All rights reserved. No part of this eHealth Source may be used or reproduced in any manner whatsoever without written permission except in the case of brief quotations embedded in articles or reviews.
Integritas Communications
55 Barr Lane, Monroe, NY 10950
www.integritasgrp.com www.exchangecme.com
FACULTY Nancy S. Reau, MD
The Richard B. Capps Chair of Hepatology
Professor, Department of Internal Medicine
Division of Digestive Diseases and Nutrition
Rush Medical College
Chief, Section of Hepatology
Associate Director, Organ Transplantation
Rush University Medical Center
Chicago, Illinois
Dr. Nancy Reau is Professor of Internal Medicine, Richard B. Capps Chair of Hepatology, Associate Director of Organ Transplantation, and Section Chief of Hepatology at Rush University Medical Center in Chicago, Illinois. She received her medical degree from The Ohio State University College of Medicine in Columbus, where she completed a residency and fellowship in gastroenterology/ hepatology, followed by a second fellowship in advanced transplant hepatology at Johns Hopkins Hospital in Baltimore, Maryland. Her primary research interests focus on viral hepatitis— from both a drug development and a clinical perspective—liver transplantation, and complications of chronic liver disease.
Dr. Reau has been an invited lecturer at numerous presentations focused on viral hepatitis, fatty liver disease, cirrhosis, and liver transplantation. She is a fellow of the American Gastroenterological Association and American Association for the Study of Liver Diseases (AASLD). Additionally, she is the current editor in chief of CLD (Clinical Liver Disease) and was an author of
the AASLD/Infectious Diseases Society of America (IDSA) hepatitis C guidance document. She was the committee chair of the AASLD public policy committee and a member of the AASLD practice guideline committee for 4 years.
Dr. Reau has authored or coauthored more than 100 peer-reviewed articles that have been published in journals such as Hepatology, Hepatitis Research and Treatment, and Clinics in Liver Disease
She is Co-chair of the American Liver Foundation Medical Advisory Committee and sits on multiple advisory boards. She is currently a member of the steering committee for the hepatitis C special interest group for the AASLD and a member of the American College of Gastroenterology training committee.
PREAMBLE Target Audience
The educational design of this activity addresses the needs of gastroenterology and hepatology specialist physicians, nurse practitioners (NPs), and physician associates (PAs) who are involved in managing patients with primary biliary cholangitis (PBC).
Program Overview
PBC is a progressive autoimmune liver disease characterized by the destruction of intrahepatic bile ducts that, if left untreated, can lead to liver failure and, ultimately, the need for liver transplantation. This multimedia eHealth Source™ has been designed to support early identification of disease, as well as the timely implementation of effective therapy for PBC. Specific topic areas covered in this activity include guideline-directed diagnostic and prognostic measures, monitoring and treatment modification procedures, data for the use of newly US Food and Drug Administration–approved peroxisome proliferator–activated receptor (PPAR) agonists, and strategies for translating evolving evidence to practice. Clinical evidence will be augmented with video commentary from expert faculty and patients with PBC, providing invaluable insight on lived experiences and actionable recommendations for practicing clinicians.
Educational Objectives
After completing this activity, the participant should be better able to:
• Describe the pathogenesis, progression, and consequent disease manifestations of PBC, including signs, symptoms, and associated comorbidities in diverse patient populations
• Utilize evidence-based diagnostic criteria and noninvasive assessment strategies to identify patients with PBC early in the disease course
• Outline data on the safety, efficacy, and placement of novel PPAR agonist therapies for the treatment of PBC
• Adopt timely monitoring schedules to assess disease symptoms, treatment response, and need to escalate pharmacotherapy
• Construct management plans informed by patient disease profiles, comorbidities, therapeutic responses, and the psychosocial aspects of PBC
Physician Accreditation Statement
Integritas Communications is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to provide continuing medical education for physicians.
Physician Credit Designation
Integritas designates this enduring activity for a maximum of 1.0 AMA PRA Category 1 Credits™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
The ANCC’s Commission on Accreditation recognizes educational activities that are approved for AMA PRA Category 1 Credits™ by
providers who are accredited by the ACCME to award credit to learners. This reciprocity agreement allows nurses and nurse practitioners to use AMA PRA Category 1 Credits™ as approved ANCC contact hours for license renewal.
The American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC)
Recognition Statement
• Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 0.5 MOC points in the American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC) program. It is the CME activity provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
• For ABIM MOC points, your information will be shared with the ABIM through Integritas’ Program and Activity Reporting System (PARS). Please allow 6-8 weeks for your MOC points to appear on your ABIM records.
• By sharing your Diplomate Board ID # and DOB, you are giving Integritas Communications permission to use this information/ data to report your participation to these Boards via the PARS.
Integritas Contact Information
For more information about the approval (ACCME credit) of this program, please contact Integritas at info@exchangecme.com.
Instructions to Receive Credit
In order to receive credit for this activity, the participant must score at least 75% on the posttest and complete the program evaluation.
Fee Information & Refund/Cancellation Policy
There is no fee for this educational activity.
Disclosures of Conflicts of Interest
Integritas and Global Education Group adhere to the policies and guidelines, including the Standards for Integrity and Independence in Accredited CE, set forth to providers by the Accreditation Council for Continuing Medical Education (ACCME) and all other professional organizations, as applicable, stating those activities where continuing education credits are awarded must be balanced, independent, objective, and scientifically rigorous. All persons in a position to control the content of an accredited continuing education program provided by Integritas are required to disclose all financial relationships with any ineligible company within the past 24 months to Integritas. All financial relationships reported are identified as relevant and mitigated by Integritas in accordance with the Standards for Integrity and Independence in Accredited CE in advance of delivery of the activity to learners. The content of this activity was vetted by Integritas to assure objectivity and that the activity is free of commercial bias.
All relevant financial relationships have been mitigated.
The faculty has the following relevant financial relationships with ineligible companies:
Nancy S. Reau, MD: Consulting Fees: Arbutus Biopharma, Gilead Sciences, Inc., Salix Pharmaceuticals, Inc., Vir Biotechnology, Inc.; Contracted Research: AbbVie, Gilead Sciences, Inc.
The planners and managers at Integritas Communications and Global Education Group have no relevant financial relationships to disclose.
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/ or investigational uses of agents that are not indicated by the US Food and Drug Administration. Integritas and Global do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of any organization associated with this activity. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings.
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed in this activity should not be
used by clinicians without evaluation of patient conditions and possible contraindications or dangers in use, review of any applicable manufacturer’s product information, and comparison with recommendations of other authorities.
This activity is provided by Integritas Communications.
This activity is supported by an educational grant from Gilead Sciences, Inc.
INTRODUCTION
VIDEO 1: Meet the Expert
Primary biliary cholangitis (PBC) is a progressive autoimmune liver disease characterized by the destruction of intrahepatic bile ducts that, if left untreated, can lead to liver failure and, ultimately, the need for liver transplantation (LT). The primary goal of this eHealth Source™ is to support the early identification and effective treatment of PBC. Specific topics that will be covered include guideline-directed diagnostic practices, timely monitoring and escalation or modification of therapy, clinical evidence for the use of newly US Food and Drug Administration (FDA)-approved peroxisome proliferator-activated receptor (PPAR) agonists, and the translation of evolving evidence to appropriate management strategies that address disease phenotype, comorbidities, patient symptoms, and unique patient needs. Lastly, the clinical data presented will be augmented with pertinent qualitative, research-
derived insights—via video commentary—to provide actionable recommendations from experts to practicing clinicians.
CHAPTER 1. PBC—SCOPE OF THE PROBLEM
Epidemiology of PBC
PBC can affect all races, ethnicities, and both sexes, but predominantly affects female individuals, with a ratio of 4 to 6:1.1 Globally, 1 in 1000 women aged >40 years are living with PBC, and in the US, the number has steadily increased over the past decade.2,3 Analysis of data from the US-based Fibrotic Liver Disease Consortium revealed a prevalence rate of 39.2 per 100,000 persons, an increase of 72% among women and 114% among men from 2006 to 2014.3
Despite the notable increase in PBC prevalence—which can largely be attributed to decreased mortality rates due to effective treatment1—PBC is persistently misdiagnosed and underrecognized. A recent US health systems–based study found that, on average, the delay between presentation of symptoms and/or abnormal lab values and diagnosis is more than 3 years.4 This delay is even more significant in Black and Hispanic patient populations, leading to progression of disease and worse outcomes.4 Due to health care disparities and a lack of disease recognition by providers in these minority patient populations, epidemiologic estimates are also hampered.
“It’s not uncommon for patients to wait 8 to 10 years to get that final diagnosis from the time they initially had an abnormal liver test, and that, obviously, can have consequences in terms of response to therapy, progression of disease.” 33
Natural History and Pathophysiology
The serologic hallmark of PBC is the presence of antimitochondrial antibodies (AMAs). AMAs are ubiquitous autoantibodies that specifically target lipoic acids found in the dehydrogenase complex of the inner mitochondrial membrane of biliary epithelial cells.1,5,6 B cells and T cells primarily target autoantigens present on bile ductular cells. The destruction of bile duct cells leads to the obstruction of bile drainage from canaliculi, resulting in cholestasis and the destruction of hepatocytes (Figure 1).7 This can lead to progressive fibrosis and cirrhosis if left untreated (Figure 2).8
Although a combination of genetic risk factors and environmental triggers is believed to be involved in the etiology of PBC,5 genetic predisposition is strongly suggested by a high concordance rate of 63% among identical twins and a relative risk of 9.13 to 10.5 in first-degree relatives.1 Daughters of index women are seen to have the highest relative risk for PBC development.9 Several human leukocyte antigen allele associations have been reported as well.9 With respect to environmental factors, bacterial infection, specifically Escherichia coli, has been the most intensively studied.10 E coli contributes to breaking immunological tolerance against the mitochondria, ultimately resulting in the production of the highly-specific AMAs.10 Other environmental factors possibly associated with insult and inducing an autoimmune reaction include cigarette smoke, hair dye, nail polish, and toxic waste9,10; however, identifying specific triggers in patients with PBC remains a challenge.
AMAs are detected in approximately 95% of patients with PBC.1,5,6 Based on AMA positivity, liver biochemistry, symptoms, and complications, 4 distinct phases of disease have been recognized (Figure 3).6 These are the 1) silent, 2) asymptomatic, 3) symptomatic, and 4) liver insufficiency phases. During the silent phase, patients are positive for AMAs but have no symptoms or abnormal liver chemistries. During the asymptomatic phase, liver enzymes are elevated but patients remain without clinical symptoms. During the symptomatic phase, patients present with symptoms of fatigue or pruritus; however, these symptoms may not correlate with disease severity and have shown no correlation with survival outcomes.1 During the last, liver insufficiency, phase, patients can present with progressive jaundice, hepatic encephalopathy, and/or liver failure. Interindividual variability, however, is high in PBC and progression is not linear or predictable; thus, patients do not necessarily pass through all phases of disease.6
Patient Burden
The most common symptoms of PBC include fatigue, pruritus, and Sicca syndrome.1 Fatigue affects up to 80% of patients and is ranked as the most debilitating of all symptoms by patients. One qualitative study aiming to capture the lived experience of patients with PBC-related fatigue defined it as “an overwhelming sustained state of exhaustion that occurs without relation to antecedent events and is unrelieved by rest or sleep.”11 The physical effects of unpredictable fatigue were found to be notable, draining patients of stamina and endurance. This consequently influences their ability to live normal lives, causing social withdrawal and disconnection.11 Despite its disabling nature, however, fatigue is often not addressed due to the lack of effective treatments.11
Pruritus, the second most common symptom, follows fatigue closely in frequency and affects up to 70% of patients. Pruritus is associated with sleep deprivation, worsening fatigue, depression, social isolation, and self-mutilation.1 The origin of PBC-related pruritus is not well understood. Patients report both local and diffuse pruritus that is worse at night and exacerbated by clothing, heat, and pregnancy. Itching often fluctuates between periods of improvement and exacerbation.1
Sicca syndrome affects up to 66% of patients.1 Patients with dry mouth are at increased risk of dental caries.5 Other symptoms
include skin complaints, such as dry skin, hyperpigmentation, xanthelasma, xanthomas, jaundice, dermatographism, and fungal infection of the feet, occurring in about 40% of patients; abdominal pain, bone, muscle or joint pain; and cirrhosis-related symptoms such as edema, ascites, or esophageal varices.9
Many studies have investigated the impact of PBC and its symptoms on quality of life (QOL). One cohort of 2353 patients with PBC found that fatigue had its greatest impact on QOL when accompanied by symptoms of social dysfunction.12 Depression was also a significant factor for impaired QOL but appeared to be a manifestation of compounding symptom burdens.12 A major reoccurring theme reported by patients is the “delegitimization” of their symptoms. Because most PBC symptoms cannot be seen and patients may look well, the impact of PBC on QOL is often minimized by family, friends, and other social contacts.13 Many patients report that they also feel a degree of stigma associated with their disease due to the association of drugs and alcohol with liver disease and cirrhosis.13
In addition to symptoms, PBC puts patients at increased risk for developing cholestatic-related complications such as the metabolic bone diseases osteopenia and osteoporosis, dyslipidemia, and liposoluble vitamin deficiencies (vitamins A, D, E, K).14 In fact, extrahepatic manifestations are seen in up to 73% of patients with PBC and can appear before, during, or after diagnosis.14 Further, and importantly, is the high incidence with which PBC coexists with other extrahepatic autoimmune diseases (EHAIDs) and conditions (Table 1).15 Although the relationship between PBC and EHAIDs is not fully understood, it is hypothesized that they share a common pathogenic pathway, thereby imposing damage not only to the liver but systemically.15
VIDEO 2: The Path to Diagnosis: Patients’ Perspectives
Why Early Diagnosis Is Critical
There is no cure for PBC, thus, patient outcomes are largely dependent on early recognition of PBC and prompt initiation of therapies that aim to halt disease progression by preventing complications of liver dysfunction. Without treatment, liver fibrosis
can progress by 1 stage every 1.5 to 2 years.1 With first-line treatment, however, patients have a 5-fold reduction in the rate of progression from early-stage disease to extensive fibrosis or cirrhosis.16
If treated at an early histologic stage, the rate of progression to extensive fibrosis or cirrhosis is reduced 5-fold.
For patients with decompensated disease and declining liver function, LT is the only curative treatment option; however, PBC recurs in 20% to 40% of post-transplant cases.1 Further, ethnic and racial disparities in the US are highly prevalent—particularly among Hispanic patients—with respect to hospitalization, inhospital mortality, LT waitlist death, and LT waitlist removal due to clinical deterioration (Table 2).17,18 Although multiple factors may contribute to such disparities, these disparities highlight the need for early identification, diagnosis, and treatment of PBC in minority populations. Also included in minority populations are male patients: PBC in these individuals is often diagnosed when they are at an older age and, therefore, the disease may have progressed to a more advanced stage. PBC in male individuals is associated with lower biochemical response to first-line pharmacotherapy, higher rate of progression to cirrhosis, higher rates of liver-related death and LT, and increased risk for hepatocellular carcinoma (HCC).1
In recent years, significant advances in the diagnosis, staging, prognostication, and treatment of PBC have occurred, making it more likely for PBC to be accurately identified in its early histologic stage, with the potential to greatly impact prognosis and improve overall patient outcomes.19
CHAPTER 2. OPTIMIZING DIAGNOSTIC PROCESSES
Keys to Early Identification and Diagnosis
Patients with PBC may present with symptoms such as pruritus, fatigue, dry eyes/mouth, and jaundice or they can be asymptomatic. Asymptomatic patients are incidentally found to have abnormal liver chemistries during routine exam or while being evaluated for other medical reasons.9 Clinical findings on physical exam vary among patients and typically correlate with the stage of disease at presentation (ie, jaundice, hepatomegaly, etc)9; however, the presence or absence of symptoms at diagnosis or on follow-up does not correlate well with survival outcomes.20 Further, some patients rapidly advance to end-stage disease and LT at a young age, whereas others can remain asymptomatic for decades.8 Because of the heterogeneity of PBC presentation, symptomatology, and clinical course, identification of the disease requires a high index of suspicion.6,9
In general, PBC should be suspected in patients with chronic elevations in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and total bilirubin, with or without PBC-specific symptoms.8 Early biochemical markers of chronic cholestasis, defined as lasting >6 months, include concurrent elevations in serum ALP and gammaglutamyl transpeptidase (GGT).7 Comprehensive evaluation includes 1) eliciting a thorough history, including current medications and personal and family history of liver or autoimmune disease, 2) imaging to rule out obstruction, and 3) laboratory work to assess for alternative liver diseases such as
hepatitis B virus and hepatitis C virus; both PBC and hepatitis, however, can co-exist. Further, metabolic dysfunction–associated steatotic liver disease and alcohol use are common in the general public and their presence does not exclude the possibility of another liver disease. Thus, it is especially important to consider treatable alternatives or concomitant injuries. Various national and international guidelines have developed diagnostic flowcharts outlining generally consistent approaches to disease identification.5,7,8,21 One example of a proposed algorithm for the diagnosis of PBC in the setting of chronic cholestasis is provided in Figure 4 1
AMA, the serologic hallmark of PBC, is positive in approximately 95% of patients.1 Other antibodies associated with PBC include antinuclear antibodies (ANAs), anti–multiple nuclear dot antibodies, anticentromere antibodies, and antinuclear envelope antibodies. In particular, PBC-specific ANAs—anti-sp100 and antigp210—are detected in about 30% of all patients with PBC, as well as in up to 50% of patients with AMA-negative disease.1 Therefore, ANA can be considered a surrogate marker and should be tested in AMA-negative patients in whom there is a high suspicion of PBC.9 Anti-gp210 has also been associated with more-severe PBC.
It is important to note that AMA positivity alone is not sufficient to make the diagnosis of PBC. At least 2 of the following 3 criteria must be present5,22:
1. Biochemical evidence of cholestasis based on ALP elevation (≥1.5 × upper limit of normal [ULN])
2. Presence of AMA (titers ≥1:40) or other PBC-specific autoantibodies, including sp100 or gp210, if AMA is negative
3. Histologic evidence of nonsuppurative destructive cholangitis and destruction of interlobular bile ducts
Although some patients may ultimately require a liver biopsy, biopsy is not necessary to establish a PBC diagnosis unless specific antibodies are absent or to exclude other concomitant diseases such as metabolic dysfunction–associated steatohepatitis or autoimmune hepatitis (AIH). Lastly, only 1 in 6 patients with a positive AMA and normal ALP will develop PBC after 5 years. Thus, if liver tests are initially normal, these patients do not require additional intervention with treatment; instead, they should be followed at 2- to 3-year intervals.5,8 An overview of the utility of laboratory tests employed during the diagnosis and follow-up of PBC is provided in Table 3 7
Staging and Risk Stratification
Baseline disease stage can be defined as early vs advanced according to the following7:
• Histology, when a biopsy is available: absent or mild fibrosis vs bridging fibrosis or cirrhosis
• Serum levels of bilirubin and albumin: both parameters normal vs at least 1 parameter abnormal
The clinical implication for staging disease is to determine prognosis (for early-stage disease) and management (for latestage disease). Prognosis, however, should be discussed within the context of response to therapy. In general, prognosis is better
for patients whose ALP remains persistently below 1.5 × ULN.8 That said, the most reliable indicators for prognosis are serum bilirubin levels and Mayo risk score (the survival probability can be predicted for up to 7 years in an untreated patient).8,23 The Mayo risk score is the most frequently used model to predict short-term survival and includes the following clinical and biochemical variables: age of the patient, serum bilirubin, serum albumin, prothrombin time, and severity of edema (https:// www.mayoclinic.org/medical-professionals/transplant-medicine/ calculators/the-updated-natural-history-model-for-primary-biliarycholangitis/itt-20434724).24
Based on bilirubin levels, prognosis can be determined as follows25:
• Two successive bilirubin values >2 mg/dL: mean survival is 4 years
• Two successive bilirubin values >6 mg/dL: mean survival is 2 years
• Two successive bilirubin values >10 mg/dL: mean survival is 1.4 years
Lastly, when considering predictors of response, one must take into account associated extrahepatic disease. Thus, collaborating across medical specialties is valuable to successfully comanaging PBC and its comorbidities in order to improve patient outcomes.26
VIDEO 3: Discussing PBC and Prognosis With Your Patient
CHAPTER 3. ESTABLISHED STANDARDS OF CARE AND EMERGING PARADIGMS FOR PBC ON-TREATMENT
MONITORING
The high heterogeneity of PBC presentation, symptomatology, and clinical course requires a personalized, lifelong approach to ensure patients’ optimal care.6 The goals of therapy in PBC are to prevent end-stage complications of liver disease and ameliorate PBCassociated symptoms7 while achieving normalization of ALP and total bilirubin levels whenever possible.
Evidence that supports these biochemical goals as treatment targets includes observations by the Global PBC Study Group, which found that a total bilirubin threshold of 0.6 × ULN had the highest ability to predict LT or death at 1 year (hazard ratio, 2.12; P<0.001). Patients with bilirubin level ≤0.6 × ULN had a 10-year survival rate of 91.3%, whereas patients above the threshold had a significantly lower survival rate, of 79.2% (P<0.001).27,28 Another observation from the Global PBC Study Group was the correlation between ALP normalization and improved 10-year survival rates. Patients with ALP ≤1 × ULN had a 10-year survival rate of 93.2% and patients with ALP between 1.0 and 1.67 × ULN had a 10-year survival rate of 86.1%.27,28
First-Line UDCA: Efficacy and Limitations
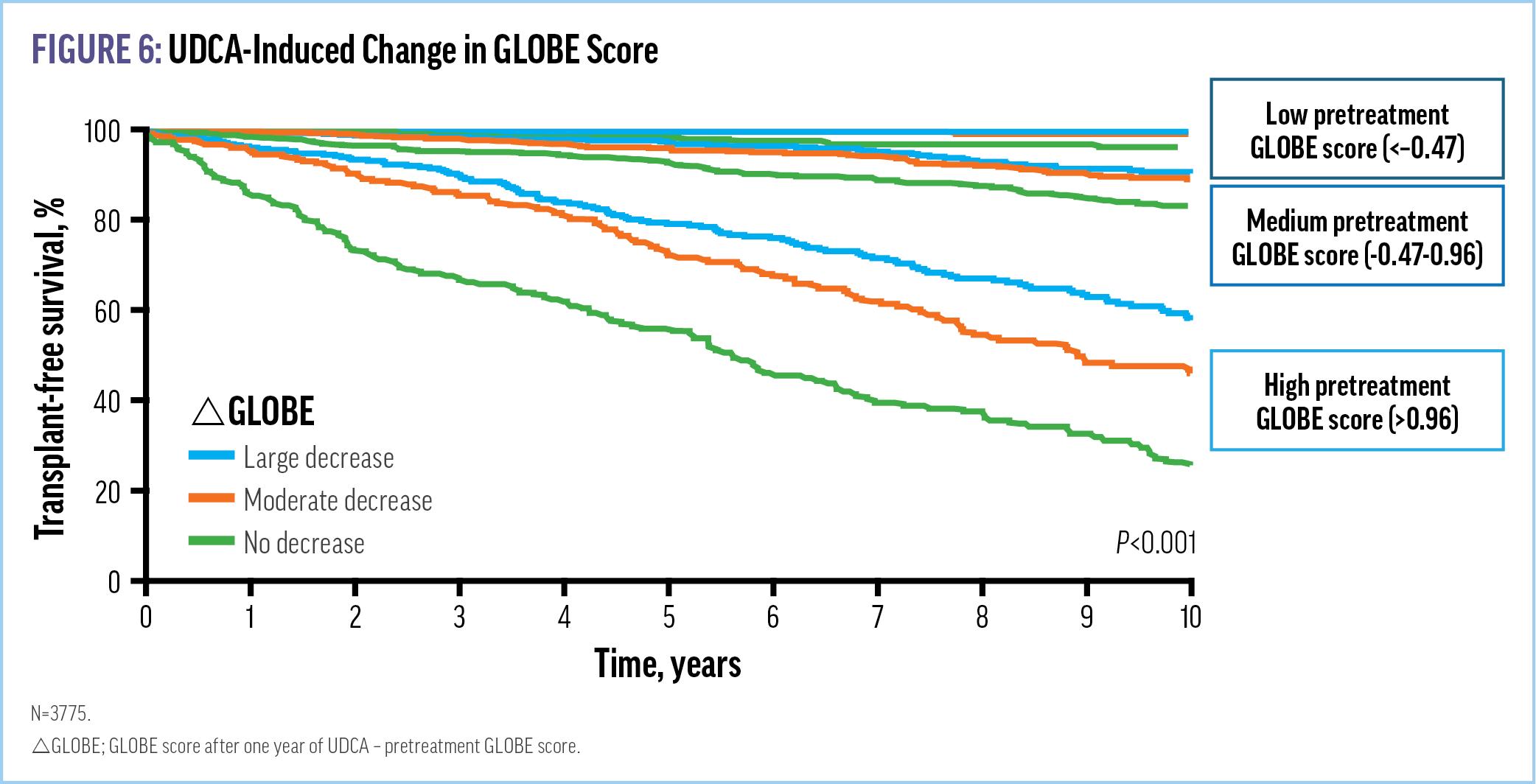
Guidelines concur that immediately following diagnosis, patients should be offered ursodeoxycholic acid (UDCA) therapy. UDCA was approved in 1997 by the FDA and is recommended by multiple international and national guidelines, as well as the American Association for the Study of Liver Diseases, as first-line treatment for all patients with PBC, irrespective of disease stage.23 UDCA is a simple, safe, and effective intervention that initiates the journey to cholestatic control and improves LT-free survival regardless of observed biochemical response (Figures 5 and 6).29-31 It is dosed in a weight-based manner, 13 to 15 mg/kg, and continued for life.23
In addition to altering the natural history of PBC, UDCA-induced changes in the Global Assessment of Liver Outcomes (GLOBE) score were found to be significantly associated with LT-free survival (Figure 5), supporting the long-term clinical gain from therapy based on its short-term impact on GLOBE score. The GLOBE score is a validated continuous prognostic model that assesses the absolute risk of LT or death among patients with PBC and was originally developed to evaluate the biochemical response to UDCA after 1 year.32 Despite UDCA’s proven efficacy, however, variability of biochemical response does exist. In fact, approximately 1 out of 3 patients has an incomplete response to UDCA, necessitating second-line therapy.33 Further, UDCA does not impact the burdensome symptoms of fatigue and pruritus commonly associated with PBC.
Monitoring Response to Therapy and Prognosis
GLOBE criteria comprise age, bilirubin, ALP, albumin, and platelets (https://www.globalpbc.com/globe).30 In addition to evaluating the
biochemical response to UDCA as mentioned, it can also be used to differentiate between low- and high-risk patients.32 Another continuous score developed by a large international consortium and externally validated by multiple studies is the United Kingdom
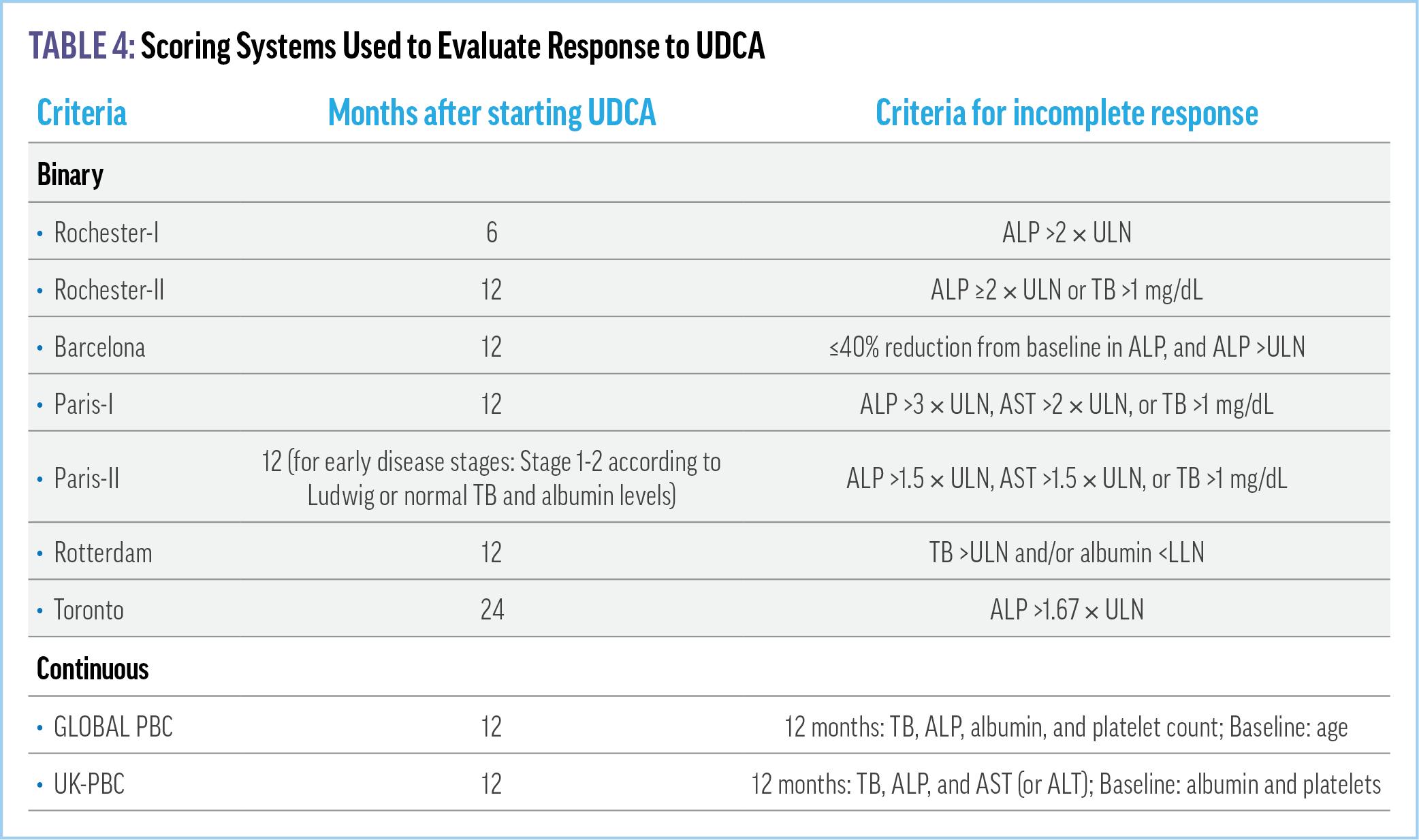
Primary Biliary Cholangitis (UK-PBC) risk score (https:// www.mdapp.co/uk-pbc-risk-score-calculator-360/). In addition to these validated criteria, there are several dichotomous tools that may be used to determine response to UDCA (Table 4).21,32,34 Among them, the primary metrics are ALP and bilirubin. Tools most frequently used in clinical practice are the Toronto and Paris-II criteria.
Patients with PBC and an insufficient response to UDCA that fulfills these criteria have poorer LT-free survival and increased rates of HCC.34 Thus, early identification of incomplete or inadequate response to UDCA is critical to ensuring timely escalation to second-line therapy and pursuing the best possible outcomes. Whereas available guidelines recommend the assessment of UDCA response at 12 months, data suggest response can be
detected within a shorter time interval. In fact, multiple recent clinical studies have sufficiently evaluated responses at 3 to 6 months after UDCA initiation. One such study, conducted by the Global PBC Study Group, investigated biochemical response patterns of 1362 UDCA-treated patients at 1 year, with the goal of determining the utility of ALP at 6 months as a predictor of insufficient UDCA response.35 The POISE criteria—defined as ALP <1.67 × ULN and normal total bilirubin at 1 year—were used to assess response to treatment at various thresholds of ALP at 6 months (Figure 7).35 Median ALP at baseline for the whole cohort was 2.22 × ULN. Results demonstrated that approximately 90% of patients with ALP >1.9 × ULN at 6 months did not meet POISE criteria at 1 year after UDCA initiation. Thus, the study concluded that an ALP threshold of 1.9 × ULN at 6 months could be used to identify patients in need of second-line therapy.
Further motivating the need for a paradigm shift in the monitoring of treatment response and early identification of partial responders who may benefit from the addition of second-line, or next-line, therapy is the speed with which response can be seen with newly approved PPAR agonist therapies—as soon as 4 weeks after treatment initiation—emphasizing the need to update scoring
systems to align with the earliest time nonresponders may be identified per clinical trial criteria.
To meet this need, new monitoring models have been proposed that take a “treat-to-succeed” approach instead of the traditional “waiting-to-fail” methodology (Figure 8).34 Thus, the decision to add a second-line agent should be prompted not only by an inadequate biochemical response to UDCA treatment but should consider the overall patient profile and risk factors.
Traditional Second-Line Treatment Options
In 2016, the FDA granted accelerated approval to obeticholic acid (OCA) based on data that demonstrated its efficacy in reducing ALP levels. OCA is a semisynthetic bile acid that acts as a farnesoid X receptor agonist and regulates bile acid metabolism. It has anti-inflammatory, anticholestatic, and antifibrotic effects.
In the 12-month phase 3 POISE study, 217 patients with PBC who had an inadequate response to or who were intolerant of UDCA
were randomly assigned to OCA 5 mg (with adjustment to 10 mg, if applicable), OCA 10 mg, or placebo.29 The primary end point of ALP <1.67 × ULN, with a reduction of ≥15% from baseline and a normal total bilirubin level,29 occurred at a higher rate with OCA 5‒10 mg and 10 mg, 46% and 47%, respectively, vs placebo, 10%. Further, the response to OCA was rapid, with a significant difference observed between treatment groups and placebo by week 2. Open-label extension studies also demonstrated consistent biochemical response rates. Pruritus was the most common adverse event, with 8 (4%) patients withdrawing from the study. Pruritus occurred at a higher incidence in the OCA 5‒10 mg and 10 mg treatment groups, 56% and 68%, respectively, vs placebo, 38%. The initiation of OCA at the lower, 5-mg, dose, however, with an adjustment to 10 mg was associated with a lower rate of discontinuation, suggesting a dose-dependent exacerbation of pruritus.
Serious adverse events also differed among OCA 5‒10 mg and 10 mg treatment groups vs placebo, 16% and 11% vs 4%, respectively. OCA was also associated with a reduction in highdensity lipoprotein cholesterol levels, an initial increase in lowdensity lipoprotein cholesterol levels, and persistent decreases in triglyceride levels.8 The use of OCA has been restricted in advanced cirrhosis—defined as cirrhosis with current or prior evidence of hepatic decompensation (eg, encephalopathy, coagulopathy) or portal hypertension (eg, ascites, gastroesophageal varices, persistent thrombocytopenia)—due to reported events of liver injury leading to hepatic decompensation or liver failure.36,37 Thus, patients should be monitored frequently for liver-related adverse reactions, ie, nausea, vomiting, diarrhea, jaundice, scleral icterus, and/or dark urine, and should permanently discontinue OCA if symptoms appear.37
In September 2024, the FDA denied full approval for OCA citing concerns regarding its clinical benefit and potential harm, requiring further clinical data to validate OCA's long-term safety and efficacy,38 a challenging feat in PBC due to the improbability of studies permanently leaving patients on placebo. Since then, a postmarketing clinical data review reported an increased risk of LT and death in patients without cirrhosis receiving OCA when compared with patients receiving placebo, HR 4.77.39 Thus, many questions centering on the potential future use of OCA remain unanswered.
Off-Label Treatment Options
Although not available in the US, bezafibrate, a pan-PPAR agonist, given in combination with UDCA has demonstrated effectiveness in improving biochemical prognostic markers of patients with highrisk PBC vs placebo, 31% vs 0 respectively.34,40 Bezafibrate in combination with OCA is also currently being studied.41 Fenofibrate, a synthetic PPAR-α agonist, has equally been associated with anticholestatic effects in a smaller body of data.42 Fibrates at therapeutic doses, however, can increase ALT and AST activity and elevate creatinine levels.34 Other adverse events include muscle-related pain (5%‒10% in bezafibrate-treated patients)34 and the risk of myopathy and rhabdomyolysis, especially in patients with renal insufficiency.8 Thus, because phase 3 data have not yet substantiated safety and efficacy in PBC, regulatory approval in PBC has yet to be granted and routine clinical use is not recommended.
VIDEO 4: Monitoring Treatment Response
CHAPTER 4. THE EMERGING ROLE OF PPAR AGONISTS AMONG SECOND-LINE THERAPIES
Although functionally similar to the fibrate class due to their mechanism of action, newly approved PPAR agonist agents for PBC are quite distinct, given their unique structure, highlyselective binding activity, and potency profiles. In 2024, two new agents in this novel class of drugs joined the treatment armamentarium.
PPAR Subtypes
PPARs are a group of nuclear receptor proteins that regulate gene transcription and play a role in many biological functions, such as inflammation and metabolism. There are 3 isotypes of PPARs— PPARα, PPARγ, PPARβ/δ—with each encoded by different genes and characterized by specific tissue allocation and actions (Figure 9).36
In hepatocytes, ligands that activate PPARα modulate bile acid metabolism through the inhibition of bile acid synthesis, increased bile acid secretion, reduction of bile acid toxicity, and detoxification of bile acid. PPARγ exerts immunomodulator activity with antiinflammatory properties, similar to PPARα. By negatively interfering with NF-κB, it suppresses production of tumor necrosis factor-α and interleukin-1β; however, this is mostly limited to Kupffer cells. In hepatocytes, ligand activation of PPAR-γ is steatogenic. PPARβ/δ is
more ubiquitous and activation of PPAR-β/δ in hepatocytes mainly results in the decrease of liver steatosis. PPARδ also has antiinflammatory effects and plays a role in the regulation of transport and absorption of bile compounds.
Given the effects on metabolism, inflammation, bile acid regulation, and liver fibrosis, the creation of selective ligands that target isoforms of PPAR have been a focus of therapeutic development in PBC.36
Clinical Trial Data for Newly Approved PPAR Agonists
Elafibrinor
The dual PPARα/δ agonist, elafibranor, is the first of 2 PPAR agonists that received accelerated approval from the FDA for
treatment of PBC in combination with UDCA or as monotherapy in patients who cannot tolerate UDCA. The elafibrinor phase 3 pivotal trial ELATIVE included 161 patients with PBC and an insufficient response to or intolerance of UDCA who were randomly assigned 2:1 to either elafibranor at a dose of 80 mg or to placebo.43 Of all patients, 95% were receiving background UDCA therapy and 35% had LSM >10 kPa or bridging fibrosis (or both) or cirrhosis. The primary end point was a composite biochemical response— defined as ALP <1.67 × ULN, reduction of ALP ≥15% from baseline, and total bilirubin ≤ULN—at week 52 (Figure 10). Key secondary end points were normalization of ALP at week 52 and change in pruritus intensity among patients with moderate to severe pruritus at baseline (defined as a Worst Itch Intensity Numeric Rating Scale [WI-NRS] score ≥4) through weeks 24 and 52 (Figures 11 and 12).
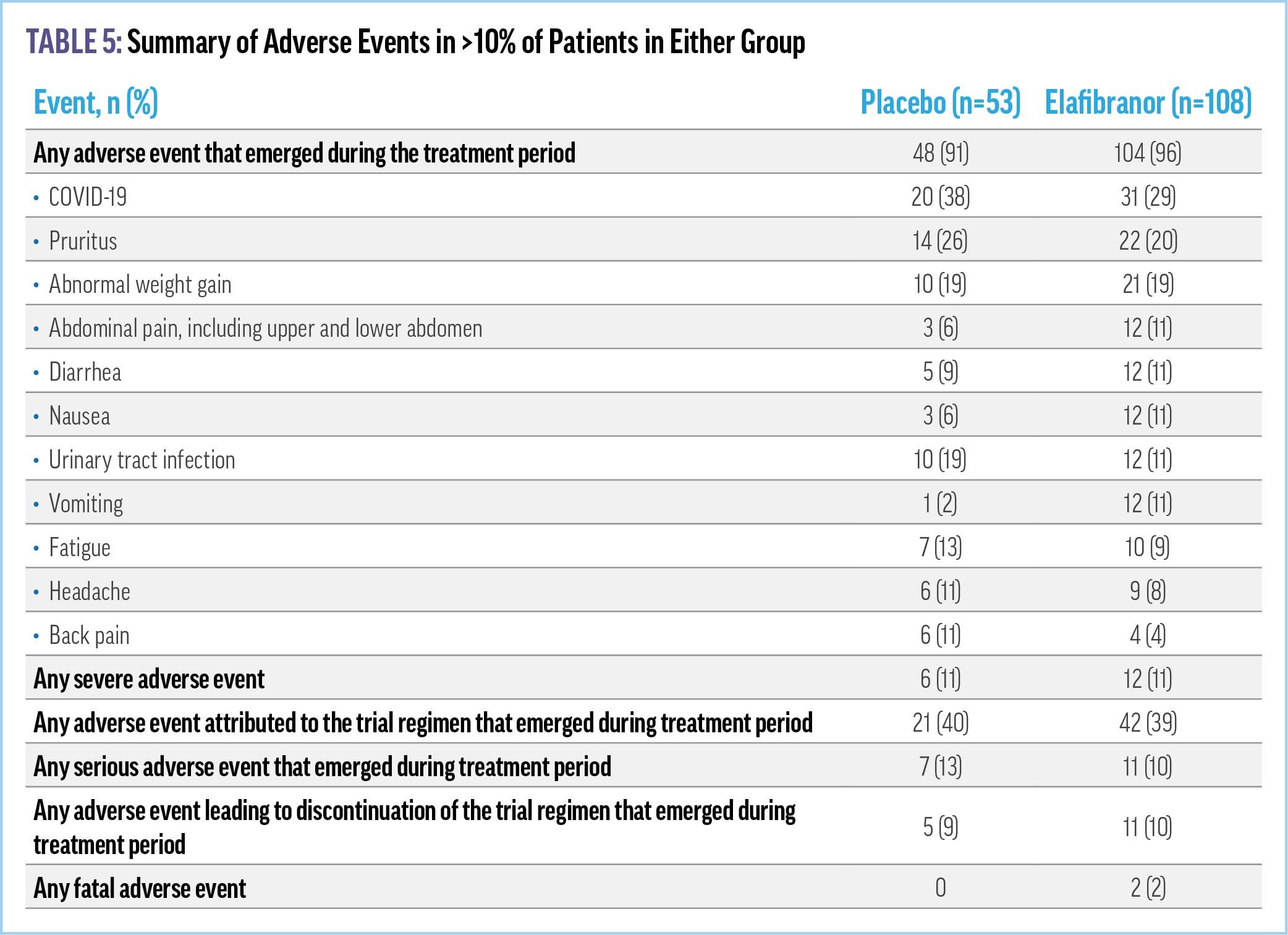
Elafibranor met the primary efficacy end point of composite biochemical response, with a rate of 51% vs 4% in placebo. ALP normalization was achieved in 15% of patients in the elafibranor group vs 0 patients in the placebo group.43 Further, reductions from baseline in ALP level were observed in the elafibranor group within 4 weeks after initiation of treatment. The predetermined secondary end point of improvement in pruritus in patients with moderate to severe pruritus was not met. Adverse events were similar between patients in the elafibranor and placebo groups, as were serious adverse events (Table 5). Elevated levels of creatine
phosphokinase led to permanent discontinuation of elafibranor in 4 (3.7%) patients in the elafibranor group, 2 of whom were receiving concomitant statin therapy. Caution was therefore noted for patients on concomitant statin therapy due to the potential risk of myalgias or rhabdomyolysis.
Seladelpar
Seladelpar, a PPARδ agonist, was the second agent in 2024 to receive fast track approval from the FDA for the treatment of PBC. The study design of the seladelpar pivotal RESPONSE trial was very similar to that of elafibranor. A total of 193 adult patients with PBC and an insufficient response to or intolerance of UDCA were randomly assigned 2:1 to seladelpar 10 mg or placebo.44 Of all patients, 93.8% were receiving background UDCA therapy and 14% had cirrhosis at baseline. The primary end point was
composite biochemical response, defined as ALP <1.67 × ULN, with ≥15% ALP decrease of 15% or more from baseline, and normal total bilirubin level up to 1.0 × ULN at 12 months (Figure 13). The 2 key secondary end points were similar to that of ELATIVE and included ALP normalization and change in pruritus NRS in patients with a baseline score ≥4 (38.3% of patients in the seladelpar group and 35.4% of patients in the placebo group).
Seladelpar met its primary end point of composite biochemical response, with a rate of 61.7% vs 20% in those receiving placebo (P<0.001).44 ALP normalization was met in 25% of patients receiving seladelpar vs 0 patients receiving placebo (Figure 14). The key secondary end point for change in pruritus in patients with moderate to severe itch was also met with statistical significance (Figure 15). Among the 49 patients with moderate to severe pruritus at baseline, reductions in pruritus NRS score were greater in the seladelpar group, 3.2 points, than in the placebo group, 1.7 points (P=0.005). Adverse events, including serious adverse events, were similar between patients in the seladelpar and placebo groups (Table 6). Creatine kinase and serum creatinine levels were also similar in the 2 groups, with no
worrisome adverse events associated with patients receiving statins.
Now, with the availability of 2 addition treatment options for patients with PBC that can safely and effectively improve biochemistries, it is imperative that attention be focused on successfully achieving the goal of disease remission and symptom control to improve patient QOL. Although real-world evidence and current guidelines have yet to provide additional guidance on the sequencing of next-line agents when 1 or more second-line therapies are not enough, we look to our experts to provide insights on these challenging questions.
VIDEO 5: Placement of Second-Line Treatment Options
CHAPTER 5. MANAGING THE WHOLE PATIENT
A Focus on Symptom Management and QOL
The complexity of symptom burden and its lack of relation to disease severity and treatment response suggest that specific stepwise approaches to symptom management are warranted that address both symptom biology and social impact.12 Thus, to address extrahepatic symptoms of disease, both pharmacologic and nonpharmacologic therapies are utilized. VIDEO 6: Addressing Patient Quality of Life Impairments in PBC
Pruritus
As previously discussed, UDCA and OCA are not used for symptom therapy. Currently, there is no FDA-approved therapy indicated for cholestatic itch associated with PBC. LT has been proven successful as treatment for intractable pruritus; however, symptom-based indications for LT are not standardized, as the current system for granting LT follows an “urgency-based” model that prioritizes patients by Model for End Stage Liver Disease score.45
That being said, the first choice for treatment of pruritus is cholestyramine, dosed at 4 to 16 g/day.34 Cholestyramine is a nonabsorbable anion exchange resin that must be given 20 minutes before meals and 2 to 4 hours before or after other medications due to its interference with intestinal absorption. Rifampicin (150‒300 mg twice daily) is a second-line agent for the treatment of pruritus refractory to cholestyramine.34 Adverse events, however, are serious, as drug-induced liver injury and renal impairment can occur. Where available, fibrates may also be used. The newer PPAR agonist therapy seladelpar has also been found to improve pruritus in patients with moderate to severe itch, as evidenced by the RESPONSE study. Whether its long-term effectiveness will result in an additional indication has yet to be determined. Lastly, a variety of clinical trials are now underway for difficult-to-treat pruritus.34 Linerixibat, an investigational targeted inhibitor of the ileal bile acid transporter, met the primary end point of improvement in itch with statistical significance in its phase 3 study.46
Fatigue
Fatigue significantly impacts patients’ QOL. Similar to pruritus, there is no FDA-approved therapy specifically indicated for the
treatment of fatigue. Further, unlike pruritus, fatigue typically persists after LT.34 Fatigue not only impedes patients’ ability to perform activities of daily living but diminishes their mental and physical capacity (Figure 16). Currently, the only management options for fatigue are structured exercise programs and education or counseling on how to cope with symptoms.34 This involves lifestyle modifications such as pacing activities, prioritizing sleep hygiene, reducing workload, and addressing any underlying contributing factors, including pruritus, that can diminish sleep quality and exacerbate fatigue.47 Other causes of fatigue, eg, depression, hypothyroidism, anemia, and sleep disorders, should be ruled out or treated if present.
Psychosocial Impairments
Psychological barriers and a sense of hopelessness can impact adherence to PBC treatment, leading to worse outcomes.48,49 Studies have shown that mental health issues associated with PBC are often overlooked by providers; meanwhile, many patients feel stigmatized by their peers, as well as by clinicians.49,50 Female patients, in particular, report that their experiences with PBC are often delegitimized, thereby exacerbating feelings of illness and interfering with social relationships.51 Moreover, issues related to
culture, social support, language, religion, and health beliefs can impair patient-clinician communication, negatively affect adherence, and exacerbate disparities in PBC treatment.17,52
Educating patients about personal psychosocial aspects of their condition supports a positive patient-provider relationship and promotes a holistic approach to reducing the chronic burden of disease. It also improves patient satisfaction and adherence. Patient support groups are another valuable resource to connect patients with peers who have similar lived experiences. Peers can provide insights on overcoming challenging aspects of chronic disease, offer a safe space to find emotional support and validation, and deliver information to those dealing with similar situations.
REFERENCES
1. Trivella J, John BV, Levy C. Primary biliary cholangitis: epidemiology, prognosis, and treatment. Hepatol Commun. 2023;7(6):e0179.
2. Jepsen P, Grønbæk L, Vilstrup H. Worldwide incidence of autoimmune liver disease. Dig Dis. 2015;33 (suppl 2):2-12.
3. Lu M, Zhou Y, Haller IV, et al; Fibrotic Liver Disease Consortium Investigators. Increasing prevalence of primary biliary cholangitis and reduced mortality with treatment. Clin Gastroenterol Hepatol. 2018;16(8):1342-1350.e1341.
4. Morales AMD, et al. Diagnostic delay in patients with primary biliary cholangitis. In: Liver Connect Annual Conference: Rush University Medical Center; 2014.
5. Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394-419.
6. Alvaro D, Carpino G, Craxi A, Floreani A, Moschetta A, Invernizzi P. Primary biliary cholangitis management: controversies, perspectives, and daily practice implications from an expert panel. Liver Int. 2020;40(11):2590-2601.
7. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol 2017;67(1):145-172.
8. Younossi ZM, Bernstein D, Shiffman ML, et al. Diagnosis and management of primary biliary cholangitis. Am J Gastroenterol. 2019;114(1):48-63.
9. Pandit S, Hrishikesh S. Primary biliary cholangitis. In: StatPearls [Internet]. StatPearls Publishing; 2024. Accessed December 31, 2024. https://www.ncbi.nlm.nih.gov/books/ NBK459209/.
10. Tanaka A, Leung PSC, Gershwin ME. Pathogen infections and primary biliary cholangitis. Clin Exp Immunol. 2019;195(1):25-34.
11. Jorgensen R. A phenomenological study of fatigue in patients with primary biliary cirrhosis. J Adv Nurs. 2006;55(6):689-697.
12. Mells GF, Pells G, Newton JL, et al; UK-PBC Consortium. Impact of primary biliary cirrhosis on perceived quality of life: the UK-PBC national study. Hepatology. 2013;58(1):273-283.
13. Corrigan M, Hirschfield G, Greenfield S, Parry J. Barriers to implementation of stratified care in primary biliary cholangitis: a scoping exercise. BMJ Open Gastroenterol 2019;6(1):e000226.
14. Chalifoux SL, Konyn PG, Choi G, Saab S. Extrahepatic manifestations of primary biliary cholangitis. Gut Liver. 2017;11(6):771-780.
15. Guo L, Zhou L, Zhang N, Deng B, Wang B. Extrahepatic autoimmune diseases in patients with autoimmune liver diseases: a phenomenon neglected by gastroenterologists. Gastroenterol Res Pract. 2017;2017:2376231.
16. Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology. 2000;32(6):1196-1199.
17. Cholankeril G, Gonzalez HC, Satapathy SK, et al. Increased waitlist mortality and lower rate for liver transplantation in Hispanic patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. 2018;16(6):965-973.e2.
18. Adejumo AC, Akhtar DH, Dennis BB, et al. Gender and racial differences in hospitalizations for primary biliary cholangitis in the USA. Dig Dis Sci. 2021;66(5):1461-1476.
19. Sohal A, Kowdley KV. Primary biliary cholangitis: promising emerging innovative therapies and their impact on GLOBE scores. Hepatic Med. 2023;15:63-77.
20. Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OFW. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53(6):865-870.
21. You H, Duan W, Li S, et al; Chinese Society of Hepatology, Chinese Medical Association. Guidelines on the diagnosis and management of primary biliary cholangitis (2021). J Clin Transl Hepatol. 2023;11(3):736-746.
22. Bowlus CL, Gershwin ME. The diagnosis of primary biliary cirrhosis. Autoimmun Rev. 2014;13(4-5):441-444.
23. Feng J, Xu JM, Fu HY, Xie N, Bao WM, Tang YM. Prognostic scores in primary biliary cholangitis patients with advanced disease. World J Gastrointest Surg. 2023;15(8):1774-1783.
24. Lammers WJ, Kowdley KV, van Buuren HR. Predicting outcome in primary biliary cirrhosis. Ann Hepatol. 2014;13(4):316-326.
25. Shapiro JM, Smith H, Schaffner F. Serum bilirubin: a prognostic factor in primary biliary cirrhosis. Gut. 1979;20(2):137-140.
26. Agrawal M, Spencer EA, Colombel JF, Ungaro RC. Approach to the management of recently diagnosed inflammatory bowel disease patients: a user’s guide for adult and pediatric gastroenterologists. Gastroenterology. 2021;161(1):47-65.
27. Murillo Perez CF, Harms MH, Lindor KD, et al; GLOBAL PBC Study Group. Goals of treatment for improved survival in primary biliary cholangitis: treatment target should be bilirubin within the normal range and normalization of alkaline phosphatase. Am J Gastroenterol. 2020;115(7):1066-1074.
28. Kowdley KV. An examination of the evidence behind biochemical markers in primary biliary cholangitis. Gastroenterol Hepatol (N Y). 2021;17(5 suppl 5):5-11.
29. Nevens F, Andreone P, Mazzella G, et al; POISE Study Group. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631-643.
30. Harms MH, van Buuren HR, Corpechot C, et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol. 2019;71(2):357-365.
31. de Veer RC, van Hooff MC, Corpechot C, et al; Global PBC Study Group. Ursodeoxycholic acid treatment-induced GLOBE score changes are associated with liver transplantation-free survival in patients with primary biliary cholangitis. Am J Gastroenterol. 2023;118(7):1196-1203.
32. Montano-Loza AJ, Corpechot C. Definition and management of patients with primary biliary cholangitis and an incomplete response to therapy. Clin Gastroenterol Hepatol 2021;19(11):2241-2251.e1.
33. Bowlus CL, Kenney JT, Rice G, Navarro R. Primary biliary cholangitis: medical and specialty pharmacy management update. J Manag Care Spec Pharm. 2016;22(10-a-s suppl):S3-S15.
34. Levy C, Manns M, Hirschfield G. New treatment paradigms in primary biliary cholangitis. Clin Gastroenterol Hepatol. 2023;21(8):2076-2087.
35. Murillo Perez CF, Ioannou S, Hassanally I, et al; Global PBC Study Group. Optimizing therapy in primary biliary cholangitis: alkaline phosphatase at six months identifies one-year non-responders and predicts survival. Liver Int. 2023;43(7):1497-1506.
36. Colapietro F, Gershwin ME, Lleo A. PPAR agonists for the treatment of primary biliary cholangitis: old and new tales. J Transl Autoimmun. 2023;6:100188.
37. Due to risk of serious liver injury, FDA restricts use of Ocaliva (obeticholic acid) in primary biliary cholangitis (PBC) patients with advanced cirrhosis. US Food and Drug Administration. May 26, 2021. Accessed December 31, 2024. https://www.fda.gov/ drugs/drug-safety-and-availability/due-risk-serious-liver-injury-fda-restricts-useocaliva-obeticholic-acid-primary-biliary-cholangitis.
38. Intercept receives complete response letter from FDA addressing OCALIVA supplemental new drug application (sNDA). News release. Intercept. November 12, 2024. Accessed December 31, 2024. https://www.interceptpharma.com/about-us/ news/?id=2979130.
39. Ocaliva (obeticholic acid) by Intercept Pharmaceuticals: drug safety communicationserious liver injury being observed in patients without cirrhosis. US Food and Drug Administration. December 12, 2024. Accessed December 31, 2024. https:// www.fda.gov/safety/medical-product-safety-information/ocaliva-obeticholic-acidintercept-pharmaceuticals-drug-safety-communication-serious-liver-injury.
40. Corpechot C, Chazouillères O, Rousseau A, et al. A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med. 2018;378(23):2171-2181.
41. Combination therapy in PBC after six months of treatment in late-breaking poster presentation at EASL Congress 2024. News release. Globe Newswire. June 5, 2024. Accessed December 31, 2024. https://www.biospace.com/intercept-presents-newdata-on-the-results-of-oca-bezafibrate-combination-therapy-in-pbc-after-sixmonths-of-treatment-in-late-breaking-poster-presentation-at-easl-congress-2024
42. Li C, Zheng K, Chen Y, et al. A randomized, controlled trial on fenofibrate in primary biliary cholangitis patients with incomplete response to ursodeoxycholic acid. Ther Adv Chronic Dis. 2022;13:20406223221114198.
43. Kowdley KV, Bowlus CL, Levy C, et al; ELATIVE Study Investigators’ Group. Efficacy and safety of elafibranor in primary biliary cholangitis. N Engl J Med 2024;390(9):795-805.
44. Hirschfield GM, Bowlus CL, Mayo MJ, et al; RESPONSE Study Group. A phase 3 trial of seladelpar in primary biliary cholangitis. N Engl J Med. 2024;390(9):783-794.
45. Khungar V, Goldberg DS. Liver transplantation for cholestatic liver diseases in adults. Clin Liver Dis. 2016;20(1):191-203.
46. Linerixibat shows positive phase III results in cholestatic pruritus (relentless itch) in primary biliary cholangitis (PBC). News release. GSK. November 19, 2024. Accessed December 31, 2024. https://www.gsk.com/en-gb/media/press-releases/linerixibatshows-positive-phase-iii-results-in-cholestatic-pruritus/.
47. Kośnik A, Wójcicki M. Fatigue in chronic liver disease patients: prevalence, pathophysiology, and management. Prz Gastroenterol. 2022;17(1):21-27.
48. Cançado GGL, Braga MH, Ferraz MLG, et al; Members of the Brazilian Cholestasis Study Group Consortium. Clinical features and treatment outcomes of primary biliary cholangitis in a highly admixed population. Ann Hepatol. 2022;27(1):100546.
49. Corrigan M, Hirschfield G, Greenfield S, Parry J. Barriers to implementation of stratified care in primary biliary cholangitis: a scoping exercise. BMJ Open Gastroenterol. 2019;6(1):e000226.
50. Mitchell C, Eddy A, Neuberger J, Mitchell-Thain R. P13 Patient experience of primary biliary cholangitis (PBC) symptom evaluation and management in clinical appointments and pathways compared to The British Society of Gastroenterology (BSG)/UK-PBC PBC treatment and management guidelines: results from PBC Foundation mobile app patient survey. Gut. 2022;71(suppl 3):A39-A41.
51. Montali L, Frigerio A, Riva P, Invernizzi P. “It’s as if PBC didn’t exist”: the illness experience of women affected by primary biliary cirrhosis. Psychol Health 2011;26(11):1429-1445.
52. Peters MG, Di Bisceglie AM, Kowdley KV, et al; PUMPS Group. Differences between Caucasian, African American, and Hispanic patients with primary biliary cirrhosis in the United States. Hepatology. 2007;46(3):769-775.
CLINICAL RESOURCE CENTER™
Guidelines
Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Lindor KD, et al. Hepatology. 2019;69(1):394-419.
Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study.
Lammers WJ, et al. Gastroenterology. 2014;147(6):1338-1349.e5;quiz e15.
https://pubmed.ncbi.nlm.nih.gov/25160979/
Efficacy and safety of elafibranor in primary biliary cholangitis.
Kowdley KV, et al. N Engl J Med. 2024;390(9):795-805. https://www.nejm.org/doi/10.1056/NEJMoa2306185
A phase 3 trial of seladelpar in primary biliary cholangitis.
Hirschfield GM, et al. N Engl J Med. 2024;390(9):783-794. https://www.nejm.org/doi/10.1056/NEJMoa2312100
Clinical Resources
American Association for the Study of Liver Disease (AASLD)
AASLD, founded in 1950, is a leading organization of scientists and health care professionals committed to preventing and curing liver disease. https://www.aasld.org/
European Association for the Study of the Liver (EASL)
EASL, founded in 1966, is a medical association dedicated to pursuing excellence in liver research, to the clinical practice of liver disorders, and to providing education to all those interested in hepatology. As of 2024, EASL serves 7000 members from 112 countries.
https://easl.eu/
Patient Resources and Advocacy
Organizations
PBCers Organization
The PBCers Organization is an online and in-person support group for people with PBC, care partners, and health care team members. https://pbcers.org/
PBC Foundation
The PBC Foundation is a UK-based charity founded on the principles of providing patients with PBC, their families, and friends with accurate, up-to-date information.
https://www.pbcfoundation.org.uk/
American Liver Foundation
ALF was created in 1976 by the AASLD to promote education, advocacy, support services, and research for the prevention, treatment, and cure of liver disease. https://liverfoundation.org/