2 minute read
J. Karl Johnson, PhD


904 Benedum Hall | 3700 O’Hara Street | Pittsburgh, PA 15261 P: 412-624-5644 C: 412-680-1783
karlj@pitt.edu http://www.engineering.pitt.edu/KarlJohnson/ William Kepler Whiteford Professor
Associate Director, Center for Research Computing
Molecular Modeling: A Tool for Development of New Materials
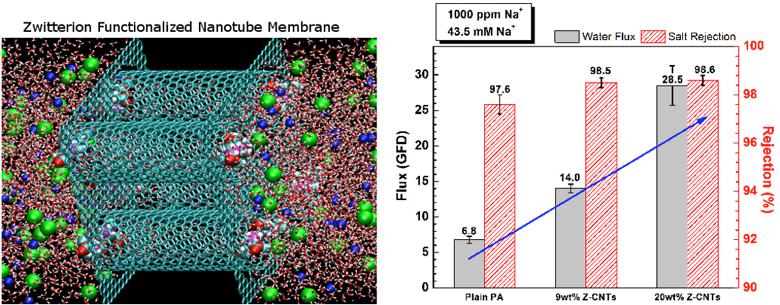
The Johnson research group uses computer simulation tools to discover, characterize, and optimize new materials from the bottom-up, starting at the levels of electrons, atoms, and molecules. Application areas studied by the Johnson group include designing new membrane materials for desalination of seawater, as shown in Figure 1, design of nanoporous materials for gas separation, CO2 capture, and conversion of CO2 to useful products, as illustrated in Figure 2, and elucidation of reaction mechanisms on surfaces and in condensed phases.
Figure 1. Molecular simulation (left) and experimental realization of a functionalized carbon nanotube membrane for desalination of seawater. Simulations first predicted that zwitterion functionalized nanotubes could be effective for desalination by imparting gatekeeper functionality to the nanotubes. The experiments verified the predictions, showing that a polyamide/carbon nanotube mixed matrix membrane has both higher flux and higher selectivity compared with plain polyamide membranes.
Molecular modeling can be used to screen a range of materials for a given application, thereby saving time and money by identifying a relatively small collection of promising candidate materials that can be investigated experimentally. For example, we have employed this strategy for screening of complex metal hydrides for hydrogen storage using a set of criteria including gravimetric, volumetric, and thermodynamic metrics. We were able to screen millions of compositions and conditions in order to identify a small subset of a few promising candidate materials that had not previously been identified. Several of these materials were investigated experimentally and some were found to outperform existing complex hydrides in many respects. This basic approach can also be applied to designing tailored sorbent and membrane materials based on metal organic framework or similar materials. We also use the techniques of molecular modeling to study complex systems involving chemical reactions on surfaces and in condensed phases. For large-scale systems this demands a hybrid approach employing both quantum mechanical modeling, to account for bond breaking and bond forming events, and classical modeling to capture long-range interactions and physical collective phenomena that are difficult to describe from a purely quantum mechanical approach.
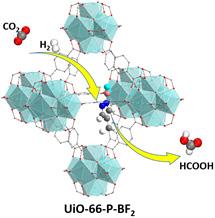
Figure 2. Porous metal organic framework, UiO-66, functionalized with a Lewis pair catalytic site for chemical reduction of CO2 to formic acid. This functional group was designed from first-principles simulations.








