division of Hematology, Department of Medicine, Mayo Clinic, Phoenix, AZ, USA
Correspondence: H.B. Alkhateeb Alkhateeb.Hassan@mayo.edu
Received: August 29, 2023.
Accepted: January 22, 2024. Early view: February 1, 2024.
https://doi.org/10.3324/haematol.2023.284185
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
The BCL6-corepressor ( BCOR ) is a tumor-suppressor gene located on the short arm of chromosome X. Data are limited regarding factors predicting survival in BCOR -mutated (m BCOR ) acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). We evaluated 138 patients with m BCOR myeloid disorders, of which 36 (26.1%) had AML and 63 (45.6%) had MDS. Sixty-six (47.8%) patients had a normal karyotype while 18 (13%) patients had complex karyotype. BCOR- mutated MDS/AML were highly associated with RUNX1 and U2AF1 co-mutations. In contrast, TP53 mutation was infrequently seen with m BCOR MDS. Patients with an isolated BCOR mutation had similar survival compared to those with high-risk co-mutations by European LeukemiaNet (ELN) 2022 criteria (median OS 1.16 vs . 1.27 years, P =0.46). Complex karyotype adversely impacted survival among m BCOR AML/MDS (HR 4.12, P <0.001), while allogeneic stem cell transplant (alloSCT) improved survival (HR 0.38, P =0.04). However, RUNX1 co-mutation was associated with an increased risk of post-alloSCT relapse (HR 88.0, P =0.02), whereas melphalan-based conditioning was associated with a decreased relapse risk (HR 0.02, P =0.01). We conclude that m BCO R is a high-risk feature across MDS/AML, and that alloSCT improves survival in this population.
The BCL6-corepressor (BCOR) is a tumor-suppressor gene located at the 11.4 locus of short arm of chromosome X.1 The BCOR gene product is predominantly involved in suppressing myeloid regulatory genes and promotes lymphopoiesis.1 Grossman et al. first demonstrated the association of somatic BCOR mutations with acute myeloid leukemia (AML).2 The recent 2022 European LeukemiaNet (ELN) recommendations on acute myeloid leukemia classifies BCOR-associated AML in the adverse risk category.3 However, the clinical outcomes of BCOR-mutated (mBCOR) AML has primarily been explored in pooled cohorts evaluating multiple other genes. For instance, Papaemmanuil et al evaluated clinical outcomes of patients with AML stratified by genetic subgroups.4 The study included mBCOR AML within the chromatin-spliceosome group, which also included patients with mutations in SRSF2, SF3B1, U2AF1,
ZRSR2, ASXL1, STAG2, BCOR, MLLPTD, EZH2, and PHF6. The study showed adverse outcomes of patients in this mutational group. Similarly, Gardin et al. evaluated the outcomes of a cohort of 471 patients with secondary AML (sAML)-like gene mutations which included 38 patients with mBCOR AML, and a trend towards worse overall survival (OS) in the presence of sAML-like mutations (HR 1.22, P=0.07) was observed.5 The outcomes of patients with mBCOR AML were not specifically evaluated in either of the studies. Damm et al. evaluated 15 patients with mBCOR myelodysplastic syndrome (MDS) and demonstrated an inferior OS compared to patients with wild-type BCOR (HR 3.3, 95% CI 1.4-8.1, P=0.008).6 Other studies evaluating outcomes of patients with mBCOR MDS are also limited by small sample size and inclusion of other gene mutations.7,8 In this study, we evaluated the genetic and clinical features, and factors predicting outcomes of patients with mBCOR AML/MDS.
The study was approved by the Mayo Clinic Institutional Review Board. Patients with WHO-defined myeloid neoplasms, including AML and MDS, and found to have a BCOR mutation on next-generation sequencing (NGS) performed between October 2015 to August 2021 on peripheral blood or bone marrow aspirate were included. 9 Patient demographics, disease characteristics at time of diagnosis and at the time of NGS testing, co-mutations, treatment-related variables and survival outcomes were extracted.
Because patients with mBCOR AML would be considered to have high-risk disease by ELN 2022 risk stratification, the disease risk for baseline characteristics was determined using the Revised International Prognostic Scoring System (IPSS-R) for MDS and the ELN 2017 risk stratification for AML.3,10,11 However, the survival outcomes of patients with m BCOR AML was compared to a control cohort by both ELN 2017 and ELN 2022 risk stratification.
Consecutive patients who had NGS testing performed from May 2015 to September 2017, and had wild-type (wt) BCOR were considered as control cohort. Data on co-mutations, cytogenetics, and disease risk stratification by ELN 2017, ELN 2022 and IPSS-R criteria were collected for patients in the control cohort. For patients undergoing alloSCT, a hematopoietic cell transplantation-comorbidity index (HCT-CI) score ≥3 was considered high. Conditioning regimen intensity was defined per the Center for International Blood and Marrow Transplant Research (CIBMTR) criteria.12 Acute graft- versus -host disease (GvHD) was graded according to Glucksberg criteria and severity of chronic GvHD was determined using the 2014 National Institute of Health consensus criteria.13,14 Relapse was defined as detection of disease after alloSCT by morphological, cytogenetic, or molecular analysis, as applicable. Relapse incidence (RI) was calculated from the time of alloSCT to the time of relapse detection.
Patients’ and disease characteristics were summarized using descriptive statistics. The statistical comparison of categorical variables was performed using χ 2 test. For continuous variables, Kruskal-Wallis test was used for comparison of medians and t test was used for comparison of means.
Comparison of co-mutations among patients with wt BCOR versus m BCOR was performed using the “epitools” package and evaluated on log10 scale to prevent skewness.15 Patients were deemed to enter the study at the time of NGS testing. Survival outcomes were analyzed in patients with AML/MDS from timepoint of study entry, unless mentioned otherwise. Kaplan-Meier and log-rank tests
were used to estimate OS.16 Median follow-up time was determined using the reverse Kaplan-Meier method.17 Cox-proportional hazard model was used to determine the effect on OS. Allogeneic stem cell transplant (alloSCT) was considered a time-dependent covariate for both univariate and multivariate Cox proportional hazard analysis.18
The cumulative incidence of non-relapse mortality (NRM) and RI in patients undergoing alloSCT was determined using the competing risks method. Fine-Gray analysis was used to determine factors influencing NRM and RI post-alloSCT.
Only those genes that were mutated in at least 5 patients (approx. 5% of the entire cohort) were included in the univariate analyses. Because BCOR gene is located on X-chromosome, the variant allele frequency (VAF) of BCOR mutation was gender-adjusted for all the analyses. Variables with P <0.20 in univariate analyses were included in multivariate analysis. R 4.2.0 (R Foundation for Statistical Computing, Vienna, Austria) was used to perform all the statistical analyses. P <0.05 was considered statistically significant.19,20
Out of 6,887 consecutive NGS tests performed, 138 (2%) patients were found to have BCOR mutation. These patients were compared to 275 patients with wt BCOR , who were considered to be the control cohort.
The control cohort consisted of 155 (82 males, 53%) patients with AML, 105 (71 males, 68%) with MDS, and 15 (5.5%) others. Median age at study entry was 74 years (interquartile range [IQR] 67-78 years) for MDS and 65 years (IQR 56-73 years) for AML. Of the 155 AML patients, 17 (11%) were favorable risk, 61 (39%) were intermediate risk, and 73 (47.1%) were adverse risk by ELN 2022 criteria. By ELN 2017 criteria, 20 (13%) patients had favorable risk, 66 (42%) had intermediate risk, and 65 (42%) patients had adverse risk disease (Table 1). Risk stratification could not be determined for 4 (2.6%) patients. The most commonly mutated gene among patients with AML was DNMT3A (N=33, 21.3%), followed by mutations in genes TET2 (N=29, 18.7%), TP53 (N=27, 17.4%), and IDH2 (N=27, 17.4%) ( Online Supplementary Table S1 ). Among the 105 MDS patients, 27 (25%) were high or very high-risk by the IPSS-R. Fourteen (13%) patients had MDS with increased blasts-type 1 (MDS-IB1), while 15 (14%) patients had MDS-IB2. The most commonly mutated gene among MDS patients was ASXL1 (N=29, 27.6%), followed by mutations in genes TET2 (N=21, 20%) and SF3B1 (N=20, 19%). TP53 gene was mutated in 15 (14.3%) patients ( Online Supplementary Table S1 ).
Table 1. Baseline characteristics of patients stratified by wild-type BCOR and mutated BCOR.
Gender, N (%)
N (%)
ELN 2017 risk stratification, N (%)
IPSS-R risk stratification, N (%)
MDS (by blasts), N (%)
Genes mutated, N (%)
wt: wild-type; m: mutated; N: number; AML: acute myeloid leukemia; MDS: myelodysplastic syndromes; ELN: European LeukemiaNet; IPSS-R: Revised International Prognostic Scoring System; IB: increased blasts.
Characteristics of patients with mBCOR acute myeloid leukemia and myelodysplastic syndromes
A total of 138 patients (96 males, 69.6%) with mBCOR hematologic disorders were evaluated. Thirty-six (26.1%) patients had AML and 63 (45.6%) had MDS (Table 2 and Online Supplementary Table S2). Among patients with AML and MDS, 64 (64.6%) had NGS testing performed before the first intervention/treatment.
Most of the patients (N=133, 96.4%) harbored only a single BCOR mutation. A total of 144 BCOR variants were seen across 138 patients. RUNX1 (50/138, 36%) and U2AF1 (38/138, 28%) were among the most commonly co-mutated genes (Figure 1A). Median VAF for BCOR mutation was 34.5% (IQR 16-61%) (Figure 1B); the median gender-adjusted VAF was 24.25% (IQR 11-37%). Mutations in the BCOR gene were present throughout the exon sequence and a particular hotspot could not be identified (Figure 1C). Frameshift variants were most common (82, 57%), followed by nonsense (45, 32%) and splice-site (16, 11%) variants, while only one (0.7%) patient had a missense mutation (Figure 1C).
Sixty-six (47.8%) patients had a normal karyotype: 16 (44.4%) among patients with AML, and 27 (42.9%) among patients with MDS (Figure 1D). None of the mBCOR AML or MDS patients had any abnormalities in chromosome 17. Monosomy 7 was found in 3 (4.8%) patients, and deletion 20q was seen in 8 (12.7%) patients with mBCOR MDS but were not seen in any patient with mBCOR AML. Other cytogenetic abnormalities among patients with mBCOR AML/MDS included trisomy 8 in 16 (16.2%) patients (7 AML, 43.8%; 9 MDS, 56.2%), deletion 5q in 8 patients (3 AML, 37.5%; 5 MDS, 62.5%), and inversion 3 in 2 (2%) patients (1 patient each with AML and MDS). One (1.59%) patient with MDS had rearrangement involving the KMT2A gene locus (11q23). Eighteen (13%) patients had complex karyotype: 7 (19.4%) among the 36 patients with AML and 6 (9.5%) among the 63 patients with MDS (Table 2, Figure 1D). Among the AML patients, one (2.8%) had favorable risk, 14 (38.9%) had intermediate risk, and 19 (52.8%) had adverse risk disease by ELN 2017 criteria; risk stratification could not be evaluated in 2 (5.6%) patients. Among the MDS patients, 43 (68.3%) had an intermediate or higher risk disease by IPSS-R; 20 (31.7%) patients had MDS-IB1, while 23 (36.5%) had MDS-IB2 (Table 2). Median white blood cell (WBC) count was 2.5x106 cells/L (range 0.9 –98.4x106) in patients with AML and 2.30x106 cells/L (range 0.8 – 14.4x106) in patients with MDS.
Among AML and MDS patients, BCOR mutations were found to be highly associated with mutations in RUNX1 gene (odds ratio [OR] 5.5; P<0.001). BCOR mutations were less commonly associated with TP53 mutations, particularly among patients with MDS (OR 0.21; P=0.02) (Figure 1E, Online Supplementary Table S3). Patients with mBCOR MDS were found to be significantly associated with U2AF1 mutation (OR 3.95; P<0.001). The U2AF1 S34F variant was the predominant mutation (20/24, 83%) in mBCOR MDS
patients (Online Supplementary Table S4). Interestingly, none of the patients with mBCOR had an NPM1 co-mutation.
Median follow-up time after study entry among patients with mBCOR AML/MDS was 3.2 years (95% CI: 2.34-3.82 years). Median survival for the entire cohort of mBCOR AML/MDS patients was 15 months. The survival was lower among patients with AML compared to MDS, but this difference was not statistically significant (median OS: 0.74 vs. 1.53 years; P=0.21) (Figure 2A, Online Supplementary Figure S1A). Complex karyotype (median OS 0.3 vs. 1.5 years; P<0.001) and age ≥70 years (median OS 0.88 vs 2.04 years; P=0.008) were associated with a worse survival (Figure 2B, C). Patients with mBCOR had similar survival regardless of the presence of additional other high-risk co-mutations by ELN 2022 criteria (median OS 1.26 vs. 1.3 years; P=0.46) (Figure 2D). The median OS from diagnosis among patients with NGS at diagnosis was similar to those who had NGS performed after treatment (median OS 2.07 vs. 2.05 years; P=0.87).
In comparison to wtBCOR patients (control cohort), survival since study entry among mBCOR patients was similar to those with adverse risk AML and numerically inferior to those with favorable or intermediate risk AML by the ELN 2017 criteria (3-year OS 29.5% vs. 24.4% vs. 41.2%; P=0.32) (Figure 2E). Survival from diagnosis among mBCOR AML patients was similar to those with adverse risk AML by ELN 2017 criteria and significantly inferior compared to those with favorable or intermediate risk AML by the ELN 2017 criteria (3-year OS 36.5% vs. 33.5% vs. 52.8%; P=0.03) (Figure 2F); similar findings were seen on evaluating survival by ELN 2022 criteria (Online Supplementary Figure S1B, C). Patients with mBCOR AML with NGS performed at diagnosis or after treatment had survival rates similar to adverse risk AML and inferior to favorable/intermediate risk AML by ELN 2017 criteria (3-year OS rate: 35.4% vs. 38.1% vs. 33.5% vs. 52.8% for mBCOR at diagnosis, mBCOR after treatment, wtBCOR with adverse risk AML and wtBCOR with favorable/intermediate risk AML, respectively; P=0.046). Survival was similar among mBCOR MDS with or without a U2AF1 mutation (P=0.89) (Online Supplementary Figure S2A) and those with mBCOR AML/MDS with or without a RUNX1 co-mutation (P=0.51) (Online Supplementary Figure S2B). Univariate analysis showed that, among patients with mBCOR, complex karyotype (HR 3.17, 95% CI: 1.69-5.96; P<0.001) and age ≥70 years at study entry (HR 2.04, 95% CI: 1.2-3.47; P=0.008) were associated with inferior survival, whereas alloSCT (HR 0.25, 95% CI: 0.11-0.57; P=0.001) was associated with an improved OS. Complex karyotype, age ≥70 years at study entry, alloSCT, and presence of EZH2 and/or KRAS co-mutations were factors included in the multivariate analysis (Online Supplementary Table S5). Multivariate analysis confirmed that complex karyotype was associated with a worse survival (HR 4.92, 95% CI:
at diagnosisin yrs,
Gender, N (%)
N (%)
2017 risk stratification, N (%)
stratification, N (%)
MDS by blasts, N (%)
N
blasts ≥ 10% at NGS, N (%)
, N (%)
AML: acute myeloid leukemia; MDS: myelodysplastic syndromes; N: number; yrs: years; IQR: interquartile range; ELN: European LeukemiaNet; IPSS-R: Revised International Prognostic Scoring System; IB: increased blasts; BM: bone marrow; Hb: hemoglobin; NGS: next-generation sequencing.
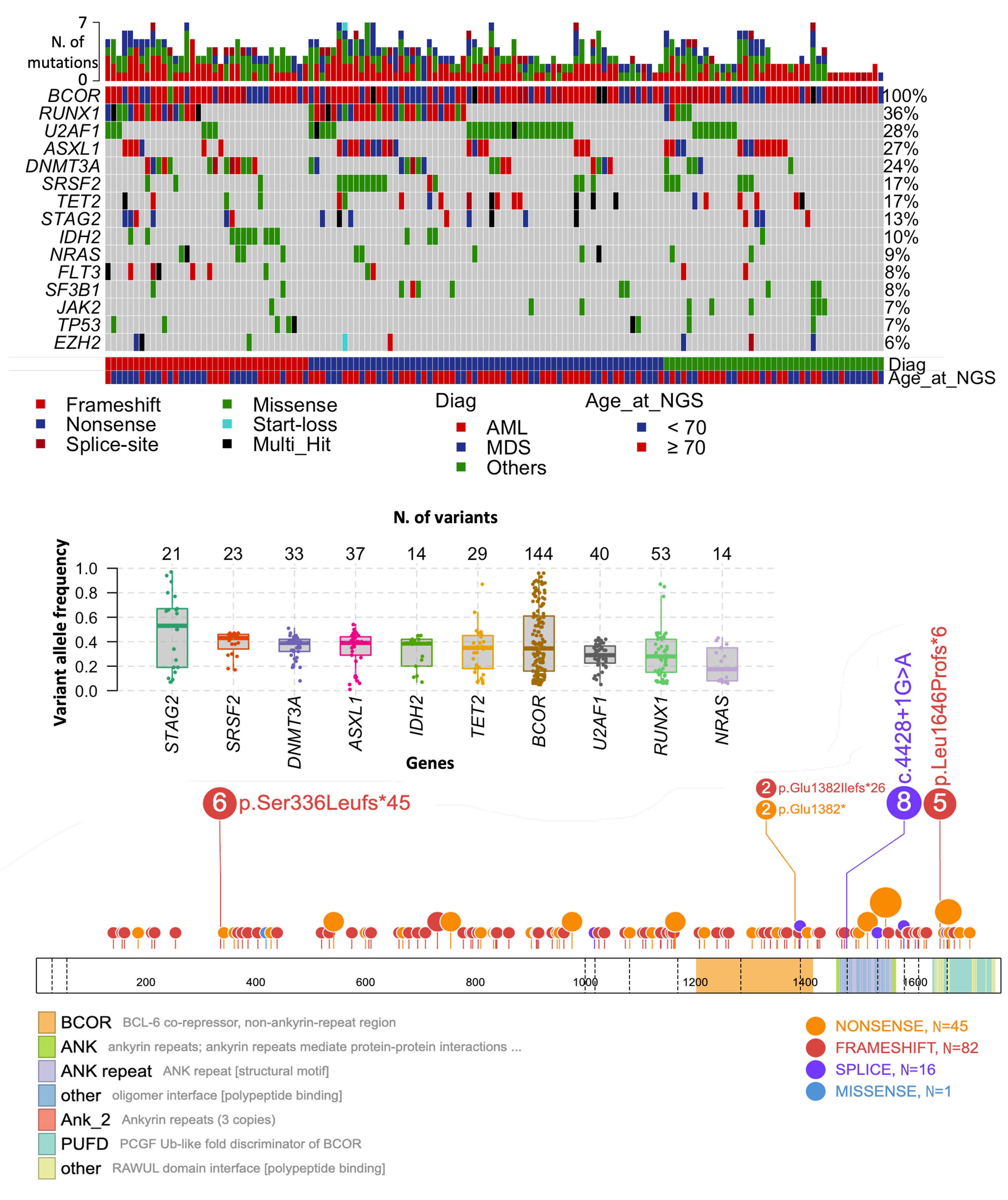
Continued on following page.
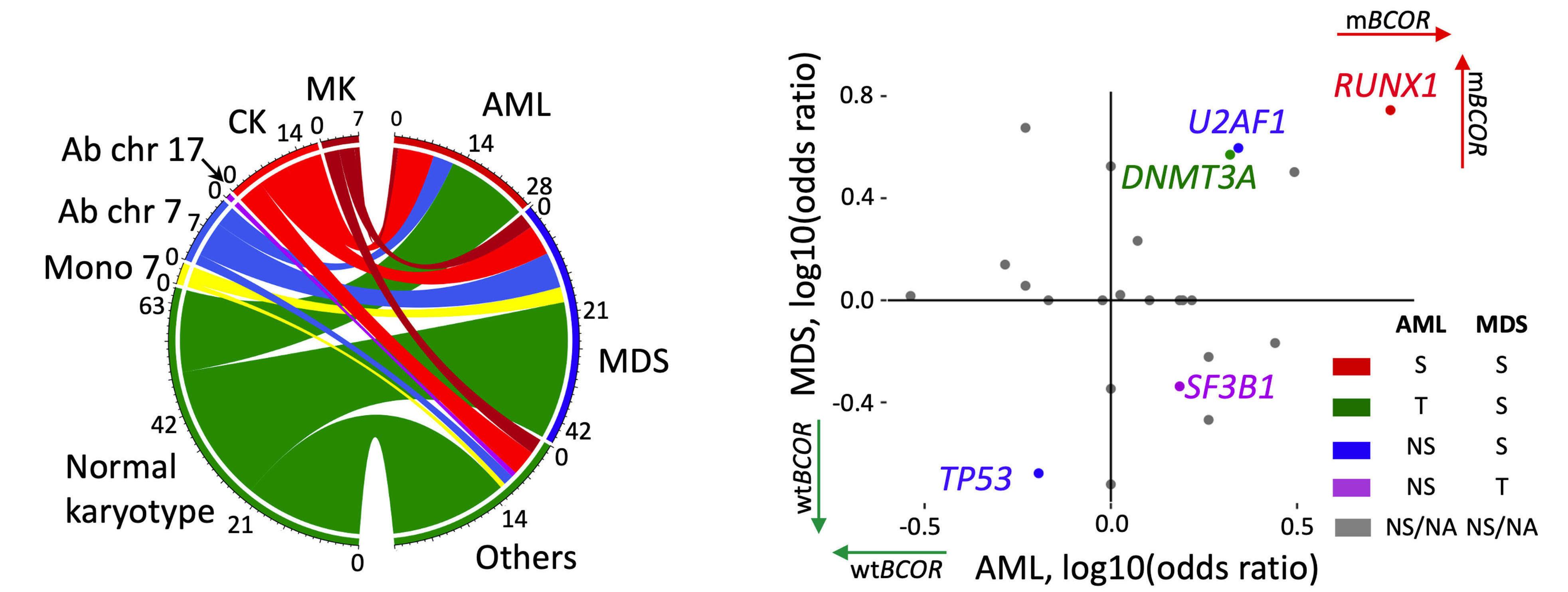
Figure 1. Genomic landscape of patients with BCOR mutations. (A) Oncoplot of patients with BCOR mutation; top 15 mutated genes are shown. (B) Top 10 genes with highest median variant allele frequency. (C) Lollipop plot showing various mutations across the BCOR gene. Mutations were most commonly frameshift or nonsense, and spread across the entire gene, without a specific hotspot region. (D) Specific cytogenetic abnormalities stratified by diagnoses: acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), or others. Approximately half of the patients had normal karyotype, whereas complex karyotype or chromosome 17 abnormalities were seen in only a minority of patients. (E) Association of BCOR mutation with other genes among patients with MDS and AML. CK: complex karyotype; MK: monosomal karyotype; Ab: abnormal; Chr: chromosome; Diag: diagnosis; Mono: monosomy; N: number; NGS: next-generation sequencing; S: significant (P<0.05); T: trend (0.10 > P≥0.05; NS: not significant (P≥0.10); NA: not applicable.
2.0811.65; P<0.001), whereas alloSCT was associated with an improved survival (HR 0.38, 95% CI: 0.14-0.996; P=0.049) (Online Supplementary Figure S1D ). Upon adjusting the BCOR VAF for gender, a trend towards inferior survival was seen among patients with BCOR VAF ≥40% (Figure 3).
A total of 30 patients (19 males, 63.3%) underwent alloSCT after study entry. Median age at alloSCT was 65 years (IQR 58-68 years). Ten patients (33.3%) had AML, 17 (56.7%) had MDS, 2 (6.7%) had myeloproliferative neoplasm, and one patient (3.3%) had chronic myelomonocytic leukemia (CMML) (Table 3).
The 27 patients with mBCOR AML/MDS were evaluated for post-alloSCT outcomes. Median follow-up time after alloSCT was 2.5 years (95% CI: 1.8-4.0 years). Median HCT-CI score was 3 (IQR 1-3); thirteen (48.1%) patients had a high HCT-CI score. Fifteen (55.6%) patients were in complete remission (CR) at the time of alloSCT, 11 (40.7%) had persistent disease, and the disease status was not known for one patient (3.7%). Among the 10 patients with AML, 5 (50%) had pre-alloSCT minimal residual disease (MRD) testing performed, 4 (40%) of whom were MRD negative and one (10%) patient was MRD positive before transplant. Median survival after alloSCT was not reached; OS rate at three years was 61.1% (Figure 4A). A total of 8 (29.6%) deaths were reported within three years after transplant, of whom 5 patients (62.5%) had NRM and 3 (37.5%) patients
died after relapse. The cumulative incidence of NRM was 23.7% and RI was 24.1% at three years after transplant (Online Supplementary Figure S3). Among the 5 patients with NRM, 2 (40%) died of infection, one (20%) died of GvHD, and the cause of death was not known for 2 (40%) patients. There was no significant difference in 3-year NRM (10% vs. 34.5%; P=0.32) and relapse (10% vs. 33.3%; P=0.24) in patients with AML or MDS (Figure 4B).
Given that BCOR is a high-risk mutation, we evaluated factors influencing post-transplant relapse. Univariate competing risk analysis showed that mutation in genes ASXL1, RUNX1 (mRUNX1), melphalan-, or busulphan-based conditioning, and reduced-intensity conditioning were factors significantly affecting post-transplant relapse ( Online Supplementary Table S6 ). Multivariate analysis showed that mRUNX1 was associated with an increased risk of post-transplant relapse (HR 88.0, 95% CI: 1.98-3918; P=0.02), and melphalan-based conditioning was associated with a decreased risk of post-transplant relapse (HR 0.02, 95% CI: 0.001-0.40; P=0.01) (Figure 4C).
Patients with mBCOR AML undergoing alloSCT had a superior post-alloSCT survival compared to those with m BCOR MDS, but it was not statistically different (3-year OS 88.9% vs. 43.2%; P=0.1) (Figure 5A), while complex karyotype was associated with an inferior post-alloSCT survival (median OS 1.24 vs. NA years; P=0.04) (Figure 5B). Multivariate analysis that included complex karyotype and diagnosis (AML vs. MDS) (Online Supplementary Table S7) did not
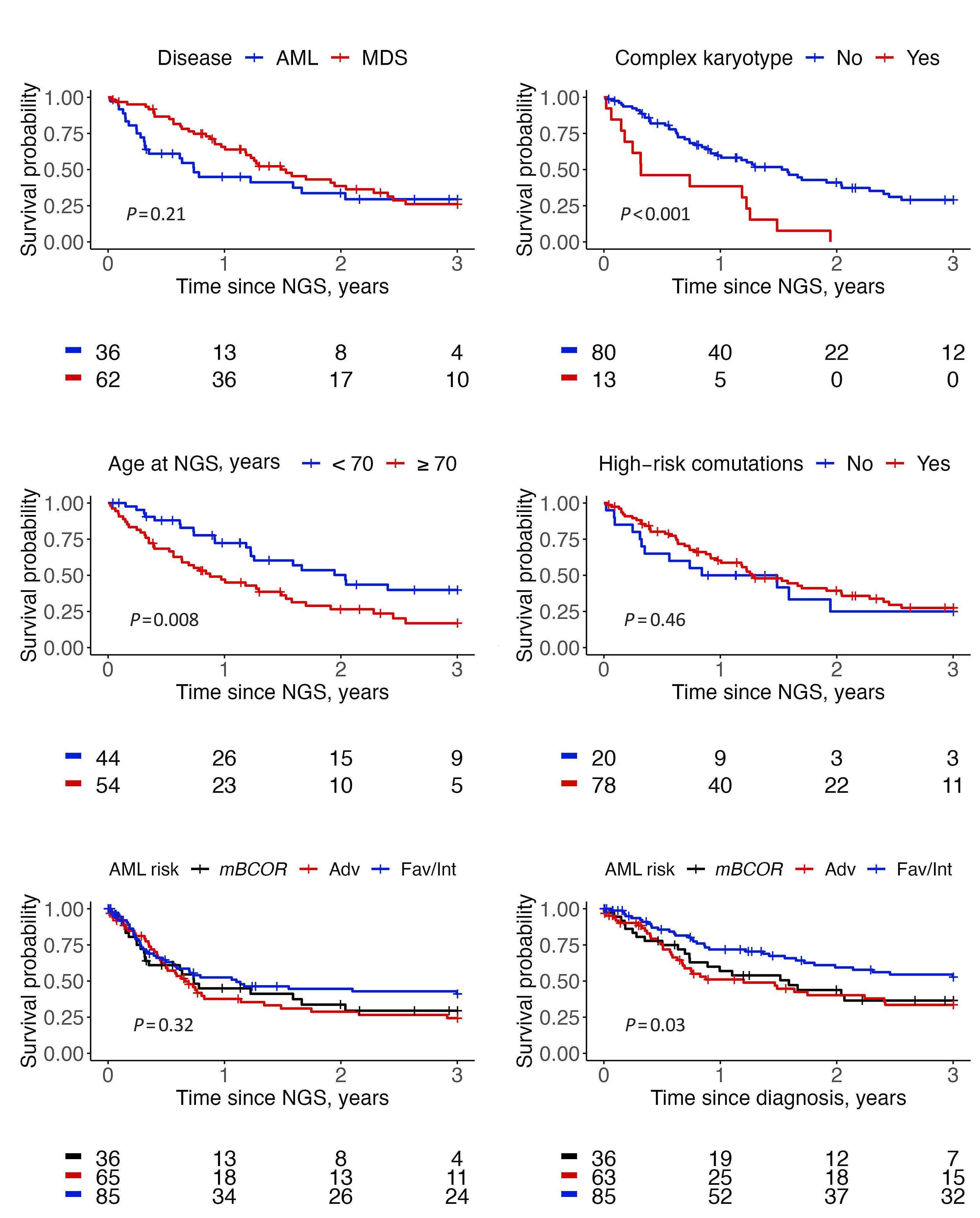
Figure 2. Clinical outcomes of patients with BCOR-mutated acute myeloid leukemia or myelodysplastic syndromes. Kaplan-Meier plots for survival since time of BCOR mutation (mBCOR) detection stratified by (A) disease type, (B) complex karyotype at next-generation sequencing (NGS), (C) age at NGS, and (D) presence/absence of high-risk co-mutations by European LeukemiaNet (ELN) 2022 criteria. (E and F) Overall survival of patients with mutated (m)BCOR versus wild-type (wt)BCOR, stratified by ELN 2017 risk category. Adv: adverse risk; AML: acute myeloid leukemia; Fav/Int: favorable / intermediate; MDS: myelodysplastic syndromes.
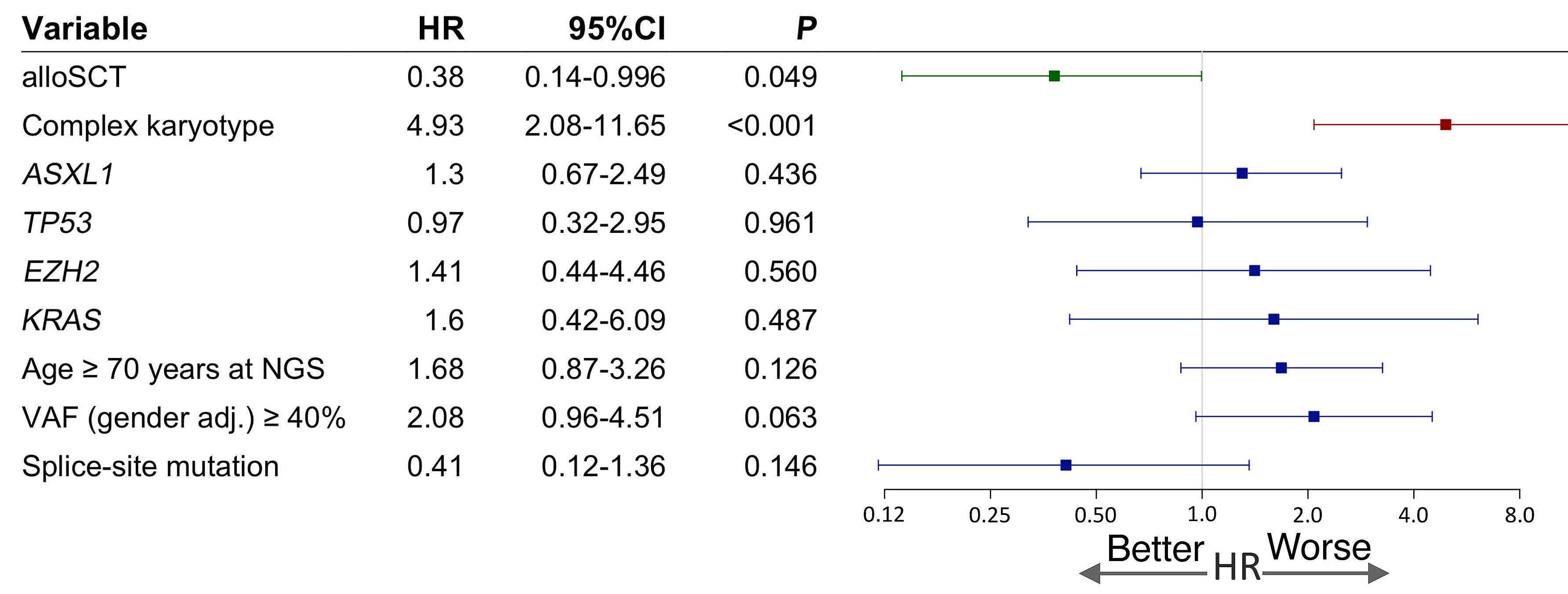
Figure 3. Multivariate analysis of factors affecting post-next-generation sequencing survival in patients with mutated BCOR adj: adjusted; HR: Hazard Ratio; NGS: next-generation sequencing; VAF: variant allele frequency.
identify either of the 2 to be independently associated with post-alloSCT survival (Figure 5C), suggesting that alloSCT was beneficial regardless of phenotype or cytogenetics.
Characteristics of mBCOR myelodysplastic syndromes / myeloproliferative neoplasms overlap syndrome
A total of 11 (7.8%) patients had myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) overlap syndrome, 6 (54.5%) of whom had CMML. The median age at diagnosis was 75 years (IQR 68-82 years). Of the 11 patients, 8 (72.7%) patients had normal karyotype while trisomy 8 was seen in 2 patients (18.2%); karyotype was not available for one patient (9.1%). ASXL1 (10 patients, 90.9%), SRSF2 (5 patients, 45.5.%) and RUNX1 (4 patients, 36.4%) were among the most commonly mutated genes. Two (18.2%) patients (1 CMML, 9.1%) harbored a mutation in the TET2 gene. Median follow-up time was 1.6 years (95% CI: 1.57-NA). Survival was higher among patients with CMML compared to other MDS/MPN disorders; however, it was not statistically significant (median OS 15 vs. 9 months; P=0.5). Survival from NGS among patients with mBCOR MDS/MPN was similar to patients with mBCOR AML (median 7.5 vs. 9 months; P=0.28), but inferior to patients with mBCOR MDS (median 7.5 vs. 18.6 months; P=0.005) (Online Supplementary Figure S4).
We described a cohort of 138 patients with m BCOR, of whom 99 (72%) had AML/MDS. Patients had BCOR mutations detected primarily in their 6th-7th decade of life and had low white blood cell (WBC) counts at diagnosis. These findings are parallel to those reported by Papaemmanuil et al. 4
Studies have shown that a majority of patients with BCOR mutation have normal karyotype.6,21 Approximately half of the patients in our cohort had normal karyotype and would have been classified in non-high-risk categories in the absence of mBCOR; complex karyotype was seen in only a minority of patients, while abnormality of chromosome 17 was not seen in any patient with mBCOR AML/MDS. We could not identify a specific hotspot of mutations and the BCOR variants were spread throughout the gene. Most of the BCOR mutations were truncating mutations, and missense mutation was a rare finding in our cohort. This is similar to the findings of Grossmann et al., who evaluated 26 patients with mBCOR AML and found 15 frameshift, 6 nonsense, and 4 splice-site mutations, and a mutation pattern that is consistent with a tumor-suppressor function.2,22,23
Mutations in the BCOR gene have previously been associated with a RUNX1 mutation.6,21 Damm et al. also showed a strong association of mBCOR MDS with mutations in the DNMT3A gene.6 Our study confirms these findings with a significant association of mutations in BCOR and DNMT3A in patients with MDS. A similar trend was also noticed in mBCOR AML in our cohort. Our study confirms that BCOR and NPM1 mutations are mutually exclusive.6,24 We also found that mBCOR MDS were highly associated with mutations in the U2AF1 gene. This is in contrast to the study by Montgomerry et al., which showed a predominant association of mBCOR with mU2AF1 in lymphoid malignancies and not in myeloid neoplasms. 25 In our study, the U2AF1 S34F was the predominant variant. The U2AF1 has been shown to portend poor outcomes in patients with MDS.26,27 Our study showed that the survival among mBCOR patients was poor, regardless of the presence or absence of the U2AF1 mutation. More importantly, the poor survival among patients with mBCOR AML/MDS was
Table 3. Characteristics and outcomes of patients with mBCOR undergoing allogeneic stem cell transplantation.
Gender, N (%)
GvHD prophylaxis, N (%)
Acute GvHD, N (%)
Chronic GvHD, N (%)
AML: acute myeloid leukemia; MDS: myelodysplastic syndromes; N: number; alloSCT: allogeneic stem cell transplantation; HCT-CI: hematopoietic cell transplantation-comorbidity index; NA: not available; CMML: chronic myelomonocytic leukemia; MPN: myeloproliferative neoplasms; ELN: European LeukemiaNet; CR: complete remission; IB: increased blasts; TBI: total body irradiation; CMV: cytomegalovirus; neg: negative; pos: positive; GvHD: graft-versus-host disease; CNI: calcineurin inhibitor; ATG: anti-thymocyte globulin; MMF: mycophenolate mofetil; PTCy: post-transplantation cyclophosphamide. *As per the European LeukemiaNet 2022 risk stratification.
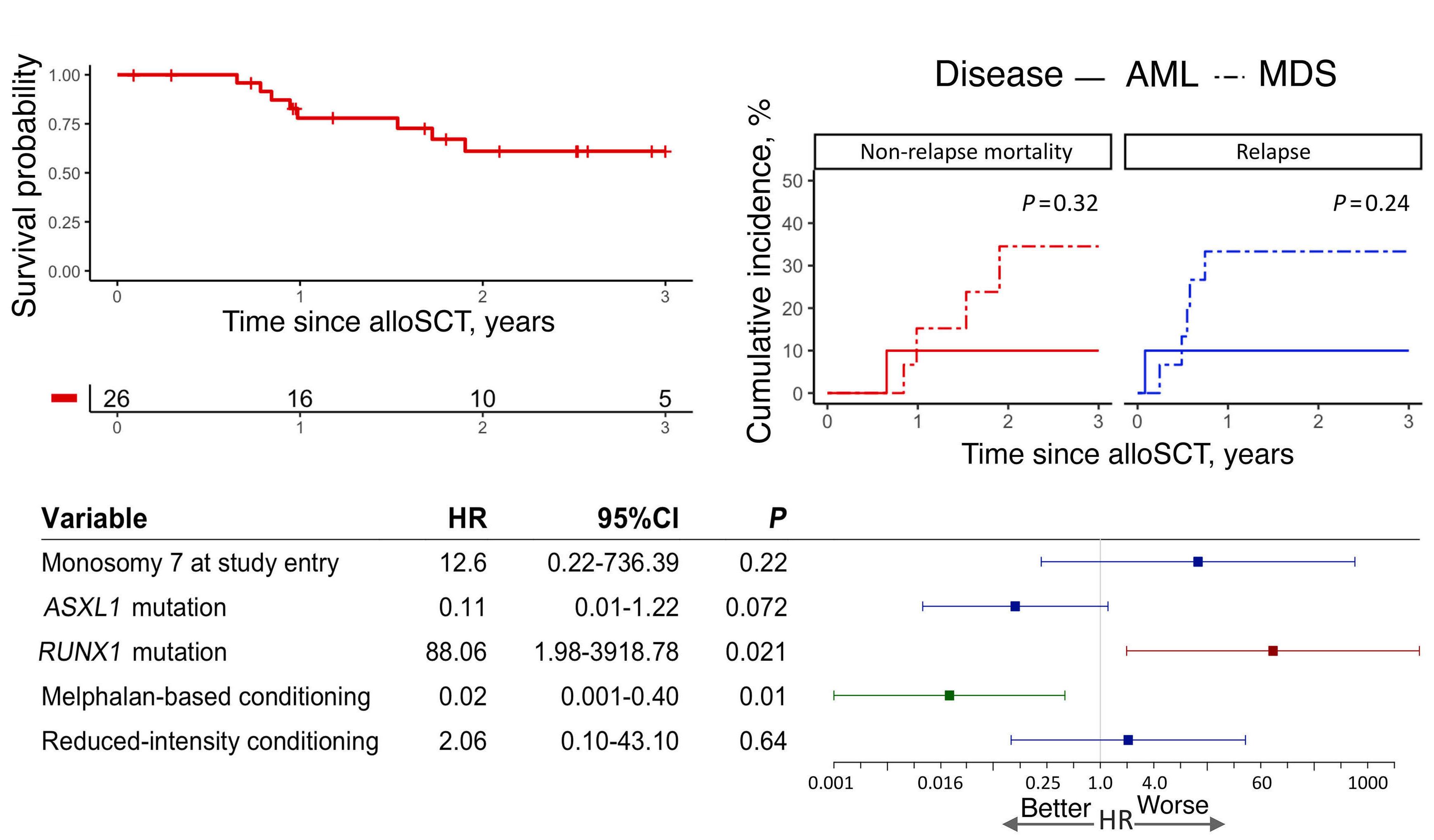
Figure 4. Outcomes of patients with BCOR-mutated acute myeloid leukemia / myelodysplastic syndromes undergoing allogeneic stem cell transplantation. (A) Overall survival after allogeneic stem cell transplantation (alloSCT). Follow-up data available in 26 patients. (B) Competing risk analysis showing cumulative incidence of non-relapse mortality (NRM) and relapse after alloSCT. (C). Multivariate competing risk analysis for relapse incidence at three years after alloSCT showing an overall adverse effect of RUNX1 mutation and a positive effect of melphalan-based conditioning. AML: acute myeloid leukemia; HR: Hazard Ratio; MDS: myelodysplastic syndromes.
in spite of association with a high-risk co-mutation such as RUNX1 or ASXL1. Our study shows that patients with m BCOR AML/MDS have poor prognosis. The poor prognosis was seen across all the subgroups of MDS and AML patients, regardless of blast percentage. A comparison with the control cohort showed that survival among mBCOR AML patients parallels those with adverse risk AML patients by both ELN 2017 and ELN 2022 criteria, confirming the adverse prognosis of patients with mBCOR AML. Our study also shows that alloSCT improves survival in patients with mBCOR AML/ MDS, and these patients should be evaluated for alloSCT at the earliest opportunity. More importantly, RUNX1 co-mutation was associated with an increased risk of post-alloSCT relapse while melphalan-based conditioning was found to have a positive effect on post-alloSCT RI. A few studies have previously evaluated the effect of alloSCT in patients with mBCOR AML.28,29 In a pooled cohort of de novo AML with secondary-type mutations including BCOR, Song et al. evaluated 15 patients undergoing alloSCT and found a cumulative RI of 33.3% at five years.28 This parallels a 3-year RI of 24.1% reported in our study. Some of the limitations of our study are that the NGS testing
was done at different time points and the relatively small sample size of patients undergoing transplant. However, the former limitation is somewhat offset by the fact that the majority of patients with mBCOR AML/MDS in our cohort had NGS testing carried out before the first intervention/treatment. The time-to-event approach from NGS testing does lead to an underestimation of survival as the duration of disease before testing is not accounted for by this methodology. However, because this bias applies to both the mBCOR and the control cohort, its effect is diminished in a comparative analysis. For the same reason, we evaluated survival outcomes from both the time points, i.e., from diagnosis and from NGS. We further compared the survival outcomes of patients found to have a BCOR mutation at diagnosis versus later in the time-course and found similar survival rates. Given the rarity of BCOR mutations, a small sample size will be a limitation of any single-institutional study.
To our knowledge, this is the largest study specifically evaluating the impact of m BCOR across patients with AML and MDS. Our study also shows that the outcomes mBCOR AML/MDS are poor regardless of the presence or absence of high-risk co-mutations, suggesting an effect of BCOR in determining disease outcomes, while complex
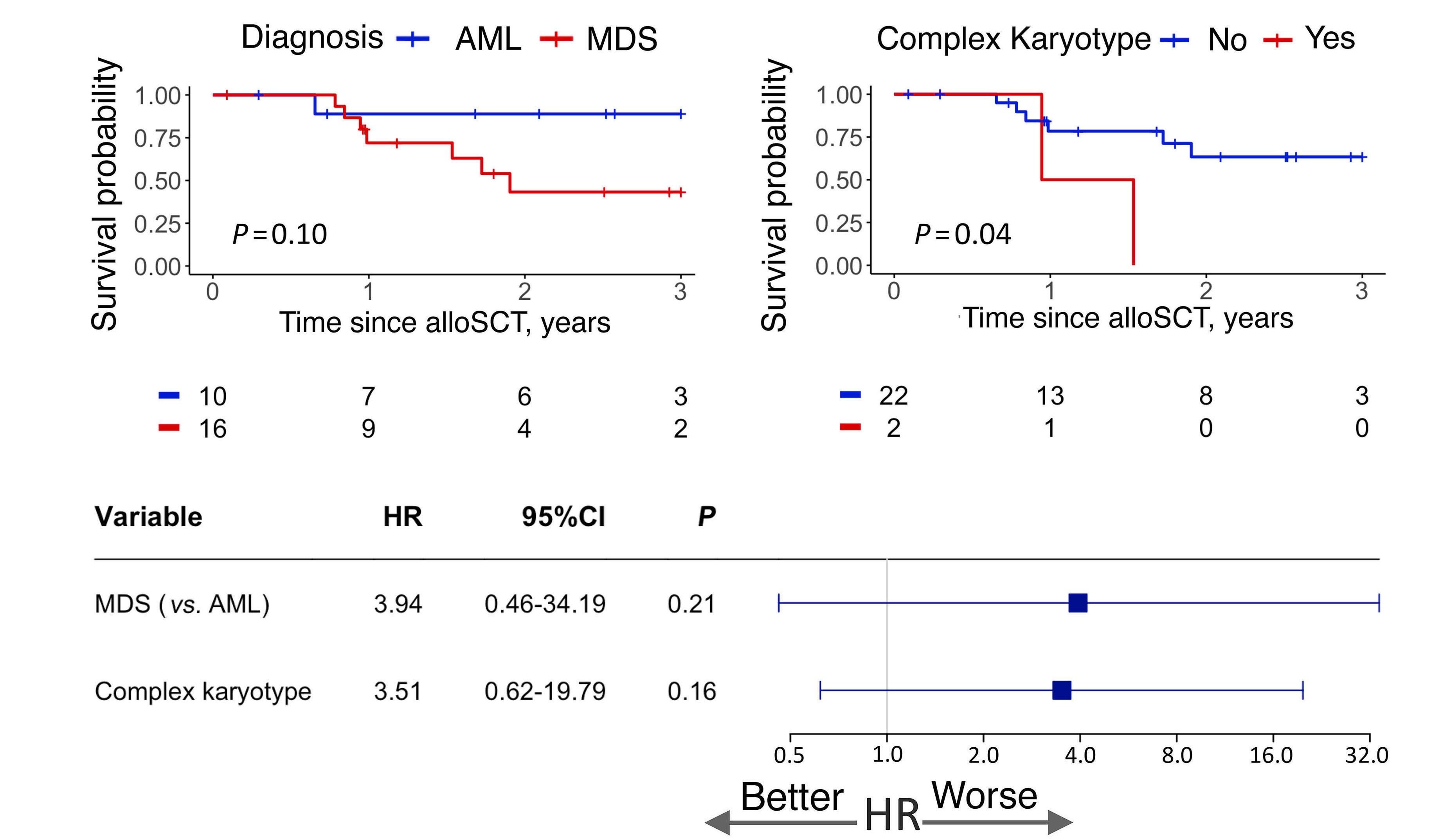
Figure 5. Factors influencing post-allogeneic stem cell transplantation survival in patients with mutated BCOR acute myeloid leukemia / myelodysplastic syndromes. Overall survival after allogeneic stem cell transplantation (alloSCT) stratified by: (A) disease type, (B) complex karyotype. (C) Multivariate analysis for 3-year post-alloSCT survival. AML: acute myeloid leukemia; HR: Hazard Ratio; MDS: myelodysplastic syndromes.
karyotype continues to have adverse outcomes. Finally, our study confirms that allogeneic transplant is associated with improved outcomes in patients with mBCOR AML/ MDS; patients harboring a RUNX1 co-mutation are at an increased risk of relapse, while melphalan-based conditioning decreases the relapse risk.
Disclosures
MVS reports research funding to the institution from Astellas, Abbvie, Celgene, and Marker Therapeutics. All of the other authors have no conflicts of interest to disclose.
Contributions
HBA, MVS and AB contributed to study design. AB, MG, RB
1. Sportoletti P, Sorcini D, Falini B. BCOR gene alterations in hematologic diseases. Blood. 2021;138(24):2455-2468.
2. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153-6163.
and BK contributed to data acquisition and analysis. AB and HBA wrote the first draft. RH, DSV and PG performed molecular testing. JF, TB, HM, CAY, JP, AAM, MMP, MRL, WJH, KB, NG, AT, AA-K, MVS and HBA contributed patients and reviewed the manuscript. All authors approved the final version of the manuscript for publication.
Acknowledgments
Figure 1C was created using ProteinPaint (https://proteinpaint.stjude.org/).
Data-sharing statement
Data may be obtained from the corresponding author upon reasonable request.
3. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345-1377.
4 Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221.
5. Gardin C, Pautas C, Fournier E, et al. Added prognostic value of secondary AML-like gene mutations in ELN intermediate-risk older AML: ALFA-1200 study results. Blood Adv.
2020;4(9):1942-1949.
6. Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169-3177.
7 Abuhadra N, Mukherjee S, Al-Issa K, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes (MDS): clonal architecture and impact on outcomes. Leuk Lymphoma. 2019;60(6):1587-1590.
8. Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241-247.
9 Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
10 Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood. 2012;120(12):2454-2465.
11. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
12. Bacigalupo A, Ballen K, Rizzo D, et al. Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant. 2009;15(12):1628-1633.
13. Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18(4):295-304.
14. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group Report. Biol Blood Marrow Transplant. 2015;21(3):389-401.e1.
15. Aragon TJ, Fay MP, Wollschlaeger D, Omidpanah A. Epitools: epidemiology tools. R package version 0.5-10.1. 2020. https:// CRAN.R-project.org/package=epitools Accessed December 2, 2023.
16. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53(282):457-481.
17 Shuster JJ. Median follow-up in clinical trials. J Clin Oncol. 1991;9(1):191-192.
18. Moore DF. Applied survival analysis using R. Cham: Springer International Publishing; 2016.
19. R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ Accessed 15 Dec 2023.
20. Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811-2812.
21. Zhang A, Liu Y, Wei S, et al. BCOR mutations in acute myeloid leukemia: clonal evolution and prognosis. Blood. 2020;136(Suppl 1):4.
22. Kumar RD, Searleman AC, Swamidass SJ, Griffith OL, Bose R. Statistically identifying tumor suppressors and oncogenes from pan-cancer genome-sequencing data. Bioinformatics. 2015;31(22):3561-3568.
23. Schroeder MP, Rubio-Perez C, Tamborero D, Gonzalez-Perez A, Lopez-Bigas N. OncodriveROLE classifies cancer driver genes in loss of function and activating mode of action. Bioinformatics. 2014;30(17):i549-i555.
24. Tiacci E, Grossmann V, Martelli MP, et al. The corepressors BCOR and BCORL1: two novel players in acute myeloid leukemia. Haematologica. 2012;97(1):3-5.
25. Montgomery ND, Galeotti J, Johnson SM, et al. Bilineal evolution of a U2AF1-mutated clone associated with acquisition of distinct secondary mutations. Blood Adv. 2021;5(24):5612-5616.
26. Tefferi A, Mudireddy M, Finke CM, et al. U2AF1 mutation variants in myelodysplastic syndromes and their clinical correlates. Am J Hematol. 2018;93(6):E146-E148.
27. Pritzl SL, Gurney M, Badar T, et al. Clinical and molecular spectrum and prognostic outcomes of U2AF1 mutant clonal hematopoiesis-a prospective Mayo Clinic cohort study. Leuk Res. 2023;125:107007.
28. Song G-Y, Kim T, Ahn S-Y, et al. Allogeneic hematopoietic cell transplantation can overcome the adverse prognosis indicated by secondary-type mutations in de novo acute myeloid leukemia. Bone Marrow Transplant. 2022;57(12):1810-1819.
29 Baranwal A, Chhetri R, Yeung D, et al. Factors predicting survival following alloSCT in patients with therapy-related AML and MDS: a multicenter study. Bone Marrow Transplant. 2023;58(7):769-776.
Roma V. Rajput,1 Vaani Shah,2 Ruba N. Shalhoub,3 Kamille West-Mitchell,4 Nu Ri Cha,4 Cathy Conry-Cantilena,4 Susan F. Leitman,4 David J. Young,5 Brian Wells,6 Georg Aue,6 Cynthia E. Dunbar,5 Bhavisha A. Patel,1 Richard W. Childs,1 Neal S. Young,1 Colin O. Wu,3 Emma M. Groarke1# and Shelley S. Kalsi2#
1Hematology Branch, National Heart, Lung, and Blood Institute, NHLBI; 2Hematology Consult and Graduate Medical Section, NHLBI; 3Office of Biostatistics Research, NHLBI; 4Department of Transfusion Medicine, Clinical Center; 5Translational Stem Cell Biology Branch, NHLBI and 6Laboratory of Transplantation Immunotherapy, NHLBI, National Institutes of Health, Bethesda, MD, USA
#EMG and SSK contributed equally as senior authors.
Correspondence: R.V. Rajput roma.rajput@nih.gov
Received: June 28, 2023. Accepted: November 30, 2023. Early view: December 7, 2023. https://doi.org/10.3324/haematol.2023.283826 ©2024 NIH (National Institutes of Health)
Patients with severe aplastic anemia (SAA) are at high risk of morbidity and mortality due to severe infections. We aimed to characterize the role of granulocyte transfusions (GT) in SAA. Primary outcomes were survival after the first GT, including overall survival (OS) at last follow up, survival to discharge, and receipt of a hematopoietic stem cell transplant (HSCT) Secondary outcomes included evaluation of clinical response at 7 and 30 days after initiation of GT, using a clinical scoring system incorporating microbiological and radiographic response. Twenty-eight SAA patients underwent 30 GT courses with a per-dose median of 1.28x109 granulocytes/kilogram (range, 0.45-4.52x109). OS from initial GT to median last follow up (551 days) was 50%, with 39% (11/28) alive at last follow up. Sixty-four percent (18/28) of all patients survived to hospital discharge. Patients with a complete or partial response, or stable infection, at 30 days had significantly better OS compared to non-responders (P=0.0004). Eighty-six percent (18/21) of patients awaiting HSCT during GT underwent a transplant and 62% (13/21) survived to post-HSCT discharge. Sex, type of infection, and percentage of days with absolute neutrophil count >0.2x109/L during the course of GT were not predictive of survival (P=0.52, P=0.7 and P=0.28, respectively). Nine of 28 (32%) patients developed new or increased human leukocyte antigen alloimmunization during their GT course. GT in SAA may have an impact on survival in those patients with improvement or stabilization of their underlying infection. Alloimmunization can occur and OS in this population remains poor, but GT may be a useful tool to bridge patients to curative treatment with HSCT.
Patients with severe aplastic anemia (SAA), a life-threatening immune-mediated bone marrow failure disorder characterized by destruction of hematopoietic stem cells, are at high risk of severe infection due to prolonged neutropenia. Hematopoietic recovery can be difficult to achieve and severe infections lead to significant morbidity and mortality. Reports, including a 10-year review of SAA patients at our institution who received granulocyte transfusions (GT), have shown a link between clinical response after initiating GT and survival, suggesting an adjunctive role for GT in managing infections in this population.1,2 Results from a prospective, multicenter, randomized controlled trial seeking to determine the efficacy of GT in patients with severe neutropenia demonstrated no significant difference in survival and microbial response
between patients who received GT with standard antimicrobial therapy and those who received standard antimicrobial therapy alone.3 The limitations of that trial included low patient accrual, leaving the study underpowered to detect a true effect of GT, and below-target GT doses, resulting in differences in success rates between those who received higher versus lower GT doses. Furthermore, only a few subjects had SAA, all of whom were assigned to the control treatment arm, limiting our interpretation of the data in the SAA population.
We aimed to provide an updated and expanded report of our institution’s experience with GT, characterizing the clinical outcomes of patients with SAA who received GT, focusing on clinical response, patients’ survival, and ability to bridge patients to curative treatment with hematopoietic stem cell transplant (HSCT).
All patients with SAA who received a minimum of one GT from 2010-2020 were included in this study. SAA patients all met modified Camitta criteria, that is, bone marrow cellularity of <30% and at least two of the following blood counts: an absolute neutrophil count (ANC) of 0.5×109/L or less, an absolute reticulocyte count of 60×109/L or less, and a platelet count of 20×109/L or less. Patients were enrolled on the following clinical trials at the time of GT: NCT00604201, NCT01174108, NCT01193283, NCT01623167, NCT03173937, and NCT03520647. The above-mentioned trials were approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute. All patients or guardians provided written informed consent to participation in the trials.
Clinical data were collected from electronic medical records or from clinical trial databases. Baseline demographic information included age, sex, initial SAA therapy, disease severity, number of rounds of immunosuppressive therapy (IST) received, ABO type, and presence of human leukocyte antigen (HLA) class I and class II antibodies prior to and after GT courses. All patients had significant infections prompting initiation of GT and type of infection, pathogen isolated, and site of infection were recorded.
Granulocyte product data included number of GT, the patient’s weight at initial GT, granulocyte dose of each product in cells, donor ABO type, donor HLA type, and donor testing for agents of transfusion-transmissible infections. ANC prior to GT, number of days that the ANC was greater than 0.2x109/L, and percentage of days with available ANC data were assessed. If multiple ANC were collected daily, the first available post-transfusion ANC drawn the morning after transfusion was used for analysis.
Patients were eligible to receive a GT if they had the following: (i) proven or probable invasive fungal disease determined by the clinical team, or (ii) a bacterial infection which, in the experience of our center, was associated with greater than 90% mortality, as well as (iii) an ANC of less than 0.2×10/L, and (iv) no response to appropriate antibiotic or antifungal therapy for 24-48 hours. The duration of a GT course was determined by multiple factors including neutrophil response to infection, clinical response or progression of infection, development of transfusion reactions, HLA alloimmunization, and the availability of granulocyte donor products for same-day administration.
Donor selection, granulocytapheresis, and transfusion
Eligible granulocyte donors were enrolled on the NCT01553214 trial. After giving informed consent, donors received a single 480 µg subcutaneous injection of filgrastim 12-24 hours prior to leukapheresis and 8 mg of oral dexamethasone 12 hours prior to leukapheresis. Granulocyte concentrates were collected using a blood separator (CS3000 Plus,
Fenwal, Deerfield, IL, USA) processing 7 L of whole blood with trisodium citrate anticoagulant (Citra Anticoagulants, Braintree, MA, USA) and 6% hetastarch (He span, Braun Medical, Irvine, CA, USA). Granulocyte concentrates were sedimented by gravity following collection to reduce red cell content if there was major ABO incompatibility. All granulocytes were irradiated and transfused within 10 hours of collection.
Outcome measures
Primary outcomes were overall survival (OS) following the first GT to last follow up, survival to hospital discharge following hospitalization for a course of GT, and percentage of patients who received an infusion of hematopoietic stem cells among those who were awaiting HSCT during the GT course. Secondary outcomes included clinical responses at 7 and 30 days after initiation of GT and association between the following variables and OS: sex, infection type, duration of ANC over 0.2×109/L during the GT course, presence of HLA alloimmunization, and granulocyte product characteristics (granulocyte dose, ABO matching).
A GT course was defined as the number of days from first transfusion to 3 days after the last transfusion to account for the effect of the last transfusion. Additional outcomes included rate of HLA alloimmunization after GT, rate of successful HSCT in patients with transplant-donor specific antibodies after GT, and rate of GT complications including GT reactions and transfusion-transmitted virus positivity in previously negative granulocyte donors.
Response was characterized using a scoring system based on response (determined by the physician and including defervescence, hemodynamic stability, and improvement in infection-related symptoms), radiographic response (decrease in size of infection on imaging), and microbiological response (resolution of bacteremia, if applicable). Complete response and partial response were defined as improvements in all three and one or two criteria, respectively. Stable disease was defined as no improvement and progressive disease was identified by evidence of a breakthrough infection or clinical deterioration. GT reactions were identified by severity and imputability based on the National Healthcare Safety Network Biovigilance Component Hemovigilance Module Protocol.4
Summary statistics are described as the median with range for continuous variables, and frequency with proportion for categorical variables. Kaplan-Meier estimators were used to compare OS distributions based on the type of 30-day response to GT. Cox proportional hazard models were used to analyze the effect of covariates on survival time. Five courses were omitted from the ANC analysis because of excessive missingness of recorded data. OS was determined from the time of the first GT. Surviving patients were censored at their last follow-up date. P
values for covariate effects were calculated using the logrank test. All statistical analyses were performed using R (version 4.0.2).
Twenty-eight SAA patients with a median age of 20 years (range, 6-65 years) underwent 30 GT courses. Initial IST included horse antithymocyte globulin/cyclosporine and eltrombopag (n=11), horse antithymocyte globulin/cyclosporine (n=8), cyclophosphamide/cyclosporine (n=6), and horse antithymocyte globulin/cyclosporine plus sirolimus (n=1). Two patients did not receive IST and went directly to HSCT. A median of eight granulocyte products per course (range, 1-39 products) were administered over a median of 23.5 days (range, 3-103 days), with a per-dose median of 1.28x10 9 cells/kg (range, 0.45-4.52x10 9 cells/ kg). Indications for GT included invasive bacterial (n=14), fungal (n=13) and mixed (n=3) infections. The patients’ characteristics are summarized in Table 1. Types of infections, organisms, and sites of infection are summarized in Online Supplementary Table S1.
Overall, 86% (25/29) of infections remained stable or responded to GT. Over a GT course, complete responses were observed in 23% (7/30) on day 7 after the initial GT, and increased to 45% (13/29) on day 30. Most of the patients with fungal infection had a partial response (6/13; 46%) or stable infection (5/13; 38%) at 7 days, with only one complete responder; these responses improved to complete (7/13; 54%), and partial (5/13; 38%) by day 30. Only one patient with a fungal infection had progressive disease, which was evident early by day 7. Patients with bacterial infections had similar numbers of responses at days 7 and 30, although three patients had worsening bacterial infection by day 30 (Table 2).
After completion of their GT course, 64% (18/28) of patients survived to hospital discharge. The rate of survival to discharge in GT recipients who achieved complete response by day 30 was 85% (11/13) (Table 2). The OS rate from initial GT was 50% at a median duration of follow up of 551 days (range, 2-4,213 days) (Figure 1A). Overall, 39% (11/28) of patients were alive at last follow up. Patients who achieved a complete or partial response, or had a stable infection at 30 days had significantly better OS than that of non-responders (P=0.0004) (Figure 1B).
Predictors
Sex, weight, type of infection, percentage of days on which the ANC was greater than 0.2×109/L, ABO donor-recipient mismatch, mean granulocyte dose, and HLA alloimmuni-
zation (negative HLA antibodies at baseline to positive HLA antibodies after GT or increased panel-reactive antibodies in those alloimmunized at baseline) were not predictive
Table 1. Demographics of the patients and characteristics of the granulocyte products.
ANC prior to GT, N
of days with ANC >0.2x109/L (median, range)
N of granulocyte courses
Granulocyte products per course, days (median, range)
(11-95)
(1-39) Granulocyte dose, cells/kg, median (range)
(0.45-4.52x109) Time from first day of documented infection to GT in days, median (range) 5 (0-20)
*Two patients did not receive immunosuppressive therapy and went directly to hematopoietic stem cell transplant. **One patient received sirolimus in addition to antithymocyte globulin and cyclosporine. ***One of the two patients who received two granulocyte courses received transplants from two matched unrelated donors. N: number; IST: immunosuppressive therapy. ATG: antithymocyte globulin; CSA: cyclosporine; EPAG: eltrombopag; HSCT: hematopoietic stem cell transplant; CMV: Cytomegalovirus; ANC: absolute neutrophil count; GT: granulocyte transfusions.
Table 2. Responses at days 7 and 30 after initiation of granulocyte infusion versus survival to hospital discharge: overall and by type of infection.
Day 7 response N (%)
Type of response
Complete
Partial
Type of response Complete
(23)
(36)
(43)
Survival to
All infections, N=30 (day 7) or 29 (day 30)*
(57)
(45) 11/13 (85)
(60)
(67)
(43)
(29)
(64)
(75)
Fungal, N=13
(46)
(38)
Mixed, N=3*
N: total granulocyte courses administered in 28 patients. Response categorized as complete: improvement in all three criteria (clinical, radiographic, and microbiological); partial: improvement in one or two criteria; stable: no improvement; and progressive: clinical decline or breakthrough infection. *One patient who died before the 30-day response assessment had mixed bacterial/fungal infection.
of survival (Table 3, Online Supplementary Table S2, Online Supplementary Figure S1).
Hematopoietic stem cell transplantation outcomes
Of 21 patients who were awaiting HSCT during their GT course, 18 (86%) successfully started conditioning. Three patients did not receive HSCT; two patients were ineligible because of their critical clinical status and ultimately died and one patient lacked a suitable donor but achieved hematopoietic recovery after IST and survived. Thirteen of the 21 (62%) awaiting HSCT survived to discharge from hospital. Of the eight patients awaiting HSCT who did not survive to hospital discharge, six received HSCT and died due to progression of the initial infection (n=5) or development of a new infection (n=1). Infections that led to death included disseminated Klebsiella infection (n=2), adenovirus (n=2), trichosporonosis (n=1), and aspergillosis (n=1). Twelve of 16 patients (75%) who were awaiting HSCT at the time of
their GT course with available engraftment data successfully engrafted (Online Supplementary Table S5). Five out of 28 patients (18%) were HLA-alloimmunized at baseline. All five (100%) demonstrated an increase in panel-reactive antibodies after GT. Of the 23 (82%) patients not alloimmunized at baseline, four (17%) developed HLA antibodies during or after GT. In total, nine of 28 (32%) patients developed new or increased HLA alloimmunization (Online Supplementary Table S3); four developed donor-specific antibodies to potential HSCT donors (Online Supplementary Table S4), three did not develop donor-specific antibodies, and two did not have available HSCT donor HLA data. All four patients who developed donor-specific antibodies to their originally planned donor successfully underwent HSCT using an alternative donor without development of graft failure; two of these patients ultimately died from progression of their initial infection. Three patients had primary or secondary graft failure; none of these patients
had HLA alloimmunization at baseline or that developed alloimmunization during the GT course (Online Supplementary Table S5).
Nineteen of the 28 (68%) patients met criteria for non-severe febrile non-hemolytic transfusion reaction during their GT course. Two patients developed an allergic reaction which was treated with antihistamines with resolution of symptoms. One patient met criteria for severe, transfusion-associated circulatory overload of probable imputability based on National Healthcare Safety Network guidelines. The patient had underlying cardiomyopathy and died 3 months after his last GT due to systemic adenovirus infection in the setting of primary graft failure after HSCT. No donor tested positive for agents of transfusion-transmissible infection.
This study is a follow up to a previous report from our institution detailing the largest clinical experience utilizing GT in SAA patients with invasive fungal and/or bacterial infections, and showed a 64% rate of survival to hospital discharge, similar to the 58% reported in the previously studied cohort, with a significantly higher OS rate in patients with a stable infection or improved response compared to that in patients with progression of infection.1 We aimed to estimate the OS based on clinical response to GT at 30 days and found a significantly higher OS in patients with a stable infection or improved response compared to those with progression of infection.
To our knowledge, this is the first analysis of long-term survival and HSCT-related outcomes in SAA patients receiving GT. Given the progressive and refractory nature of these infections prior to the initiation of GT, and that uncontrolled infections are contraindications to HSCT, we believe that the high rates of receipt of HSCT in those awaiting HSCT (86%, 18/21) and survival to hospital discharge after HSCT (75%, 13/18) are largely attributable to the effect of GT. Although some patients developed HLA antibodies, even some to their originally planned donors, all were able to undergo HSCT. Our analysis did not reveal an association between type of infection and OS, there being similar rates of survival to hospital discharge in patients with invasive fungal or bacterial infections. In our previous cohort, 44% of patients with invasive fungal or bacterial infections survived to hospital discharge compared to 62% in this current study. It has been shown that over time infection-related mortality has decreased significantly in SAA patients unresponsive to IST, likely due to improvements in supportive care and use of alternative donor transplants.5 A study evaluating the efficacy and safety of posaconazole as primary pre-
Figure 1. Overall survival of patients with severe aplastic anemia who received granulocyte transfusions. (A) Overall survival from the initial granulocyte transfusion. (B) Overall survival from the initial granulocyte transfusion based on overall response at 30 days after the granulocyte transfusions. C: complete response; P/S: partial response or stable infection; Pr: progression.
vention of invasive fungal disease in patients with SAA demonstrated a significantly lower incidence of such fungal diseases in patients treated with posaconazole compared to the incidence in a control group.6 In contrast to the prior standard of care, the emergence and prophylactic use of this broad-spectrum oral antifungal agent may be a contributing factor to the observed increase in SAA patients who survived to hospital discharge.
To date, there has been inconclusive evidence of the efficacy of GT in SAA and other at-risk populations due to several limitations including variable granulocyte collection methods, poor patient accrual in randomized controlled trials, and physician bias with randomizing patients to GT due to pre-conceived beliefs about the efficacy of these transfusions.1,3,7 A Cochrane review of ten randomized controlled trials with 587 patients evaluating efficacy of GT versus standard antimicrobial therapy alone found insufficient evidence that GT affected all-cause mortality.8 The majority of the studies included in this review did not use granulocyte colony-stimulating factor (G-CSF) for donor stimulation, whereas this is now commonly given to granulocyte donors, and thus the transfused products had substantially lower granulocyte content. The RING trial,
Table 3. Cox proportional-hazard models for time to death from first granulocyte transfusion (N=25).*
HLA-alloimmunization status
*Five courses omitted from the analysis of absolute neutrophil counts because of excessive missingness of recorded counts. ANC: absolute neutrophil count; HLA: human leukocyte antigen; GT: granulocyte transfusions.
the most recent and largest randomized controlled trial to date, studied 114 patients with severe neutropenia and suspected or confirmed invasive infection who were randomized to receive granulocytes from donors stimulated with G-CSF and dexamethasone or standard antimicrobial therapy alone. This trial was limited by lack of power due to poor patient accrual, and below-target granulocyte dose in a third of patients.3 Although an established target granulocyte dose to achieve efficacy has not been identified, a Cochrane meta-analysis derived a minimum dose of 0.1x109 cells/kg as contributing to better clinical outcomes.9 European guidelines similarly recommend a standard granulocyte apheresis product for adults to be 0.1-0.3x109 cells/kg of recipient body weight.10 Siedel et al. reviewed 59 pediatric and young adults with severe neutropenia receiving a course of GT, including several patients with SAA, and concluded that a critical minimum GT dose of 0.3x109 cells/kg was needed to support the control of infections in high-risk neutropenic patients.11 In contrast to the RING trial in which higher OS was observed in patients who received higher doses (>0.6x109 cells/kg) compared to lower doses, we did not observe an association between the mean granulocyte dose received per patient and survival.3 Again in contrast to the RING trial, only 11% (3/28) of the patients in our cohort had mean granulocyte doses less than 0.6x109 cells/kg. Furthermore, the median granulocyte dose/kg received by our patients was multifold higher than all recommended dose targets, likely crossing the threshold beyond which there is no or limited association between GT dose and clinical outcome. While Siedel et al. utilized either prednisolone or G-CSF for donor stimulation to increase granulocyte product content, the use of both G-CSF and dexamethasone produces
higher granulocyte product doses without increasing the incidence of donor side effects compared to those in donors receiving G-CSF or dexamethasone alone.12,13 Furthermore, an association between G-CSF for donor stimulation and a theoretical risk of malignancy has been refuted even for donors undergoing multiple stimulations.14 Based on these considerations concerning both donors and recipients, granulocyte collection centers should strive to achieve minimum granulocyte doses of 0.6x109/kg of recipient weight. Granulocyte apheresis product yields are maximized by using dual donor stimulation (dexamethasone plus G-CSF), and this dual stimulation regimen should be the standard to optimize the therapeutic effect of GT in patients with SAA.
One-third of our patients developed new or progressive HLA-alloimmunization during their GT course. Generally, HLA antibody testing in GT recipients is performed prior to the first transfusion, during the transfusion course if an adverse reaction occurs, and after the last transfusion.15 Our study did not reveal an association between development of HLA-alloimmunization and OS in our granulocyte-treated SAA patients, similarly to what was seen in the RING trial cohort.16 Published data have revealed the negative impact of donor-specific antibodies on engraftment of donor cells in patients who received transplants from unrelated or haploidentical donors.17,18 Ideally, granulocyte donors who share HLA alleles with potential HSCT donors should be avoided, to prevent the development of donor-specific antibodies in the transfusion recipient. However, implementation of this strategy may not always be possible because of the urgent nature of the need for GT and the limited number of granulocyte donors. Fortunately, in our cohort, all four patients who developed donor-specific
antibodies against their original hematopoietic cell donor ultimately underwent HSCT after an alternative donor was identified. Physicians will need to assess the benefit of GT versus the risk of developing donor-specific antibodies on a patient-by-patient basis.
Transfusion reactions following GT have been widely documented; febrile and allergic reactions have been reported to occur at a rate of 10-80%.3,19,20 While the rate of febrile non-hemolytic reactions in our cohort was within this reported rate range, all cases resolved after administration of antipyretics. Concomitant fever due to underlying infection may have led to an overestimate of true reactions to GT. Alternatively, the high rate of febrile non-hemolytic transfusions reactions may be related to the high dose of granulocytes in the transfused products. The rate of severe transfusion reactions in our cohort was similar to that in the most recent randomized controlled trial.3 Overall, GT in SAA recipients appears to be safe with low rates of severe reactions. As a retrospective, observational study, limited to selected SAA patients from a single institution, our findings may not be generalizable. Among SAA patients, those who were highly alloimmunized did not receive GT due to difficulty in identifying sufficient granulocyte donors who lacked the cognate antigens. Our conclusions, therefore, only apply to SAA patients who are not highly alloimmunized prior to GT. The recently published experience with GT in patients with chronic granulomatous disease at our institution described a similar response rate to GT but, in contrast to our SAA cohort, GT given prior to HSCT were associated with high rates of alloimmunization and primary graft failure.21 A successful GT strategy requires a sophisticated transfusion medicine department, including a blood collection center with access to a large pool of highly dedicated and motivated granulocyte donors, and rapid turnaround of HLA testing, which can be both cost-prohibitive and labor intensive. As our facility is research-focused and government-funded, there were adequate resources to collect, process, and administer HLA-typed granulocyte products. Our procedures for collecting and transfusing granulocyte components may not be feasible at other institutions, limiting the generalizability of our findings and application of our approach. Although GT may not be readily available at every institution, our findings can lead to centers without the capability to administer GT to consider transfer of selected patients to a specialized center, and may even lead to consideration of investment in infrastructure to support administration of GT. In the absence of randomized
1. Quillen K, Wong E, Scheinberg P, et al. Granulocyte transfusions in severe aplastic anemia: an eleven-year experience. Haematologica. 2009;94(12):1661-1668.
2. Wang H, Wu, Y, Fu R, et al. Granulocyte transfusion combined
controlled trials in SAA and using this particular technical approach to GT, our observational study provides valuable guidance for clinical practice. Statistical limitations included the low number of patients, which did not allow for multivariate analysis and may have affected the ability to detect differences in the univariate analyses of variables related to GT that affect OS.
Highlights of this updated review of SAA patients undergoing GT reveal the following. (i) GT can be offered to critically ill SAA patients with invasive infections. (ii) Adjunctive therapy with GT with continued advances in antimicrobial and antifungal therapy and other supportive care measures may help to increase OS. (iii) GT may be a particularly valuable tool in patients with life-threatening infections as a bridge to curative therapy with HSCT. (iv) HLA-alloimmunization and, specifically, donor-specific antibodies can occur and must be considered when making the decision to use GT. Avoidance of granulocyte products from donors who share HLA alleles with potential HSCT donors may mitigate this risk. (v) Transfusion reactions have been observed in SAA patients receiving GT but, in our cohort, these were generally mild and did not affect mortality.
Disclosures
NSY and CED have a cooperative research and development agreement with Novartis. The remaining authors declare that they have no competing financial interests.
RVR conceptualized the study and wrote the manuscript. VS assisted in gathering data. RS analyzed results and created figures. EMG, SSK, NSY, and COW conceptualized and performed research. KW, NC, CC, SFL, DJY, CED, BAP, BW, GA, and RWC provided clinical care and assisted in collecting data. All authors edited and approved the final manuscript.
Funding
This research was supported by the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute.
Data-sharing statement
De-identified data will be shared with other researchers upon reasonable request to the corresponding author. The sharing will require a detailed proposal to the study investigators and a data transfer agreement must be signed.
with granulocyte colony stimulating factor in severe infection patients with severe aplastic anemia: a single center experience from China. PLoS One. 2014;9(2):e88148.
3. Price TH, Boeckh M, Harrison RW, et al. Efficacy of transfusion
with granulocytes from G-CSF/dexamethasone-treated donors in neutropenic patients with infection. Blood. 2015;126(18):2153-2161.
4 Center for Disease Control and Prevention. The National Healthcare Safety Network (NHSN) Biovigilance Component Hemovigilance Module Surveillance Protocol. https://www.cdc. gov/nhsn/pdfs/biovigilance/bv-hv-protocol-current.pdf. Accessed November 20, 2023.
5. Valdez JM, Scheinberg P, Nunez O, et al. Decreased infectionrelated mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52(6):726-735.
6. Chen M, Zhuang JL, Duan MH, et al. Posaconazole as primary prevention of fungal infection in intensive immunosuppressive therapy for severe aplastic anemia. Chin J Hematol. 2018;39(2):128-131.
7 Pammi M, Brocklehurst P. Granulocyte transfusions for neonates with confirmed or suspected sepsis and neutropenia. Cochrane Database Syst Rev. 2011;2011(10):CD003956.
8. Estcourt LJ, Stanworth SJ, Hopewell S, Doree C, Trivella M, Massey E. Granulocyte transfusions for treating infections in people with neutropenia or neutrophil dysfunction. Cochrane Database Syst Rev. 2016;4(4):CD005339.
9 Stanworth SJ, Massey E, Hyde C, et al. Granulocyte transfusions for treating infections in patients with neutropenia or neutrophil dysfunction. Cochrane Database Syst Rev. 2005;20(3):CD005339.
10. Guide to the preparation, use, and quality assurance of blood components. European Committee (Partial Agreement) on Blood Transfusion. 21st ed. European Directorate for the Quality of Medicines and Healthcare. 2023.
11. Seidel MG, Minkov M, Witt V, et al. Granulocyte transfusions in children and young adults: does the dose matter? J Pediatr Hematol Oncol. 2009;31(3):166-172.
12. Stroncek DF, Yau YY, Oblitas J, Leitman SF. Administration of G-CSF plus dexamethasone produces greater granulocyte concentrate yields while causing no more donor toxicity than
G-CSF alone. Transfusion. 2001;41(8):1037-1044.
13. Liles WC, Huang JE, Llewellyn C, SenGupta D, Price TH, Dale DC. A comparative trial of granulocyte-colony-stimulating factor and dexamethasone, separately and in combination, for the mobilization of neutrophils in the peripheral blood of normal volunteers. Transfusion. 1997;37(2):182-187.
14 Shaw BE, Confer DL, Hwang W, Pulsipher MA. A review of the genetic and long-term effects of G-CSF injections in healthy donors: a reassuring lack of evidence for the development of haematological malignancies. Bone Marrow Transplant. 2015;50(3):334-340.
15. Cugno C, Deola S, Filippini P, Stroncek DF, Rutella S. Granulocyte transfusions in children and adults with hematological malignancies: benefits and controversies. J Transl Med. 2015;13(1):362.
16. Price TH, McCullough J, Strauss RG, et al. WBC alloimmunization: effects on the laboratory and clinical endpoints of therapeutic granulocyte transfusions. Transfusion. 2018;58(5):1280-1288.
17 Spellman S, Bray R, Rosen-Bronson S, et al. The detection of donor-directed, HLA-specific alloantibodies in recipients of unrelated hematopoietic cell transplantation is predictive of graft failure. Blood. 2010;115(13):2704-2708.
18. Ciurea SO, de Lima M, Cano P, et al. High risk of graft failure in patients with anti-HLA antibodies undergoing haploidentical stem-cell transplantation. Transplantation. 2009;88(8):1019-1024.
19. West KA, Conry-Cantilena C. Granulocyte transfusions: current science and perspectives. Semin Hematol. 2019;56(4):241-247.
20 Heim KF, Fleisher TA, Stroncek DF, et al. The relationship between alloimmunization and posttransfusion granulocyte survival: experience in a chronic granulomatous disease cohort. Transfusion. 2011;51(6):1154-1162.
21. Arnold DE, Chellapandian D, Parikh S, et al. Granulocyte transfusions in patients with chronic granulomatous disease undergoing hematopoietic cell transplantation or gene therapy. J Clin Immunol. 2022;42(5):1026-1035.
Yan Xu,1,2,3 Qian Zhou,1,2,3 Xiaoming Wang,4 Aijun Zhang,4 Wentao Qi,1,2,3 Yuan Li,1,2,3 Chengzu Zheng,1,2,3 Jianmin Guan,5 Tao Sun,6 Jingxin Li,7 Chunhua Lu,1,2 Yuemao Shen1,2 and Baobing Zhao1,2,3
1Key Lab of Chemical Biology (MOE), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan; 2NMPA Key Laboratory for Technology Research and Evaluation of Drug Products, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan; 3Department of Pharmacology, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan; 4Department of Pediatrics, Qilu Hospital of Shandong University, Jinan; 5Department of Hematology, Heze Municipal Hospital, Heze; 6Department of Hematology, Qilu Hospital of Shandong University, Jinan and 7Department of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China
Correspondence: B. Zhao
baobingzh@sdu.edu.cn
Received: August 6, 2023.
Accepted: November 24, 2023. Early view: December 7, 2023.
https://doi.org/10.3324/haematol.2023.284041
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Little is known about the transition mechanisms that govern early lymphoid lineage progenitors from common lymphoid progenitors (CLP). Pellino2 (PELI2) is a newly discovered E3 ubiquitin ligase, which plays important roles in inflammation and the immune system. However, the physiological and molecular roles of PELI2 in the differentiation of immune cells are largely unknown. Here, by using a conditional knockout mouse model, we demonstrated that PELI2 is required for early B-cell development and stressed hematopoiesis. PELI2 interacted with and stabilized PU.1 via K63-polyubiquitination to regulate IL-7R expression. The defects of B-cell development induced by PELI2 deletion were restored by overexpression of PU.1. Similarly, PELI2 promoted TCF3 protein stability via K63-polyubiquitination to regulate IL-7R expression, which is required for the proliferation of B-cell precursor acute lymphoblastic leukemia (BCP-ALL) cells. These results underscore the significance of PELI2 in both normal B lymphopoiesis and malignant B-cell acute lymphoblastic leukemia via the regulation of IL-7R expression, providing a potential therapeutic approach for BCP-ALL.
B-cell development arises from the commitment of hematopoietic stem cells (HSC) into common lymphoid progenitors (CLP) and subsequent B-cell lineage specification in the bone marrow (BM).1 CLP include two distinct populations: an all-lymphoid progenitor (ALP) subset that retains full lymphoid potential and early thymic seeding activity, and a B-cell-biased lymphoid progenitor (BLP) population that primarily acts as an early B-cell progenitor pool.2 Early B-cell progenitors progressively differentiate through well-defined intermediates before they migrate to peripheral lymphoid tissues for the functional activation in response to antigen exposure, including pre-pro-B cells, pro-B cells, pre-B cells, immature B cells, and mature B cells stages.1,3 This process is characterized by the sequential expression of B-cell gene program and V(D)J recombination events, and is controlled by a network of transcription factors including PU.1, Ikaros, E2A, Ebf1, and Pax5.3,4 Mutations or alterations
of these transcription factors represent an underlying cause of phenotypic features such as the developmental arrest observed in B-cell precursor acute lymphoblastic leukemia (BCP-ALL).5
Several signaling events via transmembrane receptors are critical for B-cell lineage development.6-9 Among them, IL7R signaling not only plays essential roles in B-cell lineage specification from CLP, but is also required for the survival and proliferation of early B-cell progenitors.10,11 IL-7R is a heterodimer formed by the IL-7Ra-chain (IL-7Ra) and a common γ chain (γc).8 Binding of IL-7 to IL-7R initiates phosphorylation of JAK1 and JAK3, which recruits and activates downstream signal transducer and activator of transcription (STAT) as well as PI3K/Akt/mTOR and MEKERK pathways. These signalings co-operatively activate a B-cell lineage gene expression program including Pax5, Ebf1, and BCL-2 family proteins.7 Deficiencies in the IL-7R signaling in mice or humans result in severe lymphopenia.12-15 The importance of keeping IL-7R-mediated signaling
under control is also illustrated by studies showing that IL-7 transgenic mice develop B-cell lymphomas, and that IL-7 induces proliferation of BCP-ALL cells.16 Furthermore, most recent studies have demonstrated that IL-7R mutational activation is sufficient to trigger BCP-ALL.17,18 Pellino2 (PELI2) is a newly discovered E3 ubiquitin ligase, which regulates the protein degradation, protein-protein interaction, protein translocation and signaling transduction via the ubiquitination of target proteins.19 PELI2 possesses a C-terminal RING-like domain and a phospho-threonine-binding forkhead-associated (FHA) domain that are responsible for ubiquitin ligase activity and substrate binding, respectively.20 There is growing evidence that PELI2 acts as a critical mediator for innate immunity via multiple signalings through IL-1 receptors, Toll-like receptors, and NOD-like receptors.21,22 However, the physiological and molecular roles of PELI2 in the development of immune cells are largely unknown. Here, we generated and characterized a conditional knockout mouse model in which PELI2 was specifically depleted in hematopoietic cells. We found that PELI2 was required for early B-cell development, the deficiency of which resulted in a defect of the B-cell progenitors committed from CLP. Furthermore, PELI2 promoted the proliferation of BCP-ALL cells via the expression of IL-7R.
PELI2 floxed mice (PELI2fl/fl) were generated by inserting loxp sites flanking exon 2, which when deleted results in a frame-shift and form a premature stop codon in the reading frame. All animal studies were approved by the Institutional Animal Care and Use Committees at Shandong University.
Human BCP-ALL xenograft
Nalm-6 cells xenografts were carried out as previously described.23 The 6-week-old male NSG mice (Charles River Laboratories, Beijing, China) were irradiated at 1 Gy before tail vein injection of 5x106 Nalm-6 cells infected with object virus.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as previously described.24 Cells were fixed and lysed using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) according to the manufacturer’s protocol.
Statistical analysis
Statistical analyses were performed with unpaired twotailed Student t test except where indicated otherwise using Prism (GraphPad). P<0.05 was considered statistically significant. Additional methods and detailed information are provided
in the Online Supplementary Appendix.
PELI2 was required for early B-cell development
To explore the physiological roles of PELI2 in hematopoiesis, we generated a conditional PELI2 knockout model. Mice bearing PELI2 allele with loxP-flanked exon 2 (PELI2fl/fl) were crossed with Vav-Cre transgenic mice, to generate hematopoietic-specific PELI2 knockout mice (PELI2fl/fl; Vav-cre, CKO) with a deletion of the exon 2 (Online Supplementary Figure S1A). The deletion of PELI2 expression was confirmed by qPCR assays in the mononuclear cells from BM (Online Supplementary Figure S1B). The mice with PELI2 deficiency did not differ in morphology, growth, or viability from their wild-type (WT) littermates (data not shown). However, CKO mice clearly exhibited leukopenia, as demonstrated by the reduced white blood cells but unaffected red blood cells and platelets in the peripheral blood (PB) (Figure 1A, Online Supplementary Figure S1C). In addition, the reduced numbers of lymphocytes accounted for the leukopenia phenotype in the CKO mice, which was mainly caused by the reduction in B cells (B220+) in the PB (Figure 1A, B, Online Supplementary Figure S1D).
We then examined the B-cell compartment in BM, the primary tissue in which early B-cell development occurs. The frequency and numbers of B cells (B220+) in BM was significantly decreased in CKO mice compared to that in WT mice (Figure 1C, D, Online Supplementary Figure S1E), while other lineage cells including T cells appeared normal in CKO mice (date not shown). Similarly, CKO mice also showed reduced numbers of B cells in the spleen, as well as a mild decrease in spleen weight (Online Supplementary Figure S1F, G). Although T cells were also reduced in CKO mice spleen, the early T-cell development in thymus was for the most part normal in CKO mice (Online Supplementary Figure S1H, I).
To clarify the defect in B-cell development upon the PELI2 deletion, we examined the subpopulations of B cells. The frequencies of pro-B cells and pre-B cells were slightly reduced in PELI2CKO BM while mature B cells showed a slight increase, indicating that PELI2 deficiency disrupted the sequential differentiation bias of B-cell progenitors (defined as B220+lgM-lgD-) (Online Supplementary Figure S1J). Notably, all the subpopulations of B cells were reduced in PELI2CKO BM compared to WT controls (Online Supplementary Figure S1J). Although HSC (Lin-Sca1+c-Kit+) and HPC (Lin-Sca1-c-Kit+) were not apparently compromised in CKO mice (Online Supplementary Figure S1K), CLP in CKO BM were significantly reduced compared to that of WT mice (Figure 1E, F). Using the surface marker Ly6D, we further divided CLP into ALP (Ly6D ) and BLP (Ly6D+) populations2 (Figure 1E). Compared to the equal numbers of ALP cells, the absolute BLP cells that primarily act as
early B-cell progenitors were significantly reduced in CKO mice (Figure 1G). This was further confirmed by the colony-forming assays with CKO BM Lin- cells supplemented with IL-7, which showed that the colony number and size
were significantly reduced from CKO BM cells (Figure 1H). These findings suggested that PELI2 is required for the commitment and proliferation of B-biased lymphoid progenitor cells from CLP.
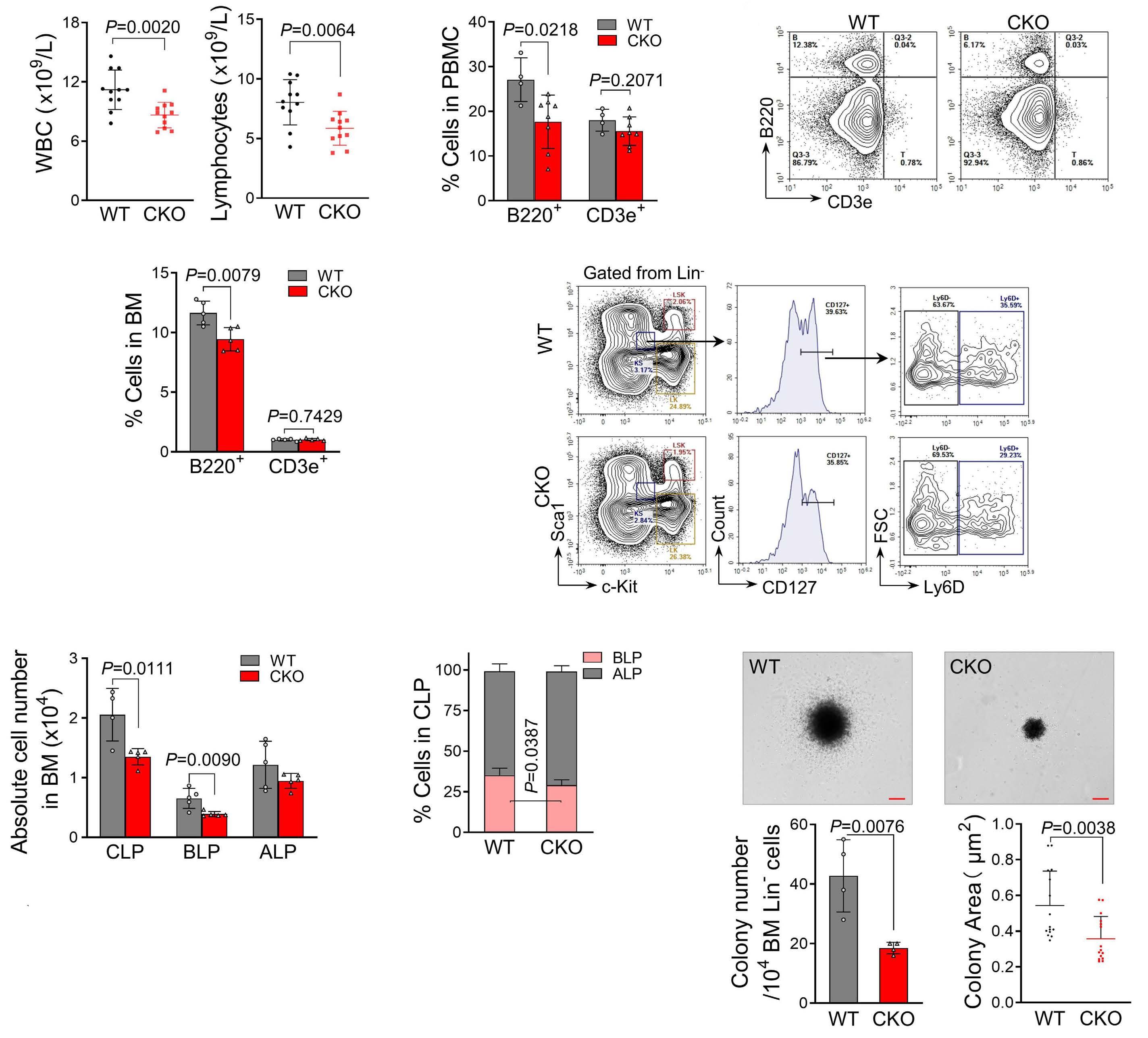
Figure 1. PELI2 deficiency impairs early B-cell development. (A) Peripheral blood analysis of wild-type (WT) and 8-week old PELI2CKO mice (CKO) (N=11). Data presented as mean±Standard Deviation (SD). (B) Quantification of B cells (B220+) and T cells (CD3e+) in the peripheral blood mononuclear cells (PBMC) from mice as in (A). Each dot represents one mouse. Data presented as mean±SD. (C) Representative flow cytometric analysis of B cells and T cells in the bone marrow (BM) of 8-week-old WT and PELI2CKO mice. (D) Quantification of the percentage of B cells and T cells in (C). Each dot represents one mouse. Data presented as mean±SD. (E) Representative flow cytometric analysis of common lymphoid progenitor (CLP) cells, B-cell-biased lymphoid progenitor (BLP) cells, and all-lymphoid progenitor (ALP) cells in the BM of 8-week-old WT and PELI2CKO mice. (F) Quantification of the numbers of CLP cells, BLP cells and ALP cells in (E). Each dot represents one mouse. Data presented as mean±SD. (G) Quantification of the percentage of BLP cells and ALP cells in the BM from indicated mice as in (E) (N=5). Data presented as mean±SD. (H) Pre-B-colony formation assays of BM lineage- cells from WT and PELI2CKO mice (N=4). Data presented as mean±SD. (Top) Representative colony morphology on day 7. Scale bar: 50µm. (Bottom) Colony number and size was quantified. All P values were determined by unpaired two-tailed Student t test unless otherwise indicated. See also Online Supplementary Figure S1 for supporting information. CKO: hematopoietic-specific PELI2 knockout mice (PELI2fl/fl; Vav-cre).
Loss of PELI2 impaired the reconstitution capacity of hematopoietic stem cells
To test whether a B-cell defect is the cell-intrinsic effect of HSC, we performed BM transplantation assays in which WT or CKO BM cells were transplanted into the lethally irradiated mice (Figure 2A). CKO-BM reconstituted mice exhibited persistently low lymphocyte counts but other blood cell values were normal at four months post transplantation (Figure 2B, Online Supplementary Figure S2A). The frequency of B cells was also significantly reduced in PB, BM and spleen of CKO-BM reconstituted mice (Figure 2C). Furthermore, CKO-BM reconstituted mice showed reduced CLP compared to WT controls (Figure 2D). Although the total LSK (Lin-Sca1+c-Kit+) cells were comparable ( data not shown), the numbers of SLAM-LSK (CD150+CD48-LSK) indicating the enriched HSC were significantly decreased in CKO-BMT mice (Online Supplementary Figure S2B). Correspondingly, PELI2 deficiency led to the increased quiescence in SLAM-LSK (Online Supplementary Figure S2C). To examine the effect of PELI2 deficiency on HSC functions, we challenged the CKO mice with 5-fluorouracil (5-FU) that induces the cell death of cycling HSPC to activate and mobilize HSC.25 CKO mice exhibited a significant decrease in LSK expansion upon 5-FU administration (Figure 2E), and succumbed to BM failure significantly earlier than their WT littermates (Figure 2F). Moreover, we further assessed the absolute number of functional HSC with limiting-dilution assays and observed an approximately 2.5-fold reduction in HSC in CKO mice (Figure 2G). These findings suggest that PELI2 is required for the self-renewal of HSC in stressed hematopoiesis.
To further confirm the functional roles of PELI2 in HSC, we performed competitive BM transplantation assays (Online Supplementary Figure S2F). PELI2CKO-derived cells showed a progressive decrease in PB in the primary transplant recipients, coinciding with a significantly impaired reconstitution in the BM (Figure 2H, I). However, PELI2CKO HSC were efficiently engrafted in the recipient BM as the WT controls indicated by homing assays (Online Supplementary Figure S2D, E). Notably, PELI2CKO HSC exhibited dramatically impaired proliferation but no relevant alteration in the lineage commitments in the recipient mice (Figure 2J, Online Supplementary Figure S2G). The competitive disadvantage of PELI2CKO HSC reconstitution was persistent in subsequent secondary recipients (Online Supplementary Figure S2H, I), indicating the defective self-renewal of HSC in PELI2CKO mice.
To rule out the possibility that the defect of PELI2CKO mice is due to a long-term accumulated consequence from the embryo stage, we also evaluated the role of PELI2 in adult hematopoiesis, using chimeric mice transplanted with PELI2fl/fl; Ubc-cre-ERT2 BM. Similar to PELI2CKO mice, PELI2 deletion after injection of tamoxifen also impaired the reconstitution capacity of HSC and early B-cell development in adult mice (Online Supplementary Figure S3).
PELI2 regulated early B-cell development through IL-7R signaling pathway
The reduced pool of B220+ cells and BLP in PELI2CKO mice prompted us to investigate the basis of impaired B-cell development. Loss of PELI2 led to a slight increase in cell death in B220+ BM cells, whereas PELI2CKO CLP showed comparable survival to WT ( Online Supplementary Figure S4A, B). Importantly, B-cell proliferation in vivo was markedly restrained upon the PELI2 deletion indicated by BrdU incorporation assays (Figure 3A). In line with this, the expression of ccnd3, a proliferation-related gene, was significantly reduced in PELI2CKO CLP and B220+ BM cells (Figure 3B). These data indicated that the B-cell defect observed in PELI2CKO mice was mainly due to the inhibition of early B-cell progenitor cell proliferation.
To understand the molecular mechanism underlying the impaired B-cell development induced by PELI2 deficiency, we performed bulk RNA sequencing of B220+ BM cells from PELI2CKO mice and their WT controls. A total of 537 differentially expressed genes (DEG) were found (≥1.5-fold, P<0.05), including transcription factors related to early B-cell development such as EBF1, FOXO1, and PAX5 (Figure 3C). These genes were also confirmed to be markedly down-regulated in the CLP as well as B220+ BM cells from PELI2CKO mice (Figure 3D, Online Supplementary Figure S4C). IL-7R, the key factor for early B-cell differentiation, was also down-regulated upon PELI2 deletion, which was further confirmed by its notably reduced expression and surface protein level in the PELI2CKO CLP (Figure 3D, E). As expected, loss of PELI2 led to the inhibition of IL-7R signaling, as indicated by the reduced phosphorylation of Stat5 and AKT (Figure 3F, G). Although signals from IL-7R were shared in B/T cell-biased lymphoid progenitors from CLP, PELI2 deletion had no effect on the T-cell specific genes, including E2AE47, HES1, and NOTCH1 (Online Supplementary Figure S4D).
To further confirm the critical role of IL-7R in the PELI2 loss-of-function phenotype, we performed rescue experiments in which PELI2CKO cKit+ BM cells transduced with lentivirus expressing IL-7R were transplanted into recipient mice. As we expected, IL-7R overexpression significantly reversed the defect of B-cell differentiation in PELI2CKO mice, indicated by the increased number of B220+ cells and BLP in BM and spleen accompanied by the high levels of lymphocytes in PB (Figure 3H, I, Online Supplementary Figure S4E, F).
PELI2 regulated IL-7R expression via PU.1 ubiquitination in early B-cell development
Several transcription factors have been seen to control the expression of IL-7R, including PU.1, RUNX1, and GA-binding protein transcription factor.26 Although the mRNA level of PU.1 was unaltered, its protein level was clearly reduced in PELI2-deficient B220+ BM cells (Figure 4A, Online Supplementary Figure S5A). In line with this, PU.1 target gene
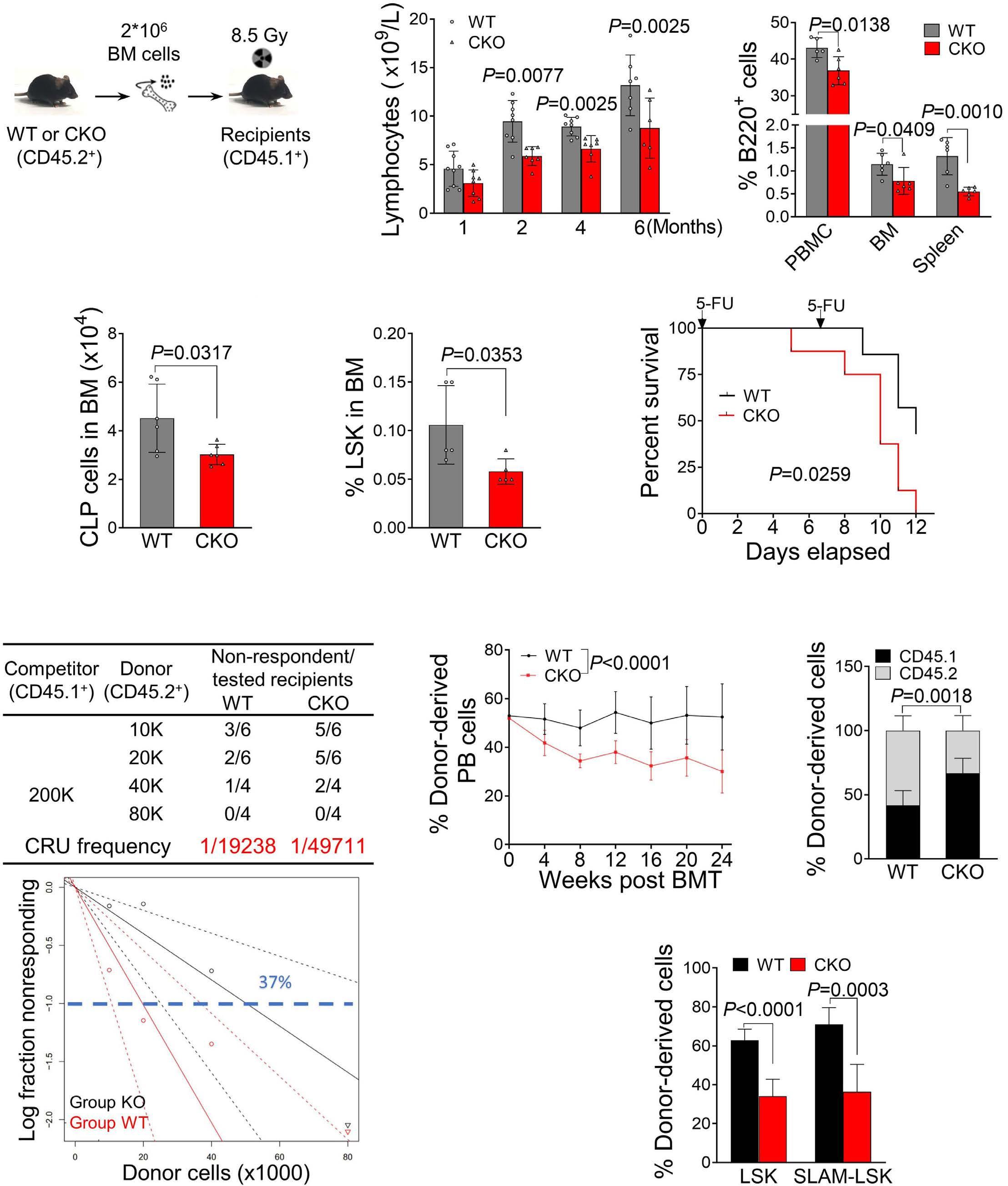
Figure 2. B lymphopenia in PELI2CKO mice was cell autonomous. (A) Schematic illustration of serial bone marrow (BM) transplantation with wild-type (WT) and hematopoietic-specific PELI2 knockout (PELI2CKO) (CKO) mice BM cells. (B) Peripheral blood (PB) chimerism analyses at the indicated time points after 1st non-competitive BM transplantation. Each dot represents one mouse. Data presented as mean±Standard Deviation (SD). (C) Percentage of B cells in the peripheral blood mononuclear cells (PBMC), BM, and spleen of indicated mice 24 weeks after BM transplantation. Each dot represents one mouse. Data presented as mean±SD. (D) Numbers of common lymphoid progenitor (CLP) cells in the BM of indicated mice as in (C). Each dot represents one mouse. Data presented as mean±SD. (E) Quantification of LSK cells in the BM of WT and PELI2CKO mice 8 days after 5-fluorouracil (5-FU, 150 mg/kg) treatment via single intraperitoneal injection. Each dot represents one mouse. Data presented as mean±SD. (F) Kaplan-Meier survival curve of indicated mice with 5-FU (150 mg/kg) via intraperitoneal injections every 7 days for two rounds. Data obtained from 8 mice in each group. P values determined by Log-rank (Mantel-Cox) test. (G) (Top) Poisson statistical analysis from the limiting dilution assays. Symbols represent the percentage of negative mice for each dose of cells. Solid lines indicate the best-fit linear model for each dosage. Dotted lines represent 95% Confidence Intervals. (Bottom) Frequencies of functional hematopoietic stem cells (HSC) were calculated according to Poisson statistics. (H) Quantification of donor-derived PB cells at the indicated time points after competitive transplantation. Data obtained from 8 mice in each group. P value determined by two-way ANOVA. (I) Quantification of donor-derived BM cells of indicated mice 24 weeks after competitive transplantation. Data presented as mean±SD from 8 mice in each group. (J) Quantification of donor-derived LSK and SLAM-LSK cells of indicated mice as in (I). Data presented as mean±SD from 8 mice in each group. All P values determined by unpaired two-tailed Student t test unless otherwise indicated. See also Online Supplementary Figures S1, S2 for supporting Information. CRU: competitive repopulating units (long-term repopulating hematopoietic stem cells).
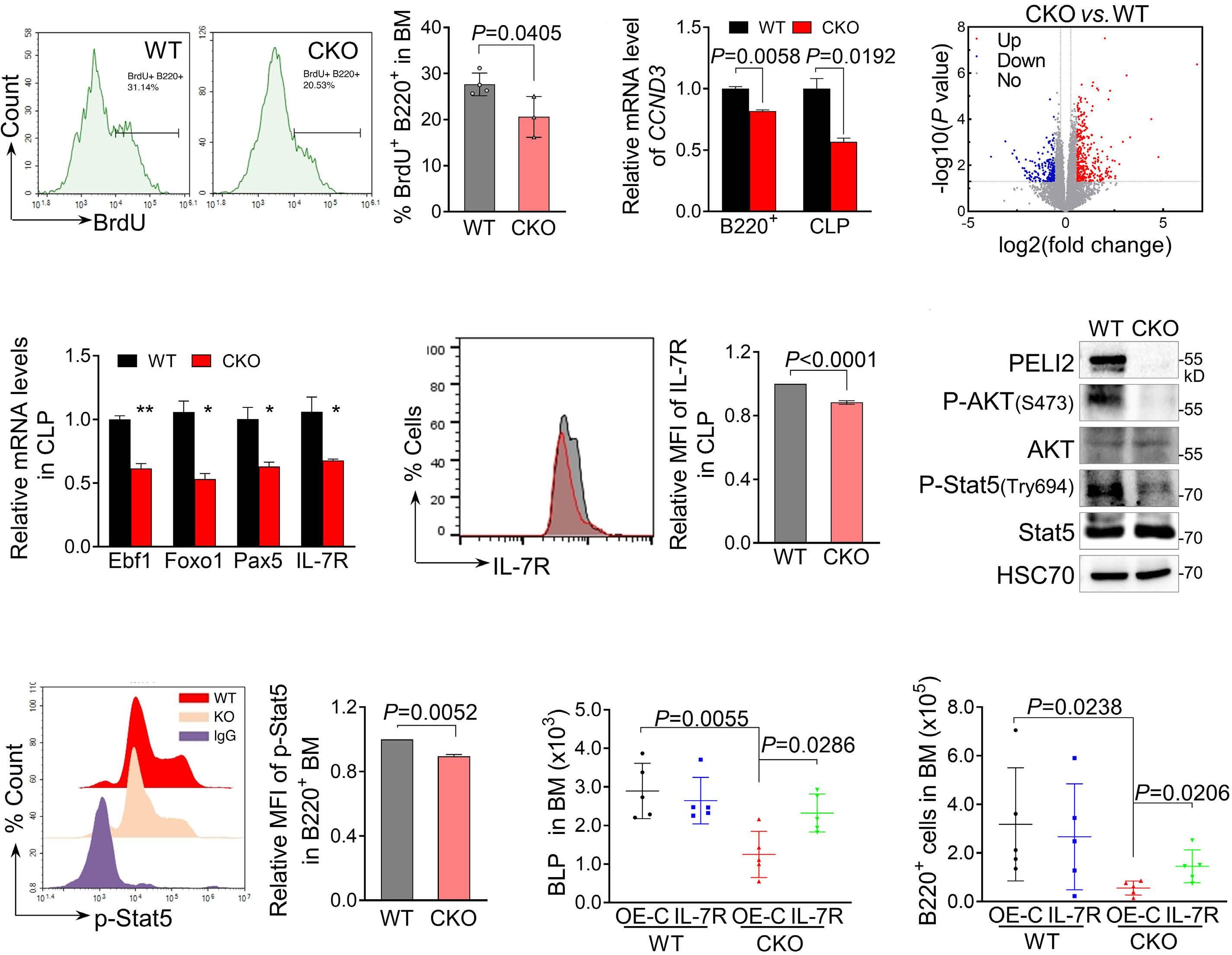
Figure 3. PELI2 regulated early B-cell development through IL-7R signaling. (A) Proliferation analysis of bone marrow (BM) B cells (B220+) from indicated mice. 8-week old mice were injected intraperitoneally with BrdU for 3 hours. BM cells were collected and stained with anti-B220 antibodies in combination with BrdU detection methodology. Percentages in the bar graph indicate BrdU-positive cells in the gated B220+ cells. Each dot represents one mouse. Data presented as mean±Standard Deviation (SD). WT: wild-type. (B) Quantification of mRNA expression of CCND3 in the common lymphoid progenitors (CLP) and B220+ cells from BM of WT and hematopoietic-specific PELI2 knockout (PELI2CKO) mice (CKO). Data presented as mean±SD from 3 independent experiments. (C) BM B220+ cells isolated from 8-week-old WT and PELI2CKO mice and analyzed by RNA sequencing. Volcano plot of the differentially expressed genes between WT and PELI2CKO BM B220+ cells are shown. (D) Quantification of mRNA expression of Ebf1, Foxo1, Pax5, and IL-7R expression in the BM CLP cells of WT and PELI2CKO mice. Data presented as mean±SD from 3 independent experiments. *P<0.05; **P<0.01. (E) Flow cytometric analysis of cell surface expression of IL-7R in the BM CLP cells of indicated mice. (Right) Quantification of mean fluorescence intensity (MFI) of IL-7R. Data presented as mean±SD from 3 independent experiments. (F) Immunoblotting analysis of indicated proteins in B220+ BM B cells from indicated mice. HSC70 was used as loading control. (G) Flow cytometric analysis of intracellular Stat5 phosphorylation in cells as in (F) except the cells were treated with IL-7 (50 ng/mL) for 30 minutes. (Right) Quantification of mean fluorescence intensity (MFI) of phosphorylated Stat5. Data presented as mean±SD from 3 independent experiments. (H and I) Quantification of B-cell-biased lymphoid progenitor (BLP) cells and B220+ cells in BM from mice transplanted with WT and PELI2CKO cKit+ BM cells transduced with lentivirus expressing IL-7R or blank vector (OE-C) at one month post transplantation. Each dot represents one mouse. Data presented as mean±SD. All P values were determined by unpaired two-tailed Student t test unless otherwise indicated. See also Online Supplementary Figure S4 for supporting information.
expression was transcriptionally repressed in PELI2CKO mice (Online Supplementary Figure S5A). In addition, PELI2 overexpression protected PU.1 from the time-dependent degradation upon cycloheximide (CHX) treatment (Online
Supplementary Figure S5B).
Considering PELI2 is an E3 ubiquitin ligase, we speculated that PELI2 promotes PU.1 stability via ubiquitination. Co-immunoprecipitation (Co-IP) assays demonstrated an
interaction between PELI2 and PU.1 (Figure 4B). To map the domains that are critical for the interaction of PELI2 and PU.1, we constructed a series of truncated forms of the two proteins (Figure 4C, E). Co-IP assays with these truncations revealed that the PELI2 FHA domain was responsible for the interaction with the PEST domain of PU.1 (Figure 4D, F). Overexpression of PELI2 promoted K63-linked ubiquitination of PU.1 (Online Supplementary Figure S5C), whereas similar approaches using K48-linked ubiquitin failed to detect any increase in ubiquitination of PU.1 (Online Supplementary Figure S5D). In line with this, a reduction in K63-linked ubiquitination but an increase in K48-linked ubiquitin of PU.1 were observed in B220+ BM cells from PELI2CKO mice (Figure 4G). These data demonstrated that PELI2 regulates PU.1 protein stability via K63-linked ubiquitination. PU.1 induced a marked increase in luciferase activity in HEK293T cells transduced with IL-7R promoter, which was further enhanced by PELI2 overexpression (Online Supplementary Figure S5E). We then analyzed chromatin occupancy of PU.1 on the IL-7R promotor region by ChIP, and found that the binding of PU.1 to the IL-7R promoter was significantly reduced in B220+ BM cells upon PELI2 deletion (Figure 4H). Importantly, PU.1 overexpression significantly restored the reduced IL-7R expression, which successfully reversed the impaired pre-B CFU formation of BM Lin- cells from PELI2CKO mice (Figure 4 I, Online Supplementary Figure S5F). Similar rescue for the defect of B-cell differentiation was also observed in CKO mice with PU.1 overexpression (Online Supplementary Figure S6). Collectively, these data demonstrated that PELI2 promotes PU.1 stability via K63linked ubiquitination to regulate IL-7R expression, which is required for early B-cell development.
PELI2 regulated cell proliferation via IL-7R signaling in B-cell precursor acute lymphoblastic leukemia cells
Given that PELI2 is essential for IL-7R expression, we assessed whether PELI2 plays important roles in the progression of BCP-ALL characterized by excessively activating IL-7R signaling.17,18 We first analyzed the published RNA sequencing data from patients with BCP-ALL, and found that PELI2 is highly expressed in parallel with IL-7R expression in PB samples obtained from 23 BCP-ALL patients compared with the corresponding normal individuals (Online Supplementary Figure S7A). Consistent with this, our independent assays with BM mononuclear cells from 7 BCP-ALL patients also revealed a significant upregulation of PELI2 expression and positive correlation with IL-7R (Figure 5A, B), which was confirmed by the increase in both protein levels in the primary BCP-ALL cells (Figure 5C).
We next utilized a BCP-ALL cell line Nalm-6 to explore the roles of PELI2 in BCP-ALL. PELI2 knockdown led to a marked reduction in IL-7R expression (Online Supplementary Figure S7B), thereby inhibiting its downstream signaling including the phosphorylation of AKT and ERK, cMyc (Figure 5D). Indeed, silencing of PELI2 dramatically inhibited
the proliferation of Nalm-6 cells and 697 cells in BCP-ALL cells (Figure 5E, Online Supplementary Figure S7C). This was further confirmed by the down-regulated expression of Ki67 and repressed DNA replication in Nalm-6 cells transduced with PELI2 shRNA (Figure 5F, Online Supplementary Figure S7D). Similarly, PELI2 knockdown also significantly reduced the colony formation ability of primary BCP-ALL CD34+ cells (Figure 5G). On the contrary, ectopic expression of PELI2 promoted the IL-7R signaling and proliferation of Nalm-6 cells (Online Supplementary Figure S7E, F), and even attenuated the effect of vincristine chemotherapy on Nalm-6 cells (Online Supplementary Figure S7G). Notably, overexpression of IL-7R effectively reverted the inhibitory proliferation of Nalm-6 cells induced by PELI2 knockdown in vitro (Figure 5H). These findings suggested that PELI2 regulates cell proliferation via IL-7R signaling in BCP-ALL cells.
TCF3 was required for the IL-7R expression in B-cell precursor acute lymphoblastic leukemia cells
Given our finding that PU.1 is relatively unaffected or undetectable in BCP-ALL (Figure 5C, Online Supplementary Figure S7A), we screened 26 transcription factors (TF) of IL-7R predicted from 3 independent databases (Online Supplementary Figure S8A). Among those highly expressed in BCP-ALL, TCF3 knockdown led to a significant reduction in IL-7R expression (Figure 6A, Online Supplementary Figure S8B), indicating that TCF3 is required for IL-7R expression in Nalm-6 cells. Similarly, TCF3 was positively correlated with the expression of PELI2 and IL-7R in BCP-ALL (Figure 6B, Online Supplementary Figure S8C, D), which is consistent with the elevated protein level in BM cells from BCP-ALL patients (Figure 5C). Furthermore, TCF3 knockdown inhibited the proliferation of Nalm-6 cells, which phenocopied the effects of PELI2 silencing (Online Supplementary Figure S8E).
Silencing of PELI2 led to a dramatic reduction in TCF3, while its ectopic expression resulted in an increase in TCF3 protein level ( Online Supplementary Figure S8F, G ). Importantly, overexpression of TCF3 effectively restored the IL-7R expression and subsequent Nalm-6 cell growth inhibited by PELI2 silencing (Figure 6C, D), suggesting that TCF3 mediated the regulation of PELI2 on the Nalm-6 cell proliferation via IL-7R expression.
To reveal the regulation of TCF3 on IL-7R expression, we performed the luciferase reporter assays in HEK293T cells driven by IL-7R promoter. Overexpression of TCF3 induced a marked increase in luciferase activity, which was enhanced by PELI2 overexpression (Online Supplementary Figure S8 I ). The binding of TCF3 on the IL-7R promoter was further confirmed by ChIP analysis in which PELI2 knockdown greatly reduced their interaction in Nalm-6 cells (Figure 6E). Collectively, these data demonstrated the PELI2-TCF3 axis regulates IL-7R expression, which is required for the proliferation of BCP-ALL cells.
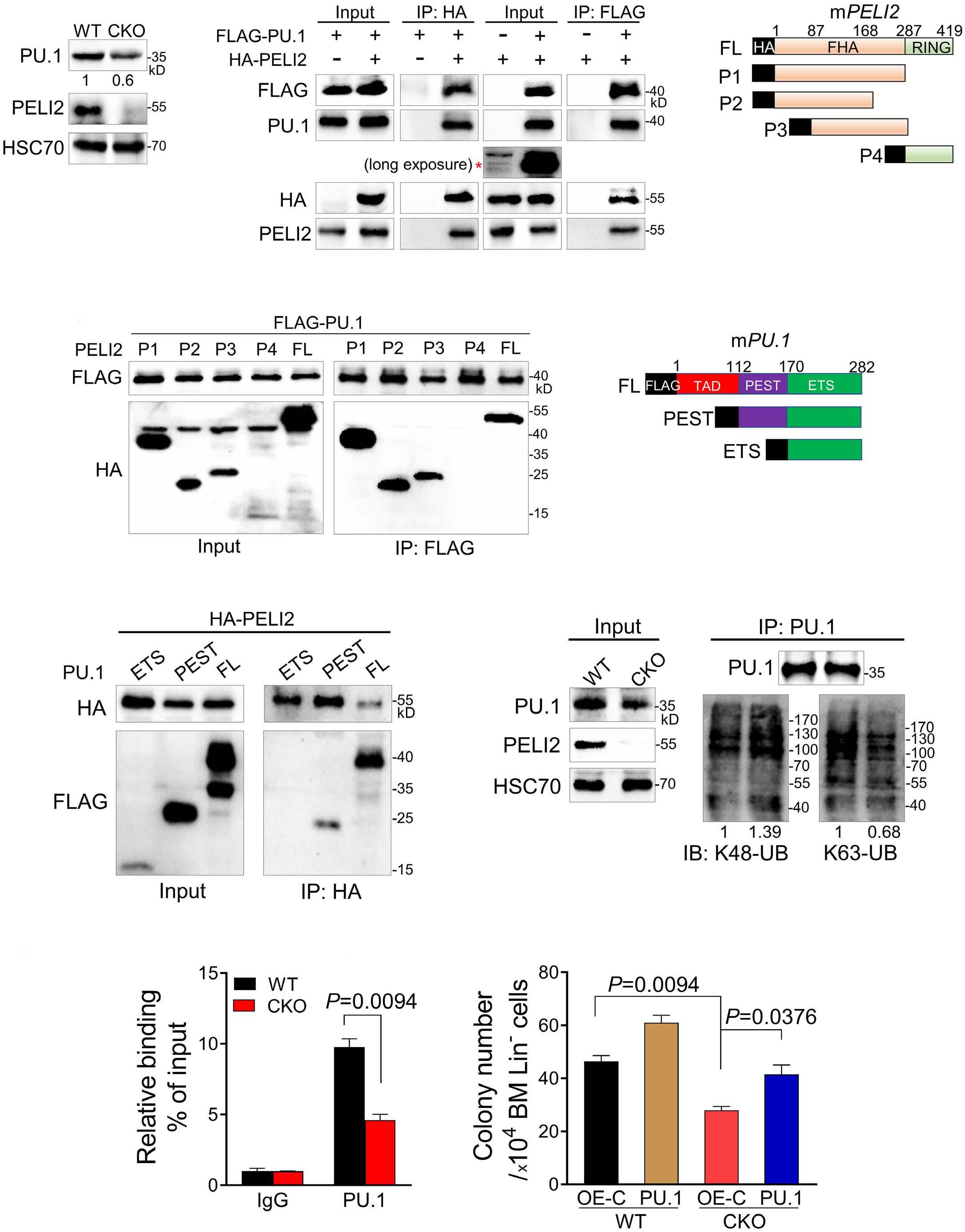
Figure 4. PELI2 regulated IL-7R expression via protecting PU.1 from degradation in normal B-cell development. (A) Immunoblotting analysis of PU.1 protein level in B cells purified from bone marrow (BM) of wild-type (WT) and hematopoietic-specific PELI2 knockout mice (PELI2CKO) mice. HSC70 was used as loading control. Numbers indicate the relative band intensity of PU.1 that normalized to HSC70. (B) Reciprocal co-immunoprecipitation (Co-IP) of exogenous FLAG-mouse PU.1 and HA-mouse PELI2 in HEK293T cells. Bands obtained from independent membranes blotted with indicated antibodies. (C) Structure of full-length and truncated mouse PELI2. (D) Co-IP analysis of PELI2 mutants binding to PU.1. HEK293T cells were co-transfected with mouse PELI2 truncations (HA tagged) and FLAG-mouse PU.1, and IP analysis was performed with anti-FLAG antibody. (E) Structure of full-length and truncated mouse PU.1. (F) Co-IP analysis of PU.1 mutants binding to PELI2. HEK293T cells were co-transfected with mouse PU.1 truncations (FLAG tagged) and HA-mouse PELI2, and IP analysis was performed with anti-HA antibody. (G) Immunoblotting analysis of ubiquitination of PU.1 immunoprecipitated from BM B cells of WT and PELI2CKO mice with K63-linkage and K48-linkage specific polyubiquitin antibodies. Numbers indicate the relative band intensity of the ubiquitin blots to WT. (H) Chromatin IP analyses of promoter binding activity of PU.1 to IL-7R in BM B220+ cells from WT and PELI2CKO mice. Data presented as mean±Standard Deviation (SD) from 3 independent experiments. (I) Pre-B colony formation assays of BM lineage- cells from WT and PELI2CKO mice transduced with lentivirus expressing PU.1 or blank vector (OE-C). Colony number was quantified. Data presented as mean±SD from 3 independent experiments. All P values determined by unpaired two-tailed Student t test unless otherwise indicated. See also Online Supplementary Figures S5, S6 for supporting information.
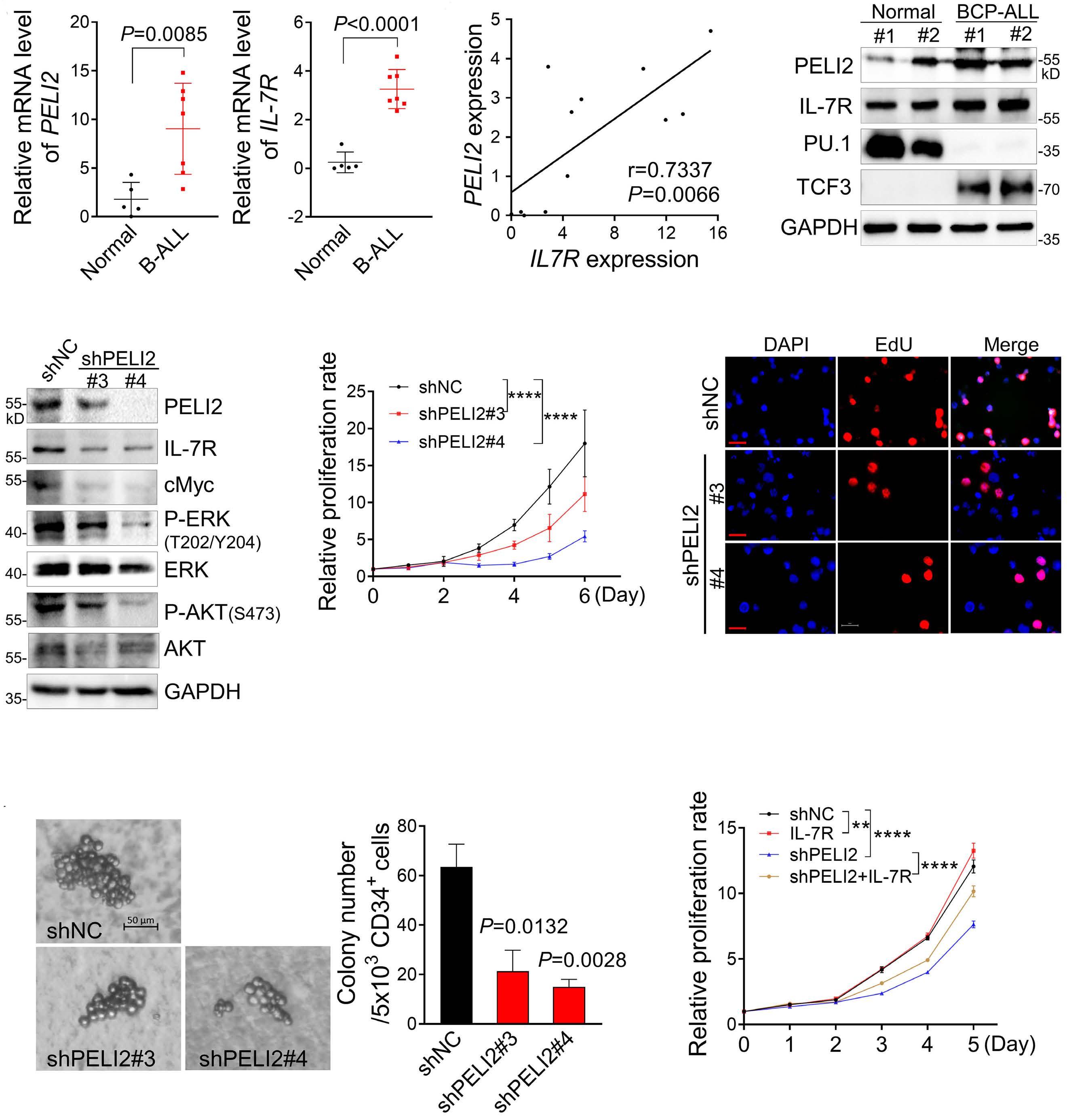
Figure 5. PELI2 was required for the cell proliferation via IL-7R signaling in B-cell precursor acute lymphoblastic leukemia cells. (A) Quantification of mRNA expression of PELI2 and IL-7R in the bone marrow mononuclear cells from primary B-cell precursor acute lymphoblastic leukemia (BCP-ALL) patients and healthy donors. Each dot represents one mouse. Data presented as mean± Standard Deviation (SD). (B) Correlation of PELI2 expression with IL-7R in (A). Pearson’s correlation coefficient (r) and paired t test P values are shown. (C) Immunoblotting analysis of indicated proteins in the bone marrow mononuclear cells from primary BCP-ALL patients and healthy donors. GAPDH was used as loading control. (D) Immunoblotting analysis of indicated proteins in Nalm-6 cells transduced with retroviruses encoding indicated shRNA. shNC represents a non-targeting shRNA. GAPDH was used as loading control. (E) Statistical analysis of cell proliferation in Nalm-6 cells as in (D). Data obtained from 3 independent experiments. P value determined by two-way ANOVA. ****P<0.0001. (F) Nalm-6 cells as in (D) were labeled with EdU for 2 hours and stained with azide-conjugated Alexa567 (red fluorescence) and DAPI (blue fluorescence). Scale bar: 20 µm. (G) Primary BCP-ALL bone marrow CD34+ cells with PELI2 knockdown were subjected to colony-forming unit assays. (Left) Representative colony morphology on day 14. Colony number was quantified. shNC represents a non-targeting shRNA. Data presented as mean±SD from 3 independent experiments. (H) Statistical analysis of cell proliferation in Nalm-6 cells transduced with retroviruses encoding indicated shRNA in the presence of IL-7R overexpression. Data obtained from 3 independent experiments. P value determined by two-way ANOVA. **P<0.01; ****P<0.0001. All P values determined by unpaired two-tailed Student t test unless otherwise indicated. See also Online Supplementary Figure S7 for supporting information.
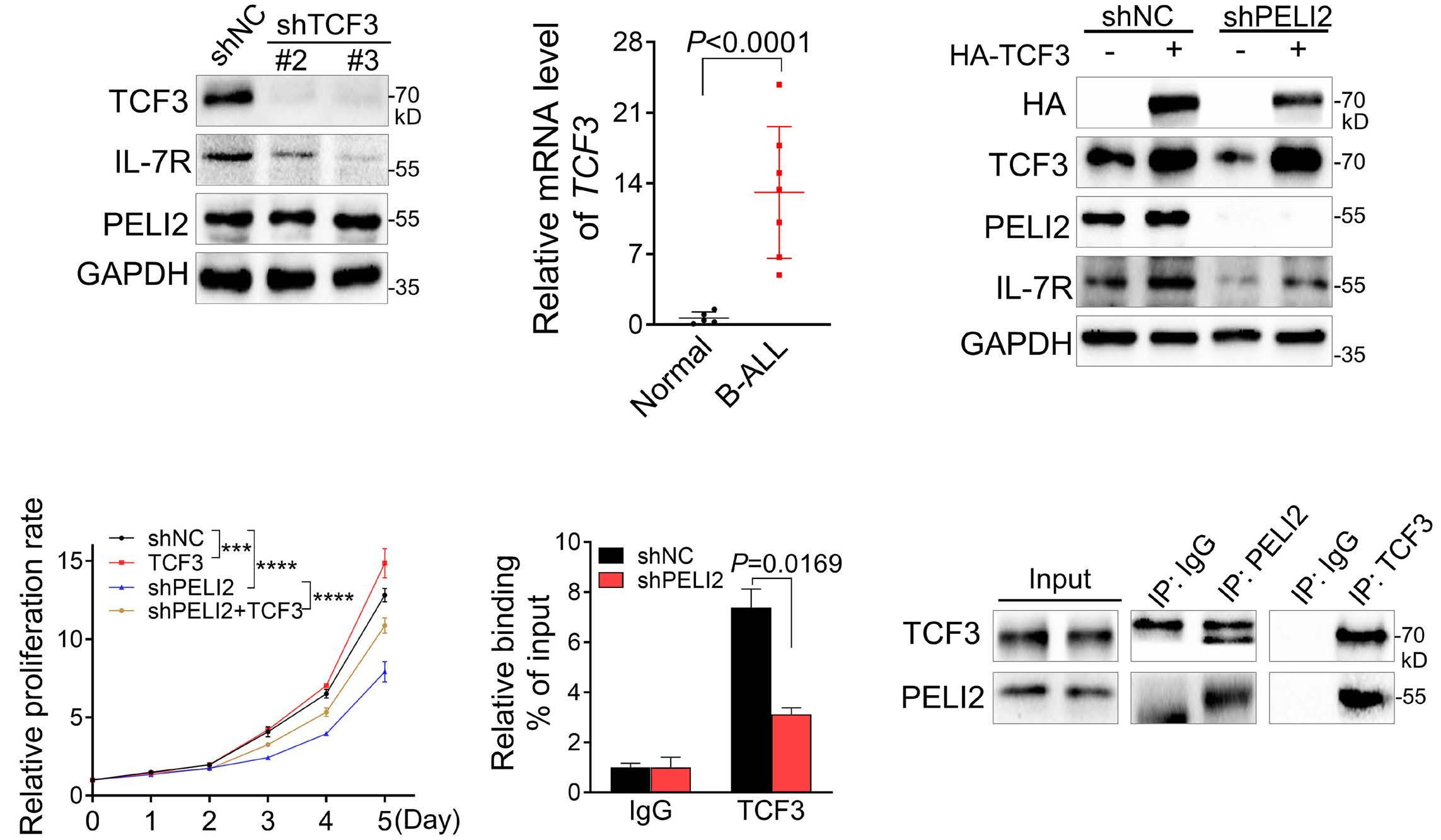
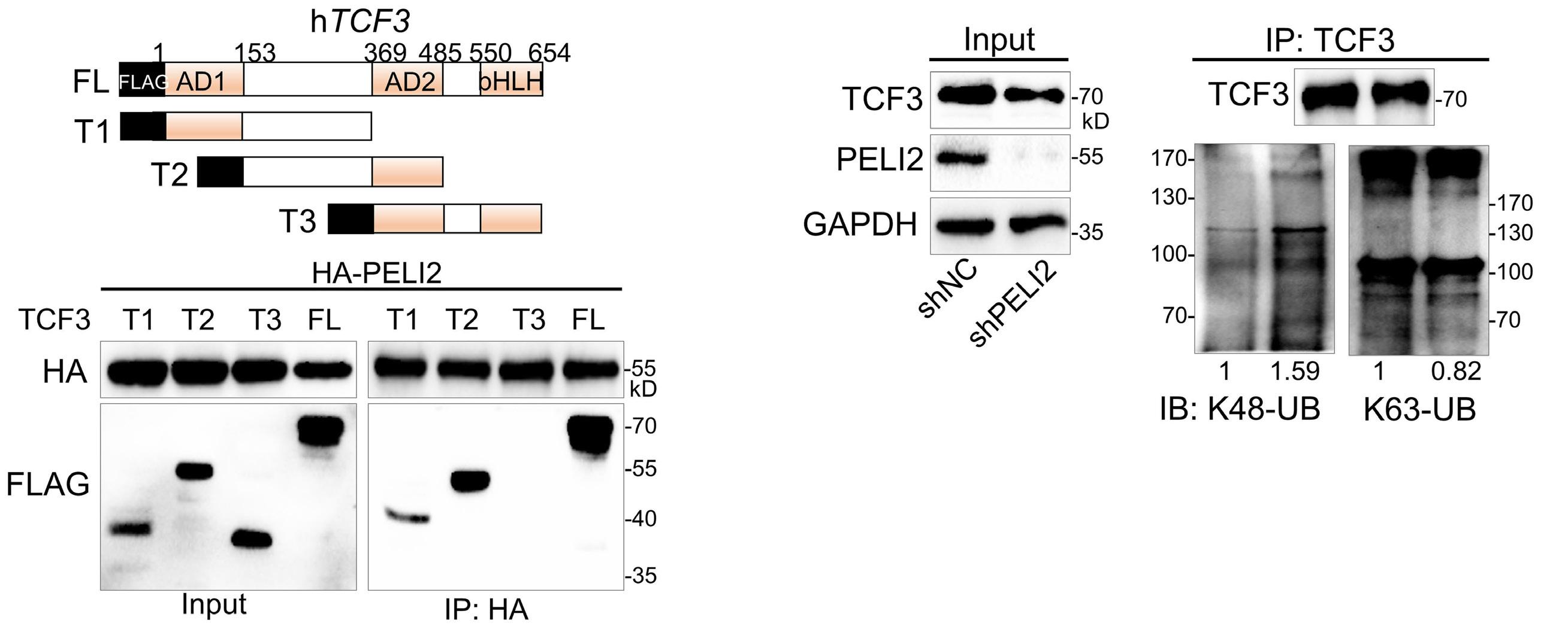
Figure 6. PELI2 stabilized TCF3 via K63-linked polyubiquitination to regulate IL-7R expression in B-cell precursor acute lymphoblastic leukemia cells. (A) Immunoblotting analysis of IL-7R and PELI2 expression in Nalm-6 cells transduced with retroviruses encoding indicated shRNA. shNC represents a non-targeting shRNA. GAPDH was used as loading control. (B) Statistical analysis of TCF3 mRNA level in the bone marrow mononuclear cells as in Figure 5A (each dot represents one sample). (C) Immunoblotting analysis of indicated proteins in Nalm-6 cells transduced with retroviruses encoding indicated shRNA in the presence of TCF3 overexpression. shNC represents a non-targeting shRNA. GAPDH was used as loading control. (D) Statistical analysis of cell proliferation in Nalm-6 cells as in (C). Data obtained from 3 independent experiments. P value determined by two-way ANOVA. ***P<0.001; ****P<0.0001. (E) Chromatin immunoprecipitation analyses of promoter binding activity of TCF3 to IL-7R in Nalm-6 cells transduced with retroviruses encoding indicated shRNA. shNC represents a non-targeting shRNA. Data presented as mean± Standard Deviation (SD) from 3 independent experiments. (F) Co-immunoprecipitation (Co-IP) analysis of endogenous PELI2 and TCF3 in Nalm-6 cells. (G) Structure of full-length and truncated human TCF3. Co-IP analysis of TCF3 mutants binding to PELI2 HEK293T cells were co-transfected with TCF3 truncations (FLAG tagged) and HA-PELI2, and IP analysis was performed with anti-HA antibody. (H) Immunoblotting analysis of ubiquitination of TCF3 immunoprecipitated from Nalm-6 cells transduced with indicated retroviruses with K63-linkage and K48-linkage specific polyubiquitin antibodies. shNC represents a non-targeting shRNA. Numbers indicate the relative band intensity of the ubiquitin blots to corresponding control. All P values determined by unpaired two-tailed Student t test unless otherwise indicated. See also Online Supplementary Figures S8, S9 for supporting information.
PELI2 promoted TCF3 protein stability via K63-linked polyubiquitination in B-cell precursor acute lymphoblastic leukemia cells
Based on the observed correlation of TCF3 protein level with PELI2 expression, we sought to determine whether PELI2 regulates TCF3 protein level via ubiquitination similar to PU.1. Indeed, upon CHX treatment, TCF3 protein stability was reduced in Nalm-6 cells with PELI2 knockdown compared to control groups (Online Supplementary Figure S9A), while enhanced protein stability was observed in Nalm-6 cells with PELI2 overexpression (Online Supplementary Figure S9B). Furthermore, the decrease in TCF3 protein induced by PELI2 knockdown was clearly abolished by the pre-treatment of MG132 (Online Supplementary Figure S9C).
Co-IP analysis demonstrated an interaction between PELI2 and TCF3 in HEK293T cells (Online Supplementary Figure S9D). Their interaction was confirmed by endogenous coIP in Nalm-6 cells (Figure 6F). Furthermore, PELI2-binding domain in TCF3 was mapped to the interval between AD1 and AD2 domains (Figure 6G), and the FHA domain of PELI2 is responsible for the TCF3-binding (Online Supplementary Figure S9E) indicated by Co-IP experiments.
Overexpression of PELI2 promoted K63-linked ubiquitination of TCF3 (Online Supplementary Figure S9F), whereas similar approaches using K48-linked ubiquitin failed to detect any increase in ubiquitination of TCF3 (Online Supplementary Figure S9G). In line with this, we also observed that K63-ubiquitination of endogenous TCF3 was increased upon the overexpression of PELI2 in Nalm-6 cells. Conversely, PELI2 knockdown led to the reduced K63-ubiquitination but increased K48-ubiquitination of TCF3 in Nalm-6 cells (Figure 6H, Online Supplementary Figure S9H).
PELI2 inhibition reduced the leukemia burden in human B-cell precursor acute lymphoblastic leukemia xenograft mice
To assess the effect of PELI2 repression on tumor progression of BCP-ALL, we established PELI2-silencing Nalm-6 cell lines using retroviral construct expressing shRNA targeting PELI2, and transplanted these cells into immunocompromised NOD scid gamma (NSG) mice (Figure 7A). All Nalm-6-bearing mice died at around 26 days, whereas the mice bearing PELI2-silencing Nalm-6 exhibited significantly prolonged median survival (Figure 7B).
As previously reported,27 Nalm-6-bearing mice exhibited rapid tumor burden and cell infiltration in spleen and BM. Compared with control Nalm-6-bearing mice, the spleen size of mice bearing PELI2-silencing Nalm-6 was significantly reduced (Figure 7C, D). Nalm-6 cell frequencies were much lower in these mice compared to controls (Figure 7E). Consistent with the role of TCF3 in mediating IL-7R expression and BCP-ALL cell proliferation in vitro, its overexpression aggravated the progression of Nalm-6-driven BCP-ALL in vivo (Figure 7B, Online Supplementary Figure S10A). Notably, replenishment of TCF3 significantly reversed
the suppression phenotypes of mice bearing PELI2-silencing Nalm-6, including the shorter survival (Figure 7B), enlarged spleen (Figure 7C, D), and aggravated infiltrating Nalm-6 cells in spleen and BM (Figure 7E). A similar reversal of PELI2 inhibition-induced suppression phenotypes of Nalm-6-driven BCP-ALL was also observed in mice bearing PELI2-silencing Nalm-6 with IL-7R overexpression (Online Supplementary Figure S10B-F).
We also performed the xenotransplantation experiment to determine the in vivo effect of PELI2 inhibition on the progress of human BCP-ALL. Primary human BCP-ALL mononuclear cells were transduced with PELI2 shRNA and then transplanted into NSG mice. Compared to the poor survival in the control group, PELI2-knockdown significantly prolonged the survival of BCP-ALL-bearing mice (Figure 7F). Furthermore, PELI2 silencing led to a significant reduction in the frequency of human leukemic blasts in the BM and spleen in recipient mice at two months post transplantation (Figure 7G). These results indicated that PELI2 inhibition reduced the human BCP-ALL burden in vivo.
IL-7R signaling mainly controls the proliferation and survival of early B-cell progenitor cells in normal B-cell development.28 Our study demonstrated that PELI2 promotes PU.1 stability via ubiquitination to regulate IL-7R expression. Loss of PELI2 leading to the degradation of PU.1, which in turn down-regulated the IL-7R expression, impaired the commitment and proliferation of early B-cell progenitors (Figure 8), whereas T-lineage differentiation was unaffected. This is consistent with the critical role of PU.1 in IL-7R expression, which specifically occurred in B-cell lineages but not T-cell lineages.26,29 Given that IL-7R is essential for lymphoid development, it is reasonable to suppose that the differing commitment of CLP relies on the available transcription factors of IL-7R. In line with this, an upstream regulatory element of PU.1 functions as a PU.1 enhancer in B cells but as a repressor in T-cell precursors.30
Our data demonstrated that PELI2 functions as a key mediator at the transition from the CLP to the earliest stage of B-cell specification via PU.1, which is consistent with the role of PU.1 in early lymphopoiesis, the deficiency of which led to the compromised differentiation of CLP into BLP.31 In addition, PU.1 is also required for the developmental progression of CLP from LMPP, the deficiency of which leads to the reduced CLP and consequent B-cell development.31,32 Given that PELI2 deletion led to reduced numbers of CLP, we cannot exclude the possibility that loss of PELI2 disrupts the transition of early lymphoid progenitors LMPP to CLP via PU.1.
Although the frequency of HSC in PELI2CKO mice BM is comparable to that of WT controls, PELI2CKO HSC exhibited notably impaired self-renewal and reconstitution upon the
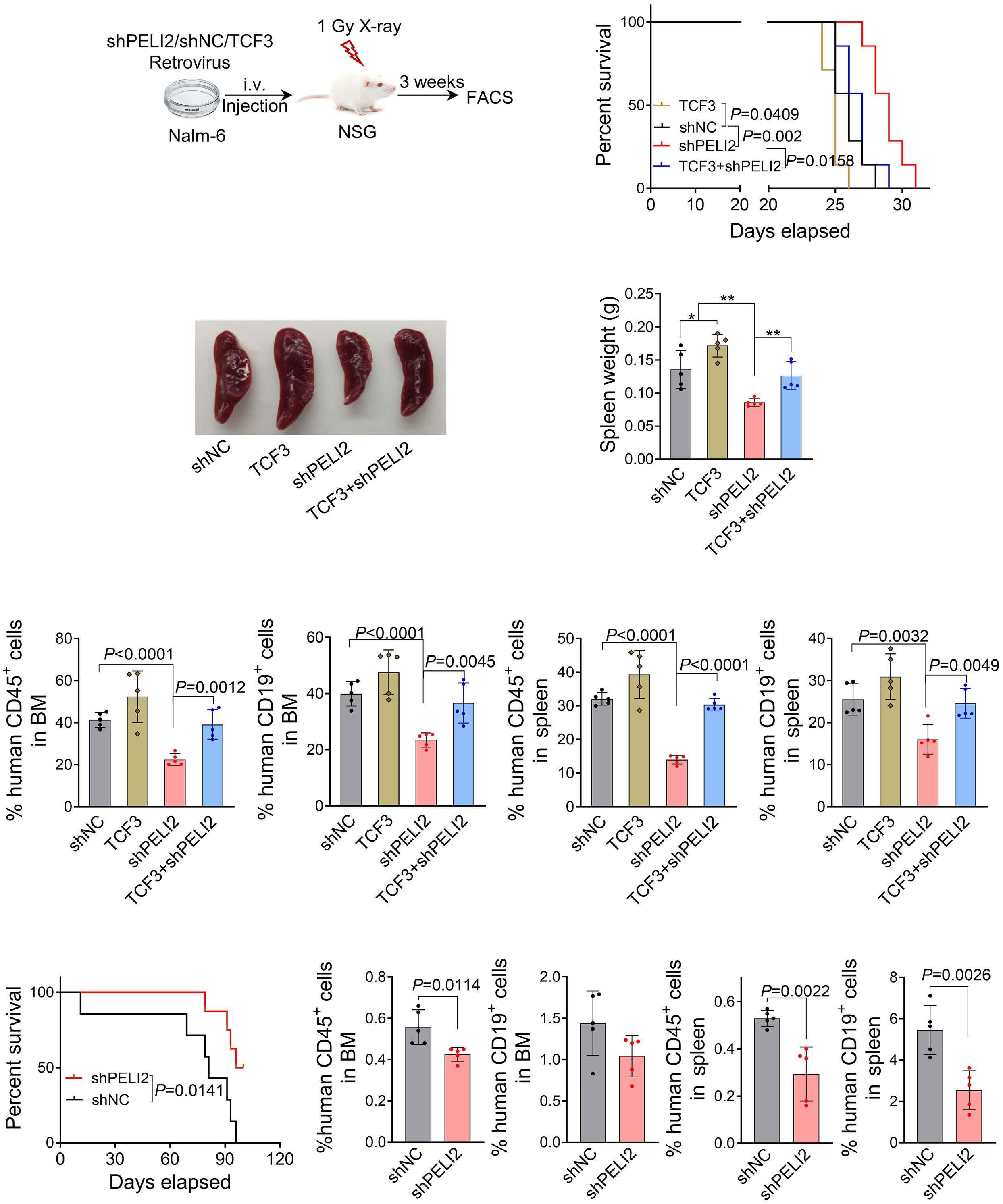
Figure 7. PELI2 inhibition reduced leukemia burden in the human B-cell precursor acute lymphoblastic leukemia xenograft. (A) Schematic representation of human BCP-ALL xenograft. Five million Nalm-6 cells, transduced with indicated retroviruses, were injected into irradiated NOD scid gamma (NSG) mice (1 Gy). shNC represents a non-targeting shRNA. (B) Kaplan-Meier survival curve of indicated Nalm-6-xenograft mice. Data obtained from 8 mice in each group. P values determined by Log-rank (Mantel-Cox) test. (C) Representative spleen of indicated mice as in (B). (D) Statistical analysis of spleen weight from mice in (C). Each dot represents one mouse. Data presented as mean±Standard Deviation (SD). *P<0.05, **P<0.01. (E) Human CD45+ and CD19+ cells from bone marrow (BM) and spleen of indicated mice were analyzed by flow cytometry. Each dot represents one mouse. Data presented as mean±SD. (F) Kaplan-Meier survival curve of indicated patient-derived xenograft (PDX) mice. Data obtained from 7 mice in each group. P values determined by Log-rank (Mantel-Cox) test. (G) Human CD45+ and CD19+ cells from BM and spleen of indicated PDX mice were analyzed at two months post transplantation by flow cytometry. Each dot represents one mouse. Data presented as mean±SD. P values determined by unpaired two-tailed Student t test unless otherwise indicated. See also Online Supplementary Figure S10 for supporting information.
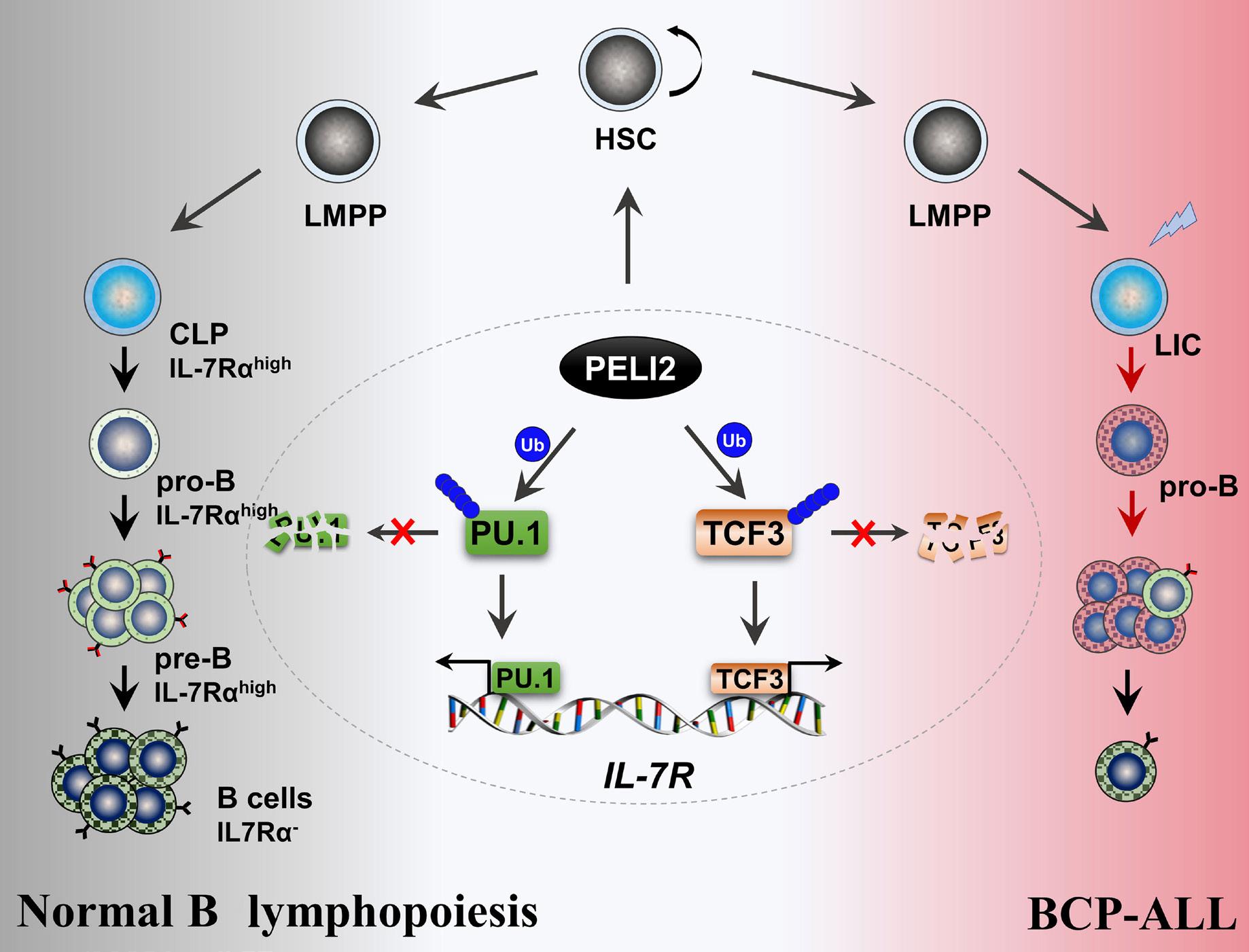
challenge of 5-FU. This was further confirmed by the defects of functional HSC in the competitive transplantation and limiting dilution assays, indicating that PELI2 is required for stressed hematopoiesis. Indeed, PU.1 is expressed in HSC and exhibits a gradual decrease during the subsequent differentiation into common lymphoid and myeloid progenitors.33 PU.1 has also been identified as a master regulator in HSC cell fate decisions and homing through the interaction with a variety of regulatory factors.34,35 Considering that PU.1 protects HSC from excessive exhaustion by controlling the transcription of multiple cell-cycle regulators,36 PELI2 may regulate HSC through PU.1 protein stability that is similar to its role in IL-7R expression during early B-cell progenitor commitment and proliferation. Given that the regulatory roles of PELI2 in immunity and potential inflammatory modulation of hematopoiesis, there is also a possibility that the defects of functional HSC in PELI2 knockout mice are feedback cues by the altered immunity induced by PELI2 deletion.
Our study also provides strong rationales for targeting IL7R for the treatment of BCP-ALL, which is characterized by a block in lymphoid differentiation leading to the accumulation of immature progenitor cells.37 There is growing evidence indicating that excessive IL-7R signaling is oncogenic, being responsible for resistance to conventional chemotherapy and targeted therapeutics.38-40 TCF3, a defined transcription factor in normal B-cell differentiation, is genetically altered via translocations, deletions or mutations in BCP-ALL,5 leading to aberrant gene expression patterns in leukemic cells. Our data demonstrated that PELI2 promotes TCF3 protein stability via ubiquitination that is required for the IL-7R expression in BCP-ALL cells
Figure 8. Schematic diagram illustrates the regulatory role of PELI2 in normal B lymphopoiesis and B-cell precursor acute lymphoblastic leukemia. PELI2 regulated early B-cell progenitor homeostasis and the progression of BCP-ALL by maintaining the IL-7R expression via the ubiquitination of PU.1 and TCF3, respectively.
(Figure 8). Indeed, TCF3 showed different expression with PU.1 during normal B-cell differentiation (Online Supplementary Figure S8H), which may account for its distinct requirement for IL-7R expression in BCP-ALL that occurs in late B-cell progenitor cells. Inhibition of PELI2 suppressed the proliferation of BCP-ALL cells in vitro and in vivo. Therefore, the high expression of PELI2 in BCP-ALL provides therapeutical benefit for targeting PELI2 in the treatment of BCP-ALL.
In summary, we demonstrate that PELI2 regulated early B-cell progenitor homeostasis and the progression of BCPALL by maintaining IL-7R expression via the ubiquitination of PU.1 and TCF3, respectively (Figure 8). Therefore, these findings provide not only a new insight into the pathogenic mechanism for B-cell precursor malignancies, but also a proof of principle that targeting PELI2 restricts B-cell precursor expansion via IL-7R inhibition.
No conflicts of interest to disclose.
BZ designed and guided research. YX, WQ and CZ performed the experiments. YX, QZ, YL, CZ and BZ analyzed the data. YX and BZ wrote the original draft. XW, AZ and JG collected the healthy donors and BCP-ALL patient samples. TS, JL, CL, YS and BZ reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
We thank Prof. Chunyan Ji (Qilu Hospital of Shandong Uni-
versity) for the gift of the Nalm-6 cell line. We thank Prof. Jianrong Wang (Suzhou University) for the gift of the 697 cell line. We also thank the Translational Medicine Core Facility of Shandong University for the availability of consultation and instruments that supported this work.
Funding
This work was supported by grants from National Natural Science Foundation of China (81874294), Taishan Scholars Program (TSQN201812015), the program for Multidisciplinary Research and Innovation Team of Young Scholars of Shandong University (2020QNQT007) and the key Program of Natural
1. Boller S GR. The regulatory network of B-cell differentiation: a focused view of early B-cell factor 1 function. Immunol Rev. 2014;261(1):102-115.
2. Inlay MA, Bhattacharya D, Sahoo D, et al. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes Dev. 2009;23(20):2376-2381.
3. Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;26(6):715-725.
4 Busslinger M. Transcriptional control of early B cell development. Annu Rev Immunol. 2004;22:55-79.
5. Somasundaram R, Prasad MAJ, Ungerbäck J, Sigvardsson M. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood. 2015;126(2):144-152.
6. Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol. 2014;14(2):69-80.
7 Corfe SA, Paige CJ. The many roles of IL-7 in B cell development; mediator of survival, proliferation and differentiation. Semin Immunol. 2012;24(3):198-208.
8. Milne CD, Paige CJ. IL-7: a key regulator of B lymphopoiesis. Semin Immunol. 2006;18(1):20-30.
9 Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131(4):959-971.
10. Peschon JJ, Morrissey PJ, Grabstein KH, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptordeficient mice. J Exp Med. 1994;180(5):1955-1960.
11. von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181(4):1519-1526.
12. Dias S, Silva HJ, Cumano A, Vieira P. Interleukin-7 is necessary to maintain the B cell potential in common lymphoid progenitors. J Exp Med. 2005;201(6):971-979.
13. Kikuchi K, Lai AY, Hsu CL, Kondo M. IL-7 receptor signaling is necessary for stage transition in adult B cell development through up-regulation of EBF. J Exp Med. 2005;201(8):1197-1203.
14. Yao Z, Cui Y, Watford WT, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci U S A. 2006;103(4):1000-1005.
15. Chou W-C, Levy DE, Lee C-K. STAT3 positively regulates an early step in B-cell development. Blood. 2006;108(9):3005-3011.
16. Barata JT, Durum SK, Seddon B. Flip the coin: IL-7 and IL-7R in
Science Foundation of Shandong Province (ZR2022LSW027). This work was also supported by the National Key Research and Development Program (2019YFA0905402) and the program for Innovative Research Team in the University of Ministry of Education of China (N. IRT_17R68).
Data-sharing statement
Supporting data are available in the Online Supplementary Appendix. Microarray data are available at GEO under accession number GSE228984. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
health and disease. Nat Immunol. 2019;20(12):1584-1593.
17 Almeida ARM, Neto JL, Cachucho A, et al. Interleukin-7 receptor a mutational activation can initiate precursor B-cell acute lymphoblastic leukemia. Nat Commun. 2021;12(1):7268.
18. Thomas KR, Allenspach EJ, Camp ND, et al. Activated interleukin-7 receptor signaling drives B-cell acute lymphoblastic leukemia in mice. Leukemia. 2022;36(1):42-57.
19 Humphries F, Bergin R, Jackson R, et al. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat Commun. 2018;9(1):1560.
20 Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21(4):301-307.
21. Xu Y, Zhao B. Research progress on E3 ubiquitin ligase Pellino proteins. Xiamen Univ Nat Sci. 2022;61(3):402-414.
22. Humphries F, Moynagh PN. Molecular and physiological roles of Pellino E3 ubiquitin ligases in immunity. Immunol Rev. 2015;266(1):93-108.
23. Bhansali RS, Rammohan M, Lee P, et al. DYRK1A regulates B cell acute lymphoblastic leukemia through phosphorylation of FOXO1 and STAT3. J Clin Invest. 2021;131(1):e135937.
24. Lu Z, Huang L, Li Y, et al. Fine-tuning of cholesterol homeostasis controls erythroid differentiation. Adv Sci (Weinh). 2022;9(2):e2102669.
25. Xiong Z, Xia P, Zhu X, et al. Glutamylation of deubiquitinase BAP1 controls self-renewal of hematopoietic stem cells and hematopoiesis. J Exp Med. 2020;217(2):e20190974.
26. DeKoter RP, Lee H-J, Singh H. PU.1 regulates expression of the interleukin-7 receptor in lymphoid progenitors. Immunity. 2002;16(2):297-309.
27. Tipanee J, Samara-Kuko E, Gevaert T, Chuah MK, VandenDriessche T. Universal allogeneic CAR T cells engineered with Sleeping Beauty transposons and CRISPR-CAS9 for cancer immunotherapy. Mol Ther. 2022;30(10):3155-3175.
28. Miller JP, Izon D, DeMuth W, Gerstein R, Bhandoola A, Allman D. The earliest step in B lineage differentiation from common lymphoid progenitors is critically dependent upon interleukin 7. J Exp Med. 2002;196(5):705-711.
29 Anderson MK, Hernandez-Hoyos G, Diamond RA, Rothenberg EV. Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development. 1999;126(14):3131-3148.
30. Rosenbauer F, Owens BM, Yu L, et al. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat Genet. 2006;38(1):27-37.
31. Pang SHM, de Graaf CA, Hilton DJ, et al. PU.1 is required for the developmental progression of multipotent progenitors to common lymphoid progenitors. Front Immunol. 2018;9:1264.
32. Iwasaki H, Somoza C, Shigematsu H, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106(5):1590-1600.
33. Staber PB, Zhang P, Ye M, et al. The Runx-PU.1 pathway preserves normal and AML/ETO9a leukemic stem cells. Blood. 2014;124(15):2391-2399.
34 Etzrodt M, Ahmed N, Hoppe PS, et al. Inflammatory signals directly instruct PU.1 in HSCs via TNF. Blood. 2019;133(8):816-819.
35. Chavez JS, Rabe JL, Loeffler D, et al. PU.1 enforces quiescence and limits hematopoietic stem cell expansion during inflammatory stress. J Exp Med. 2021;218(6):e20201169.
36. Staber PB, Zhang P, Ye M, et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell. 2013;49(5):934-946.
37. Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3407-3415.
38. Cramer SD, Aplan PD, Durum SK. Therapeutic targeting of IL-7Ralpha signaling pathways in ALL treatment. Blood. 2016;128(4):473-478.
39. Cante-Barrett K, Spijkers-Hagelstein JA, Buijs-Gladdines JG, et al. MEK and PI3K-AKT inhibitors synergistically block activated IL7 receptor signaling in T-cell acute lymphoblastic leukemia. Leukemia. 2016;30(9):1832-1843.
40 Delgado-Martin C, Meyer LK, Huang BJ, et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia. 2017;31(12):2568-2576.
Luisa Lorenzi,1 Torsten Haferlach,2 Luigi Mori,3 Matteo Simbeni,1 Wencke Walter,2 Piera Balzarini,1 Manja Meggendorfer,2 Claudia Döring,4 Silvia Lonardi,1 Mattia Bugatti,1 Claudio Agostinelli,5 Jay Mehta,6 Anita Borges,7 Abbas Agaimy,8 Ingrid Simonitsch-Klupp,9 José Cabeçadas,10 Elias Campo,11 Stefano Aldo Pileri,12 Fabio Facchetti1 Martin Leo Hansmann13 and Sylvia Hartmann4
1Pathology Unit, ASST Spedali Civili di Brescia, Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy; 2MLL Munich Leukemia Laboratory, Munich, Germany; 3Laboratory of Molecular Medicine, Department of Clinical and Experimental Science, University of Brescia, Brescia, Italy; 4Dr Senckenberg Institute of Pathology, Goethe University, Frankfurt, Germany; 5Department of Medical and Surgical Sciences, University of Bologna, Bologna, Italy; 6Neuberg Oncopath, Mumbai, India; 7Histopathology, SRL Diagnostics, Mumbai, India; 8Institute of Pathology, University Hospital, Erlangen, Germany; 9Institute of Pathology, Medical University of Vienna, Vienna, Austria; 10Department of Pathology, Portuguese Institute of Oncology, Lisbon, Portugal; 11Hematopathology Section, Hospital Clinic, IDIBAPS, University of Barcelona, Barcelona, Spain; 12Division of Hematopathology, European Institute of Oncology (IEO) IRCCS, Milan, Italy and 13Institute for General Pharmacology and Toxicology, Goethe University, Frankfurt, Germany
Correspondence: L. Lorenzi luisa.lorenzi@unibs.it
Received: June 19, 2023. Accepted: November 13, 2023. Early view: November 23, 2023.
https://doi.org/10.3324/haematol.2023.283669
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Standardized treatment options are lacking for patients with unresectable or multifocal follicular dendritic cell sarcoma (FDCS) and disease-related mortality is as high as 20%. Applying whole-genome sequencing (WGS) in one case and whole-exome sequencing (WES) in additional twelve cases, this study adds information on the molecular landscape of FDCS, expanding knowledge on pathobiological mechanisms and identifying novel markers of potential theragnostic significance. Massive parallel sequencing showed high frequency of mutations on oncosuppressor genes, particularly in RB1, CARS and BRCA2 and unveiled alterations on homologous recombination DNA damage repair-related genes in 70% (9/13) of cases. This indicates that patients with high-stage FDCS may be eligible for poly ADP ribose polymerase inhibition protocols. Low tumor mutational burden was confirmed in this study despite common PDL1 expression in FDCS arguing on the efficacy of immune checkpoint inhibitors. CDKN2A deletion, detected by WGS and confirmed by fluorescence in situ hybridization in 41% of cases (9/22) indicates that impairment of cell cycle regulation may sustain oncogenesis in FDCS. Absence of mutations in the RAS/RAF/MAPK pathway and lack of clonal hematopoiesis-related mutations in FDCS sanction its differences from dendritic cell-derived neoplasms of hematopoietic derivation. WGS and WES in FDCS provides additional information on the molecular landscape of this rare tumor, proposing novel candidate genes for innovative therapeutical approaches to improve survival of patients with multifocal disease.
Follicular dendritic cell sarcoma (FDCS) is a malignant neoplasm of mesenchymal derivation with morphological and phenotypical features of FDC, dendritic cells of mesenchymal-derivation of the B follicle.1-4 Included in the group of “histiocytic and dendritic cell neoplasms” in the revised 4th edition of World Health Organization
(WHO) classification4 as well as in the International Consensus Classification, 5 it was moved to a novel chapter of “stroma-derived neoplasms of lymphoid tissues” in the most recent WHO classification.6 FDCS can occur both at nodal and extranodal sites and can be metastatic and lethal in about a fifth of the cases.7 Complete tumor resection is the treatment of choice for localized disease while patients with multifocal or unresectable FDCS are
alternatively treated with radio- and/or chemotherapy in accordance with lymphoma, or sarcoma-like regimens with variable outcomes.7
Large, massive parallel sequencing studies performed on dendritic- and histiocytic-derived tumors have unveiled important biological pathways commonly driving some of these tumors. MAPK/ERK pathway is activated in Langerhans cell histiocytosis, Erdheim Chester disease as well as in some histiocytic sarcoma and in selected cases of Rosai-Dorfman disease.8,9 Clinical responses after targeted therapies in these settings are well documented and have changed the natural history for these diseases.9-11 In contrast, evidence indicates that the molecular mechanisms driving FDCS are different. Mutations affecting genes such as KRAS, NRAS, MAP2K1 and BRAF are rare in FDCS,7,12,13 in keeping with its non-hematopoietic, but mesenchymal derivation.12,14
Previous studies indicated an oncosuppressor-driven biology in FDCS,12,15 however, recurrent mutations of diagnostic and prognostic significance are lacking and biomarkers of theragnostic relevance are still missing.7
By whole-exome sequencing (WES) and whole-genome sequencing (WGS) approach this study aims to provide
novel molecular information on FDCS indicating innovative biomarkers of therapeutical significance.
Case selection and histopathological review
The discovery cohort consisted of 13 FDCS samples selected by tissue availability, tumor cell content and DNA quality to undergo massive parallel sequencing (MPS). One case (case #13) underwent WGS, 12 cases WES (cases #1-#12). The latter were also included in three tissue micro arrays (TMA) together with 22 additional cases, considered as validation cohort. The clinical data regarding the cohort undergoing MPS are reported in Table 1. It included seven males and six females; the average age was 66 years (range, 36-84). Diagnosis was classical FDCS in all cases but one (#3), which was diagnosed as Epstein-Barr virus–positive inflammatory follicular dendritic cell sarcoma/tumor (EBV-IFDCS/T).5,6 All cases underwent pathological revision by a consensus of expert hematopathogists (FF, SAP, MLH, SH); morphology and phenotype of the discovery cohort are detailed in Table
Table 1. Clinical features of 13 follicular dendritic cell-derived tumors/sarcoma undergoing massive parallel sequencing.
follicular dendritic cell sarcoma; EBV-IFDCS/T: Epstein-Barr virus–positive inflammatory follicular dendritic cell sarcoma/tumor;
fresh frozen; FFPE: formalin-fixed paraffin-embedded; F: female; M: male; WES: whole-exome sequencing; WGS: whole-genome sequencing; NA: not available.
2. Written informed consent was obtained from patients in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee at the University Hospital Frankfurt (157-17).
Immunohistochemistry and fluoresensce in situ hybridization studies
Immunohistochemistry was performed for the following antibodies: Serglycin and FDC-SP as previously reported,16 p16 (clone E6H4, dilution 1:4, CINtec histology kit from Roche); PD-L1 (clone 22C3, dilution 1:40; Agilent).
Structural variants (SV) and copy number variations (CNV) identified by WGS or described in the literature were tested by fluorescence in situ hybridization (FISH) with the following probes: CDKN2A/CEP9 (Vysis Abbott Molecular); SS18 Break Apart (Vysis Abbott Molecular); 1p36/1q25 and 19q13/19p13 (Vysis Abbott Molecular); CIC Break Apart (Empire Genomics). Interpretation of CDKN2A deletion was supported by counting at least 50 nuclei for each case.
Next-generation sequencing and interpretation of variants DNA was extracted from formalin-fixed paraffin-embedded (FFPE) or FF samples including at least 20% of tumor cell content without tumor enrichment or microdissection. Next-generation sequencing was performed by amplicon-based massive parallel sequencing technology on an Illumina Nova Seq 6000 (San Diego, CA, US) platform at
Munich Leukemia Laboratory, where bioinformatics analysis was carried out using the software Variant Interpreter (Illumina) and open-source databases. Tumor mutational burden and mutational signature were calculated by Base Space Variant Interpreter algorithms exclusively from WGS data. Single nucleotide variants/insertions deletions (SNV/indel) were extracted and prioritized by the variant filtering strategy detailed in Online Supplementary Figure S1 17-19 Eligibility of FDCS patients to poly ADP ribose polymerase (PARP) inhibition protocols was performed by manually interrogating the dataset of gene variants for the prevalence of mutations on genes involved in the mechanism of double strand beaks (DSB), as previously described.20
Mutations in homologous recombination-related genes mutations are common in follicular dendritic cell sarcoma SNV analysis lead to the identification of one or more mutations of known oncosuppressor genes in 12 of 13 FDCS (92%) (Table 3). CARS, RB1 and WRN genes were the most recurrently mutated in this cohort (each gene mutated in at least 3 cases). All five cases with a single or no mutation on oncosuppressor genes (#1, #3, #4, #12, #13) were unifocal at presentation, they included both nodal and extranodal cases and the EBV-IFDCS/T sample
Table 2. Cytological and phenotypical features of 13 cases of follicular dendritic cell derived tumors/sarcoma undergoing massive parallel sequencing.
CD-LF: Castleman disease-like features; Clu: clusterin; Cl4: claudin 4; FDC-SP: follicular dendritic cell-secreted protein; HPF: high power field; NA: not available; SRGN: serglycin; EBV: Epstein-Barr virus; EBER: EBV-encoded small RNA in situ hybridization.
(Online Supplementary Table S1; Table 1).
Notably, cases #2 and #10 showed the highest number of mutations in this group of genes and both showed an aggressive clinical course leading to patients’ death (Table 1). Patient #2 had a previous diagnosis of Hyaline-vascular type Castleman disease (HV-CD) and developed multiple and subsequent nodal and extranodal FDCS during more than 10 years; patient #10 presented with a multifocal extranodal
disease with rapid fatal evolution. After surgery, systemic therapy was attempted in both patients, ineffectively. In 70% of cases (9/13) (Table 4) at least one pathogenic mutation of HRD genes was found. BRCA2 and RB1 were the most mutated genes in this group; they occurred in three cases and were simultaneously found in two (#7 and #10). Mutations on TP53 and CHEK2 were also identified in two cases. Notably, in the previously mentioned aggressive
Table 3. Pathogenic mutations identified on known oncosuppressor genes in 13 follicular dendritic cell-derived tumors/sarcoma cases.
L1129Sa
CARS - A241Sa
CBFA2T3 - R697Ka
G342Sa R678*c
S1934Pa
V194Ma
G16Sa
RUNX1
STK11 - F354La
TCF3 -
TNFAIP3 - R697Ka
TP53
WRN - Y1034*c
E286Ka
R1745Ha
D604Na
G571Sa
H483Lfs*b
S424Pa
S360Ksfs*b
P195La
P213Fa G431Sa
R213*c
S1338L
K635Qa
Mutation consequence: amissense mutation; bframeshift mutation; cstop-gain mutation; *stop codon; #: case.
A693Ga
Table 4. Table of mutations identified in follicular dendritic cell-derived tumors/sarcoma on PARP inhibitor-sensitive genes.
L1137Fa G1194Va
D604Na
E286Ka
ATR - M1996Ta
BRCA1 -
I2285Va
H483Lfs*b - - S360Kfs*b
R213*c -
Q487*c
L1700*b
R258Yfs*b
I574Va
Mutation consequence: amissense mutation; bframeshift mutation; cstop-gain mutation; *stop codon; #: case; PARP: poly ADP-ribose polymerase.
cases (#2 and #10), at least one mutation of HRD was also detected (Table 4).
Structural variants and copy number variations detection in follicular dendritic cell sarcoma
WGS, which was performed on case #13, highlighted a high number of SV, including translocations, inversions, CNV including both gains and losses, affecting mainly chromosomes 3, 5, 9, 13 and chromosome 19. They are outlined in the circos plot in Figure 1 and listed in Online Supplementary Table S2. The ploidy level in case #13 was 1.94. In summary, 114 SV (76 translocations, 33 inversions and 5 deletions) and 48 CNV were identified. The karyotype of case #13 was complex, however, by comparison with previously publishes studies we could not identify analogy with previously reported SV.21-26 In order to evaluate the impact of NGS findings in identifying recurrent translocations of diagnostic significance, variations occurring in regions of interest in cancer biology were explored by FISH probes on tissues slides of case #13.
FISH probe for the 18q11.2 locus did not identify rearrangements reported on the same locus by WGS, t(16;18) (p11.2;q11.2) and t(18;22)(q11.2;q11.23), additionally, the CIC and 19q co-deletion probes confirmed the CNV gain detected by WGS but did not identify rearrangements (data not shown). The discrepancies between structural variations observed by WGS and the FISH analysis could be explained by the fact that diagnostic FISH probes are designed for specific inquiries and the size of rearrangements detected by WGS may be too small to be identified in situ. Still, FISH could indeed confirm the deletion of the CDKN2A gene, encoded in chromosome region 9p21 where three inversions were detected by WGS. FISH found homozygous deletion (gene/CEP9 ratio=0.15) and CNV gain of chromosome 9 (average CEP9/nucleus ratio=6.3) (Figure 2). Accordingly, immunostaining for p16 protein was completely negative in tumor cells (Figure 2). In order to calculate the incidence of CDKN2A deletion in FDCS, FISH was performed on a total of 22 FDCS and found deletions in 41% of cases (9/22, with gene/CEP9 ratio ranging from 0.14 to 0.53), indicating that deletion of the CDKN2A locus is a recurrent event in this tumor. P16 expression evaluated by immunohistochemistry (IHC) could not predict CDKN2A deletion: deleted cases (9) were either p16 protein-positive or negative (6/9) or focally positive (3/9); non-deleted cases were either strongly positive (3/13), partially positive (4/13) or negative (6/13).
Whole-genome sequencing in follicular dendritic cell sarcoma shows enrichment in mutational signatures SBS16, SBS8, and SBS9 and low tumor mutational burden
Mutational signatures (MS) are specific patterns of somatic mutation accumulation occurring in cancers, that can predict pathogenetic mechanisms, pathways enrichments, or cancer subtypes, with high accuracy.27 MS in cancer bi-
ology is widely applied and in continuous evolution, with inclusion of novel molecular alterations (e.g., doublet base substitution, insertions and deletions, CNV).27
MS evaluation can be reliably performed from WGS data where the incorporation of non-coding DNA allows to investigate the mutational processes involved in cancer genomics.
Therefore, by performing the first MS analysis in a case of FDCS we unveiled novel information. As shown in Figure 3, single base substitution (SBS) signature showed high contribution of SBS1 and SBS5, mutational signatures associated with age and found across many tumor subtypes. The age of the patient (79 years old) and the neoplastic nature of the analyzed tissue confirm the accuracy of this analysis. Other MS identified were SBS16, SBS8 and SBS9. Recent studies have suggested that SBS16 and SBS8 are associated with early and late replication timing, respectively,28,29 however the meaning of such information remains still uncertain. In contrast, SBS9 is recognized as associated with polymerase-η activity, known to be implicated in the B-cell germinal center reaction and somatic hypermutation. This information suggests that, despite extra-nodal localization of the disease (stomach in this case), FDCS is likely to originate from a tertiary germinal center structure. Lastly, MS of FDCS included SBS28, a signature of unknown
Figure 1. Structural and copy number variations in follicular dendritic cell sarcoma. Circos plot outlines variations identified in case #13; chromosomes are represented in the outer ring, translocations and inversions are depicted by stripes, magenta and light blue, respectively. Small deletions are shown in blue; copy number variations are indicated in the inner cycle: losses in green, gains in red.
significance associated with gastric cancer indicating that the site of disease may contribute to the mutational landscape. Any inference on MS in FDCS requires confirmation on additional cases in following studies.
Tumor mutational burden (TMB) is a novel theragnostic biomarker of response to immune-checkpoint therapies. It can be appropriately evaluated on large panels, such as WGS, while a fully standardized pipeline for its calculation on targeted panels is still missing.30 TMB, calculated by
WGS on FDCS #13, resulted low (1,66 mutations/megabase). Notably, a prevalent low TMB has been reported in most mesenchymal tumors31,32 and is in keeping with data recently obtained using a different targeted, approach in 44 cases of FDCS.12 Taken together, these results can have clinical implications since low TMB may indicate low number of neo-antigens and poor or absent response to immunotherapy. However, it is known that tumor cells in FDCS often express PD-L17,14 as confirmed also in this co-
Figure 2. Histological features of follicular dendritic cell sarcoma. On histology, case #13 shows neoplastic follicular dendritic cells (FDC) with spindle cell morphology, associated with some small, intermingled lymphocytes (A) (hematoxylin and eosin staining, 100X magnification) and characterized by unequivocal expression of FDC markers CD21 (B), CXCL13 (C) and clusterin (D) (100X magnification). PD-L1 (22C3) was strongly and diffusely positive on nearly 100% of tumor cells (E, F) (magnification 40X-200X). CDKN2A deletion evaluated by fluorescenze in situ hybridization detected large, atypical nuclei with less than 2 red signals (G, arrow) (average ± standard deviation CDKN2A/CEP9 ratio 0,15±0,63). Notably, the centromeric probe showed numerous signals (average ± standard deviation CEP9 6,30±2,85) suggesting polisomy of chromosome 9. Note the normal signal of a reactive lymphocyte (2 red and 2 green signals, arrowhead). Accordingly, p16 protein was not expressed on tumor cells (H).
hort (52%, 16/31). Based on this evidence, immunotherapy approaches have been attempted with variable results.33,34 In previous gene expression studies, CNV gain of chromosome 9p24 was proposed as mechanism of PD-L1 expression in FDCS.14,35 By WGS of case #13 we observed lack of 9p24 CNV gain despite intense and diffuse PD-L1 protein expression (Figure 2), indicating that other mechanisms than gene amplification can lead to PD-L1 expression in FDCS.
Mutations on RAS/RAF pathway genes, PDGFRB and mutations associated with clonal hematopoiesis are uncommon in follicular dendritic cell sarcoma
WES and WGS on FDCS confirmed some observations previously obtained by targeted approaches. Mutations on genes of the RAS/RAF pathway commonly mutated in hematopoietic-derived tumors such as BRAF, KRAS, MAP2K1, NF1, NRAS or PTPN11 were not detected among 13 cases of FDCS7,12,15 (Online Supplementary Table S1).
The PDGFRB p.Asn666Ser mutation, previously described in HV-CD and in FDCS12,36 was not identified in this cohort. Two missense variants of unknown pathogenicity were found on PDGFRB in two cases (p.Ser408Cys and p.Ala6Val) (Online Supplementary Table S1), one of which showing CDlike features at histology. Functional studies are needed to define the role of these mutations on stromal cells and their significance in FDCS pathogenesis.
Clonal hematopoiesis (CH) is an emerging concept and indicates the clonal expansion of mutated hematopoietic cells occurring with human aging, associated with increased risk of hematological neoplasms37 and other non-hematological disorders likely due to the involvement of mutated
Figure 3. Mutational signatures in follicular dendritic cell sarcoma. Whole-genome sequencing allowed to identify mutational signatures associated with case #13 which include, among others, signature 9, associated with immunoglobulin (IG) hypermutation.
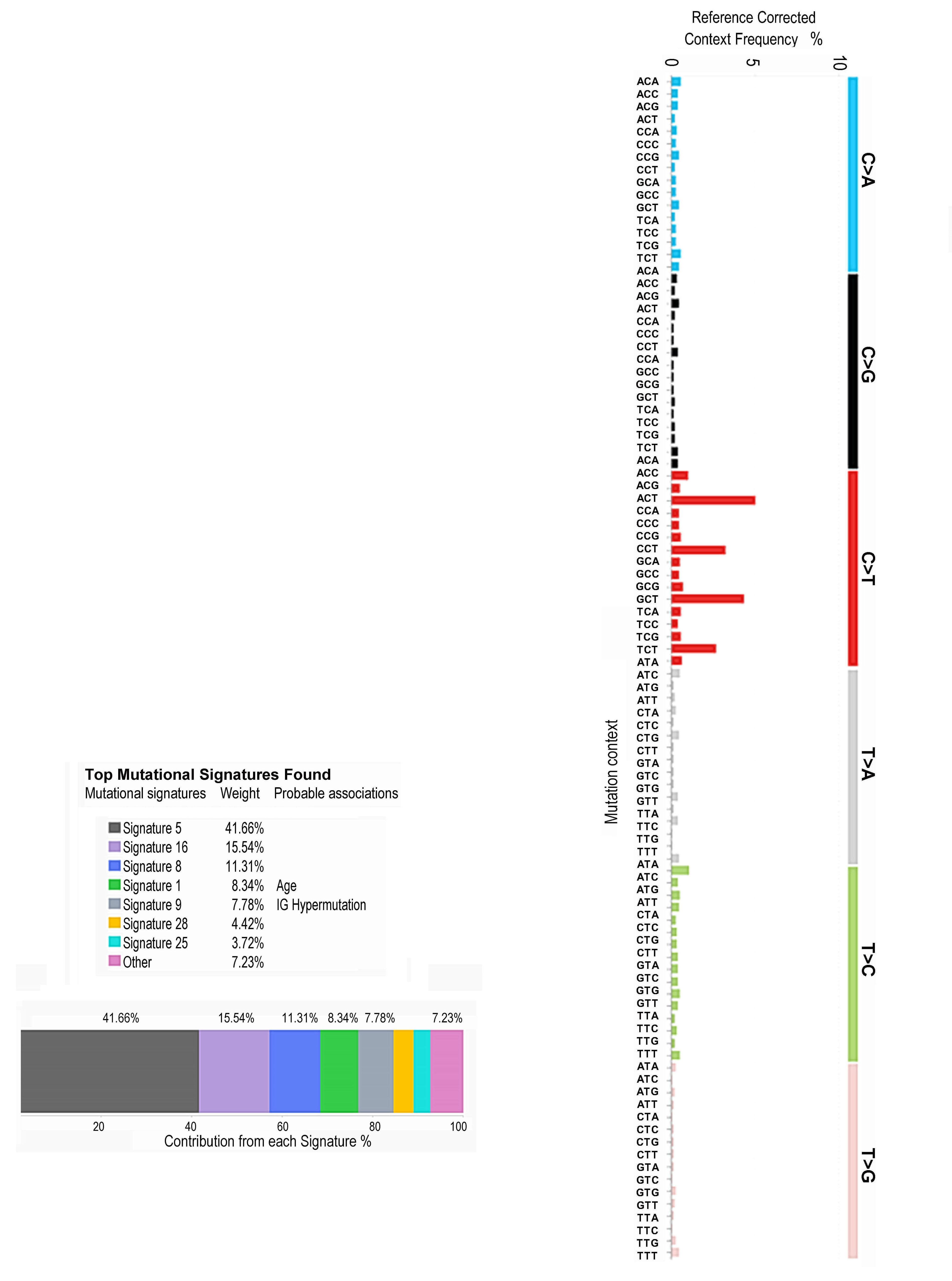
myeloid cells in the inflammatory cascade.38 Using WES and WGS we interrogated data on the incidence of CH-related mutations in 13 FDCS. No significant mutations on TET2 or DNMT3 were detected, pathogenic mutations of ASXL1 were found in case #4 and of PPM1D in case #10, both occurring on elderly patients (73 and 84 years old, respectively). Association between FDCS and lymphomas is extremely rare39,40 and immunoglobulin or T-cell receptor gene clonality was only sporadically identified in FDCS.7 Recently, mutational studies on B- and T-cell lymphomas unveiled a variety of alterations associated with lymphoma pathogenesis and diagnosis in specific settings.41 By comparing FDCS mutational landscape with lymphoma-related genes we found few pathogenic mutations in genes specifically associated with T- and germinal-center B-cell lymphomas, with the exception for genes associated with epigenetic remodeling. KMT2D and SETD2 mutations with plausible deleterious effect were found in seven of 13 (54%) and two of 13 (15%) FDCS cases, respectively. NOTCH2 gene, often mutated in marginal zone lymphoma, was mutated in three (23%). It should be noted that KMT2D and NOTCH2 mutations were detected at very low frequency, arguing on the biological significance of such alterations (Online Supplementary Table S1).
FDCS is an uncommon disease, with unpredictable outcome, occasionally lethal, in need of an effective therapeutical approach. The actual treatment protocols include surgery and chemo- and/or radiotherapy, based on the center experience. The discovery of driving alterations is a priority that could support the identification of effective drugs to cure or control metastatic disease. To this aim, this study, combining WGS and WES in a relatively large cohort of a rare sarcoma, confirms some previous data and adds novel information on its molecular landscape. In line with previous studies,12,15 a tumor suppressor driven pathobiology was observed in this cohort of FDCS, with common CDKN2A deletion and frequent mutations on RB1, BRCA2, WRN and TP53. Furthermore, accumulation of inactivating mutations on these genes was associated with multifocal disease and poor prognosis.
On the basis of this information, using WES/WGS we investigated the incidence of mutations on genes involved in the repair of double strand breaks (DSB, i.e., homologous recombination DNA damage repair, HRD). Notably, mutations on HRD-related genes were found in 70% of cases, indicating the so called “BRCAness phenotype”. It is known that HRD in cancer promotes genomic instability leading to chromosomal alterations.42 In FDCS chromosomal alterations are often reported by classical karyotyping 21-26 and were found also by WGS in this study. This suggests that chromosomal “scarring” in FDCS may be a secondary
pathogenic event, a consequence of HRD and further studies are warranted to specifically evaluate the correlation between HRD and chromosomal alterations in this setting. Still, these results prospect the opportunity for patients with unresectable disease to be candidate for PARP inhibitor therapy. Approved for breast, ovarian and recently for prostate cancer, this therapeutical approach, induces replication stall, accumulation of cytotoxic substances and formation of DSB in HRD-mutated cells by preventing the activity of PARP enzymes, leading to selected apoptosis of the neoplastic cells. Notably, the literature reports one single case of unresectable FDCS occurring in a patient carrying BRCA2 germline mutations who reached disease stabilization with PARP inhibitors.43
This could be the first targeted approach to be applied in FDCS, since as confirmed in this study, FDCS lack mutations on genes of the RAS/RAF pathway, thus excluding it from targeted therapies currently applied in histiocytic and dendritic tumors of hematopoietic derivation.7,12,15
Furthermore, the common PDL1 gene and protein expression, detected on FDCS tumor cells in this and in previous studies7,14 was proposed as a potential marker of immune checkpoint inhibition response. However, the low tumor mutational burden found in FDCS, in this and another studies,12 questions on the efficacy of this therapeutical approach.
By applying WGS in FDCS, we could investigate, for the first time, mutational signatures in this rare tumor and identified the occurrence of the germinal center (GC)-related signature SBS9. This may suggest that the neoplastic proliferation of FDC can support the mutational activity typically occurring in the B-follicle GC, even after neoplastic transformation. Alternatively, this may suggest that FDC are a target of aberrant somatic hypermutation machinery in the GC leading to their malignant transformation. This is surprising, as aberrant somatic hypermutation has been identified as leading cause of a variety of GC-derived B-cell lymphomas44,45 but has, to our knowledge, not been demonstrated to occur in FDCS so far. However, expression of activation-induced cytidine deaminase, the enzyme responsible for the somatic hypermutation process, has been well documented in FDC networks.46
When comparing the mutational landscape of FDCS with that of B- and T-cell lymphomas of GC derivation, only one gene, the epigenetic modifier KMT2D, was altered in a significant number of cases, though at low frequency. Whether this is related to aberrant somatic hypermutation or to an incipient mechanism of tumorigenesis may be further explored; however, the association of FDCS with B-cell lymphoma is an extremely rare event.7
Lastly, despite the presence of FDCS with morphological features resembling Castleman disease, including one case developing from a previous HV-CD, the CD-related mutation PDGFRB Asn666Ser was not found in this study, while two novel missense PDGFRB mutations of unknown
significance were detected in two cases, one with HV-CD features. These findings deserve further evaluation. In conclusion, this study, applying massive parallel sequencing in FDCS, confirms its molecular difference from hematopoietic-derived neoplasms, the pivotal role of oncosuppressor genes in its pathobiology and indicates a novel therapeutic approach with PARP inhibition that warrants further investigation for patients with non-resectable FDCS.
Disclosure
No conflicts of interest to disclose.
Contributions
LL and SH designed the study, interpreted the data and wrote the manuscript. TH developed the concept of the study, contributed essential material, revised the manuscript. LM, MS, PB, SL and MB performed experiments on tissue and genomic data, revised the manuscript. WW, MM and CD performed genomic data analysis and revised the
1. Monda L, Warnke R, Rosai J. A primary lymph node malignancy with features suggestive of dendritic reticulum cell differentiation. A report of 4 cases. Am J Pathol. 1986;122(3):562-572.
2. Perez-Ordonez B, Rosai J. Follicular dendritic cell tumor: review of the entity. Semin Diagn Pathol. 1998;15(2):144-154.
3. Pileri SA, Grogan TM, Harris NL, et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology. 2002;41(1):1-29.
4 Chan JKC, Pileri SA, Fletcher CDM, Weiss LM, Grogg KL. Follicular dendritic cell sarcoma. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. 2016. p. 476-478.
5. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140(11):1229-1253.
6. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720-1748.
7 Facchetti F, Simbeni M, Lorenzi L. Follicular dendritic cell sarcoma. Pathologica. 2021;113(5):316-329.
8. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6(2):154-165.
9 Facchetti F, Pileri SA, Lorenzi L, et al. Histiocytic and dendritic cell neoplasms: what have we learnt by studying 67 cases. Virchows Arch. 2017;471(4):467-489.
10 Goyal G, Tazi A, Go RS, et al. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood. 2022;139(17):2601-2621.
11. Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-DorfmanDestombes disease. Blood. 2018;131(26):2877-2890.
manuscript. JM, AB, AA, ISK, JC, EC and SAP contributed with essential material and revised the manuscript. FF and MLH developed the concept of the study, contributed essential material, revised the manuscript.
The authors are grateful to Dr Francesca Filippini for excellent support in database management, Dr Pedro Veiga for creation of circos plot in Figure 1 and Prof William Vermi for fruitful discussion.
Funding
The study was supported by Fondazione Golgi, Brescia (to FF). LL and SL are supported by Fondazione Beretta, Brescia. SH is supported by the Deutsche Forschungsgemeinschaft (grant no. HA6145/7-1).
Data-sharing statement
Original data from sequencing analysis are available in Online Supplementary Tables S1 and S2.
12. Massoth LR, Hung YP, Ferry JA, et al. Histiocytic and dendritic cell sarcomas of hematopoietic origin share targetable genomic alterations distinct from follicular dendritic cell sarcoma. Oncologist. 2021;26(7):e1263-e1272.
13. Nagy A, Bhaduri A, Shahmarvand N, et al. Next-generation sequencing of idiopathic multicentric and unicentric Castleman disease and follicular dendritic cell sarcomas. Blood Adv. 2018;2(5):481-491.
14 Laginestra MA, Tripodo C, Agostinelli C, et al. Distinctive histogenesis and immunological microenvironment based on transcriptional profiles of follicular dendritic cell sarcomas. Mol Cancer Res. 2017;15(5):541-552.
15. Frigola G, Buhler M, Marginet M, et al. MYC and TP53 alterations but not MAPK pathway mutations are common oncogenic mechanisms in follicular dendritic cell sarcomas. Arch Pathol Lab Med. 2022;147(8):896-906.
16. Lorenzi L, Doring C, Rausch T, et al. Identification of novel follicular dendritic cell sarcoma markers, FDCSP and SRGN, by whole transcriptome sequencing. Oncotarget. 2017;8(10):16463-16472.
17 Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37(Web Server issue):W305-311.
18. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24-26.
19. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557-563.
20. Heeke AL, Pishvaian MJ, Lynce F, et al. Prevalence of homologous recombination-related gene mutations across multiple cancer types. JCO Precis Oncol. 2018;2018:PO.17.00286.
21. Sander B, Middel P, Gunawan B, et al. Follicular dendritic cell sarcoma of the spleen. Hum Pathol. 2007;38(4):668-672.
22. Perry AM, Nelson M, Sanger WG, Bridge JA, Greiner TC. Cytogenetic abnormalities in follicular dendritic cell sarcoma: report of two cases and literature review. In Vivo. 2013;27(2):211-214.
23. Udayakumar AM, Al-Bahri M, Burney IA, Al-Haddabi I. Follicular dendritic cell sarcoma: cytogenetics and pathological findings. Sultan Qaboos Univ Med J. 2015;15(3):e411-414.
24. Andersen EF, Paxton CN, O’Malley DP, et al. Genomic analysis of follicular dendritic cell sarcoma by molecular inversion probe array reveals tumor suppressor-driven biology. Mod Pathol. 2017;30(9):1321-1334.
25. Della Porta M, Rigolin GM, Bugli AM, et al. Differentiation of follicular dendritic sarcoma cells into functional myeloiddendritic cell-like elements. Eur J Haematol. 2003;70(5):315-318.
26. Jones D, Amin M, Ordonez NG, Glassman AB, Hayes KJ, Medeiros LJ. Reticulum cell sarcoma of lymph node with mixed dendritic and fibroblastic features. Mod Pathol. 2001;14(10):1059-1067.
27. Alexandrov LB, Kim J, Haradhvala NJ, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94-101.
28. Yaacov A, Vardi O, Blumenfeld B, et al. Cancer mutational processes vary in their association with replication timing and chromatin accessibility. Cancer Res. 2021;81(24):6106-6116.
29. Singh VK, Rastogi A, Hu X, Wang Y, De S. Mutational signature SBS8 predominantly arises due to late replication errors in cancer. Commun Biol. 2020;3(1):421.
30. Meri-Abad M, Moreno-Manuel A, Garcia SG, et al. Clinical and technical insights of tumour mutational burden in non-small cell lung cancer. Crit Rev Oncol Hematol. 2023;182:103891.
31. Pestana RC, Beal JR, Parkes A, Hamerschlak N, Subbiah V. Impact of tissue-agnostic approvals for patients with sarcoma. Trends Cancer. 2022;8(2):135-144.
32. Vyse S, Thway K, Huang PH, Jones RL. Next-generation sequencing for the management of sarcomas with no known driver mutations. Curr Opin Oncol. 2021;33(4):315-322.
33. Lee MY, Bernabe-Ramirez C, Ramirez DC, Maki RG. Follicular dendritic cell sarcoma and its response to immune checkpoint inhibitors nivolumab and ipilimumab. BMJ Case Rep. 2020;13(4):e234363.
34. Chen N, Ba W, Zhao D, Sheng L, Zhang X. Response of tonsil follicular dendritic cell sarcoma to multimodal treatment including pembrolizumab: a case report and literature review.
Front Oncol. 2022;12:816903.
35. Griffin GK, Sholl LM, Lindeman NI, Fletcher CD, Hornick JL. Targeted genomic sequencing of follicular dendritic cell sarcoma reveals recurrent alterations in NF-kappaB regulatory genes. Mod Pathol. 2016;29(1):67-74.
36. Li Z, Lan X, Li C, et al. Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia. 2019;33(4):1035-1038.
37. Hammond D, Loghavi S. Clonal haematopoiesis of emerging significance. Pathology. 2021;53(3):300-311.
38. Jaiswal S. Clonal hematopoiesis and nonhematologic disorders. Blood. 2020;136(14):1606-1614.
39. Facchetti F, Lorenzi L. Follicular dendritic cells and related sarcoma. Semin Diagn Pathol. 2016;33(5):262-276.
40 Laurent C, Meggetto F, de Paiva GR, et al. Follicular dendritic cell tumor of the spleen associated with diffuse large B-cell lymphoma. Hum Pathol. 2008;39(5):776-780.
41. de Leval L, Alizadeh AA, Bergsagel PL, et al. Genomic profiling for clinical decision making in lymphoid neoplasms. Blood. 2022;140(21):2193-2227.
42. Stewart MD, Merino Vega D, Arend RC, et al. Homologous recombination deficiency: concepts, definitions, and assays. Oncologist. 2022;27(3):167-174.
43. Lemech CR, Williams R, Thompson SR, McCaughan B, Chin M. Treatment of breast cancer 2 (BRCA2)-mutant follicular dendritic cell sarcoma with a poly ADP-ribose polymerase (PARP) inhibitor: a case report. BMC Res Notes. 2016;9:386.
44 Pasqualucci L, Bhagat G, Jankovic M, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40(1):108-112.
45. Migliazza A, Martinotti S, Chen W, et al. Frequent somatic hypermutation of the 5’ noncoding region of the BCL6 gene in B-cell lymphoma. Proc Natl Acad Sci U S A. 1995;92(26):12520-12524.
46. Bombardieri M, Barone F, Humby F, et al. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjogren’s syndrome. J Immunol. 2007;179(7):4929-4938.
C. Cameron Yin,1 Wayne Tam,2 Serena M. Walker,3 Amandeep Kaur,4 Madhu M. Ouseph,5 Wei Xie,6 Olga K. Weinberg,7 Peng Li,8 Zhuang Zuo,1 Mark J. Routbort,1 Simon Chen,3 L. Jeffrey Medeiros,1 Tracy I. George,8 Attilio Orazi,9 Daniel A. Arber,4 Adam Bagg,3 Robert P. Hasserjian10 and Sa A. Wang1
1Department of Hematopathology, University of Texas MD Anderson Cancer Center, Houston, TX; 2Division of Hematopathology, Department of Pathology and Laboratory Medicine, Donald and Barbara Zucker School of Medicine, Hofstra/Northwell, Greenvale, NY; 3Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA; 4Department of Pathology, University of Chicago, Chicago, IL; 5Department of Pathology and Laboratory Medicine, Weill Cornell Medical Center, New York, NY; 6Department of Pathology & Laboratory Medicine, Oregon Health & Science University, Portland, OR; 7Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX; 8Department of Pathology, University of Utah, Salt Lake City, UT; 9Department of Pathology, Texas Tech University, El Paso, TX and 10Department of Pathology, Massachusetts General Hospital, Boston, MA, USA
Correspondence: S.A. Wang Swang5@mdanderson.org
C.C. Yin cyin@mdanderson.org
Received: September 17, 2023. Accepted: November 9, 2023. Early view: November 16, 2023. https://doi.org/10.3324/haematol.2023.284311
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
STAT5B has been reported as a recurrent mutation in myeloid neoplasms with eosinophilia, but its overall frequency and importance across a spectrum of myeloid neoplasms are largely unknown. We conducted a multicenter study on a series of 82 myeloid neoplasms with STAT5B mutations detected by next-generation sequencing. The estimated frequency of STAT5B mutations in myeloid neoplasms was low, <0.5%, but mutations were detected in all categories of such neoplasms, including myelodysplastic syndrome (MDS, 28%), acute myeloid leukemia (AML, 26%), myelodysplastic/myeloproliferative neoplasm (MDS/MPN, 18%), Philadelphia chromosome-negative classic MPN (12%), systemic mastocytosis (1%), and, with a notably high frequency, chronic eosinophilic leukemia, not otherwise specified (CEL-NOS, 15%). STAT5B mutations occurred preferentially in the SH2 domain (95%), involved 12 different codons, with the N642H hotspot being the most common (78%). Co-mutations were present in all cases and clonal hierarchy analysis showed that STAT5B mutations tended to be subclonal in AML, MPN, and MDS, but frequently dominant/co-dominant in CEL-NOS (83%), followed by MDS/MPN (40%). Across the group, eosinophilia and/or basophilia were common (41%), frequently observed in cases in which STAT5B mutations were detected at initial diagnosis (P<0.0001), with a high variant allele frequency (median 42.5%, P=0.0001), as a dominant/co-dominant clone (P<0.0001), involving the canonical N642H (P=0.0607), and associated with fewer co-mutations (P=0.0009). Our data show that the characteristics and importance of a STAT5B mutation differ among myeloid neoplasms, but if present as a dominant mutation and detected at initial diagnosis, it appears to be a driver mutation in a subgroup of chronic myeloid neoplasms, preferentially promoting a proliferation of eosinophils and basophils.
STAT5 is a key component of cytokine-induced signal transduction cascades, and a critical downstream mediator of transformation by oncogenic tyrosine kinases. STAT5, encoded by STAT5A and STAT5B located at chromosome 17q21.2, is fundamental for myelopoiesis, lymphoid development, macrophage functions and megakaryopoiesis, as well as basophil,
eosinophil, and mast cell functions.1 STAT5A/B activation, in most cases, is induced by upregulated function of upstream tyrosine kinases, e.g. JAK2 V617F, BCR::ABL1, FLT3-ITD, or KIT D816V.2 In humans, while STAT5A mutations have rarely been implicated in causing disease, STAT5B mutations have been linked to deregulated protein signaling and function, and hyperactivation of STAT5B3 and gain-of-function STAT5B mutations4 are associated with the development of various
hematolymphoid malignancies.
STAT5B mutations are mostly reported in T/NK-cell neoplasms, including large granular lymphocytic leukemia (CD4+ type), T-prolymphocytic leukemia, hepatosplenic T-cell lymphoma and T-lymphoblastic leukemia/lymphoma.5-9
STAT5B mutations occur mainly in the SH2 domain, with N642H being the most common. N642H is close to the phosphotyrosine-binding loop of STAT5B, and this mutation stabilizes STAT5 dimers, leading to prolonged pY-STAT5 levels and increased phosphotyrosine levels upon cytokine stimulation.10 STAT5B mutations are uncommon in myeloid neoplasms. Recently, Cross et al.11 reported STAT5B N642H in 27 of 1715 (1.6%) cases of myeloid neoplasms with eosinophilia, including seven cases with a presumed diagnosis of hypereosinophilic syndrome. It has been wondered whether STAT5B N642H is a recurrent mutation in myeloid neoplasms which may represent a specific subset of chronic myeloid neoplasms with eosinophilia. We conducted this multicenter study with three aims. First, we sought to understand the spectrum of myeloid neoplasms with STAT5B mutations. Second, we assessed the characteristics of STAT5B mutations looking for possible correlations with disease phenotype. Lastly, we examined whether STAT5B-mutated myeloid neoplasms represent a unique entity with distinct clinicopathological features.
Study group
We searched the database of eight institutions for myeloid neoplasms with STAT5B mutations tested by next-generation sequencing. Clinical and laboratory data were retrieved from the medical records. A potential concomitant T/NK-cell neoplasm carrying STAT5B mutation, including lymphocyte variant hypereosinophilic syndrome, was excluded by morphological examination, flow cytometry immunophenotyping and/or TCR gene rearrangement and, most importantly, clinical follow-up. The study was conducted according to Institutional Review Board-approved protocols of all participating institutions and in accordaancewith the Declaration of Helsinki.
Morphological evaluation
Wright-Giemsa-stained peripheral blood (PB) and bone marrow (BM) aspirate smears/touch imprints, as well as hematoxylin and eosin-stained BM clot and core biopsy specimens were reviewed and assessed for percentage of blasts, eosinophils and basophils, as well as morphological dysplasia and fibrosis. Cytochemical stains for myeloperoxidase, iron staining and histochemical stains for reticulin and collagen were performed using standard methods. The grade of myelofibrosis (MF) was assessed based on the criteria of the European Consensus on the grading of BM fibrosis.12
Immunophenotypic studies
Flow cytometry immunophenotyping was performed. The panels varied at different institutions, with the basic markers including CD2, CD3 (surface and cytoplasmic), CD4, CD5, CD7, CD13, CD14, CD15, CD19, CD25, CD33, CD34, CD36, CD38, CD45, CD56, CD64, CD117, CD123, CD133, HLA-DR, myeloperoxidase and TdT.
Cytogenetic analysis
Conventional chromosomal analysis was performed on G-banded metaphases prepared from unstimulated 24-hour and 48-hour BM cultures. Twenty metaphases were analyzed, and the results were reported using the International System for Human Cytogenetics Nomenclature, 2020.
Genomic DNA was amplified by polymerase chain reaction and subjected to mutation analysis by next-generation sequencing. The gene panels varied among different institutions, but all panels assessed for common mutations associated with myeloid neoplasms. STAT5B was assessed in all cases and the entire coding region was covered, with a limit of detection at a variant allele frequency (VAF) of 1%. Most cases were tested using an 81-gene panel that included ANKRD26, ASXL1, ASXL2, BCOR, BCORL1, BRAF, BRINP3, CALR, CBL, CBLB, CBLC, CEBPA, CREBBP, CRLF2, CSF3R, CUX1, DDX41, DNMT3A, EED, ELANE, ETNK1, ETV6, EZH2, FBXW7, FLT3, GATA1, GATA2, GFI1, GNAS, HNRNPK, HRAS, IDH1, IDH2, IKZF1, IL2RG, IL7R, JAK1, JAK2, JAK3, KDM6A, KIT, KMT2A, KRAS, MAP2K1, MPL, NF1, NOTCH1, NPM1, NRAS, PAX5, PHF6, PIGA, PML, PRPF40B, PTEN, PTPN11, RAD21, RARA, RUNX1, SETBP1, SF1, SF3A1, SF3B1, SH2B3, SMC1A, SMC3, SRSF2, STAG1, STAG2, STAT3, STAT5A, STAT5B, SUZ12, TERC, TERT, TET2, TP53, U2AF1, U2AF2, WT1, and ZRSR2. When a mutation had a VAF of ≥5%, and this VAF was within a 10% difference from the mutation with the highest VAF, we refer to the mutation as a dominant clone.13
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9. The association between categorical variables was examined using Fisher exact and Pearson χ2 square tests. The association between continuous variables was examined using the Student t test. Overall survival was calculated from the date of initial diagnosis to the date of death or last follow-up. Survival was analyzed using the Kaplan-Meier method and compared using the log-rank test. Differences between groups were considered statistically significant if P values were <0.05 in a two-tailed test.
STAT5B mutations occur in a wide spectrum of myeloid neoplasms
We identified a total of 82 cases of myeloid neoplasms
with STAT5B mutations with a VAF of ≥1%. Based on the total number of myeloid neoplasms tested by the same next-generation sequencing panel at one institution (MD Anderson Cancer Center) during the study period, we estimate that the frequency of STAT5B mutations across all myeloid neoplasms is below 0.5%. STAT5B mutations were detected at the time of initial diagnosis in 45 patients and acquired later in the course of disease in 20 patients. Initial diagnostic material was not available for assessment of STAT5B mutation in 17 patients. Of the 20 patients who acquired STAT5B mutations later in the disease course, the emergence of STAT5B was often accompanied by disease progression with an increased blast count (2 myelodysplastic syndrome [MDS] and 1 chronic myelomonocytic leukemia [CMML]) or leukemic transformation (6 acute myeloid leukemia [AML] from MDS), or relapse (2 AML).
The study group included 56 men and 26 women, with a median age of 72.7 years (range, 26.2-88.0). The cases occurred across a broad spectrum of myeloid neoplasms including MDS (n=23), AML (n=21), myelodysplastic/myeloproliferative neoplasm (MDS/MPN) (n=15), chronic eosinophilic leukemia, not otherwise specified (CEL-NOS, n=12), Philadelphia chromosome (Ph)-negative classic myeloproliferative neoplasms (MPN) (n=10), and aggressive systemic mastocytosis (SM) with eosinophilia (n=1) (Table 1). The AML group was heterogeneous but remarkably enriched by AML with myelodysplasia-related gene mutations (15/21, 71%). Genes mutated in this latter category included RUNX1 (n=8), ASXL1 (n=6), SRSF2 (n=5), U2AF1 (n=5), STAG2 (n=2) and ZRSR2 (n=1); eight cases had more than one of these mutations. Among patients with MDS/MPN, CMML was predominant (n=12, 80%). One of the two cases of MDS/MPN, NOS showed persistent eosinophilia (>10%) but slightly under 1.5x109/L (criteria for CEL-NOS). Among Ph-negative classic MPN, a case of triple-negative primary myelofibrosis (PMF) showed marked eosinophilia (>2.5x109/L) and basophilia (>1.0x109/L). The diagnoses and classifications of the patients are summarized in Table 1, and clinicopathological features are summarized in Table 2.
Morphological findings in STAT5B-mutated myeloid neoplasms
The morphological findings reported here were from the first available BM samples in which a STAT5B mutation was detected. The BM biopsy specimens were overall hypercellular (median, 80%; range, 10-100%). Dysplasia was common in all disease categories, with multilineage dysplasia (involving ≥2 lineages) observed in 12/15 (80%) patients with MDS/MPN, 18/23 (78%) with MDS, 8/13 (62%) with AML and 6/12 (50%) of those with CEL-NOS. Increased ring sideroblasts (>5%) were uncommon, being found in three cases of MDS, two cases each of AML and CEL-NOS, and one case of polycythemia vera. In addition to AML, increased blasts (≥5%) were present in 9/15 (60%) MDS/MPN, 12/23 (52%) MDS, 4/10 (40%) MPN, and 4/12 (33%) CEL-NOS patients. Significant myelofibrosis (≥ MF-2) was observed in 16/71 (23%) cases, most frequently
Table 1. Disease categories of 82 cases of myeloid neoplasms with STAT5B mutation.
AML with mutated NPM1
AML with bi-allelic CEBPA mutation
AML with myelodysplasia-related mutations
(including 1 triple-negative PMF)
MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; NOS: not otherwise specified; MPN: myeloproliferative neoplasm; PMF: primary myelofibrosis; PV: polycythemia vera; ET: essential thrombocythemia; MDS/MPN: myelodysplasia/myeloproliferative neoplasm; CEL: chronic eosinophilic leukemia.
in MPN (6/9, 67%), and less often in CEL-NOS (2/10, 20%), MDS (4/22, 18%), MDS/MPN (2/13, 15%), and AML (2/17, 12%).
The morphological findings from representative CEL-NOS cases are illustrated in Figure 1.
Eosinophilia, defined as an absolute eosinophil count of >0.5x109/L and ≥6% eosinophils in PB, was present in 27 (33%) patients (12 CEL-NOS, 6 MDS/MPN, 4 MDS, 3 MPN, 1 AML and 1 aggressive SM). Basophilia, defined as having an absolute basophil count of ≥0.2 x109/L and ≥2% basophils in PB,14 was identified in 20 (24%) patients (7 CEL-NOS, 5 MPN, 5 MDS/MPN and 3 MDS). Thirteen (16%) patients had both eosinophilia and basophilia. Increased BM eosinophils (≥6%) were observed in 17 (21%) patients, and increased BM basophils (≥2%) in 10 (12%) patients.
Other than cases classified as CMML, absolute and relative monocytosis were only observed in two cases of AML with monocytic differentiation. Relative monocytosis (≥10%) but not absolute monocytosis (≥1.0 x109/L) was present in four MDS, one PMF and none of the CEL-NOS.
Mast cells were not systemically evaluated in this study. The case of aggressive SM had large aggregates of mast cells meeting the major criteria for SM, with aberrant CD25 expression, and associated with eosinophilia. One case of CEL-NOS had increased scattered spindle mast cells with aberrant CD25 co-expression (Figure 1). Mast cells in two other cases of CEL-NOS were evaluated by flow cytometry
Table 2. Clinicopathological features of cases of myeloid neoplasms with a STAT5B mutation.
PB eosinophils >5%, N (%)
LDH, N/N
grade ≥2, N/N (%)
The single case of aggressive systemic mastocytosis is not included in this table. One patient with chronic eosinophilic leukemia, not otherwise specified had an absolute eosinophil count of 1,174 x109/L due to steroid treatment before the testing; subsequent testing showed hypereosinophilia. MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; MPN: myeloproliferative neoplasm; MDS/MPN: myelodysplasia/myeloproliferative neoplasm; CEL-NOS: chronic eosinophilic leukemia, not otherwise specified; WBC: white blood cells; AEC: absolute eosinophil count; PB: peripheral blood; ABC: absolute basophil count; LDH: lactate dehydrogenase; FU: follow-up; M: male; F: female.
and were negative for CD2, CD25 and CD30.
Cytogenetic findings of STAT5B-mutated myeloid neoplasms
Forty-six of 80 (58%) patients had a normal karyotype, 27 (34%) had a simple abnormal karyotype defined as one or two chromosomal aberrations, and seven (9%) had a complex karyotype with three or more chromosomal aberrations (Table 3). Karyotype was not available for two patients (1 AML and 1 MPN). The most common karyotypic aberrations included trisomy 8 (n=9, 11%), del7/del(7q) (n=7, 9%), del(20q) (n=6, 8%), del(11q) (n=4, 5%), del5/del(5q) (n=2, 5%), and del17/ del(17p) (n=2, 3%). There was no rearrangement of PDGFRA, PDGFRB, FGFR1, or JAK2 in cases which were assessed for these rearrangements.
Molecular characterization
STAT5B mutations were clustered in the SH2 domain in 78 (95%) cases and were missense mutations in all cases. In four (5%) remaining patients, mutations occurred in the transactivation domain (n=3) and the coiled coil domain (n=1). A total of 12 different mutations were detected, with N642H being the most common hotspot, in 64 (78%) cases, followed by Y665F (n=6, 7%), T628S (n=3, 4%) and E637K
(n=2, 2%). Other mutation codons (n=8) were observed in single cases (Online Supplementary Table S1). One patient with CEL-NOS had two STAT5B mutations, one in the SH2 domain, Y665F, and the other in the DNA binding domain, D428N. The location of STAT5B mutations is shown in Figure 2. Interpretation of non-canonical (non-N642H) STAT5B mutations, including their distribution in different disease entities, if they have been reported in the Catalogue of Somatic Mutations in Cancer (COSMIC) database, confirmed as somatic or activating mutations, and disease types in which they were reported previously, are provided in Online Supplementary Table S2. The VAF of STAT5B mutations ranged from 1.1-81.2% (median, 22.5%), with 62 (76%) cases having a VAF ≥5%.
We also evaluated the mutational profile of other genes included in the next-generation sequencing panels (Table 3). The presence of a STAT5B mutation was accompanied by other gene mutations (referred to as “co-mutations”) in all cases, with a median of four co-existing mutations per case (range, 2-10). A heatmap illustrating the co-existing mutations is shown in Figure 3. Overall, the most frequent concurrent mutations were ASXL1 and TET2, each in 29 (35%) cases, followed by SRSF2 (n=23, 28%), U2AF1 (n=19, 23%), RUNX1 (n=16, 20%) and DNMT3A (n=14, 17%). In MDS,
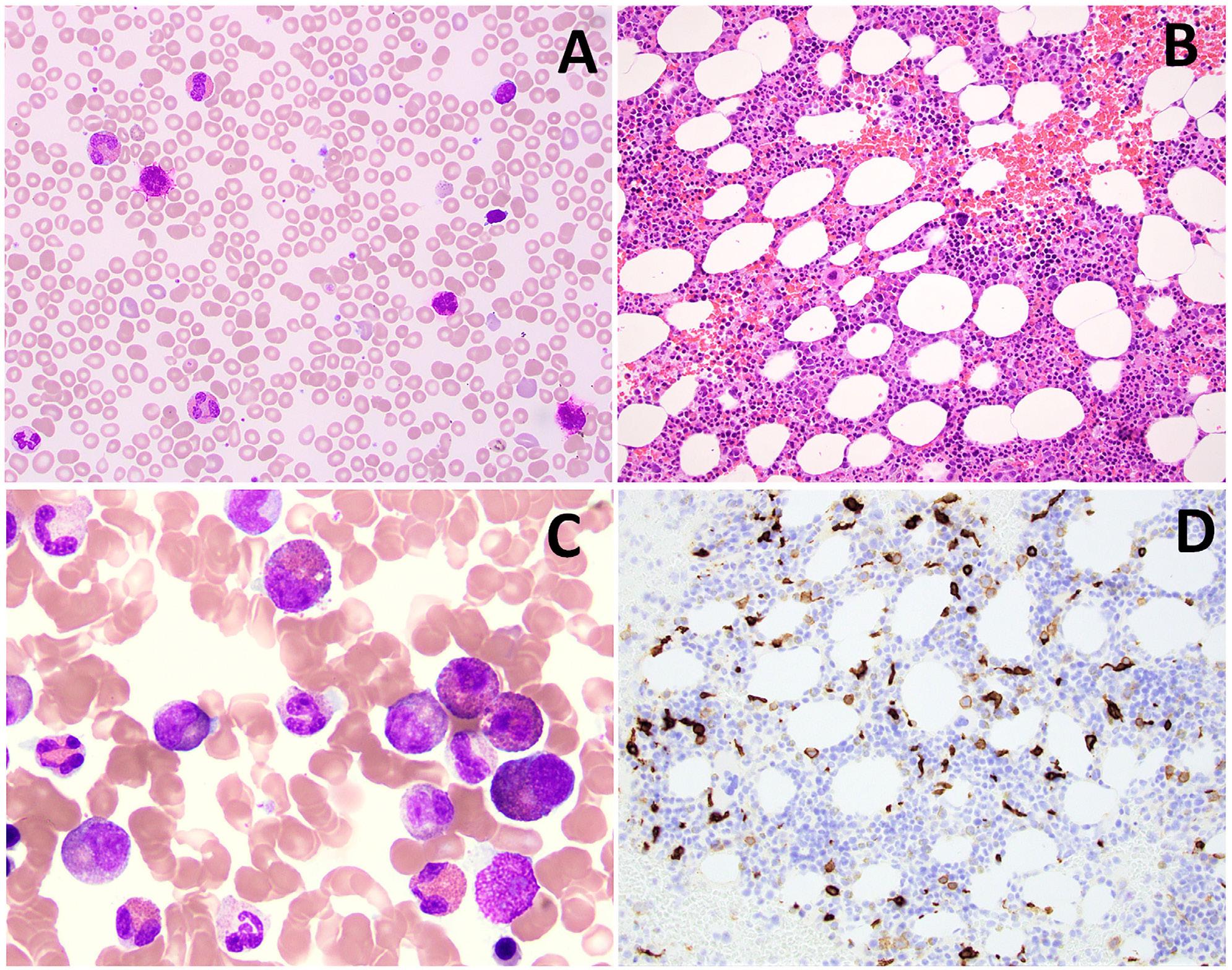
Figure 1. A representative case of chronic eosinophilic leukemia, not otherwise specified. (A) A peripheral blood smear shows eosinophilia and basophilia (Wright-Giemsa, 500x). (B) The bone marrow biopsy is hypercellular with small hypolobated dysplastic megakaryocytes (hematoxylin and eosin, 200x). (C) A bone marrow aspirate smear shows increased eosinophils and precursors, some intermediate-stage eosinophils with eo-basophilic granules (Wright-Giemsa, 500x). (D) CD117 highlights increased scattered spindle mast cells (200x). The mast cells were positive for CD25 by flow cytometry (not shown).
Table 3. Cytogenetic and molecular features of cases of myeloid neoplasms with STAT5B mutation.
Cytogenetics, N (%)
Normal or –Y only
Complex or -7/-7q, -17/-17p, -5/-5q All others
STAT5B detection, N (%)
diagnosis
MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; MPN: myeloproliferative neoplasm; MDS/MPN: myelodysplastic/myeloproliferative neoplasm; CEL-NOS: chronic eosinophilic leukemia, not otherwise specified; VAF: variant allele frequency.
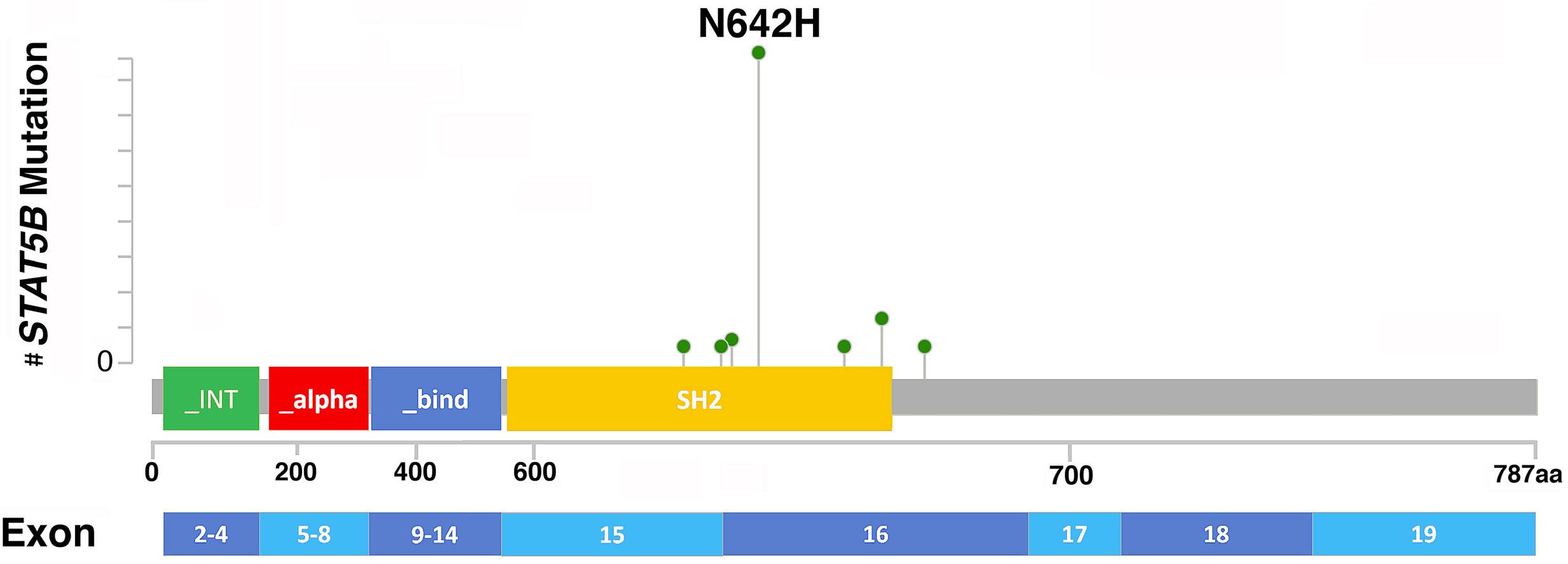
Figure 2. The location and number of STAT5B mutations. In total, 83 mutations were identified in 82 patients. One patient with chronic eosinophilic leukemia, not otherwise specified had two concurrent mutations, Y665F and D428N. STAT5B mutations clustered in the SH2 domain in 78 (95%) cases and were missense mutations in all cases, with N642H being the most common, seen in 64 (78%) cases. aa: amino acids. (Note: the number of amino acids on the X-axis is not proportionally scaled).
the most frequent co-existing mutation, present in 12/23 (52%) cases, was U2AF1, followed by ASXL1 (n=11) and TET2 (n=8). In AML, RUNX1 and TET2 were each mutated in 8/21 (38%) cases, followed by ASXL1 (n=6). In MPN, SRSF2 was mutated in 7/10 (70%) cases, followed by ASXL1 (n=3), CALR (n=3), JAK2 (n=3) and RUNX1 (n=3). In MDS/MPN, ASXL1 and TET2 mutations each occurred in 8/15 (53%) cases, followed by SRSF2 (n=5) and PHF6 (n=4) mutations. Cases of CELNOS had the lowest number of concurrent mutated genes (median 2, range 2-4), with SF3B1 mutation being most common in 4/12 (33%) cases, followed by SRSF2 (n=3) and TET2 (n=3). STAT5B mutations represented the dominant or co-dominant clone in 30 (37%) cases and a subclone in the remaining 52 (63%) cases (Table 3). Of note, in 18/20 (90%) cases in which STAT5B mutations were acquired during the course of disease, the STAT5B mutations were subclonal. Among 18 cases with mutations in codons other than N642H, 15 (83%) were non-dominant and three were co-dominant; eight (44%) were detected at initial diagnosis, four were acquired and six were unknown.
Comparison of STAT5B-mutated myeloid neoplasms with and without eosinophilia and/or basophilia
Twenty-seven (33%) patients had PB eosinophilia, 20 (24%) PB basophilia and 13 (16%) had both. Cases with eosinophilia and/or basophilia (n=34) included 12 CEL-NOS, eight MDS/MPN, seven MDS, five MPN, one AML and the case of SM. Compared to the remaining cases, cases with eosinophilia and/or basophilia tended to have STAT5B mutations detected at initial diagnosis (27/28 vs. 18/37; P<0.0001), more commonly had the canonical N642H mutation (30/34 vs. 34/48; P=0.0607), and had a higher VAF (42.5% vs. 9.8%; P=0.0001). In addition, cases with eosinophilia and/ or basophilia more often had a VAF ≥5% (30/34 vs. 32/48; P=0.0250), the mutation was a dominant or co-dominant clone (24/34 vs. 6/48; P<0.0001), and were associated
with fewer concurrently mutated genes (4 vs. 5, P=0.0009) (Table 4).
By definition, all 12 cases of CEL-NOS had hypereosinophilia, both relative (≥10%) and absolute (≥1.5x109/L).15,16 Of note, 7/12 (58%) CEL-NOS cases also had PB basophilia. The STAT5B mutations were detected at initial diagnosis in all CEL-NOS patients, with a median VAF of 44.2% (range, 7-78.5%), being a dominant clone in 10 (83%) cases. The mutation involved the N642H hotspot in 11 (92%) CELNOS cases, while one case had two mutations, Y665F and D428N.
Clinical outcome of patients with STAT5B-mutated myeloid neoplasms
Among 76 patients with treatment information available, all received disease- and risk-adapted therapies with the exception of three patients with MDS, one with CMML and one with CEL-NOS who were observed. Thirteen patients underwent allogeneic stem cell transplantation, of whom four had MDS, three had AML, three had CMML, and one each had PMF, CEL-NOS, and MDS/MPN-unclassified. With a median follow-up of 18 months (range, 1-101.5 months), AML transformation occurred in 4/12 (33%) CEL-NOS, 3/15 (20%) MDS/MPN, and 4/23 (17%) MDS patients. At the end of follow-up, 41 patients had died of disease, 17 had persistent disease, and eight had achieved complete remission, including four with MDS, two with AML, and two CMML. Fourteen patients were alive with unknown disease status. Two patients were lost to follow-up (Figure 4, Table 2).
In this study, we show that the overall frequency of STAT5B mutations in myeloid neoplasms, estimated from one of
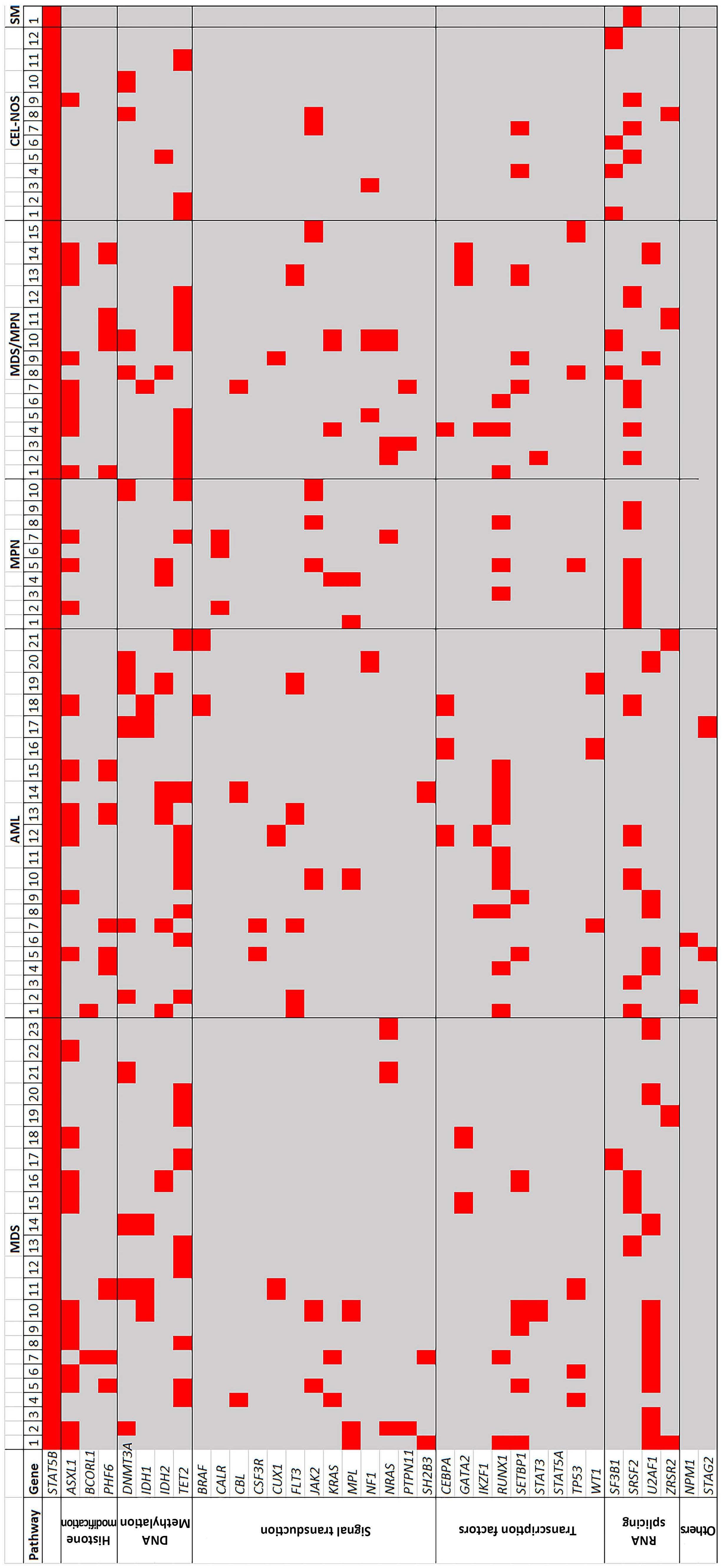
Figure 3. The STAT5B mutation profile heatmap. The cases were divided into six categories: myelodysplastic syndrome, acute myeloid leukemia, myeloproliferative neoplasm, myelodysplasia/myeloproliferative neoplasm, chronic eosinophilic leukemia, not otherwise specified, and systemic mastocytosis. Each column represents a single case. Only mutations seen in two or more cases are listed. Overall, the most frequent concurrent mutations were ASXL1 and TET2 , followed by SRSF2, U2AF1 , RUNX1 , and DNMT3A . MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; MPN: myeloproliferative neoplasm; MDS/MPN: myelodysplasia/myeloproliferative neoplasm; CEL-NOS: chronic eosinophilic leukemia, not otherwise specified; SM: systemic mastocytosis.
Table 4. Comparison between STAT5B-mutated myeloid neoplasms with and without eosinophilia/basophilia.
Eosinophilia and/or basophilia, N=34
No eosinophilia or basophilia, N=48 P
Diagnosis, N
MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; MPN: myeloproliferative neoplasm; MDS/MPN: myelodysplastic/myeloproliferative neoplasm; CEL-NOS: chronic eosinophilic leukemia, not otherwise specified; SM: systemic mastocytosis; VAF: variant allele frequency.
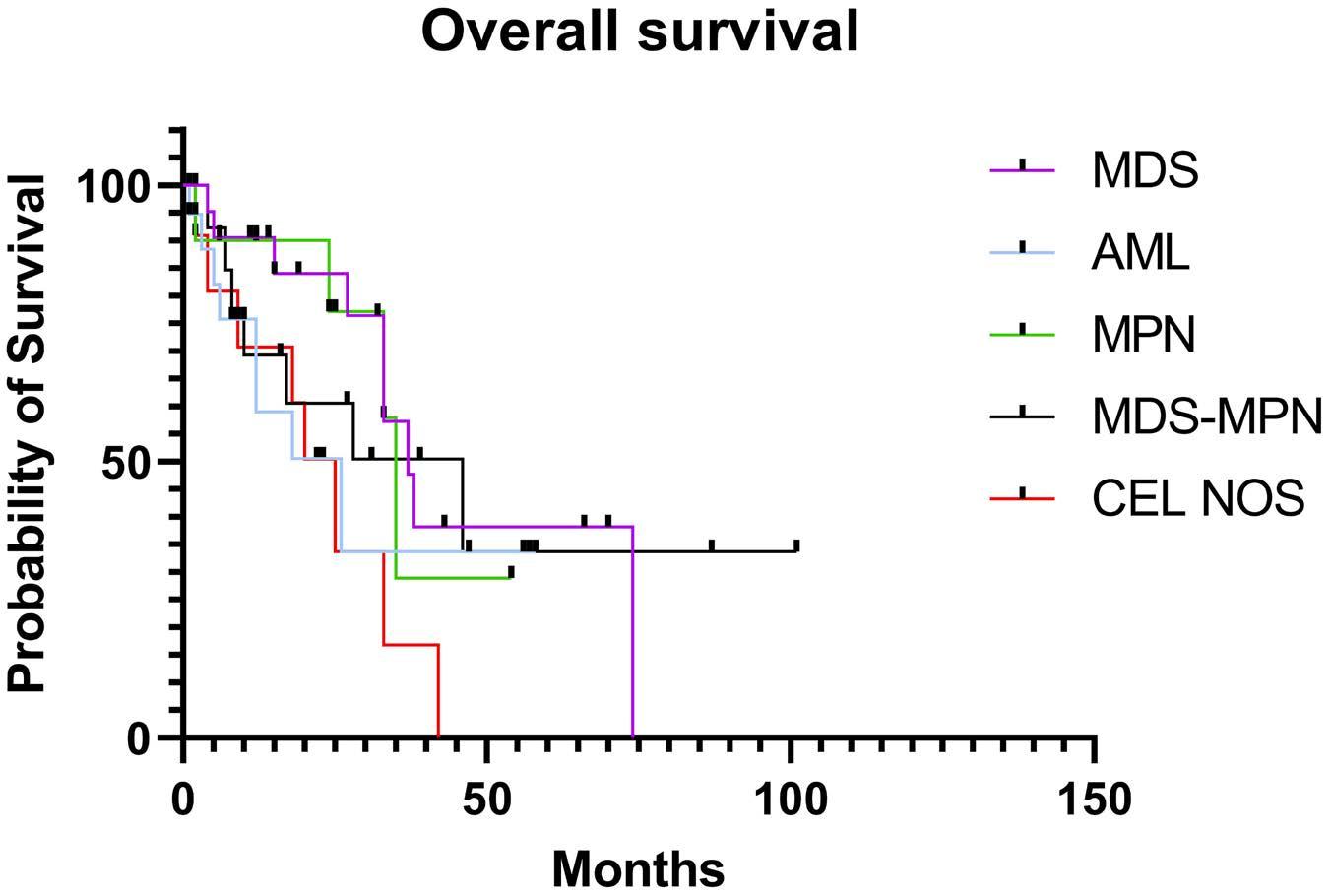
Figure 4. Overall survival of cases of STAT5B-mutated myeloid neoplasms based on disease categories. MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; MPN: myeloproliferative neoplasm; MDS-MPN: myelodysplastic/myeloproliferative neoplasm; CEL-NOS: chronic eosinophilic leukemia, not otherwise specified.
the participating institutions, is low, being below 0.5%.
This low frequency was also reported in a study by Umrau and colleagues who detected STAT5B mutations in five of 2,266 (0.22%) myeloid neoplasms including three AML, one MDS and one CMML.17 With this large cohort of cases, we show that STAT5B mutations are not confined to a specific myeloid disease category, occur most frequently at codon N642H, and are frequently associated with eosinophilia and/ or basophilia.
Unlike the study by Cross and colleagues,11 which was focused on the STAT5B N642H hotspot mutation in eosinophilic disorders, we included all myeloid neoplasms tested by
next-generation sequencing panels that covered the entire coding region of STAT5B. We showed that all STAT5B mutations were missense, predominantly occurring in the SH2 domain (95%), with occasional cases involving the transactivation domain and the coiled coil domain. A total of 12 different mutations were detected, with N642H being the most common, occurring in nearly 80% of cases, followed by Y665F (7%), T628S (4%), and one each of the others. STAT5B Y665F, the second most frequent mutation, was not detected in MDS or AML, but only seen in chronic myeloid neoplasms with proliferative features including Ph-negative classic MPN, MDS/MPN and CEL-NOS. On the other hand, except for D428N as a second STAT5B mutation in a case of CEL-NOS, other rare mutations, including T628S, were only found in MDS and AML cases. The Y665F, a somatic and activating mutation, has been reported in an aggressive variant of CD8+ T-cell large granular lymphocytic leukemia,6 and T628S, a somatic mutation but of unknown status for activating, was enriched in CD4+ large granular lymphocytic leukemia5 and T-prolymphocytic leukemia.18 T628S and V712E were previously reported in one case of MDS with ring-sideroblasts.17 Notably, these non-N642H STAT5B mutations mostly occurred as a non-dominant clone (83%) and, except for Y665F and I704L, the activating status is largely unknown (Online Supplementary Table S2); their pathogenic significance needs further studies. These findings indicate likely genotype-phenotype correlations between specific STAT5B mutation sites and affected codons and disease subtypes, but the phenomenon is probably confounded by dominant versus subclonal mutations of STAT5B STAT5B mutations can present at the time of diagnosis or be acquired during the course of disease. The proportion of STAT5B mutations detected at initial diagnosis was highest
in CEL-NOS (100%), followed by MDS/MPN in about 80% and less than 50% in AML. The median VAF of STAT5B mutations varied among different disease subtypes, being highest in CEL-NOS, followed by MDS and MDS/MPN, and lowest in Ph-negative classic MPN and AML. Concurrent gene mutations were found in all cases, but the genes mutated differed by disease categories, as expected. A similar high frequency of co-mutations with STAT5B was described in cases of myeloid neoplasms with eosinophilia,11,19 and five cases of myeloid neoplasms reported by Umrau and colleagues.17 STAT5B was a dominant or co-dominant clone in slightly over one third of the cases and was very common in CEL-NOS (>80%), followed by MDS/MPN (~40%) and lowest in AML and Ph-negative classic MPN (≤20%). Furthermore, CEL-NOS had the lowest number of concurrent gene mutations. These findings suggest that STAT5B is likely a driver gene in CEL-NOS. One of the prominent clinical features of myeloid neoplasms with STAT5B mutations was associated eosinophilia and/or basophilia, observed in nearly half of the cases, resulting in a diagnosis of CEL-NOS in 15% cases. Of note, CEL-NOS is extremely rare. According to the Surveillance, Epidemiology, and End Results (SEER) data between 2004 and 2015, CEL-NOS together with myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusion (M/LN-eo-TK) and some idiopathic hypereosinophilic syndromes occurred in only 0.4 persons per 1,000,000 population20 while the incidence of AML, MDS and MPN each was 30-40 cases per million, and CMML 4 cases per million.21 In our previous study, only 21 cases of CEL-NOS were identified in a period of 13 years, an incidence significantly below 1% of all myeloid neoplasms.22 In addition to eosinophilia, basophilia was also common, co-existing with eosinophilia in more than half of the cases of CEL-NOS. Notably, basophilia was extremely uncommon in previously published series of CEL-NOS, although the STAT5B mutation status was mostly unknown in those studies.23-25 Interestingly, basophilia was mentioned in the series of cases of eosinophilia with STAT5B mutations reported by Cross et al.11 but the authors did not provide a frequency. Other than CEL-NOS, in this case series, eosinophilia and/or basophilia was observed in around half of MDS/MPN and Ph-negative classic MPN cases but was uncommon in MDS and AML. Eosinophilia/basophilia was significantly associated with STAT5B N642H hotspot mutations, mutations detected at initial diagnosis, with a high VAF, as a dominant or co-dominant clone and having fewer co-mutations. In addition to its function in eosinophils, STAT5 is known to be critical for basophil and mast cell differentiation and maintenance through the STAT5-GATA2 pathway.26 We only had one case presenting as SM in which STAT5B N642H was detected as a dominant mutation at initial diagnosis. This case was associated with eosinophilia and lacked a KIT mutation, raising the differential diagnosis
of an eosinophilic myeloid neoplasm associated with a mast cell proliferation. One case of CEL-NOS showed increased scattered spindle mast cells with aberrant CD25 expression. The question of whether mast cell proliferation is also a common feature of STAT5B mutation, as that seen in M/LN-eo-TK,27 will require a systemic evaluation of mast cells in these cases. Aside from this case, there were several cases blurring the boundaries of classifications. A patient with triple-negative PMF showed marked eosinophilia (>2.5x109/L) and basophilia (>1.0x109/L), and a case of MDS/MPN, NOS showed persistent eosinophilia (>10%) although the count was slightly under 1.5x109/L. These cases all had a STAT5B N642H mutation detected at initial diagnosis, with a high VAF (>40%) and as a dominant clone. We question whether these cases are better considered within the spectrum of CEL-NOS given the context of STAT5B mutations.
In summary, STAT5B mutations occur across a wide spectrum of myeloid neoplasms, but show different mutational characteristics among different subtypes. In CEL-NOS, STAT5B mutations were frequently detected at initial diagnosis, with a high VAF, as a dominant clone, involving the canonical N642H hotspot, and associated with fewer co-mutations. In contrast, in MDS and AML, STAT5B mutations were more frequently present at a low VAF and as a subclone, were more likely acquired in the course of disease, often involved non-canonical mutations (i.e., other than N642H), and were usually not associated with significant eosinophilia or basophilia; a minority of MDS cases may demonstrate or develop relative eosinophilia and/or basophilia during the disease course. In Ph-negative classic MPN in which the disease phenotype is dictated by MPN-driver CALR/MPL/JAK2 mutations, STAT5B mutations preferentially occurred in PMF or fibrotic stages of polycythemia vera and essential thrombocythemia, often as a subclone. Eosinophilia and/or basophilia was seen in about half of these MPN patients, but it is difficult to attribute this entirely to STAT5B mutations because of the inherent association of increased eosinophils and/ or basophils in the fibrotic stage of MPN. These data suggest that STAT5B mutation is unlikely to be a driver in MDS, AML, and MPN with canonical JAK2/CALR/MPL mutations. STAT5B mutation features in cases classified as MDS/MPN were closer to CEL-NOS than to MDS, AML and MPN, although the median VAF was lower and STAT5B was less frequently a dominant or co-dominant clone than in CEL-NOS. It is known that STAT5B is a strong oncogenic driver in T-cell malignancies through its effect of enhancing phospho-Tyr:SH2 domain interactions and escaping negative regulatory phosphatase attack.1 We believe that STAT5B mutation in myeloid neoplasms, if occurring as a dominant clone at the time of diagnosis, is likely a driver mutation in a subgroup of chronic myeloid neoplasms lacking other genetic drivers, preferentially promoting the proliferation of eosinophils, basophils and possibly mast
cells. It will be of interest for future studies to examine whether STAT5B mutations may identify a unique subset of CEL-NOS cases with distinctive clinicopathological features. Further research is warranted to determine whether STAT5B-mutated cases currently classified as MDS/MPN or MPN (lacking canonical JAK2/CALR/MPL driver mutations) may be biologically related to STAT5B-mutated CEL-NOS and may be more appropriately classified together as a novel molecularly defined entity. As tyrosine kinase inhibitors 28 or STAT5 inhibitors are being evaluated in pre-clinical models,1 with potential future development of novel therapeutic strategies, STAT5B mutations may help to genetically define those chronic myeloid neoplasms that may benefit from targeted therapy.
1. de Araujo ED, Erdogan F, Neubauer HA, et al. Structural and functional consequences of the STAT5B(N642H) driver mutation. Nat Commun. 2019;10(1):2517.
2. Halim CE, Deng S, Ong MS, Yap CT. Involvement of STAT5 in oncogenesis. Biomedicines. 2020;8(9):316.
3. Smith MR, Satter LRF, Vargas-Hernandez A. STAT5b: a master regulator of key biological pathways. Front Immunol. 2022;13:1025373.
4 Pham HTT, Maurer B, Prchal-Murphy M, et al. STAT5BN642H is a driver mutation for T cell neoplasia. J Clin Invest. 2018;128(1):387-401.
5. Bhattacharya D, Teramo A, Gasparini VR, et al. Identification of novel STAT5B mutations and characterization of TCRbeta signatures in CD4+ T-cell large granular lymphocyte leukemia. Blood Cancer J. 2022;12(2):31.
6. Rajala HL, Eldfors S, Kuusanmaki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541-4550.
7 Wahnschaffe L, Braun T, Timonen S, et al. JAK/STAT-activating genomic alterations are a hallmark of T-PLL. Cancers (Basel). 2019;11(12):1833.
8. Kucuk C, Jiang B, Hu X, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat Commun. 2015;6:6025.
9 Bandapalli OR, Schuessele S, Kunz JB, et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica. 2014;99(10):e188-192.
10. Maurer B, Kollmann S, Pickem J, Hoelbl-Kovacic A, Sexl V. STAT5A and STAT5B - twins with different personalities in hematopoiesis and leukemia. Cancers (Basel). 2019;11(11):1726.
11. Cross NCP, Hoade Y, Tapper WJ, et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia. 2019;33(2):415-425.
12. Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128-1132.
13. Palomo L, Meggendorfer M, Hutter S, et al. Molecular landscape and clonal architecture of adult myelodysplastic/ myeloproliferative neoplasms. Blood. 2020;136(16):1851-1862.
14 Valent P, Sotlar K, Blatt K, et al. Proposed diagnostic criteria
Disclosures
No conflicts of interest to disclose.
Contributions
CCY and SAW designed the study, collected and analyzed the data, and wrote the manuscript. WT, SMW, AK, MMO, WX, OKW, PL, ZZ, MJR, SC, LJM, TIG, AO, DAA, AB, and RPH collected and analyzed data, and reviewed the manuscript.
Data-sharing statement
Data presented in this study are available upon request.
and classification of basophilic leukemias and related disorders. Leukemia. 2017;31(4):788-797.
15. Arber DA, Orazi A, Hasserjian RP, et al. International consensus classification of myeloid neoplasms and acute leukemia: integrating morphological, clinical, and genomic data. Blood. 2022;140(11):1200-1228.
16. Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703-1719.
17 Umrau K, Naganuma K, Gao Q, et al. Activating STAT5B mutations can cause both primary hypereosinophilia and lymphocyte-variant hypereosinophilia. Leuk Lymphoma. 2023;64(1):238-241.
18. Andersson EI, Putzer S, Yadav B, et al. Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and mutation profiling. Leukemia. 2018;32(3):774-787.
19 Sreedharanunni S, Jamwal M, Balakrishnan A, et al. Chronic eosinophilic leukemia with recurrent STAT5B N642H mutationan entity with features of myelodysplastic syndrome/ myeloproliferative neoplasm overlap. Leuk Res. 2022;112:106753.
20. Ruan GJ, Smith CJ, Day C, et al. A population-based study of chronic eosinophilic leukemia-not otherwise specified in the United States. Am J Hematol. 2020 ;95(10):E257-E260.
21. Srour SA, Devesa SS, Morton LM, et al. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/ myeloproliferative neoplasms in the United States, 2001-12. Br J Haematol. 2016;174(3):382-396.
22. Hu Z, Boddu PC, Loghavi S, et al. A multimodality work-up of patients with hypereosinophilia. Am J Hematol. 2018;93(11):1337-1346.
23. Wang SA, Hasserjian RP, Tam W, et al. Bone marrow morphology is a strong discriminator between chronic eosinophilic leukemia, not otherwise specified and reactive idiopathic hypereosinophilic syndrome. Haematologica. 2017;102(8):1352-1360.
24. Wang SA, Tam W, Tsai AG, et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol. 2016;29(8):854-864.
25. Kelemen K, Saft L, Craig FE, et al. Eosinophilia/ hypereosinophilia in the setting of reactive and idiopathic causes, well-defined myeloid or lymphoid leukemias, or germline disorders. Am J Clin Pathol. 2021;155(2):179-210.
26. Li Y, Qi X, Liu B, Huang H. The STAT5-GATA2 pathway is critical in basophil and mast cell differentiation and maintenance. J Immunol. 2015;194(9):4328-4338.
27. Pozdnyakova O, Orazi A, Kelemen K, et al. Myeloid/lymphoid neoplasms associated with eosinophilia and rearrangements of PDGFRA, PDGFRB, or FGFR1 or w.ith PCM1-JAK2. Am J Clin Pathol. 2021;155(2):160-178.
28. Eisenberg R, Gans MD, Leahy TR, et al. JAK inhibition in earlyonset somatic, nonclonal STAT5B gain-of-function disease. J Allergy Clin Immunol Pract. 2021;9(2):1008-1010.e2.
Thibaud Sefiane,1 Hortense Maynadié,1,2 Carmen Escurola Ettingshausen,3 Vincent Muczynski,1 Xavier Heiligenstein,4 Julien Dumont,5 Olivier D. Christophe,1 Cécile V. Denis,1 Caterina Casari1# and Peter J. Lenting1#
1Laboratory for Hemostasis, Inflammation & Thrombosis, Unité Mixed de Recherche 1176, Institut National de la Santé et de la Recherche Médicale, Université Paris-Saclay, Le Kremlin-Bicêtre, France; 2Centre de Référence de l’Hémophilie et des Maladies Hémorragiques Constitutionnelles Rares, Hôpital Bicêtre AP-HP, Université Paris-Saclay, Le Kremlin-Bicêtre, France; 3HZRM Hemophilia Center Rhine-Main, Frankfurt-Mörfelden, Mörfelden-Walldorf, Germany; 4CryoCapCell, Le Kremlin-Bicetre, France and 5Collège de France, Centre Interdisciplinaire de Recherche en Biologie (CIRB), Unité Mixed de Recherche 1050, Paris, France
#CC and PJL contributed equally as senior authors.
Abstract
Correspondence: C.V. Denis cecile.denis@inserm.fr
Received: August 23, 2023.
Accepted: November 29, 2023. Early view: December 7, 2023.
https://doi.org/10.3324/haematol.2023.284142
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Recombinant factor VIII (rFVIII), rFVIIIFc and emicizumab are established treatment options in the management of hemophilia A. Each has its unique mode of action, which can influence thrombin generation kinetics and therefore also the kinetics of thrombin substrates. Such differences may potentially result in clots with different structural and physical properties. A starting observation of incomplete wound closure in a patient on emicizumab prophylaxis led us to employ a relevant mouse model in which we noticed that emicizumab-induced clots appeared less stable compared to FVIII-induced clots. We therefore analyzed fibrin formation in vitro and in vivo In vitro fibrin formation was faster and more abundant in the presence of emicizumab than in the presence of rFVIII/rFVIIIFc. Furthermore, the time-interval between the initiation of fibrin formation and factor XIII activation was twice as long for emicizumab than as for rFVIII/rFVIIIFc. Scanning electron microscopy and immunofluorescent spinning-disk confocal microscopy of in vivo-generated clots confirmed increased fibrin formation in the presence of emicizumab. Unexpectedly, we also detected a different morphology between rFVIII/rFVIIIFcand emicizumab-induced clots. Contrary to the regular fibrin mesh obtained with rFVIII/rFVIIIFc, fibrin fibers appeared to be fused into large patches upon emicizumab treatment. Moreover, fewer red blood cells were detected in regions in which these fibrin patches were present. The presence of highly dense fibrin structures associated with a diffuse fiber structure in emicizumab-induced clots was also observed when using super-resolution imaging. We hypothesize that the modified kinetics of thrombin, fibrin and factor XIIIa generation contribute to differences in structural and physical properties between clots formed in the presence of FVIII or emicizumab.
The blood coagulation cascade is a series of highly regulated enzymatic reactions designed to generate thrombin, which is needed to produce fibrin in a timely and spatially correct manner.1 To achieve this goal, the coagulation cascade consists of (among others) thrombin-dependent feedback loops that allow for the amplification of thrombin formation. Thrombin is also needed to generate activated factor XIII (FXIIIa), which converts the freshly produced fibrin protofibrils into a co-
valently linked network, and activated thrombin-activatable fibrinolysis inhibitor, which delays fibrinolytic degradation of the fibrin network.2,3 Any disturbance of this complex process may potentially lead to an improperly formed fibrin network. Indeed, thrombin concentration has been shown to be a critical determinant of the fibrin network structure and its physical properties.4
One of the key cofactor proteins in the coagulation cascade is factor VIII (FVIII), the functional absence of which is associated with markedly reduced thrombin formation.5 The
thrombin-activated derivative of FVIII, FVIIIa, functions as a cofactor in the tenase complex, which participates in the amplifying portion of the coagulation cascade. Within the tenase complex, FVIIIa stimulates the generation of activated factor X (FXa) by the enzyme activated factor IX (FIXa). The physiological relevance of FVIII is illustrated by the severe bleeding complications associated with its deficiency, a disorder known as hemophilia A. To compensate for the deficiency of FVIII in people with hemophilia A, replacement therapy using FVIII concentrates has been the treatment of choice over the last four decades.6 Prophylactic treatment proved efficient in reducing spontaneous bleeding and subsequent joint damage. Despite its effectiveness, replacement therapy had a number of disadvantages, including the need for frequent intravenous infusions and an immunological response leading to the presence of neutralizing allo-antibodies against FVIII. These complications have led to the development of alternative treatment options, such as activated factor VII, extended half-life FVIII variants, gene therapy, and so-called non-factor therapies.7,8 One non-factor approach that has been approved for clinical use is emicizumab, a bispecific antibody that binds both the enzyme FIXa and its substrate FX, thereby mimicking part of the FVIIIa function.9,10 Emicizumab therefore allows the generation of a certain amount of thrombin without completely correcting coagulation. Clinical studies have shown marked therapeutic benefit from the use of emicizumab, with annual bleeding rates being fewer than two among patients treated with emicizumab prophylactically.11 Because of its efficacy and its subcutaneous mode of administration, increasing numbers of people with hemophilia A are using emicizumab as prophylactic therapy.12 Despite this success, it has been reported that about 5% of the patients receiving emicizumab may develop spontaneous or trauma-induced muscle bleeds, which are sometimes difficult to stop and require intensive factor replacement therapy.13 It is therefore important to better understand the molecular basis by which emicizumab, in comparison to FVIII, contributes to coagulation. Indeed, the mode of action of these two treatments is quite different.14 Both components may differ by the timing with which they start acting and when their activity stops, as well as by the location where they may act. A similar point can also be made for extended half-life variants, such as recombinant FVIIIFc (rFVIIIFc), in which the presence of the Fc portion may affect FVIII distribution and activation during coagulation. Knowing that the formation of thrombin is both time- and spatially dependent, it is of relevance to investigate whether and how emicizumab and rFVIIIFc may differ from “classic” FVIII regarding the formation of a fibrin network.
It should be noted that so far studies on fibrin formation using emicizumab have been restricted to in vitro settings.15-17 Moreover, the activity of emicizumab is often examined in global assays, such as thrombin generation assays.18-22 Although this may provide insight into the potential of emicizumab to
stimulate thrombin generation relative to FVIII, it provides little insight into the upstream processes that are required to generate a stable clot.
We have therefore explored in vivo clot formation by recombinant FVIII (rFVIII), rFVIIIFc and emicizumab using specific bleeding models and microscopic analysis, with focus on the structure of the fibrin network. We found differences in structural morphology between FVIII- and emicizumab-induced clots. Furthermore, kinetics of fibrin and FXIIIa generation are modified when using emicizumab, which could explain in part the differences in clot structure.
A description of the experimental procedures can be found in the Online Supplementary Materials.
Ethics statement
Photographs of the patient’s wound were taken during consultation, and informed consent was given by the parents for their anonymous use in this study. Animal housing and experiments were performed in accordance with French regulations and the experimental guidelines of the European Community. This project was approved by the local ethical committee of Université Paris-Saclay (Comité d’Ethique en Experimentation Animale n. 26, protocol APAFIS#26510-202007061525281 v2).
Patient
The patient was a 2-year-old boy with severe hemophilia A (FVIII <1%) on regular prophylaxis with emicizumab (6 mg/ kg every 4 weeks, subcutaneously) who experienced a deep laceration of his left foot and subsequent insufficient wound healing. A more extensive description of the patient is available in the Online Supplementary Materials
In vitro assays
A description of fibrin formation and FXIIIa generation is available in the Online Supplementary Materials.
In vivo models
A tail-vein transection model was used in this study. The model is described in the Online Supplementary Materials
Microscopy
The use of spinning-disk confocal microscopy, scanning electron microscopy and stimulated emission depletion (STED) microscopy is described in the Online Supplementary Materials
Patient
A 2-year-old boy with severe hemophilia A under regular
emicizumab prophylaxis for 2 years (6 mg/kg every 4 weeks), experienced a deep laceration of his left foot (Figure 1). The wound was treated with local application of tranexamic acid and covered with Steri-strips. Emicizumab prophylaxis continued, and bleeding stopped. However, when the Steri-strips were removed after 10 days, no wound closure or wound healing had occurred, and the covering clot appeared loose and fragile. The patient was then administered three consecutive doses of FVIII (66 IU/kg intravenously, followed by 33 IU/kg intravenously at 12-hourly intervals) on days 10 and 11 after the injury. Complete wound closure was observed at day 12, and wound healing progressed naturally in the following days.
vivo clot formation
To further explore clot formation in vivo, we compared mice given emicizumab, rFVIII or rFVIIIFc using a previously validated mouse model. In this model, mice receive emicizumab in combination with human FIX and human FX (100 IU/kg), or a single dose of rFVIII or rFVIIIFc.23 Bleeding is then assessed following transection of a tail vein.24 At the time of injury, plasma concentrations of rFVIII and rFVIIIFc
were 10 IU/dL, whereas that of emicizumab was 55 μg/mL. The FVIII concentrations were chosen based on the apparent FVIII-equivalence for emicizumab that we observed in our previous in vivo studies.23 Whereas all vehicle-treated mice started to rebleed after the initial primary stop, treatment with rFVIII, rFVIIIFc or emicizumab resulted in a permanent arrest of bleeding when the wound remained unchallenged (Figure 2A). Efficient bleeding arrest was accompanied by limited blood loss (Online Supplementary Figure S1). In contrast, when the clot covering the injury was removed 15 and 30 min after the injury, differences in response between FVIII- and emicizumab-treated mice were observed. In mice receiving rFVIII or rFVIIIFc, the bleeding readily stopped again, whereas emicizumab-treated mice displayed a pattern of frequent spontaneous re-bleeding (Figure 2B). Spontaneous re-bleeds were associated with increased blood loss ( Online Supplementary Figure S1). We considered the possibility that levels of FIX and FX had diminished below the necessary threshold to support emicizumab, since their respective half-lives are relatively short in mice (3-5 h) compared to those in humans (>20 h).23 However, re-injection of FIX and FX (100 IU/kg) 3 min
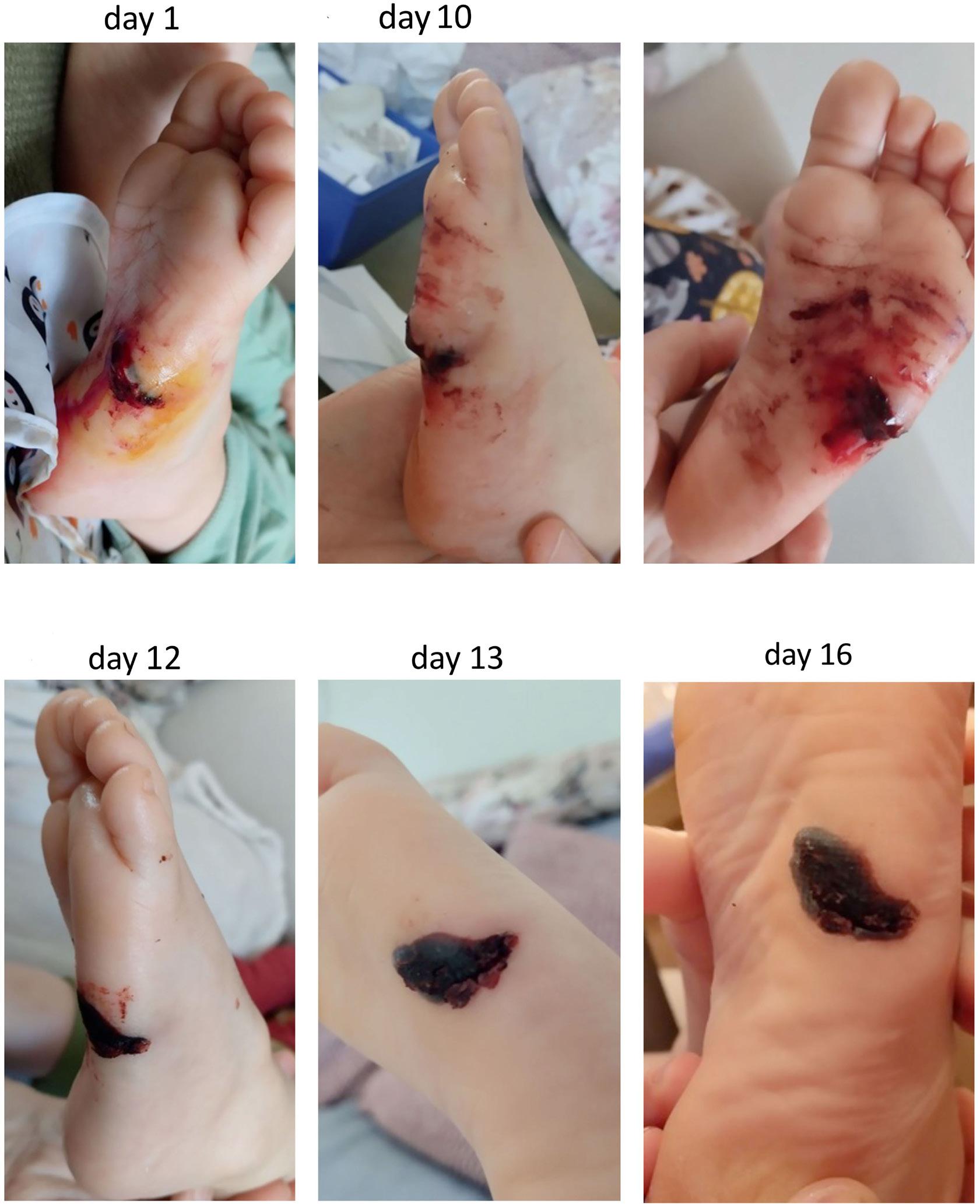
Figure 1. Incomplete wound closure. (A) A boy with hemophilia on emicizumab prophylaxis arrived at the hospital with a deep laceration in his foot (day 1). (B) The wound was treated with local application of tranexamic acid and covered with Steristrips. On day 10, the Steri-strips were removed, but the wound was still open, and no wound closure or healing was observed. The patient received treatment with factor VIII on days 10 and 11. (C) On day 12, wound closure was observed and the wound healing process progressed gradually during the following days, as shown by the photographs taken on day 13 (D) and day 16 (E).
before clot removal did not affect either bleeding profile or blood loss (Figure 2B, Online Supplementary Figure S1). We also examined whether the use of tranexamic acid (10 mg/kg) could improve the phenotype of emicizumab-treated mice. Interestingly, tranexamic acid did not prevent spontaneous re-bleeds after clot removal (Figure 2B, Online Supplementary Figure S1), suggesting that the clot instability is unrelated to increased fibrinolysis of the fibrin network. In conclusion, these in vivo data point to
potential differences in clot construction between FVIIIand emicizumab-treated mice.
Given that a stable fibrin network is key to the formation of a stable thrombus, we evaluated whether differences in the mode of action between rFVIII and emicizumab could affect upstream fibrin formation. To this end, we performed in vitro experiments using human plasma; in these
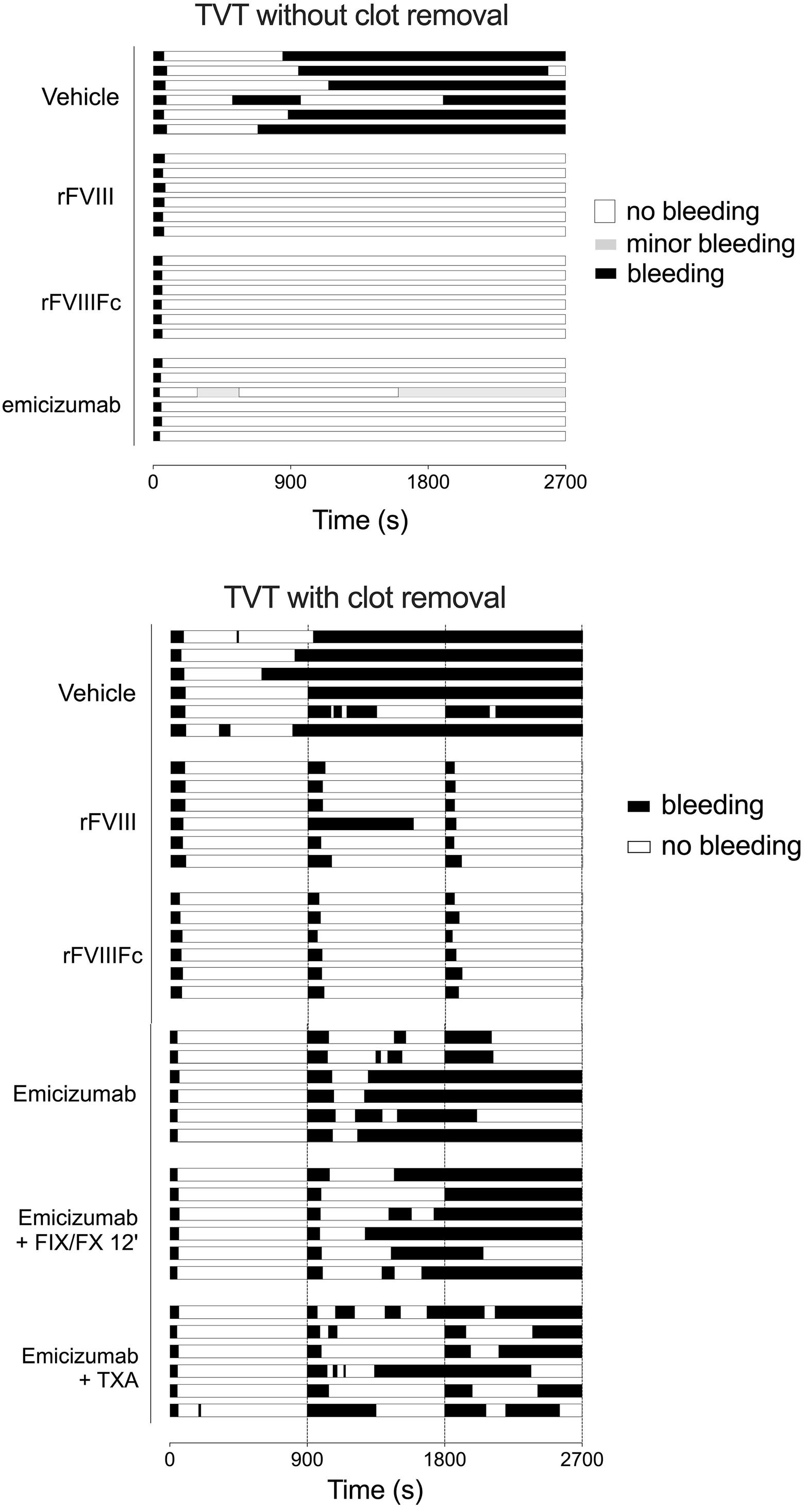
Figure 2. Hemostatic responses after tail-vein transection without and with clot removal. Where the diameter of the tail was 2.3 mm, an incision 0.5 mm deep was made in the left lateral vein. Mice receiving emicizumab also received human FIX and human FX (both at a dose of 100 IU/kg) 5 min before the injury. Both the upper and lower panels depict the bleeding profile of mice receiving vehicle, recombinant FVIII (rVIII), recombinant FVIIIFc (rFVIIIFc) or emicizumab. Plasma concentrations at the start of the procedure were 10 IU/dL for rFVIII and rFVIIIFc and 55 μg/mL for emicizumab. Black represents periods of bleeding, gray periods of minor bleeding, and white periods of non-bleeding. (A) The wound remained unchallenged after transection of the vein. (B) The dotted lines at 900 seconds and 1,800 seconds indicate the timing of clot removal in mice that were not bleeding at those instants. Amounts of blood loss measured in the mice are presented in Online Supplementary Figure S1. Emicizumab + FIX/FX 12’: re-injection of FIX and FX at 12 min in emicizumab-treated mice (i.e., 3 min before the first clot removal). Emicizumab + TXA: emicizumab-treated mice that also received tranexamic acid (10 mg/kg) 5 min before injury; TVT: tail-vein transection.
experiments rFVIII (at doses of 10 IU/dL and 100 IU/dL) or emicizumab (55 μg/mL) was added to fibrinogen-enriched FVIII-deficient plasma (Figure 3A). Fibrin formation was monitored by measuring optical density (OD) at 405 nm. In the absence of rFVIII or emicizumab, the lag-time was 32 min, and half-maximal fibrin formation was reached at 42 min (Figure 3B). The addition of rFVIII (10 and 100 IU/dL) dose-dependently shortened the lag-time to 24 and 18 min, with half-maximal fibrin formation being achieved at 33 and 26 min, respectively (Figure 3C, D). Similar data were
obtained using rFVIIIFc, indicating that coagulation proceeds similarly for both FVIII variants (Online Supplementary Figure S2). Interestingly, lag-time and time to half-maximal fibrin formation were markedly shortened in the presence of emicizumab, to 6 and 14 min, respectively (Figure 3E). In addition, the maximum OD reached with emicizumab (OD=1.17±0.03; n=6) was higher than that with either rFVIII (OD=0.95±0.06; n=6) or rFVIIIFc (0.97±0.08; n=10; P<0.0001), pointing to rapid and increased fibrin formation. Together, these data suggest that modifying the kinetics of FXa
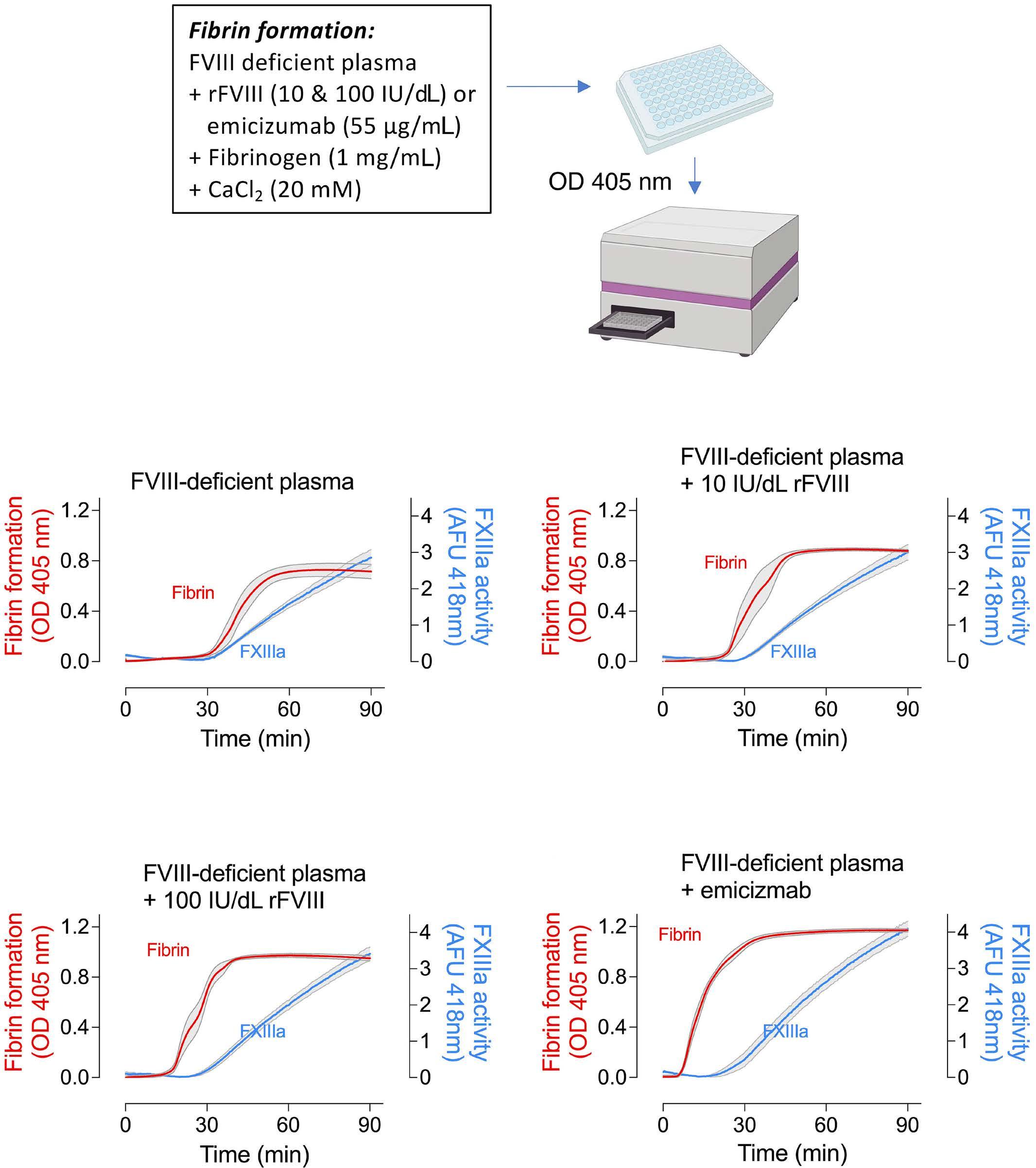
Figure 3. In vitro fibrin formation and fibrinolysis. (A) Schematic representation of in vitro fibrin formation in human FVIII-deficient plasma supplemented with recombinant FVIII (rFVIII) or emicizumab. (B-E) FVIII-deficient plasma (B) was supplemented with rFVIII at a dose of 10 IU/dL (C) or 100 IU/dL (D) or emicizumab at 55 μg/mL (E). In addition, fibrinogen was added (1 mg/mL final concentration) and the reaction was initiated by the addition of CaCl2. No other additives (tissue factor or FXIa) were used. Fibrin formation was detected by monitoring optical density values at 405 nm, while FXIII activity was measured via hydrolysis of the fluorescent A101 substrate (excitation 313 nm; emission 418 nm). Red line: fibrin formation (left Y axis) and blue line: FXIII activity (right Y axis). The mean (solid lines) and standard error (gray area around the solid line) of six to eight measurements are presented. CaCl2: calcium chloride; OD: optical density; AFU: arbitrary fluorescence units.
generation affects upstream fibrin formation.
In vitro generation of activated factor XIII
Since fibrin formation involves two distinct steps following the conversion of fibrinogen into fibrin monomers (non-covalent polymerization of fibrin monomers into protofibrils, and subsequent covalent crosslinking via FXIIIa), we decided to monitor FXIII activation in parallel experiments. FXIIIa activity was detected in all conditions (Figure 3B-E). However, the synchronization between the initiation of fibrin formation and FXIIIa generation varied between the different conditions. In particular, a longer interval was observed between the initiation of fibrin and FXIIIa formation in mice treated with emicizumab (12±2 min) compared to mice given rFVIII at a dose of 100 IU/dL (7±1 min) or 10 IU/ dL (4±1 min). Furthermore, more FXIIIa was generated in the presence of emicizumab (0.14 OD/min, 95% confidence interval [95% CI]: 0.13-0.15 OD/min) than in the presence of rFVIII 100 IU/dL (0.12 OD/min, 95% CI: 0.11-0.13; P=0.026), rFVIII 10 IU/dL (0.10 OD/min; P<0.0001) or FVIII-deficient plasma (0.11 OD/min; P<0.0001). Thus, relative to fibrin generation, activation of FXIII starts later in the presence of emicizumab than in the presence of rFVIII, while once started, more FXIIIa is formed.
In vivo generation of fibrin: scanning electron microscopy analysis
We next studied whether the in vitro differences in the kinetics of fibrin formation were also observed in vivo by applying the same tail-vein transection model described for Figure 2. Tails were collected 10 min after injury, when bleeding had stopped in all treated mice (Figure 2), and tissue sections were prepared for whole-mount scanning electron microscopy, which enables visualization of the outer portion of the clot (Figure 4A).
rFVIII-derived clots contained a fibrin network in which thin, tubular-shaped fibers of homogeneous thickness were in a regular mesh over the clot, and cellular components were evenly distributed (Figure 4B). Similar structures were also observed in tail fragments obtained from wild-type mice, whereas FVIII-deficient mice displayed a disturbed network with less fibrin and fewer fibers which had a 1.4-fold increased diameter (P=0.032) (Online Supplementary Figure S3). The emicizumab-derived clots were characterized by a completely different fibrin morphology. They consisted of thick, uneven fiber structures that seemed to fuse together, thereby forming large layered structures. Strikingly, these patch-like structures were almost devoid of cellular components, and included small and large circular-shaped holes (Figure 4C).
We analyzed these images using ImageJ-software for four different parameters: fibrin coverage, fiber diameter, number of pores and number of intersections (Figure 4D-G). In line with the in vitro fibrin generation experiments, more fibrin was detected in emicizumab-treated mice than in
rFVIII-treated ones (42±8% vs. 24±8% per field; P<0.0001) (Figure 4D). The average diameter of the fibrin fibers was also significantly increased, 1.8-fold, in emicizumab-treated mice (average of 534±90 nm vs. 292±101 nm; P<0.0001 (Figure 4E). Furthermore, there were 2-fold fewer pores in emicizumab-derived clots compared to rFVIII-derived clots (119±45 vs. 248±58 per field; P<0.0001) (Figure 4F). Finally, 1.7-fold more intersections per field of view were detected in rFVIII-induced clots compared to emicizumab-induced clots (11,078±2935 vs. 6,510±1157 per field; P<0.0001) (Figure 4G). These data indicate that clots derived from mice receiving emicizumab have a different structure from clots generated in mice receiving rFVIII.
In vivo generation of fibrin: spinning-disk confocal immunofluorescence analysis
To investigate whether differences in fibrin formation were also occurring within the interior of the injury, we first performed spinning-disk confocal imaging using anti-fibrin antibodies (Figure 5A). Five mice were analyzed for each group, with two tissue sections/mouse. Representative images are provided for each condition (wild-type mice, FVIII-deficient mice, and FVIII-deficient mice treated with rFVIII, rFVIIIFc or emicizumab) (Figure 5B-F). Similar amounts of fibrin (in terms of mean fluorescence intensity) were found in clots from rFVIII- and rFVIIIFc-treated mice compared to wild-type mice (Figure 5G). This parameter did not increase further with higher doses of rFVIII or rFVIIIFc (500 IU/dL) (Online Supplementary Figure S4).
Unexpectedly, the mean fluorescence intensity was significantly increased in clots from emicizumab-treated mice (0.66±0.30 vs. 0.14±0.05, 0.17±0.10 and 0.26±0.09 for emicizumab, rFVIII, rFVIIIFc and wild-type mice, respectively; P<0.0001) (Figure 5B-F). These data are in line with our in vitro data, which showed that fibrin formation is more abundant in the presence of emicizumab than in the presence of FVIII.
In vivo generation of fibrin: stimulated emission depletion microscopy
Although spinning-disk confocal imaging enables a quantitative analysis of fibrin formation within the interior of a clot, its resolution is insufficient to detect potential differences in structure. We therefore proceeded to perform STED microscopy, a form of super-resolution microscopy, which is less suited for quantitative analysis of large regions, but enables imaging with sub-diffraction resolution of ≤50 nm. Detailed images of fibrin structures within the interior of a clot were obtained for wild-type, FVIII-deficient and rFVIIIFc- and emicizumab-treated mice (Figure 6A). As expected, typical fiber-like structures, similar to those observed using scanning electron microscopy, were readily distinguished in the clots of wild-type, FVIII-deficient and rFVIIIFc-treated mice. The fibrin mesh was characterized by a homogeneous thickness of the fibers (Figure 6B-D).
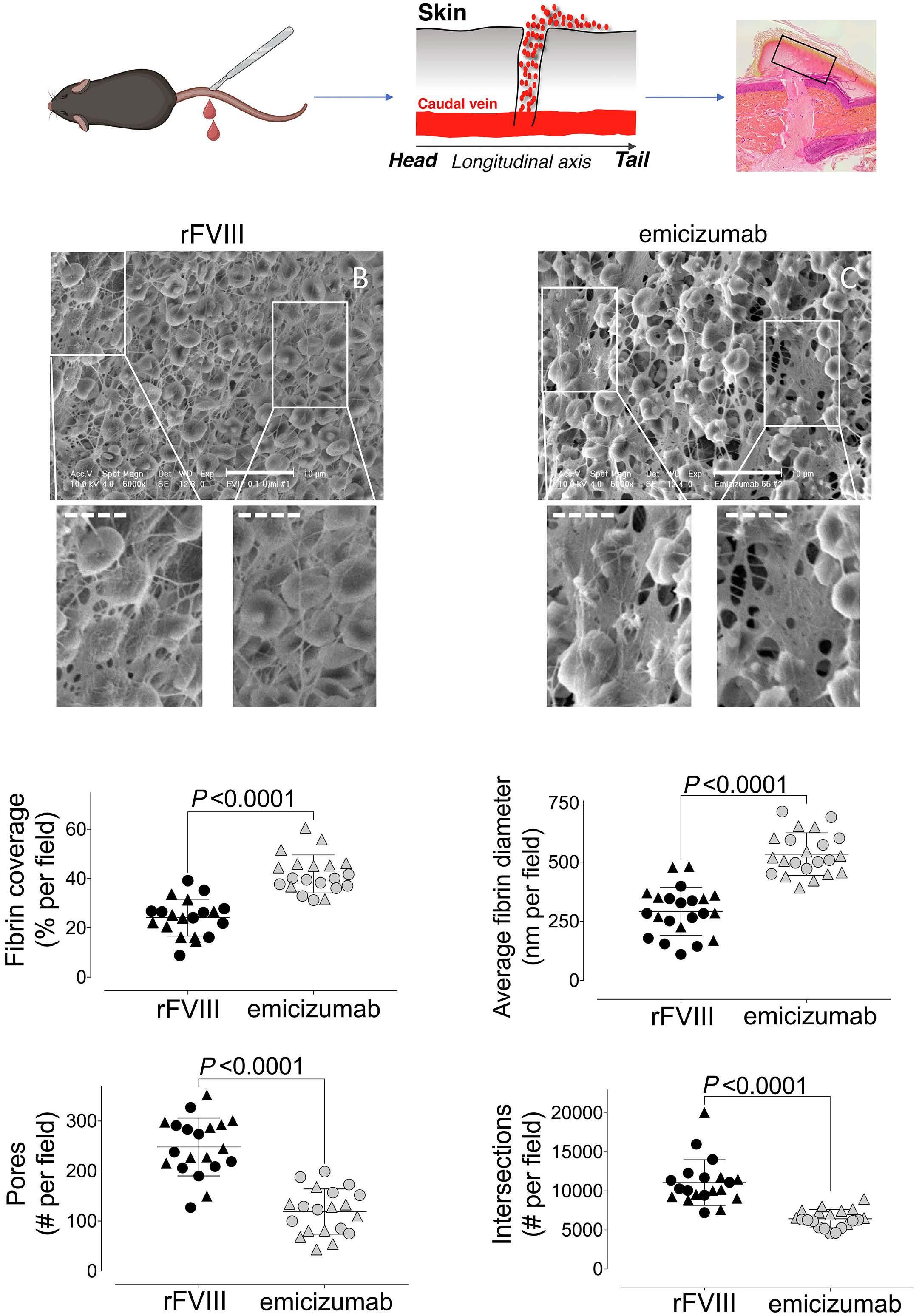
Figure 4. Scanning electron microscopy imaging of in vivo-generated fibrin networks. (A) Experimental approach for the preparation of tissue sections. The black rectangle indicates the region examined using scanning electron microscopy. (B, C) Tail fragments obtained 10 min after tail-vein transection were prepared for scanning electron microscopy using standard pre-fixation with 4% glutaraldehyde and 1% OsO4 and dehydration. Representative images of FVIII-deficient mice treated with recombinant FVIII (B) or emicizumab (C) are shown. Plasma concentrations at the start of the procedure were 10 IU/dL for rFVIII and 55 μg/mL for emicizumab. Scale bars represent 10 microns in the large image, and 5 microns in the amplified images. White boxes indicate regions of numeric zooms. (D-G) ImageJ-plugin software was used to determine fibrin coverage (D), average fibrin diameter (E), number of pores per field (F) and number of intersections per field (G). For each experiment shown in the graphs, two mice were included (round and triangle symbols) and ten fields per mouse were examined. Each symbol represents the result in a single field. The statistical analysis was performed using an unpaired Student t test. rVIII: recombinant factor VIII.
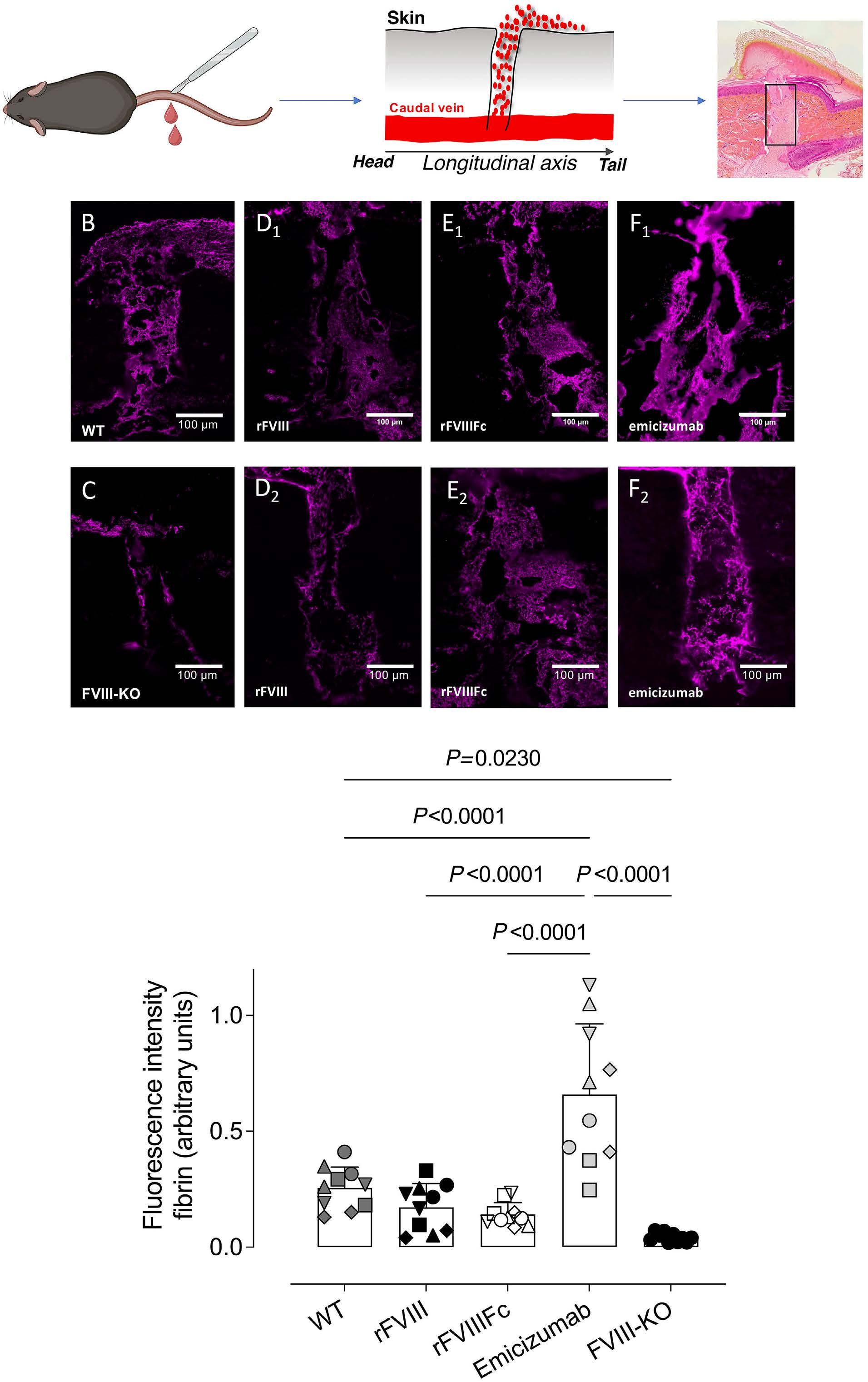
Figure 5. Fibrin content within the injured area. (A) Experimental approach for the preparation of tissue sections. The black rectangle indicates the region examined using spinning disk confocal imaging. (B-F) Sections of tail tissue obtained 10 min after tailvein transection were prepared for immunofluorescence staining using an anti-mouse fibrin antibody. Representative images are shown for wild-type mice (B), untreated FVIII-deficient mice (C); FVIII-deficient mice treated with recombinant FVIII (D1-D2), recombinant FVIIIFc (E1-E2), or emicizumab (F1-F2). Plasma concentrations at the start of the procedure were 10 IU/dL for recombinant FVIII and recombinant FVIIIFc and 55 μg/mL for emicizumab. (G) For each condition, two non-successive tissue sections from five mice (represented by square, round, diamond, up-triangle and down-triangle symbols) were analyzed using ImageJ-software for the fluorescence intensity (E). Statistical analysis was performed using one-way analysis of variance with Tukey corrections for multiple comparisons. Only statistically significant differences (P<0.05) are indicated. Each symbol represents a separate tissue section from an individual mice, and the mean ± standard deviation are indicated for each condition. WT: wild-type; rFVIII: recombinant factor VIII; rFVIIIFc: recombinant factor VIIIFc; FVIII-KO: factor VIII-knockout.
Interestingly, in each of the images (obtained from three different mice), intensely stained dot-like structures were observed, which for convenience we like to refer to as focal points. With regard to the structures observed in emicizumab-treated mice, fiber-like structures were also present (Figure 6E). However, these appeared more diffuse and their thickeness was less homogeneous. In addition, the number of focal points was considerably increased in these clots. When calculating the number of focal points using specific ImageJ-software, it was found that, on average, there were twice as many focal points in emicizumab-derived clots compared to those formed under other conditions (0.36±0.12 for emicizumab vs. 0.18±0.07, 0.21±0.08 and 0.18±0.08 for wild-type, FVIII-deficient and rFVIIIFc-treated mice, respectively (P<0.0001) (Figure 6F). Together, these findings support the concept that different fibrin structures are generated when emicizumab or FVIII is being administered.
There have been anecdotal communications on unusual bleeding episodes in patients receiving emicizumab. Here, we provide one example in which a laceration in the foot of a 2-year-old boy with severe hemophilia A on emicizumab prophylaxis showed compromised wound closure and healing. This complication was readily corrected upon treatment with FVIII. Although this single example cannot be said to be representative of the majority of people with hemophilia, it is noteworthy that Batsuli et al. reported that about 5% of patients on emicizumab prophylaxis develop spontaneous or trauma-induced muscle bleeds requiring intensive factor replacement therapy.13 These observations may originate from emicizumab being a less efficient cofactor than FVIIIa for FIXa. They might also point to fundamental differences in how clots are generated in the presence of FVIII or emicizumab.
Based on differences in functionality between FVIII and emicizumab, in this study we explored and compared fibrin formation and structure in the presence of rFVIII or rFVIIIFc and emicizumab. Our data demonstrate that there are differences not only in the amount of fibrin that is generated, but also in the structure of the fibrin network that is formed.
In order to asses clot structures in vivo consistently, we used a bleeding model that involves guided transection of the caudal vein, i.e., the tail-vein transection model. The advantage of this model lies in the reproducibility with which the injury is made using a specific template.24 FVIII-deficient mice are an established model to assess the hemostatic efficiency of FVIII and variants thereof.25,26 In contrast, there is less information regarding the use of emicizumab in FVIII-deficient mice, because emicizumab is unable to bind to murine FIXa and FX. To overcome this
limitation, we previously developed a protocol involving the co-infusion of human FIX and FX, enabling testing of the functionality of emicizumab in FVIII-deficient mice.23 Importantly, we showed that addition of human FIX and FX did not alter hemostasis in FVIII-treated mice, and that human FIX was as functional as murine FIX in in vitro clotting assays using murine plasma. In the tail-clip model, emicizumab (55 μg/mL) significantly reduced blood loss, with an efficacy that was similar to FVIII plasma concentrations of about 10 IU/dL. These concentrations were therefore used in the present study.
rFVIII, rFVIIIFc and emicizumab were similar in that they all corrected bleeding in FVIII-deficient mice in the tail-vein transection model (Figure 2). Bleeding stopped shortly after the injury, and no spontaneous re-bleeding was observed. In contrast, when clots were removed, emicizumab-treated mice displayed a pattern of frequent re-bleeds that was absent in rFVIII- or rFVIIIFc-treated mice. Control experiments verified that the re-bleeds were not due to an absence of human FIX or FX at the time of clot removal. Increased fibrinolysis was also excluded, since the use of tranexamic acid was unable to prevent these re-bleeds. Moreover, preliminary data presented by Locke et al. suggest that clot-lysis time of emicizumab-induced clots is prolonged 1.6-fold when compared to that of FVIII-induced clots.27 Our in vivo data suggest the formation of instable clots when emicizumab is used.
With fibrin being key in stabilizing wound-covering clots, we focused the rest of this study on fibrin network formation. We realize that other factors, in particular platelet activation and aggregation, are also of relevance to maintain clot stability. We are currently investigating this specific aspect as part of a separate study. When examining unbiased fibrin formation using human FVIII-deficient plasma spiked with rFVIII or emicizumab, it was clear that fibrin generation was quickest in the presence of emicizumab, and also that more fibrin was generated (Figure 3). This increased and more rapid fibrin formation is compatible with the hyper-reactivity of emicizumab in standard activated partial thromboplastin time assays, which rely on fibrin clot formation.28 One possible explanation for increased and accelerated fibrin formation is related to the different kinetics by which FVIII and emicizumab stimulate coagulation. Emicizumab will promote FXa generation as soon as FIXa becomes available. In contrast, rFVIII (or rFVIIIFc) requires feedback activation by thrombin before it will exert its cofactor function. The modified kinetics in turn alter the kinetics with which thrombin is able to activate its various substrates, including fibrinogen and FXIII.29,30 The observation that emicizumab accelerates fibrin formation is not limited to the in vitro experiments, but is also detected in vivo. Two independent microscopic approaches (scanning electron microscopy and immunofluorescent spinning-disk confocal imaging) revealed an excess of fibrin in emicizumab-treated mice compared to
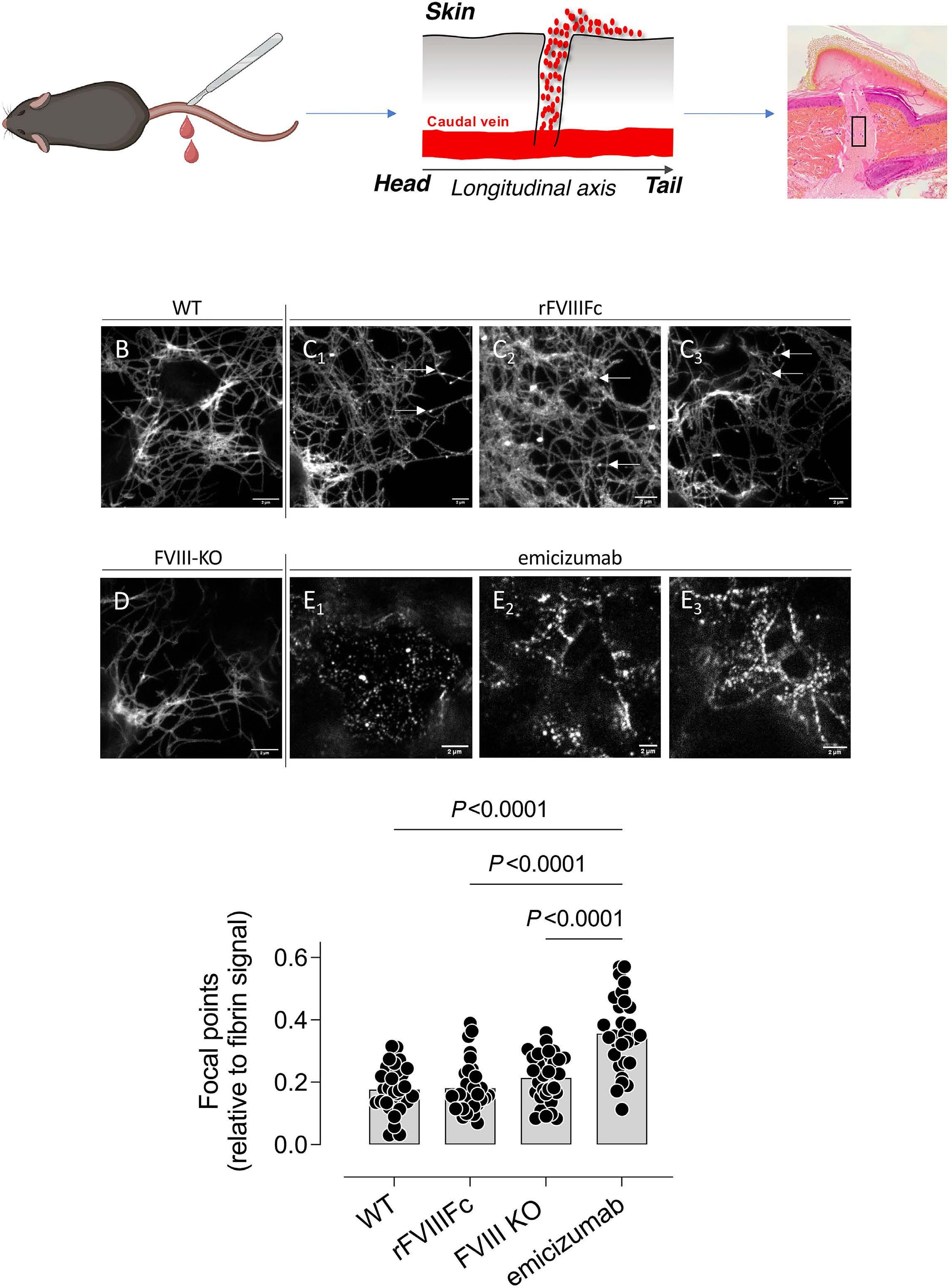
Figure 6. Fibrin structures imaged using stimulated emission depletion microscopy. (A) Experimental approach for the preparation of tissue sections. The black rectangle indicates the region examined using stimulated emission depletion microscopy (STED) microscopy. (B-E) Representative images of wild-type mice (B), FVIII-deficient mice treated with rFVIIIFc (C1-C3), untreated FVIII-deficient mice (D) or FVIII-deficient mice treated with emicizumab (E1-E3). Fibrin was detected using polyclonal goat anti-murine fibrin antibodies, and probed using STAR-RED-labeled donkey anti-goat antibodies. Plasma concentrations at the start of the procedure were 10 IU/dL for rFVIIIFc and 55 μ g/mL for emicizumab. (F) For each condition, 30 images from three different mice (10/mouse) were analyzed using ImageJ-software to calculate the presence of dot-like structures. The white arrows in panels C1-C3 indicate examples of dot-like structures. The statistical analysis was performed using one-way analysis of variance with the Dunnet test for multiple comparisons. WT: wild-type; rFIIIFc: recombinant factor VIIIFc; FVIII-KO: factor VIII-knockout.
the fibrin in FVIII-treated mice (Figures 4 and 5). Moreover, a significant correlation was observed between in vitro and in vivo fibrin formation, irrespectively of whether scanning electron microscopy or immunofluorescent spinning-disk confocal imaging was used (Figure 7).
To get insight into the structure of the fibrin network generated in vivo, we used two different microscopic techniques. With scanning electron microscopy we were able to visualize the outer region of the clot, i.e., the part that covers the injury. Unexpectedly, we observed quite an unusual fibrin network in the clots of emicizumab-treated mice. In contrast to individual intertwined fibers that are present in the structures of clots from wild-type mice and FVIII-treated mice, fibrin fibers appeared to be fused into large patches, with few individual fibers present. Consequently, average diameters were found to be excessively large (>500 nm) (Figure 4). In addition, the presence of the patches seemed to prevent the inclusion of red blood cells in these areas, resulting in a less dense clot structure.
Likewise, when super-resolution (STED) microscopy was used to study the interior of the clot, obvious differences were present between FVIII- and emicizumab-dependent clots (Figure 6). A normal fibrin mesh was found in the clots of FVIII-treated mice, whereas less recognizable fibrin staining was detected in the clots of emicizumab-treated mice. In the latter, we observed very large, diffusely stained patterns, characterized by a large number of intense spots, which we refer to as focal points. As of now, we have no information on what these focal points represent in terms of fibrin structure, but they could correspond to the patches we have seen in scanning electron microscopy images. Another possibility is that these spots represent fibrin that is accumulated at the surface of platelets which are
captured in the fibrin network. Additional studies will be performed to gain more insight into this aspect. An intriguing question is what causes the formation of such unusual fibrin networks under the influence of emicizumab. We observed that the synchronization between fibrin formation and FXIII activation was different between mice treated with emicizumab or FVIII. The interval between the initiation of fibrin and FXIIIa formation was longer in animals receiving emicizumab than in those treated with FVIII (Figure 3). This suggests that the non-covalent polymerization of fibrin profibrils continues over a longer period before FXIIIa starts making covalent connections. Furthermore, the generation of FXIIIa was more efficient in the presence of emicizumab, indicating that larger amounts of FXIIIa are available and more covalent connections can be made. These changes in kinetics could potentially contribute to the altered fibrin-network formation. The kinetics by which FXIII is activated has indeed been reported to have an important effect on the final structure of the fibrin network. For instance, the Val34Leu polymorphism in FXIII is associated with accelerated activation by thrombin, and results in a fibrin meshwork with thinner fibers and reduced porosity.31,32 It should be noted that there are discordant data regarding the formation of the fibrin network in the presence of emicizumab. Shimonishi et al. reported that the activated partial thromboplastin time reagent-triggered fibrin network generated in vitro in the presence of emicizumab was similar to that of FVIII-induced fibrin.16 In contrast, Janbain et al. found that fibrin fibers produced in the presence of emicizumab were almost 2-fold thinner than those produced in the presence of FVIII at a concentration of 10 IU/ dL.17,33 When comparing the protocols to obtain scanning
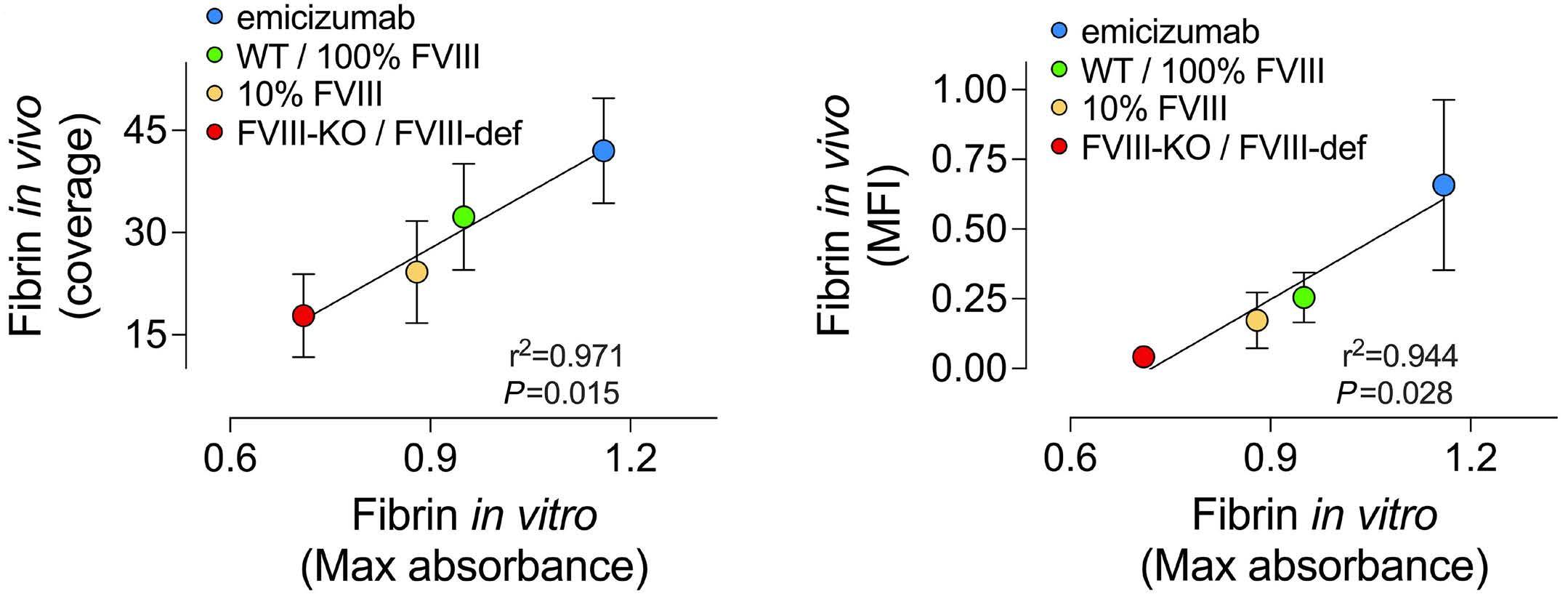
Figure 7. Correlation between in vitro and in vivo fibrin generation. (A) The amount of fibrin generated in vivo (evaluated as fibrin coverage) under different conditions (data presented in Figure 4 and Online Supplementary Figure S3) was plotted against the amount of fibrin generated in vitro (evaluated as maximal [max] absorbance; data presented in Figure 3). (B) The amount of fibrin generated in vivo (evaluated as mean fluorescence intensity [MFI] under different conditions (data presented in Figure 5) was plotted against the amount of fibrin generated in vitro (evaluated as maximal [max] absorbance; data presented in Figure 3). Red symbol: FVIII-KO mice versus FVIII-deficient plasma; orange symbol: FVIII-KO mice receiving 10 IU/dL rFVIII versus FVIII-deficient plasma spiked with 10 IU/dL rFVIII; geen symbol: wild-type mice versus FVIII-deficient plasma spiked with 100 IU/dL rFVIII; blue symbol: FVIII-KO mice receiving emicizumab (55 μg/mL) versus FVIII-deficient plasma spiked with emicizumab (55 μg/mL). WT: wild-type; FVIII: factor VIII; FVIII-KO: factor VIII knockout mice; FVIII-def: factor VIII-deficient.
electron microscopy images to analyze the fibrin structure, it appears that each of these studies used a different protocol for dehydration. Differences in dehydration protocols may affect how fibers are represented within the scanning electron microscopy images. Second, artificial activating reagents were used in the in vitro studies by Shimonishi et al. and Janbain et al., whereas the fibrin structures in our study were obtained from clots generated in vivo, a more physiological environment that also contains other blood components (red blood cells, platelets). Each of these blood components contributes to the formation of the fibrin network and its ultimate structure.34
As far as concerns the consequences of different clot architecture, it is tempting to speculate that the unstable clots originate from a modified fibrin network. Although no direct evidence is provided in this study, the presented in vivo data are compatible with this scenario. In particular, a dense fibrin structure may modify the biomechanical properties of a clot, and the unusual thick fibers may reduce the clot’s elasticity.35 Such increased rigidity seems in keeping with the re-bleeds that are observed upon clot removal in emicizumab-treated mice, but not rFVIII- or rFVIIIFc-treated mice (Figure 2).
In conclusion, rFVIII and rFVIIIFc act similarly regarding clot formation and structure. In contrast, the different mode of action of emicizumab alters the kinetics of fibrin and FXIIIa formation, resulting in different clot morphologies. We are
1. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327-358.
2. Wolberg AS, Sang Y. Fibrinogen and factor XIII in venous thrombosis and thrombus stability. Arterioscler Thromb Vasc Biol. 2022;42(8):931-941.
3. Sillen M, Declerck PJ. Thrombin activatable fibrinolysis inhibitor (TAFI): an updated narrative review. Int J Mol Sci. 2021;22(7):3670.
4. Domingues MM, Macrae FL, Duval C, et al. Thrombin and fibrinogen gamma’ impact clot structure by marked effects on intrafibrillar structure and protofibril packing. Blood. 2016;127(4):487-495.
5. Lenting PJ, van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92(11):3983-3996.
6. Mancuso ME, Mahlangu JN, Pipe SW. The changing treatment landscape in haemophilia: from standard half-life clotting factor concentrates to gene editing. Lancet. 2021;397(10274):630-640.
7 Sankar AD, Weyand AC, Pipe SW. The evolution of recombinant factor replacement for hemophilia. Transfus Apher Sci. 2019;58(5):596-600.
8. Weyand AC, Pipe SW. New therapies for hemophilia. Blood. 2019;133(5):389-398.
9. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809-818.
aware that these data represent only part of the differences that may occur in FVIII- versus emicizumab-dependent clot formation. Additional studies are in progress aiming to expand our notion of how non-factor molecules, such as emicizumab, modify clot architecture and, therefore, the process of hemostasis.
PJL receives research support (paid to the institute) from Sobi, Roche, Pfizer and Sanofi. XH is co-owner of CryoCapCell.
TS, HM, VM, XH, JD and PJL performed experiments and analyzed data. CEE provided the patient’s data; ODC, CVD, CC and PJL designed and supervised the study. All authors contributed to the interpretation of the data. PJL wrote the first draft of the manuscript, and all authors contributed to editing the final manuscript.
Funding
This study was supported by a research grant to the institute from Sobi.
Data-sharing statement
Data are available upon reasonable request to the corresponding author.
10 Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia A without Inhibitors. N Engl J Med. 2018;379(9):811-822.
11. Callaghan MU, Negrier C, Paz-Priel I, et al. Long-term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1-4 studies. Blood. 2021;137(16):2231-2242.
12. Mahlangu J, Iorio A, Kenet G. Emicizumab state-of-the-art update. Haemophilia. 2022;28 Suppl 4(Suppl 4):103-110.
13. Batsuli G, Wheeler AP, Weyand AC, Sidonio RFJr, Young G. Severe muscle bleeds in children and young adults with hemophilia A on emicizumab prophylaxis: real-world retrospective multi-institutional cohort. Am J Hematol. 2023;98(10):E285-E287.
14. Lenting PJ, Denis CV, Christophe OD. Emicizumab, a bispecific antibody recognizing coagulation factors IX and X: how does it actually compare to factor VIII? Blood. 2017;130(23):2463-2468.
15. Zong Y, Antovic A, Soutari NMH, Antovic J, Pruner I. Synergistic effect of bypassing agents and sequence identical analogue of emicizumab and fibrin clot structure in the in vitro model of hemophilia A. TH Open. 2020;4(2):e94-e103.
16. Shimonishi N, Nogami K, Ogiwara K, et al. Emicizumab improves the stability and structure of fibrin clot derived from factor VIII-deficient plasma, similar to the addition of factor VIII. Haemophilia. 2020;26(3):e97-e105.
17 Janbain M, Enjolras N, Bolbos R, Brevet M, Bordet JC, Dargaud Y. Haemostatic effect of adding tranexamic acid to emicizumab
prophylaxis in severe haemophilia A: a preclinical study. Haemophilia. 2021;27(6):1002-1006.
18. Schultz NH, Glosli H, Bjornsen S, Holme PA. The effect of emicizumab and bypassing agents in patients with hemophilia - an in vitro study. Res Pract Thromb Haemost. 2021;5(5):e12561.
19 Bravo MI, Raventos A, Perez A, Costa M, Willis T. Non-additive effect on thrombin generation when a plasma-derived factor VIII/von Willebrand factor (FVIII/VWF) is combined with emicizumab in vitro. J Thromb Haemost. 2020;18(8):1934-1939.
20 Dargaud Y, Lienhart A, Janbain M, Le Quellec S, Enjolras N, Negrier C. Use of thrombin generation assay to personalize treatment of breakthrough bleeds in a patient with hemophilia and inhibitors receiving prophylaxis with emicizumab. Haematologica. 2018;103(4):e181-e183.
21. Kizilocak H, Marquez-Casas E, Phei Wee C, Malvar J, Carmona R, Young G. Comparison of bypassing agents in patients on emicizumab using global hemostasis assays. Haemophilia. 2021;27(1):164-172.
22. Atsou S, Schellenberg C, Lagrange J, et al. Thrombin generation on vascular cells in the presence of factor VIII and/or emicizumab. J Thromb Haemost. 2024;22(1):112-125.
23. Ferriere S, Peyron I, Christophe OD, et al. A hemophilia A mouse model for the in vivo assessment of emicizumab function. Blood. 2020;136(6):740-748.
24. Johansen PB, Tranholm M, Haaning J, Knudsen T. Development of a tail vein transection bleeding model in fully anaesthetized haemophilia A mice - characterization of two novel FVIII molecules. Haemophilia. 2016;22(4):625-631.
25. Rawle FE, Lillicrap D. Preclinical animal models for hemophilia gene therapy: predictive value and limitations. Semin Thromb Hemost. 2004;30(2):205-213.
26. Lozier JN, Nichols TC. Animal models of hemophilia and related bleeding disorders. Semin Hematol. 2013;50(2):175-184.
27. Locke M, Receveur N, Kiialainen A, David T. In vitro investigation of emicizumab efficacy and mode of action in vWD type 2 and 3 samples. Res Pract Thromb Haemost. 2022;6(S1):630.
28. Adamkewicz JI, Chen DC, Paz-Priel I. Effects and interferences of emicizumab, a humanised bispecific antibody mimicking activated factor VIII cofactor function, on coagulation assays. Thromb Haemost. 2019;119(7):1084-1093.
29 Antovic A, Mikovic D, Elezovic I, Zabczyk M, Hutenby K, Antovic JP. Improvement of fibrin clot structure after factor VIII injection in haemophilia A patients treated on demand. Thromb Haemost. 2014;111(4):656-661.
30. Wolberg AS, Allen GA, Monroe DM, Hedner U, Roberts HR, Hoffman M. High dose factor VIIa improves clot structure and stability in a model of haemophilia B. Br J Haematol. 2005;131(5):645-655.
31. Ariens RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96(3):988-995.
32. Kattula S, Bagoly Z, Toth NK, Muszbek L, Wolberg AS. The factor XIII-A Val34Leu polymorphism decreases whole blood clot mass at high fibrinogen concentrations. J Thromb Haemost. 2020;18(4):885-894.
33. Janbain M, Enjolras N, Bordet JC, et al. Hemostatic effect of tranexamic acid combined with factor VIII concentrate in prophylactic setting in severe hemophilia A: a preclinical study. J Thromb Haemost. 2020;18(3):584-592.
34 Leong L, Chernysh IN, Xu Y, et al. Clot stability as a determinant of effective factor VIII replacement in hemophilia A. Res Pract Thromb Haemost. 2017;1(2):231-241.
35. Feller T, Connell SDA, Ariëns RAS. Why fibrin biomechanical properties matter for hemostasis and thrombosis. J Thromb Haemost. 2022;20(1):6-16.
Rushad Patell,1 Charles Hsu,1,2 Minggao Shi,3 Michael A. Grosso,3 Anil Duggal,3 Harry R. Büller,4 Gary Raskob5 and Jeffrey I. Zwicker2
1Division of Hematology and Hematologic Malignancies, BIDMC, Boston, MA, USA; 2Department of Medicine, Hematology Service, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 3Daiichi Sankyo Pharma Development, Edison, NJ, USA; 4Department of Vascular Medicine, Amsterdam Academic Medical Center, Amsterdam, the Netherlands and 5Hudson College of Public Health, the University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
Abstract
Correspondence: J.I. Zwicker zwickerj@mskcc.org
Received: August 28, 2023. Accepted: October 9, 2023. Early view: October 19, 2023.
https://doi.org/10.3324/haematol.2023.284192
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Thrombocytopenia occurs frequently in patients with cancer-associated thrombosis (CAT), however prospective evaluation of clinical outcomes following randomization to anticoagulants is limited. The HOKUSAI VTE Cancer study was a randomized, open-label, non-inferiority, phase III trial comparing dalteparin with edoxaban in CAT patients. This post hoc analysis of Hokusai VTE Cancer Study was performed to compare outcomes in patients with platelet count ≤100x109/L at one or more specified time points (baseline, 1-month, or 3-month) versus those without thrombocytopenia. Cumulative incidences at 180 days were calculated with death as a competing risk. The primary outcome was major bleeding; secondary outcomes were clinically relevant non-major bleeding (CRNMB), recurrent thrombosis, and survival. The analysis included 1,045 patients with primarily solid tumor malignancies (89%), median age 65 years, and 52% male. The thrombocytopenia group comprised 9.6% (N=101) of the cohort and relative to the non-thrombocytopenia cohort (N=944), experienced significantly higher major bleeding (9.0% vs. 4.0%, sub-distribution hazard ratio [SHR] =2.4; P=0.02) and CRNMB (17.9% vs. 9.6%, SHR=2.0; P=0.01). Thrombocytopenia did not impact recurrent venous thromboembolic event (VTE) (9.8% vs. 7.4%, SHR=1.3; P=0.37) nor overall mortality (21.8% vs. 26.0%, HR=0.9; P=0.48). Major bleeding was higher in patients with thrombocytopenia and gastrointestinal malignancies receiving edoxaban versus dalteparin (16.8% vs. 0; P<0.01) but similar for patients with other malignancies (P=0.30). In patients with hematologic malignances and thrombocytopenia major bleeding was higher for patients receiving dalteparin compared to edoxaban (19.0% vs. 0; P<0.01). Mild thrombocytopenia was associated with a doubling in risk of major hemorrhage in patients receiving anticoagulation for CAT. Bleeding risk for edoxaban and dalteparin varied in gastrointestinal and hematologic malignances in patients with thrombocytopenia ( clinicaltrails gov. Identifier: NCT02073682).
Thrombosis is a common complication in patients with active malignancy and has significant impact on morbidity and mortality, leading to increased health care resource utilization and financial strain.1-3 Cancer also increases the risk of bleeding which makes anticoagulation in this population challenging.4,5 Thrombocytopenia is common in patients with cancer due to either the underlying malignancy or the toxicity of cancer-directed therapies.6 Thrombocytopenia often coincides with the diagnosis of an acute venous thromboembolic event (VTE).7 Accordingly, the management of VTE in cancer patients with thrombocytopenia is challenging, as clinicians balance the benefits of anticoagulation with the likelihood of inducing a life-threatening hemorrhagic event.8 Randomized phase III trials have not been specifically
conducted in patients with cancer-associated thrombosis (CAT) and thrombocytopenia such that current guidelines are largely based on retrospective cohorts.9-11 Direct oral anticoagulants (DOAC) are replacing LMWH as the primary therapy for VTE in cancer.12 These oral medications are comparably efficacious to LMWH for VTE recurrence with similar rates of hemorrhage,13-16 and have superior patient satisfaction, quality of life, and treatment adherence compared to other anticoagulation strategies.17,15 Data from prospective clinical trials on outcomes in patients with cancer-associated thrombosis and thrombocytopenia treated with direct oral anticoagulants are lacking.
In order to assess the impact of platelet counts on clinical outcomes for acute VTE in cancer, we assessed outcomes among patients enrolled in the HOKUSAI Cancer VTE trial which was a randomized phase III trial comparing edoxaban
and dalteparin anticoagulation regimens in patients with acute VTE and cancer (clinicaltrails gov. Identifier: NCT02073682).
This study was a post hoc analysis utilizing de-identified clinical trial subject data from HOKUSAI Cancer VTE. The institutional review board at each participating center for the trial had previously approved the protocol and all pa-
tients were enrolled after written informed consent was obtained.13 The study team designed the analysis plan and this study was performed in collaboration with the sponsor (Daiichi Sankyo).
Hokusai VTE Cancer study was an open-label, non-inferiority trial, in which patients with cancer and acute symptomatic or incidental venous thromboembolism were randomized to receive either low-molecular-weight heparin for at least 5 days followed by oral edoxaban or subcutaneous dalteparin.13 The trial included adult patients with cancer with
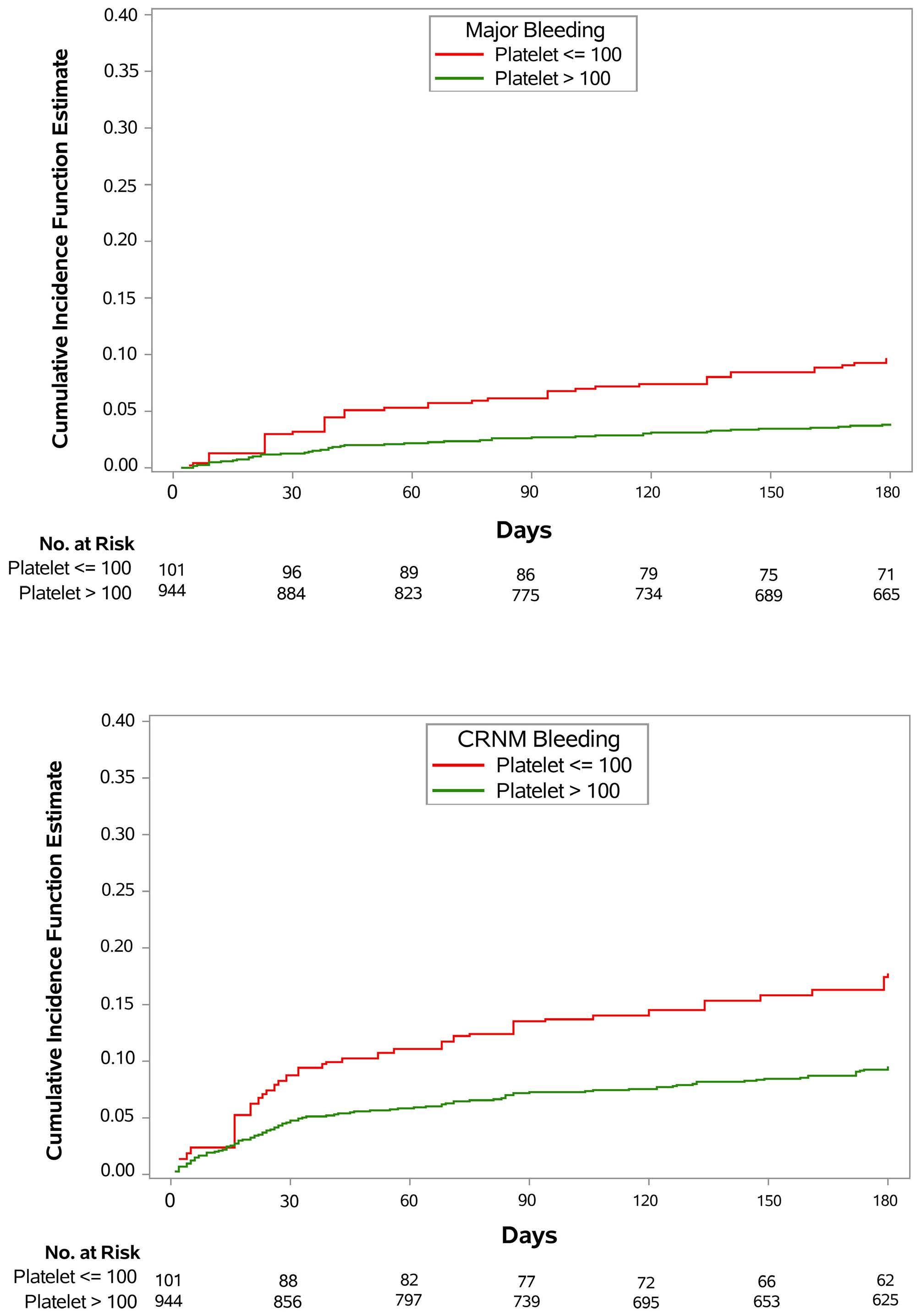
Figure 1. Cumulative incidences of major bleeding and clinically relevant non-major bleeding. (A) Major bleeding and (B) clinically relevant non-major (CRNM) bleeding. Red line represents thrombocytopenic group (patients with platelet count ≤100x109/L at 1 or more of 3 prespecified time points: baseline, 1-month, or 3-month). Green line represents the non-thrombocytopenic group (platelet count >100x109/L at all 3 time points).
acute symptomatic or incidental proximal lower extremity deep vein thrombosis or pulmonary embolism (symptomatic or incidentally detected involving segmental or more proximal pulmonary arteries). Cancer diagnosis was required to be within 2 years preceding thrombotic event and either a cancer diagnosis that was recurrent, metastatic, regionally advanced, or actively receiving cancer-directed therapy (or received treatment in the last 6 months or hematologic malignancies not in remission were eligible). Relevant exclusion criteria included platelet count <50x109/L at enrollment. All patients were treated with an anticoagulant for at least 6 months and up to 1 year. The primary composite outcome included recurrent thromboembolism, bleeding, and death. Bleeding was graded in accordance with previously published criteria by the International Society of Thrombosis and Hemostasis (ISTH).18 All outcomes were adjudicated independently by a committee as per prespecified criteria outlined in the study protocol. Per study protocol, for thrombocytopenia associated with chemotherapy in the first month of enrollment, if platelet count was 50-100x109/L, dalteparin dose was reduced by 2,500 IU until platelet recovery >100x109/L, and held if platelet count <50x109/L. Between 2 to 6 months, dalteparin dose was similarly adjusted or held, except in patients with body weight ≥99 kg, where dalteparin was reduced by 3,000 IU (instead of 2,500 IU). Edoxaban dose adjustment was based on body weight ≤60 kg, creatinine clearance between 30-50 mL/minute (min) inclusive, or concomitant use of P-glycoprotein (P-gp) inhibitors, without regard for platelet count. Dose interruption was allowed for any medical condition where continuing study drug may expose the subject to an increased hazard.
In order to assess the impact of thrombocytopenia on outcomes in these analyses, patients were grouped according to the first three time points in the trial that included blood counts (baseline, 1-month, 3-month). For the primary analysis, participants with platelet count ≤100x109/L at any of the three time points were included in the thrombocytopenic group and those without were in the non-thrombocytopenic group. The primary outcome for these analyses was major bleeding. Secondary outcomes included recurrent thrombosis, clinically relevant non-major bleeding (CRNMB), and mortality.
Statistical analysis
We estimated the cumulative incidence of the bleeding and thrombotic outcomes by identifying death as a competing risk.19 Statistical differences between the platelet cohorts (≤100x109/L or >100x109/L) were assessed using Gray test.20 We used the Fine-Gray method to construct time-to-event models and report the associated sub distribution function hazard ratios. Besides the platelet cohorts, dose-adjustment at randomization was included as a covariate in the model. Events occurring from randomization up to 180 days were included. For a given event in analysis if the event did not occur during this 180-day period, the subject was consid-
ered censored at 180 days. For overall survival the platelet cohorts (≤100x109/L or >100x109/L) and dose-adjustment at randomization were included in a Cox proportional hazard regression model and reported as hazard ratios with 95% confidence intervals.
Cumulative incidence (with death as a competing risk) of major bleeding and CRNMB was further estimated and compared statistically by Gray test within the thrombocytopenic cohort (≤100x109/L) based on treatment arm (edoxaban vs. dalteparin) for gastrointestinal (GI) cancers and hematologic malignancies separately.
This analysis included an overall cohort of 1,045 patients, with a mean age of 64 years and 48% female. One patient from the original trial was excluded due to insufficient laboratory data. Most common sites of malignancy included gastrointestinal (30%), lung (14%), genitourinary (14%), breast (12%), and gynecological (11%). Hematologic malignancies accounted for approximately 10% of the cohort (Table 1). Of patients with solid tumor diagnoses, 53% had metastatic disease and 30% had recurrent disease at enrollment. Qualifying thrombotic events included pulmonary embolism for 63% and isolated deep vein thrombosis in 37%. Thrombocytopenia (<100x109/L) was present in 101 patients (9.6%) of the total cohort. Only 14 patients had a documented platelet count <50x109/L at any of the three time points. Of the 101 patients, 52 were first noted to have platelet count <100x109/L at baseline, 28 at 1-month and 21 at 3-months. In the thrombocytopenic group, 76 (75.3%) had thrombocytopenia at only one (of 3) time point, 15 (14.9%) at two time points, and 10 (9.9%) at all three time points. The two cohorts were comparable with respect to demographics, cancer distribution, and assignment to treatment arm (Table 1). A higher proportion of patients in the thrombocytopenic cohort had hematologic malignancies (21.8% vs. 9.8%; P<0.01).
The estimated cumulative incidences at 180 days of all bleeding outcomes were higher in the thrombocytopenic group versus the non-thrombocytopenic group, including major bleeding (9.0% vs. 4.0%; SHR=2.4; P=0.02), CRNMB (17.9% vs. 9.6%; SHR=2.0; P=0.01), and major or CRNMB (24.8% vs. 12.3%; SHR=2.3; P<0.001) (Table 2). However, recurrent thrombosis were not statistically significantly different between the two groups. There was no significant difference between the two groups for death from any cause (21.8% vs. 26.0%; hazard ratio [HR]=0.9; P=0.48) or event-free survival (65.3% vs. 68.6%; HR=0.87; P=0.44) (Table 2).
Within the thrombocytopenic group, there were 33 patients with GI malignancies and 78 patients with non-GI malignancies. The thrombocytopenic group patients with non-GI malignancies experienced similar rates of major bleeding at 180 days (5.5 vs. 12.1; P=0.30) and CRNMB (22.5 vs. 16.5; P=0.48) for those assigned to edoxaban compared
Table 1. Demographic and clinical characteristics of the patients at baseline by thrombocytopenia.
Characteristics
N (%)
Met criteria to receive low dose of edoxaban based on CrCl, weight or drug interaction, N (%)
Qualifying diagnosis of venous thromboembolism, N (%)
PE with or without DVT
DVT only
Symptomatic DVT or PE
Incidental DVT or PE
Cancer site, N (%)
Frequency of thrombocytopenia, Plt ct ≤100x109/L, N (%)
At only 1 time point
At 2 time
anticoagulant, N (%)
Criteria to dose-reduce edoxaban include body weight ≤60 kg, creatinine clearance (CrCL) between 30 and 50 mL/minute inclusive, or concomitant use of P-glycoprotein (P-gp) inhibitors. Plt ct: platelet count; SD: standard deviation; DVT: deep vein thrombosis; PE: pulmonary embolism.
to dalteparin (Table 3). Of thrombocytopenic patients with GI malignancies, edoxaban was associated with higher rates of major hemorrhage (16.8 vs. 0; P<0.01) and CRNMB (25.8 vs. 0; P<0.01). These findings are consistent with previous observations of increased risk of hemorrhage with edoxaban compared with dalteparin in patients with GI malignancies.21 In the thrombocytopenic cohort, 22 patients (21.8%) had underlying hematologic malignancies and experienced higher rates of major bleeding with dalteparin compared to edoxaban (19.0% vs. 0; P<0.01).
In this post hoc analysis of the Hokusai VTE Cancer Study, thrombocytopenia was associated with a significantly in-
days.
Bleeding outcomes
Major bleeding
Clinically relevant nonmajor bleeding
Major or clinically relevant nonmajor bleeding
Thrombosis outcomes
Recurrent venous thromboembolism
Recurrent
creased risk of hemorrhage with an approximate doubling of major bleeding risk during the first 6 months after anticoagulation. To our knowledge this is the first analysis of a prospective, randomized trial dataset to evaluate the subgroup of patients with cancer-associated thrombosis and thrombocytopenia.
Thrombocytopenia, thrombosis, and bleeding are common complications in patients with active malignancy. Thrombocytopenia can result from underlying malignancy (commonly seen in hematologic malignancies) or emerge as a consequence of cancer-directed systemic therapies.6 In this randomized controlled study we found that thrombocytopenia was present in approximately one-tenth of enrolled patients with active malignancy and thrombosis. While this represents a clinically significant proportion, real-world evidence suggests this is an underestimation
Non-thrombocytopenic group, CIF (95% CI) Plt ct >100x109/L N=944
(2.9-5.4)
(5.8-9.4)
(2.6-4.8)
(3.6-5.9)
Thrombocytopenic group,
CIF: cumulative incidence function; CI: confidence interval; Plt ct: platelet count.
(5.5-17.4)
Table 3. Cumulative incidence function (in percentage) of bleeding in thrombocytopenic group, by treatment drug and cancer type.
Platelet count
All patients; Edoxaban N=48, Dalteparin N=53
Patients with GI malignancies; Edoxaban N=12, Dalteparin N=11
Patients with non-GI malignancies; Edoxaban N=36, Dalteparin N=42 ≤100x10
Patients with hematologic malignancies; Edoxaban N=12, Dalteparin N=10 ≤100x10
Patients with non-hematologic malignancies; Edoxaban N=36, Dalteparin N=43
CRNMB: clinically relevant non-major bleeding; GI: gastrointestinal.
of co-occurrence of thrombocytopenia in cancer patients with acute VTE.7 A recent study found the prevalence of thrombocytopenia (platelet count <100x109/L) in CAT to be in 22% with solid tumors diagnoses and 47% with hematologic malignancies. We attribute the difference to the inherent nature of strict inclusion criteria in a prospective randomized controlled trial (i.e., patients with platelet count <50x109/L were excluded on enrollment).
Patients with malignancies that receive anticoagulation have up to a 2-fold increased risk of major bleeding compared with anticoagulated patients without cancer.5,22 Data on the estimates of bleeding in patients with cancer thrombosis and thrombocytopenia are quite limited. A systematic review identified only two retrospective cohort studies with cancer-associated thrombosis and thrombocytopenia. In a study of 128 patients with hematologic malignancies that were diagnosed with acute thrombosis the 6-month cumulative incidence rates of hemorrhage were 6.5 (95% confidence interval [CI]: 2.2-19.5) in patients with significant thrombocytopenia (<50 K/µL) versus 1.3 (95% CI: 0.2-8.9) for those without.10 In a recent multicenter prospective study in the US that enrolled 121 patients with acute CAT and platelet count <100x109/L at time of thrombosis, 19% of patients had major bleeding in the first 60 days (95% CI: 13-27). Notably, this trial enrolled only a minority of patients with solid tumors (N=36, 30%)23 In this post hoc analysis of a randomized controlled trial we found that in patients with predominantly mild thrombocytopenia, onefourth of the patients developed clinically relevant bleeding (major bleeding or CRNMB) in the first 6 months after anticoagulation and the 180-day cumulative incidence of major bleeding was 8.9. This study represents the largest published cohort describing outcomes in patients with predominantly solid tumor diagnosis. These data highlight the importance of recognizing that even mild thrombocytopenia is a risk factor for major bleeding. The safety of DOAC in patients with thrombocytopenia is largely unknown. In the TROVE study, three of 16 patients with CAT and thrombocytopenia treated with DOAC developed CRNMB (cumulative incidence of 20%; 95% CI: 0-40).23
According to the treatment protocol in the current study, edoxaban dosing was not held or dose adjusted during periods of thrombocytopenia. Similar to the overall study findings, increased risk of hemorrhage was associated with edoxaban compared with dalteparin in patients with GI malignancies.21 When considering major bleeding, the difference between thrombocytopenic patients with GI cancers on the edoxaban arm versus the dalteparin arm is greater (16.8 vs. 0; P<0.01). This suggests that the bleeding signal previously seen in GI cancers was influenced by the thrombocytopenic population. In contrast, we found that in patients with thrombocytopenia with underlying hematologic malignancies experienced significantly higher rates of major bleeding with dalteparin compared to edoxaban (19.0% vs. 0; P<0.01). There remains a need to conduct prospective, randomized trials
to address the safety and efficacy of direct oral anticoagulants in these high-risk groups to generate quality evidence to guide clinicians.
Venous thromboembolism is associated with increased morbidity and mortality in cancer patients.3,24 The risk of recurrent VTE after a primary thrombotic event has been shown to be three to four times that of thrombosis in patients without cancer.1,5 In this study we demonstrate that although patients with thrombocytopenia had higher bleeding rates, the rate of recurrent thrombosis was not different compared to patients with platelets >100x109/L. This supports the growing evidence that thrombocytopenia is not protective for recurrent thrombosis in patients with active malignancy.25-27 Thus, the risk of hemorrhage needs to be balanced with a persistent risk of recurrent VTE when providers need to plan anticoagulation in this population. This study provides valuable insights by comparing bleeding and thrombosis rates in patients with cancer thrombosis with concomitant thrombocytopenia. The dataset is from a randomized controlled trial and thus leverages on strengths such as a prospective design, blinded validation of clinical outcomes and minimal attrition. However, we acknowledge that post hoc subgroup analyses of even high quality data has inherent limitations.28 Patients enrolled in clinical trials are often not representative of real-world scenarios.29 The HOKUSAI Cancer VTE study (as other similar trials) excluded patients with more severe thrombocytopenia at enrollment trial limiting our ability to study effects of more severe thrombocytopenia. This subgroup study had relatively few patients with thrombocytopenia (N=101). While rates of bleeding appear to be similar in the non-GI populations, we cannot conclude that outcomes are the same for DOAC versus low molecular weight heparin. In these analyses thrombocytopenia was defined based on the three earliest time points, however platelet counts in oncology populations are labile and thus we acknowledge these time points may not be reflective of platelet counts over the 180 days of the study period. Approximately half of cases were of mild-to-moderate thrombocytopenia in this study with platelet count in the 50-100x109/L range, and outcomes relative to severity of thrombocytopenia were not assessed due to limited sample sizes, and thus we are unable to make any conclusions on the safety of edoxaban in severe thrombocytopenia. These analyses were not prespecified, and patients in the two arms were not stratified by platelet count which should be recognized when evaluating the findings and the highlights the need for further prospective validation.
In conclusion, these post hoc analyses demonstrated that even mild thrombocytopenia was associated with a 2-fold risk of major bleeding. However, as consistent with prior retrospective studies, there was no concomitant decrease in recurrent thrombosis in cancer patients with thrombocytopenia. Thrombocytopenia is a frequent complication in patients with cancer and can have significant impact on outcomes of anticoagulation.
JIZ has received research funding from Incyte and Quercegen; consultancy services to Sanofi, CSL, and Calyx. CH has provided discloses consultancy services to Anthos Therapeutics. Gary Raskob reports consultancy fees or honoraria from AMAG Pharma, Anylam, Anthos Therapeutics, Bayer HealthCare Pharmaceuticals Inc., Bristol-Myers Squibb, Daiichi Sankyo Inc., Janssen Global Services LLC, Pfizer, and XaTrek; honoraria from BMS, Pfizer, Daiichi Sankyo; DSMB or advisory board membership from Anthos Therapeutics, Janssen, Bristol-Myers Squibb and Pfizer, leadership or fiduciary role in other board, society, committee or advocacy group of OU Health; stock or stock option ownership for AbbVie, Inc., Gilead Sciences Inc, GlaxoSmithKline, LLC., LLY, MRK, and Pfizer. MS, MAG and AD are employees of Daiichi. RP and HB have no conflicts of interest to disclose.
Contributions
JIZ and RP conceived the study. JIZ, RP and MS finalized
1. Levitan N, Dowlati A, Remick SC, et al. Rates of initial and recurrent thromboembolic disease among patients with malignancy versus those without malignancy. Risk analysis using Medicare claims data. Medicine (Baltimore). 1999;78(5):285-291.
2. Elting LS, Escalante CP, Cooksley C, et al. Outcomes and cost of deep venous thrombosis among patients with cancer. Arch Intern Med. 2004;164(15):1653-1661.
3. Khorana AA, Francis CW, Culakova E, et al. Thromboembolism is a leading cause of death in cancer patients receiving outpatient chemotherapy. J Thromb Haemost. 2007;5(3):632-634.
4 Al-Samkari H, Connors JM. Managing the competing risks of thrombosis, bleeding, and anticoagulation in patients with malignancy. Blood Adv. 2019;3(22):3770-3779.
5. Prandoni P, Lensing AW, Piccioli A, et al. Recurrent venous thromboembolism and bleeding complications during anticoagulant treatment in patients with cancer and venous thrombosis. Blood. 2002;100(10):3484-3488.
6. Liebman HA. Thrombocytopenia in cancer patients. Thromb Res. 2014;133 Suppl 2:S63-69.
7 Hsu C, Patell R, Zwicker JI. The prevalence of thrombocytopenia in patients with acute cancer-associated thrombosis. Blood Adv. 2023;7(17):4721-4727.
8. Al-Samkari H, Connors JM. Managing the competing risks of thrombosis, bleeding, and anticoagulation in patients with malignancy. Hematology Am Soc Hematol Educ Program. 2019;2019(1):71-79.
9 Samuelson Bannow BT, Lee A, Khorana AA, et al. Management of cancer-associated thrombosis in patients with thrombocytopenia: guidance from the SSC of the ISTH. J Thromb Haemost. 2018;16(6):1246-1249.
10. Khanal N, Bociek RG, Chen B, et al. Venous thromboembolism in patients with hematologic malignancy and thrombocytopenia. Am J Hematol. 2016;91(11):E468-E472.
11. Kopolovic I, Lee AY, Wu C. Management and outcomes of cancer-associated venous thromboembolism in patients with
data analysis plan. All authors contributed to study design and data analysis plan. Data analysis was performed by MS. The manuscript was drafted by JIZ and RP, all authors reviewed and made edits to the final manuscript.
Funding
No funding was received for the conduct of this study. JIZ is supported in part through the NIH/NCI Cancer Center Support Grant P30 CA008748. RP is supported in part by a2021 Conquer Cancer Career Development Award. MS, MG and AD are employees of Daiichi Sankyo Pharma.
Data-sharing statement
Individual participant data that underlie the results reported in this article, after de-identification may be available subject institutional review board and sponsor approval with a data use agreement on publication. Please contact zwickerj@mskcc.org for further details.
concomitant thrombocytopenia: a retrospective cohort study. Ann Hematol. 2015;94(2):329-336.
12. Ay C, Beyer-Westendorf J, Pabinger I. Treatment of cancerassociated venous thromboembolism in the age of direct oral anticoagulants. Ann Oncol. 2019;30(6):897-907.
13. Raskob GE, van Es N, Verhamme P, et al. Edoxaban for the treatment of cancer-associated venous thromboembolism. N Engl J Med. 2018;378(7):615-624.
14 Young AM, Marshall A, Thirlwall J, et al. Comparison of an oral factor Xa inhibitor with low molecular weight heparin in patients with cancer with venous thromboembolism: results of a randomized trial (SELECT-D). J Clin Oncol. 2018;36(20):2017-2023.
15. McBane RD, 2nd, Wysokinski WE, Le-Rademacher JG, et al. Apixaban and dalteparin in active malignancy-associated venous thromboembolism: the ADAM VTE trial. J Thromb Haemost. 2020;18(2):411-421.
16. Agnelli G, Becattini C, Meyer G, et al. Apixaban for the treatment of venous thromboembolism associated with cancer. N Engl J Med. 2020;382(17):1599-1607.
17 Hendriks T, McGregor S, Rakesh S, et al. Patient satisfaction after conversion from warfarin to direct oral anticoagulants for patients on extended duration of anticoagulation for venous thromboembolism - the SWAN Study. PLoS One. 2020;15(6):e0234048.
18. Schulman S, Kearon C, Subcommittee on control of anticoagulation of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost. 2005;3(4):692-694.
19 Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496-509.
20 Campigotto F, Neuberg D, Zwicker JI. Accounting for death as a competing risk in cancer-associated thrombosis studies. Thromb Res. 2012;129(Suppl 1):S85-87.
21. Kraaijpoel N, Di Nisio M, Mulder FI, et al. Clinical impact of bleeding in cancer-associated venous thromboembolism: results from the Hokusai VTE Cancer Study. Thromb Haemost. 2018;118(8):1439-1449.
22. Monreal M, Falga C, Valdes M, et al. Fatal pulmonary embolism and fatal bleeding in cancer patients with venous thromboembolism: findings from the RIETE registry. J Thromb Haemost. 2006;4(9):1950-1956.
23. Carney BJ, Wang TF, Ren S, et al. Anticoagulation in cancerassociated thromboembolism with thrombocytopenia: a prospective, multicenter cohort study. Blood Adv. 2021;5(24):5546-5553.
24. Sorensen HT, Mellemkjaer L, Olsen JH, et al. Prognosis of cancers associated with venous thromboembolism. N Engl J Med. 2000;343(25):1846-1850.
25. Samuelson Bannow BR, Lee AYY, Khorana AA, et al. Management of anticoagulation for cancer-associated thrombosis in patients with thrombocytopenia: a systematic review. Res Pract Thromb
Haemost. 2018;2(4):664-669.
26. Samuelson Bannow BT, Walter RB, Gernsheimer TB, et al. Patients treated for acute VTE during periods of treatmentrelated thrombocytopenia have high rates of recurrent thrombosis and transfusion-related adverse outcomes. J Thromb Thrombolysis. 2017;44(4):442-447.
27. Refaei M, Fernandes B, Brandwein J, et al. Incidence of catheter-related thrombosis in acute leukemia patients: a comparative, retrospective study of the safety of peripherally inserted vs. centrally inserted central venous catheters. Ann Hematol. 2016;95(12):2057-2064.
28. Bauchner H, Golub RM, Fontanarosa PB. Reporting and interpretation of randomized clinical trials. JAMA. 2019;322(8):732-735.
29. Kennedy-Martin T, Curtis S, Faries D, et al. A literature review on the representativeness of randomized controlled trial samples and implications for the external validity of trial results. Trials. 2015;16:495.
Sylvain Carras,1,2 Alexia Torroja,2 Anouk Emadali,3 Emilie Montaut,3 Nicolas Daguindau,4 Adrian Tempescul,5 Anne Moreau,6 Emmanuelle Tchernonog,7 Anna Schmitt,8 Roch Houot,9 Caroline Dartigeas,10 Sarah Barbieux,11 Selim Corm,12 Anne Banos,13 Ludovic Fouillet,14 Jehan Dupuis,15 Margaret Macro,16 Joel Fleury,17 Fabrice Jardin,18 Clementine Sarkozy,19 Ghandi Damaj,20 Pierre Feugier,21 Luc Matthieu Fornecker,22 Cecile Chabrot,23 Veronique Dorvaux,24 Krimo Bouabdallah,25 Sandy Amorim,26 Reda Garidi,27 Laurent Voillat,28 Bertrand Joly,29 Nadine Morineau,30 Marie Pierre Moles,31 Hacene Zerazhi,32 Jean Fontan,33 Yazid Arkam,34 Magda Alexis,35 Vincent Delwail,36 Jean Pierre Vilque,37 Loic Ysebaert,38 Barbara Burroni,39 Mary Callanan,40 Steven Le Gouill19 and Rémy Gressin;2 for the Lymphoma Study Association
1Université Grenoble Alpes, University Hospital, Institute For Advanced Biosciences (INSERM U1209, CNRSéUMR 5309, UGA), Molecular Biology Department, Grenoble; 2Université Grenoble Alpes, University Hospital, Institute For Advanced Biosciences (INSERM U1209, CNRS UMR 5309, UGA), Oncohematology Department, Grenoble; 3Université Grenoble Alpes, University Hospital, Institute for Advanced Biosciences (INSERM U1209, CNRS UMR 5309, UGA), Research & Innovation Unit, Grenoble; 4Hematology Department, Annecy Hospital, Annecy; 5Hematology Unit, Brest University Hospital, Brest; 6Pathology Department, University Hospital, Nantes; 7Hematology Department, University Hospital, Montpellier; 8Hematology Department, Cancerology Institute Bergonie, Bordeaux; 9Hematology Department, University Hospital, Rennes; 10Hematology Department, University Hospital, Tours; 11Hematology Department, Dunkerque Hospital, Dunkerque; 12Hematology Department, Chambery Hospital, Chambery; 13Hematology Department, Bayonne Cote Basque Hospital, Bayonne; 14Hematology Department, University Hospital, Saint Etienne; 15Lymphoid Malignancies Unit, Henri Mondor University Hospital, Assistance Publique-Hôpitaux de Paris, Créteil; 16IHBN – Hematology Department, University Hospital, Caen; 17Hematology Department, Cancerology Institute, Clermont-Ferrand; 18Hematology Department, Centre Henri Becquerel, Rouen; 19Institut Curie, Paris and Paris Saint Quentin University, UVSQ, Paris; 20Hematology Department, University Hospital, Caen; 21Hematology Department, University Hospital, Nancy; 22Hematology Department, University Hospital, Strasbourg; 23Hematology Department, University Hospital, Clermont-Ferrand; 24Hematology Department, University Hospital, Metz; 25Hematology Department, University Hospital, Bordeaux; 26Hematology & Cellular Therapy Department, Hospital Saint Vincent de Paul, Université Catholique de Lille, Lille; 27Hematology Department, Hospital Saint Quentin, Saint Quentin; 28Hematology Department, Hospital Chalon sur Saone, Chalon sur Saone; 29Hematology Department, Corbeil Hospital, Corbeil-Essonnes; 30Hematology Department, Hospital La Roche Sur Yon, La Roche Sur Yon; 31Hematology Department, University Hospital Angers, Angers; 32Hematology Department, Avignon Hospital, Avignon; 33Hematology Department, University Hospital, Besançon; 34Hematology Department, Mulhouse Hospital, Mulhouse; 35Hematology Department, Orleans Hospital, Orleans; 36OncoHematology Department, University Hospital Poitiers and INSERM, CIC 1402, University of Poitiers, Poitiers; 37Hematology Department, Cancer Center Baclesse Caen, Caen; 38Institut Universitaire du Cancer de Toulouse Oncopole, Toulouse; 39Assistance Publique – Hôpitaux de Paris (APHP), Hôpital Cochin, Department of Pathology, Centre de Recherche des Cordeliers, Sorbonne University, Inserm, UMRS 1138, Université Paris Cité, F-75006 Paris and 40Unit For Innovation in Genetics and Epigenetics and Oncology, Dijon University Hospital, Dijon, France
Correspondence: S. Carras scarras@chu-grenoble.fr
Received: June 29, 2023. Accepted: November 22, 2023. Early view: November 30, 2023. https://doi.org/10.3324/haematol.2023.283724
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Between 2011 and 2012, a phase II trial evaluated the use of the RiBVD (rituximab, bendamustine, velcade and dexamethasone) combination as first-line treatment for mantle cell lymphoma (MCL) patients over the age of 65. We have now re-examined the classic prognostic factors, adding an assessment of TP53 mutation status. Patients (N=74; median age 73 years) were treated with the RiBVD combination. Median progression-free survival (mPFS) was 79 months and median overall survival (mOS) was 111 months. TP53 mutation status was available for 54/74 (73%) patients. TP53 mutations (TP53mt) were found in 12 patients (22.2%). In multivariate analysis, among the prognostic factors (PF) evaluated, only TP53mt and an albumin level (Alb) 3.6 g/dL were independently associated with a shorter mPFS. A hazard ratio (HR) of 3.16 (1.3-9.9, P=0.014) was obtained for TP53mt versus TP53 wild-type (wt), and 3.6 (1.39-9.5, P=0.009) for Alb <3.6 g/dL versus Alb ≥3.6 g/dL. In terms of mOS, multivariate analysis identified three PF: TP53mt (HR: 5.9 [1.77-19.5, P=0.004]), Alb <3.6 g/dL (HR: 5.2 [1.4618.5, P=0.011]), and ECOG=2 (HR: 3.7 [1.31-10.6, P=0.014]). Finally, a score combining TP53 status and Alb distinguished three populations based on the presence of 0, 1, or 2 PF. For these populations, mPFS was 7.8 years, 28 months, and 2.5 months, respectively. Our prolonged follow-up confirmed the efficacy of the RiBVD regimen, comparing it favorably to other regimens. TP53mt and hypoalbuminemia emerge as strong PF that can be easily integrated into prognostic scores for older adult patients with MCL.
Mantle cell lymphoma (MCL) is a rare subtype of B-cell non-Hodgkin lymphoma characterized by the hallmark t(11;14)(q13;q32) chromosomal translocation that juxtaposes the CCND1 and IGH genes on the derivative chromosome 14, and triggers overexpression of the fusion protein.1,2 In addition to the t(11;14) translocation, the genetics of MCL are characterized by the occurrence of late complex secondary genomic events.3 Among these, TP53 alterations have emerged as a major unfavorable prognostic factor (PF) in MCL patients eligible for treatment with intensive therapies combining cytotoxic agents.4-7 However, in older patients, the impact of TP53 alterations remains less well characterized, partly due to a lack of large prospective studies.
Up to now, initial prognosis assessment has relied on clinical and biological parameters integrated into composite scores (Mantle Cell Lymphoma International Prognostic Index [MIPI], MIPIcombined, GOELAMS Index, etc.) to help define the risk of treatment failure. Nevertheless, these scores appear to work less well with older adult populations where high MIPI and MIPIb scores are over-represented, thus limiting their utility for risk stratification.8,9 Other PF have been proposed, such as complex karyotype, MYC rearrangement,10,11 alterations to KMT2D, CDKN2A, and SWI-SNF complex, or molecular signatures,7,12-14 but their usefulness with older adults remains unclear. It therefore appears important to identify new PF that can help guide therapeutic decisions related to novel, more effective, regimens for older adult subjects. No satisfactory international consensus has yet been reached for first-line management of MCL in older adults or patients who are ineligible for high-dose treatment. However, the literature provides some hints to select combination therapy that should improve progression-free
survival (PFS).8,9,15-20 We previously reported the results of a RiBVD (rituximab, bendamustine, velcade and dexamethasone) regimen without rituximab maintenance (RM) which extended median PFS (mPFS) up to five years.16 These results illustrate the significant recent improvements in clinical management of MCL compared to the 30% complete remission (CR) rate and mPFS of less than two years reported in 2000-2010 when the R-CHOP regimen was first described.
In this paper, we present an analysis of the long-term follow-up of the RiBVD phase II trial (clinicaltrials.gov identifier: 01457144) and of the PF affecting long-term survival for MCL patients, including TP53 status.
The study was approved by institutional review boards and ethics committees at all sites, and conducted in line with the Declaration of Helsinki. The study design, patients’ initial characteristics, treatment procedure, response assessment and minimal residual disease (MRD) analyses are all fully described in Gressin et al.16 Ki67 levels were assessed by immunohistochemistry (IHC) according to international recommendations.21 Staining for p53 was performed using an automated stainer (1:8000) (Automate Ventana Benchmark Ultra) with a mouse monoclonal antibody (Leica/NovoCastra clone DO7) diluted in a CC1 Tris-EDTA buffer, pH 7.8. p53 IHC was scored by visual inspection by two observers (BB and DC).22 Cases were scored on tissue microarrays (TMA) or whole slides depending on the type of sample. As the study was initially designed to monitor patients for up to five years, the updated long-term results presented here were obtained from routine practice records (comprising both clinical, morphological and biological follow-up). Data were obtained for the last consultation
or based on the recorded date of relapse, confirmed by scan or biological assessment. The final completion date for data inclusion was July 1st 2022.
For molecular analysis, using QIAprep Miniprep (Qiagen, Valencia, CA, USA), DNA was extracted from paraffin-embedded biopsy samples using a Promega A1720 kit (Promega) or from invaded bone marrow (BM) and/or peripheral blood (PB) specimens collected before initiating treatment. Samples were analyzed using a commercial Ion Ampliseq panel (TP53 panel #ID-TP5300K, Thermo Fischer Scientific, Waltham, MA, USA) including selected TP53 coding regions (exons 2 to 11, NM_000546) and splice sites (+5 bp). Ion Ampliseq libraries were prepared using 10 ng of genomic DNA. Libraries were qualified using an Agilent 4200 Tape Station System. A total of 200 base-read library templates were prepared with Ion 510 & Ion 520 & Ion 530 kits (Thermo Fischer Scientific) on an automated Ion Chef system (Thermo Fischer Scientific), according to the manufacturer’s instructions. Libraries were sequenced on an Ion GeneStudio S5 Plus system (Thermo Fischer Scientific). Torrent Suite version 5.0 software and Ionreporter pipelines (Thermo Fischer Scientific) were used independently to perform the primary analysis.
The prognostic value of all the factors included in the MIPI, MIPIc, and GOELAMS indexes were investigated by univariate analysis: lactate dehydrogenase (LDH) level (N vs. >N), Ki67 (≤30% vs. >30%), ECOG (0-1 vs. 2), B symptoms (No vs. Yes), together with absolute blood lymphocyte count (Lc) (N vs. >5 g/L [Lc>5]), cytological features (common vs. blastoid variant), Alb in g/dL (<3.6 g/dL vs. ≥3.6 g/dL) and TP53 mutational status (TP53mt vs. wild-type [TP53wt]). For Ki67 analysis, the best cut-off for survival prediction was determined by receiver operating characteristic (ROC) curve analysis (45%; data not shown). This cut-off was then applied in further analyses.
Survival functions (PFS and overall survival [OS]) were estimated based on Kaplan-Meier curves. Survival distributions for each parameter were compared using a LogRank test. Cox regression multivariate analysis was performed with parameters significantly associated with PFS or OS according to these univariate analyses (LogRank P<0.05). Statistical analyses were performed with R V4.2.1 software (CRAN).
Cohort characteristics
As described in Gressin et al.,16 74 patients were recruited (Table 1). Median age was 73 years (range: 69-77). A majority of patients had a high MIPI score (N=58, 69%), 10 (14%) had a blastoid/pleiomorphic cytology, and 34 (59%) presented Ki67 >30% (Table 1). After six cycles of RiBVD, 56/74 (75.5%) patients were in complete remission (CR).23 For the update presented here, the median follow-up for
living patients was 115 months (9.6 years) (Figure 1). Thirty-seven patients had died (50%) since the start of the study, with a median OS (mOS) of 111 months (9.3 years). The majority of deaths (21/37, 56%) were lymphoma-related. Other causes of death were: cardiovascular disease (N=4), other cancers (N= 3, Hodgkin lymphoma and adenocarcinoma of the pancreas and bladder), pneumonia (N=1), multifocal leukoencephalopathy (N=1), or unknown cause (N=6). Median PFS (mPFS) was 79 months (6.6 years), with 4 cases remaining in CR at the time of completion of the study. The median duration of response (mDOR) was 95 months (7.9 years). For blood MRD-negative patients after six cycles of RiBVD (87% of the 54 evaluable patients), the mPFS was 106 months (8.8 years).
In total, TP53 mutational status was analyzed for 54/74 (73%) patients. For 20 patients, lymphoma infiltration in BM and/or blood exceeded 15%, as assessed by flow cytometry. These samples were, therefore, suitable for TP53 analysis. In addition, tissue samples were available for 45 patients. Both tissue and informative PB/BM samples were available for 9/45 patients. A pathogenic TP53 mutation (TP53mt) was identified in 12 patients (22.2%). For the 9 patients for whom both tissue and PB/BM samples were available, the TP53 mutational status was fully concordant across both sample types (3 mt and 6 wt). Among TP53mt patients, high frequencies of allelic variants were detected in 6/12, suggesting either loss of heterozygosity and/or concomitant TP53 deletion.
Analysis of the whole cohort and the TP53 informative cohort revealed no differences in terms of baseline characteristics including MIPI scores, percentage of bulky lesions, extra-nodal involvement, or Ki67 levels (Table 1).
Univariate analysis of TP53 and other baseline prognostic factors at diagnosis
Univariate analysis revealed that patients with TP53 mutations presented a significantly shorter mPFS (7 months) and mOS (17.5 months) compared to patients with a TP53wt status (mPFS 61.5 months and mOS 95 months). TP53mt was associated with a worse response to treatment: 3/7 (43%) TP53mt MRD informative cases were positive for interim blood MRD as compared to 3/30 (10%) TP53wt MRD informative cases. In the final analysis of MRD in blood samples, there were 2/7 (28%) positive cases in the TP53mt MRD informative group versus 2/28 (7%) in the TP53wt MRD informative group. Finally, disease progression during therapy or within the six months following completion of the treatment schedule was observed in 5/12 (42%) TP53mt patients, as compared to 0/42 (0%) TP53wt patients. The following factors were also significantly associated with a shorter PFS according to univariate analysis: lymphocytes >5 g/L, Alb <3.6 g/dL, and Ki67 percentage. For OS, the following variables emerged as statistically significant: ECOG 0-1 versus 2, lymphocytes >5 g/L, Alb <3.6 g/dL, LDH >N, Ki67 >45%, and TP53mt (data not shown).
Table 1. Demographic characteristics of the TP53 informative cohort and of the whole cohort.
2 3 4
Extranodal involvement
F: female; M: male; BM: bone marrow; ECOG: Eastern Cooperative Oncology Group; int: intermediate; LDH: lactate dehydrogenase; MIPI: Mantle Cell Lymphoma International Prognostic Index. 1Median (interquartile range); N (%). 2Wilcoxon rank sum test; Pearson’s χ2 test.
Interestingly, MIPI, MIPIb, and GOELAMS Index were not significantly associated with outcome in this cohort.
Multivariate analyses of prognostic factors at diagnosis
A Cox model was applied to determine PFS, including statistically significant factors identified by univariate analysis (i.e., TP53 status, lymphocytes >5 g/L, LDH >N, Alb <3.6 g/dL and Ki67 >45%) as variables. From this analysis, only TP53mt and Alb <3.6 g/L emerged as independent prognostic factors for PFS in multivariate analysis. A hazard ratio (HR) of 3.16 (1.3-9.9, P=0.014) was found for TP53mt versus TP53wt, and 3.6 (1.39-9.5, P=0.009) for Alb <3.6 g/ dL versus Alb ≥3.6 g/dL (Figure 2A). When these two prog-
nostic markers were combined, patients with both low Alb and TP53mt (4/50, 8%) had a mPFS of 2.5 months, those with one PF (N=15/50, 30%) had a mPFS of 28 months, and finally those with Alb ≥3.6 g/dL and TP53wt (31/50, 62%) had a mPFS of 93 months (Figure 3A).
For OS, among the statistically significant factors identified by the univariate analysis, three PF appear to independently associate with an unfavorable outcome: TP53mt, ECOG=2 and Alb <3.6 g/dL. The respective HR for these PF are 5.9 (1.77-19.5, P=0.004), 5.2 (1.46-18.5, P=0.011), and 3.7 (1.31-10.6, P=0.014) (Figure 2B). When the three PF were combined, patients with two or all three PF (8/72, 10.5%) had an mOS of 15 months, those with one PF (N=21/72, 28.5%) had an
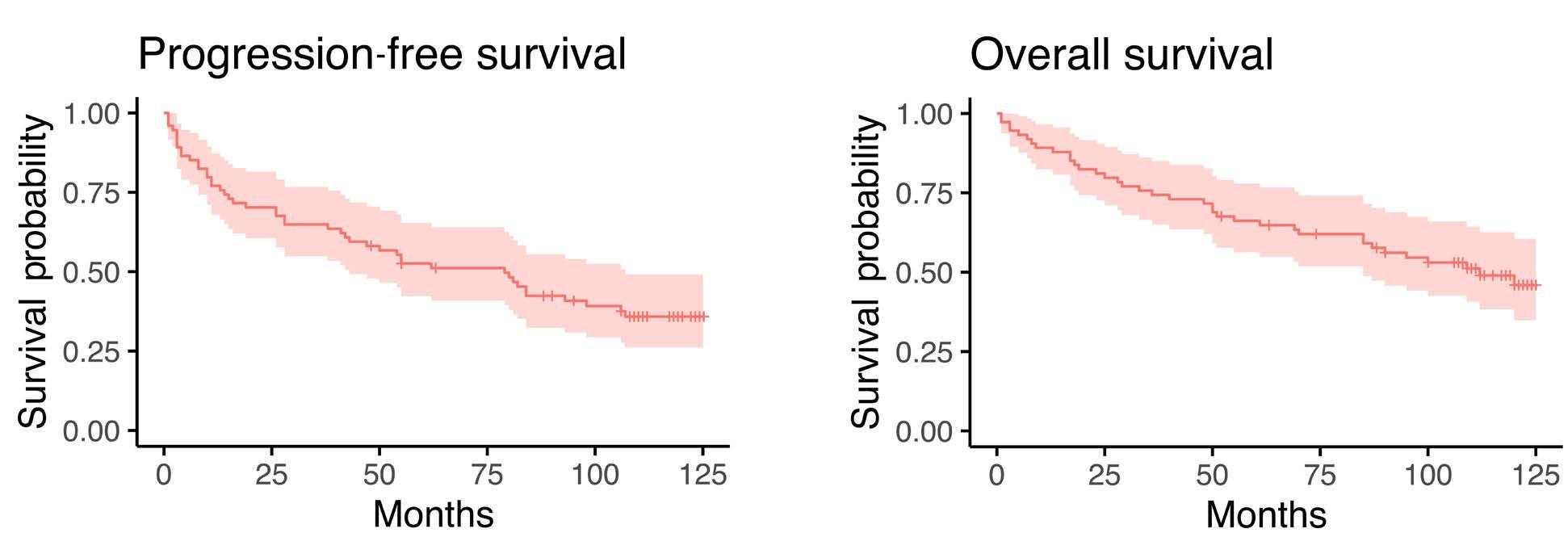
Figure 1. Outcomes for the whole population. Kaplan-Meier estimator of (A) progression-free survival and (B) overall survival for the whole population. Median follow-up was 115 months for surviving patients (N=37, 50%).
mPFS of 63 months, and, for those with no PF (45, 61%), mOS was not reached, and the estimated OS at 10 years was 61% (Figure 3B).
Correlation between TP53 status and p53 immunohistochemistry
Immunohistochemistry-based estimation of p53 expression was available for 9/12 TP53mt and 36/42 TP53wt patients. Mean percentages of positive tumoral cells were 33.6% and 8.1%, respectively (P=0.004). The level of p53 expression was above the threshold of 50% in 3/9 TP53mt cases (33.3%) versus 1/36 (2.8%) in the TP53wt group (P=0.03188) (Online Supplementary Figure S1).
We analyzed the long-term follow-up of the RiBVD phase II trial, involving a population of older adults with MCL. With a median follow-up of 9.6 years, the results confirmed the long-term efficacy of a 6-cycle RiBVD regimen without RM as first-line treatment for older adults with MCL, achieving mPFS and mOS of 5.2 years and 7.5 years, respectively. Our results suggest that this “maintenance-free regimen” is superior than previously published maintenance-free regimens for this age category (median 70-71 years), including R-CHOP, VR-CAP, RBAC500, and BR. With all these other treatment regimens, mPFS was between 22 and 35 months.8,19,24-26 In terms of mPFS, RiBVD also compared favorably with more recently published regimens, which always included at least RM. Thus, BR (+/- bortezomib) + RM (+/- lenalidomide)15,27 and BR (+/- ibrutinib) + RM (+/-ibrutinib)9 achieved complete response rates (CRR) of about 66%, and mPFS between 5.3 and 6.7 years.9,15 However, the precise comparison of these regimens is hampered by differences in inclusion criteria (e.g., median patient age 67 years for the ECOG-ACRIN E1411 study,27 and restriction of inclusion to patients with ECOG 0-1 for the SHINE study9). We, therefore, propose that the depth of response obtained after six cycles of RiBVD (with a CRR of 75.5% and blood MRD negativity in 87% of patients) could
explain the efficacy observed, even without RM. In our cohort, none of the multivariate prognostic MIPI, MIPIc or GOELAMS Index28-30 scores accurately predicted patient prognosis when treated with the RiBVD regimen. Despite the limited size of our cohort, these results suggest that these prognostic scores should be revisited, especially when dealing with populations of older adults, which is increasingly common. Interestingly, we identified mutations of TP53 and hypoalbuminemia (<3.6 g/dL) as independent unfavorable PF for both PFS and OS in this cohort of older adults with MCL. Based on our findings, TP53 status and albumin level discriminate three populations with a very different prognosis: mPFS of 7.75 years, 28 months, and 2.5 months, respectively, for populations with 0, 1, or 2 PF. Hypoalbuminemia was previously reported as an independent PF only in a few series of older adults with malignant lymphomas, including MCL.31-34 Here, it is confirmed to be an independent PF in MCL. Hypoalbuminemia is a factor of morbidity usually considered to reflect denutrition and frailty. Our results confirm that intrinsic patient characteristics should be among the considerations to review when choosing treatment strategies in older adults,33,34 especially in routine practice, as the real-world population of older adults tends to be more vulnerable and frail than patients included in clinical trials.
TP53 alterations in MCL have been reported in about 10-25% of cases, depending on whether only deletions, mutations, or both are considered.4,5,9,14,35,36 Recently, both the World Health Organization (WHO) and the International Consensus Classification of Mature Lymphoid Neoplasms (ICCN) have underlined the interest in molecular assessment of TP53 status in MCL at diagnosis.1,37 Nevertheless, whereas the unfavorable impact of TP53 alterations has been clearly demonstrated in young patients,4-7 the prognostic value of TP53 alterations in older adults ineligible for intensive strategies has only been suggested in a few studies.38,39 It, therefore, remained to be formally demonstrated. Recently, analysis of survival in the SHINE trial9 revealed a lack of benefit of adding ibrutinib to BR, suggesting that ibrutinib-based combinations cannot overcome the unfavorable impact of TP53 mutations in MCL. As yet, few data are
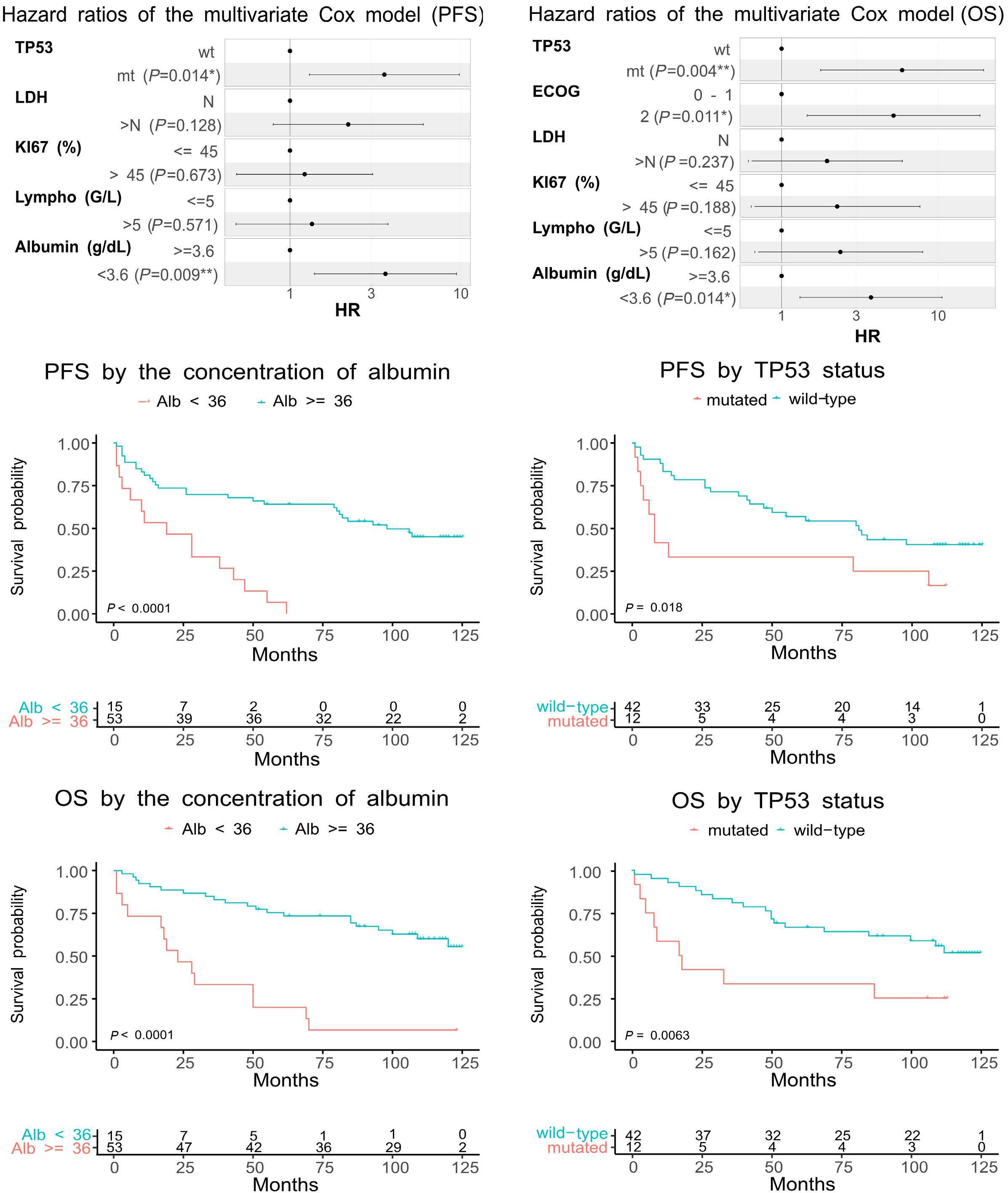
Figure 2. Multivariate analysis identifies albumin level and TP53 status as independent prognostic factors. Hazard ratios determined from multivariate Cox models of (A) progression-free survival (PFS) and (B) overall survival (OS). Only statistically significant factors, as identified by univariate analysis, were tested. Kaplan-Meier estimators for PFS (C-E) and OS (D-F), with patients classed depending on (C and D) albumin level or (E and F) TP53 status. alb: albumin; ECOG: Eastern Cooperative Oncology Group; LDH: lactate dehydrogenase; mt: mutated; N: number; wt: wild-type.
available on the efficacy of new targeted agents such as Btk inhibitors, venetoclax, or lenalidomide in patients with TP53 alterations.13,20,40,41 Nevertheless, the SHINE results9 highlight the need for a systematic assessment of TP53 status in ongoing and future trials to identify any potential benefits of treatments in this high-risk patient population.
Our results in a population of older adults are in line with previous reports; the RiBVD regimen used in this study failed to overcome the unfavorable prognostic impact of TP53 mutations. This finding reinforces the call for routine molecular testing for TP53, even in older adult patients, as proposed by the WHO and the ICCN.
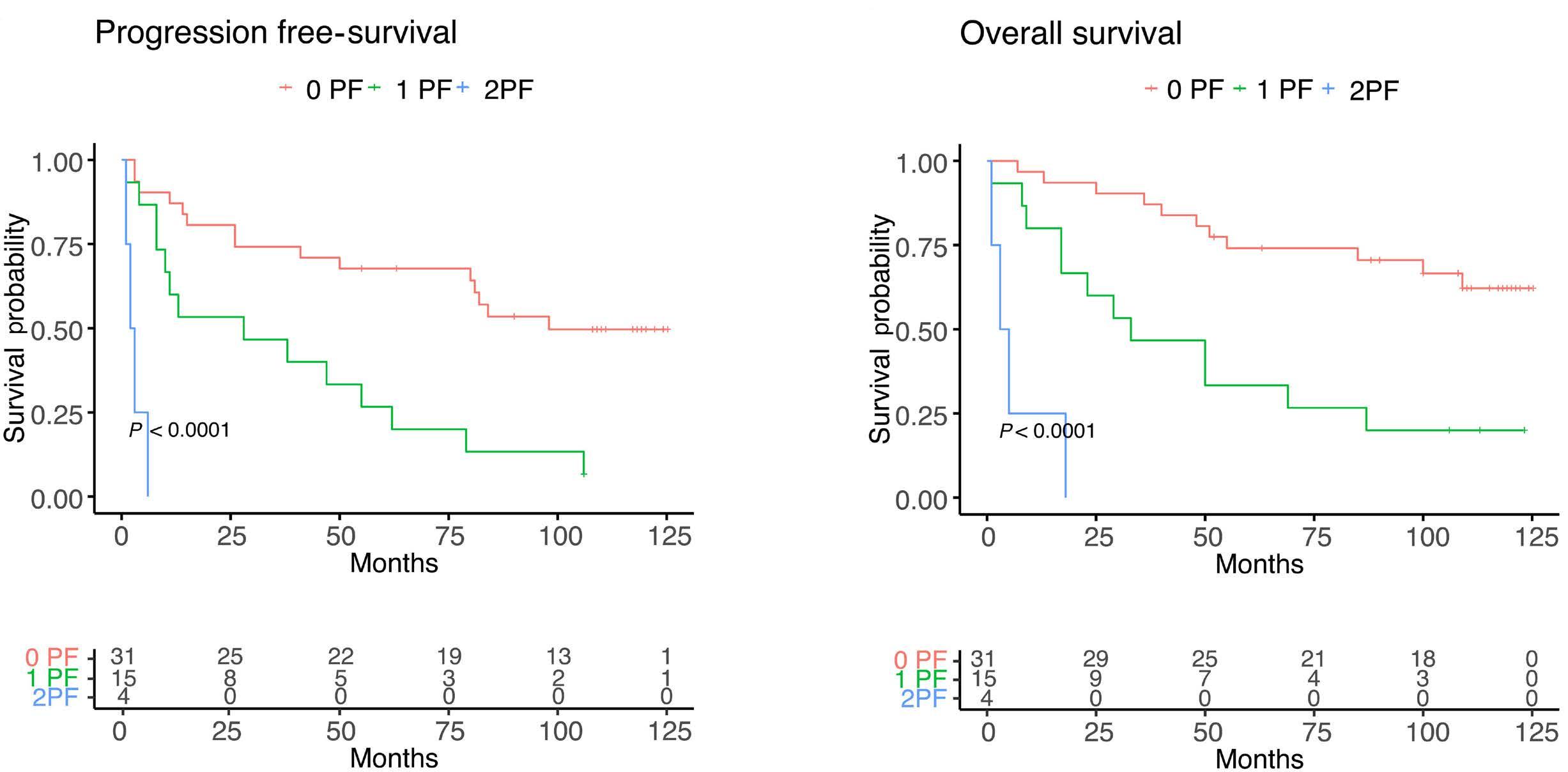
Figure 3. A composite score integrating albumin level and TP53 status is highly predictive for outcomes. Kaplan-Meier estimators for (A) progression-free survival and (B) overall survival. Patients were segregated using a composite score integrating albumin level (<3.6 g/dL or ≥3.6 g/dL) and TP53 status (mutated [mt] vs. wild-type [wt]) as prognostic factors (PF).
Previous studies have shown that high levels of p53 protein are associated with an unfavorable prognosis.22 It has, therefore, been proposed that IHC assessment of p53 expression levels be integrated into prognostic scores.42,43 However, despite differences in p53 expression levels between TP53wt and TP53mt samples in our study, IHC-estimated p53 levels do not appear to be a fully reliable surrogate marker for TP53 mutations. Considering the incidence of TP53 mutations and our results, we propose that TP53 status should be systematically assessed and integrated into prognostic indexes in routine practice and future trials, even for older adult patients.
The long-term follow-up presented here confirms the efficacy of the RiBVD regimen with respect to other non-maintenance regimens. The results also suggest that RiBVD compares favorably with more recent regimens integrating RM and iBTK alongside BR induction. Finally, TP53mt and hypoalbuminemia appear to be reliable unfavorable prognostic factors in older adults, and should, therefore, be systematically integrated in early assessments to identify very high-risk MCL patients as a guide to therapeutic de-
1. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
2. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
3. Nadeu F, Martin-Garcia D, Clot G, et al. Genomic and
cision-making.
Disclosures
No conflicts of interest to disclose.
Contributions
RG was the principal investigator of the clinical trial. MC was the biological investigator of the trial. SC and RG designed the current study, collected data, and wrote the manuscript. SC, AE and AT performed molecular analysis. BB performed histopathological analysis and analyzed IHC results. SC, RG and EM performed statistical analysis. ND, AT, AM ET, AS, RH, CD, SB, SC, AB, LF, JD, MM, JF, FJ, CS, GD, PF, LMF, CC, VD, KB, SA, RG, LV, BJ, NM, MPM, HZ, JF, YA, MA, VD, JPV, LY and SLG actively participated in the clinical trial. All authors reviewed the current manuscript.
Data-sharing statement
Clinical and biological data are available under formal request to the corresponding author.
epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood. 2020;136(12):1419-1432.
4 Delfau-Larue M-H, Klapper W, Berger F, et al. High-dose cytarabine does not overcome the adverse prognostic value of CDKN2A and TP53 deletions in mantle cell lymphoma. Blood. 2015;126(5):604-611.
5. Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify
younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903-1910.
6. Mareckova A, Malcikova J, Tom N, et al. ATM and TP53 mutations show mutual exclusivity but distinct clinical impact in mantle cell lymphoma patients. Leuk Lymphoma. 2019;60(6):1420-1428.
7 Ferrero S, Rossi D, Rinaldi A, et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high-dose therapy: a FIL study. Haematologica. 2020;105(6):1604-1612.
8. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle cell lymphoma (MCL): long-term follow-up of the Randomized European MCL Elderly Trial. J Clin Oncol. 2020;38(3):248-256.
9 Wang ML, Jurczak W, Jerkeman M, et al. Ibrutinib plus bendamustine and rituximab in untreated mantle-cell lymphoma. N Engl J Med. 2022;386(26):2482-2494.
10 Greenwell IB, Staton AD, Lee MJ, et al. Complex karyotype in patients with mantle cell lymphoma predicts inferior survival and poor response to intensive induction therapy. Cancer. 2018;124(11):2306-2315.
11. Wang L, Tang G, Medeiros LJ, et al. MYC rearrangement but not extra MYC copies is an independent prognostic factor in patients with mantle cell lymphoma. Haematologica. 2021;106(5):1381-1389.
12. Yi S, Yan Y, Jin M, et al. Genomic and transcriptomic profiling reveals distinct molecular subsets associated with outcomes in mantle cell lymphoma. J Clin Invest. 2022;132(3):e153283.
13. Agarwal R, Chan Y-C, Tam CS, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119-129.
14. Jain P, Dreyling M, Seymour JF, Wang M. High-risk mantle cell lymphoma: definition, current challenges, and management. J Clin Oncol. 2020;38(36):4302-4316.
15. Martin P, Cohen JB, Wang M, et al. Treatment outcomes and roles of transplantation and maintenance rituximab in patients with previously untreated mantle cell lymphoma: results from large real-world cohorts. J Clin Oncol. 2023;41(3):541-554.
16. Gressin R, Daguindau N, Tempescul A, et al. A phase 2 study of rituximab, bendamustine, bortezomib and dexamethasone for first-line treatment of older patients with mantle cell lymphoma. Haematologica. 2019;104(1):138-146.
17 Flinn IW, van der Jagt R, Kahl B, et al. First-line treatment of patients with indolent non-Hodgkin lymphoma or mantle-cell lymphoma with bendamustine plus rituximab versus R-CHOP or R-CVP: results of the BRIGHT 5-year follow-up study. J Clin Oncol. 2019;37(12):984-991.
18. Robak T, Jin J, Pylypenko H, et al. Frontline bortezomib, rituximab, cyclophosphamide, doxorubicin, and prednisone (VR-CAP) versus rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in transplantationineligible patients with newly diagnosed mantle cell lymphoma: final overall survival results of a randomised, open-label, phase 3 study. Lancet Oncol. 2018;19(11):1449-1458.
19 Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203-1210.
20. Yamshon S, Chen GZ, Gribbin C, et al. Nine-year follow-up of lenalidomide plus rituximab as initial treatment for mantle cell
lymphoma. Blood Adv. 2023;7(21):6579-6588.
21. Klapper W, Hoster E, Determann O, et al. Ki-67 as a prognostic marker in mantle cell lymphoma-consensus guidelines of the pathology panel of the European MCL Network. J Hematop. 2009;2(2):103-111.
22. Aukema SM, Hoster E, Rosenwald A, et al. Expression of TP53 is associated with the outcome of MCL independent of MIPI and Ki-67 in trials of the European MCL Network. Blood. 2018;131(4):417-420.
23. Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol Off J Am Soc Clin Oncol. 2007;25(5):579-86.
24. Fenske TS. Frontline therapy in mantle cell lymphoma: when clinical trial and real-world data collide. J Clin Oncol. 2023;41(3):452-459.
25. Robak T, Huang H, Jin J, et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944-953.
26. Visco C, Chiappella A, Nassi L, et al. Rituximab, bendamustine, and low-dose cytarabine as induction therapy in elderly patients with mantle cell lymphoma: a multicentre, phase 2 trial from Fondazione Italiana Linfomi. Lancet Haematol. 2017;4(1):e15-e23.
27. Smith MR, Jegede O, Martin P, et al. ECOG-ACRIN E1411 randomized phase 2 trial of bendamustine-rituximab (BR)based induction followed by rituximab (R) ± lenalidomide (L) consolidation for mantle cell lymphoma: effect of adding bortezomib to front-line BR induction on PFS. J Clin Oncol. 2021;39(15 Suppl):7503.
28. Hoster E, Dreyling M, Klapper W, et al. A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma. Blood. 2008;111(2):558-565.
29. Hoster E, Rosenwald A, Berger F, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol. 2016;34(12):1386-1394.
30 Gressin R, Caulet-Maugendre S, Deconinck E, et al. Evaluation of the (R)VAD+C regimen for the treatment of newly diagnosed mantle cell lymphoma. Combined results of two prospective phase II trials from the French GOELAMS group. Haematologica. 2010;95(8):1350-1357.
31. Peyrade F, Jardin F, Thieblemont C, et al. Attenuated immunochemotherapy regimen (R-miniCHOP) in elderly patients older than 80 years with diffuse large B-cell lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2011;12(5):460-468.
32. Mozas P, Rivero A, Rivas-Delgado A, et al. The Prognostic Nutritional Index (PNI) is an independent predictor of overall survival in older patients with follicular lymphoma. Leuk Lymphoma. 2022;63(4):903-910.
33. Akhtar OS, Anampa-Guzman A, Cortese MJ, et al. The Age, Comorbidities and Albumin (ACA) Index is independently associated with overall survival in patients with mantle cell lymphoma (MCL). Blood. 2022;140(Suppl 1):5160-5161.
34. Liu H, Zhang C-L, Feng R, et al. Validation and refinement of the Age, Comorbidities, and Albumin Index in elderly patients with diffuse large B-cell lymphoma: an effective tool for comprehensive geriatric assessment. Oncologist. 2018;23(6):722-729.
35. Shah NN, Castillo-Tokumori F, Whiting J, et al. Frontline treatment approaches in TP53-aberrant mantle cell lymphoma. Leuk Lymphoma. 2023;64(1):230-233.
36. Hill HA, Qi X, Jain P, et al. Genetic mutations and features of mantle cell lymphoma: a systematic review and meta-analysis. Blood Adv. 2020;4(13):2927-2938.
37. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140(11):1229-1253.
38. Narkhede M, Goyal G, Shea L, Mehta A, Giri S. Evaluating realworld treatment patterns and outcomes of mantle cell lymphoma. Blood Adv. 2022;6(14):4122-4131.
39 Malarikova D, Berkova A, Obr A, et al. Concurrent TP53 and CDKN2A gene aberrations in newly diagnosed mantle cell lymphoma correlate with chemoresistance and call for innovative upfront therapy. Cancers (Basel). 2020;12(8):2120.
40 Jain P, Zhao S, Lee HJ, et al. Ibrutinib with rituximab in first-
line treatment of older patients with mantle cell lymphoma. J Clin Oncol. 2022;40(2):202-212.
41. Le Gouill S, Morschhauser F, Chiron D, et al. Ibrutinib, obinutuzumab, and venetoclax in relapsed and untreated patients with mantle cell lymphoma: a phase 1/2 trial. Blood. 2021;137(7):877-887.
42. Rodrigues JM, Hassan M, Freiburghaus C, et al. p53 is associated with high-risk and pinpoints TP53 missense mutations in mantle cell lymphoma. Br J Haematol. 2020;191(5):796-805.
43. Stefancikova L, Moulis M, Fabian P, et al. Loss of the p53 tumor suppressor activity is associated with negative prognosis of mantle cell lymphoma. Int J Oncol. 2010;36(3):699-706.
Othman Al-Sawaf,1,2,3 Min-Hua Jen,4 Lisa M Hess,5 Jiewen Zhang,6 Benjamin Goebel,7 John M. Pagel,8 Sarang Abhyankar,5 Matthew S. Davids9 and Toby A. Eyre10
1University of Cologne, Faculty of Medicine and University Hospital Cologne, Department of Internal Medicine, Center for Integrated Oncology Aachen Bonn Cologne Duesseldorf, Cologne, Germany; 2Cancer Institute, University College London, London, UK; 3Francis Crick Institute, London, UK; 4Eli Lilly and Company, Bracknell, UK; 5Eli Lilly and Company, Indianapolis, IN, USA; 6TechDataServices, LLC, King of Prussia, PA, USA; 7Eli Lilly and Company, Bad Homburg, Germany; 8LOXO@Lilly, Indianapolis, IN, USA; 9Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA and 10Department of Hematology, Oxford University Hospitals NHS Foundation Trust, Oxford, UK
Correspondence: O. Al-Sawaf othman.al-sawaf@uk-koeln.de
Received: August 23, 2023.
Accepted: November 20, 2023. Early view: November 30, 2023.
https://doi.org/10.3324/haematol.2023.284150
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Venetoclax is a standard treatment for patients with chronic lymphocytic leukemia (CLL) following covalent Bruton tyrosine kinase inhibitor (cBTKi) therapy, despite relatively limited prospective data in this setting. Pirtobrutinib is a highly selective, non-covalent (reversible) BTKi that was designed to overcome the pharmacologic limitations of cBTKi and re-establish BTK inhibition. An unanchored matching-adjusted indirect comparison (MAIC) was conducted to estimate the treatment effect of pirtobrutinib versus venetoclax monotherapy in patients with cBTKi-pretreated CLL. Data from patients with CLL who were venetoclax-naïve and pretreated with cBTKi received pirtobrutinib (N=146) in the phase I/II BRUIN study were compared with the only identified trial of patients with CLL receiving venetoclax after a cBTKi (N=91), as administered as monotherapy until progression. Outcomes included progression-free survival (PFS), overall survival (OS), objective response rate (ORR), and treatment-emergent adverse events. Both unweighted and weighted analyses were conducted. PFS and OS of pirtobrutinib and venetoclax were comparable in both unweighted and weighted analyses (weighted hazard ratios for PFS: 1.01, 95% confidence interval [CI]: 0.58-1.73, P=0.98 and OS: 0.64, 95% CI: 0.25-1.67, P=0.34). ORR was significantly higher for pirtobrutinib (80.2% vs. 64.8%, P=0.01). Grade ≥3 treatment-emergent adverse events were lower in weighted analyses for pirtobrutinib versus venetoclax (all P<0.01), except for pneumonia, which was similar. These results suggest that pirtobrutinib may also be considered as an effective and well-tolerated treatment for patients with relapsed CLL following cBTKi.
Covalent Bruton tyrosine kinase inhibitor (cBTKi) therapy has increasingly become a standard of care worldwide for patients with chronic lymphocytic leukemia (CLL). Despite the marked efficacy of these agents, the majority of patients will eventually either progress or otherwise become intolerant to these agents, and as a result, the majority of patients will ultimately require additional treatment to achieve long-term disease control.1 Following progression or intolerance on cBTKi therapy, the BH3 mimetic agent and B-cell lymphoma-2 inhibitor (BCL2i) venetoclax, administered either alone or in combination with an anti-CD20 antibody, has become an important standard of care.1-4 While several retrospective studies, as well as pooled anal-
yses from early-phase clinical trials, have evaluated the effectiveness of venetoclax post-cBTKi,5-8 no randomized trials of venetoclax have been conducted exclusively in the post-cBTKi setting. The only prospective trial data of venetoclax in this setting is from a subset analysis of 91 heavily pretreated patients who had received at least one cBTKi. In the published interim analysis of these data with a median follow-up of 14 months, in which venetoclax was administered as a monotherapy continuously until progression, intolerance or withdrawal, the objective response rate (ORR) was 65% and median progression-free survival (PFS) was 24.7 months.9 As this is not feasible or desirable for all patients, alternative safe and effective treatment options for patients with CLL after failure of cBTKi therapy are warranted. While many specialists and institutions have
gained experience in the safe administration of venetoclax, careful patient selection and attention to patient care remain critical with adherence to the recommended ramp-up phase of treatment to avoid the serious adverse event (AE) of tumor lysis syndrome (TLS), which often requires administration of uric acid-lowering agents, and, less commonly, the need for hospitalization for TLS monitoring.10 Therefore, a need remains for additional safe and effective treatment options for patients with CLL after failure of cBTKi therapy. Pirtobrutinib is a highly selective, non-covalent (reversible) BTKi, that inhibits both wild-type and C481-mutant BTK with equal low nM potency and minimal in vitro off-target kinase activity. Pirtobrutinib is currently under investigation in multiple phase III trials for patients with CLL (clinicaltrials gov. Identifier: NCT05023980, NCT05254743, NCT04666038, and NCT04965493), and is approved for use in the US among patients with mantle cell lymphoma after at least two lines of therapy, including a cBTKi.11,12 Pirtobrutinib has been studied in the phase I/II BRUIN trial (clinicaltrials.gov Identifier: NCT03740529) for patients with B-cell malignancies, including 279 patients with CLL/SLL who received prior cBTKi therapy.13 In this cohort of patients who had a median of three prior lines of therapy (at least 1 containing a cBTKi), the ORR according to independent review (inclusive of partial response with lymphocytosis [PR-L]) was 73.3%, with a median PFS of 19.6 months. Among the 147 patients who had no prior BCL2i therapy, the median PFS was 22.1 months. Given these data, there are important questions regarding the comparative outcomes of single-agent pirtobrutinib and venetoclax in the post-cBTKi setting.
The primary objective of this study was to estimate the treatment effect for pirtobrutinib (BRUIN, clinicaltrials gov. Identifier: NCT03740529) versus venetoclax continuous monotherapy among patients with CLL who previously received treatment with a cBTKi in an unanchored matching-adjusted indirect comparison (MAIC).
A systematic literature review was conducted to identify published clinical trials of single-agent venetoclax among patients with relapsed/refractory CLL in the post-cBTKi setting (Online Supplementary Tables S1 and S2). One study met eligibility criteria (clinicaltrials gov. Identifier: NCT02141282).9 As only summary data were available from this trial, no selection criteria were applied to the cohort of patients treated with venetoclax; all available data were used. The analysis dataset from BRUIN was limited to patients diagnosed with CLL who had prior cBTKi exposure and excluded patients with prior BCL2i exposure, prior stem cell transplantation, or histopathological evidence of Richter transformation to more closely match the eligibility criteria for the venetoclax trial.9 The primary analysis used an informed covariate approach, which limited the covariates used in the reweighting exercise
to those with literature supporting their prognostic value. Covariates in the primary analysis included median patient age, median number of prior therapies, percent of patients who discontinued the prior cBTKi due to progression, as well as percent of patients with del(17p), del(11q), or unmutated immunoglobulin heavy variable (IGHV) gene. The following outcomes were reported in both trials and included in the MAIC: ORR by investigator assessment; PFS; OS; treatment-emergent adverse events (TEAE); and proportion of patients who discontinued treatment due to an AE. This comparison of pirtobrutinib versus venetoclax followed best practices in the identification of comparator studies and analysis of data using an unanchored MAIC.14 MAIC methods overcome limitations of naïve cross-trial comparisons14 by reducing ecological bias15 and allow for a more robust comparison between interventions that are not directly compared in a randomized trial. MAIC requires that individual patient-level data are available from at least one study, but are not available from all studies to be investigated.16
The method described by Guyot et al. 17 was used to simulate patient-level data from Kaplan-Meier charts and associated risk tables for the venetoclax trial. A lack of agreement was noted between the number at risk and the number censored in the published figures for PFS and OS in the venetoclax trial.9 As such, the digitized curve (generated using PlotDigitizer) was used to recalculate the number at risk to match the published image.
Patients in the pirtobrutinib cohort were re-weighted to match the measures of central tendency and proportion of patients for the characteristics reported for venetoclax. Since only summary baseline data were available from the venetoclax trial, the logistic regression model was estimated using the method of moments so that the weight for each individual patient was equal to the patient’s estimated odds (propensity) of being in the BRUIN study (pirtobrutinib) versus clinicaltrials gov. Identifier: NCT02141282 (venetoclax).14,16,18 Distribution of the weights applied were inspected for potential extreme values, which could be indicative of poor overlap between the study populations in the distributions of patient characteristics.19
Time-to-event outcomes were compared using Cox regression and log-rank tests; ORR and TEAE were evaluated using Fisher’s exact test. All outcomes were evaluated both as unweighted and weighted comparisons. Analyses were conducted using R4.1.2 (Posit Software PBC). Sensitivity analyses were conducted as summarized in the Online Supplementary Appendix.
The BRUIN trial began enrollment of patients to be treated with pirtobrutinib March 2019, and the study is ongoing. Data were available for analysis from the July 2022 data cut
at the time of this analysis. The venetoclax trial enrolled patients between September 2014 and November 2016, and the study was ongoing at the time of the publication of this interim analysis of the subset of patients with prior cBTKi exposure. Given the differences in time periods, a summary of the prior therapies received by patients is presented in Online Supplementary Table S3. To the best of our knowledge, no additional updates of this subset of patients treated with venetoclax have been presented. Both studies enrolled patients with CLL who had relapsed or refractory disease. For this analysis, patients in both cohorts were limited to those with prior cBTKi exposure and without prior venetoclax. After applying eligibility criteria, a total of 146 patients were available from the BRUIN trial for comparison to the venetoclax monotherapy cohort (N=91). Of note, there were no patients excluded due to having pathological evidence of Richter’s transformation.
The pirtobrutinib (N=146) and venetoclax (N=91) study cohorts included in this MAIC are presented in Table 1. Before matching, there were some differences between the trial populations studied, with patients in the pirtobrutinib study having a lower median number of prior lines of therapy, slightly older age, more patients who had discontinued the cBTKi due to progression, and a lower rate of unmutated IGHV. Median follow-up was 21.3 months and 14.0 months for the pirtobrutinib and venetoclax cohorts, respectively. After reweighting, all available characteristics were well balanced between cohorts, resulting in an effective sample size of 61. There were no significant differences observed in the unweighted or weighted comparisons of pirtobrutinib versus
venetoclax for either PFS or OS. Median PFS for pirtobrutinib was 22.1 months in unweighted and 19.4 months in weighted analyses, versus 24.7 months for venetoclax. Median OS for pirtobrutinib was not reached. The weighted HR for PFS was 1.01 (95% confidence interval [CI]: 0.58-1.73, P=0.98) and for OS was 0.64 (95% CI: 0.25-1.67, P=0.34) (Figures 1 and 2, respectively). Of note, six of the 28 (21.4%) observed deaths included in these time-to-event outcomes in the pirtobrutinib cohort were COVID19-related.
Response outcomes according to International Workshop on CLL in both unweighted and weighted analyses of pirtobrutinib versus venetoclax are presented in Table 2. ORR was 80.2% for patients treated with pirtobrutinib (inclusive of PR-L) versus 64.8% for patients treated with venetoclax (weighted odds ratio [OR]=2.22, 95% CI: 1.16-4.29, P=0.01). The rates of complete responses (CR) were 1.4% and 8.8%, respectively.
Each grade ≥3 TEAE reported in Jones et al.9 and recorded by both trials are summarized in Table 3. In both unweighted and weighted analyses, each grade ≥3 TEAE compared in this study was significantly lower for pirtobrutinib (all P<0.05), except for pneumonia, which was not significantly different between pirtobrutinib and venetoclax (weighted P=0.06). Similarly, each any grade TEAE demonstrated consistent findings for these differences between the two cohorts (Online Supplementary Table S5). There was no difference in the proportion of patients who discontinued therapy due to an AE in both unweighted and weighted analyses (weighted OR=0.44, 95% CI: 0.09-1.92, P=0.32). Each TEAE recorded in the supplemental venetoclax material that was also recorded in the pirtobrutinib trial is included in Online Supplementary Table S6, which reports details of events
aAll patients were included in the weighted analyses; however, reweighting resulted in an effective sample size of 61. bMedian number of prior lines of therapy =4 (range, 1-15). cMedian number of prior lines of therapy =3 (range, 1-9). dIncluded in sensitivity analyses only. BTKi: Bruton tyrosine kinase inhibitor; ECOG PS: Eastern Cooperative Oncology Group performance status; IGHV: immunoglobulin heavy-chain variable region gene.
such as infection, gastrointestinal disorder, metabolism and nutrition disorders, and neoplasms.
Sensitivity analyses
There were no differences between pirtobrutinib and venetoclax in the primary analysis, which limited the reweighting factors to those with known prognostic value, and sensitivity analyses, which included all baseline covariates (Online Supplementary Table S4). There were no significant differences in PFS, OS or treatment discontinuation due to adverse
events. Each grade ≥3 TEAE reported by both trials remained significantly lower for pirtobrutinib (all P<0.05), except for pneumonia, which was not also significantly different between pirtobrutinib and venetoclax (weighted P=0.06) in sensitivity analyses. There were extreme weights observed upon inspection as evidenced by the sharp drop in PFS, as a result of an event occurring for such a patient. Sensitivity analyses removing the patients with extreme weights did not change the statistical significance or direction of the HR or OR of any reported outcomes (data not shown).
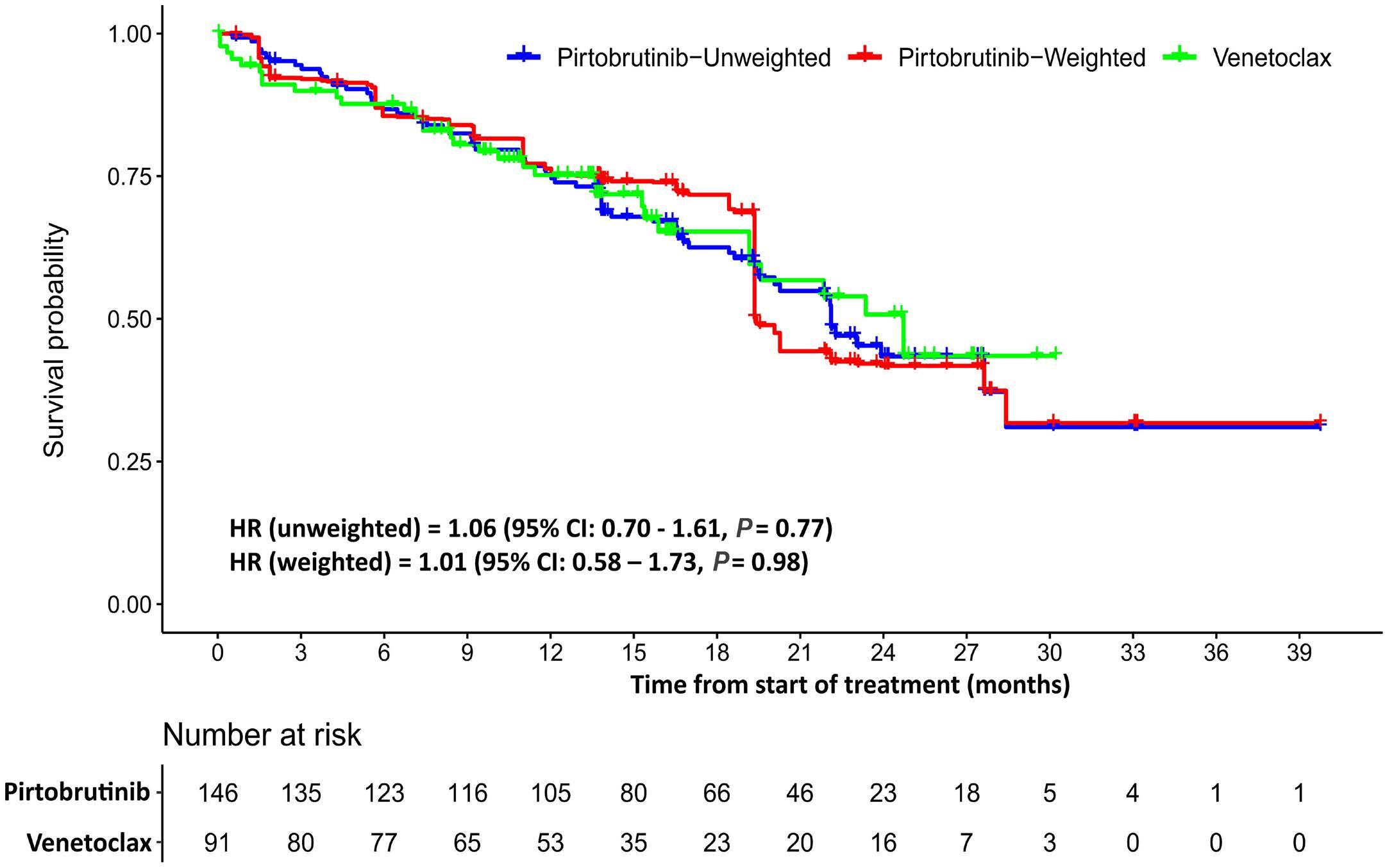
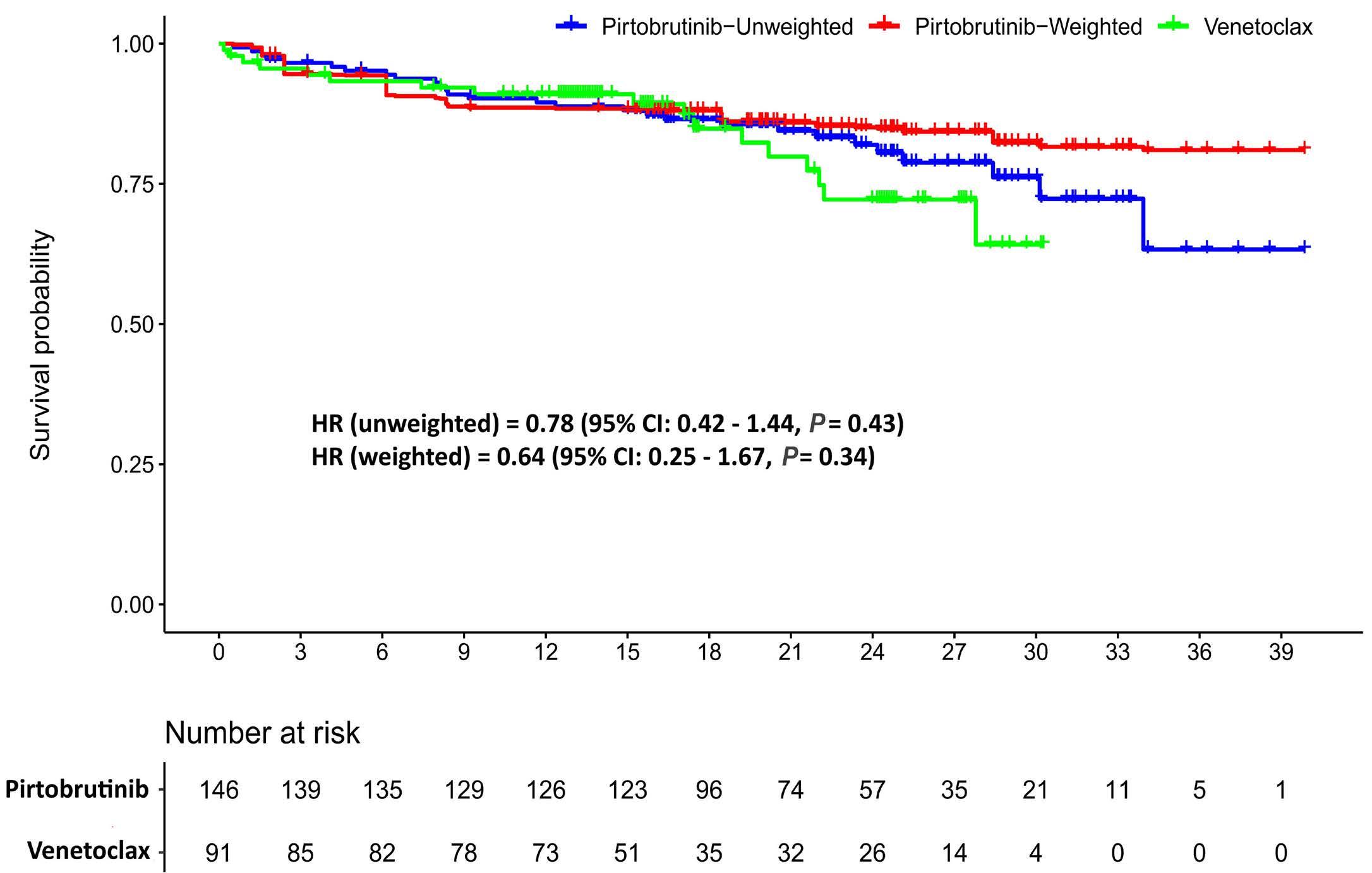
Table 2. International Workshop on Chronic Lymphocytic Leukemia response (%).
iwCLL response
aAll patients were included in the weighted analyses; however, reweighting resulted in an effective sample size of 61. IwCLL: International Workshop on Chronic Lymphocytic Leukemia; OR: odds ratio; CI: confidence interval; ORR: objective response rate; CR: complete response; Cri: CR with incomplete bone marrow recovery; PR: partial response; SD: stable disease; PD: progressive disease.
Table 3. Percent of patients with grade ≥3 adverse events.
Treatment discontinuation due to adverse events
aAll patients were included in the weighted analyses; however, reweighting resulted in an effective sample size of 61. OR: odds ratio; CI: confidence interval.
Venetoclax has become an important treatment option for patients with relapsed/refractory CLL following a cBTK inhibitor, although no randomized studies have been completed exclusively in this treatment setting. More recently, pirtobrutinib has shown promising activity in patients with relapsed/refractory CLL after cBTKi use and continues under investigation in this setting.13 No direct head-to-head data have been described between single-agent pirtobrutinib and venetoclax among these patients. Therefore, in the absence of a comparative trial, this MAIC was conducted to investigate the potential comparative outcomes of pirtobrutinib versus venetoclax in the treatment of CLL in the post-cBTKi setting. To do so required focusing on venetoclax monotherapy administered continuously until progression, as no data were identified evaluating time-limited venetoclax in combination with an anti-CD20 antibody in this treatment setting and highlights the limited published data for venetoclax post-cBTKi. While real-world data show that venetoclax monotherapy is the most common BCL2i-based therapy used post-cBTKi,20 other regimens, such as venetoclax plus rituximab or obinutuzumab, are also considered reasonable approaches in the relapsed/refractory setting. The landmark Murano trial, which studied a 24-month time-limited dura-
tion of venetoclax in addition to rituximab, only included five patients (2.5% of all patients in this arm of the trial) who had received prior B-cell receptor inhibitor-based therapy.21 There are no known trials of venetoclax plus obinutuzumab after cBTKi therapy, as this regimen was investigated in the first-line setting, limiting the ability to investigate other BCL2i-based therapies in the post-cBTKi setting.
The data from this MAIC suggest improved ORR associated with pirtobrutinib compared to venetoclax, with no differences observed in PFS and OS outcomes. ORR values reported in the venetoclax study were investigator-assessed; it is unknown if a comparison of response by independent review would have resulted in these same outcomes. Moreover, this analysis demonstrated that the comparative AE profiles of these agents potentially favored pirtobrutinib. Specifically, anemia, neutropenia, febrile neutropenia, and thrombocytopenia were each significantly lower in patients treated with pirtobrutinib; however, pneumonia and treatment discontinuations due to an AE were not different between pirtobrutinib and venetoclax.
This MAIC raises important questions about the sequencing of agents, particularly regarding the value of exhausting BTK pathway inhibition versus switching therapy based on mechanism of action. Pending the readout of upcoming randomized trials of pirtobrutinib, the placement of this agent
in the future care of patients with CLL remains an area of further evaluation. There is a need to not only rely on the results of these trials, but to proactively assess treatment sequencing of these agents in the real-world setting to optimize care for patients with CLL when a cBTKi is no longer an option. A multi-center cohort study evaluated outcomes of 63 patients with cBTKi pretreated CLL or Richter transformation (RT) who received treatment after non-covalent BTKi therapy, with more than 90% of these patients having received pirtobrutinib.22 In this cohort, eight patients with CLL and two with RT received venetoclax after the non-covalent BTKi. PFS for venetoclax for those with CLL was 14 months, and response to venetoclax was observed in seven of the ten patients.22 In a broader cohort of 247 patients enrolled the BRUIN trial with CLL who received prior cBTKi therapy, including 41% who had also received a BCL2i, the objective tumor response rate (ORR) was 73.3% and PFS was a median of 19.6 months.13 Pirtobrutinib has furthermore demonstrated efficacy in patients after both a prior cBTKi and BCL2i, with an ORR of 70.0% and median PFS of 16.8 months.13
Although the data from this MAIC further support the BRUIN trial data regarding the comparable efficacy of pirtobrutinib to venetoclax after prior non-covalent BTKi therapy, the sample size is small and the analysis only includes two trials; additional data are needed to inform treatment decision-making regarding the sequencing of care of patients with CLL. While a MAIC is an improved approach over the indirect side-by-side comparison of trials due to the reweighting algorithm, there are inherent limitations to indirect analyses that should be recognized when evaluating the findings from this study. It should be noted that in this MAIC, there were no patient-level data available for venetoclax. It is not possible to completely know if the outcomes observed would be replicated in a trial where cohorts were balanced at the individual patient level by means of randomization; while the mean/proportion of patients can be balanced, the distribution of outcomes is unknown. Prior research has shown that outcomes using MAIC methods may not always correspond to analyses where patient-level data are known for both treatment groups, but have also shown directional consistency in other studies and remain an area of uncertainty.23-25 Additionally, the reweighting exercise resulted in a smaller effective sample size; however, the effective sample size in this study is consistent with the proportion of the total sample as observed in similar analyses in CLL.26 While removing patients with extreme weights did not impact the results, there remains a limitation with lack of similarity of trials that led to these extreme weights. Therefore, these data alone preclude any definitive conclusions in the absence of randomized data and should be considered hypothesis generating findings warranting further study. Moreover, the covariates included in the analysis could not be individually evaluated due to the lack of patient-level data for venetoclax. In particular, minimal residual disease (MRD) could not be compared between trials given the lack
of baseline covariates for the subgroup assessed for MRD in the venetoclax trial. The balancing exercise was limited to those factors reported in both trials and exclude both measured and unmeasured factors that may introduce bias. For example, the venetoclax cohort was enrolled to the trial from 2014 to 2016, whereas the pirtobrutinib cohort did not begin enrollment until November 2018 and follow-up continued during the COVID19 pandemic, which can have an effect on the incidence of adverse events. Moreover, the OS outcomes could potentially be influenced by the pre- and post-protocol therapies received. While these are not evaluable due to lack of reported data, there is the possibility of more frequent use of PI3K inhibitors during the time period of the Jones et al. trial, whereas the use of PI3Ki agents has become less common due to toxicity concerns since 2018.27 Additionally, there may be some variability in the prior treatments received and other potentially prognostic variables, such as NOTCH1 mutation status, that could not be controlled by the reweighting exercise due to lack of data. The comparison of adverse events was also limited by the events reported by both trials. Furthermore, there may have been shifts in the care of patients between these non-contemporaneous trials, such as the time-limited use of venetoclax in combination with CD20 antibodies, that could have altered patient outcomes.
Despite the limitations of using a MAIC, this study provides initial insights and improves upon naïve indirect comparisons by adjusting for known cross-trial differences to suggest improved ORR, similar PFS and OS, and the favorable toxicity profile associated with pirtobrutinib. Patients who received cBTKi therapy are underrepresented in pivotal venetoclax studies, such as the MURANO trial, where less than 5% of patients had been exposed to BTK inhibitors before receiving venetoclax.3 The selection of treatment after cBTKi failure is a clinically relevant question, since the use of BTK inhibitors is widely established in most routine healthcare settings and post-BTKi salvage strategies remain understudied. This study provides data to inform treatment choice in a setting where little data exist.
In summary, this MAIC found that ORR of pirtobrutinib was higher and OS and PFS of pirtobrutinib was comparable to venetoclax monotherapy administered continuously until progression in patients with relapsed or refractory CLL previously treated with a cBTKi. Pirtobrutinib was also associated with a generally better toxicity profile compared to venetoclax, suggesting it may be an effective treatment option for patients who are venetoclax-naïve after progressing on a cBTKi.
MHJ, LMH, and BG are employees of Eli Lilly and company. JMP and SA are employees of LOXO@Lilly. JZ is an employee of DataTech Services, which receives funding from Eli Lilly and
Company for statistical and analytic data support services. MSD has received institutional research funding from AbbVie, AstraZeneca, Ascentage Pharma, Genentech, MEI Pharma, Novartis, Surface Oncology and TG Therapeutics; and personal consulting income from AbbVie, Adaptive Biosciences, Ascentage Pharma, AstraZeneca, BeiGene, BMS, Eli Lilly, Genentech, Genmab, Janssen, Merck, Mingsight Pharmaceuticals, ONO Pharmaceuticals, Secura Bio, TG Therapeutics, and Takeda. OAS reports advisory board membership at Ascentage, AstraZeneca, AbbVie, BeiGene, Eli Lilly, Gilead, Janssen and Roche; speaker honoraria from Adaptive, AstraZeneca, AbbVie, BeiGene, Eli Lilly, Gilead, Janssen and Roche; research funding from BeiGene, AbbVie, Janssen and Roche. TAE reports education honorarium, advisory board honorarium and funding for travel to scientific conferences from Roche; honorarium, research support and travel to scientific conferences from Gilead; education honorarium, advisory board honorarium from KITE; honorarium from Jansen; honorarium and travel expenses for scientific conferences from Abbvie; honorarium, research funding, travel expenses for scientific conferences from AstraZeneca; advisory board honorarium, trial steering committee membership at Loxo Oncology; advisory board honorarium, research funding from Beigene; advisory board honorarium from Incyte, Secura Bio and Autolus.
Contributions
Conceptualization, writing-review and editing was performed by OA-S. Conceptualization, methodology, formal analysis, validation, investigation, data curation, writing-review and
1. Roeker LE, Mato AR. Approaches for relapsed CLL after chemotherapy-free frontline regimens. Hematology Am Soc Hematol Educ Program. 2020;2020(1):10-17.
2. NCCN. NCCN Clinical Practice Guidelines in Oncology: CLL/SLL Version 2.2023. 2023. https://www.nccn.org/professionals/ physician_gls/pdf/cll.pdf. Accessed March 17, 2023.
3. Seymour JF, Kipps TJ, Eichhorst BF, et al. Four-year analysis of Murano study confirms sustained benefit of time-limited venetoclax-rituximab (VenR) in relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL). Blood. 2019;134(Suppl 1):355.
4 Seymour JF, Kipps TJ, Eichhorst BF, et al. Enduring undetectable MRD and updated outcomes in relapsed/ refractory CLL after fixed-duration venetoclax-rituximab. Blood. 2022;140(8):839-850.
5. Eyre TA, Kirkwood AA, Gohill S, et al. Efficacy of venetoclax monotherapy in patients with relapsed chronic lymphocytic leukaemia in the post-BCR inhibitor setting: a UK wide analysis. Br J Haematol. 2019;185(4):656-669.
6. Mato AR, Nabhan C, Barr PM, et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: a real world experience. Blood. 2016;128(18):2199-2205.
7 Hampel PJ, Rabe KG, Call TG, et al. Clinical outcomes in patients with chronic lymphocytic leukemia with disease progression on ibrutinib. Blood Cancer J. 2022;12(9):124.
8. Roberts AW, Seymour JF, Eichhorst B, et al. Pooled multi-trial
editing was performed by M-HJ. Conceptualization, methodology, validation, investigation, writing-original draft, visualization was performed by LMH. Formal analysis, validation, data curation, writing-review and editing was performed by JZ. Writing-review and editing was performed by BG and SA. Conceptualization, writing-review and editing was performed by JMP and TAE. Investigation, writing-review and editing was performed by MD.
This was an unfunded study with employee time and data resources provided by Eli Lilly and Company.
Eli Lilly and Company provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available 6 months after the indicated study has been approved in the USA and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data-sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data-sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
analysis of venetoclax efficacy in patients with relapsed or refractory chronic lymphocytic leukemia. Blood. 2016;128(22):3230.
9 Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19(1):65-75.
10 Fischer K, Al-Sawaf O, Hallek M. Preventing and monitoring for tumor lysis syndrome and other toxicities of venetoclax during treatment of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2020;2020(1):357-362.
11. Lilly. Jaypirca (pirtobrutinib) prescribing information. 2023. https://uspl.lilly.com/jaypirca/jaypirca.html#pi. Accessed March 28, 2023.
12. Wang ML, Jurczak W, Zinzani PL, et al. Pirtobrutinib in covalent BTK-inhibitor pretreated mantle cell lymphoma. J Clin Oncol. 2023;41(24):3988-3997.
13. Mato AR, Woyach JA, Brown JR, et al. Pirtobrutinib after a covalent BTK inhibitor in chronic lymphocytic leukemia. N Engl J Med. 2023;389(1):33-44.
14. Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-947.
15. Greenland S, Morgenstern H. Ecological bias, confounding, and effect modification. Int J Epidemiol. 1989;18(1):269-274.
16. Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200-211.
17 Guyot P, Ades A, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9.
18. Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials. Pharmacoeconomics. 2010;28(10):935-945.
19 Jiang Y, Ni W. Performance of unanchored matching-adjusted indirect comparison (MAIC) for the evidence synthesis of singlearm trials with time-to-event outcomes. BMC Med Res Methodol. 2020;20(1):241.
20 Eyre TA, Hess LM, Sugihara T, et al. Clinical outcomes among patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) who received treatment with a covalent BTK and BCL2 inhibitor in the United States: a realworld database study. Leuk Lymphoma. 2023;65(5):1005-1016.
21. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax–rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107-1120.
22. Thompson MC, Coombs CC, Roeker LE, et al. Outcomes of therapies and resistance mutations following non-covalent
Bruton’s tyrosine kinase inhibitor treatment for patients with chronic lymphocytic leukemia and richter transformation. Blood. 2022;140(Suppl 1):9885-9888.
23. Wong EC, Dulai PS, Marshall JK, Jairath V, Reinisch W, Narula N. Matching-adjusted Indirect comparisons vs propensity score matching with individual patient-level data to estimate treatment efficacy. Inflamm Bowel Dis. 2024;30(2):311-313.
24. Signorovitch J, Diels J, Van Sanden S, et al. Matching-adjusted indirect comparison (MAIC) results confirmed by head-to-head trials: a case study in psoriasis. J Dermatolog Treat. 2023;34(1):2169574.
25. Phillippo DM, Dias S, Elsada A, Ades A, Welton NJ. Population adjustment methods for indirect comparisons: a review of national institute for health and care excellence technology appraisals. Int J Technol Assess Health Care. 2019;35(3):221-228.
26. Davids MS, Telford C, Abhyankar S, Waweru C, Ringshausen I. Matching-adjusted indirect comparisons of safety and efficacy of acalabrutinib versus other targeted therapies in patients with treatment-naïve chronic lymphocytic leukemia. Leuk Lymphoma. 2021;62(10):2342-2351.
27. Skånland SS, Brown JR. PI3K inhibitors in chronic lymphocytic leukemia: where do we go from here? Haematologica. 2023;108(1):9-21.
concordant
1Unite de Genomique du Myelome, Institut Universitaire du Cancer Toulouse-Oncopole, Toulouse, France and 2AbbVie Inc., North Chicago, IL, USA
Correspondence: C. Hader carlos.hader@abbvie.com
Received: August 14, 2023.
Accepted: November 15, 2023. Early view: November 23, 2023.
https://doi.org/10.3324/haematol.2023.284072
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
Multiple myeloma (MM) is associated with a wide variety of recurrent genomic alterations. The most common translocation in MM is t(11;14). In this retrospective, single-center, non-interventional study, patients’ bone marrow samples were examined at diagnosis and at relapse(s) following treatment with anti-myeloma regimens to determine whether t(11;14) status was stable over time. This stability cohort consisted of 272 patients, of whom 118 were t(11;14)-positive at diagnosis and 154 were negative. All patients in the stability cohort retained the same t(11;14) status at relapse that they had at diagnosis of MM. Sixteen patients who had t(11;14)-positive MM at diagnosis had multiple longitudinal assessments by fluorescence in situ hybridization (FISH) at relapse events and remained t(11;14)-positive across all timepoints. Patients who had t(11;14)-positive disease at diagnosis of monoclonal gammopathy of unknown significance or smoldering MM also retained t(11;14) positivity through MM diagnosis and relapse. The t(11;14) fusion patterns also remained constant for 90% of patients. For detection of t(11;14), results from FISH and next-generation sequencing (NGS) were compared to determine the rate of concordance between these two methods. This concordance cohort contained 130 patients, of whom 66 had t(11;14)-positive disease and 64 were t(11;14)-negative. In this sample set, the concordance between FISH- and NGS-based detection of t(11;14) was 100%. These results strongly suggest that the t(11;14) rearrangement remains stable during the full disease course in patients with MM and can be detected by FISH- and NGS-based methodologies.
Multiple myeloma (MM) is a clonal plasma cell malignancy associated with recurrent chromosomal and genetic aberrations.1 Mutational events in the clonal evolution of MM include those considered primary and secondary genetic events, based on their immediacy during clonal development.2,3 Primary among genetic alterations are IGH translocations, including t(11;14),2 which frequently places the IGH enhancer in proximity to the CCND1 gene.4 Such primary cytogenetic abnormalities, acquired during B-cell development, facilitate transformation of normal plasma cells into pre-malignant, clonal plasma cells. Subsequent
proliferation within the bone marrow results in monoclonal gammopathy of undetermined significance (MGUS), evolving to asymptomatic smoldering MM (SMM), and finally into symptomatic MM. The t(11;14) translocation is the most common and is present in approximately 15-20% of patients;5,6 it has been associated with aberrant cyclin D1 expression and high BCL-2 expression.7 Considered as a primary genetic abnormality in plasma cell dyscrasias, t(11;14) has been shown to be present from precursor stages (MGUS and SMM) to MM and plasma cell leukemia.3,8,9 Previous studies demonstrated the relative inter-patient stability of t(11;14) status over time, but few evaluated intra-patient stability through multiple longitudinal bone
marrow assessments over the course of disease in MM.10-12
One study of 195 patients found that 25% of the patients were t(11;14)-positive at diagnosis of MM; however, 4.3% of those patients had undetectable t(11;14) at relapse.10 Conversely, 1.4% of the patients who were t(11;14)-negative at diagnosis had detectable t(11;14) at relapse.10 Similarly, a longitudinal study by Merz et al. (n=128) showed no change in IGH translocation status, including t(11;14), between primary diagnosis and relapse.11 Thus, additional studies employing longitudinal assessment of t(11;14) status across the disease spectrum of MM are necessary to confirm the stability of t(11;14).
Although fluorescence in situ hybridization (FISH) is the standard technique used to assess the presence of t(11;14), plasma cell enrichment (PCE) can be used to isolate and enrich clonal plasma cells from heterogenous bone marrow aspirate samples and enhance detection of chromosomal abnormalities by standard FISH. PCE also significantly improves the sensitivity of downstream next-generation sequencing (NGS) assays by facilitating detection of low-frequency mutations, while reducing background signal from non-clonal plasma cells.13 A recent study suggested that PCE may be more critical to accurate detection of cytogenetic abnormalities than previously believed, as using PCE improved the detection of abnormalities by direct FISH from 38.0% to 95.5% in diagnostic MM samples.14 This suggests that PCE has strong clinical value for improving FISH-based detection of genomic variability in MM. Given the genomic variability commonly associated with MM, personalized therapy targeted to individual genetic aberrations may become increasingly important in the era of targeted therapies.7 Venetoclax, a selective, orally bioavailable inhibitor of BCL-2, is the first targeted therapy for MM and, as monotherapy, has produced high response rates (40% overall response rate) in patients with detectable t(11;14).15 t(11;14) is a key predictive biomarker of response to venetoclax in relapsed and refractory (RR) MM, as observed in the BELLINI trial.15,16 Venetoclax is currently being studied in a phase III clinical trial, in combination with dexamethasone, for treatment of patients with t(11;14)-positive RRMM who have completed at least two prior lines of therapy.17 Thus, the stability of t(11;14), as well as the ability to reliably detect such aberrations through various assays, are of increasing importance in the treatment of RRMM. NGS combined with PCE has demonstrated the ability to provide equal genomic rearrangement detection as that provided by FISH,18 and may give insights into the pre- to post-treatment stability of t(11;14) in MM patients. In this study, we collected bone marrow aspirate samples from patients before and after treatment with anti-myeloma regimens to determine whether MM patients with detectable t(11;14) at diagnosis maintained t(11;14)-positive status after treatment. A NGS-based approach was used to detect t(11;14) status following PCE of bone marrow aspirates and to assess the concordance of PCE-NGS and
PCE-FISH methods. Finally, we examined the genomic landscape (mutations and copy number variations) of key selected genetic aberrations in patients with t(11;14)-positive versus t(11;14)-negative MM. This study included a total of 272 patients in the stability cohort, used to determine the stability of t(11;14) status, and 130 patients in the concordance cohort, used to determine the concordance of t(11;14) detection by the NGS- and FISH-based methodologies.
The primary objective of this retrospective, single-center, non-interventional study was to evaluate the stability of t(11;14) status at MM diagnosis and relapse using PCE-FISH on bone marrow samples collected before and after treatment with anti-myeloma regimens. The secondary objective was to assess the rate of concordance between PCE-FISH and PCE-NGS for t(11;14) detection in bone marrow samples from patients with MM.
All patients provided informed consent. Bone marrow samples were collected for both the stability cohort and the concordance cohort. Longitudinal paired samples from patients with MM at initial diagnosis and at relapse of disease were obtained only in the stability cohort. Inclusion criteria comprised age ≥18 years old and confirmation of newly diagnosed MM, MGUS, SMM, or RRMM. All samples must have had >80% plasma cells after PCE. Samples were stored in a biobank approved for research purposes at the Toulouse Cancer Institute Oncopole (Toulouse, France). Data were collected from samples that had been previously tested, as well as those that were thawed and tested explicitly for this study.
Plasma cell enrichment and t(11;14) testing
PCE was performed for all samples prior to FISH or NGS testing, using magnetic beads according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). FISH assays were performed using the Vysis IGH/CCND1 XT DF FISH Probe Kit (Abbott Molecular, Des Plaines, IL, USA). The FISH probes detected a 942 Kb region on chromosome 11 (68,363 ~ 69,305 Kb) spanning the CCND1 breakpoint region, and a 1.6 Mb IGH region on chromosome 14 (104,736 ~ 106,339 Kb). Slides were analyzed on a Zeiss Axio fluorescence microscope. For each patient, at least 100 nuclei were scored, counting the number of cells with no translocation, and the number of cells with a translocation. For cells with a translocation, the number of fusions was reported. The cutoff for determination of t(11;14) positivity was 30% of cells/nuclei with an abnormal fusion signal. NGS was performed as previously described19 using an Agilent capture panel for sequencing on the Illumina NextSeq
platform. DNA was extracted only from samples with ≥80% plasma cells in the final cell pellet after PCE. For samples stored in RLT+ buffer, DNA was extracted using the AllPrep® DNA/RNA/miRNA kit (QIAGEN, Venlo, the Netherlands). For samples stored as dry pellets, the NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany) was used. The NGS structural variant detection windows on chromosome 11 (68,800 ~ 69,800 Kb) included the CCND1 gene and upstream regulatory elements, and on chromosome 14 (105,500 ~ 106,900 Kb) included IGH. Samples with low numbers of supporting reads (≤5), suggesting that no t(11;14) translocation was detected (n=64), were identified as t(11;14)-negative. Samples with at least one structural variant within the detection window with >5 supporting reads confirming the presence of a t(11;14) translocation (n=66) were identified as t(11;14)-positive.
The frequency and characteristics of selected genetic alterations were analyzed by t(11;14) status: DIS3, MAPK pathway genes (BRAF, KRAS, NRAS, MYC, TRAF3), ATM, TP53, ATR, BIRC3, IRF4, and CYLD. 12
For analysis of the raw data, a dedicated bioinformatics pipeline (see the Online Supplementary Methods for more detail), developed especially for automatic analysis, was used.
The t(11;14) stability cohort collected paired longitudinal samples (at diagnosis and first relapse) from 272 patients; these samples were tested for t(11;14) status using FISH and the findings at diagnosis and at first relapse were compared for each patient.
The concordance cohort collected samples from 130 patients who were analyzed by both FISH and NGS. The results obtained were compared to assess concordance, sensitivity, and specificity of the testing methods. Statistical power calculations and methods can be found in the Online Supplementary Information.
Table 1. Patients’ baseline demographics.
The stability cohort consisted of 272 patients, of whom 118 were t(11;14)-positive at diagnosis and 154 were negative; this cohort had samples that were tested at diagnosis and relapse(s) for detection of t(11;14) by PCE-FISH. The concordance cohort contained 130 patients, of whom 66 were t(11;14)-positive at diagnosis and 64 were negative; this cohort had samples tested by both PCE-FISH and PCE-NGS to determine the concordance of t(11;14) detection between the two methods. Approximately 60% of all tested patients across both cohorts were male. The median age of patients included in the concordance cohort was 69 years (range, 43–91). The median age of those in the stability cohort was 60 years (range, 37-85) for t(11;14)-positive cases, and 63 years (range, 34-85) for t(11;14)-negative patients (Table 1). Patients in the concordance cohort received a variety of anti-myeloma treatment regimens; the most commonly received regimens were triplet therapy with daratumumab, lenalidomide, and dexamethasone; combination therapy with lenalidomide and dexamethasone; and triplet therapy with daratumumab, pomalidomide, and dexamethasone.
In the stability cohort, the median time between the collection of two samples for t(11;14) testing (at diagnosis and relapse) was 29.1 months (range, 1.9-149.4). In this population, stability of t(11;14) status was absolute; all patients who had detectable t(11;14) at diagnosis (n=118) retained the same status at disease relapse (Figure 1), and no t(11;14)-negative patients at diagnosis (n=154) had detectable t(11;14) at relapse. Sixteen patients with t(11;14)-positive samples at diagnosis also had samples collected at multiple relapse events (Online Supplementary Figure S1); these patients had a median of two (range, 2-3) post-diagnosis assessments, over a median of 43.3 months (range, 11.4-196.9). All these
N: number; MGUS: monoclonal gammopathy of undetermined significance; SMM: smoldering multiple myeloma; NDMM: newly diagnosed multiple myeloma; RRMM: relapsed/refractory multiple myeloma; PCL: plasma cell leukemia; FISH: fluorescence in situ hybridization; NA: not applicable.
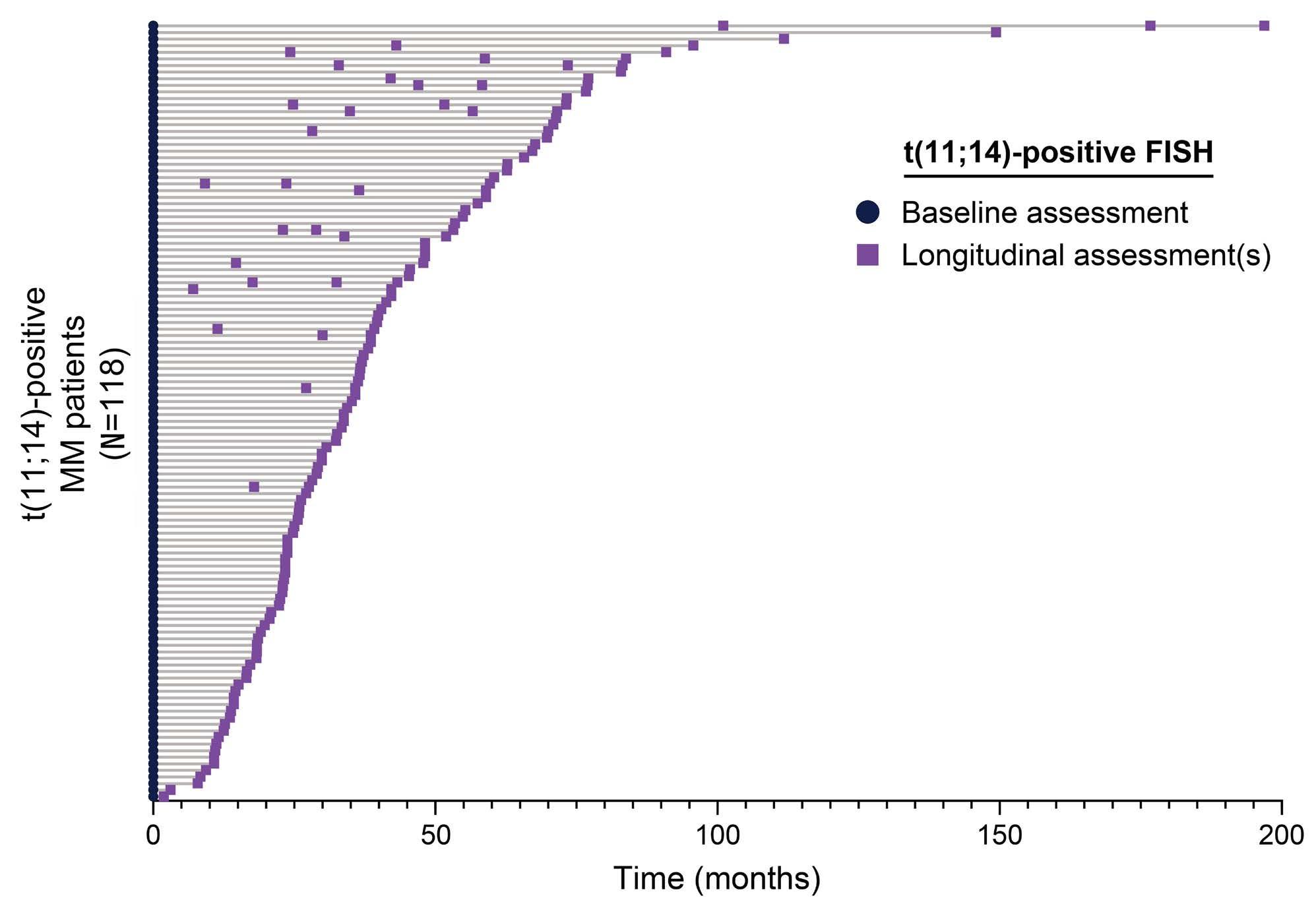
Figure 1. Timing of t(11;14) assessments, between diagnosis and at relapse, for t(11;14)-positive patients. FISH: fluorescence in situ hybridization; MM: multiple myeloma.
samples, across all timepoints, retained detectable t(11;14). When samples were positive at initial detection in patients with MGUS or SMM, the patients remained consistently t(11;14)-positive through MM diagnosis and into relapse, a median time span of 28.7 months (range, 7.1-83.2) (n=15) (Online Supplementary Figure S2). Chromosomal fusion analysis was done for t(11;14)-positive patients who transitioned from SMM/MM to RRMM on-study. The number of chromosomal fusions remained constant for 90% of patients (n=106/118), but 7% had an increase in the number of fusions from one to two, and another 3% had a decrease from two to one fusion (Figure 2).
Concordance
Among samples with at least one structural variant detection window, a total of 121 structural variants were identified. Figure 3 shows the variety of translocations found across the samples. The same 64 samples found to be t(11;14)-negative by NGS were also determined to be t(11;14)-negative by FISH. Moreover, the same 66 samples identified as t(11;14)-positive by NGS were also identified as t(11;14)-positive by FISH. This indicates 100% concordance between the two detection methods (Table 2).
Genomic profiling
Other genetic aberrations detected by NGS were highly heterogeneous and varied between t(11;14)-positive and -negative samples (Figure 4). The median total copy number aberrations was lower in t(11;14)-positive samples than in t(11;14)-negative samples (median 119 vs. 291; P<0.001). The frequency of DIS3 alterations was higher in t(11;14)-positive
samples than in negative samples (21.2% vs. 4.7%; P=0.008). MAPK pathway mutations (BRAF, KRAS, NRAS, MYC, TRAF3) were the most prevalent among all patients (n=47/130; 36%). In contrast, BRAF alterations were less common in t(11;14)-positive samples than in negative samples (6.1% vs 18.8%; P=0.034) (Figure 4).
The present study demonstrated that all patients examined for stability of t(11;14) status (i.e., the stability cohort) maintained their t(11;14) status throughout the course of the disease, from MM diagnosis to relapse, as well as during progression from MGUS/SMM to MM. Additionally, analysis of concordance for t(11;14) detection between PCE-FISH and PCE-NGS was 100%. NGS data also showed differential expression of key genetic alterations such as DIS3, BRAF, and MAPK pathway mutations between t(11;14)-positive and -negative samples.
In the 272 previously treated patients in the stability cohort, we found no instances of change in t(11;14) status between diagnosis and relapse. This is consistent with previous reports of t(11;14) status changes in only 1-6% of patients;10,11 as such, the present study in a large longitudinal cohort of patients with MM (n=272) indicates that the t(11;14) rearrangement is an early genetic event that remains stable throughout MM disease evolution, including the MGUS/ SMM-to-MM transition, as well as across lines of therapy to disease relapse. The 100% intra-patient stability of t(11;14) observed in this study could be attributed to the
well-controlled plasma cell input prior to FISH and NGS analysis, obtained after PCE (>80%), or due to improvements in technology and detection methods over the last 6-10 years. Recent studies have demonstrated that PCE is critical to optimize detection of genomic alterations;14 thus, in the present study, it is possible that t(11;14) was detected in samples in which it would have previously been determined to be undetectable. However, it should be noted that no samples in our study were excluded for having
<80% plasma cells after PCE, suggesting that consistent high-level PCE is readily achievable in clinical practice. It is notable that t(11;14) stability was also observed at the genomic level with regards to the number of fusions present. The number of t(11;14) fusions remained constant for 90% of patients (n=106/118), but an increase in the number of fusions from one to two, and a decrease from two to one fusion were observed in 7% and 3% of patients, respectively. This suggests that some genomic instability
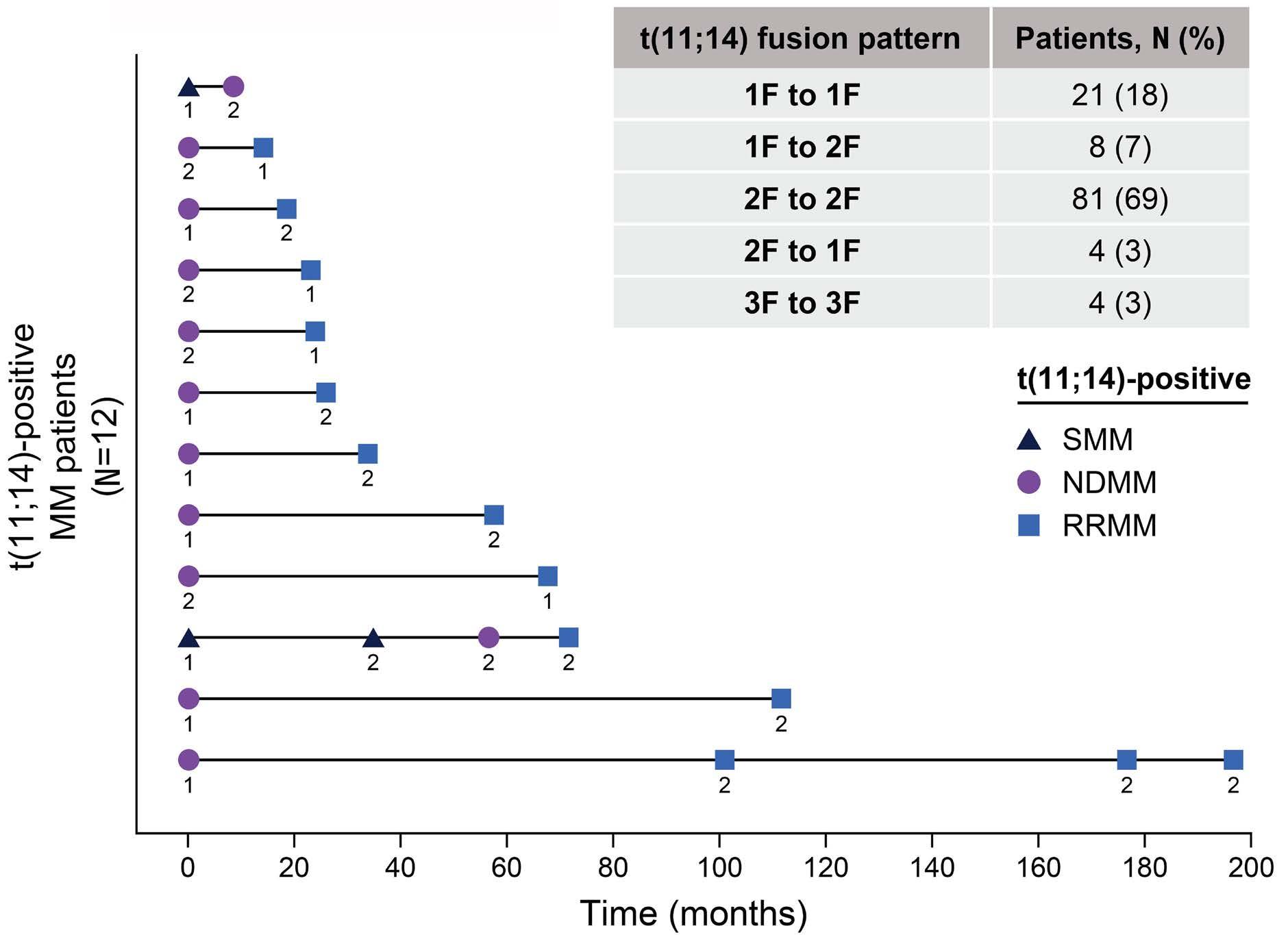
Figure 2. Disease status over time, based on chromosomal fusion analysis, for t(11;14)-positive patients who transitioned from smoldering multiple myeloma (MM) to newly diagnosed MM to relapsed/refractory MM while on-study. MM: multiple myeloma; F: fusion; SMM: smoldering multiple myeloma; NDMM: newly diagnosed multiple myeloma; RRMM: relapsed/refractory multiple myeloma.
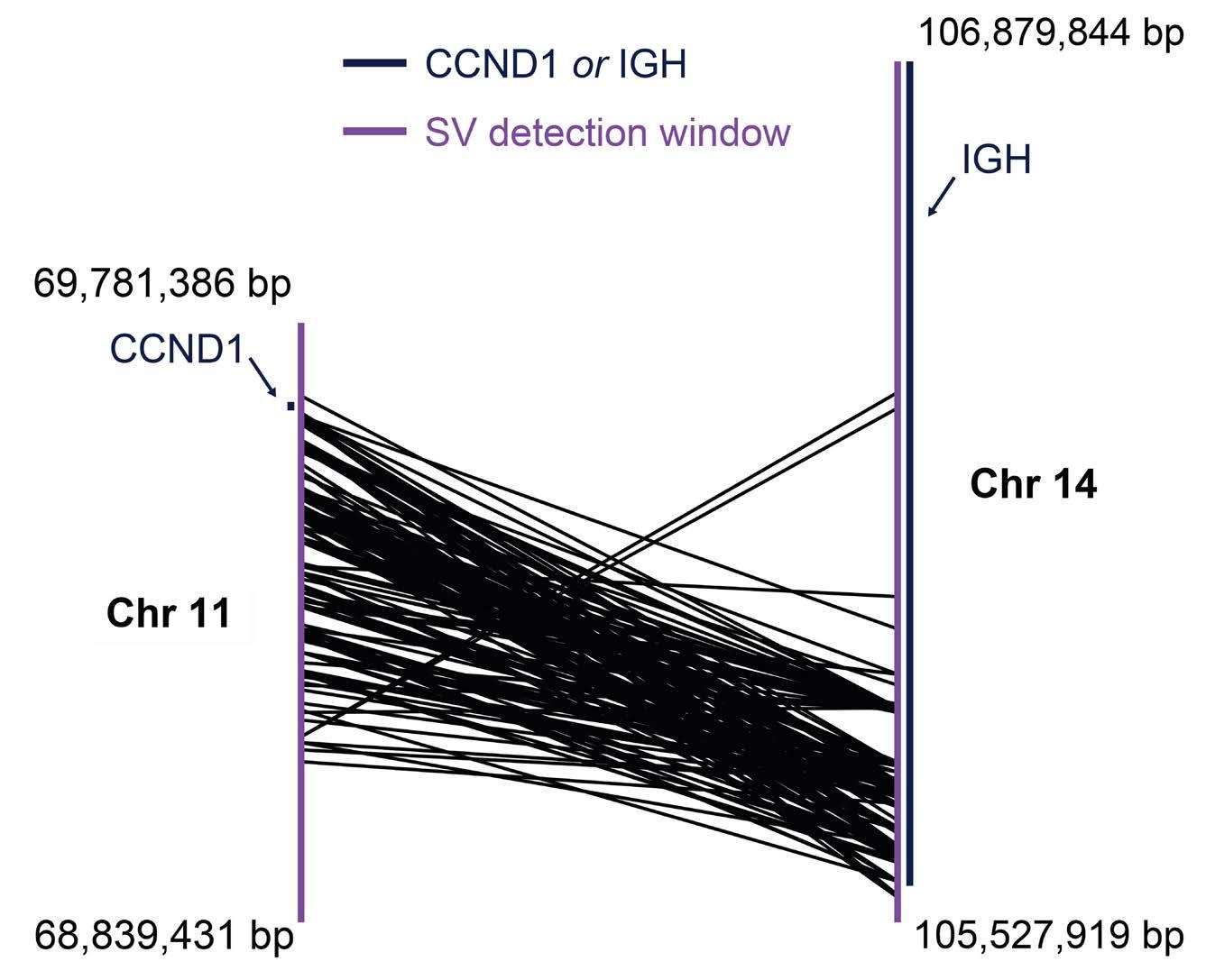
Figure 3. Genomic rearrangement (fusion) points between sites on chromosome 11 (including the location of CCND1) and chromosome 14 (including the location of IGH) in t(11;14)-positive multiple myeloma patients based on structural variant window detection. Each connecting line represents one detected rearrangement. bp: base pairs; Chr: chromosome; SV: structural variant.
is present during MM evolution, but that loss or gain of t(11;14) does not appear to be a common event that occurs outside of the primary window of transformation. The stability of t(11;14) status between diagnosis and relapse found in this study suggests that a single test for t(11;14) at diagnosis - or at any time during the patient’s journey - may be sufficient when considering targeted therapeutic approaches for patients with MM. This is particularly relevant given that post-treatment bone marrow biopsies for patients with RRMM are uncommon because they are not considered clinically relevant to justify the associated risk to patients.20 Taken together, these data indicate that targeted agents, such as venetoclax,15,16 may not require t(11;14) retesting at disease relapse when a patient has demonstrated t(11;14) positivity at a prior timepoint. These findings may also advocate for biomarker testing at diagnosis for all patients to help inform therapeutic options during the treatment journey.
Concordance in determination of t(11;14) status with FISH and NGS was 100% across 130 patients’ samples. These findings are similar to those of a recently published study of MM in which a head-to-head comparison of FISH and
NGS was performed; 78 IGH translocations, including t(11;14), were detected by both methods with a 100% concordance.21 Based on the results of this study, for the purpose of t(11;14) detection, NGS and FISH methods appear to be equally functional and accurate. The NGS workflow can be labor-intensive, and variant curation can be time-consuming and require specialized molecular geneticists; however, NGS provides additional information, such as the precise location of the chromosomal transformation in each sample and identification of other genomic alterations within these
2. Concordance rates between fluorescence in situ hybridization and next-generation sequencing methods for the detection of t(11;14).
by FISH, N
Positive by NGS, N (%) 66 (100) 0
Negative by NGS, N (%) 0
(100)
number; FISH: fluorescence in situ hybridization; NGS: next-generation sequencing.
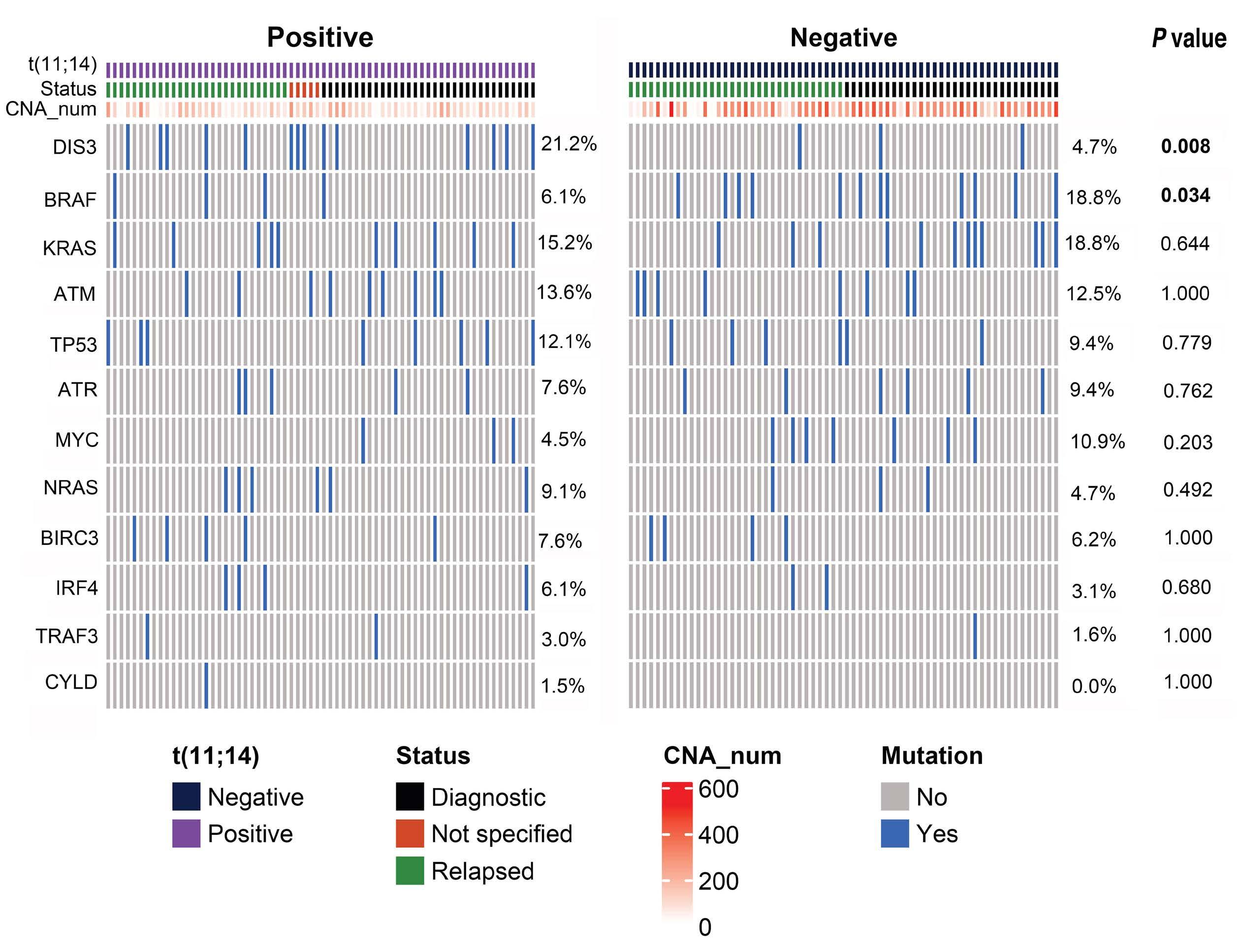
Figure 4. The frequency of recurrent genetic aberrations in patients with multiple myeloma, shown according to t(11;14) status. Patients’ disease status, t(11;14) status, and whether mutations were detected in each gene are represented above. Bold P values are statistically significant at P<0.05. CNA: copy number alteration.
samples. FISH assays are readily available in the clinical setting, and reliably and accurately detected t(11;14) status in this cohort of patients.
In this study, we found that the frequency of DIS3 alterations was significantly higher in t(11;14)-positive samples than in negative samples (P=0.008). This may have clinical implications for patients with t(11;14)-positive MM, because DIS3 mutations are associated with worse event-free survival outcomes in MM; however this must be confirmed in future studies.22 We also found that BRAF alterations were less common in t(11;14)-positive samples than in negative ones. It has been proposed that BRAF alterations may be associated with better outcomes in MM.23 By contrast, in this study, MAPK pathway mutations (BRAF, KRAS, NRAS, MYC, TRAF3) were highly prevalent overall (36%), but most tended to be less common in t(11;14)-negative samples. Regardless of specific alterations, the practicality of FISH for routine clinical testing is invaluable, while utilizing NGS to determine the presence of additional alterations will be required to provide a comprehensive assessment of the genomic heterogeneity in MM.
In conclusion, this study has demonstrated absolute stability of t(11;14) status from diagnosis to relapse, and across multiple relapses, in patients treated with anti-myeloma therapeutic regimens. Furthermore, the results demonstrate a 100% concordance in detection and determination of t(11;14) status by FISH- and NGSbased methodologies.
Disclosures
HA-L has no conflicts of interest to disclose. RT-M, XL, JAR, and CH are employees of AbbVie, and may hold stock or options in the company.
Contributions
RT-M, JAR, and CH conceived and designed the study. RT-M, JAR, and CH collected the data. All the authors were involved in the analysis and interpretation of results and in
1. Elbezanti WO, Challagundla KB, Jonnalagadda SC, BudakAlpdogan T, Pandey MK. Past, present, and a glance into the future of multiple myeloma treatment. Pharmaceuticals (Basel). 2023;16(3):415.
2. Cardona-Benavides IJ, de Ramón C, Gutiérrez NC. Genetic abnormalities in multiple myeloma: prognostic and therapeutic implications. Cells. 2021;10(2):336.
3. Chng WJ, Glebov O, Bergsagel PL, Kuehl WM. Genetic events in the pathogenesis of multiple myeloma. Best Pract Res Clin Haematol. 2007;20(4):571-596.
4 Walker BA, Wardell CP, Johnson DC, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121(17):3413-3419.
preparation of the manuscript.
Acknowledgments
Medical writing assistance was provided by Ryan J. Bourgo, PhD, and Ana Lopez, PhD, of Fishawack Facilitate Ltd., funded by AbbVie.
Venetoclax is being developed in a collaboration between AbbVie and Genentech. AbbVie and University Cancer Center of Toulouse sponsored the study and participated in the design, study conduct, collection, analysis, and interpretation of the data. No honoraria or payments were paid for authorship. This work was performed, in part, by AbbVie, Inc. and employees of AbbVie participated in the preparation, review, and approval of the publication. Medical writing assistance was funded by AbbVie.
AbbVie is committed to responsible data-sharing regarding the clinical trials it sponsors. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal, Statistical Analysis Plan (SAP), and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the US and Europe and after acceptance of this manuscript for publication. The data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/
5. Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109(8):3489-3495.
6. Fonseca R, Blood EA, Oken MM, et al. Myeloma and the t(11;14) (q13;q32); evidence for a biologically defined unique subset of patients. Blood. 2002;99(10):3735-3741.
7 Bal S, Kumar SK, Fonseca R, et al. Multiple myeloma with t(11;14): unique biology and evolving landscape. Am J Cancer Res. 2022;12(7):2950-2965.
8. Punnoose EA, Leverson JD, Peale F, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15(5):1132-1144.
9 Moreau P, Chanan-Khan A, Roberts AW, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and
dexamethasone in relapsed/refractory MM. Blood. 2017;130(22):2392-2400.
10. Hebraud B, Caillot D, Corre J, et al. Lost and gain of t(4;14) and t(11;14) in multiple myeloma patients between relapse and diagnosis: an illustration of clonal dynamic during disease course. an IFM study. Blood. 2012;120(21):196.
11. Merz M, Jauch A, Hielscher T, et al. Longitudinal fluorescence in situ hybridization reveals cytogenetic evolution in myeloma relapsing after autologous transplantation. Haematologica. 2017;102(8):1432-1438.
12. Bolli N, Avet-Loiseau H, Wedge DC, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997.
13. Lu G, Muddasani R, Orlowski RZ, et al. Plasma cell enrichment enhances detection of high-risk cytogenomic abnormalities by fluorescence in situ hybridization and improves risk stratification of patients with plasma cell neoplasms. Arch Pathol Lab Med. 2013;137(5):625-631.
14. Ha J, Cho H, Lee TG, et al. Cytogenetic testing by fluorescence in situ hybridization is improved by plasma cell sorting in multiple myeloma. Sci Rep. 2022;12(1):8287.
15. Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130(22):2401-2409.
16. Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet
Oncol. 2020;21(12):1630-1642.
17 A study of venetoclax and dexamethasone compared with pomalidomide and dexamethasone in participants with relapsed or refractory multiple myeloma. 2023 https:// clinicaltrials.gov/ct2/show/NCT03539744.
Accessed April 11, 2023.
18. Frankel D, Nanni I, Ouafik L, et al. Comparison between immunocytochemistry, FISH and NGS for ALK and ROS1 rearrangement detection in cytological samples. Int J Mol Sci. 2022;23(18):10556.
19 Bolli N, Li Y, Sathiaseelan V, et al. A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma. Blood Cancer J. 2016;6(9):e467.
20 Tschautscher MA, Jevremovic D, Buadi FK, et al. Utility of repeating bone marrow biopsy for confirmation of complete response in multiple myeloma. Blood Cancer J. 2020;10(10):95.
21. Yellapantula V, Hultcrantz M, Rustad EH, et al. Comprehensive detection of recurring genomic abnormalities: a targeted sequencing approach for multiple myeloma. Blood Cancer J. 2019;9(12):101.
22. Boyle EM, Ashby C, Tytarenko RG, et al. BRAF and DIS3 mutations associate with adverse outcome in a long-term follow-up of patients with multiple myeloma. Clin Cancer Res. 2020;26(10):2422-2432.
23. Mohamed SF, Khan M, Quesada A, et al. Disease characteristics of multiple myeloma involving BRAF mutations. Blood. 2021;138(Suppl 1):4755.
Tondre Buck,1 Monique A. Hartley-Brown,2 Yvonne A. Efebera,3 Carter P. Milner,4 Jeffrey A. Zonder,5 Paul G. Richardson,2 Taylor Salinardi6° and Megan S. Rice6°
1Spartanburg Medical Center, Center for Research and Cancer Institute, Spartanburg, SC; 2Division of Hematologic Malignancy, Department of Medical Oncology, Jerome Lipper Multiple Myeloma Center, Harvard Medical School, Dana-Farber Cancer Institute, Boston, MA; 3Division of Blood and Marrow Transplant and Cellular Therapy, OhioHealth, Columbus, OH; 4Division of Hematology and Medical Oncology, Department of Medicine, University of Mississippi Medical Center, Jackson, MS; 5Department of Oncology, Barbara Ann Karmanos Cancer Institute, Wayne State University, Detroit, MI and 6Sanofi, Cambridge, MA, USA
°Current address TS: Azurity Pharmaceuticals, Woburn, MA, USA. °Current address MSR: Vertex Pharmaceuticals Incorporated, USA.
Correspondence: T. Buck tbuck@gibbscc.org
Received: January 25, 2023. Accepted: November 17, 2023. Early view: November 30, 2023.
https://doi.org/10.3324/haematol.2023.282788
Published under a CC BY license
Examination of the impact of race and ethnicity on multiple myeloma (MM) outcomes has yielded inconsistent results. This retrospective, real-world (RW) study describes patient, disease, and treatment characteristics (and associations with survival outcomes) among newly diagnosed MM patients of non-Hispanic (NH) Black/African American (AA) and NH White race/ethnicity in the US. We included patients from the nationwide Flatiron Health electronic health record-derived de-identified database who initiated first line of therapy (LOT) for MM between January 1, 2016 and March 31, 2022. Of 4,614 patients in our study cohort, 23.3% were NH Black/AA. Non-Hispanic Black/AA patients were younger than NH White patients at diagnosis (median 68 vs. 71 years) and more likely to be female (53.4% vs. 43.5%). Rates of high-risk cytogenetics and 1q21+ were similar between races/ethnicities. The most common primary regimen used was lenalidomide-bortezomib-dexamethasone (50.1% of NH Black/AA and 48.1% of NH White patients). Receipt of stem cell transplantation during first LOT was less common among NH Black/AA (16.5%) than NH White (21.9%) patients. Unadjusted RW progression-free survival (rwPFS) and overall survival (rwOS) were similar between races/ethnicities. After multivariable adjustment, NH Black/AA race/ethnicity was associated with slightly inferior rwPFS (hazard ratio [HR]=1.13; 95% confidence interval [CI]: 1.01-1.27). The difference in rwOS (HR=1.12; 95% CI: 0.98-1.28) was not statistically significant. In general, associations between risk factors for rwPFS and rwOS were consistent between races/ethnicities. Findings from this analysis help to inform clinicians about the impact of race/ethnicity on MM treatment paradigms and outcomes in the US.
Multiple myeloma (MM) is a malignancy characterized by the proliferation of terminally differentiated plasma cells in the bone marrow. It is the second most common hematologic cancer in the US and the most common hematologic malignancy among people of Black/African American (AA) race.1 In fact, the rate of new MM cases and MM-related deaths is over two times higher among Black/AA adults than White adults in the US,1 which may be partially due to biological or genetic differences between races. According to the Surveillance, Epidemiology and End Results (SEER) data on MM prevalence, in 2020, over 34,000 Black/AA patients who
were diagnosed with MM between 1992 and 2019 were alive with the disease.2 Despite currently making up only 14.2% of the US population,3 people of Black/AA race are expected to comprise roughly 24% of the newly diagnosed MM (NDMM) population by 2034.4
With the advent of novel therapies including autologous stem cell transplant (SCT), immunomodulatory drugs (IMiD), proteasome inhibitors (PI), monoclonal antibodies (mAb), and chimeric antigen receptor T-cell (CAR T) therapies, survival has improved for MM patients in recent decades.5 Notably, survival was slower to improve among Black/AA than White patients through 2012.5,6 Several studies have shown that Black/AA patients may be less likely to receive SCT, PI, IMiD, front-line
triplet induction therapies for NDMM (and more recently approved immunotherapies for relapsed MM7-9) compared with their White counterparts.6,10-13 Interestingly, Black/AA race has also been linked, albeit inconsistently, with more favorable cytogenetic profiles than White race, including a lower prevalence of high-risk features such as del(17p) and t(4;14).11,14,15 To date, analyses of real-world (RW) datasets have yielded discrepant associations between race and survival outcomes. Analyses of US-based datasets from the SEER Program,12 Veterans Affairs (VA),16 and Flatiron Health17 have shown similar, or even improved, overall survival (OS) among Black/AA patients, particularly when there is equal access to care. However, analysis of the international Multiple Myeloma Research Foundation CoMMpass dataset11 found that Black/AA patients with MM have inferior OS to White patients, which is only partially abrogated by receipt of SCT and triplet therapies. Differences in RW findings highlight the importance of continually examining data that reflect current trends in the uptake of novel therapies and treatment strategies, as well as growing awareness of racial/ethnic disparities in cancer care. As such, the objective of this retrospective, observational cohort study was to provide an up-to-date examination of associations between patient, disease, and treatment characteristics and survival outcomes by non-Hispanic (NH) Black/AA and NH White race/ethnicity in the US.
Study design and data source
This retrospective, observational cohort study used the nationwide Flatiron Health electronic health record (EHR)-derived, de-identified database of MM patients treated in the US. The Flatiron Health database is a longitudinal database, comprising de-identified patient-level structured and unstructured data, curated via technology-enabled abstraction.18,19 During the study period, the de-identified data originated from approximately 280 US cancer clinics (~800 sites of care).
Patient selection
We included patients from the Flatiron Health MM Cohort whose race was recorded as “White” and ethnicity was not equal to “Hispanic or Latino” (NH White), whose race was recorded as “Black or African American” and ethnicity was not equal to “Hispanic or Latino” (NH Black/AA), and whose first line of therapy (LOT) was initiated between January 1, 2016 and March 31, 2022. Exclusion criteria are provided in the Online Supplementary Appendix
Assessments and outcomes
Patient, disease, and treatment characteristics were examined by race/ethnicity. RW progression-free survival (rwPFS) and rwOS, indexed to first LOT, were examined by race/ethnicity, overall and for subgroups defined by various patient,
disease, and treatment characteristics. rwPFS was defined as the time from start of front-line therapy to the date of first progression event (informed by International Myeloma Working Group criteria and incorporating abstracted M-spike values and structured free light chain values) or death. Progression events occurring within 30 days after therapy initiation were excluded, as such events are not expected to reflect progression on the therapy of interest. As the timing and frequency of laboratory tests can vary (with less frequent testing associated with longer rwPFS), a sensitivity analysis was conducted to exclude patients with a gap of more than 180 days between the date of progression event and the previous lab test. rwOS was defined as the time from start of front-line therapy to the date of death.
Patient, disease, and treatment characteristics by race/ ethnicity were summarized descriptively, using mean (standard deviation) and/or median (interquartile range [IQR]) for continuous variables and frequencies and percentages for categorical variables. The Kaplan-Meier (K-M) method was used to analyze rwPFS and rwOS for the overall populations of NH Black/AA and NH White patients, with median (95% confidence interval [CI]) reported for both outcomes. Differences in rwPFS and rwOS between races/ethnicities, overall and within subgroups were assessed using unadjusted, age-adjusted, and multivariable (MV)-adjusted Cox proportional hazards models that adjusted for age at start of first LOT, sex, practice type, region of residence, M-protein subtype at diagnosis, International Staging System (ISS) stage at diagnosis, Eastern Cooperative Oncology Group performance status (ECOG PS) at start of first LOT, cytogenetic risk, 1q21+, estimated glomerular filtration rate (eGFR) at start of first LOT, and time from diagnosis of MM to start of first LOT. Likelihood ratio tests were used to evaluate whether associations between the selected factors and rwPFS and rwOS differed by race/ethnicity. Further details of Methods are provided in the Online Supplementary Appendix.
Patient demographics and clinical characteristics
A total of 4,614 patients with MM had initiated first LOT and were included in the study cohort (Online Supplementary Figure S1); 1,077 (23.3%) patients were NH Black/AA and 3,537 (76.7%) patients were NH White. Baseline patient, disease, clinical, and first LOT characteristics are shown in Tables 1 and 2. Patients of NH Black/AA race/ethnicity were slightly younger than NH White patients on average, with a median age of 68 years versus 71 years, respectively, at both MM diagnosis and start of first LOT. A higher proportion of NH Black/AA than NH White patients were female (53.4% vs. 43.5%) and from the Southern US (63.0% vs. 36.2%),
but fewer were treated in academic practices (12.9% vs. 16.1%). Rates of commercial insurance coverage were similar between NH Black/AA and NH White patients (44.0% vs. 46.4%, respectively), as were rates of coverage under a patient assistance program (8.6% vs. 6.5%); however, rates of Medicare/Medicare+ coverage were higher among NH White patients (20.7%) than NH Black/AA patients (15.2%). Rates of high-risk cytogenetics [defined as the presence of ≥1 of del(17p), t(4;14), or t(14;16)] and 1q21+ (defined as gain [3 copies] or amplification [≥4 copies] of 1q21) were similar between NH Black/AA and NH White patients (14.7% vs. 16.1% and 18.4% vs. 21.1%, respectively). Rates of individual high-risk cytogenetic abnormalities (HRCA) were also similar between groups: t(4;14) 5.1% vs. 5.5%, t(14;16) 3.9% vs. 2.7%, and del(17p) 8.4% vs. 10.0% for NH Black/AA and NH White patients, respectively. Median time from MM diagnosis to start of first LOT was similar for NH Black/AA patients (1.06 months; IQR, 0.68-1.52) and NH White patients (1.03 months; IQR, 0.68-1.48).
Most NH Black/AA and NH White patients (52.7% in each group) received a PI + IMiD-based regimen as initial therapy during the first LOT (Table 3). Similar percentages of NH Black/AA (6.9%) and NH White (8.3%) patients received an mAb-based regimen as their initial therapy. The most common primary regimen used for both groups was lenalidomide-bortezomib-dexamethasone (50.1% of NH Black/ AA patients and 48.1% of NH White patients) (Online Supplementary Table S1). Receipt of SCT during the first LOT was less common among NH Black/AA patients (16.5%) than NH White patients (21.9%). Among those receiving SCT, rates of post-SCT consolidation (6.2% vs. 8.6%) and post-SCT maintenance therapy (64.0% vs. 63.7%) were similar between NH Black/AA and NH White patients, respectively.
Real-world progression-free survival
The rwPFS population consisted of 3,922 patients (Online Supplementary Figure S1). In K-M analyses, median rwPFS
Table 1. Baseline patient characteristics, overall and by race/ethnicity.
Characteristic, N (%) unless otherwise noted
Age in years at MM diagnosis (categorical years)
Age in years at start of first LOT (categorical years)
<65
65 - <75 ≥75
of residence
*Defined as patients from the Northeast, Midwest, West, of other part of the United States (or those with missing data). IQR: interquartile range; LOT: line of therapy; MM: multiple myeloma; NH: non-Hispanic.
Table 2. Disease, clinical, and first line of therapy characteristics, overall and by race/ethnicity.
Characteristic, N (%) unless otherwise noted
(53.5)
ISS stage at diagnosis
Stage I
Stage II
Stage III
Cytogenetic
t (4;14)
t (14;16)
1q21+† (assessed at any
eGFR‡ (mL/min/1.73 m2) at start of first LOT <60 ≥60
ECOG PS at start of first LOT 0 1
in months from MM diagnosis to first LOT start
*High-risk cytogenetics were defined as the presence of ≥1 of del(17p), t(4;14), or t(14;16). †1q21+ was defined as gain (3 copies) or amplification (≥4 copies) of 1q21. ‡Assessed using the MDRD equation. ECOG: Eastern Cooperative Oncology Group; eGFR: estimated glomerular filtration rate; Ig: immunoglobulin; IQR: interquartile range; ISS: International Staging System; LOT: line of therapy; MDRD: modification of diet in renal disease; MM: multiple myeloma; NH: non-Hispanic; PS: performance status.
from start of first LOT was similar for the overall populations of NH Black/AA (26.0 months; 95% CI: 23.2-30.2) and NH White (27.2 months; 95% CI 25.6-29.4) patients (Figure 1). Unadjusted and age-adjusted Cox proportional hazard models showed similar rwPFS among NH Black/AA and NH White patients (Online Supplementary Table S2); however, after MV adjustment, NH Black/AA race/ethnicity was associated with statistically inferior rwPFS (MV-adjusted hazard ratio [HR]=1.13; 95% CI: 1.01-1.27) when using NH
White race/ethnicity as the reference; Figure 2). MV-adjusted analyses examining associations between selected patient and disease characteristics and rwPFS by race/ethnicity are shown in Figure 2 (full subgroup analyses are shown in Online Supplementary Table S3). Age of ≥75 years at start of first LOT (compared with age of <65 years), ECOG PS of ≥2 (compared with ECOG PS of 0), ISS Stage II or III disease at diagnosis (compared with Stage I disease at diagnosis), and the presence of 1q21+ (compared with the absence
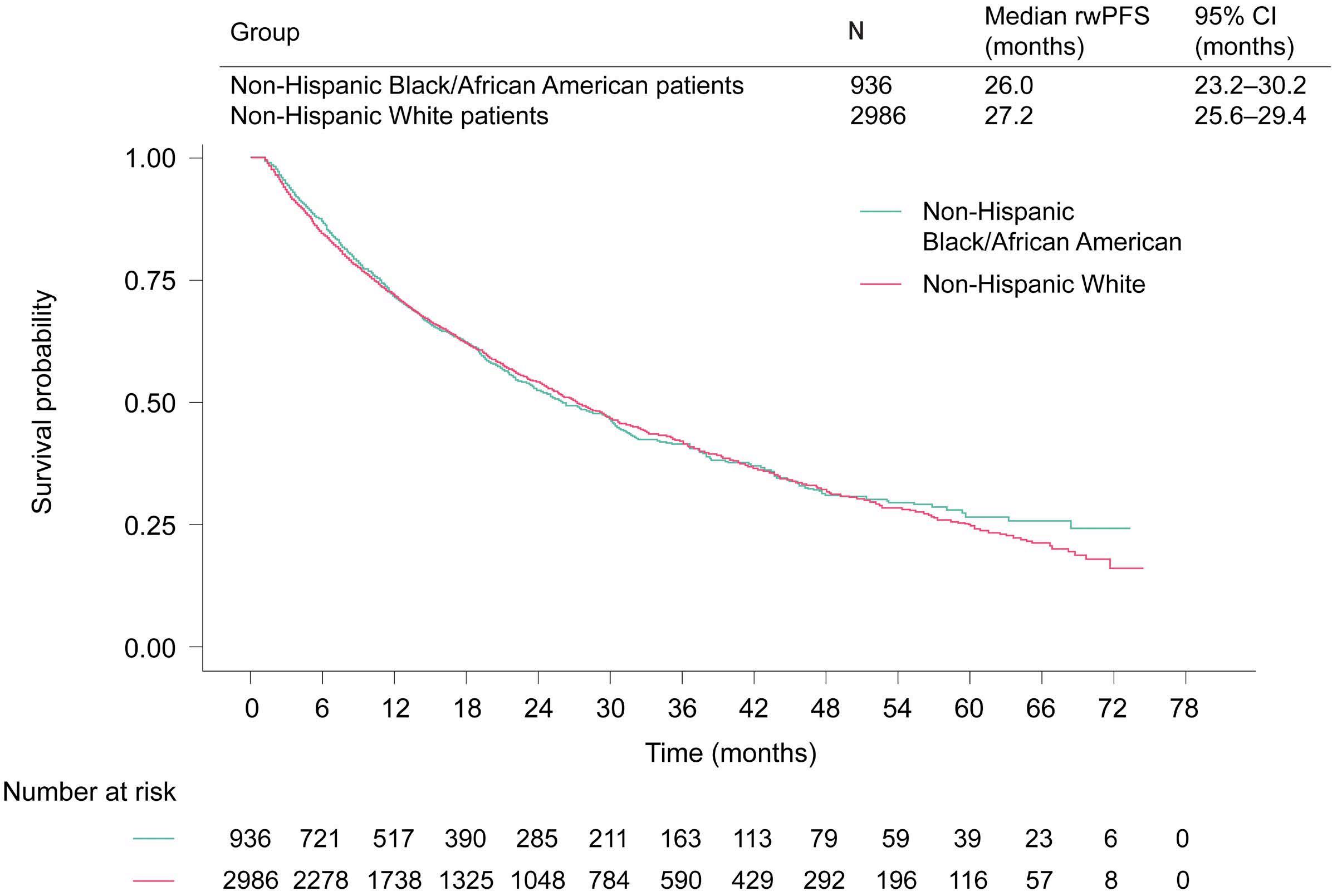
Figure 1. Kaplan-Meier analysis of real-world progression-free survival from start of first line of treatment by race/ethnicity. CI: confidence interval; LOT: line of therapy; rwPFS: real-world progression-free survival.
*Regimens containing only the drug class(es) listed (+/- steroids). †Regimens containing at least one chemotherapy agent (+/- steroids), that could also include PI and IMiD but not mAb or “other” drugs (see footnote §). ‡Regimens containing at least 1 mAb agent (+/- steroids), that could also include PI, IMiD, and chemotherapy agents but not “other” drugs (see footnote §). §Included BCMA-targeting drugs (eg, belantamab mafodotin, idecabtagene vicleucel) or those with novel mechanisms of action (e.g., panobinostat, selinexor, melflufen). BCMA: B-cell maturation antigen; IMiD: immunomodulatory drug; mAb: monoclonal antibody; NH: non-Hispanic; PI: proteasome inhibitor.
of 1q21+) were associated with statistically inferior rwPFS for both NH Black/AA and NH White patients. Age of 65-74 years (compared with age of <65 years) and the presence of high-risk cytogenetics (compared with standard-risk cytogenetics) were similarly associated with worse rwPFS among both NH Black/AA and NH White patients but were only statistically significant among NH White patients.
Likelihood ratio tests confirmed that associations between selected factors and rwPFS were not statistically different between races/ethnicities. rwPFS K-M and MV-adjusted Cox model findings were confirmed by sensitivity analyses that excluded patients with more than a 180-day gap between progression event and previous laboratory test ( Online Supplementary Figure S2; Online Supplementary Table S3).
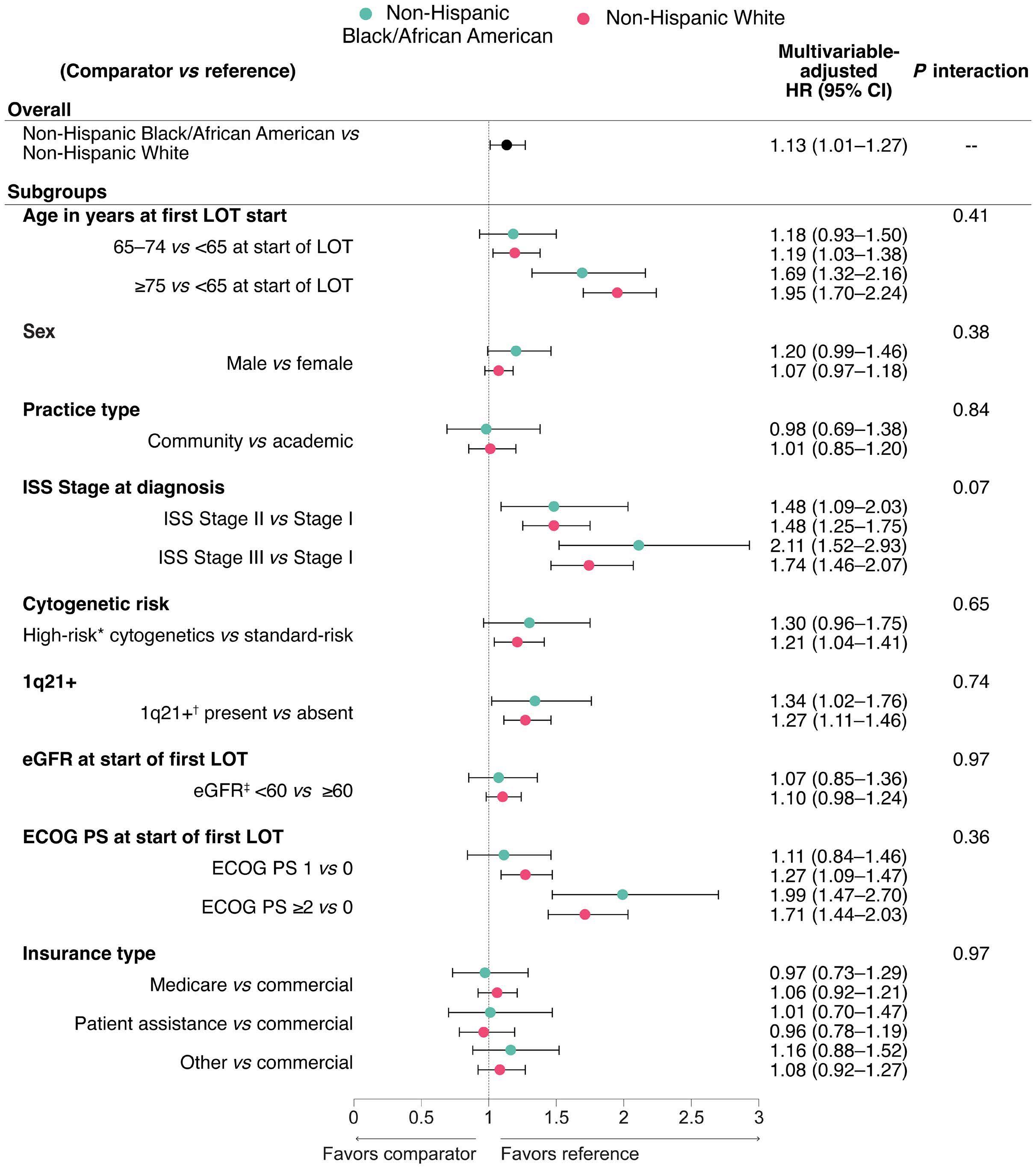
Figure 2. Multivariable-adjusted Cox proportional hazards model of real-world progression-free survival from start of first line of treatment, by race/ethnicity, overall and for select subgroups. *High-risk cytogenetics were defined as the presence of ≥1 of del(17p), t(4;14), or t(14;16). †1q21+ was defined as gain (3 copies) or amplification (≥4 copies) of 1q21. ‡Assessed using the MDRD equation; expressed as mL/min/1.73 m2. CI: confidence interval; ECOG PS: Eastern Cooperative Oncology Group performance status; eGFR: estimated glomerular filtration rate; HR: hazard ratio; ISS: International Staging System; LOT: line of therapy; MDRD: modification of diet in renal disease; MV: multivariable; PS: performance status; rwPFS: real-world progression-free survival.
In Kaplan-Meier analyses, median rwOS from the start of first LOT was similar for the overall populations of NH Black/AA (65.1 months; 95% CI: 58.2-not reached) and NH White (60.5 months; 95% CI: 58.4-66.1) patients (Figure 3). Unadjusted and age-adjusted Cox proportional hazards models showed similar rwOS among races/ethnicities (Online Supplementary Table S4). After MV-adjustment, NH Black/AA patients had inferior rwOS compared to NH White patients, however this difference was not statistically significant (HR=1.12; 95% CI: 0.98-1.28; Figure 4). MV-adjusted analyses examining associations between selected patient and disease characteristics and rwOS from start of first LOT, by race/ethnicity, are shown in Figure 4 (full subgroup analyses are shown in Online Supplementary Table S5). Age of 65 to <75 years and age of ≥75 years at start of first LOT (both compared with age of <65 years) were associated with statistically inferior rwOS for both NH Black/AA and NH White patients; this association was particularly strong for the ≥75 years age group. ECOG PS of ≥2 (compared with ECOG PS of 0), ISS Stage II or III disease at diagnosis (compared with Stage I disease at diagnosis), eGFR <60 mL/min/1.73 m2 (compared with eGFR ≥60 mL/ min/1.73 m2), and the presence of high-risk cytogenetics (compared with standard-risk cytogenetics) were also associated with statistically inferior rwOS for both races/ ethnicities. The presence of 1q21+ (compared with the absence of 1q21+) was similarly associated with worse rwOS among both groups but was only statistically significant
among NH White patients. Likelihood ratio tests confirmed that associations between selected factors and rwOS were not statistically different between races/ethnicities.
This study used recent, EHR-derived data to retrospectively analyze patient and treatment characteristics as well as survival outcomes for patients of NH Black/AA and NH White race/ethnicity in the US. Adding to findings from previous RW analyses, our study complements findings from prospective clinical trials, which may not be fully generalizable to the RW population. To our knowledge, this study is the first to evaluate both rwPFS and rwOS using robust MV-adjusted modeling among numerous subgroups of NH Black/AA and NH White patients.
Consistent with previous analyses of multiple RW data sets,6,11,17,20,21 NH Black/AA patients in our study were younger than NH White patients at diagnosis and start of first LOT. The percentage of female patients was higher among the NH Black/AA than the NH White cohort, reflective of the slight weighting of the Flatiron Health database toward patients in the Southern region of the US,19 where the most recent US Census data shows higher-than-average representation of both females and people of Black/AA race.22,23 The percentage of patients with eGFR <60 mL/min/1.73 m2 at start of first LOT was slightly higher among NH White than NH Black/AA patients (Online Supplementary Appendix; Online
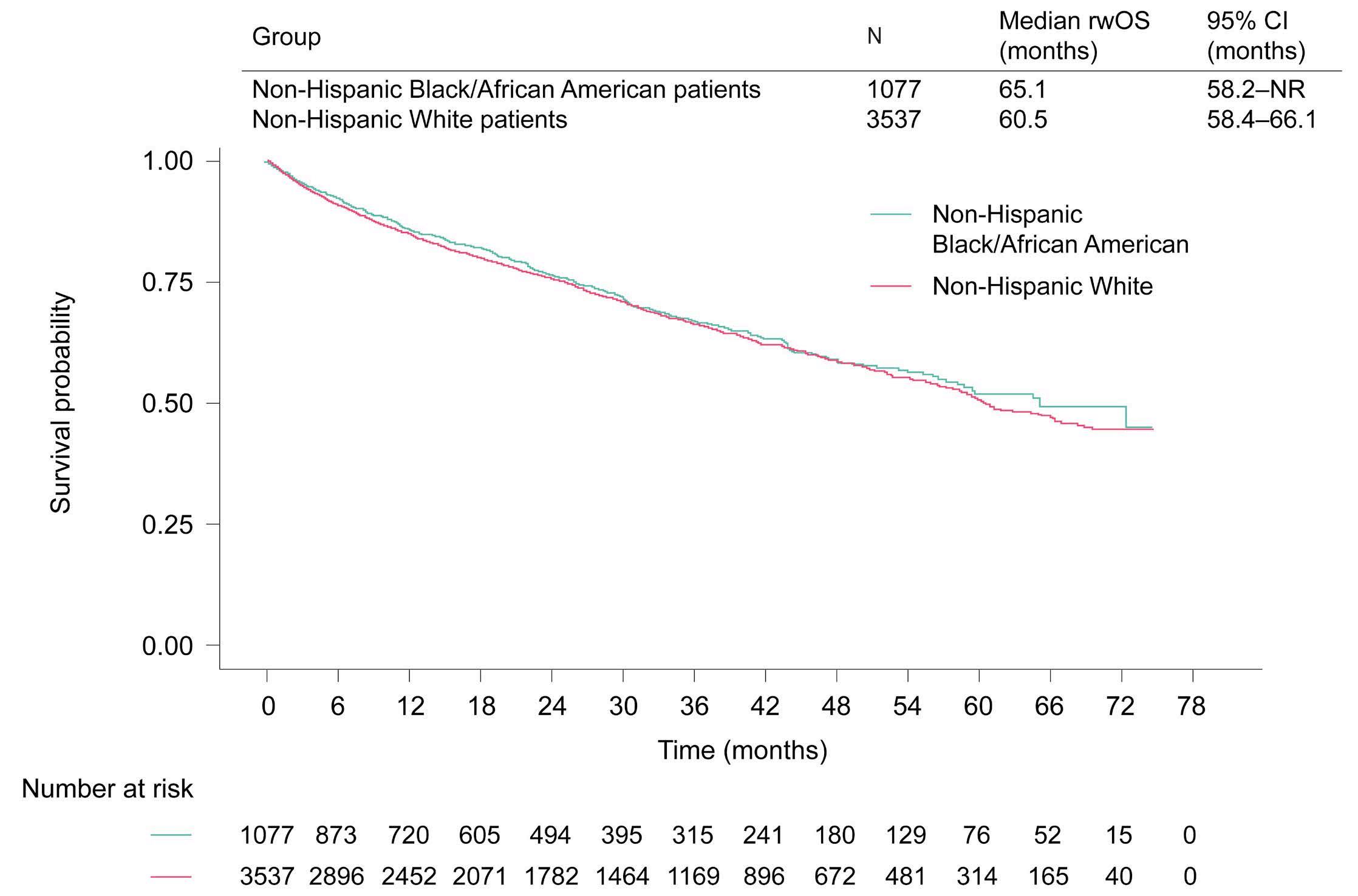
Figure 3. Kaplan-Meier analysis of real-world overall survival from start of first line of treatment by race/ethnicity. CI: confidence interval; LOT: line of therapy; NR: not reached; rwOS: real-world overall survival.
Supplementary Table S6), but NH White patients with eGFR <50 mL/min/1.73 m2 at start of first LOT were slightly more likely than their NH Black/AA counterparts to have at least one eGFR ≥60 mL/min/1.73 m2 during the first LOT. African ancestry has been associated with a significantly lower prevalence of HRCA such as del(17p) and t(4;14).14 The presence of individual HRCA including del(17p), t(4;14) and
t(14;16) was similar between NH Black/AA and NH White patients, as was the presence of 1q21+, which is also consistent with earlier analyses of the Flatiron Health MM database24 and the international CoMMpass database.11 In our study and other RW and administrative database studies, Black/AA race has been associated with lower rates of SCT use despite younger age at diagnosis.11-13,17,21 Our
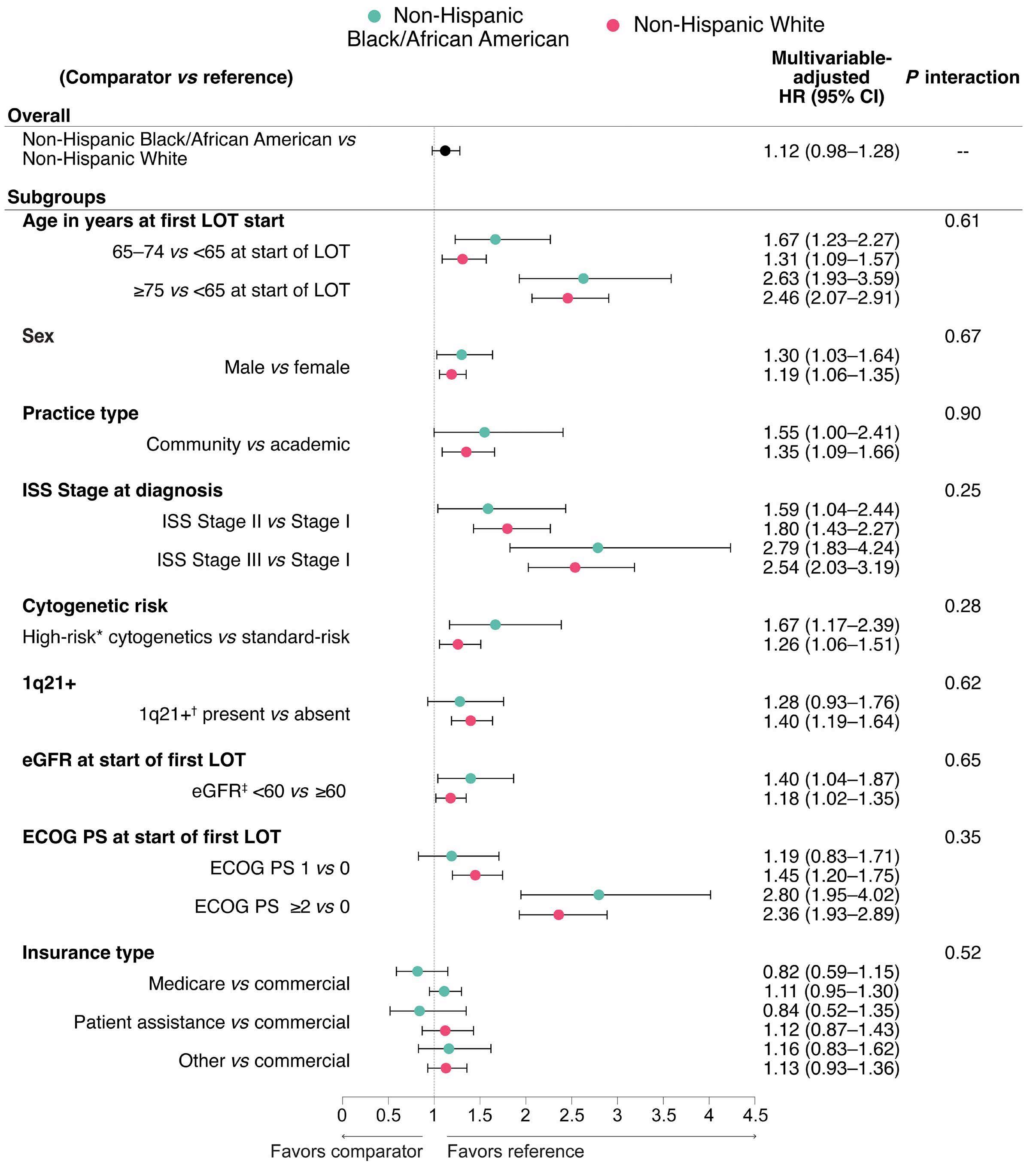
Figure 4. Multivariable-adjusted Cox proportional hazards model of real-world overall survival from start of first line of treatment, by race/ethnicity, overall and for select subgroups. *High-risk cytogenetics were defined as the presence of ≥1 of del(17p), t(4;14), or t(14;16). †1q21+ was defined as gain (3 copies) or amplification (≥4 copies) of 1q21. ‡Assessed using the MDRD equation; expressed as mL/min/1.73 m2. CI: confidence interval; ECOG PS: Eastern Cooperative Oncology Group performance status; eGFR: estimated glomerular filtration rate; HR: hazard ratio; ISS: International Staging System; LOT: line of therapy; MDRD: modification of diet in renal disease; MV: multivariable; PS: performance status; rwOS: real-world overall survival.
study was not designed to examine any differential benefit of SCT between NH Black/AA and NH White patients, though this remains an important clinical question. Black/AA patients comprised nearly 20% of the study population in the phase III DETERMINATION trial,25 which prospectively randomized patients with NDMM to lenalidomide-bortezomib-dexamethasone with and without early SCT and with all patients receiving lenalidomide maintenance until progression. In the overall population, SCT significantly improved PFS but not OS. In a preplanned subgroup analysis, PFS benefit of SCT appeared evident in the population of White patients but not among Black/AA patients. Notably, the trial was not powered to definitively evaluate PFS among subgroups, but rather to be hypothesis-generating; hence, findings among Black patients are being further evaluated to better understand how racial differences may mediate differential benefit from SCT and further analyses from this important study are anticipated with great interest.
Black/AA patients have also been reported as less likely than White patients to receive PI + IMiD-based therapies such as front-line triplet induction therapy that contains lenalidomide and bortezomib.10-13 A US-based, retrospective chart review of daratumumab users between November 2015 and May 2020 also found notable disparity in first-line daratumumab use (4.5% of Black patients vs. 9.2% of White patients).26 Rates of lenalidomide, pomalidomide, bortezomib, carfilzomib, and daratumumab use were similar between races/ethnicities in our study, suggesting that adoption of standard-of-care regimens has started to equalize across races/ethnicities within US-based community practices.
Notably, the time frame of our analysis (January 2016 through March 2022) does not fully reflect the emergence of anti-CD38 mAb into the NDMM setting. Results from MAIA27 (daratumumab-lenalidomide-prednisone vs. lenalidomide-prednisone) and ALCYONE28 (daratumumab-bortezomib-melphalan-prednisone vs. bortezomib-melphalan-prednisone), which led to Food and Drug Administration indications in the setting of newly diagnosed disease, were first published in 2019 and 2018, respectively. Completed and ongoing studies of triplet and quadruplet regimens containing anti-CD38 MAb (i.e., isatuximab, daratumumab) may lead to additional approvals in the first-line space.
Though RW endpoints inherently differ from those used in clinical trials, the use of rwPFS as a meaningful outcome is becoming more common.11,24,29-31 Utilizing Flatiron Health’s rules for PFS, the unadjusted and age-adjusted Cox proportional hazards models used in our study demonstrated similar rwPFS results for NH Black/AA and NH White patients with NDMM, whereas MV-adjusted Cox models showed slightly inferior rwPFS among NH Black/AA patients. This is an important finding, particularly given the size of our cohort and reflection of community-based practice in the US. Our analysis also suggested slightly inferior MV-adjusted rwOS for NH Black/AA patients compared with NH White patients with NDMM, though this difference was not statis-
tically significant. This lack of a significant association aligns with findings from an earlier analysis of Black/AA and White patients in the Flatirion Health MM database who initiated first-line therapy between 2011 and 2019.17 Notably, other RW studies and analyses of administrative datasets have yielded discrepant results. Analysis of the Multiple Myeloma Research Foundation’s CoMMpass data set,11 pooled from 90 sites worldwide, found that Black patients had inferior OS compared with White patients (age-adjusted HR=1.7; 95% CI: 1.2-2.4), which was only partly attenuated by receipt of triplet therapy and SCT. A VA study,32 SEER-based analysis,12 and RW analysis of the Connect MM Registry21 found that Black/ AA patients have equal, if not better, survival outcomes than their White counterparts when access to care is equal. These discrepant findings likely reflect differences in sites of care (e.g., US-based community clinics or VA system vs. international practice sites) and varying time periods of analyses, both of which may lead to important differences in available therapies or cultural awareness. In addition, population-based studies or those using administrative data (e.g., SEER data) may use different methods of data extraction and may not fully account for other factors (e.g., socioeconomic status, cultural barriers) that may impact outcomes.
We found that associations between patient and disease characteristics and survival outcomes were generally consistent between NH Black/AA and NH White patients. Importantly, the impact of high-risk cytogenetics on survival outcomes in MM patients remains less than fully understood. Alignment of our findings with those of other analyses is complicated by differences in how high-risk cytogenetics are characterized. In an analysis of patients in the Flatiron Health MM database using a slightly earlier time frame (January 2011 to May 2021), Calip et al.24 also examined the differential impact of high-risk cytogenetics on MM outcomes between races. However, they examined the association between number of HRCA (0, 1, or 2+) and rwPFS and rwOS, defining HRCA as 1q21+, del(17p), t(4;14), t(14;16) and t(14;20). Compared with patients with no HRCA, White and Black patients of any age with exactly 1 HRCA had statistically inferior rwPFS and rwOS, whereas having “double-hit MM” (2+ HRCA) was differentially predictive of poor survival across races.24 Applying these same definitions of HRCA to an analysis of the international CoMMpass database, Derman et al.11 found more widely discrepant associations between 1 and 2+ HRCA and PFS and OS among White and Black patients. Though differences in data abstraction between studies may contribute to discrepant findings, the lack of a uniform definition of high-risk cytogenetics complicates the ability to determine which cytogenetic abnormalities have the greatest impact on MM patient outcomes. Moving forward, the closer alignment in how cytogenetics are characterized within key staging/risk-defining systems (e.g., the Second Revision of the International Staging System33 and the IMWG34) may increase uniformity of these definitions across studies. Strengths of our study include adequate representation of NH Black/AA patients (23.3% of our overall study population),
which aligns with epidemiological trends and the estimated incidence of MM in Black/AA patients in the US.4,35 Calculation of P interaction values strengthens our ability to state that certain variables did not differentially impact outcomes between races/ethnicities. The Flatiron Health MM Cohort predominantly comprises patients treated within community practices in the US. As such, the resulting study cohorts may not be fully representative of patients treated at US-based academic centers or international centers. As with most EHRbased studies, our analysis is subject to potential missing or erroneous data and may not have captured all treatment received by patients. Our study was not able to precisely characterize 1q21 copy number to distinguish between gain (3 copies) or amplification (≥4 copies), which may affect the degree of risk imparted by the cytogenetic abnormality. In addition, we did not examine t(11;14) in our cohort due to a high level of missing data and variable conclusions in the literature about its prognostic effects in both the general MM population and the AA population. Indeed, efforts to better characterize the impact of gain versus amplification of 1q21 and t(11;14) in RW populations should be sought. In our study, unadjusted rwPFS and rwOS were similar between patients of NH Black/AA and NH White race/ethnicity. However, after multivariable adjustment, NH Black/AA race/ethnicity was associated with slightly inferior PFS, reflecting the need for a greater understanding of underlying factors that might contribute to survival differences between patients of different races/ethnicities, and how such factors may differ between patients seeking care at community versus academic sites. As additional therapies become available, periodic re-examination of RW data will be necessary to capture any differential use or survival impact of emerging treatment options. Strategies to improve the reliability and accuracy of abstracted data from EHR and their statistical interpretation will strengthen the ability of RW studies to meaningfully augment learnings from clinical trials. Continued efforts should be made to equalize access to care among patients of different races/ethnicities and to increase representation of patients of non-White race in clinical trials of MM. This, in turn, should impact favorably on the ability of current and future phase III study results to translate meaningfully into RW practice.36
Disclosures
TB discloses speakers bureau, advisory panels, and consultancy
1. Cancer Stat Facts: Myeloma. National Institutes of Health Surveillance, Epidemiology, and End Results Program. https:// seer.cancer.gov/statfacts/html/mulmy.html. Accessed September 25, 2022.
2. National Cancer Institute. Myeloma: People Alive with Cancer (U.S. Prevalence) on January 1, 2020. https://seer.cancer.gov/ statistics-network/explorer/application.html?site=89&data_ type=5&graph_type=12&compareBy=sex&chk_
for Sanofi and Bristol Myers Squibb. MHB discloses consultancy honorarium from AbbVie, Bristol Myers Squibb/Celgene, GSK, Janssen, Karyopharm and Sanofi; speakers honorarium from Multiple Myeloma Research Foundation and Cancer Care. YAE discloses consultancy honorarium/speakers bureau from Takeda, Oncopeptides, Janssen, GSK, Alnylam, Sanofi, Pfizer and Adaptive; advisory board membership of Takeda, Oncopeptides, Janssen, GSK, Alnylam, Sanofi and Pfizer; research support from Bristol Myers Squibb/Celgene; independent adjudication committee membership of Takeda and ORCA. CM discloses speakers bureau at AstraZeneca, Bristol Myers Squibb, Blueprint Medicine and BeiGene. JAZ discloses research support from Bristol Myers Squibb and Janssen; discloses advisory role at Bristol Myers Squibb, Janssen, Prothena, Alexion, Takeda and Regeneron; independent data safety monitoring committee chair at Bristol Myers Squibb. PGR discloses consulting for Oncopeptides, Celgene/Bristol Myers Squibb, Karyopharm, Sanofi, and GSK; research grants from Oncopeptides, Celgene/ Bristol Myers Squibb, Karyopharm and Takeda. TS and MSR are employed by Sanofi at the time of the study; may hold stock and/or stock options in the company.
TB, TS, and MSR were involved in the conception and design of the study. MSR was responsible for data analysis. TB, MHB, YAE, CM, JAZ, PGR, TS, and MSR participated in data interpretation, manuscript writing, review, and final approval of the submitted version of the manuscript.
The authors would like to thank Robert Lubwama from Sanofi for his critical review of this manuscript. Medical writing support was provided by Lindsay Gasch, PharmD, and Camile Semighini Grubor, PhD, of Envision Pharma Group, funded by Sanofi.
This investigation was supported by Sanofi.
The data that support the findings of this study have been originated by Flatiron Health, Inc. Requests for data sharing by license or by permission for the specific purpose of replicating results in this manuscript can be submitted to dataaccess@ flatiron.com.
sex_1=1&series=9&race=9&age_range=1&prev_duration=1&advopt_ precision=1&hdn_view=1. Accessed July 18, 2023.
3. American Cancer Society. Cancer facts and figures for African Americans 2022-2024. https://www.cancer.org/content/dam/ cancer-org/research/cancer-facts-and-statistics/cancer-factsand-figures-for-african-americans/2022-2024-cff-aa.pdf. Accessed September 26, 2022.
4 Rosenberg PS, Barker KA, Anderson WF. Future distribution of
multiple myeloma in the United States by sex, age, and race/ ethnicity. Blood. 2015;125(2):410-412.
5. Costa LJ, Brill IK, Omel J, Godby K, Kumar SK, Brown EE. Recent trends in multiple myeloma incidence and survival by age, race, and ethnicity in the United States. Blood Adv. 2017;1(4):282-287.
6. Waxman AJ, Mink PJ, Devesa SS, et al. Racial disparities in incidence and outcome in multiple myeloma: a populationbased study. Blood. 2010;116(25):5501-5506.
7. Ahmed N, Shahzad M, Shippey E, et al. Socioeconomic and racial disparity in chimeric antigen receptor T cell therapy access. Transplant Cell Ther. 2022;28(7):358-364.
8. Alqazaqi R, Schinke C, Thanendrarajan S, et al. Geographic and racial disparities in access to chimeric antigen receptor-T cells and bispecific antibodies trials for multiple myeloma. JAMA Netw Open. 2022;5(8):e2228877.
9. Emole J, Lawal O, Lupak O, Dias A, Shune L, Yusuf K. Demographic differences among patients treated with chimeric antigen receptor T-cell therapy in the United States. Cancer Med. 2022;11(23):4440-4448.
10 Ailawadhi S, Parikh K, Abouzaid S, et al. Racial disparities in treatment patterns and outcomes among patients with multiple myeloma: a SEER-Medicare analysis. Blood Adv. 2019;3(20):2986-2994.
11. Derman BA, Jasielec J, Langerman SS, Zhang W, Jakubowiak AJ, Chiu BC. Racial differences in treatment and outcomes in multiple myeloma: a multiple myeloma research foundation analysis. Blood Cancer J. 2020;10(8):80.
12. Dong J, Garacci Z, Buradagunta CS, et al. Black patients with multiple myeloma have better survival than White patients when treated equally: a matched cohort study. Blood Cancer J. 2022;12(2):34.
13. Fiala MA, Wildes TM. Racial disparities in treatment use for multiple myeloma. Cancer. 2017;123(9):1590-1596.
14 Greenberg AJ, Philip S, Paner A, et al. Racial differences in primary cytogenetic abnormalities in multiple myeloma: a multi-center study. Blood Cancer J. 2015;5(1):e271.
15. Kazandjian D, Hill E, Hultcrantz M, et al. Molecular underpinnings of clinical disparity patterns in African American vs. Caucasian American multiple myeloma patients. Blood Cancer J. 2019;9(2):15.
16. Fillmore NR, Yellapragada SV, Ifeorah C, et al. With equal access, African American patients have superior survival compared to White patients with multiple myeloma: a VA study. Blood. 2019;133(24):2615-2618.
17 Maignan K, Fashoyin-Aje LA, Torres AZ, et al. Exploring racial disparities in treatment patterns and outcomes for patients with multiple myeloma using real world data. Blood Cancer J. 2022;12(4):65.
18. Birnbaum B, Nussbaum N, Seidl-Rathkopf K, et al. Modelassisted cohort selection with bias analysis for generating largescale cohorts from the EHR for oncology research. arXiv. https:// doi.org/10.48550/arXiv.2001.09765. Accessed December 6, 2022.
19. Ma X, Long L, Moon S, Adamson BJS, Baxi SS. Comparison of population characteristics in real-world clinical oncology databases in the US: Flatiron Health, SEER, and NPCR. MedRxiv. https://www.medrxiv.org/content/10.1101/2020.03.16.20037143v2. Accessed December 6, 2022.
20 Ailawadhi S, Aldoss IT, Yang D, et al. Outcome disparities in multiple myeloma: a SEER-based comparative analysis of ethnic subgroups. Br J Haematol. 2012;158(1):91-98.
21. Ailawadhi S, Jagannath S, Lee HC, et al. Association between
race and treatment patterns and survival outcomes in multiple myeloma: a Connect MM Registry analysis. Cancer. 2020;126(19):4332-4340.
22. Blakeslee L, Caplan Z, Meyer JA, Rabe MA, Roberts AW. Age and sex composition: 2020. US Census Bureau. https://www2. census.gov/library/publications/decennial/2020/census-briefs/ c2020br-06.pdf. Accessed July 19, 2023.
23. Race and Ethnicity in the United States: 2010 census and 2020 census. US Census Bureau. https://www.census.gov/library/ visualizations/interactive/race-and-ethnicity-in-the-unitedstate-2010-and-2020-census.html. Accessed July 19, 2023.
24. Calip GS, Ascha MS, Wang X, et al. Racial and age-related differences in impacts of high-risk cytogenetic abnormalities on survival in multiple myeloma in a nationwide electronic health record-derived database. Blood. 2021;138(Suppl 1):4121.
25. Richardson PG, Jacobus SJ, Weller EA, et al. Triplet therapy, transplantation, and maintenance until progression in myeloma. N Engl J Med. 2022;387(2):132-147.
26. Atrash S, Thompson-Leduc P, Tai MH, et al. Patient characteristics, treatment patterns, and outcomes among black and white patients with multiple myeloma initiating daratumumab: A real-world chart review study. Clin Lymphoma Myeloma Leuk. 2022;22(8):e708-e715.
27. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115.
28. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528.
29 Bergin K, Wellard C, Augustson B, et al. Real-world utilisation of ASCT in multiple myeloma (MM): a report from the Australian and New Zealand myeloma and related diseases registry (MRDR). Bone Marrow Transplant. 2021;56(10):2533-2543.
30 Kumar L, Hussain MM, Chethan R, et al. Multiple myeloma: impact of time to transplant on the outcome. Clin Lymphoma Myeloma Leuk. 2022;22(9):e826-e835.
31. Medhekar R, Ran T, Fu AZ, Patel S, Kaila S. Real-world patient characteristics and treatment outcomes among nontransplanted multiple myeloma patients who received bortezomib in combination with lenalidomide and dexamethasone as first line of therapy in the United States. BMC Cancer. 2022;22(1):901.
32. Fillmore NR, Cirstea D, Munjuluri A, et al. Lack of differential impact of del17p on survival in African Americans compared with White patients with multiple myeloma: a VA study. Blood Adv. 2021;5(18):3511-3514.
33. D’Agostino M, Cairns DA, Lahuerta JJ, et al. Second revision of the International Staging System (R2-ISS) for overall survival in multiple myeloma: a European Myeloma Network (EMN) report within the HARMONY Project. J Clin Oncol. 2022;40(29):3406-3418.
34 Chng WJ, Dispenzieri A, Chim CS, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014;28(2):269-277.
35. Ellington TD, Henley SJ, Wilson RJ, Wu M, Richardson LC. Trends in solitary plasmacytoma, extramedullary plasmacytoma, and plasma cell myeloma incidence and myeloma mortality by racial-ethnic group, United States 20032016. Cancer Med. 2021;10(1):386-395.
36. Richardson PG, San Miguel JF, Moreau P, et al. Interpreting clinical trial data in multiple myeloma: translating findings to the real-world setting. Blood Cancer J. 2018;8(11):109.
Phyllis S. Y. Chong,1,2* Jing Yuan Chooi,1* Sze Lynn Julia Lim,2 Tae-Hoon Chung,2 Reinhard Brunmeir,2 Aaron Chung Yong Leow,2 Sabrina Hui Min Toh,2 Kalpnaa Balan,2 Muhamad Irfan Bin Azaman,2 Zhengwei Wu,2,3 Nagavidya Subramaniam,4 Leah A. Vardy4 and Wee-Joo Chng1,2,5
1Department of Medicine, Yong Loo Lin School of Medicine, National University of Singapore; 2Cancer Science Institute of Singapore, National University of Singapore; 3Genome Institute of Singapore, Agency for Science, Technology and Research (A*STAR); 4A*STAR Skin Research Labs and Skin Research Institute of Singapore, A*STAR, Immunos and 5Department of Hematology-Oncology, National University Cancer Institute of Singapore, National University Health System, Singapore
*PSYC and JYC contributed equally as first authors.
Abstract
Correspondence: W-J. Chng mdccwj@nus.edu.sg
Received: May 2, 2023.
Accepted: December 11, 2023. Early view: December 21, 2023.
https://doi.org/10.3324/haematol.2023.283467
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
REIIBP is a lysine methyltransferase aberrantly expressed through alternative promoter usage of NSD2 locus in t(4;14)-translocated multiple myeloma (MM). Clinically, t(4;14) translocation is an adverse prognostic factor found in approximately 15% of MM patients. The contribution of REIIBP relative to other NSD2 isoforms as a dependency gene in t(4;14)-translocated MM remains to be evaluated. Here, we demonstrated that despite homology with NSD2, REIIBP displayed distinct substrate specificity by preferentially catalyzing H3K4me3 and H3K27me3, with little activity on H3K36me2. Furthermore, REIIBP was regulated through microRNA by EZH2 in a Dicer-dependent manner, exemplifying a role of REIIBP in SET-mediated H3K27me3. Chromatin immunoprecipitation sequencing revealed chromatin remodeling characterized by changes in genome-wide and loci-specific occupancy of these opposing histone marks, allowing a bidirectional regulation of its target genes. Transcriptomics indicated that REIIBP induced a pro-inflammatory gene signature through upregulation of TLR7, which in turn led to B-cell receptor-independent activation of BTK and driving NFkB-mediated production of cytokines such as IL-6. Activation of this pathway is targetable using Ibrutinib and partially mitigated bortezomib resistance in a REIIBP xenograft model. Mechanistically, REIIBP upregulated TLR7 through eIF3E, and this relied on eIF3E RNA-binding function instead of its canonical protein synthesis activity, as demonstrated by direct binding to the 3’UTR of TLR7 mRNA. Altogether, we provided a rationale that co-existence of different NSD2 isoforms induced diversified oncogenic programs that should be considered in the strategies for t(4;14)-targeted therapy.
Multiple myeloma (MM) is a neoplasm of plasma cells characterized by the uncontrolled proliferation of abnormal plasma cells in the bone marrow incapable of producing functional antibodies.1 Current treatment regime involves single or combination of novel drug classes such as proteasome inhibitors (bortezomib), immunomodulatory drugs (lenalidomide) and monoclonal antibodies (daratumumab), which have significantly improved survival outcomes in patients.2-4 However, this disease still represents an important clinical challenge as it mainly affects the elderly population and frequent development of drug resistance subsequent
to initial treatment response. MM can be broadly divided into hyperdiploid and non-hyperdiploid subtypes, with the non-hyperdiploid cases identified by recurrent immnuoglobulin (Ig)G translocations such as t(4;14)(p16;q32) and t(11;14)(q13;q32) in ~15% of MM patients respectively.5,6 Such recurrent chromosomal translocations are central to the pathogenesis of MM and predicts the treatment response and clinical outcome of the patient. Patients with t(4;14) translocation, which displays a dysregulation of the NSD2 locus and its alternatively spliced variants, has one of the worst prognosis when compared to other biological subgroups, but represents an intermediate-risk group given its response towards bortezomib.7,8
We and others have sought to study the function of the protein products arising from NSD2 gene, which includes the full-length isoform NSD2, and the shorter isoforms NSD1 and REIIBP.9-19 NSD2 contains 1,365 amino acids, and harbors conserved motifs such as PWWP domain, PHDtype zinc fingers and HMG box which are typically found in proteins with chromatin-binding ability and recognition of histone marks. The C-terminal region resides a functional and catalytic SET domain, which is essential for the oncogenic activities of NSD2. In vitro and in vivo histone methyltransferase assays demonstrated that the primary activity of NSD2 is H3K36 dimethylation, which leads to a global gene activation reprogramming that drives myelomagenesis.11
Other histone modifications modulated by NSD2 includes H4K20 and H3K27 methylation, as well as H3 acetylation.12-14 Through its histone-modifying activities, NSD2 promotes cancer phenotypes such as increased cell proliferation, clonogenicity, adhesion to bone marrow stroma and tumorigenesis.11,15-17 NSD1 is identical to the N-terminus region of NSD2 spanning 647 amino acids. Despite lacking the SET domain, it regulates gene expression through binding to the promoter of target genes such as GLO1, and truncation studies indicated that the HMG box at the C-terminus of NSD1 is important for this function.18 On the other hand, the role of REIIBP, which is overexpressed independent of NSD2 breakpoint clusters, is poorly understood.9,10 This transcript arises from intron 9 of the NSD2 locus and is identical to the C-terminus of NSD2 spanning 584 amino acids, retaining the SET domain.19 Furthermore, the subcellular localization of REIIBP is found in the cytoplasm and nucleoli, which differs from NSD1 and NSD2 that reside in the nucleus.9 Hence, it is likely that REIIBP have differential histone methylation targets as well as novel functions that are not fully elucidated, and the contribution of REIIBP relative to other NSD2 isoforms as a dependency gene in t(4;14)-translocated MM remains to be evaluated. In this study, we generated a stable cell line that overexpresses REIIBP to perform an unbiased study of the histone lysine methylation and regulatory activities of REIIBP.
Plasmids, patient samples and reagents
Full-length NSD2, NSD1 and REIIBP were cloned as previously described.11,18 Dicer and TLR7 short hairpin RNA (shRNA) were constructed in pRP(shRNA)-EGFP-U6 vector and miR-26a, miR-31, and miR-203 were cloned in pRP[ncRNA]-Puro-CMV by VectorBuilder (USA). Two single guide RNA (sgRNA) targeting the coding region of eIF3E were cloned into vector backbone pRP(CRISPR)-Puro-hCas9-U6 and three sgRNA targeting its H3K4me3 TSS peak (VectorBuilder, USA). EZH2 small interfering RNA (siRNA) was purchased from Thermo Fisher Scientific (USA); EPZ-6438 and GSK-126 were purchased from Selleck Chemicals (USA). Ibrutinib, bortezomib
and loxoribine were purchased from Santa Cruz (USA). Patient samples were collected with written consent at the National University Cancer Institute Singapore with the approval from the Institutional Review Board (DSRB 2017/00196).
Histone H3 tri-methyl K27 and K4 quantification kits were purchased from Abcam and performed as per manufacturer’s instructions; 0, 2, 5, 10 or 20 µg of nuclear extracts (Thermo Fisher Scientific) were added to biotinylated substrate (unmethylated histone peptide) and anti-H3K27me3 or anti-H3K4me3 antibody was used for capture and readings taken at absorbance 450 nm with a microplate reader (Tecan). S-adenosylmethionine (SAM) methyltransferase assay was performed using 1 µM SAM as a methyl-group donor to modify 2 µg of H3 substrate by 2 µg REIIBP in a reaction buffer previously described20 at 30°C for 2 hours. Proteins were resolved on 15% SDS-PAGE and probed with indicated antibodies.
Chromatin immunoprecipitation (ChIP) sequencing was performed on isogenic cell lines RPMI8226-Vcon and RPMI8226-REIIBP (20 million cells each) using reagents obtained from Cell Signaling Technology (SimpleChIP® Enzymatic Chromatin IP Kit) according to the manufacturer’s protocol. Monoclonal antibodies used for ChIP were anti-H3K27me3 (CST, #9733) and anti-H3K4me3 (CST, #9751). DNA were extracted with MinElute PCR Purification Kit (Qiagen). More information is provided in the Online Supplementary Appendix.
In order to generate a REIIBP xenograft model, RPMI8226-REIIBP stable cells (5×106) were suspended in 0.1 mL phosphate-buffered saline and subcutaneously injected into the flanks of NOD/SCID female mice (6 weeks old, InVivos). Tumor growth was monitored using calipers every 3 days until a volume of 150 mm3 (calculated as length x width [2]/2) is reached, which developed between 2-3 weeks, and randomized into four groups: dimethyl sulfoxide (DMSO) (1% final concentration), ibrutinib (15 mg/kg), bortezomib (0.4 mg/kg) and combination (N=5 mice/group). Treatment was performed every 2 days via intraperitoneal injection for 2 weeks. Tumors were harvested for weight analysis. The responsible use of animals was approved and in accordance to protocol by Institutional Animal Care and Use Committee (IACUC; National University of Singapore).
Student’s t test was used to compare significant differences between groups using GraphPad Prism or Excel and adjusted using Benjamini-Hochberg correction method for multiple comparisons. Correlation analyses were performed between REIIBP (ENST00000382888, transcript NSD2-203
of Enesmbl annotation) and BTK in CoMMpass dataset using Pearson correlation analysis. Survival analysis was performed by Kaplan–Meier method and assessed using the log-rank test generated with MMTools. Using Wilcoxon’s test, association between REIIBP expression and bortezomib response was determined in CoMMpass (IA13a version) using the drug treatment response data that had included bortezomib.
A detailed description is available in the Online Supplementary Appendix
REIIBP is expressed in t(4;14) myeloma cells independent of other NSD2 isoforms, FGFR3 or ACA11 expression and harbored oncogenic activity
In order to elucidate the biological role of REIIBP in myeloma, we first examined the endogenous expression of REIIBP in human myeloma cell lines (HMCL). We optimized an antibody that recognizes the C-terminus region of NSD2 and detected a band that corresponded to REIIBP at ~62 kDa. Using a different N-terminus antibody, we probed for NSD1 and NSD2. Consistent with previous reports, we detected the longest isoform of NSD2 in KMS11 and KMS34 cells, and these cells harbored the highest expression for NSD1 (Figure 1A). On the contrary, REIIBP expression was lower in KMS11 and KMS34, but abundantly expressed in other t(4;14)+ cells, while the t(4;14)- cells showed little to null expression of REIIBP (Figure 1A). Next, we compared REIIBP transcript levels against other gene products that were reported to be associated with t(4;14) locus, namely FGFR3 and ACA11, 9,21 but no clear correlation were observed (Figure 1B). Additionally, in t(4;14)- cells, we could detect REIIBP mRNA that was not translated into REIIBP protein (Figure 1A, B), suggesting a post-transcriptional regulatory mechanism of REIIBP in myeloma cells. Another MM cell line commonly used to study NSD isoforms is the TKO (translocation knockout) cells generated from parental KMS11 with the exon 7 on t(4;14) translocated NSD2 allele deleted.16,22 This resulted in almost undetectable protein levels of NSD1 and NSD2, and TKO cells also lacked the protein expression of REIIBP (Online Supplementary Figure S1A). For clinical relevance, we compared the expression of REIIBP in primary patient samples with varying t(4;14) status. While REIIBP protein was detected in all t(4;14) myeloma samples, it was found in only one of three non-t(4;14) samples (Figure 1C; Online Supplementary Figure S1B). Using shRNA targeting different regions of NSD2, we found that complete abrogation of NSD2 drives a compensatory increase in REIIBP to partially rescue cell viability (Figure 1D; Online Supplementary Figure S1C). These data revealed the regulatory complexity among isoforms of NSD2 in t(4;14) myelomagenesis.
We engineered RPMI8226 to stably overexpress His-tagged
REIIBP given its low expression of all NSD2 isoform products (Figure 1E). In consideration of its proposed role as a histone methyltransferase, we checked whether REIIBP could be found in the nucleus. Ectopic and endogenous REIIBP were detected in both the nuclear and cytoplasmic compartments of the cell (Figure 1F; Online Supplementary Figure S1D). This contrasts with the exclusive expression of NSD1 and NSD2 in the nucleus. Compared to control, overexpression of REIIBP promoted myeloma cell growth in a short-term viability assay (Figure 2A) and a significant increase in soft agar clonogenic growth (Figure 2B). These were attributed to changes in cell-cycle progression (Figure 2C) with little effect on apoptosis (Online Supplementary Figure S1E). In order to exclude cell line-specific observations, we transiently overexpressed REIIBP in two other t(4;14)- cells, KMS12BM and U266, where REIIBP similarly promoted cell viability (Online Supplementary Figure S1F). Unlike NSD2,7,8 overexpression of REIIBP rendered RPMI8226 cells less sensitive towards bortezomib treatment (Figure 2D). In MMRF CoMMpass clinical dataset, REIIBP contributed to poorer response towards bortezomib-based regimen, where its expression was highest in progressive disease (PD) compared with the others in a six-level description (Figure 2E). The differences in REIIBP expression was also pronounced in a two-level segregation of negative versus positive bortezomib response groups (Figure 2F). Taken together, REIIBP conferred growth advantage to myeloma cells and modulated bortezomib response.
REIIBP is a functional histone methyltransferase with activity on histone 3 lysine 27 and lysine 4 trimethylation in vitro and in vivo
In order to determine the effects of REIIBP on histone methylation, we evaluated its substrate specificity by performing an in vitro histone methyltransferase assay using the methyl donor S-adenosylmethionine (SAM), substrate H3 and purified REIIBP (Online Supplementary Figure S2A). After incubation, the methylated products were visualized by immunoblot. We first performed the assay using bacterial cell extracts23 where we expressed a recombinant GST-tagged REIIBP, and detected specific modifications on H3K27me3 and H3K4me3 (Online Supplementary Figure S2B). As bacterially purified enzymes might not be fully activated either due to absence of post-translational modifications or other mammalian complex proteins, we repeated with 293T-purified REIIBP and confirmed the catalyzation of methylation on H3K27, H3K4 and H3K79 residues (Figure 3A). In order to rule out the possibility of contamination with other histone-modifying enzymes, we probed for EZH2 that catalyzes H3K27me324 and H3K79 methyltransferase DOT1L,25 which were both undetected in the extracts (Figure 3A). We performed further validation by directly measuring the enzymatic activity of nuclear extracts from the isogenic cells in a specific H3K27 or H3K4 histone methyltransferase reaction, where increased activity was observed with REIIBP
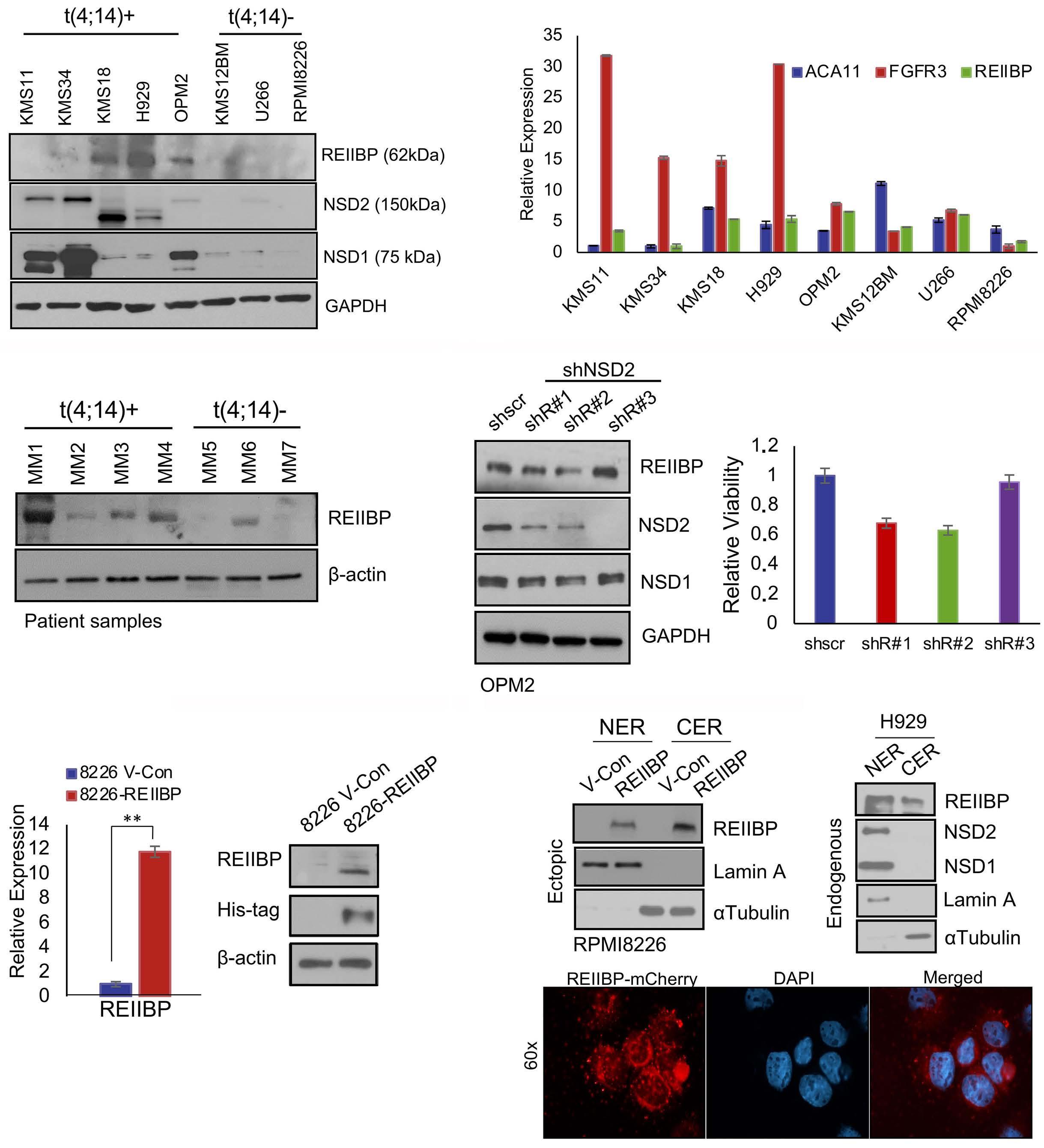
Figure 1. Expression status of REIIBP in myeloma cell lines and primary specimens. (A) Western blotting (WB) to determine the endogenous expression of NSD1, NSD2 and REIIBP in a panel of human myeloma cell lines. GAPDH was used as loading control. N=2, biological repeats, representative blots are shown. (B) Quantitative real time polymerase chain reaction (qRT-PCR) analysis of ACA11, FGFR3 and REIIBP in a panel of human myeloma cell lines. GAPDH was used as for normalization. N=3, biologically independent replicates with technical duplicates. (C) Myeloma cells from 7 patient samples with t(4;14) or non-t(4;14) status were extracted for protein and used to probe for REIIBP expression. β-actin is used for loading control. Details of patient samples can be found in Online Supplementary Figure S1B. (D) t(4;14)-positive cell line OPM2 was transiently transfected with NSD2 #1, #2 or #3 short hairpin RNA (shRNA) using the Neon transfection system. Cell viability was determined at 24 hours post-transfection (right) while immunoblotting with the indicated antibodies was performed at 48 hours post-transfection (left). Analysis from 3 biological repeats with technical octuplicates for CTG and representative images from 2 biological repeats for WB. The shRNA sequences can be found in the Online Supplementary Appendix. (E) Overexpression of REIIBP in RPMI8226 cells was determined using qRT-PCR (N=3) and WB (N=2). REIIBP expression vector contained His-tag and β-actin was used as loading control. Pairwise comparison between control and treatment group using Student’s t test (**P<0.01). (F) Ten million RPMI8226 V-Con or REIIBP cells (ectopic expression of REIIBP, left) or H929 (endogenous REIIBP, right) were lysed to extract the nuclear (NER) and cytoplasmic (CER) protein fractions. Minimal cross-contamination was confirmed with nuclear marker lamin A and cytoplasmic marker a-tubulin. NSD1 and NSD2 were found in the nucleus as previously reported. N=2, biological repeats, representative WB are shown. (Bottom) Immunoflorescence confocal microscopy images of REIIBP-mCherry expression (red florescence) in RPMI8226 cells at 60x magnification. Nuclei were stained with DAPI (blue). Image shown is a representative field.
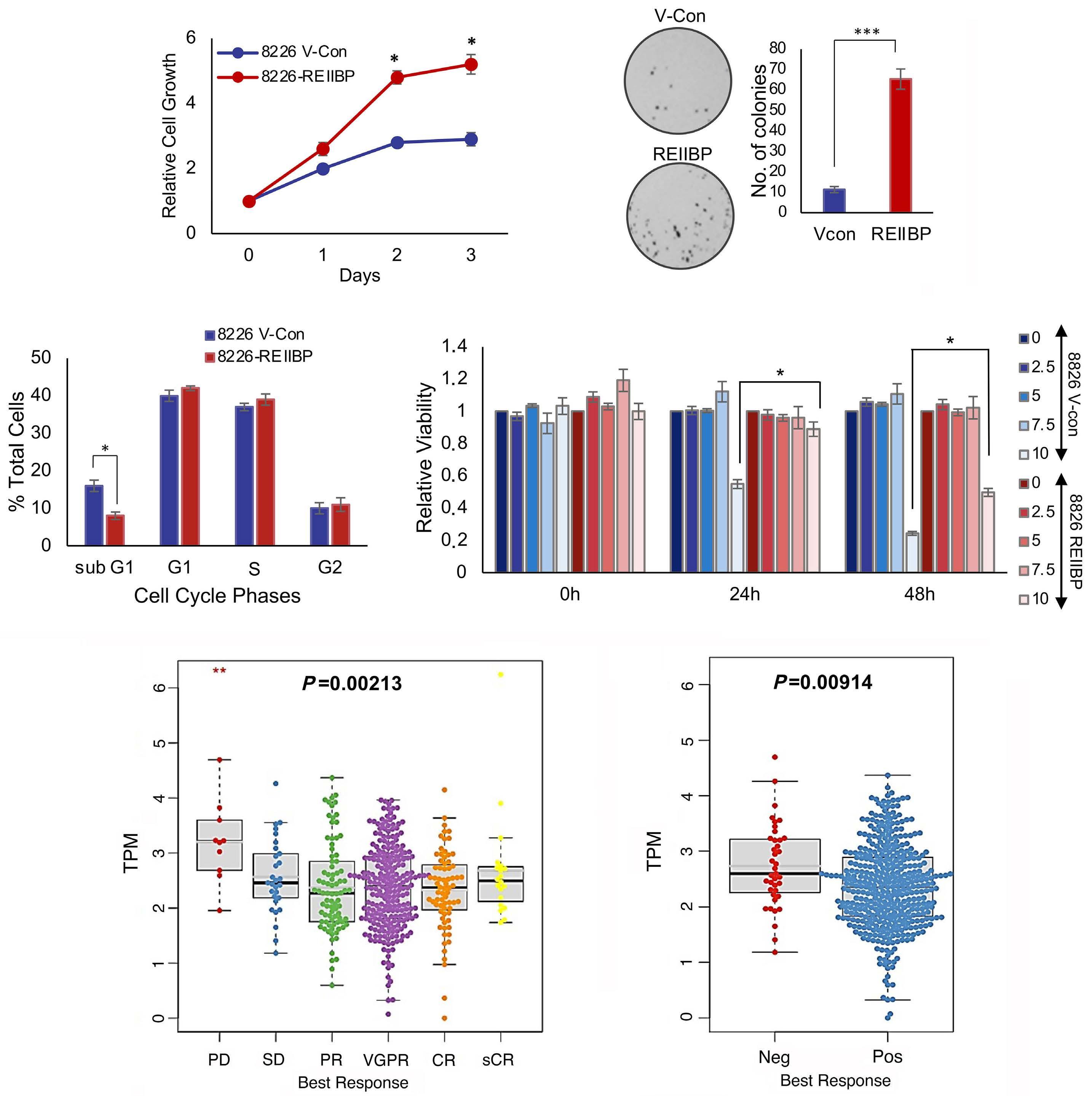
Figure 2. REIIBP is required for cell growth and conferred bortezomib resistance. (A) Cell viability was determined over 72 hours in RPMI8226 control and REIIBP-overexpressing cells and normalized against 0 hours. Three biological repeats with technical octuplicates, paired t test was performed to compare the means between REIIBP overexpressing with corresponding control (*P<0.05). (B) Colony-forming assay was performed to determine the long-term viability of RPMI8226 control and REIIBP-overexpressing cells over 2 weeks and the number of colonies was plotted. Representative images from 3 biological replicates and Student’s t test indicates significance (***P<0.001). (C) Cell cycle analysis of RPMI8226 control and REIIBP-overexpressing cells were measured using flow cytometry and the percentage of cells in sub-G1, G1, S or G2/M were indicated (N=3, independent replicates). Asterisks represent significant differences (*P<0.05; Student’s t test). (D) RPMI8226-vector control (VCon) and RPMI8226-REIIBP cells were treated with increasing concentrations of bortezomib (0, 2.5, 5, 7.5, 10 nM) and viability was determined at 24 and 48 hours using CTG assay. Viability was normalized against 0 nM and *P<0.05; Student’s t test. Experiments were performed with 3 biological repeats with technical octuplicates. (E) REIIBP expression by 6-level best response to bortezomib without t(4;14) cases in CoMMpass dataset. PD (progressive disease: 1.6%, 12/745), SD (stable disease: 6.3%, 47/745), PR (partial response: 17.4%, 130/745), VGPR (very good partial response: 51.9%, 387/745), CR (complete response: 18.7%, 139/745), sCR (stringent complete response: 4%, 30/745). (F) REIIBP expression by 2-level best response to bortezomib without t(4;14) cases in CoMMpass dataset. Negative (PD + SD: 7.9%, 59/745) and positive (PR + VGPR + CR + sCR: 92.1%, 686/745).
overexpression in a dose-dependent manner (Figure 3B). In order to complement the in vitro enzymatic assays, we did a series of in vivo immunoblot screenings of histone methylation marks. REIIBP increased the global abundance of H3K4, H3K9, H3K27 and H3K79 trimethylation, with minimal effects on dimethylation (Online Supplementary Figure S2C). In order to identify SET-dependent modifications, we transfected the cells with SET domain point mutant, which
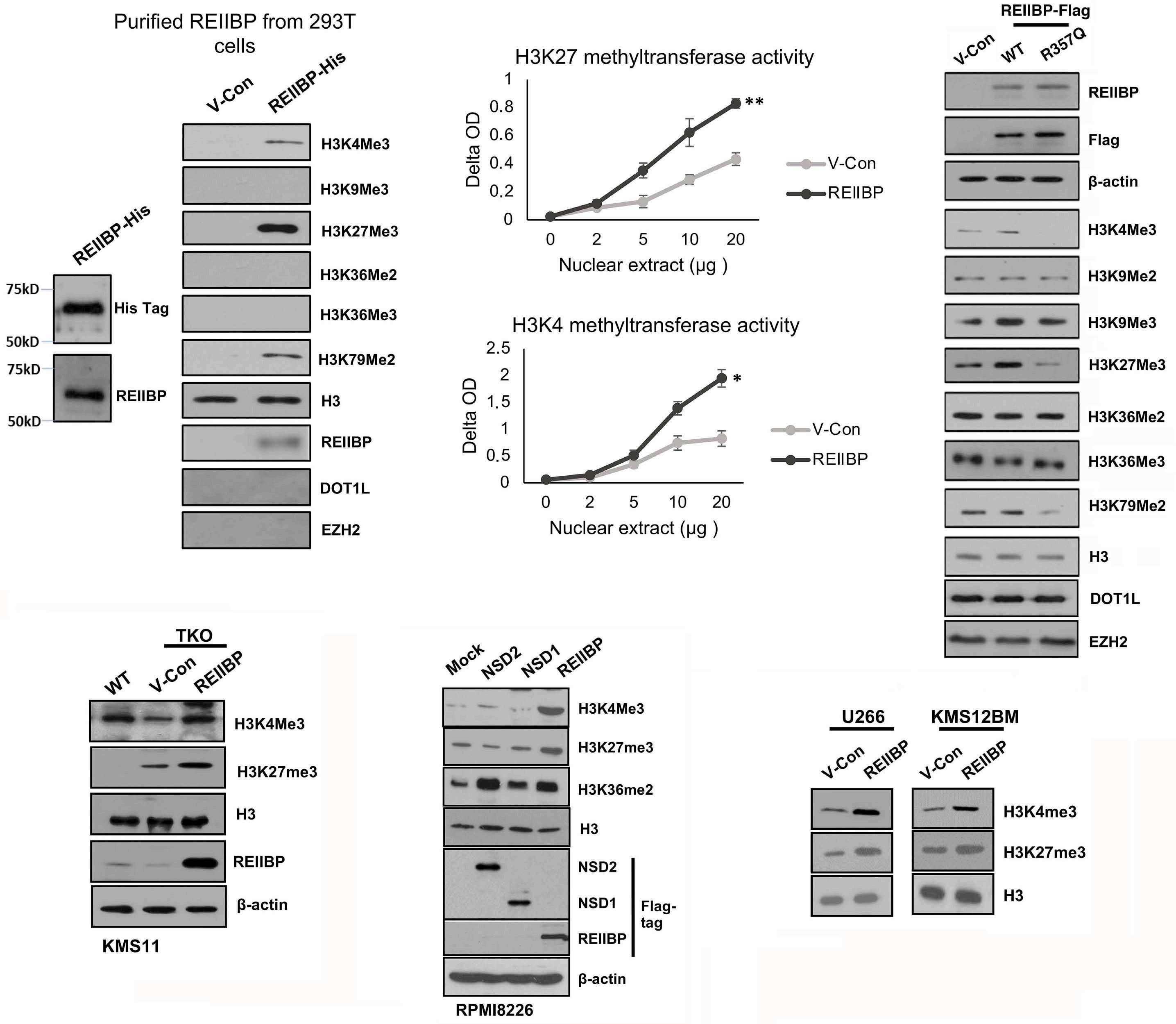
Figure 3. In vitro and in vivo histone methyltransferase assay confirm that REIIBP has a direct effect on histone methylation. (A) Purified REIIBP methyltransferase from 293T cells was added to SAM and H3 substrate in PKMT buffer. Western blotting (WB) was performed with the indicated antibodies. (B) H3K27 methyltransferase activity assay (top) was performed using increasing amount of nuclear extract from RPMI8226-vector control (VCon) or RPMI8226-REIIBP cells and readings were taken at 450 nm. N=3, biological replicates, **P<0.01, Student’s t test. H3K4 methyltransferase activity assay (bottom) was performed using increasing amount of nuclear extract from RPMI8226-VCon or RPMI8226-REIIBP cells and readings were taken at 450 nm. N=3, biological replicates, *P<0.05, Student’s t test. (C) VCon wild-type (WT) REIIBP and R357Q mutant REIIBP were transiently transfected into RPMI8226 cells and 50 µg of protein lysate were used for WB with the indicated antibodies. H3 and β-actin are the loading control. (D) KMS11 and KMS11-TKO (translocation knockout) cells transiently transfected with VCon or WT REIIBP and were used for WB with the indicated antibodies. H3 and β-actin are the loading control. (E) RPMI8226 cells were transfected with NSD1, NSD2 and REIIBP expression vectors and WB with the indicated antibodies. H3 and β-actin are the loading control. Flag-tag antibodies demonstrated equal expression of the isoforms. (F) U266 and KMS12BM transiently transfected with REIIBP expression vector from Online Supplementary Figure S1B were used to probe for histone marks. H3 is the loading control. All immunoblots were performed with 2 biological repeats and a representative experiment was shown.
saw an efficient abolishment of H3K4me3 and H3K27me3 histone marks (Figure 3C). Next, we reconstituted the expression of REIIBP in KMS11 TKO cells. It is notable that TKO cells had a higher expression of H3K27me3 than wild-type (WT), which is supported by previous findings that NSD2 induced a downregulation of H3K27me3.13 Despite this, there was a consistent increase in both H3K27me3 and H3K4me3 modifications by REIIBP (Figure 3D). As a direct comparison of the catalytic activities among NSD2, NSD1 and REIIBP, we overexpressed these proteins in parallel. We observed the most significant increase in H3K36me2 by NSD2, as expected.11 Conversely, H3K27me3 and H3K4me3 were increased by REIIBP, but not NSD1 or NSD2 (Figure 3E). These modifications by REIIBP were reproducible in other myeloma cell lines (Figure 3F; Online Supplementary
Figure S2D). Collectively, we demonstrated a SET-dependent activity of REIIBP on H3K4 and H3K27 trimethylation but not NSD2-associated H3K36me2.
EZH2 is an upstream regulator of REIIBP and is mediated through microRNA
EZH2 is a key H3K27me3 enzyme, and previous reports linked EZH2 upstream of NSD2.13,26 In order to define the relationship between REIIBP and EZH2, we first checked the expression of EZH2 in our isogenic cells. Similar levels of EZH2 suggested that the upregulation of H3K27me3 is unlikely attributed to a modulation of EZH2 levels (Online Supplementary Figure S3A). Next, we overexpressed REIIBP in a K562-EZH2 null cell line (EZH2∆/∆), which led to a restoration of H3K27me3 levels, albeit with dose-limiting effect
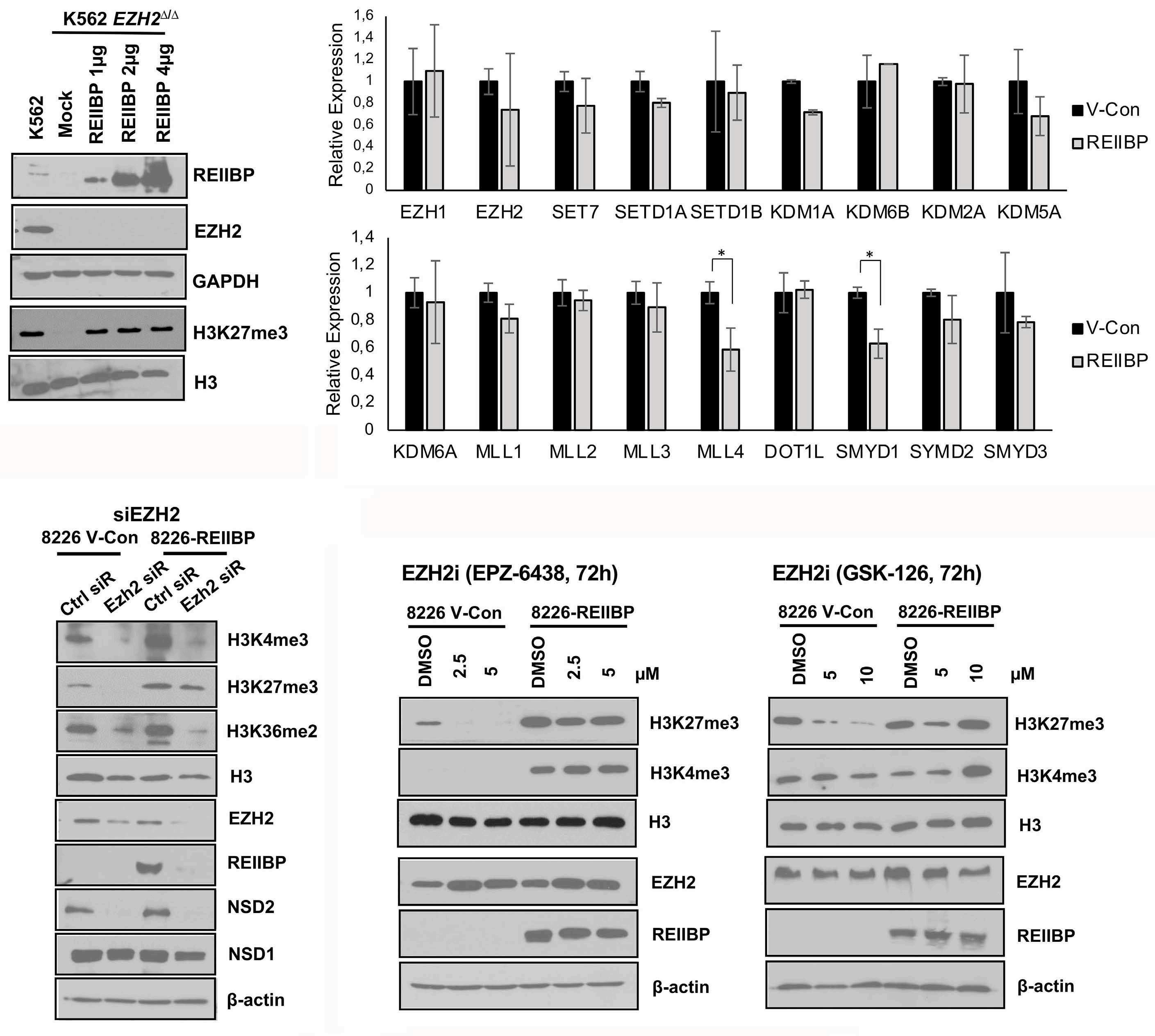
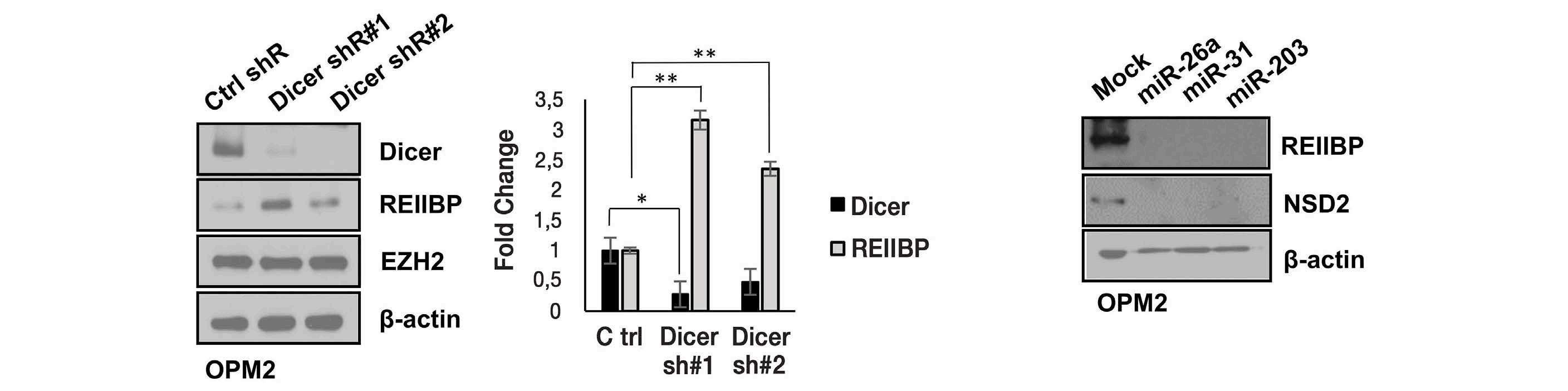
Figure 4. REIIBP regulates H3K27me3 independent of EZH2. (A) K562 EZH2∆/∆ cells were transiently transfected with increasing amounts of REIIBP plasmid and protein were extracted at 48 hours to determine the levels of H3K27me3. GAPDH and H3 are the loading controls. N=2, independent replicates. (B) Gene expression of a panel of histone methyltransferases and demethylases were determined in RPMI8226-vector control (VCon) and RPMI8226-REIIBP cells. GAPDH was used for normalization. N=3, biologically independent replicates with technical duplicates. Asterisks represent significant differences (*P<0.05) determined by Student’s t test. (C) RPMI8226-VCon and RPMI8226-REIIBP cells were transiently transfected with control or EZH2 small interfering RNA (siRNA) (100 nM) and protein harvested at 48 hours for western blotting (WB) with the respective antibodies. H3 and β-actin are the loading control (N=2, biological replicates). (D) RPMI8226-VCon and RPMI8226-REIIBP cells were treated with 2 EZH2 inhibitors, EPZ-6438 (left) and GSK-126 (right), for 72 hours before protein was harvested for WB (N=2, biological replicates, representative WB). H3 and β-actin are the loading control. (E) 1x106 OPM2 cells were transiently transfected with 2 µg of scrambled short hairpin RNA (shRNA), Dicer shRNA #1 or #2 for 48 hours and protein lysate was harvested. WB was performed with the indicated antibodies (N=2, biological replicates, representative WB). β-actin is the loading control. Transfection for 24 hours was harvested for mRNA and shown as fold change relative to scrambled shRNA control. N=3, biologically independent replicates with technical duplicates. Asterisks represent significant differences (*P<0.05; **P<0.01) determined by Student’s t test and Benjamini-Hochberg correction method was applied for multiple comparisons. (F) OPM2 cells were transiently transfected with 75 nM of miRNA-26, miRNA-31 or miRNA-203 for 48 hours and the protein levels of REIIBP and NSD2 were determined. N=2, independent replicates.
in the complete absence of EZH2 (Figure 4A). We further checked a panel of other histone methyltransferases and demethylases. Most were unchanged except for downregulation, and not upregulation, of H3K4 methyltransferases MLL4 and SMYD1 (Figure 4B). Overall, these data indicated that H3K27 and H3K4 trimethylation mediated by REIIBP were independent of other enzymes. Next, we inhibited EZH2 via two different mechanisms, siRNA-mediated abrogation of EZH2 levels, and pharmacological inhibitors (EPZ-6438 and GSK-126) known to affect EZH2-mediated H3K27me3 but leave EZH2 levels unchanged.27 Knockdown of EZH2 showed an almost complete abrogation of NSD2 and H3K36me2, thus acting as a positive control in our system. Notably, REIIBP was also abrogated but not NSD1, and REIIBP-associated H3K4me3 and H3K27me3 were reduced (Figure 4C). This indicated that EZH2 not only regulated NSD2, but REIIBP as well, and such regulation occurred at both the mRNA and protein levels (Online Supplementary Figure S3B). Treatment with EZH2 inhibitors (EZH2i) provided alternative insights as it reduced H3K27me3 levels in WT cells but not in REIIBP-overexpressing cells (Figure 4D). The residual H3K27me3 confirmed that REIIBP could modulate H3K27me3 levels that was not targetable by EZH2 inhibition. EZH2i did not affect REIIBP levels and correspondingly, H3K4me3. The observation that siEZH2 but not EZH2i affected REIIBP would suggest that EZH2 protein rather than its enzymatic activity is required for REIIBP regulation. One reported mechanism through which EZH2 regulated
NSD2 is by microRNA (miRNA).26 Given the identical 3’UTR of NSD2 and REIIBP, this prompted us to examine whether REIIBP is likewise regulated by miRNA and identify the specific miRNA that might be targeting REIIBP. For this purpose, we performed the subsequent miRNA experiments in OPM2 which harbored the 3’UTR region on endogenous REIIBP. Depletion of Dicer using two independent shRNA rescued the mRNA and protein levels of REIIBP (Figure 4E), and was reproducible in RPMI8226 (Online Supplementary Figure S3C). There were other cell lines whereby Dicer knockdown led to a downregulation of EZH2, resulting in the depletion of REIIBP (Online Supplementary Figure S3D), reinforcing our hypothesis that EZH2 was upstream of REIIBP. Lastly, we overexpressed the three EZH2 miRNA that were previously reported to target the 3’UTR of NSD2 gene,26 namely miR-26a, miR-31, and miR-203. These miRNA resulted in the abrogation of both NSD2 and REIIBP levels (Figure 4F). Altogether, we demonstrated a connection of REIIBP to EZH2 histone methyltransferases network, although REIIBP catalytic activity on H3K27me3 was independent of EZH2.
Transcriptomics identified a novel role of REIIBP in pro-inflammatory processes through activation of TLR7BTK-IL6 pathway
Previously, we reported that NSD2 is involved in the regulation of cell growth, adhesion and Wnt signaling pathways.28 In order to gain insights into the transcriptional program induced by REIIBP, we performed gene expression profiling (Figure 5A; Online Supplementary Table S1). Upon REIIBP
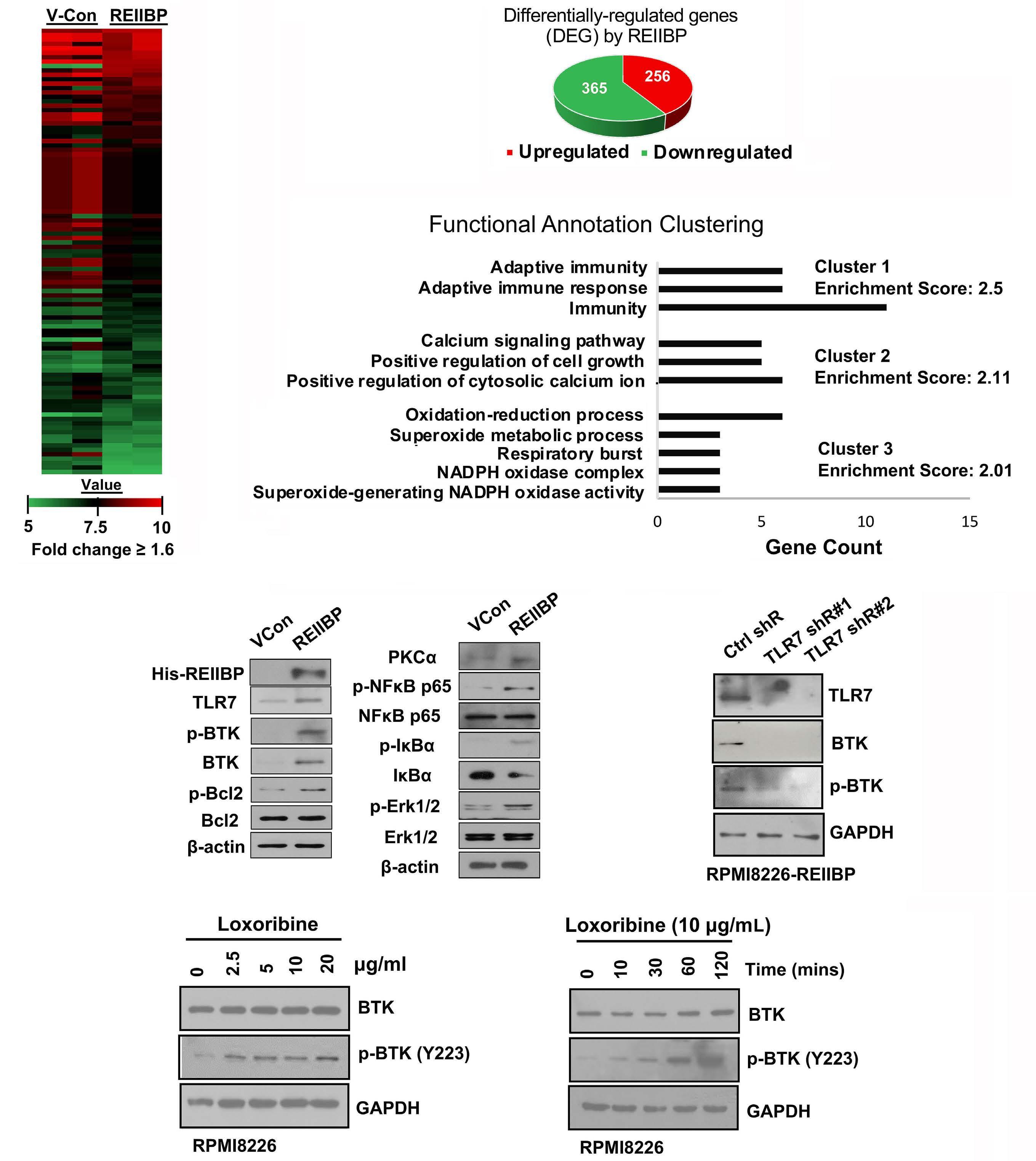
Figure 5. Global transcriptional changes in REIIBP-overexpressing cells. (A) Heat map of genes with log fold-change cutoff of 1.6 were shown. See Online Supplementary Table S1 for microarray data. N=2, independent replicates. (B) Up- and downregulated genes were plotted as a pie chart. (C) Functional annotation clustering was performed using DAVID Bioinformatics Resources49 with the differentially expressed genes (DEG). The processes with the top 3 enrichment scores are presented. The x-axis represents the number of genes, while the y-axis represents the ontology categories. (D) 50 µg of protein lysate from RPMI8226-vector control (VCon) and RPMI8226-REIIBP cells were used to probe with the indicated antibodies in TLR7-BTK-NF-kB activation pathway. β-actin is the loading control (N=2, biological repeats, representative western blot [WB] shown). (E) RPMI8226-REIIBP cells were transiently transfected with 2 TLR7-targeting short hairpin RNA (shRNA) and protein harvested 48 hours post-transfection. WB was performed with the indicated antibodies (N=2, biological repeats, representative WB shown). GAPDH is the loading control. (F) RPMI8226 cells were treated with increasing concentration of loxoribine for 60 minutes (left) or increasing duration at fixed concentration of 10 µg/mL (right). Phospho-BTK and total BTK levels were determined using WB and GAPDH is the loading control (N=2, biological repeats, representative WB shown).
overexpression, there were 365 downregulated and 256 upregulated genes (Figure 5B). The differentially expressed genes (DEG) were subjected to gene ontology analysis to reveal an enrichment in processes such as response to stimuli, cell growth and metabolism (Figure 5C), and the top five upregulated genes (CYBB, TLR7, FAIM3, BTK, PDIA2) were independently validated (Online Supplementary Figure S4A). Amongst these, Toll-like receptor 7 (TLR7) seems to be of particular relevance given its role in cytokine production to promote the survival and drug resistance of myeloma cells.29 Interestingly, we also observed the concerted upregulation of BTK, a putative downstream target of TLR,30 together with activation of its downstream effector protein phospho-NFkB31-33 (Figure 5D; Online Supplementary Figure S4B). shRNA-mediated gene silencing of TLR7 reduced both BTK and phospho-BTK (Figure 5E), while stimulation with a TLR7 agonist, loxoribine, phosphorylated BTK in myeloma cells in a dose- and time-dependent manner (Figure 5F). These suggested that in the absence of BCR in myeloma cells, TLR7 can be an alternative upstream receptor for a fully activated, phosphorylated BTK. Next, we measured the levels of pro-inflammatory cytokines and found a significant dysregulation of cytokine gene expression, particularly interleukin (IL)-6 (Figure 6A). This was coupled with increased secretion of IL-6 into the supernatant by the myeloma cells (Figure 6B). In order to assess whether REIIBP cells were dependent on BTK activation, we treated HMCL with ibrutinib.34-37 t(4;14)-positive OPM2, H929 and KMS18 with high levels of REIIBP were significantly inhibited as compared to KMS11 and KMS34 harboring lower REIIBP, while t(4;14)-negative U266 was most resistant (Figure 6C). This was confirmed in the RPMI8226 isogenic system where REIIBP expression segregated ibrutinib response (Online Supplementary Figure S5A-C). Moreover, the inhibitory effects of ibrutinib were potentiated in combination with bortezomib (Online Supplementary Figure S6A-C). In vivo, NSG mice engrafted with REIIBP cells developed tumors more efficiently than vectro control (VCon) and randomized treatment with ibrutinib-bortezomib combination demonstrated superior efficacy to single drug or DMSO control groups (Figure 6D, E). Our observations corroborated with Cancer Cell Line Encyclopedia (CCLE) and DepMap resources showing BTK dependency in MM (Online Supplementary Figure 7A, B). In patient datasets, TLR7 and BTK were associated with poor overall survival (Online Supplementary Figure 7C, D), and demonstrated correlation between their expression profiles (Online Supplementary Figure S7E).
Altered occupancy of H3K4me3 and H3K27me3 revealed the complexity in the bidirectional regulation of genes by REIIBP
In order to assemble our findings on how REIIBP histone modifications contributed to its transcriptional profile, we next performed genome-wide mapping of H3K4me3 and H3K27me3 using ChIP sequencing. Consistent with their
increased levels, the distribution of H3K4me3 was significantly enriched near the TSS upon REIIBP overexpression, while H3K27me3 signal was relatively flat across 5 kb region before and after TSS, with some focal enrichment at TSS (Figure 7A). Differential H3K4me3 and H3K27me3 peaks were subjected to gene ontology (Online Supplementary Figure S8A) and KEGG analysis to reveal enrichment in pathways such as PI3K-Akt signaling and metabolism (Online Supplementary Figure S8B), and their occupancy were analyzed with DREME motif discovery (Online Supplementary Figure S8C). Comparison of the genomic distributions indicated an expansion of H3K4me3 and H3K27me3 peaks into intergenic regions with reduced weightage on promoters (Online Supplementary Figure S8D). Next, we performed a t test of occupancy values across the whole genome to identify a list of genes whose distribution for H3K4me3 and H3K27me3 were significantly altered by REIIBP (Online Supplementary Table S2). There were 328 H3K4me3 and 1,256 H3K27me3 altered genes respectively, and 45 overlapping gene loci that were doubly marked (Online Supplementary Figure S8E). In consideration that H3K4me3 functions as a permissive histone mark that opposes H3K27me3, we sought to examine the impact of how their loss or gain affected gene expression. We found a clear association between increased H3K4me3 occupancy with upregulated transcription, but H3K27me3 peaks did not necessarily function as silencers to repress gene expression (Figure 7B). A different trend was observed with the doubly marked genes, where the effect of H3K4me3 on promoting expression is of a lesser extent. In this group, high levels of H3K27me3 could block the expression of genes despite H3K4me3 occupancy, indicating that lowering H3K27me3 is a prerequisite for expression. Accordingly, the group with H3K4me3high/H3K27me3low demonstrated the highest transcriptional activity. Overall, our data indicated a dynamic and complex control through the balance of H3K4me3 and H3K27me3 histone modifications on fine-tuning transcription.
REIIBP upregulates TLR7-BTK pathway through eIF3E We performed integrative analysis of transcriptomics and ChIP-sequencing datasets, and identified eIF3E whose elevated expression can be attributed to H3K4me3 occupancy at its transcriptional start site. To ascertain this, we designed three sgRNA and used CRISPR/Cas9 editing for precise deletion of the H3K4me3 peak, and this led to the reduction in eIF3E levels (Figure 8A). Upregulation of eIF3E by REIIBP is conserved in other HMCL (Online Supplementary Figure S9A) and associated with disease progression and adverse prognosis (Online Supplementary Figure S9B). Next, we assessed whether and if so, how eIF3E contributed to REIIBP and its pro-inflammatory phenotype. Depletion of eIF3E significantly impaired growth of REIIBP cells in viability and clonogenic assays (Online Supplementary Figure S9C-E), and suppressed the expression of TLR7 and BTK (Online Supplementary Figure S9F). Given
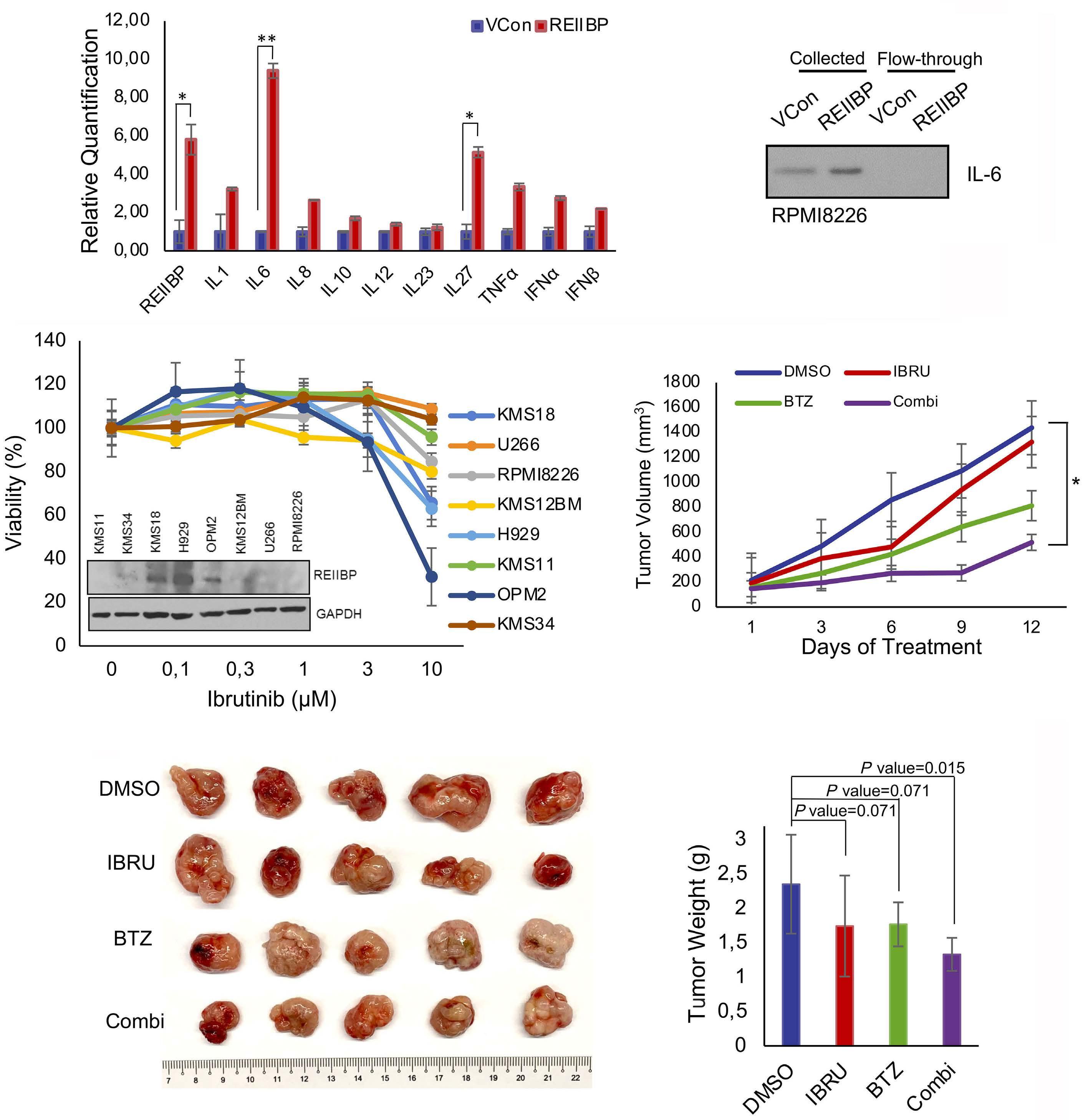
Figure 6. Ibrutinib potentiates the inhibitory effects of bortezomib. (A) The mRNA expression of multiple cytokines and chemokines were determined in RPMI8226-vector control (VCon) and RPMI8226-REIIBP cells by quantitative real time polymerase chain reaction (qRT-PCR). GAPDH was used for normalization. N=3, biologically independent replicates with technical duplicates. Asterisks represent significant differences (*P<0.05; **P<0.01) determined by Student’s t test. (B) Interleukin (IL)-6 in the cell culture supernatant was collected from filter unit and flow-through, and measured by western blotting (WB) in RPMI8226-VCon and RPMI8226-REIIBP cells (N=2, biological repeats, representative WB shown). (C) Panel of myeloma cell lines were treated with increasing concentrations of ibrutinib (IBRU) for 48 hours and viability was plotted (3 biological repeats with technical octuplicates) relative to control (100%). The expression of REIIBP from Figure 1A is included as reference. (D) Xenografts of RPMI8226-REIIBP (5 million cells) were allowed to grow for 3 weeks until desired size, and treatment with 1% dimethyl sulfoxide (DMSO), 20 mg/kg ibrutinib, 0.4 mg/kg bortezomib (BTZ) or combination proceeded for 12 days with measurement of tumors with calipers. N=5, biological repeats. Asterisks represent significant differences (*P<0.05; Student’s t test) between DMSO control and Combi group. (E) Tumors were harvested and weighed in grams. The P values were indicated in the figure by performing Student’s t test, comparing each treatment with DMSO control. Harvested tumors were photographed.
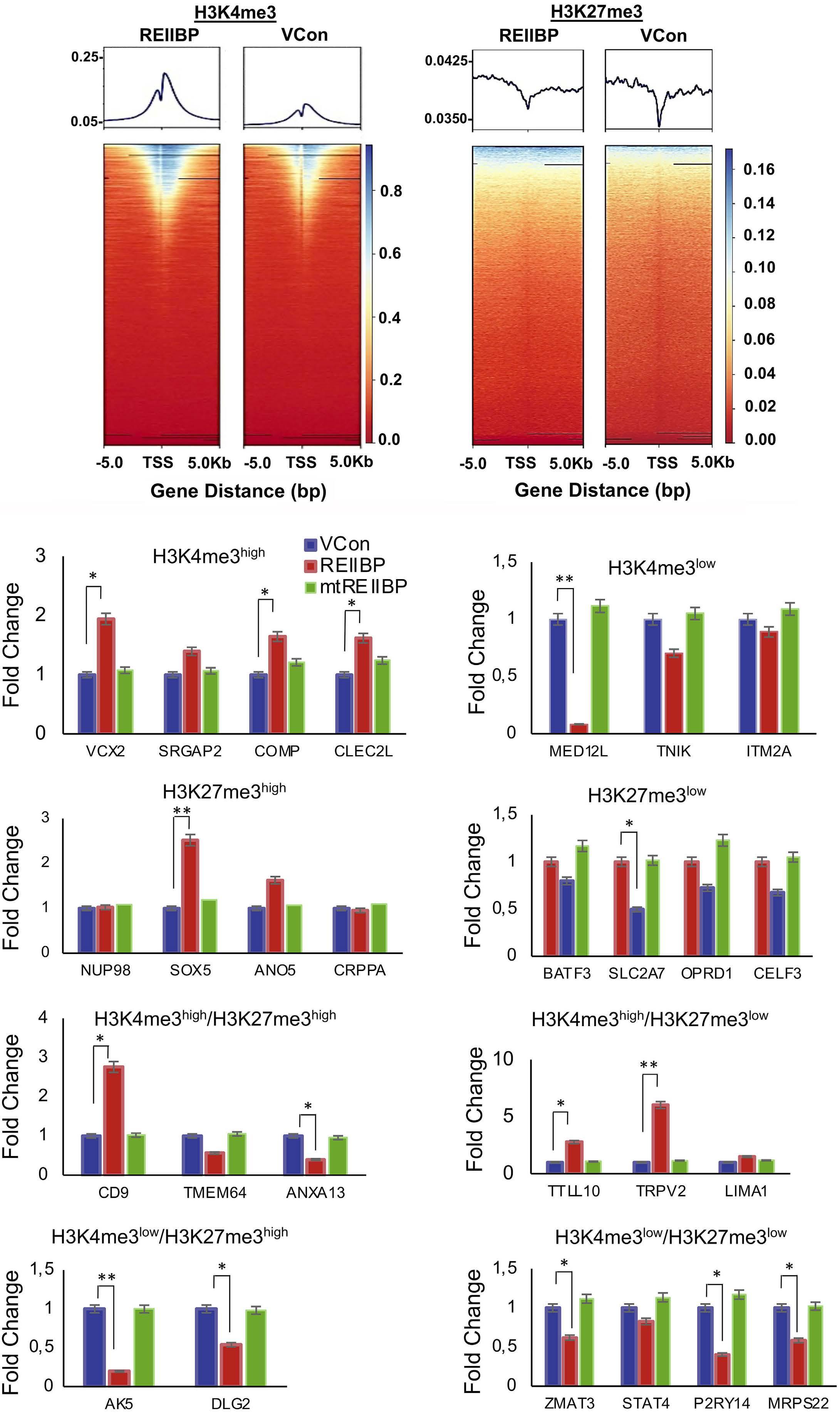
Figure 7. Chromatin immunoprecipitation sequencing of H3K4me3 and H3K27me3 revealed dynamic gene regulatory mechanisms by REIIBP. (A) Heatmap of averaged H3K4me3 and H3K27me3 signals enrichment on genomic loci at TSS from 3 biological replicates of chromatin immunoprecipitation (ChIP)-sequencing analysis was shown and normalized to the corresponding inputs. Raw ChIP-sequencing data is publicly available as GSE198026. (B) Validation of the top ranked H3K4me3, H3K27me3 and doubly-marked genes using quantitative real time polymerase chain reaction (qRT-PCR) in vector control (VCon), wildtype REIIBP and SET mutant REIIBP (R357Q). Mutant REIIBP had little changes on the expression of REIIBP differentially regulated genes when compared to control. Experiments were performed for 3 biological repeats with technical duplicates. Asterisks represent significant differences (*P<0.05; **P<0.01, Student’s t test).
that eIF3E increases TLR7 and BTK (Online Supplementary Figure S9G), we determined whether this was attributed to eIF3E canonical function as an initiator of protein synthesis. Using the analog O-propargyl-puromycin (OPP) that incorporates into newly synthesised proteins and coupled with florescence microscopy, there were no obvious differences between control and REIIBP cells in the number or rate of protein synthesis (Figure 8B). This was confirmed with the SUnSET protocol for immunoblot analysis of translation using puromycin-labeled proteins. Again, we did not detect significant differences in bulk protein translation with REIIBP (Figure 8B). More recently, eIF3 complex members have been implicated as RNA-binding protein (RBP), leading us to postulate that eIF3E might bind to the 3’UTR mRNA of oncogenic factors, known to increase mRNA stability and translation. In an RNA immunoprecipitation assay (RIP), we showed a direct interaction between eIF3E and TLR7 mRNA, and to a lesser extent, BTK (Figure 8C). Polysome profiling indicated that TLR7 was efficiently translated by associating
with polysomes found in the higher fractions in REIIBP cells as compared to control (Figure 8D; Online Supplementary Figure S10). Altogether, we revealed the mechanism of how eIF3E was epigenetically regulated by REIIBP, and its subsequent role in the translational status of TLR7.
Alternative splicing events and promoter transcription start sites are major contributors to isoform diversity, giving rise to functionally different protein products from the same gene locus, a phenomenon common for oncogenes. NSD2 is amongst the genes that has been demonstrated to produce multiple transcripts. However, the mechanism of how each transcript contributed to myeloma phenotype remains to be fully elucidated, since much of the research were focused on full-length NSD2 as the predominant isoform. Little is known about the oncogenic function and therapeutic po-
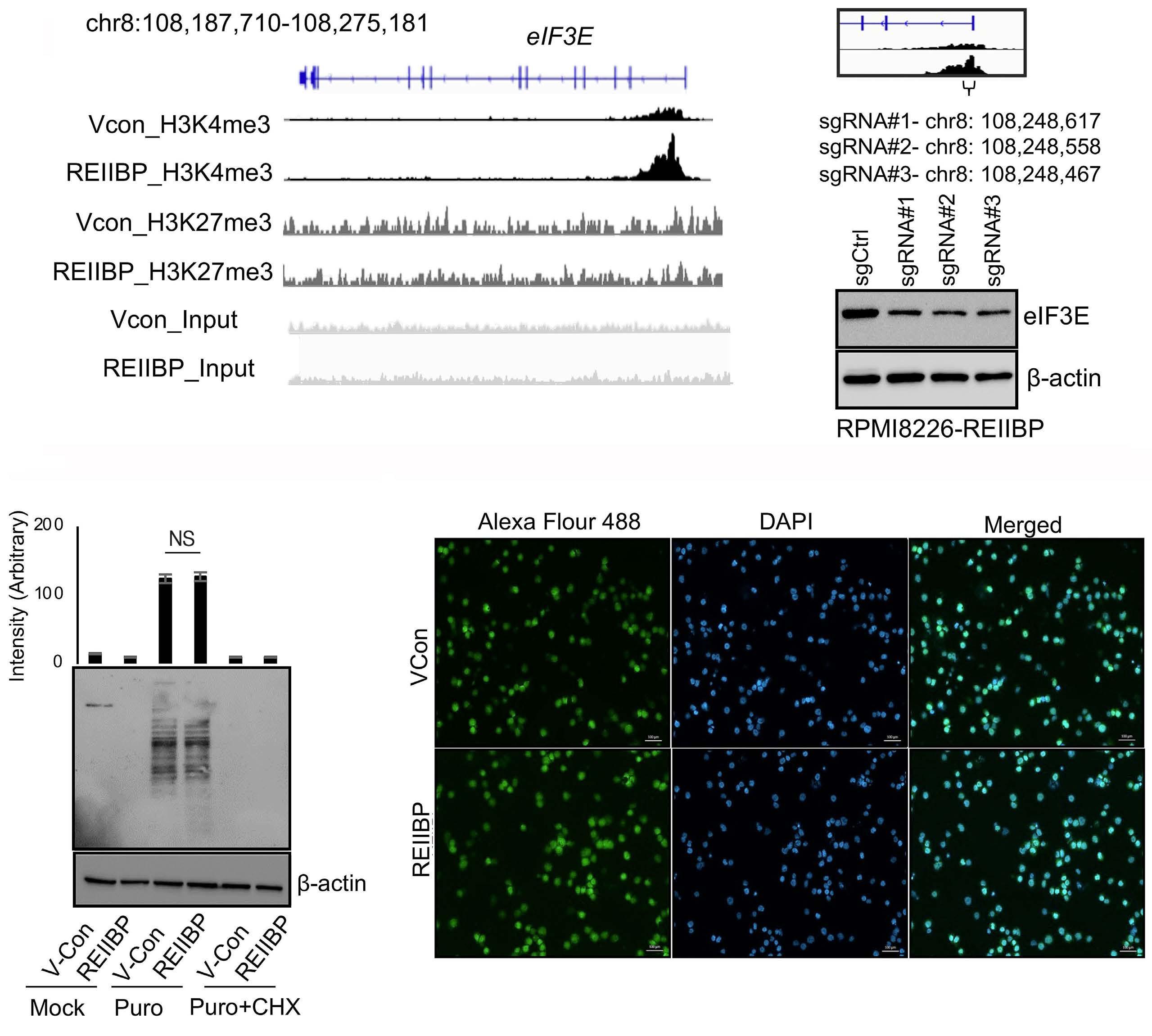
Continued on following page.
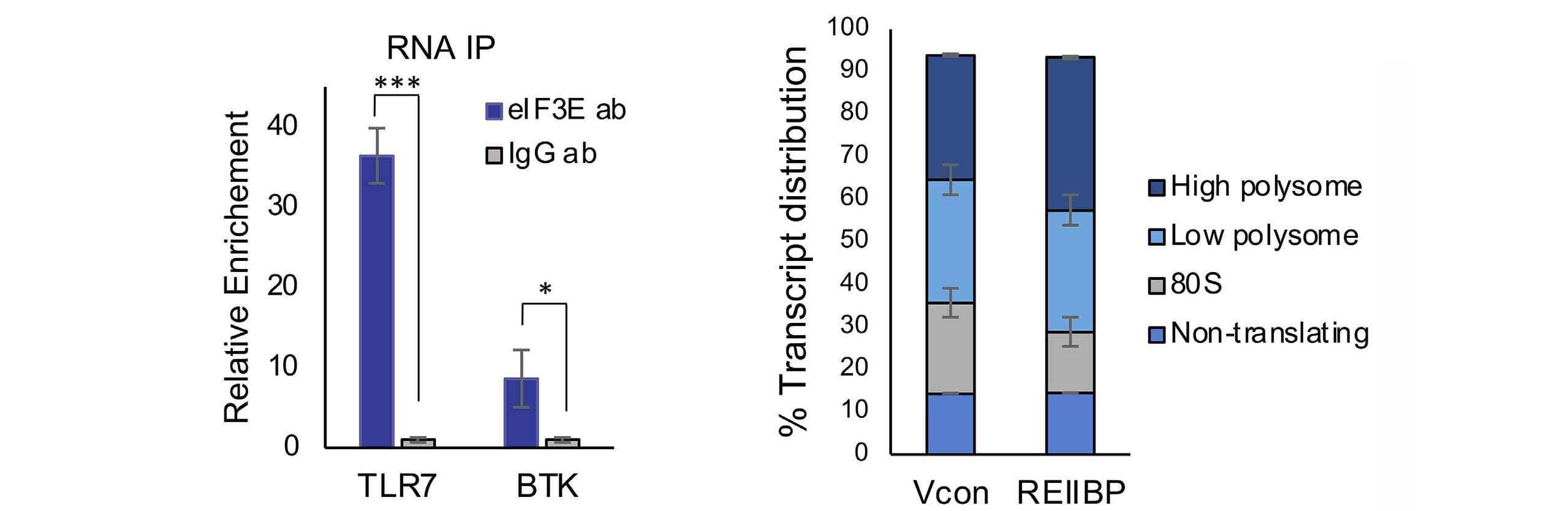
Figure 8. RNA-binding protein eIF3E was dysregulated by histone methylation and participates in REIIBP oncognesis. (A) Integrative Genomics Viewer (IGV) browser of representative gene tracks from biological triplicates of eIF3E were shown for H3K4me3 and H3K27me3 histone marks in RPMI8226-vector control (VCon) and RPMI8226-REIIBP cells. CRISPR/Cas9-mediated knockdown of H3K4me3 peak using 3 different single guide RNA (sgRNA) in RPMI8226-REIIBP cells with the positions of the sgRNA indicated. β-actin is the loading control. N=2, independent repeats, representative western blots (WB) shown. (B) Left panel: the global protein synthesis rate was determined by puromycin labeling50 coupled with immunoblot using antibody against puromycin (12D10). Lanes 1 and 2 are mock treated, lanes 3 and 4 are labeled with 10 µg/mL of puromycin for 10 minutes and lanes 5 and 6 are labeled with puromycin and treated with 100 µM cycloheximide (CHX) for 10 minutes. Right panel: O-propargyl-puromycin (OPP)-labeling coupled with immunofluorescence in RPMI8226-VCon and RPMI8226-REIIBP. Newly synthesized proteins were stained with Alexa Flour 488 (green) and nucleus stained with DAPI (blue). Image is representative field. Scale bar, 100 µm. (C) RNA immunoprecipitation (RNA IP) was performed with eIF3E antibodies or immunoglobuolin (Ig)G control, and binding with TLR7 or BTK mRNA was determined using quantitative real time polymerase chain reaction. Asterisks represent significant differences (*P<0.05; ***P<0.001, Student’s t test). (D) The % transcript distribution of TLR7 mRNA was determined in the non-translating, 80S, low polysome and high polysome after polysome profiling51 comparing between RPMI8226-VCon and RPMI8226-REIIBP; 56.2% of TLR7 mRNA were associated with polysomes as compared to 64.4% in REIIBP, indicating a higher translation efficiency in REIIBP cells.
tential of targeting REIIBP, due to the inability to perform REIIBP knockdown studies owing to a significant amount of overlapping gene sequences with NSD2, and the absence of a specific REIIBP antibody. In order to overcome these technical difficulties, we created a stable isogenic REIIBP cell line using RPMI8226, which expresses negligible levels of NSD2 isoforms. We elaborated on the molecular mechanism regulating REIIBP expression, which was not only driven by chromosomal rearrangement, but also EZH2-mediated miRNA gene silencing. Our observations corroborated a previous study that implicated EZH2 as an upstream regulator of NSD2 via its 3’UTR,26 which is identical between REIIBP and NSD2. This supports the notion that most histone modifying enzymes do not work singly, but in a concerted effort to remodel the chromatin and alter transcription.38,39 Here, we also uncovered a cooperative network consisting of several epigenetic regulators in t(4;14) MM.
One major finding is that despite homology between REIIBP and NSD2, the most prominent histone methylation activity of REIIBP is distinct from NSD2. REIIBP preferentially modifies H3K27me3 and H3K4me3, with minimal effect on H3K36me2, the primary modification by NSD2. These differences in histone substrates might undermine the efficacy of targeting SET domain specificities for clinical application. Given that the SET domain of REIIBP and NSD2 are identical, a reasonable interpretation could be differences in its subcellular localization or protein interacting partners, both of which can be caused by the absence of
N-terminus sequences in REIIBP. Our results also further emphasize the promiscuity of the SET domain, which is supported by other literature based on the observed catalytic activity of NSD2.12-15,40-41 Traditionally, EZH2 is the only known histone methyltransferase that catalyzes H3K27me3. Hence, we considered the possibility that REIIBP might indirectly regulate H3K27me3 levels through EZH2 or H3K27 demethylase JMJD3; but neither were dysregulated by REIIBP. Instead, a series of in vitro and in vivo experiments affirms that REIIBP have innate H3K27 methylation capabilities. The accumulation of H3K27me3 by REIIBP, which is associated with a closed chromatin state and transcriptional repression, was generally reflected in our microarray data where we saw more downregulated genes. H3K4me3 opposes the role of H3K27me3 by predominantly marking active promoters. In a bivalent domain, the co-occurrence of these two histone marks has been reported in embryonic stem cells as a mechanism to poise developmental genes for timely activation.42 However, we did not observe true bivalent promoters since H3K4me3 was found near the TSS, while H3K27me3 was broadly distributed throughout the intergenic region. Our genomic analyses further suggested that the presence of H3K27me3 at gene loci is generally correlated with repression of gene expression, even prevailing over genes containing a high level of H3K4me3 for transcriptional initiation by RNA polymerase II. Our data also unexpectedly revealed an epigenetic regulation of RBP in MM, which are more commonly known to be dysregulated
through mutations or gene amplifications.43 As RBP are involved in numerous RNA processing steps, their abnormalities have significant effects on post-transcriptional regulation and transcriptomes.44 Owing to the differences in histone preferences, it was within our expectations that we found little overlap of the differentially regulated genes between REIIBP and NSD2. Here, we uncovered a novel mechanism of BTK activation that bypasses BCR by REIIBP. BTK is a critical component of BCR signaling and a potent B-cell survival factor, but BCR is lost when B cell matures into plasma cells.45 Intriguingly, BTK is re-expressed in MM, and we found that upregulation of TLR7 by REIIBP could represent a novel, important mechanism of BTK reactivation. Moreover, TLR7 was not expressed in the plasma cells from healthy donors,29 making it possible to target cancer cells while sparing normal cells. We further reasoned that TLR- or BCR-mediated BTK activity could elicit differential downstream effects. Probing for all the effectors of BTK signaling identified a NF-kB-driven production and secretion of a key pro-inflammatory cytokine IL-6. Myeloma cells are highly dependent on the bone marrow tumor microenvironment and especially IL-6 for growth and survival, and the elevated expression of IL-6 is deemed a contributing factor for drug resistance.46-48 This was reflected in patients who expressed high REIIBP levels and exhibited poorer response towards bortezomib, even when t(4;14) translocation is generally perceived to be responders towards bortezomib treatment. It also rendered REIIBP cells addiction to BTK signaling and targetability using Ibrutinib, a Food and Drug Administration-approved drug, and can be considered in combinational regimes to improve bortezomib response. Altogether, this study demonstrated that REIIBP is a functional histone methyltransferase in t(4;14) myeloma, and integrative analysis unraveled a eIF3E/TLR7/BTK axis that constitutes a targetable transcriptome driven by REIIBP epigenetic reprogramming in myeloma cells.
1. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 201;364(11):1046-1060.
2. Lu S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark Res. 2013;1(1):13.
3. Quach H, Ritchie D, Stewart AK, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22-32.
4 Raedler LA. Darzalex (daratumumab): first anti-CD38 monoclonal antibody approved for patients with relapsed multiple myeloma. Am Health Drug Benefits. 2016;9(Spec Feature):70-73.
5. Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210-2221.
6. Chng WJ, Glebov O, Bergsagel PL, Kuehl WM. Genetic events in the pathogenesis of multiple myeloma. Best Pract Res Clin Haematol. 2007;20(4):571-596.
No conflicts of interest to disclose.
PSYC and JYC designed the study and performed the experiments, analyzed and interpreted data. PSYC prepared the manuscript. PSYC and THC did the bioinformatics analyses. THC and RB did the ChIP-seq analysis. LSLJ, ALCY, MIBA and ZW did the in vitro work. SHMT and KB did the mouse work. NS and LAV did polysome profiling. WJC initialized the study, provided study directions, proofread and finalized the manuscript.
We thank Dr Frank Eisenhaber for insightful discussions on HMT assays. We thank Dr Xie Zhigang for his involvement in the conceptualization of the project. We thank Dr Zhou Jianbiao for advice on the in vivo mouse work.
WJC is supported by NMRC Singapore Translational Research (STaR) Investigatorship. This research was partly supported by the National Research Foundation Singapore and the Singapore Ministry of Education under the Research Centers of Excellence initiative as well as the RNA Biology Center at the Cancer Science Institute of Singapore, NUS, as part of funding under the Singapore Ministry of Education’s Tier 3 grants, grant no. MOE2014-T3-1-006.
Data-sharing
Microarray data is provided as Online Supplementary Table S1. A list of genes with differential occupancy of H3K27me3 and H3K4me3 between RPMI8226-VCon and RPMI8226-REIIBP is provided as Online Supplementary Table S2. ChIP-sequencing data from this study were submitted to NCBI GEO (http://www. ncbi.nlm.nih.gov/geo/) under accession number GSE198026. The data analyzed in this study were obtained from MMRF CoMMpass Study at https://research.themmrf.org/.
7. Chng W, Dispenzieri A, Chim C, et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia. 2014;28(2):269-277.
8. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7(8):585-598.
9 Keats JJ, Maxwell CA, Taylor BJ, et al. Overexpression of transcripts originating from the NSD locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients. Blood. 2005;105(10):4060-4069.
10 Brito JL, Walker B, Jenner M, et al. NSD deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica. 2009;94(1):78-86.
11. Kuo AJ, Cheung P, Chen K, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44(4):609-620.
12. Pei H, Zhang L, Luo K, et al. NSD regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470(7332):124-128.
13. Popovic R, Martinez-Garcia E, Giannopoulou EG, et al. Histone methyltransferase NSD/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014;10(9):e1004566.
14. Lhoumaud P, Badri S, Rodriguez-Hernaez J, et al. NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat Commun. 2019;10(1):4843.
15. Martinez-Garcia E, Popovic R, Min D-J, et al. The NSD histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117(1):211-220.
16. Lauring J, Abukhdeir AM, Konishi H, et al. The multiple myeloma-associated NSD gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood. 2008;111(2):856-864.
17 Brito JLR, Walker B, Jenner M, et al. NSD deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica. 2009;94(1):78-86.
18. Xie Z, Chooi JY, Toh SHM, et al. NSD I acts as an oncoprotein and regulates GLO1 expression in t(4;14) multiple myeloma cells. Leukemia. 2019;33(3):739-748.
19 Garlisi CG, Uss AS, Xiao H, et al. A unique mRNA initiated within a middle intron of WHSC1/NSD encodes a DNA binding protein that suppresses human IL-5 transcription. Am J Respir Cell Mol Biol. 2001;24(1):90-98.
20. Nishioka K, Chuikov S, Sarma K, et al. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16(4):479-489.
21. Chu L, Su MY, Maggi LB Jr, et al. Multiple myeloma-associated chromosomal translocation activates orphan snoRNA ACA11 to suppress oxidative stress. J Clin Invest. 2012;122(8):2793-2806.
22. Mirabella F, Wu P, Wardell CP, et al. NSD is the key molecular target in t(4;14) myeloma. Blood Cancer J. 2013;3(5):e114.
23. Fingerman IM, Du HN, Briggs SD. In vitro histone methyltransferase assay. CSH Protoc. 2008;2008:pdb.prot4939.
24. Morey L, Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem Sci. 2010;35(6):323-332.
25. Feng Q, Wang H, Ng HH, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052-1058.
26. Asangani IA, Ateeq B, Cao Q, et al. Characterization of the EZH2-NSD histone methyltransferase regulatory axis in cancer. Mol Cell. 2013;49(1):80-93.
27. Gulati N, Béguelin W, Giulino-Roth L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk Lymphoma. 2018;59(7):1574-1585.
28. Chong PSY, Chooi JY, Lim JSL, Toh SHM, Tan TZ, Chng WJ. SMARCA2 is a novel interactor of NSD2 and regulates prometastatic PTP4A3 through chromatin remodeling in t(4;14) multiple myeloma. Cancer Res. 2021;81(9):2332-2344.
29. Bohnhorst J, Rasmussen T, Moen S, et al. Toll-like receptors mediate proliferation and survival of multiple myeloma cells. Leukemia. 2006;20(6):1138-1144.
30. Page TH, Urbaniak AM, Espirito Santo AI, et al. Bruton’s tyrosine kinase regulates TLR7/8-induced TNF transcription via nuclear factor-kB recruitment. Biochem Biophys Res Commun.
2018;499(2):260-266.
31. Shinners NP, Carlesso G, Castro I, et al. Bruton’s tyrosine kinase mediates NF-kB activation and B cell survival by B cell-activating factor receptor of the TNF-R family. J Immunol. 2007;179(6):6369.
32. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43-52.
33. Weber AN, Bittner Z, Liu X, Dang T-M, Radsak MP, Brunner C. Bruton’s tyrosine kinase: an emerging key player in innate immunity. Front Immunol. 2017;8:1454.
34. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507-516.
35. Rushworth SA, Bowles KM, Barrera LN, et al. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kB. Cell Signal. 2013;25(1):106-112.
36. Ma J, Gong W, Liu S, et al. Ibrutinib targets microRNA-21 in multiple myeloma cells by inhibiting NF-kB and STAT3. Tumour Biol. 2018;40(1):1010428317731369.
37. Murray MY, Zaitseva L, Auger MJ, et al. Ibrutinib inhibits BTKdriven NF-kB p65 activity to overcome bortezomib-resistance in multiple myeloma. Cell Cycle. 2015;14(14):2367-2375.
38. Fischle W, Wang YM, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15(2):172-183.
39 Lee JS, Smith E, Shilatifard A. The language of histone crosstalk. Cell. 2010;142(5):682-685.
40 Kim JY, Kee HJ, Choe NW, et al. Multiple-myeloma-related WHSC1/NSD isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol Cell Biol. 2008;28(6):2023-2034.
41. Woo Park J, Kim KB, Kim JY, Chae YC, Jeong OS, Seo SB. RE-IIBP methylates H3K79 and induces MEIS1-mediated apoptosis via H2BK120 ubiquitination by RNF20. Sci Rep. 2015;5:12485.
42. Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27(12):1318-1338.
43. Wang ZL, Li B, Luo YX, et al. Comprehensive genomic characterization of RNA-binding proteins across human cancers. Cell Rep. 2018;22(1):286-298.
44 Qin H, Ni H, Liu Y, et al. RNA-binding proteins in tumor progression. J Hematol Oncol. 2020;13(1):90.
45. Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14(4):219-232.
46. Chong PSY, Zhou J, Lim JSL, et al. IL6 promotes a STAT3-PRL3 feedforward loop via SHP2 repression in multiple myeloma. Cancer Res. 2019;79(18):4679-4688.
47. Chong PSY, Chng WJ, de Mel S. STAT3: a promising therapeutic target in multiple myeloma. Cancers (Basel). 2019;11(5):731.
48. Harmer D, Falank C, Reagan MR. Interleukin-6 interweaves the bone marrow microenvironment, bone loss, and multiple myeloma. Front Endocrinol (Lausanne). 2019;9:788.
49. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc. 2009;4(1):44-57.
50. Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6(4):275-277.
51. Rahim AB, Vardy LA. Analysis of mRNA translation rate in mouse embryonic stem cells. Methods Mol Biol. 2016;1341:143-155.
Sunil Lakhwani,1 Laura Rosiñol,2 Noemí Puig,3 Miguel-Angel Pico-Picos,4 Laura Medina-González,4 Joaquín Martínez-López,5 Bruno Paiva,6 María-Teresa Cedena,7 Albert Oriol,8 Rafael Ríos-Tamayo,9 María-Jesús Blanchard,10 Isidro Jarque,11 Joan Bargay,12 José-María Moraleda,13 Estrella CarrilloCruz,14 Anna Sureda,15 Isabel Krsnik,9 Esther González,16 Luis Felipe Casado,17 Josep M. Martí,18 Cristina Encinas,19 Felipe De Arriba,20 Luis Palomera,21 Antonia Sampol,22 Yolanda GonzálezMontes,23 Cristina Motlló,24 Javier De La Cruz,25 Rafael Alonso,7 María-Victoria Mateos,3 Joan Bladé,2 Juan-José Lahuerta,25 Jesús San-Miguel6# and Miguel-Teodoro Hernández1#
1Hospital Universitario de Canarias, Universidad de La Laguna, Tenerife; 2Amyloidosis and Myeloma Unit, Hospital Clínic, Barcelona; 3University Hospital of Salamanca ( IBSAL/CIC), CIBERONC, Salamanca; 4Hospital Universitario de Canarias, Tenerife; 5Hospital Universitario 12 de Octubre, Universidad Complutense, Spanish National Cancer Research Center (CNIO), Madrid; 6Cancer Center Clínica Universidad de Navarra, CIMA, IDISNA, CIBERONC, Pamplona; 7Hospital Universitario 12 de Octubre, Madrid; 8Hospital Germans Trias i Pujol, Institut Català d’Oncologia, Institut Josep Carreras, Badalona; 9Hospital Universitario Puerta de Hierro, Madrid; 10Hospital Universitario Ramón y Cajal, Madrid; 11Hospital Universitari i Politecnic La Fe, Valencia; 12Hospital Son Llàtzer, IdIsBa, Palma de Mallorca; 13Hospital Clínico Universitario Virgen de la Arrixaca, IMIB-Pascual Parrilla, University of Murcia, Murcia; 14Hospital Universitario V. Rocio, Instituto de Biomedicina de Sevilla (IBIS), CSIC, Universidad de Sevilla, Sevilla; 15Clinical Hematology Department, Institut Català d’Oncologia - L’Hospitalet, IDIBELL, Universitat de Barcelona, Barcelona; 16Hospital Universitario de Cabueñes, Gijón; 17Hospital General Universitario de Toledo, Toledo; 18Hospital Universitari Mútua de Terrassa, Terrassa; 19Hospital General Universitario Gregorio Marañon (IiSGM), Madrid; 20Hospital Morales Meseguer, IMIB-Pascual Parrilla, Universidad de Murcia, Murcia; 21Hospital Clínico Universitario “Lozano Blesa”, Zaragoza; 22Hospital Universitario Son Espases, Palma de Mallorca; 23Hospital Universitari Dr. Josep Trueta, ICO Girona, Girona; 24Hospital Althaia, Xarxa Assistencial de Manresa, Manresa and 25Hospital Universitario 12 De Octubre, Instituto de Investigación Sanitaria, Madrid, Spain
#JS-M and M-TH. contributed equally as senior authors.
Abstract
Correspondence: S. Lakhwani sunillakhwani@hotmail.com
Received: August 25, 2023.
Accepted: November 17, 2023. Early view: November 30, 2023.
https://doi.org/10.3324/haematol.2023.284154
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Immunoparesis (IP) in multiple myeloma (MM) patients can be measured by classic assessment of immunoglobulin (Ig) levels or by analysis of the uninvolved heavy/light chain pair of the same immunoglobulin (uHLC) by the Hevylite® assay. In this study we evaluate the prognostic value of recovery from IP measured by classic total Ig and uHLC assessment in newly diagnosed MM transplant-eligible (NDMM-TE) patients with intensive treatment and its association with minimal residual disease (MRD). Patients were enrolled and treated in the PETHEMA/GEM2012MENOS65 trial and continued in the PETHEMA /GEM2014MAIN trial. Total Ig (IgG, IgA and IgM) and uHLC were analyzed in a central laboratory at diagnosis, after consolidation treatment and after the first year of maintenance. MRD was analyzed by next-generation flow cytometry after consolidation (sensitivity level 2x10-6). We found no differences in progression-free survival (PFS) between patients who recovered and patients who didn’t recover from IP after consolidation when examining classic total Ig and uHLC. However, after the first year of maintenance, in contrast to patients with classic IP, patients with recovery from uHLC IP had longer PFS than patients without recovery, with hazard ratio of 0.42 (95% confidence interval [CI]: 0.21-0.81; P=0.008). Multivariate analysis with Cox proportional-hazards regression models confirmed recovery from uHLC IP after the first year of maintenance as an independent prognostic factor for
PFS, with an increase in C-statistic of 0.05 (95% CI: -0.04 to 0.14; P<0.001) when adding uHLC IP recovery. Moreover, we observed that MRD status and uHLC IP recovery affords complementary information for risk stratification. In conclusion, recovery from uHLC IP after 1 year of maintenance is an independent prognostic factor for PFS in NDMM-TE patients who receive intensive treatment. Immune reconstitution, measured as recovery from uHLC IP, provides complementary prognostic information to MRD assessment (clinicaltrials gov. Identifiers: NCT01916252 and NCT02406144).
Immunoparesis (IP) is a very common finding in multiple myeloma (MM) patients at diagnosis and it is defined as the suppression of polyclonal uninvolved immunoglobulins (Ig). Currently we can measure it in two ways. On the one hand, we can analyze classic IP by measurement of total IgG, IgA and IgM levels by nephelometry or turbidimetry. On the other hand, Hevylite® assay can detect different heavy/ light chain (HLC) pairs, so it can measure immunoparesis of the opposite pair of heavy and light chain to the one involved, which is the pair with the same heavy chain and the opposite light chain. Both types of IP are very common in MM at diagnosis and occur in 80-90% of patients.1-6
Several studies have shown that patients with classic IP at diagnosis have worse prognosis than patients without IP, in terms of progression-free survival (PFS) and overall survival (OS).1,2 Suppression of the uninvolved HLC pair of the same isotype (uHLC) and mostly severe suppression of uHLC (<50% of lower limit of normality) at diagnosis, has also been associated with poor prognosis.4-7 Recovery from classical IP has been analyzed in a few studies, mainly, but not only, after autologous stem cell transplantation (ASCT). All of these studies showed that patients who recovered from IP during or after treatment had better prognosis than patients who didn’t recover from it. However, these are observational and retrospective studies and included patients over a wide period of time, who therefore underwent non-homogeneous treatment. 8-13
Although uHLC IP recovery has not been properly studied, some papers have reported poor prognosis for patients with an abnormal ratio of involved/uninvolved HLC at follow-up or persistence of uHLC IP after treatment in heterogeneously treated patients.14-16
Minimal residual disease (MRD) in MM patients is actually the most important prognostic factor during evolution of the disease and probably will be the main driver of clinical trials in the near future.17,18 However not all MRD-negative patients have the same prognosis, and therefore other evolutive prognostic factors are needed.19
The goal of this work is to evaluate the prognostic value of IP recovery by measurement of classic total Ig and uHLC and to assess the prognostic benefit of adding IP recovery information to MRD in newly diagnosed MM transplant-eligible (NDMM-TE) patients treated with a fixed-duration approach within the GEM2012 and GEM2014 clinical trials.
We included NDMM-TE patients enrolled and treated in the PETHEMA/GEM2012MENOS65 trial (clinicaltrials gov. Identifier: NCT01916252) who continued with the PETHEMA/GEM2014MAIN clinical trial (clinicaltrials gov. Identifier: NCT02406144). In the first trial, patients received six cycles of VRD-GEM (bortezomib, lenalidomide, dexamethasone) induction, ASCT conditioned with melphalan or busulfan plus melphalan and consolidation with two cycles of VRDGEM. Afterwards, patients who achieved at least minimal response were enrolled in the second trial that randomly assigned them to maintenance with lenalidomide and lowdose dexamethasone (Rd) or Rd plus ixazomib for 2 years. Those patients who didn’t achieve MRD negativity after 2 years of maintenance received 3 more years of Rd to complete a total of 5 years of maintenance. Response assessment was performed according to International Myeloma Working Group (IMWG) criteria.20 No differences in efficacy between arms were found in the main analysis of this trial.21 Informed consent for both trials was obtained before patient participation. Each study site’s independent ethics committee reviewed and approved both protocols. The study was designed and conducted in accordance with the Declaration of Helsinki.
Analysis of classic Ig (IgG, IgA and IgM) by turbidimetry and uHLC analyzed by Hevylite® assay (The Binding Site Group Ltd, Birmingham, UK) was performed in a central laboratory in samples at diagnosis, after consolidation and after the first year of maintenance treatment. We considered IP at diagnosis to be when one or more uninvolved total Ig (classic IP) or uHLC (uHLC IP) were under the lower limit of normal (LLN). We considered recovery from classic IP to be when one or all Ig suppressed at diagnosis reached at least the LLN; recovery from uHLC IP was considered when uHLC reached at least LLN plus 10% in patients with suppressed uHLC at diagnosis.
In the PETHEMA/GEM2012MENOS65 trial, 458 patients were included of which 332 patients entered the PETHEMA/GEM2014MAIN clinical trial. We included a total of 245 patients in this study, those who had samples available at any of the three analyzed time points. Patients with light chain only MM (LCMM) were included in the analysis for classic Ig but were not included in the analysis for uHLC.
For classic total Ig, we analyzed 234 patients at diagnosis, 233 after consolidation and 173 after the first year of maintenance. For uHLC, we analyzed 202 patients at diagnosis, 207 after consolidation and 154 after the first year of maintenance (Figure 1).
Minimal residual disease analysis
MRD was evaluated after consolidation treatment by next-generation flow cytometry (NGF)17 with an estimated sensitivity level of 2x10-6
The main outcome examined in this study was PFS of patients with and without IP recovery after the first year of maintenance when measuring classic total Ig and uHLC. In addition, we explored PFS of patients according to IP recovery and MRD status.
PFS was defined as the time from the time point we are evaluating (when groups are built) until progression, relapse or death occurred.
Continuous variables are summarized with mean and range while categorical variables are summarized by number of patients and percentage. Survival analysis for PFS were conducted by Kaplan-Meier methodology and differences between survival curves were tested for statistical significance using the two-sided log-rank test. Univariate and multivariate analysis were conducted using Cox propor-
tional-hazards regression models, restricting the number of variables included according to the number of events. Concordance of each model was evaluated by Harrell’s C index and comparison between c-index values was done by t test for related samples analysis. A P value <0.05 indicated statistical significance. All statistical analyses were performed using IBM SPSS Statistics software (version 25.0).
The main characteristics of the 245 patients included (those who had samples available at any of the 3 points) are summarized in Table 1. Of note, 54.8% of patients evaluated had high-risk cytogenetics because we included those patients who had not only del17p, t(4;14), t(14;16) but also those with +1q21 cytogenetic alterations. Median PFS in these 245 patients was not reached after a median follow-up of 84 months and overall survival at 3 years was 94.6%. Number of PFS events (relapse, progression or death) was 84, two before maintenance treatment, 34 after consolidation and before first year of maintenance and 48 after the first year of maintenance.
At diagnosis we found classic IP measured by total Ig in 86.7% of patients (203/234) and uHLC IP in 94.5% of patients (191/202). No significant differences in PFS were found between patients with or without IP at diagnosis for both
Figure 1. Flowchart showing patients analyzed and not analyzed from diagnosis to post-consolidation and from diagnosis to after the first year of maintenance for classic immunglobulin G and for uninvolved heavy/light chain. IgG: immunoglobulin G; uHCL: uninvolved heavy/light chain.
methods, but it should be pointed out that the number of patients without IP was very small. For this analysis PFS was measured from diagnosis and we found hazard ratio (HR) =0.56 (95% CI: 0.26-1.22; P=0.14) for classic Ig and HR=0.49 (95% CI: 0.12-2.01; P=0.32) for uHLC.
Immunoparesis recovery
For IP recovery we analyzed only patients with IP at diagnosis, and we investigated IP recovery both after consolidation treatment and after the first year of maintenance. After consolidation we analyzed samples of 233 patients for classic total Ig levels, but we excluded patients without Ig analysis at diagnosis (10 patients) and also those who didn’t have IP at diagnosis (28 patients), so IP recovery was evaluated in the remaining 195 patients. For uHLC we analyzed samples from 207 patients, but we excluded those without information at diagnosis (15 patients) and those who didn’t have IP at diagnosis (11 patients), so we evaluated uHLC IP recovery in 181 patients. We found recovery of classic IP in 44.6% of patients (87/195) and recovery of uHLC in 49.2% of patients (89/181) who had IP at diagnosis. We found no significant differences in PFS between patients who recovered and patients who didn’t recover from IP, for both methods, at this time point (PFS for this analysis was measured from the end of consolidation). For classic Ig we found a HR=0.93 (95% CI: 0.61-1.61; P=0.99) and for uHLC we found a HR=0.82 (95% CI: 0.50- 1.36; P=0.45).
Of the 173 patients with available sample after the first year of maintenance, nine were excluded because of lack of sample at diagnosis, 21 had no IP at diagnosis and ten had an event for PFS before the end of first year of maintenance, so a total of 133 patients were investigated for classic IP recovery. A total of 124 of 154 patients were evaluated for uHCL IP recovery (10 patients without analysis of uHCL at diagnosis, 11 patients without uHCL IP at diagnosis and 9 patients who had an event for PFS before the end of the first year of maintenance were excluded). We observed recovery from classic IP in 53.8% (70/133) and recovery from uHLC immunoparesis in 63.2% of patients (77/124) who had IP at diagnosis. We found no differences in PFS between patients who recovered and patients who didn’t recover from classic IP with a HR=1.22 (95% CI: 0.65-2.30; P=0.538; Figure 2A). However, patients with recovery from uHLC IP after 1 year of maintenance had longer PFS than patients who didn’t recover from uHLC IP (P=0.008) with a HR=0.42 (95% CI: 0.21-0.81; Figure 2B). For both analyses PFS was measured from the end of the first year of maintenance. Recovery from IP for both techniques was associated with depth of response. For classic total Ig measurements, after the first year of maintenance, 57.4% of patients in complete response (CR) (58/101) had recovery from IP while only 33.3% of patients in very good partial responsse (VGPR) (9/27) recovered from classic IP (P=0.026). For uHLC after the first year of maintenance, IP recovery occurred in 65.9% of patients in CR (60/91) as compared to 53.6% of patients
in VGPR (15/28), but this difference was not statistically significant (P=0.236).
We analyzed other prognostic factors for PFS measured from the end of the first year of maintenance, but, in univariate analysis, only MRD status after consolidation and high-risk cytogenetics at diagnosis were statistically significant, in addition to recovery from uHLC IP after the first year of maintenance. We created a multivariate analysis model for PFS with uHLC IP recovery at the end of the first year of maintenance, MRD status after consolidation and high-risk cytogenetics found at diagnosis. As seen in Table 2, all three variables had independent prognostic value for PFS. Harrell’s C concordance index for this model was 0.62 (95% CI: 0.55-0.71) without uHLC IP recovery and 0.67 (95% CI: 0.58-0.76) after adding uHLC IP recovery to the model. This difference, 0.05 (95% CI: -0.04 to 0.14), was statistically significant (P<0.001).
As almost all the patients had uHLC IP at diagnosis, if we analyze uHLC values at the first year of maintenance without
Table 1. Main characteristics of patients at diagnosis.
Characteristics
Durie-Salmon stage, N (%)
High risk cytogenetics, N (%)
immunoparesis, N (%)
uHLC immunoparesis, N (%)
MM: multiple myeloma; LCMM: light chain MM; LDH: lactate dehydrogenase; ISS: international staging system; Ig: immunoglobulin; uHCL: uninvolved heavy/light chain.
taking into account uHLC levels at diagnosis (N=163), similar results are obtained. Patients with uHLC at the end of the first year of maintenance above the cutoff value (LLN plus 10%) had longer PFS than patients with uHLC below cutoff value with HR=0.45 (95% CI: 0.25-0.84; P=0.009). In a multivariate model, together with MRD status and high-risk cytogenetics, all three factors were statistically significant and Harrell’s C concordance index for this model was 0.62 (95% CI: 0.55-0.71) before including uHLC levels after the first year of maintenance and 0.67 (95% CI: 0.59-0.75) after including it, this difference was also statistically significant (0.05 [95% CI: -0.03 to 0.13); P<0.001).
Association of minimal residual disease status with immunoparesis recovery
We also analyzed the prognostic value of the association of MRD status after consolidation and uHLC IP recovery at the end of the first year of maintenance. We evaluated the association in the 122 patients where both data was available: 45 patients were MRD-negative and recovered from uHLC IP, 24 patients were MRD-negative but didn’t recover from uHLC IP, 31 patients were MRD-positive but recovered from uHLC IP and 22 patients were MRD-positive and didn’t recover from uHLC IP. PFS for this analysis was measured from the end of the first year of maintenance.
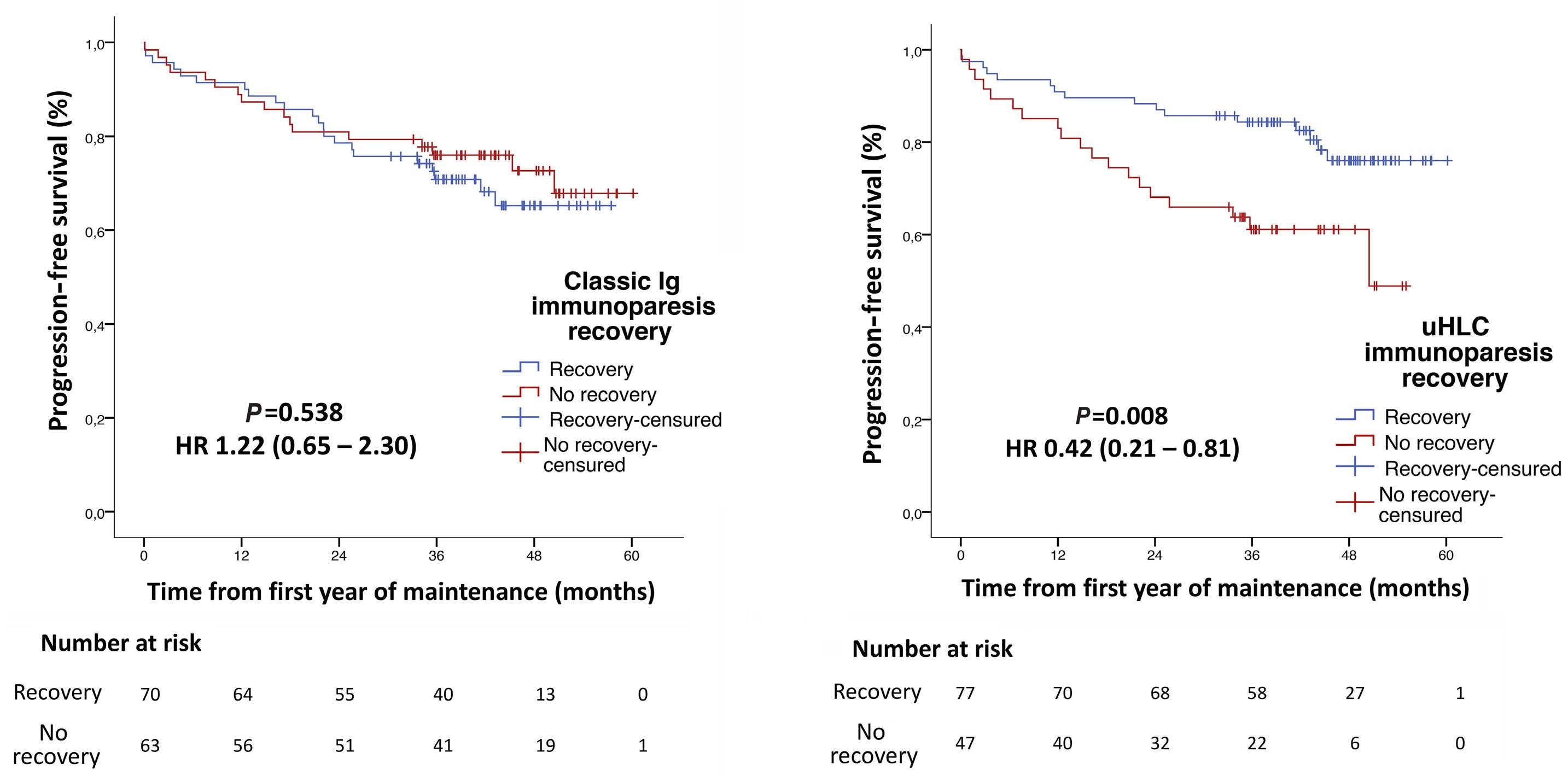
Figure 2. Progression-free survival according to immunoparesis recovery after the first year of maintenance. (A) Progression-free survival for classic immunglobulin G (IgG) immunoparesis recovery and (B) for uninvolved heavy/light chain (uHLC) immunoparesis recovery. HR: hazard ratio (95% confidence interval).
Table 2. Univariate and multivariate analysis for progression-free survival from first year of maintenance (M1).
Variables
Durie-Salmon stage(I-II vs. III), N=185
High-risk cytogenetics, N=161
LDH (normal vs. high), N=178
ISS stage (I vs. II-III), N=182
Conventional response at M1 (CR vs. VGPR), N=179
N=182
IP recovery at M1, N=124
HR: hazard ratio; CI: confidence interval; LDH: lactate dehydrogenase; ISS: international staging system; CR: complete reposnse; VGPR: very good partial response; MRD: minimal residual disease; uHCL: uninvolved heavy/light chain; IP: immunoparesis.
Patients with both favorable factors (MRD-negative and recovery from uHLC IP) had better PFS than patients with both unfavorable factors (MRD-positive without recovery from uHLC IP) with HR=0.23 (95% CI: 0.09-0.61; P=0.001; Figure 3A, B). Both groups of patients with just one favorable factor (MRD-negative without recovery from uHLC IP or MRD-positive with recovery from uHLC IP) have a similar prognosis and their prognosis is intermediate between the other two groups (Figure 3A). If these two groups of patients with just one favorable factor are joined into a single group and this is compared to the other groups, it has a HR=0.47 (95% CI: 0.19-1.15; P=0.091) compared with the “both favorable factors” group and a HR=0.48 (95% CI: 0.22-1.03; P=0.054) compared with the “both unfavorable factors” group (Figure 3B).
In this study we confirm the independent prognostic value of recovery from uHLC IP after the first year of maintenance in the setting of NDMM-TE patients treated within a clinical trial with intensive fixed-duration treatment. Furthermore, uHLC IP recovery at the end of the first year of maintenance provides complementary prognostic value to MRD detection. There was no prognostic value for recovery from classic IP after the first year of maintenance in these patients. Previous results for the prognostic value of uHLC and HLC ratio during follow-up are consistent with our findings and recovery from uHLC IP appears to be a solid prognostic marker in MM.14-16 This may be due to the important impact of dysregulation of immune system in MM patients at diagnosis that affects mainly T cells and dendritic cells, but also B cells and NK cells.22 Recovery from uHLC IP is a good marker for B-cell immune reconstitution in our population and confers good prognosis. Other prognostic markers of B-cell immune reconstitution have previously been reported such as the presence of oligoclonal bands, percentage of mature B cells in bone marrow or even recovery from classic IP.23,24 In previous studies8-13 recovery from classic IP was found to be an important prognostic factor for PFS and OS in MM. Therefore, the lack of prognostic value for recovery from classic IP in this study is surprising. A possible explanation is that previous studies included patients non-homogeneously treated, during a wide period of time and mostly with less intensive treatment. It is possible that the intensity of the treatment in this trial prevented an increase of classic total Ig levels but did not prevent uHLC recovery after the first year of maintenance. Measurement of uHLC appears to be a more sensitive method to detect immune reconstitution than measurement of classic Ig, at least in our scenario of intensive treatment. As far as we know, this is the first study to compare immunoparesis recovery of uHLC versus classic Ig. After consolidation treatment, neither recovery from classic
Ig nor uHLC IP had any prognostic value. This time point may be too early to evaluate recovery from IP. Previous studies, evaluating classic Ig in patients who underwent ASCT with less intensive induction, showed that the best time point to evaluate prognosis of recovery from IP was 1 year after transplant.9,10,12 Maintenance treatment in this trial, which is less intensive than previous treatment (induction, transplant and consolidation), allowed patients to recover from uHLC IP but not classic Ig IP after the first year. Maybe with a longer duration of maintenance treatment or maybe with a longer interval after stopping maintenance treatment, recovery from classic Ig IP will achieve a good prognostic value. After the first year of maintenance, we saw no difference in the prognostic value of recovery from uHLC or classic Ig IP between treatment arms (Rd vs. Rd plus Ixazomib), so time after transplant may be more important than the intensity of maintenance treatment when considering recovery from IP.
The proportion of patients with uHLC IP at diagnosis in our series was very high (94,6%) and we have confirmed that similar results, in terms of prognosis, are observed if we evaluate only the level of uHLC at the end of the first year of maintenance instead of uHLC IP recovery compared to baseline. Therefore, in clinical practice, if information on uHLC value at diagnosis is not available, uHLC levels after the first year of maintenance alone can be used with similar prognostic value.
Treatment-induced IP could be an important limitation for using IP recovery (classic or uHLC) as a prognostic factor. Anti-CD38 antibodies, which are now used in the first line treatment of transplant-eligible and transplant-ineligible patients, produce an important decrease of Ig levels. In addition, bispecific antibodies and chimeric antigen receptor (CAR) T cells produce a severe IP that usually needs prophylactic treatment with intravenous Ig.25,26 Most of the patients undergoing these treatments will probably not achieve recovery of Ig levels, whether measured by classic Ig or uHLC. Maybe we will need to look for small increases in uHLC levels or whether uHLC levels follow a positive trend to know which patients undergo immune reconstitution and have better prognosis. Other immune prognostic markers should also be sought to give further information. Supportive prophylactic treatment with intravenous Ig may further complicate interpretation of changes in uHLC levels, especially in IgG MM patients. Further studies to understand uHLC behavior when we use anti-CD38 antibodies, bispecific antibodies or CAR T-cell therapy are needed. In this study we can see that the prognostic value of uHLC IP recovery complements the prognostic value of an MRD result at a single time point. It is known that the prognostic value of sustained MRD negativity (for 12 or 24 months) is clearly better than the prognostic value of just one MRD-negative result.27 Some patients with a single MRD-negative result become MRD-positive after a short period of time and may suffer a relapse over the following months.19 Immune
reconstitution may play an important role in sustaining MRD negativity over time, but other factors also seem to be implicated.28 Patients with very aggressive disease, as those with two or more high-risk cytogenetic abnormalities and those with high level of circulating tumor cells at baseline lose MRD negativity more frequently.29,30 The relationship between immune reconstitution, measured
with uHLC IP recovery or other markers, evolution of MRD measured by NGF or next-generation sequencing, and aggressiveness markers of myeloma needs to be explored in future studies.
In conclusion, recovery from uHLC IP is an independent prognostic factor for NDMM-TE patients who receive intensive continuous treatment. In addition, uHLC IP recovery
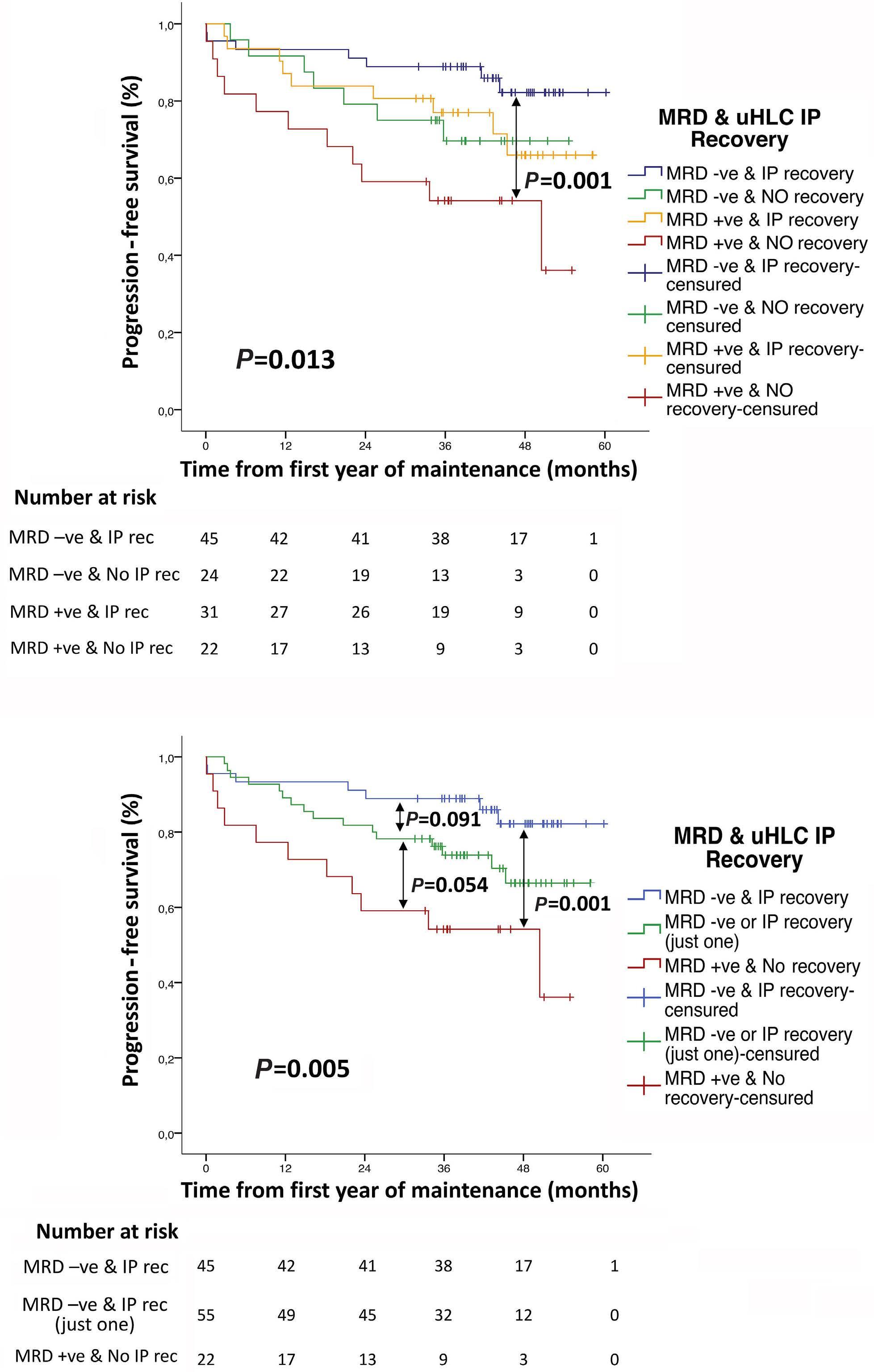
Figure 3. Progression-free survival according to minimal residual disease status after consolidation and uninvolved heavy/light chain immunoparesis recovery after the first year of maintenance. (A) Progression-free survival (PFS) of the 4 combinations of minimal residual disease MRD (positive or negative) and uninvolved heavy/ light chain (uHLC) immunoparesis (IP) (recovery or no recovery). (B) Comparison of PFS of patients with both favorable factors (MRD-negative & uHLC IP recovery), patients with only 1 favorable factor (MRD-negative or uHLC IP recovery) and patients with both unfavorable factors (MRD-positive & absence uHLC IP recovery). -ve: negative; +ve: positive, rec: recovery.
complements the prognostic value of an MRD result at a single time point, but further studies in patients treated with new therapies and longitudinal analysis of uHLC recovery alongside MRD are needed.
Disclosures
LR has received honoraria for lectures from Janssen, BMS-Celgene, Amgen, Takeda, Sanofi and GSK. NP has received honoraria for lectures and consultancy fees from The Binding Site. BP has received honoraria derived from lectures and/or participation on advisory boards from Adaptive, Amgen, Becton Dickinson, BMS-Celgene, GSK, Janssen, Roche, Sanofi and Takeda; has received research funding from BeiGene, BMS-Celgene, GSK, Roche, Sanofi and Takeda; has received consultancy fees from BMS-Celgene, Janssen, Sanofi and Takeda. RR-T has received honoraria for lectures and consultancy fees from Amgen, BMS, GSK, Janssen, Sanofi and The Binding Site; and has participated in advisory board meetings at Beckton-Dickinson, GSK and Janssen. M-VM has received honoraria derived from lectures and/or participation on advisory boards from Janssen, Celgene, Amgen, Takeda, AbbVie, GlaxoSmithKline, Oncopeptides, Sanofi, Pfizer, Roche, and Stemline; and has received consultancy fees from The Binding Site. J-JL has received consultancy fees and/or has participated on advisory board of BMS, Sanofi, Amgen and Janssen; and has received travel expenses from Celgene and Pfizer. JS-M has participated on advisory boards and consulting services for Abbvie, Amgen, BMS-Celgene, GSK, Haemalogix, Janssen, Karyopharm, MSD, Novartis, Pfizer,
1. Kastritis E, Zagouri F, Symeonidis A, et al. Preserved levels of uninvolved immunoglobulins are independently associated with favorable outcome in patients with symptomatic multiple myeloma. Leukemia. 2014;28(10):2075-2079.
2. Heaney JLJ, Campbell JP, Iqbal G, et al. Characterization of immunoparesis in newly diagnosed myeloma and its impact on progression-free and overall survival in both old and recent myeloma trials. Leukemia. 2018;32(8):1727-1738.
3. Sørrig R, Klausen TW, Salomo M, et al. Immunoparesis in newly diagnosed multiple myeloma patients: effects on overall survival and progression free survival in the Danish population. PLoS One. 2017;12(12):e0188988.
4 Ludwig H, Milosavljevic D, Berlanga O, et al. Suppression of the noninvolved pair of the myeloma isotype correlates with poor survival in newly diagnosed and relapsed/refractory patients with myeloma. Am J Hematol. 2016;91(3):295-301.
5. Boyle EM, Fouquet G, Guidez S, et al. IgA kappa/IgA lambda heavy/light chain assessment in the management of patients with IgA myeloma [published correction appears in Cancer. 2015 Mar 1;121(5):800]. Cancer. 2014;120(24):3952-3957.
6. Geng C, Yang G, Wang H, et al. Deep and partial immunoparesis is a poor prognostic factor for newly diagnosed multiple myeloma patients. Leuk Lymphoma. 2021;62(4):883-890.
7 Koulieris E, Panayiotidis P, Harding SJ, et al. Ratio of involved/ uninvolved immunoglobulin quantification by Hevylite™ assay:
Takeda, Regeneron, Roche, Sanofi, and SecuraBio. All other authors have no conflicts of interest to disclose.
SL, M-TH and JS-M conceived and designed the study. M-APP and LM-G analyzed the samples. SL and M-TH analyzed and interpreted data and performed the statistical analysis. JDC and RAF reviewed statistical analysis. SL, M-TH, JS-M, BP and JM-L reviewed and interpreted the results. SL, M-TH and JS-M wrote the paper. All authors provided patients, and reviewed and approved the manuscript.
The authors would like to thank all the patients and caregivers of this trial and all the investigators of the GEM/ PETHEMA Cooperative Study Group.
The present study was sponsored by PETHEMA (Spanish Program for the Treatment of Hematological Diseases), Madrid, Spain, and was coordinated by the Spanish Myeloma Group (GEM-PETHEMA). Transfer of samples and reagents for analysis was funded by The Binding Site Group Ltd. The contents are solely the responsibility of the authors.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
clinical and prognostic impact in multiple myeloma. Exp Hematol Oncol. 2012;1(1):9.
8. Díaz-Rodríguez R, Férnandez-González M, Martín-Martín A, et al. Resolución de la inmunoparesia como parámetro de la remisión completa real en el mieloma múltiple. LV National Congress of SEHH and XXIX National Congress of SETH, Sevilla. 2013:CO-021. https://www.sehh.es/comunicaciones-cientificasde-los-congresos-sehh-seth-2000-2017/lv-congreso-nacionalsehh-xxix-congreso-seth. Accesed November 7th, 2023.
9 González-Calle V, Cerdá S, Labrador J, et al. Recovery of polyclonal immunoglobulins one year after autologous stem cell transplantation as a long-term predictor marker of progression and survival in multiple myeloma. Haematologica. 2017;102(5):922-931.
10. Jimenez-Zepeda VH, Duggan P, Neri P, Chaudhry A, Tay J, Bahlis N. Immunoparesis and polyclonal immunoglobulin recovery after auto-SCT for patients with multiple myeloma treated at a single institution. Leuk Lymphoma. 2018;59(8):1920-1926.
11. Dávila J, González-Calle V, Escalante F, et al. Recovery of polyclonal immunoglobulins during treatment in patients ineligible for autologous stem-cell transplantation is a prognostic marker of longer progression-free survival and overall survival. Br J Haematol. 2022;198(2):278-287.
12. Ozaki S, Harada T, Yagi H, et al. Polyclonal immunoglobulin recovery after autologous stem cell transplantation is an
independent prognostic factor for survival outcome in patients with multiple myeloma. Cancers (Basel). 2019;12(1):12.
13. Eisfeld C, Eßeling E, Wullenkord R, et al. Long-term survival and polyclonal immunoglobulin reconstitution after allogeneic stem cell transplantation in multiple myeloma. Ann Hematol. 2020;99(8):1907-1915.
14 Harutyunyan NM, Vardanyan S, Ghermezi M, et al. Levels of uninvolved immunoglobulins predict clinical status and progression-free survival for multiple myeloma patients. Br J Haematol. 2016;174(1):81-87.
15. Suehara Y, Takamatsu H, Fukumoto K, et al. Abnormal heavy/ light chain ratio after treatment is associated with shorter survival in patients with IgA myeloma. Cancer Sci. 2017;108(2):187-192.
16. Ríos-Tamayo R, Puig N, Algarín M, et al. The current role of the heavy/light chain assay in the diagnosis, prognosis and monitoring of multiple myeloma: an evidence-based approach. Diagnostics (Basel). 2021;11(11):2020.
17 Paiva B, Puig N, Cedena MT, et al. Measurable residual disease by next-generation flow cytometry in multiple myeloma. J Clin Oncol. 2020;38(8):784-792.
18. Avet-Loiseau H, Ludwig H, Landgren O, et al. Minimal residual disease status as a surrogate endpoint for progression-free survival in newly diagnosed multiple myeloma studies: a metaanalysis. Clin Lymphoma Myeloma Leuk. 2020;20(1):e30-e37.
19 Mohan M, Kendrick S, Szabo A, et al. Clinical implications of loss of bone marrow minimal residual disease negativity in multiple myeloma. Blood Adv. 2022;6(3):808-817.
20 Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346.
21. Rosiñol L, Oriol A, Ríos-Tamayo R, et al. Ixazomib plus lenalidomide/ dexamethasone (IRd) versus lenalidomide/dexamethasone (Rd) maintenance after autologous stem cell transplant in patients with newly diagnosed multiple myeloma: results of the Spanish
GEM2014MAIN Trial. Blood 2021;138(Suppl 1):466.
22. Nakamura K, Smyth MJ, Martinet L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood. 2020;136(24):2731-2740.
23. Tovar N, de Larrea CF, Aróstegui JI, et al. Natural history and prognostic impact of oligoclonal humoral response in patients with multiple myeloma after autologous stem cell transplantation: long-term results from a single institution. Haematologica. 2013;98(7):1142-1146.
24. Paiva B, Cedena MT, Puig N, et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood. 2016;127(25):3165-3174.
25. Mazahreh F, Mazahreh L, Schinke C, et al. Risk of infections associated with the use of bispecific antibodies in multiple myeloma: a pooled analysis. Blood Adv. 2023;7(13):3069-3074.
26. Wang Y, Li C, Xia J, et al. Humoral immune reconstitution after anti-BCMA CAR T-cell therapy in relapsed/refractory multiple myeloma. Blood Adv. 2021;5(23):5290-5299.
27. San-Miguel J, Avet-Loiseau H, Paiva B, et al. Sustained minimal residual disease negativity in newly diagnosed multiple myeloma and the impact of daratumumab in MAIA and ALCYONE. Blood. 2022;139(4):492-501.
28. Bertuglia G, Cani L, Larocca A, Gay F, D’Agostino M. Normalization of the immunological microenvironment and sustained minimal residual disease negativity: do we need both for long-term control of multiple myeloma?. Int J Mol Sci. 2022;23(24):15879.
29 Costa LJ, Chhabra S, Medvedova E, et al. Daratumumab, carfilzomib, lenalidomide, and dexamethasone with minimal residual disease response-adapted therapy in newly diagnosed multiple myeloma. J Clin Oncol. 2022;40(25):2901-2912.
30 D’Agostino M, Oliva S, Rota-Scalabrini D, et al. Predictors of unsustained negativity in minimal residual disease (MRD)negative transplant-eligible newly diagnosed multiple myeloma (MM) patients enrolled in the FORTE trial. Clin Lymphoma Myeloma Leuk. 2022;22(Suppl):S6-7.
Alessandro Mattè,1* Enrica Federti,1* Antonio Recchiuti,2* Moayed Hamza,3 Giulia Ferri,2 Veronica Riccardi,1 Jacopo Ceolan,1 Alice Passarini,1 Filippo Mazzi,1 Angela Siciliano,1 Deepak L. Bhatt,4 David Coughlan,3 John Climax,3 Elisa Gremese,5,6 Carlo Brugnara7 and Lucia De Franceschi1
1Department of Medicine, University of Verona & AOUI Verona, Verona, Italy; 2Department of Medical, Oral, and Biotechnology Science, “G. d’Annunzio” University Chieti Pescara, Chieti, Italy; 3Afimmune Ltd., Dublin, Ireland; 4Mount Sinai Heart, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 5Division of Clinical Immunology, Fondazione Policlinico Universitario A. Gemelli-IRCCS, Università Cattolica del Sacro Cuore, Rome, Italy; 6Immunology Core Facility, Fondazione Policlinico Universitario A. Gemelli-IRCCS, Rome, Italy and 7Department of Laboratory Medicine, Boston Children’s Hospital, Department of Pathology, Harvard Medical School, Boston, MA, USA
*AM, EF and AR contributed equally as first authors.
Abstract
Correspondence: C. Brugnara carlo.brugnara@childrens.harvard.edu
Received: August 11, 2023.
Accepted: December 7, 2023. Early view: December 14, 2023.
https://doi.org/10.3324/haematol.2023.284028
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Inflammatory vasculopathy is critical in sickle cell disease (SCD)-associated organ damage. An imbalance between pro-inflammatory and pro-resolving mechanisms in response to different triggers such as hypoxia/reoxygenation or infections has been proposed to contribute to the progression of SCD. Administration of specialized pro-resolving lipid mediators may provide an effective therapeutic strategy to target inflammatory vasculopathy and to modulate inflammatory response. Epeleuton (15 hydroxy eicosapentaenoic acid ethyl ester) is a novel, orally administered, second-generation ω-3 fatty acid with a favorable clinical safety profile. In this study we show that epeleuton re-programs the lipidomic pattern of target organs for SCD towards a pro-resolving pattern. This protects against systemic and local inflammatory responses and improves red cell features, resulting in reduced hemolysis and sickling compared with that in vehicle-treated SCD mice. In addition, epeleuton prevents hypoxia/reoxygenation-induced activation of nuclear factor-kB with downregulation of the NLRP3 inflammasome in lung, kidney, and liver. This was associated with downregulation of markers of vascular activation in epeleuton-treated SCD mice when compared to vehicle-treated animals. Collectively our data support the potential therapeutic utility of epeleuton and provide the rationale for the design of clinical trials to evaluate the efficacy of epeleuton in patients with SCD.
Sickle cell disease (SCD) is a genetic disease in which a point mutation of the globin gene at the β7 position leads to the production of hemoglobin S (HbS), resulting in chronic hemolysis and anemia.1 SCD is also characterized by acute vaso-occlusive crises (VOC), a name based on the notion that occlusion of small vessels and/or capillaries by sickled cells is the triggering mechanism for the generation of inflammation and pain.1 This inflammatory vasculopathy plays a key role in the pathogenesis of both acute and chronic sickle cell-related complications, interfacing the whole blood compartment and being involved in cell-cell pro-adhesion phenomena.2 Regulatory-body approvals of new treatments such as voxelotor, an inhibitor of HbS po-
lymerization, are important recent milestones.3 However, the persistence of VOC, as well as chronic disease progression, still represent unmet therapeutic needs, which require additional approaches.
Studies in different models of inflammatory vasculopathy, such as atherosclerosis, diabetes or ischemic cardiomyopathy, have highlighted the yin-yang role of inflammatory response and pro-resolving mechanisms.4,5 Evidence from this laboratory demonstrates that the pathophysiology of SCD goes beyond the well-known role of polymerized HbS and includes imbalances in pro-inflammatory versus pro-resolving mechanisms.6 This imbalance in turn might promote disease progression and influence the severity of clinical manifestations.4,5 Consistent with this notion, multiple independent clinical studies profiling plasma, red
cells, or platelet fatty acid composition in patients with SCD demonstrate that there is an underlying relative deficiency of ω-3 fatty acids and their pro-resolving metabolites in SCD under both basal and inflammatory conditions.7-12 The observed decreases in fatty acids in SCD have also been shown to correlate with inflammation and hemoglobin in patients with SCD.7,9 An attempt to target inflammatory vasculopathy and to modulate inflammatory response through pro-resolving mechanisms has been made in other diseases, such as cardiovascular diseases, by administering ω-3 fatty acids (ω-3 polyunsaturated fatty acids [PUFA]). Indeed, supplementation with ω-3 fatty acids might act as multimodal therapy by: (i) affecting cell membrane lipid composition; (ii) modulating soluble and cellular inflammatory responses and the coagulation cascade; and (iii) favoring nitric oxide production.13,14 Previously, we reported that in the humanized mouse model for SCD, PUFA supplementation protected against acute sickle cell-related organ damage induced by hypoxia/reoxygenation (H/R) stress as a model for VOC.15 These data were complemented by evidence from human studies that the administration of ω-3 PUFA to SCD subjects reduced VOC, pain episodes, and the need for blood transfusions.16 However, the limited clinical and molecular data on PUFA administration for SCD patients still represents an obstacle to the routine use of PUFA as a therapeutic approach for patients with SCD. In addition, the specific formulation of PUFA supplements markedly affects their bioavailability and clinical outcomes, as demonstrated by the different effects of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) formulations on cardiovascular outcomes.17
Epeleuton, 15S-hydroxy eicosapentaenoic acid (15[S] HEPE) ethyl ester, is a second-generation synthetic ω-3 fatty acid derivative of C20:5 n-3 EPA.18 15(S)-HEPE, an oxylipin downstream of the 15-lipoxygenase metabolism of EPA, is endogenously produced by hypoxic vascular endothelial cells and can be further transformed to produce lipoxins A5 and B5.19,20 At the time of submitting this article for publication, epeleuton has completed an extensive toxicological evaluation and been administered to more than 340 subjects in phase I and phase II clinical trials, with a favorable safety and tolerability profile. Studies in animal models of atherosclerosis and in patients with non-alcoholic fatty liver disease show that epeleuton has protective effects against inflammatory vasculopathy and markers of systemic inflammation, suggesting its possible transferability to SCD.18 Notably, decreased concentrations of 15-HEPE were observed in humanized SCD mice under both normoxic and hypoxic conditions compared to those in healthy controls, a finding which is consistent with previously reported decreased fatty acid concentrations in patients with SCD.6 In this study, using a humanized mouse model of SCD, we found that epeleuton significantly increased organ concentrations of 15-HEPE, which has both anti-inflammatory and pro-resolving functions. We show the benefit of the oral administration of epeleuton in protecting against (H/R-induced stress in lung, liver, and kidney, known target organs for SCD.
We also found that epeleuton reprograms the functional profile of spleen macrophages towards a pro-resolving signature and prevents H/R-induced vascular endothelial overactivation. Collectively our data support the potential therapeutic utility of epeleuton, elucidate further mechanisms for the beneficial effects of ω-3 fatty acids, and provide the rationale for the design of clinical trials to evaluate the efficacy of epeleuton in patients with SCD.
Animals and design of the study
Experiments were performed on 4- to 5-month-old and sex-matched (56.7% males and 43.3% females) healthy control mice (AA: Hbatm1(HBA)Tow Hbbtm3(HBG1, HBB) Tow) and mice with SCD (SS: Hbatm1(HBA)Tow Hbbtm2(HBG1, HBB*) Tow). We chose to use 4- to 5-month-old mice since we and others have previously reported that the sickle cell-related organ damage in animals of this age is not so severe as to generate confounding factors when the impact of H/R-induced stress is evaluated.6,15,21-24 This is also corroborated by the observation of Kasztan et al ., who found that gender-related differences in kidney function of humanized SCD mice became significant after 24 weeks (6 months) of age. 22 The size of our mouse groups was established based on studies with the same mouse model for SCD generated by us and other laboratories.6,15,21-24 The animal protocol was approved by the Animal Care and Use Committee of the University of Verona (CIRSAL) and the Italian Ministry of Health (protocol # 56DC9.64), following European directive 2010/63/EU and the Federation for Laboratory Animal Science Associations guidelines and recommendations. Mice were randomly assigned to the different treatment groups and analysis. Healthy and SCD mice were treated with either vehicle (hydroxypropyl methyl cellulose, 0.5%) alone or with epeleuton (1,000 mg/kg/day) in the vehicle, administered by gavage daily for 6 weeks. The dosage of epeleuton was based on previous pharmacokinetic studies in rodents, which yielded concentrations in plasma comparable to those observed in patients.18 When indicated, mice underwent H/R stress: 10 hours of hypoxia (8% oxygen) followed by 3 hours of reoxygenation (21% oxygen) to mimic an acute VOC, as previously described.6,15 Whole blood and organs were collected from each mouse under isoflurane anesthesia. Details on organ collection, as well as evaluation of the biochemical and hematologic parameters are reported in the Online Supplementary Methods
Organ lipidomics
Details are reported in the Online Supplementary Methods
Histological and immunoblot analyses
Histological analysis of lung, kidney and liver tissue and
immunoblot analyses were carried out as previously described.6,15,21 Details are provided in the Online Supplementary Methods.
Analysis of circulating and spleen neutrophils and kidney leukocytes
Circulating and splenic total leukocytes and neutrophils were identified in freshly collected blood and spleen by flow cytometry as CD45+ cells and CD45+Ly6G+ cells.25 Details are provided in the Online Supplementary Methods
Analysis of efferocytosis and macrophage receptors
Details are reported in the Online Supplementary Methods 6
Statistical analysis
A two-tailed unpaired Student t test or two-way analysis of variance with Tukey multiple comparisons test were used for data analyses. Whenever indicated we used an unpaired Student t test with Bonferroni correction. Normality was
assessed with the Shapiro-Wilk test. Lipidomic data were analyzed using the non-parametric Kruskal Wallis test because normality was not met.
A post-hoc analysis was performed to determine the statistical power of the study (with computations carried out using G*Power 3.1.9.4). The achieved power for Figures 1A, 3A, 3B, and Online Supplementary Figure S2 was greater than or equal to 88%.
Data show values from individual mice and are presented as the mean ± standard error of mean (SEM). Differences with P<0.05 were considered statistically significant. Whisker plots show data points and mean ± SEM.
Epeleuton reduces neutrophil counts and modulates inflammatory response in mice with sickle cell disease
Epeleuton was well tolerated and did not cause major
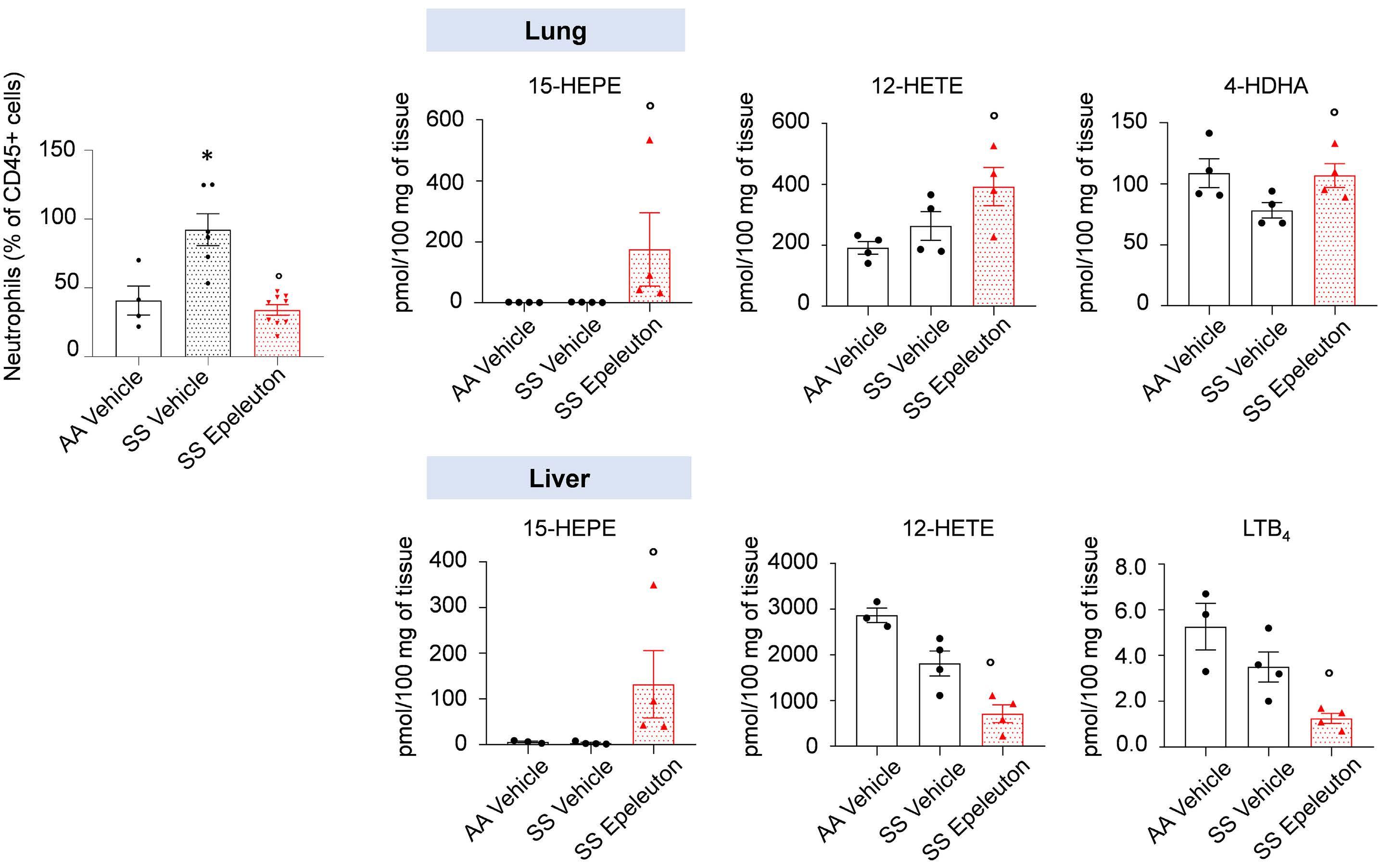
Figure 1. Epeleuton reduces circulating neutrophils and promotes a pro-resolving lipidomic profile in lung and liver of mice with sickle cell disease. (A) Circulating neutrophils identified by flow cytometric analysis as CD45+Ly6G+ cells in healthy mice (AA) and sickle cell disease (SCD) mice (SS) treated with either vehicle or epeleuton (1,000 mg/kg/day for 6 weeks). Data are presented as mean ± standard error of mean (SEM) (N=4-9). *P<0.05 compared to healthy mice; °P<0.05 compared to vehicle-treated SS animals by one-way analysis of variance. (B, C) Liquid chromatography tandem mass spectrometry-based lipidomics of oxylipins in lung (B) and liver (C) from AA, vehicle-treated SCD, or epeleuton-treated SCD mice. Results are mean ± SEM (N=3-4). °P<0.05 compared to vehicle-treated SS animals by the Kruskal Wallis test. 15-HEPE: 15-hydroxyeicosanopentaenoic acid; 12-HETE: 12-hydroxyeicosatetetraenoic acid; 4-HDHA: 4-hydroxydocosahexaenoic acid; LTB4: leukotriene B4.
changes in body weight of the mice (Online Supplementary Figure S1A). We observed a trend toward a reduction of serum creatinine in epeleuton-treated SCD mice compared to vehicle-treated mice, with no change in blood urea nitrogen (Online Supplementary Figure S1B). A significant reduction in the level of alanine aminotransferase was observed in epeleuton-treated SCD mice compared to the level in either healthy or vehicle-treated SCD mice, with no change in aspartate aminotransferase (Online Supplementary Figure S1C). We found a significant reduction in C-reactive protein, and a trend toward a reduction of pentraxin-2, two markers of systemic inflammation (Online Supplementary Figure S2A), in epeleuton-treated SCD mice. These changes were accompanied by a lower plasma level of CCL2 monocyte chemoattractant protein 1, a mediator of neutrophil recruitment and function26 (Online Supplementary Figure S2B). These data indicate a systemic anti-inflammatory effect of epeleuton in humanized SCD mice. Under normoxia, epeleuton-treated SCD mice did not show significant changes in either hemoglobin concentration or reticulocyte counts (Online Supplementary Figure S2C), whereas we observed a significant reduction in circulating neutrophils (Figure 1A, Online Supplementary Figure S3A).
Since the spleen might act as reservoir of inflammatory cells but also contribute to their clearance,27 spleen-associated neutrophils were evaluated. No major changes in spleen weight/mouse weight ratio and or in spleen neutrophils were observed between epeleuton-treated SCD mice and vehicle-treated SCD animals (Online Supplementary Figure S3B, C).
Epeleuton promotes a pro-resolving profile in target organs and prevents the activation of the nuclear factor-kB pathway in mice with sickle cell disease We carried out a lipidomic analysis of lung and liver from both mouse strains treated with either vehicle or epeleuton. In agreement with our previous study, we found abnormalities in the composition of both mono- and poly-unsaturated fatty acids in lung and liver from SCD mice compared to AA animals (Online Supplementary Table S1) despite the two mouse colonies having been fed the same diet.15 These abnormalities consisted of a significantly lower ω-3 content, mainly a linolenic acid (C18:3 n-3), docosapentaenoic acid (DPA:5 -3), eicosatetraenoic acid (C20:4 n-3), and a lower ω-6/ω-3 ratio compared to that in healthy animals (Online Supplementary Tables S1 and S2). As expected, there was a significant increase in lung and liver concentrations of 15-HEPE in epeleuton-treated SCD mice compared to vehicle-treated SCD or AA mice (Figure 1B, C). Total and differential amounts of saturated and unsaturated fatty acids in liver and spleen, as well as the ω-6/ω-3 ratio were not significantly different in epeleuton-treated SCD mice compared to vehicle-treated SCD or AA mice (Online Supplementary Tables S1 and S2). In contrast, epeleuton caused striking changes in select
oxylipins, increasing 12-HETE and 4-HDHA in lung from SCD when compared with the levels in vehicle-treated SCD mice (Figure 1B). This may have organo-protective effects, as observed in retinal tissue following H/R.28 12-HETE was significantly reduced by epeleuton in liver, which could reflect a blunted response of liver macrophages and/or platelets (which are 12-lipoxygenase-bearing cells)29 and there was a trend towards an increase of liver 4-HDHA (Figure 1C). Furthermore, significantly lower concentrations of leukotriene B4, which is a main driver of neutrophil infiltration, and of the ω-3 docosapentaenoic acid (C22:5 n-3) were measured in liver from epeleuton-treated SCD mice than in vehicle-treated SCD mice (Figure 1C). These results indicate that the changes in SCD lipid profile produced by epeleuton are specific and not related to broad changes in lipid absorption or metabolism.
The transcriptional factor NF- k B is a key regulator of inflammation, and of the expression of genes involved in initiation of inflammation with cell-specific actions such as the modulation of vascular pro-adhesion molecules, including vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1).6 We previously found that ω -3 fatty acid administration to SCD mice prevented the activation of NF- k B p65 in target organs for SCD, such as lung, kidney, and liver.15 As shown in Figure 2A-C, the activation of NF- k B was significantly reduced in lungs, kidneys, and livers from humanized SCD mice treated with epeleuton compared to the activation in SCD mice treated with vehicle (Online Supplementary Figure S4A). Consistent with these observations, in lungs, kidneys, and livers from epeleuton-treated SCD mice we found significant reductions in the expression of VCAM-1 and ICAM-1, markers of inflammatory vasculopathy and endothelial activation, and in the expression of endothelin-1 (ET-1) a potent vaso- and broncho-constrictive cytokine involved in SCD-related organ damage6 (Figure 2D, E; Online Supplementary Figures S4B and S5A). Livers from epeleuton-treated mice also displayed a marked reduction in liver protein oxidation, reaching values similar to those of healthy mice (Figure 2F, G; Online Supplementary Figure S5B).
Collectively our data indicate that epeleuton diminishes local sickle cell-related inflammatory responses and vascular endothelial activation in SCD-target organs.
Epeleuton protects against hypoxia/reoxygenationinduced hemolysis and re-programs spleen macrophages towards a pro-resolving pattern in mice with sickle cell disease
We tested epeleuton in sickle cell mice exposed to H/R stress, which is an experimental model of sickle cell-related VOC.15,30 As shown in Figure 3A, the administration of epeleuton to SCD mice prevented the H/R-induced reduction in hematocrit and hemoglobin and decreased the amount of sickled red cells (Online Supplementary Figure
S6A). This suggests that in SCD mice epeleuton protects red cells against H/R-induced stress. The improvement of red cell membrane features was also supported by a reduction in the tyrosine phosphorylation state of band 3, an integral red cell membrane protein involved in membrane stability and release of erythroid microparticles31 (Figure 3C, Online Supplementary Figure S6B), and the reduction in lactate dehydrogenase, a known marker of hemolysis (Figure 2D). In SCD mice epeleuton significantly reduced the H/R-induced increase in neutrophil counts (Figure 3E). This was associated
with lower plasma C-reactive protein and pentraxin-2 levels (Online Supplementary Figure S6C) in epeleuton-treated SCD mice than in vehicle-treated mice. We also found a significant decrease of spleen neutrophils in epeleuton-treated SCD mice exposed to H/R compared to the counts in vehicle-treated SS animals (Online Supplementary Figure S6D). This was associated with a significant reduction in classic spleen macrophage inflammatory markers (CD80, CD68) (Figure 3F) and a trend towards a reduction in the clearance of damaged red blood cells in SCD mice treated with
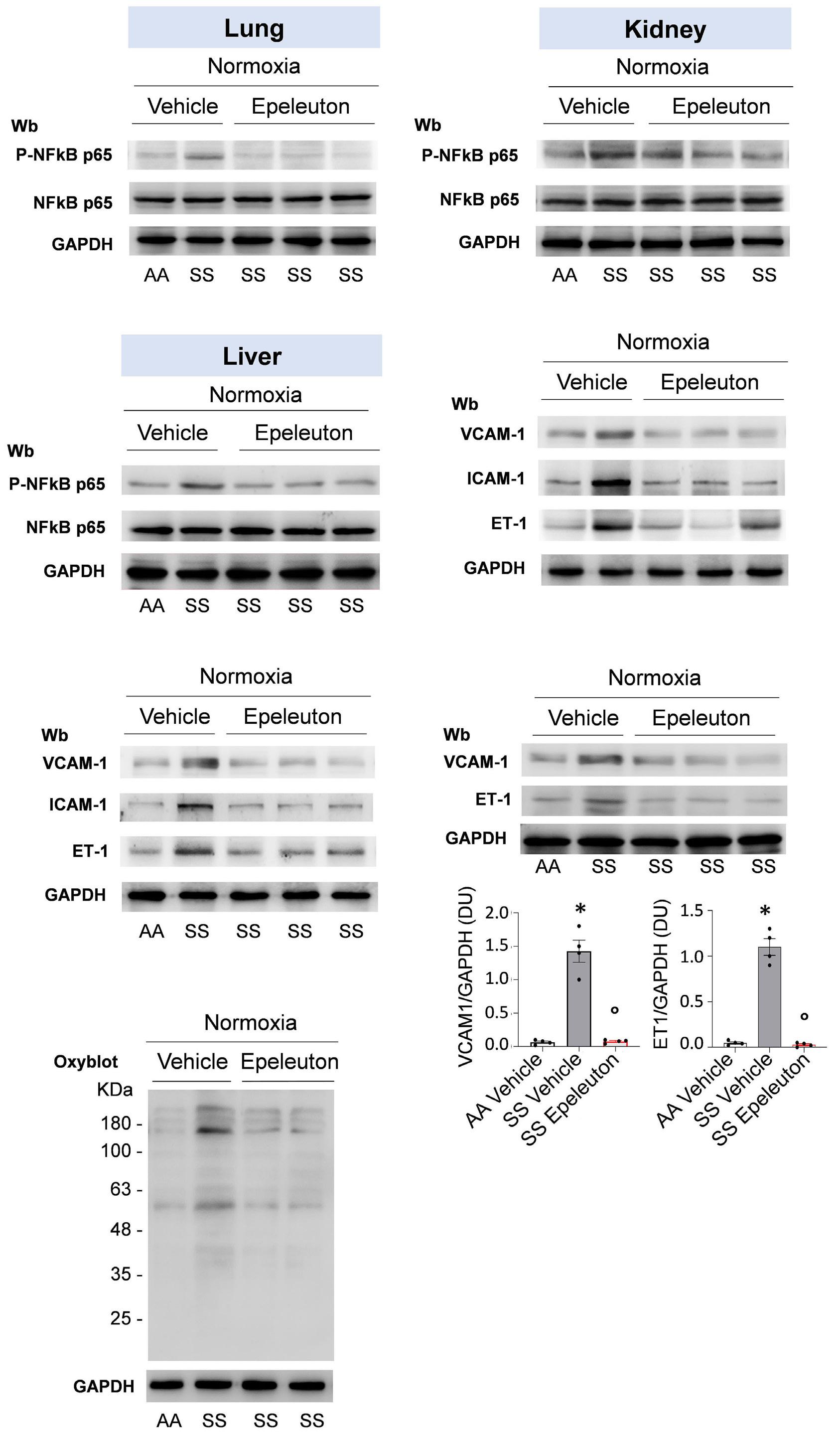
Figure 2. Epeleuton modulates inflammatory response with downregulation of markers of inflammatory vasculopathy. (A-C) Immunoblot analyses using specific antibodies against phosphorylated (p-) NF-kB p65 and NF-kB p65 in lung (A), kidney (B) and liver (C) from healthy mice (AA) and sickle cell disease (SCD) mice (SS) under normoxic conditions treated with either vehicle or epeleuton (1,000 mg/ kg/day for 6 weeks). Lane 1: vehicle-treated AA mouse; lane 2: vehicle-treated SS mouse; lanes 3-5: epeleuton-treated SCD mice (results for 3 separate animals are shown). Protein (75 µg) was loaded on an 8% T, 2.5% C polyacrylamide gel. One representative gel from four with similar results is shown. Densitometric analysis of the immunoblots is shown in Online Supplementary Figure S4A. (D, E) Immunoblot analysis using specific antibodies against VCAM-1, ICAM-1, and ET-1 in lung (D) and kidney (E) from AA and SCD mice treated as in (A). Lane 1: vehicle-treated AA mouse; lane 2: vehicle-treated SS mouse; lanes 3-5: epeleuton-treated SCD mice (results for 3 separate animals are shown). Protein (75 µg) was loaded on an 11% T, 2.5% C polyacrylamide gel. One representative gel from four with similar results is shown. Densitometric analysis of immunoblots is shown in Online Supplementary Figures S4B and S5A. (F) Upper panel. Immunoblot analysis using specific antibodies against VCAM-1 and ET-1 in liver from AA and SCD mice treated as in (A). Lane 1: vehicle-treated AA mouse; lane 2: vehicle-treated SS mouse; lanes 3-5: epeleuton-treated SCD mice (results for 3 separate animals are shown). Protein (50 µg/µL) was loaded on an 8% T, 2.5% C polyacrylamide gel. One representative gel from four with similar results is shown. Lower panel. Densitometric analysis of the immunoblots. Data are presented as means ± standard error of mean (N=4); *P<0.05 compared to AA mice; °P<0.05 compared to vehicle by one-way analysis of variance. (G) OxyBlot analysis of the soluble fractions of liver from AA and SCD mice treated as in (B). Lane 1: vehicle-treated AA mouse; lane 2: vehicle-treated SS mouse; lanes 3-5: epeleuton-treated SCD mice (results from 2 separate animals are shown). The carbonylated proteins (1 mg) were detected by treatment with 2,4-dinitrophenylhydrazine (DNP) and blotted with anti-DNP antibody. Quantification of band area is shown in Online Supplementary Figure S5B. GAPDH served as the protein loading control (A-G). Wb: western blot; NFkB: nuclear factor kB; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; VCAM-1: vascular cell adhesion molecule 1; ICAM-1: intercellular adhesion molecule 1; ET-1: endothelin-1; DU: densitometric units.
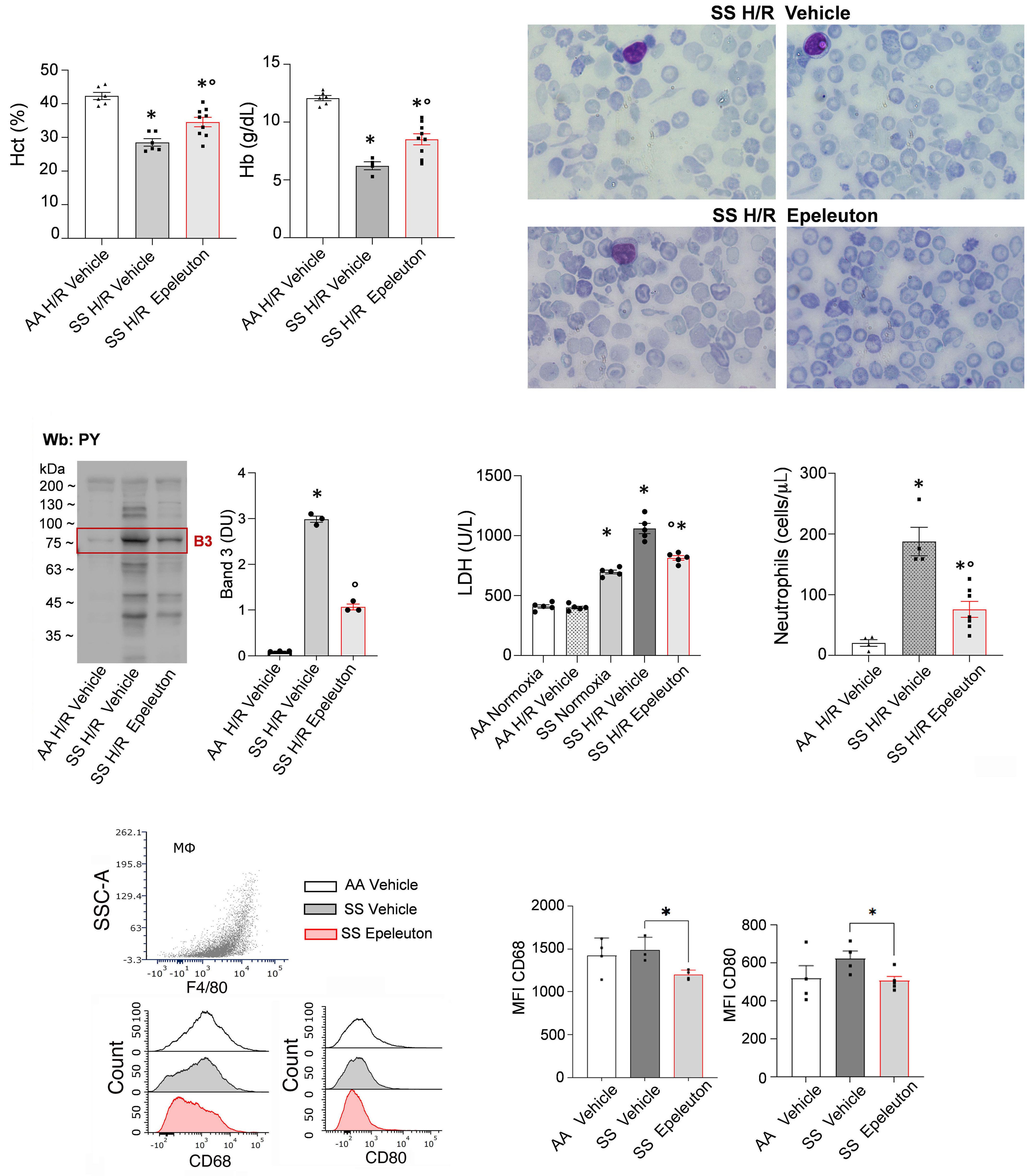
Figure 3. In sickle cell disease mice exposed to hypoxia/reoxygenation stress, epeleuton reduces the stress-induced hemolysis, modulates the inflammatory response, and reprograms spleen macrophages towards a pro-resolving pattern. (A) Hematocrit (left panel) and hemoglobin (right panel) in healthy mice (AA) and sickle cell disease (SCD) mice (SS) exposed to hypoxia/reoxygenation (H/R): stress, hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours), treated with either vehicle or epeleuton (1,000 mg/kg/day for 6 weeks). Data are presented as means ± standard error of mean (SEM) (N=4-9). *P<0.05 compared to AA mice; °P<0.05 compared to vehicle-treated mice by an unpaired t test with Bonferroni correction. (B) Erythrocyte morphology in a blood smear of SCD mice treated as in (A). One representative image is shown. Orig-
Continued on following page.
inal magnification 100×. Quantification is shown in Online Supplementary Figure S6A. (C) Immunoblot analysis using specific anti-phospho-tyrosine antibody of red cell membrane proteins from mice treated as in (A). Lane 1: vehicle-treated AA mouse exposed to H/R; lane 2: vehicle-treated SCD mouse exposed to H/R; lane 3: epeleuton-treated SS mouse exposed to H/R. Proteins (75 µ g) were loaded on an 8% T, 2.5% C polyacrylamide gel (see Online Supplementary Figure S6B for Coomassie staining, used as the loading control). Densitometric analysis of the phosphorylation of band-3 is shown on the right. Data are presented as means ± standard error of mean (SEM) (N=3). *P<0.05 compared to AA mice; °P<0.05 compared to vehicle-treated mice by an unpaired t test with the Bonferroni correction. (D) Plasma concentrations of lactate dehydrogenase in AA and SCD mice under conditions of normoxia or exposed to H/R and treated as in (A). Data are mean ± SEM (N=5). *P<0.05 compared to AA mice; °P<0.05 compared to vehicle-treated mice by one-way analysis of variance (ANOVA). (E) Circulating neutrophils identified by flow cytometric analysis as CD45+Ly6G+ cells in mice treated as in (A). Data are mean ± SEM (N=4-7), *P<0.05 compared to AA mice; °P<0.05 compared to vehicle-treated mice by one-way ANOVA. (F) Flow cytometry gating strategy and representative plots of spleen macrophages from AA or SCD mice treated with vehicle or epeleuton. M1 marker expression on spleen macrophages and red blood cell clearance from AA or SS mice fed with epeleuton, as determined by flow cytometry in F4/80+ cells. Results are means ± standard deviation (N=4 mice/group); *P<0.05 (one-way ANOVA). Hct: hematocrit; Hb: hemoglobin; Wb: western blot; PY: phosphotyrosine; B3: band 3; DU: densitometric units; LDH: lactate dehydrogenase; SSC: side scatter; MFI: mean fluorescence intensity.
epeleuton compared with that in mice treated with vehicle (Online Supplementary Figure S6E). It is noteworthy that the effects of epeleuton on neutrophils and macrophage re-programing in SCD mice are consistent with the pro-resolving actions we previously established for 17R-RvD1 in SCD mice.6
Taken together, these data indicate that epeleuton protects against H/R-induced stress, attenuating inflammation, and leading to early initiation of resolution events.
Epeleuton reduces lung injury and prevents the overactivation of nuclear factor kB and hypoxia-induced lung inflammatory vasculopathy in mice with sickle cell disease
In SCD mice treated with epeleuton, we observed a trend towards a reduction in lung inflammatory cell infiltrates compared to those in SCD mice treated with the vehicle (Figure 4A, Online Supplementary Table S3). Consistent with these observations, we found a significant decrease in the active forms of NF- k B p65 induced by H/R in SCD mice treated with epeleuton (Figure 4B). To assess whether epeleuton was protective against H/R-induced inflammation and vascular dysfunction, we evaluated lung expression of: (i) the NLRP3 inflammasome, a key player in sterile inflammation; (ii) vascular endothelial activation; and (iii) neutrophil vascular recruitment.6 As shown in Figure 4C, in lung tissue from epeleuton-treated SCD mice exposed to H/R, we found downregulation of the NLRP3 inflammasome, which has been reported to participate in inflammation in models of acute lung injury such as acute respiratory disease syndrome and asthma.32 In addition, epeleuton reduced the expression of VCAM-1, E-selectin, and thromboxane synthase 1 (TXAS) (Figure 4C). TXAS is controlled by the NF-kB p65 signaling pathway, and it has been linked to activation of both vascular endothelial cells and platelets in other models of ischemia-reperfusion damage.6,33,34
These data provide evidence that epeleuton protects SCD mice against H/R-induced lung injury by modulation of NF- k B and key mediators of vascular damage.
Epeleuton diminishes hypoxia/reoxygenation-induced kidney damage and markers of vascular dysfunction in mice with sickle cell disease
Histopathological analysis of kidney tissue from SCD mice exposed to H/R revealed no major effect of epeleuton on kidney inflammatory cell infiltrates in hematoxylin and eosin-stained preparations (Figure 5A, Online Supplementary Table S3), whereas we found a significant reduction in total leukocyte infiltrates, as determined by flow-cytometric analysis, in kidney from epeleuton-treated SCD mice compared to vehicle-treated animals (Figure 5B). We also observed a reduction of H/R-induced increases in plasma creatinine and blood urea nitrogen levels in the epeleuton-treated SCD mice compared to the levels in the vehicle-treated SCD animals (Figure 5C). This is consistent with the decreased H/R-induced activation of NF-kB p65 in kidneys from epeleuton-treated SCD mice compared to activation in vehicle-treated animals (Figure 5D). The beneficial effects of epeleuton in SCD mice were further supported by the reduction in H/R-induced increase in expression of NLRP3, VCAM-1, ET-1, and TXAS-1, which are involved in vascular activation, reduction of kidney vascular tone, and inflammation (Figure 5E, Online Supplementary Figure S7).
Taken together our data indicate that epeleuton mitigates H/R-induced acute kidney damage and modulates the related amplified inflammation and vascular dysfunction.
In sickle cell disease mice exposed to hypoxia/ reoxygenation stress, epeleuton reduces liver injury and prevents the overactivation of inflammatory and redoxrelated pathways
Compared to vehicle-treated mice, SCD mice treated with epeleuton and exposed to H/R stress had fewer inflammatory cell infiltrates and a significant reduction in thrombi formation (Figure 6A). In addition, we found a significant reduction in liver iron accumulation, mainly characterized by decreased iron accumulation in hepatocytes of SCD mice treated with epeleuton and exposed to H/R stress (Figure 6A, lower panel; Online Supplementary Figure S8A).
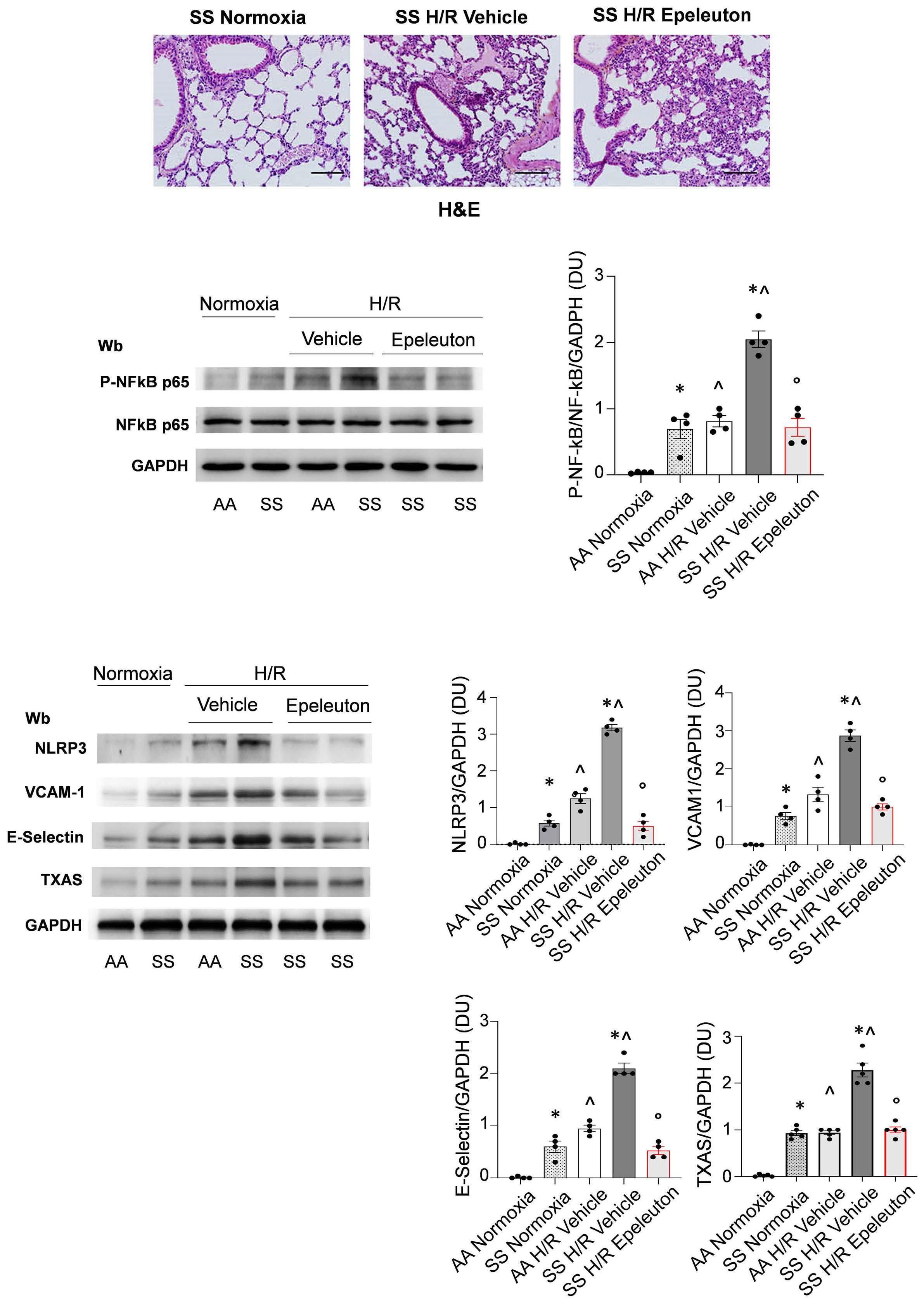
Figure 4. Epeleuton reduces lung injury, preventing the overactivation of nuclear factor kB and hypoxia-induced lung inflammatory vasculopathy in mice with sickle cell disease. (A) Representative micro-picture of hematoxylin and eosin-stained sections of lung at 200x magnification from sickle cell disease (SCD) mice (SS) in conditions of normoxia and exposed to hypoxia/reoxygenation (H/R),hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours), treated with either vehicle or epeleuton (1,000 mg/kg/day for 6 weeks) (scale bar: 50 μm) (see also Online Supplementary Table S3). (B) Left panel. Immunoblot
Continued on following page.
analysis using specific antibodies against phosphorylated (p-)NF-kB p65 and NF-kB p65 in lung from AA and SS mice treated as (A). Lane 1: AA mouse under normoxia; lane 2: SS mouse under normoxia; lane 3: vehicle-treated AA mouse exposed to H/R; lane 4; vehicle-treated SS mouse exposed to H/R; lanes 5 and 6: epeleuton-treated SS mice exposed to H/R (results from 2 separate animals are shown). Protein (75 µg) was loaded on an 8% T, 2.5% C polyacrylamide gel. GAPDH served as the protein loading control. One representative gel from four with similar results is shown. Right panel. Densitometric analysis of the immunoblots. Data are presented as means ± standard error of mean (SEM) (N=4). *P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way analysis of variance (ANOVA). (C) Left panel. Immunoblot analysis using specific antibodies against NLRP3, VCAM-1, E-selectin and TXAS in lung from AA and SS mice treated as in (A). Lane 1: AA mouse under normoxia; lane 2: SS mouse under normoxia; lane 3: vehicle-treated AA mouse exposed to H/R; lane 4: vehicle-treated SS mouse exposed to H/R; lanes 5 and 6: epeleuton-treated SS mice exposed to H/R (results for 2 separate animals are shown). Protein (75 µg) was loaded on an 8% T, 2.5% C polyacrylamide gel. GAPDH served as the protein loading control. One representative gel from four with similar results is shown. Right panels. Densitometric analyses of the immunoblot. Data are presented as means ± SEM (N=4). *P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way ANOVA. H&E: hematoxylin and eosin; Wb: western blot; NFkB: nuclear factor kB; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; DU: densitometric units; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; VCAM-1: vascular cell adhesion molecule 1; TXAS: thromboxane synthase.
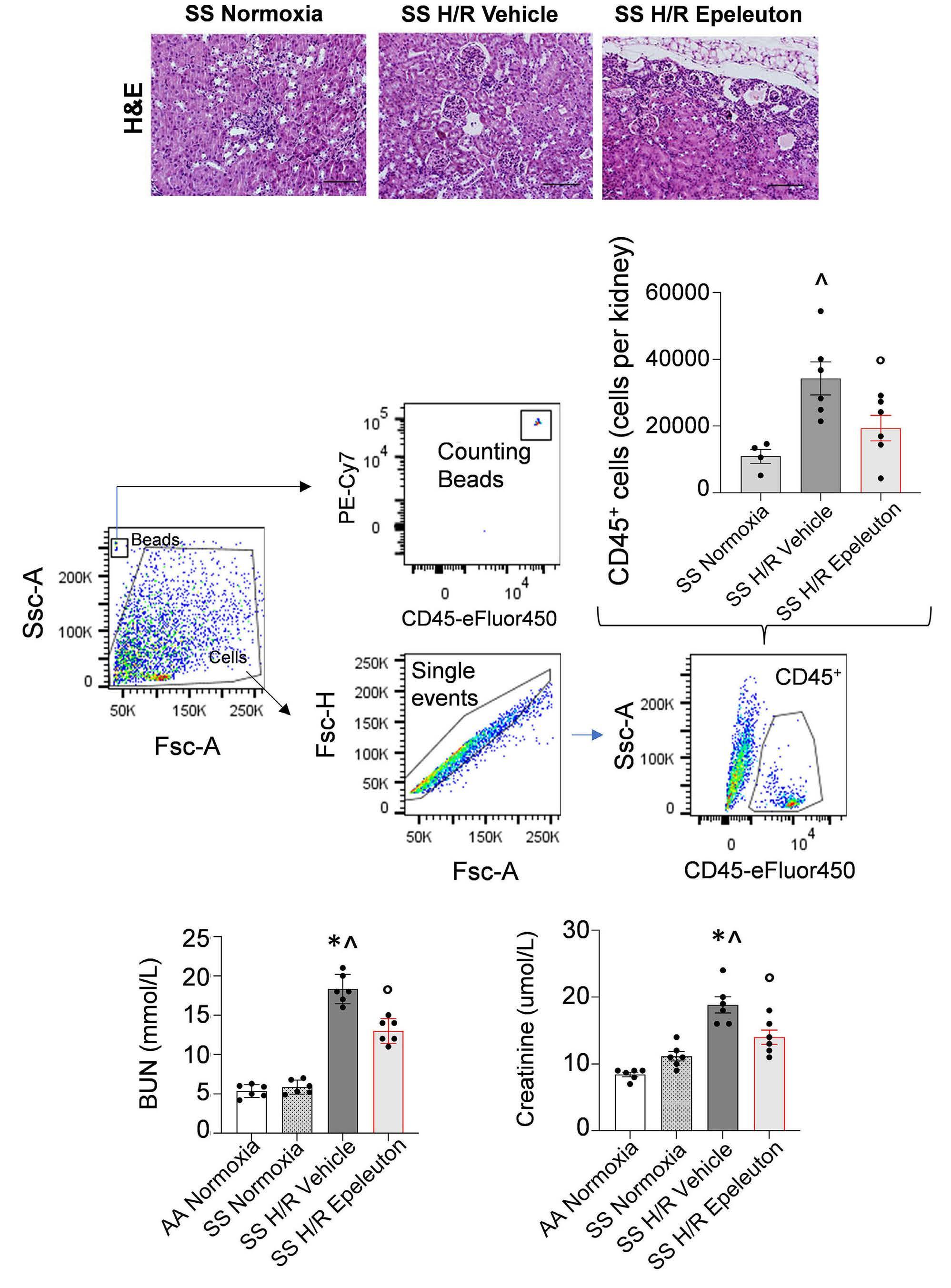
Continued on following page.
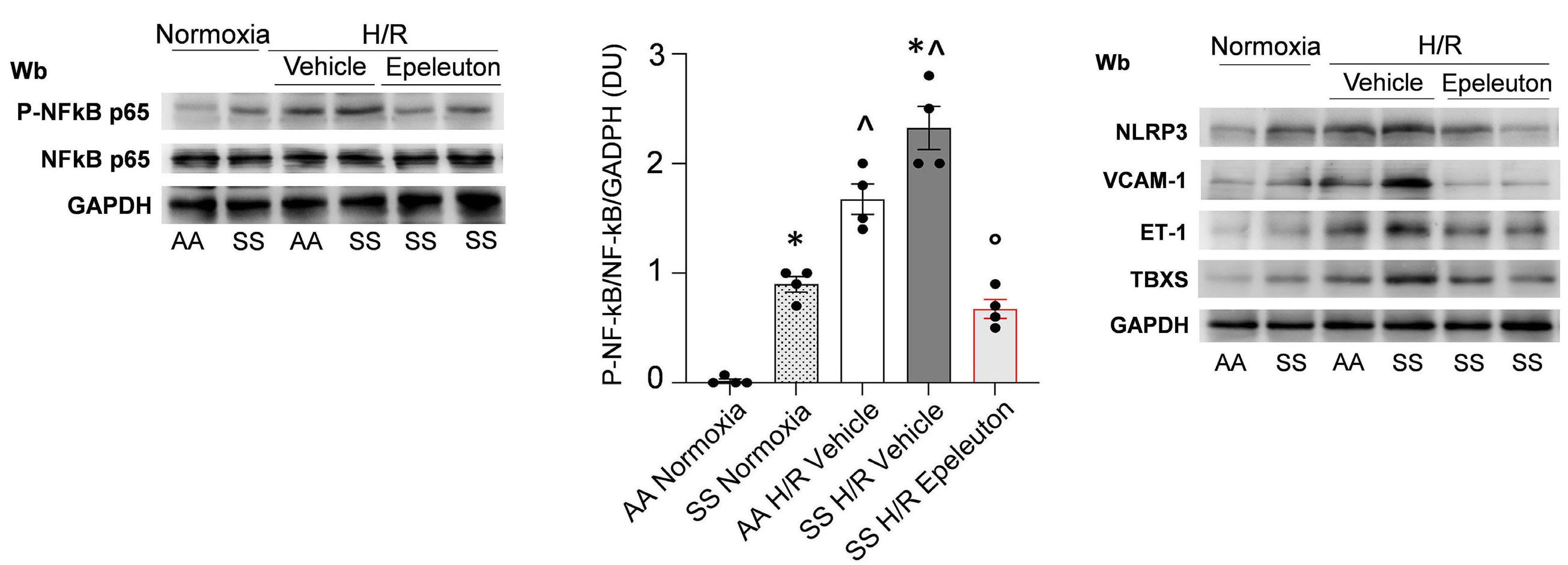
Figure 5. Epeleuton diminishes hypoxia/reoxygenation-induced kidney damage and markers of vascular dysfunction in mice with sickle cell disease. (A) Representative micro-pictures of hematoxylin and eosin-stained sections of kidney at 200x magnification from sickle cell disease (SCD) mice (SS) exposed to hypoxia/reoxygenation (H/R): hypoxia (8% oxygen; 10 hours), followed by reoxygenation (21% oxygen; 3 hours) treated with either vehicle or epeleuton (1,000 mg/kg/day for 6 weeks) (scale bar: 50 μm) (see also Online Supplementary Table S3). (B) Kidney leukocyte infiltrates determined by flow cytometry gating analysis (the gating strategy is shown in Online Supplementary Figure S8). Data are presented as means ± standard error of mean (SEM) (N=4-6). ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by an unpaired t test with Bonferroni correction. (C) Plasma blood urea nitrogen (left panel) and creatinine (right panel) in healthy (AA) and SCD (SS) mice under conditions of normoxia or exposed to H/R as in (A). Data are means ± SEM (N=6). *P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way analysis of variance (ANOVA). (D) Left panel. Immunoblot analysis using specific antibodies against phosphorylated (p-) NF-kB p65 and NF-kB p65 in kidney from AA and SS mice treated as in (A). Lane 1: AA mouse under normoxia; lane 2: SS mouse under normoxia; lane 3: vehicle-treated AA mouse exposed to H/R; lane 4: vehicle-treated SS mouse exposed to H/R; lanes 5 and 6: epeleuton-treated SS mice exposed to H/R (results from 2 separate animals are shown). Protein (75 μg) was loaded on an 8% T, 2.5% C polyacrylamide gel. One representative gel from four with similar results is shown. Right panel. Densitometric analysis of the immunoblots. Data are presented as means ± SEM (N=4). *P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way ANOVA. (E) Immunoblot analysis, using specific antibodies against NLRP3, VCAM-1, ET-1 and TBXS in kidney from AA and SS mice treated as in (A). Lane 1: AA mouse under normoxia; lane 2: SS mouse under normoxia; lane 3: vehicle-treated AA mouse exposed to H/R; lane 4: vehicle-treated SS mouse exposed to H/R; lanes 5 and 6: epeleuton-treated SS mice exposed to H/R (results for 2 separate animals are shown). Protein (75 µg) was loaded on an 11% T, 2.5% C polyacrylamide gel. One representative gel from four with similar results is shown. Densitometric analysis of immunoblots is shown on the right. Data are presented as means ± SEM (N=4). *P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way ANOVA. GAPDH served as the protein loading control (D, E). H&E: hematoxylin and eosin; BUN: blood urea nitrogen; Wb: western blot; NFkB: nuclear factor kB; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; DU: densitometric units; Ssc: side scatter; Fsc: forward scatter; PE-Cy7: phycoerythrin cyanine 7; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; VCAM-1: vascular cell adhesion molecule 1; ET1: endothelin 1; TBXS: thromboxane synthase.
In agreement with modulation of local inflammatory response and oxidation, we found reduced activation of both acute phase inflammatory and redox-related transcriptional factors NF-kB p65 and Nrf2 in epeleuton-treated SCD mice exposed to H/R (Figure 6B). This was associated with a marked reduction in liver protein oxidation (Online Supplementary Figure S8B) and a consistent downregulation of the antioxidants NQO1 and HO-1, which are regulated by Nfr235 (Online Supplementary Figure S8C). We then explored the effects of epeleuton on markers of inflammatory response and vascular activation. As shown in Figure 6C, we found lower expression of NLRP3 in livers from epeleuton-treated SCD mice exposed to H/R. Similar NLRP3 inflammasome activation has been reported in models of liver diseases, such as chronic hepatitis B or C and non-alcoholic steatohepatitis.36 In addition, epeleuton prevented the H/R-induced upregulation of VCAM-1 in liv-
ers from SCD mice exposed to H/R stress, while we found a trend towards a reduction of ET-1 in the same group of mice when compared to vehicle-treated animals (Figure 6C).
Epeleuton protects against progression of inflammatory vasculopathy related to acute hypoxia/reoxygenation stress in mice with sickle cell disease
Given the beneficial effects of epeleuton on vascular dysfunction in SCD target organs and of fatty acid supplementation on different models of vascular dysfunction and inflammatory vasculopathy,6,15,16 we evaluated the impact of epeleuton treatment on sickle cell-related inflammatory vasculopathy.
In isolated aorta from SCD mice under normoxia, epeleuton significantly downregulated the expression of ICAM-1 and ET- 1 but not of VCAM-1 compared to the expression in vehicle-treated SCD mice (Online Supplementary Figure S9)
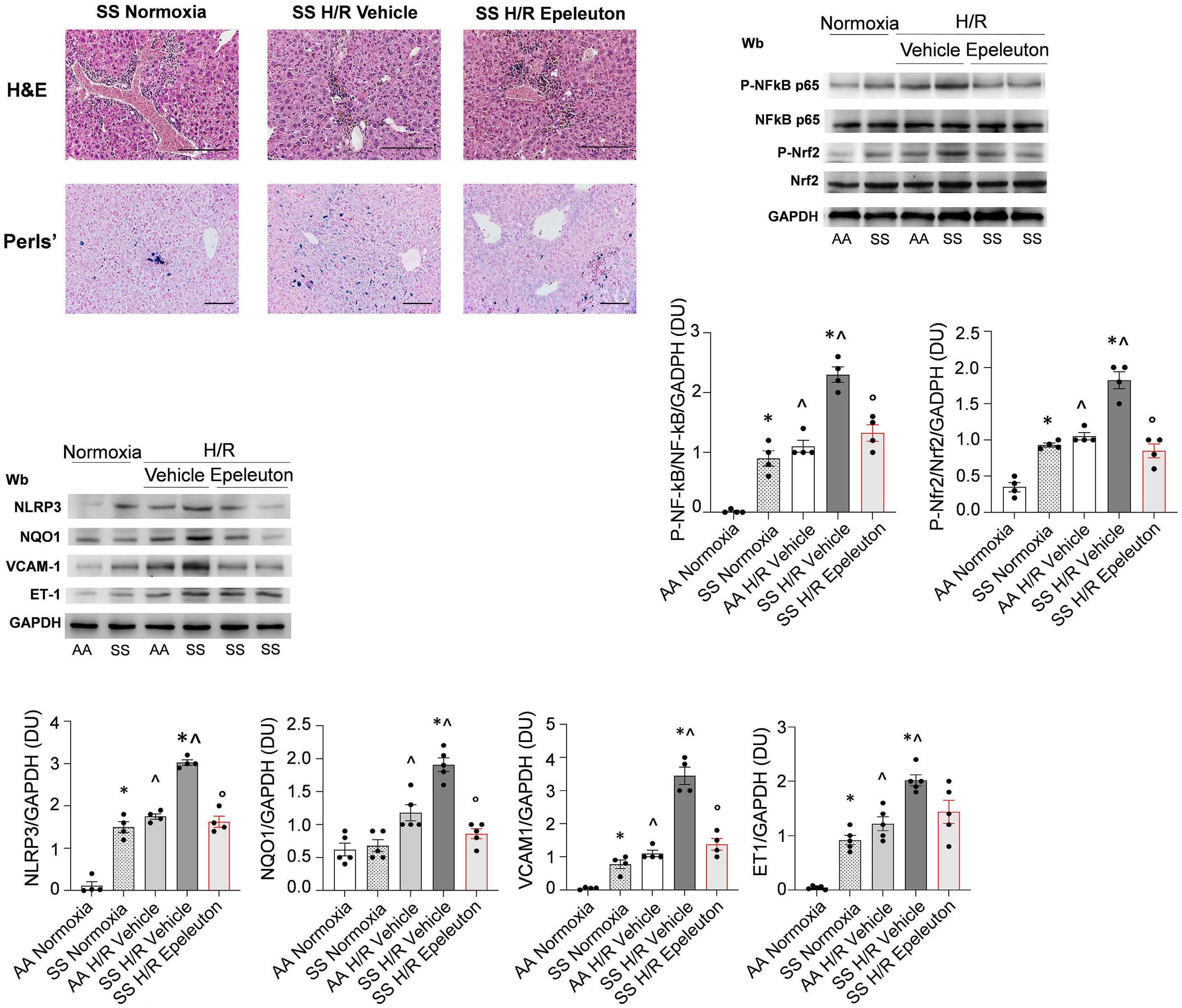
Figure 6. In sickle cell disease mice exposed to hypoxia/reoxygenation stress, epeleuton reduces liver injury and prevents the overactivation of inflammatory and redox-related pathways. (A) Representative micro-picture of hematoxylin and eosin-stained and Perls-stained sections of liver at 200x magnification from sickle cell disease (SCD) mice (SS) exposed to hypoxia/reoxygenation (H/R), hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours), treated with either vehicle or epeleuton (1,000 mg/kg/day for 6 weeks) (scale bar: 50 µm) (see also Online Supplementary Table S3). (B) Immunoblot analysis using specific antibodies against phosphorylated (p-)NF-kB p65, NF-kB p65, p-Nrf2, and Nrf2 in liver from normal (AA) and SS mice as in (A). Lane 1: AA mouse under normoxia; lane 2: SS mouse under normoxia; lane 3: vehicle-treated AA mouse exposed to H/R; lane 4: vehicle-treated SS mouse exposed to H/R; lanes 5 and 6: epeleuton-treated SS mice exposed to H/R (results from 2 separate animals are shown). Protein (75 μg) was loaded on an 8% T, 2.5% C polyacrylamide gel. One representative gel from four with similar results is shown. Densitometric analyses of the immunoblots are shown in the panels below. Data are presented as means ± standard error of mean (SEM) (N=4).*P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way analysis of variance (ANOVA). (C) Immunoblot analysis, using specific antibodies against NLRP3, NQO1, VCAM-1 and ET-1 in liver from AA and SS mice treated as in (A). Lane 1: AA mouse under normoxia; lane 2: SS mouse under normoxia; lane 3: vehicle-treated AA mouse exposed to H/R; lane 4: vehicle-treated SS mouse exposed to H/R; lanes 5 and 6: epeleuton-treated SS mice exposed to H/R (results from 2 separate animals are shown). Protein (75 µg) was loaded on an 11% T, 2.5% C polyacrylamide gel. One representative gel from four or five with similar results is shown. Densitometric analyses of the immunoblots are shown in the panels below. Data are presented as means ± SEM (N=4/5). *P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way ANOVA. GAPDH served as the protein loading control (B, C). H&E: hematoxylin and eosin; Wb: western blot; NFkB: nuclear factor kB; Nrf2: nuclear factor erythroid 2-related factor 2; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; DU: densitometric units; Ssc: side scatter; Fsc: forward scatter; PE-Cy7: phycoerythrin cyanine 7; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; NQO1: NAD(P)H quinone dehydrogenase 1; VCAM1: vascular cell adhesion molecule 1; ET1: endothelin 1.
As shown in Figure 7A, we found a significant reduction in H/R-induced expression of VCAM-1 in epeluton-treated SCD mice compared with vehicle-treated SCD mice. These data indicate that treatment with epeleuton may prevent the worsening of vascular dysfunction induced by H/R stress, with a potential to delay disease progression.
In this study, we show that epeleuton, a novel synthetic ω-3 fatty acid derivative, protects against cellular stress associated with SCD and supports early initiation of the resolution phase of inflammation. The observed effects may limit disease progression and the initiation of acute VOC, which have a negative impact on patients’ quality of life.37 Previous studies have shown the beneficial effects of dietary ω-3 fatty acid supplementation on SCD phenotype.16,38,39 How-
ever, the formulation, the route of administration and the bioavailability of ω-3 fatty acids influence the use of such supplementation in clinical practice for patients with SCD. Epeleuton has an advantageous functional profile compared to that of other formulations of ω-3 fatty acids tested in SCD such as purified ω-3 formulations including Lovaza™ (ω-3 acid ethyl esters) or SC411 and fish oils.15,39,40 Lovaza™ is a prescription esterified ω-3-acid mixture (~55.1% EPA, ~ 44.9% DHA), while SC411 is a DHA ethyl ester formulation with minimal food-drug interaction. As a second-generation ω-3 enzymatic derivative that is metabolically downstream of EPA, epeleuton has advantages as a therapeutic for SCD. From biochemical and pharmacological perspectives, therapeutics on downstream purified ω-3 derivatives are likely to be more rapidly and potently bioactive per se or upon further enzymatic reactions. The advantage conferred in SCD from bypassing the initial stages of metabolism of fatty acids such as EPA and DHA is highlighted by the previous
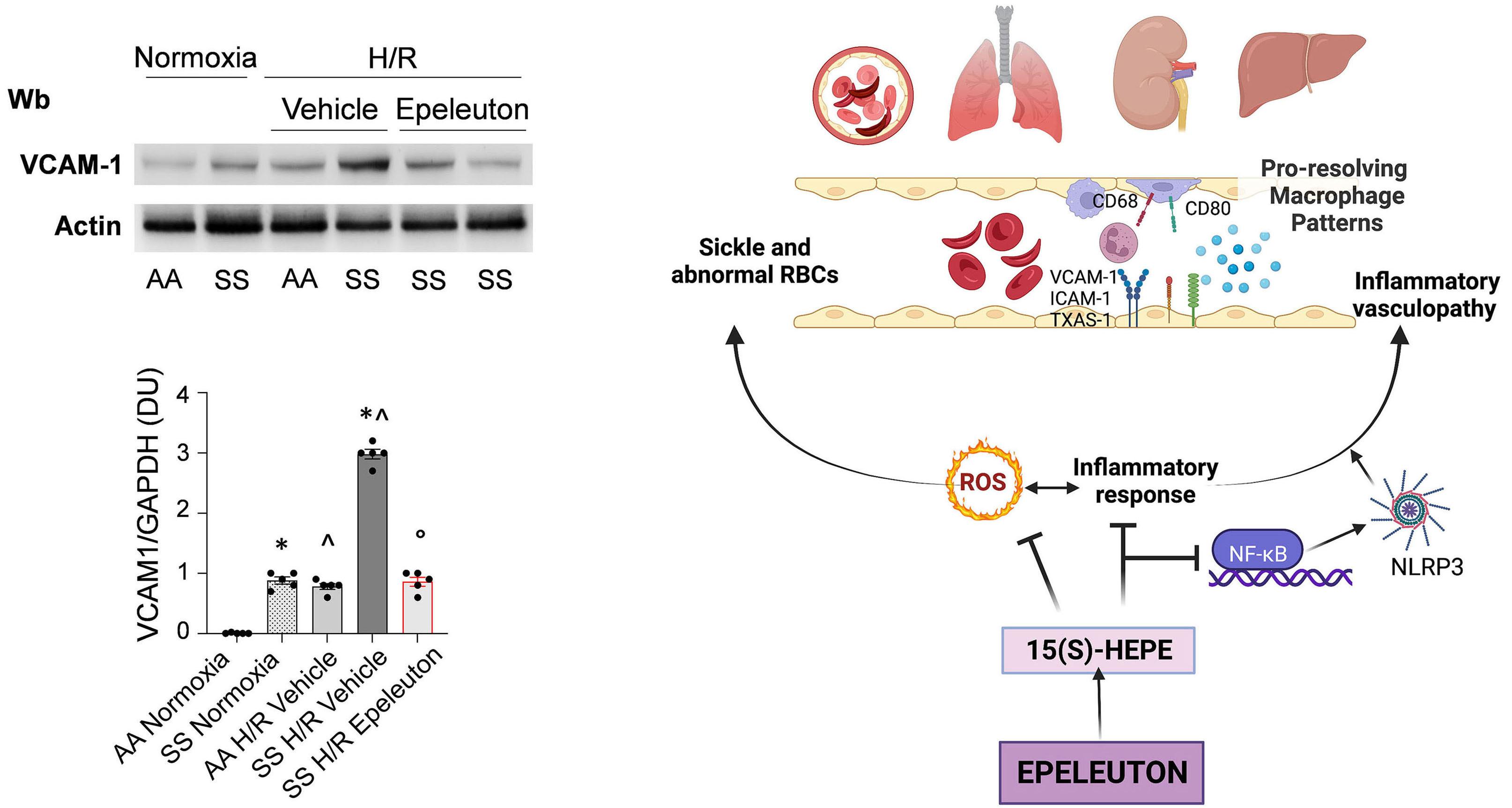
Figure 7. Epeleuton protects against progression of inflammatory vasculopathy related to acute hypoxia/reoxygenation stress in mice with sickle cell disease. (A) Immunoblot analysis, using specific antibodies against VCAM-1 (40 μg of protein loaded on an 8% T, 2.5% C polyacrylamide gel) in isolated aorta from healthy mice (AA) and sickle cell disease (SCD) mice (SS) under normoxia or exposed to hypoxia/reoxygenation (H/R) treated with either vehicle or epeleuton (1,000 mg/kg/day for 6 weeks). Lane 1: AA mouse under normoxia; lane 2: SS mouse under normoxia; lane 3: vehicle-treated AA mouse exposed to H/R; lane 4: vehicle-treated SS mouse exposed to H/R; lanes 5 and 6: epeleuton-treated SS mice exposed to H/R (results from 2 separate animals are shown). Actin served as the protein loading control. One representative gel from five with similar results is shown. Densitometric analysis of immunoblots is shown in the lower panel. Data are presented as means ± standard error of mean (N=5). *P<0.05 compared to AA mice; ^P<0.05 compared to normoxia; °P<0.05 compared to vehicle by one-way analysis of variance. (B) Schematic diagram of the dual anti-inflammatory and pro-resolution effects of epeleuton in the humanized mouse model of SCD. Epeleuton and its active moiety 15(S)-HEPE favor pro-resolving mechanisms targeting inflammation, the reactive oxygen species burst, NF-kB activation and NLRP3 inflammasome expression. This results in prevention of red blood cell sickling, inflammatory vasculopathy reduction and macrophages pro-resolving reprogramming (CD68 and CD80) in target organs for SCD. Wb: western blot; VCAM-1: vascular cell adhesion molecule 1; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; DU: densitometric units; RBC: red blood cells; ICAM-1: intercellular adhesion molecule 1; TXAS-1: thromboxane synthase 1; ROS: reactive oxygen species; NF-kB: nuclear factor kB; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; 15(S)-HEPE: 15(S) hydroxy eicosapentaenoic acid.
finding that humanized SCD mice were not as readily able to synthesize downstream fatty acid metabolites compared to healthy control mice.6 Importantly, 15(S)-HEPE, which has also been identified in resolving exudates from mice treated with ω-3 fatty acids, directly reduces polymorphonuclear leukocyte trans-endothelial migration, as does its metabolite lipoxin A5 at a higher potency.41 In SCD mice, epeleuton might attenuate the inflammatory response by favoring pro-resolving mechanisms mediated by 15(S)-HEPE. In agreement with this notion, we found increased organ content of 15-HEPE and reduction of systemic inflammation, modulation of the CCL2 chemotactic cytokine and a decrease in neutrophil counts in SCD mice treated with epeleuton. This is of particular interest since epeleuton reduces splenic neutrophils and reprograms spleen macrophages towards a pro-resolution pattern, supporting the peculiarity of epeleuton as a source of pro-resolving lipid mediators. The peculiar effect of epeleuton on lung leukotriene B4 content in SCD mice is concordant with a pro-resolving effect of epeleuton, similar to the effects described in the same model treated with exogenous 17R-RvD1.6
The dual anti-inflammatory and pro-resolution effects of epeleuton in SCD mice are highlighted by changes in lipidomic profile and a reduction in NF-kB p65 activation in target organs for SCD such as lung, liver, and kidney (Figures 1, 3, 4, and 5). This is further corroborated by decreased expression of the NLRP3 inflammasome, which activates inflammatory reactions in response to disrupted cell homeostasis in lung, kidney, and liver of SCD mice exposed to H/R stress. Growing evidence in both patients with SCD and cell/animal-based models of SCD support the importance of NLRP3 inflammasome activation in the severity of the clinical manifestations of SCD as well as in monocyte and platelet function/activation.42-44 Although the mechanism of activation of NLRP3 in SCD is still under investigation, oxidation and free heme/hemolysis have been suggested to trigger the NLRP3 inflammasome in SCD.45,46 In this model, epeleuton reduces both oxidation and hemolysis when SCD mice are exposed to H/R stress and prevents the worsening of liver iron overload and the activation of the redox-related transcription factor Nrf2. It is worth noting that previous studies have shown that the NLRP3 inflammasome might favor Nrf2 degradation, affecting total cell antioxidant power, supporting the importance of limiting reactive-oxygen species induced by activation of the NRLP3 inflammasome.47,48 Here, we found that epeleuton protects against NLRP3 inflammasome activation by: (i) preventing the activation of NF-kB p65; (ii) decreasing oxidation and the related activation of Nrf2; and (iii) reducing hemolysis. Indeed, studies in different models of acute or acute-onchronic disease of lung, kidney or liver have previously shown that inhibition of the NLRP3 inflammasome ameliorated disease outcomes.32,36,49 In addition, the observations that ω-3 fatty acids and specialized pro-resolving mediators
prevent NLRP3 inflammasome activation in different models of inflammatory and degenerative disorders further support the beneficial effects of epeleuton on sickle cell-related organ damage.50 In agreement, epeleuton prevented the expression of markers of inflammatory vasculopathy in target organs for SCD, including isolated aorta, providing a rationale for considering epeleuton as a potential, new therapeutic tool for SCD. Finally, the improvement of hemolysis in epeleuton-treated SCD mice exposed to H/R suggests a possible beneficial effect also on red cell deformability, which will be explored in future studies.
The present results support the designation of epeleuton as an orphan drug, as determined by the USA Food and Drug Administration (https://www.accessdata.fda.gov/scripts/ opdlisting/oopd/detailedIndex.cfm?cfgridkey=880022) and the European Medicines Agency (https://www.ema.europa.eu/ en/medicines/human/orphan-designations/eu-3-22-2695).18 In conclusion, we show here in humanized SCD mice that epeleuton acts as a multimodal agent targeting hemolysis, balancing inflammatory response and pro-resolving mechanisms, and vascular dysfunction. Our data support the potential therapeutic utility of epeleuton and provide a rationale for the design of a clinical trial (NCT05861453) to evaluate the efficacy of epeleuton in patients with SCD.
LDF received research funding from Afimmune. MH and DC are Afimmune employees. JC is a shareholder of Afimmune. DLB has sat on advisory boards for Angiowave, Bayer, Boehringer Ingelheim, Cardax, CellProthera, Cereno Scientific, Elsevier Practice Update Cardiology, High Enroll, Janssen, Level Ex, McKinsey, Medscape Cardiology, Merck, MyoKardia, NirvaMed, Novo Nordisk, PhaseBio, PLx Pharma, and Stasys; is a member of the Board of Directors of the American Heart Association New York City; holds stock or stock options with Angiowave (stock options), Bristol Myers Squibb (stock), DRS.LINQ (stock options), and High Enroll (stock); is a consultant for Broadview Ventures and Hims; has participated in data monitoring committees for Acesion Pharma, Assistance Publique-Hôpitaux de Paris, Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute, for the PORTICO trial, funded by St. Jude Medical, now Abbott), Boston Scientific (Chair, PEITHO trial), Cleveland Clinic, Contego Medical (Chair, PERFORMANCE 2), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo; for the ABILITY-DM trial, funded by Concept Medical), Novartis, Population Health Research Institute, Rutgers University (for the NIH-funded MINT trial); has received honoraria from the American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Chair, ACC Accreditation Oversight Committee), Arnold and Porter law firm (work related to Sanofi/Bristol-Myers Squibb clopidogrel litigation), Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute; RE-DUAL PCI
clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor-in-Chief, Harvard Heart Letter), Canadian Medical and Surgical Knowledge Translation Research Group (clinical trial steering committees), CSL Behring (AHA lecture), Cowen and Company, Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (Editor-in-Chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (Guest Editor; Associate Editor), K2P (Co-Chair, interdisciplinary curriculum), Level Ex, Medtelligence/ReachMD (CME steering committees), MJH Life Sciences, Oakstone CME (Course Director, Comprehensive Review of Interventional Cardiology), Piper Sandler, Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee, and USA national co-leader, funded by Bayer), WebMD (CME steering committees), and Wiley (steering committee); Other: Clinical Cardiology (Deputy Editor); is named on a patent for sotagliflozin assigned to Brigham and Women’s Hospital who assigned to Lexicon (neither DLB nor Brigham and Women’s Hospital receives any income from this patent); has received research funding from Abbott, Acesion Pharma, Afimmune, Aker Biomarine, Alnylam, Amarin, Amgen, AstraZeneca, Bayer, Beren, Boehringer Ingelheim, Boston Scientific, Bristol-Myers Squibb, Cardax, CellProthera, Cereno Scientific, Chiesi, CinCor, Cleerly, CSL Behring, Eisai, Ethicon, Faraday Pharmaceuticals, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Garmin, HLS Therapeutics, Idorsia, Ironwood, Ischemix, Janssen, Javelin, Lexicon, Lilly, Medtronic, Merck, Moderna, MyoKardia, NirvaMed, Novartis, Novo Nordisk, Otsuka, Owkin, Pfizer, PhaseBio, PLx Pharma, Recardio, Regeneron, Reid Hoffman
1. Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762-769.
2. Hebbel RP. The systems biology-based argument for taking a bold step in chemoprophylaxis of sickle vasculopathy. Am J Hematol. 2009;84(9):543-545.
3. Telen MJ, Malik P, Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discov. 2019;18(2):139-158.
4. Fredman G, Hellmann J, Proto JD, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859.
5. Serhan CN, Jain A, Marleau S, et al. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171(12):6856-6865.
6. Matte A, Recchiuti A, Federti E, et al. Resolution of sickle cell disease-associated inflammation and tissue damage with 17R-resolvin D1. Blood. 2019;133(3):252-265.
7 Setty BNY, Betal SG, Miller RE, et al. Relationship of omega-3 fatty acids DHA and EPA with the inflammatory biomarker hs-CRP in children with sickle cell anemia. Prostaglandins
Foundation, Roche, Sanofi, Stasys, Synaptic, The Medicines Company, Youngene, and 89Bio; has received royalties from Elsevier (Editor, Braunwald’s Heart Disease); is or has been a site co-investigator for Abbott, Biotronik, Boston Scientific, CSI, Endotronix, St. Jude Medical (now Abbott), Philips, SpectraWAVE, Svelte, and Vascular Solutions; is a trustee for the American College of Cardiology; and has performed unfunded research for FlowCo. CB has provided consultancy services for Afimmune. The other authors have no conflicts of interest to disclose.
Contributions
AM, EF contributed to the experimental design, carried out experiments, and analyzed data. AS, VR and AP carried out immunoblot analyses. JC, FM, EG, JC and DLB critically reviewed the paper. DC and MH discussed the experimental design and data and critically reviewed the data. AR and GF analyzed lipidomic data. CB and LDF designed the experiments, analyzed data and wrote the paper.
We thank Dr. Barbara Gianesin, from the For Anemia Foundation for biostatistical consultation.
Funding
This study was supported by Afimmune Ltd. under a research collaborative grant to LDF.
Data-sharing statement
All the data and protocols are stored in the Nas Synology DS216se Hard Disk, located at the University of Verona, Italy and are available on request.
Leukot Essent Fatty Acids. 2019;146:11-18.
8. Ren H, Okpala I, Ghebremeskel K, et al. Blood mononuclear cells and platelets have abnormal fatty acid composition in homozygous sickle cell disease. Ann Hematol. 2005;84(9):578-583.
9 Ren H, Obike I, Okpala I, et al. Steady-state haemoglobin level in sickle cell anaemia increases with an increase in erythrocyte membrane n-3 fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2005;72(6):415-421.
10. Ren H, Ghebremeskel K, Okpala I, et al. Abnormality of erythrocyte membrane n-3 long chain polyunsaturated fatty acids in sickle cell haemoglobin C (HbSC) disease is not as remarkable as in sickle cell anaemia (HbSS). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):1-6.
11. Enomoto TM, Isichei C, VanderJagt DJ, Fry DE, Glew RH. Decreased polyunsaturated fatty acids in sickle cell anaemia. J Trop Pediatr. 1998;44(1):28-34.
12. VanderJagt DJ, Trujillo MR, Bode-Thomas F, et al. Phase angle correlates with n-3 fatty acids and cholesterol in red cells of Nigerian children with sickle cell disease. Lipids Health Dis. 2003;2:2.
13. Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2(3):355-374.
14 Rangel-Huerta OD, Aguilera CM, Mesa MD, Gil A. Omega-3 long-chain polyunsaturated fatty acids supplementation on inflammatory biomakers: a systematic review of randomised clinical trials. Br J Nutr. 2012;107 Suppl 2:S159-170.
15. Kalish BT, Matte A, Andolfo I, et al. Dietary omega-3 fatty acids protect against vasculopathy in a transgenic mouse model of sickle cell disease. Haematologica. 2015;100(7):870-880.
16. Daak A, Rabinowicz A, Ghebremeskel K. Omega-3 fatty acids are a potential therapy for patients with sickle cell disease. Nat Rev Dis Primers. 2018;4(1):15.
17 Sherratt SCR, Libby P, Bhatt DL, Mason RP. A biological rationale for the disparate effects of omega-3 fatty acids on cardiovascular disease outcomes. Prostaglandins Leukot Essent Fatty Acids. 2022;182:102450.
18. Climax J, Newsome PN, Hamza M, et al. Effects of epeleuton, a novel synthetic second-generation n-3 fatty acid, on nonalcoholic fatty liver disease, triglycerides, glycemic control, and cardiometabolic and inflammatory markers. J Am Heart Assoc. 2020;9(16):e016334.
19 Sanak M, Levy BD, Clish CB, et al. Aspirin-tolerant asthmatics generate more lipoxins than aspirin-intolerant asthmatics. Eur Respir J. 2000;16(1):44-49.
20 Wong PY, Hughes R, Lam B. Lipoxene: a new group of trihydroxy pentaenes of eicosapentaenoic acid derived from porcine leukocytes. Biochem Biophys Res Commun. 1985;126(2):763-772.
21. Rossato P, Federti E, Matte A, et al. Evidence of protective effects of recombinant ADAMTS13 in a humanized model of sickle cell disease. Haematologica. 2022;107(11):2650-2660.
22. Kasztan M, Fox BM, Lebensburger JD, et al. Hyperfiltration predicts long-term renal outcomes in humanized sickle cell mice. Blood Adv. 2019;3(9):1460-1475.
23. Beckman JD, Abdullah F, Chen C, et al. Endothelial TLR4 expression mediates vaso-occlusive crisis in sickle cell disease. Front Immunol. 2020;11:613278.
24. Ansari J, Senchenkova EY, Vital SA, et al. Targeting the AnxA1/ Fpr2/ALX pathway regulates neutrophil function, promoting thromboinflammation resolution in sickle cell disease. Blood. 2021;137(11):1538-1549.
25. Cossarizza A, Chang HD, Radbruch A, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur J Immunol. 2019;49(10):1457-1973.
26. Reichel CA, Rehberg M, Lerchenberger M, et al. Ccl2 and Ccl3 mediate neutrophil recruitment via induction of protein synthesis and generation of lipid mediators. Arterioscler Thromb Vasc Biol. 2009;29(11):1787-1793.
27. Halade GV, Norris PC, Kain V, Serhan CN, Ingle KA. Splenic leukocytes define the resolution of inflammation in heart failure. Sci Signal. 2018;11(520):eaao1818.
28. Sapieha P, Stahl A, Chen J, et al. 5-Lipoxygenase metabolite 4-HDHA is a mediator of the antiangiogenic effect of omega-3 polyunsaturated fatty acids. Sci Transl Med. 2011;3(69):69ra12.
29 Funk CD. The molecular biology of mammalian lipoxygenases and the quest for eicosanoid functions using lipoxygenasedeficient mice. Biochim Biophys Acta. 1996;1304(1):65-84.
30 Matte A, Recchiuti A, Federti E, et al. Resolution of sickle cell disease-associated inflammation and tissue damage with 17 R-resolvin D1. Blood. 2019;133(3):252-265.
31. Noomuna P, Risinger M, Zhou S, et al. Inhibition of band 3 tyrosine phosphorylation: a new mechanism for treatment of sickle cell disease. Br J Haematol. 2020;190(4):599-609.
32. McVey MJ, Steinberg BE, Goldenberg NM. Inflammasome activation in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2021;320(2):L165-L178.
33. Amano H, Nakamura M, Ito Y, et al. Thromboxane A synthase enhances blood flow recovery from hindlimb ischemia. J Surg Res. 2016;204(1):153-163.
34 Amano H, Ito Y, Eshima K, et al. Thromboxane A2 induces blood flow recovery via platelet adhesion to ischaemic regions. Cardiovasc Res. 2015;107(4):509-521.
35. De Franceschi L, Bertoldi M, Matte A, et al. Oxidative stress and beta-thalassemic erythroid cells behind the molecular defect. Oxid Med Cell Longev. 2013;2013:985210.
36. Wu X, Dong L, Lin X, Li J. Relevance of the NLRP3 inflammasome in the pathogenesis of chronic liver disease. Front Immunol. 2017;8:1728.
37. Osunkwo I, Andemariam B, Minniti CP, et al. Impact of sickle cell disease on patients’ daily lives, symptoms reported, and disease management strategies: results from the international Sickle Cell World Assessment Survey (SWAY). Am J Hematol. 2021;96(4):404-417.
38. Ugwu A, Iloanusi N, Ugwu N, et al. Pilot assessment of omega-3 fatty acids and potassium thiocyanate in sickle cell anemia patients with conditional peak systolic cerebral artery blood velocity. Blood Cells Mol Dis. 2021;89:102564.
39 Wu CYC, Lopez-Toledano MA, Daak AA, et al. SC411 treatment can enhance survival in a mouse model of sickle cell disease. Prostaglandins Leukot Essent Fatty Acids. 2020;158:102110.
40 Daak AA, Dampier CD, Fuh B, et al. Double-blind, randomized, multicenter phase 2 study of SC411 in children with sickle cell disease (SCOT trial). Blood Adv. 2018;2(15):1969-1979.
41. Serhan CN, Clish CB, Brannon J, et al. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med. 2000;192(8):1197-1204.
42. de Freitas Dutra V, Leal VNC, Fernandes FP, et al. Genetic contribution and functional impairment of inflammasome in sickle cell disease. Cytokine. 2022;149:155717.
43. Vogel S, Arora T, Wang X, et al. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018;2(20):2672-2680.
44 Pitanga TN, Santana SS, Zanette DL, et al. Effect of lysed and non-lysed sickle red cells on the activation of NLRP3 inflammasome and LTB4 production by mononuclear cells. Inflamm Res. 2021;70(7):823-834.
45. Erdei J, Toth A, Balogh E, et al. Induction of NLRP3 inflammasome activation by heme in human endothelial cells. Oxid Med Cell Longev. 2018;2018:4310816.
46. Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol. 2010;30(5):628-631.
47. Garstkiewicz M, Strittmatter GE, Grossi S, et al. Opposing effects of Nrf2 and Nrf2-activating compounds on the NLRP3 inflammasome independent of Nrf2-mediated gene expression. Eur J Immunol. 2017;47(5):806-817.
48. Liu X, Zhang X, Ding Y, et al. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxid Redox Signal. 2017;26(1):28-43.
49. Wang Z, Hu W, Lu C, et al. Targeting NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domaincontaining-3) inflammasome in cardiovascular disorders. Arterioscler Thromb Vasc Biol. 2018;38(12):2765-2779.
50 Lopategi A, Flores-Costa R, Rius B, et al. Frontline science: specialized proresolving lipid mediators inhibit the priming and activation of the macrophage NLRP3 inflammasome. J Leukoc Biol. 2019;105(1):25-36.
Thrombocytopenia is a known risk factor for morbidity in individuals with various health conditions, including malignancy. Thrombocytopenia can lead to adverse outcomes including increased bleeding risk and mortality.1-3 Preventing and correcting thrombocytopenia may mitigate these consequences and improve patients’ outcomes.3 One proposed mechanism for remedying thrombocytopenia is to prolong the lifespan of platelets by interfering with normal platelet clearance. In addition to apoptosis, platelets are marked as aged and removed from the circulation by hepatocytes and hepatic Küpffer cells in a highly coordinated process triggered by the loss of the terminal carbohydrate moiety sialic acid (platelet desialylation).4
Neuraminidases cleave the glycosidic link and remove the terminal sialic acid. This allows the platelet to bind to the hepatic Ashwell-Morell receptor5 (likely also the macrophage galactose lectin on Küpffer cells) and be removed from the circulation.6 Thus, oseltamivir, a known neuraminidase inhibitor commonly used in the treatment of influenza, may have off-target effects by halting platelet desialylation and attenuating platelet apoptosis and phagocytosis.7 Based on this mechanistic observation, there is clinical interest in applying this finding to treat immune thrombocytopenia (ITP). Previous in vitro and in vivo studies have specifically demonstrated the important role of desialylation in the clearance of platelets in ITP, and its possibilities as a therapeutic target in ITP.8-11
Previously, only a small pilot study11 had demonstrated an increase in platelets in response to oseltamivir in patients with ITP until a recent multicenter, randomized phase II study. In this study, individuals with ITP were randomly assigned to either dexamethasone monotherapy or dexamethasone in combination with a 4-day course of oseltamivir. Researchers randomized 96 patients and reported that the group given the combination treatment achieved significantly higher response rates at both 14 days and 6 months.12
We were interested in whether we could confirm that treatment with oseltamivir is independently associated with a significant change in platelet counts before and after treatment. Using a large database, we hypothesized that receiving oseltamivir would increase platelet levels. We obtained approval for a review of electronic medical records from the Medical College of Wisconsin’s Institutional Review Board. Using TriNetX software, we extracted data from patients’ charts. In our analysis, we included patients treated in the Medical College of Wisconsin sys-
tem if they: (i) had at least one clinical encounter between January 1, 2010 and December 31, 2020; (ii) were over 18 years old; (iii) were administered oseltamivir; and (iv) had their platelet count measured at least once a maximum of 30 days before receiving oseltamivir and a post-treatment measurement a maximum of 30 days following the administration of oseltamivir. The platelet counts before and after oseltamivir administration were recorded. We interrogated patients’ charts for demographic data and variables that could affect platelet levels, including a positive influenza polymerase chain reaction test within 10 days of oseltamivir administration. We utilized codes of the tenth revision of the International Classification of Diseases (ICD10) to identify the variables of interest. We employed a multivariable nested random effects model to account for the variance of the repeated measurements within the same treatment episode and multiple episodes within the same patient. We performed analyses using SAS 9.4 (SAS Institute, Cary, NC, USA).
We identified 2,168 patients who met the enrollment criteria. Table 1 includes their demographic information as well as comorbid conditions. Some patients had multiple treatment episodes over the 10 years which were at least
Table 1. Demographics of the study population.
90 days apart, creating 2,397 patient-treatment episodes. We performed t-test analysis on the log-transformed fold change of the platelet level. On average there was a 1.14-fold increase in platelet levels after therapy with oseltamivir, with an average increase of 14%. We then broke the population down into four quartiles using baseline platelet levels. The first quartile was composed of 603 patients with a baseline platelet level equal to or less than 153x103/µL. The mean fold change for those individuals was 1.40, meaning that platelet level increased, on average, 40% after oseltamivir treatment. Compared with all patients we analyzed, the patients in the lowest quartile were found to have the highest fold change in platelets after oseltamivir administration (Figure 1). In the current circumstances of scarce blood products, novel mechanisms to prevent or ameliorate severe thrombocytopenia are critical. Similar to other studies,13,14 our results show an increase in platelet counts after oseltamivir administration, particularly in patients with the lowest baseline platelet count.
One possible explanation could be that oseltamivir increases platelets by a set amount regardless of the initial platelet count, and that the apparently more significant elevations in patients with more severe thrombocytopenia could be explained by starting with a lower platelet count from which to calculate log-fold and percentage increase. Another possibility could be differing levels of platelet desialylation between the quartiles contributing to different baseline platelet levels, as shown by research demonstrating an inverse correlation between platelet count and the extent
of specifically O-glycan desialylation in murine models.15 For example, the patients in the first quartile could have higher levels of platelet desialylation compared to those in the other quartiles, explaining both that quartile’s lower platelet baseline and its superior response to the sialidase inhibitor oseltamivir, as there are more therapeutic targets available in this group. A recent prospective cohort study showed that higher levels of desialylation can be found in certain diseases including connective tissue disease, aplastic anemia, and myelodysplastic syndromes.9 As our search did not specify these precise co-morbid conditions, it is possible that the first quartile contained more patients with these “high-desialylation” diseases, which could be responsible for the lower baseline platelet levels. More research identifying the specific mechanisms of oseltamivir’s activity at differing platelet levels would be useful.
Limitations of our work include those inherent to the retrospective cohort design, such as the inability to determine whether the effect of oseltamivir is secondary to the anti-influenza effect or to the glycan effect on platelets. Notably, however, only 27% of the cases in our database had a positive influenza test. Those patients testing positive did not have a significantly different increase in platelets compared to the overall population. This finding supports the hypothesis that the increase in platelets is not associated with recovery from the respiratory illness.
Ultimately, the large sample size in our observational study adds power to the premise that the neuraminidase inhibitor oseltamivir could benefit individuals with severe thrombo-
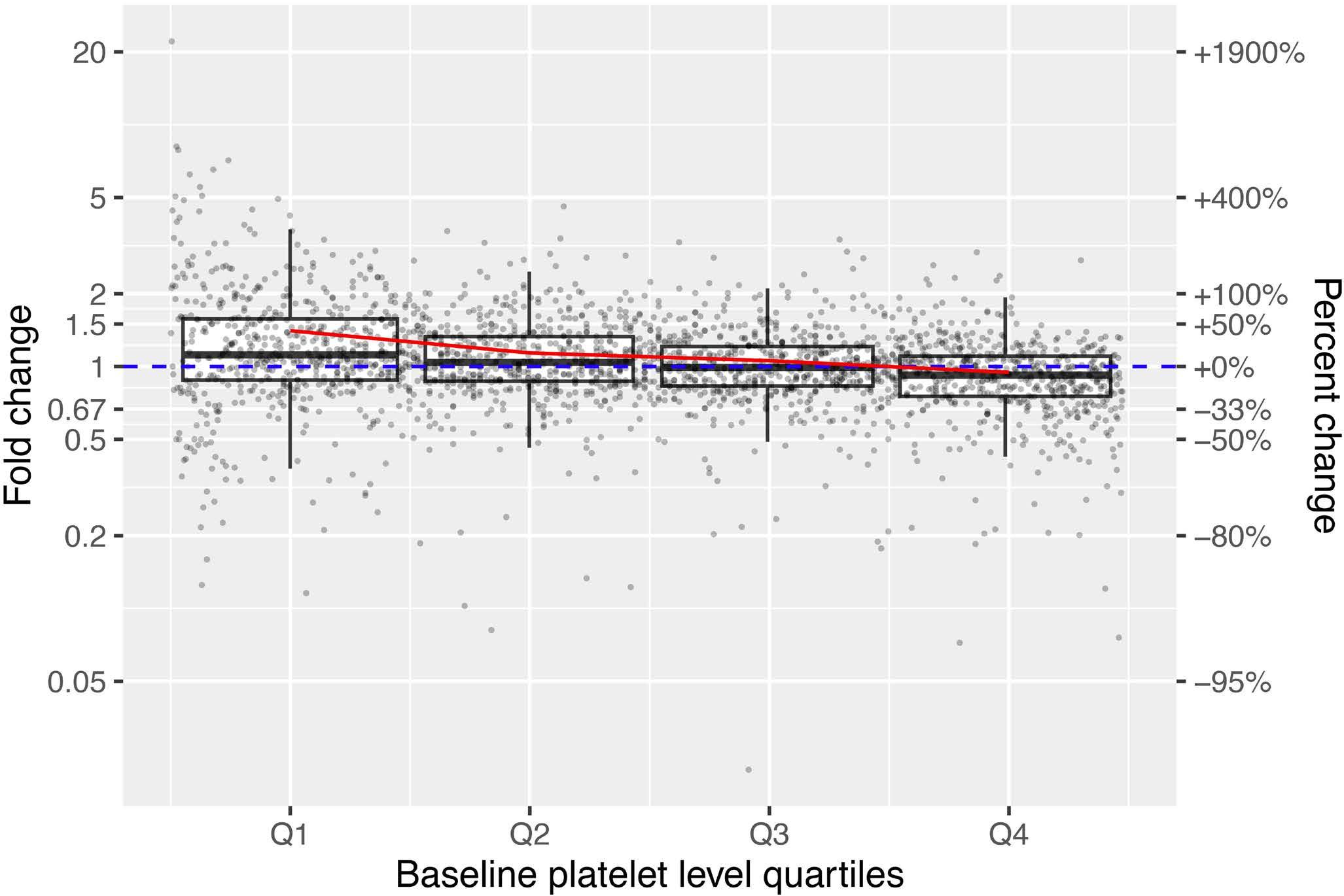
Figure 1. Fold change (left axis) and percent change (right axis) of platelet counts following oseltamivir therapy. Each point represents one subject, ordered from lowest to highest baseline platelet count along the x-axis. The box-and-whisker plots summarize the median and interquartile range from each quartile of baseline platelet count. The dashed blue line represents no change (fold-change = 1, percent-change = 0%). The solid red line connects the means within each quartile of baseline platelet count.
cytopenia even in situations other than immune-mediated thrombocytopenia. We believe that these data provide support for clinical trials to test such a hypothesis prospectively.
Authors
Chandrasekar Muthiah,1* Qinghua Lian,2* Samantha Benz,3* Aniko Szabo,2 Karin Hoffmeister,4 Juliana Perez Botero4 and Laura C. Michaelis5
1Department of Internal Medicine, Medical College of Wisconsin; 2Division of Biostatistics, Medical College of Wisconsin; 3Department of Internal Medicine, Aurora Health Care; 4Versiti and Division of Hematology/Oncology, Medical College of Wisconsin and 5Department of Medicine, Division of Hematology/Oncology, Froedtert Hospital/Medical College of Wisconsin, Milwaukee, WI, USA
*CM, QL and SB contributed equally as first authors.
Correspondence:
C. MUTHIAH - cmuthiah@mcw.edu
1. Claushuis TA, van Vught LA, Scicluna BP, et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. 2016;127(24):3062-3072.
2. Zhao X, Niu Q, Gan L, et al. Thrombocytopenia predicts mortality in Chinese hemodialysis patients- an analysis of the China DOPPS. BMC Nephrol. 2022;23(1):11.
3. Kuter DJ. Managing thrombocytopenia associated with cancer chemotherapy. Oncology (Williston Park). 2015;29(4):282-294.
4 Li R, Hoffmeister KM, Falet H. Glycans and the platelet life cycle. Platelets. 2016;27(6):505-511.
5. Grozovsky R, Hoffmeister KM, Falet H. Novel clearance mechanisms of platelets. Curr Opin Hematol. 2010;17(6):585-589.
6. Deppermann C, Kratofil RM, Peiseler M, et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. 2020;217(4):e20190723.
7 Hoffmeister KM, Falet H. Platelet clearance by the hepatic Ashwell-Morrell receptor: mechanisms and biological significance. Thromb Res. 2016;141 Suppl 2(Suppl 2):S68-72.
8. Li J, van der Wal DE, Zhu G, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun. 2015;6:7737.
https://doi.org/10.3324/haematol.2023.283731
Received: September 2, 2023.
Accepted: February 9, 2024. Early view: February 22, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
CM wrote the manuscript and performed research. QL and AS performed statistics and analyzed data. SB collected data and wrote the manuscript. LM, JPB, and KH supervised the study and edited the manuscript.
Data-sharing statement
The data that support the findings of this study are available from the corresponding author, CM, upon reasonable request.
9. Tao L, Zeng Q, Li J, et al. Platelet desialylation correlates with efficacy of first-line therapies for immune thrombocytopenia. J Hematol Oncol. 2017;10(1):46.
10. Marini I, Zlamal J, Faul C, et al. Autoantibody-mediated desialylation impairs human thrombopoiesis and platelet lifespan. Haematologica. 2021;106(1):196-207.
11. Revilla N, Corral J, Miñano A, et al. Multirefractory primary immune thrombocytopenia; targeting the decreased sialic acid content. Platelets. 2019;30(6):743-751.
12. Sun L, Wang J, Shao L, et al. Dexamethasone plus oseltamivir versus dexamethasone in treatment-naive primary immune thrombocytopenia: a multicentre, randomised, open-label, phase 2 trial. Lancet Haematol. 2021;8(4):e289-e298.
13. Shaim H, McCaffrey P, Trieu JA, DeAnda A, Yates SG. Evaluating the effects of oseltamivir phosphate on platelet counts: a retrospective review. Platelets. 2020;31(8):1080-1084.
14. Jansen AJ, Peng J, Zhao HG, Hou M, Ni H. Sialidase inhibition to increase platelet counts: a new treatment option for thrombocytopenia. Am J Hematol. 2015;90(5):E94-95.
15. Wang Y, Chen W, Zhang W, et al. Desialylation of O-glycans on glycoprotein Iba drives receptor signaling and platelet clearance. Haematologica. 2021;106(1):220-229.
Acute megakaryoblastic leukemia (AMKL) is a specific subtype of acute myelogenous leukemia (AML) with unique clinical and biological features. In sharp contrast to myeloid leukemia associated with Down syndrome (DS), AMKL of children without DS is associated with poor outcomes.1-4 Hematopoietic cell transplantation (HCT), particularly with allogeneic (allo) grafts, represents a curative treatment option for a subset of high-risk AMKL; however, currently there is no specific consensus on indications.1,5 Although some studies proposed HCT as a post-remission consolidative therapy, its clinical benefit remains controversial.5,6 Previous studies unveiled an array of recurrent genetic alterations in AMKL and demonstrated their prognostic implications.4,7-11 These genetic lesions might be utilized in risk-adopted therapies to improve the currently unsatisfactory treatment outcomes of this rare malignancy. In the search for precise risk stratification and optimal therapeutic interventions, we conducted a retrospective study of pediatric AMKL which included laboratory testing for gene fusion detection and survival analysis according to biological and clinical factors.
In the present study, we retrospectively analyzed bone marrow or blood samples containing leukemic blasts from pediatric de novo AMKL patients who were referred to study hospitals in Japan and South Korea between 1990 and 2017. AMKL diagnosis required positivity for platelet-associated antigens (CD36, CD41, CD42b, or CD61) in addition to cell morphology, and constitutional +21 was excluded by clinical phenotype and metaphase analysis.12 Reverse transcription-polymerase chain reaction (RT-PCR) was used to screen for recurrent gene fusions observed in AMKL (Online Supplementary Table S1). All samples without these lesions were analyzed with RNA sequencing to search for other gene rearrangements.13 In a final analysis, correlations among the clinical data, therapeutic interventions, cytogenetic findings, and detected gene fusions were explored. The Institutional Review Board of Nagoya University Graduate School of Medicine approved this study, and written informed consent was obtained from participants or their guardians. The probability of overall survival (OS) and leukemia-free survival (LFS) was calculated using the Kaplan-Meier method, and the distribution of groups was compared using the log-rank test. Cumulative incidence of relapse (CIR) was calculated using the Gray’s method, in which any death during remission was set to competing risk. All P values were two-sided; P<0.05 was considered statistically significant. A summary of key information of 30 patients is shown in
Figure 1A and Online Supplementary Table S2. The median age at diagnosis was 1.3 years (interquartile range [IQR], 0.3-1.8). Metaphase analysis revealed -7 in 3 (10%). All patients received AML-oriented high-dose cytarabine and anthracycline-based chemotherapy. Regarding treatment responses, 20 (67%) and 7 (23%) achieved hematologic complete remission (HCR), defined as <5% bone marrow blasts, after the first and second chemotherapy courses, respectively, whereas 2 (7%) did not respond to the initial induction therapies. The remaining one patient (3%) died soon after presentation from pneumonia.
Sixteen patients underwent HCT during the first complete remission (CR1), whereas 2 did so in the later disease stages. In the 1990s, autologous (auto) grafts were utilized in cases without HLA-matched sibling donor; in this study, allo- and auto-HCT were administrated in 14 (78%) and 4 (22%) patients, respectively. Indications of transplantation were determined by local clinicians, principally based on treatment response (e.g., not achieving remission after induction chemotherapy), donor availability (e.g., the presence of a family donor), and high-risk cytogenetics already identified at the time. Most of the patients undergoing both allo- and auto-HCT received busulfan-based myeloablative conditioning (Table 1).
The median follow-up period was 91 months (IQR, 12-145) and 20 patients were doing well at the final visit. In the study cohort, 7 and 3 died of leukemia and accompanied complications, respectively. The 5-year OS, LFS, and CIR were 66% (95% confidence interval [CI]: 47-80%), 63% (95% CI: 43-78%), and 29% (95% CI: 15-49%), respectively (Figure 1B). Univariate analysis of prognostic factors revealed that the 5-year LFS was significantly worse in patients who did not achieve HCR after the first course of induction chemotherapy than in those who did (30% [95% CI: 5.2-61%] vs. 76% [95% CI: 52-89%]; P=0.02) (Table 2). Of note, all 8 who experienced relapse died of leukemia or treatment-related complications with active disease.
Gene fusions of any type were detected by RT-PCR in 19 patients (63%). These fusions were mutually exclusive and included CBFA2T3::GLIS2, RBM15::MRTFA, NUP98::KDM5A, FUS::ERG, and KMT2A::MLLT3 in 9, 5, 3, 1, and 1 patient, respectively (Figure 1A). Comprehensive transcriptome analysis by RNA sequencing in the remaining 11 patients did not reveal any apparent driver gene rearrangements. Based on previous reports, we considered CBFA2T3::GLIS2, NUP98::KDM5A, FUS::ERG, KMT2A::MLLT3, and -7 as high-risk genetic/ cytogenetic aberrations, which were identified in 16 patients
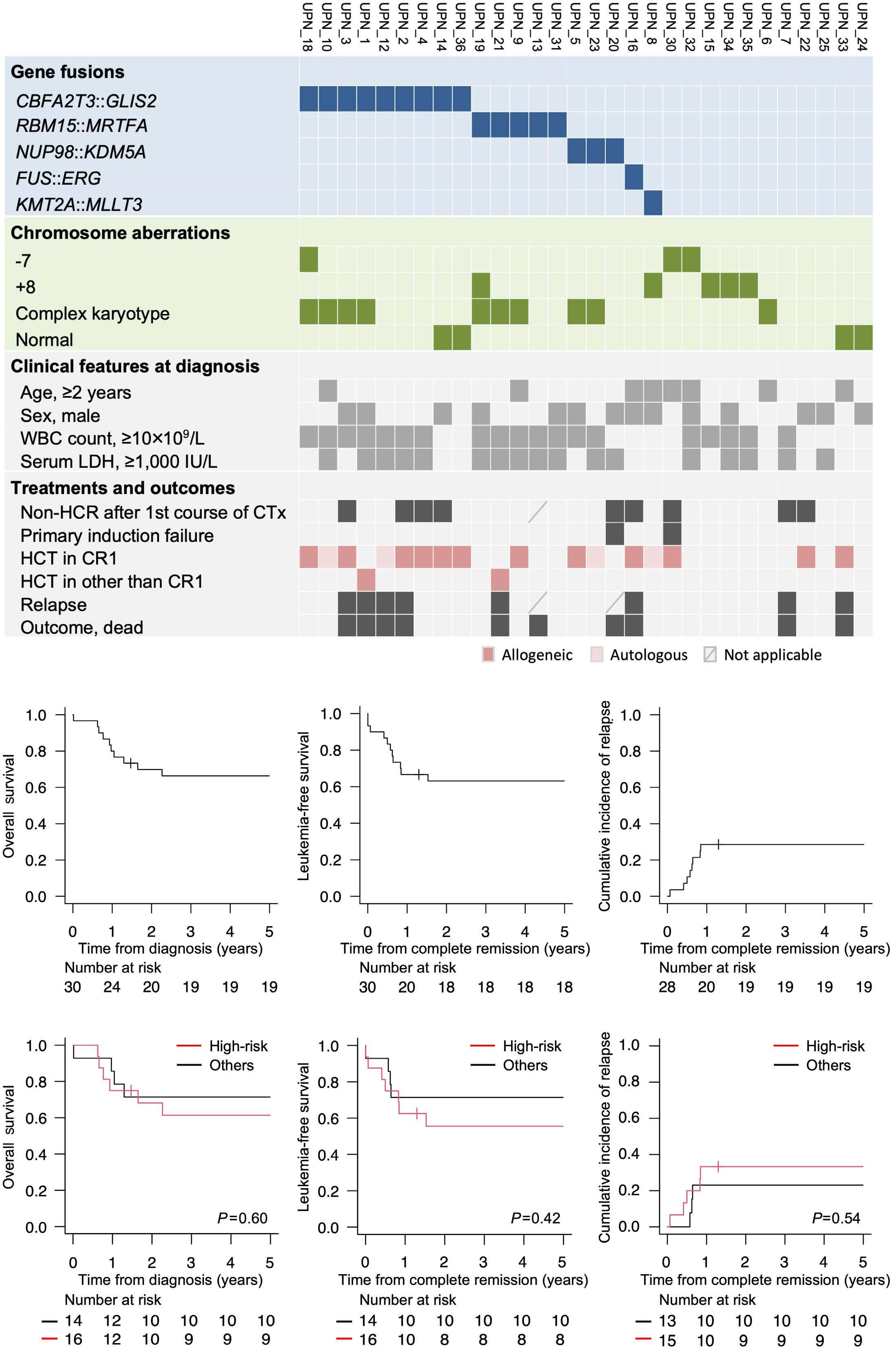
Figure 1. Clinical features and outcomes of the patients. (A) A summary of gene fusions detected, chromosome aberrations, and key clinical information on each patient. (B) Overall survival (OS), leukemia-free survival (LFS), and cumulative incidence of relapse (CIR) estimates in the overall cohort (N=30). (C) OS, LFS, and CIR estimates in patients with (red lines) or without (black lines) high-risk aberrations defined in this study: CBFA2T3::GLIS2, NUP98::KDM5A, FUS::ERG, KMT2A::MLLT3, and -7. CR1: first complete remission; CTx: chemotherapy; HCR: hematologic complete remission; HCT: hematopoietic cell transplantation; LDH: lactate dehydrogenase; WBC: white blood cell.
Table 1. Key characteristics of allogeneic and autologous hematopoietic cell transplantation.
Allogeneic, N=14
Autologous, N=4
Characteristic
CR1: first complete remission; HCT: hematopoietic cell transplantation; HLA: human leukocyte antigen; N: number; NA: not applicable; PBSC: peripheral blood stem cell.
(53%) in the study cohort.9,14,15 Five of 8 who experienced relapse had high-risk gene fusions (CBFA2T3::GLIS2, N=4; FUS::ERG, N=1), whereas another harbored RBM15::MRTFA The remaining 2 did not harbor any predefined genetic/ cytogenetic abnormalities. There was no significant difference in 5-year OS, LFS, and CIR between the high-risk and non-high-risk patients (OS 61% [95% CI: 33-81%] vs. 71% [95% CI: 41-88%], P=0.60; LFS 56% [95% CI: 29-76%] vs. 71% [95% CI: 41-88%], P=0.42; CIR 33% [95% CI: 15-63%] vs. 23% [95% CI: 8.1-56%], P=0.54) (Figure 1C). The 5-year OS, LFS, and CIR of patients with CBFA2T3::GLIS2 (N=9), which is strongly associated with adverse outcomes, were 53% (95% CI: 18-80%), 56% (95% CI: 20-81%), and 44% (95% CI: 20-80%), respectively. These rates were not statistically inferior to those of patients without CBFA2T3::GLIS2 (Table 2). In contrast to non-high-risk patients (3/14, 21%), more than half of the high-risk patients (9/16, 56%), particularly those with CBFA2T3::GLIS2 (6/9, 67%), underwent allo-HCT during CR1. In our small cohort, the survival rates of high-risk patients who underwent allo-HCT in CR1 (N=9) did not differ from those of patients who received only chemotherapy or auto-HCT as their first-line treatment (N=7) (5-year LFS, 67% [95% CI: 28-88%] vs. 57% [95% CI: 17-84%], P=0.65). Recent advances in diagnostics and intensive chemotherapy have improved outcomes in children with AMKL; however, survival rates remain extremely low in patients who experience relapse. Indeed, all 8 patients with recurrence of leukemia in the present study did not survive. Thus, identification of patients at high-risk of relapse is crucial to increase the chance of cure. Early response to conventional chemotherapy is one of the most powerful indicators, and consistent with previous reports, we demonstrated that the 5-year LFS was significantly worse in patients who were not in HCR after the first course of induction chemotherapy.3,6
On the other hand, several studies reported that a subset of patients with AMKL harbored recurrent genetic alterations which were implicated in prognosis.4,7-11 In particular, CBFA2T3::GLIS2, NUP98::KDM5A, and KMT2A rearrangements were associated with adverse outcomes, with OS rates of 14-42%, 35-36%, and 27-36%, respectively.4,9,10 Furthermore, FUS::ERG fusion was also distinctly associated with poor prognosis in pediatric AML.15
A few studies reported that HCT as post-remission consolidative treatment provided better outcomes. A representative study demonstrated that estimate of 2-year event-free survival was higher in patients who underwent allo-HCT compared to those who received chemotherapy alone (26% vs. 0%).1 Furthermore, a recent study reported that the 5-year OS was significantly higher for patients who underwent HCT in CR1 (72%) than for those who did in the second CR (23%) and non-CR (16%).16 Conversely, other studies documented no benefit of HCT, including that administrated during CR1; however, their risk stratification criteria did not include most of the recently identified genetic alterations specific to AMKL.2,3,14 While novel molecular targets in AMKL such as FOLR1 have emerged, clinically available treatments remain limited.17 Moreover, given the extremely low survival rates for relapsed patients, considering HCT as a first-line treatment for high-risk patients with AMKL is viable and reasonable. In the present study, the 5-year OS, LFS, and CIR in high-risk patients, especially in those with CBFA2T3::GLIS2, did not significantly differ from those in non-high-risk or CBFA2T3::GLIS2-negative patients. Furthermore, these rates appeared to be favorable compared to those reported in previous studies.10,11 Most of the high-risk patients (13/16 [81%], including allogeneic: 9/16 [56%]) and CBFA2T3::GLIS2-positive patients (8/9 [89%], including allogeneic: 6/9 [67%]) underwent HCT during CR1, which might be associated with better survival.
Table 2. Univariate analysis of prognostic factors for leukemia-free survival and relapse rates.
Prognostic factors
CTx: chemotherapy; HCR: hematologic complete remission; LDH: lactate dehydrogenase; N: number; WBC: white blood cell.
In a previous study, 9 of 12 patients with CBFA2T3::GLIS2 achieved HCR after induction therapy. In that cohort, 3 of 4 who received allo-HCT at CR1 survived whereas 4 of the remaining 5 who received chemotherapy alone as initial treatment died after relapse.4 The small number of patients included in the present study does not allow us to conduct detailed analysis or draw solid conclusions. Additionally, our practice did not entirely exclude rare cases of cryptic DS. Further studies in large cohorts are essential to clarify the clinical utility of AMKL-specific risk stratification that includes genetic alterations, and to assess the feasibility of allo-HCT during CR1 for high-risk patients. In conclusion, the present study identified recurrent gene rearrangements found in pediatric AMKL, with favorable survival even in patients with high-risk genetic/cytogenetic features. More precise risk stratification, incorporating treatment response and genetic features in AMKL, could help identify those who may benefit from HCT as a first-
line consolidative treatment, thereby leading to improved outcomes.
Kyogo Suzuki,1 Asahito Hama,1 Yusuke Okuno,2 Yinyan Xu,1 Atsushi Narita,1 Nao Yoshida,3 Hideki Muramatsu,1 Nobuhiro Nishio,4 Koji Kato,3 Seiji Kojima,1 Keon Hee Yoo5# and Yoshiyuki Takahashi1#
1Department of Pediatrics, Nagoya University Graduate School of Medicine, Aichi, Japan; 2Department of Virology, Nagoya City University Graduate School of Medical Sciences, Aichi, Japan; 3Department of Hematology and Oncology, Children’s Medical Center, Japanese Red Cross Aichi Medical Center Nagoya First Hospital, Aichi, Japan; 4Department of Advanced Medicine, Nagoya University Hospital, Aichi, Japan and 5Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, South Korea
#KHY and YT contributed equally as senior authors.
Correspondence:
Y. TAKAHASHI - ytakaha@med.nagoya-u.ac.jp
K.H. YOO - hema2170@skku.edu
https://doi.org/10.3324/haematol.2023.283760
Received: July 6, 2023.
Accepted: January 23, 2024. Early view: February 1, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
KS designed and performed the research, analyzed the data, and wrote the manuscript. YO and YX designed and performed the research and analyzed the data. AN, HM, NN and SK designed the research and
1. Athale UH, Razzouk BI, Raimondi SC, et al. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution’s experience. Blood. 2001;97(12):3727-3732.
2. Reinhardt D, Diekamp S, Langebrake C, et al. Acute megakaryoblastic leukemia in children and adolescents, excluding Down’s syndrome: improved outcome with intensified induction treatment. Leukemia. 2005;19(8):1495-1496.
3. Schweitzer J, Zimmermann M, Rasche M, et al. Improved outcome of pediatric patients with acute megakaryoblastic leukemia in the AML-BFM 04 trial. Ann Hematol. 2015;94(8):1327-1336.
4 Hara Y, Shiba N, Ohki K, et al. Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosomes Cancer. 2017;56(5):394-404.
5. Garderet L, Labopin M, Gorin NC, et al. Hematopoietic stem cell transplantation for de novo acute megakaryocytic leukemia in first complete remission: a retrospective study of the European Group for Blood and Marrow Transplantation (EBMT). Blood. 2005;105(1):405-409.
6. Wang Y, Lu A, Jia Y, Zuo Y, Zhang L. Outcome and prognostic features in pediatric acute megakaryoblastic leukemia without down syndrome: a retrospective study in China. Clin Lymphoma Myeloma Leuk. 2021;21(4):e301-e308.
7. Gruber TA, Larson Gedman A, Zhang J, et al. An Inv(16) (p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell. 2012;22(5):683-697.
8. de Rooij JD, Hollink IH, Arentsen-Peters ST, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia. 2013;27(12):2280-2288.
9 de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, et al. Recurrent abnormalities can be used for risk group stratification
analyzed the data. NY and KK provided samples and patient data. KHY provided samples and patient data, and supervised the project. AH and YT led the entire project, designed and performed the research, analyzed the data, and wrote the manuscript. All authors critically reviewed and approved the final version of the manuscript for publication.
The authors would like to thank all of the clinicians, patients, and their families who made this study possible with the providing of samples. The authors would also like to thank Ms. Chie Amahori, Ms. Hiroko Ono, and Ms. Yoshie Miura for their valuable assistance.
Funding
This research was supported by the Kitamura Memorial Hematological Disorder Research Fund in Nagoya University Graduate School of Medicine.
Data-sharing statement
Detailed data are available from the first author upon reasonable request: kyogo1116@gmail.com
in pediatric AMKL: a retrospective intergroup study. Blood. 2016;127(26):3424-3430.
10 de Rooij JD, Branstetter C, Ma J, et al. Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49(3):451-456.
11. Smith JL, Ries RE, Hylkema T, et al. Comprehensive transcriptome profiling of cryptic CBFA2T3-GLIS2 fusion-positive AML defines novel therapeutic options: a COG and TARGET pediatric AML study. Clin Cancer Res. 2020;26(3):726-737.
12. Bennett JM, Catovsky D, Daniel MT, et al. Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7). A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103(3):460-462.
13. Suzuki K, Okuno Y, Kawashima N, et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J Clin Oncol. 2016;34(28):3451-3459.
14. Inaba H, Zhou Y, Abla O, et al. Heterogeneous cytogenetic subgroups and outcomes in childhood acute megakaryoblastic leukemia: a retrospective international study. Blood. 2015;126(13):1575-1584.
15. Tomizawa D, Yoshida M, Kondo T, et al. Allogeneic hematopoietic stem cell transplantation for children and adolescents with high-risk cytogenetic AML: distinctly poor outcomes of FUS-ERGpositive cases. Bone Marrow Transplant. 2019;54(3):393-401.
16. Hama A, Taga T, Tomizawa D, et al. Haematopoietic cell transplantation for children with acute megakaryoblastic leukaemia without Down syndrome. Br J Haematol. 2023;201(4):747-756.
17 Tang T, Le Q, Castro S, et al. Targeting FOLR1 in high-risk CBF2AT3GLIS2 pediatric AML with STRO-002 FOLR1-antibody-drug conjugate. Blood Adv. 2022;6(22):5933-5937.
Hepatosplenic T-cell lymphoma (HSTCL) is a highly aggressive T-cell neoplasm that arises from the proliferation of gamma/delta T cells ( γδ T) infiltrating the liver, spleen, and bone marrow sinusoids.1 It commonly emerges in individuals with chronic immune suppression, predominantly affecting young adults who manifest hepatosplenomegaly, cytopenias, and systemic symptoms.2 In contrast, large granular lymphocytic leukemia (LGLL) typically manifests as an indolent proliferation of cytotoxic alpha/beta T cells ( a β T), primarily afflicting older adults with neutropenia and concurrent autoimmune disorders.3 Nevertheless, splenomegaly is frequently observed in LGLL, and leukemic cells can carry a γδT-cell receptor (TCR), complicating the differentiation from HSTCL.4,5 This study, approved by our institution’s ethics committee (number 23.34), aims to establish novel diagnostic criteria for distinguishing HSTCL from γδ T-LGLL. Herein, we report a distinctive oyster-shell morphology and identify stereotyped VD1JD1 CDR3 sequences in HSTCL cells. The morphological description of HSTCL has focused primarily on histological aspects of splenic or hepatic biopsies, with minimal emphasis on cytological features.6 We centrally reviewed 23 bone marrow aspirate smears, ten blood smears and two biopsy touch preparations (spleen and liver). Bone marrow was involved in all HSTCL cases and blood samples were infiltrated in 42% of cases. The tumor burden in bone marrow ranged from 5% to 73% and varied from sparse neoplastic cells to a diffuse involvement with large pseudometastatic aggregates. Hemophagocytosis was observed in six patients (25%). Atypical lymphoid cells were monotonous, medium-sized, with irregular nuclei displaying fine chromatin and prominent nucleoli. Their cytoplasm had a jagged outline, peripheral basophilic enhancement, and frequent cytoplasmic projections, giving the tumoral cells a distinctive oyster-shell pattern (Figure 1A-C). These cells were predominant within the neoplastic infiltrate and were observed in all the blood and bone marrow samples from HSTCL cases. In touch preparations, tumoral cells often presented with rounder blastic nuclei and reduced cytoplasm, consistent with previous descriptions (Figure 1D).6 Conversely, LGLL cells had a round nucleus with dense chromatin and pale cytoplasm with azurophilic granules (Figure 1E). Thus, the distinctive oyster-shell morphology observed in both γδ T and a β T HSTCL subtypes may provide a new criterion for
HSTCL diagnostics (Figure 1F, G). This characteristic could guide additional investigations early in this challenging diagnosis and potentially avoid more invasive procedures. The phenotypic and oncogenic characteristics of the two groups align (Table 1) with previously published data ( Online Supplementary Figure S1).2,4 All HSTCL cases were positive for the pan-T-cell markers CD3 and CD2. CD7 was positive in 14 cases (93% of tested cases). Most HSTCL cases were double negative for both CD4 and CD8 (60%). As could be expected, 18 cases (90%) exhibited a γδ -TCR while two cases expressed an a β -TCR (10%). Several markers might be useful in distinguishing HSTCL from LGLL. A complete lack of CD5 was observed in 17 HSTCL samples (15% of positive cases), whereas LGLL displayed at least weak positivity for CD5 in 81% of cases ( P<0.001). CD56 was positive in 13 HSTCL samples (87%), while only two LGLL cases were positive (13%) (P<0.001). Conversely, 14 cases of LGLL expressed CD57 (93%), while CD57 expression was detected in three of the five HSTCL cases tested. Lastly, in four bone marrow biopsies, HSTCL were positive for TIA-1 and negative for granzyme B staining, while this marker is typically expressed in cases of LGLL.3 Cytogenetic analysis was available for 14 patients with HSTCL. Isochromosome 7q was detected in ten cases (71%) and trisomy 8 in seven cases (50%). Four patients (29%) had a complex karyotype. Subsequently, we conducted a comparative mutational analysis of 21 HSTCL samples and 16 γδ T-LGLL using targeted high-throughput sequencing (Online Supplementary Table S1). In the HSTCL group, 17 patients (81%) had at least one somatic mutation. Overall, we identified 30 clinically relevant variants in 13 different genes. The most frequent event was a mutation in the SH2 domain of STAT5B, detected in eight patients (38%), with five of them carrying the N642H hotspot. DNMT3A was mutated in four cases (19%), TET2 in three patients (14%), and EZH2, TP53 and SETD2 in two patients (10%). Among γδT-LGLL, 14 (88%) had at least one significant mutation. The STAT3 gene was mutated in 12 patients (75%). A DNMT3A mutation was found in three cases (19%), and a TET2 mutation in two cases (13%). Finally, TNFAIP3, STAT5B, and IDH2 mutations were detected in one patient. Consistent with previous studies, the presence of isochromosome 7q remains the most frequent cytogenetic aberration in HSTCL, and it is retained as a diagnostic criterion in the latest revision of the World Health Organi -
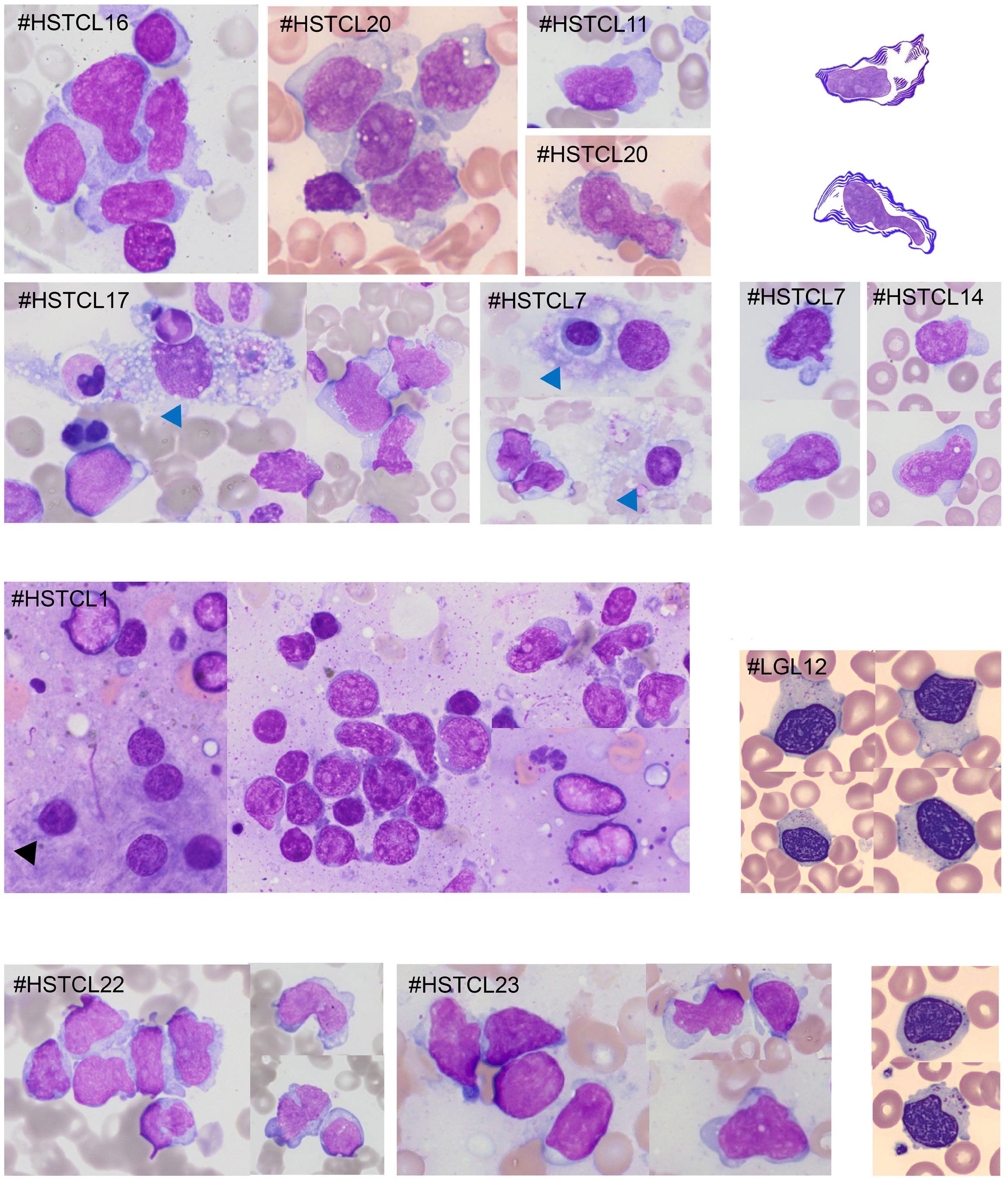
Figure 1. Comparative cytological analysis of hepatosplenic T-cell lymphoma and T-cell large granular lymphocytic leukemia. May-Grünwald-Giemsa staining, magnification x600. (A) Bone marrow aspirate smears from five representative cases of γδ hepatosplenic T-cell lymphoma (HSTCL). Blue arrows indicate images of hemophagocytosis. (B) Schematic illustration depicting the oyster shell pattern observed in HSTCL cases. (C) Tumor cells on blood smears from two γδ-HSTCL cases. (D) Liver biopsy touch preparation from one γδ-HSTCL case. The black arrow points to a Küpffer cell. (E) Large granular lymphocytes (LGL) on blood smears (CellaVision images) from one γδT-LGL leukemia case. (F) Bone marrow aspirate smears from two aβ-HSTCL cases. (G) Blood smear (CellaVision images) from one case of aβT-LGL leukemia.
zation classification.1,7 The detection of STAT5B mutations can provide an additional molecular marker for HSTCL, in contrast to STAT3 alterations which constitute the molecular hallmark of LGLL.8 Mutations in genes related to DNA methylation (TET2 and DNMT3A) were identified in both HSTCL and LGLL samples and have been commonly
reported in the context of clonal hematopoiesis, making them unsuitable as specific molecular markers for HSTCL. Conversely, SETD2 appeared to be specifically associated with HSTCL, being present with a frequency of 10% in the cases in our cohort and in up to 25% in previous studies. 9,10 Lastly, exome sequencing provided a more comprehen-
Table 1. Characteristics of patients with hepatosplenic T-cell lymphoma and γδT-cell large granular lymphocytic leukemia.
Clinical presentation, N (%)
Continuous variables are reported as the median (range) and discrete variable as a number (percentage among evaluable cases). HSTCL: hepatosplenic T-cell lymphoma; LGLL: large granular lymphocytic leukemia; NA: not available.
sive view of the genomic landscape of HSTCL, revealing alterations in other genes involved in chromatin modification such as INO8, SMARCA2, and ARID1B, as well as in other signaling pathways (e.g., PIK3CD or KRAS), offering potential therapeutic targets for this rare disease.10,11 The originality of our study stems from the analysis of TCR rearrangement specificity in a sizable cohort of HSTCL patients using high-throughput sequencing. Recently, Teramo et al. investigated the TCR repertoire profile in LGLL, revealing stereotyped TRG rearrangements associated with clinical features.12 Our study provides complementary results on the immunogenetic profile of HSTCL, highlighting
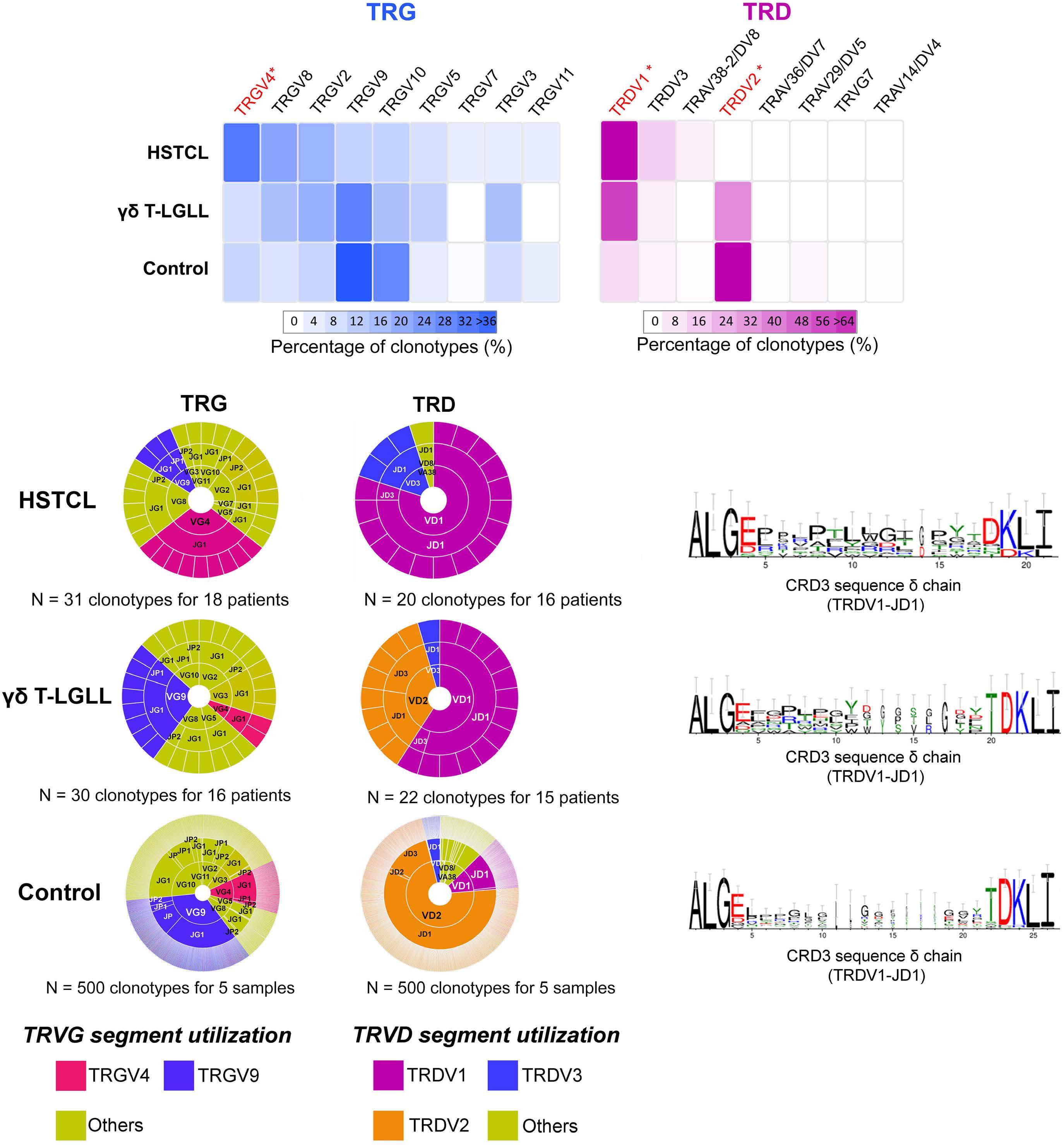
Figure 2. Comparison of TRG and TRD rearrangement profiles in dominant clonotypes from hepatosplenic T-cell lymphoma, γδT-cell large granular lymphocytic leukemia and a polyclonal control group. (A) Heatmap showing frequency of segment usage for TRG and TRD loci in the hepatosplenic T-cell lymphoma (HSTCL) group, compared to the γδT-large granular lymphocytic leukemia (LGLL) group and the polyclonal control group. *TRGV4 is significantly overrepresented in HSTCL clonotypes versus LGLL and versus control clonotypes; TRDV1 is overrepresented in HSTCL and LGLL clonotypes versus control clonotypes; TRDV2 is overrepresented in LGLL and control clonotypes versus HSTCL clonotypes (P<0.05). (B) Sunburst representation of the utilization of TRG segments in the dominant clonotypes identified in the HSTCL, LGLL and control groups. (C) Sunburst representation of the utilization of TRD segments in the dominant clonotypes identified in the HSTCL, LGLL and control groups. (D) Comparison of the CDR3 amino-acid sequences of VD1JD1 clonotypes in the HSTCL, LGLL and control groups. TRG: T-cell receptor gamma; TRD: T-cell receptor delta.
biased TRG and TRD gene usages that distinguish them from LGLL (Figure 2).
TRG and TRD gene rearrangements were sequenced according to the two-step polymerase chain reaction Euroclonality next-generation sequencing protocols on a MiSeq platform (Illumina) and analyzed with the Vidjil tool.13 We compared the dominant clonotypes from 20 HSTCL and 16 γδ T-LGLL cases with an in silico polyclonal control composed of the top 100 clonotypes from five healthy donors (Figure 2A). Regarding the TRG locus (Figure 2B), 31 clonotypes were detected in 18 HSTCL cases (90%) with a predominance of rearrangements involving the TRGV4 gene in nine cases (50%). The TRGV4 gene was significantly more prevalent in HSTCL clonotypes (29%) than in γδ TLGLL (7%; P=0.043) and the control group (9%; P=0.002). In γδ T-LGLL and the control group, the most represented gene was TRGV9, occurring in eight LGLL clonotypes (27%) and in 37% of control clonotypes, which is significantly higher than in the HSTCL group (10%) ( P=0.002). Regarding the TRD locus (Figure 2C), we identified 20 clonotypes in 16 HSTCL samples and 22 clonotypes in 15 γδ T-LGLL samples. We observed a higher number of clonotypes using the TRDV1 gene in HSTCL and γδ T-LGLL cases than in the control group (P<0.001 for both). Additionally, 13 HSTCL cases (81% of γδ T-positive cases) showed a VD1JD1 rearrangement, while this occurred in eight γδ T-LGLL cases (50%). To a lesser extent, HSCTL clonotypes used the TRDV3 gene more frequently than γδ T-LGLL did (P=0.050). In contrast, TRDV2 was more frequently represented in the control group (80% of clonotypes) and in γδ T-LGLL patients (47%) than in the HSTCL group (0%) ( P<0.001 and P=0.004, respectively). Finally, only two HSTCL patients had TRB rearrangements, corresponding to the two samples expressing aβTCR identified by flow cytometry. Overall, we demonstrate a higher prevalence of TRGV4 and TRDV1 segment utilization in HSTCL patients. This observation aligns with previous phenotyping investigations that showed a predominant expression of V δ 1 chain in HSTCL and in normal splenic γδ T cells.14,15 Moreover, tumor infiltrating V δ 1 T cells often exhibit immunosuppressive effects, in contrast to classical V γ 9V δ 2 T cells, which mainly exist in peripheral blood and have strong anti-tumor effects in various types of tumors.15 In line with the findings of Teramo et al., LGLL exhibited a higher frequency of TRGV9 and TRDV2 genes in their TRG and TRD rearrangements, respectively. Consistently, the preferential expression of the V γ 9V δ 2 phenotype in γδ T-LGLL appears to mimic the spectrum of normal T cells in the peripheral blood of healthy subjects and is associated with less symptomatic presentation in LGLL.12 In HSTCL clonotypes, we observed similar peptide amino-acid sequences within the CDR3 region of the δ chain, particularly involving VD1-JD1 rearrangements (Figure 2D and Online Supplementary Table S2). These CDR3 sequences were shorter and showed less diversity than those observed in VD1-JD1 clonotypes from
subjects with γδ T-LGLL and healthy donors, suggesting a role for antigenic recognition as an early event in HSTCL lymphomagenesis.
In conclusion, HSTCL may originate from a clonally selected γδ T population with stereotyped TCR, through the acquisition of somatic mutations leading to JAK/STAT pathway deregulation and chromatin modifications. We provide a more comprehensive characterization of HSTCL patients compared to those with γδ T-LGLL, highlighting a specific oyster-shell morphology and restricted TRG and TRD segment usages in HSTCL. Together, these findings may serve as a valuable tool for distinguishing HSTCL from other γδ T proliferations and could potentially reduce the need for more invasive procedures such as splenectomy or liver biopsy.
Anne Desmares,1,2 Simon Bouzy,2,3 Florian Thonier,4 Julien Goustille,2,5 Francisco Llamas-Gutierrez,6 Franck Genevieve,2,7 Laurane Cottin,2,7 Lucile Baseggio,2,8 Pierre Lemaire,2,9 Carinne Lecoq Lafon,2,10 Pascale Cornillet-Lefebvre,10 Anne-Cécile Galoisy,2,11 Chantal Brouzes,2,12 Emmanuelle Rault,2,13 Elodie Dindinaud,2,14 Carole Fleury,2,15 Florence Blanc-Jouvan,2,16 Soraya Wuilleme,2,3 Valérie Bardet,2,17 Thierry Fest,1,18 Thierry Lamy,18,19 Mikael Roussel,1,18 Mélanie Pannetier1,2 and Cédric Pastoret1,18
1Centre Hospitalier Universitaire de Rennes, Laboratoire d’Hématologie, Rennes; 2Groupe Francophone d’Hématologie Cellulaire, Bron; 3Centre Hospitalier Universitaire de Nantes, Laboratoire d’Hématologie, Nantes; 4INRIA (French National Research Institute), Rennes; 5Centre Hospitalier de Saint-Malo, Laboratoire de Biologie, Saint-Malo; 6Centre Hospitalier Universitaire de Rennes, Laboratoire d’Anatomopathologie, Rennes; 7Centre Hospitalier Universitaire d’Angers, Laboratoire d’Hématologie, Angers; 8Hospices Civils de Lyon – HCL, Laboratoire d’Hématologie, Bron; 9Hôpital Saint-Louis AP-HP, Laboratoire d'Hématologie, Paris; 10Centre Hospitalier Universitaire de Reims, Laboratoire d'Hématologie, Reims; 11Centre Hospitalier Universitaire de Strasbourg, Laboratoire d’Hématologie, Strasbourg; 12Hôpital Necker AP-HP, Laboratoire d'Hématologie, Paris; 13Centre Hospitalier Universitaire de Tours, Laboratoire d’Hématologie, Tours; 14Centre Hospitalier Universitaire de Poitiers, Laboratoire d'Hématologie, Poitiers; 15Hôpital Avicenne AP-HP, Laboratoire d'Hématologie, Bobigny; 16Centre Hospitalier Annecy Genevois, Laboratoire de Biologie, Epagny Metz-Tessy; 17Centre Hospitalier Universitaire Ambroise Paré AP-HP, Service d’Hématologie-ImmunologieTransfusion, Paris; 18Université de Rennes 1, INSERM UMR 1236, Rennes and 19Centre Hospitalier Universitaire de Rennes, Hématologie Clinique, Rennes, France
Correspondence: C. PASTORET - cedric.pastoret@chu-rennes.fr
https://doi.org/10.3324/haematol.2023.283856
Received: July 31, 2023.
Accepted: January 15, 2024. Early view: January 25, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
AD, SB, and CP designed the research and wrote the paper. FT performed T-cell receptor sequencing bioinformatics analysis. MP described the cytological pattern. AD, SB, JG, and MP reviewed
1. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720-1748.
2. Weidmann E. Hepatosplenic T cell lymphoma. A review on 45 cases since the first report describing the disease as a distinct lymphoma entity in 1990. Leukemia. 2000;14(6):991-997.
3. Lamy T, Moignet A, Loughran TP. LGL leukemia: from pathogenesis to treatment. Blood. 2017;129(9):1082-1094.
4 Barilà G, Grassi A, Cheon H, et al. Tγδ LGLL identifies a subset with more symptomatic disease: analysis of an international cohort of 137 patients. Blood. 2023;141(9):1036-1046.
5. Cooke CB, Krenacs L, Stetler-Stevenson M, et al. Hepatosplenic T-cell lymphoma: a distinct clinicopathologic entity of cytotoxic gamma delta T-cell origin. Blood. 1996;88(11):4265-4274.
6. Vega F, Medeiros LJ, Gaulard P. Hepatosplenic and other gammadelta T-cell lymphomas. Am J Clin Pathol. 2007;127(6):869-880.
7 Wang CC, Tien HF, Lin MT, et al. Consistent presence of isochromosome 7q in hepatosplenic T gamma/delta lymphoma: a new cytogenetic-clinicopathologic entity. Genes Chromosomes Cancer. 1995;12(3):161-164.
8. Nicolae A, Xi L, Pittaluga S, et al. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leukemia. 2014;28(11):2244-2248.
cytological data. FL-G reviewed histological data. FG, LC, LB, PL, CLL, PC-L, A-CG, CB, ER, ED, CF, FB-J, SW, and VB performed the initial investigations and addressed the cases for constitution of the cohort. TL managed the patients with large granular lymphocytic leukemia. MR provided flow cytometric data. AD, SB, TF, and CP analyzed the molecular data. All the authors critically reviewed the paper.
The authors thank the French Group of Cellular Hematology (Groupe Francophone d’Hématologie Cellulaire, Bron, France) for this collaborative work.
Data-sharing statement
Original data including metadata, images and fastq files are available upon request from cedric.pastoret@chu-rennes.fr.
9 McKinney MS, Moffitt AB, Rempel RE, et al. SETD2 functional loss through mutation or genetic deletion promotes expansion of normal and malignant γδ T cells through loss of tumor suppressor function and upregulation of oncogenic pathways. Blood. 2016;128(22):1052.
10. McKinney M, Moffitt AB, Gaulard P, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017;7(4):369-379.
11. Travert M, Huang Y, de Leval L, et al. Molecular features of hepatosplenic T-cell lymphoma unravels potential novel therapeutic targets. Blood. 2012;119(24):5795-5806.
12. Teramo A, Binatti A, Ciabatti E, et al. Defining TCRγδ lymphoproliferative disorders by combined immunophenotypic and molecular evaluation. Nat Commun. 2022;13(1):3298.
13. Brüggemann M, Kotrová M, Knecht H, et al. Standardized nextgeneration sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. 2019;33(9):2241-2253.
14 Przybylski GK, Wu H, Macon WR, et al. Hepatosplenic and subcutaneous panniculitis-like gamma/delta T cell lymphomas are derived from different Vdelta subsets of gamma/delta T lymphocytes. J Mol Diagn. 2000;2(1):11-19.
15. Li Y, Li G, Zhang J, et al. The dual roles of human γδ T cells: anti-tumor or tumor-promoting. Front Immunol. 2020;11:619954.
Hemophagocytic lymphohistiocytosis (HLH) is a rare, life-threatening condition in which immune hyperactivation and dysregulation results in a cytokine storm. HLH may be primary (due to underlying genetic mutations) or secondary to triggers such as infections, autoimmune/autoinflammatory disorders, and malignancies (HLH-M).1 In over 90% of cases, HLH-M is driven by a hematologic malignancy, with lymphoma the most common trigger.2 The clinical features of HLH-M may be driven by the hematologic malignancy itself, infection secondary to the immunosuppressed state, loss of immune homeostasis, or a combination of these. The association between acute leukemia and HLH is less well-recognized despite a few reported cases.3,4 A single case series described HLH-M with acute myeloid leukemia (AML).4 In keeping with the high mortality associated with HLH-M, the overall survival (OS) for such patients was significantly lower than age-, disease- and treatment-matched controls without HLH. Current recommended treatments for HLH-M include steroids, intravenous immunoglobulin (IVIG) and etoposide/ciclosporin-containing regimens.5 In contrast, the recommended treatment for HLH-macrophage activation syndrome (MAS), (seen in rheumatologic disease) is anti-IL1-directed therapy such as anakinra. In 2021, the UK approved the use of anakinra to treat HLH of any cause; however, the safety and efficacy data of anakinra in HLH-M remains limited.6
We report a retrospective review of all patients diagnosed with HLH and AML or acute lymphoblastic leukemia (ALL) in a single London tertiary referral center (University College London Hospitals NHS Foundation Trust [UCLH]), between 1 January 2019 and 31 December 2022, their clinical course, and successful and safe therapy with anakinra-containing regimens. Patients (or their relatives if deceased) provided written consent. The UCLH Hematology Department registered the project as a service evaluation.
To define patients with a diagnosis of HLH, we analyzed electronic records of all patients referred to the UCLH HLH multidisciplinary meeting (MDM), a national framework for case discussions among hematology, rheumatology, infectious disease, and intensive care (ICU) clinicians. We cross-referenced patients thus identified with all adult and teenage patients treated with AML and ALL on local disease-specific registries. This identified 468 patients with acute leukemia, of whom 13 (2.8%) were diagnosed with HLH. Clinical and laboratory data were collected using the electronic health records (Table 1). For diagnostic purposes, we utilized HScores (comprising laboratory and clinical parameters providing an HLH probability for each patient) using laboratory data.7 HLH was diagnosed in patients with
a clinical suspicion of HLH and an HScore >169. Median age at time of HLH diagnosis was 37 years (range 16-74). Four patients were female and 8 were male. Two patients (15%) had standard risk B-ALL. Eleven patients (85%) had AML; of these, 4 (36%) had favorable risk (inversion 16 or NPM1 mutated), whilst 7 (64%) had adverse risk (MLL rearrangement, myeloid sarcoma, therapy-related AML, and AML with myelodysplastic syndrome-related changes). Four patients were diagnosed at time of leukemia presentation, 6 during induction chemotherapy, and 3 in consolidation. Ten patients (77%) had active leukemia at the time of their HLH diagnosis; only 3 (27%) were in remission. Patients were treated with various types of immunosuppressive therapy (IST). Twelve (92%) received anakinra, 3 (17%) of whom had anakinra monotherapy whilst the remainder had anakinra with steroids (38%), steroids and IVIG (15%), or steroids, IVIG and etoposide (8%). One patient (8%) had steroids and IVIG alone. Anakinra was given intravenously in 11 patients and subcutaneously in one. The mean daily dose was 4.6 mg/kg. Eleven patients (92%) were neutropenic prior to starting anakinra; 8 patients were severely neutropenic with an absolute neutrophil count <0.5x109/L. All patients required ICU input during their HLH treatment. Eight (62%) were admitted to ICU on diagnosis with HLH and required ventilatory and/or vasopressor support; the remainder were reviewed by ICU outreach. In all, 92% patients had an initial rapid clinicopathologic response following IST, becoming afebrile, with a reduction in oxygen requirement and hemodynamic stability seen within 24-72 hours. One patient (8%) had a reduction in oxygen requirement following anakinra monotherapy but continued to spike temperatures and required oxygen for two weeks. However, this patient was positive for COVID-19 on PCR and pneumonitis on chest radiology. Another patient (8%) had an initial clinical response to anakinra, methylprednisolone and IVIG triple therapy, but displayed subsequent rapid deterioration in the context of pseudomonal infection, and required readmission to ICU and continued anakinra. The patient nevertheless died of multiorgan failure.
Nine patients (69%) had no HLH recurrence following IST. Three (23%) had an initial clinical response to IST but had subsequent biochemical and clinical relapse of HLH. These patients all had relapsed/refractory disease on HLH recurrence; no patient had relapsed HLH without contemporaneous leukemia relapse. One of these subsequently received etoposide and further steroids resulting in resolution of clinical HLH, despite underlying refractory AML. The second was re-challenged with anakinra and methylprednisolone alongside venetoclax-azacitidine as
second-line AML treatment with a clinical response to HLH treatment, although subsequently therapy was withdrawn due to refractory leukemia. The third had refractory myeloid sarcoma and continued maintenance prednisolone alongside palliative care.
Patient outcomes are shown in Figure 1. Only one (8%) patient died directly from HLH. Eight (62%) remain in remission from leukemia at the end of the study period. Three of these (23%) were in remission following chemotherapy only. Three other patients (23%) had relapsed leukemia following their HLH diagnosis but are now in remission post allogeneic stem cell transplant. Three of the 6 patients (50%) who had relapsed leukemia had contemporaneous HLH relapse. Two patients had allogeneic transplant for unfavorable risk disease; both are in remission post allogeneic transplant. Three patients (23%) died of refractory leukemia, all having unfavorable risk disease at presentation. A fourth patient (8%) has refractory therapy-related AML following initial treatment for follicular lymphoma and is currently receiving palliative care. Another patient (8%)
is in remission from leukemia but remains on dialysis for end-stage renal failure secondary to HLH. Of the 3 patients who were in remission from leukemia at the time of HLH diagnosis, one subsequently had an allograft for high-risk disease, and one died from HLH. The third patient had developed HLH secondary to acute COVID-19 infection, and remained in remission following HLH treatment. Although the link between HLH and lymphoma is well characterized, data on HLH in the context of acute leukemia are limited. In this case series, 2.8% of the patients treated with acute leukemia developed HLH, which is similar to the figure of 3% for lymphoma quoted elsewhere.8 The majority (92%) of patients had microbiologic or radiologic evidence of infection on HLH diagnosis. However, many such infections involved organisms not classically associated with HLH, e.g., rhinovirus and Streptococcus oralis 9
We hypothesize that the clinical features of HLH in these patients may be driven by a combination of the underlying leukemia, chemotherapy and infection resulting in immune dysregulation. The central role of leukemia in the
Parameter
Median H score (range)
Values
214 (181-299)
Probability of HLH, % 93-96
Peak serum ferritin, mcg/L, median (range) [reference <500] 79,889 (7,197-488,068)
Serum triglyceride, mmol/L, median (range) [reference <1.5] 3.7 (1.8-8.5)
Hepatosplenomegaly, N (%) 5 (38)
BM evidence of hemophagocytosis, N (%)
Yes
No
Not done
Evidence of infection, N (%)
Bacterial, N (%)
Viral, N (%)
Radiological, N (%)
7 (61) 4 (36) 2 (15)
12 (92)
4 (31) (E. Coli, Klebsiella, pseudomonas, streptococcus oralis) 3 (23) (COVID-19: N=2; Rhinovirus: N=1) 3 (23) (arm abscess: N=1; consolidation on HRCT: N=2)
BM: bone marrow; HLH: hemophagocytic lymphohistiocytosis; HRCT: high-resolution computed tomography; N: number.
pathogenesis of HLH-M in these cases is highlighted by the relapse of HLH occurring alongside leukemia relapse in 3 of the 6 patients who relapsed their leukemia. One patient developed HLH whilst in remission from leukemia and remained in remission from both leukemia and HLH without an allograft; however, as HLH occurred in the context of acute COVID-19 infection, which is known to be associated with HLH/MAS, it is possible that COVID-19 rather than leukemia was the key driver in this context. All patients in our cohort required ICU support. However, compared to existing literature, treatment responses and outcomes were good, with an OS rate of 62% in our cohort. Only one patient died of HLH alone; the remaining 3 patients who died during the study period died of relapsed-refractory leukemia. This contrasts with other studies with higher mortality rates; Tamamyan et al 4 reported an OS rate of 15% in 13 patients with HLH-M. Delavigne et al. 3 reported a 3-month mortality rate of 36% in AML patients presenting with HLH during induction compared to our figure of 23% for this period.
Twelve patients (92%) in our cohort received anakinra as part of their therapy for HLH-M; in 9 patients, this was used alongside steroids and/or IVIG. We recommend using intravenous anakinra since this has been shown to be more effective than subcutaneous forms in critically unwell patients.10 Anakinra use allows a reduction in the amount and duration of steroids required, which is critical given the known toxicity of high-dose steroids, including further immunosuppression and risk of opportunistic infections.11 Our data show treatment with anakinra appears safe in the context of neutropenia. We recommend early discussion of patients with possible HLH-M in an MDM setting, and early use of IV anakinra, IVIG and methylprednisolone first-line. The use of etoposide is reserved for refractory cases. We believe this is the first case series demonstrating effective treatment and high OS of HLH-M patients with concurrent leukemia diagnosis using anakinra. We hypothesize that early treatment with anakinra alongside other immunosuppression may explain the trend towards improved outcomes in our cohort. Further work is needed to characterize which patients are at increased risk of de-
1. Lehmberg K and Ehl S. Diagnostic evaluation of patients with suspected hemophagocytic lymphohistiocytosis. Br J Haematol. 2013;160(3):275-287.
2. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516.
3. Tamamyan GN, Kantarjian HM, Ning J, et al. Malignancyassociated hemophagocytic lymphohistiocytosis in adults: relation to hemophagocytosis, characteristics, and outcomes. Cancer. 2016;122(18):2857-2866.
4 Delavigne K, Bérard E, Bertoli S, et al. Hemophagocytic
veloping HLH to ensure its early detection and treatment.
Hannah Al-Yousuf,1 Jenny O’Nions,1 Andrew J. Wilson,1 Satyen Gohil,1 Jessica J. Manson1 and Elspeth M. Payne1,2
1Department of Hematology, University College London Hospitals and 2UCL Cancer institute, London, UK
Correspondence:
ELSPETH PAYNE - e.payne@ucl.ac.uk
HANNAH AL-YOUSUF - hannah.al-yousuf@nhs.net
https://doi.org/10.3324/haematol.2023.283879
Received: August 8, 2023.
Accepted: January 22, 2024.
Early view: February 1, 2024.
Published under a CC BY license
Disclosures
No conflicts of interest to disclose.
Contributions
HA carried out data collection and analysis, and wrote the manuscript. EP supervised the study and provided support in writing the manuscript. SG, JM, JO and AW provided additional supervision and support in writing the manuscript.
Funding
This work was funded by a UCLH Charity grant for HLH Service Development (reference 7253; to JM) and a CRUK Advanced Clinician Scientist Fellowship (A24873; to EMP).
Data-sharing statement
Original data and protocols can be obtained by contacting the corresponding authors.
syndrome in patients with acute myeloid leukemia undergoing intensive chemotherapy. Haematologica. 2014;99(3):474-480.
5. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477.
6. NHS England. Clinical Commissioning Policy: anakinra for hemophagocytic lymphohistiocytosis (HLH) for adults and children in all ages [210701P] (1924). https://www.england.nhs. uk/wp-content/uploads/2021/10/1924-Clinical-commissioningpolicy-anakinra-for-haemophagocytic-lymphohistiocytosis-.pdf Accessed August 1, 2023.
7 Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613-2620.
8. Daver N, McClain K, Allen CE, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123(17):3229-3240.
9 Mostaza-Fernández JL, Guerra Laso J, Carriedo Ule D, Ruiz de Morales JM. Hemophagocytic lymphohistiocytosis associated with viral infections: diagnostic challenges and therapeutic
dilemmas. Rev Clin Esp (Barc). 2014;214(6):320-327.
10 Mehta P, Cron RQ, Hartwell J, Manson JJ, Tattersall RS. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020;2(6):e358-e367.
11. Halyabar O, Chang MH, Schoettler ML, et al. Calm in the midst of cytokine storm: a collaborative approach to the diagnosis and treatment of hemophagocytic lymphohistiocytosis and macrophage activation syndrome. Pediatr Rheumatol Online J. 2019;17(1):7.
High hyperdiploidy (HeH), characterized by nonrandom chromosomal gains resulting in 51-67 chromosomes, is the most common genetic subtype of childhood B-cell precursor (BCP) acute lymphoblastic leukemia (ALL). HeH ALL typically occurs in children aged 2-4 years and overall corresponds to 25% of cases in the pediatric (<18 years) population, but, for reasons unknown, becomes less frequent with increasing age; in adult ALL it is relatively rare. Several studies have shown clear evidence of HeH frequently arising already before birth in pediatric cases.1-9 However, there are very little data on whether this also holds true for HeH arising in older children, adolescents and adults.
We previously utilized somatic single nucleotide variants (SNV) to study the “age” of the trisomies, i.e., whether their origin was recent or not in terms of clonal evolution, in 16 cases of pediatric HeH ALL.10 This was done by analyzing the mutant allele frequencies (MAF) of all somatic SNV in trisomic chromosomes to investigate whether they were present in one of two or two of three chromosomal homologues. Whereas the former could arise either before or after the chromosome became trisomic (B/ATRI SNV), the latter must have arisen before the trisomy was formed and become duplicated with the homologue (BTRI SNV) (Figure 1A). We found that BTRI SNV constituted only a small fraction of the SNV in most cases, suggesting that the chromosomal gains arose very early during clonal evolution and before the cell had acquired many passenger mutations, in line with a prenatal origin. However, the two adolescent patients in the study had much higher frequencies of BTRI SNV, indicating that older patients with HeH may differ from younger patients in this regard. Here, we have addressed this possibility further. We ascertained whole genome sequencing (WGS) data from HeH cases from local biobanks (not included in Paulsson et al. 10 and selected based on material being available except for ten cases selected based on age >16 years) (N=31), from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) Initiative (https://www.ncbi.nlm. nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000464. v21.p8) (N=33), and from the St. Jude Cloud11-14 (N=127). HeH was defined as 51-67 chromosomes without concurrent ETV6::RUNX1, TCF3::PBX1, BCR::ABL1, or KMT2A rearrangements. In addition, four cases with concurrent BCR::ABL1 and HeH were investigated (Online Supplementary Table S1). Analyses of BTRI data for 40 cases have previously been published.15 Informed consent was obtained according to the Declaration of Helsinki and the study was approved by the Swedish Ethical Review Authority. All somatic SNV
in clonal trisomies were ascertained and categorized as BTRI (MAF=~0.67) or B/ATRI SNV (MAF=~0.33) (Figure 1A), excluding somatic SNV with MAF<0.15. Cases with large subclones for trisomic chromosomes, making the distinction between BTRI and B/ATRI unclear (N=18), with less than 65% leukemic cells based on MAF (N=15), or with <100 (N=26) or >1,000 (N=10) somatic SNV in trisomic chromosomes, were excluded. The final HeH cohort included 117 pediatric (1-17 years at diagnosis; median age 4 years) and five adult cases; 99 were aged 1-9 years and 23 ≥10 years at diagnosis (25 from local biobanks, 25 from TARGET, and 72 from St. Jude Cloud) (Online Supplementary Table S1). There were no differences in age (P=0.27; Mann-Whitney two-sided test) or sex distribution (P=0.81; Fisher’s exact test) in cases excluded (36% of initial cases) and those retained. Mann-Whitney two-sided test, Spearman correlation, and Fisher’s exact test were used as indicated below and P values <0.05 were considered significant. All statistical analyses were done using R x64 v4.4.1 (https:// www.r-project.org/).
The median numbers of somatic, BTRI and B/ATRI SNV were 277.5 (range, 101-842), eight (range, 0-79), and 265 (range, 98-825), respectively, in HeH cases. To adjust for possible differences related to read depths between the cohorts and to modal number, all subsequent analyses were done on the proportion of BTRI/all SNV in trisomic chromosomes, since this should not be affected by the total number of SNV.
The median BTRI/all SNV was 3.1% (range, 0-23%; Online Supplementary Table S1), indicating that the time period after the HeH arose was always longer than the time period before, in line with previous findings.10 No sex differences were seen (Mann-Whitney two-sided test; P=0.0531). However, whereas the BTRI/all SNV percentages appeared to be constant in patients aged 1-9 years, they increased linearly in those ≥10 years at diagnosis (Spearman correlation coefficient=0.64, P=0.00092; Figure 1B, C). The latter cases also had a significantly higher percentage of BTRI SNV (median 2.6% in patients 1-9 years vs. 9.8% in patients ≥10 years; Mann-Whitney two-sided test, P=5.26x10-9; Figure 1D). That cases <10 years generally had the same proportion of BTRI SNV agrees well with a prenatal origin for the majority of these because their expected number of BTRI SNV would then be constant. In contrast, that cases ≥10 years show an increasing number of BTRI SNV by age suggests that the chromosomal gains generally arise postnatally at different time points in these cases, with the number of BTRI SNV being higher in older patients since the chromosomes that become duplicated will have acquired more passenger mutations with increasing
age. Thus, our results show that the chromosomal gains in HeH ALL arising in older patients - outside the “age peak” in early childhood – may not arise in utero.
Support for the hypothesis that pediatric HeH ALL may have a prenatal origin has previously been obtained by detection of clonotypic IGH rearrangements in Guthrie cards, observations of monozygotic twins with concurrent HeH ALL, and detection of trisomies in saved cord blood cells from children who later developed leukemia.1-9 Whereas the latter two of these methods have provided unequivocal evidence of the HeH itself being present before birth in some cases, studies of clonotypic IGH rearrangements without analysis of chromosome 14 copy number can only provide evidence for the presence of a (pre)leukemic clone, not HeH itself. In total, we found 39 reported HeH cases where prenatal origin was investigated and the patient age was given (Table 1);1-9
both positive (N=29) and negative (N=10) cases had a median age of 2 years at diagnosis. Two of three HeH cases older than 10 years at diagnosis had evidence of a preleukemic clone - albeit not conclusive for HeH since chromosomal copy numbers were not addressed - at birth.7 This agrees with our findings because the proportion of BTRI/all SNV varied also in patients ≥10 years, with some having low levels of BTRI SNV and thereby possibly a prenatal origin of the chromosomal gains.
A second question arising from our findings is whether chromosomal gains arising postnatally in older patients are the primary event or if they occur as a secondary aberration to another leukemia-initiating lesion. We have previously shown that in BCP ALL with concurrent BCR::ABL1-fusion and HeH, the chromosomal gains are most likely secondary to the fusion gene.16 Thus, such cases can serve as a model
Figure 1. The fraction of BTRI mutations, arising before the chromosomal gains, is higher in older patients with high hyperdiploid acute lymphoblastic leukemia. (A) Example of somatic single nucleotide variants (SNV) in trisomic chromosomes from case L1, showing 2 distinct peaks corresponding to B/ATRI and BTRI SNV, respectively. (B) The fraction of BTRI/all SNV in relation to age is constant in cases 1-9 years old at diagnosis but increases with age in cases 10 years or older at diagnosis. The center of the boxplot is the median and lower/upper hinges correspond to the first/third quartiles; whiskers are 1.5-times the interquartile range and the individual data points are shown as black points. The median value is shown as red points for each group. (C) Spearman correlation shows that the fraction of BTRI/all SNV increases linearly with age in patients older than 10 years at diagnosis. (D) Boxplot of the fraction of BTRI/all SNV in patients 1-9 and ≥10 years old at diagnosis, respectively, showing a higher fraction in the older age group. The center of the boxplot is the median and lower/upper hinges correspond to the first/third quartiles; whiskers are 1.5-times the interquartile range and the individual data points are shown as black points.
Table 1. High hyperdiploid acute lymphoblastic leukemia reported in the literature investigated for prenatal origin and with age information available.
for the characteristics of HeH as a secondary aberration. We investigated four ALL with concurrent BCR::ABL1 and HeH, finding very high BTRI/all SNV in all (median 22%; range 1944%), including a 4-year-old case (Online Supplementary Table S1), in line with HeH being relatively new in terms of leukemic evolution. In fact, the proportion of BTRI SNV was higher than in all but one of the HeH cases without BCR::ABL1 investigated. Thus, the analysis corroborated the view that HeH as a secondary event would indeed have high BTRI/all SNV.
We attempted to find potential other primary events in cases with high BTRI/all SNV levels, but analysis of somatic WGS data for these cases did not reveal any unusual chromosomal events, structural rearrangements, targeted deletions, or coding mutations (Online Supplementary Table S2). Furthermore, the pattern of chromosomal gains, chromosomal modal numbers, and frequencies of the most common additional somatic events were similar (Online Supplementary Table S2). Thus, the only difference we can identify between HeH ALL with high BTRI/all SNV levels and the remaining HeH cases is that the former are, on average, older at diagnosis. Importantly though, some cases with high BTRI/all SNV were found in patients <10 at diagnosis. Thus, if there is an underlying primary event in these cases, it seems to be relatively age-independent although population-based cohorts would be needed to determine this. In conclusion, we show that there is a marked difference in the percentage of BTRI/all SNV between young children and older children, adolescents, and adults in HeH ALL. We interpret this as most HeH ALL occurring in patients <10 years at diagnosis, constituting the majority of cases, have a prenatal origin of the chromosomal gains, in line with previous findings.1-9 In contrast, HeH ALL occurring in older children, adolescents, and adults generally have a later, most likely postnatal, origin of the chromosomal gains. Notably, some cases in young children may also fall into this group, suggesting a hitherto unknown heterogeneity within HeH ALL. Considering that we have recently shown that the chromosomal gains occur simultaneously and very early in the leukemogenesis of HeH ALL,15 we deem it likely that this means that also the ALL arises postnatally in these cases, although we cannot definitely exclude another, as yet hidden, primary event.
Minjun Yang,1 Rebeqa Gunnarsson,1 Nicolas Duployez,2 Marketa Zaliova,3,4 Jan Zuna,3,4 Bertil Johansson1,5 and Kajsa Paulsson1
1Department of Laboratory Medicine, Division of Clinical Genetics, Lund University, Lund, Sweden; 2Laboratory of Hematology, Centre Hospitalier Universitaire (CHU) Lille, University of Lille, INSERM Unité 1277 Canther, Lille, France; 3Department of Pediatric Hematology and Oncology, Second Faculty of Medicine, Charles University/University Hospital Motol, Prague, Czech Republic; 4Childhood Leukemia
Investigation Prague (CLIP), Prague, Czech Republic and 5Department of Clinical Genetics, Pathology, and Molecular Diagnostics, Office for Medical Services, Laboratory Medicine, Region Skåne, Lund, Sweden
Correspondence: K. PAULSON - kajsa.paulsson@med.lu.se
https://doi.org/10.3324/haematol.2023.284128
Received: August 21, 2023.
Accepted: February 8, 2024. Early view: February 15, 2024.
©2024 Ferrata Storti FoundationPublished under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
MY, RG, ND, MZ, JZ, BJ and KP performed research. MY and KP performed data analysis. KP supervised the study and wrote the manuscript with input from all authors.
Acknowledgments
The results published here are in part based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) Initiative (phs000464). This study makes use of data generated by the St. Jude Children’s Research Hospital Genomes for Kids Study.13 This study makes use of data generated by St. Jude Children’s Research Hospital.14
Funding
This study was supported by grants from the Swedish Childhood Cancer Fund, grant numbers PR2020-0033 MY, TJ2020-0024 MY, PR2021-0005 BJ, and PR2021-0016 KP; the Swedish Cancer Fund, grant numbers 23 2694 Pj (to BJ) and 22 2062 Pj (to KP); governmental funding of clinical research within the National Health Service (to BJ and KP); National Institute for Cancer Research, grant number LX22NPO5102, funded by the European Union–Next Generation EU (to MZ and JZ), Czech Health Research Council, grant number NU21-03-00128 (to MZ and JZ); and the Swedish Research Council, grant numbers 2020-01164 (to BJ) and 2020-00997 (to KP).
Data-sharing statement
The WGS data from Lund University have been deposited to the European Genome-phenome Archive (EGA) under accession number EGAS00001007052. This dataset is available under restricted access due to privacy concerns; access can be obtained for academic research by contacting the Data Access Committee via EGA. The WGS data generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) are available at https://portal. gdc.cancer.gov/projects.under accession code phs000464. WGS data for pediatric tumor samples used for analysis in this study were obtained from St. Jude Cloud.14
1. Panzer-Grümayer ER, Fasching K, Panzer S, et al. Nondisjunction of chromosomes leading to hyperdiploid childhood B-cell precursor acute lymphoblastic leukemia is an early event during leukemogenesis. Blood. 2002;100(1):347-349.
2. Maia AT, van der Velden VHJ, Harrison CJ, et al. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17(11):2202-2206.
3. Bateman CM, Alpar D, Ford AM, et al. Evolutionary trajectories of hyperdiploid ALL in monozygotic twins. Leukemia. 2015;29(1):58-65.
4. Yagi T, Hibi S, Tabata Y, et al. Detection of clonotypic IGH and TCR rearrangements in the neonatal blood spots of infants and children with B-cell precursor acute lymphoblastic leukemia. Blood. 2000;96(1):264-268.
5. Taub JW, Konrad MA, Ge Y, et al. High frequency of leukemic clones in newborn screening blood samples of children with B-precursor acute lymphoblastic leukemia. Blood. 2002;99(8):2992-2996.
6. Maia AT, Tussiwand R, Cazzaniga G, et al. Identification of preleukemic precursors of hyperdiploid acute lymphoblastic leukemia in cord blood. Genes Chromosomes Cancer. 2004;40(1):38-43.
7 Gruhn B, Taub JW, Ge Y, et al. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia. 2008;22(9):1692-1697.
8. Kacanski N, Kolarovic J, Kostic T, et al. Presence of leukemic clone-specific immunoglobulin heavy chain rearrangements in
neonatal blood spots of children with B-cell precursor acute lymphoblastic leukemia. Int J Lab Hematol. 2024;46(2):303-311.
9 Zuna J, Prouzova Z, Kalina T, et al. Backtracking of ALL to cord blood. Leuk Res. 2009;33(8):e107-108.
10. Paulsson K, Lilljebjörn H, Biloglav A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47(6):672-676.
11. Downing JR, Wilson RK, Zhang J, et al. The pediatric cancer genome project. Nat Genet. 2012;44(6):619-622.
12. McLeod C, Gout AM, Zhou X, et al. St. Jude Cloud: a pediatric cancer genomic data-sharing ecosystem. Cancer Discov. 2021;11(5):1082-1099.
13. Newman S, Nakitandwe J, Kesserwan CA, et al. Genomes for kids: the scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov. 2021;11(12):3008-3027.
14. Rusch M, Nakitandwe J, Shurtleff S, et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat Commun. 2018;9(1):3962.
15. Woodward EL, Yang M, Moura-Castro LH, et al. Clonal origin and development of high hyperdiploidy in childhood acute lymphoblastic leukaemia. Nat Commun. 2023;14(1):1658.
16. Paulsson K, Harrison CJ, Andersen MK, et al. Distinct patterns of gained chromosomes in high hyperdiploid acute lymphoblastic leukemia with t(1;19)(q23;p13), t(9;22)(q34;q22) or MLL rearrangements. Leukemia. 2013;27(4):974-977.
To discuss outcomes with patients in daily clinical practice, and to facilitate outcome comparisons between institutions and scientific research, a standardized Core Outcome Set (COS) for patients with multiple myeloma (MM) is needed.1 In the past five years, multiple COS have been developed for patients with MM across Europe.2-4 Although these COS share the same objectives and include similar outcomes, the sets also exhibit differences in the definitions and measures being used. Since incidence rates in MM are relatively low,5 standardizing these COS is warranted to accelerate data collection and data comparison. The goal of this study was, therefore, to establish a European consensus-based, standardized COS for patients with MM. This was achieved by aligning existing COS through consensus group discussions in a European and diverse group of stakeholders/experts, which resulted in an updated European consensus-based standardized COS and accompanying definitions or measures for patients with MM for use in routine clinical practice and research. The outcomes represent important indicators that are most relevant to patients with MM. With the use of this updated European consensus-based standardized COS, we expect to be able to facilitate therapeutic decision-making, allow outcome comparison across centers and countries to guide improvements in clinical practice, and accelerate scientific research.
The continuous development of new treatments for multiple myeloma (MM) provides more options to achieve increased survival, but also adds more complexity to the treatment of the disease.6 This increase in options leads to longer treatment duration and better outcome,7,8 often associated with a higher risk of adverse events such as acute renal failure, anemia, pneumonia/infections, cardiac toxicity, and polyneuropathy.9 Moreover, patients might develop long-term consequences such as fatigue, pain, cognitive problems, and depression,10 impacting patients’ health-related quality of life (HRQoL).11 Therefore, modern therapeutic strategies focus not only on improving overall survival, but also on prevention and management of acute and long-term side effects of therapy, optimizing therapy duration, and improving HRQoL.9
To monitor outcome progress, COS are needed. In the past five years, multiple COS have been developed for patients with MM across Europe,2-4 highlighting the urgent need for such a tool. Since incidence rates in MM are relatively low,5 standardizing COS is warranted to accelerate data collection and (inter)national data comparison. Also, since HRQoL becomes more pronounced as a primary or
secondary outcome in MM research, comparison of HRQoL outcomes requires standardization of patient-reported outcomes (PRO) and PRO measures (PROM).12,13
The goal of this study was, therefore, to establish a European consensus-based, standardized COS for patients with MM, which would serve as a valuable tool for clinical practice and research as a reference framework of what and how to measure in MM. This study is part of the Health Outcomes Observatory (H2O) initiative. H2O has been designed to drive value-based health care (VBHC) in Europe by improving the sustainability of health care systems and supporting health care providers to optimize care delivery by use of COS that matter to patients.14
Three existing COS were identified through a literature search of MedLine, EMBASE, and the COMET database, and through the European Myeloma Network (EMN):
1. The IMPORTA project in Spain,2 published in 2018, in which global standards for collecting outcomes (and measuring instruments) that matter most to patients were defined. The set was based on a literature review and a 2-round Delphi questionnaire among hematologists, patients, and pharmacists in Spain.
2. A VBHC MM project in the Netherlands,4 published in 2020, in which outcomes that were most relevant to patients, and instruments to measure them for use in clinical practice, were defined. This set was based on literature review and consensus by MM experts and patients from 4 Dutch hospitals, and finalized by the Dutch MM HOVON working group.
3. The HARMONY Alliance,3 an innovative medicines initiative (IMI) across Europe, in which a COS for patients with MM was defined for use in clinical trials in 2021. This set was based on 2 Delphi rounds with hematologists, patient representatives, representatives of pharmaceutical companies, and policymakers across Europe.
The alignment and update of the 3 existing COS was undertaken by a group of experts (N=17) consisting of delegates of the existing COS development teams, hematologists across Europe active in the EMN, patients and patient advocates from national and European patient organizations, and representatives from pharmaceutical companies. Relevant experts were approached through ‘snowball sampling’, and ranged with respect to age and expertise; all were based across Europe. Three online consensus meetings were held in which the 3 existing COS were used as starting point. During the meetings, the COS were discussed and compared with respect to: (1) definition of patient population,
(2) patients’ condition, (3) clinical outcomes, (4) PRO, and (5) timing and measurement process.
Inclusion of the patient conditions and outcomes was based on a predefined criterion: if the patient conditions or outcomes were included in at least 2 of the existing COS, they were deemed eligible for the updated European consensus-based standardized COS. Outcomes or patient
conditions included in one of the existing COS underwent thorough discussions and were subject to voting, by counting all meeting participants in favor for including an outcome or patient condition (>half of participants). Feasibility of measurement and data availability (e.g., in clinical records) were considered when discussing whether an outcome should be included in the updated COS or not. The updated
Exclusion of or addition to European consensus-based standardized COS
Patient conditions
Family history
Anemia at baseline
Bone lesions
Neuropathy at baseline
Transplant eligible
Induction or maintenance therapy
Reason of treatment discontinuation
Healthcare access
Ethnicity
Socio-economic status
Clinical outcomes
Completion of treatment
Place of death: hospital?
Relapse
Relapse date
Treatment adjustment: date of treatment adjustment
Second primary malignancy
Patient-reported outcomes
Pathological fractures
Fear of physical exercise
Preferences and satisfaction
Treatment adherence
Motor neuropathy
Exclude
Exclude
Exclude
Exclude
Addition
Addition
Addition
Addition, optional
Optional (not for cross country comparison)
Optional (not for cross country comparison)
Exclude
Reason for exclusion or addition
Not specific enough to be standardized
Condition of limited value and too difficult to control data registry prior to diagnosis
Condition that is too difficult to categorize in an uniform manner with uniform techniques in different centers and countries
Condition of limited value and too difficult to control data registry prior to diagnosis
Added for a more comprehensive treatment registration
Added for a more comprehensive treatment registration
Added for a more comprehensive treatment registration
Based upon concerns with respect to unequal access to care raised by patient advocates.
Legal restriction in some countries
Not informative and less reliable for cross country comparison
Measured by other treatment-related outcomes
Exclude Outcome of limited value
Addition
Addition
Addition
Addition
Needed to calculate PFS
Needed to calculate PFS
Needed for a more comprehensive treatment registration
Added as it may influence treatment strategy
Exclude Outcome of limited value
Exclude
Exclude
Exclude
Addition
Outcome of limited value
Too diverse; should be included in separate PREM
Outcome relevant to a limited group of patients with MM (only for oral medication).
Raised by patients as one of the most important outcomes
COS: Core Outcome Set; MM: multiple myeloma; PFS: progression-free survival; PREM: patient-reported experience measures.
European consensus-based standardized COS was distributed via email to the expert group for final confirmation and was approved by all.
The population definitions were slightly different across the 3 COS: the IMPORTA set was developed for patients with newly diagnosed MM, the VBHC-MM set for patients with symptomatic MM aged ≥18 years who fulfil the International Myeloma Working Group (IMWG) criteria, and the HARMONY-Alliance set for all patients with MM without any further specification. After substantial discussion, the panellists reached consensus about the following patient population definition: patients with MM aged ≥18 years of age who are newly diagnosed or had a relapse/became refractory and patients with smoldering (asymptomatic) MM (according to the IMWG criteria). Exclusion criteria were defined as: patients aged <18 years and patients with monoclonal gammopathy of undetermined significance (MGUS).
The following patient conditions: age, gender, date of diagnosis, Revised-Multiple Myeloma International Staging System (R-ISS) disease stage, comorbidities, treatment, and functioning disability and health were included in the updated European consensus-based standardized COS (Online Supplementary Table S1) because these were all part of both the IMPORTA and the VBHC-MM set (Online Supplementary Table S2); the HARMONY-Alliance set did not define patient conditions. For some of the patient conditions, definitions slightly differed across the sets and were aligned after consensus discussion. Furthermore, related to treatment, 3 items were added for a more comprehensive treatment registration, i.e., transplant eligibility, induction or maintenance therapy, and reason of treatment discontinuation. Other patient conditions that were part of one of the sets, and that were included in the updated COS after consensus discussion, were ethnicity (optional), living situation, cytogenetic risk, frailty, height, weight, and educational level (optional). Moreover, an additional item about healthcare access (access to care and barriers to accessing care) was added as an optional item based upon concerns with respect to unequal access to care raised by patient advocates. The reasons for exclusion and addition of items are listed in Table 1.
The clinical outcomes overall survival, progression-free survival, infections, neuropathy, renal failure, anemia, cardiovascular toxicities, and venous thromboembolism were included in 2 or all 3 sets, and were, therefore, included in the updated European consensus-based standardized COS. Other clinical outcomes that were part of one of the sets, and that were included in the updated European consensus-based standardized COS, were minimal residual disease, response, therapy-free interval, date of death, and active treatment <30 days before death. Furthermore, relapse, treatment adjustment, and second primary malignancy were added with the reasons for addition listed in Online Supplementary Table S2.
The PRO HRQoL, mobility, overall daily functioning, anxiety, depressive symptoms, cognitive problems, social participa-
tion and work, fatigue, dyspnea, nausea, pain, sleep problems, appetite loss, gastrointestinal problems, financial problems, body image, peripheral neuropathy, relational and sexual problems were included in 2 or all 3 sets and were, therefore, included in the updated, European consensus-based standardized COS. The EORTC QLQ-C30 and QLQ-MY20 were in all sets defined as the measures to use for the included PRO. In the HARMONY-Alliance set, mainly PROM (e.g., EORTC QLQ-C30 and QLQ-MY20) were defined, overlapping the PRO determined in the other 2 sets.
The timing of measurement in all sets was determined based on clinical relevance and feasibility. This differs between countries, and probably also between hospitals/clinics. To set a preferred standard, it is advised that all clinical- and patient-reported outcomes are to be collected at time of diagnosis and at least every three months during the 1st year, every six months during the 2nd and 3rd years, and then annually. To be able to discuss outcomes during visits, it is necessary to collect PRO-data aligned with individual clinical care. This study resulted in an updated European consensus-based standardized COS and accompanying definitions or measures for patients with MM for use in routine clinical practice and research. This COS will be implemented in Europe as part of the H2O initiative.
Simone Oerlemans,1 Belle H. de Rooij,1,2+ Christine Bennink,3,4+ Lars Bullinger,5 Annemiek Broijl,4 Mattia D’Agostino,6 Edward Laane,7 Maria Teresa Lupo-Stanghellini,8 Aurore Perrot,9 Ruth Wester,4 Viorica Cursaru,10 Hans Scheurer,11 Jan Vesseur,11 Mehul Dalal,12 Rohini Sen,13 Tanja Stamm,14 Heinz Ludwig15 and Pieter Sonneveld;4 on behalf of the Innovative Medicines Initiative - Health Outcomes Observatory (H2O) consortium
1Department of Research and Development, Netherlands Comprehensive Cancer Organisation, Utrecht, the Netherlands; 2CoRPS - Center of Research on Psychology in Somatic Diseases, Department of Medical and Clinical Psychology, Tilburg University, Tilburg, the Netherlands; 3Department of Strategy, Amphia, Breda, the Netherlands; 4Department of Haematology, Erasmus Medical Center Cancer Institute, Rotterdam, the Netherlands; 5Department of Hematology, Oncology and Cancer Immunology, Campus Virchow, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; 6Myeloma Unit, Division of Hematology, University of Torino, Azienda Ospedaliero-Universitaria Città della Salute e della Scienza di Torino, Torino, Italy; 7Hematology-Oncology Clinic, University of Tartu, Tartu, Estonia; 8Hematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy; 9Hematology Department, Cancer University Institute Oncopole, Toulouse, France; 10European Myeloma Network, Bucharest, Romania; 11Myeloma Patients Europe, Brussels, Belgium; 12Takeda Development Center Americas Inc., Lexington, MA, USA; 13Abbvie, Somerville, MA, USA; 14Institute of
Outcomes Research, Center for Medical Data Science, Medical University of Vienna and Ludwig Boltzmann Institute for Arthritis and Rehabilitation, Vienna, Austria and 15Wilhelminen Cancer Research Institute, First Department of Medicine, Center for Oncology, Hematology, and Palliative Care, Clinic Ottakring, Vienna, Austria
+BHdR and CB contributed equally.
Correspondence:
SIMONE OERLEMANS - s.oerlemans@iknl.nl https://doi.org/10.3324/haematol.2023.284282
Received: September 18, 2023. Accepted: January 24, 2024. Early view: February 1, 2024.
Published under a CC BY license
Disclosures
MDa is employed at Takeda. SR is employed at Abbvie. TS reports personal fees from AOP Health, AbbVie, Pfizer, Roche, and Takeda, outside the submitted work. LB received grants from Bayer and Jazz Pharmaceuticals outside the submitted work and personal fees from AbbVie, Amgen, Astellas, Bristol Myers Squibb, Celgene, Daiichi Sankyo, Janssen, Jazz Pharmaceuticals, Novartis, Pfizer, and Sanofi. AB has
1. Porter ME. What is value in health care? N Engl J Med. 2010;363(26):2477-2481.
2. Blade J, Calleja MA, Lahuerta JJ, Poveda JL, de Paz HD, Lizan L. Defining a set of standardised outcome measures for newly diagnosed patients with multiple myeloma using the Delphi consensus method: the IMPORTA project. BMJ Open. 2018;8(2):e018850.
3. Lang KM, Bereczky T, Geissler J, et al. Pan-stakeholder Core Outcome Set (COS) definition for hematological malignancies within the Framework of Harmony and Harmony PLUS projects. Blood. 2022;140(Suppl 1):5285-5287.
4. Oerlemans S, Bennink MC, Levin MD, et al. Development of a patient centered outcome set for patients with multiple myeloma to be used in clinical practice. Hemasphere. 2020;4(3):e366.
5. Ludwig H, Novis Durie S, Meckl A, Hinke A, Durie B. Multiple myeloma incidence and mortality around the globe; interrelations between health access and quality, economic resources, and patient empowerment. Oncologist. 2020;25(9):e1406-e1413.
6. Cavo M, Gay F, Beksac M, et al. Autologous haematopoietic stem-cell transplantation versus bortezomib-melphalanprednisone, with or without bortezomib-lenalidomidedexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/ HO95): a multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020;7(6):e456-e468.
7 Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma:
received honoraria from Janssen, BMS, Sanofi and Amgen. PS received research funding from Amgen, Celgene/GSK, Janssen, Pfizer and Sanofi.
Contributions
All authors have reviewed the manuscript, believes it represents valid work, and approved it for submission. SO, BR, CB and PS participated in the research design. All authors participated in the writing of the paper, attended the consensus meetings, and took part in the research.
H2O has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 945345-2. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation program, EFPIA, Trial Nation and JDRF. The public grant funding is matched with in-kind contributions of EFPIA partners. In addition, LB has received funding within the Innovative Medicines Initiative 2 Joint Undertaking HARMONY under grant agreement N. 116026. PS received funding from HARMONY, from the European Hematology Association, and from the Multiple Myeloma Research Foundation MMRF.
Data-sharing statement
The data dictionary that supports the European consensus-based standardized core outcome set as displayed in Online Supplementary Table S1 is available from the corresponding author upon reasonable request.
EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(3):309-322.
8. van de Donk N, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397(10272):410-427.
9. Terpos E, Mikhael J, Hajek R, et al. Management of patients with multiple myeloma beyond the clinical-trial setting: understanding the balance between efficacy, safety and tolerability, and quality of life. Blood Cancer J. 2021;11(2):40.
10 Ojo AS, Araoye MO, Ali A, Sarma R. The impact of current therapeutic options on the health-related quality of life of patients with relapse/refractory multiple myeloma: a systematic review of clinical studies. J Cancer Surviv. 2023 Jan 16. doi: 10.1007/s11764-023-01332-1. [Epub ahead of print]
11. Ramsenthaler C, Osborne TR, Gao W, et al. The impact of disease-related symptoms and palliative care concerns on health-related quality of life in multiple myeloma: a multicentre study. BMC Cancer. 2016;16:427.
12. Fragola M. Patient-reported outcomes and assessment of quality of life: a focus on multiple myeloma. J Adv Pract Oncol. 2020;11(5):513-520.
13. Salek S, Ionova T, Oliva EN, et al. The reporting, use, and validity of patient-reported outcomes in multiple myeloma in clinical trials: a systematic literature review. Cancers (Basel). 2022;14(23):6007.
14 Stamm T, Bott N, Thwaites R, et al. Building a value-based care infrastructure in Europe: the Health Outcomes Observatory. NEJM Catalyst. 2021;2(3):1148-1156.
Multiple myeloma (MM) patients suffered from high mortality during the initial waves of the COVID-19 pandemic.1 Functional studies revealed an attenuated immune response to COVID-19 infection and vaccination in MM,2 with many patients remaining seronegative and at elevated risk of breakthrough infections and severe COVID-19.3,4 Waning of immune response is well documented, but little is known about the evolution of vaccination response following successive doses and predictors of persistently poor response after 4 doses. Here, we report results of a longitudinal prospective observational study that measured COVID-19 vaccination responses after doses 2, 3 and 4 in a UK population of MM patients. The study was based on the Rare UK Diseases Study (RUDY) platform (LREC 14/SC/0126 & RUDY LREC 17/SC/0501), an established online rare disease platform with dynamic consent and participant-entered data. The study was approved by South Central / Berkshire B Research Ethics Committee. MM patients were recruited between May 2021 to September 2022. Participants self-reported clinical details, including COVID-19 vaccination doses and dates, MM disease control (by International Myeloma Working Group [IMWG] response classification) and anti-myeloma therapy at time of each dose. Participants provided serum, EDTA and heparin blood samples ≥3 weeks following dose 2, 3 and 4. Collected serum samples were analyzed for COVID-19 spike (S) and nucleocapsid (N) antibodies (IgG serology only) by turbidimetry (Abbott), as previously described.2,5 Samples producing values >50 IU/mL and >1.4 IU/mL, respectively, were considered a positive result; the assay was bound by a maximum value of 40,000 IU/mL. Peripheral blood mononuclear cells (PBMC) were isolated from heparinized samples; lymphocyte subsets were determined by immunophenotyping, and an interferon γ-release assay (Oxford Immunotec T IGRA) was used to quantify COVID-19 specific effector T cells (separately against S and N antigens), as per the manufacturer’s instructions. Positive results were defined as >8 interferon γ-releasing cells/106 PBMC; the assay was bound by a maximum value of 50 normalized counts.
A total of 141 patients provided three longitudinal samples ≥3 weeks following doses 2 (N=241), 3 (N=240), and 4 (N=229) (Online Supplementary Table S1). The median time between last vaccination and sample collection was longer after dose 4 at 105 days (vs. 66 days post-2nd and 70 days post-3rd doses) (P<0.0001). Prior exposure to natural COVID-19 infection (anti-N seropositivity) was more com-
mon after the 4th dose (12.7%) compared to earlier doses (2.9-4.6%) (P<0.0001). More patients received an adenoviral vector-based versus mRNA-based vaccine as their 2nd dose (48.1% vs. 35.3%); however, mRNA-based vaccines comprised the majority of 3rd (93.3%) and 4th (95.6%) doses (P<0.0001). At the 4th dose, 41.9% of patients reported complete response (CR) or very good partial response (VGPR), and 17.5% were receiving anti-CD38/BCMA-targeting agents. Patients with 3 serial samples were analyzed for antibody titers (N=138) and T-cell IGRA counts (N=61) against COVID-19 spike (S) and nucleocapsid (N) antigens. Median anti-S antibody titers increased between post-2nd (1,058 IU/mL; 93% seropositive) to post-3rd (5,954 IU/mL; 96% seropositive), and post-3rd to post-4th (10,995 IU/mL; 98% seropositive) doses (P<0.0001) (Figure 1A). Positive T-cell IGRA to S-antigen was observed in 62%, 56%, and 70% of patients following doses 2, 3 and 4, respectively (Figure 1B). When examining the effect of booster doses, patients in the bottom quartile of anti-S response after 2 doses had a robust increase after booster doses (median 98 vs. 4,218 IU/ mL; P=0.0013), albeit with lower titers than those in the top quartile (P<0.0001) (Figure 1C). Similarly, patients in the top 50% of T-IGRA response after 2 doses maintained stronger IGRA count values than the lower 50% after the 3rd (mean 10 vs. 22; P=0.0244) and 4th (mean 13 vs. 29; P=0.0012) doses (Figure 1D). These findings support the benefit of booster doses in augmenting immunity, but illustrate considerable variability within the MM patient cohort.
We then explored how response was associated with factors related to vaccination. Firstly, patients with a concurrent humoral response to prior natural COVID-19 exposure (anti-N sero-positivity) had greater anti-S titers (P<0.0001) after doses 2-4, respectively (Figure 2A). Secondly, anti-S titers were greater in those with a concurrently positive T-IGRA response after doses 2-4 (P<0.0001) (Figure 2B), suggesting a possible relationship between strength of humoral and cellular response. Thirdly, a greater proportion of patients achieved positive T-IGRA responses following the A-A-M-M (2 adenoviral vector-based followed by 2 mRNA-based vaccines) regimen compared with the M-M-M-M (4 mRNA-based vaccines) regimen after doses 2-4 (P<0.001) ((Figure 2C), suggesting a stronger T-cell response in patients who had received heterologous vaccine platforms. Next, we examined clinical factors associated with response. IgG anti-S titers, following dose 4, were positively correlated with total serum IgM (Spearman’s r=0.39, P<0.0001) (Figure 2D), and serum IgA (Spearman’s r=0.36, P<0.0001), but
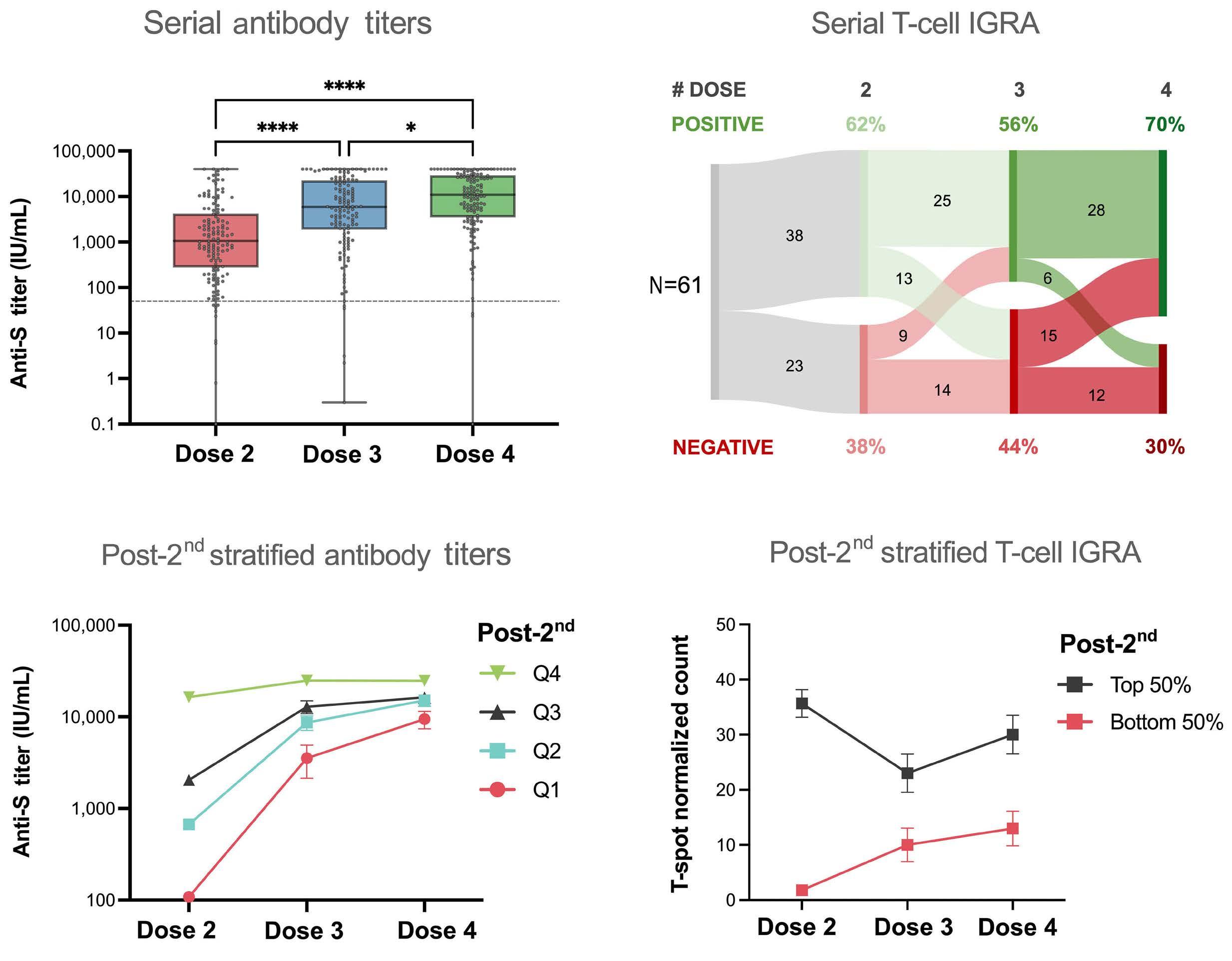
Figure 1. Longitudinal immune responses to 4 COVID-19 vaccinations in multiple myeloma patients. (A) Longitudinal change in anti-S antibody titers in uniform cohort of 138 patients providing 3 serial samples ≥3 weeks following doses 2-4. Kruskal-Wallis with Dunn’s multiple comparison test, *P<0.05, ****P<0.0001. (B) Sankey diagram showing longitudinal change in T-cell interferon γ-release assay (IGRA) positivity (normalized T-cell IGRA count ≥8) in uniform cohort of 61 patients with 3 serial T-cell assays following doses 2-4. (C) Longitudinal anti-S titers in patients stratified into 4 anti-S quartiles following 2nd dose (Q1 = bottom 25%; Q4 = top 25%) and prospectively followed after doses 3 and 4. Mean ± Standard Error of Mean (SEM). N=138 total. (D) Longitudinal normalized T-cell IGRA count to S antigen in patients stratified as top 50% (N=31) or bottom 50% (N=30) of T-cell IGRA following 2nd dose and prospectively followed after doses 3 and 4. Mean ± SEM.
not with IgG (P>0.05). Following the 4th dose, T-cell IGRA counts were positively correlated with peripheral total lymphocyte count (Spearman’s r=0.35, P<0.0001), CD4 (r=0.33, P<0.0001), CD8 (r=0.32, P<0.0001), and natural killer (NK) (r=0.27, P=0.0006) subsets (Online Supplementary Table S2). When assessing disease control and chemotherapy, patients achieving CR/VGPR at the time of dose 4 had greater median anti-S titers (24,278 IU/mL) than those with PR/stable disease (9,669 IU/mL) (P<0.01) or progressive/ relapsed (3,530 IU/mL) disease (P<0.0001) (Figure 2E); all anti-S seronegative patients had relapsed disease (N=4). Patients receiving anti-CD38 or BCMA-targeting agents at the 4th dose had lower anti-S titers (median 6,157 IU/mL) than those receiving other chemotherapy agents (median 16,102 IU/mL) (P<0.05) or no treatment (17,578 IU/mL) (P<0.05) (Figure 2E). Similarly, patients with progressive/
relapsed disease or those receiving anti-CD38/BCMA-targeting agents at the 4th dose had the lowest proportion achieving a positive T-cell IGRA (53.1% and 52.0%, respectively) (Figure 2F). Collectively, these analyses highlight immune and disease markers associated with variable vaccination-induced immunity after 4 doses. Finally, multivariate analysis identified independent predictors of persistently poor response after 4 doses (Table 1). Poor cellular response was defined by negative T-cell IGRA (below the manufacturer’s recommended cut-off). As few patients had an anti-S titer <50 IU/mL (assay positive cut-off), the World Health Organisation (WHO) threshold was used to define poor humoral response (7,352 IU/mL), as specified by the assay manufacturer. After the 4th dose, patients with anti-N seropositivity were less likely to have low anti-S (P=0.0011). Those with progressive/relapsed dis-
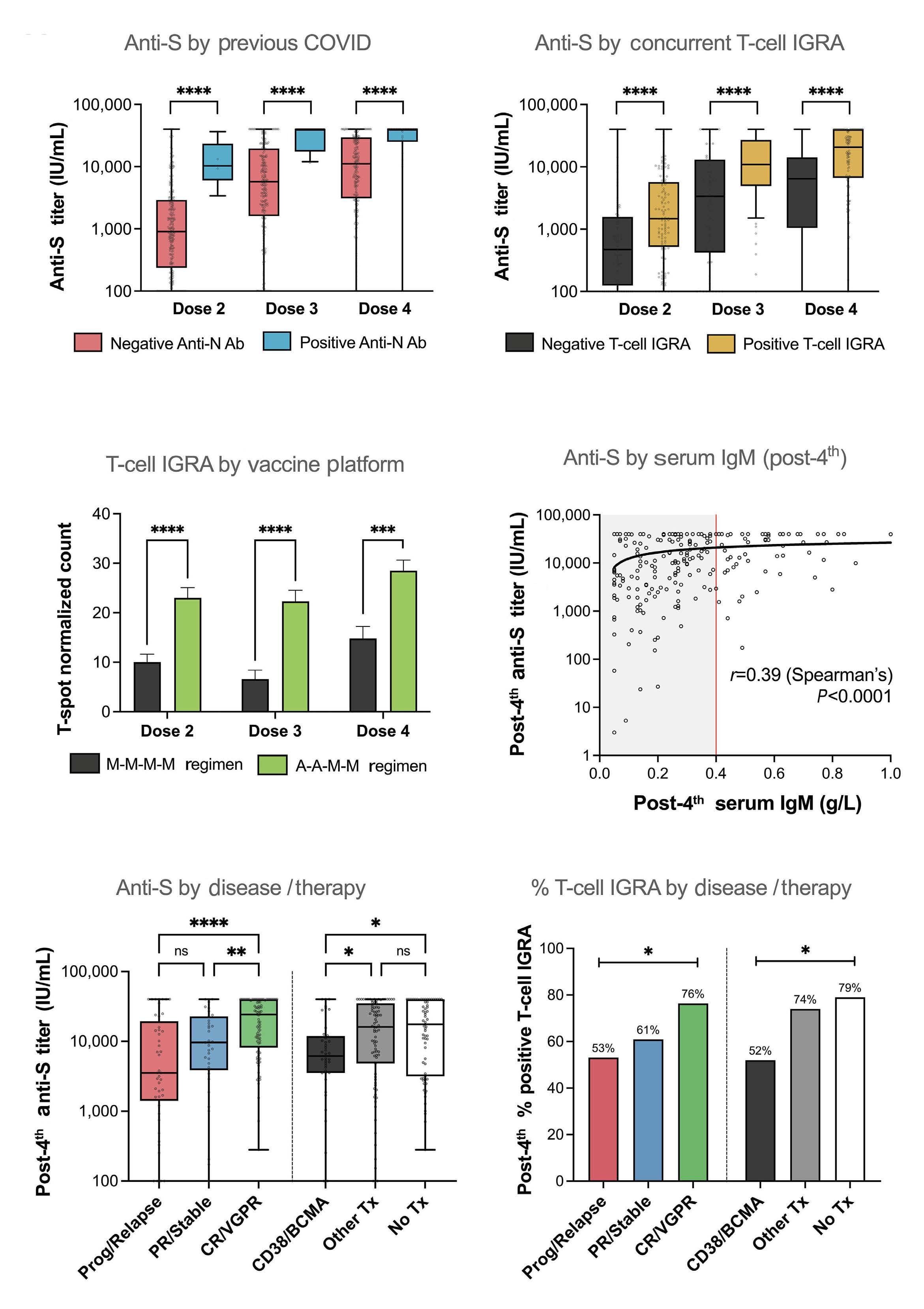
Figure 2. Vaccine and patient factors associated with variable immune response. (A) Anti-S titer in patients with versus without serological evidence of previous COVID-19 infection (defined by anti-N antibody titer ≥1.4 IU/mL), longitudinally after doses 2 (N=232 vs. 7), 3 (N=227 vs. 11) or 4 (N=196 vs. 29). Mann-Whitney test, ****P<0.0001. (B) Anti-S titer in patients with concurrently negative versus positive T-cell interferon γ-release assay (IGRA), longitudinally after doses 2 (N=77 vs. 112), 3 (N=64 vs. 79) or 4
Continued on following page.
(N=48 vs. 115). Mann-Whitney test, ****P<0.0001. (C) T-cell IGRA (normalized counts) to S antigen between cohorts of patients receiving the M-M-M-M (4 mRNA-based vaccines) versus A-A-M-M (2 adenoviral vector-based followed by 2 mRNA-based vaccines) regimens, longitudinally after doses 2 (N=65 vs. 94), 3 (N=51 vs. 72) or 4 (N=49 vs. 88). Mean ± Standard Error of Mean (SEM), Mann-Whitney test, ***P<0.001, ****P<0.0001. (D) Relationship between IgG anti-S titer and total serum IgM after 4 th dose. N=225. Spearman’s Rank correlation coefficient displayed. (E and F) Anti-S titer (E) or % positive T-cell IGRA (F) following 4th dose, by concurrent multiple myeloma disease control (International Myeloma Working Group classification of therapy response) or concurrent anti-myeloma therapy. CR: complete response; Prog: progressive; PR: partial response; TX: treatment; VGPR: very good partial response. *P<0.05, **P<0.01, ****P<0.0001, ns: not significant.
Table 1. Independent predictors of persistently poor COVID-19 vaccination-induced immunity in MM patients.
Predictors
Factor
A-A-M-M vaccines vs. M-M-M-Ma
disease vs.
vs.
Anti-CD38/BCMA Tx vs. No Txc
Other treatment vs. No Txc
Anti-N seropositivityd
Two separate binary logistic regression models were developed. Low titer is defined as COVID-19 anti-spike (Anti-S) antibody titer below World Health Organisation (WHO) cut-off threshold of 7,352 IU/mL, as per kit assay manufacturer. N=85 low anti-S vs. N=140 high anti-S. N=49 negative T-cell interferon γ-release assay (IGRA) vs. N=117 positive T-cell IGRA to COVID-19 spike antigen. aA-A-M-M (2 adenoviral vector-based followed by 2 mRNA-based vaccines) regimen, compared to M-M-M-M (4 mRNA-based vaccines) regimen. bMyeloma disease control at time of 4th dose, defined by International Myeloma Working Group (IMWG) classification of therapy response. cConcurrent anti-myeloma therapy at time of 4th dose; BCMA: B-cell maturation antigen targeting agents; No Tx: no treatment. dAnti-N seropositivity indicative of prior natural COVID-19 exposure; effect compared to those who are Anti-N seronegative. CI: Confidence Intervals; CR: complete response; OR: Odds Ratio; PR: partial response; VGPR: very good partial response.
ease were more likely (vs. CR/VGPR) to have low anti-S titers (adjusted OR 5.1, 95% CI: 2.1-13.5, P=0.0006). At borderline significance, patients taking anti-CD38 or BCMA-targeting agents at the 4th dose were more likely to have negative T-cell IGRA (adjusted OR 3.2, 95% CI: 1.0-10.7, P=0.052). Patients who had received the A-A-M-M vaccine regimen were less likely to have negative T-cell IGRA in univariate (OR 0.42, 95% CI=0.19-0.93, P=0.033) but not multivariate (P>0.05) analysis. With every 1.0x109/L increase in total lymphocyte count, the odds of negative T-cell IGRA were reduced (adjusted OR=0.26, 95% CI=0.11-0.54, P=0.0007), and for every 0.1g/L increase in serum IgM count the odds of low anti-S titer were also reduced (adjusted OR 0.65, 95% CI=0.53-0.79, P<0.0001). These findings represent clinical predictors of ongoing poor vaccine response after 4 doses in MM patients.
In this study, we report a longitudinal analysis of immune response following COVID-19 vaccinations in MM patients
and describe clinically available predictors of poor response after the 4th dose. Relative to other cohorts6 (Online Supplementary Table S3), our dataset has 3 main novelties. Firstly, we follow a large UK-wide cohort prospectively to understand how immunity evolves longitudinally. Secondly, our cohort received a mix of mRNA- and adenoviral vector-based platforms, differing from most studies that have studied exclusively mRNA-based vaccine response.6 Thirdly, we report novel routinely available predictors of poor response after 4 doses.
We confirm reported clinical associations with poor response to earlier doses (lack of prior natural infection, poor disease control, anti-CD38/BCMA therapy) hold true after the 4th dose. By univariate analyses, vaccination with 2 adenoviral vector-based and 2 mRNA-based vaccines resulted in stronger T-cell IGRA responses compared to 4 mRNA-based vaccines. This is consistent with stronger immunogenicity shown with heterologous regimens in the
general population7-10 and other MM patient cohorts.11-13 Multivariate analysis identified lower serum IgM as an independent predictor of low anti-S titer after the 4th dose, supporting an observation described after 2 doses.12 Low total lymphocyte counts predicted lack of cellular response; a similar association is noted in patients with multiple sclerosis after COVID-19 vaccination.14
There are some limitations to our analysis. Firstly, anti-S and T-cell IGRA assays had maximum values (40,000 IU/mL and 50 normalized counts, respectively), limiting predictive power as stronger responses were not distinguished. Secondly, although anti-S and T-IGRA values defining a positive antibody or T-cell response were based on historically established thresholds, the absolute values that correlate with clinical protection from COVID-19 remain unclear. Thirdly, current Omicron variants of concern (VOC) have changed; however, a recent report has found that in heavily treated MM patients, multiple doses of vaccine-induced IgG anti-S antibody cross-reacted well with a range of variants.15 Therefore, our findings remain relevant to all MM patients in the present climate with current VOC.
In conclusion, our study establishes the serial evolution of humoral and cellular immunity across doses 2-4 of COVID-19 vaccination in MM patients. Our data support the benefit of booster vaccination in augmenting robust COVID-19 immunity in MM. Additionally, we establish routinely available laboratory and clinical predictors of ongoing poor response after 4 doses, potentially enabling identification of vulnerable patients to target for booster doses or novel interventions to enhance immunity.
Gaurav Agarwal,1* Sally Moore,2* Ross Sadler,3 Sherin Varghese,3 Alison Turner,4 Lucia Y Chen,3 Jemma Larham,3 Nathanael Gray,4 Oluremi Carty,3 Joe Barrett,4 Constantinos Koshiaris,5 Jaimal Kothari,3 Stella Bowcock,6 Udo Oppermann,4 Vicky Gamble,4 Gordon Cook,7 Chara Kyriakou,8 Mark Drayson,9 Supratik Basu,10,11 Sarah McDonald,12 Shelagh McKinley,13 Sarah Gooding,3,14 Muhammad K Javaid4# and Karthik Ramasamy3#
1Division of Haematology/Oncology, Boston Children’s Hospital, Harvard Medical School, Boston, MA, USA; 2Bath Royal United Hospitals, Bath, UK; 3Oxford University Hospitals NHS Trust, Oxford, UK; 4Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, University of Oxford, Oxford, UK; 5Nuffield Department of Primary Care Health Sciences, University of Oxford, Oxford, UK; 6King’s College Hospital NHS Trust, London, UK; 7Leeds Institute of Clinical Trials Research, University of Leeds, Leeds, UK; 8University College London Hospitals NHS Trust, London, UK; 9University of Birmingham, Birmingham, UK; 10The Royal Wolverhampton NHS Trust, Wolverhampton, UK; 11University of Wolverhampton, Wolverhampton, UK; 12Blood Cancer UK, London,
UK; 13Myeloma UK, Edinburgh, UK and 14MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, Oxford, UK
*GA and SM contributed equally as first authors.
#MKJ and KR contributed equally as senior authors.
Correspondence:
K. RAMASAMY - karthik.ramasamy@ndorms.ox.ac.uk
https://doi.org/10.3324/haematol.2023.284286
Received: September 17, 2023.
Accepted: January 17, 2024.
Early view: January 25, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY license
Disclosures
LYC reports funding from the International Myeloma Society Career Development Award. NG reports grant from Kyowa Kirin, the EU commission, MRC, and NIHR, and is salaried by Jansen and Amgen for this project. UO reports grant from GSK and BMS. ChK reports a non-restricted Educational Grant for research project in QOL from Celgene/BMS. MD reports shares in Abingdon Health. SMcK reports being salaried by Myeloma UK. SG reports grants from Cancer Research UK, Innovate UK (UKRI), and Bristol Myers Squibb, honoraria from the American Society of Hematology. KMJ reports institutional grant support from Amgen. KR reports honoraria, research grants from Janssen, Celgene, Takeda, and Amgen; sits on the Advisory Board of Celgene, Takeda, Janssen, Amgen, Abbvie, Sanofi, Oncopeptides, Karyopharm, GSK, Adaptive Biotechnologies, and Pfizer; and sits on the Speaker’s Bureau of Celgene, Takeda, and Adaptive Biotechnologies. GA, SM, RS, SV, AT, JL, OC, JB, CoK, JK, StB, VG, GC, SuB and SMcD have no conflict of interests to disclose.
Contributions
MJK is responsible for study concept and design, drafting the manuscript, statistical analysis, obtained funding, and supervised the study. KR is responsible for study concept and design, drafting the manuscript, obtained funding, supervised the study, is the guarantor and accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish. RS is responsible for study concept and design, and drafting the manuscript. SM is responsible for study concept and design, and drafting the manuscript. SG is responsible for study concept and design and drafting the manuscript. SMcD and SmcK are responsible for study concept and design. GA is responsible for drafting the manuscript and statistical analysis. CoK is responsible for statistical analysis. LYC, SV, StB, ChK, MD and SuB are responsible for drafting the manuscript. VG, JB, OC, SV, AT and NG are responsible for administrative, technical, or material support. All authors are responsible for the acquisition, analysis, or interpretation of data, and critical revision of the manuscript for important intellectual content.
Funding
The work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). Funding for this study has been received from Blood Cancer Vaccine Consortium, Myeloma UK and Janssen UK. The RUDY platform has been funded by the National Institute for Health
1. Chari A, Kemal Samur M, Martinez-Lopez J, et al. Clinical features associated with COVID-19 outcome in multiple myeloma: first results from the International Myeloma Society data set. Blood. 2020;136(26):3033-3040.
2. Ramasamy K, Sadler R, Jeans S, et al. Immune response to COVID-19 vaccination is attenuated by poor disease control and antimyeloma therapy with vaccine driven divergent T-cell response. Br J Haematol. 2022;197(3):293-301.
3. Aleman A, Upadhyaya B, Tuballes K, et al. Variable cellular responses to SARS-CoV-2 in fully vaccinated patients with multiple myeloma. Cancer Cell. 2021;39(11):1442-1444.
4 Van Oekelen O, Gleason CR, Agte S, et al. Highly variable SARSCoV-2 spike antibody responses to two doses of COVID-19 RNA vaccination in patients with multiple myeloma. Cancer Cell. 2021;39(8):1028-1030.
5. Ramasamy K, Sadler R, Jeans S, et al. COVID symptoms, testing, shielding impact on patient-reported outcomes and early vaccine responses in individuals with multiple myeloma. Br J Haematol. 2022;196(1):95-98.
6. Chuleerarux N, Manothummetha K, Moonla C, et al. Immunogenicity of SARS-CoV-2 vaccines in patients with multiple myeloma: a systematic review and meta-analysis. Blood Adv. 2022;6(24):6198-6207.
7. Schmidt T, Klemis V, Schub D, et al. Immunogenicity and reactogenicity of heterologous ChAdOx1 nCoV-19/mRNA vaccination. Nat Med. 2021;27(9):1530-1535.
8. Borobia AM, Carcas AJ, Pérez-Olmeda M, et al. Immunogenicity and reactogenicity of BNT162b2 booster in ChAdOx1-S-primed participants (CombiVacS): a multicentre, open-label,
Research (NIHR). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Data-sharing statement
Data are available on request to the corresponding author.
randomised, controlled, phase 2 trial. Lancet. 2021;398(10295):121-130.
9 Andrews N, Stowe J, Kirsebom F, et al. Covid-19 Vaccine effectiveness against the Omicron (B.1.1.529) variant. N Engl J Med. 2022;386(16):1532-1546.
10 Naranbhai V, Garcia-Beltran WF, Chang CC, et al. Comparative Immunogenicity and effectiveness of mRNA-1273, BNT162b2, and Ad26.COV2.S COVID-19 Vaccines. J Infect Dis. 2022;225(7):1141-1150.
11. Goldwater MS, Stampfer SD, Sean Regidor B, et al. Third dose of an mRNA COVID-19 vaccine for patients with multiple myeloma. Clin Infect Pract. 2023;17:100214.
12. Stampfer SD, Goldwater MS, Jew S, et al. Response to mRNA vaccination for COVID-19 among patients with multiple myeloma. Leukemia. 2021;35(12):3534-3541.
13. Storti P, Marchica V, Vescovini R, et al. Immune response to SARS-CoV-2 mRNA vaccination and booster dose in patients with multiple myeloma and monoclonal gammopathies: impact of Omicron variant on the humoral response. Oncoimmunology. 2022;11(1):2120275.
14 Tortorella C, Aiello A, Gasperini C, et al. Humoral and T-cellspecific immune responses to SARS-CoV-2 mRNA vaccination in patients with MS using different disease-modifying therapies. Neurology. 2022;98(5):e541-e554.
15. Faustini SE, Hall A, Brown S, et al. Immune responses to COVID-19 booster vaccinations in intensively anti-CD38 antibody treated patients with ultra-high-risk multiple myeloma: results from the Myeloma UK (MUK) nine OPTIMUM trial. Br J Haematol. 2023;201(5):845-850.
The TENT5C/FAM46C locus on chromosome 1p12 is deleted or mutated in up to 20% of multiple myeloma (MM) patients, revealing a plasma cell (PC)-specific tumor suppressive activity.1-4 Unlike hitherto known oncosuppressors, TENT5C is not directly involved in DNA repair, cell cycle regulation or apoptosis. In fact, TENT5C is a non-canonical poly(A)polymerase that selectively polyadenylates and stabilizes mRNA encoding immunoglobulins (Ig) and other endoplasmic reticulum (ER)-targeted proteins, exerting a fundamental role in the physiology of PC as intensive antibody producing factories.5-9 We and others showed that TENT5C specificity for the secretory apparatus requires its association with ER transmembrane FNDC3 proteins, affording TENT5C localization where Ig are translated.9,10 In this way, TENT5C increases their synthesis, together with that of proteins involved in folding and trafficking along the secretory route, thereby operating as a potent booster of humoral immune response.9 Accordingly, we found that TENT5C is remarkably induced during PC differentiation (Online Supplementary Figure S1A), and that its high expression neatly discriminates MM lines from other tumors (Figure 1A). Further strengthening its suppressive role in MM pathobiology, analysis of publicly available genome-scale, pooled CRISPR-Cas9 loss-of viability screens (DepMap portal 23Q2 release) showed that TENT5C ablation results in an extremely significant, MM-specific proliferative advantage (Figure 1B). In line with a selective benefit for MM cells in reducing TENT5C expression, bone marrow-purified PC from MM patients display lower TENT5C expression than those obtained from healthy donors (Online Supplementary Figure S1B), with the subset of TENT5C-deleted MM patients showing a further decrease in TENT5C mRNA levels when compared with non-deleted MM (Online Supplementary Figure S1C). We propose that such advantage consists in restricting Ig production and the associated oxidative, metabolic, and degradative workload to save energy for proliferation. Indeed, we showed that TENT5C re-expression in mutated MM lines pushes secretory activity beyond sustainability, inducing ATP shortage, ROS accumulation, and a slower growth rate in vitro. 9 However, whether TENT5C regulates the balance between MM growth and antibody secretion in vivo remains uninvestigated. To assess TENT5C impact in vivo, we silenced or re-expressed TENT5C in ALMC-2 cells, a MM line expressing the wild-type protein and bearing a hemizygous loss in chromosome 1p,11 and then injected engineered cells into immunodeficient Rag2–/– IL2rg–/– mice. (Animal procedures
were approved by the Italian Ministry of Health and the Institutional Animal Care and Use Committee [IACUC] under protocols 1057 and 1314). Expression of FLAG-tagged TENT5C in ALMC-2 cells significantly increased the abundance of ER mRNA and proteins, and decreased cell proliferation in vitro (Figure 1C-F, Online Supplementary Figure S1D), inducing cell cycle arrest in G0/G1 phase (Online Supplementary Figure S1E). Consistently, TENT5C re-expression had the same effects on proliferation and ER proteins in 3 other TENT5C-deleted MM lines (Figure 1C, Online Supplementary Figure S1F).
Conversely, TENT5C-silenced ALMC-2 cells displayed only slight reductions in ER-associated mRNA and proteins (Figure 1D-F, Online Supplementary Figure S1G) and no differences in cell growth, viability, and cell cycle distribution (Figure 1C, Online Supplementary Figure S1D, E). In general, we found smaller effects of TENT5C silencing in ALMC-2 cells if compared to previously investigated wildtype MM lines,9 consistent with a pre-existing significant suppression of TENT5C activity in ALMC-2 cells. Importantly, TENT5C was able to promote Ig production and secretion in a dose-dependent manner, with the lowest levels in silenced ALMC-2 cells and the highest in overexpressing counterparts (Figure 1G). Together, these data support a mechanistic link between TENT5C-mediated tumor suppression and its effects on the secretory activity, whereby TENT5C acts as a natural tuner of the secretory capacity of MM cells regulating the trade-off between Ig production and PC proliferation in vitro.
To challenge this role in vivo, we first injected control, TENT5C-silenced and overexpressing cells intravenously into Rag2–/– IL2rg–/– mice. Despite higher initial circulating Ig light chain (LC) levels (Online Supplementary Figure S2A), we found reduced tumor appearance and longer survival in recipients of TENT5C-overexpressing cells (Figure 2A). Indeed, half of the mice in the overexpressing group were still alive and without any sign of disease up to 330 days after injection, while 13 out of 16 mice in the other experimental groups (mock and TENT5C-silenced) had to be sacrificed within 150 days due to the appearance of bulky abdominal plasmacytomas (Figure 2A). Moreover, in line with negative effect of high TENT5C levels on MM proliferation, mRNA and protein analyses on excised plasmacytomas showed significant selection of low TENT5C-expressing cells in the overexpressing group (Figure 2B, Online Supplementary Figure S2B). Of note, the only tumor maintaining TENT5C overexpression (lane 11 in Figure 2B) displayed the highest
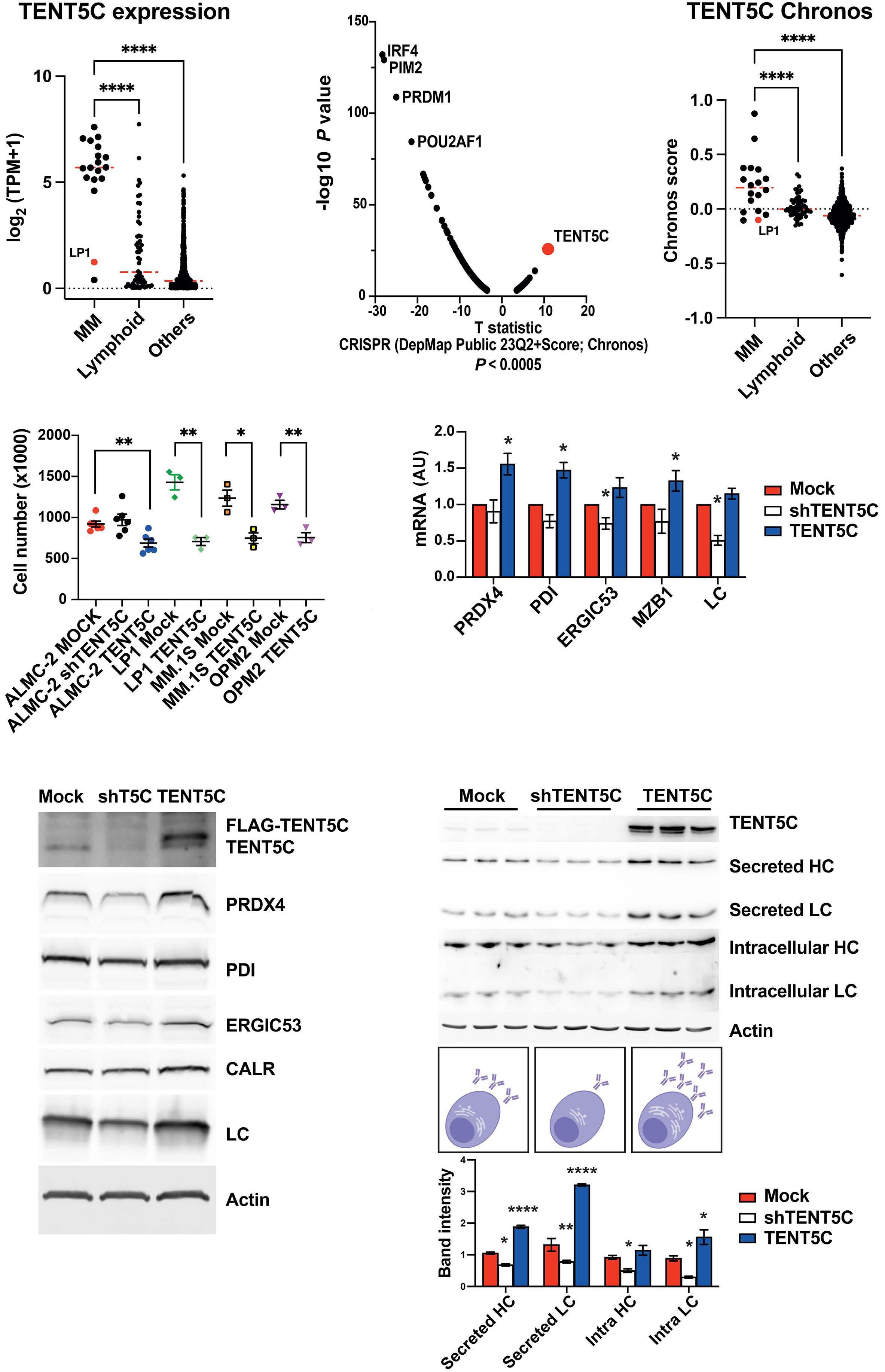
Continued on following page.
Figure 1. TENT5C is a myeloma-specific oncosuppressor that regulates endoplasmic reticulum protein expression and Ig production in a dose-dependent manner. (A) Dot-plot showing TENT5C expression in multiple myeloma (MM) versus other lymphoid versus other cancer cell lines derived from public data available in the DepMap portal (23Q2 release). The full list of 18 myeloma cell lines and of 1,001 non-MM cell lines are available at https://depmap.org/portal/download/all/; LP1 cells, known to harbor a homozygous deletion of TENT5C, are highlighted in red (median; ****P<0.0001; one-way ANOVA with Dunnett’s multiple comparison). (B) (Left) Volcano plot showing the dependencies enriched in MM cell lines versus non-MM cell lines, extracted from DepMap portal 23Q2 release. T statistic and P value for the Chronos dependency scores of the top significant genes (P<0.0005) are shown. A positive T statistic indicates a proliferative advantage of TENT5C CRISPR-KO specifically in MM cells. All genetic dependency data, statistical analyses and methods are publicly available in the DepMap portal (23Q2 release). (Right) Dot-plot showing the Chronos dependency score for TENT5C in MM versus other lymphoid versus other cancer cell lines (median; ****P<0.0001; one-way ANOVA with Dunnett’s multiple comparison). (C) Equal numbers of MM cells were seeded at 5x105 cells/ mL and counted with trypan blue staining after 2 (for ALMC-2, LP1 and MM.1S) or 3 days (for OPM2), (mean ± Standard Error of Mean [SEM]; N=3-6; *P<0.05; **P<0.01; unpaired t test vs. Mock). ALMC-2 cells were established at relapse as symptomatic MM of a patient initially diagnosed with AL amyloidosis and treated with oral dexamethasone and blood stem cell transplant. ALMC-2 cells bear a t(14;20) translocation, Myc amplification, and p53 deletion. LP1 cells bear a biallelic deletion of TENT5C while MM.1S and OPM2 cells a monoallelic deletion and a point mutation on the other allele. All the cell lines were genotyped and TENT5C gene was analyzed by Sanger sequencing to confirm wild-type TENT5C expression in ALMC-2 cells and M270V and E178A mutations in MM.1S and OPM2, respectively. Lentiviral viruses to stably express anti-TENT5C or a control shRNA were purchased by Mission shRNA (Sigma-Aldrich, SHC002 and TRCN0000166958). Human C-term FLAG-TENT5C cDNA was purchased by Genscript (OHu30151D) and was cloned in a plasmid with a hybrid bidirectional human PGK-miniCMV promoter co-expressing the protein of interest and truncated human CD271. (D) qRT-PCR analysis of mRNA of selected endoplasmic reticulum (ER)-resident proteins and Ig λ light chain (LC) in TENT5C silenced or overexpressing ALMC-2 cells (mean ± SEM normalized on H3 mRNA; N=4 for silenced, 5 for overexpressed; *P<0.05; Kruskal-Wallis one-way test with Dunn’s multiple comparison vs. Mock). Total RNA was extracted by lysis in TriFAST (Euroclone, EMR507100). 1000 ng of RNA were retro-transcribed with ImProm-II Reverse Transcriptase System (Promega, A3800). qPCR were performed using iTaq SYBR Green Supermix (Bio-Rad, 1725122) on Bio-Rad CFX96 PCR and analyzed on Bio-Rad CFX Maestro software. Primers are listed in Online Supplementary Table S1. (E) Representative immunoblots of selected ER-resident proteins and Ig λ LC in TENT5C silenced or overexpressing ALMC-2 cells. Immunoblots were performed as described by Fucci et al.9 Images were obtained using Uvitec Imager Mini HD9 (Uvitec Ltd.) for HRP-conjugated secondary Ab or FLA9000 (FujiFilm) for Alexa-Fluor conjugated secondary antibodies. Antibodies are listed in Online Supplementary Table S1. Band quantifications, performed using ImageJ software (http://rsbweb.nih.gov/ij/), are reported in Online Supplementary Figure S1G. (F) Immunoblot analyses of intracellular and secreted IgG λ light (LC) and heavy (HC) chains in TENT5C control, silenced or overexpressing ALMC-2 cells. (Top) Representative blots. (Bottom) Quantifications of intracellular and secreted IgG λ chains (N=3 normalized on actin; *P<0.05; **P<0.01; ****P<0.0001; one-way ANOVA with Dunnett’s multiple comparison vs. Mock). AU: arbitrary units.
levels of Ig and ER proteins, confirming in vivo a dose-dependent effect of TENT5C on the secretory compartment. On the contrary, no selection was detected in TENT5C-silenced tumors that maintained advantageous lower ER and Ig mRNA and protein patterns when compared with control plasmacytomas (Figure 2B, Online Supplementary Figure S2B). To timely evaluate the role of TENT5C in modulating the ratio between monoclonal protein and PC proliferation, we then injected TENT5C-overexpressing mice and control ALMC-2 subcutaneously to precisely follow tumor size in a shorter timeframe. We confirmed remarkably reduced tumor growth and longer survival in recipients of TENT5C-overexpressing cells, despite significantly higher circulating LC six days after injection (Figure 2C, D, Online Supplementary Figure S2C). At sacrifice, ex vivo qRT-PCR and immunoblot analyses showed that TENT5C-overexpressing tumors still displayed higher LC and ER mRNA and protein concentrations than control plasmacytomas (Figure 2E, Online Supplementary Figure S2D). Notably, TENT5C abundance significantly modified the ratio between tumor volume and the levels of monoclonal LC concurrently detected in sera (Figure 2F). This in vivo association of higher proliferation with decreased secretory activity clearly reveals that loss of TENT5C uncouples monoclonal protein levels from MM burden.
Notably, the impact of TENT5C on secretory products is
not restricted to Ig but also promotes the cell surface expression of calreticulin, an ER stress-induced “eat-me” signal, and CD38, an established target of monoclonal antibodies (Figure 3A, B, Online Supplementary Figure S3E), with relevant immunotherapeutic implications. Notably, attesting to biological relevance of TENT5C-mediated increase in surface calreticulin, TENT5C-overexpressing ALMC-2 cells underwent increased phagocytosis by primary macrophages in vitro (Figure 3C). Moreover, in line with an immunogenic role, qRT-PCR revealed higher expression of monocytic/macrophagic markers (i.e., CD68 and CD206) in TENT5C-overexpressing tumors, indicating larger myeloid infiltrates (Figure 3D), as further confirmed by CD68 immunoblot and immunohistochemistry analyses (Figure 3E, F). In conclusion, our data disclose a trade-off between the sustainable amount of antibodies that PC can produce and their proliferation. TENT5C acts as an “unselfish gene” whose distinctive expression, though inhibitory for PC proliferation, optimizes humoral immunity in favor of the entire organism. Conversely, MM tends to manipulate this equilibrium, blunting TENT5C expression to decrease proteosynthetic activity and favor its own growth. In keeping with this model, loss-of-function mutations have been identified in MM in key transcription factors regulating Ig production and TENT5C transcription, namely, XBP-1 and PRDM1. 12 These observations indicate that MM may undergo
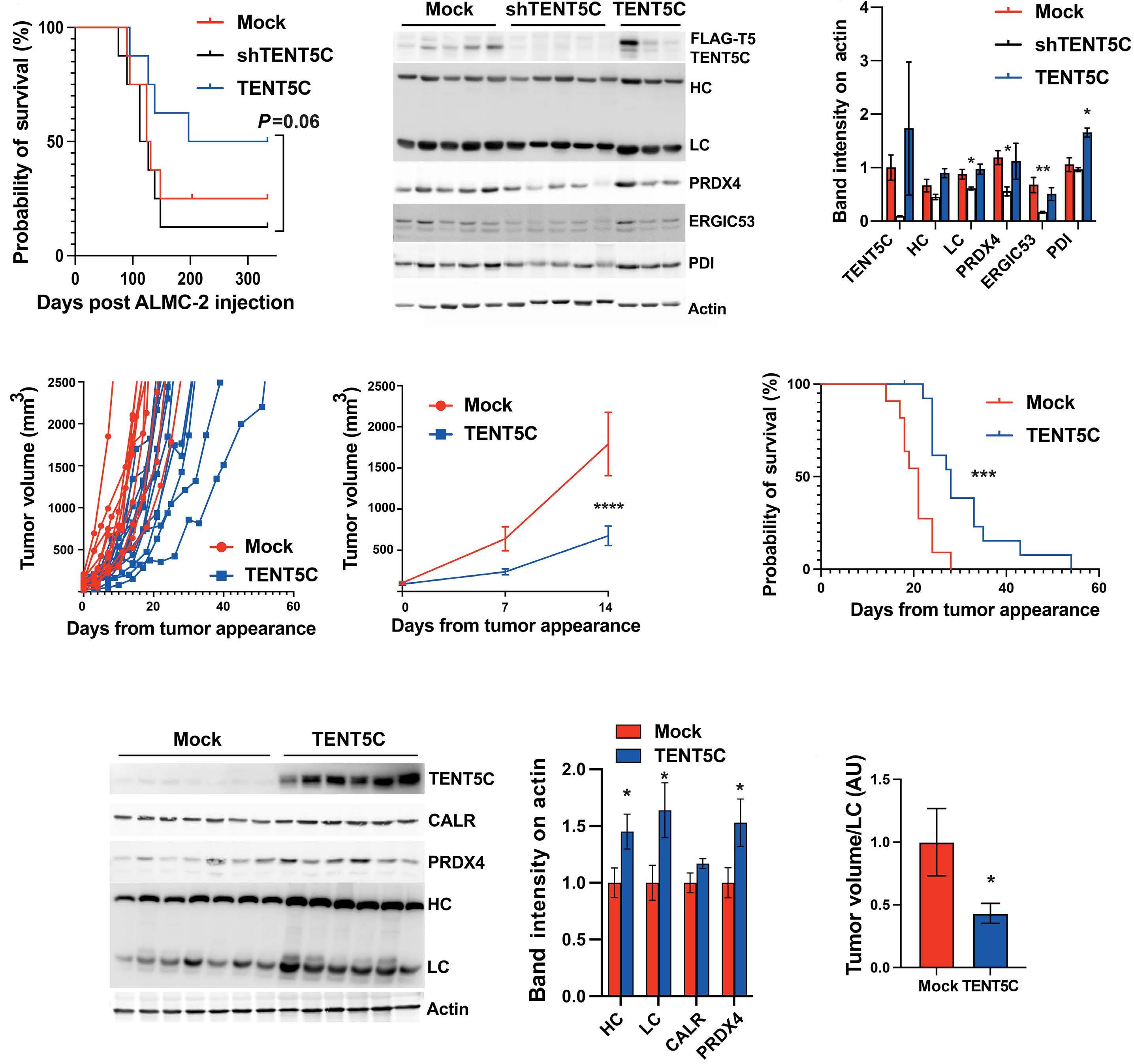
Figure 2. TENT5C regulates the trade-off between antibody secretion and multiple myeloma proliferation in vivo. (A) Kaplan-Meier survival curve of male and female 4-6-month-old BALB/c Rag2–/–γc–/– mice intravenously injected in the tail with 10x106 control, TENT5C-silenced, overexpressing or control ALMC-2 cells. General health status of the mice was monitored every 3 days and mice were sacrificed at the appearance of lower limb paralysis or abdominal plasmacytomas (N=8 per group; Log-rank Mantel-Cox test). (B) Representative immunoblots of selected ER-resident proteins and IgG λ in excised plasmacytomas formed after intravenous injection. (Left) Immunoblot images. (Right) Band quantifications (mean ± Standard Error of Mean [SEM] normalized on actin; N=5 Mock, 5 shTENT5C, 3 TENT5C; ordinary one-way ANOVA with Dunnett’s multiple comparison *P<0.05; **P<0.01; unpaired t test). (C) BALB/c Rag2–/–γc–/– mice were subcutaneously implanted with 3x106 ALMC-2 multiple myeloma (MM) cells in the lateral abdominal region. Tumor growth was monitored every 3 days by caliper assessment and mice sacrificed when tumors reached the volume of 2,500 mm3. Growth of subcutaneous tumors represented individually (left) or as an average (right) in mm3 ± SEM (N=11 mock, 14 TENT5C; ****P<0.0001; two-way ANOVA with Bonferroni’s multiple comparisons). (D) Kaplan-Meier survival curve of mice subcutaneously injected with 3x106 control or TENT5C-overexpressing ALMC-2 cells (N=11 Mock, 14 TENT5C; ***P<0.01; Log-rank Mantel-Cox test). (E) Representative immunoblots of selected ER-resident proteins and IgG λ in excised subcutaneous tumors. (Left) Immunoblot images. (Right) Band quantifications (mean ± SEM normalized on actin; N=7 Mock, 6 TENT5C; *P<0.05; unpaired t test). (F) Ratio between the tumor volume measured by caliper and Ig λ LC levels measured by immunoblot in 1 µl of serum collected at the same timepoint (mean ± SEM; N=9 Mock, 10 TENT5C, *P<0.05; unpaired t test). AU: arbitrary units; LC: light chain; HC: heavy chain.
selective pressure to lower, but not completely abolish Ig secretion, suggesting that maintaining Ig production may be beneficial through mechanisms that remain, however, elusive. Moreover, our demonstration that TENT5C modulation uncouples Ig secretion from tumor size may be extremely relevant in MM patients, where, in the presence of TENT5C inactivating mutations, monoclonal protein quantification may underestimate disease burden. In line with our observation that TENT5C loss significantly increases proliferation, inactivating mutations are among the main drivers of evolution from smoldering to active
MM.13 Moreover, deletions of locus 1p, that often include TENT5C, are an adverse prognostic factor in MM patients and predict worse responses even to the most active available regimens.2,14
Finally, our unprecedented evidence that TENT5C promotes the surface expression of calreticulin and CD38, increasing macrophagic infiltrates, suggests that TENT5C mutations may also contribute to reduce MM immunogenicity. In line with this possibility, Maura et al.15 recently found a significant correlation between low TENT5C expression and resistance to targeted immunotherapy with the anti-CD38
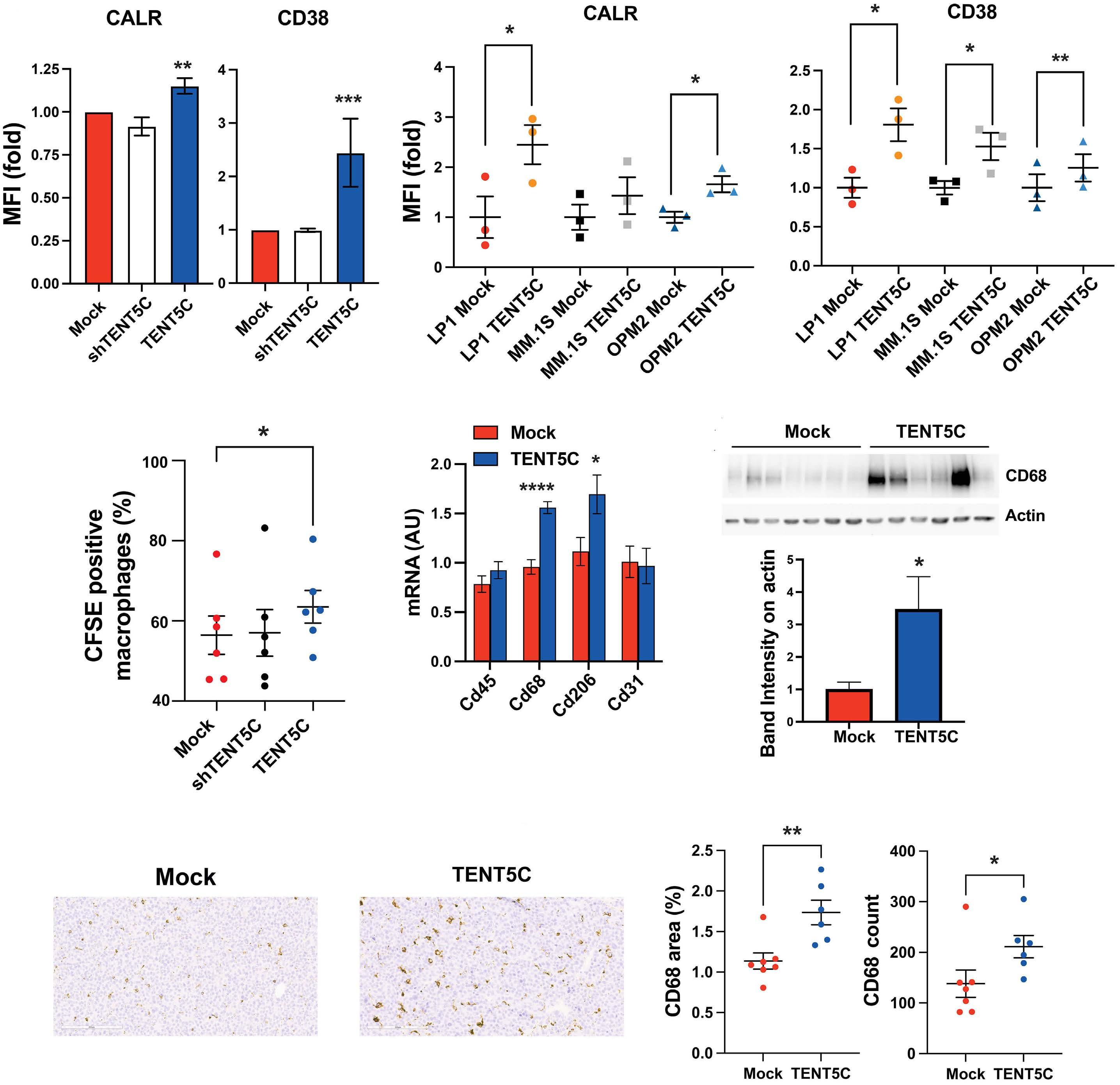
Continued on following page.
Figure 3. TENT5C promotes the expression of calreticulin and CD38 on the cell surface. (A) Cytofluorimetric analysis of CALR and CD38 surface expression in control, TENT5C-overexpressing and silenced ALMC-2 cells (average median fluorescent intensity ± Standard Error of Mean [SEM]; N=18 for CALR and 9 for CD38; **P<0.01; ***P<0.001; Kruskal-Wallis one-way test with Dunn’s multiple comparison vs. Mock). See Online Supplementary Figure S2E for representative cytofluorimetric histograms. Data were obtained with Accuri C6 Flow Cytometer (BD Biosciences) and analyzed using the FCS Express 7 Flow Research Edition (De Novo Software). (B) Cytofluorimetric analysis of CALR and CD38 surface expression in control and TENT5C-overexpressing multiple myeloma (MM) cells (average median fluorescent intensity ± SEM; N=3; *P<0.05; **P<0.01; paired t test). (C) Cytofluorimetric analysis of in vitro phagocytosis of control, TENT5C-silenced or overexpressing ALMC-2 cells by primary macrophages. Macrophages were obtained by MCSF-induced differentiation of human monocyte purified from the peripheral blood of healthy donors with Pan Monocyte Isolation kit (Miltenyi, 130-096-537). After differentiation, macrophages were stained with 2 µM CellTrace™ Far Red Cell Proliferation kit (Thermo Scientific, C34564). In parallel, TENT5C manipulated ALMC-2 cells were stained with 2 µM CellTrace™ CFSE Cell Proliferation kit (Thermo Scientific, C34544). Stained macrophages and MM cells were mixed 1:1 and incubated at 37°C for 4 hours. The percentage of double positive macrophages was then assessed by BD FACSCanto II (mean ± SEM; N=6; *P<0.05; repeated measures one-way ANOVA with Dunnett’s multiple comparisons vs. Mock). (D) qRT-PCR analysis of murine markers in excised subcutaneous tumors (mean ± SEM normalized on GAPDH mRNA; N=7 Mock, 6 TENT5C; *P<0.05; unpaired t test). (E) Immunoblots of murine CD68 in excised subcutaneous tumors. (Top) Immunoblot images. (Bottom) Band quantifications (mean ± SEM normalized on actin; N=7 Mock, 6 TENT5C; *P<0.05; unpaired t test). (F) Immunohistochemistry of murine CD68 in excised subcutaneous tumors. (Left) Representative images: scale bar 200 µm. (Right) Quantifications of CD68 area (%) and counts. Quantification of CD68+ cells was performed with ImageJ software by applying a color threshold on the DAB signal to select positive pixels and analyzing particles (>15 pixels) for area. Area of CD68+ cells was calculated on 4 images/sample (20x magnification), (mean ± SEM; N=7 Mock, 6 TENT5C; *P<0.05; **P<0.01; unpaired t test). AU: arbitrary units.
antibody daratumumab. However, since TENT5C may also promote the expression of other membrane molecules and cytokines, further investigations in immunocompetent preclinical models will be required to define the net impact of TENT5C loss on MM immunogenicity and sensitivity to immunotherapy.
To summarize, by disclosing the regulation in vivo of Ig secretion, MM growth, and the expression of PC surface markers by TENT5C our work advances the understanding of the myeloma-specific role of one of the most frequently mutated oncosuppressors in MM.
Authors
Massimo Resnati,1 Sara Pennacchio,1 Lisa Viviani,1 Tommaso Perini,1,2 Maria Materozzi,1 Ugo Orfanelli,1 Jessica Bordini, 3 Raffaella Molteni,1 Mario Nuvolone, 4,5 Matteo Da Vià,6 Francesca Lazzaroni,6 Niccolò Bolli,6,7 Simone Cenci 1,8 and Enrico Milan 1,8
1 Age Related Diseases Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Scientific Institute, Milano; 2 Hematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Scientific Institute, Milano; 3 B-cell Neoplasia Unit, Division of Experimental Oncology, IRCCS San Raffaele Scientific Institute, Milano; 4 Department of Molecular Medicine, University of Pavia, Pavia; 5 Amyloidosis Research and Treatment Center, Fondazione IRCCS Policlinico San Matteo, Pavia; 6 Hematology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano; 7 Università degli Studi di Milano, Milano and 8 University VitaSalute San Raffaele, Milano, Italy
Correspondence:
E. MILAN - milan.enrico@hsr.it
https://doi.org/10.3324/haematol.2023.284299
Received: September 18, 2023. Accepted: February 13, 2024. Early view: February 22, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
MN received honoraria from Janssen and research funding from Oncopeptides, Gate Bioscience and Pfizer, and is an inventor on a patent on immunoglobulin sequencing. MDV received funding from GSK and Jannsen (lectures and advisory boards) and Takeda and Amgen (advisory boards). NB received funding from GSK, Janssen, Amgen (honoraria for lectures) and Pfizer (advisory board). The other authors have no conflicts of interest to disclose.
Contributions
MN, SC and EM designed the research. MR, MM, UO, JB, RM, MDV, FL and NB contributed with crucial methodologies and resources. MR, SP, LV, TP and EM performed the experiments and analyzed the data. TP, SC and EM wrote the paper. MN, SC and EM provided the research funds. SC and EM supervised the research.
Acknowledgments
We are particularly grateful to Roberta Colzani for administrative assistance, Paolo Ghia, Diane F. Jelinek, Luca Rampoldi, Emilie Vénéreau, and Ineke Braakman for mice, cells, primers and antibodies, and all the members of the Cenci and Milan labs for fruitful discussions. We thank Amleto Fiocchi and the Animal Histopathology Service at IRCCS San Raffaele Scientific Institute for help with immunohistochemistry analyses. ALMC-2 cells were kindly provided by Diane F. Jelinek, Mayo Clinic, and LP1, MM.1S and OPM2 by Dr. Giovanni Tonon, San Raffaele Scientific Institute. Figure 1G was created using BioRender.com.
Funding
The work was supported by grants to EM from the International Myeloma Foundation (Brian D. Novis Junior Research Award 2019), Fondazione Cariplo Giovani Ricercatori (2018-0257), Fondazione Telethon and Fondazione Cariplo Joint Grant on rare diseases (GJC21079/2022-0577), and to EM and MN from the Italian Ministry of Health (GR-2018-12368387). Additional support came from grants to SC from Fondazione AIRC (IG 23245), the International Myeloma Society (IMS) and Paula and Rodger Riney Foundation (Translational Research Grant 2021 and 2022), and the European Union – Next Generation EU (National Recovery
1. Barbieri M, Manzoni M, Fabris S, et al. Compendium of FAM46C gene mutations in plasma cell dyscrasias. Br J Haematol. 2016;174(4):642-645.
2. Boyd KD, Ross FM, Walker BA, et al. Mapping of chromosome 1p deletions in myeloma identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as being genes in regions associated with adverse survival. Clin Cancer Res. 2011;17(24):7776-7784.
3. Bolli N, Biancon G, Moarii M, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32(12):2604-2616.
4 Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91-101.
5. Mroczek S, Chlebowska J, Kuliński TM, et al. The non-canonical poly(A) polymerase FAM46C acts as an onco-suppressor in multiple myeloma. Nat Commun. 2017;8(1):619.
6. Bilska A, Kusio-Kobiałka M, Krawczyk PS, et al. Immunoglobulin expression and the humoral immune response is regulated by the non-canonical poly(A) polymerase TENT5C. Nat Commun. 2020;11(1):2032.
7 Herrero AB, Quwaider D, Corchete LA, Mateos MV, García-Sanz R, Gutiérrez NC. FAM46C controls antibody production by the polyadenylation of immunoglobulin mRNAs and inhibits cell migration in multiple myeloma. J Cell Mol Med. 2020;24(7):4171-4182.
8. Zhu YX, Shi C-X, Bruins LA, et al. Loss of FAM46C promotes cell
and Resilience Plan, Investment Partenariato Esteso PE8 “Conseguenze e sfide dell’invecchiamento”, Project Age-ItAgeing Well in an Ageing Society). MDV was funded by the Umberto Veronesi Foundation and by Pfizer Global Medical Grants (75340503). NB is supported by the European Research Council under the European Union’s Horizon 2020 research and innovation program (817997) and Associazione Italiana Ricerca sul Cancro (IG25739).
Data-sharing statement For original data, please contact: milan.enrico@hsr.it
survival in myeloma. Cancer Res. 2017;77(16):4317-4327.
9 Fucci C, Resnati M, Riva E, et al. The interaction of the tumor suppressor FAM46C with p62 and FNDC3 proteins integrates protein and secretory homeostasis. Cell Rep. 2020;32(12):108162.
10 Manfrini N, Mancino M, Miluzio A, et al. FAM46C and FNDC3A are multiple myeloma tumor suppressors that act in concert to impair clearing of protein aggregates and autophagy. Cancer Res. 2020;80(21):4693-4706.
11. Arendt BK, Ramirez-Alvarado M, Sikkink LA, et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain–secreting human cell lines, ALMC-1 and ALMC-2. Blood. 2008;112(5):1931-1941.
12. Perini T, Materozzi M, Milan E. The immunity-malignancy equilibrium in multiple myeloma: lessons from oncogenic events in plasma cells. FEBS J. 2022;289(15):4383-4397.
13. Boyle EM, Deshpande S, Tytarenko R, et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat Commun. 2021;12(1):293.
14. Odikadze L, Joseph N, Schmidt TM, et al. Outcomes of myeloma patients with deletion 1p receiving lenalidomide, bortezomib, and dexamethasone (RVD) therapy. Blood. 2018;132(Suppl 1):1884.
15. Maura F, Boyle E, Coffey D, et al. Genomic determinants of resistance in newly diagnosed multiple myeloma treated with targeted-immunotherapy. Blood. 2022;140(Suppl 1):1137-1139.
The presented study is a randomized phase III trial with measurable residual disease (MRD) after induction therapy and event-free survival as co-primary endpoints. Patients were upfront randomized 1:1 into one of two induction schedules; gemtuzumab ozogamicin (GO) administered to intensive induction therapy on days 1, 4 and 7 (GO-147) versus GO administered once on day 1 (GO-1), as well as to glasdegib versus placebo adjunct to consolidation therapy followed by glasdegib 6-month maintenance therapy versus physician’s choice.1 All patients entering the maintenance phase were offered the opportunity to switch to oral azacitidine. The approvals of venetoclax (Venclyxto®) in unfit older patients with acute myeloid leukemia (AML) and oral azacitidine (Onureg®) as maintenance therapy in 2021 hampered recruitment considerably. Therefore, the study was closed on May 5, 2022. Based on descriptive analysis for the randomization of GO-147 versus GO-1, the numerical value of MRD negativity after induction therapy was higher in the GO-147 arm with 75% (9/12) compared to 45.5% (5/11) in the GO-1 arm. This higher rate of MRD negativity after induction therapy also translated into a numerically better median event-free survival (EFS) (296 vs. 206 days).
GO was re-approved for use in newly diagnosed AML patients by the Food and Drug Administration in 2017 and by the European Medicines Agency in 2018, after it had been withdrawn from the market in June 2010 by the marketing pharmaceutical company. In the pivotal ALFA 0701 study leading to re-approval of GO, patients in the GO arm had significantly improved median EFS (19.6 vs. 11.9 months; P=0.00018) and OS (34 vs. 19.2 months; P=0.046).2 Although the difference in OS was not statistically significant when updated data were analyzed,3 the trend to a longer OS observed in the GO arm of ALFA-0701 is consistent with the results found in a meta-analysis of individual patient data that showed a significant improvement in OS in patients treated with GO.4 Glasdegib 100 mg daily in a phase II study in older patients not fit for intensive chemotherapy in combination with low-dose cytarabine resulted in a significantly higher CR rate and better OS as compared to low-dose cytarabine alone.5 Interestingly, the beneficial effect of glasdegib on OS was not restricted to patients
achieving a CR, supporting a leukemic stem cell targeting effect of glasedib.5
Patients included in our study had newly diagnosed CD33-positive AML, were age 18 or older, and had Eastern Cooperative Oncology Group (ECOG) performance status of 2 or less. The originally planned age at inclusion was ≥60 years, however, due to unsatisfactory recruitment the age limit for inclusion was lowered to age ≥18 years via an amendment on September 28, 2021 after 9 months of recruitment. Survival endpoints were defined as recommended by the European LeukemiaNet.6 MRD negativity was defined as the absence of leukemic cells at the end of the induction therapy assessed by flow cytometry with a sensitivity of 10-4 to 10-5
The total planned sample size was 252. Patient recruitment was terminated after the inclusion of 30 patients. Of these, 26 patients were randomized to treatment, of those one never received treatment due to cardiac comorbidity and was excluded from analyses.
From the 25 patients included in the analysis only 13 patients received consolidation therapy within the trial. Therefore, efficacy was only evaluated for the first comparison, i.e., GO-1 versus GO-147. The remaining 12 patients either failed to obtain complete remission (CR) (N=7) or were censored due to early study termination (N=5).
Overall, median age at diagnosis was 64 years, 76% were male and 52% had ECOG 1 at inclusion. Secondary or therapy-related AML was present in 16% of patients. Other baseline characteristics can be found in Online Supplementary Table S1
The CR and complete remission with incomplete hematological recovery (CRi) rate was 54.5% (N=6) in the GO-1 arm and 83.3% (N=10) in the GO-147 arm (P=0.134, rate difference 28.8%, 95% confidence interval [CI]: 7.4-65). Table 1 summarizes the response to induction therapy according to therapy arm. Regarding MRD responses, there were no significant differences observable among patients achieving a CR or CRi between induction regimens. Overall MRD negativity achievement was 45.5% in the GO-1 arm and 75% in the GO-147 arm (P=0.147; rate difference 29.5%, 95% CI: 8.7-67.8). Patients treated in the GO-1 arm showed a median EFS of 206 days (95% CI: 28-206), and in the GO-147 arm
Table 1. Response to induction therapy.
GO: gemtuzumab ozogamicin; GO-1: GO administered once on day 1; GO-147: GO administered to intensive induction therapy on days 1, 4 and 7; CR: complete remission; Cri: complete remission with incomplete hematological recovery; MRD: measurable residual disease.
of 296 days (95% CI: 35-not calculable; P=0.155) (Online Supplementary Figure S1). Relapse-free survival was also in favor of GO-147 with a median of 260 days versus 176 days in the GO-1 arm (P=0.411) (Online Supplementary Figure S2). Concerning toxicity during the induction phase, 96% (24/25) of patients overall experienced at least one or more adverse events (AE). Toxicity rates were numerically higher in the GO-147 arm with a percentage of serious adverse events (SAE) of 61.5% (8/13) compared to 41.7% (5/12) in the GO-1 arm. Most frequent SAE during induction therapy were cytopenias and fever in neutropenia. During the observation periods of the consolidation and maintenance phase, rates of serious AE excluding cytopenia were 14% (1/7) in the control arm and 67% (4/6) in the glasdegib arm. Half of the patients treated with glasdegib maintenance (N=5) experienced dysgeusia and one third of the patients experienced muscle cramps as previously described.2,4 The development of clinical studies sponsored by academic centers is fraught with multifaceted challenges. Securing adequate funding, maintaining scientific and ethical standards in study design, and addressing participant recruitment and retention are formidable tasks made more complex by our constrained staffing levels. In addition to that, excessive regulatory hurdles in conducting highly complex clinical studies sponsored by academic institutions often delay the activation of well-designed trials. Not seldom, at the time point when studies are ready to be initiated, more attractive therapeutic approaches are available. During the planning and conduct of this study the approval of venetoclax for patients deemed unfit for intensive chemotherapy revolutionized the therapy of AML of those patients, and the therapy proposed in this trial ceased to be recommendable.7 Venetoclax and azacitidine are increasingly used in patients above the age of 65 years.8 Firstly, due to concerns about such patients’ ability to tolerate intensive chemotherapy regimens and secondly the limited response to intensive induction and consolidation regimens.9 Venetoclax as adjunct to low-dose hypomethylating agents showed significant improvements in remission
rates and OS compared to placebo.10,11 Success of such new therapeutic approaches with non-intensive regimens affected patient recruitment in our study negatively. Particularly during the COVID-19 pandemic, approaches that facilitate outpatient therapies were preferred.7 Furthermore, recent retrospective data analysis suggested in the same direction that azacitidine and venetoclax treatment may be equally effective to intensive chemotherapy and is associated with significantly lower infectious complications and shorter stays in hospital.12
Aiming to avoid a too early study termination the attempt was made to improve the feasibility of the trial by two consecutive amendments that reduced the patient age to 18 years. However, recruitment of the trial did not improve significantly. As a result, the study was halted, with this decision finally being supported by the previously published data concerning the broad toxicity spectrum of GO, especially in older patients.13,14 Indeed, according to the final results from the AMLSG 0909 study, the older population has obviously no benefit from the addition of GO on day 1 in any of the response or survival endpoints, whereas the rates of CR/CRi, EFS and cumulative incidence of relapse were similar between the standard and the GO arm.14 Moreover, the 30- and 60-day mortality rates were higher in the GO arm. Nonetheless, in our study, the initial hypothesis that treatment during induction therapy with GO-147 results in a higher rate of MRD negativity compared to GO-1 was at least numerically supported, and the question is still remaining whether GO administered on days 1, 4 and 7 as applied in the ALFA 0701 trial3 is in fact better as compared to GO administered only once as conducted in several other trials.4,13 In agreement with the findings of the AMLSG 0909 publication,13 we found an important amount of toxicity in both therapy arms, which was not unexpectedly higher in the GO-147 arm. Therefore, if GO therapy should be pursued during induction therapy, it should preferably be administered to a young and fit population of patients. These findings are also supported by a recent publication in which 852 older patients with AML or high-risk MDS were randomized to receive GO on day 1 (GO1) or GO on days 1 and 4 (GO2). Results showed greater reduction in MRD and improved survival in older adults with non-adverse risk genetics by GO2. This benefit from GO2 was dependent on allogeneic transplantation to translate the better leukemia clearance into improved survival.15 According to previous publications, patients harboring mutations in the NPM1 gene respond favorably to intensive induction with the “7+3” regimen plus GO, with CR rates around 85% and 5-year OS around 40-50%.14 However, impressive responses have also been observed with azacitidine and venetoclax. In the VIALE-A phase III study the overall response rate was 93% and the 2-year OS was 75% for patients (N=27) harboring a NPM1 mutation.11 The response data suggest that the less intensive combination of azacitidine and venetoclax may potentially rival inten-
sive chemotherapy in clinical outcomes for patients with NPM1-mutated AML. However, it’s crucial to acknowledge the limitations of these observations, given the absence of randomized trials. The open question is whether there are indications to start gemtuzumab ozogamicine during induction therapy in newly diagnosed AML or if it is time to let it go?
Sonia Jaramillo,1 Johannes Krisam,2 Lucian Le Cornet,3 Markus Kratzmann,3 Lukas Baumann,2 Olga Eissymont,2 Martina Crysandt,4 Martin Görner,5 Sabine Kayser,3,6,7 Stefan Krause,8 Christoph Schliemann,9 Tobias Gaska,10 Martin Kaufmann,11 Jens Chemnitz,12 Markus Schaich,13 Alexander Hoellein,14 Uwe Platzbecker,7 Meinhard Kieser,2 Carsten Müller-Tidow1 and Richard F. Schlenk1,3
1Department of Internal Medicine V, Heidelberg University Hospital, Heidelberg; 2Institute of Medical Biometry, University of Heidelberg, Heidelberg; 3NCT-Trial Center, National Center of Tumor Diseases, Heidelberg University Hospital and German Cancer Research Center, Heidelberg; 4Department of Medicine IV, Aachen University Hospital, Aachen; 5Department of Hematology, Oncology and Palliative Medicine, Community Hospital Bielefeld, Bielefeld; 6Institute of Transfusion Medicine and Immunology, Medical Faculty Mannheim, Heidelberg University, Mannheim; 7Department of Medicine IHematology and Cell Therapy, University Hospital Leipzig, Leibzig; 8Department of Medicine V, Erlangen University Hospital, Erlangen; 9Department of Medicine A, Münster University Hospital, Münster; 10Department of Hematology and Oncology, St. Josef Brothers’ Hospital Paderborn, Paderborn; 11Department of Hematology, Oncology and Palliative Medicine, Robert-Bosch Hospital Stuttgart, Stuttgart; 12Department of Internal Medicine, Hematology, Oncology and Palliative Medicine, Prot. Monastery Hospital St. Jakob Koblenz, Koblenz; 13Department of Hematology, Oncology and Palliative Medicine, Winnenden Hospital, Winnenden and 14Department of Internal Medicine III - Hematology and Oncology, Red Cross Hospital Munich, Munich, Germany
1. Jaramillo S, Krisam J, Le Cornet L, et al. Rationale and design of the 2 by 2 factorial design GnG-trial: a randomized phase-III study to compare two schedules of gemtuzumab ozogamicin as adjunct to intensive induction therapy and to compare double-blinded intensive postremission therapy with or without glasdegib in older patients with newly diagnosed AML. Trials. 2021;22(1):765.
2. Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508-1516.
3. Lambert J, Pautas C, Terre C, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, phase III ALFA-0701 trial.
Correspondence:
R.F. SCHLENK - richard.schlenk@nct-heidelberg.de https://doi.org/10.3324/haematol.2023.284346
Received: September 22, 2023.
Accepted: February 9, 2024.
Early view: February 22, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
RFS discloses consulting for or advisory board membership with Daiichi Sankyo, Pfizer, Astellas, and Novartis; research funding from PharmaMar, AstraZeneca, Pfizer, Roche, Boehringer Ingelheim, and Daiichi Sankyo; travel, accommodations, and expenses covered by Daiichi Sankyo. The other authors declare no competing financial interests.
Contributions
RFS developed the concept and designed the study. SJ provided study materials. All authors collected and assembled data, analyzed and interpreted data, wrote the manuscript, and gave their final approval of the manuscript.
Acknowledgments
The authors thank the participating centers of the Study Alliance of Leukemia (SAL, www.sal.aml.org) for their commitment in the trial. Study drug was provided free-of-charge by Pfizer Pharma GmbH.
Funding
The trial was co-financed by funds of the German Research Organization (DFG- SCHL 2118/2-1). The study was supported by Pfizer Pharma GmbH.
Data-sharing statement
The datasets used and/or analyzed during the current study are available from the last author on reasonable request.
Haematologica. 2019;104(1):113-119.
4 Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
5. Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33(2):379-389.
6. Dohner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 ELN recommendations from an international expert panel. Blood.
2022;140(12):1345-1377.
7 Jaramillo S, Schlenk RF. Update on current treatments for adult acute myeloid leukemia: to treat acute myeloid leukemia intensively or non-intensively? That is the question. Haematologica. 2023;108(2):342-352.
8. Othman J, Amer M, Amofa R, et al. Venetoclax with azacitidine or low dose cytarabine as an alternative to intensive chemotherapy in fit adults during the COVID19 pandemic: real world data from the UK National Health Service. Blood. 2021;138(Suppl 1):2321.
9 Juliusson G, Billstrom R, Gruber A, et al. Attitude towards remission induction for elderly patients with acute myeloid leukemia influences survival. Leukemia. 2006;20(1):42-47.
10 DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7-17.
11. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N
Engl J Med. 2020;383(7):617-629.
12. Matthews AH, Perl AE, Luger SM, et al. Real-world effectiveness of CPX-351 vs venetoclax and azacitidine in acute myeloid leukemia. Blood Adv. 2022;6(13):3997-4005.
13. Schlenk RF, Paschka P, Krzykalla J, et al. Gemtuzumab ozogamicin in NPM1-mutated acute myeloid leukemia: early results from the prospective randomized AMLSG 09-09 phase III study. J Clin Oncol. 2020;38(6):623-632.
14. Dohner H, Weber D, Krzykalla J, et al. Intensive chemotherapy with or without gemtuzumab ozogamicin in patients with NPM1-mutated acute myeloid leukaemia (AMLSG 09-09): a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2023;10(7):e495-e509.
15. Freeman SD, Thomas A, Thomas I, et al. Fractionated vs singledose gemtuzumab ozogamicin with determinants of benefit in older patients with AML: the UK NCRI AML18 trial. Blood. 2023;142(20):1697-1707.
Higher-grade bone marrow (BM) fibrosis is associated with worse survival in patients with myeloproliferative neoplasms.1,2 Moreover, fibrotic changes in myelofibrosis (MF) progressively remodel the BM niche, resulting in impaired hematopoiesis and progressive worsening of anemia and thrombocytopenia, which are associated with reduced quality of life and poor prognosis.3-5 Zinpentraxin alfa (previously PRM-151) is a recombinant form of human pentraxin-2, an endogenous regulator of the tissue damage inflammatory response, and a natural inhibitor of fibrosis.6-8 A twostage phase II trial (NCT01981850) evaluated the efficacy and safety of zinpentraxin alfa in patients with MF. In the open-label stage 1, zinpentraxin alfa showed evidence of clinical activity and tolerable safety as monotherapy and in combination with ruxolitinib in patients with primary or secondary MF.9 Here we report the findings of stage 2 of this trial, which suggested signs of clinical activity of zinpentraxin alfa in patients with difficult-to-treat MF. This randomized, double-blind, phase II trial (NCT01981850) evaluated the efficacy and safety of three different doses of zinpentraxin alfa as monotherapy in patients aged ≥18 years with intermediate-1/2 and high-risk primary or secondary MF who were anemic or thrombocytopenic and ineligible for, intolerant of, or had an inadequate prior response to ruxolitinib. Eligible patients had MF grade ≥2 BM fibrosis and had had a BM biopsy within 4 weeks prior to treatment initiation to establish the baseline fibrosis score. The trial comprised three periods: a 4-week screening period, the main phase (9×4-week treatment cycles; total of 36 weeks), and a 4-week follow-up (Online Supplementary Figure S1). Patients without disease progression or discontinuation due to toxicity and with potential clinical benefit could continue zinpentraxin alfa 10 mg/kg treatment in an open-label extension phase. In the main phase, patients stratified by baseline hematologic status (anemia and/or thrombocytopenia) were randomized 1:1:1 using an interactive response system to receive zinpentraxin alfa 0.3 mg/ kg (group 1), 3 mg/kg (group 2), or 10 mg/kg (group 3) on days 1, 3, and 5 of cycle 1, and day 1 of each subsequent 28-day cycle. The patients, investigators, assessors, and sponsor were blinded to study treatment. Patients provided written informed consent before enrollment. The primary endpoint was BM response rate, defined as the percentage of patients with reduction from baseline in BM fibrosis by ≥1 grade per European Consensus criteria10 at any time, as determined by a central adjudication panel. Secondary and exploratory endpoints included hemoglobin concentration,
platelet count, Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS), spleen size improvements, and best overall response per International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) criteria. Adverse events, serious adverse events, and infusion-related reactions were recorded.
In stage 2 of this study, 98 patients were randomized to group 1 (n=33), group 2 (n=32), or group 3 (n=33) between November 23, 2015, and November 11, 2016. Online Supplementary Figure S2 provides full details of the patients’ disposition in the study. The patients’ baseline characteristics are summarized in Online Supplementary Table S1 The population of patients had a poor prognosis: 85.6% had intermediate-2 or high-risk disease, and 64.2% had centrally determined BM fibrosis grade 3 at baseline. Most patients (83.5%) had a baseline hemoglobin <100 g/L and 39.2% were dependent on red blood cell transfusions at baseline. Overall, 60.8% of patients had severe thrombocytopenia (platelets ≤50×109/L), and 15.5% were dependent on platelet transfusions at baseline.
In total, 28/97 patients (28.9%; 95% confidence interval: 19.85-37.88) had a BM response (group 1: n=10, 30.3%; group 2: n=10, 31.3%; group 3: n=8, 25.0%) (Figure 1A). Logistic regression analysis of pairwise comparisons between the three groups showed no statistically significant differences (P=0.58-0.93). Of the 28 patients with a BM response in the main phase, 26 (26.8% of all patients) had a best shift of 1-grade improvement, and two (2.1% of all patients) had a 2-grade improvement per European Consensus/World Health Organization criteria (Figure 1B). In the main phase, hemoglobin improvements were observed in 12/97 (12.4%) patients (group 1: n=5, 15.2%; group 2: n=5, 15.6%; group 3: n=2, 6.3%), and platelet count improvements were observed in 32/97 (33.0%) patients (group 1: n=9, 27.3%; group 2: n=11, 34.4%; group 3: n=12, 37.5%). Packed red blood cell and platelet transfusion requirements and changes in platelet count are shown in Figure 1C and D, respectively. During the combined main phase and open-label extension period, of the 15 patients with hemoglobin improvement, eight (53%) had a BM response, and of the 37 patients with a platelet count improvement, 12 (32%) had a BM response (Figure 2). The duration of hemoglobin and platelet improvements among BM responders and non-responders is shown in Figure 2. Hemoglobin and platelet count trajectories among BM responders and non-responders indicated relatively stable hemoglobin and platelet levels over time in most patients.
Figure 1. Efficacy outcomes in the main study phase. (A) Bone marrow (BM) response rate by central review at any time. (B) Best shifts in BM fibrosis score during the main phase of the study. (C) Improvement in hemoglobin level during the main phase of the study. (D) Improvement in platelet count during the main phase of the study. *Baseline BM fibrosis data were missing for two patients. BM response rate was defined as the percentage of patients with a reduction from baseline in BM fibrosis by ≥1 grade per European Consensus criteria.10 The primary endpoint of BM response rate was analyzed using logistic regression, with BM response at any time as the response variable and treatment group as the explanatory variable. The analysis was adjusted on a randomized stratum. Two pairwise comparisons (group 2 vs group 1 and group 3 vs. group 1) were computed with the aim of demonstrating superiority and, consequently, an adjusted two-sided level of significance of 0.025 was used. A third comparison (group 3 vs. group 2) was not expected to have enough power to demonstrate any difference with the planned sample size. This comparison was considered exploratory and was conducted using an unadjusted two-sided level of significance of 0.05. Baseline red blood cell transfusion dependency was defined as ≥2 units packed red blood cells every 4 weeks for 12 weeks prior to day 1 of cycle 1, regardless of baseline hemoglobin level. Baseline platelet transfusion dependency was defined as ≥2 platelet transfusions in any 12 weeks prior to day 1 of cycle 1, regardless of baseline platelet level. BM: bone marrow; PRBC: packed red blood cell.
Figure 2. Duration of hemoglobin and platelet count improvements. (A, B) Duration of improvement in hemoglobin concentration (A) and platelet count (B) during the main study phase and the open-label extension period combined. The x axis shows the duration of response and does not necessarily start from week 0 of the study period. Each bar represents an individual patient; the patients’ identities are shown as consecutive numbers, with patients who had both hemoglobin and platelet improvements in red text. “BM” indicates patients who had a bone marrow response. Hemoglobin improvement for red blood cell transfusion-dependent patients was defined as either an absence of any red blood cell transfusion during any consecutive 12-week interval with a hemoglobin level of ≥80 g/L, or a ≥50% reduction from baseline in red blood cell transfusions for 12 consecutive weeks with a hemoglobin level of ≥80 g/L. For red blood cell transfusion-independent patients, improvement was defined as a ≥10 g/L increase in hemoglobin level for 12 consecutive weeks without transfusions. Platelet improvement for platelet transfusion-dependent patients was defined as either becoming transfusion independent for 12 consecutive weeks, or a reduction in transfusion need from baseline of ≥50% for 12 consecutive weeks. For platelet transfusion-independent patients with a baseline platelet count <100×109/L, improvement was defined as a ≥50% increase in the number of platelets for 12 consecutive weeks or normalization of platelet count (above lower limit of normal). BM response was defined as a reduction from baseline in bone marrow fibrosis by ≥1 grade per European Consensus criteria. BM: bone marrow; PRBC: packed red blood cell; RBC: red blood cell; Hgb: hemoglobin.
MPN-SAF TSS, spleen size, and best overall response per IWG-MRT criteria were evaluated across the combined main phase and open-label extension. Of evaluable patients, 32/94 (34%) had a ≥50% reduction in MPN-SAF TSS compared to the baseline score at any time, 32/76 (42.1%) had any reduction in spleen volume at any time, and no patients had ≥35% reduction in spleen volume at week 36.
Clinical improvement was seen in 16/97 (16.5%) patients, and 67 (69.1%) had stable disease.
Safety results are summarized in Table 1. Generally, zinpentraxin alfa was well tolerated across all doses; 97 patients experienced ≥1 treatment-emergent adverse event. In total, 77 serious treatment-emergent adverse events were reported in 39 patients, most frequently pneumonia (n=5;
Table 1. Summary of treatment-emergent adverse effects in the safety population during the main phase, by cohort and overall.
Patients, N (%)
TEAE (≥10% of
leading to discontinuation of zinpentraxin alfa
aGroup 1 was treated with zinpentraxin alfa 0.3 mg/kg Q4W. bGroup 2 was treated with zinpentraxin alfa 3 mg/kg Q4W. cGroup 3 was treated with zinpentraxin alfa 10 mg/kg Q4W. dFatal treatment emergent adverse events were reported as the following preferred terms: acute myeloid leukemia, malignant neoplasm progression, myelofibrosis, primary myelofibrosis, transformation to acute myeloid leukemia, death, pancytopenia, cardiopulmonary failure, obstructive femoral hernia, cachexia, cerebral hemorrhage (all N=1), pneumonia (N=2), disease progression (N=2). Q4W: every 4 weeks; TEAE: treatment-emergent adverse event; AE: adverse event.
5.2%) and epistaxis (n=3; 3.1%). Because of the small sample size and the low number of serious treatment-emergent adverse events, conclusions cannot be drawn regarding potential differences in the safety of different doses. Of the 15 fatal treatment-emergent adverse events (15.5%), one death was reported as related to the study treatment; however, the investigator reported that the death was likely related to underlying thrombocytopenia due to MF, leading to a bleed. Infusion-related reactions were reported in four (4.1%) patients; all these reactions were grade 1/2, except for grade 3 urticaria in one patient in group 2. No new safety signals were reported during the open-label extension phase of the study. Genetic analysis revealed similar mutational profiles across all treatment groups. No notable changes were identified in variant allele frequency in any treatment group during the study and most patients had changes of ±5%, which could be due to variation or background noise. Overall, zinpentraxin alfa treatment showed some improvements in BM fibrosis and hematologic parameters across all doses, with reduction in BM fibrosis at any time observed in approximately 30% of patients. Despite the lack of a clear dose–response relationship observed within the tested dose range, the lack of a control arm, and the fact that only around half of patients had biopsy results available at all three post-baseline timepoints, responses in patients with advanced BM failure are suggestive of clinical activity of zinpentraxin alfa in MF. Furthermore, some of these patients with very poor prognosis were treated with zinpentraxin alfa for a prolonged period, up to 46.7 months, which was also somewhat unexpected. Despite advanced and high-risk disease, improvements in hemoglobin levels and platelet counts were reported across all treatment groups. Reductions in red blood cell transfusion dependence in various populations of patients have been observed previously with other treatments; however, to our knowledge, only pacritinib has achieved notable results in patients with severe thrombocytopenia (platelet counts ≤50×109/L) and red blood cell transfusion dependence, albeit in the setting of limited or no prior JAK inhibitor exposure.11 Ruxolitinib discontinuation leads to a poor prognosis and progressive worsening of anemia and thrombocytopenia.12,13 However, most patients in the current study, of whom 76.3% had previously received ruxolitinib, had stable or improved hemoglobin levels and platelet counts. The hematologic improvements observed with zinpentraxin alfa are important because analysis of recent momelotinib trials suggests that hematologic improvement may serve as a surrogate endpoint predictive of improved overall survival.14 The current study did not assess overall survival and numbers of patients were small; however, some patients with high-risk features and poor prognosis following ruxolitinib discontinuation had long-lasting treatment (median 7.5 months; range, 0.2-46.7). Furthermore, since transfusion dependence is
burdensome to patients, the reported reductions in red blood cell and platelet transfusions in transfusion-dependent patients are also important from a quality-of-life perspective.3
Limitations of this study include the lack of a placebo arm, a heterogeneous population of patients, and a small sample size, which make it difficult to interpret trends. Advanced disease stage and negative prognostic factors may also have reduced the likelihood of observing effects on fibrosis. Finally, it is unclear whether a 35% threshold for reduction in spleen size is appropriate in the setting of relapsed/refractory patients with advanced disease, and in several studies few or no patients have achieved ≥35% spleen volume reduction in this setting.15
In summary, zinpentraxin alfa treatment showed signs of clinical activity, including improvements in fibrosis, disease-related hematologic parameters, and symptoms, in difficult-to-treat patients with MF who were ineligible for, intolerant of, or had inadequate response to ruxolitinib. Results should be interpreted with caution because of the small sample sizes and lack of a placebo arm. The potential for additional clinical benefit in newly diagnosed patients and those with less fibrosis remains a hypothesis to be examined in future clinical trials. The results from stage 1 and stage 2 of this trial will inform future investigations of zinpentraxin alfa in patients with MF.
Srdan Verstovsek,1° Moshe Talpaz,2 Martha Wadleigh,3 Alessandro Isidori,4 Peter te Boekhorst,5 Michael R. Savona,6 Prithviraj Bose,1 Olga Pozdnyakova,7 Ruben Mesa,8° Tarec C. El-Galaly,9° Jennifer O’Sullivan,10 Katia Gamel,9 Brian Higgins,11 Sudhakar Katakam,9 Boyan Todorov,9 Kerstin Trunzer9 and Claire N. Harrison10
1The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Michigan Medicine - The University of Michigan, Ann Arbor, MI, USA; 3Dana-Farber Cancer Institute, Boston, MA, USA; 4Hematology and Stem Cell Transplant Center, AORMN Hospital, Pesaro, Italy; 5Erasmus Medical Center, Rotterdam, the Netherlands; 6VanderbiltIngram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN, USA; 7Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA; 8Mays Cancer Center at UT Health San Antonio MD Anderson, San Antonio, TX, USA; 9F. Hoffmann-La Roche, Ltd., Basel, Switzerland; 10Guy’s & St Thomas’ NHS Foundation Trust, London, UK and 11Genentech Inc, South San Francisco, CA, USA
°Current address of SV: Kartos Therapeutics, Redwood City, CA, USA
°Current address of RM: Wake Forest Baptist Comprehensive Cancer Center, Winston-Salem, NC, USA
°Current address of TCE-G: Department of Hematology, Aalborg University Hospital, Aalborg, Denmark
Correspondence: C.N. HARRISON - claire.harrison@gstt.nhs.uk
https://doi.org/10.3324/haematol.2023.284410
Received: October 3, 2023.
Accepted: January 18, 2024. Early view: January 25, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY license
Disclosures
SV has received research support from BMS, Constellation, CTI BioPharma, Galecto, Geron, Incyte, Kartos, Novartis, NS Pharma, Protagonist, PharmaEssentia, Roche, and Sierra, and consulting fees from BMS, Celgene, Constellation, Incyte, and Novartis. MT has served as a member on a board or advisory committee for BMS, Novartis, and Sumitomo; has received support for attending meetings from Sumitomo; has received research support from Promedior and Roche; and has a leadership role with the Society of Hematologic Oncology. MRS has received royalties or licenses from Boehringer Ingelheim; has received consulting fees from Geron, Karyopharm, and Ryvu; has received support for attending meetings from Ryvu and Taiho; holds patents from Boehringer Ingelheim; has served as a member on a board or advisory committee for AbbVie, Bristol Myers Squibb, CTI, Geron, GSK, Karyopharm, Novartis, Rigel, Ryvu, Taiho, and Treadwell; has stock or stock options in Karyopharm and Ryvu; and has received research funding from ALX Oncology, Astex, Incyte, Takeda, and TG Therapeutics. PB has received research support from Blueprint, BMS, Cogent, CTI, Disc, Geron, Incyte, Ionis, Janssen, Kartos, Karyopharm, MorphoSys, Sumitomo, and Telios, and has received honoraria/consulting fees from AbbVie, Blueprint, BMS, Cogent, CTI, GSK, Incyte, Ionis, Jubilant, Karyopharm, Morphic, MorphoSys, Novartis, PharmaEssentia, and Sumitomo. RM has received consulting fees and honoraria from AbbVie, Blueprint, BMS, CTI BioPharma, Genentech, Geron, GSK, Incyte, MorphoSys, Novartis, Sierra, Sierra Oncology, and Telios. TCE-G was employed by Roche at the time this work was performed. KG is an employee of Roche. BH is an employee of Roche/Genentech and owns stocks in the company. BT was contracted by Roche at the time this work was performed. KT is an employee of Roche and owns stocks in the company. CNH has
1. Lekovic D, Gotic M, Perunicic-Jovanovic M, et al. Contribution of comorbidities and grade of bone marrow fibrosis to the prognosis of survival in patients with primary myelofibrosis. Med Oncol. 2014;31(3):869.
2. Guglielmelli P, Rotunno G, Pacilli A, et al. Prognostic impact of bone marrow fibrosis in primary myelofibrosis. A study of the AGIMM group on 490 patients. Am J Hematol. 2016;91(9):918-922.
3. Naymagon L, Mascarenhas J. Myelofibrosis-related anemia: current and emerging therapeutic strategies. Hemasphere. 2017;1(1):e1.
4 Hernández-Boluda JC, Correa JG, Alvarez-Larrán A, et al.
received consulting fees from AbbVie, AOP, BMS, Constellation Pharmaceuticals, CTI BioPharma, Galecto, GSK, Karyopharm, Keros, MorphoSys, Novartis, Promedior, and Roche; has received honoraria from AbbVie, BMS, GSK, and Novartis; has advisory roles for Galecto and Keros; has received support from Novartis for attending meetings; and has a leadership or fiduciary role with the European Hematology Association and MPN Voice. MW, AI, PtB, OP, JO’S, and SK have no conflicts of interest to disclose.
SV and CNH conceived and designed the work. SV, AI, PtB, MRS, KT, MT, MW, JO’S, TCE-G, and CNH acquired data. SV, PtB, MRS, BH, KT, OP, SK, KG, JO’S, and TCE-G analyzed the data. SV, AI, PtB, MRS, BH, BT, KT, OP, SK, MW, KG, and JO’S interpreted the results. All authors were involved in reviewing/revising the manuscript, approved the final version, and vouch for the accuracy of the content included in the manuscript.
The authors would like to thank the patients, their family members, participating staff at all of the study centers, Dr Dao Wang for her contribution to the safety analysis, and the Promedior study team.
This study was supported by Promedior (original sponsor) and F. Hoffmann-La Roche, Ltd. (current sponsor). Medical writing support, under the direction of the authors, was provided by Fiona Scott, PhD, on behalf of CMC Affinity, a division of IPG Health Medical Communications, funded by F. Hoffmann-La Roche, Ltd, in accordance with Good Publication Practice (GPP 2022) guidelines.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org). Further details on Roche’s criteria for eligible studies are available at https://vivli.org/members/ourmembers. For further details on Roche’s global policy on the sharing of clinical information and how to request access to related clinical study documents, visit https://www.roche. com/research_and_development/who_we_are_how_we_work/clinical_ trials/our_commitment_to_data_sharing.htm.
Clinical characteristics, prognosis and treatment of myelofibrosis patients with severe thrombocytopenia. Br J Haematol. 2018;181(3):397-400.
5. Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285-299.
6. Castaño AP, Lin S-L, Surowy T, et al. Serum amyloid P inhibits fibrosis through FcγR-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009;1(5):5ra13.
7 Verstovsek S, Manshouri T, Pilling D, et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J Exp
Med. 2016;213(9):1723-1740.
8. Zahr AA, Salama ME, Carreau N, et al. Bone marrow fibrosis in myelofibrosis: pathogenesis, prognosis and targeted strategies. Haematologica. 2016;101(6):660-671.
9 Verstovsek S, Foltz L, Gupta V, et al. Safety and efficacy of zinpentraxin alfa as monotherapy or in combination with ruxolitinib in myelofibrosis: stage I of a phase II trial. Haematologica. 2023;108(10):2730-2742.
10. Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90(8):1128-1132.
11. Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with
myelofibrosis: a randomized cinical trial. JAMA Oncol. 2018;4(5):652-659.
12. Mascarenhas J, Mehra M, He J, Potluri R, Loefgren C. Patient characteristics and outcomes after ruxolitinib discontinuation in patients with myelofibrosis. J Med Econ. 2020;23(7):721-727.
13. Sastow D, Mascarenhas J, Tremblay D. Thrombocytopenia in patients with myelofibrosis: pathogenesis, prevalence, prognostic impact, and treatment. Clin Lymphoma Myeloma Leuk. 2022;22(7):e507-e520.
14 Mesa R, Harrison C, Oh ST, et al. Overall survival in the SIMPLIFY-1 and SIMPLIFY-2 phase 3 trials of momelotinib in patients with myelofibrosis. Leukemia. 2022;36(9):2261-2268.
15. Tremblay D, Mascarenhas J. Next generation therapeutics for the treatment of myelofibrosis. Cells. 2021;10(5):1034.
Lymphocytic-variant hypereosinophilic syndrome (L-HES) is an indolent T-cell lymphoproliferative disorder characterized by chronic blood hypereosinophilia and eosinophil-related organ damage 1,2 secondary to the production of eosinophilopoietic cytokines (including interleukin-5 [IL-5]) by clonal T cells, among which CD3 -CD4 + T cells are the most frequent subset. 3,4 L-HES is usually sensitive to oral corticosteroids (OCS), but high maintenance doses are frequently required. OCS-sparing treatment (including mepolizumab) has variable often transient efficacy.1,3,5,6 Moreover, up to 5% to 10% of L-HES patients will ultimately develop an angioimmunoblastic T-cell lymphoma (AITL). 3,7 Overall, both high OCS exposure, failure and/or toxicity of OCS-sparing therapies and the risk of progression to AITL underscore the unmet need for novel treatments targeting clonal T cells and not only eosinophils. We previously reported that peripheral CD3CD4 + T cells express C-C chemokine receptor 4 (CCR4). 8 Here, we first report on the use of off-label mogamulizumab, a defucosylated monoclonal antibody targeting CCR4-positive cells through antibody-dependent cellular cytotoxicity 9,10 in refractory L-HES.
We assessed expression of CCR4 by clonal CD3- CD4 + T cells in the skin and lymph nodes i.e., the two main organs involved in L-HES. We found significant CCR4 expression on tissue CD4 + T cells (Figure 1A). We then confirmed the ability of mogamulizumab to induce antibody-dependent cellular cytotoxicity (ADCC) in vitro on sorted CD3 -CD4 + T cells from two L-HES patients, using homologous sorted natural killer (NK) cells as effector cells. As expected, NK cells induced the lysis of CD3 -CD4 + T cells coated with mogamulizumab (10 µ g/mL) (Figure 1B). We then treated four refractory L-HES patients with off-label mogamulizumab, administered intravenously at 1.0 mg/kg weekly for the first 28-day cycle, then on days 1 and 15 of subsequent cycles. Blood response was defined as complete if the subset of CD3 -CD4 + T cells was <0.5% of all lymphocytes, or as partial if the number of CD3 -CD4 + T cells decreased by >50% without reaching complete response at the time of the last perfusion. Patients provided informed consent before receiving mogamulizumab as off-label treatment. Data were retrospectively collected from the French registry LYMPHEO (clinical and pathological characteristics of patients with LYMPhocyte-variant HyperEOsinophilic syndrome). As provided by the French legislation on the use of health data, the four patients were informed by their referring physician about the possibility of using their data and none objected. This study was conducted in accordance
with the Declaration of Helsinki and was approved by an Ethics Committee (IRB00012437).
Case #1 was a 60-year-old female complaining of severe refractory pruritus despite treatment with OCS and pegylated interferon a (pINF- a ) (Table 1). Mogamulizumab was administered for 20 infusions and stopped after sustained response was achieved. Blood response was complete (CD3 -CD4 +: 0.003x10 9/L vs . 4.624x10 9/L at baseline) with no eosinophilia. Six months after stopping mogamulizumab, she was asymptomatic under 5 mg/day of OCS. Nevertheless, a clinical relapse occurred, preceded by an increase in T-cell clone size and absolute eosinophil count (AEC). She was successfully treated by another short course of 4 weekly infusions of mogamulizumab. No clinical relapse has been reported since then with 12 months of follow-up.
Case #2 was a 59-year-old male complaining of refractory pruritus and enlarged lymph nodes despite treatment with OCS and pINF- a . Lymph node biopsy showed an infiltration by CCR4 +CD4 + T cells without features of AITL. Eight weeks after starting mogamulizumab, positron emission tomography/computed tomography showed an improvement of lymph node enlargement (Figure 1C). After 27 perfusions, blood response was partial (CD3CD4 +: 0.050x10 9/L vs . 0.336x10 9/L at baseline) without eosinophilia. While remaining asymptomatic on lowdose OCS, grade 3 CD3 +CD4 + lymphopenia (yet without opportunistic infection) was reported. Mogamulizumab was discontinued due to personal convenience and no relapse has been reported since then with 12 months of follow-up.
Case #3 was a 41-year-old female presenting with pruritus, episodic angioedema, and enlarged lymph nodes. Mogamulizumab was initiated due to persistent episodic angioedema and a rapid increase of both CD3 -CD4+ T cells (up to 1.824x10 9/L) and eosinophils (7.8x10 9/L), despite treatment with both OCS and pINF- a . Mogamulizumab was discontinued after the seventh infusion due to recurrent grade 2 infusion-related systemic reactions. Blood response was complete (CD3 -CD4 +: 0.006x10 9/L vs . 1.824x10 9/L at baseline) with persistent moderate eosinophilia (1.0x10 9/L). Grade 3 CD3 +CD4 + lymphopenia was reported with no opportunistic infection during follow-up. She remained asymptomatic (yet with persistent moderate eosinophilia) during the next 18 months, yet a relapse ultimately occurred preceded by an increase of both T-cell clone size and AEC.
Case #4 was a 47-year-old female complaining of chronic refractory urticaria and arthralgia. Seven years after
disease onset, she complained of erythroderma accompanied by skin ulcerations, lymph node enlargement and a drastic increase in the number of CD3 -CD4 + T cells (up to 55.586x10 9/L). Clonal T cells were found in both skin and lymph node biopsies yet without signs of AITL. Mogamulizumab was started with OCS. Two hours after the second infusion, she was admitted to the intensive care unit for acute respiratory distress. Bronchoalveolar lavage showed mixed eosinophilic and lymphocytic (including 70% of CD3 -CD4 + T cells: 1.332x10 9/L) alveolitis. AEC increased to 15.0x10 9/L and the serum level of IL-5 was high (162.0 pg/mL; normal range, 0.1-0.6). She received high-dose steroids and antibiotics, and her condition improved. Meanwhile, the CD3 -CD4 + T-cell count sharply plummeted to 10.780x10 9/L within a few days.
Nevertheless, lymph node enlargement increased, and erythroderma worsened over the following weeks. Mogamulizumab was resumed at a lower dose (1/10), with good tolerance and efficiency. After the fourth infusion, blood response was partial (CD3 -CD4 +: 9.035x10 9/L vs 55.586x10 9/L at baseline) with moderate eosinophilia (0.7x10 9/L). Unfortunately, the following weeks were marked by multiple severe infectious events which delayed the mogamulizumab infusions. Despite initial improvement, erythroderma further worsened, an increase of clonal T cells was evidenced and mogamulizumab was ultimately withdrawn due to these adverse events (AE). Grade 3 CD3 +CD4 + lymphopenia was also reported. Overall, blood responses (complete, N=2 or partial, N=2) and a decrease in AEC were reported in all cases (Figure 2).
Figure 1. Histologic findings, antibody-dependent cellular cytotoxicity results, and lymph node response to mogamulizumab. (A) Lymph node biopsy showing a benign infiltration of CCR4+CD4+ T cells. Left: hematoxylin and eosin staining x400 magnification, scale bar=50 µm. Right: immunostaining x100 magnification, scale bar=200 µm, anti-CD4 (upper panel), anti-CD8 (middle panel) and anti-CCR4 (lower panel). (B) Ability of mogamulizumab to induce antibody-dependent cellular cytotoxicity (ADCC) on sorting CD3-CD4+ T cells by conducting an ADCC in vitro assay in 2 lymphocytic-variant hypereosinophilic syndrome (L-HES) patients using sorted CD3-CD4+ T cells as targeted cells (T) and homologous sorted natural killer (NK) cells as effector cells (E). NK cells kill CD3-CD4+ T cells coated with mogamulizumab (10 µg/mL) in a dose-dependent manner. No significant lysis was observed without mogamulizumab (data not shown). (C) Positron emission tomography/computed tomography scan showing a large lymph node enlargement (arrows) in case #2: 8 weeks (W8) before starting mogamulizumab (left), at baseline (middle) and W8 after treatment onset (right). E/T: effector/target ratio.
In terms of clinical response, two patients experienced lymph node enlargement, which subsequently improved rapidly. Despite substantial tapering in OCS doses, all cases showed clinical response. Although prolonged blood and clinical responses have been observed after discontinuation of mogamulizumab, relapses still occurred suggesting that mogamulizumab has a suspensive effect. In terms of safety, mogamulizumab was discontinued in two patients due to AE. CD3 +CD4 + T-cell lymphopenia was reported in three of four cases but was pre-existing in three cases. Safety profile was in line with published data in patients treated with mogamulizumab for mycosis fungoides.12 Here, infectious events were reported in case 4, who otherwise had additional risk factors for infection e.g., treatment with high doses of OCS and multiple skin ulcerations. She also presented a serious AE related to tumor lysis syndrome (likely triggered by NK-mediated ADCC induced by mogamulizumab) leading to the massive release of IL-5 and subsequent rebound of AEC. Hence lower doses of mogamulizumab, transient use of OCS and/or mepolizumab should be considered in future patients with very high number of CD3 -CD4 + T
cells which are about to start mogamulizumab. Overall, we suggest that in L-HES patients the first OCS-sparing agent should remain pINF- a (i.e., the drug with the most available evidence; starting with a low weekly dose to improve tolerability) 11,12 or mepolizumab (i.e., the only drug licensed in FIP1L1::PDGFRA -negative HES; 300 mg subcutaneously every 4 weeks),6,13,14 bearing in mind that failures have been reported with both drugs and that comparative data are lacking. Janus kinase inhibitors are other promising therapies for treating HES-related symptoms.15 Bearing in mind the risk of both tumoral lysis syndrome and infections (as well as the fact that relapses may occur after discontinuation of the drug), mogamulizumab represents a new therapeutic option for the subgroup of patients with severe multi refractory-treatment disease, especially with clonal T-cell-related symptoms or earlier in cases of significant tumor syndrome and/or large or rapidly increasing clonal T-cell count. In patients who achieve complete remission under mogamulizumab, whether fix or on-demand (e.g., tailored by circulating CD3-CD4 + T cells and/or AEC maintenance infusions could be beneficial to prevent
1. Patient baseline characteristics at the time of refractory lymphocytic-variant hypereosinophilic syndrome diagnostic.
DNMT3A exon 15
c.1814T>C: p.L605P (VAF 1.41%) Lymph node Pruritus Lymph node enlargement
Splenomegaly
F
DNMT3A exon 14
c.1667+1G>A (VAF 2%)
DNMT3A exon 22
c.2578T>C: p.W860R (VAF 2%)
SMC1A exon 4
c.571A>T: p.N191Y (VAF 56%)
Skin
Medullar
Lymph node BAL
Splenomegaly
Urticaria
Erythroderma
Skin ulceration
Arthralgia
Lymph node enlargement
Splenomegaly
Interstitial lung disease
OCS pINF-a Omalizumab
Mepolizumab
#Next generation sequencing (NGS) panel: ABL1, ANKRD26, ARID1A, ASXL1, ASXL2, ATM, ATRX, B2M, BAX, BCL2, BCOR, BCORL1, BIRC3, BRAF, BRCA1, BRCA2, BTK, CALR, CARD11, CBL, CCND1, CD274, CD28, CD58, CD79A, CD79B, CDKN2A, CEBPA, CHEK2, CIITA, CREBBP, CRLF2, CSF2RA, CSF2RB, CSF3R, CUX1, CXCR4, DDX3X, DDX41 DHX15 DHX34 DNMT3A, EGR2, EP300, ETNK1, ETV6, EZH2, FBXW7, FGFR1, FLT3, FOXO1, GATA1, GATA2, GNA13, GNAS, GNB1, HRAS, ID3, IDH1, IDH2, IKZF1, IL2RG, IL3RA, IL5RA, IL7R, IRF4, ITPKB, JAK1, JAK2, JAK3, KDM6A, KIT, KLF2, KMT2A, KMT2D, KRAS, LUC7L2, MAP2K1, MAP3K14, MBD4, MEF2B, MPL, MSC, MYC, MYD88, NF1, NFE2, NIPBL, NOTCH1, NOTCH2, NPM1, NRAS, NSD2, PAX5, PDGFRA, PDGFRB, PHF6, PIGA, PIM1, PLCG1, PLCG2, PPM1D, PRDM1, PRPF8, PTEN, PTPN11, PTPRD, RAD21, RB1, RHOA, RIT1, RPS15, RUNX1, SAMD9, SAMD9L, SETBP1, SETD2, SF3B1, SH2B3, SMC1A, SMC3, SOCS1, SPI1, SRP72, SRSF2, STAG2, STAT1, STAT3, STAT5A, STAT5B, STAT6, TBL1XR1, TCF3, TCL1A, TERC, TERT, TET2, TNFAIP3, TNFRSF14, TP53, TYK2, U2AF1, UBA1, UBTF, VAV1, WT1, XPO1, ZBTB7A, ZRSR2. *All biopsies showed an infiltration of eosinophils and CCR4+CD4+ T cells. AEC: absolute eosinophil count; BAL: bronchoalveolar lavage; F: female; pINF-a: pegylated-interferon a; M: male; OCS: oral corticosteroids; TCR rear.: clonal T-cell receptor rearrangement in blood; VAF: variant allele frequency.
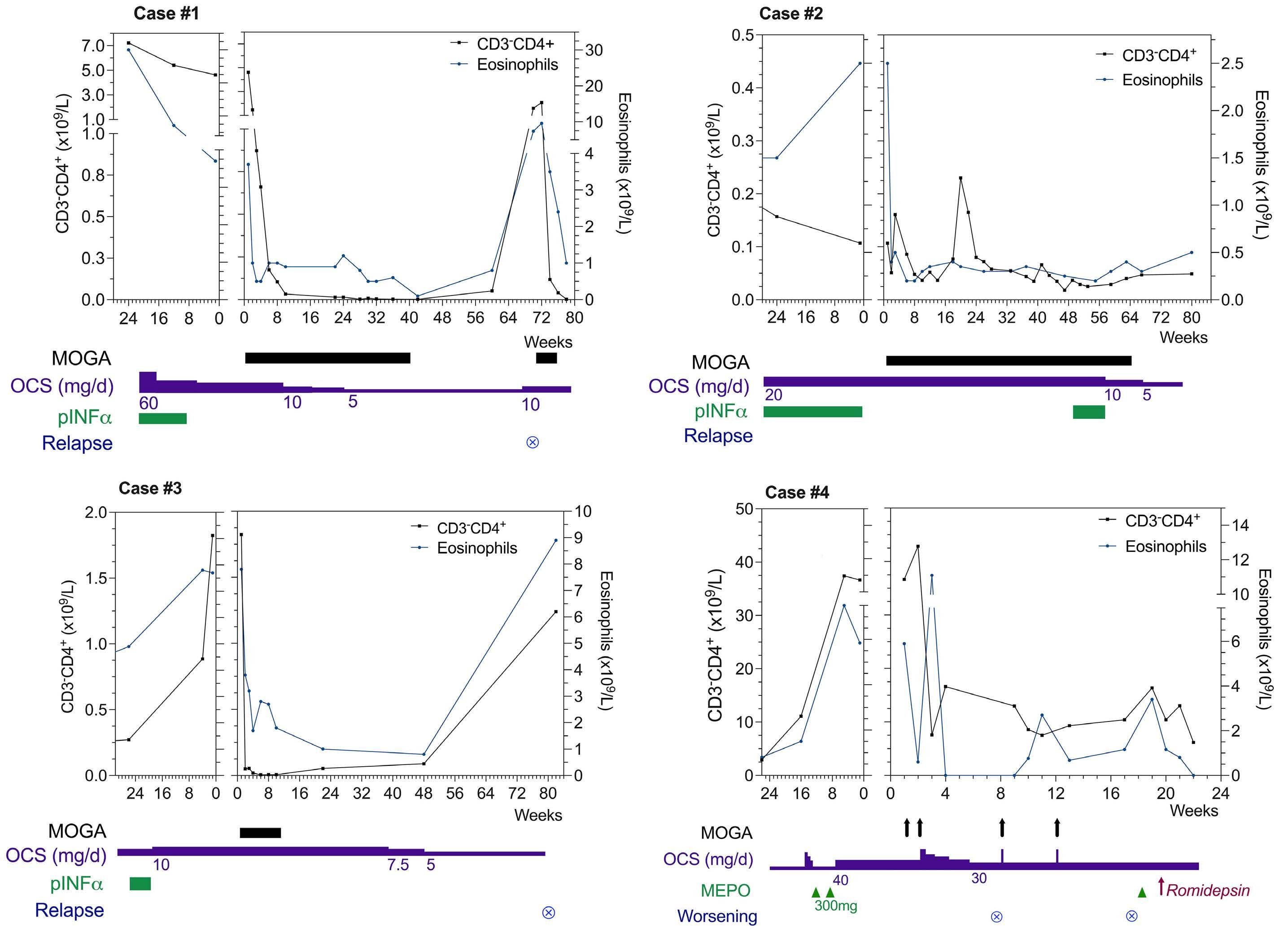
Figure 2. Hematological response to mogamulizumab in four refractory lymphocytic-variant hypereosinophilic syndrome patients. pINF-a: pegylated interferon a; MEPO: mepolizumab; MOGA: mogamulizumab; OCS: oral corticosteroids, d: day.
relapses and transformation to high-grade lymphoma remains to be investigated.
Au tho rs
Emmanuel Ledoult 1,2, Matthieu Groh 2,3, Bertrand Meresse, 2 Romain Dubois,4 Jacques Trauet, 2,5 Elise Toussaint,6 Marie Delbeke, 2 Eric Hachulla,1,2 Louis Terriou,1 Adèle De Masson,7 Michele Vasseur, 8 Myriam Labalette, 2,5 David Launay,1,2 Jean-Emmanuel Kahn 9 and Guillaume Lefevre 1,2,5
1Service de Médecine Interne et d’Immunologie Clinique, Centre de Référence des Syndromes Hyperéosinophiliques (CEREO), CHU Lille, Lille; 2INFINITE - Institute for Translational Research in Inflammation, Université de Lille, Lille; 3Service de Médecine Interne, Centre de Référence des Syndromes Hyperéosinophiliques (CEREO), Hôpital Foch, Suresnes; 4Institut de Pathologie, CHU Lille, Lille; 5Laboratoire d’Immunologie, CHU Lille, Lille; 6Service d’Hématologie, Institut de Cancérologie
Strasbourg Europe, Strasbourg; 7INSERM U976, Service de Dermatologie, Hôpital Saint-Louis, Paris; 8Pharmacie Centrale, CHU Lille, Lille and 9Université Paris Saclay, Service de Médecine Interne, Hôpital Ambroise Paré, APHP, Boulogne Billancourt, France
Correspondence: E. LEDOULT - emmanuel.ledoult2@chu-lille.fr https://doi.org/10.3324/haematol.2023.284429
Received: October 9, 2023. Accepted: January 31, 2024. Early view: February 8, 2024.
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
EL, MG, J-EK and GL designed the research and wrote the paper. BM and MD performed ADCC in vitro assay. RD analyzed tissues from the patient. JT performed flow cytometry phenotyping. EL, MG, ET, EH, LT, AD, MV, ML, DL, J-EK and GL collected and
1. Lefèvre G, Copin M-C, Staumont-Sallé D, et al. The lymphoid variant of hypereosinophilic syndrome: study of 21 patients with CD3-CD4+ aberrant T-cell phenotype. Medicine (Baltimore). 2014;93(17):255-266.
2. Valent P, Klion AD, Roufosse F, et al. Proposed refined diagnostic criteria and classification of eosinophil disorders and related syndromes. Allergy. 2023;78(1):47-59.
3. Carpentier C, Verbanck S, Schandené L, et al. Eosinophilia associated with CD3−CD4+ T cells: characterization and outcome of a single-center cohort of 26 patients. Front Immunol. 2020;11:1765.
4. Shi Y, Wang C. What we have learned about lymphocytic variant hypereosinophilic syndrome: a systematic literature review. Clin Immunol. 2022;237:108982.
5. Lefevre G, Copin MC, Roumier C, Aubert H, 2015. CD3-CD4+ lymphoid variant of hypereosinophilic syndrome: nodal and extranodal histopathological and immunophenotypic features of a peripheral indolent clonal T-cell lymphoproliferative disorder. Haematologica. 2015;100(8):1086-1095.
6. Kuang FL, Fay MP, Ware J, et al. Long-term clinical outcomes of high-dose mepolizumab treatment for hypereosinophilic syndrome. J Allergy Clin Immunol Pract. 2018;6(5):1518-1527.
7 Roufosse F, Leval L de, Krieken H van, Deuren M van. Lymphocytic variant hypereosinophilic syndrome progressing to angioimmunoblastic T-cell lymphoma. Leuk Lymphoma. 2015;56(6):1891-1894.
8. Ledoult E, Groh M, Kahn JE, et al. Assessment of T-cell polarization on the basis of surface marker expression: diagnosis and potential therapeutic implications in lymphocytic
analyzed data. EL made the figures.
Data-sharing statement
For original data, please contact the corresponding author.
variant hypereosinophilic syndrome. J Allergy Clin Immunol Pract. 2019;8(3):1110-1114.
9. Kim YH, Bagot M, Pinter-Brown L, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2018;19(9):1192-1204.
10 Beylot-Barry M, Quereux G, Nardin C, et al. Effectiveness of mogamulizumab in patients with Mycosis Fungoides or Sézary syndrome: a multicentre, retrospective, real-world French study. J Eur Acad Dermatol Venereol. 2023;37(9):1777-1784.
11. Choi C, Moller D, Tan J, et al. Pegylated interferon alpha 2a is an effective and well-tolerated treatment option for lymphocytevariant hypereosinophilic syndrome. Br J Haematol. 2020;188(5):e68-72.
12. Groh M, Rohmer J, Etienne N, et al. French guidelines for the etiological workup of eosinophilia and the management of hypereosinophilic syndromes. Orphanet J Rare Dis. 2023;18(1):100.
13. Roufosse F, Lavareille A de, Schandené L, et al. Mepolizumab as a corticosteroid-sparing agent in lymphocytic variant hypereosinophilic syndrome. J Allergy Clin Immunol. 2010;126(4):828-835.
14 Rothenberg ME, Klion AD, Roufosse FE, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med. 2008;358(12):1215-1228.
15. Faguer S, Groh M, Vergez F, et al. JAK inhibition for CD3− CD4+ lymphocytic-variant hypereosinophilic syndrome. Clin Immunol. 2023;251:109275.
Splanchnic vein thrombosis (SVT) include portal vein thrombosis (PVT), Budd-Chiari syndrome (BCS), and splenic and/ or mesenteric vein thrombosis (S/MVT). They are frequently associated with Philadelphia-negative myeloproliferative neoplasms (MPN),1 with up to 50% of idiopathic SVT cases exhibiting the JAK2 V617F mutation.2 Diagnosing MPN in the context of SVT is challenging because the resultant portal hypertension and subsequent splenomegaly and hypersplenism can lower blood cell counts, obscuring the elevated counts typical of MPN. The routine inclusion of JAK2 V617F mutation screening in SVT patient evaluations has enabled the identification of MPN that lack the usual hematological features, representing about 15% of BCS and PVT cases.2 These patients, who otherwise would remain undiagnosed, are categorized as having unclassified MPN (MPN-U), leaving the decision to initiate cytoreductive therapy unresolved. Prior to the discovery of the JAK2 V617F mutation, demonstrating an increased red cell mass (RCM) through isotopic methods was a key
diagnostic criterion for polycythemia vera (PV). According to the 2008 World Health Organization (WHO) criteria, hemoglobin levels above 18.5 g/dL in males and 16.5 g/dL in females were considered indicative of increased RCM, leading to the discontinuation of RCM measurement in many facilities.3 We hypothesized that for patients with JAK2 V617F-positive SVT and normal hemoglobin levels, RCM measurement could facilitate the diagnosis of PV, thereby guiding the initiation and adjustment of cytoreductive therapy.
A retrospective study was conducted on 71 patients diagnosed with SVT, carrying the JAK2 V617F mutation but presenting normal hemoglobin and hematocrit levels initially. Data were gathered from eight French medical centers. Patients gave written consent and the study was conducted in accordance with the Declaration of Helsinki. Key characteristics are detailed in Table 1. The median age at diagnosis was 44 years, with females comprising 48% of the cohort. The median interval between SVT diagnosis and JAK2 V617F
Palpable splenomegaly, N/N
BM biopsy, hyperplasia, N/N 0-2 lineage
Type of splanchnic vein thrombosis, N/N (%)
Portal
Budd-Chiari
Porto-sinusoidal vascular disease 55/70 (78) 12/70 (17) 3/70 (4)
(77) 7/31 (23) 0/31 (0) 31/39 (80) 5/39 (12) 3/39 (8)
RCM: red cell mass ; PV: polycythemia vera; BM: bone marrow; EP: erythopoietin.
mutation detection was 1 month (range, 0-144). Coincident diagnosis of SVT and JAK2 V617F mutation was observed in 64% of cases within a 3-month margin. The mutation was detected within 2 years post-SVT in 14 patients, beyond 2 years in nine patients, with one case’s timeline unspecified. Increased RCM values over 125%, suggesting masked PV, were found in 56% of patients. Notably, average hemoglobin levels and hematocrits were comparable between the masked PV subgroup and non-PV patients. The total plasma volume value was available in 61 cases and was increased in 91% masked PV versus 46% non-PV, with a mean value of 140% versus 114%, respectively (P<0.001). Splenomegaly was palpable in a higher proportion of masked PV patients. PVT was the most common SVT, found in 78% of patients, while BCS affected 17%, and porto-sinusoidal vascular disease was present in 4%. JAK2 V617F allele burden at diagnosis and erythropoietin (EPO) levels were not significantly different between masked PV and non-PV patients. A bone marrow biopsy was performed and contributive in 32 patients (45%), i.e., 18 of 40 masked PV and 14 of 31 non-PV. Panmyelosis, the histology criterion of PV in the WHO 2016 classification, was found in eight of 18 masked PV patients and, interestingly, in two of 14 other JAK2 V617F patients. The final diagnoses were masked PV in 40 patients, ET in ten, PMF in three, and MPN-U in 18 cases. Upon diagnosis of SVT, therapeutic-dose anticoagulation was initiated for all but four patients. At the time the JAK2 V617F mutation was identified, three patients were prescribed aspirin, and one patient with masked PV did not receive any antithrombotic treatment due to low platelet counts. After the diagnosis of MPN, 87% of patients began cytoreductive treatment after a median period of 6 months from their SVT diagnosis (range, 0-239): treatment started within 1 year of thrombosis for 37 patients, and after 1 year for 25 patients. The cytoreductive regimen included hydroxyurea for 43 patients, interferon for 18 patients, and ruxolitinib for one patient. Eight patients did not undergo any cytoreductive therapy; this group included two masked PV patients who were treated solely with phlebotomies. The treatment details for one patient remained undisclosed. The average follow-up duration was 77 months, with a range from 3 to 358 months (interquartile range, 45-130 months). During this period, five patients initially diagnosed with conditions other than PV - two with ET and three with MPN-U - progressed to PV 1, 5, 9, and 15 years after their first diagnosis. In two cases, this progression was marked by increased red cell values, meeting the criteria for overt PV. The remaining three patients (two MPN-U and one ET) showed signs of evolving into masked PV as indicated by routine RCM assessments. Secondary myelofibrosis developed in two patients 17 and 19 years post-detection of the JAK2 V617F mutation. Venous thrombosis recurred in five patients, involving the portal vein in three and the spleen in two, occurring at various times ranging from 1 to 21 years after their initial SVT diagnosis. At the time of these throm-
botic events, three patients were diagnosed with masked PV with normal red cell values, one had progressed from MPN-U to overt PV, and another had MPN-U with normal red cell values. Three of them were receiving cytoreductive therapy, and four anticoagulation therapy at the time of the thrombosis recurrence. Eight of 40 patients with a masked PV diagnosis underwent a second RCM assessment after at least 1 year of cytoreductive treatment. RCM levels remained stable in two patients, decreased but stayed above 125% in two others (1 of whom was re-evaluated due to SVT recurrence), and slightly increased in two. Only two patients achieved normal RCM levels. The total plasma volume remained elevated across the board (mean 139%; range, 125-150%). Those with persistently high RCM were considered under-treated, prompting significant therapeutic adjustments, including the addition of phlebotomies for two patients, increased dosages of cytoreductive agents for another two, and a switch in cytoreductive agents for the remaining two. The progression of red cell values, RCM, and associated treatment modifications for these eight patients are detailed in Figure 1A.
Our study presents the largest known cohort of patients with JAK2-mutated SVT evaluated for RCM. A key finding is that over half of these patients were diagnosed with masked PV, despite normal hemoglobin and hematocrit levels. Among 18 patients with masked PV who underwent bone marrow biopsy, only eight exhibited panmyelosis, which is a major WHO 2016 criterion for PV. Bone marrow biopsies are particularly challenging for JAK2 V617F patients with SVT due to the need for therapeutic-dose anticoagulation and possible thrombocytopenia from portal hypertension. Therefore, we advocate for routine RCM evaluation in JAK2 V617F-mutated SVT patients to accurately classify the underlying MPN and to ensure appropriate cytoreductive treatment.
Concerns have been raised in several studies about the reliability of hemoglobin and hematocrit as indicators of RCM in PV.5-9 The notion of masked PV emerged upon observing patients with JAK2 V617F mutation, increased hematocrit and hemoglobin values below WHO 2008 criteria, and heightened RCM. Hemodilution, indicated by a total plasma volume exceeding 110%, is far more prevalent in masked PV than in overt PV.10 Absence of cytoreductive therapy can lead to the undertreatment of masked PV, heightening thrombosis risks compared to overt PV.11 These insights contributed to the revision of the WHO 2016 PV criteria, lowering the required hemoglobin levels to 16.5 g/dL for men and 16 g/dL for women.12 However, data indicates that 25-50% of PV patients defined by an RCM greater than 125% still present with normal red cell counts, posing a risk of PV underdiagnosis and subsequent undertreatment.9,13 This is especially concerning in JAK2 V617F-positive SVT, where thrombosis occurrence requires cytoreductive treatment and hemodilution is frequent.
In our patient group, 91% of those with masked PV had
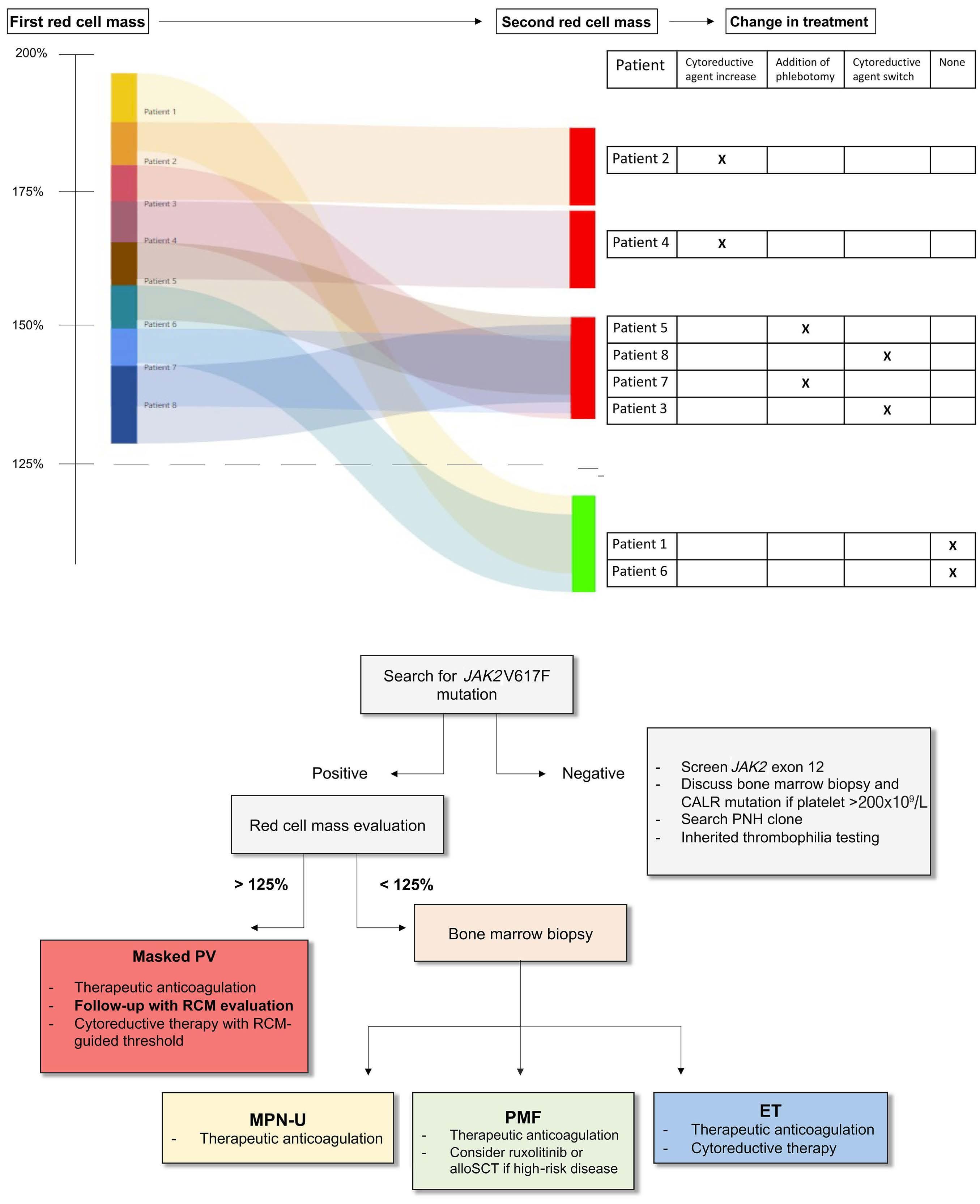
Figure 1. Role of red cell mass in myeloproliferative neoplasm with splanchnic vein thrombosis and normal red cell values. (A) Evolution of red cell mass (RCM) under cytoreductive therapy for 8 patients. The therapeutic adaptations that were made after second RCM evaluation are reported on the right. (B) Proposed strategy for diagnosis and follow-up of non-cirrhotic splanchnic vein thrombosis (SVT) with normal red cell value at diagnosis. ET: essential thrombocythemia; MPN-U: myeloproliferative neoplasm - undetermined; PMF: primary myelofibrosis; PV: polycythemia vera.
increased plasma volume, significantly higher than that of non-PV patients, suggesting that hemodilution may play a key role in differentiating between masked and overt PV post-SVT. This aligns with a previous cohort of JAK2-mutated SVT patients, where less than 10% had raised red cell counts per WHO 2008, yet about 60% had RCM over 125 %.14 In our follow-up, RCM remained high in six of eight evaluated masked PV patients despite cytoreductive therapy and normal red cell values, indicating that red cell counts alone may not suffice for adjusting cytoreductive therapy dosages. Furthermore, three of five SVT recurrence cases occurred in patients with masked PV under anticoagulation therapy, who had normal red cell values at recurrence. Of these, one patient had subsequent RCM evaluation which exceeded the mean normal predicted value by 25%. Additionally, three of four patients with normal RCM at MPN diagnosis who had a second evaluation showed progression toward masked PV. Collectively, these findings emphasize that red cell values are not reliable indicators of RCM at diagnosis or during the follow-up of SVT-associated MPN, owing to the prevalent hemodilution. Current treatment guidelines, which do not recommend cytoreduction for patients with normal hemoglobin and advise keeping hematocrit below 45%, may inadvertently result in the undertreatment of masked PV within the SVT context.15 A limitation of our study is the accessibility of isotopic RCM evaluation; however, the CO rebreathing technique, which shows promising correlation with isotopic methods, may broaden the feasibility of RCM assessment globally.16 In summary, our findings advocate for routine RCM evaluation in patients diagnosed with JAK2 V617F-mutated SVT who have normal red cell values to detect masked PV. Monitoring RCM during treatment may also inform more personalized hematocrit thresholds (as depicted in Figure 1B). Further research is necessary to solidify RCM’s role in managing SVT. Results from an ongoing FIM study on the effects of cytoreductive agents in MPN with normal blood counts (clinicaltrials gov. identifier: NCT04539678) are forthcoming.
Authors
Jean Galtier,1 Louis Drevon,2 Yannick Le Bris,3 Stephane Giraudier,2 Mathieu Wemeau,4 Laurence Legros,5 Damien Luque Paz,6 François Girodon,7 Jean-Jacques Kiladjian,2 Charles Mesguich,8 Marie Parrens,9
1. De Stefano V, Qi X, Betti S, Rossi E. Splanchnic vein thrombosis and myeloproliferative neoplasms: molecular-driven diagnosis and long-term treatment. Thromb Haemost. 2016;115(2):240-249.
2. Smalberg JH, Arends LR, Valla DC, Kiladjian J-J, Janssen HLA, Leebeek FWG. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood. 2012;120(25):4921-4928.
Clémence Mediavilla,1 Lydia Roy,10 Alexandre Guy,11 Olivier Mansier,11,12 Jean-Christophe Ianotto13 and Chloe James11,12
1Service d’Hématologie et Thérapie Cellulaire, CHU de Bordeaux, Bordeaux; 2Service d’Hématologie Clinique, Hôpital Saint-Louis, APHP, Saint-Louis; 3Service d’Hématologie Biologique, CHU de Nantes, Nantes; 4Service d’Hématologie Clinique, CHRU de Lille, Lille; 5Service d’Hématologie Clinique, APHP-Hôpital Bicêtre, Bicêtre; 6Service d’Hématologie Biologique, CHU d’Angers, Angers; 7Service d’Hématologie Biologique, CHU de Dijon, Dijon; 8Service de Médecine Nucléaire, CHU de Bordeaux, Bordeaux; 9Service d’Anatomie et Cytologie Pathologique, CHU de Bordeaux, Bordeaux; 10Service d’Hématologie Clinique, Hôpital Universitaire Henri Mondor, AP-HP & Faculté de Santé, UPEC, Crétei; 11 Service d’Hématologie Biologique, CHU de Bordeaux, Bordeaux; 12Université Bordeaux, INSERM, Biologie des Maladies Cardiovasculaires, U1034, Pessac and 13Service d’Hématologie et d’Hémostase Clinique, CHU de Brest, Brest, France
Correspondence:
J. GALTIER - jean.galtier@chu-bordeaux.fr
https://doi.org/10.3324/haematol.2023.284488
Received: October 27, 2023. Accepted: January 26, 2024. Early view: February 8, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of intererest to disclose.
Contributions
JG, LD and YLB performed the research. JG and CJ designed the research study. JG analyzed the data. JG and CJ wrote the paper. All author made substantial contributions to research design and interpretation of data.
Acknowledgments
We thank the France Intergroupe des syndrome Myéloprolifératifs (FIM) for the support in the conception of the study.
Data-sharing statement
Data can be available on direct request to the corresponding author.
3. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951.
4 Sant’Antonio E, Guglielmelli P, Pieri L, et al. Splanchnic vein thromboses associated with myeloproliferative neoplasms: An international, retrospective study on 518 cases.
Am J Hematol. 2020;95(2):156-166.
5. Silver RT, Chow W, Orazi A, Arles SP, Goldsmith SJ. Evaluation of WHO criteria for diagnosis of polycythemia vera: a prospective analysis. Blood. 2013;122(11):1881-1886.
6. Alvarez-Larrán A, Ancochea A, Angona A, et al. Red cell mass measurement in patients with clinically suspected diagnosis of polycythemia vera or essential thrombocythemia. Haematologica. 2012;97(11):1704-1707.
7. Johansson PL, Safai-Kutti S, Kutti J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: a study of patients with polycythaemia vera and apparent polycythaemia. Br J Haematol. 2005;129(5):701-705.
8. Grenier M, Callegarin D, Nughe M, Gardie B, Riedinger JM, Girodon F. Can absolute polycythaemia be identified without measurement of the red cell mass? Br J Haematol. 2020;190(2):e107-e110.
9. Hasselbalch HC. Time for revival of the red blood cell count and red cell mass in the differential diagnosis between essential thrombocythemia and polycythemia vera? Haematologica. 2019;104(11):2119-2125.
10 Maslah N, Soret J, Dosquet C, et al. Masked polycythemia vera: analysis of a single center cohort of 2480 red cell masses.
Haematologica. 2020;105(3):e95-e97.
11. Lussana F, Carobbio A, Randi ML, et al. A lower intensity of treatment may underlie the increased risk of thrombosis in young patients with masked polycythaemia vera. Br J Haematol. 2014;167(4):541-546.
12. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
13. Silver RT, Krichevsky S. Distinguishing essential thrombocythemia JAK2V617F from polycythemia vera: limitations of erythrocyte values. Haematologica. 2019;104(11):2200-2205.
14 Kiladjian J-J, Cervantes F, Leebeek FWG, et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood. 2008;111(10):4922-4929.
15. Finazzi G, De Stefano V, Barbui T. Splanchnic vein thrombosis in myeloproliferative neoplasms: treatment algorithm 2018. Blood Cancer J. 2018;8(7):64.
16. Maaziz N, Georges M, Basille D, et al. PB2165: The corebreathing method is a reliable test in determining the red cell mass: the old pots make the best soups. Hemasphere 2023;7(Suppl):e993538d.
Reactivation from latency of the human cytomegalovirus (CMV) continues to negatively impact hematopoietic stem cell transplant (HCT) outcomes.1 Due to treatment-related lymphopenia and T-cell dysfunction, up to 70% CMV-seropositive recipients can experience CMV reactivation, and if left untreated, end-organ disease may develop in up to 30% of these patients, leading to life-threatening complications.2,3 Current antiviral strategies to treat or prevent CMV reactivation are effective, though there are many drawbacks. Pre-emptive antiviral therapy (PET)4 is associated with drug-related organ toxicities, delays in immune reconstitution, and subsequent late-onset CMV disease. Food and Drug Administration (FDA)-approved (November 2017) 100-day letermovir prophylaxis reduces the incidence of clinically significant CMV infection (CMV disease or CMV viremia leading to PET) to 37.5% at 24 weeks in CMV-seropositive HCT recipients, and showed an acceptable safety profile.5 However, when letermovir is withdrawn, approximately 30% of patients develop CMV viremia, consistent with delayed immune reconstitution, associated with pharmacologic suppression of CMV.6 Recent data demonstrate continued efficacy and safety with extended letermovir use up to day 200 post transplant in high-risk patients, though the effect of prophylaxis was lost after the drug was stopped.7 Alternative strategies to suppress both early and late CMV reactivation and its sequelae by enhancing and sustaining protective CMV-specific cellular immune reconstitution remain of great interest. Therapeutic vaccination of CMV seropositive HCT recipients can bolster immune control of CMV viremia post HCT, mitigating the risk of severe CMV sequelae.8 A phase I trial of PepVax,9 an investigational CMV vaccine showed safety, immunogenicity, and reduced CMV reactivation and usage of antivirals, paving the way to the current phase II trial to assess its efficacy in reducing CMV reactivation of ≥1,250 IU/mL and disease before day 100 post HCT (CMV events, primary endpoint). Developed at City of Hope, PepVax is a peptide-based vaccine, composed of the immunodominant HLA-A*0201 restricted CMV pp65495-503 CD8 T-cell epitope, linked to a tetanus T-helper epitope and co-administered with PF-03512676 adjuvant.9 The HLA A*0201 allele is most frequently represented in Caucasian individuals (approx. 46%) as well as other ethnicities, and is much less common in African and Asian Americans (approx. 16%).10 In
this multicenter, placebo-controlled, randomized phase II trial (clinicaltrials.gov 02396134), patients were enrolled before HCT, reassessed for eligibility on day 28 post HCT, and randomized 1:1 to PepVax or placebo. Injections were administered subcutaneously on days 28 and 56 post HCT. Enrollment was halted when a planned interim analysis indicated that PepVax did not achieve the study primary endpoint. We herein report and discuss the complete 1-year outcomes from eligible recipients (N=61) who were randomized to PepVax (N=32) and placebo (N=29), until enrollment was terminated. There was no difference in CMV events between arms (P=0.15). PepVax was confirmed to be well tolerated and immunogenic. Significantly higher levels of pp65495-503 specific T cells were measured in non-viremic participants of the PepVax compared to the placebo arm (P=0.015). Furthermore, higher frequency of pp65495–503 CD8 T cells, displaying an effector phenotype with antiviral role was observed in PepVax compared to placebo recipients (P=0.034).
The FDA and institutional regulatory board authorities at each participating site approved the PepVax study protocol. From June 3, 2015, to November 11, 2017, 76 eligible patients were approached at 4 US cancer centers (City of Hope, Fred Hutchinson Cancer Center, The Ohio State University and University of Minnesota) for study participation (Online Supplementary Figure S1). All study patients provided written informed consent, were CMV-seropositive, HLA A*0201, 18-75 years old, and were undergoing HCT using myeloablative conditioning or reduced intensity conditioning from a CMV-seropositive or seronegative matched related or unrelated donor with 8/8 allele matching (Table 1). They were excluded from receiving the study treatment (PepVax or placebo injections) if engraftment had failed or if they relapsed, had grade III-IV acute graft-versus-host disease (aGvHD) (according to the Keystone Consensus grading system), experienced CMV reactivation ≥1250 IU/mL, had received >1 mg/kg of body weight steroids per day within seven days, or had any ongoing non-hematologic toxicity of grade ≥3 (according to the Common Terminology Criteria for Adverse Events, version 4.03). This trial was designed to have 90% power to detect a reduction of CMV events in the PepVax arm from 40% to 15%, at a one-sided 0.10 level of significance with a sample size of 48 patients randomized to each arm. With the expected dropout rate of 10-15% pri-
Table 1. Patients’ characteristics at baseline.
Race/ethnicity, N (%)
Primary diagnosis, N (%)
Disease Risk Index, N (%)
performance score, N (%)
N (%)
Donor CMV-serostatus, N (%)
N (%)
Graft source, N (%)
GvHD prophylaxis, N (%)
Tacrolimus/sirolimus
Tacrolimus or CSA/MTX
Tacrolimus or CSA/MMF
CSA/sirolimus/MMF
CD34+ infused cell dose, x106cells/kg
(range)
CD3+ infused cell dose, x106cells/kg
ALC on day 28 post-HCT, cells/µl
CD3+CD8+ on day 28 post HCT, %
Median (range)
(7.36-60.86)
(12.68-61.19)
ALC: absolute lymphocyte count; ALL: acute lymphoblastic leukemia; AML: acute myelogenous leukemia; CML: chronic myeloid leukemia; CMML: chronic myelomonocytic leukemia; CMV: cytomegalovirus; CSA: cyclosporin A; GvHD: graft-versus-host disease; HCT CI: hematopoietic cell transplantation-specific comorbidity index; MDS: myelodysplastic syndromes; MMF: mycophenolate mofetil; MPN: myeloproliferative neoplasms; MTX: methotrexate; N: number; PA: placebo arm; VA: vaccine arm.
or to randomization, the total accrual was expected to be of 106-115 participants. For the primary efficacy outcome, we assessed if PepVax reduced the cumulative incidence rate of CMV events compared to placebo. An interim analysis was performed per protocol once half (N=48) of the planned study participants (N=96) reached day 100 post HCT. Data analyzed showed that the futility boundary was crossed, since in the PepVax arm a higher number of CMV events (5 in 24 patients, 21%) was observed compared to the placebo arm (3 in 24 patients, 12.5%).
Table 1 shows characteristics of all randomized participant (N=61) until study closure to further accrual, following the interim analysis outcome. HCT variables were closely balanced between arms. The primary outcome of CMV events (Table 2, Online Supplementary Figure S2A) occurred in 8 patients of the PepVax arm (25.1%) and in 4 of the placebo
Table 2. Outcomes by assigned treatment arm.*
Outcomes
Primary outcome
arm (13.8%). By day 180, the CMV event rate increased to 33% for the PepVax arm (total, 10 CMV events), and remained at 13.8% for the placebo arm. In agreement with previous findings,2,11 a higher number of CMV events (34.8%) were observed in recipients who received an HCT from a CMV-seronegative negative donor compared to those (15.6%) who received a transplant from a CMV-seropositive donor. Also, in the CMV-serostatus subgroup analysis, CMV events rates were higher in the PepVax compared to the placebo arm (Online Supplementary Figure S2B). Secondary outcomes (Table 2) of PepVax including safety, impact on GvHD, measures of transplant success, antiviral usage, and immunogenicity were favorable.
An explanation for the difference between the promising pilot trial findings,9 and the current phase II trial was terminated because lack of efficacy could not be clearly
Cellular immunity (see Figure 3)
*Values are numbers of patients (%) unless otherwise indicated; CI: Confidence Interval; GvHD: graft-versus-host disease; HCT: hematopoietic stem cell transplantation; HR: hazard ratio; N: number; NA: not applicable; PA: placebo arm; pts: patients; RD: risk difference; VA: vaccine arm. 1Cytomegalovirus (CMV) reactivation of ≥1,250 IU/ml or CMV disease prior to day 100 post HCT. 2No grade IV acute GvHD (according to the Keystone Consensus grading system) was observed. 3No grade 4 probably or definitely related (according to the Common Terminology Criteria for Adverse Events, CTCAE v.4.03) to vaccination within 2 weeks from first and second injection was observed. 4Defined as number of days from HCT to post HCT day with ≥1,250 IU/ml; one PA patient was treated for gastrointestinal CMV disease and did not develop CMV viremia. 5P-value from Wilcoxon two-sample test. Test used: ‡Gray’s; §Fisher’s exact; †Log-rank. All P values for comparing primary, secondary or exploratory endpoints by arm were two-sided. P=0.05 was considered significant. SAS version 9.4 (SAS Institute) was used.
identified. PepVax was produced using the same manufacturing process and met the same release criteria. The main difference was that this phase II trial enrolled patients from 4 US cancer centers, whereas the phase I study was a single center study.9 It is possible that heterogeneity in transplant regimens (conditioning, GvHD prophylaxis), and institutional standard of care for CMV monitoring and intervention among centers may have affected the efficacy endpoint. The lack of a stratified randomization by center, which could have reduced imbalances among centers and increased statistical power,12 is a limitation of this study. Furthermore, the CMV event rate in the phase I was 38.8% for the untreated observational arm, which is within the historical CMV reactivation range.11 In contrast, in the current phase II trial, the CMV event rate in the placebo arm was 13.8% and in the PepVax arm was 25.1%. Hence, the observed rate was unexpectedly lower than that assumed in the protocol design (approx. 40%).11
PepVax vaccinated patients reconstituted significantly higher levels of pp65495–503-specific CD8 T cells compared to the placebo arm (P=0.015) (Figure 1A) in HCT recipients who were not experiencing a CMV event. In recipients with a CMV-seropositive HCT donor, the vaccine effect in augmenting post HCT levels of pp65495–503-specific CD8 T cells was substantial (P=0.008) among PepVax vaccinated recipients compared to placebo recipients. In contrast, it
Awas modest in HCT recipients with a CMV-seronegative donor (P=not assessable due to insufficient data) ( Online Supplementary Figure S3). This interesting outcome suggests that in some viremic patients primary immune responses to other MHC class I restricted CMV epitopes could have been preferentially generated, possibly leading to the expansion of different CMV-specific T-cell subsets during viral reactivation. Several groups have reported that vaccinia virus immunized volunteers expressing the same HLA class I molecule recognized different vaccinia virus-derived epitopes, suggesting a highly variable pattern of T-cell epitope recognition in the human population.13 Hence, there is the need to re-evaluate current strategies for protective T-cell epitope discovery.
Similarly to the phase I findings,9,14 PepVax not only increased frequency of pp65495–503-specific CD8 T cells, but also impacted their functional immune profiles. Effector subsets increased more vigorously during immune reconstitution in non-viremic patients from the PepVax arm compared to the placebo arm (P=0.034) (Figure 1B). Of note, the functionality and antiviral role of CMV-specific T cells have been linked to immune reconstitution profiles characterized by pools of differentiated effector memory T cells (TEM) and large subsets that, as in TEM, have lost membrane expression of the co-stimulatory molecule CD28, but also re-express the RA isoform of CD45 (TEMRA).14 T-central memory and
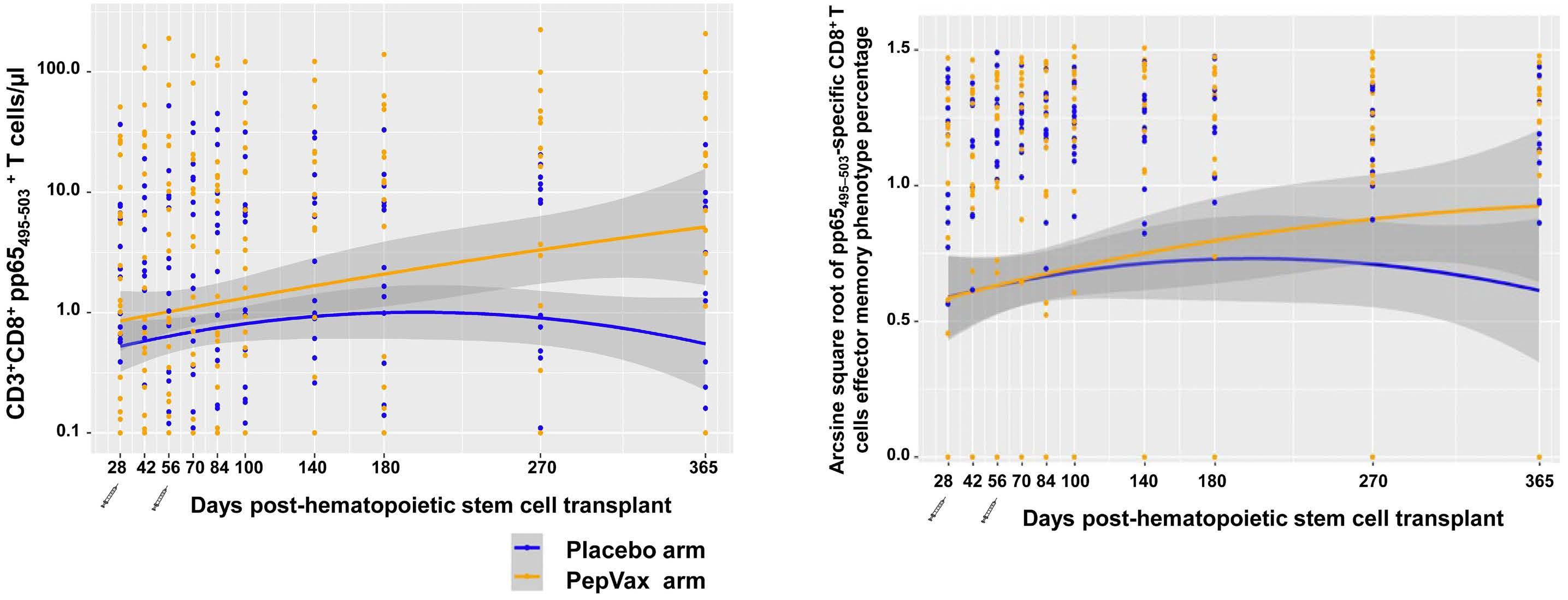
Figure 1. Levels and memory phenotype of pp65495–503-specific CD8 T cells. pp65495–503-specific CD3+ CD8+ T cells were monitored by measuring binding to HLA-A*0201 pp65495–503 and HIVgag77-85 (control) multimers (Immudex Dextramers), as previously described.9,14 (A) Longitudinal levels as T cells/µl and (B) memory phenotype frequency of pp65495–503-specific CD3+ CD8+ T cells were computed using the (A) LOESS scatter plot smoother providing the marginal geometric mean concentrations through time for each arm (as identified in the color legend). A 95% confidence band is shown in gray, and individual measurement trajectories are shown for each participant up to 7 days before the protocol-defined cytomegalovirus event. Logarithmic spacing of both scales is used to aid visualization. Distribution of pp65495–503 specific CD8 T-cell levels and memory phenotypes (%) were approximately normal after log 10-transformation and arcsine square root-transformation, respectively. Generalized estimating equation models were used to assess the effect of vaccines on immunological responses. All analyses were performed using SAS v.9.4 (SAS Institute). (B) pp65495–503-specific CD3+ CD8+ effectors included CD45RA- CD28- effector memory T cells (TEM) and effector ‘revertant’ memory T cells, re-expressing the RA isoform of the CD45 surface marker (TEMRA).14 The syringe symbol indicates the post-hematopoietic stem cell transplant day of PepVax injections.
naïve T-cell subsets were comparable between arms (data not shown). Using Fine-Gray regression model, no significant associations were found between CMV events and pp65495–503-specific CD8 T-cell levels or memory. In the development of CMV subunit vaccines, choice of the vaccine technology can play a critical role in promoting robust immune response.8 The ASP0113 CMV vaccine, which has been stopped following the phase III failure, used a DNA-based platform. It has been shown that both DNA and peptide vaccines may induce limited immunogenicity even if used in combination with adjuvants, and to date no such vaccines have been licensed for human use.15,16 In contrast, viral vector-based vaccines, can mimic a natural infection, resulting in strong, long-lasting immune responses against the pathogen antigens.8 Triplex, a candidate CMV vaccine, uses the modified vaccinia virus Ankara (MVA) expressing pp65, IE1-exon4, and IE2-exon5 CMV genes. It is currently the only CMV vaccine for transplant indication, which met its primary endpoints in phase II trials.8
Owing to the lack of efficacy, further development of PepVax has been paused. Nonetheless, based on its favorable tolerability and immunogenicity, it could find future application as an immunotherapeutic to be used in combination with letermovir prophylaxis. Vaccinating HCT donor and/or recipient8 with PepVax in a combination regimen strategy with letermovir may overcome the immune impairment and delay in CMV-specific cellular reconstitution related to the drug-induced decrease in CMV antigen exposure. Though limited to HLA A*0201 recipients, this easy to implement combination therapy could benefit transplant patients in whom lack of CMV-specific T-cell polyfunctionality may increase the risk for late CMV infection sequalae after letermovir prophylaxis discontinuation.
Ryotaro Nakamura,1* Corinna La Rosa,1* Dongyun Yang,2* Joshua A. Hill,3 Armin Rashidi,3,4 Hannah Choe,5 Qiao Zhou,1 Chetan Raj Lingaraju,1 Teodora Kaltcheva,1 Jeffrey Longmate,2 Jennifer Drake,6 Cynthia Slape,7 Lupe Duarte,1 Monzr M. Al Malki,1 Vinod A. Pullarkat,1 Ahmed Aribi,1 Steven Devine,5 Michael R. Verneris,8 Jeffrey S. Miller,4 Stephen J. Forman,1 Ibrahim Aldoss1 and Don J. Diamond1
1Department of Hematology and Hematopoietic Cell Transplantation, City of Hope National Medical Center, Duarte, CA; 2Department of Computational and Quantitative Medicine, City of Hope National Medical Center, Duarte, CA; 3Fred Hutchinson Cancer Center, Seattle, WA; 4Department of Medicine, Division of Hematology, Oncology, and Transplantation, University of Minnesota, Minneapolis, MN; 5Division of Hematology, Department of Internal Medicine, The Ohio State University, Columbus, OH; 6Department of Shared Resources-Clinical Trial Office, City of Hope National Medical Center, Duarte, CA; 7Department of Clinical Research, City of Hope National Medical Center, Duarte, CA and 8University of Colorado School of
Medicine, Children’s Hospital of Colorado, Aurora, CO, USA
*RN, CLR and DY contributed equally as first authors.
Correspondence:
D.J. DIAMOND - ddiamond@coh.org
https://doi.org/10.3324/haematol.2023.284544
Received: November 7, 2023. Accepted: January 31, 2024. Early view: February 8, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
CLR reports receiving consulting fees and research funding from Helocyte Inc. DJD reports receiving fees for consulting fees, patent royalties, research funding, and fees for serving on the Advisory Board of Helocyte Inc., has two patents 8,580,276 and 9,675,689 that are licensed to Helocyte, is a co-inventor of the Patent Cooperation Treaty (PCT) application that covers the development of a COVID-19 vaccine (PCT/US2021/032821) licensed to GeoVax Labs Inc., and receives consulting fees and research support from GeoVax Labs Inc., is co-inventor on a patent application covering the design and construction of the synthetic modified vaccinia Ankara platform (PCT/US2021/016247), is co-inventor on a patent application covering the development of a COVID-19 vaccine (PCT/ US2021/032821), is an employee of City of Hope National Medical Center (Duarte, CA, USA), which developed and funded the vaccine trial. DJD and CLR are co-inventors on a patent covering the “VACCINATION OF HEMATOPOIETIC STEM CELL DONORS WITH CYTOMEGALOVIRUS TRIPLEX COMPOSITION” (PCT/US2023/032052).
DJD, QZ, CRL and TK were supported by grants from the National Cancer Institute (NCI) 1R01 CA18045, NCI-SAIC-Frederick 28XS061 and Helocyte Inc. RN is a consultant for Omeros, Bluebird, Viracor Eurofins, Magenta Therapeutics, Kadmon, Napajen Pharma, received research funding from Helocyte Inc., Miyarisan Pharmaceutical, and received travel, accommodations, expenses from Kyowa Hakko Kirin, and Alexion Pharmaceuticals. All of the other authors have no conflicts of interest to disclose.
Contributions
RN, JAH, AR, HC, MMAM, VAP, AA, SD, MRV, JSM, SJF and IA treated and clinically assessed the patients for adverse events and graftversus-host disease. DJD and JL designed the study. QZ and CRL performed specimen processing, reagent preparation, and immunological assays. CLR and CRL analyzed the immune monitoring data. DY and JL performed the statistical analyses. CLR, RN and DJD wrote the initial manuscript, and all authors approved the final version.
We thank the patients, participating institutions and their clinical
teams, research nurses and research coordinators without whose support this trial could not have been conducted. We acknowledge Dr. Thai M. Cao as a participating physician with involvement in patient trial enrollment and care, before, during and after transplant.
Funding
Supported by grants from the National Cancer Institute (NCI) 1R01
1. Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol Oncol Clin North Am. 2011;25(1):151-169.
2. Teira P, Battiwalla M, Ramanathan M, et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood. 2016;127(20):2427-2438.
3. Einsele H, Ljungman P, Boeckh M. How I treat CMV reactivation after allogeneic hematopoietic stem cell transplantation. Blood. 2020;135(19):1619-1629.
4 Green ML, Leisenring W, Stachel D, et al. Efficacy of a viral loadbased, risk-adapted, preemptive treatment strategy for prevention of cytomegalovirus disease after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012;18(11):1687-1699.
5. Marty FM, Ljungman P, Chemaly RF, et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017;377(25):2433-2444.
6. Zamora D, Duke ER, Xie H, et al. Cytomegalovirus-specific T-cell reconstitution following letermovir prophylaxis after hematopoietic cell transplantation. Blood. 2021;138(1):34-43.
7 Bansal R, Gordillo CA, Abramova R, et al. Extended letermovir administration, beyond day 100, is effective for CMV prophylaxis in patients with graft versus host disease. Transpl Infect Dis. 2021;23(2):e13487.
8. La Rosa C, Diamond DJ. Triplex, a viral vectored CMV vaccine for transplant indications: clinical trial updates. Vaccine Insights. 2023;2(7):287-308.
9 Nakamura R, La Rosa C, Longmate J, et al. Viraemia,
CA181045; NCI-SAIC-Frederick 24XS044, and Helocyte Inc. to DJD. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health (NIH).
Data-sharing statement
The clinical trial protocol and the anonymized data presented in this study can be made available upon reasonable request.
immunogenicity, and survival outcomes of cytomegalovirus chimeric epitope vaccine supplemented with PF03512676 (CMVPepVax) in allogeneic haemopoietic stem-cell transplantation: randomised phase 1b trial. Lancet Haematol. 2016;3(2):e87-98.
10. Longmate J, York J, La Rosa C, et al. Population coverage by HLA class-I restricted cytotoxic T-lymphocyte epitopes. Immunogenetics. 2001;52(3-4):165-173.
11. Zhou W, Longmate J, Lacey SF, et al. Impact of donor CMV status on viral infection and reconstitution of multifunction CMV-specific T cells in CMV-positive transplant recipients. Blood. 2009;113(25):6465-6476.
12. Schulz KF, Grimes DA. Unequal group sizes in randomised trials: guarding against guessing. Lancet. 2002;359(9310):966-970.
13. Gilchuk P, Hill TM, Wilson JT, Joyce S. Discovering protective CD8 T cell epitopes--no single immunologic property predicts it! Curr Opin Immunol. 2015;34:43-51.
14 La Rosa C, Longmate J, Lingaraju CR, et al. Rapid acquisition of cytomegalovirus-specific T cells with a differentiated phenotype, in nonviremic hematopoietic stem transplant recipients vaccinated with CMVPepVax. Biol Blood Marrow Transplant. 2019;25(4):771-784.
15. Li L, Petrovsky N. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert Rev Vaccines. 2016;15(3):313-329.
16. Malonis RJ, Lai JR, Vergnolle O. Peptide-based vaccines: current progress and future challenges. Chem Rev. 2020;120(6):3210-3229.
Allogeneic hematopoietic stem cell transplantation (HSCT) remains a first-line therapeutic option for younger patients with severe aplastic anemia (SAA).1 Elderly age has been proven to be associated with a relatively higher mortality following allogeneic HSCT for the treatment of SAA, partly due to poorer organ function.2 Data from the European Society for Blood and Marrow Transplant (EBMT) and the Center for International Blood and Marrow Transplant Research (CIBMTR) demonstrated that allogeneic HSCT led to a 3-year overall survival of 56% among 499 SAA patients older than 50 years.3 In this large study, all transplants were from matched sibling donors (MSD) or matched unrelated donors (MUD) but did not include haploidentical donors (HID).3 In recent decades, HID HSCT for SAA has made great advances, and the upper limit of age for such transplants among SAA recipients has been continuously broken.4,5 A retrospective study indicated comparable survival outcomes between transplantation from HID and MSD or unrelated donors (URD) for SAA patients aged 40 years and older, with a median age of 43-48.5 years: the 3-year overall survival ranged from 86.7-100% in the three groups.6 However, the outcomes of allogeneic HSCT in SAA patients older than 50 years, especially including HID HSCT, have rarely been reported.
Based on data from the Chinese Blood and Marrow Transplant Registry (CBMTR), we analyzed the outcomes of SAA patients older than 50 years, most of whom had undergone HID HSCT. A total of 76 patients who underwent a first allogeneic HSCT between January 2014 and December 2022 were enrolled from 25 transplant centers. The study protocol was approved by the institutional review board. All patients gave their written informed consent to the procedure. The details of conditioning regimens for transplants from different types of donors are summarized in Online Supplementary Table S1. The details of graft-versus-host disease (GvHD) prophylaxis have been described previously.6,7 As shown in Table 1, 16 (21.1%) patients received HSCT from MSD, 55 (72.4%) from HID, and five (6.5%) from URD. The median time from diagnosis to HSCT was 4.2 months (range, 0.2-279.8) in the entire cohort. With regard to HSCT timing, 41 patients underwent salvage transplantation after the failure of immunosuppressive therapy, and 22 received immunosuppressive therapy including anti-thymocyte globulin. The last follow-up for all living patients was September 30, 2023. The median follow-up for surviving patients following HSCT was 821 days (range, 278-3,434). The comparisons
Disease type, N (%)
timing, N (%)
Disease course in months, median (range)
Previous treatment, N (%)
Previous transfusion
units, median (range)
units, median (range)
Donor type, N (%)
sibling donor
donor
Donor-recipient blood type, N (%)
Matched
Minor mismatched
N (%)
Mononuclear cells, x109/kg, median (range)
CD34 cells, 106/kg, median (range) 4.7 (0.6-15.3)
HSCT: hematopoietic stem cell transplantation; RBC: red blood cell; HCT-CI: Hematopoietic Cell Transplantation Comorbidity Index. A unit of a blood component means the component extracted from one unit of a whole blood donation (approximately 200 mL).
of basic characteristics among different donor groups are provided in Online Supplementary Table S1
Seventy-one patients survived for more than 28 days. A total of 70 (98.6%) patients achieved myeloid engraftment, and all of them showed complete donor chimerism at 1 month after transplantation. The cumulative incidence of myeloid engraftment was 92.0±0.1% in the entire cohort, with engraftment occurring at the median time of 14 days (range, 8-28). Fifty-nine patients achieved platelet engraftment within a median time of 15 days (range, 8-100). The cumulative incidence of platelet recovery was 77.6±0.2%. The cumulative incidences of myeloid engraftment and platelet recovery in the three groups are indicated in Online Supplementary Figure S1A, B.
With regard to acute GvHD, none of the patients in the cohort of MSD recipients experienced grade II-IV acute GvHD. The cumulative incidences of grade II-IV and grade III-IV acute GvHD at 100 days were 10.9±0.2% vs. 20.0±4.0% (P=0.295), 5.5±0.1% vs. 20.0±4.0% (P=0.222) after HID and URD transplants, respectively (Online Supplementary Figure S1C, D). Patients who survived longer than 100 days were evaluable for chronic GvHD based on 2014 National Institutes of Health criteria. None of the patients in the groups transplanted from URD suffered chronic GvHD. HID transplant recipients had a seemingly higher 3-year cumulative incidence of chronic GvHD compared to MSD transplant recipients, but the difference was not statistically significant (24.8±0.7% vs. 6.3±0.4%; P=0.230) (Online Supplementary Figure S1E). The MSD and HID groups had similar 3-year incidences of moderate chronic GvHD (6.3±0.4% vs 6.2±0.2%; P=0.896) (Online Supplementary Figure S1F), and no severe chronic GvHD occurred.
In terms of virus reactivation within 100 days, the cumulative incidences for cytomegalovirus and Epstein-Barr virus were 43.4±0.3% and 17.1±0.2%, respectively, in the entire cohort. The MSD, HID and URD groups had similar incidences of cytomegalovirus (43.8±1.7%, 43.6±0.5%, and 40.0±6.8%, respectively; P=0.896) (Online Supplementary Figure S1G) and of Epstein-Barr virus (18.8±1.0%, 18.2±0.3%, and 0%, respectively; P=0.584) (Online Supplementary Figure S1H).
One patient in the HID group suffered an Epstein-Barr virus-related post-transplant lymphoproliferative disorder. A total of 17 patients died of transplant-related causes, at a median of 51 days (range, 4-384) after transplantation (Online Supplementary Table S1). The overall survival at 3 years was 77.2±4.9% in the whole cohort. In univariate analysis (Online Supplementary Table S2), the 3-year overall survival of patients in the MSD, HID, and URD groups was 100%, 71.8±6.2%, and 60.0±21.9%, respectively (P=0.053) (Figure 1). We also observed that older age of patients, female sex, and higher Hematopoietic Cell Transplantation-specific Comorbidity Index (HCT-CI) score were associated with worse survival. In addition, a trend was observed for ABO blood type incompatibility, with overall survival probability being worse for incompatible donor-recipient pairs. The above
potentially significant factors for survival were included in the multivariate analysis. The multivariate analysis showed that the hazard ratio was increased for older patients (≥55 years, relative risk [RR]=4.539, 95% confidence interval [95% CI]: 1.590-12.963; P=0.005), those with a higher HCT-CI score (≥2, RR=7.726, 95% CI: 2.761-21.620; P<0.001), and recipients with ABO blood type incompatible donors (RR=5.629, 95% CI: 1.808-17.532; P=0.003) when predicting overall survival. Combining these three parameters, a predictive risk model for the outcome of allogeneic HSCT in elderly SAA patients was established: low risk (0 factors, n=19), intermediate risk (1 factor, n=41), and high risk (2-3 factors, n=16). The probabilities of overall survival at 3 years after allogeneic HSCT were 100%, 82.9±5.9%, and 31.3±13.2% for the low-, intermediate-, and high-risk groups, respectively (P<0.001) (Figure 2).
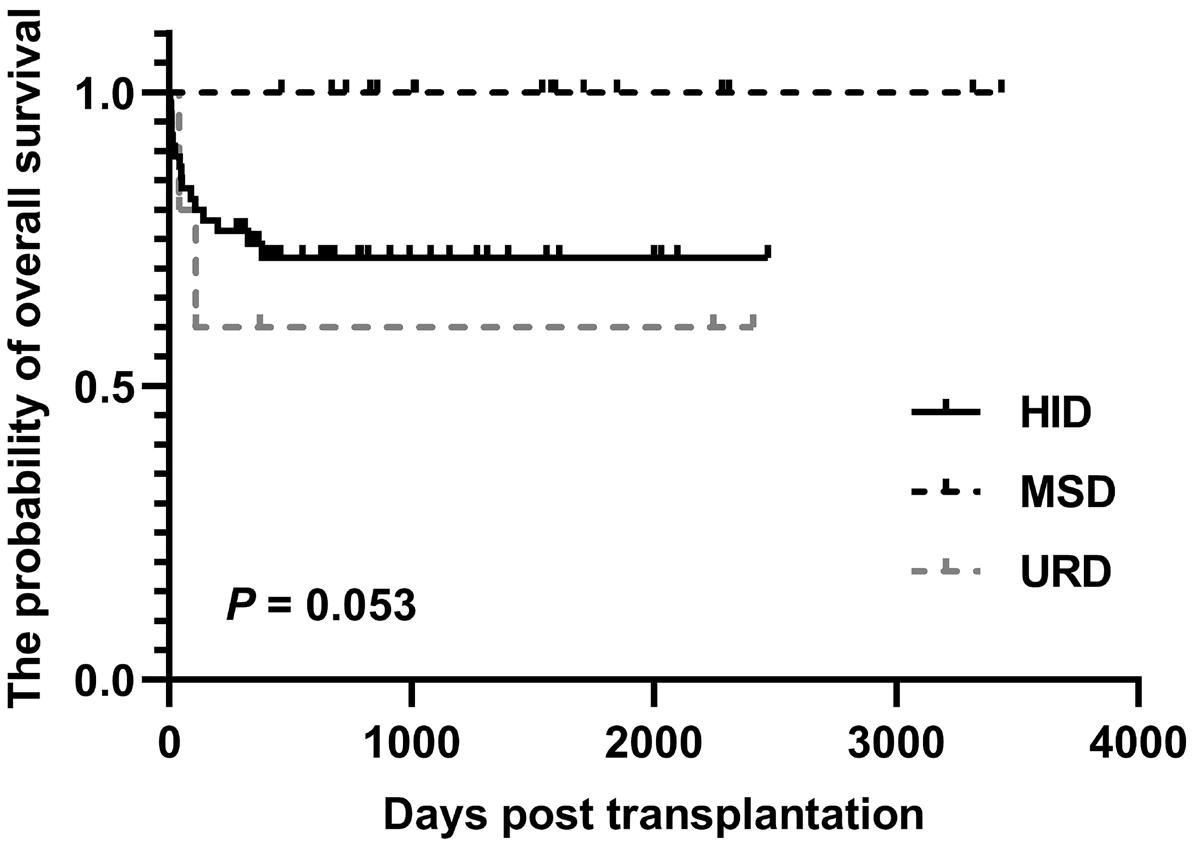
Figure 1. The overall survival of patients with severe aplastic anemia older than 50 years who underwent allogeneic hematopoietic stem cell transplantation from different donors. HID: haploidentical donor; MSD: matched sibling donor; URD: unrelated donor.
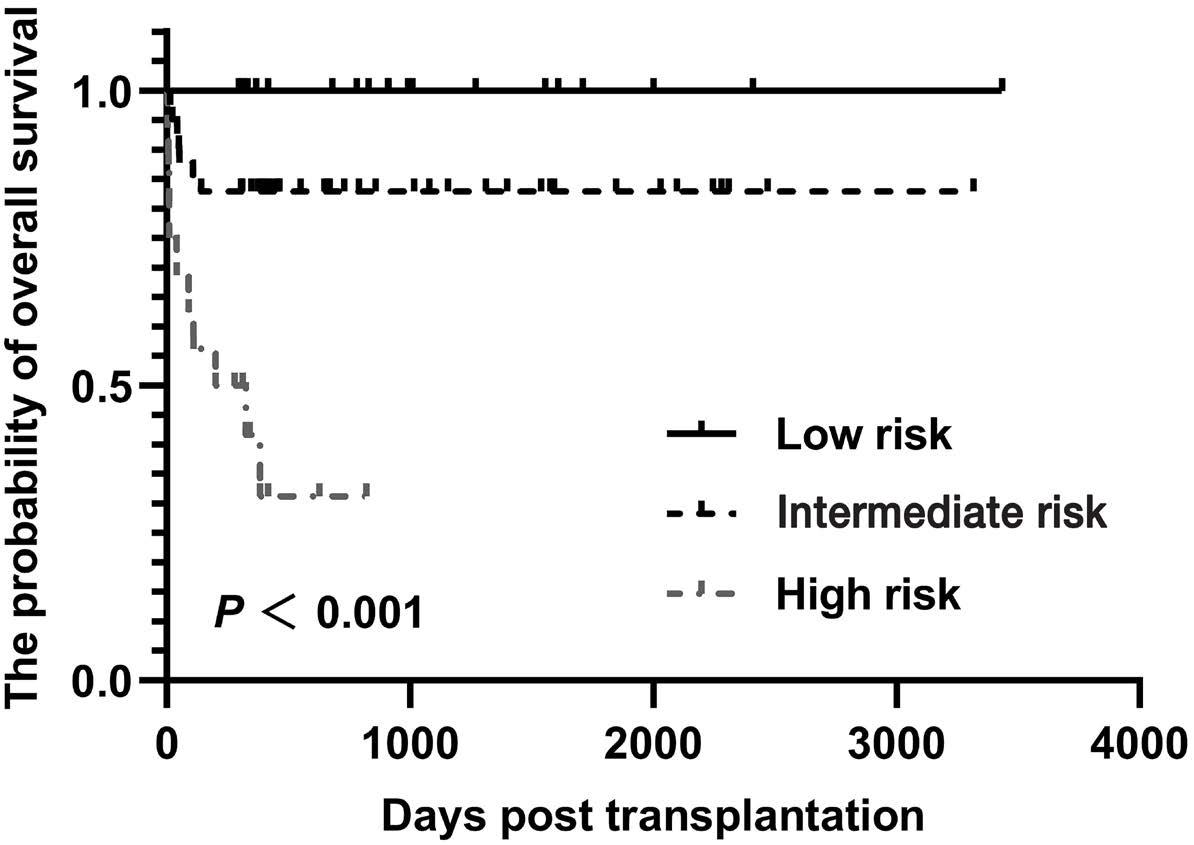
Figure 2. The probabilities of overall survival for low-risk, intermediate-risk, and high-risk groups among patients with severe aplastic anemia older than 50 years of age. Older age (≥55 years), higher Hematopoietic Cell Transplantation Comorbidity Index score (≥2), and ABO blood type incompatibility were factors predictive of poorer survival. Based on these three parameters, a predictive risk model was established: low risk (0 factors), intermediate risk (1 factor), and high risk (2-3 factors).
The data from the CBMTR indicated that allogeneic HSCT led to a 3-year survival of 77.2% among SAA patients older than 50 years. Currently, patients have estimated 3-year overall survival rates of 100% and 60% after transplantation from MSD and MUD, respectively; the corresponding 3-year overall survival rates of MSD and MUD HSCT recipients from the EBMT and CIBMTR were 57% and 48%, respectively.3 It should be noted, first, that patients in the CBMTR were younger than those from the EBMT or CIBMTR, with a median age of 54 years versus 57.8 years, respectively, at transplantation. Second, the transplants by the EBMT and CIBMTR were performed between 2005 and 2016, while those from the CBMTR were performed between 2014 and 2022. Improvements in transplantation techniques and supportive care are factors that cannot be ignored when comparing transplants performed in different periods. In this study, 55 patients undergoing allogeneic HSCT had HID, and their transplants resulted in a 3-year overall survival of 71.8%. Previously, the efficacy of haploidentical transplantation had been proven among younger recipients.4,5,8 Excluding the early deaths, 49 of 50 patients (98%) achieved myeloid engraftment. The GvHD incidences were similar to those in a previous study. The incidences of grade II-IV acute GvHD and 3-year chronic GvHD were 10.9% and 24.8%, respectively, in our cohort. DeZern et al. observed grade II-IV acute GvHD and 1-year chronic GvHD incidences of 16% and 26% following haploidentical transplantation to treat SAA.8 When the causes of mortality were analyzed, severe infection and regimen-related toxicities were the most common causes, which might be attributable to weak immune function and fragile organ function among elderly SAA patients.
We observed that older age, higher HCT-CI scores and ABO blood type incompatibility were obvious adverse factors. Consistently, Giammarco et al. found that the 5-year survival rates of patients aged 50 to 59 years and those aged over 60 years were 58% and 45% for SAA patients undergoing MSD or URD transplantation.9 Besides, two previous studies showed that higher HCT-CI scores were associated with inferior survival among SAA patients after haploidentical transplantation, meaning that higher comorbidity burdens resulted in poorer survival.10,11 There are conflicting data on the impact of ABO incompatibility on survival in different disease categories.12,13 Previously, minor ABO incompatibility was found to increase the rate of grade III-IV acute GvHD but not to affect survival in a cohort of SAA patients undergoing haploidentical transplantation.13
Recently, the addition of eltrombopag to standard immunosuppressive therapy was shown to improve the rate of hematologic response,14 thus the comparison of HSCT versus triple immunosuppressive therapy would be essential among the elderly. This retrospective study had a small number of patients, especially in the group with unrelated donors, which may have weakened the statistical power of the study. The predictive model has limitations, due to the lack of a validation
set and limited sample size. Large-scale prospective studies are, therefore, needed to validate these results. In summary, allogeneic HSCT deserves consideration as a treatment option among elderly SAA patients, especially for those younger than 55 years. For those older than 55 years, patients with lower comorbidity burdens might benefit from allogeneic HSCT, and an ABO-compatible donor should be recommended. In the future, prospective data are essential to forward the position of allogeneic HSCT among elderly SAA patients as a potentially curative approach to the treatment of this disease.
Zheng-Li Xu,1,2* Lan-Ping Xu,1,2* Yi-Cheng Zhang,3-5* Yu-Hong Zhou,6 Er-Lie Jiang,7,8 Jian-Ping Zhang,9 Bin Fu,10 Gui-Fang Ouyang,11 Xian-Min Song,12 Xue-Jun Zhang,13 Yu-Jun Dong,14 Nai-Nong Li,15 Ling Wang,16 Xi Zhang,17 Peng-Cheng He,18 Fan-Sheng Kong,19 Hui-Xia Liu,20 Li Liu,21 Lin Liu,22 Tai-Wu Xiao,23 Wen-Wei Xu,24 Xiao-Jun Xu,25 Guo-Lin Yuan,26 Hai Yi,27 Dan Yu,28 Li Yu29 and Xiao-Jun Huang1,2,30,31
1Peking University People’s Hospital, Peking University Institute of Hematology, Beijing; 2National Clinical Research Center for Hematologic Disease, Beijing; 3Department of Hematology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan; 4Key Laboratory of Organ Transplantation, Ministry of Education, NHC Key Laboratory of Organ Transplantation, Key Laboratory of Organ Transplantation, Chinese Academy of Medical Sciences, Wuhan; 5Immunotherapy Research Center for Hematologic Diseases of Hubei Province, Wuhan; 6Department of Hematology, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Hangzhou; 7State Key Laboratory of Experimental Hematology, National Clinical Research Center for Blood Diseases, Haihe Laboratory of Cell Ecosystem, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin; 8Tianjin Institutes of Health Science, Tianjin; 9Hebei Yanda Lu Daopei Hospital, Langfang; 10Department of Hematology, Xiangya Hospital of Central South University, National Clinical Research Center for Geriatric Diseases, Changsha; 11Department of Hematology, the First Affiliated Hospital of Ningbo University, Ningbo; 12Department of Hematology, Shanghai General Hospital, Shanghai Jiaotong University School of Medicine, Shanghai; 13Department of Hematology, The Second Hospital of Hebei Medical University, Hebei Key Laboratory of Hematology, Shijiazhuang; 14Department of Hematology, Peking University First Hospital, Beijing; 15Department of Hematology, Hematopoietic Stem Cell Transplantation Center, Fujian Institute of Hematology, Fujian Provincial Key Laboratory on Hematology, Fujian Medical University Union Hospital, Translational Medicine Center on Hematology of Fujian Medical University, Fuzhou; 16Department of Hematology, Affiliated Qingdao Central Hospital of Qingdao University, Qingdao; 17Medical Center of Hematology, Xinqiao Hospital, State Key Laboratory of Trauma and Chemical Poisoning, Army Medical University, Chongqing; 18Department of Hematology, The
First Affiliated Hospital of Xi’an Jiaotong University, Xi’an; 19The Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan; 20Department of Hematology, Shanghai Zhaxin Integrated Traditional Chinese and Western Medicine Hospital, Shanghai; 21Department of Hematology, Tangdu Hospital, The Air Force Medical University, Xi’an; 22Department of Hematology, The First Affiliated Hospital of Chongqing Medical University, Chongqing; 23Department of Hematology, Liaocheng People’s Hospital, Liaocheng; 24Department of Hematology, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Jinan; 25Department of Hematology, The Seventh Affiliated Hospital, Sun Yet-sen University, Shenzhen; 26Department of Hematology, Xiangyang Central Hospital, The Affiliated Hospital of Hubei University of Arts and Science, Xiangyang; 27Department of Hematology, The General Hospital of Western Theater Command, Chengdu; 28Department of Hematology, Wuhan No. 1 Hospital, Wuhan; 29Department of Hematology, the Second Affiliated Hospital of Nanchang University, Nanchang; 30Beijing Key Laboratory of Hematopoietic Stem Cell Transplantation, Beijing and 31PekingTsinghua Center for Life Sciences, Beijing, China.
+Z-LX, L-PX and Y-CZ contributed equally as first authors.
Correspondence:
X. HUANG - xjhrm@medmail.com.cn
https://doi.org/10.3324/haematol.2023.284581
Received: October 31, 2023.
Accepted: January 23, 2024. Early view: February 1, 2024.
1. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018;2(15):2020-2028.
2. Devillier R, Dalle JH, Kulasekararaj A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the EBMT Severe Aplastic Anemia Working Party. Haematologica. 2016;101(7):884-890.
3. Rice C, Eikema DJ, Marsh JCW, et al. Allogeneic hematopoietic cell transplantation in patients aged 50 years or older with severe aplastic anemia. Biol Blood Marrow Transplant. 2019;25(3):488-495.
4 Xu LP, Xu ZL, Wang SQ, et al. Long-term follow-up of haploidentical transplantation in relapsed/refractory severe aplastic anemia: a multicenter prospective study. Sci Bull (Beijing). 2022;67(9):963-970.
5. Xu ZL, Xu LP, Wu DP, et al. Comparable long-term outcomes between upfront haploidentical and identical sibling donor transplant in aplastic anemia: a national registry-based study. Haematologica. 2022;107(12):2918-2927.
6. Zhang YY, Mo WJ, Zuo YY, et al. Comparable survival outcome between transplantation from haploidentical donor and
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
X-JH designed the research. Z-LX, L-PX, and X-JH analyzed the data and wrote the manuscript. All authors provided data on patients and gave final approval of the manuscript.
Acknowledgments
We acknowledge all the patients included in the study. The authors would like to thank American Journal Experts for assistance with editing the English.
Funding
This work was supported by National Key Research and Development Program of China (n. 2022YFA1103300), Major Program of the National Natural Science Foundation of China (n. 82293630), Key Program of the National Natural Science Foundation of China (n. 81930004), the National Natural Science Foundation of China (n. 82100227 & 81873446), the National High Technology Research and Development Program of China (n. 2021YFA1101504), and the Key Research and Development Program of Hubei Province (n. 2022BCA017).
Data-sharing statement
The data that support the findings of this study are available upon reasonable request from the corresponding author.
matched related donor or unrelated donor for severe aplastic anemia patients aged 40 years and older: a retrospective multicenter cohort study. Clin Transplant. 2020;34(3):e13810.
7 Li Y, Wang N, Li L, et al. Haploidentical transplantation with modified post-transplantation cyclophosphamide for patients with primary aplastic anemia: a multicenter experience. Transplant Cell Ther. 2021;27(4):331.e1-331.e7.
8. DeZern AE, Eapen M, Wu J, et al. Haploidentical bone marrow transplantation in patients with relapsed or refractory severe aplastic anaemia in the USA (BMT CTN 1502): a multicentre, single-arm, phase 2 trial. Lancet Haematol. 2022;9(9):e660-e669.
9 Giammarco S, Peffault de Latour R, Sica S, et al. Transplant outcome for patients with acquired aplastic anemia over the age of 40: has the outcome improved? Blood. 2018;131(17):1989-1992.
10 Xu LP, Yu Y, Cheng YF, et al. Development and validation of a mortality predicting scoring system for severe aplastic anaemia patients receiving haploidentical allogeneic transplantation. Br J Haematol. 2022;196(3):735-742.
11. Xu LP, Xu ZL, Wang FR, et al. Unmanipulated haploidentical transplantation conditioning with busulfan, cyclophosphamide and anti-thymoglobulin for adult severe aplastic anaemia. Bone
Marrow Transplant. 2018;53(2):188-192.
12. Canaani J, Savani BN, Labopin M, et al. Impact of ABO incompatibility on patients’ outcome after haploidentical hematopoietic stem cell transplantation for acute myeloid leukemia - a report from the Acute Leukemia Working Party of the EBMT. Haematologica. 2017;102(6):1066-1074.
13. Ma YR, Wang WJ, Cheng YF, et al. Impact of ABO incompatibility
on outcomes after haploidentical hematopoietic stem cell transplantation for severe aplastic anemia. Bone Marrow Transplant. 2020;55(6):1068-1075.
14 Peffault de Latour R, Kulasekararaj A, Iacobelli S, et al. Eltrombopag added to immunosuppression in severe aplastic anemia. N Engl J Med. 2022;386(1):11-23.
Primary central nervous system (CNS) lymphoma (PCNSL) represents an uncommon aggressive type of non-Hodgkin lymphoma exclusively affecting the brain, spinal cord, eyes, and cerebrospinal fluid, with no systemic non-CNS involvement. The overall prognosis is generally poor despite major advances in the last two decades. High-dose methotrexate (HD-MTX) is considered the mainstay of induction therapy. Autologous hematopoietic stem cell transplantation (ASCT) and whole-brain radiotherapy (WBRT) are similarly effective consolidation treatments, but the latter brings a high risk of significant irreversible neurotoxicity.1,2 In the long-term follow-up analysis, patients treated with methotrexate, cytarabine, thiotepa, and rituximab (MATRix) induction treatment and ASCT consolidation had a 7-year overall survival (OS) rate of 70%.3 However, elderly patients account for nearly 70% of all PCNSL and many patients are transplant-ineligible. In a real-world analysis in the modern era, only 23% of patients <60 years old and 2% of patients ≥60 years old received ASCT consolidation treatment.4 The overall prognosis of those patients without ASCT consolidation is poor. So far, the optimal therapeutic options are limited for transplant-ineligible patients. Thus, an effective and well-tolerated treatment option for those patients ineligible for ASCT is dearly needed. Lenalidomide is an orally administered small-molecule drug with direct anti-tumor and immunomodulatory effects. Available evidence indicates the effectiveness of lenalidomide maintenance in CNS lymphoma and its tolerability in older individuals.5 In addition, preclinical findings demonstrated that lenalidomide and rituximab enhance antibody-dependent cell-mediated cytotoxicity to exert synergistic antitumor effects.6 Lenalidomide, rituximab, and chemotherapy administered in combination in refractory/ relapsed PCNSL showed promising anti-tumor effects.7 Nevertheless, the combination of lenalidomide, rituximab, and methotrexate R2-MTX regimen as first-line therapy in transplant-ineligible PCNSL deserves further verification. Based on these premises, a multicenter, single-arm, open-labeled, phase II clinical trial was conceived and conducted to evaluate the efficacy and safety of R2-MTX for individuals with newly diagnosed PCNSL. All patients were transplant-ineligible or unwilling to receive upfront ASCT. The study was registered with clinicaltrials gov. Identifier: NCT04934579. The trial was approved by the ethics review committee of the Second Affiliated Hospital, Zhejiang University. Signed informed consent was provided by each patient.
Induction treatment was performed with six cycles of lenalidomide (25 mg/day [d], orally, d1-10), rituximab (375 mg/m2, intravenously [IV], d0), and methotrexate (3.5 g/ m2, IV, d1) every 3 weeks, followed by four cycles of maintenance treatment with lenalidomide (25 mg/day, orally, d1-14, d29-42) and rituximab (375 mg/m2, IV, d1) (R2 regimen) every 8 weeks. Patients who did not achieve partial response (PR) after four cycles of induction treatment or who experienced disease progression (PD) at any time point were withdrawn from the study. Salvage WBRT was performed for individuals without complete remission (CR) before maintenance treatment. WBRT was performed at 30 Gy (2 Gy/dose, 5 doses/week for 3 weeks) plus local tumor field-size irradiation up to 45 Gy. The primary endpoint was the objective response rate (ORR) at the end of induction therapy. Secondary endpoints were best ORR recorded during induction therapy, progression-free survival (PFS), OS, and safety. As previously reported, the ORR of methotrexate monotherapy for newly diagnosed PCNSL cases varies between 40% and 60%. Meanwhile, the ORR of R2 regimen for refractory/relapse PCNSL was 35.6%.8 The ORR of R+MTX ± chemotherapy varies between 50% and 89%. Therefore, the standard treatment ORR was assumed to be 0.50 (null hypothesis, P0), and the expected ORR of R2-MTX regimen was 0.85 (P1). Assuming a 2-sided a (type I error) of 0.05 and a power of 0.80, the study required at least 14 evaluable patients. Considering a 15% dropout rate, the minimal enrollment goal was 17 participants in this study. Between Jan 1, 2020 and May 16, 2022, this study screened 20 cases of newly diagnosed PCNSL, of whom 17 were finally included. The full study flow diagram is shown in the Online Supplementary Figure S1. Baseline clinicodemographic features are summarized in Table 1. The median patient age at presentation was 60 (range, 44-75) years. Seven cases (41.1%) were male and nine cases (52.9%) were of Eastern Cooperative Oncology Group score >2. Fifteen (88.2%) participants had activated B-cell-like DLBCL. Treatment response was determined by central neuroradiology review with contrast-enhanced brain magnetic resonance imaging every two cycles during induction therapy and every cycle during maintenance therapy, according to the International Primary CNS Lymphoma Collaborative Group (IPCG) criteria. At the end of induction therapy, 13 (76.5%) patients achieved CR, and two (11.8%) patients achieved PR, indicating an ORR of 88.2% (95% confidence interval [CI]: 63.6-98.5) (Figure 1A). The best ORR during induction therapy was 94.1% (95% CI: 71.3-99.9) (Figure 1B). The last
Table 1. Baseline characteristics.
Factors
Age in years, median (range)
Male, N (%)
Previous surgery, N (%)
Craniotomy resection Stereotactic biopsy 5 (29.4) 12 (70.6)
*Deep lesions were located in the thalamus, corpus callosum, brainstem, cerebellum, and ventricles. ECOG: Eastern Cooperative Oncology Group; IELSG: International Extranodal Lymphoma Study Group; MSKCC: Memorial Sloan-Kettering Cancer Center.
follow-up date was September 15, 2023. Following a median follow-up of 23 months (range, 1-41), the median PFS was 29.0 months (Figure 1C), and the median OS was not reached (Figure 1D). The estimated 2-year PFS rate was 58.8% and the estimated 3-year OS rate was 76.0%. In subgroup analysis, participants with low-to-intermediate IELSG risk had better PFS ( P =0.0035) and OS ( P =0.01) than the high-risk group 9 ( Online Supplementary Figure S2A, B ). PFS and OS were similar among different risk subgroups based on the MSKCC scoring system 10 ( P =0.99 and P =0.093, respectively) ( Online Supplementary Figure S2C, D ).
All participants experienced treatment-related adverse events (TRAE). The commonest all-grade TRAE included leukopenia (94.1%), neutropenia (82.4%), increased alanine aminotransferase (ALT) (58.9%), increased aspartate aminotransferase (AST) (52.9%), anemia (47.1%), and constipation (41.2%). Grade ≥3 TRAE included neutropenia (47.1%), leukopenia (23.5%), thrombocytopenia (11.8%), increased ALT (5.9%), and increased AST (5.9%) (Table 2). Severe AE (liver dysfunction) was reported in
one patient. No treatment discontinuation occurred due to AE. No unexpected toxicities were observed. This multicenter, phase II trial first reported the effectiveness of the R2-MTX regimen with a relatively high ORR of 88.2%. The IELSG32 trial applied MATRix for first-line treatment with an ORR of 87%, compared with relatively lower ORR for methotrexate, cytarabine with rituximab group (74%) and methotrexate with cytarabine group (51%).11 Furthermore, other regimens combining drugs with MTX + rituximab had lower ORR, e.g., temozolomide (MT-R regimen) in the Alliance 50202 study (77%),12 and procarbazine with/without lomustine in the PRIMAIN study (49.5%).13 Regarding long-term survival, consolidation with either WBRT or ASCT, achieved 2-year PFS and OS rates of 61% and 69%, respectively in the IELSG32 study.11 In the PRECIS study,1 consolidation therapy with either WBRT or ASCT, induced 2-year PFS of 63% and 87%, respectively. In the Alliance 50202 study,12 consolidation therapy with etoposide plus cytarabine conferred 2-year PFS and OS of 57% and 70%, respectively. No maintenance therapy was applied in the above clinical trials. In this study, R2 maintenance therapy without intended consolidation, produced an estimated 2-year PFS rate of 58.8% and an estimated 3-year OS rate of 76.0%. The advantage of R2-MTX on the anti-tumor effect might be associated with the pharmacological features of lenalidomide, especially its capability to cross the blood-brain barrier. Furthermore, the molecular abnormalities in PCNSL include NF- k B pathway activation, which makes it possible to treat PCNSL with lenalidomide.14 More specifically, lenalidomide increases the activation and proliferation of natural killer (NK) and T cells, and enhances the generation of immune synapses, causing apoptosis in lymphoma cells, while rituximab induces NK-cell-mediated antibody-dependent cellular cytotoxicity in lymphoma cells, showing a synergistic activation of anti-tumor immunity by combining lenalidomide and rituximab.6 The immunomodulatory effects of lenalidomide and rituximab make this combination a good prospect for the maintenance of the anti-lymphoma effect.15 Nevertheless, the survival benefit of R2-MTX followed by R2 maintenance needs to be further verified. In subgroup analysis, we found that participants with low-to-intermediate IELSG risk had better PFS and OS than the high-risk group. It may suggest that low-to-intermediate-risk patients benefit most from such less intensified strategies. Here, the AE of R2-MTX appeared manageable with no additional safety concerns. Although all patients had TRAE, relatively fewer grade ≥3 TRAE were observed. The grade ≥3 TRAE of R2-MTX in the present study were mainly hematological toxicity (neutropenia: 47.1% and leukopenia: 23.5%). In the IELSG 32 trial,11 the widely used MATRix regimen resulted in 67% grade ≥3 neutropenia, while HD-MTX with cytarabine induced 52%
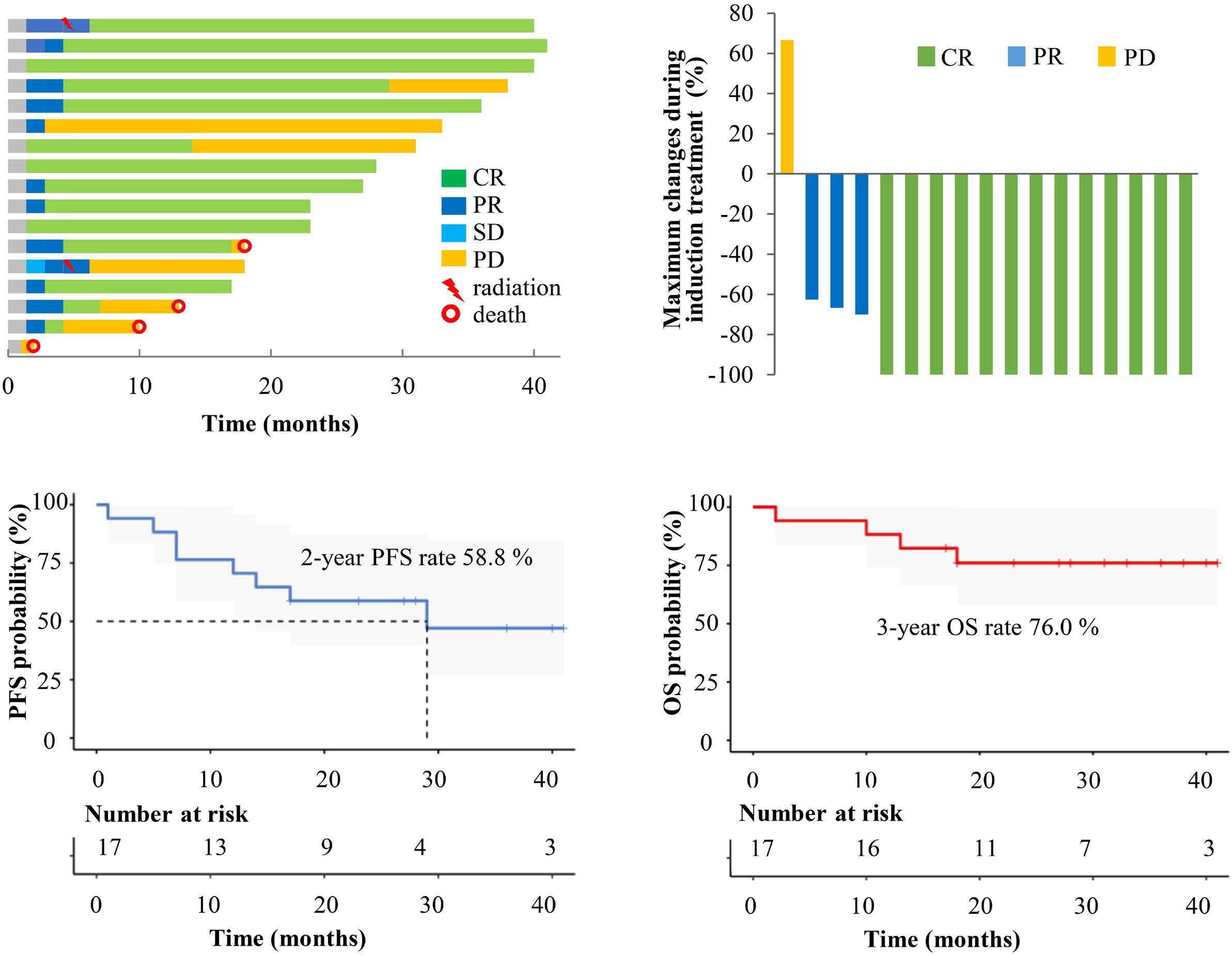
Table 2. Adverse events.
Leukopenia
Anemia 8 (47.1) 0
Thrombocytopenia 4 (23.5) 2 (11.8)
Increased alanine aminotransferase 10 (58.8) 1 (5.9)
Increased aspartate aminotransferase elevation
Increased bilirubin 0 0
Renal insufficiency 1 (5.9) 0
Skin rash 1 (5.9) 0
Deep vein thrombosis 3 (17.6) 0
Sinus bradycardia 3 (17.6) 0
Numbness of extremities 2 (11.8) 0
Constipation 7 (41.2) 0
Hypotension 1 (5.9) 0
Non-infectious fever 3 (17.6) 0
Urinary tract infection 3 (17.6) 0
grade ≥3 leukopenia. In the PRIMAIN study,13 R-MP with/ without lomustine regimens resulted in 56.7% grade ≥3 leukopenia. Numerically, the R2-MTX regimen showed possible better tolerability.
This study had several limitations. Firstly, among the 17 patients enrolled, 12 patients were transplant-ineligible because of HCT-CI score ≥3 (N=6) or age ≥65 years old (N=6), and the remaining five transplant-eligible patients were unwilling to receive upfront ASCT. The main reasons for refusing ASCT in China included the low acceptability of ASCT, the inaccessibility of thiotepa (a conditioning drug for ASCT) at the initial stage of this study, and unaffordable high costs of ASCT. Secondly, the sample size was small, and the follow-up was short, leading to immature PFS and OS data. Further studies are warranted to address these limitations. In conclusion, the R2-MTX regimen appears to be effective and well-tolerated in newly diagnosed transplant-ineligible PCNSL, especially for those patients with low-to-intermediate IELSG risk and lack of access to ASCT. Based on its favorable tolerability and good anti-tumor effect, R2-MTX might provide a promising option for newly diagnosed PCNSL.
Authors
Xianggui Yuan,1* Yaping Xie,2* Nengwen Xu,3* Hui Liu,1 Panpan Chen,1 Aiqi Zhao,1 Yun Liang1 and Wenbin Qian1,4
1Department of Hematology, the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou; 2Department of Hematology, the Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine, Hangzhou; 3Department of Hematology, the Lishui Hospital, Zhejiang University School of Medicine, Lishui and 4National Clinical Research Center for Hematologic Diseases, the First Affiliated Hospital of Soochow University, Suzhou, China
*XY, YX and NX contributed equally as first authors.
Correspondence:
W.-B. QIAN - qianwb@zju.edu.cn
Y. LIANG - liangyun@zju.edu.cn https://doi.org/10.3324/haematol.2023.284834
Received: December 8, 2023.
Accepted: February 2, 2024.
1. Houillier C, Taillandier L, Dureau S, et al. Radiotherapy or autologous stem-cell transplantation for primary CNS lymphoma in patients 60 years of age and younger: results of the Intergroup ANOCEF-GOELAMS randomized phase II PRECIS study. J Clin Oncol. 2019;37(10):823-833.
2. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Whole-brain radiotherapy or autologous stem-cell transplantation as consolidation strategies after high-dose methotrexate-based chemoimmunotherapy in patients with primary CNS lymphoma: results of the second randomisation of the International Extranodal Lymphoma Study Group-32 phase 2 trial. Lancet Haematol. 2017;4(11):e510-e523.
3. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Long-term efficacy, safety and neurotolerability of MATRix regimen followed by autologous transplant in primary CNS lymphoma: 7-year results of the IELSG32 randomized trial. Leukemia. 2022;36(7):1870-1878.
4. Houillier C, Soussain C, Ghesquières H, et al. Management and outcome of primary CNS lymphoma in the modern era. Neurology. 2020;94(10):e1027-e1039.
5. Vu K, Mannis G, Hwang J, Geng H, Rubenstein JL. Low-dose lenalidomide maintenance after induction therapy in older patients with primary central nervous system lymphoma. Br J Haematol. 2019;186(1):180-183.
6. Wu L, Adams M, Carter T, et al. lenalidomide enhances natural killer cell and monocyte-mediated antibodydependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res. 2008;14(14):4650-4657.
7 Yang C, Cui Y, Ren X, et al. Orelabrutinib combined with lenalidomide and immunochemotherapy for relapsed/
Early view: February 15, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interests to disclose.
Contributions
WBQ and YL conceived and designed the protocol. XGY, YPX, and NWX collected and analyzed the data. PPC performed data analysis. XGY, HL and AQZ drafted the manuscript. All authors reviewed the manuscript.
Funding
The present study was funded by the National Natural Science Foundation of China (No. 82170219), and Zhejiang province commonweal projects (No. BY24H080013).
Data-sharing statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
refractory primary central nervous system lymphoma: a retrospective analysis of case series. Front Oncol. 2022;12:901797.
8. Ghesquieres H, Chevrier M, Laadhari M, et al. Lenalidomide in combination with intravenous rituximab (REVRI) in relapsed/ refractory primary CNS lymphoma or primary intraocular lymphoma: a multicenter prospective ‘proof of concept’ phase II study of the French Oculo-Cerebral lymphoma (LOC) Network and the Lymphoma Study Association (LYSA)†. Ann Oncol. 2019;30(4):621-628.
9 Ferreri AJM, Blay J-Y, Reni M, et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol. 2003;21(2):266-272.
10. Abrey LE, Ben-Porat L, Panageas KS, et al. Primary central nervous system lymphoma: the Memorial Sloan-Kettering Cancer Center prognostic model. J Clin Oncol. 2006;24(36):5711-5715.
11. Ferreri AJ, Cwynarski K, Pulczynski E, et al. Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol. 2016;3(5):e217-227.
12. Rubenstein JL, Hsi ED, Johnson JL, et al. Intensive chemotherapy and immunotherapy in patients with newly diagnosed primary CNS lymphoma: CALGB 50202 (Alliance 50202). J Clin Oncol. 2013;31(25):3061-3068.
13. Fritsch K, Kasenda B, Schorb E, et al. High-dose methotrexate-based immuno-chemotherapy for elderly
primary CNS lymphoma patients (PRIMAIN study). Leukemia. 2017;31(4):846-852.
14 Hernández-Verdin I, Kirasic E, Wienand K, et al. Molecular and clinical diversity in primary central nervous system lymphoma. Ann Oncol. 2023;34(2):186-199.
15. Yilmaz U, Salihoglu A, Soysal T. An overview of lenalidomide in combination with rituximab for the treatment of adult patients with follicular lymphoma: the evidence to date. Drug Des Devel Ther. 2021;15:3809-3820.
Cerebral venous sinus thrombosis (CVST) is a rare cerebrovascular disorder predominantly observed in adults, but can pose unique challenges in pediatric patients.1 The underlying etiologies of CVST are diverse, encompassing hypercoagulable states, infections, and trauma. While concomitant thrombocytopenia is uncommon in CVST cases,2 instances of CVST and thrombocytopenia have been reported following administration of adenoviral vector-based vaccines, ChAdOx1 nCoV-19 and Ad26.COV2.S.3-5 This condition has been termed vaccine-induced immune thrombotic thrombocytopenia (VITT), which is caused by antibodies against platelet factor 4 (PF4). In this paper, we report on a pediatric case of severe CVST and thrombocytopenia, emerging 1 week after an adenovirus infection. Recurrent occlusion and profound thrombocytopenia, which responded to intravenous immunoglobulin (IVIG) therapy, was observed. Platelet-activating anti-PF4/heparin antibodies were identified in patient’s serum. Informed signed consent was obtained from the parents of the patient to publish the case. Our patient is a 7-year-old girl who presented to the family physician with a sudden onset of severe headache and vomiting. One week prior, she had fever and conjunctivitis. There was no cranial trauma in the medical history. She was initially hospitalized in a regional hospital where computed tomography (CT) detected bilateral frontal subdural hematoma. Laboratory investigation showed isolated thrombocytopenia (11x109/L). After 2 days of hospitalization, she was transferred to the pediatric intensive care unit at our university hospital. At admission, D-dimer was 54 µg/mL fibrinogen equivalent units (FEU) and fibrinogen (40 mg/dL) as well as factor XIII (33%) were decreased. Infection-associated immune thrombocytopenia was suspected. Anti-platelet antibody presence, which was tested using the monoclonal antibody immobilization of platelet antigens assay, and anti-phospholipid antibodies were not detected. Adenovirus infection was detected in the throat swab by polymerase chain reaction (PCR). PCR for following viruses were negative: SARS-CoV-2, Influenza A, Influenza B, respiratory syncytial virus, human papillomavirus, rhinovirus, herpes simplex virus-1 (HSV-1), HSV-2, enterovirus, cytomegalovirus, and Epstein-Barr virus. Bacterial and fungal cultures were also negative. Magnetic resonance imaging with angiography revealed an extensive thrombosis in the superior sagittal sinus, partially affecting the sigmoid sinus and cortical veins. On day 4, interventional thrombectomy was successfully performed, restoring orthograde drainage from the anterior through the middle and distal segments (Figure 1A, B). After the proce-
dure, the patient experienced retroperitoneal bleeding and subsequently hemorrhagic shock, and received platelet and erythrocyte concentrates. Interventional coiling of the inferior epigastric artery was required to stop the bleeding. After thrombectomy, D-dimer reduced to 3.3 µg/mL. Cranial CT was conducted due to the deterioration of neurological symptoms, revealing re-thrombosis of a similar extent to the initial presentation, along with congestive bleeding. Thrombocytopenia, elevated D-Dimer and decreased fibrinogen levels indicated uncontrolled hypercoagulation. Therapeutic anticoagulation with unfractionated heparin (UFH) was started, and a second mechanical thrombectomy was performed on day 5. Platelet count remained below 50x109/L despite several platelet transfusions. Given the challenging course with bleeding complications and the suspicion of immune thrombocytopenia, the patient received high-dose intravenous immunoglobulin (IVIG) (a total of 45g IVIG) on days 9 and 10, leading to a swift normalization of the platelet count (>100x109/L) in the following days (Figure 1C).
On day 12, hematoma evacuation and decompressive craniectomy were performed due to progressive subdural hematoma. In the following course, the patient showed gradual improvement. Sonographic monitoring showed stable retroperitoneal hematoma. Coagulation parameters upon IVIG and under anticoagulation showed significant improvement. Discharged on day 33, the patient demonstrated a cooperative demeanor, mild word-finding difficulties, rest tremor, and normal muscle tone and strength. However, concentration and perseverance were still clearly reduced. Anticoagulation was switched to low molecular weight heparin at discharge. The rare co-occurrence of CVST with thrombocytopenia has prompted our consideration of a rare variant of heparin-induced thrombocytopenia (HIT), known as spontaneous HIT, which can manifest without prior heparin exposure. Additionally, heightened awareness in our laboratory driven by the emergence of VITT cases 2 years ago, which also often present with thrombocytopenia and CVST, led us to consider an anti-PF4 antibody related disorder. The retrospective investigation of the sample from day 5 of hospitalization showed a strong immunoglobulin (Ig)G PF4/heparin enzyme immunosorbent assay reaction (Zymutest HIA IgG, Hyphen Biomed, Neuville-sur-Oise, France, optical density (OD) 2.8; normal range, 0-0.5). A modified heparin-induced platelet activation (HIPA) assay was performed with addition of exogenous PF4 as previously described.6 The HIPA assay was negative with low concentration of heparin making HIT very unlikely (Figure 2A). In contrast, the modified HIPA was
positive (platelet aggregation within 5 minutes [min] in the presence of PF4 and within 15-35 min without exogenous PF4), which is a serological pattern that mimics VITT (Figure 2A).3 Antibody-induced procoagulant platelets, determined by expression of P-selectin and phosphatidylserine (PS) externalization on the platelet surface, were analyzed using flow cytometer as previously described.3 Antibody-mediated procoagulant platelet formation was observed in the
absence of heparin and was decreased in the presence of low concentration of heparin but completely inhibited by high concentration of heparin (Figure 2B). Additionally, serum-induced procoagulant phenotype was inhibited with IV.3 (Fcγ receptor IIa blocking monoclonal antibody anti-CD32 [moAb IV.3; StemcellTM technologies, Vancouver, Canada], 20 µg/mL) or IVIG (30 µg/mL), indicating FcγRIIA dependency (Figure 2B). Increased PS externalization can

Figure 1. Radiological images and course of the platelet counts throughout the hospitalization. (A) Digital subtraction angiography was performed showing thrombosis of the superior sagittal sinus (black arrows). (B) Angiography after mechanical recanalization shows the recanalized cerebral superior sagittal sinus (black arrows). (C) Platelet count and other laboratory findings throughout hospital admission labeled with relevant interventions. Abbreviations and normal range of laboratory parameters: platelet count (normal range: 150450x103/µL), D-dimer (normal range: 0-0.5 μg/mL fibrinogen equivalent units), fibrinogen (normal range: 170-410 mg/dL), activated partial thromboplastin time (aPTT; normal range: <40 seconds), factor XIII (normal range: 70-140%). IVIG: intravenous immunoglobulin; EIA: enzyme immunosorbent assay; Ig: immunglobulin; HIPA: heparin-induced platelet activation; PF4: platelet factor 4.
initiate plasmatic coagulation and subsequent increased thrombin generation on the platelet surface.8 The ability of patient’s serum to induce thrombosis was investigated using a novel ex vivo model for antibody-mediated thrombosis (utilizing the BioFlux 200 system from Fluxion Biosciences, Alameda, USA).7 Patient’s serum induced significant thrombus formation with increased fibrin deposition compared to healthy control (mean cumulative area of thrombus [%SAC] ± standard error of mean (SEM): 2.3±0.36 vs. 0.9±0.2; P<0.01; Figure 3). Thrombus formation was markedly inhibited by IV.3 (%SAC±SEM: 0.02±0.02) and IVIG (%SAC±SEM: 1.29±0.21). Our data suggest that anti-PF4 antibodies can be generated after adenovirus infection even without a previous aberrant exposure to heparin or COVID-19 vaccine. These antibodies seem to harbor the ability of inducing thrombosis in a mechanistically similar way as VITT antibodies. Most importantly,
our ex vivo data emphasizes the advantage of IVIG to prevent antibody-mediated thrombosis and thrombocytopenia. Antibodies against PF4 lead to HIT and VITT, in which the immune tolerance to PF4 is disrupted, resulting in clonal expansion of B cells and subsequent secretion of anti-PF4 antibodies.8 Recently, platelet-activating anti-PF4 antibodies were detected in an unvaccinated patient with monoclonal gammopathy and multiple thrombotic complications.9 Similar to our case, Warkentin et al. published very recently two cases (a pediatric and an adult patient) developing anti-PF4 antibodies almost 1 week after adenovirus infection.10 Both patients had thrombocytopenia and thrombotic events (fatal CVST in the child and multiple arterial and venous thrombosis in the adult). Our patient did not have previous heparin or COVID-19 vaccine exposure. HIT could also be ruled out. No additional risk factors for CVST were identified. The trigger
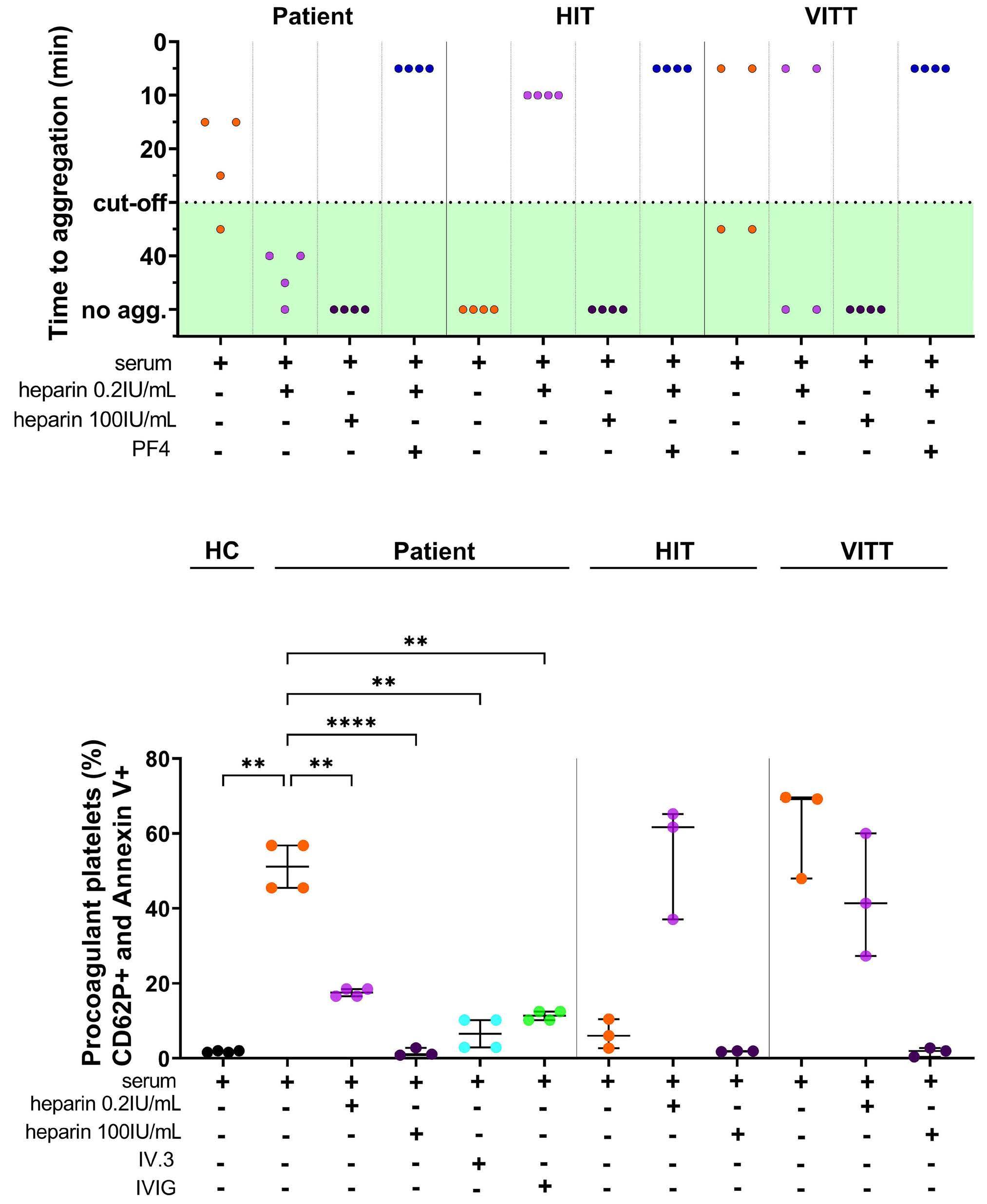
Figure 2. Patient sera induces platelet activation and procoagulant platelet phenotype. (A) Heparin-induced platelet activation (HIPA): patient serum activated platelets with exogenous platelet factor 4 (PF4) in HIPA assay, however, patient’s sera did not activate platelets in the presence of low (0.2 IU/mL) or high concentration (100 IU/mL) of heparin. Historical representative samples from a heparin-induced thrombocytopenia (HIT) and a vaccine-induced immune thrombotic thrombocytopenia (VITT), patient are also depicted in the figure. (B) Flow cytometer: procoagulant platelet phenotype, determined by co-expression of P-selectin and phosphatidylserine (PS) on platelet surface, was analyzed after incubation with patient’s sera. Where indicated, platelets were pretreated with IV.3 (Fc γ receptor IIA blocking monoclonal antibody) or immunoglobulin (IVIG). Patient’s serum was tested with washed platelets from 4 healthy donors. Historical samples of patients with HIT (N=3) and VITT (N=3) was also shown on the figure. HC: healthy control; **P<0.01, ****P<0.0001; IVIG: intravenous immunoglobulin.
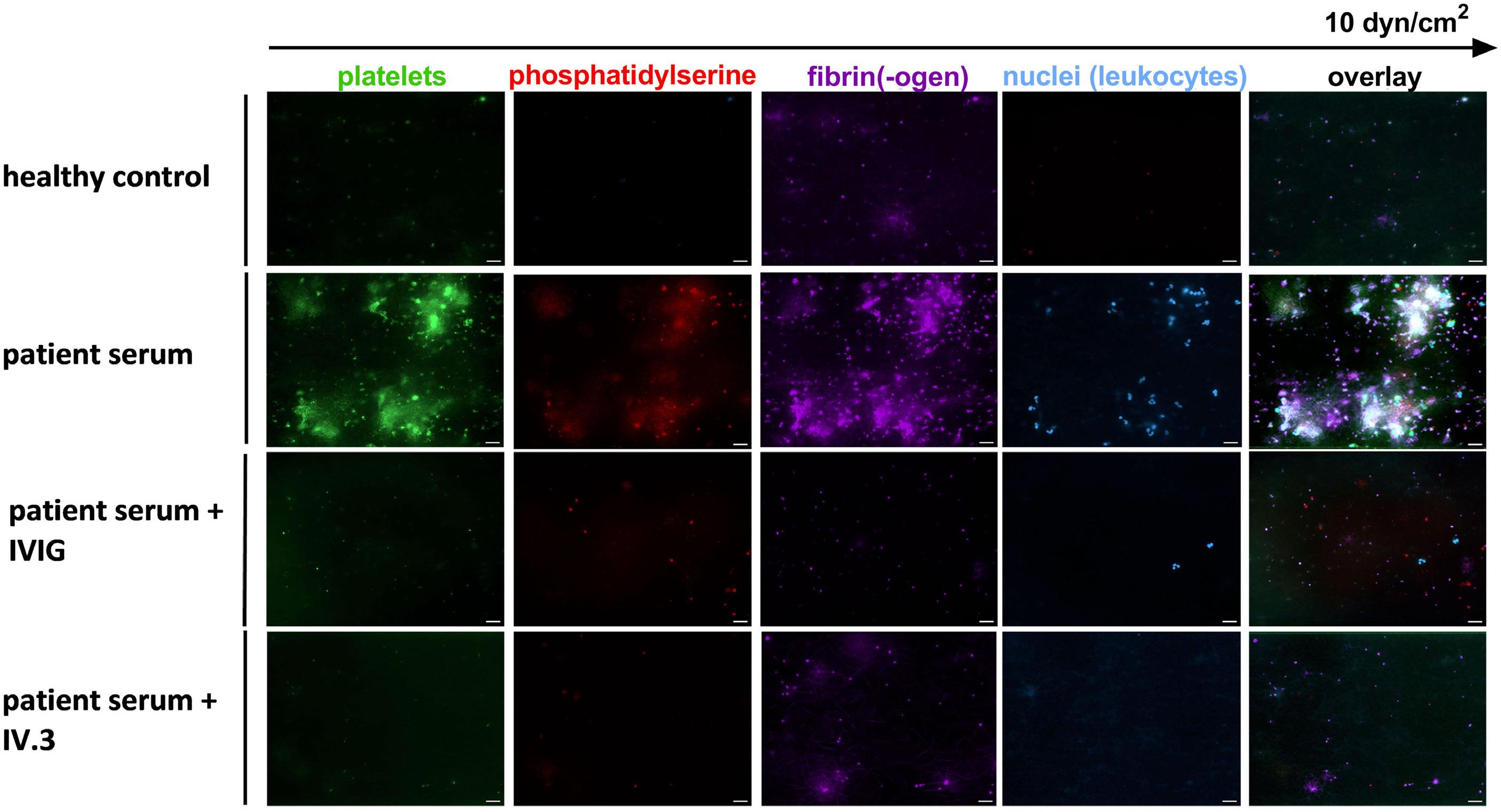
Figure 3. Patient sera induces thrombus formation in vitro. Platelet-rich plasma (PRP) obtained from healthy individuals in a volume of 37.5 µL was subjected to a 60-minute incubation with serum (5 µL) from the patient, all while under rotation. Following this incubation period, the samples were labeled using 3,3’-dihexyloxacarbocyaniniodid (DiOC6, 2.5 µM; Sigma Aldrich, Saint Louis, USA), Alexa Fluor (AF) 647-Annexin A5 (at a 1:200 dilution), AF 546-labeled human fibrinogen (at a concentration of 8.5 µg/mL), and Hoechst 33342 (at a concentration of 3 µg/mL; Thermo Scientific, Carlsbad, USA). When specified, the PRP was preincubated with IV.3 (20 µg/mL) or intravenous immunoglobulin (IVIG) (30 µg/mL). After the labeling procedure, the samples were reconstituted into autologous whole blood. Subsequently, the samples were recalcified and subjected to perfusion through microfluidic channels (BioFlux 200, Fluxion Biosciences, Alameda, USA) at a venous shear rate set at 250 s-1 (equivalent to 10 dyne/cm2) for a duration of 10 minutes. Images were acquired at x40 magnification in different fluorescence channels using a Zeiss Axio Observer 7 microscope. The acquired images were uniformly processed using adjusted threshold settings and the exclusion of any image artefacts using of Fiji image processing software. Representative fluorescence images are shown (N=3). Scale bar 20µm.
for the development of anti-PF4/heparin antibodies in our patient is not clear. It was suggested that adenovirus or components of vaccine might be responsible for the development of anti-PF4 antibodies in VITT patients.11 Anti-PF4 antibodies from VITT patients recognizes complexes of adenovirus hexon proteins and PF4. Furthermore, ChAdOx1 can bind to PF4 as well as coxsackievirus and adenovirus receptor (CAR), which also supports the procoagulant situation of the platelets in the case of an adenovirus infection analogous to VITT.12 Concurrent pro-inflammatory factors may be the link to an enhanced immune response to PF4 and thus the formation of anti-PF4 antibodies in adenovirus infection. Anticoagulation in CVST is challenging due to bleeding risk, with 40% presenting with hemorrhagic infarct at diagnosis, making immediate heparinization difficult. In our patient, the presence of intracranial hemorrhage and severe thrombocytopenia led to a cautious approach and delayed initiation of heparin. Due to high procoagulant state, patients with HIT and VITT require therapeutic anticoagulation. Heparin is, however, contraindicated in HIT. Nonetheless, successful heparin use has been reported in VITT13 and therapeutic dose
heparin can disrupt the interaction between VITT antibodies and PF4 in vitro. 14 Importantly, our patient, despite high anti-PF4/heparin antibodies, didn’t develop new thrombosis after heparin treatment and platelet count remained stable. IVIG is a well-established first-line treatment for patients with ITP, but more recently it has been of increasing interest in the treatment of HIT and VITT.15 IVIG mitigates platelet activation via competitive FcγRIIA binding.16 In VITT patients with CVST, IVIG therapy correlated with lower mortality.13 We observed a rapid increase of platelet count after IVIG therapy (Figure 1C), supporting an immune-mediated platelet activation as the underlying mechanism of the thrombocytopenia. IVIG can be considered in patients with antibody-mediated platelet activation and thrombocytopenia.
Our current study confirms the recent findings10 that anti-PF4 antibodies may be responsible for a severe thromboembolic complication such as CVST and thrombocytopenia after adenovirus infection in the absence of prior exposure to heparin or COVID-19 vaccine. This case underscores the importance of studying the role of anti-PF4 antibodies in thrombotic events beyond HIT and VITT. Further research
is required to elucidate the underlying mechanisms, which could potentially impact the management of patients with unexplained thrombosis and thrombocytopenia.
Günalp Uzun,1,2 Jan Zlamal,1,2 Karina Althaus,1,2 Andrea Bevot,3 Florian Hennersdorf,4 Nina Wolska,1 Anna Jock,5 Jan Kern,5 Vanya Icheva,5 Sven Poli,6,7 Ulrike Ernemann,4 Andreas Neu8 and Tamam Bakchoul1,2
1Institute for Clinical and Experimental Transfusion Medicine, University Hospital of Tübingen; 2Center for Clinical Transfusion Medicine; 3Department of Neuropediatrics and Developmental Medicine, University Children’s Hospital, University of Tübingen; 4Department of Diagnostic and Interventional Neuroradiology, University Hospital Tübingen; 5Department of Pediatric Cardiology, Pulmonology and Intensive Care Medicine, University Children’s Hospital, University of Tübingen; 6Department of Neurology & Stroke, University Hospital of Tübingen; 7Hertie Institute for Clinical Brain Research, Eberhard-Karls University and 8Department of Pediatric Endocrinology and Diabetes, University Children’s Hospital, University of Tübingen, Tübingen, Germany
Correspondence:
T. BAKCHOUL - tamam.bakchoul@med.uni-tuebingen.de
https://doi.org/10.3324/haematol.2023.284127
Received: August 21, 2023. Accepted: October 19, 2023. Early view: October 26, 2023.
Published under a CC BY license
Disclosures
TB has received research funding from CoaChrom Diagnostica GmbH, DFG, Robert Bosch GmbH, Stiftung Transfusionsmedizin und Immunhämatologie e.V., Ergomed, Surrey, DRK Blutspendedienst,
1. Ropper AH, Klein JP. Cerebral venous thrombosis. N Engl J Med. 2021;385(1):59-64.
2. Payne AB, Adamski A, Abe K, et al. Epidemiology of cerebral venous sinus thrombosis and cerebral venous sinus thrombosis with thrombocytopenia in the United States, 2018 and 2019. Res Pract Thromb Haemost. 2022;6(2):e12682.
3. Althaus K, Möller P, Uzun G, et al. Antibody-mediated procoagulant platelets in SARS-CoV-2-vaccination associated immune thrombotic thrombocytopenia. Haematologica. 2021;106(8):2170-2179.
4 Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med. 2021;384(22):2092-2101.
Deutsche Herzstiftung, Ministerium für Wissenschaft, Forschung und Kunst Baden-Württemberg; has received lecture honoraria from Aspen Germany GmbH, Bayer Vital GmbH, CSL Behring, Bristol-Myers Squibb GmbH & Co., Doctrina Med AG, Meet The Experts Academy UG, StreamedUp, Stago GmbH, Mitsubishi Tanabe Pharma GmbH and Novo Nordisk Pharma GmbH; has provided consulting services to Terumo; has provided expert witness testimony relating to heparin-induced thrombocytopenia (HIT) and non-HIT thrombocytopenic and coagulopathic disorders; has a pending patent for a flow cytometrybased assay to detect HIT and VITT (all of these are outside the current work). SP is a steering committee member of the International Cerebral Venous Thrombosis Consortium; received research support from BMS/ Pfizer, Boehringer-Ingelheim, Daiichi Sankyo, the European Union, the German Federal Joint Committee Innovation Fund, and the German Federal Ministry of Education and Research, Helena Laboratories and Werfen as well as speakers’ honoraria/consulting fees from Alexion, AstraZeneca, Bayer, Boehringer-Ingelheim, BMS/Pfizer, Daiichi Sankyo, Portola and Werfen (all outside the submitted work). All other authors have no conflicts of interest to disclose.
Contributions
GU, KA and TB designed the study. AB, FH, AJ, JK, VI, UE and AN were responsible for the treatment of the patient. GU and KA collected and analyzed the clinical data. JZ and NW performed the experiments. GU, JZ, KA, NW, SP and TB analyzed the data, interpreted the results and wrote the manuscript. All authors read and approved the manuscript.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) and from the Herzstiftung to TB (BA5158/4 and TSG-Study). GU (CS 30140-0) and JZ (CS 3015-0-0) take part in the MINT-Clinician Scientist Program of the Medical Faculty Tübingen, funded by DFG (493665037).
Data-sharing statement
Data may be requested for academic collaboration from the corresponding author.
6. Uzun G, Althaus K, Hammer S, et al. Diagnostic performance of a particle gel immunoassay in vaccine-induced immune thrombotic thrombocytopenia. Hamostaseologie. 2023;43(1):22-27.
7 Zlamal J, Singh A, Weich K, et al. Platelet phosphatidylserine is the critical mediator of thrombosis in heparin-induced thrombocytopenia. Haematologica. 108(10):2690-2702.
8. Wang JJ, Armour B, Chataway T, et al. Vaccine-induced immune thrombotic thrombocytopenia is mediated by a stereotyped clonotypic antibody. Blood. 2022;140(15):1738-1742.
9. Greinacher A, Langer F, Schonborn L, et al. Platelet-activating anti-PF4 antibodies mimic VITT antibodies in an unvaccinated patient with monoclonal gammopathy. Haematologica.
5. See I, Su JR, Lale A, et al. US case reports of cerebral venous sinus thrombosis with thrombocytopenia after Ad26.COV2.S vaccination, March 2 to April 21, 2021. JAMA. 2021;325(24):2448-2456.
2022;107(5):1219-1221.
10 Warkentin TE, Baskin-Miller J, Raybould AL, et al. Adenovirusassociated thrombocytopenia, thrombosis, and VITT-like antibodies. N Engl J Med. 2023;389(6):574-577.
11. Greinacher A, Selleng K, Palankar R, et al. Insights in ChAdOx1 nCoV-19 vaccine-induced immune thrombotic thrombocytopenia. Blood. 2021;138(22):2256-2268.
12. Baker AT, Boyd RJ, Sarkar D, et al. ChAdOx1 interacts with CAR and PF4 with implications for thrombosis with thrombocytopenia syndrome. Sci Adv. 2021;7(49):eabl8213.
13. Perry RJ, Tamborska A, Singh B, et al. Cerebral venous
thrombosis after vaccination against COVID-19 in the UK: a multicentre cohort study. Lancet. 2021;398(10306):1147-1156.
14 Singh A, Toma F, Uzun G, et al. The interaction between antiPF4 antibodies and anticoagulants in vaccine-induced thrombotic thrombocytopenia. Blood. 2022;139(23):3430-3438.
15. Warkentin TE. High-dose intravenous immunoglobulin for the treatment and prevention of heparin-induced thrombocytopenia: a review. Exp Rev Hematol. 2019;12(8):685-698.
16. Uzun G, Althaus K, Singh A, et al. The use of IV immunoglobulin in the treatment of vaccine-induced immune thrombotic thrombocytopenia. Blood. 2021;138(11):992-996.
Multicentric Castleman disease (MCD) is characterized by enlarged lymph nodes in two or more lymph node stations, characteristic features on microscopic analysis of enlarged lymph node tissue, and a variety of clinical symptoms.1,2 It is divided into three subtypes: i) human Herpes virus 8 (HHV8)-associated MCD which usually occurs in HIV-positive individuals, ii) POEMS-MCD (polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes) and iii) idiopathic MCD (iMCD).1-3 iMCD is subclassified into iMCD–TAFRO ([thrombocytopenia, anasarca, fever, reticulin fibrosis and organomegaly] or iMCD–NOS [not otherwise specified]).1,2 The histopathological spectrum of iMCD is delineated as regressed germinal centers and prominent vascularization on the hypervascular end of the spectrum, hyperplastic germinal centers with prominent plasmacytosis on the plasmacytic end of the spectrum and overlapping features of both would represent mixed histopathology.2
Interleukin (IL)-6 has been implicated in the pathophysiology of MCD especially if its levels are elevated.2,4 HHV8 can replicate in plasmablasts and produce the viral homolog of IL-6 which along with other cytokines produces symptoms and pathology found in HHV MCD.3 HHV8 MCD usually occurs in HIV-positive patients.3 We hereby delineate an interesting case of severe idiopathic MCD which masqueraded as atypical hemolytic uremic syndrome (aHUS) and showed excellent response to rituximab after refractoriness to IL-6 antagonist therapy.
A 39-year-old female with no prior medical history initially presented with abdominal pain and body aches. Within a few days, her clinical status worsened, and she developed severe anemia, thrombocytopenia, and acute renal failure (ARF). Peripheral smear showed few schistocytes (approximately 2% of 1,000 red blood cells) and target cells. She had elevated lactate dehydrogenase (LDH) levels (1,202 IU/L) and indirect hyperbilirubinemia (3.3 mg/dL). Retic count and haptoglobin were normal. She became unstable and was transferred to the intensive care unit. She was intubated, required pressors for shock and was started on continuous renal replacement therapy (CRRT). She underwent daily plasma exchange for 5 days. However, her platelet count and hemoglobin continued to drop. Moreover, the ADAMTS13 level returned normal. The patient was started on eculizumab, an anti-complement C5 protein antibody for suspected aHUS. In addition, she received intravenous immunoglobulin and steroids. The platelet count improved with eculizumab, climbing from 18x109/l to 92x109/L after 2 weeks of therapy. She improved clinically, was extubated and transferred out of the intensice care untit.
The platelet counts subsequently started to decline. An aHUS panel was sent to an outside laboratory. It showed
a modest increase in serum C5b-9 above reference intervals. Her dose of eculizumab was increased and she was restarted on steroids. Bone marrow biopsy was negative for leukemia, lymphoma, myelodysplasia, or hemophagocytosis and mutational analysis was unrevealing for BCR-ABL, JAK 2 mutations, MPL or CALR mutation. She received intermittent transfusions with platelet, packed red blood cells, and fresh frozen plasma. Her kidney function recovered after a few weeks, and she was eventually discharged with a platelet count of 230x10 9/L and hemoglobin of 7.6 g/dL
She was admitted again 4 months later with abdominal pain and evaluation showed a right adnexal lesion. Diagnostic laparoscopy revealed intra-abdominal hemoperitoneum. She developed ARF requiring hemodialysis (HD) and had persistent anemia and thrombocytopenia requiring multiple transfusions. Peripheral blood smear showed no evidence of schistocytes. Haptoglobin and LDH were normal. She was eventually discharged with outpatient HD. She was kept on eculizumab maintenance therapy every 2 weeks for around 2 months. She was again admitted with abdominal pain and was found to have a large left adnexal ovarian mass with a hemorrhagic component as well as pelvic lymphadenopathy. She underwent a left perirenal lymph node biopsy which showed non-specific findings like paracortical acute inflammatory infiltrates, hemorrhage, and rare fibrin thrombi. Bone marrow biopsy was repeated and showed only mildly hypercellular marrow with multilineage hematopoiesis. Flow cytometry on the marrow aspirate was unremarkable. In the interim, she was given romiplostim due to persistent severe thrombocytopenia. Given the suspicion of an autoimmune process of unknown etiology, she was also given rituximab 375 mg/ m² weekly for 4 weeks. The patient stabilized over the next few weeks and was discharged. She received romiplostim 10 mcg/kg weekly for almost 3 months. Her platelet counts slowly rose while she was on the drug. She underwent an axillary lymph node biopsy 3 months later for evaluation of persistent lymphadenopathy which showed non-specific extramedullary hematopoiesis. She was once again admitted 6 months later with worsening anemia, thrombocytopenia and renal failure. She also had new cervical and supraclavicular lymphadenopathy. Parvovirus, Epstein-Barr virus and Cytomegalovirus were negative. Repeat bone marrow biopsy showed only hypercellular marrow with myeloid and megakaryocytic hyperplasia. She underwent an excisional biopsy of an occipital lymph node which showed an enlarged lymph node with marked expansion of paracortex by sheets of plasma cells, hyaline vascular change, with regressed germinal centers, onion-skinning of mantle zones and vascular proliferation (Figures 1-3). This was consistent with Castleman disease, plasma cell variant. IL-6 levels
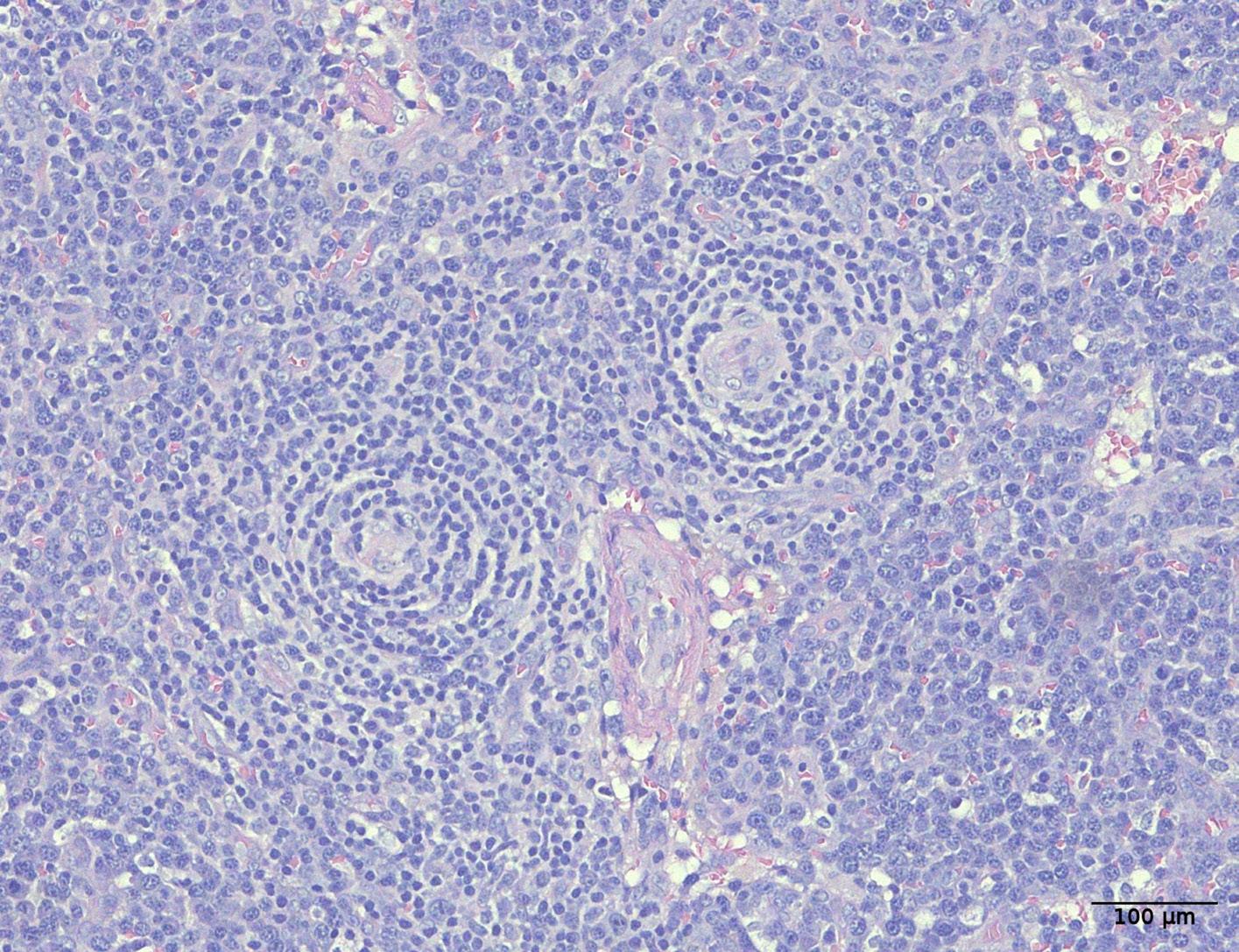
1. Characteristic histopathologic findings of Castleman disease. Hematoxylin and eosin image showing a regressed germinal center doublet with onion skinning and radially penetrating vessel.
came back significantly elevated at 51.1 pg/mL (normal is 0-15.5 pg/mL). HIV and HHV8 testing were negative. Thus, she was diagnosed with idiopathic MCD. Based on the clinical findings, it was more inclined toward the idiopathic TAFRO subtype. She was started on the IL-6 antagonist siltuximab. She improved with normalization of creatinine and hemoglobin and partial recovery of platelet count and was discharged. The patient was admitted again 5 months later with chills, myalgias and right upper quadrant pain. Imaging of the chest, abdomen and pelvis showed no change in her existing lymphadenopathy and no new findings. Positron-emssion tomography (computed tomography showed no evidence of highly fluorodeoxyglucose-avid disease and no significant change in the size of the previously existing adenopathy. She developed increasing lethargy, rising uric acid, elevated CRP, and acute kidney injury consistent with Castleman relapse. She was started on methylprednisolone and rituximab 375 mg/ m² intravenously weekly. Methylprednisolone was later changed to prednisone. Her kidney function recovered. The hemoglobin and platelet count improved with a new baseline of 10.5-11.5 g/dL and 200-250x109/L respectively. The patient was thereafter continued on maintenance rituximab 375 mg/m² every 8 weeks. The patient has been relapse-free for over 2 years.
Our patient was diagnosed with iMCD as HHV8 and HIV were negative and clinical signs/symptoms were parallel with the TAFRO subtype. The initial suspicion of atypical HUS was made due to anemia, thrombocytopenia, renal dysfunction and signs of hemolysis like schistocytes on peripheral smear, increased LDH and indirect bilirubin. This suspicion was somewhat reinforced due to a partial response to eculizumab therapy. However, there was un-
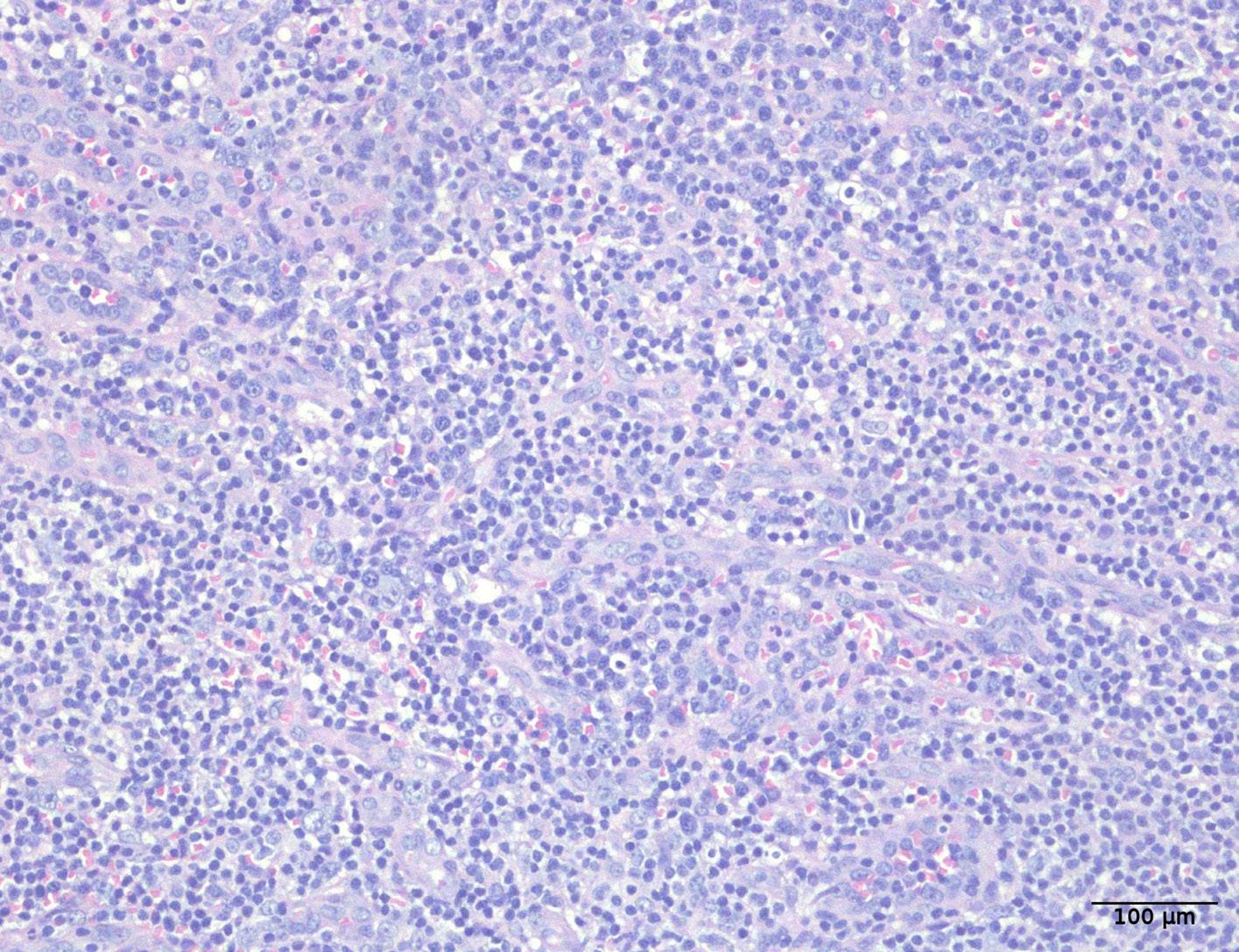
2. Characteristic histopathologic findings of Castleman disease. Hematoxylin and eosin image showing an expanded paracortex with hyaline vascular proliferation and increased plasma cells.
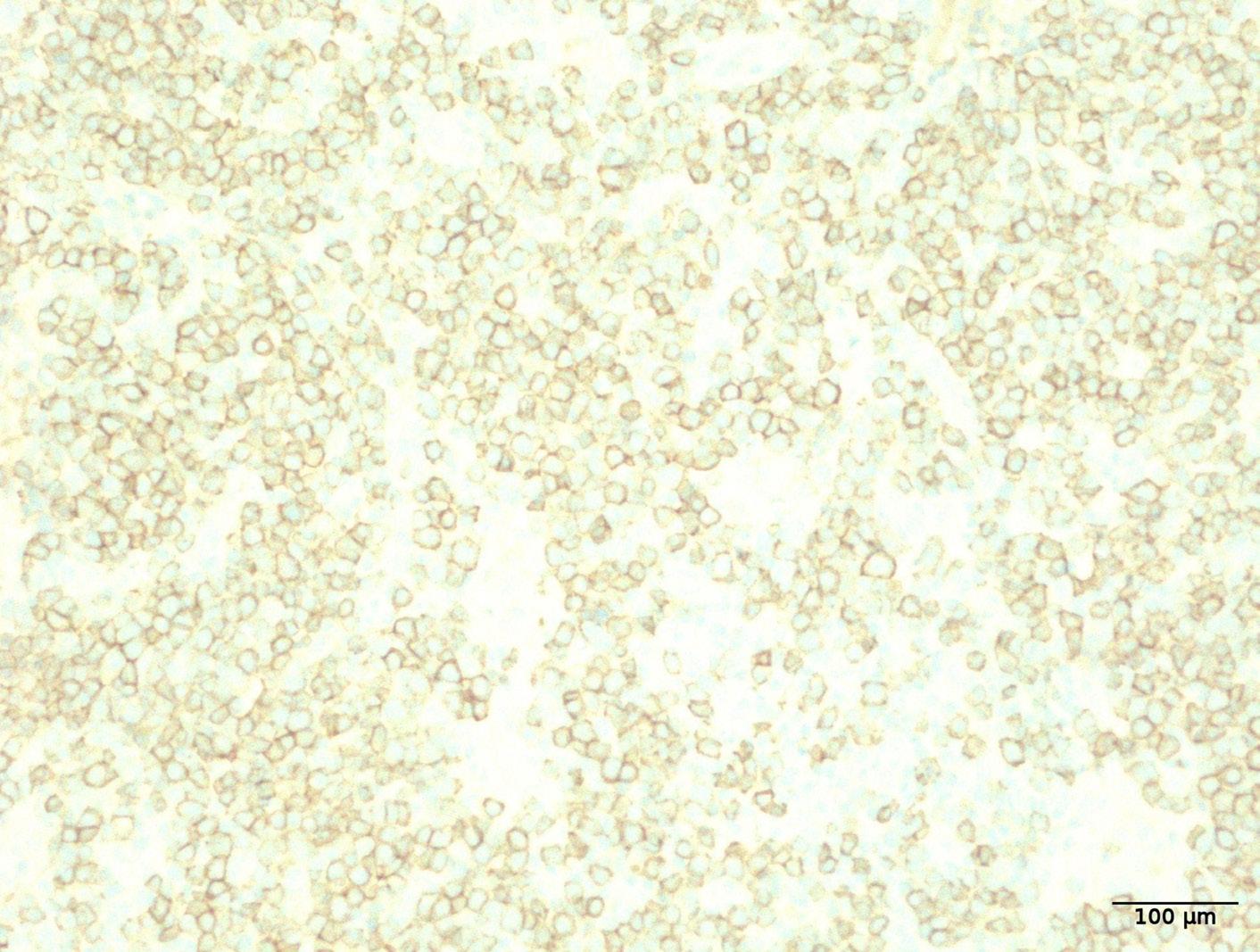
Figure 3. Immunostaining for plasma cells. CD138 immunostaining highlighting the sheets of plasma cells.
certainty as the patient continued developing repeated episodes of anemia, thrombocytopenia, and kidney failure despite being on eculizumab. Multiple bone marrow and lymph node biopsies were unrevealing. Lymph node biopsy for the third time finally showed characteristic features of MCD. In patients presenting with symptoms that could explain MCD with no alternate diagnosis, a single unremarkable lymph node cannot rule out MCD. Julie et al. also described a case with idiopathic multicentric disease in which initial lymph node biopsy showed only reactive changes and repeat biopsy discovered characteristic changes of MCD.5 In iMCD cases, IL-6 plays a significant role in driving symp-
toms and pathology in many cases of iMCD.4 A randomized clinical trial of IL-6 antagonist siltuximab versus placebo showed durable responses with siltuximab in almost one of three patients versus no response to placebo.6 However, not all iMCD patients have IL-6 elevation and many patients either may not respond to siltuximab or may relapse after initial response.6
For iMCD, further treatment is based on severity.7 Rituximab is as an alternative first-line agent for more indolent cases of iMCD to avoid giving toxic chemotherapy to such patients.7 Rituximab eliminates CD20+ B cells and plasmablasts.7 A very small number of case reports/series have described the use of rituximab in iMCD.7 One report described 25 cases of iMCD, who were treated with rituximab as firstline therapy and the CR and PR rates were 20% and 48%, respectively.8 In a study with 61 patients with histologically confirmed MCD, 49 patients were treated with rituximab. The overall survival was 94% at 2 years and 90% at 5 years with rituximab compared with 42% and 33% in patients who did not receive rituximab.9
For more severe cases, combination chemotherapy with/ without rituximab is generally preferred.7,10 CHOP or CVAD (cyclophosphamide, vincristine, adriamycin, etoposide) regimens have shown good responses in patients with severe iMCD.7 One study showed sustained remission rates of 27% with combination chemotherapy in patients with severe iMCD.7 However, these chemotherapy regimens have serious side effects like bone marrow suppression.7 Our patient had severe iMCD manifestations and required several red blood cell and platelet transfusions and CRRT/ HD sessions. A rituximab-based regimen without chemotherapy was considered due to concerns for tolerability. The patient responded well with recovery of counts and kidney function. The response has been sustained with maintenance rituximab for over 2 years. Prospective studies evaluating the efficacy of rituximab in iMCD may be conducted to better comprehend its efficacy and safety.
1. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3(4):e163-175.
2. Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353-1364.
3. Suda T, Katano H, Delsol G, et al. HHV-8 infection status of AIDS-unrelated and AIDS-associated multicentric Castleman’s disease. Pathol Int. 2001;51(9):671-679.
4 Yoshizaki K, Murayama S, Ito H, Koga T. The Role of interleukin-6 in Castleman disease. Hematol Oncol Clin North Am. 2018;32(1):23-36.
5. Semenchuk J, Merchant A, Sakhdari A, Kukreti V. Five biopsies, one diagnosis: challenges in idiopathic multicentric Castleman disease. BMJ Case Rep. 2020;13(11):e236654.
6. Fajgenbaum DC, Kurzrock R. Siltuximab: a targeted therapy for
1Department of Internal Medicine, University of Missouri - Kansas City, Kansas City, MO; 2Department of Hematology/Oncology, Mayo Clinic in Florida, Jacksonville, FL; 3Department of Hematology/ Oncology, University of Missouri - Kansas City, Kansas City, MO and 4Department of Hematology/Oncology, Saint Luke’s Cancer Institute, Saint Luke’s Hospital of Kansas City, Kansas City, MO, USA
Correspondence: F. COSSOR - fcossor@saint-lukes.org
https://doi.org/10.3324/haematol.2023.284309
Received: October 20, 2023. Accepted: January 16, 2024.
Early view: January 25, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
FC and HM were involved in the conception and design. HM was involved in the data acquisition and drafting of the article. AO and FC did critical revision for important intellectual content. All authors have given their final approval and agree to be accountable for all aspects of the work.
Data-sharing statement
The patient’s data is available through the Saint Luke’s Hospital electronic medical records. More information can be obtained by contacting the corresponding author.
idiopathic multicentric Castleman disease. Immunotherapy. 2016;8(1):17-26.
7 van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115-2124.
8. van Rhee F, Greenway A, Stone K. Treatment of idiopathic Castleman disease. Hematol Oncol Clin North Am. 2018;32(1):89-106.
9 Bower M, Newsom-Davis T, Naresh K, et al. Clinical features and outcome in HIV-associated multicentric Castleman’s disease. J Clin Oncol. 2011;29(18):2481-2486.
10 Fajgenbaum DC. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood. 2018;132(22):2323-2330.
Immune-mediated thrombotic thrombocytopenic purpura (iTTP) is an autoimmune thrombotic microangiopathy caused by inhibitory autoantibodies against the von Willebrand factor (VWF)-cleaving protease ADAMTS13, resulting in microthrombosis, thrombocytopenia and hemolytic anemia.1 According to international guidelines, standard treatment for iTTP includes therapeutic plasma exchange (TPE), immunosuppressive therapy (glucocorticoids and/or rituximab) and caplacizumab.2 TPE is a life-saving procedure aimed to remove anti-ADAMTS13 autoantibodies and ultra-large VWF multimers and to give donor ADAMTS13; however, it carries substantial risks related to the central venous catheter (e.g., bleeding risk and infections), the exposure to donor plasma (e.g., infusion reactions) and electrolytes disturbances. Caplacizumab is a nanobody that binds to the A1 domain of the VWF blocking its interaction with the GP1b glycoprotein on the platelets’ surface, halting the formation of small vessel microthrombi and the consequent consumptive anemia and thrombocytopenia. When added to standard treatment, a lower incidence of TTP-related complications, earlier platelet count normalization, a re-
duced number of TPE procedures, and a lower incidence of exacerbations were observed.3 Given this evidence and the risks associated with TPE, the possibility of treating acute iTTP minimizing or avoiding the use of TPE is of particular interest.
Herein we report three cases of relapsed iTTP treated without TPE at our center (Table 1; Figure 1). Written informed consent was obtained from all participating subjects. Case 1: A 67-year-old man with a previous episode of acute iTTP, presented at the follow-up visit with neurological manifestations (confusion and aphasia). He was on aspirin due to previous non-TTP-related recurrent ischemic strokes. The laboratory exams showed mild anemia and thrombocytopenia and >1% schistocytes per high power field at the peripheral blood smear. The suspect of iTTP relapse was confirmed by evidence of severe ADAMTS13 activity deficiency (<3 IU/dL; normal range, 45-138 IU/dL) and high anti-ADAMTS13 autoantibodies titer (50 U/mL, negative values <12 U/mL). A brain computed tomography showed several recent ischemic lesions. Daily treatment with glucocorticoids and caplacizumab was started. TPE
and aphasia
rituximab, azathioprine, mycophenolate mofetil
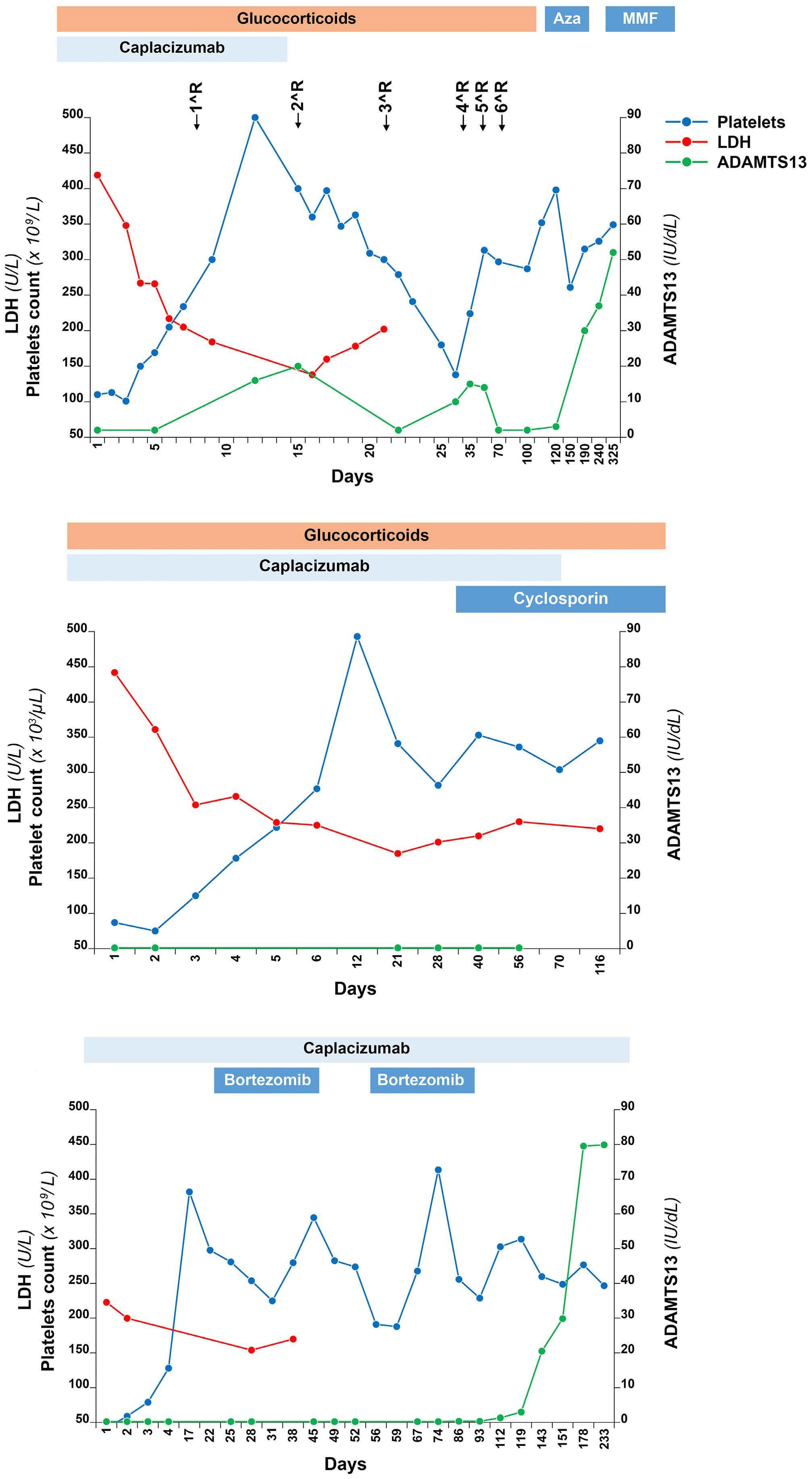
Figure. Graphical representation of the clinical and laboratory features of the reported patients. Panel (A) shows the course of the platelet count, hemoglobin and lactate dehydrogenase (LDH) levels over time in the first patient, while panel (B) shows the course in the second patient and panel (C) in the third patient. Aza: azacytidine; MMF: mycophenolate mofetil.
was omitted based on platelet count above 100x109/L. A prompt improvement of the neurologic manifestations and platelet count was observed. The clinical response, defined as sustained (at least 2 consecutive days) platelet count normalization (>150x109/L) and LDH levels <1.5 ULN, without new or progressive ischemic organ damage,4 was achieved on day 5. However, ADAMTS13 activity remained undetectable, so that on day 8 weekly 375 mg/kg rituximab was started, achieving a partial ADAMTS13 remission (20 IU/ dL) on day 15. On day 16 the patient developed an episode of melena with severe anemia due to active bleeding from arteriovenous intestinal malformations and a duodenal ulcer that were promptly treated. Caplacizumab and aspirin were stopped. After a total of six rituximab infusions and 10 weeks of steroid treatment, ADAMTS13 became again undetectable. Therefore, a third-line immunosuppressive treatment with azathioprine was started, subsequently replaced by mycophenolate mofetil because of liver toxicity. This treatment was well tolerated and induced a complete response with normalization of ADAMTS13 activity, which was maintained at the last follow-up visit (30 months after iTTP relapse).
Case 2: A 64-year-old woman with a history of relapsing iTTP and a persistent severe ADAMTS13 deficiency despite multiple immunosuppressive therapies, was diagnosed for a new relapse when new bruising appeared and the laboratory exams showed a platelet count decrease to 85x109/L. She was admitted to the hospital and immediately treated with glucocorticoids plus caplacizumab, without TPE because of the mild thrombocytopenia and the paucisymptomatic presentation. She achieved a clinical response on day 4 and was discharged continuing her treatment with prednisone and caplacizumab. Since ADAMTS13 activity remained undetectable and the titer of anti-ADAMTS13 autoantibodies high, on day 27 an immunosuppressive treatment with cyclosporin was started and caplacizumab was slowly discontinued. A sustained clinical response was obtained until the last control (4 months after the iTTP relapse).
Case 3: A 34-year-old man had the first acute episode of iTTP in 2022 when he was treated with TPE, glucocorticoids and caplacizumab. Caplacizumab was discontinued on day 37 (ADAMTS13 activity was undetectable) and 7 days later the patient had a clinical exacerbation which was treated with the same regimen plus rituximab. A second attempt in discontinuing caplacizumab was made on day 122, despite ADAMTS13 activity being still undetectable; 9 days later the patient experienced a second clinical exacerbation, therefore caplacizumab alone was promptly restarted, with normalization of the platelet count after 3 days. Since ADAMTS13 activity remained undetectable, on day 153 an off-label immunosuppressive therapy with bortezomib was started and caplacizumab was slowly discontinued. The patient showed a progressive normalization of ADAMTS13 activity and the negativization of anti-ADAMTS13 autoantibodies, and caplacizumab was safely discontinued 9
months after the onset of acute iTTP. At the last follow-up, 12 months after the iTTP event, the patient was still in clinical and complete ADAMTS-13 remission.
In the last years, few cases of acute iTTP treated without TPE have been reported. Eight different reports of iTTP in Jehovah’s Witness patients showed the use of multiple immunosuppressive therapies without TPE due to their religious beliefs. All patients had a good clinical and laboratory response, although most likely slower (median time to platelet count normalization, 14 days) than they would have had with standard treatment; ADAMTS13 response was not reported.5 Another report described a patient with relapsed iTTP treated with low-dose rituximab plus corticosteroids without TPE due to difficulties related to the COVID-19 pandemic, obtaining complete hematological response in 6 weeks.5
More recently, few cases of iTTP successfully treated with immunosuppressive therapy together with caplacizumab and without TPE have been published.6-9 One report described a patient with a first episode of iTTP successfully treated with rituximab and caplacizumab, because of the patient’s request not to be hospitalized.6 Another case of relapsing iTTP was reported in a Jehovah’s Witness patient treated with caplacizumab, steroids and rituximab: also this patient rapidly recovered with platelet count and ADAMTS13 activity normalization.7 The case of another patient with a first episode of iTTP treated with caplacizumab, steroids and rituximab due to an anaphylactic reaction to plasma after the first TPE has been reported, with sustained clinical and ADAMTS13 response.8 Finally, German authors reported a case series of seven episodes of acute iTTP treated with caplacizumab and steroids (4 with rituximab) confirming the TPE-free approach to be effective. All patients obtained a clinical remission with a median time to platelet count normalization of 3.5 days and a median time to partial ADAMTS13 remission of 25 days. The decision of omitting TPE was based on the patient’s clinical presentation, on poor venous access and on patient’s preference.9
In our three patients, considering the not severe clinical presentation and the relatively mild thrombocytopenia, we decided for a TPE-free approach. Platelet count normalization was achieved after a median of 5 days, while partial ADAMTS13 remission was obtained after 14 days in the first patient, never in the second, and after 9 months in the third. The better response observed in the first patient suggests the importance of achieving an early immunosuppression, supporting the use of caplacizumab together with an anti-CD20 monoclonal antibody upfront when using a TPE-free approach.
This case series adds evidence to previous ones on the efficacy of caplacizumab in addition to steroids in acute iTTP. According to the results of a Spanish study showing a higher efficacy in achieving the clinical response when caplacizumab is started within the first 3 days (median time to platelet count normalization 4 days [interquartile
range (IQR), 3-5] compared to 14.5 days [IQR, 10-26.5] when delayed after 7 days),10 a sudden start of caplacizumab, especially in TPE-sparing regimens, seems to be of pivotal importance.
Even though the median time to ADAMTS13 remission is 28 days11 when caplacizumab is used as frontline treatment together with steroids and TPE, also considering the absence of data on efficacy outcome in TPE-free regimens, we precautionary decided to add rituximab in the first patient and cyclosporin in the second due to the absence of an ADAMTS13 remission, and we added bortezomib in the third patient because of the dependence on caplacizumab. All three reported patients showed a protracted clinical course with the necessity of many lines of immunosuppressive treatment. Even though this may be due to their previous iTTP clinical history, we cannot exclude a detrimental role of TPE omission in the frontline treatment of acute iTTP episodes on ADAMTS13 response induction and consequently on the risk of exacerbation or relapse.
Treatment with caplacizumab was well tolerated except for the first patient who showed major gastrointestinal bleeding due to gastric ulcers and concomitant treatment with anti-platelet drugs. Even though randomized controlled trials and the most recent real-world studies are reassuring on caplacizumab safety, our case series together with previous ones12 highlights the importance of paying more attention to patients with concomitant risk factors for bleeding. In conclusion, our case series shows the efficacy of TPEfree regimens in patients with acute iTTP. Only a few case reports and retrospective series have been published on such regimens and a clinical trial on the use of caplacizumab together with immunosuppressive therapy without TPE (MAYARI study, clinicaltrials gov. Identifier: NCT05468320) is currently recruiting. Despite the good outcome of our patients, to date TPE-free regimens should not be routinely used until most robust data from clinical trials will be published. However, currently available data reassure clinicians forced not to use TPE. Moreover, our case series highlights the importance of carefully balancing the risk/benefit ratio of continuing treatment with anti-platelet agents while on caplacizumab, especially in the presence of concomitant risk factors for bleeding, in view of a comprehensive and personalized approach.
1. Page EE, Kremer Hovinga JA, Terrell DR, Vesely SK, George JN. Thrombotic thrombocytopenic purpura: diagnostic criteria, clinical features, and long-term outcomes from 1995 through 2015. Blood Adv. 2017;1(10):590-600.
2. Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for treatment of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2496-2502.
3. Scully M, Cataland SR, Peyvandi F, et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura.
Marco Capecchi,1,2 Giada Gazzola,1,3 Pasquale Agosti,4 Pasqualina De Leo,1 Ilaria Mancini,1 Barbara Ferrari,1 Juri Alessandro Giannotta,1 Andrea Artoni1 and Flora Peyvandi1,4
1Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Angelo Bianchi Bonomi Hemophilia and Thrombosis Center, Milan, Italy; 2Gruppo Ospedaliero Moncucco - Clinica Moncucco, Division of Hematology, Lugano, Switzerland; 3Università degli Studi di Milano, Department of Oncology and Onco-hematology, Milan, Italy and 4Università degli Studi di Milano, Department of Pathophysiology and Transplantation, and Fondazione Luigi Villa, Milan, Italy
Correspondence:
F. PEYVANDI - flora.peyvandi@unimi.it
https://doi.org/10.3324/haematol.2023.284438
Received: October 12, 2023.
Accepted: February 13, 2024.
Early view: February 22, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
PA, BF, AA received honoraria for participating as a speaker at educational meetings organized by Sanofi. IM received honoraria for participating as a speaker at educational meetings organized by Instrumentation Laboratory and Sanofi. FP has received honoraria for participating as a speaker in education meetings organized by Grifols, Roche and Sanofi, and she is member of scientific advisory boards of Biomarin, Roche, Sanofi, Sobi, Takeda. The other authors have no conflicts of interest to disclose.
Contributions
MC, GG, IM and JAG collected the data. MC and GG analyzed the data. MC and GG wrote the manuscript. All authors interpreted the data and carefully revised the manuscript.
Data-sharing statement
For original data, please contact the corresponding author.
N Engl J Med. 2019;380(4):335-346.
4 Falanga A, Chiaramonte M, De Silvestro G, et al. Guidelines for the diagnosis and treatment of thrombotic thrombocytopenic purpura. Italian Society of Hematology (SIE), March 2021 version; https://snlg.iss.it/?cat=6 (document in italian). Accessed on January, 4 2024.
5. Galindo-Calvillo CD, Rodríguez-Roque CS, Gómez-De León A, Tarín-Arzaga L, Gómez-Almaguer D. Treating thrombotic thrombocytopenic purpura without plasma exchange during the
COVID-19 pandemic. A case report and a brief literature review. Transfus Apher Sci. 2021;60(3):103107.
6. Sukumar S, George JN, Cataland SR. Shared decision making, thrombotic thrombocytopenic purpura, and caplacizumab. Am J Hematol. 2020;95(4):E76-E77.
7 Chander DP, Loch MM, Cataland SR, George JN. Caplacizumab therapy without plasma exchange for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2019;381(1):92-94.
8. Irani MS, Sanchez F, Friedman K. Caplacizumab for treatment of thrombotic thrombocytopenic purpura in a patient with anaphylaxis to fresh-frozen plasma. Transfusion. 2020;60(8):1666-1668.
9 Völker LA, Brinkkoetter PT, Knöbl PN, et al. Treatment of acquired thrombotic thrombocytopenic purpura without plasma
exchange in selected patients under caplacizumab. J Thromb Haemost. 2020;18(11):3061-3066.
10 Pascual Izquierdo MC, Mingot-Castellano ME, Kerguelen Fuentes AE, et al. Real-world effectiveness of caplacizumab vs standard of care in immune thrombotic thrombocytopenic purpura. Blood Adv. 2022;6(24):6219-6227.
11. Coppo P, Bubenheim M, Azoulay E, Galicier L, et al. A regimen with caplacizumab, immunosuppression, and plasma exchange prevents unfavorable outcomes in immune-mediated TTP. Blood. 2021;137(6):733-742.
12. Capecchi M, Mocellin C, Abbruzzese C, Mancini I, Prati D, Peyvandi F. Dramatic presentation of acquired thombotic thrombocytopenic purpura associated with COVID-19. Haematologica. 2020;105(10):e540.

Journal of the Ferrata Storti Foundation