Journal of the Ferrata Storti Foundation


Journal of the Ferrata Storti Foundation

Much cited Journal

Impact Factor 2022: 10.1
CiteScore 2022: 13.3
Fast review process

Submission ® 1st decision (submit to peer review or quick rejection): 3 days
Submission ® 2nd decision for peer-reviewed papers (accept, reject or make changes): 24 days
Low publication cost
The publisher is a non-profit Foundation that keeps the cost for authors as low as possible
Journal of the Ferrata - Storti Foundation
Editor-in-Chief
Jacob M. Rowe (Jerusalem)
Deputy Editors
Carlo Balduini (Pavia), Jerry Radich (Seattle)
Associate Editors
Michael Deininger (Milwaukee), Shai Izraeli (Tel Aviv), Pier Mannuccio Mannucci (Milan), Jessica Okosun (London), Pavan Reddy (Ann Arbor), David C. Rees (London), Paul G. Richardson (Boston), Francesco Rodeghiero (Vicenza), Gilles Salles (New York), Kerry Savage (Vancouver), Aaron Schimmer (Toronto), Richard F. Schlenk (Heidelberg)
Statistical Consultant
Catherine Klersy (Pavia)
AI Consultant
Jean Louis Raisaro (Lausanne)
Editorial Board
Walter Ageno (Varese), Sarit Assouline (Montreal), Andrea Bacigalupo (Roma), Taman Bakchoul (Tübingen), Pablo Bartolucci (Créteil), Katherine Borden (Montreal), Marco Cattaneo (Milan), Corey Cutler (Boston), Kate Cwynarski (London), Ahmet Dogan (New York), Mary Eapen (Milwaukee), Francesca Gay (Torino), Ajay Gopal (Seattle), Alex Herrera (Duarte), Martin Kaiser (London), Marina Konopleva (Houston), Nicolaus Kröger (Hamburg), Austin Kulasekararaj (London), Shaji Kumar (Rochester), Ann LaCasce (Boston), Matthew J. Mauer (Rochester) Neha Mehta-Shah (St. Louis), Moshe Mittelman (Tel Aviv), Alison Moskowitz (New York), Yishai Ofran (Haifa), Farhad Ravandi (Houston), John W. Semple (Lund), Liran Shlush (Toronto), Sarah K. Tasian (Philadelphia), Pieter van Vlieberghe (Ghent), Ofir Wolach (Haifa), Loic Ysebaert (Toulouse)
Managing Director
Antonio Majocchi (Pavia)
Editorial Office
Lorella Ripari (Office & Peer Review Manager), Simona Giri (Production & Marketing Manager), Paola Cariati (Graphic Designer), Giulia Carlini (Graphic Designer), Debora Moscatelli (Graphic Designer), Igor Poletti (Graphic Designer), Diana Serena Ravera (Peer Review), Laura Sterza (Account Administrator), Andrew Sturgeon (Peer Review)
Assistant Editors
Luca Arcaini (Scientific Consultant), Luk Cox (Graphic Artist), Britta Dost (English Editor), Anne Freckleton (English Editor), Rosangela Invernizzi (Scientific Consultant), Marianna Rossi (Scientific Consultant), Massimo Senna (Information Technology), Rachel Stenner (English Editor)
Haematologica (print edition, pISSN 0390-6078, eISSN 1592-8721) publishes peer-reviewed papers on all areas of experimental and clinical hematology. The journal is owned by a non-profit organization, the Ferrata Storti Foundation, and serves the scientific community following the recommendations of the World Association of Medical Editors (www. wame.org) and the International Committee of Medical Journal Editors (www.icmje.org).
Haematologica publishes Editorials, Original articles, Review articles, Perspective articles, Editorials, Guideline articles, Letters to the Editor, Case reports & Case series and Comments. Manuscripts should be prepared according to our guidelines (www.haematologica.org/information-for-authors), and the Uniform Requirements for Manuscripts Submitted to Biomedical Journals, prepared by the International Committee of Medical Journal Editors (www.icmje.org).
Manuscripts should be submitted online at http://www.haematologica.org/.
Conflict of interests. According to the International Committee of Medical Journal Editors (http://www.icmje. org/#conflicts), “Public trust in the peer review process and the credibility of published articles depend in part on how well conflict of interest is handled during writing, peer review, and editorial decision making”. The ad hoc journal’s policy is reported in detail at www.haematologica.org/content/policies.
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articles to the Ferrata Storti Foundation. No formal permission will be required to reproduce parts (tables or illustrations) of published papers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with commercial intent will require written permission and payment of royalties.
Subscription. Detailed information about subscriptions is available at www.haematologica.org. Haematologica is an open access journal and access to the online journal is free. For subscriptions to the printed issue of the journal, please contact: Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, E-mail: info@haematologica.org).
Rates of the printed edition for the year 2022 are as following:
Institutional: Euro 700
Personal: Euro 170
Advertisements. Contact the Advertising Manager, Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, e-mail: marketing@haematologica.org).
Disclaimer. Whilst every effort is made by the publishers and the editorial board to see that no inaccurate or misleading data, opinion or statement appears in this journal, they wish to make it clear that the data and opinions appearing in the articles or advertisements herein are the responsibility of the contributor or advisor concerned. Accordingly, the publisher, the editorial board and their respective employees, officers and agents accept no liability whatsoever for the consequences of any inaccurate or misleading data, opinion or statement. Whilst all due care is taken to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this journal, should only be followed in conjunction with the drug manufacturer’s own published literature.
Direttore responsabile: Prof. Carlo Balduini; Autorizzazione del Tribunale di Pavia n. 63 del 5 marzo 1955. Printing: Press Up, zona Via Cassia Km 36, 300 Zona Ind.le Settevene - 01036 Nepi (VT)
Associated with USPI, Unione Stampa Periodica Italiana. Premiato per l’alto valore culturale dal Ministero dei Beni Culturali ed Ambientali
Image taken from the Editorial by Meritxell Alberich-Jorda and Libor Macurek in this issue.
2024 Immunosuppression with posttransplant cyclophosphamide for allogeneic hematopoietic cell transplantation
M. Eapen
https://doi.org/10.3324/haematol.2024.285749
2026 Type I interferons: leukemia’s old foe in the limelight again
A. Kumar and S. Swaminathan
https://doi.org/10.3324/haematol.2024.285079
2029 Hematopoietic stem cell fate under the influence of Ser/Thr protein phosphatases
M. Alberich-Jorda and L. Macurek
https://doi.org/10.3324/haematol.2024.285040
2032 Indirect treatment comparisons: how to MAIC it right?
E. Bachy
https://doi.org/10.3324/haematol.2023.284534
2035 High-risk stays high-risk: Bruton tyrosine kinase inhibitors in B-cell malignancies
O. Al-Sawaf
https://doi.org/10.3324/haematol.2024.285029
2038 The clonal hydra: neoantigen-specific T-cell response in germ cell tumors
S. Mehra and J. Taylor
https://doi.org/10.3324/haematol.2023.284862
2041 Hemostasis and endothelial functionality: the double face of coagulation factors
C. Olgasi et al.
https://doi.org/10.3324/haematol.2022.282272
2049 Role of minimal residual disease assessment in multiple myeloma
R. Szalat et al.
https://doi.org/10.3324/haematol.2023.284662
2060 CXCL8 and its cognate receptors CXCR1/CXCR2 in primary myelofibrosis
G. Vermeersch et al.
https://doi.org/10.3324/haematol.2023.284921
Acute Lymphoblastic Leukemia
2073 B-cell precursor acute lymphoblastic leukemia elicits an interferon-α/b response in bone marrowderived mesenchymal stroma
M.W.E. Smeets et al.
https://doi.org/10.3324/haematol.2023.283494
Acute Lymphoblastic Leukemia
2085 Evaluation of the genetic basis of familial-associated early-onset hematologic cancers in an ancestral/ ethnically diverse population
Q. Feng et al.
https://doi.org/10.3324/haematol.2023.284224
Acute Myeloid Leukemia
2092 Targeting TNF/IL-17/MAPK pathway in hE2A-PBX1 leukemia: effects of OUL35, KJ-Pyr-9, and CID44216842
H. Luo et al.
https://doi.org/10.3324/haematol.2023.283647
Acute Myeloid Leukemia
2111 TLR3 agonism augments CD47 inhibition in acute myeloid leukemia
H.E. Ramsey et al.
https://doi.org/10.3324/haematol.2023.283850
Acute Myeloid Leukemia
2122 Matched unrelated donor transplantation versus haploidentical transplantation with post-transplant cyclophosphamide in children with acute myeloid leukemia: a PDWP-EBMT study
A. Ruggeri et al.
https://doi.org/10.3324/haematol.2023.284445
Cell Therapy & Immunotherapy
2131 Harnessing the cytotoxic granule exocytosis to augment the efficacy of T-cell-engaging bispecific antibody therapy
M. Casey et al.
https://doi.org/10.3324/haematol.2023.284435
Hematopoiesis
2144 Phosphatase, Mg2+/Mn2+ dependent 1B regulates the hematopoietic stem cell homeostasis via the Wnt/b-catenin signaling
Z. Lu et al.
https://doi.org/10.3324/haematol.2023.284305
Myelodysplastic Syndromes
2157 Patterns of lower risk myelodysplastic syndrome progression: factors predicting progression to highrisk myelodysplastic syndrome and acute myeloid leukemia
A.G. Jain et al.
https://doi.org/10.3324/haematol.2023.283661
Non-Hodgkin Lymphoma
2165 Phase I study of the Syk inhibitor sovleplenib in relapsed or refractory mature B-cell tumors
Y. Song et al.
https://doi.org/10.3324/haematol.2022.282401
Non-Hodgkin Lymphoma
2177 Matching-adjusted indirect comparison from the Lymphoma Epidemiology of Outcomes Consortium for Real World Evidence (LEO CReWE) study to a clinical trial of mosunetuzumab in relapsed or refractory follicular lymphoma
M.J. Maurer et al.
https://doi.org/10.3324/haematol.2023.283737
Non-Hodgkin Lymphoma
2186 Autologous stem cell transplant in fit patients with refractory or early relapsed diffuse large B-cell lymphoma that responded to salvage chemotherapy
A.M. Tun et al.
https://doi.org/10.3324/haematol.2023.284704
Non-Hodgkin Lymphoma
2196 Longitudinal, natural history study reveals the disease burden of idiopathic multicentric Castleman disease
M. Sarmiento Bustamante et al.
https://doi.org/10.3324/haematol.2023.283603
Plasma Cell Disorders
2207 Elucidation of molecular basis of osteolytic bone lesions in advanced multiple myeloma
D. Shin et al.
https://doi.org/10.3324/haematol.2023.283784
Plasma Cell Disorders
2219 Selinexor, daratumumab, bortezomib and dexamethasone for the treatment of patients with relapsed or refractory multiple myeloma: results of the phase II, non-randomized, multicenter GEMSELIBORDARA study
V. González-Calle et al.
https://doi.org/10.3324/haematol.2023.284089
Plasma Cell Disorders
2229 Carfilzomib, thalidomide, and dexamethasone are safe and effective in relapsed and/or refractory multiple myeloma: final report of the single-arm, multicenter, phase II ALLG MM018/AMN002 study
S. Ninkovic et al.
https://doi.org/10.3324/haematol.2023.284238
Plasma Cell Disorders
2239 Isatuximab-pomalidomide-dexamethasone versus pomalidomide-dexamethasone in patients with relapsed and refractory multiple myeloma: final overall survival analysis
P.G. Richardson et al.
https://doi.org/10.3324/haematol.2023.284325
Plasma Cell Disorders
2250 Prior cancer and risk of monoclonal gammopathy of undetermined significance: a population-based study in Iceland and Sweden
S. Rögnvaldsson et al.
https://doi.org/10.3324/haematol.2023.284365
Platelet Biology & its Disorders
2256 GPIbα CAAR T cells function like a Trojan horse to eliminate autoreactive B cells to treat immune thrombocytopenia
J. Zhou et al.
https://doi.org/10.3324/haematol.2023.283874
2271 Pubertal development of transfusion-dependent thalassemia patients in the era of oral chelation with deferasirox: results from the French registry
M. Veneziano Broccia et al.
https://doi.org/10.3324/haematol.2023.283610
2277 Characterization of zanubrutinib safety and tolerability profile and comparison with ibrutinib safety profile in patients with B-cell malignancies: post hoc analysis of a large clinical trial safety database
J.R. Brown et al.
https://doi.org/10.3324/haematol.2023.283846
2284 Genomic landscape of patients in a phase II study of zanubrutinib in ibrutinib- and/or acalabrutinibintolerant patients with B-cell malignancies
L. Xu et al.
https://doi.org/10.3324/haematol.2023.283861
2290 Proteogenomic profiling uncovers differential therapeutic vulnerabilities between TCF3::PBX1 and TCF3::HLF translocated B-cell acute lymphoblastic leukemia
L. Blümel et al.
https://doi.org/10.3324/haematol.2023.283928
2297 Bendamustine and rituximab as first-line treatment for symptomatic splenic marginal zone lymphoma: long-term outcome and impact of early unmeasurable minimal residual disease attainment from the BRISMA/IELSG36 phase II study
E. Iannitto et al.
https://doi.org/10.3324/haematol.2023.284109
2303 Cytomegalovirus Triplex vaccine in pediatric hematopoietic stem cell transplant patients at high risk for cytomegalovirus complications: evaluation of vaccine safety, immunogenicity and impact on viremia requiring antivirals
C. La Rosa et al.
https://doi.org/10.3324/haematol.2023.284256
2309 Efficacy of DYRK1A inhibitors in novel models of Down syndrome acute lymphoblastic leukemia
S.L. Carey-Smith et al.
https://doi.org/10.3324/haematol.2023.284271
2316 Selection of dormant cells during treatment of T-lineage lymphoblastic leukemia and CREB as a therapeutic target
D. Masic et al.
https://doi.org/10.3324/haematol.2023.284335
2321 Mycophenolate mofetil is associated with inferior overall survival in cytomegalovirus-seropositive patients with acute myeloid leukemia undergoing hematopoietic cell transplantation
R.M. Saliba et al.
https://doi.org/10.3324/haematol.2023.284501
2326 Validation of LymphGen classification on a 400-gene clinical next-generation sequencing panel in diffuse large B-cell lymphoma: real-world experience from a cancer center
M.L. Zhu et al.
https://doi.org/10.3324/haematol.2023.284565
2331 Health-related quality of life in relapsed/refractory multiple myeloma treated with melflufen and dexamethasone: analyses from the phase III OCEAN study
F.H. Schjesvold et al.
https://doi.org/10.3324/haematol.2023.284635
2337 Belantamab mafodotin: an important treatment option for vulnerable patients with triple class exposed relapsed and/or refractory multiple myeloma
M.V. Mateos et al.
https://doi.org/10.3324/haematol.2023.284694
2341 Evidence for a cytoplasmic proplatelet promoting factor that triggers platelet production
J.E. Italiano et al.
https://doi.org/10.3324/haematol.2023.284755
2346 Incidence and outcome of central nervous system relapse after hematopoietic stem cell transplantation in patients suffering from acute myeloid leukemia and acute lymphoblastic leukemia: a study from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation
S. Blum et al.
https://doi.org/10.3324/haematol.2023.284858
2351 High annualized bleeding rates in pediatric patients with inherited platelet function disorders
S. Saini et al.
https://doi.org/10.3324/haematol.2024.284996
2355 Efficacy and safety of daratumumab plus bortezomib and dexamethasone in newly diagnosed Mayo 2004 stage IIIA or IIIB light-chain amyloidosis: a prospective phase II study
K. Shen et al.
https://doi.org/10.3324/haematol.2024.285145
2359 Elderly long-term survivors in the Nordic phase II study with first-line maintenance temozolomide for primary central nervous system lymphoma: a 10-year follow-up
E.J. Pulczynski et al.
https://doi.org/10.3324/haematol.2024.285207
2364 Neoantigen-specific T-cell response after donor lymphocyte infusion associates with favorable outcome in a patient with i(12p) germ cell tumor, acute leukemia and sarcoma of the same clonal origin
V. Genoud et al.
https://doi.org/10.3324/haematol.2023.284318
2368 Phenomenon of tumor flare with talquetamab in a patient with extramedullary myeloma
M. Forsberg et al.
https://doi.org/10.3324/haematol.2023.284436
2372 Defibrotide improves COVID-19-related acute respiratory distress syndrome in myeloma patients after chimeric antigen receptor T-cell treatment without compromising virus-specific and anti-myeloma T-cell responses
M.H. Kocoglu et al.
https://doi.org/10.3324/haematol.2023.284793
2378 Erratum to: Ultra-deep mutational landscape in chronic lymphocytic leukemia uncovers dynamics of resistance to targeted therapies
D.W. Woolston et al.
https://doi.org/10.3324/haematol.2024.285151
2379 Erratum to: Genomic breakpoint-specific monitoring of measurable residual disease in pediatric non-standard risk acute myeloid leukemia
M. Maurer-Granofszky et al.
https://doi.org/10.3324/haematol.2024.285153
Mary Eapen
Medical College of Wisconsin, Milwaukee, WI, USA
E-mail: meapen@mcw.edu
https://doi.org/10.3324/haematol.2024.285749
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license

TITLE
AUTHORS
JOURNAL

Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplant cyclophosphamide.
Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ.
Blood. 2001;98(12):3456-3464. doi: 10.1182/blood.v98.12.3456.
Allogeneic hematopoietic cell transplant for hematologic malignancies can be curative but until about two decades ago was limited to those with a human leukocyte antigen (HLA)-matched sibling or an HLA-matched or mismatched adult donor. Increased access to transplantation in the early 2000s was in part brought about by transplanting bone marrow from a haploidentical relative. The landmark publication from the Johns Hopkins University School of Medicine in 2001 showed that in mouse models a combination of fludarabine, total body irradiation and cyclophosphamide is sufficient conditioning for engraftment with 10x106 major histocompatibility complex (MHC)-incompatible bone marrow cells and that fludarabine can be substituted for T-cell specific antibodies.1 Posttransplant cyclophosphamide decreased the incidence and severity of acute graft-versus-host disease (GvHD) after transplantation of MHC-incompatible bone marrow cells and this was not associated with global immunosuppression, which is consistent with cyclophosphamide’s selective toxicity to T cells activated by antigen recognition (i.e., selective toxicity to proliferating alloreactive T cells instead of non-proliferating, non-alloreactive T cells). Reducing the dose of total body irradiation to 200 cGy further minimized the risk of GvHD. The safety and efficacy of the nonmyeloablative conditioning and high-dose posttransplant cyclophosphamide at preventing graft rejection and GvHD after transplantation of bone marrow from a haploidentical relative were studied in patients with advanced hematologic malignancies at the Johns Hopkins and Fred Hutchinson Cancer Research Center (Figure 1).2 Sixty-eight consecutive patients for whom standard allogeneic or autologous transplantation was unavailable or
inappropriate were enrolled between 1999 and 2006. The trial2 concluded that posttransplant immunosuppression with high-dose cyclophosphamide, tacrolimus and mycophenolate mofetil was associated with low incidences of fatal graft rejection, severe acute GvHD and extensive chronic GvHD, and a suggestion of effective clinical immune reconstitution as evidenced by low rates of severe opportunistic infections. Recurrent disease was the major cause of treatment failure. Nevertheless, the 2-year survival was 36%.
During the same period, at the University of Minnesota John Wagner led a phase II trial of transplantation of two HLA-mismatched unrelated umbilical cord blood units after a nonmyeloablative conditioning regimen and posttransplant immunosuppression with cyclosporine and mycophenolate mofetil, which paved the way for another alternative source of donor cells. In 2006, the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) in the USA conducted two parallel phase II trials. Using identical inclusion and exclusion criteria and a common study design, the trials evaluated the effectiveness of haploidentical donor and double umbilical cord blood transplantation for adults with high-risk leukemia or lymphoma who lacked a suitable matched related donor. These multicenter trials confirmed that transplantation of both haploidentical donor grafts and double umbilical cord blood was effective and set the stage for a multicenter randomized trial to assess the relative efficacy of these two alternative sources of donor cells.3 Unlike the timely accrual to the phase II parallel trials, the randomized trial failed to accrue as planned and was closed to further entry of patients by its Data Safety Monitoring Board after having
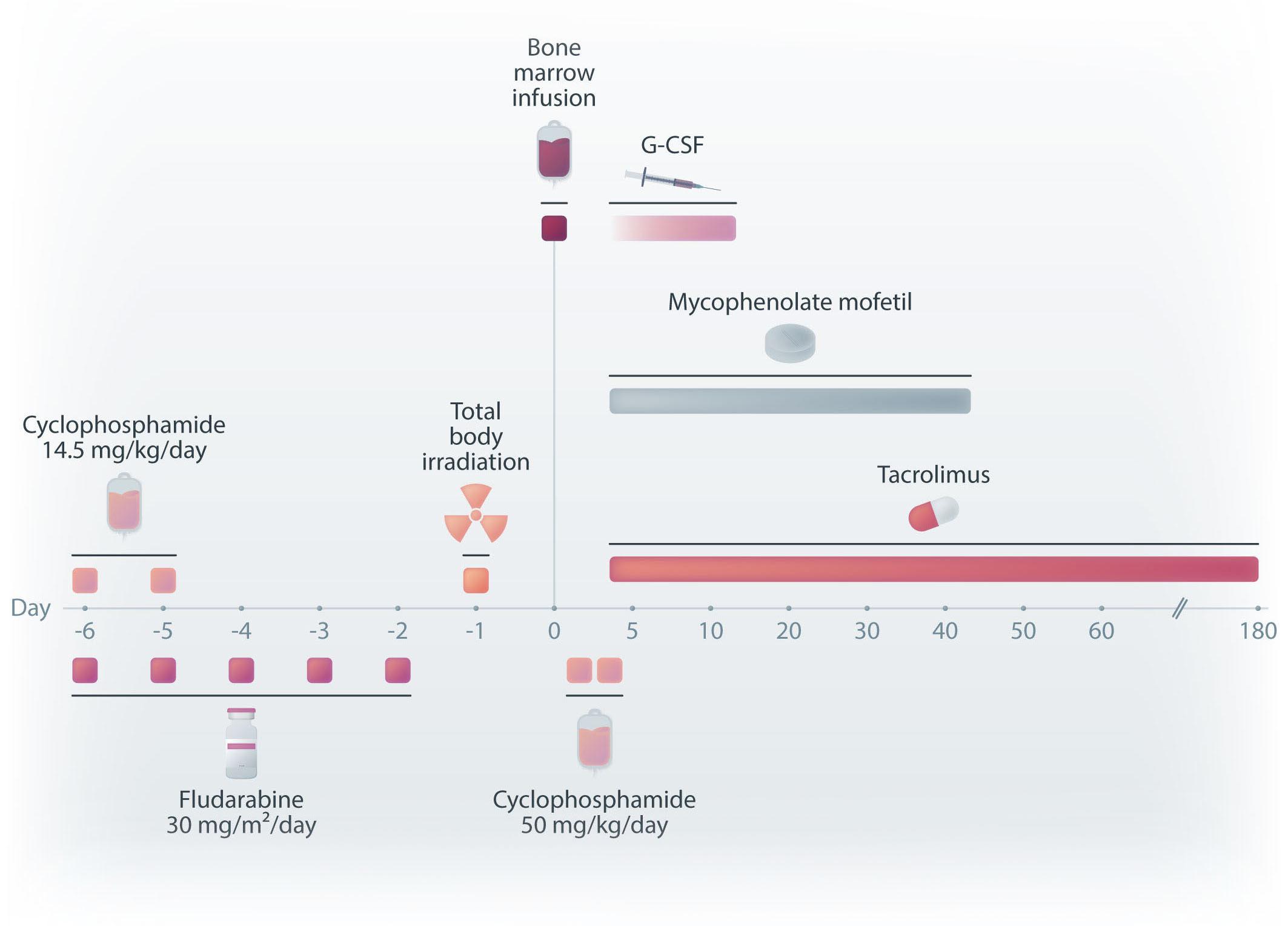
Figure 1. Nonmyeloablative haploidentical bone marrow transplant conditioning and immunosuppressive regimens. G-CSF: granulocyte colony-stimulating factor. Adapted with permission from Blood. 2021;137(3):420-428.
included 90% of the planned accrual. This trial concluded that both alternative sources of donor cells extend access to transplantation. Lower non-relapse mortality and higher 2-year survival favored haploidentical transplantation.3 Consequently, there are substantially more haploidentical transplants and very few umbilical cord blood transplants performed in the USA now.
Control of GvHD is one of the most important determinants of a successful outcome after allogeneic hematopoietic cell transplantation and, for approximately four decades, a calcineurin inhibitor with methotrexate became the standard regimen for GvHD prevention. Immunosuppression with posttransplant high-dose cyclophosphamide, tacrolimus and mycophenolate mofetil compared to tacrolimus and methotrexate was recently studied in a randomized trial for adults
1. Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood. 2001;98(12):3456-3464.
with hematologic malignancy.4 Patients underwent transplantation from an HLA-matched sibling or HLA-matched or mismatched unrelated donor. This trial concluded that the rate of GvHD-free, relapse-free survival was higher after immunosuppression with posttransplant high-dose cyclophosphamide, tacrolimus and mycophenolate mofetil and extends the application of this approach to GvHD prevention. In conclusion, immunosuppression with posttransplant, high-dose cyclophosphamide, tacrolimus and mycophenolate mofetil increased access to transplantation and has set a new standard for GvHD prevention after hematopoietic cell transplantation.
Disclosures
No conflicts of interest to disclose.
2. Luznik L, O’Donnell PV, Symons HJ, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14(6):641-650.
3. Fuchs EJ, O’Donnell PV, Eapen M, et al. Double unrelated umbilical cord blood vs. HLA-haploidentical bone marrow transplantation: BMT CTN 1101 trial. Blood. 2021;137(3):420-428.
4 Bolanos-Meade J, Hamadani M, Wu J, et al. Posttransplantation cyclophosphamide-based graft-versus-host disease prophylaxis. N Engl J Med. 2023;388(25):2338-2348.
Anil Kumar1 and Srividya Swaminathan1-3
1Department of Systems Biology, 2Department of Pediatrics and 3Department of Hematologic Malignancies, Beckman Research Institute of City of Hope, Duarte, CA, USA
Correspondence: S. Swaminathan sswaminathan@coh.org
Received: March 1, 2024. Accepted: March 14, 2024. Early view: March 21, 2024.
https://doi.org/10.3324/haematol.2024.285079
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Smeets et al 1 shed new light on the regulation of the classical immunomodulatory cytokine family, type I interferons (IFN-I), in the ETV6-RUNX1 subgroup of acute lymphoblastic leukemia (ALL). Discovered in 1957 by Isaacs and Lindenmann, IFN-I gained prominence as critical regulators of antiviral innate immune responses. IFN-I production in response to viral infection or DNA damage induces the expression of IFN-stimulated genes (ISG). These ISG mediate the anti-proliferative, pro-apoptotic, and pro-inflammatory functions of IFN-I, a process that ultimately results in the elimination of the infected and/or damaged cells either directly or by host immune cells.2 Immune responses in cancers mimic those seen during viral infections.2 Consistent with this, Dunn and colleagues made the landmark discovery that IFN-I restrict solid tumorigenesis by activating both innate and adaptive arms of anticancer host immune defenses.3 IFN-I are thus widely regarded as ‘anticancer’ cytokines. IFN-α2, a member of the IFN-I family, was the first immunotherapeutic agent to be approved by the US Food and Drug Administration in 1986 for the treatment of hairy cell leukemia.4 The remarkable response rates observed in patients with hairy cell leukemia led to the expanded use of this cytokine in the treatment of other hematopoietic malignancies, including ALL.5 In ALL, IFN-I were shown to increase relapse-free survival in patients who received an allogeneic bone marrow hematopoietic stem cell transplant.5 Despite the remarkable improvements in clinical outcomes of patients treated with IFN-I, IFN-I gradually lost their charm as ‘wonder drugs’ due to off-target toxicity associated with their administration.2 Nevertheless, their strong anticancer function could never be refuted. IFN-I were unfortunately used in clinics at a time when their mode of therapeutic action was not completely understood. However, recently identified mechanisms of action of IFN-I in human ALL by Smeets et al.1 and us6 rekindle the interest of the scientific community in harnessing the therapeutic
potential of this age-old cytokine family in treating ALL. In 2005, Einav et al.7 discovered that patients with the most common and treatable form of childhood B-cell precursor (BCP) ALL, the ETV6-RUNX1+ subtype, were approximately three times more likely to exhibit an enhanced expression of ISG in comparison to children with high-risk BCP ALL subtypes such as those with MLL, E2A-PBX1, or BCR-ABL1 rearrangements and hypodiploidy. However, they did not delve into why ETV6-RUNX1 BCP ALL were associated with interferonopathy, which cells in the leukemia microenvironment cause such interferonopathy, and which class(es) of IFN were induced.
In the current issue of Haematologica, Smeets et al.1 answer some questions arising from the publication by Einav and colleagues.7 They found that in pediatric patients with BCP ALL, bone marrow mesenchymal stromal cells (MSC) are a critical source of interferons, specifically the IFN-I α and b Among childhood BCP ALL subtypes, they found that ETV6RUNX1+ ALL most profoundly induces ISG in co-cultured bone marrow MSC derived from healthy donors and from children with ETV6-RUNX1+ and other ALL (hyperdiploid and those with DUX4, CRLF2, and EPOR translocations). They show that induction of ISG in MSC co-cultured with ETV6RUNX1+ ALL cells occurs partly by direct contact between leukemic cells and MSC via tunneling nanotubes (Figure 1). Overall, the findings of Smeets and colleagues suggest that ETV6-RUNX1 could be a direct inducer of the IFN-I pathway in normal and ALL patient-derived MSC. Mechanistically how expression of ETV6-RUNX1 in leukemic blasts triggers the IFN-I signature in surrounding non-malignant MSC remains to be delineated. Together, the studies by Einav et al. and Smeets et al. raise interest in studying these biological mechanism(s).
Smeets and colleagues also observed that IFN-I from MSC co-cultured with ETV6-RUNX1+ BCP ALL cells do not directly impact the viability of the leukemic cells or their sensitivity to chemotherapeutic agents.1 This observation
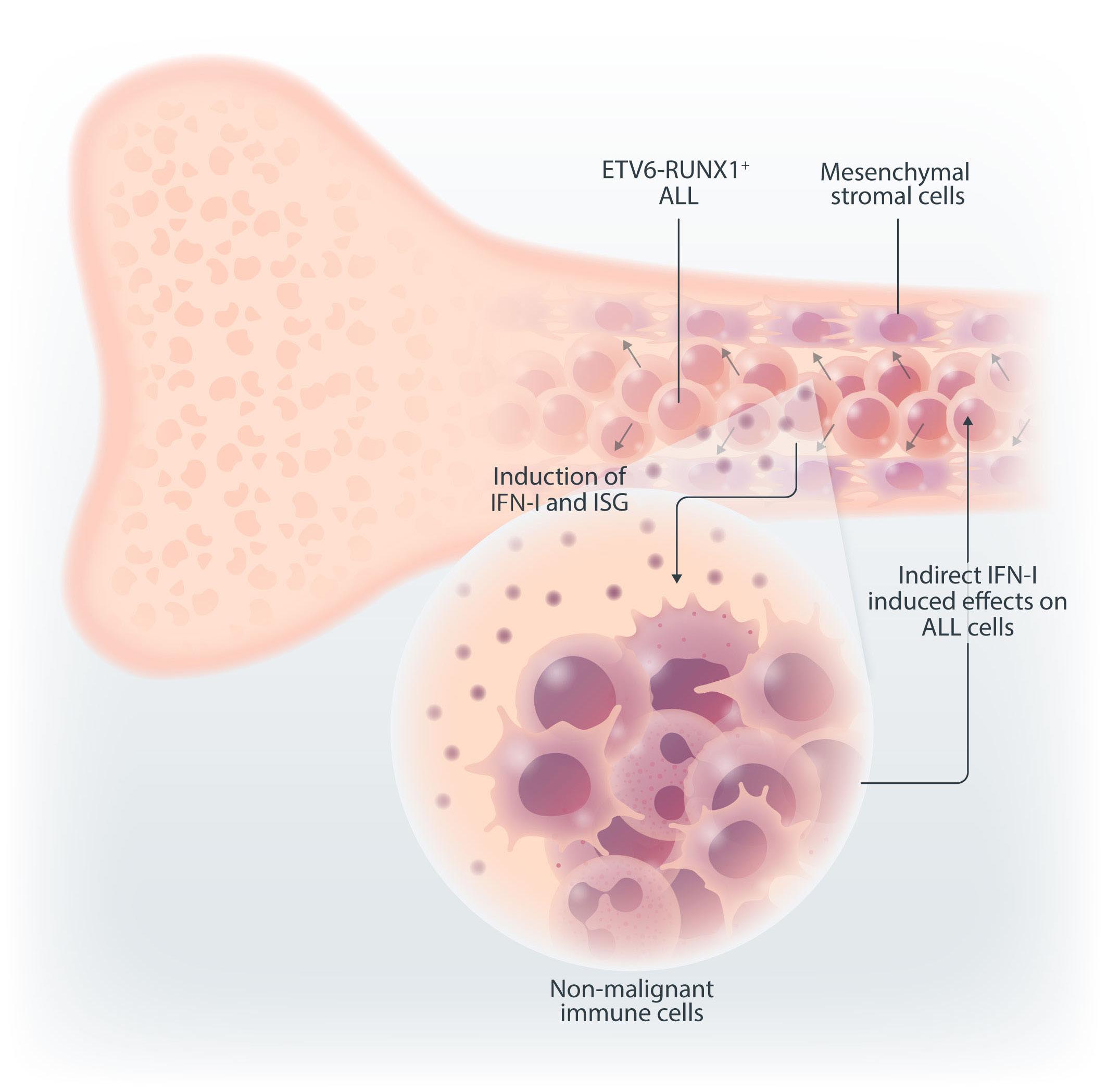
Figure 1. Type I interferons in B-cell precursor acute lymphoblastic leukemia. Smeets et al. found that acute lymphoblastic leukemia (ALL) cells, primarily the ETV6-RUNX1+ subtype, induce the paracrine expression of type I interferons (IFN-I) and IFN-I-stimulated genes (ISG) in co-cultured bone marrow mesenchymal stromal cells (MSC). IFN-I pathway induction in MSC is partially mediated via direct contact between leukemia cells and MSC. Induction of ISG and IFN-I production from MSC has an indirect effect on the ALL. This indirect effect of MSC-derived IFN-I may be potentially mediated by non-malignant host immune cells.
is consistent with Dunn et al.’s seminal finding that only cells of the hematopoietic lineage mediate the anticancer effects of IFN-I. Dunn et al. made their discovery in a solid tumor model of fibrosarcoma that is very different from leukemia in which malignancy arises in the hematopoietic cells themselves.3 Studying the role of IFN-I in BCP ALL, we showed that IFN-I mediate their anti-leukemic effects indirectly by activating host immune defenses. We found that IFN-I enhance the production and maturation of the non-malignant, innate immune cytotoxic natural killer cells in the ALL microenvironment by stimulating the production of interleukin-15, the IFN-I-induced cytokine critical for natural killer-cell homeostasis.6 We found significantly higher expression of interleukin-15 in patients with ETV6-RUNX1+ BCP ALL than in higher risk ALL subgroups, suggesting more intact IFN-I-induced immune responses in the former.6 The work by Smeets
et al. thus complements ours by demonstrating that: (i) MSC-derived IFN-I mediate their effects on ALL indirectly; and (ii) ETV6-RUNX1+ BCP ALL have a distinctly higher IFN-I-driven immune response signature in comparison to other ALL subtypes (Figure 1).
The publication by Smeets and colleagues is topical and opens additional avenues for research. The ETV6-RUNX1 subgroup of BCP ALL is unique in terms of its clinical outcome and etiology. Children with this form of ALL have one of the most favorable clinical outcomes with eventfree survival for these patients after standard therapies exceeding 90%.8 The increased IFN-I pathway signature in ETV6-RUNX1-driven BCP ALL as compared to other ALL subtypes and the heightened ability of ETV6-RUNX1+ ALL cells to induce ISG in surrounding MSC 1 could explain the favorable clinical outcomes of patients with this form of ALL. Another interesting feature of ETV6-RUNX1+ BCP ALL
is that not all individuals who acquire this translocation in utero go on to develop overt leukemia.9 The extent of IFN-I-mediated antileukemic responses by MSC may determine the risk of development of overt leukemia in individuals born with the ETV6::RUNX1 rearrangement.10 The above theories require experimental testing. Lessons learnt from the role of IFN-I in ETV6-RUNX1+ ALL will also inform the development of safer therapeutic alternatives
1. Smeets MWE, Orsel J, Stalpers F, et al. B-cell precursor acute lymphoblastic leukemia elicits an interferon α/b response in bone marrow-derived mesenchymal stroma. Haematologica. 2024;109(7):2073-2084.
2. Kumar A, Khani AT, Swaminathan S. Type I interferons: one stone to concurrently kill two birds, viral infections and cancers. Curr Res Virol Sci. 2021;2:100014.
3. Dunn GP, Bruce AT, Sheehan KC, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6(7):722-729.
4 Asmana Ningrum R. Human interferon alpha-2b: a therapeutic protein for cancer treatment. Scientifica (Cairo). 2014;2014:970315.
5. Arellano ML, Langston A, Winton E, Flowers CR, Waller EK. Treatment of relapsed acute leukemia after allogeneic transplantation: a single center experience. Biol Blood Marrow Transplant. 2007;13(1):116-123.
6. Kumar A, Khani AT, Duault C, et al. Intrinsic suppression of type
to direct IFN-I administration for treating higher risk BCP ALL subgroups.
Disclosures
No conflicts of interest to disclose.
Contributions
Both authors contributed equally.
I interferon production underlies the therapeutic efficacy of IL-15-producing natural killer cells in B-cell acute lymphoblastic leukemia. J Immunother Cancer. 2023;11(5):e006649.
7 Einav U, Tabach Y, Getz G, et al. Gene expression analysis reveals a strong signature of an interferon-induced pathway in childhood lymphoblastic leukemia as well as in breast and ovarian cancer. Oncogene. 2005;24(42):6367-6375.
8. Schrappe M, Bleckmann K, Zimmermann M, et al. Reducedintensity delayed intensification in standard-risk pediatric acute lymphoblastic leukemia defined by undetectable minimal residual disease: results of an international randomized trial (AIEOP-BFM ALL 2000). J Clin Oncol. 2018;36(3):244-253.
9 Wiemels J, Cazzaniga G, Daniotti M, et al. Prenatal origin of acute lymphoblastic leukaemia in children. Lancet. 1999;354(9189):1499-1503.
10. Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6(3):193-203.
Meritxell Alberich-Jorda and Libor Macurek
Institute of Molecular Genetics of the Czech Academy of Sciences, Videnska, Prague, Czech Republic
Correspondence: L. Macurek libor.macurek@img.cas.cz
Received: February 22, 2024. Accepted: February 27, 2024. Early view: March 7, 2024. https://doi.org/10.3324/haematol.2024.285040
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
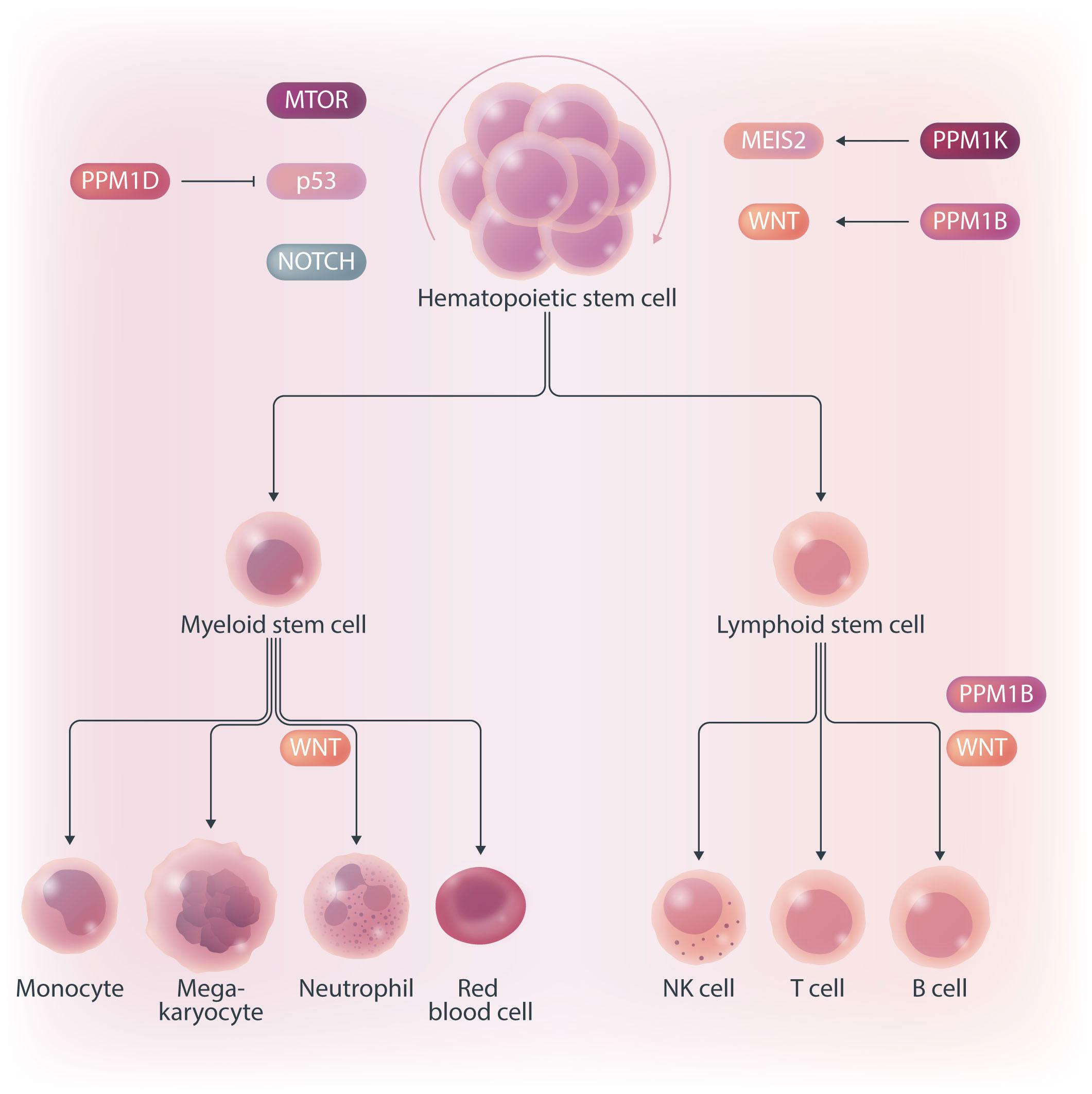
Hematopoietic stem cells (HSC) are multipotent cells capable of unlimited self-renewal and are essential for production of blood and immune cells throughout life. HSC reside in a quiescent state in the bone marrow and proliferate only after certain stimuli. Failure in wakening these quiescent cells may result in hematologic defects, and, therefore, this process is tightly regulated by multiple signaling pathways. Recent research suggests that Ser/ Thr protein phosphatases may be involved in HSC biology more than previously anticipated. In this issue, Lu and colleagues show that protein phosphatase PPM1B controls homeostasis of HSC through regulation of the Wnt/b-catenin signaling pathway. Using a transgenic Ppm1bCKO mouse model with VAV-Cre mediated conditional deletion of exon 2 of the Ppm1b gene in hematopoietic cells, they showed that PPM1B is necessary for proliferation of HSC.1 Impaired functionality of HSC in Ppm1bCKO animals was further demonstrated by limiting dilution assays and serial transplantation experiments. Data from the animal model were recapitulated in vitro using the small molecule inhibitor of PPM1B (HN2522), as well as by depletion of PPM1B by RNA interference. In addition, Ppm1bCKO mice also exhibited alterations in common lymphoid progenitors, which resulted in B-cell leukocytopenia, whereas the myeloid lineage was unaffected. Furthermore, RNAseq analysis from Lineage- Sca-1+ c-Kit+ (LSK) hematopoietic stem and progenitor cells revealed that several signaling pathways, including WNT, were dysregulated in Ppm1bCKO animals. In particular, several downstream targets of b-catenin, including Fzd1, Jun, Camk2b, Lrp5, Ccnd1 and Gpc4, were down-regulated upon the deletion of Ppm1b suggesting that the defect in HSC may be caused by suppression of WNT signaling. Indeed, LSK cells from Ppm1bCKO animals showed increased levels of the inactive form of b-catenin, which is phosphorylated at Ser33/37/Thr41. Finally, the Authors nicely demonstrated that stimulation of the WNT
pathway by BML-284 rescued the phenotypes in Ppm1bCKO mice, supporting the conclusion that PPM1B controls HSC through stimulation of the WNT pathway.
PPM1B belongs to a conserved PP2C family of Ser/Thr phosphatases that require binding of manganese/magnesium ions for their activity. They function as single subunit enzymes and their substrate specificity is influenced by internal linker regions. Interestingly, other members of this conserved phosphatase family have also been implicated in hematopoiesis. In particular, mitochondrial PPM1K was proposed to regulate branched amino acid catabolism in HSC and loss of PPM1K impaired maintenance of the HSC pool.3 In addition, loss of the nuclear PPM1D (also called WIP1) phosphatase caused mTORC-dependent expansion of the HSC compartment in Ppm1d-/- animals, whereas truncating gain-of-function mutations in PPM1D reduced self-renewal of HSC.4,5 Interestingly, the truncated PPM1D stimulates HSC survival after genotoxic stress by inhibiting the p53 pathway and can promote therapy-induced acute myeloid leukemia.5,6 Altogether, these observations point at the crucial role of Ser/Thr phosphatases in the regulation of HSC properties and their potential contribution to the development of hematologic disorders. In fact, several phosphatases have been implicated in hematologic malignancies as well as in solid tumors, and may represent attractive pharmacological targets in future clinical interventions. The development of small molecule inhibitors to protein phosphatases is challenging, and few compounds show satisfactory specificity and efficiency in cellular and animal models. For example, the selective PPM1D inhibitor GSK2830371 suppressed the growth of p53-positive cancer cells in vitro and in animal models.5-7 In the present study, Lu and colleagues used a new PPM1B inhibitor (HN252), proved its efficiency in animal models, and elegantly validated its specificity in the Ppm1bCKO animals.1,2 This tool will be invaluable for exploring the functions of PPM1B in several systems. Additionally, consid-
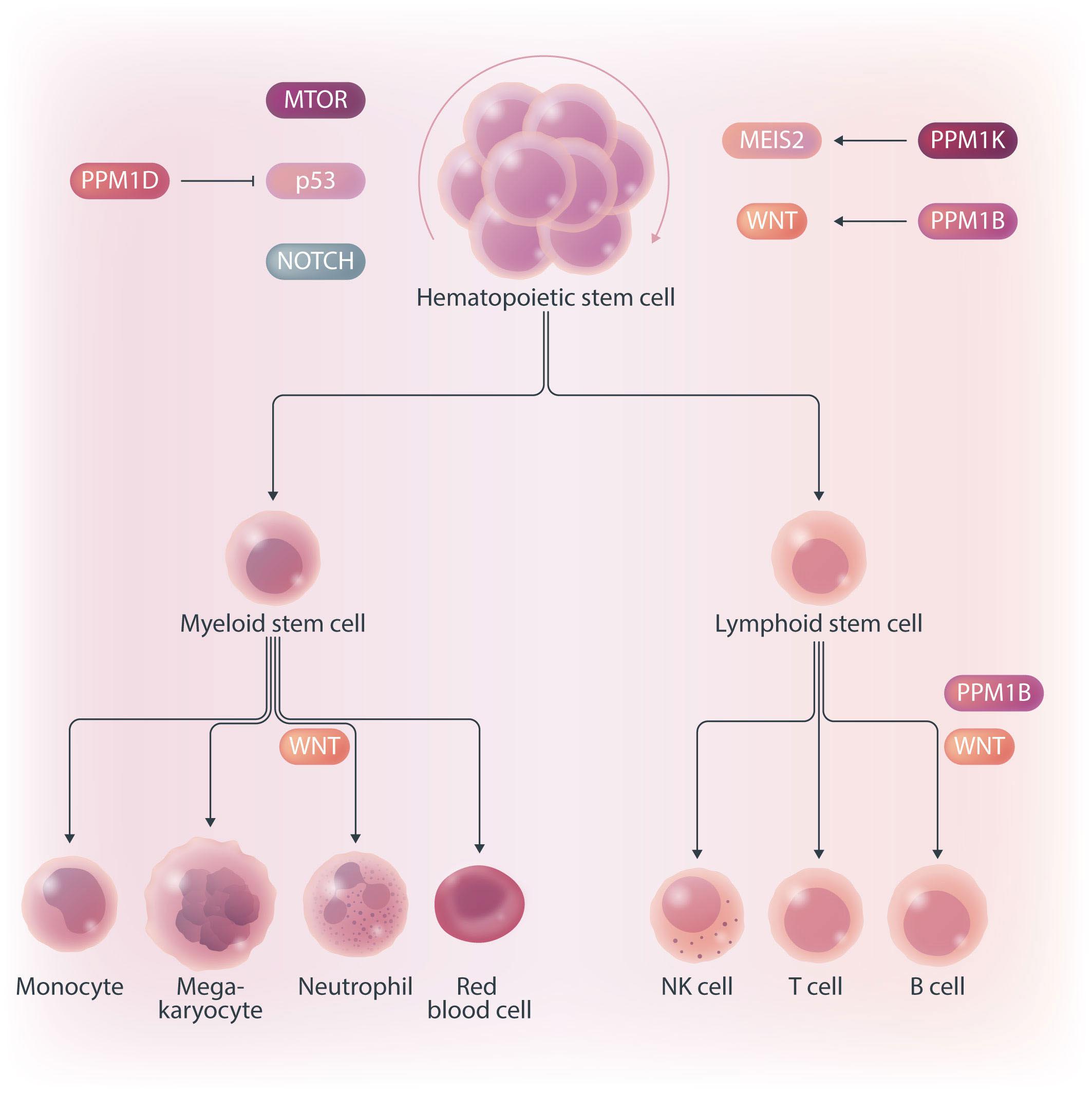
Figure 1. PP2C family protein phosphatases regulating the hematopoiesis. PPM1B and PPM1K promote self-renewal of hematopoietic stem cells (HSC) through activating WNT/b-catenin and MEIS2 pathways, respectively. In contrast, PPM1D promotes differentiation of HSC by modulating MTOR, p53 and NOTCH pathways. In addition, PPM1B regulates production of B cells but, surprisingly, is not needed in myeloid lineage.
ering the B-cell leukocytopenia observed in Ppm1bCKO mice, it might be interesting to investigate the potential of PPM1B inhibitor in suppressing B-cell leukemias. Mechanistically, Lu and colleagues point at deregulation of the Wnt/b-catenin signaling pathway as the underlying cause of the B-cell phenotype present in Ppm1bCKO mice. Nevertheless, the role of the Wnt/b-catenin signaling pathway in the hematopoietic system is controversial, and discrepancies are justified by the use of different models and approaches.8 Here, Lu and colleagues bring into the field a new player, PPM1B, which by its dephosphorylating action can promote the active form of b-catenin, and thus enhance the activity of this pathway. However, while b-catenin is critical for T-cell,9 B-cell,10 and granulocytic development,11,12 they report alterations only in the B-cell compartment. Thus, we can hypothesize that PPM1B does not act in myeloid progenitors or T cells, or that it does not regulate b-catenin phosphory-
lation uniformly in all hematopoietic cells. Ultimately, since the Wnt/b-catenin signaling pathway is regulated at multiple levels and is subjected to spatiotemporal regulations, in the future it will be interesting to investigate the impact of PPM1B on this signaling pathway also in other tissues besides the bone marrow.
In summary, Ser/Thr protein phosphatases of the PP2C family are now emerging as new regulators of hematopoiesis and potential pharmacological targets. Their possible clinical use will depend on the development and careful validation of selective small molecule inhibitors.
Disclosures
No conflicts of interest to disclose.
Contributions
LM wrote the manuscript with the input from MAJ.
1. Lu Z, Yu H, Li Y, et al. Phosphatase, Mg2+/Mn2+ dependent 1B regulates the hematopoietic stem cells homeostasis via the Wnt/ b-catenin signaling. Haematologica. 2024;109(7):2144-2156.
2. Lu Z, Xiao P, Zhou Y, et al. Identification of HN252 as a potent inhibitor of protein phosphatase PPM1B. J Cell Mol Med. 2020;24(22):13463-13471.
3. Liu X, Zhang F, Zhang Y, et al. PPM1K regulates hematopoiesis and leukemogenesis through CDC20-mediated ubiquitination of MEIS1 and p21. Cell Rep. 2018;23(5):1461-1475.
4 Chen Z, Yi W, Morita Y, et al. Wip1 deficiency impairs haematopoietic stem cell function via p53 and mTORC1 pathways. Nat Commun. 2015;6:6808.
5. Burocziova M, Danek P, Oravetzova A, Chalupova Z, AlberichJorda M, Macurek L. Ppm1d truncating mutations promote the development of genotoxic stress-induced AML. Leukemia. 2023;37(11):2209-2220.
6. Miller PG, Sperling AS, Mayerhofer C, et al. PPM1D modulates hematopoietic cell fitness and response to DNA damage and is a therapeutic target in myeloid malignancy. Blood.
2023;142(24):2079-2091.
7. Gilmartin AG, Faitg TH, Richter M, et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol. 2014;10(3):181-187.
8. Carpenter KA, Thurlow KE, Craig SEL, Grainger S. Wnt regulation of hematopoietic stem cell development and disease. Curr Top Dev Biol. 2023;153:255-279.
9. Xu Y, Banerjee D, Huelsken J, Birchmeier W, Sen JM. Deletion of beta-catenin impairs T cell development. Nat Immunol. 2003;4(12):1177-1182.
10 Ranheim EA, Kwan HCK, Reya T, Wang Y-K, Weissman IL, Francke U. Frizzled 9 knock-out mice have abnormal B-cell development. Blood. 2005;105(6):2487-2494.
11. Danek P, Kardosova M, Janeckova L, et al. b-Catenin-TCF/LEF signaling promotes steady-state and emergency granulopoiesis via G-CSF receptor upregulation. Blood. 2020;136(22):2574-2587.
12. Hérault A, Binnewies M, Leong S, et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature. 2017;544(7648):53-58.
Emmanuel Bachy
Hospices Civils de Lyon, Hematology Department, Lyon, France
As more phase II non-comparative clinical trial data are generated with new emerging treatment options, indirect cross-trial and real-life comparisons have grown exponentially. Sometimes they aim to provide anticipated information while waiting for ongoing phase III trials, while most often they try to substitute for head-to-head comparisons that will never be performed by pharma companies or academic consortia. In this issue of Haematologica, Maurer and colleagues performed a matching-adjusted indirect comparison (MAIC) between patients from the Lymphoma Epidemiology for Outcomes (LEO) Consortium for Real World Evidence (CReWE) and from a phase I/II study of mosunetuzumab, a CD20xCD3 T-cell engaging bispecific antibody, as a single agent (GO29781 – clinicaltrials.gov 02500497).1,2 Phase III randomized studies are of the highest quality standards for treatment comparison because random allocation can control both for measured confounders (e.g., age, sex, disease stage, treatment line, performance status) and potential unmeasured confounders (e.g., unquantifiable belief by the physician that a particular patient would benefit from a given drug or unmeasured socio-economic determinants). Amidst both end of the spectrum ranging from basic unadjusted or unmatched comparisons and the phase III gold standard stand various statistical approaches trying to mitigate these potential biases (Figure 1A). A MAIC is usually considered when individual patient data (IPD) are available for one group of patients while only aggregated data are available for the comparator (e.g., median or mean values, interquartile range). This is usually the case when trial sponsors (usually pharma companies) want to compare the outcome for patients treated with the product they develop with a competitor drug for which IPD are not available. This is also the case here where an academic consortium compares real-world IPD from a large cohort of patients to a given trial. Basically, a MAIC relies on selecting patients and pondering outcome according to their IPD characteristics to render them as close as possible to aggregated data available from a trial publication
Correspondence: E. Bachy emmanuel.bachy@chu-lyon.fr
Received: January 11, 2024. Accepted: February 8, 2024. Early view: February 15, 2024.
https://doi.org/10.3324/haematol.2023.284534
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
(Figure 1B).3,4 MAIC is usually considered as providing a poor confounding control as opposed to comparisons where IPD are available for both treatment groups (e.g., using propensity score-based matched comparisons or adjustments) but is usually the only option when only aggregated data are available for one treatment group. Furthermore, and like any comparison except randomized trials, MAIC cannot control for unmeasured biases that could confound the comparison of outcomes. In follicular lymphoma, a disease still considered as incurable for most patients, overall survival is now believed to extend beyond a median 20 years with a significant fraction of patients dying from non-lymphoma-related cause.5,6 However, disease outcome is highly heterogeneous with some patients experiencing poor survival despite the theoretical indolent nature of the disease.7,8 Therefore, taking into account disease heterogeneity between FL patient cohorts is critical for cross-trial and other treatment comparisons. The study conducted by Maurer and colleagues shows that after careful weighting of patient data from the LEO CReWE to match clinical characteristics with those from the GO29781 study, overall (80% vs. 73%) and complete (60% vs. 53%) responses rates (ORR and CRR) were found slightly higher in the trial cohort. Despite these higher response rates, no obvious difference was observed regarding 12-month progression-free survival (PFS) (60% vs. 58% in the LEO vs. trial cohorts, respectively). However, and as stated by the authors, response status assessment differences between trial (with frequent imaging exams) and routine practice are a major bias for PFS measurement possibly penalizing response duration in the mosunetuzumab trial. Furthermore, 40% of patients in the LEO cohort were treated as part of a clinical trial for the selected index therapy, making it quite different from standard of care (SOC) strategies outside of specific tertiary care centers. Altogether, on one hand, prognosis is likely overestimated in the LEO cohort in contrast with routine practice because of patient selection. But, on the other hand, 40% of them have probably been followed more closely to what
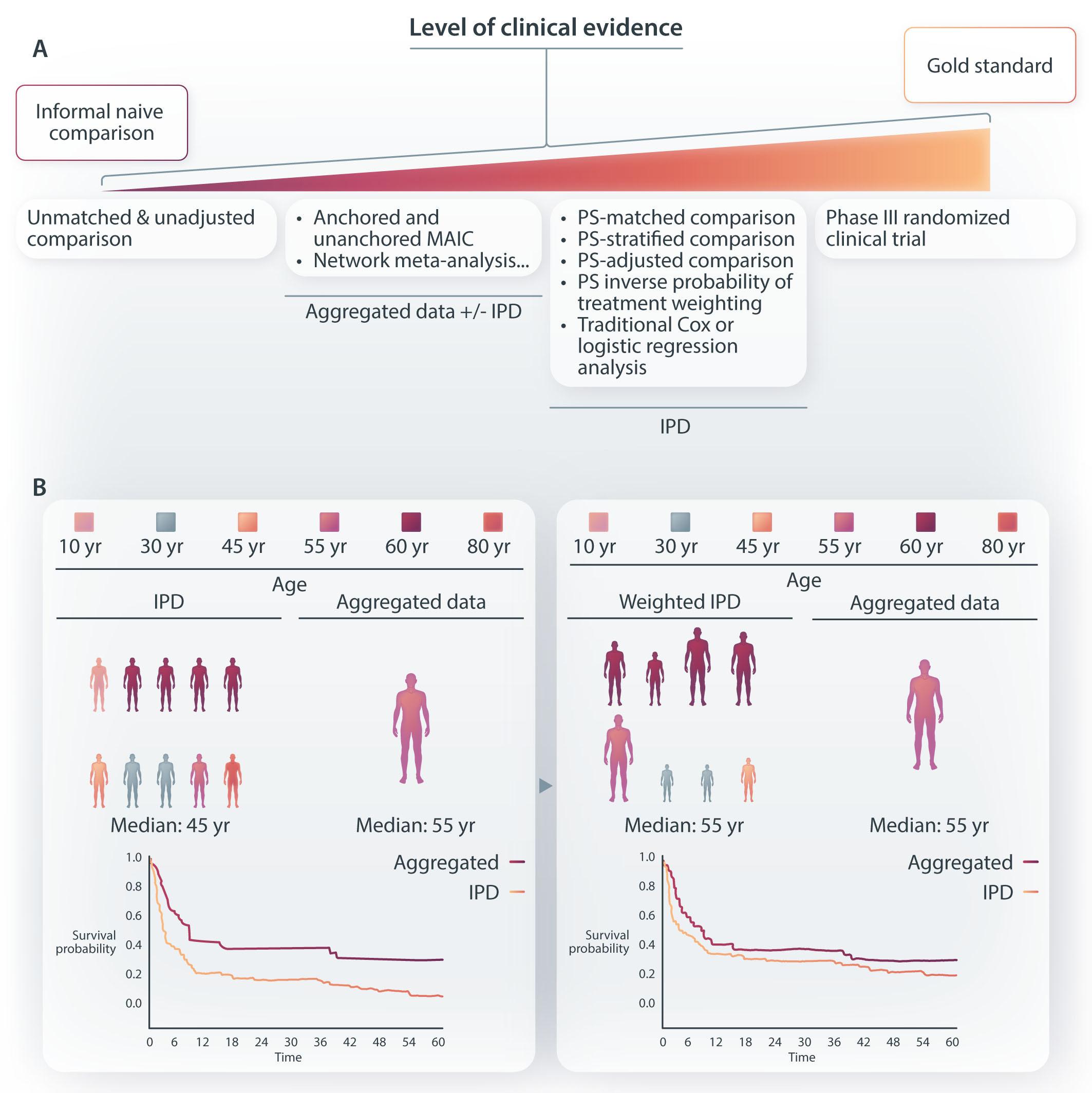
Figure 1. Principle and level of clinical evidence of matching-adjusted indirect comparison. (A) Level of evidence of various treatment comparisons from unadjusted and unmatched comparison (poorest level of evidence) to randomized treatment allocation (gold standard). The list of various statistical approaches to make patient population as comparable as possible is not exhaustive. (B) Matching-adjusted indirect comparison (MAIC) allows for the comparison between aggregated patient data (e.g., based on patient characteristics from a trial publication) and individual patient data (from another trial or a real-life cohort or any other source of individual data). Basically, by removing or pondering patient characteristics to closely match final aggregated data (e.g., patient median age depicted here), the final group of patients from the cohort with available individual patient data (IPD) is rendered as similar as possible to the cohort for which only aggregated data are available (e.g., here, similar median age). This is performed for all variables that are considered as critical confounders for treatment comparison. Finally, outcome is compared in the 2 matched populations; here, the prognosis of the IPD is depicted as better after matching (blue line), while, by definition, the survival of the aggregated data population is left unchanged after matching (red line). Depicted data and survivals are for illustration only and are not based on true or relevant values or weights. PS: propensity score; yr: year.
was performed in the GO29781 trial and more accurately than what is usually performed outside a trial setting. It is also important to notice that among those patients from the LEO cohort, 11 received either another bispecific antibody or a chimeric antigen receptor T-cell therapy and 38 another novel agent as monotherapy or in combination with an anti-CD20 monoclonal antibody. This explains, at least in part, the good outcome of patients from the LEO cohort and is critical to appreciate the small differences observed
in the MAIC according to the various scenarios tested. Beyond information about efficacy of this bi-specific antibody, many lessons are to be learned from this study for any hematologist aiming at developing a critical appraisal of MAIC conclusions as they become increasingly used to support early phase study results for therapies still under development. First, results are strongly dependent upon which scenario and constraints are applied to the model. Second, a careful analysis of which variables have
been selected (or omitted), and the reason why they have been incorporated or not, should be thoroughly examined. Third, inclusion/exclusion criteria, outcome definition, balance in the average baseline characteristics after matching and sensitivity analyses should be key in interpreting results of MAIC. Finally, extreme caution should always be applied when drawing conclusions based on such indirect comparisons and one should remember that only well-controlled randomized study can balance unmeasured confounders. Regarding
1. Maurer MJ, Casulo C, Larson MC, et al. Matching-adjusted indirect comparison from the Lymphoma Epidemiology of Outcomes Consortium for Real World Evidence (LEO CReWE) study to a clinical trial of mosunetuzumab in relapsed or refractory follicular lymphoma. Haematologica. 2024;109(7):2177-2185.
2. Budde LE, Sehn LH, Matasar M, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. 2022;23(8):1055-1065.
3. Signorovitch JE, Wu EQ, Yu AP, et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics. 2010;28(10):935-945.
4. Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative
all these critical points, this academy-concepted study will be highly informative to the reader beyond results of the comparison itself and despite all limitations of the statistical approach.
Honorarium/Ad Board from Kite, a Gilead Company, Bristol Myers Squibb, Novartis, Pfizer, Incyte, ADC Therapeutics, Roche, Takeda; fees from Kite, a Gilead Company, Bristol Myers Squibb, Novartis, Pfizer; funding from Amgen, BMS.
effectiveness research. Value Health. 2012;15(6):940-947.
5. Batlevi CL, Sha F, Alperovich A, et al. Follicular lymphoma in the modern era: survival, treatment outcomes, and identification of high-risk subgroups. Blood Cancer J. 2020;10(7):74.
6. Sarkozy C, Maurer MJ, Link BK, et al. Cause of death in follicular lymphoma in the first decade of the rituximab era: a pooled analysis of French and US cohorts. J Clin Oncol. 2019;37(2):144-152.
7 Casulo C, Byrtek M, Dawson KL, et al. Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the National LymphoCare Study. J Clin Oncol. 2015;33(23):2516-2522.
8. Maurer MJ, Bachy E, Ghesquieres H, et al. Early event status informs subsequent outcome in newly diagnosed follicular lymphoma. Am J Hematol. 2016;91(11):1096-1101.
Othman Al-Sawaf
University of Cologne, Faculty of Medicine and University Hospital Cologne, Department I of Internal Medicine, Center for Integrated Oncology Aachen Bonn Cologne Duesseldorf, Germany, Cancer Institute, University College London, UK and Francis Crick Institute, London, UK
Correspondence: O. Al-Sawaf othman.al-sawaf@uk-koeln.de
Received: February 14, 2024.
Accepted: February 29, 2024. Early view: March 7, 2024.
https://doi.org/10.3324/haematol.2024.285029 ©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Xu et al. present an overview of the genomic landscape of patients with B-cell malignancies who received the Bruton tyrosine kinase (BTK) inhibitor zanubrutinib after prior intolerance to ibrutinib and/or acalabrutinib.1 Based on a 106-gene targeted next-generation sequencing panel, they characterized baseline and relapse samples from 71 patients with relapsed/refractory B-cell malignancies. In line with previous reports on relapsed/ refractory B-cell malignancies, a high rate of mutations in genes such as TP53, SF3B1 and ATM was observed prior to the start of zanubrutinib. Patients with these alterations had a shorter progression-free survival under zanubrutinib therapy than patients without these alterations. The authors also looked at the incidence and impact of BTK and PLCG2 mutations, which are known contributors to treatment resistance, particularly in the context of chronic lymphocytic leukemia. Three patients had BTK C481S mutations at different variant allele frequencies prior to starting zanubrutinib and had relatively short responses. Of nine patients with progression while receiving treatment with zanubrutinib, four had BTK C481S mutations, mostly accompanied by PLCG2 mutations. Overall, the authors concluded that the mutational landscape and particularly its prognostic relevance in terms of response and duration of response are important in the context of BTK inhibitors. The study contained a particular cohort of patients with intolerance to ibrutinib and/or acalabrutinib, who constitute up to 40% of patients with relapsed/refractory chronic lymphocytic leukemia.2 Given the availability of several covalent and non-covalent BTK inhibitors, questions regarding their optimal sequencing are becoming increasingly relevant also for routine care. National and international treatment guidelines primarily recommend a switch to a different class of agents, e.g. Bcl-2 inhibitors, for patients with disease progression on a continuous covalent BTK inhibitor.3 However, it is currently unclear which factors need to be considered for patients who develop intolerance to
a BTK inhibitor, e.g. due to toxicity, and therefore have to discontinue treatment. It was previously demonstrated that a switch from ibrutinib or acalabrutinib to zanubrutinib in patients who had intolerance, mostly due to fatigue, hypertension, arthralgia, rash or atrial fibrillation, is feasible and can reduce or avoid recurrence of toxicity.4 A general caveat regarding such analyses, which have also been conducted for acalabrutinib,5 is the lack of a standardized definition of intolerance, which can lead to uncontrolled biases that can influence clinical decisions, such as re-imbursement considerations, availability of studies or frailty of patients. Patients with relapsed/refractory B-cell malignancies commonly present with a complex and heterogeneous mutational background, as the disease has been exposed to various selective pressures by treatments. The study by Xu et al. confirms this by demonstrating a high frequency of TP53, SF3B1, ATM and NOTCH1 mutations in up to a third of patients who had discontinued ibrutinib or acalabrutinib. Interestingly, when looking at the prognostic relevance of these alterations, apart from NOTCH1, they were associated with a significantly shorter progression-free survival under zanubrutinib, suggesting similar molecular vulnerabilities as with other covalent BTK inhibitors (Figure 1). Recent studies have shed light on the functional impact of BTK mutations in the context of BTK inhibitor resistance. The BTK C481S mutation is the most frequent BTK mutation arising in patients undergoing continuous BTK inhibition.6 Functionally, this variant is associated with an altered binding site at the kinase domain of BTK, thereby leading to reduced affinity of covalent BTK inhibitors. Multiple other BTK mutations, such as T474I or T316A, have been described, which also confer treatment resistance and reduced binding affinity. In addition, other mutations, such as L528W, affect the activity of BTK and confer a kinase-dead state, despite an unaltered binding site. Several studies have reported L528W mutations occurring particularly after zanubrutinib treatment, possibly more commonly than after
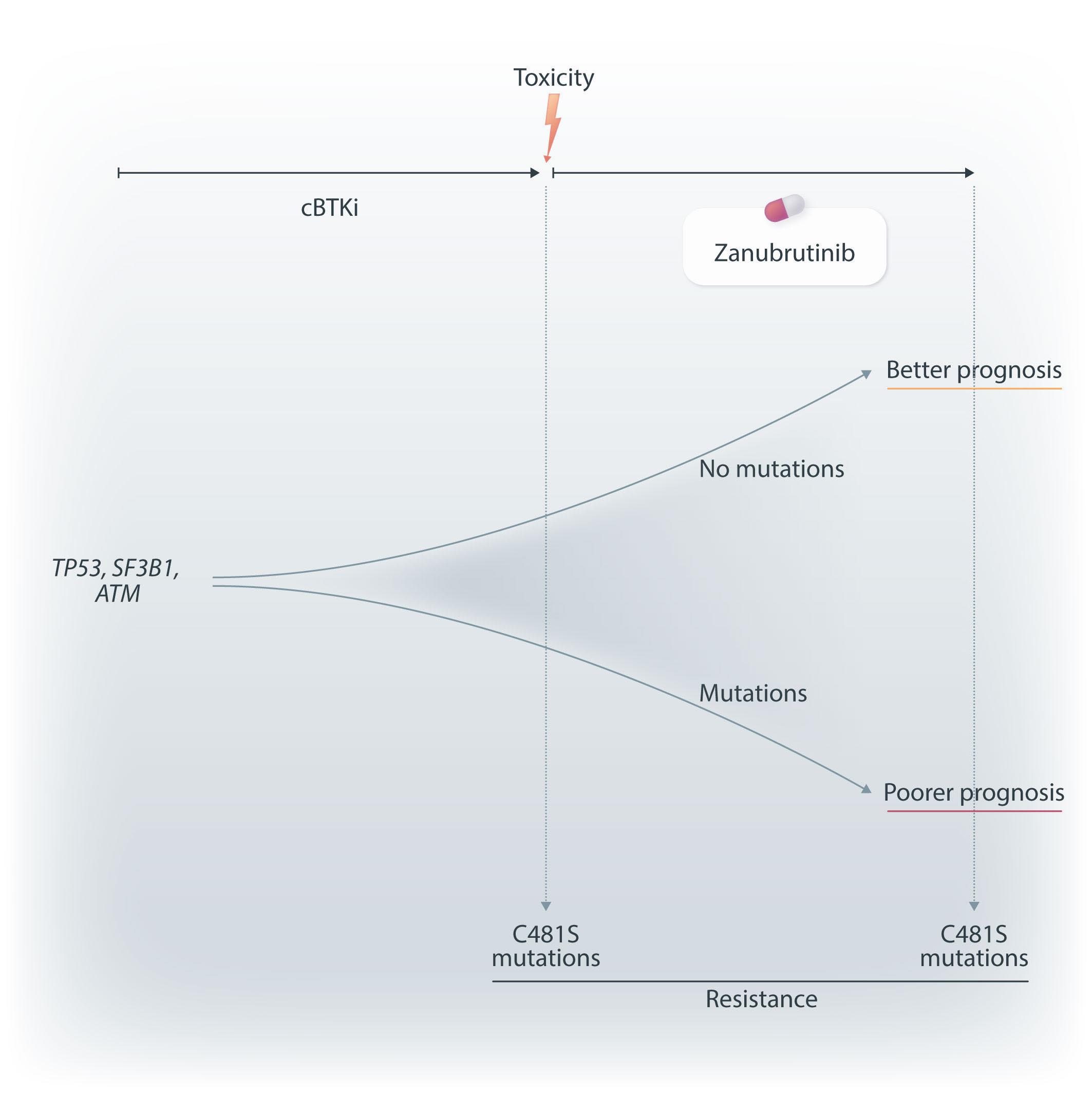
the use of ibrutinib or acalabrutinib.7 This observation is of high clinical relevance, as non-covalent BTK inhibitors, which have demonstrated efficacy also in the presence of C481S mutations, could be adversely affected in the context of L528W mutations.8 Thus, the mutational pattern can have clinical implications regarding the sequence of giving zanubrutinib before or after pirtobrutinib. In the study by Xu et al. no L528W mutations were found in patients receiving zanubrutinib after prior ibrutinib or acalabrutinib therapy. With the caveat that the number of relapse samples sequenced was limited, this study at least does not suggest a particular over-enrichment of this variant in a cohort treated with ibrutinib/acalabrutinib followed by zanubrutinib.
Several open questions remain. As the number of patients progressing on zanubrutinib was limited in this cohort and the follow-up was relatively short, statistical analyses of prognostic factors were still heavily underpowered. Fur-
1. Xu L SM, Flinn IW, et al. Genomic landscape of patients in a phase II study of zanubrutinib in ibrutinib- and/or acalabrutinib-intolerant patients with B-cell malignancies. Haematologica. 2024;109(7):2284-2289.
thermore, our understanding of the clonal evolutionary patterns for patients discontinuing a BTK inhibitor due to toxicity and then undergoing treatment with a different BTK inhibitor is still incomplete. Most insights generated in this study are derived from the patients with chronic lymphocytic leukemia, from whom material for sequencing could be obtained from peripheral blood. However, the mutational patterns in nodal entities, such as mantle cell lymphoma and marginal zone lymphoma, are still understudied. Finally, given limited sample size and the heterogeneity of reasons for discontinuing treatment, further studies are required to better understand clinical and genomic features that might be associated with toxicity and intolerance to targeted agents.
Disclosures
No conflicts of interest to disclose.
2. Muñoz J, Sarosiek S, Castillo JJ. Managing ibrutinib-intolerant patients with B-cell malignancies. Oncologist. 2023;28(4):309-318.
3. Stephens DM. NCCN guidelines update: chronic lymphocytic
leukemia/small lymphocytic lymphoma. J Natl Compr Canc Netw. 2023;21(5.5):563-566.
4. Shadman M, Flinn IW, Levy MY, et al. Zanubrutinib in patients with previously treated B-cell malignancies intolerant of previous Bruton tyrosine kinase inhibitors in the USA: a phase 2, open-label, single-arm study. Lancet Haematol. 2023;10(1):e35-e45.
5. Rogers KA, Thompson PA, Allan JN, et al. Phase II study of acalabrutinib in ibrutinib-intolerant patients with relapsed/ refractory chronic lymphocytic leukemia. Haematologica. 2021;106(9):2364-2373.
6. Wang H, Zhang W, Yang J, Zhou K. The resistance mechanisms and treatment strategies of BTK inhibitors in B-cell lymphoma. Hematol Oncol. 2021;39(5):605-615.
7 Blombery P, Thompson ER, Lew TE, et al. Enrichment of BTK Leu528Trp mutations in patients with CLL on zanubrutinib: potential for pirtobrutinib cross-resistance. Blood Adv. 2022;6(20):5589-5592.
8. Wang E, Mi X, Thompson MC, et al. Mechanisms of resistance to noncovalent Bruton’s tyrosine kinase inhibitors. N Engl J Med. 2022;386(8):735-743.
Shefali Mehra1 and Justin Taylor2
1Miller Medical School and 2Sylvester Comprehensive Cancer Center, Miami, FL, USA
Correspondence: J. Taylor
Jxt1091@miami.edu
Received: January 9, 2024.
Accepted: January 15, 2024. Early view: January 25, 2024.
https://doi.org/10.3324/haematol.2023.284862
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Germ cell tumors (GCT) occur in men aged 15-35 years and are the most prevalent malignancies within this demographic. Over the past decade, introduction of platinum-based therapeutics have proven to effectively treat GCT and improve outcomes.1 GCT, while predominantly occurring in the testes, can be found to originate in the mediastinum, accounting for 10% of all mediastinal neoplasms. 2 Mediastinal GCT pose a unique challenge to clinicians due to their propensity for the development of other malignancies. One in every 17 patients with a mediastinal GCT will develop a hematologic malignancy. 3,4 Apart from hematologic malignancies, sarcomas and carcinomas can develop in patients with mediastinal GCT. Despite the success of platinum-based regimens in testicular GCT, primary mediastinal GCT have proven to be resistant to platinum-based agents, necessitating alternative treatment strategies.5 Interestingly, it has been shown that GCT and hematologic malignancies arising in the same individual share the presence of isochromosome 12p [i(12p)].6,7 Using whole exome sequencing, Taylor and colleagues definitively showed that these malignancies arise from a progenitor that can differentiate into GCT, sarcomas, or hematologic malignancies. 8 Despite the improved understanding of their origin, the outcomes for mediastinal GCT with secondary somatic tumors is around 6 months, making discovery of novel treatments for these patients imperative. Genoud et al. report results of a case study consisting of a 22-year-old male with a mediastinal PLAP-positive mixed GCT with CD61 + large cells. 9 The patient’s bone marrow was infiltrated with CD34-, CD61+, and CD43+ cells with multilobulated nucleus and eosinophilic cytoplasm showing polyploidy, +der(3), and i(12p) on cytogenic analysis. Following a diagnosis of synchronous acute myeloid leukemia (AML) and primary mixed GCT, the patient was initially treated with three cycles of cytarabine, mitoxantrone, etoposide, and cisplatin. Intrathecal therapy con-
sisting of methotrexate, cytarabine, and methylprednisone was administered during the second cycle. Moreover, two additional cycles of etoposide and cisplatin were given. The patient achieved complete leukemic remission and metabolic activity of the mediastinal mass was shown to have decreased. Following conditioning, the patient underwent an allogeneic hematopoietic stem cell transplant (HSCT) from a mismatched unrelated donor with CD34+ harvested donor cells. Six months after HSCT, the patient was found to have 60% increased mass of residual and metabolic activity with biopsy showing a fusiform sarcoma with angiomatous differentiation. Ten months after HSCT, the patient was given two donor lymphocyte infusions (5x105 and 6x106 CD3+ cells/kg). Ultimately, complete donor chimerism was obtained and persistent complete hematologic remission was achieved such that no neoplastic disease was found even 10 years after HSCT. Somatic mutations from the three tumors were used in order to identify mutation-specific neoantigens by filtering through the following criteria: major histocompatibility complex binding affinity, strong binding affinity compared to normal proteins, and protein expression in cancer. Identification of 84 common neoepitopes that played a role in recognition of mutation-specific proteins revealed the sarcoma-specific neo-peptide IL36G16-24YPSMCKPIT, which elicited a positive response when tested in interferon-γ ELISPOT assays with the patient’s peripheral blood mononuclear cells. Furthermore, the neoantigen response to IL36G16-24YPSMCKPIT was, remarkably, detected in the patient’s peripheral blood mononuclear cells 7 years postHSCT. Tumor exon sequencing revealed 48 somatic mutations detected in the GCT, 17 in the leukemia, and 331 in the sarcoma with phylogenic reconstruction revealing 13 shared events between the three malignancies confirming a common clonal origin. A TP53 driver splice site mutation, chromosome 17 splice site mutation involving CDK12, and five loss of heterozygosity events including PTEN loss of
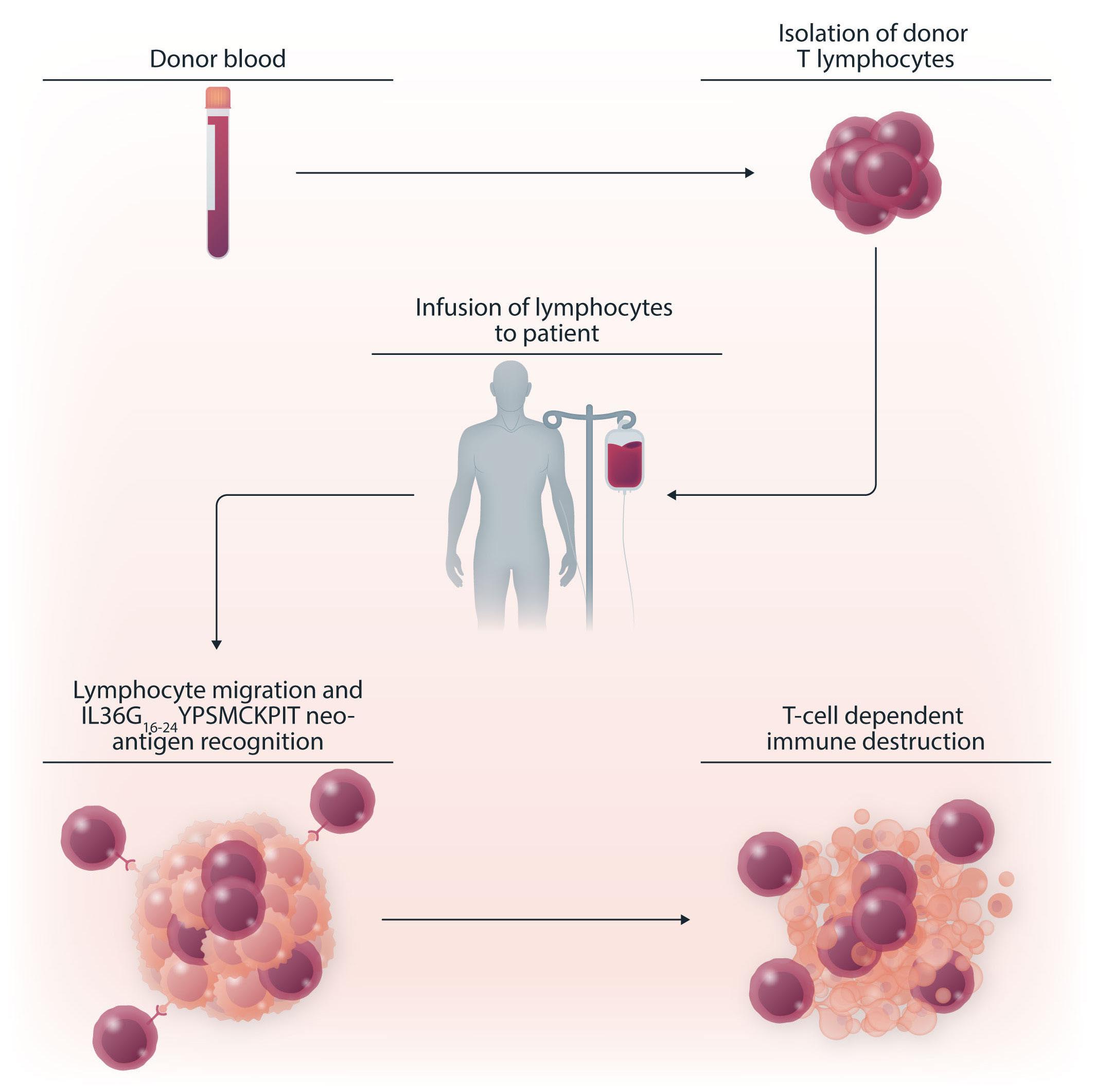
germ cell tumors.
heterozygosity were all seen amongst the GCT, hematologic malignancy, and sarcoma. Like the multi-headed hydra monster from Greek mythology, mediastinal GCT with secondary somatic tumors tend to come back stronger when the heads are chopped off. However, the key to killing the hydra, and possibly to overcoming this often fatal malignancy, is to strike a blow to the common part (the body, or in this case, shared neoantigen).
Despite Genoud et al. successfully demonstrating the potential of donor T-lymphocyte infusions in this case study, there are some limitations to the present findings. Firstly, the study’s reliance on a single patient and lack of a comparative group limits its ability to establish broad conclusions or generalize findings to a larger population. Currently, high-dose chemotherapy and autologous SCT have been used to treat relapsed GCT.10 However, such a treatment regimen has been shown to have patients recurrently relapsing with hematologic disease, all of which have demonstrated common mutations.8 The findings in this
study argue for the role of allogeneic SCT and also open the doors to further exploration of novel treatments. For example, with the identification of the sarcoma-specific neo-peptide IL36G16-24YPSMCKPIT, there is potential for the development of chimeric antigen receptor (CAR) T-cell therapy which may prove to have efficacy against development of malignancies in patients with GCT. Additionally, a concern with usage of donor lymphocyte infusions is that patients are at high risk for developing graft-versus-host disease, which has further potential to evolve into chronic disease. Development of alternative therapies that can elicit a neoantigen-specific T-cell response, such as CAR T-cell therapy, which have predominantly acute adverse events, may provide clinicians and patients with better alternatives with regards to therapeutic options. In conclusion, the study by Genoud et al. provides hope of successful treatment options for patients with mediastinal GCT refractory to general therapy by using neoantigen-specific T-cell response following a donor lym-
phocyte infusion post allogeneic HSCT. The authors have demonstrated a favorable outcome in a patient 10 years after HSCT which if shown in a larger number of patients, could ultimately help patients with achieving persistent and complete remission from mediastinal GCT and secondary somatic malignancies.
1. Cheng L, Albers P, Berney DM, et al. Testicular cancer. Nat Rev Dis Primers. 2018;4(1):29.
2. Dulmet EM, Macchiarini P, Suc B, Verley JM. Germ cell tumors of the mediastinum. A 30-year experience. Cancer. 1993;72(6):1894-1901.
3. DeMent SH, Eggleston JC, Spivak JL. Association between mediastinal germ cell tumors and hematologic malignancies. Report of two cases and review of the literature. Am J Surg Pathol. 1985;9(1):23-30.
4 Hartmann JrT, Nichols CR, Droz J-P, et al. Hematologic disorders associated with primary mediastinal nonseminomatous germ cell tumors. J Natl Cancer Inst. 2000;92(1):54-61.
5. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol. 1997;15(2):594-603.
6. Oshrine BR, Olsen MN, Heneghan M, et al. Acquired
No conflicts of interest to disclose.
Contributions
SM and JT conceived, wrote and edited the manuscript.
isochromosome 12p, somatic TP53 and PTEN mutations, and a germline ATM variant in an adolescent male with concurrent acute megakaryoblastic leukemia and mediastinal germ cell tumor. Cancer Genet. 2014;207(4):153-159.
7. Lu C, Riedell P, Miller CA, et al. A common founding clone with TP53 and PTEN mutations gives rise to a concurrent germ cell tumor and acute megakaryoblastic leukemia. Cold Spring Harb Mol Case Stud. 2016;2(1):a000687.
8. Taylor J, Donoghue MTA, Ho C, et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. J Clin Invest. 2020;130(12):6668-6676.
9 Genoud V, Dutoit V, Tran Thang N-N, et al. Neoantigen-specific T-cell response after donor lymphocyte infusion associates with favorable outcome in a patient with i(12p) germ cell tumor, acute leukemia and sarcoma of the same clonal origin. Haematologica. 2024;109(7):2364-2367.
10 Zhao GQ, Dowell JE. Hematologic malignancies associated with germ cell tumors. Expert Rev Hematol. 2012;5(4):427-437.
Cristina Olgasi,1* Simone Assanelli,2* Alessia Cucci2* and Antonia Follenzi2,3
1Department of Translational Medicine, Università del Piemonte Orientale, Novara; 2Department of Health Sciences, Università del Piemonte Orientale, Novara and 3Dipartimento Attività Integrate Ricerca Innovazione, Azienda Ospedaliero-Universitaria SS. Antonio e Biagio e C. Arrigo, Alessandria, Italy
*CO, SA and AC contributed equally as first authors.
Correspondence: A. Follenzi antonia.follenzi@med.uniupo.it
Received: October 2, 2023. Accepted: February 15, 2024. Early view: February 29, 2024.
https://doi.org/10.3324/haematol.2022.282272
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
Hemostasis is a sophisticated sequence of events aimed at repairing vessel injury. This process occurs in combination with angiogenesis, which leads to new blood vessel formation, helping in wound repair and facilitating tissue healing. The fine mechanisms that regulate hemostasis and angiogenesis are well described, but for a long time, coagulation factors (CF) have been considered merely players in the coagulation cascade. However, evidence from several experiments highlights the crucial functions of these CF in regulating endothelial functionality, especially in the angiogenic process. Some of these CF (e.g., thrombin and tissue factor) have been widely investigated and have been described as triggering intracellular signaling related to endothelial cell (EC) functionality. For others (e.g., factor VIII and thrombomodulin), potential receptors and molecular mechanisms have not been fully elucidated but some data show their potential to induce EC response. This review focuses on the emerging roles of selected CF in regulating EC functions, highlighting in particular their ability to activate signaling pathways involved in angiogenesis, migration, proliferation and endothelial barrier stability.
Coagulation factors (CF) are a well-known class of proteins essential for hemostasis. They can be divided into proand anti-CF according to their function in promoting or arresting coagulation, respectively. Pro-CF work together in a cascading sequence, initiating a series of enzymatic reactions that lead to the formation of a blood clot; this process is counterbalanced by anti-CF for a correct homeostasis. CF participate in the regulation of the clotting process by interacting with platelets, vessel walls, and other proteins to form a stable clot, thus preventing excessive bleeding when blood vessels are injured.1 Beyond hemostasis, angiogenesis is a physiological mechanism involved in the repair of vessel injury. The vascular sprouting and the new blood vessel formation are required for wound closure and co-operate with the hemostatic system maintaining blood flow and regulating platelet adherence and fibrin deposition. During vascular injury, initial vessel constriction occurs to control blood flow and to reduce hemorrhage, followed by sub-endothelial matrix exposure where platelets can adhere.2 Vascular endothelial
cells are first required to bind and anchor the clot and then, from the clot margins, they invade the fibrin structure to form a new vessel wall. Many proteins released by endothelial cells (EC) and present in the blood are required to finely control this process. Indeed, by adhering to EC, platelets regulate angiogenesis releasing pro-angiogenic molecules, like vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), epidermal growth factor (EGF), angiopoietin-1 (Ang1), insulin-like growth factor-1 and -2 (IGF-1 and IGF-2), platelet-derived growth factor (PDGF), and sphingosine 1-phosphate. On the other hand, platelets can also release proteins that inhibit vessel formation, such as endostatin, thrombospondin-1 (TSP-1), plasminogen activator inhibitor-1 (PAI-1), and angiostatin.3 Recent evidence supports the notion that angiogenesis and broader EC functions can be influenced by some specific CF. These factors have been shown to have a variety of roles, encompassing both pro- and anti-angiogenic features, and influencing EC permeability. Their multifaceted action extends beyond simple hemostatic roles, contributing to the intricate modulation of angiogenic processes and the regulation of EC barrier function.2
Thus, studying the crossroads between hemostasis and angiogenesis would promote better understanding of vascular dynamics. This review aims to explore this intricate interplay, deciphering the dual facets of some CF, not merely as hemostatic agents, but also as regulators of EC functionality.
Thrombin
Thrombin, a multifaceted enzyme pivotal in hemostasis, plays a critical role in forming clots by cleaving fibrinogen into fibrin, generating the structural backbone of clots that entrap platelets and blood cells to arrest blood flow from injured vessels. It amplifies the coagulation cascade by activating the platelets, further helping clot formation at injury sites. Its influence extends beyond coagulation, triggering inflammation and specific signaling pathways through G protein-coupled receptors (GPCR), particularly protease-activated receptor 1 (PAR1), across various cell types.1
Interestingly, thrombin has been demonstrated to impact on EC in several different ways, including increasing their permeability. Indeed, thrombin disrupts the endothelial barrier, influencing the organization and disassembly of cell-cell adhesion proteins.4 By binding to GPCR, thrombin decreases cyclic adenosine 3′,5′-monophosphate (cAMP) and increases Ca2+ levels through specific secondary mediators, resulting in endothelial cytoskeleton rearrangement and regulation of Ras homologous (Rho) family GTPases, ultimately leading to increased permeability.5 Recent phospho-proteomic analyses of thrombin-stimulated EC unveiled a novel non-canonical thrombin-mediated pathway through p38 mitogen-activated protein (MAP) kinase, promoting EC barrier disruption.6 Furthermore, corroborating this hypothesis, some studies indicate that thrombin induces capillary tube regression akin to other pro-inflammatory agents and is responsible for endothelial dysfunction while the inhibition of its activity partially restores physiological EC homeostasis.7
Consequently, extensive research has been carried out to probe into the angiogenic role of thrombin and its impact on tumor development. Notably, thrombin elevates the expression of various angiogenic factors like angiopoietin-2, growth-regulated oncogene (GRO-α) and VEGF through c-FOS transcriptional regulation in in vitro EC culture.8 It has also been described as increasing cancer invasion enhancing the expression of matrix metalloproteinase-9 and integrin beta1 (b1) on the cell surface9 and promoting vasculogenic mimicry, a process which transforms tumor cells into EC, through PAR1 and nuclear factor kappaB
(NF-κB) signaling.10 Its angiogenic potential is also evident in rat brains, where it enhances new vessel formation after intracranial hemorrhage, and its inhibition preserves blood-brain barrier integrity.11 In agreement with this, a role in murine embryonic vascular development has been shown for PAR1, and its inhibition results in reduced angiogenesis and attenuated permeability in in vivo models.12,13 Interestingly, thrombin has been observed to control EC functions not only through PAR, but also directly binding integrin alpha-v-beta3 (αvb3) and promoting EC attachment, migration and survival.14 The binding of thrombin with integrins should not be surprising, considering the pivotal role of these receptors in finely regulating EC functions. However, the role of thrombin on the in vitro formation of vascular tubule networks remains a topic of debate, contingent upon the quantity of thrombin employed for EC stimulation: high concentrations enhance EC permeability through great PAR1 activation, while lower doses exhibit a protective effect, mainly cleaving PAR3.15 Additionally, thrombin has been associated with both venoconstriction and arterial vasodilation, highlighting the complexity of its role in endothelium biology.16
Current data suggest that thrombin induces unique but conflicting responses within the human vasculature (Table 1), emphasizing the need for new studies to confirm its action on EC.
Factor VIII and von Willebrand factor
Factor VIII (FVIII) and its partner, von Willebrand factor (vWF), engage in a tightly co-ordinated interplay within the coagulation cascade, forming a complex that is pivotal for hemostasis. vWF acts as a carrier protein for FVIII, safeguarding it from premature degradation in the bloodstream. Upon vascular injury, vWF adheres to exposed collagen at the injury site, facilitating platelet adhesion and aggregation. This localization of vWF-bound FVIII primes the coagulation process, enabling FVIII to interact with activated platelets and initiate the coagulation cascade, increasing the catalytic activity of factor IX (FIX) and, thus, activating factor X (FX).17
Given the primary production of FVIII by EC,18 recent studies have been investigating the link between FVIII and EC functionality. Specifically, FVIII has been shown to induce transcriptional and functional changes in EC leading to a decrease in in vitro adherence and increased permeability, with paxillin as main mediator of these changes.19 An altered and uncontrolled joint vascular remodeling has also been shown in FVIII-deficient mice after induction of hemarthrosis, suggesting a non-physiological angiogenic mechanism which could be a contributing cause of prolonged and repetitive bleedings.20 Indeed, hemophilia A (HA) patients show reduced flow-mediated dilation (FMD) and hyperemic velocity time integral (VTI) compared to healthy controls,21 pointing to alterations of both macrovascular and microvascular endothelial functions for
Table 1. Summary of endothelial cell functions regulated by selected pro-coagulation factors.
Pro-CF Study
In vitro or in vivo models
Rabiet et al.4 HUVEC and EA.hy926
Hirano et al.5 Porcine aortic EC
Lin et al.6 EA.hy926
Koller et al.7 HUVEC and mice
Catar et al.8 HMEC-1
Radjabi et al.9 U2-OS osteosarcoma cells
Zhao et al.10 A549 cells and Lewis cells
Thrombin
Factor VIII
Von Willebrand factor
Zhou et al.11 Rat
Griffin et al.12 PAR1-deficient mice
Li et al.13 BMX-KO mice
Tsopanoglou et al.14 HUVEC
Zolotoff et al.15 HBEC-5i EC
Guðmundsdóttir et al.16 Human subjects
Cadé et al.19 HUVEC
Bhat et al.20 FVIII-deficient mice
Sun et al.21 HA patients
Manon-Jensen et al.22 HA patients
Starke et al.24 HUVEC and healthy and vWD BOEC
Randi et al.25 vWF-deficient mice
Xu et al.26 vWF-deficient and anti-vWF antibody-treated mice
de Vries et al.27 vWF-deficient mice
Ishihara et al.28 vWF-deficient mice and HUVEC
Giannarelli et al.33 HUVEC and human aortic EC
Van Den Berg et al.34 ECRF cell line and HUVEC
Tissue factor
Kocatürk et al.35 MCF-7
Sluka et al.36 asTF knock-in mice
Regulated EC function Identified receptor
Permeability -
Permeability -
Permeability PAR1
Capillary tube regression -
Angiogenesis PAR1
Invasion PAR1
Vasculogenic mimicry PAR1
Angiogenesis -
High embryonic lethality PAR1
Permeability PAR1
Attachment, migration and survival Integrin αvb3
Alteration of BBB depending on the dose PAR1 PAR3
Vasoconstriction and vasodilation PAR1
Permeability
Angiogenesis
FMD and VTI
ECM production
Proliferation, migration and angiogenesis
Angiogenesis
Angiogenesis -
Angiogenesis -
Angiogenesis -
Angiogenesis Integrins
Angiogenesis and migration
Proliferation
Integrin α6b1
Integrin αVb3
Integrin b1
Embryonic lethality due to vascular instabilityCarmeliet et al.37 flTF-deficient mice
Zhu et al.38 pCMVEC
Embryonic lethality due to vascular instability -
Angiogenesis PAR2
Pro-CF: pro-coagulation factor; HUVEC: human umbilical vein endothelial cells; EA.hy926: human umbilical vein cell line; EC: endothelial cells; HMEC-1: human microvascular endothelial cells; U2-OS: osteosarcoma cell line; A549: adenocarcinomic human alveolar basal epithelial cells; Lewis cells: non-small-cell lung cancer (NSCLC) of mice; BMX: bone marrow kinase on the X chromosome; HBEC-5i: cerebral microvascular endothelial cell line; ECRF cell line: immortalized vascular endothelial cells (EC); MCF-7: human breast cancer cell line; pCMVEC: porcine cerebral microvascular endothelial cells; asTF: alternative splicing tissue factor; flTF: full length TF; PAR: protease-activated receptor; BBB: blood barrier brain; HA: hemophilia A; FMD: flow-mediated dilation; VTI: velocity time integral; ECM: extracellular matrix; BOEC: blood outgrowth endothelial cell; vWD: von Willebrand disease.
which no clear culprit has yet been identified; we could speculate that the absence of FVIII itself might induce these impairments. This hypothesis is further sustained by the fact that treatment of HA patients with FVIII results in a reduced extracellular matrix dysfunction,22 likely responsible for the increased vessel permeability in these individuals. The potential role of FVIII in EC hemostasis needs to be further characterized and the mechanism(s) triggered by FVIII should be elucidated. Many receptors control the bioavailability of FVIII, but none of them have been described as transducing a signaling related to EC functionality after FVIII binding. There is some evidence to show that these receptors are involved in the regulation of
EC; in particular, low-density lipoprotein receptor-related protein 1 (LRP-1) has been demonstrated to have a role in vascular permeability and angiogenesis, but its expression in EC is still controversial.23
Despite playing a significant role in blood coagulation, vWF is typically not classified as a CF in the traditional sense. However, it not only has a pivotal function during coagulation and hemostasis, but it has also been described to have several direct effects on EC stability. Indeed, vWF has been shown to negatively control vascular formation, decreasing EC migration, proliferation, and angiogenesis.24 Moreover, mice lacking vWF display an elevated vessel formation and a large vascular network in the ear.25 These data suggest
that vWF could be an anti-angiogenic factor and this is supported by the demonstration that hypoxia induced an increased vessel formation in the brain microvasculature of both vWF-/- and anti-vWF antibody-treated mice.26 In contrast, another study showed that vWF-/- mice have reduced angiogenesis after ischemia27 and, in line with these results, vWF was found to play a pro-angiogenic function, binding and recruiting several growth factors thanks to its heparin-binding domain.28 Although it is well-known that vWF binds integrin αvb3 on the EC surface, little is understood about how it regulates vascular stability through this receptor,24 and the downstream mediators activated by vWF in EC have not yet been clarified, with the exception of MAP kinase p38, found activated in EC after vWF treatment, and resulting in an increase in mesenchymal cell adhesion to EC.29
The available data on FVIII controlling EC functions are still not complete, and vWF effects on EC appear, in some cases, controversial, suggesting that it may display both pro- and anti-angiogenic roles (Table 1). Thus, further investigations are needed to elucidate their role in EC homeostasis.
Following damage to the blood vessel, tissue factor (TF) joins with activated factor VII (FVII) in inducing the activation of FX, leading to fibrin deposition. TF is mainly expressed by platelets, neutrophils, eosinophils, fibroblasts, pericytes, keratinocytes, while monocytes increase its expression after stimulation with lipopolysaccharide (LPS) through activator protein 1 (AP-1) and NF-κB activation.30 This protein has two isoforms: one is full-length (fl-TF), bound to the cell membrane, and the other one is the soluble alternatively spliced (as-TF) isoform. Both isoforms have been demonstrated to play major roles beyond coagulation; specifically, they are involved in the regulation of angiogenesis, tumor growth, metastasis, and inflammation.31 Indeed, most of the present data suggest that TF is a pro-metastatic and angiogenic factor,32 and that the main isoform involved in the angiogenic process is the as-TF, which can exert its action through the hypoxia-inducible factor-1 (HIF1)/VEGF pathway.33 Moreover, as-TF has been shown to increase tubulogenesis, binding integrin α6b1 and activating MAP kinase p42/p44, while it enhances EC migration through integrin αvb3 and MAP kinase p38.34 Congruently, as-TF promotes cancer proliferation through integrin b1 signaling, further highlighting the pivotal interconnection of CF with integrins.35 However, as-TF alone is not enough to promote the correct formation of the vasculature during embryogenesis,36 suggesting that both isoforms are necessary to regulate blood vessel formation and maintenance. Indeed, already in 1996, it was demonstrated that fl-TF−/− mice die during embryonic development due to vascular failure.37 Moreover, it was described that fl-TF can induce EC migration through PAR2.38
Overall, these data show that both TF isoforms can directly bind integrins and GPCR, regulating EC functionality (Table 1).
The intricate system of coagulation within the bloodstream is a finely tuned process and a delicate equilibrium is maintained by the interplay of pro- and anti-CF. These proteins with anti-coagulant activity act as essential regulators, counteracting the clot-forming cascade to prevent excessive blood clotting.
One of the major anti-CF is antithrombin (AT), which inhibits most pro-coagulant proteases of the coagulation cascade, with thrombin and activated FX and factor IX (FIX) as main targets of its action.39 It also displays an anti-inflammatory role, binding heparan sulfate proteoglycans (HSPG) on the EC surface (especially syndecan-4) and increasing prostacyclin.40 Notably, cleaved and latent forms of AT have been demonstrated to be potent anti-angiogenic factors down-regulating the expression of genes related to extracellular matrix assembly in EC, such as perlecan, byglycan and syndecans.41 Its anti-proliferative function has been demonstrated in EC as well as in tumorigenic cell lines42 and, specifically, its heparin-binding site was shown to inhibit proliferation, migration, capillary-like tube formation, and perlecan expression.41 HSPG have been described as interacting with integrins to regulate many EC functions; thus, it has been hypothesized that the interplay between the AT-HSPG complex and the extracellular matrix (ECM) organization could be orchestrated through integrin signaling.40
Interestingly, also thrombomodulin (TM) seems to be implicated in angiogenesis regulation. Indeed, TM has been shown to be a pro-angiogenic factor, activating focal adhesion kinase and binding fibronectin.43 This observation was strengthened by the demonstration of the in vitro and in vivo activity of recombinant EGF-like domain plus the O-glycosylation site-rich domain of TM.44 In contrast, recombinant lectin-like domain of TM has been shown to interact with Lewis-Y carbohydrate antigen, inhibiting angiogenesis.45 Recently, its role has also been assessed in TM-deficient EC and, several assays have shown it to tightly regulate EC quiescence.46 Taken together, these findings reveal that TM exhibits a dual role as both a proand an anti-angiogenic factor.
Importantly, another well-known anti-CF, activated protein C (APC), has been described to protect the endothelial barrier binding angiopoietin-1 receptor (Tie2)47 or PAR1, inducing a signaling in which b-arrestin-2 and dishevelled-2 are involved.48 Further investigations have revealed that APC induces anti-apoptotic signaling in EC in a caveolin-1-dependent manner,49 and increases phosphorylation of proteins mainly related to gene expression and actin binding, corroborating the role of APC in stabilizing the EC barrier.6
Table 2. Summary of endothelial cell functions regulated by selected anti-coagulation factors.
Anti-CF Study In vitro or in vivo models Regulated function Identified receptor
Antithrombin
Thrombomodulin
Activated protein C
Panicker et al.40 EA.hy926 and HDMEC
Zhang et al.41 HUVEC
O’Reilly et al.42 EC (unspecified)
Hsu et al.43 HUVEC
Shi et al.44 HUVEC
Kuo et al.45 HUVEC
Apoptosis HSPG
Angiogenesis -
Angiogenesis -
EC adhesion, migration and angiogenesis -
Angiogenesis -
Angiogenesis Lewis Y Ag
Giri et al.46 TM-deficient EA.hy926 Quiescence -
Minhas et al.47 HUVEC and mice
Soh et al 48
Molinar-Inglis et al.49
Lin et al 6
EA.hy926
EA.hy926
EA.hy926
Permeability Tie2
Permeability PAR1
Apoptosis PAR1
Permeability PAR1
Anti-CF: anti-coagulation factor; EA.hy926: human umbilical vein cell line; EC: endothelial cells; HDMEC: human dermal microvascular EC; HUVEC: human umbilical vein EC; HSPG: heparan sulfate proteoglycans; Lewis Y Ag: Lewis-Y carbohydrate antigen; TM: thrombomodulin; Tie2: angiopoietin 1 receptor; PAR: protease-activated receptor.
Overall, as described above for the pro-CF, even the anti-CF exert pivotal roles in EC maintenance (Table 2), but the way they do this requires further studies in order for these mechanisms to be fully defined.
New evidence has recently emerged on the complex interplay between coagulation and vascular homeostasis; CF possess a different role beyond their conventional involvement in coagulation cascade. Indeed, these factors intricately regulate the stability and the functionality of EC, suggesting a profound relationship between hemostasis and endothelial functions. Some observations also highlight the important effect of these CF, not only in mature EC, but also in vascular development during embryogenesis. Indeed, the absence of some CF (e.g., TF) or their receptors (e.g., PAR1) results in embryo lethality.12,37 Our review has explored specific pro-CF, including thrombin, FVIII, TF, and a few anti-CF, as the primary focus was to elucidate their roles in orchestrating endothelial functionality. While spotlighting these critical CF, it is important to acknowledge the relevance of other factors, such as factor V (FV) and factor XIII (FXIII), which have been recognized as enhancing EC migration and maintaining the vascular homeostasis, 50,51 and FIX, described as controling EC permeability. 52 Additionally, factor I (FI), FVII, and factor XII (FXII), have been shown to play a pro-angiogenic role. 53 On the other hand, FX angiogenic potential activity is still a subject of debate and it is not fully understood if it plays a pro- or an anti-angiogenic role. 53 This evidence further corroborates the hypothesis that CF can be main players both in coagulative and angiogenic processes.
The intricate pathways through which CF influence EC stability mainly involve direct receptor binding, triggering signaling pathways regulating angiogenesis, proliferation, migration, and permeability (Table 1, 2). Notably, CF demonstrate versatility by binding to diverse receptors such as GPCR, receptor tyrosine kinases (RTK), and especially integrins (Figure 1). The CF-receptor interactions underscore the significance of these factors in modulating various physiological endothelial functions. Yet, for certain CF, such as FVIII, whose specific receptors remain elusive, there is speculation as to how they act: is it through known EC surface receptors? Or is it by affecting thrombin generation, which in turn impacts EC functionality? Interestingly, integrins consistently emerge as being crucial membrane transmembrane receptors in mediating CF activity on EC. The persistent prominence of integrins as the principal receptors for CF among various studies reinforces their critical role in regulating endothelial responses to these factors, establishing them as the possible, but not the only, key players in the intricate landscape of EC functionality.
Despite their diverse effects on EC stability, those pathways triggered by CF that have been studied mainly converge on the regulation of EC angiogenesis through the expression of angiogenic factors (e.g., thrombin and TF), and in controlling EC permeability through ECM remodeling (e.g., FVIII and AT). As the ECM profoundly impacts tissue health, further investigation into how CF guide ECM organization remains critical to validate the initial findings. Additionally, elucidating whether CF act individually or collaborate in finely tuned regulatory networks to regulate EC, as in the coagulation cascade, remains an intriguing path for future exploration. Importantly, these findings would help define a more holistic treatment strategy, primarily for coagulation disorders (e.g., coagulopathies), but also
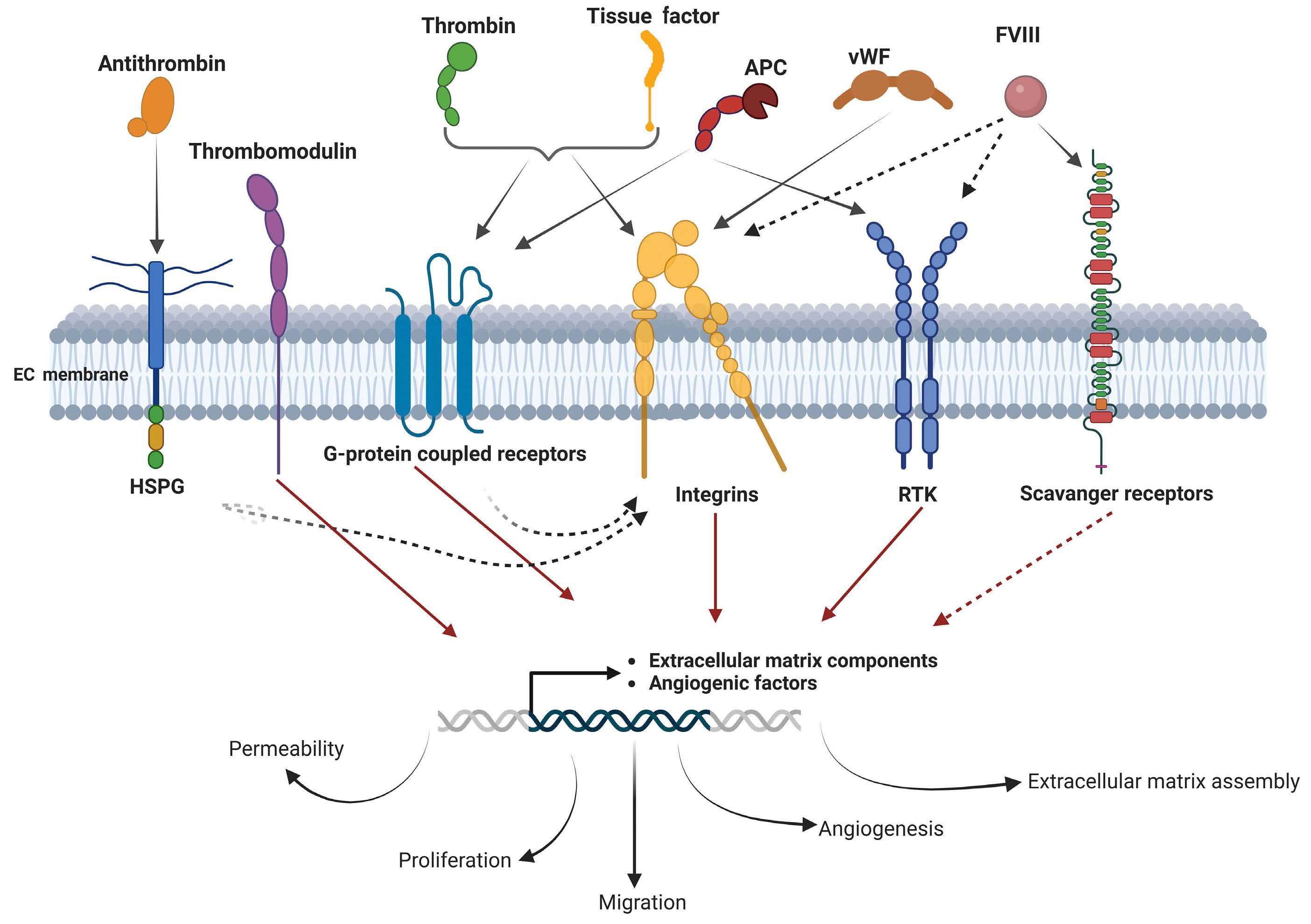
Figure 1. Coagulation factors binding to receptors involved in endothelial cell functions. Antithrombin binds to heparan sulfate proteoglycans (HSPG) to exert its function in endothelial cells (EC). Thrombomodulin can induce the expression of genes involved in extracellular matrix and angiogenesis. Thrombin and tissue factor bind G-protein coupled receptors and integrins. von Willebrand factor (vWF) binds integrin on EC and activated protein C (APC) binds G-protein coupled receptors and receptor tyrosine kinases (RTK). Factor VIII (FVIII) binds to known scavenger receptors, but its involvement with integrins or RTK in the regulation of EC functionality has never been fully explored. The activation of these receptors induces the expression of genes involved in extracellular matrix organization and angiogenesis modulating EC functions.
for conditions where both coagulation and angiogenesis are impaired, such as vascular diseases and cancer. For example, FVIII is expressed by several tumors independently of vWF expression, suggesting a possible unique role of FVIII in cancer physiopathology.54
Revealing the multifaceted roles of CF uncovers novel physiological mechanisms, shedding light on their functions beyond conventional hemostasis. While selected CF have already been the subject of initial studies, others await deeper exploration, suggesting an exciting trajectory in understanding their involvement in endothelial biology.
Disclosures
No conflicts of interest to disclose.
Contributions
CO, SA, AC and AF wrote and revised the manuscript. CO generated the figure using BioRender tool (Agreement N: HV26FVNYFH). AF conceived, wrote, and revised the manuscript.
The authors thank Dr. Chiara Borsotti and Saicharan Akula for their help in critically reading and revising the manuscript.
Funding
This work was partially supported by a Telethon grant (N. GGP19201 to AF) and Bando per la Ricerca Roche to CO.
1. Madhusudhan T, Kerlin BA, Isermann B. The emerging role of coagulation proteases in kidney disease. Nat Rev Nephrol. 2016;12(2):94-109.
2. Neubauer K, Zieger B. Endothelial cells and coagulation. Cell Tissue Res. 2022;387(3):391-398.
3. Italiano JE, Richardson JL, Patel-Hett S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet α granules and differentially released. Blood. 2008;111(3):1227-1233.
4 Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani MG, Dejana E. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol. 1996;16(3):488-496.
5. Hirano M, Hirano K. Critical role of Rho proteins in myosin light chain di-phosphorylation during early phase of endothelial barrier disruption. J Physiol Sci. 2022;72(1):1-13.
6. Lin Y, Wozniak JM, Grimsey NJ, et al. Phosphoproteomic analysis of protease-activated receptor-1 biased signaling reveals unique modulators of endothelial barrier function. Proc Natl Acad Sci U S A. 2020;117(9):5039-5048.
7. Koller GM, Schafer C, Kemp SS, et al. Proinflammatory mediators, IL (interleukin)-1b, TNF (tumor necrosis factor) α, and thrombin directly induce capillary tube regression. Arterioscler Thromb Vasc Biol. 2020;40(2):365-377.
8. Catar R, Moll G, Hosp I, et al. Transcriptional regulation of thrombin-induced endothelial VEGF induction and proangiogenic response. Cells. 2021;10(4):910.
9 Radjabi AR, Sawada K, Jagadeeswaran S, et al. Thrombin induces tumor invasion through the induction and association of matrix metalloproteinase-9 and beta1-integrin on the cell surface. J Biol Chem. 2008;283(5):2822-2834.
10 Zhao B, Wu M, Hu Z, et al. Thrombin is a therapeutic target for non-small-cell lung cancer to inhibit vasculogenic mimicry formation. Signal Transduct Target Ther. 2020;5(1):117.
11. Zhou HJ, Tang T, Cui HJ, et al. Thrombin-triggered angiogenesis in rat brains following experimental intracerebral hemorrhage. J Neurosurg. 2012;117(5):920928.
12. Griffin CT, Srinivasan Y, Zheng YW, Huang W, Coughlin SR. A role for thrombin receptor signaling in endothelial cells during embryonic development. Science. 2001;293(5535):1666-1670.
13. Li Z, Yin M, Zhang H, et al. BMX represses thrombin-PAR1mediated endothelial permeability and vascular leakage during early sepsis. Circ Res. 2020;126(4):471-485.
14 Tsopanoglou NE, Andriopoulou P, Maragoudakis ME. On the mechanism of thrombin-induced angiogenesis: involvement of αvb3-integrin. Am J Physiol Cell Physiol. 2002;283(5):C1501-1510.
15. Zolotoff C, Puech C, Roche F, Perek N. Effects of intermittent hypoxia with thrombin in an in vitro model of human brain endothelial cells and their impact on PAR-1/PAR-3 cleavage. Sci Rep. 2022;12(1):1-14.
16. Gumundsdottir IJ, Lang NN, Boon NA, et al. Role of the endothelium in the vascular effects of the thrombin receptor (protease-activated receptor type 1) in humans. J Am Coll Cardiol. 2008;51(18):1749-1756.
17 Terraube V, O’Donnell JS, Jenkins P V. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia. 2010;16(1):3-13.
18. Follenzi A, Benten D, Novikoff P, Faulkner L, Raut S, Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest.
2008;118(3):935-945.
19. Cadé M, Muñoz-Garcia J, Babuty A, et al. FVIII regulates the molecular profile of endothelial cells: functional impact on the blood barrier and macrophage behavior. Cell Mol Life Sci. 2022;79(3):145.
20 Bhat V, Olmer M, Joshi S, et al. Vascular remodeling underlies rebleeding in hemophilic arthropathy. Am J Hematol. 2015;90(11):1027-1035.
21. Sun H, Yang M, Fung M, et al. Adult males with haemophilia have a different macrovascular and microvascular endothelial function profile compared with healthy controls. Haemophilia. 2017;23(5):777-783.
22. Manon-Jensen T, Tangada S, Bager C, et al. Evaluation of collagen turnover biomarkers as an objective measure for efficacy of treatment with rurioctocog alfa pegol in hemophilia A patients: a secondary analysis of a randomized controlled trial. J Thromb Haemost. 2024;22(1):90-100.
23. Hossain A, Tauhid L, Davenport I, et al. LRP-1 pathway targeted inhibition of vascular abnormalities in the retina of diabetic mice. Curr Eye Res. 2017;42(4):640-647.
24. Starke RD, Ferraro F, Paschalaki KE, et al. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117(3):1071-1080.
25. Randi AM, Smith KE, Castaman G. von Willebrand factor regulation of blood vessel formation. Blood. 2018;132(2):132-140.
26. Xu H, Cao Y, Yang X, et al. ADAMTS13 controls vascular remodeling by modifying VWF reactivity during stroke recovery. Blood. 2017;130(1):11-22.
27. de Vries MR, Peters EAB, Quax PHA, Nossent AY. von Willebrand factor deficiency leads to impaired blood flow recovery after ischaemia in mice. Thromb Haemost. 2017;117(7):1412-1419.
28. Ishihara J, Ishihara A, Starke RD, et al. The heparin binding domain of von Willebrand factor binds to growth factors and promotes angiogenesis in wound healing. Blood. 2019;133(24):2559-2569.
29 Ruf W, Yokota N, Schaffner F. Tissue factor in cancer progression and angiogenesis. Thromb Res. 2010;125(Suppl 2):S36-S38.
30 Mackman N, Brand K, Edgington TS. Lipopolysaccharidemediated transcriptional activation of the human tissue factor gene in THP-1 monocytic cells requires both activator protein 1 and nuclear factor kappa B binding sites. J Exp Med. 1991;174(6):1517-1526.
31. Unruh D, Horbinski C. Beyond thrombosis: the impact of tissue factor signaling in cancer. J Hematol Oncol. 2020;13(1):1-14.
32. Ruf W, Yokota N, Schaffner F. Tissue factor in cancer progression and angiogenesis. Thromb Res. 2010;125(Suppl 2):S36-S38.
33. Giannarelli C, Alique M, Rodriguez DT, et al. Alternatively spliced tissue factor promotes plaque angiogenesis through the activation of hypoxia-inducible factor-1α and vascular endothelial growth factor signaling. Circulation. 2014;130(15):1274-1286.
34 Van Den Berg YW, Van Den Hengel LG, Myers HR, et al. Alternatively spliced tissue factor induces angiogenesis through integrin ligation. Proc Natl Acad Sci U S A. 2009;106(46):19497-19502.
35. Kocatürk B, Van Den Berg YW, Tieken C, et al. Alternatively spliced tissue factor promotes breast cancer growth in a b1 integrin-dependent manner. Proc Natl Acad Sci U S A. 2013;110(28):11517-11522.
36. Sluka SHM, Akhmedov A, Vogel J, et al. Alternatively spliced tissue factor is not sufficient for embryonic development. PLoS One. 2014;9(5):e97793.
37. Carmeliet P, Mackman N, Moons L, et al. Role of tissue factor in embryonic blood vessel development. Nature. 1996;383(6595):73-75.
38. Zhu T, Mancini JA, Sapieha P, et al. Cortactin activation by FVIIa/ tissue factor and PAR2 promotes endothelial cell migration. Am J Physiol Regul Integr Comp Physiol. 2011;300(3):R577-585.
39 Rezaie AR, Giri H. Anticoagulant and signaling functions of antithrombin. J Thromb Haemost. 2020;18(12):3142-3153.
40 Panicker SR, Biswas I, Giri H, Cai X, Rezaie AR. PKC (protein kinase C)-δ modulates AT (antithrombin) signaling in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2020;40(7):1748-1762.
41. Zhang W, Chuang YJ, Jin T, et al. Antiangiogenic antithrombin induces global changes in the gene expression profile of endothelial cells. Cancer Res. 2006;66(10):5047-5055.
42. O’Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999;285(5435):1926-1928.
43. Hsu Y-Y, Shi G-Y, Wang K-C, et al. Thrombomodulin promotes focal adhesion kinase activation and contributes to angiogenesis by binding to fibronectin. Oncotarget. 2016;7(42):68122-68139.
44. Shi CS, Shi GY, Chang YS, et al. Evidence of human thrombomodulin domain as a novel angiogenic factor. Circulation. 2005;111(13):1627-1636.
45. Kuo CH, Chen PK, Chang BI, et al. The recombinant lectin-like domain of thrombomodulin inhibits angiogenesis through
interaction with Lewis Y antigen. Blood. 2012;119(5):1302-1313.
46. Giri H, Panicker SR, Cai X, Biswas I, Weiler H, Rezaie AR. Thrombomodulin is essential for maintaining quiescence in vascular endothelial cells. Proc Natl Acad Sci U S A. 2021;118(11):e2022248118.
47. Minhas N, Xue M, Jackson CJ. Activated protein C binds directly to Tie2: possible beneficial effects on endothelial barrier function. Cell Mol Life Sci. 2017;74(10):1895-1906.
48. Soh UJK, Trejo JA. Activated protein C promotes proteaseactivated receptor-1 cytoprotective signaling through b-arrestin and dishevelled-2 scaffolds. Proc Natl Acad Sci U S A. 2011;108(50):E1372-E1380.
49 Molinar-Inglis O, Birch CA, Nicholas D, et al. aPC/PAR1 confers endothelial anti-apoptotic activity via a discrete, b-arrestin-2mediated SphK1-S1PR1-Akt signaling axis. Proc Natl Acad Sci U S A. 2021;118(49):e2106623118.
50. Cui J, O’Shea KS, Purkayastha A, Saunders TL, Ginsburg D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature. 1996;384(6604):66-68.
51. Dardik R, Loscalzo J, Inbal A. Factor XIII (FXIII) and angiogenesis. J Thromb Haemost. 2006;4(1):19-25.
52. Mamiya A, Kitano H, Kokubun S, Hidai C. Activation peptide of coagulation factor IX regulates endothelial permeability. Transl Res. 2016;177:70-84.
53. Reinhardt C, Manukyan D, Ruf W. The role of coagulation factor signaling in angiogenesis and vascular remodeling. Endothel Sign Develop Dis. 2015;233-256.
54 Walker GE, Merlin S, Zanolini D, et al. Factor VIII as a potential player in cancer pathophysiology. J Thromb Haemost. 2022;20(3):648-660.
Raphael E. Szalat,1 Kenneth C. Anderson2 and Nikhil C. Munshi2
1Section of Hematology and Medical Oncology, Boston University School of Medicine and Boston Medical Center and 2Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
Correspondence: R. Szalat raphael.szalat@bmc.org
Received: November 10, 2023. Accepted: January 31, 2024. Early view: February 8, 2024.
https://doi.org/10.3324/haematol.2023.284662 ©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Multiple myeloma (MM) is a hematologic malignancy characterized by clonal proliferation of plasma cells. MM is a heterogeneous disease, featured by various molecular subtypes with different outcomes. With the advent of very efficient therapies including monoclonal antibodies, bispecific T-cell engagers and chimeric antigen receptor T cells (CAR T cells), most MM patients now have a prolonged survival. However, the disease remains incurable, and a subgroup of high-risk patients continue to have early relapse and short survival. Novel and highly sensitive methods have been developed allowing the detection of minimal residual disease (MRD) during or after treatment. Achievement of MRD negativity is a strong and independent prognostic factor in both prospective randomized clinical trials and in the real-world setting. While MRD assessment is now a validated endpoint in clinical trials, its incorporation in clinical practice is not yet established and its potential impact on guiding therapy remains under in-depth evaluation. Here we discuss the different methods available for MRD assessment and the role of MRD evaluation in MM management.
Multiple myeloma (MM) is a heterogeneous disease characterized by more than 10 distinct molecular subtypes associated with variable outcomes.1 The therapeutic landscape of MM has dramatically changed over the last five years. The incorporation of monoclonal antibodies, first in the relapse setting, and more recently in front-line treatment in a triplet or quadruplet regimen, the approval of chimeric antigen cell (CAR) T-cell therapy, and the recent approval of bi-specific monoclonal antibodies or T-cell engagers have revolutionized MM treatment and prognosis.2-4 With more than 14 drugs approved by the US Food and Drug Administration (FDA), there are now various treatment options and most MM patients now have prolonged survival.2 However, MM remains incurable and therefore the ability to identify high-risk patients, and to appropriately sequence therapy based on disease characteristics and response to treatment is critical. Along with plasma cell molecular and cytogenetic characteristics, response to treatment is another major prognostic factor, and its assessment is an essential part of patient care. The definition of hemato-
logic response has evolved in the past 20 years with the incorporation of novel highly sensitive methods to allow comparison of treatment strategies in clinical trials. International consensus criteria defining hematologic response in MM were first established in 1998 and revised in 2016, especially to incorporate the free light chain (FLC) dosage. The original definition of a complete response (CR) only required bone marrow (BM) to have <5% plasma cells, irrespective of their clonal nature, while the 2016 criteria defined CR as negative immunofixation on serum and urine, disappearance of any soft tissue plasmacytomas, and <5% plasma cells in BM aspirates. Stringent CR was defined as CR plus normal FLC ratio and absence of clonal cells in BM biopsy by immunohistochemistry (κ/λ ratio ≤4:1 or ≥1:2 for κ and λ patients, respectively, after counting ≥100 plasma cells). Overall, achievement of CR is associated with a significantly prolonged period of progression-free survival (PFS) and overall survival (OS).5 However, not all patients achieving CR have a good prognosis as some patients achieving CR unfortunately have early relapse or progression. The concept of minimal residual disease (MRD) which refers to the ability to detect a very small
number of malignant plasma cells during or after treatment was adopted in the International Myeloma Working Group (IMWG) consensus criteria in 2015 to provide more accurate hematologic response assessment. Since then, MRD evaluation has been shown to significantly improve hematologic response evaluation and to improve prognostic stratification after therapy in newly diagnosed transplant eligible and transplant non-eligible patients and in relapsed disease. The prognostic role of MRD is now well documented and has been demonstrated in several retrospective and prospective studies, and MRD negativity is now an established criteria in MM clinical trials. However, the impact of MRD assessment on treatment decisions remains to be determined and is currently under in-depth investigation in several randomized clinical trials.6-16 In addition to BMbased MRD evaluation, novel techniques utilizing whole body imaging (WBI) and blood-based evaluation have been developed and will likely improve MRD evaluation in MM patients. Here, we describe the different methods available to assess MRD, and discuss the clinical applications and challenges of using MRD in clinical practice.
First MRD evaluation was performed by multiparametric flow cytometry (MFC), a technique available worldwide able to identify monotypic plasma cells in the BM even if present at a low level.17,18 Conventional MFC MRD approaches usually use 4-10 cell markers (colors) but are limited by relatively low sensitivity, absence of standardization, and lack of reproducibility. Therefore, next-generation flow cytometry (NGF), a more sensitive method, was developed and standardized by EuroFlow to overcome most of the conventional MFC limitations.19-21 NGF is based on a more efficient sample preparation protocol for acquisition of up to 10 million BM cells and uses 8-12 colors characterizing most cellular subtypes and normal plasma cells (CD138 and CD38) and aberrant plasma cell markers (CD20, CD56, CD19, CD45, CD27, CD28, CD33, and CD117). Additionally, intra-cytoplasmic markers for κ or λ immunoglobulin light chains are used to confirm monotypic or clonal cells. Importantly, NGF high sensibility allows detecting one abnormal plasma cell out of 10^-6 cells and does not require a sample at the time of diagnosis.21 It is also adapted to patients receiving anti-CD38 therapy, despite the potential interference with plasma cell detection.21 Standardized data analysis methods allow for an increased sensitivity and reliability. The clinical impact of high-sensitivity MRD detection by NGF has been validated in randomized clinical trials and in real-world patients with MM.19,22 In the GEM/PETHEMA trials, only 7% of patients achieving MRD-negativity (MRD <2 10^-6 cells) were
reported to have disease progression with half of those patients progressing with extra-medullary disease (EMD). Achievement of MRD negativity was associated with an 82% and 88% reduction in the risk of progression and death (Hazard Ratios [HR] of 0.18 and 0.12; P<0.001), respectively. Importantly, MRD negativity overcame the poor prognostic value of high-risk cytogenetics at diagnosis.19 The EuroFlow NGF approach has now been validated by the IMWG as the reference flow cytometry method to evaluate MRD negativity after therapy.5 The EuroFlow process, which uses 2-tube 8-color methodology, is widely used in Europe, Asia and the US. However, several groups, especially in the US, have developed other methods, using single-tube, 10- or 12-color methods that are similarly efficient, more cost-effective, and conform to the IMWG and National Comprehensive Cancer Network guidelines.23
High-throughput DNA sequencing methods developed to study B- or T-cell receptor repertoire have been applied to MM. These methods can identify one malignant cell in 1 million analyzed cells (10^-6). The Adaptive Biotechnologies (clonoSEQ) NGS assay is currently the only assay cleared by the FDA for MRD evaluation in BM from patients with MM. NGS can detect clonotypes that are defined by sharing identical immunoglobulin gene sequence reads with a frequency ≥5%. This strategy requires an initial BM sample to identify the predominant clone, and allows the myeloma clone to be detected in 90-92% of myeloma patients.16 The prognosis value of achieving NGS-based MRD negativity has been demonstrated in several randomized trials. In the Intergroupe Francophone du Myélome 2009 trial, MRD negativity was a strong prognostic factor for both PFS (adjusted HR: 0.22; 95% Confidence Interval [CI]: 0.15-0.34; P<0.001) and OS (adjusted HR: 0.24; 95% CI: 0.11-0.54; P=0.001). Patients who were MRD negative had a higher probability of prolonged PFS than patients with MRD-positive disease and a cytogenetic risk profile, regardless of the treatment arm or International Staging System disease stage at diagnosis. The level of MRD correlated with outcome, and the deeper the level of MRD (<10^-6), the better the prognosis.16 A pooled analysis searching for associations between patients achieving CR or better (≥CR) with MRD-negative status and PFS from 4 randomized clinical trials, confirmed that relapsed/ refractory MM (RRMM) and transplant ineligible newly diagnosed MM (TIE NDMM) patients achieving ≥CR with MRD negativity had a significant PFS benefit (NDMM and RRMM HR: 0.20; P<0.0001; TIE NDMM and RRMM ≤2 PL HR 0.20, P<0.0001).14 Remarkably, achievement of MRD negativity is, independently of the study arm, associated with best and similar outcome in newly diagnosed MM patients who are transplant eligible and transplant ineligible, as well as in relapse refractory disease.14,15 These data strongly support the concept that achieving MRD negativity may be more important than how it was actually achieved.
Bone marrow MRD assessment requires repeated invasive procedures, and is limited by the known patchy nature of MM and the possibility of EMD that are inherent to the disease. Therefore, alternative approaches including blood or imaging-based MRD evaluation assessment have been developed.24
Circulating plasma cells
The presence of circulating plasma cells (CPC) in peripheral blood (PB) can be detected in most MM patients and is associated with poor prognosis. Different methods have been used to assess the presence of CPC. The standard EuroFlow NGF is reliable and requires a small volume of blood. Other methods are available that use a plasma cell enrichment method, which requires a larger blood sample, and is more sensitive but also more complex. NGF has been used to identify and track CPC in MM patients with interesting results. However, while CPC detection appears to be a powerful prognostic factor, CPC is unlikely to be a good MRD marker. Indeed, a comparison between NGF in BM and PB after therapy in a real-world case series of 137 patients showed that 40% of patients achieving bloodbased MRD negativity had BM MRD-positive disease, strongly suggesting that blood NGF-based MRD evaluation is a less sensitive MRD marker than BM MRD.22,25,26
Circulating cell-free DNA (cfDNA)-based methods, often referred to as ‘liquid biopsy’, allow tracking genomic aberrations such as tumor mutations, copy number aberration or translocation present in circulating cfDNA isolated from blood plasma.27-29 Multiple studies showed a high concordance of somatic mutations and copy-number alterations between BM and cfDNA of patients with MM.30-33 However, the low level of circulating tumor DNA is a significant challenge and most current methods are not sensitive enough. Ultradeep targeted sequencing has significantly improved the detection of cfDNA, but its sensitivity relates to the number of tumor mutations available to track and has so far only been evaluated in few clinical studies. In a study which compared blood and BM evaluation with NGS and cfDNA in 42 patients, there was only 49% consistency and poor correlation between the two methods. Similar to CPC detection, BM MRD was more often positive and suggested lack of sensitivity of the cfDNA approach.34 Novel and more sensitive methods are needed before cfDNA can be utilized as a standard approach.
Single-cell RNA sequencing (scRNA-seq) is another powerful technology widely used in research. It allows transcriptomic analysis at a single cell level and can detect rare malignant cells.35 Ongoing research is investigating whether this approach could even allow the selection of therapy based on transcriptomic features and clonal heterogeneity.36,37 However, its availability, its relative complex workflow, reproducibility and cost are significant challenges that need to be addressed before it can be considered for use in clinical practice.38 This approach is also limited by the fact that it can currently only evaluate a certain number of cells, far fewer than with flow- or NGS MRD-based assessment methods. Therefore, the lack of detection of malignant cells would not necessarily correspond to negative MRD, and scRNAseq appears more as a potential complimentary method that may help tailor therapy to target MRD positive cells rather than to determine MRD status.
Instead of tracking the malignant residual plasma cell, mass spectrometry (MS) methods have been applied to detect the monoclonal immunoglobulin produced by the malignant plasma cells. This very sensitive method can detect the presence of a monoclonal immunoglobulin at a much lower concentration than standard serum immunofixation. MS can also distinguish therapeutic monoclonal antibodies from myeloma monoclonal immunoglobulin, and can identify post-translational modifications relevant for patients with monoclonal gammopathy of clinical significance, including light chain (AL) amyloidosis.39-41 Several MS methods have been developed. Matrix-assisted laser desorption/ionization MS (MALDI-TOF MS) and the MASS-FIX assay have been shown to be particularly efficient, and both more sensitive and more specific than serum protein electrophoresis and immunofixation.40,42,43 Another MS approach was initially developed by the Binding Site company in collaboration with the Mayo Clinic. The Mayo Clinic went on to develop their own approach using a commercial assay, while the Binding Site assay, which has more selective probes, was sold to ThermoFisher and is used in individual labs. Methods using liquid chromatography-tandem mass spectrometry (LC-MS/MS) are currently under clinical investigation and are not yet FDA-approved. Other MS methods focusing on quantifying unique clonotypic peptides (MS-MRD) derived from the variable region of the monoclonal immunoglobulin by enzymatic digestion followed by LC-MS/MS have been developed and provide great sensitivity. MS-MRD demonstrated a 1,000-fold higher sensitivity compared to serum protein electrophoresis (SPEP) and can be used to monitor patients with MRD.40,44-46 While some preliminary reports suggest that MS may be a strong predictor of PFS,47 additional studies are needed to incorporate these assays into clinical practice. In particular, the persistence of the monoclonal immunoglobulin in the serum in the context
of BM MRD negativity detected by serum immunofixation or MS has been reported in different studies and may be related to the immunoglobulin half-life rather than to an MRD-positive disease. Therefore, incorporation of MS needs to be clarified.
imaging and minimal residual disease assessment
While imaging studies do not allow the detection of active disease at the single cell resolution, relatively novel WBI techniques including positron emission tomography with computed tomography (PET/CT) and magnetic resonance imaging (MRI) allow a better characterization of bone lesions and EMD. PET/CT and whole-body MRI have been evaluated to assess residual disease after therapy.48-51 Both methods are very sensitive and have been shown to complement BM-based MRD assessment considering the patchy nature of MM and its spatial heterogeneity. Patients presenting with EM lesions or with para-medullary plasmacytomas are at higher risk of developing EMD even in the context of BM MRD-negativity.52 18Fluorodeoxyglucose (18F-FDG)PET is a very sensitive method to identify active disease, and several studies showed that PET-positive lesions after completion of therapy is associated with poorer outcome, while FDG-PET/CT negativity after autologous stem cell transplantation (ASCT) in patients achieving CR predicts a lower risk of progression or death.53-56 Patients obtaining PET/CT normalization upon therapy have comparable prognosis to patients without baseline increased metabolism, suggesting the value of treating until suppression of glucose metabolism.56 In the FORTE trial, a high concordance between PET/CT and NGS (84%) and between PET/CT and MFC (93%) at 10^−5 in the identification of BM residual disease was reported. By contrast, there was a discrepancy in the assessment of MRD in patients with focal lesions in approximately 33-37% of cases, suggesting that PET/ CT alone might not be accurate enough.57 Similarly, in the CASSIOPET study, a significant concordance between BM and PET/CT-based MRD assessment was reported in 109 patients, but the data suggested a higher sensitivity for the BM-based MRD method.58 A standardized definition of PET/CT complete metabolic response has been proposed considering the uptake of the liver as threshold and is currently under confirmation in independent prospective series of patients.48 A significant challenge related to 18FDG PET/CT relates to the 10-15% of MM patients with no FDG-avid lesions due to lack of hexokinase enzyme, which is responsible for FDG trapping in the myeloma cells or to the absence of identified lesions.59 New PET/CT tracers including CD38, radiolabeled antibodies and VLA-4 (clinicaltrials.gov 03804424) represent new, potentially more sensitive methods that are under investigation.13,60,61 Along these lines, conjugating daratumumab with the positron emitting radioisotopes Copper-64 (64Cu) and Zirconium-89 (89Zr) has allowed for the creation of immunoPET tracers.
89Zr-Daratumumab has demonstrated an ability to detect MM cells or lesions when not detected by 18FDG-PET/CT and other clinically standard imaging methods.62,63 However, the lesser availability of these newer tracers, interpatient heterogeneity regarding specific targets, and lack of prospective data remain important challenges to be addressed. Similar to PET/CT, presence of residual lesions after highdose chemotherapy followed by autologous stem cell transplant (HDC-SCT) identified by whole body MRI (wbMRI) is associated with adverse prognostic significance.64-66 MRI seems to be more sensitive in diagnostic methods to identify myeloma lesions than PET/CT. In a prospective study comparing PET/CT and wbMRI in 60 patients, wbMRI showed significantly higher detection of focal lesions at all anatomic sites (except ribs, scapulae, and clavicles) and of diffuse disease at all sites. However, MRI is not able to differentiate between vital and necrotic tissue within pre-existing osteolytic lesions64 and therefore PET/CT positivity may be more accurate to assess MRD and to predict patient outcome.67-69 A more sensitive MRI-based method called diffusion weighted imaging (DWI) is a promising alternative allowing more accurate detection of active lesions and is under investigation.70,71 Whether MRD is assessed with PET/CT or wbMRI, the complementary role of imaging studies to BM-based MRD evaluations is significant and strategies to include both are being evaluated. Based on the increasing amount of MRD data, and the availability, reproducibility and standardization of MRD methods, MRD assessment has become an important and validated criterion in clinical trials. MRD assessment is now used for patient selection, risk stratification or enrichment of clinical trial subgroups, and as an endpoint. MRD assessment will likely contribute to expedite drug development.13 However, using MRD assessment to guide therapy and MRD incorporation in clinical practice is not yet validated. We here discuss some of the most significant challenges that have been or need to be addressed.
As discussed above, MRD assessment using BM-based methods remains the gold standard with increasing data regarding WBI. Availability, cost, prognostic power, and consistency are important factors to consider. Regarding BM-based MRD, NFC and NGS are the 2 methods of choice to evaluate BM MRD and both have been shown to constitute a strong prognostic marker in MM. Table 1 summarizes the main characteristics of these methods. Comparison between flow cytometry and NGS methods has been performed in randomized clinical trials. In the phase II multicenter randomized FORTE trial, 86% of correlation with MRD at a sensitivity of 10^−5 in patients ≥CR was reported.72 In the phase III CASSIOPEIA trial, MFC and NGS were consistent in 83.5% with a sensitivity of 10^−5 . 73 A direct comparison between NGF and NGS (not Clonoseq platform) was also reported in a study of 106 patients
Table 1. Available methods to assess bone marrow minimal residual disease in multiple myeloma.
BM-based MRD NGF
NGS
BM evaluation Yes Yes
Standardization Euroflow
Evaluation required at diagnosis
Not required
Clonoseq
Required
Fresh sample Yes No
Cost + ++
Applicability Universal ~90% of patients
Sensitivity 10^-5-10^-6 10^-6
BM: bone marrow; MRD: minimal residual disease; NGF: next-generation flow cytometry; NGS: next-generation sequencing.
Table 2. Available whole body imaging methods to complement bone marrow minimal residual disease assessment in multiple myeloma.
BM evaluation No No
Standardization Yes Yes
Evaluation required at diagnosis Not required*
Negative in ~10% of MM patients Not required*
Cost ++ ++
Applicability ++ + Sensitivity ++ +++
BM: bone marrow; CT: computed tomography; MM: multiple myeloma; PET: positron emission tomography; WBI: whole body imaging; wbMRI: whole body magnetic resonance imaging. *Useful to have WBI at diagnosis to evaluate response. Important to confirm if PET/CT positive at diagnosis.
showing a high correlation (R2 = 0.905).74 As NGS and NGF are comparable, each method should be considered based on local availability. Complementary imaging methods to assess bone and EM MRD currently include wbMRI and PET/CT imaging, and provide additional information, while BM- and WB imaging-based methods have relatively good concordance and provide additional information, particularly regarding EMD and high-risk MM. WBI should, therefore, be used in combination with BM-based MRD in patients with high-risk or EM disease. Table 2 summarizes the characteristics of WB imaging methods. Some studies now support the benefit of combining both MRD methods in patient care.71
When should minimal residual disease assessment be performed?
Another important challenge regarding MRD utilization relates to its timeline. A first question is whether MRD evaluation should be done early during front-line treatment (for example, before SCT) or later on (after consolidation or during maintenance). Several trials have evaluated each of these time points, and because of the dynamic nature of MRD which implies transition from MRD positivity to negativity and vice-versa, it has become clear that several evaluations are, in fact, more informative. These observations led to define the concept of ‘sustained’ MRD negativity which appears to be the critical prognostic factor in MM.75 Having defined this concept led to another important
question, which is how much time should pass between the 2 time points. It seems that longer time points will likely be associated with better outcome, and studies are investigating if six months, one year or two years of MRD negativity are more relevant to potentially impact treatment decision. For example, in chronic myeloid leukemia (CML), strategies to stop therapy after two years of sustained MRD negativity have been developed.76,77 However, CML and MM are very different diseases biologically, and large phase III clinical trials are needed and are, indeed, ongoing to address this question. In future clinical practice, we anticipate that MRD assessment will be useful to evaluate the efficacy of a particular treatment strategy, before or after consolidation therapy or during maintenance in newly diagnosed myeloma and relapse/refractory disease. It is important to mention that the advent of cellular therapies including bi-specific antibodies and CAR T-cell therapy have revolutionized patient outcome and are associated with dynamic and often dissociated patterns of MRD and serological residual disease. Therefore, the role of MRD evaluation in the context of these novel therapies remains to be fully validated.78
How far is minimal residual disease evaluation useful in high-risk myeloma patients?
High-risk MM is defined by the presence of del17p, t(4;14), t(14;16), low albumin, high b2 microglobulin and elevated lactate dehydrogenase (LDH).79 MM patients experiencing
early relapse after front-line therapy have also the poorest outcome.80 Important studies are ongoing to improve treatment strategies in this subgroup of patients. Different results regarding clinical impact of MRD status in highrisk patients have been reported.81,82 While achievement of MRD negativity is associated with clinical improvement in HR MM, it does not overcome its poor prognosis and HR MM may still have early progression. In addition, data from the large phase III trial (Myeloma XI) showed that high-risk molecular features had an adverse effect on PFS and OS even for those patients achieving MRD-negative status.83 BM MRD evaluation only is likely to be insufficient to fully assess MRD, in the context of patchy disease and EMD, as discussed above. Indeed, in a study comparing BM and imaging-based MRD assessment, 12% of patients who achieved BM MRD negativity by flow cytometry had positive PET/CT or whole-body diffusion-weighted MRI (WB-DWI-MRI) and had a shorter PFS in comparison to patients with both BM and imaging MRD negativity.56 Further data are needed to better interpret and use MRD status, especially in high-risk patients and patients with EMD. Sustained MRD and combined BM and imaging-based MRD assessment are important parameters to consider. Both sustained MRD negativity and combined BM and imaging MRD studies (PET/CT or wbMRI) appear to be the most valuable approaches in HR MM patients. Results from randomized trials are expected to address this important question. Similarly, in the context of RR MM, while achievement of MRD negativity is associated with better outcome, most of the patients do experience relapse. This is well illustrated by the KarMMa trial that evaluated the efficacy and safety of Idecabtagene vicleucel (ide-cel) in patients with RR myeloma, for example, in which 26% of the patients achieved MRD negativity, including 79% of patients achieving CR. However, only 40% of patients achieving at least CR were in remission at 20 months of follow-up.84 It seems that sustained MRD negativity combined with WBI will be more relevant in that context.
Should minimal residual disease assessment be made only in patients achieving complete hematologic response?
Another important point relates to which patients should be evaluated for MRD. In clinical trials, MRD investigations were performed either in MM patients achieving CR or stringent CR or at specific timepoints of a given therapeutic protocol (e.g., before SCT, before or during maintenance). It was showed that patients achieving CR with MRD positivity had significantly worse outcome than patients achieving both CR and MRD negativity. However, several studies have reported MRD negative rates in MM patients achieving very good partial response (VGPR), and indeed, up to 25% of patients achieving MRD negativity assessed by either flow cytometry or NGS have persistent positive immunofixation and are, therefore, classified as
VGPR.85 Importantly, retrospective and prospective studies showed that patients with positive IF and MRD negativity have similar outcome to patients with negative IF and MRD negativity. The discrepancy between positive IF and MRD negativity may be related to several reasons, including EM disease, BM sample not representative of full BM or long half-life of the monoclonal immunoglobulin.85 This is further highlighted in studies utilizing MS (the sensitive assay to detect monoclonal immunoglobulin described above). In one study, the monoclonal immunoglobulin was still detectable by matrix-assisted laser desorption/ ionization-time-of-flight mass spectrometer (MALDI-TOF) in 69% of patients who achieved a conventionally defined CR and were BM-based NGF-MRD-negative after 100 days from ASCT.86 Finally, discordant MRD and IF results are frequently observed after CAR T-cell therapy, with low rates of CR observed in patients achieving MRD negativity, particularly in the first six months after treatment, suggesting that MRD evaluation which reflects the clearance of myeloma cells in the BM could be an independent prognostic marker in that setting.78 Therefore, assessing MRD in patients achieving at least VGPR or better is very relevant and informative.
Should minimal residual disease status impact therapeutic decision-making?
Minimal residual disease negativity is a very strong and now established prognostic marker. However, its impact on therapeutic decision-making remains to be determined. Several randomized clinical trials are currently ongoing to address this question (Table 3). The goals of these trials are to determine if treatment should be adapted based on MRD status: intensification of treatment in case of MRD positivity, stop maintenance therapy in case of sustained MRD negativity or treatment change in case of MRD status conversion from negativity to positivity. The MASTER trial pioneered this strategy and evaluated the role of BM-based MRD in treatment during consolidation. Patients received combined daratumumab, carfilzomib, lenalidomide and dexamethasone (Dara-KRd) as induction therapy and BM MRD was performed by NGS at different time points (end of induction, after HDC-SCT, and every 4 cycles of consolidation) to inform the use and duration of treatment with Dara-KRd. Treatment was stopped in patients who achieved 2 consecutive MRD-negative assessments. Among 123 included patients, 43% had none, 37% had one, and 20% had 2 high-risk cytogenetic abnormalities (HRCA), and 96% had BM MRD trackable by NGS. With a median follow-up of 25.1 months, 80% of patients reached MRD negativity (78%, 82%, and 79% for patients with 0, 1, and 2+ HRCA, respectively), including 66% who reached MRD <10-6, and 71% who reached 2 consecutive MRD negative assessments during therapy, entering treatment-free surveillance. Two-year PFS was 87% (91%, 97%, and 58% for patients with 0, 1, and 2 HRCA, respectively).
Table 3. Ongoing prospective clinical trials evaluating minimal residual disease-adapted therapy.
UMCC 2018.056 (NCT04140162)
MIDAS (NCT04934475)
PERSEUS (NCT03710603)
DRAMMATIC (NCT04071457)
Patient population
Phase II study with MRD-driven adaptive strategy in treatment for newly diagnosed MM with upfront daratumumab-based therapy
Phase III clinical trials in newly diagnosed MM patients
Phase III clinical trial daratumumab, VELCADE (bortezomib), lenalidomide and dexamethasone compared to VELCADE, lenalidomide and dexamethasone in subjects with previously untreated MM
Phase III clinical trial lenalidomide +/daratumumab/rHuPh20 as post-ASCT maintenance for MM w/MRD to direct therapy duration (DRAMMATIC)
EMN20 (NCT04096066)
MASTER-2 (NCT05231629)
Phase III clinical trial. A trial that compares two treatments in newly diagnosed myeloma patients not eligible for transplant (KRd vs. Rd)
RADAR (EudraCT 2019-001258-25)
Phase II clinical trial. Sequential therapy in MM guided by MRD assessments (MASTER-2)
Phase III clinical trial. Risk-adapted therapy directed according to response comparing treatment escalation and de-escalation strategies in NDMM suitable for stem cell transplant
Treatment scheme
This phase II trial will test whether the combination of DaraRd as induction therapy, followed by DRVd consolidation therapy, if needed, will result in more patients achieving MRD-negative status, relative to the standard of care. Consolidation therapy will be administered only to those patients with MRD-positive status after induction therapy.
IFM 2020-02 will enroll patients eligible for ASCT aged <66 years. All patients will receive induction based on 6 cycles (28-day) of Isa-KRd, in order to achieve deep responses and high MRD negativity rates. Patients will be classified at diagnosis according to cytogenetics (standard vs high-risk cytogenetics defined by the LP score including 17p deletion, t(4;14), del(1p32), gain 1q, trisomy 21 and trisomy 5.
A phase II study comparing D-VRd vs. VRd in subjects with previously untreated multiple myeloma who are eligible for high-dose therapy. MRDnegative subjects will stop daratumumab after sustained MRD negativity for 12 months and after a min. of 24 months of maintenance. Daratumumab should be restarted at recurrence of MRD or confirmed loss of CR without disease progression.
In this trial, patients who received HDC-SCT are randomized between lenalidomide for 2 years and lenalidomide + daratumumab. After 2 years of maintenance, MRD is assessed to guide further therapy. MRD-positive patients will continue with the assigned treatment. MRD-negative patients will be further randomized to either continue or discontinue the assigned treatment.
This protocol is a randomized, multicenter study designed to determine the MRD negativity and the PFS of KRd treatment regimen. Patients will be randomized in a 1:1 ratio to receive KRd (Arm A) or Rd (Arm B). Patients will be stratified basing on ISS and fitness status using a web-based procedure completely concealed to study participants. Patients will be treated until disease progression or intolerance to the therapy. The only exception is for patients enrolled in KRd arm who achieve at least a VGPR during the 1st year of treatment and in sustained MRD negativity (MRD negative at least at 10-5 after 1 and 2 years of therapy): these patients will stop carfilzomib administration after 2 years, whereas treatment with lenalidomide and dexamethasone will be continued.
This research study will determine the proportion of patients with lowest MRD response obtainable after receiving 6 cycles of study treatment. MRD is MM cells below the level of 1 cancer cell out of 100,000 in the BM. For patients who become MRD ‘negative’ (i.e. less than 1 cancer cell out of 100,000) at the end of 6 cycles of therapy, this study will evaluate if that good response can be maintained with 3 additional cycles of treatment instead of use of ASCT. For patients who are MRD ‘positive’ at the end of 6 cycles of therapy, this study will answer whether more patients can become and remain MRD ‘negative’ with ASCT plus teclistamab in combination with daratumumab when compared with patients who undergo ASCT followed by lenalidomide plus daratumumab.
All participants will receive the same initial induction treatment and during this time will have genetic tests to determine whether they have ‘standardrisk’ or ‘high-risk’ disease. Following this chemotherapy treatment participants will receive ASCT. After induction treatment participants will be allocated to a second stage treatment group based on their genetic risk, high-risk or standard-risk, and on how well the myeloma has responded to the initial treatment. Each treatment group will then receive different combinations of medication to investigate their benefit. Treatment will comprise of combinations of isatuximab, bortezomib, cyclophosphamide, lenalidomide and dexamethasone.
ASCT: autologous stem cell transplanation; BM: bone marrow; CR: complete remission; DaraRd: daratumumab + lenalidomide + dexamethasone; DRVd: daratumumab + lenalidomide + bortezomib + dexamethasone; D-Vrd: daratumumab, VELCADE (bortezomib), lenalidomide, and dexamethasone; HDC: high-dose chemotherapy; IFM: Intergroupe Francophone du Myélome; Isa-KRd: KRd-isatuximab; ISS: International Staging System; KRd: carfilzomib, lenalidomide and dexamethasone; MIDAS: Minimal Residual Disease Adapted Strategy; NDMM: newly diagnosed patients with multiple myeloma (MM); PFS: progression-free survival; Rd: lenalidomide-dexamethasone; Vrd: VELCADE, lenalidomide, and dexamethasone; VGPR: very good partial response.
Cumulative incidence of MRD resurgence or progression 12 months after cessation of therapy was 4%, 0%, and 27% for patients with 0, 1, or 2 HRCA, respectively.82 Similarly, a phase III clinical trial (GEM2012MENOS65) evaluated lenalidomide and dexamethasone maintenance with or without ixazomib in newly diagnosed myeloma patients, with treatment stopping after 24 cycles in case of BMbased MRD-negativity. Patients achieving MRD negativity after 24 months of maintenance therapy had a low progression rate (17.2%) at four years, strongly suggesting that the duration of maintenance therapy can be tailored based on MRD negativity. 87 Accordingly, data regarding achievement of MRD negativity and early relapse have been reported but not yet published in high-risk MM patients included in the CASSIOPEIA trial.73 Although longer follow-up is needed, these trials already suggest that cessation of treatment may be feasible in patients with standard risk cytogenetics but not for patients with HRCA. Improving MRD assessment by combining BM and imaging evaluation may be more relevant in high-risk cytogenetic patients in front-line. Another important observation has been reported in a phase II study evaluating MRD dynamics during lenalidomide maintenance. Patients who lost MRD negativity were more likely to progress than patients with sustained MRD negativity (HR infinite; P <0.0001) as expected, but also and worse than patients with persistent MRD positivity (HR 5.88, 95% CI: 1.18-33.33; P =0.015) at the 2-year landmark. These data suggest that the dynamic of the disease is another very important parameter to consider when evaluating the use of MRD as a therapeutic guide. 88
The ongoing MIDAS trial ( clinicaltrials.gov 04934475 ) is designed to evaluate the role of HDC-SCT on the basis of MRD status after induction in newly diagnosed MM patients. In this trial, patients are treated with 6 cycles of quadruplet regimen induction with combined isatuximab, carfilzomib, lenalidomide and dexamethasone (Isa-KRd) and evaluated for BM-based MRD (with a threshold of 10 −5 cells) post induction. Patients are next stratified into standard risk (MRD negativity < 10 −5) or high risk (MRD positivity> 10 −5). Patients achieving MRD negativity following induction are randomized to receive 6 additional cycles of Isa-KRd followed by maintenance or HDC-SCT, followed by 2 cycles of Isa-KRd, and maintenance with lenalidomide for three years. High-risk patients defined by MRD positivity post induction are randomly assigned to receive HDC-SCT followed by 2 cycles of Isa-KRd versus tandem HDC-SCT followed by isatuximab-iberdomide maintenance for three years. This ambitious trial will address whether MRD status can be used to guide therapy, and if HDC-SCT remains the gold standard in patients achieving early MRD negativity after induction. Several other randomized clinical trials are evaluating
MRD-based treatment decision. The PERSEUS trial (clinicaltrials.gov 03710603 ) will evaluate the possibility of stopping daratumumab during maintenance in patients achieving sustained MRD negativity for 12 months and after a minimum of 24 months of maintenance, and the benefit of restarting daratumumab in case of MRD conversion (from negative to positive) or confirmed loss of CR without IMWG disease progression criteria.89 The AURIGA trial ( clinicaltrials.gov 03901963), randomly assigns patients who have achieved VGPR but who are MRD-positive to receive daratumumab and lenalidomide versus lenalidomide maintenance for the primary endpoint of MRD conversion at 12 months from initiation of maintenance.
The DRAMMATIC trial (clinicaltrials.gov 04071457) by the Southwest Oncology Group (SWOG; S1803) randomly assigns patients to receive daratumumab and lenalidomide versus lenalidomide maintenance post HDC-SCT. After two years of maintenance, MRD is assessed to guide further therapy. MRD positive patients will continue with the assigned treatment while MRD negative patients will be further randomized to either continue or discontinue the assigned treatment. The OPTIMUM trial (clinicaltrials.gov 03941860) by the Eastern Cooperative Oncology Group (ECOG; EAA171) will randomly assign MRD positive patients who are receiving lenalidomide maintenance after HDC-SCT to receive ixazomib or placebo in addition to continuing lenalidomide. In conclusion, MRD assessment methods have significantly improved in the past two decades and allow identification of patients with deep hematologic response. BM-based methods using NGF and NGS are to date the most available, standardized, and sensitive methods. WBI includes wbMRI, and PET/CT are also very interesting, and when combined with BM MRD assessment provide better evaluation especially in the setting of high-risk cytogenetics and EMD. Achievement of MRD negativity is a very strong prognostic factor that is now an established endpoint in myeloma clinical trials. Persistent or sustained MRD negativity portends better outcome in newly diagnosed and RR disease, including after CAR T-cell therapy in myeloma and may allow discontinuation of therapy in patients without high-risk cytogenetics. Several clinical trials are currently ongoing to establish whether MRD can be used to guide therapy and to monitor disease activity.
No conflicts of interest to disclose.
Contributions
All authors made a significant contribution to this article. RES, NCM and KCA took part in the conception of the manuscript, in drafting, revising and critically reviewing the article, and gave final approval of the version to be published.
1. Broyl A, Hose D, Lokhorst H, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood. 2010;116(14):2543-2553.
2. Moreau P, Kumar SK, San Miguel J, et al. Treatment of relapsed and refractory multiple myeloma: recommendations from the International Myeloma Working Group. Lancet Oncol. 2021;22(3):e105-e118.
3. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394(10192):29-38.
4 Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115.
5. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346.
6. Rawstron AC, Gregory WM, de Tute RM, et al. Minimal residual disease in myeloma by flow cytometry: independent prediction of survival benefit per log reduction. Blood. 2015;125(12):1932-2935.
7. Rawstron AC, Child JA, de Tute RM, et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX study. J Clin Oncol. 2013;31(20):2540-2547.
8. Paiva B, Gutierrez NC, Rosinol L, et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood. 2012;119(3):687-691.
9 San Miguel JF, Almeida J, Mateo G, et al. Immunophenotypic evaluation of the plasma cell compartment in multiple myeloma: a tool for comparing the efficacy of different treatment strategies and predicting outcome. Blood. 2002;99(5):1853-1856.
10 Paiva B, Cedena MT, Puig N, et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood. 2016;127(25):3165-3174.
11. Munshi NC, Avet-Loiseau H, Rawstron AC, et al. Association of minimal residual disease with superior survival outcomes in patients with multiple myeloma: a meta-analysis. JAMA Oncol. 2017;3(1):28-35.
12. Landgren O, Devlin S, Boulad M, Mailankody S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: a meta-analysis. Bone Marrow Transplant. 2016;51(12):1565-1568.
13. Anderson KC, Auclair D, Adam SJ, et al. Minimal residual disease in myeloma: application for clinical care and new drug registration. Clin Cancer Res. 2021;27(19):5195-5212.
14. Cavo M, San-Miguel J, Usmani SZ, et al. Prognostic value of minimal residual disease negativity in myeloma: combined analysis of POLLUX, CASTOR, ALCYONE, and MAIA. Blood. 2022;139(6):835-844.
15. Munshi NC, Avet-Loiseau H, Anderson KC, et al. A large metaanalysis establishes the role of MRD negativity in long-term survival outcomes in patients with multiple myeloma. Blood Adv. 2020;4(23):5988-5999.
16. Perrot A, Lauwers-Cances V, Corre J, et al. Minimal residual
disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. 2018;132(23):2456-2464.
17 Rawstron AC, Orfao A, Beksac M, et al. Report of the European Myeloma Network on multiparametric flow cytometry in multiple myeloma and related disorders. Haematologica. 2008;93(3):431-438.
18. Paiva B, Almeida J, Perez-Andres M, et al. Utility of flow cytometry immunophenotyping in multiple myeloma and other clonal plasma cell-related disorders. Cytometry B Clin Cytom. 2010;78(4):239-252.
19 Paiva B, Puig N, Cedena MT, et al. Measurable residual disease by next-generation flow cytometry in multiple myeloma. J Clin Oncol. 2020;38(8):784-792.
20 Paiva B, van Dongen JJ, Orfao A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood. 2015;125(20):3059-3068.
21. Flores-Montero J, Sanoja-Flores L, Paiva B, et al. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia. 2017;31(10):2094-2103.
22. Sanoja-Flores L, Flores-Montero J, Puig N, et al. Blood monitoring of circulating tumor plasma cells by next generation flow in multiple myeloma after therapy. Blood. 2019;134(24):2218-2222.
23. Roshal M, Flores-Montero JA, Gao Q, et al. MRD detection in multiple myeloma: comparison between MSKCC 10-color single-tube and EuroFlow 8-color 2-tube methods. Blood Adv. 2017;1(12):728-732.
24. Cescon DW, Bratman SV, Chan SM, Siu LL. Circulating tumor DNA and liquid biopsy in oncology. Nat Cancer. 2020;1(3):276-290.
25. Bertamini L, Oliva S, Rota-Scalabrini D, et al. High levels of circulating tumor plasma cells as a key hallmark of aggressive disease in transplant-eligible patients with newly diagnosed multiple myeloma. J Clin Oncol. 2022;40(27)3120-3131.
26. Garces JJ, Cedena MT, Puig N, et al. Circulating tumor cells for the staging of patients with newly diagnosed transplant-eligible multiple myeloma. J Clin Oncol. 2022;40(27)3151-3161.
27. Burgener JM, Rostami A, De Carvalho DD, Bratman SV. Cell-free DNA as a post-treatment surveillance strategy: current status. Semin Oncol. 2017;44(5):330-346.
28. Scherer F, Kurtz DM, Diehn M, Alizadeh AA. High-throughput sequencing for noninvasive disease detection in hematologic malignancies. Blood. 2017;130(4):440-452.
29 Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20(2):71-88.
30 Kis O, Kaedbey R, Chow S, et al. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat Commun. 2017;8:15086.
31. Mithraprabhu S, Sirdesai S, Chen M, Khong T, Spencer A. Circulating tumour DNA analysis for tumour genome characterisation and monitoring disease burden in extramedullary multiple myeloma. Int J Mol Sci. 2018;19(7):1858.
32. Manier S, Park J, Capelletti M, et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat Commun. 2018;9(1):1691.
33. Rustad EH, Coward E, Skytoen ER, et al. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica. 2017;102(7):1266-1272.
34 Mazzotti C, Buisson L, Maheo S, et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018;2(21):2811-2813.
35. Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet. 2019;20(7):404-416.
36. Jang JS, Li Y, Mitra AK, et al. Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J. 2019;9(1):2.
37. Ledergor G, Weiner A, Zada M, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med. 2018;24(12):1867-1876.
38. Shalek AK, Benson M. Single-cell analyses to tailor treatments. Sci Transl Med. 2017;9(408):eaan4730.
39. Thoren KL. Mass spectrometry methods for detecting monoclonal immunoglobulins in multiple myeloma minimal residual disease. Semin Hematol. 2018;55(1):41-43.
40. Martins CO, Huet S, Yi SS, et al. Mass spectrometry-based method targeting Ig variable regions for assessment of minimal residual disease in multiple myeloma. J Mol Diagn. 2020;22(7):901-911.
41. Chapman JR, Thoren KL. Tracking of low disease burden in multiple myeloma: using mass spectrometry assays in peripheral blood. Best Pract Res Clin Haematol. 2020;33(1):101142.
42. Mills JR, Kohlhagen MC, Dasari S, et al. Comprehensive assessment of M-proteins using nanobody enrichment coupled to MALDI-TOF mass spectrometry. Clin Chem. 2016;62(10):1334-1344.
43. Barnidge DR, Dispenzieri A, Merlini G, Katzmann JA, Murray DL. Monitoring free light chains in serum using mass spectrometry. Clin Chem Lab Med. 2016;54(6):1073-1083.
44 Liyasova M, McDonald Z, Taylor P, et al. A personalized mass spectrometry-based assay to monitor M-protein in patients with multiple myeloma (EasyM). Clin Cancer Res. 2021;27(18):5028-5037.
45. Zajec M, Jacobs JFM, Groenen P, et al. Development of a targeted mass-spectrometry serum assay to quantify M-protein in the presence of therapeutic monoclonal antibodies. J Proteome Res. 2018;17(3):1326-1333.
46. Wijnands C, Langerhorst P, Noori S, et al. M-protein diagnostics in multiple myeloma patients using ultra-sensitive targeted mass spectrometry and an off-the-shelf calibrator. Clin Chem Lab Med. 2023;62(3):540-550.
47. Derman BA, Stefka AT, Jiang K, et al. Measurable residual disease assessed by mass spectrometry in peripheral blood in multiple myeloma in a phase II trial of carfilzomib, lenalidomide, dexamethasone and autologous stem cell transplantation. Blood Cancer J. 2021;11(2):19.
48. Zamagni E, Nanni C, Dozza L, et al. Standardization of (18) F-FDG-PET/CT according to Deauville criteria for metabolic complete response definition in newly diagnosed multiple myeloma. J Clin Oncol. 2021;39(2):116-125.
49 Zamagni E, Tacchetti P, Barbato S, Cavo M. Role of imaging in the evaluation of minimal residual disease in multiple myeloma patients. J Clin Med. 2020;9(11):3519.
50 Hillengass J, Usmani S, Rajkumar SV, et al. International myeloma working group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 2019;20(6):e302-e312.
51. Zamagni E, Tacchetti P, Cavo M. Imaging in multiple myeloma: How? When? Blood. 2019;133(7):644-651.
52. Cavo M, Terpos E, Nanni C, et al. Role of (18)F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: a consensus statement by the International Myeloma Working Group. Lancet Oncol. 2017;18(4):e206-e217.
53. Moreau P, Attal M, Caillot D, et al. Prospective evaluation of magnetic resonance imaging and [(18)F]fluorodeoxyglucose positron emission tomography-computed tomography at diagnosis and before maintenance therapy in symptomatic patients with multiple myeloma included in the IFM/DFCI 2009 trial: results of the IMAJEM study. J Clin Oncol. 2017;35(25):2911-2918.
54 Zamagni E, Nanni C, Mancuso K, et al. PET/CT improves the definition of complete response and allows to detect otherwise unidentifiable skeletal progression in multiple myeloma. Clin Cancer Res. 2015;21(19):4384-4390.
55. Rasche L, Alapat D, Kumar M, et al. Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia. 2019;33(7):1713-1722.
56. Davies FE, Rosenthal A, Rasche L, et al. Treatment to suppression of focal lesions on positron emission tomographycomputed tomography is a therapeutic goal in newly diagnosed multiple myeloma. Haematologica. 2018;103(6):1047-1053.
57 Zamagni E, Nanni C, Gay F, et al. Impact of imaging FDG-PET/CT minimal residual disease assessment on outcomes and matching with bone marrow techniques in newly diagnosed transplant eligible multiple myeloma (MM) patients: results of the phase II randomized Forte trial. Blood. 2020;136(Suppl 1):27-28.
58. Moreau P, Zweegman S, Perrot A, et al. Evaluation of the prognostic value of positron emission tomography-computed tomography (PET-CT) at diagnosis and follow-up in transplanteligible newly diagnosed multiple myeloma (TE NDMM) patients treated in the phase 3 Cassiopeia study: results of the Cassiopet Companion study. Blood. 2019;134(Suppl 1):692.
59 Rasche L, Angtuaco E, McDonald JE, et al. Low expression of hexokinase-2 is associated with false-negative FDG-positron emission tomography in multiple myeloma. Blood. 2017;130(1):30-34.
60. Pandit-Taskar N. Functional imaging methods for assessment of minimal residual disease in multiple myeloma: current status and novel immunoPET based methods. Semin Hematol. 2018;55(1):22-32.
61. Anderson KC, Auclair D, Kelloff GJ, et al. The role of minimal residual disease testing in myeloma treatment selection and drug development: current value and future applications. Clin Cancer Res. 2017;23(15):3980-3993.
62. Ulaner GA, Sobol NB, O’Donoghue JA, et al. CD38-targeted immuno-PET of multiple myeloma: from xenograft models to first-in-human imaging. Radiology. 2020;295(3):606-615.
63. Ulaner GA, Landgren CO. Current and potential applications of positron emission tomography for multiple myeloma and plasma cell disorders. Best Pract Res Clin Haematol. 2020;33(1):101148.
64 Hillengass J, Ayyaz S, Kilk K, et al. Changes in magnetic resonance imaging before and after autologous stem cell transplantation correlate with response and survival in multiple myeloma. Haematologica. 2012;97(11):1757-1760.
65. Mosebach J, Shah S, Delorme S, et al. Prognostic significance of tumor burden assessed by whole-body magnetic resonance imaging in multiple myeloma patients treated with allogeneic stem cell transplantation. Haematologica. 2018;103(2):336-343.
66. Hillengass J, Merz M, Delorme S. Minimal residual disease in multiple myeloma: use of magnetic resonance imaging. Semin Hematol. 2018;55(1):19-21.
67. Messiou C, Giles S, Collins DJ, et al. Assessing response of myeloma bone disease with diffusion-weighted MRI. Br J Radiol. 2012;85(1020):e1198-1203.
68. Messiou C, Kaiser M. Whole body diffusion weighted MRI-a new view of myeloma. Br J Haematol. 2015;171(1):29-37.
69. Messiou C, Porta N, Sharma B, et al. Prospective evaluation of whole-body MRI versus FDG PET/CT for lesion detection in participants with myeloma. Radiol Imaging Cancer. 2021;3(5):e210048.
70. Belotti A, Ribolla R, Cancelli V, et al. External validation of diffusion weighted whole body MRI (DW-MRI) response assessment category (RAC) criteria proposed by the myeloma response assessment and diagnosis system (MY-RADS) imaging recommendations: prognostic role of imaging response after transplant in multiple myeloma and comparison with MRD evaluation by flow cytometry. Blood. 2020;136(Suppl 1):41-42.
71. Bockle D, Tabares P, Zhou X, et al. Minimal residual disease and imaging-guided consolidation strategies in newly diagnosed and relapsed refractory multiple myeloma. Br J Haematol. 2022;198(3)515-522.
72. Oliva S, Genuardi E, Petrucci MT, et al. Impact of minimal residual disease (MRD) by multiparameter flow cytometry (MFC) and next-generation sequencing (NGS) on outcome: results of newly diagnosed transplant-eligible multiple myeloma (MM) patients enrolled in the Forte trial. Blood. 2020;136(Suppl 1):44-45.
73. Avet Loiseau H, Sonneveld P, Moreau P, et al. Daratumumab (DARA) with bortezomib, thalidomide, and dexamethasone (VTd) in transplant-eligible patients (Pts) with newly diagnosed multiple myeloma (NDMM): analysis of minimal residual disease (MRD) negativity in Cassiopeia Part 1 and Part 2. Blood. 2021;138(Suppl 1):82.
74. Medina A, Puig N, Flores-Montero J, et al. Comparison of nextgeneration sequencing (NGS) and next-generation flow (NGF) for minimal residual disease (MRD) assessment in multiple myeloma. Blood Cancer J. 2020;10(10):108.
75. Mohan M, Kendrick S, Szabo A, et al. Clinical implications of loss of bone marrow minimal residual disease negativity in multiple myeloma. Blood Adv. 2022;6(3):808-817.
76. Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029-1035.
77 Cortes J, Rea D, Lipton JH. Treatment-free remission with
first- and second-generation tyrosine kinase inhibitors. Am J Hematol. 2019;94(3):346-357.
78. Paiva B, Manrique I, Rytlewski J, et al. Time-dependent prognostic value of serological and measurable residual disease assessments after idecabtagene vicleucel. Blood Cancer Discov. 2023;4(5):365-373.
79 Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863-2869.
80 Corre J, Montes L, Martin E, et al. Early relapse after autologous transplant for myeloma is associated with poor survival regardless of cytogenetic risk. Haematologica. 2020;105(9):e480-483.
81. Jakubowiak AJ, Kumar S, Medhekar R, et al. Daratumumab improves depth of response and progression-free survival in transplant-ineligible, high-risk, newly diagnosed multiple myeloma. Oncologist. 2022;27(7):e589-e596.
82. Costa LJ, Chhabra S, Medvedova E, et al. Daratumumab, carfilzomib, lenalidomide, and dexamethasone with minimal residual disease response-adapted therapy in newly diagnosed multiple myeloma. J Clin Oncol. 2022;40(25):2901-2912.
83. de Tute RM, Pawlyn C, Cairns DA, et al. Minimal residual disease after autologous stem-cell transplant for patients with myeloma: prognostic significance and the impact of lenalidomide maintenance and molecular risk. J Clin Oncol. 2022;40(25):2889-2900.
84. Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705-716.
85. Paiva B, San-Miguel JF, Avet-Loiseau H. MRD in multiple myeloma: does CR really matter? Blood. 2022;140(23):2423-2428.
86. Abeykoon JP, Murray DL, Murray I, et al. Implications of detecting serum monoclonal protein by MASS-fix following stem cell transplantation in multiple myeloma. Br J Haematol. 2021;193(2):380-385.
87. Rosinol L, Oriol A, Rios R, et al. Lenalidomide and dexamethasone maintenance with or without ixazomib, tailored by residual disease status in myeloma. Blood. 2023;142(18):1518-1528.
88. Diamond B, Korde N, Lesokhin AM, et al. Dynamics of minimal residual disease in patients with multiple myeloma on continuous lenalidomide maintenance: a single-arm, singlecentre, phase 2 trial. Lancet Haematol. 2021;8(6):e422-e432.
89 Sonneveld P, Dimopoulos MA, Boccadoro M, et al. Daratumumab, bortezomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2024;390(4):301-313.
Gaël Vermeersch,1,2 Paul Proost,2 Sofie Struyf,2 Mieke Gouwy2 and Timothy Devos1,2
1Department of Hematology, University Hospitals Leuven and 2Laboratory of Molecular Immunology, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, University of Leuven, Leuven, Belgium
Correspondence: T. Devos timothy.devos@uzleuven.be
Received: December 27, 2023. Accepted: February 16, 2024. Early view: February 29, 2024.
https://doi.org/10.3324/haematol.2023.284921 ©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
BCR::ABL1 negative myeloproliferative neoplasms (MPN) form a distinct group of hematologic malignancies characterized by sustained proliferation of cells from multiple myeloid lineages. With a median survival of 16-35 months in patients with high-risk disease, primary myelofibrosis (PMF) is considered the most aggressive entity amongst all BCR::ABL1 MPN. Additionally, for a significant subset of patients, MPN evolve into secondary acute myeloid leukemia (AML), which has an even poorer prognosis compared to de novo AML. As the exact mechanisms of disease development and progression remain to be elucidated, current therapeutic approaches fail to prevent disease progression or transformation into secondary AML. As each MPN entity is characterized by sustained activation of various immune cells and raised cytokine concentrations within bone marrow (BM) and peripheral blood (PB), MPN may be considered to be typical inflammation-related malignancies. However, the exact role and consequences of increased cytokine concentrations within BM and PB plasma has still not been completely established. Up-regulated cytokines can stimulate cellular proliferation, or contribute to the development of an inflammation-related BM niche resulting in genotoxicity and thereby supporting mutagenesis. The neutrophil chemoattractant CXCL8 is of specific interest as its concentration is increased within PB and BM plasma of patients with PMF. Increased concentration of CXCL8 negatively correlates with overall survival. Furthermore, blockage of the CXCR1/2 axis appears to be able to reduce BM fibrosis and megakaryocyte dysmorphia in murine models. In this review, we summarize available evidence on the role of the CXCL8-CXCR1/2 axis within the pathogenesis of PMF, and discuss potential therapeutic modalities targeting either CXCL8 or its cognate receptors CXCR1/2.
BCR::ABL1 negative myeloproliferative neoplasms (MPN) constitute a distinct group of hematologic malignancies characterized by sustained proliferation of cells from multiple myeloid lineages. Within MPN, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF) are the 3 most common entities. PV is characterized by panmyelosis, ET by thrombocytosis, while PMF can present with various changes in blood cell count and is characterized by extensive formation of fibrous tissue within the bone marrow (BM). Most patients with MPN harbor mutually exclusive somatic mutations, which constitutively activate signal transducing pathways resulting in uncontrolled cellular proliferation. The genes Janus kinase 2 (JAK2), myeloproliferative leukemia virus onco-
gene (MPL), and calreticulin (CALR) are the most affected with mutational frequencies varying amongst different MPN subtypes. Within all subtypes, JAK2V617F is the most common mutation with a reported frequency of approximately 95% in patients with PV, 60% in ET, and 50% in PMF. Additionally, roughly 30% of patients with PMF harbor mutations in the CALR gene and 10% in the MPL gene. A small percentage of patients with PMF are considered triple negative, which indicates the absence of mutated JAK2, CALR or MPL 1 While the majority show slow progression, for a subset of patients, MPN rapidly evolve into BM failure or they develop secondary acute myeloid leukemia (AML) (frequency 10-15%), also called MPN blast phase.2 Patients with PMF show highly variable survival rates, ranging from several decades to a median survival of 16-35 months for patients with high-risk disease.3
While in the past they were considered as separate entities, it is currently well accepted that MPN form a continuum wherein entities can evolve into each other. However, the exact mechanisms of disease development, transformation, and progression remain to be elucidated. MPN may be considered to be a model of inflammation-related cancer development as each MPN entity is characterized by sustained activation of various immune cells and tends to show a unique cytokine expression pattern within BM and peripheral blood (PB). Expression of the pro-inflammatory chemokine CXCL8 (also known as interleukin-8 [IL-8]) is increased in PB and BM plasma of patients with myelofibrosis and its concentration negatively correlates with overall survival (OS).4-6 Here we discuss the potential role of CXCL8 and its cognate receptors CXCR1/2 in the pathogenesis of PMF.
As mentioned above, the JAK2, CALR and MPL genes frequently carry acquired MPN-restricted driver mutations. The JAK2 protein is a member of the JAK family and is characterized by two kinase domains amongst which one is catalytically active while the other functions as a pseudokinase preventing self-activation. JAK2 is intracellularly connected with receptors such as the erythropoietin receptor (EPOR), MPL, and granulocyte-colony stimulating factor receptor (G-CSFR). Activation by the appropriate ligands induces a conformational change and then results in activation of JAK2 through phosphorylation. Phosphorylated JAK2 functions as a docking station for signaling molecules, such as signal transducer and activator of transcription (STAT), which eventually initiates further downstream signaling resulting in cellular proliferation.7 Independently from STAT, JAK2 may also initiate other signaling pathways, e.g., mitogen activated protein-kinase (MAPK), AKT (protein kinase B) or phosphoinositide 3 (PI3)-kinase (Figure 1). The MPL gene codes for the thrombopoietin (TPO) receptor, which activates JAK2 upon binding of its ligand. Within MPN, gain-of-function-mutations of MPL typically occur at amino acid W515 causing activation of the MPL receptor, and downstream JAK-STAT signaling, independently from TPO binding.5-7
In contrast to the genes mentioned above, the CALR encoded protein is not directly involved in cellular proliferation but is a chaperone contributing to calcium storage and structural control of N-glycosylated proteins. In its mutated form, CALR interacts with the TPO receptor and induces constitutive activation of JAK2 and STAT proteins without binding of TPO. CALR mutations are described as type 1 or 2 depending on the presence of a 52-base pair
deletion or 5-base pair insertion in exon 9, respectively.7-9 Type 1, which is more prevalent in PMF, is associated with greater phenotypic changes, including BM hypocellularity and megakaryocytic lineage amplification.10 Instead of being monoclonal, MPN may possibly be an oligoclonal disease characterized by the existence of several molecular distinct clones at once. Previously it has been proposed that patients with MPN may generally show two distinct patterns of acquiring mutations. Firstly, those who acquire mutations in a driver gene followed by additional mutations. Secondly, those acquiring driver mutations on a background of mutations already present in non-driver genes. Many of these affected non-driver genes, such as Tet methylcytosine dioxygenase 2 (TET2) and DNA methyltransferase 3 (DNMT3A), are frequently involved in the age-related phenomenon clonal hematopoiesis of intermediate potential (CHIP). CHIP is characterized by the acquisition of somatic mutations resulting in the expansion of clonal hematopoietic progenitor cells. Several genes predict worse prognosis or are associated with blast phase when mutated; amongst these are TET2, ASXL1, and TP53 11,12 The role of inherited variants in these genes is still not completely understood and concerns a growing area of research within MPN.11 Germline polymorphisms may contribute or predispose a person to the development of a chronic inflammatory state, characterized by increased cytokine production or myeloid response, and thus genetic instability or even MPN development.5,11
In recent years, researchers studying MPN pathophysiology expanded their focus from hematopoietic stem and progenitor cells (HSPC) to the whole microenvironment surrounding these cells, called ‘the bone marrow niche’.5 The BM is one of the most complex tissues within the human body and comprises multiple cell types, such as endothelial cells, multipotent mesenchymal stromal cells, osteoblasts, and adipocytes. As such, one cell type may influence the functioning of another and vice versa. It is well known that the composition and functioning of the BM niche is extensively influenced by changing conditions, such as inflammation or infection.13-15 Megakaryocytes play a central role within MPN pathogenesis. The mutually exclusive driver mutations mentioned above result in constitutive activation of the JAK2 signaling pathway, which initially results in megakaryocyte hyperplasia and subsequently dysplasia.7 Aberrant megakaryopoiesis is a pathological hallmark of MPN, as megakaryocytes in myelofibrosis display morphologic abnormalities such as hypolobulated nuclei and clustering. Next to this, higher proliferative capacities and decreased rates of apoptosis are observed. Single cell analysis revealed aberrant molecular signatures and differentiational bias
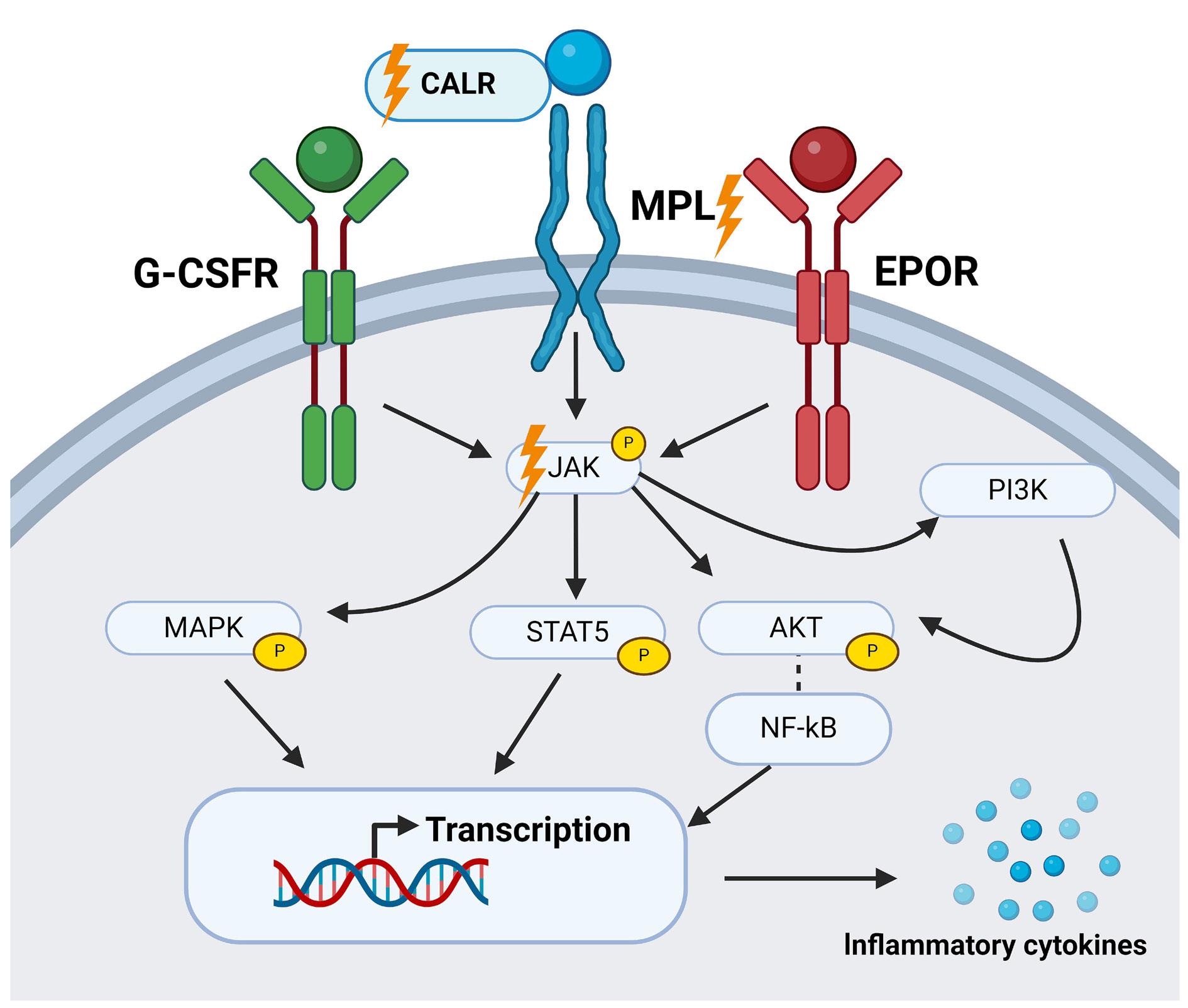
Figure 1. Overview of signaling pathways in myeloproliferative neoplasms. The Janus Kinase 2 (JAK2) protein is intracellularly connected with receptors such as the granulocyte-colony stimulating factor receptor (G-CSFR), the thrombopoietin receptor (myeloproliferative leukemia virus oncogene [MPL]), and erythropoietin receptor (EPOR). Activation of these receptors by their appropriate ligands (G-CSF, thrombopoietin [TPO] and EPO, respectively) induces phosphorylation of JAK2. Phosphorylated JAK2 serves as a docking station for signal transducer and activator of transcription 5 (STAT5) which initiates further downstream signaling pathways. Activated JAK2 may also stimulate the activation of other pathways such as mitogen activated protein-kinase (MAPK), phosphoinositide 3-kinase (PI3K) or the nuclear factor kappa B pathway (NF-κB) through activation of AKT (protein kinase B). NF-κB activation can also be mediated through activation of toll-like receptors (TLR) (not shown). Mutations in JAK2, calreticulin (CALR) or MPL genes result in uncontrolled activation of these proliferative pathways, enhanced cellular survival and production of various inflammatory cytokines, together promoting development of hematologic malignancies. Lightning symbol indicates occurrence of mutations in myeloproliferative neoplasm-associated driver genes (MPL, CALR and JAK2) coding for the corresponding proteins.
towards megakaryocyte characteristics in hematopoietic stem cells of patients with MPN.16 MPN-associated megakaryocytes express low levels of the GATA1 transcription factor, which is associated with increased production of transforming growth factor (TGF)-b. TGF-b is a pleiotropic cytokine with anti-inflammatory but profibrotic properties, and stimulates production of collagen, fibronectin and extracellular matrix. In addition to TGF-b, megakaryocytes in MPN show increased secretion of other cytokines, such as CXCL8, IL-6, and platelet-derived growth factor (Figure 2).17
Furthermore, histological analysis of BM from MPN patients shows an increased incidence of megakaryocytes enclosing neutrophilic granulocytes, a phenomenon called emperipolesis. Emperipolesis appears to be preserved amongst mammalian species and is increased in conditions associated with systemic inflammation and high platelet demand. The phenomenon of megakaryocytes engulfing neutrophils was first described by Larsen in 1970, but the
exact biological role and molecular mechanism is still not fully understood.18,19 Emperipolesis is most likely mediated through multiple ligand-receptor interactions. Reduced in vitro emperipolesis is observed in megakaryocytes derived from mice deficient in intracellular adhesion molecule-1 (ICAM-1) and CD18.20 CD18 (also known as lymphocyte function-associated antigen 1 [LFA-1]) is a b2-integrin expressed on neutrophils and is, through various interactions including with ICAM-1, a primary receptor involved in neutrophil recruitment to inflamed environments.21 P-selectin, or CD62P, which is normally restricted to the α-granules, shows aberrant expression on the demarcation system of megakaryocytes within GATA1low mice. GATA1low mice function as a murine model of PMF, recapitulating the hyperactivation of the TPO/MPL/JAK2 axis. Interestingly, within these mice, the deletion of CD62P disrupts interactions between neutrophils and megakaryocytes, and results in reduced concentrations of TGF-b and fibrosis.22-25 Moreover, in GA-
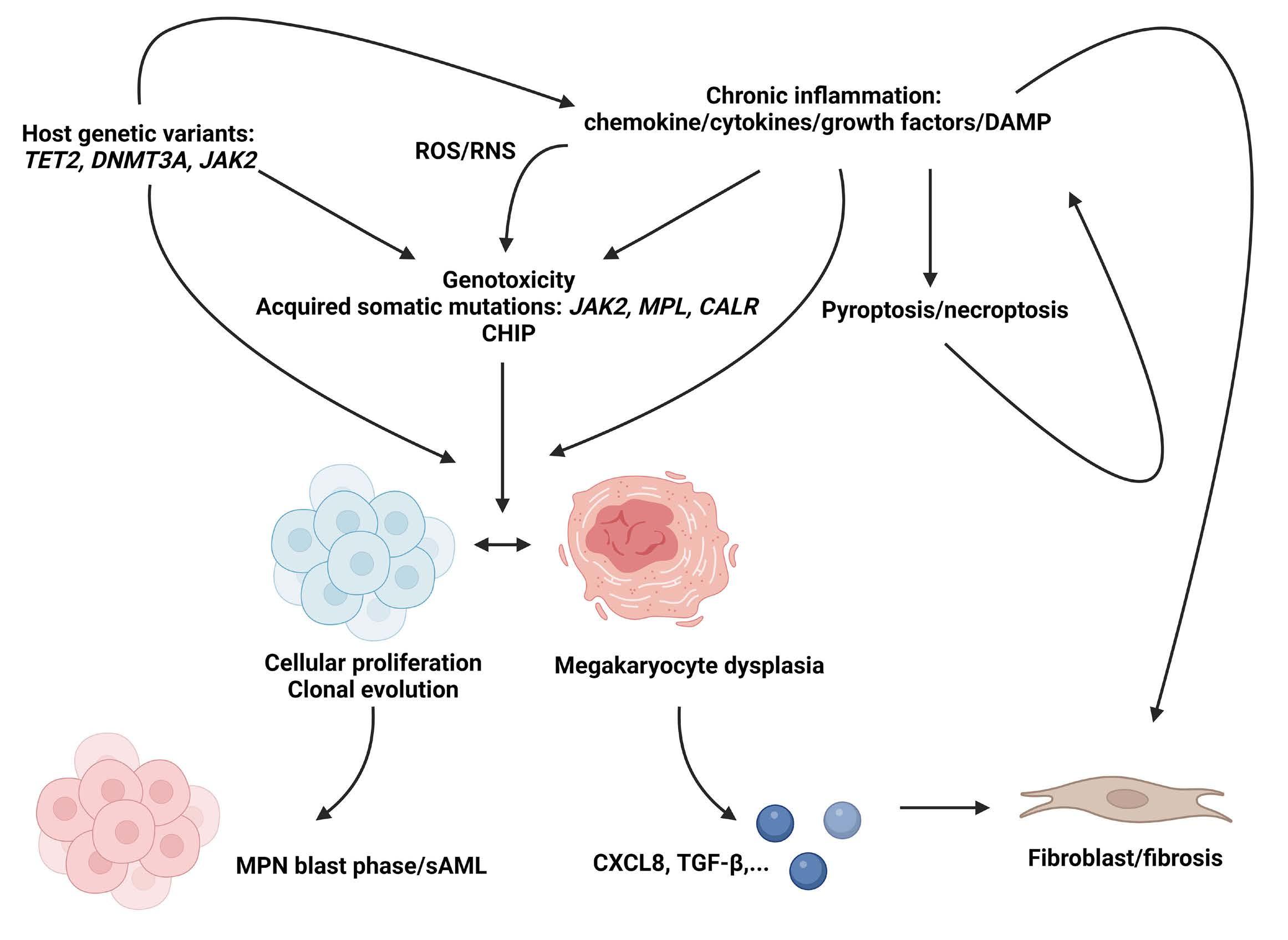
Figure 2. Overview of primary myelofibrosis pathophysiology. The role of inherited genetic variants is incompletely understood in primary myelofibrosis (PMF) and other BCR::ABL1 negative myeloproliferative neoplasms (MPN) but may contribute to an increased susceptibility of acquiring somatic mutations or chronic inflammatory states. Acquired somatic mutations may occur in key driver genes encoding Janus Kinase 2 (JAK2), the thrombopoietin receptor (MPL) or calreticulin (CALR) or may contribute to the development of clonal hematopoiesis of intermediate potential (CHIP), of which the pathophysiological role in MPN is also not completely understood. PMF, like other MPN, is associated with a chronic hyperinflammatory state characterized by increased concentrations of chemokines, cytokines, such as tumor necrosis factor-alpha (TNF-α) and growth factors. These may contribute to cellular proliferation, clonal evolution and megakaryocyte dysplasia, or may directly stimulate fibrosis through interaction with fibroblasts/mesenchymal cells. Direct immunologic cytotoxicity through release of reactive oxygen/nitrogen species (ROS/RNS) by activated leukocytes (e.g., downstream of TLR sensing of damage associated molecular pattern molecules [DAMP]) may be another pathway resulting in genotoxic stress or megakaryocyte dysplasia. Whether neutrophil engulfment by megakaryocytes (i.e., emperipolesis) directly contributes to megakaryocyte dysplasia remains to be elucidated. Clonal evolution may eventually result in blast transformation or secondary acute myeloid leukemia (sAML). Aberrant megakaryocytes show increased secretion of cytokines such as CXCL8 (interleukin-8 [IL-8]) or transforming growth factor-b (TGF-b) which contribute to the development of PMF.
TA1low mice, the use of reparixin, which acts as an inhibitor of the CXCL8 receptors CXCR1/CXCR2, or anti-CD62P antibodies combined with ruxolitinib resulted in reduced chemotaxis of neutrophils and decreased emperipolesis between neutrophils and megakaryocytes.26,27 Within mouse models not mimicking PMF pathophysiology, the use of CD18 antibodies also reduced neutrophil-megakaryocyte emperipolesis, while blocking antibodies against other membrane targets such as CD62P and CXCR2 appeared to have no effect.20,28 It is important to mention that, in 2019, the ADORE trial investigated the clinical efficacy of 5 different agents, amongst which the monoclonal anti-CD62P antibody crizanlizumab, in combination with ruxolitinib. Unfortunately, the study was suspended in 2022 after an interim-analysis.29
Myeloproliferative neoplasms may be considered to be typical inflammation-related malignancies, with, notably, PMF as the subtype associated with the highest inflammatory burden. Previous research tried to identify whether specific cytokine signatures correlate with MPN subtypes. However, most of those studies provided heterogenous results and primarily focused on PB plasma.4 Nonetheless, as cytokine functionality may be dose-dependent, some cytokines may be relevant at the BM level, whereas their concentration within PB plasma may be less relevant. Focusing solely on PB concentrations may thus result in the incorrect neglect of potential cytokines contributing within BM pathophys-
iology. There is increasing evidence that the presence of an inflammatory cytokine storm within the BM niche may trigger the development of myelofibrosis or even stimulate transformation into secondary AML. Only a select number of studies evaluated BM cytokine profiles in myelofibrosis compared to BM from other MPN subtypes or healthy controls. Previous studies demonstrated significantly increased levels of CXCL8, CXCL10 (interferon γ-induced protein 10 [IP-10]), IL-6Ra, IL-18, and TGF-b in BM of patients with PMF compared to healthy controls.4,30,31 Others measured considerably different cytokine concentrations in BM compared to PB. By example, one study investigating cytokine profiles in BM versus PB of 24 MPN patients reported significantly higher concentrations of 10 cytokines (IL-1ra, IL-1b, IL-7, IL-12p40, IL-15, IL-16, CXCL9 [monokine induced by γ interferon/MIG], macrophage colony-stimulating factor [M-CSF], granulocyte colony-stimulating factor [G-CSF], platelet-derived growth factor-BB [PDGF-BB]) and tissue inhibitor of metallopeptidase inhibitor 1 (TIMP-1) in the BM niche. Compared to PB plasma from healthy controls, CXCL8 was significantly elevated in both PB plasma and BM of patients with MPN. However, no statistically significant differences in CXCL8 concentrations were observed between BM and PB from patients. As this study included only a limited number of patients (i.e., 4 with PMF), further studies are needed.32 Although constitutive activation of JAK-STAT appears to be a major player in the pathogenesis of MPN, current therapeutic approaches inhibiting JAK2, such as ruxolitinib, seem to be ineffective in preventing evolution of the disease or avoiding transformation into secondary AML. Therefore, a role of other downstream signaling pathways in the hyperproliferative state associated with MPN is suspected. This hypothesis is supported by other findings, amongst which the long latency between acquiring JAK2 mutational status and development of the disease, as well as the different observed disease phenotypes and kinetics despite identical underlying mutation.12 Currently, allogeneic stem cell transplantation remains the only potentially curative treatment option for PMF. In addition, the often higher age of patients with PMF frequently limits the ability to use full intensity conditioning. However, it has to be mentioned that reduced-intensity regimens still offer significant survival advantages in these patients.33
Recent research shows persistent hyperactive nuclear factor kappa-B (NF-κB) and MAPK signaling in patients with myelofibrosis treated with the JAK2-inhibitor ruxolitinib. Interestingly, the concentration of cytokines, including that of CXCL8, appears to be only minimally influenced by treatment with ruxolitinib.34 NF-κB hyperactivation was not only confined to CD34+ cells, but was observed throughout different myeloid and lymphoid cell populations. It is hypothesized that, through production of NF-κB-activating cytokines, NF-κB hyperactivation may be transmitted from malignant clones to non-malignant cells.35 NF-κB is a central transcriptional regulator of various inflammatory cytokines
aberrantly expressed in PMF, including CXCL8, TGF-b, and tumor necrosis factor-alpha (TNF-α). In general, 2 distinct NF-κB activation pathways, known as the classical and alternative pathways, can be distinguished. The classical, or canonical, NF-κB pathway is activated downstream of toll-like receptors (TLR), for example, activated by S100A8 and S100A9, or by cytokines (e.g., IL-1b and TNF-α) in an autocrine loop.5,36-38 Activation of the canonical pathway is associated with myeloproliferation in situations such as emergency hematopoiesis and myeloid malignancies. TLR are part of the innate immune system and function as pattern recognition receptors that recognize pathogen-associated molecular patterns (PAMP) from microbial organisms and damage-associated molecular patterns (DAMP), such as S100A8/9 resulting from cellular damage.36 Release of TNF-α or DAMP may result in pyroptosis and necroptosis, which are different forms of programmed cell death and may further stimulate local inflammation through release of additional cytokines and DAMP (Figure 2).39,40 TNF-α can activate various downstream signaling pathways through binding with its receptors TNFRSF1a and TNFRSF1b (also known as TNFR1 and TNFR2). These receptors are, respectively, associated with either apoptosis or proliferation, both activating NF-κB pathways in their target cells. The dual functioning of TNF-α resulted in the hypothesis that TNF-α may promote clonal dominance by simultaneously inhibiting benign hematopoiesis while stimulating myeloproliferation of the malignant clones.5 In mice, it has been shown that release of S100A8/9 results in genotoxic stress, and transcriptional activation of the S100A8/9-TLR pathway predicts leukemic evolution and progression-free survival in myelodysplastic syndromes (MDS).41 Basiorka et al. recently showed that the formation of large, filamentous clusters of apoptosis-associated speck-like protein containing a CARD (also known as PYCARD or ASC) adaptor protein might serve as a biomarker for pyroptotic cell death in MDS and correlates with S100A8/9 concentration. These clusters are called ASC specks and are released upon cytolysis. Within this study, no statistical differences were observed in patients with PMF; however, only 3 patients were included.42 Release of IL-1b has a direct, stimulatory effect on megakaryopoiesis, promotes polyploidization, and results in increased levels of profibrotic TGF-b. Pharmacological inhibition of IL-1b reduced myelofibrosis in a Jak2V617F mouse model and combination with ruxolitinib even resulted in complete reversal of fibrosis.43
The CXCL8 gene, composed of 4 exons and 3 introns, is located on chromosome 4 and codes for a precursor CXCL8 protein of 99 amino acids.44 This precursor protein is eventually cleaved into a 77 amino acid (CXCL8(1-77)), or
less frequently a 79 amino acid (CXCL8(-2-77)), protein and can be produced by almost every cell type. CXCL8 is part of the CXC-chemokine family, which contains low molecular mass proteins (~8-10 kDa) that guide leukocyte migration during homeostasis and inflammatory states. The chemokine subfamily classification in CXC or CC chemokines is based on conserved cysteines along the protein structure. While CXC chemokines generally bind CXC receptors (CXCR) and CC chemokines bind CC receptors (CCR), chemokine redundancy is observed (i.e., several chemokine ligands attract the same leukocyte subtype because they bind to the same receptor).45
CXCL8 interacts with its chemokine receptors CXCR1 and CXCR2, previously known as IL-8RA and IL-8RB respectively. The human IL8RA and IL8RB genes are located on chromosome 2.46 Both receptors are distinguished by their ligand selectivity. CXCR1 shows high affinity for CXCL6 (granulocyte chemotactic protein-2 [GCP-2]) and CXCL8. In addition to these ligands, CXCR2 also binds CXCL1 (growth-related oncogene- α [GRO- α ]), CXCL2 (GRO- b ), CXCL3 (GRO- γ ), CXCL5 (epithelial cell-derived neutrophil-activating peptide-78 [ENA-78]), and CXCL7 (neutrophil-activating peptide-2 [NAP-2]).47 Both receptors are predominantly expressed on neutrophils but also appear on other myeloid or lymphoid immune cells, such as basophils, monocytes, and CD8+ T-lymphocytes.48 Aberrant CXCL8 signaling is present in various hyperinflammatory and fibrosis-related diseases, amongst which idiopathic pulmonary fibrosis.45,49 The production of CXCL8 may be increased in response to pro-inflammatory cytokines, such as IL-1 and TNF- α , which stimulate CXCL8 production by binding on their cognate receptors and activating the NF- κ B pathway.46,48,50,51 CXCL8 activity is also influenced by post-translational changes, such as truncation by proteases. By example, truncation of CXCL8 (1-77) to CXCL8 (7-77) by gelatinase B, a matrix metalloproteinase (MMP-9) mainly produced by neutrophils, results in a 10- to 27-fold higher potency in neutrophil activation. 52,53
The variable quaternary structures of chemokines (existing potentially as monomers, (hetero)dimers, multimers, or in association with soluble or cell-bound glycosaminoglycans) adds an extra complexity to the research into their functionalities and receptor interactions. These variables further explain why divergent effects may be observed with the same chemokine. In vitro experiments suggest CXCL8, which may exist as a monomer or dimer, to be more potent as a monomer. Nonetheless, the exact effect of its quaternary structure on functionality is not completely understood. 53,54 It was recently shown that CXCL8 mainly tends to bind with CXCR2 as a dimer, whereas CXCR1 strongly binds CXCL8 as a monomer. In the case of the CXCL8 dimers, one monomer interacts with the chemokine recognition site 1 (CRS1) of CXCR2. CRS1 is located at the NH 2-terminus of the receptor and originates from a conserved Pro-Cys (PC) motif. While
CRS1 is responsible for the initial recruitment of CXCL8, another region called CRS2 appears to be essential for activation of CXCR2 and interacts with the conserved GluLeu-Arg motif (ELR) of CXCL8. The ELR motif is located at the NH2-terminus of CXCL8 and is highly conserved amongst all CXC chemokines with neutrophil-activating characteristics. 55 Contrary to other ELR + chemokines (CXCL1/2/3/5/7), CXCL8 also binds to CXCR1. This specificity of CXCL8 for CXCR1 can be explained by the higher number of polar residues within the CRS1 region of CXCR1 and the charged residues in the NH 2-terminal regions of CXCL8; for example, a salt bridge is formed between D26 in CXCR1 and K16 in CXCL8(1-77). For the reader interested in structural biology, we refer to the articles from Ishimoto et al. and Liu et al. wherein the structural basis of, respectively, CXCR1 and CXCR2 activation is presented by using cryo-electron microscopy. 55,56
Classical chemokine receptors, including CXCR1 and CXCR2, are G protein-coupled receptors (GPCR). Activation of GPCR results in the dissociation of the G α and G b / γ subunit.47 The separated G b / γ subunit activates phospholipase C b 2 (PLC b 2), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) and subsequently forms the secondary messenger molecules diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3). The formation of IP3 eventually results in the release of Ca2+ from the endoplasmic reticulum and activates protein kinases, such as protein kinase C (PKC), which is crucial for cellular migration, degranulation, and adhesion.48 Besides PLC b 2, activation of CXCR1/CXCR2 may also initiate other pathways such as activation of phosphoinositide 3-kinase γ (PI3K γ ) and phospholipase D (PLD). PI3K γ phosphorylates PIP2 into phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and activates kinases such as AKT (protein kinase B), resulting in increased cellular proliferation and survival. Activation of PLD is specifically linked to CXCR1 and is associated with the production of reactive oxygen/nitrogen species (ROS/RNS), as well as the release of neutrophil extracellular traps (NET) (Figure 3).48,57-59 CXCL8 is also able to bind the Duffy antigen/receptor for chemokines (DARC), also known as atypical chemokine receptor ACKR1, which functions as a scavenging ‘sink’ receptor on red blood cells (RBC) and influences the plasma concentration of various chemokines. It is believed that DARC may play a critical role in preventing oncogenesis by reducing the load of protumorigenic/proangiogenic chemokines. As such, DARC status may provide a potential explanation for the higher incidence rates and more aggressive characteristics of breast cancer in Black/African-American women, who generally carry the Duffy null allele on RBC with higher frequency, compared to White/European-American women.47,60,61 Although DARC status was not investigated, Peseski et al. previously reported significantly reduced OS of Non-white compared to White patients with MPN.62 Contrary to humans, mice do not express CXCL8 but ex-
press lipopolysaccharide-induced CXC chemokine (LIX) or GCP-2, which is the murine homolog of human CXCL5 and CXCL6, as most potent neutrophil-attracting chemokine. As LIX/GCP-2 is able to bind both CXCR1 and CXCR2, it is also considered to be a functional homolog of human CXCL8.48 While the functional characteristics of murine CXCR2 have been well characterized, those of murine CXCR1 remain largely unknown. Consequently, most of our knowledge is derived from studies focusing on CXCR2. Interestingly, human CXCL8 is able to bind both murine CXCR1 and CXCR2.63
CXCL8 concentrations are increased independently from mutational status within PB plasma of patients with PMF.64,65 Similarly, within BM CXCL8, concentrations are increased amongst all MPN subtypes (PV, ET, and PMF) and no significant association between cytokine levels and mutational status is observed.31 Within MPN, increased concentration of CXCL8 correlates with adverse outcomes, including reduced OS. Nonetheless, the exact role of CXCL8 and its cognate receptors in myelofibrosis are still unknown. Single-cell cytokine

Figure 3. Molecular properties of CXCL8 and its cognate receptors CXCR1 and CXCR2. CXCL8 interacts with its cognate receptors CXCR1 and CXCR2, which are predominantly expressed on neutrophils but may also appear on lymphoid immune cells or other myeloid cells such as megakaryocytes. In contrast with CXCR1, the CXCR2 receptor appears over-expressed in CD34+ cells from patients with myelofibrosis compared to healthy controls. CXCR1 and CXCR2 are G protein-coupled receptors and CXCL8 mainly tends to bind CXCR2 as a dimer, while CXCR1 strongly binds CXCL8 as a monomer. After activation of the GPCR, the G-protein dissociates into Gα and Gb/γ subunits. The separated Gb/γ subunits activate phospholipase C b2 (PLCb2) which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) and then forms the secondary messenger molecules diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3). IP3 stimulates the release of Ca2+ from the endoplasmic reticulum. Release of Ca2+ activates kinases, such as protein kinase C (PKC), which play a role in cellular migration or degranulation. Besides PLCb2, the Gb/γ subunit may also activate phosphoinositide 3-kinase γ (PI3Kγ) which phosphorylates PIP2 into phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 may activate kinases such as AKT (protein kinase B) and hereby stimulate cellular proliferation. Activation of phospholipase D (PLD) is mediated by CXCR1 activation and plays a role in the release of reactive oxygen-nitrogen species (ROS/RNS) and neutrophil extracellular traps (NET). The activity of CXCL8 may be reduced by heterodimerization with CXCL4 (also known as platelet factor-4 [PF-4]) or interaction with the Duffy antigen/receptor for chemokines (DARC) which functions as a scavenger receptor. CXCL8 properties may be influenced by post-translational changes, such as truncation by proteases, amino-acid side-chain modifications (e.g., citrullination or tyrosine nitration) or variations in quaternary structure (i.e., formation of monomers/dimers). AKT: protein kinase B; CXCL4: chemokine (CXC motif) ligand 4 (also known as platelet factor-4); CXCL8: CXC chemokine ligand 8 (also known as interleukin-8 [IL-8]), DAG: diacylglycerol; DARC: Duffy antigen/receptor for chemokines; IP3: inositol 1,4,5-triphosphate; PI3Kγ: phosphoinositide 3-kinase γ; PIP2: phosphatidylinositol 4,5-bisphosphate; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; KC: protein kinase C; PLD: phospholipase D.
assays revealed an increased proportion of CXCL8 secreting CD34+ cells within patients with myelofibrosis compared to other MPN-subtypes. Patients with expanded CXCL8-secreting clones showed higher leukocytosis and higher-grade reticulin fibrosis compared to patients without these clones.6,66 CXCL8 negatively regulates healthy hematopoiesis, including megakaryopoiesis, through mechanisms that are still not completely understood.67-70 However, contradictory observations were made as other researchers showed enhanced cellular proliferation and fitness of MF-derived CD34+ cells co-cultured with exogenous CXCL8. It is still not known whether these differences might be explained by dose- or time-dependent mechanisms.6 The effects of CXCL8 on megakaryopoiesis are most likely mediated through CXCR1/2 signaling, as expression of both receptors was previously shown in megakaryocytes and megakaryocyte progenitor cells.70,71 In contrast with CXCR1, the CXCR2 receptor appears over-expressed in CD34+ cells from patients with myelofibrosis compared to healthy controls.6,66 Interestingly, the use of neutralizing antibodies against either CXCL8, CXCR1 or CXCR2 resulted in increased megakaryocyte maturation and reduced ploidy.66 Recent findings also indicate a selective advantage of pre-malignant hematopoietic stem cell clones aberrantly expressing CXCL8 through increased interactions with the endothelial niche.72 Among 30 tested cytokines within PB of patients with PMF, increased CXCL8 concentrations predicted inferior leukemia-free survival and CXCL8 was the only cytokine associated with ≥1% circulating blasts.64 One of the mechanisms preventing CXCL8-mediated activation of CD34+ progenitor cells might be the formation of heterodimers with CXCL4. CXCL4, also known as platelet factor-4 (PF-4), is a CXC chemokine and abundant α-granule protein within BM. The functional consequences of this heterodimerization vary; CXCL8 and CXCL4 synergize in the attraction of neutrophils, whereas the angiostatic activity of CXCL4 prevails above the angiogenic activity of CXCL8, likewise the binding of CXCL4 to CXCL8 inhibits CXCL8-mediated signaling in CD34+ progenitor cells.73,74 It has been proposed that high intramedullary concentrations of CXCL4 and CXCL8 might promote extramedullary hematopoiesis, which is extensively present in PMF. Extramedullary hematopoiesis notably involves the mobilization of hematopoietic, mesenchymal, and endothelial cells to so-called ‘new’ vascular niches within involved organs such as the spleen and liver. Although the exact mechanisms contributing to mobilization of these cells are still not fully understood, these extramedullary hematopoietic niches tend to play an important role in MPN progression.66,75,76 For example, in contrast to BM progenitor cells, it was previously shown that blood-derived CD34+ progenitors expanded and differentiated better when co-cultured with fibroblasts derived from myelometaplasic spleen compared to fibroblasts derived from normal BM.77 Within the GATA1low model, it has also been suggested that CD62P-dependent interac-
tion between neutrophils and megakaryocytes within the spleen mediates local production of TGF-b and thus the formation of a splenic environment supporting the proliferation of hematopoietic stem cells.22
The complex interplay between chemokines and hematopoiesis in these different hematopoietic niches is far from completely understood but forms an essential field of research. It is important to emphasize that chemokines might act differently within these microenvironments, as chemokines tend to show context-dependent functionalities. Indeed, (hetero)dimerization, processing, synergy and/or antagonism may drastically affect chemokine activity and chemokines known as ‘inhibitory’ may become ‘stimulatory’.71,78
Angiogenesis and expression of proangiogenic factors, such as vascular endothelial growth factor (VEGF) are increased within the BM of MPN patients, especially in PMF. The JAK2 pathway tends to play a central role in PMF-associated angiogenesis, as a strong positive correlation between BM microvessel density and JAK2V617F mutant allele burden (≥55% mutant alleles) was found. Nonetheless, similar to hematopoiesis, angiogenesis in MPN involves multiple pathways, as microvessel density is increased in JAK2 negative cases as well, and mutated JAK2 is only present in approximately 50% of patients with PMF.4,79-81 Contrary to microvessel density, BM VEGF expression does not clearly correlate with JAK2V617F mutant allele burden.79 Besides these proangiogenic factors, chemokines such as CXCL8 are also known inducers of angiogenesis. All ELR+ CXC chemokines stimulate endothelial cell migration and proliferation, whereas CXCR3 binding chemokines that lack this ELR motif are angiostatic. CXCL8 stimulates angiogenesis through its interaction with both CXCR1 and CXCR2 on endothelial cells, resulting in a 2-phase process, characterized by an early phase with the formation of actin stress fibers, and a later phase with cortical actin accumulation and cell retraction.82 Elevated cytokines in PMF, such as IL-1b induce CXCL8 and thus angiogenesis, while others, including interferon-α (IFN-α), IFN-b, and IFN-γ, up-regulate angiostatic CXCR3 ligands (CXCL9, CXCL10 and CXCL11).55,83
CXCR1 and CXCR2 are key receptors mediating activation and chemotaxis of neutrophils. Researchers previously tried to reveal discriminating characteristics of both receptors through investigation of their downstream signaling pathways. CXCR1 plays a crucial role in the chemotaxis of neutrophils, as well as in the release of ROS and NET.45,84 The CXCL8-CXCR1/2 axis could thus play an important role in the increased NETosis observed in patients with MPN and its association with thrombosis.85 Nonetheless, current data on the role of NETosis in MPN-associated thrombosis is conflicting and beyond the scope of this review.85-87 Naïve
neutrophils show higher CXCR1 expression compared to cells in an activated state. Indeed, CXCR1 expression is down-regulated by increased concentrations of cytokines, such as TNF- α, or through the activation of TLR2 and TLR4. Like CXCR1, CXCR2 is a major chemokine receptor in regulating neutrophil mobility and appears to be more responsive to lower CXCL8 concentrations. Activation of CXCR2 tends to stimulate CXCL8 signaling through CXCR1 as it increases its expression. In contrast, activation of CXCR1 results in downregulation of CXCR2 surface expression.45,84,88,89 In physiological circumstances, the release of maturated neutrophils from the BM is mediated by the activation of CXCR2, which antagonizes the effects of the CXCL12 (stromal cell-derived factor 1α [SDF-1α])/CXCR4 chemokine axis.90 Interaction between CXCR4 and its ligand CXCL12 retains CXCR4 expressing cells within the BM niche. CXCR4 is down-regulated on mature neutrophils by cytokines, e.g., G-CSF. Similar to CXCR1, the expression of CXCR2 is influenced by the cellular state of activation, and stimulation of the cells with TNF-α results in downregulation of CXCR2. Nonetheless, it should be emphasized that altered receptor expression does not necessarily result in altered functional responses, and the opposite is also true.84,91,92 As mentioned earlier, another important note is that mice lack CXCL8, and that most of our knowledge on CXCR1/2 signaling pathways is derived from murine models. Therefore, extrapolation of murine experiments concerning CXCR1/2 biology to humans is difficult.45,48,63 In cancer biology, it is well known that CXCL8 plays a crucial role in the recruitment of neutrophils (tumor associated neutrophils [TAN]) to the tumor microenvironment. TAN show N1 or N2 phenotypes; N1 show anti-tumor activity through the release of inflammation-associated cytokines stimulating immune surveillance and local inflammation, whereas N2 show immunosuppressive and pro-angiogenic characteristics. N2 also stimulate remodeling of the extracellular matrix by the release of proteases. In solid malignancies, TAN attracted by CXCL8 are associated with poor clinical outcome and metastasis.93 MDS is characterized by sustained elevation of CXCL8 concentrations, and neutrophils tend to show decreased migration capacities towards CXCL8 gradients.94,95 Moreover, as impaired mobility correlates with inferior prognosis, migration analysis of PB neutrophils was previously proposed as a prognostic tool within MDS.96 The functional and phenotypic characteristics of BM neutrophils in PMF are currently unknown.
As mentioned, dysregulated inflammatory signaling is a key feature in the pathophysiology of myeloproliferative disorders, and especially PMF. The exact effects of multiple elevated cytokines within MPN are far from completely
understood. This review focuses on the role of CXCL8, as there is extensive interest in its role in oncogenesis due to its angiogenic and proinflammatory characteristics.93 In AML and MDS, inhibition of CXCR2 selectively inhibited immature hematopoietic cell lines due to higher expression of CXCR2 in CD34+ cells compared to healthy controls. Additionally, CXCL8 was identified as one of the few genes significantly over-expressed in different stem and progenitor subsets.94 Previously, researchers had already expressed their interest in CXCL8 as a therapeutic target in PMF. Dunbar et al. showed that hematopoietic progenitor cells from patients with myelofibrosis carry an enriched CXCL8-CXCR2 pathway signature and exhibit increased proliferation after exposure to exogenous CXCL8.6 To date, multiple classes of CXCR1/2 inhibitors have been characterized. In PMF, most evidence has been gathered with the CXCR1/2 inhibitor reparixin, which is an R-ibuprofen derivative. Treatment with reparixin in aged-matched GATA1low mice reduces BM fibrosis. In addition, GATA1low mice treated with reparixin express lower levels of TGF-b, whereas expression of CXCR1/2 remains unchanged and expression of GATA1 increases.97 Genetic deletion of Cxcr2 abrogates fibrosis and improves OS in the hMPLW515L fibrosis mouse model. Interestingly, administration of reparixin to human myelofibrosis-derived megakaryocytes reduces levels of both CXCL8 and VEGF in vitro.6 In June 2023, a phase II clinical trial with reparixin in patients with PMF was initiated (clinicaltrials.gov 05835466). The estimated study completion date is in March 2026.98 Other classes of CXCR1/2 inhibitors include the diaryl urea class and boronic acid-containing molecules, such as danirixin and SX-682, respectively. Danirixin is CXCR2-selective, and was tested to reduce neutrophil activation and NET production in patients with chronic obstructive pulmonary disease (COPD), but appeared effective in only a subset of individuals. Although these clinical trials with danirixin were stopped due to insufficient efficacy, the results suggest CXCR2-independent neutrophil activation was not negligible in a subset of patients.99,100 SX-682 is an oral dual allosteric inhibitor and was recently successfully tested in patients with hypomethylating agent failure MDS as part of a phase I trial.101
Besides its receptors, CXCL8 itself may also be a therapeutic target. BMS-986253 (previously known as HuMax-IL8) is a humanized monoclonal antibody against CXCL8. CXCL8 became a therapeutic target in various cancers as it tends to promote the acquisition of mesenchymal features, immune escape, and the recruitment of protumoral immune cells, e.g., myeloid-derived suppressor cells to the tumor environment. Blocking CXCL8 prevented acquisition of mesenchymal features by tumor cells and reduced treatment resistance. Various clinical trials with BMS-986253 in combination with antibodies targeting programmed death-1 (PD-1)/cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) in advanced tumors such as melanoma are
ongoing.102,103 In November 2022, a phase I/II clinical trial of BMS-986253 monotherapy or in combination with DNA methyltransferase inhibitors within patients with MDS was initiated (clinicaltrials.gov 05148234). The estimated study completion date is in July 2025.104 We refer to the review of Tremblay et al. for an extensive description of other therapeutic targets beyond the CXCL8-CXCR1/2 axis such as TGF-b1 (AVID200) or PI3K (parsaclisib) in PMF.105 Whether CXCL8-CXCR1/2 inhibition is superior compared to these therapeutic targets is unknown.
With a median survival of 16-35 months for high-risk patients, PMF shows the most aggressive characteristics amongst all MPN. Current therapeutic approaches such as JAK-inhibitors are ineffective in reducing progression of PMF or avoiding transformation into secondary AML. Aberrant megakaryopoiesis is a pathological hallmark within MPN, and megakaryocytes in myelofibrosis show higher proliferative capacities and morphologic abnormalities such as hypolobulated nuclei and clustering. As multiple cytokines are increased in PB and BM of patients with PMF, various pathways may concomitantly contribute to its pathogenesis. The chemokine CXCL8 is of particular interest within PMF, and MPN in general, as patients show increased concentrations within BM and PB independently of mutational status. Moreover, an increased concentration is associated with reduced OS and higher rates of secondary AML. The CXCL8-CXCR1/2 axis might play a central role within PMF pathogenesis as blockage
1. Constantinescu SN, Vainchenker W, Levy G, Papadopoulos N. Functional consequences of mutations in myeloproliferative neoplasms. Hemasphere. 2021;5(6):e578.
2. Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood. 2020;136(1):61-70.
3. Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416-1430.
4 Masselli E, Pozzi G, Gobbi G, et al. Cytokine profiling in myeloproliferative neoplasms: overview on phenotype correlation, outcome prediction, and role of genetic variants. Cells. 2020;9(9):2136.
5. Fisher DAC, Fowles JS, Zhou A, Oh ST. Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front Immunol. 2021;12:683401.
6. Dunbar AJ, Kim D, Lu M, et al. CXCL8/CXCR2 signaling mediates bone marrow fibrosis and is a therapeutic target in myelofibrosis. Blood. 2023;141(20):2508-2519.
7 Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667-679.
of the CXCR1/2 receptors in murine models results in increased megakaryocyte maturation and reduces both megakaryocyte ploidy and BM fibrosis. Interestingly, a phase II clinical trial with reparixin, a CXCR1/2 inhibitor, was initiated in June 2023 with estimated study completion date in March 2026. Although we have learned much more about PMF and MPN pathophysiology, further in-depth research will still be needed to fully disentangle the exact consequences of altered cytokine expressions. In addition, a particular focus on the characteristics of the CXCL8-CXCR1/2 axis within PV and ET evolving into post-PV/ET myelofibrosis may add crucial knowledge to our understanding of the biological continuum of these diseases. A better understanding of the spatiotemporal and concentration-dependent signaling of chemokines/ cytokines will hopefully further increase our treatment armamentarium in PMF, and MPN in general.
No conflicts of interest to disclose.
The authors met the criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). GV, SS and MG contributed to writing the manuscript. PP, SS, MG and TD critically revised the manuscript.
Acknowledgments
Figures created with BioRender.com.
Funding
No funding was received for this review.
8. Marty C, Pecquet C, Nivarthi H, et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood. 2016;127(10):1317-1324.
9. Elf S, Abdelfattah NS, Chen E, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov. 2016;6(4):368-381.
10. Benlabiod C, Cacemiro MDC, Nédélec A, et al. Calreticulin del52 and ins5 knock-in mice recapitulate different myeloproliferative phenotypes observed in patients with MPN. Nat Commun. 2020;11(1):4886.
11. Masselli E, Pozzi G, Carubbi C, Vitale M. The genetic makeup of myeloproliferative neoplasms: role of germline variants in defining disease risk, phenotypic diversity and outcome. Cells. 2021;10(10):2597.
12. Maslah N, Benajiba L, Giraudier S, Kiladjian JJ, Cassinat B. Clonal architecture evolution in myeloproliferative neoplasms: from a driver mutation to a complex heterogeneous mutational and phenotypic landscape. Leukemia. 2023;37(5):957-963.
13. Ho YH, Del Toro R, Rivera-Torres J, et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem
Cell. 2019;25(3):407-418.
14 Haas S, Hansson J, Klimmeck D, et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell. 2015;17(4):422-434.
15. Johnson CB, Zhang J, Lucas D. The role of the bone marrow microenvironment in the response to infection. Front Immunol. 2020;11:585402.
16. Psaila B, Wang G, Rodriguez-Meira A, et al. Single-cell analyses reveal megakaryocyte-biased hematopoiesis in myelofibrosis and identify mutant clone-specific targets. Mol Cell. 2020;78(3):477-492.
17 Melo-Cardenas J, Migliaccio AR, Crispino JD. The role of megakaryocytes in myelofibrosis. Hematol Oncol Clin North Am. 2021;35(2):191-203.
18. Larsen TE. Emperipolesis of granular leukocytes within megakaryocytes in human hemopoietic bone marrow. Am J Clin Pathol. 1970;53(4):485-489.
19. Cunin P, Nigrovic PA. Megakaryocyte emperipolesis: a new frontier in cell-in-cell interaction. Platelets. 2020;31(6):700-706.
20 Cunin P, Bouslama R, Machlus KR, et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. Elife. 2019;8:e44031.
21. Ding ZM, Babensee JE, Simon SI, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163(9):5029-5038.
22. Spangrude GJ, Lewandowski D, Martelli F, et al. P-selectin sustains extramedullary hematopoiesis in the Gata1low model of myelofibrosis. Stem Cells. 2016;34(1):67-82.
23. Schmitt A, Jouault H, Guichard J, Wendling F, Drouin A, Cramer EM. Pathologic interaction between megakaryocytes and polymorphonuclear leukocytes in myelofibrosis. Blood. 2000;96(4):1342-1347.
24. Zingariello M, Fabucci M, Bosco D, et al. Differential localization of P-selectin and von Willebrand factor during megakaryocyte maturation. Biotech Histochem. 2010;85(3):157-170.
25. Centurione L, Di Baldassarre A, Zingariello M, et al. Increased and pathologic emperipolesis of neutrophils within megakaryocytes associated with marrow fibrosis in GATA-1low mice. Blood. 2004;104(12):3573-3580.
26. Arciprete F, Verachi P, Martelli F, et al. Inhibition of CXCR1/2 reduces the emperipolesis between neutrophils and megakaryocytes in the Gata1low model of myelofibrosis. Exp Hematol. 2023;121:30-37.
27. Verachi P, Gobbo F, Martelli F, et al. Preclinical studies on the use of a P-selectin blocking monoclonal antibody to halt progression of myelofibrosis in the Gata1low mouse model. Exp Hematol. 2023;117:43-61.
28. Tanaka M, Aze Y, Fujita T. Adhesion molecule LFA-1/ICAM-1 influences on LPS-induced megakaryocytic emperipolesis in the rat bone marrow. Vet Pathol. 1997;34(5):463-466.
29 Perkins AC, Burbury K, Lehmann T, et al. Adore: a randomized, open-label, phase 1/2 open-platform study evaluating safety and efficacy of novel ruxolitinib combinations in patients with myelofibrosis. Blood. 2020;136(Suppl 1):52-53.
30. Campanelli R, Rosti V, Villani L, et al. Evaluation of the bioactive and total transforming growth factor b1 levels in primary myelofibrosis. Cytokine. 2011;53(1):100-106.
31. Cominal JG, Cacemiro M da C, Berzoti-Coelho MG, et al. Bone marrow soluble mediator signatures of patients with Philadelphia chromosome-negative myeloproliferative neoplasms. Front Oncol. 2021;11:665037.
32. Chen P, Wu B, Ji L, et al. Cytokine consistency between bone marrow and peripheral blood in patients with Philadelphianegative myeloproliferative neoplasms. Front Med. 2021;8:598182.
33. McLornan D, Szydlo R, Koster L, et al. Myeloablative and reduced-intensity conditioned allogeneic hematopoietic stem cell transplantation in myelofibrosis: a retrospective study by the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2019;25(11):2167-2171.
34 Fisher DAC, Miner CA, Engle EK, et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia. 2019;33(8):1978-1995.
35. Fisher DAC, Malkova O, Engle EK, et al. Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia. 2017;31(9):1962-1974.
36. Mascarenhas J, Hoffman R. A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Blood. 2013;121(24):4832-4837.
37. Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72(5):847-855.
38. Zhao JL, Ma C, O’Connell RM, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell. 2014;14(4):445-459.
39. Ketelut-Carneiro N, Fitzgerald KA. Apoptosis, pyroptosis, and necroptosis-Oh my! The many ways a cell can die. J Mol Biol. 2022;434(4):167378.
40. Longhitano L, Li Volti G, Giallongo C, et al. The role of inflammation and inflammasome in myeloproliferative disease. J Clin Med. 2020;9(8):2334.
41. Zambetti NA, Ping Z, Chen S, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19(5):613-627.
42. Basiorka AA, McGraw KL, Abbas-Aghababazadeh F, et al. Assessment of ASC specks as a putative biomarker of pyroptosis in myelodysplastic syndromes: an observational cohort study. Lancet Haematol. 2018;5(9):e393-e402.
43. Rai S, Grockowiak E, Hansen N, et al. Inhibition of interleukin-1b reduces myelofibrosis and osteosclerosis in mice with JAK2V617F driven myeloproliferative neoplasm. Nat Commun. 2022;13(1):5346.
44 Modi WS, Dean M, Seuanez HN, Mukaida N, Matsushima K, O’Brien SJ. Monocyte-derived neutrophil chemotactic factor (MDNCF/IL-8) resides in a gene cluster along with several other members of the platelet factor 4 gene superfamily. Hum Genet. 1990;84(2):185-187.
45. Rajarathnam K, Schnoor M, Richardson RM, Rajagopal S. How do chemokines navigate neutrophils to the target site: dissecting the structural mechanisms and signaling pathways. Cell Signal. 2019;54:69-80.
46. Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. 2014;10(5):593-619.
47. Bachelerie F, Ben-Baruch A, Burkhardt AM, et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev. 2014;66(1):1-79.
48. Cambier S, Gouwy M, Proost P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol. 2023;20(3):217-251.
49 Yang L, Herrera J, Gilbertsen A, et al. IL-8 mediates idiopathic pulmonary fibrosis mesenchymal progenitor cell fibrogenicity. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L127-L136.
50 Matsushima K, Morishita K, Yoshimura T, et al. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of MDNCF mRNA by interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167(6):1883-1893.
51. Schröder JM, Sticherling M, Henneicke HH, Preissner WC, Christophers E. IL-1 alpha or tumor necrosis factor-alpha stimulate release of three NAP-1/IL-8-related neutrophil chemotactic proteins in human dermal fibroblasts. J Immunol. 1990;144(6):2223-2232.
52. Van den Steen PE, Proost P, Wuyts A, Van Damme J, Opdenakker G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood. 2000;96(8):2673-2681.
53. Mortier A, Berghmans N, Ronsse I, et al. Biological activity of CXCL8 forms generated by alternative cleavage of the signal peptide or by aminopeptidase-mediated truncation. PloS One. 2011;6(8):e23913.
54 Nasser MW, Raghuwanshi SK, Grant DJ, Jala VR, Rajarathnam K, Richardson RM. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J Immunol. 2009;183(5):3425-3432.
55. Liu K, Wu L, Yuan S, et al. Structural basis of CXC chemokine receptor 2 activation and signalling. Nature. 2020;585(7823):135-140.
56. Ishimoto N, Park JH, Kawakami K, et al. Structural basis of CXC chemokine receptor 1 ligand binding and activation. Nat Commun. 2023;14(1):4107.
57 Teijeira Á, Garasa S, Gato M, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856-871.
58. Alfaro C, Teijeira A, Oñate C, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. 2016;22(15):3924-3936.
59. Yalavarthi S, Gould TJ, Rao AN, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015;67(11):2990-3003.
60 Van Alsten SC, Aversa JG, Santo L, et al. Association between ABO and Duffy blood types and circulating chemokines and cytokines. Genes Immun. 2021;22(3):161-171.
61. Jinna N, Rida P, Su T, et al. The DARC side of inflamm-aging: Duffy antigen receptor for chemokines (DARC/ACKR1) as a potential biomarker of aging, immunosenescence, and breast oncogenesis among high-risk subpopulations. Cells. 2022;11(23):3818.
62. Peseski AM, Saliba AN, Althouse SK, Sayar H. Does race play a role in complications and outcomes of Philadelphia chromosome-negative myeloproliferative neoplasms? Hematol Oncol Stem Cell Ther. 2022;15(2):30-38.
63. Fan X, Patera AC, Pong-Kennedy A, et al. Murine CXCR1 Is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J Biol Chem. 2007;282(16):11658-11666.
64 Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10):1356-1363.
65. Øbro NF, Grinfeld J, Belmonte M, et al. Longitudinal cytokine profiling identifies GRO-α and EGF as potential biomarkers of disease progression in essential thrombocythemia. Hemasphere. 2020;4(3):e371.
66. Emadi S, Clay D, Desterke C, et al. IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood. 2005;105(2):464-473.
67. Daly TJ, LaRosa GJ, Dolich S, Maione TE, Cooper S, Broxmeyer HE. High activity suppression of myeloid progenitor proliferation by chimeric mutants of interleukin 8 and platelet factor 4. J Biol Chem. 1995;270(40):23282-23292.
68. Broxmeyer HE, Cooper S, Cacalano G, Hague NL, Bailish E, Moore MW. Involvement of interleukin (IL) 8 receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. J Exp Med. 1996;184(5):1825-1832.
69 Sanchez X, Suetomi K, Cousins-Hodges B, Horton JK, Navarro J. CXC chemokines suppress proliferation of myeloid progenitor cells by activation of the CXC chemokine receptor 2. J Immunol. 1998;160(2):906-910.
70. Adeli EK, Abolghasemi H, Ebtekar M, Pourpak Z, Kheirandish M. Effects of CXCR1 and CXCR2 inhibition on expansion and differentiation of umbilical cord blood CD133(+) cells into megakaryocyte progenitor cells. Cytokine. 2011;55(2):181-187.
71. Gewirtz AM, Zhang J, Ratajczak J, et al. Chemokine regulation of human megakaryocytopoiesis. Blood. 1995;86(7):2559-2567.
72. Binder V, Li W, Faisal M, et al. Microenvironmental control of hematopoietic stem cell fate via CXCL8 and protein kinase C. Cell Rep. 2023;42(5):112528.
73. Gouwy M, Struyf S, Catusse J, Proost P, Van Damme J. Synergy between proinflammatory ligands of G protein-coupled receptors in neutrophil activation and migration. J Leukoc Biol. 2004;76(1):185-194.
74. Nesmelova IV, Sham Y, Dudek AZ, et al. Platelet factor 4 and interleukin-8 CXC chemokine heterodimer formation modulates function at the quaternary structural level. J Biol Chem. 2005;280(6):4948-4958.
75. Dudek AZ, Nesmelova I, Mayo K, Verfaillie CM, Pitchford S, Slungaard A. Platelet factor 4 promotes adhesion of hematopoietic progenitor cells and binds IL-8: novel mechanisms for modulation of hematopoiesis. Blood. 2003;101(12):4687-4694.
76. Lataillade JJ, Pierre-Louis O, Hasselbalch HC, et al. Does primary myelofibrosis involve a defective stem cell niche? From concept to evidence. Blood. 2008;112(8):3026-3035.
77 Brouty-Boyé D, Briard D, Azzarone B, et al. Effects of human fibroblasts from myelometaplasic and non-myelometaplasic hematopoietic tissues on CD34+ stem cells. Int J Cancer. 2001;92(4):484-488.
78. Broxmeyer HE. Chemokines in hematopoiesis. Curr Opin Hematol. 2008;15(1):49-58.
79 Medinger M, Skoda R, Gratwohl A, et al. Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2-V617F mutational status. Br J Haematol. 2009;146(2):150-157.
80 Boveri E, Passamonti F, Rumi E, et al. Bone marrow microvessel density in chronic myeloproliferative disorders: a study of 115 patients with clinicopathological and molecular correlations. Br J Haematol. 2008;140(2):162-168.
81. Wróbel T, Mazur G, Surowiak P, Wolowiec D, Jelen M, Kuliczkowsky K. Increased expression of vascular endothelial growth factor (VEGF) in bone marrow of patients with myeloproliferative disorders (MPD). Pathol Oncol Res. 2003;9(3):170-173.
82. Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2001;280(6):L1094-L1103.
83. Strieter RM, Polverini PJ, Kunkel SL, et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270(45):27348-27357.
84. Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol. 2020;17(5):433-450.
85. Schmidt S, Daniliants D, Hiller E, Gunsilius E, Wolf D, Feistritzer C. Increased levels of NETosis in myeloproliferative neoplasms are not linked to thrombotic events. Blood Adv. 2021;5(18):3515-3527.
86. Marin Oyarzún CP, Carestia A, Lev PR, et al. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci Rep. 2016;6:38738.
87. Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436):eaan8292.
88. Hauser CJ, Fekete Z, Goodman ER, Kleinstein E, Livingston DH, Deitch EA. CXCR2 stimulation primes CXCR1 [Ca2+]i responses to IL-8 in human neutrophils. Shock. 1999;12(6):428-437.
89 Sabroe I, Jones EC, Whyte MKB, Dower SK. Regulation of human neutrophil chemokine receptor expression and function by activation of Toll-like receptors 2 and 4. Immunology. 2005;115(1):90-98.
90 Martin C, Burdon PCE, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19(4):583-593.
91. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120(7):2423-2431.
92. Kim HK, De La Luz Sierra M, Williams CK, Gulino AV, Tosato G. G-CSF down-regulation of CXCR4 expression identified as a
mechanism for mobilization of myeloid cells. Blood. 2006;108(3):812-820.
93. Liu Q, Li A, Tian Y, et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016;31:61-71.
94 Schinke C, Giricz O, Li W, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015;125(20):3144-3152.
95. Fuhler GM, Knol GJ, Drayer AL, Vellenga E. Impaired interleukin-8- and GROalpha-induced phosphorylation of extracellular signal-regulated kinase result in decreased migration of neutrophils from patients with myelodysplasia. J Leukoc Biol. 2005;77(2):257-266.
96. Schuster M, Moeller M, Bornemann L, et al. Surveillance of myelodysplastic syndrome via migration analyses of blood neutrophils: a potential prognostic tool. J Immunol. 2018;201(12):3546-3557.
97. Verachi P, Gobbo F, Martelli F, et al. The CXCR1/CXCR2 inhibitor reparixin alters the development of myelofibrosis in the Gata1low mice. Front Oncol. 2022;12:853484.
98. National Cancer Institute (NCI). A phase II study of reparixin in patients with myelofibrosis Myeloproliferative Neoplasms Research Consortium [MPN-RC 120]. https://www.clinicaltrials. gov/study/NCT05835466?cond=NCT05835466&rank=1 Accessed January 26, 2024.
99. Sitaru S, Budke A, Bertini R, Sperandio M. Therapeutic inhibition of CXCR1/2: where do we stand? Intern Emerg Med. 2023;18(6):1647-1664.
100. Keir HR, Richardson H, Fillmore C, et al. CXCL-8-dependent and -independent neutrophil activation in COPD: experiences from a pilot study of the CXCR2 antagonist danirixin. ERJ Open Res. 2020;6(4):00583-02020.
101. Sallman DA, DeZern AE, Gayle AA, et al. Phase 1 results of the first-in-class CXCR1/2 inhibitor SX-682 in patients with hypomethylating agent failure myelodysplastic syndromes. Blood. 2022;140(Suppl 1):2070-2072.
102. Schalper KA, Carleton M, Zhou M, et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat Med. 2020;26(5):688-692.
103. Ramachandra N, Gupta M, Schwartz L, et al. Role of IL8 in myeloid malignancies. Leuk Lymphoma. 2023;64(11):1742-1751.
104 National Cancer Institute (NCI). A phase I/II trial of BMS-986253 in myelodysplastic syndromes. clinicaltrials.gov; 2022. https:// clinicaltrials.gov/study/NCT05148234. Accessed December 22, 2023.
105. Tremblay D, Mesa R. Novel treatments for myelofibrosis: beyond JAK inhibitors. Int J Hematol. 2022;115(5):645-658.
Mandy W. E. Smeets,1,2* Elisabeth M. P. Steeghs,1* Jan Orsel,2 Femke Stalpers,2 Myrthe M. P. Vermeeren,2 Christina H. J. Veltman,2 Lotte Slenders,2 Stefan Nierkens,2,3 Cesca van de Ven2 and Monique L. den Boer1,2
1Department of Pediatrics, Erasmus MC-Sophia, Rotterdam; 2Princess Máxima Center for Pediatric Oncology, Utrecht and 3Center for Translational Immunology, University Medical Center, Utrecht, the Netherlands
*MWES and EMPS contributed equally as first authors.
Abstract
Correspondence: M.L. den Boer
m.l.denboer@prinsesmaximacentrum.nl
Received: May 13, 2023.
Accepted: February 15, 2024. Early view: February 29, 2024.
https://doi.org/10.3324/haematol.2023.283494
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) can hijack the normal bone marrow microenvironment to create a leukemic niche which facilitates blast cell survival and promotes drug resistance. Bone marrow-derived mesenchymal stromal cells (MSC) mimic this protective environment in ex vivo co-cultures with leukemic cells obtained from children with newly diagnosed BCP-ALL. We examined the potential mechanisms of this protection by RNA sequencing of flow-sorted MSC after co-culture with BCP-ALL cells. Leukemic cells induced an interferon (IFN)-related gene signature in MSC, which was partially dependent on direct cell-cell signaling. The signature was selectively induced by BCP-ALL cells, most profoundly by ETV6-RUNX1-positive ALL cells, as co-culture of MSC with healthy immune cells did not provoke a similar IFN signature. Leukemic cells and MSC both secreted IFNα and IFNb, but not IFNγ. In line, the IFN gene signature was sensitive to blockade of IFNα/b signaling, but less to that of IFNγ. The viability of leukemic cells and level of resistance to three chemotherapeutic agents was not affected by interference with IFN signaling using selective IFNα/b inhibitors or silencing of IFN-related genes. Taken together, our data suggest that the leukemia-induced expression of IFNα/b-related genes by MSC does not support survival of BCP-ALL cells but may serve a different role in the pathobiology of BCP-ALL.
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common pediatric malignancy, and is characterized by malignant transformation and clonal expansion of B-precursor cells in the bone marrow.1-3 The 5-year event-free survival rate of pediatric patients with BCP-ALL is currently ~90%.4-8 However, despite a relatively high cure rate, BCP-ALL still represents a major cause of cancer-related mortality in children, which can be attributed to treatment-related death or relapse of the leukemia.6,9
Several studies have shown that the bone marrow microenvironment facilitates leukemogenesis and contributes to cellular drug resistance.9-15 Bone marrow-derived mesenchymal stromal cells (MSC) are a major component of the bone marrow microenvironment.16 Ex vivo, these cells provide a survival benefit to co-cultured leukemic cells and induce resistance to chemotherapeutic drugs. We previously showed that leukemic cells become re-sensitized to
chemotherapeutics ex vivo when the interaction with MSC is disrupted, e.g., by interfering with tunneling nanotube formation.10 Leukemic cells use tunneling nanotubes as an effective mechanism to communicate with MSC. Transfer of mitochondria, autophagosomes, and transmembrane proteins towards MSC has been established.14 Disruption of tunneling nanotubes resulted in an altered profile of cytokines secreted by MSC and a reduced survival benefit for leukemic cells.10,14 These findings indicate that MSC play an important role in the maintenance of BCP-ALL, although the mechanism is yet unknown. To elucidate potential mechanisms, we investigated the effect of leukemic cells on the gene expression profile of MSC. Subsequently, we studied whether differentially expressed genes contributed to the viability and drug responsiveness of BCP-ALL. Our study shows that the interferon (IFN) α/b pathway is selectively activated in MSC upon interaction with BCP-ALL cells but not with normal, healthy immune cells. However, interference with this pathway did not affect either the viability or the
level of drug resistance, suggesting that the IFNα/b response may serve a different role in the pathobiology of BCP-ALL.
More details on the methods involved in this research are provided in the Online Supplementary Methods.
Mesenchymal stromal cells and primary B-cell precursor acute lymphoblastic leukemia cells
MSC were obtained from bone marrow aspirates taken from pediatric BCP-ALL patients and healthy donors as specified in Online Supplementary Table S1, using previously described procedures.17
Primary BCP-ALL cells were collected from bone marrow aspirates taken from children with newly diagnosed BCP-ALL (<18 years) which were left over after diagnostic procedures, and for which written informed consent was given by patients, parents, or guardians in line with institutional review board guidelines. The patients’ characteristics are presented in Online Supplementary Table S2. All samples were archived in liquid nitrogen and showed >90% leukemic blasts after having been thawed.
Healthy immune cells
Mononuclear cells were isolated from umbilical cord blood, with the approval of the ethics committee of Erasmus University Medical Center and the scientific research committee of Franciscus Vlietland Hospital. Antibodies against B-, T-, and natural killer (NK)-cell, monocyte, granulocyte, and dendritic cell markers were purchased for immune cell characterization using flow cytometry (Cytoflex LX; Beckman Coulter).
Co-culture of leukemic cells, healthy immune cells and mesenchymal stromal cells
MSC (0.225x106) were seeded in six-well plates with 3 mL of medium and cultured for 24 hours. This medium was replaced with 3 mL medium containing 5.0x106 freshly thawed primary BCP-ALL cells or healthy immune cells, or 0.9x106 cells in the case of BCP-ALL cell lines. Cells were cultured for 40 hours before supernatants were taken to measure cytokine levels by Luminex multiplex immunoassay.18 In parallel, BCPALL and MSC were fractionated by flow sorting on a SH800S Cell Sorter (Sony, San Jose, CA, USA) and used as input for RNA sequencing. Both mono-cultured and co-cultured BCPALL cells and MSC were processed in the same way. Purity and viability checks were performed after sorting (Online Supplementary Figure S1, Online Supplementary Table S3).
Direct cell-cell contact-mediated interferon response
Dye transfer from Vybrant DiI-labeled (Invitrogen) ALL cells to MSC was determined in direct co-cultures and indirect co-cultures using a transwell setup (0.4 μm pore-size; Corning, New York, NY, USA). Accumulation of dye in MSC was
measured after 40 hours of exposure to BCP-ALL cells by flow cytometry (Cytoflex S). Cells in the upper and lower compartments of the transwell were flow-sorted for RNA sequencing as mentioned above.
RNA sequencing
Total RNA (RNA integrity number >6.8) from sorted MSC and leukemic cells was used for ribonucleotide-depleted long-noncoding RNA-sequencing (NovaSeq 6000; Illumina, San Diego, CA, USA). EdgeR (version 3.28.1) was used to perform differential gene expression analysis. Pathway analysis was performed using Pathvisio software.19
Lentiviral silencing
Primary BCP-ALL cells (875,000; 1.0x106 cells/mL) were cultured for 72 hours with and without IFN signature gene-silenced MSC (50,000). The viability of BCP-ALL cells was determined by flow cytometry.
Inhibition of interferon signaling
Inhibitors of IFNα/b (1.8 ng/mL), and IFNγ (R&D Systems; 3.0 ng/mL) were used in unstimulated co-cultures of MSC and BCP-ALL cells for 40 and 120 hours. Cell viability and the IFN-related gene expression signature were assessed with flow cytometry and quantitative reverse transcriptase polymerase chain reaction (RT-qPCR), respectively.
Drug exposure experiments
The sensitivity of leukemic cells to L-asparaginase, daunorubicin, and prednisolone in the presence of IFNα/b inhibitors was evaluated by flow cytometry after 120 hours’ exposure. The drug concentration most discriminative for MSC-induced resistance was determined for each individual BCP-ALL sample prior to the inhibitor experiments (Online Supplementary Figure S2).
Quantitative reverse transcriptase polymerase chain reaction
RT-qPCR was performed using a QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific) (Online Supplementary Methods)
Statistical analysis
One-way analysis of variance tests and one-sided (un)paired t tests were performed using GraphPad Prism. Correction for multiple testing was included when appropriate. A (paired EdgeR) P value <0.05 was considered statistically significant.
B-cell precursor acute lymphoblastic leukemia cells induce an interferon-related gene signature in mesenchymal stromal cells
We performed RNA sequencing on sorted fractions of MSC
after exposure to primary BCP-ALL patients’ samples (n=15; Figure 1A) for 40 hours, a co-culture condition for which we previously established that a leukemic niche is created by the interaction of BCP-ALL cells with MSC.14 Pathway analysis of RNA-sequencing data revealed enrichment for IFN-pathway-activated genes in the MSC after co-culture with BCP-ALL cells (Online Supplementary Figure S3). The
induction of IFN-related genes could not be attributed to other cell types, e.g., T-lymphocytes, present in the samples (Online Supplementary Table S2). All three tested origins of bone marrow-derived MSC (MSC#1-3) had similar levels of increased IFN-related gene expression in co-culture with BCP-ALL cells, suggesting that the leukemia-induced change in expression is independent from the origin of
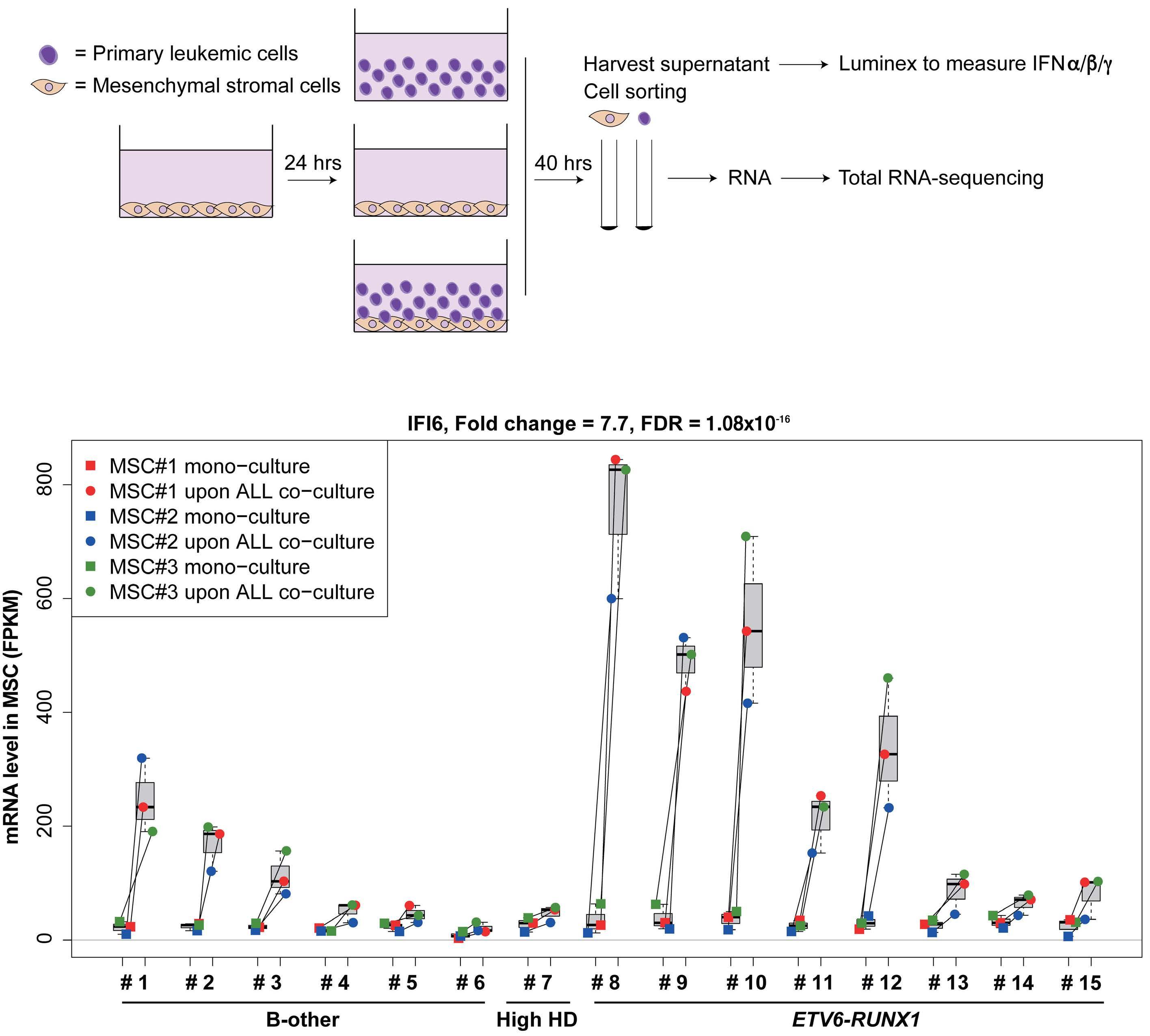
Figure 1. B-cell precursor acute lymphoblastic leukemia cells induce an interferon-related gene signature in mesenchymal stromal cells. (A) Diagram of the experimental setup. RNA sequencing was performed on sorted viable mesenchymal stromal cells (MSC) and acute lymphoblastic leukemia (ALL) fractions after mono-culture and after MSC/ALL co-culture; for the gating strategy see Online Supplementary Figure S1. Secreted interferon levels were measured in equal volumes of supernatant of mono-cultures and MSC/ALL co-cultures (Figure 6A). (B) IFI6 mRNA levels expressed in paired samples of MSC sorted after mono-culture (squares; MSC#1-3 indicated in red, blue, and green, respectively.) and MSC sorted after co-culture with BCP-ALL cells from 15 individual patients (#1, ETV6-RUNX1-like; #2-6, B-other; #7, high hyperdiploid; #8-15, ETV6-RUNX1; circles). Boxplots represent the interquartile range, the median is depicted by a line. IFN: interferon; FDR: P value for the false discovery rate; FPKM: fragments per kilobase million; HD: high hyperdiploid.
MSC (Figure 1B and Online Supplementary Figure S4). This is strengthened by the fact that an ETV6-RUNX1-positive case induced a similar upregulation of IFN-related genes IFI6 and IFITM1 in ETV6-RUNX1-derived MSC as well as in B-other derived and healthy control MSC (Online Supplementary Figure S5). In addition to IFI6 and IFITM1, other
IFN-related genes were also highly upregulated in MSC after co-culture with ETV6-RUNX1-positive cells (ALL#17; Online Supplementary Figure S6).
The IFN-related genes including IFI6, MX1, IFI27, and OAS3, were 4.3- to 7.7-fold upregulated in MSC upon co-culture with BCP-ALL compared to MSC kept in mono-cul-
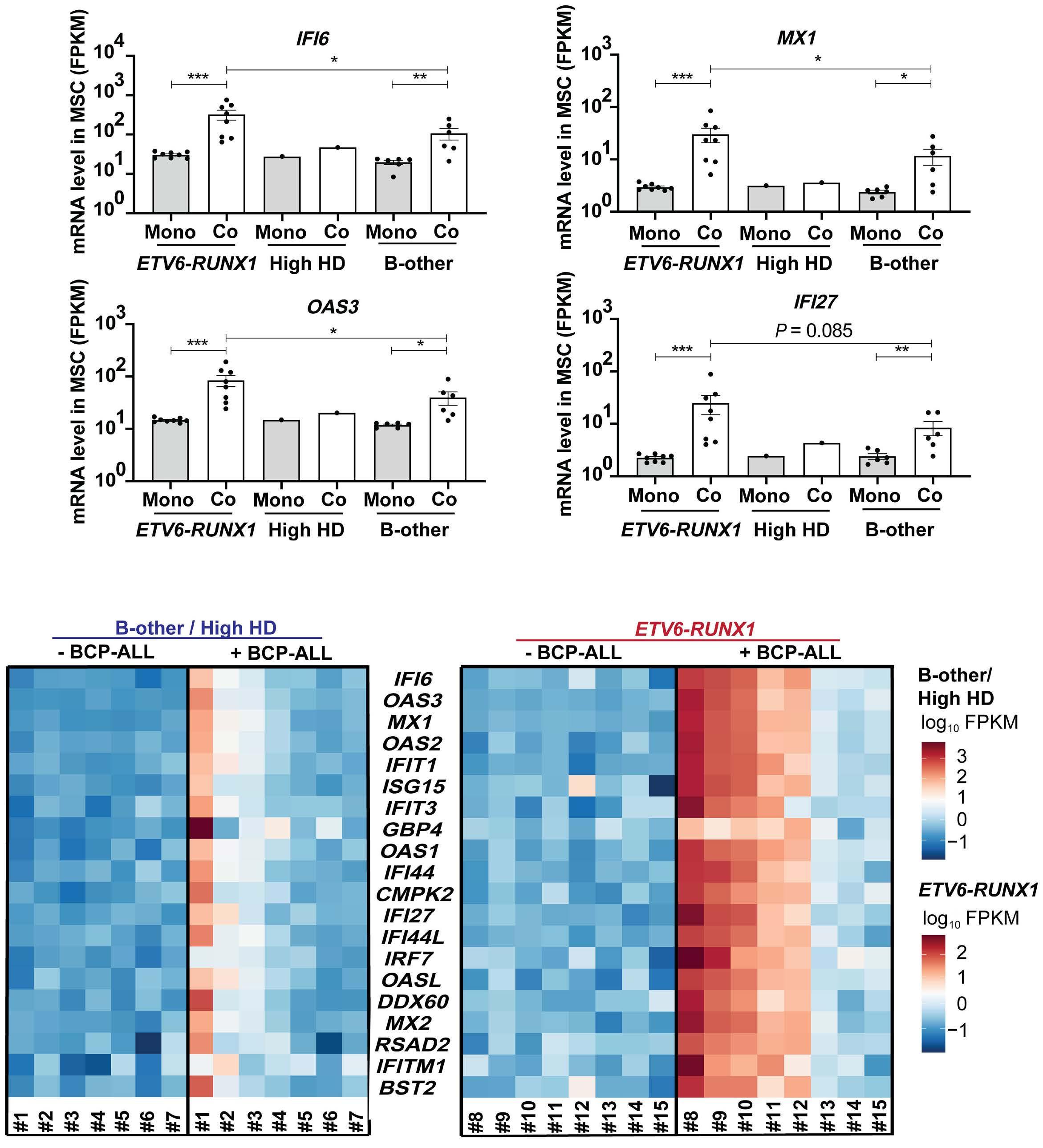
Figure 2. ETV6-RUNX1 B-cell precursor acute lymphoblastic leukemia cells most potently induce an interferon-related gene signature in mesenchymal stromal cells. (A) mRNA expression levels in mesenchymal stromal cells (MSC) of IFI6, MX1, OAS3 and IFI27 upon mono-culture (gray bars) and co-culture (white bars) summarized according to B-cell precursor acute lymphoblastic leukemia (BCP-ALL) subtype (N=8 for ETV6-RUNX1, N=1 for high hyperdiploid, N=6 for B-other). Each dot represents the average expression of interferon (IFN)-related genes in three MSC per sort. The mean ± standard error of mean is indicated. (B) Heatmap of expression levels of IFN-related genes in MSC sorted after mono-culture (- BCP-ALL) and those after co-culture with 15 different ALL cases (+ BCP-ALL). The heatmap is grouped by BCP-ALL subtype: high hyperdiploid (HD) and B-other ALL cases (#1-7) or ETV6-RUNX1 ALL cases (#8-15). Blue, low expression. Red, high expression. *P<0.05, **P<0.01, ***P<0.001. MSC: mesenchymal stromal cells; FPKM: fragments per kilobase million; Mono: mono-culture; Co: co-culture; HD: high hyperdiploid; BCP-ALL: B-cell precursor acute lymphoblastic leukemia.
ture (false discovery rate [FDR]=1.08x10-16, FDR=2.23x10-15, FDR=3.81x10-11, and FDR=1.44x10-15, respectively.) (Figure 1B and Online Supplementary Figure S7 ). Other IFN-related genes such as IFITM1, IFI44L, IFIT1 and ISG15 were 3.1- to 4.3-fold upregulated (FDR=1.08x10-6, FDR=5.21x10-11, FDR=2.99x10-12, and FDR=4.73x10-12, respectively (Online Supplementary Figure S7 ). STAT1, IRF7 , and IRF9 were also upregulated in MSC (FDR=9.61x10-06, FDR=1.47x10-10, and FDR=0.00137, respectively), but to a lesser extent (1.5- to 1.9-fold) compared to the previously mentioned genes (Online Supplementary Figure S7). Vice versa, no MSC-induced change in expression of IFN-signature genes in BCP-ALL cells was found (Online Supplementary Figures S8 and S9). We observed, however, that the intrinsic IFN-related gene profile was slightly higher in mono-cultured ETV6-RUNX1 ALL cells compared to B-other or high hyperdiploid ALL samples (Online Supplementary Figures S8 and S9). Expression of IFN-related genes was remarkably higher in MSC after co-culture with ETV6-RUNX1-positive BCP-ALL cells (n=8) compared to their expression in MSC sorted after co-culture with B-other BCP-ALL cells (n=6) as exemplified for IFI6 (3-fold increase; P=0.031), MX1 (2.6-fold increase; P=0.048) and OAS3 (2.1-fold increase; P=0.04) in Figure 2A and for a set of 20 IFN signature genes in Figure 2B. Of particular interest is case ALL#1, which induced a similar IFN-related gene signature in MSC as ETV6-RUNX1 cases. ALL#1 turned out to be an ETV6-RUNX1-like case as defined by gene expression.20 In correspondence with
the observation for patients’ ALL cells (Figure 2A, B), we noticed that MSC exposed to the ETV6-RUNX1-positive cell line REH consistently upregulated IFN α / b genes whereas this was more variable for the non- ETV6-RUNX1 cell lines (Online Supplementary Figure S10). Exposure of MSC to healthy immune cells did not induce the typical IFN signature (n=5) (Figure 3A-C). However, we did observe induction of CXCL10 expression (P=0.018), which was most likely due to an interaction with monocytes since this induction was mainly observed in samples containing larger numbers of monocytes. No other IFN signature genes were induced. Thus, we found that the IFN signature was selectively induced by BCP-ALL cells, most profoundly by ETV6-RUNX1-positive ALL cells, as co-culture of MSC with healthy immune cells did not provoke a similar IFN signature.
In previous studies we showed that the viability of leukemic cells depends on a close and direct contact with MSC mediated via tunneling nanotubes,10 as can be visualized by reduced DiI dye transfer from pre-labeled ALL cells in the upper compartment to MSC in the bottom compartment of a transwell setting (87.6% decrease, P<0.0001) (Figure 4A, B). Although preventing the formation of tunneling nanotubes significantly reduced the expression levels of CXCL10 (3.3-fold, P=0.016), IFI44L (1.57-fold, P=0.009) and IFITM1 (1.5-fold, P=0.005), not all tested genes were affected (i.e., IFI6, IFITM1, MX1, IFI27 ) (Figure 4C). This suggests that tunneling nanotube-dependent signaling
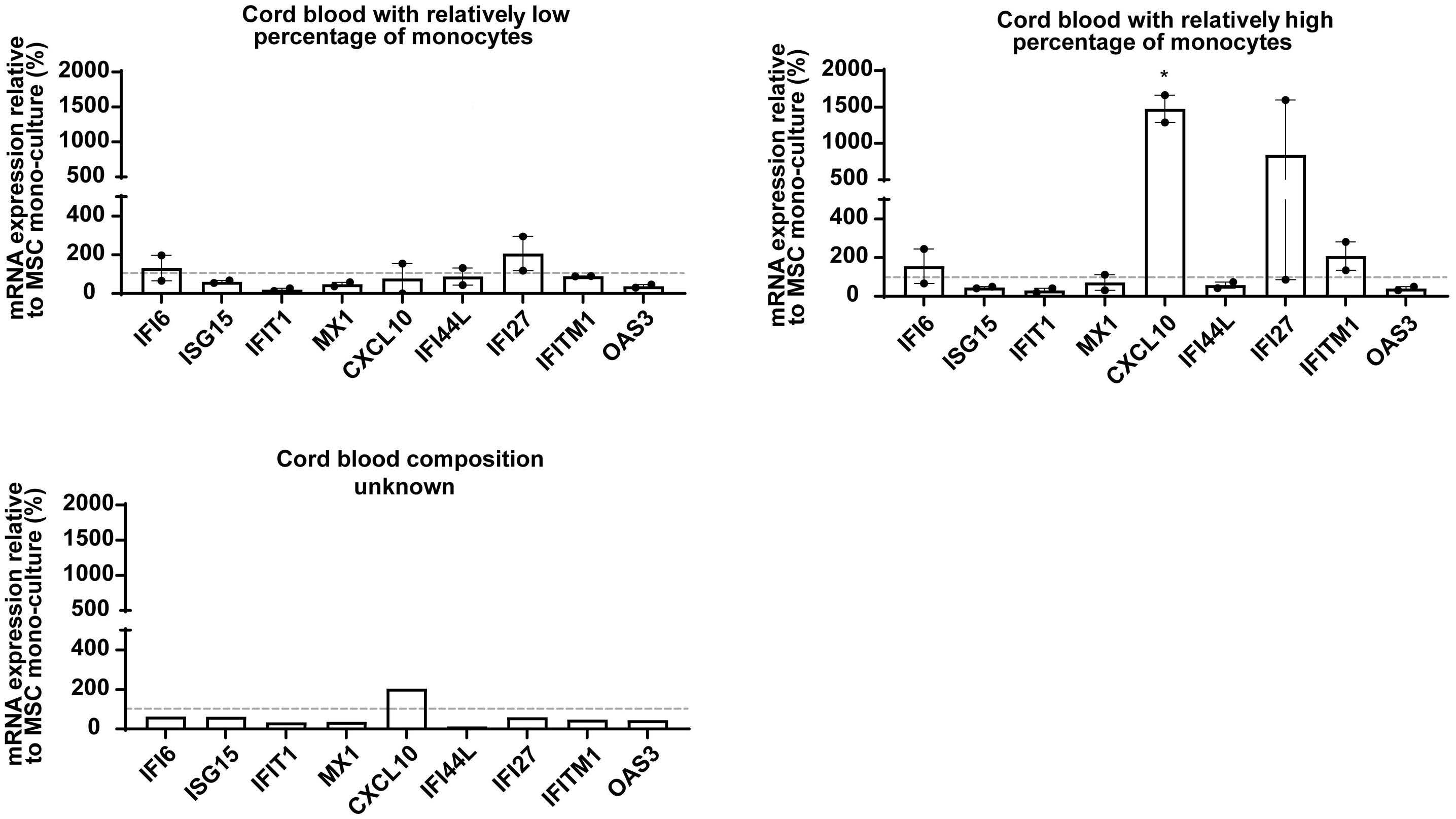
Figure 3. Normal cord blood, with the exception of monocyte-containing preparations, has a limited effect on expression levels of interferon-related genes in mesenchymal stromal cells. IFI6, ISG15, IFIT1, MX1, CXCL10, IFI44L, IFI27, IFITM1, and OAS3 gene expression levels measured by reverse transcriptase quantitative polymerase chain reaction in mesenchymal stromal cells (MSC) (MSC#2) sorted after co-culture with mononuclear cells from five different cord blood samples: cord blood with relatively (A) low (N=2) or (B) high (N=2) percentages of monocytes (>20%), or (C) an unknown cellular composition (N=1). Bars are means of triplicate measurements ± standard error of mean for two independent co-culture experiments. The dashed line (---) indicates mRNA expression levels of MSC after mono-culture, set at 100%. P<0.05 MSC: mesenchymal stromal cells.
only partially contributes to the BCP-ALL-induced gene expression changes in MSC.
To examine whether the upregulation of IFN-related genes in MSC had a causal effect on the viability of primary BCP-ALL cells, IFI6, MX1, IFI27, IFI44L and ISG15 were lentivirally silenced resulting in an efficient knockdown of 67-93% for individual genes (Online Supplementary Figure
S11A-E). The viability of MSC upon knockdown of several IFN-related genes was largely unaffected compared to controls, except for shIFI27-treated MSC (P<0.0001) (Online Supplementary Figure S11F). Efficient knockdown of CXCL10 in MSC could not be achieved by four different short hairpin constructs tested (data not shown). Silencing of the strong IFN gene signature observed in MSC upon
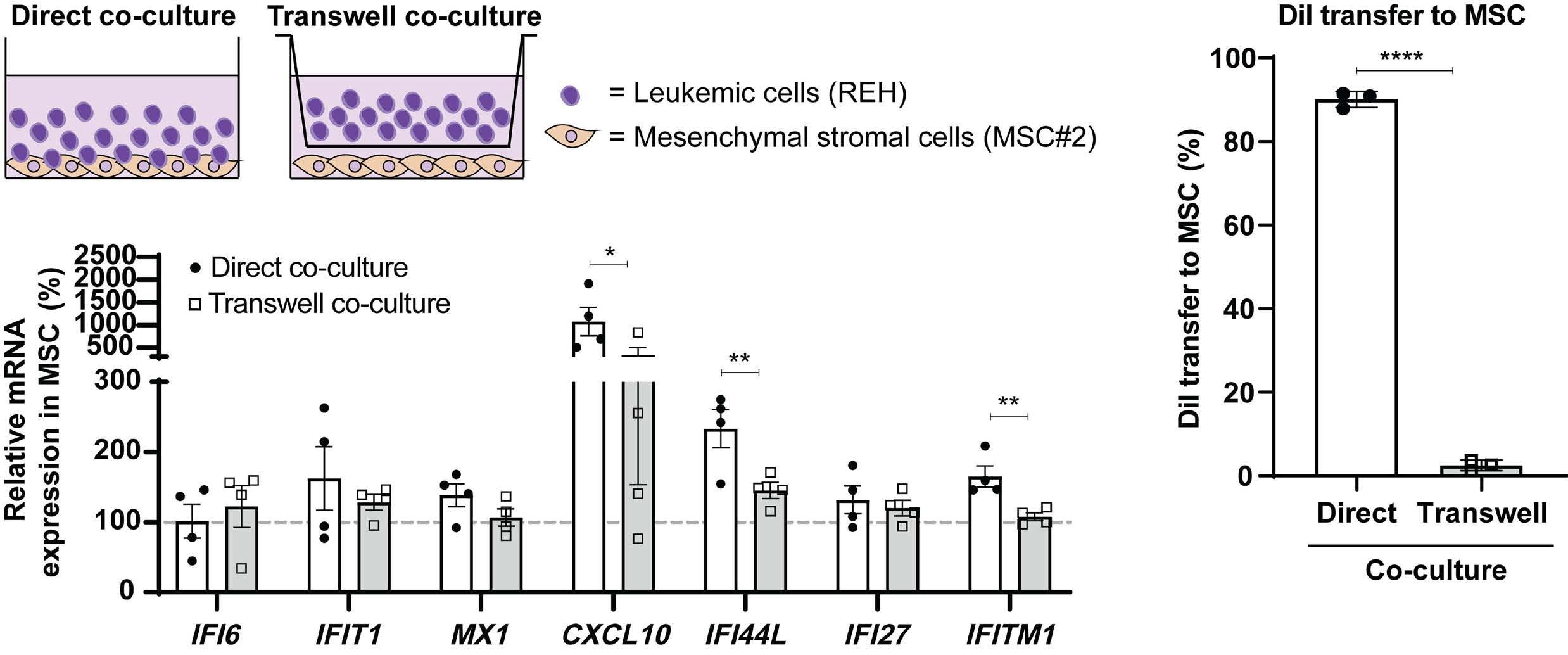
Figure 4. Direct cell-cell contact contributes to induction of Interferon-related gene expression in mesenchymal stromal cells. (A) Schematic overview of the experimental setup of direct co-culture and transwell co-culture of REH cells with mesenchymal stromal cells (MSC) (MSC#2) using 0.4 µm pore-size transwells. (B) Percentage of DiI-positive MSC after direct (circles, white bars) and transwell (squares, gray bars) co-culture with REH leukemic cells for 40 hours. Dots represent averaged duplicate measurements; bars represent means ± standard error of mean (SEM) for three independent experiments. (C) Interferon-related gene expression levels, measured by reverse transcriptase quantitative polymerase chain reaction, in MSC sorted after direct (circles, white bars) or transwell (squares, gray bars) co-culture with REH cells for 40 hours. Dots represent averaged triplicate measurements; bars represent means ± SEM for four independent experiments. The dashed line (---) indicates mRNA expression levels of MSC after mono-culture, set at 100%. *P<0.05, **P<0.01, ****P<0.0001. MSC: mesenchymal stromal cells.
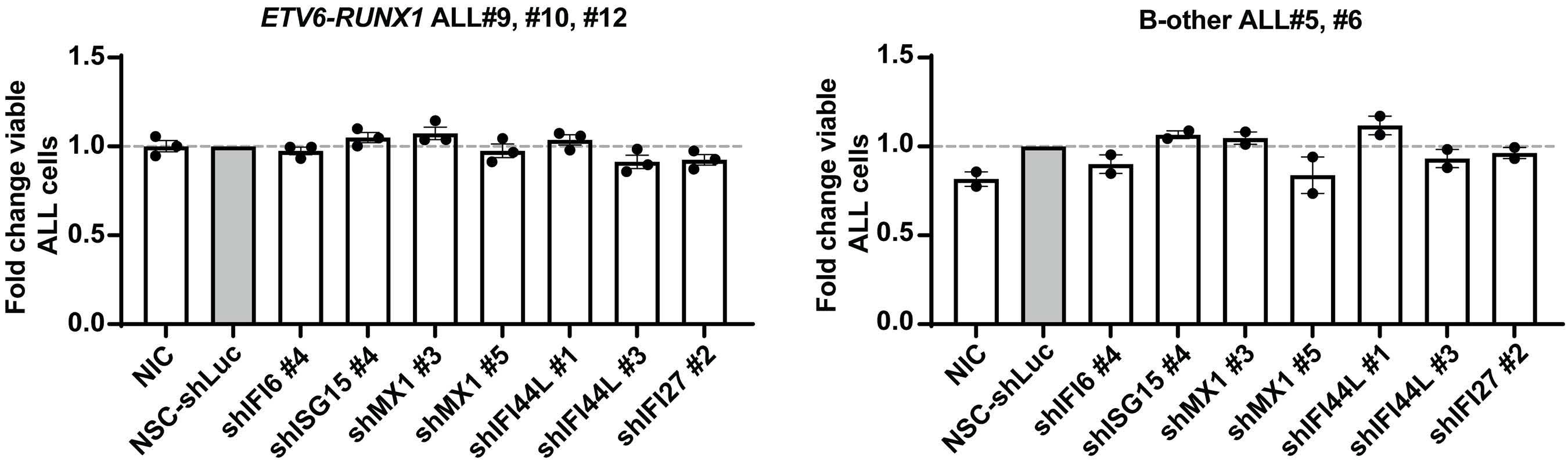
Figure 5. Silencing of interferon-related genes in mesenchymal stromal cells does not affect the viability of primary B-cell precursor acute lymphoblastic leukemia cells. Fold-change in percentage of viable leukemic cells after 72 hours of co-culture with transduced mesenchymal stromal cells (MSC) (MSC#2). A non-silencing control - short-hairpin luciferase (NSC-shLuc) is used as reference. (A) ETV6-RUNX1 (N=3), and (B) B-other (N=2) B-cell precursor acute lymphoblastic leukemia (BCP-ALL) cells. Five shRNA per gene were tested for knockdown efficiency; only shRNA resulting in >60% knockdown (shIFI6 #4, shISG15 #4, shMX1 #3/5, shIFI44L #1/3, and shIFI27 #2, see Online Supplementary Methods) were used. Bars represent means ± standard error of mean of duplicate measurements for three independent experiments. A dashed line (---) indicates that viability of ALL cells is equal to the viability of ALL cells co-cultured with NSC-shLuc-MSC (fold-change = 1; gray bars). ALL: acute lymphoblastic leukemia; NIC: non-infected control; NSC-shLuc: non-silencing control - short-hairpin-luciferase; sh: short hairpin.
exposure to ETV6-RUNX1-positive cells did not markedly decrease the viability of primary ETV6-RUNX1 (nor B-other) BCP-ALL cells in 3-day culture assays (Figure 5). Moreover, silencing the key regulator of the IFN-signaling pathway, i.e., STAT1, in MSC also did not result in altered ALL via-
bility after 40 or 120 hours of co-culture, while there was a reduction of ≥60% in STAT1 protein expression (Online Supplementary Figure S12). Thus, the viability of leukemic cells was not affected by interference with IFN signaling via silencing of individual IFN-related genes.
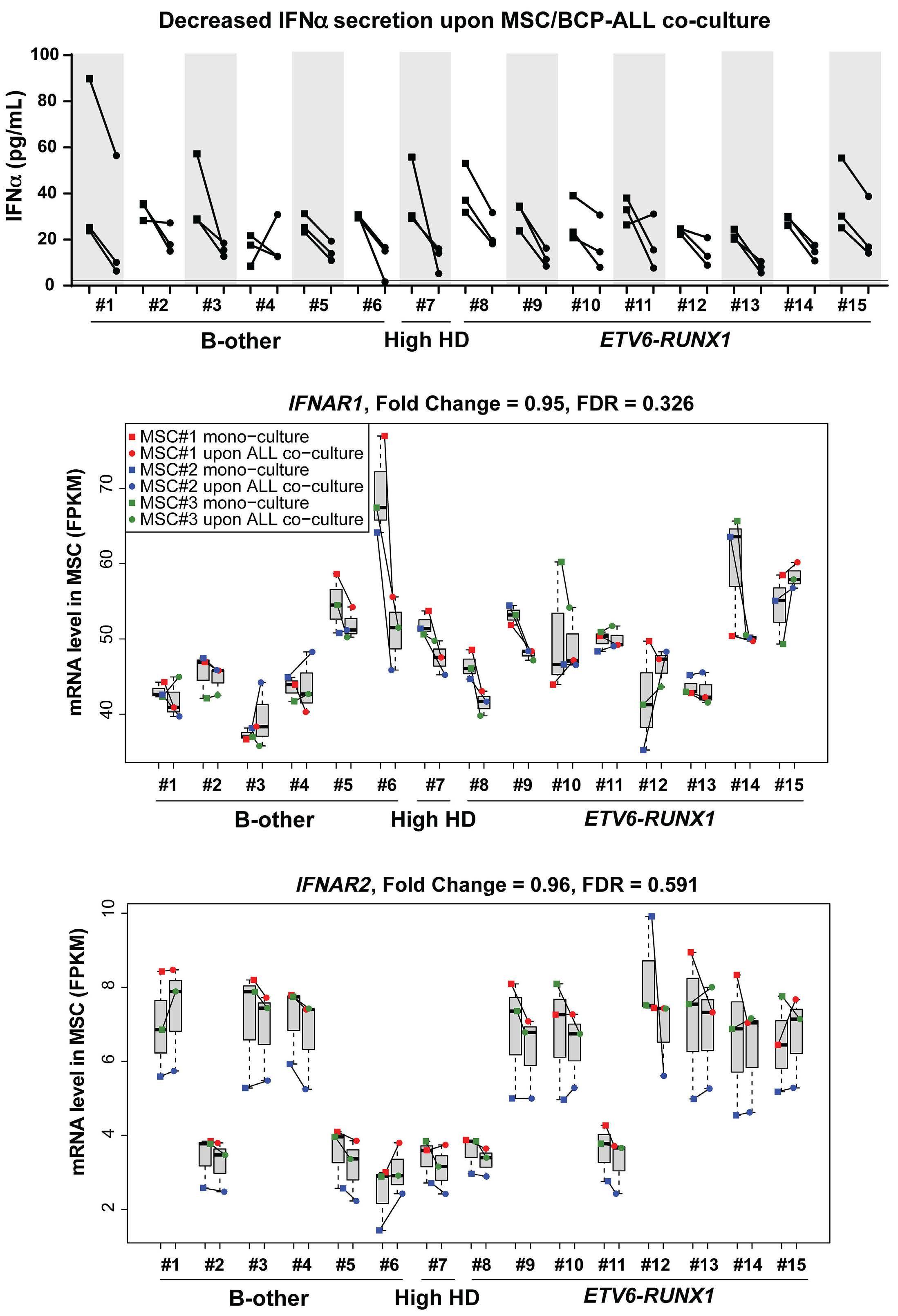
Figure 6. Interferon-α secretion and IFNAR1 expression decreased in mesenchymal stromal cells after co-culture with primary B-cell precursor acute lymphoblastic leukemia cells. (A) Interferon-alpha (IFNα) concentration in supernatants collected after 40 hours from 15 mesenchymal stromal cells (MSC) (MSC#13) and B-cell precursor acute lymphoblastic leukemia (BCP-ALL) (ALL#1-15) mono- and co-cultures. IFN α concentration is corrected for medium background level. Squares and circles indicate IFNα secretion level in the sum of MSC and BCP-ALL mono-cultures and their co-cultures, respectively. The limit of detection is indicated by a solid line. The range of detection for IFNα is 0.5-2,503 pg/mL. (B) IFNAR1 and (C) IFNAR2 mRNA levels expressed in paired samples of MSC sorted after mono-culture (squares; MSC#1-3 indicated in red, blue, and green, respectively.) and MSC sorted after co-culture with BCP-ALL cells from 15 individual patients (#1-15; circles). Boxplots represent the interquartile range, the median is depicted by a line. IFN: interferon; MSC: mesenchymal stromal cells; BCP-ALL: B-cell precursor acute lymphoblastic leukemia; HD: high hyperdiploid; FDR: P-value false discovery rate; FPKM: fragments per kilobase million.
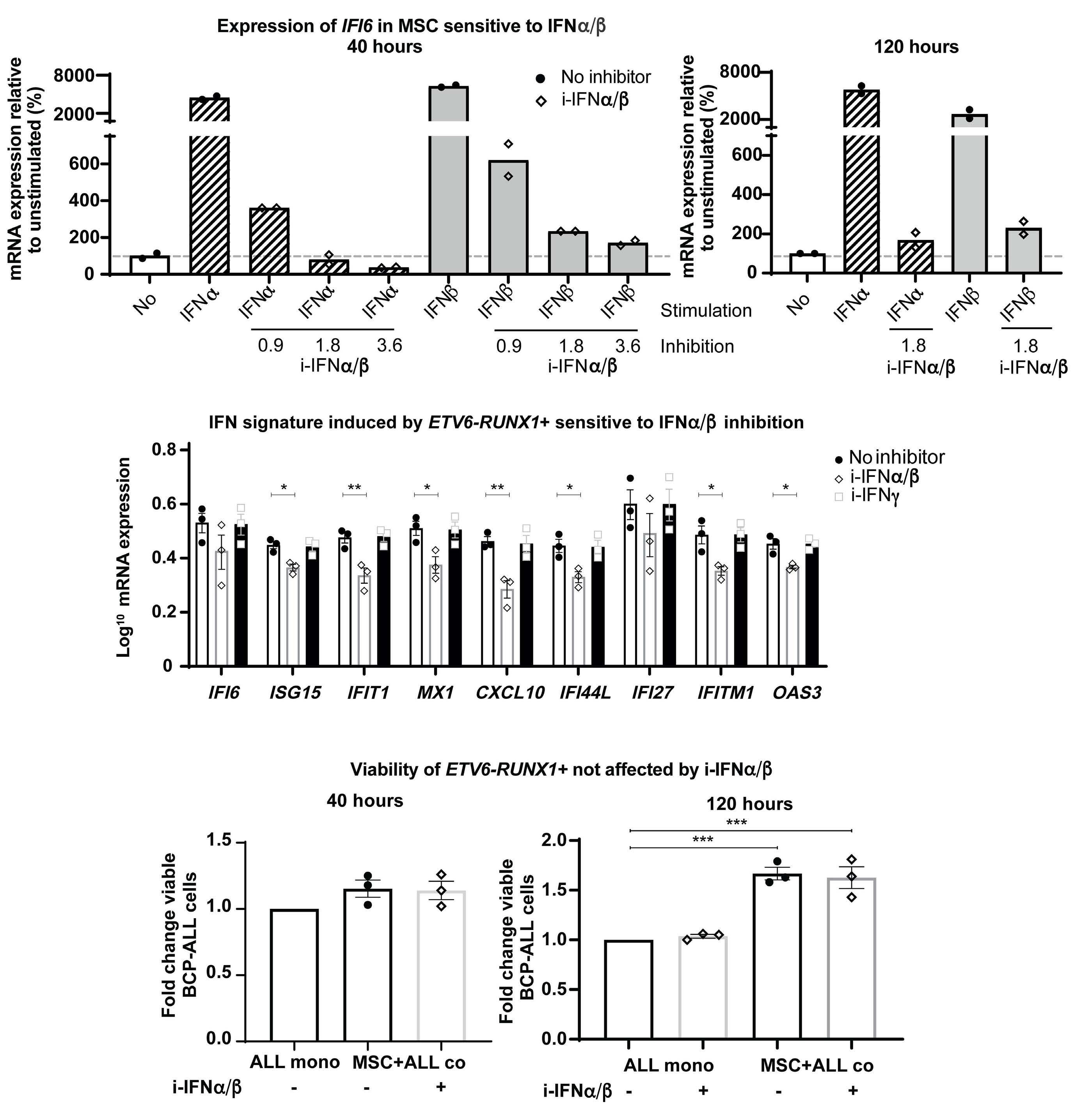
Figure 7. Interferon-mediated response in mesenchymal stromal cells does not affect primary ETV6-RUNX1 B-cell precursor acute lymphoblastic leukemia viability. (A) mRNA expression levels of IFI6 in mesenchymal stromal cells (MSC) (MSC#2) after 40 hours (left graph) and 120 hours (right graph) exposure to recombinant human interferon (IFN)α (3.0 U/mL; striped bars), IFNb (60 pg/mL; gray bars) and inhibitors thereof (i-IFNα/b; 0.9, 1.8 and/or 3.6 ng/mL) (open diamonds). Unstimulated MSC served as the control (white bars; filled circles). The dashed line (---) indicates baseline mRNA expression levels in mono-cultures of MSC set at 100%. (B) Log10-transformed IFN-related gene expression levels, measured by reverse transcriptase quantitative polymerase chain reaction, in MSC exposed to primary ETV6-RUNX1-positive B-cell precursor acute lymphoblastic leukemia (BCP-ALL) cells for 40 hours with and without inhibitors of IFNα/b (1.8 ng/mL; open diamonds) or IFNγ (3 ng/mL; open squares). Bars represent means ± standard error of mean (SEM) of triplicate measurements for three ETV6-RUNX1-positive samples (ALL#9-11). (C) Fold change in viability of ETV6-RUNX1 BCP-ALL cells after 40 hours (left graph) and 120 hours (right graph) of mono-culture with i-IFNα/b (1.8 ng/mL), MSC co-culture without i-IFNα/b, or MSC co-culture with i-IFNα/b (1.8 ng/mL), normalized to mono-culture without i-IFNα/b. Bars represent means ± SEM of triplicate measurements for three independent experiments. *P<0.05, **P<0.01, ***P<0.001. MSC: mesenchymal stromal cells; i-IFNα/b: inhibitors of interferon α/b; i-IFNγ: inhibitor of interferon γ; BCP-ALL: B-cell precursor acute lymphoblastic leukemia; mono: mono-culture; co: co-culture.
B-cell precursor acute lymphoblastic leukemia cells trigger an IFNα/b but not IFNγ response in mesenchymal stromal cells
Remarkably, the amount of secreted IFNα (and IFNb) was decreased in the co-culture setting compared with the levels detected in mono-culture of BCP-ALL cells and MSC, an effect unrelated to ETV6-RUNX1 status (Figure 6A and Online Supplementary Figure S13A, left). This is remarkable since the ETV6-RUNX1-positive cells clearly induced the expression of IFN-related genes in MSC (Figures 1 and 2).
The expression level of the IFNα/b-binding IFNAR1, but not IFNAR2 receptor, was often decreased in MSC after co-culture with BCP-ALL, independently of ETV6-RUNX1 status (Figure 6B). Secreted levels of IFNγ were below the level of quantifiable detection in virtually all samples and (mono/ co)-culture conditions (Online Supplementary Figure S13A, right). Expression levels of the IFNγ receptor genes IFNGR1 and IFNGR2 were variable and not related to ETV6-RUNX1 status (Online Supplementary Figure S13C).
The gene regulation in MSC is sensitive to IFNα/b since addition of recombinant IFNα and IFNb to mono-cultures of MSC for 40 and 120 hours clearly induced the expression of the (most significantly upregulated) IFN-index gene IFI6 in MSC. In correspondence, the ETV6-RUNX1-mediated induction of IFI6 expression was prevented by simultaneous addition of inhibitors of IFNα/b signaling, resulting in 56-fold
and 27-fold reductions, respectively, after 40 hours of incubation and 35-fold and 12-fold reductions, respectively, after 120 hours of incubation (Figure 7A). The ETV6-RUNX1 BCP-ALL-induced expression of IFN-related genes in MSC was also reduced upon exposure to inhibitors of IFNα/b but not to inhibitors of IFNγ (Figure 7B). However, addition of these inhibitors did not affect the viability of BCP-ALL cells in co-culture with MSC, after either 40 hours or 120 hours of incubation, whereas MSC clearly provided a survival benefit to ETV6-RUNX1 BCP-ALL cells at a prolonged incubation of 120 hours (Figure 7C). This MSC-induced survival benefit for ETV6-RUNX1 BCP-ALL was not sensitive to IFNα/b inhibition (Figure 7C, right). Furthermore, inhibitors of IFNα/b did not modulate MSC-induced resistance of ETV6-RUNX1 BCP-ALL cells to L-asparaginase, daunorubicin and prednisolone (Figure 8). In conclusion, the level of resistance of BCP-ALL cells to three chemotherapeutic agents was unaffected by blockade of IFN signaling in MSC.
Leukemic cells communicate with components of the bone marrow microenvironment in such a way that protection against chemotherapy and survival of leukemic cells is stimulated.9-13,21 In this study, we showed that ETV6-RUNX1 BCP-
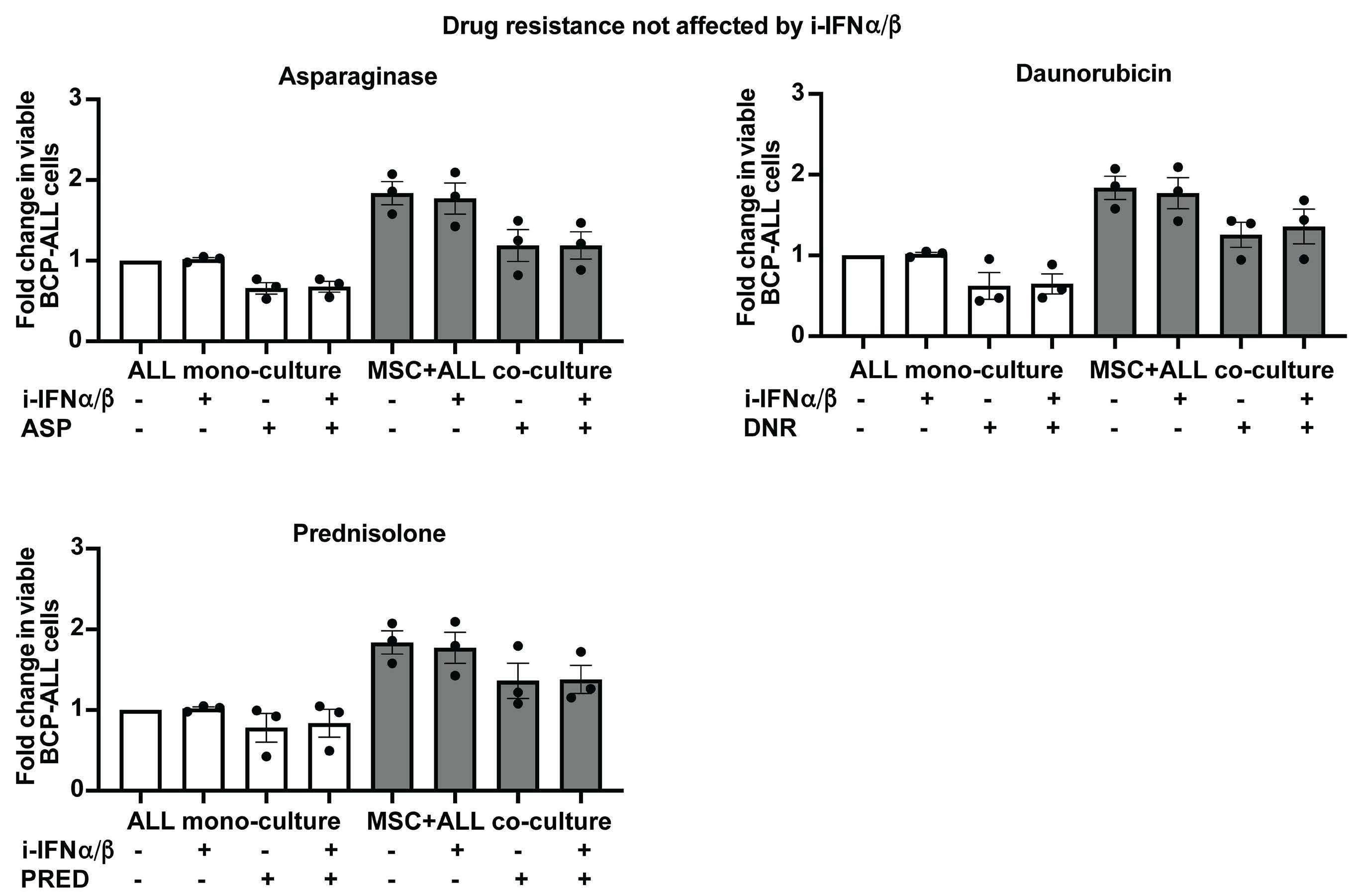
Figure 8. The interferon-mediated response in mesenchymal stromal cells does not affect the sensitivity of primary ETV6RUNX1 B-cell precursor acute lymphoblastic leukemia cells to drugs. Fold change in percentages of viable ETV6-RUNX1 B-cell precursor acute lymphoblastic leukemia cells in 120-hour mono-culture or mesenchymal stromal cell co-culture upon exposure to (A) L-asparaginase, (B) daunorubicin, or (C) prednisolone (concentration as determined in Online Supplementary Figure S2) and/or inhibitors of interferon α/b (1.8 ng/mL), normalized to values in mono-culture without drug exposure or interferon inhibitors. Bars represent means ± standard error of mean of triplicate measurements for three independent experiments. i-IFNα/b: inhibitors of interferon α/b; BCP-ALL: B-cell precursor acute lymphoblastic leukemia; MSC: mesenchymal stromal cells; ASP: L-asparaginase; DNR: daunorubicin; PRED: prednisolone.
ALL cells induce an IFN-related gene signature in MSC, which was dependent on IFNα/b signaling, but independent of IFNγ The IFN gene signature of MSC was leukemia-dependent since a mixed population of normal cord blood cells had a limited effect on expression levels of these genes in MSC, with an exception for monocyte-containing samples which induced, among others, CXCL10 expression. Monocytes, however, were absent in our BCP-ALL samples, strengthening the leukemia-driven origin of the IFN signature in MSC.
BCP-ALL samples with an ETV6-RUNX1 translocation were the most potent inducers of the IFN-related gene expression signature in MSC. This was true for both primary BCP-ALL samples as well as an ETV6-RUNX1 cell line. In correspondence, we observed that addition of IFNα/b inhibitors (but not an IFNγ inhibitor) to co-cultures of ETV6-RUNX1 BCPALL and MSC reduced the expression levels of IFN-related genes in the MSC compared to those in MSC that were co-cultured in the absence of these inhibitors.
ETV6 is known to be a transcriptional activator of a variety of IFN genes,22 and in correspondence higher baseline expression levels of IFN-related genes have been found in ETV6-RUNX1-driven leukemia.23 We also noticed some higher baseline levels in ETV6-RUNX1-positive ALL than in B-other ALL cases, but more remarkable was the upregulation of IFN-related genes in MSC upon exposure to ETV6-RUNX1-positive cells. This upregulation was partially dependent on close cell-cell contact, since we noticed that the expression was reduced when BCP-ALL cells and MSC were physically separated in transwell experiments. In line with this, Dander et al. showed that the ETV6-RUNX1 fusion can trigger modifications in adhesion molecule expression and adherence capacity of B-precursor cells.24
Members of the IFN family are known to combat viral infections, modulate immune responses, and stimulate antitumor activities.25-27 We noted that BCP-ALL cells, irrespective of ETV6-RUNX1 status, downregulated expression of the MSC-IFNα/b receptor (IFNAR1) and secreted IFNα/b levels in co-culture with MSC. The most likely explanation is that this occurs due to receptor internalization upon ligand binding, which is part of the functional process of IFN-pathway activation.28 Internalization of the IFNAR1/2-IFNα/b complex may be visualized by a fluorescence-based imaging method using a pH-sensitive dye that binds the receptor and will only fluoresce in acidic environments inside the cell.29,30 Recently, interferons were shown to elicit immune suppressive mechanisms in cancer, which may promote cancer progression and induce therapy resistance.31,32 Several studies have investigated the expression profile of IFN-related genes in cells derived from distinct cancer types.33-36 A subset of IFN-related genes, including the genes we found upregulated in MSC, i.e., IFI27, OAS1, OAS3, MX1 and ISG15, was persistently overexpressed in tumor cells resistant to chemotherapy or radiotherapy.33,34,36 We here observed that these same genes were induced in non-malignant
stromal support cells, i.e., the MSC, upon interaction with ETV6-RUNX1-positive ALL cells. MSC provide a survival benefit to BCP-ALL cells and protect these cells against chemotherapeutic drugs.9-12 However, we did not observe that the IFN-induced gene signature causally contributed to these processes since neither silencing of individual IFN-related genes or the key pathway regulator STAT1 in MSC nor blockade of IFNα/b signaling counteracted this MSC-mediated benefit. Our data therefore imply that the ETV6-RUNX1 BCP-ALL-induced IFNα/b signature in MSC serves a different role, e.g., attracting other cells, which are beneficial in maintaining leukemia, to the leukemic niche. IFNα/b are involved in recruitment, function, maturation and/ or activation of immune cells, such as NK cells, monocytes and dendritic cells.37,38 Cytokine and chemokine production can be induced by IFNα/b and may help to recruit inflammatory monocytes and NK cells to the site of inflammation.31,38 In contrast, prolonged activation of IFNα/b responsive genes can also suppress normal immune cell functions. In solid tumors, migration of NK cells towards the tumor site was observed. Trapped in the tumor microenvironment these NK cells downregulated NKG2D, the receptor that is important in defense against cancer by the immune system.39 This network of trapped, but not functional (innate) immune cells, resulted in a condition promoting tumor cell proliferation, survival, and metastasis.39,40 However, increased IFN-related gene expression in MSC after contact with leukemic cells may also reflect an anti-leukemic response of MSC in the tumor microenvironment to attract other immune cells.31,41,42
A recent study by Kumar et al. showed that reduced production of IFNα/b, leading to decreased NK-cell surveillance, promotes development of B-ALL in vivo 42 Restoring this IFNα/b production allows for NK effector cells to reduce leukemia progression in mice that are prone to MYC-driven B-ALL. In high-risk acute myeloid leukemia, IFNα/b administration after hematopoietic stem cell transplantation may be effective in preventing relapse.43 As interferons are often described as cytokines with a dual role,26,31,38 it still needs to be determined whether the observed IFNα/b response in our study is advantageous or detrimental to leukemic cells. In conclusion, our study reveals that IFNα/b but not IFNγ contribute to an IFN signature elicited by BCP-ALL in MSC. The induction of IFN-related genes in MSC did not affect the viability and level of drug resistance of BCP-ALL cells observed upon exposure to MSC. Our data warrant further studies into the role of the BCP-ALL-induced IFNα/b response, especially in the context of ETV6-RUNX1-positive ALL. This may increase our understanding of how leukemic cells manipulate the immune microenvironment, as recently suggested for IFNα/b-dependent activation of NK cells and dendritic cell-presenting leukemia-specific antigens. This knowledge is important in the emerging field of cellbased immunotherapies that are being applied ever more frequently in BCP-ALL.3,42
Disclosures
No conflicts of interest to disclose.
Contributions
MWES performed experiments, collected data, analyzed data, and wrote the paper for this study. EMPS performed Affymetrix gene studies and lentiviral silencing experiments. MWES, EMPS, MLdB, and CvdV designed the study and finalized this manuscript. JO, FS, MMPV, CHJV, and CvdV performed experiments. LS analyzed the RNA sequencing data. SN was responsible for the Luminex assays. All authors reviewed the final manuscript.
Acknowledgments
We would like to thank all group members of the Erasmus Medical Center in Rotterdam, and Princess Máxima
1. Bürgler S, Nadal D. Pediatric precursor B acute lymphoblastic leukemia: are T helper cells the missing link in the infectious etiology theory? Mol Cell Pediatr. 2017;4(1):6.
2. Kato M, Manabe A. Treatment and biology of pediatric acute lymphoblastic leukemia. Pediatr Int. 2018;60(1):4-12.
3. Pastorczak A, Domka K, Fidyt K, Martyna P, Firczuk M. Mechanisms of immune evasion in acute lymphoblastic leukemia. Cancers (Basel). 2021;13(7):1536.
4 Steeghs EMP, Boer JM, Hoogkamer AQ, et al. Copy number alterations in B-cell development genes, drug resistance, and clinical outcome in pediatric B-cell precursor acute lymphoblastic leukemia. Sci Rep. 2019;9(1):4634.
5. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541-1552.
6. Schwab C, Harrison CJ. Advances in B-cell precursor acute lymphoblastic leukemia genomics. Hemasphere. 2018;2(4):e53.
7 Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105(11):2524-2539.
8. Pieters R, De Groot-Kruseman H, Van Der Velden V, et al. Successful therapy reduction and intensification for childhood acute lymphoblastic leukemia based on minimal residual disease monitoring: study ALL10 from the Dutch Childhood Oncology Group. J Clin Oncol. 2016;34(22):2591-2601.
9 Naderi EH, Skah S, Ugland H, et al. Bone marrow stromaderived PGE2 protects BCP-ALL cells from DNA damageinduced p53 accumulation and cell death. Mol Cancer. 2015;14(1):1-12.
10 Polak R, De Rooij B, Pieters R, Den Boer ML. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood. 2015;126(21):2404-2414.
11. Usmani S, Sivagnanalingam U, Tkachenko O, Nunez L, Shand JC, Mullen CA. Support of acute lymphoblastic leukemia cells by nonmalignant bone marrow stromal cells. Oncol Lett. 2019;17(6):5039-5049.
12. Duan CW, Shi J, Chen J, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell. 2014;25(6):778-793.
13. Portale F, Cricrì G, Bresolin S, et al. ActivinA: a new leukemiapromoting factor conferring migratory advantage to B-cell
Center in Utrecht for their project input and help in (leukemic) sample processing. Special acknowledgment for the bio-informatics support from Alex Q. Hoogkamer. We also thank the Franciscus Vlietland Hospital in Schiedam for providing cord blood samples, and the MultiPlex Core Facility in the Center for Translational Immunology, University Medical Center Utrecht for performing Luminex experiments.
Funding
This study was funded by the Pediatric Oncology Foundation Rotterdam (SKOCR) and the Oncode Institute in Utrecht.
Data-sharing statement
Prof. Monique L. den Boer (m.l.denboer@prinsesmaximacentrum.nl) can be contacted for original data.
precursor-acute lymphoblastic leukemic cells. Haematologica. 2019;104(3):533-545.
14 De Rooij B, Polak R, Stalpers F, Pieters R, Den Boer ML. Tunneling nanotubes facilitate autophagosome transfer in the leukemic niche. Leukemia. 2017;31(7):1651-1654.
15. Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861-1865.
16. Baryawno N, Przybylski D, Kowalczyk MS, et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. 2019;177(7):1915-1932.
17. van den Berk LCJ, van der Veer A, Willemse ME, et al. Disturbed CXCR4/CXCL12 axis in paediatric precursor B-cell acute lymphoblastic leukaemia. Br J Haematol. 2014;166(2):240-249.
18. Smids C, Horje CSHT, Nierkens S, et al. Candidate serum markers in early Crohn’s disease: predictors of disease course. J Crohns Colitis. 2017;11(9):1090-1100.
19. van Iersel MP, Kelder T, Pico AR, et al. Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics. 2008;9:399.
20. Zaliova M, Stuchly J, Winkowska L, et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica. 2019;104(7):1396-1406.
21. Pastorczak A, Domka K, Fidyt K, Martyna P, Firczuk M. Mechanisms of immune evasion in acute lymphoblastic leukemia. Cancers (Basel). 2021;13(7):1536.
22. Mostafavi S, Yoshida H, Moodley D, et al. Parsing the interferon transcriptional network and its disease associations. Cell. 2016;164(3):564-578.
23. Einav U, Tabach Y, Getz G, et al. Gene expression analysis reveals a strong signature of an interferon-induced pathway in childhood lymphoblastic leukemia as well as in breast and ovarian cancer. Oncogene. 2005;24(42):6367-6375.
24. Dander E, Palmi C, D’Amico G, Cazzaniga G. The bone marrow niche in B-cell acute lymphoblastic leukemia: the role of microenvironment from pre-leukemia to overt leukemia. Int J Mol Sci. 2021;22(9):4426.
25. Schreiber G. The molecular basis for differential type I
interferon signaling. J Biol Chem. 2017;292(18):7285-7294.
26. Boukhaled GM, Harding S, Brooks DG. Opposing roles of type I interferons in cancer immunity. Annu Rev Pathol. 2021;16:167-198.
27. Chmiest D, Sharma N, Zanin N, et al. Spatiotemporal control of interferon-induced JAK/STAT signalling and gene transcription by the retromer complex. Nat Commun. 2016;7:13476.
28. Zanin N, Viaris de Lesegno C, Lamaze C, Blouin CM. Interferon receptor trafficking and signaling: journey to the cross roads. Front Immunol. 2021;11:615603.
29 Adie EJ, Francis MJ, Davies J, et al. CypHer 5: a generic approach for measuring the activation and trafficking of G protein-coupled receptors in live cells. Assay Drug Dev Technol. 2003;1(2):251-259.
30 Nath N, Godat B, Zimprich C, et al. Homogeneous plate based antibody internalization assay using pH sensor fluorescent dye. J Immunol Methods. 2016;431:11-21.
31. Budhwani M, Mazzieri R, Dolcetti R. Plasticity of type I interferon-mediated responses in cancer therapy: from antitumor immunity to resistance. Front Oncol. 2018;(8):322.
32. Provance OK, Lewis-Wambi J. Deciphering the role of interferon alpha signaling and microenvironment crosstalk in inflammatory breast cancer. Breast Cancer Res. 2019;21(1):59.
33. Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol. 2014;41(2):156-173.
34 Weichselbaum RR, Ishwaran H, Yoon T, et al. An interferonrelated gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105(47):18490-18495.
35. Duarte CW, Willey CD, Zhi D, et al. Expression signature of IFN/
STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS One. 2012;7(1):e29653.
36. Erdal E, Haider S, Rehwinkel J, Harris AL, McHugh PJ. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017;31(4):353-369.
37. Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res. 2011;17(9):2619-2627.
38. Lee AJ, Ashkar AA. The dual nature of type I and type II interferons. Front Immunol. 2018;9:2061.
39 Wennerberg E, Pfefferle A, Ekblad L, et al. Human anaplastic thyroid carcinoma cells are sensitive to NK cell-mediated lysis via ULBP2/5/6 and chemoattract NK cells. Clin Cancer Res. 2014;20(22):5733-5744.
40 Gun SY, Lee SWL, Sieow JL, Wong SC. Targeting immune cells for cancer therapy. Redox Biol. 2019;25:101174.
41. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405-414.
42. Kumar A, Taghi Khani A, Duault C, et al. Intrinsic suppression of type I interferon production underlies the therapeutic efficacy of IL-15-producing natural killer cells in B-cell acute lymphoblastic leukemia. J Immunother Cancer. 2023;11(5):e006649.
43. Magenau JM, Peltier D, Riwes M, et al. Type 1 interferon to prevent leukemia relapse after allogeneic transplantation. Blood Adv. 2021;5(23):5047-5056.
Qianxi Feng,1 Keren Xu,1 Mancy Shah,2 Shaobo Li,1 Andrew D. Leavitt,3 Lucy A. Godley,2 Adam J. de Smith1 and Joseph L. Wiemels1
1Center for Genetic Epidemiology, Department of Population and Public Health Sciences, University of Southern California, Los Angeles, CA; 2Division of Hematology/Oncology, Department of Medicine, and the Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL and 3Departments of Medicine and Laboratory Medicine, University of California, San Francisco, CA, USA
Abstract
Correspondence: J.L. Wiemels wiemels@usc.edu
Received: September 20, 2023. Accepted: December 29, 2023. Early view: January 11, 2024.
https://doi.org/10.3324/haematol.2023.284224
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Genetic predisposition to hematologic malignancies has historically been addressed utilizing patients recruited from clinical trials and pedigrees constructed at major treatment centers. Such efforts leave unexplored the genetic basis of variations in risk by race/ethnic group shown in population-based surveillance data where cancer registration, compulsory by law, delivers universal enrollment. To address this, we performed exome sequencing on DNA isolated from newborn bloodspots derived from sibling pairs with early-onset cancers across California in which at least one of the siblings developed a hematologic cancer, using unbiased recruitment from the full state population. We identified pathogenic/likely pathogenic (P/ LP) variants among 1,172 selected cancer genes that were private or present at low allele frequencies in reference populations. Within 64 subjects from 32 families, we found 9 LP variants shared between siblings, and an additional 7 such variants in singleton children (not shared with their sibling). In 8 of the shared cases, the ancestral origin of the local haplotype that carries P/LP variants matched the dominant global ancestry of study participant families. This was the case for Latino sibling pairs on FLG and CBLB, non-Latino White sibling pairs in TP53 and NOD2, and a shared GATA2 variant for a non-Latino Black sibling pair. A new inherited mutation in HABP2 was identified in a sibling pair, one with diffuse large B-cell lymphoma and the other with neuroblastoma. Overall, the profile of P/LP germline variants across ancestral/ethnic groups suggests that rare alleles contributing to hematologic diseases originate within their race/ethnic origin parental populations, demonstrating the value of this discovery process in diverse, population-based registries.
Hematologic malignancies (including leukemias, lymphomas, and multiple myeloma) are the most commonly diagnosed early-onset cancers among children, adolescents, and young adults.1 Inherited and de novo mutation of genes within critical cell development and growth/signaling pathways are central oncogenic events in the pathogenesis of hematologic cancers.2 Pathogenic/likely pathogenic (P/LP) germline variants were identified in approximately 10% of pediatric hematologic cancer patients regardless of family history.3-6 The risk of early-onset cancers (of any type) diagnosed under 26 years of age is 2.97 times higher among siblings and mothers who have a proband with hematologic cancer in the same family,7 indicating that inherited germline variants may contribute to this excessive cancer risk. This risk varies among ancestral/
ethnic groups,7 suggesting that predisposition variants may vary by identity or frequency among groups. Among early-onset hematologic malignancies, acute lymphoblastic leukemia is the most common, accounting for approximately 25% of all cancers diagnosed in children; and other lymphoid malignancies account for an additional 10% of all cancers.1,8 In our examination of linked population registries in California, the relative early-onset cancer risk is 2.87 times higher among siblings and mothers given a proband with leukemia, and 4.66 times higher given a proband with lymphoma.7 Acute lymphoblastic leukemia (ALL) is the most common leukemia in children and young adults. Recent studies have identified deleterious germline variants in TP53, PAX5, IKZF1, and ETV6 as risk factors for ALL9-11 but have not assessed the spectrum of deleterious germline variants among ancestral groups or in individuals
with a family history identified in a population-based registry. Concerning the elevated familial risk of lymphoid malignancies other than leukemia that our group has observed in California,7 previous studies have reported similar results: that first-degree relatives of patients with non-Hodgkin lymphoma (NHL), Hodgkin lymphoma (HL), or chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) among European origin populations have approximately 1.7 times, 3.1 times, and 8.5 times higher risk of developing these malignancies, respectively.12-17 Rare variants associated with familial-associated lymphoma risk, however, have not been studied extensively.18
The risk of hematologic cancers varies by ancestral/ethnic group. In California, Latino individuals have a higher incidence of ALL compared to those of other ancestral/ethnic groups across all ages;19 however, the highest familial risk (identified by families with 2 or more independent sibling cancers) was found among non-Latino Asian/Pacific Islanders (NLAPI).7 For lymphomas diagnosed under 19 years of age, the incidence among non-Latino White (NLW) subjects is higher than subjects of other ancestral/ethnic groups.7 However, the variation in germline predisposition that may drive this difference in risk by ancestral/ethnic group has not been formally addressed. Standard US state-based population registries may be useful to study rare genetic predispositions in an objective, “real world” population-based manner when biological samples are available. Here, we utilize linked cancer population registries in California along with the California Biobank to identify the nature and ancestral origin of rare P/LP germline variants that may explain this variation in cancer risk.
Our study methods were reviewed and approved by the Committee for Protection of Human Subjects (IRB) of the State of California. Cancer patients were derived from linked population-based registries in California, as previously described.7 Briefly, the dataset was created by linking information from the California Cancer Registry (CCR) and California Birth Statistical Master File. The linked dataset encompassed all cancer cases comprehensively diagnosed in individuals aged 0-26 years old (since 26 is the oldest age for which it was possible to perform this linkage at the time of this work), and their sibling’s and their mother’s cancers also captured within this registry. To study the rare genetic predispositions in association with hematologic cancers, we first identified all sibling pairs with cancer in the database. For all individuals with hematologic cancers (leukemia or lymphoma) and a sibling with cancer (of any type, excluding any iatrogenic cancers), we processed the pair for sequencing. Overall, 70 patients from 35 families were eligible for inclusion. DNA preparation and sequencing, and mapping and variant identification were performed as explained in the Online Supplementary Methods. The GATK pipeline for germline
short variant calling was performed, with variant filtering as appropriate. Presence of Down syndrome (DS) was assessed by chromosome 21 copy number normalized to all other chromosomes. Pathogenicity of variants in 1,172 genes (Online Supplementary Table S1) was initially assessed using informatic tools (Varsome, PeCanPie), followed by strict manual curation of all identified alleles by ACMG/ AMP guidelines.
The ancestral identity of each individual was assessed by matching local haplotypes with one of 5 Earth superpopulations: Latino (LAT), European (EUR), East Asian (EAS), African (AFR), South Asian (SAS). The informatic tool RFMix was used for this purpose, and the global ancestry of each individual was calculated by summing up the ancestral components of each of the haplotypes for the entire genome.
Among the 70 patients eligible for inclusion, 67 samples had adequate DNA for genotyping, and 64 of the 67 samples were sibling pairs from the same family and were included in the genetic analyses. The 64 subjects were from 32 families, among which 12 (38%) were LAT and 10 (31%) were non-Latino White (NLW) families (Table 1). Including parents and all healthy or diseased children, the average size of each LAT family was 4.92 people/family and the average size of NLW families was 6.10 people/family. One out of the 32 families had a mother diagnosed with cancer (desmoplastic small round cell tumor) in her twenties; we note that given the recruitment period of 1989-2015 we have a limited available follow-up time to track adult cancer in parents. We do not report age of onset of any cancer case to protect confidentiality of the families evaluated in this study. One subject (#534, family #2) was identified to have DS (# chr21 reads= 917228; population mean and Standard Deviation [SD] of chr21 = 651691+/-128812).
The mean sequencing coverage across subjects was 69.1 (range: 37.2-98.7) (Online Supplementary Table S2). Our initial software-guided curation yielded 14 unique P/LP variants. Upon further refined manual curation, 5 P/LP variants were reclassified as variants of uncertain significance (VUS), leaving 9 rare P/LP variants shared between siblings among 8 sibling pairs (Online Supplementary Table S3), and 105 rare VUS that are shared between siblings among the 32 families (Online Supplementary Table S4). Notably, along with demoting 5 LP variants to VUS, manual curation rescued one VUS to be LP (HABP2), and reclassified another 23 VUS to be LB (likely benign; Online Supplementary Table S4). Among the P/LP variants, one (11%) was found in LAT families, 2 (22%) were found in NLW families, 2 (22%) were found in non-Latino
Black (NLB) families, 2 (33%) were found in non-Latino Asian/Pacific Islander (NLAPI) families, and one (11%) was found in a non-Latino American Indian/Alaskan Native (NLAIAN) family. Among the VUS, 41 (39%) were found in LAT families, 21 (22%) were found in NLW families, 10 (10%) were found in NLB families, 27 (28%) were found in NLAPI families, and one (1%) was found in a NLAIAN family (Table 2). In the LAT families, P/LP variants were detected in ATM (NM_000051.3:c.3158dup; one sibling had T-cell leukemia and the other had diffuse large B-cell lymphoma) in family #27. For NLW families, P/LP variants were detected on TP53 (family #6, NM_000546.5:c.586C>T; one sibling with acute leukemia, not otherwise specified [NOS], and the other with signet ring carcinoma), and NOD2 (family #25, NM_022162.2:c.1515dup; both siblings diagnosed with HL NOS). For NLB families, P/LP variants were detected on GATA2 (family #15, NM_001145661.1:c.1009C>T; one sibling AMML-M4 and second sibling, AMKL-M7) and NOD2 (family #35, NM_022162.2:c.1515dup; both siblings with Burkitt cell leukemia). In separate NLAPI families, P/LP variants were detected on FLG (family #2, NM_002016.1:c.3321del; HL and AMKL-M7), and CBLB (family #21, NM_170662.4:c.2307del; large B-cell lymphoma and neuroblastoma). The siblings in this family also shared an additional nonsense variant in HABP2. For one NLAIAN family (#29), a P/LP variant was detected on SBF2 (NM_030962.3:c.4400del; one sibling with acute lymphoblastic leukemia NOS and the other sibling
with hepatoblastoma) (Online Supplementary Table S4). In addition, we identified 7 rare P/LP variants that are not shared between the 2 siblings. In 4 LAT subjects without any P/LP variants shared with siblings, these included a nonsense variant in AMER1 (NM_152424.3:c.28C>T), a frameshift deletion in HLTF (NM_003071.3:c.1967del), and missense variants in GJB2 (NM_004004.5:c.596C>T) and PINK1 (NM_032409.2:c.1040T>C) (Online Supplementary Table S5). In one NLW subject diagnosed with a malignant teratoma, we found a non-shared nonsense variant in SMARCA4 (NM_001128849.1:c.4038G>A). In an NLB subject who developed acute megakaryoblastic leukemia (AMKL), and who also shared a P/LP variant in GATA2 with their sibling who developed acute myeloid leukemia (family #15), a non-shared nonsense variant was identified in FAT1 (NM_005245.3:c.10957G>T). Finally, in a NLAPI subject who developed AMKL, and shared a P/LP variant in FLG with their sibling who developed HL (family #2), a nonshared rare P/LP variant was identified in exon 2 of GATA1 (NM_002049.3:c.115G>T) (Online Supplementary Table S5), in which somatic mutations are known drivers of AMKL. Of note, this GATA1 variant had an allele fraction of 71.4% and was detected in the subject identified with trisomy 21 (subject #534; see above); thus, it is likely to be a somatic GATA1 mutation that would be indicative of transient abnormal myelopoiesis (TAM), which occurs frequently in newborns with DS and is a precursor to AMKL in some cases.20
Table 1. Demographics of the 32 proband-sibling pairs, California Cancer Registry, 1989-2015.
Self-reported race/ethnicity, N (%)
Latino, all races
NLW
NLB
NLAPI
NLAIAN
Cancer site, N (%)
Leukemias
Lymphomas
Brain tumors
Neuroblastoma
Retinoblastoma
Renal tumors
Hepatic tumors
Bone tumors
Sarcomas
Germ cell tumors
Neoplasms and melanomas
N: number; NLW:
American Indian/ Alaskan Native; SD: Standard Deviation.
Ancestry of pathogenic/likely pathogenic germline variants
For the one LAT family with a shared sibling variant, the P/LP variant in ATM was shared on an SAS haplotype. For NLW families, the P/LP variants on TP53 and NOD2 shared EUR ancestry, which match their predicted dominant global ancestry. For one NLB family, the P/LP variant on GATA2 shared AFR ancestry on local haplotypes and this
matches their predicted dominant global ancestry. For NLAPI families, 2 pathogenic variants were classified as AMR and one (the novel HABP2 variant) as EAS. For the one NLAIAN family, the P/LP variant on SBF2 has shared local EUR ancestry, which matched their predicted dominant global ancestry (Figure 1, Online Supplementary Table S3 ).
Figure 1. Predicted global ancestries of study subjects and local ancestries of putatively pathogenic variants, California Cancer Registry, 1989-2015. *Predicted local ancestry matches the predicted dominant global ancestry. LW: Latino White; NLW: non-Latino White; NLB: non-Latino Black; NLAPI: non-Latino Asian/Pacific Islander; NLAIAN: non-Latino American Indian/Alaskan Native; AFR: African; AMR: Admixed American; EAS: East Asian; SAS: South Asian; EUR: European.
class, N (%)
VUS: variants of uncertain significance; NLW: non-Latino White; NLB: non-Latino Black; NLAPI: non-Latino Asian/Pacific Islander; NLAIAN: non-Latino American Indian/Alaskan Native.
This is the first study to our knowledge using purely population-based, no patient contact resources to examine genetic causes of family-based cancer clustering. We demonstrate that deleterious variants shared between family members contribute to early-onset hematologic cancers and that these can be revealed using California’s linked birth and cancer registries and neonatal blood spot Biobank. This establishes the utility of these population-based resources to study familial cancer predisposition successfully, as well as to identify germline inherited risk alleles among varied ancestral groups. The profile of germline P/LP variants across ancestral/ethnic groups share similarities as well as distinctive characteristics. Consistent with previous findings, we note that individuals with familial early-onset hematologic cancers harbor deleterious germline variants in TP53, GATA2, and ATM 9,21 Pathogenic variants in TP53 have been associated with many hematologic cancers including lymphoblastic,22 myeloid leukemias,23 and lymphomas.24-26 Deleterious germline variants in GATA2 have been associated with myeloid malignancies,27,28 and variants in ATM have been associated with T-cell prolymphocytic leukemia,29 mantle cell lymphoma,30 and gliomas.31 The non-shared SMARCA4 germline P/ LP variant that we identified in a subject with teratoma is consistent with previous reports that have described germline mutations in SMARCA4 for teratomas.32,33 Some P/LP variants exhibit novelty. The shared SBF2 in one NLAIAN family is a novel discovery in childhood cancers. SBF2 is associated with cancer by a newly identified long non-coding RNA, SBF2-AS1. SBF2-AS1 was initially characterized in non-small cell lung cancer,34 and has recently been reported to be over-expressed in multiple adulthood cancers in an East Asian population.35-46 P/LP variants in NOD2 is also a novel discovery in childhood cancers, and, intriguingly, one variant was found in 2 siblings who both developed HL. NOD2 is a gene that is involved in immune response,47 and has been associated with colorectal cancer.48 In addition, we also reported a shared FLG mutation in association with childhood hematologic cancers. Somatic mutations of this gene have been reported to be associated with autoimmune diseases.49 A HABP2 mutation was shown to be shared between siblings with malignant large-B-cell lymphoma and neuroblastoma. HABP2 encodes a serine protease, and an inactivating mutational variant (G534E) distinct from the one found here was previously linked to extramedullary thyroid neoplasia.50 The nonsense mutation found here in this gene (C290*) has not been reported previously and likely represents a private familial mutation.
There are several strengths and limitations of this study. A major strength is the linkage of population-based cancer registries to identify subjects for sequencing. Thus, we have established a novel perspective on genetic predisposition that drives the excessive familial risk of early-onset hematologic cancers without selection or recruitment bias that may af-
fect clinic or referral-based studies. We found P/LP alleles in about a quarter (25%) of families, as well as many suspect VUS alleles in addition, and an additional 7 families (22%) that have a P/LP mutation in one child only. This compares with the approximately 5-10% frequency of such alleles in sample series chosen without regards to family history.3-6 A study that examined predisposition in the context of multiple sibling myeloid cancers in families using anecdotal clinical referral recruitment of families and liberal mutation identification criteria found candidate mutations in 83 of 86 families.51 While we also similarly found VUS and P/LP mutations in nearly every subject, we emphasize the reporting of highly curated L/LP alleles, which in our 64 subjects yielded 23 (36%) mutations. Given the large size of our base population and definition of “cancer families” as those with a minimum of only 2 cases, it is likely that some families will be afflicted by multiple cancers by chance without predisposing alleles or be influenced via familial sharing of an environmental exposure. Still, our 36% yield compares with a 4.4% yield in an assessment of leukemia-associated P/LP mutations in a large study of sporadic patients,4 attesting to the power of population-based recruitment in the current investigation. Benefiting from the highly diverse population in California, we are the first research group that has assessed the variation in genetic predisposition in association with familial hematologic cancer risk in multiple ancestral/ethnic groups over a defined population, appraising cancer predisposition in a selection-bias free, consent-bias free population. However, limited by the length of time in data collection and the rarity of childhood cancers, we have only identified a selection of mutations that are unique to an ancestral/ ethnic group. In addition, we have no way to evaluate or confirm the impact of non-shared P/LP alleles inherited by chance by those siblings that did not share a P/LP allele. Also, limited to analysis of SNV and indels, we did not assess variation in copy number or epigenetic alterations that may be potentially pathogenic. Cancer risk is increased by both genetic and environmental factors. Family members are generally exposed to the same environments and therefore those in-common environments may contribute to the observed familial cancer clustering. To understand such clustering better, concomitant assessments of environmental factors in coordination with germline genetics should be considered for future studies. Such analyses will help to establish the role of strong cancer predisposition variants more definitively among different ancestral/ethnic groups, and whether such mutations harbor varied penetrance in the context of different ancestral backgrounds and environments. This type of evaluation is increasingly critical in the intermixed population of California and other world regions, and has profound implications for family genetic counseling in the context of improved cancer patient survival.
Disclosures
No conflicts of interest to disclose.
Contributions
QF and KX performed primary data analysis, with help from SL. MS performed manual variant classifications. AL, LAG and JLW conceived the study and acquired funding. AJdS and JLW supervised statistical analysis, with help from MS and LAG. QF wrote the first draft, and JLW, LAG, and AJdS completed the writing team. All authors read and approved the final draft.
Funding
This study was funded by the V Foundation for Cancer Re-
1. Siegel RL, Miller KD, Jemel A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7-30.
2. Rampersaud E, Ziegler DS, Iacobucci I, et al. Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv. 2019;3(7):1039-1046.
3. Saletta F, Dalla Pozza L, Byrne JA. Genetic causes of cancer predisposition in children and adolescents. Transl Pediatr. 2015;4(2):67-75.
4 Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336-2346.
5. Plon SE, Lupo PJ. Genetic predisposition to childhood cancer in the genomic era. Annu Rev Genomics Hum Genet. 2019;20:241-263.
6. Ripperger T, Bielack SS, Borkhardt A, et al. Childhood cancer predisposition syndromes-a concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A. 2017;173(4):1017-1037.
7 Feng Q, Nickels E, Muskens IS, et al. Increased burden of familial-associated early-onset cancer risk among minority Americans compared to non-Latino Whites. Elife. 2021;10:e64793.
8. Cerhan JR, Slager SL. Familial predisposition and genetic risk factors for lymphoma. Blood. 2015;126(20):2265-2273.
9 Stieglitz E, Loh ML. Genetic predispositions to childhood leukemia. Ther Adv Hematol. 2013;4(4):270-290.
10 Trevino LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001-1005.
11. Churchman ML, Qian M, Te Kronnie G, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. 2018;33(5):937-948.
12. Goldin LR, Landgren O, McMaster ML, et al. Familial aggregation and heterogeneity of non-Hodgkin lymphoma in populationbased samples. Cancer Epidemiol Biomarkers Prev. 2005;14(10):2402-2406.
13. Goldin LR, Bjorkholm M, Kristinsson SY, Turesson I, Landgren O. Elevated risk of chronic lymphocytic leukemia and other indolent non-Hodgkin’s lymphomas among relatives of patients with chronic lymphocytic leukemia. Haematologica. 2009;94(5):647-653.
14. Goldin LR, Bjorkholm M, Kristinsson SY, Turesson I, Landgren O. Highly increased familial risks for specific lymphoma subtypes. Br J Haematol. 2009;146(1):91-94.
15. Kristinsson SY, Bjorkholm M, Goldin LR, et al. Risk of lymphoproliferative disorders among first-degree relatives of
search (FP067172; to ADL, LAG, JLW). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Data-sharing statement
Sharing of raw and processed whole exome sequencing data from California Biobank specimens is restricted by California state statutory laws and IRB (see Online Supplementary Appendix for further details). Results from specific genes or regions may be requested from wiemels@usc.edu.
lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia patients: a population-based study in Sweden. Blood. 2008;112(8):3052-3056.
16. Goldin LR, Pfeiffer RM, Gridley G, et al. Familial aggregation of Hodgkin lymphoma and related tumors. Cancer. 2004;100(9):1902-1908.
17 Chang ET, Smedby KE, Hjalgrim H, et al. Family history of hematopoietic malignancy and risk of lymphoma. J Natl Cancer Inst. 2005;97(19):1466-1474.
18. Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10(4):241-251.
19 Feng Q, de Smith AJ, Vergara-Lluri M, et al. Trends in acute lymphoblastic leukemia incidence in the US from 2000-2016: an increased risk in Latinos across all age groups. Am J Epidemiol. 2021;190(4):519-527.
20. Roy A, Roberts I, Norton A, Vyas P. Acute megakaryoblastic leukaemia (AMKL) and transient myeloproliferative disorder (TMD) in Down syndrome: a multi-step model of myeloid leukaemogenesis. Br J Haematol. 2009;147(1):3-12.
21. Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21(2):122-137.
22. Salmoiraghi S, Rambaldi A, Spinelli O. TP53 in adult acute lymphoblastic leukemia. Leuk Lymphoma. 2018;59(4):778-789.
23. Barbosa K, Li S, Adams PD, Deshpande AJ. The role of TP53 in acute myeloid leukemia: challenges and opportunities. Genes Chromosomes Cancer. 2019;58(12):875-888.
24. Jain P, Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am J Hematol. 2019;94(6):710-725.
25. Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903-1910.
26. Deng M, Xu-Monette ZY, Pham LV, et al. Aggressive B-cell lymphoma with MYC/TP53 dual alterations displays distinct clinicopathobiological features and response to novel targeted agents. Mol Cancer Res. 2021;19(2):249-260.
27. Pasquet M, Bellanne-Chantelot C, Tavitian S, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. 2013;121(5):822-829.
28. Kennedy AL, Shimamura A. Genetic predisposition to MDS: clinical features and clonal evolution. Blood. 2019;133(10):1071-1085.
29. Kanagal-Shamanna R, Bao H, Kearney H, et al. Molecular characterization of novel ATM fusions in chronic lymphocytic
leukemia and T-cell prolymphocytic leukemia. Leuk Lymphoma. 202263(4):865-875.
30 Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(45):18250-18255.
31. Lin L, Cai J, Tan Z, et al. Mutant IDH1 enhances temozolomide sensitivity via regulation of the ATM/CHK2 pathway in glioma. Cancer Res Treat. 2021;53(2):367-377.
32. Genestie C, Blanc-Durand F, Auguste A, et al. Clinical utility of SMARCA4 testing by immunohistochemistry in rare ovarian tumours. Br J Cancer. 2020;122(4):564-568.
33. Dong A, Lu Y, Lu B. Genomic/epigenomic alterations in ovarian carcinoma: translational insight into clinical practice. J Cancer. 2016;7(11):1441-1451.
34. Lv J, Qiu M, Xia W, et al. High expression of long non-coding RNA SBF2-AS1 promotes proliferation in non-small cell lung cancer. J Exp Clin Cancer Res. 2016;35:75.
35. Chen G, Gu Y, Han P, et al. Long noncoding RNA SBF2-AS1 promotes colorectal cancer proliferation and invasion by inhibiting miR-619-5p activity and facilitating HDAC3 expression. J Cell Physiol. 2019;234(10):18688-18696.
36. Chen R, Xia W, Wang X, et al. Upregulated long non-coding RNA SBF2-AS1 promotes proliferation in esophageal squamous cell carcinoma. Oncol Lett. 2018;15(4):5071-5080.
37. Gao F, Feng J, Yao H, et al. LncRNA SBF2-AS1 promotes the progression of cervical cancer by regulating miR-361-5p/FOXM1 axis. Artif Cells Nanomed Biotechnol. 2019;47(1):776-782.
38. He M, Feng L, Qi L, Rao M, Zhu Y. Long noncoding RNASBF2-AS1 promotes gastric cancer progression via regulating miR-545/ EMS1 axis. Biomed Res Int. 2020;2020:6590303.
39. Hua YQ, Zhu YD, Xie GQ, et al. Long non-coding SBF2-AS1 acting as a competing endogenous RNA to sponge microRNA-142-3p to participate in gemcitabine resistance in pancreatic cancer via upregulating TWF1. Aging (Albany NY). 2019;11(20):8860-8878.
40 Li Y, Liu G, Li X, et al. Long non-coding RNA SBF2-AS1 promotes hepatocellular carcinoma progression through regulation of miR-140-5p-TGFBR1 pathway. Biochem Biophys Res Commun.
2018;503(4):2826-2832.
41. Tian YJ, Wang YH, Xiao AJ, et al. Long noncoding RNA SBF2-AS1 acts as a ceRNA to modulate cell proliferation via binding with miR-188-5p in acute myeloid leukemia. Artif Cells Nanomed Biotechnol. 2019;47(1):1730-1737.
42. Xia W, Liu Y, Cheng T, et al. Down-regulated lncRNA SBF2-AS1 inhibits tumorigenesis and progression of breast cancer by sponging microRNA-143 and repressing RRS1. J Exp Clin Cancer Res. 2020;39(1):18.
43. Yu H, Zheng J, Liu X, et al. Transcription factor NFAT5 promotes glioblastoma cell-driven angiogenesis via SBF2-AS1/miR-3383p-mediated EGFL7 expression change. Front Mol Neurosci. 2017;10:301.
44 Zha W, Li X, Tie X, et al. The molecular mechanisms of the long noncoding RNA SBF2-AS1 in regulating the proliferation of oesophageal squamous cell carcinoma. Sci Rep. 2021;11(1):805.
45. Zhang Q, Pan X, You D. Overexpression of long non-coding RNA SBF2-AS1 promotes cell progression in esophageal squamous cell carcinoma (ESCC) by repressing miR-494 to up-regulate PFN2 expression. Biol Open. 2020;bio.048793.
46. Zhang Z, Yin J, Lu C, et al. Exosomal transfer of long non-coding RNA SBF2-AS1 enhances chemoresistance to temozolomide in glioblastoma. J Exp Clin Cancer Res. 2019;38(1):166.
47. Caruso R, Warner N, Inohara N, Nunez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity. 2014;41(6):898-908.
48. Branquinho D, Freire P, Sofia C. NOD2 mutations and colorectal cancer - where do we stand? World J Gastrointest Surg. 2016;8(4):284-293.
49 Ross KA. Coherent somatic mutation in autoimmune disease. PLoS One. 2014;9(7):e101093.
50 Gara SK, Jia L, Merino MJ, et al. Germline HABP2 mutation causing familial nonmedullary thyroid cancer. N Engl J Med. 2015;373(5):448-455.
51. Rio-Machin A, Vulliamy T, Hug N, et al. The complex genetic landscape of familial MDS and AML reveals pathogenic germline variants. Nat Commun. 2020;11(1):1044.
Haiping Luo,1* Qiqi Li,1* Jiaxin Hong,1 Zhibin Huang,1 Wenhui Deng,1 Kunpeng Wei,1 Siyu Lu,1 Hailong Wang,2 Wenqing Zhang1 and Wei Liu1
1Division of Cell, Developmental and Integrative Biology, School of Medicine, South China University of Technology and 2KingMed School of Laboratory Medicine, Guangzhou Medical University, Department of Basic Research, Guangzhou Laboratory, Guangzhou, China
*HL and QL contributed equally as first authors.
Abstract
Correspondence: W. Liu liuwei7@scut.edu.cn
W. Zhang mczhangwq@scut.edu.cn
Received: June 13, 2023. Accepted: February 12, 2024. Early view: February 22, 2024.
https://doi.org/10.3324/haematol.2023.283647
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
t(1;19)(q23;p13) is one of the most common translocation genes in childhood acute lymphoblastic leukemia (ALL) and is also present in acute myeloid leukemia (AML) and mixed-phenotype acute leukemia (MPAL). This translocation results in the formation of the oncogenic E2A-PBX1 fusion protein, which contains a trans-activating domain from E2A and a DNA-binding homologous domain from PBX1. Despite its clear oncogenic potential, the pathogenesis of E2A-PBX1 fusion protein is not fully understood (especially in leukemias other than ALL), and effective targeted clinical therapies have not been developed. To address this, we established a stable and heritable zebrafish line expressing human E2A-PBX1 (hE2A-PBX1) for high-throughput drug screening. Blood phenotype analysis showed that hE2A-PBX1 expression induced myeloid hyperplasia by increasing myeloid differentiation propensity of hematopoietic stem cells (HSPC) and myeloid proliferation in larvae, and progressed to AML in adults. Mechanistic studies revealed that hE2A-PBX1 activated the TNF/IL-17/MAPK signaling pathway in blood cells and induced myeloid hyperplasia by upregulating the expression of runx1. Interestingly, through high-throughput drug screening, three small molecules targeting the TNF/IL-17/MAPK signaling pathway were identified, including OUL35, KJ-Pyr-9, and CID44216842, which not only alleviated the hE2A-PBX1-induced myeloid hyperplasia in zebrafish but also inhibited the growth and oncogenicity of human pre-B ALL cells with E2A-PBX1. Overall, this study provides a novel hE2APBX1 transgenic zebrafish leukemia model and identifies potential targeted therapeutic drugs, which may offer new insights into the treatment of E2A-PBX1 leukemia.
t(1;19)(q23;p13) is the most common chromosomal translocation in leukemia, causing 5-7% of childhood ALL1,2 and also AML/MPAL. 3,4 In ALL, E2A-PBX1 is not a primary ALL prognostic factor, but it is associated with poor response to therapy and short remission. 5 ALL patients with E2A-PBX1 translocation appear to have a high risk of relapse, and this translocation is also associated with known high-risk clinical features, including elevated white blood cell counts at diagnosis and central nervous system leukemia.6,7 Furthermore, a previous study has shown that the E2A-PBX1 fusion gene can serve as an effective minimal residual disease (MRD) marker following induction chemotherapy, the relapse rate of patients with rapid early conversion to negative for this gene is relatively low. 5,8 Reports based on sporadic cases have indicated that E2A-PBX1-positive patients also exhibit
high-risk clinical features, including high cell counts, high serum lactate dehydrogenase levels, and central nervous system (CNS) involvement. 3 Although recent intensified treatment regimens and allogeneic hematopoietic stem cell transplantation have improved the prognosis of adult ALL patients with E2A-PBX1, 9 its systemic toxicity and high cost should not be overlooked.
The E2A-PBX1 protein resulting from t(1;19)(q23;p13) fusion combines the N-terminal transactivation domain of E2A (also known as TCF3) and the C-terminal DNA-binding homologous domain of PBX1.10 As a major regulator of B-lymphocyte generation, E2A determines the differentiation of B-cell lineages,11 also plays an important role in maintaining the hematopoietic stem cell pool and promoting the maturation of myelo-lymphoid and myelo-erythroid progenitors.12 PBX1, a TALE (3-amino-acid loop extension) transcription factor, interacts with other members of the HOX family to enhance their DNA-binding
specificity and affinity.13 PBX1 not only plays an important role in regulating morphologic patterning, organogenesis, and hematopoiesis, but is also an important component of the protein complex that regulates the expression of developmental genes.14
The oncogenic role of E2A-PBX1 has been reported in both the myeloid and lymphoid systems of mice and cell lines. For example, the transduction of E2A-Pbx1 into mouse myeloid progenitor cells via retroviral vectors results in AML in mice.14,15 Adding granulocyte-macrophage colony-stimulating factor (GM-CSF), E2A-Pbx1 can also immortalize myeloid progenitor cells in vitro . 16 On the other hand, the specific induction of E2A-PBX1 expression in the mouse lymphoid system leads to T/B-ALL,17-19 and the injection of E2A-PBX1 pro-T cells into mice leads to T-ALL and AML. 20 Although the oncogenic potential of E2A-PBX1 is clear, its pathogenic mechanism has not been fully elucidated (especially in leukemias other than ALL). Currently, there are three main explanations for the mechanism underlying E2A-PBX1-induced leukemia: i) E2A-PBX1 oncoprotein may deregulate the expression of critical genes normally controlled by PBX/HOX/MEINOX complexes; ii) it alters the function of transcriptional co-activators; and iii) it impairs the function of wildtype (WT) E2A. 21 Recently, a study using two pre-B ALL cell lines found that E2A-PBX1 can induce pre-B ALL by directly interacting with RUNX1 and co-activating the expression of oncogenes, including RUNX1 . 1 This study strongly supports the gain-of-function oncogenic role of E2A-PBX1.1 However, the mechanism by which E2A-PBX1 mediates AML oncogenesis is still unclear. More importantly, although studies have reported the development of potential drugs for E2A-PBX1 leukemia, 22,23 clinically effective targeting drugs for E2A-PBX1 leukemia are still lacking.
In order to better understand the mechanisms behind E2A-PBX1-induced leukemia and develop effective targeted therapies, we constructed an inducible human-derived E2A-PBX1 leukemia model using zebrafish. In zebrafish, just like in mammals, definitive hematopoietic stem cells (HSC) arise from the ventral wall of the dorsal aorta (VDA), which is equivalent to the aorta-gonad-mesonephros (AGM) in mammals. 24 These HSC then migrate into the caudal hematopoietic tissue (CHT), where they expand and subsequently colonize the kidney marrow, equivalent to the mammalian bone marrow, to generate hematopoietic cells throughout adulthood. 24 Zebrafish have proven to be an excellent model for studying hematopoietic disorders, particularly in extensive therapeutic compound screenings, due to their transparency, small size, short lifespan (2-3 years), high fecundity and genetic similarity to humans. 25 In recent years, the successful establishment of zebrafish patient-derived xenograft (zPDX) models has further advanced the use of zebrafish in biomedical research applications. 26,27
In our research, this hE2A-PBX1 transgenic zebrafish exhibited a phenotype of aberrant myeloid expansion and lymphoid dysplasia in larvae and eventually progressed to AML-like disease with age. Further mechanistic analysis revealed that hE2A-PBX1 activated the TNF/IL-17/MAPK signaling pathway in blood cells and induced myeloid hyperplasia by upregulating runx1 expression. Importantly, through drug screening, we found that the small molecule compounds OUL35, KJ-Pyr-9 and CID44216842, which target the TNF/IL-17/MAPK signaling pathway, not only significantly alleviated the AML-like phenotype in the h E2A-PBX1 zebrafish model, but also blocked the oncogenicity of human E2A-PBX1 pre-B cells. In conclusion, our study provides insights into the mechanisms by which E2A-PBX1 causes AML-like disease and identifies several promising compounds for targeted therapy.
Generation of pT2AL- hsp70-hE2A-PBX1 construct and of Tg(hsp70:E2A-PBX1-EGFP) transgenic zebrafish
The pT2AL- hsp70-E2A-PBX1 -EGFP plasmid is composed of hsp70 promoter, a h E2A-PBX1 cDNA fragment obtained via polymerase chain reaction (PCR) from human samples, the Tol2 element and the SV40 poly A sequence. The E2A and PBX1 cDNA fragments were linked together through overlap PCR. Following AgeI/SphI digestion, the h E2A-PBX1 sequence was inserted downstream of hsp70 promoter and subcloned into the pT2AL plasmid. This construct, named pT2AL- hsp70-hE2A-PBX1 -EGFP, was used for microinjection into zebrafish embryos at the one-cell stage along with Tol2 transposon mRNA to generate the Tg(hsp70:E2A-PBX1-EGFP ) transgenic zebrafish.
Abdominal tissue (above the pelvic fin) from euthanized adult fish was fixed in 4% paraformaldehyde (PFA) and subsequently processed into frozen sections, 28 permeabilized with 0.3% triton-X-100, and blocked with 5% fetal bovine serum. The sections were then incubated with rabbit-anti-Lcp1 monoclonal antibody (1:200), 29 followed by Alexa Fluor 488-anti-rabbit antibody (Invitrogen; 1:400) and DAPI (Invitrogen) for fluorescent visualization.
Bromodeoxyuridine labeling
Sibling and Tg(hsp70:E2A-PBX1-EGFP) embryos at 5-6 days post-fertilization (dpf) were incubated in 10 mM bromodeoxyuridine (BrdU) (Sigma-Aldrich) for 4 hours. The embryos were then stained with goat-anti-DsRed (Abcam; 1:400) and rabbit-anti-Lcp1 monoclonal antibody (1:200), followed by incubation with Alexa Fluor 488-anti-goat antibody (Invitrogen; 1:400) and Alexa Fluor 488-anti-rabbit antibody (Invitrogen; 1:400) for fluorescent visualization, according to the provided instructions. 30
Continued on following page.
Figure 1. Generation and characterization of transgenic zebrafish expressing human E2A-PBX1 (hE2A-PBX1). (A) Schematic representation of Tg(hsp70:E2A-PBX1-EGFP) expression vector, and Sanger sequencing confirmed the presence of the hE2A-PBX1 sequence. FP: forward primer; pA: poly A termination sequence. (B) F4 generation hE2A-PBX1-transgenic embryos exhibited EGFP expression at 32 hours post-fertilization (hpf). EGFP- (sibling) and EGFP+ (Tg(hsp70:E2A-PBX1-EGFP)) embryos obtained by crossing the stable line Tg(hsp70:E2A-PBX1-EGFP) with wild-type (WT) fish. Tg(hsp70:E2A-PBX1-EGFP) embryos exhibited strong EGFP fluorescence (white arrows). (C) Specific polymerase chain reaction (PCR) amplification of a 231 bp fragment within the hE2A-PBX1 fusion region confirmed the integration of hE2A-PBX1 cDNA sequence into the genomes of Tg(hsp70:E2A-PBX1-EGFP). (D) Real time quantitative PCR (RT-qPCR) analysis showed hE2A-PBX1 mRNA high expression in Tg(hsp70:E2A-PBX1-EGFP) compared to the siblings at 3 days post-fertilization (dpf). The black asterisks indicate statistical difference (Student t tests, mean ± standard error of the mean; ****P<0.0001) (E) Wright-Giemsa staining of whole blood cells from siblings and Tg(hsp70:E2A-PBX1-EGFP) at 5 dpf. Myelocytes are indicated by red arrows in the image. (E’) Statistical analysis of the cell counts in panel (E). The black asterisks indicate statistical difference (one-way ANOVA, mean ± standard error of the mean; *P<0.05). N>150, number of zebrafish larvae. (F) Survival curves of Tg(hsp70:E2A-PBX1-EGFP) and sibling larvae up to 15 days after heat shock. The black asterisks indicate statistical difference (log-rank [Mantel-Cox] test; ****P<0.0001).
TUNEL and immunofluorescence analyses
Transferase dUTP nick end labeling (TUNEL) assay (In Situ Cell Death Detection Kit; Roche) was performed as instructed. The samples were stained with goat-anti-DsRed (Abcam; 1:400) and rabbit-anti-Lcp1 antibody (1:200), followed by incubation with Alexa Fluor 488-anti-goat antibody (Invitrogen; 1:400) and Alexa Fluor 488-anti-rabbit antibody (Invitrogen; 1:400) for fluorescent visualization.
RNA-sequencing analysis
Whole kidney marrow (KM) cell suspensions were isolated from adult WT and Tg(hsp70:E2A-PBX1-EGFP). RNA extraction was performed using TRIZOL (Invitrogen),30 and the extracted RNA samples were sent for library construction and RNA sequencing at Guangzhou Jidio Biotechnology.
Drug and inhibitor treatment
The small molecule inhibitors we screened were randomly selected from TargetMol’s Bioactive Compound Library (Online Supplementary Excel S1). These compounds were all diluted to 20 mM as storage solution. KJ-Pyr-9, OUL35, A-484954, LRRK2-IN-1, CID44216842 were dissolved in dimethyl sulfoxide (DMSO), while L-arginine hydrochloride was dissolved in ddH2O.
Animal care and xenograft procedure
Animal studies were performed in accordance with the animal research advisory committee of South China University of Technology. At 3 dpf, zebrafish larvae (runx1w84x) were injected with 600 human leukemic cells per embryo through the yolk sac into the posterior cardinal vein (PCV) or dorsal aorta (DA).31-33 The embryos were maintained at 35°C. On 1 day post-injection (dpi), zebrafish larvae were treated with the specified drug concentrations. Confocal microscopy was employed from 1 dpi to 4 dpi for image examination.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 6.000. Survival analysis utilized Kaplan-Meier (K-M) curves and the log-rank (Mantel-Cox) test. Student t tests compared two groups, and one-way analysis of variance
(one-way ANOVA) with Tukey’s adjustment was used for multiple group comparisons. A significance level of P<0.05 was applied, and all data were presented as mean ± standard error of the mean.
Generation of an inducible humanized E2A-PBX1 transgenic zebrafish
In order to investigate the effects of humanized E2A-PBX1 on hematopoiesis, we created transgenic zebrafish with hE2A-PBX1 fusion gene expression controlled by the hsp70 promoter. The pT2AL-hsp70-E2A-PBX1-EGFP plasmid with correct sequence (Figure 1A) was obtained after sequencing and microinjected into WT zebrafish to obtain the F0 generation (Online Supplementary Figure S1A). We obtained a diverse range of progeny by crossing the F0 with WT fish and identified a transgenic line (named: Tg(hsp70:E2A-PBX1EGFP)) in the F4 generation, which stably inherited the transgene and exhibited strong EGFP fluorescence. After inducing the transgenic zebrafish with heat shock at 39.5°C, EGFP expression was observed in the whole body of 32 hpf embryos (Figure 1B). In addition, PCR detected a 231 bp human-derived PBX1 target fragment in Tg(hsp70:E2A-PBX1EGFP) transgenic zebrafish (Figure 1C), real time quantitative PCR (RT-qPCR) showed higher hE2A-PBX1 mRNA expression in Tg(hsp70:E2A-PBX1-EGFP) at 3 dpf (Figure 1D). These results demonstrate that we successfully constructed a heritable and stable Tg(hsp70:E2A-PBX1-EGFP) transgenic zebrafish lineage with induced expression of the hE2A-PBX1. Furthermore, qPCR analysis showed consistent expression of endogenous tcf3a and tcf3b (equivalent to human E2A) in both the embryonic tail hematopoietic tissue (CHT) and adult peripheral blood of Tg(hsp70:E2A-PBX1-EGFP) zebrafish (Online Supplementary Figure S1B-D). This suggests that Tg(hsp70:E2A-PBX1-EGFP) zebrafish maintain normal expression of endogenous e2a while overexpressing human E2A-PBX1. Next, to examine the potential effects of the hE2A-PBX1 oncoprotein on blood development in zebrafish, we collected whole blood from zebrafish at 5 dpf after heat shock and performed Giemsa staining. It was found that
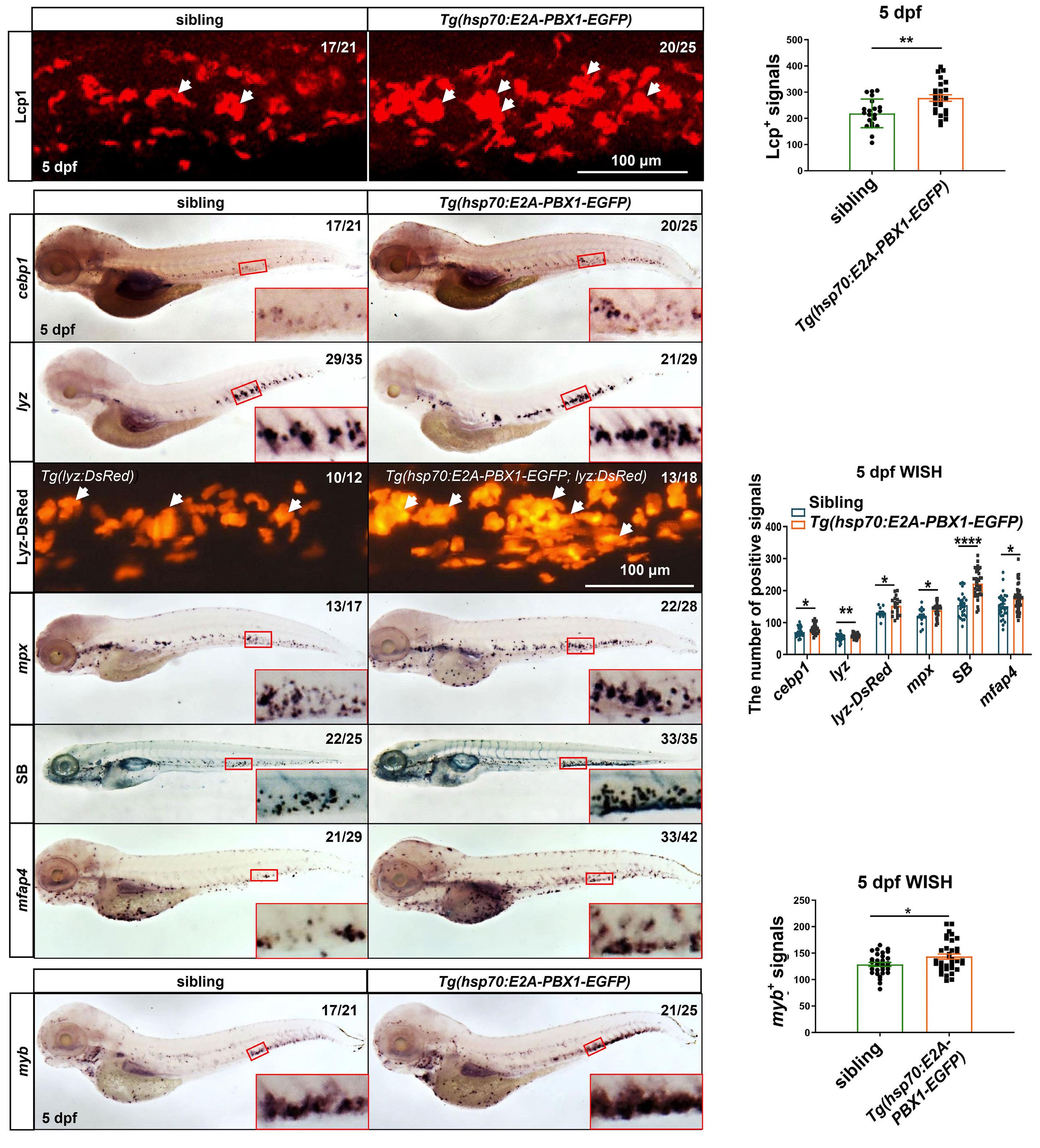
Figure 2. Induction of hE2A-PBX1 expression in zebrafish larvae leads to myeloid cell and hematopoietic stem cell expansion. (A) Immunofluorescence staining of Lcp1+ cells in siblings (left panel) and Tg(hsp70:E2A-PBX1-EGFP) (right panel) at 5 days post-fertilization (dpf). (A’) Statistical analysis of the Lcp1+ signals in panel (A). The black asterisks indicate statistical difference (Student t tests, mean ± standard error of the mean [SEM]; **P<0.01) (B) Whole mount in situ hybridization (WISH) of cebp1, lyz, mpx and mfap4 expressions in Tg(hsp70:E2A-PBX1-EGFP) (right panel) were higher than siblings (left panel) at 5 dpf. The number of lyzDsRed+ cells (caudal hematopoietic tissue [CHT] region and Sudan Black B-positive [SB+] cells in Tg(hsp70:E2A-PBX1-EGFP) (right) was higher than siblings (left) at 5 dpf. The CHT is enlarged in the red box (original magnification ×200). (B’) Statistical analysis of the positive signals (cebp1, lyz, lyz-DsRed, mpx, SB and mfap4) in panel (B). The black asterisks indicate statistical difference
Continued on following page.
(Student t tests, mean ± SEM; *P<0.05, **P<0.01, ****P<0.0001) (C) WISH of myb expressions in Tg(hsp70:E2A-PBX1-EGFP) (right panel) were higher than siblings (left panel) at 5 dpf. (C’) Statistical analysis of the myb+ signals in panel (C). The black asterisks indicate statistical difference (Student t tests, mean ± SEM; *P<0.05). N/N: number of zebrafish larvae showing representative phenotype/total number of zebrafish larvae examined.
the number of myeloid cells was significantly increased (Figure 1E, E’), indicating that transient induction of hE2APBX1 expression may disrupt the hematopoiesis in zebrafish. Additionally, survival analysis of Tg(hsp70:E2A-PBX1-EGFP) fish revealed a significant increase in mortality within 15 days following the induction of hE2A-PBX1 expression, in comparison to their siblings (Figure 1F).
hE2A-PBX1 induces abnormal expansion of myeloid cells in zebrafish larvae
In order to further explore the effect of hE2A-PBX1 expression on the development of different hematopoietic lineages, we detected the marker genes of each hematopoietic lineage in Tg(hsp70:E2A-PBX1-EGFP) transgenic zebrafish after heat shock by whole mount in situ hybridization (WISH) and specific staining. The results showed a significant increase in myeloid cells (l-plastin: a myeloid gene that marks granulocytes and macrophages, also known as lcp1) (Figure 2A, A’), which was consistent with the hemogram changes in zebrafish larvae (Figure 1E). Further assays of myeloid terminally differentiated granulocytes and macrophages revealed a significant increase in the number of immature granulocytes (cebp1), mature granulocytes (lyz, mpx, Sudan Black B [SB]) and macrophages (mfap4) in both 3 dpf and 5 dpf (Figure 2B, B’; Online Supplementary S2A, A’). As myeloid cells originate from HSPC;34 we aimed to investigate whether the increased number of myeloid cells was caused by the tendency of HSPC toward the myeloid lineage or if there was an overall increase in HSPC leading to increased differentiation into various lineages? In order to address this question, we examined the changes of HSPC (myb), lymphocytes (rag1, rag2), and erythrocytes (be1) in Tg(hsp70:E2A-PBX1-EGFP) after heat shock. The results showed that myb+ HSPC increased at 5 dpf (Figure 2C, C’), while the number of rag1+ and rag2+ lymphocytes decreased significantly (Online Supplementary Figure S2B, B’), and be1+ erythrocytes showed no significant change (Online Supplementary Figure S2C, C’). These data suggest that hE2A-PBX1 expression may lead to myeloid expansion in zebrafish by increasing HSPC with myeloid differentiation potential.
In addition, it has been reported that the expression of PBX1 maintains cell proliferation,21 while the E2A protein promotes cell differentiation and has anti-proliferation effect.35 Therefore, we investigated whether hE2A-PBX1 influences the proliferation of myeloid and lymphocytes in Tg(hsp70:E2A-PBX1-EGFP). The proliferation and apoptosis of myeloid/lymphocytes in Tg(hsp70:E2A-PBX1-EGFP) fish were examined after heat shock by co-staining with BrdU/
TUNEL and Lcp1/Rag2, respectively. The results showed a significant increase in the proportion of proliferating cells among Lcp1+ myeloid cells in 5 dpf Tg(hsp70:E2A-PBX1EGFP) compared to the siblings (Figure 3A, 3A’), while the proportion of apoptotic cells showed a significant decrease (Figure 3B, B’). In contrast, Rag2+ lymphocytes showed a significant decrease in proliferation (Online Supplementary Figure S3A, A’), with no significant change in apoptotic cells (Online Supplementary Figure S3B, B’). These data suggest that expression of hE2A-PBX1 leads to abnormal myeloid expansion by increasing the myeloid differentiation potential of HSPC on the one hand and inducing myeloid hyperplasia on the other hand.
In order to investigate the potential relationship between myeloid expansion and early mortality in hE2A-PBX1 zebrafish, we treated heat-shocked hE2A-PBX1 fish with drugs capable of reducing myeloid proliferation, namely cytarabine36-38 and flavopiridol.39,40 The results showed that, compared to the DMSO-treated control group, both cytarabine and flavopiridol treatments significantly reduce the population of SB+ myeloid cells in hE2A-PBX1 fish (Online Supplementary Figure S3C, C’), and increase their survival rate (Online Supplementary Figure S3D). This suggests that myeloid cell proliferation may be one of the contributing factors to the elevated mortality in hE2A-PBX1 zebrafish.
Induction hE2A-PBX1 expression in adult zebrafish leads to an acute myeloid leukemia-like phenotype
In order to analyze whether induced expression of hE2A-PBX1 leads to blood tumors in adult zebrafish, we administered 1-month continuous heat shock to 3-month-old adult fish and collected peripheral blood (PB) cells and kidney marrow (KM) blood cells for Giemsa staining to detect hematological changes. Although 1 month of hE2A-PBX1 induction did not alter the hemogram in PB, it resulted in a significant increase in myeloid cells in KM (Online Supplementary Figure S4A, A’). We then extended the heat shock time to 3 months to determine whether the degree of myeloid expansion induced by hE2A-PBX1 is related to the induction time. The results showed that the longer expression of hE2APBX1 not only further exacerbated the myeloid expansion and lymphoid reduction in KM, but also resulted in mild myeloid hyperplasia in PB (Online Supplementary Figure S4B, B’). Since myeloid leukemia is commonly associated with the patient’s age, we induced 3-month continuous expression of hE2A-PBX1 in 1-year-old zebrafish. We found that the AML-like phenotype was more pronounced in aged zebrafish after 3 months of hE2A-PBX1 induced compared to young zebrafish. In addition to the significant increase
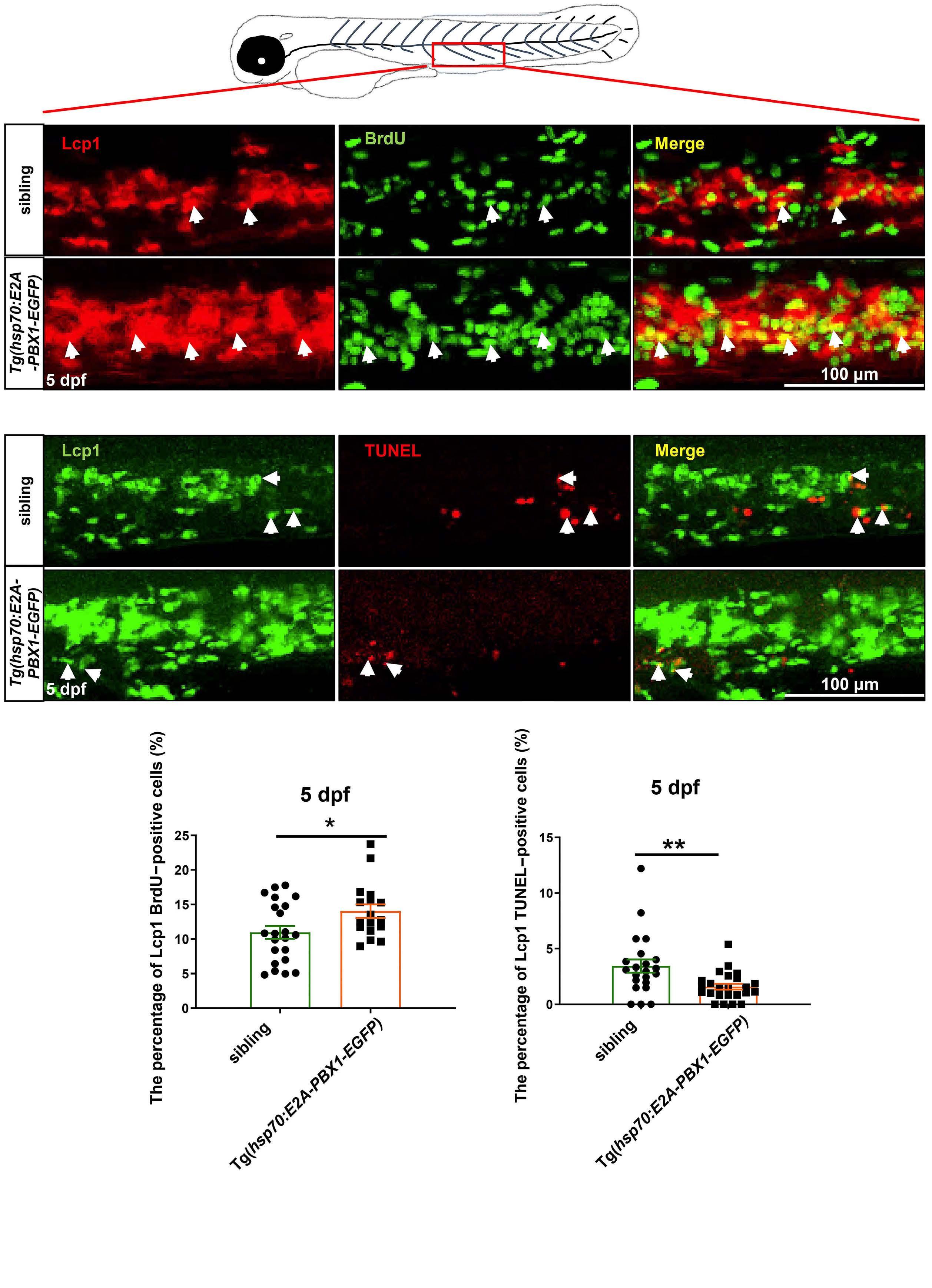
Figure 3. Abnormal myeloid cell expansion in Tg(hsp70:E2A-PBX1-EGFP) fish caused by proliferation and apoptosis perturbation. (A) Immunofluorescence double staining of Lcp1 and bromodeoxyuridine (BrdU) antibodies reveals a significant increase in myeloid cell proliferation in the caudal hematopoietic tissue (CHT) region of 5 days post-fertilization (dpf) Tg(hsp70:E2A-PBX1-EGFP) larvae (N=22) compared with the siblings (N=17). Lcp1/BrdU double-positive cells are indicated by white arrows. (A’) Statistical analysis of percentage of Lcp1+ BrdU+ cells in panel (A). The black asterisks indicate statistical difference (Student t tests, mean ± standard error of the mean; *P<0.05). (B) Co-staining of Lcp1 and transferase dUTP nick end labeling (TUNEL) was used to detect the apoptosis in the CHT region of 5 dpf Tg(hsp70:E2A-PBX1-EGFP) larvae (N=22) compared with the siblings (N=24). Lcp1/TUNEL double-positive cells are indicated by white arrows. (B’) Statistical analysis of percentage of Lcp1+ TUNEL+ cells in panel (B). The black asterisks indicate statistical difference (Student t tests, mean ± standard error of the mean; **P<0.01).
in myeloid cells and a further decrease in lymphocytes in KM, there was also a notable rise in myeloid blasts, and myeloid cells in PB showed a further increase (Figure 4A, A’; Online Supplementary Figure S4C). Furthermore, we ob-
served that the kidneys of Tg(hsp70:E2A-PBX1-EGFP) were significantly enlarged, and the enlargement worsened with the increase of hE2A-PBX1 expression and age (data not shown), especially in 1-year-old zebrafish with continuous
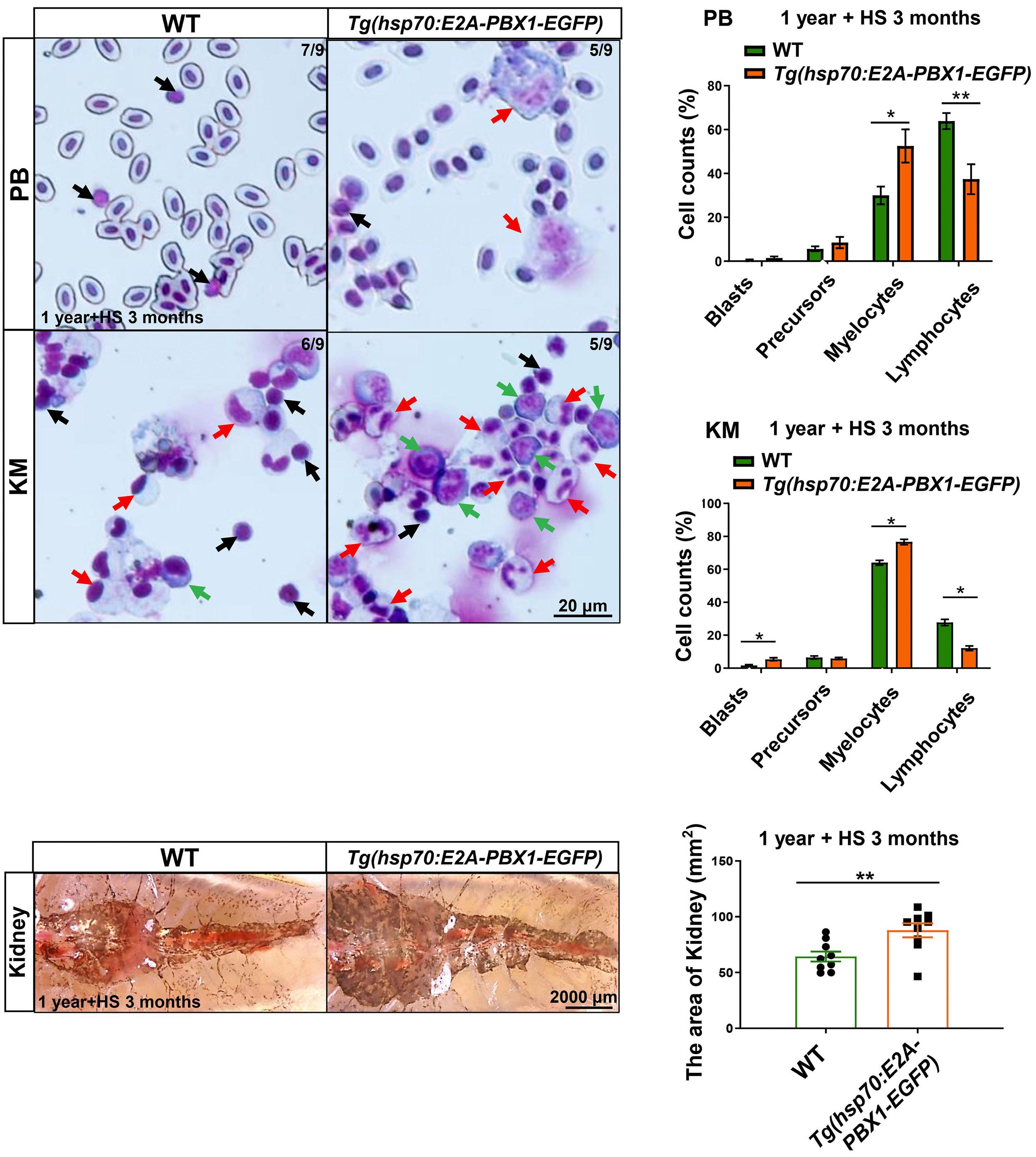
Figure 4. Tg(hsp70:E2A-PBX1-EGFP) adult fish exhibit abnormal myeloid cell expansion which resembles acute myeloid leukemia-like phenotypes. (A) May-Grunwald-Giemsa staining of peripheral blood (PB) cells (upper panels) and kidney marrow (KM) blood cells (lower panels) in 1-year-old wild-type (WT) (left panel) and Tg(hsp70:E2A-PBX1-EGFP) (right panel) adult fish after 3-month heat shock. Red arrows indicate myelocytes, black arrows indicate lymphocytes and green arrows indicate blasts. Original magnification ×400. Blood cell counts of PB and KM were calculated manually based on their morphology. (A’) Statistical analysis of cell counts in panel (A). The black asterisks indicate statistical difference (N=9, one-way ANOVA, mean ± standard error of the mean; *P<0.05, **P<0.01). (B) Kidneys from 1-year-old transgenic fish were enlarged to 1.36-fold in area compared with siblings after 3-month heat shock. (B’) Statistical analysis of the area of kidney in panel (B). The black asterisks indicate statistical difference (N=9, Student t tests, mean ± standard error of the mean; **P<0.01).
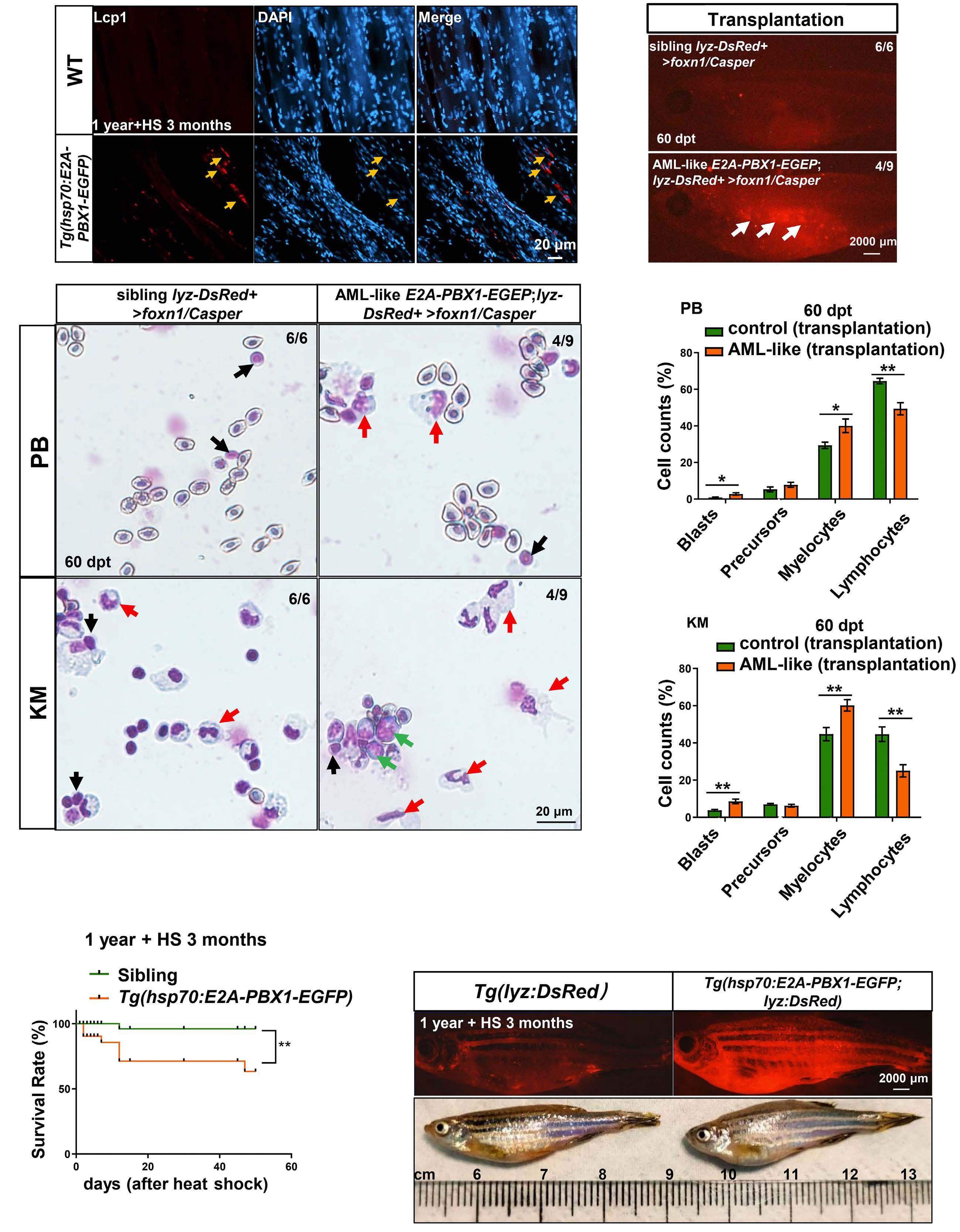
Figure 5. Tg(hsp70:E2A-PBX1-EGFP;lyz:DsRed) transgenic cells with induced acute myeloid leukemia-like disease are transplantable. (A) Immunofluorescent staining of the myeloid-specific marker Lcp1 in frozen sections confirmed myeloid cell invasion of the skeletal musculature in 1-year-old wild-type (WT) (left panel) and Tg(hsp70:E2A-PBX1-EGFP) (right panel) adult fish after 3-month heat shock (yellow arrows show invasion areas of myeloid cells into muscle tissue). (B) Direct observation of engrafted acute myeloid leukemia (AML)-like cells. At 60 days post-transplantation (dpt), AML-like E2A-PBX1 Lyz-DsRed+ cells could be visualized in foxn1/Casper-recipient kidney marrow (KM). The white arrow indicates the engrafted acute myeloid
Continued on following page.
leukemia (AML)-like E2A-PBX1 Lyz-DsRed+ cells. N/N, number of zebrafish larvae showing representative phenotype/total number of zebrafish larvae examined. (C) Wright-Giemsa staining of blood cells in recipient KM after AML-like E2A-PBX1 and WT Lyz-DsRed+ transplantation. The red arrows indicate the AML-like myelocytes, the green arrows indicate the blasts and the black arrows indicate lymphocytes in recipients. (C’) Statistical analysis of cell counts in panel (C). The black asterisks indicate statistical difference (one-way ANOVA, mean ± standard error of the mean; *P<0.05, **P<0.01). (D) Survival curves of Tg(hsp70:E2APBX1-EGFP) and WT fish up to 90 day after heat shock. The black asterisks indicate statistical difference (log-rank [Mantel-Cox] test; **P<0.01). (E) Imaging of dying Tg(hsp70:E2A-PBX1-EGFP;lyz:DsRed) at 15-month-old after 3-month heat shock, reveals severe myeloid cell invasion.
3-month induction of hE2A-PBX1 (Figure 4B, B’). In order to clarify the malignancy of myeloid cells in Tg(hsp70:E2A-PBX1-EGFP), we assessed the invasion of myeloid cells in the muscle after 3 month of heat shock in 3-month-old and 1-year-old zebrafish. The results showed a significant invasion of Lcp1+ myeloid cells in adult fish muscle after induction of hE2A-PBX1 expression compared with the control group (Figure 5A; Online Supplementary Figure S5A). Consistent with this, by using transgenic zebrafish, we observed pronounced systemic invasion of Lyz-DsRed+ myeloid cells in 1-year-old fish after 3 months of heat shock (Online Supplementary Figure S5B). We also detected substantial Lyz-DsRed+ myeloid cell invasion in the kidneys, liver, and spleen upon dissection of fresh tissues from adult zebrafish (Online Supplementary Figure S5C). In order to further confirm the transplantability of hE2A-PBX1-derived myeloid cells, we transplanted whole KM blood cells from Tg(hsp70:E2A-PBX1-EGFP;lyz:DsRed) zebrafish into immunocompromised adult host foxn1/Casper (Online Supplementary Figure S5D). The results revealed that 2 months after transplantation, compared to Tg(lyz:DsRed) control donors, hE2A-PBX1-derived AML-like donor cells successfully repopulated lyz-dsred+ cells in the foxn1/Casper recipients (Figure 5B). Blood smears stained with Giemsa also showed a significant increase in myeloid cells and blasts in both peripheral blood (PB) and KM in recipients following AML-like fish KM transplants (Figure 5C, C’). Furthermore, survival analysis indicates that with the prolonged duration of heat shock, there is a gradual increase in mortality among 1-year-old hE2A-PBX1 zebrafish, accompanied by more severe systemic granulocytic invasion (Figure 5D, E). Taken together, our findings indicate that the induced expression of hE2A-PBX1 leads to myeloid expansion in adult fish and has the potential to progress to AML-like disease. Additionally, the degree of disease is positively correlated with the expression level of hE2A-PBX1 and the age of the animal.
hE2A-PBX1 induces myeloid expansion and activates TNF/IL-17/MAPK signaling pathway through upregulation of runx1 expression
A recent in vitro study has shown that hE2A-PBX1 can activate oncogenes (including RUNX1 ), through direct interaction with RUNX1 via PBX1, leading to the development of pre-B ALL.1 In addition, mutations in RUNX1 have been found to be a frequent cause of AML, T-ALL,
B-ALL, and myelodysplastic syndrome in clinical.41 In order to investigate whether hE2A-PBX1 causes AML-like symptoms are related to its regulation of runx1 , we examined the expression level of runx1 in Tg(hsp70:E2A-PBX1EGFP) after heat shock by WISH and qPCR. The results showed that the expression level of runx1 in transgenic zebrafish at 3 dpf and 5 dpf dpf after heat shock was significantly increased compared to the control group (Figure 6A, A’, B). In order to further investigate the role of Runx1 in hE2A-PBX1-induced myeloid expansion, we crossed Tg(hsp70:E2A-PBX1-EGFP) with the runx1 lossof-function mutant ( runx1 w84x ) to obtain Tg(hsp70:E2APBX1-EGFP) ; runx1 w84x embryos and detected the number of lyz + granulocytes. The results showed a significant reduction in the number of granulocytes in both the sibling and h E2A-PBX1 transgenic zebrafish after runx1 deletion ( Online Supplementary Figure S6A, A’ ). Additionally, to investigate the dose-dependent relationship between the expression level of runx1 and the number of granulocytes, we used the reported runx1 morpholino (MO) to knock down runx1 gradient (injection concentration: 0.7 mM, 0.5 mM, 0.2 mM) and detected the number of lyz + granulocytes by WISH. Our results showed that the reduction in granulocytes was similar to that of runx1 w84x when injected at a concentration of 0.7 mM ( Online Supplementary Figure S6B, B’ ), while restoring granulocyte numbers to sibling levels when injected at 0.2 mM (Figure 6C, C’), with intermediate levels observed at 0.5 mM ( data not shown ). These results suggest that hE2A-PBX1 induces myeloid expansion by upregulating Runx1 expression. Further, we found that deletion of one allele of runx1 in hE2A-PBX1 zebrafish (Tg(hsp70:E2APBX1; runx1 w84x/+ ) increased survival to a level similar to that of siblings, whereas overexpression ( Tg(hsp70:E2APBX1-EGFP; hsp70:myc-runx1) ) did not further exacerbate mortality in hE2A-PBX1 zebrafish ( Online Supplementary Figure S6C ). This is possibly because hE2A-PBX1 transgenic zebrafish already have extremely high levels of runx1 expression, and further elevating runx1 levels may not lead to additional mortality.
In order to further explore the effect of upregulation of runx1 expression on hematopoietic transcription regulation in the h E2A-PBX1 transgenic line, we performed RNA sequencing analysis on blood cells of KM from 3-month-old Tg(hsp70:E2A-PBX1-EGFP) after 3-month continuous heat shock. The results showed that the TNF/
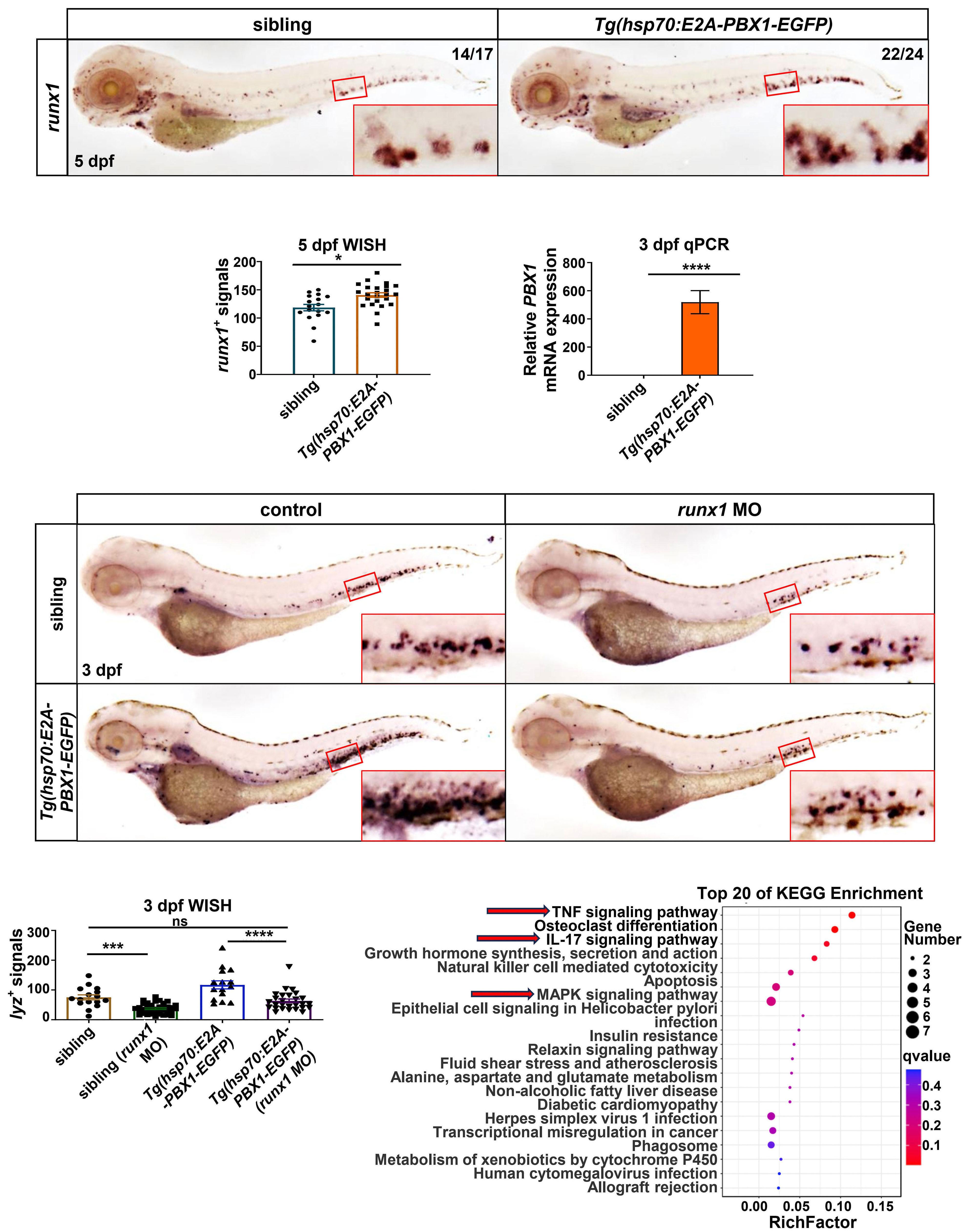
Continued on following page.
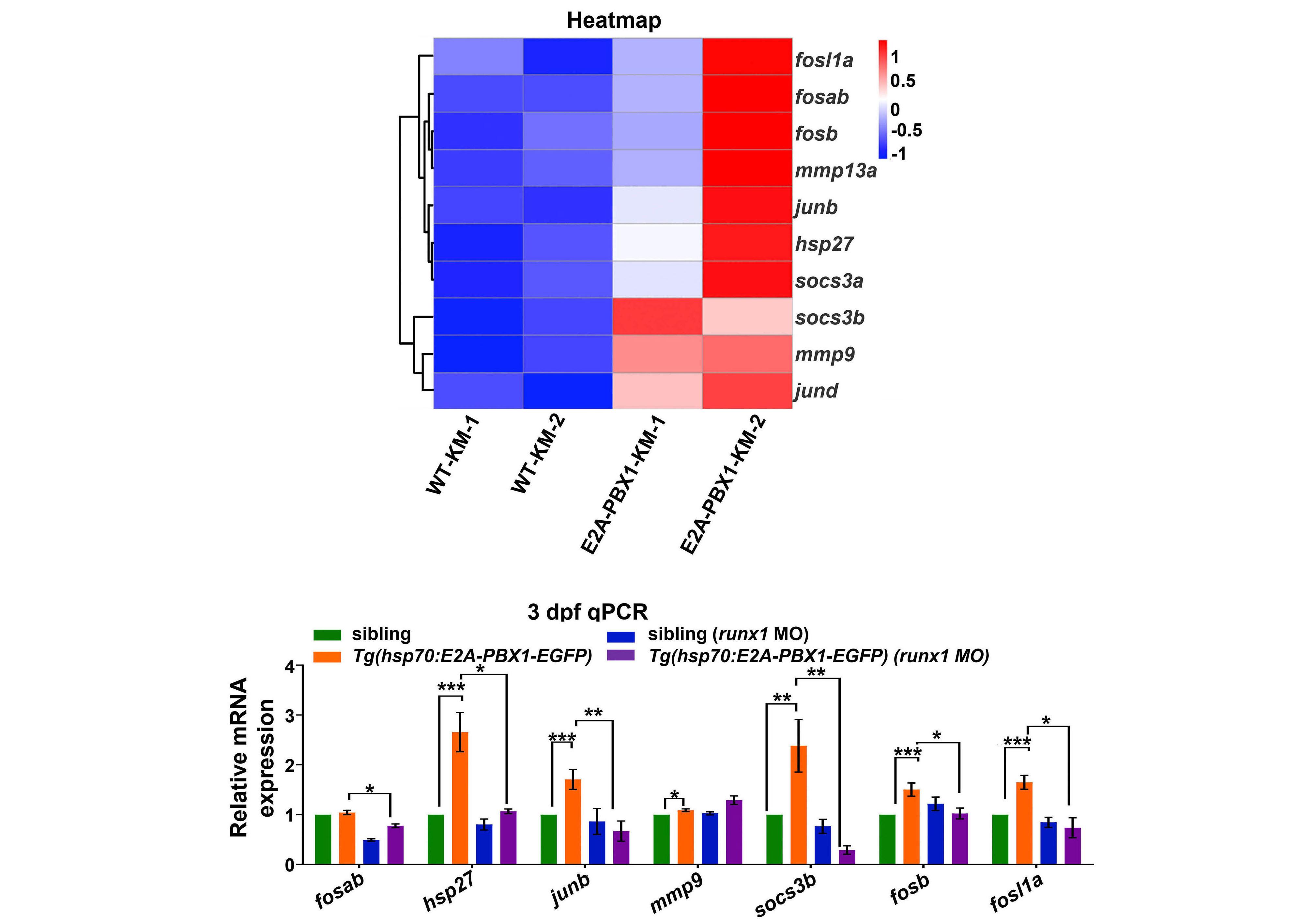
Figure 6. hE2A-PBX1 activate the TNF/IL-17/MAPK signaling pathway through upregulation of runx1 expression. (A) Whole mount in situ hybridization (WISH) of runx1 expressions in Tg(hsp70:E2A-PBX1-EGFP) (right panel) were higher than siblings (left panel) at 5 days post-fertilization (dpf). The caudal hematopoietic tissue (CHT) is enlarged in the red box (original magnification ×200). (A’) Statistical analysis of the runx1+ signals in panel (A). The black asterisks indicate statistical difference (Student t tests, mean ± standard error of the mean [SEM]; *P<0.05). N/N: number of zebrafish larvae showing representative phenotype/total number of zebrafish larvae examined. (B) Real time quantitative polymerase chain reaction (RT-qPCR) analysis showed increased runx1 mRNA expression in Tg(hsp70:E2A-PBX1-EGFP) compared to the sibling controls at 3 dpf. The black asterisks indicate statistical difference (Student t tests, mean ± SEM; ****P<0.0001) (C) Decreased number of lyz+ neutrophils in sibling and Tg(hsp70:E2APBX1-EGFP) larvae after injecting 0.2 mM runx1 morpholino (MO) at 3 dpf. The control groups were treated with 0.2 mM random sequence MO. The CHT is enlarged in the red box (original magnification ×200). (C’) Statistical analysis of the lyz+ signals shown in panel (C). The black asterisks indicate statistical difference (N≥15, one-way ANOVA, mean ± SEM; ***P<0.001, ****P<0.0001). (D) Top 20 enriched KEGG pathways identified in the analysis of differentially expressed genes (DEG) in KM cells from 6-monthold Tg(hsp70:E2A-PBX1-EGFP) compared to control groups after 3-month heat shock. The TNF/IL-17/MAPK signaling pathway is highlighted by the red arrow. (E) Heatmap of DEG involved in the TNF/IL-17/MAPK signaling pathway between WT and hE2A-PBX1 adult fish. Red and blue represent an increase and decrease gene expression levels, respectively; P<0.05. (F) RT-qPCR analysis showing mRNA expression of fosab, hsp27, junb, mmp9, socs3b, fosb and fosl1a in Tg(hsp70:E2A-PBX1-EGFP) compared to siblings after injecting 0.2 mM runx1 MO at 3 dpf. The control groups were treated with 0.2 mM random sequence MO (one-way ANOVA, mean ± SEM; *P<0.05, **P<0.01, ***P<0.001). TNF: tumor necrosis factor; IL-17: interleukin-17; MAPK: mitogen-activated protein kinase.
IL-17/MAPK signaling pathway was enriched in h E2A-PBX1 zebrafish (Figure 6D), with ten genes ( fosl1a, fosab, fosb, mmp13a, junb, hsp27, socs3a, socs3b, mmp9 and jund ) were significantly upregulated in these pathways (Figure 6E). Additionally, we microinjected sibling and hE2A-PBX1 transgenic zebrafish with 0.2 mM runx1 MO, and detected the changes of significantly upregulated genes in the TNF/
IL-17/MAPK signaling pathway after knockdown of runx1. The results showed that knockdown of runx1 could significantly reverse the high expression of hsp27, junb, socs3b, fosb and fosl1a in hE2A-PBX1 transgenic line (Figure 6F). These data suggest that hE2A-PBX1 can induce myeloid expansion and activate the TNF/IL-17/MAPK signaling pathway through upregulation of runx1 expression.
KJ-Pyr-9, OUL35, and CID44216842 effectively alleviate hE2A-PBX1-induced myeloid expansion in zebrafish
In order to identify small molecule compounds that can target hE2A-PBX1, we screened 560 small molecule inhibitors using Tg(hsp70:E2A-PBX1-EGFP) zebrafish. After 3 days of heat shock, embryos of Tg(hsp70:E2A-PBX1-EGFP) were collected and treated with drugs on day 4 (initial screening concentration was 12 μM), and then fixed at 6 dpf to detect the number of SB+ granulocytes (Figure 7A). From the initial screening, we obtained seven small molecule inhibitors that significantly reduced the number of SB+ granulocytes in a concentration gradient of 8 μM, 14 μM, 20 μM and 24 μM (Online Supplementary Table S1). Interestingly, these seven drugs not only include L-arginine hydrochloride targeting inflammatory pathways, A-484954 and GLPG1837 targeting autophagy pathways, LRRK2-IN-1 targeting apoptosis pathways, but also include CID4421684, OUL35 and KJPyr-9 targeting TNF/IL-17/MAPK, which is consistent with our previous finding that hE2A-PBX1 can activate the TNF/ IL-17/MAPK signaling pathway in zebrafish (Figure 6D). In order to compare the effects of these three drugs with the existing chemotherapeutic drug Ara-C (cytarabine), we treated hE2A-PBX1 zebrafish with 6.17 mM Ara-C, 8 μM KJ-Pyr-9, 20 μM CID44216842 and 24 μM of OUL35, respectively. The results showed that all three inhibitors reduced the number of SB+ granulocytes to the same ex-
tent as Ara-C (Figure 7B, B’), but OUL35 and CID44216842 exhibited a lower embryo deformity rate (Online Supplementary Figure S7A). Consistently, intraperitoneal injection of adult fish with cytarabine (8,000 mg/kg), CID44216842 (125 mg/kg), KJ-Pyr-9 (90 mg/kg), and OUL35 (150 mg/kg) for 5 consecutive days significantly reduced the proportion of myeloid cells in the KM (Online Supplementary Figure S7B, B’). Additionally, KJ-Pyr-9, OUL35, and CID44216842 also lowered the blast percentage in the KM. These findings indicate that KJ-Pyr-9, OUL35, and CID44216842 can alleviate the myeloid expansion in hE2A-PBX1 fish. Further, by long-term drug treatment (administration for 7 consecutive days) of hE2A-PBX1 zebrafish at 5 dpf, we found that cytarabine, CID44216842 and OUL35 could significantly improve the survival rate of hE2A-PBX1 zebrafish (Online Supplementary Figure S7C). KJ-Pyr-9 did not improve the death of hE2A-PBX1 zebrafish, which may be due to other side effects caused by long-term treatment of KJ-Pyr-9. It has been reported that TNF42-44 and IL-1745-47 can activate the MAPK pathway. In order to investigate whether TNF/IL-17 can similarly activate the MAPK signaling in Tg(hsp70:E2APBX1-EGFP), we treated 4 dpf hE2A-PBX1 embryos with TNF-α inhibitors pomalidomide and lenalidomide, as well as IL-17 inhibitor Y-320. After 48 hours, we examined the activation of the MAPK pathway, expression of downstream target genes, and changes in SB+ myeloid cell numbers.
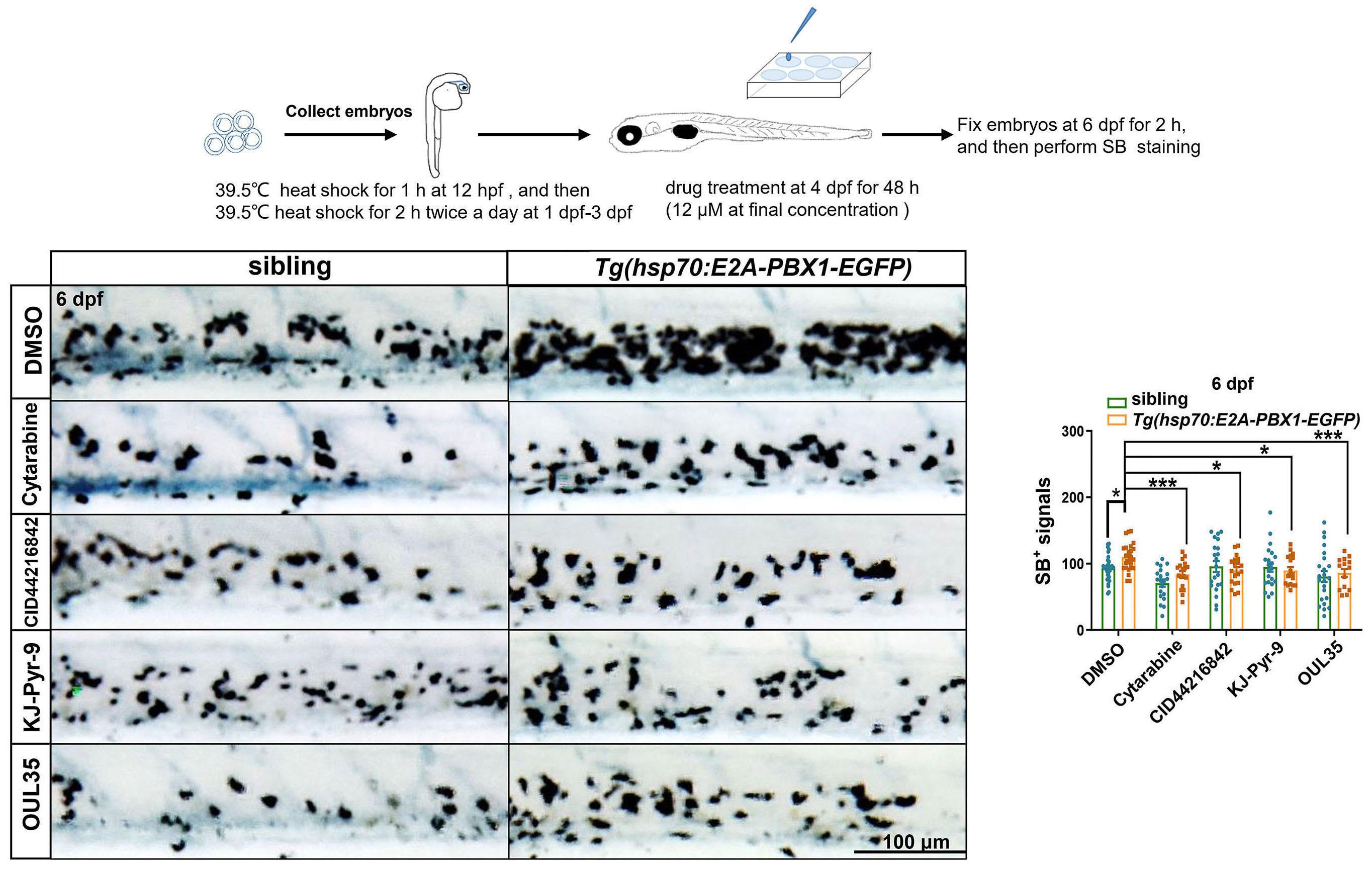
Continued on following page.
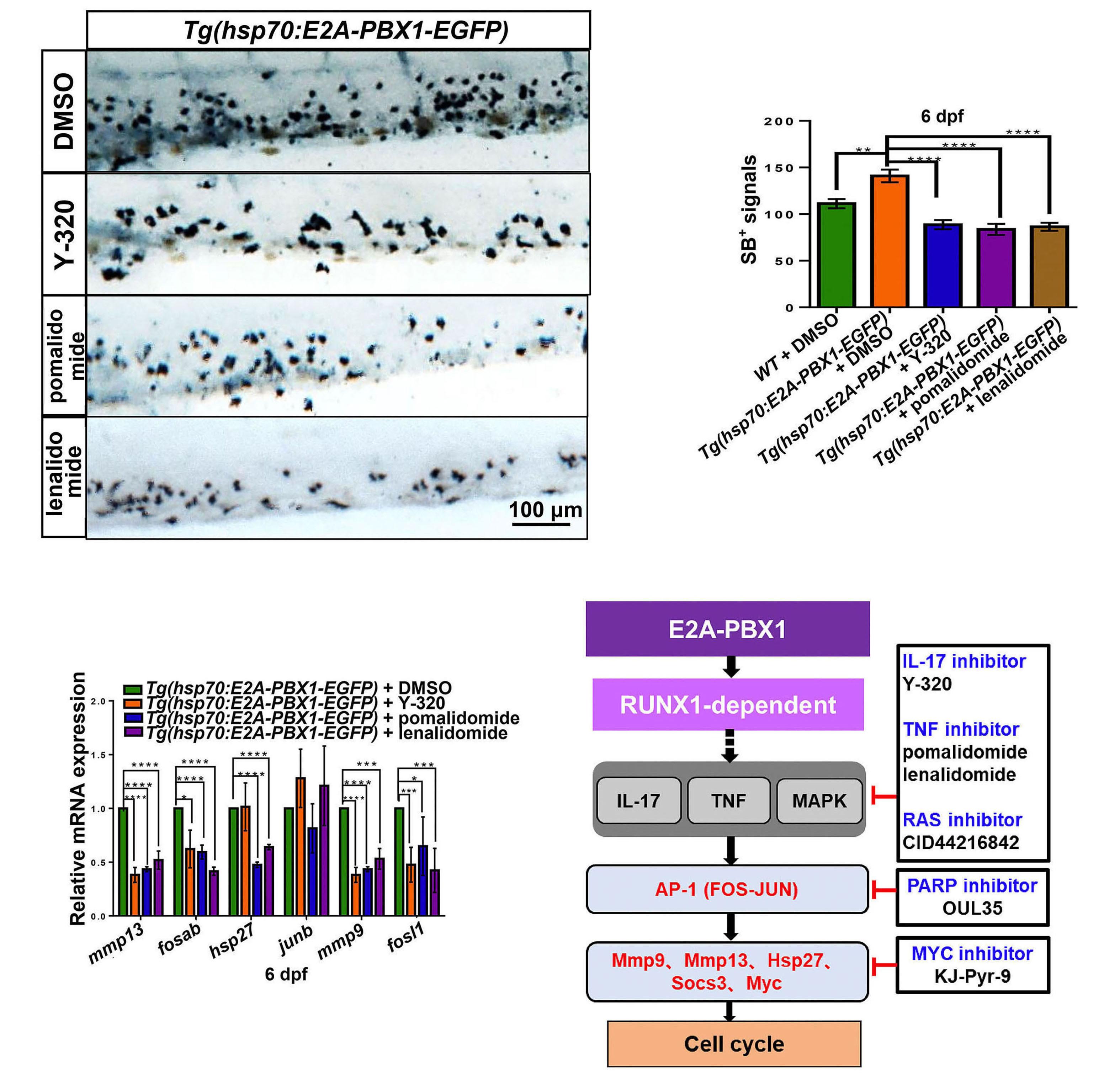
Figure 7. KJ-Pyr-9, OUL35, and CID44216842 effectively alleviate hE2A-PBX1-induced myeloid expansion in zebrafish larvae. (A) Flowchart illustrating the high-throughput drug screening process conducted in this study. Briefly, embryos were collected and subjected to a 1-hour heat shock at 12 hours post-fertilization (hpf), followed by twice daily 2-hour heat shocks from 1 to 3 days post-fertilization (dpf). Subsequently, zebrafish larvae were soaked in a solution containing small molecule compounds (initial screening concentration was 12 μM), the number of neutrophils was quantified using Sudan Black B (SB) staining at 6 dpf. (B) SB staining of sibling (left panel) and Tg(hsp70:E2A-PBX1-EGFP) (right panel) larvae after treatment with dimethyl sulfoxide (DMSO), 6.17 mM Ara-C (cytarabine), 8 μM KJ-Pyr-9, 20 μM CID44216842 and 24 μM of OUL35 at 6 dpf. (B’) Statistical analysis of the SB+ signals shown in panel (B). The black asterisks indicate statistical difference (N≥18, one-way ANOVA, mean ± standard error of the mean [SEM]; *P<0.05, ***P<0.001). (C) SB staining of hE2A-PBX1 zebrafish larvae after inhibitors treatment (DMSO, 500 µM pomalidomide, 500 μM lenalidomide, and 0.75 μM Y-320 inhibitor, respectively). The caudal hematopoietic tissue (CHT) is enlarged in the red box (original magnification ×200). (C’) Statistical analysis of the SB+ signals in panel (C). The black asterisks indicate statistical difference (N≥18, one-way ANOVA, mean ± SEM; **P<0.01, ****P<0.0001). (D) Real time quantitative polymerase chain reaction (RT-qPCR) analysis showing mRNA expression of mmp13, fosab, hsp27, junb, mmp9, and fosl1a in Tg(hsp70:E2A-PBX1-EGFP) compared to siblings after inhibitor treatment (one-way ANOVA, mean ± SEM; *P<0.05, ***P<0.001, ****P<0.0001). (E) Schematic representation of inhibitors targeting TNF/IL-17/MAPK signaling pathway. TNF: tumor necrosis factor; IL-17: interleukin-17; MAPK: mitogen-activated protein kinase. The AP-1 (activator protein 1) transcription factor is a dimeric complex that contains members of the JUN, FOS, ATF and MAF protein families.
The results showed that TNF and IL-17 inhibitors did not activate phosphorylation levels of Erk, Akt, p-38, and Jnk in the MAPK pathway (Online Supplementary Figure S7D, D’). However, they did decrease the expression of the target gene
mmp13, fosab, mmp9 and fosl1a, and finally alleviate myeloid cell proliferation in hE2A-PBX1 zebrafish (Figure 7C, D). These data suggest that TNF and IL-17 regulate myeloid development by upregulating the expression of target genes, not through
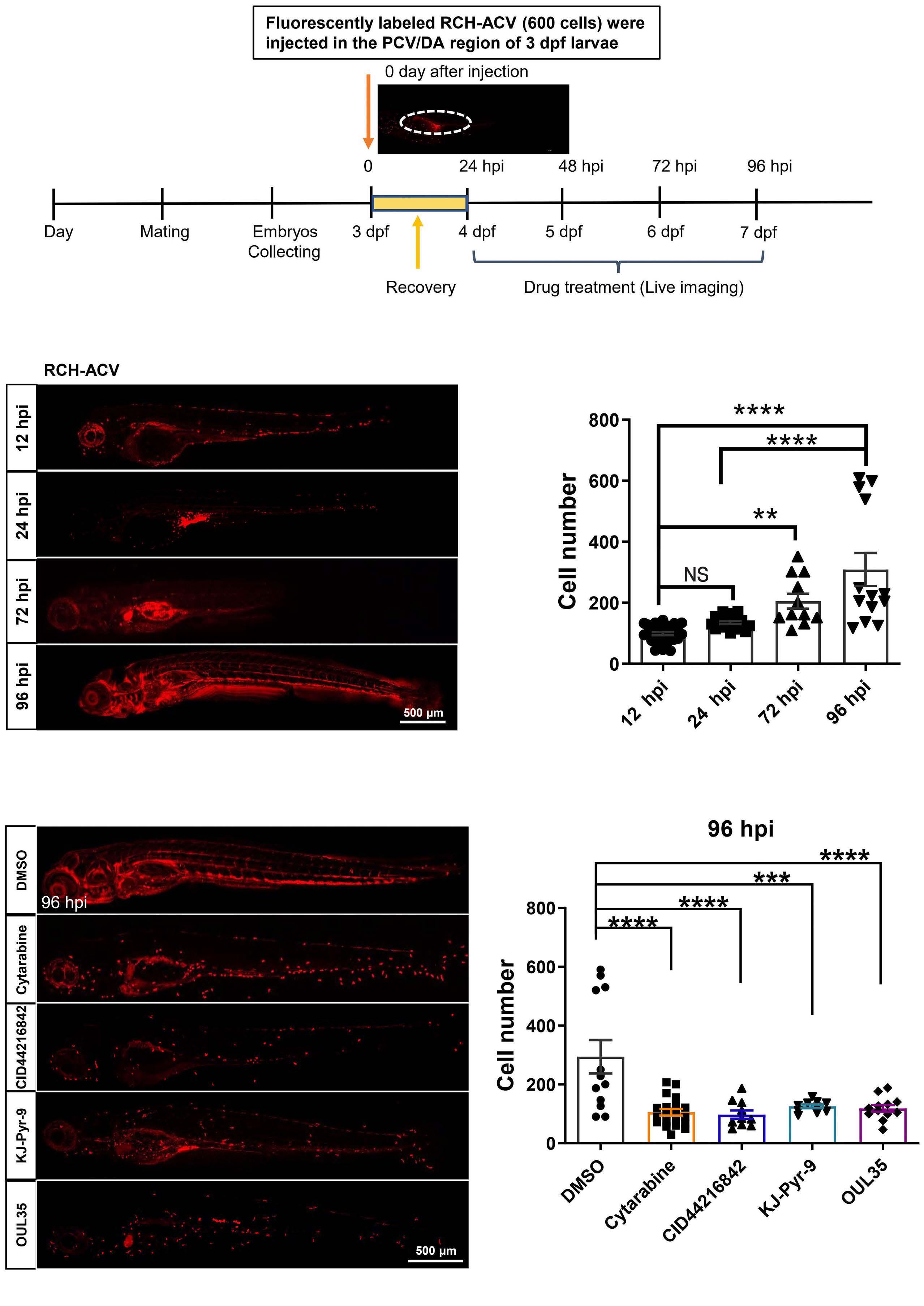
Continued on following page.
Figure 8. KJ-Pyr-9, OUL35 and CID44216842 effectively inhibit the oncogenicity of human E2A-PBX1-associated pre-B-lineage acute lymphoblastic leukemia. (A) Schematic representation of xenograft development using human B-lineage acute lymphoblastic leukemia (B-ALL) E2A-PBX1(+) RCH-ACV cells in zebrafish larvae. Briefly, embryos were injected with 600 fluorescently labeled RCH-ACV cells into the posterior cardinal vein (PCV) or dorsal aorta (DA) at 3 days post-fertilization (dpf), followed by treatment with small molecules at 24 hours post-injection (hpi). The pre-B ALL cells injected in runx1w84x embryos were monitored by imaging at 12 hpi, 24 hpi, 72 hpi and 96 hpi. (B) RCH-ACV cells were calculated after xenografting into runx1w84x zebrafish at 12 hpi, 24 hpi, 72 hpi and 96 hpi. (B’) Statistical analysis of the RCH-ACV cell number after injecting in panel (B). The black asterisks indicate statistical difference (N≥11, one-way ANOVA, mean ± standard error of the mean; **P<0.01, ****P<0.0001). (C) RCH-ACV cells were calculated after xenografting into runx1w84x zebrafish, followed by treatment with dimethyl sulfoxide (DMSO), 6.17 mM Ara-C (cytarabine), 8 μM KJ-Pyr-9, 20 μM CID44216842 and 24 μM OUL35 at 96 hpi. (C’) Statistical analysis of the RCH-ACV cell number with drug treatment after injecting in panel (C). The black asterisks indicate statistical difference (N≥10, one-way ANOVA, mean ± standard error of the mean; ***P<0.001, ****P<0.0001).
the activation of the MAPK signaling pathway (Figure 7E).
KJ-Pyr-9, OUL35 and CID44216842 effectively inhibit the oncogenicity of human E2A-PBX1-associated pre-B lineage acute lymphoblastic leukemia
Considering that hE2A-PBX1 can promote pre-B ALL by upregulating RUNX1 expression, we further verified whether KJ-Pyr-9, OUL35, and CID44216842 could also alleviate human pre-B-ALL with E2A-PBX1. We treated RCH-ACV cells with cytarabine, KJ-Pyr-9, OUL35, and CID44216842 for 60 hours, followed by assessing cell proliferation using the CCK8 assay and apoptosis with the TUNEL assay. The results demonstrated that cytarabine, as well as these three small molecule compounds, significantly inhibited the proliferation of RCH-ACV cells and increased apoptosis in these cells (Online Supplementary Figure S8A, B). This suggests that the E2A-PBX1+ B-ALL cell line (RCH-ACV) is sensitive to these three small molecule inhibitors. We stained RCH-ACV with the lipophilic dye Dil and microinjected it at a concentration of 600 cells per embryo into posterior cardinal vein (PCV) or DA sites in immunodeficient runx1w84x fish at 3 dpf (Figure 8A). Drug treatment was performed on the day after injection (24 hpi), and the number of leukemia cells was counted at 12-96 hpi. Our findings revealed that DMSO-treated zebrafish showed rapid expansion of RCH-ACV cells at 24-96 hpi (Figure 8B), whereas treatment with OUL35, KJ-Pyr-9, and CID44216842 inhibited the high-speed expansion of RCH-ACV cells (Figure 8C). These data suggest that KJPyr-9, OUL35 and CID44216842 can effectively inhibit the oncogenicity of human E2A-PBX1-associated pre-B ALL.
In summary, we developed a hE2A-PBX1 transgenic zebrafish line that can induce myeloid expansion and eventual progression to AML-like hemogram. We found that hE2A-PBX1 induces upregulation of the runx1-activated TNF/IL-17/MAPK signaling pathway. Through high-throughput drug screening and validation, we identified compounds KJ-Pyr-9, OUL35 and CID44216842 that can alleviate hE2A-PBX1-induced AML-like disease in zebrafish as well as the human pre-B ALL. Our study highlights the potential of these compounds as targeted
drugs for the future clinical treatment of E2A-PBX1 leukemia. Clinically, hE2A-PBX1 primarily induces ALL, but in our hE2A-PBX1 zebrafish model, it causes myeloid leukemia. There are two possible explanations for this: i) impaired E2A function: E2A plays a critical role in regulating early B-cell development and can enhance B-cell-specific gene transcription even in non-lymphoid cells.1,48 The fusion gene hE2A-PBX1 disrupts the normal structure and function of WT E2A, resulting in E2A dosage and functional defects. This disruption leads to aberrant B-cell development and differentiation. Notably, in our hE2A-PBX1 transgenic fish, the endogenous e2a structure remains intact with normal expression levels, indicating a distinction from human cases. ii) Expression distribution of hE2A-PBX1 in different blood lineages: in patients, the expression of hE2A-PBX1 is controlled by the endogenous E2A promoter, implying that lymphocytes may exhibit higher expression levels of hE2A-PBX1 compared to other blood cells. The lymphoid lineage, with relatively higher expression of hE2A-PBX1, might be more prone to leukemic transformation. In our transgenic fish, hE2A-PBX1 was expressed at the initial stage of hematopoiesis (HSPC cells) through heat shock induction. This led to elevated expression of Runx1 in HSPC, ultimately allowing HSPC to acquire a propensity for myeloid differentiation and the potential for myeloid expansion. It is important to note that the induction of both lymphoid and myeloid leukemogenesis by hE2A-PBX1 is strongly correlated with heightened Runx1 activity, as supported by our findings and previous studies.1,49 Inhibiting the abnormal activation of RUNX1 and its downstream pathways shows promise as a potential shared therapeutic strategy for treating E2A-PBX1 multitype leukemia. Based on previous structural and molecular functional studies, there are three main explanations for the potential mechanisms underlying hE2A-PBX1-induced pre-B leukemia:21 i) The fusion gene hE2A-PBX1 impairs the function of WT E2A; ii) hE2A-PBX1 oncoprotein may deregulate the expression of critical genes normally controlled by PBX/ HOX/MEINOX complexes; and iii) hE2A-PBX1 alters the function of transcriptional co-activators, such as histone acetyltransferases p300 and CREB-binding protein (CBP). These three factors may coordinate and regulate the occurrence of hE2A-PBX1 pre-B leukemia. On the one hand,
the insufficient dose of WT E2A affects the regulation of lymphangiogenesis development.50,51 On the other hand, hE2A-PBX1 induces oncogenic properties, such as cell proliferation, by altering the expression of downstream genes or transcriptional co-activators of PBX1.52 In our study, the induction of hE2A-PBX1 expression, along with the regular expression of the endogenous E2A gene, leads to the development of myeloid leukemia. This suggests that the acquired function of hE2A-PBX1, rather than a deficiency of WT E2A, is the primary cause of its carcinogenic potential. However, disruption of the E2A gene may contribute to alterations in lymphoid development. Consistent with this, an unbiased analysis of E2A-PBX1 cistrome in pre-B ALL cells reveals that E2A-PBX1 can interact with RUNX1, resulting in transcriptome alterations, including activation of RUNX1 itself.1 Moreover, our study revealed aberrant activation of Runx1 in hE2A-PBX1 transgenic fish. Notably, inhibiting Runx1 and its downstream pathway demonstrated significant alleviation in the expansion of leukemic cells in both zebrafish and human cell lines. These findings emphasize that hE2A-PBX1 exerts a significant impact on tumorigenesis by regulating the genome through direct binding with RUNX1. Whether this effect is synergistically facilitated by the inappropriate activation of target genes through the fusion of PBX1 DNA binding motifs remains to be further investigated. Currently, there are no clinically targeted drugs for the treatment of E2A-PBX1 leukemia, and there are relatively few studies on the treatment of E2A-PBX1-associated AML-like disease and MPAL. In this study, we show that the small molecule compounds KJ-Pyr-9, OUL35 and CID44216842 obtained by hE2A-PBX1 zebrafish leukemia model have great potential. OUL35, recognized as a selective PARP inhibitor that targets PKCδ, a PARP substrate that activates MAPK,53,54 has demonstrated the ability to protect HeLa cells from ARTD10-induced cell death.55 KJ-Pyr-9 is an inhibitor of MYC,56 a transcriptional regulatory factor encoded by the common proto-oncogene MYC, which mainly exerts its regulatory effects through binding with MAX protein.57 Notably, the expression of max was significantly increased in hE2A-PBX1 zebrafish (data not shown). Literature reports indicate that KJ-Pyr-9 can target MYC to inhibit pleomorphic glioblastoma multiforme (GBM) and significantly reduce the viability of squamous cell carcinoma (SCC) and adenocarcinoma (AC)-derived cells.58 CID44216842 acts as an inhibitor of RAS, a key regulator of signaling pathways controlling normal cell growth and malignant transformation. Additionally, CID44216842 has been demonstrated to inhibit stimulus-induced Cdc42 activation
References
1. Pi W-C, Wang J, Shimada M, et al. E2A-PBX1 functions as a coactivator for RUNX1 in acute lymphoblastic leukemia. Blood. 2020;136(1):11-23.
in Swiss 3T3 fibroblasts.59 While in vitro cellular experiments confirm their efficacy in tumor cell therapy, it is essential to conduct experiments using animal tumor models before progressing to clinical trials. In our study, these three small molecule compounds exhibit the ability to alleviate hE2APBX1-induced AML-like disease in zebrafish as well as pre-B ALL associated with hE2A-PBX1, by targeting the TNF/IL-17/ MAPK pathway downstream of Runx1. However, it is not clear whether these small molecule compounds are applicable to other blood diseases with abnormal RUNX1 activity, and more preclinical animal studies are needed to confirm this. Overall, our findings shed new light on the treatment of E2A-PBX1 and other Runx1-related disorders. Although there is much work to be done, we anticipate that our research will contribute to the development of innovative therapeutic strategies for these diseases.
No conflicts of interest to disclose.
WL and WZ conceived and supervised the study and edited the article. HL and QL performed experiments and wrote, reviewed, and edited the article. JH, WD, KW, SL, and HW performed the experiments. ZH reviewed and edited the article.
The authors would like to thank Dr. Huafeng Xie for providing RCH-ACV cells. The authors would like to thank Dr. Kailun Li, Dr. Xiao Fang, Dr. Panpan Zhu, Dr Shuang Zhao and Dr. Zhibin Huang for their helpful suggestions.
Funding
This work was supported by the National Natural Science Foundation of China (82070163), Guangdong Basic and Applied Basic Research Foundation (2020A1515011218) and Special Fund of Science and Technology Innovation Cultivation for College Students in Guangdong Province (pdjh2020b0048).
Data-sharing statement
The data supporting the findings of this study are available within the article and its Online Supplementary Appendix. Data produced in this manuscript are available on Gene Expression Omnibus (GEO) with accession number GSE234222 (Go to https://www.ncbi.nlm.nih.gov/geo/query/ acc.cgi?acc=GSE234222, enter the token qjebmoqwprsjtkn into the box).
2. Duque-Afonso J, Lin CH, Han K, et al. E2A-PBX1 remodels oncogenic signaling networks in B-cell precursor acute lymphoid leukemia. Cancer Res. 2016;76(23):6937-6949.
3. Amor Vigil AM, Fernández Martínez L, Díaz Alonso CA, et al. Transcripto E2A-PBX1 en paciente pediátrico con leucemia aguda híbrida linfoide b/mieloide. Rev Cubana Hematol Inmunol Hemoter. 2017;33(3):95-101.
4 Troussard X, Rimokh R, Valensi F, et al. Heterogeneity of t(1;19) (q23;p13) acute leukaemias. French Haematological Cytology Group. Br J Haematol. 1995;89(3):516-526.
5. Hong Y, Zhao X, Qin Y, et al. The prognostic role of E2A-PBX1 expression detected by real-time quantitative reverse transcriptase polymerase chain reaction (RQ-PCR) in B cell acute lymphoblastic leukemia after allogeneic hematopoietic stem cell transplantation. Ann Hematol. 2018;97(9):1547-1554.
6. Irving JAE, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016;128(7):911-922.
7 Hunger SP. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood. 1996;87(4):1211-1224.
8. Zhao W, Li Z-g, Gao C. Detection of E2A/PBX1 fusion gene and its clinical significance in childhood acute lymphoblastic leukemia. Zhonghua Er Ke Za Zhi. 2008;46(7):550-551.
9 Jia M, Hu B-F, Xu X-J, Zhang J-Y, Li S-S, Tang Y-M. Clinical features and prognostic impact of TCF3-PBX1 in childhood acute lymphoblastic leukemia: a single-center retrospective study of 837 patients from China. Curr Probl Cancer. 2021;45(6):100758.
10. Diakos C, Xiao Y, Zheng S, Kager L, Dworzak M, Wiemels JL. Direct and indirect targets of the E2A-PBX1 leukemia-specific fusion protein. PLoS One. 2014;9(2):e87602.
11. Murre C. Regulation and function of the E2A proteins in B cell development. In: Gupta S, Alt F, Cooper M, Melchers F, Rajewsky K, eds. Mechanisms of lymphocyte activation and immune regulation Xi. B Cell Biology. Boston, MA: Springer; 2007. p. 1-7.
12. Semerad CL, Mercer EM, Inlay MA, Weissman IL, Murre C. E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc Natl Acad Sci U S A. 2009;106(6):1930-1935.
13. Malouf C, Ottersbach K. Molecular processes involved in B cell acute lymphoblastic leukaemia. Cell Mol Life Sci. 2018;75(3):417-446.
14 Aspland SE, Bendall HH, Murre C. The role of E2A-PBX1 in leukemogenesis. Oncogene. 2001;20(40):5708-5717.
15. Kamps MP, Baltimore D. E2A-Pbx1, the t(1;19) translocation protein of human pre-B-cell acute lymphocytic leukemia, causes acute myeloid leukemia in mice. Mol Cell Biol. 1993;13(1):351-357.
16. Kamps MP, Wright DD. Oncoprotein E2A-Pbx1 immortalizes a myeloid progenitor in primary marrow cultures without abrogating its factor-dependence. Oncogene. 1994;9(11):3159-3166.
17 Dedera DA, Waller EK, LeBrun DP, et al. Chimeric homeobox gene E2A-PBX1 induces proliferation, apoptosis, and malignant lymphomas in transgenic mice. Cell. 1993;74(5):833-843.
18. Bijl J, Sauvageau M, Thompson A, Sauvageau G. High incidence of proviral integrations in the Hoxa locus in a new model of E2a-PBX1-induced B-cell leukemia. Genes Dev. 2005;19(2):224-233.
19 Sera Y, Yamasaki N, Oda H, et al. Identification of cooperative genes for E2A-PBX1 to develop acute lymphoblastic leukemia. Cancer Sci. 2016;107(7):890-898.
20 Sykes DB, Kamps MP. E2a/Pbx1 induces the rapid proliferation of stem cell factor-dependent murine pro-t cells that cause acute T-lymphoid or myeloid leukemias in mice. Mol Cell Biol.
2004;24(3):1256-1269.
21. Hyndman BD, LeBrun DP. E2A-PBX1. In: Schwab, M. (eds) Encyclopedia of cancer. Springer, Berlin, Heidelberg; 2009. p. 935-936.
22. van de Ven C, Boeree A, Stalpers F, Zwaan CM, Den Boer ML. Ibrutinib is not an effective drug in primografts of TCF3-PBX1. Transl Oncol. 2020;13(10):100817.
23. Eldfors S, Kuusanmaki H, Kontro M, et al. Idelalisib sensitivity and mechanisms of disease progression in relapsed TCF3-PBX1 acute lymphoblastic leukemia. Leukemia. 2017;31(1):51-57.
24. Rasighaemi P, Basheer F, Liongue C, Ward AC. Zebrafish as a model for leukemia and other hematopoietic disorders. J Hematol Oncol. 2015;8(1):1-10.
25. Ali S, Champagne DL, Spaink HP, Richardson MK. Zebrafish embryos and larvae: a new generation of disease models and drug screens. Birth Defects Res C Embryo Today. 2011;93(2):115-133.
26. Bentley VL, Veinotte CJ, Corkery DP, et al. Focused chemical genomics using zebrafish xenotransplantation as a pre-clinical therapeutic platform for T-cell acute lymphoblastic leukemia. Haematologica. 2015;100(1):70-76.
27. Haney MG, Moore LH, Blackburn JS. Drug screening of primary patient derived tumor xenografts in zebrafish. J Vis Exp. 2020(158):10.
28. Chauvigné F, Yilmaz O, Ferré A, et al. The vertebrate Aqp14 water channel is a neuropeptide-regulated polytransporter. Commun Biol. 2019;2(1):462.
29 Jin H, Sood R, Xu J, et al. Definitive hematopoietic stem/ progenitor cells manifest distinct differentiation output in the zebrafish VDA and PBI. Development. 2009;136(4):647-654.
30 Jin H, Li L, Xu J, et al. Runx1 regulates embryonic myeloid fate choice in zebrafish through a negative feedback loop inhibiting Pu.1 expression. Blood. 2012;119(22):5239-5249.
31. Pruvot B, Jacquel A, Droin N, et al. Leukemic cell xenograft in zebrafish embryo for investigating drug efficacy. Haematologica. 2011;96(4):612-616.
32. Zeng Y, Zhang X, Lin D, et al. A lysosome-targeted dextrandoxorubicin nanodrug overcomes doxorubicin-induced chemoresistance of myeloid leukemia. J Hematol Oncol. 2021;14(1):1-6.
33. Khan N, Mahajan NK, Sinha P, Jayandharan GR. An efficient method to generate xenograft tumor models of acute myeloid leukemia and hepatocellular carcinoma in adult zebrafish. Blood Cells Mol Dis. 2019;75:48-55.
34. Xu J, Du L, Wen Z. Myelopoiesis during zebrafish early development. J Genet Genomics. 2012;39(9):435-442.
35. LeBrun DP. E2A basic helix-loop-helix transcription factors in human leukemia. Front Biosci. 2003:8:s206-s222.
36. Baker WJ, Royer GL, Jr., Weiss RB. Cytarabine and neurologic toxicity. J Clin Oncol. 1991;9(4):679-693.
37. Ota K. [Cytarabine]. Gan To Kagaku Ryoho. 1987;14(10):2983-2987. Japanese.
38. Magina KN, Pregartner G, Zebisch A, et al. Cytarabine dose in the consolidation treatment of AML: a systematic review and meta-analysis. Blood. 2017;130(7):946-948.
39 Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res. 2015;39(12):1312-1318.
40 Chen KTJ, Militao GGC, Anantha M, Witzigmann D, Leung AWY, Bally MB. Development and characterization of a novel flavopiridol formulation for treatment of acute myeloid leukemia. J Control Release. 2021;333:246-257.
41. Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017;129(15):2070-2082.
42. Yan Z, Dai J, Wang J, et al. RNF167-mediated ubiquitination of Tollip inhibits TNF-α-triggered NF-κB and MAPK activation. FASEB J. 2023;37(8):e23089.
43. Niu L, Fang Y, Yao X, et al. TNFα activates MAPK and Jak-Stat pathways to promote mouse Muller cell proliferation. Exp Eye Res. 2021;202:108353.
44 Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26(3):237-245.
45. Aslani MR, Sharghi A, Boskabady MH, et al. Altered gene expression levels of IL-17/TRAF6/MAPK/USP25 axis and proinflammatory cytokine levels in lung tissue of obese ovalbuminsensitized rats. Life Sci. 2022;296:120425.
46. Roussel L, Houle F, Chan C, et al. IL-17 Promotes p38 MAPKdependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol. 2010;184(8):4531-4537.
47. Hata K, Andoh A, Shimada M, et al. IL-17 stimulates inflammatory responses via NF-κB and MAP kinase pathways in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol. 2002;282(6):G1035-G1044.
48. Sloan SR, Shen CP, McCarrick-Walmsley R, Kadesch T. Phosphorylation of E47 as a potential determinant of B-cellspecific activity. Mol Cell Biol. 1996;16(12):6900-6908.
49. Licht JD. Oncogenesis by E2A-PBX1 in ALL: RUNX and more. Blood. 2020;136(1):3-4.
50 Huang A, Zhao H, Quan Y, Jin R, Feng B, Zheng M. E2A Predicts prognosis of colorectal cancer patients and regulates cancer cell growth by targeting miR-320a. Plos One. 2014;9(1):e85201.
51. Xu W, Carr T, Ramirez K, McGregor S, Sigvardsson M, Kee BL.
E2A transcription factors limit expression of Gata3 to facilitate T lymphocyte lineage commitment. Blood. 2013;121(9):1534-1542.
52. Lu Q, Kamps MP. Selective repression of transcriptional activators by Pbx1 does not require the homeodomain. Proc Natl Acad Sci U S A. 1996;93(1):470-474.
53. Mandal JP, Shiue C-N, Chen Y-C, et al. PKCδ mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression. Free Radic Biol Med. 2021;163:69-87.
54 Tian Y, Korn P, Tripathi P, et al. The mono-ADP-ribosyltransferase ARTD10 regulates the voltage-gated ion channel Kv1.1 through protein kinase C delta. BMC Biol. 2020;18(1):143.
55. Murthy S, Desantis J, Verheugd P, et al. 4-(Phenoxy) and 4-(benzyloxy)benzamides as potent and selective inhibitors of mono-ADP-ribosyltransferase PARP10/ARTD10. Eur J Med Chem. 2018;156:93-102.
56. Hart JR, Garner AL, Yu J, et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proc Natl Acad Sci U S A. 2014;111(34):12556-12561.
57 Lourenco C, Resetca D, Redel C, et al. MYC protein interactors in gene transcription and cancer. Nat Rev Cancer. 2021;21(9):579-591.
58. Windmoeller BA, Beshay M, Helweg LP, et al. Novel primary human cancer stem-like cell populations from non-small cell lung cancer: inhibition of cell survival by targeting NF-κB and MYC signaling. Cells. 2021;10(5):1024.
59. Hong L, Kenney SR, Phillips GK, et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem. 2013;288(12):8531-8543.
Haley E. Ramsey,1,2 Agnieszka E. Gorska,1 Brianna N. Smith,1 Andrew J. Monteith,1 Londa Fuller,1 Maria P. Arrate1 and Michael R. Savona1,3,4
1Department of Medicine, 2Program in Cancer Biology, 3Center for Immunobiology and 4Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN, USA
Correspondence: M.R. Savona michael.savona@vanderbilt.edu
Received: June 29, 2023.
Accepted: December 15, 2023. Early view: December 28, 2023.
https://doi.org/10.3324/haematol.2023.283850
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
CD47-SIRPa is a myeloid check point pathway that promotes phagocytosis of cells lacking markers for self-recognition. Tumor cells can overexpress CD47 and bind to SIRPa on macrophages, preventing phagocytosis. CD47 expression is enhanced and correlated with a negative prognosis in acute myeloid leukemia (AML), with its blockade leading to cell clearance. ALX90 is an engineered fusion protein with high affinity for CD47. Composed of the N-terminal D1 domain of SIRPα genetically linked to an inactive Fc domain from human immunoglobulin (Ig) G, ALX90 is designed to avoid potential toxicity of CD47-expressing red blood cells. Venetoclax (VEN) is a specific B-cell lymphoma-2 (BCL-2) inhibitor that can restore apoptosis in malignant cells. In AML, VEN is combined with azanucleosides to induce superior remission rates, however treatment for refractory/relapse is an unmet need. We questioned whether the anti-tumor activity of a VENbased regimen can be augmented through CD47 inhibition (CD47i) in AML and how this triplet may be enhanced. Human AML cell lines were sensitive to ALX90 and its addition increased efficacy of a VEN plus azacitidin (VEN+AZA) regimen in vivo. However, CD47i failed to clear bone marrow tumor burden in PDX models. We hypothesized that the loss of resident macrophages in the bone marrow in AML reduced efficiency of CD47i. Therefore, we attempted to enhance this medullary macrophage population with agonism of TLR3 via polyinosinic:polycytidylic acid (poly(I:C)), which led to expansion and activation of medullary macrophages in in vivo AML PDX models and potentiated CD47i. In summary, the addition of poly(I:C) can enhance medullary macrophage populations to potentiate the phagocytosis merited by therapeutic inhibition of CD47.
CD47 is a ubiquitous cell surface protein that serves as myeloid checkpoint and is the ligand for the macrophage receptor, SIRPα. The immune system relies on the binding of CD47-SIRP α to recognize cells as ‘self’ leading to the moniker of the ‘don’t eat me’ signal.1,2 The CD47-SIRPα is a myeloid check point and is highly conserved in mammalian species. Cancer cells aberrantly overexpress CD47 to evade immune detection and destruction. As CD47 expression is prevalent in hematologic malignancies, trials targeting CD47 with monoclonal antibodies have been a recently explored strategy in the clinic.3,4 Targeting CD47 has led to immune response against tumor tissues, but also led to opsonization of RBC, as older RBC with less CD47 are more susceptible to premature clearance by CD47 antagonists, leading to anemia. Previous
groups have tried to address CD47 antagonist-induced hematotoxicity using dual targeting bispecific antibodies to CD47 and anti-SIRPα antibodies.5-7 The novel CD47 inhibitor evorpacept (ALX148) was engineered with an inactive Fc binding subunit and a high affinity CD47 binding domain. The inactive Fc domain prevents a pro-phagocytic signal and consequent clearance of RBC to improve the overall tolerability of CD47-targeted therapy.8 ALX90 is a small molecule inhibitor of CD47, with nanomolar level selectivity that serves as a tool compound for ALX148.8-10 ALX90 did not exhibit hemagglutination activity when compared to other CD47 antibody clones in human, monkey and murine blood samples. Similarly, the clinical analog, ALX148, led to no changes to the hematological parameters in treated mice.9 Acute myeloid leukemia (AML) is hallmarked by the acquisition of somatic mutations and chromosomal structural abnormalities, the arrest of normal cellular differentiation,
and clonal expansion of immature myeloid cells, leading to ineffective hematopoiesis. In the past, standard of care therapy has been limited to cytotoxic chemotherapy such as cytarabine and anthracycline. However, since 2017 several targeted therapies were approved by the Food and Drug Administration as viable therapeutic options. The BCL-2 inhibitor VEN has demonstrated particular success, which has emerged as a critical component to the standard of care in multiple lymphoid and myeloid neoplasms. As a single agent, VEN demonstrates modest activity in relapsed/refractory AML,11 yet when used in combination with a DNA methyltransferase inhibitor (DNMTi) 12 or low-dose cytosine arabinoside (LDAC) led to remission in 73% of newly diagnosed elderly patients.13-15 Unfortunately, despite these encouraging response rates, most patients treated with VEN will eventually relapse. Previous studies have suggested mechanisms of resistance for both primary and acquired BCL-2 resistance include increased levels of MCL-1 as well as mutations in TP53 . 12,16,17 Increasing and prolonging VEN efficacy in hematologic malignancies remains a priority. We first sought to test CD47 inhibition in the context of an AML VEN-based regimen to determine if efficacy could be increased. We added ALX90 to a standard of care VEN/azacitidine (AZA) regimen in multiple AML in vivo models. In cell line-derived xenografts (CDX) models where medullary tumor burden is low (<50% cell of bone marrow [BM] cell volume at moribund) and native macrophages are present at endpoint found that CD47 inhibition led to a significant decrease in tumor burden as a monotherapy, and augments standard of care therapy with VEN/AZA. However, in patient-derived xenograft (PDX) models with high tumor burden and low native medullary macrophage constituency, CD47i failed to decrease medullary tumor burden as a monotherapy, despite almost complete clearance of tumor cells within the peripheral blood and spleen. Within the paucity of macrophages present in the marrow of PDX models, almost all were polarized toward M2. This implied that the small population of tumor-supportive M2 macrophages, and lack of M1 tumor-suppressive macrophages limited the potential of CD47 inhibition. Thus, in an attempt to increase medullary macrophage function in mice, we co-treated with poly(I:C), a toll-like receptor 3 (TLR3) agonist which mimics viral double stranded RNA and is known to stimulate macrophage differentiation and polarization to M1 macrophages.18-20 As expected, in naïve NSGS mice, poly(I:C) transiently increased M1 macrophage populations in bone marrow, splenic, and blood compartments. Surprisingly, when poly(I:C) was combined with a VEN/AZA + ALX90 in PDX models it led not only to significant decreases in tumor burden in all compartments, but allowed for return to normal murine hematopoiesis and healthy BM. Poly(I:C) combined with ALX90 likewise reduced patient-derived leukemia in the
BM of immunocompromised mice. These data suggest that in the setting of AML, the presence of active BM M1 macrophage populations is required to induce a meaningful response to CD47i. We demonstrate here that by increasing BM macrophage function in murine models of AML, a more effective immune microenvironment may be restored, thereby potentiating CD47i in AML.
Experiments were conducted on primary patient samples which were provided by the Vanderbilt-Ingram Cancer Center Hematopoietic Malignancies Repository and are in accordance with the tenets of the Declaration of Helsinki and approved by the Vanderbilt University Medical Center Institutional Review Board.
AML cell lines MV-4-11 and THP-1 were purchased from the American Type Culture Collection (Manassas, VA). The MOLM-13 cell line was purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). ATCC and DSMZ cell lines are authenticated by short tandem repeat profiling and cytochrome C oxidase gene analysis. Cells were used for the experiments presented here within 10-30 passages from thawing. MV-4-11 cell line was grown in IMDM, and all other cell lines were cultured in RPMI and supplemented with 10-20% fetal bovine serum and 100 U/mL penicillin and 100 ug/mL streptomycin. Cells were kept at 37°C in a 5% CO2 incubator.
All animal studies were conducted in accordance with guidelines approved by the IACUC at Vanderbilt University Medical Center. Female NSGS [NOD/SCID/Tg(hSCF/hGMCSF/hIL3)] mice, 6-8 weeks old were irradiated with 1 Gy microwave radiation. Twenty-four hours later, mice were injected with 1x106 MV-4-11, 3x103 MOLM-13 or 2x106 AML leukapheresis primary patient cells via tail vein injection. Leukapheresis sample description is included in the Online Supplementary Appendix. After establishing microchimerism, 1-2 weeks post-transplant, mice were treated with either VEN and AZA (VEN 20 mg/kg 5 days [D] on 2 D off, AZA upfront Monday/Wedneday/Friday [M/W/F] 1.5 mg/kg), with and without ALX 90 (30 mg/kg M/W/F), the combination, or vehicle. Poly(I:C) (Invivogen) was dosed at 15 mg/kg M/W/F every 2 weeks, with 1 week off between cycles, for up to 8 weeks. Peripheral blood draws were taken weekly to assess chimerism.
For flow cytometry of murine blood and tissues, red blood cells were lysed with EL Buffer on ice (Qiagen), with remain-
ing cells washed and resuspended in 1x phosphate-buffered saline (PBS) with 1% bovine serum albumin and stained for 15 minutes with the following antibodies: human CD45APC, human CD33-PE-Cy7, murine CD45-PE and DAPI (Biolegend). For CD47 measurement in cell line experiments, human CD47-FITC (Biolegend) was used. As a background control, positive mean fluorescence intensity (MFI) from either control human IgG4-FITC or human IgG1-FITC was subtracted from CD47 MFI to estimate final value. Cells were washed and submitted for flow cytometric analysis using a 3-laser LSRII (Becton Dickinson).
Immunohistochemistry
Tissues were fixed in 4% paraformaldehyde for 48 hours and stored in 70% ethanol before being embedding in paraffin and sectioned at 5 µm. The bone tissue was decalcified prior to being embedded in paraffin. Sections were dewaxed in Xylene and rehydrated in ethanol. Standard Mayer’s Hematoxylin and Eosin staining was performed. Antigen retrieval using a standard pH 6 sodium citrate buffer (BioGenex) was performed and sections were stained with monoclonal mouse anti-human CD45 (Dako, M0701, dilution 1:200) using M.O.M. Kit (Vector).
Unless otherwise noted, data were summarized using the mean (+/- standard deviation). Per group sample sizes are presented in figures and results reported from two separate experiments, unless stated otherwise. In order to avoid normality assumptions, pairwise group comparisons were made using the non-parametric Mann-Whitney U test. The non-parametric Spearman correlation was used to assess pairwise variable associations. Data were analyzed using Graph Pad Prism 6.0 for Windows (GraphPad Software, La Jolla California USA, www.graphpad.com). *P<0.05, **P<0.01 and ***P<0.001.
ALX90 significantly conceals surface expression of CD47 on multiple acute myeloid leukemia cell lines
In order to ensure CD47 specificity of the CD47i ALX90, we incubated AML cell lines MV-4-11, MOLM-13, and THP1 with ALX90 or magrolimab and stained for CD47. Flow cytometry revealed a significant decrease in detectable surface expression of CD47 MFI in the presence of ALX 90
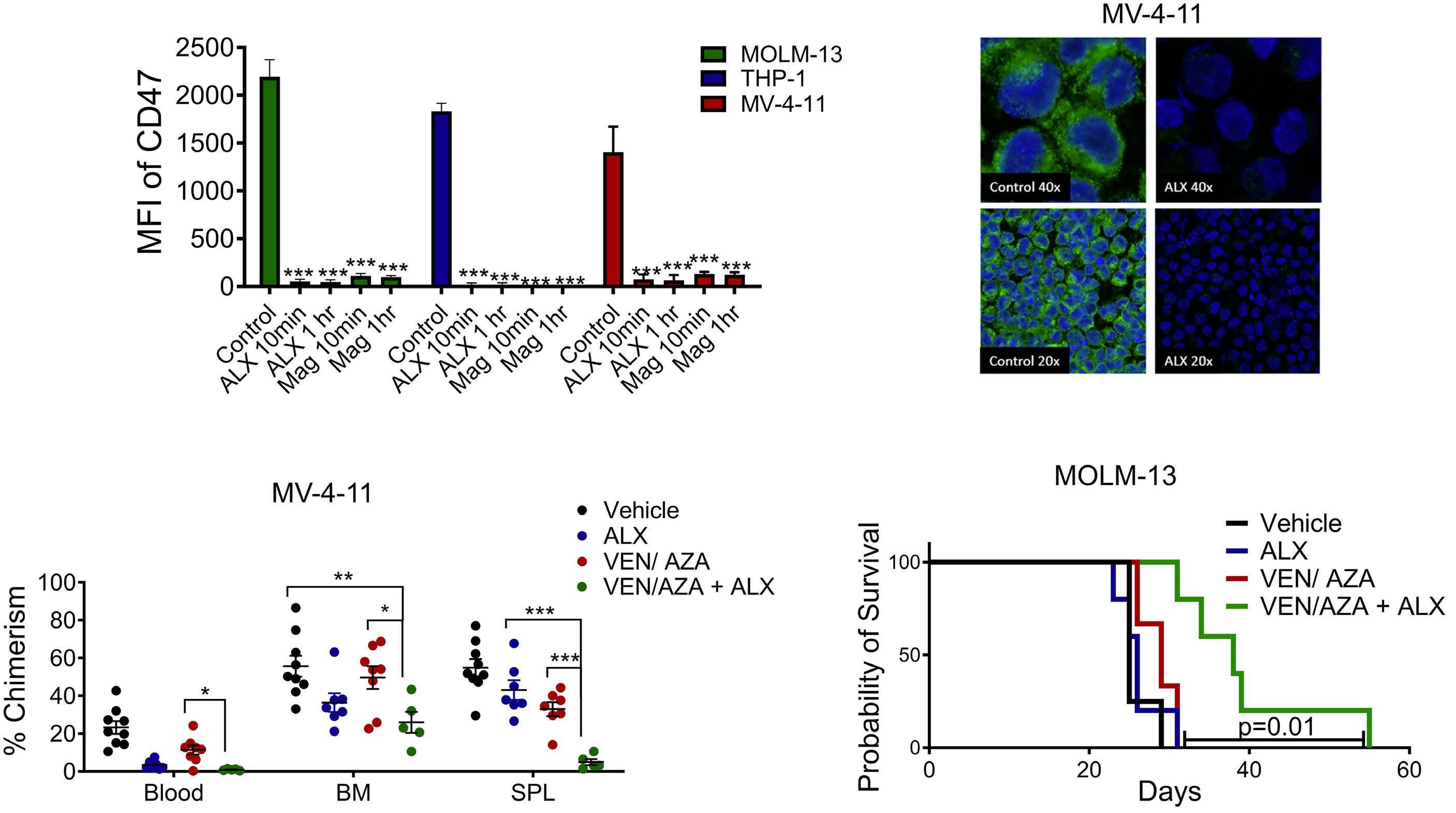
Figure 1. ALX90 conceals CD47 both in vivo and in vitro. In vitro incubation of MV-4-11, MOLM-13 and THP-1 with CD47 inhibitors ALX90 (100 nM) or magrolimab (10 ug/mL) conceals surface expression of CD47 in acute myeloid leukemia (AML) cell lines when analyzed via (A) flow cytometry and (B) in the MV-4-11 cell line by confocal microscopy (green: hCD47, blue: DAPI). (C) The addition of ALX90 to a venetoclax/azacitidine (VEN/AZA) regimen leads to decreased tumor burden in an MV-4-11 as shown through flow cytometric analysis. (D) Survival was increased in a MOLM13 cell line-derived xenograft (CDX) when ALX90 was added to VEN/AZA (P=0.01). (D). MFI: mean fluorescence intensity; Mag: magrolimab; BM: bone marrow; SPL: spleen. *P<0.05, **P<0.01 and ***P<0.001.
and magrolimab, representing coverage of CD47 protein (Figure 1A). Complete inhibition of CD47 by ALX90 was confirmed using immunofluorescence in MV-4-11 (Figure 1B) as well as MOLM-13 and THP-1 (Online Supplementary Figure S1A).
The addition of CD47 inhibition to venetoclax/azacitidine treatment regimen leads to decreased tumor burden and lifespan prolongation in multiple in vivo cell line derived models of acute myeloid leukemia
In order to determine the efficacy of CD47 inhibition in vivo, NSGS mice were treated with a VEN/AZA, ALX90 or a combination. On day 28, when vehicle mice became moribund, tissues were collected for tricompartmental analysis, and human CD45/CD33+ AML cells were counted relative to the amount of healthy murine CD45+ cells. As shown in Figure 1C the addition of CD47i to a VEN/AZA regimen led to a significant decrease in AML when compared to a VEN/ AZA alone. Furthermore, CD47i amended splenic weight, a result of reduced leukemia within the spleen and decreased extramedullary hematopoiesis, which is reflective of healthy BM (Online Supplementary Figure S1B), as well as body weight (Online Supplementary Figure S1C). Bone marrow and splenic tissues were stained for human CD45,
and immunohistochemistry revealed a significant decrease of tumor cells in both bone marrow and spleen (Online Supplementary Figure S2A). In an additional MOLM-13 cell line xenograft, the addition of CD47 inhibition led to significant increases in lifespan compared to ALX or VEN/AZA treatments alone (Figure 1D).
CD47i leads to decreased tumor burden in extramedullary compartments of multiple acute myeloid leukemia patient-derived xenograft models, but not within bone marrow
Given our observation that AML PDX models confluently involved the BM, we began measuring the expression of CD47 in various AML PDX models in an attempt to correlate surface protein expression as a biomarker. As shown in Figure 2A, varying levels of CD47 were noted between patient samples, which were not dependent on any detected mutations (Online Supplementary Table S1). In order to further elucidate the role of CD47 as a targetable biomarker, we explored the efficacy of monotherapy in PDX models with both high “++” (18-12-001) and low “+” (18-10-009) CD47 expression. Post-transplantation, peripheral chimerism was tested weekly in these models in search of circulating tumor cells, and leukemic engraftment was established
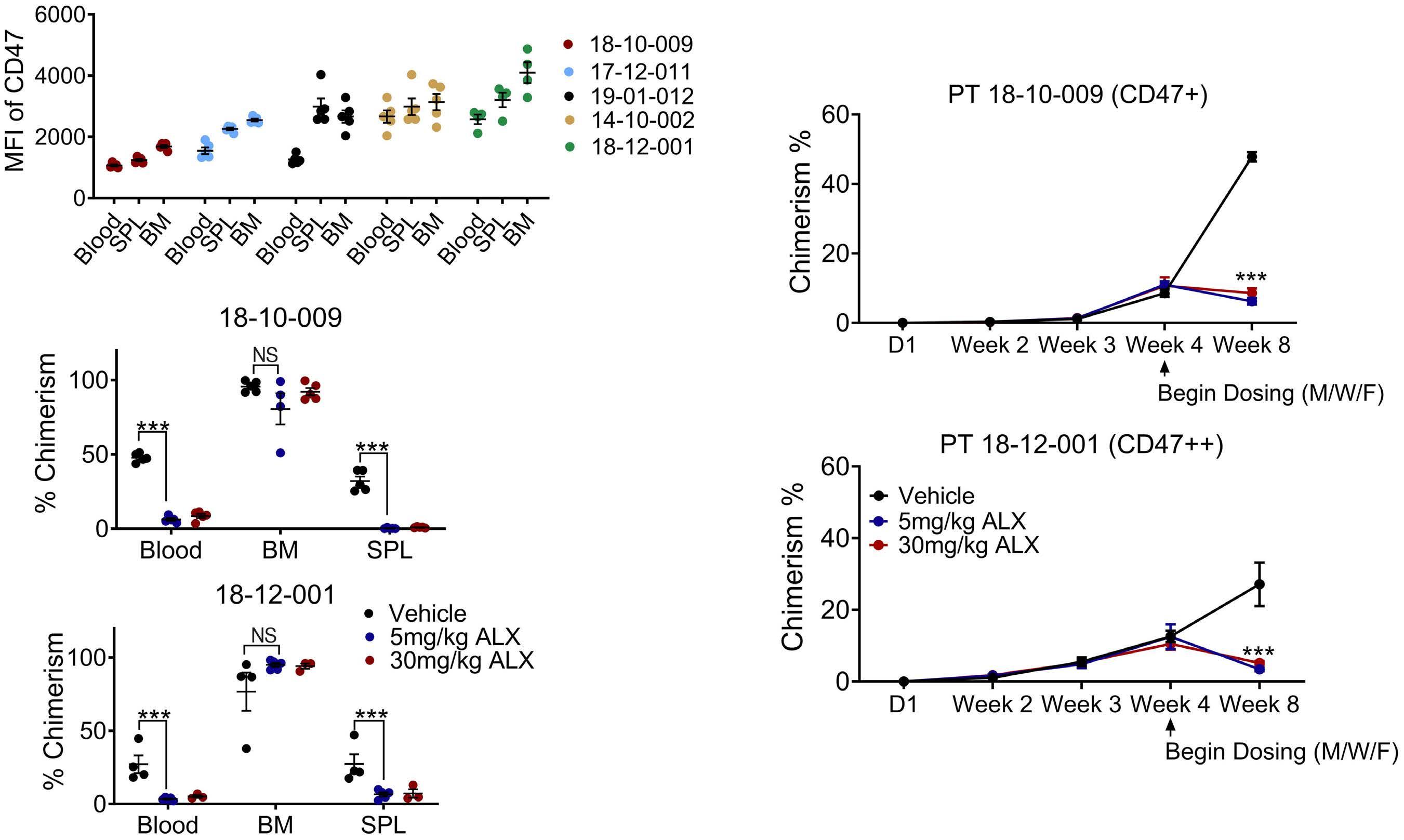
Figure 2. CD47 inhibition monotherapy reveals minimal tumor clearance from bone marrow in a patient-derived xenograft model. (A) Surface expression of hCD47 was measured by flow cytometry in multiple patient-derived xenograft (PDX) models. (B) CD47 inhibition leads to decreased leukemia in peripheral blood of PDX models with significant differences shown between 30 mg/mL and vehicle groups. (C) Chimeric analysis of PDX models reveals the effects of ALX in blood, spleen (SPL), and bone marrow (BM). MFI: mean fluorescence intensity; M/W/F: Monday/Wednesday/Friday; NS: not significant. *P<0.05, **P<0.01 and ***P<0.001.
at 4 weeks post-transplant. At this juncture, mice were treated with ALX90 monotherapy at a low 5 mg/kg and high 30 mg/kg dose (Figure 2B). Four weeks after treatment, mice were euthanized, and tissues were harvested for tricompartmental analysis. CD47i monotherapy led to a significant decrease in tumor burden in both the blood and spleen (Figure 2C). Contrary to CDX models, much of the BM remained unaffected in these PDX (Figure 2C). In order to confirm that ALX90 bound to CD47 antigen on PDX cells, we harvested human cells from the blood, BM and spleen from these mice, stained them for CD47, and quantified ALX90 bound human hematopoietic CD45+/CD33+ cells in a dose-dependent fashion (Figure 3A).
In order to determine if VEN/AZA increased efficacy of CD47i monotherapy, we treated the CD47-low AML PDX (18-10009), with VEN/AZA and ALX90. Tumor kinetics were monitored peripherally through weekly measurements in blood chimerism, showing a significant decrease in chimerism in a regimen of all three drugs (Online Supplementary Figure S2B). Mice were sacrificed at 8 weeks, and VEN/AZA+ALX was found to lead to significant decreases of AML in both the blood and spleen of mice, with a more modest effect is more modest in the bone marrow (Figure 3B); however, the addition of CD47i failed to prolong the lifespan of VEN/ AZA treated mice in this experiment (Figure 3C).
CD47 inhibition in the bone marrow may be dependent upon the number and type of macrophages residing within the bone marrow during treatment CD47 inhibition was detected on BM resident tumor cells in a PDX model of AML (Figure 3A), yet tumor chimerism
levels remained unchanged in that compartment (Figure 3B). In order to understand this, we examined the resident macrophages responsible for phagocytosis of cells treated with CD47i. Using the remaining cells from our PDX models from the experiment in Figure 3, we calculated the number of macrophages residing within the compartments and compared with the number of macrophages found within a healthy, naïve NSGS mouse. In all treatment groups, macrophages were increased in the blood and significantly decreased in the other tissues, with the largest diminishment observed in the BM (Figure 4A-C).
Leukemia-associated macrophages (LAM) are a type of tissue-resident macrophages noted in the presence of leukemia. LAM are largely noted to be CD206+ ‘pro-tumoral’, M2 macrophages. M2 macrophages are considered pro-tumoral due to their decreased phagocytotic ability and production of immunosuppressive cytokines.21,22 Interestingly, CD206 expression has been noted as a negative prognostic factor in AML.21 By contrast, M1 (CD11b+) macrophages are associated with pro-inflammatory/anti-tumoral activity with increased phagocytosis and the production of inflammatory cytokines.23 Treatments such as lipopolysaccharide and interferon-γ (IFN-γ) have been shown to promote proliferation and polarization of macrophages to an M1 status, supporting TH1 activation and tumoricidal activity.24,25 While little is known about the effect that AML blasts have on the tissue-resident macrophages in murine PDX models, we began to look at these populations. To this end, we began characterizing the F4/80+ resident macrophage populations in two additional PDX models 8 weeks post-transplantation (20-09-002 and 19-03-007),
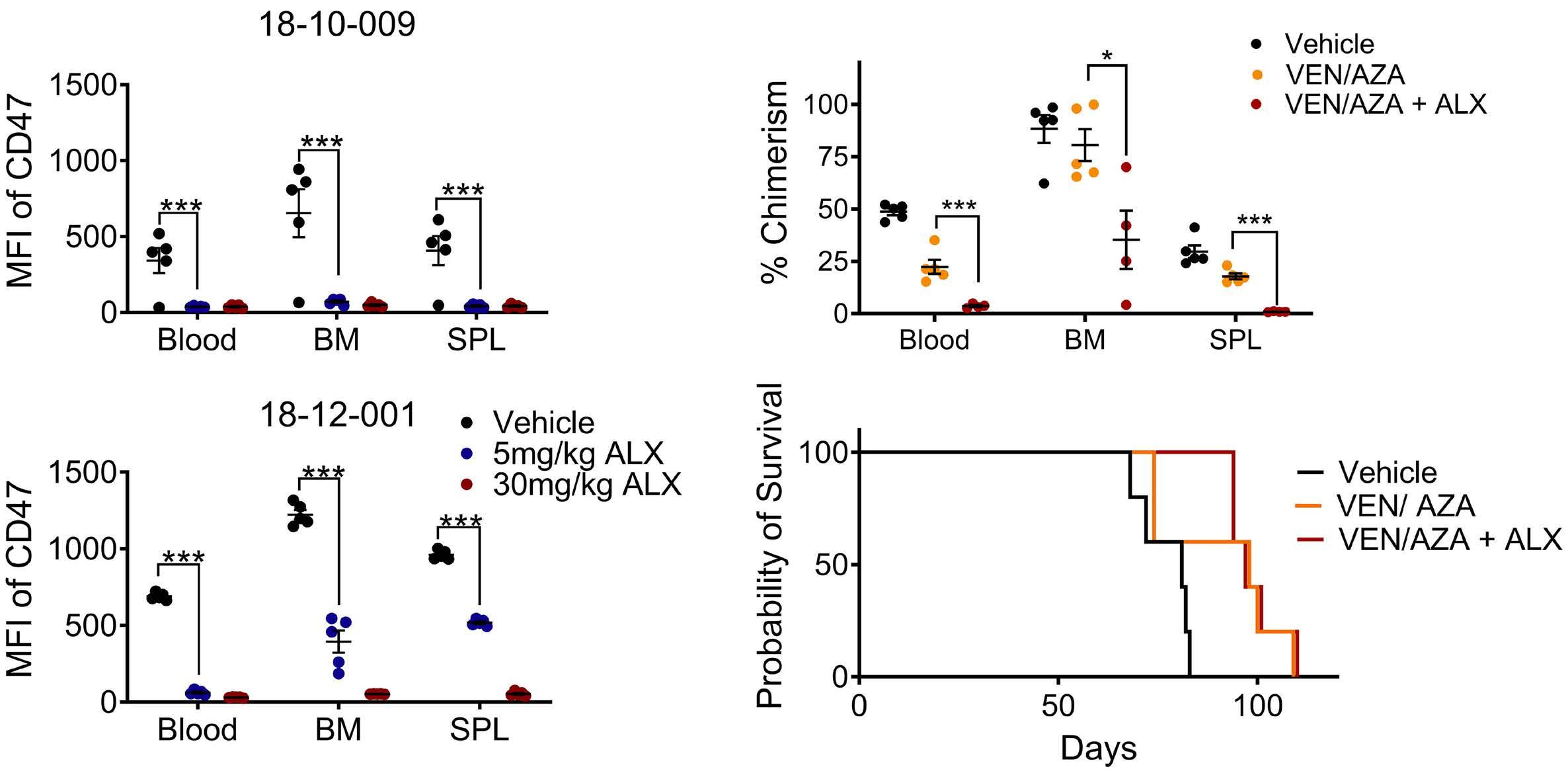
Figure 3. The activity of CD47i in the bone marrow. (A) Flow cytometric analysis denoting coverage of hCD47 epitope on human acute myeloid leukemia (AML) cells of 2 AML patient-derived xenograft (PDX) models. The addition of ALX90 to a venetoclax/ azacitidin (VEN/AZA)-based regimen in AML PDX (18-10-009), leads to decreases in intercompartmental tumor burden (B) with Kaplan-Maier analysis of an VEN/AZA and VEN/AZA + ALX regimen showing failure to increase lifespan (C). MFI: mean fluorescence intensity; BM: bone marrow; SPL: spleen. *P<0.05, **P<0.01 and ***P<0.001.
comparing them to naïve NSGS mice. In the BM, the more tumor permissive M2 phenotype was dominant in PDX and undifferentiated M0 phenotypic macrophages were more common in native NSGS mice (Online Supplementary Figure S3A). These findings imply that the presence of AML blasts within the BM affects the relative number of macrophages, and polarizes those resident macrophages toward a tumor-permissive phenotype.
TL3 agonism leads to the expansion of F4/80 macrophages in the bone marrow, spleen and blood of treated mice
Given to the historical application of IFN- γ in myeloid malignancies, and the role IFN-γ plays in macrophage differentiation, IFN-γ was proposed as potential therapeutic to artificially increase macrophage populations within the BM.26-28 However, IFN-γ has only been shown to increase or activate macrophages with the addition of LPS.29-31 Poly(I:C) is a TLR3 agonist which mimics viral double-stranded RNA in a sterile immune response known to stimulate macrophage differentiation and polarization to M1 macrophages.18 Systemic administration of poly(I:C) in a TLR3-deficient mouse resulted in significant reductions in both macrophage activation and cytokine production compared to TLR3 wild-type.32 Additionally, poly(I:C) has synergized previously with CD47 blockade in colorectal cancer models.33 As variations of poly(I:C) are in use in over 13 clinical trials currently as an immunoadjuvant, we adapted use of
this TLR3 agonist in our murine models.34-37 Administering poly(I:C) over a 2-week period revealed significant shifts in macrophage phenotype, most notably the additional presence or increase in M1 macrophage populations (Figure 5A). Furthermore, the ratio of M1/M2 macrophages changed significantly between the treatment time points in these naïve NSGS mice, with dramatic shifts being noted after 2 weeks of poly(I:C) (Figure 5B). Additionally, levels of macrophages in of poly(I:C)-treated mice increased while receiving treatment (Figure 5C).
TLR3 agonism potentiates CD47i by increasing active macrophages in a patient-derived xenograft model Given the response of naïve NSGS mice to TLR3 agonism via poly(I:C) treatment, we attempted to increase BM macrophage populations to augment the effect of ALX90 in PDX models. In a AML PDX (18-12-001) mice were treated with ALX90 and or poly(I:C) to illustrate effects of TLR3 stimulation on CD47 inhibition. We found that poly(I:C) augmented the use of ALX90, but had negligible effects on the BM chimerism when used as a single agent ( Online Supplementary Figure S3B). Neither PDX patient samples (18-12-001, 18-10-009, an additional sample 21-12-019) nor cell lines (MV-4-11, MOLM-13) were affected by poly(I:C) alone, suggesting that poly(I:C) is not inherently toxic to blast cells (Online Supplementary Figure S4A). In order to illustrate if addition of TLR3 agonism could improve upon triplet therapy (Figure 2), we transplanted PDX 18-10-009
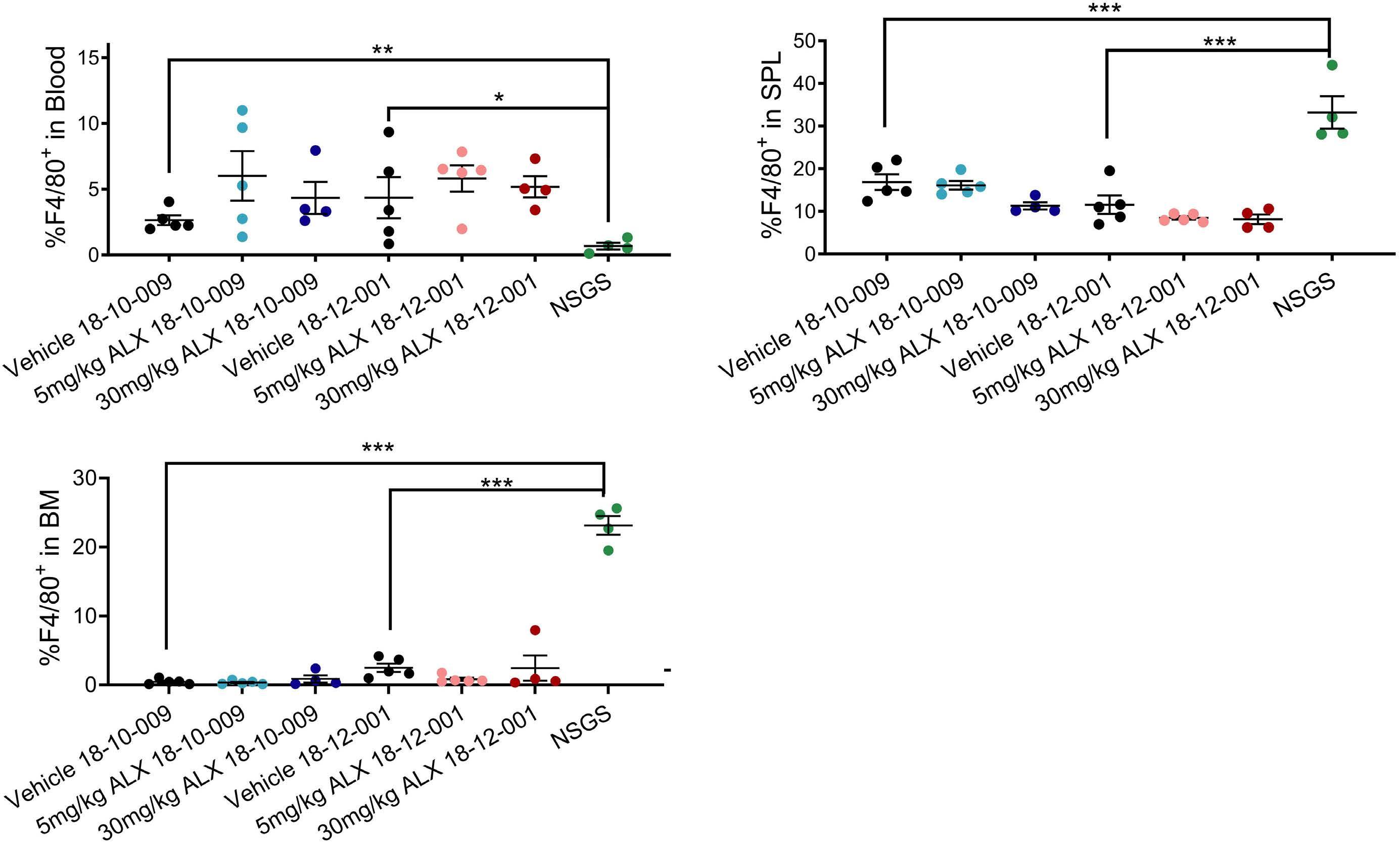
Figure 4. Analysis of acute myeloid leukemia patient-derived xenograft resident murine macrophage populations. (A-C) Murine macrophage (F4/80) levels were measured by flow cytometry in both patient-derived xenograft (PDX) models under ALX treatment, and within a naïve untreated NSGS mouse in peripheral blood, splenic (SPL) and bone marrow (BM) compartments. *P<0.05, **P<0.01 and ***P<0.001.
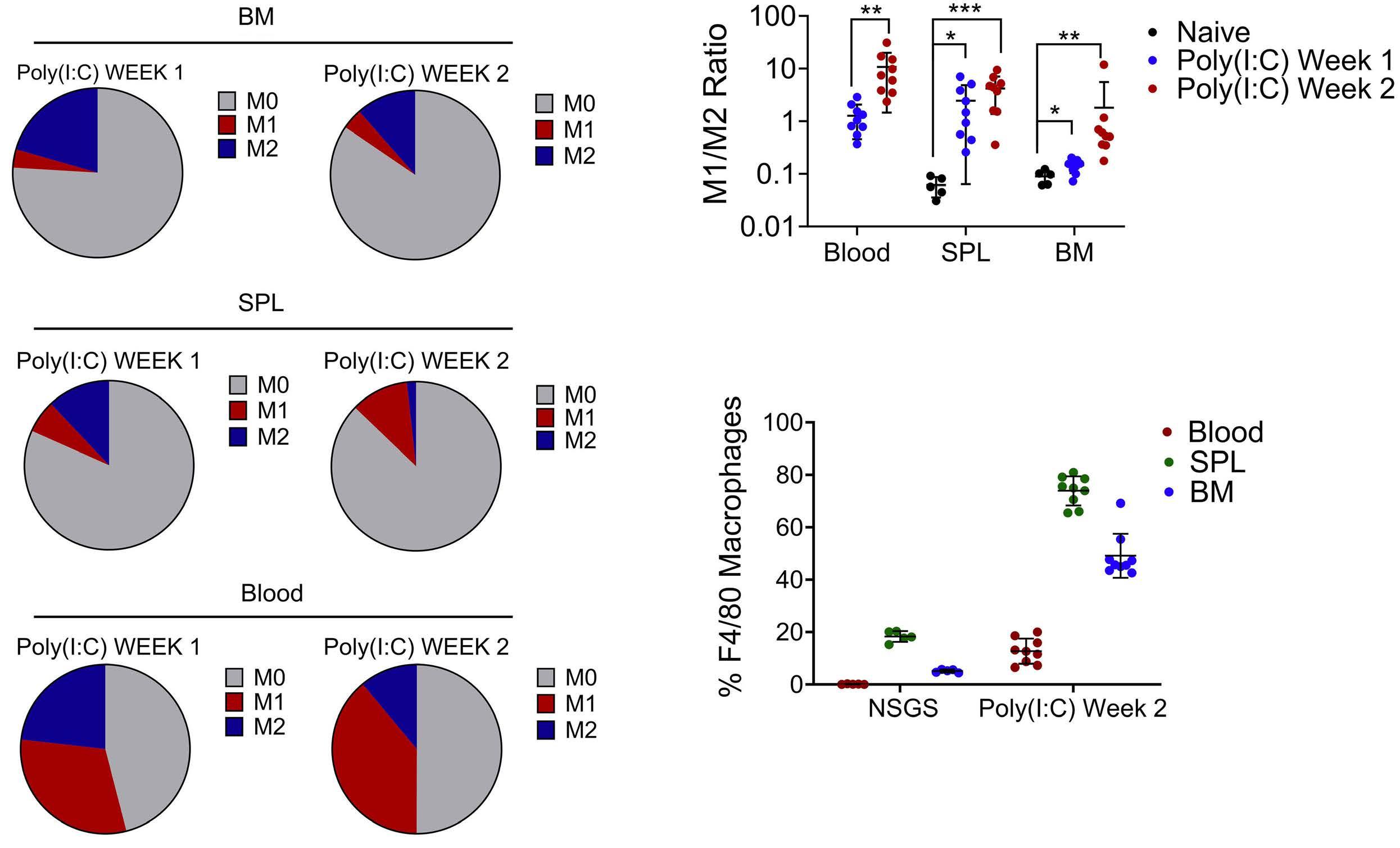
Figure 5. The activity of polyinosinic:polycytidylic acid on murine macrophages. (A) Polyinosinic:polycytidylic acid (poly(I:C)) leads to overall shifts in macrophage populations in the bone marrow (BM), spleen (SPL) and peripheral blood of treated mice. Shifts in M1/M2 ratio develop across compartments (B) while macrophage numbers increase (C) upon poly(I:C) administration. *P<0.05, **P<0.01 and ***P<0.001.
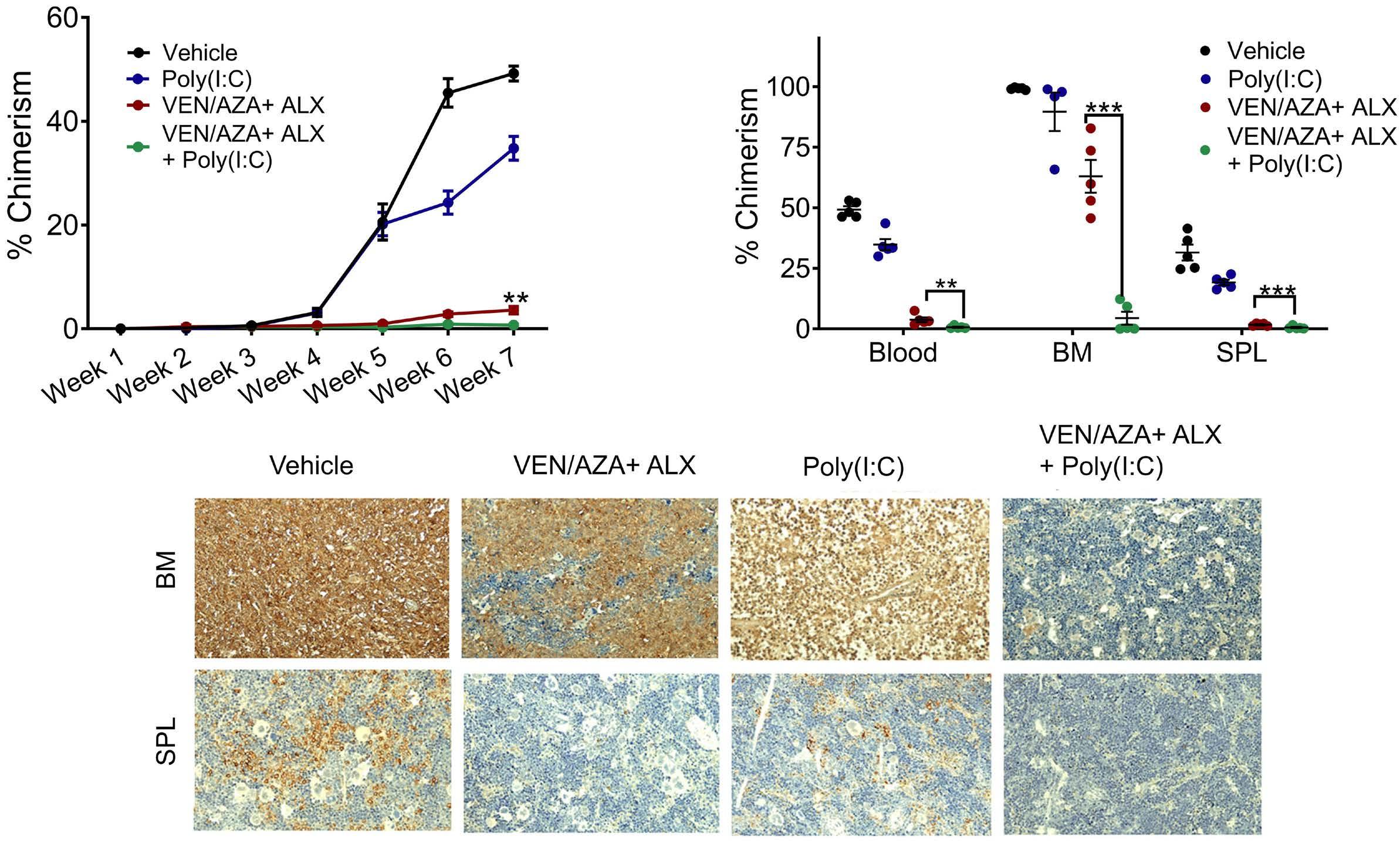
Figure 6. The addition of polyinosinic:polycytidylic acid to a venetoclax/azacitidin regimen augments CD47 inhibition in a patient-derived xenograft model. (A) Peripheral chimerism and (B) endpoint analysis of a venetoclax/azacitidin plus ALX (VEN/AZA + ALX) regimen with the addition of polyinosinic:polycytidylic acid (poly(I:C)). (C) Immunohistochemistry for hCD45 reveals visible decreases in human cells with the bone marrow (BM) and spleen (SPL) of patient-derived xenograft (PDX) mice treated with VEN/AZA + ALX + poly(I:C). **P<0.01 and ***P<0.001.
and began treatment with VEN/AZA + ALX + poly(I:C). The “quadruplet therapy” of poly(I:C) together with VEN/AZA + ALX led to a drop in peripheral chimerism (Figure 6A). Again, at 8 weeks, mice were sacrificed and a significant decrease in leukemia was seen in the blood, spleen and, notably, BM in instances in which poly(I:C) was added to a Ven/ AZA + ALX regimen (Figure 6B, C). We analyzed the quality of the remaining murine BM in this model for functional hematopoiesis. Erythropoiesis was noted through higher counts of TER119+ cells with the BM, along with higher levels of myeloid (CD11b+) and neutrophil (CD11b+/Ly6g+) levels (Figure 7A). Furthermore, the macrophage population within the BM of VEN/AZA+ALX-treated mice versus VEN/AZA + ALK + poly(I:C)-treated mice was significantly diminished. The resident macrophages within the BM of these VEN/AZA + ALX + poly(I:C)-treated mice included a greater portion with a M1 phenotype (Figure 4B). In cytospins, the the return to more normal hematopoiesis was evident (Figure 7B). Furthermore, the addition of poly(I:C) to a VEN/AZA + ALX regimen significantly extended survival in treated mice, even after ceasing treatment (Figure 7C). Finally, to confirm active phagocytosis we conducted an ex vivo phagocytosis assay with two additional AML primary patient samples. For this, naïve NSGS mice were again
treated with poly(I:C) or control vehicle and after 2 weeks of treatment, the two BM groups were harvested and incubated with pHrodo-labeled patient samples pre-incubated with or without ALX90. Target cells labeled with pHrodo will only fluoresce when under the highly acidic condition of the phagolysosome. This fluorescence, when emitted from a macrophage, ensures that the engulfed cell was successfully phagocytosed and further incorporated into a functional phagolysosome. As shown in Figure 8A, F4/80+ bone marrow macrophages displayed increased phago-lysosomal acidification following phagocytosis of patient samples when ALX is employed in combination with poly(I:C) treated BM versus BM from vehicle-treated mice. In order to similarly validate the cooperativity of ALX and poly(I:C) in human cells, peripheral blood monocytes were isolated from healthy donors, differentiated into macrophages and further subjected to macrophage stimulation with poly(I:C). These macrophages were incubated with pHrodo-treated AML cells which were then subsequently treated with CD47i (ALX90 or magrolimab). Baseline phagocytosis in these assays was measured in cultures where AML target cells were incubated with drug corresponding IgG antibodies. While there was expected variation between patients, consistently the greatest fold changes in phagocytosis were
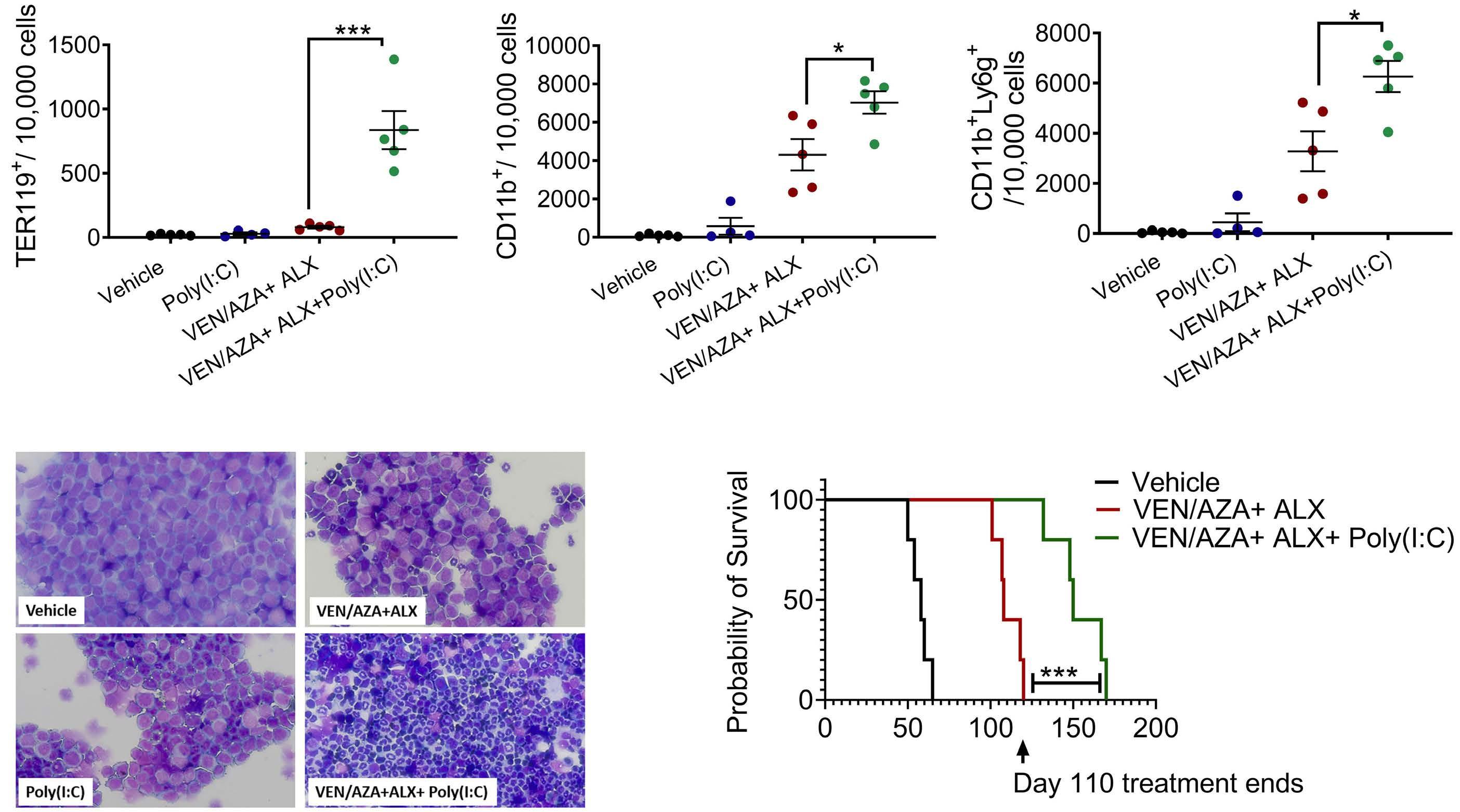
Figure 7. Murine hematopoiesis and lifespan after a venetoclax/azacitidin plus ALX plus polyinosinic:polycytidylic acid regimen. (A) Flow cytometry of remaining murine bone marrow (BM) in patient-derived xenograft (PDX) models reveals significant differences in erythroid, myeloid, and neutrophil populations. (B) Wright Giemsa staining of BM cytospins reveal decreased acute myeloid leukemia (AML) blast populations in venetoclax/azacitidin plus ALX plus polyinosinic:polycytidylic acid (VEN/AZA + ALX + poly(I:C))-treated PDX mice. (C) Kaplan Mayer analysis affirms increased lifespan when poly(I:C) is added to a VEN/AZA + ALX regimen.
noted in the assay populations where macrophages had been pretreated with poly(I:C) and AML samples had been treated with ALX90 or magrolimab (Figure 8B; Online Supplementary Figure S5). These data show that the capacity of CD47i is significantly enhanced when the appropriate macrophage populations are available.
The majority of clinically investigated immunotherapies in AML have been focused on the adaptive immune system, such as chimeric antigen receptor T-cell and T-cell immune checkpoint inhibitors. However, focusing on macrophage and innate immunity is proving a meaningful adjunct of immunotherapy. CD47, which functions as an anti-phagocytic signal, allows for CD47 expressing cells to escape phagocytosis by allowing for its cognate binding to SIRP-α on macrophages. However, many cancers overexpress CD47 as an evolutionary mechanism thereby evading phagocytosis. CD47 inhibition has real clinical potential, with CD47 inhibitors showing benefit in solid tumors and hematological cancers in early clinical trials. While the solid tumor microenvironment typical contains a plethora of macrophages,38-40 limitations of clinical benefit in hematologic malignancies may be rooted in the availability of macrophages, or at least M0/ M1 macrophages in the bone marrow microenvironment. As shown in our studies, CD47 blockade greatly augments the clearance of peripheral AML blasts, but has less success in the BM despite sufficient CD47 present on the cell surface of BM blasts. Although peripheral blood in PDX models contains low numbers of macrophages, there are enough M0/M1 macrophages to clear the engrated AML
from the blood with CD47i. We suspected that a loss of the majority of resident medullary macrophages may be responsible for this lack of phagocytic activity in the BM and while the resident macrophages were diminished in PDX models, they were also polarized as tumor-permissive M2 macrophages/LAM. With the induction of macrophage populations via poly(I:C), inhibition of CD47 was not only potentiated as a monotherapy for medullary reduction of tumor, but allowed for better response when combined with standard of care.
It has been previously shown that CD47 expression, while significantly higher on AML cells than in comparison to normal BM, was variable between AML patients.41 Such findings might lead to the belief that high and low CD47 expression might determine drug efficacy, however, we found that CD47 intensity was not a biomarker of efficacy. The inter-patient variability of CD47 expression on AML blasts made no difference in effectivity of the drug in in vitro phagocytosis assays. Additionally, while MFI in our study reflected the quantity of CD47 expressed, the capacity to clear leukemia was no different between PDX models of high and low intensity CD47.
The concept of M2 macrophages driving tumor progression by sustaining an immunosuppressive environment is a phenomenon of growing interest. Additional studies are beginning to bring to light the role of tumor suppressive M2 macrophages in hindering necessary therapies such as immune checkpoint blockade, radiotherapy and chemotherapy.42,43
T cells interact with LAM to directly or indirectly effect response to tumor.44 While the NSGS mice used in our modeling have negligible levels of T cells due to mutation in interleukin 2 receptor γ, M2 macrophages are dominant
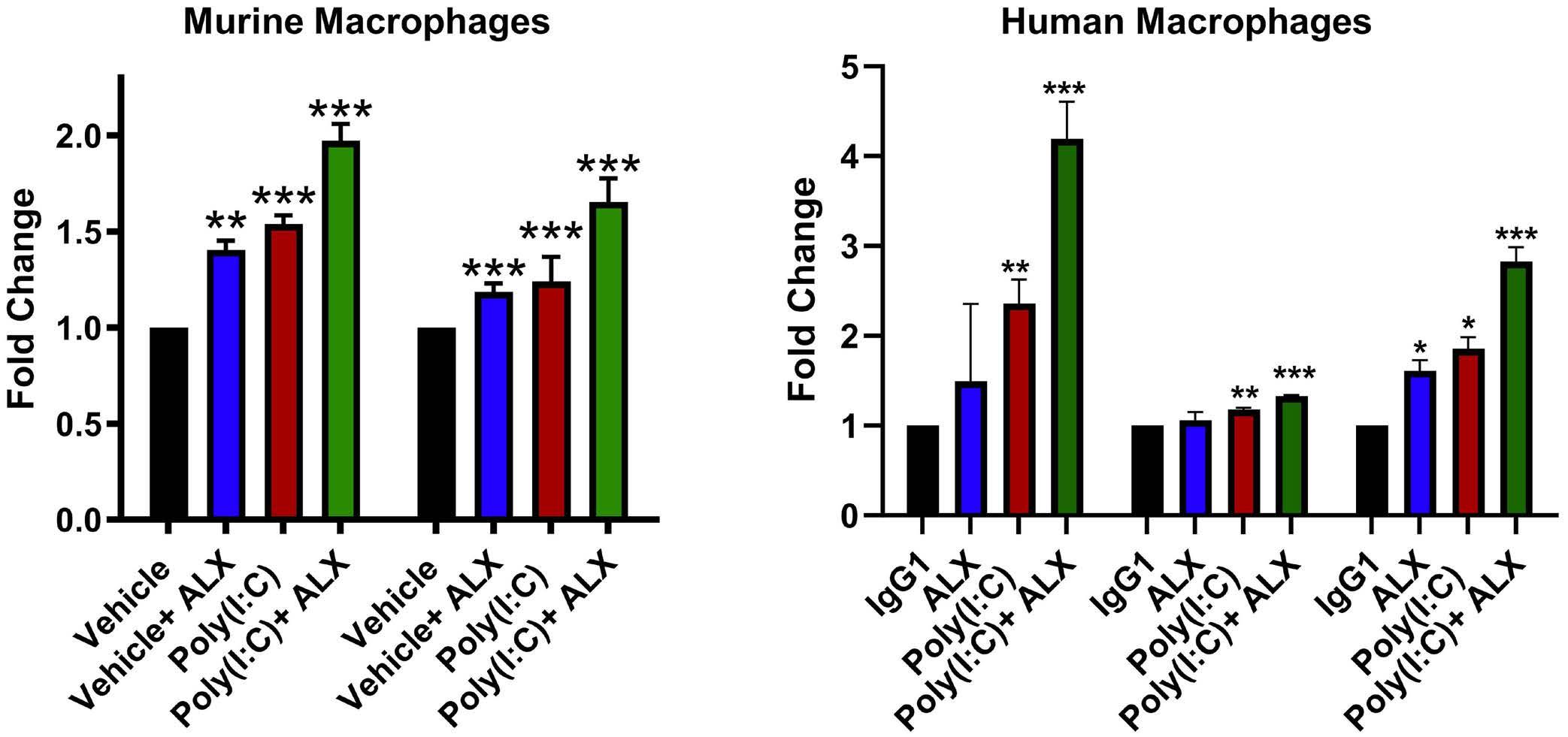
Figure 8. Polyinosinic:polycytidylic acid enhances CD47 inhibitor-induced phagocytosis. (A) Phagocytosis assays using macrophages from vehicle- or polyinosinic:polycytidylic acid (poly(I:C))-treated mouse bone marrow (BM) incubated with pHrodo-labeled cells from patient acute myeloid leukemia (AML) 18-12-001 and 19-03-007 reveal increases in phagocytosis in poly(I:C)-treated BM. (B) Healthy donor macrophages, when pretreated with poly(I:C) reveal increases in phagocytosis of CD47-treated AML (18-12001) cells. Statistical comparisons in 3 individual donors shown here as ALX monotherapy, poly(I:C) monotherapy and combination against IgG1 control. Ig: immunoglobulin.
in AML xenografts. Our study may provide greater insight into the relationship between AML cells and monocyte/ macrophage polarization which can be studied in maturational and mutational subsets for better understanding of how to utilize this type of immunotherapy for AML. Furthermore, we suggest that this increased phagocytic ability of poly(I:C)-treated BM results from the concomitant increase of M1-primed macrophages after treatment. In previous studies, CD11c activation in macrophages through IFN-γ use was shown to increase broad spectrum phagocytosis, resulting in an enhancement of anti-tumor immunity via promotion of phagocytosis.45 The increases of CD11c-positive macrophages in our study are consistent with this IFN-γ induced M1 priming, and does not require LPS administration required for response with IFN. The ability to increase active macrophage populations within the BM of AML patients could potentiate the use of antiCD47-based therapy, particularly when clonal leukemia has physically displaced functional hematopoietic cells, and polarized macrophages to a tumor permissive M2 subtype. Further, it may argue for incorporation of CD47 inhibition into maintenance therapy after establishing remission with VEN/AZA so normal macrophage constituency may reconstitute a healthy BM.
Disclosures
MRS receives research funding from ALX Oncology, Astex, Incyte, Takeda and TG Therapeutics; has stock in Karyopharm and Ryvu; serves on advisory boards or consults or BMS, CTI, Forma, Geron, GSK, Karyopharm, Rigel, Ryvu, Taiho and Treadwell.
Contributions
Conception and design by HR and MS. Development of methodology by HR, AM, AG, PA, BNS and MS. Acquisition of data (provided animals, acquired and managed patient samples, provided facilities, etc.) by HR, BNS, AG, PA, LF and MS.
1. Kelley SM, Ravichandran KS. Putting the brakes on phagocytosis: “don’t-eat-me” signaling in physiology and disease. EMBO Rep. 2021;22(6):e52564.
2. Jaiswal S, Jamieson CHM, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271-285.
3. Sikic BI, Lakhani N, Patnaik A, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J Clin Oncol. 2019;37(12):946-953.
4 Advani R, Flinn I, Popplewell L, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711-1721.
5. Wang Y, Ni H, Zhou S, et al. Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis) by HR, AM, AG, PA and MS. Writing, review, and/or revision of the manuscript by all authors. Administrative, technical, or material support (i.e., reporting or organizing data, constructing database) by all authors. Study supervision by MS.
The Vanderbilt-Ingram Cancer Center (VICC) Hematopoietic Malignancies Tissue Repository, the Vanderbilt University Medical Center Translational Pathology Shared Resource, and other VICC shared Core Services were critical in completion of this work. The authors would like to acknowledge Vanderbilt SWERV writing core for the editorial guidance.
Funding
This study was supported by ALX Oncology. MRS is a Leukemia and Lymphoma Clinical Scholar, and receives funding from the Adventure Alle Fund, Beverly and George Rawlings Endowment, Biff Ruttenberg Foundation, and the NIH RO1 CA262287-01 and U01 OH012271-01. The Vanderbilt-Ingram Cancer Center is supported by a NIH P30 CA068485–19. The REDCap database tool is supported by grant UL1 TR000445 from NCATS/NIH. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. Confocal microscopy was performed though the use of the Vanderbilt Cell Imaging Shared Resource (CISR) supported by NIH grants CA68485, DK20593, DK58404, DK59637, and EY08126. The Zeiss LSM880 confocal microscope was acquired through NIH S10 OD021630 equipment grant.
Data-sharing statement
Protocols and original data from this manuscript are available upon request by contacting the corresponding author.
Immunother. 2021;70(2):365-376.
6. Chen SH, Dominik PK, Stanfield J, et al. Dual checkpoint blockade of CD47 and PD-L1 using an affinity-tuned bispecific antibody maximizes antitumor immunity. J Immunother Cancer. 2021;9(10):e003464.
7. Dheilly E, Moine V, Broyer L, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dualtargeting bispecific antibodies. Mol Ther. 2017;25(2):523-533.
8. Lakhani NJ, Chow LQM, Gainor JF, et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): a first-in-human, open-label, multicentre, phase 1 dose-escalation and doseexpansion study. Lancet Oncol. 2021;22(12):1740-1751.
9 Kauder SE, Kuo TC, Harrabi O, et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a
favorable safety profile. PLoS One. 2018;13(8):e0201832.
10 Kuo TC, Chen A, Harrabi O, et al. Targeting the myeloid checkpoint receptor SIRPalpha potentiates innate and adaptive immune responses to promote anti-tumor activity. J Hematol Oncol. 2020;13(1):160.
11. Konopleva M, Pollye DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106-1117.
12. Nechiporuk T, Kurtz SE, Nikolova O, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019;9(7):910-925.
13. Wei AH, Strickland Jr SA, Hou J-Z, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II Study. J Clin Oncol. 2019;37(15):1277-1284.
14 DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7-17.
15. Ivanov V, Yeh, S-P, Mayer J, et al. Design of the VIALE-M phase III trial of venetoclax and oral azacitidine maintenance therapy in acute myeloid leukemia. Future Oncol. 2022;18(26):2879-2889.
16. Kim K, Maiti A, Loghavi S, et al. Outcomes of TP53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer. 2021;127(20):3772-3781.
17 Ramsey HE, Fischer MA, Lee T, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566-1581.
18. Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O. The Toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. J Neurosci. 2010;30(48):16091-16101.
19 Wang A, Kang X, Wang J, Zhang S. IFIH1/IRF1/STAT1 promotes sepsis associated inflammatory lung injury via activating macrophage M1 polarization. Int Immunopharmacol. 2023;114:109478.
20. Anfray C, Mainini F, Digifico E, et al. Intratumoral combination therapy with poly(I:C) and resiquimod synergistically triggers tumor-associated macrophages for effective systemic antitumoral immunity. J Immunother Cancer. 2021;9(9):e002408.
21. Xu ZJ, Gu Y, Wang CZ, et al. The M2 macrophage marker CD206: a novel prognostic indicator for acute myeloid leukemia. Oncoimmunology. 2020;9(1):1683347.
22. Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophagebased approaches for cancer immunotherapy. Cancer Res. 2021;81(5):1201-1208.
23. Madeddu C, Gramignano G, Kotsonis P, et al. Microenvironmental M1 tumor-associated macrophage polarization influences cancerrelated anemia in advanced ovarian cancer: key role of interleukin-6. Haematologica. 2018;103(9):e388-e391.
24. Vidyarthi A, Khan N, Agnihotri T, et al. TLR-3 stimulation skews M2 macrophages to M1 through IFN-alphabeta signaling and restricts tumor progression. Front Immunol. 2018;9:1650.
25. Xie C, Liu C, Wu B, et al. Effects of IRF1 and IFN-beta interaction on the M1 polarization of macrophages and its antitumor function. Int J Mol Med. 2016;38(1):148-160.
26. DeKelver RC, et al. RUNX1-ETO induces a type I interferon response which negatively effects t(8;21)-induced increased self-renewal and leukemia development. Leuk Lymphoma. 2014;55(4):884-891.
27. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788-792.
28. Alsufyani A, Alanazi R, Woolley JF, Dahal LN. Old Dog, New trick: type I IFN-based treatment for acute myeloid leukemia. Mol Cancer Res. 2021;19(5):753-756.
29 Muller E, Christopoulos PF, Halder S, et al. Toll-like receptor ligands and interferon-gamma synergize for induction of antitumor M1 macrophages. Front Immunol. 2017;8:1383.
30 Munoz-Rojas AR, Kelsey I, Pappalardo JL, Chen M, Miller-Jensen K. Co-stimulation with opposing macrophage polarization cues leads to orthogonal secretion programs in individual cells. Nat Commun. 2021;12(1):301.
31. Lu S, Li D, Xi L, Calderone R. Interplay of interferon-gamma and macrophage polarization during Talaromyces marneffei infection. Microb Pathog. 2019;134:103594.
32. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732-738.
33. Zhong C, Wang L, Hu S, et al. Poly(I:C) enhances the efficacy of phagocytosis checkpoint blockade immunotherapy by inducing IL-6 production. J Leukoc Biol. 2021;110(6):1197-1208.
34 Ewel CH, Urba WJ, Kopp WC, et al. Polyinosinic-polycytidylic acid complexed with poly-L-lysine and carboxymethylcellulose in combination with interleukin 2 in patients with cancer: clinical and immunological effects. Cancer Res. 1992;52(11):3005-3010.
35. Feldman S, Hughes WT, Darlington RW, Kim HK. Evaluation of topical polyinosinic acid-polycytidylic acid in treatment of localized herpes zoster in children with cancer: a randomized, double-blind controlled study. Antimicrob Agents Chemother. 1975;8(3):289-294.
36. Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29(3):330-336.
37. Pollack IF, Jakacki R, Butterfield LH, et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol. 2014;32(19):2050-2058.
38. Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. 2017;9(3):289-302.
39. Li X, Liu R, Su X, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18(1):177.
40 Yang X, Feng W, Wang R, et al. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. Oncoimmunology. 2018;7(4):e1412910.
41. Yan X, Lai B, Zhou X, et al. The differential expression of CD47 may be related to the pathogenesis from myelodysplastic syndromes to acute myeloid leukemia. Front Oncol. 2022;12:872999.
42. Anfray C, Ummarino A, Andon FT, Allavena P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells. 2019;9(1):46.
43. Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495-499.
44 Petty AJ, Li A, Wang X, et al. Hedgehog signaling promotes tumorassociated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. J Clin Invest. 2019;129(12):5151-5162.
45. Tang Z, Davidson D, Li R, et al. Inflammatory macrophages exploit unconventional pro-phagocytic integrins for phagocytosis and anti-tumor immunity. Cell Rep. 2021;37(11):110111.
Annalisa Ruggeri,1* Nicole Santoro,2* Jacques-Emmanuel Galimard,3 Krzysztof Kalwak,4 Mattia Algeri,5,6 Ludmila Zubarovskaya,7 Krzysztof Czyzewski,8 Elena Skorobogatova,9 Petr Sedlacek,10 Caroline Besley,11 Adriana Balduzzi,12,13 Yves Bertrand,14 Julia Peristeri,15 Franca Fagioli,16 Mariane Ifversen,17 Jolanta Gozdzik,18 Christina Peters,19 Birgitta Versluijs,20 Alessandra Biffi,21 Arcangelo Prete,22 Maura Faraci,23 Ibrahim Ghemlas,24 Ivana Bodova,25 Olga Aleinikova,26 Arnaud Dalissier,27 Vanderson Rocha28 and Selim Corbacioglu29
1IRCCS San Raffaele Scientific Institute, Milano, Italy; 2Hematology Unit, Department of Oncology and Hematology, Santo Spirito Hospital, Pescara, Italy; 3EBMT Statistical Unit, Paris, France; 4Department of Pediatric Hematology, Oncology and Bone Marrow Transplantation, Wroclaw Medical University, Wroclaw, Poland; 5Department of Pediatric Hematology Oncology, IRCCS Bambino Gesu Children’ s Hospital, Rome, Italy; 6Department of Health Sciences, Magna Graecia University, Catanzaro, Italy; 7RM Gorbacheva Research Institute, Pavlov University, St. Petersburg, Russia; 8Department of Pediatric Hematology and Oncology, Collegium Medicum, Nicolaus Copernicus University Torun, Bydgoszcz, Poland; 9The Russian Children’ s Research Hospital, Department of Bone Marrow Transplantation, Moscow, Russia; 10University Hospital Motol, Department of Pediatric Hematology and Oncology, Prague, Czech Republic; 11Bristol Royal Hospital for Children, Department of Pediatric Oncology/BMT, Bristol, UK; 12Hematopoietic Stem Cell Transplantation Unit, Fondazione IRCCS San Gerardo dei Tintori, Monza, Italy; 13Department of Medicine and Surgery, Milano-Bicocca University, Monza, Italy; 14Institut d`Hematologie et d`Oncologie Pediatrique, Lyon, France; 15St Sophia Children’s Hospital Oncology Center, Athens, Greece; 16Onco-Ematologia Pediatrica Centro Trapianti Cellule Staminali, Torino, Italy; 17Copenhagen University Hospital, Rigshospitalet, Department of Children and Adolescents Medicine, Copenhagen, Denmark; 18Department of Clinical Immunology and Transplantation, Jagiellonian University Medical College, Children’s Hospital, Krakow, Poland; 19St. Anna Children’s Hospital, Department of Pediatrics, Medical University of Vienna, Vienna, Austria; 20Princess Maxima Center, Utrecht, the Netherlands; 21Clinica di Oncoematologia Pediatrica, Dipartimento di Pediatria, Padova, Italy; 22IRCCS-Azienda Ospedaliero Universitaria, Bologna, Italy; 23HSCT Unit, Department Hemato-Oncology, IRCSS Istituto G. Gaslini, Genova, Italy; 24Pediatric Hematology/Oncology, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia; 25Pediatric University Teaching Hospital BMT Unit, II Children`s Clinic Bratislava, Slovakia; 26Belorussian Center for Pediatric Oncology and Hematology, Minsk, Russia; 27EBMT Paris Study Unit, Paris, France; 28BMT Unit, Sao Paolo University, Sao Paolo, Brasil and 29University of Regensburg, Regensburg, Germany
*AR and NS contributed equally as first authors.
Correspondence: A. Ruggeri ruggeri.annalisa@hsr.it
Received: October 26, 2023.
Accepted: December 28, 2023. Early view: January 4, 2024.
https://doi.org/10.3324/haematol.2023.284445
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
In children with acute myeloid leukemia (AML) who lack a human leukocyte antigen (HLA) identical sibling, the donor can be replaced with an HLA-matched unrelated donor (MUD) or a haploidentical donor (haplo). We compared outcomes of patients <18 years with AML in first and second complete remission (CR1 & CR2) undergoing a hematopoietic stem cell transplantation (HCT) either with a MUD with anti-thymocyte globulin (ATG) (N=420) or a haplo HCT with post-transplant cyclophosphamide (PT-CY) (N=96) after a myeloablative conditioning regimen (MAC) between 2011 and 2021, reported to the
European Society for Blood and Marrow Transplantation. A matched pair analysis was performed to adjust for differences among groups. The final analysis was performed on 253 MUD and 95 haplo-HCT. In the matched cohort, median age at HCT was 11.2 and 10 years and median year of HCT was 2017 and 2018, in MUD and haplo-HCT recipients, respectively. The risk of grade III-IV acute graft-versus-host disease (aGVHD) was significantly higher in the haplo group (hazard ratio [HR]=2.33, 95% confidence interval [CI]: 1.18-4.58; P=0.01). No significant differences were found in 2 years overall survival (OS; 78.4% vs. 71.5%; HR=1.39, 95% CI: 0.84-2.31; P=0.19), leukemia-free survival (LFS; 72.7% vs. 69.5%; HR=1.22, 95% CI: 0.76-1.95; P=0.41), CI of relapse (RI; 19.3% vs. 19.5%; HR=1.14, 95% CI: 0.62-2.08; P=0.68) non-relapse-mortality (NRM; 8% vs. 11%; HR=1.39, 95% CI: 0.66-2.93; P=0.39) and graft-versus-host free relapse-free survival (GRFS; 60.7% vs. 54.5%, HR=1.38, 95% CI: 0.95-2.02; P=0.09) after MUD and haplo-HCT respectively. Our study suggests that haplo-HCT with PT-CY is a suitable option to transplant children with AML lacking a matched related donor.
Childhood acute myeloid leukemia (AML) is a rare and heterogeneous disease, with an incidence of seven cases per million children younger than 15 years.1 Improvements in genomic characterization, disease response monitoring,2 as well as the introduction of new drugs3 and advances in allogeneic hematopoietic cell transplantation (HCT) techniques4 have resulted in improved outcomes with survival rates reaching 70% in high-income countries. First line treatment approaches for pediatric AML include four or five cycles of myelosuppressive chemotherapy followed by HCT for high-risk patients,5 while HCT is offered in second clinical remission for other cases.5,6 In recent years, the development of HCT from alternative donors, preferably from matched unrelated donors (MUD), has provided the means to offer HCT, when no human leukocyte antigen (HLA)-matched family donor is available. However, in up to 40% of patients a MUD cannot be identified in international donor registries.7 Recently, HCT from relatives sharing only one HLA-haplotype with the recipient (haplo-HCT) has emerged as a suitable alternative, with the great advantage that haplo-donors may be available for almost all patients, in due time, with a flexible schedule and additional cellular therapies being readily available. In haplo-HCT, different techniques to overcome the HLA barrier8-12 have been developed for improving immune reconstitution and graft-versus-leukemia with no excess of graft-versus-host disease (GVHD). In the last years, the use of T-cell replete haplo-HCT with post-transplant cyclophosphamide (PT-CY),8 has rapidly increased across the world, showing identical clinical outcomes to matched donors (MD) in several retrospective studies in adults.8 Many studies demonstrated that PT-CY given after graft infusion can eliminate alloreactive T cells while preserving hematopoietic cells as well as memory and regulatory T cells.13,14 In pediatric patients affected by acute leukemia, promising clinical results have been reported,13,15-19 but specific data on AML are lacking. Here, we compared the results of a haplo-HCT with PTCY to a MUD HCT using anti-thymocyte globulin (ATG) for the treatment of children affected by AML reported to the
European Society for Blood and Marrow Transplantation (EBMT) registry.
Study design and definitions
This multicenter retrospective study was conducted on behalf of the Pediatric Diseases Working Party (PDWP) of EBMT after approval by the institutional review board of the PDWP. Data were collected according to EBMT rules. Patients or legal guardians provided written informed consent for data collection and analysis in accordance with the Declaration of Helsinki. Included were pediatric patients (age <18 years) diagnosed with AML, transplanted in first or second complete remission from 2011 to 2021, who underwent a first allo-HCT using either a 10/10 MUD with ATG or a haploidentical family donor (≥2 recipient-donor HLA mismatch number) in a haplo-HCT with PT-CY. Transplants were performed in 117 EBMT centers. All patients received a myeloablative conditioning (MAC) regimen, and the graft source was bone marrow (BM) or peripheral blood, according to the transplant center policy. In patients receiving ex vivo T-cell depletion, alemtuzumab or a combination of ATG and PT-CY was excluded.
The primary objective was to compare the leukemia-free survival (LFS) of AML patients receiving either a haplo-PTCY or a MUD-ATG HCT. The secondary objectives were the comparison of overall survival (OS), non-relapse mortality (NRM), relapse incidence (RI), incidence of both acute GVHD (aGVHD) and chronic GVHD (cGVHD), GVHD-free/ relapse-free survival (GRFS) in both groups. LFS was defined as time from HCT to first event of relapse or death. OS was defined as the time from HCT to death from any cause. RI was defined as the time from HCT to the first event of leukemia recurrence. Non-relapse mortality (NRM) was defined as death without evidence of relapse. RI and NRM were mutually competing events. GRFS was defined as the time from HCT to the first event among grade 3-4 aGVHD, extensive cGVHD, relapse, and death.20 Grade 2-4
aGVHD and cGVHD were assigned and graded using standard criteria.21, 22 Competing events of aGVHD and cGVHD were relapse and death. Cytogenetics abnormalities were classified according to the genetically defined prognostic stratification of the 2017 European Leukemia Net cytogenetic classification system.1 Neutrophil recovery was defined as the time from HCT to the first of 3 consecutive days with neutrophil counts above 0.5x109/L; platelet recovery was defined as independence from platelet transfusion for at least 7 days with a platelet count of more than >20×109/L. Death and consecutive HCT were competing events.
The overall population included 420 MUD and 96 haplo PTCY. A matched pair analysis was performed to reduce or eliminate confounding factors: the matching included exact match on disease status, cell source and age group at HCT, and nearest match on year of HCT, age at diagnosis, female donor to male recipient, Lansky score and cytomegalovirus match. A maximum of three MUD matched patients was allowed for one haplo PT-CY. There were three controls for 73, two for 12 and one for ten haplo PT-CY. For one haplo PT-CY it was impossible to find a control. Finally, a match could be identified for 253 MUD and 95 haplo PT-CY. Median values with respective interquartile ranges (IQR), were used to express quantitative variables while frequencies and percentages were used for categorical variables. On the unmatched population, differences between MUD and haplo PT-CY on quantitative and qualitative variables have been tested using χ2 and Wilcoxon tests respectively. LFS, OS and GRFS were estimated using the Kaplan-Meier estimator. All outcomes with competing events were estimated using the cumulative incidence function. Median follow-up was estimated using the reverse of the Kaplan-Meier method. All outcomes were censored at last follow-up or at 2 years due to a different follow-up between the groups. Differences in outcomes were tested using a Cox model including a cluster term for each pair. Results are expressed as hazard ratio (HR). Outcomes and HR are presented with their 95% confidence interval (CI). All tests are two-sided with an error rate fixed at 5%. Analyses were done using R statistical software version 4.0.2 (http://www.R-project.org), and matching was performed using the MatchIt package.
Patients and transplant characteristics
Patient and transplant characteristics are summarized in Table 1. Overall, 96 and 420 patients receiving a haplo PTCY or MUD HCT, respectively, fulfilled the inclusion criteria. Median age at transplant was 9.8 (IQR, 3.0-15.2) years for haplo PT-CY and 7.7 (IQR, 2.8-13.6) years for MUD (P=0.03). Patients in the haplo group were transplanted more recently (median 2018 for haplo vs. 2016 for MUD, P<0.01). In both
groups the majority of patients were in CR1 at transplant (72.9% haplo vs. 70.5% MUD; P=0.63).
Cytogenetic risk at diagnosis was not significantly different in the two groups (good: 21.3% vs. 13.6%, intermediate: 48% vs. 44.3%, poor: 30.7% vs. 42.1% in haplo PT-CY and MUD respectively).
Donors were older in the haplo PT-CY than in the MUD cohort: 35.7 years (IQR, 29.9-42.7) versus 27.7 years (IQR, 23.3-34.2; P<0.01). BM was the most frequently used stem cell source in both groups (65.6% for haplo vs. 63.6% for MUD; P=0.71).
Conditioning regimen was MAC, mainly based on busulfan (Bu), with Bu/fludarabine (66.7%) based in the haplo PT-CY and Bu/CY (28.8%) or Bu/CY/melphalan (38.3%) being the most common in MUD.
Post-HCT immunosuppression consisted mainly of cyclosporine (CSA) plus mycophenolate mofetil (MMF) in the haplo PT-CY (52.1%) and CSA plus methotrexate (MTX) in the MUD cohort (68.6%).
The baseline characteristics of the matched cohort are described in Table 2.
Engraftment, acute and chronic graft-versus-host disease
The CI of day 60 neutrophils and day 180 platelet recovery for haplo PT-CY was 90.4% (95% CI: 82-95) and 92.2% (95% CI: 82.6-96.6) while for MUD it was 97.1% (95% CI: 93.9-98.6) and 93.8% (95% CI: 89.1-96.4) respectively.
The 100 day grade 2-4 aGVHD was 36.7% (95% CI: 26.8-46.6) for haplo PT-CY and 28.7% (95% CI: 23.2-34.4) for MUD and CI of grade 3-4 aGvHD was 14.4% (95% CI: 8.1-22.5) versus 6.4% (95% CI: 3.8-9.9), respectively.
2-year CI of cGVHD was 22.4% (95% CI: 13.4-32.8) and 18.5% (95% CI: 13.5-24.1) for haplo PT-CY and MUD respectively; 2-year CI of extensive cGVHD was 6.6% (95% CI: 2.4-13.8) and 8.6% (95% CI: 5.3-13) for haplo PT-CY and MUD (Table 3). According to donor type, there were no statistically significant differences between groups on the incidence of grade 2-4 aGVHD (HR=1.26, 95% CI: 0.84-1.89; P=0.27), cGVHD (HR=1.81, 95% CI: 0.75-4.37; P=0.19) and extensive cGVHD (HR=1.01, 95% CI: 0.25-4.03; P=0.99). The risk of grade 3-4 aGvHD was significantly higher in haplo PT-CY (HR=2.33, 95% CI: 1.18-4.58; P=0.03) (Table 4).
Results of the punctual estimation of outcomes after matched-pair analysis are summarized in Table 3.
The 2-year CI of RI was 19.5% (95% 11.4-29.2) for haplo PTCY versus 19.3% (95% CI: 14.4-29.4) for MUD. The 2-year NRM was 11% (95% CI: 5.2-19.1) versus 8% (95% CI: 5-11.9) after haplo PT-CY and MUD respectively.
The 2-year OS was 71.5% (95% CI: 59.1-80.7) and 78.4% (95% CI: 72.2-83.4), the 2-year LFS was 69.5% (95% CI: 57.7-78.6) and 72.7% (95% CI: 66.3-78.1) and the 2-year GRFS was 54.5% (95% CI: 42.5-65) and 60.7% (95% CI: 53.8-66.9) for
Table 1. Patients and transplant characteristics.
MUD: matched unrelated donors; haplo: haploidentical; PT-CY: post-transplant cyclophosphamide, HCT: hematopoietic stem cell transplantation; IQR: interquartile range; CR: complete remission; BM: bone marrow; PB: peripheral blood; KPS: Karnofsky Performance status, CMV: cytomegalovirus; GVHD: graft-versus-host disease; CSA: cyclosporin; MTX: methotrexate, MMF: mycophenolate mofetile; Bu: busulfan, Flu: fludarabine; Cy: cyclophosphamide; Mel: melphalan; Treo: treosulfan; Pos: positive; Neg: negative.
Table 2. Patients and transplant characteristics in the matched pair cohort.
MUD: matched unrelated donors; haplo: haploidentical; PT-CY: post-transplant cyclophosphamide, HCT: hematopoietic stem cell transplantation; IQR: interquartile range; CR: complete remission; BM: bone marrow; PB: peripheral blood; KPS: Karnofsky Performance status, CMV: cytomegalovirus; CSA: cyclosporin; MTX: methotrexate, MMF: mycophenolate mofetile; Bu: busulfan; Flu: fludarabine; Cy: cyclophosphamide; Mel: melphalan; Treo: treosulfan; Pos: positive; Neg: negative.
Table 3. Survival outcomes after matched-pair analysis.
MUD: matched unrelated donors; haplo: haploidentical; CI: confidence interval; OS: overall survival; LFS: leukemia-free survival; RI: relapse incidence; NRM: non-relapse mortality; a: acute, c: chronic; GVHD: graft-versus-host disease; ext: extensive; GRFS: graft-versus-host free, relapse-free survival.
haplo PT-CY and MUD respectively (Figure 1).
The most common causes of death were disease recurrence (61.9% in the haplo and 59.6% in the MUD group, respectively), infections (28.6% vs. 19.1%), and GVHD (9.5% vs. 8.5%).
There were no statistically significant differences between groups on RI (HR=1.14, 95% CI: 0.62-2.08; P=0.68), NRM (HR=1.39, 95% CI: 0.66-2.93; P=0.39), OS (HR=1.39, 95% CI: 0.84-2.31; P=0.19), LFS (HR= 1.22, 95% CI=0.76-1.95; P=0.41) and GRFS (HR=1.38, 95% CI: 0.95-2.02; P=0.09) (Table 4).
Allogeneic HCT from MUD remains the standard of care in patients who lack a MSD, especially for pediatric patients in which transplant-related toxicities and late effects are particularly relevant with regard to a long life expectancy.23-28 The recent emergence of HLA haplo-HCT extended the availability of donors to most pediatric patients without a MD.29 Significant improvements of supportive care and the technical development of highly effective T-cell depleted and T-cell replete HLA-haploidentical platforms, have resulted in improved outcomes and reduced transplant-related mortality for patients undergoing haplo-HCT,30 independently of the haplo platform used.31,36,37 T-cell-depleted haploidentical platforms have been successfully used despite high costs for graft processing and specialized expertise.11,32,33 Of particular interest, Locatelli et al 10 reported the outcome of a cohort of 80 children with acute leukemia transplanted from a haplo donor after αb T-cell and CD19+ B-cell depletion. All children received a myeloablative conditioning and ATG for GVHD prophylaxis
Table 4. Impact of donor type for outcomes censored at 2 years in the matched cohort.
2-4 1.26 (0.84-1.89) 0.27 aGVHD 3-4 2.33 (1.18-4.58) 0.01 cGVHD 1.81 (0.75-4.37) 0.19
cGVHD 1.01 (0.25-4.03) 0.99
1.38 (0.95-2.02) 0.09
HR: hazard ratio; CI: confidence interval; OS: overall survival; LFS: leukemia-free survival; RI: relapse incidence; NRM: non-relapse mortality; a: acute, c: chronic; GVHD: graft-versus-host disease; ext: extensive; GRFS: graft-versus-host free, relapse-free survival.
and graft rejection; no post-transplant GVHD prophylaxis was given. The 5-year OS was 72% and LFS 71% with no differences in AML and ALL. TRM was 5% and RI was 24%. No patients developed grade 2-4 aGVHD with visceral involvement, grade 3-4 aGVHD or cGVHD. Bertaina et al 12 compared the outcomes of 98 children treated with αb T-cell depletion compared with 127 MUD and 118 mismatched UD (MMUD). Five-year LFS was not significantly different in the three groups (67%, 55%, and 62%, respectively), while a lower incidence of aGVHD was reported in patients treated with T-cell depleted haplo-HCT, compared with MUD and MMUD (2-4 aGVHD was 35% vs. 44% vs. 16%, respectively). On the other hand, T-cell-replete haplo-HCT approaches are mainly based on PT-CY, pioneered by the John Hopkins
group in the adult setting.8,34 This platform is now the most widely adopted strategy because it is cost-effective and easily replicated. In recent years, emerging evidence also supports the use of this platform in the pediatric population with hematological malignancies.19,35 Saglio et al. 15 retrospectively compared the outcome of 23 pediatric patients undergoing haplo-HCT with PT-CY for acute leukemia with patients undergoing HCT from MUD (N=41) and HLA-mismatched unrelated donor (MMUD) (N=26) from a single institution. Five-year OS, NRM and RI were not different for the three groups, confirming that haplo PT-CY is a suitable clinical option for pediatric patients. More recently, Srinivasan et al 16 compared outcomes of haplo-HCT PT-CY with peripheral blood (N=26) to matched sibling donor (MSD) (N=31) and MUD HCT (N=47), both with
BM as stem cell source. Results showed that haplo-HCT PT-CY with peripheral blood had comparable outcomes to BM MSD and MUD HCT. Hong et al. 17 compared the outcomes of children and adolescents with high-risk acute leukemia who underwent haplo-HCT PT-CY (N=35) or MUD HCT (N=45) after a busulfan-based myeloablative conditioning. No differences were observed in the main outcomes. In a subgroup analysis of patients with AML (haplo, N=16; MUD, N=16), the 3-year GRFS, LFS, and OS rates in haplo-HCT and MUD groups were 80.8% versus 61.9%, 87.1% versus 73.9%, and 93.8% versus 85.6%, respectively. Here, we report the largest series of children with AML treated with haplo PT-CY reported to the EBMT registry. Our results are in line with the previous single center reports and confirmed the overall feasibility of the PT-CY approach
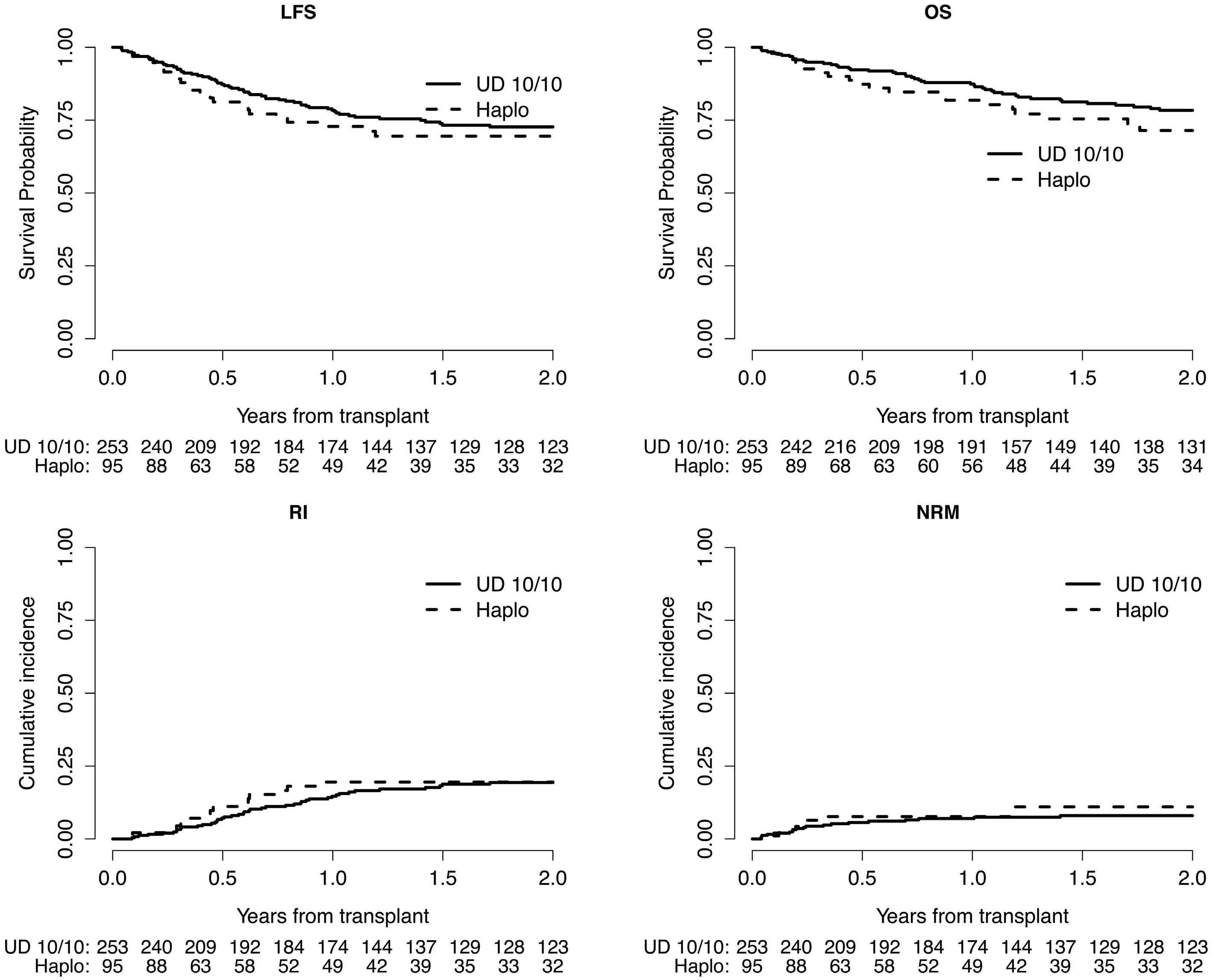
Figure 1. Two-years survival outcomes after matched unrelated donor and haploidential hematopoietic cell transplantation after matched pair analysis. Two-year leukemia-free survival (LFS), overall survival (OS), relapse incidence (RI) and non-relapse mortality (NRM) after matched unrelated donor (MUD) and haploidential hematopoietic cell transplantation in children with acute myleoid leukemia after matched pair analysis.
in pediatric AML. Importantly, in our cohort haplo HCT was associated with higher incidence of grade 3-4 aGVHD and this could be related to the graft source itself, namely the content of CD3+ cells in the poeripheral blood stem cells as well to the differences in the overall GVHD prophylaxis in the two group. Importantly MUD recipients received ATG which is a key factor associated with reduced risk of GVHD. Whenever the combination of ATG and PTCY could be considered in the pediatric setting deserves further investigation. Overall, no differences in pivotal outcome parameters were found between haplo PT-CY and MUD. The results were consistent across the different disease status when we checked for interaction between the disease status and the donor type. We are aware of the limitations of this study, namely the retrospective nature, the short follow-up, due to the recent use of PT-CY in this setting and the limited data on infection and immune reconstitution due to the registry based analysis. However, we believe that our results are important and highlight the feasibility of this approach also in children. How the PTCY approach could be adopted also in the unrelated donor setting in the pediatric HCT deserves further investigation. Eligibility criteria for HCT for each cooperative group are beyond the purpose of this multinational retrospective study. Nevertheless, eligibility for haplo-HCT may be either the same as for MUD or more restricted to higher risk patients only. Therefore, in case any unknown prognostic feature could not be adjusted within the matched pair analysis, the worst risk distribution would affect the outcome of the haplo PT-CY more than the MUD cohort. In the pair match analysis, the year of transplant was not considered as exact matching but using a propensity score. This resulted in a median year of transplant of 2017 and 2018 for MUD and haplo respectively. Consequently, the median follow-up
1. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
2. Segerink WH, de Haas V, Kaspers GJL. Measurable residual disease in pediatric acute myeloid leukemia: a systematic review. Expert Rev Anticancer Ther. 2021;21(4):451-459.
3. Egan G, Chopra Y, Mourad S, Chiang KY, Hitzler J. Treatment of acute myeloid leukemia in children: a practical perspective. Pediatr Blood Cancer. 2021;68(7):e28979.
4 Reinhardt D, Antoniou E, Waack K. Pediatric acute myeloid leukemia - past, present, and future. J Clin Med. 2022;1(3):504.
5. Rubnitz JE, Kaspers GJL. How I treat pediatric acute myeloid leukemia. Blood.2021;138(12):1009-1018.
6. Horan JT, Alonzo TA, Lyman GH, et al. Impact of disease risk on efficacy of matched related bone marrow transplantation for pediatric acute myeloid leukemia: the Children’s Oncology Group. J Clin Oncol. 2008;26(35):5797-5801.
was different in the two groups. In order to solve this issue and make the groups more comparable, we censored the outcomes at 2 years preventing us to provide results at a longer period to avoid imbalance between the groups. Such a haplo platform may enlarge access to HCT to virtually all eligible pediatric patients with AML. Furthermore, the prompt availability and the flexibility of a family member may be crucial in the challenging HCT scheduling of rapidly evolving pediatric malignancies, such as high-risk AML. Albeit MUD HCT remains the standard of care, our study confirmed in a large international analysis the comparable results of haplo-HCT with PT-CY and MD HCT, so that pediatric patients with AML who either lack a MD or cannot afford a MUD HCT, can be safely transplanted without delay.
No conflicts of interest to disclose.
Contributions
AR and SC designed the study. NS and AR wrote the manuscript. JEG performed the statistical analysis. KK, MA, LZ, KC, ES, PS, CB, AB, YB, JP, FG, MI, JG, CP, BV, AB, AP, MF, IG, IB, OA, AD, and VR provided cases for the study. All authors edited and approved the manuscript.
We thank the clinical staff and investigators of the EBMT centers involved in this research and especially thank the patients who participated in the study.
Data-sharing statement
Data cannot be shared unless a specific request is sent to the EBMT.
7 Gragert L, Eapen M, Williams E, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. Registry. N Engl J Med. 2014;371(4):339-348.
8. Luznik L, O’Donnell PV, Symons HJ, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-iose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14(6):641-650.
9 Di Bartolomeo P, Santarone S, De Angelis G, et al. Haploidentical, unmanipulated, G-CSF-primed bone marrow transplantation for patients with high-risk hematologic malignancies. Blood. 2013;121(5):849-857.
10 Locatelli F, Merli P, Pagliara D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after ab T-cell and B-cell depletion. Blood. 2017;130(5):677-685.
11. Massei MS, Capolsini I, Perruccio K, et al. HLA-haploidentical transplantation with regulatory and conventional T-cells adoptive immunotherapy in pediatric patients with high-risk acute leukemia. Transplant Cell Ther. 2021;58(5):526-533.
12. Bertaina A, Zecca M, Buldini B, et al. Unrelated donor vs HLAhaploidentical a/b T-cell- and B-cell-depleted HSCT in children
with acute leukemia. Blood. 2018;132(24):2594-2607.
13. Symons HJ, Zahurak M, Cao Y, et al. Myeloablative haploidentical BMT with posttransplant cyclophosphamide for hematologic malignancies in children and adults. Blood Adv. 2020;4(16):3913-3925.
14 Bolaños-Meade J, Hamadani M, Wu J, et al. Posttransplantation cyclophosphamide-based graft-versus-host disease prophylaxis. N Engl J Med. 2023;388(25):2338-2348.
15. Saglio F, Berger M, Spadea M, et al. Haploidentical HSCT with post transplantation cyclophosphamide versus unrelated donor HSCT in pediatric patients affected by acute leukemia. Bone Marrow Transplant. 2021;56(3):586-595.
16. Srinivasan A, Raffa E, Wall DA, et al. Outcome of haploidentical peripheral blood allografts using post-transplantation cyclophosphamide compared to matched sibling and unrelated donor bone marrow allografts in pediatric patients with hematologic malignancies: a single-center analysis. Transplant Cell Ther. 2022;28(3):158.e1-158.e9.
17 Hong KT, Park HJ, Kim BK, An HY, Choi JY, Kang HJ. Posttransplantation cyclophosphamide-based haploidentical versus matched unrelated donor peripheral blood hematopoietic stem cell transplantation using myeloablative targeted busulfanbased conditioning for pediatric acute leukemia. Transplant Cell Ther. 2022;28(4):195.e1-195.e7.
18. Berger M, Lanino E, Cesaro S, et al. Feasibility and outcome of haploidentical hematopoietic stem cell transplantation with post-transplant high-dose cyclophosphamide for children and adolescents with hematologic malignancies: an AIEOP-GITMO retrospective multicenter study. Biol Blood Marrow Transplant. 2016;22(5):902-909.
19. Ruggeri A, Galimard JE, Paina O, et al. Outcomes of unmanipulated haploidentical transplantation using posttransplant cyclophosphamide (PT-Cy) in pediatric patients with acute lymphoblastic leukemia. Transplant Cell Ther. 2021;27(5):424.e1-424.e9.
20 Ruggeri A, Labopin M, Ciceri F, Mohty M, Nagler A. Definition of GvHD-free, relapse-free survival for registry-based studies: an ALWP-EBMT analysis on patients with AML in remission. Bone Marrow Transplant. 2016;51(4):610-611.
21. Martino R, Romero P, Subirá M, et al. Comparison of the classic Glucksberg criteria and the IBMTR Severity Index for grading acute graft-versus-host disease following HLA-identical sibling stem cell transplantation. Bone Marrow Transplant. 1999;24(3):283-287.
22. Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 Diagnosis and Staging Working Group Report. Biol Blood Marrow Transplant. 2015;21(3):389-401.
23. Willemze AJ, Geskus RB, Noordijk EM, Kal HB, Egeler RM, Vossen JM. HLA-identical haematopoietic stem cell transplantation for acute leukaemia in children: less relapse with higher biologically effective dose of TBI. Bone Marrow Transplant. 2007;40(4):319-327.
24. Peters C, Matthes-Martin S, Poetschger U, et al. Stem-cell transplantation in children with acute lymphoblastic leukemia: a prospective international multicenter trial comparing sibling donors with matched unrelated donors-The ALL-SCT-BFM-2003
trial. J Clin Oncol. 2015;33(11):1265-1274.
25. Saarinen-Pihkala UM, Gustafsson G, Ringdén O, et al. No disadvantage in outcome of using matched unrelated donors as compared with matched sibling donors for bone marrow transplantation in children with acute lymphoblastic leukemia in second remission. J Clin Oncol. 2001;19(14):3406-3414.
26. Gu Z, Wang L, Yuan L, et al. Similar outcomes after haploidentical transplantation with post-transplant cyclophosphamide versus HLA-matched transplantation: a meta-analysis of case-control studies. Oncotarget. 2017;29;8(38):63574-63586.
27. Shaw PJ, Kan F, Ahn KW, et al. Outcomes of pediatric bone marrow transplantation for leukemia and myelodysplasia using matched sibling, mismatched related, or matched unrelated donors. Blood. 2010;116(20):4360-4367.
28. Leung W, Campana D, Yang J, et al. High success rate of hematopoietic cell transplantation regardless of donor source in children with very high-risk leukemia. Blood. 2011;118(2):223-230.
29 Passweg JR, Baldomero H, Chabannon C, et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: monitoring of activities and trends over 30 years. Bone Marrow Transplant. 2021;56(7):1651-1664.
30 Shah RM. Contemporary haploidentical stem cell transplant strategies in children with hematological malignancies. Bone Marrow Transplant. 2021;56(7):1518-1534.
31. Algeri M, Merli P, Locatelli F, Pagliara D. The role of allogeneic hematopoietic stem cell transplantation in pediatric leukemia. J Clin Med. 2021;10(17):3790.
32. Bertaina A, Merli P, Rutella S, et al. HLA-haploidentical stem cell transplantation after removal of αb+ T and B cells in children with nonmalignant disorders. Blood. 2014;124(5):822-826.
33. Merli P, Algeri M, Li Pira G, et al. Alpha/beta T-cell and B-cell depletion HLA-haploidentical hematopoietic stem cell transplantation is an effective treatment for children/young adults with acute leukemia. Blood. 2018;132(Suppl 1):2169.
34 Bashey A, Zhang MJ, McCurdy SR, et al. Mobilized peripheral blood stem cells versus unstimulated bone marrow as a graft source for T-cell–replete haploidentical donor transplantation using post-transplant cyclophosphamide. J Clin Oncol. 2017;35(26):3002-3009.
35. Jaiswal SR, Chakrabarti A, Chatterjee S, et al. Haploidentical peripheral blood stem cell transplantation with posttransplantation cyclophosphamide in children with advanced acute leukemia with fludarabine-, busulfan-, and melphalanbased conditioning. Biol Blood Marrow Transplant. 2016;22(3):499-504.
36. Pérez-Martínez A, Ferreras C, Pascual A, et al. Haploidentical transplantation in high-risk pediatric leukemia: a retrospective comparative analysis on behalf of the Spanish working Group for bone marrow transplantation in children (GETMON) and the Spanish Grupo for Hematopoietic Transplantation (GETH). Am J Hematol. 2020;95(1):28-37.
37. Shelikhova L, Ilushina M, Shekhovtsova Z, et al. αb T celldepleted haploidentical hematopoietic stem cell transplantation without antithymocyte globulin in children with chemorefractory acute myelogenous leukemia. Biol. Blood Marrow Transplant. 2019;25(5):e179-e182.
Mika Casey,1 Carol Lee,1 Sharon M. Hoyte,1 Rebecca L. Johnston,1 Wing Yu Kwok,1 Soi Cheng Law,2 Maher K. Gandhi,2 Simon J. Harrison3,4 and Kyohei Nakamura1
1Cancer Research Program, QIMR Berghofer Medical Research Institute, Brisbane, Queensland; 2Mater Research, University of Queensland, Brisbane, Queensland; 3Clinical Hematology, Peter MacCallum Cancer Center and Royal Melbourne Hospital, Melbourne, Victoria and 4Sir Peter MacCallum, Department of Oncology, University of Melbourne, Parkville, Victoria, Australia
Correspondence: K. Nakamura
kyohei.nakamura@qimrberghofer.edu.au
Received: October 30, 2023.
Accepted: January 12, 2024. Early view: January 25, 2024.
https://doi.org/10.3324/haematol.2023.284435
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
T-cell-engaging bispecific antibody (T-BsAb, also known as BiTE) therapy has emerged as a powerful therapeutic modality against multiple myeloma. Given that T-BsAb therapy redirects endogenous T cells to eliminate tumor cells, reinvigorating dysfunctional T cells may be a potential approach to improve the efficacy of T-BsAb. While various immunostimulatory cytokines can potentiate effector T-cell functions, the optimal cytokine treatment for T-BsAb therapy is yet to be established, partly due to a concern of cytokine release syndrome driven by aberrant interferon (IFN)-γ production. Here, we functionally screen immunostimulatory cytokines to determine an ideal combination partner for T-BsAb therapy. This approach reveals interleukin (IL)-21 as a potential immunostimulatory cytokine with the ability to augment T-BsAb-mediated release of granzyme B and perforin, without increasing IFN-γ release. Transcriptome profiling and functional characterization strongly support that IL-21 selectively targets the cytotoxic granule exocytosis pathway, but not pro-inflammatory responses. Notably, IL-21 modulates multiple steps of cytotoxic effector functions including upregulation of co-activating CD226 receptor, increasing cytotoxic granules, and promoting cytotoxic granule delivery at the immunological synapse. Indeed, T-BsAb-mediated myeloma killing is cytotoxic granule-dependent, and IL-21 priming significantly augments cytotoxic activities. Furthermore, in vivo IL-21 treatment induces cytotoxic effector reprogramming in bone marrow T cells, showing synergistic anti-myeloma effects in combination with T-BsAb therapy. Together, harnessing the cytotoxic granule exocytosis pathway by IL-21 may be a potential approach to achieve better responses by T-BsAb therapy.
Multiple myeloma, a plasma cell neoplasm, has historically been recognized as an incurable disease. Over the past decade, the treatment landscape of multiple myeloma has dramatically evolved with T-cell redirection immunotherapies such as T-cell-engaging bispecific antibody (T-BsAb, also known as BiTE) therapy and chimeric antigen receptor (CAR) T-cell therapy.1-3 T-BsAb therapy can redirect endogenous polyclonal T cells to recognize and eliminate tumor cells by simultaneously engaging target antigens on tumor cells and CD3 on T cells. As the cancer-immunity cycle is frequently dysregulated in multiple myeloma,4 T-BsAb therapy is recognized as a potential approach to stimulate adaptive immunity to control multiple myeloma. Recently, T-BsAb therapies targeting B-cell maturation antigen (BCMA), G-protein coupled receptor family C group 5 member
D, and Fc Receptor-Like 5 have shown impressive clinical responses in heavily pretreated patients with relapsed/ refractory multiple myeloma.1,2,5
Despite impressive clinical responses, treatment failure and relapse remain major obstacles. Still, it remains largely unknown how we can overcome therapeutic resistance mediated by loss of target antigen, immunosuppression, and T-cell hyporesponsiveness.4,6,7 Given that T-BsAb stimulate endogenous T cells and that T cells are functionally impaired in the myeloma bone marrow (BM),4,8-10 reprogramming dysfunctional T cells might be necessary to achieve better disease control by T-BsAb therapy. Indeed, in a recent preclinical study, we showed that priming of cytotoxic lymphocytes by innate immune activation can improve T-BsAb efficacy, supporting the importance of harnessing effector T-cell functions prior to T-BsAb therapy.11 Immunostimulatory cytokines including common γ-chain
cytokines (IL-2, IL-7, IL-15, and IL-21) and interferon (IFN)-γ-inducing cytokines (IL-12 and IL-18) are recognized as potent adjuvants for cancer immunotherapy by their ability to support either T-cell proliferation or modulating effector functions.12-14 Currently, therapeutic administration of these recombinant cytokines is widely being tested in combination with immune checkpoint inhibitors and other immunotherapy.12 While cytokine-based therapy can be a possible combination partner for T-cell redirection therapy, overstimulation of T cells can drive activation-induced cell death and increase the risk of severe immune-related adverse events, such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome. Activated T cells release pro-inflammatory cytokines including IFN-γ, tumor necrosis factor (TNF)-α, and granulocyte-macrophage colony-stimulating factor (GMCSF), all of which are known to fuel the vicious cycle of CRS by stimulating monocytes/macrophages to produce IL-1b and IL-6.15-17 Above all, IFN-γ plays a crucial role in the immunopathology of CRS, as blockade or deletion of IFN-γ can ameliorate cytokine-related toxicities in CAR T-cell therapy.18,19 By contrast, the perforin/granzyme-mediated cytotoxic granule pathway negatively regulates macrophage activation syndrome (MAS) and hemophagocytic lymphohistiocytosis (HLH) by killing activated monocytes/ macrophages. Indeed, familial HLH is seen in patients with mutations in the perforin gene (PRF1) or genes related to cytotoxic granule exocytosis.20,21 More recently, Ishii et al. showed that adoptive transfer of perforin-deficient CAR T cells, but not wild-type CAR T cells, triggers the MAS/ HLH-like preclinical phenotype with the excessive release of IL-1 family cytokines.22 These results provide evidence that the cytotoxic granule pathway negatively regulates aberrant macrophages inflammatory responses during T-cell redirection immunotherapy. Importantly, accumulated evidence supports that IFN-γ is dispensable for the efficacy of CAR T therapy against hematological malignancies, but not solid malignancies.18,19,23 Thus, we hypothesize that a cytokine-based therapy selectively harnessing the cytotoxic granule pathway might be an ideal strategy to achieve better responses by T-BsAb therapy. By performing a functional screening of immunostimulatory cytokines, this study aims to determine the optimal approach for cytokine-mediated priming of effector T cells and to establish proof-of-concept for improving the efficacy of T-BsAb therapy against multiple myeloma.
Details on experimental procedures and materials are included in the Online Supplementary Appendix
Multiple myeloma cell lines
Human multiple myeloma cell lines (JJN-3 cells, KMS-11
cells, and RPMI8226)24 were cultured in RPMI1640 media supplemented with sodium pyruvate, non-essential amino acids, penicillin/streptomycin, and 10% heat-inactivated fetal bovine serum (complete RPMI [cRPMI]) Luciferase was transduced by retrovirus transfection of the MSCVmCherry-IRES-luc2 vector, as described previously.25 Mycoplasma negativity was confirmed by the Mycoplasma Detection Kit. The transplantable Vk14451 myeloma cells expressing enhanced green fluorescent protein (EGFP) were maintained as previously described.11,26
Primary cells and clinical samples
Peripheral blood mononuclear cells (PBMC) were isolated from buffy coats from healthy donors using LymphoprepTM density gradient medium. CD8 T cells were magnetically isolated by EasySepTM Human CD8+ T Cell Isolation Kit. Clinical samples from patients with newly diagnosed multiple myeloma were collected at the Princess Alexandra Hospital, Brisbane. The study was approved by the QIMR Berghofer Human Research Ethics Committee (P2125) and by the Metro South Hospital and Health Service Human Research Ethics Committee (9448).
Screening assays
PBMC (4×105) from healthy donors and patients with newly diagnosed multiple myeloma were co-cultured with JJN-3 myeloma cells (1×105) in the presence of anti-BCMA T-BsAb (0.1 μg/mL) and one of the following recombinant human immunostimulatory cytokines: rIL-2 (500 U/mL), rIL-7, rIL-12, rIL-15, rIL-18 and rIL-21 (50 ng/mL). The levels of granzyme B, perforin, IFN-γ, TNF-α, and GM-CSF in culture supernatants were determined by enzyme-linked immunosorbant assay (ELISA) 3 days after co-culture. Results were calculated as fold changes relative to control (T-BsAb stimulation in the absence of immunostimulatory cytokines).
T-cell-engaging bispecific antibody assays
For T-cell proliferation assays, PBMC were labeled with CellTraceTM Violet (CTV) and co-cultured with JJN-3 myeloma cells (1×105) in cRPMI supplemented with recombinant IL-2 (rIL-2, 100 U/mL) at the effector-to-target ratio (E: T ratio) of 4:1. These cells were stimulated with the indicated concentrations of rIL-21 and anti-BCMA T-BsAb for 3 days, and CTV-dilution in live CD8 T cells was measured by flow cytometry. In some experiments, unlabeled PBMC were used to examine expression levels of CD226 and CD28 in CD8 T cells. The levels of cytotoxic granules (granzyme B and perforin) and cytokines (IFN- γ , TNF- α , and GM-CSF) in culture supernatants were measured by ELISA. For ex vivo stimulation in primary samples, bulk bone marrow (BM) mononuclear cells were reconstituted in cRPMI supplemented with rIL-2 (50 U/mL) (8×105/mL). These cells were stimulated with anti-BCMA T-BsAb (0.2 µg/mL) in the presence or absence of rIL-21 (100 ng/mL) for 3 days.
C57BL/6 wild-type mice were bred and maintained in house. Mice were intravenously challenged with 2×106 Vk14451 cells expressing EGFP. Tumor-bearing mice with paraproteinemia were intraperitoneally (i.p.) treated with recombinant murine IL-21 (100 μg, i.p.), according to a dosing schedule reported previously.27,28 In some experiments, tumor-bearing mice were treated with anti-mouse BCMA T-BsAb (25 μg, i.p.) either alone or in combination with rIL-21. Mice were monitored over time for clinical endpoints. All experiments were approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee (P3533).
Statistical differences were tested with GraphPad Prism 9 software. For comparisons between two groups, a Mann-Whitney U test or paired t test was used. For multiple comparisons, a Friedman test, a repeated measures ANOVA, or Kruskal-Wallis test was used, followed by indicated post hoc tests. Differences in survival were evaluated with a Mantel-Cox test. P<0.05 was considered to be statistically significant (*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001).
Functional screening of immunostimulatory cytokines to determine an ideal combination partner for T-cellengaging bispecific antibody therapy
We sought to identify an immunostimulatory cytokine regulating the cytotoxic granule pathway with negligible impacts on the production of IFN-γ. To this end, PBMC from five healthy donors and five patients with newly diagnosed multiple myeloma were co-cultured with JJN-3 myeloma cells in the presence of anti-BCMA T-BsAb and one of the following immunostimulatory cytokines: rIL-2, rIL-7, rIL-12, rIL-15, rIL-18, or rIL-21 (Figure 1A). After 3 days of co-culture, levels of effector molecules in culture supernatants were determined. The release of granzyme B was enhanced by the addition of rIL-12, rIL-18, or rIL-21, whereas a significant increase in perforin levels was only seen by rIL-21 (Figure 1B, C). Consistent with the ability to drive type 1 immune responses,29 both rIL-12 and rIL-18 dramatically augmented the release of IFN-γ (Figure 1D). However, notably, treatment with rIL-21 showed a negligible impact on IFN-γ production (Figure 1D). Moreover, treatment with rIL-21 did not increase levels of TNF-α and GM-CSF (Figure 1E, F). These results suggest that rIL-21 might be a potential adjuvant for enhancing the cytotoxic granule pathway, but not pro-inflammatory responses.
Dose-dependent effects of IL-21 on CD8 T cells stimulated with T-cell-engaging bispecific antibody
In order to validate the results from the immunostimulatory cytokine screening, we examined the dose-dependent
effects of rIL-21 on T-BsAb-induced cytokine production from eight different healthy donor-derived PBMC. Indeed, treatment with rIL-21, at 20 ng/mL or higher, significantly increased the release of granzyme B and perforin without increasing levels of pro-inflammatory cytokines IFN- γ, TNF, and GM-CSF (Figure 2A). Intriguingly, levels of IFN-γ and GM-CSF were modestly but significantly reduced in the presence of rIL-21 (Figure 2A), further supporting the differential impact of IL-21 on T-BsAb-induced release of cytotoxic granules and pro-inflammatory cytokines. Next, we addressed whether rIL-21 could modulate the activation of CD8 T cells. The upregulation of an early activation marker CD69 was detectable on CD8 T cells 5 hours after T-BsAb stimulation, especially at high concentrations of T-BsAb (Figure 2B). Frequencies of CD69-expressing CD8 T cells significantly increased in the presence of rIL-21, even at suboptimal concentrations of T-BsAb, suggesting that treatment with rIL-21 lowered the activation threshold in CD8 T cells (Figure 2B). By contrast, treatment with rIL-21 showed negligible impacts on T-BsAb-mediated proliferation (Figure 2C). Intriguingly, treatment with rIL21 upregulates the expression levels of CD226 and CD28, key receptors for tumor recognition and co-stimulation, respectively (Figure 2D).30-32 Together, these results suggest that IL-21 modulates selective effector functions, rather than broadly stimulates CD8 T cells.
Treatment with IL-21 transcriptionally regulates the cytotoxic granule pathway in CD8 T cells stimulated with T-cell-engaging bispecific antibody
It is appreciated that inflammatory cytokines act as a third signal for functional maturation of CD8 T cells.29 In order to obtain a comprehensive profiling of effector programs regulated by IL-21, we performed bulk RNA sequencing (RNA-seq) of T-BsAb-stimulated CD8 T cells from six healthy donors in the absence (control) or presence of rIL-21 using a paired design (Figure 3A; Online Supplementary Figure 1A; Online Supplementary Table S1). Principal components analysis (PCA) revealed samples clustered by donor along principal component (PC) 1 and PC2, whilst PC2 versus PC3 separated control CD8 T cells and IL-21-primed CD8 T cells (Figure 3B). Differential expression analysis between IL-21primed and control CD8 T cells identified 2,609 differentially expressed genes, of which 1,280 were upregulated, and 1,329 were downregulated (false discovery rate <0.05; Online Supplementary Table S2). Reactome pathway analysis of the genes upregulated in IL-21-primed CD8 T cells revealed enrichment of neutrophil degranulation (granule exocytosis) as well as Rho and other GTPase pathways (Figure 3C; Online Supplementary Table S3), key signaling pathways for cytoskeletal reorganization in cytotoxic T cells.33 By contrast, pro-inflammatory pathways such as IFN, TNF, and IL-1 signaling pathways were enriched based on Reactome pathway analysis of the genes downregulated in IL-21-primed CD8 T cells (Online Supplementary Figure
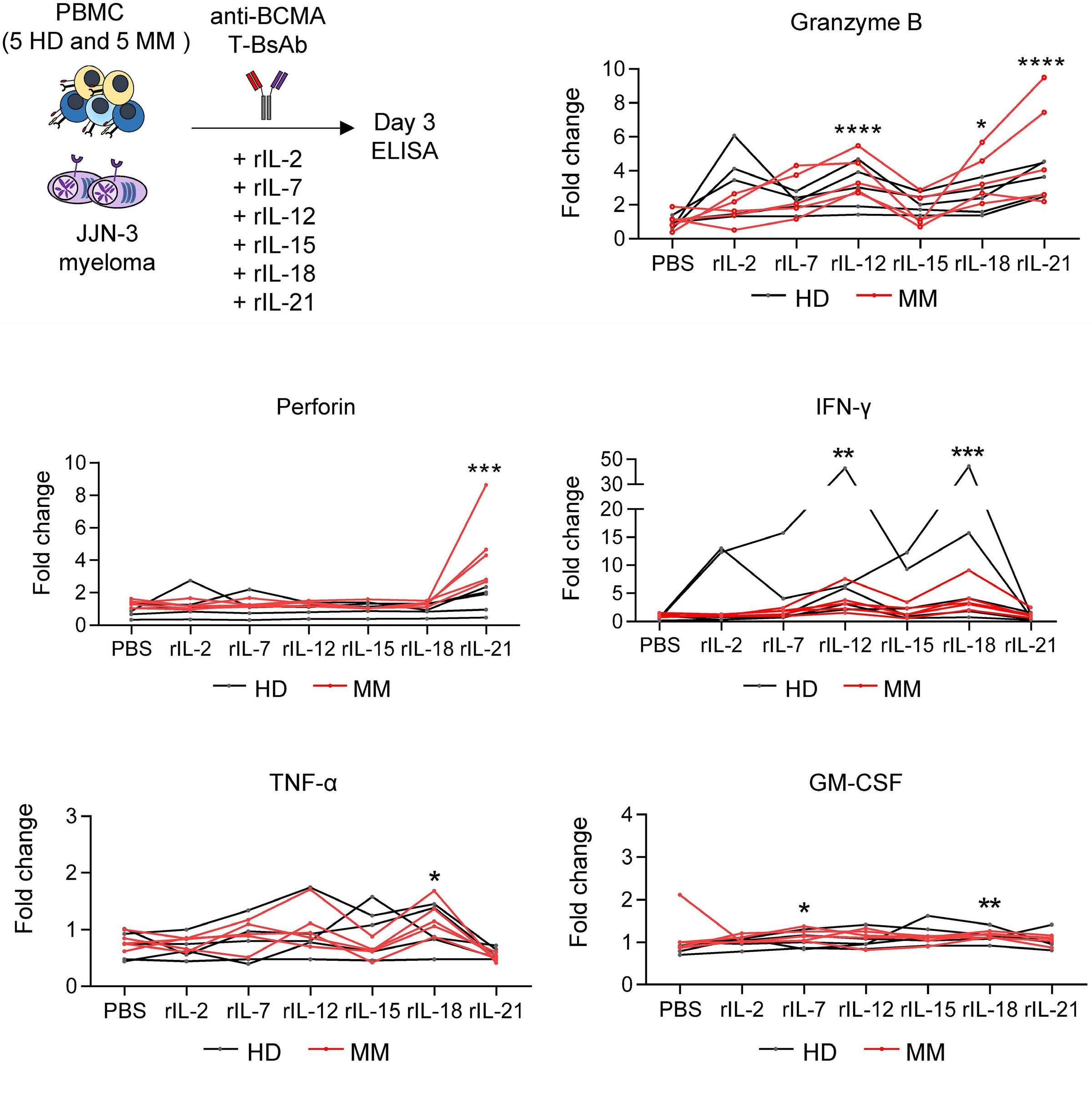
Figure 1. Functional screening of immunostimulatory cytokines to determine an ideal combination partner for T-cell-engaging bispecific antibody therapy. (A) Schematic illustrates the screening of immunostimulatory cytokines. Peripheral blood mononuclear cells (PBMC, 4×105) from healthy donors (HD, N=5) and patients with multiple myeloma (MM, N=5) were co-cultured with JJN-3 myeloma cells (1×105) in the presence of T-cell-engaging bispecific antibody targeting B-cell maturation antigen (anti-BCMA T-BsAb, 0.1 µg/mL) and each of following recombinant immunostimulatory cytokines: recombinant interleukin-2 (rIL-2) (500 U/mL), rIL-7, rIL-12, rIL-15, rIL-18 and rIL-21 (50 ng/mL). (B-F) Individual graphs showing the levels of granzyme B (B), perforin (C), interferon-γ (IFN-γ) (D), tumor necrosis factor-α (TNF-α) (E), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (F) in culture supernatants 3 days after stimulation, determined by enzyme-linked immunosorbant assay (ELISA). Data are shown as fold changes, relative to T-BsAb stimulation without cytokine treatment. Results from 1 experiment are shown, and differences were tested for statistical significance using a Friedman test with a Dunn’s multiple comparisons test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
S1B; Online Supplementary Table S4), supporting the cytotoxic granule pathway as a key target of IL-21. Indeed, genes encoding cytotoxic granules (GZMB, GZMH, NKG7, and GNLY) were significantly increased in IL-21-primed CD8 T cells (Figure 3D; Online Supplementary Table S2). Together, IL-21 transcriptionally induces the cytotoxic effector program in CD8 T cells.
IL-21 priming augments cytotoxic granule exocytosis and myeloma-killing activity by T-cell-engaging bispecific antibody
The results from transcriptome profiling strongly suggest that IL-21 priming could enhance cytotoxic granule-dependent tumor-killing activities. Thus, we assessed T-BsAb-mediated cytotoxic activities of CD8 T cells primed with or without rIL-21 (Figure 4A). Indeed, IL-21 priming markedly augmented T-BsAb-mediated cytotoxicity against three
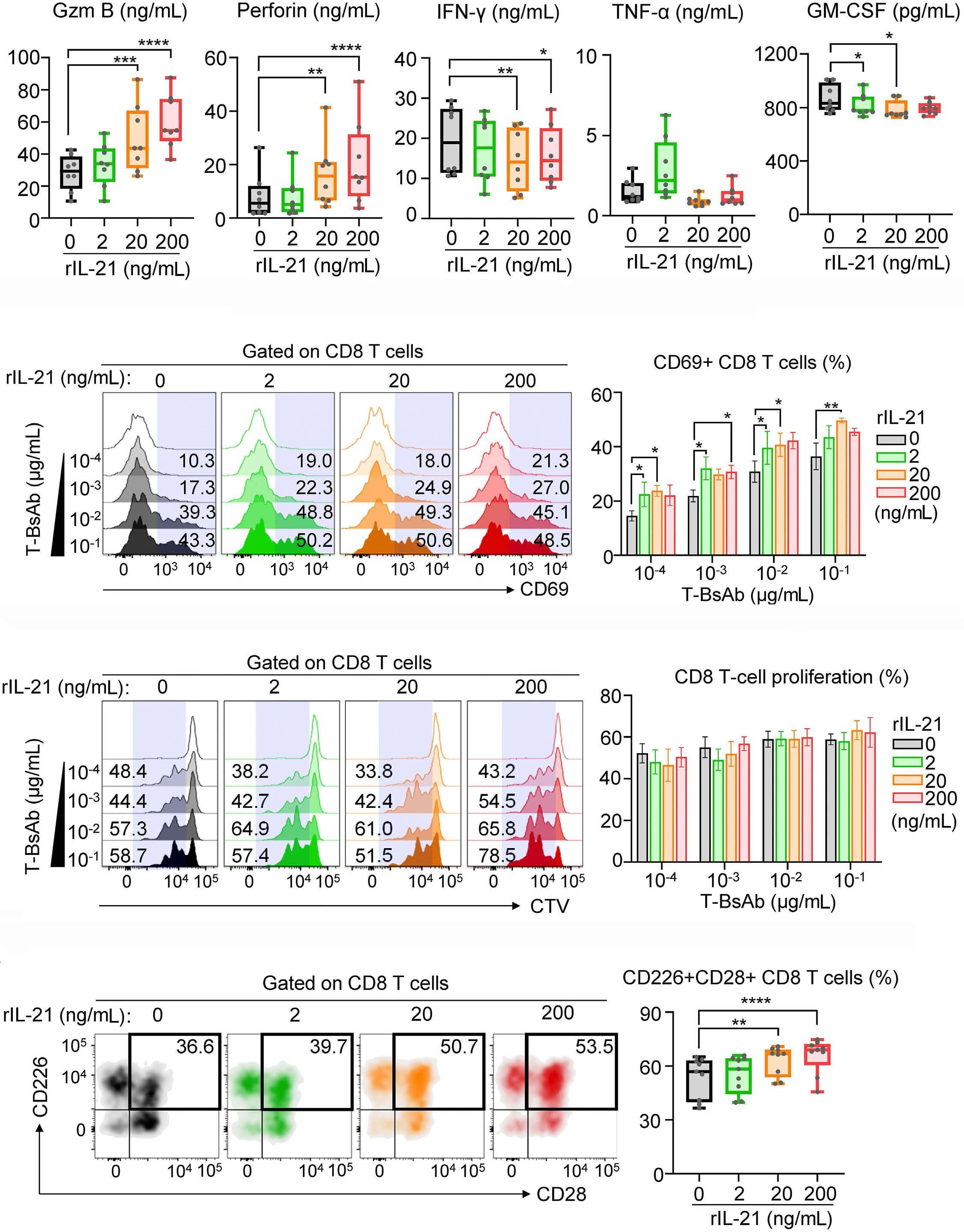
Figure 2. Treatment with recombinant IL-21 augments cytotoxic granule release without affecting the production of pro-inflammatory cytokines. (A) Peripheral blood mononuclear cells from healthy donors and JJN-3 myeloma were co-cultured for 3 days in the presence of T-cell-engaging bispecific antibody targeting B-cell maturation antigen (anti-BCMA T-BsAb) and indicated concentrations of recombinant interleukin-21 (rIL-21). Box-and-whisker plots showing levels of indicated effector proteins and cytokines in culture supernatants (N=8). (B) Representative histograms (left) and bar graphs (right), showing expression levels of CD69 in CD8 T cells 5 hours after stimulation with indicated concentrations of anti-BCMA T-BsAb and rIL-21. Data are shown as mean ± standard error of mean (N=5). (C) CTV-labeled peripheral blood mononuclear cells and JJN-3 myeloma cells were co-cultured for 3 days in the presence of indicated concentrations of anti-BCMA T-BsAb and rIL-21. Representative histograms (left) and bar graphs (right) showing CD8 T-cell proliferation (N=4). Data are shown as mean ± standard error of mean. (D) Representative histograms (left) and box-and-whisker plots (right) showing expression levels of CD28 and CD226 in CD8 T cells after 3 days of co-culture in the presence of T-BsAb (0.1 µg/mL) and indicated concentrations of IL-21 (N=9). Data were pooled from 2 experiments, and differences were tested for statistical significance using a repeated measures ANOVA with a post hoc Dunnett’s multiple comparisons test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. CTV: CellTrace™ Violet; Gzm B: granzyme B; INF-γ: interferon-γ; TNF-α: tumor necrosis factor-α
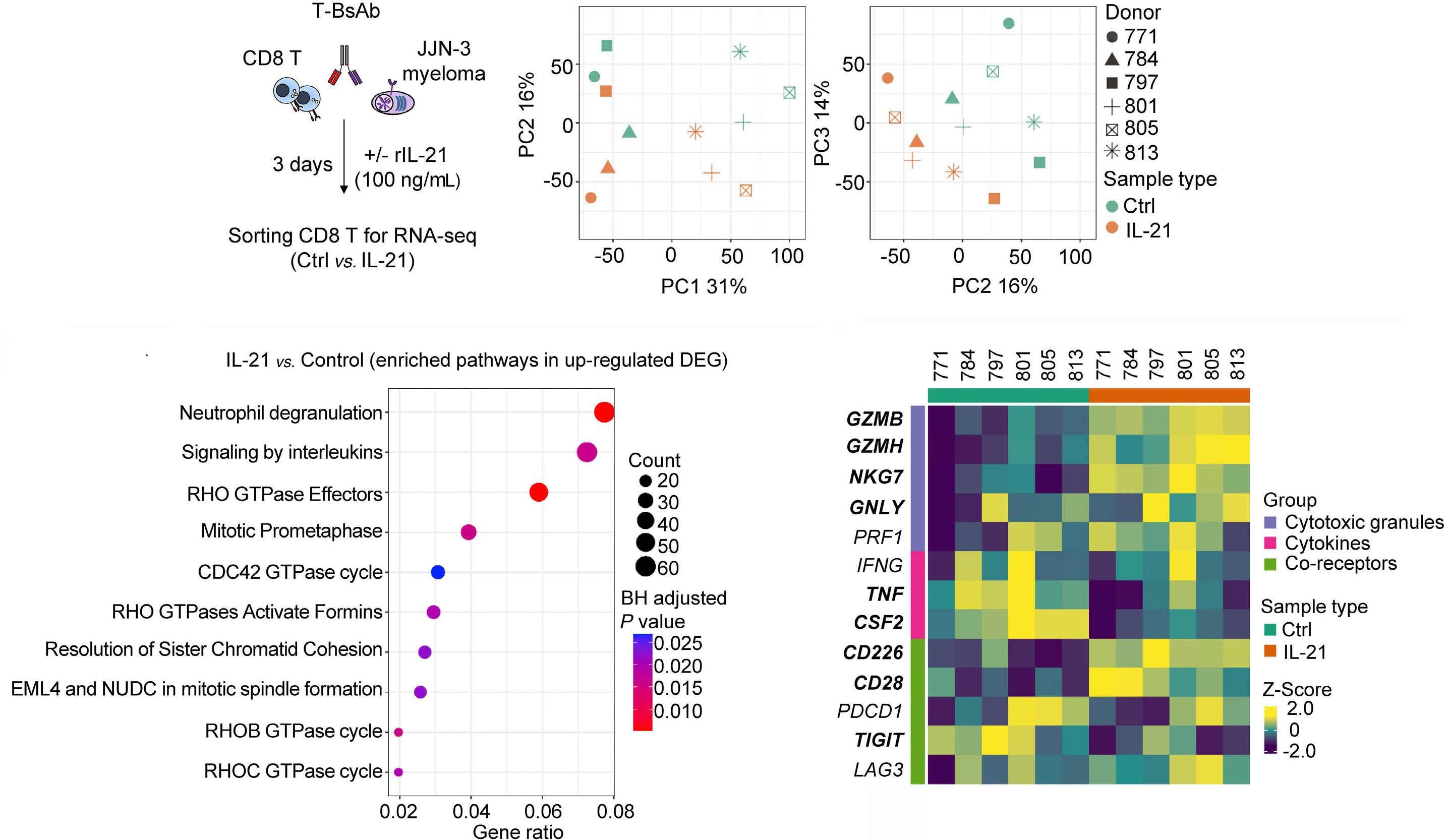
Figure 3. IL-21 transcriptionally modulates the cytotoxic granule exocytosis pathway. (A) Isolated CD8 T cells were co-cultured with JJN-3 in the presence of T-cell-engaging bispecific antibody targeting B-cell maturation antigen (anti-BCMA T-BsAb, 0.1 µg/ mL) with or without recombinant interleukin-21 (rIL-21, 100 ng/mL), followed by sorting of CD8 T cells for RNA sequencing (RNAseq). A schematic illustrates the sample preparation. (B) Principal component analysis (PCA) of log2-transformed normalized protein-coding genes across all samples. Principal component (PC) 1 versus PC2 (left) and PC2 versus PC3 (right) of the scaled and centered protein-coding genes from 6 different donors colored by sample type. (C) Enriched Reactome pathways in differentially expressed genes (DEG) significantly upregulated in IL-21-primed CD8 T cells compared with control CD8 T cells. (D) Heatmap showing z-scored mRNA abundances of genes related to cytotoxic granules (GZMB, GZMH, NKG7, GNLY, and PRF1), pro-inflammatory cytokines (IFNG, TNF, and CSF2), and co-receptor (CD226, CD28, PCDC1, TIGIT, LAG3). DEG are indicated in bold text. Ctrl: control.
myeloma cell lines, JJN-3, RPMI8226, and KMS-11 (Figure 4B). Target myeloma cells expressed IL-21 receptor; however, treatment with rIL-21 in these cells showed negligible impacts on expression levels of BCMA and ligands for co-receptors in tumor cells (Online Supplementary Figure S2). While we observed that the expression level of CD226 was upregulated by rIL-21 (Figure 2D), anti-CD226 blocking monoclonal antibody (mAb) did not inhibit T-BsAb-mediated cytotoxicity (Figure 4C), suggesting that the enhanced in vitro cytotoxic activity was explained by other mechanisms. Importantly, T-BsAb-mediated tumor-killing activities were not ameliorated by anti-IFN-γ neutralizing mAb, regardless of IL-21 treatment (Figure 4C). By contrast, pretreatment with a perforin inhibitor, concanamycin A, largely abrogated cytotoxic activities in both control CD8 T cells and IL-21primed CD8 T cells, indicating that the T-BsAb-mediated myeloma-killing activity was cytotoxic granule-dependent (Figure 4C). Additionally, IL-21-primed CD4 T cells showed upregulation of cytotoxic granules and enhanced myeloma-killing activities (Online Supplementary Figure S3). Thus,
IL-21 induces the cytotoxic program in both CD8 T cells and CD4 T cells.
The cytotoxic immunological synapse acts as a molecular platform, triggering dynamic cytoskeletal reorganization and delivery of cytotoxic granules via exocytosis.34 Given that granule exocytosis and signaling pathways related to cytoskeletal reorganization were transcriptionally upregulated by IL-21 (Figure 3C), we next evaluated whether treatment with rIL-21 could promote immunological synapse formation and degranulation. Indeed, IL-21-primed CD8 T cells more rapidly expressed the degranulation marker CD107a, compared to control CD8 T cells (Figure 4D). The polarization of microtubule-organizing center (MTOC) toward the contact site is recognized as a crucial step for delivering cytotoxic granules.34 In IL-21-primed CD8 T cells, rapid MTOC polarization was observed after T-BsAb stimulation (Figure 4E, F). Together, IL-21 priming promotes the cytotoxic granule exocytosis pathway.
IL-21 treatment enhances the in vivo efficacy of T-cellengaging bispecific antibody therapy by modulating cytotoxic T-cell phenotypes in the myeloma bone marrow In order to further understand the impact of rIL-21 treatment on T-cell phenotypes in vivo, we used the Vk14451 myeloma preclinical model.11 Tumor-bearing mice with paraproteinemia were treated with rIL-21, according to a published dosing schedule27 (Figure 5A). Treatment with rIL-21 showed negli-
gible impacts on tumor burden (Figure 5B) and the number of immune subsets in the myeloma BM (Figure 5C; Online Supplementary Figure S4). However, circulating levels of granzyme B, but not IFN-γ, were significantly increased by rIL-21 treatment (Figure 5D). The downregulation of co-receptors CD226 has been recognized as a key feature of dysfunctional T cells in patients with multiple myeloma.30,31 Importantly, rIL-21 treatment significantly increased BM CD8 T cells and
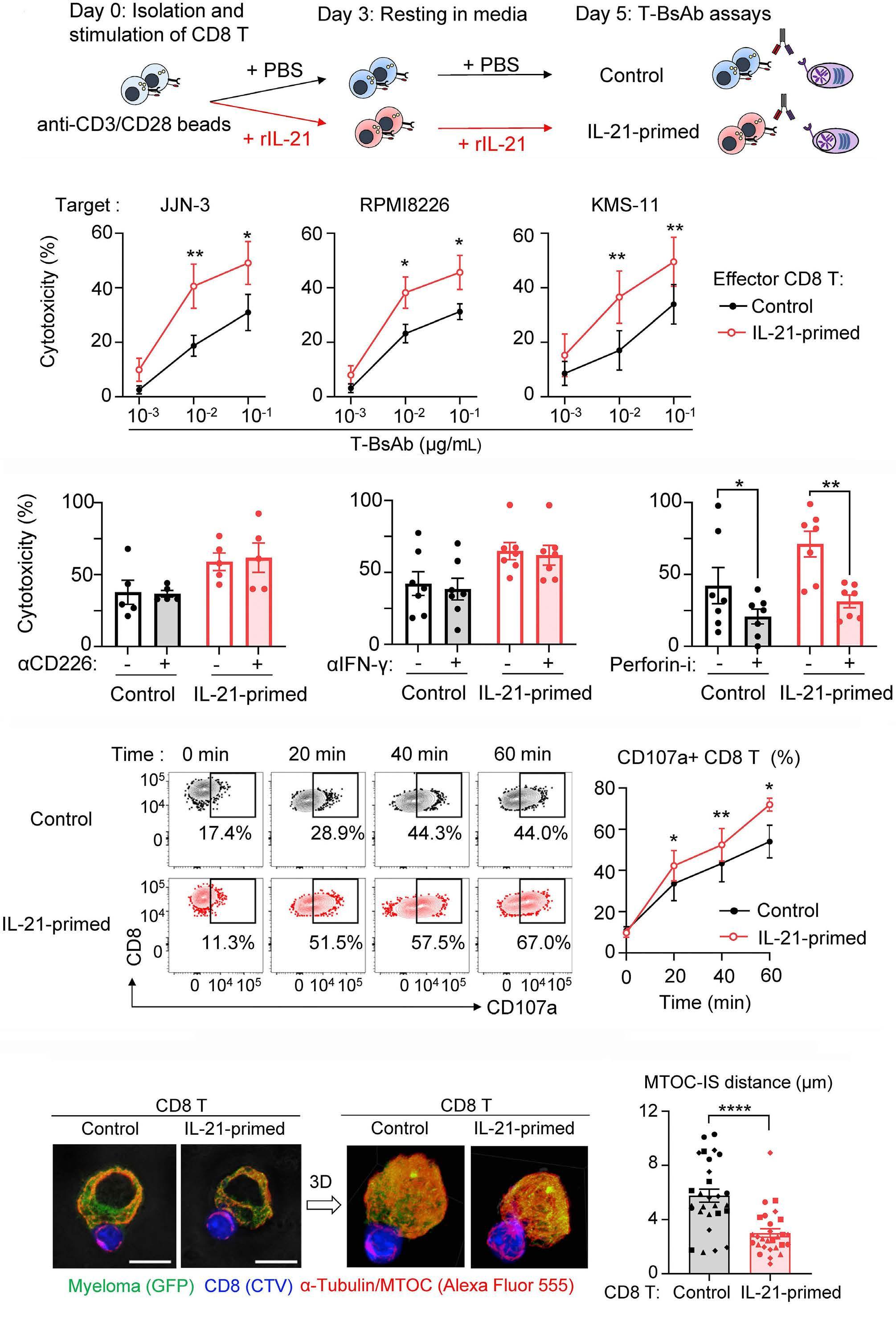
Continued on following page.
Figure 4. Priming by IL-21 augments granule-dependent myeloma-killing activity. (A) Isolated CD8 T cells were stimulated with anti-CD3/CD28 beads in the presence or absence of recombinant interleukin-21 (rIL-21), followed by maintenance for 2 days without anti-CD3/CD28 beads. After the washout of rIL-21, these effector CD8 T cells were used for assays. Schematic illustrates the preparation of control and IL-21-primed CD8 T cells. (B) CD8 T cells were co-cultured with target myeloma cells expressing luciferase at a 2:1 effector to target cell ratio (E: T ratio) for 4 hours. Graphs showing target cytotoxicity with different concentrations of T-cell-engaging bispecific antibody targeting B-cell maturation antigen (anti-BCMA T-BsAb) (N=7). (C) CD8 T cells were co-cultured with RPMI8226 cells at a 3:1 E: T ratio in the presence of anti-BCMA T-BsAb (0.1 µg/mL). Graphs showing the impact of anti-CD226 blocking monoclonal antibody (mAb, 5 µg/mL) (left), anti-interferon-γ (IFN-γ) neutralizing mAb (5 µg/mL) (middle), or pretreatment with a perforin inhibitor (Perforin-I, 50 nM) (right) on target cytotoxicity (N=5-7). (D) Representative plots (left) and graphs (right) showing frequencies of CD107a+ cells in CD8 T cells after co-culture with RPMI8226 myeloma cells for indicated periods in the presence of anti-BCMA T-BsAb (N=7). (E, F) CTV-labeled CD8 T cells were co-cultured with RPMI8226-GFP myeloma cells for 15 minutes at a 1:1 E: T ratio in the presence of anti-BCMA T-BsAb. Representative confocal images showing polarization of the microtubule-organizing center (MTOC) to the immunological synapse (IS) with 3D-reconstituted images (E). Scale bars indicate 10 µm. Graphs showing the distance between the MTOC and IS (right), with each symbol representing 1 donor (N=4) (F). Data are shown as mean ± standard error of mean. Results were pooled from 2 experiments. Differences were tested for statistical significance using a paired t test (B, D), a repeated measures ANOVA with a post hoc Holm-Sidak’s multiple comparisons test (C), and a Mann-Whitney U test (F). *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. Perforin-I: perforin inhibitor; GFP: green fluorescent protein; CTV: CellTrace™ Violet.
CD4 T cells expressing CD226 and granzyme B (Figure 5E, F). These results support that rIL-21 treatment can enhance the cytotoxic lymphocyte program in the myeloma BM. We next asked whether rIL-21 can enhance the efficacy of anti-BCMA T-BsAb therapy in the Vk14451 myeloma preclinical model (Figure 6A). Consistent with in vitro results, pretreatment with rIL-21 did not enhance T-BsAb-mediated IFN-γ release, while the combination approach significantly increased plasma levels of granzyme B (Figure 6B). Strikingly, the combination therapy reduced serum paraprotein levels (Figure 6C), and significantly prolonged survival compared to T-BsAb monotherapy (Figure 6D). Together, IL-21 priming is a potential approach to enhance the efficacy of T-BsAb against multiple myeloma.
Treatment with IL-21 modulates effector functions in primary bone marrow CD8 T cells from patients with multiple myeloma
Exhausted CD8 T cells are frequently observed in BM, compared to peripheral blood in patients with multiple myeloma.35 Thus, we addressed whether rIL-21 treatment could augment cytotoxic effector phenotypes in myeloma BM T cells. To this end, patient-derived BM mononuclear cells were cultured in the presence of T-BsAb with or without rIL-21 for 3 days (Figure 7A). In line with our results from PBMC, treatment with rIL-21 significantly upregulated CD226 in T-BsAb-stimulated myeloma BM CD8 T cells, while the upregulation of CD69 and CD28 was mainly driven by T-BsAb (Figure 7B-D). Importantly, rIL-21 treatment markedly enhanced the release of granzyme B and perforin, but not pro-inflammatory cytokines in T-BsAb-stimulated myeloma BM mononuclear cells (Figure 7E). Taken together, IL-21 can augment cytotoxic effector functions in patient-derived BM CD8 T cells.
In this study, we demonstrated that harnessing the cy-
totoxic granule exocytosis pathway through IL-21 can augment T-BsAb-mediated anti-myeloma effects in vitro and in vivo. Unlike other immunostimulatory cytokines, IL-21 treatment did not enhance the release of IFN-γ and other pro-inflammatory cytokines, indicating that IL-21 priming preferentially harnesses cytotoxic effector functions, and can augment granule-dependent myeloma-killing activities by T-BsAb therapy.
Recent results from single-cell immune landscape profiling revealed an accumulation of terminally differentiated, functionally impaired T cells in the myeloma BM, especially in patients with relapsed/refractory multiple myeloma.9,36 Since the effector mechanism of T-BsAb therapy largely depends on endogenous T cells, functional impairment of T cells may limit T-BsAb efficacy against multiple myeloma. Indeed, Friedrich et al. recently demonstrated that the presence of exhausted-like CD8 T cell clones was associated with treatment failure in patients treated with anti-BCMA T-BsAb therapy,7 providing a rationale for reprogramming dysfunctional CD8 T cells to achieve better clinical responses. Over the past few years, new approaches have been developed to improve the functionality of CAR T cells during ex vivo manufacturing.37,38 By contrast, to improve the efficacy of T-BsAb therapy, systemic therapy will be inevitable to reinvigorate endogenous T cells in the myeloma BM. However, the optimal therapeutic approach remains to be established due to the risk of CRS exacerbation. In this context, results from this study reveal IL-21 as an ideal partner for T-BsAb therapy, providing important translational implications. We showed that treatment with rIL-21 did not enhance IFN-γ release both in vitro and in vivo. Given that IFN-γ is a key driver for CRS and MAS/HLH,18,19,39 but dispensable for eliminating malignant B cells by immunotherapy,18,23 avoiding the excess release of IFN-γ will be an important consideration for T-BsAb therapy against hematological malignancies. Of note, conflicting results have been reported on whether or not IL-21 can induce Th1 responses and IFN-γ production, possibly due to context-dependent effects of IL-21 on T-cell
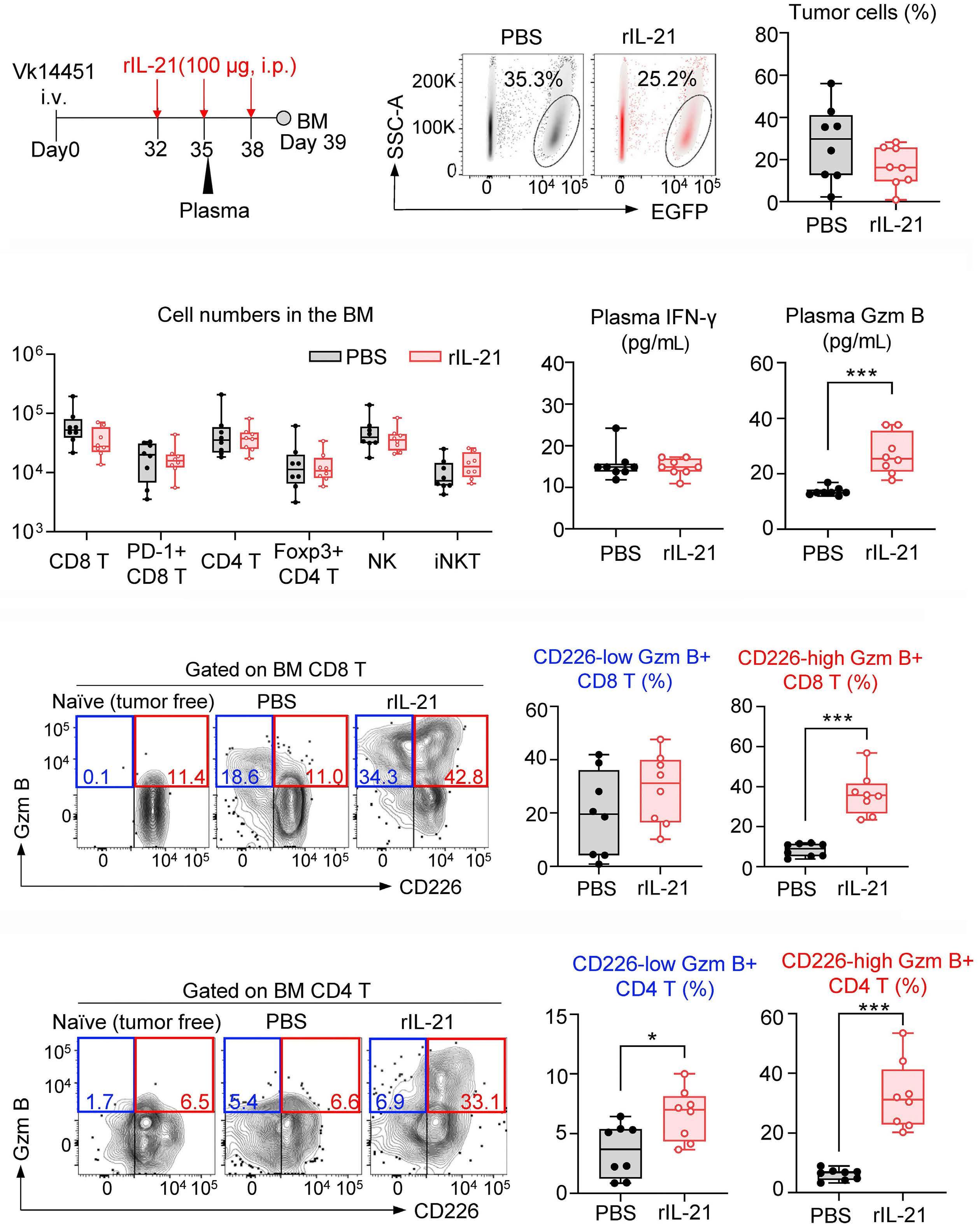
Figure 5. Treatment with recombinant IL-21 drives cytotoxic T-cell programming in the myeloma bone marrow. (A) Schematic illustrates the experimental design for murine recombinant interleukin-21 (rIL-21) treatment in the preclinical Vk14451 myeloma model. (B) Representative flow cytometry plots (left) and graphs showing frequencies (right) of EGFP+ tumor cells in the bone marrow (BM). (C) Graphs showing numbers of indicated immune subsets in the myeloma BM. (D) Graphs showing plasma levels of interferon-γ (IFN-γ) (left) and granzyme B (Gzm B) (right) 6 hours after the second injection of rIL-21. (E, F) Representative flow cytometry plots (Ieft) and graphs (right) showing frequencies of CD8 T cells (E) and CD4 T cells (F) expressing CD226 and Gzm B in the myeloma BM. Data are shown as box-and-whisker plots pooled from 2 experiments (N=8 per group). Differences were tested for statistical significance using a Mann-Whitney U test. *P<0.05; ***P<0.001. SSC: side scatter; EGFP: enhanced green fluorescent protein; PBS: phosphate-buffered saline.
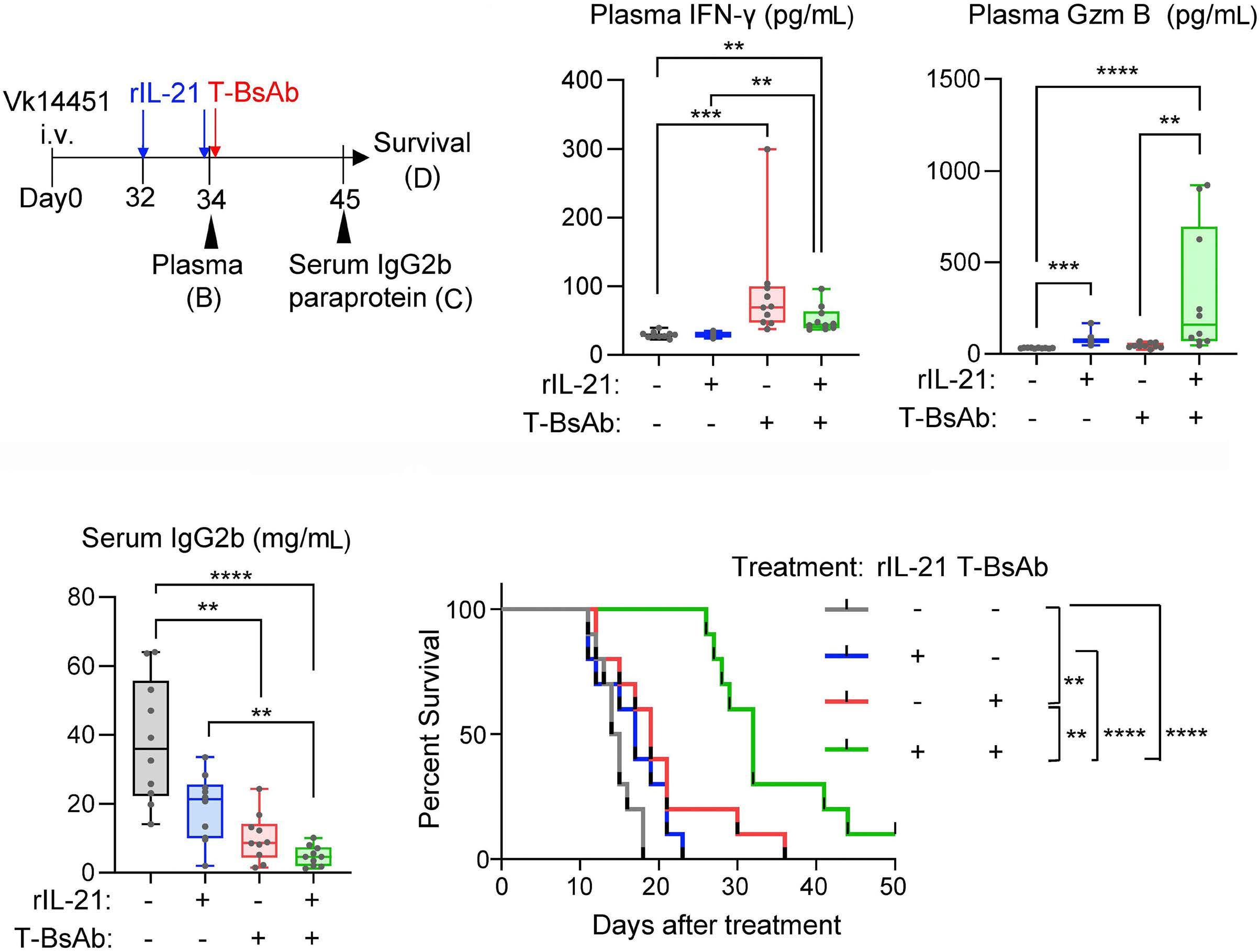
Figure 6. Pretreatment with recombinant IL-21 dramatically improves the efficacy of T-cell-engaging bispecific antibody therapy in the preclinical myeloma model. (A) C57BL6 wild-type mice were challenged with Vk14451 myeloma cells, and intraperitoneally (i.p.) treated with 2 shots of murine recombinant interleukin-21 (rIL-21, 100 µg), followed by T-cell-engaging bispecific antibody targeting mouse B-cell maturation antigen (anti-mouse BCMA T-BsAb, 25 µg). A schematic illustrates the experimental design. (B) Box-and-whisker plots showing plasma levels of interferon-γ (IFN-γ) and granzyme B (Gzm B) 6 hours after treatment. (C) Box-and-whisker plots showing serum levels of immunoglobulin (Ig)G2b paraprotein 11 days after treatment. (D) Kaplan-Meier survival curves of mice after indicated treatment. Data are pooled from 2 experiments (N=7-11 per group). Differences were tested for statistical significance using a Kruskal-Wallis test with post hoc Dunn’s multiple comparisons test (B, C) and a Mantel-Cox test (D). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
differentiation and effector functions.40-42 However, our comprehensive profiling strongly supports the cytotoxic granule pathway as a key target of IL-21. Various immunostimulatory cytokines are known to improve anti-tumor functions by natural killer cells and CD8 T cells.14,43 Although the upregulation of cytotoxic granules by IL-21 has been reported,42,44 our results indicate that the multiple steps of the cytotoxic granule pathway are modulated by IL-21. It is noteworthy that priming CD8 T cells by IL-21 transcriptionally and functionally promoted cytoskeletal reorganization and granule degradation at the immunological synapse. Overall, IL-21 has unique roles in modulating effector functions. We provided preclinical proof-of-concept for IL-21 priming as a potential strategy to improve T-BsAb efficacy against multiple myeloma. Intriguingly, lenalidomide can induce IL-21 production from T cells, raising the possibility that IL21 signaling might be implicated in its immunostimulatory mechanisms.45 However, results from a recent preclinical
study have shown that the addition of pomalidomide to anti-BCMA T-BsAb therapy only modestly improved the efficacy due to exhaustion of T cells in the myeloma BM,46 suggesting that reprogramming of dysfunctional T cells by exogenous IL-21 might be necessary to fully augment T-cell functions. We observed that IL-21-primed human and mouse CD4 T cells markedly upregulated granzyme B expression levels. Given that T-BsAb-mediated generation and activation of regulatory T cells counteract anti-myeloma immunity elicited by T-BsAb,24 the IL-21-driven cytotoxic program in CD4 T cells might contribute to overcome this negative feedback regulation. Alternatively, the activation of natural killer cells, natural killer T cells, and γδ T cells by IL-21 might also contribute to anti-myeloma immunity, as these cells are frequently exhausted in patients with relapsed/refractory myeloma.9 Recently, Li et al. showed that IL-21 can enhance T-cell infiltration in solid malignancies.47 Thus, IL-21 might be beneficial for controlling immunologically “cold” tumor
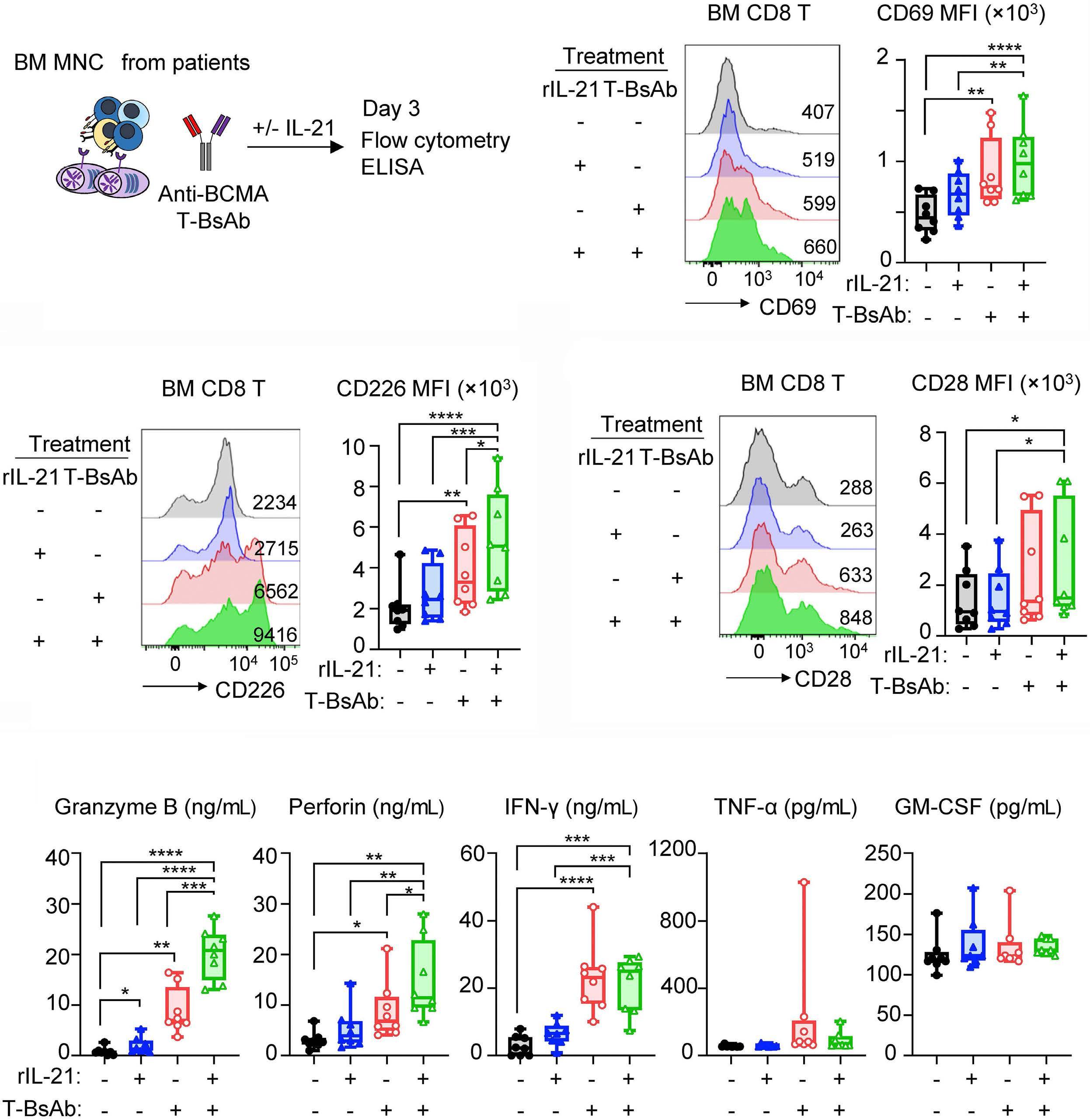
Figure 7. IL-21 improves effector functions in myeloma bone marrow CD8 T cells stimulated with T-cell-engaging bispecific antibody. (A) Primary bone marrow mononuclear cells (BM MNC, 8×105) from 8 patients with newly diagnosed multiple myeloma were stimulated with T-cell-engaging bispecific antibody targeting B-cell maturation antigen (anti-BCMA T-BsAb, 0.2 µg/mL) in the presence or absence of recombinant interleukin-21 (rIL-21, 100 ng/mL) for 3 days. A schematic illustrates the experimental design. (B-D) Representative histograms and box-and-whisker plots showing the expression levels of CD69 (B), CD226 (C), and CD28 (D) on bone marrow (BM) CD8 T cells. Numbers indicate mean fluorescence intensity (MFI). (E) Box and whisker plots showing levels of indicated effector proteins and cytokines in culture supernatants. Data are pooled from 2 experiments (N=8). Differences were tested for statistical significance using a repeated measures ANOVA with a post hoc Holm-Sidak’s multiple comparisons test. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. TNF-α: tumor necrosis factor-α; GM-CSF: granulocyte-macrophage colony-stimulating factor.
lesions such as extramedullary disease, though further studies are necessary to understand the impact of IL-21 on the myeloma immune microenvironment. For clinical translation, rIL-21 treatment has been already tested in combination with rituximab in patients with relapsed chronic lymphocytic leukemia and low-grade B-cell
lymphomas, and that weekly injection of rIL-21 (100 μg/kg) was tolerable in patients.48 Besides, a recent preclinical study has also shown no major immune-mediated toxicities either alone or in combination with immune checkpoint inhibitors.27 One of the limitations of our study is that we could not address whether or not the addition of rIL-21 has an
impact on CRS, as the CRS-like phenotype is not observed in preclinical T-BsAb models. Another limitation of the current study is that this study exclusively evaluated the impact of rIL-21 on anti-BCMA T-BsAb against multiple myeloma. It is reported that IL-21 can directly induce apoptosis in B-cell lymphomas, but not in multiple myeloma, possibly by upregulating BIM (a member of the pro-apoptotic BH3-only members protein) in B-lymphoma cells.49-51 In this context, beyond the IL-21-driven cytotoxic program, rIL-21 treatment may be able to lower the apoptotic threshold in B-lymphoma cells, leading to enhancement of anti-CD20 T-BsAb efficacy. Further studies are warranted to understand the tumor type-specific effect of rIL-21. Lastly, it is important to address whether IL-21 can improve T-cell immunity in patients with relapsed/refractory myeloma, although T-BsAb therapy is being tested as an earlier line of therapy (clinicaltrials gov. Identifier: NCT05469893). Despite these limitations, results from this study provide important translational implications for improving the efficacy of T-BsAb therapy against multiple myeloma and other hematological malignancies.
MKG reports research support unrelated to this work from Janssen and Beigene. SJH reports support unrelated to this work from AbbVie (consultancy, advisory board, investigator on studies); Amgen, Celgene, Janssen Cilag and Novartis (consultancy, honoraria, advisory board, research funding, investigator on studies); GSK (consultancy, research funding, advisory board); Roche/Genentech (consultancy, honoraria, advisory board, investigator on studies); Takeda (consultancy, honoraria, advisory board); Haemalogix (scientific advisory board, research funding, investigator); Sanofi (consultancy/
1. Moreau P, Garfall AL, van de Donk N, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495-505.
2. Chari A, Minnema MC, Berdeja JG, et al. Talquetamab, a T-cellredirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med. 2022;387(24):2232-2244.
3. Munshi NC, Anderson LD, Jr., Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705-716.
4 Casey M, Nakamura K. The cancer-immunity cycle in multiple myeloma. Immunotargets Ther. 2021;10:247-260.
5. Cohen AD, Harrison SJ, Krishnan A, et al. Initial clinical activity and safety of BFCR4350A, a FcRH5/CD3 T-cell-engaging bispecific antibody, in relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):42-43.
6. Lee H, Ahn S, Maity R, et al. Mechanisms of antigen escape from BCMA- or GPRC5D-targeted immunotherapies in multiple myeloma. Nat Med. 2023;29(9):2295-2306.
7 Friedrich MJ, Neri P, Kehl N, et al. The pre-existing T cell landscape determines the response to bispecific T cell engagers in multiple myeloma patients. Cancer Cell. 2023;41(4):711-725.
advisory role). KN received honorariums from Janssen, Sanofi, and Bristol Myers Squibb. The other authors have no conflicts of interest to disclose.
KN designed the research and wrote the manuscript. MC, CL, WYK and KN performed experimental work and analyzed the data. SMH and RLJ performed data analysis. SCL and MKG provided key materials. SCL, MKG and SJH contributed to critical discussion and data interpretation. KN conceived and supervised the study. All authors read and approved the final version of the manuscript.
The authors wish to thank Bristol Myers Squibb for providing anti-mouse T-BsAb. The authors also thank technical support from the QIMR flow cytometry facility. We thank Ross Koufariotis and the QIMR Genome Informatics team for their help processing RNA-sequencing data.
The authors deeply appreciate funding support from the Play for a Cure Foundation. KN is supported by the NHMRC project grant (1159593) and the Naito Foundation. This project was supported by grant 2000538 awarded through the 2020 Priority-driven Collaborative Cancer Research Scheme and funded by the Leukemia Foundation with the support of Cancer Australia.
Data-sharing statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
8. Nakamura K, Smyth MJ, Martinet L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood. 2020;136(24):2731-2740.
9. Tirier SM, Mallm J-P, Steiger S, et al. Subclone-specific microenvironmental impact and drug response in refractory multiple myeloma revealed by single-cell transcriptomics. Nature Commun. 2021;12(1):6960.
10 Dhodapkar MV. The immune system in multiple myeloma and precursor states: lessons and implications for immunotherapy and interception. Am J Hematol. 2023;98(Suppl 2):S4-S12.
11. Casey M, Tu C, Harrison SJ, Nakamura K. Invariant NKT cells dictate antitumor immunity elicited by a bispecific antibody cotargeting CD3 and BCMA. Blood Adv. 2022;6(17):5165-5170.
12. Berraondo P, Sanmamed MF, Ochoa MC, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. 2019;120(1):6-15.
13. Nakamura K, Bald T, Smyth MJ. Cancer-killing, decoy-resistant interleukin-18. Immunol Cell Biol. 2020;98(6):434-436.
14 Waldmann TA. Cytokines in cancer immunotherapy. Cold Spring Harb Perspect Biol. 2018;10(12):a028472.
15. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer
immunotherapy. Nat Rev Immunol. 2022;22(2):85-96.
16. Li J, Piskol R, Ybarra R, et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med. 2019;11(508):eaax8861.
17 Sterner RM, Sakemura R, Cox MJ, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133(7):697-709.
18. Bailey SR, Vatsa S, Larson RC, et al. Blockade or deletion of IFNγ reduces macrophage activation without compromising CAR T-cell function in hematologic malignancies. Blood Cancer Discov. 2022;3(2):136-153.
19 Manni S, Del Bufalo F, Merli P, et al. Neutralizing IFNγ improves safety without compromising efficacy of CAR-T cell therapy in B-cell malignancies. Nat Commun. 2023;14(1):3423.
20 Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15(6):388-400.
21. La Rosée P, Horne A, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133(23):2465-2477.
22. Ishii K, Pouzolles M, Chien CD, et al. Perforin-deficient CAR T cells recapitulate late-onset inflammatory toxicities observed in patients. J Clin Invest. 2020;130(10):5425-5443.
23. Larson RC, Kann MC, Bailey SR, et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nature. 2022;604(7906):563-570.
24. Casey M, Lee C, Kwok W, et al. Regulatory T cells hamper the efficacy of T cell-engaging bispecific antibody therapy. Haematologica. 2024;109(3):787-789.
25. Casey M, Segawa K, Law SC, et al. Inhibition of CD39 unleashes macrophage antibody-dependent cellular phagocytosis against B-cell lymphoma. Leukemia. 2023;37(2):379-387.
26. Nakamura K, Kassem S, Cleynen A, et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell. 2018;33(4):634-648.
27. Lewis KE, Selby MJ, Masters G, et al. Interleukin-21 combined with PD-1 or CTLA-4 blockade enhances antitumor immunity in mouse tumor models. Oncoimmunology. 2017;7(1):e1377873.
28. Mittal D, Caramia F, Michiels S, et al. Improved treatment of breast cancer with anti-HER2 therapy requires interleukin-21 signaling in CD8+ T cells. Cancer Res. 2016;76(2):264-274.
29 Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. 2010;22(3):333-340.
30 Weulersse M, Asrir A, Pichler AC, et al. Eomes-dependent loss of the co-activating receptor CD226 restrains CD8(+) T cell antitumor functions and limits the efficacy of cancer immunotherapy. Immunity. 2020;53(4):824-839.
31. Braun M, Aguilera AR, Sundarrajan A, et al. CD155 on tumor cells drives resistance to immunotherapy by inducing the degradation of the activating receptor CD226 in CD8(+) T cells. Immunity. 2020;53(4):805-823.
32. Chung DJ, Pronschinske KB, Shyer JA, et al. T-cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunol Res. 2016;4(1):61-71.
33. Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol. 2009;9(9):630-644.
34 Dieckmann NM, Frazer GL, Asano Y, Stinchcombe JC, Griffiths GM. The cytotoxic T lymphocyte immune synapse at a glance. J Cell Sci. 2016;129(15):2881-2886.
35. Zelle-Rieser C, Thangavadivel S, Biedermann R, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. 2016;9(1):116.
36. Bailur JK, McCachren SS, Doxie DB, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight. 2019;5(11):e127807.
37. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346-351.
38. Wilkens AB, Fulton EC, Pont MJ, et al. NOTCH1 signaling during CD4+ T-cell activation alters transcription factor networks and enhances antigen responsiveness. Blood. 2022;140(21):2261-2275.
39. Rainone M, Ngo D, Baird JH, et al. Interferon-γ blockade in CAR T-cell therapy-associated macrophage activation syndrome/ hemophagocytic lymphohistiocytosis. Blood Adv. 2023;7(4):533-536.
40 Monteleone G, Monteleone I, Fina D, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology. 2005;128(3):687-694.
41. Suto A, Wurster AL, Reiner SL, Grusby MJ. IL-21 inhibits IFNgamma production in developing Th1 cells through the repression of Eomesodermin expression. J Immunol. 2006;177(6):3721-3727.
42. Tian Y, Zajac AJ. IL-21 and T cell differentiation: consider the context. Trends Immunol. 2016;37(8):557-568.
43. Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov. 2014;13(5):379-395.
44 White L, Krishnan S, Strbo N, et al. Differential effects of IL-21 and IL-15 on perforin expression, lysosomal degranulation, and proliferation in CD8 T cells of patients with human immunodeficiency virus-1 (HIV). Blood. 2007;109(9):3873-3880.
45. Browning RL, Byrd WH, Gupta N, et al. Lenalidomide induces interleukin-21 production by T cells and enhances IL21-mediated cytotoxicity in chronic lymphocytic leukemia B cells. Cancer Immunol Res. 2016;4(8):698-707.
46. Meermeier EW, Welsh SJ, Sharik ME, et al. Tumor burden limits bispecific antibody efficacy through T cell exhaustion averted by concurrent cytotoxic therapy. Blood Cancer Discov. 2021;2(4):354-369.
47. Li Y, Cong Y, Jia M, et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat Commun. 2021;12(1):951.
48. Timmerman JM, Byrd JC, Andorsky DJ, et al. A phase I dosefinding trial of recombinant interleukin-21 and rituximab in relapsed and refractory low grade B-cell lymphoproliferative disorders. Clin Cancer Res. 2012;18(20):5752-5760.
49 Brenne AT, Ro TB, Waage A, Sundan A, Borset M, Hjorth-Hansen H. Interleukin-21 is a growth and survival factor for human myeloma cells. Blood. 2002;99(10):3756-3762.
50 Sarosiek KA, Malumbres R, Nechushtan H, Gentles AJ, Avisar E, Lossos IS. Novel IL-21 signaling pathway up-regulates c-Myc and induces apoptosis of diffuse large B-cell lymphomas. Blood. 2010;115(3):570-580.
51. Gowda A, Roda J, Hussain SR, et al. IL-21 mediates apoptosis through up-regulation of the BH3 family member BIM and enhances both direct and antibody-dependent cellular cytotoxicity in primary chronic lymphocytic leukemia cells in vitro. Blood. 2008;111(9):4723-4730.
Zhiyuan Lu,1,2,3* Hanzhi Yu,4* Yanxia Li,1,2 Guangsen Xu,1,2 Xiaoxun Li,1,2 Yongjun Liu,1,2 Yuemao Shen,1,2 Zhigang Cai4 and Baobing Zhao1,2,5
1Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan; 2NMPA Key Laboratory for Technology Research and Evaluation of Drug Products, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan; 3School of Pharmaceutical Sciences & Institute of Materia Medica, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan; 4The Province and Ministry Co-Sponsored Collaborative Innovation Center for Medical Epigenetics, Department of Pharmacology, School of Basic Medical Science, Tianjin Medical University, Tianjin and 5Department of Pharmacology, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, China
*ZL and HY contributed equally as first authors.
Correspondence: Baobing Zhao baobingzh@sdu.edu.cn
Received: September 18, 2023.
Accepted: January 31, 2024. Early view: February 8, 2024.
https://doi.org/10.3324/haematol.2023.284305
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Hematopoietic stem cells (HSC) are primarily dormant in a cell-cycle quiescence state to preserve their self-renewal capacity and long-term maintenance. How HSC maintain the balance between activation and quiescence remains largely unknown. Herein, we found that phosphatase, Mg2+/Mn2+ dependent 1B (Ppm1b) is required for the expansion of phenotypic HSC in vitro. By using a conditional knockout mouse model in which Ppm1b was specifically depleted in hematopoietic cells, we demonstrated that loss of Ppm1b impaired the HSC homeostasis and hematopoietic reconstitution. Ppm1b deficiency mice also exhibited B-cell leukocytopenia, which is due to the compromised commitment and proliferation of B-biased lymphoid progenitor cells from common lymphoid progenitors. With the aid of a small molecular inhibitor, we confirmed the roles of Ppm1b in adult hematopoiesis that phenocopied the effects with loss of Ppm1b. Furthermore, transcriptome profiling of Ppm1b-deficient HSC revealed the disruptive quiescence of HSC. Mechanistically, Ppm1b interacted with b-catenin and mediated its dephosphorylation. Loss of Ppm1b led to the decrease in the active b-catenin (non-phosphorylated) that interrupted the Wnt/b-catenin signaling in HSC, which consequently suppressed HSC expansion. Together, our study identified an indispensable role for Ppm1b in regulating HSC homeostasis via the Wnt/b-catenin pathway.
Hematopoietic stem cells (HSC) have the potential for both self-renewal and multipotent differentiation into all blood cell lineages during the lifespan.1 They are primarily dormant in a cell-cycle quiescence state to preserve their self-renewal capacity and long-term maintenance, which is responsible for maintaining and rebuilding hematopoiesis.2,3 Functional HSC homeostasis is tightly regulated by multiple factors including cell cycle-associated factors and cell metabolism. Intrinsic programs, cell-autonomous and extrinsic signals from the microenvironment comprise a complicated network, which preserves the balance between
activation and quiescence in HSC.4,5
The reversible phosphorylation of protein regulated by kinase and phosphatase is one of the main post-translational modifications that is involved in an extremely wide range of intracellular processes including metabolism, cell death, cell proliferation and differentiation.6,7 Recent studies have demonstrated that protein kinase plays a crucial role in the functional regulation of HSC, such as receptor tyrosine kinases (FLT3,8 c-KIT,9 and TIE210), glycogen synthase kinase 3b, 11 sphingosine kinase 2,12 and Janus kinase 2.13 Despite the equally important biological significance, our understanding of protein phosphatases in hematopoietic regeneration still lags behind that of kinases due to their complicated
active centers and lower substrate specificity. Emerging evidence of phosphatases in controlling HSC homeostasis and functions has been gradually elucidated in recent years. Src homology 2 domain-containing phosphatase 2 (Shp2) plays a critical role in controlling the survival and maintenance of HSC.14 Ptpn21 has been reported to control HSC homeostasis.15 STS1/STS2 functions in normal and stress hematopoiesis via the regulation of HSC fitness.16
The protein phosphatase 2C (PP2C) family is a member of the metal-dependent protein phosphatases (PPM) of serine/ threonine phosphatases. PP2C family members are known to be negative regulators of cell stress response pathways, and involved in cell cycle control via the dephosphorylation of cyclin-dependent kinases. PP2C family members such as PPM1K and PPM1D, have been demonstrated to play functional roles in maintaining the stemness of HSC.17,18
Protein phosphatase, Mg2+/Mn2+ dependent 1B (Ppm1b, also known as PP2Cb), is involved in the regulation of multiple signaling pathways by targeting substrate protein dephosphorylation,19,20 including MAPK signaling, TGF- b signaling, TNF signaling, AMPK signaling, and cell cycle. Ppm1b showed high expression in embryonic stem cells,21 and its deficiency led to early pre-implantation lethality.22 Our previous study suggested that Ppm1b may participate in the regulation of hematopoietic processes such as B-/T-cell receptor signaling, FOXO signaling, and the pathogenesis of leukemia.23 It had consistently been reported that Ppm1b regulates terminal erythropoiesis by interacting with an erythroid Kruppel-like factor.24 However, the role of Ppm1b in hematopoiesis is still largely unknown. In this study, we generated and characterized a conditional knockout (KO) mouse model in which Ppm1b was specifically depleted in hematopoietic cells, and demonstrated that loss of Ppm1b impaired the normal HSC pool and B-cell development. Furthermore, Ppm1b regulates the functional expansion of HSC via Wnt/b-catenin signaling.
Ppm1b floxed mice (Ppm1bfl/fl) were generated by inserting loxp sites flanking exon 2, which, when deleted, results in a frame-shift and forms a premature stop codon in the reading frame. Vav-cre mice were purchased from the Jackson Laboratory. The animals were kept in individually ventilated cages with filtered germ-free air, and maintained with sterilized water and irradiated food. The Institutional Animal Care and Use Committees at Shandong University (#19026) approved all animal experiments in our study.
Flow cytometric assays
Single-cell suspensions of tissues, cultured cells, or peripheral blood (PB) were prepared and stained as previously described.25,26 Detailed information for antibodies
is provided in the Online Supplementary Appendix. Flow cytometric analysis was performed with an ACEA NovoCyte flow analyzer, and the results were further analyzed with FlowJo and NovoExpress software.
The purified lineage-negative cells were cultured in StemSpan expansion medium (StemCell) supplemented with 10 ng/mL mouse TPO (Peprotech) and 10 ng/mL mouse SCF (Peprotech) for different time periods as indicated in the figure legends (Figures 1-7).
Non-competitive and competitive bone marrow transplantation (BMT) was performed as described previously.27 Briefly, for non-competitive BMT, 2x106 BM mononuclear cells (BMMC) (CD45.2) from Ppm1bCKO or Ppm1bfl/fl mice were injected retro-orbitally into lethally irradiated wild-type (WT) recipient mice (CD45.1). For competitive BMT, 1x106 BMMC cells from Ppm1bCKO or Ppm1bfl/fl mice (CD45.2) mixed with an equal number of WT CD45.1 BMMC were injected retro-orbitally into lethally-irradiated CD45.1 recipient mice. For serial competitive BMT, 2x106 donor BMMC from primary transplant recipient mice were transplanted into lethally irradiated CD45.1 congenic recipients. PB was analyzed every four weeks by flow cytometry to assess engraftment.
Different doses (1x104, 2x104, 4x104, 8x104) of BMMC from Ppm1bCKO or Ppm1bfl/fl mice (CD45.2) plus 2x105 competitor BMMC (CD45.1) were transplanted retro-orbitally into lethally irradiated WT recipient mice (CD45.1). Sixteen weeks after transplantation, the frequency of donor-derived cells (CD45.2) was evaluated by flow cytometry. ELDA (bioinf. wehi.edu.au/software/elda/) and L-Calc software (StemCell) were used to analyze HSC frequency.
Proximity ligation assay
Proximity ligation assay (PLA) was performed using the DuoLink reagents as previously described.28 Briefly, LSK cells were sorted by an Aria III flow cytometer and plated in 8-mm glass coverslips. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 separately, followed by incubating with blocking solution. Cells were then incubated with Ppm1b and b-catenin overnight at 4°C. After incubation with PLA probes, probe ligation, and amplification separately, cells were visualized using an Axio Observer fluorescent A1 microscope.
Ppm1b inhibition led to the compromised proliferation of phenotypic hematopoietic stem cells By qRT-PCR, we confirmed that Ppm1b is highly expressed
in LSK (Lin Sca-1+c-Kit+) cells that represent relatively enriched HSC, compared to that of hematopoietic progenitor cell (Lin Sca-1-c-Kit+, LSK) and lineage-negative BM cells (Online Supplementary Figure S1A). To explore the role of Ppm1b in HSC function, we first treated the HSC isolated from mouse BM with Ppm1b inhibitor HN252 which we developed in our previous study.23 The cells in the LSK fraction exhibited a dose-dependent decrease after 48 hours (hr) of culture with HN252, accompanied by the reduced LSK cells (Figure 1A, Online Supplementary Figure
S1B). HN252 treatment also led to a significant increase in apoptotic cells in the LSK fraction (Online Supplementary Figure S1C), but the overall apoptosis rate was relatively low (<5%), suggesting that cell death is not the main cause of impaired HSC expansion induced by HN252. We then examined the proliferation of HSC via CFSE staining, in which cell division was tracked via the changes in intracellular fluorescence.29,30 Interestingly, the relative mean fluorescence intensity (MFI) of CFSE in LSK was 2-fold higher than that of the control group after 48 hr of culture with
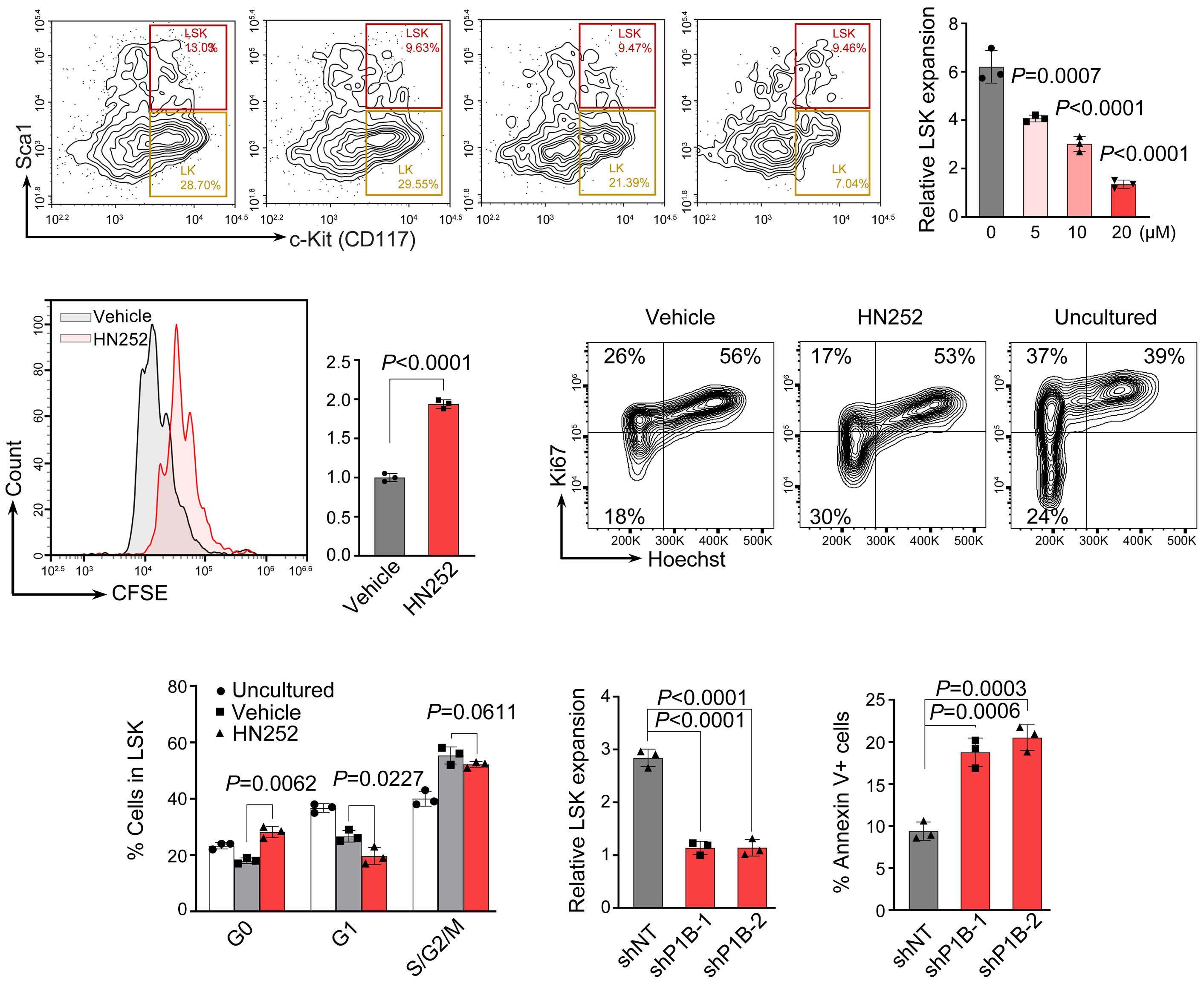
Figure 1. Ppm1b inhibition led to the repressed proliferation of phenotypic hematopoietic stem cells. (A) Lineage negative cells were cultured with HN252 at multiple concentrations (0, 5, 10, and 20 μM) for 48 hours (hr). The relative proliferation ratio of Lin Sca-1+c-Kit+ (LSK) cells was quantified by flow cytometry, and normalized to the uncultured cells. (B) CFSE labeled lineage negative cells were cultured with HN252 (10 µM) or vehicle (control) for 48 hr, and the mean fluorescence intensity (MFI) of CFSE in LSK cells was quantified by flow cytometry. (C) Representative cell cycle profile of LSK cells after in vitro culture with control or HN252 (10 μM) for 48 hr. Cell cycle was analyzed using Hoechst 33342 and Ki67 staining by flow cytometry. (D) Quantification of cells in indicated phases in (C). (E and F) Lineage negative cells were transduced with retroviruses encoding Ppm1b shRNA (shP1B-1 or shP1B-2) and cultured for 48 hr; the cell number (E) and apoptosis (F) were analyzed by flow cytometry. P values were determined by one-way ANOVA with Tukey’s multiple comparisons test (A) or by unpaired two-tailed Student t test (B-F). Data are presented as mean ± Standard Deviation from 3 independent experiments. See also Online Supplementary Figure S1.
HN252, indicating that Ppm1b inhibition led to the proliferative suppression of HSC (Figure 1B). Compared to the freshly isolated LSK from BM (uncultured), the frequency of HSC at the G0-phase (Ki67-) was obviously decreased, which represents quiescent cells, while active cycling cells (Ki67+) were dramatically increased. This is consistent with previous findings that HSC undergoes active cell cycling stimulated by supplemented hematopoietic cytokines in in vitro culture.31,32 Notably, HN252 treatment led to a significant increase in G0 cells coupled with the reduced G1 and G2/M-phase frequency in LSK, suggesting that Ppm1b is required for HSC to switch from quiescence to an active state (Figure 1C, D). In line with the findings of HN252 treatment, Ppm1b knockdown significantly inhibited the proliferation of LSK in in vitro culture, accompanied by a remarkable increase in cell death in LSK (Figure 1E, F). These data indicate that Ppm1b plays functional roles in the proliferation of HSC.
Ppm1b deficiency mice exhibited B-cell leukocytopenia
To explore the role of Ppm1b in the hematopoietic system, we generated hematopoietic-specific Ppm1b KO mice (conditional KO, CKO) by crossing mice bearing Ppm1b allele with loxP-flanked exon 2 with Vav-Cre transgenic mice (Online Supplementary Figure S2A). The ablation efficiency of Ppm1b was confirmed in mononuclear cells of BM and PB by qRT-PCR (Online Supplementary Figure S2B ). CKO mice exhibited a dominant defect in white blood cells (WBC) that was mainly due to the reduction of lymphocytes in PB, whereas red blood cells (RBC) and platelets (PLT) were unaffected (Figure 2A, Online Supplementary Figure S2C). Interestingly, the low lymphocyte attributed to the significantly reduced B cells (B220+) but not T cells (CD3+) in PB from CKO mice compared to that of control mice (Online Supplementary Figure S2D). Similar defective B lymphocytes were also observed in CKO BM (Figure 2B). We next examined the B-cell compartment in the BM according to the well-established surface markers (Online Supplementary Figure S2E ), which is characterized by pre-pro B cell, pro-B cell, pre-B cell, immature B cell, and mature B cell.33 Loss of Ppm1b resulted in a remarkable decrease in cell number in all subpopulations of B lymphocytes compared to that of WT controls, although there was no difference in their relative frequencies between the two groups of mice (Online Supplementary Figure S2E-I). Furthermore, common lymphoid progenitor (CLP) cells that commit into B-lineage progenitor cells also exhibited a decrease in CKO mice (Figure 2C, D). However, loss of Ppm1b did not disrupt the T lymphogenesis that also starts with the commitment of CLP, evidenced by the equal thymus and subpopulations of T lymphocytes (Online Supplementary Figure S2J-L). These findings suggest that Ppm1b is required for the commitment and proliferation of B-biased lymphoid progenitor cells from CLP, which is further supported by the markedly reduced colony number
and size from CKO lineage-negative BM cells supplemented with IL-7 (Figure 2E, F, Online Supplementary Figure S2M).
Loss of Ppm1b led to the suppression of phenotypic hematopoietic stem cell expansion in vivo
Ppm1bCKO mice exhibited a significantly diminished frequency and absolute number of HSC (lineage-Sca1+c-Kit+, LSK) in BM (Figure 3A, Online Supplementary Figure S3A), while the committed HPC (lineage-Sca1-c-Kit+, LK) were comparable between these two groups of mice (Online Supplementary Figure S3B). To further explore the impact of Ppm1b deficiency on the HSC, we examined its subpopulations according to the different identifications.8,34 Interestingly, Ppm1bCKO mice exhibited the obviously decreased long-term HSC (CD34 CD135 LSK, LT-HSC) and short-term HSC (CD34 +CD135 LSK, ST-HSC) (Figure 3B, Online Supplementary Figure S3C). A similar reduction was also observed in SLAM-HSC (CD150+CD48 LSK) representing the quiescent HSC in Ppm1bCKO mice (Online Supplementary Figure S3D).35 To further determine the number of functional HSC of Ppm1bCKO mice in the BM, we performed limiting dilution assays and found that the frequency of competitive repopulation units (CRU) in Ppm1bCKO mice was significantly lower than that of control mice (Figure 3C). Indeed, Ppm1bCKO HSC showed comparable homing activity to WT ones (Online Supplementary Figure S3E). Moreover, Ppm1b deletion had an undetectable effect on the HSC survival (data not shown). Cell cycle analysis by Ki67 staining demonstrated that Ppm1b-deficient HSC showed more quiescence than that of WT control (Figure 3D). This is confirmed by the low cell division of ex vivo cultured Ppm1b-deficient LSK indicated by the relatively high CFSE staining ( Online Supplementary Figure S3F ). These findings indicate that loss of Ppm1b impaired the active proliferation of HSC.
To confirm the regulatory role of Ppm1b in functional HSC, we challenged CKO mice with 5-fluorouracil (5-FU) that eliminates cycling cells and triggers the hematopoietic repopulation.36 All the Ppm1bCKO mice died at 20 days of continuous 5-FU injection, whereas 40% of WT mice survived after 3 rounds of administration (Figure 3E), indicating that Ppm1b is required for the stressed reconstitution of HSC. Consistently, the frequencies of HSC at the G0 and G1 phase in CKO mice also showed a more dramatic decrease after 5-FU administration compared with the steady state (Online Supplementary Figure S3G), suggesting that Ppm1b deletion impaired the transition between G0 to G1 phase under stress hematopoiesis.
To further assess the function of Ppm1b-deficient HSC, we performed competitive serial transplantation assays (Online Supplementary Figure S3H). Ppm1bCKO-derived cells showed a successive reduction in the primary recipients and subsequent recipients (Online Supplementary Figure S3 I), indicating the impaired reconstitution of CKO HSC. Notably, the frequencies of LSK and SLAM-LSK derived
from Ppm1b CKO BM were also significantly reduced in primary and subsequent recipients (Figure 3F, Online Supplementary Figure S3J ). In addition, B cells derived from Ppm1b CKO BM also showed a progressive decrease in the recipients ( Online Supplementary Figure S3K, L ), which is consistent with the defect of B cells in primary CKO mice.
Ppm1b regulated the hematopoietic stem cell pool and B-cell differentiation in adult hematopoiesis
Given that the defects of Ppm1b CKO mice may be due to a consequence of embryo stage, we analyzed whether Ppm1b also play roles in adult hematopoiesis. To this end, WT mice were pretreated with Ppm1b inhibitor HN252 and then challenged with a sublethal dose of 5-FU (Figure
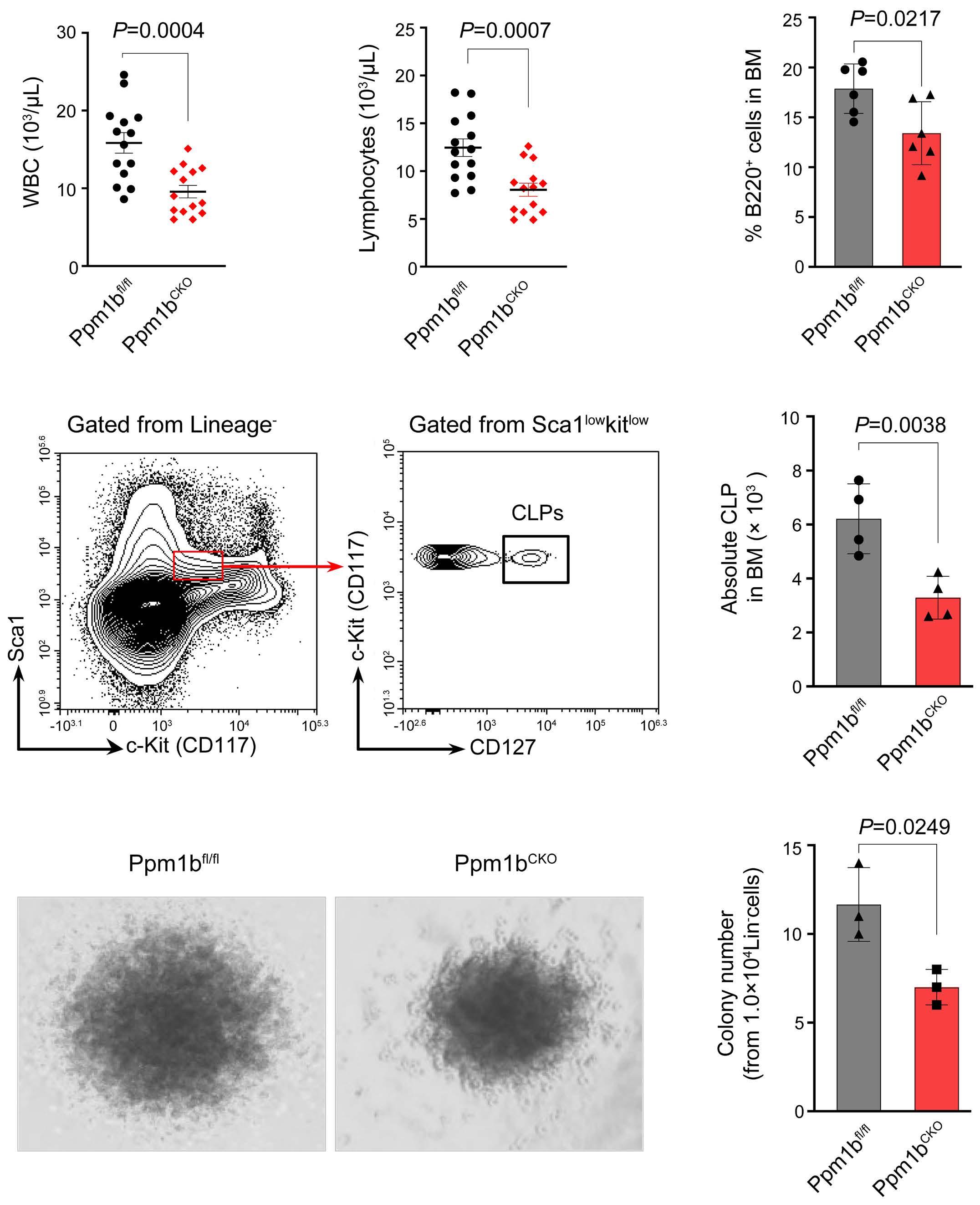
Figure 2. Ppm1b deficiency mice exhibited B-cell leukocytopenia. (A) Absolute number of white blood cells (WBC) and lymphocytes in peripheral blood from Ppm1bCKO and Ppm1bfl/fl mice at the age of 8 weeks (N=14). (B) Statistical analysis of the percentage of B cells in bone marrow (BM) from indicated mice (N=6). (C) Representative flow cytometric profiles of the strategy for common lymphoid progenitors (CLP), Lin Sca-1lowc-KitlowCD127+ cells. (D) Quantification of the cell number of CLP cells in BM from indicated mice (N=4). (E) Representative profiles of the colonies after 7 days of culture from indicated mice. (F) Quantification of the colony number in (I) (N=3). All P values were determined by unpaired two-tailed Student t test. See also Online Supplementary Figure S2.
4A). Compared to control group mice, HN252 treatment led to a greater decrease and subsequently slower recovery of WBC and lymphocytes but not PLT and RBC in PB (Figure 4B, Online Supplementary Figure S4A). HN252-treated mice
showed markedly reduced B220+ cells in PB and BM after 12 days of 5-FU injection when the blood cells had completely recovered in control mice (Figure 4C, D). Moreover, it is notable that the proportion of LSK cells was also
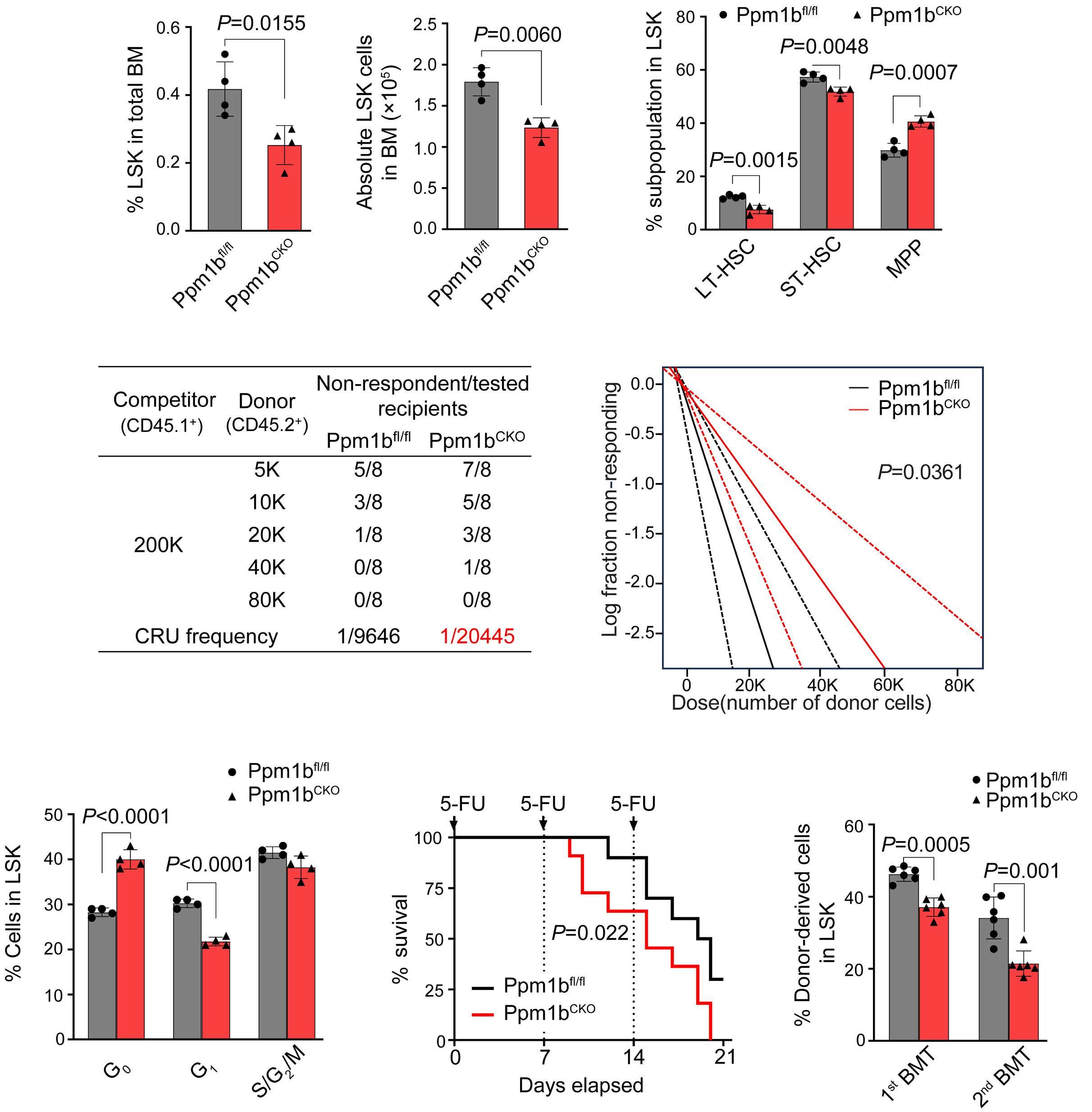
Figure 3. Loss of Ppm1b leads to the suppression of phenotypic hematopoietic stem cell expansion in vivo. (A) Statistical analysis of the percentage (left) and number (right) of Lin Sca-1+c-Kit+ (LSK) cells in 8-week-old Ppm1bCKO and Ppm1bfl/fl mice. (A) N=4. (B) Statistical analysis of the percentage of LT-HSC, ST-HSC and MPP cells (all gated from LSK) (N=4). (C) Limiting dilution transplantation assays were performed by transplanting different doses of bone marrow mononuclear cells (BMMC) from indicated mice. (Left) The number of functional hematopoietic stem cell (HSC) in bone marrow (BM) of Ppm1bCKO and Ppm1bfl/fl mice was measured by flow cytometry after 16 weeks of transplantation. Recipients with <2% donor-derived cells in BM were defined as non-respondent (N=8). (Right) Fraction of HSC in BM of Ppm1bCKO and Ppm1bfl/fl mice by Poisson statistical analysis of the data using L-Calc software. Solid lines indicate the best-fit linear model for each dataset. Dashed lines for 95% Confidence Intervals; χ2 test=4.39; P=0.0361. (D) Cell cycle analysis of LSK cells from indicated mice by flow cytometry using Hoechst 33342 and Ki67 staining (N=4). (E) Kaplan-Meier survival curves of Ppm1bCKO and Ppm1bfl/fl mice treated thrice with 5-FU (150 mg/kg) (N=12). (F) Percentage of donor-derived LSK cells in the BM of the primary and secondary recipient mice (N=6). Twotailed unpaired Student t test was used to generate the P values. The significance for survival analyses was calculated by logrank (Mantel-Cox) test. See also Online Supplementary Figure S3. BMT: bone marrow transplantation; CRU: competitive repopulating units.
significantly reduced in HN252-treated mice BM upon the challenge of 5-FU (Figure 4E). These findings phenocopied the defect of the HSC pool and B-cell differentiation in Ppm1bCKO mice. To avoid the off-target effect of HN252, we performed experiments to confirm the specificity of HN252 on Ppm1b. There were no significant changes in WBC and lymphocytes in PB observed in Ppm1b deficient mice with or without HN252 treatment after 5-FU injection (Online Supplementary Figure S4B), as well as the cell number of B220+ cells and the proportion of LSK cells in the BM
(Online Supplementary Figure S4C, D). In addition, HN252 treatment showed no notable effect on the relative mean fluorescence intensity of CFSE in Ppm1b-deficient LSK (Online Supplementary Figure S4E). These results indicate that HN252 affects the HSC pool and B-cell differentiation in adult hematopoiesis mainly via targeting Ppm1b.
Transcriptome profiling of Ppm1b-deficient hematopoietic stem cells reveals altered stem cell property
To explore the basis of abnormal hematopoiesis upon Ppm1b
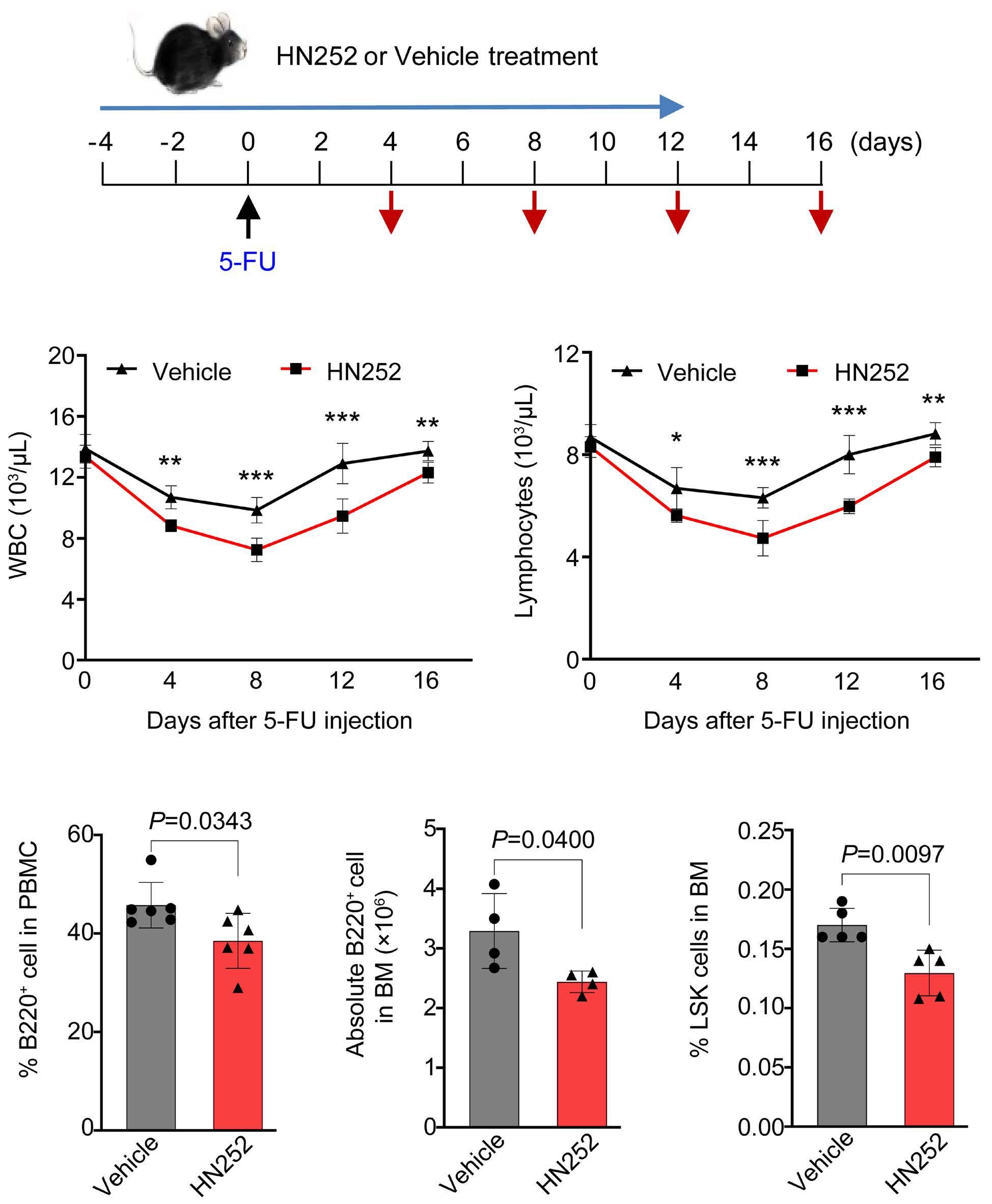
Figure 4. Ppm1b regulates hematopoietic stem cell pool and B-cell differentiation in stressed hematopoiesis. (A) Experimental design for HN252-treated mice in response to 5-FU-induced emergency hematopoiesis. Long blue arrow indicates HN252 or Vehicle injection every 2 days. Black arrow indicates the time point for 5-FU injection. Red arrows indicates the bleeding for blood assays. (B) Quantification of white blood cells (WBC) and lymphocytes in peripheral blood at indicated time points during HN252 injection (N=6). (C) The mice were treated with HN252 as in (A), and the ratio of B cells in peripheral blood was analyzed by flow cytometry (N=6). (D and E) The number of B cells (D) and the percentage of Lin Sca-1+c-Kit+ (LSK) cells (E) from indicated mice in (C) were measured by flow cytometry (N=6). Data are presented as mean ± Standard Deviation. All P values were determined by unpaired two-tailed Student t test. See also Online Supplementary Figure S4. BM: bone marrow; GO: gene ontology; PBMC: peripheral blood mononuclear cells.
deficiency, we performed RNA sequencing analysis with purified lineage negative cells from Ppm1bCKO and the corresponding mice (Online Supplementary Figure S5A). Enriched gene sets were notably related to B-cell activation and immunoglobulin production (Online Supplementary Figure S5B). Given that transcription factors are crucial in controlling B-cell development,37 we next examined the expression of the multiple transcriptional regulators in B220+ BM cells. Compared to B220+ cells from the corresponding mice, Ppm1bCKO B cells showed significant downregulation of these transcription factors (Online Supplementary Figure S5C), in which flow cytometry further confirmed Forkhead Box O1 (FOXO1) protein levels to be reduced (Online Supplementary Figure S5D).
Moreover, KEGG enrichment analysis revealed that Wnt signaling pathway was substantially enriched in the Ppm1bCKO group (Figure 5A, Online Supplementary Table S1). Several Wnt signaling-related genes were down-regulated upon the deletion of Ppm1b, including Fzd1, Jun, Camk2b, Lrp5, Ccnd1, and Gpc4 (Online Supplementary Figure S6A). The reduced pool of HSC in Ppm1bCKO mice prompted us to investigate the basis of impaired HSC. We then performed single-cell RNA sequencing with sorted LSK cells from Ppm1bCKO mice and their corresponding controls. A total of 3,651 CKO and 3,713 WT cells were divided into 24 clusters with unsupervised clustering, which was visualized with uniform manifold approximation and projection (UMAP) (Online Supplementary Figure S6A, B). Four subpopulations referring to
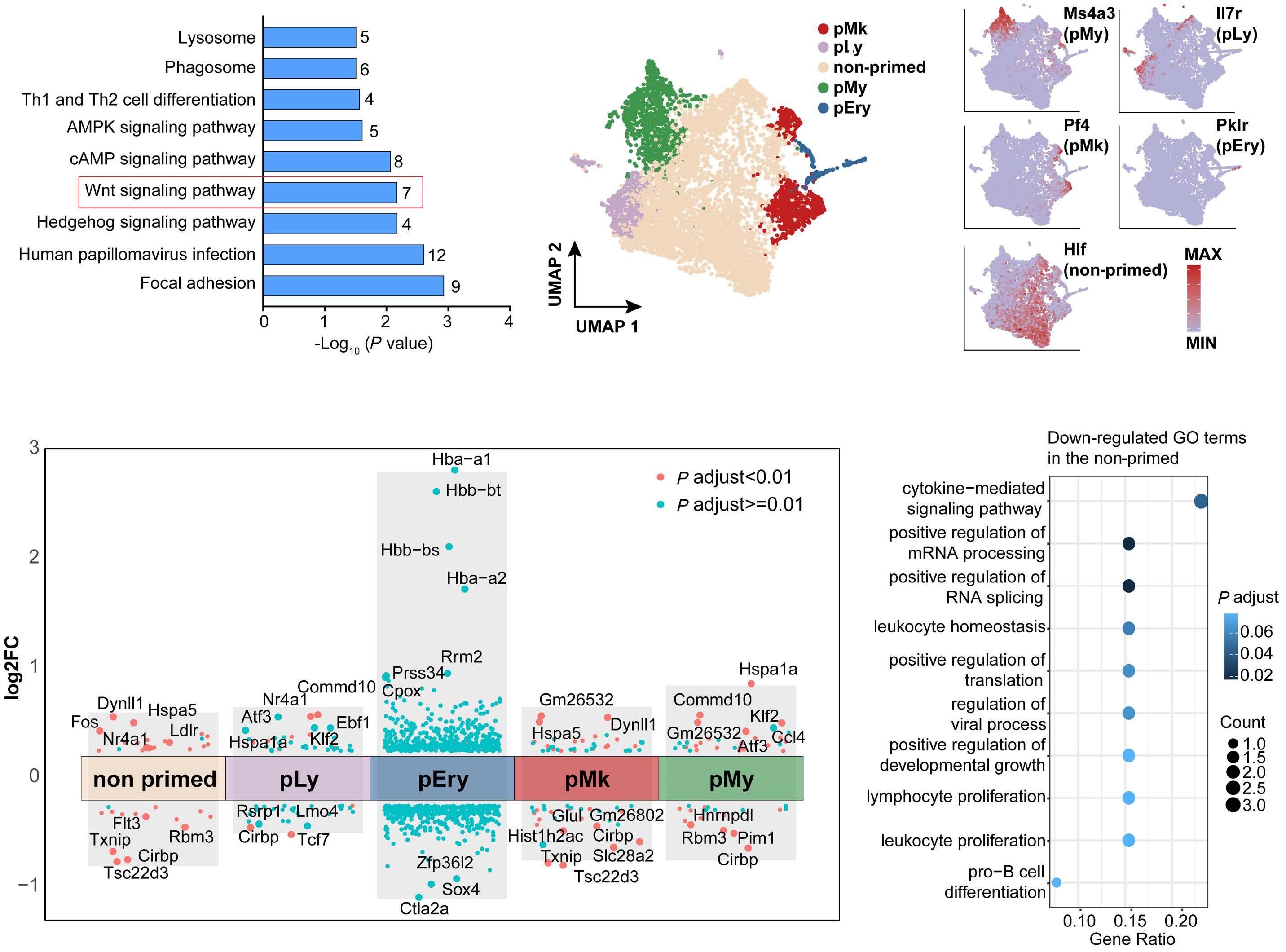
Figure 5. Transcriptome profiling of Ppm1b-deficient hematopoietic stem cells reveals altered stem cell properties. (A) KEGG pathway analysis indicating altered signaling pathways including Wnt signaling between Ppm1bCKO and Ppm1bfl/fl LSK cells (P<0.05). Numbers shown in each bar indicate the number of enriched proteins in relative terms. (B) A uniform manifold approximation and projection (UMAP) plot of 7,364 Lin Sca-1+c-Kit+ (LSK) cells (3,651 for pm1bfl/fl, 3,713 for Ppm1bCKO) categorized into 5 clusters based on distinct differentiation potentials. (C) UMAP plots illustrating principal identifiers (expression of marker genes) of various LSK sub-populations. (D) Volcano plot showing differential expressed genes (DEG) between Ppm1bCKO and Ppm1bfl/fl within each cluster (P.adjust<0.01). (E). Gene ontology (GO) analysis of down-regulated DEG between Ppm1bCKO and Ppm1bfl/fl in the non-primed cluster (P<0.05). See also Online Supplementary Figures S5 and S6.
the commitment gene markers were identified, including myeloid-biased (pMy), lymphoid-biased (pLy), megakaryocytic-biased (pMk), and erythroid-biased (pEry) clusters (Figure 5B). The remaining cell populations, characterized by the maintenance of stemness and lacking a specific propensity for lineage differentiation according to enrichment analysis, were annotated as “non-primed”. This cluster was further confirmed by the elevated expression of Hlf, a master transcription factor predominantly expressed in an HSC subpopulation with extensive self-renewal capacity (Figure 5C).38 Consistent with the findings in flow cytometric analysis, the relative frequency of the “non-primed” cluster was also reduced in the Ppm1bCKO group (Online Supplementary Figure S6C).
Several genes associated with HSC homeostasis were identified in the non-primed cluster between CKO and WT groups (Figure 5D, Online Supplementary Table S2). Nf4a1 has been reported to restrict HSC proliferation in part through activation of a C/EBPα-driven antiproliferative network by directly binding to a hematopoietic-specific Cebpa enhancer, a member of the Nr4a subfamily of nuclear receptors.39 Fos serves as a gatekeeper in the cell cycle progression of dormant HSC, whose sustained expression inhibits HSC entry into the cell cycle.40 These two genes showed obvious upregulation in CKO HSC compared to that of the corresponding controls (Figure 5D). On the other hand, it has been demonstrated that down-regulated Txnip is required for the hematopoietic reconstitution and HSC self-renewal capacity.41 In line with the previous report that low FLT3 expression led to a blockage at G0 phase in HSC,42 CKO HSC showed reduced FLT3 expression and enhanced quiescence. Furthermore, GO enrichments with these differentially expressed genes revealed that the physiological activity was down-regulated in CKO HSC, such as cytokine-mediated signaling, mRNA metabolism, and translation, which phenocopies the quiescent status (Figure 5E). These transcriptomic changes further confirmed the impact of Ppm1b on HSC expansion.
Loss of Ppm1b impaired the functional expansion of hematopoietic stem cells through Wnt/b-catenin pathway
Given that the phosphorylation of b-catenin functions as a downstream key node of Wnt signaling, and it has been previously identified as a potential subtract of PPM1B, we speculated that Ppm1b regulates HSC via Wnt/b-catenin signaling. Indeed, the phosphorylation of b-catenin was dramatically increased in Ppm1bCKO HSC compared to that of WT controls (Figure 6A). The target genes of Wnt/b-catenin signaling such as Cylin D1 and Myc, were also down-regulated upon Ppm1b deletion (Figure 6B). Proximity ligation assay in sorted LSK cells indicated that Ppm1b interacted with b-catenin (Figure 6C). Moreover, Ppm1b inhibitor HN252 treatment led to a considerable decrease in the active b-catenin (non-phosphorylated) in the LSK cells (Online Supplementary Figure S7A, B), which is further confirmed by the downregulation
of its target genes (Online Supplementary Figure S7C). These data suggest that Ppm1b mediates the b-catenin activity via the dephosphorylation in HSC.
To confirm Wnt/b-catenin signaling is responsible for the impaired expansion of HSC with Ppm1b deficiency, we performed rescue experiments with a potent Wnt signaling activator BML-284 (BML)43 and found that BML could activate the expression of Wnt-targeted genes in Ppm1b deficient cells (Online Supplementary Figure S7D). Lineage negative BM cells were sorted from CKO and WT mice and labeled with CFSE, followed by in vitro culture with BML treatment. Compared to the high MFI of CFSE in Ppm1bCKO LSK cells after 48 hr ex vitro culture, BML treatment led to a significant reduction in CFSE staining (Figure 6D). This was further confirmed by the increase in LSK cells derived from Ppm1bCKO mice upon BML treatment (Online Supplementary Figure S7E), indicating that BML reversed the suppressed HSC expansion with Ppm1b deficiency.
To verify the functional roles of Ppm1b/Wnt/b-catenin axis in HSC expansion in vivo, we pretreated Ppm1bCKO and WT mice with BML for four days and then challenged them with a sublethal dose of 5-FU. Similar to the findings in ex vitro culture, the frequencies and cell number of HSC in Ppm1bCKO mice were remarkably increased by BML treatment, whereas the HSC in Ppm1bfl/fl mice remained unchanged (Figure 6E, Online Supplementary Figure S7F). Furthermore, BML treatment led to a significant decrease in LSK cells at G0 phases but a notable increase at the G2/S/M-phase in Ppm1bCKO mice, compared to that of the control group (Figure 6F). However, BML had no detectable impact on the cell cycle of LSK cells in WT control mice (Figure 6F). These data confirm that loss of Ppm1b impaired the HSC homeostasis via the blockage of Wnt/b-catenin signaling.
In stable state conditions, most HSC are in a state of quiescence with only a small proportion undergoing self-renewal or differentiation.44,45 The balance between the quiescence and activation of HSC is tightly regulated to protect the HSC pool from exhaustion, which is also required for the capabilities of long-term hematopoiesis.46 In this study, we identified the critical role of Ppm1b in the regulation of HSC homeostasis and B-cell development in vitro and in vivo. Ppm1b inhibition suppressed the expansion of HSC due to the blockage of the cell cycle at G0 phase. Moreover, Ppm1b deficiency led to a reduction in the early B-cell progenitors committed from CLP, which consequently impaired B-cell differentiation (Figure 7). Altogether, our work demonstrated that Ppm1b emerges as a key regulator in normal hematopoiesis. The canonical Wnt/b-catenin signaling has been documented to be crucial for the self-renewal and differentiation of HSC.47 However, its regulatory role in HSC remains a subject of debate.48 Weissman et al. found that constitutive
activation of Wnt/b-catenin signaling led to the enhanced proliferation and repopulation of HSC via overexpression of active b-catenin.49 In line with this finding, Kincade et al. also confirmed that constitutively active b-catenin accelerates the expansion of HSPC in vitro 50 However, other research groups demonstrated that constitutive activation of b-catenin inhibited the repopulation and differentiation of HSC by using transgenic mice.51,52 These inconsistent re-
sults indicated that the role of Wnt/b-catenin signaling in HSC fate decisions is dependent on its degree. Our finding underscored the essential roles of Wnt/b-catenin signaling in HSC homeostasis. Loss of Ppm1b led to the decrease in active b-catenin (non-phosphorylated state) and its target genes’ expression in HSC, resulting in a blockage of self-renewal in Ppm1b-deficient HSC. This was efficiently reversed by activating Wnt signaling (Figure 7).
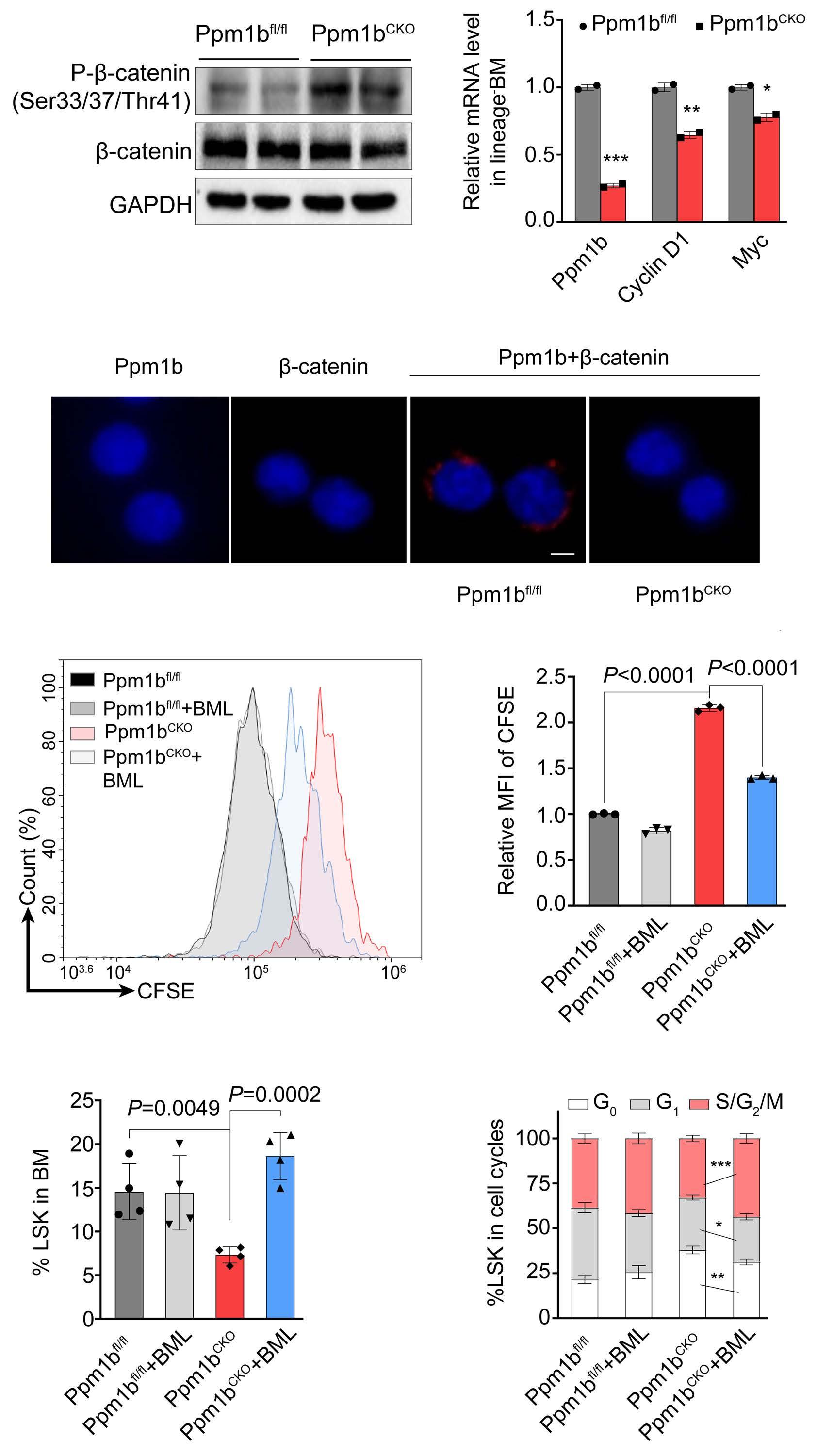
Figure 6 Loss of Ppm1b impaired the function of hematopoietic stem cells through Wnt/b-catenin pathway. (A) Western blot analysis of indicated proteins in purified lineage negative cells from Ppm1bCKO and Ppm1bfl/fl mice. (B) Quantification of mRNA expression of Ppm1b, cyclin D1 and Myc in cells from (A). 18S ribosomal RNA was used as an internal control. (C) Representative confocal images of proximity ligation assay between Ppm1b and b-catenin in Lin Sca1+c-Kit+ (LSK) cells. Sorted LSK cells from 3 independent Ppm1bCKO and Ppm1bfl/fl mice were stained with indicated antibodies separately, and were visualized using a fluorescent microscope. Scale bar: 5 μM. (D) Lineage negative cells purified from indicated mice were labeled with CFSE, and then the cells were cultured with or without BML-284 (10 nM) for 48 hours (hr) in vitro. The fluorescence intensity of CFSE of LSK cells was quantified by flow cytometry. (E) Ppm1bCKO and Ppm1bfl/fl mice were treated with BML-284 (5 mg/kg) or not for 4 days, followed by a single dose injection of 5-FU for another 7 days. The frequency of LSK cells was determined from indicated mice by flow cytometry (N=4). (F) Cell cycles of LSK cells in (E) were analyzed by flow cytometry using Hoechst 33342 and Ki67 staining (N=4). *P<0.05, **P<0.01, ***P<0.001. All P values were determined by unpaired twotailed Student t test. Data are presented as mean ± Standard Deviation from 3 independent experiments. See also Online Supplementary Figure S7. BM: bone marrow; MFI: mean fluorescence intensity.
We demonstrated that Ppm1b interacted with b-catenin, and Ppm1b deletion led to the inhibition of Wnt/b-catenin signaling via the dephosphorylation in HSC. Although the role of Wnt/b-catenin signaling in B-cell development has been revealed by LEF1 KO mice that led to a reduction in B cells,53 we did not observe any changes in the phosphorylation of b-catenin and downstream target genes’ expression in Ppm1b-deficient B cells (data not shown). This is consistent with our findings that Ppm1b is required for the commitment and proliferation of B-biased lymphoid progenitor cells from CLP but not the consequent B-cell differentiation. Given that B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is characterized by uncontrolled proliferation of early B-cell progenitors, our work identified Ppm1b as the potential target for the clinical treatment of BCP-ALL. Our data showed that loss of Ppm1b led to the defect in B-cell but not T-cell development. Indeed, lymphopoiesis is an intricate process that gives rise to all lymphocytes which starts with the commitment of HSC to CLP.54 Even though
CLP generate T and B cells when properly stimulated,55 it seems that CLP are mainly progenitors for B cells, owing to the limited or negligible T-cell generation from CLP.56,57 By the expression of Ly6D, CLP can be defined as all-lymphoid progenitors (Ly6D ), which have the potential to generate T and natural killer cells, and B-cell biased lymphocyte progenitors (Ly6D+).58 Moreover, there are still CLP-independent pathways of innate T-cell development which are more efficient than CLP.59 Importantly, the changes in lymphocyte counts in the first two weeks after 5-FU were most likely not driven by differentiation from HSC but rather due to lymphocyte death and subsequent homeostatic proliferation, as well as differentiation from more committed progenitors (such as BM CLP or Flt3+ MPP) (Figure 4). Given that PLT and RBC in HN252-treated mice showed mild changes compared to that of the control group, a single 5-FU treatment is probably not sufficient to impair the function of HSC pretreated with the PPM1B inhibitor. These data would rather seem to point toward a CLP-intrinsic effect of PPM1B. However, these
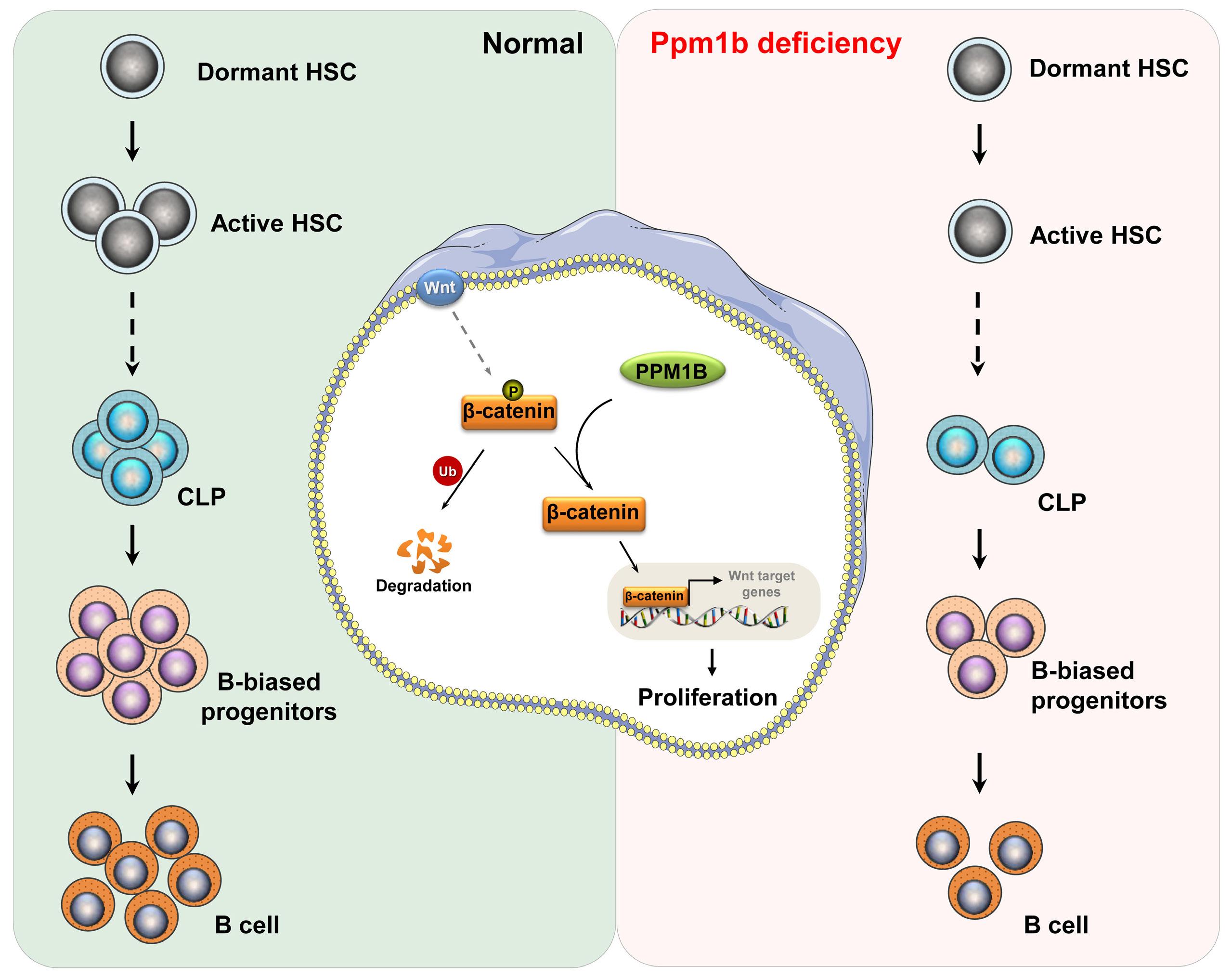
Figure 7. Schematic diagram illustrates the regulatory role of Ppm1b in hematopoietic stem cell self-renewal and B-cell development. Ppm1b is required for hematopoietic stem cell (HSC) proliferation and early B-cell progenitor differentiation. Loss of Ppm1b led to the decrease of the active b-catenin (non-phosphorylated) that interrupted the Wnt/b-catenin signaling in HSC, which consequently suppressed HSC expansion. CLP: common lymphoid progenitors.
findings were not invalidated by the serial transplant or CRU data, suggesting that there might be two at least partially independent stages of PPM1B activity. Consequently, the impact of Ppm1b deletion on B-cell development may take place at the level of stem and progenitor cells, even in populations lacking an apparent inclination for lineage differentiation. In summary, our study identified an essential role for Ppm1b in HSC homeostasis via Wnt/b-catenin signaling. Our findings may provide novel insights into the understanding of HSC self-renewal and B-cell development, and also offer clues for the pathogenesis of their related hematologic malignancies.
Disclosures
No conflicts of interest to disclose.
Contributions
BZ designed and guided research. ZL, YH, GX, XL and YL performed the experiments. ZL, YH, ZL, CZ and BZ analyzed the data. ZL and BZ wrote the original draft. ZL, YS, CZ and BZ reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
We thank the Translational Medicine Core Facility and Core
1. Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125(17):2605-2613.
2. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342(6161):1242974.
3. de Haan G, Lazare SS. Aging of hematopoietic stem cells. Blood. 2018;131(5):479-487.
4 Verovskaya EV, Dellorusso PV, Passegue E. Losing sense of self and surroundings: hematopoietic stem cell aging and leukemic transformation. Trends Mol Med. 2019;25(6):494-515.
5. Wilkinson AC, Igarashi KJ, Nakauchi H. Haematopoietic stem cell self-renewal in vivo and ex vivo. Nat Rev Genet. 2020;21(9):541-554.
6. Humphrey SJ, James DE, Mann M. Protein phosphorylation: a major switch mechanism for metabolic regulation. Trends Endocrinol Metab. 2015;26(12):676-687.
7 Kamada R. Metal-dependent Ser/Thr protein phosphatase PPM family: evolution, structures, diseases and inhibitors. Pharmacol Ther. 2020;215:107622.
8. Adolfsson J, Mansson R, Buza-Vidas N, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythromegakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121(2):295-306.
9. Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, Suda T. In vivo and in vitro stem cell function of c-kit- and Sca-1-positive murine hematopoietic cells. Blood. 1992;80(12):3044-3050.
10. Ito K, Turcotte R, Cui JH, et al. Self-renewal of a purified Tie2(+) hematopoietic stem cell population relies on mitochondrial clearance. Science. 2016;354(6316):1156-1160.
11. Holmes T, O’Brien TA, Knight R, et al. Glycogen synthase kinase-3 beta inhibition preserves hematopoietic stem cell activity and inhibits leukemic cell growth. Stem Cells.
Facility of the School of Basic Medical Sciences of Shandong University for consultation and instrument availability that supported this work. This work was supported by grants from the National Natural Science Foundation of China (81874294, 32300957), Taishan Scholars Program (TSQN201812015) and the key Program of Natural Science Foundation of Shandong Province (ZR2022LSW027), Qilu Young Scholars Program (to BZ), and the Multidisciplinary Research and Innovation Team of Young Scholars (2020QNQT007) of Shandong University. This work was also supported by the program for Innovative Research Team at University of the Ministry of Education of China (IRT_17R68), the academic promotion program of Shandong First Medical University (No.2019LJ003), the Natural Science Foundation of Shandong Province (ZR2023QH427), and the Innovation Team of Shandong Higher School Youth Innovation Technology Program (2022KJ197).
Data-sharing statement
Supporting data are available in the Online Supplementary Appendix. RNA sequencing data that support the findings of this study have been deposited in NCBI SRA under accession number GSE23774. The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
2008;26(5):1288-1297.
12. Li CZ, Wu BH, Li YS, et al. Loss of sphingosine kinase 2 promotes the expansion of hematopoietic stem cells by improving their metabolic fitness. Blood. 2022;140(15):1686-1701.
13. Zhang XF, Wang JF, Matczak E, Proper J, Groopman JE. Janus kinase 2 is involved in stromal cell-derived factor-1 alphainduced tyrosine phosphorylation of focal adhesion proteins and migration of hematopoietic progenitor cells. Blood. 2001;97(11):3342-3348.
14. Chan G, Cheung LS, Yang WT, et al. Essential role for Ptpn11 in survival of hematopoietic stem and progenitor cells. Blood. 2011;117(16):4253-4261.
15. Ni F, Yu WM, Wang X, et al. Ptpn21 controls hematopoietic stem cell homeostasis and biomechanics. Cell Stem Cell. 2019;24(4):608-620
16. Zhang J, Vakhrusheva O, Bandi SR, et al. The phosphatases STS1 and STS2 regulate hematopoietic stem and progenitor cell fitness. Stem Cell Reports. 2015;5(4):633-646.
17 Liu XY, Zhang FF, Zhang YP, et al. PPM1K regulates hematopoiesis and leukemogenesis through CDC20-mediated ubiquitination of MEIS1 and p21. Cell Rep. 2018;23(5):1461-1475.
18. Chen ZY, Yi WW, Morita YH, et al. Wip1 deficiency impairs haematopoietic stem cell function via p53 and mTORC1 pathways. Nat Commun. 2015;6:6808.
19 Marley AE, Kline A, Crabtree G, Sullivan JE, Beri RK. The cloning expression and tissue distribution of human PP2C beta. FEBS Lett. 1998;431(1):121-124.
20 Li ZY, Chen RY, Li YX, et al. A comprehensive overview of PPM1B: from biological functions to diseases. Eur J Pharmacol. 2023;947:175633.
21. Skottman H, Mikkola M, Lundin K, et al. Gene expression
signatures of seven individual human embryonic stem cell lines. Stem Cells. 2005;23(9):1343-1356.
22. Sasaki M, Ohnishi M, Tashiro F, et al. Disruption of the mouse protein Ser/Thr phosphatase 2Cbeta gene leads to early preimplantation lethality. Mech Dev. 2007;124(6):489-499.
23. Lu ZY, Xiao P, Zhou Y, et al. Identification of HN252 as a potent inhibitor of protein phosphatase PPM1B. J Cell Mol Med. 2020;24(22):13463-13471.
24. Yien YY, Bieker JJ. Functional Interactions between erythroid Kruppel-like factor (EKLF/KLF1) and protein phosphatase PPM1B/ PP2C beta. J Biol Chem. 2012;287(19):15193-15204.
25. Lu ZY, Huang LX, Li YX, et al. Fine-tuning of cholesterol homeostasis controls erythroid differentiation. Adv Sci. 2022;9(2):2102669.
26. Lu ZY, Xu GS, Li YX, Lu CH, Shen YM, Zhao BB. Discovery of N-arylcinnamamides as novel erythroblast enucleation inducers. Bioorg Chem. 2022;128:106105.
27. Mei Y, Han X, Liu YJ, Yang J, Sumagin R, Ji P. Diaphanous-related formin mDia2 regulates beta2 integrins to control hematopoietic stem and progenitor cell engraftment. Nat Commun. 2020;11(1):3172.
28. Blanchard EL, Loomis KH, Bhosle SM, et al. Proximity ligation assays for in situ detection of innate immune activation: focus on in vitro-transcribed mRNA. Mol Ther Nucleic Acids. 2019;14:52-66.
29 Tario JD, Humphrey K, Bantly AD, Muirhead KA, Moore JS, Wallace PK. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. J Vis Exp. 2012;70:e4287.
30 Filby A, Begum J, Jalal M, Day W. Appraising the suitability of succinimidyl and lipophilic fluorescent dyes to track proliferation in non-quiescent cells by dye dilution. Methods. 2015;82:29-37.
31. Wilkinson AC, Ishida R, Nakauchi H, Yamazaki S. Long-term ex vivo expansion of mouse hematopoietic stem cells. Nat Protoc. 2020;15(2):628-648.
32. Wilkinson AC, Nakauchi H. Stabilizing hematopoietic stem cells in vitro. Curr Opin Genet Dev. 2020;64:1-5.
33. Klein C, Zwick A, Kissel S, et al. Ptch2 loss drives myeloproliferation and myeloproliferative neoplasm progression. J Exp Med. 2016;213(2):273-290.
34. Yang L, Bryder D, Adolfsson J, et al. Identification of Lin(-)Sca1(+) kit(+)CD34(+)Flt3- short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood. 2005;105(7):2717-2723.
35. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109-1121.
36. Takebe N, Zhao SC, Ural AU, et al. Retroviral transduction of human dihydropyrimidine dehydrogenase cDNA confers resistance to 5-fluorouracil in murine hematopoietic progenitor cells and human CD34(+)-enriched peripheral blood progenitor cells. Cancer Gene Ther. 2001;8(12):966-973.
37. Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;26(6):715-725.
38. Lehnertz B, Chagraoui J, MacRae T, et al. HLF expression defines the human hematopoietic stem cell state. Blood. 2021;138(25):2642-2654.
39 Freire PR, Conneely OM. NR4A1 and NR4A3 restrict HSC proliferation via reciprocal regulation of C/EBPα and inflammatory signaling. Blood. 2018;131(10):1081-1093.
40 Okada S, Fukuda T, Inada K, Tokuhisa T. Prolonged expression of
c-fos suppresses cell cycle entry of dormant hematopoietic stem cells. Blood. 1999;93(3):816-825.
41. Jeong M, Piao ZH, Kim MS, et al. Thioredoxin-interacting protein regulates hematopoietic stem cell quiescence and mobilization under stress conditions. J Immunol. 2009;183(4):2495-2505.
42. Gotze KS, Ramirez M, Tabor K, Small D, Matthews W, Civin CI. Flt3(high) and Flt3(low) CD34(+) progenitor cells isolated from human bone marrow are functionally distinct. Blood. 1998;91(6):1947-1958.
43. Liu J, Wu X, Mitchell B, Kintner C, Ding S, Schultz PG. A smallmolecule agonist of the Wnt signaling pathway. Angew Chem Int Ed Engl. 2005;44(13):1987-1990.
44 Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195(5):709-720.
45. Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. 2014;141(24):4656-4666.
46. Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9(2):115-128.
47. Richter J, Traver D, Willert K. The role of Wnt signaling in hematopoietic stem cell development. Crit Rev Biochem Mol Biol. 2017;52(4):414-424.
48. Carpenter KA, Thurlow KE, Craig SEL, Grainger S. Wnt regulation of hematopoietic stem cell development and disease. Curr Top Dev Biol. 2023;153:255-279.
49 Reya T, Duncan AW, Ailles L, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409-414.
50 Baba Y, Yokota T, Spits H, Garrett KP, Hayashi SI, Kincade PW. Constitutively active beta-catenin promotes expansion of multipotent hematopoietic progenitors in culture. J Immunol. 2006;177(4):2294-2303.
51. Kirstetter P, Anderson K, Porse BT, Jacobsen SEW, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7(10):1048-1056.
52. Scheller M, Huelsken J, Rosenbauer F, et al. Hematopoietic stem cell and multilineage defects generated by constitutive betacatenin activation. Nat Immunol. 2006;7(10):1037-1047.
53. Reya T, O’Riordan M, Okamura R, et al. Wnt signaling regulates B lymphocyte proliferation through a LEF-1 dependent mechanism. Immunity. 2000;13(1):15-24.
54 Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91(5):661-672.
55. Porritt HE, Rumfelt LL, Tabrizifard S, et al. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity. 2004;20(6):735-745.
56. Allman D, Sambandam A, Kim S, et al. Thymopoiesis independent of common lymphoid progenitors. Nat Immunol. 2003;4(2):168-174.
57. Schwarz BA, Bhandoola A. Circulating hematopoietic progenitors with T lineage potential. Nat Immunol. 2004;5(9):953-960.
58. Inlay MA, Bhattacharya D, Sahoo D, et al. Ly6d marks the earliest stage of B-cell specification and identifies the branchpoint between B-cell and T-cell development. Genes Dev. 2009;23(20):2376-2381.
59. Ghaedi M, Steer CA, Martinez-Gonzalez I, Halim TYF, Abraham N, Takei F. Common-lymphoid-progenitor-independent pathways of innate and T lymphocyte development. Cell Rep. 2016;15(3):471-480.
Akriti G. Jain,1 Somedeb Ball,2 Luis Aguirre,3 Najla Al Ali,4 David Kaldas,5 Sara Tinsley-Vance,4 Andrew Kuykendall,4 Onyee Chan,4 Kendra Sweet,4 Jeffrey E. Lancet,4 Eric Padron,4 David A. Sallman4 and Rami Komrokji4
1Leukemia Division, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH; 2VanderbiltIngram Cancer Center, Nashville, TN; 3Dana-Farber Cancer Institute, Boston, MA; 4Department of Malignant Hematology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL and 5Department of Internal Medicine, University of South Florida, Tampa, FL, USA
Correspondence: R. Komrokji Rami.Komrokji@moffitt.org
A.G. Jain Jaina9@ccf.org
Received: June 1, 2023.
Accepted: January 22, 2024. Early view: February 1, 2024. https://doi.org/10.3324/haematol.2023.283661
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
The patterns of low-risk myelodysplastic syndrome (MDS) progression and the clinical and molecular features of those patterns have not been well described. We divided our low-risk (LR) MDS patients (N=1,914) into 4 cohorts: 1) patients who remained LR-MDS (LR-LR; N=1,300; 68%), 2) patients who progressed from LR to high-risk (HR) MDS (LR-HR) without transformation into acute myeloid leukemia (AML) (N=317; 16.5%), 3) patients who progressed from LR to HR MDS and then AML (LR-HR-AML; N=124; 6.5%), and 4) patients who progressed from LR MDS directly to AML (LR-AML; N=173; 9%). Risk factors for progression included: male gender, low absolute neutrophil count (ANC), low platelet count, high bone marrow (BM) blasts, ferritin >1000 mcg/L, albumin <3.5 g/dL, multi-lineage dysplasia (MLD), and lack of ring sideroblasts. Among patients with marked BM fibrosis (N=49), 18% progressed directly to AML. Somatic mutations (SM) associated with an increased risk of direct or indirect AML progression included SRSF2 and NRAS. SM in IDH1, IDH2 and NPM1 were more common in patients with direct AML transformation. SM associated with progression to higher risk disease only, without AML transformation, were ASXL1, TP53, RUNX1, and CBL. SF3B1 mutation was associated with less progression. About 171 patients (13.1% of all LR-LR patients) died within two years of diagnosis of LR-MDS without disease progression. Among the 61 cases with documented cause of death, 18 patients (29.5%) died from cytopenia and MDS-related complications. Identifying patterns of disease progression of LR MDS patients and their predictive factors will be crucial to be able to tailor therapy accordingly.
Myelodysplastic syndromes (MDS) are heterogeneous stem cell neoplasms that primarily affect the elderly population. The approach to treatment of MDS includes management of symptoms or cytopenias, and treatments to alter the natural history of the disease in order to prevent progression to acute myeloid leukemia (AML). Management is largely risk-adapted based on various prognostic scoring systems. The International Prognostic Scoring System (IPSS) was introduced in 1997 and was an important standard to assess prognosis in patients with MDS at initial diagnosis.1 The IPSS divides patients into 4 risk categories: low, intermediate-1, intermediate-2, and high risk.1 Bone marrow (BM) blasts, karyotype and number of cytopenias
are the only 3 variables comprising the IPSS. The Revised International Prognostic Scoring System (IPSS-R) was introduced in 2012. This added a refined cytogenetic classification, degree of cytopenia, and improved BM blast percentage as categories, and introduced 5 subgroups of MDS: very low (VL) risk, low risk (LR), intermediate, high, and very high risk.2 Some other tools that have been developed and widely used for risk stratification include the Global MD Anderson Model, the lower risk MD Anderson model (LR-MDAS), and the WHO-based prognostic scoring system (WPSS).3-5 In low-risk disease (LR MDS), the aim of therapy is to improve cytopenias in patients with anemia, neutropenia and/ or thrombocytopenia in order to improve quality of life and prevent complications from cytopenias in an attempt to prolong overall survival (OS). Cytopenia-related complications
and exacerbation of co-existing comorbidities significantly affect the disease burden, and affect quality of life and OS in low-risk disease.6,7 The LR-MDAS model suggested that almost a third of the LR MDS patients stratified by the IPSS progress or die within a short time after diagnosis. It incorporated age, hemoglobin, severity of thrombocytopenia, karyotype, and BM blasts ≥4% as variables, and divided patients into 3 groups, upstaging 25% of LR MDS patients into the higher risk disease category. Historically, 30-40% of MDS patients progress to AML; however, unfortunately, the majority of patients are thought to succumb from disease complications, namely cytopenia or an interplay of cytopenia with comorbidities. The patterns of LR MDS disease progression and causes of mortality are not well delineated in literature. Disease progression can be in the form of further BM failure associated with more severe cytopenias or increased myeloblasts with transformation to higher risk disease and/or AML. The molecular phenotypes associated with different patterns of disease progression and clonal evolution have not been well studied. Hence, we undertook this study to better delineate factors that can help identify patients with LR MDS at risk for progression and/or death early in their disease course who may benefit from early intervention rather than the “wait and watch” approach.
Patient selection
We identified 1,914 patients with an established diagnosis of very low- and low-risk MDS as per the IPSS-R from the Moffitt Cancer Center MDS database. Laboratory values and prognostic scores were determined at the time of diagnosis or referral prior to treatment. The study was approved by the Institutional Review Board. We divided our patients into 4 cohorts based on the pattern of their disease progression: 1) patients who remained in the low-risk MDS category throughout their period of follow-up (LR-LR); 2) patients who progressed from LR to HR MDS (LR-HR) without AML transformation; 3) patients who progressed from LR to HR MDS and then AML (LR-HR-AML); and 4) patients who directly progressed from LR MDS to AML (LR-AML).
Characterization of mutational profile
Next generation sequencing (NGS) was performed on peripheral blood or BM as a part of standard work up during patients’ initial presentation. The NGS panels targeted up to 406 genes across multiple platforms: FoundationOne Heme, Genoptix Myeloid Molecular Profile, Genoptix NexCourse Complete, Illumina TruSight Myeloid-54, and the Moffitt 98-gene Myeloid Action Panel.
Definitions of survival
Overall survival (OS) was measured from time of diagnosis
until death or censored at time of last patient follow-up. For patients that progressed to AML, leukemia-free survival (LFS) was calculated from the time of diagnosis to development of AML.
Statistical methods
Categorical variables were compared using Fisher’s exact and χ2 tests, and quantitative data were compared using the Mann-Whitney U test. Time-to-event analyses were assessed using the Kaplan-Meier method. The log-rank test was used to compare OS between groups. Statistical analyses were performed using IBM SPSS statistics version 26.
We divided the patients with low-risk and very low-risk MDS into the 4 cohorts described above. The majority of the patients, 68% (N=1,300) remained in LR-MDS without progressing to high-risk MDS or AML, 16.5% patients (N=317) progressed from low-risk to high-risk MDS (LR-HR) without AML transformation, 6.5% patients (N=124) progressed from low-risk to high-risk MDS and then AML (LR-HR-AML), and 9% patients (N=173) progressed from low-risk MDS directly to AML (LR-AML). At a median follow-up of 99 months, the median OS for the LR-LR, LR-HR, LR-HR-AML, and LR-AML groups was 80.7, 61.2, 48.3, and 42.8 months, respectively (P<0.0001) (Figure 1). The median time to progression to AML in the LR-HR-AML and LR-AML groups was 29 months, and the median time to progress from LR to HR-MDS was
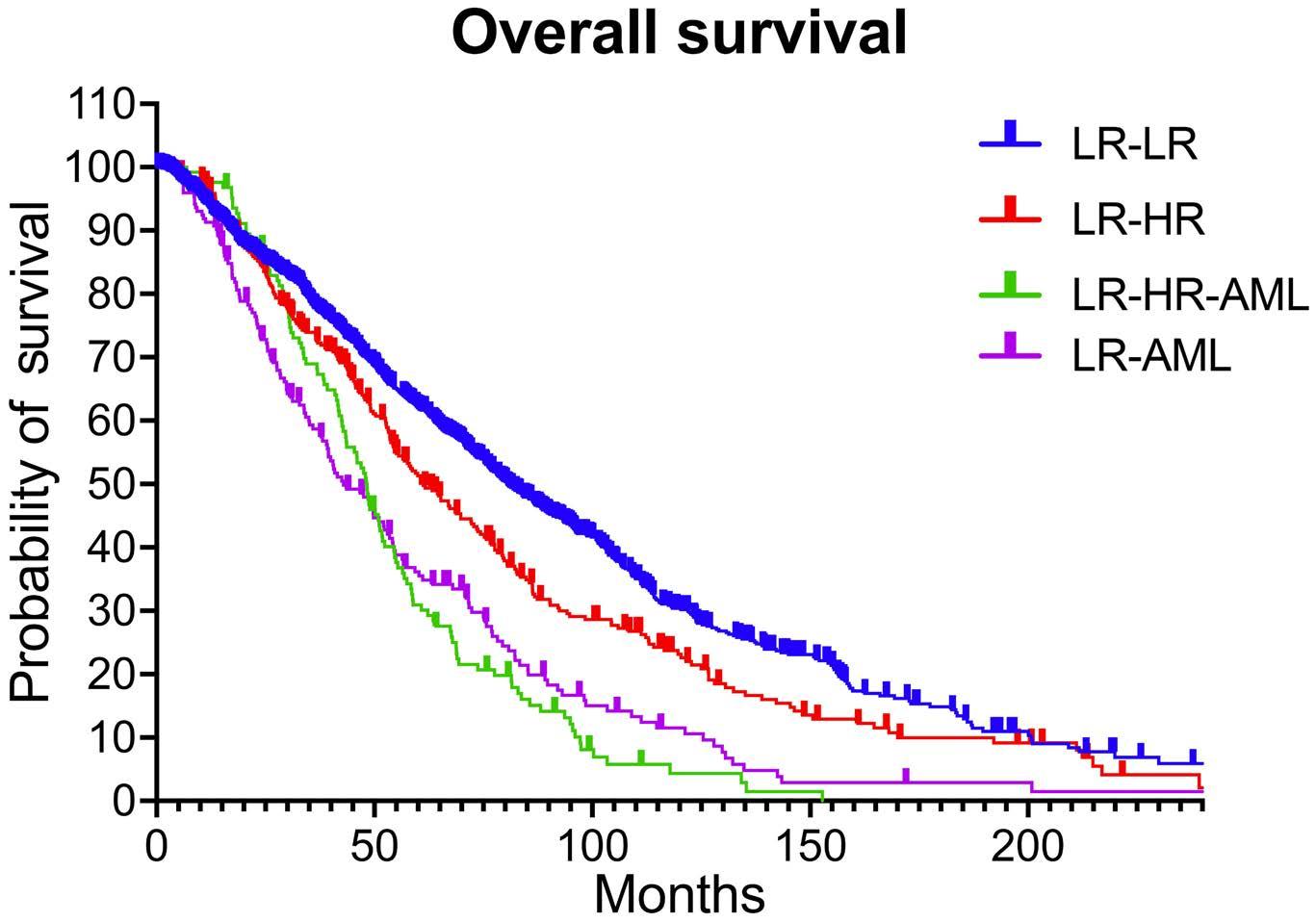
Figure 1. Overall survival of the 4 cohorts. At a median follow up of 99 months, the median overall survival (OS) for the patients who remained in the low-risk (LR) myelodysplastic syndromes (MDS) category throughout their period of follow-up (LR-LR), patients who progressed from LR to high-risk (HR) MDS without acute myeloid leukemia (AML) transformation (LR-HR), patients who progressed from LR to HR MDS and then AML (LRHR-AML), and patients who directly progressed from LR MDS to AML (LR-AML) was 80.7, 61.2, 48.3, and 42.8 months, respectively.
22 months.
Table 1 shows baseline characteristics of the 4 cohorts. The median age at diagnosis of patients in the 4 cohorts was similar (70, 68, 69, and 70 years respectively; P=0.484). Clinical and laboratory risk factors for progression to higher risk or AML included male gender (P<0.0001), low absolute neutrophil count (ANC) (P=0.04), low platelet count (P<0.001), high BM blasts (P<0.001), ferritin >1000 mcg/L (P<0.0001), albumin <3.5 g/dL (P=0.012). Pathologic risk factors for progression included multi-lineage dysplasia (MLD) (P<0.001) and lack of ring sideroblasts (P<0.0001). Patients with any del5q abnormality had a higher rate of progression to HRMDS (P=0.001). Among patients with grade 2-3 BM fibrosis
(N=49), 18% progressed directly to AML (P=0.009). There was no difference in rates of progression when patients were stratified by the lower risk MD Anderson model (LR-MDAS). Table 1 summarizes treatment patterns among the cohorts. As expected, more patients in the LR-AML (53.8%) and LRHR-AML (87.9%) cohorts were treated with hypomethylating agents, while more patients in the LR-LR (24.6%) and LR-HR (30%) cohorts were treated with lenalidomide. As expected, the rate of allogeneic stem cell transplantation (alloSCT) was significantly lower in patients that remained low-risk compared to the other 3 cohorts that progressed to high-risk disease and/or AML: N=63 (4.8%) in the LR-LR cohort; N=53 (16.7%) in the LR-HR cohort; N=18 (14.5%) in the LR-HR-AML
Lab values, mean (range) WBC x109/L
x109/L Hb g/dL
Platelets x109/L
Ferritin
<3.5 g/dL
3.5-4 g/dL >4 g/dL
(0.3-175.2)
(0.09-152.4)
0 (N=1,000)
1 (N=327)
2 (N=158)
3 (N=49)
Continued on following page.
Lower risk MD Anderson Model
Deaths, N (%)
<2 years
LR-LR: patients who remained in the low-risk (LR) myelodysplastic syndrome (MDS) category throughout their period of follow up; LR-HR: patients who progressed from LR to high-risk (HR) MDS without acute myeloid leukemia (AML) transformation; LR-HR-AML: patients who progressed from LR to HR MDS and then AML; LR-AML: patients who directly progressed from LR MDS to AML; WBC: white blood cell count; ANC: absolute neutrophil count; Hb: hemoglobin; BM: bone marrow; t-MDS: therapy-related MDS; SLD: single lineage dysplasia; MLD: multilineage dysplasia; RS: ring sideroblasts; EB: excess blasts; MPN: myeloproliferative neoplasm: RS-T: ring sideroblasts and thrombocytosis; ATG: antithymocyte globulin; ICT: iron chelation therapy; alloSCT: allogeneic stem cell transplant.
cohort; and N=27 (15.6%) in the LR-AML cohort (P<0.0001). Gene mutations associated with an increased risk of direct or indirect AML progression included SRSF2 (P<0.0001) and NRAS (P=0.003). Mutations in IDH1 (P=0.0001), IDH2 (P=0.029), and NPM1 (P<0.0001) were more common in patients with direct AML transformation from lower risk. Mutations associated with progression to both higher risk disease only, without AML and/or direct/indirect AML were ASXL1 (P=0.009), TP53 (P<0.0001), RUNX1 (P<0.0001), and CBL (P=0.0074). The presence of an ETV6 mutation was associated with progression to HR-MDS but not AML (P<0.0001). PHF6 mutations were associated with progression to HR-MDS and then AML (P=0.0093). SF3B1 mutation was associated with less progression (P<0.0001) (Table 2, Figure 2).
We divided the patients in the LR-LR cohort (i.e., those who remained low-risk; N=1,300) into 3 subgroups based on the number of patients that died in <2, 2-5, and >5 years, and found that about 171 (13.1%) patients of all LR-LR patients died <2 years after diagnosis of LR-MDS without disease progression to HR-MDS and/or AML. Among the 61 cases with documented cause of death, 18 patients (29.5%) died from cytopenia and MDS-related complications, including 2 from infection and 16 from anemia.
To the best of our knowledge, this is the first and one of the largest cohorts to explore in detail the different patterns of low-risk MDS disease progression, including progressing to
HR-MDS without AML transformation (16.5%), HR-MDS to AML (6.5%), and directly to AML (9%). The majority of LRMDS patients do not actually progress during their life span. Our overall rate of progression to HR-MDS and/or AML (32%) was higher than the 8% reported by de Swart et al. in the VL and LR IPSS-R cohort in their study validating the IPSS-R in LR-MDS patients.8 We noted that male gender was one of the factors associated with disease progression. Similar to our observation, female gender has been shown to be associated with better survival.8
The LR-MDAS model was put forth by Garcia-Manero et al. in 2007 to identify patients with LR MDS who may benefit from early intervention.4 They included 856 patients with low or intermediate-1 risk disease based on the IPSS. They reported that low platelets, anemia, older age (≥ 60 years), high BM blasts (≥ 4%), and poor-risk cytogenetics were associated with worse survival. The LR-MDAS model was also validated by us in 1,288 LR-MDS patients using the IPSS.9 However, in our study, the LR-MDAS model was not able to predict for progression to HR-MDS or AML. This shows that other parameters, such as ferritin, albumin, pathologic characteristics like MLD, BM fibrosis, and, most importantly, mutational profile need to be incorporated to provide a better prognosis in LR MDS patients.
In the study by Greenberg et al. that introduced the IPSS-R, some other factors were reported that predicted survival, but their impact on the prognostic score was low. Among them were variables including performance status, serum ferritin, and lactate dehydrogenase.2 In the past, serum ferritin had been linked to worse survival, but not to progression.
Table 2. Mutational profile of the 4 cohorts.
LR-LR: patients who remained in the low-risk (LR) myelodysplastic syndrome (MDS) category throughout their period of follow-up; LR-HR: patients who progressed from LR to high-risk (HR) MDS without acute myeloid leukemia (AML) transformation; LR-HR-AML: patients who progressed from LR to HR MDS and then AML; LR-AML: patients who directly progressed from LR MDS to AML.
In a study on 107 LR MDS patients, Kawabata et al. showed that the 3-year OS for the high ferritin group (ferritin >210 ng/mL) was significantly shorter than the low ferritin group (66% vs. 79%), while the rates of leukemic progression were similar between the 2 groups.10 In contrast, we noted higher ferritin in patients that progressed to HR-MDS, HR-AML, and directly to AML, but this may reflect the severity of the anemia as the disease progresses, and is most likely the result of progression rather than its cause. Considering high ferritin levels, iron chelation therapy (ICT) has been suggested to improve EFS and OS.11,12 In our study, the LR-HR cohort had the most patients that were treated with ICT, probably because this group included patients that could tolerate ICT, while the other two groups, LR-HR-AML and LR-AML, were patients in a poorer medical condition, with progression to AML, and so a smaller number of patients were treated with ICT.
Mutations that were associated with increased risk of progression to AML in our study were SRSF2 and NRAS. Mutations associated with progression to higher risk disease only, without AML and/or direct/indirect AML, were ASXL1, TP53, RUNX1, and CBL. The presence of an ETV6 mutation was associated with progression to HR-MDS, but not to
AML. Similar to our study, Bejar et al., in their study validating LR-MDAS in lower risk patients, reported that ASXL1, U2AF1, SRSF2, RUNX1, NRAS, and CBL were over-represented in the highest-risk LR-PSS category.13 In their study, ASXL1, RUNX1, EZH2, SRSF2, U2AF1, and NRAS mutations were also associated with shorter OS.13 We also found that IDH1, IDH2, and NPM1 were more common in patients with direct AML transformation. Similar to our study, Bejar et al. reported that SF3B1 mutation was significantly under-represented in the higher risk LR-MDS category and showed a non-significant trend toward longer OS.13 Recently, the molecular IPSS (IPSS-M) was introduced, which combines mutations with other hematologic and cytogenetic parameters.14 Using the IPSS-M, Bernard et al. were able to re-stratify 46% patients. We validated the IPSS-M at our center and found that 45% of patients were re-stratified, including approximately 75% of patients that the IPSS-R classified as very-low (VL) who were upstaged (the majority to IPSS-M low and ML/MH).15 In our study, at a median follow up of 99 months, the median OS for the LR-LR, LR-HR, LR-HR-AML, and LR-AML groups was 80.7, 61.2, 48.3, and 42.8 months, respectively. In the study by Greenberg et al. on the IPSS-R, the OS for the VL and LR IPSS-R categories was 8.8 and 5.3 years, which is
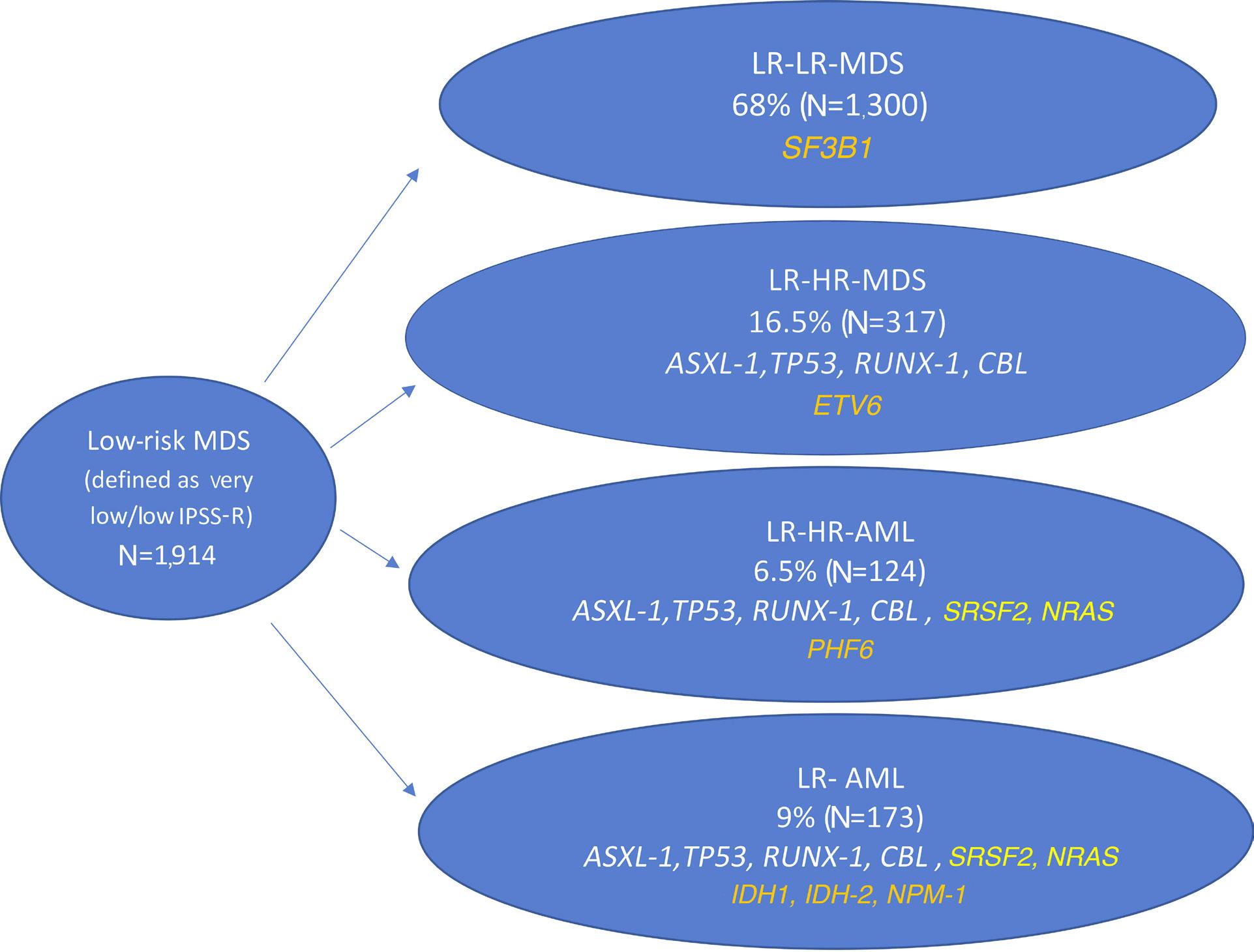
comparable to the OS of our LR-LR (6.7 years) and LR-HR (5.1 years) cohorts.2 The median time to progression to AML in our LR-HR-AML and LR-AML groups was 29 months, and the median time to progress from LR to HR-MDS was 22 months. Greenberg et al. reported the time for 25% of patients to develop AML (AML/25%) to be around 10.8 years for the LR group and not reached for the VL risk group.2 In addition, in the study reporting the LR-MDAS model, 90% of patients died without transforming to AML.4 When we looked at patients that remained in the LR-LR cohort, we found that 13.1% of patients died within two years of diagnosis, with 29% of the patients dying from cytopenias and MDS-related complications without progressing to HR disease or to AML. Greenberg et al. also described that 27% and 40% of patients belonging to the VL and the LR categories died, with 87% and 83% of patients not transforming to AML.2 In another similar study on outcomes of 531 patients with LR MDS by the Connect Myeloid Disease Registry by Komrokji et al., 54% of the 144 patients who had a follow-up BM autopsy available had a 5% increase in BM blasts. The overall rate of disease progression was about 15% in the Connect LR-MDS cohort. About 213 patients died from the 531 LR MDS patients, and 56.3% deaths were attributed to disease-related complications, followed by 14.6% related to cardiovascular disease (CVD).16 In another recent study on 2,396 LR MDS patients by Madry et al., infection (17.8%) and CVD (9.8%) were reported as the main causes of death other than disease progression. They also noted that the relative survival of patients with LR MDS decreases from 94.3% at the first year to 59.6% at five years, hence implying that >40% of patients with LR-MDS die, either directly
Figure 2. Algorithm showing patterns of progression and of low-risk myelodysplastic syndromes. IPSS-R: Revised International Prognostic Scoring System; LR-LR: patients who remained in the low-risk (LR) myelodysplastic syndromes (MDS) category throughout their period of follow-up; LR-HR: patients who progressed from LR to high-risk (HR) MDS without acute myeloid leukemia (AML) transformation; LR-HR-AML: patients who progressed from LR to HR MDS and then AML; LR-AML: patients who directly progressed from LR MDS to AML.
or indirectly, from MDS-related causes within five years of diagnosis.17 This further affirms our belief that there is a need to better prognosticate these LR-MDS patients that might need earlier intervention in certain cases. Allogeneic stem cell transplantation (alloSCT) is the only cure for MDS. AlloSCT is generally recommended at the time of disease progression and is avoided for LR disease due to early transplant-related mortality, in contrast to the long OS for patients with LR MDS.18-20 If we can identify the patients with LR-MDS who die early, i.e., within two years, without progression clearly related to disease, we might be able to offer them an alloSCT, and significantly improve their survival and quality of life.
Despite being limited by the retrospective design, our study provides important conclusions among a large, predominantly treated cohort. We delineated and identified the clinical and molecular factors associated with different patterns of disease progression. For patients who remained in LR MDS, 13% died in <2 years, and for those with documented cause of death, one-third of deaths within two years were directly MDS-related. Other causes of mortality reported in LR MDS largely include cardiac events. The interplay of cytopenia and such comorbidities may be crucial, as cytopenia may exacerbate those conditions and indirectly contribute to mortality. In fact, several models incorporate comorbidities in MDS risk stratification, but do not detail the interplay between cytopenia and those concomitant diseases.21,22 There is increasing evidence that certain clonal hematopoietic events, such as TET-2 somatic mutation, are associated with other comorbidities such as atherosclerosis.23,24 The impact of treating cytopenias among patients with comorbidities,
alleviating transfusion burden and/or achieving hematologic improvement should be further studied. Identifying those LR MDS patients with higher risk features of disease progression or disease-related death within two years will be crucial to tailor therapy accordingly.
Disclosures
AK reports research support from Protagonist, Blueprint Medicines Corporation, GSK - Sierra Oncology, Prelude Pharmaceuticals, Bristol Myers Squibb, and Morphosys; consultancy fees from Imago Biosciences, Incyte, Blueprint Medicines Corporation, Novartis, Abbvie, GSK - Sierra Oncology, Bristol Myers Squibb, Pharmaessentia, and CTI Biopharma; honoraria from Imago Biosciences, Incyte, Blueprint Medicines Corporation, Novartis, Abbvie, GSK - Sierra Oncology, Bristol Myers Squibb, Pharmaessentia, and CTI Biopharma; and has been a member of the Speakers Bureau for Imago Biosciences, Incyte, Blueprint Medicines Corporation, Novartis, Abbvie, GSK - Sierra Oncology, Bristol Myers Squibb, Pharmaessentia, and CTI Biopharma. OC has received research funding from Syntrix Pharmaceuticals. KS reports consultancy fees from Pfizer, Mablytics, berGenBio, Curis, Novartis, Gilead Sciences, Inc., AROG, Astellas, and Bristol Myers Squibb; has sat on the Advisory Board of Pfizer, berGenBio, Curis, Novartis, Gilead Sciences, Inc., AROG, Astellas, and Bristol Myers Squibb; has received research funding from Pfizer, Syntrix Pharmaceuticals, and Incyte; and honoraria from Mablytics, berGenBio, Curis, Novartis, Gilead Sciences, Inc., AROG, Astellas, and Bristol Myers Squibb. JEL has received consultancy fees from Dedham Group, Jasper Therapeutics, Astellas, Dava Oncology, Novartis, Agios/Servio, Boxer Capital, Jazz, BerGenBio, Millenium Pharma/Takeda, ElevateBio Management, Daiichi Sankyo, AbbVie, and Servier; and research funding from Syntrix
1. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079-2088.
2. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465.
3. Kantarjian H, O’Brien S, Ravandi F, et al. Proposal for a new risk model in myelodysplastic syndrome that accounts for events not considered in the original International Prognostic Scoring System. Cancer. 2008;113(6):1351-1361.
4 Garcia-Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22(3):538-543.
5. Malcovati L, Germing U, Kuendgen A, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25(23):3503-3510.
Pharmaceuticals, and Celgene/ Bristol Myers Squibb. EP has received research funding from Bristol Myers Squibb, Syntrix Pharmaceuticals, Kura, and Incyte; and honoraria from Taiho, Stemline, and Blueprint. DAS has sat on the Advisory Board of Intellia, Aprea, Syndax, Bristol Myers Squibb, Nemucore, Agios, AbbVie, Kite, Shattuck Labs, and Novartis; has received consultancy fees from Takeda, Novartis, and Magenta; has sat on the Speakers Bureau of Bristol Myers Squibb and Incyte; has received research funding from Aprea and Syntrix Pharmaceuticals; and Patents & Royalties from Lixte. RK has received consultancy fees from Servier, AbbVie, Geron, Acceleron Pharma, Taiho Oncology, Novartis, Jazz Pharmaceuticals, and Bristol Myers Squibb; honoraria from Servier, AbbVie, CTI BioPharma, Innovent, PharmaEssentia, Takeda, Jazz Pharmaceuticals, and Bristol Myers Squibb; has sat on the Speakers Bureau of Servier, AbbVie, Jazz Pharmaceuticals and Bristol Myers Squibb; has sat on the Advisory Board of CTI BioPharma, Taiho Oncology, PharmaEssentia, Takeda, Novartis, Bristol Myers Squibb; and has received research funding from Bristol Myers Squibb. AGJ, SB, LA, NAA and DK have no conflicts of interest to disclose.
AGJ, SB, and LA analyzed the data. NAA collected the data. AGJ wrote the manuscript. RK conceptualized the retrospective review and performed a thorough revision of the manuscript. DK, ST, AK, OC, EP, KS, JEL and DAS reviewed the manuscript.
No funding was received for this work.
Data-sharing agreement
The MDS database related to this study is not available.
6. Komrokji RS, Zhang L, Bennett JM. Myelodysplastic syndromes classification and risk stratification. Hematol Oncol Clin North Am. 2010;24(2):443-457.
7 Volpe VO, Garcia-Manero G, Komrokji RS. Myelodysplastic syndromes: a new decade. Clin Lymphoma Myeloma Leuk. 2022;22(1):1-16.
8. de Swart L, Smith A, Johnston TW, et al. Validation of the revised international prognostic scoring system (IPSS-R) in patients with lower-risk myelodysplastic syndromes: a report from the prospective European LeukaemiaNet MDS (EUMDS) registry. Br J Haematol. 2015;170(3):372-383.
9 Komrokji R, Ramadan H, Al Ali N, et al. Validation of the LowerRisk MD Anderson Prognostic Scoring System for Patients With Myelodysplastic Syndrome. Clin Lymphoma Myeloma Leuk. 2015;15 Suppl:S60-63.
10 Kawabata H, Usuki K, Shindo-Ueda M, et al. Serum ferritin levels at diagnosis predict prognosis in patients with low blast count myelodysplastic syndromes. Int J Hematol. 2019;110(5):533-542.
11. Angelucci E, Greenberg P, Izquierdo M, Garcia-Manero G. Iron chelation in transfusion-dependent patients with low- to
intermediate-1-risk myelodysplastic syndromes. Ann Intern Med. 2020;173(7):595-596.
12. Leitch HA, Parmar A, Wells RA, et al. Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient-related factors and measuring from time of first red blood cell transfusion dependence: an MDS-CAN analysis. Br J Haematol. 2017;179(1):83-97.
13. Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol. 2012;30(27):3376-3382.
14. Bernard E, Tuechler H, Greenberg Peter L, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evidence. 2022;1(7):EVIDoa2200008.
15. Aguirre LE, Al Ali N, Ball S, et al. Validation of the Molecular International Prognostic Scoring System (IPSS-M) Risk Stratification Model for Myelodysplastic Syndromes. Blood. 2022;140(Suppl 1):1125-1127.
16. Komrokji RS, George TI, Erba HP, et al. Treatment patterns and outcomes of patients with lower-risk myelodysplastic syndromes in the connect ® myeloid disease registry. Blood. 2021;138(Suppl 1):3686.
17 Mądry K, Lis K, Fenaux P, et al. Cause of death and excess mortality in patients with lower-risk myelodysplastic syndromes (MDS): a report from the European MDS registry. Br J Haematol. 2023;200(4):451-461.
18. Cutler CS, Lee SJ, Greenberg P, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for lowrisk myelodysplasia is associated with improved outcome. Blood. 2004;104(2):579-585.
19 Jain AG, Elmariah H. BMT for Myelodysplastic syndrome: when and where and how. Front Oncol. 2021;11:771614.
20 de Witte T, Bowen D, Robin M, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood. 2017;129(13):1753-1762.
21. Della Porta MG, Malcovati L, Strupp C, et al. Risk stratification based on both disease status and extra-hematologic comorbidities in patients with myelodysplastic syndrome. Haematologica. 2011;96(3):441-449.
22. Naqvi K, Garcia-Manero G, Sardesai S, et al. Association of comorbidities with overall survival in myelodysplastic syndrome: development of a prognostic model. J Clin Oncol. 2011;29(16):2240-2246.
23. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111-121.
24. Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355(6327):842-847.
Yuqin Song,1* Junning Cao,2* Qingyuan Zhang,3* Caixia Li,4 Lugui Qiu,5 Junyuan Qi,5 Huilai Zhang,6 Wenyu Li,7 Lihong Liu,8 Hongmei Jing,9 Keshu Zhou,10 Weijing Zhang,11 Liling Zhang,12 Daqi Li,13 Liqun Zou,14 Haiyan Yang,15 Wenbin Qian,16 Hui Zhou,17 Jianda Hu,18 Hongyan Yin,19 Sisi Fu,19 Songhua Fan,19 Qian Xu,19 Jian Wang,19 Xiaoyun Jia,19 Guangxiu Dai,19 Weiguo Su19 and Jun Zhu1
1Peking University Cancer Hospital & Institute, Beijing; 2Fudan University Shanghai Cancer Center, Shanghai; 3The Affiliated Tumor Hospital of Harbin Medical University, Harbin; 4The First Affiliated Hospital of Soochow University, Suzhou; 5Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences, Tianjin; 6Tianjin Medical University Cancer Institute & Hospital, Tianjin; 7Guangdong Provincial Peoples Hospital, Guangzhou; 8The Fourth Hospital of Hebei Medical University, Shijiazhuang; 9Peking University Third Hospital, Beijing; 10Henan Cancer Hospital, Zhengzhou; 11Beijing Shijitan Hospital Affiliated to Capital Medical University, Beijing; 12Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan; 13Jinan Central Hospital Affiliated to Shandong First Medical University, Jinan; 14West China Hospital of Sichuan University, Chengdu; 15Zhejiang Cancer Hospital, Hangzhou; 16The First Affiliated Hospital of School of Medicine, Zhejiang University, Hangzhou; 17Hunan Cancer Hospital, Changsha; 18Fujian Medical University Union Hospital, Fuzhou and 19HUTCHMED Limited, Shanghai, China
*YS, JC and QZ contributed equally as first authors.
Correspondence: J. Zhu zhujun@csco.org.cn
Received: November 9, 2022.
Accepted: January 4, 2024. Early view: January 18, 2024.
https://doi.org/10.3324/haematol.2022.282401
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Sovleplenib (HMPL-523) is a selective spleen tyrosine kinase (Syk) inhibitor with anti-tumor activity in preclinical models of B-cell malignancy. We conducted a dose-escalation and dose-expansion phase I study of sovleplenib in patients with relapsed/ refractory mature B-cell tumors. Dose escalation followed a 3+3 design; patients received oral sovleplenib (200-800 mg once daily [q.d.] or 200 mg twice daily [b.i.d.], 28-day cycles). During dose expansion, patients were enrolled into four cohorts per lymphoma classification and treated at the recommended phase II dose (RP2D) (clinicaltrials gov. Identifier: NCT02857998). Overall, 134 Chinese patients were enrolled (dose escalation, N=27; dose expansion, N=107). Five patients experienced dose-limiting toxicities: one each of amylase increased (200 mg q.d.), febrile neutropenia (800 mg q.d.), renal failure (800 mg q.d.), hyperuricemia and blood creatine phosphokinase increased (200 mg b.i.d.) and blood bilirubin increased and pneumonia (200 mg b.i.d.). RP2D was determined as 600 mg (>65 kg) or 400 mg (≤65 kg) q.d.. The primary efficacy end point of independent review committee-assessed objective response rate in indolent B-cell lymphoma was 50.8% (95% confidence interval: 37.564.1) in 59 evaluable patients at RP2D (follicular lymphoma: 60.5%, marginal zone lymphoma: 28.6%, lymphoplasmacytic lymphoma/Waldenström macroglobulinemia, 0%). The most common (≥10% patients) grade ≥3 treatment-related adverse events in the dose-expansion phase were decreased neutrophil count (29.9%), pneumonia (12.1%) and decreased white blood cell count (11.2%). Pharmacokinetic exposures increased dose-proportionally with ascending dose levels from 200-800 mg, without observed saturation. Sovleplenib showed anti-tumor activity in relapsed/refractory B-cell lymphoma with acceptable safety. Further studies are warranted.
Non-Hodgkin lymphomas (NHL) are the 12th most common cancer globally, with more than 544,000 diagnoses per annum. 1,2 In China, approximately 92,800 NHL cases are diagnosed annually, 64% of which are mature B-cell malignancies.1,3 A fraction of patients with mature B-cell tumors are resistant to or relapse after current first-line therapies; therefore, an unmet need exists for new treatment options.4,5
Spleen tyrosine kinase (Syk) is a cytoplasmic protein tyrosine kinase that is ubiquitously expressed in hematopoietic cells, including B cells.6 Syk mediates B-cell receptor (BCR) signaling, which is pivotal in the pathogenesis of B-cell lymphoma7,8 and has been identified as an oncogenic driver in B-cell malignancies.8 Targeting Syk signaling, which is upstream of both Bruton tyrosine kinase (BTK) and phosphoinositide 3-kinase (PI3Kδ), may overcome drug resistance to BTK inhibitors (BTKi) or PI3Kδ inhibitors in chronic lymphocytic leukemia (CLL).9 Blocking Syk activity, using either small interfering RNA or a chemical inhibitor, leads to cell cycle arrest in diffuse large B-cell lymphoma (DLBCL) cells.10 Syk inhibition also blocks cellular proliferation in follicular lymphoma (FL), mantle cell lymphoma (MCL) and Burkitt lymphoma.11 Targeting the Syk pathway has demonstrated promise in the treatment of B-cell malignancies given the clinical activities of several Syk inhibitors in clinical trials;9,12-22 however, to date, no Syk inhibitors are approved for lymphoma. Sovleplenib (HMPL-523) is a novel, small-molecule, potent and highly selective oral Syk inhibitor.23 Sovleplenib demonstrated a superior selectivity compared to R406 (the active metabolite of fostamatinib), especially against KDR and RET. In vitro, sovleplenib could effectively inhibit the phosphorylation of Syk and its downstream signaling molecule BLNK, ERK, AKT, PLCγ 1 and P38. Sovleplenib potently inhibited B-cell activation in human whole blood with a half maximal effective concentration (EC50) of 0.157 μM and anti-IgD antibody induced B-cell activation in rat and mouse whole blood with EC50 of 0.546 and 1.000 μM, respectively. Moreover, sovleplenib inhibited cell survival in a panel of B-cell lymphoma cell lines, with a lower half maximal inhibitory concentration (IC50) and an increased apoptotic rate in REC-1 cells compared with other Syk inhibitors.24 We conducted a phase I study to determine the safety, tolerability, efficacy and pharmacokinetics of sovleplenib in patients with relapsed/refractory mature B-cell malignancies in China.
This multicenter, open-label phase I study with doseescalation and dose-expansion phases was conducted in China between December 2016 and April 2021. Dose escalation employed a conventional 3+3 study design with doses from 200-800 mg once daily (q.d.). One additional regimen with a 200 mg twice-daily (b.i.d.) starting dose was explored. Dose expansion at the recommended phase II dose (RP2D) was conducted using a basket design for four cohorts: (A) CLL/small lymphocytic lymphoma (SLL), (B) aggressive B-cell lymphoma, (C) MCL and (D) indolent B-cell lymphoma. Patients received oral sovleplenib in 28-day cycles until progressive disease (PD), intolerable toxicity or consent
withdrawal. Prophylactic antibiotics were not permitted. The primary objective of the dose-escalation phase was to evaluate safety and determine the RP2D by the safety review committee (SRC). The primary objective of the dose-expansion phase was to evaluate safety and preliminary efficacy.
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Ethical approval was obtained by the ethics committees. A list of ethics committees that approved the study is included in the Online Supplementary Appendix. All patients provided written informed consent.
Eligible adult patients had pathologically confirmed mature B-cell tumors that progressed or were ineligible for standard treatment (see Online Supplementary Appendix), including CLL/SLL, aggressive B-cell lymphoma (DLBCL, Richter syndrome and grade 3b FL), MCL or indolent B-cell lymphoma (marginal zone lymphoma [MZL], lymphoplasmacytic lymphoma/Waldenström macroglobulinemia [LPL/ WM], grades 1, 2, 3a FL) in dose expansion; an Eastern Cooperative Oncology Group performance status of 0-1; and bidimensionally measurable disease at baseline in dose expansion (except CLL or LPL/WM patients). Patients with protocol-defined laboratory abnormalities or a history of clinically significant liver disease were excluded. Detailed eligibility criteria are listed in the Online Supplementary Appendix
Treatment-emergent adverse events (TEAE) were assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03. Tumor response was assessed according to modified IWCLL 2008 criteria for CLL/ SLL, IWM-7 for LPL/WM and modified Cheson 2014 criteria for other B-cell lymphomas. Responses were assessed with enhanced computed tomography (CT) every 8 weeks for the first 24 weeks, and every 12 weeks thereafter. For DLBCL, MCL, FL, and other fluorodeoxyglucose-sensitive lymphoma, responses were assessed by positron emission tomography-computed tomography (PET-CT) with enhanced CT according to Cheson 2014 criteria. For pharmacokinetic analyses, plasma samples were collected at multiple time points (Online Supplementary Appendix). Plasma concentrations of sovleplenib and its metabolites were assessed using liquid chromatography-tandem mass spectrometry (LabCorp Drug Development, Shanghai, China), and biomarker concentrations were measured by MILLIPLEX human cytokine/chemokine panel assay (Millipore).
The study planned to enroll 217-232 patients, with 27-42 during dose escalation and 190 during dose expansion based on adverse event (AE) assumptions (Online Supple-
mentary Appendix).
Patients who received ≥1 dose of sovleplenib were included in the safety set (SS). The efficacy-evaluable analysis set (EEAS) included all patients in the SS who had ≥1 valid postbaseline tumor evaluation. Objective response rate (ORR) and 95% confidence interval (CI) were calculated using the Clopper-Pearson method. Time to event was estimated using the Kaplan-Meier method. Analyses of time to response (TTR) and duration of response (DOR) were based on the EEAS, and analyses of time to progression (TTP) and progression-free survival (PFS) were based on the SS. Post hoc analyses of investigator-assessed tumor response were performed in CLL/SLL or FL according to prior treatment status.
The pharmacokinetic analysis set included patients who received ≥1 dose of sovleplenib with concentration data
available. Pharmacokinetic parameters were analyzed with a non-compartmental model using Phoenix WinNonlin software (v8.1). Pharmacokinetic and biomarker analyses were summarized using descriptive statistics.
Patients and demographics
Between December 15, 2016, and September 3, 2020, 166 patients were screened at 18 centers across China; 134 were enrolled and received sovleplenib (Figure 1).
Dose escalation
During dose escalation, 27 patients were enrolled (Figure 1). Among them, 23 (85.1%) were classified as Ann Arbor
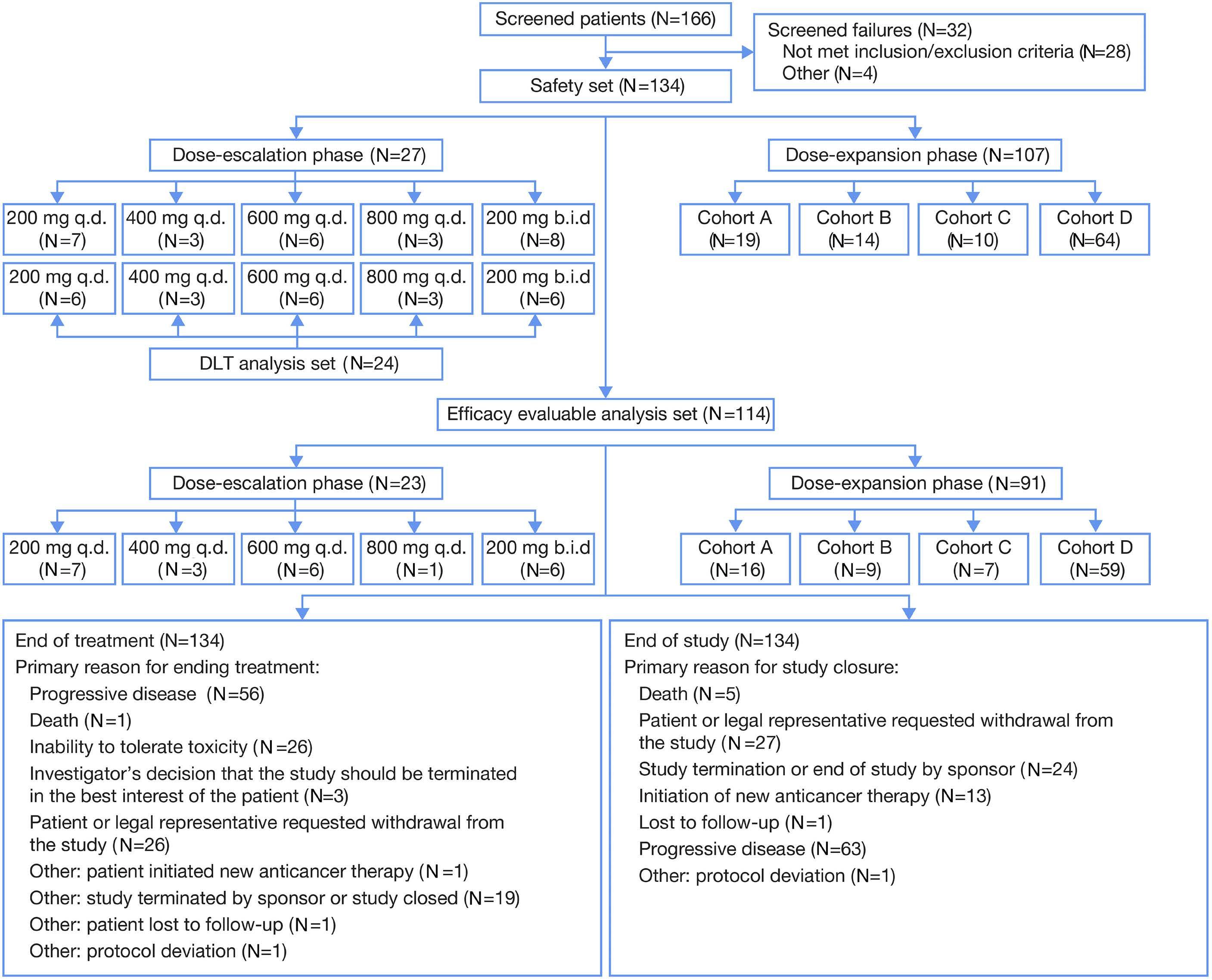
Figure 1. Patient disposition. The safety set (SS) includes all patients who have taken at least 1 dose of study drug. The efficacy-evaluable analysis set (EEAS) includes all patients in the SS who have at least 1 valid postbaseline tumor evaluation. b.i.d.: twice daily; DLT: dose-limiting toxicity; q.d.: once daily.
stage III or IV. The median time from diagnosis to first dose was 32.9 months (range, 5.4-104.3) (Online Supplementary Table S1). All patients had received previous anti-tumor treatment, with 13 (48.1%) having received ≥3 lines of treatment. Twenty-one (77.8%) patients had received previous anti-CD20 antibody treatment. All patients received ≥1 dose of sovleplenib, as follows: 200 mg q.d. (N=7), 400 mg q.d. (N=3), 600 mg q.d. (N=6), 800 mg q.d. (N=3), 200 mg b.i.d. (N=8). At data cutoff (April 30, 2021), all patients had discontinued treatment, most of them discontinued because of PD (N=12; 44.4%).
Dose expansion
During dose expansion, 107 patients were enrolled in co-
horts A (N=19), B (N=14), C (N=10) or D (N=64). The number of enrolled patients being less than the planned sample size was due to difficulties in enrolling patients with CLL/ SLL and indolent B-cell lymphoma. The median time from diagnosis to first dose was 37.3 months (range, 1.9-152.4) (Table 1). Of patients enrolled, 83 (77.6%) were classified as Ann Arbor stage III or IV. All patients received prior anti-tumor treatment, and 48 (44.9%) received ≥3 lines of treatment. Eighty-seven (81.3%) patients had received previous anti-CD20 antibody treatment. In cohort A, eight (42.1%) patients had received a prior BTKi, and two (10.5%) patients had a received prior BCL-2 inhibitor (Table 1). Other baseline demographic and disease characteristics are presented in Online Supplementary Table S2. One (0.9%)
phase.
Baseline ECOG score, N (%)
since last relapse or disease progression
Lines of last anti-tumor drug treatment, N (%)
1 line
2 lines
≥3 lines
Median time in months from last systemic therapy to sovleplenib treatment (range)
Chemotherapy, N (%)
Alkylating agents
Anthracycline
Vinblastines
Purine analogs
Rituximab-based therapy, N (%)
ASCT: autologous stem cell therapy; BCL-2: B-lymphocytoma-2; BCR: B-cell receptor; BTK: Bruton tyrosine kinase; CLL: chronic lymphocytic leukemia; DLBCL: diffuse large B-cell lymphoma; ECOG: Eastern Cooperative Oncology Group; IPI: International Prognostic Index; MCL: mantle cell lymphoma; Pi3K: phosphatidylinositol 3-kinase; SLL: small lymphocytic lymphoma.
patient had received autologous hematopoietic stem cell transplant. As of data cutoff (April 30, 2021), all patients had discontinued treatment; most of them discontinued because of PD (N=44; 41.1%).
Safety/adverse events
Dose escalation
The median duration of exposure was 3.2 months and 92.6% of patients had a relative dose intensity between 80% and 110%.
One patient, two patients, and two patients treated with 200 mg q.d., 200 mg b.i.d., and 800 mg q.d., respectively, experienced dose-limiting toxicity (DLT) events (definitions of DLT are stated in the Online Supplementary Appendix): grade 3 amylase increased (200 mg q.d., dose interruption, resolved without supportive or concomitant treatment), grade 4 hyperuricemia and blood creatine phosphokinase increased (200 mg b.i.d., dose interruption, unknown outcome), grade 3 blood bilirubin increased and pneumonia (200 mg b.i.d., treatment discontinuation due to pneumonia, both events resolved with supportive or concomitant treatment), grade 3 febrile neutropenia (800 mg q.d., dose interruption, resolved with supportive or concomitant treatment), and grade 3 renal failure (800 mg q.d., treatment discontinuation, resolved with supportive or concomitant treatment). The maximum tolerated dose was determined as 600 mg q.d., and the SRC determined this as the recommended dose for dose expansion on December 6, 2017. As of July 30, 2018, of 28 patients treated with 600 mg q.d., nine (32.1%) experienced grade ≥3 treatment-related AE (TRAE) during the first cycle of study treatment. In the exploratory analysis, patients who experienced treatment-related grade 3 AE had higher plasma exposure of sovleplenib and tended to have lower body weight (≤65 kg) versus those who experienced no drug-related grade ≥3 AE. In addition, drug exposure was body weight-dependent, evidenced by higher plasma sovleplenib concentration detected in patients with lower body weight. Therefore, weight-based dosing was introduced in the expansion cohort to reduce the risk of grade ≥3 AE in patients with lower body weight. The SRC recommended the RP2D of 600 mg q.d. in patients with a weight of >65 kg and 400 mg q.d. in those with a weight of ≤65 kg. If patients did not experience grade ≥2 AE at the starting dose of 400 mg q.d., after one or two treatment cycles the dose could be increased to 500 mg q.d. to maximize treatment efficacy. All (100%) patients experienced TEAE and, among them, 17 (63.0%) reported grade ≥3 TEAE. No fatal TEAE were reported. TEAE leading to treatment discontinuation occurred in five (18.5%) patients, including pneumonia in two (7.4%) patients (1 in 200 mg q.d. and 1 in 200 b.i.d.) and renal failure, malignant pleural effusion and anemia in one patient each. Pneumonia (1 patient in 200 mg b.i.d.), renal failure and anemia were considered possibly related to treatment, and the other TEAE were unlikely related. The event of malignant
pleural effusion was resolved with closed thoracentesis drainage. All (100%) patients had TRAE (Table 2); 13 (48.1%) patients had grade ≥3 TRAE. Five patients experienced treatment-related serious AE, including pneumonia (N=2; 200 mg q.d. and 200 mg b.i.d.), upper respiratory infection (N=1; 200 mg q.d.), blood bilirubin increased (N=1; 200 mg b.i.d.), interstitial lung disease (N=1; 600 mg q.d.), renal failure (N=1; 800 mg q.d.) and febrile neutropenia (N=1; 800 mg q.d.). All treatment-related serious AE were grade 3 and recovered/resolved with symptomatic treatment, dose modification and/or dose discontinuation. Thirteen (48.1%) patients reported TEAE leading to dose interruption or reduction; most common (≥2 patients) were neutrophil count decreased (18.5%), white blood cell count decreased (11.1%), blood bilirubin increased (7.4%), pneumonia (7.4%) and febrile neutropenia (7.4%).
The median duration of exposure was 3.7 months (range, 0.1-35.7 months); 91.6% of patients had a relative dose intensity between 80% and 110%.
During dose expansion, 106 (99.1%) patients reported TEAE; among them, 105 (98.1%) were related to study drug (Table 2). The most common (≥30%) TRAE included neutrophil count decreased (60.7%), aspartate aminotransferase increased (51.4%), white blood cell count decreased (50.5%), alanine aminotransferase increased (44.9%), anemia (32.7%) and platelet count decreased (31.8%). The common grade ≥3 TRAE with an incidence of ≥5% included neutrophil count decreased (29.9%), pneumonia (12.1%), white blood cell count decreased (11.2%) and platelet count decreased (9.3%). Four (3.7%) patients (1 patient in cohort B and 3 patients in cohort D) experienced grade 1-2 blood lactate dehydrogenase increased; among these, two events were considered as treatment related. TEAE leading to death were reported in five (4.7%) patients, including one case each of sudden cardiac death, platelet count decreased, interstitial lung disease, pneumonia and acute kidney injury; all were considered treatment-related (possibly related). In addition, 20 (18.7%) patients experienced TEAE that led to treatment discontinuation, among which pneumonia (N=5; 4.7%) and interstitial lung disease (N=3; 2.8%) were most common. No notable difference was observed among cohorts for the incidence of TEAE leading to treatment discontinuation.
Dose escalation
Based on the EEAS (N=23), the overall ORR during dose escalation was 21.7% (95% CI: 7.5-43.7), where five patients (21.7%; 4 FL and 1 SLL) achieved partial response (PR), at 400 mg q.d. (1 PR), 600 mg q.d. (3 PR) and 200 mg b.i.d. (1 PR), and none achieved complete response (CR). In addition, nine (39.1%) achieved stable disease (SD). Of five responders, median TTR (mTTR) was 3.6 months (95% CI:
Table 2. Common treatment-emergent adverse events that related to study drug (any grade ≥20%) in the dose-escalation and dose-expansion phases.
Dose-escalation phase, N (%)
1.9-not calculable [NC]) and median DOR (mDOR) was 11.0 months (95% CI: 4.7-NC). The median PFS (mPFS) was 8.3 months (95% CI: 1.8-16.5), and median TTP (mTTP) was 8.3 months (95% CI: 1.8-16.5).
Dose expansion
Cohort A: chronic lymphocytic leukemia/small lymphocytic lymphoma
Among CLL/SLL patients in the EEAS (N=16), ORR was 56.3% (95% CI: 29.9-80.2) per investigator assessments (Table 3; Online Supplementary Figure S1). The mean best percentage change in tumor burden from baseline in CLL/SLL patients was -66.58 (± 30.80%) (Online Supplementary Figure S1). mTTR was 1.9 months (95% CI: 1.8-1.9) and mDOR was 13.1 months (95% CI: 1.8-NC). In the SS (N=16), mPFS was 14.9 months (95% CI: 3.7-NC; Online Supplementary Figure S2).
Cohort B: aggressive B-cell lymphomas
Among aggressive B-cell lymphoma patients in the EEAS (N=9), ORR was 33.3% (95% CI: 7.5-70.1) per investigator assessments (Table 3; Online Supplementary Figure S1). The mean best percentage change in tumor burden from baseline in aggressive B-cell lymphoma patients was 21.90% (± 87.407%) (Online Supplementary Figure S1). In the SS (N=14), mTTP was 1.9 months (95% CI: 0.8-9.1) and mPFS was 1.9 months (95% CI: 0.8-9.1; Online Supplementary Figure S2).
Cohort C: mantle cell lymphoma
Among MCL patients in the EEAS (N=7), the ORR was 28.6% (95% CI: 3.7-71.0) per investigator assessments, and two
(28.6%) patients achieved PR (Table 3; Online Supplementary Figure S1). The mean best percentage change in tumor burden from baseline in MCL patients was -9.8% (± 28.265%) (Online Supplementary Figure S1). Kaplan-Meier plots of PFS are provided in Online Supplementary Figure S2
Cohort D: indolent B-cell lymphoma
Per independent review committee assessments, eight (13.6%) patients in the EEAS for cohort D (N=59) achieved CR and 22 (37.3%) patients achieved PR; the overall ORR was 50.8% (95% CI: 37.5-64.1; Table 3; Figure 2). In addition, 26 (44.1%) achieved SD. mTTR was 2.7 months (95% CI: 1.8-3.7), and mDOR was 15.7 months (95% CI: 7.4-NC). In the SS (N=64), mTTP was 12.0 months (95% CI: 8.2-NC), and mPFS was 12.0 months (95% CI: 6.3-NC). FL patients (grade 1, 2, 3a; EEAS, N=43) achieved an IRC-assessed ORR of 60.5% (95% CI: 44.4-75.0); with seven (16.3%) patients achieved CR and 19 (44.2%) patients achieved PR. The mDOR was not reached and 14 (32.6%) maintained SD. Among FL patients in the SS (N=47), mTTP was 12.0 months (95% CI: 8.2-NC) and mPFS was 11.0 months (95% CI: 6.3-NC). Among 14 MZL patients in the EEAS, the IRC-assessed ORR was 28.6% (95% CI: 8.4-58.1).
Tumor response per investigator assessments (Table 3; Online Supplementary Figures S1-3) was consistent with that per IRC assessments in cohort D.
The mean best percentage change in tumor burden from baseline based on investigator assessment was similar to that based on IRC assessment (Figure 2). Among 57 patients with change in tumor burden from baseline, the
aIRC: tumor response in the indolent B-cell lymphoma cohort was assessed by the independent review committee (IRC) and investigators (INV), and in the other cohorts was assessed by the INV. bIncluding 1 patient with minor response. cMedian progression-free survival (PFS) was evaluated from the safety set. CI: confidence interval; CLL: chronic lymphocytic leukemia; CR: complete response; DOR: duration of response; MCL: mantle cell lymphoma; NC: not calculable; ORR: objective response rate; PD: progressive disease; PR: partial response; PR-L: partial response with increased lymphocyte count; SD: stable disease; SLL: small lymphocytic lymphoma; TTR: time to response.
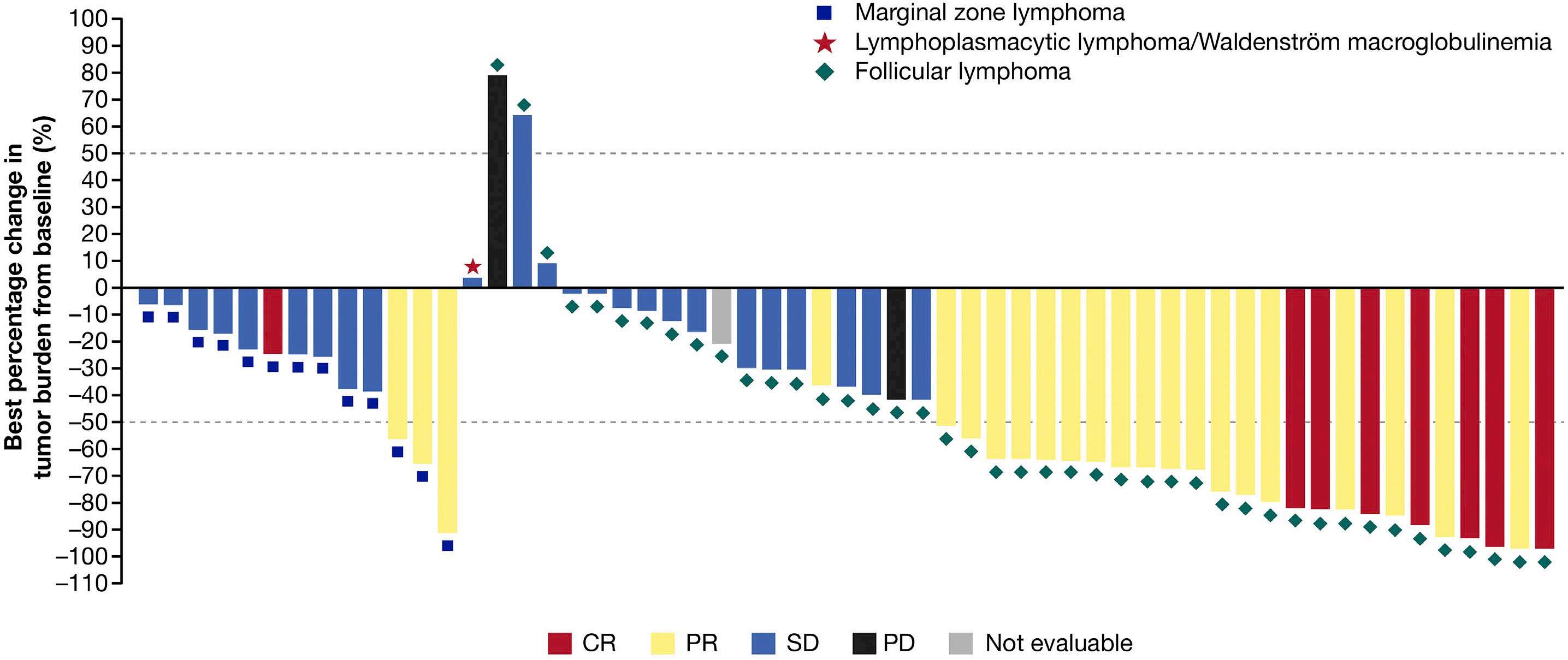
Figure 2. Best percentage change in tumor burden from baseline according to independent review committee assessments based on the efficacy-evaluable analysis set in the indolent B-cell lymphoma cohort. CR: complete response; PD: progressive disease: PR: partial response; SD: stable disease.
mean best percentage change was -44.08% (±37.748%).
Chronic lymphocytic leukemia/small lymphocytic lymphoma and follicular lymphoma subgroup analysis
Consistent anti-tumor activity was observed across CLL/ SLL subgroups defined by prior treatment with BTKi or lines of prior treatment (Online Supplementary Table S3 ). CLL/SLL patients who had received prior BTKi (EEAS, N=8) and those who were BTKi-naive (EEAS, N=8) had ORR of 50% (95% CI: 15.7-84.3) and 62.5% (95% CI: 24.5-91.5), respectively, with mDOR of 14.7 months (95% CI: 1.9-NC) and 13.1 months (95% CI: 1.8-NC; Figure 3). mPFS was 16.5 months (95% CI: 1.6-NC) in CLL/SLL patients who had received prior BTKi and 13.8 months (95% CI: 3.5-NC) in those who were BTKi-naive. In patients with prior BTKi or ≥3 prior lines (EEAS, N=10), five achieved PR/PR with increased lymphocyte count (PR-L), with an ORR of 50% (95% CI: 18.7-81.3), mPFS of 16.5 months (95% CI: 1.6-NC) and mDOR of 14.7 months (95% CI: 1.8-NC).
Among 43 efficacy-evaluable FL patients, the ORR was 60.5% (95% CI: 44.4-75.0), with an mDOR of 14.8 months (95% CI: 6.5-NC), mTTR of 1.9 months (95% CI: 1.9-3.6) and mPFS of 11.0 months (95% CI: 6.3-16.6; Online Supplementary Table S3; Figure 4). Subgroup analysis revealed that patients who had received ≥2 lines (including anti-CD20) of treatment (EEAS, N=27) had an ORR of 59.3% (95% CI: 38.8-77.6), mPFS of 8.2 months (95% CI: 5.5-16.6) and mDOR of 9.0 months (95% CI: 3.6-NC). Anti-CD20-naive FL patients (EEAS, N=4) reported a higher ORR (75%, 95% CI: 19.4-99.4) than those without prior anti-CD20 treatment (59%, 95% CI: 42.1-74.4). An mDOR of 14.8 months (95% CI: 5.6-NC) and mTTR of 1.9 months (95% CI: 1.9-3.6)
was recorded in FL patients with anti-CD20 treatment history. mPFS in prior anti-CD20–treated FL patients was 8.3 months (95% CI: 5.5-16.6).
Pi3K inhibitor–treated FL patients (EEAS, N=6) had an ORR of 66.7% (95% CI: 22.3-95.7) versus Pi3K inhibitor-naive patients (EEAS, N=37, 59.5% [95% CI: 42.1-75.3]). Those previously treated with a Pi3K inhibitor had an mDOR of 5.5 months (95% CI: 1.9-NC), and mTTR of 1.9 months (95% CI: 1.8-NC). mPFS in Pi3K inhibitor-naive patients was 12.0 months (95% CI: 6.3-NC) versus 5.5 months (95% CI: 1.8NC) in patients previously treated with a Pi3K inhibitor.
After continuous q.d. oral administration (200-800 mg), the median T max for sovleplenib was between 3.00 and 5.97 hours, arithmetic mean t1/2 was 7.91-14.4 hours and the mean accumulation ratio range was 1.51-2.65 (Online Supplementary Table S4). Exposures increased with dose increase in the dose range of 200-800 mg q.d., but linear relationship between dose and Cmax/AUC0-t from dose escalation was not concluded. The steady state of sovleplenib was reached within 7 days in most dose cohorts. The geometric mean (geometric% CV) of Cmax on day 15 was 221 (46.6%) and 320 (47.3%) ng/mL at 400 and 600 mg q.d., respectively; and the geometric mean (geometric% CV) of AUC0-tau at 400 and 600 mg q.d. on day 15 was 2,910 (43.8%) and 4,320 (51.6%) h·ng/mL, respectively. The mean concentration-time profiles of sovleplenib at steady state are presented in Online Supplementary Figure S3 Pharmacokinetic parameters of sovleplenib and its metabolites are presented in Online Supplementary Tables S5 and S6, respectively.
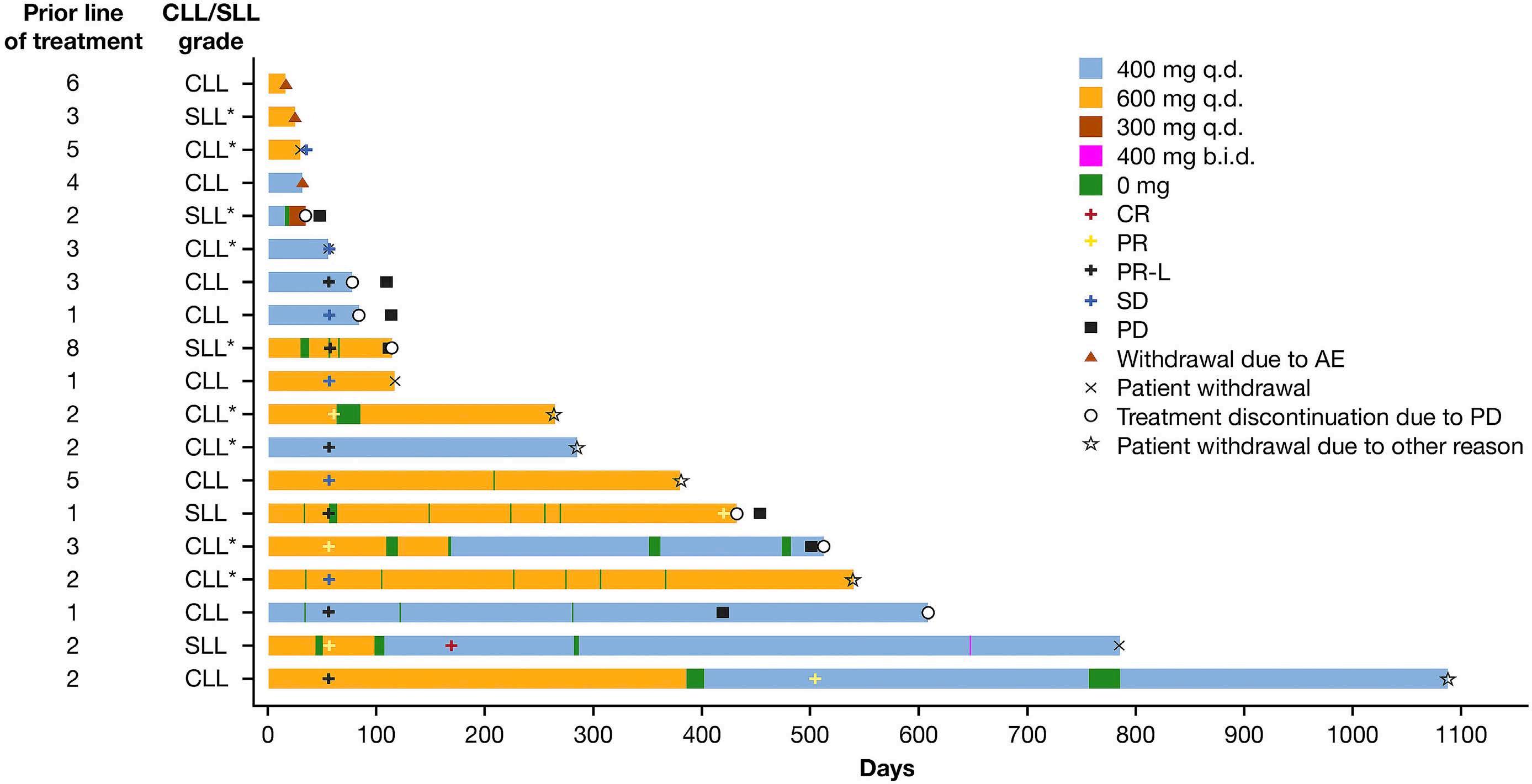
Figure 3. Duration of treatment in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma in the dose-expansion phase. CLL: chronic lymphocytic leukemia; SLL: small lymphocytic lymphoma; b.i.d.: twice daily; AE: adverse event; CR: complete response; PD: progressive disease; PR: partial response; PR-L: PR with increased lymphocyte count; q.d.: once daily; SD: stable disease.
Biomarker analysis
During dose expansion, sovleplenib induced reductions in plasma levels of CCL22, CCL17 and CXCL13 chemokines compared with baseline (Online Supplementary Figure S4). A similar trend was observed in separate analysis with cohorts A, B and D (Online Supplementary Figure S5). Consistent reduction was also observed with sovleplenib treatment during dose escalation (data not shown). Moreover, decreased levels of CCL3 and CCL4 in three of four CLL patients were observed, but not in other NHL subtypes (data not shown).
This phase I study examined safety, tolerability and preliminary efficacy of sovleplenib in relapsed/refractory B-cell tumors. The RP2D was determined to be 600 mg q.d. in patients >65 kg and 400 mg q.d. in patients ≤65 kg. Clinical responses were observed in CLL/SLL, DLBCL, MCL, MZL, LPL/WM and FL, including in heavily pretreated patients. The safety profile of sovleplenib was consistent with other Syk inhibitors; most toxicities were manageable, with no new safety concerns.12,14,18,25 Most common TEAE included neutropenia, leukopenia, anemia and platelet count decreased, with similar toxicities across cohorts. Some of the common toxicities reported for other Syk inhibitors, such as diarrhea, nausea, dyspepsia,
headache, hypertension and fatigue, were less frequent in our study.12,16,21 Although in patients treated with fostamatinib diarrhea was a DLT event (21%) or one of the most common TRAE (41%),12,16 it was not a DLT of sovleplenib. The incidence of diarrhea was 3.7% in the dose escalation and 9.3% in the dose expansion. Pneumonia and interstitial lung disease were reported in the current study; pulmonary toxicities were also seen with entospletinib.17,18,26 The safety profile may be attributable to the high target specificity of sovleplenib, which leads to low off-target adverse effects. Five patients who experienced probably sovleplenib-related TEAE leading to death had other confounding factors, including previous primary diseases and disease progression. A challenge in treating B-cell malignancies is the heterogeneity of pathogenesis due to the complex nature of B-cell signaling and diversity of aberrant B-cell constitutive activation.27 This study showed that sovleplenib has anti-tumor activity in lymphomas of different histology, including FL, CLL, SLL, indolent B-cell lymphoma, DLBCL and MCL. In a heavily pretreated population, the ORR in FL (grade 1, 2, 3a) was 60.5%. ORR in other cohorts ranged from 28.6% in both MCL and DLBCL cohorts to 56.3% in the CLL/SLL cohort.
The anti-tumor activity of sovleplenib was particularly noted in cohort D, where the overall IRC-assessed ORR was 50.8%, mDOR was 15.7 months, and mPFS was 12.0 months. More specifically, significant anti-tumor activity was demonstrated in FL patients with an IRC-assessed ORR of 60.5%,
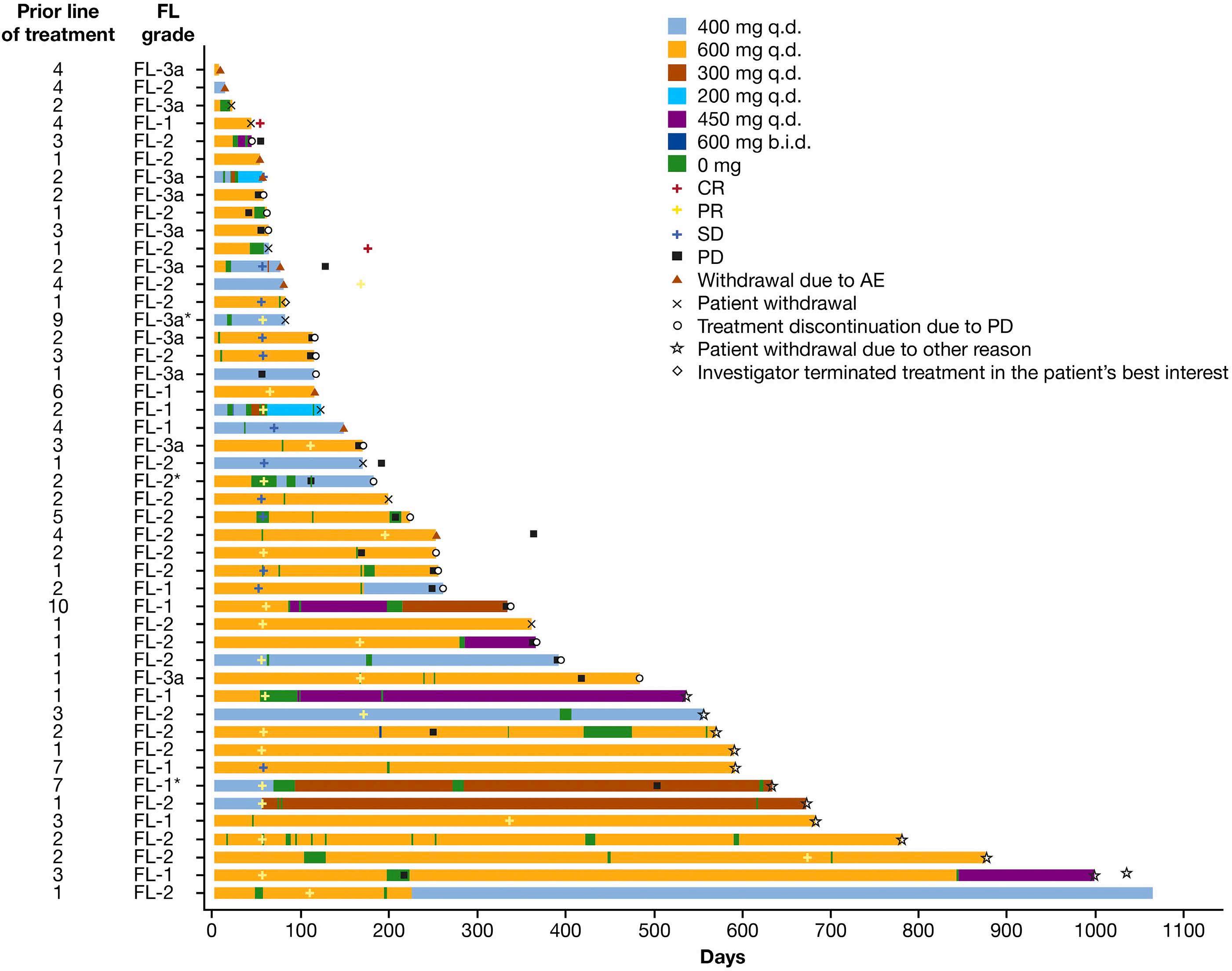
4. Duration of treatment in patients with follicular lymphoma in the dose-expansion phase. FL: follicular lymphoma; b.i.d.: twice daily; AE: adverse event; CR: complete response; PD: progressive disease; PR: partial response; PR-L: PR with increased lymphocyte count; q.d.: once daily; SD: stable disease.
mPFS of 11.0 months and mDOR not reached, indicating that this malignancy is distinctly susceptible to Syk inhibition. The Syk/FLT3 inhibitor TAK-659 has also been shown to achieve a high ORR of 89%, albeit in a small cohort (N=14) of FL patients.12,18,25 In contrast, modest anti-tumor activity in FL patients treated with entospletinib or fostamatinib was noted, with ORR of 10% and 17%, respectively, and mPFS of 4.6 and 5.7 months, respectively. The differences in ORR may be due to different prior treatment regimens, such as 96.9% versus 33.3% of patients with MZL, LPL/WM and FL (grades 1, 2, 3a) receiving prior anthracycline in our study versus that in entospletinib.18 Half of the FL patients in the study of TAK-659 had underwent prior autologous transplant.12,18,25 Our subgroup analysis indicated that FL patients with prior Pi3K inhibitor treatment also experienced an encouraging tumor response (ORR, 66.7%; mDOR, 5.5 months; mPFS, 5.5 months), as did patients who received
prior anti-CD20 monoclonal antibody therapy (ORR, 59%; mDOR, 14.8 months; mPFS, 8.3 months). This could indicate that sovleplenib may be an option after anti-CD20, or indeed Pi3K inhibitor, treatment failure given that Syk signaling is upstream of Pi3K during BCR activation.28 Sovleplenib-mediated anti-tumor activity was also encouraging in CLL/SLL, with an ORR of 56.3%, mDOR of 13.1 months and mPFS of 14.9 months. Previous Syk inhibitor studies with entospletinib, cerdulatinib and fostamatinib demonstrated ORR results of 33-61% in CLL/SLL patients, with an mPFS of 5.6-13.8 months.12,17,27 Current CLL/SLL first-line therapy includes chemotherapy, anti-CD20 immunotherapy, BTKi and BCL-2 inhibitors. In CLL/SLL patients previously treated with BTKi, or or received ≥3 lines of treatments, sovleplenib showed a notable anti-tumor activity, with an ORR of 50%, mDOR of 14.7 months and mPFS of 16.5 months; therefore, sovleplenib may be explored as
a potential subsequent treatment option in patients who cannot tolerate or become resistant to BTKi.
The target inhibition based on the IC50 indicated that the onset dose of sovleplenib was 400 mg q.d.. At 400 and 600 mg q.d., the coverage time of target inhibition was over 24 hours according to the IC50.
Microenvironment-related biomarkers may predict clinical outcomes.29 BCR activation and co-stimulation by CD40+ T cells promote the secretion of CCL17 and CCL22, where these two chemokines could potentially induce trans-endothelial migration of activated T cells.30 Monocyte-derived nurse-like cells attract and protect leukemic CLL cells through secretion of chemokines, such as CXCL12 and CXCL13, thus promoting CLL disease progression, as demonstrated in CLL animal models.30,31 While in our study, a distinct reduction in CCL22, CCL17 and CXCL13 was observed in cohorts A, B and D, which indicates the contribution of sovleplenib to the tumor microenvironment.17,32,33 Sovleplenib reduced biomarker levels consistently during dose escalation; however, a statistically significant dose-dependent correlation was not observed, similar to previous results seen in a dual SYK/JAK inhibitor.34
Limitations of this study include its single-arm design, small sample size and enrollment of Chinese patients only. Additionally, long-term overall survival data were not collected.
In conclusion, sovleplenib demonstrated acceptable tolerability in B-cell lymphomas patients; encouraging anti-tumor activities were observed, especially in CLL/SLL and FL. Further research is warranted to determine potential biomarkers to select patients that may be primed to benefit from Syk inhibitors in relapsed/refractory B-cell
1. International Agency for Research on Cancer. Cancer fact sheets. https://gco.iarc.fr/today/fact-sheets-cancers Accessed 12 February 2022.
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209-249.
3. Sun J, Yang Q, Lu Z, et al. Distribution of lymphoid neoplasms in China: analysis of 4,638 cases according to the World Health Organization classification. Am J Clin Pathol. 2012;138(3):429-434.
4 Casulo C, Nastoupil L, Fowler NH, Friedberg JW, Flowers CR. Unmet needs in the first-line treatment of follicular lymphoma. Ann Oncol. 2017;28(9):2094-2106.
5. Stegemann M, Denker S, Schmitt CA. DLBCL 1L-what to expect beyond R-CHOP? Cancers (Basel). 2022;14(6):1453.
6. Goodman PA, Wood CM, Vassilev A, Mao C, Uckun FM. Spleen tyrosine kinase (Syk) deficiency in childhood pro-B cell acute lymphoblastic leukemia. Oncogene. 2001;20(30):3969-3978.
7 Chen L, Monti S, Juszczynski P, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse
malignancies.
HY, SFu, SFan, QX, JW, XJ, GD, WS are employees of HUTCHMED Limited.
HY, SFan and JZ conceptualized and designed the study. HY, SFu, SFan, QX, JW, XJ, GD and WS provided administrative support for study conduct. YS, JC, QZ, CL, LQ, JQ, HZhang, WL, LL, HJ, KZ, WZ, LZhang, DL, LZou, HYang, WQ, HZ and JH provided provision of study material or patients. YS, JC, QZ, CL, LQ, JQ, HZhang, WL, LL, HJ, KZ, WZ, LZhang, DL, LZou, HYang, WQ, HZ and JH performed the research and collected the data. QX analyzed the data. HY, SFu, SFan, QX, JW, XJ and GD interpreted the results. All authors had full access to study data, drafted, reviewed and approved the manuscript for submission.
The authors would like to thank all patients and their families, the investigators, and all staff who participated in this study. Medical writing support was provided by Deirdre Kelly (Parexel), in accordance with Good Publication Practice 3 guidelines.
This study was sponsored by HUTCHMED Limited.
Data-sharing statement
Individual data will not be made available.
large B-cell lymphoma. Blood. 2008;111(4):2230-2237.
8. Efremov DG, Laurenti L. The Syk kinase as a therapeutic target in leukemia and lymphoma. Expert Opin Investig Drugs. 2011;20(5):623-636.
9 Awan FT, Thirman MJ, Patel-Donnelly D, et al. Entospletinib monotherapy in patients with relapsed or refractory chronic lymphocytic leukemia previously treated with B-cell receptor inhibitors: results of a phase 2 study. Leuk Lymphoma. 2019;60(8):1972-1977.
10 Cheng S, Coffey G, Zhang XH, et al. SYK inhibition and response prediction in diffuse large B-cell lymphoma. Blood. 2011;118(24):6342-6352.
11. Leseux L, Hamdi SM, Al Saati T, et al. Syk-dependent mTOR activation in follicular lymphoma cells. Blood. 2006;108(13):4156-4162.
12. Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578-2585.
13. Currie KS, Kropf JE, Lee T, et al. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine
kinase. J Med Chem. 2014;57(9):3856-3873.
14 Bussel J, Arnold DM, Grossbard E, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: results of two phase 3, randomized, placebo-controlled trials. Am J Hematol. 2018;93(7):921-930.
15. Bussel JB, Arnold DM, Boxer MA, et al. Long-term fostamatinib treatment of adults with immune thrombocytopenia during the phase 3 clinical trial program. Am J Hematol. 2019;94(5):546-553.
16. Flinn IW, Bartlett NL, Blum KA, et al. A phase II trial to evaluate the efficacy of fostamatinib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). Eur J Cancer. 2016;54:11-17.
17 Sharman J, Hawkins M, Kolibaba K, et al. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2015;125(15):2336-2343.
18. Andorsky DJ, Kolibaba KS, Assouline S, et al. An open-label phase 2 trial of entospletinib in indolent non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2019;184(2):215-222.
19 Hamlin PA, Flinn IW, Wagner-Johnston N, et al. Efficacy and safety of the dual SYK/JAK inhibitor cerdulatinib in patients with relapsed or refractory B-cell malignancies: results of a phase I study. Am J Hematol. 2019;94(4):e90-e93.
20 Coffey G, Betz A, DeGuzman F, et al. The novel kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-cell cancer. J Pharmacol Exp Ther. 2014;351(3):538-548.
21. Liu D, Mamorska-Dyga A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol. 2017;10(1):145.
22. Lam B, Arikawa Y, Cramlett J, et al. Discovery of TAK-659 an orally available investigational inhibitor of spleen tyrosine kinase (SYK). Bioorg Med Chem Lett. 2016;26(24):5947-5950.
23. Cai Y, He J, Wu Z, et al. Preclinical pharmacology characterization of sovleplenib (HMPL-523), an orally available Syk inhibitor. J Pharmacol Exp Ther. 2024;388(1):156-170.
24. Yang N, Deng W, Sun Q, et al. HMPL-523, a novel SYK inhibitor
showed anti-tumor activities in vitro and in vivo. Blood. 2016;128(22):3970-3970.
25. Gordon LI, Kaplan JB, Popat R, et al. Phase I study of TAK-659, an investigational, dual SYK/FLT3 inhibitor, in patients with B-cell lymphoma. Clin Cancer Res. 2020;26(14):3546-3556.
26. Burke JM, Shustov A, Essell J, et al. An open-label, phase II trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18(8):e327-e331.
27. Efremov DG, Turkalj S, Laurenti L. Mechanisms of B cell receptor activation and responses to B cell receptor inhibitors in B cell malignancies. Cancers (Basel). 2020;12(6):1396.
28. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
29 Sun R, Medeiros LJ, Young KH. Diagnostic and predictive biomarkers for lymphoma diagnosis and treatment in the era of precision medicine. Mod Pathol. 2016;29(10):1118-1142.
30 Ghia P, Strola G, Granziero L, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32(5):1403-1413.
31. Reinart N, Nguyen PH, Boucas J, et al. Delayed development of chronic lymphocytic leukemia in the absence of macrophage migration inhibitory factor. Blood. 2013;121(5):812-821.
32. Quiroga MP, Balakrishnan K, Kurtova AV, et al. B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406. Blood. 2009;114(5):1029-1037.
33. Coffey GP, Feng J, Betz A, et al. Cerdulatinib pharmacodynamics and relationships to tumor response following oral dosing in patients with relapsed/refractory B-cell malignancies. Clin Cancer Res. 2019;25(4):1174-1184.
34. Hamlin PA, Flinn I, wagner-Johnston N, et al. Clinical and correlative results of a phase 1 study of cerdulatinib (PRT062070) a dual SYK/JAK inhibitor in patients with relapsed/ refractory B cell malignancies. Blood. 2015;126(23):3929.
Matthew J. Maurer,1,2 Carla Casulo,3 Melissa C. Larson,1 Thomas M. Habermann,2 Izidore S. Lossos,4 Yucai Wang,2 Loretta J. Nastoupil,5 Christopher Strouse,6 Dai Chihara,5 Peter Martin,7 Jonathon B. Cohen,8 Brad S. Kahl,9 W. Richard Burack,3 Jean L. Koff,8 Yong Mun,10 Anthony Masaquel,10 Mei Wu,10 Michael C. Wei,10 Ashwini Shewade,10 Jia Li,10 James R. Cerhan,1 Brian K. Link6 and Christopher R. Flowers5
1Department of Quantitative Health Sciences, Mayo Clinic, Rochester, MN; 2Division of Hematology, Mayo Clinic, Rochester, MN; 3Department of Medicine, Wilmot Cancer Institute, University of Rochester, Rochester, NY; 4Department of Medicine, Comprehensive Sylvester Cancer Center, University of Miami, Miami, FL; 5University of Texas, MD Anderson Cancer Center, Houston, TX; 6Department of Medicine, University of Iowa, Iowa City, IA; 7Department of Medicine, Weill Cornell Medical College, New York, NY; 8Department of Hematology and Medical Oncology, Winship Cancer Institute, Emory University, Atlanta, GA; 9Department of Medicine, Washington University in St Louis, St Louis, MO and 10Genentech, Inc., South San Francisco, CA, USA
Correspondence: M.J. Maurer maurer.matthew@mayo.edu
Received: June 14, 2023. Accepted: November 17, 2023. Early view: November 30, 2023.
https://doi.org/10.3324/haematol.2023.283737
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Mosunetuzumab is a novel bispecific antibody targeting epitopes on CD3 on T cells and CD20 on B cells with the goal of inducing T-cell mediated elimination of malignant B cells. A recent pivotal phase I/II clinical trial (GO29781) demonstrated that mosunetuzumab induced an overall response rate (ORR) of 80%, complete response (CR) rate of 60%, and a median progression-free survival (PFS) of 17.9 months in patients with relapsed/refractory (R/R) follicular lymphoma (FL) following at least two prior lines of systemic therapy, including alkylator and anti-CD20 antibody-based therapy. Historical data from cohorts receiving therapy for R/R FL can provide some context for interpretation of single-arm trials. We compared the results from the mosunetuzumab trial to outcomes from a cohort of patients with R/R FL from the LEO Consortium for Real World Evidence (LEO CReWE). We applied clinical trial eligibility criteria to the LEO CReWE cohort and utilized matching-adjusted indirect comparison weighting to balance the clinical characteristics of the LEO CReWE cohort with those from the mosunetuzumab trial. ORR (73%, 95% CI: 65-80%) and CR rates (53%, 95% CI: 45-61%) observed in the weighted LEO CReWE cohort were lower than those reported on the mosunetuzumab trial (ORR=80%, 95% CI: 70-88%; CR=60%, 95% CI: 49-70%, respectively). PFS at 12 months was similar in the weighted LEO CReWE (60%, 95% CI: 51-69%) and the mosunetuzumab (58%, 95% CI: 47-68%) trial. Sensitivity analyses examining the impact of matching variables, selection of line of therapy, and application of eligibility criteria provide context for best practices in this setting.
Follicular lymphoma (FL) is a highly heterogeneous disease often characterized as indolent in behavior requiring intermittent systemic therapy over time.1 Although most patients with FL will experience a life expectancy com-
parable to that of the general population, a subset will have early disease-related mortality, often preceded by early relapse following initial immunochemotherapy (IC), refractoriness to alkylator therapies, or transformation to aggressive lymphoma.2 Patients with relapsed or refractory (R/R) FL have several therapeutic options available without
an agreed-upon standard.3 As such, selection of therapy in the third line or later requires thoughtful consideration of patient characteristics, prior therapy, expected toxicity, and disease behavior. Several reports of longitudinal FL patient cohorts illustrate that, on average, patients experience progressively shorter response durations or treatment-free intervals over sequential treatment lines.4-7 Thus, patients needing treatment after two or more prior therapies (especially if refractory to prior agents) represent a population with unmet needs that may be addressed by novel strategies.
Mosunetuzumab is a novel T-cell engaging bispecific antibody targeting epitopes on CD3 on T cells and CD20 on B cells with the goal of inducing T-cell-mediated elimination of malignant B cells. A recent pivotal phase I/II clinical trial (GO29781; clinicaltrials.gov identifier: NCT02500407) demonstrated that mosunetuzumab induced an overall response rate (ORR) of 80%, a complete response (CR) rate of 60%, and a median progression-free survival (PFS) of 17.9 months in patients with R/R FL following at least two prior lines of systemic therapy, including alkylator and anti-CD20 antibody-based therapy.8 Single-arm clinical trial designs such as GO29781 result in limited ability to determine how well unmet clinical needs have been addressed due to lack of a control cohort. Historical data from cohorts receiving therapy for R/R FL can provide some context for interpretation,4,9 but may suffer from a composition of patients that differ substantially from the trial cohort of interest in any of several relevant characteristics such as prior treatment histories, drug class refractoriness, and evolving patterns of care. We have recently demonstrated that, in expert academic US practices, treatment selection for R/R FL is quite variable and reflects disease heterogeneity.9 The aims of the present study were to provide a focused comparison of outcomes in a comparator real-world population to the GO29781 trial and evaluate potential best practices for implementing these types of comparison studies in the setting of R/R FL.
The Lymphoma Epidemiology of Outcomes Consortium for Real World Evidence (LEO CReWE) was used to build a real-world evidence cohort of patients with R/R FL who received at least two prior lines of systemic therapy; full details can be found in our previous publication.9 The study was approved by the Mayo Clinic Institutional Review Board. The population for the primary analysis comprised patients who met all of the following eligibility criteria: i) received systemic therapy for FL grade 1-3A after at least two prior lines of systemic therapy that included an anti-CD20-directed therapy and an alkylating agent; ii) met all key eligibility criteria from the GO29781 trial (listed in Online Supplementary Appendix) for a potential index line
of therapy with no missing eligibility or matching data; and iii) did not have transformed disease prior to a potential index line of therapy (Figure 1). Matching variables were as follows: age (years) at index therapy (mean, Standard Deviation [SD]); prior lines of therapy (mean, SD); progression of disease within 24 months (POD24) following front-line IC (yes vs. no vs. did not receive IC); double-refractory to anti-CD20 and alkylator therapy (yes vs. no); and elevated lactate dehydrogenase (LDH) at index therapy (yes vs. no). Matching-adjusted indirect comparison10 (MAIC) weighting was performed on the LEO CReWE dataset to select the index line for each patient and generate weights for comparison to the GO29781 study. The primary outcome measure for this study was ORR, defined as the proportion of patients with best response as CR or partial response (PR) during the available follow-up beginning from the start of therapy (index line) to the earliest of the following: documented progression of disease, initiation of a new line of anti-lymphoma therapy, transformation to an aggressive lymphoma, death, or end of follow-up as per the pre-specified clinical cut-off date. Secondary outcome measures were CR rate and PFS. Further details are provided in the Online Supplementary Appendix.
The statistical analysis plan (SAP) was jointly developed and finalized prior to the primary analysis. All analyses were performed by members of the LEO Cohort Statistics and Informatics Core (MJM, MCL). MAIC,10 a form of propensity score weighting, was applied to individual patient data (IPD) from the LEO CReWE study.11 A series of sensitivity analyses was performed to evaluate the impact of study outcomes when utilizing alternate approaches to the following: i) inclusion/exclusion criteria; ii) choice of matching variables; and iii) method for selecting which line of therapy to use for a given patient when multiple lines of therapy meet study eligibility. Continuous variables were summarized using descriptive statistics (median, interquartile range [IQR], or mean, SD); categorical variables, including response rates, were summarized as proportions and/or rates. Time-to-event variables were summarized using Kaplan-Meier curves and 95% Confidence Intervals (CI). Associations between groups and categorical endpoints (e.g., ORR and CR) were assessed using logistic regression and summarized with Odds Ratios and 95% CI. Further details on the study analyses can be found in the SAP (Online Supplementary Appendix).
LEO CReWE cohort
The starting population for the study consisted of 441 patients who received systemic therapy for FL grade 1-3A following at least two prior lines of systemic therapy that
included an anti-CD20-directed therapy and an alkylating agent. Seventy-three patients (17%) were excluded due to presence of variables that did not meet the inclusion / exclusion criteria based on the GO29781 clinical trial (Online Supplementary Table S1), and 157 patients (36%) were excluded due to missing data on one or more variables from inclusion / exclusion criteria and inability to confirm all GO29781 eligibility criteria, yielding 211 patients for the application of the MAIC analysis. An additional 9 patients were missing one or more of the matching variables, resulting in 202 LEO CReWE patients for the primary analysis. Full details on the 202 LEO CreWE patients prior to weighting can be found in Online Supplementary Table S2. The index therapy utilized in the primary analysis was 3rd line for 116 patients (57%), 4th line for 48 patients (24%), and 5th line or later for 38 patients (19%). Median age at index therapy was 60 years (IQR: 53-68), and 118 patients (58%) were male. Most patients (N=157, 78%) had stage III/IV disease at index therapy; 59 patients (29%) had elevated LDH at index therapy and 31 (15%) had bulky disease (>7 cm). Eighty-six patients (43%) experienced POD24 following first-line IC, 63 patients (31%) had disease progression after 24 months to front-line IC, and 53 patients (26%) did not receive firstline IC. One hundred and twenty-three patients (61%) had received prior anthracycline, and 26 patients (13%) had received a prior autologous stem cell transplant. One hundred and forty-one patients (70%) had FL refractory to previous CD20 antibody therapy, with 38% refractory to alkylating agents, and 69% refractory to their most recent prior line of therapy. The median time from diagnosis to index line was 62 months (IQR 41-93), and the median time from start of prior therapy to start of index line was 11 months (IQR 4-15). The most common class of therapy at index line was CD20 antibody-based IC (N=57, 28%). Additional therapy class included novel therapies with or without CD20 antibody (N=47, 23%), CD20 antibody and lenalidomide-based therapy (N=28, 14%), platinum-based salvage chemotherapy (N=23, 11%), and CD20 antibody monotherapy (N=18, 9%) (Online Supplementary Table S3). Twenty-eight patients (14%) received a stem cell transplant as index therapy (N=18 autologous and N=10 allogeneic), 7 received chimeric antigen receptor (CAR) T-cell therapy, and 4 received a bi-specific antibody; 80 patients (40%) received index therapy on a clinical trial (Online Supplementary Table S4). Most patients (N=171, 85%) received their index therapy at one of the eight LEO institutions. Response assessment on index therapy was positron emission tomography (PET)- or PET/computed tomography (CT)-based in 109 patients (54%), CT-based in 73 patients (36%), and clinical or unspecified in 20 patients (10%). At a median follow-up of 58.0 months from start of index therapy (range 0.03-252), 107 patients (53%) had had a PFS event after index therapy, and 47 patients (23%) had died. The unweighted CR and ORR in the 202 patients were 58% (95% CI: 51-65) and 78% (95% CI: 71-83), respectively; the unweighted 12-month PFS was 65% (95% CI: 59-72).
Details on the GO29781 trial have been previously reported.8 Most relevant to this analysis, median age at study entry in the 90 patients enrolled was 60 years (IQR 53-67), and 61% were male. LDH was elevated in 39% at study entry. Median number of previous lines of therapy was 3 (IQR 2-4), with 38% receiving 2 previous lines, 31% receiving 3 previous lines, and 31% receiving more than 3 previous lines. Thirty-eight patients (42%) had POD24 to front-line immunochemotherapy, and 48 (53%) had double-refractory disease to previous anti-CD20 and alkylator therapies.
Primary comparison of LEO CReWE to GO29781 by matching-adjusted indirect comparison weighted analysis
Prior to weighting, significant differences were observed in the clinical characteristics of the primary analysis cohort selected from the LEO CReWE (N=202) and the GO29781 trial (Table 1). LEO CReWE patients were less heavily pre-treated and had fewer cases of double refractory disease. LEO CReWE patients also had a longer time from previous treatment to their index therapy. Application of the MAIC utilizing the 5 matching variables resulted in an effective sample size (ESS; the number of independent non-weighted individuals that would be required to give an estimate with the same precision as the weighted sample estimate) of 127.3 and a weighted N of 167; the distribution of the weights is shown in Online Supplementary Figure S1. MAIC weighting rebalanced the distributions of all 5 matching variables, including number of prior lines of therapy. Other key clinical variables were also better aligned between the LEO CReWE and GO29781 studies after application of weights (Table 1). ORR observed in the weighted LEO CReWE cohort (73%, 95% CI: 65-80%) was lower than the ORR (80%, 95% CI: 70-88%) reported on the GO29781 trial (Table 2). Similarly, CR in the weighted LEO CReWE cohort (53%, 95% CI: 45-61%) was lower than the GO29781 trial (60%, 95% CI: 49-70%). PFS at 12 months was similar in the LEO CReWE cohort (12-month PFS 60%, 95% CI: 5169%) and the GO29781 trial (12-month PFS 58%, 95% CI: 47-68%) (Figure 2).
We performed a series of pre-specified sensitivity analyses to determine the impact of inclusion/exclusion criteria, matching variables, and index therapy selection on the outcomes in the study (Table 3). Application of GO29781based trial criteria and the requirement of non-missing data in the LEO CReWE cohort resulted in exclusion of over half of potential patients (230 out of 441 potential patients, 52%) from the primary analysis. Utilizing an alternative set of clinical trial inclusion / exclusion criteria had little impact on sample size or study results compared to the primary analysis. Ignoring the clinical trial eligibility criteria would have greatly increased the overall (N=357) and ef-
Table 1. Comparison of key patient characteristics in GO29781 versus LEO CReWE cohort (unweighted and matching-adjusted indirect comparison weighted).
N: number; MAIC: matching-adjusted indirect comparison; ESS: effective sample size; LDH: lactate dehydrogenase; POD24: progression of disease in 24 months; 1L: first-line; IC: immunochemotherapy; LOT: line of therapy; SCT: stem cell transplant.
Table 2. Primary results: comparison of GO29781 to LEO CReWE cohort.
N: number; ORR: overall response rate; CI: confidence interval; CR: complete response; PFS12: progression-free survival at 12 months.
fective (N=297) sample size of the study but made only a small impact on weighted estimates (ORR=74%, CR=56%, 12-month PFS 63%). Utilizing different sets of matching variables and/or variable types (e.g., dichotomous vs. continuous) in the MAIC implementation also had little effect on the ORR, CR, and PFS estimates. The greatest impact was seen when changing the algorithm for selecting the index line of therapy for patients with multiple eligible lines of therapy. Using a randomly selected treatment line in the LEO CReWE Cohort yielded a lower ESS (N=84.7) (Online Supplementary Figure S2), though clinical characteristics remained well balanced after weighting. Outcomes in the LEO CReWE cohort were also inferior when using a random line (ORR=72%, CR=42%, 12-month PFS=54%) compared to the primary analysis. Utilizing the first eligible line for a patient resulted in an essentially unusable analysis, with an ESS of 20.1 and most patients having near-zero weights (Online Supplementary Figure S3). Selecting the last eligible line yielded nearly identical results to the primary analysis, due to the preferential weighting of later lines of therapy in LEO CReWE. Summary details of the sensitivity analysis results can be found in Table 3.
In a post-hoc subset analysis of LEO CReWE by receipt of index therapy on clinical trial, response rates (ORR=76%, 95% CI: 65-85%; CR=56%, 95% CI: 44-67%) were higher and PFS12 was lower (57%, 95% CI: 44-73%) on trials compared to index therapy received off-trial (ORR=70%, 95% CI: 59-79%; CR=50%, 95% CI: 40-61%; PFS12=62%, 95% CI: 51-75%) (Online Supplementary Table S5).
In this study, we evaluated response rates and PFS in an observational cohort of patients with FL treated in the third line or later after prior alkylator and anti-CD20-based therapy. The LEO CReWE cohort was restricted to those meeting key eligibility criteria in the GO29781 trial and further re-weighted to align with the clinical characteristics of the patients treated on GO29781. Strengths of the study include the assembly of a large observational cohort of patients with R/R FL and detailed clinical annotation and outcomes from diagnosis through all lines of therapy. LEO CreWE patients with R/R FL were treated in medical cen-
ters and clinical contexts similar to those for the GO29781 trial, with 40% of patients receiving index therapy on a clinical trial, which is distinct from observational studies involving cohorts of patients with R/R FL from general practices.12-14 MAIC rebalanced the LEO CReWE cohort to align with the clinical characteristics of patients enrolled on the GO29781 trial. Standard limitations of these types of comparative effectiveness studies apply, including potential bias by both observed and unobserved differences in the patients between the two cohorts. Adverse event data were not available in the LEO CReWE cohort, and thus this study does not provide clinical comparisons of toxicity or tolerability. Safety profiles and quality of life should be considered when making clinical decisions regarding therapy in this clinical space.
Individual patient-level data (IPD) from GO29781 were not available for this analysis, and thus the weighting of the LEO CReWE data was based on the summary statistics for the GO29781 trial data as opposed to matching or propensity weighting approaches utilizing IPD. The MAIC approach utilized in this study weighted the individuals in the LEO CReWE cohort such that the key clinical characteristics used for matching were balanced between the LEO CReWE and GO29781. Using this approach, all eligible patients from the LEO CReWE cohort were utilized in the analysis, but LEO CReWE patients with disparate clinical characteristics compared to the GO29781 trial participants contributed less information to the analysis compared to LEO CReWE patients with similar clinical characteristics as the GO29781 trial. Methods for checking fit and calibration in a MAIC weighting approach are limited compared to traditional propensity score weighting, in which IPD is available. Additionally, residual confounding may remain after MAIC weighting.
Response rates observed in the LEO CReWE cohort (weighted ORR=73%, CR=53%) were encouraging for R/R FL, yet remained lower than those observed in the GO29781 study (ORR=80%, CR=60%). PFS at 12 months was similar between the LEO CReWE cohort and GO29781 trial based on the reported duration of follow-up used in this analysis. Notably, response rates (ORR=76%, CR=56%) were higher and PFS12 (57%) was lower for LEO CReWE index therapies received on clinical trial, though this may be confounded by patient selection and varying response rates of different treatment classes. Response and progression are assessed differently between clinical trials and routine clinical practice, which needs to be considered in the interpretation of these results. Clinical trials feature frequent and standardized disease assessment that includes imaging at regular intervals. In routine clinical care, intervals for response assessment are not stringently dictated, and imaging may not necessarily be performed on a regular schedule. Further, clinicians may not universally confirm response with repeat bone marrow biopsy (BMB) in patients who had prior bone marrow involvement, and clinical plans to proceed to
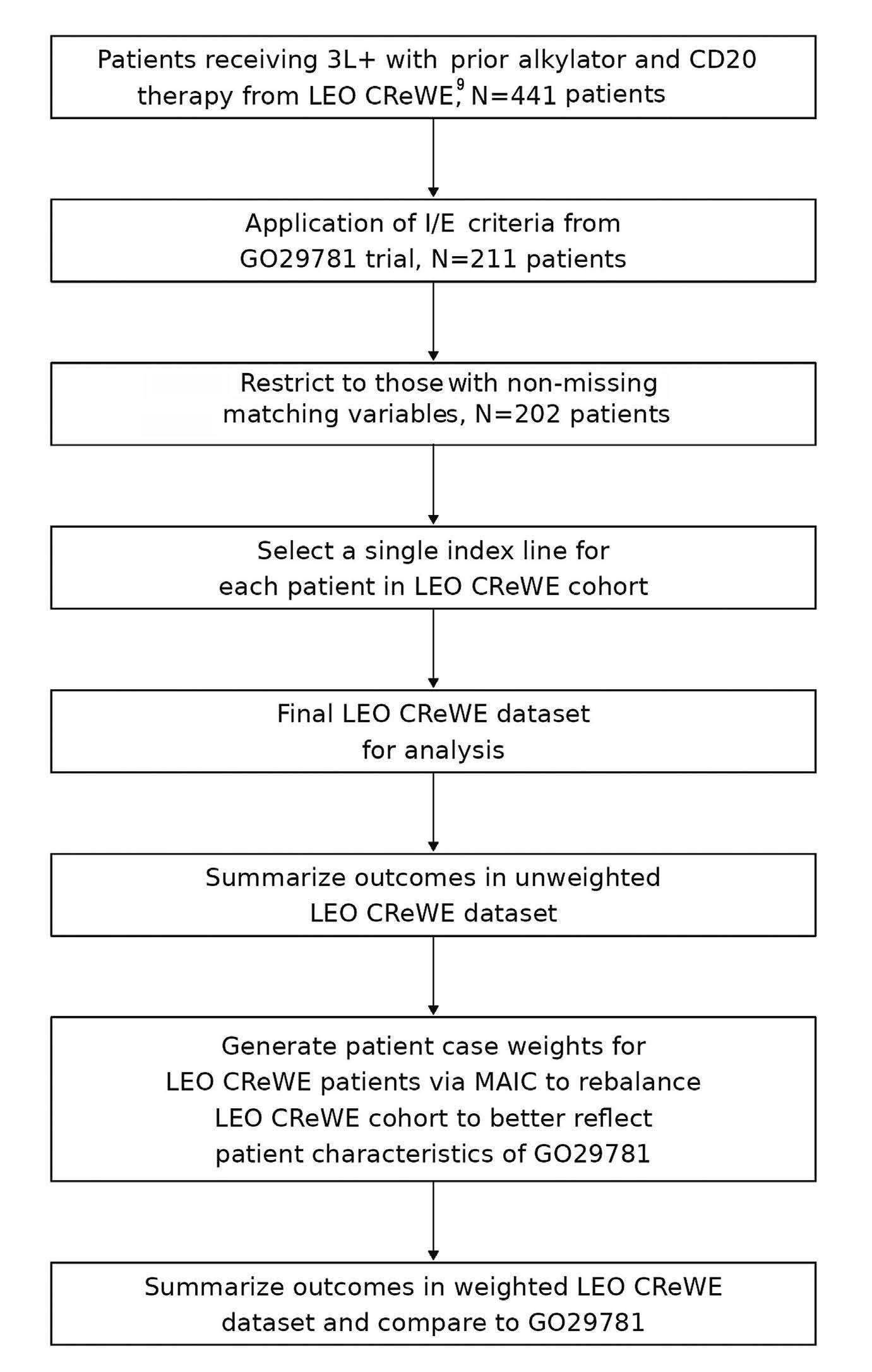
Figure 1. Analysis flow chart. I / E: inclusion / exclusion; MAIC: matching-adjusted indirect comparison; N: number; PFS: progression-free survival.
transplantation based on response may influence decision making. In the GO29781 trial, a BMB was required at study entry with repeat BMB to confirm complete response if bone marrow involvement was present at baseline. This was not required in the LEO CReWE cohort and only 41% of patients had bone marrow biopsied at the start of index therapy (data not shown). However, in an analysis of 580 patients pooled from 7 National Clinical Trials Network (NCTN) clinical trials of FL, Rutherford and colleagues identified that BMB were irrelevant to assessing complete response in 99% of patients.15 Thus it is unlikely that differences in routine BMB between trial and observational cohorts has a significant impact on the interpretation of results. Unlike aggressive lymphoma, where progression often manifests clinically prior to planned imaging,16 routine imaging on clinical trials may contribute to shorter PFS in the setting of FL (as compared to observational studies without routine imaging) when an increase in tumor size may not correspond to a clinical indication for evaluation or subsequent therapy. A prior study from three centers
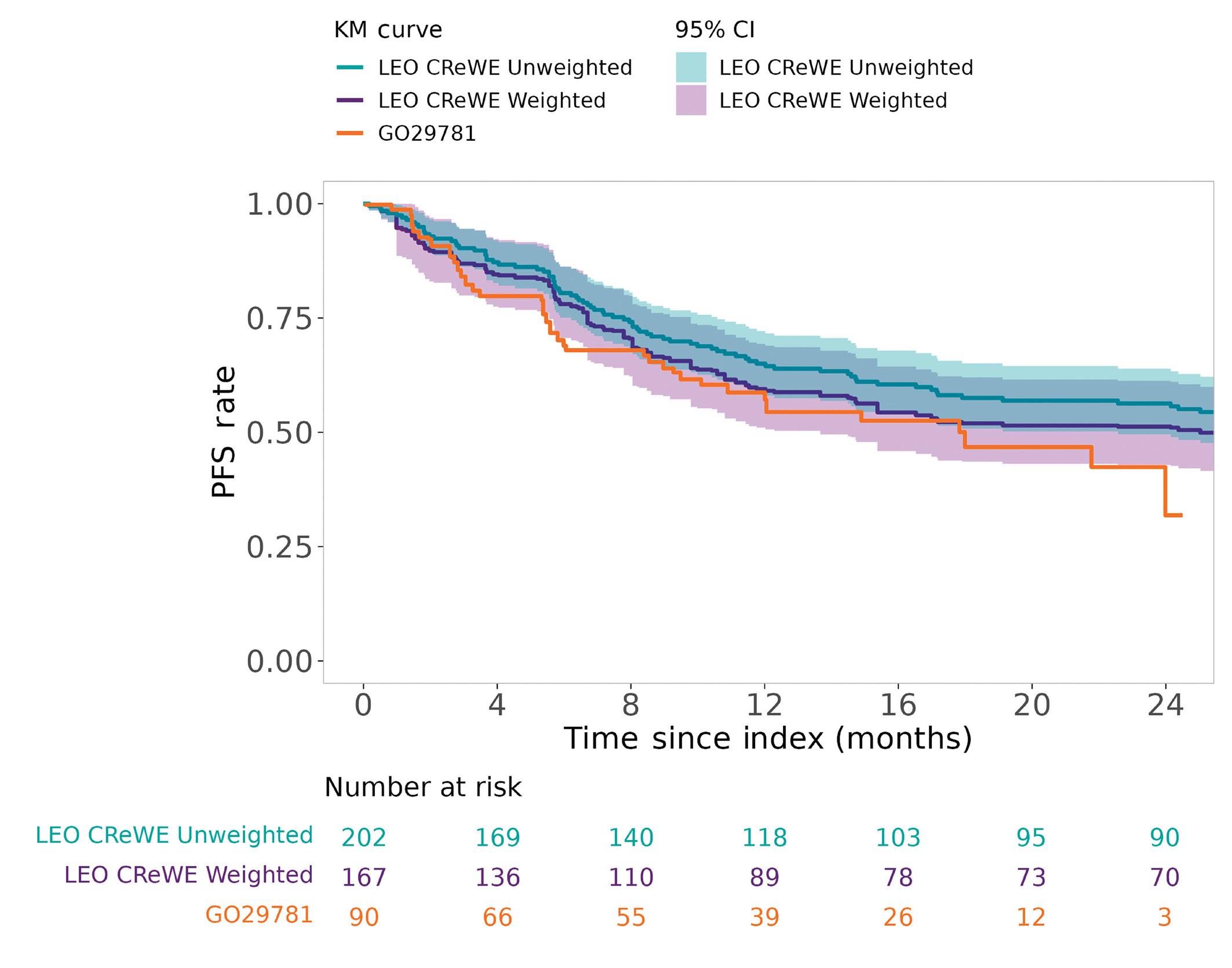
within LEO CReWE identified that 54-64% of relapses to front-line therapy for FL were detected clinically.17 A distinct advantage of the LEO consortium is the involvement of experts in lymphoma clinical trials and patient management, including leaders in the NCTN and several members of the NCI Lymphoma Steering Committee, who have expertise in lymphoma response assessment. Despite these commonly cited limitations for synthetic cohorts, several recent studies18-20 have used this strategy to support the impact of phase II trials in lymphoma. Our study differs notably from the others by demonstrating modest differences between the measured outcomes of the phase II trial results and the synthetic cohort. There are several possible explanations for this difference, including the unique choice of MAIC methodology to accommodate lack of IPD from GO29781, the line of index therapy or variables chosen for matching, improved assessment of outcomes and/or improved outcomes in an observational cohort managed by lymphoma experts, or the true relative activity of mosunetuzumab compared to other options in this patient setting. The generation of a synthetic cohort and/or implementation of a matching-based analysis requires a series of decisions that may influence the results. Analytical decisions such as eligibility criteria, selection of index therapy, and matching variables are then applied to the cohort selected for the analysis. We performed a series of sensitivity analyses to
address the uncertainty in estimation of outcomes introduced by such decisions. Selecting the cohort and index line is especially critical given the heterogeneity in treatment selection and expected outcomes for patients with R/R FL. The index line of therapy for LEO CReWE was chosen based on the line of therapy most like the clinical characteristics of the GO29781 trial based on MAIC weights. Our initial publication of the LEO CReWE FL cohort primarily focused on outcomes in patients at initial time of eligibility for the GO29781 trial (i.e., third line or beyond with prior anti-CD20 and alkylator therapy), with 94% of patients achieving this in the third line setting.9 However, utilizing the first eligible line in our sensitivity analysis resulted in essentially unusable results due largely to imbalances in the number of prior lines of therapy. This highlights the importance of careful examination of clinical characteristics and thoughtful alignment of a synthetic cohort with clinical trial criteria. In contrast, alternative collections of matching variables and trial inclusion / exclusion criteria had little impact on effective sample size or weighted outcomes.
These results support our previously published data showing that, despite multiple recurrences, patients
Table 3. Sensitivity analyses of matching-adjusted indirect comparison weighting scenarios in LEO CReWE cohort.
N: number; MAIC: matching-adjusted indirect comparison; ESS: effective sample size; CI: confidence interval; POD24: progression of disease in 24 months; 1L: first-line; LOT: line of therapy; ORR: overall response rate; CR: complete response; PFS12: progression-free survival at 12 months; I/E: inclusion / exclusion; ECOG PS: Eastern Cooperative Oncology Group Performance Status; FLIPI: Follicular Lymphoma International Prognostic Index.
with R/R FL respond favorably to therapy, albeit of limited duration. These data suggest that the encouraging response rates observed in this novel class of bispecific therapy in a heavily pre-treated population yield similar PFS to our comparison cohort with the current study follow-up. Although the methodology utilized in this analysis has limitations relative to a randomized clinical trial, it helps to provide comparative context for clinical outcomes and patterns of care. Differences in response and progression assessment methodology should be taken into consideration when making direct comparisons between clinical trials and observational cohorts of patients treated in routine clinical practice. Development of a set of best practices by clinical expert consensus for these types of comparative effectiveness analyses in the R/R FL space may be beneficial for more consistency in future studies. Comprehensive data that include safety, tolerability, quality of life, as well as efficacy, should be considered when evaluating treatment options for patients with R/R FL.
MJM declares advisory board (Adaptive Biotechnologies, AstraZeneca, GenMab); consulting (BMS); research funding (Genmab, Genentech/Roche, BMS); spouse employment and
stock (Exact Sciences). CC declares research funding (SecuraBio, Genmab, Gilead, Genentech, Inc.). MCL declares research funding (Genentech, Genmab). TMH declares research funding (Genentech, Sorrento); consulting or advisory role (Eli Lilly & Co., Morphosys, Incyte, Biegene, Loxo Oncology); data monitoring committee (Seagen, Tess Therapeutics, Eli Lilly & Co.). ISL declares current employment (University of Miami); honoraria (Adaptive); consulting or advisory role (LRF); research funding (NCI); provided compensation teaching (Kyowa Kirin). YW declares research funding (Incyte, InnoCare, LOXO Oncology, Eli Lilly, MorphoSys, Novartis, Genentech, Genmab); advisory board (Eli Lilly, LOXO Oncology, TG Therapeutics, Incyte, InnoCare, Kite, Jansen, BeiGene); consultancy (Innocare, AbbVie); honorarium (Kite). LJN declares research funding (BMS, Caribou Biosciences, Epizyme, Genentech, Gilead/Kite, Genmab, Janssen, IGM Biosciences, Novartis, Takeda); honoraria (Abbvie, ADC Therapeutics, BMS, Caribou Biosciences, Epizyme, Genentech/Roche, Gilead/ Kite, Genmab, Incyte, Janssen, MEI, Novartis, Takeda); DSMC (Genentech/Roche, MEI, Takeda, Denovo). CS declares current employment (University of Iowa); honoraria (Iowa Oncology Society, Curio Science); consulting or advisory role (Pfizer, GSK). DC declares research funding (Janssen, Epizyme, BMS, MorphoSys). PM declares consulting or advisory role (Abbvie, AztraZeneca, Beigene, BMS, Daiichi, Genentech, Janssen). JBC
declares consulting or advisory role (BeiGene, ADCT, Lilly, AstraZeneca, Janssen, Genentech, Inc., HutchMed, Kite/Gilead); research funding (Genentech, Inc., Celgene, Novartis, Takeda, AstraZeneca, Loxo/Lilly, Lam Therapuetics, BioInvent); Expert Testimony (Hawaii). BSK declares consultancy (Astrazeneca, ADCT Therapeutics, Roche, Genetech, Abbvie, MEI, AcertaPharma,Pharmacyclics, Celgene/BMS, Beigene, Kite, Janssen, Incyte, Hutchmed, TG Therapeutics, BeiGene, Genmab, Seattle Genetics); research funding (Genetech, Abbvie, Celgene/ BMS, BeiGene Astra Zeneca, Hutchmed); speaker’s bureau (Research to Practice). JLK declares consulting or advisory role (Gamida Cell, MorphoSys, BeiGene USA, TG Therapeutics); research funding (Viracta Therapeutics). YM declares current employment (Genentech/Roche); stock ownership (Genentech/Roche). AM declares current holder of individual stocks in a privately-held company (Roche); current holder of stock options in a privately-held company (Roche). MCW declares current employment (Genentech); stock ownership (Roche); travel, accommodation, and expenses (Genentech/Roche). AS declares current employment (F. Hoffmann-La Roche Ltd.); stock ownership (F. Hoffmann-La Roche Ltd). JL declares stock ownership (F. Hoffmann-La Roche Ltd.). JC declares consulting or advisory role (BMS, Protagonist Therapeutics SMC). BKL declares consulting or advisory role (Genentech/ Roche, MEI Pharma Inc.); research funding (Genentich/Roche, Genmab, Celgene, AstraZeneca); expert testimony (Amgen). CRF declares consulting or advisory role (Bayer, Gilead, Spectrum Pharmaceuticals, AbbVie, Celgene, Denovo Biopharma, BeiGene, Karyopharm Therapeutics, Pharmacyclics/Janssen, Genentech/Roche, Epizyme, Genmab, Seattle Genetics, Fore-
1. Jacobsen E. Follicular lymphoma: 2023 update on diagnosis and management. Am J Hematol. 2022;97(12):1638-1651.
2. Sarkozy C, Maurer MJ, Link BK, et al. Cause of death in follicular lymphoma in the first decade of the rituximab era: a pooled analysis of French and US cohorts. J Clin Oncol. 2019;37(2):144-152.
3. Qualls D, Salles G. Prospects in the management of patients with follicular lymphoma beyond first-line therapy. Haematologica. 2022;107(1):19-34.
4 Link BK, Day BM, Zhou X, et al. Second-line and subsequent therapy and outcomes for follicular lymphoma in the United States: data from the observational National LymphoCare Study. Br J Haematol. 2019;184(4):660-663.
5. Rivas-Delgado A, Magnano L, Moreno-Velázquez M, et al. Response duration and survival shorten after each relapse in patients with follicular lymphoma treated in the rituximab era. Br J Haematol. 2019;184(5):753-759.
6. Batlevi CL, Sha F, Alperovich A, et al. Follicular lymphoma in the modern era: survival, treatment outcomes, and identification of high-risk subgroups. Blood Cancer J. 2020;10(7):74.
7 Salles G, Schuster SJ, Fischer L, et al. A retrospective cohort study of treatment outcomes of adult patients with relapsed or refractory follicular lymphoma (ReCORD-FL). Hemasphere.
sight Diagnostics, BMS, Curio Science, AstraZeneca, MorphoSys); stock ownership (Foresight Diagnostics, N Power); research funding (Acerta Pharma, Janssen, Gilead, Celgene, TG Therapeutics, Genentech/Roche, Pharmacyclics, AbbVie, Millennium, Alimera Sciences, Xencor, 4D Pharma, Adaptimmune, Amgen, Bayer, Cellectis, EMD Serono, Guardant Health, Iovance Biotherapeutics, Kite/Gilead, MorphoSys, Nektar, Novartis, Pfizer, Sanofi, Takeda, Ziopharm Oncology). WRB has no conflicts of interest to disclose.
MM, CC, ML, JL, TH, IL, YW, LN, CS, DC, PM, JC, BK, WB, JK, YM, AM, MW, MCW, AS, JL, JC, BL and CF are responsible for the conception and/or design of the study/research, for analysis and/or interpretation of the data, and wrote the manuscript. MM, CC, ML, JL, TH, IL, YW, LN, CS, DC, PM, JC, BK, WB, JK, JC, BL and CF are responsible for the collection and/or assembly of the data.
This work was supported by a grant from the National Cancer Institute: Lymphoma Epidemiology of Outcomes (U01CA195568) and a research grant from Genentech. No medical writer was used for this manuscript.
Data-sharing statement
The data in the study are not publicly available. Data sharing policies and the process to request the LEO CReWE data that support the findings of this study can be found on the LEO Cohort website: https://leocohort.org/
2022;6(7):e745.
8. Budde LE, Sehn LH, Matasar M, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. 2022;23(8):1055-1065.
9 Casulo C, Larson MC, Lunde JJ, et al. Treatment patterns and outcomes of patients with relapsed or refractory follicular lymphoma receiving three or more lines of systemic therapy (LEO CReWE): a multicentre cohort study. Lancet Haematol. 2022;9(4):e289-e300.
10 Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940-947.
11. Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Making. 2018;38(2):200-211.
12. Albarmawi H, Nagarajan M, Onukwugha E, et al. Follicular lymphoma treatment patterns between 2000 and 2014: a SEER-Medicare analysis of elderly patients. Future Oncol. 2020;16(8):353-365.
13. Xie C, Li R, Huang X, Chihara D, Flowers CR. The impact of
sequence of therapy for older patients with follicular lymphoma: SEER-Medicare analysis. Clin Lymphoma Myeloma Leuk. 2022;22(10):e938-e946.
14 Ayers EC, Margolis D, Landsburg DJ. Real world outcomes in patients with relapsed/refractory diffuse large B-cell lymphoma receiving palliative intent therapies. Clin Lymphoma Myeloma Leuk. 2020;20(10):661-667.
15. Rutherford SC, Yin J, Pederson L, et al. Relevance of bone marrow biopsies for response assessment in US National Cancer Institute National Clinical Trials Network Follicular Lymphoma Clinical Trials. J Clin Oncol. 2023;41(2):336-342.
16. Thompson CA, Ghesquieres H, Maurer MJ, et al. Utility of routine post-therapy surveillance imaging in diffuse large B-cell lymphoma. J Clin Oncol. 2014;32(31):3506-3512.
17 Goldman ML, Mao JJ, Strouse CS, et al. Surveillance imaging during first remission in follicular lymphoma does not impact overall survival. Cancer. 2021;127(18):3390-3402.
18. Neelapu SS, Locke FL, Bartlett NL, et al. Comparison of 2-year outcomes with CAR T cells (ZUMA-1) vs salvage chemotherapy in refractory large B-cell lymphoma. Blood Adv. 2021;5(20):4149-4155.
19 Salles G, Schuster SJ, Dreyling M, et al. Efficacy comparison of tisagenlecleucel vs usual care in patients with relapsed or refractory follicular lymphoma. Blood Adv. 2022;6(22):5835-5843.
20 Ghione P, Palomba ML, Patel A, et al. Comparative effectiveness of ZUMA-5 (axi-cel) vs SCHOLAR-5 external control in relapsed/ refractory follicular lymphoma. Blood. 2022;140(8):851-860.
Aung M. Tun,1,2* Yucai Wang,1* Seth Maliske,3 Ivana Micallef,1 David J. Inwards,1 Thomas M. Habermann,1 Luis Porrata,1 Jonas Paludo,1 Jose Villasboas Bisneto,1 Allison Rosenthal,4 Mohamed A. Kharfan-Dabaja,5 Stephen M. Ansell,1 Grzegorz S. Nowakowski,1 Umar Farooq3 and Patrick B. Johnston1
1Division of Hematology, Mayo Clinic, Rochester, MN; 2Division of Hematologic Malignancies and Cellular Therapeutics, The University of Kansas, Kansas City, KS; 3Division of Hematology, Oncology, and Blood & Marrow Transplantation, University of Iowa, Iowa City, IA; 4Internal Medicine, Division of Hematology/Oncology, Mayo Clinic Arizona, Scottsdale, AZ and 5Division of Hematology-Oncology and Blood and Marrow Transplantation and Cellular Therapy Program, Mayo Clinic, Jacksonville, FL, USA
*AMT and YW contributed equally as first authors.
Correspondence: A.M. Tun atun@kumc.edu
P.B. Johnston johnston.patrick@mayo.edu
Received: November 18, 2023. Accepted: January 11, 2024. Early view: January 18, 2024.
https://doi.org/10.3324/haematol.2023.284704
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Chimeric antigen receptor T-cell therapy is the new standard of care in fit patients with refractory or early relapsed diffuse large B-cell lymphoma (DLBCL). However, there may still be a role for salvage chemotherapy (ST) and autologous stem cell transplant (ASCT) in certain circumstances (e.g., lack of resources for chimeric antigen receptor T-cell therapy, chemosensitive relapses). We retrospectively studied 230 patients with refractory or early relapsed DLBCL who underwent ST and ASCT. The median line of ST was one (range, 1-3). Best response before ASCT was complete response in 106 (46%) and partial response in 124 (54%) patients. The median follow-up after ASCT was 89.4 months. The median progression-free (PFS) and overall survival (OS) were 16.1 and 43.3 months, respectively. Patients relapsing between 6 to 12 months after frontline therapy had a numerically better median PFS (29.6 months) and OS (88.5 months). Patients who required one line of ST, compared to those requiring more than one line, had a better median PFS (37.9 vs. 3.9 months; P=0.0005) and OS (68.3 vs. 12.0 months; P=0.0005). Patients who achieved complete response had a better median PFS (71.1 vs. 6.3 months; P<0.0001) and OS (110.3 vs. 18.9 months; P<0.0001) than those in partial response. Patients who achieved complete response after one line of ST had the most favorable median PFS (88.5 months) and OS (117.2 months). Post-ASCT survival outcomes of patients with refractory or early relapsed DLBCL appeared reasonable and were particularly favorable in those who required only one line of ST to achieve complete response before ASCT, highlighting the role of this procedure in select patients with chemosensitive disease.
Diffuse large B-cell lymphoma (DLBCL), the most common type of aggressive non-Hodgkin lymphoma, constitutes approximately 30% of newly diagnosed cases of non-Hodgkin lymphoma each year in the United States.1,2 Approximately 30-40% of patients encounter relapsed or refractory disease after frontline immunochemotherapy, and their survival outcomes are generally unfavorable – particularly in those with refractory disease or an early relapse occurring within 12 months of initial diagnosis (or frontline treatment), as reported by multiple studies including those by the Center for International Blood and Marrow Transplant Research
(CIBMTR), the Molecular Epidemiology Resource of the University of Iowa/Mayo Clinic Lymphoma Specialized Program of Research Excellence, as well as the CORAL study.3-14 To improve outcomes of such patients, three phase III randomized clinical trials were conducted in patients with refractory disease or an early relapse within 12 months of treatment completion, comparing chimeric antigen receptor T-cell (CAR-T) products (axicabtagene ciloleucel in ZUMA-7, tisagenlecleucel in BELINDA, lisocabtagene maraleucel in TRANSFORM) to the then standard-of-care salvage chemotherapy (ST) followed by high-dose chemotherapy (HDT) and autologous stem cell transplant (ASCT), which established axicabtagene ciloleucel and lisocabtagene maraleucel as the
new standard of care in the second-line setting.15-17 However, there are still significant challenges and barriers to timely delivery of CAR-T therapy in routine clinical practice. CAR-T therapy, to date, is mainly offered in major referral centers, and patients often need to travel long distances for evaluation for such therapy. Patients also need to spend weeks to months for eligibility and fitness assessment, insurance approval, CAR-T manufacturing, and other logistical planning.18,19 A substantial number of patients experience symptomatic, life-threatening progressive disease before receiving CAR-T therapy and often require urgent systemic chemotherapy in the form of a salvage or bridging therapy.19 Moreover, clinical activities of CAR-T therapy remain modest in the second-line setting, with the median event-free survival being only 8.3 months with axicabtagene ciloleucel in ZUMA-7 and 10.1 months with lisocabtagene maraleucel in TRANSFORM; the majority of patients treated with CAR-T in both studies experienced a relapse.15,17 Furthermore, CAR-T therapy is associated with significant morbidities such as cytokine release syndrome, neurotoxicity, and cytopenia, and its long-term complications remain a major concern.19 Given the challenges and barriers as well as the modest clinical activities of CAR-T therapy, there is a need to explore alternative strategies and identify patients who can truly benefit from HDT and ASCT. It is well understood that ASCT (or rescue) enables the use of HDT, in patients with proven chemosensitive disease, to eradicate residual lymphoma cells that have evaded ST.20 Autologous CAR-T therapy utilizes genetically modified patient’s T cells, directed against CD19 antigen on lymphoma cells, for its therapeutic effect.15,17 Accordingly, fit patients with chemosensitive disease can benefit from consolidation with HDT and ASCT, unlike those with chemoresistant disease.11,21 We, therefore, hypothesized that HDT and ASCT would be beneficial and preferable in patients with proven chemosensitive disease after one line of ST, and those with a later relapse 6-12 months after completing frontline immunochemotherapy. To verify this hypothesis, we conducted a retrospective study of patients with refractory disease or relapse within 12 months of completing frontline immunochemotherapy, using information from the Mayo Clinic and the University of Iowa lymphoma and transplant databases.
The institutional review boards at the Mayo Clinic and the University of Iowa approved this study. Data were abstracted on consecutive adult patients who underwent ASCT for DLBCL between July 2000 and December 2017 at the Mayo Clinic or between April 2003 and April 2020 at the University of Iowa. Clinical, pathological, molecular characteristics, cell of origin by Hans algorithm, as well as treatment course, clinical response to treatment (as determined by the treat-
ing physician), and clinical outcome data were extracted by chart reviews.
For this study, eligible patients were those who were treated with frontline R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, prednisolone) or R-CHOP-like immunochemotherapy, had refractory disease (i.e., disease progression during frontline immunochemotherapy or persistent disease after completion of frontline therapy) or an early relapse occurring within 12 months of completing frontline immunochemotherapy, and underwent ST and ASCT. Patients whose response to ST was assessed by positron emission tomography and computed tomography scan and achieved a complete response (CR) or partial response (PR), as determined by treating physicians, were included. DLBCL was classified as per hematopathologists in the Mayo Clinic and the University of Iowa. Patients with MYC and BCL2 and/or BCL6 rearrangements (i.e., double-hit or triple-hit lymphoma) and transformed indolent lymphoma at diagnosis were also included, unless previously treated with immunochemotherapy. Patients who did not receive frontline rituximab and those who underwent hematopoietic stem cell transplant in the frontline (after achieving complete remission to frontline immunochemotherapy) were excluded.
Descriptive statistics are used to report baseline characteristics, treatment information, and response status. The reverse Kaplan-Meier method was used to estimate median follow-up time.22,23 Duration of response (DOR) was defined as time from initial response after ASCT to disease progression, relapse, or death. Post-ASCT progression-free survival (PFS) was defined as time from ASCT until progression, relapse, or death from any cause. Post-ASCT overall survival (OS) was defined as time from ASCT to death from any cause. Post-ASCT PFS and OS were plotted using the Kaplan-Meier method. Log-rank tests and Cox proportional hazards models were used to evaluate the impact of clinico-pathological characteristics, treatment variables, and response status on PFS and OS. P values <0.05 were considered statistically significant. Cumulative incidences of relapse and non-relapse mortality as well as causes of death were analyzed with competing risk models.24 Causes of death were categorized as a result of lymphoma progression, as a result of treatment-related toxicities, non-lymphoma-related causes (other causes), and unknown causes. Statistical analyses were performed in JMP v16 and XLSTAT v2021.2.
Clinicopathological characteristics of the study population and treatment
A total of 230 eligible patients were included in the study (Online Supplementary Figure S1). The baseline charac-
teristics of patients at initial diagnosis are summarized in Online Supplementary Table S1. Clinical characteristics at relapse are described in Table 1. Of the total 230 patients, 157 (68%) had refractory disease or relapsed within 6 months of completing frontline therapy and 73 (32%) had a relapse between 6 to 12 months after frontline therapy. The median age at relapse/ST was 60 years (range, 19-78), and 107 (47%) patients were aged >60 years. The Eastern Cooperative Oncology Group performance status was ≤1 in 181 (97%) patients. Fifty-six patients (39%) had an elevated level of lactate dehydrogenase, 21 (12%) had involvement of more than one extranodal site, and 109 (61%) had advanced stage disease. Treatment patterns and responses to therapy are presented in Online Supplementary Table S2. First-line ST consisted of platinum or high-dose cytarabine-containing chemotherapy in 201 (87%) cases. A median of one line (range, 1-3) of ST was required. The number of lines of ST was one in 178 (77%) and more than one in 52 (23%) patients. Response before ASCT was CR in 106 (46%) and PR in 124 (54%) patients. The median age at ASCT was 60 years (range, 19-78). A BEAM (carmustine, etoposide, cytarabine, and melphalan) conditioning regimen was used in 213 (93%) patients. Following ASCT, 123 (56%) patients achieved CR, and 18 (8%) patients received post-ASCT consolidative radiation therapy.
Outcomes following autologous stem cell transplantation
The median follow-up after ASCT was 89.4 months (95% confidence interval [95% CI]: 73.5-99.2), and post-ASCT outcomes at 12 months, 24 months, and 60 months are summarized in Online Supplementary Table S3. The median PFS and OS after ASCT were 16.1 months (95% CI: 9.3-43.3) and 43.3 months (95% CI: 24.2-75.5), respectively. The 24-month PFS and OS rates were 47% and 57%, respectively (Figure 1A, B). The median DOR in patients who achieved CR or PR after ASCT was 96.4 months (95% CI: 61.6-160.6), with a 24-month DOR rate of 72%. The cumulative incidences of post-ASCT relapse and non-relapse mortality rate were 43.4% and 3.9%, respectively, at 12 months (Figure 1C). One hundred eighteen patients had a relapse (see Online Supplementary Table S4 for their subsequent management), with a median post-relapse OS of 6.0 months (95% CI: 3.8-8.3) (Online Supplementary Figure S2), and 136 died during the follow-up (see Online Supplementary Table S5 for causes of death). Lymphoma was the primary cause of death after ASCT, with a 12-month estimated death rate of 28.5% due to lymphoma, 2.1% due to therapies, 1.3% due to other causes, and 0.9% of unknown causes (Figure 1D).
Outcomes according to clinicopathological characteristics
No statistically significant differences in PFS and OS were seen based on age at relapse, sex, Eastern Cooperative Oncology Group performance status, serum lactate de-
hydrogenase level, extranodal site involvement, and stage at relapse (Table 2). Time to first relapse/refractory status (relapse between 6-12 months vs. refractory or relapse <6 months after frontline therapy) was not associated with a significant difference in PFS (median PFS 29.6 vs. 10.1 months; P=0.47) (Figure 2A, Table 2), but there was a trend for improvement in OS (median OS 88.5 vs. 28.0 months; P=0.07) (Figure 2B, Table 2). Patients who required one line of ST, compared to those who required more than one line of ST, had significantly better PFS (median PFS 37.9 vs. 3.9 months; P=0.0005) (Figure 2C, Table 2) and OS (median OS 68.3 vs. 12.0 months; P=0.0005) (Figure 2D, Table 2). In addition, patients who achieved a CR prior to ASCT, compared to those who achieved a PR, had significantly better PFS (median PFS 71.1 vs. 6.3 months; P<0.0001) (Figure 3A, Table 2) and OS (median OS 110.3 vs 18.9 months; P<0.0001) (Figure 3B, Table 2). Patients who achieved CR after one line of ST had the most favorable PFS and OS, with median PFS of 88.5 vs. 9.1 vs. 12.0 vs. 3.2 months (P<0.0001) and median OS of 117.2 vs. 28.8 vs. 32.5 vs. 7.1 months (P<0.0001) in cases with CR after one line of ST vs. PR after one line of ST vs. CR after more than one line of ST vs. PR after more than one line of ST (Figure 3C,
Table 1. Characteristics of patients with relapsed or refractory diffuse large B-cell lymphoma at relapse/salvage therapy.
†Primary refractory diffuse large B-cell lymphoma (N=106) and relapsed in <6 months (N=51). ASCT: autologous stem cell transplant; ECOG PS: Eastern Cooperative Oncology Group performance status.
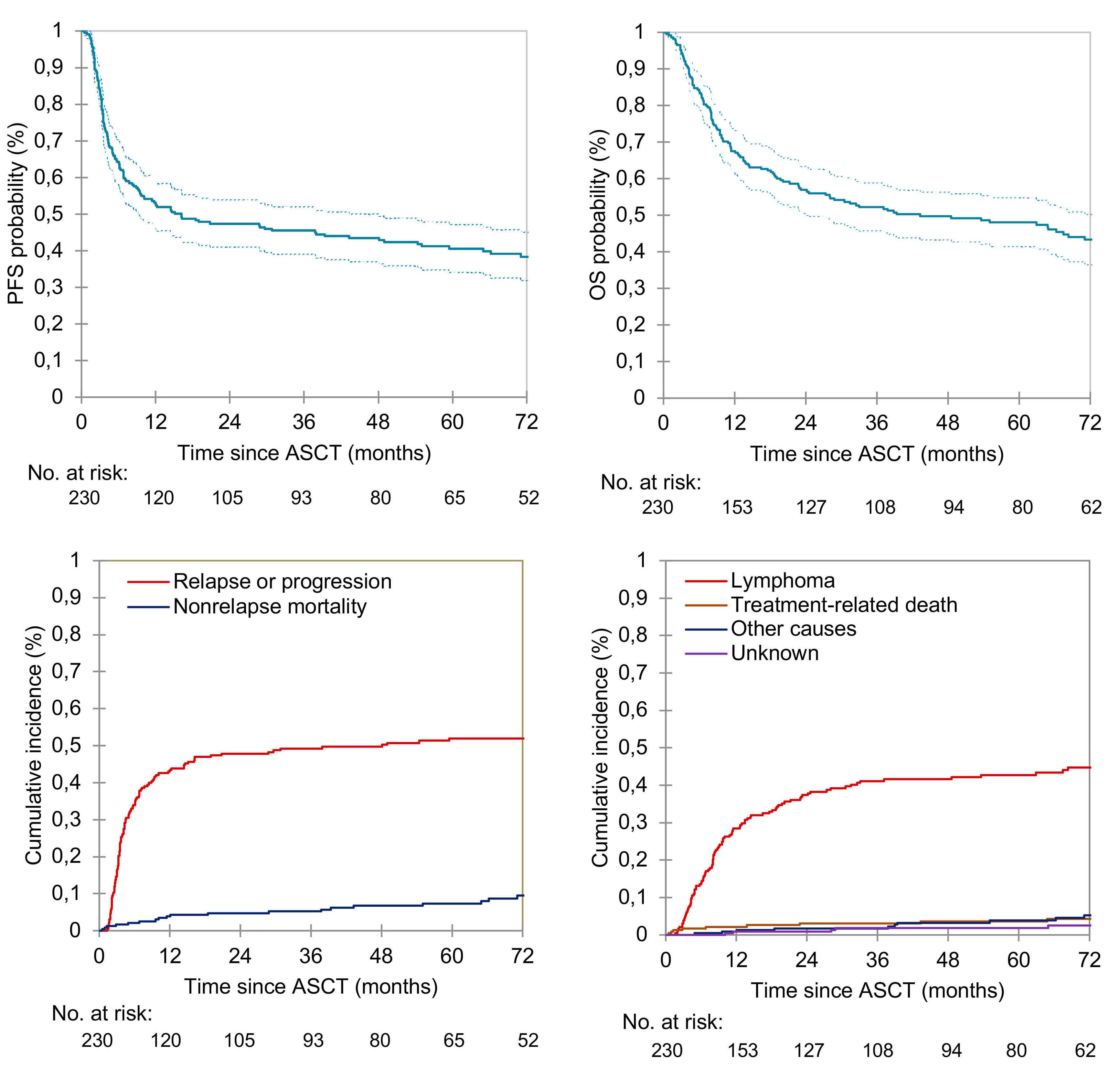
Figure 1. Outcomes after autologous stem cell transplantation in patients with refractory or early relapsed diffuse large B-cell lymphoma. (A) Progression-free survival. (B) Overall survival. (C) Cumulative incidence of relapse and non-relapse mortality. (D) Cumulative incidence of deaths divided by cause. PFS: progression-free survival; ASCT: autologous stem cell transplant; OS: overall survival.
D, Table 2). In multivariate Cox regression models adjusted for age at ASCT and sex, lines of ST and response to ST before ASCT remained prognostic for PFS (1 line of ST: hazard ratio [HR]=0.53, 95% CI: 0.36-0.77; P=0.0008 and CR: HR=0.49, 95% CI: 0.35-0.69; P<0.0001) and OS (1 line of ST: HR=0.51, 95% CI: 0.35-0.74; P=0.0005 and CR: HR=0.46, 95% CI: 0.32-0.66; P<0.0001). Additionally, in this multivariate model, time to first relapse/refractory status (relapse between 6-12 months vs. refractory or relapse <6 months after frontline therapy) showed a trend for improvement in PFS (HR=0.83, 95% CI: 0.59-1.18; P=0.31) and a statistically significant improvement in OS (HR=0.67, 95% CI: 0.46-0.98; P=0.04) (Online Supplementary Table S6).
Note that patients with a MYC rearrangement, compared to those without, had significantly inferior PFS (median PFS 3.1 vs. 43.3 months; P=0.0001) and OS (median OS 6.2
vs. 67.4 months; P<0.0001) (Table 2, Online Supplementary Figure S3).
The new standard of care in fit patients with primary refractory or early relapsed DLBCL occurring within 12 months of completing frontline immunochemotherapy is CAR-T therapy with axicabtagene ciloleucel or lisocabtagene maraleucel according to the results of the contemporary ZUMA-7 and TRANSFORM studies.15,17 Our study of such patients, treated with ST and ASCT, documented reasonable survival outcomes with a median PFS of 16.1 months and the 24-month PFS of 47%. The results from our study are in keeping with those of the CIBMTR that
Table 2. Survival after autologous stem cell transplantation by clinicopathological characteristics and response status.
<6 months‡, N=157
N=123
N=56 7.7
N=109
N=9 3.1 (1.9-7.7)
N=53
N=52
N=106
Lines of ST and response status
CR after 1 line of ST, N=90 88.5 (43.3-NR) 65 (55-75) 117.2 (75.5-NR) 70 (61-80)
PR after 1 line of ST, N=88
(5.4-16.2)
(19.1-163.8)
(4.7-14.4) 28 (13-43)
(32-81) PR after >1 line of ST, N=36 3.2 (2.8-5.4) 25 (11-39)
†Time to relapse from completion of first-line therapy. ‡This category included patients with primary refractory disease. PFS: progression-free survival; OS: overall survival; 95% CI: 95% confidence interval; NR: not reached; ECOG PS: Eastern Cooperative Oncology Group performance status; ST: salvage chemotherapy; CR: complete response; PR: partial response.
reported a 3-year post-ASCT PFS rate of 44% despite early immunochemotherapy failure within 12 months of initial diagnosis and another study from the CIBMTR, in patients with primary refractory DLBCL, which reported a 3-year post-ASCT PFS rate of 46.8%.10,25 The median DOR in our patients who achieved CR or PR after ASCT was 96.4 months (24-month DOR, 72%). These results indicate that patients with chemosensitive relapsed or refractory disease, despite having primary refractory disease to frontline immunochemotherapy or an early relapse, can expect durable disease
control with ASCT consolidation. These findings are further supported by durable responses seen in patients assigned to the standard-of-care group of the ZUMA-7 and TRANSFORM studies, with the former resulting in a median DOR of 8.9 months in patients who had a response (i.e., CR or PR) and the latter reporting a median DOR of 14.5 months (12-month DOR rate, 54.7%) in patients achieving CR.15,17 More favorable survival outcomes were observed in our patients who required only one line of ST and those who achieved CR, with a median PFS of 37.9 months (24-month
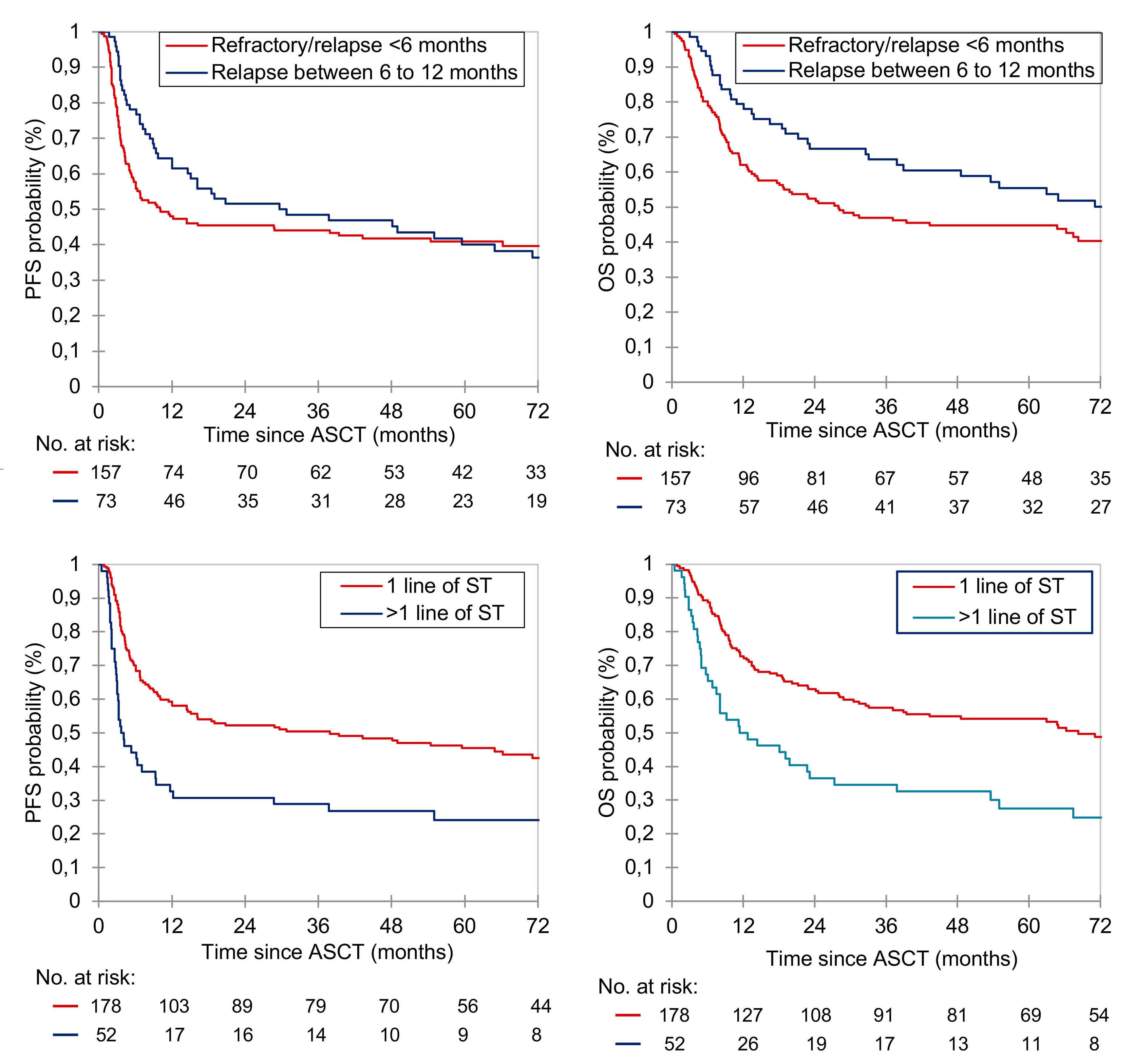
Figure 2. Post-autologous stem cell transplant outcomes according to refractory and/or time to relapse status and line of salvage chemotherapy. (A) Progression-free survival and (B) overall survival by refractory and/or time to relapse status. (C) Progression-free survival and (D) overall survival by line of salvage chemotherapy. PFS: progression-free survival; ASCT: autologous stem cell transplant; ST: salvage chemotherapy; OS: overall survival.
PFS, 52%) and 71.1 months (24-month PFS, 62%), respectively. Most importantly, survival outcomes were excellent in patients who had only one line of ST and resulted in CR, with their median PFS being 88.5 months (24-month PFS, 65%). These findings imply that HDT, followed by ASCT rescue, confers complete eradication of lymphoma cells that are biologically sensitive to chemotherapy. In patients who achieved PR after one line of ST, our study showed that the median PFS was 9.1 months, with the 24-month PFS rate being 39%. The CIBMTR study found a 5-year post-ASCT PFS rate of 41% in patients with failure of early frontline immunochemotherapy (i.e. primary refractory disease or relapse within 12 months of diagnosis).12 A similar result was documented in the MD Anderson Cancer Center with a 5-year post-ASCT PFS rate of 40% in patients with relapsed or refractory DLBCL who had residual
disease before ASCT.26 Variations in survival rates among these studies are due, in part, to different study populations, study eras, patterns of relapse, and management approaches.12,26 In addition, the definition of PR by treating physicians is subjective, and varies depending on imaging modality (i.e., computed tomography alone or with positron emission tomography).27 Nevertheless, despite achieving only PR, their survival outcomes after ASCT are reasonable, with a long-term remission rate of ~40%, supporting the role of ASCT consolidation.12,26 In the CAR-T therapy era, the role of ASCT consolidation in patients achieving PR to ST was reported by the CIBMTR.28 In this study, the efficacy of ASCT was compared with that of axicabtagene ciloleucel in a total of 411 patients of whom 266 undergoing ASCT consolidation and 145 receiving axicabtagene ciloleucel.28 The study found that ASCT consolidation, compared to
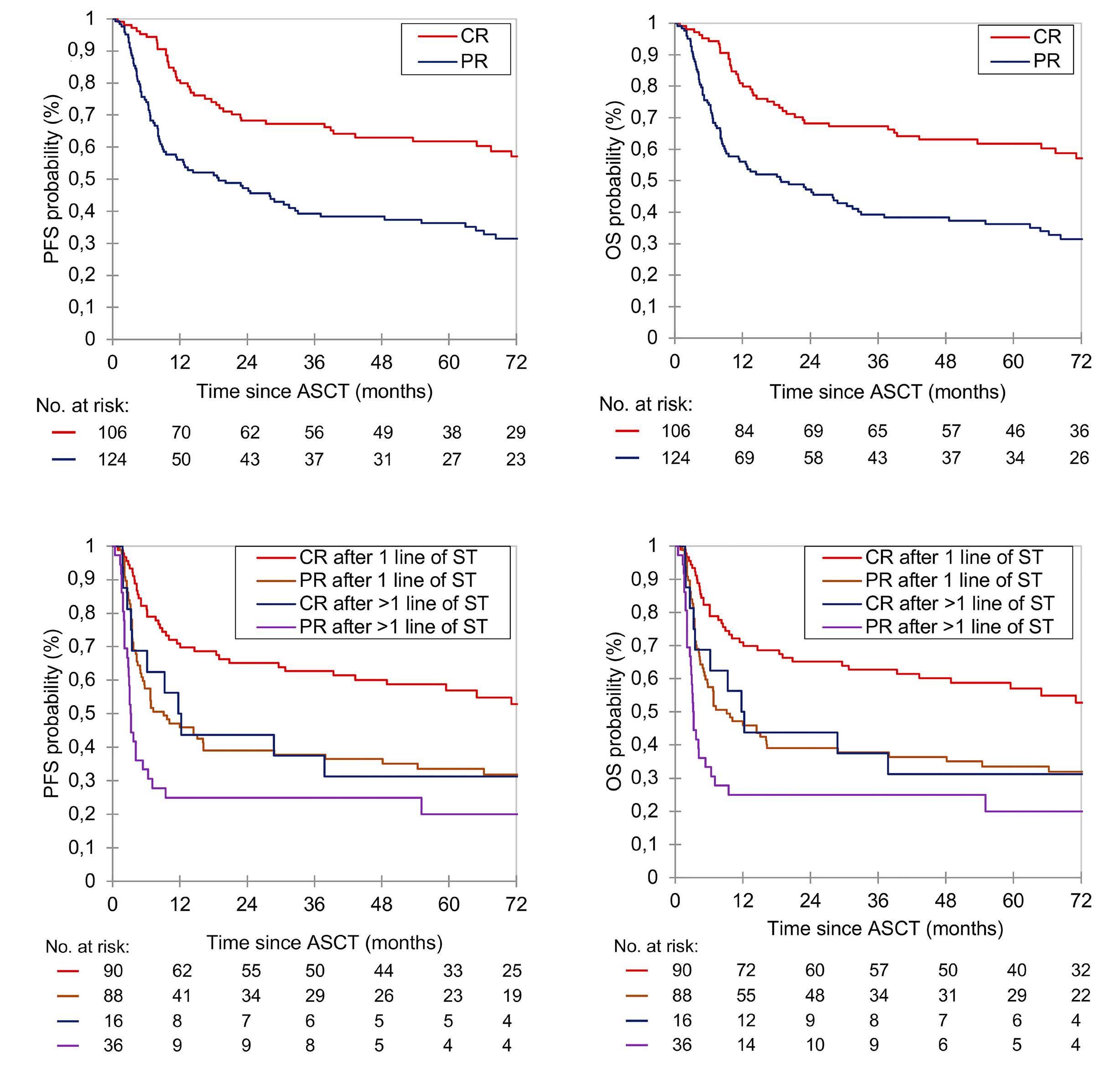
Figure 3. Post-autologous stem cell transplant outcomes according to line of slavage chemotherapy and response status. (A) Progression-free survival and (B) overall survival by response status. (C) Progression-free survival and (D) overall survival by line of salvage therapy and response status. PFS: progression-free survival; OS: overall survival; ASCT: autologous stem cell transplant; CR: complete response; PR: partial response; ST: salvage chemotherapy.
axicabtagene ciloleucel, was associated with a lower rate of relapse/progression (2-year rate, 40% vs. 53%; P=0.05), a trend for superior PFS (2-year rate, 52% vs. 42%; P=0.1), and a superior OS rate (2-year rate, 69% vs. 47%; P=0.004).28 The caveat regarding this study was that the median line of therapy was higher in patients receiving axicabtagene ciloleucel, and the difference was no longer seen when the analysis was done in patients who received two or fewer lines of therapy (1-year PFS, 59% in the ASCT group vs. 65% in the axicabtagene ciloleucel group; P=0.5).28
There is no consensus among lymphoma clinicians when defining early treatment failure. Most historical studies, such as the CIBMTR, Molecular Epidemiology Resource, and CORAL studies, identified patients with refractory disease
or relapse within 12 months of initial diagnosis as having an unfavorable risk.7,10,12,13 In contrast, the three randomized clinical trials (ZUMA-7, TRANSFORM, and BELINDA) and the NCIC-CTG LY.12 study defined patients with refractory disease or relapse within 12 months of completing frontline therapy as high risk.15–17,29 Consequently, selection of the optimal treatment strategy becomes challenging for patients who relapse between 6 to 12 months after completing frontline therapy.19 It is noteworthy that these patients achieve a remission lasting at least 6 months after completing frontline immunochemotherapy, indicating chemosensitive biology, and are more likely to respond to ST and be able to proceed with HDT and ASCT.7 In our study, patients who relapsed between 6 to 12 months
after frontline therapy, compared to those with refractory or relapse within 6 months, did not have a statistically significant improvement in PFS after ASCT (P=0.47), although PFS was numerically better in the former (median PFS 29.6 vs. 10.1 months). Additionally, there was a trend for improvement in OS in the univariate analysis (P=0.07), and a statistically significant improvement in OS, after adjustment for age at ASCT and sex, in the multivariate model (P=0.04). Therefore, after establishing the chemosensitivity of the disease, careful consideration between HDT followed by ASCT and CAR-T is warranted in such patients due to their favorable survival rate after ASCT (24-month PFS, 52%), although the findings from our study population, receiving care at the Mayo Clinic and the University of Iowa, may not be broadly generalizable. There remain logistical and financial barriers that prevent timely access to CAR-T therapy.30 Patients often need to wait weeks to months for frailty and fitness assessments, overcoming logistical and financial barriers, as well as CAR-T manufacturing (although a significantly shorter wait time is anticipated with the use of allogeneic CAR-T products).16,30,31 Moreover, patients with relapsed or progressive disease are often symptomatic requiring immediate intervention. Thus, it is reasonable to initiate ST, especially in resource-limited settings. Those who achieve PR can consider proceeding with ASCT based on reasonable survival outcomes, and this practice is supported by the report from the CIBMTR favoring ASCT consolidation, as discussed earlier.12,28 For those achieving CR, ASCT consolidation seems favored over CAR-T in patients with early treatment failure, as per the recent study from the CIBMTR that reported a lower 2-year relapse rate (22.8% vs. 45.9%; P<0.001) and a superior 2-year PFS rate (70.9% vs. 48.3%; P<0.001), although it is difficult to draw definitive conclusions due to its retrospective study design.32 Similar survival outcomes were reported for patients who received CAR-T therapy while in CR, with a 2-year PFS rate of 44% in the MD Anderson Cancer Center cohort and a 1-year PFS rate of 59.6% in a study from eight academic centers in the USA.33,34 Notably, patients relapsing or progressing after CAR-T therapy have unfavorable outcomes due to the limited availability and efficacy of subsequent ST.19 After CAR-T therapy, cytopenias and patients’ intolerance of intensified therapy and the potential of stem cell mobilization failure make ASCT less feasible.19,35 Although ASCT consolidation is generally favored in patients with established chemosensitive disease, post-ASCT outcomes of those with a MYC rearrangement are poor, despite them having chemosensitive disease, with a median PFS of only 3.1 months; CAR-T therapy may be preferred in such patients.36,37
It is, however, worth noting that only about half of patients with relapsed or refractory DLBCL treated with intensified therapy are able to proceed with ASCT consolidation, although higher response rates can be expected with the
incorporation of novel therapeutic agents.7,13,15,17,29,38,39 In the CORAL study, ~63% of patients achieved CR or PR to ST, namely RICE (rituximab, ifosfamide, carboplatin, and etoposide) and R-DHAP (rituximab, dexamethasone, highdose cytarabine, and cisplatin), and ~53% of patients subsequently proceeded with ASCT. In NCIC-CTG LY.12, ~45% of patients had responses to the ST regimens GDP (gemcitabine, dexamethasone, and cisplatin) and DHAP.7,29 The real-world analysis of the Molecular Epidemiology Resource database found that 40% of patients treated aggressively with second-line ST for relapsed or refractory DLBCL were able to proceed with ASCT.13 In participants of the ZUMA-7 and TRANSFORM trials, who were then randomized to the ST group, ASCT consolidation was done in 38% and 46% of patients, respectively.15,17 However, response to ST can be improved with the incorporation of novel therapy; notably, polatuzumab vedotin added to RICE produced an overall response rate (i.e., CR or PR) of 92%, with an acceptable toxicity profile, in a multicenter phase II study.39 Response rates could also improve with incorporation of bispecific CD20xCD3 monoclonal antibodies to ST,40,41 and these strategies are being evaluated by currently ongoing clinical studies such as those investigating glofitamab plus RICE (NCT05364424) and epcoritamab plus GDP (NCT05852717). The strengths of our study include systematic review of a large number of patients with relapsed or refractory DLBCL following failure of frontline R-CHOP or R-CHOP-like immunochemotherapy who then underwent ASCT after the first, early relapse or in a refractory state, with the availability of long-term follow-up data. However, our study has limitations due to its retrospective study design with potential selection bias, geographical bias with limited generalizability, and lack of centralized histopathology review, as well as some missing information and a long study period that spans over two decades with potential heterogeneity in management approaches.
In conclusion, survival outcomes after ASCT are favorable in at least a subset of patients with primary refractory or early relapsed DLBCL. Post-ASCT survival outcomes are more favorable in patients who require only one line of ST and those who achieved CR to ST. Furthermore, survival outcomes after ASCT are excellent in patients achieving CR after one line of ST. In patients who relapse between 6 to 12 months after completing frontline therapy, the choice between CAR-T therapy and ST followed by HDT and ASCT should be considered carefully. These data support the role of ST and ASCT consolidation as a second-line treatment strategy in select patients with primary refractory or early relapsed DLBCL in an appropriate clinical context.
Disclosures
No conflicts of interest to disclose.
Contributions
AMT, YW, and PBJ conceived and designed the study and
prepared the first draft of the manuscript. AMT and SM collected and assembled data. All authors analyzed and interpreted data, reviewed and revised the manuscript, and approved the final version.
1. Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107(1):265-276.
2. Al-Hamadani M, Habermann TM, Cerhan JR, Macon WR, Maurer MJ, Go RS. Non-Hodgkin lymphoma subtype distribution, geodemographic patterns, and survival in the US: A longitudinal analysis of the National Cancer Data Base from 1998 to 2011. Am J Hematol. 2015;90(9):790-795.
3. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235-242.
4 Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116(12):2040-2045.
5. Sehn LH, Donaldson J, Chhanabhai M, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol. 2005;23(22):5027-5033.
6. Habermann TM, Weller EA, Morrison VA, et al. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol. 2006;24(19):3121-3127.
7 Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184.
8. Pfreundschuh M, Kuhnt E, Trümper L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol. 2011;12(11):1013-1022.
9 Maurer MJ, Ghesquières H, Jais J-P, et al. Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol. 2014;32(10):1066-1073.
10 Hamadani M, Hari PN, Zhang Y, et al. Early failure of frontline rituximab-containing chemo-immunotherapy in diffuse large B cell lymphoma does not predict futility of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2014;20(11):1729-1736.
11. Tun AM, Maliske S, Wang Y, et al. Progression-free survival at 24 months as a landmark after autologous stem cell transplant in relapsed or refractory diffuse large B-cell lymphoma. Transplant Cell Ther. 2022;28(9):610-617.
12. Shah NN, Ahn KW, Litovich C, et al. Is autologous transplant in relapsed DLBCL patients achieving only a PET+ PR appropriate in the CAR T-cell era? Blood. 2021;137(10):1416-1423.
Data-sharing statement
This article includes the data supporting the findings and interpretation of the results. Please contact the corresponding authors for additional data.
13. Farooq U, Maurer MJ, Thompson CA, et al. Clinical heterogeneity of diffuse large B cell lymphoma following failure of front-line immunochemotherapy. Br J Haematol. 2017;179(1):50-60.
14 Wang Y, Farooq U, Link BK, et al. Late relapses in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. J Clin Oncol. 2019;37(21):1819.
15. Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. 2022;386(7):640-654.
16. Bishop MR, Dickinson M, Purtill D, et al. Second-line tisagenlecleucel or standard care in aggressive B-cell lymphoma. N Engl J Med. 2022;386(7):629-639.
17 Kamdar M, Solomon SR, Arnason J, et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as secondline treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysi. Lancet. 2022;399(10343):2294-2308.
18. Mikhael J, Fowler J, Shah N. Chimeric antigen receptor T-cell therapies: barriers and solutions to access. JCO Oncol Pract. 2022;18(12):800-807.
19 Perales M-A, Anderson LD, Jain T, et al. Role of CD19 chimeric antigen receptor T cells in second-line large B cell lymphoma: lessons from phase 3 trials. An expert panel opinion from the American Society for Transplantation and Cellular Therapy. Transplant Cell Ther. 2022;28(9):546-559.
20. Philip T, Guglielmi C, Hagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive nonHodgkin’s lymphoma. N Engl J Med. 1995;333(23):1540-1545.
21. Wudhikarn K, Johnson aM, Inwards DJ, et al. Outcomes of older adults with non-Hodgkin lymphoma undergoing autologous stem cell transplantation: a Mayo Clinic cohort analysis. Transplant Cell Ther. 2023;29(3):176.e1-176.e8.
22. Shuster JJ. Median follow-up in clinical trials. J Clin Oncol. 1991;9(1):191-192.
23. Schemper M, Smith TL. A note on quantifying follow-up in studies of failure time. Control Clin Trials. 1996;17(4):343-346.
24. Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141-1154.
25. Bal S, Costa LJ, Sauter C, Litovich C, Hamadani M. Outcomes of autologous hematopoietic cell transplantation in diffuse large B cell lymphoma refractory to firstline chemoimmunotherapy. Transplant Cell Ther. 2021;27(1):55.e1-55.e7.
26. Cherng HJ, Xu G, Feng L, et al. Positron emission tomography derived metrics in relapsed or refractory large B-cell lymphoma with residual disease before autologous stem cell transplant. Br J Haematol. 2023;200(1):35-44.
27. Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3068.
28. Shadman M, Pasquini M, Ahn KW, et al. Autologous transplant
vs chimeric antigen receptor T-cell therapy for relapsed DLBCL in partial remission. Blood. 2022;139(9):1330-1339.
29. Crump M, Kuruvilla J, Couban S, et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J Clin Oncol. 2014;32(31):3490-3496.
30. Hoffmann MS, Hunter BD, Cobb PW, Varela JC, Munoz J. Overcoming barriers to referral for chimeric antigen receptor T-cell therapy in patients with relapsed/refractory diffuse large B-cell lymphoma. Transplant Cell Ther. 2023;29(7):440-448.
31. Mailankody S, Matous J V, Chhabra S, et al. Allogeneic BCMAtargeting CAR T cells in relapsed/refractory multiple myeloma: phase 1 UNIVERSAL trial interim results. Nat Med. 2023;29(2):422-429.
32. Shadman M, Wooahn K, Kaur M, et al. Autologous transplant (auto-HCT) is associated with improved clinical outcomes compared to CAR-T therapy in patients (pts) with large B-cell lymphoma (LBCL) achieving a complete remission. Blood. 2023;142(Suppl 1):781.
33. Jallouk AP, Gouni S, Westin J, et al. Axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma patients in complete metabolic response. Haematologica. 2023;108(4):1163-1167.
34 Wudhikarn K, Tomas AA, Flynn JR, et al. Low toxicity and excellent outcomes in DLBCL patients without residual lymphoma at the time of CD19 CAR T cell therapy. Blood Adv.
2023;7(13):3192-3198.
35. Di Blasi R, Le Gouill S, Bachy E, et al. Outcomes of patients with aggressive B-cell lymphoma after failure of anti-CD19 CAR T-cell therapy: a DESCAR-T analysis. Blood. 2022;140(24):2584-2593.
36. Costa LJ, Maddocks K, Epperla N, et al. Diffuse large B-cell lymphoma with primary treatment failure: ultra-high risk features and benchmarking for experimental therapies. Am J Hematol. 2017;92(2):161-170.
37. Herrera AF, Mei M, Low L, et al. Relapsed or refractory doubleexpressor and double-hit lymphomas have inferior progressionfree survival after autologous stem-cell transplantation. J Clin Oncol. 2017;35(1):24-31.
38. Tun AM, Ansell SM. Immunotherapy in Hodgkin and nonHodgkin lymphoma: innate, adaptive and targeted immunological strategies. Cancer Treat Rev. 2020;88:102042.
39 Herrera AF, Chen L, Crombie JL, et al. Polatuzumab vedotin combined with R-ICE (PolaR-ICE) as second-line therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2022;140(Suppl 1):1065-1067.
40 Dickinson MJ, Carlo-Stella C, Morschhauser F, et al. Glofitamab for relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2022;387(24):2220-2231.
41. Thieblemont C, Phillips T, Ghesquieres H, et al. Epcoritamab, a novel, subcutaneous CD3xCD20 bispecific T-cell–engaging antibody, in relapsed or refractory large B-cell lymphoma: dose expansion in a phase I/II trial. J Clin Oncol. 2022;41(12):2238-2247.
Mateo Sarmiento Bustamante,1* Sheila K. Pierson,1* Yue Ren,2 Adam Bagg,3 Joshua D. Brandstadter,4 Gordan Srkalovic,5 Natalie Mango,1 Daisy Alapat,6 Mary Jo Lechowicz,7 Hongzhe Li,2 Frits van Rhee,8 Megan S. Lim9 and David C. Fajgenbaum1
1Center for Cytokine Storm Treatment & Laboratory, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA; 2Department of Biostatistics, Epidemiology, and Informatics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA; 3Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA; 4Division of Hematology/Oncology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA; 5Sparrow Herbert-Herman Cancer Center, Michigan State University College of Human Medicine, Lansing, MI; 6Department of Pathology, College of Medicine, University of Arkansas for Medical Sciences, Little Rock, AR; 7Department of Hematology and Medical Oncology, Emory University School of Medicine and Winship Cancer Institute, Atlanta, GA; 8Myeloma Center, Winthrop P. Rockefeller Cancer Institute, University of Arkansas for Medical Sciences, Little Rock, AR and 9Department of Pathology and Laboratory Medicine, Memorial Sloan Kettering Cancer Center, New York City, NY, USA.
*MSB and SKP contributed equally as first authors.
Correspondence: D.C. Fajgenbaum davidfa@pennmedicine.upenn.edu
Received: June 13, 2023.
Accepted: December 29, 2023.
Early view: January 11, 2024.
https://doi.org/10.3324/haematol.2023.283603
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Idiopathic multicentric Castleman disease (iMCD) is a rare hematologic disorder with heterogeneous presentations ranging from moderate constitutional symptoms to life-threatening multiorgan system involvement. There are vastly different clinical subtypes, with some patients demonstrating thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly (TAFRO) and others having milder/more moderate symptoms with potential for severe disease (not otherwise specified, NOS). Due to its rarity and heterogeneity, the natural history and long-term burden of iMCD are poorly understood. We investigated real-world medical data from ACCELERATE, a large natural history registry of patients with Castleman disease, to better characterize the long-term disease burden experienced by these patients. We found that iMCD-TAFRO patients face a significant hospitalization burden, requiring more time in the hospital than iMCDNOS patients during the year surrounding diagnosis (median [interquartile range]: 36 [18-61] days vs. 0 [0-4] days; P<0.001). In addition, we found life-sustaining interventions, such as mechanical ventilation (17%) and dialysis (27%), were required among iMCD patients, predominantly those with iMCD-TAFRO. iMCD-NOS patients, however, spent a significantly greater proportion of time following disease onset in a state of disease flare (median 52.3% vs. 18.9%; P=0.004). Lastly, we observed severe iMCD-related morbidities, such as acute renal failure, sepsis and pneumonia, among others, arising after iMCD diagnosis, impairing the patients’ quality of life. These data demonstrate a substantial disease burden experienced by iMCD patients and emphasize the importance of ongoing research into iMCD to aid disease control.
Idiopathic multicentric Castleman disease (iMCD) is a rare lymphoproliferative disorder characterized by widespread lymphadenopathy and a systemic inflammatory syndrome. While the pathophysiology of the disease remains poorly understood, existing data have implicated interleukin 6 and the mTOR and JAK-STAT pathways.1-5 As with other inflammatory disorders, iMCD is characterized by constitu-
tional symptoms and systemic inflammation that can lead to multiorgan failure or death.6 Several different clinical subtypes of iMCD have been identified. Patients with the most severe subtype who demonstrate thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly are classified as having the TAFRO subtype.7 Other patients who do not meet TAFRO criteria are said to have iMCD not otherwise specified (NOS) and typically present with a milder clinical
phenotype.7 A subset of NOS patients have thrombocytosis and hyper-gammaglobulinemia and have historically been described as having idiopathic plasmacytic lymphadenopathy.8
A recent epidemiological study based on USA insurance claims found that approximately 1,000 patients were diagnosed with iMCD each year.9 Making a diagnosis of iMCD is challenging because it is based on non-specific clinical features and characteristic lymph node histopathology. Underdiagnosis is likely as diagnostic criteria were not developed until 2017, and there is no known diagnostic serum biomarker.10 Challenges with diagnosis and limited understanding of the disease burden may contribute to poor outcomes. Data from the USA before 2012 suggested 5- and 10-year mortality rates of 35% and 60% for iMCD patients whereas more recent data from electronic medical records suggest 25% mortality at 5 years.11,12 Three-year survival estimates based on a recent large cohort of patients in China were 65.7% for TAFRO patients, 87.2% for NOS patients without the characteristics of idiopathic plasmacytic lymphadenopathy, and 98.5% for NOS patients with the features of idiopathic plasmacytic lymphadenopathy.13 In the USA and Europe, siltuximab, a monoclonal antibody directed against interleukin 6, is the only approved treatment and first-line recommended therapy.14,15 Tocilizumab, a monoclonal antibody directed against the interleukin 6 receptor, is recommended when siltuximab is not available and is approved for use in iMCD in Japan.16 For patients with severe disease who do not respond to first-line treatment with interleukin 6 blockade, multiagent cytotoxic chemotherapy is recommended.15
Due to its rarity and heterogeneous nature, the natural history and long-term burden of iMCD are not well understood. In light of the limited understanding of the burden of disease and the significant consequences of underestimating the risks of iMCD,17 we sought to investigate the burden of iMCD on patients and the healthcare system. ACCELERATE (NCT02817997), a longitudinal natural history study of Castleman disease, is ideally positioned to characterize the natural history and burden associated with iMCD. Herein, we present data from ACCELERATE to demonstrate that iMCD patients face a high burden of disease with severe multisystem organ involvement, the onset of life-threatening iMCD-related morbidities, long periods of hospitalizations during which interventions are frequently required, and extensive periods of time spent with flares of active disease.
Study population
Patients with a pathology report suggestive of Castleman disease were invited to enroll into ACCELERATE beginning in October 2016.18 Enrollment is open to patients in the USA and globally. Given that the registry website is written
in English and records from institutions in English-speaking countries are more feasibly obtained, there is a bias towards enrollment of English-speaking patients. After enrollment, all available medical data from the time of symptom onset to the time of analysis were collected from each patient’s treating institution(s). Data were reviewed and abstracted into the study database, and each case underwent a rigorous, systematic review by three hematopathologists and four hematologist-oncologists to confirm the accuracy of the iMCD diagnosis. While it is impossible to know the exact number of patients in this study who have been investigated previously, it is possible that some patients have been included in prior case reports/small series.
Diease severity at diagnosis was classified according to the iMCD treatment guidelines and at least two criteria had to be met for a diagnosis of severe iMCD: an Eastern Cooperative Oncology Group performance status ≥2 or hospitalization, fluid retention, hemoglobin ≤8.0 g/dL, pulmonary involvement/interstitial pneumonitis with dyspnea, or stage IV renal dysfunction within 90 days of the diagnosis made by lymph node biopsy.15
To ensure standardization, we defined a new flare as the onset of at least two new iMCD disease symptoms or the clinical worsening of previously stable signs or symptoms. Flares also required at least one laboratory finding from the iMCD diagnostic minor criteria that was previously normal to become abnormal. A flare ended when a patient achieved at least 50% reduction of symptoms (minor criteria from the iMCD diagnostic criteria).10
The Systematized Nomenclature of Medicine – Clinical Terms (SNOMED-CT) ontology was used to categorize iMCD-related morbidities and comorbidities.
Patient-reported outcomes
Patients enrolled in ACCELERATE were given outcome surveys every 3 months throughout the duration of their enrollment. Data were analyzed from patients who completed the survey at least once. Response data from EuroQoL five-dimension, five-level (EQ-5D-5L), a validated quality-of-life (QOL) questionnaire, and from the MCD symptom score, a validated MCD symptom survey developed for the phase II siltuximab clinical trial, were analyzed for each patient’s most recent survey completion date.14
The EQ-5D-5L captures health on the day of the survey, as well as ease with each of mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. The MCD symptom survey captures 16 symptoms on a scale scored from 1 to 6 with 1 being the least severe (did not experience) and 6 being the most severe (very severe). The sum of the responses yields the total MCD symptom score. The 16 symptoms assessed included cough, shortness of breath, loss of appetite, fatigue, lack of energy,
feeling weak, sores or rash on skin (skin lesions), itching, numbness or tingling, pain, fever, swollen lymph nodes, swelling or edema in other body areas, night sweats, and excessive daytime sweating.
A χ 2 test was used to compare proportions, and Wilcoxon rank sum was used to test for differences between continuous data. To compare hospitalization rates between iMCD patients and the general population, a matched case-control analysis was conducted using public data from the 2018 National Health Information Survey (NHIS), a cross-sectional household survey and the principal source of information on the health of the non-institutionalized population in the USA.19,20 For each case, four controls were matched for age (within 1 year), sex, and race.19,20 Kruskal-Wallis followed by Wilcoxon rank sum testing with the Bonferroni correction were used to compare controls with iMCD subtypes. Spearman rank correlation test was used to determine the correlation between QOL score and MCD symptom score. Analyses were performed using R v 4.04.
The ACCELERATE natural history registry has received ethical approval from the University of Pennsylvania Institutional Review Board, with the most recent approval being given on March 9, 2023 (protocol: 824758).
Patients with idiopathic multicentric Castleman disease present with severe clinical and laboratory abnormalities
Of the 136 patients suspected to have iMCD who were reviewed, the ACCELERATE expert panel confirmed that 102 (75%) met both clinical and pathological criteria for iMCD.10 The cohort with a confirmed diagnosis had a slight predominance of males (n=58, 56.9%), a median (interquartile range [IQR]) age of 35.3 (22.2-47.5) years, and 65% were identified as White (Table 1). The median (IQR) follow-up time after iMCD diagnosis was 3.4 (1.3-6.1) years for the full cohort, 3.3 (1.2-5.3) years for TAFRO patients, and 3.6 (1.6-8.1) years for iMCD-NOS patients. Eight patients were
Table 1. Characteristics of the cohort of patients with idiopathic multicentric Castleman disease.
Severity at diagnosis, N (%)
Severe
Race, N (%)
White
to answer
Country of enrollment, N (%)
Histopathological subtype, N (%)
Hypervascular/hyaline vascular
iMCD: idiopathic multicentric Castleman disease; TAFRO: thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/ renal failure, and organomegaly; NOS: not otherwise
dead at the time of data collection (Online Supplementary Table S1). Sixty-one (60%) patients met TAFRO criteria7 and 41 (40%) were classified as having iMCD-NOS. TAFRO patients tended to be more commonly male (TAFRO 62% compared to NOS 49%) and younger (mean [standard deviation]: 33.0 [17.4] vs. 40.1 [13.9] years) than NOS patients. First, we investigated disease severity at diagnosis and across age ranges. Of the 100 patients with sufficient information to determine disease severity at diagnosis, 77 (77%) initially presented with severe disease. Notably, we found iMCD in individuals of all ages, with the youngest patient in the cohort diagnosed at 1.8 years and the oldest at 74.4 years. We examined severity by age range visually and observed that the majority of patients <30 or >60 years of age presented with severe disease (Figure 1A). When we examined this trend statistically, we found a significant difference among the groups ( P =5.3x10-4). Patients <30 and patients >60 years old were more likely to have severe disease (94.6% and 90.0%, respectively) as compared to patients between 30 and 60 years old (62.2%) ( Online Supplementary Table S2). All but one patient >60 years
old presented with severe disease and all but two of the patients <30 years presented with severe disease, while a large proportion of patients in the 30- to 60-year-old age group presented with mild-moderate disease, which likely affected this result (Figure 1A).
We next investigated the clinical and laboratory abnormalities present at diagnosis ( Online Supplementary Table S3). Specifically, we focused on the degree of anemia and hypoalbuminemia as markers of disease activity at diagnosis in this cohort. Across all patients, the median (IQR) hemoglobin concentration was 7.8 (6.6-10.5) g/dL and the median (IQR) albumin level was 2.2 (1.8-2.9) g/dL; patients in both TAFRO and NOS subgroups had median levels below the lower limits of normal. Eighty-seven (85.3%) patients had anemia and 81 (79.4%) had hypoalbuminemia at diagnosis (Figure 1B). We also determined the proportions of patients overall and by subtype who had clinical abnormalities (Figure 1C, Online Supplementary Table S3 ). A large majority of patients presented with fluid retention (84%), splenomegaly (72%), and/or hepatomegaly (60%).
While TAFRO patients accounted for most patients with
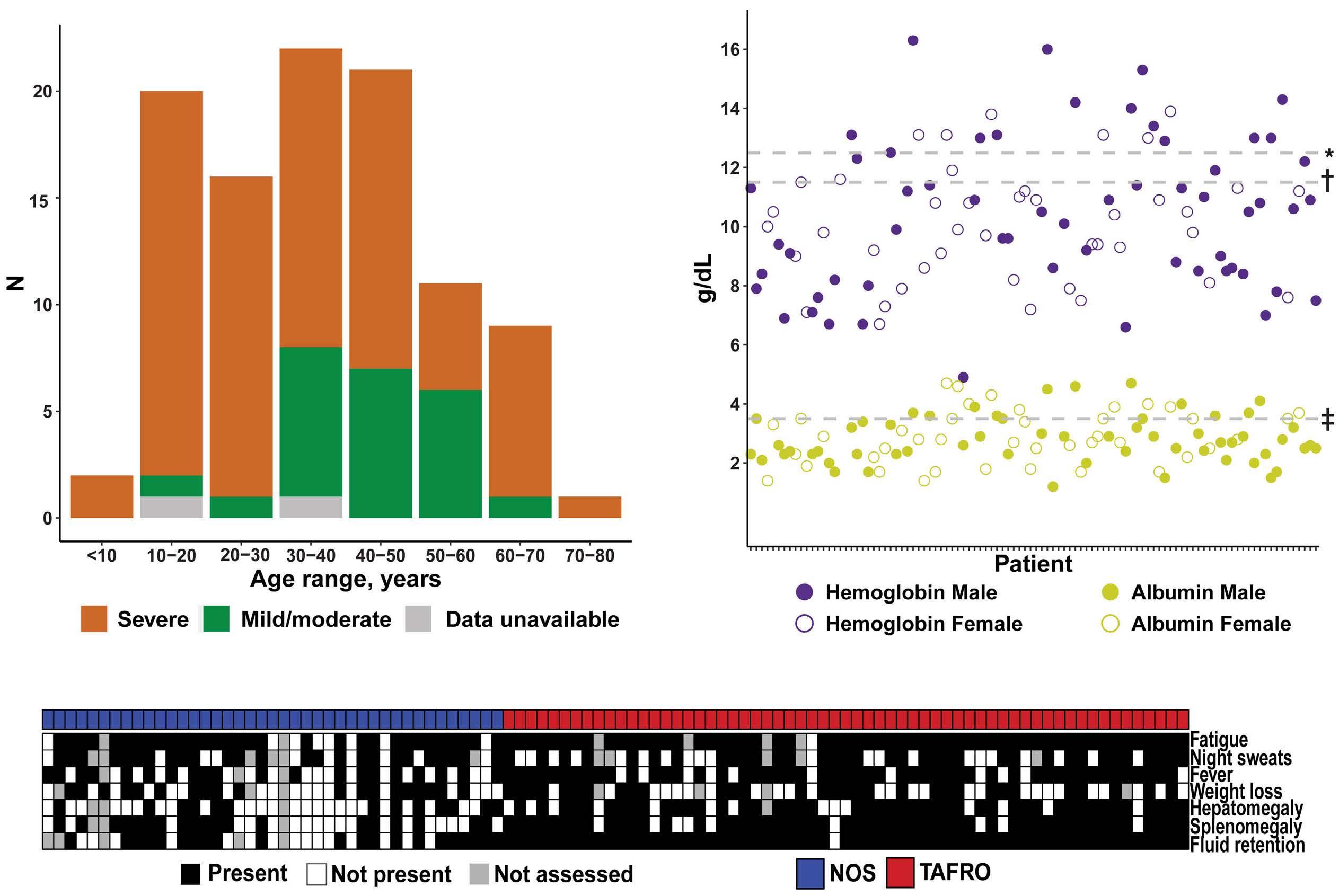
1. Patients with idiopathic multicentric Castleman disease demonstrate severe disease at diagnosis. (A) A large proportion of patients with idiopathic multicentric Castleman disease (iMCD) across all ages had severe disease. Notably, 95% of patients under the age of 30 years presented with severe disease at diagnosis. (B) Within the cohort of 102 iMCD patients, 87 (85.3%) had anemia and 81 (79.4%) had hypoalbuminemia at diagnosis. *Lower limit of normal for hemoglobin in males (12.5 g/dL). †Lower limit of normal for hemoglobin in females (11.5 g/dL). ‡Lower limit of normal for albumin (3.5 g/dL). (C) A large majority of patients presented with clinical symptoms, which ranged from mild to severe. Patients with both clinical subtypes of iMCD (TAFRO and NOS) demonstrated clinical abnormalities. TAFRO: thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly; NOS: not otherwise specified.
these clinical signs, we found a substantial subset of NOS patients with the same severe clinical features. Among the 102 patients, 77 (76%) presented with severe disease at diagnosis, including 57 (93%) TAFRO patients and 20 (51%) NOS patients (Table 1). These data demonstrate that iMCD patients of any age and subtype can have severe and burdensome disease.
Idiopathic multicentric Castleman disease patients with TAFRO face significant hospitalization burden
We examined the number of hospitalized days due to iMCD symptoms as another measure of disease burden. In the year surrounding diagnosis (6 months preceding and after the diagnostic biopsy), iMCD patients were hospitalized for a median (IQR) of 18 (0.5-38.0) days. When we stratified
by days hospitalized prior to and following diagnosis, we found that patients were hospitalized a median (IQR) of 5 (0-13.8) days in the 6 months preceding their diagnosis (Figure 2A). In the 6 months following diagnosis, patients were hospitalized a median (IQR) of 7.5 (0-25.0) days. Eleven patients (10.8%) spent more than 60 days in hospital in the year surrounding their diagnosis, highlighting the morbidity of this disease.
Next, we examined hospitalizations by subtype of iMCD to evaluate any differences in burden of hospitalizations. We found that TAFRO patients spent significantly more days in hospital than did NOS patients. The median (IQR) days in hospital in the year surrounding diagnosis was 36 (1861) for patients with TAFRO and 0 (0-4) for NOS patients (W=153.5, P=1.3x10-13) (Figure 2B). In order to understand
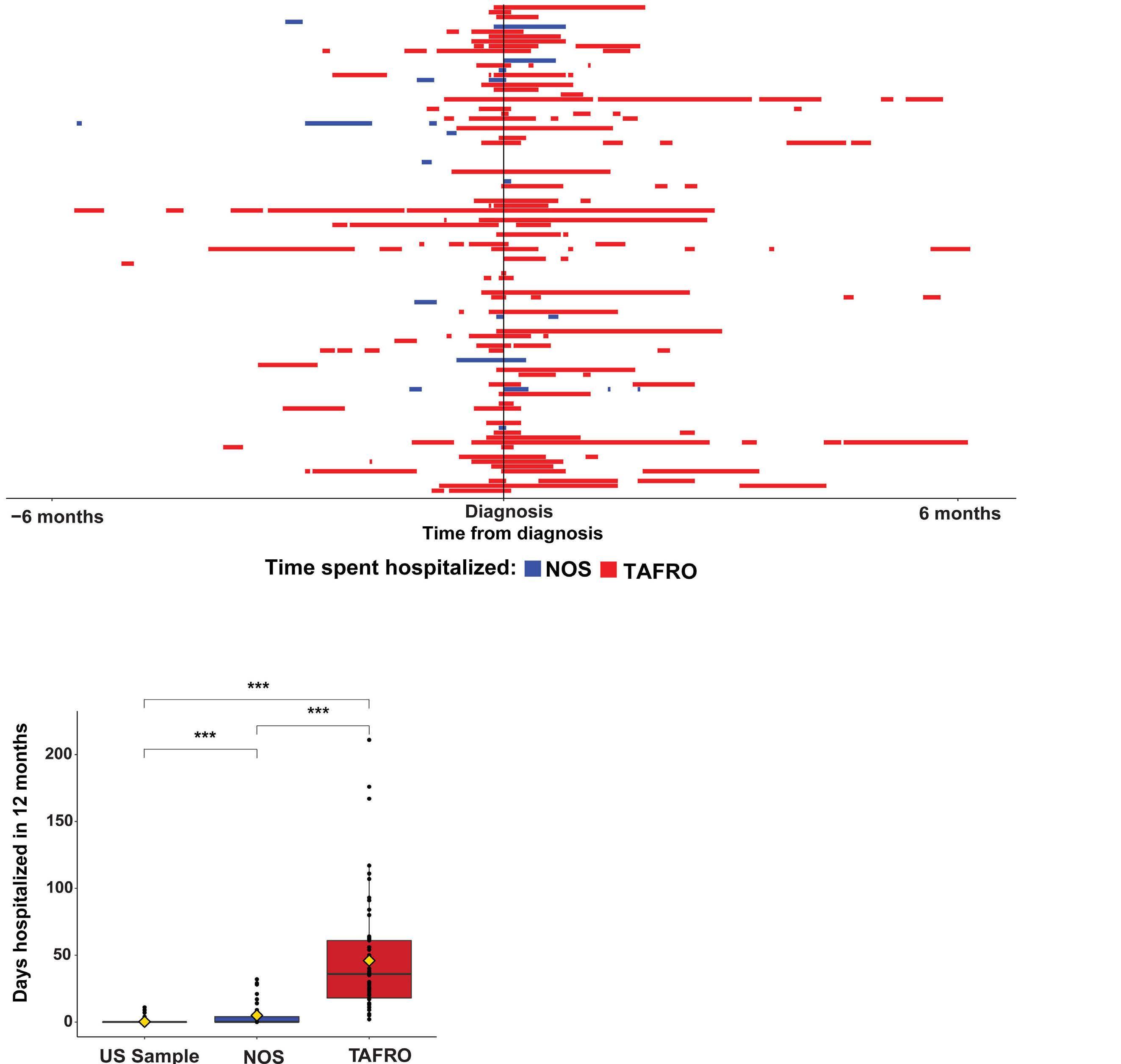
Figure 2. Patients with idiopathic multicentric Castleman disease face a high burden of hospitalizations. (A) Patients with idiopathic multicentric Castleman disease (iMCD) are hospitalized a large proportion of time in the 6 months leading up to their diagnosis and the 6 months following diagnosis (year around diagnosis). The graph depicts each patient on the Y axis with the time hospitalized prior to diagnosis and the number of days hospitalized after diagnosis. Red indicates the time that iMCD patients with TAFRO (thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly) spent in hospital, and blue indicates the time that patients with iMCD not otherwise specified (NOS) were hospitalized. In the 6 months prior to diagnosis, patients were hospitalized a mean (standard deviation [SD]) of 10.6 (17.5) days and a median (interquartile range [IQR]) of 5 (0-13.8) days, while in the year following diagnosis, patients were hospitalized a mean (SD) of 18.9 (28.2) days and a median (IQR) of 7.5 (0-25.0) days. (B) A sample of the USA population from the 2018 National Health Information Survey (NHIS), a proxy for the general population, spent significantly less time hospitalized in a 12-month period (median [IQR]: 0 [0-0], mean [SD]: 0.24 [1.3] days) than either TAFRO patients (median [IQR]: 35 [18-61], mean [SD]: 46.0 [42.0] days; P<2.2x10-16) or NOS patients (median [IQR]: 0 [0-4], mean [SD]:4.9 [9.0] days; P=8.3x10-13) spent hospitalized in the year around diagnosis. TAFRO patients were hospitalized significantly more days than NOS patients within the year of diagnosis (P=1.3x10-13). ***P<0.001.
both subtypes in the context of a general population and given that most of our cohort were from the USA, we accessed the NHIS 2018 survey data 20 and identified a control population of 408 individuals matched for age (±1 year), sex, and race. In a 12-month period, control patients were hospitalized for a median (IQR) of 0 (0-0) days. The number of days hospitalized differed between the general population and both TAFRO (W=47.5; P<2.2x10-16) and NOS (W=5,638; P=8.3x10-13) patients. This demonstrates that compared to the general population, patients with iMCD require substantially greater use of the healthcare system and frequently need to be admitted to hospital. We hypothesized that initiating a course of treatment for iMCD would result in a reduction in the time spent in hospital. To test this hypothesis, we first compared the proportion of patients hospitalized at the initiation of the first iMCD treatment regimen, excluding corticosteroid monotherapy, to the proportion hospitalized 4 weeks after initiating a treatment regimen. Four weeks after initiation was selected to allow sufficient time for the respective treatment regimen to have an effect. Among the 99 patients who received treatment with at least one regimen, 49 (49.5%) were hospitalized at the time of starting the treatment regimen. After 4 weeks of treatment, the number hospitalized had been reduced to 20 (20.2%) patients (X=17.4; P=3.0x10-5). We stratified the patients by type of treatment regimen, considering the regimens most frequently administered as first-line therapy, including siltuximab ± corticosteroids, tocilizumab ± corticosteroids, rituximab ± corticosteroids, chemotherapy-based regimens, and immunomodulator(s) ± corticosteroids. Corticosteroid monotherapy was not included in this analysis given that this approach is not recommended and patients often receive a short course while awaiting a correct diagnosis. We found that among the 32 patients treated with siltuximab ± corticosteroids as first-line therapy, 11 (34.3%) were hospitalized at the time of regimen initiation, and two (6.3%) were hospitalized 4 weeks after starting the treatment regimen (P=0.01). While fewer patients were hospitalized 4 weeks after starting treatment with each of the other regimens, we did not find any other statistically significant differences, although small sample sizes limit interpretation (Online Supplementary Table S4). Overall, these data demonstrate the clear burden of hospitalizations early in the course of iMCD and the importance of administering targeted therapy at the time of diagnosis.
Patients with idiopathic multicentric Castleman disease often require urgent interventions and demonstrate multisystem organ involvement
Considering the degree of hospitalization burden, we next investigated the distribution of organ system involvement among iMCD patients ± 365 days from diagnosis (Online Supplementary Methods). Cytopenias were the most common laboratory abnormalities, experienced by 97.1% (n=99)
of the cohort. This most frequent abnormality was anemia, which was present in 87 (85.3%) patients. We also found large proportions of patients experiencing symptoms or laboratory abnormalities related to other organ systems, such as respiratory (n=90, 88.2%), gastrointestinal (n=86, 84.3%), hepatic (n=76, 74.5%), neurological (n=68, 66.7%), renal (n=63, 61.8%), cardiovascular (n=63, 61.8%), and ocular (n=27, 26.5%) systems (Figure 3A, Online Supplementary Tables S5 and S6).
Next, we looked at life-sustaining interventions related to organ system involvement. We found that 27 (26.5%) patients required dialysis and 17 (16.7%) patients required a ventilator during at least one iMCD-related hospitalization. Additionally, 47 (46.0%) patients required paracentesis, 42 (41.1%) patients received a red blood cell transfusion, and 22 (21.6%) patients received a platelet transfusion (Figure 3B). These interventions, including all platelet transfusions, were mostly required for TAFRO patients.
We also quantified the proportion of time patients spent in a state of flare. We found that NOS patients spent a significantly greater proportion of time in flare from presentation until last known follow-up (median [IQR]: 52.3% [21.0-99.6]) compared to TAFRO patients (18.9% [10.8-52.5], W=1,673; P=0.004) (Figure 3C, D). Of the eight deceased patients in this cohort, six had TAFRO and five of these TAFRO patients died within 2 years of diagnosis. These data suggest that while TAFRO patients experience a greater degree of organ failure and more hospital interventions, NOS patients experience a longer continuation of milder, chronic symptoms.
Patients with idiopathic multicentric Castleman disease develop severe disease-related morbidities and comorbidities following diagnosis
We next sought to identify and quantify the morbidities and comorbidities occurring both before and after the diagnosis of iMCD. Prior to the iMCD diagnosis, we found that the most common conditions diagnosed among the full cohort were hypertension (n=26, 25.5%), obesity (n=23, 22.5%), asthma (n=21, 20.6%), gastroesophageal reflux disease (n=14, 13.7%), and depression (n=11, 10.8%) (Figure 4A). Stratification by iMCD subtype did not reveal apparent differences in conditions identified before the diagnosis of iMCD (Online Supplementary Figure S1).
Following the iMCD diagnosis, 48% (n=49) of the cohort developed acute renal failure, 15.7% (n=16) developed chronic kidney disease, 13.7% (n=14) developed iron deficiency anemia, 10.7% (n=11) developed pneumonia, 6.9% (n=7) developed sepsis, and 6.9% (n=7) developed thrombotic microangiopathy (Figure 4B). Acute renal failure was the most common iMCD-related morbidity diagnosed following disease onset in both subtypes (Online Supplementary Figure S2). Sepsis and thrombotic microangiopathy were, however, primarily diagnosed in TAFRO patients. We also stratified the top three morbidities arising for the full
cohort following iMCD onset by organ systems, revealing other serious conditions such as atypical hemolytic uremic syndrome (n=5, 4.9%), acute cholecystitis (n=5, 4.9%), and congestive heart failure (n=4, 3.9%) (Online Supplementary Table S7). Few malignancies were identified following diagnosis; the most common malignancy was papillary thyroid carcinoma, which occurred in two patients 1.3 and 3.6 years after iMCD diagnosis (Online Supplementary Table S8).
Quality of life is inversely correlated with symptoms in idiopathic multicentric Castleman disease
We questioned patients about their experiences living with iMCD by asking about QOL and the presence of ongoing iMCD-related symptoms. Fifty-nine (57.8%) patients responded. At a median (IQR) of 3.9 (2.3-8.0) years after diagnosis, iMCD patients reported a median (IQR) QOL score of 80 (71.5-90.0) in the EQ-5D-5L, on a scale of 0 to 100
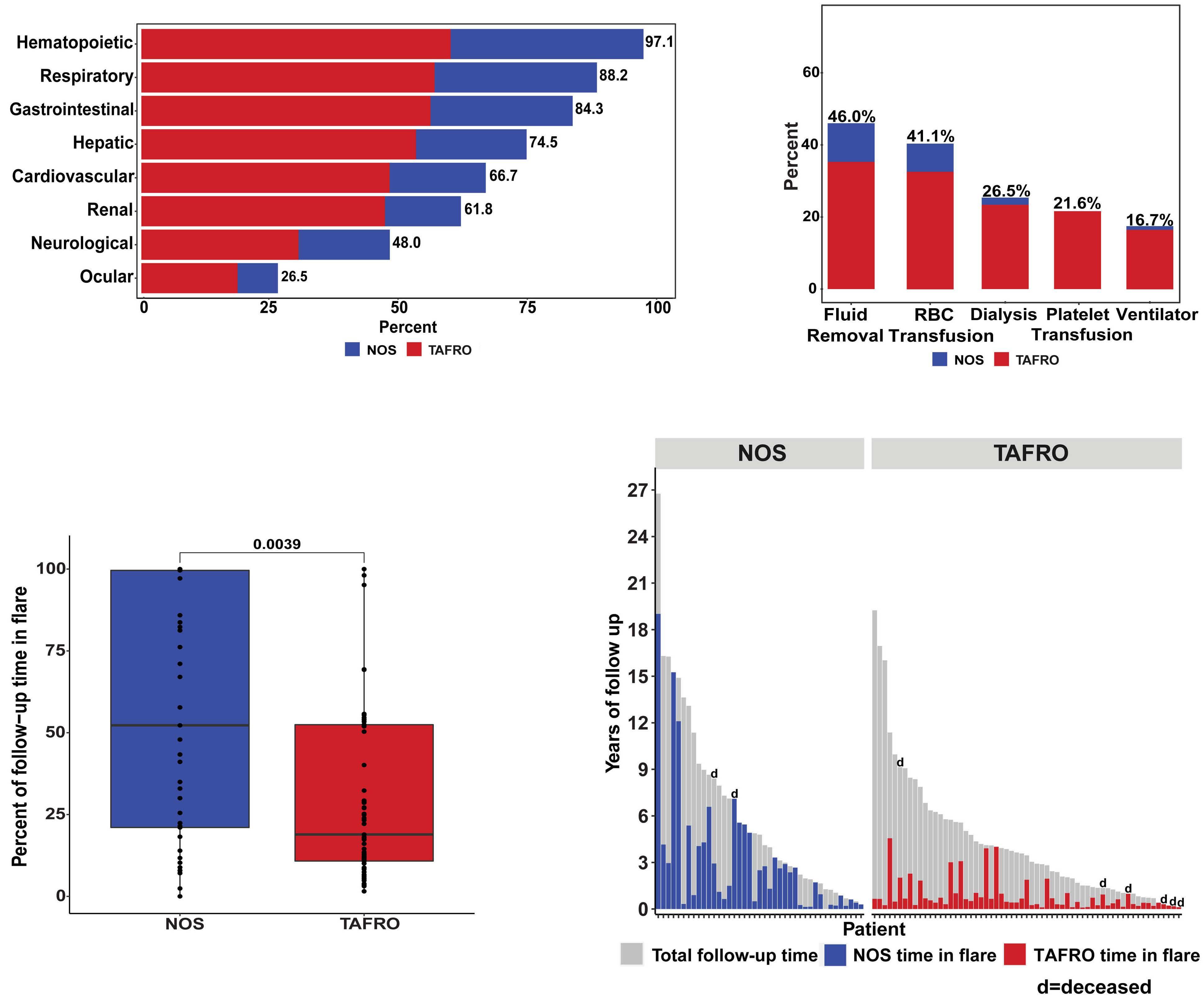
Figure 3. Patients with idiopathic multicentric Castleman disease experience a range of organ system involvement, require various hospital interventions, and demonstrate ongoing flares. (A) Organ system involvement in idiopathic multicentric Castleman disease (iMCD) ranked from most frequently observed to least frequently observed. Over 97% of the iMCD cohort experienced some hematopoietic dysfunction. Notably, both patients with TAFRO (thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly) and those with iMCD not otherwise specified (NOS) experienced significant organ system involvement and dysfunction. (B) The severity of organ dysfunction is reflected by the degree of healthcare intervention(s) required. Over one-quarter of patients (N=27 [26.5%]) required the use of a ventilator, and 17 (16.7%) patients required dialysis. Additionally, 47 (46.0%) patients required fluid removal (paracentesis), 42 (41.1%) patients received a red blood cell transfusion, and 22 (21.6%) patients received a platelet transfusion. (C) NOS patients spent a significantly greater proportion of time in flare from presentation until last known information (median [interquartile range]: 52.3% [21.0-99.6]) compared to TAFRO patients (18.9% [10.8-52.5], W=1673; P=0.004). (D) Each patient is represented by a vertical bar. The bar extends the length of follow-up from the start of the first flare. NOS patients are represented on the left, and TAFRO patients are represented on the right. The blue bars represent the proportion of time NOS patients spent in flare. The red bars represent the proportion of time TAFRO patients spent in flare. The designation ‘d’ indicates a deceased patient. RBC: red blood cells.
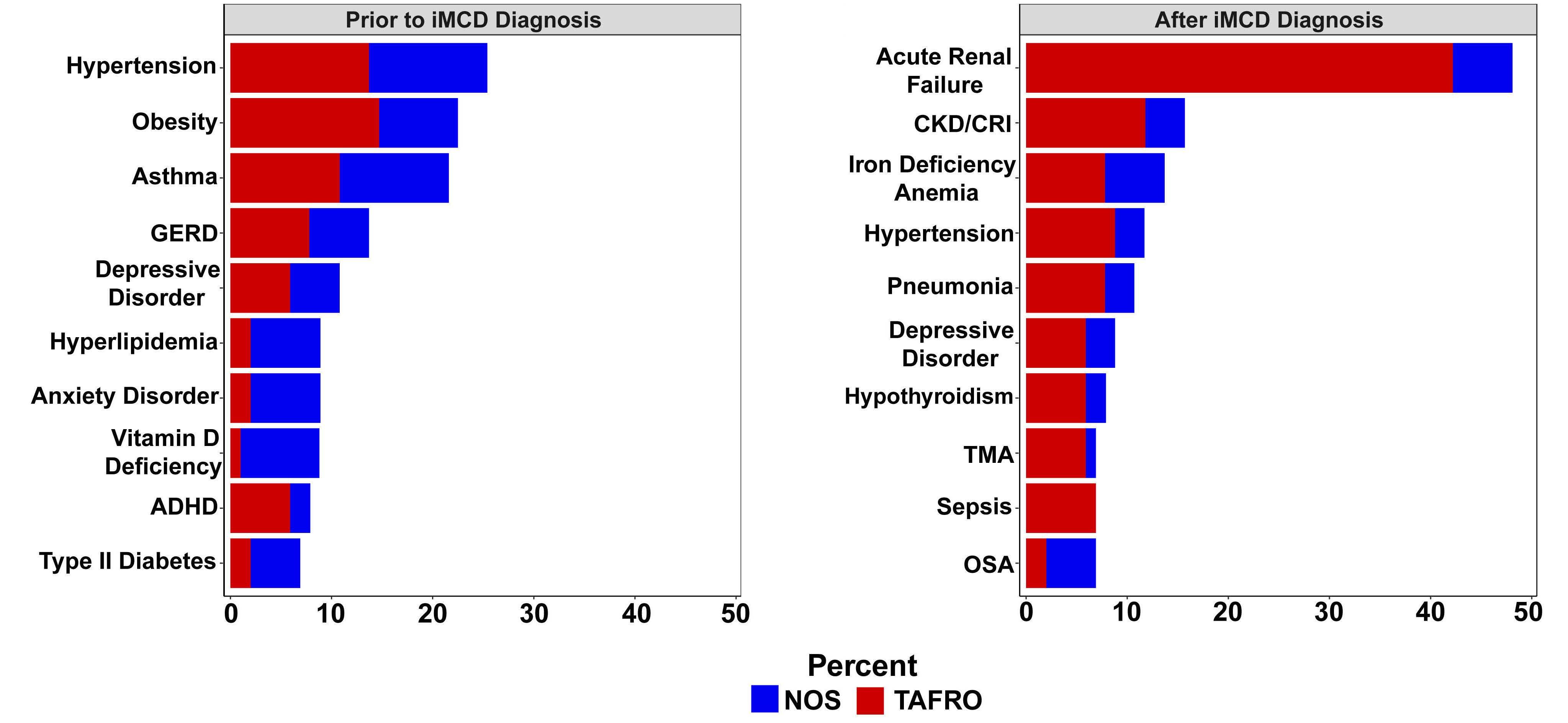
Figure 4. Patients with idiopathic multicentric Castleman disease have numerous comorbid and morbid conditions contributing to the burden of the disease. (A) Prior to diagnosis, the most commonly diagnosed comorbidities among the full cohort of patients with idiopathic multicentric Castleman disease (iMCD) mirrored common comorbidities among the USA population and included hypertension (N=26, 25.5%), obesity (N=23, 22.5%), asthma (N=21, 20.6%), gastroesophageal reflux disease (N=14, 13.7%), and depression (N=11, 10.8%). (B) Following the diagnosis of iMCD, patients experienced an array of burdensome comorbidities and morbidities including acute renal failure (N=49, 48.0%), chronic kidney disease/chronic renal insufficiency (N=16, 15.7%) and iron deficiency anemia (N=11, 10.8%) among others. GERD: gastroesophageal reflux disease; ADHD: attention-deficit hyperactivity disorder; CKD/CRI: chronic kidney disease/chronic renal insufficiency; TMA: thrombotic microangiopathy; OSA: obstructive sleep apnea; NOS: not otherwise specified; TAFRO: thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/ renal failure, and organomegaly.
with 0 being the worst health imaginable and 100 being the best health imaginable. This compares to a mean score of 80.4 in the general USA population.21 That same day, the patients reported a median MCD symptom score of 29.5 (IQR, 20-37; min-max: 16-96) with 16 being the best possible score (no iMCD symptoms) and 96 being the worst possible score. We investigated whether there was any relationship between a patient’s QOL score and number of days hospitalized prior to completing the survey, but did not found a correlation (R=0.038; P=0.8) (Online Supplementary Figure S3). We also looked for differences in subsequent QOL scores between patients who presented with severe disease and mild/moderate disease at baseline as well as between TAFRO and non-TAFRO patients and found no significant differences between groups (TAFRO vs. NOS: W=481, P=0.31; severe vs. mild/moderate at baseline: W=298.5, P=0.61). We determined the correlation between QOL score and MCD symptom score based on data collected a median (IQR) of 3.9 (2.3-8.0) years after diagnosis. QOL scores were inversely correlated with MCD symptom score (R=-0.69; P<0.001) (Figure 5). This negative correlation appeared to hold for patients with both NOS and TAFRO subtypes. This suggests that several years after diagnosis, patients reporting active iMCD symptoms may be having a decrease in their QOL from those symptoms.
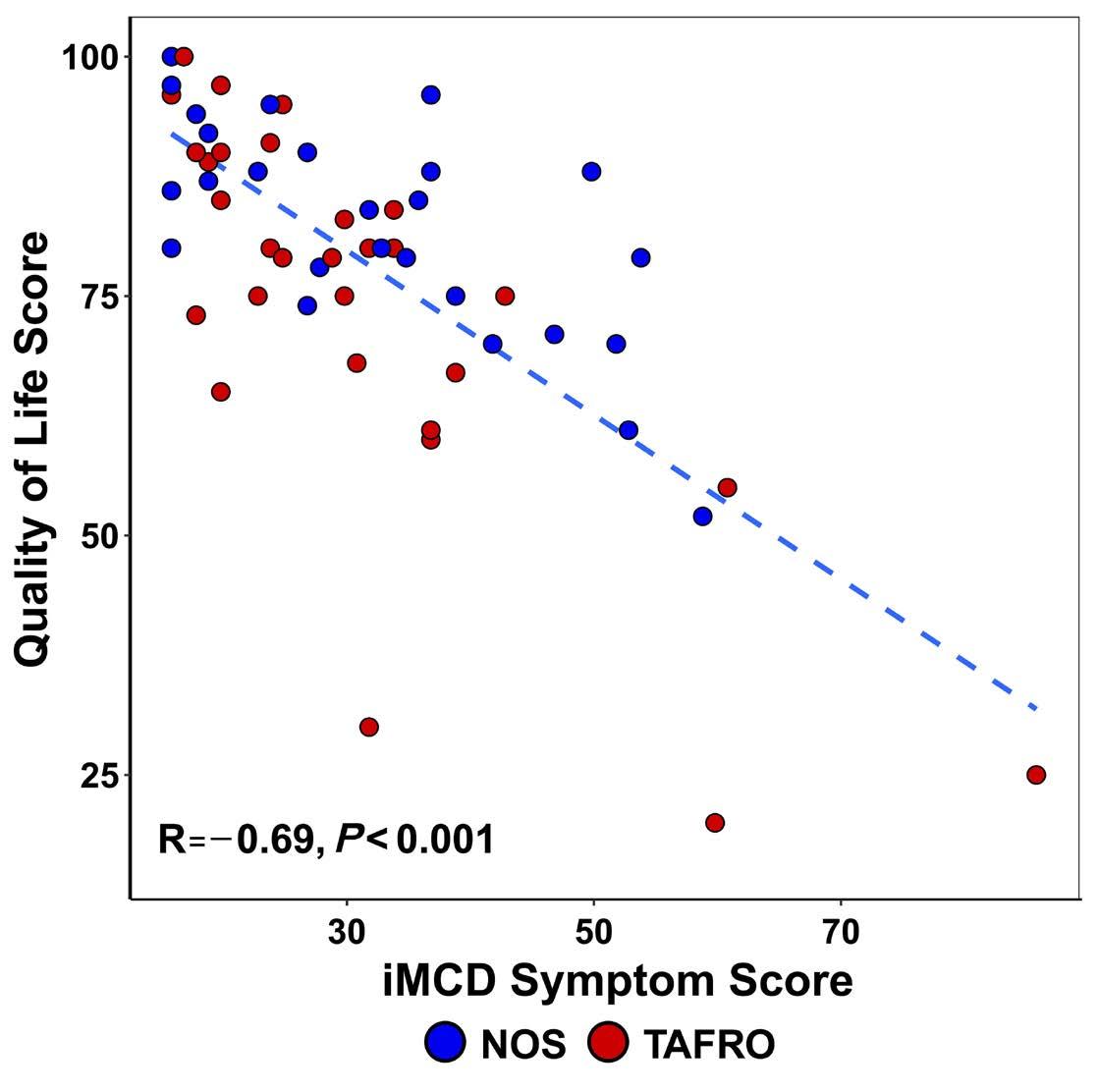
Figure 5. Quality of life of patients with idiopathic multicentric Castleman disease is inversely correlated with degree of symptoms. There is a correlation between quality-of-life score and idiopathic multicentric Castleman disease (iMCD) symptom score. Lower quality of life correlates with higher iMCD symptom score and higher quality of life correlates with lower iMCD symptom score (R=-0.69, P <0.001). NOS: not otherwise specified; TAFRO: thrombocytopenia, anasarca, fever/elevated C-reactive protein, reticulin fibrosis/renal failure, and organomegaly.
iMCD is a rare, heterogeneous disorder, and its full impact on patients’ lives is still not well understood. Since the consensus diagnostic criteria for iMCD are relatively new, long-term epidemiological and medical data from patients with confirmed iMCD are sparse. The ACCELERATE natural history registry is ideally suited to better understand the burden of disease as the most extensive source of longitudinal clinical data (median 3.4 years of follow-up after diagnosis) for these patients. ACCELERATE also provides independent adjudication about the accuracy of diagnosis for each patient through a panel of clinicians and hematopathologists who are experienced in diagnosing and treating iMCD.18 We found that patients with iMCD require extensive use of the healthcare system and experience long-term effects in terms of iMCD-related morbidities and comorbidities, time in disease flare, and QOL. There are distinct clinical subtypes of iMCD and, importantly, we found differences in the burden of disease between these subtypes. Patients with TAFRO required significantly longer stays in hospital and disproportionately required interventions such as dialysis, mechanical ventilation, and transfusions, illustrating the severe, life-threatening nature of this disease. NOS patients, however, spent a significantly greater proportion of time following diagnosis in a state of flare compared to TAFRO patients. These data reveal the challenges iMCD patients experience across the spectrum of clinical subtypes and severity. The patient-reported outcome data from our study indicate that patients with both subtypes continue to experience an impact on their QOL even years after diagnosis and that persistent iMCD-related symptoms lower QOL significantly. Notably, QOL scores reflect the state of patients who have thus far survived their disease and may, therefore, be biased towards a healthier subset of the disease population. Although NOS patients spent significantly less time in hospital, we found that 51.3% met criteria for severe disease at presentation. This remains lower than the 93.4% of TAFRO patients who presented with severe disease. A recent study from China characterizing 418 iMCD-NOS patients applied the same definition of severity and found that only 87 (21%) patients met criteria for severe disease at presentation.13 The researchers also noted a lower 3-year overall survival rate among NOS patients with severe (76%) compared to mild/moderate (94%) disease and found that severity at diagnosis was associated with death. Notably, our study showed high proportions of patients in both younger (<30 years) and older (>60 years) age groups who presented with severe disease. In our study, patients under 30 years old were significantly more likely to present with severe disease, and nearly all patients <18 years presented with severe disease. This may be associated with the median age of the TAFRO subtype of patients being 31 years, compared to 39 years for patients with iMCD-NOS, and with the fact
that 93% of TAFRO patients presented with severe disease. The relatively young age at diagnosis, burden of disease, and availability of disease-controlling therapies highlight the importance of rapid diagnosis and treatment. This study found that iMCD patients experienced a greater degree of hospitalization than the general population experiences in a given year, with this being primarily due to the increased rates of hospitalization among iMCD-TAFRO patients.20 Our observation that patients required extensive hospital care in the year surrounding diagnosis is consistent with the findings of a recent claims-based study.17 We found that 4 weeks after the first-administered treatment regimen, the proportion of hospitalized patients decreased significantly, including patients treated with siltuximab ± corticosteroids (34% vs. 6%), which is the current consensus first-line recommendation.15
We also found that the conditions most frequently diagnosed in our cohort prior to the diagnosis of iMCD were similar to those among the general USA population. A report from the Agency for Healthcare Research and Quality found that hypertension, diabetes, and chronic respiratory disease were among the most common conditions associated with inpatient stays and that depression was present in 10% of patients requiring admission to hospital,22 which is similar to the findings in our cohort prior to the diagnosis of iMCD. We did not identify a condition present in a large proportion of iMCD patients prior to the iMCD diagnosis that might suggest a predisposition or trigger to iMCD, which has an as-yet-unknown etiology. Following the diagnosis of iMCD, patients in both the TAFRO and NOS subtypes developed life-threatening morbidities, most commonly acute renal failure. Consistent with previous reports, we found a high degree of both renal and hepatic dysfunction.17
In this cohort, few patients developed a malignancy following the diagnosis of iMCD. In fact, only one patient developed a myeloid malignancy (myelodysplastic syndrome), which contrasts with a key finding from a systematic literature review and a claims-based study that found an increase in myeloid and solid malignancies.17,23 No patients developed diffuse large B-cell lymphoma despite previous reports of its association with MCD, though predominantly in human herpes virus 8-positive MCD.24-26 We found that our rate of malignancies was comparable to that in the control population in the claims-based study.17 This discrepancy may be due to insufficient follow-up time to detect subsequent malignancies or to more rigorous validation of the iMCD diagnosis in our cohort and recent formalization of exclusion criteria for iMCD. Specifically, iMCD diagnostic criteria preclude patients from being concurrently diagnosed with iMCD and a number of malignancies, given the overlapping histopathology and the fact that malignancies can cause reactive lymph node changes that resemble iMCD. It is, therefore, possible that some patients in the claims-based study who should have only been classified as having a malignancy were incorrectly classified as
having both. Conversely, our strict inclusion criteria might have inadvertently excluded true iMCD patients who also developed malignancies.
This study has several limitations. First, 13 patients with the NOS subtype met the criteria for idiopathic plasmacytic lymphadenopathy (thrombocytosis and hypergammaglobulinemia) and may represent a distinct subtype from other iMCD-NOS patients.27 Due to the low number of patients and the similar, more moderate presentation, these patients were combined with other non-TAFRO patients in the NOS category.13 Second, our study included a larger proportion of TAFRO patients than NOS patients, so may not be representative of the whole iMCD population. To address this, we stratified our analyses between TAFRO and NOS subtypes. It is possible that the higher number of TAFRO patients in this study is a result of patients with more severe disease being more likely to seek resources online and thus enroll into the ACCELERATE registry. However, the study design for our registry is biased towards patients who survive and who are able to enroll themselves into a registry compared to patients who die shortly after diagnosis and would need a family member to enroll them. Ultimately, our data demonstrate that an iMCD diagnosis imparts a long-term burden of disease on patients which results in high rates of hospitalization, the development of life-threatening iMCD-related morbidities and comorbidities requiring intensive interventions, and reduced QOL from active symptoms. These data demonstrate the importance of ongoing research into iMCD, the need to recognize and diagnose iMCD sooner, and the relevance of focused efforts to identify diagnostic and/or disease biomarkers to aid in disease management.
DCF has received research funding for the ACCELERATE registry, consulting fees from EUSA Pharma and study drug with no associated research funding for the clinical trial of sirolimus from Pfizer (NCT03933904); he has two provisional patents pending related to the diagnosis and treatment of iMCD. GS has received speakers’ bureau fees from Takeda, Janssen Pharmaceuticals, Foundation Medicine, and EUSA Pharma. AB has served on a EUSA Pharma Castleman disease advisory board. DA has served on the Castleman disease medical advisory board for EUSA Pharma and from 03/2023 to present serves as the advisory board’s co-chair. JDB has received consulting fees from EUSA Pharma. FvR has received consulting fees from EUSA Pharma, GlaxoSmithKline, Karyopharm, and Takeda and has received research funding
1. Beck JT, Hsu SM, Wijdenes J, et al. Brief report: alleviation of systemic manifestations of Castleman’s disease by monoclonal anti-interleukin-6 antibody. N Engl J Med. 1994;330(9):602-605.
from Janssen Pharmaceuticals and Bristol Myers Squibb. MJL serves on advisory boards for EUSA Pharma and Secura Bio, Inc., serves as an honorarium speaker for Southeastern Lymphoma Symposium, and receives consulting fees from the University of Pennsylvania. All other authors report no conflicts of interest.
MSB was responsible for the formal analysis, writing the original draft as well as reviewing and editing the paper and visualization. SKP conceived the study and was responsible for the formal analysis, writing the original draft as well as reviewing and editing the paper and visualization. YR was responsible for the formal analysis, and wrote, reviewed and edited the paper. AB, JDB, GS, DA, MJL, HL, FvR, and MSL provided supervision and were involved in writing, reviewing and editing the paper. NM conducted investigations and was involved in writing, reviewing and editing the paper. DCF conceived and supervised the study, acquired funding and was involved in writing, reviewing and editing the paper.
The authors thank all the patients and their families for their participation in the ACCELERATE registry. We also thank the Castleman Disease Collaborative Network (CDCN) and the ACCELERATE Registry team for their support. We thank the volunteers for the CDCN who have supported this research, including Mary Zuccato and Mileva Repasky. We also thank Shawnee Bernstein, Nathan Hersh, Gerard Hoeltzel, and Jeremy Zuckerberg for their contributions to this study. Finally, we thank Faizaan Ahkter, Erin Napier, Eric Haljasmaa, Katherine Floess, Mark-Avery Tamakloe, Victoria Powers, Alexander Gorzewski, Johnson Khor, Reece Williams, Jasira Ziglar, Amy Liu, Saishravan Shyamsundar, Criswell Lavery, and Bridget Austin.
The ACCELERATE natural history registry has received funding from Janssen Pharmaceuticals (2016-2018), EUSA Pharma, LLC (USA), which has merged with Recordati Rare Diseases Inc. (2018-2022), and the U.S. Food & Drug Administration (R01FD007632) (2022-Present). DCF also receives funding from the National Heart, Lung, and Blood Institute (R01HL141408) (2018-present).
Data-sharing statement
All source data reported in this study are available by contacting the ACCELERATE team at accelerate@uphs.upenn.edu.
2. Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989;74(4):1360-1367.
3. Fajgenbaum DC, Langan RA, Japp AS, et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL-6-blockaderefractory idiopathic multicentric Castleman disease. J Clin Invest. 2019;129(10):4451-4463.
4 Arenas DJ, Floess K, Kobrin D, et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood. 2020;135(19):1673-1684.
5. Pierson SK, Shenoy S, Oromendia AB, et al. Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease. Blood Adv. 2021;5(17):3445-3456.
6. Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383(23):2255-2273.
7 Nishimura Y, Fajgenbaum DC, Pierson SK, et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol. 2021;96(10):1241-1252.
8. Takeuchi K. Idiopathic plasmacytic lymphadenopathy: a conceptual history along with a translation of the original Japanese article published in 1980. J Clin Exp Hematop. 2022;62(2):79-84.
9 Mukherjee S, Martin R, Sande B, Paige JS, Fajgenbaum DC. Epidemiology and treatment patterns of idiopathic multicentric Castleman disease in the era of IL-6-directed therapy. Blood Adv. 2022;6(2):359-367.
10 Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646-1657.
11. Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997-1002.
12. Cohen AB, Swaminathan A, Wang XL, et al. Clinical characteristics, treatment patterns, and overall survival of real-world patients with idiopathic multicentric Castleman disease. J Clin Oncol. 2021;39(15 suppl):7048.
13. Zhang L, Dong YJ, Peng HL, et al. A national, multicenter, retrospective study of Castleman disease in China implementing CDCN criteria. Lancet Reg Health West Pac. 2023;34:100720.
14 van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966-974.
15. van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115-2124.
16. Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized antiinterleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627-2632.
17 Mukherjee S, Kanhai K, Kauffman D, et al. Organ dysfunction, thrombotic events and malignancies in patients with idiopathic multicentric Castleman disease: a population-level US health claims analysis. Leukemia. 2022;36(10):2539-2543.
18. Pierson SK, Khor JS, Ziglar J, et al. ACCELERATE: a patientpowered natural history study design enabling clinical and therapeutic discoveries in a rare disorder. Cell Rep Med. 2020;1(9):100158.
19. CDC. Hospital utilization (in non-Federal short-stay hospitals). 2018. https://www.cdc.gov/nchs/fastats/hospital.htm#print. Accessed June 2023.
20. NHIS. About the National Health Interview Survey. 2019. https:// www.cdc.gov/nchs/nhis/about_nhis.htm. Accessed June 2023.
21. Jiang R, Janssen MFB, Pickard AS. US population norms for the EQ-5D-5L and comparison of norms from face-to-face and online samples. Qual Life Res. 2021;30(3):803-816.
22. Owens PL, Liang L, Barrett ML, Fingar KR. Comorbidities associated with adult inpatient stays, 2019. Healthcare Cost and Utilization Project (HCUP) statistical briefs. Rockville (MD), 2006.
23. Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3(4):e163-175.
24. Payera E, Zofia M, Zsofia S, et al. Diagnostic and therapeutic difficulties in diffuse large B-cell lymphoma arising from HHV8 positive Castleman disease. J Hematol. 2012;1(2):65-69.
25. Sukswai N, Lyapichev K, Medeiros LJ, Khoury JD. Unusual case of human herpesvirus 8-positive large B-cell lymphoma associated with Castleman disease. Clin Case Rep. 2019;7(3):587-588.
26. Whooley M, Keirns D, Wu XX, Luker A, Silberstein PT, Hsia B. Diffuse large B-cell lymphoma in HHV8+ multicentric Castleman disease: a national analysis of demographic features. J Clin Oncol. 2023;41(16 suppl):e19535.
27. Nishikori A, Nishimura MF, Nishimura Y, et al. Idiopathic plasmacytic lymphadenopathy forms an independent subtype of idiopathic multicentric Castleman disease. Int J Mol Sci. 2022;23(18):10301.
Dongyeop Shin,1* Myung-Jin Kim,2* Soyeon Chun,3* Dongchan Kim,4 Chansu Lee,5 Kwang-Sung Ahn,6 Eunyoung Jung,2 Dayeon Kim,2 Byung-Chul Lee,2 Daehee Hwang,3,7 Yonghwan Kim2 and Sung-Soo Yoon1,4
1Department of Internal Medicine, Seoul National University Hospital; 2Department of Biological Sciences, Research Institute of Women’s Health and Digital Humanity Center, Sookmyung Women’s University; 3School of Biological Sciences, Seoul National University; 4Cancer Research Institute, Seoul National University College of Medicine; 5Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine; 6Functional Genome Institute, PDXen Biosystem Inc. and 7Bioinformatics Institute, Seoul National University, Seoul, South Korea
*DS, M-JK and SC contributed equally as first authors.
Correspondence: Y. Kim yhkim@sookmyung.ac.kr
D. Hwang daehee@snu.ac.kr
Sung-Soo Yoon ssysmc@snu.ac.kr
Received: June 19, 2023. Accepted: January 2, 2024. Early view: January 11, 2024.
https://doi.org/10.3324/haematol.2023.283784
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Osteolytic bone lesion is a major cause of lower quality of life and poor prognosis in patients with multiple myeloma (MM), but molecular pathogenesis of the osteolytic process in MM remains elusive. Fms-like tyrosine kinase 3 ligand (FLT3L) was reported to be elevated in bone marrow (BM) and blood of patients with advanced MM who often show osteolysis. Here, we investigated a functional link of FLT3L to osteolytic process in MM. We recruited 86, 306, and 52 patients with MM, acute myeloid leukemia (AML), and acute lymphoblastic leukemia (ALL), respectively. FLT3L levels of patients with hematologic malignancies were measured in BM-derived plasma and found to be significantly higher in MM than in AML or ALL, which rarely show osteolysis. FLT3L levels were further elevated in MM patients with bone lesion compared with patients without bone lesion. In vitro cell-based assays showed that the administration of FLT3L to HEK293T, HeLa, and U2OS cells led to an increase in the DKK1 transcript level through STAT3 phosphorylation at tyrosine 705. WNT reporter assay showed that FLT3L treatment reduced WNT signaling and nuclear translocation of b-catenin. These results collectively show that the FLT3L-STAT3-DKK1 pathway inhibits WNT signaling-mediated bone formation in MM, which can cause osteolytic bone lesion. Finally, transcriptomic profiles revealed that FLT3L and DKK1 were predominantly elevated in the hyperdiploidy subtype of MM. Taken together, FLT3L can serve as a promising biomarker for predicting osteolytic bone lesion and also a potential therapeutic target to prohibit the progression of the osteolytic process in MM with hyperdiploidy.
Multiple myeloma (MM) is the second most common hematologic malignancy involving malignant plasma cells. MM is characterized by the presence of clonal plasma cells producing fragmented monoclonal immunoglobulins of various heavy and light chain subtypes. In the US, the overall 5-year survival rate for patients with MM is 55%,1,2 despite combination chemotherapy with potent target agents, and even high-dose chemotherapy and autologous hematopoietic stem cell transplantation. MM causes various complications, including osteolytic bone lesion, anemia, renal failure, hypercalcemia, and hyperviscosity syndrome.3
Osteolytic bone lesions at multiple sites are a unique feature of MM, which differs from other hematologic cancers
such as acute and chronic leukemia or lymphoma. The chief complaint in about two-thirds of MM patients is bone pain due to the direct bone involvement of myeloma tumor cells or indirect effect of myeloma tumor cells or tumor microenvironment-secreting cytokines on bony skeleton. Skeletal-related events (SRE) in MM patients include intractable bone pain, fracture, and paralysis due to spinal cord compression,4 which radically lower patients’ quality of life (QoL) and place a substantial burden on healthcare resources.5,6 Recent advances in treatment for MM have remarkably improved the median overall survival of patients with MM up to over 5 years.7 Accordingly, QoL has become a more important issue for long-term MM survivors. Furthermore, bone osteolysis has a negative impact on the survival of MM.8 Osteolytic bone lesions, a major cause of morbidity
and mortality in MM patients, are induced by an imbalance in bone remodeling characterized by the suppressed osteoblast formation and function, increased osteoclast formation and activity, as well as direct cell interactions between MM cells and osteoclasts, and the release of cytokines and growth factors.9 One of the key mechanisms contributing to the development of osteolytic bone lesions is the activation of osteoclasts resulting in enhanced bone resorption and subsequent bone loss.10 However, the molecular mechanisms underlying the pathogenesis of MM-induced osteolytic bone lesions are still not fully understood. The putative mechanism of bone osteolysis in intercellular interactions between myeloma cells and osteoclast has been extensively studied. Cytokines of IL-6, IL-1b, TNF-α, MIP1α, and receptor activator of nuclear factor κ-B ligand (RANKL)11 generated in the bone marrow (BM) microenvironment accentuate the interactions between myeloma cells and BM stromal cells, which enhance osteoclastic activity. In contrast, Dick Kopf-related protein 1 (DKK1), an extracellular antagonist of WNT signaling, is known to contribute to the formation of osteolytic bone lesion by depressing osteoblastic activity.12 Specifically, the high expression levels of DKK1 in the BM microenvironment have been associated with osteolytic bone lesions in MM patients13 and regulation of the WNT signaling pathway. Recent studies have investigated the molecular mechanisms underlying osteolytic bone lesions in MM, providing critical insights into the disease’s pathogenesis. The cross-talk between MM cells and the BM microenvironment closely modulates the WNT/b-catenin signaling pathway, which promotes osteoblast activity and suppresses osteoclast differentiation.14 Many studies have demonstrated the importance of the WNT/b-catenin pathway in healthy bone development through canonical WNT signaling participation in the regulation of osteoblast differentiation.15 MM cells secrete WNT antagonists that disrupt BM regulation of osteoblastic differentiation, leading to osteolytic lesions.16 Dysregulation of WNT pathway by DKK1, secreted from MM cells, affects the BM microenvironment, leading to altered bone homeostasis and osteolytic bone disease.14 Several studies have investigated the expression pattern and functions of DKK1 in MM, revealing significantly higher DKK1 expression in MM patients compared to healthy controls, which is associated with poor prognosis.17 Additionally, preclinical studies have demonstrated that targeting DKK1 using monoclonal antibodies or small molecule inhibitors can inhibit MM cell proliferation and induce apoptosis both in vitro and in vivo. 18
Recently, Fms-like tyrosine kinase 3 ligand (FLT3L), a cytokine that plays a crucial role in hematopoiesis and immune regulation, has been reported to be elevated in BM and blood of patients with MM,19 and correlated with vascular endothelial growth factor (VEGF) which may reflect the increased proliferative tumor activity based on the association between FLT3L level and Ki-67 positivity in MM.20 FLT3L binds to the FLT3 receptor, which is expressed on hematopoiet-
ic stem cells and myeloid progenitor cells.21 In MM, FLT3L has been shown to promote the proliferation and survival of malignant plasma cells,20 as well as the recruitment of immunosuppressive cells to the tumor microenvironment.22 Inhibiting FLT3L using a small molecule inhibitor has reduced proliferation and increased apoptosis in MM cells.23 Similarly, targeting FLT3L with antibody-based therapy has demonstrated significant antitumor activity in MM.24 While emerging evidence highlights the crucial role of FLT3L in MM pathogenesis and its potential as a therapeutic target, further studies are needed to fully understand the underlying mechanisms of FLT3L function in MM and develop more effective FLT3L-targeted therapies for the disease. Therefore, we investigated the link of FLT3L to bone osteolysis in MM and explored the molecular mechanism in view of the osteolytic phenomenon in MM.
The patients who had been diagnosed with hematologic malignancies at Seoul National University Hospital from March 2004 to December 2012 were enrolled in this study. BM-derived blood samples from patients with leukemia or MM were used to measure FLT3L. Primary samples were obtained with informed consent (Seoul National University Hospital [SNUH] Institutional Review Board [IRB] N. 1902047-1008). The study was approved by the SNUH IRB (N. 1902-047-1008).
HEK293T, HeLa, U2OS, and MOLP8 cells were used to investigate the effect of FLT3L on intracellular signaling. HEK293T, HeLa, and U2OS cells were grown in high-glucose DMEM, and MOLP8 cells were cultured in RPMI. Culture media were supplemented with 10% FBS and 100 U/mL penicillin-streptomycin (all Gibco; Grand Island, NY, USA). Cells were maintained in a humidified 5% CO2 at 37°C. Recombinant human FLT3L ligand (rhFLT3L) was purchased from R&D SYSTEMS (Minneapolis, MN, USA). Stattic (Sigma-Aldrich; St. Louis, MO, USA), a STAT3 inhibitor, was pre-treated for one hour before treatment with rhFLT3L in HEK293T cells.
Plasma was extracted from patients’ BM-derived blood by gradient centrifugation, and used to measure FLT3L. FLT3L level was measured using human FLT3L pre-coated enzyme-linked immunosorbent assay (ELISA) kit (Biogems, Westlake Village, CA, USA) according to the manufacturer’s protocol. The standard curve was created by generating a 4-parameter logistic (4-PL) regression for the concentrations versus measured intensities. The concentration in the sample was taken as an estimate of the measured intensity using the standard curve.
Clinical variables of patients with MM included age, sex, disease status, serum M-protein, and osteolytic lesion; these were reviewed in the electronic medical records and included in the description of patients’ characteristics. MM-related variables of stage, subtype, the presence of anemia, hypercalcemia, azotemia, and osteolytic bone lesion were converted into categorical variables and used for survival analysis in view of prognostic factors.
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA of the cells was extracted using RNeasy Mini Kit and QIAshredder (GIAGEN; Valencia, CA, USA) and synthesized into cDNA through the SuperScript III First-Strand Synthesis System (Invitrogen; Carlsbad, CA, USA). mRNA level was determined by 2XqPCRBIO SyGreen Blue Mix LoROX (PCR Biosystems; Wayne, PA, USA) and normalized by that of an internal control GAPDH. All reactions were carried out in LightCycler® 96 (Roche; Indianapolis, IN, USA). Primer sequences used were as follows: hFLT3L sense: 5’-ACCTATCTCCTCCTGCTGCT-3’, antisense: 5’-GGTAGTCAGACAGCTCACGG-3’; hDKK1 sense: 5’-GTCCAAGATCTGTAAACCTGTCCT-3’, antisense: 5’-AGCCTAGAAGAATTACTGGCTTGA-3’, hGAPDH sense: 5’-GCAAATTCCATGGCACCGTC-3’, antisense: 5’-TCGCCCCACTTGATTTTGGA-3’; mDkk1 sense: 5’-TCCGTCTGCCTCCGATCATC-3’, antisense: 5’-GCCTTTCCGTTTGTGCTTGG-3’; mGapdh sense: 5’- CATGTTCCAGTATGACTCCACTC-3’, antisense: 5’-GGCCTCACCCCATTTGATGT-3’. SpectraMax i3x (NFEC-2017-12-241146) was used for quantitative real-time polymerase chain reaction (RT-qPCR) analysis at the Core Facility Center for Chronic and Metabolic Diseases at Sookmyung Women’s University.
Statistical analysis
Statistical analyses for clinical parameters were performed using Stata version 13 (Stata Corp.; College Station, TX, USA) and GraphPad Prism v 5.00 (GraphPad Software Inc.; San Diego, CA, USA). Data were presented as mean ± standard deviation (SD). Statistical significance (P-values) was determined by one-way ANOVA followed by Dunnett’s post hoc correction or two-way ANOVA with Tukey’s post hoc correction. The Mann-Whitney test was used to compare the level of FLT3L between two groups. Interquartile range (IQR) was calculated to estimate the distribution of FLT3L level in each group. Linear regression analysis was used to investigate the association between FLT3L level and myeloma tumor cell burden in BM. Overall survival (OS) was defined as the interval between the initial diagnosis of MM and death due to any cause or the last clinical follow-up. Survival probabilities were estimated using the Kaplan-Meier method and analyzed using Cox proportional hazard model.
We obtained mRNA expression data (GSE2658) from the Gene
Expression Omnibus (GEO) database together with patient subtype (PAM cluster) and clinical information. As instructed in the original study,22 we used the data from 414 samples in the dataset after removing the samples contaminated with myeloid or normal plasma cells. The probe intensities were first converted into log2-intensities and then normalized using quantile normalization.23 To identify expressed genes, we fitted a Gaussian mixture model to the distribution of the normalized log2-intensities and selected the genes whose intensities were higher in half of the samples than the threshold intensity at which the 2 fitted Gaussian probability density functions meet. Next, we identified molecular signatures that define individual subtypes of patients with MM using the previously reported statistical method.24 In brief, for the selected expressed genes, we calculated their log2-fold-changes with respect to their median values, and the log2-fold-changes were then normalized using quantile normalization. For each gene, we calculated an observed t statistic value in the comparison of one subtype versus the others. We then estimated an empirical null distribution of the t statistic value by performing random permutations of the samples 1,000 times, calculating t statistic values for
Variables
Disease status at time of FLT3L measurement
BM: bone marrow; FLT3L: fms-like tyrosine kinase 3 ligand; N: number; PB: peripheral blood.
all expressed genes using each permuted dataset, and estimating a Gaussian kernel density function for the resulting t statistic values. Using the estimated empirical distribution, we computed the adjusted P-values for the observed t statistic value for each gene by performing two-sided testing. Finally, we identified molecular signatures as those that had t test P<0.05 and log2-fold-changes >1.5.
Enrichment analysis of cellular pathways and gene ontology biological processes
To identify cellular processes and pathways represented by the molecular signatures for each patient subtype, we performed the enrichment analysis of pathways and gene ontology biological processes (GOBP) for the selected molecular signatures using ConsensusPathDB.25 The cellular pathways and GOBP represented by the molecular signatures for each subtype were selected as those with P<0.05, and the number of molecules involved in the pathway or GOBP ≥3.
FLT3L reflects osteolytic bone lesion and prognosis in multiple myeloma FLT3L level was reported to be elevated in BM and blood of patients with MM. To confirm this finding in Korean MM patients, and to further examine the association of FLT3L level with bone osteolytic bone lesion, we collected BM-derived plasma samples from 86, 306, and 52 patients with MM, acute myeloid leukemia (AML), and acute lymphoblastic leukemia (ALL), respectively. FLT3L level of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM) was included (N=42), as MGUS/ SMM involves no osteolytic bone lesion.25 Patients’ clinical characteristics are presented in Table 1. FLT3L levels were significantly (P<0.05) elevated in Korean patients with MM (median 161.8 pg/mL, interquartile range [IQR] 73.96-233.50) compared to those in AML (median 28.16, IQR 8.48-107.30) and ALL (median 46.11, IQR 12.52-172.80) (Figure 1A). Consid-
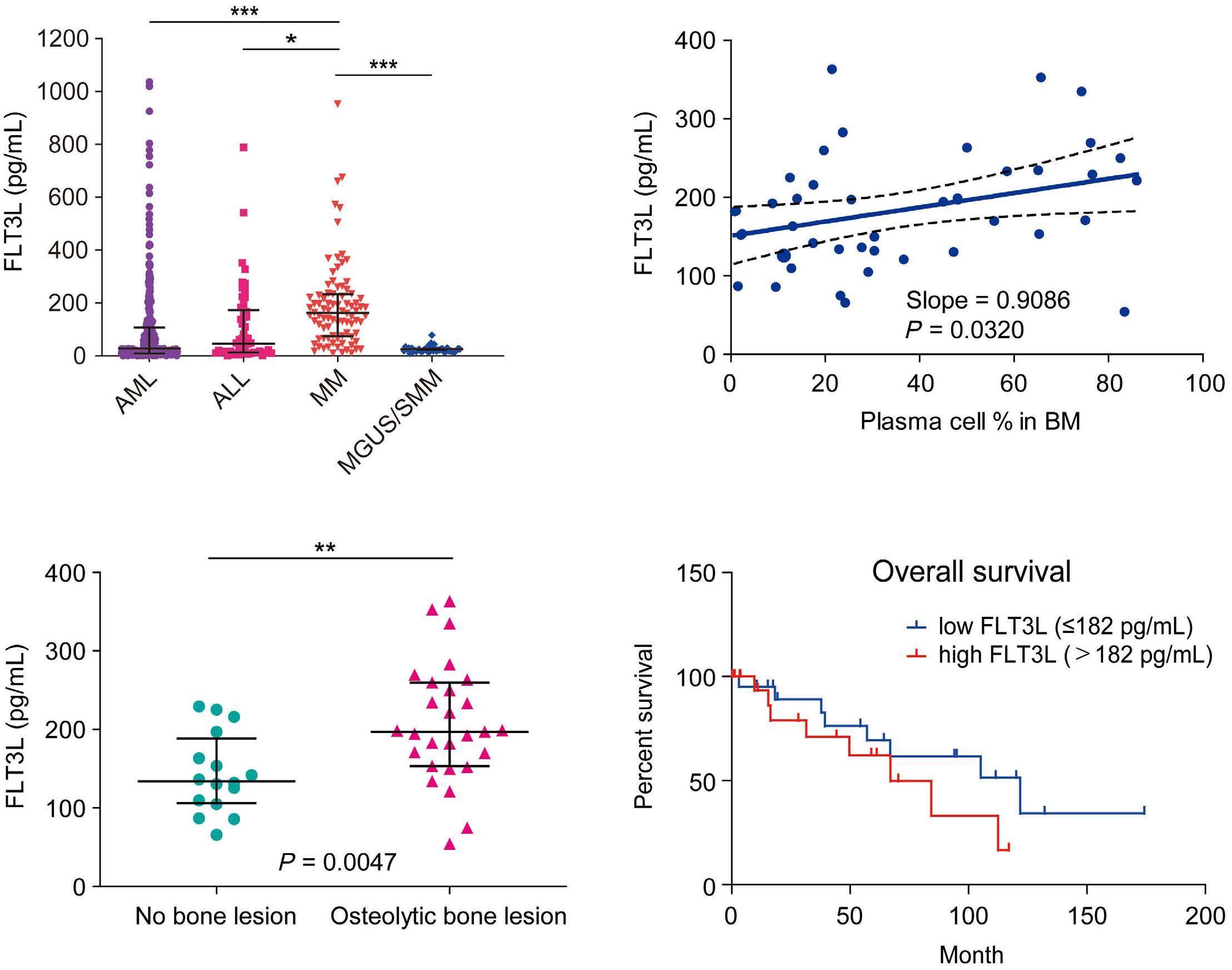
Figure 1. Plasma expression of Fms-like tyrosine kinase 3 ligand is elevated in multiple myeloma patients. (A) The plasma level of Fms-like tyrosine kinase 3 ligand (FLT3L) in bone marrow (BM) is significantly higher in multiple myeloma (MM) (N=86; median 161.8 pg/mL, interquartile range [IQR] 73.96-233.5) than in acute myeloid leukemia (AML) (N=306; median 28.16, IQR 8.48-107.3), acute lymphoblastic leukemia (ALL) (N=52; median 46.11, IQR 12.52-172.8), and monoclonal gammopathy of undetermined significance (MGUS) / smoldering multiple myeloma (SMM) (N=42). (B) The plasma level of FLT3L in BM of MM shows a linear correlation with the percentage of plasma cell / total nucleated cells in BM aspirates at the time of initial diagnosis (N=43). (C) The plasma level of FLT3L in BM is higher in MM with osteolytic bone lesion (N=27, median 194.70, IQR, 152.52-242.27) than in MM without bone lesion (N=16, median 141.62 pg/mL, IQR, 109.57-215.98) at the time of initial diagnosis (P=0.0047). (D) Kaplan-Meier survival curves for MM patients with low and high FLT3L expression levels. *P<0.05, **P<0.01, ***P<0.001, ns: not significant.
ering the infrequent occurrence of osteolytic bone lesions in ALL and AML, these findings indicate a potential association between FLT3L and bone osteolysis in MM.
FLT3L levels in BM aspirates and plasma were also reported to increase through MM progression and reach the maximal in patients with relapsed/refractory MM.19 However, we were not able to determine a statistically meaningful increase in FLT3L in relapse/refractory MM or in disease status in our cohort (Online Supplementary Figure S1A-C). The observed heterogeneity in FLT3L levels could be due to a possible discrepancy in the sample collection (collection time and sample status). As an alternative measure to reflect tumor burden and aggressiveness of tumor microenvironment, we thus determined the percentage of plasma cells among total
nucleated cells and then examined the association of the plasma cell percentage with FLT3L levels (N=43). The percentage of plasma cells showed a significant (P=0.032) positive correlation with the FLT3L level (Figure 1B), supporting the view that the FLT3L level increases through MM progression in Korean patients. To further investigate the association of the FLT3L level with bone osteolysis, we next grouped the 43 MM patients for whom the FLT3L levels were measured into 27 patients with osteolytic bone lesion and 16 patients without the lesion. The FLT3L level was significantly (P<0.01) elevated in patients with osteolytic bone lesion (median 194.70, IQR, 152.52-242.27) compared with those in patients without the bone lesion (median 141.62 pg/mL, IQR, 109.57215.98) (Figure 1C). Moreover, we performed a multivariate
Table 2. Prognostic factors for overall survival of treatment-naïve multiple myeloma (N=43)
Anemia
Hypercalcemia
Azotemia
Hypoalbuminemia
Osteolytic
logistic regression analysis using FLT3L as a bi-categorical dependent variable (≤182 pg/mL vs. >182 pg/mL) and clinical factors as independent variables in the treatment-naïve MM patients (N=43). This analysis showed that the percentages (%) of BM plasma cell and hyperdiploidy were significantly associated with FLT3L in MM (Online Supplementary Table S1), consistent with the finding in Figure 1B. This analysis suggests the associations of serum M-protein level and the presence of osteolytic lesions with FLT3L levels (P=0.071, 0.073, and 0.069, respectively). These data thus imply that these 4 factors may collectively contribute to FLT3L levels. We next examined the association of the FLT3L level with the OS of the 43 patients. The high level of FLT3L in BM (>182 pg/ mL) was significantly (P=0.018) associated with poor survival
in multivariate analysis (Hazard Ratio [HR]=65.96, 95% Confidence Interval [CI]: 2.06-2109.60), as indicated by the median survivals of 121.8 months in the low FLT3L group (N=22, ≤182 pg/mL) and 67.03 months in the high FLT3L group (N=21, >182 pg/mL) (Figure 1D). Interestingly, many clinical parameters, such as age, subtype of heavy and light chain, cytogenetics of hypo- and hyperdiploidy, anemia, hypercalcemia, azotemia, and hypoalbuminemia, were not significantly associated with OS (Table 2). On the other hand, sex tended to show a marginal (P=0.078) correlation with survival in multivariate analysis, and International Staging System (ISS) advanced stage (III) was a poor prognostic factor compared to stage I (HR=3.85 and 813.31, 95% CI: 0.83-17.88 and 6.60-100259.2; P=0.086 and 0.006 in univariate and multivariate analyses,
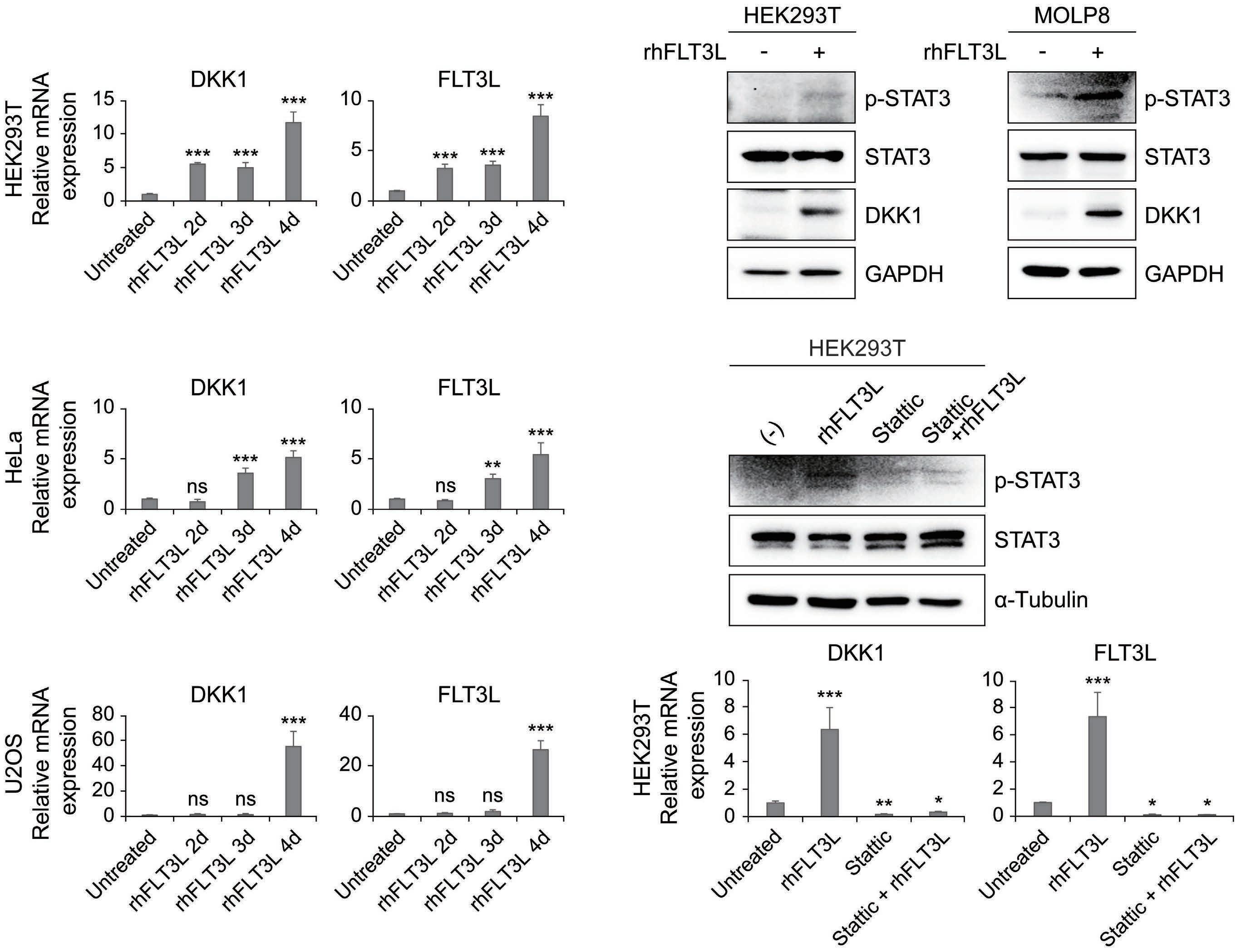
Figure 2. Fms-like tyrosine kinase 3 ligand enhances Dick Kopf-related protein 1 expression via STAT3 signaling. (A) The mRNA expression of Dick Kopf-related protein 1 (DKK1) and Fms-like tyrosine kinase 3 ligand (FLT3L) obtained from HEK293T, HeLa, and U2OS cells treated with 40 ng/mL rhFLT3L for from two to approximately four days, as analyzed by quantitative real-time polymerase chain reaction (RT-qPCR) (N=4). (B) Western blot analysis of phosphorylation of STAT3, total STAT3 and DKK1 under treatment with rhFLT3L (40 ng/mL) in HEK293T and MOLP8 cells for three days. (C) In HEK293T on treatment with rhFLT3L (40 ng/ mL), Stattic (10 μM), or both for three days, protein level of phosphorylated STAT3 and total STAT3 and mRNA level of DKK1 and FLT3L were analyzed by western blotting and RT-qPCR, respectively (N=8). Statistical analysis was performed by one-way ANOVA with Dunnett’s multiple comparison. Error bars indicate the standard deviation of 3 independent replicates. *P<0.05, **P<0.01, ***P<0.001, ns: not significant (P>0.05).
respectively). Taken together, these data suggest that the FLT3L level can serve as a promising factor that reflects both osteolytic bone lesion and prognosis in MM.
FLT3L enhances DKK1 expression via STAT3 signaling
It has been reported that expression of the DKK1 gene, a soluble inhibitor for WNT signaling, is significantly elevated in plasma cells from patients presenting with MM, and the enhanced DKK1 expression in turn leads to defects in osteoblast differentiation.12 However, the molecular basis of how DKK1 expression is enhanced in MM has not been determined. As both FLT3L and DKK1 are increased in MM patients, we hypothesized that expression of DKK1 might be positively regulated by FLT3 signaling. To test this hypothesis, we treated FLT3L to HEK293T, HeLa, and U2OS cells and measured the DKK1 transcription levels using RT-qPCR. Intriguingly, cells treated with FLT3L exhibited the increased DKK1 transcript levels (Figure 2A). The DKK1 transcript level increased over time during the culture of up to four days. Consistent with previous reports,26,27 we confirmed that FLT3L transcript is also enhanced upon FLT3L treatment (Figure 2A, right).28 Next, we attempted to elucidate the molecular basis of the increased DKK1 expression through FLT3L signaling. FLT3L signaling was reported to be mediated by STAT3 phosphorylation.29,30 We consistently found that STAT3 was activated by phosphorylation at tyrosine 705 in HEK293T and MOLP8, an MM cell line, upon treatment of FLT3L (Figure 2B). Again we were able to observe enhanced DKK1 expression in response to FLT3L treatment by western blot analysis (Figure 2B). In order to confirm that the enhanced DKK1 expression is mediated by STAT3 phosphorylation, we treated the cell with STAT3 signaling inhibitor, Stattic, and found that treatment of Stattic significantly reduced FLT3L-mediated STAT3 phosphorylation (Figure 2C, top). RT-qPCR showed that the inhibition of STAT3 results in a significant reduction in DKK1 expression (Figure 2C, bottom). These results suggest that FLT3L signaling activates the DKK1 transcription through STAT3 phosphorylation.
FLT3L-induced DKK1 attenuates WNT signaling to reduce osteogenesis
We next tested whether the enhanced DKK1 expression impairs WNT signaling, which can result in reduced osteogenic activity. To this end, we examined the b-catenin activity in HEK293T cells in response to DKK1 and/or FLT3L. It was seen that DKK1 actually reduced the b-catenin protein level, which became further exacerbated when FLT3L was added (Figure 3A). Correspondingly, FLT3L treatment led to reduced nuclear translocation of b-catenin (Figure 3B). To further confirm this FLT3L-mediated reduction of WNT signaling at the cellular level, we performed a WNT reporter assay using the construct in which the expression of luciferase was under the promoter containing b-catenin responsible elements. As expected, the reporter activity was reduced on FLT3L treatment, which was much further reduced when
DKK1 was added (Figure 3C). In order to validate the impact of FLT3L on osteoblast differentiation at the cellular level, we performed alkaline phosphatase (ALP) staining analysis, which has been widely used for evaluating osteogenesis. We found a significant reduction in ALP staining and activity in the mouse pre-osteoblast cell line, MC3T3-E1, following treatment with FLT3L or DKK1, while the addition of BMP2 enhanced the osteoblast differentiation (Figure 3D, E). In addition, RT-qPCR with the cells in the same setting showed that treatment of FLT3L significantly enhanced DKK1 expression at the transcriptional level (Figure 3F), demonstrating the detrimental effect of FLT3L on osteogenesis. To further explore the relationship of the FLT3L-DKK1 pathway with osteolytic bone lesions, we next measured DKK1 level by ELISA in a subgroup of MM patients whose BM plasma samples were available (N=35). FLT3L and DKK1 levels tended to show a positive correlation in MM patients with osteolytic bone lesion (Adjusted [Adj] R2 0.0922, P=0.1809), but a negative correlation in MM patients without osteolytic bone lesion (Adj R2 = -0.0183) (Online Supplementary Figure S2A, B). Next, we explored whether FLT3L would be also implicated in osteoclastogenesis, which could lead to bone loss. To this end, we established a TRAP assay using mouse BM and found that FLT3L treatment does not affect osteoclastogenesis (Online Supplementary Figure S3A, B). Taken together, these findings demonstrate that osteolytic bone lesions mediated by the FLT3L-DKK1 pathway is due to the reduced osteogenesis through the inhibited WNT signaling, and not to the enhanced osteoclastogenesis.
FLT3L and DKK1 are highly expressed in malignant plasma cells with hyperdiploidy
It has been reported that patients with hyperdiploidy (HY) showed the elevated level of DKK1, but only about 59% of these patients had osteolytic bone lesions.12 This suggests that only a subset of patients show osteolytic bone lesion mediated by the FLT3L-DKK1 pathway. To explore this subset of patients, we obtained the previously reported mRNA expression profiles of plasma cells from 414 patients with MM (GSE265822 in GEO database). When the gene expression profiles from all patients were used, we found virtually no correlation (Spearman’s correlation = -0.02) in mRNA expression levels of FLT3L and DKK1 (Online Supplementary Figure S4A). We then examined mRNA expression patterns of FLT3L and DKK1 across the previously defined 7 subtypes of MM patients and found that the HY subtype showed the highest median mRNA expression level of both FLT3L and DKK1 (Figure 4A). Moreover, the HY subtype showed the largest percentage (43.10%) of patients showing high (≥ median expression level in all samples) expression levels of both FLT3L and DKK1 compared with the other subtypes (Figure 4B, Online Supplementary Figure S4B). Correspondingly, the HY subtype had the smallest percentage (6.03%) of patients with low expression levels of FLT3L and DKK1. These results suggest that HY subtype may represent a subset of patients
with FLT3L-DKK1 pathway-mediated bone osteolysis. To understand the characteristics of the HY subtype, we next identified 662 genes predominantly up-regulated in the HY subtype (Online Supplementary Figure S5A) and then examined cellular pathways represented by these genes using ConsensusPathDB31 (Figure 4C). These genes defining the HY subtype were significantly (P<0.05) associated with FLT3 and JAK-STAT signaling pathways, consistent with our
findings (see Figure 2). We further divided the HY subtype patients into those with high levels of both FLT3L and DKK1 (FLT3LHDKK1H in Figure 4B) and the others (FLT3LHDKK1L, FLT3LLDKK1H and FLT3LLDKK1L), and identified 984 differentially expressed genes (DEG; 471 up-regulated and 513 down-regulated in FLT3LHDKK1H) between the 2 subgroups. The up-regulated genes most strongly represented cytokine response, and also, significantly, other pathways previously
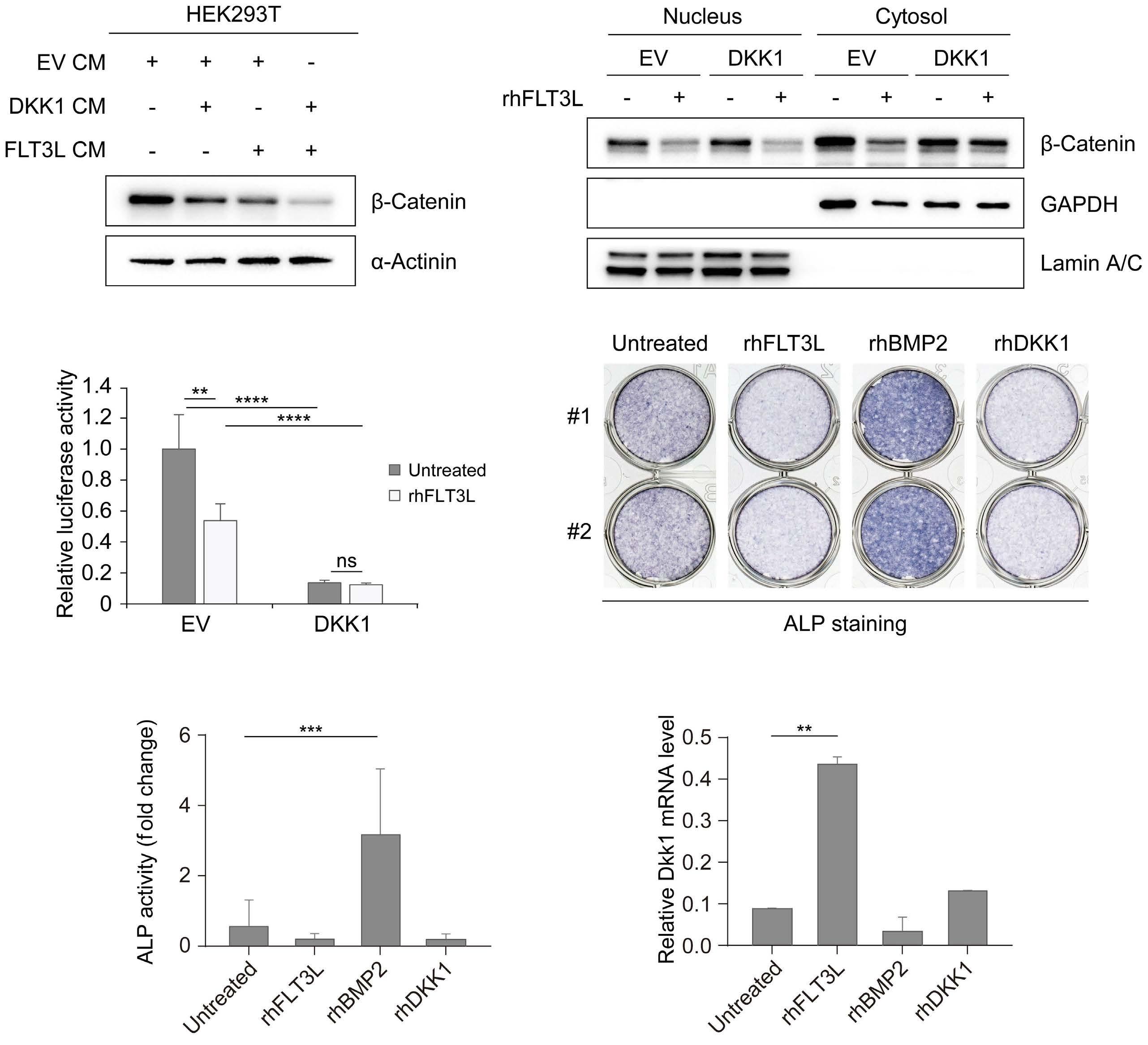
Figure 3. Fms-like tyrosine kinase 3 ligand-induced Dick Kopf-related protein 1 attenuates WNT/b-catenin signaling. (A) Conditioned media (CM) obtained from HEK293T transiently transfected with DNA construct, such as Dick Kopf-related protein 1 (DKK1), Fms-like tyrosine kinase 3 ligand (FLT3L), or both were treated into newly HEK293T cells for three days. Expression of b-catenin protein from cell lysates was analyzed by western blot assay. (B) EV (empty vector) and DKK1 transiently expressing HEK293T cells were treated with rhFLT3L (40 ng/mL) for three days. Nuclear and cytoplasmic fraction from the cell pellets was separated by Extraction kit. Protein levels of b-catenin and GAPDH (cytosol) and Lamin A/C (Nucleus) as loading controls were determined by western blotting. (C) Reporter gene assay for b-catenin-mediated transcriptional activation was conducted in HEK293T transiently transfected with plasmids carrying EV or DKK1 under treatment of rhFLT3L (40 ng/mL) for three days (N=4). (D) Alkaline phosphatase staining and (E) activity were evaluated with MC3T3-E1 cells treated with mock, rhFLT3L, rhBMP2 or rhDKK1 for seven days (N=6). (F) Relative mRNA expression level of DKK1 was determined by qRT PCR. mRNA was prepared from the MC3T3-E1 cells treated with mock, rhFLT3L, rhBMB2 or rhDKK1 for seven days (n=3). Error bars indicate the standard deviation of at least 3 independent replicates. ALP: alkaline phosphatase **P<0.01, ***P<0.001, ****P<0.0001, ns: not significant (P>0.05)
reported to be associated with bone lesion or loss (Figure 4D, left: VEGF,32 mTOR,33 TNF10 and MAPK34 signaling pathways).
In contrast, the down-regulated genes were associated with bone growth and its associated pathways (Figure 4D, right: cell development, response to EGF, ECM organization, calcium signaling, and bone resorption).
To focus on the cytokine response, we identified 9 secretory proteins among the DEG and found that BMP5, NPY, and
POSTN were up-regulated in FLT3LHDKK1H while CALCA, COMP, CTSK, CRYAB, NTF3, and TNC35 were down-regulated (Online Supplementary Figure S5B). Among them, 4 down-regulated genes (CALCA, 36 CTSK, 37 CRYAB, 38 and TNC35) are known to be associated with WNT signaling and osteogenesis, and showed the lowest expression levels in FLT3LHDKK1H across all subtypes of MM, suggesting that they may serve as potential effector cytokines leading to reduced bone osteogenesis by
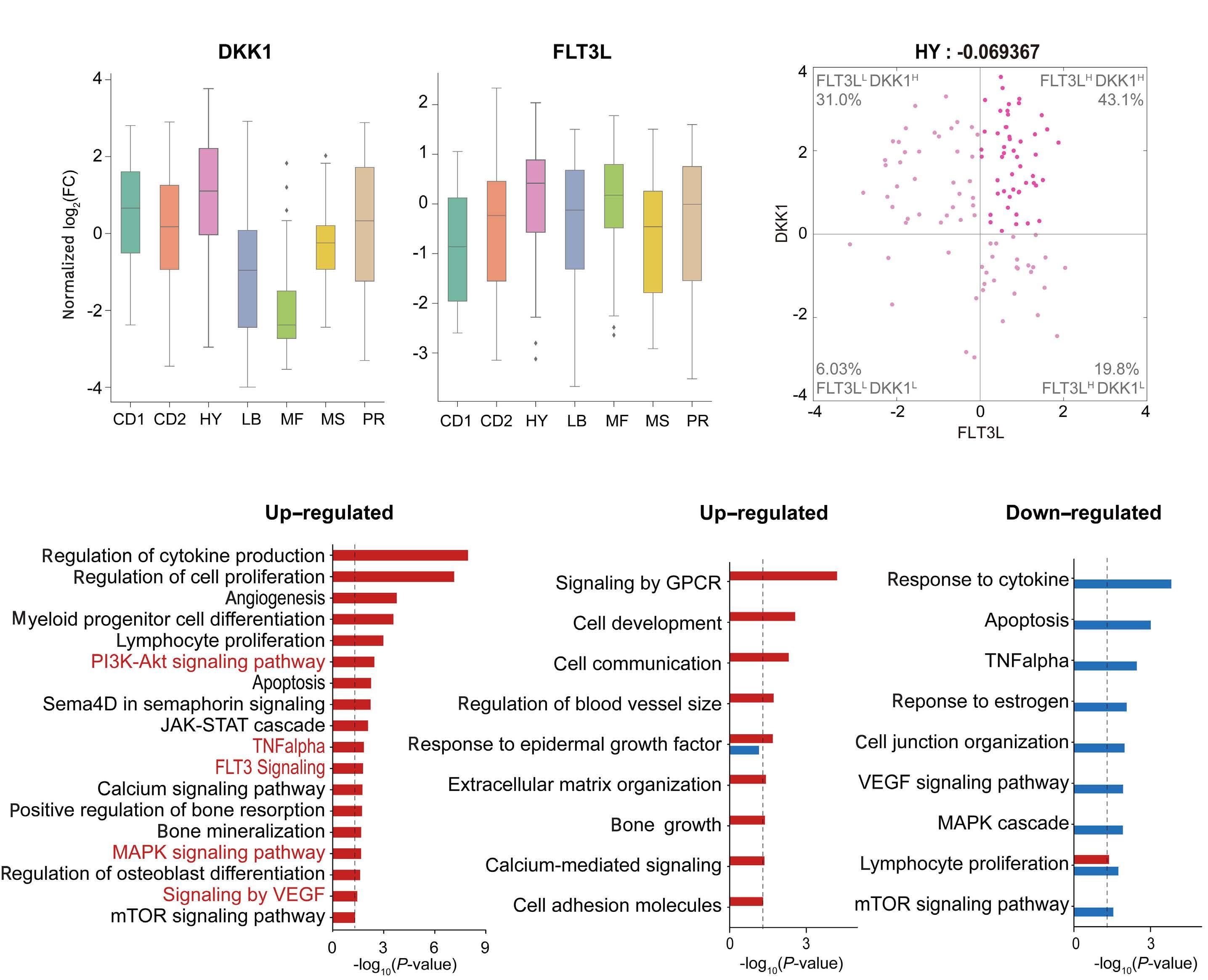
Figure 4. Fms-like tyrosine kinase 3 ligand and Dick Kopf-related protein 1 are highly expressed in plasma cells of multiple myeloma patients with hyperdiploidy subtype. (A) Boxplots showing distributions of log2-fold-changes of mRNA expression levels of Fms-like tyrosine kinase 3 ligand (FLT3L) and Dick Kopf-related protein 1 (DKK1) across 7 subtypes (CD1, CD2, HY, LB, MF, MS and PR) reported by Zhan et al 50 Log2-fold changes were computed with respect to the median mRNA expression value in all samples. (B) Scatterplot showing log2-fold changes of mRNA expression levels between FLT3L (x-axis) and DKK1 (y-axis) in patients (N=116) of the hyperdiploidy (HY) subtype. Spearman’s correlation between FLT3L and DKK1 (top) and the percentage of patients in each quadrant are displayed in the HY subtype. Median expression value for each gene was used to define high expression of the gene. (C) Gene ontology biological processes (GOBP) and pathways represented by the signature genes for the HY subtype. (D) GOBP and pathways represented by the up-regulated (left) or down-regulated (right) genes in the HY subgroup with high expression of both FLT3L and DKK1 (FLT3LHDKK1H) compared to the other subgroups. Significance (P-value) of the GOBP and pathways being enriched by the genes is displayed as -log10(P-value). Cut-off of enrichment P-value (0.05) is indicated by the line. Pathways used to build a network model are highlighted. GPCR: G protein coupled receptors.
FLT3L-DKK1 pathway. In summary, a network model (Online Supplementary Figure S6) shows that FLT3L binds to its receptor FLT3, and FLT3 then activates STAT3 signaling to induce DKK1, which inhibits WNT signaling to decrease its target genes involved in bone formation.
Osteolytic bone lesions greatly diminish the QoL for advanced MM patients. Unfortunately, the molecular mechanisms underlying the development of these bone lesions have not yet been determined, which poses a challenge for the development of effective therapeutic strategies. In this study, we aimed to address this knowledge gap by examining plasma samples derived from the BM of 86 MM patients, 306 AML patients, and 52 ALL patients. Our investigation revealed that the expression of FLT3L in MM patients was significantly higher compared to AML and ALL patients. Notably, elevated FLT3L levels were found to be positively correlated with a poorer prognosis in MM patients. In vitro analysis demonstrated that FLT3L-mediated STAT signaling pathways contribute to the upregulation of DKK1 expression, a soluble antagonist for the WNT signal cascade. Given the role of WNT signaling in osteoblastic differentiation, these findings suggest that the FLT3L-STAT3-DKK1 axis is involved in the development of osteolytic bone lesions in MM patients. Furthermore, our analysis of previously collected transcriptome data revealed that the activation of the FLT3L-STAT3-DKK1 pathway predominantly occurs in the HY subtype of MM. This finding holds significant promise for improving the accuracy of bone osteolysis prediction and treatment, as it provides a potential diagnostic marker to identify the involvement of the FLT3L-STAT3-DKK1 pathway. Taken together, these findings provide novel insights into the molecular mechanisms underlying osteolytic bone lesions in MM patients. By uncovering the FLT3L-STAT3-DKK1 axis as a key pathway, these studies shed light on the new opportunities for the diagnosis of MM and the development of potential therapeutic interventions for MM patients. Hyperdiploidy has been seen to be a good prognostic factor in MM,39,40 while, in our study, high FLT3L was associated with osteolytic bone lesion and poor survival. To explore the relationship between FLT3L, HY, and FLT3L-DKK1-mediated bone osteolysis, we performed multivariate logistic regression analysis. In this analysis, FLT3L served as a dependent variable, while clinical factors including HY and osteolytic bone lesions were treated as independent variables. We noticed that FLT3L was not only associated with hyperdiploidy, which is known as a good prognostic factor (P=0.044), but also associated with plasma cell fraction in BM, a surrogate marker of disease stage (P=0.029) (Online Supplementary Table S1). These findings may appear inconsistent with the claim in the current study that HY-MM subtype presumably represented FLT3L-DKK1 pathway-mediated pathological
features, leading to subsequent deterioration in the QoL of the affected patients. However, recent studies have revealed that even among patients with the HY-MM subtype there is a considerable cytogenetic heterogeneity, resulting in distinct clinical outcomes and diverse prognoses.39,41,42 Indeed, in our study, the enrichment of the FLT3LHDKK1H group among HYMM subtype patients led to discernible differences in transcriptomic profiles compared to the other HY-MM patients (Figure 4F). Therefore, detailed functional relationships of FLT3L-DKK1 pathway with the previously established MM subtypes, along with its potential use as biomarkers for MM prognosis prediction, still have to be clarified. Phase III randomized clinical trials proved that pamidronate and zoledronate, the 2 most commonly used bisphosphonates in the clinic, reduce bone osteolysis in MM.28,43 However, the prolonged use of these bisphosphonates can cause the serious complication of bisphosphonate-related osteonecrosis of jaw (BRONJ) or subtrochanteric femoral insufficiency.44,45 Although denosumab, a RANKL inhibitor recently approved for bone-directed adjunctive therapy for MM, was proven to be more effective than the bisphosphonates,46 the side effect of BRONJ has also been reported in patients treated with denosumab. The addition of bisphosphonate46,47 or RANKL-targeted monoclonal antibody to the standard chemotherapy regimens can lessen bone resorption by suppressing osteoclastic activity, but these agents cannot fully inhibit the progression of the bone osteolytic process. Furthermore, the recovery of osteolytic lesions during treatment is rarely observed, which means that the current strategy to target osteoclastic activity in MM patients with bone involvement has still not been successfully achieved. Thus, it is important to develop alternative or complementary therapies that can target reduced osteoblastic activity that can, therefore, be used together with existing therapeutic options to improve the treatment of bone osteolysis in patients with MM.
New effective bone-directed therapeutic strategies need to be developed to overcome the adverse effect of the current standard therapies and improve MM patients’ QoL. Targeting the FLT3L-STAT3-DKK1 pathway that primarily reduces the osteoblastic activity via suppression of WNT signaling might be a promising therapeutic approach because it can restore the reduced osteoblastic activity. Inhibition of FLT3L can thus provide a complementary therapeutic axis to the existing bisphosphonates and RANKL inhibitors that mainly target the osteoclastic activity. Targeting both osteoblastic and osteoclastic activities using a combined therapy of FLT3L/ FLT3 inhibitors,48 bisphosphonates, RANKL inhibitor and/or the standard chemotherapy are expected to effectively improve therapeutic outcomes for MM patients with osteolytic bone lesions. As a downstream regulator of FLT3 signaling, DKK1 induces inhibition of WNT signaling in osteoblastic differentiation in MM.49 DKK1 inhibitors have been proposed as a promising therapeutic strategy to target the reduced osteoblastic activity in MM. The benefits and disadvantag-
es of FLT3L/FLT3 inhibitors over DKK1 inhibitors should be further investigated in a large cohort of patients. We enrolled patients diagnosed with MM from 2004 to 2012, a period in which the ISS still had to gain popularity among clinicians, so we could not collect information from the ISS in our cohort. Nevertheless, we think that our information aligns well, not with the ISS, but with the old Durie-Salmon staging system, since the ISS does not include information about bone osteolysis, while the Durie-Salmon staging system does. Enrichment of patients having high expression of both FLT3L and DKK1 in the HY subtype of MM suggested that FLT3L-mediated bone osteolysis might be prominent in the HY subtype. Accordingly, therapies to inhibit the FLT3L-mediated bone osteolysis might be most effective for patients in the HY subtype. These subtype-associated predictions should be verified in plasma cells derived from a large cohort of patients with MM.
Disclosures
No conflicts of interest to disclose.
Contributions
YK, SSY and DH designed and supervised the studies. DS recruited, treated, and followed the enrolled patients together with SSY. DS collected BM-derived plasma cells from patients.
1. Turesson I, Bjorkholm M, Blimark CH, Kristinsson S, Velez R, Landgren O. Rapidly changing myeloma epidemiology in the general population: Increased incidence, older patients, and longer survival. Eur J Haematol. 2018;101(2):237-244.
2. Sneyd MJ, Gray AR, Morison IM. Trends in survival from myeloma, 1990-2015: a competing risks analysis. BMC Cancer. 2021;21(1):821.
3. Dimopoulos MA, Delimpasi S, Katodritou E, et al. Significant improvement in the survival of patients with multiple myeloma presenting with severe renal impairment after the introduction of novel agents. Ann Oncol. 2014;25(1):195-200.
4. Denaro V, Denaro L, Albo E, Papapietro N, Piccioli A, Di Martino A. Surgical management of spinal fractures and neurological involvement in patients with myeloma. Injury. 2016;47(Suppl 4):S49-S53.
5. Body JJ, Pereira J, Sleeboom H, et al. Health resource utilization associated with skeletal-related events: results from a retrospective European study. Eur J Health Econ. 2016;17(6):711-721.
6. Kim C, Bhatta S, Cyprien L, Fonseca R, Hernandez RK. Incidence of skeletal-related events among multiple myeloma patients in the United States at oncology clinics: observations from realworld data. J Bone Oncol. 2019;14:100215.
7 Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: a report from International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863-2869.
8. Morgan GJ, Davies FE, Gregory WM, et al. Long-term follow-up of MRC Myeloma IX trial: survival outcomes with
DS, DK, CL and KSA measured cytokines with DK, CL and KSA. MJK performed most of the in vitro experiments. EJ, BCL and DK performed the TRAP assay. SC analyzed transcriptome database. YK, DS, DH and SSY wrote the draft of the manuscript and all authors revised the final version for publication.
Acknowledgments
We are grateful to the patients and their families for their participation in this study. The authors would like to thank the members of Genome Maintenance at Sookmyung Women’s University, Seoul, South Korea, for their invaluable comments and suggestions.
Funding
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIP) (NRF-2023R1A2C3007266, NRF-2021R1A6A1A03038890 to YK and RS-2023-00239194 to MJK). This research was partially supported by Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Education (N. 2021R1A6C101A564).
Data-sharing statement
All the original data, reagents and protocols are available upon request.
bisphosphonate and thalidomide treatment. Clin Cancer Res. 2013;19(21):6030-6038.
9. Musolino C, Allegra A, Innao V, Allegra AG, Pioggia G, Gangemi S. Inflammatory and anti-inflammatory equilibrium, proliferative and antiproliferative balance: the role of cytokines in multiple myeloma. Mediators Inflamm. 2017;2017:1852517.
10 Terpos E, Ntanasis-Stathopoulos I, Gavriatopoulou M, Dimopoulos MA. Pathogenesis of bone disease in multiple myeloma: from bench to bedside. Blood Cancer J. 2018;8(1):7.
11. Raje N, Roodman GD. Advances in the biology and treatment of bone disease in multiple myeloma. Clin Cancer Res. 2011;17(6):1278-1286.
12. Tian E, Zhan F, Walker R, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349(26):2483-2494.
13. Dun X, Jiang H, Zou J, et al. Differential expression of DKK-1 binding receptors on stromal cells and myeloma cells results in their distinct response to secreted DKK-1 in myeloma. Mol Cancer. 2010;9:247.
14 Yuan Y, Guo M, Gu C, Yang Y. The role of Wnt/b-catenin signaling pathway in the pathogenesis and treatment of multiple myeloma (review). Am J Transl Res. 2021;13(9):9932-9949.
15. Glass DA 2nd, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751-764.
16. van Andel H, Kocemba KA, Spaargaren M, Pals ST. Aberrant Wnt signaling in multiple myeloma: molecular mechanisms and targeting options. Leukemia. 2019;33(5):1063-1075.
17 Politou MC, Heath DJ, Rahemtulla A, et al. Serum
concentrations of Dickkopf-1 protein are increased in patients with multiple myeloma and reduced after autologous stem cell transplantation. Int J Cancer. 2006;119(7):1728-1731.
18. Yu F, Yu C, Li F, et al. Wnt/b-catenin signaling in cancers and targeted therapies. Signal Transduct Target Ther. 2021;6(1):307.
19 Steiner N, Hajek R, Sevcikova S, et al. High levels of FLT3-ligand in bone marrow and peripheral blood of patients with advanced multiple myeloma. PLoS One. 2017;12(7):e0181487.
20. Kokonozaki M, Kanellou P, Pappa CA, et al. Serum levels of soluble FLT3 ligand in patients with active multiple myeloma constitute marker of bone marrow plasma cell proliferative activity. Crit Rev Oncog. 2017;22(3-4):255-262.
21. Anandasabapathy N, Breton G, Hurley A, et al. Efficacy and safety of CDX-301, recombinant human Flt3L, at expanding dendritic cells and hematopoietic stem cells in healthy human volunteers. Bone Marrow Transplant. 2015;50(7):924-930.
22. Uckun FM. Overcoming the immunosuppressive tumor microenvironment in multiple myeloma. Cancers (Basel). 2021;13(9):2018.
23. Hayakawa F, Sugimoto K, Harada Y, et al. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013;3(11):e166.
24. Chérel M, Gouard S, Gaschet J, et al. 213Bi radioimmunotherapy with an anti-mCD138 monoclonal antibody in a murine model of multiple myeloma. J Nucl Med. 2013;54(9):1597-1604.
25. Cho H. Diagnosis and management of monoclonal gammopathy of clinical significance. Blood Res. 2022;57(S1):20-26.
26. Zheng R, Levis M, Piloto O, et al. FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood. 2004;103(1):267-274.
27. Zhou J, Bi C, Janakakumara JV, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009;113(17):4052-4062.
28. Berenson JR, Lichtenstein A, Porter L, et al. Efficacy of pamidronate in reducing skeletal events in patients with advanced multiple myeloma. Myeloma Aredia Study Group. N Engl J Med. 1996;334(8):488-493.
29 Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3Ldependent dendritic cell differentiation. Immunity. 2003;19(6):903-912.
30 Chauvistré H, Küstermann C, Rehage N, et al. Dendritic cell development requires histone deacetylase activity. Eur J Immunol. 2014;44(8):2478-2488.
31. Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB--a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37(Database issue):D623-628.
32. Gau YC, Yeh TJ, Hsu CM, Hsiao SY, Hsiao HH. Pathogenesis and treatment of myeloma-related bone disease. Int J Mol Sci. 2022;23(6):3112.
33. Liu Z, Liu H, Li Y, et al. Adiponectin inhibits the differentiation and maturation of osteoclasts via the mTOR pathway in multiple myeloma. Int J Mol Med. 2020;45(4):1112-1120.
34 Liu H, He J, Yang J. Tumor cell p38 MAPK: a trigger of cancer bone osteolysis. Cancer Cell Microenviron. 2015;2(1):e464.
35. Beiter K, Hiendlmeyer E, Brabletz T, et al. beta-Catenin regulates the expression of tenascin-C in human colorectal tumors. Oncogene. 2005;24(55):8200-8204.
36. Zhou R, Yuan Z, Liu J, Liu J. Calcitonin gene-related peptide promotes the expression of osteoblastic genes and activates the WNT signal transduction pathway in bone marrow stromal stem cells. Mol Med Rep. 2016;13(6):4689-4696.
37. Bonnet N, Brun J, Rousseau JC, Duong LT, Ferrari SL. Cathepsin K controls cortical bone formation by degrading periostin. J Bone Miner Res. 2017;32(7):1432-1441.
38. Zhu B, Xue F, Li G, Zhang C. CRYAB promotes osteogenic differentiation of human bone marrow stem cells via stabilizing b-catenin and promoting the Wnt signalling. Cell Prolif. 2020;53(1):e12709.
39. Chng WJ, Kumar S, Vanwier S, et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007;67(7):2982-2989.
40. Smadja NV, Bastard C, Brigaudeau C, Leroux D, Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood. 2001;98(7):2229-2238.
41. Weinhold N, Kirn D, Seckinger A, et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101(3):e116-119.
42. Chretien ML, Corre J, Lauwers-Cances V, et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood. 2015;126(25):2713-2719.
43. Musto P, Petrucci MT, Bringhen S, et al. A multicenter, randomized clinical trial comparing zoledronic acid versus observation in patients with asymptomatic myeloma. Cancer. 2008;113(7):1588-1595.
44 Raje N, Woo SB, Hande K, et al. Clinical, radiographic, and biochemical characterization of multiple myeloma patients with osteonecrosis of the jaw. Clin Cancer Res. 2008;14(8):2387-2395.
45. Park-Wyllie LY, Mamdani MM, Juurlink DN, et al. Bisphosphonate use and the risk of subtrochanteric or femoral shaft fractures in older women. JAMA. 2011;305(8):783-789.
46. Raje N, Terpos E, Willenbacher W, et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, doubledummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018;19(3):370-381.
47. Himelstein AL, Foster JC, Khatcheressian JL, et al. Effect of longer-interval vs standard dosing of zoledronic acid on skeletal events in patients with bone metastases: a randomized clinical trial. JAMA. 2017;317(1):48-58.
48. Gebru MT, Wang HG. Therapeutic targeting of FLT3 and associated drug resistance in acute myeloid leukemia. J Hematol Oncol. 2020;13(1):155.
49 Qiang YW, Barlogie B, Rudikoff S, Shaughnessy JD Jr. Dkk1induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone. 2008;42(4):669-680.
50 Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020-2028.
Verónica González-Calle,1* Paula Rodríguez-Otero,2* Anna Sureda,3 Felipe de Arriba,4 Marta Reinoso,5 Paz Ribas,6 Ana Pilar González-Rodríguez,7 Yolanda González,8 Albert Oriol,9 Joaquín Martínez-López,10 Marta Sonia González,11 Miguel T. Hernández,12 Maialen Sirvent,13 Teresa Cedena,10 Noemí Puig,1 Bruno Paiva,2 Joan Bladé,14 Juan José Lahuerta,10 Jesús F. San-Miguel2 and María-Victoria Mateos1
1Department of Hematology, University Hospital of Salamanca, CAUSA, IBSAL and Center for Biomedical Research in Network of Cancer, CIBERONC, Salamanca; 2Cancer Center Clínica Universidad de Navarra, CIMA, IDISNA, CIBERONC, Pamplona; 3Serve d’Hematologia Clínica, Institut Català D’Oncologia-L’Hospitalet. IDIBELL, Universitat de Barcelona, Barcelona; 4Hospital Morales Meseguer, IMIB-Pascual Parrilla, Universidad de Murcia, Murcia; 5Hospital Universitario Virgen del Rocío, Sevilla; 6Hospital Universitario Dr. Peset, Valencia; 7Hospital Universitario Central de Asturias, Oviedo; 8Institut Catalá D’Oncologia Girona, Girona; 9Institut Catalá D’Oncologia Badalona, Badalona; 10Hospital Universitario 12 de Octubre, Madrid; 11Complejo Hospitalario Universitario de Santiago, La Coruña; 12Hospital Universitario de Canarias, Tenerife; 13Hospital Donostia, San Sebastián and 14Hospital Clinic i Provincial de Barcelona, IDIBAPS, Barcelona, Spain
*VG-C and PR-O contributed equally as first authors.
Correspondence: P. Rodriguez-Otero paurodriguez@unav.es
Received: September 20, 2023. Accepted: February 2, 2024. Early view: February 15, 2024.
https://doi.org/10.3324/haematol.2023.284089
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
The treatment landscape for multiple myeloma has significantly evolved in the last decade. Notwithstanding, a large proportion of patients continue to relapse and novel combinations continue to be needed. In this phase II study, selinexor, a first-in-class inhibitor of exportin-1 was evaluated in combination with standard daratumumab-bortezomib-dexamethasone (DVd), for the treatment of relapsed and refractory multiple myeloma (RRMM). The aim of the trial was to assess the efficacy and safety of the combination of selinexor with DVd (S-DVd).
A total of 57 patients were enrolled in the two parts of the study. Part 1 enrolled a heavily pretreated population with at least three prior lines (PL) of therapy and part 2 enrolled an early relapse population with at least one PL of therapy. The primary endpoint was complete response (CR) rate in part 2 and overall response rate (ORR) in part 1. In the latter, 24 patients were treated with a median of three PL. Overall response rate (ORR) was 50% with two CR. Median progression-free survival (PFS) was 7 months. In part 2, 33 patients were enrolled, with a median of one PL. ORR was 82% and CR or better was 33%. Median PFS was 24 months. In lenalidomide-refractory patients, a median PFS of 22.1 months was observed. Thrombocytopenia was the most common hematological adverse event (69%; grade 3-4: 34%) and nausea, the most frequent non-hematological adverse event (38%; grade 3-4: 6%). Sixty-two percent of the patients required dose modifications. In summary, although the primary endpoint of the study was not met, the combination of S-DVd showed encouraging clinical efficacy with a generally manageable safety profile representing a potential option for the treatment of RRMM patients.
The translation of innovative drugs from bench to patient has allowed continued improvement in survival in multiple myeloma (MM) patients.1 Despite this benefit, many patients with MM will eventually relapse and become multi-drug resistant highlighting the need of developing new drugs and combinations based on drugs with novel mechanisms of action.
Selinexor is a first-in-class oral inhibitor of a nuclear export protein called exportin 1 (XPO-1). By blocking this protein, most tumor suppressor proteins remain in the nucleus, resulting in cell cycle arrest and death of malignant plasma cells.2,3 Selinexor showed anti-MM activity in preclinical studies leading to the development of several clinical trials evaluating selinexor in combination with different antimyeloma drugs.4 Currently, selinexor is approved in different settings and combinations. Selinexor in combination with dexamethasone (Sd) is approved for penta-refractory MM adult patients who have received at least four prior lines (PL) of therapies, based on the positive results of the STORM trial.5 In addition, selinexor is approved in combination with bortezomib and dexamethasone (SVd) for relapsed and refractory MM (RRMM) patients who have already received at least one PL of treatment based on efficacy and safety results of the BOSTON trial.6
Current treatment guidelines for RRMM patient, particularly in those lenalidomide (len)-refractory, recommended the use of proteasome inhibitors (PI)-based combinations together with anti-CD38 monoclonal antibodies, such as daratumumab or isatuximab.7,8 Mechanisms of action of daratumumab include induction of apoptosis, immune-mediated actions and immunomodulatory functions, resulting in deep responses and prolonged survival when combined with immunomodulatory drugs (IMID), or PI, allowing even its combination in quadruplets with an acceptable toxicity in the upfront setting.9,10 One of the approved combinations for RRMM after one PL is daratumumab plus bortezomib and dexamethasone (DVd), based on results of the phase III CASTOR trial, where an improvement in the progression-free survival (PFS) as compared with bortezomib and dexamethasone (Vd) was demonstrated.11 Based on this background, we hypothesize that the addition of selinexor may improve the efficacy of DVd in a well tolerated manner. Here we present the results
Table 1. Treatment schedule in the clinical trial GEM-Selibordara.
of a phase II clinical trial conducted by the Spanish myeloma group (GEM/PETHEMA) to investigate the efficacy and safety of the quadruplet selinexor plus DVd (S-DVd) in RRMM patients.
An open-label, non-randomized, multicenter, phase II study to evaluate the efficacy and safety of S-DVd in RRMM patients (GEM-SELIBORDARA trial). The study was designed by GEM/PETHEMA and carried out in 14 Spanish hospitals from July 2018 to March 2021. The cut-off date was February 6, 2023. The study had two different parts. In part 1, eligible patients had to have received at least three PL of therapy, including a PI (bortezomib) and any IMID and to be refractory to the last line of therapy or double refractory as per IMWG definition.12 In part 2, eligible patients had received at least one PL. Prior treatment with PI was allowed if prior response, no grade ≥3 related-toxicity, and a washout of at least 6 months. Prior therapy with selinexor or anti-CD38 monoclonal antibodies was not permitted (see protocol in the Online Supplementary Appendix).
Patients received DVd (with intarvenous daratumumab) at the approved dose and schedule, in combination with selinexor days 1, 8, 15 and 22 (100 mg in part 1 and 60 mg in part 2) (Table 1). Treatment was given until disease progression or unacceptable toxicity. Recommended supportive treatment to minimize nausea was 5-HT3 receptor antagonists (Online Supplementary Appendix).
All patients provided written informed consent. The protocol was approved by the Spanish Health Authorities and a central ethical committee. The study was conducted according to the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice (ICHGCP). The trial was registered at clinicaltrials gov. Identifier: NCT03589222.
The primary endpoint of the trial was to evaluate the efficacy of the combination S-DVd in terms of overall response rate (part 1) and complete response rate (CR) or better in part 2, according to IMWG criteria.13 Additional study endpoints are included in Online Supplementary Appendix.
Daratumumab (16 mg/kg IV/SC) weekly C1 and C2, C3-C6 Q2W, C7+ Q4W
Bortezomib (1.3 mg/m2 SC): d1, 8, 15 & 22 (C1-C8), d1 & 15 (C9+).
Selinexor 100 mg d1, 8, 15 & 22
4-week duration cycles
Dexamethasone 40 mg d1, 8, 15 & 22
Selinexor 60 mg d1, 8, 15 & 22
5-week duration cycles
Treatment was continued until disease progression or unacceptable toxicity
IV: intravenously; SC: subcutaneously, C: cycle; Q2W: every 2 weeks; Q4W: every 4 weeks; d: day.
Statistical analysis
The sample size was estimated based on the primary endpoint of the study. For part 1, the hypothesis was that we would be able to increase the ORR from 30% with daratumumab monotherapy14 and 45% with SVd in patients refractory to bortezomib15 to 70% with the quadruplet combination of SDVd. With 57 patients we would obtain statistical power of approximately 90% with a one-sided α error of 0.05. For part 2, the primary endpoint was CR rate. The CR rate reported with the SVd in patients with RRMM after one to three PL was 13%. Our hypothesis was that by adding daratumumab the CR rate could increase up to 35%. With 52 patients we would obtain a statistical power of approximately 90% to reject the null hypothesis, with a predefined α error of 0.05 (one-sided). Considering
a percentage of withdrawals of 10%, the final sample size would be 57 patients. The response rate was determined using an intent-to-treat (ITT) analysis. The statistical analysis done is detailed in the Online Supplementary Appendix.
Efficacy in part 1
Twenty-four patients were included in part 1 (Figure 1). Median age was 66 years (range, 44-76). Seventeen percent of the patients had an Eastern Cooperative Oncology Group (ECOG) performance status score of 2. Eight (35%) patients had an International Staging System (ISS) staging score III, three (16%) a R-ISS staing score III, and five of 21
Table 2. Patient baseline characteristics.
Part 1 N=24 Part 2 N=33
Age in years, median (range) 66 (44-76) 69 (33-81)
>75, N (%) 2 (8.3) 9 (27.3)
Male, N (%) 13 (54.2) 20 (60.6)
ECOG, N (%)
0 12 (50.0) 16 (48.5)
1 8 (33.3) 16 (48.5)
2 4 (16.7) 1 (3.0)
MM subtype, N (%)
IgG 13 (54.2) 21 (63.6)
IgA 6 (25.0) 8 (24.2)
IgD 0 1 (3.0)
Bence-Jones 5 (20.8) 3 (9.1) ISS, N (%)
I 4 (17.4) 5 (18.5)
II 11 (47.8) 15 (55.6) III 8 (34.8) 7 (25.9)
Unknown 1 6
R-ISS, N (%)
I 0 2 (8.0) II 16 (84.2) 19 (76.0) III 3 (15.8) 4 (16.0)
Unknown 5 8 High LDH, N (%) 4 (28.6) 4 (14.8)
HRCA positive/studied, N/N (%) 5/19 (26.3) 5/26 (19.2) del(17p) 5/21 (23.8) 4/26 (15.4)
t(4;14) 0/20 1/26 (3.7)
t(14;16) 0/19 0/26
1q gain 13/20 (65.0) 18/29 (62.1)
1p loss 1/19 (5.0) 2/24 (8.3)
Extramedullary disease, N (%) 3 (12.5) 3 (9.1)
Median number of PL (range) 3 (2-3) 1 (1-3)
1 (%) - 20 (60.6)
2 (%) 7 (29.2) 5 (15.1)
3 (%) 17 (70.8) 8 (24.2)
Prior IMID, N (%)
Yes 24 (100) 27 (81.8)
No 0 4 (18.2)
Type of prior IMID, N (%)
Thalidomide 13 (54.2) 15 (45.5)
Len 24 (100) 23 (71.8)
Pomalidomide 9 (37.5) 6 (18.7)
Prior PI, N (%)
Yes
24 (100) 30 (90.9)
No 0 3 (9.1)
Type of prior PI, N (%)
Bortezomib 24 (100) 30 (90.9)
Carfilzomib 11 (45.8) 4 (12.1)
Ixazomib 0 (0) 0 (0)
Refractory to the last line of therapy, N (%) 24 (100) 12 (36.4)
Refractory to len, N (%) 23 (95.8) 15 (45.5)
Refractory to PI, N (%) 17 (70.8) 5 (15.1)
Double refractory len/PI, N (%) 17 (70.8) 4 (12.1)
Prior ASCT, N (%) 7 (29.2) 15 (45.5)
MM: multiple myeloma; ISS: International Staging System; R-ISS: revised International Staging System; LDH: lactate dehydrogenase; HRCA: highrisk cytogenetic abnormalities; IMID: immunomodulatory drugs; ECOG: Eastern Cooperative Oncology Group; PL: prior line; PI: proteasome inhibitor; len: lenalidomide; ASCT: autologous stem cell transplantation.
patients had del(17p). The median number of PL was three (range, 2-3). Twenty-three (96%) patients were refractory to len and 17 (71%), double refractory. All patients were exposed to both PI and IMID and refractory to the last line of treatment (Table 2).
In the ITT population, CR or better was achieved in two patients (8%). ORR was 50% (Figure 2). Median time to response was 1.8 months (range, 1.3-7.3) and median DoR was 13.5 (range, 2.8-48.2). No differences in responses according to PL of therapy were detected (Figure 3A). At the time of data cutoff, 21 (87.5%) patients had discontinued treatment (Figure 1). The main reason for treatment discontinuation was disease progression (17 patients). One patient discontinued due to adverse events (AE) and another patient withdrawn informed consent after one cycle. After a median follow-up of 39.7 months (range, 12.2-51.0), 22 (91.7%) progressed or died with a median PFS of 7.2 months (95% confidence interval [CI]: 3.6-10.8) (Figure 4A). Fourteen (58.3%) patients died, and median OS was 28.5 months (95% CI: 0.0-57.9) (Figure 4B). No differences were observed in PFS or OS according to number of PL.
In part 2, 33 patients were included, with a median age of 69 years (range, 33-81). Forty-eight percent had an ECOG of 0 or 1. Seven (26%) patients had an ISS III, four (16%) R-ISS III, and five (19%) patients had high-risk cytogenetic abnormalities. Median number of PL was one (range, 1-3). Twenty-seven (82%) were previously exposed to IMID; and 30 (91%) to PI. Fifteen patients (45%) were refractory to len, and 12 (36%) were refractory to the last line of treatment (Table 2). Regarding efficacy of the ITT population, 11 patients (33%) achieved CR. Measurable residual disease (MRD) negativity was reported in nine patients (27%). ORR was 82% (Figure 2). Median time to response was 1.8 months (range, 1.2-4.1) and median DOR was 22.4 (range, 3.3-30.8). Best responses were achieved after a median of three cycles (range, 1-13). Numerically higher CR rates were found in patients after one PL or two PL as compared to three PL (45%, 40%, 0%, respectively) (P=not significant [NS]) (Figure 3A). CR rates were consistent among several subgroups analyzed including len-refractory (27% vs. 39%; P=NS), R-ISS III or HR-CA.
At the time of the data cutoff and with a median follow-up of 27.0 months (range, 19.7-32.5), 19 (57.6%) patients had progressed or died and the median PFS was 25.1 months (95% CI: 16.0-34.2) (Figure 4A). Eleven (33.3%) patients died, and median OS was not reached i.e., not estimable (NE) (95%
Figure 2. Response rates in part 1 and part 2 of the study. ITT: intention-to-treat; ORR: overall response rate; CR: complete response; VGPR: very good partial response; PR: partial response.
Figure 3. Detail of responses in the study. Panel (A) represents response rate in the intention-to-treat (ITT) population in part 1 and 2 of the study according to the number of prior lines (PL) of treatment. Panel (B) represents responses according to refractoriness to lenalidomide (len) (note: only 1 patient in part 1 was not refractory to len (len-ref), best response in this patient was minor response). ORR: overall response rate; CR: complete response; VGPR: very good partial response; PR: partial response.
Figure 4. Kaplan-Meier survival curves in part 1 and 2 of the study. Panel (A) shows progression-free survival for (PFS) the intention-to-treat (ITT) population in part 1 and part 2. Panel (B) shows overall survival for the ITT population in part 1 and part 2. Panel (C) shows PFS for patients according to number of prior lines (PL) of therapy; Panel (D) shows PFS according to lenalidomide refractoriness (len-ref) in part 2 of the trial.
CI: NE-NE), with a 2-year OS rate of 71.7% (Figure 4B). In the post hoc subgroup analysis, median PFS for those relapsing after one PL, two PL and three PL were 27.5 months (95% CI: 22.3-32.7), 31.6 months (95% CI: NE-NE), and 12.3 months (95% CI 3.2-21.3), respectively (Figure 4C). Median OS for these patients according to PL of treatment was not reached (95% CI: NE-NE), 31.6 months (95% CI: NE-NE) (P=0.8), and 22.8 months (95% CI: 15.7-29.9) (P=0.1), respectively. Notably, PFS (22.8 months vs. 31.6 months, hazard ratio [HR] =1.5; 95% CI: 0.6-3.7; P=0.4) and OS (30.0 months vs. not reached, HR=1.4; 95% CI: 0.4-4.8; P=0.6) were comparable between patients refractory and not refractory to len, respectively (Figure 4D). Importantly, no differences were found across other subgroups like ISS staging score, R-ISS or the presence of high-risk cytogenetic abnormalities but this comparison is limited due to small sample size.
Hematological AE were the most frequently reported AE in both parts of the study, overall, 47 (82.4%) of patients had at least one hematological AE. Thrombocytopenia was the most common AE present in 70.2% of patients (grade 3-4 in 45.6%) followed by neutropenia in 38.6% of the patients (grade 3-4 in 29.8%). Infection was the most frequent non-hematological AE and occurred in 42 (73.6%) of patients (grade 3-4 in 31.6%). Regarding gastrointestinal AE, diarrhea was reported in 38.6% (grade 3-4: 3.5%) and nausea or vomiting in 35.1% (grade 3-4: 8.8%). Peripheral neuropathy occurred in ten (17.5%) patients, without any grade 3 or 4 events. No differences in safety have been observed between part 1 or 2 of the study. There was a trend toward a higher percentage of patients with thrombocytopenia (83.3% vs. 60.6%, respectively; P=0.06) and
neutropenia (50.0% vs. 30.3%; P=0.1) in part 1. Similar rates of infection were observed, with seven cases with grade 3-4 infection in part 1 and nine in part 2, with two fatal cases occurred in the study (1 patient in each part due to septic shock) (Table 3). Overall, one patient in part 1 discontinued the treatment due to toxicity (severe cytopenias persisting beyond the cycle 1).
Median number of cycles received in part 1 of the study
was 7 (range, 1-47) and 17 (range, 1-27) in part 2. Cumulative doses of selinexor, daratumumab, bortezomib and dexamethasone delivered are depicted in Table 4, and no statistically significant differences among these cumulative doses administered between part 1 and part 2 of the study were found. Regarding dose modifications, selinexor was the drug most frequently modified and was reduced in 30 (52.7%) patients, (10 dose reductions in part 1 and 20
Table 3. Incidence of most common treatment-related adverse events.
ITT: intention-to-treat population; AE: adverse events.
Table 4. Cycles, cumulative doses of selinexor, daratumumab, bortezomib and dexamethasone (S-DVd) and dose modifications in GEM Selibordara trial.
Part 1: weekly selinexor dose 100 mg in 4-week cycles. Part 2: weekly selinexor dose
in part 2; P=0.2) and discontinued in eight (14%) patients (5 in part 1 and 3 in part 2; P= 0.2). Bortezomib was the second drug most commonly modified and was reduced in 15 (26.3%) and withdrawn in four (7.0%) (3 in part 1 and 1 in part 2; P=0.3), patients. Dexamethasone was reduced in ten (17.5%) patients (5 in each part) and withdrawn in two (3.5%) patients belonging to part 1 of the study. No patient discontinued daratumumab permanently. Only one patient discontinued daratumumab treatment temporarily due to a grade 4 cytopenia and restarted therapy after recovery (Table 4).
The addition of selinexor to DVd (S-DVd) resulted in encouraging clinical efficacy with a safety profile generally consistent with known safety profile of selinexor. Higher efficacy was observed when S-DVd was administrated in earlier lines of treatment and, also in len-refractory patients. The primary endpoint of the study was CR rate. If we analyze the ITT population, as a whole, the CR rate was 22%, although, it is important to acknowledge that 22 of 57 patients enrolled were heavily pretreated and 52% previously exposed to bortezomib. Thus, although the trial did not meet its primary endpoint potentially due to the changes in the study design and mixed patient populations, the CR rate in part 2 here presented seems encouraging. In part 1, the population included was consistent with the population enrolled in the pivotal studies that the led to the approval of daratumumab monotherapy (mostly double refractory and daratumumab-naïve).16,17 In this trial, increased efficacy was observed with the addition of selinexor plus bortezomib to daratumumab (median PFS 7,2 months as compared with the 3.7 months PFS reported for daratumumab single agent). However, the major interest of our trial regards part 2 since the clinical characteristics of the patients were closer to those treated with DVd in the phase III CASTOR trial (almost half [49%] of patients received only 1 PL of therapy, and one third were refractory to last line of treatment).18,19 In this context, the addition of selinexor to the backbone DVd in the present trial may have led to a deepening in the responses as compared to CASTOR in terms of MRD-negative rate in the ITT population (27% vs. 15%,20 respectively), translating into potentially longer PFS for S-DVd compared to the DVd-treated population in the CASTOR trial (median PFS of 25.1 vs. 16.7 months, respectively, or 18 months for patients with one or more PL in the CASTOR trial).19,21 Furthermore, 45% of the S-DVd-treated patients were len-refractory, whereas only 24% in the DVd arm of the CASTOR trial, and interestingly, in these subsets the PFS also appeared to be longer with S-DVd versus DVd (median PFS of 22.8 vs. 7.8 months).21 As previously mentioned, len-refractory patients remained a difficult to treat pop-
ulation and carfilzomib + anti-CD38 monclonal antibodies (MoAb) are becoming standard of care in this setting based on positive results of the phase III CANDOR and IKEMA studies, respectively. These studies were also conducted in a similar RRMM population after one to three PL of therapy, apparently obtaining a similar ORR (84-87%) and CR rates (29-40%) as in part 2 of GEM Selibordara trial, but with a slightly underrepresented population of len-refractory patients in both CANDOR and IKEMA studies (33%).22–25 Still, PFS was longer in both IKEMA and CANDOR trial as compared to the trial here presented. The safety profile is different, and the intravenous formulation of carfilzomib has its drawbacks. In this sense, the availability of regimens such as S-DVd could be an alternative for patients with cardiovascular comorbidities, when there is a preference for subcutaneous and oral administration or in places where the combination of anti-CD38 plus carfilzomib and dexamethasone is not yet available.
In addition, it is worth mentioning that in the phase III BOSTON trial the SVd combination was investigated for patients with RRMM after one PL based.6 In this scenario, the addition of daratumumab to SVd may also appear to improve the depth of responses compared to SVd alone (CR or better: 33% vs. 17%; MRD negativity: 82% vs. 17%), and, as a result, also to prolong PFS (median PFS: 25.1 months vs. 13.9 months), despite a high proportion of prior bortezomib-exposed patients in part 2 of GEM Selibordara (91%) as compared to the BOSTON trial (69%). Moreover, focusing on len-refractory patients, deeper responses were also seen in the current trial as compared to the BOSTON study (ORR: 73% vs. 68%, and CR or better: 27% vs. 9%,), as well as longer PFS (median 22.8 vs. 10.2 months), suggesting a potential role of this quadruplet combination in this setting.
Regarding safety, incidence of treatment related AE were generally consistent with other studies with regimens containing selinexor, daratumumab, bortezomib and dexamethasone, such as SVd and DVd. A similar incidence of hematological AE such as thrombocytopenia or anemia was observed, although a higher incidence of neutropenia was reported in this trial compared to the BOSTON trial, likely related to the addition of daratumumab, but overall grades 3-4 were similar.6,11 With regard to non-hematological AE, infections were the most frequently observed with S-DVd, although it is true that not only respiratory infections have been considered, which in the BOSTON and CASTOR studies reached an incidence of 48-68%. Asthenia or fatigue were similar to that reported in the BOSTON trial. Regarding gastrointestinal toxicity, it was consistent with what was reported with SVd in the BOSTON trial, and, of note, loss of appetite was observed in only 5.3% of participants, probably due to the prophylactic measures implemented in the GEM Selibordara protocol. Importantly the discontinuation rate due to AE was lower although a significant proportion of patients did require dose modifications to
manage treatment related AE.
Our study has several limitations. First, it is a phase II non-randomized trial, with a limited number of patients, and, some of the prior comparisons are limited by different confounders such as differences in study design, methods, or patient’s characteristics. Indeed, the weekly bortezomib schedule may explain the lower incidence of peripheral neuropathy seen in the GEM-Selibordara trial (21%, grade 3-4: 0%) as compared to the CASTOR trial (50%, grade 3-4: 4.5%).11 Regarding selinexor dosing, in the part 2 the schedule and dose was adjusted (60 mg every week in a 5-week cycle) to improve tolerability. As a result, there were fewer discontinuations as compared to part 1, suggesting that 60 mg weekly of selinexor might be the optimal dose for the combination with DVd in a well balanced manner between efficacy and toxicity.26
In summary, encouraging efficacy and generally manageable safety were observed in this phase II multicenter trial evaluating S-DVd in RRMM patients, including in patients who were len-refractory, suggesting this combination could be an attractive option for len-refractory but daratumumab-sensitive patients if cardiovascular comorbidities are a concern or oral formulations are preferred.
VG-C discloses consulting for or advisory role at Janssen; speakers’ bureau at Janssen, GlaxoSmithKline, Pfizer, Bristol Myers Squibb/Celgene, travel and accommodation expenses from Janssen and GlaxoSmithKline. PR-O discloses honoraria derived from consulting or advisory board role at Celgene-BMS, Janssen, Roche, Abbvie, Pfizer, GSK, Sanofi and H3Biomedicine; steering committee membership at Celgene-BMS, Regeneron and Janssen; speaker’s bureau at Janssen, Celgene-BMS, GSK, Sanofi and Abbvie; travel grant from Pfizer. FA discloses honoraria for lectures and advisory board from Janssen, BMS-Celgene, Amgen, GSK, Sanofi and Takeda. AO discloses honoraria from consultant activities for Amgen, Celgene, BMS, GSK, Janssen and Sanofi. JM-L discloses honoraria from consulting activities for Astellas Pharma, BMS, F.Hoffman-La Roche, Janssen, Novartis and Sanofi; research grant from BMS. MVM discloses honoraria derived from lectures and advisory role at Amgen, Celgene, BMS, GSK, Janssen, Pfizer, Regeneron, Sanofi and Takeda. M-TH discloses consulting or advisory role at Janssen, Sanofi/ Aventis, Celgene/Bristol Myers Squibb and GlaxoSmithKline; speakers’ bureau at Janssen, Celgene/Bristol Myers Squibb
1. Puertas B, González-Calle V, Sobejano-Fuertes E, et al. Novel agents as main drivers for continued improvement in survival in multiple myeloma. Cancers (Basel). 2023;15(5):1558.
2. Vogl DT, Dingli D, Cornell RF, et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol. 2018;36(9):859-866.
and GlaxoSmithKline; research funding from Celgene/Bristol Myers Squibb (Inst). M-TC discloses honoraria from Janssen. NP discloses honoraria from Amgen, Celgene, Janssen, Takeda and The Binding Site; consulting or advisory role at Amgen, Celgene, Janssen and Takeda; speakers’ bureau of Celgene; research funding from Celgene, Janssen, Amgen and Takeda; travel and accommodation expenses from Amgen, Celgene, Janssen and Takeda. BP discloses honoraria for lectures and advisory boards from Adaptive, Amgen, BMS-Celgene, Gilead, GSK, Janssen, Oncopeptides, Roche, Sanofi and Takeda; unrestricted grants from Beigene, BMS-Celgene, EngMab, GSK, Roche, Sanofi and Takeda; consultancy for BMS-Celgene, Janssen and Sanofi. JB discloses honoraria for lectures and advisory boards from Janssen, BMS-Celgene, Amgen, Takeda and Oncopeptides. JJL discloses consulting or advisory role at Celgene, Amgen, Janssen-Cilag and Sanofi; travel and accommodation expenses from Celgene and Pfizer. JSM discloses consulting activies at Abbvie, Amgen, BMS, Celgene, F.Hoffman-La Roche, GSK, HaemaLogiX, KaryoPharm, Merck, Novartis, Pfizer, Regeneron, Sanofi-Aventis, Takeda and SecuraBio. MVM discloses honoraria for lectures and advisory boards from Janssen, Celgene, Takeda, Amgen, GSK, Abbvie, Pfizer, Regeneron, Roche, Sanofi and Oncopeptides. The remaining authors have no conflicts of interest to disclose.
MVM and JSM designed the study and wrote the protocol. VG-C and PR-O made the statistical analysis and wrote the manuscript. All authors contributed to enrolling patients in the clinical trial and provided data. All author reviewed and approved the manuscript.
The Spanish Myeloma Group is open to the possibility of sharing the data used in this study for research projects as long as they do not interfere with the present or future objectives of the clinical trial. The interest and feasibility of any clinical or biological research proposal based on the data from this study must be approved by the board of directors of the Spanish Myeloma Group. In such a case, the data, deposited in REDCap, will be presented in anonymized CSV format. This availability is subject to the laws and provisions in force that regulate the development of clinical trials both in Spain and in the European Union. Data are available on request from the corresponding author.
3. Syed YY. Selinexor: first global approval. Drugs 2019;79(13):1485-1494.
4. Chen C, Siegel D, Gutierrez M, et al. Safety and efficacy of selinexor in relapsed or refractory multiple myeloma and Waldenstrom macroglobulinemia. Blood. 2018;131(8):855-863.
5. Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor-
dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019;381(8):727-738.
6. Grosicki S, Simonova M, Spicka I, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-perweek bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. Lancet. 2020;396(10262):1563-1573.
7 Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma: EHA-ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021;32(3):309-322.
8. Moreau P, Kumar SK, San Miguel J, et al. Treatment of relapsed and refractory multiple myeloma: recommendations from the International Myeloma Working Group. Lancet Oncol. 2021;22(3):e105-e118.
9 Mateos M V, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528.
10 Voorhees PM, Sborov DW, Laubach J, et al. Addition of daratumumab to lenalidomide, bortezomib, and dexamethasone for transplantation-eligible patients with newly diagnosed multiple myeloma (GRIFFIN): final analysis of an open-label, randomised, phase 2 trial. Lancet Haematol. 2023;10(10):e825-e837.
11. Sonneveld P, Chanan-Khan A, Weisel K, et al. Overall survival with daratumumab, bortezomib, and dexamethasone in previously treated multiple myeloma (CASTOR): a randomized, open-label, phase III trial. J Clin Oncol. 2023;41(8):1600-1609.
12. Rajkumar S V, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-548.
13. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346.
14 Usmani SZ, Nahi H, Weiss BM, et al. Safety and efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed and refractory multiple myeloma: final results from GEN501 and Sirius. Blood. 2017;130(Suppl 1);3107.
15. Grosicki S, Simonova M, Spicka I, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-perweek bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. Lancet. 2020;396(10262):1563-1573.
16. Usmani SZ, Nahi H, Plesner T, et al. Daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma: final results from the phase 2 GEN501 and SIRIUS trials. Lancet Haematol. 2020;7(6):e447-e455.
17 Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. The Lancet 2016;387(10027):1551-1560.
18. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
19 Spencer A, Lentzsch S, Weisel K, et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of CASTOR. Haematologica. 2018;103(12):2079-2087.
20 Weisel KC, Sonneveld P, Mateos M-V, et al. Efficacy and safety of daratumumab, bortezomib, and dexamethasone (D-Vd) versus bortezomib and dexamethasone (Vd) in first relapse patients (pts) with multiple myeloma (MM): four-year update of Castor. Blood. 2019;134(Suppl 1):3192.
21. Mateos M-V, Sonneveld P, Hungria V, et al. Daratumumab, bortezomib, and dexamethasone versus bortezomib and dexamethasone in patients with previously treated multiple myeloma: three-year follow-up of CASTOR. Clin Lymphoma Myeloma Leuk. 2020;20(8):509-518.
22. Dimopoulos M, Quach H, Mateos M-V, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197.
23. Usmani SZ, Quach H, Mateos M-V, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2022;23(1):65-76.
24. Moreau P, Dimopoulos M-A, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet. 2021;397(10292):2361-2371.
25. Martin T, Dimopoulos M-A, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in patients with relapsed multiple myeloma: updated results from IKEMA, a randomized Phase 3 study. Blood Cancer J. 2023;13(1):72.
26. Mikhael J, Noonan KR, Faiman B, et al. Consensus recommendations for the clinical management of patients with multiple myeloma treated with selinexor. Clin Lymphoma Myeloma Leuk. 2020;20(6):351-357.
Carfilzomib, thalidomide, and dexamethasone are safe
refractory
Slavisa Ninkovic,1,2 Simon J. Harrison,3,4 Je-Jung Lee,5,6 Nick Murphy,7 Jae Hoon Lee,8 Jane Estell,9 Vivien M. Chen,9,10 Noemi Horvath,11 Kihuyn Kim,12 Richard Eek,13 Bradley Augustson,14 Soo-Mee Bang,15 Shang-Yi Huang,16 Rajeev Rajagopal,17 Ferenc Szabo,18 Daniel Engeler,19 Belinda E. Butcher,20,21 Peter Mollee,22,23 Brian Durie,24 Wee Joo Chng25# and Hang Quach1,2#
1Department of Hematology, St. Vincent’s Hospital Melbourne, Melbourne, Australia; 2Faculty of Medicine, University of Melbourne, St. Vincent’s Hospital Melbourne, Melbourne, Australia; 3Department of Hematology, Peter MacCallum Cancer Center and Royal Melbourne Hospital, Melbourne, Australia; 4Sir Peter MacCallum Department of Oncology, University of Melbourne, Melbourne, Australia; 5Department of Hematology-Oncology, Chonnam National University Hwasun Hospital, Hwasun, South Korea; 6Chonnam National University Medical School, Hwasun, South Korea; 7Department of Hematology, The Royal Hobart Hospital, Hobart, Australia; 8Department of Hematology, Gachon University Gil Medical Center, Incheon, South Korea; 9Department of Hematology, Concord Repatriation General Hospital, Concord, Australia; 10Faculty of Medicine and Health, University of Sydney, Sydney, Australia; 11Department of Hematology, Royal Adelaide Hospital, Adelaide, Australia; 12School of Medicine, Samsung Medical Center, Seoul, South Korea; 13Border Medical Oncology Research Unit, Albury Wodonga Regional Cancer Center, Albury, Australia; 14Hematology Cancer Care, Sir Charles Gairdner Hospital, Perth, Australia; 15Department of Internal Medicine, Seoul National University Bundang Hospital, Seongnam, South Korea; 16Department of Medicine, National Taiwan University, Taipei, Taiwan; 17Department of Hematology, Middlemore Hospital, Auckland, New Zealand; 18Department of Hematology, Royal Darwin Hospital, Darwin, Australia; 19Australian Leukemia and Lymphoma Group, Melbourne, Australia; 20Biostatistics and Medical Writing, WriteSource Medical Pty Ltd, Sydney, Australia; 21School of Biomedical Sciences, University of New South Wales, Sydney, Australia; 22Department of Hematology, Princess Alexandra Hospital, Brisbane, Australia; 23School of Medicine, University of Queensland, Brisbane, Australia; 24Samuel Oschin Comprehensive Cancer Institute, CedarsSinai Outpatient Cancer Center, Los Angeles, CA, USA and 25Department of HematologyOncology, National University Cancer Institute, Singapore, Singapore
#WJC and HQ contributed equally as senior authors.
Correspondence: H. Quach hang.quach@svha.org.au
Received: September 19, 2023.
Accepted: January 5, 2024. Early view: January 18, 2024.
https://doi.org/10.3324/haematol.2023.284238
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
This multicenter, phase II study of the Australasian Lymphoma and Leukemia Group and the Asian Myeloma Network investigated fixed-duration (18-month) treatment with carfilzomib (K), thalidomide (T), and dexamethasone (d) (KTd) in patients with relapsed and/or refractory multiple myeloma who had received one to three prior lines of therapy. Patients received induction with up to 12 28-day cycles of carfilzomib (20 mg/m2 intravenously in cycle 1 on days 1 and 2, then 56 mg/m2 [36 mg/m2 for patients ≥75 years] from day 8 onwards), thalidomide 100 mg orally in the evening and weekly dexamethasone 40 mg (20 mg for patients ≥75 years). During maintenance, thalidomide was omitted, while carfilzomib was continued on days 1, 2, 15, and 16 with fortnightly dexamethasone. The primary endpoint was progression-free survival. Secondary endpoints were overall response rate, overall survival, duration of response, safety, and tolerability. Ninety-three patients (median age 66.3 years [range, 41.9-84.5]) were enrolled and followed up for a median of 26.4 months (range, 1.6-54.6). The median pro-
gression-free survival was 22.3 months (95% confidence interval: 15.7-25.6) and the 2-year progression-free survival was 46.3% (95% confidence interval: 35.1-52.8). The median overall survival was not reached and the 2-year overall survival was 73.8% (95% confidence interval: 62.9-81.9). The overall response rate was 88% (73% had a very good partial response or better). There was no difference in the depth of response, progression-free survival or overall survival comparing Asian and non-Asian cohorts (P=0.61). The safety profile of KTd was consistent with that of each individual drug. KTd is well tolerated and effective in patients with relapsed and/or refractory multiple myeloma irrespective of Asian or non-Asian ethnicity and provides an alternative treatment option, particularly in circumstances in which the use of carfilzomib, lenalidomide, and dexamethasone (KRd) is limited by access, cost, or renal impairment.
Multiple myeloma is a neoplasm of clonal plasma cells, characterized by a vicious cycle of response and relapse with ultimate development of resistance to therapy in most patients. The steadfast introduction and approval of novel agents, including first- and second-generation proteasome inhibitors, immunomodulatory drugs and monoclonal antibodies, in both the newly diagnosed and relapsed/refractory settings, has translated into progressive improvements in survival, especially for the elderly and patients with favorable-risk disease.1-3 We are currently entering an even more promising era of cellular- and immune-based therapies with remarkable responses seen even in heavily pre-treated patients.4,5 In parallel, however, the associated healthcare costs are steadily increasing and while treatment-related drug costs account for less than a third of the total, the increased dependency on inpatient and outpatient services required for delivery of novel therapies is expected to further jeopardize their affordability and accessibility for all patients.2,6
The management of relapsed or refractory multiple myeloma (RRMM) is complex, taking into consideration prior drug exposure, response, and toxicity profile, as well as disease- and patient-related factors. The challenge of managing RRMM is compounded by limited access to novel agents outside of clinical trials. In the late 2000s, the combination of lenalidomide and dexamethasone (Rd) was a well-established and utilized standard of care in early RRMM.7 With contemporary practice, however, most patients receive upfront lenalidomide as part of either a triplet or even quadruplet therapy with continuous or maintenance treatment until disease progression.8 For patients who are not refractory to lenalidomide, re-treatment at relapse with the Rd backbone in combination with novel agents including carfilzomib, ixazomib, daratumumab, or elotuzumab is effective, and was instrumental in the registrational approval of these agents.9-13 Most patients at first relapse will have been exposed to bortezomib, but are not necessarily refractory to the drug. However, the ENDEAVOR study demonstrated that for these patients, switching to a second-generation proteasome inhibitor, carfilzomib, is superior to re-treatment with bortezomib across a variety of subgroups of patients.14
The phase III ASPIRE study compared fixed-duration carfilzomib, lenalidomide, and dexamethasone (KRd) followed by Rd against Rd until disease progression in a cohort of patients who had received one to three prior lines of therapy, including 20% lenalidomide-exposed patients. The triplet KRd significantly improved overall response rate (87.1 vs. 66.7%; P<0.001) and progression-free survival (PFS; 26.3 vs 17.6 months; hazard ratio [HR]=0.69; 95% confidence interval [95% CI]: 0.57-0.83; P=0.0001) compared to Rd alone. At the time of this study design, in certain jurisdictions including the Asia-Pacific region, thalidomide, a first-generation immunomodulatory drug, was a more affordable alternative to lenalidomide. This single-arm, multicenter, phase II study conducted jointly by the Australasian Leukemia and Lymphoma Group (ALLG) and the Asian Myeloma Network (AMN) evaluated the safety and efficacy of fixed-duration carfilzomib, thalidomide, and dexamethasone (KTd) in patients with RRMM who had received one to three prior lines of therapy. The study was registered with the Australian New Zealand Clinical Trials Registry (identifier: ACTRN12615000818538) and ClinicalTrials.gov (identifier: NCT03140943)
Patients
Adults (≥18 years of age from the ALLG or ≥21 years of age from the AMN) with RRMM, evidence of measurable disease and a history of one to three prior lines of therapy were eligible. Measurable disease was considered as a serum M-protein ≥5 g/L, or urine M-protein ≥200 mg/24 h or, in patients without detectable serum or urine M-protein, serum free light chains >100 mg/L (involved light chain) and an abnormal κ/λ ratio or for IgA patients, whose disease can only be reliably measured with serum quantitative IgA immunoglobulin, a value ≥7.5 g/L. Induction therapy followed by stem cell transplant and consolidation/maintenance therapy was considered as one line of treatment. Patients with prior exposure to any immunomodulatory drug (thalidomide, lenalidomide or pomalidomide) or bortezomib, but not carfilzomib, were eligible. All patients had an Eastern Cooperative Oncology Group performance status 0 to 2, with adequate hematologic function (absolute neutrophil
count ≥1.0x109/L independent of growth factor support for at least 1 week, platelet count ≥50x109/L or ≥30x109/L if >50% plasma cell burden on bone marrow biopsy), renal function (calculated or measured creatinine clearance of ≥15 mL/min), and hepatic function (serum bilirubin <1.5 times the upper limit of normal, aspartate aminotransferase and alanine aminotransferase <3 times the upper limit of normal) at the time of screening. Patients with New York Heart Association class III or IV cardiac failure, or grade 3 or 4 peripheral neuropathy (or grade 2 with pain) were excluded. The study protocol was approved by the relevant human research ethics committee at all participating institutions. All patients provided written informed consent.
Patients were treated on a 28-day cycle for a pre-defined period of 18 months, consisting of 12 induction and six maintenance cycles, unless prior disease progression, unacceptable adverse events, or withdrawal of consent. The first ten patients from each of the ALLG and AMN cohorts were treated on a lead-in safety phase. Intravenous carfilzomib was given at a dose of 20 mg/m2 in cycle 1 on days 1 and 2, followed by dose escalation to 27 mg/m2 on days 8, 9, 15, and 16 (i.e., carfilzomib 20/27 mg/m2). Providing that four or fewer patients in each cohort experienced grade 4 treatment emergent adverse events attributed by investigators to carfilzomib exposure during the first two cycles, all subsequent patients <75 years were escalated to 56 mg/m2 from day 8 onwards (i.e., carfilzomib 20/56 mg/m2), while those ≥75 years were escalated to 36 mg/m2 from day 8 onwards (i.e., carfilzomib 20/36 mg/m2). Additionally, patients not achieving at least a partial response after the first two cycles during the lead-in safety phase and providing they did not experience ≥ grade 3 carfilzomib-related toxicity, were escalated to either carfilzomib 20/56 mg/m2 (<75 years) or 20/36 mg/m2 (≥75 years). For comprehensive insights into both hematologic and non-hematologic toxicities necessitating dose adjustments, further details are provided in Online Supplementary Table S1. Oral dexamethasone 40 mg was administered on days 1, 8, 15, and 22 (20 mg for patients ≥75 years) together with oral thalidomide 100 mg on day 1 to day 28. During the maintenance phase of treatment, carfilzomib was administered on days 1, 2, 15, and 16, dexamethasone on days 1 and 15, while thalidomide was omitted. Concomitant medication and supportive care, including venous thromboembolism prophylaxis, were prescribed at the discretion of investigators, following institutional practices, which included options such as low dose aspirin (100 mg daily), low molecular weight heparin, direct acting oral anticoagulants or vitamin K antagonists. Local response assessments were performed in National Association of Testing Authorities (NATA)/Royal College of Pathologists Australasia (RCPA) accredited laboratories prior to day 1 of each treatment cycle, and interpreted, according to the International Myeloma Working Group
Uniform Response Criteria, by local investigators with automated sponsor oversight and resolution of discrepancies in response assessment by the coordinating principal investigator.15 Patients were followed up monthly for disease progression and survival until 1 year after the completion of the last patient’s last cycle of treatment or induction therapy, whichever occurred earlier. The primary endpoint was to assess the PFS in patients with RRMM, who had received one to three prior lines of therapy, when treated with the fixed-duration KTd. The secondary endpoints were overall response rate, OS, duration of response, time to progression and safety and tolerability. Additionally, peripheral blood, bone marrow aspirate and trephine samples were collected at the time of screening, after 6 months of KTd and at the time of either disease progression or complete response, or both, with a view to interrogate changes in the immune system and the bone marrow tumor microenvironment and correlate findings to treatment outcomes. These translational, exploratory endpoints will be reported on at a later date.
The sample size for this study was estimated using a Simon minimax two-stage design, with a minimum of 37 patients per jurisdiction required in order to have an 80% power with a two-sided α of 0.05 to reject the null hypothesis of ≤50% PFS at 6.5 months (based on results from the OPTIMUM study), compared to the alternative hypothesis of ≥70% PFS at 6.5 months.16 PFS and overall survival (OS) were defined as the time, in consecutive days from the start of treatment (day 1 of cycle 1) to disease progression or death from any cause, whichever came first (PFS) or death from any cause (OS). Duration of response was defined as the time in consecutive days from date of first response (partial response or better) to the date of progression or death from any cause. Time to progression was defined as the time from day 1 of cycle 1 to the date of progression with deaths due to causes other than progression censored. Time-to-event analyses were censored by the closeout date or the date of last follow-up for patients lost to follow-up. The influence of prognostic factors (age, cytogenetic abnormalities, lines of prior therapy, previous thalidomide resistance, progression within 6 months vs. >6 months, baseline b2-microglobulin and International Prognostic Scoring System score at baseline) on PFS, time to progression, OS and response were explored using Cox proportional hazard regression or multiple logistic regression, as appropriate. Given the sample size, these analyses are considered hypothesis-generating. Although not specifically powered, comparisons between ethnically Asian and nonAsian populations were pre-specified, post-hoc analyses in the statistical analysis plan which was developed and approved prior to database lock. All hypothesis testing was two-sided, with values of P<0.05 considered statistically significant. Analyses were conducted using Stata MP for Mac v17 (Statacorp, College Station, TX, USA).
Between March 2017 and May 2020, 93 patients were screened of whom three patients were ineligible and excluded from the final analysis set (Australia n=49; Asia n=41). The patients’ disposition in the study is summarized in Figure 1. The patients’ baseline characteristics according to ethnic background (Asian [n=44] vs. non-Asian [n=46]) are presented in Table 1.
The cut-off date for final analysis was May 31, 2022. The median duration of follow-up was 26.4 months (range, 1.6-54.6 months) for non-Asians and 26.2 months (0.7-52.7 months) for Asians. A total of 64 primary events were recorded with a median PFS of 22.3 months (95% confidence interval [CI] 15.7-25.6) (Figure 2A), a 6.5-month PFS of 80.4% (95% CI: 70.4-87.3%) and a 2-year PFS of 46.3% (95% CI: 35.1-56.8%). The median PFS for patients who had received one prior line of therapy (n=48) was 22.3 months (95% CI: 12.9-26.0%), whereas it was 20.5 months for patients who had received two prior lines (n=20; 95% CI: 5.95-27.2%), and 20.0 months for patients who had received three prior lines (n=22; 95% CI: 13.9-28.6%).
A total of 29 deaths occurred with a median OS not reached and a 2-year OS of 73.8% (95% CI: 62.9-81.9%) (Figure 2C). The overall response rate was 88% with 73% patients
achieving a very good partial response or better and 32% attaining a complete response or better. No differences were seen in the depth of response (P=0.69), PFS (P=0.18) or OS (P=0.61) when comparing the Asian and non-Asian cohorts of patients (Figure 2B, D; Table 1). The median time to first response was 0.92 months (range, 0.92-0.95) while the median time to best response was 3.65 months (range, 2.53-4.57). The median duration of response for patients achieving a partial response or better was 22.6 months (95% CI: 18.2-25.4) and the median time to progression was 23.4 months (95% CI: 18.9-26.2) with a 2-year time to progression of 49.7% (95% CI: 37.9-60.4%).
Figure 3 shows hazard ratios and 95% confidence intervals for PFS in pre-specified subgroups according to baseline characteristics including age (18-64 vs. 65-74 vs. ≥75 years), cytogenetic risk as determined by fluorescence in situ hybridization (high risk denoted by the presence of t(4;14), t(14;16) or del(17p) vs. standard risk), creatinine clearance (<30 vs. 30-60 vs. ≥60 mL/min), serum b2-microglobulin (≤3.5 vs. 3.5-5.5 vs. >5.5 mg/mL), Revised International Scoring System stage at screening (stage I vs. stage II vs stage III), prior lines of therapy (1 vs. >1), prior bortezomib exposure and prior thalidomide exposure.
A total of 90 patients received at least one dose of study treatment. The median duration of treatment was 14.2 months (0.2-20.6 months). Forty patients (44%) discontin-
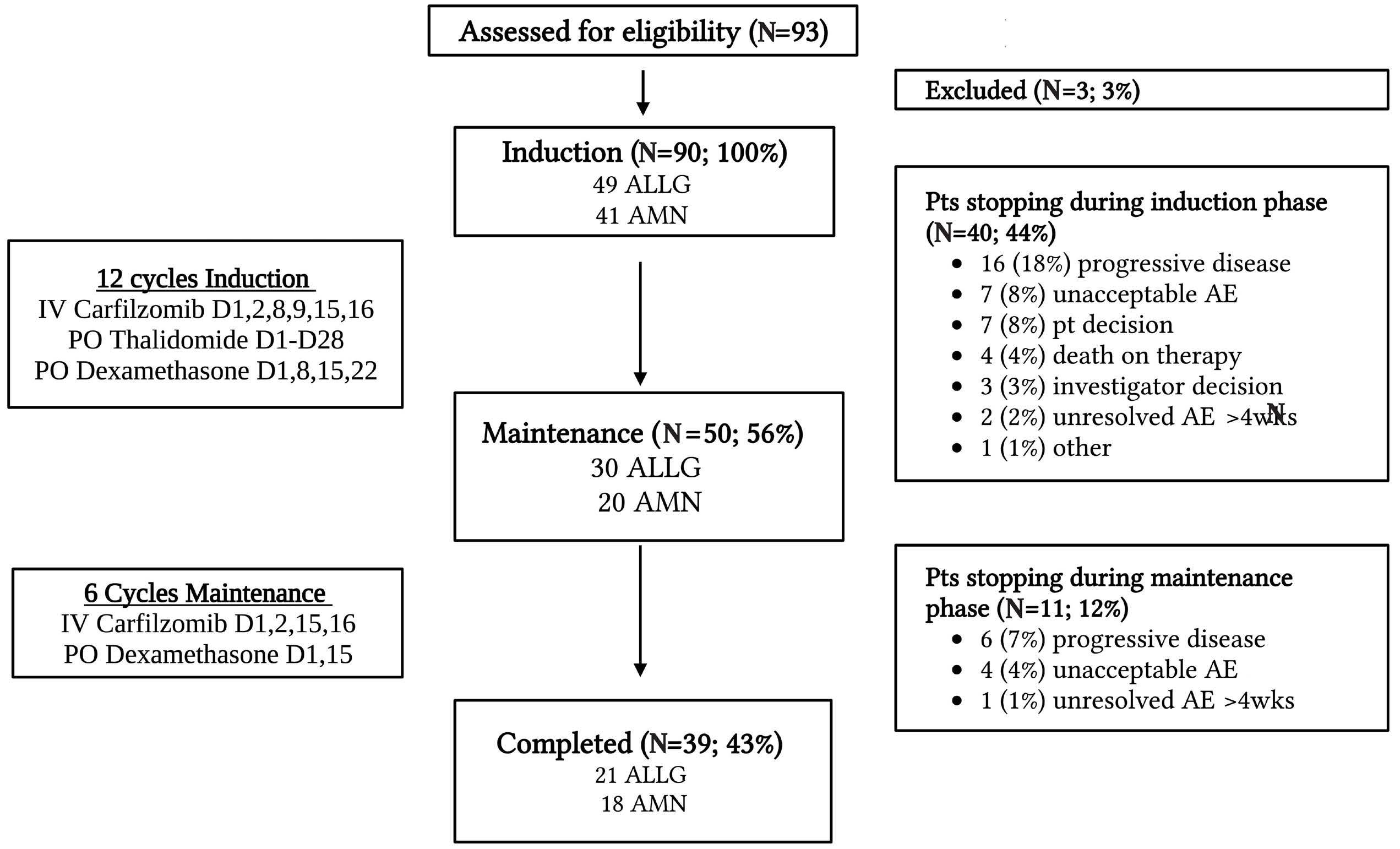
1. Patients’ disposition in the study. ALLG: Australian Lymphoma and Leukemia Group; AMN: Asian Myeloma Network; IV: intravenous; PO: per os; D: day; pt: patient; AE: adverse effect; wks: weeks.
ued treatment during the induction phase (most commonly due to progressive disease [n=16; 18%]; unacceptable adverse events [n=7; 8%] and patients’ decision [n=7; 8%]) while 11 patients (12%) discontinued treatment during the maintenance phase (most commonly due to progressive disease [n=6; 7%], unacceptable adverse events [n=4, 4%] or unresolved adverse events [n=1; 1%]). Thirty-nine patients (43%) completed the pre-defined 18 cycles of therapy. The relative dose intensity (RDI), defined as the proportion of the intended carfilzomib dose, decreased significantly with increasing dose levels (the median RDI was 98.8% for the 20/27 mg/m2 dose [n=21] vs. 93.3% for the 20/36 mg/m2 dose [n=6] and 89.9% for the 20/56 mg/m2 dose [n=60]; P<0.001) although there was no difference in the
median RDI observed between the non-Asian and Asian cohorts of patients (91.5% vs. 95.6%; P=0.17). Almost all patients (99%) experienced at least one adverse event; 74% of patients experienced at least one event attributed to carfilzomib, 66% experienced at least one event related to dexamethasone while 76% of patients reported events related to thalidomide. Carfilzomib-related adverse events triggered a dose delay for 42% of patients, dose delay and reduction for 13% and carfilzomib discontinuation in 17% of patients. Thalidomide was discontinued due to adverse events in 21% of patients.
The most common adverse events of any grade are summarized in Table 3 and include dyspnea (38.9%), upper respiratory tract infection (36.7%), peripheral sensory neu-
Age distribution, N of patients (%)
years old
years old
years old
Characteristic
Serum LDH, N of patients (%)
R-ISS stage, N of patients (%)
Distribution of prior therapy, N of patients (%)
prior line
Previous therapies, N of patients (%)
Prior peripheral neuropathy, N of patients (%)
ANZ: Australia and New Zealand; ECOG PS: Eastern Cooperative Oncology Group performance status; CrCl: creatinine clearance; LDH: lactate dehydrogenase; ULN: upper limit of normal; R-ISS: Revised International Staging System; AutoSCT: autologous stem cell transplant.
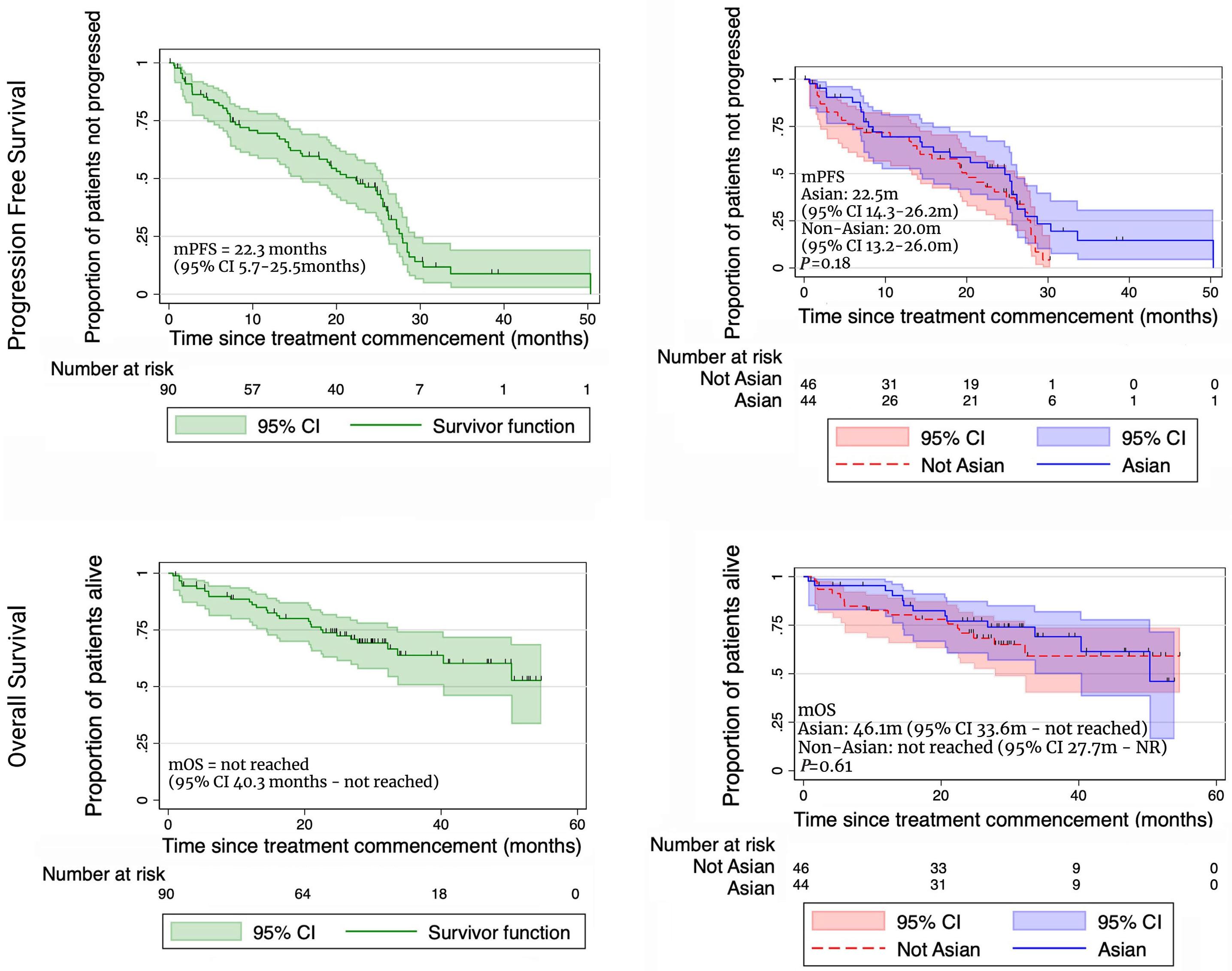
Figure 2. Progression-free survival and overall survival. (A, B) Progression-free survival is displayed for the entire cohort (A) and separated by ethnicity (B). (C, D) Overall survival is displayed for the entire cohort (C) and separated by ethnicity (D). mPFS: median progression-free survival; mOS: median overall survival; 95% CI: 95% confidence interval; NR: not reached.
Response by ethnicity.
N
Overall survival
Median (95% CI) in months 2-year
ORR: overall response rate; VGPR: very good partial response; CR: complete response; PFS: progression-free survival; 95% CI: 95% confidence interval; OS: overall survival; NR: not reached.
ropathy (31.1%), fatigue (26.7%) and peripheral edema (23.3%). No difference was observed in the rate of adverse events between Asian and non-Asian patients. Adverse events of ≥ grade 3 were reported in 76% of patients and at least one serious adverse event was recorded in 61% of patients. The most common hematologic adverse events were neutropenia (11.1%), thrombocytopenia (11.1%), anemia (8.9%), hemolysis (7.8%) and thrombotic thrombocytopenic purpura (2.2%). Hematologic adverse events of ≥ grade 3 included neutropenia (8.9%), thrombocytopenia (4.4%), anemia (2.2%), and thrombotic thrombocytopenic purpura (2.2%). Other adverse events of special interest were hypertension (22.2%), muscle weakness (20.0%), thromboembolic event (grouped term; 11.1%), cardiac failure (grouped term; 2.2%) and thrombotic thrombocytopenic purpura (2.2%). A total of 29 patients (32%) died during treatment or within 30 days of receiving the last dose of study treatment; 23 deaths (79%) were due to multiple myeloma, four were due to infective causes (including one case of severe acute respiratory syndrome coronavirus 2 infection), one was a cardiac death and one was the result of a road traffic incident.
The combination of a second-generation proteasome inhibitor, carfilzomib, with a first-generation immunomodulatory drug, thalidomide, and dexamethasone, irrespective of prior exposure to proteasome inhibitors or immunomodulatory drugs, is well tolerated and efficacious in patients with relapsed myeloma who have received one to three prior lines of therapy. Despite a fixed duration of treatment of 18 months, the combination of KTd led to a median PFS of 22.3 months. Granted the limitations of cross-trial comparisons, these results for KTd are comparable to those for KRd, with a reported median PFS of 26.3 months in the phase III ASPIRE study, and better than the 18.7 months that was reported for the Kd doublet in the ENDEAVOR study.10,14 For patients who had received only one prior line of therapy, the median PFS with KTd was not dissimilar to that reported in the ENDEAVOR study; 22.3 months and 22.2 months, while patients who had received two or three prior lines who were treated with KTd appear to have derived a benefit with a median PFS of 20.5 months and 20.0 months, respectively, whereas patients who had received two or more prior lines treated with Kd on the ENDEAVOR study had a median PFS of 14.9 months.17 Considering the notable prevalence of very good partial responses or better (73%) and complete responses or better (32%) with the KTd combination, there would have been compelling interest in conducting a more comprehensive evaluation of depth of response; however, local infrastructure to perform routine minimal residual disease assessment by multiparametric flow cytometry was limited at the time of study setup, representing a limitation of the study.
Carfilzomib was given for 18 months in both our study and the ASPIRE study; however, unlike the latter study in which the backbone of immunomodulatory drugs (Rd) was continued until disease progression, due to concern about peripheral neuropathy, thalidomide in our MM018/AMN0002 study was only continued for 12 months. Indeed, the motor neuropathy rate was minimal (6.6% any grade, 2.2% grade ≥3), in contrast to the more notable sensory neuropathy (31.1% any grade; 11.1% grade ≥3). Among the 32 patients reporting any-grade sensory or motor neuropathy, only six (18.8%) had pre-existing peripheral neuropathy (grade 1 or 2 without associated pain), likely stemming from prior anti-myeloma therapy having resulted in residual deficits.
Continuous therapy has been shown to improve PFS.18 A landmark analysis of the ASPIRE study performed at the 18-month mark after randomization, when carfilzomib was discontinued, demonstrated a lower PFS hazard ratio for KRd versus Rd (HR=0.58 [95% CI: 0.46-0.74]) compared to that for the overall study cohort (HR=0.69 [95% CI: 0.570.83]), begging the question of whether PFS would have been improved further had carfilzomib been continued until progressive disease in the KRd arm.19 Similarly, in our study we noted a sharp drop-off in the PFS curve within months of cessation of carfilzomib-dexamethasone maintenance after the protocol-defined 18 months of treatment
Table 3. Adverse events.
Other adverse events of special interest
(Figure 2A). Combined, these observations suggest that carfilzomib ought to be used until disease progression, a strategy which was shown to be safe and effective in the ENDEAVOR study.14
Consistent with previous reports on pre-specified subgroup analyses, albeit acknowledging limited numbers of patients within these subgroups, the KTd combination appears equally successful irrespective of age or cytogenetic risk group of the patients. Similarly, Revised International Staging System score, prior stem cell transplant or prior exposure to bortezomib or thalidomide did not have an impact on outcomes.20-22 Although 32% of patients had previously been treated with thalidomide, with exposure rates of 15% in the non-Asian and 50% in the Asian study cohorts, this disparity appears to align with regional frontline therapy practices at the time. It is important to note that data regarding refractoriness to prior treatments were not collected in this study, posing a minor limitation in interpreting the results of these sub-group analyses. A less favorable outcome compared to that of the overall group was still seen in patients with elevated b2-microglobulin with a trend towards increased efficacy of KTd in patients
having second- or third-line therapy, similarly consistent with evidence that carfilzomib remains efficacious whether used early or late in relapse.23 In our study, patients with poor renal function still did poorly compared to the overall cohort, and while the sample size is too small to make definitive remarks, impaired renal function is known to be a poor prognostic factor in myeloma.24 Strong evidence already exists that carfilzomib is safe and efficacious irrespective of renal function, with no starting dose adjustments required even in patients with end-stage renal failure.25,26 Given that thalidomide, as opposed to lenalidomide, is more practical in patients with renal impairment, KTd could be an effective combination when lenalidomide cannot be used.
Of interest, both the impressive overall response rate and the benefit to PFS were similarly observed in both the Asian and non-Asian cohorts of patients. This is consistent with previous reports of efficacy of carfilzomib in Asian patients and a subgroup analysis of the ENDEAVOR and A.R.R.O.W. trials which specifically reported on outcomes in Asian patients.27-29 This report, while cognizant of the smaller sample size, highlighted increased rates of ≥ grade 3 adverse events, especially grade ≥3 cardiac failure, in the Asian pop-
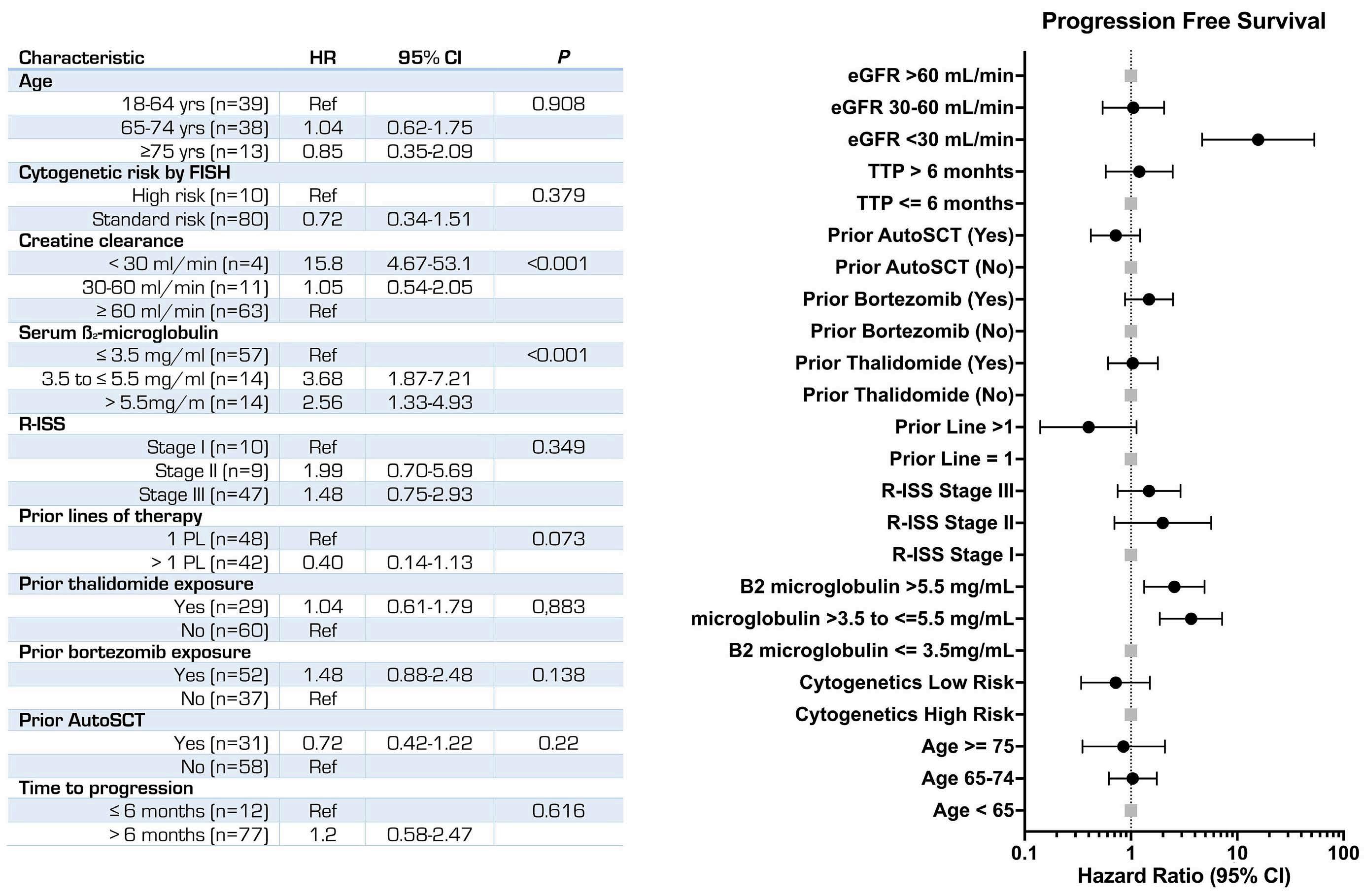
Figure 3. Hazard ratios and 95% confidence intervals for progression-free survival in pre-specified subgroups according to baseline characteristics. HR: hazard ratio; 95% CI: 95% confidence interval; yrs: years; FISH: fluorescence in situ hybridization; R-ISS: Revised International Staging System; PL: prior line; autoSCT: autologous stem cell transplant; eGFR: estimated glomerular filtration rate; TTP: thrombotic thrombocytopenic purpura.
ulation compared to the overall population of the ENDEAVOR and A.R.R.O.W. trials. In our cohort of patients, while dyspnea was the most commonly reported adverse event of any grade and the most common ≥3 grade event, documented cardiac failure was reported in a single, non-Asian patient. One explanation for the reduced rates of cardiac failure seen in our study may be the benefit of developed clinical experience with carfilzomib at the time of study initiation and routine measures to mitigate risks associated with carfilzomib therapy including strict monitoring and management of systemic hypertension, fluid balance and symptom-driven carfilzomib dose delays and reductions. Indeed, carfilzomib dose reduction rates in the ENDEAVOR, A.R.R.O.W. and ASPIRE studies were comparable to those in our study, and while these did not routinely report on carfilzomib dose delay, 42% of our cohort of patients experienced an adverse event-triggered carfilzomib dose delay.10,14,30 Another adverse event of special interest, carfilzomib-induced thrombotic thrombocytopenic purpura, while rare has been reported in association with carfilzomib previously.31 Both of our patients who developed this adverse event were Asian and developed a non-immune thrombocytopenia with a nadir platelet count of <30x109/L, blood film features of microangiopathic hemolysis with red cell fragmentation, and ADAMTS-13 levels >10%, thus excluding a diagnosis of de novo thrombotic thrombocytopenic purpura. Both were on the 56 mg/m2 dose, developed features early in treatment (first and third cycle) and responded to immunosuppression and plasma exchange.
In conclusion, KTd demonstrates favorable tolerability with commonly encountered toxicities that require proactive management in routine clinical practice. KTd is efficacious in patients with RRMM irrespective of Asian or non-Asian ethnicity, and irrespective of prior exposure to immunomodulatory drugs or proteasome inhibitors in first-line treatment of multiple myeloma. This combination may be an alternative to KRd in circumstances in which delivery of lenalidomide is limited by cost, access, or renal impairment. The use of carfilzomib until disease progression may be considered to further improve the
1. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122-1128.
2. Fonseca R, Abouzaid S, Bonafede M, et al. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia. 2017;31(9):1915-1921.
3. Binder M, Nandakumar B, Rajkumar SV, et al. Mortality trends in multiple myeloma after the introduction of novel therapies in the United States. Leukemia. 2022;36(3):801-808.
4. Zanwar S, Nandakumar B, Kumar S. Immune-based therapies in the management of multiple myeloma. Blood Cancer J. 2020;10(8):84
5. Mailankody S, Landgren O. T-cell engagers — modern immunebased therapies for multiple myeloma. N Engl J Med.
PFS as this has been shown to be safe in the ENDEAVOR study.14
Disclosures
SJH reports consulting or advisory roles for Celgene, Hoffman-La Roche AG, Genetech USA, HaemaLogiX, Janssen Global Services, and Novartis and research support from HaemaLogiX and Janssen Global Services. HQ reports consulting or advisory roles for Amgen, Antengene, Bristol-Myers Squibb/Celgene, Celgene, GSK, Janssen-Cilag, Karyopharm Therapeutics, Pfizer, Roche, and Sanofi and research support from Amgen, Bristol-Myers Squibb/Celgene, Celgene, GSK, Karyopharm Therapeutics and Sanofi. All other authors have no conflicts of interest to disclose.
Contributions
WJC, HQ, PM, and BD developed the concept and design of the study. SN, SJH, JJL, NM, JHJ, JE, VMC, NH, KK, RE, BA, SMB, SYH, RR, WJC, and HQ managed patients and participated in the collection of clinical data. DE managed the data collection and assembly. BB performed the statistical analyses. SN and HQ interpreted the data and wrote the manuscript. All authors critically revised the manuscript and reviewed and approved the final version.
The authors thank all the patients, families, caregivers, research nurses, study coordinators, sub-investigators and support staff who contributed to this study.
This study was supported by a grant from Amgen which also provided the carfilzomib used in this study, and by funding from the CASS (Contributing to Australian Scholarship and Science) foundation.
Data-sharing statement
Original data and protocols may be obtained upon written request.
2022;387(6):558-561.
6. Seefat MR, Cucchi DGJ, Dirven S, Groen K, Zweegman S, Blommestein HM. A sstematic review of cost-effectiveness analyses of novel agents in the treatment of multiple myeloma. Cancers. 2021;13(22):5606.
7 Dimopoulos MA, Chen C, Spencer A, et al. Long-term follow-up on overall survival from the MM-009 and MM-010 phase III trials of lenalidomide plus dexamethasone in patients with relapsed or refractory multiple myeloma. Leukemia. 2009;23(11):2147-2152.
8. Musto P, Montefusco V. Are maintenance and continuous therapies indicated for every patient with multiple myeloma? Exp Rev Hematol. 2016;9(8):743-751.
9 Kastritis E, Roussou M, Gavriatopoulou M, et al. Impact of last lenalidomide dose, duration, and IMiD-free interval in patients with myeloma treated with pomalidomide/dexamethasone.
Blood Adv. 2019;3(23):4095-4103.
10 Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372(2):142-152.
11. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621-1634.
12. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
13. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621-631.
14 Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38.
15. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346.
16. Kropff M, Baylon HG, Hillengass J, et al. Thalidomide versus dexamethasone for the treatment of relapsed and/or refractory multiple myeloma: results from OPTIMUM, a randomized trial. Haematologica. 2012;97(5):784-791.
17 Moreau P, Joshua D, Chng W-J, et al. Impact of prior treatment on patients with relapsed multiple myeloma treated with carfilzomib and dexamethasone vs bortezomib and dexamethasone in the phase 3 ENDEAVOR study. Leukemia. 2017;31(1):115-122.
18. Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med. 2012;366(19):1759-1769.
19 Dimopoulos M, Wang M, Maisnar V, et al. Response and progression-free survival according to planned treatment duration in patients with relapsed multiple myeloma treated with carfilzomib, lenalidomide, and dexamethasone (KRd) versus lenalidomide and dexamethasone (Rd) in the phase III ASPIRE study. J Hematol Oncol. 2018;11(1):49.
20 Dimopoulos MA, Stewart AK, Masszi T, et al. Carfilzomib, lenalidomide, and dexamethasone in patients with relapsed multiple myeloma categorised by age: secondary analysis from the phase 3 ASPIRE study. Br J Haematol. 2017;177(3):404-413.
21. Weisel K, Mateos M-V, Gay F, et al. Efficacy and safety profile of deep responders to carfilzomib-based therapy: a subgroup
analysis from ASPIRE and ENDEAVOR. Leukemia. 2021;35(6):1732-1744.
22. Avet-Loiseau H, Fonseca R, Siegel D, et al. Efficacy and safety of carfilzomib, lenalidomide, and dexamethasone vs lenalidomide and dexamethasone in patients with relapsed multiple myeloma based on cytogenetic risk status: subgroup analysis from the phase 3 atudy ASPIRE (NCT01080391). Blood. 2015;126(23):731.
23. Mateos M-V, Goldschmidt H, San-Miguel J, et al. Carfilzomib in relapsed or refractory multiple myeloma patients with early or late relapse following prior therapy: a subgroup analysis of the randomized phase 3 ASPIRE and ENDEAVOR trials. Hematol Oncol. 2018;36(2):463-470.
24. Mellors PW, Binder M, Ketterling RP, et al. Metaphase cytogenetics and plasma cell proliferation index for risk stratification in newly diagnosed multiple myeloma. Blood Adv. 2020;4(10):2236-2244.
25. Dimopoulos M, Siegel D, White DJ, et al. Carfilzomib vs bortezomib in patients with multiple myeloma and renal failure: a subgroup analysis of ENDEAVOR. Blood. 2019;133(2):147-155.
26. Quach H, White D, Spencer A, et al. Pharmacokinetics and safety of carfilzomib in patients with relapsed multiple myeloma and end-stage renal disease (ESRD): an open-label, single-arm, phase I study. Cancer Chemother Pharmacol. 2017;79(6):1067-1076.
27. Iida S, Watanabe T, Matsumoto M, et al. Carfilzomib monotherapy in Japanese patients with relapsed or refractory multiple myeloma: a phase 1/2 study. Cancer Sci. 2019;110(9):2924-2932.
28. Quach H, Yoon S-S, Kim K, et al. Carfilzomib use among patients with relapsed/refractory multiple myeloma in the Asia Pacific region: characteristics and outcomes by regimen from a prospective, real-world study. Blood. 2022;140(Suppl 1):4264-4266.
29. Dimopoulos MA, Moreau P, Iida S, et al. Outcomes for Asian patients with multiple myeloma receiving once- or twiceweekly carfilzomib-based therapy: a subgroup analysis of the randomized phase 3 ENDEAVOR and A.R.R.O.W. trials. Int J Hematol. 2019;110(4):466-473.
30 Moreau P, Mateos M-V, Berenson JR, et al. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018;19(7):953-964.
31. Eigbire-Molen O, Hermelin D, Blackall D. Carfilzomib-induced thrombotic microangiopathy: focus on pathogenesis. J Med Cases. 2022;13(6):274-280.
Isatuximab-pomalidomide-dexamethasone versus pomalidomide-dexamethasone
Paul G. Richardson,1 Aurore Perrot, 2 Jesus San Miguel, 3 Meral Beksac,4 Ivan Spicka, 5,6 Xavier Leleu,7 Fredrik Schjesvold, 8,9 Philippe Moreau,10 Meletios A. Dimopoulos,11 Shang-Yi Huang,12 Jiri Minarik,13 Michele Cavo,14 H. Miles Prince,15 Sandrine Macé,16 Rick Zhang,17 Franck Dubin,16 Mony Chenda Morisse 17 and Kenneth C. Anderson 1
1Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, USA; 2CHU de Toulouse, IUCT-O, Université de Toulouse, UPS, Service d’Hématologie, Toulouse, France; 3Clínica Universidad de Navarra, Navarra, CCUN, CIMA, IDISNA, CIBER-ONC, Pamplona, Spain; 4Department of Hematology, Ankara University, Ankara, Turkey; 5General Faculty Hospital, Prague, Czech Republic; 6First Faculty of Medicine, Charles University, Prague, Czech Republic; 7Service d’Hématologie et Thérapie Cellulaire, CHU and CIC INSERM 1402, Poitiers Cedex, France; 8Oslo Myeloma Center, Department of Hematology, Oslo University Hospital, Oslo, Norway; 9KG Jebsen Center for B Cell Malignancies, University of Oslo, Oslo, Norway; 10Hematology Department, CHU Nantes, Nantes, France; 11Department of Clinical Therapeutics, School of Medicine, National and Kapodistrian University of Athens School of Medicine, Athens, Greece; 12Department of Hematology, National Taiwan University Hospital, Taipei, Taiwan; 13Department of Hemato-Oncology, Faculty of Medicine and Dentistry, Palacky University and University Hospital Olomouc, Olomouc, Czech Republic; 14IRCCS Azienda Ospedaliero-Universitaria di Bologna, Seràgnoli Institute of Hematology, and the Department of Medical and Surgical Sciences, Bologna University School of Medicine, Bologna, Italy; 15Immunology and Molecular Oncology, Epworth Healthcare, University of Melbourne, Melbourne, Victoria, Australia; 16Sanofi, Vitry-sur-Seine, France and 17Sanofi, Cambridge, MA, USA
Correspondence: P. Richardson paul_richardson@dfci.harvard.edu
Received: October 4, 2023. Accepted: January 24, 2024. Early view: February 1, 2024. https://doi.org/10.3324/haematol.2023.284325
Published under a CC BY license

The primary and prespecified updated analyses of ICARIA-MM (clinicaltrial gov. Identifier: NCT02990338) demonstrated improved progression-free survival (PFS) and a benefit in overall survival (OS) was reported with the addition of isatuximab, an anti-CD38 monoclonal antibody, to pomalidomide-dexamethasone (Pd) in patients with relapsed/refractory multiple myeloma. Here, we report the final OS analysis. This multicenter, randomized, open-label, phase III study included patients who had received and failed ≥2 previous therapies, including lenalidomide and a proteasome inhibitor. Between January 10, 2017, and February 2, 2018, 307 patients were randomized (1:1) to isatuximab-pomalidomide-dexamethasone (Isa-Pd; N=154) or Pd (N=153), stratified based on age (<75 vs. ≥75 years) and number of previous lines of therapy (2-3 vs. >3). At data cutoff for the final OS analysis after 220 OS events (January 27, 2022), median follow-up duration was 52.4 months. Median OS was 24.6 months (95% confidence interval [CI]: 20.3-31.3) with Isa-Pd and 17.7 months (95% CI: 14.426.2) with Pd (hazard ratio=0.78; 95% CI: 0.59-1.02; 1-sided P=0.0319). Despite subsequent daratumumab use in the Pd group and its potential benefit on PFS in the first subsequent therapy line, median PFS2 was significantly longer with Isa-Pd versus Pd (17.5 vs. 12.9 months; log-rank 1-sided P=0.0091). In this analysis, Isa-Pd continued to be efficacious and well tolerated after follow-up of approximately 52 months, contributing to a clinically meaningful, 6.9-month improvement in median OS in patients with relapsed/refractory multiple myeloma.
Multiple myeloma (MM) primarily remains an incurable disease, and although novel agents have improved response and survival rates, almost all patients relapse either on or after these treatments.1-4 Treatment choice for relapsed and refractory MM (RRMM) is determined by refractoriness and exposure to prior drugs.5
Due to the need for novel treatments for RRMM, monoclonal antibodies targeting CD38 have emerged, such as daratumumab and isatuximab.6-9 Isatuximab is a monoclonal antibody targeting a specific epitope of the human cell-surface antigen CD38, which is widely and uniformly expressed on myeloma cells.10-12
Isatuximab was investigated in combination with pomalidomide and dexamethasone (Pd) in a phase III, randomized, multicenter, open-label trial (ICARIA-MM; clinicaltrials gov. Identifier: NCT02990338).13 After a median follow-up of 11.6 months, median progression-free survival (PFS) was 11.5 (95% confidence interval [CI]: 8.9-13.9) months with isatuximab–pomalidomide–dexamethasone (Isa-Pd) and 6.5 (95% CI: 4.5-8.3) months with Pd alone.13 Median overall survival (OS) was not reached in either treatment arm (hazard ratio [HR]=0.687; 95% CI: 0.461-1.023; P=0.0631).13
Results from a prespecified updated analysis at 24 months after the primary analysis reported a median OS of 24.6 months (95% CI: 20.3-31.3) with Isa-Pd versus 17.7 months (95% CI: 14.4-26.2) months with Pd (HR=0.76, 95% CI: 0.57-1.01).14
Based on the primary results of ICARIA-MM, isatuximab has been approved in several countries in combination with Pd for adult patients with RRMM who have received ≥2 previous therapies, including lenalidomide and a proteasome inhibitor (PI).11,15
This analysis of ICARIA-MM reports the final OS, conducted when 220 deaths occurred.
Study design and participants
ICARIA-MM is a prospective, multicenter, randomized, open-label, parallel-group phase III study conducted at 102 sites in 24 countries (Online Supplementary Appendix, page 2-3). Detailed inclusion and exclusion criteria, study design, randomization and masking, and procedures have been previously described and are detailed in the Online Supplementary Appendix, page 4-7.13,14
Briefly, eligible patients were ≥18 years old, had RRMM, received ≥2 previous therapies, and had failed therapy with lenalidomide and a PI (alone or in combination). Failure to therapy included progression on or within 60 days, intolerance to lenalidomide or the PI, or disease progression within 6 months after achieving at least a partial response (PR). Patients refractory to previous anti-CD38 therapy, with
prior pomalidomide exposure, with an Eastern Cooperative Oncology Group Performance Status >2, an ongoing toxic effect grade >2 from previous therapy (grade according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03), with active primary amyloid light-chain amyloidosis, or concomitant plasma cell leukemia were excluded.
The trial complied with the International Conference on Harmonization Good Clinical Practice Guideline and the Declaration of Helsinki. All participating institutional review boards and ethics committees approved the study protocol, and patients provided written informed consent.
Following eligibility confirmation, patients were randomized using an interactive response technology (IRT) system in a 1:1 ratio to the Isa-Pd (isatuximab) arm or to the Pd (control) arm. Randomization was stratified by age (<75 vs. ≥75 years) and number of previous lines of therapy (2 or 3 vs. >3).
Intravenous isatuximab 10 mg/kg was administered on days 1, 8, 15, and 22 of cycle 1 and days 1 and 15 for subsequent cycles. Intravenous or oral dexamethasone 40 mg (20 mg if age ≥75 years) was administered on days 1, 8, 15, and 22, and oral pomalidomide 4 mg was administered on days 1-21. Premedication and isatuximab preparation were described previously.13,14
The primary endpoint of ICARIA-MM was PFS as determined by the IRC. No PFS per IRC update is provided based on this additional follow-up. Key secondary endpoints include overall response rate (ORR) and OS. ORR was defined as the proportion of patients with complete response (CR), stringent CR, very good partial response (VGPR), and PR as best overall response, assessed by IRC using the International Myeloma Working Group criteria. No updated ORR per IRC is provided. OS was defined as the time from the date of randomization to date of death from any cause. If death was not observed before the analysis data cutoff date, OS was censored at the last date that the patient was known to be alive or at the cutoff date, whichever was first. Exploratory endpoints included PFS2, ORR on further therapy (best overall response reported by the investigator), PFS on the first line of further therapy, and time to next treatment (TTNT). Additional definitions and methods are detailed in the Online Supplementary Appendix, page 4-7 and have been defined previously.14,16
Based on the primary efficacy endpoint (PFS) using the assumptions that the control arm had a median PFS of 4.0 months; the Isa-Pd arm was assessed to have 40%
risk reduction in HR versus control, corresponding to an improvement in the true median PFS time from 4.0 to 6.67 months; and a log-rank test at a 1-sided 2.5% significance level, a total of 162 PFS events were calculated to be needed to achieve 90% power for the study. Based on these assumptions, 220 deaths were needed to achieve 80% power for the study.
For the final analysis, a data cutoff date of March 14, 2022, was selected, and data were included up to this date (last patient last visit). Per protocol, the cutoff date for the final OS analysis was to occur when 220 OS events had been observed. The 220th OS event occurred on January 27, 2022, and this date was considered to be the OS cutoff date for the primary OS analysis.
Efficacy analyses were conducted using the intention-to-treat (ITT) population, defined as patients who provided signed informed consent and were allocated a randomization number by the IRT. Safety was assessed in all patients from the ITT population who received at least a partial dose of study treatment. Patients were considered lost to follow-up if the last contact was ≥8 weeks prior to the data cutoff.
OS was analyzed using the Kaplan-Meier method and corresponding 95% CI, which was calculated with log-log transformation of survival function and the method devised by Brookmeyer and Crowley. HR and corresponding 95% CI are calculated from a Cox proportional hazards model, stratified by age and number of previous lines of therapy. Prespecified analyses were completed for updated PFS by investigator assessment (sensitivity analysis), PFS on subsequent therapy or death, and PFS on the first line of subsequent therapy (separate summaries for subsequent therapy with/without daratumumab-based therapy), and TTNT using OS analysis methods. P values for exploratory endpoints are provided for descriptive purposes only. Additional statistical methods have been described previously.14,16 All statistical analyses were performed with SAS (version 9.4; SAS Institute, Cary, NC) or R (version 3.4.3; R Foundation, Vienna, Austria). Since the updated analysis, no additional amendments to the protocol have been made. This study is registered with clinicaltrials gov. Identifier: NCT02990338
Between January 10, 2017, and February 1, 2018, 387 patients were screened and 307 were randomized (Isa-Pd, 154; control, 153) at 102 sites in 24 countries. Cutoff for the first OS interim analysis, at the time of the primary PFS analysis, was October 11, 2018 (reported previously).13 Cutoff for the preplanned second interim analysis was October 1, 2020 (reported previously).14 Cutoff for the final OS analysis was January 27, 2022, per protocol (Figure 1). Data cutoff for last patient last visit was March 14, 2022. Baseline characteristics were similar in both arms, as pre-
viously published and briefly shown in Table 1. All patients previously received PI and IMiD agents.
Median treatment duration was 11.0 (range, 2.6-12.4) months among patients receiving Isa-Pd and 5.5 (range, 4.4-21.8) months among patients receiving Pd. At data cutoff (March 14, 2022), 16 (10.4%) patients receiving Isa-Pd and three (2.0%) patients receiving Pd remained on treatment. The most frequent reason for definitive discontinuation was progressive disease (isatuximab, 65.6%; control, 76.5%).
After a median follow-up of 52.4 months, median PFS per investigator assessment (ignoring symptomatic deterioration) showed consistent improvement with longer follow-up (Isa-Pd: 11.1 months, 95% CI: 7.8-13.8; Pd: 5.9 months, 95% CI: 4.5-7.9; HR=0.57, 95% CI: 0.44-0.73; 1-sided P<0.0001; Online Supplementary Figure S1).
The prespecified required number of 220 OS events occurred on January 27, 2022, which was the analysis cutoff date. Of the 220 deaths, 106 (68.8%) occurred in the Isa-Pd arm and 114 (74.5%) in the Pd arm. Median OS was 24.6 months (95% CI: 20.3-31.3) in the Isa-Pd group and 17.7 months (95% CI: 14.4-26.2) in the Pd group (HR=0.78, 95% CI: 0.59-1.02; 1-sided P=0.0319; Figure 2). Early separation was observed in the OS curves between arms; however, because the 1-sided P value for statistical significance was set to 0.02 based on the previous interim analysis, the current analysis did not cross the level of significance. Censored patients (Isa-Pd: 48/154 [31%]; Pd: 39/153 [26%]) remained alive at data cutoff (Isa-Pd: 39/48 [81%]; Pd: 31/39 [80%]), were alive at the last contact before the cutoff date (1/48 [2%]; 0/39), or were lost to follow-up (8/48 [17%]; 8/39 [21%]). Subgroup analyses of OS are shown in Online Supplementary Figure S2
Among patients receiving subsequent anti-myeloma therapy, daratumumab was given to 23 (23%) of 102 patients in the Isa-Pd group and 71 (60%) of 119 patients in the Pd group (Online Supplementary Table S3). In order to estimate the treatment effect in the absence of a switch to subsequent anti-cancer therapy with daratumumab, sensitivity analyses using the RPSFT model were performed, with overall similar results (HR=0.706, 95% CI: 0.538-0.926) to the ITT estimate, in favor of the Isa-Pd arm (HR=0.776, 95% CI: 0.594-1.015; Online Supplementary Appendix, page 17). An OS sensitivity analysis was conducted to assess the impact of death due to coronavirus disease 2019 infection, with similar results to the ITT estimate (Online Supplementary Appendix, page 18).
Overall, 102 (66%) of 154 patients in the Isa-Pd group and 119 (78%) of 153 patients in the Pd group received subsequent anti-myeloma therapy. Median TTNT was longer with isatuximab (15.5 months, 95% CI: 12.1-19.8) versus Pd (8.9 months, 95% CI: 6.3-11.5; 1-sided P<0.0001; Figure 3). Median PFS on subsequent therapy or death (PFS2) was longer in the Isa-Pd group (17.5 months, 95% CI: 14.9-19.2) versus Pd (12.9 months, 95% CI: 10.1-16.6; 1-sided P=0.0091; Figure 2).
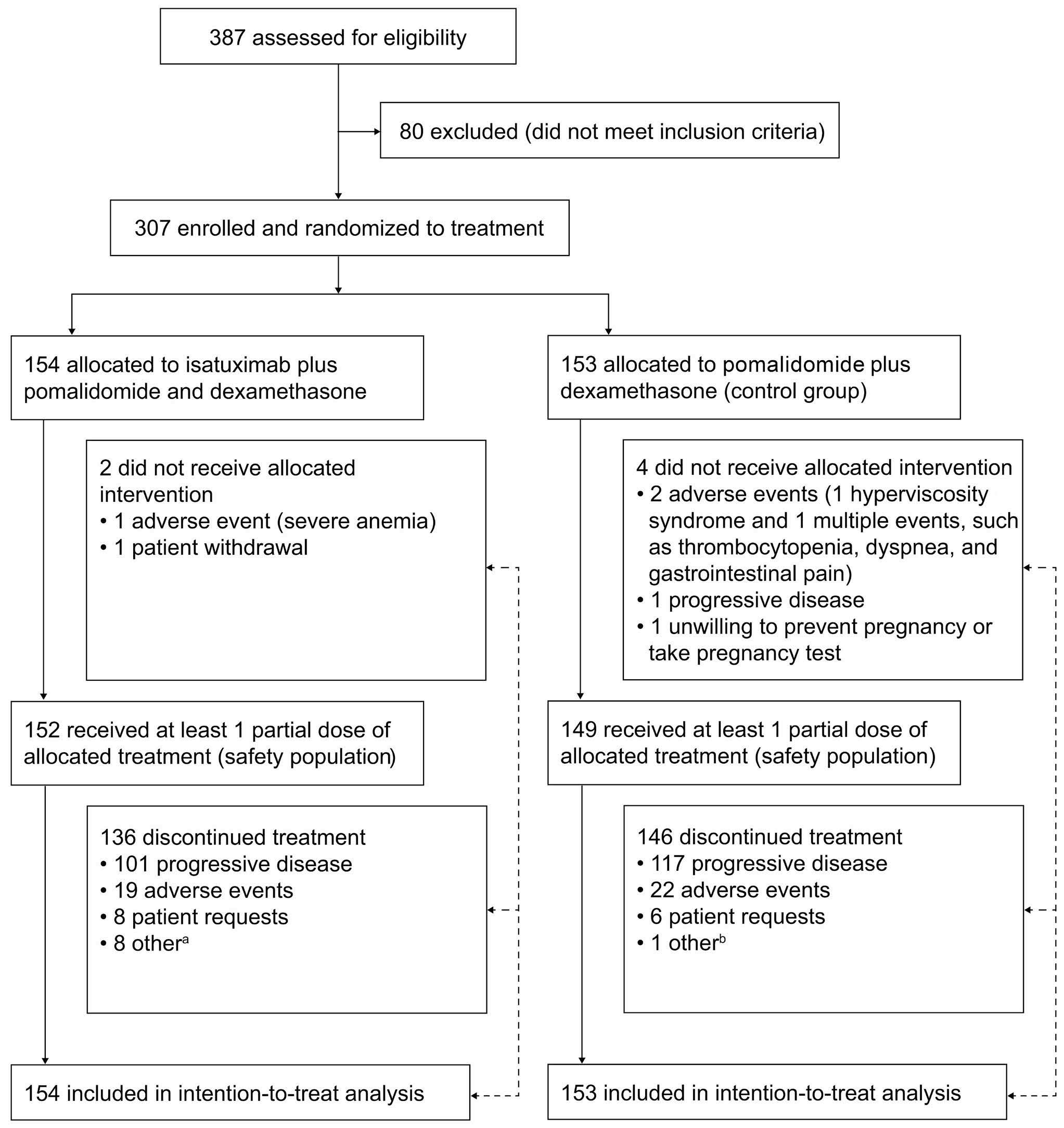
Figure 1. Trial profile as of data cutoff for the final overall survival analysis. aInvestigator decision due to free light chain increase (N=3), physician’s decision (suspected progression, N=1), unconfirmed progression (N=1), poor compliance to protocol (N=1), investigator decided to switch treatment to daratumumab-pomalidomide-dexamethasone (N=1), investigator kept the same isatuximab-pomalidomide-dexamethasone (Isa-Pd) combination off protocol, as the product is available commercially (N=1). bPhysician decision. OS: overall survival.
The proportional hazard assumption was met (Schoenfeld residuals test) for the following Cox models: PFS by investigator (P=0.89), OS (P=0.28), TTNT (P=0.44), and PFS2 (P=0.42).
The ORR was higher with Isa-Pd versus Pd, consistent with the primary analysis and second interim analysis (Online Supplementary Table S2; Online Supplementary Figure S3).
Deeper responses were also observed with Isa-Pd versus Pd. Minimal residual disease negativity was observed in ten (6%) patients in the Isa-Pd group at the 10-5 sensitivity level (18 patients with data available) but in no patients in the Pd group (3 patients with data available; data not shown).
More patients in the Isa-Pd group versus the Pd group received subsequent alkylating agents, PI, corticosteroids, and other treatments (i.e., investigational anti-neoplastic drugs, cisplatin, etoposide, and stem cells), whereas IMiD
agents were administered to a similar proportion of patients in both groups (Online Supplementary Table S3).
Exploratory analysis of ORR, CR, VGPR, or PR on selected subsequent therapies with/without daratumumab are shown in Figure 4 and Online Supplementary Figure S4. Among non-daratumumab-based regimens, non-IMiDbased regimens versus IMiD-based regimens led to better response rates with Isa-Pd (28/71 [39%] vs. 3/27 [11%]). Per the inclusion criteria, patients previously failed treatment with lenalidomide and a PI.
ORR with subsequent daratumumab in any subsequent line was lower for patients in the Isa-Pd group (5/21 [24%]) vs. the Pd group (23/57 [40%]); however, rates of ≥VGPR were similar (3/21 [14%] vs. 10/57 [18%]). Daratumumab in combination led to improvements in both arms (Isa-Pd: 4/14 [29%]; Pd: 13/29 [45%]) versus daratumumab monotherapy
Table 1. Baseline demographic and patient characteristics of the intention-to-treat population.
Characteristic
Age in years
Median (IQR)
<65, N (%)
65–75, N (%)
≥75, N (%)
Sex, N (%)
Ethnicity, N (%)
History of asthma or COPD, N (%)
eGFR <60 mL/min/1.73 m2, N/N (%)
autologous stem-cell transplantation, N (%)
Time since initial diagnosis in years, median (IQR)
of myeloma at diagnosis, N (%)
(κ+λ)
ISS stage at study entry, N (%)
Cytogenetic risk at baseline,a N (%)
of previous lines of therapy, median (IQR)
Previous therapy, N (%)
Alkylating agent
Refractory to treatment, N (%)
Last line of therapy
Immunomodulatory agent
Lenalidomide
Proteasome inhibitor
Lenalidomide and proteasome inhibitor
Lenalidomide last line
(21)
(10)
(97)
(95)
(94)
(77)
(72)
(60)
(99)
(94)
(92)
Cytogenetic analysis was performed by fluorescence in situ hybridization by a central laboratory with cutoff of 50% for del(17p) and 30% for t(4;14) and t(14;16). aHigh-risk cytogenetic status was defined as the presence of at least del(17p), t(4;14), or t(14;16) chromosomal abnormalities. COPD: chronic obstructive pulmonary disease; eGFR: estimated glomerular filtration rate; IgA: immunoglobulin A; IgG: immunoglobulin G; Isa-Pd: isatuximab-pomalidomide-dexamethasone; ISS: International Staging System (ISS staging is derived based on the combination of serum b2-microglobulin and albumin); Pd: pomalidomide-dexamethasone.
± steroids (Online Supplementary Figure S4). Some patients receiving daratumumab, alone or in combination, as a first subsequent line of therapy achieved ≥VGPR, even after receiving prior isatuximab study therapy (2/8 patients [25%]) with a short washout period (median, 13 days) (Figure 4).
Among patients receiving non-daratumumab–based therapy, median PFS on the first line of subsequent therapy was similar in the Isa-Pd group (4.6 months, 95% CI: 3.1-6.6 in 69/93 [74%] patients receiving subsequent non-daratumumab therapy) versus the Pd group (5.2 months, 95%
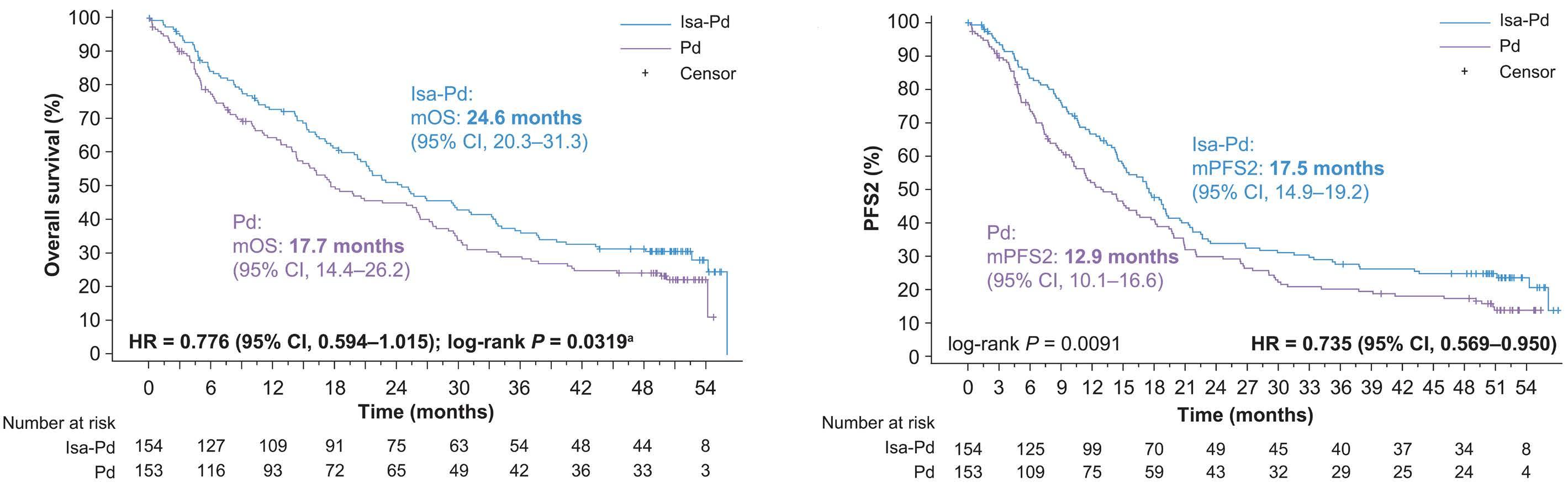
Figure 2. Overall survival and progression-free survival on subsequent therapy or death in the intention-to-treat population. (A) Kaplan–Meier analysis of overall survival (OS) after 220 events. Patients who were alive at the cutoff date (January 27, 2022), alive at the last contact before the cutoff date, or lost to follow-up were censored. (B) Kaplan-Meier analysis of time from randomization to disease progression on subsequent therapy or death, as assessed by investigators. aThe 1-sided log-rank P value is provided for descriptive purposes. Patients who did not experience an event were censored (denoted by crosses). Hazard ratio (HR) and corresponding 95% confidence intervals (CI) are from a Cox proportional hazard model, stratified by age and number of previous lines of therapy. Isa-Pd: isatuximab-pomalidomide-dexamethasone; mOS: median overall survival; mPFS2: median progression-free survival on subsequent therapy or death; OS: overall survival; Pd: pomalidomide-dexamethasone; PFS: progression-free survival; PFS2: progression-free survival on subsequent therapy or death.
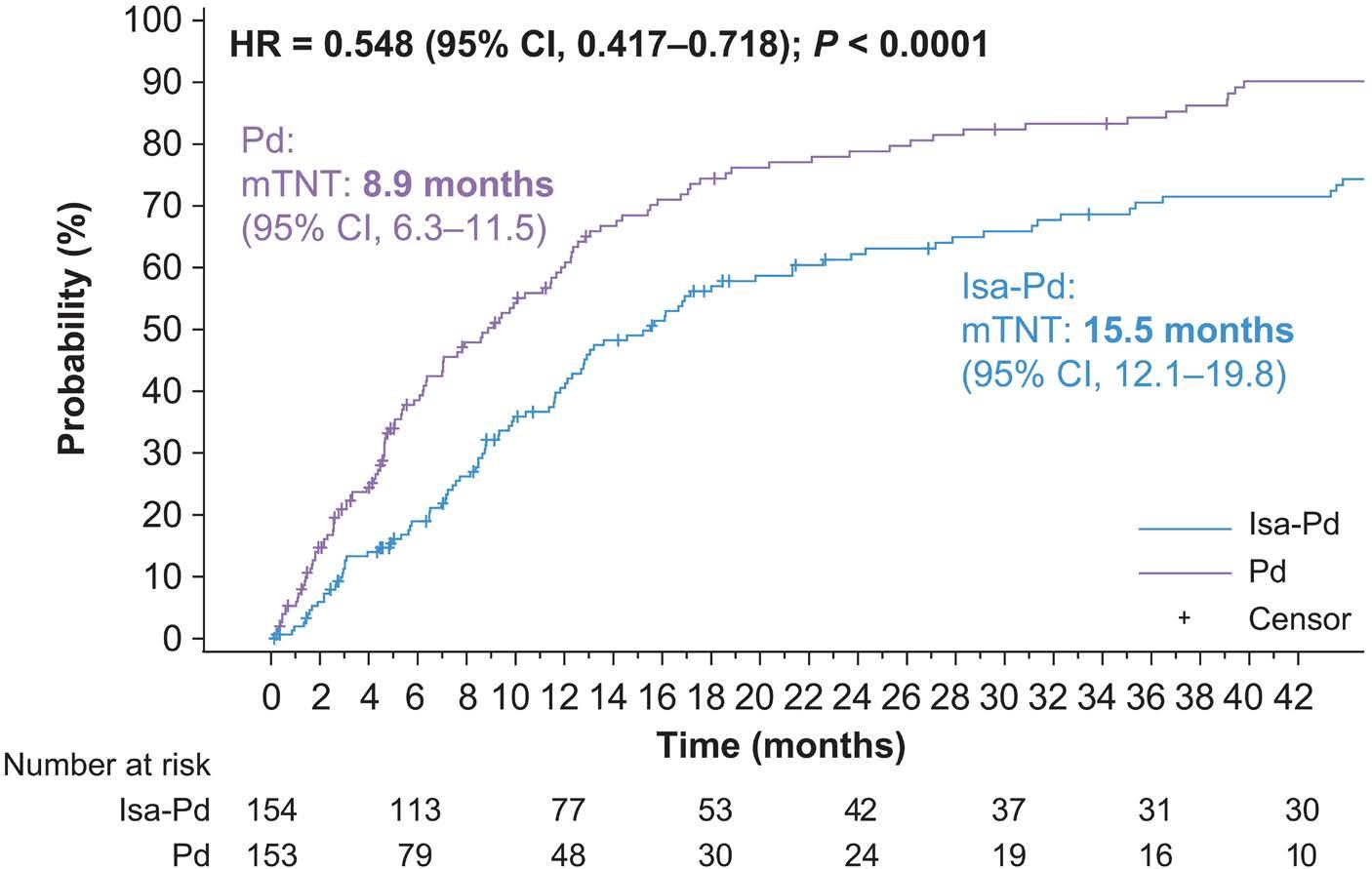
Figure 3. Time to next treatment (intention-to-treat population). Kaplan-Meier analysis of time to next treatment, as reported by investigators. Patients who did not proceed to subsequent anti-myeloma treatment before the cutoff date were censored. Median follow-up was 52.4 months. Hazard ratios (HR) and corresponding 95% confidence intervals (CI) are from a Cox proportional hazard model stratified by age and number of previous lines of therapy. Isa-Pd: isatuximab-pomalidomide-dexamethasone; mTNT: median time to next treatment; Pd: pomalidomide-dexamethasone.
CI: 3.8-7.3 in 43/67 [64%] patients receiving subsequent non-daratumumab therapy) (Online Supplementary Figure S5). However, among patients receiving daratumumab-based therapy as the first subsequent line (N=9; Pd: N=52), median PFS was lower with Isa-Pd (Isa-Pd: 2.2 months, 95% CI: 0.03-7.4 in 7/9 [78%] patients receiving subsequent daratumumab) versus Pd (5.7 months, 95% CI: 3.8-10.8 in 39/52 [75%] patients receiving subsequent daratumumab)
(Online Supplementary Figure S5). The median time between the last dose of investigational medicinal product and the first subsequent therapy with daratumumab was shorter in the Isa-Pd group (N=9; 13 days; range, 2-100 days) versus the Pd group (N=52; 22 days; range, 1-822 days; data not shown; post hoc).
With longer follow-up, no new safety concerns were identified with Isa-Pd. The overall safety summary is in Online Supplementary Table S4, and the most frequently reported treatment-emergent adverse events (TEAE) are in Table 2.
Compared with the Pd group, the Isa-Pd group, respectively, had longer cumulative exposure to treatment (216.1 vs. 137.3 patient-years) and a larger median number of cycles started per patient (11; interquartile range [IQR], 4-22.5 vs. 6; IQR, 3-13; Online Supplementary Table S1). In the safety population, 70 (46%) of 152 patients had pomalidomide dose reductions and 62 (41%) had dexamethasone dose reductions in the isatuximab group versus 40 (27%) of 149 patients with pomalidomide dose reductions and 45 (30%) with dexamethasone reductions in the Pd group (Online Supplementary Table S1). The most frequently reported grade ≥3 TEAE in the Isa-Pd and Pd groups were neutropenia (77 [51%] of 152 vs. 52 [35%] of 149), pneumonia (35 [23%] vs. 31 [21%]), and thrombocytopenia (20 [13%] vs. 18 [12%]; Online Supplementary Table S6). TEAE reported in ≥10% of patients and ≥5% higher with isatuximab are shown in Online Supplementary Table S5. TEAE reported in ≥5% of patients are shown in Online Supplementary Table S7. Infusion reactions (IR) were reported with Isa-Pd (2 patients in the Pd group experienced IR upon subsequent daratumumab therapy), and all were reversible; only one IR has been reported since the primary analysis (cycle 19;
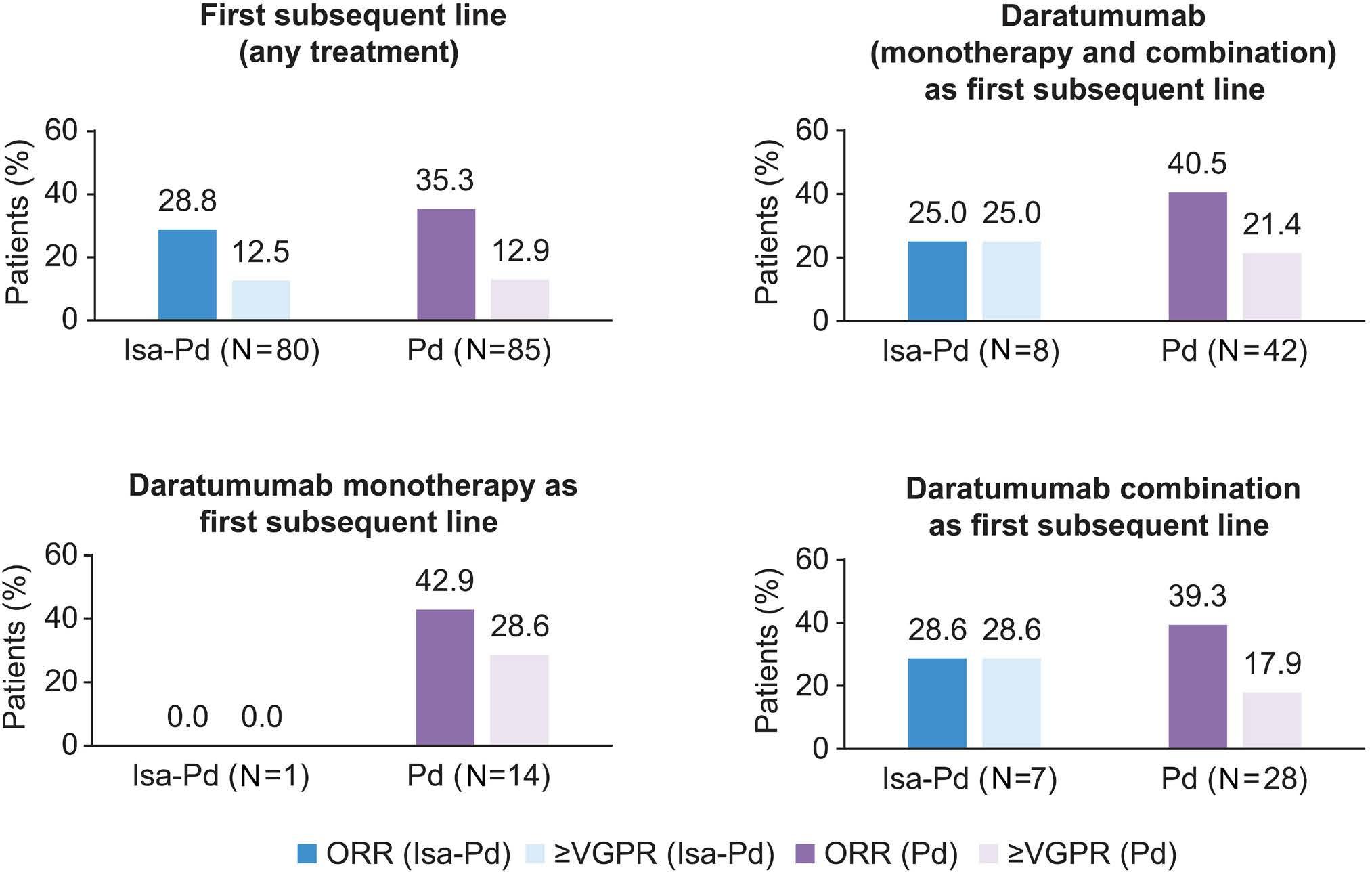
Figure 4. Exploratory analysis of response rate on first subsequent therapy,* intention-to-treat population. *Median washout period between therapies was 13 (range, 2-100) days for the isatuximab group and 22 (range, 1-822) days for the control group. Isa-Pd: isatuximab-pomalidomide-dexamethasone; ORR: overall response rate; Pd: pomalidomide-dexamethasone; VGPR: very good partial response or better.
data not shown), whereas all others were reported in the first three infusions. Grade 3/4 IR were reported in four (3%) of 152 patients. Hematologic laboratory abnormalities are shown in Online Supplementary Table S8. SPM occurred in ten (7%) of 152 patients in the Isa-Pd group and three (2%) of 149 in the Pd group (Online Supplementary Appendix page 21). Of these, three (Isa-Pd) occurred during the post-treatment period.
Treatment-emergent serious AE (SAE) occurred in 112 (74%) of 152 patients in the Isa-Pd group and 91 (61%) of 149 in the Pd group (Online Supplementary Table S9). Pneumonia was the most frequent SAE (all grades, both groups), reported in 35 (23%) of 152 patients in the Isa-Pd group and 31 (21%) of 149 in the Pd group. TEAE with a fatal outcome were reported in 23 (15%) of 152 patients in the Isa-Pd group and 19 (13%) of 149 in the Pd group. There were two (1%) treatment-related deaths in the Isa-Pd group (sepsis, 1; cerebellar infarction, 1) and two (1%) in the Pd group (pneumonia, 1; urinary tract infection, 1; data not shown). Overall, 108 (71%) patients in the Isa-Pd group and 113 (76%) in the Pd group died during the on- or post-treatment period due to disease progression (Isa-Pd: 76 [50%]; Pd: 81 [54%]), AE (7 [5%] vs. 8 [5%]), or other causes (25 [16%] vs. 24 [16%]; data not shown).
Dose reductions for pomalidomide and dexamethasone due to TEAE were more frequent in the Isa-Pd arm versus the Pd arm (pomalidomide reductions in 115/152 [76%] patients vs. 70/149 [47%] patients; dexamethasone reductions in 104 [68%] patients vs. 76 [51%] patients) and were primarily due to infections and neutropenia (data not shown). Definitive
treatment discontinuation due to TEAE was infrequent and occurred at similar rates in both treatment arms (19/152 [13%] patients in the Isa-Pd group vs. 22/149 [15%] patients in the Pd group; Online Supplementary Table S10).
This final OS analysis of ICARIA-MM showed a clinically meaningful benefit with a 6.9-month improvement in median OS with the addition of isatuximab to Pd (HR=0.78; log-rank 1-sided P=0.0319). Because the 1-sided P value for statistical significance was set to 0.02 due to α level spent at previous interim analyses, the current analysis did not cross the level of significance; however, early separation was observed in the OS curves between arms. The more frequent use of subsequent daratumumab in the control group (59.7% vs. 22.5% [isatuximab group]) may have led to a diminished treatment effect between the two arms after daratumumab use and affected the power to detect statistically significant OS in the ITT analysis, given the planned number of events and sample size, which was supported by sensitivity analyses to estimate the OS treatment effect in the absence of daratumumab therapy using the RPSFT model that demonstrated a better estimate versus the ITT estimate, with overall similar results (HR=0.706, 95% CI: 0.538-0.926) in favor of the Isa-Pd arm (described in the Online Supplementary Appendix, page 17).
The RPSFT analysis estimated a counterfactual factor and indicated there was a gain in survival time of the patients
Table 2. Treatment-emergent adverse events, safety population.
Isatuximab
TEAE, N (%)
Adverse events (AE) occurring in 20% or more of patients in any group are reported. TEAE: treatment-emergent AE.
in the control arm after switching to daratumumab.
Long-term outcome measures showed continuous overall benefit for patients randomized to isatuximab versus control treatment, suggesting the prolonged benefit of isatuximab use in earlier lines without inducing more resistant disease refractory to subsequent treatments. There was a significant TTNT delay in the Isa-Pd group versus the Pd group (15.5 vs. 8.9 months). Despite subsequent daratumumab use in the Pd group and its potential benefit on PFS in the first subsequent therapy line, median PFS2 was significantly longer with Isa-Pd versus Pd (17.5 vs. 12.9 months; log-rank 1-sided P=0.0091). Based on these data, anti-CD38 therapy should be used as early as possible during the treatment continuum.
Extended follow-up to approximately 52 months revealed continued efficacy and tolerability of the isatuximab regimen, representing an important option for patients who are refractory to other treatments, such as lenalidomide. At study entry, approximately 95% of patients were refractory to IMiD agents (93% to lenalidomide, specifically). Looking at all non-daratumumab-based therapies in any subsequent line, non-IMiD- versus IMiD-based regimens appear to have better response rates after isatuximab. Similar rates of ≥VGPR were seen between groups regardless of subsequent regimens.
More patients in the control group received subsequent therapy and subsequent daratumumab therapy, as more patients in the Isa-Pd arm were still receiving study treatment at data cutoff. As expected, ORR were higher with subsequent daratumumab therapy after Pd versus Isa-Pd; however, ≥VGPR rates were similar. Daratumumab-based combination therapy led to improved responses in both arms versus monotherapy. Patients receiving daratumumab
alone or in combination as first subsequent line of therapy achieved ≥VGPR, even after Isa-Pd therapy with a short washout period. Patients who received daratumumab after Isa-Pd had shorter PFS than those who did not receive subsequent daratumumab; however, only nine patients received daratumumab after the isatuximab combination. No cross-resistance to regimens without daratumumab was observed in the isatuximab group versus the control group. In a study examining the use of isatuximab following daratumumab, improved responses were observed among those patients with longer intervals from the last daratumumab dose to the first isatuximab dose, with a disease control rate of 58.3% (last dose ≥6 months) versus 26.4% (last dose <6 months).17 Although limited by small sample size and open-label nature of the study, these results provide initial information on potential efficacy differences related to treatment sequencing. Additional studies are needed to better understand the optimal sequencing and timing of isatuximab and daratumumab in patients with RRMM. After a follow-up of approximately 52 months, more patients remained on treatment in the Isa-Pd versus Pd group. The proportion of patients discontinuing due to AE was similar in both arms, indicating no increased risk with the addition of isatuximab resulting from longer exposure in the isatuximab group. Importantly, the overall safety profile observed with Isa-Pd was not different from previous analyses of ICARIA-MM.13,14 At this final analysis, SPM were reported in 7% of patients in the isatuximab group, consistent with the second interim analysis,14 similar to the cumulative incidence of SPM in patients with MM at 10 years (7.4%),18 and with ranges reported in RRMM registry studies.19 Furthermore, SPM occurrence did not have a detrimental impact on OS in patients receiving
supporting the overall favorable benefit of the regimen. Although cross-trial comparisons must be conducted with caution, final OS analysis of the phase II ELOQUENT-3 trial demonstrated that elotuzumab, a monoclonal antibody targeting signaling lymphocytic activation molecule F7, in combination with Pd, significantly improved OS after a minimum follow-up of 45 months.20 The proportion of patients with International Staging System Stage 3 at study entry (12% vs. 22%) in the triplet combination groups was substantially lower in ELOQUENT-3 than in ICARIA-MM. Among relevant adverse prognostic factors, there was a trend toward more patients with gain(1q21) in the Isa-Pd (49.4%) versus Pd arm (39%) in ICARIA-MM; the proportion of patients with gain(1q21) in the elotuzumab and Pd arms was similar in ELOQUENT-3.21 There were more patients aged >65 years in the Isa-Pd (64.9%) versus Pd arm (54.2%), whereas it was balanced between the elotuzumab and Pd arms (63% vs. 61%).21 The use of subsequent anti-CD38 therapy was balanced between the two arms in ELOQUENT-3 (53.3% in the elotuzumab arm vs. 49.2% in the Pd arm), which may have impacted OS. Results from the phase III APOLLO trial investigating daratumumab–pomalidomide–dexamethasone versus Pd demonstrated clinically meaningful OS benefit with the daratumumab combination (34.4 vs. 23.7 months; HR=0.82, 95% CI: 0.61-1.11; not statistically significant), with a HR similar to that observed in ICARIA-MM (HR=0.78, 95% CI: 0.59-1.02).22 Patients in APOLLO received fewer prior lines of therapy and were less refractory versus patients in the ICARIA-MM study (median prior lines, 2 vs. 3; 80% vs. 94% refractory to lenalidomide; 48% vs. 77% refractory to a PI; 42% vs. 72% refractory to both lenalidomide and a PI). Long-term follow-up of the OPTIMISMM trial investigating pomalidomide-bortezomib-dexamethasone versus bortezomib-dexamethasone demonstrated a slight trend toward OS benefit (35.6 vs. 31.6 months, HR=0.94, 95% CI: 0.77-1.15; not statistically significant).23,24 These findings combined with the clinically meaningful OS benefit demonstrated in ICARIA-MM with the Isa-Pd combination indicate the value of treating patients with RRMM using a regimen that includes pomalidomide and a monoclonal antibody. Limitations of the current study include its open-label nature, the absence of patients refractory to previous daratumumab therapy, and the imbalance of the subsequent use of daratumumab that may have affected the power to detect statistically significant OS. Other limitations of OS analyses in general include the decreased number of deaths compared with cases of disease progression at any time point and the resulting decreased power of OS analysis; the influence of competing risks of death and crossover therapy on patient survival time; and the fact that median OS is approximately 1 year longer than median PFS, wherein several lines of therapy can impact OS with lesser effects of the initial experiment.25 In summary, the final OS analysis of this large, multicenter study confirmed that Isa-Pd continued to be efficacious and well tolerated after follow-up of approximately
52 months, contributing to a clinically meaningful benefit with a 6.9-month improvement in median OS, further supporting its position as a standard-of-care therapy for patients with RRMM and informing real-world practice.26
PGR reports research funding from Bristol Myers Squibb/Celgene, Karyopharm, Oncopeptides, and Takeda and participation on an entity’s board of directors or advisory committee for AstraZeneca, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Karyopharm, Oncopeptides, Protocol Intelligence, Regeneron, Secura Bio, Sanofi, and Takeda. AP reports honoraria from AbbVie, Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Sanofi, and Takeda. JS-M reports honoraria from AbbVie, Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Haemalogix, Janssen, Karyopharm, Merck Sharp & Dohme, Novartis, Regeneron, Roche, Sanofi, SecuraBio, and Takeda. MB reports honoraria from Amgen, Celgene, Janssen, Sanofi, and Takeda and participation on an entity’s board of directors or advisory committee for Amgen, Janssen, Oncopeptides, and Takeda. IS reports honoraria from Amgen, Bristol Myers Squibb, Celgene, Janssen-Cilag, Novartis, PharmaMar, Sanofi, and Takeda. FS reports research funding from Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Sanofi, and Targovax; honoraria from AbbVie, Amgen, Bristol Myers Squibb, Daiichi Sankyo, GlaxoSmithKline, Janssen, Novartis, Oncopeptides, Pfizer, Sanofi, SkyliteDX, and Takeda; and participation on an entity’s board of directors or advisory committee for AbbVie, Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Sanofi, and Takeda. PM reports honoraria from AbbVie, Amgen, Celgene, GlaxoSmithKline, Janssen, and Sanofi. MAD reports honoraria from Amgen, BeiGene, Bristol Myers Squibb, Janssen, and Takeda. JM reports honoraria from Amgen, Bristol Myers Squibb/Celgene, Takeda, Janssen, and Sanofi and participation on an entity’s board of directors or advisory committees for Amgen, Bristol Myers Squibb/Celgene, Janssen, Oncopeptides, Sanofi, and Takeda. MC reports consulting fees from Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, and Sanofi and honoraria from Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, and Sanofi. SM, FD, RZ, and MCM are employees of Sanofi and may hold stock and/or stock options. KCA reports consulting fees from AstraZeneca, Janssen, Pfizer, and Precision Biosciences. XL, S-YH, and HMP have no conflicts of interest to disclose.
Contributions
PGR, AP, JS-M, MB, IS, XL, FS, PM, MAD, S-YH, JM, MC, and HMP were investigators in the study and contributed to data acquisition. PGR and AP were co-primary investigators of the ICARIA-MM study. PGR, SM, RZ, FD, and MCM contributed to the analysis, verification, and interpretation of data for the work. KCA was the chairman of the study Steering Committee. All authors revised the work for important intellectual content and assume responsibility for data integrity and the
decision to submit this manuscript for publication, had full access to the study data, edited and reviewed manuscript drafts, and approved the final version for submission.
Acknowledgments
We thank the participating patients and their families and the study centers and investigators for their contributions to the study. Coordination of the development of this manuscript, facilitation of author discussion, and critical review were provided by Aidee Ayala Camargo, PhD, Sci Comms Director, at Sanofi. The authors thank Laure Malinge for statistical contributions to the manuscript. Medical writing support was provided by Erin Burns-Tidmore, PhD, of Envision Pharma Group, funded by Sanofi.
1. Abramson HN. The multiple myeloma drug pipeline-2018: a review of small molecules and their therapeutic targets. Clin Lymphoma Myeloma Leuk. 2018;18(9):611-627.
2. Legarda MA, Cejalvo MJ, de la Rubia J. Recent advances in the treatment of patients with multiple myeloma. Cancers (Basel). 2020;12(12):3576.
3. Ninkovic S, Quach H. Shaping the treatment paradigm based on the current understanding of the pathobiology of multiple myeloma: an overview. Cancers (Basel). 2020;12(11):3488.
4 Rajkumar SV, Kumar S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020;10(9):94.
5. Cornell RF, Kassim AA. Evolving paradigms in the treatment of relapsed/refractory multiple myeloma: increased options and increased complexity. Bone Marrow Transplant. 2016;51(4):479-491.
6. van de Donk N, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. 2018;131(1):13-29.
7. Munshi C, Aarhus R, Graeff R, Walseth TF, Levitt D, Lee HC. Identification of the enzymatic active site of CD38 by sitedirected mutagenesis. J Biol Chem. 2000;275(28):21566-21571.
8. Malavasi F, Deaglio S, Funaro A, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88(3):841-886.
9. Chillemi A, Quarona V, Antonioli L, Ferrari D, Horenstein AL, Malavasi F. Roles and modalities of ectonucleotidases in remodeling the multiple myeloma niche. Front Immunol. 2017;8:305.
10 Jiang H, Acharya C, An G, Zhong M, Feng X, Wang L, et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia. 2016;30(2):399-408.
11. Sanofi. SARCLISA [Package Insert]. Bridgewater, NJ; 2021. https://products.sanofi.us/Sarclisa/Sarclisa.pdf. Accessed August 28, 2023.
12. Tai YT, Anderson KC. Targeting CD38 alleviates tumor-induced immunosuppression. Oncotarget. 2017;8(68):112166-112167.
13. Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet.
This study was sponsored by Sanofi.
Qualified researchers can request access to patient-level data and related study documents, including the clinical study report, study protocol with any amendments, blank case report forms, statistical analysis plan, and dataset specifications. Patient-level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data-sharing criteria, eligible studies, and process for requesting access are at: https://www.vivli.org.
2019;394(10214):2096-2107.
14. Richardson PG, Perrot A, San-Miguel J, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): follow-up analysis of a randomised, phase 3 study. Lancet Oncol. 2022;23(3):416-427.
15. European Medicines Agency. Sarclisa. https://www.ema.europa. eu/en/documents/smop/chmp-post-authorisation-summarypositive-opinion-sarclisa-ii-03_en.pdf. Accessed Aug 28, 2023.
16. Richardson PG, Attal M, Campana F, et al. Isatuximab plus pomalidomide/dexamethasone versus pomalidomide/ dexamethasone in relapsed/refractory multiple myeloma: ICARIA Phase III study design. Future Oncol. 2018;14(11):1035-1047.
17 Mikhael J, Belhadj-Merzoug K, Hulin C, et al. A phase 2 study of isatuximab monotherapy in patients with multiple myeloma who are refractory to daratumumab. Blood Cancer J. 2021;11(5):89.
18. Wang J, Lv C, Zhou M, Xu JY, Chen B, Wan Y. Second primary malignancy risk in multiple myeloma from 1975 to 2018. Cancers (Basel). 2022;14(19):4919.
19 Musto P, Anderson KC, Attal M, et al. Second primary malignancies in multiple myeloma: an overview and IMWG consensus. Ann Oncol. 2017;28(2):228-245.
20 Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for relapsed/refractory multiple myeloma: final overall survival analysis from the randomized phase II ELOQUENT-3 trial. J Clin Oncol. 2023;41(3):568-578.
21. Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N Engl J Med. 2018;379(19):1811-1822.
22. Dimopoulos MA, Terpos E, Boccadoro M, et al. Subcutaneous daratumumab plus pomalidomide and dexamethasone (D-Pd) versus pomalidomide and dexamethasone (Pd) alone in patients with relapsed or refractory multiple myeloma (RRMM): overall survival results from the phase 3 Apollo Study. Blood. 2022;140(Suppl 1):7272-7274.
23. Richardson PG, Oriol A, Beksac M, et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with
lenalidomide (OPTIMISMM): a randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(6):781-794.
24. Beksac M, Richardson P, Oriol A, et al. Pomalidomide, bortezomib, and dexamethasone vs bortezomib and dexamethasone in relapsed or refractory multiple myeloma (OPTIMISMM): final survival outcomes from a randomized, open-label, phase 3 trial. Clin Lymphoma Myeloma Leuk. 2023;23:S27-S28.
25. Saad ED, Buyse M. Statistical controversies in clinical research: end points other than overall survival are vital for regulatory approval of anticancer agents. Ann Oncol. 2016;27(3):373-378.
26. Richardson PG, San Miguel JF, Moreau P, et al. Interpreting clinical trial data in multiple myeloma: translating findings to the real-world setting. Blood Cancer J. 2018;8(11):109.
Sæmundur Rögnvaldsson,1,2 Sigrun Thorsteinsdóttir,1,3 Elisavet Syriopoulou,4,5 Ingigerdur Sverrisdottir,1,6 Ingemar Turesson,7 Elias Eythorsson,1,2 Jon Thorir Oskarsson,1 Thorir Einarsson Long,1,7 Brynjar Vidarsson,2 Pall Torfi Onundarson,1,2 Bjarni A. Agnarsson,1,2 Margret Sigurdardottir,2 Isleifur Olafsson,2 Ingunn Thorsteinsdottir,2 Thor Aspelund,1 Gauti Kjartan Gislason,1 Andri Olafsson,1 Jon Kristinn Sigurdsson,1 Malin Hultcrantz,8 Brian G. M. Durie,9 Stephen Harding,10 Magnus Bjorkholm,4,11 Ola Landgren,12 Thorvardur Jon Love1,2 and Sigurdur Yngvi Kristinsson1,2
1Faculty of Medicine, University of Iceland, Reykjavík, Iceland; 2Landspítali – The National University Hospital of Iceland, Reykjavík, Iceland; 3Rigshospitalet, Copenhagen, Denmark; 4Karolinska Institutet, Stockholm, Sweden; 5Red Door Analytics AB, Stockholm, Sweden; 6Sahlgrenska University Hospital, Gothenburg, Sweden; 7Skåne University Hospital, Lund, Sweden; 8Myeloma Service, Department of Medicine, Memorial Sloan-Kettering Cancer Center, New York, NY, USA; 9Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Outpatient Cancer Center, Los Angeles, CA, USA; 10Binding Site Group Ltd., Birmingham, UK; 11Karolinska University Hospital, Stockholm, Sweden and 12Myeloma Program, Department of Medicine, University of Miami, Sylvester Comprehensive Cancer Center, Miami, FL, USA
Correspondence: S. Rögnvaldsson srognvald@hi.is
Received: September 25, 2023. Accepted: January 4, 2024. Early view: January 11, 2024.
https://doi.org/10.3324/haematol.2023.284365
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
There is some evidence that a prior cancer is a risk factor for the development of multiple myeloma (MM). If this is true, prior cancer should be associated with a higher prevalence or increased progression rate of monoclonal gammopathy of undetermined significance (MGUS), the precursor of MM and related disorders. Those with a history of cancer might therefore constitute a target population for MGUS screening. This two-part study is the first study to evaluate a relationship between MGUS and prior cancers. First, we evaluated whether prior cancers were associated with having MGUS at the time of screening in the Iceland Screens Treats or Prevents Multiple Myeloma (iStopMM) study that includes 75,422 individuals screened for MGUS. Next, we evaluated the association of prior cancer and the progression of MGUS to MM and related disorders in a population-based cohort of 13,790 Swedish individuals with MGUS. A history of prior cancer was associated with a modest increase in the risk of MGUS (odds ratio=1.10; 95% confidence interval: 1.00-1.20). This excess risk was limited to prior cancers in the year preceding MGUS screening. A history of prior cancer was associated with progression of MGUS, except for myeloid malignancies which were associated with a lower risk of progression (hazard ratio=0.37; 95% confidence interval: 0.16-0.89; P=0.028). Our findings indicate that a prior cancer is not a significant etiological factor in plasma cell disorders. The findings do not warrant MGUS screening or different management of MGUS in those with a prior cancer.
Monoclonal gammopathy of undetermined significance (MGUS) is the asymptomatic precursor of multiple myeloma (MM), Waldenström macroglobulinemia (WM), and other lymphoproliferative disorders (LPD).1,2 MGUS is common in the general population with a prevalence of 4.2% in subjects over the age of 50 years, but only in 0.5-1.5% per year does the MGUS progress to active malignancy.2-4 The
causes of MM, WM and other LPD are poorly understood but both genetic5 and environmental6,7 causes have been implicated. Importantly, because MGUS is the precursor of MM, WM, and related LPD, the etiology of these malignancies would be expected to involve the development or progression of MGUS.
Having cancer increases the risk of a second primary malignancy8 but MM and WM are usually not considered as second primary malignancies. Theoretically, various factors
could lead to the emergence of MM and WM or other LPD as second primary malignancies. These comprise therapy- and disease-related carcinogenic effects,9 including immune dysfunction which is critical to the progression of MGUS.10 Furthermore, there may be shared genetic or environmental factors. There is limited epidemiological evidence that having a solid or myeloid cancer decreases or increases the risk of MM, respectively.11 Similar, conflicting results have been found in the patterns of cancers in family members of those who have had WM.12 In contrast, other LPD, in particular chronic lymphocytic leukemia, have been associated with prior cancer. However, this may be due to biased detection of asymptomatic chronic lymphocytic leukemia during follow-up.13 We are not aware of any studies that have examined the risk of MGUS and its progression in those with a prior cancer. We were motivated to assess the relationship of prior cancers and the development of MGUS in the unique population-based and screened Iceland Screens, Treats, or Prevents Multiple Myeloma (iStopMM) study cohort and the progression of MGUS in a large population-based MGUS registry from Sweden with up to 36 years of follow-up. By doing so we aimed to improve our understanding of the etiology of MGUS and its progression, and thereby the pathways that lead to MM, WM, and other LPD. Furthermore, cancer is common in the general population with more than 40% of individuals estimated to develop cancer during their lifetime.14 If those who have prior cancer have elevated risks of MGUS or its progression, they may be a target population for MGUS screening.
The study included two cohorts. The first cohort was acquired from the iStopMM study, a population-based screening study for MGUS. In total, 75,422 Icelanders ≥40 years old (51% of the total eligible population) were screened by serum protein electrophoresis and free light chain assay. Those with a prior history of MM, WM or other LPD were excluded. The study has been described elsewhere.15 The second cohort was acquired from a population-based MGUS registry including 13,790 subjects diagnosed in Sweden between 1987 and 2013. This registry has also been described in greater detail elsewhere.16 Prior and subsequent cancer diagnoses were acquired from the Icelandic and Swedish cancer registries, which have both been shown to have high accuracy and timeliness.17,18 Additional diagnoses of subsequent WM and chronic lymphocytic leukemia were acquired from the Swedish Patient Registry.19 Prior cancers were analyzed as any cancer, solid cancers, non-melanoma skin cancer, and myeloid cancer (Online Supplementary Table S1). The timing of prior cancers was grouped as <1, 1-5, 5-10, and >10 years before screening.
For the statistical analysis, we first assessed the risk of MGUS at screening after a prior cancer diagnosis in the iStopMM study cohort using a case-control design. Logistic regression was used to estimate odds ratios (OR) for MGUS for those with a prior cancer diagnosis with stratification by cancer subtype, by the timing of the prior cancer, and by MGUS isotype, adjusting for sex and age as a non-linear variable using four-knot restricted cubic splines. Second, we assessed the association of a prior cancer before MGUS diagnosis in the Swedish cohort, and subsequent MGUS progression. Because MGUS can be a temporary diagnosis before the diagnosis of MM or related disorders, participants were followed from 3 months after the diagnosis of MGUS to the MGUS progression or censoring at death and end of follow-up. To account for the competing risk of death we used Fine-Grey survival models to estimate sub-distribution hazard ratios (sHR) and adjusted for age and sex.
The study was approved by the Icelandic Science Ethics Committee and the Regional Ethics Board of Stockholm.
A total of 74,654 participants in the iStopMM study were included, of whom 3,579 had MGUS. At total of 9,891 participants had at least one of a total of 10,598 prior cancer diagnoses, of whom 70%, 29%, and 1% had solid, non-melanoma skin, and myeloid cancer, respectively (Table 1). A prior history of cancer was associated with a 10% increased risk of developing MGUS overall (OR=1.10; 95% confidence interval (CI): 1.00-1.20) (Figure 1). This difference was only observable for prior cancer within a year before screening for MGUS overall and non-IgM MGUS (OR=1.36; 95% CI: 1.071.74 and OR=1.57; 95% CI: 1.18-2.08) (Figure 2). No specific prior cancer category or solid cancer subtype was found to be associated with MGUS at screening except the combined category of hepatic, biliary, and pancreatic cancer as well as cancers of the urinary tract (OR=2.59; 95% CI: 1.39-4.48 and OR=1.27; 95% CI: 1.00-1.61, respectively) (Online Supplementary Table S2). A sensitivity analysis was performed to rule out that this effect was driven by those who had previously known MGUS before screening, for whom the date of MGUS screening was moved to the date of MGUS diagnosis. This did not affect the results (data not shown). Of the 13,790 participants in the Swedish cohort, 1,834 (13%) had a prior cancer diagnosis before MGUS and 1,658 (12%) subsequently experienced MGUS progression (Table 2). Those who had any prior cancer did not have a significantly increased risk of MGUS progression overall (sHR=1.14; 95% CI: 0.97-1.34; P=0.11) but those with a prior myeloid cancer had a significantly lower risk of MGUS progression (sHR=0.37; 95% CI: 0.16-0.89; P=0.028) (Figure 3). However, only five individuals with a prior history of myeloid cancer progressed during the study period. There was no statis-
Prior cancer,* N (%)
Solid cancer#
Non-melanoma skin cancer#
Myeloid cancer#
Time from prior cancer, N (%)
years
(12.9) 6,837 (9.4) 2,854 (3.9)
Note that some participants had more than one prior cancer of different subtype. *Number of individuals with at least one prior cancer and as a ratio of all individuals in that group. #Number of prior cancer diagnoses of that subtype and as a ratio of all cancers. iStopMM: Iceland Screens, Treats, or Prevents Multiple Myeloma; MGUS: monoclonal gammopathy of undetermined significance; IQR: interquartile range.
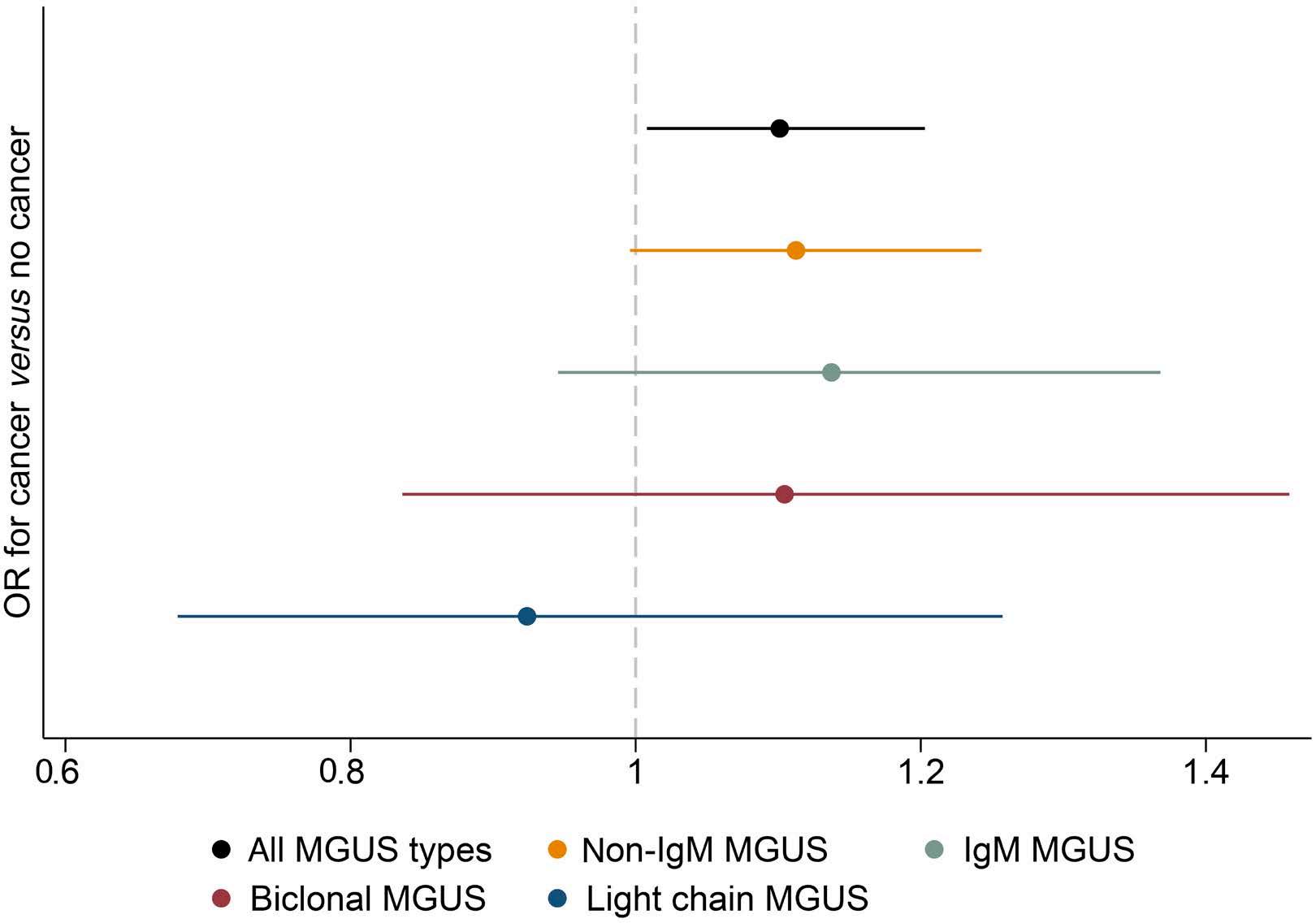
tically significant difference in progression risk overall for those with any subtype of solid or myeloid cancer, except in the case of myeloproliferative neoplasms for which those with a prior history had a lower risk of MGUS progression (Online Supplementary Table S3).
In this study based on two high-quality large population-based cohorts including close to 100,000 individuals, we found that a prior cancer is associated with a 10% increase in the risk of having MGUS later in life. Prior cancer is therefore not a clinically significant risk factor for MGUS.
Figure 1. The odds ratios of monoclonal gammopathy of undetermined significance and its subtypes for those with a prior cancer diagnosis as compared to those without a prior cancer diagnosis. OR: odds ratio; MGUS: monoclonal gammopathy of undetermined significance; IgM: immunoglobulin M.
Furthermore, this increased risk of MGUS was only observed for prior cancers less than a year before screening and because MGUS is often present for decades2,3 without progressing, it is unlikely that this is a causative association. Potential explanations include common risk factors, cytotoxic therapy lowering the polyclonal background and revealing an M protein, or reverse causality. Another potential explanation is exogenous monoclonal antibodies. However, the pattern of prior cancer types associated with MGUS is not consistent with that hypothesis. These findings indicate that MGUS is not an etiological factor in the development of MGUS and that MGUS screening in individuals with prior cancer is not warranted. A prior history of cancer was not associated with an in-
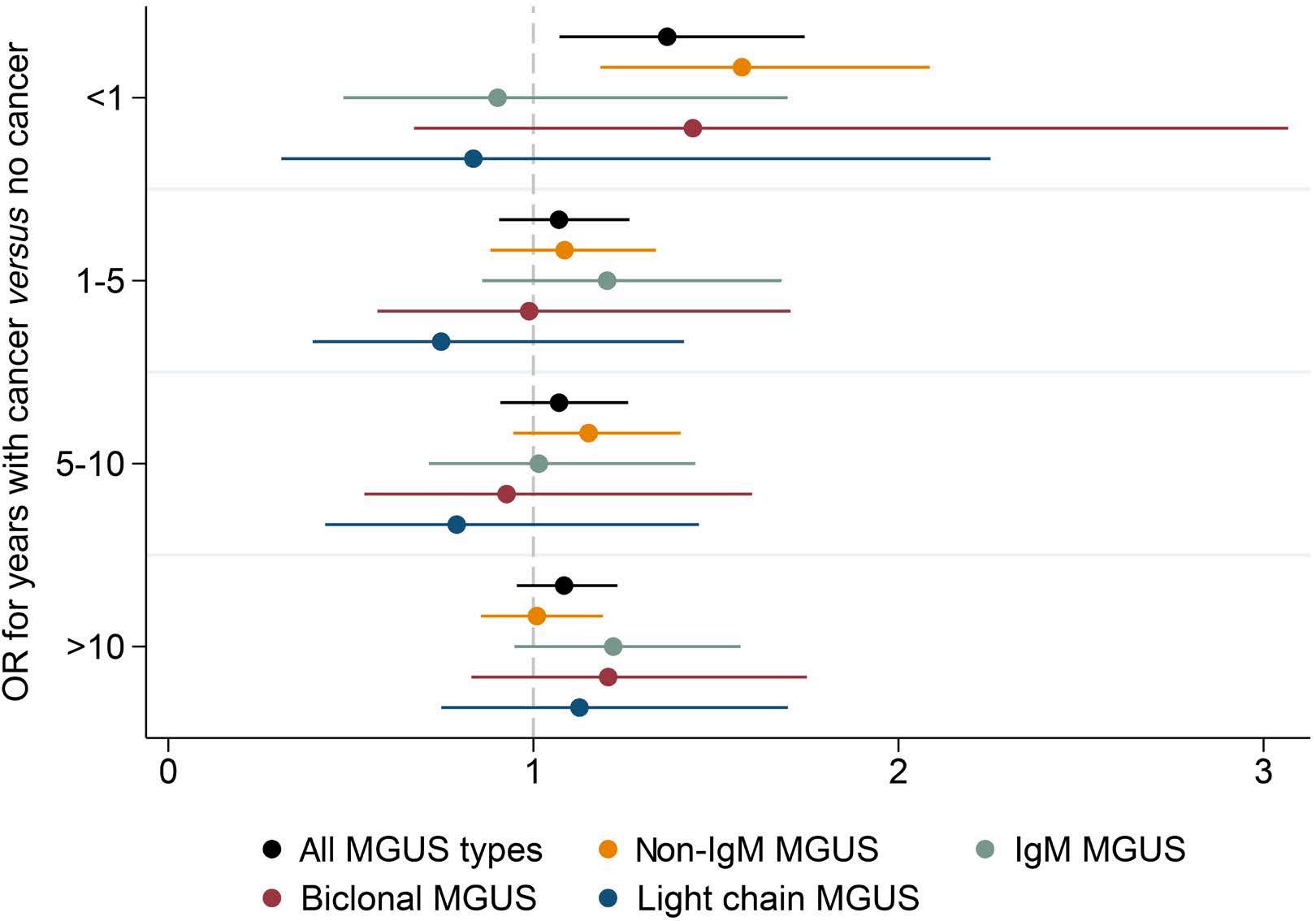
Figure 2. The odds ratios of having monoclonal gammopathy of undetermined significance for those with a prior cancer compared to those without a prior cancer by the number of years passed since the diagnosis of the prior cancer and the subtype of gammopathy. OR: odds ratio; MGUS: monoclonal gammopathy of undetermined significance; IgM: immunoglobulin M.
the affected individual.
Progression, N (%)
Multiple myeloma
Waldenström macroglobulinemia
Amyloidosis
Other LPD
*As determined by the Kaplan-Meier estimator with censoring as the outcome. IQR: interquartile range; LPD: lymphoproliferative disorders.
creased risk of MGUS progression overall. However, surprisingly, a prior myeloid cancer was associated with a significant reduction in the risk of MGUS progression. It is important to note that many myeloid cancers, particularly myeloproliferative disorders, often do not require therapy20 and that serum protein electrophoresis is often performed during the work-up of hematologic disorders in Sweden. Those individuals with MGUS and a prior myeloid cancer may therefore have had a higher rate of low-risk MGUS. Our interpretation of the results is that a prior history of cancer is not meaningfully associated with MGUS progression. These findings indicate that a prior history of cancer should not affect the management of MGUS, except in the setting of goals-of-care in relation to the general health of
No prior cancer subtype appeared to drive the increased risk of MGUS observed but an increased risk was seen in those with hepatic, biliary, and pancreatic cancers and cancer of the urinary tract. Importantly, this was in the setting of multiple comparisons and these associations may have been spurious. The findings are in contrast to those of a population-based study by Langseth et al. from Norway in which the authors found that solid tumors were associated with a lower risk of MM. Furthermore, they found myeloid cancer to be associated with a higher risk of MM, the opposite finding of our study.11 In contrast to this prior study, the current study is based on a screened cohort and long-term follow-up of a large MGUS cohort providing an unbiased epidemiological view of the pathogenesis of MM and related disorders. Furthermore, the prior study was affected by important potential biases including detection of low-grade myeloproliferative disorders in the work-up leading to the diagnosis of MM (a phenomenon we have observed in diabetes21) and misclassification of MM bone lesions as solid tumor metastasis. It is therefore likely that the current study reflects the true underlying association of prior cancers and MM more accurately.
This study has several strengths. First, the risk of MGUS was assessed using screening in the iStopMM study cohort. This avoided the selection bias that affects most other MGUS cohorts in which MGUS has been detected during the work-up for other disorders.22 Secondly, the risk of MGUS progression was assessed in a large population-based MGUS cohort with up to 36 years of follow-up. Finally, the Icelandic and Swedish cancer registries are very high quality leading to accurate determination of prior cancers and subsequent MGUS progression.17-19
The study also has important limitations. Firstly, because MGUS is asymptomatic and can be present for decades without developing into malignancy, the direction of the
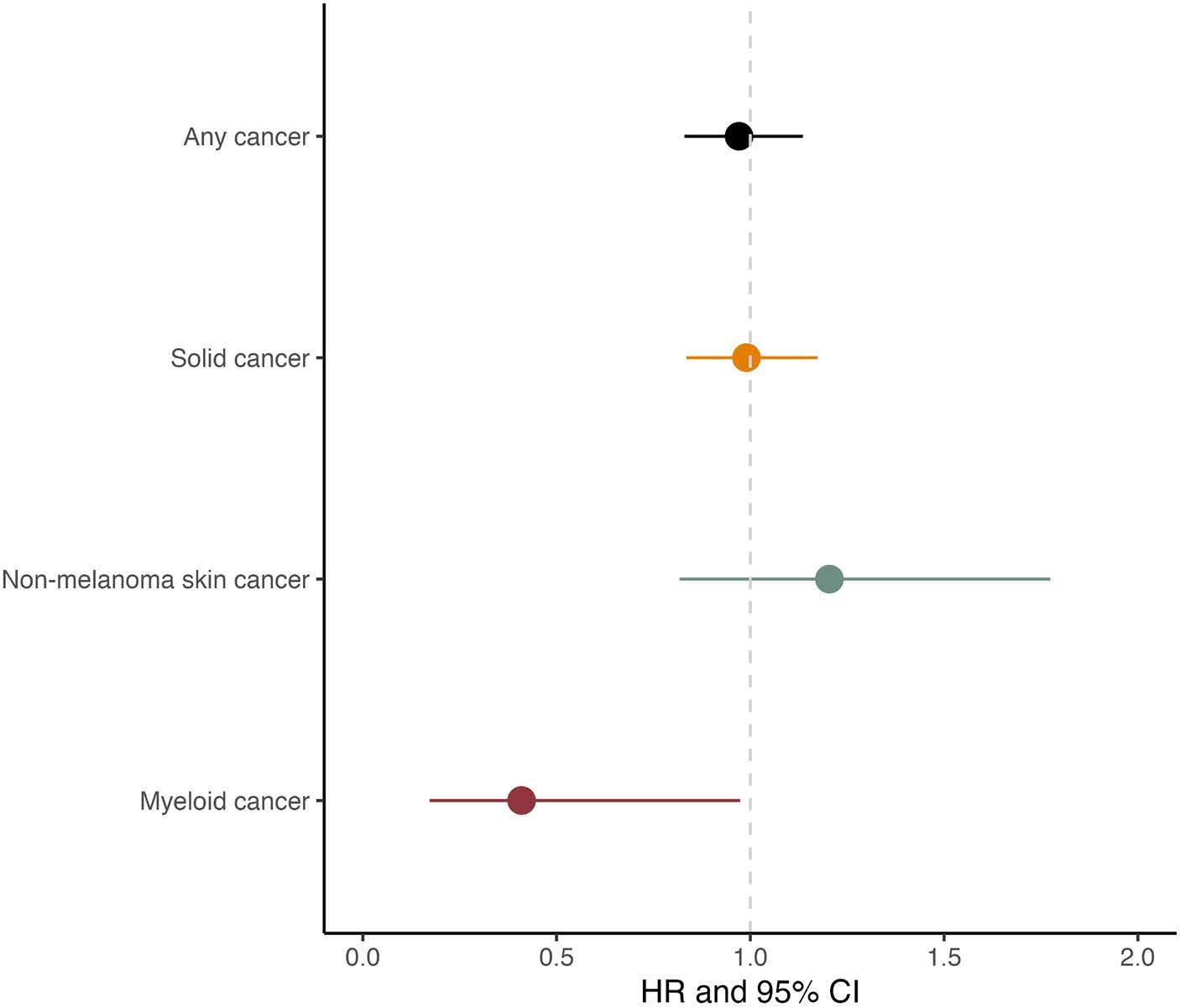
observed association is unclear. Secondly, although the total number of prior cancers was high, some cancer subtypes remained rare, precluding the detection of any rare cancer subtype-specific risks. However, due to the size of the study such associations are unlikely to hold much clinical significance. Thirdly, the study data did not include many prior cancers after the advent of immuno-oncology and targeted therapies23,24 which may affect the risk of MGUS or its progression in ways different from those of conventional therapies. Fourthly, the study populations are genetically and ethnically homogenous which may affect the nature of the prior cancers and may limit the generalizability of the results. Finally, all participants in the iStopMM study had to consent to MGUS screening and those with a prior cancer might be more or less likely to want to undergo screening; however, effect is non-differential between the groups in the analysis and should not have affected the overall result.
In conclusion, in this large study based on two complementary population-based cohorts, we found that a recent history of cancer was associated with a modest increase in the prevalence of MGUS. However, prior cancer was not associated with the risk of MGUS progression. This association is unlikely to be causal or to be of clinical significance. Based on these findings we conclude that MM, WM, and other LPD preceded by MGUS are not significant second primary malignancies. The findings do not warrant surveillance of those with prior cancer for plasma cell disorders or differential management of MGUS in those with a prior cancer. The study provides robust evidence against the hypothesis that prior cancers and associated therapies cause plasma cell disorders and can help guide the attention of investigators towards other potential risk factors that could improve our understanding of the pathogenesis of plasma cell disorders and lead to potential avenues of prevention.
Figure 3. A forest plot of hazard ratios for risk of progression of monoclonal gammopathy of undetermined significance for those with any type of prior cancer and by prior cancer subtype. HR: hazard ratio; CI: confidence interval.
MB has been part of a grant committee for Incyte; sat on educational program committees for Roche, Pfizer, and Bristol-Myers Squibb; acted as a consultant or advisor for Janssen-Cilag and Schain Research; and received a research grant from Takeda. MH has received research grants from Amgen, GlaxoSmithKline and Daiichi Sankyo Company; and has acted as a consultant or advisor for Bristol-Meyers Squibb, Curio Science, GlaxoSmithKline, and Intellisphere. SH is currently employed at The Binding Site Ltd. OL has received research funding from the National Cancer Institute/ National Institutes of Health, Food and Drug Administration, Leukemia & Lymphoma Society, Rising Tide Foundation, Multiple Myeloma Research Foundation, International Myeloma Foundation, Paula and Rodger Riney Foundation, Tow Foundation, Myeloma Solutions Fund, Perelman Family Foundation, Amgen, Celgene, Janssen, Takeda, Glenmark, Seattle Genetics, and Karyopharm; has received honoraria for scientific talks or participated in advisory boards for Adaptive, Amgen, Binding Site, Bristol-Meyers Squibb, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer; and has sat on independent Data Monitoring Committees for Takeda, Merck, and Janssen. SYK has received research funding from Amgen and Celgene; and has sat on an independent Data Monitoring Committee for Jansen.
SR conceived the study and its design with further input from ST, IS, EE, JTO, TEL, and SYK. Data were analyzed by SR and ES. ITu, MB, and SYK collected data from Sweden. GKG, AO, and JKS curated data with supervision from TA. SYK supervised the project and the iStopMM study. SR, ST, IS, EE, JTO, TEL, BV, PTO, BAA, MS, IO, ITh, MH, BGMD, SH, OL, TJL, and SYK are part of the iStopMM team of investigators. All co-authors had access to and edited the manuscript.
The work of this study was supported by research funding from Landspítali-the National University Hospital of Iceland, the International Myeloma Foundation, the European Research Council, Rannís – the Icelandic Center for Research, the Swedish Cancer Society, and Thorsman’s Foundation. OL is supported by a Sylvester Comprehensive Cancer Center NCI core grant (P30 CA 240139), Riney Family Multiple My-
1. Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412-5417.
2. Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood. 2003;102(10):3759-3764.
3. Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362-1369.
4 Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet. 2010;375(9727):1721-1728.
5. Canzian F, Piredda C, Macauda A, et al. A polygenic risk score for multiple myeloma risk prediction. Eur J Hum Genet. 2022;30(4):474-479.
6. Thordardottir M, Lindqvist EK, Lund SH, et al. Obesity and risk of monoclonal gammopathy of undetermined significance and progression to multiple myeloma: a population-based study. Blood Adv. 2017;1(24):2186-2192.
7. Landgren O, Shim YK, Michalek J, et al. Agent orange exposure and monoclonal gammopathy of undetermined significance: an Operation Ranch Hand veteran cohort study. JAMA Oncol. 2015;1(8):1061-1068.
8. Zheng X, Li X, Wang M, et al. Second primary malignancies among cancer patients. Ann Transl Med. 2020;8(10):638.
9. van den Boogaard WMC, Komninos DSJ, Vermeij WP. Chemotherapy side-effects: not all DNA damage is equal. Cancers (Basel). 2022;14(3):627.
10. Lopes R, Caetano J, Ferreira B, Barahona F, Carneiro EA, João C. The immune microenvironment in multiple myeloma: friend or foe? Cancers (Basel). 2021;13(4):625.
11. Langseth ØO, Myklebust TÅ, Johannesen TB, Hjertner Ø, Waage A. Patterns of previous and secondary malignancies in patients with multiple myeloma. Eur J Haematol. 2021;106(4):529-536.
12. Hanzis C, Ojha RP, Hunter Z, et al. Associated malignancies in patients with Waldenström’s macroglobulinemia and their kin. Clin Lymphoma Myeloma Leuk. 2011;11(1):88-92.
13. van den Broek EC, Liu L, Posthuma EFM, Janssen-Heijnen MLG, Coebergh JWW, Soerjomataram I. Increased risk of chronic lymphocytic leukaemia among cancer survivors in the Netherlands: increased detection, causal factors or both? Ann
eloma Research Program Fund, Tow Foundation, Myeloma Solutions Fund, and Perelman Family Foundation.
Data-sharing statement
Current approvals do not allow for sharing of the underlying study data. However, data may be shared with other investigators pending the review of the iStopMM investigators and the Icelandic Scientific Ethics Committee.
Hematol. 2014;93(1):157-162.
14 Sasieni PD, Shelton J, Ormiston-Smith N, Thomson CS, Silcocks PB. What is the lifetime risk of developing cancer?: the effect of adjusting for multiple primaries. Br J Cancer. 2011;105(3):460-465.
15. Rögnvaldsson S, Love TJ, Thorsteinsdottir S, et al. Iceland Screens, Treats, or Prevents Multiple Myeloma (iStopMM): a population-based screening study for monoclonal gammopathy of undetermined significance and randomized controlled trial of follow-up strategies. Blood Cancer J. 2021;11(5):94.
16. Kristinsson SY, Björkholm M, Goldin LR, McMaster ML, Turesson I, Landgren O. Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/ Waldenström macroglobulinemia patients: a population-based study in Sweden. Blood. 2008;112(8):3052-3056.
17 Sigurdardottir LG, Jonasson JG, Stefansdottir S, et al. Data quality at the Icelandic Cancer Registry: comparability, validity, timeliness and completeness. Acta Oncol. 2012;51(7):880-889.
18. Smith Jervelund S, De Montgomery CJ. Nordic registry data: value, validity and future. Scand J Public Health. 2020;48(1):1-4.
19 Turesson I, Linet MS, Björkholm M, et al. Ascertainment and diagnostic accuracy for hematopoietic lymphoproliferative malignancies in Sweden 1964-2003. Int J Cancer. 2007;121(10):2260-2266.
20. Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29(5):573-582.
21. Shah UA, Rögnvaldsson S, Derkach A, et al. Diabetes mellitus and risk of plasma cell and lymphoproliferative disorders in 94,579 cases and 368,348 matched controls. Haematologica. 2022;107(1):284.
22. Sigurbergsdóttir AÝ, Rögnvaldsson S, Thorsteinsdottir S, et al. Disease associations with monoclonal gammopathy of undetermined significance can only be evaluated using screened cohorts: results from the Iceland Screens, Treats, or Prevents Multiple Myeloma (iStopMM) study. Haematologica. 2023;108(12):3392-3398.
23. Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395(10229):1078-1088.
24. Yu JX, Hubbard-Lucey VM, Tang J. Immuno-oncology drug development goes global. Nat Rev Drug Discov. 2019;18(12):899-900.
Jie Zhou,1,2* Yanyan Xu,3* Jinhui Shu,1,2 Haojie Jiang,3 Linlin Huang,1,2 Min Xu,1,2 Junling Liu,3 Yu Hu1,2 and Heng Mei1,2
1Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei; 2Hubei Clinical Medical Center of Cell Therapy for Neoplastic Disease, Wuhan, Hubei and 3Department of Biochemistry and Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, China
*JZ and YX contributed equally as first authors.
Abstract
Correspondence: H. Mei hmei@hust.edu.cn
Y. Hu
dr_huyu@126.com
Received: July 2, 2023.
Accepted: January 23, 2024. Early view: February 1, 2024.
https://doi.org/10.3324/haematol.2023.283874
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Breakthrough treatment for refractory and relapsed immune thrombocytopenia (ITP) patients is urgently needed. Autoantibody-mediated platelet clearance and megakaryocyte dysfunction are important pathogenic mediators of ITP. Glycoprotein (GP) Ibα is a significant autoantigen found in ITP patients and is associated with poor response to standard immunosuppressive treatments. Here, we engineered human T cells to express a chimeric autoantibody receptor (CAAR) with GPIbα constructed into the ligand-binding domain fused to the CD8 transmembrane domain and CD3ζ-4-1BB signaling domains. We performed cytotoxicity assays to assess GPIbα CAAR T-cell selective cytolysis of cells expressing anti-GPIbα B-cell receptors in vitro. Furthermore, we demonstrated the potential of GPIbα CAAR T cells to persist and precisely eliminate GPIbα-specific B cells in vivo. In summary, we present a proof of concept for CAAR T-cell therapy to eradicate autoimmune B cells while sparing healthy B cells with GPIbα CAAR T cells that function like a Trojan horse. GPIbα CAAR T-cell therapy is a promising treatment for refractory and relapsed ITP patients.
Primary immune thrombocytopenia (ITP) is a bleeding disorder mainly mediated by pathogenic anti-platelet autoantibodies secreted by autoreactive B cells and plasma cells, eventually leading to accelerated platelet destruction and megakaryocyte dysfunction.1-3 Platelet autoantibodies are predominantly directed against the platelet glycoproteins (GP) IIb/IIIa (CD41/CD61) and GPIb/IX (CD42b/CD42c/CD42a) in ITP.4,5 B-cell-targeted therapies in ITP patients appear to be well tolerated short term but lack long-term tolerance, and many patients relapse after drug withdrawal. Only 20% to 30% of patients treated with CD20-targeted B-cell depletion therapy (rituximab) can achieve a long-term response (5 years).6-8 A possible reason for this therapeutic failure is the presence of rituximab-resistant splenic memory B cells and long-lived plasma cells (LLPC) secreting high-affinity autoantibodies.9-12 Failure of splenectomy therapy may be due to the presence of peripheral memory B cells and bone marrow LLPC in ITP patients.9,13 Thus, novel therapeutic strategies that eradicate autoreactive B cells (including
memory B cells) while sparing healthy B cells would significantly advance ITP treatment.
Chimeric antigen receptor (CAR) T-cell therapy has achieved remarkable success in neoplastic hematologic disorders, especially B-cell malignancies, with the cost of eradicating normal B cells. Based on CAR T-cell therapy, the concept of chimeric autoantibody receptor T cells (CAAR T) was proposed and applied to the treatment of pemphigus14 and muscle-specific tyrosine kinase myasthenia gravis.15 The extracellular ligand-binding domain of the CAAR structure was designed to contain autoantigens, thus mediating the specific cytolysis of autoreactive B cells by T cells in the above studies.
GPIbα, a surface membrane protein of platelets, initiates signaling events within platelets by binding to the A1 domain of von Willebrand factor (VWF).16,17 In ITP, platelet-associated anti-GPIb/IX antibodies are associated with a lower platelet count18,19 and inadequate responses to corticosteroids, intravenous immunoglobulin (IVIg), rituximab, and recombinant human thrombopoietin (rhTPO) compared with anti-GPIIb/IIIa antibodies.20-24 Epitope mapping of ITP
sera containing anti-GPlb/lX antibodies revealed that most autoepitopes are in different sites in GPIbα 25 In addition, anti-GPIbα antibodies can induce profound irreversible thrombocytopenia by an Fc-independent mechanism.26 As the intermembrane distance of the immunologic synapse is a critical design parameter of the ligand-binding domain,27 various truncated forms of GPIbα were used as the targeting domain in the study. In this work, we proposed a new strategy for immunotherapy for ITP in which autoantigen-modified T cells function like a Trojan horse to trap autoreactive B cells and perform specific killing. We constructed the GPIbα fragment into the second-generation CAR structure and verified that GPIbα CAAR T cells function as expected in vitro and in vivo. GPIbα CAAR T is a precision cellular immunotherapy with potential to induce complete and durable remission of refractory and relapsed ITP patients.
Animal experiments were approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (IACUC no. 3284). Experiments involving humans were conducted per the Declaration of Helsinki and were approved by the Ethics Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (IEC-J (487)).
GPIbα chimeric autoantibody receptor plasmid construction
The native GPIbα ectodomain of various lengths was constructed into a second-generation CAR structure containing a CD8α hinge/transmembrane region, 4-1BB co-stimulatory domain, and intracellular CD3ζ domain. Lentiviral vectors with or without green fluorescent protein (GFP) expression were used. GFP was linked with the CAAR fragment to facilitate the detection of GPIbα CAAR expression. The GPIbα mutant plasmid was constructed using the QuikChange Site-Directed Mutagenesis Kit.
Modified light transmission analysis
CAAR3 and GPIbα residue 233-mutated CAAR3 (CAAR3-mutg233k) were expressed on the Jurkat T-cell surface with lentivirus transduction. Jurkat-CAAR3-T and Jurkat-CAAR3-mutg233k T cells were resuspended to a concentration of 107 cells/mL, and 300 μL of cell suspension was added to a colorimetric cup. Then, 5 μg/mL human VWF was added in the presence of 0.25 mg/mL ristocetin. Under the shear force and the induction of ristocetin,16 the VWF protein binds to Jurkat T cells expressing extracellular domains of GPIbα (CAAR3) on the surface, eventually triggering an aggregation reaction of the cells, which can be measured as an increase in light transmission collected by a platelet aggregation analyzer.
In vitro cytotoxicity assays
Donor-matched non-transduced T (NTD-T) cells or GPIbα CAAR T cells were co-incubated with control hybridoma (PE) and the target anti-GPIbα hybridoma cells (APC) at an effector to target (E:T) ratio of 5:1, while the ratio of target hybridoma cells to control hybridoma cells was 3:1. Cytotoxicity was evaluated at 24 hours using flow cytometry based on the changes in the ratio of target hybridoma cells to control hybridoma cells.
Anti-GPIbα hybridoma xenograft models
Six-to-eight-week-old NSG mice (NOD-scid IL2Rγnull) were intravenously inoculated with anti-GPIbα B cells (105 cells per mouse), followed 2 days later by injection with LBD-mutg233k T cells or NTD T cells (107 cells per mouse), and monitored for engraftment and therapeutic response by the IVIS® system every few days.
Cytometric bead array
The microbeads carrying different fluorescence (APC) intensities were separately coated with monoclonal antibodies: anti-GPIX (clone: sz-1), anti-granule membrane protein (GMP) 140 (clone: sz-51), anti-GPIb (clone: sz-2), anti-GPIIb (clone: sz-22), and anti-GPIIIa (clone: sz-21). Platelets were isolated from ITP patients or healthy controls, then platelet lysate was incubated with the coated microbeads, followed by FITC-conjugated goat-anti-human IgG polyclonal antibodies, and finally analyzed with flow cytometry.
GPIbα enzyme-linked immunospot assay
GPIbα CAAR T-cell-specific cytolysis of anti-GPIbα antibody-producing human B cells was measured using an enzyme-linked immunospot (ELISpot) assay. peripheral blood mononucelar cells isolated from patients and healthy control were stimulated and then incubated with GPIbα CAAR T cells, anti-CD19 CAR T cells, or NTD T cells on the polyvinylidene difluoride-bottomed microplate coated with human GPIbα protein, anti-human IgG antibody, or BSA. Specific antibodies secreted by B cells will form spots on the microplates, which can be counted. Details on these protocols and additional methods are provided in the Online Supplementary Appendix.
Development of GPIbα chimeric autoantibody receptor plasmids
GPIbα is the largest subunit of the GPIb-IX complex (Figure 1A), and 282 N-terminal membrane-distal residues, which are composed of seven leucine-rich repeats (LRR), make up the ligand-binding domain (LBD), which binds to VWF and other ligands. The VWF binding region is followed by a heavily O-glycosylated macroglycopeptide domain and a mechanosensory domain (MSD).28 Anti-LBD antibodies are
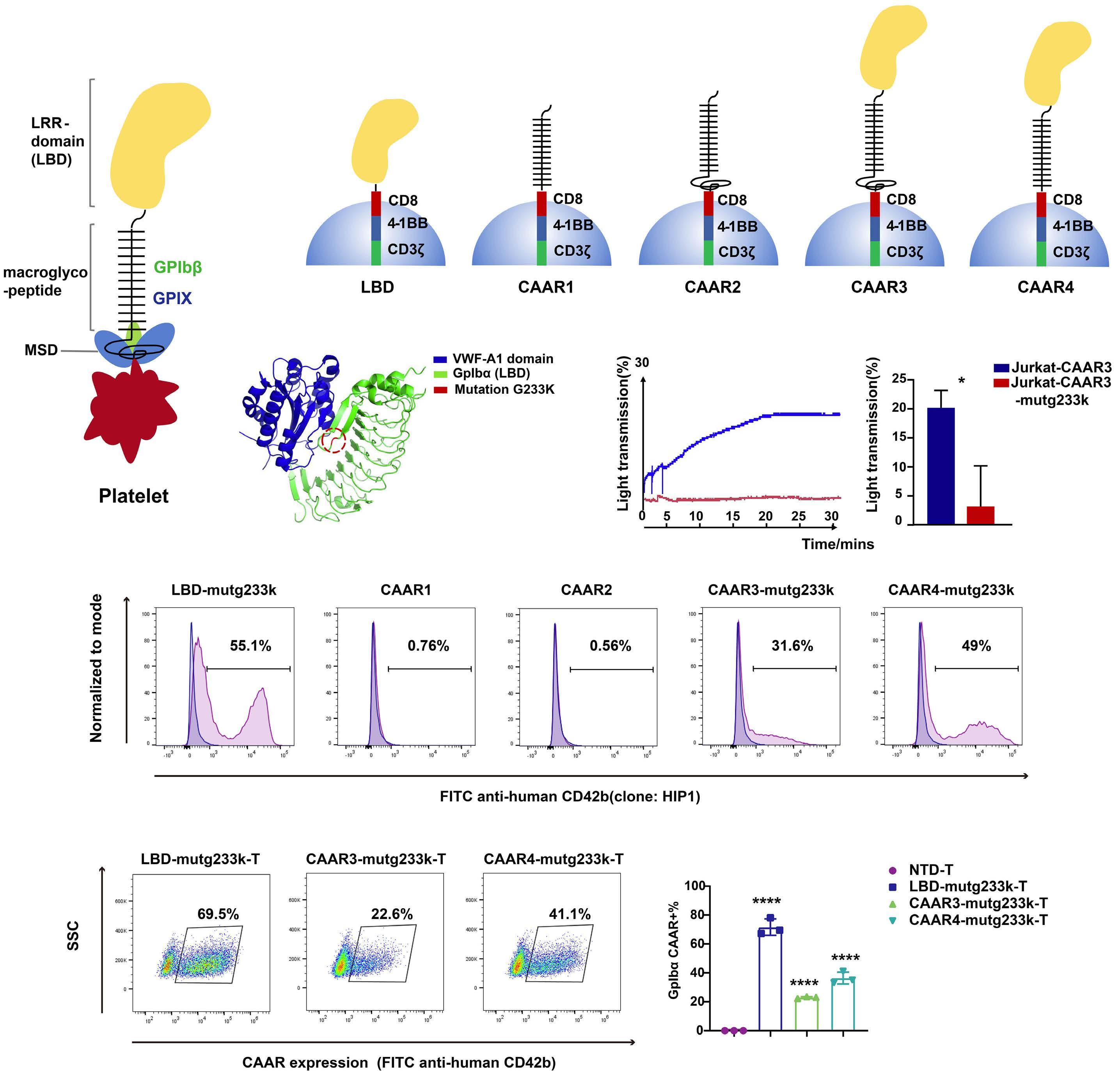
Figure 1. Construction and validation of the GPIbα chimeric autoantibody receptor structure. (A) The GPIb-IX complex contains GPIbα, GPIbb (green), and GPIX (blue); GPIbα is the largest subunit. GPIbα is composed of seven leucine-rich repeats (LRR), make up the ligand-binding domain (LBD), followed by a heavily O-glycosylated macroglycopeptide domain and a mechanosensory domain (MSD). (B) Schematic of GPIbα chimeric autoantibody receptor (CAAR) constructs comprising diverse lengths of the native GPIbα ectodomain, followed by the CD8α transmembrane domain and 4-1BB-CD3ζ intracellular co-stimulatory and activation domains, named LBD, CAAR1, CAAR2, CAAR3, and CAAR4. (C) Crystal structures (Protein data bank ID: 1m10) of the NH2-terminal domain of GPIbα (green, residues 1 to 290) and its complex with the von Willebrand Factor (VWF) A1 domain (blue, residues 498 to 705) are shown, and the loss-of-function mutation at residue 233 of GPIbα (red, G233K) is indicated. (D) Ristocetin-induced VWF binding to GPIbα fragments expressed on Jurkat T cells. CAAR3 and residue 233-mutated CAAR3 (CAAR3-mutg233k) were expressed on the surface of Jurkat T cells, and cell aggregation was measured as an increase in light transmission (%) (N=3; *P<0.05). (E) In order to identify the mature conformational epitopes, mutated-GPIbα CAAR plasmids were transfected into HEK293T cells, and then the cells were stained with an anti-human CD42b (GPIbα) antibody (clone: HIP1). (F) At a multiplicity of infection (MOI) of 25, primary human T cells were transduced with LBD-mutg233k-CAAR, CAAR3-mutg233k-CAAR, and CAAR4mutg233k-CAAR lentivirus, and CAAR expression was determined using an anti-human CD42b antibody (clone: HIP1). The transduction efficiency of GPIbα CAAR+ in T cells from 3 healthy donors was calculated. SSC: side scatter; NTD T: non-transduced T cells. ****P<0.0001.
associated with refractoriness to IVIg or steroids in ITP patients,23,26,29 and the MSD juxtamembrane portion of GPIbα is related to ligand binding and signal transduction.30 Native GPIbα ectodomains of various lengths (LBD/CAAR1/CAAR2/ CAAR3/CAAR4) were constructed into a second-generation CAR structure containing a CD8α hinge/transmembrane region, 4-1BB co-stimulatory domain, and intracellular CD3ζ domain, as shown in Figure 1B and Online Supplementary Figure S1. LBD CAAR contains the LRR domain; CAAR1 contains the macroglycopeptide domain; CAAR2 contains the macroglycopeptide domain plus MSD; CAAR3 comprises the LRR domain, the macroglycopeptide domain, and MSD; and CAAR4 consists of the LRR domain and the macroglycopeptide domain. The five CAAR structures were first expressed on the surface of HEK293T cells, and the proper conformation of the CAAR structures was verified by the anti-human platelet GPIbα antibody (clone: HIP1). As shown in Online Supplementary Figure S2A, CAAR expression was successfully detected in LBD CAAR, CAAR3, and CAAR4 but not in CAAR structures that did not contain LBD (CAAR1, CAAR2), probably because this antibody recognizes a site in the LBD. The above results preliminarily proved that the conformational epitope of GPIbα CAARs is consistent with that of native human platelet GPIbα. CAAR expression in primary human T cells was also detected. T cells were transduced with GPIbα-CAAR lentivirus (multiplicity of infection 25 to 100), and the transduction efficiencies of GPIbα CAAR T cells were determined with an anti-human platelet GPIbα antibody (clone: HIP1) by flow cytometry (Online Supplementary Figure S2B).
GPIbα G233K mutation inhibited von Willebrand factor binding to GPIbα chimeric autoantibody receptor T cells The platelet surface membrane protein GPIbα participates in platelet plug formation by binding to the A1 domain of VWF, which is already attached to the subendothelium.16 The b-switch region (AAs 227-241) of GPIbα plays an essential role in forming the GPIbα-VWF complex, and residue 233 plays a critical role in regulating VWF binding.31-33 The crystal structures of the NH2-terminal domain of GPIbα (residues 1 to 290) and its complex with the VWF-A1 domain (residues 498 to 705) are shown in Figure 1C. In order to avoid affecting the regular role of the GPIbα-VWF complex in the process of hemostasis after CAAR T-cell infusion, we chose residue 233 (G233K) of GPIbα for mutation in the CAAR structure. CAAR3 and GPIbα residue 233-mutated CAAR3 (CAAR3-mutg233k) were expressed on the Jurkat T-cell surface with lentivirus transduction. As shown in the aggregometry measurements in Figure 1D, under shear force and induction by ristocetin, the VWF protein bound to Jurkat-CAAR3 T cells, triggering a cell aggregation reaction, and an increase in light transmission was observed, which was not detected in the Jurkat-CAAR3-mutg233k T-cell group. The results demonstrated that the G233K mutation of GPIbα expressed on the cell surface strongly
inhibits its binding to VWF, which could alleviate possible off-target effects after CAAR T-cell infusion in vivo. The mutated GPIbα CAAR structures were named LBD-mutg233k, CAAR3-mutg233k, and CAAR4-mutg233k, and the integrity of the CAAR epitopes was confirmed with HIP1 (Figure 1E). The transduction efficiencies of the mutated GPIbα CAAR T cells are presented in Figure 1F. At a multiplicity of infection (MOI) of 25, LBD-mutg233k T cells exhibited the highest transduction efficiency, followed by CAAR4-mutg233k T and CAAR3-mutg233k T cells. The size of the CAAR fragment was an essential factor affecting the transfection efficiency as indicated, and the results from three healthy donor T cells are shown (Figure 1F).
Generation of four anti-GPIbα hybridomas with different chimeric autoantibody receptor binding sites
In order to generate hybridomas and antibodies that bind to human platelet GPIbα, washed human platelet lysate was used as the antigen for mouse immunization, and mouse serum was screened by GPIbα enzyme-linked immunsorbant assay (ELISA). The four selected anti-GPIbα hybridomas were named Gvb1, Gvb2, Gvb3, and Gvb4. Human platelets were incubated with each APC-conjugated hybridoma antibody (Gvb1/Gvb2/Gvb3/Gvb4) and FITC-anti-human CD42b antibody simultaneously to analyze the ability of the acquired antibodies to bind to human platelets. The flow cytometry results (Figure 2A) showed that the four anti-GPIbα antibodies could bind to human platelets, demonstrating their ability to bind with the native conformation epitope of GPIbα. As target cell surface antigen density might affect CAR T-cell cytolytic efficiency,27 the density of BCR on the surface of each hybridoma was then measured with an anti-mouse IgG antibody (APC-conjugated). The BCR mean fluorescence intensity (MFI) of the four hybridomas is presented in Figure 2B, varying among the anti-GPIbα hybridomas, and the Gvb3 hybridoma showed the highest surface BCR density, followed by the Gvb1, Gvb2, and Gvb4 hybridomas.
In order to verify the binding ability of the hybridoma antibodies to mutated GPIbα CAAR and identify the binding sites between them, we further expressed mutated GPIbα CAAR in HEK293T cells. The proportion of cells that bound to the hybridoma antibodies in total GPIbα CAAR-GFP-expressing cells (Q2/(Q2+Q3)) was determined and represented the binding force between the hybridoma antibodies and CAAR. As shown in Figure 2C-F, Gvb1, Gvb2, and Gvb3 could bind to LBD-mutg233k-CAAR, CAAR3-mutg233k-CAAR, and CAAR4-mutg233k-CAAR, while Gvb4 could bind to all GPIbα CAAR. The locus recognized by Gvb4 is thus in the overlapping part of LBD-mutg233k-CAAR and CAAR1/CAAR2. The findings revealed that though the binding epitopes of each hybridoma antibody to the CAAR should be similar, the binding efficiencies varied. Gvb4 could bind to all GPIbα CAAR, proving that its binding site is the same site shared by the above CAAR but exhibited different binding effi -
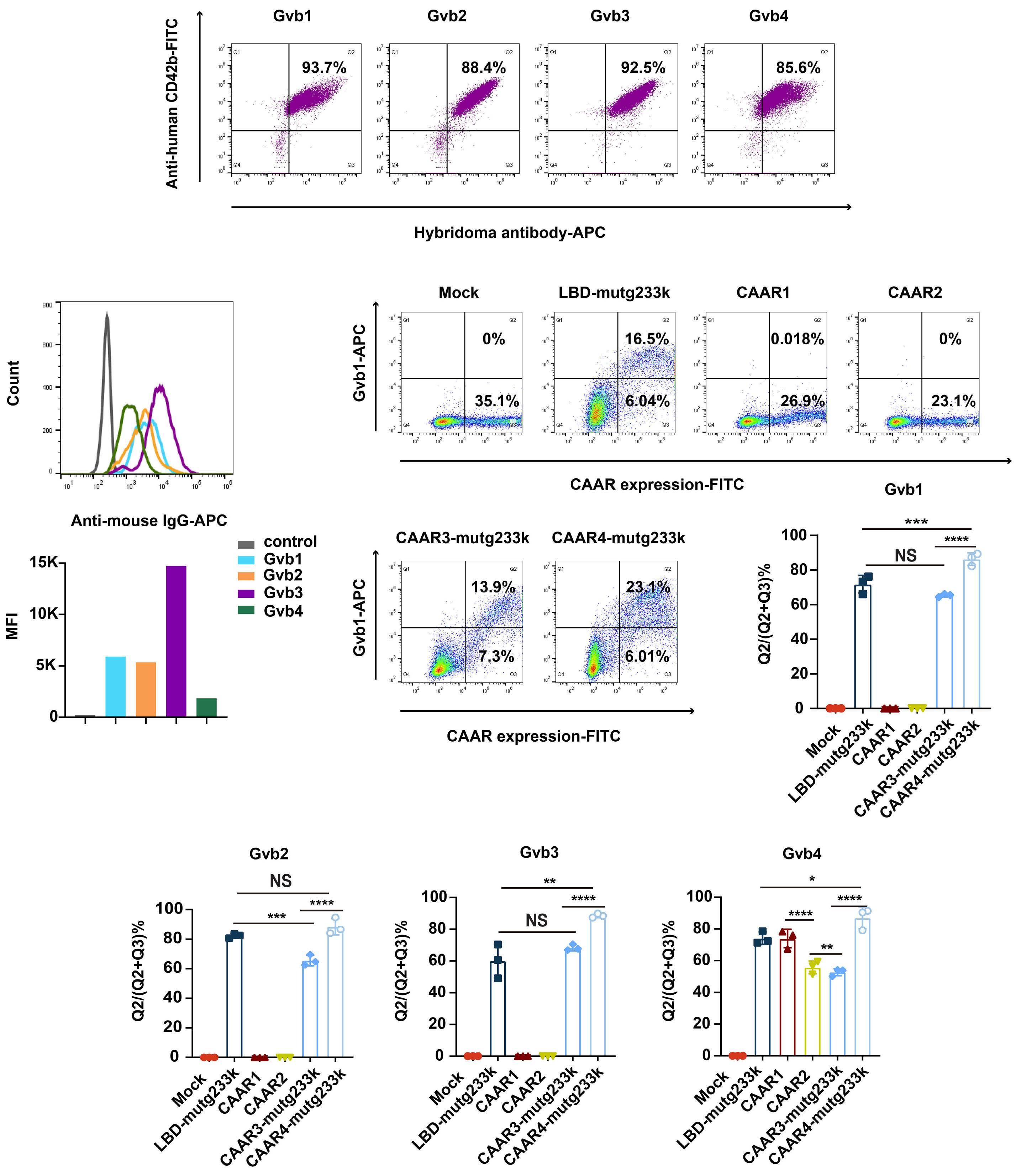
Figure 2. Generation of four anti-GPIbα hybridoma antibodies and validation of their binding epitopes with GPIbα chimeric autoantibody receptors. (A) Binding detection of the 4 anti-GPIbα hybridoma antibodies (Gvb1/Gvb2/Gvb3/Gvb4) to human platelets using flow cytometry. Platelets were incubated with anti-human CD42b (GPIbα) antibody (clone: HIP1) and each anti-GPIbα hybridoma antibody (APC-conjugated) simultaneously. (B) B-cell receptor (BCR) density quantification for the 4 anti-GPIbα hybridoma cells. Hybridoma cells were stained with goat anti-mouse IgG antibodies (APC-conjugated). The mean fluorescence intensity (MFI) of BCR expression is shown in a histogram and bar graph. (C-F) Identification of the different binding epitopes of the hybridoma antibodies Gvb1, Gvb2, Gvb3, and Continued on following page.
Gvb4 to mutated GPIbα chimeric autoantibody receptor (CAAR) structures. GPIbα CAAR were expressed on the surface of HEK293T cells, which were incubated with each hybridoma antibody (APC-conjugated). Green fluorescent protein (GFP) was linked with the CAAR fragment to facilitate the detection of GPIbα CAAR expression. The binding force between each hybridoma antibody and the CAAR was calculated as the ratio of cells bound to hybridoma antibodies in total GPIbα CAAR-GFP-expressing cells (Q2/(Q2+Q3)). NS: not significant P>0.05; *P<0.05; **P<0.01; ***P<0.005; ****P< 0.0001.
ciencies to these CAAR; this suggested that the different CAAR structures had different spatial conformations that may affect the binding force of the hybridoma antibody. In summary, we obtained four hybridomas with different binding sites that could simulate B cells from ITP patients with different anti-GPIbα epitopes for further CAAR T-cell cytolytic assays. As Gvb4 could bind to all the CAAR (Figure 2F), the transduction efficiencies of GPIbα CAAR T cells were determined using the APC-conjugated hybridoma antibody Gvb4, as shown in Online Supplementary Figure S2C
In vitro, GPIbα chimeric autoantibody receptor T cells exhibited specific cytolysis of autoreactive B cells In order to verify the specific lysis of autoreactive B cells by GPIbα CAAR T cells, NTD T or GPIbα CAAR T cells were incubated with each anti-GPIbα hybridoma various at ratios ranging from 1:1 to 10:1. At 16 hours postincubation, the medium supernatants were collected and measured immediately for LDH activity. GPIbα CAAR T cells demonstrated specific lysis of autoreactive B cells (Gvb1/Gvb2/ Gvb3/Gvb4) targeting different GPIbα domains in a manner dependent on the E:T ratio and NTD T cells showed no cytotoxicity (Figure 3A). Notably, the pernicious effects of GPIbα CAAR T cells varied due to variations in binding epitopes of the target cell to CAAR, and LBD-mutg233k T cells exhibited the most robust cytolysis function against the four hybridomas. We also performed cytotoxicity assays with flow cytometry to further prove the specific cytolytic function of the CAAR T cells. NTD T or LBD-mutg233k T cells were coincubated with hybridoma control and the target anti-GPIbα hybridoma cells. The original ratio of target hybridoma cells to control hybridoma cells was 3:1. As shown in Figure 3B-E, due to the specific lysis of target hybridoma cells by LBD-mutg233k T cells, the ratio of target cells to control cells in the co-culture system decreased significantly compared with that in the NTD T group. Similar results for CAAR3-mutg233k T, CAAR4-mutg233k T, CAAR1 T, and CAAR2 T cells were obtained and are presented in Online Supplementary Figure S3. High levels of the pro-inflammatory cytokines (Figure 3F) IL-2 and interferon (IFN)-γ were secreted by T cells after stimulation with each target hybridoma and varied among different GPIbα CAAR T cells and tested extremely low in the NTD T groups. Consistent with the binding assay results (Figure 2C-F), even though the binding sites of GPIbα CAAR and each hybridoma were similar, the difference brought about by the spatial conformation of the CAAR eventually resulted in differences in the binding forces and cytolytic functions of the GPIbα CAAR T cells. LBD-mutg233k T cells showed
a stable spatial conformation and robust cytolytic ability. CAAR4-mutg233k T cells, comprising the LBD and a heavily O-glycosylated macroglycopeptide domain, exhibited the most potent binding with hybridoma antibodies, and the cytolytic efficiency was inferior to that of LBD-mutg233k T cells. CAAR3-mutg233k-CAAR has an extra MSD structure and a poorer killing effect than CAAR4-mutg233k-CAAR. The MSD structure’s instability28 may influence the binding of CAAR3-mutg233k T cells and anti-GPIbα hybridomas, possibly accounting for the lower killing efficiency.
Soluble anti-GPIbα antibodies slightly affect GPIbα chimeric autoantibody receptor T-cell cytotoxicity Soluble autoantibodies can either prevent CAAR contact with anti-CAAR BCR or enhance cytotoxicity by activating CAAR T cells; therefore, their impact on CAAR T-cell cytotoxicity is erratic.14,15 In order to determine soluble anti-GPIbα antibody effects on GPIbα CAAR T-cell activity, we performed cytotoxicity assays in the presence of a wide range of concentrations (0-40 μg/mL) of monoclonal antibodies (mAb) or mixed mAb, as no quantitative data on plasma anti-GPIbα IgG concentration in ITP patients were currently available. GPIbα CAAR T-cell cytotoxicity against hybrid target cells (Gvb1/Gvb2/Gvb3/Gvb4) was mildly increased with higher effector-to-target ratios in the presence of soluble anti-GPIbα polyclonal IgG (a mixture of Gvb1, Gvb2, Gvb3, and Gvb4, and the concentration of each antibody is 5 μg/mL) (Figure 4A), as were IFN-γ levels (Figure 4B). Then, NTD T or LBD-mutg233k T cells and mixed target cells were incubated with each anti-GPIbα mAb at various concentrations (0, 10, 20, or 40 μg/mL). Cytotoxicity (Figure 4C) and IFN-γ levels (Figure 4D) were slightly affected in a concentration-related manner and varied among the anti-GPIbα mAb. The cytolytic efficiency of CAAR T cells and the IFN-γ levels were marginally enhanced or somewhat reduced with increasing antibody concentration in different groups. Therefore, we conclude that the impact of soluble anti-GPIbα mAb on CAAR T-cell function generally varies and is minimal.
GPIbα chimeric autoantibody receptor T cells demonstrated in vivo persistence and specific cytolytic activity with no apparent organ toxicity
A xenograft model was developed to assess the cytolytic efficiency and safety of GPIbα CAAR T cells in vivo. Luciferase (Luc)/GFP-expressing anti-GPIbα hybridoma cells (105 cells per mouse) and LBD-mutg233k T cells (107 cells per mouse) were intravenously injected into 6–8-week-old NSG mice, which were examined for engraftment and therapeutic re-
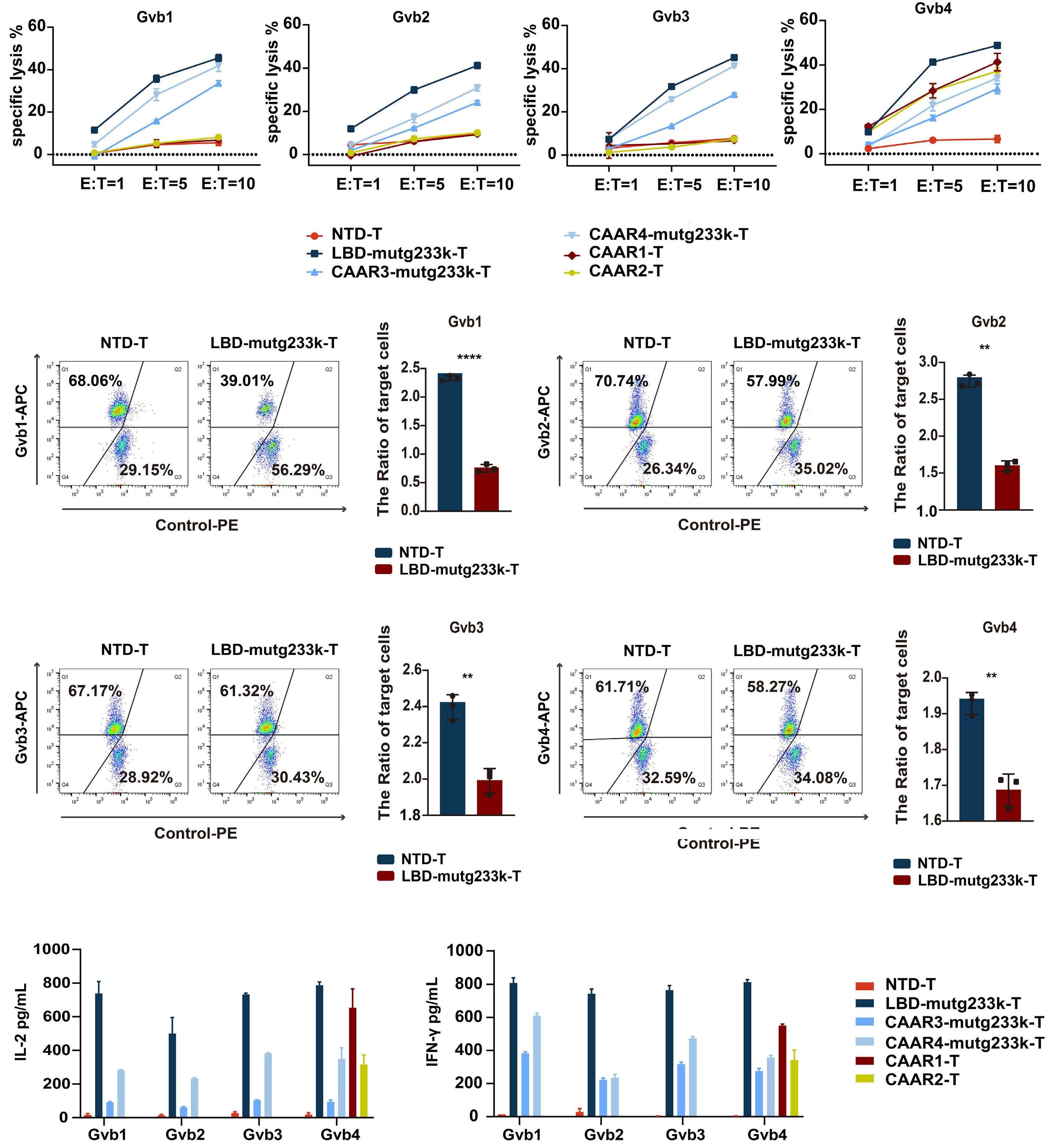
Figure 3. GPIbα chimeric autoantibody receptor expression on primary human T cells directs specific cytolysis of anti-GPIbα B cells in vitro. (A) Hybridoma cells (Gvb1/Gvb2/Gvb3/Gvb4) were co-incubated with non-transduced T cells (NTD T) or GPIbα chimeric autoantibody receptor (CAAR) T cells at the indicated effector to target (E:T) ratios for 16 hours. Specific lysis was measured by lactate dehydrogenase (LDH) release into the supernatant. (B-E) NTD T or LBD-mut223k T cells were co-incubated with control hybridoma (PE) and anti-GPIbα hybridomas (APC) at an E:T ratio of 5:1. The ratio of the target hybridoma to total control hybridoma cells was 3:1. Cytotoxicity was evaluated at 24 hours using flow cytometry, as indicated by the changes in the ratio of the target hybridoma to the control hybridoma. **P<0.01; ****P<0.0001. (F) Cytokines (interleukin [IL]-2, interferon [IFN]-γ) produced by NTD T or GPIbα CAAR T cells after 5:1 co-culture with each anti-GPIbα hybridoma cell for 16 hours, as measured in the supernatant by enzyme-linked immunosorbant assay.
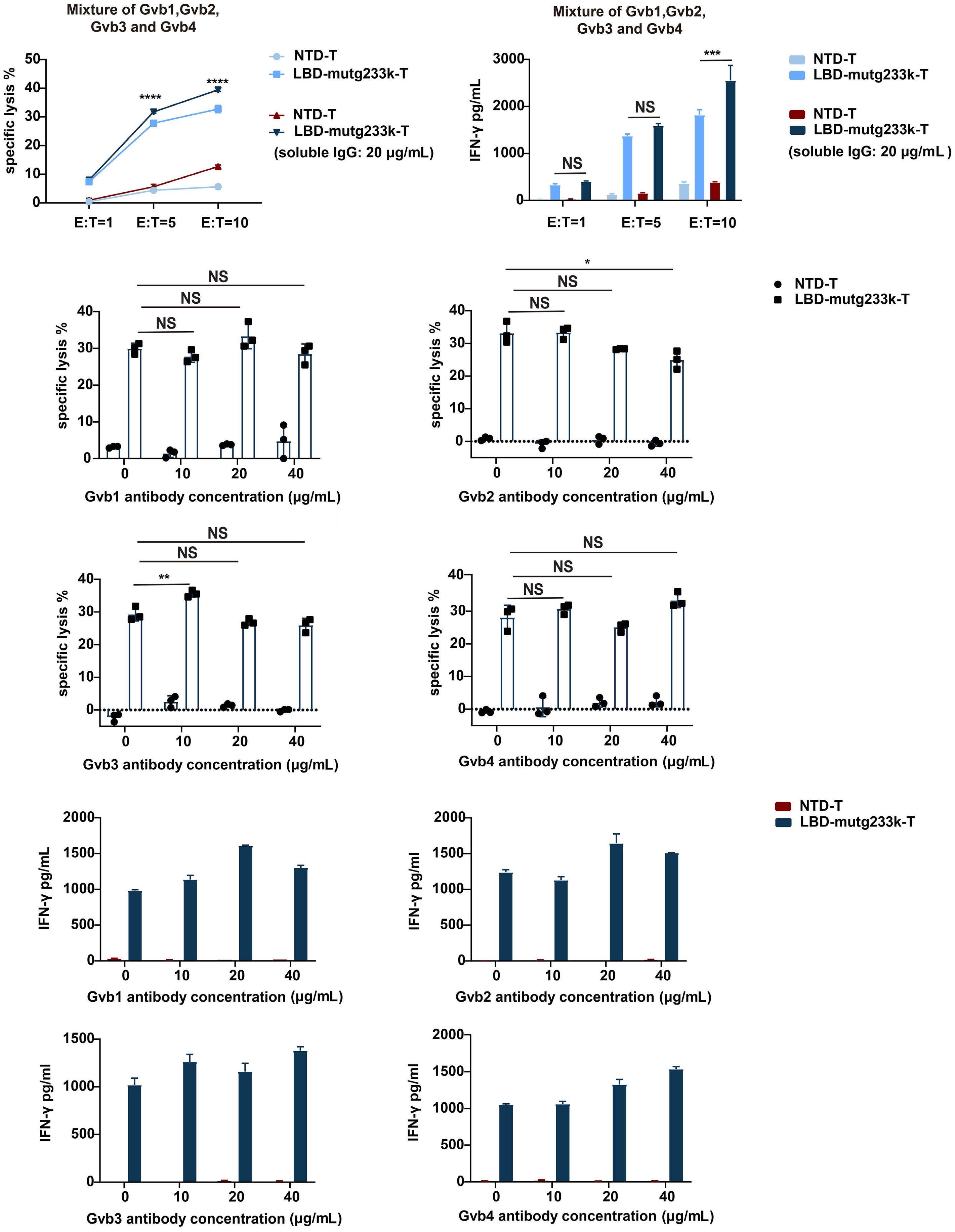
Continued on following page.
Figure 4. Evaluation of soluble anti-GPIbα antibody effects on GPIbα chimeric autoantibody receptor T-cell cytotoxicity. (A) Non-transduced T cells (NTD T) or LBD-mut223k T cells were co-incubated with a mixture of the 4 anti-GPIbα target cells at the indicated effector to target (E:T) ratios in the presence or absence of soluble anti-GPIbα immunoglobulin (Ig)G polyclonal antibody (a mixture of Gvb1, Gvb2, Gvb3, and Gvb4, and the concentration of each antibody is 5 µg/mL). Cytotoxicity was evaluated at 16 hours by lactate dehydrogenase (LDH) release assay. Meanwhile, human interferon (IFN)-γ (B) in cell culture supernatants was quantitated by enzyme-linked immunosorbant assay (ELISA) after 16 hours. (C-D) NTD T or LBD-mutg233k T cells were incubated with each anti-GPIbα IgG monoclonal antibody (0, 10, 20, or 40 μg/mL) at an E:T ratio of 5. Cytotoxicity was evaluated at 16 hours by LDH release assay, and human IFN-γ in cell culture supernatants was quantitated by ELISA after 16 hours. NS: not significant P>0.05; *P<0.05; **P<0.01; ***P<0.005; ****P<0.0001.
sponse. Mice treated with NTD T cells were used to monitor the allogenic effects. Bioluminescence imaging indicated that GPIbα CAAR T cells significantly reduced anti-GPIbα hybridoma outgrowth compared to NTD T cells (Figure 5A). From the 14th day to the 21st day, the autoreactive B-cell burden (mean ROI) of the LBD-mutg233k T-treated mice 3 and 4 (T3 and T4) decreased from 4,096.406 to 1,028.801 and 28,029.28 to 2,671.87 (photons/s/cm2/sr), respectively (Figure 5B). Total human T cells and CAAR T cells in peripheral blood (PB) were monitored every few days (Figure 5C).
PB T cells of the CAAR T-treated mice proliferated rapidly from day 18 to day 24, far exceeding the proliferation of the NTD T group, which encounters with target cells can explain. Control and LBD-mutg233k T-cell-treated mice were euthanized 21-28 days after hybridoma cell/T-cell injection, and T-cell persistence and penetration in bone marrow, spleen, and blood samples were assessed by flow cytometry. Consistent with the PB results, T cells of the CAAR T-treated mice isolated from the spleen and bone marrow exhibited a more potent ability to persist and proliferate in vivo than T cells in the control group, as shown in Figure 5D, E. Immunofluorescence imaging (Online Supplementary Figure S4A) also indicated lymphocyte infiltration and persistence in the liver and spleen. The plasma anti-GPIbα antibody titer increased in mice treated with NTD T cells from day 7 to 21. In contrast, the titers in GPIbα CAAR T-cell-treated mice were significantly reduced compared to those in NTD T-cell-treated mice by day 21 after T-cell injection (Figure 5F). The reduced serum anti-GPIbα ELISA results also reflected hybridoma control in LBD-mutg233k T-treated mice. Off-target cytotoxic effects of GPIbα-CAAR T cells on mice were not observed, as hematoxylin and eosin (H&E) staining revealed that the morphology of tissues and organs in mice did not change significantly, and serum biochemical levels were normal (Online Supplementary Figure S4B, C). In conclusion, GPIbα CAAR T cells demonstrated in vivo persistence and specific cytolytic capacity with no apparent organ toxicity.
GPIbα chimeric autoantibody receptor T cells showed potential in eradicating autoreactive B cells of immune thrombocytopenia patients
In order to test the ability of GPIbα CAAR T cells to interact with native GPIbα autoantibodies from ITP patients, a plasma antibody binding assay (Figure 6A; Online Supplementary Figure S5A) was first applied. Patients diagnosed
with primary ITP were included and tested for specific platelet autoantibodies using a cytometric bead array (Figure 6B; Online Supplementary Figure S5B). Sera from three ITP patients with anti-GPIb antibodies and healthy controls were diluted at 1:10 and 1:20 and then incubated with HEK293T cells expressing the LBD-mutg233k-CAAR structure tagged with GFP. After incubation, the cells were stained with APC-anti-human immunoglobulin (Ig)G antibodies. The flow cytometry results (Figure 6A) showed that the serum anti-GPIbα antibodies of patient 3 could react with LBD-mutg233k-CAAR, but no binding of patient 1 and patient 2 plasma antibodies to LBD-mutg233k-CAAR was detected. A possible explanation for this negative result is that platelet lysates were used to screen patients with anti-GPIb antibodies, and the level of platelet antibodies in the lysates is much higher than the platelet antibodies in the plasma. At the same time, we examined the binding reaction of plasma from patient 3 to CAAR3-mutg233kCAAR and CAAR4-mutg233k-CAAR. Interestingly, although CAAR3-mutg233k-CAAR and CAAR4-mutg233k-CAAR contained fragments of LBD-mutg233k-CAAR, they did not react with serum antibodies from patient 3, and the results are presented in Online Supplementary Figure S5A. The ELISpot assay was employed to verify GPIbα CAAR T-cell potential in eradicating autoreactive B cells in ITP patients. ELISpot analysis (Figure 6C) showed that anti-GPIbα IgG B cells, but not total IgG B cells from ITP patients, were depleted by GPIbα CAAR T cells. Meanwhile, anti-CD19 CAR T cells eliminated all IgG B cells from ITP patients and healthy controls. In summary, we confirmed the viability of the “Trojan hypothesis” for the treatment of ITP patients. GPIbα CAAR T cells function like a “Trojan horse”, trapping autoreactive B cells and performing specific killing. (Figure 6D)
Our study presents a novel concept for the treatment of refractory and relapsed ITP patients with autoantigen-modified T cells based on CAR T cells. Since patients with anti-GPIbα antibodies have a poor response to standard immunosuppressive therapy, GPIbα was constructed into the ligand-binding domain of the CAAR structure in this study. Anti-GPIbα antibodies can cause platelet desialylation, mediate Fc-independent platelet eradication,34,35 affect thrombopoietin production in the liver, and account for
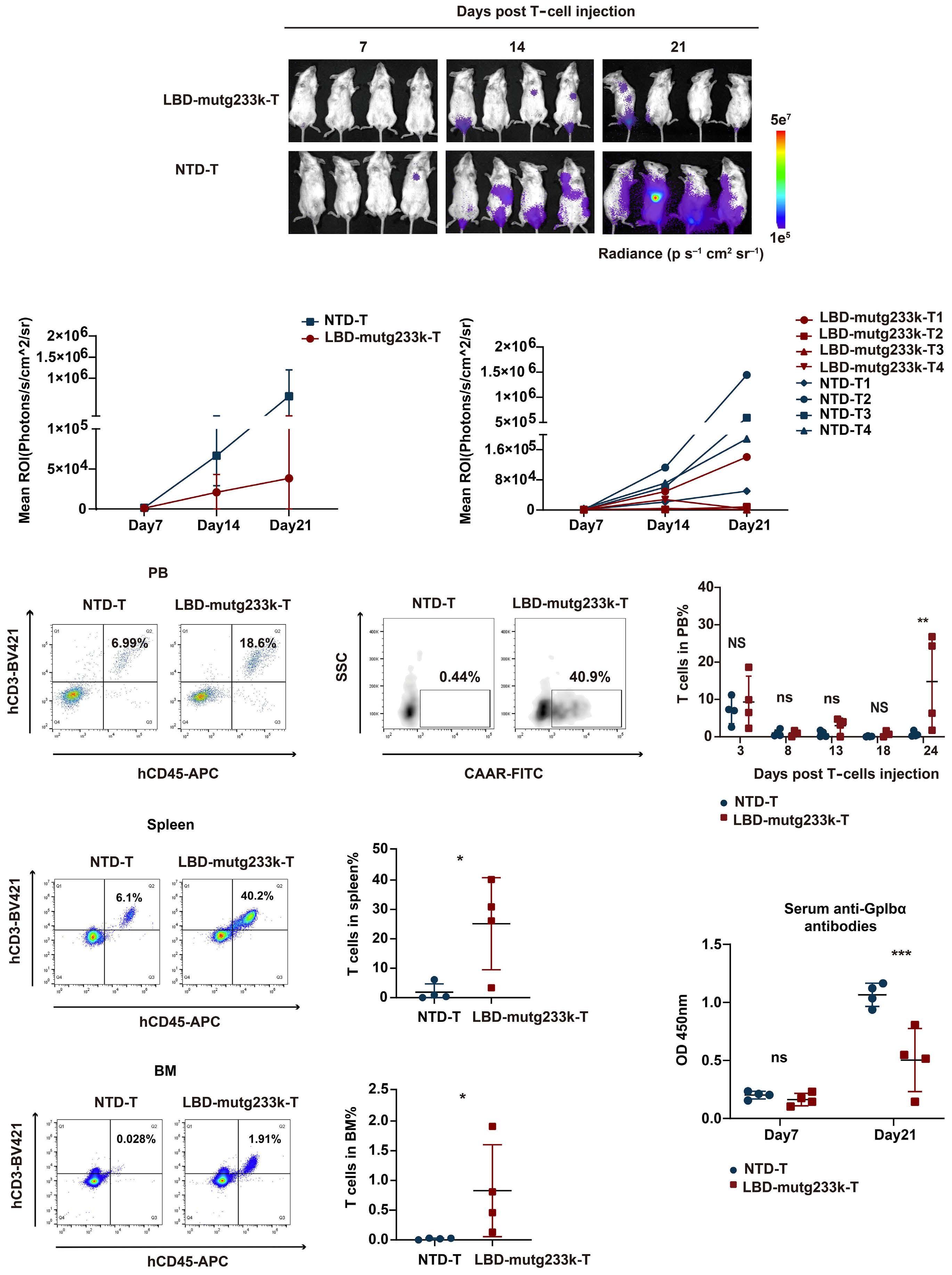
Continued on following page.
Figure 5. GPIbα chimeric autoantibody receptor T cells mediated autoreactive B-cell depletion in vivo. (A) Anti-GPIbα B cells and ligand-binding domain LBD-mutg233k T cells were intravenously inoculated into 7-week-old female NSG mice. Autoreactive B-cell burden was monitored via bioluminescence imaging every few days. (B) Quantification of the autoreactive B-cell burden indicated by the radiance detected in the region of interest with group values or individual values. T1, 2, 3, 4 mean mice 1, 2, 3, 4. (C) Flow cytometry analysis of total human T cells in the peripheral blood (PB) of non-transduced T cell (NTD T)-treated mice (N=4) and LBD-mutg233k T-treated mice (N=4) monitored every few days. T cells were stained with anti-human CD45 antibody (APC) and anti-human CD3 antibody (BV421). Chimeric autoantibody receptor (CAAR) expression in T cells was detected with an anti-human CD42b antibody (FITC). T-cell penetration and persistence in the spleen (D) and bone marrow (BM) (E) of the mice were analyzed with flow cytometry. T cells were stained with anti-human CD45 antibody (APC) and anti-human CD3 antibody (BV421). (F) The serum anti-GPIbα antibody titer of the mice on day 7 and day 21 was detected by enzyme-linked immunosorbant assay at a dilution ratio of 20. NS: not significant P>0.05; *P<0.05; **P<0.01; ***P<0.005. ROI: region of interest; SSC: side scatter.
the inactivated negative feedback mechanism of megakaryocyte-generated platelets that leads to peripheral platelet destruction.30,36 In this work, site-mutated GPIb α ectodomain of varying lengths were incorporated into second-generation CAR structures to direct specific T-cell cytolysis against autoreactive B cells. In ITP, no cell immunotherapy targeting B cells is currently used. Compared to CAR T-cell therapy, CAAR T-cell therapy ultimately and precisely removes autoreactive B cells and has fewer side effects due to a lower “tumor” burden.14 This work demonstrated the in vitro cytolytic capacity and persistence of GPIbα CAAR T cells and their ability to eliminate GPIb α-specific B cells in vivo precisely. B-cell depletion therapy not only reduces autoantibody production but also reduces splenic CD8+ T-cell proliferation in vitro, as well as the ability of CD8+ T cells to activate and mediate ITP 37 and T-follicular helper cells in both the spleen and the blood.38 It is anticipated that B-cell clearance mediated by CAAR T-cell therapy may have an additional impact on moderating immunological dysregulation in ITP patients.
The N-terminus of GPIb α contains binding sites for VWF, and anti-GPIbα antibodies can interfere with normal platelet function by inhibiting GPIb α -VWF-mediated platelet aggregation, which may increase the severity of the patient’s hemorrhage at the same time.39 In order to prevent the off-target effect caused by the competitive binding of GPIb α CAAR T cells with VWF, we generated mutations (Figure 1B; Online Supplementary Figure S6) at residues 231, 232, and 233 of GPIb α (K231V/Q232V/G233k/G233D). The results demonstrated that GPIb α mutations strongly inhibited the binding of GPIb α to VWF, which could alleviate possible off-target effects after CAAR T-cell infusion in vivo.
The binding force between CAR T cells and cancer cells and the size of the CAR antigen are related to CAR T-cell efficacy, which may explain the varied cytolytic capacity of the CAAR,40,41 as shown by the results of the hybridoma antibody binding assay (Figure 2) and in vitro cytotoxic assay (Figure 3) in our study. GPIb α CAAR T cells with various truncated forms of GPIb α all exhibited cytotoxicity against anti-GPIb α hybridomas. Meanwhile, the epitopes of autoantibodies are also diverse in ITP patients, so it is feasible to choose the most suitable GPIb α CAAR T-cell
therapy for different patients. The MSD of GPIb α is critical in sensing shear stress and converts this mechanical information into a protein-mediated signal in platelets, 28 the role and structural changes of which were also evaluated in this work. CAAR4-mutg233k-CAAR did not contain the MSD; CAAR4-mutg233k T cells showed a better response to anti-GPIb α autoantibodies and exhibited better killing efficiency than CAAR3-mutg233k T cells, which contain MSD. MSD does affect the spatial conformation of the CAAR and negatively affects subsequent binding and cytolytic functions, which did not seem to be necessary in the CAAR constructs. The same results were also confirmed in cytolytic assays of CAAR1 and CAAR2 (Figure 3F; Online Supplementary Figure S3).
In the in vivo study, a hybridoma xenograft model was used, referring to existing research.14 The four hybridomas were obtained by immunizing BALB/C mice with human platelets. The hybridoma antibodies could only target human GPIb α and could not bind to mouse platelets to mediate platelet destruction in mice; thus, we could not assess whether platelets increase after infusion of CAAR T cells. The humanized mouse model was considered, but previous research42,43 has shown that human platelets are low in the PB of humanized mouse models, limiting their applicability. Although we were unable to monitor the therapeutic effect of the CAAR T cells, we did show that the CAAR T cells could selectively lyse target cells, reduce autoantibody titers, and result in a lower human platelet clearance rate ( Online Supplementary Figure S4D ) in an in vivo xenograft mouse model. In addition, control of the anti-GPIb α hybridoma burden in LBD-mutg233k T-treated mice validated CAAR T-cell persistence in vivo In order to verify the potential of GPIb α CAAR T cells in clinical applications, we first demonstrated their reactivity with sera from ITP patients with anti-GPIb antibodies. We found that anti-GPIb antibodies from patient 3 could bind to the LBD of GPIb α (LBD-mutg233k-CAAR) but did not react with CAAR3-mutg233k-CAAR or CAAR4-mutg233kCAAR, which also contained the LBD. We hypothesized that the expression of the LBD is more stable than that of other extracellular domains of GPIb α , as it can mediate rapid downstream action.29 In addition, studies have shown that the macroglycopeptide domain can affect the binding of LBD to ligands.44-46 We anticipated that
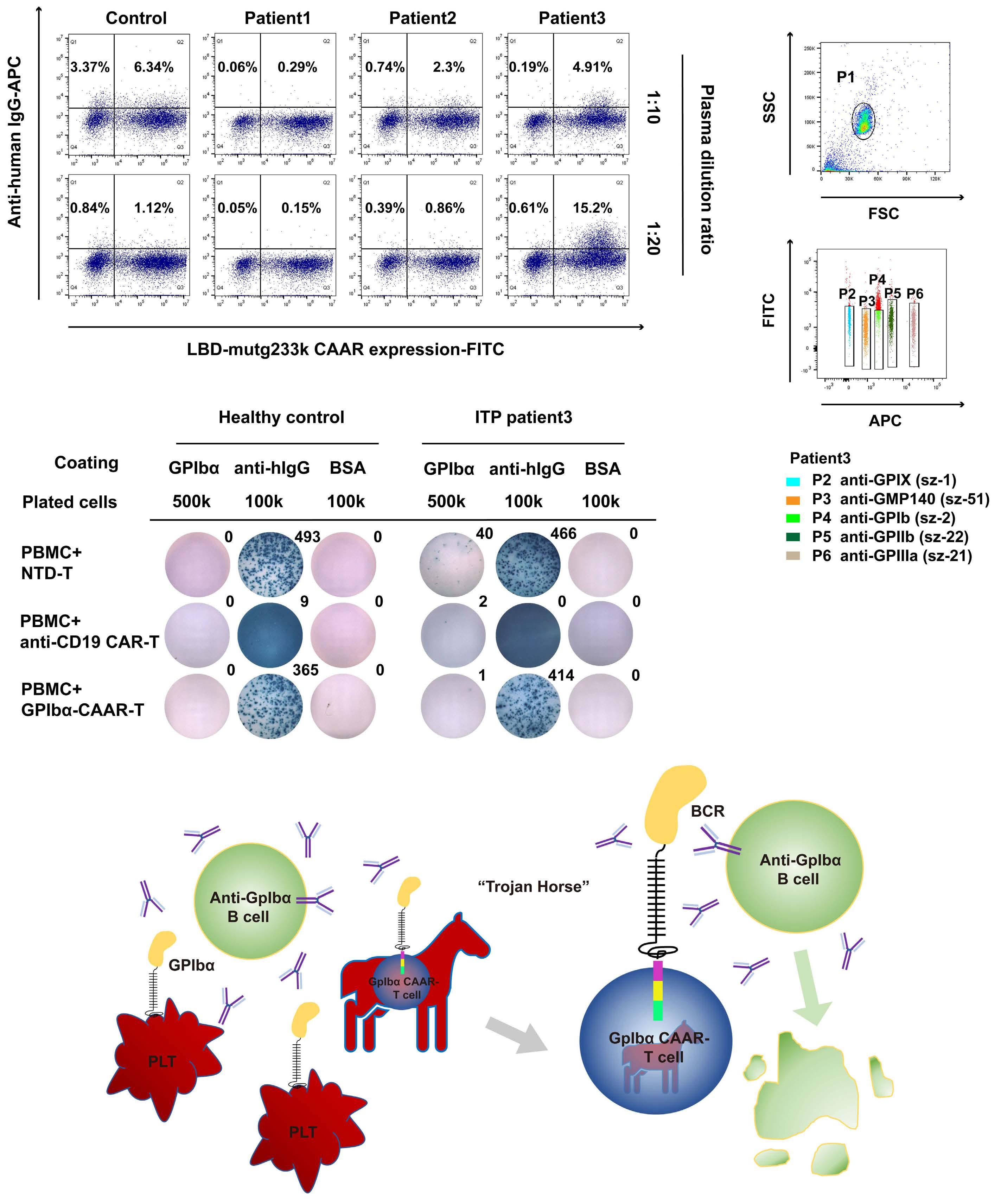
Figure 6. GPIbα chimeric autoantibody receptor T cells recognized and eliminated autoreactive B cells from immune thrombocytopenia patients. (A) Plasma from 3 immune thrombocytopenia (ITP) patients with anti-GPIb antibodies and healthy controls was diluted at 1:10 and 1:20 and incubated with HEK293T cells expressing ligand-binding domain LBD-mutg233k-chimeric autoantibody receptor (CAAR)-tagged with green fluorescent protein (GFP). The cells were then stained with anti-human immuno-
globulin (Ig)G antibodies (APC). (B) A cytometric bead array was applied to detect platelet-specific antibodies in ITP patient 3. Cutoff values were determined with reference to negative controls (mean + 3 standard deviations). Autoantibodies targeting human platelet GPIX/GPIb/GPIIb/GPIIIa/GMP140 were detected. GMP140, granule membrane protein 140. (C) Peripheral blood mononucelar cells (PBMC) from ITP patient 3 or healthy controls were stimulated with interleukin (IL)-2 and R848 for 2 days and then co-cultured with GPIbα CAAR T, anti-CD19CAR T, or non-transduced T (NTD T) cells for 1 day on plates coated with GPIbα protein and anti-human IgG antibody. Bovine serum albumin (BSA) served as a negative control antigen. One spot represents IgG antibodies secreted by a single B cell. The number of spots detected in each well is shown in the top right corner. (D) The conceptual graph of GPIbα CAAR T therapy in ITP. GPIbα CAAR T cells act like a “Trojan horse”, trapping autoreactive B cells and performing specific killing. PLT: platelet; BCR: B-cell receptor.
macroglycopeptide sequences could form large steric hindrances, interfering with antibodies’ binding to CAAR structures (CAAR3-mutg233k-CAAR and CAAR4-mutg233kCAAR). Furthermore, the structure of MSD is unstable,28 and we can’t tell whether the MSD expressed in the CAAR structure is folded or expanded, which may also interfere with the binding of CAAR3-mutg233k-CAAR and autoantibodies. The changes that occurred in the O-glycosylated macroglycopeptide domain and MSD in the CAAR expressed in the cells are worth exploring. An exciting and gratifying result was that GPIbα CAAR T cells successfully eliminated anti-GPIbα B cells from a patient with refractory ITP, and normal B cells were not destroyed compared to anti-CD19 CAR T cells, supporting further clinical application. As anti-GPIIb/IIIa(αIIb/b3)5,47 is the most commonly detected autoantibody in ITP patients, we are also trying to construct GPIIb/IIIa-CAAR T cells. However, platelet-associated anti-GPIIb/IIIa autoantibodies from chronic ITP patients mainly depend on conformationally intact GPIIb/IIIa and divalent cations for maximal binding.5,48 The N-terminal globular head of GPIIb-IIIa seems to play an essential role as a hot spot for autoantigenic epitopes in chronic ITP.49 Integrating both αIIb and b3 into the CAAR structure is difficult, and it is challenging to ensure that the conformational epitopes created by the two subunits are consistent with the native epitopes. Though difficult, related work is still being done by our team. In summary, a novel GPIbα CAAR was constructed, and we demonstrated GPIbα CAAR T-cell’s efficacy and safety in vitro and in vivo models. GPIbα CAAR T-cell therapy is a
1. Cooper N, Ghanima W. Immune thrombocytopenia. N Engl J Med. 2019;381(10):945-955.
2. Stockelberg D, Hou M, Jacobsson S, Kutti J, Wadenvik H. Evidence for a light chain restriction of glycoprotein Ib/IX and IIb/ IIIa reactive antibodies in chronic idiopathic thrombocytopenic purpura (ITP). Br J Haematol. 1995;90(1):175-179.
3. Roark JH, Bussel JB, Cines DB, Siegel DL. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood. 2002;100(4):1388-1398.
4. Hou M, Stockelberg D, Kutti J, Wadenvik H. Immunoglobulins targeting both GPIIb/IIIa and GPIb/IX in chronic idiopathic thrombocytopenic purpura (ITP): evidence for at least two different IgG antibodies. Br J Haematol. 1997;98(1):64-67.
5. McMillan R. Autoantibodies and autoantigens in chronic
viable treatment option for patients with refractory and relapsed ITP.
No conflicts of interest to disclose.
Contributions
HM, JL, JZ and YX designed the experiments. HM, JL and YH analyzed the data. ZJ and HM wrote the paper. JZ and YX performed the experiments. JS, HJ, LH and MX helped with the experiments.
The authors thank Jun Peng and Ming Hou from Qilu Hospital, Shandong University, for providing specimens and for their helpful comments and suggestions.
This work was supported by the National Natural Science Foundation of China (grant 82070124 to HM, and grant 82330005 to HM), the Technology Innovation Plan key research and development projects of Hubei Province (grant 2023BCB019 to HM), and the Basic Research Support Programs Foundation of Huazhong University of Science and Technology (grant 2023BR033 to HM).
Data-sharing statement
Data that support the findings of this study are available from the corresponding author upon reasonable request.
immune thrombocytopenic purpura. Semin Hematol. 2000;37(3):239-248.
6. Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med. 2007;146(1):25-33.
7 Deshayes S, Khellaf M, Zarour A, et al. Long-term safety and efficacy of rituximab in 248 adults with immune thrombocytopenia: results at 5 years from the French prospective registry ITP-ritux. Am J Hematol. 2019;94(12):1314-1324.
8. Tjønnfjord E, Holme PA, Darne B, et al. Long-term outcomes of patients treated with rituximab as second-line treatment for adult immune thrombocytopenia - follow-up of the RITP study. Br J Haematol. 2020;191(3):460-465.
9. Canales-Herrerias P, Crickx E, Broketa M, et al. High-affinity autoreactive plasma cells disseminate through multiple organs
in patients with immune thrombocytopenic purpura. J Clin Invest. 2022;132(12):e153580.
10 Yu TS, Wang HY, Zhao YJ, et al. Abnormalities of bone marrow B cells and plasma cells in primary immune thrombocytopenia. Blood Adv. 2021;5(20):4087-4101.
11. Mahévas M, Patin P, Huetz F, et al. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. J Clin Invest. 2013;123(1):432-442.
12. Crickx E, Chappert P, Sokal A, et al. Rituximab-resistant splenic memory B cells and newly engaged naive B cells fuel relapses in patients with immune thrombocytopenia. Sci Transl Med. 2021;13(589):eabc3961.
13. Kuwana M, Okazaki Y, Kaburaki J, Kawakami Y, Ikeda Y. Spleen is a primary site for activation of platelet-reactive T and B cells in patients with immune thrombocytopenic purpura. J Immunol. 2002;168(7):3675-3682.
14. Ellebrecht CT, Bhoj VG, Nace A, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353(6295):179-184.
15. Oh S, Mao X, Manfredo-Vieira S, et al. Precision targeting of autoantigen-specific B cells in muscle-specific tyrosine kinase myasthenia gravis with chimeric autoantibody receptor T cells. Nat Biotechnol. 2023;41(9):1229-1238.
16. Kroll MH, Harris TS, Moake JL, Handin RI, Schafer AI. von Willebrand factor binding to platelet GpIb initiates signals for platelet activation. J Clin Invest. 1991;88(5):1568-1573.
17. Du X. Signaling and regulation of the platelet glycoprotein Ib-IX-V complex. Curr Opin Hematol. 2007;14(3):262-269.
18. Nomura S, Yanabu M, Soga T, et al. Analysis of idiopathic thrombocytopenic purpura patients with antiglycoprotein IIb/ IIIa or Ib autoantibodies. Acta Haematol. 1991;86(1):25-30.
19 Hou M, Stockelberg D, Kutti J, Wadenvik H. Antibodies against platelet GPIb/IX, GPIIb/IIIa, and other platelet antigens in chronic idiopathic thrombocytopenic purpura. Eur J Haematol. 1995;55(5):307-314.
20. Peng J, Ma SH, Liu J, et al. Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: a multicenter cohort study. J Thromb Haemost. 2014;12(4):497-504.
21. Go RS, Johnston KL, Bruden KC. The association between platelet autoantibody specificity and response to intravenous immunoglobulin G in the treatment of patients with immune thrombocytopenia. Haematologica. 2007;92(2):283-284.
22. Feng R, Liu X, Zhao Y, et al. GPIIb/IIIa autoantibody predicts better rituximab response in ITP. Br J Haematol. 2018;182(2):305-307.
23. Webster ML, Sayeh E, Crow M, et al. Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbalpha antibodies. Blood. 2006;108(3):943-946.
24. Yu Y, Hou Y, Zhao Y, et al. Platelet autoantibody specificity and response to rhTPO treatment in patients with primary immune thrombocytopenia. Br J Haematol. 2021;194(1):191-194.
25. He R, Reid DM, Jones CE, Shulman NR. Extracellular epitopes of platelet glycoprotein Ib alpha reactive with serum antibodies from patients with chronic idiopathic thrombocytopenic purpura. Blood. 1995;86(10):3789-3796.
26. Nieswandt B, Bergmeier W, Rackebrandt K, Gessner JE, Zirngibl H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood. 2000;96(7):2520-2527.
27. Jayaraman J, Mellody MP, Hou AJ, et al. CAR-T design: elements and their synergistic function. EBioMedicine. 2020;58:102931.
28. Zhang W, Deng W, Zhou L, et al. Identification of a juxtamembrane mechanosensitive domain in the platelet mechanosensor glycoprotein Ib-IX complex. Blood. 2015;125(3):562-569.
29. Quach ME, Dragovich MA, Chen W, et al. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood;131(7):787-796.
30. Xu M, Li J, Neves MAD, et al. GPIbα is required for plateletmediated hepatic thrombopoietin generation. Blood. 2018;132(6):622-634.
31. Huizinga EG, Tsuji S, Romijn RA, et al. Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain. Science. 2002;297(5584):1176-1179.
32. Matsubara Y, Murata M, Sugita K, Ikeda Y. Identification of a novel point mutation in platelet glycoprotein Ibalpha, Gly to Ser at residue 233, in a Japanese family with platelet-type von Willebrand disease. J Thromb Haemost. 2003;1(10):2198-2205.
33. Dong J, Schade AJ, Romo GM, et al. Novel gain-of-function mutations of platelet glycoprotein IBalpha by valine mutagenesis in the Cys209-Cys248 disulfide loop. Functional analysis under statis and dynamic conditions. J Biol Chem. 2000;275(36):27663-27670.
34 Li J, van der Wal DE, Zhu G, et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun. 2015;6:7737.
35. Tao L, Zeng Q, Li J, et al. Platelet desialylation correlates with efficacy of first-line therapies for immune thrombocytopenia. J Hematol Oncol. 2017;10(1):46.
36. Kosugi S, Kurata Y, Tomiyama Y, et al. Circulating thrombopoietin level in chronic immune thrombocytopenic purpura. Br J Haematol. 1996;93(3):704-706.
37. Guo L, Kapur R, Aslam R, et al. CD20+ B-cell depletion therapy suppresses murine CD8+ T-cell-mediated immune thrombocytopenia. Blood. 2016;127(6):735-738.
38. Audia S, Rossato M, Trad M, et al. B cell depleting therapy regulates splenic and circulating T follicular helper cells in immune thrombocytopenia. J Autoimmun. 2017;77:89-95.
39 Li J, Callum JL, Lin Y, Zhou Y, Zhu G, Ni H. Severe platelet desialylation in a patient with glycoprotein Ib/IX antibodymediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica. 2014;99(4):e61-63.
40 Xiao Q, Zhang X, Tu L, Cao J, Hinrichs CS, Su X. Size-dependent activation of CAR-T cells. Sci Immunol. 2022;7(74):eabl3995.
41. Leick MB, Silva H, Scarfò I, et al. Non-cleavable hinge enhances avidity and expansion of CAR-T cells for acute myeloid leukemia. Cancer Cell. 2022;40(5):494-508.
42. Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108(2):487-492.
43. Hu Z, Yang YG. Full reconstitution of human platelets in humanized mice after macrophage depletion. Blood. 2012;120(8):1713-1716.
44 Othman M, Notley C, Lavender FL, et al. Identification and functional characterization of a novel 27-bp deletion in the macroglycopeptide-coding region of the GPIBA gene resulting in platelet-type von Willebrand disease. Blood. 2005;105(11):4330-4336.
45. López JA, Ludwig EH, McCarthy BJ. Polymorphism of human glycoprotein Ib alpha results from a variable number of tandem repeats of a 13-amino acid sequence in the mucin-like macroglycopeptide region. Structure/function implications. J Biol Chem. 1992;267(14):10055-10061.
46. Li CQ, Dong JF, López JA. The mucin-like macroglycopeptide region of glycoprotein Ibalpha is required for cell adhesion to immobilized von Willebrand factor (VWF) under flow but not for static VWF binding. Thromb Haemost. 2002;88(4):673-677.
47. Brighton TA, Evans S, Castaldi PA, Chesterman CN, Chong BH. Prospective evaluation of the clinical usefulness of an antigenspecific assay (MAIPA) in idiopathic thrombocytopenic purpura
and other immune thrombocytopenias. Blood. 1996;88(1):194-201.
48. Fujisawa K, Tani P, McMillan R. Platelet-associated antibody to glycoprotein IIb/IIIa from chronic immune thrombocytopenic purpura patients often binds to divalent cation-dependent antigens. Blood. 1993;81(5):1284-1289.
49 Tomiyama Y, Kosugi S. Autoantigenic epitopes on platelet glycoproteins. Int J Hematol. 2005;81(2):100-105.
Over the last decades, advances in transfusion regimen, chelation therapy and iron overload monitoring have allowed a steady increase in life expectancy of patients with transfusion-dependent thalassemia (TDT), mainly related to a decrease in cardiac mortality.1,2 However, endocrinopathies remain the most frequent and early complications of iron overload in TDT patients, the first being hypogonadism with a prevalence ranging from 30 to 60% in numerous registries and cohorts.3-5 Early diagnosis and treatment are essential to achieve normal puberty, target height, and reduce the risk of long-term infertility and sexual dysfunction. It has been established that well-managed chelation therapy and its early initiation, allowing good control of iron overload, reduce the risk of hypogonadism.6,7 However, definitions of pubertal disorders and hypogonadism vary according to studies, and data from the literature regarding pubertal development are often incomplete.
The French National Thalassemia Registry (NaThalY) has recently initiated a study on the health status of TDT children aged <15 years, mostly (80%) under deferasirox (DFX) therapy, reporting an excellent probability of survival (98.3%) and a very low rate of iron-related complications.8 In the context of the global improvement in endocrinopathies already described under DFX,9 here we report the remaining pubertal disorders and hypogonadism in TDT adolescents and young adults included in the French Thalassemia Registry who received oral chelation with DFX before puberty onset. This study respects the French ethical rules. The French registry was approved by the Comission Nationale de l’Informatique et des Libertés (CNIL) (declaration N. 903064V1, authorization N. 04-1396).
A total of 50 patients, treated in 24 registry-participating centers, accumulated at least three years of DFX therapy before puberty in their history of chelation therapy and met the inclusion criteria of this study (Figure 1). Their median age at last visit was 20 years for girls (N=25, range 14-24) and 18 years for boys (N=25, range 14-24). Regular transfusions and chelation therapy were initiated early, at 1.3 years old (range 0.3-4) and 3 years old (range 1-6), respectively. The median duration of DFX chelation before 13 years in girls and 14 years in boys was seven years (range 3-12).
All 50 patients were evaluable for pubertal delay, defined as lack of pubertal onset by 13 years in girls and 14 years in boys. All had spontaneous puberty onset, at a median age of 12.5 years in girls (range 10.5-14) and 13.5 years in boys (range 12.5-15). Eight patients (16%) experienced delayed puberty, mostly (7/8) simple pubertal delay, with a delay
ranging from 6 to 12 months (Table 2). Among the 7 patients with simple pubertal delay, 5 were boys.
Only one patient, suffering from a pubertal delay, presented with primary amenorrhea out of the 24 girls who had either menstruated or who were >15 years old (4.2%). Median age at first menstruation was 13.5 years (range 12-17). Two girls had their first menstruation delayed, at 16 and 17 years old, both after a simple pubertal delay.
Twenty-three patients were assessable for secondary amenorrhea (menstruated girls). Three developed secondary amenorrhea (13%) 2, 3, and 7 years after their menarche. Mean follow-up duration after menarche for the 20 other girls without secondary amenorrhea was 6.3 years.
The 4 patients with primary or secondary amenorrhea had hypogonadism with low sexual hormone levels (estradiol <25 pg/mL). Three showed hypogonadotropic hypogonadism (HH) (Online Supplementary Table S1). Despite existing HH,
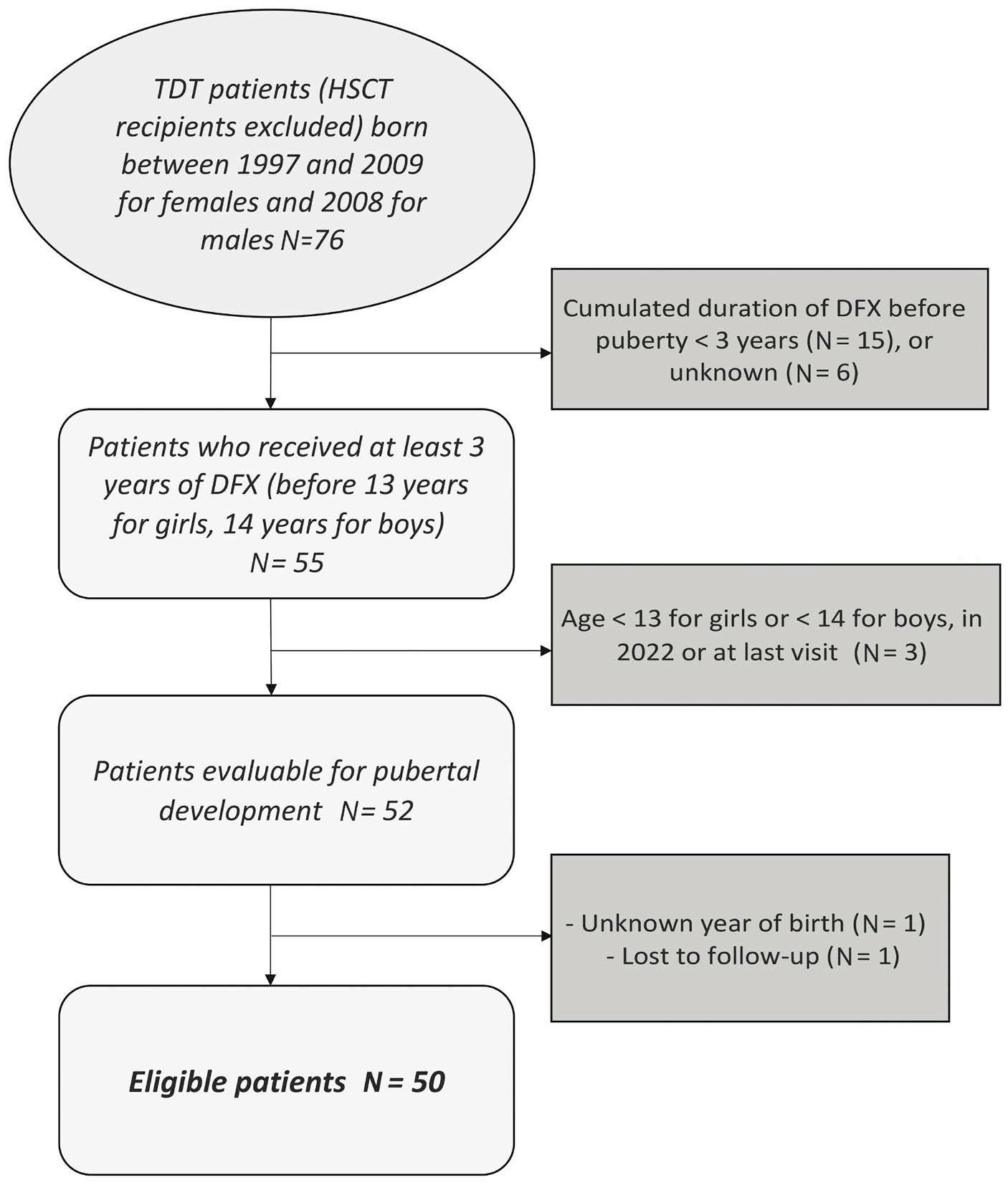
Figure 1. Study population. DFX: deferasirox; HSCT: hematopoietic stem cell transplantation; TDT: transfusion-dependent thalassemia.
Table 1. Clinico-biological characteristics of study population.
Characteristic
Sex, N (%)
N (%)
Place of birth, N (%)
France Outside
(IQR)
(IQR)
First chelator
N (%)
Chelator at 13 (girls) or 14 (boys) years, N (%)
DFX accumulated duration before puberty in years, median (IQR)
Other endocrine disorder, N (%)
Ferritin, microg/L,** median (IQR)
Mean at 10 years old
NA, N (%)
Mean between 10 years old and puberty
NA, N (%)
History of ferritin >2,500 microg/L twice or more*, N (%)
Liver iron concentration LIC, mg/g dw,** median (IQR)
Mean at 10 years old
ND, N (%)
NA, N (%)
Mean between 10 years old and puberty
ND, N (%)
NA, N (%)
History of LIC >15 mg/g, N (%)
NA, N (%) 6.3 (3.9-12.2)
(643-1,859)
(5.1) 7 (17.9) 3 (7.7)
IQR: interquartile range; DFO: deferoxamine; DFP: deferiprone; DFX: deferasirox; dw: dry weight; GH: growth hormone; LIC: liver iron concentration; NA: not available; ND: not done. *At least 12 months apart. **No statistically significant difference was noted in clinical/biological characteristics of patients with and without disorders.
one patient experienced a spontaneous pregnancy, the only one reported in the cohort. The fourth female had hypogonadism of mixed origin, with a suspected premature ovarian failure (low anti-Mullerian hormone: 1.14 pmol/L) and only moderate elevation of follicle-stimulating hormone (FSH) (15 UI/L), probably due to pituitary overload. No paternity was reported.
No puberty abnormalities other than simple pubertal delay were noted in boys. Completion of puberty for the 18 male patients aged >17 years was clinically assessed (Table 2), biological data not being routinely collected in the registry. Finally, a total of 11 patients (22%) experienced simple de-
layed puberty (N=7) or hypogonadism (N=4). No significant difference was noted between these patients and the rest of the cohort concerning the age at first transfusion, the age at chelation onset or the cumulative duration of DFX before puberty (Table 1). Among these 11 patients, 7 had a history of severe iron overload before puberty (serum ferritin levels >2,500 µg/L twice at least 12 months apart or liver iron content >15 mg/g dry weight) compared with 13/39 patients who did not experience pubertal development disorders or hypogonadism (P=0.1). All 4 patients with hypogonadism already had a history of severe iron overload before the onset of puberty.
Characteristic
Age at evaluation in years, median (IQR) Range
Boys, N=25
14-24 Age at pubertal onset in years, median (IQR)
Age at menarche in years, median (IQR)
Range
NE (Non-menstruating <15 years), N (%)
Age at end of puberty in years, median (IQR)
Evaluated, N (%)
NE (<17 years at assessment), N (%)
NA, N (%)
Disorders of pubertal development°, N (%)
Pubertal delay
Including simple pubertal delay
Primary amenorrhea*
Secondary amenorrhea**
Arrested puberty
Height, SD°°, median (IQR)
At 10 years
NA, N (%)
Pre-pubertal height
NA, N (%)
Final height†
Evaluated, N (%)
NE (< 17 years at assessment), N (%)
(22)
(14)
NA, N (%) -0.7 (-1.3 to 0) 4 (8) -1.2 (-1.7 to -0.3) 3 (10)
Pubertal spurt in cm, median (IQR)
Evaluated, N (%)
NE (<17 years at assessment), N (%)
NA, N (%)
(-1.2 to 0)
(70)
(22) 4 (8)
20.5 (17-24.5)
(68)
(22) 5 (10)
(-1.5 to -0.1) 2 (8) -1.2 (-1.7 to -0.2) 1 (4) -0.3 (-1.1 to 0) 19 (76) 4 (16) 2 (8) -0.7 (-1.2 to 0) 2 (8) -1.2 (-1.8 to 0.8) 2 (8) -0.8 (-1.2 to -0.5) 16 (64) 7 (28) 2 (8)
(16.5-22)
(76)
(16) 2 (8)
(20.5-26) 15 (60) 7 (28) 3 (12)
IQR: interquartile range; NA: not available; NE: not evaluable; SD: standard deviation. *Evaluable patients N=24. **Evaluable patients N=23. °Pubertal delay: lack of testicular development (testis volume <4 mL) in boys aged 14 years (Tanner stage <G2) or absence of thelarche in 13-year old girls (Tanner stage <S2). Simple pubertal delay: puberty with delayed onset but then proceeding normally, without the need for any treatment. Arrested puberty: failure to continue pubertal progression over a period of two years after spontaneous onset of puberty, or a delay between thelarche and menarche of four years or more. End of puberty: Tanner stage 5 and growth completed. °°Pre-pubertal height: last height available in the year of puberty onset. Final height: height reached at the end of puberty, no difference between two visits at least 12 months apart. Pubertal spurt: final height - pre-pubertal height. Height in cm were converted to SD according to French Growth Charts. †As patients originated from various countries, heights were also converted to SD according to the WHO growth chart; median final height was -0.6 SD (-0.2 SD in girls, -0.9 SD in boys).
Although the age at onset of puberty and at first menstruation remained delayed in TDT patients compared with the general French population (i.e., 10.5 years in girls and 12 in boys, 12 years for menarche),10 the frequency of delayed puberty in this study was lower than previously reported under well-conducted deferoxamine (DFO) treatment,5,7,11 and rarely evolved into hypogonadism. Height was moderately reduced at the age of 10 years (-0.8 Standard Deviation [SD] in girls, -0.7 SD in boys), then going down to -1.2 SD for both genders reflecting pre-pubertal inflection. Most patients then experienced pubertal growth spurt, with a median of 18 cm in girls and 22 cm in boys. Final height, reached in 35/50 patients, was only moderately reduced (-0.7 SD), with a median height of 161 cm in girls (-0.3 SD) and 168.5 cm in boys (-0.8 SD) (Online Supplementary Figure S1). Patients with impaired pubertal development or hypogonadism had a median final height of -0.3 SD (Table 2). Six patients (4 boys, 2 girls) had growth hormone (GH) deficiency, of whom 3 had a history of severe iron overload before puberty. Age at GH treatment onset ranged from 12 to 15 years. There was no difference in final height when GH-treated patients were excluded. Despite pubertal spurts lower than those reported for the general population (25 cm in girls and 28 cm in boys),10 final heights have improved in comparison with those previously reported under DFO in the French registry (-1.2 SD).4 Another study found that final height was -1.9 SD in patients exclusively chelated by DFO, compared with -1.2 SD in patients who had been chelated with DFX for at least six years.5 This difference could be due in part to the well-documented direct deleterious effect of DFO on growth in young children. Regarding other endocrine disorders, one patient developed central hypothyroidism during adolescence and one patient had type 1 diabetes, which was not related to iron overload as it occurred early in childhood (7 years old) with evidence of autoantibodies. No cardiac or hepatic disorders were reported. One patient with severe iron overload and HH died at 17 years from septic shock; this was the only death reported in the cohort.
Deferasirox was first marketed in the European Union in 2007 and since then has been the most widely prescribed iron chelator for TDT patients in Europe and the United States. This oral chelation therapy has been associated with decreased frequency of endocrine complications including hypogonadism in adult patients.12 Scarce published data (few patients studied) document the frequency of pubertal delay in TDT adolescents receiving DFX. An American study found a rate of pubertal disorders (both simple delay and hypogonadism) in 4 out of 15 adolescents.13 In another study, incidence of simple pubertal delay was evaluated at 15%, i.e., 3 out of the 20 TDT patients who had been pre-pubertal at the start of the study and had received at least three consecutive years of DFX.9
It remains difficult to distinguish between an improved global control of iron overload and a possible specific effect
of DFX on the prevention of pubertal disorders secondary to iron overload. Indeed, the once-daily oral administration of DFX improved adherence with the daily lifelong chelation therapy prescribed.14 Additionally, DFX, with its prolonged half-life and permanent blood exposure could allow better pituitary protection, secondary to continuous chelation of non-transferrin-bound iron.
To our knowledge, our study is the first to provide detailed information on puberty development and to document its improvement in a large cohort of 50 adolescents treated with DFX. This is a retrospective multicenter registry study and data concerning family history of growth and puberty or adherence to chelation are missing, and there is no systematic uniform endocrinological follow-up of puberty. Thus, while in girls, objective events such as the occurrence of the first menstruation and the regularity of cycles provide a simple means to monitor puberty, assessment is more difficult in boys. This less accurate monitoring means that the rate of arrested puberty or early hypogonadism in boys is probably underestimated. Given the impact of pubertal and growth disorders on patients’ quality of life and on future fertility, the systematic integration of an endocrinologist in the healthcare team of TDT children and adolescents is currently recommended in national and international guidelines. Additionally, the patients studied are still young and cannot yet be assessed for the occurrence of hypogonadism in adulthood, the frequency of which increases with age.4,7,15 It is, therefore, important in the near future to accurately evaluate the effect of long-term DFX chelation therapy on the prevention of hypogonadism in TDT young adults.
Mathilde Veneziano Broccia,1,2 Julia Vergier,3 Audrey Benoit,4 Yoann Huguenin,5 Anne Lambilliotte,6 Marie Pierre Castex,7 Stephanie Gourdon,8 Ghislaine Ithier,9 Kamila Kebaili,10 Pierre Rohrlich,11 Corinne Pondarre,12 Abdourahim Chamouine,13 Pauline Simon,14 Kokou Placide Agbo Kpati,15 Slimane Allali,16 Sandrine Baron-Joly,17 Sophie Bayart,18 Nicolas Billaud,19 Valentine Brousse,20 Cecile Dumesnil,21 Nathalie Garnier,10 Isabelle Guichard,22 Laure Joseph,23 Annie Kamdem,24 Julie Maitre,25 Catherine Mathey,1 Catherine Paillard,26 Aurelie Phulpin,27 Cecile Renard,10 Cecile Stoven,28 Mohamed Touati,29 Capucine Trochu,30 Suzanne Mathieu Nafissi,31 Catherine Badens,4 Sarah Szepetowski1,2 and Isabelle Thuret1,2
1Service d’Hematologie, Immunologie et Oncologie Pediatrique, Hopital La Timone Enfants, AP-HM, Marseille; 2Centre de Reference MCGRE, Service d’Hematologie, Immunologie et Oncologie Pediatrique, Hopital La Timone Enfants, AP-HM, Marseille; 3Service de Pediatrie Multidisciplinaire, Hopital de la Timone Enfants, AP-HM, Marseille; 4National Thalassemia Registry (NaThalY), Service de Genetique, Hopital La Timone Enfants, AP-HM, Marseille; 5Service d’Oncologie et d’Hematologie Pediatrique, Hopital Pellegrin, CHU de
Bordeaux, Bordeaux; 6Service d’Hematologie et d’Oncologie Pediatrique, Centre Hospitalo-Universitaire de Lille, Lille; 7Service d’Hemato-Oncologie Pediatrique, Hopital des Enfants, Toulouse; 8Service d’Oncologie et Hematologie Pediatrique, Centre Hospitalier Universitaire de La Reunion, Saint Denis; 9Centre de Reference MCGRE, Unite d’Hematologie, Hopital Robert Debre, Assistance Publique-Hopitaux de Paris (AP-HP), Paris; 10Institut d’Hematologie et d’Oncologie Pediatrique, Hospices Civils de Lyon, Lyon; 11Service d’Hematologie Pediatrique, CHU de Nice, Nice; 12Centre de Reference MCGRE, Service de Pediatrie, Centre Hospitalier Intercommunal de Creteil, Creteil, Inserm U955, Universite ParisXII, Creteil; 13Centre de Reference MCGRE, Service de Pediatrie, Centre Hospitalier de Mayotte, Mamoudzou, Mayotte; 14Service d’Hematologie Pediatrique, CHU de Besançon, Besançon; 15Service de Pediatrie-Neonatologie-Medecine de l’Adolescent, GHEF-Site de Marne La Vallee, Jossigny; 16Centre de Reference MCGRE, Service de Pediatrie Generale, Hopital Necker-Enfants Malades, AP-HP, Paris; 17Service de Pediatrie, CHU de Nimes, Nimes; 18Service d’Hematologie Pediatrique, CHU de Rennes, Rennes; 19Service de Pediatrie, CHR Metz-Thionville, Metz; 20Centre de Référence MCGRE, Service d’Hematologie-Immunologie, Hopital Robert Debre, AP-HP, Paris; 21Service d’Hematologie Pediatrique, CHU de Rouen, Rouen; 22Service de Medecine Interne, Hopital Nord, Saint-Etienne; 23Centre de Reference MCGRE, CIC Biotherapie, Hopital NeckerEnfants Malades, AP-HP, Paris; 24Centre de Reference MCGRE, Service de Pediatrie, Centre Hospitalier Intercommunal de Creteil (CHIC), Creteil; 25Service de Pediatrie, CHR d’Orleans, Orleans; 26Service d’Hematologie et d’Oncologie Pediatrique, Hopital Hautepierre, Strasbourg; 27Service d’Onco-Hematologie Pediatrique, CHRU Nancy, Vandoeuvre les Nancy; 28Service de Pediatrie, CHU La Reunion, Groupe Hospitalier Sud Reunion, Saint Pierre; 29Service d’Hematologie Clinique, CHU Limoges, Limoges; 30Service de Pediatrie, Hopital d’Enfants, Roubaix and 31Hopitaux de Brabois, CHRU de Nancy, Vandoeuvre les Nancy, France
1. Ballas SK, Zeidan AM, Duong VH, DeVeaux M, Heeney MM. The effect of iron chelation therapy on overall survival in sickle cell disease and b-thalassemia: a systematic review. Am J Hematol. 2018;93(7):943-952.
2. Forni GL, Gianesin B, Musallam KM, et al. Overall and complication-free survival in a large cohort of patients with b-thalassemia major followed over 50 years. Am J Hematol. 2023;98(3):381-387.
3. De Sanctis V, Eleftheriou A, Malaventura C; Thalassaemia International Federation Study Group on Growth and Endocrine Complications in Thalassaemia. Prevalence of endocrine complications and short stature in patients with thalassaemia major: a multicenter study by the Thalassaemia International Federation (TIF). Pediatr Endocrinol Rev. 2004;2 Suppl:2249-255.
4. Thuret I, Pondarré C, Loundou A, et al. Complications and treatment of patients with b-thalassemia in France: results of the National Registry. Haematologica. 2010;95(5):724-729.
5. Soliman AT, Yassin MA, De Sanctis V. Final adult height and endocrine complications in young adults with b-thalassemia major
Correspondence:
M. VENEZIANO BROCCIA - Mathilde.veneziano-broccia@ap-hm.fr
https://doi.org/10.3324/haematol.2023.283610
Received: September 20, 2023. Accepted: February 13, 2024. Early view: February 22, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
MVB and AB performed the research. MVB, JV and IT wrote the manuscript. SS and IT supervised the study. All authors critically revised the manuscript and approved the final version for publication.
Acknowledgments
We are very grateful to all physicians listed in Appendix A (Online Supplementary Appendix) who continually contribute to the NaThalY registry.
Funding
This study did not benefit from any specific funding. The National NaThalY Registry receives fundings from the National Network for constitutional diseases of red blood cells and erythropoiesis (filière MCGRE) and from private pharmaceutical companies.
Data-sharing statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
(TM) who received oral iron chelation (OIC) in comparison with those who did not use OIC. Acta Biomed. 2018;89(2-S):27-32.
6. Bronspiegel-Weintrob N, Olivieri NF, Tyler B, Andrews DF, Freedman MH, Holland FJ. Effect of age at the start of iron chelation therapy on gonadal function in beta-thalassemia major. N Engl J Med. 1990;323(11):713-719.
7 Borgna-Pignatti C, Rugolotto S, De Stefano P, et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica. 2004;89(10):1187-1193.
8. Donze C, Benoit A, Thuret I, et al. b-thalassemia in childhood: current state of health in a high-income country. Br J Haematol. 2023;201(2):334-342.
9 Casale M, Citarella S, Filosa A, et al. Endocrine function and bone disease during long-term chelation therapy with deferasirox in patients with b-thalassemia major. Am J Hematol. 2014;89(12):1102-1106.
10 Moniez S, Pienkowski C, Cartault A. [Puberté normale et pathologique.] J Pédiatrie Puéric. 2023;36(3):94-106.
11. De Sanctis V, Soliman AT, Yassin MA, et al. Hypogonadism in
male thalassemia major patients: pathophysiology, diagnosis and treatment. Acta Bio-Medica Atenei Parm 2018;89(2-S):6-15.
12. Casale M, Forni GL, Cassinerio E, et al. Risk factors for endocrine complications in transfusion-dependent thalassemia patients on chelation therapy with deferasirox: a risk assessment study from a multi-center nation-wide cohort. Haematologica. 2022;107(2):467-477.
13. Noetzli LJ, Panigrahy A, Mittelman SD, et al. Pituitary iron and volume predict hypogonadism in transfusional iron overload.
Am J Hematol. 2012;87(2):167-171.
14 Trachtenberg FL, Gerstenberger E, Xu Y, et al. Relationship among chelator adherence, change in chelators, and quality of life in thalassemia. Qual Life Res. 2014;23(8):2277-2288.
15. Albu A, Barbu CG, Antonie L, Vladareanu F, Fica S. Risk factors associated with hypogonadism in b-thalassemia major patients: predictors for a frequent complication of a rare disease. Postgrad Med. 2014;126(5):121-127.
Zanubrutinib is a next-generation Bruton tyrosine kinase inhibitor (BTKi) designed to minimize off-target effects associated with toxicities that have limited long-term treatment with ibrutinib, a first-generation BTKi. A previous pooled safety analysis of zanubrutinib monotherapy using data from six clinical trials (N=779) found that treatment was generally well tolerated,1 with infections, hemorrhage, and neutropenia the most commonly reported categories of treatment-emergent adverse events (TEAE) of special interest (AESI). Rates of cardiovascular toxicities with zanubrutinib, including atrial fibrillation (afib)/flutter and hypertension, were considerably lower than those observed previously with ibrutinib. Here, we expanded on these findings and combined updated data from six studies examined in a prior pooled analysis1 with data from four additional studies (Online Supplementary Table S1). A comparative analysis of zanubrutinib versus ibrutinib was also conducted using data from two of these ten studiesthe randomized phase III trials ALPINE (relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma2,3) and ASPEN cohort 1 (Waldenström macroglobulinemia4). The findings for the pooled zanubrutinib population (N=1,550) were consistent with those of the prior analysis, and the comparative analysis demonstrated the favorable safety profile of zanubrutinib 160 mg twice daily (N=425) compared with ibrutinib 420 mg once daily (N=422) (clinicaltrials gov. Identifiers: NCT03189524, NCT03206918, NCT03206970, NCT03332173, NCT03846427, NCT02343120, NCT03053440, NCT03336333, NCT03734016, NCT04170283).
Studies were approved by the independent ethics committees/institutional review boards at each participating institution and conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. All patients provided written informed consent. The median age of the pooled zanubrutinib population was 66.2 years, and the majority of patients were male (66.3%) (Table 1). Most patients had chronic lymphocytic leukemia/small lymphocytic lymphoma (60.5%), and approximately two-thirds had relapsed/refractory disease (68.9%). In the comparative analysis using data from ALPINE3 and ASPEN (cohort 1),5 baseline characteristics were generally similar between zanubrutinib- and ibrutinib-treated patients. In the total pooled zanubrutinib population, 45.0% of patients received zanubrutinib for ≥36 months (median, 34.4 months [range, 0.1-90.0 months]), and 56.5% of patients remained on zanubrutinib as of the data cutoff. In the comparative
analysis, median treatment duration was 32.6 months (range, 0.4-68.7 months) for zanubrutinib versus 25.7 months (range, 0.1-59.3 months) for ibrutinib. Relative dose intensity was comparable between treatments, but a greater percentage of patients were on zanubrutinib versus ibrutinib treatment for ≥36 months (29.4% vs. 25.4%; median time to discontinuation by Kaplan-Meier estimate, 63.3 vs. 42.2 months). In the comparative analysis, zanubrutinib-treated patients were more likely to still be on treatment at data cutoff than those treated with ibrutinib (69.9% vs. 45.0%).
TEAE leading to treatment discontinuation were reported in 13.6% of patients in the total pooled zanubrutinib population (Online Supplementary Table S2). In the comparative analysis, TEAE leading to treatment discontinuation were less common with zanubrutinib versus ibrutinib (14.1% vs. 22.0%). Infections were the most common TEAE leading to treatment discontinuation in the pooled zanubrutinib and comparative analysis populations (total zanubrutinib, 4.5%; ASPEN/ALPINE zanubrutinib, 5.4%; ASPEN/ALPINE ibrutinib, 6.6%). In the comparative analysis, ibrutinib-treated patients were more likely than zanubrutinib-treated patients to experience cardiac disorder (MedDRA system organ class) TEAE that led to discontinuation (4.3% [N=18; most common, afib, N=7] vs. 0.5% [N=2; cardiomegaly and ventricular extrasystoles, each N=1]).
Deaths attributed to TEAE occurred in 7.3% of patients in the total pooled zanubrutinib population and 8.7% and 10.2% of patients treated with zanubrutinib and ibrutinib, respectively, in the comparative analysis (Online Supplementary Table S2). Infections were the most common TEAE leading to death (total pooled zanubrutinib, 3.7%; ASPEN/ALPINE zanubrutinib, 5.2%; ASPEN/ALPINE ibrutinib, 6.2%). Cardiac disorder TEAE leading to death occurred in seven patients (1.7%) treated with ibrutinib versus one patient (0.2%) treated with zanubrutinib (see footnotes to Online Supplementary Table S2).
In this pooled analysis, 97.9% of patients who received zanubrutinib monotherapy had ≥1 TEAE (grade ≥3, 66.9%), and 49.2% had serious TEAE (Online Supplementary Table S2). TEAE considered treatment-related by the investigator were reported in 79.4% of patients (grade ≥3, 35.7%). The most common (any grade in ≥10% of patients; grade ≥3 in ≥5%) non-hematologic TEAE reported are shown in Figure 1A. No grade ≥3 non-hematologic TEAE were reported in ≥10% of patients; the most common were pneumonia (8.4%; treatment-related, 4.1%) and hypertension (8.1%; treatment-re-
Table 1. Demographics and baseline characteristics.
Characteristics
<65, N (%)
≥65 to <75, N (%)
N (%)
Sex, N (%)
Race, N (%)
Geographic region, N (%)c
performance status, N (%)
history, N (%)f
Concomitant
CLL/SLL: chronic lymphocytic leukemia/small lymphocytic lymphoma; ECOG: Eastern Cooperative Oncology Group. aIncludes patients with Waldenström macroglobulinemia from ASPEN cohort 1 (N=101) and patients with CLL/SLL from ALPINE (N=324). bIncludes patients with Waldenström macroglobulinemia from ASPEN cohort 1 (N=98) and patients with CLL/SLL from ALPINE (N=324). cLocation of study site enrollment. Asia includes China (Mainland and Taiwan) and South Korea; Europe includes Austria, Belgium, Belarus, Bulgaria, Czech Republic, France, Germany, Greece, Italy, the Netherlands, the Russian Federation, Poland, Spain, Sweden, Turkey, and the UK; and North America includes the United States and Canada. dIncludes patients with Richter transformation (N=13), hairy cell leukemia (N=11), B-lineage lymphoma (N=1), and indolent lymphoma (N=1). ePatients with Waldenström macroglobulinemia from ASPEN cohort 1. fPercentages are expressed using the number of patients with available medical history (all zanubrutinib, N=1,477; ASPEN/ALPINE zanubrutinib, N=409; ASPEN/ALPINE ibrutinib, N=411). gSystem organ class. hIndividual preferred term. iConcomitant medications are defined as medications that started before the first dose of study treatment and were continuing at the time of the first dose of study treatment or started on or after the date of the first dose of zanubrutinib treatment up to the last zanubrutinib dose date + 30 days or initiation of a new anticancer therapy. Patients with >1 medication within a class level and preferred name were counted only once within that class level and preferred name. Medication class was designated per the Anatomical Therapeutic Chemical classification system. jExcluding acetylsalicylic acid.
lated, 3.4%). Pneumonia (8.2%) was the only serious TEAE in ≥5% of patients. In summary, these findings were consistent with those for the prior pooled safety analysis,1 even with a median treatment duration ≈9 months longer. Select TEAE preferred terms were grouped as AESI (“opportunistic infections” included preferred terms under the narrow standardized MedDRA query “opportunistic infections”; for all other AESI preferred terms, see Tam et al.1). In order to account for differing treatment exposures across the trials, exposure-adjusted incidence rates (EAIR) of these AESI were determined for the total pooled zanubrutinib population (Figure 1B; see legend for EAIR calculation and assumption) and comparative analysis populations (Figure 1C).
Infections, hemorrhage, and neutropenia were the most
frequently reported AESI in the total pooled zanubrutinib population, even after adjusting for dose exposure (Figure 1B). Despite a longer median treatment duration, the EAIR of the cardiovascular AESI were comparable to those of the earlier analysis (hypertension, 6.81 in the present analysis vs. 6.87 persons per 100 person-years [PY] in Tam et al.;1 afib/flutter, 1.74 vs. 1.45 persons per 100 PY, respectively). ALPINE had a greater hypertension EAIR than SEQUOIA and ASPEN; exclusion of data from ALPINE decreased the hypertension EAIR to 5.73 persons per 100 PY. In ALPINE, the hypertension rate was similar between the zanubrutinib and ibrutinib arms; however, the incidence of cardiac disorders such as afib/flutter was higher in the ibrutinib arm,3 whereas incidence in the zanubrutinib arm remained low and comparable to that observed in SEQUOIA and ASPEN.
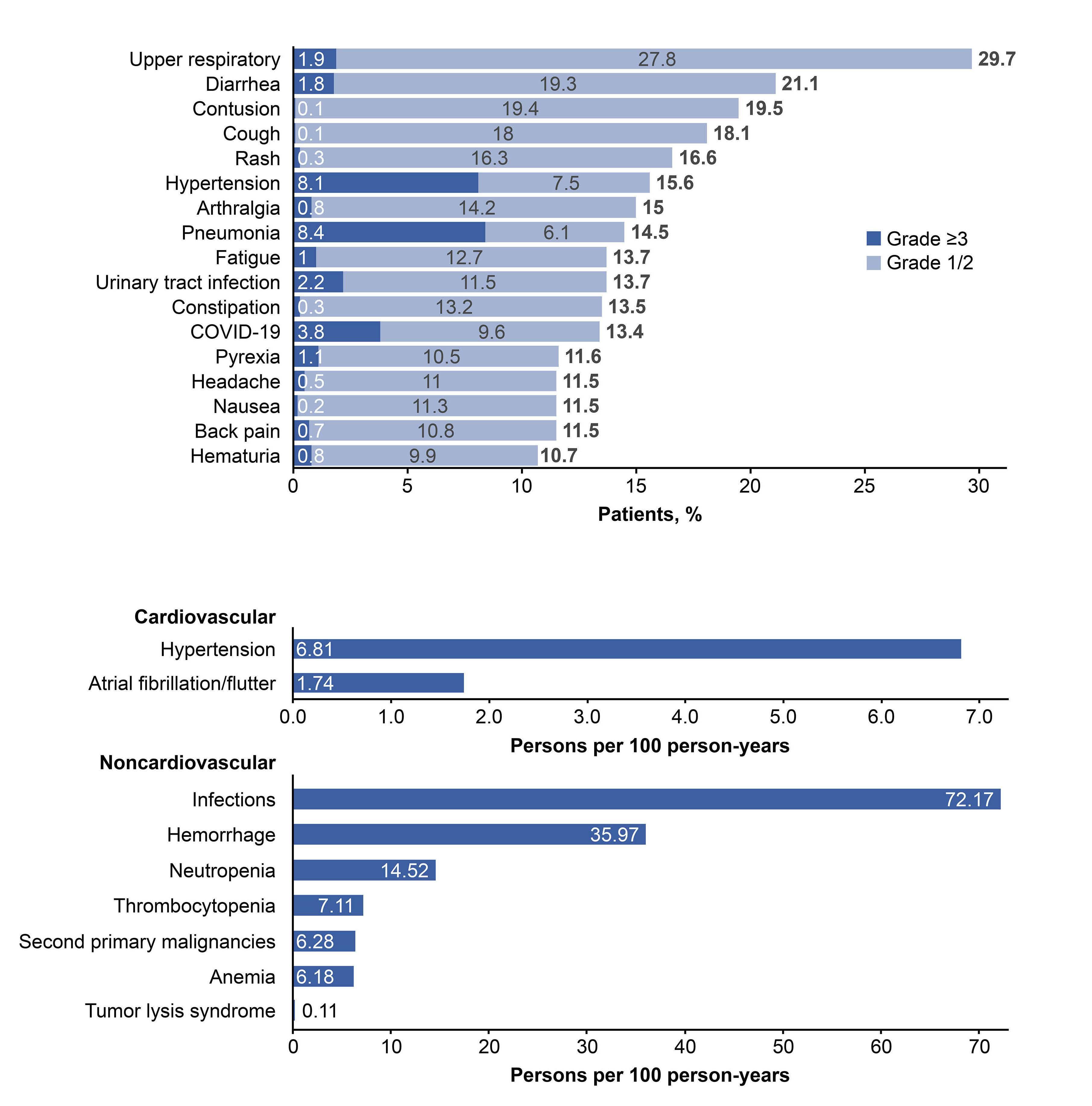
Continued on following page.
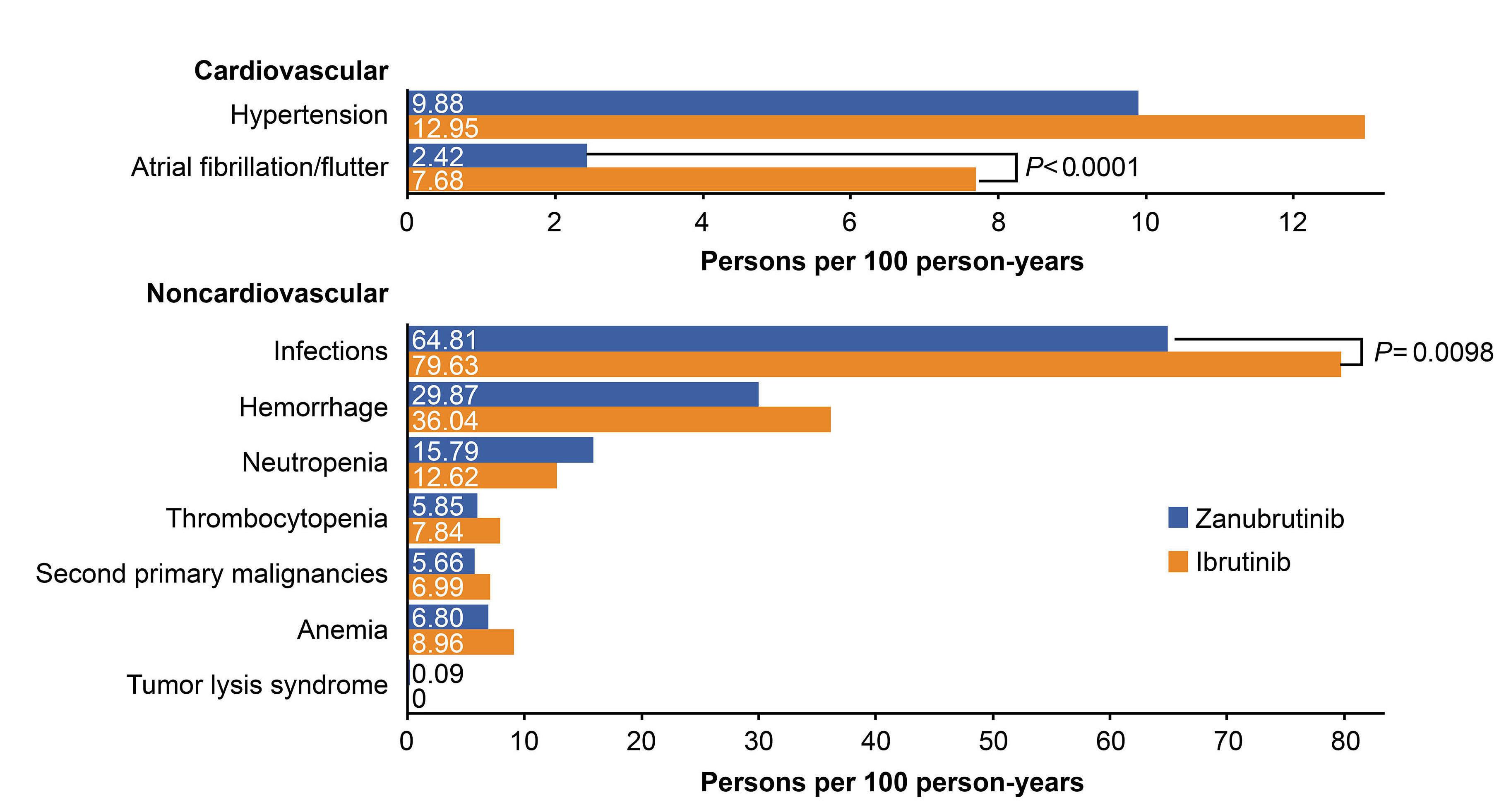
Figure 1. Incidence of any-grade non-hematologic treatment-emergent adverse events and exposure-adjusted incidence rates of adverse events of special interest. (A) Treatment-emergent adverse events (TEAE) reported in ≥10% or grade ≥3 TEAE in ≥5% of patients treated with zanubrutinib (N=1,550) are shown. TEAE were defined as adverse event (AE) preferred terms with an onset date or worsening in severity from baseline (prior to treatment) at or after the first dose of zanubrutinib and up to the last zanubrutinib dose date + 30 days or initiation of new anticancer therapy, whichever occurred first. Worsening of an event to grade 5 beyond the last zanubrutinib dose date + 30 days and prior to initiation of new anticancer therapy was also considered treatment emergent. (B, C) The AE of special interest (AESI) shown are grouped terms. The preferred terms for the TEAE included in each AESI category are as previously published,1 except for “opportunistic infections,” which included preferred terms under the narrow standardized MedDRA query “opportunistic infections.” Exposure-adjusted incidence rates (EAIR) were calculated as the number of patients who experienced a specific AESI divided by the total exposure time (i.e., the first dose date to the first event date or to the treatment-emergent period end date if there was no event) in years for all patients and then multiplied by 100 to express as persons per 100 person-years. Of note, EAIR assumes that the risk of an event occurring is constant over time and serves as an additional means for evaluating safety events. In (B), data are shown for the total pooled zanubrutinib population (N=1,550). In (C), data are shown for the comparative analysis of patients treated with zanubrutinib (N=425) or ibrutinib (N=422) as part of the randomized studies ASPEN (cohort 1) or ALPINE. The Poisson regression model was used to compare EAIR between treatment groups, with the number of patients who experienced events as the dependent variable and log(exposure time) as the offset. The P value based on χ2 test was reported. All statistical tests were two-sided, with P<0.05 considered significant; no adjustments for multiple comparisons were made. COVID-19: coronavirus disease 2019.
Importantly, across all ten trials, no zanubrutinib-treated patients discontinued due to hypertension. In the comparative analysis, all EAIR of AESI, except for neutropenia, were numerically lower in patients treated with zanubrutinib versus ibrutinib (Figure 1C). Although the neutropenia EAIR was slightly higher with zanubrutinib, the infection EAIR was significantly lower (64.81 vs. 79.63 persons per 100 PY; P=0.0098) with zanubrutinib, even after excluding COVID-19-related infection terms (54.48 vs. 69.96 persons per 100 PY; P=0.0029). The EAIR for afib/flutter was also significantly lower with zanubrutinib versus ibrutinib (P<0.0001). The hypertension EAIR was also reduced in patients receiving zanubrutinib versus ibrutinib (P=0.0610). AESI EAIR analyzed over time were relatively constant or decreased with zanubrutinib (Figure 2; time to first event data, Online Supplementary Figure S1). In the comparative analysis, AESI EAIR over time were numerically lower with zanubrutinib versus ibrutinib, except for neutropenia, which
was higher in the first 12 months of treatment and is considered an on-target effect of BTK inhibition.6 However, this was not accompanied by an elevated infection EAIR nor was neutropenia a substantial cause of discontinuation (7.1% [3/42]). Increases of >10 persons per 100 PY in the EAIR for anemia and hemorrhage were observed with ibrutinib between the >24-month exposure intervals. In contrast, the greatest increase between consecutive intervals with zanubrutinib was 4.1 persons per 100 PY (hemorrhage). At all treatment intervals evaluated, the EAIR for afib/ flutter was 6.7 to 13.6 persons per 100 PY higher with ibrutinib than with zanubrutinib. In the present analysis, the afib/flutter EAIR was relatively constant in the first 2 years of ibrutinib exposure but steadily increased with each subsequent year of treatment. In contrast, the EAIR in patients who received zanubrutinib was much lower at all intervals, with only slight increases observed after 2 to 3 years of exposure. This relatively stable incidence of
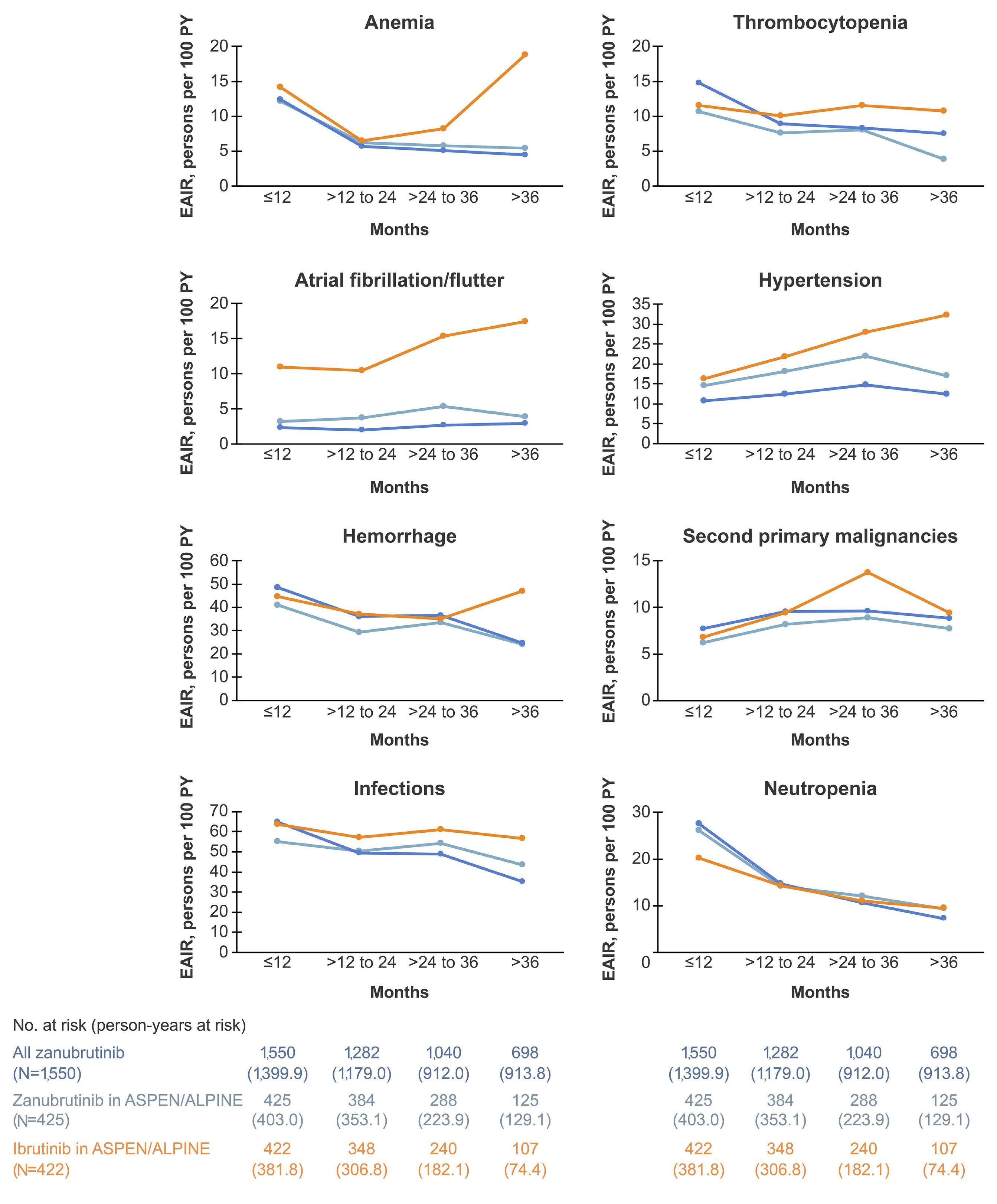
Figure 2. Exposure-adjusted incidence rates of select adverse events of special interest over time. Adverse events of special interest (AESI) are grouped terms as defined in the legend for Figure 1. Exposure-adjusted incidence rates (EAIR) at each time interval were calculated as the number of patients who experienced a specific AESI during that time interval divided by the total exposure time in years at the corresponding time interval. This value was then multiplied by 100 to express as persons per 100 person-years (PY). Data are shown for the total pooled zanubrutinib population (N=1,550) and the comparative analysis patient populations (patients treated with zanubrutinib [N=425] or ibrutinib [N=422] as part of the randomized studies ASPEN cohort 1 or ALPINE; each treatment group is labeled as ASPEN/ALPINE).
afib/flutter with zanubrutinib, despite extended exposure, is important for long-term treatment. Additionally, a lower incidence of afib may minimize the need for supportive care (e.g., anticoagulants, antiplatelet agents) that can further increase the bleeding risk associated with BTKi. Finally, although hypertension in patients receiving ibrutinib has been associated with increased incidence of major cardiovascular AE,7 the incidence of cardiac disorder TEAE was comparable for zanubrutinib across ALPINE, ASPEN, and SEQUOIA despite the higher hypertension EAIR observed in ALPINE.
Due to the continuous dosing of BTKi in most B-cell malignancies, low treatment discontinuation rates and long-term tolerability are key considerations, particularly in patients with B-cell malignancies such as chronic lymphocytic leukemia/small lymphocytic lymphoma who tend to be aged >65 years and have other (e.g., cardiovascular) comorbidities.8,9 The first-in-class BTKi ibrutinib has drastically improved treatment of numerous B-cell malignancies, but cardiac arrhythmias and their associated outcomes are a frequently cited concern10-12 and are possibly due to off-target inhibition of kinases such as TEC and CSK.13,14 Such toxicities can limit the duration and, consequently, the benefit15 of treatment. Zanubrutinib was designed with greater selectivity to minimize off-target effects. In this analysis, zanubrutinib remained well tolerated, consistent with the previous analysis,1 with no emergence of new safety signals, even at a median treatment duration of approximately 3 years. In the comparative analysis, zanubrutinib exhibited a more favorable safety profile than ibrutinib, as demonstrated by the longer median treatment duration and lower frequency of TEAE, including cardiac disorders, that led to treatment discontinuation or death. These analyses support zanubrutinib as an appropriate long-term treatment option for patients with B-cell malignancies.
Jennifer R. Brown,1 Paolo Ghia,2 Wojciech Jurczak,3 Brad S. Kahl,4 Nicole Lamanna,5 Tadeusz Robak,6 Mazyar Shadman,7 Constantine S. Tam,8 Lugui Qiu,9 Jason Paik,10 Tommi Salmi,11 Liping Wang,12 Jun Zhang,10 Meng Zhang,10° Aileen Cohen,10 Han Ma10 and Alessandra Tedeschi13
1Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA; 2Università Vita-Salute San Raffaele and IRCCS Ospedale San Raffaele, Milan, Italy; 3Maria Skłodowska-Curie National Research Institute of Oncology, Kraków, Poland; 4Siteman Cancer Center, Washington University School of Medicine, St Louis, MO, USA; 5Herbert Irving Comprehensive Cancer Center, Columbia University, New York, NY, USA; 6Medical University of Łódź, Łódź, Poland; 7Fred Hutchinson Cancer Research Center, Seattle, WA, USA; 8Alfred Hospital and Monash University, Melbourne, Victoria, Australia; 9State Key Laboratory of Experimental Hematology,
National Clinical Medical Research Center for Blood Diseases, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China; 10BeiGene USA, Inc, San Mateo, CA, USA; 11BeiGene International GmbH, Basel, Switzerland; 12BeiGene (Shanghai) Co, Ltd, Shanghai, China and 13ASST Grande Ospedale Metropolitano Niguarda, Milan, Italy
°Current address: ProKidney Corp., Winston-Salem, NC, USA.
Correspondence:
J.R. BROWN - Jennifer_Brown@dfci.harvard.edu
https://doi.org/10.3324/haematol.2023.283846
Received: September 5, 2023. Accepted: February 14, 2024. Early view: February 29, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
JRB discloses consultancy for AbbVie, Acerta/AstraZeneca, Alloplex Biotherapeutics, BeiGene, Galapagos NV, Genentech/Roche, Grifols Worldwide Operations, InnoCare Pharma Inc, iOnctura, Kite, Loxo/ Lilly, Merck, Numab Therapeutics, Pfizer and Pharmacyclics; research funding from BeiGene, Gilead, iOnctura, Loxo/Lilly, MEI Pharma and TG Therapeutics. PG discloses honoraria from AbbVie, ArQule/MSD, AstraZeneca, BeiGene, Celgene/Juno/Bristol Myers Squibb, Janssen, Lilly/Loxo, MEI Pharma, Roche and Sanofi; research funding from AbbVie, AstraZeneca, Janssen and Sunesis. WJ discloses consultancy for Janssen, AstraZeneca, MEI Pharma, Lilly, Takeda, Roche, AbbVie and BeiGene; research funding from AbbVie, Bayer, BeiGene, Celgene, Janssen, Roche, Takeda, TG Therapeutics, AstraZeneca, MEI Pharma and Lilly. BSK discloses research funding from BeiGene to Washington University School of Medicine (St Louis, MO, USA); consulting fees from AbbVie, AstraZeneca, BeiGene, Janssen and Pharmacyclics. NL discloses consultancy for AbbVie, AstraZeneca, BeiGene, Lilly/Loxo, Genentech, Janssen and Pharmacyclics; research funding from AbbVie, AstraZeneca, BeiGene, Lilly/Loxo, Genentech, Octapharma, Oncternal, MingSight and TG Therapeutics. TR discloses research funding from BeiGene, Octapharma, AstraZeneca, Janssen, Regeneron and GSK; honoraria from AstraZeneca, BeiGene, Janssen, AbbVie, Octapharma, Regeneron and GSK; travel, accommodations, expenses from AstraZeneca. MS discloses consultancy for AbbVie, Genentech, AstraZeneca, Sound Biologics, Pharmacyclics, BeiGene, Bristol Myers Squibb, MorphoSys/Incyte, TG Therapeutics, Innate Pharma, Kite Pharma, Adaptive Biotechnologies, Epizyme, Lilly, Adaptimmune, Mustang Bio, Regeneron, Merck, Fate Therapeutics, MEI Pharma and Atara Biotherapeutic; research funding from Mustang Bio, Celgene, Bristol Myers Squibb, Pharmacyclics, Gilead, Genentech, AbbVie, TG Therapeutics, BeiGene, AstraZeneca, Sunesis, Atara
Biotherapeutics, Genmab, MorphoSys/Incyte and Vincerx. CST discloses research funding from Janssen, AbbVie and BeiGene; honoraria from Janssen, AbbVie, BeiGene, Loxo and AstraZeneca. LQ discloses consultancy for and speakers bureau of Janssen, AstraZeneca, Takeda, Roche, AbbVie and BeiGene. TS discloses employment at BeiGene Switzerland GmbH; is an equity holder of BeiGene Ltd. MZ, JP, LW, JZ and HM disclose employment at BeiGene. AC discloses employment at BeiGene; is an equity holder of BeiGene; discloses travel, accommodations, expenses from BeiGene. AT discloses consultancy for BeiGene, AstraZeneca, AbbVie and Janssen; honoraria from BeiGene, AstraZeneca, AbbVie and Janssen; speakers bureau of BeiGene, AstraZeneca, AbbVie and Janssen; travel, accommodations, expenses from BeiGene, AstraZeneca, AbbVie and Janssen.
Contributions
JRB, PG, WJ, BSK, NL, TR, MS, CST, LQ, and AT enrolled patients, performed research, and contributed to data collection, analysis, and interpretation. JP, TS, LW, MZ, AC, and HM contributed to this study’s conceptualization and design, data curation, formal analysis, data interpretation, methodology, and validation. All authors contributed to the writing, review, editing, and final approval of this manuscript.
Acknowledgments
The authors thank the patients and their families, investigators, co-investigators, and the study teams at each of the participating
1. Tam CS, Dimopoulos M, Garcia-Sanz R, et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B-cell malignancies. Blood Adv. 2022;6(4):1296-1308.
2. Hillmen P, Eichhorst B, Brown JR, et al. Zanubrutinib versus ibrutinib in relapsed/refractory chronic lymphocytic leukemia and small lymphocytic lymphoma: interim analysis of a randomized phase III trial. J Clin Oncol. 2023;41(5):1035-1045.
3. Brown JR, Eichhorst B, Hillmen P, et al. Zanubrutinib or ibrutinib in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2023;388(4):319-332.
4 Tam CS, Garcia-Sanz R, Opat S, et al. ASPEN: long-term followup results of a phase 3 randomized trial of zanubrutinib versus ibrutinib in patients with Waldenström macroglobulinemia. J Clin Oncol. 2022;40(s16):7521.
5. Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenstrom macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038-2050.
6. Honda F, Kano H, Kanegane H, et al. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol. 2012;13(4):369-378.
7 Estupiñán HY, Berglof A, Zain R, et al. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front Cell Dev Biol. 2021;9:630942.
centers for the clinical trials included in this analysis. This analysis was sponsored by BeiGene Co, Ltd. Medical writing and editorial assistance were provided by Jenna M. Gaska, PhD, of Nucleus Global, an Inizio Company, and supported by BeiGene.
Funding
This work was supported by funding from BeiGene USA, Inc. Data analyses were performed by biostatisticians at BeiGene USA, Inc, and BeiGene (Beijing) China.
Data-sharing statement
BeiGene voluntarily shares anonymous data on completed studies responsibly and provides qualified scientific and medical researchers access to anonymous data and supporting clinical trial documentation for clinical trials in dossiers for medicines and indications after submission and approval in the United States, China, and Europe. Clinical trials supporting subsequent local approvals, new indications, or combination products are eligible for sharing once corresponding regulatory approvals are achieved. BeiGene shares data only when permitted by applicable data privacy and security laws and regulations. In addition, data can only be shared when it is feasible to do so without compromising the privacy of study participants. Qualified researchers may submit data requests/research proposals for BeiGene review and consideration through BeiGene’s Clinical Trial Webpage at https://www.beigene.com/our-science-andmedicines/our-clinical-trials/.
8. Thandra KC, Barsouk A, Saginala K, et al. Epidemiology of nonHodgkin’s lymphoma. Med Sci (Basel). 2021;9(1):5.
9. Hester LL, Park SI, Wood WA, et al. Cause-specific mortality among Medicare beneficiaries with newly diagnosed nonHodgkin lymphoma subtypes. Cancer. 2019;125(7):1101-1112.
10. Coutre SE, Byrd JC, Hillmen P, et al. Long-term safety of singleagent ibrutinib in patients with chronic lymphocytic leukemia in 3 pivotal studies. Blood Adv. 2019;3(12):1799-1807.
11. Gordon MJ, Jones JE, George B, et al. Long-term outcomes in patients with chronic lymphocytic leukemia treated with ibrutinib: focus on hypertension and cardiovascular toxicity. Cancer. 2023;129(14):2192-2200.
12. Brown JR, Moslehi J, O’Brien S, et al. Characterization of atrial fibrillation adverse events reported in ibrutinib randomized controlled registration trials. Haematologica. 2017;102(10):1796-1805.
13. Fleming MR, Xiao L, Jackson KD, et al. Vascular impact of cancer therapies: the case of BTK (Bruton tyrosine kinase) inhibitors. Circ Res. 2021;128(12):1973-1987.
14 McMullen JR, Boey EJ, Ooi JY, et al. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood. 2014;124(25):3829-3830.
15. Barr PM, Brown JR, Hillmen P, et al. Impact of ibrutinib dose adherence on therapeutic efficacy in patients with previously treated CLL/SLL. Blood. 2017;129(19):2612-2615.
Bruton tyrosine kinase (BTK) inhibitors have shown remarkable efficacy in the treatment of B-cell malignancies, yet many patients develop intolerance to these drugs leading to treatment discontinuation. Ibrutinib, the first-in-class BTK inhibitor, can cause adverse effects including cardiotoxicities, bleeding, and cytopenias leading to discontinuation in up to 16% of patients, largely due to off-target activity.1 The second-generation, more selective BTK inhibitor, acalabrutinib, also leads to adverse effects and treatment discontinuation in up to 23% of patients.2 Zanubrutinib is a next-generation covalent, irreversible BTK inhibitor that has been approved world-wide for the treatment of patients with B-cell malignancies including chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), previously treated mantle cell lymphoma (MCL), Waldenström macroglobulinemia, and relapsed or refractory marginal zone lymphoma. Preclinical and clinical studies have demonstrated that zanubrutinib has superior potency, selectivity, efficacy, and a more favorable toxicity profile compared to ibrutinib.3-6 Results from BGB-3111-215 (NCT04116437), an ongoing phase II study evaluating the safety and efficacy of zanubrutinib monotherapy, demonstrate that zanubrutinib could be a safe treatment option for patients with B-cell malignancies who exhibited intolerance to prior treatment with ibrutinib (cohort 1) or to ibrutinib and/or acalabrutinib (cohort 2).6
To date, it is unclear whether the genomic profile of patients with B-cell malignancies who are intolerant to ibrutinib and/ or acalabrutinib is associated with intolerance or response to subsequent therapy. In this post-hoc analysis, a highly sensitive, targeted, next-generation sequencing panel (PredicineHEMETM; Predicine, Hayward, CA, USA) with full exon coverage of 106 genes commonly mutated in hematologic malignancies was used to explore the genetic profiles of patients enrolled in study BGB-3111-215.7 This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. Written informed consent was obtained from each patient and institutional review board approval was obtained at each study site. Samples were sequenced to a median depth of >20,000 reads, with a validated sensitivity of 0.25% mutant allele frequency for all genomic regions and 0.1% for mutational hotspots. Variant allele frequency (VAF) <0.1% for hotspot mutations and VAF <0.25% for non-hotspot mutations were excluded from analysis. Germline and clonal hematopoiesis of indeterminate potential mutations were also excluded from analysis. Baseline blood samples from 95.9% (71/74)
of all patients enrolled in the study and from 77.8% (7/9) of patients with disease progression (CLL: n=5; SLL: n=1; MCL: n=1) at data cutoff were available for analysis. Baseline demographics and disease characteristics were similar between cohorts and are summarized in Online Supplementary Table S1. Of note, most patients enrolled in this study had CLL or SLL (n=54).
We identified mutations in 91.5% (65/71) of baseline samples and in all (7/7) samples from patients with progressive disease (median = 4 mutations per sample; range, 1-14). The types of mutations identified as well as mutation frequencies are shown in Figure 1. Across all patients intolerant to BTK inhibitors, the most common baseline mutations were in TP53 (32%), SF3B1 (23%), ATM (18%), NOTCH1 (17%), and CHEK2 (15%) (Figure 1B). The mutation spectra of these genes are visualized as lollipop plots (Online Supplementary Figure S1) and are similar to those that have been observed in other studies.8 As expected, mutation prevalence at baseline differed among diseases and was mostly consistent with the findings in previous studies of relapsed or refractory patients with various B-cell malignancies (Figure 1B).8 In patients with CLL or SLL, the most frequently mutated genes were TP53 (16/54, 30%), SF3B1 (15/54, 28%), ATM (13/54, 24%), and NOTCH1 (11/54, 20%) – all within cell signaling pathways (e.g., DNA damage response and NOTCH signaling) known to be associated with disease susceptibility and/or poor prognosis in these patients. Observed rates of baseline TP53 (5/8 vs. 11/46, P=0.04), ATM (4/8 vs. 9/46, P=0.08), and SF3B1 (5/8 vs. 10/46, P=0.03) mutations were higher in patients who later developed progressive disease (n=8) than in patients who did not (n=46) (Figure 2A). Patients with these mutations also had a shorter progression-free survival compared to that of patients without mutations in these genes, as evidenced by the observed hazard ratios (and 95% confidence intervals): TP53 3.2 (0.84-11.8), SF3B1 5.9 (1.48-23.77), or ATM 5.5 (1.47-20.72) (Figure 2B-D), which is consistent with previous reports.9,10 However, the mutation frequency of NOTCH1 was similar in patients with and without disease progression (Figure 2A) and NOTCH1 mutation status was not correlated with progression-free survival (Figure 2E), a finding inconsistent with that of a previous study of patients with relapsed or refractory CLL treated with ibrutinib in whom NOTCH1 mutations were strongly associated with shorter progression-free survival (P=0.00002) and overall survival (P=0.0001).11 This discrepant finding was not due to the increased sensitivity of the PredicineHeme panel as patients with NOTCH1 VAF <1% (45.5% [5/11]) exhibited a similar
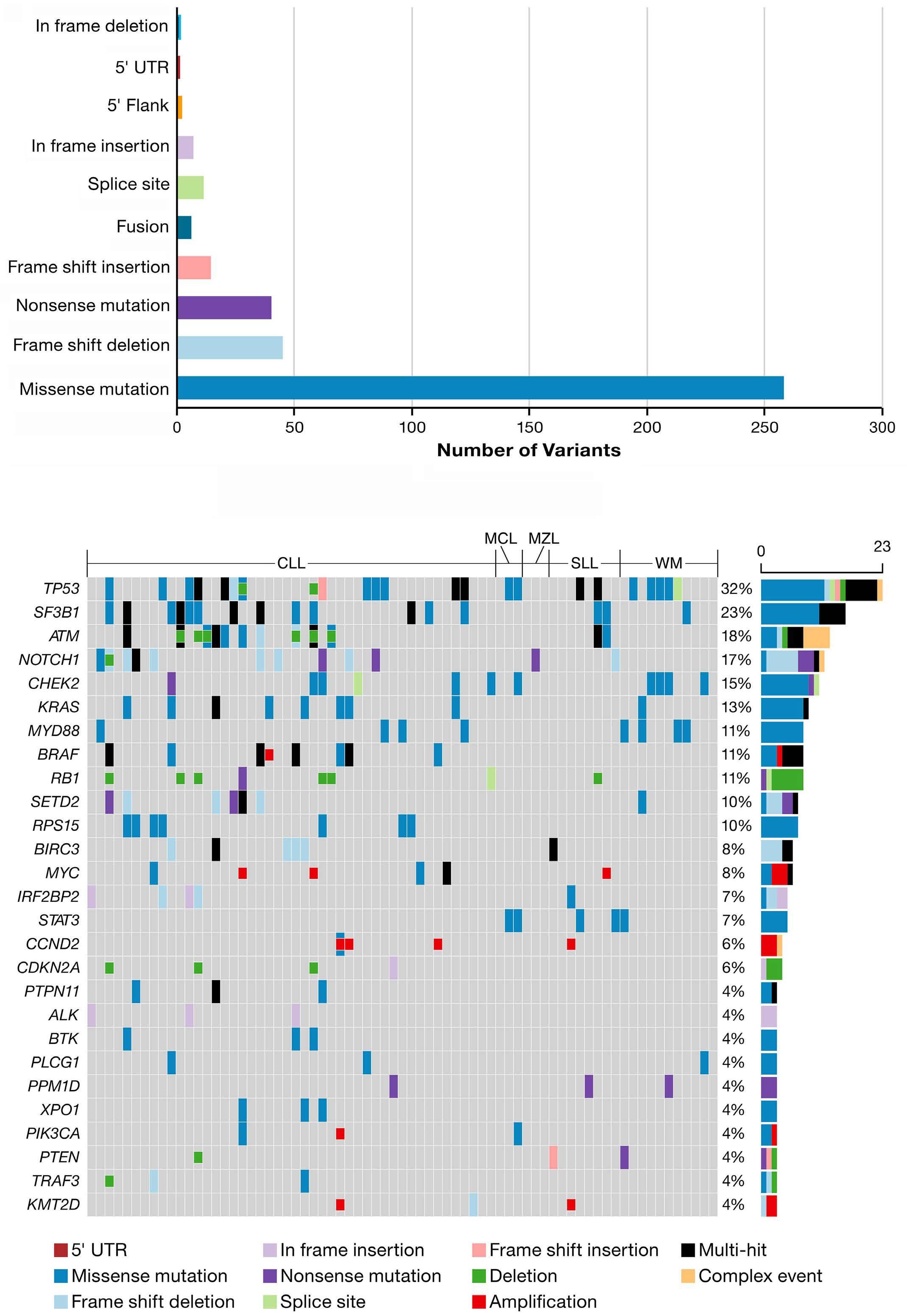
Figure 1. Variant classification and heatmap representation of mutations detected at baseline in at least three patients with B-cell malignancies. Samples were sequenced to a median depth of >20,000 reads, with a validated sensitivity of 0.25% mutant allele frequency for all genomic regions, and 0.1% for mutational hotspots. Variant allele frequency (VAF) <0.1% for hotspot mutations and VAF <0.25% for non-hotspot mutations were excluded from the analysis. Germline and clonal hematopoiesis of indeterminate potential mutations were also excluded from the analysis. (A) The number of variants is shown on the X-axis. (B) DNA mutation profile of patients and the distribution of mutations among different study cohorts by mutation type and treatment status. Each column represents one patient, and each row represents one gene (indicated by the gene symbol on the left). Mutation rates of each gene are shown on the right. Mutation type is color-coded as shown in the figure. UTR: untranslated region; CLL: chronic lymphocytic leukemia; MCL: mantle cell lymphoma; MZL: marginal zone lymphoma; SLL: small lymphocytic lymphoma; WM: Waldenström macroglobulinemia.
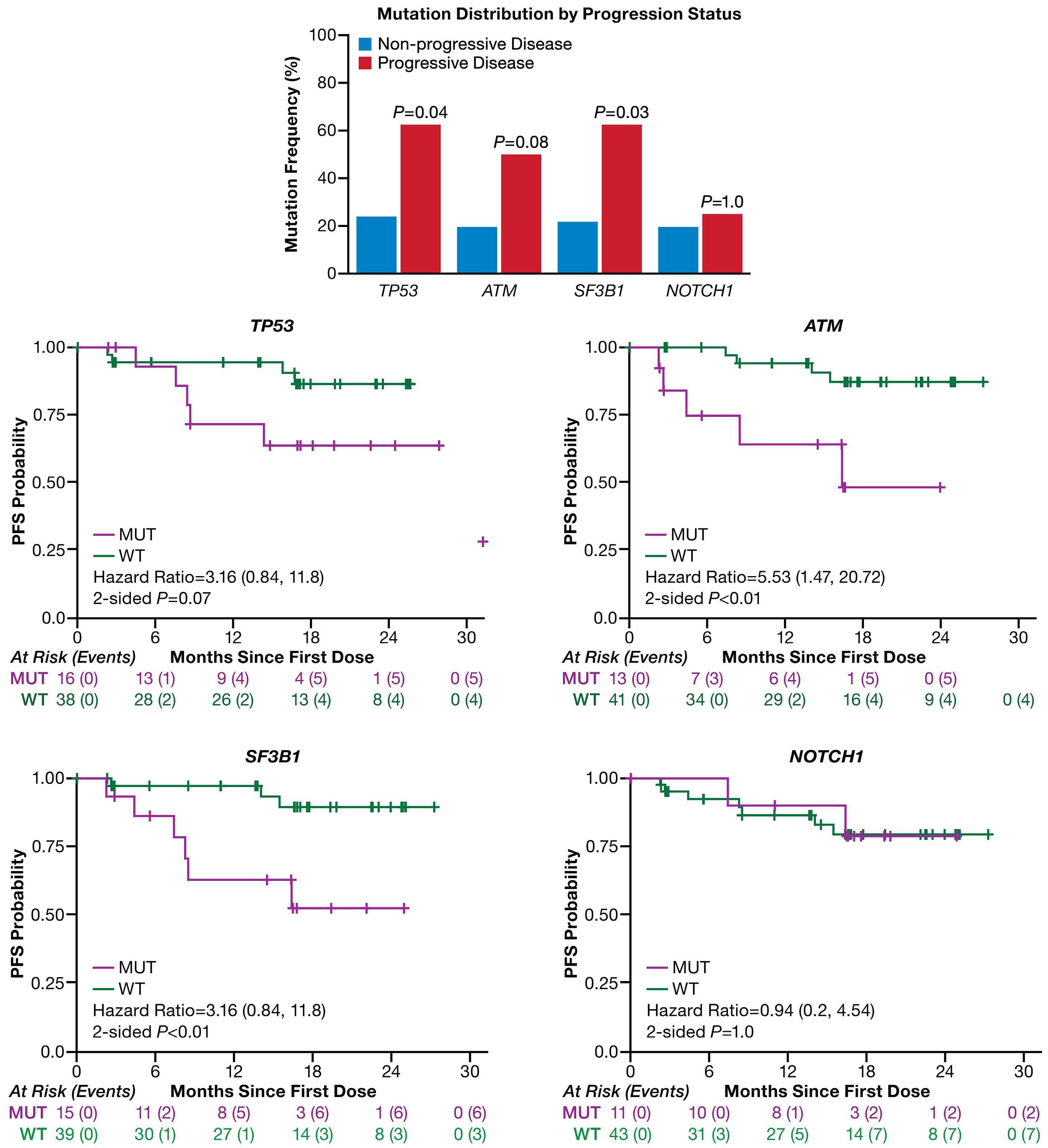
Figure 2. The presence of TP53, SF3B1, or ATM mutations is associated with inferior outcomes in patients with chronic lymphocytic leukemia or small lymphocytic lymphoma. (A) Distributions of baseline mutations between patients who developed progressive disease and those who did not were compared using proportions. (B-E) The association between genetic mutations and progression-free survival (PFS, defined as time from the date of first zanubrutinib dose to date of first progression of disease or death) was quantified by the log-rank test and hazard ratio and summarized by the Kaplan-Meier method. As of June 6, 2022, the data cutoff, patients were censored for PFS if: (i) they had no documented disease progression or death; (ii) they initiated subsequent anti-cancer therapy prior to progressive disease; or (iii) they progressed or died after more than one consecutive missed disease assessment. All analyses were performed using either R or SAS (version 9.4). Patients with mutations in TP53, SF3B1, and ATM, but not NOTCH1, showed shorter PFS. MUT: mutant; WT, wild type; PFS: progression-free survival.
progression-free survival to that of patients with NOTCH1 VAF >1%. Instead, the finding suggests the possibility that, in contrast to ibrutinib, zanubrutinib may uniquely suppress the outgrowth of clones with NOTCH1 mutations. Although the numbers of patients with Waldenström macroglobulinemia, MCL, and marginal zone lymphoma enrolled in our study were small and association analyses could not be performed, mutation profiles of these patients are reported for reference (Figure 1).
At the data cutoff for this analysis, nine patients (12.7%; 9/71) who were intolerant to ibrutinib and/or acalabrutinib and who were subsequently treated with zanubrutinib developed progressive disease. We assessed BTK inhibitor resistance mutations in BTK and PLCG2 in these patients (Table 1). At baseline, BTK C481S mutations were detected in 4.2% (3/71) of patients (Figure 1B). Of these, two patients progressed on zanubrutinib; patients n. 3 and n. 9 progressed at 4.6 months and 17.7 months of zanubrutinib treatment, respectively (Table 1). A third patient (data not shown) with a baseline BTK C481S mutation died from COVID-19 shortly after enrollment before disease assessments could be made. Patient n. 3, with CLL, had a high frequency (VAF=60.9%) of BTK C481S mutations at baseline. This patient had long exposure to ibrutinib treatment (~65 months) prior to enrolling in the study and had disease progression after 4.6 months of zanubrutinib treatment (Table 1). Patient n. 9, with CLL, who also had long prior exposure to
ibrutinib (51.9 months), initially presented with a low frequency BTK C481S mutation (VAF=0.9%) that increased in frequency at the time of disease progression (VAF=20.4%) (Table 1). This patient also had new PLCG2 mutations (L845F and D993H; VAF<1% for both mutations) at disease progression (Table 1). It is worth noting that this patient stayed on zanubrutinib treatment for 17.7 months before disease progression, with stable disease as best overall response. This suggests that BTK C481S mutations at low VAF did not prevent zanubrutinib efficacy in this patient. At the time of disease progression, patients n. 1 (CLL) and n. 2 (SLL) had developed new BTK and PLCG2 mutations. Patient n. 1 acquired high frequencies of BTK C481S mutations (VAF=19.2%) as well as low frequency mutations in PLCG2 (L845F, N750D, and R665W; VAF<1% for all three PLCG2 mutations) that had not been observed at baseline (Table 1). This patient was on zanubrutinib treatment for 9.2 months before disease progression. Patient n. 2 acquired BTK mutations (C481S: VAF=3.8%; C481Y: VAF=14.0%) and PLCG2 mutations (S707F, L845V, M1141K, and E1139del; VAF <6% for all PLCG2 mutations) at disease progression (Table 1). This patient was on zanubrutinib treatment for 17.9 months prior to disease progression. All BTK mutations detected in this study were located at the BTK-inhibitor binding site (C481S or C481Y). The remaining five patients with disease progression had no BTK or PLCG2 mutations detected at baseline or at disease progression. Patient n. 5, with MCL, had a CCND1-IGH
L845F, 2535A>C (1.0%)
N750D, 2248A>G (0.8%) R665W, 1993C>T (0.3%) 2
C481S, 1442G>C (0.3%) C481S, 14421T>A (3.8%) C481Y, 1442G>C (14.0%)
S707F, 2120C>T (5.8%)
L845V, 2533T>G (1.7%) E1139del, 3417_3419del (4.7%) M1141Lys, 3422T>A (0.9%)
L845F, 2535A>C (0.4%) D993H, 2977G>C (0.6%)
aInitial sample collected on study day 87. bInitial sample collected on study day 141. BTK: Bruton tyrosine kinase gene; VAF: variant allele frequency; PD: progressive disease; PLCG2: phospholipase C gamma 2 gene; CLL: chronic lymphocytic leukemia; MCL: mantle cell lymphoma; SLL: small lymphocytic lymphoma; NA: not applicable; ND: not detected.
fusion mutation at both baseline and disease progression; CCND1-IGH fusions have been reported to be associated with ibrutinib resistance in MCL patients.13 Four other patients with CLL harbored mutations in genes associated with poor prognosis, including TP53, ATM, and SF3B1 (Online Supplementary Table S2).9,10
Here we show that the gene mutational profile of patients with B-cell malignancies who were intolerant to ibrutinib and/or acalabrutinib is comparable to that of patients with relapsed or refractory disease. For example, patients with mutations in TP53, SF3B1, or ATM genes had a less favorable prognosis in this study and similar to that previously observed in patients treated with BTK inhibitors.9,10 Furthermore, progression-free survival was comparable between zanubrutinib-treated patients with or without NOTCH1 mutations. Lastly, four of seven intolerant patients who progressed on zanubrutinib acquired new BTK mutations and/or had an increase in the frequency of BTK mutations. Although there are limitations to this study (e.g., small sample size, short follow-up times, and a lack of direct comparison to non-intolerant patients), this is the first study to describe the genomic landscape of patients with B-cell malignancies who were intolerant to ibrutinib and/or acalabrutinib and were switched to treatment with zanubrutinib.
Linlin Xu,1 Mazyar Shadman,2 Ian W. Flinn,3 Moshe Y. Levy,4 Ryan Porter,5 John M. Burke,6 Syed F. Zafar,7 Jennifer L. Cultrera,8 Jamal Misleh,9 Edwin C. Kingsley,10 Habte A. Yimer,11 Benjamin Freeman,12 Arvind Chaudhry,13 Praveen K. Tumula,14 Mitul D. Gandhi,15 Rocco Crescenzo,1 Kunthel By,1 Aileen Cohen,1 Dih Yih Chen,1 Adam Idoine,1 Sudhir Manda,16 Jeff P. Sharman17 and Vanitha Ramakrishnan1
1BeiGene (Beijing) Co., Ltd., Beijing, China and BeiGene USA, Inc., San Mateo, CA, USA; 2Fred Hutchinson Cancer Research Center, University of Washington, Seattle, WA, USA; 3Sarah Cannon Research Institute/Tennessee Oncology, Nashville, TN, USA; 4Texas Oncology Baylor Charles A. Sammons Cancer Center, Dallas, TX, USA; 5SSM Health Dean Medical Group, Madison, WI, USA; 6Rocky Mountain Cancer Centers, Aurora, CO, USA; 7Florida Cancer Specialists & Research Institute, Fort Myers, FL, USA; 8Florida Cancer Specialists & Research Institute, Leesburg, FL, USA; 9Medical Oncology Hematology Consultants PA, Newark, DE, USA; 10Comprehensive Cancer Centers of Nevada, Las Vegas, NV, USA; 11Texas Oncology, Tyler, TX, USA; 12Summit Medical Group, Florham Park, NJ, USA; 13Summit Cancer Centers, Spokane, WA, USA; 14Texas Oncology, Amarillo, TX, USA; 15Virginia Cancer Specialists, Gainesville, VA, USA; 16Arizona Oncology/US Oncology Research, Tucson, AZ, USA and 17Willamette Valley Cancer Institute and Research Center, Eugene, OR, USA
Correspondence: L. XU - Linlin.Xu@beigene.com
https://doi.org/10.3324/haematol.2023.283861
Received: July 10, 2023.
Accepted: January 18, 2024. Early view: January 25, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
The study protocol was developed by BeiGene, Ltd. in collaboration with the study investigators. BeiGene was also involved in data collection, analysis, and interpretation of results. Statistical analyses were performed by statisticians at BeiGene. LX, RC, KB, AC, AI, and VR are employees of and hold stock/shares in BeiGene Co. MS is a consultant for AbbVie, Genentech, AstraZeneca, Sound Biologics, Pharmacyclics, BeiGene, Bristol Myers Squibb, Morphosys/Incyte, TG Therapeutics, Innate Pharma, Kite Pharma, Adaptive Biotechnologies, Epizyme, Eli Lilly, Adaptimmune, Mustang Bio, Regeneron, Merck, Fate Therapeutics, MEI Pharma, and Atara Biotherapeutics, and receives research funding from Mustang Bio, Celgene, Bristol Myers Squibb, Pharmacyclics, Gilead, Genentech, AbbVie, TG Therapeutics, BeiGene, AstraZeneca, Sunesis, Atara Biotherapeutics, Genmab, and Morphosys/Incyte. IF is a member of a board of directors or advisory committees at Vincerx. ML receives consulting fees from AbbVie, Amgen, AstraZeneca, BeiGene, Bristol Myers Squibb, Genmab, GSK, Incyte, Janssen, Karyopharm, Kite, Lilly, Sanofi, Seagen, and Takeda, payment or honoraria from AbbVie, Amgen, AstraZeneca, BeiGene, Bristol Myers Squibb, Genmab, GSK, Incyte, Janssen, Karyopharm, Kite, Lilly, Sanofi, Seagen, and Takeda, travel support from AbbVie, Amgen, AstraZeneca, BeiGene, Bristol Myers Squibb, Genmab, GSK, Incyte, Janssen, Karyopharm, Kite, Lilly, Sanofi, Seagen, and Takeda, and has a leadership or fiduciary role from Sellas. JMB receives consulting fees from AbbVie, Adaptive Biotechnologies, AstraZeneca, BeiGene, Bristol Myers Squibb, Constellation, Eli Lilly, Epizyme, Foresight, Genentech, Genmab, Kura, Kymera, Morphosys, Novartis, Nurix, TG Therapeutics, Verastem, and X4 Pharmaceuticals and receives payments/honoraria from Seagen and BeiGene. SFZ receives honorarium from Bristol Myers Squibb, Immunosome, and AbbVie. MDG receives honoraria from Karyopharm, TG Therapeutics, Janssen, and GSK. RC has equity with Pfizer and GSK and individual stocks in SAGA Diagnostics. D-YC has equity with BeiGene. JPS receives consulting fees from TG Therapeutics, Genentech, AbbVie, AstraZeneca, BeiGene, Bristol Myers Squibb, and Merck, and research funds from Genentech, Celgene, Gilead Sciences, TG Therapeutics, Merck, and Takeda.
Contributions
MS, IWF, MYL, RP, JMB, SFZ, JLC, JM, ECK, HAY, BF, AC, PKT, MDG, SM, and JPS enrolled patients and collected clinical data. LX, RC, KB, AC, D-YC, AI, and VR contributed to the intellectual content, conception, and design of the study. LX and KB processed data, analyzed statistics, generated figures, and wrote the manuscript. All authors contributed to data interpretation and revised the manuscript.
The authors wish to thank the study patients, their supporters, the investigators, and clinical research staff at the study centers. Assistance with medical writing and editorial support, under the direction of the authors, was provided by Lisa McGraw, PhD, and Elizabeth Hermans, PhD.
Funding
This study was sponsored by BeiGene, Ltd.
Data-sharing statement
BeiGene voluntarily shares anonymous data on completed studies responsibly and provides qualified scientific and medical researchers
1. Munir T, Brown JR, O’Brien S, et al. Final analysis from RESONATE: up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol. 2019;94(12):1353-1363.
2. Ghia P, Pluta A, Wach M, et al. Acalabrutinib versus investigator’s choice in relapsed/refractory chronic lymphocytic leukemia: final ASCEND trial results. Hemasphere. 2022;6(12):e801.
3. Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851-859.
4 Hillmen P, Eichhorst B, Brown JR, et al. Zanubrutinib versus ibrutinib in relapsed/refractory chronic lymphocytic leukemia and small lymphocytic lymphoma: interim analysis of a randomized phase III trial. J Clin Oncol. 2022;41(5):1035-1045.
5. Brown JR, Eichhorst B, Hillmen P, et al. Zanubrutinib or ibrutinib in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2023;388(4):319-332.
6. Shadman M, Flinn IW, Levy MY, et al. Zanubrutinib in patients with previously treated B-cell malignancies intolerant of previous Bruton tyrosine kinase inhibitors in the USA: a phase 2, open-label, single-arm study. Lancet Haematol. 2023;10(1):e35-e45.
access to anonymous data and supporting clinical trial documentation for clinical trials in dossiers for medicines and indications after submission and approval in the USA, China, and Europe. Clinical trials supporting subsequent local approvals, new indications, or combination products are eligible for sharing once corresponding regulatory approvals are achieved. BeiGene shares data only when permitted by applicable data privacy and security laws and regulations. In addition, data can only be shared when it is feasible to do so without compromising the privacy of study participants. Qualified researchers may submit data requests/research proposals for BeiGene review and consideration through BeiGene’s clinical trial webpage at https://www.beigene.com/our-science-and-medicines/ our-clinical-trials/.
7 Predicine. PredicineHEME CLIA Validated NGS Assay for Hematologic Malignancies. https://www.predicine.com. Accessed July 2023.
8. Gaidano G, Rossi D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematology Am Soc Hematol Educ Program. 2017;2017(1):329-337.
9 Nadeu F, Delgado J, Royo C, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127(17):2122-2130.
10 Bomben R, Rossi FM, Vit F, et al. Clinical impact of TP53 disruption in chronic lymphocytic leukemia patients treated with ibrutinib: a campus CLL study. Leukemia. 2023;37(4):914-918.
11. Del Poeta G, Biagi A, Laurenti L, et al. Impaired nodal shrinkage and apoptosis define the independent adverse outcome of NOTCH1 mutated patients under ibrutinib therapy in chronic lymphocytic leukaemia. Haematologica. 2021;106(9):2345-2353.
12. Nakhoda S, Vistarop A, Wang YL. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and nonHodgkin lymphoma. Br J Haematol. 2023;200(2):137-149.
13. Mohanty A, Sandoval N, Das M, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget. 2016;7(45):73558-73572.
Although therapy escalation has led to improved 5-year overall survival rates for patients with B-cell acute lymphoblastic leukemia (B-ALL), few effective treatment options are available for relapsed and treatment-resistant disease. This applies particularly to specific subtypes of B-ALL, such as patients harboring TCF3 (formerly E2A) fusions. TCF3, encoding members of the E protein (class I) family of helix-loop-helix transcription factors, is a master regulator of B-cell development and is involved in several chromosomal translocations associated with lymphoid malignancies, such as the translocation t(1;19)(q23;p13.3), resulting in the TCF3::PBX1 fusion (5% of pediatric B-ALL) or the translocation t(17;19)(q22;p13) generating the TCF3::HLF fusion (~0.5% of pediatric B-ALL).2 Omics research for the discovery of novel treatment strategies in hematological cancer is still based largely on transcriptomics, although it is increasingly recognized that this does not translate well into the expression of proteins, which are the main targets of drugs and functional entities of biological processes. In this study, we comprehensively analyzed the proteomic landscapes of TCF3::HLF+ (N=6) and TCF3::PBX1+ (N=5) B-ALL employing primary patient-derived xenografts (PDX), liquid chromatography tandem mass spectrometry and data-dependent acquisition. Approval for the study reported here was granted by the Ethics Committee of the Medical Faculty of the Christian-Albrechts-University, Kiel, Germany (vote D508/13). We detected 6,863 proteins (6,123 without ≥2 missing values; Online Supplementary Table S1), which allowed a clear distinction between TCF3::HLF+ and TCF3::PBX1+ leukemia by unsupervised hierarchical clustering and principal component analysis (Figure 1A, B). Proteomic profiling proved a useful tool for prioritizing drug targets, as only 8.45% of the significantly differentially expressed genes (N=119/1,409; P<0.05 and minimal log2 fold change of ±1) previously detected by RNA sequencing2 showed differential expression on protein level confirmed by our proteomic analysis (Online Supplemenary Figure S1A). In contrast, 34.8% (N=119/342) of differentially regulated proteins detected by proteomics were also differentially expressed on RNA level. As a proof-of-concept, we examined overlap of differentially expressed genes (cutoffs: P<0.05 and minimal log2 fold change of ±1) from RNA sequencing and proteomic analysis obtained from a previously published dataset of ETV6::RUNX1+ (N=9) and high hyperdiploid (N=18) primary ALL patient samples.3 While only 3.63% (N=82/2,262) of differentially expressed genes detected via RNA sequencing showed differential expression on protein
level, 92.13% (N=82/89) of differentially regulated proteins were also differentially expressed on RNA level (Online Supplementary Figure S1B).
In order to identify protein classes presenting specific therapeutic vulnerabilities, we performed gene set enrichment analysis (GSEA). We identified several gene sets enriched in either of the two subgroups (Figure 1C). RNA biology, mitochondrial translation and cellular respiration were the most prominent enriched gene sets for TCF3::HLF+ leukemia. In addition, strongly increased MYC expression and enrichment in MYC targets (Figure 1D, E) were detected, consistent with TCF3::HLF-driven activation of a MYC enhancer cluster previously shown using extensive functional genomics.4 For TCF3::PBX1+ leukemia, immune response/ cell cycle, actin cytoskeleton, cell morphogenesis and RTK signaling were among the most prominent enriched gene sets (Figure 1C). We validated therapeutic vulnerabilities indicated by GSEA using high-throughput drug screening. To this end, we tested the sensitivity of leukemic cell lines (TCF3::HLF+: HAL-01; TCF3::PBX1+: 697 and RCH-ACV) and mononuclear cells from peripheral blood of three healthy donors against a drug library of over 600 Food and Drug Administration-approved or clinical trial phase I-IV anti-cancer drugs. TCF3::HLF+ and TCF3::PBX1+ leukemic cells showed a differential response towards 109 drugs based on the area under the curve (AUC) as response parameter (Figure 2A; Online Supplementary Table S2; AUC<0.8 and >1.2 as a cutoff). Compared to our previous screening of bioactive compounds (N=98) employing the PDX samples,2 the cell lines showed similarly increased sensitivity towards compounds, such as BCL2 and mTOR inhibitors for TCF3::HLF+, and aurora kinase and polo-like kinase inhibitors for TCF3::PBX1+ B-ALL (Figure 2A). In addition, we identified novel potential drug targets. These included MDM2 and DNA/RNA synthesis for TCF3::HLF+ and microtubule/tubulin and cyclin-dependent kinases (CDK) for TCF3::PBX1+ leukemic cells (Figure 2A). In order to confirm these findings, we chose drugs from those groups, which did not affect normal peripheral blood cells (Figure 2B-E), and treated TCF3::PBX1+ (RCH-ACV) and TCF3::HLF+ (HAL-01) cells with half half-maximal inhibitory concentration (IC50), IC50 and double IC50 concentrations to investigate apoptosis induction. We demonstrated increased caspase 3/7 activity and apoptotic subG1 cells in TCF3::PBX1+ B-ALL in response to the microtubule/tubulin inhibitor ixabepilone and the CDK inhibitor SNS-032. For TCF3::HLF+ leukemic cells, we verified increased apoptotic cell death upon idasanutlin
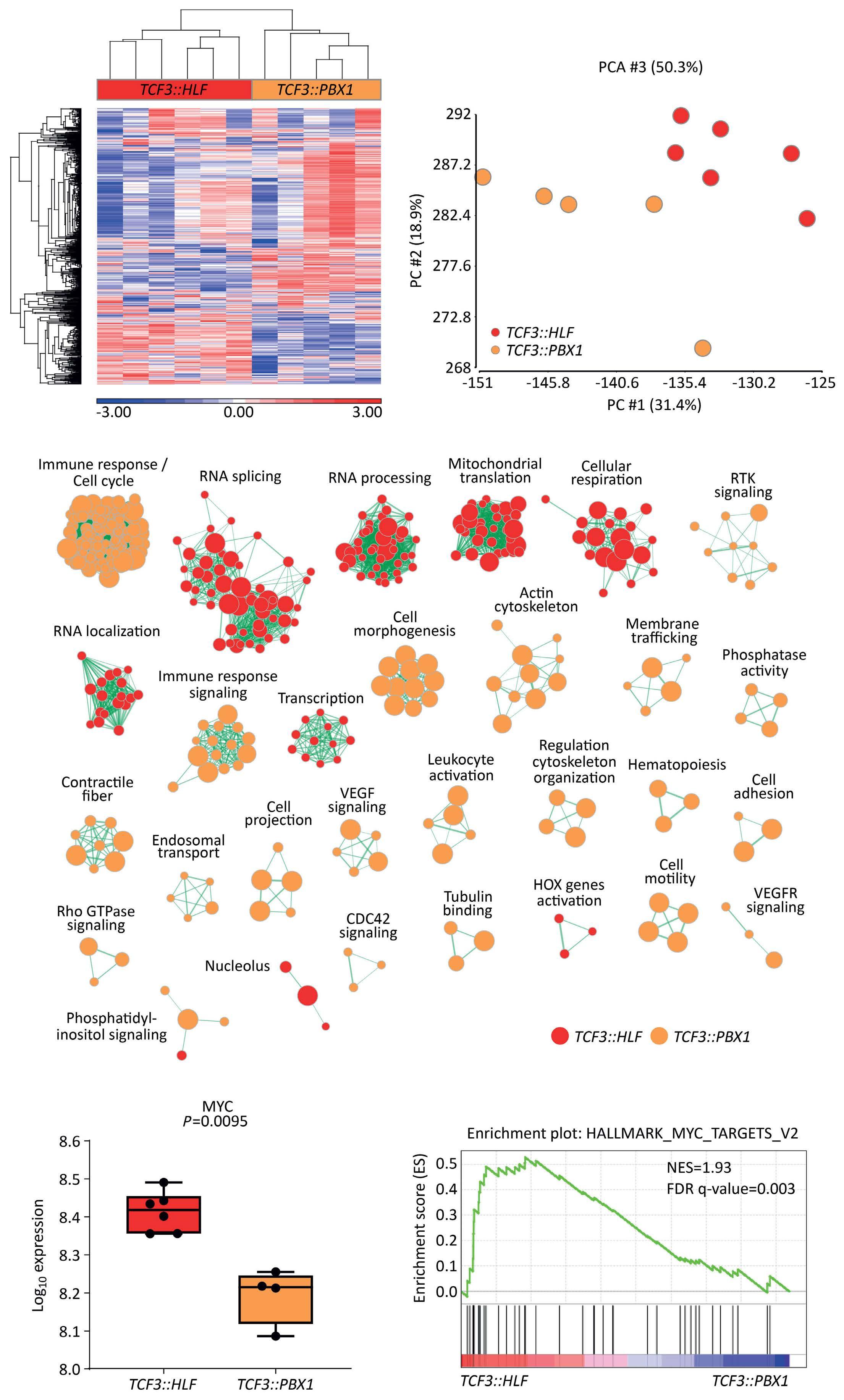
Figure 1. Proteomic profiling distinguishes TCF3::HLF + and TCF3::PBX1 + leukemia and uncovers therapeutic vulnerabilities for both subtypes. Unsupervised hierarchical clustering (A), principal component analysis (PCA) (B) and gene set enrichment analysis (GSEA) (C) was performed on the proteomic data of 6 TCF3::HLF+ (in red) and 5 TCF3::PBX1 + (in orange) B-cell acute lymphoblastic leukemia (B-ALL) patient-derived xenograft samples. For unsupervised hierarchical clustering, the 10% most variable proteins were used based on the standard deviation. PCA was performed on all proteins. Both subtypes clearly segregate into distinct clusters suggesting highly distinct proteomic landscapes. (C) GSEA is based on all proteins and identifies several gene sets enriched in either of the two subgroups. (D) Proteomic data shows enrichment of MYC protein expression in TCF3::HLF + versus TCF3::PBX1+ leukemic samples in this study. In order to determine differential expression, non-parametric Mann-Whitney t test (twotailed) was used. (E) Gene set enrichment plot of MYC targets showing a positive correlation with TCF3::HLF+ leukemia.
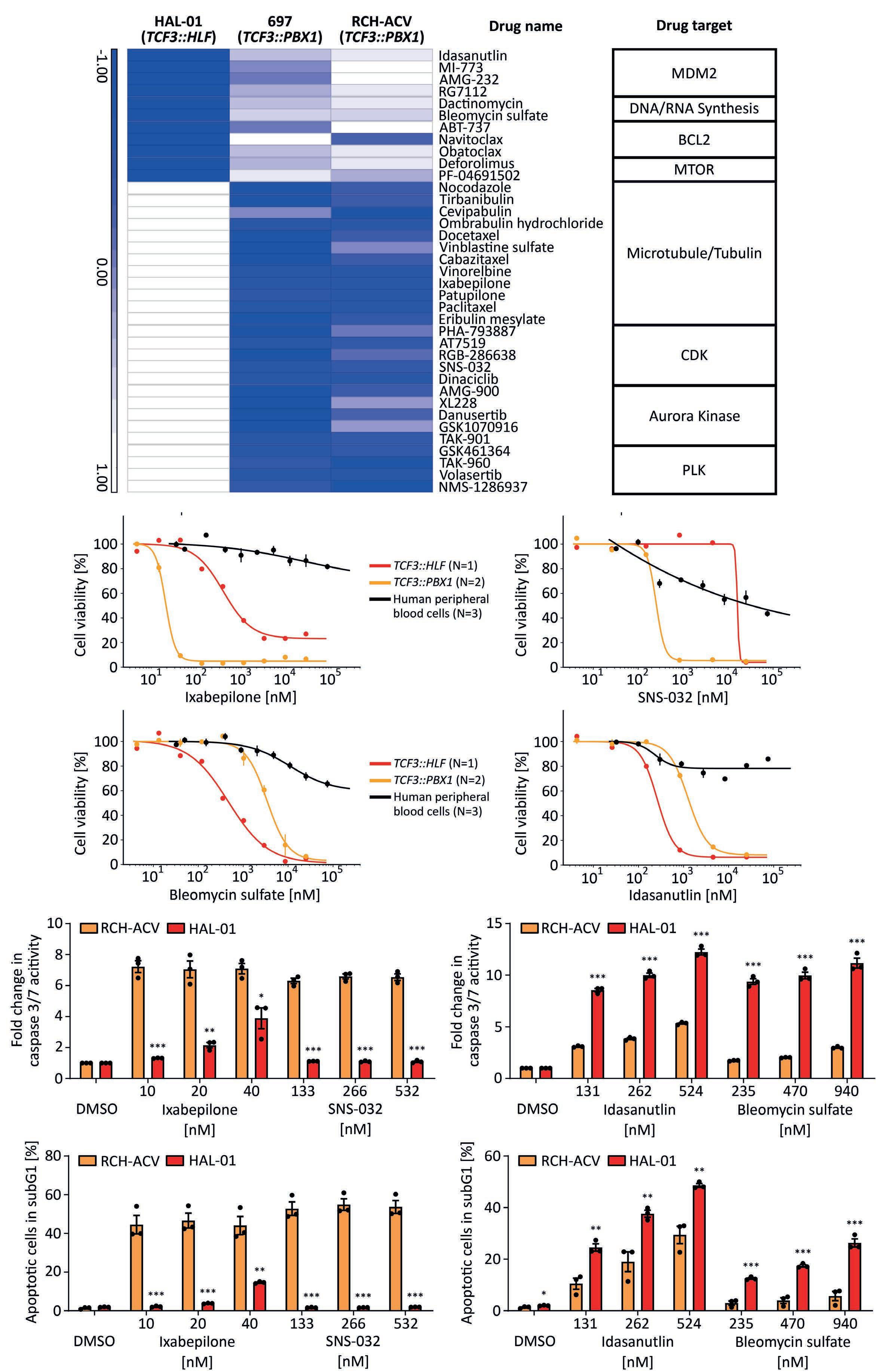
Continued on following page.
Figure 2. High-throughput drug screening and functional analysis confirm therapeutic vulnerabilities for TCF3::HLF+ and TCF3::PBX1+ leukemic cells. (A) HAL-01 (TCF3::HLF+), 697 and RCH-ACV (both TCF3::PBX1+) were exposed to a drug library, consisting of over 600 compounds. The supervised heatmap is based on the area under the curve (AUC, minimal fold change of <0.8 or >1.2 as a cutoff) as response parameter. Presented are groups of drugs with a differential response between TCF3::PBX1+ and TCF3::HLF+ leukemic cells. High sensitivity is shown in blue and low sensitivity is shown in white. (B-E) Presented are the dose-response curves for ixabepilone (B), SNS-032 (C), bleomycin sulfate (D) and idasanutlin (E), showing a differential response of TCF3::HLF+ versus TCF3::PBX1+ leukemic cells. Human peripheral blood cells from 3 healthy donors (in black) served as control cells (B-E). (F-I) RCHACV and HAL-01 were treated with ixabepilone (10 nM, 20 nM, 40 nM), SNS-032 (133 nM, 266 nM, 532 nM), idasanutlin (131 nM, 262 nM, 524 nM) or bleomycin sulfate (235 nM, 470 nM, 940 nM) dissolved in dimethyl sulfoxide (DMSO) or DMSO as negative control. (F, G) Bar graphs representing the fold change in caspase 3/7 activity 24 hours after treatment. Caspase 3/7 activity was determined using the Caspase-Glo 3/7 Assay (Promega). (H, I) Bar graphs represent the proportion of apoptotic cells (subG1) 48 hours after treatment. Cell cycle profiles were generated by flow cytometric measurement of propidium iodide intercalation into DNA after partial cell lysis in hypotonic buffer (0.1% sodium citrate, 0.1% Triton X-100, 0.5 mg/mL RNase A containing 40 µg/mL propidium iodide). Values shown in (F-I) represent mean ± standard error of the mean of 3 biologically independent replicates. NS: not significant; *P<0.05; **P<0.01; ***P<0.001 (t test).
(MDM2 inhibitor) and bleomycin sulfate (DNA/RNA synthesis inhibitor) treatment (Figure 2F-I).
Besides the detection of vulnerabilities to specific drug classes, we aimed to identify novel targets for drug development. In our proteomic analyses, the B-lymphoid tyrosine kinase (BLK) was the most significantly upregulated protein for the TCF3::PBX1+ subtype (Figure 3A; Online Supplementary Figure S1C; Online Supplementary Table S1; minimal log2 fold change of ±1 and significance level of P<0.05 as cutoffs). BLK encodes a non-receptor tyrosine kinase of the src family of proto-oncogenes that plays an important role in precursor (pre) B-cell receptor (BCR) signaling and early B-cell development.5 RNA-sequencing and expression microarray data by us and others supported this finding (Figure 3B-D). We examined human gene expression data derived from four independent data sets of >3,000 leukemia cases6-9 available at the R2: genomics analysis and visualization platform (http://r2.amc.nl). These data indicated a subpopulation of leukemia samples that highly co-expresses BLK and PBX1 (Figure 3B; Online Supplementary Figure S1D-F). In two of the data sets, information on chromosomal translocations was available. There, the BLK and PBX1 co-expressing subpopulation was specifically associated with the TCF3::PBX1 fusion (Figure 3B; Online Supplementary Figure S1D).8,9 In the Microarray Innovations in LEukemia (MILE) study8 all 36 cytogenetically identified TCF3::PBX1+ cases were BLKhigh expressing (N=1,897 other leukemia or myelodysplastic syndrome [MDS], N=71 normal controls). Similarly, in another study9 all six TCF3::PBX1+ cases and none of the other samples (N=185 other B-ALL, N=3 normal controls) were both PBX1 and BLK high expressing. RNA-sequencing data of our cohort showed high RNA expression of BLK in all TCF3::PBX1+ leukemia cases (N=5 at diagnosis, N=8 after transplantation into NSG mice)2 compared to TCF3::HLF+ cases (N=5 at diagnosis, N=22 after transplantation) (Figure 3C, D).
Thus, we hypothesized that interference with BLK signaling might present a potential treatment strategy for TCF3::PBX1 -rearranged B-ALL in particular. In order to test this, we treated TCF3::PBX1 + BLK high (RCH-ACV) and TCF3::HLF + BLK low (HAL-01) cells with a first selective ir-
reversible BLK inhibitor BLK-IN-2.10 BLKhigh cells responded in a dose-dependent manner starting at nanomolar concentrations (IC50=0.2169 μM), whereas BLKlow cells showed little or no response (≥167-fold less, IC50=36.20 μM) (Figure 3E; Online Supplementary Figure S1G-I). We further tested the impact of BLK-IN-2 on other genetic subtypes of B-ALL and noticed preferential sensitivity of only the TCF3::PBX1+ subtype to BLK-IN-2 (Online Supplementary Figure S1I). In order to test if BLK inhibition synergizes with the specific vulnerabilities identified in our proteomic screen, we performed combined treatment with ixabepilone (microtubule/tubulin inhibitor). Indeed, both drugs synergized in TCF3::PBX1+ cell lines, but not in the TCF3::HLF+ cell line HAL-01 (Figure 3F-H). We further tested, if interference with pre-BCR signaling including BTK inhibitors would have the same impact. To this end, we tested the response of TCF3::PBX1+ B-ALL cell lines to ibrutinib and other BTK-targeting drugs (acalabrutinib, spebrutinib, LFM-A13). The response, however, was low and did not differ from cells lacking pre-BCR expression.
As previously reported by Geng et al., BLK is a signature gene of adult TCF3::PBX1+ B-ALL.9 Combining chromatin immoprecipitation sequencing, DNA methylation and expression profiling, the authors identified hypomethylation and overexpression of BLK in adult TCF3::PBX1+ B-ALL. In this study, upregulated genes targeted by TCF3::PBX1 included pre-BCR components and pre-BCR downstream signaling molecules.11 Ligand-independent autonomous tonic preBCR activation via self-aggregation is a main mechanism for pre-BCR activation and leads to constitutive activation of BLK11 (indicated by phosphorylation of the activating tyrosine Y388) observed in several TCF3::PBX1+ cell lines and primary B-ALL.11 Pre-BCR function induces activation of the transcription factor BCL6, which further increases pre-BCR signaling in a self-enforcing positive feedback loop and directly activates BLK transcription. More than 90% of TCF3::PBX1+ leukemia cases are pre-BCR+ and critically rely on pre-BCR-dependent signaling for proliferation.5 Thus, targeting BLK to abrogate pre-BCR downstream signaling presents an attractive approach for therapeutic intervention in TCF3::PBX1+ B-ALL.
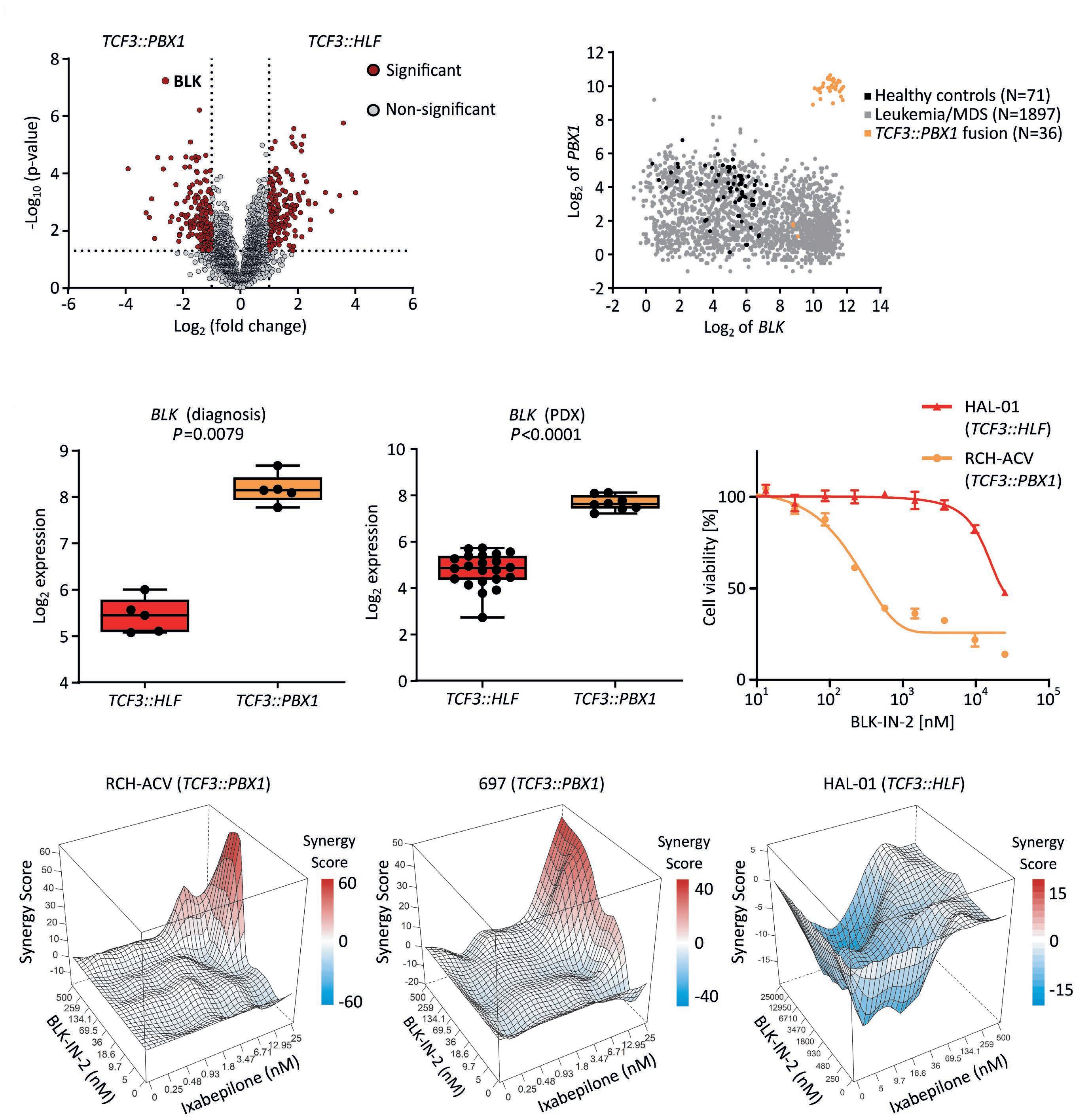
Figure 3. Proteogenomic profiling detects BLK as a marker of TCF3::PBX1+ B-cell acute lymphoblastic leukemia targetable by the selective BLK inhibitor BLK-IN-2. (A) The volcano plot presents significantly (red) or non-significantly (grey) dysregulated proteins in TCF3::PBX1+ (left) or TCF3::HLF+ (right) leukemias. BLK is the most significantly dysregulated protein for the TCF3::PBX1+ subtype (minimal log2 fold change of ±1 and significance level of P<0.05 as cutoffs). (B) Gene expression data of human patient samples is derived from data sets available at the R2: genomics analysis and visualization platform (http://r2.amc.nl). Dot plot presents PBX1 and BLK expression in healthy controls (black), TCF3::PBX1+ (orange) and other unstratified leukemic samples (grey). Data of the Mixed Leukemia - MILE - 2004 - MAS5.0 - u133p2 study8 is shown. Three further studies are presented in the Online Supplementary Figure S1D-F. (C,D) BLK RNA expression in TCF3::HLF+ (N=5) and TCF3::PBX1+ (N=5) B-ALL at the time of diagnosis (C) and after transplantation into NSG mice (TCF3::HLF+: N=22; TCF3::PBX1+: N=8). (D). Data is derived from our previous study.2 In order to determine differential expression, non-parametric Mann-Whitney t test (two-tailed) was used. (E) Dose-response curves for BLK-IN-2 show a differential response of TCF3::HLF+ (red) versus TCF3::PBX1+ (orange) leukemic cells. (F-H) Synergy drug screening of BLK-IN-2 and Ixabepilone reveals a strong synergistic effect in TCF3::PBX1+ cell lines, while no such effect is detected in the TCF3::HLF+ cell line HAL-01. Representative synergy plots of 3 independent experiments are shown. Drug concentration ranges were chosen according to the predetermined half-maximal inhibitory concentration (IC50) values of each cell line (RCH-ACV. and 697: 5-500 nM BLK-IN-2, 0.25-25 nM ixabepilone; HAL-01: 0.25-25 μM BLK-IN-2, 5-500 nM ixabepilone). Dimethyl sulfoxide (DMSO) was used as a negative control. ZIP synergy score analysis was conducted using the synergyfinder package version 3.0.14 with additional baseline correction.
Previously, interference with pre-BCR signaling has been suggested as a therapeutic option for TCF3-rearranged ALL12 and the inhibitors ibrutinib, dasatinib and idelalisib to be effective against TCF3::PBX1+ B-ALL.5,12,13 Inhibition of BTK, a downstream signaling kinase of the BCR, by ibrutinib is clinically beneficial in BCR+ B-cell malignancies such as non-Hodgkin lymphomas and multiple myeloma. However, in our analyses, the response of TCF3::PBX1+ B-ALL cell lines to BTK-targeting drugs was low and not corresponding to pre-BCR expression. This is in line with the observation that ibrutinib exerts a cytostatic rather than a cytotoxic effect on pre-BCR+ B-ALL cells5 and is further supported by the lack of in vivo effectivity against TCF3::PBX1+ primogafts.14 However, TCF3::PBX1+ PDX samples responded well to the tyrosine kinase inhibitor dasatinib.2 Still, high doses might be required for targets other than BCR::ABL1 and might be limited in the relapse setting due to toxicity.13
Taken together, proteomic-based profiling is a powerful tool to discover highly specific and sensitive cancer biomarkers and oncogenic pathway activation.15 Here, we uncovered proteomic alterations associated with TCF3::HLF+ or TCF3::PBX1+ B-ALL and revealed potential therapeutic options for these subtypes. These include previously known sensitivities for TCF3::HLF+ (e.g., BCL2 and mTOR) and TCF3::PBX1+ B-ALL (e.g., aurora kinase and polo-like kinase), as well as potential novel drug targets, such as MDM2 and DNA/RNA synthesis for TCF3::HLF+ or microtubule/tubulin and CDK for TCF3::PBX1+ leukemic cells. Our data suggest that TCF3::PBX1+ B-ALL might be sensitive to treatment with selective BLK inhibitors, especially in combination with microtubule/tubulin targeting drugs, such as ixabepilone. Due to high BLK expression in TCF3::PBX1+ B-ALL cells, such inhibitors could selectively eradicate leukemic cells at doses eliciting less side-effects on normal tissue. A limitation of our study is that this was not tested in mouse models. In future studies, it would be interesting to apply BLK inhibition to suitable mouse models of TCF3::PBX1+ leukemia and to test synergism with other drugs. Larger numbers of patient samples need to be tested to show the applicability for TCF3::PBX1+ leukemia.
Authors
Lena Blümel,1,2 Flavia Bernardi,3,4 Daniel Picard,1,2 Jacob Torrejon Diaz,3,4 Vera H. Jepsen,1,5 Rebecca Hasselmann,1,5 Julian SchlieheDiecks,1,5 Jasmin Bartl,1,2,6 Nan Qin,1,2 Beat Bornhauser,7 Sanil Bhatia,1,5 Blerim Marovka,7 Veronique Marsaud,3,4 Florent Dingli,8 Damarys Loew,8 Martin Stanulla,9# Jean-Pierre Bourqin,7# Arndt Borkhardt,1,5# Marc Remke,1,2# Olivier Ayrault3,4# and Ute Fischer1,5#
1Department of Pediatric Oncology, Hematology and Clinical Immunology, Heinrich Heine University Düsseldorf, Medical Faculty, and University Hospital Düsseldorf, Düsseldorf, Germany; 2Division of Pediatric Neuro-Oncogenomics, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), partner site Essen/
Düsseldorf, Düsseldorf, Germany; 3Institut Curie, CNRS UMR, INSERM, PSL Research University, Orsay, France; 4CNRS UMR 3347, INSERM U1021, Université Paris Sud, Université Paris-Saclay, Orsay, France; 5German Cancer Consortium (DKTK), partner site Essen/ Düsseldorf, Düsseldorf, Germany; 6Group for Interdisciplinary Neurobiology and Immunology - INI-research, Institute of Zoology, University of Hamburg, Hamburg, Germany; 7Department of Oncology and Children’s Research Center, University Children’s Hospital Zurich, Zurich, Switzerland; 8Institut Curie, PSL Research University, Centre de Recherche, CurieCoreTech Spectrométrie de Masse Protéomique, Paris, France and 9Department of Pediatric Hematology and Oncology, Hannover Medical School, Hannover, Germany
#MS, J-PB, AB, MR, OA and UF contributed equally as senior authors.
Correspondence:
U. FISCHER - ute.fischer@med.uni-duesseldorf.de
https://doi.org/10.3324/haematol.2023.283928
Received: July 17, 2023.
Accepted: February 15, 2024.
Early view: February 29, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
LB, JB, MS, J-PB, AB, MR, OA and UF planned and directed the study. Patient-derived xenograft models were provided by BB, BM and J-PB. FB, JTD, VM, FD, DL and OA conducted proteomic profiling and analyzed proteomic data. DP provided bioinformatic analyses. LB designed and performed the in vitro experiments, supported by VJ, JS-D and RH. NQ generated dose-response curves. SB provided intellectual contributions to the project and to the interpretation of the results. LB, VJ and UF wrote the manuscript. Figures were designed and drafted by LB, DP, NQ, VJ, JS-D and UF. All authors edited and contributed to the final manuscript.
Acknowledgments
Frauke-Dorothee Meyer, Sarah Göbbels and Bianca Killing are acknowledged for excellent technical assistance.
Funding
This work was supported by grants from the German José-Carreras Leukemia Foundation (DJCLS 07 R/2019 [to AB], DJCLS 18R/2021 [to UF], DJCLS 21R/2019 [to MR and UF]), the Elterninitiative Kinderkrebsklinik e.V. (to MR and JB), the Katharina-Hardt-Stiftung (to AB), the parents’ initiative Löwenstern e.V. (to AB and UF), the Gert-und-Susanna-Meyer Foundation (to MR), the Research Commission of the Medical Faculty of the Heinrich Heine University (to MR and NQ), the German Research Foundation (HE 8807/3-1 [to
UF], RE 2857/2-1 and KFO 337 [to MR]), the German Cancer Aid (Deutsche Krebshilfe, grant no. 111537 [to MR]) and the “Cancer Prevention - Graduate School” by German Cancer Aid, grant no. 70114736 and coordinated by the German Cancer Research Center (DKHZ), grant no. 70114766 (to UF), and the Saint Baldrick’s Robert J. Arceci Innovation Award (to OA).
Data-sharing statement
The data set generated and analyzed during the current study will be made available at PRIDE.1
1. Perez-Riverol Y, Bai J, Bandla C, et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50(D1):D543-D552.
2. Fischer U, Forster M, Rinaldi A, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet. 2015;47(9):1020-1029.
3. Yang M, Vesterlund M, Siavelis I, et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat Commun. 2019;10(1):1519.
4 Huang Y, Mouttet B, Warnatz HJ, et al. The Leukemogenic TCF3-HLF complex rewires enhancers driving cellular identity and self-renewal conferring EP300 vulnerability. Cancer Cell. 2019;36(6):630-644.
5. Kim E, Hurtz C, Koehrer S, et al. Ibrutinib inhibits pre-BCR(+) B-cell acute lymphoblastic leukemia progression by targeting BTK and BLK. Blood. 2017;129(9):1155-1165.
6. Polak R, Bierings MB, van der Leije CS, et al. Autophagy inhibition as a potential future targeted therapy for ETV6RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Haematologica. 2019;104(4):738-748.
7 Loh ML, Zhang J, Harvey RC, et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children’s Oncology Group TARGET Project. Blood. 2013;121(3):485-488.
8. Kohlmann A, Kipps TJ, Rassenti LZ, et al. An international
standardization programme towards the application of gene expression profiling in routine leukaemia diagnostics: the Microarray Innovations in LEukemia study prephase. Br J Haematol. 2008;142(5):802-807.
9 Geng H, Brennan S, Milne TA, et al. Integrative epigenomic analysis identifies biomarkers and therapeutic targets in adult B-acute lymphoblastic leukemia. Cancer Discov. 2012;2(11):1004-1023.
10 Fu T, Zuo Y, Zhong Z, Chen X, Pan Z. Discovery of selective irreversible inhibitors of B-Lymphoid tyrosine kinase (BLK). Eur J Med Chem. 2022;229:114051.
11. Geng H, Hurtz C, Lenz KB, et al. Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell. 2015;27(3):409-425.
12. van der Veer A, van der Velden VH, Willemse ME, et al. Interference with pre-B-cell receptor signaling offers a therapeutic option for TCF3-rearranged childhood acute lymphoblastic leukemia. Blood Cancer J. 2014;4(2):e181.
13. Eldfors S, Kuusanmaki H, Kontro M, et al. Idelalisib sensitivity and mechanisms of disease progression in relapsed TCF3-PBX1 acute lymphoblastic leukemia. Leukemia. 2017;31(1):51-57.
14. van de Ven C, Boeree A, Stalpers F, Zwaan CM, Den Boer ML. Ibrutinib is not an effective drug in primografts of TCF3-PBX1. Transl Oncol. 2020;13(10):100817.
15. Meissner F, Geddes-McAlister J, Mann M, Bantscheff M. The emerging role of mass spectrometry-based proteomics in drug discovery. Nat Rev Drug Discov. 2022;21(9):637-654.
Splenic marginal zone lymphoma (SMZL) is a rare histotype of non-Hodgkin lymphoma, accounting for only 2% of all cases.1 At diagnosis, up to 30% of patients may be asymptomatic, while others may present with cytopenia(s), abdominal lymph node swelling, splenomegaly, B-symptoms, and secondary autoimmune diseases.2 SMZL follows an indolent course that, similarly to other indolent lymphomas, can be complicated by the histological evolution into diffuse large B-cell lymphoma (DLBCL).3 After the diagnosis of SMZL, the risk of lymphoma-related death increases significantly, especially in patients who experience progression within the first 24 months.4 However, the 5-year conditional survival of the remaining patients is comparable to that of the general population,3 and the overall survival (OS) exceeds 10 years. Rituximab immunotherapy is a commonly used first-line treatment due to its effectiveness and mild toxicity.5-6 Although splenectomy is an effective procedure, it can lead to severe and potentially fatal acute and late complications.5 Additionally, the role of bendamustine-rituximab chemoimmunotherapy has yet to be defined, and the preferred first-line treatment is undetermined due to the lack of randomized studies.
It is unclear if the quality of clinical response, complete versus partial (CR vs. PR), correlates with the time-related outcomes. Additionally, insufficient evidence prevents evaluating the prognostic role of acquiring an undetectable minimal residual disease (MRD-) status.7-8 Given this context, the IELSG conducted the BRISMA-IELSG36 phase II study (EudraCT number: 2011-000880-28; clinicaltrails gov. Identifier: NCT02853370). Approval by local ethics committees and written informed consent by all participants before study entrance was required. Through a series of 56 SMZL patients, the primary study objective aimed to assess the efficacy and toxicity of combining bendamustine with rituximab (BR) as a first-line therapy.9 Herein, we present the updated results integrated with MRD data (median follow-up [FU]: 69 months; 95% confidence interval [CI]: 67-72).
Eligible patients needed to exhibit active/symptomatic disease, and the clinical responses scored according to the criteria described below, proposed by the Splenic Lymphoma Study Group (SLSG)10 for non-splenectomized patients: partial response (PR): ≥50% improvement in the disease manifestations including resolution or decrease in spleen
size, improvement on cytopenias and resolution or decrease in lymphadenopathy. Bone marrow (BM) should show a reduction in lymphoid infiltration; complete response (CR): resolution of organomegaly, normalization of the blood counts (Hb>12 g/dL, platelets >100x109/L; neutrophils >1.5x109/L and no evidence of circulating clonal B cells); no evidence of lymphoma in BM through immunohistochemistry. Online
Supplementary Table S1 compares the SLSG and Lugano response criteria for non-PET-avid NHL.
In addition, we adopted the term “unconfirmed complete response” (CRu).6;9 CRu describes patients who still exhibit some degree of cytopenia and splenomegaly at the end of treatment (EOT) but who subsequently meet the criteria for CR at the first FU visit.9 The MRD analysis was performed centrally in a EuroMRD standardized laboratory (https:// euromrd.org/) by droplet digital polymerase chain reaction (ddPCR) with ASO primers targeting IGH rearrangements in the BM and peripheral blood (PB) samples, expressed as copies out 250 ng of genomic DNA as previously described.11 The MRD assay was considered reliable when reaching a sensitivity of at least 1x10-4, calculated considering the tumor infiltration at baseline. Accordingly, in each ddPCR experiment, the “10-1” (as control) and the “10-4” dilution points were included.
Patients with a molecular marker underwent MRD assessment after three cycles (early restaging [ER]) at EOT, and yearly at each annual FU assessment.
Clinical information has been updated for all living patients except for two. Presenting features are shown in Online Supplementary Table S2. The median age was 66 years, and 41 patients (73%) were older than 60 years. Fifty-one patients (91%) achieved a major response (34 CR; 7 CRu; 10 PR). All the CRu patients and one in stable disease (SD) at EOT improved slowly and steadily, meeting the criteria for CR without additional treatment. Progression-free survival (PFS) events included three progressions documented in patients scored in SD at EOT, six relapses, and one death (due to sepsis at 1,6 months). According to the intention-to-treat analysis, the 5-year PFS was 83% (95% CI: 71-91), and the OS was 93% (95% CI: 82-97) (Figure 1A, B). These results are nearly identical to those achieved with six weekly infusions of rituximab followed by 1-2 years of maintenance therapy.6 None of the ten patients in PR progressed during
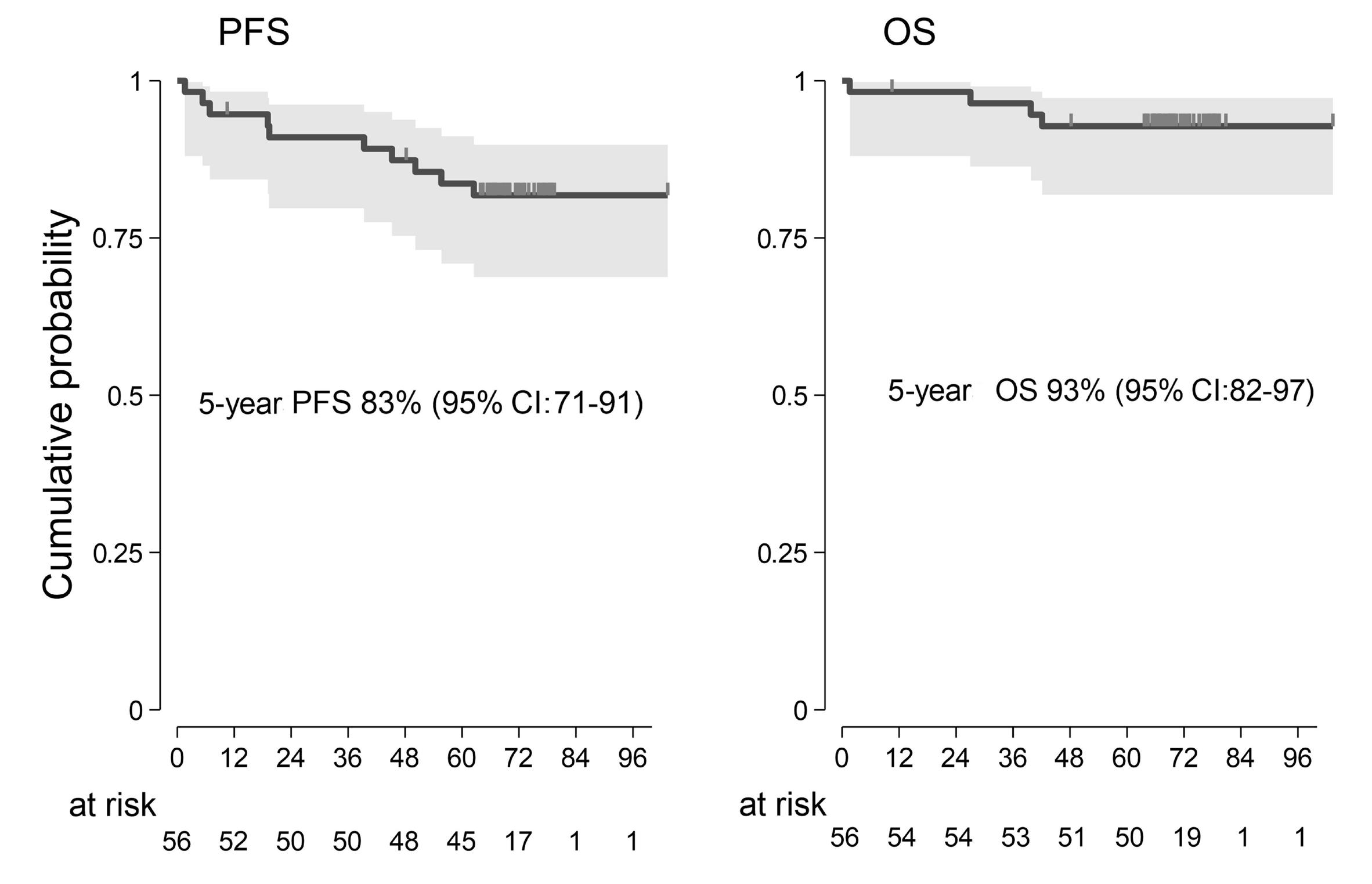
Figure 1. Progression-free survival and overall survival at 5 years with a median observation time of 69 months. (A) Progression-free survival (PFS). (B) Overall survival (OS). CI: confidence interval.
the median observation time of 69 months (range 64-103).
At baseline, 53 of 56 patients assessable for MRD had at least one FU sample and were screened for a monoclonal IGH rearrangement. A reliable MRD molecular marker was found in 42 of 53 (79%) patients, consistent with the literature.8 The ASO assay achieved adequate sensitivity for MRD analysis (at least 10-4) in 37 of 53 (70%) cases. Among these 37 cases, 14 were identified in both BM and PB samples, 18 in PB only, and five in BM only (Figure 2B).
MRD data were available in five of ten PR patients; at EOT, four were MRD-, and one was MRD+. Four of six CR patients who relapsed had MRD EOT assessment: three were MRD+ and one MRD-. Figure 2A shows the clinical response, MRD status at different time points, and outcome for each of the 37 evaluable patients. The baseline MRD burden was not associated with presenting features, PFS, and OS (P>0.5).
Four patients died: one due to sepsis and three due to lymphoma at 1.6, 27, 40 and 42 months, respectively. Two cases of G>2 infections occurred during treatment,9 and three cases (2 acute bronchitis, 1 lung tuberculosis) occurred during the follow-up. Two hundred and eighty-four cycles were administered during the study, with 85% given at full dose. Bendamustine dose was reduced in four patients in cycles 4-6. Relative dose intensity was 0.99 for bendamustine and 0.98 for rituximab.
Seven patients developed a second primary cancer (SPC); the SPC includes one follicular thyroid cancer, one DLBCL, one renal-cell carcinoma, one basal cell carcinoma, one
5q- myelodysplastic syndrome, one malignant peripheral nerve sheath tumor, and one prostate adenocarcinoma. The 5-year cumulative incidence rate of SPC was 11%. BR treatment resulted in high rates of undetectable MRD: 40% at the ER (17/37), 54% (14/26) at the EOT, and 65% (20/31) after 1 year (Figure 2B). Interestingly, a significant reduction in MRD had already been attained at ER assessment (median 4.7x10-5 in BM and below the quantitative level in PB, respectively). The additional BR courses (1/3 CR patients and 3/9 PR patients) did not contribute to a significant further reduction in residual MRD levels (median 1.4x10-5 in BM and BQL in PB, respectively) (Online Supplementary Figure S1A, B). MRD+ predicted a statistically significant inferior PFS at each evaluated timepoint (ER, EOT, and FU1) (Figure 3A-C). Notably, acquiring early MRD- status in non-invasive peripheral blood samples was associated with a significant improvement in PFS (MRD- vs. MRD+ 100% vs. 68%; 95% CI: 35-86; P=0.006) (Figure 3D). Our analysis of MRD assessment aligns with published data for rituximab-chemotherapy-treated SMZL patients.7-8 Cervetti et al. generated MRD data assessing by real-time quantitative PCR (RQ-PCR) IGH rearrangements in a retrospective series of 50 SMZL (EOT MRD- 48%; PFS: 100% vs. 73%; P=0.023).8 Lyu et al. investigated MRD status by employing multi-color flow cytometry (MFC) in a prospective series of 71 patients (EOT MRD- 77%; PFS: 74,8±6,5% vs. 31,4±12,6%; P<0.001).7 In our study, the EOT MRD status in a landmark analysis was significantly associated with
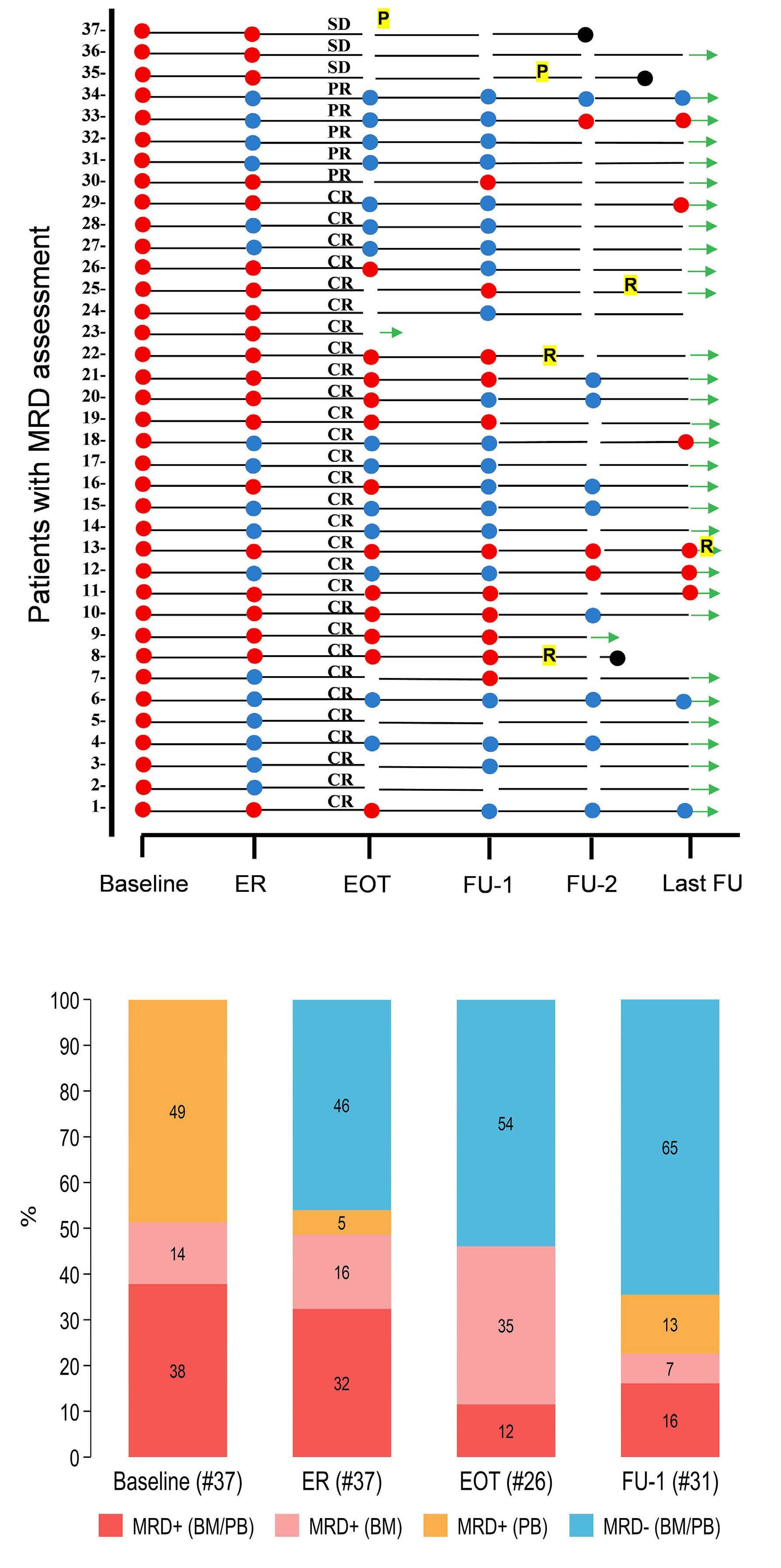
Figure 2. Clinical response and undetectable minimal residual disease rate at different time points. (A) Individual minimal residual disease (MRD) status, clinical response, and outcome at different times. (B) Undetectable MRD rate at different time points. ER: early restaging at 4 months; EOT: end of treatment at 6 months; FU-1: 1-year follow-up; FU2: 2-year follow-up; Last-FU: last available control. Red dot: MRD+ (at least 1 MRD-positive sample, either bone marrow [BM] or peripheral blood [PB]); blue spot: MRD- (unmeasurable MRD); black dot: dead; CR: complete remission; PR: partial remission; SD: stable disease; R: relapse; P: progression; green arrow: alive at last FU.
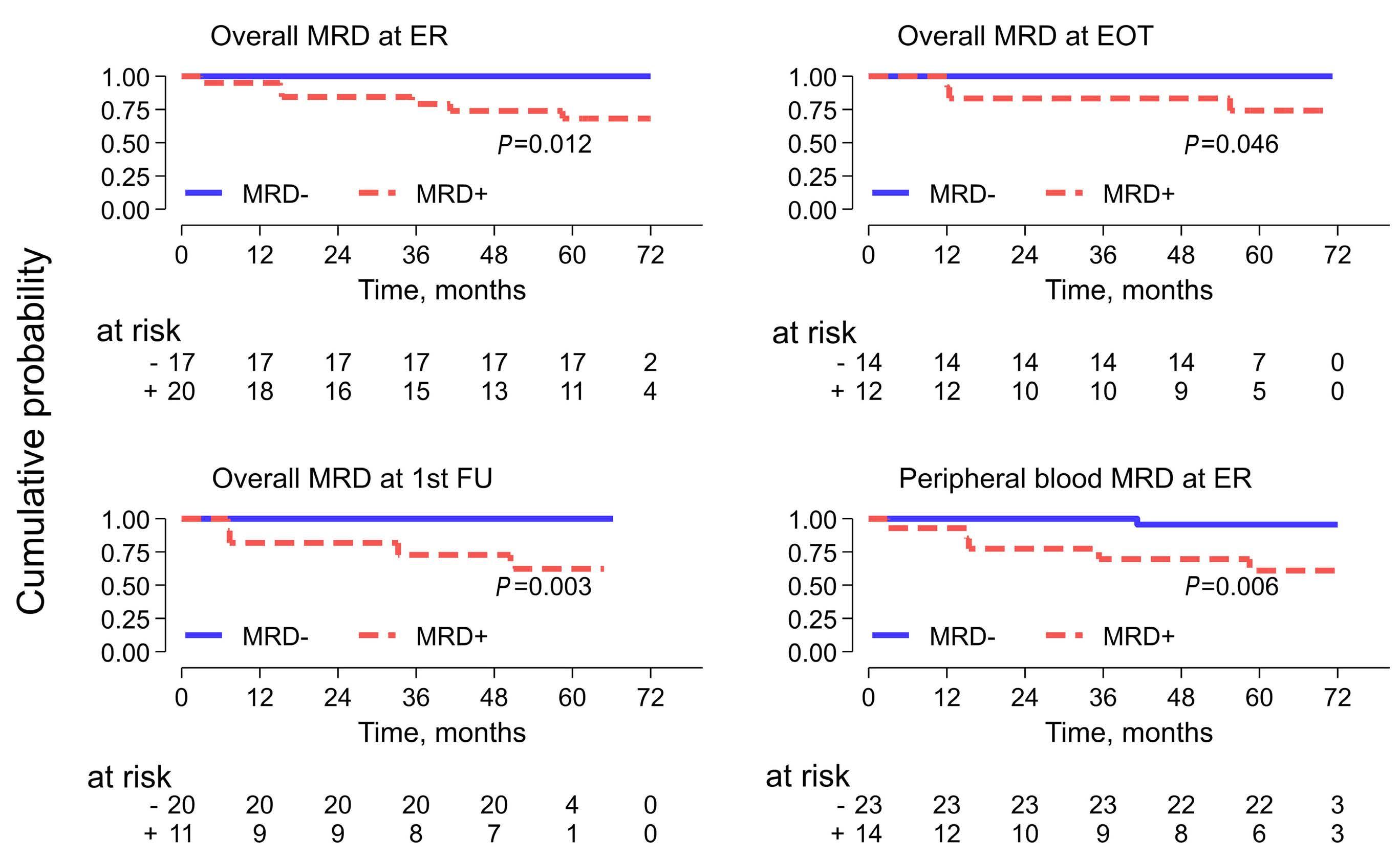
Figure 3. Landmark progression-free survival analysis stratified by minimal residual disease evaluated at different time points. (A) Overall minimal residual disease (MRD) at early restaging after 3 bendamustine rituximab courses (ER). (B) Overall MRD at end of treatment (EOT). (C) Overall MRD at 1-year follow-up (FU-1). (D) Peripheral blood MRD at ER. PFS: progression-free survival; overall MRD: at least 1 MRD-positive sample (either bone marrow or peripheral blood); MRD-: unmeasurable MRD.
PFS (Figure 3B), while no difference in PFS was observed between CR and PR groups (P=0.438; data not shown). Three MRD+ patients slowly converted to unmeasurable MRD- status, and seven CRu and one SD patient eventually met the criteria for CR at FU1 12 months after completion of treatment. None of the patients received off-protocol treatments or maintenance therapy. A speculative explanation for the spontaneous conversion to MRD- might be the gradual elimination of minimal subsets of residual neoplastic cells by the restored activity of the immune system after EOT. Accordingly, the MRD levels of these three patients were below 1x10-4 (namely 2, 2, and 3 copies/50,000 cells). In conclusion, the BRISMA study substantiates that BR chemoimmunotherapy accomplishes a swift and significant reduction in tumor bulk for patients with symptomatic SMZL, thereby facilitating notably high 5-year PFS and OS rates. Other retrospective studies have reported comparable PFS and OS rates with less toxic treatments.6;13 However, this phase II study with extended FU provides compelling evidence for considering bendamustine-rituximab as an effective treatment for symptomatic SMZL patients.
Three other informative results come from the MRD analysis: i) our findings support the assertion that attaining MRD- after just three courses of treatment is a promising indicator
of favorable long-term outcomes, even when monitored in PB.7-8,12 Though data are not yet mature enough to use MRD status in clinical decisions, it could help stratify patients in future trials. Furthermore, ii) integrating MRD results with response criteria is worth exploring due to the imaging limitations in detecting the disease in the spleen.14 Finally, iii) the incidence of additional cancers in this series aligns well with the cumulative incidence rate documented in existing literature.15 Consequently, we recommend conducting an SPC assessment before initiating a BR treatment and throughout the FU period, particularly in elderly patients.
Emilio Iannitto,1,2 Simone Ferrero,3 Côme Bommier,4,5 Daniela Drandi,3 Martina Ferrante,3 Krimo Bouabdallah,6 Sylvain Carras,7 Guido Gini,8 Vincent Camus,9 Salvatrice Mancuso,10 Luigi Marcheselli,11 Angela Ferrari,12 Michele Merli,13 Benoit Tessoulin,14 Caterina Stelitano,15 Kheira Beldjord,4 Giovanni Roti,16 Fabrice Jardin,8 Barbara Castagnari,17 Francesca Palombi,18 Lucile Baseggio,19 Alexandra Traverse-Glehen,20 Claudio Tripodo,2 Anna Marina Liberati,21 Margherita Parolini,22 Sara Usai,23 Caterina Patti,24 Massimo Federico,25 Maurizio Musso,1 Marco Ladetto,26 Emanuele Zucca27 and Catherine Thieblemont4,5
1La Maddalena Department of Oncology, Oncohematology and Bone Marrow Transplantation Unit, Palemo, Italy; 2Tumor Immunology Unit, Department of Health Sciences, University of Palermo School of Medicine, Istituto di Patologia Generale, Palermo, Italy; 3Division of Hematology, Department of Molecular Biotechnologies and Health Sciences, University of Torino, Torino, Italy; 4Universitè Paris Citè, Paris, France; 5AP-HP, Hôpital Saint-Louis, Hemato-oncologie, Paris, France; 6CHU de Bordeaux - Hôpital Haut Lèvêque and Centre François Magendie, Service d’Hématologie Clinique et Thérapie Cellulaire, Pessac, France; 7Universitè Grenoble Alpes, Institute for Advanced Biosciences, INSERM U1209, CNRS UMR 5309, University Hospital, Molecular Biology and Hematology Departments, Grenoble, France; 8Ematologia AOU delle Marche, Ancona, Italy; 9Department of Hematology, Centre Henri Becquerel, Rouen, France; 10Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialties (PROMISE), Hematology Unit, University of Palermo, Palermo, Italy; 11Fondazione Italiana Linfomi ETS, Modena, Italy; 12Azienda Unità Sanitaria Locale-IRCCS Arcispedale Santa Maria Nuova, Reggio Emilia, Italy; 13Hematology, Ospedale di Circolo e Fondazione Macchi - ASST Sette Laghi, Varese, Italy; 14Nantes University Hospital and CRC2 NA, Nantes, France; 15UO Ematologia AO Bianchi-Melacrino-Morelli, Reggio Calabria, Italy; 16Dipartimento di Medicina e Chirurgia, Università di Parma, AOU di Parma, Parma, Italy; 17Department of Hematology, Ravenna Hospital, Ravenna, Italy; 18Hematology Unit, IRCCS Regina Elena National Cancer Institute, Roma, Italy; 19Laboratoire d’Hèmatologie Biologique, Groupement Hospitalier Lyon-Sud/Hospices Civils de Lyon, Lyon, France; 20Service d’Anatomie Pathologique, Groupement Hospitalier LyonSud/Hospices Civils de Lyon, Lyon, France; 21Oncohematology Unit, University of Perugia, Azienda Ospedaliera S.Maria, Terni, Italy; 22Ematologia Istituto Romagnolo Per Lo studio dei Tumori Dino Amadori, Meldola, Italy; 23Ospedale Oncologico A. Businco, Cagliari, Italy; 24U.O.C. di Oncoematologia Ospedali Riuniti Villa SofiaCervello, Palermo, Italy; 25CHIMOMO Department, University of Modena and Reggio Emilia, Modena, Italy; 26University of Eastern Piedmont Amedeo Avogadro, S.S. Antonio and Biagio and Cesaro Arrigo Hospital, Alessandria, Italy and 27IOSI, Oncology Institute of Southern Switzerland and Institute of Oncology Research (IOR), International Extranodal Lymphoma Study (IELSG), Bellinzona, Switzerland
Correspondence: E. IANNITTO - emilio.iannitto@gmail.com
https://doi.org/10.3324/haematol.2023.284109
Received: November 2, 2023. Accepted: March 7, 2024. Early view: March 14, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
SF reports research funding from Janssen, Morphosys, Gilead and Beigene; consultancy for EusaPharma, Janssen, Sandoz and Abbvie; advisory board at EusaPharma, Janssen, Clinigen, Incyte and Italfarmaco; speakers honoraria from Janssen, EusaPharma, Servier and Gentili. ML reports institutional research funding from Roche, Celgene, Abvie, Millenium, Janssen, Sanofi, Verastem, Novartis, Morphosis, GSK, Oncopeptides, Karipharm, Onconova, Archigen, Beigene and BMS; honoraria for lectures, presentations, bureaus, travel accommodation from IQVIA, Incite, Celgene, Abbvie, Janssen and BMS. VC dislcoses honoraria from Kite, a Gilead Company, Bristol Myers Squibb, Novartis, Incyte, Kyowa Kirin, Abbvie, Ideogen and Takeda; travel fees from Pfizer, Kite, Gilead, Bristol Myers Squibb and Novartis; research funding paid to institution from Astra Zeneca, Bristol Myers Squibb, Novartis and Ideogen. BC discloses honoraria from Kite, Gilead, Bristol Myers Squibb, Novartis, Incyte, Kyowa Kirin, Abbvie, Ideogen and Takeda; travel fees from Pfizer, Kite, a Gilead Company, Bristol Myers Squibb and Novartis; research funding paid to institution from Astra Zeneca, Bristol Myers Squibb, Novartis and Ideogen. EZ received research support to institution from AstraZeneca, Beigene, Celgene/BMS, Incyte, Janssen, and Roche; discloses honoraria for medical educational events from Abbvie (advisory boards) from BeiGene, BMS, Curis, Eli/Lilly, Incyte, Ipsen, Janssen, Merck, Miltenyi Biomedicine and Roche; travel grants from BeiGene, Janssen, Gilead, and Roche. ML dislcoses consultancy, participation in advisory boards, an invitation to scientific meetings, institutional research support, and contracts with AbbVie, Acerta, Amgen, ADC Therapeutics, BeiGene, Celgene/BMS, Eusapharma, GSKI, Gentili, Gilead/Kite, Lilly, Novartis, Incyte J&J, Jazz, Regeneron, Roche and Sandoz. CT reports advisory/consultancy roles at and travel accommodations/expenses from Roche, Incyte, Novartis, Kite/Gilead, Amgen, Takeda and BMS.
EI developed the study concept and design, analyzed and interpreted the data, and wrote the manuscript. CT, EZ and MF developed the study concept and design and interpreted the data. SF performed MRD investigations data analysis and wrote the manuscript. DD and MF performed MRD investigations and contributed to data analysis. LB, ATG, and CLTR contributed to the study design and supervised the diagnostic process. LM performed statistical analysis. All authors provided materials or patients, collected, assembled, analyzed and interpreted the data, gave the final approval of the manuscript and are accountable for all aspects of the work.
This research received an unrestricted contribution from Mundipharma.
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
1. Piris MA, Isaacson PG, Swerdlow SH, et al Splenic marginal zone lymphoma. In: Swerdlow SH, Campo E, Harris NL, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th ed. Lyon: IARC Press; 2017. p. 223-225.
2. Arcaini L, Rossi D, Paulli M. Splenic marginal zone lymphoma: from genetics to management. Blood. 2016;127(17):2072-2081.
3. Kalashnikov I, Tanskanen T, Viisanen L, et al. Transformation and survival in marginal zone lymphoma: a Finnish nationwide population-based study. Blood Cancer J. 2023:13(62):1-8.
4 Luminari S, Merli M, Rattotti S, et al. Brief report early progression as a predictor of survival in marginal zone lymphomas: an analysis from the FIL-NF10 Study. Blood. 2019;134(10):798-801.
5. Zucca E, Arcaini L, Buske C, et al. Marginal zone lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(1):17-29.
6. Kalpadakis C, Pangalis GA, Sachanas S, et al. Rituximab monotherapy in splenic marginal zone lymphoma: prolonged responses and potential benefit from maintenance. Blood. 2018;132(6):666-670.
7 Lyu R, Wang T, Wang Y, et al. Undetectable minimal residual disease is an independent prognostic factor in splenic marginal zone lymphoma. Br J Haematol. 2021;194(5):862-869.
8. Cervetti G, Galimberti S, Pelosini M, Ghio F, Cecconi N, Petrini M. Significant efficacy of 2-chlorodeoxyadenosine and rituximab
in the treatment of splenic marginal zone lymphoma (SMZL): extended follow-up. Ann Oncol. 2013;24(9):2434-2438.
9 Iannitto E, Bellei M, Amorim S, et al. Efficacy of bendamustine and rituximab in splenic marginal zone lymphoma: results from the phase II BRISMA/IELSG36 study. Br J Haematol. 2018;183(5):755-765.
10 Matutes E, Oscier D, Montalban C, et al. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia. 2008;22(3):487-495.
11. Drandi D, Alcantara M, Benmaad I, et al. Droplet digital PCR quantification of mantle cell lymphoma follow-up samples from four prospective trials of the European MCL Network. Hemasphere. 2020;4(2):e347.
12. Bommier C, Lambert J, Nowakowski G, Zucca E, Thieblemont C. What prognostic markers should be evaluated in marginal zone lymphoma? A survey among leading international experts. Hemasphere. 2022;6(2):e680.
13. Else M,Marín-Niebla A, de la Cruz F, et al. Rituximab, when used alone or in combination, is superior to other treatment modalities in splenic marginal zone lymphoma. Br J Haematol. 2012;159(3):322-328.
14 Albano D, Giubbini R, Bertagna F. 18F-FDG PET/CT in splenic marginal zone lymphoma. Abdom Radiol. 2018;43(10):2721-2727.
15. Iannitto E, Minardi V, Callea V, et al. Assessment of the frequency of additional cancers in patients with splenic marginal zone lymphoma. Eur J Haematol. 2006;76(2):134-140.
Cytomegalovirus (CMV) reactivation remains a leading clinical concern, contributing to substantial morbidity and mortality following allogeneic hematopoietic cell transplantation (HCT), in both adult and pediatric highrisk CMV-seropositive recipients.1,2 Pre-emptive antiviral therapy (PET) greatly reduces the risk of CMV disease and mortality post-transplant, but it is associated with significant economic burden, toxicity and rates of failure.3,4 In the case or pediatric patients, many PET antivirals have intolerable toxicity or have been insufficiently evaluated.5,6 US Food and Drug Administration approved letermovir prophylaxis up day 200 post-transplant7 is effective with minimal myelosuppression, and it is tolerated in pediatric patients.8 Currently its use remains off-label in these patients, pending outcomes of an ongoing phase II trial ( clinicaltrials gov. Identifier: NCT03940586 ). The main drawback of letermovir is the observed increase in clinically significant CMV infection and disease requiring antivirals, when prophylaxis is discontinued.7,9 The decreased CMV reactivation and the consequent decreased CMV antigen exposure during letermovir prophylaxis may delay CMV-specific immune reconstitution leading to breakthrough CMV viremia requiring antivirals. Notably, polyfunctional T-cell responses specific for CMV pp65 and IE-1 proteins, which are involved in controlling CMV viremia are significantly decreased in HCT patients receiving letermovir prophylaxis, compared with HCT recipients receiving PET.10 Additionally, there is no difference in mortality after 1 year between letermovir and PET-treated T-cell replete HCT recipients.9 Infusion of CMV-specific T cells can promote restoration of durable, functional antiviral immunity. Despite the success of this strategy, ready availability of adoptive T-cell therapy remains difficult to achieve.11 Furthermore, in December 2023 phase III trials of posoleucel,12 an off-the-shelf, multi-virus specific T-cell therapy, including CMV were halted for futility. Consequently, immunotherapeutic strategies to suppress both early and late clinical reactivation, through enhancing and sustaining protective immune reconstitution against CMV remain an unmet need in the letermovir era. Triplex is a modified vaccinia Ankara (MVA) vectored vaccine which expresses three immunodominant CMV proteins; pp65, IE1-exon4, and IE2-exon5. It has been designed to
accelerate reconstitution of protective T-cell CMV-specific immunity. Triplex demonstrated tolerability and immunogenicity in healthy adults, autologous and allogeneic HCT recipients, in whom it reduced CMV viremia.13 Based on Triplex favorable tolerability, immunogenicity and efficacy outcomes we investigated whether vaccinating HCT pediatric recipients, with Triplex safely promoted and expanded CMV-specific T cells protecting from clinical viremia requiring PET in the presence of letermovir.
We designed a non-randomized, open label, phase Ib pilot trial to evaluate safety, optimal dose, and immunogenicity of Triplex vaccine in CMV-seropositive pediatric allogeneic HCT recipients. The study was conducted in accordance with the Helsinki Declaration at City of Hope National Medical Center (COH), was approved by the local Institutional Review Board, and registered as clinicaltrials gov. Identifier: NCT 03354728
Fifteen pediatric (age 1-21 years, as defined by the US Federal Food, Drug, and Cosmetic Act) CMV-seropositive allogeneic HCT transplant recipients were consented to be enrolled in the study, from August 2018 through January 2023. Several factors were associated with the trial slow accrual, including reassigning the study to a new principal investigator (PI), as the initial PI transferred to a different Institution; the significant COVID-19 pandemic disruption on cancer clinical trials and constrained financial resources. Clinical characteristics of the study population including Karnofsky performance score, conditioning regimen and stem cell source are described in Table 1. Exclusion criteria for administration of Triplex injection on day 28 post-HCT included: diagnosis of relapse, engraftment failure, acute graft-versus-host disease (GVHD) of grade 3 or 4 (according to the Keystone Consensus grading system), administration of steroids with a dosage above 1.0 mg/kg per day within 7 days, ongoing non-hematologic toxicity of grade 3 or higher (according to the Common Terminology Criteria for Adverse Events, version 4.03) and development of CMV viremia with quantitative polymerase chain reaction (qPCR) >500 copies/mL.
Six patients did not meet inclusion and did not receive the planned study intervention (Online Supplementary Figure 1S). Triplex injections were administered to nine eligible recipients on days 28 and 56 post-transplant, consistent
with timing of vaccination in our previous patient studies.13 Two Triplex vaccine dose levels (DL1=108 PFU and DL2=5x 108 PFU) were evaluated in two age groups (11-21 and 1-10 years old). DL2 is the Triplex dose administered to healthy adults and HCT patients in our published trials.13 Triplex-related adverse event (AE) assessment, including GVHD, non-relapse mortality, relapse, CMV reactivation requiring PET and CMV disease were prospectively monitored, recorded according to institutional standard of care, and summarized in Table 1 and Online Supplementary Table 1S. The impact of Triplex on cellular immunity was investigated and focused on magnitude, function, and quality of CMV-specific CD4 and CD8 T-cell expansion. Longitudinal immune monitoring was performed measuring 4-1BB (CD137) surrogate marker of T-cell activation by surface staining of peripheral blood mononuclear cells (PBMC), following 24 hours of stimulation with either pp65, IE1 or IE2 peptide libraries and medium as control. The CD137 assay was combined with the monitoring of CD28, CD45RA memory phenotype markers,14 to achieve a comprehensive immunological assessment of the Triplex vaccine effect on quantity and quality of CMV-specific T-cell functional expansion. PBMC from blood draws obtained before Triplex vaccination at day 28 and afterwards on days 42, 56, 70, 84, 100, 140, 180, and 270 were analyzed by fluorescence-activated cytometry in the research laboratory setting (FC; Gallios™, Beckman Coulter with Kaluza analysis software, Brea, CA).2 Concentrations of pp65, IE1- or IE2-specific CD3+CD4+CD137+ and CD3+CD8+CD137+ T cells were longitudinally measured using multiparameter (6 colors) flowcytometry. When either CMV-specific CD3+CD8+CD137+ T-cell or CD3+CD4+CD137+ T-cell populations were ≥0.2%, a further analysis for CD28 and CD45RA memory membrane markers could be performed. CD45RA+CD28+ cells were classified as naïve; CD45RA- CD28+ cells, as central memory (TCM); and CD28- cells, as effector T cells. Within the effector T-cell group, two subpopulations were identified: CD45RA-CD28- cells (TEM) and CD45RA+CD28effector “revertant” T cells, re-expressing the RA isoform of the CD45 surface marker (TEMRA).
From August 2018 to April 2023, nine eligible patients each received two injections of Triplex with no safety concerns and no dose limiting toxicity. Triplex was well tolerated at both DL and no AE grade ≥2 possibly or probable related to the vaccine was recorded (Table 1). Enrollment for DL1 vaccinees included three transplant 11-21-year-old recipients and three 1-10-year-old recipients. In the DL2, three transplant 11-21-year-old recipients were vaccinated. All nine participants completed the two injection Triplex vaccination regimen. All except one patient (COH002) received letermovir prophylaxis. CMV reactivation was treated with PET on day 44 for COH002 and on day 216 for COH011 following >1.0 mg/kg steroid treatment for chronic GVHD. COH014 withdrew from study on day 140 post-HCT, to return to their home country; no history of CMV reactivation was observed through 1-year post-HCT. COH013 experienced
relapse on day 158 post-HCT and further research
draws were suspended. We found high levels of functional CMV-specific T cells fol-
Table 1. Characteristics and outcomes of the Triplex-vaccinated pediatric hematopoietic stem cell transplant recipients.
CMV viremia or disease requiring antiviral treatment1
mortality1
(grade 3-4)2
CMV: cytomegalovirus; IQR: interquartile range; AE: adverse event; GVHD: graft-versus-host disease. 1Through day 100 after transplant; 2according to the Keystone Consensus grading system; 3within 2 weeks from each vaccination; 4on the basis of Common Terminology Criteria for Adverse Events (CTCAE), version 4.03 at least probably or definitely related to the Triplex vaccine; 5>100 days after transplant; as per standard of care, pre-emptive antiviral therapy (PET) was started in high-risk hematopoietic stem cell transplant; patients receiving letermovir prophylaxis when 2 consecutive CMV quantitative polymerase chain reaction (qPCR) were >500 copies/mL or when one qPCR was >500 copies/mL, if letermovir prophylaxis was not used. Low-risk patients started PET with CMV qPCR ≥1,500 copies/mL or with rising qPCR regardless of prophylaxis (1 copy/mL ≈0.93 IU/mL). *Values are numbers of patients (percentages) unless otherwise indicated.
lowing Triplex vaccination, which had a similar longitudinal profiles and often surpassed those observed in healthy adults and HCT recipients from previous Triplex vaccination studies13 (Figure 1; Online Supplementary Figure S2). High levels of functional CMV-specific T-cell response were measured in patients treated at both Triplex DL, in both age groups, in transplant recipients with either CMV-seropositive or CMV-seronegative donors undergoing matched related, unrelated or haploidentical HCT, using bone marrow or peripheral stem cell sources. The elevated response may have contributed to restoring protective antiviral cellular immunity after transplant in the Triplex-vaccinated pediatric recipients, who did not receive PET to control CMV reactivation. However, a randomized study is required to confirm such a hypothesis. A possible explanation of the vigorous response to Triplex in pediatric recipients may be linked to the effective activity of the thymus, which is highly functional in children, protecting against serious infections and leading to good vaccine responses.15 High-dose post-transplant cyclophosphamide used in haploidentical HCT pediatric recipients to prevent acute GVHD, did not induce CMV-specific T-cell depletion, in agreement with previous studies in adult recipients.16 As often reported in the adult transplant population, use of high dosage corticosteroids for GVHD reduced CMV-specific T-cell responses3,14 and in COH011 led to late PET treatment. As visualized by the gray bands in Figure 2A, functionally activated pp65, IE1, IE2-specific T cells increased post-transplant during immune-reconstitution. Linear mixed models were used to evaluate the longitudinal changes on log10-transformed
concentration of CMV-specific T cells. The analysis showed a significant increase in pp65-specific T cells/µL (left plot) after day 28 first Triplex injection through day 270 post-HCT (P=0.013). As previously found in healthy adults and in HCT recipients vaccinated with Triplex,13 longitudinal pp65-specific T-cell expansions were the most consistent and the highest in magnitude among the current recipient population, while enhanced levels of both IE1- and IE2-specfic T cells were observed in some pediatric patients (Figure 1). The memory phenotype analysis (Figure 2B) focused on pp65-specific T-cell responses, and showed that functionally activated pp65-specific CD137+CD8+ T cells had a predominant TCM profile early post-transplant, and reduced naïve T-cell subsets.13 In the vaccinated recipients, pp65-specific CD8 T cells showed a memory phenotype pattern mainly consisting of antigen-experienced TCM, immediately after transplant. By linear mixed models, the TCM subset significantly decreased following Triplex first injection (P=0.043) acquiring enhanced effector (TEMRA + TEM) functions (P=0.026), through day 270 post-HCT. Strong predominance of long-lived and functional effector T cells has been reported during primary CMV infection, since these cells are critical for viremia control.17
This is the first-in-pediatric HCT recipient study of Triplex. The early post-HCT administration of a biologic vaccine has few precedents in pediatric clinical trial history. Though clinical research is of considerable importance in developing safe medications, pediatric trials pose many ethical and clinical challenges. Our pilot trial outcomes are of interest in the transplant context, as they demonstrated
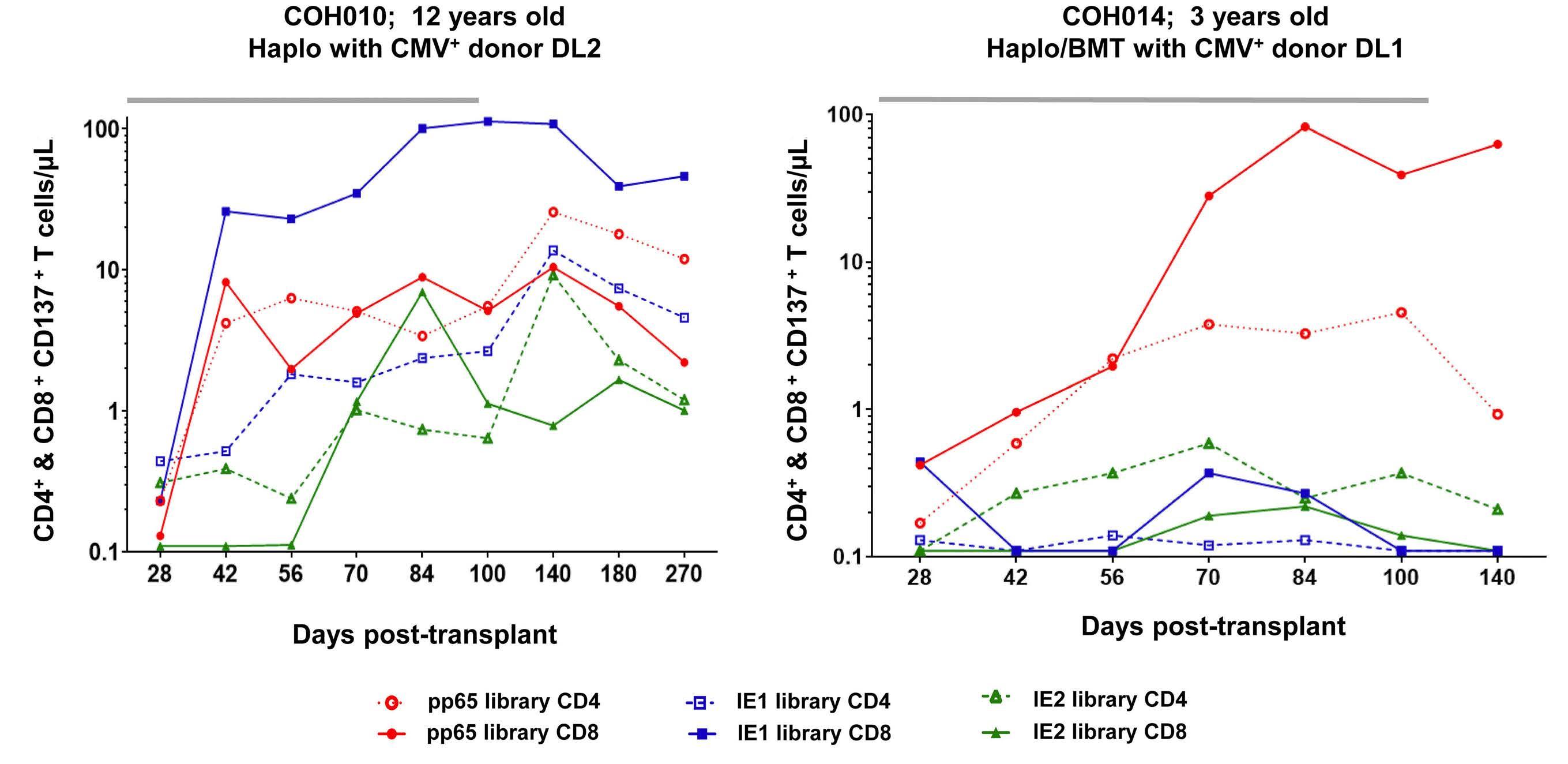
Figure 1. Cytomegalovirus-specific T-cell responses. Longitudinal immune profiles of COH010 and COH014 pediatric haploidentical (Haplo) recipients in response to Triplex vaccination. Cytomegalovirus (CMV)-specific T cells/μL are shown by distinct colors (pp65, IE1-, and IE-2) and line types (CD4 and CD8), as indicated in legend. Letermovir duration is shown by the black line on plot top (Haplo). BMT: bone marrow transplant; DL: dose level.
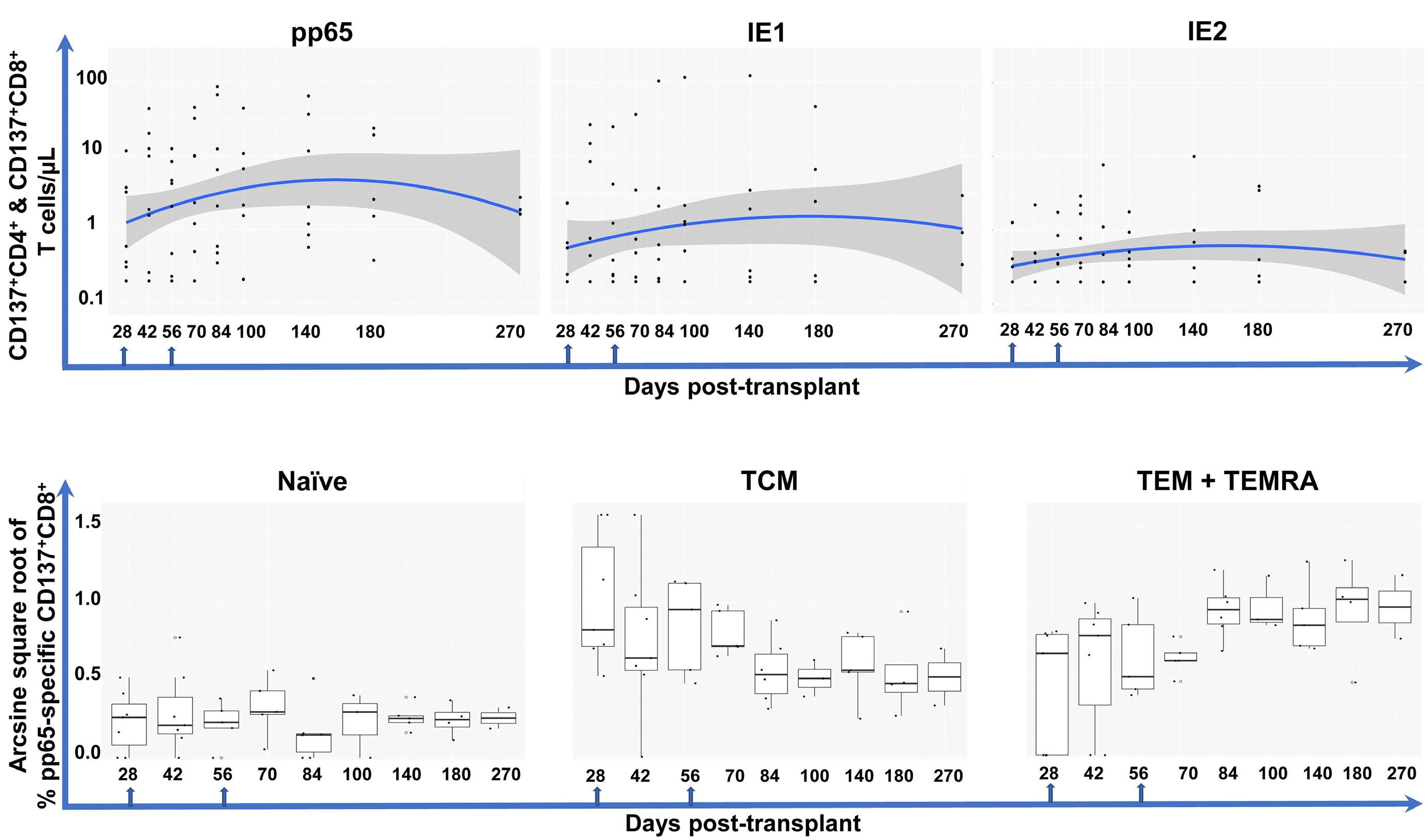
Figure 2. Levels and memory phenotypes of functional cytomegalovirus-specific T cells in the Triplex-vaccinated pediatric recipients. (A) Longitudinal levels of combined CD137+CD3+ CD4+ and CD137+CD3+ CD8+ T cells specific for pp65, IE1 and IE2 antigens (as specified at the top of each plot). The band shown was computed using the loess scatterplot smoother providing the marginal geometric mean concentrations through time. A 95% confidence band is shown in gray, and individual measurement trajectories are shown for each participant up to 7 days before cytomegalovirus (CMV) reactivation, requiring antivirals (COH002 and COH011). Linear mixed models were used to evaluate the changes on log10-transformed concentration of CMV-specific T cells from day 28 through day 270. Both linear and quadratic terms of study days were included in the models on CMV-specific T cells. An autoregressive covariance matrix was used to account for the correlation in participant’s T-cell responses over time. Logarithmic spacing is used to aid visualization. Arrows indicate Triplex injections on days 28 and 56 post-transplant. (B) Box plots showing percentages on the arcsine scale of pp65-specific, CD137+CD8+ naïve, T central (TCM) and effector (TEM + TEMRA) memory phenotype subset (as indicated at the top of each plot). The box spans the interquartile range, the central bar shows the median, and the whiskers extend to 1.5-times the interquartile range. Percentages of memory subsets from day 28 though day 270 were evaluated using linear mixed models and study day was included as a categorical predictor.
feasibility and safety of vaccinating immunosuppressed pediatric patients with Triplex. The encouraging preliminary results suggest that in HCT pediatric patients receiving letermovir prophylaxis, Triplex safely expands large numbers of long lasting functional CMV-specific T cells early post-transplant, which may prevent subsequent CMV reactivation requiring PET. The current key outcomes laid the groundwork for the planning of a multicenter, randomized placebo-controlled phase II trial. Though Triplex vaccination can overcome the letermovir-induced immune impairment,10 our long-term goal is to eliminate this daily oral medication for young patients who already have polypharmacy burdens. Novel successful strategies of vaccinating the immunocompetent HCT donor and possibly also the recipient with Triplex may suffice to prevent CMV reactivation by promoting early post-HCT sustained, protective CMV-immune reconstitution.
Corinna La Rosa,1 Yoonsuh Park,1 Dongyun Yang,1 Qiao Zhou,1 Teodora Kaltcheva,1 Nicole Karras,2 Jerry Cheng,2 Weili Sun,3 Don J. Diamond1# and Anna Pawlowska2#
1Department of Hematology and Hematopoietic Cell Transplantation, City of Hope National Medical Center, Duarte, CA; 2Department of Pediatrics, City of Hope National Medical Center, Duarte, CA and 3The Janssen Pharmaceutical Companies of Johnson & Johnson, Los Angeles, CA, USA
#AP and DJD contributed equally as senior authors.
Correspondence: C. LA ROSA - clarosa@coh.org
https://doi.org/10.3324/haematol.2023.284256
Received: September 8, 2023.
Accepted: February 23, 2024. Early view: February 29, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
CLR reports receiving consulting fees and research funding from Helocyte Inc. DJD reports receiving fees for royalties, research funding, and fees for serving on the advisory board of Helocyte Inc. DJD is co-inventor on a patent application covering the design and construction of the synthetic modified vaccinia Ankara platform (PCT/US2021/016247); is co-inventor on a patent application covering the development of a COVID-19 vaccine (PCT/ US2021/032821); is an employee of City of Hope National Medical Center (Duarte, CA, USA), which developed and funded the vaccine trial; receives consulting fees, patent royalties, research funding, and fees for serving on the advisory board of Helocyte Inc.; in addition, DJD has two patents 8,580,276 and 9,675,689 that are licensed to Helocyte; is a co-inventor of the Patent Cooperation Treaty (PCT) application that covers the development of a COVID-19 vaccine (PCT/US2021/032821) licensed to GeoVax Labs Inc.; receives consulting fees and research support from GeoVax Labs Inc. DJD and CLR are co-inventors on a patent covering the “Vaccination of hematopoietic stem cell donors with cytomegalovirus Triplex composition” (PCT/US2023/032052).
Contributions
AP, WS, NK, and JC treated and clinically assessed the patients for
1. Khandelwal P, Millard HR, Thiel E, et al. Hematopoietic stem cell transplantation activity in pediatric cancer between 2008 and 2014 in the United States: a Center for International Blood and Marrow Transplant research report. Biol Blood Marrow Transplant. 2017;23(8):1342-1349.
2. Czyzewski K, Styczynski J, Giebel S, et al. Age-dependent determinants of infectious complications profile in children and adults after hematopoietic cell transplantation: lesson from the nationwide study. Ann Hematol. 2019;98(9):2197-2211.
3. Green ML, Leisenring W, Xie H, et al. Cytomegalovirus viral load and mortality after haemopoietic stem cell transplantation in the era of pre-emptive therapy: a retrospective cohort study. Lancet Haematol. 2016;3(3):e119-127.
4 Zavras P, Su Y, Fang J, et al. Impact of preemptive therapy for cytomegalovirus on toxicities after allogeneic hematopoietic cell transplantation in clinical practice: a retrospective singlecenter cohort study. Biol Blood Marrow Transplant. 2020;26(8):1482-1491.
5. Abate D, Cesaro S, Cofano S, et al. Diagnostic utility of human cytomegalovirus-specific T-cell response monitoring in predicting viremia in pediatric allogeneic stem-cell transplant patients. Transplantation. 2012;93(5):536-542.
6. Laberko A, Bogoyavlenskaya A, Shelikhova L, et al. Risk factors for and the clinical impact of cytomegalovirus and Epstein-Barr
adverse events and acute graft-versus-host disease. DJD and WS designed the study. YP, QZ, and TK performed specimen processing, reagent preparation, and immunological assays. CLR and YP analyzed the immune monitoring data. DY performed the statistical analyses. CLR and DJD wrote the initial manuscript; and all authors approved the final version.
We thank the patients and their parents, nurses, technicians, research coordinators and clinicians, and the staff at the City of Hope (COH) without whose support this trial could not have been conducted. MVA was provided under a Material Transfer Agreement to COH from the National Institute of Allergy and Infectious Diseases (NIAID), Laboratory of Viral Diseases (Dr. Bernard Moss, Director). The following reagents were obtained through the National Institute of Health (NIH) AIDS Reagent Program, Division of AIDS, NIAID, NIH: HCMV pp65 Peptide Pool (referred as pp65 library in the manuscript). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The study was supported by grants from the National Cancer Institute (NCI) 5R01 CA077544; NCI P30 CA033572; P50 SPORE CA107399; 1R01 CA181045; NCI-SAIC-Frederick 28XS061; 1R01 CA266783 and Helocyte Inc. (to DJD).
Data-sharing statement
The clinical trial protocol and the anonymized data presented in this study can be made available upon reasonable request.
virus infections in pediatric recipients of TCR-alpha/beta- and CD19-depleted grafts. Biol Blood Marrow Transplant. 2017;23(3):483-490.
7 Russo D, Schmitt M, Pilorge S, et al. Efficacy and safety of extended duration letermovir prophylaxis in recipients of haematopoietic stem-cell transplantation at risk of cytomegalovirus infection: a multicentre, randomised, doubleblind, placebo-controlled, phase 3 trial. Lancet Haematol. 2023;11(2):e127-e135.
8. N PD, Cirincione A, Siver M, et al. Letermovir for cytomegalovirus prevention in adolescent patients following hematopoietic cell transplantation. J Pediatric Infect Dis Soc. 2022;11(7):337-340.
9 Su Y, Stern A, Karantoni E, et al. Impact of letermovir primary cytomegalovirus prophylaxis on 1-year mortality after allogeneic hematopoietic cell transplantation: a retrospective cohort study. Clin Infect Dis. 2022;75(5):795-804.
10. Zamora D, Duke ER, Xie H, et al. Cytomegalovirus-specific T-cell reconstitution following letermovir prophylaxis after hematopoietic cell transplantation. Blood. 2021;138(1):34-43.
11. Shafat MS, Mehra V, Peggs KS, Roddie C. Cellular therapeutic approaches to cytomegalovirus infection following allogeneic stem cell transplantation. Front Immunol. 2020;11:1694.
12. Pfeiffer T, Tzannou I, Wu M, et al. Posoleucel, an allogeneic,
off-the-shelf multivirus-specific T-cell therapy, for the treatment of refractory viral infections in the post-HCT setting. Clin Cancer Res. 2023;29(2):324-330.
13. La Rosa C, Diamond DJ. Triplex, a viral vectored CMV vaccine for transplant indications: clinical trial updates. Vaccine Insights. 2023;2(7):287-308.
14 Aldoss I, La Rosa C, Baden LR, et al. Poxvirus vectored cytomegalovirus vaccine to prevent cytomegalovirus viremia in transplant recipients: a phase 2, randomized clinical trial. Ann Intern Med. 2020;172(5):306-316.
15. Semmes EC, Chen JL, Goswami R, Burt TD, Permar SR, Fouda GG. Understanding early-life adaptive immunity to guide interventions for pediatric health. Front Immunol.
2020;11:595297.
16. Cesaro S, Crocchiolo R, Tridello G, et al. Comparable survival using a CMV-matched or a mismatched donor for CMV+ patients undergoing T-replete haplo-HSCT with PT-Cy for acute leukemia: a study of behalf of the infectious diseases and acute leukemia working parties of the EBMT. Bone Marrow Transplant. 2018;53(4):422-430.
17 Day EK, Carmichael AJ, ten Berge IJ, Waller EC, Sissons JG, Wills MR. Rapid CD8+ T cell repertoire focusing and selection of high-affinity clones into memory following primary infection with a persistent human virus: human cytomegalovirus. J Immunol. 2007;179(5):3203-3213.
Despite significant advances, outcomes for children with Down syndrome (DS, trisomy 21) who develop acute lymphoblastic leukemia (ALL) remain poor. Reports of large DSALL cohorts have shown that children with DS have inferior event-free survival (EFS) and overall survival (OS) compared to children without DS.1-3
Children with DS also exhibit increased treatment-related mortality due to infections and toxicities following chemotherapy, higher cumulative risk of relapse and inferior outcomes following relapse.4 This situation highlights the dire need for the development of more potent and targeted therapies to improve the survival and quality of care for these vulnerable children, who often have additional co-morbidities linked to trisomy 21 that complicates their clinical management. Targeted approaches and immunotherapies have shown promising results for pediatric leukemia.5 Hence, the development of new models of DS-ALL are needed to rapidly advance drug discovery and refine existing treatment strategies. We recently established DS-ALL patient-derived xenografts (PDX) and demonstrated that targeting somatic alterations found in DS-ALL using MEK inhibitors combined with conventional treatment has the potential to improve outcomes for these children.6 Targeting the dosage-sensitive mechanisms resulting from the extra copy of chromosome 21 is also an area of intense investigation.7-9 As such, inhibition of the chromosome 21 kinase DYRK1A using EHT1610 or of its direct targets FOXO1 and STAT3, has shown promising cytotoxic effects both in vitro and in vivo.8 In this study, we developed novel clinically relevant models of DS-ALL to facilitate the assessment of new therapeutic agents. First, we modeled oncogenic cooperation seen in DS-ALL in vitro 10 To this end, we transduced wild-type (WT) and trisomic (Tc1) 8-10-week-old bone marrow cells with retroviruses encoding the frequently observed mutant KRASG12D or the more rarely seen BCR-ABL fusion11 (animal experiments were approved by institutional ethics committee and followed Australian guidelines for the care and use of animals). Of note, the bone marrow stroma and hematopoietic stem cell and progenitor compartment composition did not significantly differ between disomic and trisomic mice, except from an increased proportion of multipotent progenitors MPP2 at the expense of the less committed MPP1 (Online Supplementary Figure S1A-C). Trisomic Tc1 progenitors exhibited increased capacity to form colony-forming unit (CFU) pre-B colonies in vitro compared to WT (Online Supplementary Figure S1D), as seen previously in the partially trisomic Ts1Rhr (Ts1) model.7 Ectopic expression of KRASG12D or BCR-ABL enhanced the number and replating capacity of both WT and Tc1 CFU pre-B colonies compared to the
empty vector MIC (MSCV-IRES-mCherry) (Online Supplementary Figure S1E, F). Next, we established two murine DS-ALL cell lines (Tc1-KRASG12D and Tc1-BCR-ABL), disomic controls (WT-KRASG12D and WT-BCR-ABL), as well as an independent Ts1/Cdkn2a-KRASG12D cell line (established from triple transgenic Ts1Rhr, Mb1-Cre, Cdkn2afl/fl donor mice); attempts to develop CRLF2-rearranged/JAK2 mutant murine DS-ALL cell lines were unsuccessful. Interestingly, although KRASG12D led to a constitutive phosphorylation of Erk1/2, the highest levels of Erk1/2 phosphorylation and cytokine independence for cell proliferation were only observed in WT-KRASG12D cells (Figure 1A, B). All KRASG12D-expressing murine cell lines engrafted in sub-lethally irradiated C57Bl/6J mice in primary and secondary recipients and ultimately succumbed to leukemia with complete penetrance, with recipients displaying mCherry-positive cells in the peripheral blood, bone marrow, and in the spleen (data not shown). Engrafted recipient mice exhibited splenomegaly associated with a leukemia-driven disorganized architecture (Online Supplementary Figure S1G, H). We also confirmed that the engrafted cell lines exhibit a phenotype similar to the cell lines cultured in vitro; although we noted clonal selection of a CD24+/BP1- pro-B population from the WT-KRASG12D cells in vivo, which was retained in secondary recipients (Figure 1C, D; and data not shown). Altogether, we developed novel murine models of DS-ALL, providing a unique platform suitable for testing targeted therapies.
In order to validate the clinical relevance of these murine cells, we focused on the therapeutically targetable chromosome 21 kinase DYRK1A, as recent reports have emphasized its role in childhood leukemia, regardless of Down syndrome.8 First, we used short hairpin RNA (shRNA) interference to show that all five cell lines, disomic (N=2) or trisomic (N=3) and expressing either KRASG12D or BCR-ABL oncogenes, were sensitive to Dyrk1a knock-down (KD) (Figure 2A, B; Online Supplementary Figure S1I). We next assessed the efficacy of new potent DYRK1A inhibitors using a DYRK1A-focused library which included EHT1610 used as control, Leucettinib-21 and its inactive isomer (compounds inspired by Leucettines and Leucettamine B, a natural substance produced by the marine sponge Leucetta microraphis),12,13 and three additional DYRK1A inhibitors whose chemical structure is based on the 7-azaindole scaffold, AM28, AM30 and AM45.14 In dose-response experiments, we showed that AM30 and Leucettinib-21 were cytotoxic in both WT-KRASG12D and Tc1-KRASG12D cell lines (Figure 2C). We also observed that Leucettinib-21, AM30 and AM45 were more potent than EHT1610 in decreasing cellular growth in all cell lines tested, and that Tc1-KRASG12D cells always exhibited lower half-maximal inhibitory concentra-
Figure 1. Establishment of novel Down syndrome acute lymphoblastic leukemia models for preclinical testing. (A) Constitutive phosphorylation of Stat5 and Erk1/2 in KRASG12D and BCR-ABL-expressing murine cells (starved for 6 hours). (B) Growth of murine wild-type (WT)-KRASG12D, Tc1-KRASG12D and Ts1/Cdkn2a-KRASG12D cells with or without cytokines (Il-7, Scf and Flt3-L, 10 ng/mL) over 6 days. (C) Kaplan-Meier analysis comparing survival of primary (1R) and secondary (2R) sub-lethally irradiated recipient mice engrafted with 1-2x106 Tc1-KRASG12D (1R N=4, 2R N=6), WT-KRASG12D (1R N=5, 2R N=6) and Ts1Rhr/Cdkn2a-KRASG12D (1R N=5, 2R N=6) cell lines; **P<0.01. (D) Phenotype of the murine cell lines assessing surface expression of BP1 and CD24 (phenotype of the WTBCR-ABL and Tc1-BCR-ABL cell lines are in the Online Supplementary Figure 1F), and representative flow plots showing phenotype of mCherry-positive cell lines in primary and secondary recipients. w/: with; w/o: without. Cyt: cytokine.
Figure 2. Genetic and pharmacological inhibition of DYRK1A decreases growth of Down syndrome acute lymphoblastic leukemia cells. (A) Ratio of wild-type (WT)-KRASG12D and Tc1-KRASG12D transduced with GFP-expressing Banshee vectors encoding two shDyrk1a compared to empty Banshee-U6 counterparts over 9 days (N=4 replicates); ***P<0.001. (B) Validation of Dyrk1a knockdown at the protein level 48 hours after transduction (GFP-sorted). Dyrk1a band intensities were quantified and normalized as a ratio of shDyrk1a-transduced to control U6-transduced WT-KRASG12D cells. (C) Cytotoxic effect of increasing doses (in μM) of the DYRK1A inhibitors EHT1610, AM30, Leucettinib-21 (LCTB-21) and its inactive isomer iso-Leucettinib-21 (Iso LCTB-21) at 48 hours in murine WT-KRASG12D and Tc1-KRASG12D cells assessed by flow cytometry (Annexin V staining). (D) Dose-response curves assessing efficacy of EHT1610, LCTB-21 and Iso LCTB-21 at 72 hours by alamarBlue cell viability assay in murine WT-KRASG12D and Tc1-KRASG12D cells. (E) Heatmap integrating relative half-maximal inhibitory concentration (IC50) values obtained for the DYRK1A inhibitors tested in our murine cell lines.
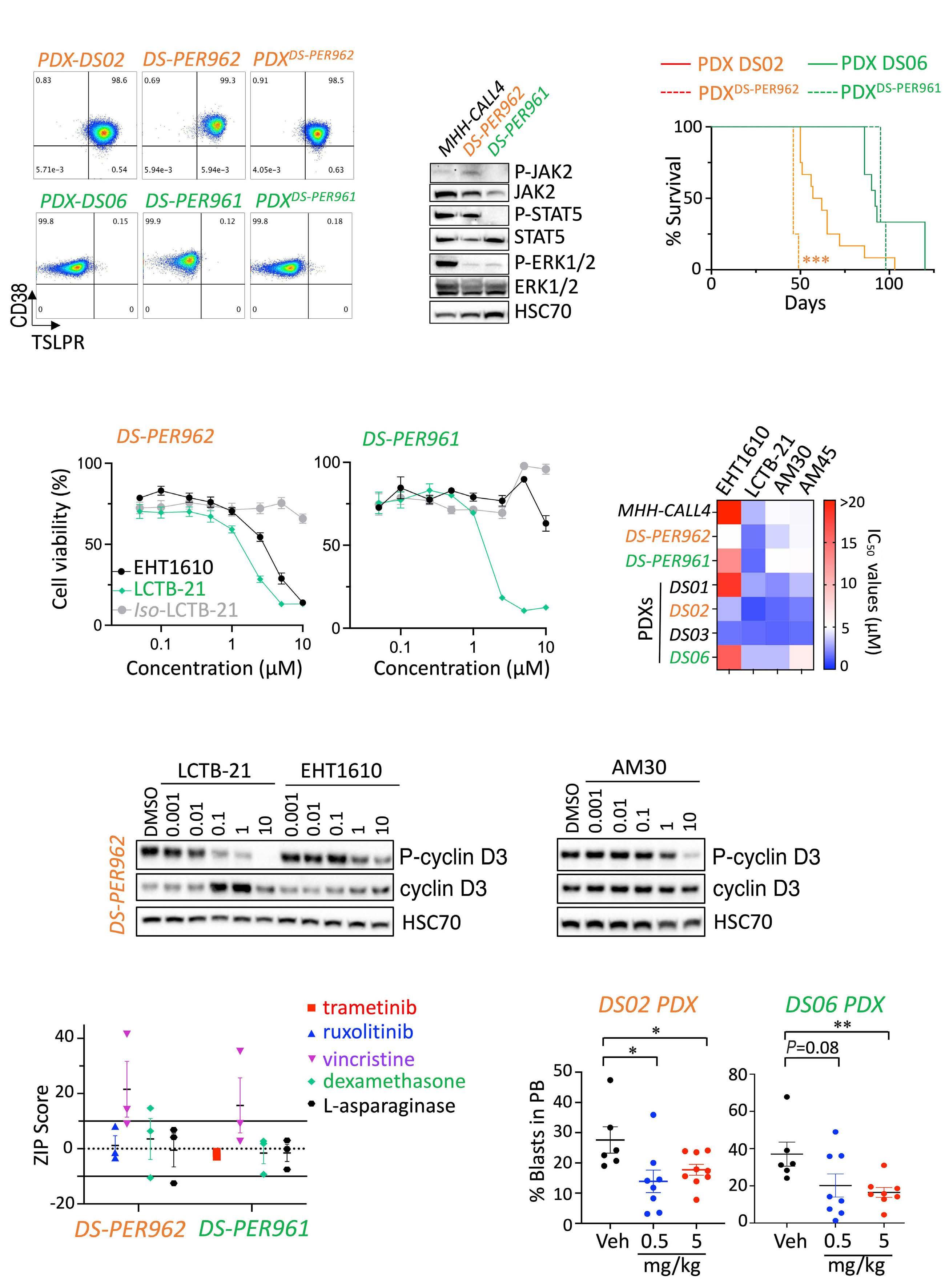
Continued on following page.
Figure 3. Efficacy of DYRK1A inhibition in novel human Down syndrome acute lymphoblastic leukemia cell lines and patient-derived xenograft models. (A) Representative flow plots assessing CD38 and TSLPR expression of the human DS-PER962 and DSPER961 Down syndrome acute lymphoblastic leukemia (DS-ALL) cell lines compared to their corresponding DS06 and DS02 patient-derived xenografts (PDX), and to NSG recipients engrafted with 1x106 DS-ALL cell lines (PDXcells). (B) Western blots assessing the constitutive phosphorylation of JAK2, STAT5 and ERK1/2 in DS-PER962 and DS-PER961 cell lines following a 6-hour starvation; the non-DS MHH-CALL4 (CRLF2-rearranged/JAK2 mutant) ALL cell line was used as control. (C) Kaplan-Meier curves comparing the survival of the DS-PER962 and DS-PER961 PDX (N=3-4) to their corresponding DS02 and DS06 PDX (N=9-12); ***P<0.001. (D) Efficacy of EHT1610, Leucettinib-21 (LCTB-21) and its inactive isomer iso-Leucettinib-21 (Iso LCTB-21) in the human DS-PER962 and DS-PER961 cell lines (72 hours). (E) Heatmap representing the relative half-maximal inhibitory concentration (IC50) values obtained in the non-DS-ALL, DS-ALL human cell lines and DS-ALL cells freshly harvested from PDX models. (F) Western blot comparing the effect of EHT1610, LCTB-21 and AM30 on phospho-cyclin D3 and total cyclin D3 stability after 6 hours of treatment in the DS-PER962 cell line. (G) ZIP scores obtained from combining Leucettinib-21 (10 doses from 0.01 to 10 μM) with trametinib (0.0001-20 μM), ruxolitinib (0.0001-20 μM), vincristine (0.0005-0.5 μM), dexamethasone (0.00025-20 μM) and L-asparaginase (0.1-20 μM) in the human DS-ALL cell lines. ZIP scores <-10 =antagonism; -10 to 10 =additive; >10 =synergy. (H) In vivo effect of Leucettinib-21 on leukemia burden in the peripheral blood (PB) of the DS02 and DS06 PDX models (oral gavage, 2 weeks and 4 weeks of treatment respectively); *P<0.05, **P<0.01. Veh: vehicle.
tion (IC50) values than its disomic counterpart WT-KRASG12D (Figure 2D, E; Online Supplementary Figure S2A). In order to expand our observations, we next established two human DS-ALL cell lines: DS-PER961 and DS-PER962 from our previously reported DS06 (KRASG12S-positive) and DS02 (CRLF2-rearranged/JAK2I682F-positive) PDX.6 These unique models represent the first human cell lines for DSALL, with comprehensive characterization at the genomic, transcriptomic and phenotypic levels, confirming their resemblance to the PDX models from which they originated (Figure 3A; Online Supplementary Figure S3A-E). Western blot analyses confirmed phosphorylation of ERK1/2 and STAT5 downstream of KRASG12S and JAK2I682F mutants in DS-PER961 and DS-PER962 respectively (Figure 3B). Using NOD-SCID-γc-/- (NSG) mice, we also showed that both DSALL cell lines engrafted into immunocompromised recipients with DS-PER962 cells being more aggressive than their PDX counterparts (Figure 3C). Using these human cells, we confirmed that Leucettinib-21, AM30 and AM45 were potent inhibitors in human DS-ALL cells and in MHH-CALL4 (a nonDS CRLF2-rearranged and JAK2 mutant ALL cell line known to be sensitive to DYRK1A inhibition) (Figure 3D, E; Online Supplementary Figure S2B-D).8 No significant effect was seen for AM28 nor for the inactive isomer of Leucettinib-21 (Figure 3D; Online Supplementary Figure S2B, C). Importantly, efficacy of these DYRK1A inhibitors was demonstrated in DSALL blasts freshly harvested from four DS-ALL PDX models (described in 6), validating the suitability of our DS-ALL cells to assess efficacy of new therapies (Figure 3E). Compared to EHT1610 and AM30, Leucettinib-21 was the most potent compound in inhibiting phosphorylation of the known DYRK1A target cyclin D3 in a dose-dependent manner shown in DS-PER962 (Figure 3F), DS-PER961 and in murine cells (Online Supplementary Figure S2E), but had limited effect on FOXO1-phosphorylation (Online Supplementary Figure S2F-I). Next, we evaluated in vitro drug combinations of Leucettinib-21 with targeted and conventional therapies and identified synergy with vincristine and an additive effect with dexamethasone and L-asparaginase (Figure 3G; Online Supplementary Figure S2J). An additive effect between Leu-
cettinib-21 and the targeted therapies ruxolitinib (a JAK1/2 inhibitor) and trametinib (a MEK1/2 inhibitor) was also seen in DS-PER962 (CRLF2-positive/JAK2I682F mutant) and in the DS-PER961 (KRASG12S) cell lines, respectively (Figure 3G). Finally, we assessed the efficacy of Leucettinib-21 in vivo in the DS06 and DS02 PDX models and observed that in vivo treatment with Leucettinib-21 decreased leukemia burden but did not fully eradicate leukemia (Figure 3H). Together, this data demonstrates the suitability and clinical relevance of the novel murine and human models we have established and emphasizes the key role of DYRK1A in DS-ALL. Compared to other children, higher sensitivity to treatment-related toxicity in children with DS-ALL remains a major clinical challenge. This has significantly limited the development of novel targeted therapies for this patient population, ultimately delaying translation into clinical trials. Recently, integration of immunotherapeutic approaches has offered promise in reducing toxicity. Indeed, Laetsch et al. reported comparable outcomes between DS- and nonDS children with relapsed/refractory B-ALL who received CD19-directed chimeric antigen receptor T-cell therapy, including similar rates of CD19-negative relapse,15 and an ongoing clinical trial is currently assessing whether blinatumomab can replace two blocks of consolidation chemotherapy for treatment of de novo DS-ALL (clinicaltrial gov. Identifier: NCT03911128). As an alternative approach, identifying key vulnerabilities in DS-ALL blasts could provide the molecular basis for development of novel targeted therapies. While resources such as the recent in-depth characterization of the genetic landscape of DS-ALL can provide such insight,10 we have developed novel models of DS-ALL and shown that inhibiting dosage-sensitive mechanisms altered by trisomy 21 may also represent a new avenue to integrate agents with low toxicity and ultimately improve outcomes and quality of care for children with DS-ALL. We demonstrated that a reduction in DYRK1A expression is sufficient to decrease the growth of DS-ALL cell lines, confirmed the sensitivity of human and murine cells to DYRK1A inhibition and showed that the leading candidate, Leucettinib-21, potentiates the cytotoxic effect of other chemotherapeutic and targeted
agents and delayed leukemia expansion in vivo, with no detectable toxicity identified in the peripheral blood (Online Supplementary Figure S2K). Leucettinib-21 has recently completed regulatory preclinical safety studies and is primed for early phase clinical assessment. Strikingly, studies have demonstrated the preclinical impact of targeting DYRK1A activity in different subtypes of childhood leukemia,8 further emphasizing the potential benefit for investigating Leucettinib-21 or other new potent DYRK1A inhibitors in clinical trials for ALL.
Altogether, this study has established and comprehensively characterized the first DS-ALL cell lines, providing suitable and clinically relevant cellular models to identify new molecular weaknesses in DS-ALL and test the efficacy of novel targeted therapies (as exemplified here with DYRK1A inhibition), alone or in combination with standard of care, to ultimately develop new, less toxic treatments to improve the outcome for children with DS-ALL.
Shannon L. Carey-Smith,1,2* Maryam H. Simad,1* Kunjal Panchal,1,2 Carlos Aya-Bonilla,1 Hannah Smolders,1 Sang Lin,1 Jesse D. Armitage,1 Vivien T. Nguyen,1 Kathryn Bentley,1 Jette Ford,1 Sajla Singh,1 Joyce Oommen,1 Anouchka P. Laurent,3 Thomas Mercher,3 John D. Crispino,4 Andrew P. Montgomery,5 Michael Kassiou,5 Thierry Besson,6 Emmanuel Deau,7 Laurent Meijer,7 Laurence C. Cheung,1,2,8 Rishi S. Kotecha1,2,9,10 and Sébastien Malinge1,2,10
1Telethon Kids Cancer Center, Telethon Kids Institute, Perth, Western Australia, Australia; 2Curtin Medical School, Curtin University, Perth, Western Australia, Australia; 3U1170 INSERM, Gustave Roussy, Villejuif, France; 4Department of Hematology, St Jude Children’s Hospital, Memphis, TN, USA; 5School of Chemistry, University of Sydney, New South Wales, Australia; 6University Rouen Normandie, INSA Rouen Normandie, CNRS, COBRA UMR 6014, Rouen, France; 7Perha Pharmaceuticals, Perharidy Peninsula, Roscoff, France; 8Curtin Health Innovation Research Institute, Curtin University, Perth, Western Australia, Australia; 9Department of Clinical Hematology, Oncology, Blood and Marrow Transplantation, Perth Children’s Hospital, Perth, Western Australia, Australia and 10University of Western Australia, Perth, Western Australia, Australia
*SLC-S and MHS contributed equally as first authors.
Correspondence:
S. MALINGE - sebastien.malinge@telethonkids.org.au
https://doi.org/10.3324/haematol.2023.284271
Received: September 13, 2023. Accepted: February 19, 2024. Early view: February 29, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
JDC is a member of the scientific advisory board for Alethiomics, a consultant for Cellarity, and receives research funding from Syndax. LM is a founder of Perha Pharmaceuticals. ED and LM are coinventors in the Leucettinib patents. RSK discloses advisory board participation from Jazz Pharmaceuticals, Amgen and Link Healthcare. All other authors have no conflicts of interest to disclose.
Contributions
SLCS, MHS and SM conceived and designed the experiments. SLCS, MHS, KP, CAB, HS, SL, JDA, VTN, KB, JF, SS, JO and SM performed the experiments. APL, TM, JDC, TB, APM, MK, ED and LM provided reagents, materials and technical insight. SLCS, MHS, KP, CAB, LCC and RSK significantly contributed to analysis and interpretation of the results. SM supervised the project. SLCS and SM wrote the manuscript. All authors edited and approved the final version of the manuscript for submission.
We thank L. Munoz, T. Johns, G. Chua, P. Kumar, T. Lassmann, and staff members of the Bioresources facility at Telethon Kids Institute for their support in obtaining reagents, processing samples and housing/monitoring animals related to this study.
This research was supported by Australian Government Research Training Program (RTP) Scholarships (to SLCS and KP). CAB is supported by a Fellowship from the Jerome Lejeune and Sisleyd’Ornano Foundations. TB is supported by the University of Rouen Normandy, INSA Rouen Normandy, the Centre National de la Recherche Scientifique (CNRS), European Regional Development Fund (ERDF), Labex SynOrg (ANR-11-LABX-0029), Carnot Institute I2C, the XL-Chem Graduate School of Research (ANR-18-EURE-0020 XL CHEM), and by Region Normandie. APM is supported by a postdoctoral research fellowship from the University of Sydney’s Drug Discovery Initiative. MK is a recipient of a National Health and Medical Research Council Principal Research Fellowship (APP1154692). LM is supported by grants from the Jerome Lejeune Foundation, l’Agence Nationale de la Recherche (ANR) (DYRK-DOWN), BpiFrance (i-Nov, vague 9), the European Union’s Horizon 2020 research and innovation programme (Grant #848077) (GO-DS21), and the European Innovation Council (EIC) Accelerator Down-Autonomy project (190138295). SM is supported by a fellowship from the Cancer Council Western Australia (CCWA, Grant #877). This study was supported by project grants from the Child Cancer Research Foundation (RSK, LCC and SM), Cancer Council Western Australia (Grant #1068) and the Jerome Lejeune Foundation (Grant #1806).
Data-sharing statement
RNA-sequencing files are available via the Gene Expression Omnibus (GEO) database under the accession number GSE245056.
1. Buitenkamp TD, Izraeli S, Zimmermann M, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014;123(1):70-77.
2. Ceppi F, Stephens D, den Hollander BS, et al. Clinical presentation and risk factors of serious infections in children with Down syndrome treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 2016;63(11):1949-1953.
3. Rabin KR, Devidas M, Chen Z, et al. Outcomes in children, adolescents, and young adults with Down syndrome and ALL: a report from the Children’s Oncology Group. J Clin Oncol. 2024;42(2):218-227.
4 Meyr F, Escherich G, Mann G, et al. Outcomes of treatment for relapsed acute lymphoblastic leukaemia in children with Down syndrome. Br J Haematol. 2013;162(1):98-106.
5. Rafei H, Kantarjian HM, Jabbour EJ. Targeted therapy paves the way for the cure of acute lymphoblastic leukaemia. Br J Haematol. 2020;188(2):207-223.
6. Laurent AP, Siret A, Ignacimouttou C, et al. Constitutive activation of RAS/MAPK pathway cooperates with trisomy 21 and is therapeutically exploitable in Down syndrome B-cell leukemia. Clin Cancer Res. 2020;26(13):3307-3318.
7 Lane AA, Chapuy B, Lin CY, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet. 2014;46(6):618-623.
8. Bhansali RS, Rammohan M, Lee P, et al. DYRK1A regulates B cell acute lymphoblastic leukemia through phosphorylation of
FOXO1 and STAT3. J Clin Invest. 2021;131(1):e135937.
9. Page EC, Heatley SL, Eadie LN, et al. HMGN1 plays a significant role in CRLF2 driven Down Syndrome leukemia and provides a potential therapeutic target in this high-risk cohort. Oncogene. 2022;41(6):797-808.
10 Li Z, Chang TC, Junco JJ, et al. Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood. 2023;142(2):172-184.
11. O’Doherty A, Ruf S, Mulligan C, et al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309(5743):2033-2037.
12. Tahtouh T, Durieu E, Villiers B, et al. Structure-activity relationship in the Leucettine family of kinase inhibitors. J Med Chem. 2022;65(2):1396-1417.
13. Lindberg MF, Deau E, Miege F, et al. Chemical, biochemical, cellular, and physiological characterization of Leucettinib-21, a Down syndrome and Alzheimer’s disease drug candidate. J Med Chem. 2023;66(23):15648-15670.
14 Zhou Q, Phoa AF, Abbassi RH, et al. Structural optimization and pharmacological evaluation of inhibitors targeting dualspecificity tyrosine phosphorylation-regulated kinases (DYRK) and CDC-like kinases (CLK) in glioblastoma. J Med Chem. 2017;60(5):2052-2070.
15. Laetsch TW, Maude SL, Balduzzi A, et al. Tisagenlecleucel in pediatric and young adult patients with Down syndromeassociated relapsed/refractory acute lymphoblastic leukemia. Leukemia. 2022;36(6):1508-1515.
The slower clearance of acute lymphoblastic leukemia (ALL) cells during induction therapy is associated with an increased risk of relapse. Insight into the mechanism(s) of the multidrug resistance, which these cells clearly display, may lead to more specific, minimal residual disease (MRD)-directed therapies. We have recently reported on the use of single-cell, high dimensional mass cytometry to functionally characterize B-lineage ALL cells in both drug-naïve cells at disease presentation and in those persisting during induction.1 The large panel of more than 30 antibodies allowed for the discrimination of leukemia cells from normal cells, based on the identification of a leukemia-associated immunophenotype (LAIP), and provided information on cell cycle, apoptosis and the activation state of key signaling pathways, known to be recurrently activated in ALL. We demonstrated the presence of a strongly activated CREB pathway across a broad spectrum of cytogenetic groups and, using high dimensional anal-
Table 1. Details of patients used in the study.
yses, showed that activated CREB was prominent in all viable cell clusters at both presentation and in MRD. We also showed that minor subpopulations with hyperactive CREB visible at presentation had a selective advantage during induction therapy. Furthermore, using a specific inhibitor of CREB transcriptional function, we validated CREB as a therapeutic target in ALL cells. Here, we expand on our previous study using a mass cytometric antibody panel specific for T-lineage ALL (T-ALL) and similarly show the presence of activated CREB in almost all viable cell clusters during therapy, their sensitivity to CREB inhibition and importantly show that MRD is made up of non-cycling cells that appear to be selected for during therapy. These data suggest that MRD-directed therapies should be cell cycle-independent and that the CREB pathway is a valid therapeutic target in T-ALL and, as we have previously shown, B-lineage ALL.
Bone marrow samples were accessed through the New-
LK350
LK350
Minimal residual disease (MRD) was assessed using flow cytometry following the principles of our standardized method for B-lineage acute lymphoblastic leukemia (ALL) and involved 5 panels of 6 antibody combinations, along with Syto 41 to exclude red cell contamination.10 *Sample was from a mediastinal mass. **There was inadequate sample for Flow MRD but polymerase chain reaction (PCR) MRD was reported as >0.01%. ***Patient had been exposed to 2 days of dexamethasone. UN: unknown; CyTOF: cytometry time of flight; WBC: white blood cell count.
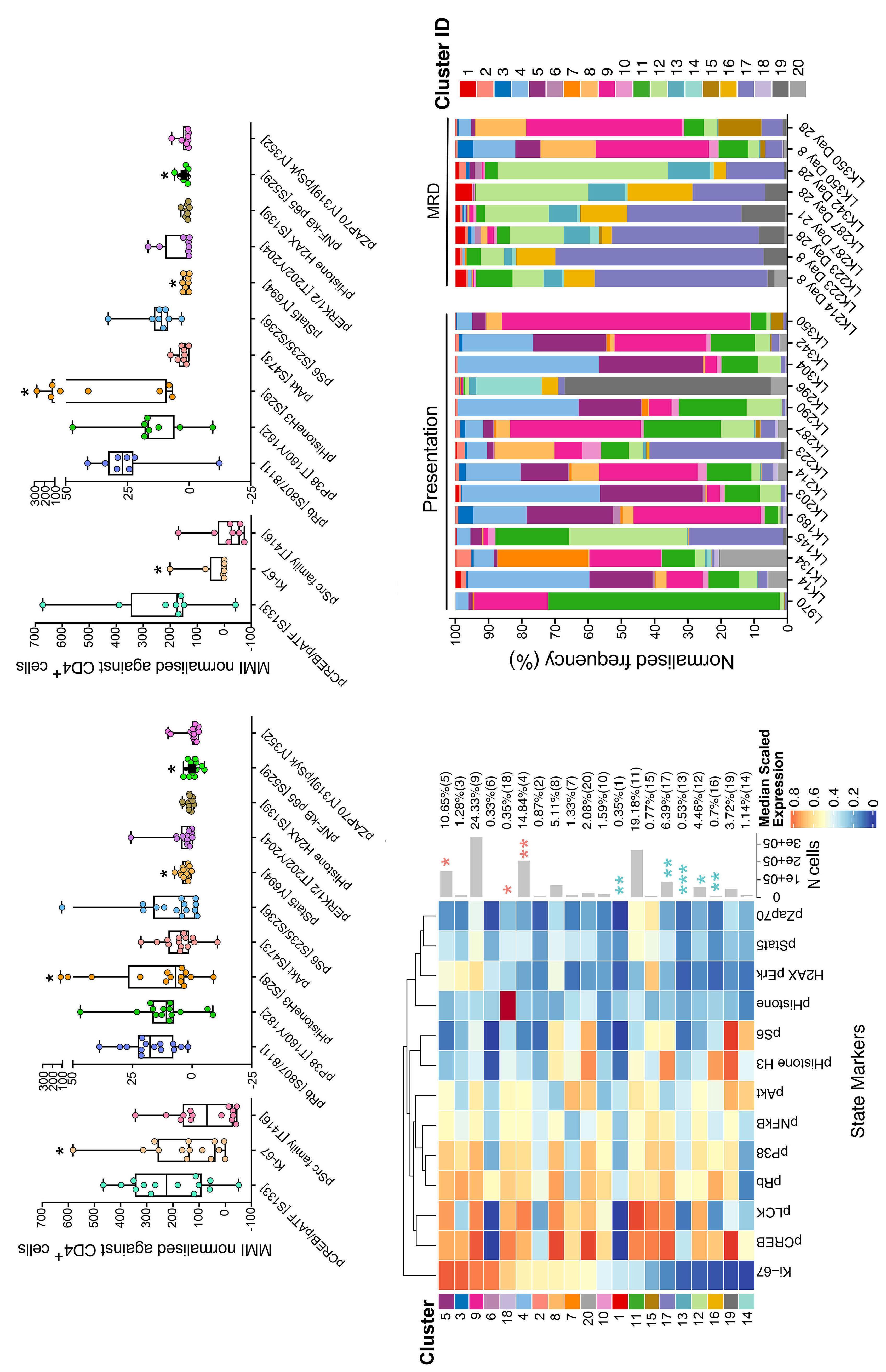
Figure 1. Ubiquitous activation of pCREB in acute lymphoblastic leukemia cell clusters and dormancy of minimal residual disease. Mean mass intensity (MMI) of phospho-markers and Ki67 in presentation T-lineage acute lymphoblastic leukemia (T-ALL) cells (A) and minimal residual disease (MRD) cells (B) normalized against internal CD4 + T cells. MMI signals are shown across 2 graphs to better represent data. Heatmap showing median marker expression of 13 phospho-markers across 20 populations generated with FlowSOM after metaclustering with ConsensusClusterPlus using gated ALL cells (C). Heatmap colour shows the median of the arcsinh, 0-1 transformed marker expression calculated from all gated leukemic cells. The column dendogram shows hierarchical similarity been phospho-markers while the row dendogram shows the hierarchical similarity between the 20 clusters, generated using the Euclidean distance metric and average linkage. Cluster proportions are shown by barplots and percentages on the right. Asterisk over the barplots denote clusters showing significantly different proportions between Presentation and MRD samples. Pink asterisks are clusters more prominent in Presentation ALL cells and blue asterisks are more prominent in MRD. *Represents values which were considered statistically significant P <0.05, ** for P <0.01 and *** for P <0.001. FlowSOM generated cluster composition by percentage in all patients analysed in study, showing differing trends between presentation and MRD time points (D).
castle Hematology Biobank, after appropriate consent (reference numbers 2002/111 and 07/H0906) from children treated with a with a standard three or four drug induction ALL regimen and who had slow clearance of disease (MRD levels ranging from 0.30% to 56.70% with a mean of 19.99% +/- standard deviation [SD] of 18.50). We analyzed 14 bone marrow samples at presentation of T-ALL and eight samples from five patients during follow-up, taken at day 8 (N=3), day 21 (N=1) and day 28 of induction therapy (N=4) using mass cytometry with previously published methods (antibody panel is shown in Online Supplementary Table S1).1 Patient details are shown in Table 1. Live, single, non-apoptotic ALL cells were identified by their specific LAIP and the mean mass intensity (MMI) of all antibody signals expressed relative to those from normal CD4-positive cells present in the same sample. The most prominent signals in presentation samples were pCREB/ pATF1 [S133] (mean 214.55; range, -50.82 to 466.60), Ki67 (mean 158.97; range, 0.67-582.60) and pSRC family [T416] (mean 80.44; range, -43.90 to 344.16) (Figure 1A; Online Supplementary Table S2). The latter having binary separation with high and low activation and likely to reflect levels of pLCK.2,3 Western blotting confirmed that the prominent activated form was pCREB, with minimal signal from pATF1 (Online Supplementary Figure S1). Compared to presentation ALL cells, MRD cells had a significant decrease in Ki67 levels with a mean of 158.97 falling to 34.62 and an increase in pHH3, with a mean of 21.20 increasing to 87.91 in MRD (P<0.05) (Figure 1B; Online Supplementary Table S2). There were also more subtle changes in pSTAT5, a signal of 2.96 at presentation decreasing to 1.27 at MRD and an increase in pNFKB p65, -0.30 at presentation increasing to 2.06 in MRD cells (P<0.05).
We next used cytometry by time of flight (CyTOF) workflow R pipeline to perform cluster analyses using phospho-signals (and Ki67) from ALL cells to investigate cluster composition in drug naïve cells and in those after therapy. We generated 20 unique ALL cell clusters (Figure 1C, D; Online Supplementary Table S2). ALL populations were complex heterogeneous, showing multiple different clusters at both presentation and in MRD cells, but there were clear differences in cluster abundance (Figure 1C). The closely related clusters, 4 and 5, as well as cluster 18, were significantly more represented at presentation (P<0.05) and were characterized by multiple signaling nodes including pLCK, pCREB, pRb, pp38 and high Ki67. There were a number of clusters more prevalent in MRD cells including clusters 1, 12, 13, 16 and 17 (P<0.05). All of these MRD clusters had strikingly low levels of Ki67, with the more abundant ones also expressing higher levels of pHH3 and pRb. Uniform manifold approximation and projection visualization of MRD and presentation clusters revealed MRD to be enriched in the left and right hand areas of the map. Visualization of pCREB confirmed its almost ubiquitous expression across the UMAP in both presentation
and MRD ALL cells. Clusters 1 and 6 were exceptions in that they expressed low pCREB but represented less than 1% of all cells but cluster 1 was more prominent in MRD (Figures 2A, B and 1C, D). Visualization of Ki67 clearly showed MRD-enriched areas to have significantly lower expression (Figure 2C; Online Supplementary Figure S3). This was even more apparent in UMAP projections from individual patients (Figure 2D) and, for patients with multiple time points, there was a further selection of Ki67-negative cells as treatment progressed (LK287, LL223 and LK350). Notably, only one of five patients had MRD cells had both Ki67-negative and positive cell islands (LK360). Clusters 12, 13, 16 and 17 were visible in presentation samples but were clearly more prominent in the MRD (Figure 2E, F).
We next assessed the sensitivity of four patient-derived xenografts (PDX) T-ALL co-cultured with mesenchymal stromal cell support to 666-15, a selective CREB inhibitor, and showed a mean ± sd, Half maximal inhibitory concentration (IC50) of 3.6±0.2 μM (Figure 2G). Robust, dose-dependent apoptosis was shown with 666-15 (N=3; P=0.01) with almost 100% of cells in the early stages of apoptosis after 48 hours exposure to IC50 concentrations (Figure 2H). At the same time point, cell cycle analyses corroborated the cytotoxic action of 666-15 by the increased proportion of cells with subG1 DNA content, though this did not attain statistical significance (N=3; P=0.07), and also demonstrated the majority of cells to be in G0/G1 (Figure 2I).
The advancement in cytometric methodologies has provided a stepwise increase in the number of analytic parameters that are enabling extensive functional analyses of rare cell populations, such as clinical MRD. Here, we confirm the presence of activated CREB in almost all T ALL cell clusters at both presentation and MRD and show that MRD is non cycling and, similar to B ALL, is derived from cell populations evident at diagnosis that are enriched during induction therapy. Dormancy is a common mechanism of cancer cells to achieve/acquire multi-drug resistance, at least to therapies reliant upon cell division,4 and our data support others in suggesting that ALL MRD cells are dormant.5 Eliminating MRD cells at the end of induction might be best achieved with targeted drugs that are not dependent on cycling cells such as the CREB pathway or alternatively re-engaging cell cycling, prior to more standard chemotherapeutics.
CREB overexpression and hyperactivation is reported in a number of cancer types and is considered a therapeutic target as it appears to be a vital signallng node downstream of many oncogenes.6 An earlier study by van der Sligte et al ., demonstrated that both knockdown and pharmacological inhibition of CREB induced cell cycle arrest and apoptosis in T ALL cells by impacting on genes involved in apoptosis, cell cycle and glucose metabolism.7 Here, we confirm the dependency of T-ALL cells on CREB activity with a next generation CREB inhibitor,
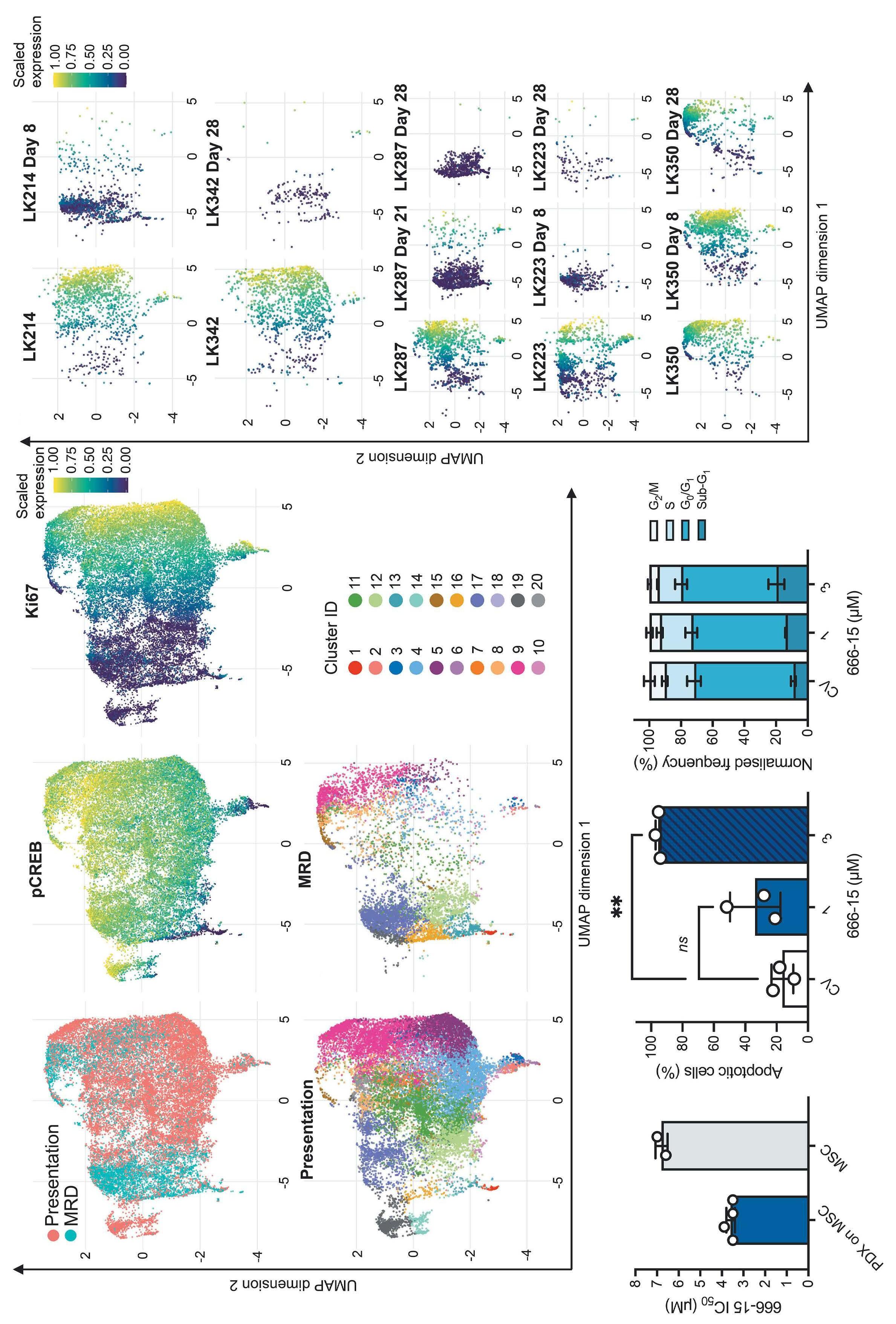
Figure 2. Dormant minimal residual disease cells arise from subpopulations present at diagnosis and the CREB inhibitor 666-15 is cytotoxic in T-cell acute lymphoblastic leukemia cells. Uniform manifold approximation and projection (UMAP) of acute lymphoblastic leukemia (ALL) cells generated using intracellular antibody signals showing cluster segregation in presentation (red) and minimal residual disease (MRD) (blue) samples (A). UMAP of ALL cells showing scaled expression of pCREB (B) and Ki67 (C). UMAP of ALL cells in individual patients showing Ki67 levels in paired presentation and MRD time points (D). UMAP of ALL cells showing cluster distribution in presentation (E) and MRD samples (F). Histogram of 666-15 half maximal inhibitory concentration (IC 50 ) in patient-derived xenografts (PDX) on MSC and MSCs alone (mean ± standard deviation [SD] are shown (G). Histogram of percentage of apoptotic cells as measured by annexin V binding in cells dosed for 48 hours with control vehicle (CV) or 2 different doses of 666-15 in triplicate PDX (H). Histogram showing the normalized frequency of cells in various stages of the cell cycle in cells dosed for 48 hours with CV or 2 different doses of 666-15 in triplicate PDX. Mean ± SD are shown. **Denotes statistical significance where P <0.01.
666-15. Cytotoxicity was observed at drug concentrations, which reduced tumor growth without overt toxicity in mice models, suggesting a potential therapeutic window of CREB inhibition.8,9 Together, these data suggest that MRD-directed therapies should be cell-cycle independent and that the CREB pathway is a valid therapeutic target for both T- and B-lineage ALL cells.
Authors
Dino Masic,1 Hayden L Bell,1 Frederik W van Delft,1,2 and Julie Anne Elizabeth Irving1
1Wolfson Childhood Cancer Research Center, Newcastle University Center for Cancer and 2Department of Pediatric Hematology and Oncology, Great North Children’s Hospital, Newcastle upon Tyne, UK
Correspondence: J. IRVING - Julie.Irving@newcastle.ac.uk https://doi.org/10.3324/haematol.2023.284335
Received: September 26, 2023.
Accepted: February 14, 2024. Early view: February 22, 2024.
1. Masic D, Fee K, Bell H, et al. Hyperactive CREB subpopulations increase during therapy in pediatric B-lineage acute lymphoblastic leukemia. Haematologica. 2023;108(4):981-992.
2. Gocho Y, Liu J, Hu J, et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat Cancer. 2021;2(3):284-299.
3. Shi Y, Beckett MC, Blair HJ, et al. Phase II-like murine trial identifies synergy between dexamethasone and dasatinib in T-cell acute lymphoblastic leukemia. Haematologica. 2021;106(4):1056-1066.
4. Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci. 2019;40(2):128-141.
5. Ebinger S, Ozdemir EZ, Ziegenhain C, et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell. 2016;30(6):849-862.
6. Sapio L, Salzillo A, Ragone A, Illiano M, Spina A, Naviglio S.
©2024 Ferrata Storti Foundation
Published under a CC BY license

Disclosures
JAEI has received funding from Hoffmann-La Roche Ltd for work unrelated to this manuscript. All other authors have no conflicts of interest to disclose.
Contributions
DM and HLB performed research. JAEI, DM and HLB designed the research study. DM, JAEI, HLB and FWvD analyzed the data. DM and JAEI wrote the first draft of the paper and all authors critically reviewed and approved the final draft.
Acknowledgments
The authors are indebted to Andy Filby and David McDonald from the Newcastle Flow Facility for their expertise in mass cytometric analyses.
Funding
The authors would like to gratefully acknowledge Children with Cancer UK, Newcastle Hospitals NHS Foundation Trust and JGW Paterson for funding this study.
Data-sharing statement
Data is available on request.
Targeting CREB in cancer therapy: a key candidate or one of many? An update. Cancers. 2020;12(11):3166.
7. van der Sligte NE, Kampen KR, ter Elst A, et al. Essential role for cyclic-AMP responsive element binding protein 1 (CREB) in the survival of acute lymphoblastic leukemia. Oncotarget. 2015;6(17):14970-14981.
8. Xie F, Li BX, Kassenbrock A, et al. Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J Med Chem. 2015;58(12):5075-5087.
9 Li BBX, Gardner R, Xue CH, et al. Systemic Inhibition of CREB is Well-tolerated in vivo. Sci Rep. 2016;6:34513.
10. Irving J, Jesson J, Virgo P, et al. Establishment and validation of a standard protocol for the detection of minimal residual disease in B lineage childhood acute lymphoblastic leukemia by flow cytometry in a multi-center setting. Haematologica. 2009;94(6):870-874.
A combination of a calcineurin-inhibitor (CNI) with mycophenolate mofetil (MMF) or methotrexate (MTX) is commonly used as graft-versus-host disease (GvHD) prophylaxis for HLA-matched donor hematopoietic cell transplantation (HCT). However, the use of MMF, which targets both T- and B-lymphocytes, is associated with a higher risk of cytomegalovirus (CMV) reactivation as compared to MTX.1 Moreover, CMV reactivation is associated with an increased risk of non-relapse mortality (NRM) and worse overall survival (OS).2 We, therefore, postulated that the use of MMF as compared to MTX may increase these risks, especially in CMV-seropositive recipients. To investigate this hypothesis, we compared the HCT outcomes of four groups: MTX/ CMV– (n=916), MTX/CMV+ (n=1,527), MMF/CMV– (n=267), and MMF/CMV+ (n=395).
We used an existing Center for International Blood and Marrow Transplant Research (CIBMTR) publicly available dataset3 from a previous publication.4 Our study population included patients ≥18 years with acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), or myelodysplastic neoplasm (MDS) who underwent HCT with peripheral blood grafts from 10/10 HLA-matched unrelated donors between 2008 and 2017. All patients received tacrolimus with either MMF or MTX. Patients who received ex-vivo T-cell-depleted/CD34+ cell-selected grafts or post-transplant cyclophosphamide (PTCy) were excluded. Our primary outcome of interest was OS. Secondary outcomes included grades III-IV acute GvHD, chronic GvHD, relapse, and NRM. The Kaplan-Meier method was used to estimate OS probability. The cumulative incidence method accounting for competing risks was used to estimate the incidence of GvHD, NRM, and relapse. Competing risks considered were death or disease relapse for GvHD; disease relapse or relapse-related deaths for NRM; and death before relapse (NRM) for relapse. Predictors in univariate and multivariable analyses were evaluated using Cox proportional hazards regression for OS, and Fine-Gray regression accounting for competing risks for GvHD, NRM, and relapse. Factors significant at the 5% level (P≤0.05) were retained in the final model, except the main effect (GvHD prophylaxis/ recipient CMV serostatus), which was retained in the final model irrespective of the level of statistical significance. Bonferroni-adjusted P values are reported for multivariate analyses for the main effect to account for multiple testing.
Interaction effects between the main effect and other statistically significant co-variables were tested and accounted for as indicated in multivariable regression analyses. All reported outcomes are at 3 years, except acute GvHD which is at day 180. The proportionality of hazards assumption was tested graphically and statistically. All statistical analyses were performed using STATA/IC 16.1 (StataCorp LLC, College Station, TX, USA). As the data analysis was carried out at The MD Anderson Cancer Center, the local Institutional Review Board approved this study (protocol: 2022-0684), which was conducted in accordance with the Declaration of Helsinki. The dataset is publicly available3 for data sharing, in accordance with CIBMTR guidelines. The study population’s baseline characteristics are shown in Table 1. In patients with AML (n=1,572), OS was significantly (P<0.001) inferior in CMV+ recipients who received MMF prophylaxis. Overall survival at 3 years was only 31% (95% confidence interval [95% CI]: 25-37) in the MMF/CMV+ group, compared with 54% (95% CI: 45-63) in the MMF/ CMV– group, 51% (95% CI: 48-55) in the MTX/CMV+ group, and 58% (95% CI: 53-63) in the MTX/CMV– group (Figure 1A). This effect persisted in multivariate analysis: compared to the overall mortality in the MTX/CMV– group, the risk was 1.8-fold higher in the MMF/CMV+ group, while it did not differ significantly in other groups (Table 2, Online Supplementary Table S1). Inferior OS in the MMF/CMV+ group was driven by a higher risk of NRM (Figure 1B). The risks of relapse (Figure 1C) and grade III-IV acute and chronic GvHD (Figure 1D) did not differ significantly between the groups.
In MDS patients (n=1,080), GvHD prophylaxis/recipient CMV serostatus had no significant association with OS, or the risk of relapse, but NRM varied by the Hematopoietic Cell Transplantation-specific Comorbidity Index (HCT-CI) score (Table 2, Online Supplementary Figure S1A). Among those with an HCT-CI score ≥3, the MTX/CMV+ group had a higher risk of NRM than had the MTX/CMV– group, while no statistically significant differences were seen in patients with an HCT-CI score 0-2. The risk of chronic GvHD was higher in the MMF/CMV– group than in the MTX groups.
In ALL patients (n=453), GvHD prophylaxis/recipient CMV serostatus had no significant associations with OS, NRM, or relapse (Table 2, Online Supplementary Figure S1B). The risk of chronic GvHD was higher in the MMF/CMV– group than in the MTX groups.
There were no significant predictors of grade III-IV acute GvHD in any disease type. Next, we tested whether the use of PTCy prophylaxis altered these observations. We used a separate CIBMTR matched unrelated donor peripheral blood HCT cohort 5 of patients with AML (n=136), ALL (n=42) or MDS (n=64) who received PTCy/CNI/MMF prophylaxis ( Online Supplementary Table S2 ). In this population too, in multivariate analysis, CMV + recipients had a significantly worse OS
Table 1. Baseline characteristics.
Recipient age in years
(range)
N (%)
N
intensity, N (%)
vivo T-cell depletion, N (%)
N (%)
Disease status at HCT, N (%)
HCT-CI, N (%)
Donor age, N (%) >35
Donor CMV, N (%)
than CMV– recipients, but only if they had AML (hazard ratio [HR]=2.7, 95% CI: 1.1-6.4; P =0.03) and not if they had ALL (HR=0.4, 95% CI: 0.1-1.5; P =0.2), or MDS (HR=0.7, 95% CI: 0.3-1.5; P =0.3). The risk of relapse and NRM did not differ by CMV serostatus in any disease group, although these analyses were limited by the small number of events.
In summary, in the CNI study population of patients who all received tacrolimus for GvHD prophylaxis with either
in
MTX: methotrexate; CMV: cytomegalovirus; MMF: mycophenolate mofetil; AML: acute myeloid leukemia; ALL: acute lymphoblastic leukemia; MDS: myelodysplastic neoplasms; MAC: myeloablative conditioning; RIC: reduced-intensity conditioning; NMA: non-myeloablative conditioning; HCT: hematopoietic cell transplantation; HCT-CI: Hematopoietic Cell Transplantation-specific Comorbidity Index; IQR: interquartile range.
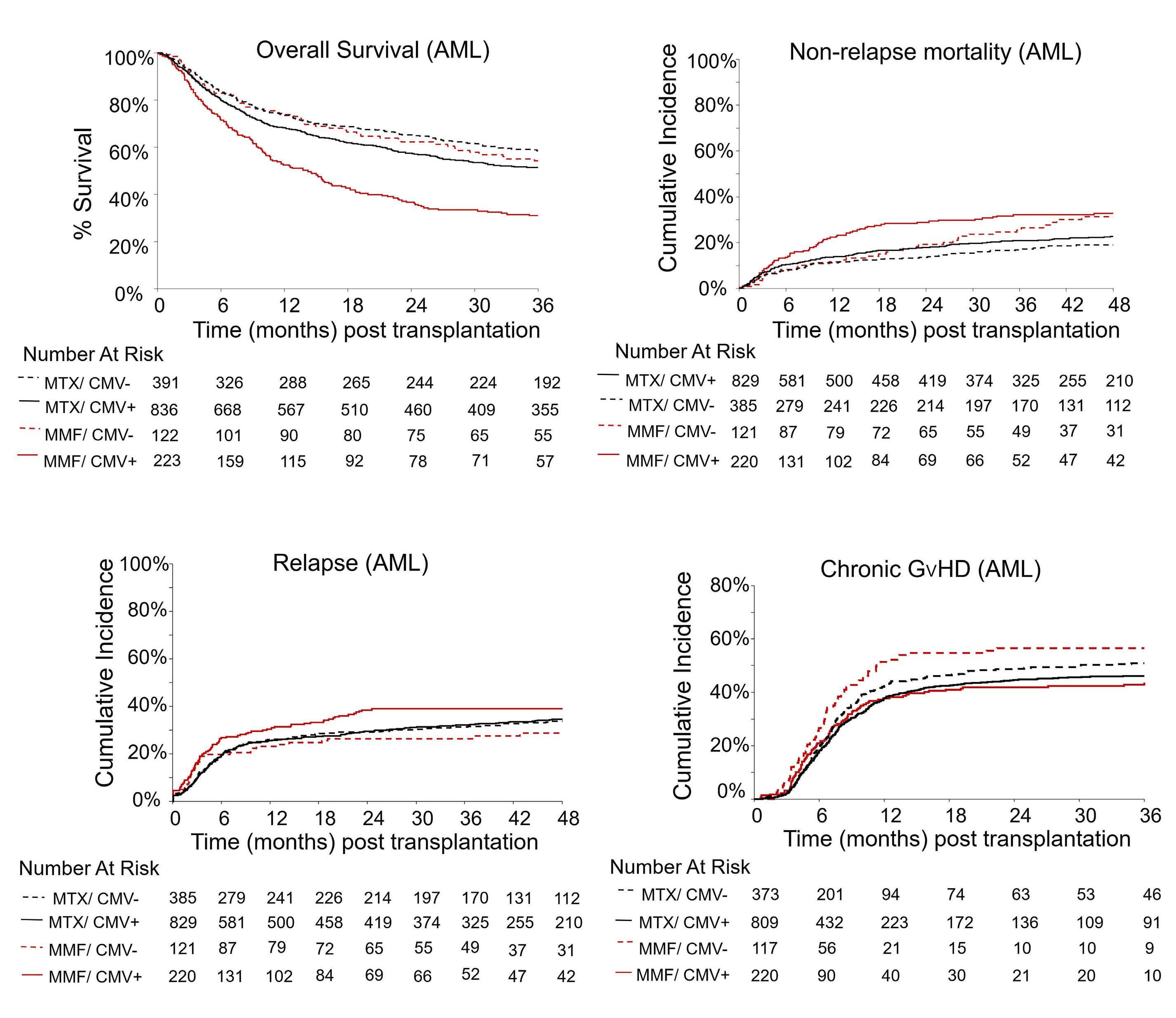
Figure 1. Outcomes of patients according to their cytomegalovirus status and whether they received methotrexate or mycophenolate mofetil for graft-versus-host disease prophylaxis. Among patients with acute myeloid leukemia, four groups were studied: patients given methotrexate (MTX) who were cytomegalovirus (CMV) seronegative (MTX/CMV–, dotted black), patients given MTX who were CMV seropositive (MTX/CMV+, solid black), patients given mycophenolate mofetil (MMF) who were CMV seronegative (MMF/CMV–, dotted maroon) and those given MMF who were CMV seropositive (MMF/CMV+, solid maroon). Outcomes include (A) overall survival, (B) non-relapse mortality, (C) relapse, and (D) chronic graft-versus-host disease. AML: acute myeloid leukemia; GvHD: graft-versus-host disease.
MMF or MTX and underwent matched unrelated donor HCT with peripheral blood grafts, we showed that MMF was associated with a significantly higher risk of NRM and worse OS in CMV + individuals but only in AML patients, and not in those with MDS or ALL. The underlying mechanism of this observation is unclear, but one hypothesis is as follows. It is known that CMV induces an exaggerated proliferation of natural killer cells, γ /δ T cells, and cytotoxic T cells.6,7 As MMF inhibits both B- and T-lymphocytes,1 it is associated with broader immunosuppressive effects than MTX, and CMV-induced lymphocytic expansion 8,9 is
suppressed by MMF more than by MTX.10 Moreover, MMF is administered for an extended period after HCT, while MTX is given for a short course. It is, therefore, conceivable that MMF may thwart CMV-driven expansion of both innate and adaptive immune cells, which may not only negate any potential protective effects of CMV on relapse but may also increase the risk of infections and NRM. Several studies have shown that CMV is associated with a reduced risk of relapse, but predominantly in AML patients 11-13 and not in those with lymphoid malignancies,11,13,14 or even MDS.12,13 The additional inhibition
*Full models are shown in Online Supplementary Table S1. There were no significant predictors of grade III-IV acute GvHD in any disease type. **In patients with a Hematopoietic Cell Transplantation-specific Comorbidity Index (HCT-CI) score ≥3. Among those with an HCT-CI 0-2, taking the MTX/CMV– group as the reference, the hazard ratios (with 95% confidence intervals) were 0.9 (0.6-1.3) for the MTX/CMV+ group (P=0.5), 0.9 (0.4-1.7) for the MMF/CMV– group (P=0.7), and 1.6 (0.9-2.6) for the MMF/CMV+ group (P=0.09). OS: overall survival; NRM: non-relapse mortality; GvHD: graft-versus-host disease; CI: confidence interval; HR: hazard ratio; AML: acute myeloid leukemia; ALL: acute lymphoblastic leukemia; MTX: methotrexate; CMV: cytomegalovirus; MMF: mycophenolate mofetil; MDS: myelodysplastic neoplasms.
of humoral and T-cell immunity in the presence of MMF would be predicted to promote CMV reactivation with its associated highly suppressive immunological imprinting and infectious sequelae.15
The effect of MMF when used with PTCy remains unknown. In our ad hoc analysis of the PTCy cohort, our findings were similar to those in the CNI cohort (not given PTCy), i.e., increased mortality in CMV + recipients with AML, but not in those with ALL or MDS. However, the independent effects of MMF and CMV on this association could not be determined as all patients received PTCy/CNI/MMF. These findings are hypothesis-generating and provide foundational data for further studies to assess the interaction between MMF and CMV serostatus in patients with AML, versus other hematologic malignancies, receiving PTCy-based prophylaxis.
Limitations of our study are the lack of data on CMV reactivation and causes of death. Because we lacked these data, we do not know the precise reason for the worse OS in the MMF/CMV + group. Of note, almost all patients in the CNI cohort and a majority in the PTCy cohort underwent HCT in the era before letermovir was approved by the US Food and Drug Administration. Whether or not the use of letermovir alters these conclusions also remains to be investigated.
In conclusion, our data suggest that the use of MMF with CNI prophylaxis should be avoided in CMV-seropositive AML patients undergoing matched unrelated donor HCT. Further studies are needed to assess the interaction
between MMF and CMV serostatus when PTCy is used for GvHD prophylaxis, and in the setting of letermovir prophylaxis.
Rima M. Saliba,1 Stephanie J. Lee,2 Paul A. Carpenter,2,3 Geoffrey R. Hill,4,5 Catherine J. Lee,2 Amin Alousi,1 May Daher,1 George Chen,1 Richard E. Champlin,1 Katayoun Rezvani,1 Elizabeth J. Shpall1 and Rohtesh S. Mehta2
1Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX; 2Clinical Research Division, Fred Hutchinson Cancer Center, Seattle, WA; 3Division of Pediatric Hematology, Oncology, Bone Marrow Transplant & Cellular Therapy, Seattle Children’s Hospital, Seattle, WA; 4Translational Science and Therapeutics Division, Fred Hutchinson Cancer Center, Seattle, WA and 5Clinical Research Division, Fred Hutchinson Cancer Center, Houston, TX, USA
Correspondence: R.S. MEHTA - rmehta@fredhutch.org
https://doi.org/10.3324/haematol.2023.284501
Received: October 19, 2023.
Accepted: February 16, 2024. Early view: February 29, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
RMS performed the statistical analysis, interpreted data, and wrote the statistical section of the manuscript. SJL, PAC, JRH, CJL, AA, GC, MD, REC, KR, and EJS reviewed and interpreted the data, reviewed the manuscript, and provided critical feedback. RSM and RMS conceptualized the study design and interpreted data. RSM wrote the manuscript with the help of RMS. RMS and RSM had full access to the raw data which is publicly available. All authors approved the manuscript. The corresponding author had the final responsibility for submitting the manuscript for publication.
1. Ueda Oshima M, Xie H, Zamora D, et al. Impact of GVHD prophylaxis on CMV reactivation and disease after HLAmatched peripheral blood stem cell transplantation. Blood Adv. 2023;7(8):1394-1403.
2. Teira P, Battiwalla M, Ramanathan M, et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood. 2016;127(20):2427-2438.
3. CIBMTR. https://cibmtr.org/CIBMTR/Resources/PubliclyAvailable-Datasets#. Accessed 9/6/2023.
4 Arrieta-Bolanos E, Crivello P, He M, et al. A core group of structurally similar HLA-DPB1 alleles drives permissiveness after hematopoietic cell transplantation. Blood. 2022;140(6):659-663.
5. Gooptu M, Romee R, St Martin A, et al. HLA-haploidentical vs matched unrelated donor transplants with posttransplant cyclophosphamide-based prophylaxis. Blood. 2021;138(3):273-282.
6. Nachbaur D, Bonatti H, Oberaigner W, et al. Survival after bone marrow transplantation from cytomegalovirus seropositive sibling donors. Lancet. 2001;358(9288):1157-1159.
7 Elmaagacli AH, Koldehoff M. Cytomegalovirus replication reduces the relapse incidence in patients with acute myeloid leukemia. Blood. 2016;128(3):456-459.
8. Beziat V, Liu LL, Malmberg JA, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood. 2013;121(14):2678-2688.
Acknowledgments
We thank the Center for International Blood and Marrow Transplant Research (CIBMTR) staff for providing the dataset used in this study.
Funding
This dataset was collected by the CIBMTR which is supported primarily by the Public Health Service U24CA076518 from the National Cancer Institute; the National Heart, Lung, and Blood Institute; the National Institute of Allergy and Infectious Diseases; 75R60222C00011 from the Health Resources and Services Administration; N00014-21-1-2954 and N00014-23-1-2057 from the Office of Naval Research; the National Marrow Donor Program/Be The Match; and the Medical College of Wisconsin.
Data-sharing statement
The data are publicly available and accessible at https://cibmtr.org/ CIBMTR/Resources/Publicly-Available-Datasets.
9 Foley B, Cooley S, Verneris MR, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood. 2012;119(11):2665-2674.
10 Ohata K, Espinoza JL, Lu X, Kondo Y, Nakao S. Mycophenolic acid inhibits natural killer cell proliferation and cytotoxic function: a possible disadvantage of including mycophenolate mofetil in the graft-versus-host disease prophylaxis regimen. Biol Blood Marrow Transplant. 2011;17(2):205-213.
11. Ruggeri L, Capanni M, Casucci M, et al. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94(1):333-339.
12. Green ML, Leisenring WM, Xie H, et al. CMV reactivation after allogeneic HCT and relapse risk: evidence for early protection in acute myeloid leukemia. Blood. 2013;122(7):1316-1324.
13. Manjappa S, Bhamidipati PK, Stokerl-Goldstein KE, et al. Protective effect of cytomegalovirus reactivation on relapse after allogeneic hematopoietic cell transplantation in acute myeloid leukemia patients is influenced by conditioning regimen. Biol Blood Marrow Transplant. 2014;20(1):46-52.
14 Mariotti J, Maura F, Spina F, et al. Impact of cytomegalovirus replication and cytomegalovirus serostatus on the outcome of patients with B cell lymphoma after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2014;20(6):885-890.
15. Degli-Esposti MA, Hill GR. Immune control of cytomegalovirus reactivation in stem cell transplantation. Blood. 2022;139(9):1277-1288.
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non-Hodgkin lymphoma and is characterized by high clinicopathologic heterogeneity.1 This has fueled several efforts at subclassification aiming to identify prognostic and clinically relevant subgroups. Currently, the most widely used classification is based on gene expression profiles, stratifying DLBCL according to the cell-of-origin (COO) into activated B-cell (ABC) and germinal center B-cell (GCB) subtypes.2-4 Patients with ABC-DLBCL have been reported to have less favorable responses to standard therapy than those with GCB-DLBCL. Unfortunately, clinical studies based on this classification have repeatedly failed to improve the outcome of DLBCL patients, suggesting that this simplified scheme is not sufficiently effective for clinical trial design.5 To address this problem, several recent studies have utilized high-resolution genomic analysis to subcategorize patients based on genetic alteration profiles.6-8 This approach has uncovered broader biological heterogeneity than the ABC/GCB paradigm. In a recent National Cancer Institute (NCI) study, observations have been applied to develop a probabilistic classification algorithm, LymphGen, that enables the subclassification of DLBCL into 6 molecularly distinct groups: MCD, BN2, EZB, ST2, A53, and N1, with prognostic correlations.9,10 Biomolecular clusters defined by other studies have overlapped with those of the NCI study.11 However, all large-scale studies have primarily been performed using data derived by whole genome, whole exome sequencing, or a large comprehensive target next-generation sequencing (NGS) panel carried out as part of wider research endeavors. In contrast, in routine clinical practice, sequencing is necessarily targeted to limited sets of genes with a key role for immediate clinical management. In this study, therefore, we aimed to apply the published LymphGen algorithm to a validated clinical NGS assay: the Memorial Sloan Kettering Cancer Center’s (MSKCC) Integrated Mutation Profiling of Actionable Cancer Targets for Hematologic malignancies NGS panel (MSK-IMPACT HEME, herein referred to as IMPACT), which is currently utilized in the management of patients with DLBCL in the clinical setting. IMPACT is a clinical validated hybridization capture-based assay designed to detect genetic alterations, including single nucleotide variants (SNV), small insertions and deletions (INDEL), and copy number alterations (CNA), implicated in the oncogenesis of hematopoietic malignancies (Online Supplementary Figure S1). The custom designed DNA probes target all protein-coding exons and the ad-
jacent 20 bp of intronic sequence of 400 key oncogenes and tumor suppressor genes. IMPACT uses either saliva or nail clippings as a source of germline DNA to confidently identify somatic mutations in hematologic tumor cells. Details of the methodology and analytical code have been published previously.12,13 Published data from the NCI DLBCL cohort was used to evaluate the performance of LymphGen with only the genes in the IMPACT panel. Applying the LymphGen cluster allocation to the limited set of 400 genes on cases from the NCI cohort by filtering the NCI variant calling data into genes on the IMPACT panel demonstrated an overall accuracy of 92%, with 86% sensitivity and 98% specificity, using cluster allocation based on the original panel as ground truth (Figure 1A, B). In total, 58% of cases were ultimately classified into one of the 6 subtypes (or composite subtypes), which was only marginally below the 63% reported with the use of comprehensive methods, including whole exon, deep amplicon and RNA-sequencing in the original NCI report. The cases that were successfully classified included 11.7% MCD type, 12.7% BN2 type, 13.7% EZB type, 9.2% A53 type, 5.3% ST2 type, and 0.8% N1 type, compared to the original NCI LymphGen study which included 13.9% MCD type, 16.1% BN2 type, 13.2% EZB type, 6.6% A53 type, 4.7% ST2 type, and 2.8% N1 type (Figure 1C). Misclassification primarily encompassed cases deemed as unclassified (“Other”) by either the comprehensive NCI panel or the IMPACT panel, as shown in the confusion matrix (Figure 1A). Similar to the NCI study, most cases of EZB subtypes were of the GCB COO; most cases of MCD and N1 subtypes were of ABC COO; and BN2 and A53 subtypes carried both GCB and ABC types, while cases in the ST2 subtype contained more GCB type than ABC type (Figure 1D). In our classification using the genes on the targeted IMPACT panel approach, within 58% of classified cases, 34% of cases were called with high confidence (“Core” group, >90% probability), while 19% of cases were called with lower confidence (“Extended” group, 50-90% probability) (Figure 1C). Compared to the original NCI allocation (47% “Core” and 10% “Extended” group cases), there is a slight reduction in calling confidence from the narrower IMPACT panel. The percentage of cases being classified in the “Core” group varies in subtypes, with the highest in N1 subtype and the lowest in ST2, which represents the confidence of classification in these subtypes (Figure 1E). In the “Core” group, classification accuracy by the IMPACT gene panel increased to 96%; sensitivity and specificity
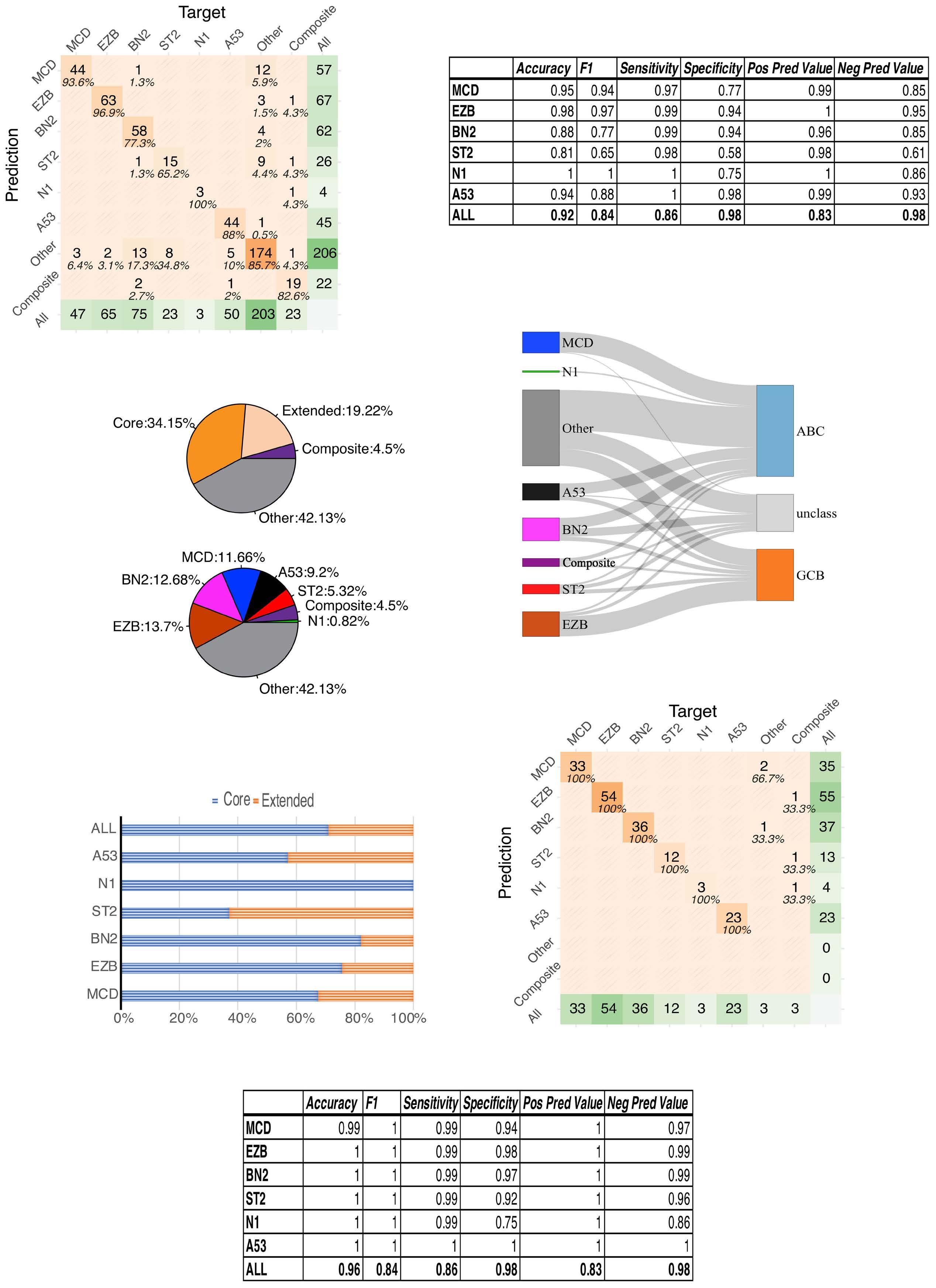
Figure 1. LymphGen classification on National Cancer Institute cohort data filtered by IMPACT panel. (A) Confusion matrix of IMPACT panel compared to National Cancer Institute (NCI) panel (all cases). The target represents the classification by NCI panel; the prediction represents the classification by IMPACT panel. The center number shows the number of cases; the percentage
Continued on following page.
represents the proportion of cases classified by the IMPACT panel for a certain subtype in the cases classified by the NCI panel for the same subtype (considered as ground truth in this comparison). (B) Performance of the classification. (C) Cases from the NCI cohort filtered into the IMPACT panel were classified by LymphGen into 6 subclasses (MCD, EZB, BN2, N1, ST2, A53). The cases with more than one assigned subclass are indicated as “Composite”. The cases that cannot be classified into these subclasses are labeled as “Other”. Regarding categories of the subclassification: tumors with subtype probabilities of >90% or 50-90% were defined as ‘‘Core’’ or ‘‘Extended’’ subtype members, respectively. (D) Correlation of LymphGen classification and cell-of-origin by Han’s algorithm. (E) Composition of “Core” and “Extended” group in each subtypes. (F) Confusion matrix of the IMPACT panel compared to the NCI panel for “Core” predicted group only. The target represents the classification by the NCI panel; the prediction represents the classification by the IMPACT panel. The center number shows the number of cases; the percentage represents the proportion of cases classified by IMPACT for a certain subtype in the cases classified by the NCI panel for the same subtype (considered as ground truth in this comparison). (G) Performance of the IMPACT panel compared to the NCI panel within the “Core” group. ABC: activated B-cell subtype; GCB: germinal center B-cell subtype.
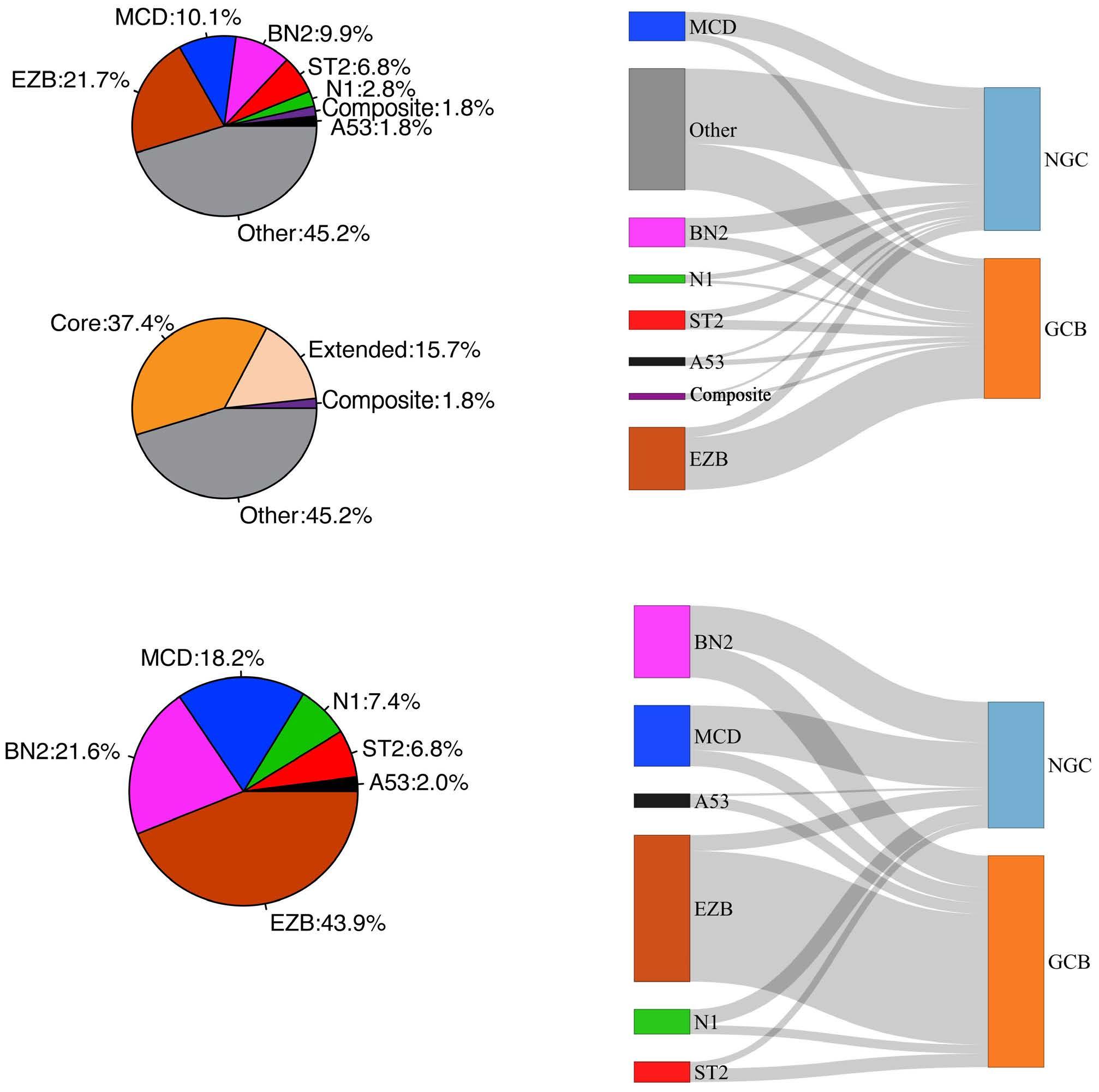
were 86% and 98%, respectively. These results suggested that we could confidently classify cases into subtypes if defined by the “Core” (Figure 1F, G).
Copy number alteration was included in LymphGen input data to use the full algorithm. However, in the clinical setting, the copy number calling accuracy might be compromised by poor sample quality or low tumor content. To explore the accuracy of classification using the IMPACT gene panel by LymphGen when CNA is not available, we applied only SNV, small INDEL, and fluorescence in situ hybridization (FISH) for BCL2 and BCL6 translocation in-
Figure 2. LymphGen classification on the Memorial Sloan Kettering cohort. (A) 396 diffuse large B-cell lymphoma cases sequenced by IMPACT were classified by LymphGen into 6 subclasses: MCD, EZB, BN2, N1, ST2, A53. Cases with more than one assigned subclass are indicated as “Composite”. Cases that cannot be classified into these subclasses are labeled as “Other”. (B) Correlation of LymphGen classification and cell-of-origin (COO) by Han’s algorithm. (C) Categories of the subclassification: tumors with subtype probabilities of >90% or 50-90% were defined as ‘‘Core’’ or ‘‘Extended’’ subtype members, respectively. (D) Composition of the subtypes in the cases from the “Core” group (classified based on probability >90%). (E) Correlation of LymphGen classification and COO by Han’s algorithm in the cases from the “Core” group.
formation to the algorithm. The overall balanced accuracy was 81%, with 83% sensitivity and 89% specificity overall (Online Supplementary Figure S2A ). The A53 subtype was omitted as expected, because the classification of the A53 subtype mainly relied on CNA. Twenty-three cases (4.7%) were misclassified into the “Other” (unclassified) group, suggesting that the lack of CNA reduces the algorithm’s ability to classify cases into subclasses (Online Supplementary Figure S2B ). These results showed that the optimized CNA information is essential for classifying the A53 subtype and further helps increase the overall
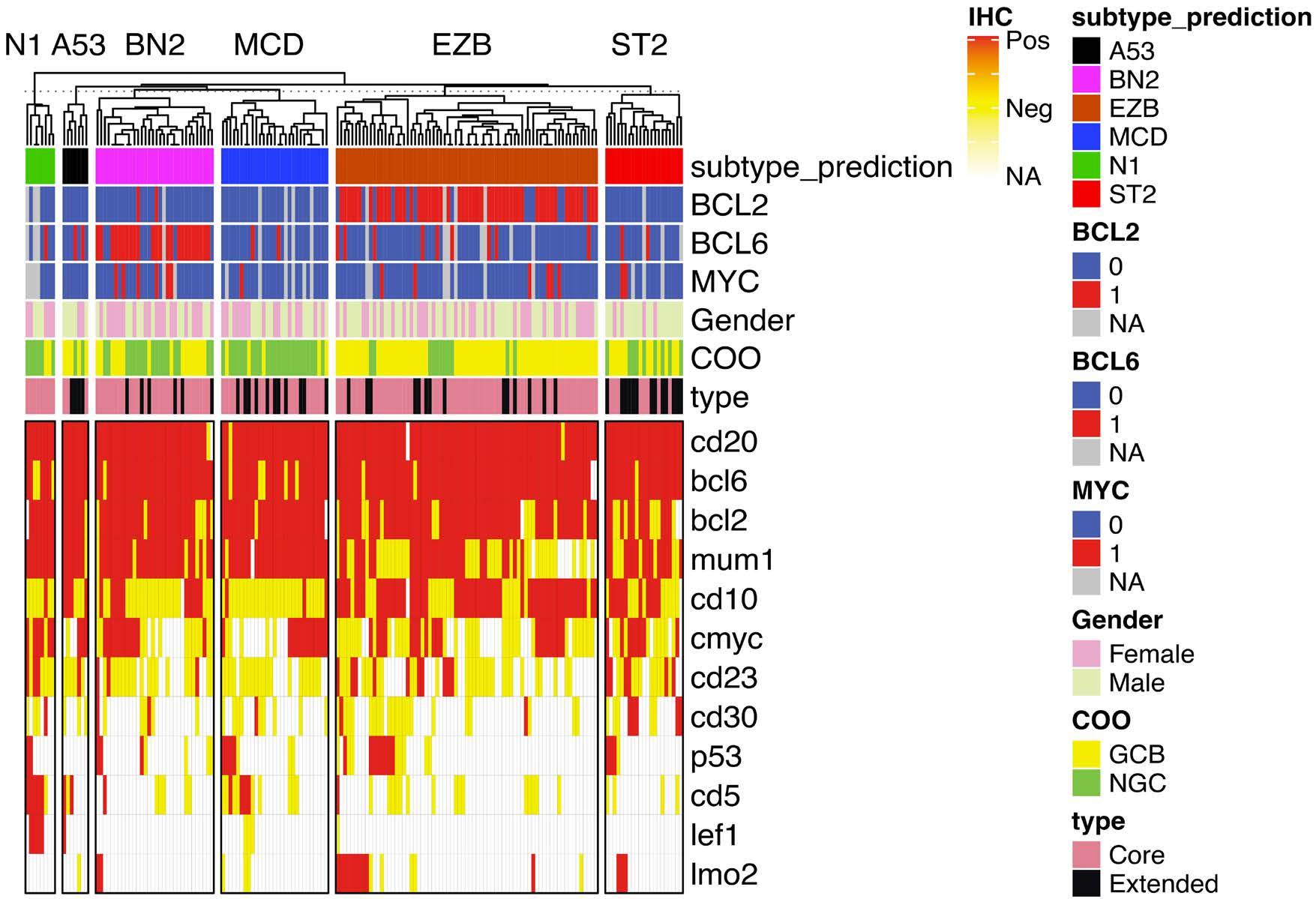
accuracy of the classification method. We then applied LymphGen to the MSK DLBCL clinical cohort of 396 cases (320 cases of the cohort have both BCL2 and BCL6 clinical cytogenetic results available) sequenced by IMPACT (data available at https://www.cbioportal.org/ study/summary?id=mbn_msk_2024). When the LymphGen algorithm was applied to the MSK DLBCL cohort, 55% of the cases were assigned into one of the subtypes (MCD: 10%, EZB: 22%, BN2: 10%, N1: 3%, ST2: 7%, A53: 2%, composite: 2%) (Figure 2A). Within these cases, about two-thirds were in the “Core” group, and one-third were in the “Extended” group (Figure 2C). The correlation of genetic subtypes and COO based on Hans’ algorithm was similar to the NCI cohort. Most EZB subtypes were GCB type, while most MCD and N1 subtypes were non-GCB type; BN2, ST2, and A53 subtypes carry a mixture of GCB and non-GCB types (Figure 3B). We further investigated the composition of each subclass and its relationship with COO classification in the “Core” group; 148 (37.4% of total cases) cases classified as “Core” group were assigned into one of the 6 classes (MCD: 18.2%, EZB: 44%, BN2: 22%, N1: 7%, ST2: 7%, A53: 2%). The correlation of genetics subtypes and COO was similar to that observed in all cases. (Figure 2D, E). Essential genes in the IMPACT panel which significantly distinguish each subtype were identified by Benjamini-Hochberg procedure and χ2 test (Online Supplementary Figure S3A).
In the MSK DLBCL cohort, cytogenetics and immunophenotypes observed in each genetic subtype were similar to those identified in the NCI cohort. As expected, BCL2 translocations were enriched in the EZB subtype (54 out of 67 cases, 81%), and BCL6 translocations were enriched in the BN2 subtype (24 out of 31 cases, 77%). MYC translocations were seen more frequently (6 out of 39 cases, 15.3%) in BN2 subtypes when co-existing with BCL6 translocation. MYC translocations were also seen in a small proportion of EZB
Figure 3. Heatmap of immunophenotype and cytogenetic features in subtypes. All cases classified into one of the 6 classes are clustered by the subclass. The immunohistochemistry status of each maker is shown in a color block (red: positive; yellow: negative; white: not available / not performed). Fluorescence in situ hybridization results for BCL2, BCL6, and MYC translocations are shown in the color block as indicated here. Cell-of-origin (COO) and classification type (“Core” vs. “Extended”) are also annotated by color block.
(7 out of 86 cases, 8.1%), MCD (2 out of 40 cases, 5.0%), and ST2 (2 out of 27 cases, 7.4%) subtypes. Immunophenotypes were evaluated by immunohistochemical stains in all cases as part of the diagnostic work-up. Consistent with the germinal center phenotype, CD10, CD23, and LMO2 were more frequently found expressed in the EZB and ST2 subtypes, while MUM1 was seen more frequently in the BN2, MCD, N1, and A53 subtypes. Interestingly, 50% (4 out of 8 cases) of N1 subtypes expressed CD5 and LEF1 by immunohistochemistry, indicating that some of the N1 subtype cases might represent Richter’s transformation of chronic lymphocytic leukemia. Only 2 cases with BCL2 translocation fell in the unclassified group, suggesting that BCL2 translocation is a robust classifier for the EZB subtype (Figure 3A). Furthermore, survival analyses within the MSK cohort have demonstrated trends consistent with those observed in other studies. However, statistical significance was not achieved, likely due to the inherent heterogeneity of the cohort and the relatively short duration of follow-up (data not shown).
In conclusion, in this study, we validated genetic classification for DLBCL by LymphGen on our clinically validated 400-gene targeted NGS panel, IMPACT, with high sensitivity and specificity and showed that the classification can be applied to routine clinical practice. To note, in our practice, over 99% of DLBCL cases were successfully sequenced, thanks to detailed pre-analytical checks that identified and excluded inadequate samples. Success rates, however, may differ with other workflows. The findings also indicate the accuracy of the classification can be further enhanced if detection of CNA can be improved. Our study supports the view that genomic classifications of DLBCL using high complexity testing such as LymphGen can be readily translated into routine clinical care using well-designed smaller targeted NGS panels such as IMPACT. Such panels offer opportunities for clinical decision-making
and selection of patients for risk-adapted and biomarker selected clinical trials.
Meng-Lei Zhu,1 Esther Drill,2 Erel Joffe,3 Gilles Salles,3 Alfredo Rivas Delgado,3 Andrew Zelenetz,3 Maria Lia Palomba,3 Maria Arcila1# and Ahmet Dogan1#
1Department of Pathology and Laboratory Medicine; 2Department of Epidemiology & Biostatistics and 3Lymphoma Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, USA
#MA and AD contributed equally as senior authors.
Correspondence:
M.L. ZHU - zhum1@mskcc.org https://doi.org/10.3324/haematol.2023.284565
Received: October 31, 2023.
Accepted: March 1, 2024.
Early view: March 14, 2024.
1. Ennishi D, Hsi ED, Steidl C, Scott DW. Toward a new molecular taxonomy of diffuse large B-cell lymphoma. Cancer Discov. 2020;10(9):1267-1281.
2. Boltezar L, Prevodnik VK, Perme MP, Gasljevic G, Novakovic BJ. Comparison of the algorithms classifying the ABC and GCB subtypes in diffuse large B-cell lymphoma. Oncol Lett. 2018;15(5):6903-6912.
3. Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275-282.
4 Rosenwald A, Wright G, Chan WC, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(25):1937-1947.
5. Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125(1):22-32.
6. Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396-1407.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
MZ designed the research, analyzed and interpreted the data, wrote and revised the manuscript. ED and EJ, GS, ARD analyzed and interpreted the data, and edited the manuscript. AZ and MLP collected the study cohort and edited the manuscript. MA and AD designed the research, analyzed and interpreted the data, edited the manuscript, and supervised the project.
Funding
We thank the MSK Cancer Center for their support (Grant/Core Grant: P30 CA008748). ARD was funded by a grant from Fundación Española de Hematología y Hemoterapia.
Data-sharing statement
NGS sequencing data of this study is available on https://www. cbioportal.org
7 Chapuy B, Stewart C, Dunford AJ, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679-690.
8. Lacy SE, Barrans SL, Beer PA, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood. 2020;135(20):1759-1771.
9. Runge HFP, Lacy S, Barrans S, et al. Application of the LymphGen classification tool to 928 clinically and geneticallycharacterised cases of diffuse large B cell lymphoma (DLBCL). Br J Haematol. 2021;192(1):216-220.
10 Wright GW, Huang DW, Phelan JD, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37(4):551-568.
11. Morin RD, Arthur SE, Hodson DJ. Molecular profiling in diffuse large B-cell lymphoma: why so many types of subtypes? Br J Haematol. 2022;196(4):814-829.
12. Ptashkin RN, Ewalt MD, Jayakumaran G, et al. Enhanced clinical assessment of hematologic malignancies through routine paired tumor and normal sequencing. Nat Commun. 2023;14(1):6895.
13. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703-713.
Relapsed or refractory multiple myeloma (RRMM) is associated with severe symptoms, some of which have been strongly linked to impairments in health-related quality of life (HRQoL), notably pain, fatigue, and a decline in physical and emotional functioning.1 Furthermore, HRQoL deteriorates with each subsequent line of therapy in RRMM.2 Hence, treatment goals, particularly in later lines of therapy, should include preserving HRQoL. Melphalan flufenamide (melflufen) is a first-in-class peptide-drug conjugate that utilizes increased peptidase expression to selectively release potent alkylating agents inside tumor cells. Melflufen is approved in Europe for the treatment of patients with triple-class refractory RRMM with ≥3 prior lines of therapy and time to progression (TTP) >36 months after prior autologous stem cell transplant (ASCT), if received. Approval was based on the results of the phase II HORIZON study and further supported by those of the phase III OCEAN study.3-5 OCEAN met its primary endpoint with melflufen plus dexamethasone demonstrating superior progression-free survival compared with pomalidomide plus dexamethasone in RRMM.4 Across trials, the safety profile of melflufen plus dexamethasone has been characterized primarily by hematologic adverse events that are clinically manageable, with infrequent grade 3/4 non-hematologic adverse events.3,4,6 HRQoL over time was preserved with melflufen plus dexamethasone in patients with advanced RRMM in the HORIZON trial..7 Pomalidomide plus dexamethasone has also been shown to be safe and effective without negatively affecting HRQoL, including in later lines of therapy.8 In this letter, we report HRQoL based on patient-reported outcomes (PRO) in a subset of patients from OCEAN receiving either melflufen plus dexamethasone or pomalidomide plus dexamethasone. Overall, melflufen plus dexamethasone treatment resulted in HRQoL comparable to that of pomalidomide plus dexamethasone, further supporting the use of melflufen plus dexamethasone in heavily pretreated patients with RRMM.
The OCEAN study design has been previously published.4 The study was conducted in compliance with the ethical principles set forth in the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice Guidelines and was approved by national regulatory authorities and independent ethics committees/ review boards. All patients provided written consent to participate. PRO assessments were added as an exploratory endpoint on May 24, 2019 (protocol v4.1), approximately 2 years after the study started.4 Only patients enrolled on/
after protocol v4.1 and who completed ≥1 PRO questionnaires were included in this analysis.. PRO questionnaires were administered before dosing at baseline and day 1 of each treatment cycle, at the end-of-treatment visit, disease progression, and the start of a new treatment. Three PRO assessments were used: the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-Core 30 (EORTC QLQ-C30) version 3, scored from 0-100, with higher scores indicating better functional status/ QoL and functional scales, and an increase in severity of symptoms for the symptom scales;9,10 the EORTC Quality of Life Questionnaire-Multiple Myeloma module (EORTC MY20), scored from 0-100 with higher scores indicating greater symptom severity;10 and the European Quality of Life 5 Dimensions 3-Level (EQ-5D-3L) questionnaire that evaluates generic health status, also with scores ranging from 0-100 (death=0; perfect health=100) with higher scores indicating a better health state.11 PRO results were compared between the treatment groups (melflufen and pomalidomide), and also within the melflufen group – between patients with TTP >36 months after prior ASCT or without prior ASCT (target population) and those with TTP <36 months after ASCT (non-target population). Mean scores at baseline, at each treatment cycle up to cycle 6, and mean change from baseline through cycle 6 were analyzed. Responder analysis was conducted, defined as the proportion of patients with improved, stable, or worsening EORTC QLQ-C30 and MY20 scores at cycle 6. We used a cutoff of a 10-point change for improvement or worsening scores, since this is the minimal important difference resulting in clinical benefit.12
Of the 495 patients enrolled and randomized in OCEAN, 158 with PRO assessments were included in the pre-specified PRO analysis (melflufen group, n=77; pomalidomide group, n=81). Forty-four patients in the melflufen group had not received previous ASCT or had TTP >36 months after previous ASCT, and 33 had TTP <36 months after previous ASCT. Baseline characteristics were generally well matched between treatment groups for patients with PRO data. Both treatment groups showed similar PRO scores at baseline with respect to general health and well-being, as measured by the EORTC QLQ-C30 multi-item scales, including global health status/QoL and physical and emotional functioning; symptom burden, as measured by the EORTC QLQ-C30 fatigue and pain scales; EORTC MY20 disease symptom scale and side effects of treatment score; and “health today,” as measured by the EQ-5D-3L visual analog scale (VAS) score (Table 1). Importantly, mean
PRO scores remained generally consistent with baseline ones through cycle 6 in both treatment groups (Figure 1). Consistent with this, mean change from baseline in PRO scores through cycle 6 was also largely unchanged (Online Supplementary Figure S1).
Overall, there was no difference in the proportion of patients with improvements or stable or worsening PRO measures over time between treatment groups, as exemplified by a snapshot at cycle 6 (Figure 2). Most patients had improved or stable PRO measures at cycle 6. The proportions of patients with improved PRO measures at cycle 6 ranged from 20% to 39% in the melflufen group and 11% to 33% in the pomalidomide group, and the proportions with stable PRO measures ranged from 26% to
72% for melflufen and from 37% to 67% for pomalidomide.
In contrast, the proportion of patients with worsening PRO measures was generally low, particularly for physical functioning (melflufen: 13% and pomalidomide: 15%), emotional functioning (8% and 20%), disease symptoms (10% and 20%), and side effects of treatment (13% and 26%).
Because a TTP <36 months after previous ASCT has been shown to be associated with worse outcomes in patients treated with melflufen and dexamethasone,5 PRO assessments were compared between the target and non-target populations within the melflufen group.
Mean PRO scores at baseline with melflufen and dexamethasone were similar between the target and non-target populations and trended similar to those in the overall
Table 1. Baseline characteristics of patients with and without patient-reported outcomes assessments.a
EORTC QLQ-C30 multi-item scales, mean score (range)c
EORTC QLQ-C30 symptom scales, mean score (range)d
EORTC MY20 score, mean (range)e
aPatients from OCEAN enrolled after protocol v4.1 who did not complete the PRO questionnaire were excluded from the analysis. bBaseline characteristics in the PRO analysis population and in patients not included in the PRO analysis population were comparable, except for a higher proportion of patients in the PRO analysis population having an ECOG PS score of 0 (44% vs. 34%). cEORTC QLQ-C30 global health status/QoL, emotional functioning, and physical functioning scores range from 0 to 100; a higher score indicates better function.9 dEORTC QLQ-C30 fatigue and pain symptoms range from 0 to 100; a higher score indicates greater severity of symptoms.10 eEORTC MY20 disease symptoms and side effects of treatment scores range from 0 to 100; a higher score indicates greater severity of symptoms.10 fEQ-5D-3L VAS scores range from 0 to 100; a higher score indicates a better health state (0 equals death; 100 equals perfect health).11 PRO: patient-reported outcome; melflufen: melphalan flufenamide; ECOG PS: Eastern Cooperative Oncology Group performance status; EORTC: European Organization for Research and Treatment of Cancer; QLQ-C30: Core Quality of Life Questionnaire; QoL: quality of life; MY20: Multiple Myeloma Module; EQ-5D-3L: European Quality of Life 5 Dimensions 3 Level Version; VAS: visual analog scale.
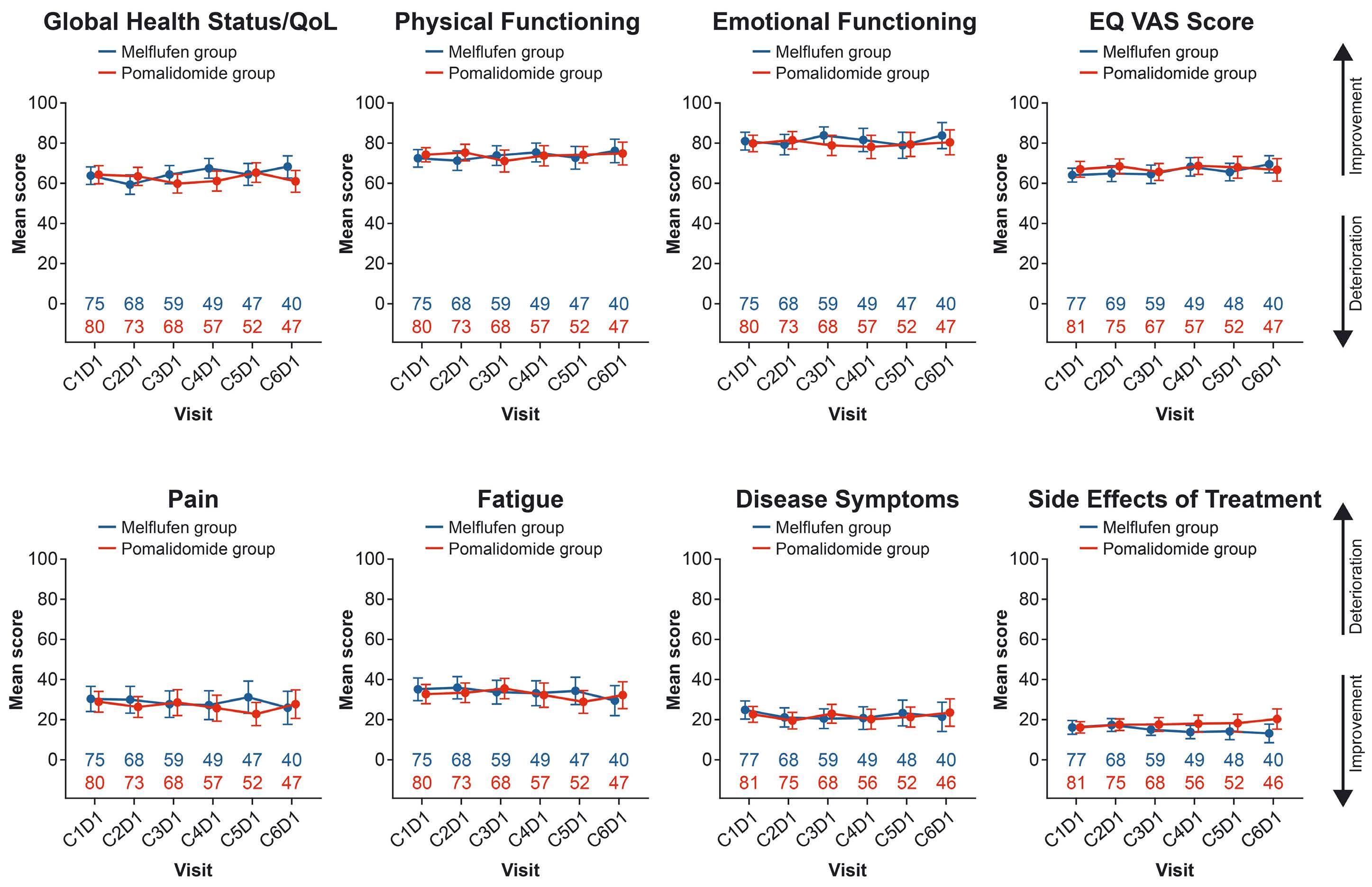
1. Mean scores at baseline through cycle 6 of treatment with melflufen plus dexamethasone or pomalidomide plus dexamethasone. C: cycle; D: day; dex: dexamethasone; melflufen: melphalan flufenamide; QoL: quality of life; VAS: visual analog scale.
population (Online Supplementary Table S1). Mean baseline global health status/QoL scores were 65.3 in the target population and 61.9 in the non-target population, physical functioning scores were 73.2 and 71.5, and emotional functioning scores were 83.3 and 78.0. Similarly, mean baseline scores for fatigue (30.7 and 40.7), pain (26.2 and 35.4), MM-specific disease symptoms (23.0 and 27.1), side effects of treatment (15.9 and 16.3), and EQ-5D-3L VAS scores (64.8 and 62.8) were similar between the target and non-target populations. Likewise, mean PRO scores remained generally consistent through cycle 6 of treatment with melflufen and dexamethasone between the target and non-target populations ( Online Supplementary Figure S2).
These results are consistent with the preserved HRQoL seen with melflufen plus dexamethasone in the HORIZON study.7 Although progression in <36 months after prior ASCT is a negative prognostic factor for overall survival,5 PRO measures of the target and non-target populations within the melflufen group were similar at baseline and remained consistent through cycle 6 of therapy. In patients with RRMM, HRQoL tends to decrease with each subsequent
line of therapy.2 Currently available therapeutic options for RRMM mostly maintain HRQoL but do not achieve clinically meaningful improvements.13 Comparisons across trials, however, are limited by differences in reporting of clinically meaningful improvements, and differences in patient populations and trial design. In the phase III APOLLO study of pomalidomide plus dexamethasone with or without daratumumab, HRQoL measured using the EORTC QLQ-C30 and EORTC QLQ-MY20 questionnaires remained generally consistent with baseline in both arms, although between-group differences did not favor the pomalidomide-dexamethasone arm for most QoL scales.14
In the phase II DREAMM-2 study, HRQoL measured by the EORTC QLQ-MY20 scale was maintained with long-term follow-up with belantamab mafodotin, with only up to 38% of evaluable patients (n=45) experiencing improvements in pain in different locations at week 7.15
Results from our analysis are limited by the fact that PRO assessments were collected only from patients who were randomized after protocol amendment 4.1.
Subgroup analyses for the target and non-target populations within the melflufen group were further limited by small pa-
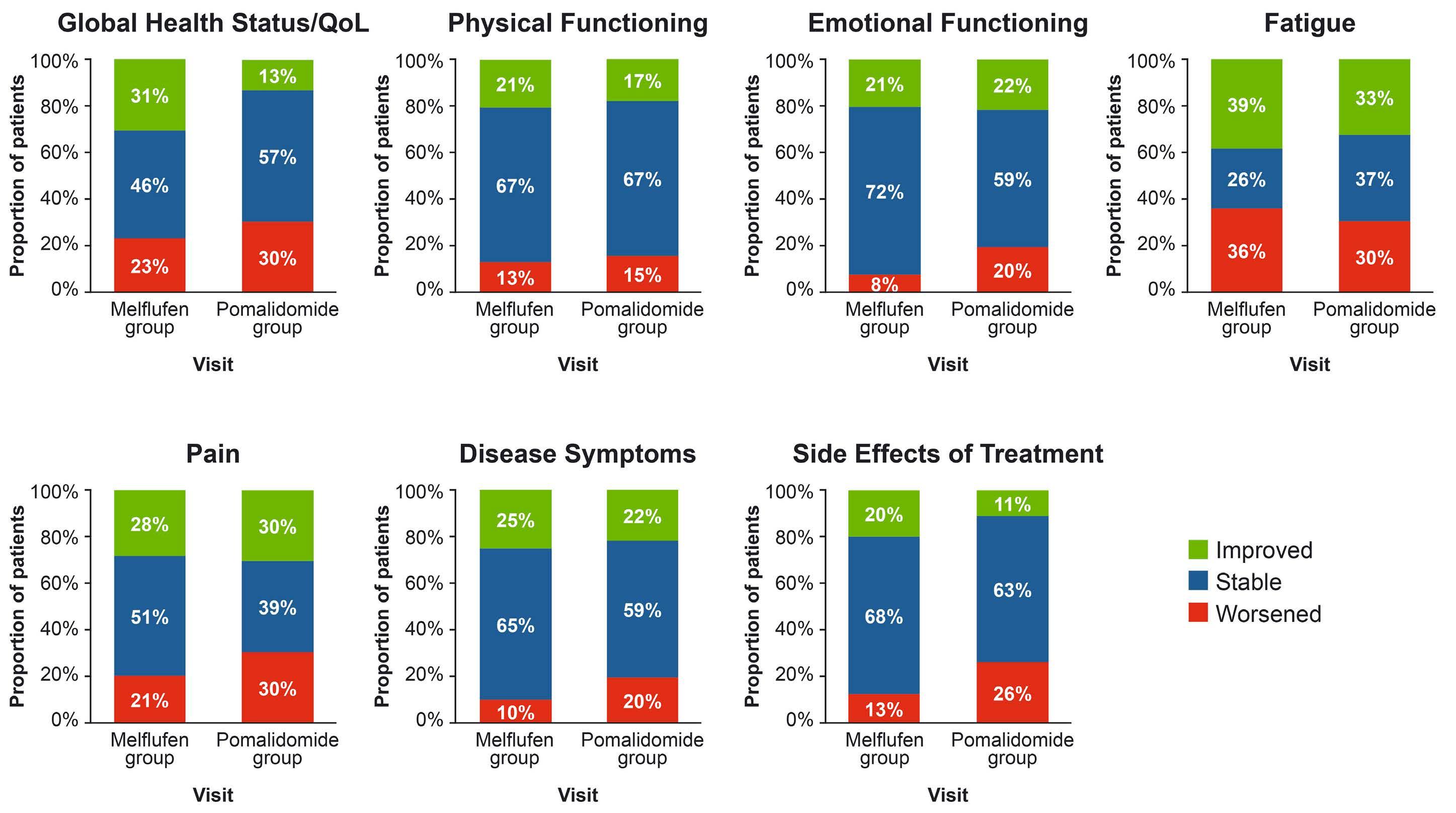
Figure 2. Proportion of patients with improved, stable, or worsened patient-reported outcome measures at cycle 6. An improvement or worsening of an outcome was defined as a change of ≥10 points. QoL: quality of life; melflufen: melphalan flufenamide.
tient numbers. Nevertheless, in both groups, more than half of the patients for whom PRO assessments were available were still on study at cycle 6. Although not a statistically powered comparison, 31% of patients receiving melflufen and 13% receiving pomalidomide showed improvement (≥10 points’ change) in global health status/QoL scores at cycle 6, whereas 46% of patients receiving melflufen and 57% receiving pomalidomide showed stable global health status/ QoL scores at cycle 6. These results from later timepoints likely suggest a continuous selection of patients benefiting from therapy and being able to tolerate it. Pomalidomide plus dexamethasone was the standardof-care for patients with RRMM when the OCEAN study was initiated, and the combination does not have a negative impact on HRQoL. Despite the higher frequency of hematologic adverse events in the melflufen group than in the pomalidomide group in OCEAN,4 these adverse effects had limited impact on patients’ HRQoL in the melflufen group. A greater proportion of patients were stable or even experienced improvements than those with worsening PRO measures with both treatment regimens, suggesting that, in terms of impact on HRQoL, melflufen plus dexamethasone is comparable to pomalidomide plus dexamethasone, despite the different routes of administration (intravenous for melflufen and oral for pomalidomide). These findings aid in meaningful
translation of melflufen plus dexamethasone treatment to real-world practice.
Fredrik H. Schjesvold,1 Heinz Ludwig,2 Sossana Delimpasi,3 Pawel Robak,4 Daniel Coriu,5 Waldemar Tomczak,6 Ludek Pour,7 Ivan Spicka,8 Meletios-Athanasios Dimopoulos,9 Tamas Masszi,10 Natalia G. Chernova,11 Anna Sandberg,12 Marcus Thuresson,12 Stefan Norin,12 Nicolaas A. Bakker,12 Maria-Victoria Mateos,13 Paul G. Richardson14 and Pieter Sonneveld15
1Oslo Myeloma Center, Department of Hematology, Oslo University Hospital and KG Jebsen Center for B Cell Malignancies, University of Oslo, Oslo, Norway; 2Department of Medicine I, Center for Medical Oncology and Hematology with Outpatient Department and Palliative Care, Wilhelminen Cancer Research Institute, Vienna, Austria; 3Bone Marrow Transplantation Unit and Department of Hematology, Evangelismos Hospital, Athens, Greece; 4Department of Hemato-Oncology, University Hospital Ostrava, Ostrava, Czech Republic; 5University of Medicine and Pharmacy “Carol Davila”, Center of Hematology and Bone Marrow Transplantation, Fundeni Clinical Institute, Bucharest, Romania; 6Department of HematoOncology and Bone Marrow Transplantation, Medical University of Lublin, Lublin, Poland; 7Department of Internal Medicine,
Hematology and Oncology, University Hospital Brno, Babak Myeloma Group, Faculty of Medicine, Masaryk University, Brno, Czech Republic; 8First Department of Medicine - Department of Hematology, First Faculty of Medicine, Charles University and General Hospital in Prague, Prague, Czech Republic; 9Department of Clinical Therapeutics, National and Kapodistrian University of Athens School of Medicine, Athens, Greece; 10Department of Internal Medicine and Hematology, Semmelweis University, Budapest, Hungary; 11National Research Center for Hematology, Moscow, Russia; 12Oncopeptides AB, Stockholm, Sweden; 13Hospital Clínico Universitario de Salamanca/IBSAL/CIC, Salamanca, Spain; 14DanaFarber Cancer Institute, Boston, MA, USA and 15Erasmus MC Cancer Institute, Rotterdam, the Netherlands
Correspondence:
F.H. SCHJESVOLD - fredrikschjesvold@gmail.com
https://doi.org/10.3324/haematol.2023.284635
Received: December 21, 2023. Accepted: February 19, 2024. Early view: February 29, 2024.
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Disclosures
FHS has received grants or contracts from Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Targovax, and Sanofi; has received payment or honoraria for lectures or speakers’ bureau participation from AbbVie, Amgen, Bristol Myers Squibb, Daiichi Sankyo, GlaxoSmithKline, Janssen, Novartis, Oncopeptides, Pfizer, Sanofi, SkylineDX, and Takeda; has served on a data safety monitoring or advisory board for AbbVie, Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Sanofi, and Takeda. HL has received research grants from Sanofi and Amgen; has served on data monitoring committees for GSK, Janssen, and Takeda; and has received payment or honoraria for speakers’ bureau participation from Pfizer, BMS, Janssen, Amgen, and Sanofi. SD has received payment or honoraria for speakers’ bureau participation from and served on an advisory board for Amgen, GlaxoSmithKline, Janssen, Pfizer, Sanofi, Sobi, and Takeda. DC has received travel support from Genesis Biopharma and Accord Healthcare. IS has received consulting fees from and served on a data monitoring or advisory board for Amgen, Bristol Myers Squibb, Celgene, Janssen, Novartis, PharmaMar, Sanofi, and Takeda; has received payment or honoraria for lectures or speakers’ bureau participation from Amgen, Bristol Myers Squibb, Celgene, Janssen, Sanofi, and Takeda; and has received meeting and/or travel support from Amgen, Bristol Myers Squibb, Celgene, and Janssen. M-AD has received payment or honoraria for advisory board participation from Amgen, BeiGene, Bristol Myers Squibb, BeiGene, Inc, GSK, Janssen, Menarini, Regeneron, Sanofi, and Takeda. TM has received payment or honoraria for advisory board participation from AbbVie, JanssenCilag, Novartis, Pfizer, and Sanofi. AS, SN, and NAB are employees of
and receive stock or stock options from Oncopeptides. MT is a consultant of and receives stock or stock options from Oncopeptides. M-VM has received payment or honoraria for lectures or speakers’ bureau participation from Amgen, Bristol Myers Squibb/ Celgene, GlaxoSmithKline, Janssen, Pfizer, Sanofi, and Takeda and has served on a data safety monitoring or advisory board for Amgen, bluebird bio, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Pfizer, Regeneron, Sanofi, and Takeda. PGR has served on advisory committees for Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Karyopharm, Oncopeptides, Regeneron, and Sanofi and has received research grants from Bristol Myers Squibb/Celgene Karyopharm, Oncopeptides, and Takeda. PS has received grants or contracts from Amgen, Celgene, Janssen, Takeda, and SkylineDx. PR, NGC, LP, and WT have no competing interests to disclose.
The study sponsor, Oncopeptides AB, conceptualized and designed the OCEAN study in collaboration with FSH, M-AD, M-VM, PGR, and PS. Patients’ data were collected by study site investigators. Data were analyzed by AS, MT, and SN. All authors had access to the data, participated in the interpretation of the data, took part in drafting and revising the manuscript, and approved the final version before submission.
The authors would like to thank the patients and their families for participating in this trial, and the trial investigators and coordinators for their contributions to the trial. We thank Jayasri Srinivasan, MD, MBA, and Swati Ghatpande, PhD, of Team 9 Science for providing medical writing assistance under the guidance of the authors, which was funded by Oncopeptides AB in accordance with Good Publications Practice (GPP) 2022 guidelines.
Funding
Funding for the study (ClinicalTrials.gov identifier: NCT03151811) and for editorial assistance was provided by Oncopeptides AB.
Data-sharing statement
Oncopeptides is committed to sharing clinical study data with qualified researchers to enable enhancement of public health. As such, Oncopeptides will share anonymized patient-level data on request or if required by law or regulation. Qualified scientific and medical researchers can request patient-level data for studies of Oncopeptides’ pharmaceutical substances listed on ClinicalTrials. gov and approved by health authorities in the USA and the European Union. Patient-level data for studies of newly approved pharmaceutical substances or indications can be requested 9 months after approval by the US Food and Drug Administration and European Medicines Agency. Such requests are assessed at Oncopeptides’ discretion, and the decisions depend on the scientific merit of the proposed request, data availability, and the purpose of the proposal. The applicants should be willing to submit both positive and negative findings to a scientific journal. If Oncopeptides agrees to share clinical data for research purposes,
the applicant is required to sign an agreement for data sharing before data release to ensure that the patients’ data are deidentified. In case of any risk of re-identification on anonymized data despite measures to protect patients’ confidentiality, the data will not be shared. The patients’ informed consent will always be respected. If the anonymization process will provide futile data, Oncopeptides has the right to refuse the request. Oncopeptides will
1. LeBlanc MR, Hirschey R, Leak Bryant A, LeBlanc TW, Smith SK. How are patient-reported outcomes and symptoms being measured in adults with relapsed/refractory multiple myeloma? A systematic review. Qual Life Res. 2020;29(6):1419-1431.
2. Engelhardt M, Ihorst G, Singh M, et al. Real-world evaluation of health-related quality of life in patients with multiple myeloma from Germany. Clin Lymphoma Myeloma Leuk. 2021;21(2):e160-e175.
3. Richardson PG, Oriol A, Larocca A, et al. Melflufen and dexamethasone in heavily pretreated relapsed and refractory multiple myeloma. J Clin Oncol. 2021;39(7):757-767.
4 Schjesvold FH, Dimopoulos MA, Delimpasi S, et al. Melflufen or pomalidomide plus dexamethasone for patients with multiple myeloma refractory to lenalidomide (OCEAN): a randomised, head-to-head, open-label, phase 3 study. Lancet Haematol. 2022;9(2):e98-e110.
5. Sonneveld P, Richardson PG, Ludwig H, et al. Benefit versus risk assessment of melflufen and dexamethasone in relapsed/ refractory multiple myeloma: analyses from longer follow-up of the OCEAN and HORIZON studies. Clin Lymphoma Myeloma Leuk. 2023;23(9):P687-696.
6. Richardson PG, Bringhen S, Voorhees P, et al. Melflufen plus dexamethasone in relapsed and refractory multiple myeloma (O-12-M1): a multicentre, international, open-label, phase 1-2 study. Lancet Haematol. 2020;7(5):e395-e407.
7 Larocca A, Leleu X, Touzeau C, et al. Patient-reported outcomes in relapsed/refractory multiple myeloma treated with melflufen plus dexamethasone: analyses from the phase II HORIZON study. Br J Haematol. 2022;196(3):639-648.
8. Song KW, Dimopoulos MA, Weisel KC, et al. Health-related quality of life from the MM-003 trial of pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone in relapsed and/or refractory multiple myeloma. Haematologica. 2015;100(2):e63-67.
provide access to patient-level clinical trial analysis datasets in a secured environment upon execution of the data-sharing agreement. Oncopeptides will also provide the protocol, statistical analysis plan, and the clinical study report synopsis if needed. For additional information or requests for access to Oncopeptides’ clinical trial data for research purposes, please contact us at: medinfoglobal@oncopeptides.com.
9. Fayers PM, Aaronson NK, Bjordal K, Groenvold M, Curran D, Bottomley A, on behalf of the EORTC Quality of Life Group. The EORTC QLQ-C30 Scoring Manual. 3rd ed. Brussels: European Organisation for Research and Treatment of Cancer, 2001. https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual. pdf. Accessed October 20, 2023.
10 Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365-376.
11. EuroQol G. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208.
12. Osoba D, Rodrigues G, Myles J, Zee B, Pater J. Interpreting the significance of changes in health-related quality-of-life scores. J Clin Oncol. 1998;16(1):139-144.
13. Ojo AS, Araoye MO, Ali A, Sarma R. The impact of current therapeutic options on the health-related quality of life of patients with relapse/refractory multiple myeloma: a systematic review of clinical studies. J Cancer Surviv. 2024; 18: 673-697
14 Terpos E, Dimopoulos MA, Boccadoro M, et al. Health-related quality of life in patients with relapsed/refractory multiple myeloma treated with pomalidomide and dexamethasone +/subcutaneous daratumumab: patient-reported outcomes from the APOLLO trial. Am J Hematol. 2022;97(4):481-490.
15. Popat R, Lonial S, Lee HC, et al. DREAMM-2: belantamab mafodotin effect on disease symptoms and health-related quality of life in patients with relapsed/refractory multiple myeloma (RRMM). Presented at the European Hematology Association (EHA) Virtual Congress, 2020; Abstract EP1746. https://library.ehaweb.org/eha/2020/eha25th/294226/rakesh. popat.dreamm-2.belantamab.mafodotin.effect.on.disease. symptoms.and.html. Accessed October 18, 2023.
We, as leaders in the European myeloma clinical research community and from nine countries across the European Union, are writing in response to the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) recommendation to not renew the conditional marketing authorization of belantamab mafodotin issued on September 15, 2023.
Multiple myeloma (MM) is the second most frequent hematological cancer with four to five new cases per 100,000 inhabitants/year and although remarkable progress has occurred in the last years, it does remain for most patients an incurable disease.1
There are three main drug classes used for the treatment of MM: proteasome inhibitors, immunomodulatory drugs and CD38 monoclonal antibodies, all of which are now part of the standard of care for patients with MM in early lines of therapy. However, when patients become triple class exposed and/or refractory to these treatments, they typically have poor outcomes,2 thus representing an unmet medical need with a lack of new standards of care options in this population.
New targets and new approaches have emerged to address this unmet need including new targets, like BCMA3,4 or GPRC5D5 and new modalities, like chimeric antigen receptor T-cell (CAR T) therapies or bispecific monoclonal antibodies and all of them have shown to be effective in the triple class refractory population resulting in their approval. However, despite their proven benefit, safety concerns such as risk of serious infections and burden of administration often makes these agents less suitable for elderly patients or those with other comorbidities. Additionally, accessibility represents a significant hurdle for most patients in Europe, leaving many patients without viable options and without proven effective therapies.
Belantamab mafodotin is a BCMA-targeted therapy in the modality of an antibody drug conjugate and it was the first drug, in this category, approved for the treatment of triple class refractory MM patients. Although belantamab mafodotin is a BCMA-targeted therapy, its mechanism of action is different6-8 and makes it suitable for some MM patients not eligible for either CAR T cells or bispecific antibodies. The rationale for the approval was the significant clinical benefit observed for those patients included in the DREAMM-29 clinical trial (overall response rate [ORR], 32%) and especially those who experienced a partial response or better with a durability of response of 12.5 months (median duration of responses [DoR], 95% confidence interval [CI]:
4.2-19.3 months), and a tolerable safety profile. We acknowledge that the phase III DREAMM-3 study 10 failed to meet its primary endpoint, but it is important to note that belantamab mafodotin is not indicated to replace pomalidomide in this current label but is a useful addition to the therapeutic armamentarium for patients with pomalidomide failure. Indeed, it has been shown to be effective in the pomalidomide-exposed patients as was demonstrated in the DREAMM-2 trial. Other trials assessing new agents such as venetoclax (CANOVA trial11) and melflufen (OCEAN trial)12 have shown the challenges of doublet comparisons in the relapsed/refractory setting. However, the unmet medical need in specific sub-types of patients and especially in those who are not candidates for the currently approved therapies justify the possibility of having beneficial alternatives available to them, such as belantamab mafodotin and targeting BCMA as an antibody drug conjugate. Indeed, melflufen as a peptide drug conjugate, is currently fully approved in Europe given the results of the HORIZON study with the supportive results of OCEAN and based upon a similar premise.13 Moreover, as with belantamab mafodotin both venetoclax and melflufen have been shown to be effective in pomalidomide-exposed patients and to be especially active in combination. Despite not meeting its primary endpoint in the DREAMM-3 trial,10 belantamab mafodotin demonstrated numerical improvement with a median progression-free survival (mPFS) of 11.2 months versus pomalidomide-dexamethasone at 7 months, and an improved hazard ratio (HR) of 0.90 after 10 months more of follow-up. Overall response rate (41%, 95% CI: 34.2-47.7 vs. 36%, 95% CI: 26.5-45.4), depth of response as measured by VGPR or better (25% vs. 8%) and DoR (25.6 months, 95% CI: 20.7-not reached [NR] vs. 10.4 months, 95% CI: 7.6-21.1) were markedly superior in the belantamab mafodotin arm versus pomalidomide-dexamethasone, supporting a meaningful treatment effect and potential clinical benefit.10
As investigators and physicians managing MM patients, we believe belantamab mafodotin provides an important treatment option for an important subgroup of patients, such as the elderly and/or frail patients who may not tolerate the rigors of intensive therapies, as well as for individuals with renal impairment where other more intensive treatments targeting BCMA can be especially challenging, not least as this is a frequent complication of advanced MM. Moreover, patients who are unable to adhere to the demanding administration of bispecific antibodies and wish
to avoid step-up dosing and in-patient hospitalization can benefit significantly from the more manageable dosing regimen of belantamab mafodotin, at a minimum of every 3 to 6 weeks or longer. In addition, in cases of aggressive relapses where treatment should be initiated promptly, belantamab mafodotin in combination with other therapies can provide a rapid and successful alternative, bypassing the long delays as-
sociated with prolonged manufacturing process required for CAR T-cell therapies, as one example. Considering the safety profile, belantamab mafodotin has been shown to be manageable in most patients in both the investigational9,10 and real-world14,15 settings. Eye-related side effects are proving better tolerated and reversible with a low rate of treatment discontinuation due to ocular adverse events now being reported (for example, 2% of the
Table 1. Baseline characteristics and outcomes of the patients treated with belantamab mafodotin in DREAMM-2 trial and in the published real-world experience.
Basal characteris tics of patients receiving belantamab mafodotin
N (%)
High-risk cytogenetics, N (%)
(42) del17p, 17 t(4;14), 15 1q21+, 28 t(14;20), 1
Adapted from Ntanasis-Stathopoulus_Int. J. Mol. Sci. 202315. ASCT: autologous stem cell transplant; ISS: international staging system; m: median; NR: not reported; OS: overall survival; ORR: overall response rate; PFS: progression-free survival; poma: pomalidomide; PR: partial response; tx: treatment; (s)CR: (stringent) complete response; VGPR: very good partial response.
217 patients entered in the DREAMM-3 trial).10
This safety profile is crucial when we manage heavily pretreated MM patients with severe immunosuppression and a previous history of infections because the other alternatives, like CAR T cells or bispecific antibodies, have reported a high incidence of severe infections, including those requiring hospitalizations.16 Other toxicities like cytokine release syndrome (CRS) and neurotoxicity, although manageable in most patients, are not associated with belantamab mafodotin, which further facilitates outpatient management of patients in clinical practice.
Furthermore, patients residing in remote areas, distant from academic centers where advanced therapies are administered, often face formidable barriers to treatment access. Patients lacking a robust caregiver or family support may face challenges with treatments that require significant monitoring and staying away from home for several weeks. Belantamab mafodotin, with its more manageable administration requirements, offers an option for these under-served populations alleviating some of these burdens. In conclusion, we as authors and treating physicians endorsing this letter and firmly consider that belantamab mafodotin is a vital addition to treatment armamentarium of MM, particularly for our triple class exposed refractory patients with limited treatment options. The results published on real-world practice14,15 also support this conclusion (Table 1). Its unique attributes address the specific needs of patients who have exhausted conventional, available treatments and who may not find suitability with other recently approved therapies. It is important to keep in mind that despite the approvals of some of novel options mentioned before, their accessibility represents a significant hurdle for most patients in Europe, leaving many patients without viable options.
As we strive for more inclusive and effective treatments, the accessibility, and clinical benefits of belantamab mafodotin should remain an option for this vulnerable patient population, as with other more convenient outpatient options in this setting.
Maria Victoria Mateos,1,2 Katja Weisel,3 Evangelos Terpos,4 Sossana Delimpasi,5 Efstathios Kastritis,4 Elena Zamagni,6 Michel Delforge,7 Enrique Ocio,8,9 Eirini Katodritou,10 Francesca Gay,11,12 Alessandra Larocca,11,12 Xavier Leleu,13 Paula Rodriguez Otero,14-17 Fredik Schjesvold,18-20 Michele Cavo21,22 and Meletios A. Dimopoulos4
1Hospital Universitario de Salamanca, Salamanca, Spain; 2University of Salamanca, Salamanca, Spain; 3University Hospital HamburgEppendorf, Hamburg, Germany; 4National and Kapodistrian University of Athens, Medicine School, Athens, Greece; 5Evangelismos General Hospital, Athens, Greece; 6University of Bologna, Bologna, Italy;
7University Hospital Leuven, Leuven, Belgium; 8Hospital Universitario Marqués de Valdecilla (IDIVAL), Santander, Spain; 9University of Cantabria, Santander, Spain; 10Theageneion Cancer Hospital, Thessaloniki, Greece; 11University of Torino, Turin, Italy; 12AOU Città della Salute e della Scienza of Turin, Turin, Italy; 13Hopital La Mileterie, Poitiers, France; 14Clinica Universidad de Navarra, Pamplona, Spain; 15Centro de Investigación Médica Aplicada (CIMA), Pamplona, Spain; 16Instituto de Investigación Sanitaria de Navarra IDISNA, Pamplona, Spain; 17Centro de Investigación Biomédica en Red en Cáncer (CIBERONC) Pamplona, Spain; 18Oslo Myeloma Center, Oslo, Norway; 19Oslo University Hospital, Oslo, Norway; 20KG Jebsen Center for B Cell Malignancies, University of Oslo, Oslo, Norway; 21IRCCS Azienda Ospedaliero-Universitaria di Bologna, Istituto di Ematologia “Seràgnoli”, Bologna, Italy and 22Università di Bologna, Bologna, Italy
Correspondence:
M. V. MATEOS - mvmateos@usal.es
https://doi.org/10.3324/haematol.2023.284694
Received: November 17, 2023.
Accepted: February 13, 2024.
Early view: February 22, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
MVM has received honoraria derived from lectures and participation in boards from Janssen, Celgene-BMS, GSK, Sanofi, Pfizer, AbbVie, Regeneron and Novartis. KW has received research funding (to institution) from Abbvie, Amgen, BMS/Celgene, Janssen, GSK, Sanofi and Takeda; honoraria form Abbvie, Amgen, Adaptative Biotech, Astra Zeneca, BMS/Celgene, BeiGene, Janssen, GSK, Karyopharm, Novartis, Oncopeptides, Pfizer, Roche Pharma, Sanofi, Stemline, Takeda and Menarini; and consulting fees from Abbvie, Amgen, Adaptative Biotech, BMS/Celgene, BeiGene, Janssen, GSK, Karyopharm, Oncopeptides, Pfizer, Roche Pharma, Sanofi, Takeda and Menarini. ET has received research funding from Amgen, GSK, Janssen, Sanofi and Takeda; honoraria from Amgen, BMS, Astra/ Zeneca, EUSA Pharma, GSK, Janssen, Menarini/Stemline, Pfizer, Sanofi and Takeda; and travel expenses from Amgen, Astra/Zeneca, EUSA Pharma, Sanofi and Takeda. SD has not conflict of interest to disclose. EK has received honoraria derived from lectures and participation in boards from Janssen, GSK and Pfizer. EZ has received honoraria and AB member from Janssen, BMS, Sanofi, Amgen, GSK, Pfizer, Oncopeptides and Menarini/Stemline. MD has received speaker honoraria from BMS, GSK, Janssen and Stemline. EO discloses honoraria from Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Oncopeptides, Pfizer, Regeneron, Sanofi, and Takeda; consulting/advisory role from AbbVie, Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Menarini/Stemline Therapeutics, Oncopeptides, Pfizer, Sanofi, and Takeda; speakers’ bureau from Janssen; and travel/accommodation expenses from
Bristol Myers Squibb, GlaxoSmithKline, Janssen, and Lilly. Eirini K discloses research support/principal investigator from Amgen, Janssen, Takeda, Sanofi, Karyopharm, GSK, Abbvie, Pfizer and GSK; honoraria from Amgen, Genesis-Pharma, Janssen, Takeda, IntegrisPharma, Sanofi, Abbvie and GSK; and scientific advisory board from Amgen, Janssen, Takeda and Sandoz. FG discloses honoraria/advisory board from Janseen, BMS, Takeda, Amgen, Sanofi, Roche, Abbvie and GSK; and advisory board from Pfizer and Oncopeptides. AL discloses honoraria and scientific advisory board from Janssen-Cilag, BMS, Amgen, Takeda, Oncopeptides, GSK, Sanofi and Karyopharm. XL has received honoraria from Janssen, Takeda, Sanofi, Pfizer, Roche, BMS, GSK, Iteos, Abbvie, Regeneron and Amgen. PR-O discloses receiving honoraria from consulting activities from BMS, Janssen, Sanofi, Kite Pharma, Abbvie, Oncopeptides, Takeda, Pfizer, Roche, Pfizer and GSK, and from lectures from BMS, Janssen, Sanofi, GSK, Amgen, Regeneron and Takeda. FS has received honoraria derived from lectures and participation in boards from Abbvie, GSK, Celgene, Takeda, Janssen, Oncopeptides, Sanofi, BMS, Amgen, BMS, Novartis,
1. Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up(†). Ann Oncol. 2021;32(3):309-322.
2. Gandhi UH, Cornell RF, Lakshman A, et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33(9):2266-2275.
3. Lee L, Bounds D, Paterson J, et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br J Haematol. 2016;174(6):911-922.
4 O’Connor BP, Raman VS, Erickson LD, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199(1):91-98.
5. Nath K, Costa BA, Mailankody S. GPRC5D as a novel immunotherapeutic target in multiple myeloma. Nat Rev Clin Oncol. 2023;20(5):281-282.
6. Montes de Oca R, Alavi AS, Vitali N, et al. Belantamab mafodotin (GSK2857916) drives immunogenic cell death and immunemediated antitumor responses in vivo. Mol Cancer Ther. 2021;20(10):1941-1955.
7 Tai YT, Anderson KC. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy. 2015;7(11):1187-1199.
8. Tai YT, Mayes PA, Acharya C, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123(20):3128-3138.
9. Nooka AK, Cohen AD, Lee HC, et al. Single-agent belantamab mafodotin in patients with relapsed/refractory multiple myeloma: final analysis of the DREAMM-2 trial. Cancer. 2023;129(23):3746-3760.
10 Dimopoulos MA, Hungria VTM, Radinoff A, et al. Efficacy and safety of single-agent belantamab mafodotin versus
SkyliteDX, Pfizer and Daiki-Sankyo. MC received honoraria from Janssen, Celgene/Bristol Myers Squibb, Sanofi, Takeda, Amgen, AbbVie, Adaptive, GSK, Pfizer and Menarini/Stemline; and served on a speakers bureau for Janssen, Celgene/Bristol Myers Squibb and Sanofi. MAD has received honoraria from participation in advisory boards and satellite symposia from Amgen, Sanofi, Regeneron, Menarini, Takeda, GSK, BMS, Janssen and Beigene.
Contributions
All authors contributed to writing the letter. All authors critically revised and approved the final version of the letter and agree with presented format.
Data-sharing statement
The data that support this manuscript are not openly available because they have not been generated by the auhtors but can be available from the corresponding author of each study, here referenced, and after a reasonable request.
pomalidomide plus low-dose dexamethasone in patients with relapsed or refractory multiple myeloma (DREAMM-3): a phase 3, open-label, randomised study. Lancet Haematol. 2023;10(10):e801-e812.
11. Mateos M-V, Beksac M, Dimopoulos M, et al. Results from the randomized, open-label phase 3 CANOVA study of venetoclaxdexamethasone versus pomalidomide- dexamethasone in patients with t(11;14)-positive relapsed/refractory multiple myeloma. Presented at International Myeloma Society Annual Meeting; September 27-30, 2023; Athens, Greece. Abstract OA-52.
12. Schjesvold FH, Dimopoulos MA, Delimpasi S, et al. Melflufen or pomalidomide plus dexamethasone for patients with multiple myeloma refractory to lenalidomide (OCEAN): a randomised, head-to-head, open-label, phase 3 study. Lancet Haematol. 2022;9(2):e98-e110.
13. Richardson PG, Oriol A, Larocca A, et al. Melflufen and dexamethasone in heavily pretreated relapsed and refractory multiple myeloma. J Clin Oncol. 2021;39(7):757-767.
14. de la Rubia J, Alonso R, Clavero ME, et al. Belantamab mafodotin in patients with relapsed/refractory multiple myeloma: results of the compassionate use or the expanded access program in Spain. Cancers (Basel). 2023;15(11):2964.
15. Ntanasis-Stathopoulos I, Malandrakis P, Fotiou D, et al. Realworld effectiveness and safety of belantamab mafodotin monotherapy in triple-class refractory multiple myeloma. Int J Mol Sci. 2023;24(14):11829.
16. Markouli M, Ullah F, Unlu S, et al. Toxicity profile of chimeric antigen receptor T-cell and bispecific antibody therapies in multiple myeloma: pathogenesis, prevention and management. Curr Oncol. 2023;30(7):6330-6352.
Megakaryocytes are specialized cells that enlarge and become polyploid through repeated cycles of DNA replication without cell division. During development, megakaryocytes become full of platelet-specific granules, expand their cytoplasmic content of cytoskeletal proteins, and develop a demarcation membrane system. Megakaryocytes then generate platelets by remodeling their cytoplasm into proplatelet extensions, which serve as assembly lines for platelet production.1-3 Several laboratories have made fundamental discoveries in the mechanics of platelet biogenesis, including identifying the cytoskeletal forces that power proplatelet elongation, defining the mechanics of organelle transport and packaging, and establishing new mechanisms of the final stages of platelet production.4-7 Despite this progress, our understanding of the cellular and molecular basis of the process by which megakaryocytes trigger proplatelet production has clearly lagged behind. Identification of a master regulator that triggers platelet production from megakaryocytes has the potential to reveal novel targets for treating thrombocytopenia. Here we report a rigorous set of microinjection experiments that reveal a cytoplasmic factor, designated proplatelet-promoting factor (PPF), that triggers platelet production in megakaryocytes. PPF was first detected when cytoplasm from proplatelet-producing megakaryocytes was isolated and microinjected back into round megakaryocytes lacking proplatelets, and this led to the immediate initiation of proplatelet production. Furthermore, the frequency with which proplatelet production was initiated was proportional to the volume of injected cytoplasm. Using various agents and treatments, we show that PPF was inactivated by protein modification procedures but resistant to inactivation by methods that modify nucleic acids. Specifically, PPF remained active after treatment with UV, nucleases, weak detergents, and dialysis, but was inactivated by treatment with proteases, sodium dodecylsulfate (SDS), phenol extraction, and heat. These observations provide compelling evidence that there is a protein, or multiple proteins, expressed during proplatelet formation that serve as an internal molecular trigger for proplatelet production.
We hypothesized that megakaryocytes undergoing proplatelet production may contain a cytoplasmic agent responsible for the initiation of proplatelet formation. This led us to ask if the microinjection of cytosol from proplatelet-producing megakaryocytes is sufficient to trigger round megakaryocytes to start proplatelet production. To test this, we adapted a microinjection approach that was initially used to identify mitosis promoting factor (Figure 1A).8 We first separated proplatelet-producing megakaryo -
cytes from round megakaryocytes (Figure 1B) and generated separate supernatant (S100) from both populations to use as the starting material for microinjection into round non-proplatelet-producing megakaryocytes (Figure 1C). Following injection of cytosol from proplatelet-producing megakaryocytes into round megakaryocytes, 83 + 6% of recipient megakaryocytes formed proplatelets within 1 hour (hr) (Figure 1D, E). Conversely, cytoplasm prepared from round, non-proplatelet-producing donor megakaryocytes had almost no effect when injected into recipient round megakaryocytes (3.8+2.1% of megakaryocytes made proplatelets). Proplatelet initiation was also minimally observed in control non-injected and sham saline-injected recipient round megakaryocytes at 1 hr (Figure 1E), demonstrating the mechanical force of microinjection did not cause proplatelet initiation. To ensure that round megakaryocytes injected with PPF did not just initiate proplatelet formation, but also continued to elongate and elaborate proplatelets, we examined these cells again after 12 hr. Round megakaryocytes microinjected with cytosol containing PPF continued to progress beyond proplatelet initiation and formed highly developed proplatelets with the hallmark beads-on-a-string appearance, emphasizing the physiological significance of this assay (Figure 2A, B). Of note, recipient cells injected with cytosol from round megakaryocytes had still not initiated proplatelet formation after 12 hr. Proplatelet initiation was also minimally observed in control non-injected and sham saline-injected recipient round megakaryocytes at the 12-hr time point (Figure 2B). However, cells injected with cytosol from proplatelet-producing megakaryocytes, round megakaryocytes, non-injected and saline-injected controls had all formed proplatelets at similar rates by 24 hr, suggesting our observations were not likely to be due to injection of an inhibitor in the cytosol from round donor megakaryocytes (Figure 2C). In order to confirm that the initiation of proplatelet formation was due to PPF, we performed a dose response, which revealed that the frequency of proplatelet initiation was directly proportional to the volume of injected cytosol from proplatelet-producing megakaryocytes (Figure 2D). To test the need for the cytosol being injected into cells versus megakaryocytes being incubated with the cytosol, we added S100 into the culture medium of round megakaryocytes. Cytosol at dilutions of 1:10 and 1:100 from both round and proplatelet-producing megakaryocytes was cultured with round (day 4) megakaryocytes (Figure 2E). Cytoplasm prepared from proplatelet-producing donor megakaryocytes had little effect when cultured with round megakaryocytes for 1 hr (1:10 dilution: 4.7+1.5% of megakaryocytes made
proplatelets; 1:100 dilution: 4.3+2.5% of megakaryocytes made proplatelets) or 12 hr (1:10 dilution: 4.3+1.5% of megakaryocytes made proplatelets; 1:100 dilution: 5.3+2.5% of megakaryocytes made proplatelets) suggesting PPF does not have an effect from an extracellular environment. Proplatelet
initiation was also minimally observed in controls, in which cytoplasm prepared from round non-proplatelet-producing megakaryocytes was cultured with round megakaryocytes at 1 hr (1:10 dilution: 6.0+2.6% of megakaryocytes made proplatelets; 1:100 dilution: 3.2+6.1% of megakaryocytes
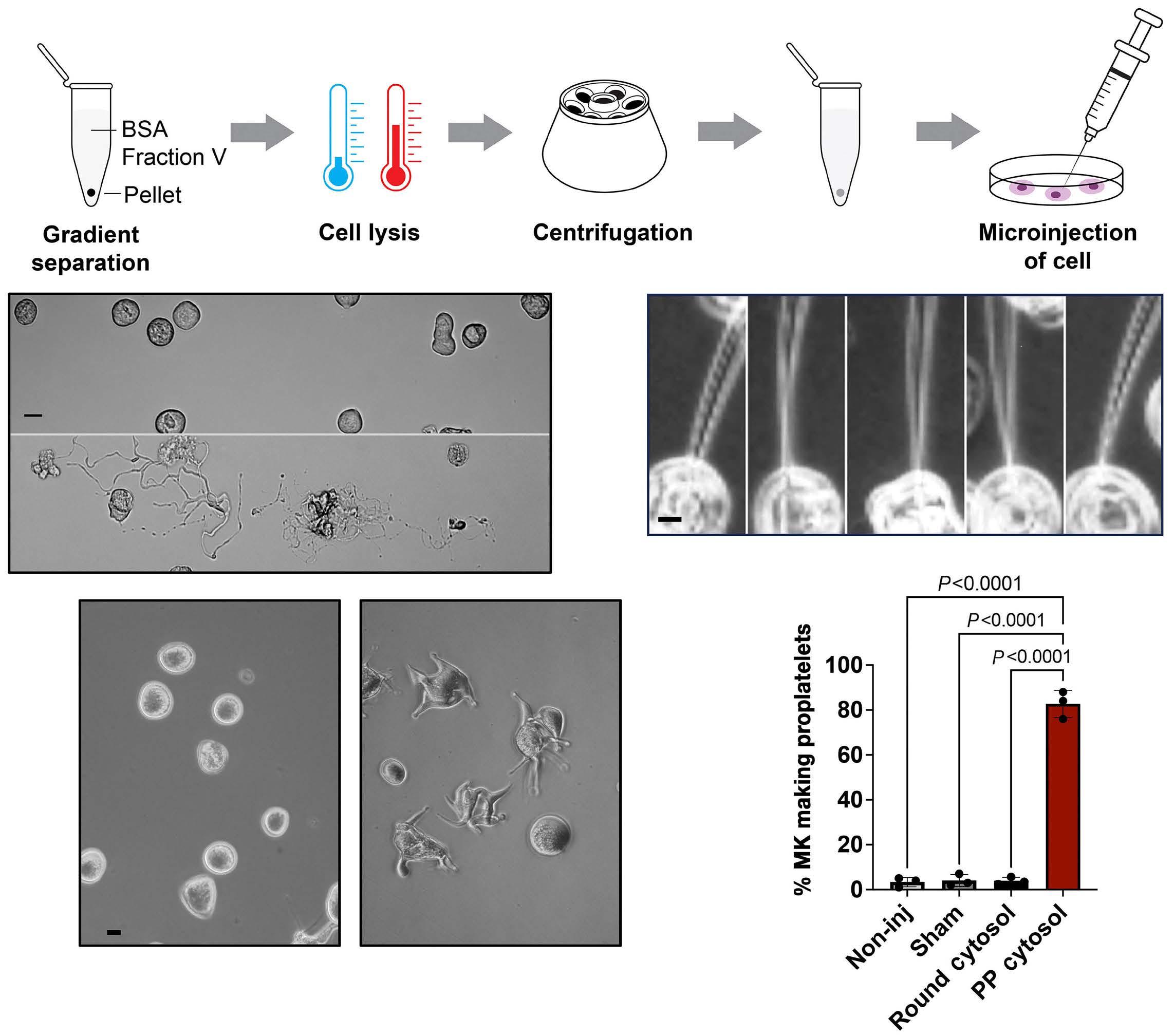
Figure 1. Evidence supporting a proplatelet-promoting factor. (A) Overview of approach to identify proplatelet-promoting factor starting with separation of proplatelet-producing megakaryocytes (MK) from round MK. Murine fetal liver cells were recovered from wild-type CD1 mice (Charles River Laboratories) on embryonic day 13.5 and cultured as previously described.11 Fetal liver cell cultures (15,000 MK per fetal liver) were layered on a single-step gradient (1.5-3.0% BSA) on day 4, and MK were allowed to sediment for 1 hour (hr). The MK pellet was resuspended in media and cultured for an additional 24 hr, during which proplatelet production was readily observed. MK cultures were layered on a second single-step gradient (1.5-3.0%) on day 5 and intermediate stages in platelet production were resolved within different fractions of the gradient. Proplatelet-producing MK localized to the BSA fraction, whereas round, non-proplatelet-producing MK localized to the pellet. Isolated MK fractions were washed with PBS and then lysed by 3 freeze-thaw cycles and then centrifuged at 100,000 g for 1 hr at 4°C. The supernatant (S100) was recovered and used immediately as the starting material (at 45 mg/mL protein concentration) for microinjection or stored at -80°C until use. A Harvard apparatus PII-100A injector was used for microinjection. Needles were purchased from World Precision Instruments. For each experiment, at least 40 round MK were microinjected with untreated S100 or S100 that was treated as indicated in each experiment. Experiments were repeated in biological triplicates. To determine the percentage of MK making proplatelets, MK were observed on a Nikon Eclipse TS2 microscope with either a 20x/0.40 Ph1 Adl or 40x/0.55 Ph2 Adl objective using an Olympus O-color 3 camera using QCapture Pro 7 software and categorized as round or proplatelet-producing (with cellular extensions) by eye. Data are expressed as percentage of proplatelet-producing MK. Cells were injected with 0.075 nL unless noted otherwise. All data are presented as mean + Standard Deviation (SD). N=3 biological replicates. (B) Representative images showing isolation of round (top) and proplatelet-producing (bottom) MK. Scale bar = 20 μm. (C) Representative images showing microinjection of round MK. Scale bar = 10 μm. (D) Representative images of 1-hr time-point showing microinjection of round MK with cytosol from round MK (left) and cytosol from proplatelet-producing MK (right). Scale bar = 20 μm. (E) Percentage of MK making proplatelets 1 hr post microinjection. N=3 biological replicates. Data are presented as mean + SD. Non-inj: not injected. P<0.0001.
made proplatelets) or 12 hr (1:10 dilution: 4.0+2.6% of megakaryocytes made proplatelets; 1:100 dilution: 3.5+2.0% of megakaryocytes made proplatelets).
To begin to define PPF, we carried out a detailed characterization of the proplatelet-promoting activity contained in the S100 cytosol. We adapted a strategy used to characterize prions9 and systematically added chemical agents and detergents to PPF to determine whether they altered its ability to induce proplatelet formation (Figure 3A). We found that the proplatelet-triggering activity of the crude S100 extract was stable for months at -80°C, and for at least three weeks at 4°C. However, it was unstable at higher temperatures; at 25°C and 37°C, the activity was lost after 5 hr. Further, PPF lost its ability to stimulate proplatelet production when exposed to protein-destroying treatments such as phenol, proteinase K, or trypsin for 1 hr at 25°C. However, pre-incubation of proteinase K with PMSF restored PPF activity. Notably, there was no loss in PPF activity after nucleic acid-destroying treatments including UV radiation, ribonuclease, or deoxyribonuclease. Non-denaturing, ionic detergents (at 2.5%) such as Triton X-100, Nonidet P-40,
octyl glucoside, and Tween-20 also did not inactivate the agent. In contrast, the denaturing detergent SDS inactivated PPF at a concentration of 1%. High-speed centrifugation to remove membranes from the S100 also failed to inactivate PPF. There was also no loss in PPF activity when S100 was dialyzed (molecular weight cutoff 5,000 kDa), suggesting PPF is not a small molecule.
Given that our previous studies demonstrated that inhibitors of protein synthesis blocked proplatelet initiation,10 we wondered whether injection of PPF could rescue this phenotype and overcome the loss of proplatelet formation with protein synthesis inhibition. Indeed, injection of PPF caused megakaryocytes treated with puromycin to avidly begin proplatelet production (Figure 3B-D). Together, these data provide evidence for a megakaryocyte-intrinsic protein or multiple proteins, PPF, that is a master trigger for platelet production. Further, they suggest that PPF is present in megakaryocytes as they undergo proplatelet formation and justify future studies examining its potential existence in platelets. While PPF is sufficient to trigger proplatelet formation, we recognize that external factors in the bone
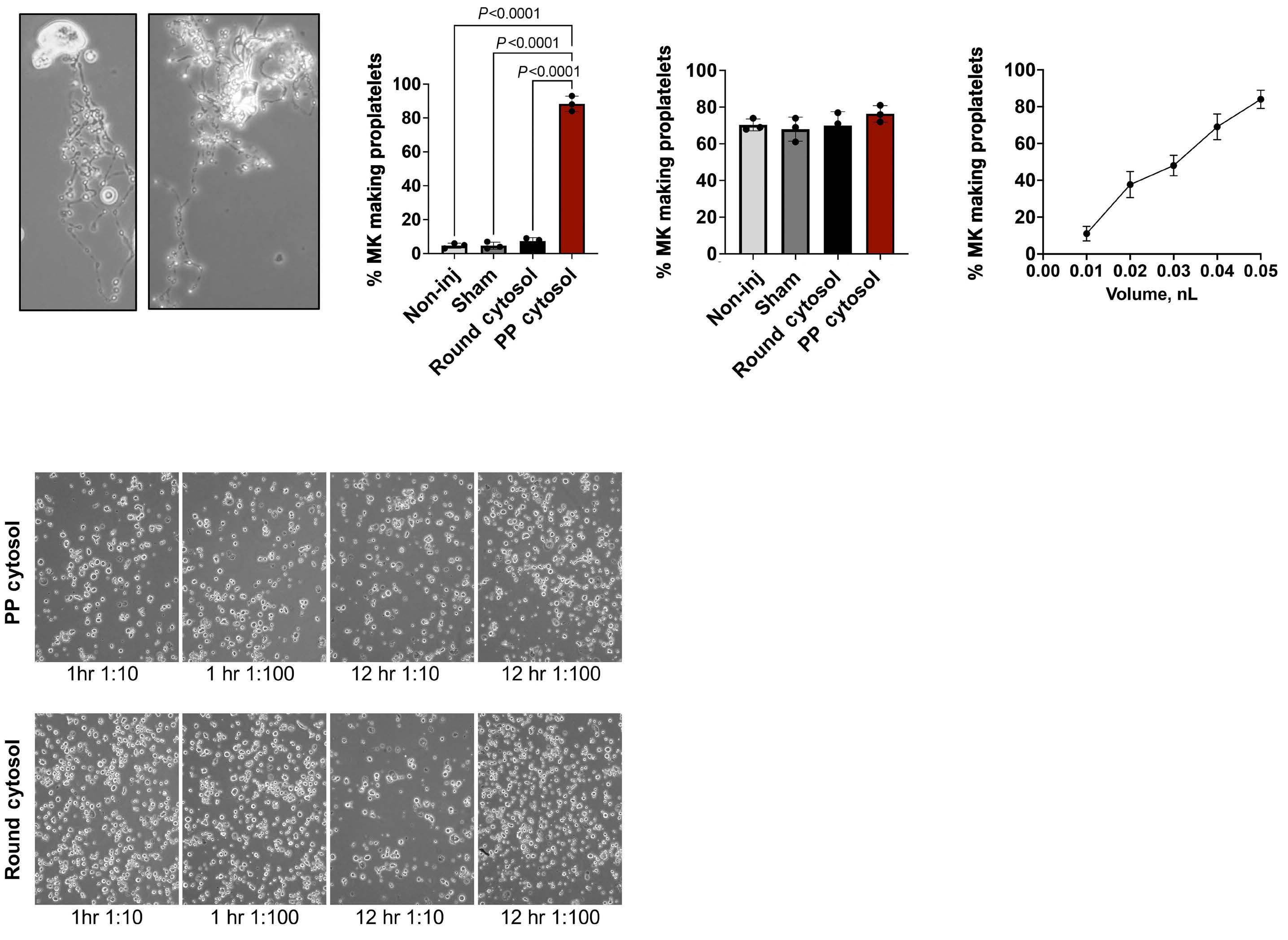
Figure 2. Data substantiating a proplatelet-promoting factor. (A) Representative images of megakaryocytes (MK) 12-hours (hr) post microinjection showing that round MK injected with S100 from proplatelet-producing (PP) MK continue to develop classic beadson-string proplatelets. Scale bar = 10 μm. Original magnification = 40x. (B) Percentage of MK making proplatelets 12-hr post microinjection. N=3 biological replicates. Data are presented as mean + Standard Deviation (SD). (C) Percentage of MK making proplatelets 24-hr post microinjection. N=3 biological replicates. Data are presented as mean + SD. (D) Graph showing that the frequency of proplatelet initiation is proportional to the volume of injected S100 from PP MK. Volume is in nL and cells assayed 1 hr after injection. N=3 biological replicates. Data are presented as mean + SD. Round MK were injected with indicated volume of S100 from PP MK. (E) Representative images showing the effect of the addition of cytosol on round MK. Cytosol (S100 supernatant) was placed in the culture medium at dilutions of 1:10 and 1:100. (Top) Representative images of round MK cultured with cytosol from PP MK. (Bottom) Representative images of round MK cultured with cytosol from round MK. N=3 biological replicates. Scale bar = 80 μm. Original magnification = 10x. P<0.0001.
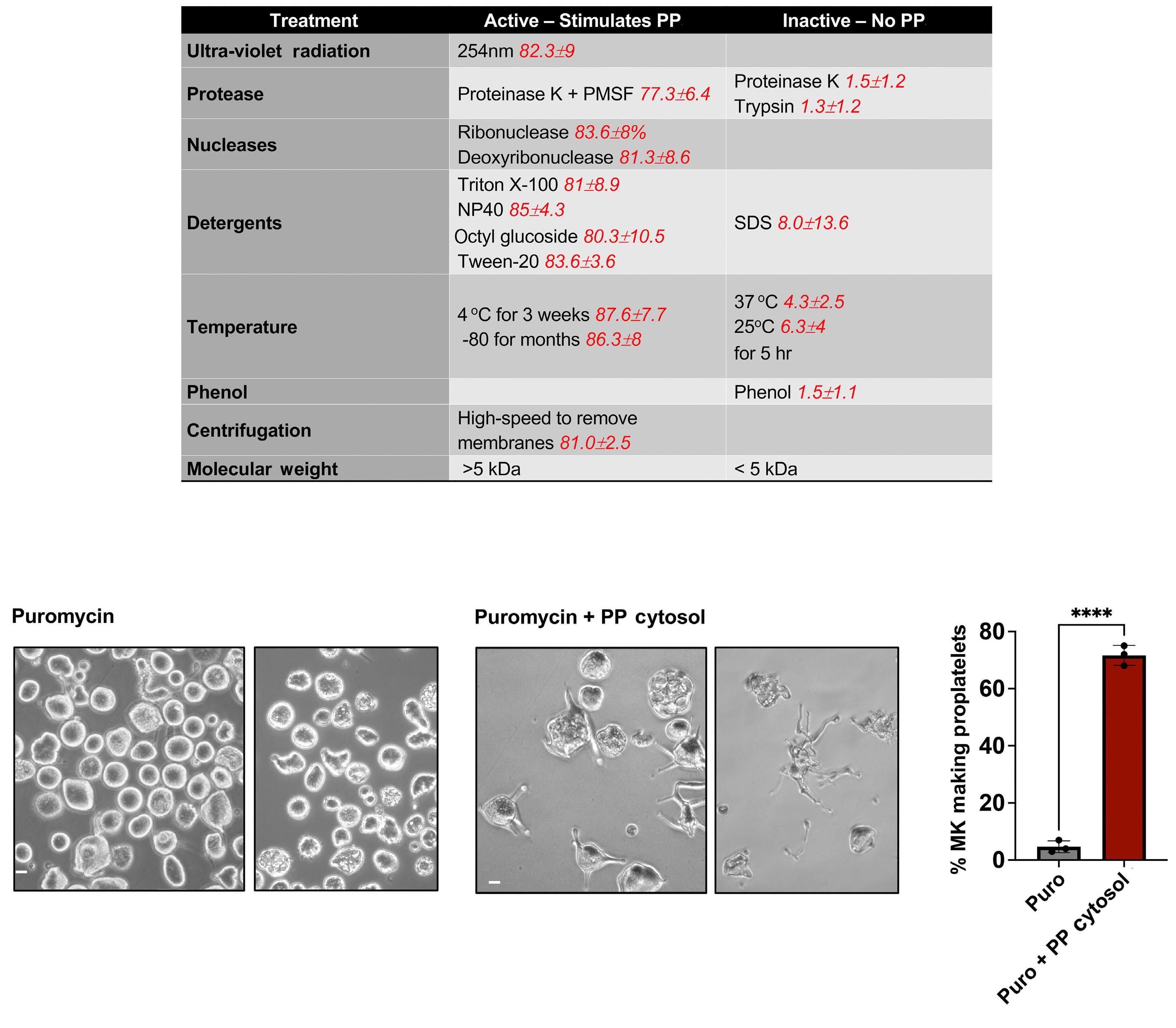
Figure 3. Molecular qualities of proplatelet-promoting factor. (A) Table showing molecular treatments done to S100 derived from proplatelet-producing megakaryocytes (MK), and corresponding percentage of MK making proplatelets after 1 hour (hr) when treated S100 was microinjected into round MK (indicated in red). UV irradiation was carried out at 254 nm. S100 was irradiated on ice and placed beneath GE germicidal lamps (emax = 254 nm) at approximately 5 cm. A Spectroline UV dosimeter was used to measure the delivered doses, calculated at 7.2 + 1.2 J/m2/sec. The calculated dose on the S100 was 10,000 J/m2. For high-speed centrifugation, S100 was centrifuged at 18,000 g for 90 minutes (min) in a Thermo Scientific Sorvall Lynx 4000 centrifuge. For size separation, 100 μL of S100 was placed in a Millipore filter with a molecular weight cut off at 5 kDa and centrifuged for 2 hr. The lower layer was collected, and upper layer reconstituted to the original volume in 10 mM KH2HPO4 (pH 6.8) buffer. Proteinase K, trypsin, PMSF, sodium dodecyl sulfate, puromycin, Triton X-100, Nonidet P-40, octyl glucoside, and Tween-20 were all obtained from Sigma. Proteinase K digestion was carried out at 100 μg/mL for 1 hr at 37°C. PhMeSO2F (PMSF) was used at 0.1 mM to inhibit proteinase K. Digestion with RNase A or DNase I was carried out at 50 μg/mL for 1 hr. Trypsin was used at 100 μg/mL for 1 hr. For phenol extraction, prior to extraction with phenol, cytosol was suspended in 30 mM Tris-acetate, pH 8.3. Equal volumes of cold phenol were used to extract the samples at 4°C. Phase separation was accomplished by centrifugation at 4,000 g for 10 min at 4°C. The aqueous phase was recovered. N=3 biological replicates. Data are presented as mean + SD. (B) Puromycin treatment (250 μg/mL) caused a block in proplatelet formation10 (4.6+1.7% proplatelet production). Representative images of 8-hr time-point after puromycin treatment (original magnification 40x). (C) S100 from proplatelet-producing MK was microinjected into puromycin-treated MK, which triggered proplatelet production. Representative images of 1-hr time-point (original magnification 40x) after injection of S100 obtained from proplatelet-producing MK (71.6+8.2% proplatelet initiation). Scale bars = 20 μm. (D) Proplatelet production in the presence of puromycin and after microinjection with S100 from proplatelet-producing megakaryocytes. N=40 cells per condition per replicate. N=3 biological replicates. Data are presented as mean + SD. ****P=0.0001. PPF: proplatelet-promoting factor.
marrow microenvironment may provide signaling to regulate this internal factor during proplatelet production.
Authors
Joseph E. Italiano Jr1,2 and Kellie R. Machlus1,2
1Vascular Biology Program, Department of Surgery, Boston Children’s Hospital and 2Harvard Medical School, Boston, MA, USA
Correspondence:
J. ITALIANO - joseph.italiano@childrens.harvard.edu
https://doi.org/10.3324/haematol.2023.284755
Received: November 25, 2023.
Accepted: February 19, 2024.
Early view: February 29, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
JEI has a financial interest in and is a founder of StellularBio, a biotechnology company focused on making donor-independent
1. Becker RP, De Bruyn PP. The transmural passage of blood cells into myeloid sinusoids and the entry of platelets into the sinusoidal circulation; a scanning electron microscopic investigation. Am J Anat. 1976;145(2):183-205.
2. Radley JM. Ultrastructural aspects of platelet production. Progr Clin Biol Res. 1986;215:387-398.
3. Radley JM, Scurfield G. The mechanism of platelet release. Blood. 1980;56(6):996-999.
4 Italiano JE Jr, Lecine P, Shivdasani RA, Hartwig JH. Blood platelets are assembled principally at the ends of proplatelet processes produced by differentiated megakaryocytes. J Cell Biol. 1999;147(6):1299-1312.
5. Richardson JL, Shivdasani RA, Boers C, Hartwig JH, Italiano JE Jr. Mechanisms of organelle transport and capture along proplatelets during platelet production. Blood. 2005;106(13):4066-4075.
6. Thon JN, Macleod H, Begonja AJ, et al. Microtubule and cortical
platelet-like cells at scale, and has a financial interest in and is a founder of SpryBio, a biotechnology company focused on using shelf-stable platelets to treat osteoarthritis. His interests are managed by the Boston Children’s Hospital. KRM has no conflicts of interest to disclose.
Contributions
JEI conceptualized and designed the research, conducted the experiments, analyzed and interpreted the data, and wrote the manuscript. KRM designed and interpreted the research, and edited and reviewed the manuscript.
We thank Dani Freire, Emma Nikols, Clementine Payne and Karen Guo for critical reading of the manuscript.
Funding
JEI is supported by the National Institute of Health, National Heart, Lung, and Blood Institute (R01HL68130 and R35HL161175). KRM is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R03DK124746) and the National Heart, Lung, and Blood Institute (R01HL151494).
Data-sharing statement
Please contact the corresponding author for access to the original data related to this manuscript.
forces determine platelet size during vascular platelet production. Nat Commun. 2012;3:852.
7 Thon JN, Montalvo A, Patel-Hett S, et al. Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191(4):861-874.
8. Masui Y, Markert CL. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J Exp Zool. 1971;177(2):129-145.
9 Prusiner SB. Prions. Sci Am. 1984;251(4):50-59.
10. Machlus KR, Wu SK, Stumpo DJ, et al. Synthesis and dephosphorylation of MARCKS in the late stages of megakaryocyte maturation drive proplatelet formation. Blood. 2016;127(11):1468-1480.
11. Vijey P, Posorske B, Machlus KR. In vitro culture of murine megakaryocytes from fetal liver-derived hematopoietic stem cells. Platelets. 2018;29(6):583-588.
Central nervous system (CNS) relapses in acute leukemia (AL) are serious disease recurrences, especially after allogeneic hematopoietic stem cell transplantation (allo-HSCT). The incidence and overall survival (OS) after treatment and best treatment strategies remain largely unknown.
A study on 2,045 patients reports the incidence of CNS relapse after allo-HSCT for acute myeloid leukemia (AML) to be 1.81%.1 Pre-allo-HSCT CNS involvement was an independent risk factor for CNS relapse post-allo-HSCT, with a lower 3-year OS (60.3 vs. 81.5%). In AML, screening for CNS disease is unsystematic and varies: it is sometimes routine, sometimes only in patients with hyperleukocytosis or monoblasts, and irregular for the elderly. The definition of CNS involvement is not clear: most only consider cytologically visible leukemic cells, others consider minor infiltration in flow cytometry. A study on 48 patients (monocytic phenotype, hyperleukocytosis, FLT3-ITD+ or CNS symptoms) demonstrated a CNS involvement in 52%,2 with unknown incidence of CNS relapse post-allo-HSCT. Another study on 103 patients demonstrated 32% had CNS positivity, screened by either cytology, or flow cytometry.3 In acute lymphoblastic leukemia (ALL), CNS involvement is an established risk factor and routinely screened for; it is treated therapeutically and prophylactically. CNS relapse is the most important risk factor for relapse and mortality; CNS prophylaxis is integrated into treatment protocols.4 Around 5% of patients show cytological CNS involvement, but cytology negative CNS infiltration is frequent, demonstrated by animal and post-mortem studies.5
Central nervous system relapse after allo-HSCT is life-threatening; however, the exact incidence in a large leukemic cohort is not known. CNS is among the most frequent sites of extramedullary disease progression, especially in ALL.6 In AML, initial CNS involvement is present in 2-4% of patients.7-9 The incidence, OS after treatment, and best treatment strategies remain unknown. Literature is scarce and mostly reported as single case reports. One study on CNS relapse in ALL describes 457 patients after first allo-HSCT in first or second complete remission (CR), 15% of whom had a CNS involvement pre-allo-HSCT.10 Forty-eight
percent received post-allo-HSCT CNS prophylaxis. Overall, 18 patients (4%) developed CNS relapse, with pre-allo-HSCT CNS involvement as the only identified risk factor. Fifty percent of these patients had CNS involvement prior to allo-HSCT, regardless of post-allo-HSCT CNS prophylaxis or the conditioning regimen used. A study on allo-HSCT in 71 AML patients with CNS involvement at diagnosis, 52 of whom received intrathecal chemotherapy alone and 19 in combination with irradiation, demonstrated a worse relapse-free survival than a control group without CNS involvement and a disease-free survival and OS benefit from additional irradiation.11 A study on 1,226 AML, ALL or CML patients described the incidence of CNS relapse after allo-HSCT in a 10-year observation study as 2.3% (29 patients).12 Risk factors were ALL, non-remission at allo-HSCT, prior CNS disease and prophylactic intrathecal chemotherapy post-allo-HSCT. Three-year OS after CNS relapse was 18% and 1-year OS 42%. Long-term survival was observed only in few patients, and only without systemic disease. We performed the current retrospective, multicenter analysis based on the registry of the Acute Leukemia Working Party (ALWP) of the European Society for Blood and Marrow Transplantation (EBMT, more than 600 transplant centers, mainly in Europe) to define the incidence and outcome of CNS relapse after HSCT in patients suffering from AML and ALL.
The study was approved by the ALWP and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Data from allo-HSCT performed between 1996 and 2016 were screened and patients with the following criteria were included: ≥18 years, with AML or ALL, first allo-HSCT in first complete remission (CR), all donor types. We identified patients with and without CNS relapse. We also collected information on type of relapse, relapse site, treatment, and outcome after CNS relapse. The primary endpoint of this study was OS after CNS relapse. Secondary endpoints were: cumulative incidence of CNS relapse after allo-HSCT, CR after CNS relapse and incidence of acute and chronic graft-versus-host disease (GvHD) after CNS relapse.
Univariate comparisons were performed with χ2 and Fisher’s exact tests for categorical variables and Mann-Whitney test for continuous variables. Probability of OS was calculated using the Kaplan-Meier estimator. Probabilities of CNS relapse and GvHD were estimated using cumulative incidence curves. CNS relapse, death and relapse in other sites were competing events. Concerning GvHD, both death and relapse were considered as competing events. Univariate analyses were performed with the log-rank test for OS and Gray’s test for cumulative incidence estimates. Results were expressed as estimates with a 95% confidence interval (95% CI). All tests were two-sided with a type 1 error rate fixed at 0.05. Statistical analyses were performed with SPSS 27.0 and R 4.1.1.
Study inclusion criteria were met by 7,991 patients: 5,724 (71.6%) AML, and 2,267 (28.4%) ALL. Patients were transplanted between 1996 and 2016.
Median age was 44 years (interquartile range [IQR]: 31.655.4), 6,014 (75.3%) were in first, 1,800 (22.5%) in second, and 177 (2.2%) in third CR. Ninety-one patients of the 7,991 (1.1%) experienced CNS relapse after allo-HSCT; of those, we have information for 88 patients at diagnosis: 4 patients (4.5%) showed initial CNS involvement (see Online Supplementary Table S1 for patients and transplant characteristics, and patients with and without CNS relapse).
Incidence of CNS relapse was 0.9% (95% CI: 0.7-1.2 ), 1.2% (95% CI: 1-1.5 ) and 1.4% (95% CI: 1.1-1.7 ) at two, five and ten years post allo-HSCT (Figure 1A).
Patients with ALL had higher risk for CNS relapse (1.9% vs. 0.9% in AML at 5 years, P=0.002) (Figure 1B). The risk of developing a CNS relapse after allo-HSCT was higher after 2010 (median time of transplant, P=0.013) (Figure 2A). No other risk factors such as age, donor type, conditioning,
use of total body irradiation, patient or donor sex were identified (Online Supplementary Table S2).
Median age of patients with CNS relapse was 41 years (IQR: 30.8-50.4). Sixty-one patients (67%) were transplanted in CR1, 27 (29.7%) in CR2, and 3 (3.3%) in CR3. Thirty-six (39.6%) showed an isolated CNS relapse, 30 (33%) a combination of CNS and bone marrow (BM) relapse, 23 (25.3%) suffered a CNS relapse secondary to a hematologic relapse, and 2 (2.2%) after a molecular relapse. Of all CNS relapse patients, 72.5% did not show GvHD before the CNS relapse while 27.5% did. Median time from transplant to CNS relapse was 396 days (IQR: 197-737).
Fifty-two patients had AML (57.1%), 39 ALL (42.9%) (see Online Supplementary Table S3 for patients’ and transplant characteristics). ALL patients with CNS relapse were younger: 33.3 years versus 46.1 years in AML (P<0.01). Time from transplant to CNS relapse was 401 days in AML (IQR: 247.8-834.2) and 343 days in ALL (IQR: 128.5-665.5) patients (P=0.092). The median follow-up after CNS relapse was 88.3 months.
For 66 patients (72.5%), CNS relapse was the first relapse, 25.3% already had a hematologic relapse, and 2.2% already had a molecular relapse. Cumulative incidence of CNS relapse at five years was 4% (95% CI: 2.2-6.5) for AML CBF patients versus 0.7% (95% CI: 0.5-1) for patients with intermediate or adverse risk AML (P<10-3). All 13 CBF AML patients who relapsed have received cytarabine before transplant; however, we do not have information on exact dosages, neither do we have information on the chemotherapy regimen in CBF AML patients who did not relapse. Thirty-six AML and 30 ALL patients developed CNS relapse as first relapse post HSCT. Thirty-one (48.6%) achieved CR after the CNS relapse (41.2% CR in AML and 56.7% in ALL,
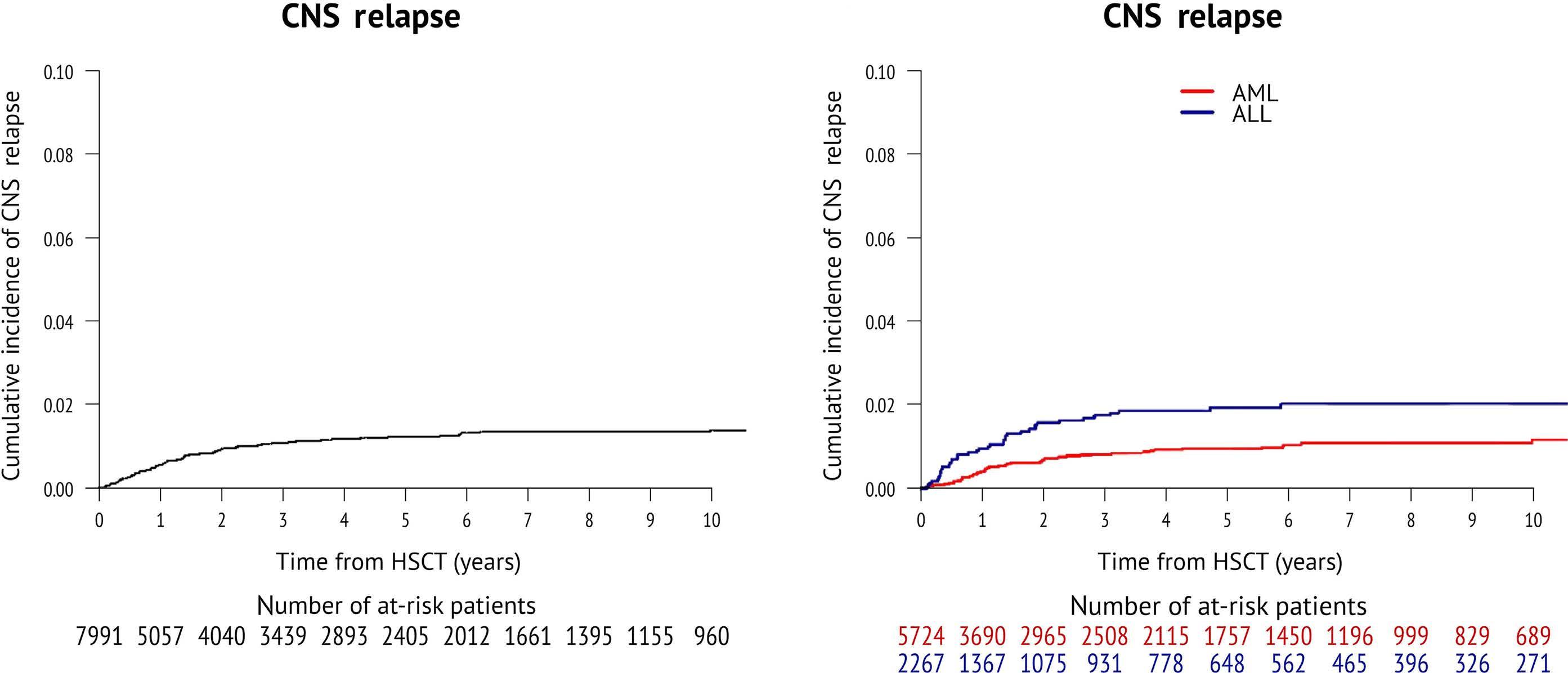
P=0.22). Second CNS relapse after the first was detected in 15 (22.7%) of these patients: 5 in AML (13.9%) and 10 (33.3%) in ALL patients.
The OS for the 66 patients was 24% at two years and 12% at five years (Figure 2B). There was no difference in OS between AML and ALL patients (AML: 28.8% and 8.2% at 2 and 5 years; ALL: 19.4% and 15.6% at 2 and 5 years, P=0.66) (Figure 3A). The 2-year OS was better in isolated CNS relapse versus CNS and concomitant BM relapse (37.5% vs. 6.7%, P=0.004) (Figure 3B).
This study is the largest multicenter study on CNS relapse in leukemia after allo-HSCT to date. We demonstrate that CNS relapse after allo-HSCT is a rare event (1.1%), but is more frequent in ALL than in AML; the prognosis, however, is dismal. Around half the patients could achieve CR, but the OS remains low (8.2% in AML, 15.5% in ALL at 5 years). OS was better in isolated CNS (37.5% vs. 6.7% at 2 years). Only 7 of the 91 patients were alive five years after CNS relapse, 6 patients were censored alive before five years, so only 13 of 91 patients were alive at their last follow-up
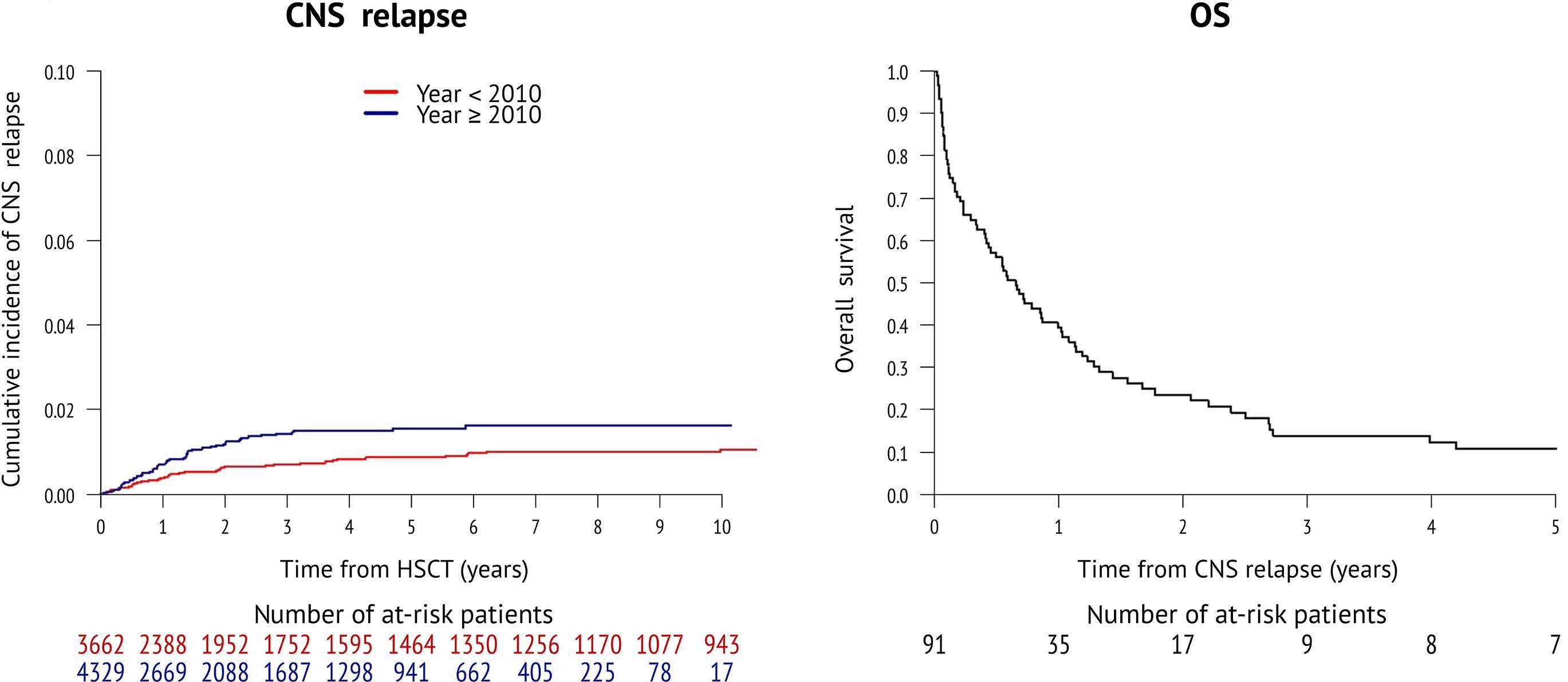
2. Central nervous system relapse by year and overall survival after relapse. (A) Central nervous system (CNS) relapse by year. (B) Overall survival (OS) after relapse for the entire population. HSCT: hematopoietic stem cell transplantation.
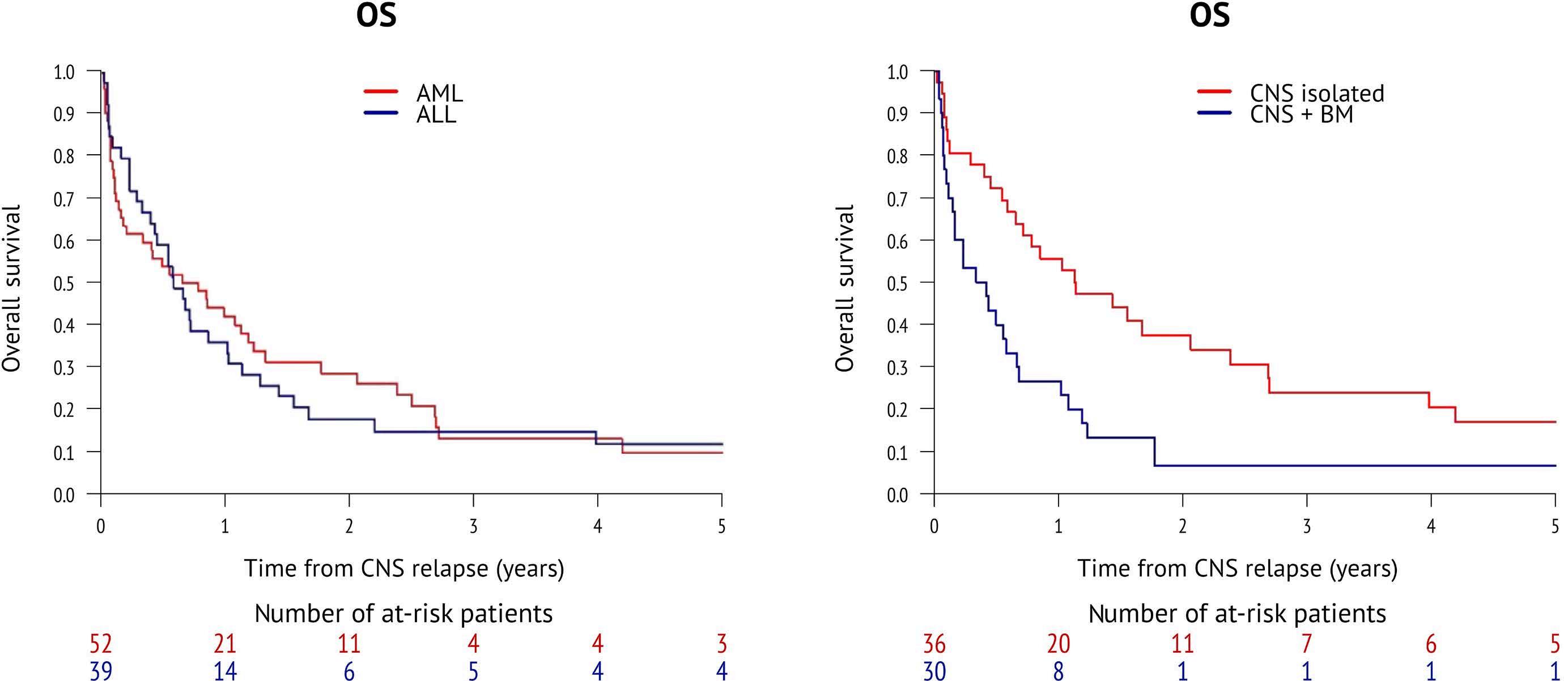
Figure 3. Overall survival by diagnosis and central nervous system isolated versus combined with bone marrow. (A) Overall survival (OS) after relapse year of diagnosis. (B) OS after isolated central nervous system (CNS) relapse versus bone marrow (BM) and CNS relapse. HSCT: hematopoietic stem cell transplantation.
with a range of 1-22 years after relapse. In conclusion, CNS relapse after allo-HSCT still has a dismal prognosis with few long-term survivors.
Our study has several limitations: no routine screening for CNS disease at diagnosis at all participating centers, and heterogeneous treatment and conditioning for allo-HSCT. The higher incidence in recent years is probably due to better reporting of CNS relapses. With current treatment protocols with routine intrathecal chemotherapy and/or prophylaxis, it is difficult to lower relapse rates after allo-HSCT in ALL. In AML, screening for concomitant CNS disease at diagnosis is not routinely performed, and future studies are needed to investigate a possible correlation between CNS infiltration at diagnosis and relapse after allo-HSCT. Currently, early detection and treatment of CNS relapse after allo-HSCT seems to be the best and only measure to take for this otherwise dismal complication, as our study demonstrates a better outcome in patients with isolated CNS as compared to a combined CNS and BM relapse. There does not seem to be the need to adapt the approach of screening or adding post-transplant prophylaxis based on leukemia subtypes as no difference was detected. However, due to the retrospective nature of our study, no definite recommendation can be made.
Sabine Blum,1* Yves Chalandon,2* Myriam Labopin,3 Jürgen Finke,4 Tobias Gedde-Dahl,5 Tarek Ben Othman,6 Jan J. Cornelissen,7 Pavel Jindra,8 Hélène Labussière-Wallet,9 Matthew Collin,10 Stig Lenhoff,11 Guido Kobbe,12 Norma C. Gutiérrez,13 Arnon Nagler14 and Mohamad Mohty15
1Hematology Service, University Hospital and University of Lausanne, Lausanne, Switzerland; 2Division of Hematology and Faculty of Medicine, Geneva University Hospitals and University of Geneva, Geneva, Switzerland; 3Sorbonne University, Department of Hematology, EBMT Paris Study Office, Hôpital Saint Antoine, and INSERM UMRs 938, Paris, France; 4University Hospital of Freiburg, Freiburg, Germany; 5University Hospital Oslo, Oslo, Norway; 6Centre National de Greffe de Moelle, Tunis, Tunisia; 7Erasmus MC Cancer Institute, University Rotterdam, Rotterdam, The Netherlands; 8Charles University Hospital, Pilsen, Czech Republic; 9Hôpital Lyon Sud, Pierre Bénite, France; 10Freeman Hospital, Newcastle Upon Tyne, UK; 11Skanes University Hospital, Lund, Sweden; 12University Hospital Duesseldorf, Heinrich
1. Chen Q, Zhu XL, Zhao X, et al. Prognosis and risk factors for central nervous system relapse after allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia. Ann Hematol. 2021;100(2):505-516.
Heine University, Medical Faculty, Duesseldorf, Germany; 13University Hospital of Salamanca, IBSAL, Cancer Research Center-IBMCC (USALCSIC), CIBERONC (CB16/12/00233), Salamanca, Spain; 14Chaim Sheba Medical Center, Tel-Hashomer, Israel and 15Sorbonne University, SaintAntoine Hospital, AP-HP, INSERM UMRs 938, Paris, France
*SB and YC contributed equally as first authors.
Correspondence:
S. BLUM - sabine.blum@chuv.ch
Y. CHALANDON - yves.chalandon@hcuge.ch
https://doi.org/10.3324/haematol.2023.284858
Received: December 15, 2023. Accepted: February 27, 2024. Early view: March 7, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
YC reports consulting fees from MSD, Novartis, Incyte, BMS, Pfizer, Abbvie, Roche, Jazz, Gilead, Amgen, Astra-Zeneca, and Servier, and travel support from MSD, Roche, Gilead, Amgen, Incyte, Abbvie, Janssen, Astra-Zeneca, and Jazz. GK reports an Advisory Role or Speaker Honoraria from MSD, Pfizer, Amgen, Novartis, Gilead, BMSCelgene, Abbvie, Biotest, Takeda, Eurocept, and JAZZ, and Financing of Scientific Research from BMS-Celgene, Amgen, Abbvie, Medac and Eurocept. None of the other authors have any conflicts of interest to disclose.
Contributions
SB, YC and ML wrote the manuscript. ML performed statistical analyses. AN and MM revised the manuscript. SB, YC, JF, TGD, TBO, JJC, PJ, HLW, MC, SL, GK, NCG, AN and MM provided patients and patient follow-up. All authors approved the manuscript before submission.
Funding
This study was supported by a grant from the Ministry of Health of the Czech Republic – Conceptual Development of Research Organization (Faculty Hospital in Pilsen – FNPl, 00669806).
Data-sharing statement
Enquiries for data sharing can be made to the EBMT Acute Leukemia Working Party & Lymphoma Working Party.
2. Tatarian J, Byrd K, Male HJ, Lin TL. Central nervous system involvement in adult acute myeloid leukemia patients. Leuk Res. 2022;118:106882.
3. Del Principe MI, Buccisano F, Soddu S, et al. Involvement of
central nervous system in adult patients with acute myeloid leukemia: incidence and impact on outcome. Semin Hematol. 2018;55(4):209-214.
4 Pinkel D, Simone J, Hustu HO, Aur RJ. Nine years’ experience with “total therapy” of childhood acute lymphocytic leukemia. Pediatrics. 1972;50(2):246-251.
5. Williams MT, Yousafzai YM, Elder A, et al. The ability to cross the blood-cerebrospinal fluid barrier is a generic property of acute lymphoblastic leukemia blasts. Blood. 2016;127(16):1998-2006.
6. Poon LM, Hamdi A, Saliba R, et al. Outcomes of adults with acute lymphoblastic leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19(7):1059-1064.
7 Bassan R, Barbui T. Remission induction therapy for adults with acute myelogenous leukemia: towards the ICE age? Haematologica. 1995;80(1):82-90.
8. Holmes R, Keating MJ, Cork A, et al. A unique pattern of central nervous system leukemia in acute myelomonocytic leukemia
associated with inv(16)(p13q22). Blood. 1985;65(5):1071-1078.
9 Rees JK, Gray RG, Swirsky D, Hayhoe FG. Principal results of the Medical Research Council’s 8th acute myeloid leukaemia trial. Lancet. 1986;2(8518):1236-1241.
10 Hamdi A, Mawad R, Bassett R, et al. Central nervous system relapse in adults with acute lymphoblastic leukemia after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2014;20(11):1767-1771.
11. Mayadev JS, Douglas JG, Storer BE, Appelbaum FR, Storb R. Impact of cranial irradiation added to intrathecal conditioning in hematopoietic cell transplantation in adult acute myeloid leukemia with central nervous system involvement. Int J Radiat Oncol Biol Phys. 2011;80(1):193-198.
12. Oshima K, Kanda Y, Yamashita T, et al. Central nervous system relapse of leukemia after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2008;14(10):1100-1107.
Platelet function disorders are common inherited bleeding disorders.1,2 While severe inherited platelet function disorders (IPFD) such as Glanzmann thrombasthenia and Bernard Soulier syndrome are easier to diagnose, diagnosing milder phenotypes of IPFD remains complex. Up to 9% of healthy children can have epistaxis, and 12% of infants in the general pediatric population demonstrate bruising, complicating the ability to diagnose IPFD in otherwise healthy children.3,4 Structured bleeding assessment tools (BAT), such as those developed by the International Society of Thrombosis and Hemostasis (ISTH), are useful as screening instruments to guide testing for bleeding disorders.5,6 Our group has previously established an IPFD incidence of 4.5% in an unselected, prospective cohort of adolescents referred for heavy menstrual bleeding (HMB) using ISTH-BAT and systematic testing for IPFD.7 We aimed to determine the prevalence and types of IPFD in children referred for bleeding, examine bleeding events and annualized bleeding rates (ABR), and investigate the predictive ability of the ISTH-BAT for IPFD diagnosis, treatment for bleeding events, and severe bleeding events. We hypothesized that bleeding scores (BS) are higher in pediatric patients with IPFD than those without, and that the ISTH-BAT would predict bleeding events and receipt of treatment in IPFD.
Our retrospective study, approved by the institutional review board, included patients aged 0 to 18 years referred for bleeding symptoms between 2019 and 2021 to The University of Texas Southwestern Medical Center (UTSW) Pediatric Hemostasis and Thrombosis Program. A standardized hemostatic evaluation is undertaken for every patient with high suspicion for primary hemostatic defect. The minimum standardized evaluation includes a complete blood count
with mean platelet volume, blood smear, prothrombin time, activated partial thromboplastin time, fibrinogen activity, von Willebrand panel, and platelet aggregation using whole blood impedance aggregometry (WBA). Additional testing is individualized and may include clotting factor assays, evaluation for disorders of hyperfibrinolysis, platelet flow cytometry, and/or genetic testing. To be eligible, we included patients who completed more than one clinic visit and, either had an ISTH-BAT documented in the medical chart or sufficient bleeding history captured to calculate the ISTH-BAT, and underwent the minimum standardized hemostatic evaluation. Only patients with reproducible (≥2) abnormal WBA findings were included. We excluded patients without documented bleeding histories, those with anticoagulant-associated bleeding, or those with additional causes of increased bleeding risk.
Demographic and clinical information from each patient’s electronic medical record was recorded. IPFD was diagnosed when there was impaired aggregation to ≥2 agonists (excluding collagen or ristocetin) and/or impaired ATP secretion on ≥2 occasions using WBA and luminescence as previously described.7,8 von Willebrand disease (vWD) was defined as quantitative vWF levels <0.50 IU/dL (vWF antigen, vWF:RCo or vWF:GP1bM) on ≥2 occasions. All patients met Type 1 vWD (vWD-1) criteria based on previously established guidelines.9 Controls had a bleeding phenotype but no identifiable bleeding disorder on comprehensive laboratory evaluation. A total of 193 participants (67% female) with a median age of 13 years (interquartile range [IQR]: 7-15) were included; 18% (N=34) were diagnosed with IPFD, 24% (N=46) with vWD-1, and in 58% (N=113) the bleeding disorder evaluation did not reveal an identified hemostatic defect (Table 1). Of
of bleed , N (%)
N: number; vWD: von Willebrand disease; IPFD: inherited platelet function disorders; IQR: interquartile range; ISTH-BAT: International Society of Thrombosis and Hemostasis Bleeding Assessment Tools; ABR: annualized bleeding rates. Controls had a bleeding phenotype but no identifiable bleeding disorder on comprehensive laboratory evaluation.
the IPFD participants, 12 (35%) had aspirin-like defect, one (3%) had Bernard Soulier, and in the remaining 20 (59%), the identified defect was non-specific (reduced aggregation and/or secretion to ≥1 agonist).
Annualized bleeding rate was calculated by dividing the number of clinically significant episodes one year before the first evaluation by history and/or emergency department, doctor’s surgery, or hospital visits for bleeding. Median ABR was 12 (IQR: 2-13) for IPFD, 12 (IQR: 4.5-14) for vWD-1, and 12 (IQR: 1-13) for controls (P=0.13) (Figure 1A). The most frequent bleeding event in all groups was HMB (N=96, 48.2%). The subsequent most frequent bleeding event in the IPFD cohort was surgical bleeding, while in vWD-1 and controls, it was epistaxis (Online Supplementary Figure S1A).
Severe bleeding events, defined as any bleeding event requiring medical or surgical interventions including high-dose oral hormonal therapy every 6 or 8 hours/day for several days, intravenous estrogen, blood transfusion, and surgical interventions to stop bleeding, occurred in 18 (53%) IPFD, 20 (43%) vWD-1, and 50 (44%) control participants. The median number of bleeds requiring treatment was 2 (IQR: 1-12) for IPFD, 12 (IQR: 1-13) for vWD-1, and 3 (IQR 0-12) for controls (P=0.16) (Figure 1B). Receipt of treatment was defined as any bleeding requiring systemic therapy or surgical intervention for bleeding symptoms, but excluding topical agents (i.e., oxymetazoline for epistaxis). Hormonal therapy was the most utilized treatment in vWD-1 (N=26; 43%) and controls (N=56; 48%), whereas surgical therapy, including packing, suturing, and cauterization, was highest in IPFD (N=14; 33%) (Online Supplementary Figure S1B).
Median BS was 3 (IQR: 2.75-4) in the IPFD group, 4 (IQR: 3-5) in vWD-1, and 3 (IQR: 3-4) in controls (Figure 1C). BS between the vWD-1 and control groups varied significantly (P=0.012), while no significant difference was found between IPFD and vWD-1 or controls. Clinically significant bleeding events were defined as bleeding involving all sites but ex-
cluding bleeding lasting <10 minutes, events not requiring intervention, and all cutaneous bleeding except for wound bleeds lasting >10 minutes. In all study groups, median BS was significantly higher in those with clinically significant bleeding events than in those without (P<0.05) (Figure 2).
A BS >4 in IPFD (area under the curve [AUC]=0.85) and vWD-1 (AUC=0.85) groups predicted clinically significant bleeding events with very good accuracy (Online Supplementary Figure S2A). For bleeding events requiring treatment, a BS >3 in the IPFD group (AUC=0.95) predicted treatment with excellent accuracy, while a BS >2 in the vWD-1 group (AUC = 0.71) predicted treatment with good accuracy (Online Supplementary Figure S2B). Severe bleeding events were predicted at BS >7 in the IPFD group (AUC=0.89) with very good accuracy and BS >5 in the vWD-1 group (AUC=0.62) with sufficient accuracy (Online Supplementary Figure S2C). The likelihood of clinically significant bleeding events, bleeds requiring treatment, and severe bleeding events were significantly higher in the IPFD and vWD-1 groups when the BS was above the respective identified cut-off values, except for severe bleeding in vWD (Online Supplementary Figure S3). We show that IPFD is common among pediatric patients referred to a tertiary care center, with 18% of our participants diagnosed with IPFD. Varying incidence seen in other studies, such as 5% by Bidlingmaier et al.10 and 21% by Adler et al., 11 can be explained by differences in sample size, prospective nature, and different methods of testing platelet function. We demonstrate high ABR in IPFD participants that were not significantly different than those with vWD-1. Gresele et al 12 found higher bleeding rates in IPFD than vWD patients, but once patients with Glanzmann thrombasthenia and Bernard Soulier were removed, similar to our study population, no significant differences remained. A higher proportion (20%) of IPFD participants suffered surgical bleeding events compared to vWD-1 and required more invasive interventions, such as surgery. This is in stark contrast to the data
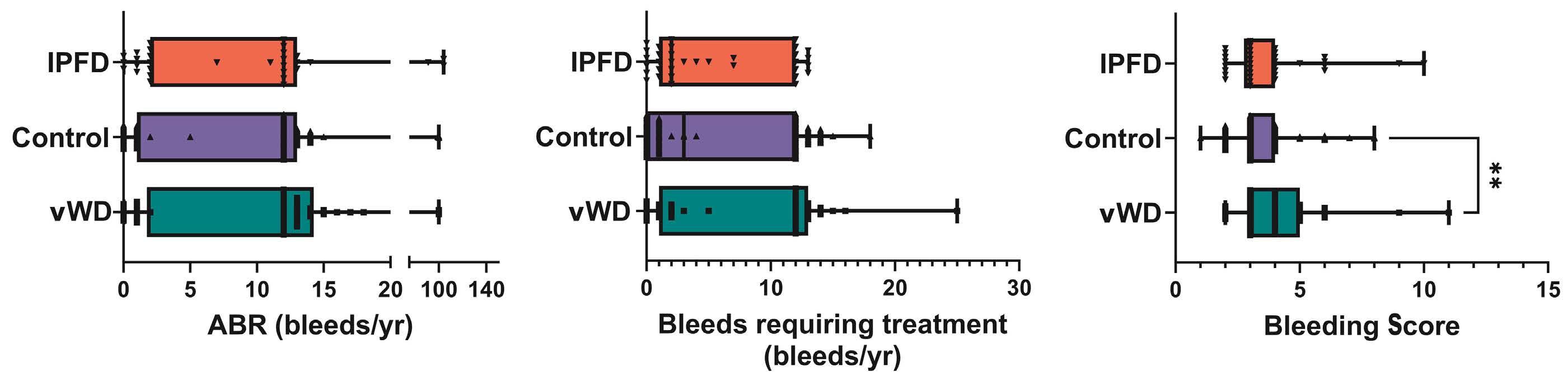
Figure 1. International Society of Thrombosis and Hemostasis Bleeding Assessment Tool Bleeding Score, annual bleeding rate, and bleeds requiring treatment among study population. (A) Annual bleeding rate (ABR), (B) bleeding events requiring treatment, and (C) International Society of Thrombosis and Hemostasis Bleeding Assessment Tools Bleeding Score between study group participants. Data are presented as medians and interquartile ranges. **P<0.01. vWD: von Willebrand disease; IPFD: inherited platelet function disorder; yr: years. Controls had a bleeding phenotype but no identifiable bleeding disorder on comprehensive laboratory evaluation.
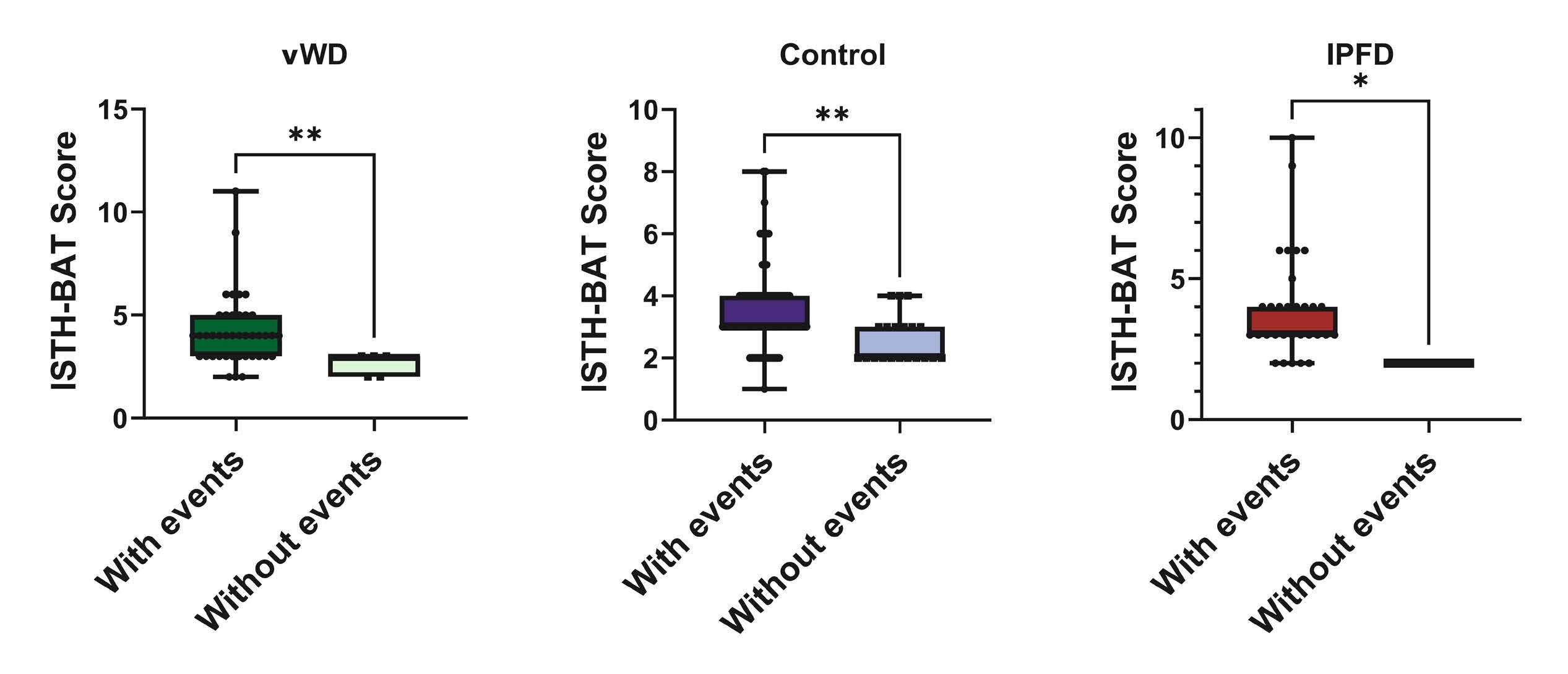
Figure 2. International Society of Thrombosis and Hemostasis Bleeding Assessment Tool Bleeding Score between participants with and without clinically significant bleeding events in each study group. Data are presented as medians and interquartile ranges. *P<0.05; **P<0.01. ISTH-BAT: International Society of Thrombosis and Hemostasis Bleeding Assessment Tools; vWD: von Willebrand disease; IPFD: inherited platelet function disorder. Controls had a bleeding phenotype but no identifiable bleeding disorder on comprehensive laboratory evaluation.
provided by Gresele et al., which reported <5% of pediatric IPFD bleeding events to be surgical, likely explained by the prospective nature of the study where IPFD diagnosis was already established and early intervention possible.12 ISTH-BAT scores were comparable for IPFD and vWD-1 participants. Gresele et al. found a significantly higher bleeding score in pediatric IPFD versus vWD-1 participants (8 vs. 4, respectively).12 However, Bidlingmaier et al. found no difference in BS, with both pediatric IPFD and vWD-1 participants having a BS of 3.10 This is likely driven by the inclusion of severe platelet disorders in the Gresele study, whereas the Bidlingmaier study included a similar cohort of IPFD as ours, leading to comparable results.
For our IPFD participants, BS >4 predicted subsequent bleeding events, BS >3 predicted bleeding events requiring treatment, and BS >7 predicted severe bleeding events. Our study predicted bleeding events at a lower BS than Gresele et al. (BS >7), likely due to higher ABR within our study population.12 Gresele et al. found that 75% of adult IPFD patients with a BS >2 required treatment, but did not comment on the ability of ISTH-BAT to predict treatment requirements.13 Gresele et al. found that a BS >6 was predictive of severe bleeding events in adult IPFD participants;12 their inclusion of severe bleeding disorders, such as Glanzmann thrombasthenia and Type 2 and 3 vWD, likely increased their ability to predict severe bleeding events at lower bleeding scores.
In summary, IPFD is common in children referred for bleeding and characterized by high ABR. IPFD presentation overlaps with children without an identified bleeding disorder and vWD-1. The ISTH-BAT alone was not discriminatory for IPFD in our cohort, highlighting the need for platelet aggregation testing in children referred for bleeding. Patients with
high ISTH-BAT scores at IPFD diagnosis are more likely to experience severe bleeding events and require treatment. While HMB was the most common cause of bleeding in all cohorts, IPFD participants received more surgical interventions compared to vWD-1 and controls, whose most common treatment was hormonal therapy; as such, HMB was undertreated in IPFD. Our results underscore the importance of high diagnostic suspicion for IPFD in children referred for a bleeding disorder evaluation. Further, we confirm the utility of the ISTH-BAT for IPFD and bleeding prognosis in pediatric patients.
Shelly Saini,1 Song Zhang,2 Sean Yates,3 Jessica Garcia,1,4 Ruchika Sharma,1,4 Joseph Formella,1 Ravindra Sarode3 and Ayesha Zia1,4
1Department of Pediatrics; 2O’Donnell School of Public Health; 3Department of Pathology and 4Division of Hematology/Oncology, The University of Texas Southwestern Medical Center, Dallas, TX, USA
Correspondence: A. ZIA - Ayesha.zia@utsouthwestern.edu
https://doi.org/10.3324/haematol.2024.284996
Received: January 7, 2024. Accepted: March 19, 2024. Early view: March 28, 2024.
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
SS and AZ conceptualized, designed, and performed research. SS, SZ and AZ analyzed the data. SY and RS blindly and independently adjudicated abnormal whole blood aggregation results for all participants. SS and AZ wrote, and all other authors critically edited the manuscript.
1. Podda G, Femia EA, Pugliano M, Cattaneo M. Congenital defects of platelet function. Platelets. 2012;23(7):552-563.
2. Nurden AT, Freson K, Seligsohn U. Inherited platelet disorders. Haemophilia. 2012;18(Suppl 4):154-160.
3. McGarry GW. Recurrent epistaxis in children. BMJ Clin Evid. 2013;2013:0311.
4 Carpenter RF. The prevalence and distribution of bruising in babies. Arch Dis Child. 1999;80(4):363-366.
5. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063-2065.
6. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20(6):831-835.
7 Zia A, Jain S, Kouides P, et al. Bleeding disorders in adolescents with heavy menstrual bleeding in a multicenter prospective US cohort. Haematologica. 2020;105(7):1969-1976.
8. Zhou L, Schmaier AH. Platelet aggregation testing in plateletrich plasma: description of procedures with the aim to develop standards in the field. Am J Clin Pathol. 2005;123(2):172-183.
Funding
This work was supported by the National Institutes of Health (grant N. 1R01HL153963), the American Heart Association (grant N. 20IPA35320263), and the National Heart, Lung, and Blood Institute (grant N. R38HL150214).
Data-sharing statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
9 James PD, Connell NT, Ameer B, et al. ASH ISTH NHF WFH 2021 guidelines on the diagnosis of von Willebrand disease. Blood Adv. 2021;5(1):280-300.
10 Bidlingmaier C, Grote V, Budde U, Olivieri M, Kurnik K. Prospective evaluation of a pediatric bleeding questionnaire and the ISTH bleeding assessment tool in children and parents in routine clinical practice. J Thromb Haemost. 2012;10(7):1335-1341.
11. Adler M, Kaufmann J, Alberio L, Nagler M. Diagnostic utility of the ISTH bleeding assessment tool in patients with suspected platelet function disorders. J Thromb Haemost. 2019;17(7):1104-1112.
12. Gresele P, Falcinelli E, Bury L, et al. The ISTH bleeding assessment tool as predictor of bleeding events in inherited platelet disorders: communication from the ISTH SSC Subcommittee on Platelet Physiology. J Thromb Haemost. 2021;19(5):1364-1371.
13. Gresele P, Orsini S, Noris P, et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: a communication from the Platelet Physiology SSC. J Thromb Haemost. 2020;18(3):732-739.
Immunoglobulin light-chain (AL) amyloidosis has shown a rapid and high rate of response to daratumumab, but Mayo stage IIIB patients are excluded from most trials.1 We aimed to prospectively evaluate the efficacy of daratumumab plus bortezomib and dexamethasone (Dara-VD) in Mayo stage III, especially IIIB, AL through this phase II study (clinicaltrials gov. Identifier: NCT04474938). The treatment scheme was intravenous daratumumab at 16 mg/kg weekly in cycles 1-2, every 2 weeks for cycles 3-6 and every 4 weeks for cycles 7-12, 1.3 mg/m2 bortezomib and 20 mg dexamethasone once weekly for six cycles of 28 days each. The primary outcome was hematologic response ≥ very good partial response (VGPR) at 3 months. A total of 40 patients were enrolled, including 20 with IIIB disease. The percentage of patients with response ≥ VGPR at 3 months was 67.5%, including 47.5% complete response (CR). The percentage who had a cardiac response at 6 months was 47.5%. After a median follow-up of 24.0 months, the 2-year overall survival (OS) estimate reached 69.8%. The response ≥ VGPR at 3 months (70.0%) and cardiac response at 6 months (50.0%) in the IIIB subgroup were comparable to those in the IIIA subgroup. The 2-year OS rate was 65.0% in IIIB patients. Grade ≥3 treatment-related adverse events occurred in 40.0% of patients. Dara-VD had favorable efficacy and safety in advanced AL amyloidosis, including IIIB disease. As is known, the benefits of bortezomib in severely advanced AL amyloidosis have not been demonstrated definitively.2,3 In the ANDROMEDA trial, the combination of daratumumab with bortezomib, cyclophosphamide and dexamethasone (Dara-VCD) achieved a deeper hematologic response and longer survival across cardiac stages.4,5 Unfortunately, patients with stage IIIB disease were excluded from the above study. Agents targeting the deposited amyloid fibrils on affected organs are appealing, but not ready for routine clinical practice.6,7 Thus, the management of advanced cardiac AL amyloidosis remains a major unmet medical need. The ongoing EMN22 study is evaluating daratumumab monotherapy in stage IIIB patients.8 Yet past experience has indicated that there might be only one chance to start effective treatment in patients with advanced heart involvement, who have low probability of receiving salvage therapy. Aside from monotherapy, a combination regimen followed by de-escalation is also worth prospective investigation. Therefore, we conducted this phase II, single-arm study to evaluate the efficacy and safety of front-line Dara-VD in both IIIA and IIIB AL amyloidosis patients (Online Supplementary Figure S1). The exclusion criteria included a
co-diagnosis of multiple myeloma (patients whose only myeloma defining event was serum free light chain ratio ≥100 could be included) or Waldenström’s macroglobulin-
Table 1. Baseline demographic and clinical characteristics of the patients (N=40).
Characteristics
Male, N (%)
(75.0)
(range)
Mayo 2004 stage IIIB, N (%)
(50.0) Mayo 2012 stage, N (%)
NYHA stage, N (%)
Symptom-diagnosis interval in months, median (range) 8 (2-43) Involved organs
Number of organs involved, N (%) 2 (1-4)
Laboratory values, median (range) cTnI, μg/L NT-proBNP, ng/L IVS, mm LVEF, % 24hUP, g eGFR, mL/min/1.73 m2 ALP, U/L* dFLC, mg/L BMPC, %
0.17 (0.07-3.07) 7,801 (803-35,000) 15 (11-23) 56 (32-83) 0.24 (0.03-12.57) 77 (12-124) 106 (38-643) 274 (72-2,966) 5 (0-25)
FISH, N (%) t(11;14), N†=39 Gain 1q21, N†=29 Del 17p, N†=29 14 (35.9) 4 (13.8) 2 (6.9)
*The upper limit of normal is 135 U/L. †Number of patients evaluated. 24hUP: 24-hour urine protein; ALP: alkaline phosphatase; BMPC: bone marrow plasms cell; cTnI: cardiac troponin-I; dFLC: difference between involved and uninvolved free light chain; eGFR: estimated glomerular filtration rate; FISH: fluorescence in situ hybridization; IVS: intraventricular septum; LVEF: left ventricular ejection fraction; NT-proBNP: N-terminal pro-brain natriuretic peptide; NYHA: New York Heart Association.
emia; severely compromised hematologic function; and inadequate liver function or estimated glomerular filtration rate < 40 mL/min/1.73 m2 (those with renal dysfunction due to renal involvement could be included). All patients gave written informed consent prior to inclusion in this study, which was approved by the Institutional Ethics Committee of Peking Union Medical College Hospital (HS-2414) on June 23, 2020. Hematologic and organ responses were defined according to the current validated criteria.9-12 Patients who died before response assessment and did not have a post-baseline assessment were categorized under the no-response group. All analyses were performed based on the intention-to-treat principle.
From May 2021 to March 2022, 40 patients were included (Online Supplementary Figure S1). Baseline demographic and clinical characteristics are displayed in Table 1. One patient whose cardiac troponin-I was 0.07 µg/L was categorized as stage III since cardiac troponin-T was higher than 0.035 µg/L. The hematologic responses are presented in Table 2.
The percentage of patients who had a hematologic VGPR or better at 3 months after treatment initiation was 67.5% (95% confidence interval [CI]: 52.3-82.7), including 47.5% (95% CI: 31.3-63.7) with CR and 20.0% (95% CI: 7.0-33.0) with VGPR, and the overall response rate was 82.5% (95% CI: 70.2-94.8).
Considering the best hematologic response at any time point, the intention-to-treat estimate of the CR rate was 57.5%.
Twenty-eight (70.0%) patients achieved difference between involved and uninvolved free light chain (dFLC) <10 mg/L or involved free light chain ≤20 mg/L. One patient experienced
hematologic progression and discontinued treatment after eight cycles. The median time to first hematologic response was 7 days (range, 7-28 days). The median time to hematologic CR was 60 days (range, 7-200 days).
A summary of the cardiac, renal and hepatic response assessments is shown in Online Supplementary Table S1. The percentage who had a cardiac response at 6 months was 47.5%, including 2.5% CR, 25.0% VGPR and 20.0% partial response. Cardiac progression at 6 months was observed in four (10.0%) patients. The median time to first cardiac response was 3 months (range, 1-11 months). The percentages of patients who had a renal response and a hepatic response were 41.7% and 20.0%. The median follow-up duration was 24.0 months. As of the last follow-up, eight patients had discontinued treatment due to cardiac sudden death, and two patients had died due to cardiac sudden death several months after withdrawal. Two patients died because of heart failure exacerbation from infection. The early mortality rates (within 1 month and 3 months from treatment initiation) were 15.0% and 17.5%, respectively. The 2-year OS estimate was 69.8% (95% CI: 53.0-81.6). Treatment-related adverse events (TRAE) are listed in Online Supplementary Table S2. Grade ≥3 TRAE occurred in 16 (40.0%) patients. The most common grade 3 TRAE were diarrhea (10.0%). Grade 4 diarrhea was noted in one patient and improved after symptomatic treatment. Grade 3 or higher pulmonary infection were noted in three patients, including bacterial pneumonia in two patients and COVID-19 pneumonia in one patient. Grade 5 bacterial pneumonia
Table 2. Summary of overall confirmed hematologic responses (N=40).
stage IIIA
occurred in one patient, who died of infection-induced heart failure exacerbation afterward. Four patients, one patient and one patient suspended bortezomib due to diarrhea, intestinal obstruction and acute cholecystitis, respectively. One patient reduced the dose of bortezomib because of diarrhea.
The proportions of hematologic CR, VGPR or better in Mayo stage IIIB patients were comparable to those in Mayo stage IIIA patients (Table 2). Although the early mortality rate within 1 month was higher in the IIIB subgroup (25.0% and 5.0%, respectively), the percentages of cardiac response at 6 months were similar between IIIA and IIIB (45.0% and 50.0%, respectively). The 2-year OS rates were 74.3% (95% CI: 48.7-88.4) and 65.0% (95% CI: 40.3-81.5) in the IIIA and IIIB subgroups, respectively (Figure 1).
To our knowledge, this is the first study in AL patients with stage III disease, including those with N-terminal pro-brain natriuretic peptide >8,500 ng/L, that prospectively explored the efficacy and safety of a daratumumab-based regimen. In the current study, the hematologic and cardiac responses of Dara-VD compared favorably with those reported with bortezomib-based therapy. Median OS was significantly extended even in the IIIB subgroup.13 Along with pursuing a rapid and deep hematologic response, minimizing side effects is equally important in the management of advanced cardiac AL patients. Thus, the EMN22 study tried to explore the feasibility of daratumumab monotherapy in newly diagnosed stage IIIB patients and reported early results in 40 patients.8 The overall hematologic response rate was 70.0% at 3 months and cardiac response rate was 27.5% at 6 months,8 lower than the estimates in IIIB patients from our study. Their median OS was 10.3 months,8 inferior to the prognosis in the current study. The early mortality rate within 1 month was 7.5%, lower than the estimate from our study. As far as we know, single-agent daratumumab may compromise the rapidity and depth of the hematologic response, which is strongly associated with the cardiac response and long-term survival. Before the routine application of anti-fibrillary antibodies in AL amyloidosis, our findings highlight the importance of upfront combination treatment in late-stage patients.
Given the strong anti-plasma cell effect of daratumumab combined with bortezomib, we wondered whether it was necessary to include alkylating drugs in first-line therapy. Thus, when we designed the current trial, cyclophosphamide was abandoned to reduce myelosuppression. Chakraborty et al. recently published a retrospective analysis of 19 IIIB patients treated with Dara-VCD front-line therapy.14 The proportions of patients achieving ≥ VGPR at 3 months were similar.14 Cardiac response was achieved by 56% of Chakraborty et al.’s patients, and their 1-year OS was 67.5%,14 similar to the estimates in our study. The early mortality rate within 1 month was 10.5%. Grade ≥3 infections were noted in 21.1% of their patients,14 while the percentage in our study was 15.0%. The comparable response and survival further support the advantage of the upfront combination
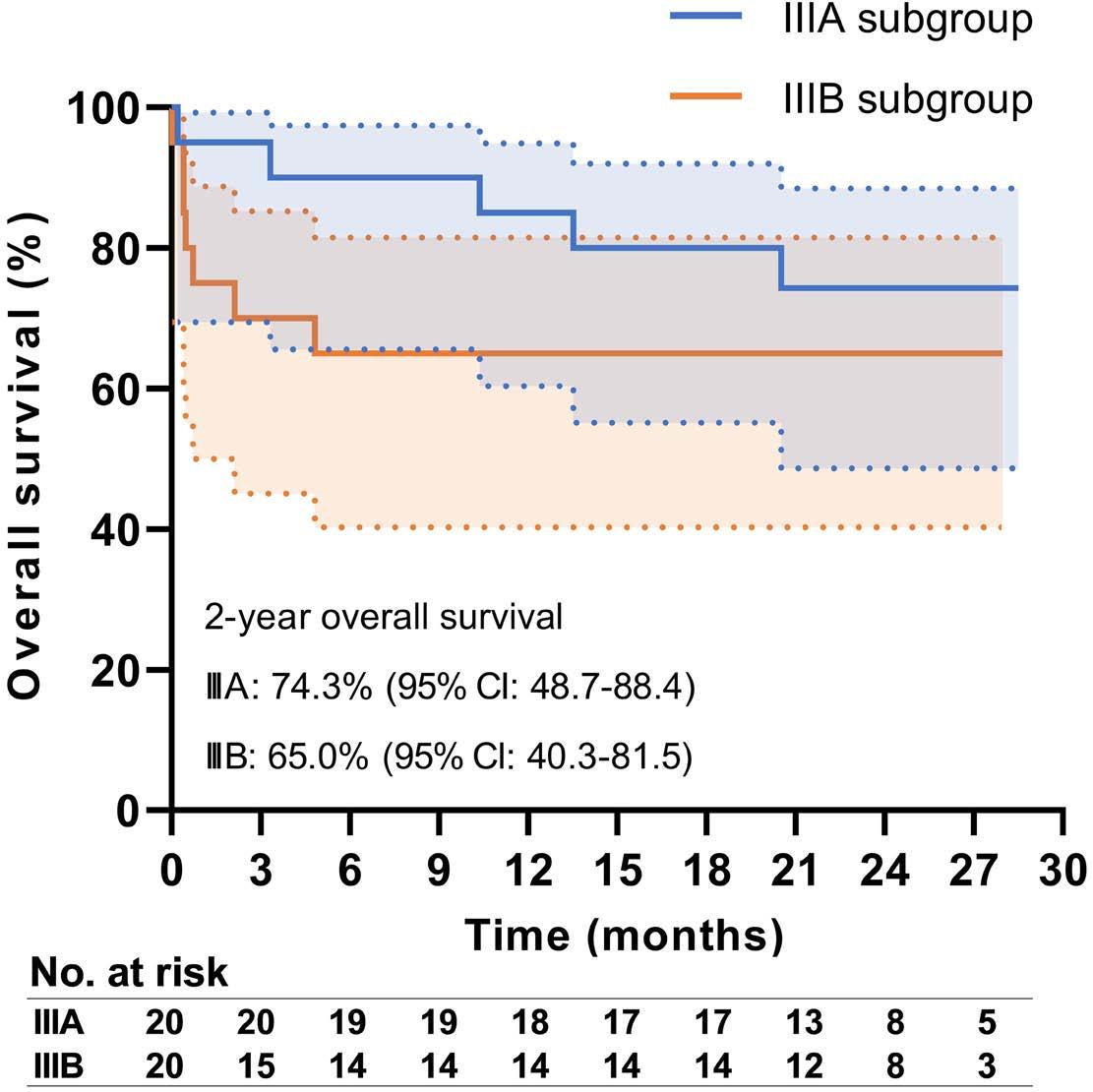
Figure 1. Kaplan-Meier graph of overall survival by Mayo 2004 stage. The blue and orange areas indicate the 95% confidence interval (CI) of overall survival.
regimen in IIIB patients. As we assumed, alkylating agents do not need to be used initially.
Of note, bortezomib-related diarrhea was prominent in this group of patients with advanced cardiac damage.4 We did record grade 3-4 diarrhea in 12.5% of cases. Therefore, close monitoring and thorough patient education are key to avoiding volume deletion and electrolyte disturbances from diarrhea. It was noteworthy that five early cardiac deaths occurred in stage IIIB patients. One limitation of current study was that the starting dose of bortezomib was not reduced for these patients with very advanced disease. In a recently published retrospective study on IIIB patients treated with daratumumab either with or without bortezomib, the percentage of patients who experienced early mortality within the first 2 months after initiating therapy was also as high as 22%.15 It remains uncertain whether a dose titration of bortezomib and dexamethasone could have potentially reduced arrhythmic deaths while maintaining a satisfactory impact on treatment response quality and rate in stage IIIB patients. Only a further comparative study might answer this question.
In conclusion, our trial demonstrates that the Dara-VD regimen is well tolerated and has promising efficacy even in AL patients with very advanced cardiac involvement.
Kai-ni Shen,1* Ya-juan Gao,1* Long Chang,1 Lu Zhang,1 Xin-xin Cao,1 Zhuang Tian,2 Yi-ning Wang,3 Dao-bin Zhou1 and Jian Li1,4
1Department of Hematology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union
Medical College; 2Department of Cardiology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College; 3Department of Radiology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College and 4State Key Laboratory of Common Mechanism Research for Major Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
*KNS and YJG contributed equally as first authors.
Correspondence: J. LI - lijian@pumch.cn
https://doi.org/10.3324/haematol.2024.285145
Received: February 3, 2024.
Accepted: March 15, 2024.
Early view: March 28, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
1. Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013;121(17):3420-3427.
2. Palladini G, Schonland S, Merlini G, et al. The management of light chain (AL) amyloidosis in Europe: clinical characteristics, treatment patterns, and efficacy outcomes between 2004 and 2018. Blood Cancer J. 2023;13(1):19.
3. Shen KN, Zhang CL, Tian Z, et al. Bortezomib-based chemotherapy reduces early mortality and improves outcomes in patients with ultra-high-risk light-chain amyloidosis: a retrospective case control study. Amyloid. 2019;26(2):66-73.
4. Kastritis E, Palladini G, Minnema MC, et al. Daratumumabbased treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385(1):46-58.
5. Minnema MC, Dispenzieri A, Merlini G, et al. Outcomes by cardiac stage in patients with newly diagnosed AL amyloidosis: phase 3 ANDROMEDA trial. JACC CardioOncol. 2022;4(4):474-487.
6. Edwards CV, Rao N, Bhutani D, et al. Phase 1a/b study of monoclonal antibody CAEL-101 (11-1F4) in patients with AL amyloidosis. Blood. 2021;138(25):2632-2641.
7. Shen KN, Fu WJ, Wu Y, et al. Doxycycline combined with bortezomib-cyclophosphamide-dexamethasone chemotherapy for newly diagnosed cardiac light-chain amyloidosis: a multicenter randomized controlled trial. Circulation. 2022;145(1):8-17.
Disclosures
No conflicts of interest to disclose.
Contributions
KNS and JL contributed to the concept and design of the study. KNS, YJG, LC, LZ, XXC, ZT, YNW, DBZ and JL collected data, KNS and YJG further contributed to data analysis and drafted the manuscript. All authors contributed to data interpretation, critically reviewed the manuscript and approved the final version.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 82200230, to KNS); the National High Level Hospital Clinical Research Funding (grant no. 2022-PUMCH-A-260, to KNS); the CAMS Innovation Fund for Medical Sciences (grant no. 2021I2M-1-019, to JL); and the National High Level Hospital Clinical Research Funding (grant no. 2022-PUMCH-C-051, to JL); and funding from Xi’an Janssen Pharmaceutical Ltd., Beijing, China.
Data-sharing statement
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
9. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541-4549.
10 Palladini G, Schonland SO, Sanchorawala V, et al. Clarification on the definition of complete haematologic response in lightchain (AL) amyloidosis. Amyloid. 2021;28(1):1-2.
11. Muchtar E, Dispenzieri A, Wisniowski B, et al. Graded cardiac response criteria for patients with systemic light chain amyloidosis. J Clin Oncol. 2023;41(7):1393-1403.
12. Palladini G, Hegenbart U, Milani P, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood. 2014;124(15):2325-2332.
13. Gao YJ, Shen KN, Miao HL, et al. A retrospective study of ultrahigh-risk (Mayo 2004 stage IIIb) AL amyloidosis and identification of predictors related to extremely early death. Leuk Lymphoma. 2022;63(7):1758-1761.
14 Chakraborty R, Rosenbaum C, Kaur G, et al. First report of outcomes in patients with stage IIIb AL amyloidosis treated with Dara-VCD front-line therapy. Br J Haematol. 2023;201(5):913-916.
15. Oubari S, Hegenbart U, Schoder R, et al. Daratumumab in first-line treatment of patients with light chain amyloidosis and Mayo stage IIIb improves treatment response and overall survival. Haematologica. 2024;109(1):220-230.
8. Kastritis E, Minnema M, Dimopoulos MA, et al. Efficacy and safety of daratumumab monotherapy in newly diagnosed patients with stage 3B light chain amyloidosis: a phase 2 study by the European Myeloma Network. Blood. 2023;142(Suppl 1):539.
Primary central nervous system lymphoma (PCNSL) is a separate group of lymphomas confined to the central nervous system (CNS) including the eyes. More than 95% of the cases represent aggressive CD20+ diffuse large B-cell lymphoma (DLBCL) with distinct clinical and biological features and poor prognosis.1
The outcome of PCNSL is particularly poor among elderly patients, and the improvements achieved in survival during recent decades appear to be restricted to younger PCNSL patients.2,3 The main feature of our Nordic phase II PCNSL study was its focus on elderly patients. The treatment for the elderly subgroup in our study consisted of de-escalated induction immunochemotherapy followed by maintenance temozolomide. We have previously reported the 22-month results with 2-year overall survival (OS) rates of 60.7% and 55.6% (P=0.40) and progression-free survival (PFS) rates of 33.1% and 44.4% (P=0.74), among younger and elderly patients, respectively.4 In our study, we found an astonishing good result for the elderly patients with a median duration of response (DOR) not reached among elderly patients, compared to a median DOR of 10 months among younger patients treated with more intensive immunochemotherapy but without maintenance temozolomide. One of our hypotheses is that the improved results among the elderly subgroup were due to maintenance temozolomide therapy. In PCNSL, late relapses occur but long-term follow-up data is scarce. Herein, we present long-term outcomes and neurocognitive performance at 10 years after diagnosis in the Nordic phase II first-line PCNSL study (clinicaltrials gov. Identifier: 01458730).
In this prospective multicenter phase ll study, immunocompetent, newly diagnosed, and histologically confirmed PCNSL patients up to 75 years of age at diagnosis were treated with an age-adjusted multi-agent immunochemotherapy regimen. The induction regimen for the younger subgroup (18-65 years at diagnosis) included high-dose methotrexate (HD-MTX) and HD-cytarabine combined with rituximab, ifosfamide, cyclophosphamide, dexamethasone, intrathecally administered liposomal cytarabine, vindesine, and vincristine. In the de-escalated induction regimen for the elderly subgroup (66-75 years at diagnosis), cyclophosphamide and vincristine were replaced with temozolomide, followed by temozolomide maintenance in patients responding to induction therapy.4
For baseline evaluation, response assessment, and statistical design, see the original article.4 All patients were intended to be followed for 120 months after the end of treatment, which
in the younger subgroup was the end of induction therapy and, in the elderly subgroup, was the end of maintenance therapy. Patients with refractory disease, progressive disease (PD), and patients not achieving a complete response (CR) at the end of planned therapy went off-study and were followed for OS only. OS, PFS, and DOR were estimated by using the Kaplan-Meier method. Crude differences between the groups of patients were tested using the log-rank test. Median follow-up was calculated using the reverse Kaplan-Meier method.
The study was approved by the local ethics committees and conducted in agreement with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Sixty-six patients (35 males and 31 females) from 12 centers in Denmark, Norway, Sweden, and Finland were enrolled between May 2007 and October 2010. Baseline characteristics, treatment, responses, and adverse events have been previously reported.4 The overall response rate was 73.8% for the entire cohort, 69.9% in the younger and 80.8% in the elderly subgroup. Myelosuppression after HD-cytarabine cycles was the most common grade 3-4 adverse event. After a median follow-up of 10 years, the 10-year OS rate was 29.8% (95% confidence interval [CI]: 20.5-43.4), the 10-year PFS rate was 13.3% (95% CI: 6.6-26.8), and median DOR was 13.2 months (95% CI: 7.7-72.4) for the entire cohort. Among the younger patients, the 10-year OS rate was 30.4% (95% CI: 18.8-49.1), and the 10-year PFS rate was 8.5% (95% CI: 2.6-27.8). Likewise, the 10-year OS rate was 29.6% (95% CI: 16.6-53.0), and the 10-year PFS rate was 21.3% (95% CI: 8.646.9), for the elderly patients. Median PFS was 13 months for the entire cohort (range, 1,2-162,8 months), 11 months in the younger (range, 1.8-168.5 months), and 57 months (range, 0-140 months) in the elderly subgroups (Figure 1)..
Seventeen of 66 patients (25.8%) were known to be alive at 10 years after diagnosis. Long-term survivors were three younger male patients, eight younger female patients, two elderly male patients, and four elderly female patients. Seven patients had primary refractory disease, six younger patients, and one elderly patient. Early relapses less than 1 year after the end of treatment were observed in 21 patients. Of these, 13 patients belonged to the younger subgroup. Later relapse occurred in 14 patients, at 15-168 months after the completion of therapy.
The number of deaths at the end of the study was 46 (69.7% of the 66 patients enrolled), and three patients were lost to follow-up at around 8 years. The cause of death was PCNSL in 25 patients. Four patients (6%) aged 64, 66, 73, and 74
years at diagnosis experienced treatment-related death due to neutropenia and sepsis-induced multi-organ failure,
all after the first HD-cytarabine dose. The cause of death was either unknown or not indicated in 14 patients. In the
Figure 1. Overall survival, progression-free survival, and duration of response after complete remission. Long-term survival of patients and duration of complete remissions (CR). Demonstrative Kaplan-Meier curves depict (A) overall survival in the entire study population, (B) progression-free survival in the entire study population, (C) duration of response in the entire study population and differences in overall survival, progression-free survival, and duration of response between elderly and younger subgroups (D, E, and F, respectively).
remaining three cases, the cause of death was general deterioration, second malignancy, and neurotoxicity after radiotherapy at relapse.
In the entire study population, Eastern Cooperative Oncology Group performance score (ECOG PS), mini-mental state examination (MMSE), and functional independence measure (FIM) improved or remained unchanged in the majority of the patients with similar results among long-term survivors. For the long-term survivors, patient characteristics, ECOG PS, neurocognition, number, and localization of tumor(s) at diagnosis and relapse are shown in Table 1.
Here, we report the long-term results of the Nordic phase II study on newly diagnosed PCNSL patients treated with a multiagent immunochemotherapy and a de-escalated induction regimen followed by temozolomide maintenance only for elderly patients. In this study, we found a significant proportion of elderly long-term survivors, with a 10-year OS rate of 29.8%. We also report the improvement in both neurocognitive status as well as functional independence of the patients treated in our study.
With the longest follow-up among all published randomized trials in PCNSL, Ferreri et al. 6 have reported a 7-year OS rate of 70% with a median follow-up of 88 months in
patients treated in IELSG32 trial with MATRix followed by consolidation therapy. However, elderly patients (age 66-70 years at diagnosis) were eligible for that trial only if their PS was considerably good (ECOG PS 2 or less) whereas in our study, patients up to the age of 75 years at diagnosis were eligible for the trial also in poor PS (ECOG PS 0-4). Moreover, 44% of the patients in our elderly subgroup were older than 70 years at diagnosis and 26% of the patients had a poor PS (ECOG PS 3-4) leading it impossible to compare the results of these two studies.
The intergroup ANOCEF-GOELAMS study published by Omuro et al. 7 was the first randomized trial specifically designed for elderly PCNSL patients. In that study, two different HD-MTX-based regimens were studied with one of the regimens containing temozolomide, which was also a part of the regimen for elderly patients in our study.
In ANOCEF-GOELAMS, 48 patients were assigned to the MTX-temozolomide arm, with a 2-year OS rate of 39% in this group but no long-term follow-up data published. In our study, the 2-year OS rate for elderly patients was notably higher, 55.6%.4
Two phase II studies including elderly PCNSL patients have published long-term results with 10-year follow-up data.8,9
Table 1. Patient characteristics, Eastern Cooperative Oncology Group performance score, neurocognitive data, number and localization of the tumor, localization of tumor at relapse for the confirmed long-term survivors. F: female; M:
Seidel et al.8 reported a Bonn group’s study with a 10-year OS rate of 9% in patients above 60 years of age at diagnosis whereas there were no long-term survivors at 10 years in the other phase II trial MATILDE reported by Ferreri et al. 9 Compared to these results, the long-term survival of elderly patients in our trial is significantly higher. With the median OS of 33.3 months among elderly patients in our study, the results remain more favorable also compared to other studies specifically designed for elderly PCNSL patients (with a median OS ranging from 14 to 17.5 months).7,10-12
In summary, the Nordic PCNSL study was finalized as planned 10 years after the end of treatment. Seventeen of 66 patients were alive and in good health based on their PS (ECOG PS of 0-1 in most of the long-term survivors), MMSE, and FIM scores (at or close to the maximum level in almost all longterm survivors). We conclude that, in our study, consolidation with maintenance temozolomide has been beneficial regarding survival in vulnerable elderly PCNSL patients. Based on our results, we suggest future clinical trials for elderly PCNSL patients to study novel agents in combination with a HD-MTX-based induction immunochemotherapy regimen without HD-cytarabine, followed by maintenance temozolomide consolidation. Additional, preferably randomized, studies, are warranted to clarify whether consolidation with maintenance therapy could be of benefit to sustain remission and improve survival also in younger PCNSL patients, particularly those that are not fit for dose-intensive consolidation therapy. Having the potential for late relapses and neurotoxicity in mind, we find that all prospective PCNSL clinical trials should routinely report at least 10-year follow-up results in the future.
Elisa Jacobsen Pulczynski,1,2 Mikkel Runason Simonsen,3,4 Outi Kuittinen,5,6 Unn-Merete Fagerli,7,8 Martin Erlanson,9 Øystein Fluge,10 Sirpa Leppä,11,12 Bjørn Østenstad,13 Alexander Fosså,13,14 Mikael Eriksson,15,16 Tarec El-Galaly,17 Hanne Kuitunen,18 Karin Papworth,9 Maria Ljungqvist,19,20 Martin B. Pedersen1 and Marjukka Pollari11,21
1Department of Hematology, Aarhus University Hospital, Aarhus, Denmark; 2Department of Hematology, Region Hospital Goedstrup, Herning, Denmark; 3Department of Hematology, Clinical Cancer Research Center, Aalborg University Hospital, Aalborg, Denmark; 4Department of Mathematical Sciences, Aalborg University, Aalborg, Denmark; 5Department of Oncology, Kuopio University Hospital Cancer Center, Kuopio, Finland; 6Faculty of Medicine, Institute of Clinical Medicine, University of Eastern Finland, Kuopio, Finland; 7Department of Oncology, St Olav University Hospital, Trondheim, Norway; 8Department of Clinical and Molecular Medicine, Norwegian University of Science and Technology (NTNU), Trondheim, Norway; 9Department of Radiation Sciences, Umeå University, Umeå, Sweden; 10Department of Oncology and Medical Physics, Haukeland University Hospital, Bergen, Norway; 11Research Programs Unit/Applied Tumor
Genomics Research Program, University of Helsinki, Helsinki, Finland; 12Department of Oncology, Helsinki University Hospital Comprehensive Cancer Center, Helsinki, Finland; 13Department of Oncology, Oslo University Hospital, Oslo, Norway; 14KG Jebsen Center for B-cell Malignancies, University of Oslo, Oslo, Norway; 15Department of Oncology, Skane University Hospital, Lund, Sweden; 16Medical Oncology, Lund University, Lund, Sweden; 17Department of Hematology, Aalborg University Hospital, Aalborg, Denmark; 18Cancer Center, Oulu University Hospital, Oulu, Finland; 19Department of Clinical Science and Education, Södersjukhuste, Karolinska Insitutet, Stockholm, Sweden; 20Hematology Center, Karolinska University Hospital, Stockholm, Sweden and 21Department of Oncology, Tays Cancer Center, Tampere University Hospital, Tampere, Finland
Correspondence:
M. POLLARI - marjukka.pollari@pirha.fi
https://doi.org/10.3324/haematol.2024.285207
Received: February 1, 2024.
Accepted: March 19, 2024.
Early view: March 28, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
EJP was the principal investigator (PI) of the study, the national PI in Denmark, responsible for the design of the study, data management, accrual of patients in Denmark, and wrote the manuscript together with MP. MRS performed data analysis. OK was involved in protocol planning, was the national PI in Finland, and responsible for accrual of patients in Finland. UMF was involved in protocol planning, was the national PI in Norway, and responsible for accrual of patients in Norway. ME was involved in protocol planning, was the national PI in Sweden, and responsible for accrual of patients in Sweden. ØF, BØ, and AF recruited and treated patients in Norway. SL recruited and treated patients in Finland. ME recruited and treated patients in Sweden. TEG, HK, KP, ML, and MBP are current members of the Nordic Primary Central Nervous System Lymphoma Group and were involved in writing the manuscript. MP is the chair of the Nordic Primary Central Nervous System Lymphoma Group, the corresponding author of this study, performed data analysis together with MRS, and wrote, revised as well as reformated the manuscript together with EJP.
Funding
This work was funded by grants from Nordic Cancer Union, Norpharma, Roche, Schering-Plough, and Inge og Jørgen Larsens Mindelegat.
Data-sharing statement
No shared data are available.
1. Braggio E, Van Wier S, Ojha J, et al. Genome-wide analysis uncovers novel recurrent alterations in primary central nervous system lymphomas. Clin Cancer Res. 2015;21(17):3986-3994.
2. Houillier C, Soussain C, Ghesquieres H, et al. Management and outcome of primary CNS lymphoma in the modern era: An LOC network study. Neurology. 2020;94(10):e1027-e1039.
3. Mendez JS, Ostrom QT, Gittleman H, et al. The elderly left behind-changes in survival trends of primary central nervous system lymphoma over the past 4 decades. Neuro Oncol. 2018;20(5):687-694.
4. Pulczynski EJ, Kuittinen O, Erlanson M, et al. Successful change of treatment strategy in elderly patients with primary central nervous system lymphoma by de-escalating induction and introducing temozolomide maintenance: results from a phase II study by the Nordic Lymphoma Group. Haematologica. 2015;100(4):534-540.
5. Ferreri AJ, Blay JY, Reni MA, et al. Prognostic scoring system for primary CNS lymphomas: the International Extranodal Lymphoma Study Group experience. J Clin Oncol 2003;21(2):266-272.
6. Ferreri AJM, Cwynarski K, Pulczynski E, et al. Long-term efficacy, safety and neurotolerability of MATRix regimen followed by autologous transplant in primary CNS lymphoma: 7-year results of the IELSG32 randomized trial. Leukemia. 2022;36(7):1870-1878.
7 Omuro A, Chinot O, Taillandier L, et al. Methotrexate and temozolomide versus methotrexate, procarbazine, vincristine, and cytarabine for primary CNS lymphoma in an elderly population: an intergroup ANOCEF-GOELAMS randomised phase 2 trial. Lancet Haematol. 2015;2(6):e251-259.
8. Seidel S, Pels H, Schlomer S, et al. Twenty-year follow-up of a pilot/phase II trial on the Bonn protocol for primary CNS lymphoma. Neurology. 2020;95(23):e3138-e3144.
9 Ferreri AJ, Ciceri F, Brandes AA, et al. MATILDE chemotherapy regimen for primary CNS lymphoma: results at a median followup of 12 years. Neurology. 2014;82(15):1370-1373.
10 Fritsch K, Kasenda B, Hader C, et al. Immunochemotherapy with rituximab, methotrexate, procarbazine, and lomustine for primary CNS lymphoma (PCNSL) in the elderly. Ann Oncol. 2011;22(9):2080-2085.
11. Hoang-Xuan K, Taillandier L, Chinot O, et al. Chemotherapy alone as initial treatment for primary CNS lymphoma in patients older than 60 years: a multicenter phase II study (26952) of the European Organization for Research and Treatment of Cancer Brain Tumor Group. J Clin Oncol. 2003;21(14):2726-2731.
12. Illerhaus G, Marks R, Muller F, et al. High-dose methotrexate combined with procarbazine and CCNU for primary CNS lymphoma in the elderly: results of a prospective pilot and phase II study. Ann Oncol. 2009;20(2):319-325.
A 22-year-old man was admitted to our hospital for bleeding diathesis with anemia and severe thrombocytopenia. He reported dry cough, fever, weight loss, and asthenia. Rare blast cells were observed on the blood smear. A 8.7 cm anterior mediastinal mass was seen on computed tomography (CT) scan. The levels of α-fetoprotein and lactate dehydrogenase were 45.7 μg/L and 7,380 U/L, respectively and the b-human chorionic gonadotropin level was normal. Biopsy through mediastinotomy revealed a placental alkaline phosphatase (PLAP)-positive mixed germ cell tumor (GCT), in which atypical large cells, later determined to be CD61-positive, were focally identified (Figure 1A-C). In addition to the seminoma, there were mature and immature teratomatous components represented by disorganized respiratory epithelium and a poorly differentiated mesenchymatous tissue, respectively. The bone marrow was massively infiltrated by similar, CD34-negative, CD61- and CD43- positive cells, with multilobulated nucleus and eosinophilic cytoplasm (Figure 1D, E). Staining for CD45, PLAP as well as lymphocytic and epithelial markers was negative. Cytogenetic analysis of the bone marrow showed a complex karyotype including polyploidy, +der(3) and an isochromosome i(12)p. Given these results, synchronous acute megakaryocytic leukemia (AML) and primary mediastinal mixed GCT was diagnosed, an association carrying a dismal prognosis.1 Inspired by a report of combined chemotherapy targeting both tumoral components in a pediatric patient,2 we designed a regimen based on cytarabine, mitoxantrone, etoposide phosphate, and cisplatin. After the first cycle of chemotherapy, bone marrow was seen to be leukemia-free, with a normal karyotype. The patient received three cycles of this chemotherapy regimen, with an intrathecal chemotherapy (methotrexate, cytarabine, and methylprednisolone) during the second cycle. Two additional cycles of etoposide phosphate and cisplatin were given at reduced doses due to hematotoxicity and infectious complications. After completion of chemotherapy, the leukemia was in complete remission, the mediastinal mass had decreased in metabolism (SUV max 5.1 vs. 7.2) and size (4.7x.4.7cm vs 8.6x5.8cm), and the α-fetoprotein level was normal. The patient underwent hematopoietic stem cells transplantation (HSCT) from a mismatched unrelated donor (human leukocyte antigen [HLA] compatibility 8/10, with incompatibility to 1 HLA-A and 1 DQB1 allele), with a dose of 0.37x106/kg CD34+ donor cells harvested from bone marrow (1.3x108/kg
nucleated cells). Conditioning consisted of anti-thymocyte globulins 10 mg/kg/d for 5 days, cyclophosphamide 60 mg/ kg/d for 2 days, etoposide phosphate 34 mg/kg, methylprednisolone 1 g for 2 days, and total body irradiation (total dose 12 Gy). The patient required numerous blood transfusions and experienced hemorrhagic cystitis but returned home 2 months after transplantation. A medullary biopsy performed at 3 months showed no residual leukemia. Six months after HSCT, in the context of recurrent chest pain, a 18F-fluoro-2-deoxy-D-glucose positron emssion tomography (PET)/CT showed a 60% increase in size of the residual mediastinal mass with abnormal metabolic activity. α-fetoprotein and b-human chorionic gonadotropin levels were normal. Histopathological examination of the surgically resected mass showed a fusiform sarcoma with angiomatous differentiation (Figure 1F). The cells were negative for both CD61 and PLAP. The post-operative course was complicated by mediastinitis, pleural effusion, and lung edema. Due to the appearance of mixed chimerism 10 months after HSCT, we administered two donor lymphocyte infusions (DLI) (5x105 and 6x106 CD3+ cells/kg). Complete donor chimerism was obtained and persistent complete hematological remission was confirmed. Almost 10 years after HSCT, the patient has no evidence of neoplastic disease, and an excellent performance status without any limitation in daily activities. In order to explore the possible relation between the three tumors, we performed tumor exome sequencing. Forty-eight somatic mutations were detected in the GCT, 17 in the leukemia and 331 in the sarcoma. Phylogenetic reconstruction revealed 13 shared genetic events, suggesting a common clonal origin (Figure 2A). One of these was a driver splice site mutation in TP53 (c.920-1 G>A), reported as COSM6917 in the COSMIC v76 database. The variant allele frequency (VAF) of this mutation was >2 times higher than median VAF of all other somatic mutations in the tumors (Figure 2B), suggesting a probable loss of the other allele and the lack of wild-type TP53 in tumor cells. Another splice site mutation, also on chromosome 17, involved CDK12. Other shared genetic alterations included five loss of heterozygosity (LOH) events, including PTEN LOH.
Given the relatively high number of mutations detected in the sarcoma, we sought to identify whether mutation-specific anti-tumor T-cell responses played a role in the extraordinary course of this patient. We used somatic mutations from the three tumors to predict HLA class I-restricted
neoepitopes. These neoantigens were prioritized based on predicted major histocompatibility complex (MHC) binding affinity (absolute and quantile rank), excess affinity compared with the non-mutated protein, and protein expression in cancer (Protein Atlas). After filtering with these criteria, we retained 84 neo-peptides for further analysis. As expected, the number of potential neo-peptides was much higher in sarcoma than in AML and GCT. Some of these neo-peptides were shared by two tumors, and ten were common to the GCT, AML, and sarcoma. These 84 peptides were synthetized and tested in pools in interferon (IFN)-γ ELISPOT assays with the patient’s peripheral blood mononuclear cells (PBMC) after 1 week of in vitro amplification. PBMC were obtained after complete donor chimerism. Positive response in two pools containing the common interleukin (IL) 36G16-24 YPSMCKPIT peptide was confirmed using the corresponding peptide (Figure 3). This neoantigen was restricted by HLA-B*54:01, an HLA allele shared between the donor and the patient and was detected in the sarcoma but neither in the GCT nor AML. The neoepitope-specific T-cell response was detected
again in the patient’s PBMC 7 years after HSCT. Association between GCT and hematologic malignancies is a rare condition, occurring in about 2-5% of patients with primary mediastinal non-seminomatous GCT.1 The median time from diagnosis of GCT to hematologic malignancy is 5 months but both tumors are diagnosed simultaneously in 33% of cases, with median survival from GCT diagnosis ranging from 1 to 6 months.1,3 Sarcomas in patients with GCT are also rare. They are observed mainly in the context of primary mediastinal GCT, bear a poor prognosis and are thought to derive from yolk sac tumors.4 We describe here a case of simultaneous AML and mediastinal GCT, followed by a mediastinal sarcoma. Treatment with combined chemotherapy, allogeneic HSCT/ DLI and surgery resulted in a durable complete remission, possibly maintained through immunosurveillance.
A recent study analyzing 15 patients with GCT and associated hematological malignancies demonstrated that these malignancies evolve independently from a common precursor and not from further differentiation of the GCT:1 Indeed, germ cells and hematopoietic cells are both em-
Figure 1. Histological analysis of the patient’s tumors. (A-C) Seminomatous component of the mediastinal tumor: (A) hematoxilin and eosin (H&E) staining, (B) placental alkaline phosphatase (PLAP) staining, (C) CD61 staining showing the megakaryocytic differentiation. (D, E) Bone marrow biopsy at initial presentation: (D) H&E staining, (E) CD61 staining showing the megakaryocytic differentiation. (F) H&E staining of the sarcoma that developed in the mediastinum following bone marrow transplantation. The vascular differentiation can be seen by the presence of red blood cells in the cytoplasm of certain cells and was confirmed by ERG staining (data not shown). Magnification x400.
bryologically derived from the yolk sac and aorta-gonado-mesonephros. The timing of the hematologic malignancy diagnosis, nearly always during the year after GCT diagnosis and sometimes simultaneously, argues against secondary leukemia. Clues for a common clone are the presence of hematologic malignant cells in the yolk sac component of the GCT,5 and the description of shared karyotype alterations, such as an isochromosome i(12p)6 which was observed in the present case and typically occurs in GCT, but never in de novo hematological malignancies.1 Our data also support a common origin since the three patient’s tumors carried a rare TP53 somatic variant and PTEN LOH, as previously described in similar situations.7,8 Together, they may play a critical role in pathogenesis and sarcoma differentiation. If TP53 inactivation is frequent in cancer, splice variants are far less common than missense variants.9 No germline TP53 alteration was found in this patient, although this variant has been reported in a hereditary syndrome. In addition, some of the 13 shared somatic variants detected, including TP53
and CDK12 splice site mutations, are considered putative driver mutations, which induce genomic instability and defective DNA repair, rendering tumor cells prone to further genetic alterations promoting tumor progression. Altogether, our molecular analysis confirmed the common origin of the three cancers and identified additional drivers that may contribute to the unusual switch to sarcoma phenotype. The extraordinary clinical outcome described here can potentially be explained by the combination of (i) an aggressive polychemotherapy regimen, (ii) the anti-tumor effect of the allogenic HSCT and subsequent DLI, and (iii) a neoantigen-specific T-cell response, for which the direct causality with clinical outcome could not be tested because of unavailability of viable tumor material. Debulking of AML/GCT was initially obtained with a chemotherapy regimen combining compounds targeting AML (cytarabine, mitoxantrone TD, etoposide) and GCT (etoposide and cisplatin), that may deserve further investigation in other patients with this rare entity. A beneficial role of allogenic HSCT and DLI, which
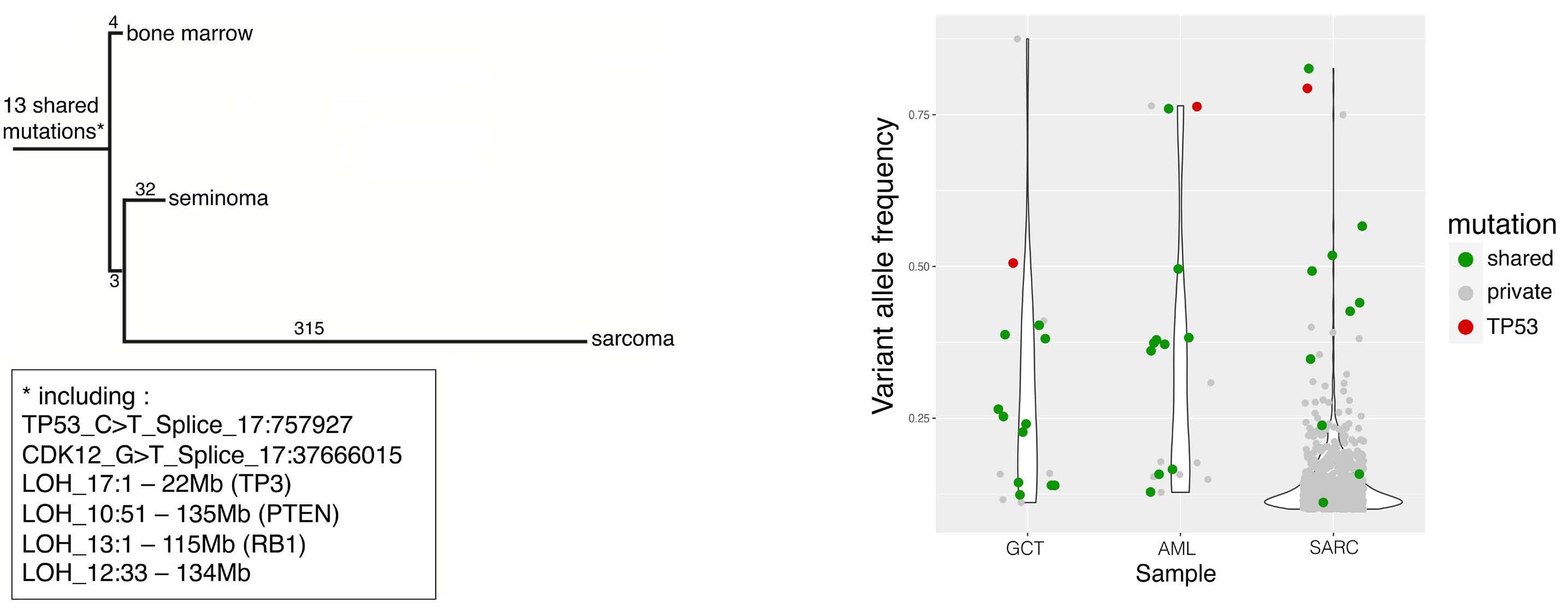
Figure 2. Molecular analysis of the patient’s tumors. (A) Phylogenetic tree based on shared and private exomic mutations in different tumors from the patient. Putative driver events are marked in boxes. (B) Violin plots of variant allele frequency (VAF) of mutations in tumors. Red - TP53 driver mutation. Green - common mutations. GCT: germ cell tumor; AML: acute megakaryocytic leukemia; SARC: sarcoma.
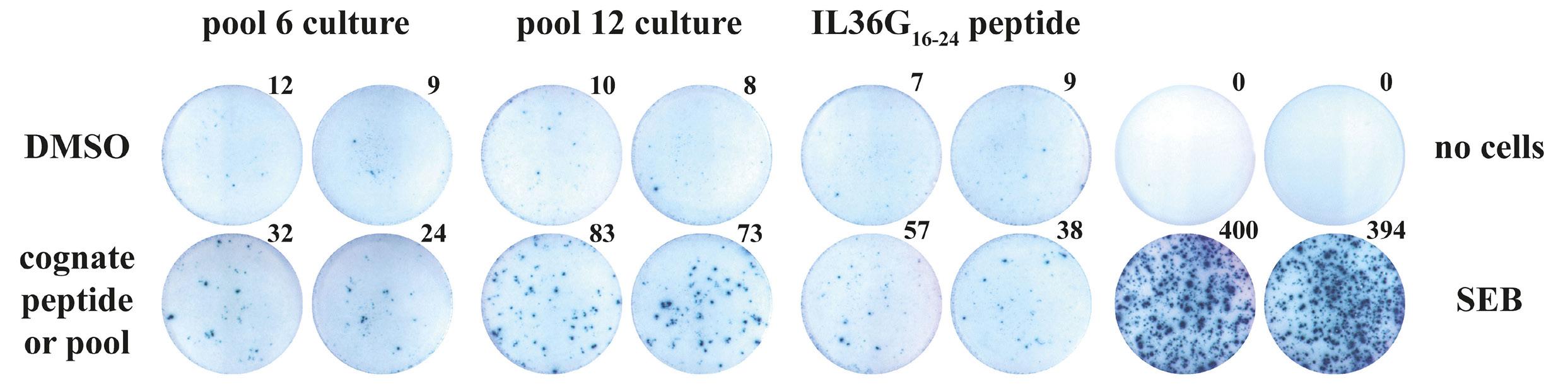
Figure 3. Interferon-γ ELISPOT assay showing recognition of peptide pools 6 and 12, and of the interleukin 36G16-24 peptide common to the two pools. Displayed number above the spot is per 200,000 peripheral blood mononulcear cells. DMSO: dimethyl sulfoxide; IL: interleukin; SEB: Staphylococcal enterotoxin B.
mediated the recovery of a full complete chimerism, is highly probable and in accordance with the series reported by Taylor and colleagues1 where the only patient with long-term survival had received HSCT. In addition to the anti-tumor effect mediated by the allogenic reaction, we were able to identify a neoantigen-specific T-cell response restricted by an HLA allele shared between the donor and the patient (HLA-B*54), detectable at two time points. The long-term persistence of this neoantigen-specific T-cell response suggests a potential role in the favorable outcome of this patient with an extremely poor prognosis and provides an additional clinical observation supporting the importance of genomic instability, mutational load and neoantigens in both anti-tumor immunity and response to immunotherapy.10
Vassilis Genoud,1* Valérie Dutoit,2,3* Nhu-Nam Tran Thang,1*° Andrew Janowczyk,4° Thomas McKee,3,5 Yves Chalandon,1,3 Petros Tsantoulis1,3 and Pierre-Yves Dietrich1,2,3°
1Department of Oncology, Geneva University Hospital, Geneva; 2Laboratory of Tumor Immunology, Faculty of Medicine and Center for Translational Research in Onco-Hematology, University of Geneva, Geneva; 3Swiss Cancer Center Léman, Lausanne; 4SIB Swiss Institute of Bioinformatics, Lausanne and 5Clinical Pathology Service, Geneva University Hospital, Geneva, Switzerland
*VG, VD and N-NTT contributed equally as first authors.
P-YD current address: Hirslanden Clinique des Grangettes, Chêne Bougeries, Switzerland
°N-NTT current address: Clinique de la Source, Lausanne, Switzerland °AJ current addresses: Department of Biomedical Engineering, Emory
1. Taylor J, Donoghue MT, Ho C, et al. Germ cell tumors and associated hematologic malignancies evolve from a common shared precursor. J Clin Invest. 2020;130(12):6668-6676.
2. Hiramatsu H, Morishima T, Nakanishi H, et al. Successful treatment of a patient with Klinefelter’s syndrome complicated by mediastinal germ cell tumor and AML(M7). Bone Marrow Transplant. 2008;41(10):907-908.
3. Spencer Chapman M, May PC, Olavarria E, Nadal Melsio E. Three distinct hematological malignancies from a single germ cell tumor: a case report. J Med Case Rep. 2020;14(1):222.
4 Magers MJ, Kao CS, Cole CD, et al. “Somatic-type” malignancies arising from testicular germ cell tumors: a clinicopathologic study of 124 cases with emphasis on glandular tumors supporting frequent yolk sac tumor origin. Am J Surg Pathol. 2014;38(10):1396-1409.
5. Orazi A, Neiman RS, Ulbright TM, Heerema NA, John K, Nichols CR. Hematopoietic precursor cells within the yolk sac tumor component are the source of secondary hematopoietic malignancies in patients with mediastinal germ cell tumors.
University, Atlanta, GA, USA; Department of Clinical Pathology, Geneva University Hospital, Geneva, Switzerland and Department of Oncology, Lausanne University Hospital, Lausanne, Switzerland
Correspondence:
P-Y. DIETRICH - pierre-yves.dietrich@hirslanden.ch
https://doi.org/10.3324/haematol.2023.284318
Received: September 20, 2023. Accepted: December 27, 2023. Early view: January 4, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
VG, VD and NT conceived and wrote the article. AJ and PT analyzed and interpreted the patient molecular data. TMK performed the histological examination. YC and PYD managed the patient. PYD conceived the treatment protocol. All authors read and approved the final manuscript.
Acknowledgments
Written consent from the patient was obtained to publish this case report.
Data-sharing statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Cancer. 1993;71(12):3873-3881.
6. Yu N, Kim HR, Cha YJ, Park EK, Kim JW. Development of acute megakaryoblastic leukemia with isochromosome (12p) after a primary mediastinal germ cell tumor in Korea. J Korean Med Sci. 2011;26(8):1099-1102.
7 Akizuki K, Sekine M, Kogure Y, et al. TP53 and PTEN mutations were shared in concurrent germ cell tumor and acute megakaryoblastic leukemia. BMC Cancer. 2020;20(1):5.
8. Lu C, Riedell P, Miller CA, et al. A common founding clone with TP53 and PTEN mutations gives rise to a concurrent germ cell tumor and acute megakaryoblastic leukemia. Cold Spring Harb Mol Case Stud. 2016;2(1):a000687.
9. Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat. 2014;35(6):672-688.
10. McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463-1469.
Multiple myeloma (MM) is the second most common hematological malignancy; it is incurable, leading to a disease course with multiple relapses.1 There have been significant improvements in survival over the past two decades, driven by the development of novel targeted agents and combination therapies. Bispecific T-cell engagers (BiTE) are the latest addition to the therapeutic armamentarium with response rates in triple-class refractory approximately double that of other treatments (apart from chimeric antigen receptor [CAR] T-cell therapy). The development of BiTE has been one of the most novel and promising developments in MM in the last 2 years.2
Recently, talquetamab, a humanized antibody that targets CD3, a receptor present in T cells and the G protein-coupled receptor class 5 member D (GPRC5D), an orphan receptor on malignant plasma cells3 has received Food and Drug Administration approval for relapsed MM after four prior lines of therapy. This first-in-class BITE was studied in a phase I clinical trial for R/R MM that showed response rates of 64% to 75% and a duration of response of 7.8 to 10.2 months based on the dose given.3 The response rate for extramedullary disease (EMD) was lower at 40% to 45%. While exceptionally promising, the full efficacy and treatment course of this drug is still being explored in larger studies. In the past two decades, a new phenomenon of rapid temporary onset, or pseudo-disease progression before disease response was identified as a side effect of immunotherapy.4
First identified in chronic lymphocytic leukemia (CLL) this phenomenon now known as tumor flare reaction (TFR)4 has also been seen in MM and has even been reported in solid tumors treated with checkpoint inhibitors.5 It has been identified as a low-level (4%) adverse event of BITE use in lymphoma.6 Despite its identification, this phenomenon remains poorly understood, and its incidence is likely underreported. TFR has been associated with morbidity and mortality. In this case, we report an episode of TFR associated with talquetamab in a patient with MM presenting with extramedullary manifestations. We present a 75-year-old male patient with a medical history of end-stage renal disease requiring hemodialysis (secondary to his myeloma). He also had a history of atrial fibrillation, and a prior stroke with no residual deficits. He was diagnosed in May 2020. Initial diagnosis revealed 70% plasma cells in the bone marrow and a high-risk 1q21 gain in cytogenetics. The patient had received multiple prior lines of therapy including two prior immunomodulatory agents (lenalidomide and pomalidomide), two proteasome inhibitors (bortezomib and carfilzomib), daratumumab, alkylating agents (cyclophosphamide, melphalan). He pro-
gressed quickly through multiple therapies and developed extramedullary skin lesions that were biopsy proven to be involved with myeloma. The patient also developed plasmacytomas in the form of ulcerated lesions in his stomach (biopsy proven) that led to melena that was stabilized with local interventions on endoscopy. In May 2023, the patient was admitted for a teclistamab step-up cycle. Initial doses of teclistamab were accompanied by worsening pain in the skin lesions as well as the development of new skin lesions. In the setting of new skin lesions, it was not clear whether the patient was responding to teclistamab. However, light chains were improving, and therapy was continued with palliative radiotherapy (RT) to the painful skin lesions. After three full doses of teclistamab, the patient presented with melena due to a plasmacytoma eroding into the gastric mucosa. This was accompanied by progression of disease as seen by the continued development of numerous skin lesions and a rise in involved light chains. The patient was given palliative RT to his stomach and taken off teclistamab. The patient was then given hyperfractionated cyclophosphamide over 2 days. There were neither significant changes in skin lesions nor in light chain levels. He was subsequently treated with talquetamab with ramp-up dosing on days 1, 3 and 5 and full-dose administration on day 7. On the second day of talquetamab treatment, the patient reported an increase in the growth of fungating lesion behind the left knee. On physical exam the patient had a 6 cm by 4 cm fungating mottled gray mass independent of bone on the popliteal fossa. He had enumerable smaller 1-3 cm masses over the surface of his body. No other physical exam findings such as tenderness, or other rashes were noted on exam. By the third day, these lesions appeared more erythematous. The patient also reported pain in these lesions. The pain was managed with supportive medications such as gabapentin and opioids.
On 6 day of treatment, the patient developed a high-grade fever and chills, suspected to be symptoms of cytokine release syndrome (CRS). Administration of tocilizumab led to symptom resolution. A full dose of talquetamab was administered on day 7 without complications, and by that time, the patient’s pain had improved. On day 8 since initiating talquetamab, the patient was discharged with reduced pain in the lesions. Follow-up investigations showed resolved skin lesions (Figure 1), a significant drop in κ/λ ratios, and decreased lactate dehydrogenase (LDH) levels (Table 1) suggesting complete response (CR). Inflammatory markers a month prior to talquetamab ferritin was 1,564 ng/mL and C-reactive protein (CRP) was 1.9 mg/dL both normalized 3 months after therapy.
The initial increase in size, erythema around these skin lesions and worsening pain followed by resolution of these lesions was concluded to be an example of TFR secondary to talquetamab. The patient was only on prophylactic antimicrobials and there was no discharge from these fungating lesions and clinically we did not suspect cellulitis. The patient’s lesions were consistent with prior biopsy proven EMD. Most significantly there was a clear temporal relationship to treatment, and a correspondence with serological myeloma markers. The rise in LDH (a frequent surrogate for disease activity and tumor lysis) and even a slight rise in the κ light chains corresponded with the expansion of skin lesions. The skin lesions resolved completely as the light chain ratio normalized, LDH fell and patient achieved a biochemical CR. The CR in this penta-refractory, unfavorable cytogenetics patient continues to affirm the effectiveness of novel BiTE but this rare and poorly understood phenomenon is important as it could impact patient outcomes in several ways. Initially, when the patient was given teclistamab, the patient had worsening skin lesions and pain. Given the possibility of TFR, we continued
treatment for four full doses with palliative measures such as local RT. However, with a clear increase in light chains on progressive assessments and development of melena, we decided to discontinue teclistamab. We were certain at that point that the patient had progression of disease. When the patient was given talquetamab, pain as well as worsening erythema at the site of skin lesions occurred. This was short-lived (a few days) as compared to progression of disease on teclistamab. With the first outpatient visit for the patient about a week after the first full dose of talquetamab, it was clear that the patient was responding both in terms of skin lesions as well as involved light chains. We confirmed his response with positron emission tomography/computed tomography (Figure 2)
To our knowledge only one other case of TFR has been reported with talquetamab though more cases in the setting of anti-BCMA directed therapies have been reported.8,9 None of these cases involved dermatological progression, and the other case of talquetamab induced TFR was noted on positron emission tomography scan only. Furthermore, the flare in tumor markers corresponding with our patient’s
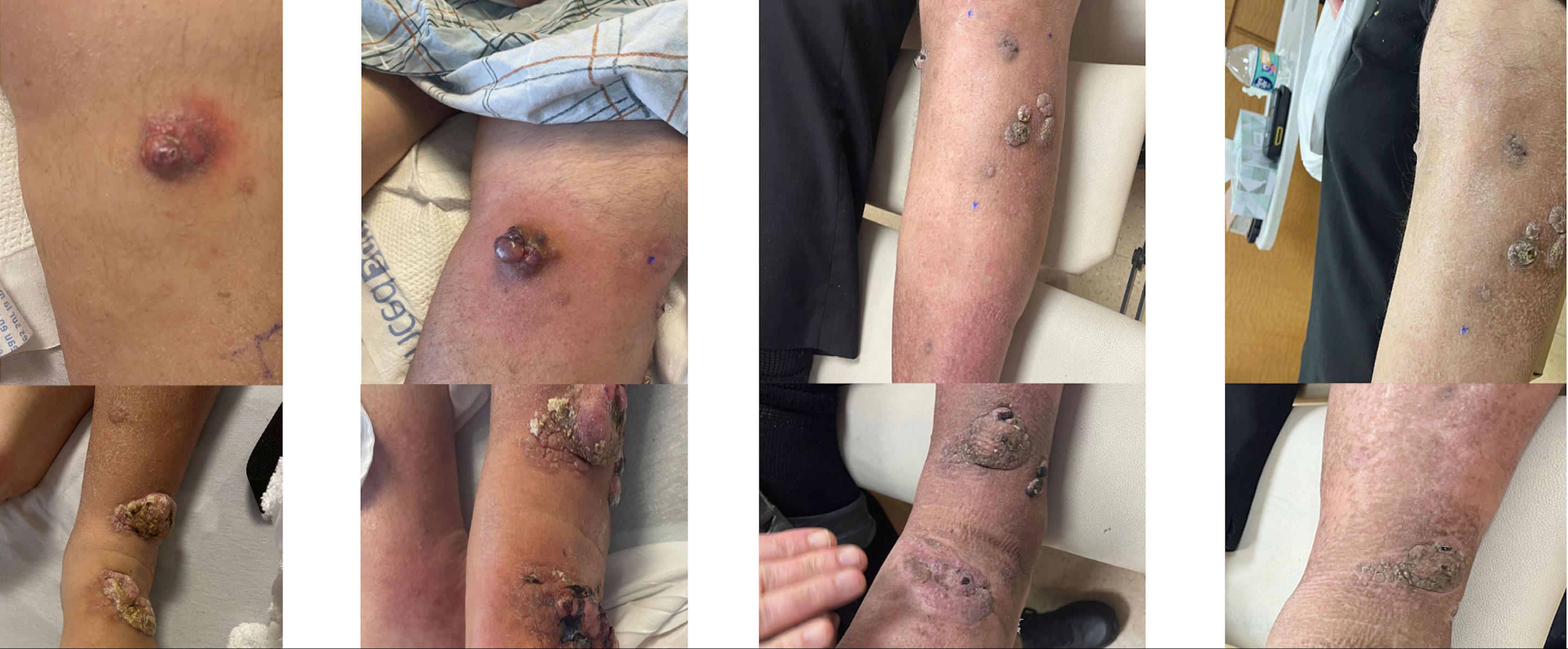

Figure 2. Timeline comparing positron emission tomography scans from April 2023 and August 8, 2023. Scan show interval improvement in left proximal thigh mass and left subcutaneous nodularity, alongside treatment dates.
flare in skin lesions provides a biochemical timeline not documented previously. In these other cases, patients were also managed symptomatically and monitored with resolution of TFR at 6 weeks. As such awareness of the timing and nature of the phenomenon appears to be key to preventing adverse outcomes. While poorly understood, the phenomenon of TFR has been linked to an immune response and T-cell infiltration leading to inflammation and disease activity.7 This is important to consider for EMD patients as they have traditionally been considered to have a higher risk disease and a lower response rate to the BiTE. Given the intrinsic immunomodulatory and T-cell engaging nature of BiTE, TFR may prove to be a greater adverse event in BITE therapies than prior therapies and will remain an ongoing challenge to differentiate from a true progression. It is for these reasons that improving the identification, management, and the decision of therapy continuation in patients with TFR is a matter of clinical importance. Importantly, both talquetamab and teclistamab were safely given to this patient with end-stage renal disease on hemodialysis, a patient population that was excluded in the pivotal clinical trials. Finally, our case shows that patients resistant to one T-cell redirecting therapy may respond to a second agent with a different target. The effectiveness of talquetamab in this teclistamab-refractory patient provides further data for BiTE sequencing.
As further immunotherapies are introduced for myeloma patients, the development of guidelines for BiTE sequencing, and TFR monitoring will be beneficial to develop a uniform
approach for the management of patients with EMD that are treated with BiTE.
Mark Forsberg,1 Santiago Beltran,2 Mendel Goldfinger,1 Murali Janakiram,3 Deepak Kalbi,4 Amit Verma,1 Dennis Cooper1 and Nishi Shah1
1Department of Oncology, Montefiore Medical Center, New York, NY; 2Division of Internal Medicine, Montefiore Medical Center-Wakefield Campus, New Yor, NY; 3Department of Hematology and Hematopoietic Cell Transplantation, Cit of Hope Cancer Center, Duarte, CA and 4Department of Radiology, Montefiore Medical Center, New Yor, NY, USA
Correspondence: N. SHAH - nisshah@montefiore.org
https://doi.org/10.3324/haematol.2023.284436
Received: October 20, 2023.
Accepted: February 13, 2024. Early view: February 22, 2024.
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
Disclosures No conflicts of interest to disclose.
Contributions
NS, DC, MG were responsible for patient care and finalized the manuscript. DK was responsible for the radiology images. MF and SB wrote the manuscript. MJ, MG and AM supervised the study.
1. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review (CSR) 1975-2018. Cronin KA eds. 2020.
2. Kegyes D, Constantinescu C, Vrancken L. Patient selection for CAR T or BiTE therapy in multiple myeloma: which treatment for each patient? J Hematol Oncol. 2022;15(1):78.
3. Chari A, Minnema MC, Berdeja JG, et al. Talquetamab, a T-cell–redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med. 2022;387(24):2232-2244.
4 Chanan-Khan A, Miller KC, Takeshita K, et al. Results of a phase 1 clinical trial of thalidomide in combination with fludarabine as initial therapy for patients with treatment-requiring chronic lymphocytic leukemia CLL. Blood. 2005;106(10):3348-3352.
5. Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412-7420.
6. Budde LE, Assouline S, Sehn LH, et al. Single-agent
Data-sharing statement
Not applicable as no datasets were generated and/or analyzed for this study.
mosunetuzumab shows durable complete responses in patients with relapsed or refractory B-cell lymphomas: phase I dose-escalation study. J Clin Oncol. 2022;10;40(5):481-491.
7. Aue G, Njuguna N, Tian X, et al. Lenalidomide-induced upregulation of CD80 on tumor cells correlates with T-cell activation, the rapid onset of a cytokine release syndrome and leukemic cell clearance in chronic lymphocytic leukemia. Haematologica. 2009;94(9)1266-1273.
8. Leipold AM, Werner RA, Düll J, et al. Th17.1 cell driven sarcoidosis-like inflammation after anti-BCMA CAR T cells in multiple myeloma. Leukemia. 2023;37(3):650-658.
9 Jamet B, Bodet-Milin C, Moreau P, Kraeber-Bodéré F, Touzeau C. 2-[18F]FDG PET/CT flare-up phenomena following T-cell engager bispecific antibody in multiple myeloma. Clin Nucl Med. 2023;48(5):e230-e231.
The COVID-19 pandemic has caused significant morbidity and mortality, especially in patients with pre-existing comorbidities. Although the introduction of vaccines has decreased both the incidence and severity of the infection, protection is suboptimal in immunocompromised patients. We and others have shown previously that in patients with hematologic malignancies who received chimeric antigen receptor T-cell (CAR T) treatment, antibody responses to currently available vaccines are severely impaired while T-cell responses are largely maintained.1-3. Until we have broader coverage vaccines with stronger, durable immunogenicity, we will need treatments that can suppress tissue-damaging viral-induced inflammation in patients with CAR T-cell-induced immunosuppression and in particular target the endothelial pathobiology of the virus and its related endotheliitis.
Here, we describe the clinical course and correlative studies of two fully SARS-CoV2-vaccinated patients with relapsed refractory myeloma (RRMM) who had severe COVID-19 infection shortly after their CAR T treatments. Both remained in the intensive care unit (ICU) for prolonged periods requiring
high-flow oxygen despite multipronged therapy. However, their clinical conditions improved remarkably shortly after initiation of defibrotide which was given after obtaining informed consent per institutional guidelines and which appropriately suppressed the harmful SARS-CoV-2-induced non-specific inflammatory response and related cytokine release syndrome (CRS). Importantly, our correlative studies suggested no negative impact either on the mounting of adaptive virus-specific antibody and/or T-cell responses or on the in vivo expansion and persistence of CAR T cells. Both patients recovered well from their infection and their myeloma remains in deep and sustained remission. These data suggest that defibrotide has the potential to suppress the damaging effects of the anti-COVID-19 inflammatory response while maintaining adaptive antiviral and anti-tumor immune responses.
A 67-year-old man with a 13-year history of immunoglobulin (Ig)G-λ RRMM including upfront autologous stem cell transplantation (ASCT) (Table 1) received BCMA targeting
1 Lenalidomide, bortezomib, dexamethasone (RVd) - autologous stem cell transplantation - lenalidomide maintenance
Carfilzomib, pomalidomide, dexamethasone (KPd)
3 Elotuzumab, pomalidomide, dexamethasone (EPd)
4 Daratumumab, pomalidomide, dexamethasone (DPd)
5 Panobinostat, bortezomib, dexamethasone (PanoVd)
6 Daratumumab, lenalidomide, bortezomib, dexamethasone (Dara-RVd)
7 Daratumumab, cyclophosphamide, bortezomib, lenalidomide, dexamethasone
Patient 2 Line
1 Cyclophosphamide, bortezomib, dexamethasone (CyBorD) - autologous stem cell transplantation - RVd maintenance
2 Daratumumab, dexamethasone doublet
3 Daratumumab, pomalidomide, dexamethasone
4 Daratumumab, carfilzomib, dexamethasone
5 Belantamab, dexamethasone
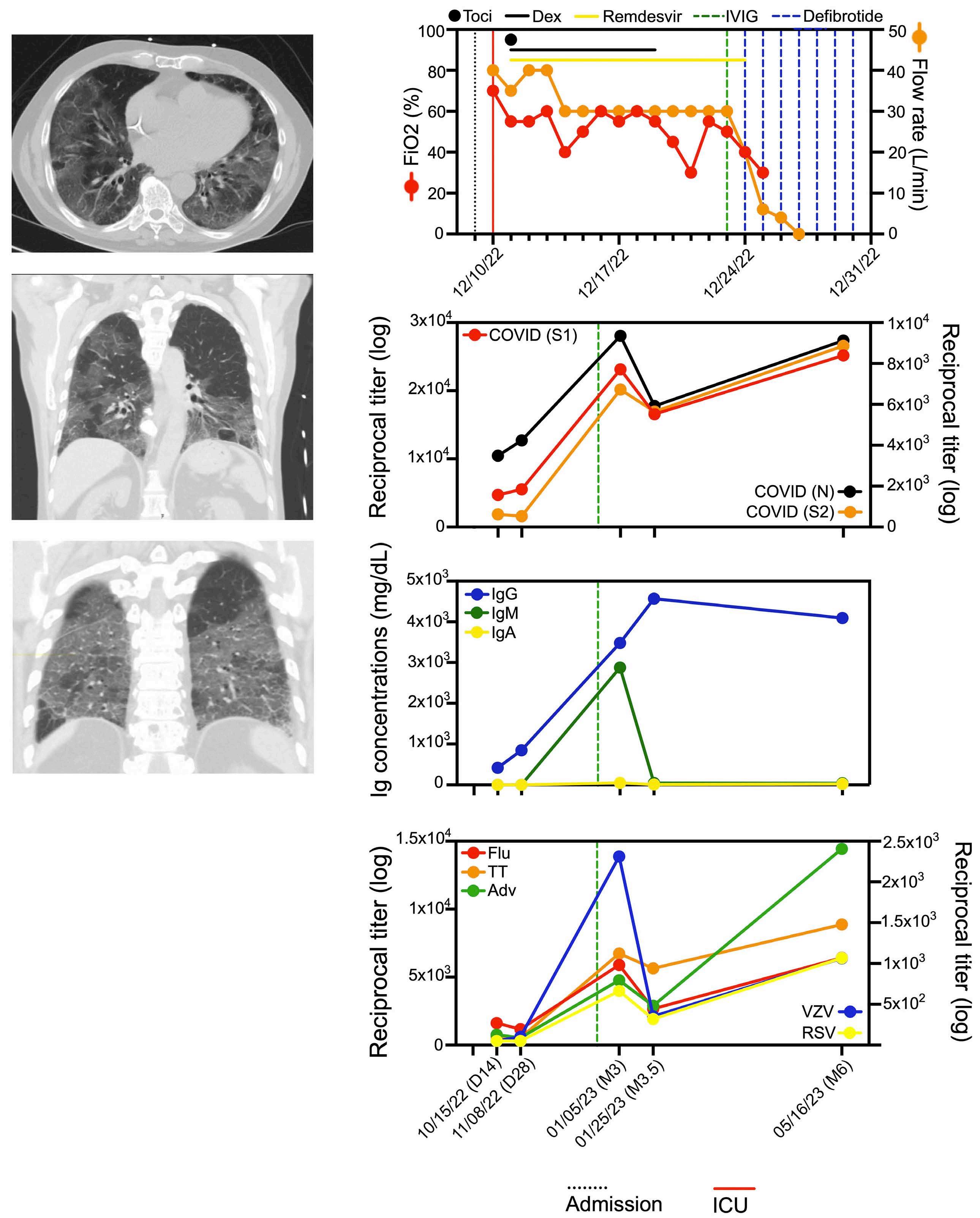
Figure 1. Clinical course and antiviral antibody titers of a myeloma patient #1 with COVID-19-related acute respiratory distress syndrome. (A) Computed tomographic (CT) images of chest for Patient 1. Axial and coronal sections display diffuse and extensive alveolar damage showing ground-glass consolidations that persist after 1 week of intensive care unit (ICU) treatment including high-dose steroids and remdesivir. (B) Supplemental O2 requirement of our patient while admitted to the ICU. Both fraction of inspired oxygen (FIO2) (red) and O2 flow rates (orange) are indicated. Dosing of defibrotide (blue) and intravenous immunoglobulin (IVIG) (green) are indicated by vertical dashed lines. Administration of both tocilizumab (toci; black dot) and dexamethasone (dex; black line) for cytokine release syndrome (CRS) are indicated as is administration of remdesvir (yellow line). (C) Reciprocal immungolbulin (Ig)G antibody endpoint titers against SARS-CoV-2 proteins spike (S)1 (red), S2 (orange), and nucleocapsid protein
Continued on following page.
(N; black) as determined by enzyme-linked immunosorbent assay (ELISA) are shown. The temporal relationship to IVIG infusion is indicated by a green dashed vertical line. (D) Quantitative immunoglobulins (IgG, IgM, IgA) over time and temporal relationship to IVIG infusion (green dashed vertical line). Absolute serum concentrations of the different immunoglobulins were measured using human IgG, IgM, and IgA ELISA kits (Invitrogen, Cat. No. BMS2091, BMS2098, BMS2096) as per the manufacturer’s instructions. (E) Reciprocal IgG antibody endpoint titers against microbial proteins influenza A nucleoprotein (Flu; red), tetanus toxoid (TT; orange), adenovirus type 5 hexon protein (Adv; green), varicella zoster virus envelope glycoprotein E (gE) protein (VZV; blue) and respiratory syncytial virus nucleoprotein (RSV; yellow) as determined by ELISA are shown. The temporal relationship to IVIG infusion is indicated by a green dashed vertical line. Serum antibody responses against recombinant, full-length SARS-CoV-2 proteins or viral control proteins were determined by ELISA as previously described.
ciltacabtagene autoleucel (ciltacel) CAR T for relapsed, penta-refractory disease after standard cyclophosphamide and fludarabine lymphodepleting chemotherapy in October 2022. His course was notable for grade 2 CRS which was treated with single-dose tocilizumab. After discharge, he had neutropenia that responded to granulocyte colony-stimlating factor (G-CSF) and achieved full count recovery. Approximately 1.5 months after CAR T-cell infusion, he developed fever, chills, cough, and exertional dyspnea and was readmitted with severe SARS-CoV-2 infection. He refused nirmatrelvir-ritonavir (paxlovid), but quickly became hypoxemic, and was transferred to the ICU. Computed tomography (CT) imaging showed diffuse ground-glass consolidations and ruled out pulmonary embolism (Figure 1A). For the majority of his 21-day admission, he remained symptomatic with cough and on high-flow O2 (15 days) despite a prolonged course of high-dose steroids and remdesivir but did not require intubation. After a 10-day course of intravenous (i.v.) steroids, he received intravenous immuoglobulin (IVIG), and 1 day later (on his 14th day of ICU stay), he was started on i.v. defibrotide for 7 days. Although there was some variation in fraction of inspired oxygen (FIO2) levels before the administration of defibrotide, a marked drop in FiO2, O2 flow rate, and most importantly a rapid clinical improvement only occurred after the application of the treatment (Figure 1B). After his discharge, he had bacteremias (pseudomonas and rothria) and cytomegalovirus (CMV) viremia which were successfully treated with appropriate anti-microbial therapy. Immediately before his clinical recovery from COVID-19 infection, he was treated with defibrotide which is most likely what led to the improvement in clinical status. Before his CAR T treatment, he had been receiving monthly IVIG infusions, and his pre-IVIG serum IgG level was within normal limits, making this a less likely contributor. We do not use tocilizumab routinely for the treatment of COVID; however, two of the studies leading to an emergency use authorization (EUA) by the Food and Drug Administration (FDA) reported different medians of 6 and 14 days for time to significant clinical improvement. Our patient did not have a meaningful improvement and remained on high-flow oxygen during his 14-day ICU stay despite tocilizumab. Another possible explanation for his clinical improvement may be immune reconstitution. However, at 2 months, although he was not neutropenic, he still had low T-cell counts (CD4+ 138 cells/ μL) and at 4 months, he only had partial immune reconsti-
tution (CD4+ 800 cells/μL, IgA <8 mg/dL). His bone marrow biopsy at 5.5 months was normocellular with trilineage hematopoiesis and no monoclonal plasma cell population was identified by morphology, immunohistochemistry or measurable residual disease (next generation sequencing [NGS]). His SARS-CoV-2 polymerase chain recation (PCR) was positive at 6 months but subsequently cleared. Defibrotide is a multifunctional endothelial stabilizing agent that received US regulatory approval for severe hepatic sinusoidal obstruction syndrome/veno-occlusive disease (VOD/ SOS) in 2016, and European Medicines Agency approval in 2013.4 Given the high mortality rate of severe VOD/SOS, it is now broadly used in real-world practice.5 The postulated primary mechanism of action is the stabilization and/or reversal of endothelial cell dysfunction. Moreover, defibrotide has also been shown to abrogate the deleterious effects of CD8+ and regulatory T cells on human microvascular endothelial cells (MVEC).6 These findings paved the way for the initial clinical trials in allogeneic transplant settings for VOD/SOS. A large CIBMTR study reported a 22.1% difference in the resolution of severe VOD/SOS at day +100.7 Given its favorable effects on endothelial dysfunction,8 defibrotide was also investigated in non-VOD/SOS conditions that complicate infectious and non-infectious inflammation characterized by endotheliopathy, or so-called endotheliitis.9,10 In a preclinical study, defibrotide mitigated the endothelial cell injury induced by plasma from patients with COVID-19-related vasculopathies.11 Specifically, MVEC were exposed to primary samples with either acute COVID-19 infection or acute TMA in the presence and absence of defibrotide. Caspase-8 which was used as an endothelial cell activation surrogate marker was suppressed with defibrotide and signaling pathway alterations were noted.11 In acute malaria, defibrotide interfered with the coagulation/inflammation cycle, inhibited TLR-mediated dendritic cell activation, and reduced interferon-γ production.9 One proposed mechanism for SARS-CoV-2-related pulmonary organ damage is the formation of neutrophil extracellular traps (NET) associated with an abundance of neutrophils in autopsy specimens from COVID-19 patients.12 Interestingly, in the setting of antiphospholipid syndrome, defibrotide was demonstrated to inhibit NET formation and venous thrombosis.13-15 An open-label, single-center phase I study investigated the safety and efficacy of a 7-day course of defibrotide in patients with COVID-19-related acute respiratory distress syndrome (ARDS).16 Of 12 patients treated

Continued on following page.
Figure 2. Chimeric antigen receptor T-cell expansion and anti- SARS-CoV-2 T cells in two myeloma patients with COVID-19-related acute respiratory distress syndrome. Panels (A-D) are the correlative studies of the first patient in the report. Panel (E) shows chimeric antigen receptor (CAR) T-cell expansion characteristics of the second patient. (A) Blood samples were collected under Institutional Review Board (IRB)-approved protocol 2043GCCC (IRB HP-00091736). Peripheral blood plasma was generated by centrifugation at 400 G and frozen immediately at -80°C. Peripheral blood mononuclear cells (PBMC) were isolated using density gradient centrifugation and cryopreserved in liquid nitrogen. Proportions of CD4+ and CD8+ BCMA-targeted CAR T cells were determined in the peripheral blood of our patient at different time points using flow cytometry. Dot plots show percentage of CAR-expressing T cells versus all T cells. CAR T cells were identified by staining of the expression of the CAR on the cell surface using a recombinant BCMA protein as CAR detection reagent and co-staining with anti-CD3 and other T-cell markers (Online Supplementary Table S1). (B) CAR T-cell CD4+ and CD8+ subtypes were quantified at the different time points and CAR T-cell memory subtypes (naïve, central memory [CM], effector-memory [EM], effector-type [EFF]) were determined by co-staining for CD45RA and CD62L. T cells specific for the S (C) and the N (D) proteins of the SARS-CoV-2 virus were identified ex vivo after short-term stimulation of total PBMC using libraries of overlapping peptides covering the complete sequence of the respective proteins. Intracellular staining of cytokines followed by flow cytometry served as a read-out assay. SARS-CoV-2-specific CD4+ T cells (upper rows) were defined as TNFα/CD40L (CD154)-double positive CD3+CD4+ T cells and SARS-CoV-2-specific CD8+ T cells (lower rows) were defined as IFNγ/TNFα-double positive CD3+CD8+ T cells. (E) CAR T-cell expansion kinetics in the second patient. Dot plots show percentage of CAR-expressing T cells versus all T cells. CAR T cells were identified by staining for the expression of the CAR on the cell surface using a recombinant BCMA protein as CAR detection reagent and co-staining with anti-CD3.
(10 on mechanical ventilation), nine (75%) survived all of whom had a baseline pO2/FIO2 ratio (PFR) of >125 whereas three patients with baseline PFR <125 died.16 Although the primary mechanism of action in this setting appears to be endothelial stabilization, there is likely a multi-pronged suppression of non-specific inflammation and endotheliitis, which is suggested by the inhibition of both donor neutrophil and T-cell trafficking resulting in reduced acute graft-versus-host disease severity in a murine mismatched allogeneic transplantation model, as well as other recently derived hypotheses for modulating activated endothelium and disrupting viral infection.17, 18 Our steroid- and remdesivir-refractory patient showed rapid clinical improvement following initiation of defibrotide treatment (Figure 1B). Correlative studies demonstrated COVID-specific antibody responses which were probably enhanced by IVIG administrations (Figure 1C-E). Importantly, we observed robust CAR T-cell expansion favoring a central memory phenotype at month 3 (Figure 2A, B). Similarly, despite the absence of full immune reconstitution at month 2 (CD4+ <200), there were clear CD4+ and CD8+ T-cell responses against the spike (S) and nucleocapsid (N) proteins at month 3 (Figure 2C, D).
A 79-year-old man with an 8-year history of IgG-κ RRMM including upfront ASCT (Table 1) presented with cough and profound hypoxemia 45 days after ciltacel with SARS-CoV-2 infection. He was treated with paxlovid and was refractory to two separate inpatient courses of high-dose steroids and remdesivir administered 9 days apart. During 21-day ICU course, after persistent hypoxemia requiring 7-day high-flow oxygen support, he was started on defibrotide which led to a 30% drop in FIO2 immediately after the first dose. Unlike patient 1, he never received tocilizumab and IVIG was not given until 48 hours after administration of defibrotide, a time point when FIO2 was already reduced by 30%. Correlative studies also confirmed robust CAR T expansion (Figure 2E). He was
discharged on 2 lpm flow regulator. At 3 months, he did not require supplemental O2 and his myeloma was in remission with negative bone marrow biopsy and positron emission tomography (PET)/CT imaging.
Notably, both patients shared a historical triad of ASCT, CAR T treatment and SARS-CoV-2 infection all of which are well-established risk factors for endothelial activation and subsequent vascular damage. Although there are limited data on the impact of defibrotide on immune response in acute infectious inflammatory states, our clinical observations and correlative data suggest a prompt and clinically meaningful suppression of COVID-induced inflammatory responses with well-preserved SARS-CoV-2 specific antibody and T-cell immunity as well as sustained and effective anti-myeloma CAR T-cell kinetics. Notably, no hemorrhagic events were observed in either of these patients with defibrotide treatment. Patient 1 is 1 year out from CAR T-cell treatment, and remains with minimal exertional symptoms, without supplemental oxygen. He is PCR-negative for SARS-CoV-2 and his myeloma is in MRD-negative (NGS) remission. Patient 2 is 5.5 months out from CAR T, his myeloma remains in remission and he does not require O2 support, as well as being PCR-negative for SARS-CoV-2. Given the complexity of CAR T-cell treatments, their unique adverse effect profiles such as CRS and potential infection profile including COVID-19, further investigation of defibrotide as a promising therapeutic option is clearly warranted.
Mehmet H. Kocoglu,1,2,3 Paul G. Richardson,4 Clifton C. Mo,4 Aaron P. Rapoport1,2,3 and Djordje Atanackovic1,2,3,5
1University of Maryland Greenebaum Comprehensive Cancer Center, Baltimore, MD; 2Department of Medicine, University of Maryland School of Medicine, Baltimore, MD; 3Transplant and Cellular Therapy Program, University of Maryland Greenebaum Comprehensive Cancer Center, Baltimore, MD; 4Dana Farber Cancer Institute, Boston, MA
and 5Department of Microbiology and Immunology, University of Maryland, Baltimore, MD, USA
Correspondence: M. H. KOCOGLU - mkocoglu@som.umaryland.edu https://doi.org/10.3324/haematol.2023.284793
Received: December 5, 2023. Accepted: February 15, 2024. Early view: February 22, 2024.
©2024 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
PGR has received research funding from Jazz pharmaceuticals. The remaining authors have no conflicts of interest to disclose.
1. Atanackovic D, Luetkens T, Omili D, et al. Vaccine-induced T-cell responses against SARS-CoV-2 and its Omicron variant in patients with B cell-depleted lymphoma after CART therapy. Blood. 2022;140(2):152-156.
2. Dahiya S, Luetkens T, Lutfi F, et al. Impaired immune response to COVID-19 vaccination in patients with B-cell malignancies after CD19 CAR T-cell therapy. Blood Adv. 2022;6(2):686-689.
3. Atanackovic D, Kreitman RJ, Cohen J, et al. T cell responses against SARS-CoV-2 and its Omicron variant in a patient with B cell lymphoma after multiple doses of a COVID-19 mRNA vaccine. J Immunother Cancer. 2022;10(7):e004953.
4 Richardson PG, Carreras E, Iacobelli M, Nejadnik B. The use of defibrotide in blood and marrow transplantation. Blood Adv. 2018;2(12):1495-1509.
5. Nauffal M, Kim HT, Richardson PG, et al. Defibrotide: real-world management of veno-occlusive disease/sinusoidal obstructive syndrome after stem cell transplant. Blood Adv. 2022;6(1):181-188.
6. Eissner G, Hartmann I, Kesikli A, et al. CD4+CD25+FoxP3+ regulatory T cells enhance the allogeneic activity of endothelialspecific CD8+/CD28-CTL. Int Immunol. 2011;23(8):485-492.
7 Strouse C, Richardson P, Prentice G, et al. Defibrotide for treatment of severe veno-occlusive disease in pediatrics and adults: an exploratory analysis using data from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2016;22(7):1306-1312.
8. Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M. What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood. 2016;127(13):1719-1727.
9. Francischetti IM, Oliveira CJ, Ostera GR, et al. Defibrotide interferes with several steps of the coagulation-inflammation
Contributions
MK designed the study, analyzed the data, prepared figures, and wrote the manuscript. PGR helped to design the study, analyze data and write the manuscript. CCM and APR analyzed data and helped to write the manuscript. DA performed experiments, analyzed the data, and prepared figures.
Funding
This study was funded by two grants from the Kahlert Foundation (to DA), by the Maryland Department of Health’s Cigarette Restitution Fund Program (to DA) and by the National Cancer Institute - Cancer Center Support Grant (CCSG) P30CA134274, and was in part supported by the Paula and Rodger Riney Foundation (to PGR and CCM).
Data-sharing statement
The data presented in this report is available upon request.
cycle and exhibits therapeutic potential to treat severe malaria. Arterioscler Thromb Vasc Biol. 2012;32(3):786-798.
10 Fernandez S, Palomo M, Molina P, et al. Progressive endothelial cell damage in correlation with sepsis severity. Defibrotide as a contender. J Thromb Haemost. 2021;19(8):1948-1958.
11. Elhadad S, Redmond D, Tan A, et al. Defibrotide mitigates endothelial cell injury induced by plasmas from patients with COVID-19 and related vasculopathies. Thromb Res. 2023;225:47-56.
12. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med. 2020;217(6):e20200652.
13. Shi H, Gandhi AA, Smith SA, et al. Endothelium-protective, histone-neutralizing properties of the polyanionic agent defibrotide. JCI Insight. 2021;6(17):e149149.
14 Ali RA, Estes SK, Gandhi AA, et al. Defibrotide inhibits antiphospholipid antibody-mediated neutrophil extracellular trap formation and venous thrombosis. Arthritis Rheumatol. 2022;74(5):902-907.
15. Onuora S. Defibrotide inhibits NET-mediated thrombosis in APS models. Nat Rev Rheumatol. 2022;18(2):63.
16. Frame D, Scappaticci GB, Braun TM, et al. Defibrotide therapy for SARS-CoV-2 ARDS. Chest. 2022;162(2):346-355.
17. Garcia-Bernal D, Palomo M, Martinez CM, et al. Defibrotide inhibits donor leucocyte-endothelial interactions and protects against acute graft-versus-host disease. J Cell Mol Med. 2020;24(14):8031-8044.
18. Richardson E, Garcia-Bernal D, Calabretta E, et al. Defibrotide: potential for treating endothelial dysfunction related to viral and post-infectious syndromes. Expert Opin Ther Targets. 2021;25(6):423-433.
David W. Woolston,1* Nathan D. Lee,2* Mazyar Shadman,1,2* Elena Latorre-Esteves,2 Xin Ray Tee,2 Jeanne Fredrickson,2 Brendan F. Kohrn,2 Chaitra Ujjani,1,2 Ashley Eckel,2 Brian Till,1,2 Min Fang,1,2 Jerald Radich,1,2 Ivana Bozic,1,2# Rosa Ana Risques2# and Cecilia C.S. Yeung1,2#
1Fred Hutchinson Cancer Center and 2University of Washington, Seattle, WA, USA
*DWW, NDL, and MS contributed equally as first authors. #IB, RAR, and CCSY contributed equally as senior authors.
Correspondence: C.S. Yeung_Cecilia cyeung@fredhutch.org
Received: January 25, 2024. Accepted: January 25, 2024.
https://doi.org/10.3324/haematol.2024.285151
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
On page 837 of the article by Woolston et al., 1 in the sentence reproduced below, “ibrutinib” should be replaced with “acalabrutinib”:
Incorrect sentence:
“Sample R002-A was collected after 3 months of combination therapy with ibrutinib and venetoclax.”
Correct sentence:
“Sample R002-A was collected after 3 months of combination therapy with acalabrutinib and venetoclax.”
References
1. Woolston DW, Lee ND, Shadman M, et al. Ultra-deep mutational landscape in chronic lymphocytic leukemia uncovers dynamics of resistance to targeted therapies. Haematologica. 2024;109(3):835-845.
Margarita Maurer-Granofszky,1,2* Stefan Köhrer,1,2* Susanna Fischer,1,2 Angela Schumich,1 Karin Nebral,1,2 Patrizia Larghero,3 Claus Meyer,3 Astrid Mecklenbräuker,1,2 Nora Mühlegger,1 Rolf Marschalek,3 Oskar A. Haas,1 Renate Panzer-Grümayer1 and Michael N. Dworzak1,2,4
1St. Anna Children’s Cancer Research Institute (CCRI), Vienna, Austria; 2Labdia Labordiagnostik, Vienna, Austria; 3Institute of Pharmaceutical Biology/Diagnostic Center of Acute Leukemia (DCAL), Goethe University, Frankfurt/Main, Germany and 4St. Anna Children’s Hospital, Department of Pediatrics, Medical University of Vienna, Vienna, Austria
*MMG and SK contributed equally as first authors.
Correspondence: N. Dworzak_Michael dworzak@stanna.at
Received: January 25, 2024. Accepted: January 25, 2024.
https://doi.org/10.3324/haematol.2024.285153
©2024 Ferrata Storti Foundation Published under a CC BY-NC license
In the article pre-published online and in the March 2024 issue of Haematologica, 1 we have to correct that:
- Since the percentage minimal residual disease for patient 8 was 0.0100 at the first induction timepoint (day 28), the polymerase chain reaction minimal residual disease result should be “positive” instead of “negative” (Page 5, Table 1, P8, Column 6). In the main text on Page 5 the sentence “Three of these samples were positive by gDNA-PCR but below the threshold of 0.1% (Table 1)” should be replaced by “All of these samples were positive by gDNA-PCR but
below the threshold of 0.1% (Table 1).”
- In Table 2, the numbers in column 3 (PCRpos/FCMpos) apply to column 4 (PCRneg/FCMneg) and vice versa
- No threshold was applied for the results shown in Figure 5, as described correctly in the main text (Page 8). Figure 5 itself contained an error, stating that a cut-off of 0.1% was used. The corrected Table 1, Table 2 and Figure 5 are shown below.
The authors apologize for the errors and state that these do not change any scientific conclusions or interpretations of the data.
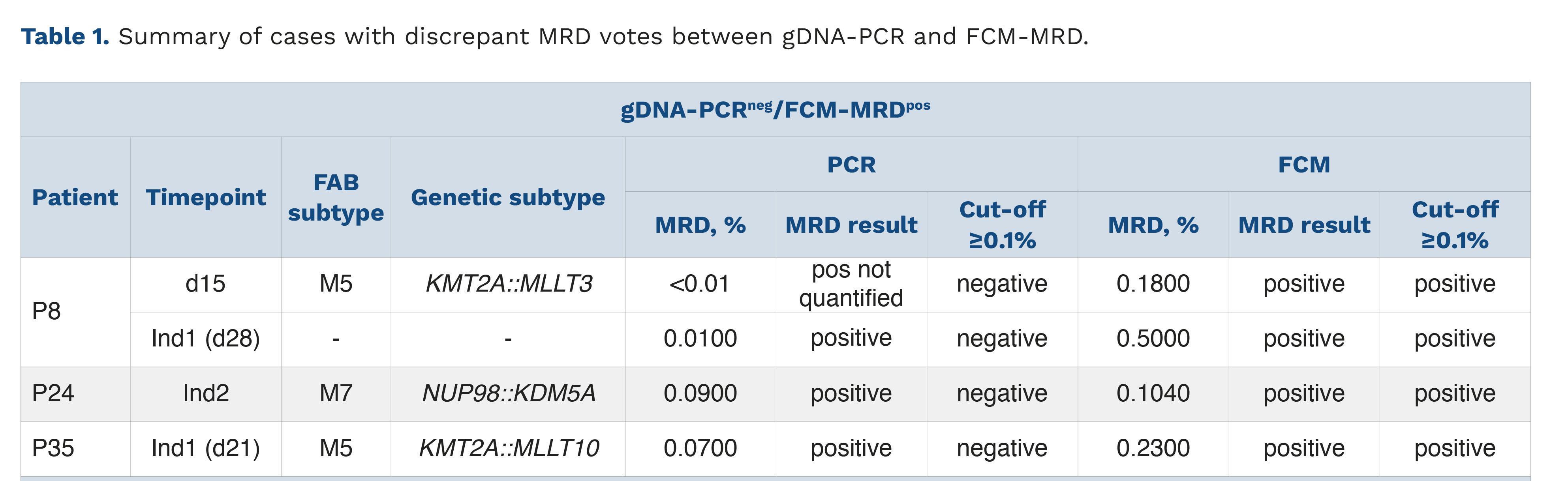
Continued on following page.
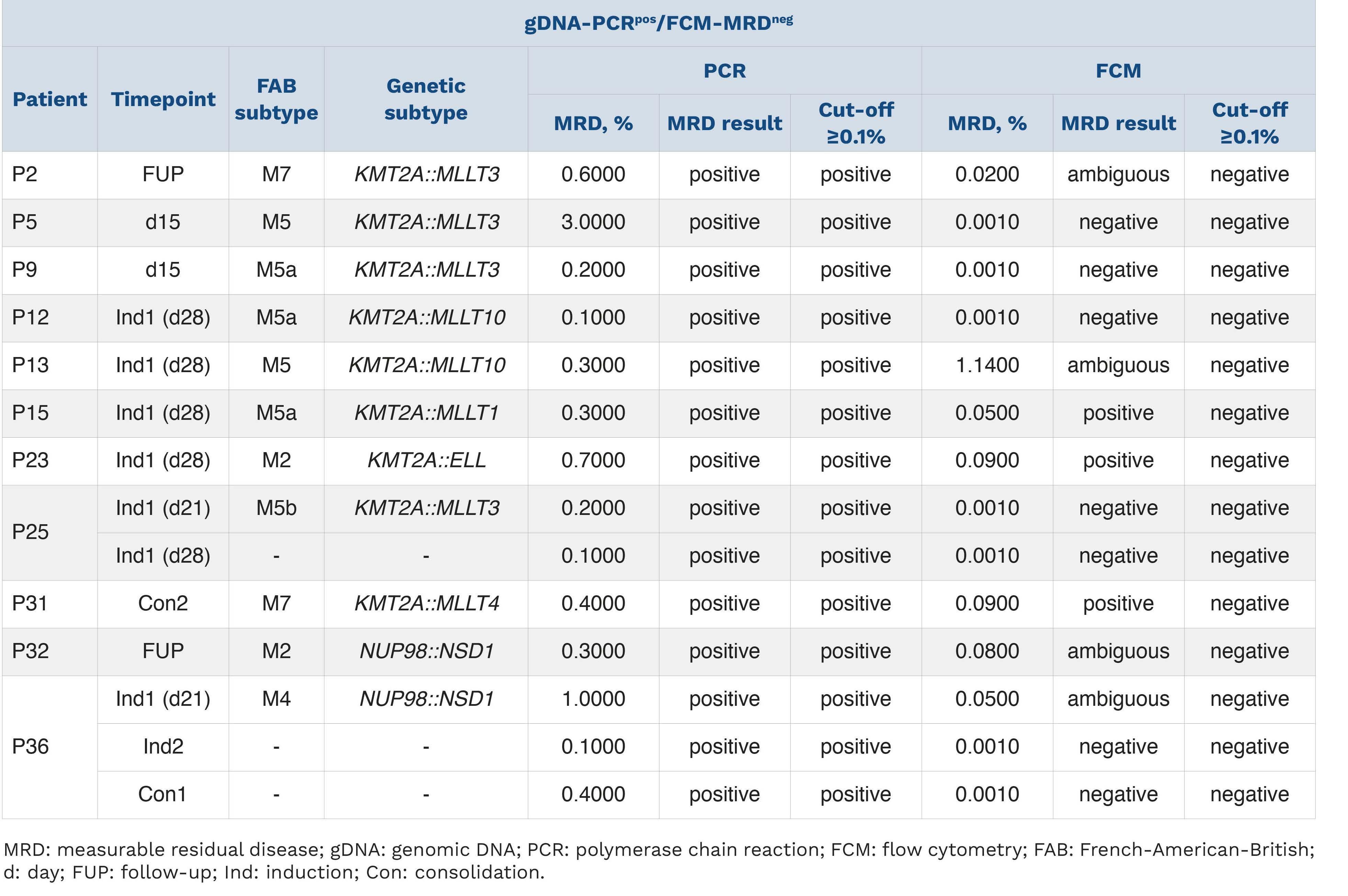
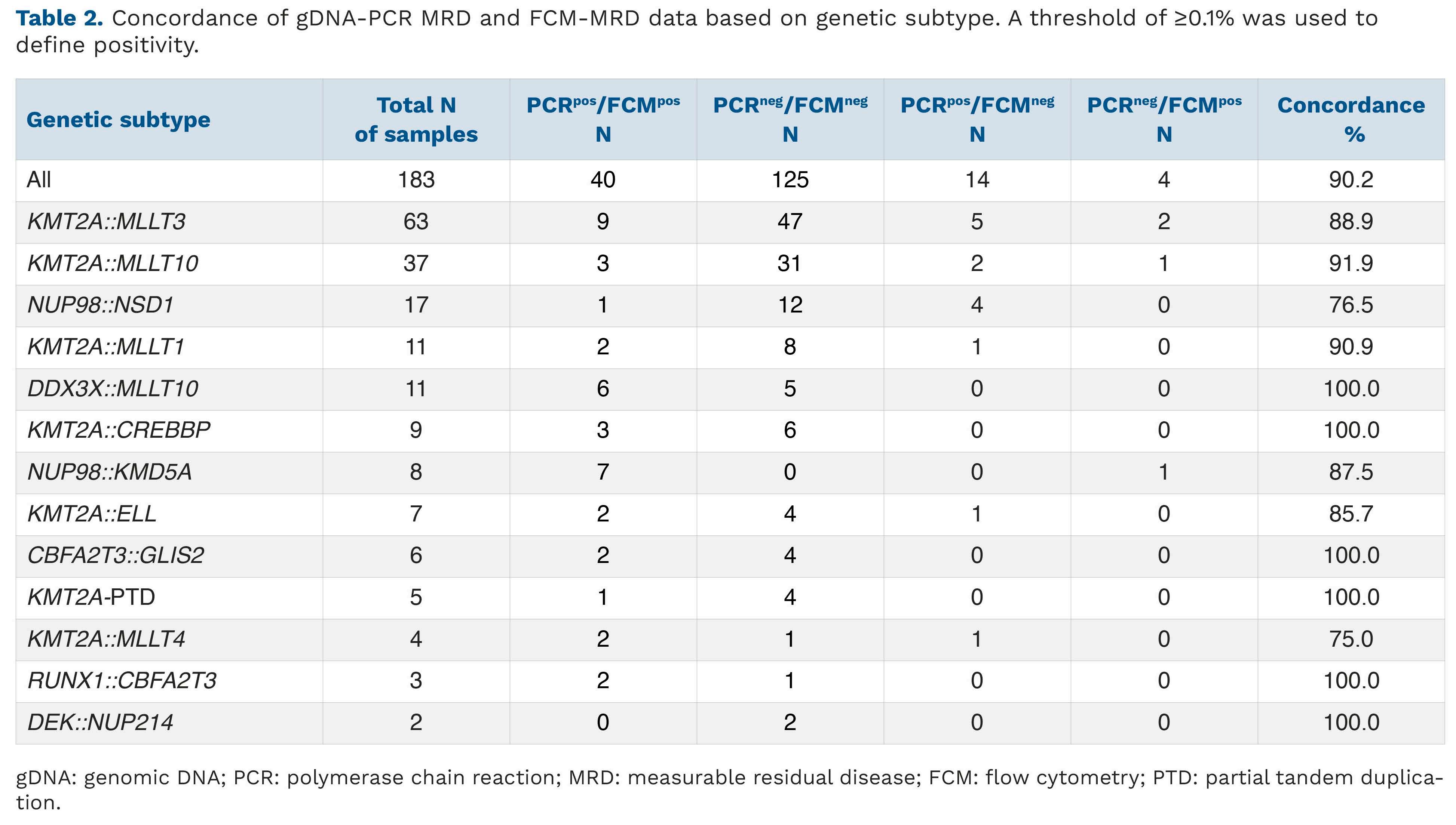
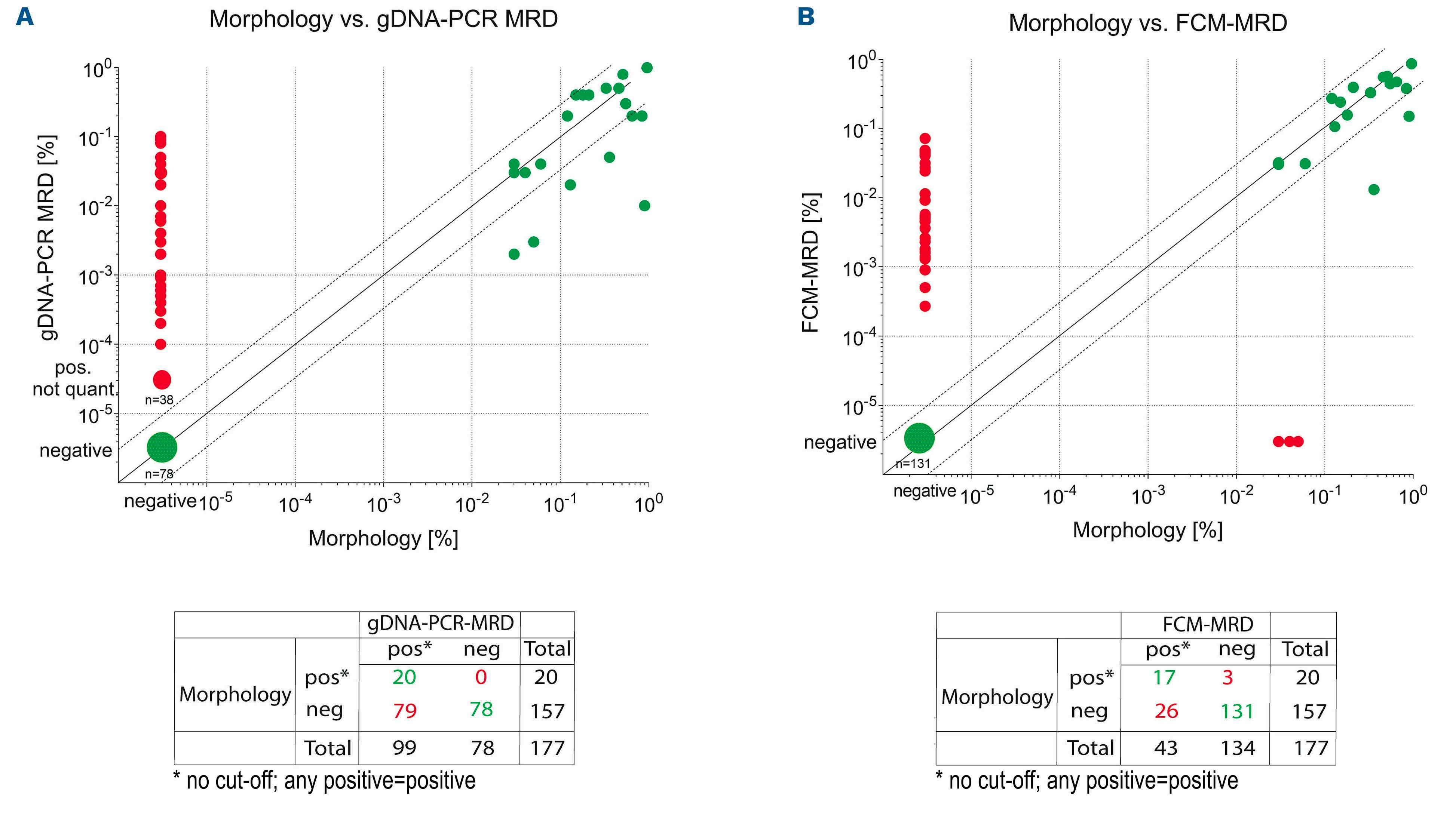
Figure 5. Concordance of gDNA-PCR MRD and FCM-MRD with conventional morphological assessment. Each symbol represents one MRD estimate. Values that are MRD-positive or MRD-negative using both methodologies are considered concordant (green dots), whereas discordant samples are negative with one methodology but positive with another (red dots). In addition, dashed lines above/below the x=y line mark the range of variance according to Dworzak et al., 35 i.e. between 3x larger or smaller till 1/3 of the x=y value. Statistics performed using GraphPad Prism. gDNA: genomic DNA; PCR: polymerase chain reaction; MRD: measurable residual disease; FCM: flow cytometry.
1. Maurer-Granofszky M, Köhrer S, Fischer S, et al. Genomic breakpoint-specific monitoring of measurable residual disease in pediatric non-standard risk acute myeloid leukemia. Haematologica. 2024;109(3):740-750.

Journal of the Ferrata Storti Foundation