

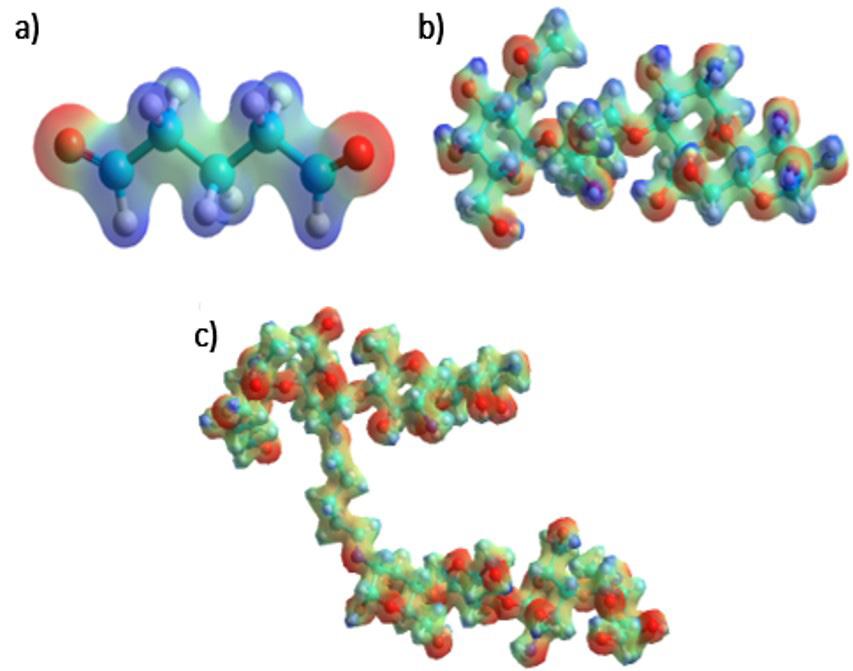
Electrostatic potential map of (a) glutaraldehyde, (b) chitosan, and (c) hydrogel, showing the highest (blue) and lowest (red) electron density sites.


Electrostatic potential map of (a) glutaraldehyde, (b) chitosan, and (c) hydrogel, showing the highest (blue) and lowest (red) electron density sites.






















































∙ Tecnologia de ponta: apresentando os nossos mais recentes instrumentos para análise térmica e reologia, projetados com incomparável precisão e confiabilidade. Aprimorando as capacidades de investigação: Soluções personalizadas para síntese de polímeros, caracterização e muito mais, facilitando descobertas inovadoras.


















Aplicações versáteis: Ideal para uma grande variedade de áreas de pesquisa de polímeros, que pode ir desde biomateriais e reciclagem à fabricação de aditivos e soluções energéticas.























Para mais informações, por favor entre em contato com: NETZSCH do Brasil Ind. Com. Ltda.











Rua Samuel Morse, 120 cj.123 – CEP:04576-060 – São Paulo/SP Contato: João Mesquita, Telefone: 011.2134.0306, Celular: 011.9.8690.9299
joao.mesquita@netzsch.com, https://analyzing-testing.netzsch.com/pt-BR

e d I tor I al C ou NCI l
Antonio Aprigio S. Curvelo (USP/IQSC) - President m ember S
Ailton S. Gomes (UFRJ/IMA), Rio de Janeiro, RJ (in memoriam)
Alain Dufresne (Grenoble INP/Pagora)
Artur José Monteiro Valente (UC/DQ)
Bluma G. Soares (UFRJ/IMA)
César Liberato Petzhold (UFRGS/IQ)
Cristina T. Andrade (UFRJ/IQ)
Edson R. Simielli (Simielli - Soluções em Polímeros)
Edvani Curti Muniz (UEM/DQI)
Elias Hage Jr. (UFSCar/DEMa)
José Alexandrino de Sousa (UFSCar/DEMa)
José António C. Gomes Covas (UMinho/IPC)
José Carlos C. S. Pinto (UFRJ/COPPE)
Júlio Harada (Harada Hajime Machado Consutoria Ltda)
Luiz Antonio Pessan (UFSCar/DEMa)
Luiz Henrique C. Mattoso (EMBRAPA)
Marcelo Silveira Rabello (UFCGU/AEMa)
Marco Aurelio De Paoli (UNICAMP/IQ)
Nikos Hadjikristidis (KAUST/ PSE)
Osvaldo N. Oliveira Jr. (USP/IFSC)
Paula Moldenaers (KU Leuven/CIT)
Raquel S. Mauler (UFRGS/IQ)
Regina Célia R. Nunes (UFRJ/IMA)
Richard G. Weiss Washington (GU/DeptChemistry) (in memoriam)
Roberto Pantani, (UNISA/DIIn)
Rodrigo Lambert Oréfice (UFMG/DEMET)
Sebastião V. Canevarolo Jr. (UFSCar/DEMa)
Silvio Manrich (UFSCar/DEMa)
Financial support:
Available online at: www.scielo.br

e d I tor I al C omm I ttee
Sebastião V. Canevarolo Jr. – Editor-in-Chief
a SS o CI ate e d I tor S
Alain Dufresne
Artur José Monteiro Valente
Bluma G. Soares
César Liberato Petzhold
José António C. Gomes Covas
José Carlos C. S. Pinto
Marcelo Silveira Rabello
Paula Moldenaers
Richard G. Weiss (in memoriam)
Roberto Pantani
Rodrigo Lambert Oréfice
d e S kto P P ubl IS h IN g www.editoracubo.com.br
“Polímeros” is a publication of the Associação Brasileira de Polímeros
São Paulo 994 St. São Carlos, SP, Brazil, 13560-340 Phone: +55 16 3374-3949
emails: abpol@abpol.org.br / revista@abpol.org.br http://www.abpol.org.br
Date of publication: September 2024
Polímeros / Associação Brasileira de Polímeros. vol. 1, nº 1 (1991) -.- São Carlos: ABPol, 1991-
Quarterly v. 34, nº 3 (September 2024)
ISSN 0104-1428
ISSN 1678-5169 (electronic version)
1. Polímeros. l. Associação Brasileira de Polímeros.

Website of the “Polímeros”: www.revistapolimeros.org.br
E E E E E E E E E E E E E E
o r I g IN al a rt IC le
Thermal and mechanical properties of filaments for additive manufacturing
Iago Rodrigues de Abreu, Renato de Sousa Nascimento Junior, Allef Gabriel Da Silva Fortes, Rudy Folkersma, Luigi Veloso Leitão, Fabio Delano Penha Marques, Arthur Antônio Sousa Sampaio, Layara Lorrana Ribeiro Leite de Castro, Daniella Stepheny Carvalho Andrade, Tatianny Soares Alves and Renata Barbosa e20240025
Potential biodegradable materials containing oat hulls, TPS, and PBS by thermoplastic injection
Samuel Camilo da Silva, Fabiola Azanha de Carvalho and Fabio Yamashita e20240026
Evaluation of graphene incorporation for mechanical properties of polypropylene composites
Júlio Harada, Claudio Aparecido Marcondes, Juliana Arquinto, Maria da Conceição Costa Pereira and Leonardo Gondim de Andrade e Silva e20240027
Properties of regenerated cellulose films with silver nanoparticles and plasticizers
Lays Furtado de Medeiros Souza Kataoka, Maria del Pilar Hidalgo Falla and Sandra Maria da Luz e20240028
Organocatalyzed polymerization of diblock copolymer based on poly(3-hexylthiophene) and poly(furfurylmethacrylate)
Hai Le Tran, Chau Duc Tran, Cam Hong Thi Nguyen, Thao Phuong Le Nguyen, Le-Thu Thi Nguyen, Thiet-Quoc Nguyen, Mai Ha Hoang, Tam Huu Nguyen and Ha Tran Nguyen e20240029
Development of nano-antimicrobial material based on bacterial cellulose, silver nanoparticles, and ClavaninA
Glícia Maria Oliveira, Alberto Galdino Silva-Junior, Octávio Luiz Franco, José Lamartine de Andrade Aguiar, Flávia Cristina Morone Pinto, Reginaldo Gonçalves de Lima-Neto, Maria Danielly Lima de Oliveira and César Augusto Souza de Andrade e20240030
Biodegradable hybrid PLA composites incorporating coffee husks and mineral fillers
Riquelme Gomes da Silva, Laysa Silva Barboza, Pedro Henrique Poubel Mendonça da Silveira, Marceli do Nascimento da Conceição, Roberto Carlos da Conceição Ribeiro and Daniele Cruz Bastos e20240031
Poly(vinyl alcohol) membranes produced by portable electrospinning device for in situ applications
André Luiz dos Santos and Sérgio Henrique Pezzin e20240032
Effect of synthesis conditions on the properties of magnetic crosslinked polystyrene microspheres
Thiago Alexandre de Oliveira Bouças, Marcos Antonio da Silva Costa, Henrique Almeida Cunha, Manoel Ribeiro da Silva, Wang Shu Hui and Jacira Aparecida Castanharo e20240033
Greener waterborne epoxy coatings with optimized UV-resistance
Mauro Sergio da Silva, Alana Gabrieli de Souza, Derval dos Santos Rosa, Ticiane Sanches Valera and Hélio Wiebeck e20240034
Analysis of the adsorption of Hg2+, Ni2+ and Cu2+ on chitosan hydrogels
Billy Alberto Ávila-Camacho and Norma Aurea Rangel-Vázquez e20240035
Evaluation of recycled polypropylene properties with banknote-reinforced
Ingrid Amélia dos Santos Matusinho, Ítalo Rocha Coura, Patterson Patricio de Souza and Patrícia Santiago de Oliveira Patrício e20240036
In Cassandra Callmann and her team’s proof-ofconcept, the sticky materials removed heavy metals including cadmium and lead.
Water polluted with heavy metals can pose a threat when consumed by humans and aquatic life. Sugarderived polymers from plants remove these metals but often require other substances to adjust their stability or solubility in water. Now, researchers from The University of Texas at Austin report on a sugar-like polymer that traps heavy metals within insoluble clumps for easy removal. In proof-of-concept tests, the polymer removed ionic cadmium and lead from river water spiked with these persistent contaminants.
Some heavy metal ions can be toxic at high levels in drinking water. Methods for removing these contaminants, such as filtration, can be energy intensive and rely on metal-capturing membranes that clog quickly and must be replaced.
To improve water purification, researchers have turned to plants. Plants defend their cells with a barrier of polysaccharides, made of macromolecules with repeating sugar units, that trap metal ions. For example, in a recent study, researchers used sticky polysaccharide extracts from okra and aloe to remove microplastics from wastewater. However, some polysaccharides dissolve in water, requiring additives to form insoluble gels for metal capture and removal. So, Cassandra Callmann and her research team set out to design a single material with sugar-like structures and controllable water solubility to remove heavy metals from water.
The team constructed several polymers, each having a water-insoluble backbone with different water-soluble carbohydrates dangling from the repeating units like charms on a bracelet. In initial tests, the carbohydrate “charm” that attracted and bound ionic cadmium most efficiently contained a carboxylic acid group. Next, in tests of water spiked with ionic cadmium, the polymer with carboxylic acid formed visible clumps after three minutes, and the clumps could be filtered out. The clumps also redissolved, releasing the cadmium, by adjusting the acidity of the water. After three cycles of binding, clumping and redissolving, the polymer maintained the same metal-trapping efficiency, demonstrating its potential as a recyclable material.
As a proof-of-concept, the team next tested the carbohydrate-containing polymer on Colorado River water spiked with ionic cadmium and lead. The river sample contained substantially more ionic calcium, sodium and magnesium than the added metals. Over a 24-hour period, the polymer captured up to 20% and 45% of the added cadmium and lead, respectively, and minimal amounts of the other metal ions. The researchers say their new material is a promising step towards more efficient, reusable and selective materials for water purification.
Source: The University of Texas at Austin – cns.utexas.edu
Researchers have innovated polymer synthesis using a Tesla coil to initiate polymerization through a remote spark discharge, successfully synthesizing high-purity polymers.
Researchers at the University of Tsukuba have devised a novel method for synthesizing widely used polymers, such as polystyrene, through radical polymerization. This approach utilizes a remote spark discharge from a Tesla coil—a high-voltage, high-frequency generator— enabling the reaction to occur without the need for traditional catalysts or polymerization initiators. This breakthrough represents a significant advancement in the field of synthetic polymer chemistry.
Polymers like polystyrene and acrylic resins, found in products such as food containers, packaging, and thermal cases, are traditionally synthesized using metal catalysts and radical polymerization initiators.
In their latest study, they introduced a novel method for polymer synthesis which involves generating monomer radicals through a remote spark discharge treatment using a Tesla coil, which is a high-voltage, high-frequency generator. These radicals then act as polymerization initiators.
The Tesla coil’s capability to generate a powerful discharge without a counter electrode enables the spark discharge treatment to be conducted externally to the reaction vessel. Using this method, the researchers have successfully synthesized high-purity polystyrene and polymethyl methacrylate (acrylate).
Moreover, the team has discovered a new method for synthesizing polymers by applying a similar spark discharge treatment to conjugated polymers. They use the “soliton” generated by the treatment as an initiator. This innovative approach is a first in synthetic polymer chemistry and opens new avenues for material synthesis using electromagnetic waves. “.
Source: SciTechDaily - Science, Space and Technology –scitechdaily.com



DATA: 19 A 23 DE OUTUBRO DE 2025
LOCAL: CAMPOS DO JORDÃO/SP

RESERVE ESTA DATA!
O CBPol é o principal evento científico da América Latina dedicado à área de polímeros, reunindo renomados pesquisadores e especialistas da academia e da indústria, tanto do Brasil quanto do exterior. O congresso oferece um espaço privilegiado para a discussão das mais recentes pesquisas, avanços tecnológicos e inovações no campo dos polímeros. Aproveite a oportunidade de interagir com os maiores especialistas do setor e contribuir para o futuro dos materiais poliméricos!




Inovação em Polímeros, Natureza e Gastronomia em um só lugar!
Inovação em Polímeros, Natureza e Gastronomia em um só lugar! Acesse nosso


October
International Conference on Polymers and Advanced Materials - Polymat 2024
Date: October 20-25, 2024
Location: Huatulco, Oaxaca
Website: iim.unam.mx/polymat
Plastics Recycling Technology
Date: October 23-24, 2024
Location: Vienna, Austria
Website: ami-events.com/event/bc7e919f-830f-4c9b-9963e5ca9f7f7b47/summary?RefId=Website_Home
Global Research Conference on Polymer Science, Composite Materials and its Application Date: October 24-26, 2024
Location: Barcelona, Spain
Website: https://polymerresearch2024.com/
November
Performance Polyamides
Date: November 12, 2024
Location: Cleveland, Ohio, United States of America
Website: ami-events.com/event/41d53af0-26aa-44f2-b060a2304697f534/summary?RefId=Website_AMI
Plastics Extrusion World Expo North America
Date: November 13-14, 2024
Location: Cleveland, Ohio, United States of America
Website: na.extrusion-expo.com/
5th World Expo on Biopolymers and Bioplastics
Date: November 13-14, 2024
Location: Dubai, United Arab Emirates
Website: biopolymers.materialsconferences.com/
Plastic Waste Free World Conference & Expo 2024
Date: November 13-14, 2024
Location: Cologne, Germany
Website: plasticfree-world.com
Fire Resistance in Plastics Europe
Date: November 18-20, 2024
Location: Cologne, Germany
Website: ami-events.com/event/64e3be94-9d51-4437-855188a45f3faa39/summary?RefId=Website_AMI
25th Brazilian Congress of Materials Engineering and Science - 25th CBECiMat
Date: November 24-28, 2024
Location: Fortaleza, Ceará, Brazil Website: cbecimat.com.br
December
XVIII Simposio Latinoamericano de Polímeros, XV Congreso Iberoamericano de Polímeros y XI Coloquio de Macromoléculas – SLAP 2024
Date: December 2-6, 2024
Location: Puerto Varas, Chile
Website: www.slap2024.cl
Polymer Engineering for Energy
Date: December 3-4, 2024
Location: London, United Kingdom
Website: ami-events.com/event/535774b9-c2a2-432d-be1fb7864beed551/summary?RefId=Website_AMI
Polymers in Hydrogen and CCUS Infrastructure
Date: December 5-6, 2024
Location: London, United Kingdom
Website: ami-events.com/event/a90b3d2c-b31f-4ffa-91a80dd7dddca53b/summary?RefId=Website_AMI
EBC 24 – The European Bioplastics Conference 2024
Date: December 10-11, 2024
Location: Berlin, German
Website: european-bioplastics.org/events/ebc/ Polymers — 5th International Congress and Exhibition
Polymers Azerbaijan: Construction and modernisation of plants
Date: December 11-13, 2024
Location: Baku, Azerbaijan
Website: https://polymerscongress.com/
January
9th Int’l Conference on Polymer Materials Science(PMS 2025)
Date: January 4-6, 2025
Location: Sanya, China
Website: www.academicx.org/PMS/2025
Thermoplastic Concentrates Forum
Date: January 20-22, 2025
Location: Tampa, Florida, United States of America
Website: ami-events.com/LXL0m2?locale=en&Refid=Website_AMI
February
Polyethylene Films
Date: February 17-19, 2025
Location: Tampa, Florida, United States of America
Website: ami-events.com/event/44f91be8-b556-4ec1-8fc208ce6f94dcbf/websitePage:8ce5942f-1379-4ec2-92e6db27048f1d3a?Refid=Website_AMI
March
ANTEC® 2025
Date: March 3-6, 2025
Location: Philadelphia, Pennsylvania, United States of America
Website: 4spe.org/antec
Conference Mechanics of Bond Breaking: from Single Molecules to Polymer Networks (MBB 2025)
Date: March 10-12, 2025
Location: Brussels, Belgium
Website: mbb2025.org
National Graduate Research Polymer Conference
Date: March 18-21, 2025
Location: Arizona State University, Tempe, Arizona, United States of America Website: 2025ngrpc.asu.edu/
April
40th International Conference of the Polymer Processing Society
Date: April 22-25, 2025
Location: Auckland, New Zealand Website: www.pps-40.org/
May
Polymer Sourcing and Distribution
Date: May 19-21, 2025
Location: Malaga, Spain
Website: ami-events.com/event/f82e6fcc-bfc5-4deb-b4814f20b4c03717/summary?RefId=Website_AMI
Plastic Closure Innovations
Date: May 19-21, 2025
Location: Malaga, Spain
Website: ami-events.com/event/2738902d-d7c8-4610-8199ec58a3f6f9d8/summary?RefId=Website_AMI
June
Plastics Recycling Technology
Date: June 10-11, 2025
Location: Long Beach, California, United States of America Website: ami-events.com/event/8663c550-b6d5-496e-9ca315f9fd54094e/summary?RefId=Website_AMI
International Conference on Macromolecular and Supramolecular Chemistry - ICMSC 2025
Date: June 21-22, 2025
Location: Vienna, Austria
Website: waset.org/macromolecular-and-supramolecularchemistry-conference-in-june-2025-in-vienna
EPF 2025 — European Polymer Congress 2025
Date: June 22-25, 2025
Location: Groningen, Netherlands Website: www.epf2025.org
Fluoropolymers 2025
Date: June 22-25, 2025
Location: Savannah, Georgia, United States of America Website: polyacs.org/25fluoropolymers

Iago Rodrigues de Abreu1 , Renato de Sousa Nascimento Junior1 , Allef Gabriel Da Silva Fortes1 , Rudy Folkersma2 , Luigi Veloso Leitão3 , Fabio Delano Penha Marques3 , Arthur Antônio Sousa Sampaio3 , Layara Lorrana Ribeiro Leite de Castro1 , Daniella Stepheny Carvalho Andrade4 , Tatianny Soares Alves1,3 and Renata Barbosa1,3*
1Laboratório de Polímeros e Materiais Conjugados – LAPCON, Programa de Pós-graduação em Ciência e Engenharia dos Materiais - PPGCM, Universidade Federal do Piauí – UFPI, Teresina, PI, Brasil
2NHL Stenden University of Applied Sciences, Emmen, Netherlands
3Curso de Engenharia de Materiais – CT, Universidade Federal do Piauí – UFPI, Teresina, PI, Brasil
4Curso de Engenharia de Materiais – CT, Universidade Federal de Pernambuco – UFPE, Recife, PE, Brasil
*rrenatabarbosa@yahoo.com
aOhis paper has been partially presented at the 17th Brazilian Polymer Oongress, held in Joinville, OO, 29/Oct - 02/Oov/2023
Obstract
This study explores the development of biodegradable filaments for additive manufacturing, aiming to create membranes for oil and water separation. Using a mixture of poly (lactic acid) and poly (butylene adipate co-terephthalate) (PLA/ PBAT) with additives such as zinc oxide, biocide and carnauba wax. The research evaluates the potential of these materials in membrane prototyping for this application. Characterization techniques such as FTIR, XRD, DSC and tensile strength were employed. The results showed that the addition of the additives did not induce the formation of new bands in the FTIR. However, new diffraction peaks appeared in the composites indicating the presence of the zinc oxide. DSC revealed a double peak in melting and crystallization temperatures and the mechanical tests showed significant influence of additives on tensile strength. The composite filaments proved to be suitable for 3D printing, suggesting their applicability in the manufacture of filter membranes for oil and water separation.
Keywords: additive manufacturing, material extrusion, zinc oxide, biocide, carnauba wax.
How to cite: Abreu, I. R., Nascimento Junior, R. S., Fortes, A. G. D. S., Folkersma, R., Leitão, L. V., Marques, F. D. P., Sampaio, A. A. S., Castro, L. L. R. L., Andrade, D. S. C., Alves, T. S., & Barbosa, R. (2024). Thermal and mechanical properties of filaments for additive manufacturing. Polímeros: Ciência e Tecnologia, 34(3), e20240025. https://doi. org/10.1590/0104-1428.20240034
The distribution and supply of drinking water is indispensable in society, but the ongoing contamination of aquatic environments remains a constant challenge and a significant environmental problem that requires efficient solutions. Sources of pollution arise from various sectors, including agriculture, steel production, textiles, food and petrochemical industries, responsible for the emission of a range of pollutants, from water-soluble toxic compounds to insoluble oily substances[1,2]. Several technologies have been used in the industrial sphere to treat and/or mitigate the effects of this pollution. Processes including centrifugation, chemical coagulation, membrane filtration, physical adsorption, and ionic filtration are often criticized for their high operating costs and the generation of additional waste that can harm the environment[3-5]
Recently, the adoption of microfiltration membranes has emerged as an effective solution, both as a primary treatment
and as a complement to conventional purification methods, especially those that have specific wettability properties. They are mainly used due to characteristics such as efficiency, low energy consumption and structure[6]. However, challenges such as membrane sizing caused by particle aggregation represent a point and a gap to be addressed in research and development aimed at improvements[7]
Among the manufacturing technologies currently available, additive production, commonly known as 3D printing, emerges as a notable innovation, offering the ability to produce complex parts at reduced costs. This technology is particularly valued for its versatility and design freedom, allowing the development of functional devices with different architectures[8,9]
The literature reports the manufacture of materials with superhydrophilic and superoleophobic characteristics by 3D printing for oil/water separation, mainly with surface
Abreu, I. R., Nascimento Junior, R. S., Fortes, A. G. S., Folkersma, R., Leitão, L. V., Marques, F. D. P., Sampaio, A. A. S., Castro, L. L. R. L., Andrade, D. S. C., Alves, T. S., & Barbosa, R.
functionalization. Li et al.[10] produced special superhydrophilic and superhydrophobic membranes by 3D inkjet printing (Binder Jetting) using cellulose acetate, polyvinyl alcohol and silica nanoparticles, achieving high efficiency in oil/water separation.
Furthermore, nanostructured materials have currently received attention in wastewater treatment we can mention some materials such as: Zeolite Minerals[11], Biochar[12] , graphene oxide – GO[13,14], titanium dioxide - TiO2 [13,15,16] , iron oxide – FeO[17]and zinc oxide – ZnO[9] .
Oil-water separation techniques face several technical challenges, from pore size specificity to membrane surface characteristics such as roughness and ability to repel or attract water (wetting). One of the main problems encountered is the clogging of the membrane pores by oil incrustation, significantly reducing filtration efficiency. To overcome these obstacles, solutions such as antifouling coatings stand out; they often combine biocides with other compounds to prevent oil accumulation in membranes[18-20]
Natural materials emerge in this field of research mainly due to their physical-chemical properties, cost-benefit and positive environmental impact, offering sustainable alternatives for water treatment[21,22]. For example, carnauba wax, obtained from the leaves of the Copernicia prunifera palm, is notable for its applications on self-cleaning and antifouling surfaces, as well as possessing anti-fog and anti-corrosion Properties[23,24] In addition, the incorporation of biocidal agents provides an effective solution to the challenge of microbial fouling, a common problem in industrial and aquatic environments. In this way, these materials are able to prevent the adhesion and growth of harmful organisms on their surfaces, particularly valuable in pipelines and water purification systems to maintain operational efficiency and safety[25] .
In addition to this, the adoption of biodegradable materials, such as the Ecovio® blend, composed of polylactic acid (PLA) and (PBAT), represents an environmentally responsible strategy. These materials have been used in the production of filters with antimicrobial properties, highlighting the commitment to sustainability in water treatment[26]
Based on the above, the objective of this study was to produce biodegradable polymeric filaments by extrusion, intended for use in additive manufacturing processes that employ the filament extrusion technique for prototyping filter membranes with specific wettability properties, intended to optimize oil separation treatment. and water.
2.1 Materials
The commercial blend Ecovio® (EC), produced by BASF SA, and formed by Poly (lactic acid) - PLA and Poly (butylene adipate-co-terephthalate) - PBAT, was used as the polymeric matrix. The following additives were used to form the composites: Sanitized Commercial Biocide MB E 22-70 (BCD), Zinc Oxide (ZnO) and Carnauba Wax (CW).
2.2 Preparation of the composites and filaments
The materials were previously mixed using concentrations of 3 e 5% for zinc oxide and 1% for the biocide and carnauba wax, respectively, in relation to the polymeric
blend. Subsequently, the compositions were processed in a single-screw extruder model AX-16 from AX plastics (L/D = 26) under a temperature profile of 170, 175 and 180 °C and screw speed of 40 rpm. The composite obtained was cooled in water and granulated in a pelletizer.
The pelletized composites obtained were dried in an oven at 80 °C for 4h and soon after the filaments were prepared using the same single screw extruder under the same processing conditions. The cooling was carried out by bathing in water and aid of a chillers of the brand Filmaq3D. The dimensions of the filaments were controlled by means of a reel, of the same brand, acting in the process of pulling the filaments, allowing adjustment of diameter of 1.75 ± 0.10 mm during the process.
2.3.1 Fourier transform infrared spectroscopy - FTIR
FTIR spectra were obtained on a Nicolet Summit FTIR spectrometer operating in attenuated total reflectance (ATR) mode. The scan was used in the range of 4000 to 500 cm-1 , totaling 16 scans in the analysis. The analyzes were carried out in the GREENPAC laboratory (Polymer Application Center - NHL Stenden), Netherlands.
2.3.2 X-ray Diffraction - XRD of Composite Filaments
The filaments were evaluated in a Bruker X-ray diffractometer, model D6 phaser. The incident radiation used was the Cu-Kα (λ = 1.5406 Å) scan between 5 and 75° (2θ), at a speed of 2/min at a power of 40 kV/30mA. The analysis was conducted at the University of Groningen (Groningen - Netherlands) in the Department of Macromolecular Chemistry & New Polymer Materials.
2.3.3 Differential Scanning Calorimetry - DSC
The DSC analysis of the filaments was carried out using a TA Instruments® DSC25 under a nitrogen atmosphere, in aluminum crucibles, and a heating ramp of 10°C/min with a scanning range between -20 and 200°C. The analyzes were carried out at the Green PAC – Polymer Application Center located at the NHL Stenden University of Applied Sciences (Emmen – Netherlands).
2.3.4
Tensile strength testing was performed on the filaments in accordance with ASTM D3379. Five samples of each composition were selected, and the test was carried out at a speed of 5 mm/min, with a 5 kN load cell on an EMIC universal machine, model DL 30000, belonging to the Materials Engineering Course at the Federal University of Piauí.
2.3.5
The data obtained from the tests tensile strength were statistically analyzed. For this, we used the analysis of variance (ANOVA) through the OriginPro software. Additionally, the Tukey test was applied to identify significant differences between the data sets, adopting a significance level of 5%. This methodology allowed to verify relevant changes with p > 0.05.
The FTIR spectra of the composite filaments (EC/3ZnO; EC/5ZnO; EC/3ZnO/BCD; EC/5ZnO/BCD; EC/3ZnO/BCD/ CW and EC/5ZnO/BCD/CW) and the polymer matrix (EC) are presented in Figure 1
The FTIR spectra of the pure mixture (EC) filaments showed characteristic bands of the two polymers that compose it. Thus, bands can be observed in the region of 2923 cm-1 and 2849 cm-1 associated with symmetric and asymmetric stretching vibrations of the CH2 and CH3 groups[27,28]. Bands at 1712 cm-1 and 1754 cm-1 referring to the C=O stretching vibrations (ester group) present in PLA and PBAT[28,29]. The bands observed at 1455 cm-1 and 1270 cm-1 refer, respectively, to the asymmetric bending vibration of the CH3 group of the PLA phase and the symmetric stretching vibration of the CO of the PBAT phase[30,31]. At 1180 and 1080 cm-1, the bands are characteristic of C=O stretching vibration[29]. The bands at 872 and 757 cm-1 are attributed to the absorption of the ester group and the vibration absorption of the α-methyl group, in addition, they can also be attributed to the amorphous and crystalline phases of PLA, respectively (Malinowski et al., 2020)[32]. The bending vibration band of the CH plane of the benzene ring of PBAT is represented at 728 cm-1[29,30]. It was not possible to observe modification or emergence of new absorption bands with the incorporation of additives.
In relation to zinc oxide, the main filler in the composite, it was also not possible to identify the formation of new bands due to the insertion of this filler. The literature reports Zn-O stretching in the regions of 906 - 644 cm-1 and 1507 - 1378 cm-1 attributed to the asymmetric stretching of the oxygen present in the composition[33,34]. The insertion of ZnO may have caused greater degradation of the PBAT phase as reported in the literature in bands that are identified by the absorption of carboxylic groups or carboxylates at 1660, 1550, 1424 and 969 cm-1 [35]
The literature reports bands related to the bonds CH, C=O, C≡C, CN, NH at 1848, 1468, 1515, 1161 and 722 cm-1 , respectively, in carnauba wax[36]. Likewise, the biocidal compound encapsulated with EVA did not induce changes in the composites, possibly this behavior refers to the chemical groups being from the same region as the Blend, with characteristic peaks at 1736, 1370, 1238, 1020 and 608 cm-1 [37]. From this discussion, the absence of new bands in the composites compared to the polymeric blend indicates that there was no interaction with the incorporation of additives.
Figure 2 shows the diffractograms of the composite filaments. The XRD curves for the blend filaments showed peaks related to the phases that make up the blend. Due to the semi-crystalline profile of the polymer, it is possible to observe a large amorphous halo and some well-defined peaks. Peaks were detected at 15.6°, 17.4°, 20.4°, 23° and 24.8° referring to the PBAT phase, respectively, corresponding to the planes (010), (111), (100) and (111)[38,39]
The crystalline phase of PLA is observed in peaks close to those found in the PBAT phase. The literature reports peaks

Figure 1. Infrared spectrum for the compositions EC, EC/3ZnO, EC/5ZnO, EC/3ZnO/BCD, EC/5ZnO/BCD, EC/3ZnO/BCD/CW and EC/5ZnO/BCD/CW

Figure 2. Diffractography of filaments EC, EC/3ZnO, EC/5ZnO, EC/3ZnO/BCD, EC/5ZnO/BCD, EC/3ZnO/BCD/CW and EC/5ZnO/BCD/CW
2θ = 16.76° and 19.26° referring to the crystallographic planes (200) and (201)[40,41] .
For zinc oxide, the main diffraction peaks were found at 2θ of 31.8°, 34.5°, 36.3°, 47.6°, 56.6°, 62.9° and 68° characteristic of this compound, with these peaks referring to planes (100), (002), (101), (102), (110), (103) and (112), respectively, corroborating the card (JCPDS 36-1451) and literature[42-45]. Carnauba wax has a semi-crystalline characteristic that can be visualized, with main peaks of greater intensity observed at angles of approximately 21.5°, 24.1°, as also reported by[46-49]. As previously mentioned, the biocide is a compound formed by zinc pyrithione loaded into EVA that presents characteristic peaks in at 21.9° and 23.8° referring to the orthorhombic system of the EVA unit cell, respectively for o plans (110) and (200)[45,50]. Possibly the pyrithione peaks were overlapped by the EVA peaks or it was not possible to read them, the literature reports two diffraction peaks at 11.1° and 22.5° for this compound[51]
Abreu, I. R., Nascimento Junior, R. S., Fortes, A. G. S., Folkersma, R., Leitão, L. V., Marques, F. D. P., Sampaio, A. A. S., Castro, L. L. R. L., Andrade, D. S. C., Alves, T. S., & Barbosa, R.
The thermal behavior by determination of glass transition temperature (Tg), cold crystallization temperature (Tcc) and crystalline melting temperature (Tm) is presented in Figure 3. The main function of the first heating is to quench the heat history in the material, but it can be of fundamental importance in the study of the aging of the material[52]
The curves indicated that the glass transition temperatures in all filaments showed values close to 65 °C in both the first and second heating. The literature reports that such thermal event is related to the PLA phase present in the blend. The insertion of additives in the PLA/PBAT matrix did not imply the change of this thermal transition[52-55]
It is also possible to observe an exothermic event after the glass transition temperature of the PLA, at approximately 102 C, and prior to the crystalline melting temperature. This behavior may be related to the crystalline melting temperature (Tm) of PBAT as reported in the literature and thermal relaxation and cold crystallization of PLA molecules, because the increase in temperature implies the increase of energy in the polymer chains between the amorphous and crystalline domains[53] .
For the blend curve and compositions with 3 and 5% ZnO, it was possible to observe that the crystalline melting temperature showed the same behavior. However, two crystalline fusion peaks were formed with the insertion of 1% in biocide and 1% in carnauba wax, even in lower percentage. The same behavior was recorded in the cooling stage, where the biocide and carnauba wax acted as nucleating agents in the recrystallization of the material[54,56]. This fact can be observed by the variation in the crystallinity percentage of the PLA phase with the incorporation of these additives.
The 2nd heating curves (b) showed the same behavior of the curves for the first heating, however, unlike the first ramp, only a fusion peak was detected for the compositions containing 1% of biocide and 1% of wax. Through the cooling (c) it was possible to observe that the nucleant effect of the biocide and the wax, resulted in the reduction of the crystalline melting temperature in approximately 3 °C.
Pascual-González et al.[57] produced PLA filaments with zinc oxide and observed the formation of a double crystalline melting peak and a reduction in the crystalline melting temperature for compositions with values above 7.5% of said additive.

Figure 3. DSC curves for the first heating (a), second heating (b) and cooling (c) for the compositions EC, EC/3ZnO, EC/5ZnO, EC/3ZnO/ BCD, EC/5ZnO/BCD, EC/3ZnO/BCD/CW, and EC/5ZnO/BCD/CW.
Thermal and mechanical properties of filaments for additive manufacturing
Increased melting and crystallization temperatures facilitate consistent solidification in the extrusion printing process, resulting in better interlayer adhesion and preventing delamination during printing. However, the DSC curves obtained during the first heating suggest that a secondary crystallization process may occur after printing, due to the additional time available for accommodation and ordering of polymer chains and the polycrystalline character with the insertion of these nucleant agents, such as carnauba wax[56] The results presented, especially in compositions with carnauba wax, suggest a controlled crystallinity for the 3D printing process, being beneficial in reducing defects such as warping, resulting from high residual stresses, indicating a good printability.
The ability of filaments to resist tensile strength is crucial for their processability in the extrusion process. It is essential that the filament has adequate resistance to preserve its integrity during transport to the extruder as well as at the time of transport by pulleys[58]. Table 1 illustrates the mechanical behavior in terms of maximum tension and elastic modulus of filaments derived from the blend and its composites.
It was observed that the EC composition presented a higher Maximum Stress value (31.50 MPa), while the EC/3ZnO/BCD/CW sample presented a higher Elasticity Modulus (916.47 MPa). Furthermore, the behavior when tensioned presented a completely different profile for each composition. According to the results, the pure polymer filament composition generally presented the highest values of mechanical properties among the other compositions. The results indicated a slight decrease in values that can be attributed to poor filler dispersion in the filament extrusion process, resulting in poor interfacial adhesion of the additive particles with the polymer matrix[33,58]
It was also observed that when comparing the values of the modulus of elasticity of the filament blended with the EC/3ZnO composition, a reduction in the stiffness of the material was noted, however, with the incorporation of the biocide and mainly carnauba wax, the values increased to be higher than those reported in previous compositions. In this sense, the incorporation of the biocide and carnauba wax increased the rigidity of the filament. Mathew et al. [29] evaluated the tensile strength properties of filaments
Table 1. Tensile strength and modulus of elasticity for the compositions EC, EC-3ZnO, EC/5ZnO, EC/3ZnO/BC, EC/5ZnO/ BCD, EC/3ZnO/BC/CW and EC/5ZnO/BCD/CW.
Compositions
EC 31.5 ± 1.69 a 779.19 ± 35.78 b
EC/3ZnO 26.10 ± 1.20 a,d 719.64 ± 42.65 b
EC/5ZnO 27.35 ± 1.21 a,c 793.55 ± 67.88 a,b
EC/3ZnO/BCD 26.49 ± 0.69 a,b,c 824.08 ± 37.75 a
EC/5ZnO/BCD 25.76 ± 0.63 a,b,c,d 851.96 ± 55.043 a
EC/3ZnO/BCD/CW 27.72 ± 0.58 a,b 916.47 ± 54.49 a
EC/5ZnO/BCD/CW 26.90 ± 0.66 b,c,d 756.51 ± 121.32 a,b
a,b,c,d Results of groups marked with the same letter show no statistical difference (p>0.05) between the means according to the Tukey test.
composed of the PLA/PBAT blend in different quantities and observed a reduction in tensile strength, flexion and modulus of elasticity with an increase in the percentage of PBAT, however, at 40% a reduction in elongation was noted due to the increase in the dispersed phase. The modulus of elasticity showed an increase in its values in relation to the little variation detected in the tensile strength. It is important to highlight that filaments produced in the laboratory on a small scale are subject to having their properties affected by factors such as humidity, variation in diameter, as well as low homogeneity in composition[59] .
The results of the statistical analysis revealed that the incorporation of loads does not have significant effect when the filament is evaluated in relation to maximum strength. For the modulus of elasticity, there was a significant increase in the compositions EC/3ZnO/BCD, EC/5ZnO/BCD, EC/3ZnO/ BCD/CW, EC/3ZnO/BCD/CW demonstrating greater tension in the composite, impacting the tensile strength of the filaments. This increase in stiffness is beneficial in the transport of the filament by the pulleys since the polymeric blend (EC) is a more plastic material due to the percentage of PBAT in the composition.
Polymeric filaments for additive manufacturing by filament extrusion were successfully produced and had their chemical, thermal and mechanical properties evaluated. The FTIR spectra indicated the absence of new bands with the insertion of the biocide and carnauba wax into the polymeric blend, showing no chemical changes in the composites. Regarding the XRD analysis, peaks relating to the ECOVIO, ZnO, wax phase were recorded and the peaks corresponding to the biocide phase may have been overlapped by the amorphous region of the blank. DSC analysis showed double peak formation of Tm and Tcc during the first heating and cooling for compositions containing biocide and carnauba wax, as well as changes in Tm values on the second heating. The evaluation of the tensile strength properties indicated that the incorporation of additives modified the mechanical resistance. The composition EC/3ZnO/BCD/CW exhibited higher modulus of elasticity resulting in greater stiffness justified by the insertion of the biocide and wax. The filaments produced showed satisfactory properties, enabling their application as functionalized membranes in water/oil treatment. Furthermore, more studies and characterizations on the viability of the product will be necessary. Thus, with the innovative nature of the work, new perspectives are opened for the production of membranes without the need for extra surface treatment.
• Conceptualization – Iago Rodrigues de Abreu.
• Data curation – Iago Rodrigues de Abreu.
• Formal analysis – Iago Rodrigues de Abreu; Fabio Delano Penha Marques; Arthur Antônio Sousa Sampaio.
• Funding acquisition – Renata Barbosa; Tatianny Soares Alves; Rudy Folkersma.
• Investigation – Iago Rodrigues de Abreu.
Abreu, I. R., Nascimento Junior, R. S., Fortes, A. G. S., Folkersma, R., Leitão, L. V., Marques, F. D. P., Sampaio, A. A. S., Castro, L. L. R. L., Andrade, D. S. C., Alves, T. S., & Barbosa, R.
• Methodology – Iago Rodrigues de Abreu; Fabio Delano Penha Marques; Arthur Antônio Sousa Sampaio; Luigi Veloso Leitão; Renato de Sousa Nascimento Junior; Allef Gabriel da Silva Fortes.
• Project administration – Iago Rodrigues de Abreu; Renata Barbosa.
• Resources – Renata Barbosa.
• Software – NA.
• Supervision – Renata Barbosa; Rudy Folkersma.
• Validation – Renato de Sousa Nascimento Junior; Luigi Veloso Leitão; Fabio Delano Penha Marques; Arthur Antônio Sousa Sampaio; Allef Gabriel da Silva Fortes.
• Visualization – Layara Lorrana Ribeiro Leite de Castro; Daniella Stepheny Carvalho Andrade.
• Writing – original draft – Iago Rodrigues de Abreu
• Writing – review and editing – Iago Rodrigues de Abreu; Renata Barbosa.
6. Acknowledgements
The authors would like to thank the Federal University of Piauí; the Laboratory of Polymers and Conjugated Materials (LAPCON); NHL Stenden University of Applied Sciences; GREENPAC Laboratory (Polymer Application Center); Piauí State Research Support Foundation (FAPEPI); National Council for Scientific and Technological Development (CNPq) and Funding: This work was supported by CNPq [process number: 308446/2018-6] and [process number: 308309/2022-7].
7. References
1. Luo, X., He, Z., Gong, H., & He, L. (2022). Recent advances in oil-water separation materials with special wettability modified by graphene and its derivatives: a review. Chemical Engineering and Processing, 170, 108678 http://doi.org/10.1016/j. cep.2021.108678
2 Abu-Thabit, N. Y., Uwaezuoke, O. J., & Abu Elella, M. H. (2022). Superhydrophobic nanohybrid sponges for separation of oil/ water mixtures. Chemosphere, 294, 133644. http://doi. org/10.1016/j.chemosphere.2022.133644 PMid:35065181.
3 Zhu, Y., Liu, Y., Mohamed, H. F., Zheng, X., He, J., & Lin, L. (2022). Rigid, eco-friendly and superhydrophobic SiO2Polyvinyl alcohol composite sponge for durable oil remediation. Chemosphere, 307(Pt 4), 135990 http://doi.org/10.1016/j. chemosphere.2022.135990 PMid:35977562.
4 Malczewska, B., Farnood, R. R., & Tabe, S. (2022). Natural organic matter removal by electrospun nanofiber membranes coated with heated aluminum oxide particles. Journal of Water Process Engineering, 45, 102498. http://doi.org/10.1016/j. jwpe.2021.102498
5 Dias, R., Daam, M. A., Diniz, M., & Maurício, R. (2023). Drinking water treatment residuals, a low-cost and environmentally friendly adsorbent for the removal of hormones - a review. Journal of Water Process Engineering, 56, 104322 http://doi. org/10.1016/j.jwpe.2023.104322
6 Rao, L., You, X., Chen, B., Shen, L., Xu, Y., Zhang, M., Hong, H., Li, R., & Lin, H. (2022). A novel composite membrane for simultaneous separation and catalytic degradation of oil/water emulsion with high performance. Chemosphere, 288(Pt 1), 132490 http://doi.org/10.1016/j.chemosphere.2021.132490 PMid:34624347.
7 Liu, L., Xiao, Z., Liu, Y., Li, X., Yin, H., Volkov, A., & He, T. (2021). Understanding the fouling/scaling resistance of superhydrophobic/omniphobic membranes in membrane distillation. Desalination, 499, 114864 http://doi.org/10.1016/j. desal.2020.114864
8 Yan, C., Ma, S., Ji, Z., Guo, Y., Liu, Z., Zhang, X., & Wang, X. (2019). 3D printing of an oil/water mixture separator with in situ demulsification and separation. Polymers, 11(5), 774 http://doi.org/10.3390/polym11050774 PMid:31052425.
9 Kumbhakar, P., Ambekar, R. S., Mahapatra, P. L., & Tiwary, C. S. (2021). Quantifying instant water cleaning efficiency using zinc oxide decorated complex 3D printed porous architectures. Journal of Hazardous Materials, 418, 126383 http://doi. org/10.1016/j.jhazmat.2021.126383 PMid:34329007.
10. Li, X., Shan, H., Zhang, W., & Li, B. (2020). 3D printed robust superhydrophilic and underwater superoleophobic composite membrane for high efficient oil/water separation. Separation and Purification Technology, 237, 116324 http:// doi.org/10.1016/j.seppur.2019.116324.
11 Kennedy, A. J., Ballentine, M. L., Das, A., Griggs, C. S., Klaus, K., & Bortner, M. J. (2021). Additive manufacturing for contaminants: ammonia removal using 3D printed polymerzeolite composites. ACS ES&T Water, 1(3), 621-629. http:// doi.org/10.1021/acsestwater.0c00131
12 Tengku Yasim-Anuar, T. A., Yee-Foong, L. N., Lawal, A. A., Farid, M. A. A., Yusuf, M. Z. M., Hassan, M. A., & Ariffin, H. (2022). Emerging application of biochar as a renewable and superior filler in polymer composites. RSC Advances, 12(22), 13938-13949 http://doi.org/10.1039/D2RA01897G PMid:35558839.
13. Zhong, Q., Shi, G., Sun, Q., Mu, P., & Li, J. (2021). Robust PVA-GO-TiO2 composite membrane for efficient separation oil-in-water emulsions with stable high flux. Journal of Membrane Science, 640, 119836 http://doi.org/10.1016/j. memsci.2021.119836
14 Zeng, T., Yu, Y., Li, Z., Zuo, J., Kuai, Z., Jin, Y., Wang, Y., Wu, A., & Peng, C. (2019). 3D MnO2 nanotubes@reduced graphene oxide hydrogel as reusable adsorbent for the removal of heavy metal ions. Materials Chemistry and Physics, 231, 105-108 http://doi.org/10.1016/j.matchemphys.2019.04.019
15 Vidales, M. J. M., Nieto-Márquez, A., Morcuende, D., Atanes, E., Blaya, F., Soriano, E., & Fernández-Martínez, F. (2019). 3D printed floating photocatalysts for wastewater treatment. Catalysis Today , 328, 157-163 http://doi.org/10.1016/j. cattod.2019.01.074
16 Wang, D., Zhi, T., Liu, L., Li, Y., Yan, W., Tang, Y., He, B., Hu, L., Jing, C., & Jiang, G. (2022). 3D printing of TiO2 nano particles containing macrostructures for As(III) removal in water. The Science of the Total Environment, 815, 152754 http://doi. org/10.1016/j.scitotenv.2021.152754 PMid:34995588.
17. Yang, Z.-F., Li, L.-Y., Hsieh, C.-T., Juang, R.-S., & Gandomi, Y. A. (2018). Fabrication of magnetic iron Oxide@Graphene composites for adsorption of copper ions from aqueous solutions. Materials Chemistry and Physics, 219, 30-39 http:// doi.org/10.1016/j.matchemphys.2018.07.053
18 Thomas, C. M., Kumar, D., Scheel, R. A., Ramarao, B., & Nomura, C. T. (2022). Production of medium chain length polyhydroxyalkanoate copolymers from agro-industrial waste streams. Biocatalysis and Agricultural Biotechnology, 43, 102385 http://doi.org/10.1016/j.bcab.2022.102385
19 Baig, U., Al-Kuhaili, M. F., & Dastageer, M. A. (2023). Photoresponsive Zinc Oxide-coated alumina ceramic membrane with super-wettable and self-cleaning features fabricated by single step RF magnetron sputtering for oily water treatment. Process Safety and Environmental Protection, 175, 541-553 http://doi.org/10.1016/j.psep.2023.05.044
Thermal and mechanical properties of filaments for additive manufacturing
20 Zhao, X., Zhang, R., Liu, Y., He, M., Su, Y., Gao, C., & Jiang, Z. (2018). Antifouling membrane surface construction: chemistry plays a critical role. Journal of Membrane Science, 551, 145-171 http://doi.org/10.1016/j.memsci.2018.01.039
21 al-Shaeli, M., Al-Juboori, R. A., Al Aani, S., Ladewig, B. P., & Hilal, N. (2022). Natural and recycled materials for sustainable membrane modification: recent trends and prospects. The Science of the Total Environment, 838(Pt 1), 156014 http:// doi.org/10.1016/j.scitotenv.2022.156014 PMid:35584751.
22. Li, S., Huang, L., Wang, D., Zhou, S., Sun, X., Zhao, R., Wang, G., Yao, T., Zhao, K., & Chen, R. (2023). A review of 3D superhydrophilic porous materials for oil/water separation. Separation and Purification Technology, 326, 124847 http:// doi.org/10.1016/j.seppur.2023.124847
23 Bashari, A., Koohestani, A. H. S., & Salamatipour, N. (2020). Eco-friendly Dual-functional textiles: green water-repellent & anti-bacterial. Fibers and Polymers, 21(2), 317-323 http:// doi.org/10.1007/s12221-020-9568-6
24 AbdulKadir, W. A. F. W., Ahmad, A. L., & Boon Seng, O. (2021). Carnauba wax/halloysite nanotube with improved antiwetting and permeability of hydrophobic PVDF membrane via DCMD. Membranes (Basel), 11(3), 228 http://doi.org/10.3390/ membranes11030228 PMid:33807017.
25 Ali, A., Jamil, M. I., Jiang, J., Shoaib, M., Amin, B. U., Luo, S., Zhan, X., Chen, F., & Zhang, Q. (2020). An overview of controlled-biocide-release coating based on polymer resin for marine antifouling applications. Journal of Polymer Research, 27(4), 85 http://doi.org/10.1007/s10965-020-02054-z
26. Facchi, D. P., Facchi, S. P., Souza, P. R., Bonafé, E. G., Popat, K. C., Kipper, M. J., & Martins, A. F. (2022). Composite filter with antimicrobial and anti-adhesive properties based on electrospun poly(butylene adipate-co-terephthalate)/poly(acid lactic)/Tween 20 fibers associated with silver nanoparticles. Journal of Membrane Science , 650 , 120426 http://doi. org/10.1016/j.memsci.2022.120426
27 Yang, J., Li, W., Mu, B., Xu, H., Hou, X., & Yang, Y. (2022). Simultaneous toughness and stiffness of 3D printed nanoreinforced polylactide matrix with complete stereo-complexation via hierarchical crystallinity and reactivity. International Journal of Biological Macromolecules, 202, 482-493 http:// doi.org/10.1016/j.ijbiomac.2022.01.090 PMid:35051500.
28 Hao, Y., Chu, Y., Zhang, M., Shi, W., Chen, Y., Li, D., & Li, L. (2022). Preparation of functional degradable antibacterial film and application in fresh-keeping of grass carp. Journal of Agriculture and Food Research, 9, 100341 http://doi. org/10.1016/j.jafr.2022.100341
29 Mathew, J., Das, J. P., Tp, M., & Kumar, S. (2022). Development of poly (butylene adipate-co-terephthalate) PBAT toughened poly (lactic acid) blends 3D printing filament. Journal of Polymer Research, 29(11), 474 http://doi.org/10.1007/s10965022-03320-y
30. Shankar, S., & Rhim, J.-W. (2022). Effect of types of zinc oxide nanoparticles on structural, mechanical and antibacterial properties of poly(lactide)/poly(butylene adipate-co-terephthalate) composite films. Food Packaging and Shelf Life, 21, 100327. http://doi.org/10.1016/j.fpsl.2019.100327
31 Pascoalino, L. A., Souza, R. L., Marques, N. N., & Curti, P. S. (2020). Caracterização e avaliação do comportamento termorresponsivo de fibras de Ecovio®/ PNIPAAm eletrofiadas. Matéria (Rio de Janeiro), 25(3), e-12830 http://doi.org/10.1590/ s1517-707620200003.1130
32 Malinowski, R., Moraczewski, K., & Raszkowska-Kaczor, A. (2020). Studies on the uncrosslinked fraction of PLA/PBAT blends modified by electron radiation. Materials (Basel), 13(5), 1068 http://doi.org/10.3390/ma13051068 PMid:32121084.
33 Jamnongkan, T., Jaroensuk, O., Khankhuean, A., Laobuthee, A. , Srisawat , N. , Pangon , A. , Mongkholrattanasit , R. , Phuengphai, P., Wattanakornsiri, A., & Huang, C.-F. (2022). A comprehensive evaluation of mechanical, thermal, and antibacterial properties of PLA/ZnO nanoflower biocomposite filaments for 3D printing application. Polymers, 14(3), 600 http://doi.org/10.3390/polym14030600 PMid:35160589.
34 Valerio, T. L., Maia, G. A. R., Gonçalves, L. F., Viomar, A., Banczek, E. P., & Rodrigues, P. R. P. (2019). Study of the Nb2O5 insertion in ZnO to dye-sensitized solar cells. Materials Research, 22(suppl 1), e20180864 http://doi.org/10.1590/19805373-mr-2018-0864
35 Anžlovar, A., Kržan, A., & Žagar, E. (2018). Degradation of PLA/ZnO and PHBV/ZnO composites prepared by melt processing. Arabian Journal of Chemistry, 11(3), 343-352. http://doi.org/10.1016/j.arabjc.2017.07.001
36 Ge, C., Xu, X., Ma, F., Zhou, J., & Du, C. (2022). Biomimetic modification of water-borne polymer coating with carnauba wax for controlled release of urea. International Journal of Molecular Sciences, 23(13), 7422 http://doi.org/10.3390/ ijms23137422
37 Park, K., Sadeghi, K., Panda, P. K., Seo, J., & Seo, J. (2022). Ethylene vinyl acetate/low-density polyethylene/oyster shell powder composite films: Preparation, characterization, and antimicrobial properties for biomedical applications. Journal of the Taiwan Institute of Chemical Engineers, 134, 104301 http://doi.org/10.1016/j.jtice.2022.104301
38 Yan, D., Wang, Z., Guo, Z., Ma, Y., Wang, C., Tan, H., & Zhang, Y. (2020). Study on the properties of PLA/PBAT composite modified by nanohydroxyapatite. Journal of Materials Research and Technology, 9(5), 11895-11904 http://doi.org/10.1016/j. jmrt.2020.08.062
39 Thiyagu, T. T., Gokilakrishnan, G., Uvaraja, V. C., Maridurai, T., & Arun Prakash, V. R. (2022). Effect of SiO2/TiO2 and ZnO Nanoparticle on Cardanol Oil Compatibilized PLA/PBAT Biocomposite Packaging Film. Silicon, 14(7), 3795-3808 http://doi.org/10.1007/s12633-021-01577-4
40 Laput, O., Vasenina, I., Salvadori, M. C., Savkin, K., Zuza, D., & Kurzina, I. (2019). Low-temperature plasma treatment of polylactic acid and PLA/HA composite material. Journal of Materials Science, 54(17), 11726-11738 http://doi.org/10.1007/ s10853-019-03693-4
41 Cao, M., Cui, T., Yue, Y., Li, C., Guo, X., Jia, X., & Wang, B. (2023). Preparation and Characterization for the Thermal Stability and Mechanical Property of PLA and PLA/CF Samples Built by FFF Approach. Materials (Basel), 16(14), 5023 http://doi.org/10.3390/ma16145023 PMid:37512297.
42 Benabid, F. Z., Kharchi, N., Zouai, F., Mourad, A.-H. I., & Benachour, D. (2019). Impact of co-mixing technique and surface modification of ZnO nanoparticles using stearic acid on their dispersion into HDPE to produce HDPE/ZnO nanocomposites. Polymers & Polymer Composites, 27(7), 389-399 http://doi.org/10.1177/0967391119847353
43 Li, Y., Sun, H., Zhang, Y., Xu, M., & Shi, S. Q. (2019). The three-dimensional heterostructure synthesis of ZnO/cellulosic fibers and its application for rubber composites. Composites Science and Technology, 177, 10-17 http://doi.org/10.1016/j. compscitech.2019.04.012
44 Aida, M. S., Alonizan, N. H., Hussein, M. A., Hjiri, M., Abdelaziz, O., Attaf, R., & Zarrad, B. (2022). Facile synthesis and antibacterial activity of bioplastic membrane containing in doped ZnO/cellulose acetate nanocomposite. Journal of Inorganic and Organometallic Polymers and Materials, 32(4), 1223-1233. http://doi.org/10.1007/s10904-021-02171-2.
45 Barakat, M. A.Y., & El-Wakil, A. E.-A. (2021). Preparation and characterization of EVA/ZnO composites as piezoelectric
Abreu, I. R., Nascimento Junior, R. S., Fortes, A. G. S., Folkersma, R., Leitão, L. V., Marques, F. D. P., Sampaio, A. A. S., Castro, L. L. R. L., Andrade, D. S. C., Alves, T. S., & Barbosa, R.
elements for ultrasonic transducers. Materials Research Express, 8(10), 105304 http://doi.org/10.1088/2053-1591/ac29fb
46 Liu, S., Li, L., Li, B., Zhu, J., & Li, X. (2022). Size effect of carnauba wax nanoparticles on water vapor and oxygen barrier properties of starch-based film. Carbohydrate Polymers, 296, 119935 http://doi.org/10.1016/j.carbpol.2022.119935 PMid:36088025.
47 Magno, J. A. P., Da, S., Neto, V. P., Cavalcante, C. E. C., Queiroz, S. K. S. S., Queiroz, I. S., Jr., Fraga, F. E. N., Andrade, H. D., & Melo, R. R. (2021). Synthesis and characterization of organic substrate in the S band for application in microstrip antennas. Journal of Materials Science Materials in Electronics, 32(2), 1829-1841. http://doi.org/10.1007/s10854-020-04951-x.
48 Milanovic, J., Levic, S., Manojlovic, V., Nedovic, V., & Bugarski, B. (2011). Carnauba wax microparticles produced by melt dispersion technique. Chemical Papers, 65(2), 213220 http://doi.org/10.2478/s11696-011-0001-x
49. Villalobos-Hernández, J. R., & Müller-Goymann, C. C. (2006). Sun protection enhancement of titanium dioxide crystals by the use of carnauba wax nanoparticles: the synergistic interaction between organic and inorganic sunscreens at nanoscale. International Journal of Pharmaceutics, 322(1-2), 161-170 http://doi.org/10.1016/j.ijpharm.2006.05.037 PMid:16824709.
50 Ning, X., Song, X., Zhang, S., Wang, Y., & Feng, Y. (2022). Insights into flow improving for waxy crude oil doped with EVA/SiO2 nanohybrids. ACS Omega, 7(7), 5853-5863. http:// doi.org/10.1021/acsomega.1c05953 PMid:35224346.
51 Jian, W., Jin, Z., Yang, J., Meng, G., Liu, H., & Liu, H. (2022). Anticorrosion and antibiofouling performance of in-situ prepared layered double hydroxide coating modified by sodium pyrithione on aluminum alloy 7075. Journal of Industrial and Engineering Chemistry, 113, 419-430 http://doi.org/10.1016/j. jiec.2022.06.017
52. Rebelo, R. C., Gonçalves, L. P. C., Fonseca, A. C., Fonseca, J., Rola, M., Coelho, J. F. J., Rola, F., & Serra, A. C. (2022). Increased degradation of PLA/PBAT blends with organic acids and derivatives in outdoor weathering and marine environment. Polymer, 256, 125223 http://doi.org/10.1016/j. polymer.2022.125223
53 Çoban, O., Bora, M. Ö., Kutluk, T., & Özkoç, G. (2018). Mechanical and thermal properties of volcanic particle
filled PLA/PBAT composites. Polymer Composites, 39(S3), E1500-E1511 http://doi.org/10.1002/pc.24393
54 Ding, Y., Zhang, C., Luo, C., Chen, Y., Zhou, Y., Yao, B., Dong, L., Du, X., & Ji, J. (2021). Effect of talc and diatomite on compatible, morphological, and mechanical behavior of PLA/PBAT blends. E-Polymers, 21(1), 234-243 http://doi. org/10.1515/epoly-2021-0022.
55 Pietrosanto, A., Scarfato, P., Di Maio, L., Nobile, M. R., & Incarnato, L. (2020). Evaluation of the suitability of poly(lactide)/ poly(butylene-adipate-co-terephthalate) blown films for chilled and frozen food packaging applications. Polymers, 12(4), 804 http://doi.org/10.3390/polym12040804 PMid:32260170.
56 Prasong, W., Muanchan, P., Ishigami, A., Thumsorn, S., Kurose, T., & Ito, H. (2020). Properties of 3D printable poly(lactic acid)/ poly(butylene adipate-co-terephthalate) blends and nano talc composites. Journal of Nanomaterials, 8040517, 1-16 http:// doi.org/10.1155/2020/8040517
57 Pascual-González, C., de la Vega, J., Thompson, C., FernándezBlázquez, J. P., Herráez-Molinero, D., Biurrun, N., Lizarralde, I., Sánchez del Río, J., & González, C. (2022). Processing and mechanical properties of novel biodegradable poly-lactic acid/ Zn 3D printed scaffolds for application in tissue regeneration. Journal of the Mechanical Behavior of Biomedical Materials, 132, 105290. http://doi.org/10.1016/j.jmbbm.2022.105290. PMid:35671668.
58 Spoerk, M., Sapkota, J., Weingrill, G., Fischinger, T., Arbeiter, F., & Holzer, C. (2017). Shrinkage and warpage optimization of expanded-perlite-filled polypropylene composites in extrusionbased additive manufacturing. Macromolecular Materials and Engineering, 302(10), 1700143 http://doi.org/10.1002/ mame.201700143
59 Kodali, D., Umerah, C. O., Idrees, M. O., Jeelani, S., & Rangari, V. K. (2021). Fabrication and characterization of polycarbonate-silica filaments for 3D printing applications. Journal of Composite Materials, 55(30), 4575-4584 http:// doi.org/10.1177/00219983211044748
Received: Mar. 23, 2024
Revised: June 01, 2024 Accepted: June 06, 2024
Samuel Camilo da Silva1* , Fabiola Azanha de Carvalho1 and Fabio Yamashita1
1Laboratório de Tecnologia, Departamento de Ciência e Tecnologia de Alimentos, Universidade Estadual de Londrina – UEL, PR, Brasil *samuelcamiloutfpr@outlook.com
Obstract
Fossil-origin plastics have raised great concerns due to their non-biodegradable nature. Biodegradable polymers can be an alternative for these materials’, however they have higher cost. The use of agro-industrial waste in blends with biopolymers can provide cheaper materials’ with improved properties. This study aims to develop low-cost biodegradable materials by extrusion and thermoplastic injection using oat hulls, polybutylene succinate (PBS), and starch. Six formulations with different concentrations of oat hulls (0-56% w/w) were extruded in a single-screw extruder, and then the materials were produced by thermoplastic injection. The extrusion aligned the oat hull fibers, making the material dimensionally stable. The oat hulls enhanced stiffness and reduced material density compared to non-hull counterparts. Besides that, the oat hulls are a low-cost agro-industrial byproduct, and it was possible to produce biodegradable materials with up to 56% hulls and only 20% PBS. These biodegradable materials are environmentally friendly and non-toxic.
Keywords: biodegradable materials, blending, extrusion, natural fibers, mechanical properties.
Part of this paper was published as pre-print at https://www.researchsquare.com/article/rs-3315119/v1
How to cite: Silva, S. C., Carvalho, F. A., & Yamashita, F. (2024). Potential biodegradable materials containing oat hulls, TPS, and PBS by thermoplastic injection. Polímeros: Ciência e Tecnologia , 34 (3), e20240026. https://doi.org/10.1590/0104-1428.20240019
The escalating production of fossil-origin materials, currently amounting to 350 million tons annually, has sparked growing concerns due to their non-biodegradable nature and significant impact on the ecosystem[1].In response to these challenges, biodegradable polymers have emerged as an alternative to fossil-origin materials. According to ASTM standards[2], biodegradable polymers undergo decomposition in natural aerobic environments (composting) through the metabolic activity of microorganisms capable of metabolizing their molecular structures. Despite their potential, biodegradable polymers still face limitations compared to synthetic polymers, particularly mechanical properties, water vapor barriers, and production costs, making large-scale adoption challenging[3-5]. The utilization of blends, are well known for reducing material costs and improving specific properties[5-7]. Extensive research has investigated the incorporation of different components, such as starch[8,9] and fibers[10,11], particularly agro-industrial residues or byproducts[3,12]
Polybutylene succinate (PBS) is a biopolymer from the family of polyesters that can be processed through injection or extrusion to produce rigid or flexible materials comparable to polypropylene/polyethylene. PBS presents excellent biodegradation properties in soil and water[13]. Additionally, starch, which is widely available from diverse sources (corn, potato, and cassava), can also be effectively processed through
injection or extrusion to produce biodegradable materials at a lower cost compared to petroleum-derived or aliphatic polyesters[7,14-18]. A significantly underutilized agro-industrial residue with high potential is oat hulls, a byproduct of oat milling processing, yielding approximately 25-36% during milling[19,20]. Despite their versatility, oat hulls are primarily adopted as biomass for generating electricity and steam[21] , overlooking its potential for other valuable applications, such as in biodegradable blends. The low density and weight of these natural fibers can be attractive in applications where lighter and more rigid materials are desired, such as in the coating of cups, straws, spoons, single-use trays for some foods, in the automotive industry, tissue engineering, and marine industry, among others[10,22,23]
Combining oat hull, PBS, and starch can ensure biodegradability, lower cost, and reasonable material properties. This could guarantee the production of a new material with many possible applications, such as single-use trays, food packaging, among others, using a by-product considered to be waste in the oats industry, while making it cheaper and more environmentally friendly. To do this, it is necessary to study the behavior of oat hulls at different concentrations to understand its effect as a fiber-reinforced agent. This study aims to develop low-cost biodegradable materials by extrusion and thermoplastic injection using oat hulls, PBS, and starch.
Silva, S. C., Carvalho, F. A., & Yamashita, F.
2.1 Material
The materials used in this study included corn starch (14% moisture) (APTI™,Brazil), glycerol (technical grade glycerin, Dinâmica, Brazil), polybutylene succinate (PBS) (TK-BIO®, China) (Table 1), and oat hull (SL-Alimentos, Brazil). Before material production, oat hulls were grounded (Mill An 11, Brazil) and sieved (28 mesh) to enhance their homogeneity.
2.2 Production of biodegradable materials by thermoplastic injection
Preliminary tests were conducted using different concentrations of PBS, starch, and oat hulls to evaluate their processability in a pilot single-screw extruder (model EL-75, BGM, Brazil) and pilot injector AX16-lll (AX-Plasticos, Brazil). Three formulations (T20: 20 PBS %-w/w; 44,8 Starch %-w/w; 24 Glycerol %-w/w; 11,2 Oat hulls %-w/w; T60: 20 PBS %-w/w; 22,4 Starch %-w/w; 24 Glycerol %-w/w; 33,6 Oat hulls %-w/w; T100: 20 PBS %-w/w; 0 Starch %-w/w; 24 Glycerol %-w/w; 56 Oat hulls %w/w.) were produced in a pilot single screw extruder (90/120/120/110°C) using a temperature profile from heating zone 1 to zone 4 at a screw speed of 40 RPM. After extrusion, they were processed in the pilot injector to produce a dog bone-shaped specimen type IV[24]. The temperature profile was set at 120/120/110°C from the feeder to the nozzle. Based on the preliminary tests, it was decided to set the concentration of PBS and glycerol (plasticizer) constant and vary the concentrations of starch and oat hulls, as detailed in Table 1. All material was manually mixed and processed as described in the preliminary tests.
Mechanical tensile tests (Young’s modulus, tensile strength, and elongation at break) were performed according to ASTM 638-14[24] using a universal testing machine (EMIC, model DL 2000, Brazil). Before the analysis, the specimens were conditioned at room temperature, maintaining a relative humidity of 53 ± 2% for one week as per ASTM 638-14[24] standard procedures.
The microstructure images of the samples were captured using a scanning electron microscope (Philips, model FEI Quanta 200, USA). Before imaging, the specimens were subjected to cryogenic fracture by immersion in liquid
nitrogen and metalized with a thin layer of gold using a metallizer [Bal-Tec, model SCD-050, Germany]. All samples were analyzed using a 20 kV voltage accelerator and 2000x magnification.
The crystallinity of the biodegradable materials was determined using X’PertPRO equipment (Panalytical, Philips, Netherlands) with copper Ka radiation (α = 1.5418 A) operating at room temperature at 40 kV. The relative crystallinity index (RCI) was calculated as the ratio between the area of the crystalline region and the sum of crystalline and amorphous regions (Equation 1)[9,25]
Where Ca is the crystalline area, and Aais the amorphous area.
2.6 Density
Density was calculated by weighing the mass and measuring the volume using a digital caliper (Starrett, Brazil). Five specimens from each formulation were placed in a desiccator for one week under a relative humidity of 53%.Then, the specimens were weighed and measured to obtain thickness, length, and width.
After production, ten specimens from each formulation were measured using a digital caliper (Starrett, Brazil). These specimens were subsequently placed in desiccators at a relative humidity of 53% and a temperature of 25°C for one week. The linear contraction index (LCI) was determined using Equation 2[26].
Where Lcm is the initial specimen length and Lcp is the length after one week.
Approximately 0.5 to 0.8 g of the biodegradable material was placed in an Aquasorp isotherm generator (Decagon Devices, USA), and the experimental moisture sorption data was fitted by the Guggenhein-Anderson-de-Boer (GAB)
Potential biodegradable materials containing oat hulls, TPS, and PBS by thermoplastic injection
model (Equation 3) using the Nonlinear Regression Module (Statistica Software 7.0,StatSoft, USA).
Where aw = water activity, Xw = equilibrium moisture content (kg/kg dry solid), m0 = monolayer moisture content (kg/kg dry solid), and C and k = GAB constants.
2.9 Color
The CIELabcolor parameters (L*,a*, and b*) of five specimens of each formulation were measured with a colorimeter (Minolta CR 400, Japan) with a visual angle of 10°, according to ASTM D2244-09B[27]
2.10 Statistical analysis
All data obtained were evaluated by analysis of variance (ANOVA) and Tukey’s test at a 5% significance level (p < 0.05) using Statistica software, version 7.0 (StatSoft, USA).
3.Results and Discussions
3.1 Mechanical properties
The results of the mechanical properties of the biodegradable materials are shown in Table 2
The F0 formulation had the highest tensile (TS) strength (1.8 MPa), approximately double that of F100 (0.9 MPa). The higher the oat hull concentration, the lower the TS, probably due to the limited interfacial adhesion between the oat hulls and the polymeric matrix. The interaction between the polymeric matrix (PBS) and the oat hull is difficult due to the hydrophilic nature of the hull[28]. According to Mochane et al.[10], the hydrophilic characteristics of natural fibers may adversely affect interfacial adhesion, mainly when the polymeric matrix is hydrophobic, as the PBS, resulting in poor mechanical properties[29,30]. Other factors, such as fiber length, fiber type, extraction process, and moisture absorption, can decrease the TS[30] .
According to Aydemir and Gardner[31], in blends of polyhydroxybutyrate and polylactic acid reinforced with cellulose nanofibrils, increasing the fiber concentration did not improve the mechanical properties, and there was a greater agglomeration and clogging of the fibers in the matrix structure, enhancing the stiffness and ductility of the material. Ayu et al.[32] produced sheets of PBS, starch, and empty fruit buncher fiber (EFB), and according to the
authors, adding fiber fillers up to 8 wt% decreased tensile and flexural strength due to a lack of interfacial adhesion and poor dispersion of the fibers in the PBS matrix. As the literature indicates, incorporating natural fibers in blends to improve mechanical properties can present some difficulties due to fiber dispersion, polar and non polar phases, fiber concentration, and extrusion process
The F0 material had the highest flexibility, possibly due to its high thermoplastic starch content, which increases its elasticity[33]. Thermoplastic starch (TPS), when in the presence of a plasticizer such as glycerol, has its flexibility increased by lowering the glass transition temperature (Tg),which can be beneficial for some applications such as food packaging and films[34]. The material with 20% oat hull content (F20) had approximately 7% lower elongation than F0. Calabia et al.[35] produced sheets of PBS and cotton fiber and reported a decrease in the elongation at break of approximately 8% in formulations containing fiber. This decrease in elongation was attributed to the reduction in PBS chain movement, leading to higher stiffness in the materials. Similarly, Yang et al.[36] produced injected materials using bamboo fiber and polypropylene, and they found that as the fiber concentration increased, the elongation at break (%) decreased. This decrease resulted from difficulties maintaining fiber dispersion in the blend, which adversely affected material flexibility. The same occurred in this study, where the material became more stiff and less flexible.
The images of the F0 material (Figure 1a, Figure 1b) showed starch granules (circular shape) and a plasticized superficial area, characteristic of blends containing thermoplastic starch[37,38]. Cracks were also observed due to the poor compatibility between PBS and starch, forming a heterogeneous phase (immiscible blend). Similar characteristics can create a strong bond, resulting in dimensional stability and improved mechanical and barrier properties[39,40] .
The F20 (Figure 2a, Figure 2b), F40 (Figure 3a, Figure 3b), F60 (Figure 4a, Figure 4b), F80 (Figure 5a, Figure 5b), and F100 (Figure 6a, Figure 6b) materials’ surface and fracture images showed oat hull fibers (cylindrical shape), most of which were agglomerated and aligned, possibly due to extrusion orientation. Many cavities and pores were also observed in the formulations containing oat hulls, which might explain the decrease in the mechanical properties of these materials. Similar observations were reported by Calabia et al.[35] for PBS and cotton fiber composites and Ayu et al.[32] for sheets containing empty fruit bunches, PBS,
a,b,c,d,eMeans at the same column with different letters represent a significant difference (p ≤ 0.05) between theformulations, according to Tukey’s test.
Silva, S. C., Carvalho, F. A., & Yamashita, F.




Potential biodegradable materials containing oat hulls, TPS, and PBS by thermoplastic injection


and starch. In both studies, it was possible to observe the presence of long fibers and voids in the structure, indicating weak interfacial adhesion.
Mechanical and barrier properties highly depend on morphology status[10,32]. Some methods can improve the interfacial surface between the components, such as treatments with silanes, alkalis, or acetones, reducing incompatibility[12] However, the work aimed to produce biodegradable and nontoxic materials, and chemical or surface cleaning methods can harm the environment.
The X-ray diffractograms and the respective relative crystallinity index (RCI) of the biodegradable materials are presented in Figure 7.
Two peaks (19.9° and 22.3°) were identified in all blend formulations, and the oat hull concentration did not influence the crystallinity of the materials. Hu et al.[41] produced blends containing PBS and different types of cellulose, and all diffractograms showed reduced peaks, suggesting that the natural fibers used had low crystallinity. Liu et al.[38] produced materials with starch, PBS, and ionic liquid; varying starch content did not modify the PBS crystalline phase.
PBS showed peaks at approximately 19.6°, 22.3°, and 28.8°, and peaks near 19.5° and 22.5° are characteristic of the crystalline phase of PBS[42,43]. PBS also showed a larger peak at 22.3° when compared to the other formulations because

Figure 7. X-ray diffractograms and relative crystallinity index (RCI) of the biodegradable materials.
the addition of starch, oat hulls, and glycerol impaired the crystallinity properties of PBS during the production/extrusion process of the materials. This same behavior was observed by Xu et al.[43] for blends produced with PBS and corn starch, possibly as starch particles obstruct PBS segments. The low relative crystallinity indexes (RCIs) of the biodegradable materials containing starch, ranging from 10.57% to 15.24%, can be attributed to the destruction of the semicrystalline
Silva, S. C., Carvalho, F. A., & Yamashita, F.
structure of starch during the extrusion process, leading to the formation of higher amorphous zones[8,44]
The linear contraction indexes (LCIs) of the biodegradable materials are presented in Table 3. F0 (1.56%) and F20 (1.07%) had the highest LCI values. LCI of the other materials was not significantly different and ranged from 0.70 to 0.78%. Materials produced by injection molding can contract as they change from melting to solid under atmospheric pressure[45]
Increasing the fiber concentration resulted in a lower contraction of the injected material because the fibers can act as fillers, i.e., occupying spaces in the blend structure and making it less capable of shrinking or expanding[46,47] . This can be advantageous since it avoids the appearance of marks on thicker parts of the materials, provides good geometrical stability,avoids deformation after injection, and reduces material shrinkage[47]. The SEM analysis of the materials (item 3.2) with oat hulls showed that the fibers were oriented and agglomerated in a cylindrical shape,
Table 3. Linear Contraction Index (LCI) of the biodegradable materials.
Formulation
F0 1.56 ± 0.06a
F20 1.07 ± 0.06b
F40 0.78 ± 0.04c
F60 0.75 ± 0.05c
± 0.03c
± 0.04c
*a,b,c Means with different letters represent a significant difference (p ≤ 0.05) between theformulations, according to Tukey’s test.
Table 4. Density of the biodegradable materials produced by injection extrusion.
Formulation
± 0.02b
± 0.04b
1.35 ± 0.04b F60 1.32 ± 0.04b F80 1.35 ± 0.03b
F100 1.28 ± 0.02a
a,b Means with different letters represent a significant difference (p ≤ 0.05) between the formulations, according to Tukey’s test .
suggesting that these aligned and clogging fibers decreased the materials’ LCI. The LCI can also be influenced by temperature, pressure, injection flow rate, equipment design, and material composition[45,48]
F100 had the lowest density, and the other formulations did not show significant differences (Table 4) because of the lower density of the PBS compared to thermoplastic starch (TPS). Additionally, the high oat hull content in the F100 formulation contributed to decreased material density, as fibers have lower densities than TPS. One of the main advantages of adding natural fibers in polymeric structures is to reduce the material density[10,49,50]. This can lead to applications where lighter and less flexible materials are desired, such as food packaging, single-trays, cups, and the automotive industry[22,23]. Aslan et al.[51] produced composites of polypropylene and sisal fiber, fiberglass, and carbon fiber, and increasing sisal fiber concentration reduced the material’s densities. The density of the material decreases with the addition of natural fibers in the blend in most cases.
The Guggenheim-Anderson-de-Boer (GAB) parameter values of the moisture sorption isotherms of the biodegradable materials are presented in Table 5.
The m0 of the F0 material (16.11 g 100 g-1) was the highest among all formulations, mainly due to its higher proportion of thermoplastic starch (TPS) and the absence of oat hull because TPS is known for its high hydrophilicity[8]. As the oat hull content increased (F20, F40, F60, F80, F100), the m0 values decreased compared with F0 material due to the reduction of TPS concentration in the blend. The F100 material had no starch in its composition and had an intermediary m0 value between F0 and F80 because the oat hull is less hydrophobic than PBS but more hydrophobic than TPS. The k and C values were similar for all formulations.
The CIELabcolor parameters L*, a*, and b* of the biodegradable materials are presented in Table 6.
F0 had the highest luminosity (62.69), and the formulations containing oat hulls had lower values, ranging between 41 and 45. The decrease in luminosity can be attributed to the opaque nature of oat hulls, which can significantly influence the color parameters. For the a* parameter F20 presented the highest value (4.09). The b* parameter of the F0 material
Table 5. GAB model parameters of the moisture sorption isotherms of the biodegradable materials.
Formulations containing F - Oat Hulls in different concentrations.
Potential biodegradable materials containing oat hulls, TPS, and PBS by thermoplastic injection
Table 6. CIELabcolor parameters of the biodegradable materials.
± 2.03
The L* parameter is luminosity; a* red/green coordinate; b* yellow/blue coordinate.
(9.79) was higher than that of the others because it did not contain an oat hull in its formulation, only PBS and starch, resulting in a yellowish color.
In this study, all the materials with oat hulls had a brownish color. The oat hulls are composed of pentosans (30-35%) and protein (4%)[20], which are components that can contribute to the Maillard reaction during the extrusion process at high temperatures (90-110°C). This Maillard reaction could explain the color alteration observed in the materials with oat hulls.
4. Conclusions
Biodegradable materials of oat hulls, starch, and polybutylene succinate presented excellent processability at the extrusion, to produce the pellets, and at the thermoplastic injection, to produce the dog bone-shaped specimens. The extrusion aligned the oat hull fibers, making the material dimensionally stable, i.e., without the shrinkage observed in materials without fiber. Furthermore, incorporating oat hulls enhanced stiffness and reduced material density compared to non-hull counterparts. These materials’ are adequate to produce lighter and stiffer materials for commercial uses, such as fast-food packaging and single-use utensils like trays, spoons, and cups. Besides that, the oat hulls area low-cost agro-industrial byproduct, and it was possible to produce biodegradable materials with up to 56% hulls and only 20% PBS. Their potential for large-scale manufacturing is highlighted by their compatibility with existing equipment and processes in the plastics industry. For future projects, new studies using FTIR spectroscopy can be carried out to better understand the interactions between oat hulls, TPS and PBS.
5.
• Conceptualization – Fabio Yamashita.
• Data curation – NA.
• Formal analysis – Samuel Camilo da Silva; Fabiola Azanha de Carvalho.
• Funding acquisition – Fabio Yamashita.
• Investigation – Samuel Camilo da Silva; Fabiola Azanha de Carvalho.
• Methodology – Fabio Yamashita.
• Project administration – Fabio Yamashita.
• Resources – Fabio Yamashita.
• Software – NA.
• Supervision – Fabio Yamashita.
• Validation – Samuel Camilo da Silva; Fabiola Azanha de Carvalho.
• Visualization – Samuel Camilo da Silva; Fabiola Azanha de Carvalho; Fabio Yamashita.
• Writing – original draft – Samuel Camilo da Silva; Fabio Yamashita.
• Writing – review & editing – Fabio Yamashita.
6. Acknowledgements
We thank all the colleagues who helped develop different parts of this manuscript. This research was funded by FINEP- Financier of Studies and Projects (Brazil), grant number BIOMATS- 2021 (0128/21).
7.
1 Bratovcic, A. (2019). Degradation of micro-and nano-plastics by photocatalytic methods. Journal of Nanoscience and Nanotechnoly Applications, 3(3), 304 http://doi.org/10.18875/2577-7920.3.304
2 American Society for Testing and Materials – ASTM. (2019). ASTM D6400-19: standard specification for labeling of plastics designed to be aerobically composted in municipal or industrial facilities West Conshohocken: ASTM
3 Samir, A., Ashour, F. H., Hakim, A. A., & Bassyouni, M. (2022). Recent advances in biodegradable polymers for sustainable applications. Npj Materials Degradation, 6(1), 68. http://doi. org/10.1038/s41529-022-00277-7
4 Rai, P., Mehrotra, S., Priya, S., Gnansounou, E., & Sharma, S. K. (2021). Recent advances in the sustainable design and applications of biodegradable polymers. Bioresource Technology, 325, 124739 http://doi.org/10.1016/j.biortech.2021.124739 PMid:33509643.
5 Zhong, Y., Godwin, P., Jin, Y., & Xiao, H. (2020). Biodegradable polymers and green-based antimicrobial packaging materials: a mini-review. Advanced Industrial and Engineering Polymer Research, 3(1), 27-35 http://doi.org/10.1016/j.aiepr.2019.11.002
6 Wu, F., Misra, M., & Mohanty, A. K. (2021). Challenges and new opportunities on barrier performance of biodegradable polymers for sustainable packaging. Progress in Polymer Science, 117, 101395 http://doi.org/10.1016/j.progpolymsci.2021.101395
7 Bulatović, V. O., Mandić, V., Kučić Grgić, D., & Ivančić, A. (2021). Biodegradable polymer blends based on thermoplastic starch. Journal of Polymers and the Environment, 29(2), 492508 http://doi.org/10.1007/s10924-020-01874-w
8 Mali, S., Grossmann, M. V. E., & Yamashita, F. (2010). Filmes de amido: produção, propriedades e potencial de utilização. Semina: Ciências Agrárias, 31(1), 137-155 http:// doi.org/10.5433/1679-0359.2010v31n1p137
Silva, S. C., Carvalho, F. A., & Yamashita, F.
9 Müller, C. M. O., Laurindo, J. B., & Yamashita, F. (2011). Effect of nanoclay incorporation method on mechanical and water vapor barrier properties of starch-based films. Industrial Crops and Products, 33(3), 605-610 http://doi.org/10.1016/j. indcrop.2010.12.021
10 Mochane, M. J., Magagula, S. I., Sefadi, J. S., & Mokhena, T. C. (2021). A review on green composites based on natural fiber-reinforced polybutylene succinate (PBS). Polymers, 13 ( 8 ), 1200 http://doi.org/10.3390/polym13081200 PMid:33917740.
11 Praveena, B. A., Buradi, A., Santhosh, N., Vasu, V. K., Hatgundi, J., & Huliya, D. (2022). Study on characterization of mechanical, thermal properties, machinability and biodegradability of natural fiber reinforced polymer composites and its applications, recent developments and future potentials: a comprehensive review. Materials Today: Proceedings, 52(Pt 3), 1255-1259 http://doi.org/10.1016/j.matpr.2021.11.049
12 Ilyas, R. A., Zuhri, M. Y. M., Aisyah, H. A., Asyraf, M. R. M., Hassan, S. A., Zainudin, E. S., Sapuan, S. M., Sharma, S., Bangar, S. P., Jumaidin, R., Nawab, Y., Faudzi, A. A. M., Abral, H., Asrofi, M., Syafri, E., & Sari, N. H. (2022). Natural fiber-reinforced polylactic acid, polylactic acid blends and their composites for advanced applications. Polymers, 14(1), 202 http://doi.org/10.3390/polym14010202 PMid:35012228.
13 Rafiqah, S. A., Khalina, A., Harmaen, A. S., Tawakkal, I. A., Zaman, K., Asim, M., Nurrazi, M. N., & Chimg, H. L. (2021). A review on properties and application of bio-based poly (butylene succinate). Polymers, 13(9), 1436 http://doi. org/10.3390/polym13091436 PMid:33946989.
14 Jiang, T., Duan, Q., Zhu, J., Liu, H., & Yu, L. (2020). Starchbased biodegradable materials: challenges and opportunities. Advanced Industrial and Engineering Polymer Research, 3(1), 8-18 http://doi.org/10.1016/j.aiepr.2019.11.003
15 Cheng, H., Chen, L., McClements, D. J., Yang, T., Zhang, Z., Ren, F., Miao, M., Tian, Y., & Jin, Z. (2021). Starch-based biodegradable packaging materials: a review of their preparation, characterization and diverse applications in the food industry. Trends in Food Science & Technology, 114, 70-82 http://doi. org/10.1016/j.tifs.2021.05.017
16 . Lopez-Gil, A., Rodriguez-Perez, M. A., De Saja, J. A., Bellucci, F. S., & Ardanuy, M. (2014). Strategies to improve the mechanical properties of starch-based materials: plasticization and natural fibers reinforcement. Polímeros: Ciência e Tecnologia, 24(spe), 36-42 http://doi.org/10.4322/polimeros.2014.054
17 Combrzyński, M., Oniszczuk, T., Kupryaniuk, K., Wójtowicz, A., Mitrus, M., Milanowski, M., Soja, J., Budziak-Wieczorek, I., Karcz, D., Kamiński, D., Kulesza, S., Wojtunik-Kulesza, K., Kasprzak-Drozd, K., Gancarz, M., Kowalska, I., Ślusarczyk, L., & Matwijczuk, A. (2021). Physical properties, spectroscopic, microscopic, x-ray, and chemometric analysis of starch films enriched with selected functional additives. Materials (Basel), 14(10), 2673 http://doi.org/10.3390/ma14102673 PMid:34065230.
18. Combrzyński, M., Matwijczuk, A., Wójtowicz, A., Oniszczuk, T., Karcz, D., Szponar, J., Niemczynowicz, A., Bober, D., Mitrus, M., Kupryaniuk, K., Stasiak, M., Dobrzański, B., & Oniszczuk, A. (2020). Potato starch utilization in ecological loose-fill packaging materials Sustainability and characterization. Materials (Basel) , 13 (6 ), 1390. http://doi.org/10.3390/ ma13061390 PMid:32204364.
19 Paschoal, G. B., Muller, C. M. O., Carvalho, G. M., Tischer, C. A., & Mali, S. (2015). Isolation and characterization of nanofibrillated cellulose from oat hulls. Quimica Nova, 38(4), 478-482. http://doi.org/10.5935/0100-4042.20150029.
20 Girardet, N., & Webster, F. H. (2011). Oat milling: specifications, storage, and processing. In F. H. Webster, & P. J. Wood (Eds.),
Oats: chemistry and technology (pp. 301-309). USA: AACC International, Inc. http://doi.org/10.1094/9781891127649.014
21 Holtzapple, M. T. (2003) Hemicelluloses, In B. Caballero, L. Trugo, & P. Finglas (Eds.), Encyclopedia of food sciences and nutrition (pp. 3060-3071). London: Academic press http:// doi.org/10.1016/B0-12-227055-X/00589-7.
22 Ferreira, D. C. M., Molina, G., & Pelissari, F. M. (2020). Biodegradable trays based on cassava starch blended with agroindustrial residues. Composites. Part B, Engineering, 183, 107682 http://doi.org/10.1016/j.compositesb.2019.107682
23 Huang, A., Peng, X., Geng, L., Zhang, L., Huang, K., Chen, B., Gu, Z., & Kuang, T. (2018). Electrospun poly (butylene succinate)/cellulose nanocrystals bio-nanocomposite scaffolds for tissue engineering: Preparation, characterization and in vitro evaluation. Polymer Testing, 71, 101-109 http://doi. org/10.1016/j.polymertesting.2018.08.027
24 American Society for Testing and Materials – ASTM. (2014). ASTM D638-14: standard test method for tensile properties of plastics West Conshohocken: ASTM
25 Köksel, H., Sahbaz, F., & Özboy, Ö. (1993). Influence of wheat-drying temperatures on the birefringence and X-ray diffraction patterns of wet-harvested wheat starch. Cereal Chemists, 70(4), 481-483. Retrieved in 2024, March 4, from https://www.cerealsgrains.org/publications/cc/backissues/1993/ Documents/70_481.pdf
26 American Society for Testing and Materials – ASTM. (2000). ASTM D955-00: standard test method of measuring shrinkage from mold dimensions of thermoplastics West Conshohocken: ASTM
27 American Society for Testing and Materials – ASTM. (2009). ASTM D2244-09b: standard practice for calculation of color tolerances and color differences from instrumentally measured color coordinates West Conshohocken: ASTM
28 Mohammed, L., Ansari, M. N. M., Pua, G., Jawaid, M., & Islam, M. S. (2015). A review on natural fiber reinforced polymer composite and its applications. International Journal of Polymer Science, 2015(1), 243947 http://doi.org/10.1155/2015/243947
29. Kamarudin, S. H., Mohd Basri, M. S., Rayung, M., Abu, F., Ahmad, S., Norizan, M. N., Osman, S., Sarifuddin, N., Mat Desa, M. S. Z., Abdullah, U. H., Tawakkal, I. S. M. A., & Abdullah, L. C. (2022). A review on natural fiber reinforced polymer composites (NFRPC) for sustainable industrial applications. Polymers, 14(17), 3698 http://doi.org/10.3390/ polym14173698 PMid:36080773.
30 Khalid , M. Y. , Al Rashid , A. , Arif , Z. U. , Ahmed , W. , Arshad, H., & Zaidi, A. A. (2021). Natural fiber reinforced composites: sustainable materials for emerging applications. Results in Engineering, 11, 100263. http://doi.org/10.1016/j. rineng.2021.100263
31 Aydemir, D., & Gardner, D. J. (2020). Biopolymer blends of polyhydroxybutyrate and polylactic acid reinforced with cellulose nanofibrils. Carbohydrate Polymers, 250, 116867 http://doi.org/10.1016/j.carbpol.2020.116867 PMid:33049817.
32. Ayu, R. S., Khalina, A., Harmaen, A. S., Zaman, K., Isma, T., Liu, Q., Ilyas, R. A., & Lee, C. H. (2020). Characterization study of empty fruit bunch (EFB) fibers reinforcement in poly (butylene) succinate (PBS)/starch/glycerol composite sheet. Polymers, 12(7), 1571. http://doi.org/10.3390/polym12071571. PMid:32679865.
33 Ferri, J. M., Garcia‐Garcia, D., Carbonell‐Verdu, A., Fenollar, O., & Balart, R. (2018). Poly (lactic acid) formulations with improved toughness by physical blending with thermoplastic starch. Journal of Applied Polymer Science, 135(4), 45751 http://doi.org/10.1002/app.45751
34 Khan, B., Niazi, M. B. K., Samin, G., & Jahan, Z. (2017). Thermoplastic starch: A possible biodegradable food packaging
Potential biodegradable materials containing oat hulls, TPS, and PBS by thermoplastic injection
material—A review. Journal of Food Process Engineering, 40(3), e12447 http://doi.org/10.1111/jfpe.12447
35 Calabia, B. P., Ninomiya, F., Yagi, H., Oishi, A., Taguchi, K., Kunioka, M., & Funabashi, M. (2013). Biodegradable poly (butylene succinate) composites reinforced by cotton fiber with silane coupling agent. Polymers, 5(1), 128-141 http:// doi.org/10.3390/polym5010128
36 Yang, F., Long, H., Xie, B., Zhou, W., Luo, Y., Zhang, C., & Dong, X. (2020). Mechanical and biodegradation properties of bamboo fiber‐reinforced starch/polypropylene biodegradable composites. Journal of Applied Polymer Science, 137(20), 48694. http://doi.org/10.1002/app.48694.
37 Ayu, R. S., Khalina, A., Harmaen, A. S., Zaman, K., Jawaid, M., & Lee, C. H. (2018). Effect of modified tapioca starch on mechanical, thermal, and morphological properties of PBS blends for food packaging. Polymers, 10(11), 1187 http://doi. org/10.3390/polym10111187 PMid:30961112.
38 Liu, D., Qi, Z., Zhang, Y., Xu, J., & Guo, B. (2015). Poly (butylene succinate)(PBS)/ionic liquid plasticized starch blends: preparation, characterization, and properties. Stärke, 67(9-10), 802-809 http://doi.org/10.1002/star.201500060
39 Taib, M. N. A. M., & Julkapli, N. M. (2019). Dimensional stability of natural fiber-based and hybrid composites. In M. Jawaid, M. Thariq, & N. Saba (Eds.), Mechanical and physical testing of biocomposites, fibre-reinforced composites and hybrid composites (pp. 61-79). UK: Woodhead Publishing http://doi. org/10.1016/B978-0-08-102292-4.00004-7
40. Raj, S. S. R., Dhas, J. E. R., & Jesuthanam, C. P. (2021). Challenges on machining characteristics of natural fiberreinforced composites–A review. Journal of Reinforced Plastics and Composites , 40 ( 1-2 ), 41 - 69 http://doi. org/10.1177/0731684420940773
41 Hu, X., Su, T., Pan, W., Li, P., & Wang, Z. (2017). Difference in solid-state properties and enzymatic degradation of three kinds of poly (butylene succinate)/cellulose blends. RSC Advances, 7(56), 35496-35503 http://doi.org/10.1039/C7RA04972B
42 Zhao, Z., Lei, B., Du, W., Yang, Z., Tao, D., Tian, Y., Xu, J., & Zhang, X. (2020). The effects of different inorganic salts on the structure and properties of ionic liquid plasticized starch/poly (butylene succinate) blends. RSC Advances, 10(7), 3756-3764 http://doi.org/10.1039/C9RA08218B. PMid:35492637.
43 Xu, J., Chen, Y., Tian, Y., Yang, Z., Zhao, Z., Du, W., & Zhang, X. (2021). Effect of ionic liquid 1-buyl-3-methylimidazolium halide on the structure and tensile property of PBS/corn starch blends. International Journal of Biological Macromolecules,
172, 170-177 http://doi.org/10.1016/j.ijbiomac.2021.01.062 PMid:33450339.
44 Phetwarotai , W. , Potiyaraj , P. , & Aht‐Ong , D. (2012 ). Characteristics of biodegradable polylactide/gelatinized starch films: effects of starch, plasticizer, and compatibilizer. Journal of Applied Polymer Science, 126(S1), E162-E172 http://doi. org/10.1002/app.36736
45 Kowalska , B. ( 2007 ). Injection moulding contraction and the pressure-volume-time relation. International Polymer Science and Technology, 34(8), 41-48. http://doi. org/10.1177/0307174X0703400810
46 Sykacek, E., Hrabalova, M., Frech, H., & Mundigler, N. (2009). Extrusion of five biopolymers reinforced with increasing wood flour concentration on a production machine, injection moulding and mechanical performance. Composites. Part A, Applied Science and Manufacturing, 40(8), 1272-1282 http:// doi.org/10.1016/j.compositesa.2009.05.023
47. Mougin, G., Magnani, M., & Eikelenberg, N. (2009). Naturalfibres composites for the automotive industry: challenges, solutions and applications. International Journal of Materials & Product Technology, 36(1-4), 176-188. http://doi.org/10.1504/ IJMPT.2009.027829
48 Araújo, C., Pereira, D., Dias, D., Marques, R., & Cruz, S. (2023). In-cavity pressure measurements for failure diagnosis in the injection moulding process and correlation with numerical simulation. International Journal of Advanced Manufacturing Technology, 126(1-2), 291-300 http://doi.org/10.1007/s00170023-11100-1
49 Kannan, G., & Thangaraju, R. (2022). Recent progress on natural lignocellulosic fiber reinforced polymer composites: a review. Journal of Natural Fibers, 19(13), 7100-7131 http:// doi.org/10.1080/15440478.2021.1944425
50 Yusof, F. M., Wahab, N. A., Rahman, N. L. A., Kalam, A., Jumahat, A., & Taib, C. F. M. (2019). Properties of treated bamboo fiber reinforced tapioca starch biodegradable composite. Materials Today: Proceedings, 16(Pt 4), 2367-2373 http://doi. org/10.1016/j.matpr.2019.06.140.
51 Aslan, M., Tufan, M., & Küçükömeroğlu, T. (2018). Tribological and mechanical performance of sisal-filled waste carbon and glass fibre hybrid composites. Composites. Part B, Engineering, 140, 241-249 http://doi.org/10.1016/j.compositesb.2017.12.039
Received: Mar. 04, 2024
Revised: May 20, 2024
Accepted: June 07, 2024
Júlio Harada1* , Claudio Aparecido Marcondes2 , Juliana Arquinto1 , Maria da Conceição Costa Pereira1 and Leonardo
Gondim de Andrade e Silva1
1Comissão Nacional de Energia Nuclear – CNEN, Instituto de Pesquisas Energéticas e Nucleares – IPEN, São Paulo, SP, Brasil
2Universidade Estadual de Campinas – UNICAMP, Campinas, SP, Brasil
*h.julio729@gmail.com
Obstract
Graphene nanoparticles (GnP) were selected as an alternative reinforcing nanofillers, which enhances the mechanical properties of polypropylene (PP-homo and PP-block copolymer). It may play the role of compatibilizer in polypropylenegraft-maleic anhydride (PP-g-MA) from a double-screw extruder. The effect of GnP loading and the use of the PP-gMA compatibilizer were studied. The influence on the mechanical properties of tensile strength, elongation at break, IZOD impact strength, and melting temperature in PP-homo and PP-block copolymer were investigated and compared. Their improvement was observed at to a certain amount of 0.3% (by mass) of GnP depending on the substrate nature: homopolymer or block copolymer. The increase from 69.3 J.m-1 to 176.0 J.m-1 corresponds to a 2.5 times rising in IZOD impact resistance for block copolymer. The PP block copolymer has a significant affinity in respect of the presence of graphene and the compatibilizer due to the elastomeric co-monomer present in the block copolymer.
Keywords: graphene nanoparticles, polypropylene, homopolymer, block copolymer
How to cite: Harada, J., Marcondes, C. A., Arquinto, J., Pereira, M. C. C., & Silva, L. G. A. (2024). Evaluation of graphene incorporation for mechanical properties of polypropylene composites. Polímeros: Ciencia e Tecnologia, 34(3), e20240027. https://doi.org/10.1590/0104-1428.20240016
Graphene nanoparticle (GnP) is a single layer of carbon atoms packed into a honeycomb structure, as it is depicted in Figure 1 where the graphene structure is illustrated. Since the independent discovery of graphene in 2004, the extensive research has been conducted explaining its exceptional properties[1-7]. GnPs are graphene nanocrystals layered in platelet structures held together by Van der Waals forces. The addition of GnPs in the conventional polyolefin demonstrates the promising potential for various applications due to their outstanding thermal conductivity (3000-5000 W.m-1.K-1), 4 times greater in comparison to copper which presents a value of 401 W.m-1.K-1 in high crystallization temperature. While the tensile strength of typical steel is 80,000 psi, the impressive mechanical tensile strength (18.85 million psi) is 36,000 times greater for this material. The comparison of other properties such as Young modulus is 150 million psi, when for typical steel it is 32 million, 5 times lower. A superior electrical conductivity may be noticed in respect to copper. The electronic mobility of graphene is 200,000 cm2.Vs-1 , while the capacitance of 550 F.g-1 was determined. These properties make polyolefin with incorporated GnP valuable for several applications such as nucleating agents, UV protection, flow auxiliary, antimicrobial and bacteriostatic uses, gas barriers, composite density improvement, flame retardants, and various other applications.
Polypropylene (PP) is one of the most widely used thermoplastics due to its excellent physical and mechanical properties, chemical resistance, recyclability, high processability, and low cost[8,9]. Its composites can be also employed in the fields of electronics, automotive, aerospace, sensors, packaging, textiles (ropes and mats) and much more.

Harada, J., Marcondes, C. A., Arquinto, J., Pereira, M. C. C., & Silva, L. G. A.
The researchers have extensively studied the various properties of these materials due to the high potential of PP composites incorporating the GnPs. Accordingly, they have found that the performances of these composites are highly dependent on the physical characteristics of the GnPs. For example, Liang et al.[10,11] showed that the tensile modulus of pure PP (1.25 GPa) was doubled reaching up the value of 2.5 GPa in the presence of GnP particles with diameters smaller than 10 µm and with the thickness smaller than 5 nm. Chunhui et al.[12] also revealed that the flexural strength is inversely proportional to graphite size. The crystallization behavior of this polymer matrix is influenced, when GnPs are present in the polymer matrix. Therefore, the size of GnPs must be chosen carefully to maximize the performance of material. However, various properties of composites[13] were systematically investigated the influences of a wide range of GnP sizes ranging from a some to hundreds of microns.
Several physical properties, such as the number of particles per unit volume and surface area, vary with the size of the GnPs. Therefore it must be taken in consideration, when the various properties of composites are discussed. When the tensile properties of PP-GnP as the graphene concentration increased, the tensile strength and modulus increased, as well as the elongation at break decreased simultaneously[14]. The mechanical properties of these composites decreased at higher temperatures, 165 °C and 170 °C, due to the more intensive melting. Therefore, a melting temperature of 160 °C was identified as the ideal temperature. Furthermore, the compaction time is also a significant parameter in the preparation of PP-GnP. During the determination of the thermal properties, the analyzed single melting temperature decreased with 0.3% GnP (loss of 6 °C) and 0.5% GnP (loss of 4 °C). Other thermal properties will be analyzed in a subsequent work. It was observed that the use the simultaneity presence of GnP and the selected compatibilizer significantly influence the melting temperature of pure PP. The melting temperatures and crystallinity of PP-GnP increase as the amount of graphene increased, indicating that graphene acts as a nucleation agent. The mechanical properties of the PP-GnP composites are improved with increasing their graphene content, which indicates an effective charge transfer between the PP and GnP matrix (graphene). The melting composition can affect the dispersion of the composite PP-GnP and the interfacial interaction between graphene and PP chains[15]. The heat causes to the orientation of molecular chains and graphene to the hot stretching. The graphene agglomeration can interfere with the interfacial adhesion among PP composites, worsening the interfacial interaction between graphene and PP matrix by inducing the molecular orientation of the composite. Chinnasamy et al.[16] studied the reactive compatibilization effect of butyl rubber nanocomposites reinforced with graphene oxide, concluding that the GO content increased above 1.6% by weight in the butyl rubber matrix leading to the agglomeration of material, this behaviors is accompanied by the deterioration of mechanical properties.
The PP homopolymer type H 103 and the PP block copolymer type CP241 were purchased from BRASKEM, São Paulo - Brazil. The selected graphene (GnP) was obtained from ProCene™, from USA, as well as the compatibilizer PP-g-MA, from USA (Sigma - Aldrich). The materials were extruded in a twin-screw extruder with a diameter of 20 mm and L/D 40 at a temperature of 160 ºC to, 250 ºC at 70 rpm on equipment from AX Plásticos, São Paulo - Brazil, the resulting composites grains at levels of 0.3% and 0.5% of GnP and 0.015% of PP-g-MA, by mass. The composite specimens were injected into the Battenfeld, Germany, injection machine, clamping force 160 tons and injection capacity 200 g.
The manufacturing parameters are: temperature profile: 250/ 250/ 260/ 270 ºC, from feed zone to injection nozzle; injection pressure: 150 to 160 bar; mold temperature 60 ºC; injection speed 22 mm/min; cooling time 20 s, total cycle 35 s. to evaluate the following characteristics: tensile strength, Izod impact, hardness, and melting temperature, according to the ASTM/ISO standards.
Mechanical and thermal properties were characterized according to the corresponding ASTM/ISO standards.
▪ Tensile strength and elongation (ASTM D638)
The tests were carried out using a model 5567 universal mechanical testing machine with a grip-to-grip distance of 50 mm, a grip speed of 50 mm/min, and a load cell of 50 kN. The tensile strength properties at yield and break, as well as elongation at yield and break, were determined according to ASTM D638 standard.
▪ IZOD impact resistance (ASTM D256)
The tests were carried out at UNIGEL. In the Izod impact resistance test with notch and without notch, a CEAST impact machine was used according to ASTM D256 standard.
▪ Surface hardness (ASTM D785)
The tests were carried out at Thermoblend using a Shore D durometer according to ASTM D785 standard.
▪ Melting temperature (ISO 11357)
The tests were carried out at Thermoblend according to ISO 11357 standard.
▪ Statistical significance (ISO-ABNT-NBR 3534-1)
Statistical tests were performed in accordance with the ISO-ABNT-NBR 3534-1 standard.
3.1 PP-homopolymer
The influence of the compatibilizer (PP-g-MA) and graphene (GnP) on the mechanical properties and melting
Evaluation of graphene incorporation for mechanical properties of polypropylene composites
temperatures of PP homopolymer composites with levels of 0.3% and 0.5% GnP +PP-g-MA by mass, are analyzed. Table 1 and Table 2 show the characteristic properties of the pure PP-homopolymer and the PP-homopolymer + GnP (0.3% and 0.5%).
After incorporating of 0.3% GnP into PP, the tensile strength increased by 0.2 MPa. However, the incorporation of 0.5% GnP led to a decrease in the value of tensile strength by 0.3 MPa. Moreover. The addition of 0.3% GnP causes a decrease in the sample elongation by 9.5%, whereas the amount of 0.5% GnP, the produces a more pronounced decrease at 13.3%. The IZOD impact (with notch) increases by 26 J.m-1 with 0.3% GnP and by 11 J.m-1 with 0.5% GnP. In contrast, IZOD impact without a notch resulted in no break (NB*). Simultaneity, did not observe a modification in the values of hardness. Regarding the melting temperature, the addition of 0.3% GnP caused a decrease of 6 °C (from
180 °C to 174 °C), while the addition of 0.5% GnP produced a decrease of 4 °C (from 180 °C to 176 °C). Both concentrations of graphene and the compatibilizer significantly influenced the melting temperature compared to pure homo PP.
The impact of the compatibilizer (PP-g-MA) and the combination effect of compatibilizer and graphene (GnP) on the mechanical properties of PP-block copolymer composites were assessed. Similarly, levels of 0.3% and 0.5% GnP by mass were analyzed as in previous tests.
Table 3 and Table 4 show the characterization of the pure PP-block copolymer and those of the PP-block copolymer with added GnP (0.3% and 0.5%).
The tensile strength increased by 2.0 MPa upon incorporation of 0.3% GnP and by 2.4 MPa with 0.5% GnP.
Table 1. Results of the characterization (melting point, tension and elongation) of the PP-homopolymer. PP-homopolymer + GnP (0.3% and 0.5%)
Material
Table 2. Results of the characterization (Hardness Shore D, IZOD with notch and IZOD without notch) of the PP-homopolymer.
PP-homopolymer + GnP (0.3% and 0.5%)
Material
NB* - no break; W notch = with notch; WO notch = without notch
Table 3. Results of the characterization (melting point, tension and elongation) of the PP-block copolymer.
PP-block copolymer pure, and block copolymer plus GnP (0.3% and 0.5%).
Table 4. Results of the characterization (Hardness Shore D, IZOD with notch and IZOD without notch) of the PP-block copolymer.
PP-block copolymer pure, and block copolymer plus GnP (0.3% and 0.5%).
Material
Characterization Graphene
NB* - no break; W notch = with notch; WO notch = without notch
Harada, J., Marcondes, C. A., Arquinto, J., Pereira, M. C. C., & Silva, L. G. A.
Additionally, tensile strength increased by 11.2% when the samples contain 0.3% GnP and increased by 0.8% when 0.5% GnP is present. Regarding the IZOD impact strength, with notches an increase of 106.7 J.m-1 (increment of 150%) was observed with 0.3% GnP. While the loading is 0.5% GnP, the impact strength increased by 97.4 J.m-1. Incorporating up to 0.3% GnP is deemed optimal, as beyond this threshold, there isn’t a significant modification in the mechanical properties of tested materials. Concerning thermal properties, the melting temperature rose by 3°C (from 168°C to 171°C) with 0.3% GnP and by 4°C (from 168°C to 172°C) with 0.5% GnP. It was noted that both the GnP loading and the presence of compatibilizer significantly influence the melting temperature of pure PP block copolymer.
PP-homopolymer composites containing GnP and PP-g-MA, evidenced that loading of 0.3% GnP is optimal concentration. Beyond this threshold, there was found a decrease in the mechanical properties due to the agglomeration and slipping effects of GnP, along with increased costs for PP composites with GnP levels exceeding 0.3%. The presence of 0.3% GnP in PP increases the cost.
It was observed that in the samples consisting of PP-block copolymer composites containing GnP and PP-g-MA both the loading of GnP and the presence of the compatibilizer had a significantly positive impact on the mechanical properties and melting temperature of the pure PP block copolymer. The PP block copolymer exhibits a high affinity in respect to graphene due to the ethylene elastomeric co-monomer. The compatibilizer in the PP block copolymer facilitates its interaction with graphene. The proficiency in the operation and utilization of equipment for dispersing and plasticizing graphene in polymers are essential for achieving optimal polymer composites. This study provides an effective strategy for the producing PP-GnP composites. The incorporation of graphene, the possessing high intrinsic mechanical characteristics, can enhance the mechanical properties significantly, with the IZOD impact strength increasing by over 150% in polymer composites containing 0.3% graphene. This finding is of particular interest to industries such as automotive and packaging, among others, as the graphene phase offers ample space for interaction with polymer molecules.
• Conceptualization – Júlio Harada; Claudio Aparecido Marcondes.
• Data curation – Maria da Conceição Costa Pereira; Juliana Arquinto; Leonardo Gondim de Andrade e Silva.
• Formal analysis – Julio Harada; Claudio Aparecido Marcondes; Leonardo Gondim de Andrade e Silva.
• Funding acquisition – NA.
• Investigation – Julio Harada; Claudio Aparecido Marcondes.
• Methodology – Julio Harada; Juliana Arquinto; Maria da Conceição Costa Pereira; Leonardo Gondim de Andrade e Silva.
• Project administration – Julio Harada; Maria da Conceição Costa Pereira; Leonardo Gondim de Andrade e Silva.
• Resources – Maria da Conceição Costa Pereira; Leonardo Gondim de Andrade e Silva.
• Software – NA.
• Supervision – Maria da Conceição Costa Pereira; Leonardo Gondim de Andrade e Silva.
• Validation – Julio Harada; Leonardo Gondim de Andrade e Silva; Maria da Conceição Costa Pereira.
• Visualization – Julio Harada; Claudio Aparecido Marcondes; Juliana Arquinto.
• Writing – original draft – Julio Harada; Claudio Aparecido Marcondes; Juliana Arquinto.
• Writing – review & editing – Julio Harada; Claudio Aparecido Marcondes; Maria da Conceição Costa Pereira; Juliana Arquinto; Leonardo Gondim de Andrade e Silva.
6. Acknowledgements
The authors thank UNIGEL and Thermoblend for their support in this work and CNPq, CAPES, and FAPESP for financial support.
1 Rao, C. N. R., Sood, A. K., Subrahmanyam, K. S., & Govindaraj, A. (2009). Graphene: the new two-dimensional nanomaterial. Angewandte Chemie International Edition in English , 48(42), 7752-7777 http://doi.org/10.1002/anie.200901678 PMid:19784976.
2 Geim, A. K., & Novoselov, K. S. (2007). The rise of graphene. Nature Materials, 6(3), 183-191 http://doi.org/10.1038/ nmat1849 PMid:17330084.
3 Geim, A. K. (2009). Graphene: status and prospects. Science, 324(5934), 1530-1534. http://doi.org/10.1126/science.1158877. PMid:19541989.
4 Singh, V., Joung, D., Zhai, L., Das, S., Khondaker, S. I., & Seal, S. (2011). Graphene based materials: past, present and future. Progress in Materials Science, 56(8), 1178-1271 http:// doi.org/10.1016/j.pmatsci.2011.03.003.
5 Kuilla, T., Bhadra, S., Yao, D., Kim, N. H., Bose, S., & Lee, J. H. (2010). Recent advances in graphene based polymer composites. Progress in Polymer Science, 35(11), 1350-1375 http://doi.org/10.1016/j.progpolymsci.2010.07.005
6 Young, R. J., Kinloch, I. A., Gong, L., & Novoselov, K. S. (2012). The mechanics of graphene nanocomposites: a review. Composites Science and Technology, 72(12), 1459-1476 http:// doi.org/10.1016/j.compscitech.2012.05.005
7 Potts, J. R., Dreyer, D. R., Bielawski, C. W., & Ruoff, R. S. (2011). Graphene-based polymer nanocomposites. Polymer, 52(1), 5-25 http://doi.org/10.1016/j.polymer.2010.11.042
8 Kalaitzidou, K., Fukushima, H., & Drzal, L. T. (2007). Mechanical properties and morphological characterization of exfoliated graphite–polypropylene nanocomposites. Composites. Part A, Applied Science and Manufacturing, 38(7), 1675-1682 http:// doi.org/10.1016/j.compositesa.2007.02.003
9 An, J.-E., Jeon, G. W., & Jeong, Y. G. (2012). Preparation and properties of polypropylene nanocomposites reinforced with exfoliated graphene. Fibers and Polymers, 13(4), 507-514 http://doi.org/10.1007/s12221-012-0507-z
10 Liang, J. Z., Du, Q., Wei, L. Y., Tsui, C. P., Tang, C. Y., Law, W. C., & Zhang, S. D. (2015). Melt extrudate swell behavior
Evaluation of graphene incorporation for mechanical properties of polypropylene composites
of graphene nano-platelets filled-polypropylene composites. Polymer Testing, 45 , 179-184 http://doi.org/10.1016/j. polymertesting.2015.06.007
11 Liang, J.-Z., Du, Q., Tsui, G. C.-P., & Tang, C.-Y. (2016). Tensile properties of graphene nano-platelets reinforced polypropylene composites. Composites. Part B, Engineering, 95, 166-171 http://doi.org/10.1016/j.compositesb.2016.04.011
12 Chunhui, S., Mu, P., & Runzhang, Y. (2008). The effect of particle size gradation of conductive fillers on the conductivity and the flexural strength of composite bipolar plate. International Journal of Hydrogen Energy, 33(4), 1035-1039 http://doi. org/10.1016/j.ijhydene.2007.11.013
13 Inuwa, I. M., Hassan, A., Samsudin, S. A., Kassim, M. H. M., & Jawaid, M. (2014). Mechanical and thermal properties of exfoliated graphite nanoplatelets reinforced polyethylene terephthalate/polypropylene composites. Polymer Composites, 35(10), 2029-2035 http://doi.org/10.1002/pc.22863
14 . Wang, J., Song, F., Ding, Y., & Shao, M. (2020). The incorporation of graphene enhances the mechanical properties of polypropylene
self-reinforced polymer composites. Materials & Design, 195, 109073 http://doi.org/10.1016/j.matdes.2020.109073
15 Natarajan, S., Rathanasamy, R., Palaniappan, S. K., Velayudham, S., Subburamamurthy, H. B., & Pal, K. (2020). Comparison of MA-g-PP effectiveness through mechanical performance of functionalised graphene reinforced polypropylene. Polímeros: Ciência e Tecnologia, 30(3), e20200035 http:// doi.org/10.1590/0104-1428.05620
16 Chinnasamy, S. , Rathanasamy, R. , Kumar, H. K. M. , Jeganathan, P. M., Palaniappan, S. K., & Pal, S. K. (2020). Reactive compatibilization effect of graphene oxide reinforced butylrubber nanocomposites. Polímeros: Ciência e Tecnologia, 30(3), e2020032 http://doi.org/10.1590/0104-1428.05920
Received: Feb. 08, 2024
Revised: May 20, 2024
Accepted: Jun. 24, 2024
Lays Furtado de Medeiros Souza Kataoka1* , Maria del Pilar Hidalgo Falla2 and Sandra Maria da Luz1,2
1Programa de Pós-graduação em Ciências Mecânicas, Departamento de Engenharia Mecânica, Universidade de Brasília – UnB, Brasília, DF, Brasil
2Programa de Pós-graduação em Integridade de Materiais da Engenharia, Faculdade do Gama, Universidade de Brasília – UnB, Brasília, DF, Brasil
*lays.furtado94@gmail.com
aOhis paper has been partially presented at the 17th Brazilian Polymer Oongress, held in Joinville, OO, 29/Oct - 02/Oov/2023.
Obstract
Nanocomposite films from cellulose jute fibers can be promising for application in electronic devices, mainly when silver nanoparticles (AgNPs) are added, which increases their conductivity. This work studied the transmittance, conductivity, and thermal stability of regenerated cellulose films (RCF) by adding AgNPs and plasticizers. Carboxymethylcellulose (CMC), sorbitol, and glycerol plasticizers were incorporated at 15 wt.% and 0.5 wt.% of AgNPs. The resulting materials were studied using Ultraviolet-visible and Fourier Transform Infrared Spectroscopy, electrical analysis using the Van der Pauw method, and thermogravimetry. As a result, an average size of 9 nm for AgNPs was verified. Incorporating AgNP in the films decreased the transmittance by a minimum of 34% compared to pure RCF and reduced thermal stability by at least 10 ºC. Nanocomposites containing plasticizers showed an improved conductivity after incorporating these components, from 10-2 to 10-1 (S/cm).
Keywords: regenerated cellulose films, silver nanoparticles, transmittance, conductivity, thermal stability.
How to cite: Kataoka, L. F. M. S., Falla, M. D. P. H., & Luz, S. M. (2024). Properties of regenerated cellulose films with silver nanoparticles and plasticizers. Polímeros: Ciência e Tecnologia, 34(3), e20240028. https://doi.org/10.1590/0104-1428.20240025
Cellulose can be applied in various forms, such as fibers, films, and composite materials, for several industrial uses[1]. Among these forms, conductive regenerated cellulose films are studied primarily for their applications in highvalue electronic devices such as sensors, transistors, solar cells, supercapacitors, electromagnetic shielding, and other elements[2]. To obtain cellulose, plants composed of lignocellulose containing a significant proportion of cellulose have attracted interest due to their abundant, renewable, and naturally occurring polymers. Jute (Corchorus capsularis) is among the natural fibers that stand out as one of the greatest and cheapest sources of cellulose[3], with several applications worldwide, and it is only surpassed by cotton[4]
Regenerated cellulose films are obtained in such a way that the cellulose’s fibrous and semi-crystalline structure must be broken down in a dissolution process and then undergo a subsequent coagulation step[2]. Systems that use alkaline solutions such as sodium hydroxide (NaOH) for cellulose dissolution are considered more cost-effective, and toxic substances are not used or produced throughout the process. As a result, water becomes a potential coagulant in the NaOH aqueous dissolution system because it is easily accessible and presents controlled results, such as porosity[5]
Conductive materials such as metals, inorganic oxides, conductive polymers, and carbon materials can be physically or chemically introduced into their structure[2]. Among these, metal nanoparticles can be applied in polymeric films due to their potential to enhance the overall properties of polymeric materials, such as conductivity[6-8]. These conductive films can be promising candidates for the manufacture of electronic components, as seen in previous works, where silver nanoparticles provided increased conductivity of regenerated cellulose films[9]. A homogeneous morphology was also observed, without agglomerates, which indicates an increase in the degree of molecular orientation of the films obtained by the regenerated process of the samples, probably due to the plasticizing effect of carboxymethylcellulose (CMC) used to stabilize the silver nanoparticle solution. This effect can improve the physical and chemical properties of the films and allows for more application options for these conductive films[9]
The appropriate selection of the plasticizer is influenced by several factors, such as the compatibility between the polymers used and the desired final properties[10]. Plasticizers are components that occupy intermediate positions between long polymeric chains, effectively increasing the distance between them and reducing secondary intermolecular
Kataoka, L. F. M. S., Falla, M. D. P. H., & Luz, S. M.
interactions, making them suitable for applications requiring flexibility and ductility, such as sheets or thin films[11-13] Various properties, such as tensile strength, compression, toughness, dimensional and thermal stability, surface aspects, and optical properties, can be modified by incorporating plasticizers into regenerated cellulose films[12]
Plasticizers derived from renewable sources such as CMC, sorbitol, and glycerol are compatible with RCF films that can improve the mechanical flexibility of films and modify other physicochemical properties, such as thermal stability improvement[14]. CMC is a water-soluble derivative obtained through cellulose, sodium hydroxide, and monochloroacetic acid suspension reaction. It is widely recognized as an important source of cellulose used to enhance the properties of biofilms[15]. Sorbitol is a polyol commonly used in producing films using polysaccharides as raw materials[16]. Glycerol is one of the most common plasticizers used, mainly in biodegradable films[17]. When used as a plasticizer, glycerol interacts with cellulose chains, increasing molecular mobility and, consequently, the hydrophilicity and flexibility of the plasticized films[18] These regenerated cellulose conductive films can be seen as a more sustainable option than petrochemical polymers, given the use of renewable raw materials in their manufacture and their biodegradable characteristics.
In regenerated cellulose conductive films, the insertion of plasticizers such as CMC, sorbitol, and glycerol is still understudied. Various works on the insertion of plasticizers in regenerated cellulose films are found in the literature, but these do not involve films with the insertion of conductors. Therefore, there is a study gap in the literature. In addition to conductivity, factors such as transmittance, chemical composition, and application temperature range should be studied. Then, in this work, regenerated cellulose films with the incorporation of silver nanoparticles (AgNsP) and plasticizers were evaluated for transmittance, conductivity, and thermal stability.
2.1 Materials
The bleached jute cellulose was obtained in previous work[9]. The following chemicals of reagent grade were used: sodium borohydrate (NaBH4, 98%, Dynamic), silver nitrate (AgNO3, 99.9%, Neon), sodium carboxymethyl cellulose (CMC, Sigma–Aldrich), sodium hydroxide (NaOH, 97%, Greentec), sorbitol (98%, Dynamic), and glycerol (98%, Synth).
2.2 Synthesis and characterization of silver nanoparticles
To obtain the nanoparticle solution, 450 mL of an 18.0 x 10-6 mol/L NaBH4 solution was kept below 3 °C in an ice bath and added to an Erlenmeyer flask under vigorous magnetic stirring. Through a burette, 150 mL of a 9.0 x 10-6 mol/L solution of AgNO3 was added at an additional rate of 1 drop/s. The chemical reaction between NaBH4 and AgNO3 in those concentrations resulted in AgNPs at 250 ppm. To avoid silver nanoparticle aggregation, the solution was stabilized with 0.6 g of CMC, which has the function of coating the
nanoparticles. The silver nanoparticle solution, prepared by drying a small drop on a copper grid, was characterized by Transmission Electronic Microscopy (TEM) in the JEOL JEM 1011 equipment, with a voltage of 40 kV. The ImageJ software took size measurements and statistical analysis of silver nanoparticle dimensions from TEM images.
2.3
The bleached jute cellulose was ground in an automatic analytical mill Quimis Q298A for 30 seconds. Depending on the sample, as shown in Table 1, each solution was prepared as follows: To obtain the RCF film, 2.2 g of cellulose was dissolved in 100 mL of a pre-cooled solution (-12°C for 18 h) containing 8% (w/v) NaOH solution. The solution was kept in a freezer for at least 18 h to reach around -12oC equilibrium to achieve the cooling conditions. Also, to obtain the RCF with the AgNPs, 26 mL of nanoparticle solution at 250 ppm was added to achieve 0.5 wt.% of silver nanoparticle content before the pre-cooling. To prepare the RCF films with the plasticizers, the CMC, sorbitol, and glycerol, in amounts corresponding to 15 wt.% for cellulose, were added to the pure RCF solution. The same process was performed for the nanocomposites, which resulted in nanocomposites containing 15 wt.% of plasticizer and 0.5 wt.% of silver nanoparticle. To dissolve the solution, all samples were submitted under vigorous stirring at 3000 rpm and room temperature every 7 min.
To mold the films and regenerate the cellulose, the mixture containing cellulose, and nanoparticles was subjected to centrifugation at 4000 rpm for 10 min to exclude the undissolved part and perform degassing. Then, 7 mL of the resulting solution was poured onto a polypropylene Petri dish with a diameter of 85 mm. The RCF and nanocomposite films were pre-dried at room temperature for 18 h until gelation. Afterward, the sample was immersed in a water bath (distilled water) at room temperature for 15 min to coagulate and regenerate the cellulose. Finally, the films were dried at room temperature for 48 h before the characterization.
The RCF and nanocomposite films were assessed with Evolution 220 Thermo Scientific equipment to determine their transmittance. The films’ transmittance spectra were analyzed across 200 to 800 nm wavelengths.
Table 1. Composition of regenerated cellulose films.
-RCF - AgNP - 0.5%
RCF - CMC 15% CMC -
RCF - CMC - AgNP 15% CMC 0.5%
RCF - Sorbitol 15% Sorbitol -
RCF - Sorbitol - AgNP 15% Sorbitol 0.5%
RCF - Glycerol 15% Glycerol -
RCF - Glycerol - AgNP 15% Glycerol 0.5%
2.5 Fourier Transform Infrared (FTIR) spectroscopy
FTIR spectra of RCF and nanocomposite films were obtained using a Fourier-transform infrared spectrometer (NICOLET IS10, Thermo Scientific) by transmittance accessory from 500 to 4000 cm−1 with 64 scans collected at an interval of 4 cm−1
2.6 Electrical measurements of films by the Van der Pauw method
Using the Van der Pauw method to characterize thin films, the sheet resistance measurements of nanocomposite films were obtained through four-point measurements using the Agilent B1500A Semiconductor Device Analyzer. The samples were subjected to a current range from 100 uA to 1 mA with a 10 uA interval, up to a limit of 1 V. To calculate resistivity and conductivity, film thicknesses were measured with a Mitutoyo digital micrometer (error of 0.001mm), taken at 10 points across the material.
From the sheet resistance measurements ( ) s R , expressed in units of ohms per square ( / Ω ) e and the uniform thickness of the films ( ) t , the sheet resistivity s () ρ can be obtained using Equation 1. ssR.t .cm ρ=Ω
Where the sheet conductivity ( sσ ) can then be calculated using Equation 2:
s 1 S/cm σ=
2.7 Thermogravimetric Analysis (TGA)
The RCF and nanocomposite films were characterized via thermogravimetry analysis (TGA: SDT Q600 TA Instruments). Approximately 10 mg of each sample was analyzed in alumina pans. The analysis was conducted under a nitrogen atmosphere (flow rate 50 mL/min) and a heating rate of 10 °C min-1 from 30 to 600 °C.
3.1 Morphological characterization of silver nanoparticles (AgNP) and transmittance characterization of films
Figure 1 shows the transmission micrographs of the AgNP solution. The micrographs show that the nanoparticles are homogeneous in size. The average size found for the nanoparticles was 9.0 ± 2.0 nm, with spherical characteristics within the range typically obtained in the literature using the synthesis method from silver nitrate and sodium borohydride solutions[19].
Figure 2 shows the appearance of each studied film, according to Table 1. The incorporation of AgNPs changed the color of the films, which caused the samples to darken due to the yellowish color of the nanoparticle solution. Visually, the RCF—CMC—AgNP and RCF—Glycerol— AgNP films are opaquer, similar to the RCF—Glycerol film. Subsequently, UV-Vis spectroscopy reveals the effect of incorporating plasticizers and AgNPs on the optical properties of these films.
The transmittance of conductive films is an important factor to be evaluated, as the mutually opposing properties of conductivity and transparency using metallic conductive materials in films are challenging. This challenge may restrict their application in electronic devices like optoelectronics, including solar cells and touch screens[20,21]. Additionally, incorporating plasticizers into regenerated cellulose films can increase or decrease the transmittance of these films[22] . Therefore, UV-Vis investigated the effect of plasticizer and AgNP incorporation on the transmittance of regenerated cellulose films. The UV-Vis results and the transmittance values for the films at 550 nm are presented in Figure 3. The transmittance curve for all RCF films was observed throughout the wavelength range between 200 and 800 nm[9] Figure 3 reveals low transmittance of the RCF - CMC - AgNP, RCF – Glycerol, and RCF - Glycerol - AgNP films in the UV region, suggesting that these samples have anti-UV


properties. In the visible region of the spectra, we noticed an increase in transmittance in all films. In addition, the transmittance of silver nanocomposites decreased with the incorporation of plasticizers since these components provide increased absorption, as seen in the literature[14]
The RCF sample obtained a high transmittance value, as reported in some studies with similar applications for regenerated cellulose films[2]. Incorporating sorbitol into RCF - Sorbitol increased the transmittance value to greater than pure RCF. However, containing plasticizers in RCFCMC and RCF-Glycerol samples decreased the transmittance concerning RCF, with the latter experiencing a significant decrease upon adding glycerol.
The presence of a transmittance band, near 300-400 nm for the nanocomposite films containing AgNPs, indicates the existence of this component in the samples. The bands in this wavelength range are related to these spherical metal nanoparticles. As observed in the spectra, AgNPs with smaller sizes exhibit an absorption peak at shorter wavelengths[19]. Regarding the nanocomposite films, the RCF - Sorbitol - AgNP sample had lower transmittance than the RCF – AgNP. However, the plasticizer had the best result compared to the other plasticizers, in which the RCF - CMC - AgNP had the highest transmittance loss.
To identify the functional groups within the structure of the films and the chemical modifications resulting from the incorporation of plasticizers, the FTIR spectra of the controlled regenerated cellulose and nanocomposite films are shown in Figure 4. The absorption bands around 3440 cm-1 indicate the presence of cellulose due to the stretching of the hydrogenbonded O-H, and around 2900 cm-1 corresponds to the stretching of the C-H functional group[14]
For nanocomposite films, a shift was observed in the C=O stretching from 1636 cm-1 to 1642 cm-1, possibly due to the interaction between silver nanoparticles and the carbonyl group,


Properties of regenerated cellulose films with silver nanoparticles and plasticizers
which suggests coordination between them[9,23]. The absorption band around 1410 cm-1 is associated with the symmetrical bending of CH2 in cellulose, indicating correspondence with the cellulose II polymorph. A doublet at 1333-1311 cm-1 and a band at 1365 cm-1 were observed, corresponding to the angular bending of the COH and HCC bonds, ordinarily present in crystalline cellulose[24]. The peaks between 997 and 1200 cm-1 are also characteristic of cellulose. The peaks at 1170 cm-1, 1040-1070 cm-1, and 894 cm-1 are related to the stretching of the C-O vibrations and C-H deformation of the skeletal pyranose ring of cellulose[9]. At 997 cm-1, there is a CO stretching peak. No chemical bond between cellulose and NaOH was identified from the FTIR [9].
Polyols, such as sorbitol and glycerol, exhibit bands ranging from 1500 to 1200 cm-1 attributed to the overlap of C-H in-plane and O-H bending in polyol molecules, coinciding with cellulose peaks[25]. Similarly, CMC also features characteristic peaks of cellulose, which it derives from. In films plasticized with CMC, the presence of an absorption band at 1590 cm-1 is observed, corresponding to the stretching vibration of carboxyl groups (COO-)[26,27] , where the deprotonation of these groups is linked to the cellulose structure[28]. In glycerol-plasticized films, a peak around 850 cm-1 corresponds to the vibrations of the C-H groups[29].
The chemical changes in regenerated cellulose films can lead to physical alterations as the plasticizer connects to the biopolymer molecules, promoting greater mobility in the polymer chains and reducing the density between the molecules, affecting the film’s optical properties. Therefore, investigating the transmittance of films is important to classify future applications of these films. The chemical changes in regenerated cellulose films can lead to physical changes as the plasticizer connects to the biopolymer molecules, which promotes greater mobility in the polymer chains and reduces the density between the molecules, affecting the film’s optical properties. Therefore, investigating the transmittance of films is important to classify future applications of these films.
The concentration of AgNPs can affect the conductivity of regenerated cellulose films[9]. However, few studies have been conducted on the analysis of these films involving the incorporation of plasticizers. Therefore, given their two-dimensional nature, the Van der Pauw method allows the measurement of the electrical properties of these nanocomposite-regenerated cellulose films, reducing experimental errors such as those generated by contact misalignment[30]. Table 2 shows the conductivity values of the nanocomposite films.
Nanocomposites with plasticizers achieved a significant improvement in conductivity after incorporation of these components and ranged from 10-2 to 10-1 in relation to RCF - AgNP with the same nanoparticle content, with an increase of 292.3% for RCF - CMC - AgNP, 159.50% for RCFSorbitol - AgNP and 230.6% for RCF - Glycerol - AgNP. The CMC plasticizer provided better conductivity values in the nanocomposite, followed by glycerol and sorbitol. The difference between the plasticized films RCF - CMC - AgNP with higher conductivity and RCF - Sorbitol -
Table 2. Conductivity data for nanocomposites. Sample
RCF - AgNP 1 0.1014 0.06836 ±0.0230
2 0.0536
3 0.0501
RCF - CMC - AgNP 1 0.2898 0.2682 ± 0.0504
2 0.1934
3 0.3087
RCF - Sorbitol - AgNP 1 0.2060 0.1774 ± 0.0359
2 0.1267
3 0.1994
RCF - Glycerol - AgNP 1 0.2069 0.2260 ± 0.0506
2 0.2953
3 0.1758
RCF *2 [9] 3.28 x 10-2
RCF – 0.290% AgNP *2 [9] 5.32 x 10-2
*1 Three films of each nanocomposite studied were analyzed. For each film, electrical measurements were carried out in quadruplicate.
*2 Literature data.
AgNP with lower conductivity was 51.2%. The difference between RCF - CMC - AgNP and RCF - Glycerol - AgNP was smaller at 18.7%.
The electrical data for the pure RCF and the nanocomposite containing 0.290% AgNP described in Table 2, shows that increasing the content of silver nanoparticles from 0.290% to 0.5% in the RCF – AgNP (without plasticizer) provided an increase of 28.5% of conductivity. We also observed an increase of 717.8% in the conductivity of RCF - CMC –AgNP in relation to the conductivity of pure RCF[9]
Other works show the effect of increasing conductivity with plasticizer incorporation. Composite films of regenerated cellulose nanoparticles (RCNs) with poly(3,4— ethylenedioxythiophene) (PEDOT) achieved increased conductivity with the incorporation of RCNs that acted as a plasticizer, which improved the films’ mechanical properties[31]. In blends of polyaniline and nitrile rubber, there was an increase in conductivity using the properties of imidazolium-based ionic liquids as plasticizers[32] .
Conductive regenerated cellulose films in the order of magnitude of 10-2 to 10-1 S/cm, found for the nanocomposite films in this work, are promising for applications such as supercapacitors[33-35], strain sensors[36], electromagnetic interference shielding[37,38], anti-static cellulose films[39]
The AgNP are good electrical conductors but can also be good thermal conductors because they are metals. Therefore, it is important to study the thermal stability of films when incorporating this material and plasticizers.
TG and DTG investigated the effect of incorporating plasticizers and AgNP on the thermal stability of regenerated cellulose films, and the curves are represented in Figure 5 Table 3 shows the thermal stability, peak temperatures of RCFs and nanocomposites, and weight loss from 100 °C to 600 °C.
A thermal event occurs at temperatures below 100°C for all samples due to moisture loss[9]. We also include information regarding the thermal stability of the samples and the temperature of the main degradation peak corresponding to cellulose, which ranged from approximately 253°C to 334°C.
The least thermally stable samples where cellulose degradation peaks shifted to lower temperatures were those with glycerol, probably due to this component degrading at lower temperatures, as was previously observed in the literature[14], with a difference of 81.2 ºC from the RCFGlycerol - AgNP for RCF. Therefore, the main degradation of these samples plasticized with glycerol occurred below 330 ºC and above 385 ºC for the others.
The thermal stability of all films with plasticizer incorporation was lower than pure RCF. The incorporation of plasticizer reduces the rigidity of the polymer chains. As a result, the thermal stability of the films was eventually reduced. Thus, incorporating plasticizer also reduced all films’ initial and final decomposition temperature, possibly due to the mixing and interactions of the regenerated cellulose with the plasticizers[40]
Incorporating AgNP in all nanocomposite films shifted the cellulose degradation peak to lower temperatures. This can be correlated to the catalytic action of AgNPs on the cellulose polymer matrix, as they act as initiators and facilitators of thermal degradation of the polymer matrix due to their thermal conductivity, as seen in other works on polymer applications[41,42] .
Optoelectronic applications are limited because the films studied in this work do not have high transmittance. However, the thermal stability together with the conductivity values in the order of magnitude of 10-2 to 10-1 S/cm, found for the nanocomposite films, are promising for applications such as supercapacitors[33-35], strain sensors[36], electromagnetic interference shielding[37,38], anti-static cellulose films[39], as seen in other papers.
Incorporating AgNPs and plasticizers into nanocomposite films decreased the transmittance compared to RCF films, making it difficult for light to pass through and limiting it to optical applications. AgNP nanocomposite films were less thermally stable than RCF without nanoparticles but did not impact large temperature differences. The FTIR spectra made it possible to visualize the film’s functional groups, characteristics of cellulose, and plasticizers used. Furthermore, it also showed band shift, the possible interaction between AgNPs, and the carbonyl group in films. Nanocomposites containing plasticizers showed a significant improvement in conductivity after these components were incorporated, which ranged from 10-2 to 10-1 compared to RCF - AgNP for the same nanoparticle content. Therefore, incorporating plasticizers considerably improved the mobility of electrons in the cellulose polymer chain, making the material more conductive and potentially increasing application possibilities. The CMC plasticizer provided better conductivity values in the nanocomposite, followed by glycerol and sorbitol.
5. Author’s Contribution
• Conceptualization – Lays Furtado de Medeiros Souza Kataoka; Sandra Maria da Luz.
• Data curation – Lays Furtado de Medeiros Souza Kataoka.
• Formal analysis – Lays Furtado de Medeiros Souza Kataoka; Maria del Pilar Hidalgo Falla.
• Funding acquisition – Sandra Maria da Luz.
• Investigation – Lays Furtado de Medeiros Souza Kataoka.
• Methodology – Lays Furtado de Medeiros Souza Kataoka.
• Project administration – Lays Furtado de Medeiros Souza Kataoka; Sandra Maria da Luz.
• Resources – Sandra Maria da Luz; Maria del Pilar Hidalgo Falla.
• Software – NA.
• Supervision – Sandra Maria da Luz; Maria del Pilar Hidalgo Falla.
• Validation – Lays Furtado de Medeiros Souza Kataoka; Sandra Maria da Luz.
• Visualization – Lays Furtado de Medeiros Souza Kataoka.
• Writing – original draft – Lays Furtado de Medeiros Souza Kataoka.
• Writing – review & editing – Lays Furtado de Medeiros Souza Kataoka; Sandra Maria da Luz.
6. Acknowledgements
The authors thank Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Universidade de Brasília (DPI, DPG, and BCE) for the financial support.
7. References
1 Abdul Khalil, H. P. S., Bhat, A. H., & Ireana Yusra, A. F. (2012). Green composites from sustainable cellulose nanofibrils: a review. Carbohydrate Polymers, 87(2), 963-979. http://doi. org/10.1016/j.carbpol.2011.08.078
2 Liu, X., Xiao, W., Ma, X., Huang, L., Ni, Y., Chen, L., Ouyang, X., & Li, J. (2020). Conductive regenerated cellulose film and its electronic devices – A review. Carbohydrate Polymers, 250, 116969 http://doi.org/10.1016/j.carbpol.2020.116969 PMid:33049865.
3 Kataoka, L. F. M. S., Hidalgo Falla, M. D. P., & Luz, S. M. (2021). The influence of potassium hydroxide concentration and reaction time on the extraction cellulosic jute fibers. Journal of Natural Fibers, 19(13), 6889-6901 http://doi.org/ 10.1080/15440478.2021.1934934
4 Erdoğan, U. H., Seki, Y., Aydoğdu, G., Kutlu, B., & Akşit, A. (2016). Effect of different surface treatments on the properties of jute. Journal of Natural Fibers, 13(2), 158-171 http://doi. org/10.1080/15440478.2014.1002149
5 Budtova, T., & Navard, P. (2016). Cellulose in NaOH–water based solvents: a review. Cellulose (London, England), 23(1), 5-55 http://doi.org/10.1007/s10570-015-0779-8
6 Meera, K., & Ramesan, M. T. (2023). Development of high-performance biopolymer nanocomposites derived from carboxymethyl chitosan/boehmite via green synthesis. Polymer Composites, 44(3), 1135-1148 http://doi.org/10.1002/pc.27159
7 Meera, K., Arun, K., & Ramesan, M. T. (2023). High performance biopolymer blend nanocomposites derived from cashew gum/ polyvinyl alcohol/boehmite for flexible electronic devices. Journal of Applied Polymer Science, 140(7), e54300 http:// doi.org/10.1002/app.54300
8 Ramesan, M. T., Subburaj, M., Mathew, G., & Bahuleyan, B. K. (2023). Utilization of copper sulphide nanoparticles for the development of cashew tree gum/chitin biopolymer blend nanocomposites. Journal of Thermoplastic Composite Materials, 36(7), 984-1003 http://doi.org/10.1177/08927057211046282
9 Kataoka, L. F. M. S., Leão, R. M., Gontijo, A. B., Falla, M. D. P. H., & Luz, S. M. (2022). Regenerated cellulose films from jute fibers applied in conductive nanocomposites. Materials Today. Communications, 33, 104645 http://doi.org/10.1016/j. mtcomm.2022.104645
10. Cheremisinoff, N. P. (1998). Advanced polymer processing operations. USA: Noyes Publications.
11 Vanin, F. M., Sobral, P. J. A., Menegalli, F. C., Carvalho, R. A., & Habitante, A. M. Q. B. (2005). Effects of plasticizers and their concentrations on thermal and functional properties of gelatin-based films. Food Hydrocolloids, 19(5), 899-907 http://doi.org/10.1016/j.foodhyd.2004.12.003
12 Callister, W. D., Jr. (2007). Materials science and engineering: an introduction USA: John Wiley & Sons
13 Meera, K., & Ramesan, M. T. (2023). Tailoring the performance of boehmite nanoparticles reinforced carboxymethyl chitosan/ cashew gum blend nanocomposites via green synthesis. Polymer, 268, 125706 http://doi.org/10.1016/j.polymer.2023.125706
14 Pang, J., Liu, X., Zhang, X., Wu, Y., & Sun, R. (2013). Fabrication of cellulose film with enhanced mechanical properties in ionic liquid 1-allyl-3-methylimidazolium chloride (AmimCl). Materials (Basel), 6(4), 1270-1284 http://doi.org/10.3390/ ma6041270 PMid:28809209.
15. Caraschi, J. C., & Campana, S. P., Fo. (1999). Influência do grau de substituição e da distribuição de substituintes sobre as propriedades de equilíbrio de carboximetilcelulose em solução aquosa. Polímeros: Ciência e Tecnologia, 9(2), 70-77 http:// doi.org/10.1590/S0104-14281999000200015
16 McHugh, T. H., & Krochta, J. M. (1994). Sorbitol- vs glycerolplasticized whey protein edible films: integrated oxygen permeability and tensile property evaluation. Journal of Agricultural and Food Chemistry, 42(4), 841-846 http://doi. org/10.1021/jf00040a001.
17 Pérez, O. E., Sánchez, C. C., Pilosof, A. M. R., & Patino, J. M. R. (2008). Dynamics of adsorption of hydroxypropyl methylcellulose at the air-water interface. Food Hydrocolloids, 22(3), 387-402 http://doi.org/10.1016/j.foodhyd.2006.12.005
18 Mali, S., Sakanaka, L. S., Yamashita, F., & Grossmann, M. V. E. (2005). Water sorption and mechanical properties of cassava starch films and their relation to plasticizing effect. Carbohydrate Polymers, 60(3), 283-289 http://doi.org/10.1016/j. carbpol.2005.01.003.
19 Leopold, N., & Lendl, B. (2003). A new method for fast preparation of highly surface-enhanced Raman scattering (SERS) active silver colloids at room temperature by reduction of silver nitrate with hydroxylamine hydrochloride. The Journal of Physical Chemistry B, 107(24), 5723-5727 http:// doi.org/10.1021/jp027460u
Kataoka, L. F. M. S., Falla, M. D. P. H., & Luz, S. M.
20 Wassei, J. K., & Kaner, R. B. (2010). Graphene, a promising transparent conductor. Materials Today, 13(3), 52-59 http:// doi.org/10.1016/S1369-7021(10)70034-1
21 Ma, X., Deng, Q., Wang, L., Zheng, X., Wang, S., Wang, Q., Chen, L., Huang, L., Ouyang, X., & Cao, S. (2019). Cellulose transparent conductive film and its feasible use in perovskite solar cells. RSC Advances, 9(17), 9348-9353 http://doi. org/10.1039/C9RA01301F PMid:35520713.
22 Kaco, H., Zakaria, S., Chia, C. H., & Zhang, L. (2014). Transparent and printable regenerated kenaf cellulose/PVA film. BioResources, 9(2), 2167-2178 http://doi.org/10.15376/ biores.9.2.2167-2178.
23 Yang, Q., Wang, F., Tang, K., Wang, C., Chen, Z., & Qian, Y. (2003). The formation of fractal Ag nanocrystallites via γ-irradiation route in isopropyl alcohol. Materials Chemistry and Physics, 78(2), 495-500 http://doi.org/10.1016/S02540584(02)00379-6
24 Ludueña, L. N., Vecchio, A., Stefani, P. M., & Alvarez, V. A. (2013). Extraction of cellulose nanowhiskers from natural fibers and agricultural byproducts. Fibers and Polymers, 14(7), 1118-1127. http://doi.org/10.1007/s12221-013-1118-z.
25 Pourfarzad, A., Ahmadian, Z., & Habibi-Najafi, M. B. (2018). Interactions between polyols and wheat biopolymers in a bread model system fortified with inulin: a Fourier transform infrared study. Heliyon, 4(12), e01017 http://doi.org/10.1016/j. heliyon.2018.e01017 PMid:30560212.
26. Su, J.-F., Huang, Z., Yuan, X.-Y., Wang, X.-Y., & Li, M. (2010). Structure and properties of carboxymethyl cellulose/ soy protein isolate blend edible films crosslinked by Maillard reactions. Carbohydrate Polymers, 79(1), 145-153 http://doi. org/10.1016/j.carbpol.2009.07.035
27 Mondal, M. I. H., Yeasmin, M. S., & Rahman, M. S. (2015). Preparation of food grade carboxymethyl cellulose from corn husk agrowaste. International Journal of Biological Macromolecules, 79 , 144-150 http://doi.org/10.1016/j. ijbiomac.2015.04.061 PMid:25936282.
28 Tavares, K. M., Campos, A., Luchesi, B. R., Resende, A. A., Oliveira, J. E., & Marconcini, J. M. (2020). Effect of carboxymethyl cellulose concentration on mechanical and water vapor barrier properties of corn starch films. Carbohydrate Polymers, 246, 116521 http://doi.org/10.1016/j.carbpol.2020.116521 PMid:32747230.
29 Ayala, G., Agudelo, A. C., & Vargas, R. (2012). Effect of glycerol on the electrical properties and phase behavior of cassava starch biopolymers. Dyna, 79(171), 138-147. Retrieved in 2024, March 24, from http://www.scielo.org.co/pdf/dyna/ v79n171/a18v79n171.pdf
30 Van Der Pauw, L. J. (1958). A method of measuring specific resistivity and Hall effect of discs of arbitrary shapes. Philips Research Reports, 13(1), 1-9
31 Choi, S. M., Han, S. S., & Shin, E. J. (2020). Highly stretchable conductive nanocomposite films using regenerated cellulose nanoparticles. ACS Applied Polymer Materials, 2(10), 43874398 http://doi.org/10.1021/acsapm.0c00294
32 Prudêncio, L., Camilo, F. F., & Faez, R. (2014). Líquidos iônicos como plastificantes em blendas de borracha nitrílica/polianilina. Quimica Nova, 37(4), 618-623 http://doi.org/10.5935/01004042.20140103
33. Liu, S., Yu, T., Wu, Y., Li, W., & Li, B. (2014). Evolution of cellulose into flexible conductive green electronics: A smart strategy to fabricate sustainable electrodes for supercapacitors. RSC Advances, 4(65), 34134-34143 http://doi.org/10.1039/ C4RA07017H
34. Zhao, D., Chen, C., Zhang, Q., Chen, W., Liu, S., Wang, Q., Liu, Y., Li, J., & Yu, H. (2017). High performance, flexible, solidstate supercapacitors based on a renewable and biodegradable mesoporous cellulose membrane. Advanced Energy Materials, 7(20), 1700739 http://doi.org/10.1002/aenm.201700739
35 Tian, J., Peng, D., Wu, X., Li, W., Deng, H., & Liu, S. (2017). Electrodeposition of Ag nanoparticles on conductive polyaniline/cellulose aerogels with increased synergistic effect for energy storage. Carbohydrate Polymers, 156, 19-25 http:// doi.org/10.1016/j.carbpol.2016.09.005 PMid:27842813.
36 Mun, S., Zhai, L., Min, S.-K., Yun, Y., & Kim, J. (2016). Flexible and transparent strain sensor made with silver nanowire-coated cellulose. Journal of Intelligent Material Systems and Structures, 27(8), 1011-1018 http://doi.org/10.1177/1045389X15577651
37 Chen, J., Xu, J., Wang, K., Qian, X., & Sun, R. (2015). Highly thermostable, flexible, and conductive films prepared from cellulose, graphite, and polypyrrole nanoparticles. ACS Applied Materials & Interfaces, 7(28), 15641-15648 http:// doi.org/10.1021/acsami.5b04462 PMid:26135618.
38 Lee, T.-W., & Jeong, Y. G. (2015). Regenerated cellulose/ multiwalled carbon nanotube composite films with efficient electric heating performance. Carbohydrate Polymers, 133, 456-463 http://doi.org/10.1016/j.carbpol.2015.06.053 PMid:26344302.
39 Huang, H.-D., Liu, C.-Y., Zhang, L.-Q., Zhong, G.-J., & Li, Z.-M. (2015). Simultaneous reinforcement and toughening of carbon nanotube/cellulose conductive nanocomposite films by interfacial hydrogen bonding. ACS Sustainable Chemistry & Engineering, 3(2), 317-324. http://doi.org/10.1021/sc500681v.
40 Khan, A., Niazi, M. B. K., Naqvi, S. R., & Farooq, W. (2018). Influence of plasticizers on mechanical and thermal properties of methyl cellulose-based edible films. Journal of Polymers and the Environment, 26(1), 291-300 http://doi.org/10.1007/ s10924-017-0953-1.
41 Silva, W. T. A. (2021). Espumas de poliestireno impregnadas com nanopartículas de prata para a redução catalítica de p-nitrofenol em água (Master’s thesis). Universidade Estadual da Paraíba, Campina Grande
42. Costa, F. S. (2016). Desenvolvimento de nanocompósitos de poli(3-hidroxibutirato-co-3-hidroxivalerato) com nanopartículas de prata (Master’s thesis). Universidade Federal de São Paulo, São José dos Campos.
Received: Mar. 18, 2024
Revised: June 18, 2024
Accepted: June 24, 2024
Hai Le Tran1,2,3,4 , Chau Duc Tran2,4 , Cam Hong Thi Nguyen1 , Thao Phuong Le Nguyen1 , Le-Thu Thi Nguyen2,4 , Thiet-Quoc Nguyen5 , Mai Ha Hoang6 , Tam Huu Nguyen1,4 and Ha Tran Nguyen1,2,4*
1National Key Laboratory of Polymer and Composite Materials, Ho Chi Minh City University of Technology – HCMUT, Ho Chi Minh City, Vietnam
2Ho Chi Minh City University of Technology – HCMUT, Ho Chi Minh City, Vietnam
3University of Science, VNU-HCM, Ho Chi Minh City, Vietnam
4Vietnam National University Ho Chi Minh City – VNU-HCM, Ho Chi Minh City, Vietnam
5Institute of Applied Materials Science, Vietnam Academy of Science and Technology, Ho Chi Minh City, Vietnam
6Institute of Chemistry, Vietnam Academy of Science and Technology, Ha Noi, Vietnam
*nguyentranha@hcmut.edu.vn
Obstract
A novel conjugated rod–coil diblock copolymer poly(3-hexylthiophene)-block-poly(furfuryl methacrylate) (P3HT-bPFMA) has been successfully synthesized for the first time using photoinduced organocatalyzed atom transfer radical polymerization (O-ATRP). This process utilized an organic photoredox catalyst of N-aryl phenoxazine, namely 10-(Perylene-3-yl-10H-Phenoxazine, under 365 nm UV irradiation. The diblock copolymer P3HT-b-PFMA was produced efficiently in a controlled manner, resulting in designed average molecular weights and a narrow polydispersity index. Notably, the furfurylmethacrylate (FMA) monomer derived from biomass-based furfuryl compounds was applied for this controlled polymerization, leading to the formation of conjugated diblock copolymers. The synthesized P3HT-b-PFMA was characterized through 1H-NMR, FT-IR, and GPC methods. Furthermore, the optical and hydrophilic-hydrophobic properties of P3HT-b-PFMA were also evaluated through UV-Vis spectroscopy and contact angle measurements.
Keywords: diblock copolymer, poly(3-hexylthiophene), organic photocatalyst, biomass-based monomer, atom transfer radical polymerization.
How to cite: Tran, H. L., Tran, C. D., Nguyen, C. H. T., Nguyen, T. P. L., Nguyen, L. T. T., Nguyen, T. Q., Hoang, M. H., Nguyen, T. H., & Nguyen, H. T. (2024). Organocatalyzed polymerization of diblock copolymer based on poly(3-hexylthiophene) and poly(furfurylmethacrylate). Polímeros: Ciência e Tecnologia , 34(3), e20240029. https://doi.org/10.1590/0104-1428.20240003
1. Introduction
Over the past decades, the conjugated polymer has become a promising organic material for optoelectronic applications[1-5] . This is because they have numerous excellent properties, including high conductive properties in the oxidization state, good solubility in typical solvents, low toxicity, and strong chemical stability in normal environmental conditions. Among conjugated polymers, poly(3-hexylthiophene) (P3HT) has been intensively researched for widespread electronic applications, including organic solar cells, organic field effect transistors, the electrode in lithium batteries, and protective coating[6-10]. Additionally, there have been many reports showing that rod-coil block copolymers based on poly(3-hexylthiophene) are potentially utilized in organic electronics[11-14]. Such block copolymers based on P3HT have contributed advanced properties by improving
mechanical strength and forming self-organized structures in various morphologies on the nanometer scale, such as lamellar, cylindrical and spherical structures[15-17]. Moreover, the incorporation of functionalized coil block segments in the P3HT-based block copolymers can pave the way for the development of optical sensor organic materials[18,19]
The controlled/living radical polymerization methods have been used as primary techniques for synthesizing rod-coil diblock copolymers[12]. Among these methods, atom transfer radical polymerization (ATRP) has been extensively utilized for the synthesis of rod-coil diblock copolymers. ATRP has been proven successful in creating various advanced polymers with welldefined structures and controlled architecture[20-23]. In traditional ATRP, metal catalysts are employed in the polymerization
Tran, H. L., Tran, C. D., Nguyen, C. H. T., Nguyen, T. P. L., Nguyen, L. T. T., Nguyen, T. Q., Hoang, M. H., Nguyen, T. H., & Nguyen, H. T.
process, which archived a control over the molecular weight, the narrow polydispersity index, and controlled end-groups of the resulting polymers. However, such polymers produced through traditional ATRP with transition metal catalysts may retain traces of metal, which causes tremendous damage in subsequent uses of the polymeric products[24]. This poses challenges for applications in biomedicine and optoelectronic fields. To address this issue, organic photocatalyst (O-ATRP/ metal-free ATRP) has been explored and developed for controlled radical polymerization, gradually replacing traditional ATRP with transition metal catalysts[25-29] .
On the other hand, biomass-derived furfuryl methacrylate (FMA), prepared from cellulosic-biomass-derived furfural, emerges as a potential substitute for the current petroleumbased methacrylate monomers, such as methyl methacrylate (MMA) and iso-butyl methacrylate[30]. The FMA monomer owns a reactive functional furfuryl group, making it a valuable material for practical applications such as coatings, sealing, and adhesives[31,32]. However, when FMA monomer undergoes conventional radical polymerization and classical anionic polymerization, it results in insoluble poly(furfuryl methacrylate) (PFMA). This outcome is attributed to excessive chain transfer related to the reactive furfuryl group present in both the polymer and the monomer. To overcome this challenge, atom-transfer radical polymerization (ATRP) of FMA has been employed to produce gel-free polymers with controlled average molecular weight[33]. Pérez et al.[34] reported diblock copolymers consisting of poly(trimethylene carbonate) and poly(furfuryl methacrylate) PFMA end blocks. These copolymers were synthesized through sequential organocatalytic ring-opening polymerization (ROP) and photoinitiated metal-free atom-transfer radical polymerizations (O-ATRP) of furfuryl methacrylate. These copolymers exhibited thermoreversible properties. Additionally, Raffa et al. synthesized a series of copolymers of styrene and furfuryl methacrylate, applied for self-healing materials due to the thermally reversible Diels–Alder (DA) reaction[35] . However, there is a lack of reports on the synthesis and investigation of diblock copolymers based on a coil segment of poly(furfuryl methacrylate) with P3HT as a rod segment.
To address this, we reported the synthesis of rod-coil diblock copolymer utilizing regioregular poly(3-hexylthiophene) (P3HT) as the rod segment and poly(furfuryl methacrylate) (PFMA) polymerized from furfuryl methacrylate as the coil segment. The polymerization of the FMA monomer was conducted through photoinitiated organocatalyzed atom transfer radical polymerization under UV irradiation, employing 10-(Perylene-3-yl-10H-Phenoxazine (PPOZ) as a photocatalyst. The resulting diblock copolymer, termed poly(3-hexylthiophene)-block-poly(furfuryl methacrylate) (P3HT-b-PFMA), was characterized using 1H NMR, FTIR spectroscopies, and GPC analysis. Furthermore, we explored the optical properties of the diblock copolymer through UV-Vis spectroscopy.
2.1 Materials
3-Hexylthiophene, (Diacetoxyiodo)benzene, 3-Bromo perylene, 10H-phenoxazine, furfuryl alcohol,
1,3-Bis(diphenylphosphino)propane nickel(II) chloride, iodine, chloro 1-methylethyl magnesium solution (2 mol/L in THF), methacryloyl chloride, potassium carbonate (K2CO3, 99.5%) and N-bromosuccinimide were purchased from Fisher and Acros Organics. These chemicals were preserved in a glove box at ambient temperature. Anhydrous tetrahydrofuran (THF, 99.8%), 2-2-Bromo-2-methylpropionyl bromide (Br-iBuBr), triethylamine (NEt3, 99%), sodium borohydride (NaBH4, 99%), copper(I) bromide (CuBr, 98%), Palladium(II) acetate (Pd(OAc)2, 98%), tri-tert-butylphosphine (P(t-Bu)3, 97%), sodium tert-butoxide (NaOtBu, 97%), and phosphoryl chloride (POCl3, 99%) were obtained from Sigma-Aldrich. Methanol (99.8%), chloroform (CHCl3, 99.5%), toluene (99.5%), n-hexane (99%), ethyl acetate (99%), and diethyl ether (99%) were acquired from Fisher and Acros Organics. All the solvents were used as received.
TLC analysis was collected on glass surface coated by a fluorescent indicator F-254. FT-IR analysis was carried out on a Tensor 27 Bruker instrument, involving 264 scans with a resolution set at 4 cm-1. For 1H NMR spectra, a Bruker Avance 500 MHz instrument was utilized, employing a solvent of deuterated chloroform (CDCl3) and an internal reference of Tetramethylsilane. GPC analysis was executed by using gel permeation chromatography (Varian Polymer PL-GPC 50) with an RI detector. The GPC experiments employed anhydrous THF as the eluent, with a flow rate of 1.0 mL.min-1. Polystyrene (PS) standards were used to evaluate the average molecular weight and the molecular weight distribution of the resulting polymers. The optical properties of the polymers were assessed through UV–Vis spectroscopy using Agilent UV-Vis 8453 diode array, covering a wavelength range from 190 nm to 1100 nm.
The synthesis procedure involved dissolving furfuryl alcohol (5 mL, 1 eq) in 20 mL of THF, followed by an addition of triethylamine (10 mL, 1.25 eq) to the solution. The mixture was then cooled to 5 °C, and methacryloyl chloride (8.4 mL, 1.5 eq) was dropwise added over 1 h. The reaction proceeded for 16 h at room temperature. Subsequently, the mixture was diluted with 30 mL of dichloromethane and subjected to multiple washes by deionized water (50 mL). Then, the organic phase was dried using anhydrous K2CO3 and subsequently filtrated to collect the anhydrous organic mixture. The solvent was then removed under reduced pressure to obtain the crude product. Purification of the crude product was achieved through silica gel column chromatography using ethyl acetate: n-hexane (1:50) as the eluent, resulting in the isolation of a light-yellow oil with a yield of 95%.
1H NMR (500 MHz, CDCl3): δ (ppm): 1.95 (s, 3H), 5.14 (s, 2H), 5.57 (s, 1H), 6.13 (s, 1H), 6.36 (d, 1H), 6.42 (d, 1H), 7.42 (s, 1H). Elemental Analysis for C9H10O3: Calculated C, 65.05; H, 6.07; O, 28.88. Found: C 66.20; H, 5.96; O, 27.84.
2.4 Synthesis of photocatalyst 10-(perylen-yl)-10Hphenoxazine (PPOZ)
PPOZ was synthesized with a modified synthetic procedure from our previous article[36]. A magnetic stir bar was introduced
Organocatalyzed polymerization of diblock copolymer based on poly(3-hexylthiophene) and poly(furfurylmethacrylate)
into a 25 mL round-bottom flask, which was then evacuated and filled with nitrogen. Subsequently, 8 mL of toluene was added to the reaction flask. The 10H-phenoxazine (160 mg, 0.81 mmol), Pd(OAc)2 (3.61 mg, 0.016 mmol), NaOtBu (116 mg, 1.20 mmol), and P(t-Bu)3 (6.50 mg, 0.032 mmol) were sequentially added to the solution. Following this, 3-Bromo perylene (297 mg, 0.90 mmol) was introduced to the reaction. The reaction proceeded at 110 °C for 4 h. Then, the temperature of the flask was reduced to ambient temperature. Afterwards, the mixture was diluted with CHCl3 (50 mL), washed with deionized water, and dried over K2CO3. The organic layer was then filtered, and the solvent evaporated using a rotary evaporator to obtain the crude product. Purification of the crude product was achieved through column chromatography using a mixture of ethyl acetate: n-hexane (5:95), resulting in the isolation of a white solid powder. The product was further dried at 70 °C under vacuum for 24 h, yielding 85%. 1H NMR (500 MHz, CDCl3): δ (ppm): 5.88 (d, 2H), 6.53 (t, 1H), 6.64 (t, 2H), 6.73 (d, 2H), 7.48 (t, 1H), 7.54 (t, 3H), 7.75 (t, 2H), 7.91 (d, 1H), 8.25 (t, 3H), 8.35 (d, 1H). Elemental Analysis for C32H19NO: Calculated C, 88.66; H, 4.42; N, 3.23; O, 3.69. Found: C, 87.92; H, 4.63; N, 3.36; O, 4.09.
2.5 Synthesis of rod-coil diblock copolymer poly(3hexylthiophene)-block-poly(furfuryl methacrylate) (P3HT-b-PFMA)
P3HT-b-PFMA was synthesized through metal-free atom transfer radical polymerization (ATRP) utilizing the P3HT-macroinitiator and PPOZ as a photoredox catalyst according to our previous report[37]. In a 25 mL flask, 100 mg of P3HT-macroinitiator (Mn 1H NMR = 9000 g/mol, 0.011 mmol) was combined with 5 mL of THF solvent using a syringe, and the solution was stirred
until homogeneous. Then, a solution of FMA (36.5 mg, 0.22 mmol) and PPOZ (2.38 mg, 0.005 mmol) was added. The mixture underwent three freeze-pump-thaw cycles for degassing, followed by continuous stirring until homogeneity was achieved. Subsequently, the solution was inserted in a UV-box (365 nm) for 24 h at room temperature. Finally, the resulting polymer solution was extracted with CHCl3, precipitated into cold methanol, and dried under vacuum, yielding 125 mg of the desired product with a conversion rate of 68%. FT-IR (cm-1): 795, 1014, 1453, 1509, 1561, 1728, 2853, 2922, 2953. 1H NMR (500 MHz, CDCl3), δ (ppm): 6.96 (s, 1H), 3.60 (s, 3H), 2.80 (t, 2H), 1.69 (sex, 2H), 1.49 (q, 6H), 0.89 (t, 3H). GPC: Mn = 12400 g/mol, Ð = 1.42. M n estimated by 1H NMR = 12650 g/mol.
Scheme 1 presents the preparation of the monomer FMA and the synthesis of diblock copolymer poly(3hexylthiophene)-block-poly(furfuryl methacrylate) (P3HTb-PFMA) through O-ATRP using a photocatalyst PPOZ. The P3HT macroinitiator was synthesized using the GRIM method, providing end groups for the macroinitiator[37] The FMA monomer was obtained from an esterification reaction between furfuryl alcohol and methacryloyl chloride in the presence of triethylamine, yielding 95%. On the other hand, the photocatalyst PPOZ was synthesized from 10H-phenoxazine with 3-Bromo perylene through a C-N cross-coupling reaction, resulting in an 85% yield. The PPOZ exhibited maxima absorption at 254 nm, 350 nm, and 440 nm. The obtained results of 1H NMR and gel permeation chromatography (GPC) determined the M n of the P3HT macroinitiator was approximately 9000 g/mol[38]. Subsequently, the P3HT macroinitiator

Tran, H. L., Tran, C. D., Nguyen, C. H. T., Nguyen, T. P. L., Nguyen, L. T. T., Nguyen, T. Q., Hoang, M. H., Nguyen, T. H., & Nguyen, H. T.
was utilized to polymerize of FMA monomer in the presence of the PPOZ with various contents under UV irradiation to produce the diblock copolymers. To achieve a controlled metal-free ATRP, the molar ratio of monomers/ macroinitiator/PPOZ was established as follows: ([FMA]/ [P3HT-macroinitiator]/[PPOZ]) = 100/1/0.01-0.5. The polymerization was conducted at room temperture for 24 h using anhydrous tetrahydrofuran. The resulting diblock copolymers P3HT-b -PFMA were precipitated in cold methanol, followed by filtration under vacuum. Subsequently, the diblock copolymers P3HT- b-PFMA were dried at 70 °C for 24 hours.
Based on the principles of conventional ATRP established by the Pearson et al.[39], and Matyjaszewski group[40], the proposed mechanism for the O-ATRP using the PPOZ as the photocatalyst can be illustrated in Scheme 2. Upon UV irradiation, PPOZ is excited to a state capable of reducing the P3HT-macroinitiator through an oxidation
process, generating an activated radical for polymerization propagation. Additionally, the oxidation of the PPOZ•+Br complex creates a deactivation, leading to the regeneration of P3HT-macroinitiator and the neutral state of PPOZ. This activation and deactivation process maintains control over the radical polymerization. When conducting the O-ATRP for furfuryl methacrylate with a low content of photocatalyst ([FMA]:[P3HT- macroinitiator]:[PPOZ] = 30:1:0.01) in THF sovent, the polymerization conversion was low, which accounted for 10% (Entry 1, Table 1). Increasing the catalyst content to 0.05 equivalents while maintaining other reaction parameters, the polymerization conversion significantly increased to 68% (Entry 2, Table 1). Conversely, at a catalyst content of 0.1 equivalent, the polymerization conversion decreased to 54% (Entry 3, Table 1). The result could be due to the high loading of PPOZ, which generates many radicals, leading to the quenching of polymerization propagation and resulting in polymers with lower molecular

aConversion was calculated by gravimetric method, as follow: Conv = (m - mI - mPPOZ)/mM wherein, m is the weight of product, mI is the weights of the macroinitiator, mPPOZ and mM are the weights of the PPOZ catalyst and monomer, respectively; bNumber–average molecular weight (Mn) and polydispersity index (Đ) as determined by GPC in THF at 35 °C.
Organocatalyzed polymerization of diblock copolymer based on poly(3-hexylthiophene) and poly(furfurylmethacrylate)
weight compared to the theoretical number–average molar mass of the polymer.
The obtained diblock copolymers from the O-ATRP were analyzed via 1H NMR after 24 h. In Figure 1, the peak at 6.97 ppm corresponds to the methine proton of thiophene moieties, while the other peaks at 0.91, 1.35, 1.71, and 2.8 ppm are attributed to the alkyl chain of 3-hexylthiophene. Additionally, the peak at 4.91 ppm is assigned to the methylene of furfuryl methacrylate units, and the peaks at 6.35 ppm and 7.43 ppm correspond to the protons of furfuryl ring moieties, revealing signals corresponding to the PFMA block (peaks c, d, e, and f). Consistent with the results of 1H NMR characterization, the GPC traces of the diblock copolymer P3HT-b-PFMA were found to shift toward higher molecular weights over reaction time (Figure 2). This confirms the successful synthesis of diblock copolymer P3HT-b-PFMA. The determination of the polymerization degree of the PFMA block was conducted on 1H NMR spectroscopy, calculated based on the relative intensities of the methine group of P3HT (peak


7, δ = 6.97 ppm) and that of furfuryl (peak f, 7.43 ppm). The estimated molecular weight (Mn) of the coil polymer block PFMA was calculated to be approximately 3650 g/mol, corresponding to 22 repeating units of furfuryl methacrylate. Consequently, the Mn of diblock copolymers P3HT-b-PFMA was estimated to be around 12650 g/mol. The estimated M n for the P3HT-b-PFMA diblock copolymers aligns reasonably well with the experimental molecular weights evaluated by GPC, although GPC molecular weights are not absolute values.
To investigate the influence of UV light on the organic photocatalyst polymerization, controlled polymerization experiments were conducted by switching on/off the UV light. The experiments used a ratio of [FMA]:[P3HTMacroinitiator]:[PPOZ] = 30:1:0.05 under a nitrogen condition. The polymerization flask experienced replicated cycles of UV illumination exposure for 2 h, followed by periods of darkness for 1 h at each interval to isolate the copolymer product and calculate the polymerization conversion. The copolymers acquired at each time point were analyzed using GPC for a determination of the average molecular weight. The results indicated that monomers were not consumed when the UV light turned off ( Figure 3A ). Additionally, the molecular weight increased when exposed to UV light during the reaction. Furthermore, a linear plot of conversion versus polymerization time confirmed that the FMA polymerization followed first-order kinetics throughout the reaction (Figure 3B). In Figure 3C, the GPC traces of copolymers clearly illustrate a gradual increase in molecular weight with reaction time.
The solubility of the diblock copolymer P3HT-b-PFMA was investigated in various solvents using UV-Vis spectroscopy. The UV-Vis absorption spectrum of P3HT-b-PFMA in various solvents and as a thin film is presented in Figure 4 The results indicate that the diblock copolymers P3HT-bPFMA exhibited good solubility in chloroform (CHCl3), dichloromethane (CH2Cl2), tetrahydrofuran (THF), and toluene. However, in ethyl acetate, the diblock copolymers P3HT-b-PFMA was not completely soluble and precipitated, leading to P3HT chain aggregation. Additionally, the solid thin film of P3HT-b-PFMA showed a red-shift with a maximum absorption at 520 nm and a shoulder absorption at 610 nm. The absorption spectrum of the thin film P3HT-b-PFMA is similar to that of the homopolymer rr-P3HT, which has an optical band gap of 1.9 eV.
The incorporation of a coil polymer into the rod segment could change the hydrophilicity of P3HT-based materials. Therefore, the surface wettability of diblock copolymers P3HT-b-PFMA was examined through contact angle measurements. Figure 5 shows the water contact angle when water was deposited on the surfaces of homopolymer P3HT and diblock copolymers P3HT-b-PFMA. The homopolymer P3HT exhibited a contact angle value of 98.8o throughout the experiment, indicative of hydrophobic properties. In contrast, water contact angles on diblock copolymers P3HT-b-PFMA exhibited a value of 71.8o, indicating a more hydrophilic nature. This result proves that the incorporation of the PFMA segment into the hydrophobic P3HT changes the hydrophilicity of P3HT from hydrophobic to amphiphilic properties.
Tran, H. L., Tran, C. D., Nguyen, C. H. T., Nguyen, T. P. L., Nguyen, L. T. T., Nguyen, T. Q., Hoang, M. H., Nguyen, T. H., & Nguyen, H. T.

Figure 3. (A) Plot of molecular weight of copolymer P3HT-b-PFMA vs time demonstrating the control over polymerization propagation through irradiation ([FMA]:[P3HT-Macroinitiator]:[PPOZ] = 30:1:0.05; (B) First-order kinetic plot of monomer conversion vs time; (C) GPC traces of P3HT-b-PFMA diblock copolymers vs reaction time.

4. The UV-Vis absorption spectra of P3HT-b-PFMA in different solvents and in thin film.
Organocatalyzed polymerization of diblock copolymer based on poly(3-hexylthiophene) and poly(furfurylmethacrylate)

4. Conclusion
In the present work, we have successfully synthesized a new rod-coil diblock copolymer, P3HT-b-PFMA, through O-ATRP using the organic photocatalyst polymerization of furfuryl methacrylate monomers The P3HT-b-PFMA was characterized by using 1H NMR, GPC, FT-IR, and UV-vis methods to evaluate their chemical structure and optical properties. Additionally, the hydrophilicity of diblock copolymer P3HT-b-PFMA was assessed through contact angle measurements. The obtained P3HT-b-PFMA exhibited amphiphilic properties, making them soluble in various organic solvents. This result holds potential significance for electronic circuit printing using novel organic semiconductors.
5. Author’s Contribution
• Conceptualization – Hai Le Tran; Ha Tran Nguyen.
• Data curation – Hai Le Tran; Thiet Quoc Nguyen; Mai Ha Hoang.
• Formal analysis – Hai Le Tran.
• Funding acquisition – Hai Le Tran.
• Investigation – Hai Le Tran; Chau Duc Tran.
• Methodology – Cam Hong Thi Nguyen; Thao Phuong Le Nguyen; Le-Thu Thi Nguyen; Thiet-Quoc Nguyen; Mai Ha Hoang; Tam Huu Nguyen; Ha Tran Nguyen.
• Project administration – Ha Tran Nguyen.
• Resources – Hai Le Tran; Ha Tran Nguyen.
• Software – Thiet-Quoc Nguyen; Mai Ha Hoang.
• Supervision – Ha Tran Nguyen.
• Validation – Ha Tran Nguyen.
• Visualization – Hai Le Tran.
• Writing – original draft – Hai Le Tran; Ha Tran Nguyen.
• Writing – review & editing – Ha Tran Nguyen.
6. Acknowledgements
The research is funded by Viet Nam National University Ho Chi Minh City under grant number C2021-20-17. The authors acknowledge the support of time and facilities from
Ho Chi Minh City University of Technology (HCMUT), VNU-HCM for this study.
1 Pankow, R. M., & Thompson, B. C. (2020). The development of conjugated polymers as the cornerstone of organic electronics. Polymer, 207, 122874 http://doi.org/10.1016/j. polymer.2020.122874.
2 Hildner, R., Köhler, A., Müller-Buschbaum, P., Panzer, F., & Thelakkat, M. (2017). π-conjugated donor polymers: structure formation and morphology in solution, bulk and photovoltaic blends. Advanced Energy Materials, 7(16), 1700314 http:// doi.org/10.1002/aenm.201700314
3 Guo, X., Baumgarten, M., & Müllen, K. (2013). Designing π-conjugated polymers for organic electronics. Progress in Polymer Science, 38(12), 1832-1908 http://doi.org/10.1016/j. progpolymsci.2013.09.005
4 Li, B., Yang, X., Li, S., & Yuan, J. (2023). Stable block copolymer single-material organic solar cells: progress and perspective. Energy & Environmental Science, 16(3), 723-744 http://doi.org/10.1039/D2EE03082A
5 Lipomi, D. J., & Bao, Z. (2017). Stretchable and ultraflexible organic electronics. MRS Bulletin, 42(2), 93-97 http://doi. org/10.1557/mrs.2016.325
6 Agbolaghi, S., & Zenoozi, S. (2017). A comprehensive review on poly(3-alkylthiophene)-based crystalline structures, protocols and electronic applications. Organic Electronics, 51, 362-403 http://doi.org/10.1016/j.orgel.2017.09.038
7 Dang, M. T., Hirsch, L., Wantz, G., & Wuest, J. D. (2013). Controlling the morphology and performance of bulk heterojunctions in solar cells: lessons learned from the benchmark poly(3-hexylthiophene):[6,6]-phenyl-c61-butyric acid methyl ester system. Chemical Reviews, 113(5), 3734-3765 http://doi. org/10.1021/cr300005u PMid:23347135.
8 Wadsworth, A., Hamid, Z., Bidwell, M., Ashraf, R. S., Khan, J. I., Anjum, D. H., Cendra, C., Yan, J., Rezasoltani, E., Guilbert, A. A. Y., Azzouzi, M., Gasparini, N., Bannock, J. H., Baran, D., Wu, H., Mello, J. C., Brabec, C. J., Salleo, A., Nelson, J., Laquai, F., & McCulloch, I. (2018). Progress in poly(3-hexylthiophene) organic solar cells and the influence of its molecular weight on device performance. Advanced Energy Materials, 8(28), 1801001. http://doi.org/10.1002/ aenm.201801001
9 Marrocchi, A., Lanari, D., Facchetti, A., & Vaccaro, L. (2012). Poly(3-hexylthiophene): synthetic methodologies and properties
in bulk heterojunction solar cells. Energy & Environmental Science, 5(9), 8457-8474 http://doi.org/10.1039/c2ee22129b
10 Kleinschmidt, A. T., Root, S. E., & Lipomi, D. J. (2017). Poly(3hexylthiophene) (P3HT): fruit fly or outlier in organic solar cell research? Journal of Materials Chemistry. A, Materials for Energy and Sustainability, 5(23), 11396-11400 http://doi. org/10.1039/C6TA08317J
11 Kang, S., Kim, G.-H., & Park, S.-J. (2022). Conjugated block copolymers for functional nanostructures. Accounts of Chemical Research, 55(16), 2224-2234 http://doi.org/10.1021/acs. accounts.2c00244 PMid:35921179.
12 Liu, C.-L., Lin, C.-H., Kuo, C.-C., Lin, S.-T., & Chen, W.-C. (2011). Conjugated rod–coil block copolymers: Synthesis, morphology, photophysical properties, and stimuli-responsive applications. Progress in Polymer Science, 36(5), 603-637. http://doi.org/10.1016/j.progpolymsci.2010.07.008
13 Yassar, A., Miozzo, L., Gironda, R., & Horowitz, G. (2013). Rod-coil and all-conjugated block copolymers for photovoltaic applications. Progress in Polymer Science, 38(5), 791-844 http://doi.org/10.1016/j.progpolymsci.2012.10.001
14 Dang, M. T., Hirsch, L., & Wantz, G. (2011). P3HT:PCBM, best seller in polymer photovoltaic research. Advanced Materials, 23(31), 3597-3602. http://doi.org/10.1002/adma.201100792. PMid:21936074.
15 Lee, K. S., Lee, J., Choi, C., Seo, Y., Moon, H. C., & Kim, J. K. (2018). Vertically oriented nanostructures of poly(3dodecylthiophene)-containing rod–coil block copolymers. Macromolecules, 51(13), 4956-4965 http://doi.org/10.1021/ acs.macromol.7b02739
16 Cativo, M. H. M., Kim, D. K., Riggleman, R. A., Yager, K. G., Nonnenmann, S. S., Chao, H., Bonnell, D. A., Black, C. T., Kagan, C. R., & Park, S.-J. (2014). Air-liquid interfacial self-assembly of conjugated block copolymers into ordered nanowire arrays. ACS Nano, 8(12), 12755-12762 http://doi. org/10.1021/nn505871b PMid:25486546.
17 Jin, S.-M., Hwang, J. H., & Lee, E. (2023). Crystallizationdriven solution-state assembly of conjugated block copolymers in materials science. Macromolecules, 56(10), 3474-3496 http://doi.org/10.1021/acs.macromol.3c00374
18 Hu, Y.-Y., Su, M., Ma, C.-H., Yu, Z., Liu, N., Yin, J., Ding, Y., & Wu, Z.-Q. (2015). Multiple stimuli-responsive and white-light emission of one-pot synthesized block copolymers containing poly(3-hexylthiophene) and poly(triethyl glycol allene) segments. Macromolecules, 48(15), 5204-5212 http:// doi.org/10.1021/acs.macromol.5b01120
19 Le Nguyen, T. P. L., Bui, T. T., Nguyen, C. H. T., Le, D. T., Nguyen, T. H., Nguyen, L.-T. T., Nguyen, Q.-T., Hoang, M. H., Yokozawa, T., & Nguyen, H. T. (2023). Diblock copolymers poly(3-hexylthiophene)-block-poly(2-(dimethylamino)ethyl methacrylate-random-1-pyrenylmethyl methacrylate), controlled synthesis and optical properties. Journal of Polymer Research, 30(8), 292. http://doi.org/10.1007/s10965-023-03684-9.
20 Moad, G., Rizzardo, E., & Thang, S. H. (2008). Radical additionfragmentation chemistry in polymer synthesis. Polymer, 49(5), 1079-1131 http://doi.org/10.1016/j.polymer.2007.11.020
21 Hawker, C. J., Bosman, A. W., & Harth, E. (2001). New polymer synthesis by nitroxide mediated living radical polymerizations. Chemical Reviews, 101(12), 3661-3688 http://doi.org/10.1021/ cr990119u PMid:11740918.
22. Braunecker, W. A., & Matyjaszewski, K. (2007). Controlled/ living radical polymerization: features, developments, and perspectives. Progress in Polymer Science, 32(1), 93-146 http://doi.org/10.1016/j.progpolymsci.2006.11.002
23 Matyjaszewski, K., & Tsarevsky, N. V. (2014). Macromolecular engineering by atom transfer radical polymerization. Journal
of the American Chemical Society, 136(18), 6513-6533 http:// doi.org/10.1021/ja408069v PMid:24758377.
24 Lorandi, F., Fantin, M., & Matyjaszewski, K. (2022). Atom transfer radical polymerization: a mechanistic perspective. Journal of the American Chemical Society, 144(34), 1541315430. http://doi.org/10.1021/jacs.2c05364. PMid:35882005.
25 Discekici, E. H., Anastasaki, A., Alaniz, J. R., & Hawker, C. J. (2018). Evolution and future directions of metal-free atom transfer radical polymerization. Macromolecules, 51(19), 7421-7434 http://doi.org/10.1021/acs.macromol.8b01401
26 Theriot, J. C., Lim, C.-H., Yang, H., Ryan, M. D., Musgrave, C. B., & Miyake, G. M. (2016). Organocatalyzed atom transfer radical polymerization driven by visible light. Science, 352(6289), 1082-1086 http://doi.org/10.1126/science.aaf3935 PMid:27033549.
27 Treat, N. J., Sprafke, H., Kramer, J. W., Clark, P. G., Barton, B. E., Read de Alaniz, J., Fors, B. P., & Hawker, C. J. (2014). Metal-free atom transfer radical polymerization. Journal of the American Chemical Society, 136(45), 16096-16101. http:// doi.org/10.1021/ja510389m. PMid:25360628.
28 Corbin, D. A., & Miyake, G. M. (2022). Photoinduced organocatalyzed atom transfer radical polymerization (O-ATRP): precision polymer synthesis using organic photoredox catalysis. Chemical Reviews, 122(2), 1830-1874 http://doi.org/10.1021/ acs.chemrev.1c00603 PMid:34842426.
29 Gonçalves, S. A., Rodrigues, P. R., & Vieira, R. P. (2021). Metal-free organocatalyzed atom transfer radical polymerization: synthesis, applications, and future perspectives. Macromolecular Rapid Communications, 42(15), e2100221 http://doi.org/10.1002/ marc.202100221 PMid:34223686.
30. Thakur, S., Chaudhary, J., Singh, P., Alsanie, W. F., Grammatikos, S. A., & Thakur, V. K. (2022). Synthesis of Bio-based monomers and polymers using microbes for a sustainable bioeconomy. Bioresource Technology , 344 (Pt A), 126156 http://doi. org/10.1016/j.biortech.2021.126156 PMid:34695587.
31 Wang, Z., Ganewatta, M. S., & Tang, C. (2020). Sustainable polymers from biomass: bridging chemistry with materials and processing. Progress in Polymer Science, 101, 101197 http://doi.org/10.1016/j.progpolymsci.2019.101197
32 Wang, C.-G., Surat’man, N. E., Wang, S., Li, Z., & Lim, J. Y. C. (2023). Functional polymers from biomass-based monomers. In Z. Li, J. Y. C. Lim, & C.-G. Wang (Eds.), Circularity of plastics (pp. 173-208). London: Elsevier http://doi.org/10.1016/ B978-0-323-91198-6.00009-7.
33 Wang, J., Yuan, L., Wang, Z., Rahman, M. A., Huang, Y., Zhu, T., Wang, R., Cheng, J., Wang, C., Chu, F., & Tang, C. (2016). Photoinduced metal-free atom transfer radical polymerization of biomass-based monomers. Macromolecules, 49(20), 77097717 http://doi.org/10.1021/acs.macromol.6b01997
34 Pérez, A., Lligadas, G., Ronda, J. C., Galià, M., & Cádiz, V. (2022). Thermoreversible poly(trimethylene carbonate)-based block copolymers containing reactive furfuryl groups via sequential ROP and ATRP. European Polymer Journal, 162, 110873 http://doi.org/10.1016/j.eurpolymj.2021.110873
35 Raffa, P., Kassi, A., Gosschalk, J., Migliore, N., Polgar, L. M., & Picchioni, F. (2021). A structure-properties relationship study of self-healing materials based on styrene and furfuryl methacrylate cross-linked via diels–alder chemistry. Macromolecular Materials and Engineering, 306(4), 2000755 http://doi.org/10.1002/mame.202000755
36 Nguyen, C. H. T., Nguyen, T. H., Nguyen, T. P. L., Tran, H. L., Luu, T. H., Tran, C. D., Nguyen, Q.-T., Nguyen, L.-T. T., Yokozawa, T., & Nguyen, H. T. (2023). Aerobic direct arylation polycondensation of N-perylenyl phenoxazine-based fluorescent conjugated polymers for highly sensitive and selective TNT
Tran, H. L., Tran, C. D., Nguyen, C. H. T., Nguyen, T. P. L., Nguyen, L. T. T., Nguyen, T. Q., Hoang, M. H., Nguyen, T. H., & Nguyen, H. T. Polímeros, 34(3), e20240029, 2024
Organocatalyzed polymerization of diblock copolymer based on poly(3-hexylthiophene) and poly(furfurylmethacrylate)
explosives detection. Dyes and Pigments, 219, 111613 http:// doi.org/10.1016/j.dyepig.2023.111613
37. Nguyen, T. H., Nguyen, L.-T. T., Nguyen, V. Q., Phan, L. N. T., Zhang, G., Yokozawa, T., Phung, D. T. T., & Nguyen, H. T. (2018). Synthesis of poly(3-hexylthiophene) based rod-coil conjugated block copolymers via photoinduced metal-free atom transfer radical polymerization. Polymer Chemistry, 9(18), 2484-2493 http://doi.org/10.1039/C8PY00361K
38 Bui, T. T., Nguyen, T. H., Tran, H. L., Tran, C. D., Le, D. T., Dao, D. N., Nguyen, T. P. L., Nguyen, L. T., Nguyen, L.-T. T., Nguyen, T. Q., Cu, S. T., Hoang, M. H., Yokozawa, T., & Nguyen, H. T. (2023). Synthesis of rod–coil conjugated diblock copolymers, poly(3-hexylthiophene)-block-poly(2(4,6-dichlorotriazin-2-yl]oxy)ethyl methacrylate) and click chemistry. Chemicke Zvesti, 77(8), 4439-4456. http://doi. org/10.1007/s11696-023-02793-5
39 Pearson, R. M., Lim, C.-H., McCarthy, B. G., Musgrave, C. B., & Miyake, G. M. (2016). Organocatalyzed atom transfer radical polymerization using N-aryl phenoxazines as photoredox catalysts. Journal of the American Chemical Society, 138(35), 11399-11407. http://doi.org/10.1021/jacs.6b08068. PMid:27554292.
40 Pan, X., Fang, C., Fantin, M., Malhotra, N., So, W. Y., Peteanu, L. A., Isse, A. A., Gennaro, A., Liu, P., & Matyjaszewski, K. (2016). Mechanism of photoinduced metal-free atom transfer radical polymerization: experimental and computational studies. Journal of the American Chemical Society, 138(7), 2411-2425 http://doi.org/10.1021/jacs.5b13455 PMid:26820243.
Received: Mar. 20, 2024
Revised: June 20, 2024
Accepted: July 10, 2024
Glícia Maria Oliveira1 , Alberto Galdino Silva-Junior1,2 , Octávio Luiz Franco3 , José Lamartine de Andrade Aguiar4 , Flávia Cristina Morone Pinto4 , Reginaldo Gonçalves de Lima-Neto5 , Maria Danielly Lima de Oliveira1,2 and César Augusto Souza de Andrade1,2*
1Programa de Pós-graduação em Inovação Terapêutica, Universidade Federal de Pernambuco – UFPE, Recife, PE, Brasil
2Laboratório de Biodispositivos Nanoestruturados, Departamento de Bioquímica, Universidade Federal de Pernambuco – UFPE, Recife, PE, Brasil
3Programa de Pós-graduação em Ciências Genômicas e Biotecnologia, Centro de Análises Proteômicas e Bioquímica de Brasília, Universidade Católica de Brasília – UCB, Brasília, DF, Brasil
4Programa de Pós-graduação em Cirurgia, Departamento de Cirurgia, Universidade Federal de Pernambuco – UFPE, Recife, PE, Brasil
5Laboratório de Pesquisa e Diagnóstico em Doenças Tropicais, Centro de Ciências Médicas, Universidade Federal de Pernambuco – UFPE, Recife, PE, Brasil
*csrandrade@gmail.com
Obstract
This study presents a novel approach to obtaining nano-antimicrobial hybrid material by integrating electrospun nanofibers based on cellulosic biopolymer (BP) associated with antimicrobial agents, specifically silver nanoparticles (AgNPs) and Clavanin A (ClavA), an antimicrobial peptide obtained from the marine tunicate Styela clava. The electrospinning technique produced the blended polyvinyl alcohol:BP nanofibers. Chemical crosslinking was performed to ensure the stability of the nanofibers. The nanofibers had an average diameter of 568 nm for PVA nanofibers and 648 nm for PVA nanofibers functionalized with silver nanoparticles. The nanohybrid material demonstrates significant inhibition zones against Gram-positive (Bacillus subtilis, Staphylococcus aureus) and Gram-negative (Pseudomonas aeruginosa, Klebsiella pneumoniae) bacteria. P. aeruginosa exhibits a substantial inhibition zone of 15 mm. Thus, the nanohybrid material was effective against this challenging pathogen. Combining electrospun nanofibers, bacterial cellulose hydrogel, and antimicrobial agents establishes a solution that could combat microbial threats in wound care.
Keywords: antimicrobial peptide, Clavanin A, electrospinning, silver nanoparticles, sugarcane biopolymer.
How to cite: Oliveira, G. M., Silva-Junior, A. G., Franco, O. L., Aguiar, J. L. A., Pinto, F. C. M., Lima-Neto, R. G., Oliveira, M. D. L., & Andrade, C. A. S. (2024). Development of nano-antimicrobial material based on bacterial cellulose, silver nanoparticles, and ClavaninA. Polímeros: Ciência e Tecnologia, 34(3), e20240030. https://doi.org/10.1590/01041428.20240029
1. Introduction
Chronic or acute wounds create a conducive environment for microbial colonization, posing a severe threat to healing processes. Microorganisms in wounds amplify the risk of infections, complicating treatment and potentially leading to systemic issues[1]. The increase in healthcare expenditures arises from the need for specialized wound care, antibiotics, and surgical interventions[2]. Chronic wounds demand prolonged treatment, escalating healthcare costs and contributing to productivity losses as individuals grapple with persistent health challenges. Additionally, the economic impact is related to increased antibiotic resistance, requiring more resources for research and development of alternative treatments[3] Effectively managing wound-related microbial issues is
imperative for the public health and alleviating the strain on healthcare systems and global economies[2]
Current wound dressings, while effective, often face limitations in addressing diverse wound types and promoting optimal healing. Traditional dressings may lack specificity in managing infections, controlling inflammation, or facilitating tissue regeneration[4]. The need for new alternatives stems from the increasing prevalence of complex wounds, such as chronic ulcers and traumatic injuries, which demand advanced materials and technologies for personalized care. Innovative wound dressings, incorporating bioactive compounds, nanomaterials, and smart polymers, are essential to enhance therapeutic outcomes, reduce healing time, and
L.,
L.
minimize the economic burden associated with prolonged treatments[5]
The electrospinning technique, when associated with synthetic polymers such as polyvinyl alcohol (PVA) and natural polymers such as bacterial cellulose biopolymer, results in biocompatible and biodegradable nanofibers with broad biomedical potential[6]. PVA is a semi-crystalline hydrophilic, biocompatible, non-toxic polymer with remarkable chemical and thermal stability that favors the polymer in multiple medical, cosmetic, food, and pharmaceutical applications[7]
Electrospun nanofibers, with their high surface area and structural similarity to the extracellular matrix (ECM), provide an ideal platform for wound healing applications[8] Mimicking the ECM architecture with a nanometer scale of 50-500 nm in diameter, these nanofibers offer a biomimetic environment that promotes cell adhesion, migration, and tissue regeneration[6]. As healing dressings, electrospun nanofibers facilitate efficient moisture management, enhance drug delivery, and accelerate wound closure, making them promising candidates for advanced wound care solutions[9]
The cellulosic biopolymer (BP), synthesized through the enzymatic action of the bacterium Zoogloea sp. on a sugarcane molasses substrate, demonstrates remarkable potential for biomedical applications[10,11]. Cellulose is a linear homopolysaccharide composed of units of β-D-glucopyranose joined in long unbranched chains with β-glycosidic compounds (1→4). This sustainable and bio-derived material offers versatility in drug delivery systems, tissue engineering, and wound healing due to its biocompatibility and unique mechanical properties. The synthesis process aligns with the growing demand for eco-friendly alternatives in the biomedical field[12] .
In recent decades, nanomaterials have attracted the interest of the biomedical industry due to their applications against various diseases. In this sense, silver nanoparticles (AgNPs) are among the most studied nanomaterials mainly due to their highly efficient antimicrobial characteristics[13-16] Their mechanism of antimicrobial action originated by binding to the negatively charged bacterial cell wall, causing subsequent destabilization of the cell envelope and alteration of the membrane permeability[17].
The innate immune system has key components, such as antimicrobial peptides (AMPs), which form the first line of defense against pathogens. Clavanin A (ClavA) is an antimicrobial peptide (AMP) extracted from hemocytes of the tunicate of the invertebrate Styela Clava, revealed to be an excellent alternative against antimicrobial resistance[18] . The association of the antimicrobial properties of the peptides with AgNPs yields a synergistic approach to combating infections and enhancing wound healing[19]. Antimicrobial peptides exhibit broad-spectrum activity against bacteria, fungi, and viruses, while AgNPs provide sustained release of antimicrobial ions. Incorporating these components into healing dressings, such as electrospun nanofibers, creates a multifaceted solution that prevents infection and accelerates the healing process. Their synergistic action offers a promising avenue for developing advanced wound dressings with improved efficacy and reduced antibiotic resistance concerns[20]
ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) are frequently associated with infections. In addition, these species are known to be drug-resistant[21]. Furthermore, damaged tissue from an injury can acquire infection caused by several bacterial species, such as Pseudomonas spp., Staphylococcus aureus, Klebsiella spp., Escherichia coli, and Proteus spp[22]
The present study focuses on the development, characterization, and evaluation of antimicrobial polymeric electrospun nanofibers based on polyvinyl alcohol (PVA), AgNPs, BP, and ClavA peptide. It focuses on antimicrobial activity against Gram-positive (B. subtilis and S. aureus) and Gram-negative (K. pneumoniae, E. coli, and P. aeruginosa) bacteria.
Silver nitrate (AgNO3), sodium borohydride (NaBH4), potassium ferricyanide (K3[Fe(CN)6]), and potassium ferrocyanide (K4[Fe(CN)6]) were purchased from Sigma Aldrich (St. Louis, MO, USA). Polyvinyl alcohol (PVA) hydrolysis: Mol % = 87-89%; viscosity (20 °C, 4% CP) 40-48 and trisodium citrate were obtained from Dinâmica (Brazil). All chemicals were of analytical grade and used as received. Ultrapure water (18.2 Ω.cm-1) used in the experiments was obtained using a Millipore Milli-Q plus purification system (Billerica, USA).
BP hydrogel from sugarcane molasses and synthesized by Zoogloea sp. through a flotation process[23] was obtained by the Sugarcane Biopolymers Research Group (POLISA, Brazil - https://www.polisa.ind.br). Clavanin A (ClavA) peptide (amino acid sequence VFQFLGKIIHHVGNFVHGFSHVFNH2) was synthesized by Aminotech (São Paulo, Brazil) using the fluorenylmethyloxycarbonyl (F-moc) technique, purified by high-performance liquid chromatography (purity > 95%) and lyophilized.
2.2 Preparation of PVA nanofibers hybrid composite membranes
Initially, 10% PVA was prepared in deionized water, remaining under constant stirring for 2h at 80 °C until a homogeneous solution was obtained. After that, the solution passes through an ultrasonic bath to remove bubbles. Then, the solution was poured into a 25 mL syringe with a metal needle (gauge 18) at room temperature. The needle was fixed at a distance of 15 cm from a grounded collector plate covered with aluminum foil. A voltage of 24 kV was applied to the polymer solution through an electrode connected to the needle. In addition, a fixed flow rate of 0.75 µL.min-1 was used.
The electrospinning process was carried out for ~5h. After, the fiber was removed from the collector and dried in air. Finally, the fibers were subjected to chemical crosslinking in a solution containing glutaraldehyde and hydrochloric acid[24]. After 5 min, the nanofibers were washed thoroughly
Development of nano-antimicrobial material based on bacterial cellulose, silver nanoparticles, and ClavaninA
with deionized water. After drying, the nanofibers were stored at 5 °C until use.
2.3
AgNPs were prepared according to Acharya et al.[25] with some modifications. NaBH4 and tri-sodium citrate were used as reducing and stabilizing agents, respectively. Initially, the nanofibers were cut (1×1 cm2) and inserted into a beaker containing 10 mL of 1 mM AgNO3 and 0.5 mM trisodium citrate and were then subjected to magnetic stirring for 30 min. Then, 1 mL NaBH4 (2 mM) was dripped into the solution, which immediately changed color, indicating the formation of nanoparticles impregnated into the fiber structure. The solution remained stirring for another 30 min. Finally, the fibers were washed three times in deionized water, dried, and stored.
2.4 Functionalization of nanofibers with BP and Clav
The functionalization of the nanofibers was carried out through absorption, with 1mL of BP applied to each disc, and the remaining submerged for 1 hour, then the excess was removed. Peptide incorporation was achieved by adding 10 µL of Clav A peptide (100 µM) to each disc.
2.5 Characterization
Scanning electron microscopy (SEM) was performed using a Tescan MIRA3 microscope 200 V - 30 kV (Czech Republic). The nanofibers were deposited on a sheet and placed directly on a double-sided carbon tape fixed to the sample port. To obtain SEM images, a thin layer of gold was deposited over the samples using an SC-701 Quick Coater metallizer (Sanyu, USA). The diameter distribution of the nanofibers was obtained using ImageJ software. Histograms were prepared using OriginPro9.
Differential scanning calorimetry (DSC) curves were obtained using a calorimeter model DSC-50 (Shimadzu®) at a heating rate of 20 °C.min-1. The experiments were performed under a dynamic nitrogen atmosphere (50 mL.min-1), and a temperature range from 25 °C to 250 °C. The tests were carried out using a sample mass of 2.00±0.10 mg in closed aluminum crucibles. Blank curves were obtained to evaluate the system’s baseline. Indium metal (Tfusion=156.6 °C; Δfusion=28.7 J.g-1) with a purity of 99.99% was used for DSC cell calibration.
Fourier Transform Infrared Spectroscopy (FTIR) measurements were performed using an Agilent Cary 630 FTIR spectrometer (Agilent Technologies, Australia) connected to a diamond-attenuated total reflectance (ATR) sampling accessory. Spectra were recorded between 1.0004.000 cm-1 with a resolution of 2 cm-1 .
For electrochemical analysis, the samples were immobilized in a corrosion cell, with the nanofiber as the working electrode, Ag/AgCl saturated with 3M KCl as the reference electrode, and platinum wire as the auxiliary counter electrode. The electrodes were immersed in the electrochemical cell containing a solution of 10 mM K4[Fe(CN)6]/K3[Fe(CN)6] (1:1, v/v) in phosphate buffer (PBS, pH 7.4), used as a redox probe. The experiments were carried out inside a Faraday cage. Square Wave Voltammetry (SWV) was performed
with a sweep potential between 0 V and 1.2 V with a sweep rate of 50 mV.s-1, step potential 0.010 V, amplitude 0.025 V, and frequency 5 Hz.
The antimicrobial activity was evaluated against S. aureus ATCC 25923, K. pneumoniae ATCC 700603, P. aeruginosa ATCC 27853, E. coli ATCC 35218, and B. subtilis ATCC 6633, the inoculum concentration being used is 108 cells.mL-1. The antibiotics used in the control discs were gentamicin 10µg for gram-positive bacteria, meropenem 10µg for gram-negative bacteria, and 300µg polymyxin. The inhibition zones were evaluated by the disk diffusion method[26]. Initially, the bacterial inoculum was distributed on the surface of Mueller-Hinton Agar and allowed to rest for 3 min. After, the nanofibers were cut into discs and equally distributed on the plate surface, which were incubated at 35 ± 1 °C for 24 h. The growth inhibition halo was measured using a millimeter ruler, and unmodified nanofiber disks were used as a control.
3.1
Figure 1 shows the ATR-FTIR of pure PVA and its mixtures with BP, Ag, and ClavA peptide. The spectrum of pure PVA electrospun nanofibers exhibited bands of O-H stretching vibrations at 3332 cm-1 and C-H stretching vibrations at 2925 cm-1 [26]. A particularity is the existence of close absorption bands at 1740 cm-1 for PVA and 1718 cm-1 for PVA-BP fibers that are generally associated with the stretching vibration of CO carbonyl groups of PVA chains[27]. The increase in the concentration of the other components results in a PVA peak decrease, such as occurring at 1435 cm-1 to 1425 cm-1 in the PVA-BP fibers. Also, a decrease in the intensity of the 840 cm-1 peak of the sample containing PVA to 820 cm-1 corresponding to PVA-BP-Ag-ClavA samples[28]
An intense absorption band was observed at 1376 cm-1 , associated with the presence of the Ag+NO3 ion. The vibration

Figure 1. ATR-FTIR spectra of the tested samples at wavenumbers ranging from 4.000 cm-1 to 1.000 cm-1
Oliveira, G. M., Silva-Junior, A. G., Franco, O. L., Aguiar, J. L. A., Pinto, F. C. M., Lima-Neto, R. G., Oliveira, M. D. L., & Andrade, C. A. S.
peak detected at 1164 cm-1 in samples containing the biopolymer is related to the bending vibration of the C-H and C-O bonds in the aromatic rings of polysaccharides[29]

The peak at 1036 cm -1 can be attributed to the C-O-C and C-O-H stretching vibration of the sugar ring[30]
The morphology of the synthesized nanofibers (Figure 2) was analyzed by SEM (Figure 3). The micrographs showed that the nanofibers in different compositions formed a homogeneous three-dimensional matrix. Agglomerates were not observed for all samples[31] Figure 3 shows SEM images and histograms with the diameter distributions corresponding to the functionalized PVA nanofibers. As expected, the average diameters of the nanofibers varied slightly. The PVA nanofibers had a diameter of 568 nm, while the PVA-Ag nanofibers had the largest diameter measuring 648 nm. The morphology of the AgNPs-containing nanofiber (Figure 3B) shows surface changes suggesting the incorporation of the particles[32]
The images obtained from energy dispersive X-ray spectroscopy (Figure 4) demonstrated the distribution of AgNPs in the nanofibers.
The EDS spectrum presents the elemental analysis of carbon and oxygen peaks corresponding to their binding energies. As predicted, cellulose mainly contains carbon, oxygen, and hydrogen[29]. Elemental mapping revealed the homogeneous dispersion of silver nanoparticles on the surface of the nanofibers[33]
PVA nanofibers favor a decrease in the melting peak due to morphological modification[34]. The thermal behavior of the samples was investigated by measuring endothermic melting events (Figure 5).
The highest melting peak of PVA-Ag-Bp-ClavA nanofibers was 207.03 °C. A similar melting temperature was previously found in a study on the preparation and characterization of PVA microfibers associated with titanium dioxide, where thermograms of the synthesized fibers indicated the melting temperature at 223.7 °C[35]. Another


4. X-ray energy dispersive spectroscopy (EDS) of PVA-Ag-BP nanofibers shows the main chemical elements identified in the sample: C
Ag (green), and O (red). On the right, the elemental map and its respective energy peaks are shown.
study developed electrospun nanofibers with cephalexin prepared from PVA associated with different biopolymers presented a melting temperature of pure PVA nanofibers of 176.50 °C; when associated with carboxymethylcellulose, there was an increase in the temperature of melting to 195.47 °C[31]. A previous study that evaluated the thermal behavior of PVA showed a similar result, with a peak melting temperature of 189 °C[36]. The melting peak of pure PVA nanofibers was 130.92 °C since the presentation of nanofibers favors a decrease in the melting peak due to morphological modification and is associated with changes resulting from crosslinking[34]. According to the melting peak value of the sample containing BP, Ag, and ClavA, it is evident that the thermal stability was significantly higher than that of pure PVA. The increase in thermostability from 130.92 °C to 207.03 °C may be caused by the formation of hydrogen bonds between PVA and the biopolymer[31]
The electrochemical behavior of PVA, Ag, and BP-embedded nanofibers was studied. SWV technique provided values related to surface modification, facilitating the monitoring of current responses as a function of the applied potential and allowing the characterization of the sample’s charge transfer rates[37]. PVA nanofibers have insulating properties[36]. PVA nanofibers have improved electron transport properties when associated with metal nanoparticles[38]. The modifications of the PVA fibers with Ag and BP showed increased peaks (Figure 6), indicating a greater concentration or activity of redox species.
A recent study showed that the electrical conductivity of bacterial cellulose increases when associated with metal nanoparticles[39]
After adding the compounds to the fibers, the degree of interaction can be evaluated through the percentage of relative deviation of the anodic current variation ΔI, using the following Equation 1[40,41]:

Figure 5. Thermal analysis of the PVA, PVA-Ag, PVA-Ag-BP, and PVA-Ag-BP-ClavA.
Table 1 shows the ΔI results for the studied systems. ΔI values revealed an increase in the peak current of fibers containing Ag and BP, when compared to PVA nanofibers.
The antimicrobial activity of the pure PVA fiber and functionalized nanofibers was investigated against Grampositive and Gram-negative bacteria using the disc diffusion method. The results are shown in Figure 7 and summarized in Table 2. PVA fiber did not reveal inhibitory activity against all microorganisms evaluated. On the other hand, functionalized nanofibers showed significant antibacterial
Oliveira, G. M., Silva-Junior, A. G., Franco, O. L., Aguiar, J. L. A., Pinto, F. C. M., Lima-Neto, R. G., Oliveira, M. D. L., & Andrade, C. A. S.
activity. The nanofibers composed of PVA-Ag, PVA-AgClavA, and PVA-Ag-BP showed satisfactory inhibitory action against gram-positive bacteria (S. aureus and B. subtilis). There was a formation of an inhibition zone for P. aeruginosa, with a larger halo of 15mm for PVA-Ag, PVA-Ag-ClavA, and PVA-Ag-BP-ClavA systems. In addition, PVA-Ag-BP showed a halo of 13mm against S. aureus and 12mm for B. subtilis.
PVA-Ag and PVA-Ag-BP-ClavA demonstrated inhibitory activity against K. pneumoniae. Of note, the most common

bacterial species in sepsis are S. aureus (30% of cases, 14% of which are methicillin-resistant) and Pseudomonas spp. (14% of cases)[42]. Of note, the antimicrobial action of AgNPs is linked to the surface area of the nanomaterial. The highest concentrations of Ag+ ions released were identified in AgNPs with greater surface area. Favorably, the low release of Ag + ions was found for AgNPs with low surface area, resulting in failures of antimicrobial characteristics[43]. It is well known that cellulose compounds utilized to functionalize the AgNps-impregnated PVA nanofiber are poorly soluble in agar and water. On the other hand, AgNPs coated in substances that dissolve in water demonstrate advantageous antibacterial properties. It is worth mentioning that bacterial cellulose acts as a physical barrier to prevent infections but does not have any antibacterial features. Therefore, adding recognized bacteriostatic and bactericidal substances to surfaces, like AgNPs and antimicrobial peptides, enables the BC to exhibit antimicrobial activity as therapeutic materials. Thus, these agents were adsorbed by BP matrices due to their large surface area and the presence of hydroxyl groups that can interact chemically and intermolecularly[44,45] .
Table 1. Variation in the anodic current (iPA) of the nanofibers.
Samples iPA (µA) Δi

Development of nano-antimicrobial material based on bacterial cellulose, silver nanoparticles, and ClavaninA
Table 2. Evaluation of the antibacterial activity of the nanofibers, as follows: absence of inhibition halo PVA, PVA-Ag, PVA-Ag-ClavA, PVA-Ag-BP, PVA-Ag-BP-ClavA, Gentamicin (letter G), Polymyxin B (letter P), Meropenem (letter M), and not applicable (-).
Sample
Staphylococcus aureus ATCC 25923
Microorganisms
Pseudomonas aeruginosa ATCC 27853
Klebsiella pneumoniae ATCC 700603
Bacillus subtilis ATCC 6333
PVA 0 0 0 0
PVA-Ag 13mm 15mm 8mm 12mm
PVA-Ag-Clav A 13mm 15 mm 0 12mm
PVA-Ag-BP 13mm 14mm 0 12mm
PVA-Ag-BP-ClavA 12mm 15mm 8mm 10mm
G 20mm - - 17mm
M 30mm 28mm 30mm
P - 14mm 13mm 11mm
Subtitle: 0 = absence of inhibition halo; G = Gentamicin; P = Polymyxin B; M = Meropenem; (-) = Not applicable.
No significant results were obtained for E. coli. The low antibacterial activity against E. coli can be justified by the presence of lipopolysaccharides that defend against antibacterial agents[46]. Gopiraman et al.[34] Investigating the antimicrobial activity of anionic cellulose nanofiber composites coated with silver revealed an absence of halo formation in the studied samples.
Clavanin A is highly effective against Gram-positive bacteria, such as methicillin-resistant Staphylococcus aureus, Gram-negative bacteria and fungi[42]. However, in this study, there was no significant increase in the antibacterial action of the nanofibers functionalized with the peptide; there was a slight increase in the inhibition halo for Pseudomonas aeruginosa.
This study presents a new and eco-friendly alternative for preparing nanofibers with antimicrobial activity based on PVA associated with BP obtained from sugar cane molasses. AgNPs and ClavA peptide were effectively incorporated into the polymeric nanofibers. Modified nanofibers revealed electrical variation with the increase in charge transfer. The nanofibers showed inhibitory activity against microorganisms frequently associated with wound infections such as B. subtilis, S. aureus, P. aeruginosa, and K. pneumoniae. Nanofibers containing ClavA peptide were more effective against P. aeruginosa. The developed nanofibers stand out as potential candidates in the biomedical field, such as wound dressings and surgical instrumentation coatings, environments known to be favorable for the colonization of microorganisms.
• Conceptualization – Glícia Maria Oliveira; César Augusto Souza de Andrade.
• Data curation – Glícia Maria Oliveira; Alberto Galdino Silva-Junior.
• Formal analysis – César Augusto Souza de Andrade, Maria Danielly Lima de Oliveira
• Funding acquisition – César Augusto Souza de Andrade; Maria Danielly Lima de Oliveira.
• Investigation – Glícia Maria Oliveira; Alberto Galdino Silva-Junior.
• Methodology – César Augusto Souza de Andrade; Maria Danielly Lima de Oliveira; Alberto Galdino Silva-Junior; Octávio Luiz Franco; José Lamartine de Andrade Aguiar; Flávia Cristina Morone Pinto; Reginaldo Gonçalves de Lima-Neto.
• Project administration – César Augusto Souza de Andrade; Maria Danielly Lima de Oliveira.
• Resources – César Augusto Souza de Andrade; Octávio Luiz Franco; José Lamartine de Andrade Aguiar; Flávia Cristina Morone Pinto; Reginaldo Gonçalves de Lima-Neto.
• Software – NA.
• Supervision – César Augusto Souza de Andrade.
• Validation – César Augusto Souza de Andrade; Maria Danielly Lima de Oliveira; Alberto Galdino Silva-Junior.
• Visualization – César Augusto Souza de Andrade; Maria Danielly Lima de Oliveira; Alberto Galdino Silva-Junior.
• Writing – original draft – Glícia Maria de Oliveira.
• Writing – review & editing – César Augusto Souza de Andrade; Maria Danielly Lima de Oliveira; Alberto Galdino Silva-Junior.
6. Acknowledgements
The authors are grateful for the support from the Coordination for the FACEPE and the Brazilian National Council of Scientific and Technological Development/ CNPq (grant numbers 304678/2021-0 and 304680/2021-4).
7. References
1 Caldwell, M. D. (2020). Bacteria and antibiotics in wound healing. The Surgical Clinics of North America, 100(4), 757776. http://doi.org/10.1016/j.suc.2020.05.007. PMid:32681875.
2 Sen, C. K., Gordillo, G. M., Roy, S., Kirsner, R., Lambert, L., Hunt, T. K., Gottrup, F., Gurtner, G. C., & Longaker, M. T. (2009). Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair and Regeneration, 17(6), 763-771 http://doi.org/10.1111/j.1524475X.2009.00543.x PMid:19903300.
Oliveira, G. M., Silva-Junior, A. G., Franco, O. L., Aguiar, J. L. A., Pinto, F. C. M., Lima-Neto, R. G., Oliveira, M. D. L., & Andrade, C. A. S.
3 Hurlow, J., & Bowler, P. G. (2022). Acute and chronic wound infections: microbiological, immunological, clinical and therapeutic distinctions. Journal of Wound Care, 31(5), 436-445. http://doi.org/10.12968/jowc.2022.31.5.436 PMid:35579319.
4 Shalaby, M. A., Anwar, M. M., & Saeed, H. (2022). Nanomaterials for application in wound healing: current state-of-the-art and future perspectives. Journal of Polymer Research, 29(3), 91. http://doi.org/10.1007/s10965-021-02870-x
5 Kolimi, P., Narala, S., Nyavanandi, D., Youssef, A. A. A., & Dudhipala, N. (2022). (Year). Innovative treatment strategies to accelerate wound healing: trajectory and recent advancements. Cells, 11(15), 2439 http://doi.org/10.3390/cells11152439 PMid:35954282.
6 Singh, B. K., & Dutta, P. K. (2015). Chitin, chitosan, and silk fibroin electrospun nanofibrous scaffolds: A prospective approach for regenerative medicine. In S. Kalia (Ed.), Chitin and chitosan for regenerative medicine (pp. 151-189). New Delhi: Springer http://doi.org/10.1007/978-81-322-2511-9_7
7. Zhang, W., Ronca, S., & Mele, E. (2017). Electrospun nanofibres containing antimicrobial plant extracts. Nanomaterials, 7(2), 42 http://doi.org/10.3390/nano7020042 PMid:28336874.
8 Gao, C., Zhang, L., Wang, J., Jin, M., Tang, Q., Chen, Z., Cheng, Y., Yang, R., & Zhao, G. (2021). Electrospun nanofibers promote wound healing: theories, techniques, and perspectives. Journal of Materials Chemistry. B, Materials for Biology and Medicine, 9(14), 3106-3130 http://doi.org/10.1039/ D1TB00067E. PMid:33885618.
9 Nadaf, A., Gupta, A., Hasan, N., Fauziya, Ahmad, S., Kesharwani, P., & Ahmad, F. J. (2022). Recent update on electrospinning and electrospun nanofibers: current trends and their applications. RSC Advances, 12(37), 23808-23828. http://doi.org/10.1039/ D2RA02864F PMid:36093244.
10 Oliveira, G. M., Gomes, A. O., Fo., Silva, J. G. M., Silva, A. G., Jr., Lins, E. M., Oliveira, M. D. L., & Andrade, C. A. S. (2023). Bacterial cellulose biomaterials for the treatment of lower limb ulcers. Revista do Colégio Brasileiro de Cirurgiões, 50(1), e20233536 http://doi.org/10.1590/0100-6991e-20233536-en PMid:37222383.
11. Silva, J. G. M., Pinto, F. C. M., Oliveira, G. M., Silva, A. A., Campos, O., Jr., Silva, R. O., Teixeira, V. W., Melo, I. M. F., Paumgartten, F. J. R., Souza, T. P., Carvalho, R. R., Oliveira, A. C. A. X., Aguiar, J. L. A., & Teixeira, Á. A. C. (2020). Nonclinical safety study of a sugarcane bacterial cellulose hydrogel. Research, Society and Development, 9(9), e960997932 http:// doi.org/10.33448/rsd-v9i9.7932
12 Pinto, F. C. M., De-Oliveira, A. C. A. X., De-Carvalho, R. R., Gomes-Carneiro, M. R., Coelho, D. R., Lima, S. V. C., Paumgartten, F. J. R., & Aguiar, J. L. A. (2016). Acute toxicity, cytotoxicity, genotoxicity and antigenotoxic effects of a cellulosic exopolysaccharide obtained from sugarcane molasses. Carbohydrate Polymers, 137, 556-560 http://doi. org/10.1016/j.carbpol.2015.10.071. PMid:26686163.
13 Gurunathan, S. (2019). Rapid biological synthesis of silver nanoparticles and their enhanced antibacterial effects against Escherichia fergusonii and Streptococcus mutans Arabian Journal of Chemistry, 12(2), 168-180. http://doi.org/10.1016/j. arabjc.2014.11.014
14 Behravan, M., Panahi, A. H., Naghizadeh, A., Ziaee, M., Mahdavi, R., & Mirzapour, A. (2019). Facile green synthesis of silver nanoparticles using Berberis vulgaris leaf and root aqueous extract and its antibacterial activity. International Journal of Biological Macromolecules, 124, 148-154 http:// doi.org/10.1016/j.ijbiomac.2018.11.101 PMid:30447360.
15 Kalaivani, R., Maruthupandy, M., Muneeswaran, T., Hameedha Beevi, A., Anand, M., Ramakritinan, C. M., & Kumaraguru, A. K. (2018). Synthesis of chitosan mediated silver nanoparticles
(Ag NPs) for potential antimicrobial applications. Frontiers in Laboratory Medicine, 2(1), 30-35 http://doi.org/10.1016/j. flm.2018.04.002.
16 Singh, H., Du, J., Singh, P., & Yi, T. H. (2018). Extracellular synthesis of silver nanoparticles by Pseudomonas sp. THG-LS1.4 and their antimicrobial application. Journal of Pharmaceutical Analysis, 8(4), 258-264 http://doi.org/10.1016/j.jpha.2018.04.004 PMid:30140490.
17 Guzmán, M. G., Dille, J., & Godet, S. (2009). Synthesis of silver nanoparticles by chemical reduction method and their antibacterial activity. International Journal of Chemical and Biomolecular Engineering, 2(3), 104-111
18. Pasupuleti, M., Schmidtchen, A., & Malmsten, M. (2012). Antimicrobial peptides: key components of the innate immune system. Critical Reviews in Biotechnology, 32(2), 143-171 http://doi.org/10.3109/07388551.2011.594423 PMid:22074402.
19. Xu, J., Li, Y., Wang, H., Zhu, M., Feng, W., & Liang, G. (2021). Enhanced antibacterial and anti-biofilm activities of antimicrobial peptides modified silver nanoparticles. International Journal of Nanomedicine, 16, 4831-4846 http://doi.org/10.2147/IJN. S315839 PMid:34295158.
20 Browne, K., Chakraborty, S., Chen, R., Willcox, M. D., Black, D. S., Walsh, W. R., & Kumar, N. (2020). A new era of antibiotics: the clinical potential of antimicrobial peptides. International Journal of Molecular Sciences, 21(19), 7047 http://doi.org/10.3390/ijms21197047 PMid:32987946.
21 Santajit, S., & Indrawattana, N. (2016). Mechanisms of antimicrobial resistance in ESKAPE pathogens. BioMed Research International, 2016, 2475067 http://doi.org/10.1155/2016/2475067 PMid:27274985.
22. Kalan, L. R., & Brennan, M. B. (2019). The role of the microbiome in nonhealing diabetic wounds. Annals of the New York Academy of Sciences, 1435(1), 79-92 http://doi. org/10.1111/nyas.13926 PMid:30003536.
23 Paterson-Beedle, M., Kennedy, J. F., Melo, F. A. D., Lloyd, L. L., & Medeiros, V. (2000). A cellulosic exopolysaccharide produced from sugarcane molasses by a Zoogloea sp. Carbohydrate Polymers, 42(4), 375-383 http://doi.org/10.1016/ S0144-8617(99)00179-4
24 Ullah, S., Hashmi, M., Hussain, N., Ullah, A., Sarwar, M. N., Saito, Y., Kim, S. H., & Kim, I. S. (2020). Stabilized nanofibers of polyvinyl alcohol (PVA) crosslinked by unique method for efficient removal of heavy metal ions. Journal of Water Process Engineering, 33, 101111 http://doi.org/10.1016/j. jwpe.2019.101111
25 Acharya, D., Mohanta, B., Pandey, P., Singha, M., & Nasiri, F. (2017). Optical and antibacterial properties of synthesised silver nanoparticles. Micro & Nano Letters, 12(4), 223-226 http://doi.org/10.1049/mnl.2016.0666
26 İspir, E., Toroğlu, S., & Kayraldız, A. (2008). Syntheses, characterization, antimicrobial and genotoxic activities of new Schiff bases and their complexes. Transition Metal Chemistry, 33(8), 953-960. http://doi.org/10.1007/s11243008-9135-2
27 Abdul Khalil, H. P. S., Davoudpour, Y., Islam, M. N., Mustapha, A., Sudesh, K., Dungani, R., & Jawaid, M. (2014). Production and modification of nanofibrillated cellulose using various mechanical processes: a review. Carbohydrate Polymers, 99, 649-665 http://doi.org/10.1016/j.carbpol.2013.08.069 PMid:24274556.
28 Qashou, S. I., El-Zaidia, E. F. M., Darwish, A. A. A., & Hanafy, T. A. (2019). Methylsilicon phthalocyanine hydroxide doped PVA films for optoelectronic applications: FTIR spectroscopy, electrical conductivity, linear and nonlinear optical studies. Physica B, Condensed Matter , 571 , 93 -100 http://doi. org/10.1016/j.physb.2019.06.063
Development of nano-antimicrobial material based on bacterial cellulose, silver nanoparticles, and ClavaninA
29 Bai, J., Li, Y., Yang, S., Du, J., Wang, S., Zheng, J., Wang, Y., Yang, Q., Chen, X., & Jing, X. (2007). A simple and effective route for the preparation of poly(vinylalcohol) (PVA) nanofibers containing gold nanoparticles by electrospinning method. Solid State Communications, 141(5), 292-295 http:// doi.org/10.1016/j.ssc.2006.10.024
30 Sofla, M. R. K., Brown, R. J., Tsuzuki, T., & Rainey, T. J. (2016). A comparison of cellulose nanocrystals and cellulose nanofibres extracted from bagasse using acid and ball milling methods. Advances in Natural Sciences: Nanoscience and Nanotechnology, 7(3), 035004. http://doi.org/10.1088/20436262/7/3/035004
31 Ghozali, M., Meliana, Y., & Chalid, M. (2021). Synthesis and characterization of bacterial cellulose by Acetobacter xylinum using liquid tapioca waste. Materials Today: Proceedings, 44(Part 1), 2131-2134 http://doi.org/10.1016/j.matpr.2020.12.274
32 Hameed, M. M. A., Khan, S. A. P. M., Thamer, B. M., AlEnizi, A., Aldalbahi, A., El-Hamshary, H., & El-Newehy, M. H. (2021). Core-shell nanofibers from poly(vinyl alcohol) based biopolymers using emulsion electrospinning as drug delivery system for cephalexin drug. Journal of Macromolecular Science, Part A: Pure and Applied Chemistry, 58(2), 130-144 http://doi.org/10.1080/10601325.2020.1832517
33 Nasikhudin, D., Diantoro, M., Kusumaatmaja, A., & Triyana, K. (2016). Preparation of PVA/Chitosan/TiO2 nanofibers using electrospinning method. AIP Conference Proceedings, 1755(1), 150002 http://doi.org/10.1063/1.4958575
34 Gopiraman, M., Deng, D., Saravanamoorthy, S., Chung, I.M., & Kim, I. S. (2018). Gold, silver and nickel nanoparticle anchored cellulose nanofiber composites as highly active catalysts for the rapid and selective reduction of nitrophenols in water. RSC Advances, 8(6), 3014-3023 http://doi.org/10.1039/ C7RA10489H PMid:35541203.
35 Blanes, M., Gisbert, M. J., Marco, B., Bonet, M., Gisbert, J., & Balart, R. (2010). Influence of glyoxal in the physical characterization of PVA nanofibers. Textile Research Journal, 80(14), 1465-1472 http://doi.org/10.1177/0040517509357654
36 Oliveira, A. H. P., Moura, J. A. S., & Oliveira, H. P. (2013). Preparação e caracterização de microfibras de poli(álcool vinílico)/ dióxido de titânio. Polímeros: Ciência e Tecnologia, 23(2), 196-200 http://doi.org/10.1590/S0104-14282013005000013
37 van Etten, E. A., Ximenes, E. S., Tarasconi, L. T., Garcia, I. T. S., Forte, M. M. C., & Boudinov, H. (2014). Insulating characteristics of polyvinyl alcohol for integrated electronics. Thin Solid Films, 568, 111-116 http://doi.org/10.1016/j. tsf.2014.07.051
38. Abeykoon, S. W., & White, R. J. (2022). Continuous square wave voltammetry for high information content interrogation of conformation switching sensors. ACS Measurement Science Au, 3(1), 1-9. http://doi.org/10.1021/acsmeasuresciau.2c00044. PMid:36817008.
39 Katouah, H. A., El-Sayed, R., & El-Metwaly, N. M. (2021). Solution blowing spinning technology and plasma-assisted oxidation-reduction process toward green development of electrically conductive cellulose nanofibers. Environmental Science and Pollution Research International, 28(40), 56363-56375. http://doi.org/10.1007/s11356-021-14615-w PMid:34050912.
40 Khamwongsa, P., Wongjom, P., Cheng, H., Lin, C. C., & Ummartyotin, S. (2022). Significant enhancement of electrical conductivity of conductive cellulose derived from bamboo and polypyrrole. Composites Part C: Open Access, 9, 100314 http://doi.org/10.1016/j.jcomc.2022.100314
41 Meirinho, S. G., Dias, L. G., Peres, A. M., & Rodrigues, L. R. (2017). Electrochemical aptasensor for human osteopontin detection using a DNA aptamer selected by SELEX. Analytica Chimica Acta, 987, 25-37 http://doi.org/10.1016/j.aca.2017.07.071 PMid:28916037.
42 Miranda, J. L., Oliveira, M. D. L., Oliveira, I. S., Frias, I. A. M., Franco, O. L., & Andrade, C. A. S. (2017). A simple nanostructured biosensor based on clavanin A antimicrobial peptide for gram-negative bacteria detection. Biochemical Engineering Journal, 124, 108-114. http://doi.org/10.1016/j. bej.2017.04.013
43 Silva, O. N., Fensterseifer, I. C. M., Rodrigues, E. A., Holanda, H. H. S., Novaes, N. R. F., Cunha, J. P. A., Rezende, T. M. B., Magalhães, K. G., Moreno, S. E., Jerônimo, M. S., Bocca, A. L., & Franco, O. L. (2015). Clavanin A improves outcome of complications from different bacterial infections. Antimicrobial Agents and Chemotherapy, 59(3), 1620-1626 http://doi. org/10.1128/AAC.03732-14 PMid:25547358.
44 Villarreal-Gómez, L. J., Pérez-González, G. L., Bogdanchikova, N., Pestryakov, A., Nimaev, V., Soloveva, A., Cornejo-Bravo, J. M., & Toledaño-Magaña, Y. (2021). Antimicrobial effect of electrospun nanofibers loaded with silver nanoparticles: influence of Ag incorporation method. Journal of Nanomaterials, 2021, e9920755 http://doi.org/10.1155/2021/9920755
45 Gromovykh, T. I., Vasil’kov, A. Yu., Sadykova, V. S., Feldman, N. B., Demchenko, A. G., Lyundup, A. V., Butenko, I. E., & Lutsenko, S. V. (2019). Creation of composites of bacterial cellulose and silver nanoparticles: evaluation of antimicrobial activity and cytotoxicity. International Journal of Nanotechnology, 16(6-10), 408-420 http://doi.org/10.1504/IJNT.2019.106615
46 Garza-Cervantes, J. A., Mendiola-Garza, G., Macedo de Melo, E., Dugmore, T. I. J., Matharu, A. S., & Morones-Ramirez, J. R. (2020). Antimicrobial activity of a silver-microfibrillated cellulose biocomposite against susceptible and resistant bacteria. Scientific Reports, 10(1), 7281 http://doi.org/10.1038/ s41598-020-64127-9 PMid:32350328.
Received: Mar. 20, 2024
Revised: Jul. 08, 2024
Accepted: Jul. 17, 2024
Riquelme Gomes da Silva1 , Laysa Silva Barboza1 , Pedro Henrique Poubel Mendonça da Silveira1,2 , Marceli do Nascimento da Conceição3 , Roberto Carlos da Conceição Ribeiro3 and Daniele Cruz Bastos1*
1Faculdade de Ciências Exatas e Engenharias, Universidade do Estado do Rio de Janeiro – UERJ, Rio de Janeiro, RJ, Brasil
2Programa de Pós-graduação em Ciência e Engenharia de Materiais, Instituto Militar de Engenharia –IME, Rio de Janeiro, RJ, Brasil
3Coordenação de Rochas Ornamentais, Centro de Tecnologia Mineral – CETEM, Rio de Janeiro, RJ, Brasil
*daniele.bastos@uerj.br
Obstract
This article details the development of hybrid composites with a PLA matrix filled with coffee husks, potassium feldspar, and Bahia Beige marble. Comprehensive analysis included FTIR, hardness, contact angle, density tests, SEM for microstructural insights, and XRF for optimizing raw material compositions. Also, variance analysis was applied in all results. The study revealed that these biodegradable composites hold promise for sustainable applications. Density variations were noted due to particle compaction, and hardness slightly decreased with coffee husks, attributed to uneven component distribution. Increased hydrophilicity was observed with filler addition. SEM confirmed strong interfacial adhesion, and color consistency was maintained. Notably, coffee husks significantly enhanced the degradation rate of PLA, achieving a 100% higher rate compared to pure PLA. The presence of calcium and potassium minerals offers additional benefits for soil health. The study suggests that thermoformed, multi-layered composite capsules can be fully biodegradable, promoting environmental sustainability in coffee capsule production.
Keywords: biodegradability, poly(lactic acid), polymer composite sustentability.
How to cite: Silva, R. G., Barboza, L. S., Silveira, P. H. P. M., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C. (2024). Biodegradable hybrid PLA composites incorporating coffee husks and mineral fillers. Polímeros: Ciência e Tecnologia, 34(3), e20240031. https://doi.org/10.1590/0104-1428.20240020
Coffee husks among the most consumed beverages worldwide, offering a wide range of products and consumption opportunities. Over the past decades, coffee has evolved from a simple commodity into a specialized product, ushering in a new era of consumption where quality, flavor, and aesthetics are the primary aspects sought after in the quest for the perfect brew. With advancements in technology, new methods of coffee preparation have emerged, including the popular coffee capsule, which enables the rapid and convenient production of hot coffee[1,2] .
However, the rapid growth in coffee capsule consumption has given rise to a series of environmental issues stemming from improper disposal of these capsules. Coffee capsules are composed of a mixture of materials, including plastics, aluminum, paper, and organic materials, which pose significant recycling challenges. In Brazil, despite the potential capacity to recycle 60% to 75% of these capsules, the actual return rate is only 11%[3,4]. These environmental concerns have compelled coffee companies to implement alternative recycling programs, with industry leaders such as Nespresso establishing numerous collection points for used capsules. Nonetheless, a substantial portion of these
capsules often ends up in landfills after the aluminum is recovered for recycling purposes.
Plastic capsules are primarily made from polypropylene (PP), a low-cost material typically combined with ethylene vinyl alcohol and coated with a layer of aluminum/ polyethylene to enhance oxygen barrier properties. This packaging plays a crucial role in preserving the aroma of coffee during its storage period, minimizing the loss of highly volatile compounds and the ingress of oxygen[5-7] . To address environmental concerns, the use of biodegradable plastics in specific applications, such as soil cover films, trash bags, and disposable packaging, has been proposed as an advancement in the field of bioeconomic, reducing the carbon footprint. A notable example of a bioplastic that is both renewable-based and biodegradable is polylactic acid (PLA)[8-13]
However, PLA exhibits some limitations compared to petroleum-derived plastics, such as low thermal stability and inadequate barrier properties, particularly in hightemperature environments. To overcome these challenges, researchers have explored the development of composites
Silva, R. G., Barboza, L. S., Silveira, P. H. P. M., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C.
and nanocomposites using biodegradable polymers like PLA, reinforced with materials from renewable sources. Additionally, hybrid composites have been developed by blending natural, synthetic, or a combination of fibers in a single matrix to enhance the physical, mechanical, and thermal properties of these materials[14-16]. The hybridization of high-aspect-ratio fibers with low-aspect-ratio particulate mineral fillers is an established industrial technique for minimizing anisotropic effects, with common examples of low-aspect-ratio particulate fillers used to modify thermoplastics including calcium carbonate, talc, mica, and glass beads[16-21].
Furthermore, coffee husks, representing 12% to 18% of the dry weight of a coffee cherry, contain significant quantities of carbohydrates, fibers (cellulose, hemicellulose, and lignin), and proteins[22]. They are considered renewable, biodegradable materials and have a lower environmental impact when compared to synthetic fibers. These components are akin to those found in most lignocellulosic fillers used in the production of polymer-based composites, making coffee husks a promising alternative to conventionally employed materials[23-25]. Simultaneously, stone processing industries generate solid waste and stone slurry during their dimensioning process, which often ends up in landfills or other disposal sites, causing significant environmental impacts[16,26-28]
In this context, the novelty of this study is the production of biodegradable hybrid PLA composites PLA as a matrix incorporating coffee husks waste and minerals rich in potassium and calcium. These composites have the potential to be utilized in food packaging materials, such as coffee capsules, providing an eco-friendly alternative to conventional options. Additionally, recycled minerals could be employed as soil seedling fixatives and as sources for releasing micronutrients. This research aims to contribute to environmental sustainability and foster innovative solutions in the food and packaging industries.
2.1 Raw materials
The PLA used in this work as a polymeric matrix was purchased from Minnesota (USA, grade 2003D, Mn 88,500 Da and Mw/Mn 1.8; density 1.24 g.cm-3, and used as received, in pellet form. The organic waste, Arabic coffee husks (CH), with density of 1.4554 ± 0.0007 g.cm3, were provided by the company Unique Cafés Especiais (São Lourenço, MG, Brazil). Coffee husks were ground with industrial knives to a particle size of less than 2 mm. The mineral samples were donated by CETEM (Rio de
Janeiro, Brazil). To enrich the composite with calcium, waste from the cutting of Bahia beige marble (BB), with density of 2.832 ± 0.0005 g.cm-3 (Ourolândia, Bahia), was used, with granulometry of less than 20 μm. Also, potassium from a potassium feldspar (PF) (Serra Negra, Sergipe, Brazil), with granulometry of less than 20 μm and density of 2.8608 ± 0.0015 g.cm-3, was used.
The materials were previously dried in an oven with forced air circulation at 100 °C until constant weight for 24 h, and stored in a desiccator for another 24 h before processing. The materials were mixed manually, and the composites were compounded with PLA/coffee husks/ potassium feldspar/Bahia beige marble. (PLA/CH/PF/ BB) in different proportions, as described in Table 1. Each formulation was fed into a twin-screw extruder (TeckTril, DCT model, L/D=40, D=20) equipped with ten heating zones, ranging from 95 to 190 °C, from the feed to die, and rotating at 47 rpm. The specimens for characterization were obtained by pressing 10 g of pelletized samples from the extrusion at a temperature of 190 °C for 300 s, with pressure of 70 KPa for film formation, and then cooled in a cold press for 60 s.
2.3
The mineralogical composition of Bahia beige, potassium feldspar and coffee husks were evaluated by the X-Ray fluorescence (XRF). This analysis was carried out with an Axios Max Panalytical (WDS-2) spectrometer operating at 4 kW. The calcination loss was determined with a Leco TGA-701 thermogravimetric analyzer with two heating ramps, one from 25-107 °C at 10 °C/min and the second from 107-1000 °C at 40 °C/min. The test ended after three identical sequential weights.
2.4
The morphological analysis (SEM) of raw materials and processed composites was conducted using a Tescan Vega 3 microscope. The samples were previously cryogenically fractured into transverse sections and then coated with gold in a sputtering chamber. Images were obtained using an electron beam power of 15kV and magnifications of 200 and 1500x.
Fourier-transform infrared spectra were acquired using a Nicolet 6700 FTIR spectrometer (Thermo Scientific). The samples were mounted on an attenuated total reflectance (ATR) accessory equipped with a ZnSe crystal prior to scanning. The spectra were obtained with an accumulation
Biodegradable hybrid PLA composites incorporating coffee husks and mineral fillers
of 120 scans. Density analyses were conducted at 23 °C following the guidelines of ASTM D792[29]. A Gehaka DSL910 densimeter was employed. Five samples for accurate density determination represented each group. The Shore D hardness tests were conducted in accordance with ASTM D2240-15[30], utilizing a GS-702 durometer. The arithmetic mean of five measurements was calculated for each sample. The wettability of the materials surface was examined through water contact angle measurements using a Ramé-Hart NRL A-100-00 goniometer. The evolution of the droplet shape was recorded with a CCD camera every 10 s for a period of 90 s for each sample, at room temperature.
A spectrophotometer (Delta Vista 450G) was used to measure the color parameters of the specimens using a highefficiency LED source and D65, A, C and F7 illuminants at 10º angle and 16 mm aperture. The color measurement of PLA and composites was performed according to the CIElab method (Commission Internationale de L’Eclairage) which allows obtaining the parameters L*, referring to luminosity, which varies from black (0) to white (100); a*, which is the intensity of the red(+)/green(-) color; and b*, the intensity of the yellow(+)/blue(-) color[26]. Laboratory-accelerated soil degradation studies were conducted at 25 °C in microbially active soil beds (garden soil with 2% humus content, 2224% water, pH 5.6, stored in glass containers). PLA and composite samples (mass 0.322-0.340 g and thickness 0.8-1.1 mm) were removed from containers, washed with distilled water, cleaned by sonication, dried and weighed. Aliquots of the acid solution were collected to monitor the levels of potassium and calcium by inductively coupled plasma (ICP) analysis, initially and after 30 days.
Variance analysis (ANOVA) was applied in all results to verify, with a 95% confidence level, any significant differences between the averages. In positive cases, the mean values of the results were then compared using the Tukey’s test, also called honestly significant difference (HSD). This is calculated by Equation 1, in which “q” is a tabulated constant, “EMS” is the error mean square and “r” is the repetitions number for each condition [31].
3.1 XRF results
Table 2 provides the results of XRF analysis for samples of coffee husk, potassium feldspar, and Bahia Beige marble. The following data reveal information about the chemical composition of these samples. XRF analysis of Bahia Beige, as shown in Table 2, indicated that calcium constitutes approximately one third of the total sample composition, also accounting for about 47 wt.% during calcination, which is related to carbonates. This result suggests that the material naturally occurs in the form of calcium carbonate, known as calcite.
Table 2. Chemical composition (wt.%) of the raw materials used, as determined by XRF.
CH = coffee husks; PF = potassium feldspar; BB = Bahia beige.
As for potassium feldspar, which is used as a source of potassium, the analysis revealed that it contains approximately 4 wt.% K2O and 62.6 wt.% SiO2 in its composition. These values are consistent with the use of this mineral as a source of potassium in various applications. Coffee husks, on the other hand, exhibit a composition characterized by a high concentration of carbon, accounting for 90 wt.% of the sample. Additionally, the analysis reveals that they contain approximately 2 wt.% of SiO2 and 4.30 wt.% of K2O. The presence of 0.83 wt.% of CaO contributes to the overall structure of the husks. The high value of Loss on Ignition (LOI) of 89.9 wt.% is indicative of typical thermal decomposition of organic materials[32]
Regarding potassium feldspar, its composition is marked by a high concentration of SiO2 at 62.6%. Other significant oxides include Al2O3 and K2O, representing 14.30 and 4.00 wt.%, respectively. Oxides such as Fe2O3 and CaO are also present, with fractions of 6.90 and 4.90 wt.%, respectively. Other oxides, such as Na2O, are found in smaller quantities. The LOI of 0.34 wt.% is attributable to the ceramic characteristics of potassium feldspar.
Figure 1 shows the FTIR-ATR spectra of coffee husk, potassium feldspar and bahia beige raw materials.
Figure 1 presents the FTIR spectra of the starting materials in the range of 500 to 4000 cm-1, encompassing all relevant absorption bands. In the infrared spectrum of potassium feldspar, a series of overlapping bands between 650 and 1000 cm-1 are prominent. The absorption band in the region of 1250 to 830 cm-1 can be attributed to the asymmetric stretching vibration of Si-O groups, with a peak maximum at 998 cm-1, the symmetric stretching at 800 and 780 cm-1, and the symmetric and asymmetric bending modes
Silva, R. G., Barboza, L. S., Silveira, P. H. P. M., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C.


of Si-O at 650 cm-1, respectively[33,34]. The bands found at 2846 and 2915 cm-1, both in potassium feldspar and bahia beige, are related to the vibrations corresponding to CH2 and CH3, and have been previously recorded in polypropylene composites reinforced with bahia beige. The peak at 987 and 1047 cm−1 corresponds to C=C bending, while the peak at 1081 cm−1 corresponds to C-O stretching. These three bands are commonly found in marbles[35]
The obtained infrared spectrum for coffee husk exhibits similarities with previously documented records in literature[36,37]. The analysis has revealed the presence of absorption peaks attributable to various chemical compounds, such as caffeine, carbohydrates, and proteins, all of which have been previously explored in publications related to the coffee theme. The spectral range situated between 3600 and 3000 cm-1 reflects the presence of water and the presence of OH (hydroxyl) groups on the surface of coffee husk, with a peak observed at 3301 cm-1. Such behavior is reported by Marchi et al.[38] for epoxy matrix composites reinforced with ubim fibers (Geonoma Baculífera), but it occurs generally in all lignocellulosic fibers. The peak located at 2935 cm-1 is associated with the presence of lignocellulosic components, such as in coffee husk[39]. Caffeine also exhibits characteristic absorptions in the spectral regions of 1731 and 1596 cm-1 . It is noteworthy that wavenumbers in the range of 17001600 cm-1 have a known correlation with the concentration of chlorogenic acid in coffee samples, as discussed by Capek et al.[40]. In addition to these observations, notable absorbance peaks were detected at 1241, 1014, and 771 cm-1 , which may be associated with components such as sucrose in the 1242-1218 cm-1 range, and arabinogalactans at 10651020 cm-1 [41-43] Table 3 summarizes the absorption bands related to the functional groups of each of the reinforcement materials used.
FTIR spectra of PLA showed characteristic absorption bands of asymmetric and symmetric C=O and C-O 1747 and 1079 cm-1, respectively (Figure 2). A peak found at 1747 cm-1 in PLA is related to C=O stretching. The addition of coffee husk may contain functional groups capable of forming hydrogen bonds with the carbonyl group of PLA. These hydrogen bonds can modify the molecular structure of PLA,
Material Absorption Range Functional Group/ Vibration
Coffee Husk 3600-3000 O-H; Water
Coffee Husk 2935 Caffeine
Coffee Husk 1731, 1596 Caffeine
Coffee Husk 1700-1600 Chlorogenic acid
Coffee Husk 1241 Sucrose
Coffee Husk 1014 Arabinogalactans
Coffee Husk 771 Arabinogalactans
Potassium Feldspar 1250-830 Asymmetric stretching of Si-O
Potassium Feldspar 998 Asymmetric stretching of Si-O
Potassium Feldspar 800, 780 Asymmetric stretching of Si-O
Potassium Feldspar 655 Symmetric and asymmetric bending of Si-O
Potassium Feldspar 2846, 2915 CH2 and CH3 vibrations
Bahia Beige 2846, 2915 CH2 and CH3 vibrations
Bahia Beige 987, 1047 C=C bending
Bahia Beige 1081 C-O stretching
altering the spectroscopic characteristics of the C = O band, consequently reducing its intensity in the composites[44] Such behavior is also observed in the peaks at 1380 and 1349 cm−1 of PLA, where these peaks represent C-H stretching. These peaks lost intensity with the addition of coffee husk. The bands at 867 and 755 cm-1, were attributed to the crystal and amorphous phases of neat[45,46]. The infrared bands of BB displayed results consistent with the characteristic bands of limestone, namely 1454 - 1380/867 - 755 cm-1 (carbonate ion stretching). The composites exhibited a physical interface as no changes were observed in the infrared peaks[47,48] A decrease at peak strength occurred with increasing amount of natural fiber in composites, suggesting physical interaction. Table 4 summarizes the absorption bands found in PLA and the composites, along with their relationship with the functional groups present in each band.
Table 5 presents the results obtained in the density, hardness, and contact angle tests.
The results obtained for the studied composites were compared with the values of the traditional coffee capsule made of Polypropylene (PP) found in the market. The density values of the composites ranged from 1.138 to 1.259 g/ cm3, with density decreasing as the filler content in the composites increased. Similar density results were found by Chagas et al.[16] when studying hybrid thermoplastic composites based on polypropylene with additions of Bahia Beige and coconut fiber. In their study, the authors found density values ranging from 0.829 to 1.146 g/cm3. Density variations are attributed to differences in particle compaction and particle wall roughness[49,50]
Material Absorption
PLA 1747 C=O stretching
PLA 1079 C-O stretching
PLA 1380, 1349 C-H stretching
PLA 867, 755 Crystalline and amorphous phases
Composites 1746 C=O stretching (reduced)
Composites 1380, 1349 C-H stretching (reduced)
Composites 1454 Carbonate ion stretching
Composites 867-755 Carbonate ion stretching
Table 5. Results of density, hardness and contact angle of formulations.
Based on the density results presented in Table 5, Table 6 provides the analysis of variance for the density values of the composites.
The equality hypothesis, with a confidence level of 95%, was confirmed as the calculated F value was lower than the tabulated Fc value. This indicates that there was no significant difference in the inclusion of coffee husk, potassium feldspar, and bahia beige reinforcements in the PLA matrix. For a more detailed analysis, a comparison of means was conducted using the Tukey test, as shown in Table 7
From the results of the Tukey test, it is observed that groups A and B are statistically different from group C, as they do not share the same letter designation for a certain statistical equality. Within groups A and B, there are no significant differences in mean density, as they share the same letter designation. However, group B contains various compositions of PLA and coffee husk, with slightly decreasing densities as the percentage of coffee husk increases. This trend indicates that the incorporation of coffee husk affects the density of the composite materials. This Tukey analysis is conducted as a complementary approach to ANOVA, as it allows for the comparison of means and, based on the minimum significant difference, determines statistical equality/ difference from this difference in mean values. However, this increase will exert little influence in practical applications such as coffee capsules. The low density inherent to plastic materials renders them advantageous in applications where weight reduction is imperative, particularly in industries such as packing, automotive and aerospace.
Capsule = traditional coffee capsule made of PP.
The results of the Shore D hardness test showed that the hardness of the coffee capsules is higher than that of PLA and the composites. The capsules exhibited hardness values of 80.67 Shore D, while PLA had an average value of 78.67 Shore D. The addition of 10 wt.% coffee husk did not result in changes in the hardness of the composite, remaining the same as PLA. Additions of 20, 30, and 40 wt.% coffee husk, combined with potassium feldspar and Bahia beige, led to a reduction in the hardness of the hybrid composites due to the possible uneven distribution of reinforcements in the polymer matrix. The increase in filler content in the matrix decreases the hardness of the composite, as the interaction of
Table 6. ANOVA results for the density of Coffee Capsule and the studied composites.
Table 7. Comparison of means by Tukey test for the density values of the composites and coffee capsule.
Silva, R. G., Barboza, L. S., Silveira, P. H. P. M., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C.
studied composites.
polymer molecules is hindered, mainly due to the presence of organic fibers from coffee husks that are further away from the PLA chains. However, the incorporation of these organic reinforcements at high levels facilitates the degradation of the composites in agricultural soils, as expected, since the aim is to facilitate biodegradation[16,51,52] .
Table 8 provides the ANOVA results for the hardness values of the composites.
The equality hypothesis, with a confidence level of 95%, was confirmed as the calculated F value was lower than the tabulated Fc value. This indicates that there was no significant difference in the inclusion of coffee husk, potassium feldspar, and bahia beige reinforcements in the PLA matrix. The mean comparison of hardness values by Tukey test is shown in Table 9
The results of the Tukey Test indicate that all analyzed conditions have very similar means. The grouping designation “A” suggests that there are no statistically significant differences between the means of these groups. The decrease in hardness of PLA and composites compared to the coffee capsule is not very significant when observed through statistical analysis.
The contact angle results revealed that the values obtained for PLA are consistent with those reported by Manju et al.[34], and are similar to the contact angle observed in the commercial coffee capsule. The variation in the contact angle of the composites was attributed to the rough surface texture caused by the presence of fillers. According to Kadea et al.[53], a rough surface could facilitate the diffusion of moisture or water into the material, thereby accelerating degradation. Regarding the contact angle, values below 90º were recorded, indicating the hydrophilicity of the composites, similar to the commercial capsule, that probably receive additives during processing, reducing the contact angle if compared to neat PP contact anlge. Additives can impact the biodegradability of polymers[54]
However, the addition of fillers resulted in a decrease of approximately 10º in the contact angle values, enhancing the hydrophilic characteristic. Siakeng et al.[54] demonstrated that a PLA and pineapple leaf fiber biocomposite degraded faster than pure PLA. This implies that these materials, when in contact with soil, are likely to degrade more readily, as they readily absorb water and consequently decompose more quickly, releasing micronutrients (calcium, potassium, and carbon) into the soil.
Table 10 provides the ANOVA results for the contact angle values of the composites.
The equality hypothesis, with a confidence level of 95%, was confirmed as the calculated F value was lower than the tabulated Fc value. This indicates that there was no significant difference in the inclusion of coffee husk, potassium feldspar, and bahia beige reinforcements in the
Table 9. Comparison of means by Tukey test for the hardness values of the composites and coffee capsule.
Mean Groups
Coffee Capsule 80.67 A
80/10/5/5 78.67 A PLA 78.67 A
70/20/5/5 74.33 A
60/30/5/5 71.33 A
50/40/5/5 69.33 A
PLA matrix. The mean comparison of contact angle values by Tukey test is shown in Table 11
Based on the comparison of means, it is concluded that there is a statistically significant difference between groups A and B. The composites in group A tend to have higher contact angles compared to the composites in group B. This may indicate greater hydrophobicity in the composites of group A compared to those in group B. These findings are important for understanding how different composite compositions can affect surface contact properties, which may have implications in specific applications such as coatings or packaging materials.
Due to the lower water contact angle of the Biocomposites, moisture or water in the soil can more easily diffuse into the material, accelerating degradation[53]
Figure 3 shows the SEM micrographs of coffee husk, potassium feldspar and bahia beige.
Such findings are reported in the work of Santos et al.[47] , who developed polypropylene matrix composites reinforced with Bahia Beige, obtaining a visual grain distribution similar to that presented in the current study.
The SEM micrographs of coffee husk reveal particles formed from agglomerated fibers of irregular lengths, along with some smaller particles remaining after the blade milling process. These results indicate that coffee husk exhibits a tendency to exfoliate when subjected to mechanical forces, leading to the formation of smaller fibers and particles[55] . The micrograph at 1500x magnification provides a detailed view of the surface of various coffee husk fibers, varying in shapes and sizes, that have clustered together. The rough surface displays a pattern of valleys and peaks distributed across its entire extent. This surface irregularity is particularly advantageous when seeking effective mechanical adhesion to the polymeric matrix, especially in the absence of chemical fiber treatment or the use of compatibilizing agents[56]
The SEM image of potassium feldspar reveals the particle distribution. It can be observed that smaller-sized particles tend to agglomerate, while larger-sized ones do

Table 10. ANOVA results for the contact angle of Coffee Capsule and the studied composites.
not exhibit this tendency[57]. In general, potassium feldspar particles have dimensions on the order of a few tens of micrometers. The images of Bahia Beige showcase the grain morphology, highlighting an irregular distribution with the presence of larger grains, suggesting incomplete grain comminution. Figure 4 shows the fracture micrograph of the coffee capsule and the analyzed composites.
In the SEM micrograph of the coffee capsule, we observe a slightly rough surface on the polypropylene due to processing. This behavior was previously observed in the study by Silveira et al.[58], where polypropylene composites reinforced with hemp fibers functionalized with maleic anhydride were processed. The processing of the pure polymer resulted in a structure with low porosity and high uniformity, but the addition of fibers and the compatibilizing agent led to the formation of porosity.
The images of the PLA show a surface with little roughness, with some stress propagation marks in the fractured region. The addition of reinforcements increased the surface roughness of the analyzed composites. In the images of the 80/10/5/5 composite, we can observe particles attached to the material at the fracture region. This indicates good interfacial adhesion in the composite, as there was little material detachment during fracture. The increase in
Table 11. Comparison of means by Tukey test for the hardness values of the composites and coffee capsule.
70/20/5/5 76.70 A
PLA 74.80 A
Coffee Capsule 74.10 A
60/30/5/5 65.85 B
80/10/5/5 64.25 B
50/40/5/5 64.02 B
coffee husk concentration not only increased the roughness of the composites but also the porosity. This is evident in the images of the 70/20/5/5, 60/30/5/5, and 50/40/5/5 composites, where we can see the presence of pores.
The color parameters in Table 12 show L*, a* and b* values of the composites. The L* values of PLA film without particles were 85.42 (D65), 85.6 (A), 85.41 (C) and 85.36 (F7).
The lightness was greater in the PLA composites with all illuminants used. The L* value decreased, because distributed
Silva, R. G., Barboza, L. S., Silveira, P. H. P. M., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C.

particles act as a light barrier in PLA composite films, as shown in the SEM image in Figure 4. The morphology of composites can be explained by the good distribution of particles in the PLA matrix. The PLA composites had a light brown color because a* and b* values increased compared to pure PLA, which is related to the increased amount of coffee husks. The analysis of samples using different illuminants (D65, A, C and F) enabled evaluating variations in the color of the material under routine daily conditions.
The morphology of composites can significantly influence the results of a colorimetric analysis in various ways. The distribution, size, and shape of the filler particles affect color uniformity, light reflection, and absorption. Rougher or more complex surfaces can diffuse light differently, altering the perception of color. The opacity and transparency of the composite, influenced by particle concentration, also impact color intensity and hue. The color of the filler particles directly modifies the final color of the
material, and internal light reflection can intensify certain colors or cause iridescence[59]. Color consistency pertains to the extent to which the color of a product or material remains uniform across different batches, manufacturing runs, or viewing conditions. It denotes the degree of adherence to a specified standard or reference in terms of color accuracy over time and under varying circumstances. Maintaining color consistency is imperative in numerous industries, including packing, printing, textiles, and cosmetics, to ensure that products consistently meet stringent quality standards and satisfy customer expectations. Taking D65 as the standard reading, a color difference not exceeding 5 CIElab units for each parameter is acceptable, so the samples showed good color consistency[5,60]
3.6 Biodegradability results
Figure 5 shows the biodegradability results for PLA and composites.
Biodegradable hybrid PLA composites incorporating coffee husks and mineral fillers

After a 30-day incubation period at 25 °C, the PLA film deteriorated by 68.95%. Similar results were achieved by Mitchell and Hirt[61], for PLA fibers in diferente diameters (32 and 118 mm). The PLA fibers subjected to conditions of 80 °C and 100% relative humidity experienced a daily weight reduction of 2% up to day 30. On day 30, the authors related that the average weight loss reached 69% for the PLA32 samples and 65% for the PLA118 samples, respectively. According to their reults, following the 30-day degradation period, there was an insufficient amount of material remaining from either diameter for further analysis.
In our work, the addition of coffee husk resulted in an increase in the biodegradation of the composites, leading to complete degradation of the PLA composites for the 80/10/5/5, 60/30/5/5, and 50/40/5/5 composites.
According to Vasile et al.[62] and Kadea et al.[53] the degradation rate of the biocomposites in soil can be influencied by time, humidity, temperature, the soil mocroorganism, interactions between PLA and fiber, and the tickness of composite. Lignocellulosic fibers have been widely studied due to their unique properties, including biodegradability, mechanical strength, and low cost. When incorporated into plastic composite materials, these fibers can enhance the material’s biodegradation, facilitating microbial decomposition and reducing environmental impact.
The 70/20/5/5 composite exhibited behavior similar to that of PLA. This demonstrates that it’s possible to reduce the percentage of PLA by using waste in the formulation, which can not only decrease the price of the final product but also contribute to sustainability. Additionally, the levels of calcium and potassium increased from 200 to 800 mg/L and from 22 to 46 mg/L, respectively, after 30 days. These minerals can be used as seedling fixatives in soil and as sources of micronutrient release.
The 60/30/5/5 and 50/40/5/5 composites exhibited a higher biodegradation rate in the first 17 days. After this period, all composites showed an exponential increase in the biodegradation rate. PLA and the 70/20/5/5 composite showed less deterioration at the end of the test, with 68.95% and 69.24%, respectively. After 30 days, the 80/10/5/5, 60/30/5/5, and 50/40/5/5 composites showed a final deterioration of 100%.
In this article, we developed hybrid composites of PLA matrix filled with coffee husks, potassium feldspar, and Bahia Beige marble. We conducted a comprehensive assessment that included the analysis of the composition of the reinforcing materials, the identification of components and evaluation of chemical reactions through FTIR, and the investigation of physical and mechanical properties through hardness, contact angle, and density tests, in addition to microstructural analysis by SEM. Also, variance analysis was performed in all results.
Based on the results obtained, it is evident that biodegradable hybrid composites made from PLA exhibit significant potential for sustainable applications. The XRF analysis provided information about the composition of the raw materials, helping to optimize the compositions used in the manufacturing process of the composites. Density results showed variations due to particle compaction and particle wall roughness. Although there was a slight reduction in hardness with the addition of coffee husks, this change was attributed to the uneven distribution of components in the matrix.
Contact angle analysis demonstrated the hydrophilic nature of the composites, similar to that of commercial capsules, but the addition of fillers increased hydrophilicity. SEM images confirmed a strong interfacial adhesion of the components due to melting during processing. Furthermore, color parameters indicated consistency in coloration among the formulations.
Biodegradation tests revealed that the addition of coffee husks significantly increased the degradation rate of PLA composites. After 30 days of incubation at 25°C, the PLA film without coffee husks deteriorated by 68.94%, while composites with coffee husks showed a 100% higher degradation rate. Additionally, the presence of calcium and potassium minerals in the composites could be beneficial, as these minerals can serve as soil seedling fixatives and sources of micronutrient release.
It is important to emphasize that the final product’s creation will involve several processing stages. As a result, it will be manufactured using the thermoforming process with multi-layered composite sheets, resulting in capsules with multiple layers.
These results are promising, suggesting that coffee capsules containing poly(lactic acid) enriched with coffee husks and calcium and potassium minerals can be 100% biodegradable, contributing to a reduction in environmental impact. The combination of increased hydrophilicity and an accelerated degradation rate makes these composites ideal for sustainable applications, facilitating decomposition in the soil after consumption. This study paves the way for an eco-friendlier approach to coffee capsule manufacturing and related products.
5. Author’s Contribution
• Conceptualization – Daniele Cruz Bastos; Roberto Carlos da Conceição Ribeiro.
Silva, R. G., Barboza, L. S., Silveira, P. H. P. M., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C.
• Data curation – Pedro Henrique Poubel Mendonça da Silveira.
• Formal analysis – Laysa Silva Barboza.
• Funding acquisition – Daniele Cruz Bastos; Roberto Carlos da Conceição Ribeiro.
• Investigation – Riquelme Gomes da Silva; Laysa Silva Barboza.
• Methodology – Daniele Cruz Bastos; Roberto Carlos da Conceição Ribeiro.
• Project administration – Daniele Cruz Bastos; Roberto Carlos da Conceição Ribeiro.
• Resources – Riquelme Gomes da Silva; Laysa Silva Barboza; Pedro Henrique Poubel Mendonça da Silveira.
• Software – Pedro Henrique Poubel Mendonça da Silveira.
• Supervision – Daniele Cruz Bastos; Roberto Carlos da Conceição Ribeiro.
• Validation – Daniele Cruz Bastos; Roberto Carlos da Conceição Ribeiro.
• Visualization – Daniele Cruz Bastos; Pedro Henrique Poubel Mendonça da Silveira.
• Writing – original draft – Pedro Henrique Poubel Mendonça da Silveira; Marceli do Nascimento da Conceição.
• Writing – review & editing – Daniele Cruz Bastos; Pedro Henrique Poubel Mendonça da Silveira.
6. Acknowledgements
The authors acknowledge to CNPq, and Faperj.
7. References
1 Samoggia, A., & Busi, R. (2023). Sustainable coffee capsule consumption: understanding Italian consumers’ purchasing drivers. Frontiers in Sustainable Food Systems, 7, 1-13 http:// doi.org/10.3389/fsufs.2023.1088877
2 Samoggia, A., & Riedel, B. (2019). Consumers’ perceptions of coffee health benefits and motives for coffee consumption and purchasing. Nutrients, 11(3), 653. http://doi.org/10.3390/ nu11030653 PMid:30889887.
3 Domingues, M. L. B., Bocca, J. R., Fávaro, S. L., & Radovanovic, E. (2020). Disposable coffee capsules as a source of recycled polypropylene. Polímeros , 30 (1), e2020009 http://doi. org/10.1590/0104-1428.05518
4 Nanni, A., Colonna, M., & Messori, M. (2022). Fabrication and characterization of new eco-friendly composites obtained by the complete recycling of exhausted coffee capsules. Composites Science and Technology, 222, 109358 http://doi. org/10.1016/j.compscitech.2022.109358
5. Suaduang, N., Ross, S., Ross, G. M., Pratumshat, S., & Mahasaranon, S. (2019). Effect of spent coffee grounds filler on the physical and mechanical properties of poly(lactic acid) bio-composite films. Materials Today: Proceedings, 17(Part 4), 2104-2110 http://doi.org/10.1016/j.matpr.2019.06.260
6 Otoukesh, M., Vera, P., Wrona, M., Nerin, C., & Es’haghi, Z. (2020). Migration of dihydroxyalkylamines from polypropylene coffee capsules to Tenax® and coffee by salt-assisted liquid–liquid extraction and liquid chromatography-mass spectrometry. Food Chemistry , 321 , 126720 http://doi.org/10.1016/j. foodchem.2020.126720 PMid:32276149.
7 Cincotta, F., Tripodi, G., Merlino, M., Verzera, A., & Condurso, C. (2020). Variety and shelf-life of coffee packaged in capsules. Lebensmittel-Wissenschaft + Technologie, 118, 108718 http:// doi.org/10.1016/j.lwt.2019.108718
8 Garlotta, D. (2001). A literature review of poly(lactic acid). Journal of Polymers and the Environment, 9(2), 63-84. http:// doi.org/10.1023/A:1020200822435.
9 Taib, N.-A. A. B., Rahman, M. R., Huda, D., Kuok, K. K., Hamdan, S., Bakri, M. K. B., Julaihi, M. R. M. B., & Khan, A. (2023). A review on poly lactic acid (PLA) as a biodegradable polymer. Polymer Bulletin, 80(2), 1179-1213 http://doi. org/10.1007/s00289-022-04160-y
10 Ashothaman, A., Sudha, J., & Senthilkumar, N. (2023). A comprehensive review on biodegradable polylactic acid polymer matrix composite material reinforced with synthetic and natural fibers. Materials Today: Proceedings, 80(Part 3), 2829-2839 http://doi.org/10.1016/j.matpr.2021.07.047
11. Swetha, T., Bora, A., Mohanrasu, K., Balaji, P., Raja, R., Ponnuchamy, K., Muthusamy, G., & Arun, A. (2023). A comprehensive review on polylactic acid (PLA): synthesis, processing and application in food packaging. International Journal of Biological Macromolecules, 234, 123715 http:// doi.org/10.1016/j.ijbiomac.2023.123715 PMid:36801278.
12 Wu, Y., Gao, X., Wu, J., Zhou, T., Nguyen, T. T., & Wang, Y. (2023). Biodegradable polylactic acid and its composites: Characteristics, processing, and sustainable applications in sports. Polymers, 15(14), 3096 http://doi.org/10.3390/ polym15143096 PMid:37514485.
13 Di Bartolo, A., Infurna, G., & Dintcheva, N. T. (2021). A review of bioplastics and their adoption in the circular economy. Polymers, 13(8), 1229 http://doi.org/10.3390/polym13081229 PMid:33920269.
14 Lima, A. M., Santos, M. C. C., Bastos, D. C., Libano, E. V. D. G., & Pereira, P. S. C. (2021). Polypropylene/sugarcane bagasse composites: influence of processing. Brazilian Journal of Development, 7(12), 110053-110065 http://doi.org/10.34117/ bjdv7n12-002
15 Chandgude, S., & Salunkhe, S. (2021). In state of art: mechanical behavior of natural fiber-based hybrid polymeric composites for application of automobile components. Polymer Composites, 42(6), 2678-2703 http://doi.org/10.1002/pc.26045
16 Chagas, G. N., Barros, M. M., Leão, A. G., Tapanes, N. L. C. O., Ribeiro, R. C. C., & Bastos, D. C. (2022). A hybrid green composite for automotive industry. Polímeros, 32(2), e2022017. http://doi.org/10.1590/0104-1428.20220027.
17 Morelli, C. L., Pouzada, A. S., & Sousa, J. A. (2009). Influence of hybridization of glass fiber and talc on the mechanical performance of polypropylene composites. Journal of Applied Polymer Science, 114(6), 3592-3601 http://doi.org/10.1002/ app.30980
18 Hashemi, S. (2008). Hybridization effect on flexural properties of single- and double-gated injection moulded acrylonitrile butadiene styrene (ABS) filled with short glass fibres and glass beads particles. Journal of Materials Science, 43(14), 4811-4819 http://doi.org/10.1007/s10853-008-2683-1
19. Carvalho, G. B., Canevarolo, S. V., Jr., & Sousa, J. A. (2020). Influence of interfacial interactions on the mechanical behavior of hybrid composites of polypropylene/short glass fibers/ hollow glass beads. Polymer Testing, 85, 106418 http://doi. org/10.1016/j.polymertesting.2020.106418
20 Lapčík, L., Maňas, D., Lapčíková, B., Vašina, M., Staněk, M., Čépe, K., Vlček, J., Waters, K. E., Greenwood, R. W., & Rowson, N. A. (2018). Effect of filler particle shape on plasticelastic mechanical behavior of high density poly(ethylene)/ mica and poly(ethylene)/wollastonite composites. Composites.
Biodegradable hybrid PLA composites incorporating coffee husks and mineral fillers
Part B, Engineering, 141, 92-99 http://doi.org/10.1016/j. compositesb.2017.12.035
21 Gerardo, C. F., França, S. C. A., Santos, S. F., & Bastos, D. C. (2020). A study of recycled high-density polyethylene with mica addition: influence of mica particle size on wetting behavior, morphological, physical, and chemical properties. International Journal of Developmental Research, 10(6), 37223-37228
22 Hejna, A., Formela, K., & Saeb, M. R. (2015). Processing, mechanical and thermal behavior assessments of polycaprolactone/ agricultural wastes biocomposites. Industrial Crops and Products, 76, 725-733 http://doi.org/10.1016/j.indcrop.2015.07.049
23. Sung, S. H., Chang, Y., & Han, J. (2017). Development of polylactic acid nanocomposite films reinforced with cellulose nanocrystals derived from coffee silverskin. Carbohydrate Polymers, 169, 495-503 http://doi.org/10.1016/j.carbpol.2017.04.037 PMid:28504172.
24 Sanjay, M. R., Madhu, P., Jawaid, M., Senthamaraikannan, P., Senthil, S., & Pradeep, S. (2018). Characterization and properties of natural fiber polymer composites: a comprehensive review. Journal of Cleaner Production, 172, 566-581 http:// doi.org/10.1016/j.jclepro.2017.10.101
25. Hejna, A. (2021). Potential applications of by-products from the coffee industry in polymer technology: current state and perspectives. Waste Management, 121(3), 296-330 http://doi. org/10.1016/j.wasman.2020.12.018 PMid:33406477.
26 Sharma, R. K., Lakhani, R., & Tomar, P. (2018). A simple novel mix design method and properties assessment of foamed concretes with limestone slurry waste. Journal of Cleaner Production, 171, 1650-1663 http://doi.org/10.1016/j. jclepro.2017.10.073
27 Barros, M. M., Oliveira, M. F. L., Ribeiro, R. C. C., Bastos, D. C., & Oliveira, M. G. (2020). Ecological bricks from dimension stone waste and polyester resin. Construction & Building Materials, 232, 117252 http://doi.org/10.1016/j. conbuildmat.2019.117252.
28 Malaquias, E. O., Pereira, A. A. M., Jr., Silveira, P. H. P. M., Altoé, L., & Teles, C. R. (2021). Uso de cálculo estequiométrico para avaliação da produção de biogás obtido da água residuária do café. Brazilian Journal of Production Engineering, 7(2), 48-58 http://doi.org/10.47456/bjpe.v7i2.34548
29 American Society for Testing and Materials – ASTM. (2020). ASTM D792-20: standard test methods for density and specific gravity (relative density) of plastics by displacement. West Conshohocken: ASTM
30 American Society for Testing and Materials – ASTM. (2021). ASTM D2240-15(2021): standard test method for rubber property-durometer hardness. West Conshohocken: ASTM
31 Ribeiro, M. P., Neuba, L. M., Silveira, P. H. P. M., Luz, F. S., Figueiredo, A. B.-H. S., Monteiro, S. N., & Moreira, M. O. (2021). Mechanical, thermal and ballistic performance of epoxy composites reinforced with Cannabis sativa hemp fabric. Journal of Materials Research and Technology, 12, 221-233 http://doi.org/10.1016/j.jmrt.2021.02.064
32. Acchar, W., Dultra, E. J. V., & Segadães, A. M. (2013). Untreated coffee husk ashes used as flux in ceramic tiles. Applied Clay Science, 75-76, 141-147 http://doi.org/10.1016/j. clay.2013.03.009
33 Saikia, B. J., Parthasarathy, G., & Sarmah, N. C. (2008). Fourier transform infrared spectroscopic estimation of crystallinity in SiO2 based rocks. Bulletin of Materials Science, 31(5), 775779. http://doi.org/10.1007/s12034-008-0123-0.
34 Manju, P., Krishnan, P. S. G., & Nayak, S. K. (2021). Effect of morphology and hydroxyl groups of different nanoparticles on the properties of PLA bionanocomposites. Materials Today: Proceedings, 41(Part 5), 1169-1174 http://doi.org/10.1016/j. matpr.2020.09.508
35 Khan, A., Patidar, R., & Pappu, A. (2021). Marble waste characterization and reinforcement in low density polyethylene composites via injection moulding: towards improved mechanical strength and thermal conductivity. Construction & Building Materials, 269, 121229 http://doi.org/10.1016/j. conbuildmat.2020.121229
36. Wang, N., Fu, Y., & Lim, L. (2011). Feasibility study on chemometric discrimination of roasted Arabica coffees by solvent extraction and Fourier transform infrared spectroscopy. Journal of Agricultural and Food Chemistry, 59(7), 3220-3226 http://doi.org/10.1021/jf104980d PMid:21381653.
37 Wang, N., & Lim, L.-T. (2012). Fourier transform infrared and physicochemical analyses of roasted Coffee. Journal of Agricultural and Food Chemistry, 60(21), 5446-5453 http:// doi.org/10.1021/jf300348e. PMid:22563854.
38 Marchi, B. Z., Silveira, P. H. P. M., Bezerra, W. B. A., Nascimento, L. F. C., Lopes, F. P. D., Candido, V. S., Silva, A. C. R., & Monteiro, S. N. (2023). Ballistic performance, thermal and chemical characterization of ubim fiber (Geonoma baculifera) reinforced epoxy matrix composites. Polymers, 15(15), 3220 http://doi.org/10.3390/polym15153220 PMid:37571114.
39 Reis, N., Franca, A. S., & Oliveira, L. S. (2013). Performance of diffuse reflectance infrared Fourier transform spectroscopy and chemometrics for detection of multiple adulterants in roasted and ground coffee. Lebensmittel-Wissenschaft + Technologie, 53(2), 395-401 http://doi.org/10.1016/j.lwt.2013.04.008
40. Capek, P., Paulovičová, E., Matulová, M., Mislovičová, D., Navarini, L., & Suggi-Liverani, F. (2014). Coffea arabica instant coffee: chemical view and immunomodulating properties. Carbohydrate Polymers, 103, 418-426 http://doi.org/10.1016/j. carbpol.2013.12.068 PMid:24528749.
41 Ribeiro, J. S., Ferreira, M. M. C., & Salva, T. J. G. (2011). Chemometric models for the quantitative descriptive sensory analysis of Arabica coffee beverages using near infrared spectroscopy. Talanta, 83(5), 1352-1358 http://doi.org/10.1016/j. talanta.2010.11.001 PMid:21238720.
42 Craig, A. P., Botelho, B. G., Oliveira, L. S., & Franca, A. S. (2018). Mid infrared spectroscopy and chemometrics as tools for the classification of roasted coffees by cup quality. Food Chemistry, 245, 1052-1061 http://doi.org/10.1016/j. foodchem.2017.11.066 PMid:29287322.
43 Barrios-Rodríguez, Y., Collazos-Escobar, G. A., & GutiérrezGuzmán , N. (2021 ). ATR-FTIR for characterizing and differentiating dried and ground coffee cherry pulp of different varieties (Coffea arabica L.). Engenharia Agrícola, 41(1), 70-77 http://doi.org/10.1590/1809-4430-eng.agric.v41n1p70-77/2021
44. Chieng, B. W., Ibrahim, N. A., Yunus, W. M. Z. W., & Hussein, M. Z. (2013). Poly(lactic acid)/poly(ethylene glycol) polymer nanocomposites: effects of graphene nanoplatelets. Polymers, 6(1), 93-104 http://doi.org/10.3390/polym6010093
45. Wang, N., Zhang, X., Ma, X., & Fang, J. (2008). Influence of carbon black on the properties of plasticized poly (lactic acid) composites. Polymer Degradation & Stability, 93(6), 10441052 http://doi.org/10.1016/j.polymdegradstab.2008.03.023
46. Angin, N., Caylak, S., Ertas, M., & Cavdar, A. D. (2022). Effect of alkyl ketene dimer on chemical and thermal properties of polylactic acid (PLA) hybrid composites. Sustainable Materials and Technologies, 32 , e00386 http://doi.org/10.1016/j. susmat.2021.e00386
47 Santos, R. S., Silveira, P. H. P. M., Bastos, B. C., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C. (2023). Development of environmentally ecofriendly composites based on polypropylene/ Bahia Beige Waste: effect of reinforcement content on physical, mechanical, chemical, and microstructural properties. Recent Progress in Materials, 5(3), 27 http://doi.org/10.21926/ rpm.2303027
Silva, R. G., Barboza, L. S., Silveira, P. H. P. M., Conceição, M. N., Ribeiro, R. C. C., & Bastos, D. C.
48 Bakshi, P., Pappu, A., Bharti, D. K., & Patidar, R. (2021). Accelerated weathering performance of injection moulded PP and LDPE composites reinforced with calcium rich waste resources. Polymer Degradation & Stability, 192, 109694 http://doi.org/10.1016/j.polymdegradstab.2021.109694.
49 Ou, R., Xie, Y., Wolcott, M. P., Sui, S., & Wang, Q. (2014). Morphology, mechanical properties, and dimensional stability of wood particle/high density polyethylene composites: effect of removal of wood cell wall composition. Materials & Design, 58, 339-345 http://doi.org/10.1016/j.matdes.2014.02.018
50 Silveira, P. H. P. M., Conceição, M. N., Pina, D. N., Paes, P. A. M., Monteiro, S. N., Tapanes, N. L. C. O., Ribeiro, R. C. C., & Bastos, D. C. (2024). Impact of different mineral reinforcements on HDPE COMPOSITES: effects of melt flow index and particle size on physical and mechanical properties. Polymers, 16(14), 2063. http://doi.org/10.3390/polym16142063. PMid:39065380.
51 Borsoi, C., Berwig, K. H., Scienza, L. C., Zoppas, B. C. D. A., Brandalise, R. N., & Zattera, A. J. (2014). Behavior in simulated soil of recycled expanded polystyrene/waste cotton composites. Materials Research, 17(1), 275-283. http://doi. org/10.1590/S1516-14392013005000167
52 Coelho, K. V. S., Líbano, E. V. D. G., Ramos Filho, F. G., Santos, S. F., Pereira, P. S. C., & Bastos, D. C. (2021). Development of wood plastic composite with reduced water absorption. International Journal of Developmental Research, 11(2), 44547-44551
53 Kadea, S., Kittikorn, T., & Hedthong, R. (2024). Sustainable laminate biocomposite of wood pulp/PLA with modified PVAMFC compatibilizer: weathering resistance and biodegradation in soil. Industrial Crops and Products, 218, 118913 http://doi. org/10.1016/j.indcrop.2024.118913.
54 Siakeng, R., Jawaid, M., Asim, M., & Siengchin, S. (2020). Accelerated weathering and soil burial effect on biodegradability, colour and texture of coir/pineapple leaf fibres/PLA biocomposites. Polymers, 12(2), 458 http://doi.org/10.3390/polym12020458 PMid:32079111.
55 Jaramillo, L. Y., Vásquez-Rendón, M., Upegui, S., Posada, J. C., & Romero-Sáez, M. (2021). Polyethylene-coffee husk eco-composites for production of value-added consumer products. Sustainable Environment Research, 31(1), 34 http:// doi.org/10.1186/s42834-021-00107-6
56 Petinakis, E., Yu, L., Simon, G. P., Dai, X. J., Chen, Z., & Dean, K. (2014). Interfacial adhesion in natural fiber‐reinforced polymer composites. In V. K. Thakur (Ed.), Lignocellulosic polymer composites: processing, characterization, and properties (pp. 17-39). Massachusetts: Scrivener Publishing LLC. http:// doi.org/10.1002/9781118773949.ch2.
57 Almeida, P. O., Gerardo, C. F., de Leão, A. G., França, S. C. A., Santos, S. F., & Bastos, D. C. (2021). Sustainable composites based on recycled high-density polyethylene/mica. Materials Research, 24(2), e20200418 http://doi.org/10.1590/19805373-mr-2020-0418
58 Silveira, P. H. P. M., Santos, M. C. C., Chaves, Y. S., Ribeiro, M. P., Marchi, B. Z., Monteiro, S. N., Gomes, A. V., Tapanes, N. L. C. O. , Pereira , P. S. C. , & Bastos , D. C. (2023 ). Characterization of thermo-mechanical and chemical properties of polypropylene/hemp fiber biocomposites: impact of maleic anhydride compatibilizer and fiber content. Polymers, 15(15), 3271 http://doi.org/10.3390/polym15153271 PMid:37571165.
59 Acosta, A. P., Amico, S. C., Delucis, R. A., Missio, A. L., Rodrigues, M. B. B., Ribeiro, A. C. R., Goularte, M. P., & Gatto, D. A. (2024). Surface analysis of different wood polymer composites exposed to artificial weathering. Journal of Wood Chemistry and Technology, 44(2), 88-101 http://doi.org/10.1 080/02773813.2023.2299814
60 Melgosa, M., Ruiz-López, J., Li, C., García, P. A., Della Bona, A., & Pérez, M. M. (2020). Color inconstancy of natural teeth measured under white light-emitting diode illuminants. Dental Materials, 36(12), 1680-1690 http://doi.org/10.1016/j. dental.2020.10.001 PMid:33172605.
61. Mitchell, M. K., & Hirt, D. E. (2015). Degradation of PLA fibers at elevated temperature and humidity. Polymer Engineering and Science, 55(7), 1652-1660 http://doi.org/10.1002/pen.24003
62 Vasile, C., Pamfil, D., Rapa, M., Darie-Nit, R. N., Mitelut, A. C., Popa, E. E., Popescu, P. A., Draghici, M. C., & Popa, M. E. (2018). Study of the soil burial degradation of some PLA/ CS biocomposites. Composite B Engineering, 142, 251-262 http://doi.org/10.1016/j.compositesb.2018.01.026
Received: Feb. 26, 2024 Revised: July 11, 2024 Accepted: July 17, 2024
André Luiz dos Santos1* and Sérgio Henrique Pezzin2
1Programa de Pós-graduação em Ciência e Engenharia de Materiais, Universidade do Estado de Santa Catarina – UDESC, Joinville, SC, Brasil
2Departamento de Química, Universidade do Estado de Santa Catarina – UDESC, Joinville, SC, Brasil *andrelusantos11@gmail.com
Obstract
Portable electrospinning devices have some advantages over conventional benchtop equipment, such as lower cost, better transportability, ease of operation and flexibility in size, shape and deposition surface, especially for in situ applications, such as wound dressings, drug release and cosmetics. In this work, poly(vinyl alcohol) membranes were produced from aqueous solutions, with different viscosities and at different electric field values, using a portable electrospinning device prototype. Field-emission scanning electron microscopy was used to characterize the morphology of the membranes. The results showed that the prototype allowed the electrospinning of membranes, presenting fiber diameter of 147±11 nm and deposition width of 4,6±0.3 cm, similar to the values obtained with the benchtop device.
Keywords: electrospinning, portable devices, PVA, nanofibers, in situ applications.
How to cite: Santos, A. L., & Pezzin, S. H. (2024). Poly(vinyl alcohol) membranes produced by portable electrospinning device for in situ applications. Polímeros: Ciência e Tecnologia, 34(3), e20240032. https://doi.org/10.1590/01041428.20230107
To bring electrospinning technology closer to end users, especially for in situ applications, such as wound dressings, miniaturized, portable and more affordable electrospinning equipment has been researched and presented commercially[1-7] The possibility for the nanofibrous membrane to be deposited directly on the desired surface, reduces patient care time, prevents infection, eliminates the need for packaging and transporting the membranes, reduces pain and would make it possible to produce membranes in different shapes and sizes, depending on the skin lesion[1,8-10]. In addition to the potential medical applications, especially in outdoor activities, such as disaster rescues and battlefields, where it can be used to deposit a functional dressing directly on wounds, this technology can be also applied in the cosmetic and drug delivery areas, by adding active ingredients or drugs, such as anti-inflammatories and analgesics, to the polymeric solution[11]. Therefore, portable devices are more convenient for hospitals ambulances and possibly for everyday use by the end consumer[8,12-14]. Several authors have reported the development of these portable devices, using common commercial components, such as a miniature high voltage converters, which can supply voltages in the range of 1 to 10 kV. Some are battery operated and current limiting measures are employed for safer operation. These portable devices can be operated with just one hand, some can be placed in the pocket, and are operated at distances of 3 to 5 cm from the deposition surface[10,11,15,16]. Studies related to portable in situ electrospinning reported small, lightweight devices capable of electrospin submicron fibers
made from various polymers with similar quality to those made on a conventional device presenting good performance and real-time control, demonstrating that these portable electrospinning devices can be an attractive alternative to existing benchtop machines[17]
However, a factor of concern in the electrospinning of polymeric solutions is the use of toxic solvents, not only due to the possibility of residual solvent present in the nanofibers produced, but also regarding to their handling during the spinning process[18]. Due to their cytotoxicity, organic solvents cannot be used for in situ electrospinning, as they can compromise wound healing, nor in cellular electrospinning (solutions with the addition of live cells)[5,19]. In this sense, following the concept of “Green Electrospinning”, the use of toxic solvents can be avoided by using water-soluble polymers, even though only a small range of polymers are water-soluble[20]. Poly(vinyl alcohol), PVA, is one of these few vinyl polymers that is water-soluble, non-toxic, biocompatible, susceptible to biodegradation in the presence of microorganisms and considered a green polymer[21-24] It is a synthetic, semi-crystalline polymer, produced from the hydrolysis of poly(vinyl acetate), with a degradation temperature starting at 150 °C[25,26]. It is commercially available in variations regarding its molecular weight and degree of hydrolysis (DH, from 70% to 99%)[23,27,28]
Therefore, the aim of this work is to use a lab-made portable device prototype for electrospinning membranes from aqueous PVA solutions, employing different electric field
A. L., & Pezzin, S. H.
configurations (voltages of 6 and 8 kV and needle-to-collector distances of 5 and 6 cm) and solution viscosities (for three different ranges of molecular weights and concentrations of 10 and 9% m/m), to evaluate the viability of this device in obtaining membranes and analyze the material and process parameters that influence their morphology (fiber diameter and deposition width). To the best of our knowledge, there is no literature that had reported on the combined use of this specific material, process parameters and electrospinning aparattus, which gives an innovative aspect to this study. It allowed a better understanding of electrospinning parameters, like the deposition width behavior, which is especially relevant for applications that require greater precision and smaller deposition area, such as portable devices.
2.1 Materials
Polyvinyl alcohol (PVA, Selvol™, Sekisui, China) partially hydrolyzed (88% DH), in three different molecular weight ranges: PVA1 (‘Low Mw’, 31,000-50,000 g/mol), PVA2 (‘Intermediary Mw’, 85,000-124,000 g/mol) and PVA3 (‘High Mw’, 146,000-186,000 g/mol), kindly provided by Vetta Quimica (Brazil), in powder form. The aqueous solutions were prepared with deionized water (Milli-Q) by magnetically stirring the systems at 80 °C for 12 h.
2.2 Electrospinning device
A lab-made prototype of a portable electrospinning apparatus, composed of a siringe support, a collector made of a flat aluminum plate with distance adjustment, and an adjustable miniature high voltage electrical supply (10x10x5.2 cm), which converts 220 V (AC) to 10 kV (DC), is shown in Figure 1a. An Eletrotech Lab EF 2B CRT 0212 (DBM Eletrotech Ltd., Brazil) benchtop electrospinning equipment, consisting of a high voltage supply (0 to 20 kV) QSC2020 (Inergiae, Brazil), a syringe pump (0.6 to 600 ml/h) and an adapted flat aluminum plate collector, Figure 1b, was used for comparison with the results obtained using the portable device. When using both devices, a 10 ml disposable syringe, with a stainless steel needle 0.55x20 mm (24G ¾”), was used as a reservoir for the polymeric solutions.
The collector was covered with a 7x7 cm aluminum foil on which all membranes were deposited. All experiments were carried out in a laboratory with controlled temperature and humidity of approximately 22 °C and 45%, respectively. The spinning time was 3 min for all membranes.
After preliminary experiments to identify a range of viable process parameter values, a PVA2 polymeric solution at 9% (m/m) was electrospun under the action of four different electric fields (1; 1.2; 1.3 and 1.6 kV/cm), generated by varying voltage (6 and 8 kV) and needle-to-collector distance (5 and 6 cm), using the portable device prototype. In a second experiment, using the bench-top electrospinning device at 8 kV and needle-to-collector distance of 6 cm, four PVA solutions of different viscosities, in two different molecular weight ranges (PVA2 and PVA3) and concentrations (9 and 10% m/m) were electrospun in order to evaluate the effect of these parameters on the morphology of the membranes and also to compare and validate the results obtained with the portable device. All the electrospinning experiments were performed in triplicate.
The viscosities of the polymeric solutions were measured using a MVD-5 digital rotational viscometer (Marte, Brazil). The viscosity values were obtained by an average of 6 measurements for each solution, at 12, 30 and 60 RPM, according to the viscosity range of each solution. The intrinsic viscosity [η] was calculated using the Mark Houwink relationship ([η] = 6.51.10−4 Mw0.628) for PVA dissolved in water[29]. The conductivities of the polymeric solutions were measured using a NI CVM (Nova Instruments) benchtop conductivity meter at an ambient temperature of approximately 24 °C.
A field emission gun – scanning electron microscope (FEG-SEM) JSM-6710F (JEOL, Japan) was used to analyze the morphology of the membranes, which were covered with gold for 90 s at 40 mA, using a Denton Vacuum (U.S.A.) Desk V equipment. The fiber diameters were estimated by

Poly(vinyl alcohol) membranes produced by portable electrospinning device for in situ applications
manually measuring the diameter of 20 fibers homogeneously distributed in the micrographs at 15,000X magnification, using the ImageJ® software. The percentage pore area on the surface of the membranes was measured by methodology described by Wijayanti et al.[30], with the aid of the ImageJ® software. For the determination of deposition widths, the diameters of the electrospun membranes deposited on the collector surface were photographed and measured, following the methodology proposed by Chui et al.[31] .
The statistical software MINITAB17 was used to process the fiber diameter values measured in each membrane, making it possible to obtain an average diameter value, with standard deviation and graphical representation through a histogram of its diameter distribution. Pearson’s P correlation coefficient (-1 ≤ P ≤ 1) was calculated to statistically analyze the direction and strength of the relationship between the variables (for instance, electric field and fiber diameter). Values of between 0 and 0.19 indicate a very weak correlation, from 0.2 to 0.39 a weak correlation, 0.4 to 0.69 a moderate correlation, 0.7 to 0.89 a strong correlation and 0.9 to 1.0 indicate a very strong correlation.
3.1 Solution viscosities and conductivities
The viscosity and conductivity values of polymeric solutions, composed of PVA with different molecular weights and concentrations, are presented in Table 1. It is observed that the viscosity of the PVA solution increases with the molecular weight and concentration, which can be attributed to the greater entanglement between the polymer chains and intermolecular interactions between hydroxyl groups, respectively[32] .
The properties of nanofibrous membranes are significantly affected by the physicochemical properties of the polymeric solutions, such as rheological behavior. When analyzing PVA membranes, Morais et al.[32] observed that there is a direct relationship between the viscosity of the formulation and the entanglement of the chains in the electrospun nanofibers, as there was an increase in viscosity between polymeric solutions with 7 and 8% (m/m) PVA in water. The concentration of the solution plays an important role in stabilizing the fibrous structure of electrospun nanofibers and it was observed that a fibrous structure could not be stabilized for [η]C < 4, where [η] is the intrinsic viscosity and C is the concentration. For the formation of stable nanofibers, the [η]C values must be between 5 and 12 (5<[η]C< 12)[27]
The electrical conductivity can also limit the electrospinning process. The formation of defect-free fibers will not occur at very low conductivity values, where charge migration to the surface of electrospinning formulations is almost completely avoided, and also at excessively high conductivity values[33] , i.e. there is an ideal range of conductivity values in which electrospinning is viable, generating micro- or nano-metric fibers. The fiber diameter tends to decrease, and the diameter distribution to increase, with electrical conductivity[32] . Conductivity values varying from 277.6 to 313.6 µS/cm for 7 - 8% (m/m) aqueous PVA (Mw 85,500 g/mol – 89.5% DH) solutions, close to the values shown in Table 1, has been recenty reported[32]
Preliminary studies using the portable device prototype were carried out to identify a range of viable process and material parameter values. It was observed that for PVA2 solutions at 10% (m/m), voltages up to 8 kV and needle-to-collector distance of 6 cm, only one Taylor cone is formed at the tip of the needle, projecting a single visible jet (in the stable stage), which allowed the deposition of a circular-shaped membrane with greater deposition precision, Figure 2a. At voltages above 8 kV, the formation of multiple jets was observed.
For solutions of PVA1 and PVA3 at 10% (m/m), the formation of fibers was not possible at these conditions (8 kV and 6 cm) and, thus, they were discarded for further experiments. In the case of PVA1, it was observed the projection of solution drops on the collector, forming a beaded structure, Figure 2b, which characterizes an electrospray process instead of electrospinning[1]. This behavior can be explained by the very low viscosity of the solution[19] and high conductivity[34], leading to greater instability. PVA3 solutions presented difficulty in forming the Taylor cone, due to the higher viscosity and an insufficient needle-to-collector distance to form the zone of instability and thinning of the jet, not allowing proper evaporation of the solvent until its deposition on the collecting surface. This fact caused the accumulation of the polymeric solution in the collector, Figure 2c. In all cases, it was observed dripping of solutions on the support table, as well as projection of some drops onto the collecting surface, which can be justified by the low volatility of the solvent (water), decreasing with the increase in the molecular weight of PVA.
Figure 3 presents the FEG-SEM images of the four membranes electrospun from a 9% (m/m) PVA2 solution using the portable device, with the variation of electric field values (1; 1.2; 1.3 and 1.6 kV/cm), by a combination of two electrical voltages (6 and 8 kV) and two needle-to-collector
A. L., & Pezzin, S. H.


distances (5 and 6 cm). Table 2 presents the values of average fiber diameter and deposition width (membrane diameter) of the PVA2 membranes.
It is observed the formation of membranes with fibers in the nanometric scale, randomly oriented and without the presence of defects. There was a decrease in the average diameter values of the fibers with the increase in the electric field value, which is characteristic of a strong negative correlation between these two parameters, confirmed by a Pearson correlation coefficient P=-0.844. This behavior has been also reported for the electrospinning of 2.5% PEO aqueous solutions[35]. It is interesting to mention that the value of the electric field, determined by dividing the applied electrical voltage by the distance between the surface of the drop of solution pending at the tip of the needle and
the deposition collecting surface, is generally about 1 kV/ cm for conventional polymer electrospinning[36]
In summary, surface charges on the fluid surface increase with increasing applied electric field, and the elongation of the
Poly(vinyl alcohol) membranes produced by portable electrospinning device for in situ applications
electrospun jet increases with increasing jet surface charges and time of flight[35]. Furthermore, it is observed that the fiber diameters do not vary so significantly by changing the applied electric field as by changing the material parameters, such as conductivity and viscosity.
Figure 4 shows the photographs of electrospun PVA2 membranes deposited on the aluminum collecting surfaces.
Considering its approximately circular shape, it is possible to observe a decrease in diameter with the increase of the electric field intensity, as shown in Table 2, characterizing a decrease in the area of the membrane deposited on the collecting surface, which is also characteristic of a very strong negative correlation, according to a Pearson correlation coefficient P= -0.934. In the same way that the unstable region of whipping depends on the intensity of the applied electric field, influencing the stretching of the fiber, in this region, due to cone formation and the jet path, a variation in the formation of the deposition area or spreading diameter is observed, which can be measured by the diameter of the membrane collected on the deposition surface. Therefore, the controllability of nanofiber deposition is also affected by instabilities caused at this stage, with the length of the straight jet in the first stable stage increasing with the increase in the applied electric field and, consequently, the whipping time decreases with increasing applied electric field[35]
To obtain a more precise and controllable fiber deposition, an electrospinning technique with modified electric fields, using a metal cone attached to the capillary and/or an electrode positioned in the jet path has been proposed[37]
In order to validate the results obtained in the previous experiment using the portable miniature electrospinning device, electrospun PVA membrane were also obtained by using a conventional benchtop electrospinning equipment. Figure 5 presents the photograph and the FEG-SEM image of the membrane electrospun from PVA2 at 9% m/m, using the same electric field (1.3 kV/cm, at 8 kV and 6 cm) of the previous portable electrospinning experiment. In this experiment, however, a 1.2 ml/h flow rate was employed. It was observed the formation of a membrane with fibers in the nanometric scale, randomly oriented, with an average diameter of 134.2±10 nm and deposition width of 6.3±1.5 cm.
In Table 3, the values of average fiber diameter and deposition width are presented for PVA2 membranes electrospun from a 9% (m/m) solution by the portable and the benchtop electrospinning devices. Although it shows a diference of 1.7 cm for the depositon width, it can be observed that there was no significant variation in the mean
Table 3. Comparative analysis of fiber diameter and deposition width results between portable and benchtop devices.
Spinning device


A. L., & Pezzin, S. H.
fiber diameter obtained between the two devices, using the same material and process parameters, suggesting that the electrospinning performance and quality of the electrospun membranes produced by the portable device are similar to those obtained using the commercial benchtop equipment.
Another relevant difference between both devices was the use of an uncontrolled/uniform flow rate, as a pressure was mannualy applied to the piston of the syringe by the operator, varying according to the formation or decrease of the drop of solution at the tip of the needle in the portable electrospinning device. This was due to the fact that lower viscosity values were employed, together with the use of water, which presents a relatively low volatility. The formation of a drop of polymeric solution at the tip of the needle and its control during the spinning process was thus easier, as no greater variation in fiber diameter was observed, due to the balance between the rates at which the solution is dispensed/extracted from the capillary[38] .
Membranes composed of fibers with random orientation morphology, presenting fiber diameters in the range of 110 to 520 nm, correspond to the ECM of human skin and a porosity range of 63 to 71% facilitates cell adhesion and proliferation[39]. A similar study reported the electrospinning with a portable device at 10 kV of a 8% PVA (Mw 186,000 g/ mol) solution with the addition of stem cells and it was observed that the cells were evenly distributed on the filaments of the membrane and that the low voltage of the handheld device did not affect cell viability[19] .
It is concluded that the developed prototype of a portable electrospinning device is suitable for the electrospinning of aqueous PVA solutions into membranes composed of randomly oriented fibers with nanometric diameter, similar to those obtained by laboratory benchtop equipment. Additionally, this study enabled a better understanding of the effects of process and material parameters on the fiber morphology, deposition width and surface pore area of the membranes. It was observed that the use of lower voltages provides more stable electrospinning, with the formation of only one Taylor cone and polymer jet, resulting in greater deposition precision, which is advantageous for both in situ deposition applications and for cell electrospinning, which requires precisely lower values of high electrical voltage.
The use of a biocompatible polymer and a non-toxic solvent provided both a lower environmental impact and greater suitability for medical applications, thus being viable for potential applications such as wounds dresings or for transdermal administration systems. The use of water as a solvent prevented the clogging effect, since no solidification of the solution at the tip of the needle was observed and made possible the manual control of the solution flow rate, which contribute for the elimination of a flow control system and reduction in the cost of an eventual commercial portable device.
5. Author’s Contribution
• Conceptualization – André Luiz dos Santos; Sérgio Henrique Pezzin.
• Data curation – André Luiz dos Santos.
• Formal analysis – André Luiz dos Santos.
• Funding acquisition - Sérgio Henrique Pezzin.
• Investigation – André Luiz dos Santos.
• Methodology – André Luiz dos Santos; Sérgio Henrique Pezzin.
• Project administration – André Luiz dos Santos; Sérgio Henrique Pezzin.
• Resources – André Luiz dos Santos; Sérgio Henrique Pezzin.
• Software – NA.
• Supervision – André Luiz dos Santos; Sérgio Henrique Pezzin.
• Validation – André Luiz dos Santos.
• Visualization – André Luiz dos Santos.
• Writing – original draft – André Luiz dos Santos.
• Writing – review & editing – André Luiz dos Santos; Sérgio Henrique Pezzin.
6. Acknowledgements
Thanks to FAPESC for the financial support (2021TR928), to CAPES for the scholarship (88882.447332/2019-01), the Multi-User Facility infrastructure from CCT/UDESC and Vetta quimica for the PVA samples.
7. References
1 Ding, B., Wang, X., & Yu, J. (Eds.) (2019). Electrospinnin: nanofabrication and applications Amsterdam: Elsevier
2 Long, Y.-Z., Yan, X., Wang, X. X., Zhang, J., & Yu, M. (2019). Electrospinning: the setup and procedure. In B. Ding, X. Wang, & J. Yu (Eds.), Electrospinning: nanofabrication and applications (pp. 21-52). Amsterdam: Elsevier. http://doi. org/10.1016/B978-0-323-51270-1.00002-9
3 Keirouz, A., Wang, Z., Reddy, V. S., Nagy, Z. K., Vass, P., Buzgo, M., Ramakrishna, S., & Radacsi, N. (2023). The history of electrospinning: past, present, and future developments. Advanced Materials Technologies, 8(11), 2201723 http://doi. org/10.1002/admt.202201723
4 Huston, L. G., Kooistra-Manning, E. A., Skinner, J. L., & Andriolo, J. M. (2019). Combined electrostatic and air driven electrospinning for biomedical applications. Journal of Vacuum Science and Technology. B, Nanotechnology & Microelectronics : Materials, Processing, Measurement, & Phenomena : JVST B, 37(6), 062002 http://doi.org/10.1116/1.5122659
5 Yan, X., Yu, M., Ramakrishna, S., Russell, S. J., & Long, Y.-Z. (2019). Advances in portable electrospinning devices for: in situ delivery of personalized wound care. Nanoscale, 11(41), 19166-19178. http://doi.org/10.1039/C9NR02802A. PMid:31099816.
6 Ji, D., Lin, Y., Guo, X., Ramasubramanian, B., Wang, R., Radacsi, N., Jose, R., Qin, X., & Ramakrishna, S. (2024). Electrospinning of nanofibres. Nature Reviews Methods Primers, 4(1), 1 http://doi.org/10.1038/s43586-023-00278-z
7. Xue, J., Wu, T., Dai, Y., & Xia, Y. (2019). Electrospinning and electrospun nanofibers: methods, materials, and applications. Chemical Reviews, 119(8), 5298-5415 http://doi.org/10.1021/ acs.chemrev.8b00593 PMid:30916938.
8 Brako, F., Luo, C., Craig, D. Q. M., & Edirisinghe, M. (2018). An inexpensive, portable device for point-of-need generation
Poly(vinyl alcohol) membranes produced by portable electrospinning device for in situ applications
of silver-nanoparticle doped cellulose acetate nanofibers for advanced wound dressing. Macromolecular Materials and Engineering, 303(5), 1700586. http://doi.org/10.1002/ mame.201700586
9 Liu, X.-F., Zhang, J., Liu, J.-J., Zhou, Q.-H., Liu, Z., Hu, P.-Y., Yuan, Z., Ramakrishna, S., Yang, D.-P., & Long, Y.-Z. (2020). Bifunctional CuS composite nanofibers via in situ electrospinning for outdoor rapid hemostasis and simultaneous ablating superbug. Chemical Engineering Journal, 401, 126096 http://doi.org/10.1016/j.cej.2020.126096.
10 Zhou , T. , Wang , Y. , Lei , F. , & Yu , J. ( 2020 ). In-situ electrospinning for intestinal hemostasis. International Journal of Nanomedicine, 15, 3869-3875 http://doi.org/10.2147/IJN. S241909 PMid:32764920.
11 Xu, S.-C., Qin, C.-C., Yu, M., Dong, R.-H., Yan, X., Zhao, H., Han, W.-P., Zhang, H.-D., & Long, Y.-Z. (2015). A batteryoperated portable handheld electrospinning apparatus. Nanoscale, 7(29), 12351-12355 http://doi.org/10.1039/C5NR02922H PMid:26154994.
12 Hu, P.-Y., Zhao, Y.-T., Zhang, J., Yu, S.-X., Yan, J.-S., Wang, X.-X., Hu, M.-Z., Xiang, H.-F., & Long, Y.-Z. (2020). In situ melt electrospun polycaprolactone/Fe3O4 nanofibers for magnetic hyperthermia. Materials Science and Engineering C, 110, 110708 http://doi.org/10.1016/j.msec.2020.110708 PMid:32204020.
13 Sofokleous, P., Stride, E., Bonfield, W., & Edirisinghe, M. (2013). Design, construction and performance of a portable handheld electrohydrodynamic multi-needle spray gun for biomedical applications. Materials Science and Engineering C, 33(1), 213-223 http://doi.org/10.1016/j.msec.2012.08.033 PMid:25428065.
14 Bhattarai, N., Edmondson, D., Veiseh, O., Matsen, F. A., & Zhang, M. (2005). Electrospun chitosan-based nanofibers and their cellular compatibility. Biomaterials, 26(31), 61766184 http://doi.org/10.1016/j.biomaterials.2005.03.027 PMid:15885770.
15 Revia, R. A., Wagner, B. A., & Zhang, M. (2019). A portable electrospinner for nanofiber synthesis and its application for cosmetic treatment of alopecia. Nanomaterials (Basel, Switzerland), 9(9), 1317 http://doi.org/10.3390/nano9091317 PMid:31540131.
16 Liu, G.-S., Yan, X., Yan, F.-F., Chen, F.-X., Hao, L.-Y., Chen, S.-J., Lou, T., Ning, X., & Long, Y.-Z. (2018). In Situ electrospinning iodine-based fibrous meshes for antibacterial wound dressing. Nanoscale Research Letters, 13(1), 309 http:// doi.org/10.1186/s11671-018-2733-9 PMid:30284048.
17 Mouthuy, P.-A., Groszkowski, L., & Ye, H. (2015). Performances of a portable electrospinning apparatus. Biotechnology Letters, 37(5), 1107-1116 http://doi.org/10.1007/s10529-014-1760-6 PMid:25549609.
18 Fadil, F., Affandi, N. D. N., Misnon, M. I., Bonnia, N. N., Harun, A. M., & Alam, M. K. (2021). Review on electrospun nanofiber-applied products. Polymers, 13(13), 2087 http:// doi.org/10.3390/polym13132087 PMid:34202857.
19 Xu, S., Lu, T., Yang, L., Luo, S., Wang, Z., & Ye, C. (2022). In situ cell electrospun using a portable handheld electrospinning apparatus for the repair of wound healing in rats. International Wound Journal, 19(7), 1693-1704 http://doi.org/10.1111/ iwj.13769 PMid:35142063.
20 Lv, D., Zhu, M., Jiang, Z., Jiang, S., Zhang, Q., Xiong, R., & Huang, C. (2018). Green electrospun nanofibers and their application in air filtration. Macromolecular Materials and Engineering, 303(12), 1800336 http://doi.org/10.1002/ mame.201800336.
21 Ngadiman, N. H. A., Noordin, M. Y., Idris, A., Shakir, A. S. A., & Kurniawan, D. (2015). Influence of polyvinyl alcohol
molecular weight on the electrospun nanofiber mechanical properties. Procedia Manufacturing, 2, 568-572 http://doi. org/10.1016/j.promfg.2015.07.098
22 Park, J.-C., Ito, T., Kim, K.-O., Kim, K.-W., Kim, B.-S., Khil, M.-S., Kim, H.-Y., & Kim, I.-S. (2010). Electrospun poly(vinyl alcohol) nanofibers: effects of degree of hydrolysis and enhanced water stability. Polymer Journal, 42(3), 273-276 http://doi. org/10.1038/pj.2009.340
23 Chiellini, E., Corti, A., D’Antone, S., & Solaro, R. (2003). Biodegradation of poly (vinyl alcohol) based materials. Progress in Polymer Science, 28(6), 963-1014 http://doi.org/10.1016/ S0079-6700(02)00149-1
24 Araújo, E. S. (2015). Otimização de micro/nanofibras de polímeros biocompatíveis sintetizadas pela técnica de eletrofiação para aplicações farmacológicas (Doctoral thesis). Universidade Federal da Bahia, Salvador
25. Peppas, N. A., & Merrill, E. W. (1976). Differential scanning calorimetry of crystallized PVA hydrogels. Journal of Applied Polymer Science, 20(6), 1457-1465 http://doi.org/10.1002/ app.1976.070200604.
26 Greiner, A., & Wendorff, J. H. (2007). Electrospinning: a fascinating method for the preparation of ultrathin fibers. Angewandte Chemie International Edition, 46(30), 5670-5703. http://doi.org/10.1002/anie.200604646 PMid:17585397.
27 Koski, A., Yim, K., & Shivkumar, S. (2004). Effect of molecular weight on fibrous PVA produced by electrospinning. Materials Letters, 58(3-4), 493-497 http://doi.org/10.1016/S0167577X(03)00532-9
28 Jones, J. I. (1973). Polyvinyl alcohol. Properties and applications Chichester: John Wiley
29 Tacx, J. C. J. F., Schoffeleers, H. M., Brands, A. G. M., & Teuwen, L. (2000). Dissolution behavior and solution properties of polyvinylalcohol as determined by viscometry and light scattering in DMSO, ethyleneglycol and water. Polymer, 41(3), 947-957 http://doi.org/10.1016/S0032-3861(99)00220-7
30 Wijayanti, I. D., Saputra, A. K., Ibrahim, F., Rasyida, A., Suwarta, P., & Sidharta, I. (2022). An ultra-low-cost and adjustable in-house electrospinning machine to produce PVA nanofiber. HardwareX, 11, e00315 http://doi.org/10.1016/j. ohx.2022.e00315 PMid:35592726.
31 Chui , C.-Y. , Mouthuy, P.-A. , & Ye , H. ( 2018 ). Direct electrospinning of poly(vinyl butyral) onto human dermal fibroblasts using a portable device. Biotechnology Letters, 40(4), 737-744 http://doi.org/10.1007/s10529-018-2522-7 PMid:29464571.
32 Morais, M. S., Bonfim, D. P. F., Aguiar, M. L., & Oliveira, W. P. (2022). Electrospun poly (vinyl alcohol) nanofibrous mat loaded with green propolis extract, chitosan and nystatin as an innovative wound dressing material. Journal of Pharmaceutical Innovation, 18(2), 704-718 http://doi.org/10.1007/s12247022-09681-7 PMid:36061220.
33 Sill, T. J., & Von Recum, H. A. (2008). Electrospinning: applications in drug delivery and tissue engineering. Biomaterials, 29(13), 1989-2006 http://doi.org/10.1016/j.biomaterials.2008.01.011 PMid:18281090.
34 Jia, L., & Qin, X.-H. (2013). The effect of different surfactants on the electrospinning poly(vinyl alcohol) (PVA) nanofibers. Journal of Thermal Analysis and Calorimetry, 112(2), 595-605 http://doi.org/10.1007/s10973-012-2607-9
35 Angammana, C. J. (2011). Analysis of the effects of solution conductivity on electrospinning process and fiber morphology (Doctoral thesis). University of Waterloo, Canada
36 Reneker, D. H., Yarin, A. L., Fong, H., & Koombhongse, S. (2000). Bending instability of electrically charged liquid jets of polymer solutions in electrospinning. Journal of Applied Physics, 87(9), 4531-4547 http://doi.org/10.1063/1.373532
A. L., & Pezzin, S. H.
37 Luo, W.-L., Zhang, J., Qiu, X., Chen, L.-J., Fu, J., Hu, P.-Y., Li, X., Hu, R.-J., & Long, Y.-Z. (2018). Electric- Field-Modified In Situ Precise Deposition of Electrospun Medical Glue Fibers on the Liver for Rapid Hemostasis. Nanoscale Research Letters, 13(1), 278 http://doi.org/10.1186/s11671-018-2698-8 PMid:30203107.
38 Zargham, S., Bazgir, S., Tavakoli, A., Rashidi, A. S., & Damerchely, R. (2012). The effect of flow rate on morphology and deposition area of electrospun nylon 6 nanofiber. Journal of Engineered Fibers and Fabrics, 7(4), 155892501200700414. http://doi.org/10.1177/155892501200700414.
39 Hartatiek , H. , Yudyanto , Y. , Rahim , L. F. , Amalia , S. , Nurhuda, M., Masruroh, M., & Santjojo, D. J. D. H. (2023). Morphology, porosity, and biodegradation of PVA/PEG/ chitosan nanofiber scaffolds for skin tissue engineering. AIP Conference Proceedings, 2634(1), 020015. http://doi. org/10.1063/5.0111554
Received: Jan. 29, 2024
Revised: May 25, 2024
Accepted: Jul. 01, 2024
Thiago Alexandre de Oliveira Bouças1 , Marcos Antonio da Silva Costa1 , Henrique Almeida Cunha1 , Manoel Ribeiro da Silva2 , Wang Shu Hui3 and Jacira Aparecida Castanharo1*
1Laboratório de Química de Polímeros, Instituto de Química, Universidade do Estado do Rio de Janeiro –UERJ, Rio de Janeiro, RJ, Brasil
2Laboratório de Física, Instituto de Ciências, Universidade Federal de Itajubá – UNIFEI, Itajubá, MG, Brasil
3Departamento de Engenharia Metalúrgica de Materiais, Escola Politécnica, Universidade de São Paulo –USP, São Paulo, SP, Brasil *jaciracastanharo@gmail.com
Obstract
In this work, magnetic polymeric microspheres were prepared through the copolymerization of styrene and divinylbenzene in the presence of magnetite synthesized in our laboratory. The effects of magnetite modification with oleic acid, stirring speed, initiator concentration and stabilizer concentration on the particles’ properties were studied. The magnetic microspheres were characterized according to morphology, thermal stability, particle size and distribution, and magnetic properties. The microspheres had spherical shape, micrometric size and superparamagnetic properties. Particle size was mainly affected by stirring speed, while particle size dispersion was strongly affected by initiator and stabilizer concentrations. The saturation magnetization of the microspheres was similar to the values reported in the literature for microspheres with the same content of magnetic material. The modification with oleic acid promoted the incorporation of magnetic material.
Keywords: magnetic properties, magnetite, suspension polymerization, styrene-divinylbenzene copolymer.
How to cite: Bouças, T. A. O., Costa, M. A. S., Cunha, H. A., Silva, M. R., Hui, W. S., & Castanharo, J. A. (2024). Effect of synthesis conditions on the properties of magnetic crosslinked polystyrene microspheres. Polímeros: Ciência e Tecnologia, 34(3), e20240033. https://doi.org/10.1590/0104-1428.20230113
The production of non-magnetic polymeric microspheres has undergone major development during the last four decades, gaining a wide range of applications, from column packing to enzyme immobilization. These applications are based on the phenomenon of adsorption. Porous particles have been developed for this purpose, but they have drawbacks such as fouling, slow mass transfer and difficult pore cleaning. Microspheres with magnetic properties can overcome these problems, since they can be designed to be non-porous with high contact surface, and can be easily agglomerated or separated by the use of an electromagnetic field[1,2]
There are three common ways of imparting magnetic properties to polymeric microspheres: the particles are designed to have polymeric shells and magnetic cores[3,4]; the magnetic material is dispersed onto polymeric particles[5-7]; or the magnetic material is formed in situ on the surface of the polymeric microspheres[8]. The magnetic materials can be iron oxides[9-12], ferrites[13] and several other metal compounds and alloys. In some applications, the magnetic material is modified to be compatible with the polymeric matrix, which is produced by a large range of monomers and polymerization methods[1]. Magnetic styrene-divinylbenzene
copolymers produced via suspension polymerization have been extensively studied regarding their ease of preparation, mechanical, thermal and chemical properties; porous structure and degree of particle size control[4,6,14-16]
Classic suspension polymerization is generally freeradical initiated and involves two phases: an aqueous phase and an organic phase. The organic phase is a mixture of monomers, an initiator and sometimes diluents, while the aqueous phase contains a stabilizer and an inorganic salt to avoid coalescence and fragmentation of the monomer droplets. Mechanical stirring is used to create and maintain the suspension. Particle size and size distribution depend mainly on the stirring speed, and the nature and relative concentrations of the stabilizer and initiator. The proportion between the aqueous and organic phase and nature of the monomers and inorganic salt concentration also affect the particles’ size[15,17]
Magnetic polymer particles have wide applications in biomedical treatment and diagnosis and molecular and environmental separation. These applications require the magnetic polymer particles to have controlled size and size distribution for precise targeting and reproducibility[18] .
Bouças, T. A. O., Costa, M. A. S., Cunha, H. A., Silva, M. R., Hui, W. S., & Castanharo, J. A.
Conceição et al.[15] prepared microspheres based on styrene and divinylbenzene (STY-DVB) containing carbonyl iron powder OX coated with 10% wt-Fe2O3 without diluents. They observed that the initiator concentration had an important influence on the microspheres’ size distribution, in particular that increased initiator concentration assured narrower particle polydispersity. Ramos et al.[16] also studied the variation of the initiator on magnetic poly(styrene-co-divinylbenzene) (P(STY-DVB)-M), but in this case diluents and maghemite were used. They concluded that the amount of initiator was the most important factor for the yield of resins, where higher concentrations of benzoyl peroxide (BPO) were associated with greater yield. In the present work, we used different percentages of BPO as initiator and magnetite as magnetic material. Magnetic microspheres based on the same monomers were also prepared by Santa Maria et al. [14]. In their case, the initiator used was different than BPO, the magnetic material was carbonyl iron powder OX coated with 10% wt-Fe2O3, and different suspension agents were used. According to the authors, the composites showed good magnetic properties and the suspending agents and diluent system employed strongly influenced the microstructure and distribution of iron particles on the surface of the composite granules. Poly(vinyl alcohol) (PVA) was one of the suspending agents used in their study, but at a fixed concentration that was lower than in the present work. Formiga et al.[4] also synthetized P(STY-DVB)-M with the main objective of producing core shells in two steps. In the first step, they employed a high stirring speed (800 rpm) to decrease the particle size of the cores and facilitate the increase in particle size of the subsequent shells.
Many other researchers have also investigated P(STYDVB)-M. However, we did not find any study of P(STYDVB)-M involving the behavior of microspheres in terms of particle size produced by variation of stirring speed and concentrations of suspending agent (PVA) and initiator (BPO) (4 and 6%), seeking to improve the size distribution in the suspension polymerization system.
2.1 Materials
Commercial styrene (STY) and divinylbenzene (DVB) were supplied by Sigma-Aldrich; commercial grade polyvinyl alcohol (PVA) was purchased from Air Products (São Paulo, Brazil), with hydrolysis degree = 85% and molecular weight = 80,000 to 125,000; and benzoyl peroxide (BPO) and other chemicals were purchased from Vetec Quimica Fina Ltda. (Rio de Janeiro, Brazil). All were used as received.
Magnetite was produced according to the Massart method[19], namely co-precipitation of iron (II) and iron (III) in a basic medium. FeSO4 and FeCl3 were the iron salts used, and NH4OH assured the basic property of the solution. Magnetic precipitation was used to separate the magnetite produced. Magnetite modified with oleic acid was produced using the same method, but before magnetic precipitation oleic acid was mixed with magnetite by mechanical stirring and heating for 30 minutes[20,21]
Microspheres were produced via suspension polymerization. The aqueous phase was a water solution of polyvinyl alcohol (PVA), 1, 2 or 3% w/v, and NaCl, 1% w/v. The organic phase was a mixture of styrene and divinylbenzene, in 9:1 molar proportion, containing 2, 4 or 6 mol% benzoyl peroxide (BPO) and 10% w/v magnetite modified with oleic acid. The aqueous to organic phase proportion was 4:1 by volume. The polymerization system was heated to 90 °C and mechanically stirred for 24 hours at 480, 600 or 720 rpm, respectively. The microspheres were washed and separated by filtration, and dried for 48 hours at 60 °C.
Dried microspheres were segregated by size in an automatic sieve shaker (IPAS Retsch®) and size ranges from 500 to 75 micrometers were classified. The ranges with greatest percentage by mass were used in the characterizations. Microspheres selected from the most significant size ranges were analyzed using scanning electronic microscopy (Philips SEM, model XL-30). Samples were coated with a thin gold layer and submitted to acceleration voltage of 20 kV, high vacuum, and presence of secondary and backscattered electrons. Magnification of 60X, 200X, 800X or 5,000X was used.
2.5 Magnetic and thermal properties
Magnetic properties were analyzed with a Lake Shore model 7400 vibrating sample magnetometer (VSM) using cycle time of 1 second and hysteresis cycle time of 10 minutes. The magnetic field varied from 12,000 to -12,000 Oe. Thermogravimetric analysis was performed with a TA Instruments Q500 analyzer, where 20 to 30 grams of sample was heated in a nitrogen atmosphere to 600 °C at a rate of 10 °C/min (equipment error ± 4 °C).
This work presents the results of analyzing the influence of several parameters on the final properties of the microspheres. Thus, this section describes each parameter separately.
3.1
It is known that stirring speed directly affects particle size (PS) and size distribution (PSD) in suspension polymerization systems[17]. In this study, we evaluated stirring speeds of 480, 600 and 720 rpm while maintaining PVA and BPO concentrations at 1% and 2%, respectively. Figure 1 shows the influence of stirring speeds on particle size distribution of the polymer. The particle sizes of sample TR02 were mostly between 88 and 180 µm, while the other samples had predominant particle sizes smaller than 75 µm. As can be also observed in Figure 1, in sample TR02 most of the particles had two sizes (bimodal distribution). In suspension polymerization, the evolution of particle size comprises four stages. First there is the “transition stage”, during which the average droplet size decreases according to the droplet breakup rate. The second is the “quasi-steady state”, where

the droplet rupture rate is equal to that of coalescence, resulting in a constant average droplet size. The fourth is the “growth or sticky stage”, in which the droplet breakup rate lags behind the coalescence rate due to increased droplet interference. This stage leads to larger droplets. Finally, there is the “point of identification”, where the viscous droplets resulting from the previous phase can no longer be broken or undergo coalescence. The sequence of these four stages is what often causes the size distribution of the droplets to become wider than that of the initial monomer droplets[22] Therefore, the size of the final droplets is determined by the contribution of rupture and coalescence. Both processes are related to the surface energy of the droplet, which is proportional to the interfacial tension. For low coalescence systems, bimodality is observed due to the droplet size distribution being attributed to the drop breakup process[23] Brown and Pitt[24] also attributed bimodality in suspension systems to a breakup process that involves removal of small droplets whose sizes are approaching the maximum stable value. Thus, the production of a bimodal distribution from a rupture process must be assigned to erosive rupture involving the removal of a number of small drops to a larger one. This gives rise to two overlapping distributions with modes corresponding to daughter or satellite droplets, depending on the volume ratio[23]. The TR17 and TR10 samples had significant yields and increased particle sizes smaller than 150 µm, with no significant variations. The concentration of particles gradually increased until it reached the smallest particle size range detectable by the analyzer.
In sample TR02, more than half of the concentrated mass of particles was between 88-180 µm, whereas sample TR17 had a significantly smaller amount in the range below 75 µm. Sample TR10 had a larger amount of particles concentrated in the smaller size range than sample TR02. These results indicate that increasing the stirring rate decreased the particle sizes. This can be attributed to the fact that stronger stirring produced smaller droplets in the suspension because the kinetic energy ruptured larger particles. They also were more uniform, since they were better distributed in the dispersed phase.
The particle size distribution and consequently the size of the polymer beads formed depend on the balance between the rates of breakage and coalescence of the
droplets. One of the factors that influences this mechanism is the stirring speed of the reaction mixture[17]. Therefore, as can be seen in Figure 2, the SEM analyses showed that all particles had spherical morphology and no agglomerations. It can be concluded that the agitation mechanism remained balanced. In addition, these results corroborate the sieving result presented in Figure 1. It is evident that increasing the stirring speed during synthesis produced particle size distributions with narrower ranges. It can also be noted that sample TR02 (Figure 2a) had larger particle sizes than TR10 (Figure 2b) and TR17 (Figure 2c). It is also noteworthy that some material adhered to their surfaces (Figures 2a, 2b and 2c, highlighted), probably due to the presence of added magnetic material. Similar results were reported in the literature[25]
Thermogravimetric analysis (Figure 3a) showed a single stage of thermal degradation of all the samples. There also was no significant variation in initial degradation temperatures (TONSET), which were approximately 310 °C; 300 °C and 317 °C for TR02, TR10 and TR17, respectively. In all cases, the residue content (magnetite) of the three samples was around 3%. The degradation temperatures varied by about 17 °C (5%) among the three samples values considered acceptable by the method. The TMAX values also did not present significant variation (466 °C, 450 °C and 455 °C of TR02, TR10 and TR17, respectively). The residue was mainly due to the degradation of inorganic material present in the sample, in turn related to the content of Fe3O4 incorporated. The residue level was around 3%, showing that the incorporation of magnetite in these samples was much lower than expected (10% of the total mass of magnetic material added). These results are in agreement with previous reports in the literature[15].
The analysis of the magnetic properties (Figure 3b) showed that samples TR10 and TR17 had slightly higher saturation magnetization than TR02, probably associated with the fact that smaller particle sizes mean a higher number of particles in the same mass of microspheres, and a greater amount of magnetite contained in that mass (considering constant content and size of magnetite particles). Therefore, a sample with smaller particle size has better magnetic properties, which are measured in relation to mass[19]. The samples had low hysteresis (residual magnetization less than 0.1 emu/g) and saturation magnetization levels between 1.5 and 2.2 emu/g. Lee et al.[26] obtained saturation magnetization values in the range of 1.2 to 4.0 emu/g. According to the authors, those results were adequate for the use of these materials as ionexchange resins with magnetic properties.
In this study, the PVA was evaluated at 1, 2 and 3%w/v, maintaining stirring speed and BPO concentrations at 480 rpm and 2%, respectively.
Suspension agent concentration has been found to have a strong influence on suspension stability, thus being directly responsible for the average size and size distribution of monomer droplets (organic phase) inside the aqueous phase[13]. When an inadequate amount of stabilizer is used, the suspension will be unstable and the monomer/polymer droplets will coalesce and become large[27]. The suspension
Bouças, T. A. O., Costa, M. A. S., Cunha, H. A., Silva, M. R., Hui, W. S., & Castanharo, J. A.

agent used in this work was PVA, whose behavior depends on the extent to which the acetate groups are hydrolyzed[28] In this case, the hydrolysis degree was 85%, considered good for droplet stabilization in aqueous media, since the droplets can retain their integrity even when stirring speeds are increased[29]. This could explain why none of the synthesized samples had collapsed spheres. As can be seen in Figure 4, SEM micrographs of the samples revealed particles with spherical shape as well as agglomeration, with
some microspheres having non-uniform surfaces (highlighted in Figures 4a and 4b) where 1 and 2% w/v PVA were used. Also, some of them were rough and some were smooth.
Figure 5 shows that the particle size distribution narrowed with increasing concentration of suspending agent to a certain point. In sample TR05, more than two-thirds of the particle size was in the 88 to 150 µm range, and had 2% PVA, while TR02, with 1% PVA, had two substantial peaks and less than 60% of the sizes in the same range. However, sample

TR18, containing 3% PVA, had a wider distribution than the others, with two sample yield peaks and practically a third of the yield concentrated in the 180 to 250 μm range (the range with the best performance). The distribution followed the bimodal pattern of TR02, with particle size concentration in even larger ranges.
We observed an optimal PVA content, since overly high or low concentration of the suspension agent led to broader particle size distribution and larger particles[30]. However, the presence of large amounts of polymeric material can have an opposite effect when the PVA solution is saturated. Excessive amounts of PVA can lead to aggregation of particles. This fact can explain why sample TR18, with higher PVA content, exhibited a more random particle size profile and higher yields of larger size fractions. This hypothesis is corroborated by Figure 4d, where agglomerated microspheres can be seen in sample TR18, but are not found in samples TR02 (Figure 4a) and TR05 (Figure 4b).
As can be seen in Figure 6a, thermogravimetric analysis showed a single stage of thermal degradation for all the samples. There was no significant variation between the degradation onset temperature of TR02 and TR05. Their TONSET values were approximately 280 °C and the residue content (magnetite) was around 5%. However, the TONSET of TR18 was around 325 °C (3% PVA). It is known that the nature of the sample (mass, volume or physical form) can affect TGA results. Therefore, it is possible that the significant polydispersion and also the greater volume of

agglomerations in TR18 may have influenced this result in relation to the other samples. The TMAX value also did not vary significantly between samples TR02 and TR05 (both approximately 415 °C). Sample TR18, on the other hand, presented a slightly different TMAX, of 420 °C. The TGA graphs also showed that TR18, with the greatest suspension agent content, contained significantly lower solid residue (3%) than the other samples (5%), with lower PVA content. The residue was mainly attributed to degradation of the inorganic material present in the sample, and hence was

related to the content of Fe3O4 incorporated. The residue values showed that the incorporation of magnetite in all the samples was much lower than we expected (10% of the total mass of magnetic material added). These results are in agreement with another study[26]. There are two possible causes of this: either the agglomeration of particles reduced the amount of magnetite inside the spheres; or the sampling and selection of microspheres was poor and the relative number of non-magnetic particles was higher. Samples TR02 and TR05 had higher saturation magnetization (1.5 and 1.7 emu/g, respectively) than sample TR18 (1.1 emu/g) (Figure 6b). This can be an indication that TR18 had lower magnetic material content than the other two. Again, the samples showed typical superparamagnetic behavior at room temperature with any hysteresis loop.
In this study, the BPO was evaluated at 2, 4 and 6% mol, maintaining stirring speed and PVA concentrations at 480 rpm and 1% w/v, respectively. The content of initiator affects the number of threads started in the reaction medium, thus influencing the particles’ sizes and their distribution[15,31] The higher the number of threads initiated, the smaller and more uniform the particles produced will be. We verified that similar to the suspension agent, the initiator content also had optimal values. High concentrations of BPO led to particle agglomeration and broader particle size distribution. Figure 7 shows that increasing the initiator concentration to a certain point narrowed the particle size distribution. Sample TR07, with 4% BPO, had a much narrower distribution than TR02, with 2%. More than three-quarters of the particle mass was concentrated in the 88-150 µm size range. Sample TR19, however, although having higher initiator content, had a much wider distribution, with less than half of the yield concentrated in the 88-150 µm range (the range containing the highest yield). Even so, the distribution gradually increased, both in larger and smaller ranges, unlike the bimodal pattern of sample TR02. According to the literature, increasing the initiator concentration causes an instantaneous increase in the concentration of oligomeric radicals, which consequently increases the rate of association of oligomers and the rate of coagulation of unstable particles, resulting

7. Polymer particle size distribution obtained by sieving with initiator content: 2% mol BPO (TR02); 4% mol BPO (TR07); and 6% mol BPO (TR19).
in larger particle sizes[30]. The particle size distribution in this study, however, decreased when the initiator content increased from 2% to 4%, but the distribution increased with 6% initiator content. This is in line with the theory proposed by Liu et al.[30]. The narrower distribution in TR07 can be attributed to the fact that 4% is the optimal initiator content for the system in question, below which there may be few radicals, so that chain junction termination is the most common termination mechanism, explaining the greater aggregation in sample TR02 (2% BPO) in relation to TR07 (4% BPO). Conceição et al.[15], studying magnetic STY-DVB, reported a tendency for the polydispersion to decrease with increasing initiator concentration. We did not observe this behavior.
SEM micrographs of the samples (Figure 8) showed particles with spherical shape as well as agglomeration (surface highlight in Figures 8a, 8b and 8c). However, there was no uniformity of the microspheres’ surface appearance. Some had rougher surfaces than others. The smallest and most uniform particles were produced with 4% BPO.
As shown in Figure 9a, all the samples had a single thermal degradation stage. The degradation onset values
Effect of synthesis conditions on the properties of magnetic crosslinked polystyrene microspheres


were around 280, 275 and 270 °C for TR02, TR07 and TR19, respectively, while their maximum degradation temperatures were 420 °C, 418 °C and 400 °C, respectively. Ramos et al.[16] reported that a higher percentage of initiator caused a decrease in the degradation onset temperature. Only large differences in initiator concentration affected the thermal stability of the polymers formed in this study (TR02 compared to TR19). The VSM results (Figure 9b) showed no relationship between the increase in BPO concentration and saturation magnetization. This may be due to the selection of samples analyzed, reinforcing the previously described heterogeneous behavior of iron
incorporation in polymeric particles[25]. The MS values were 1.5, 0.7 and 2.0 emu/g for TR02, TR07 and TR19, respectively. Although the saturation magnetization results presented different values for all samples, the residues observed by TGA indicated no difference, reinforcing once again the heterogeneity of the incorporation of magnetic material in the microspheres.
The stirring speed was the main influence on the size of the obtained polymeric particles. At higher speeds,
Bouças, T. A. O., Costa, M. A. S., Cunha, H. A., Silva, M. R., Hui, W. S., & Castanharo, J. A.
smaller particles and narrower particle size distribution were obtained. The contents of initiator and suspending agent also influenced the particle size distribution of the synthesized copolymers. The initiator did not have a significant effect on the thermal stability of the polymers formed or on the amount of inorganic material incorporated. It was possible to obtain magnetic polymeric microspheres with behavior close to superparamagnetism for possible use in separation processes.
5. Author’s Contribution
• Conceptualization – Thiago Alexandre de Oliveira Bouças; Marcos Antonio da Silva Costa.
• Data curation – NA.
• Formal analysis – Wang Shu Hui; Manoel Ribeiro da Silva; Thiago Alexandre de Oliveira Bouças.
• Funding acquisition - Marcos Antonio da Silva Costa.
• Investigation – Thiago Alexandre de Oliveira Bouças.
• Methodology – Thiago Alexandre de Oliveira Bouças; Marcos Antonio da Silva Costa.
• Project administration – Marcos Antonio da Silva Costa.
• Resources – Marcos Antonio da Silva Costa; Wang Shu Hui; Manoel Ribeiro da Silva.
• Software – NA.
• Supervision – Marcos Antonio da Silva Costa.
• Validation – NA.
• Visualization – Thiago Alexandre de Oliveira Bouças; Henrique Almeida Cunha; Jacira Aparecida Castanharo.
• Writing – original draft – Thiago Alexandre de Oliveira Bouças; Henrique Almeida Cunha.
• Writing – review & editing – Marcos Antonio da Silva Costa; Jacira Aparecida Castanharo; Wang Shu Hui; Manoel Ribeiro da Silva.
6. Acknowledgements
The authors thank the National Council for Scientific and Technological Development (CNPq), Office to Coordinate Improvement of Higher Education Personnel (CAPES), and Carlos Chagas Filho Research Foundation of the State of Rio de Janeiro (FAPERJ) (Proc. nº E-26010.000982/2019, Cooperative Research Network on Nanostructured Materials and Device Engineering) for financial support.
7. References
1 Philippova, O., Barabanova, A., Molchanov, V., & Khokhlov, A. (2011 ). Magnetic polymer beads: recent trends and developments in synthetic design and applications. European Polymer Journal, 47(4), 542-559 http://doi.org/10.1016/j. eurpolymj.2010.11.006
2. Martin, C., & Cuellar, J. (2004). Synthesis of a novel magnetic resin and the study of equilibrium in cation exchange with amino acids. Industrial & Engineering Chemistry Research, 43(2), 475-485 http://doi.org/10.1021/ie0302239
3 Castanharo, J. A., Ferreira, I. L. M., Silva, M. R., & Costa, M. A. S. (2018). Core-shell magnetic particles obtained by
seeded suspension polymerization of acrylic monomers. Polímeros: Ciência e Tecnologia, 28(5), 460-467 http://doi. org/10.1590/0104-1428.10517.
4 Formiga, W. J. F., Mello, I. L., Costa, M. A. S., Silva, M. R., & Oliveira, M. G. (2013). Microesferas poliméricas magnéticas à base de estireno e divinilbenzeno com morfologia casca e núcleo. Polímeros: Ciência e Tecnologia, 23(2), 262-269. http://doi.org/10.4322/polimeros.2013.075
5 Formiga, W. J. F., Silva, M. R., Cunha, H. A., Castanharo, J. A., Ferreira, I. F. M., & Costa, M. A. S. (2023). Influence of benzoyl peroxide and divinylbenzene concentrations on the properties of poly(glycidyl methacrylate-co-divinylbenzene) magnetic microspheres. Macromolecular Reaction Engineering, 17(4), 2200070 http://doi.org/10.1002/mren.202200070
6. Souza, F. S., Costa, M. A. S., Santa Maria, L. C., Mello, I. L., Silva, M. R., & Wang, S. H. (2013). Síntese e caracterização de copolímeros reticulados à base de estireno, divinilbenzeno e metacrilato de metila com propriedades magnéticas. Polímeros: Ciência e Tecnologia, 23(1), 82-90. http://doi.org/10.1590/ S0104-14282013005000004
7 Queiroz, C. N., Cunha, H. A., Silva, M. R., Oliveira, M. G., Castanharo, J. A., Ferreira, I. L. M., & Costa, M. A. S. (2023). Influence of synthesis parameters on the magnetic, thermal, and morphological properties of poly(glycidyl methacrylate-co-divinylbenzene)/magnetite. Macromolecular Reaction Engineering, 17(4), 2200073 http://doi.org/10.1002/ mren.202200073
8 Santa Maria, L. C., Costa, M. A. S., Soares, J. G. M., Wang, S. H., & Silva, M. R. (2005). Preparation and characterization of manganese, nickel and cobalt ferrites submicron particles in sulfonated crosslinked networks. Polymer, 46(25), 1128811293 http://doi.org/10.1016/j.polymer.2005.09.055
9 Pastukhov, A. V. (2021 ). Magnetic sorbents based on hypercrosslinked copolymers of styrene and divinylbenzene with immobilized iron oxides. Reactive & Functional Polymers, 160, 104823-104829 http://doi.org/10.1016/j. reactfunctpolym.2021.104823
10 Chae, H. S., Piao, S. H., Han, W. J., & Choi, H. J. (2018). Core/ shell polystyrene/magnetite hybrid nanoparticles fabricated by pickering emulsion polymerization and their magnetorheological response. Macromolecular Chemistry and Physics, 219(5), 1700408 http://doi.org/10.1002/macp.201700408
11 Yu, P., Sun, Q., Pan, J., Tan, Z., Dai, J., Yan, Y., & Cheng, F. (2013). Performance of poly(styrene–divinylbenzene) magnetic porous microspheres prepared by suspension polymerization for the adsorption of 2, 4-dichlorophenol and 2, 6-dichlorophenol from aqueous solutions. Adsorption Science and Technology, 31(7), 641-656. http://doi.org/10.1260/0263-6174.31.7.641.
12 Mendes, M. S. L., Araujo, A. B., Neves, M. A. F. S., & Pedrosa, M. S. (2022). Advances in magnetic polymeric styrene-divinylbenzene nanocomposites between magnetite and maghemite nanoparticles: an overview. Current Applied Polymer Science, 5(1), 3-14 http://doi.org/10.2174/2452271 605666220304091807
13 Qin, Z., Ma, Z.-H., Zhi, J.-K., & Fu, Y.-L. (2019). A facile synthesis of magnetite single-crystal particles by employing graphene oxide sheets as template for promising application in magnetic fluid. Rare Metals, 38(8), 764-769 http://doi. org/10.1007/s12598-018-1197-5
14 Santa Maria, L. C., Leite, M. C. A. M., Costa, M. A. S., Ribeiro, J. M. S., Senna, L. F., & Silva, M. R. (2004). Characterization of magnetic microspheres based on network styrene and divinylbenzene copolymers. Materials Letters, 58(24), 30013006. http://doi.org/10.1016/j.matlet.2004.05.028.
15 Conceição, B. M., Costa, M. A. S., Santa Maria, L. C., Silva, M. R., & Wang, S. H. (2011). A study of the initiator concentration’s
Effect of synthesis conditions on the properties of magnetic crosslinked polystyrene microspheres
effect on styrene- divinylbenzene polymerization with iron particles. Polímeros: Ciência e Tecnologia, 21(5), 409-415
16 Ramos, G. S. M., Mendes, M. S. L., Neves, M. A. F. S., Pedrosa, M. S., & Silva, M. R. (2020). Experimental design to evaluate the efficiency of maghemite nanoparticles incorporation in styrene-divinylbenzene copolymers. Journal of Applied Polymer Science, 138(18), 50318 http://doi.org/10.1002/app.50318
17 Machado, F., Lima, E. L., & Pinto, J. C. (2007). Uma revisão sobre os processos de polimerização em suspensão. Polímeros: Ciência e Tecnologia, 17(2), 166-179 http://doi.org/10.1590/ S0104-14282007000200016
18. Yuan, Q. E., & Williams, R. A. (2007). Large scale manufacture of magnetic polymer particles using membranes and microfluidic devices. China Particuology , 5 (1-2 ), 26 -42 http://doi. org/10.1016/j.cpart.2007.02.001
19 Massart, R. (1981). Preparation of aqueous magnetic liquids in alkaline and acidic media. IEEE Transactions on Magnetics, 17(2), 1247-1248 http://doi.org/10.1109/TMAG.1981.1061188
20 Guo, Z., Bai, S., & Sun, Y. (2003). Preparation and characterization of immobilized lipase on magnetic hydrophobic microspheres. Enzyme and Microbial Technology, 32(7), 776-782 http://doi. org/10.1016/S0141-0229(03)00051-6
21 Koneracká, M., Kopčanský, P., Antalík, M., Timko, M., Ramchand, C. N., Lobo, D., Mehta, R. V., & Upadhyay, R. V. (1999). Immobilization of proteins and enzymes to fine magnetic particles. Journal of Magnetism and Magnetic Materials, 201(1-3), 427-430 http://doi.org/10.1016/S03048853(99)00005-0
22 Alroaithi, M., Jahanzad, F., & Sajjadi, S. (2018). Suppressing coalescence and improving uniformity of polymer beads in suspension polymerization using a two-stage stirring protocol. Industrial & Engineering Chemistry Research, 57(35), 1188311892 http://doi.org/10.1021/acs.iecr.8b01599
23 Chatzi, E. G., Boutris, C. J., & Kiparissides, C. (1991). Monitoring of drop size distributions in agitated bessels. 2. Effect of stabilizer concentration. Industrial & Engineering Chemistry Research, 30(6), 1307-1313 http://doi.org/10.1021/ ie00054a035
24 Brown, D. E., & Pitt, K. (1972). Drop size distributions of stirred non-coalescing liquid-liquid systems. Chemical Engineering Science, 27(3), 557-583 http://doi.org/10.1016/00092509(72)87013-1
25. Castanharo, J. A., Ferreira, I. L. M., Costa, M. A. S., Silva, M. R., Costa, G. M., & Oliveira, M. G. (2015). Microesferas magnéticas à base de poli(metacrilato de metila-co-divinilbenzeno) obtidas por polimerização em suspensão. Polímeros: Ciência e Tecnologia, 25(2), 192-199
26. Lee, Y., Rho, J., & Jung, B. (2003). Preparation of magnetic ion-exchange resins by the suspension polymerization of styrene with magnetite. Journal of Applied Polymer Science, 89(8), 2058-2067. http://doi.org/10.1002/app.12365.
27 Brooks, B. (2010). Suspension polymerization processes. Chemical Engineering & Technology, 33(11), 1737-1744 http://doi.org/10.1002/ceat.201000210
28 Atanase, L. I., & Riess, G. (2010). Poly(vinyl alcohol-co-vinyl acetate) complex formation with anionic surfactants particle size of nanogels and their disaggregation with sodium dodecyl sulfate. Colloids and Surfaces. A, Physicochemical and Engineering Aspects, 355(1-3), 29-36. http://doi.org/10.1016/j.colsurfa.2009.11.024.
29 Zerfa, M., & Brooks, B. W. (1996). Vinyl chloride dispersion with relation to suspension polymerization. Chemical Engineering Science, 51(14), 3591-3611 http://doi.org/10.1016/00092509(96)00002-4
30. Liu, X.-Y., Ding, X.-B., Zheng, Z.-H., Peng, Y.-X., Long, X.-P., Wang, X.-C., Chan, A. S. C., & Yip, C. W. (2003). Synthesis of novel magnetic polymer microspheres with amphiphilic structure. Journal of Applied Polymer Science, 90(7), 18791884 http://doi.org/10.1002/app.12860
31. Chaudhary, V., & Sharma, S. (2019). Suspension polymerization technique: parameters affecting polymer properties and application in oxidation reactions. Journal of Polymer Research, 26(5), 102. http://doi.org/10.1007/s10965-019-1767-8.
Received: Nov. 25, 2023
Revised: Mar. 28, 2024
Accepted: July 08, 2024
Mauro Sergio da Silva1 , Alana Gabrieli de Souza2 , Derval dos Santos Rosa2* , Ticiane Sanches Valera1 and Hélio Wiebeck1
1Departamento de Engenharia Metalúrgica e de Materiais – PMT, Escola Politécnica – POLI, Universidade de São Paulo – USP, São Paulo, SP, Brasil
2Centro de Engenharia, Modelagem, e Ciências Sociais Aplicadas – CECS, Universidade Federal do ABC – UFABC, Santo André, SP, Brasil
*derval.rosa@ufabc.edu.br
Obstract
The UV-resistance performance of waterborne epoxy coatings with different aminic-curable substances was evaluated to solve the widely known problem of poor weathering resistance of coatings. Samples were evaluated by color and gloss changes, FTIR, thermal properties, and macroscopic morphology. Results indicated that higher aminic values in curing agents degraded faster than lower aminic values (increases of ~3% in delta b and delta E between 5 and 20%). All samples showed a gloss variation varying between 40 and 70 G.U. FTIR and Tg indicated the resin degradation when using curing agents of unmodified aliphatic amine because of its rapid chemical degradation under a UV environment, as confirmed by microscopy images. The curing agents with aminic values between 200 and 300 KOH/g and the equivalent weight of amine hydrogen higher than 120 g/eq showed the best performance in epoxy coatings and good stability against accelerated aging, being promising options for future applications.
Keywords: epoxy coatings, curing agent, yellowing, weathering.
How to cite: Silva, M. S., Souza, A. G., Rosa, D. S., Valera, T. S., & Wiebeck, H. (2024). Greener waterborne epoxy coatings with optimized UV-resistance. Polímeros: Ciência e Tecnologia, 34(3), e20240034. https://doi.org/10.1590/01041428.20230133.
Epoxy resins play an important role as protective coatings due to their chemical inertness that allows good corrosion protection, besides their chemical resistance, electrical insulation properties, and adhesion to heterogeneous substrates. Two-component epoxy/amine systems are widely used in civil construction and industrial maintenance. However, the currently used systems generally contain high levels of volatile organic compounds (VOCs) that are harmful to human health and the environment. Thus, to improve the environmentally friendly character of this class of materials, there is a movement to shift the solvent-borne system to waterborne ones[1]. As an example, the European Union in the Directive 2004/42/CE of the European Parliament and of the Council of 21 April 2004 stated the maximum rates of volatile organic compounds for several types of coatings.
Waterborne coatings are widely used as an effective method of protecting against corrosion on metallic substrates, and several works have reported the importance and effectiveness of these materials. Wang et al. (2019)[1] prepared waterborne epoxy coatings with graphene modified with lignin for anticorrosive applications and reported that nanocomposite coatings showed an anti-corrosion effect due to the labyrinth effect caused by the fillers. Sheng et al.[2] prepared waterborne polyurethane composites containing
MXene nanosheets and reported low corrosion current and UV-blocking properties.
Despite the excellent applicability and economic interest, epoxy systems are sensitive to exposure to UV radiation, not only as a function of the incidence of sunlight but also of the visible light spectrum[3]. The polymeric structure undergoes photochemical damage on the surface of the applied paint film, resulting in its degradation[4]. The use of additives that block and absorb UV radiation is common to delay this effect but results in high costs and relatively low effectiveness for long periods of exposure during the working time of the painted substrate[2,5]. As a result of the degradation process, the coatings are subjected to chalking, gloss loss, and yellowing, decreasing the coating capacity to shield the substrate, and implying a complete loss of function[6,7]. Liu et al.[8] prepared waterborne epoxy resin containing emulsified asphalt to improve mechanical properties and reported waterproof performance, aging, and wear resistance. Li et al.[7] prepared epoxy coating with polyetheramine-functionalized graphene oxide and reported that the filler was important to avoid the yellowing of the coating, quantified by color parameters.
Substantial work has been done to improve the weathering resistance of waterborne epoxy coatings to guarantee long
Silva, M. S., Souza, A. G., Rosa, D. S., Valera, T. S., & Wiebeck, H.
shelf-life for these materials without property loss, such as color, mechanical resistance, and others[9-12]. Among the main properties, several additives are usually incorporated, such as nanoparticles, graphene, etc. However, even with a wide range of possibilities, there is still a knowledge gap in efficient additives that delay or prevent the degradation of epoxy coatings during application, which is a technicalscientific challenge. Aiming to expand the possibilities of use and application materials, this work proposes the development of waterborne epoxy coatings using aminiccurable substances available on the market to reduce color deterioration and property loss, ensuring good properties for coatings performance.
2.1 Materials
Westlake Epoxy (Texas, USA) kindly donated the waterbased epoxy resin dispersion and the five different types of curing agents (Table 1). The EPI-REZ Resin 7521-WH57 (resin) has solids of 55-59%, equivalent epoxy weight of 480-600 g/eq, and viscosity at 25 °C of 600-6000 cP. Momentive Performance Materials (New York, USA) donated the leveling additive and the deionized water, and the co-solvent was acquired by The Dow Chemical Company (Michigan, USA). The chemical structures of the curing agents are confidential. Table S1 – Supplementary
Raw material
EPI-REZ Resin 7521-WH-57 (Resin)
EPIKURE 8530-W-75
EPIKURE 6870-W-53
EPIKURE 3223
EPIKURE 3234
EPIKURE 8546-W-55
Dowanol DPnB (co-solvent)
Material shows the main properties of all the catalysts. CU abbreviation refers to curing agent.
2.2 Formulation development
The formulations were done based on the stoichiometry of the system considering the ammine hydrogen proportion related to the epoxy content. Usually, the stoichiometry ratio is 1:1 to maximize the crosslinking density, as reported in previous works[11,12], and it is calculated according to Equation 1, where EWa is the equivalent weight of ammine and EWe is the epoxydic equivalent weight, and PHR is the per hundred resin based on the solids.
The result represents the amount of curing agent necessary to react with 100 g of epoxydic resin for the proportion 1:1[13,14]
After initial tests to adjust the raw materials quantities (water, additive, and solvent), 12 formulations were designed to evaluate the curing agent efficiency. The preparation process was divided into two parts: 1—resin preparation and 2—curing agent addition. Before the mixture of parts 1 and 2, the raw materials were weighted separately, according to Table 2
Description
Waterborne dispersion of epoxydic resin
Amine curing agent
Amine curing agent
Amine curing agent
Amine curing agent
Amine curing agent
COATOSIL 28161 (Additive) Silicon-based additive
Characteristics
Film-forming agent
Reaction catalyst for epoxy systems
Reaction catalyst for epoxy systems
Reaction catalyst for epoxy systems
Reaction catalyst for epoxy systems
Reaction catalyst for epoxy systems
for waterborne systems
for waterborne systems
Additive to improve paint spread
Developed formulations aiming to determine the best proportion between resin and curing agent for the development of waterborne epoxy coatings.
Greener waterborne epoxy coatings with optimized UV-resistance
After the weighting, the parts A and B were mixed following the system stoichiometry for ten minutes and sprayed into rectangular panels using a spray gun HVLP – High Volume Low Pressure, and then left in a chamber free of light, at 25 °C, for seven days, for complete coatings curing, as schematically illustrated in Figure 1
After the samples had completely cured, accelerated weathering tests were conducted in a QUV-A chamber (Adexim-Comexim, São Paulo, Brazil) with 500 hours of exposure, following the ASTM G154-23[15]. Throughout the exposure time, the samples were evaluated seven times: 39, 109, 162, 207, 375, 439, and 500 hours.
2.3 Characterization
2.3.1 Appearance changes
The differences in the samples’ appearance were quantified by color and gloss measurements. The color was
spectrophotometrically measured in the CIELAB space, in triplicate, similar to the Yari and Rostami work[16], using a Spectro 2 Guide spectrophotometer (BYK Gardner, Wesel, Germany). The gloss values were measured using a MicroTRI-Gloss glossmeter (BYK Gardner, Wesel, Germany).
2.3.2 Fourier-transform infrared spectroscopy
FTIR spectra of coatings before and after the weathering were recorded in Spectrum 2 Equipment (PerkinElmer, Connecticut, USA), equipped with an attenuated total reflectance (ATR) accessory. Data acquisition was performed in a range of 500–4000 cm-1, 62 scans, and a spectral resolution of 4 cm-1 .
2.3.3 Differential scanning calorimetry
The glass transition temperature (Tg) was analyzed by differential scanning calorimetry (DSC) using a TA

Silva, M. S., Souza, A. G., Rosa, D. S., Valera, T. S., & Wiebeck, H.
Instrument Series Q20 (TA Instruments, New Castle, USA). The specimens were heated from -20 °C to 180 °C at a rate of 10 °C/min.
2.3.4 Macroscopic morphology
The coatings’ surface morphology was analyzed using a microscope TESCAN VEGA3 from Tescan do Brasil (São Paulo, Brazil). The equipment is equipped with a heated tungsten filament cathode operating at 30 keV. Images were recorded at 500x.
3. Results and Discussions
3.1 Appearance changes - color
Epoxy coatings have a wide range of applications in coatings’ field due to their high range of attainable properties and versatility. However, under severe conditions of oxidation, sunlight, and other external agents, the coatings’ tend to degrade, resulting in chemical and physical changes. CIELab delta b and delta E parameters during the exposure testing were measured and are presented in Tables 3 and 4. Accelerated weathering usually changes the sample appearance because it changes the reflectance spectra, altering color shade and lightness. Changes in color parameters are usually associated with resin degradation, in this case, the epoxy waterborne resin, by the oxidation and evolution of gaseous and soluble products[17]. In this case, the aromatic moiety presence absorbs UV radiation, resulting in discoloration and chalking.
The b* parameter is attributed to the yellow-blue axis, and the b* increase in the positive direction indicates the yellowing of coating samples. According to Verma, the overall color difference (∆E) in PU coatings is best represented by ∆b since the epoxy resin absorbs more UV light due to the benzene ring structure[18]. According to Rivaton et al., the photo-oxidation of epoxy resin involves reactions with the methylene groups in the α-position to the ether groups related to secondary hydroxyl groups dehydration[19]
In the first weathering hours, experiments 8, 9, and 10 showed the most prominent yellowness (Table 3); these samples were prepared with CU-3 and CU-4, the curing agents with the higher aminic values. After the complete exposure time, it was observed that most experiments
Table 4. T g results of the investigated coatings before and after aging (0 and 500 h of exposure). Sample
showed significant color changes over time, and samples with higher b* values for both the negative and positive axis became darker due to oxidation of the degradation products[20]. According to Ghasemi-Kahrizsangi et al., the color change is associated with light absorption by exciting an electron from the ground state into an excited state by chromophore groups, such as C=C and C=O. After the UV radiation absorption, some discoloration or color change can be observed[21]. The delta E was evaluated to evaluate the color change in a more detailed way (Table S2 and Figure 2).
Considering the ∆E results, it is observed values between 1 and 16 after 500 h of UV-light exposition. The literature shows a broad range of values that depend on the curing agent and several types of resin. Ghrohrodi et al. prepared epoxy coatings containing nano-porous Zr(IV) and reported ∆E between 5.3 and 7.1. According to the authors, the epoxy coating shows intrinsic low UV resistance (~7), and the use of nanoparticles can decrease the coating damage after UV exposure[22]. Li et al.[23] prepared epoxy graphene coatings aiming for weather and corrosion resistance and found ∆E between 4.1 and 15.6. The authors attributed higher delta E values to the absence of graphene, resulting in composites with high sensitivity to color changes, and the smallest values, i.e., most resistant ultraviolet aging samples, were reported as those with 0.5% graphene.
Greener waterborne epoxy coatings with optimized UV-resistance
According to DIN 55987[24,25], the UV stability of coatings is acceptable and highly stable if ∆E ≤ 2, but samples that slightly exceed this value can be considered stable if more tests are conducted to confirm the coating performance. In this work, after 20 days of exposure (500 h), only formulation 3 showed acceptable results, followed by 4 and 9, i.e., curing agents CU-2 and CU-4 (both curing agents are unmodified aliphatic amines). The chromophoric groups are expected to react with the curing agents, reducing the chemical groups available to interact with the UV light and decreasing the color changes. On the other hand, samples 6, 7, and 8 showed the strong degradation of the polymeric material, indicating that CU-3 and CU-5 (unmodified aliphatic amine and modified polyamine, respectively) are UV-light unstable and not adequate for the coatings that aim for UV-resistance.
3.2 Appearance changes - gloss
Generally, exposure to weathering results in a marked decrease in gloss due to resin degradation and surface roughness[11]. Figure 3 shows the gloss 60° evolution at times 0 h and 500 h, and all samples showed a high initial gloss (>90 GU). The gloss values of all coatings, except for sample 8, were almost the same, revealing that the curing agents didn’t alter the surface gloss.


As expected, all samples showed a significant decrease in gloss values associated with photo-degradation after accelerated aging[12]. According to Rezig et al.[26], after the photodegradation, the coating surface results in nonhomogeneous surfaces with the formation of pits, resulting in an increase in roughness and decrease in thickness, which affects the light reflection and, consequently, gloss values. Another possible reason is that during degradation, organic material loss occurs because of photooxidation, changing the molecular bonds and resulting in microscopic changes in the coatings[27,28]. Most experiments showed decreases in the gloss higher than 60% after 500 h of weathering, except for samples 1, 2, and 4, suggesting that these samples are more resistant to the weathering conditions than the others. These results agree with the delta E values since the change in surface roughness influences the light interactions with the substrate by decreasing the gloss and affecting the color perception, i.e., high delta E values[29]. These results are consistent with those presented in SEM results in the next sections.
Samples 1, 2, and 4 were cured with CU-1 and CU-4, and the curing agents have in common the aminic value (between 200 and 300 KOH/g) and the equivalent weight of amine hydrogen (higher than 120 g/eq)[30]. Besides, according to Klippstein et al.[13], the curing agent for waterborne formulations can result in incompatibility with the epoxy resin, resulting in several problems, such as the tendency to exude the curing agent to the surface, a decrease of stability, changes in molecular weight and reduced reactivity. In this way, there is a possibility that the CU-3 (unmodified aliphatic amines), CU-4 (unmodified aliphatic amines), and CU-5 (modified polyamine with 55 wt% solids) have a slight incompatibility with the matrix that, at accelerated weathering, reduces the coating resistance. Another fact in agreement with the presented hypothesis is that the amines are more hydrophilic than the epoxy resin, and a possible incompatibility could result in concentrations of amine in localized domains, which, under severe aging conditions, accelerate the film’s degradation.
FTIR analysis was conducted to evaluate the chemical structure of the coatings before and after the accelerated weathering (Figure 4). The epoxy resin characteristic peaks include a broad band centered at 3383 cm-1 (extra hydroxyl groups generated during the curing reaction), and another between 3018 and 2770 cm-1 containing three minor peaks centered at 2973, 2914, and 2868 cm-1, attributed to C-H stretching vibrations from methylene and methyl groups, and C-H bonds of aliphatic and aromatic methine groups[29] These peaks were found for all samples after curing.
Other peaks were found at 1740 cm-1 (carbonyl groups stretching vibrations), 1605 and 1580 cm-1 (C-C stretching of aromatic rings), 1507 cm-1 (N-H bending vibrations), 1548 cm-1 (stretching vibrations of aromatic rings), 1383, 1359, 1293 cm-1 and 1233 cm-1 (C-O-C stretch from ether groups), 1180 cm-1 (O-H bend), 1106 and 1083 cm-1 (C-O vibrations), 939 cm-1 (epoxide group C-O-C), 823, 768, and 728 cm-1 (C-H out of plane bending), and 552 cm-1 (aromatic ring bending)[22,30-34]. Before the weathering, the samples
Silva, M. S., Souza, A. G., Rosa, D. S., Valera, T. S., & Wiebeck, H.

Figure 4. FTIR spectra of samples before (0h) and after (500h) accelerated weathering for the experiments with different curing agents (CU): a) 1 and 2 (CU-1), b) 3 and 4 (CU-2), c) 5 and 6 (CU-5), d) 7 and 8 (CU-1 + CU-3), e) 9 and 10 (CU-1 + CU-4), and f) 11 and 12 (CU-1 + CU-5).
showed very similar FTIR spectra with the main characteristic peaks at the same wavenumber for the different curing agents, as shown in Figure 4
The curing agent significantly influenced the chemical structure of the films after degradation, highlighting the changes in the region 1800-1550 cm-1, identified in the insert of all the graphs in Figure 4.
The bands in the region 3500-2900 cm-1 showed a decrease in intensity attributed to photooxidation after accelerated aging and the peak located at 1507 cm-1[35] All coatings showed the disappearance of peak 1740 cm-1 and a new band at ~1650 cm -1 related to aldehyde and peracid formation and the carbonyl formation during oxidation, respectively. The changes in these peaks agree with the photooxidative mechanism proposed by Bellinger and Verdu[26,36]. During the oxidation process, a ketone formation occurs derived from the secondary OH groups, and amide groups are generated from the abstraction of hydrogen from the methylene groups adjacent to the crosslink.
All the chemical changes are a consequence of epoxy chain scission that results in the formation of carbonyl, hydroperoxide, and other radical groups that are associated with photoreactions. Awad et al. reported that the appearance of carbonyl and hydroxyl groups after degradation results from the oxidation reactions and represents the polymeric scission[37]. The degradation generally induces surface cracking and loss of mass because of the bioactive transformation products formed during the degradation, which can be released into the environment as gas or leached compounds[38] Additionally, after the degradation, the coating becomes more brittle and rigid, increasing the stress absorption and generating microcracks[39]
Analyzing the FTIR graphs of Figure 4, the chemical structure loss of Experiments 8 and 10 is visible, which is associated with the complete degradation of the samples on a molecular level. Even with different curing agents, it is possible to note that all experiments showed, at least, a slight degradation of the matrix, as highlighted in the Figure 4 inserts, indicating that the coatings maintained a
UV-sensitive characteristic. It is interesting to note that the FTIR data corroborate a degradation of the coating resin in all samples but is more intense for samples 8 and 10, which is coherent with the degradation chemistry since both samples contain unmodified aliphatic amines from the curing agents. This result is related to the large variation in brightness and color observed for these samples, indicating the unfeasibility of using these coatings – and curing agents with aliphatic amines - in applications that require resistance to weathering. According to Bellinger and Verdu, these results can be attributed to the depletion of the oxidizable groups, and the loss of oxidized products formed during the degradation[36].
DSC was conducted to evaluate the effect of different curing agents on the coatings’ thermal properties by glass transition temperature analysis. The Tg of coatings is mainly influenced by the crosslink density, being a proportional property, i.e., the greater the crosslink density, the higher the T g [37,40]. T g values of the coatings before and after aging are reported in Table 4. Experiments 7, 8, 9, 10, 11, and 12 showed T g values higher than 40 °C before the accelerated aging, indicating that the mixture of CU-1 with CU-4 and CU-5 resulted in higher crosslink density when compared
to samples 1-6 that were cured with only one type curing agent, i.e., not a mixture. Samples with high Tg values are expected to have a slower ion diffusion rate and better barrier properties. Besides, the CU mixture probably produces a superior ability to emulsify liquid epoxy resin, resulting in more compact and uniform coatings. Lower Tg values indicate lower free volumes, i.e., less rigid and highly flexible coating.
After the accelerated weathering, all samples showed higher Tg values, which can be associated with the photolysis of the epoxy resin and the QUV temperature that can promote curing reactions or the exudation of some compounds, as previously discussed in FTIR[28,36]. In this case, if the samples were not completely cured before the test, the completion will occur during the aging, i.e., there are chemical changes. These results indicate that after aging, all samples are harder. According to Xiong and Li, the increase in the Tg values can be a result of dehydration condensation between hydroxide radicals inside the coatings, which can be the key to higher stability[41]
Optical micrographs of the coatings surface of all the samples after the accelerated aging are shown in Figure 5 (400x). The experiments showed different

Silva, M. S., Souza, A. G., Rosa, D. S., Valera, T. S., & Wiebeck, H.
morphological aspects depending on the weathering behavior. Samples 6-10 and 12 showed the worst appearance with a high number of fractures, voids, and a high number of defects. These samples also showed high Tg values, making the coating more rigid and brittle and allowing the materials to absorb stresses. According to Onn et al.[42], the changes in the surface morphology are associated with blistering, surface segregation, inhomogeneity, and swelling because of polymer network degradation. The observed changes are also associated with surface color change – yellowing, as discussed before, because of chromophoric group generation due to polymer degradation[9]. Additionally, the gloss reduction is attributed to the high number of protrusions on the surface samples and the increase in light diffusion due to the changes in the microstructures[4]
Samples 2-5 (one curing agent used), 7 (CU-1+CU-3), 10 (CU-1+CU-4), and 11 (CU-1+CU-5) showed minor visual changes, indicating moderate degradation with fewer weathering products, probably due to the formulations’ UV absorption and scattering ability[30,38,43]. However, macroscopically, samples 7 and 10 showed many defects, which is not possible for future applications. Considering all the results presented in this work, the samples with the best UV-resistance performance are 2 (CU-1), 3 (CU-2), and 4 (CU-2), i.e., the curing agents CU-3 and CU-4 (unmodified aliphatic amines) probably are not adequate for the studied system.
This work presented the development and UV-resistance performance of waterborne epoxy coatings with aminiccurable substances. Color and gloss changes, FTIR, thermal properties, and macroscopic morphology evaluated the coatings. Results indicated that samples with higher aminic values tended to degrade faster than those with lower aminic values, as confirmed by higher yellowness and darkness –caused by the oxidation of the polymer and the hydrophilic nature of these groups. Additionally, all samples showed a decrease in gloss values associated with surface roughness and polymer degradation changes. FTIR results confirm the scission of polymer chains with significant changes and the disappearance of the main peaks associated with the epoxy structure. The T g increased values after weathering might be ascribed to the photo-oxidative products of the coupling reactions due to the photolysis of the resin. The results were confirmed by morphological images that indicated degradation characteristics and loss of structure of some samples and indicated the most suitable samples for future studies and applications. Additionally, comparing the curing agents, those unmodified aliphatic amines showed an inferior performance with a high tendency to degrade under UV environment. Finally, the curing agents with aminic values between 200 and 300 KOH/g and the equivalent weight of amine hydrogen higher than 120 g/eq showed the best performance in epoxy coatings and good stability against accelerated aging, being promising options for future applications.
• Conceptualization – Mauro Sergio da Silva; Ticiane Sanches Valera; Hélio Wiebeck.
• Data curation – Mauro Sergio da Silva; Ticiane Sanches Valera; Hélio Wiebeck.
• Formal analysis – Mauro Sergio da Silva; Alana Gabrieli de Souza.
• Funding acquisition – Derval dos Santos Rosa; Ticiane Sanches Valera; Hélio Wiebeck.
• Investigation – Mauro Sergio da Silva; Alana Gabrieli de Souza.
• Methodology – Mauro Sergio da Silva; Ticiane Sanches Valera; Hélio Wiebeck.
• Project administration – Ticiane Sanches Valera; Hélio Wiebeck.
• Resources – Derval dos Santos Rosa; Ticiane Sanches Valera; Hélio Wiebeck.
• Software – NA.
• Supervision – Derval dos Santos Rosa; Ticiane Sanches Valera; Hélio Wiebeck.
• Validation – Mauro Sergio da Silva; Ticiane Sanches Valera; Hélio Wiebeck.
• Visualization – Mauro Sergio da Silva; Alana Gabrieli de Souza.
• Writing – original draft – Mauro Sergio da Silva; Alana Gabrieli de Souza; Derval dos Santos Rosa; Ticiane Sanches Valera; Hélio Wiebeck.
• Writing – review & editing – Mauro Sergio da Silva; Alana Gabrieli de Souza; Derval dos Santos Rosa; Ticiane Sanches Valera; Hélio Wiebeck.
This research was funded by CNPq (308053/2021-4 and 403934/2021-4) and FAPESP (2022/01382-3, 2021/147141, 2020/13703-3). The authors thank the CAPES (Code 001), UFABC, USP, IPT, IPEN, and REVALORES Strategic Unit.
7. References
1. Wang, S., Hu, Z., Shi, J., Chen, G., Zhang, Q., Weng, Z., Wu, K., & Lu, M. (2019). Green synthesis of graphene with the assistance of modified lignin and its application in anticorrosive waterborne epoxy coatings. Applied Surface Science, 484, 759770 http://doi.org/10.1016/j.apsusc.2019.03.229
2. Sheng, X., Li, S., Huang, H., Zhao, Y., Chen, Y., Zhang, L., & Xie, D. (2020). Anticorrosive and UV-blocking waterborne polyurethane composite coating containing novel two-dimensional Ti3C2 MXene nanosheets. Journal of Materials Science, 56(6), 4212-4224 http://doi.org/10.1007/s10853-020-05525-2
3 Rashvand, M., Ranjbar, Z., & Rastegar, S. (2011). Nano zinc oxide as a UV-stabilizer for aromatic polyurethane coatings. Progress in Organic Coatings, 71(4), 362-368 http://doi. org/10.1016/j.porgcoat.2011.04.006
4. Colonetti, E., Rovani, R., Westrup, J. L., Cercená, R., Cargnin, M., Peterson, M., & Dal-Bó, A. G. (2022). Effects of resin/curing agent stoichiometry and coalescence of emulsion particles on the properties of waterborne epoxy coatings upon accelerated weathering. Materials Chemistry and Physics, 275, 125228125238. http://doi.org/10.1016/j.matchemphys.2021.125228.
5 Wu, Y., Wu, X., Yang, F., & Ye, J. (2020). Preparation and Characterization of Waterborne UV Lacquer Product Modified
Greener waterborne epoxy coatings with optimized UV-resistance
by Zinc Oxide with Flower Shape. Polymers, 12(3), 668-679 http://doi.org/10.3390/polym12030668 PMid:32192083.
6 Li, Z., Zhu, L., Xie, X., Zhou, M., Fu, C., & Chen, S. (2023). High-Hardness, Water-Stable, and UV-Resistant Conductive Coatings Based on Waterborne PEDOT:PSS/Epoxy/(KH560/ SiO2) Composite. Journal of Composites Science, 7(2), 51-68. http://doi.org/10.3390/jcs7020051.
7 Li, K., Shan, W., Cui, J., Qiu, H., Yand, G., Zheng, S., & Yang, J. (2020). Enhanced corrosion resistance and weathering resistance of waterborne epoxy coatings with polyetheraminefunctionalized graphene oxide. Journal of Coatings Technology and Research, 17(1), 171-180 http://doi.org/10.1007/s11998019-00252-z
8 Liu, F., Zheng, M., Fan, X., Li, H., & Wang, F. (2021). Performance evaluation of waterborne epoxy resin-SBR compound modified emulsified asphalt micro-surfacing. Construction & Building Materials, 295, 123588 http://doi. org/10.1016/j.conbuildmat.2021.123588
9. Nikafshar, S., McCracken, J., Dunne, K., & Nejad, M. (2021). Improving UV-stability of epoxy coating using encapsulated halloysite nanotubes with organic UV-stabilizers and lignin. Progress in Organic Coatings, 151 , 105843 http://doi. org/10.1016/j.porgcoat.2020.105843
10 Woo, R. S. C., Chen, Y., Zhu, H., Li, J., Kim, J.-K., & Leung, C. K. Y. (2007). Environmental degradation of epoxy–organoclay nanocomposites due to UV exposure. Part I: photo-degradation. Composites Science and Technology, 67(15-16), 3448-3456 http://doi.org/10.1016/j.compscitech.2007.03.004
11 Huo, S., Liu, Z., Li, C., Wang, X., Cai, H., & Wang, J. (2019). Synthesis of a phosphaphenanthrene/benzimidazole-based curing agent and its application in flame-retardant epoxy resin. Polymer Degradation & Stability, 163, 100-109. http:// doi.org/10.1016/j.polymdegradstab.2019.03.003.
12 Wang, C., Huo, S., Ye, G., Song, P., Wang, H., & Liu, Z. (2023). A P/Si-containing polyethylenimine curing agent towards transparent, durable fire-safe, mechanically-robust and tough epoxy resins. Chemical Engineering Journal, 451(Part 2), 138768-138771 http://doi.org/10.1016/j.cej.2022.138768
13 Klippstein, A., Cook, M., & Monaghan, S. (2012). Water-based epoxy systems. In K. Matyjaszewski, & M. Möller (Eds.), Polymer science: a comprehensive reference (pp. 519-539), Netherlands: Elsevier http://doi.org/10.1016/B978-0-44453349-4.00281-8
14. Asada, C., Honjo, K., & Nakamura, Y. (2021). Utilization of steam-treated and milling-treated lignin from moso bamboo as curing agent of epoxy resin. Waste and Biomass Valorization, 12(11), 6261-6272 http://doi.org/10.1007/s12649-021-01444-8
15 American Society for Testing and Materials – ASTM. (2023). ASTM G154-23 -Standard Practice for Operating Fluorescent Ultraviolet (UV) Lamp Apparatus for Exposure of Materials USA: ASTM International
16 Yari, H., & Rostami, M. (2020). Enhanced weathering performance of epoxy/ZnO nanocomposite coatings via functionalization of ZnO UV blockers with amino and glycidoxy silane coupling agents. Progress in Organic Coatings, 147, 105773 http://doi. org/10.1016/j.porgcoat.2020.105773
17. Yang, X. F., Tallman, D. E., Bierwagen, G. P., Croll, S. G., & Rohlik, S. (2002). Blistering and degradation of polyurethane coatings under different accelerated weathering tests. Polymer Degradation & Stability, 77(1), 103-109 http://doi.org/10.1016/ S0141-3910(02)00085-X
18 Verma, G. (2019). Weathering, salt spray corrosion and mar resistance mechanism of clay (nano-platelet) reinforced polyurethane nanocomposite coatings. Progress in Organic Coatings, 129, 260-270 http://doi.org/10.1016/j.porgcoat.2019.01.028
19 Rivaton, A., Moreau, L., & Gardette, J.-L. (1997). Photooxidation of phenoxy resins at long and short wavelengths: II. Mechanisms of formation of photoproducts. Polymer Degradation & Stability, 58(3), 333-339 http://doi.org/10.1016/ S0141-3910(97)00088-8
20. Herrera, R., Muszynska, M., Krystofiak, T., & Labidi, J. (2015). Comparative evaluation of different thermally modified wood samples finishing with UV-curable and waterborne coatings. Applied Surface Science, 357(Pt B), 1444-1453 http://doi. org/10.1016/j.apsusc.2015.09.259.
21 Ghasemi-Kahrizsangi, A., Neshati, J., Shariatpanahi, H., & Akbarinezhad, E. (2015). Improving the UV degradation resistance of epoxy coatings using modified carbon black nanoparticles. Progress in Organic Coatings, 85, 199-207 http://doi.org/10.1016/j.porgcoat.2015.04.011
22 Ghohrodi, A. R., Ramezanzadeh, M., & Ramezanzadeh, B. (2022). Investigating the thermo-mechanical and UV-shielding properties of a nano-porous Zr(IV)-type metal-organic framework (MOF) incorporated epoxy composite coating. Progress in Organic Coatings, 164, 106693 http://doi.org/10.1016/j. porgcoat.2021.106693.
23 Li, Z.-J., Wang, F.-S., Lai, Y.-C., Shi, Z.-E., & Yu, Y.-H. (2021). Flexible epoxy graphene thermoset with excellent weather and corrosion resistance. Progress in Organic Coatings, 151, 106052 http://doi.org/10.1016/j.porgcoat.2020.106052
24 Zeng, W., Zhou, Q., Zhang, H., & Qi, X. (2018). One-coat epoxy coating development for the improvement of UV stability by DPP pigments. Dyes and Pigments, 151, 157-164. http://doi. org/10.1016/j.dyepig.2017.12.058
25 Erznožnik, H., Razboršek, T., & Gunde, M. K. (2016). Characterization of orange pigments in decorative outdoor coatings and their weather fastness. Progress in Organic Coatings, 99, 47-54 http://doi.org/10.1016/j.porgcoat.2016.05.007
26 Rezig, A., Nguyen, T., Martin, D., Sung, L., Gu, X., Jasmin, J., & Martin, J. W. (2006). Relationship between chemical degradation and thickness loss of an amine-cured epoxy coating exposed to different UV environments. Journal of Coatings Technology and Research, 3(3), 173-184 http://doi. org/10.1007/BF02774507.
27 Bano, H., Khan, M. I., & Kazmi, S. A. (2011). Structure and microstructure studies of epoxy coating after natural exposure testing. Journal of the Chemical Society of Pakistan, 33(4), 454-463. Retrieved in 2024, March 25, from https://jcsp.org. pk/ArticleUpload/3126-14641-1-PB.pdf
28 Yang, J.-W., Cho, H.-J., & Gong, Y.-D. (2023). Analytical approach to degradation structural changes of epoxydicyandiamide powder coating by accelerated weathering. Progress in Organic Coatings, 175 , 107357 http://doi. org/10.1016/j.porgcoat.2022.107357
29 Amrollahi, S., Yari, H., & Rostami, M. (2022). Investigating the weathering performance of epoxy silicone nanocomposite coatings containing various loadings of Glycidoxypropyltrimethoxysilanemodified Zinc oxide nanoparticles. Progress in Organic Coatings, 172, 107094 http://doi.org/10.1016/j.porgcoat.2022.107094
30 Wang, J., Ma, L., Ding, X., Xu, H., Wang, Y., Zhao, M., Ren, C., & Zhang, D. (2023). Tea polyphenol radical scavenger loaded UV absorber for corrosion resistant and weathering resistant epoxy coating fabrication. Progress in Organic Coatings, 180, 107553 http://doi.org/10.1016/j.porgcoat.2023.107553
31 Soleimani, M., Bagheri, E., Mosaddegh, P., Rabiee, T., Fakhar, A., & Sadeghi, M. (2021). Stable waterborne epoxy emulsions and the effect of silica nanoparticles on their coatings properties. Progress in Organic Coatings, 156, 106250 http:// doi.org/10.1016/j.porgcoat.2021.106250.
32 Li, R., Leng, Z., Zhang, Y., & Ma, X. (2019). Preparation and characterization of waterborne epoxy modified bitumen
Silva, M. S., Souza, A. G., Rosa, D. S., Valera, T. S., & Wiebeck, H.
emulsion as a potential high-performance cold binder. Journal of Cleaner Production, 235, 1265-1275 http://doi.org/10.1016/j. jclepro.2019.06.267
33 Udoh, I. I., Shi, H., Liu, F., & Han, E.-H. (2020). Microcontainerbased waterborne epoxy coatings for AA2024-T3: effect of nature and number of polyelectrolyte multilayers on active protection performance. Materials Chemistry and Physics, 241, 122404 http://doi.org/10.1016/j.matchemphys.2019.122404
34 Wärnheim, A., Edvinsson, C., Sundell, P.-E., Heydari, G., Deltin, T., & Persson, D. (2022). Depth-resolved FTIR-ATR imaging studies of coating degradation during accelerated and natural weathering: influence of biobased reactive diluents in polyester melamine coil coating. ACS Omega, 7(27), 23842-23850. http://doi.org/10.1021/acsomega.2c02523 PMid:35847300.
35 Bellinger, V., Bouchard, C., Claveirolle, P., & Verdu, J. (1981). Photooxidation of epoxy resins cured by non-aromatic amines. Photochemistry, 1(1), 69-80 http://doi.org/10.1016/01442880(81)90016-6
36 Bellinger, V., & Verdu, J. (1985). Oxidative skeleton breaking in epoxy amine networks. Journal of Applied Polymer Science, 30(1), 363-374. http://doi.org/10.1002/app.1985.070300132.
37 Awad , S. A. , Mahini , S. S. , & Fellows , C. M. (2019 ). Modification of the resistance of two epoxy resins to accelerated weathering using calcium sulfate as a photostabilizer. Journal of Macromolecular Science – Part A, 56(4), 316-326 http:// doi.org/10.1080/10601325.2019.1578179.
38 Bell, A. M., Keltsch, N., Shweyen, P., Reifferscheid, G., Ternes, T., & Buchinger, S. (2021). UV aged epoxy coatings Ecotoxicological effects and released compounds. Water Research X, 12, 100105 http://doi.org/10.1016/j.wroa.2021.100105 PMid:34189451.
39 Fernández-Álvarez, M., Velasco, F., & Bautista, A. (2021). Performance of ultraviolet exposed epoxy powder coatings functionalized with silica by hot mixing. Journal of Materials Research and Technology, 10, 1042-1057 http://doi.org/10.1016/j. jmrt.2020.12.094
40 Levin, J. R., Daisey, G., Elbert, K. C., Mallardi, J., Westmeyer, M., & Williams, D. (2023). Acrylic binder and formulation design for more sustainable Elastomeric Cool Roof Coatings (ERCs). In H. N. Cheng, & R. A. Gross (Eds.), Sustainable green chemistry in polymer research (Vol. 2, pp. 203-218). USA: American Chemical Society.
41 Xiong, A., & Li, J. (2023). Constructing stable transparent hydrophobic POSS@epoxy-group coatings for waterproofing protection of decorative-painting surfaces. Polymer Bulletin, 81(2), 1403-1419 http://doi.org/10.1007/s00289-023-04780-y
42. Onn, M., Ahmad, Z., Zainuddin, A., & Iliyas, S. M. M. (2022). Morphology and characterization study on effective microorganism (EM) water based epoxy coatings. Materials Today: Proceedings , 66 (Pt 10 ), 4026 -4032 . http://doi. org/10.1016/j.matpr.2022.05.334
43 Gao, T., He, Z., Hihara, L. H., Mehr, H. S., & Soucek, M. D. (2019). Outdoor exposure and accelerated weathering of polyurethane/polysiloxane hybrid coatings. Progress in Organic Coatings, 130, 44-57 http://doi.org/10.1016/j. porgcoat.2019.01.046
Received: Mar. 25, 2024
Revised: June 18, 2024
Accepted: June 24, 2024
Greener waterborne epoxy coatings with optimized UV-resistance
Supplementary material accompanies this paper.
Table S1. Characteristics of the catalysts (curing agents) and the main differences considering their properties.
Table S2. Delta E evolution during the weathering tests at the 7 different times.
This material is available as part of the online article from https://doi.org/10.1590/0104-1428.20230133
Billy Alberto Ávila-Camacho1 and Norma Aurea Rangel-Vázquez1*
1Departamento de Posgrado e Investigación, Instituto Tecnológico de Aguascalientes – TecNM, Aguascalientes, AGS, México *norma.rv@aguascalientes.tecnm.mx
Obstract
Chitosan-based adsorbents have high efficiency in removing heavy metals from water. In this study, the adsorption of Hg2+, Ni2+ and Cu2+ onto chitosan/glutaraldehyde hydrogels were analyzed using the semi-empirical method PM3. Based on thermodynamic analysis of these systems, the adsorption processes were spontaneous, exothermic and highly stable, because all the values of the Gibbs free energy, the enthalpy of formation and the binding energy were negative, on the other hand, each of the systems were analyzed using electrostatic potential maps, where it was observed that the functional groups amino (NH2) and hydroxyl (OH) are the main active sites of the adsorbent. Through an FTIR analysis, the correct cross-linking between chitosan and glutaraldehyde was confirmed, as well as the union of the different cations on the surface of the adsorbent.
Keywords: adsorption, chitosan, glutaraldehyde, heavy metals, hydrogel.
How to cite: Ávila-Camacho, B. A., & Rangel-Vázquez, N. A. (2024). Analysis of the adsorption of Hg2+, Ni2+ and Cu2+ on chitosan hydrogels. Polímeros: Ciência e Tecnologia, 34(3), e20240035. https://doi.org/10.1590/0104-1428.20240053
Treatment of polluted water is a very important issue for the society because it is currently required to improve its quality to minimize the negative impacts on these ecosystems and human health. Water quality has been mainly affected by the industrialization of developed countries, as well as a significant increase in the world’s population. There are different types of contaminants as, dyes, organic matter, pharmaceutical compounds, heavy metals, among others[1]. However, heavy metals are the contaminants that cause the greatest concern, since they are highly toxic even in low concentrations, in addition, they are not biodegradable, so their persistence in water could last for a long time[2]. Heavy metals such as mercury (Hg), nickel (Ni), copper (Cu), zinc (Zn), lead (Pb) and iron (Fe) have densities greater than 5 gr/cm3[3]. They have different applications at industrial level[4]. However, heavy metals can cause different toxicity symptoms[5]. For this reason, it is necessary to remove heavy metals from water.
Adsorption processes is a physicochemical process in which a substance present in a mobile phase adheres to the surface of a solid material. Different studies have confirmed the efficiency of this process in the single and multicomponent removal of heavy metals[6,7]. Chitosan is an insoluble linear polycationic polymer due to its bonds formed by hydrogen bonds. This polymer is highly biocompatible, low cost, biodegradable and can be easily chemically modified, which is why it has received great attention as a promising material for various processes including as adsorbent in the treatment of heavy metals[8,9]. Hydrogels are defined as three-dimensional networks composed by polymers
(e.g., chitosan) that can swell and absorb a certain volume of water into their structure. These materials are made up of various polymeric molecules of a hydrophilic nature, which can be cross-linked through chemical bonds, ionic interactions, or hydrogen bonds.
The swelling capacity of hydrogels is mainly due to the hydrophilic functional groups found within the polymeric network that constitutes the hydrogel, which allows it to absorb and retain large amounts of water, up to 400 times its original weight[10,11]. There are several techniques adopted for the preparation of hydrogels. Specifically, chemical cross-linking involves the use of a cross-linking agent to join two polymer chains. Chemical crosslinking can be applied to natural polymers (chitosan, alginate, carboxymethylcellulose), which is achieved through the reaction of their functional groups (OH, COOH, NH2), with cross-linkers such as aldehyde (glutaraldehyde), so this type of cross-linking is permanent[11,12]
Several investigations have used chitosan hydrogel as an adsorbent in monocomponent systems or in the form of beads[13], which leads to several disadvantages, for example, a smaller adsorption surface, difficulty in controlling the size, less flexibility and difficulty in scaling up. On the other hand, the addition of graphene oxide[14] or carbon nanotubes functionalized with chitosan[15] increase the synthesis costs of chitosan-based adsorbents. This study proposes the use of chitosan hydrogels cross-linked only with glutaraldehyde to simultaneously remove 3 heavy metals from aqueous solutions, reducing the high costs in the synthesis of the
B. A., & Rangel-Vázquez, N. A.
material by drying it at room temperature, reducing the risk of degradation and eliminating the formation of secondary products such as aldehydes and ketones by avoiding the use of sodium hydroxide in the synthesis.
On the other hand, simulation and computational modeling are used for the prediction and optimization of molecular structures, to obtain binding energies and thermodynamic and biological properties, using semi-empirical methods with the objective to optimize the synthesis of materials[16] .
The semiempirical PM3 method has been used to identify evidence for the proposed kinetic models, as well as to explain the adsorption of heavy metals on polymeric materials. On the other hand, quantitative structure-activity relationship (QSAR) parameters are a useful tool to describe the relationships between the biological activity of a molecule and its physicochemical characteristics through its molecular structure[17]. So, the objective of this work was, the thermodynamic analysis, modeling, simulation, and the experimental adsorption of single and multicomponent systems of heavy metals as, Hg2+, Ni2+ and Cu2+ in chitosan hydrogels crosslinked with glutaraldehyde.
2.1
The computational analyzes were determined on a DELL brand PC with an i7 processor and 16 Gb of RAM. The molecules were analyzed individually and after the adsorption of the ions in the hydrogel using the Hyperchem software. Semi-empirical models combine quantum theory with empirical approximations to describe the electronic structure and properties of molecules, specifically the Parametric method 3 (PM3) was used to optimize and minimize the geometric energy based on the position of each heavy metal ion in the hydrogel. This model is applicable to organic and inorganic molecules with multiple bonds and functional groups and is more computationally efficient than AB initio quantum theory methods, making it suitable for studying large and complex molecular systems. This model is essentially a reparameterization of AM1 using a different parameterization strategy and only two Gaussian functions. The Polak-Ribiere algorithm and 2250 iterations were used with a convergence level of 0.01 kcal/Å mol.
The molecular structures were built according to the Hyperchem software operating manual using the “build” option; then, using the “model” option, the angles and bond distances of each of the complexes were corrected to obtain the optimization geometry, thermodynamic parameters and QSAR. For chitosan, four glucosamine units were studied, where one unit was acetylated to simulate a deacetylation degree of 75%.
The FTIR spectra were obtained from 200 to 4000 cm-1 after the optimization geometry, where an analysis of the rotations and vibrations of the systems at room temperature was carried out. The calculated thermodynamic properties were Gibbs free energy (ΔG), binding energy, Enthalpy of formation (ΔHf) and dipole moment. On the other hand, the calculated QSAR properties were surface area, volume, and polarizability. The electrostatic potential map (EPM) provides a contour plot of a molecule, indicating the sites of highest and lowest electron density.
Table 1 shows the thermodynamic properties of glutaraldehyde, chitosan, and the hydrogel, where a negative Gibbs free energy is observed for all cases, specifically in the hydrogel of -464.183 Kcal/mol, indicating the spontaneous nature of the crosslinking through the primary amino groups of chitosan and the aldehyde groups of glutaraldehyde.
The values of the binding energy and the enthalpy of formation of the hydrogel were -19,469.414 and -1,470.360 Kcal/mol respectively, which indicated that the hydrogel is highly stable and exothermic[18]. On the other hand, the dipole moment is higher in the hydrogel, due to the difference in the electronegativities of the C-H, N-H, C-C, C-O and O-H bonds, respectively.
The QSAR properties of glutaraldehyde, chitosan, and the hydrogel are shown in Table 2, where the surface area and volume in the hydrogel is significantly higher, due to chitosan/glutaraldehyde crosslinking from the Schiff bases reaction. On the other hand, the polarizability of the hydrogel is 130.28 Å3, that is, the tendency to generate induced electric dipole moments is greater, causing a fluctuation in the dipole moment of the hydrogel.
Figure 1 shows the FTIR spectra of glutaraldehyde, chitosan, and chitosan/glutaraldehyde hydrogel, respectively. Figure 1a) show the FTIR of glutaraldehyde where, the band show between 3103-2999 cm-1 corresponded to the asymmetric stretching of the H-C-H bonds, to 2058 cm-1 it was attributed to the stretching of the C=O bonds. The H-C-H scissoring was appreciated at 1375 cm-1 and at 1168 cm-1 was assigned to the asymmetric stretching of the C-C bonds. The bands located between 784 and 770 cm-1 were attributed to the swinging of the CH2 bonds and finally, the bands at 600-574 and 226 cm-1 corresponded to the swinging in and out of plane of the C=O bonds, respectively.
Analysis of the adsorption of Hg2+, Ni2+ and Cu2+ on chitosan hydrogels

shown. In 1874 and 1872 cm-1 was assigned to the stretching of the C=N bond of the imine groups, which were formed from the Schiff base reactions due to the crosslinking of glutaraldehyde and chitosan[20]
The FTIR of chitosan can be seen in Figure 1b) where the stretching of the O-H bonds of chitosan were located between 3717 and 3598 cm-1, the symmetrical stretching of the amino groups (NH2) was appreciated at 3490-3483 cm-1 . The range of 2951-2911 cm-1 was assigned to the symmetric and asymmetric stretching of CH3 of the residual acetyl group, in the range of 2836 to 2706 cm-1 the stretching of the CH2 bonds of hydroxymethyl and of the CH bonds of chitosan was observed. At 1793 cm-1 was attributed to the stretching of the C=O bond of the residual acetyl, while in the region of 1686 and 1673 cm-1 was assigned to the NH2 scissoring. At 1572 cm-1 was attributed to the stretching of the acetyl C-N-C bonds. The peak with the highest intensity located between 1297 and 1245 cm-1 was assigned to the swinging of the C-O and C-C bonds, and at 1086 cm-1 corresponded to the swinging of C-H. At 875 cm-1 the stretching of the chitosan C-C bonds was observed and, finally, in the region of 626-600, 400 and 320 cm-1 were attributed to the out-of-plane swinging of the OH, NH and NH2 bonds, respectively[19]
The FTIR of the hydrogel is observed in Figure 1c), where a displacement and increase in the intensity of the characteristic peak between 3907-3796 cm-1 of the stretching of the O-H bonds due to the hydroxyl groups of chitosan is
The electrostatic potential is related to the electronic density, which represent an important descriptor to determine the sites of electrophilic attack and nucleophilic reactions, as well as the interactions of hydrogen bonds. Figure 2 shows the electronic distribution of chitosan, glutaraldehyde, and hydrogel where the electrophilic and nucleophilic regions are red and blue, respectively. Specifically in the hydrogel, the regions with negative electrostatic potential (-0.132 au) were in the oxygen atoms of the chitosan hydroxyl groups (red spheres), which are the most reactive sites for adsorbate binding.
On the other hand, the nitrogen atoms (blue color) of the amino groups also presented a slightly negative electrostatic potential, so they can also undergo an electrophilic attack. Chitosan is highly chelating[21], that is, it can interact or form coordinate bonds with other ions or ligands. This phenomenon depends on the availability of empty molecular orbitals and the pH of the solution due to the competition of protons with other ions[22]. For example, in chitosan, amino and hydroxyl groups represent the most reactive functional groups that are responsible for metal ion chelation due to the presence of a lone pair of electrons on the nitrogen and oxygen atoms[22,23]
Table 3 shows the thermodynamic properties of the adsorption systems. The Gibbs free energy (ΔG) was negative for all the systems, where the adsorption of the different

cations was spontaneous, in addition, the adsorption occurred due to the lone pair of electrons shared by the NH2 and OH functional groups generating covalent bonds[24]. The binding energy and enthalpy determined that the different adsorptions were stable and exothermic.
Finally, the dipole moment was related to electronegativity, that is, the greater the difference in electronegativities between two atoms in a molecule, the greater the dipole moment and, therefore, the polarity. Thus, in the different adsorptions, a higher dipole moment was observed due to the C-O, C-N, O-H and N-H bonds, which presented higher electronegativities differences compared to the rest of the
bonds. Specifically, in the CG-Ni single system, a dipole moment of 10.56 debyes was generated, higher than that of the CG-Cu and CG-Hg systems with 9.047 and 8.655 debyes respectively, in the same way it occurred with the binary systems that contained Ni where the magnitude of the dipole moment was higher, so adsorption may also be higher in such systems[25]
Table 4 shows the QSAR properties of the single and multicomponent adsorption. In the CG-Hg system was observed that both the surface area and the volume are greater (1,320.61 Å2 and 3,493.96 Å3 respectively) than the other 2 monocomponent systems CG-Ni and CG-Cu, due to the
Table 4. QSAR properties.
volume of the Hg cation of 96.74 Å3, while the volume of Ni and Cu is 33.05 and 36.83 Å3, respectively[26]. On the other hand, the polarizability values indicated that there may be a temporary fluctuation in the dipole moment of the systems in the presence of an electric field.
The binding energies, the dipole moment, the geometry of the heavy metals, as well as the polarizability of each complex allowed to predict the trend described in the study. Based on the previous results it can be inferred that the possible adsorption tendency would be Ni > Cu > Hg in any of the systems, however, the electronegativity of the heavy metal ions must be considered (2, 1.91 and 1.9, for Hg, Ni and Cu respectively)[27]. If the electronegativity of heavy metal ions is high, the affinity towards the functional groups of the adsorbent will be greater, adsorbing more easily[28], therefore, the trend could be the following: Hg > Cu > Ni. Hg has an electronegativity of 2, therefore, it will have a greater attraction for the electron pairs donated by the amine and hydroxyl groups of the chitosan structure. [29] On the other hand, the stability constants of the amine complexes for Hg, Cu and Ni are approximately 1.3x1023 , 1.3x1013 and 1.1x108 respectively, the higher the value of the constant, the greater the affinity between the metal and the amine group, which represents a greater stability of the complex so, the values are consistent with the predicted trend.[30] Besides, both Cu and Ni are considered intermediate acids, the difference is that Cu tends to form complexes with hard bases such as oxygen, while Ni forms complexes with hard bases such as nitrogen, this behavior is decisive in the adsorption capacities of each heavy metal[31]. The adsorption of Cu is in the middle because the amount of hydroxyl groups is greater in the chitosan structure, while Ni, by preferring the union with the groups containing nitrogen, is adsorbed less, due to the amount of amine groups present in the chitosan chains is lower because a significant part of said groups are converted into imine groups due to cross-linking.
There is no published information yet on the adsorption of these 3 heavy metals in a ternary system using a chitosan hydrogel, however, there are some others in binary systems using chitosan with similar deacetylation degrees, for example, Boddu et al, carried out the adsorption of Ni and Cu using a chitosan-based adsorbent, said polymer had a deacetylation degree ≥75%, the adsorption capacities they obtained were 86.2 and 78.1 mg/g for Cu and Ni respectively where the trend was Cu>Ni.[32] Kalyani et al synthesized a low molecular weight chitosan-based adsorbent for the removal of Cu and Ni, where the adsorption capacities were
for Cu and Ni of 196.07 and 114.94 mg/g respectively, that is, the trend was Cu>Ni.[33] Finally, Vieira et al, investigated the adsorption mechanisms of Hg, Cu and Cr using glutaraldehyde cross-linked chitosan hydrogels with a cross-linking degree of 85%, where the adsorption trend obtained was Hg>Cu>Cr.[34]
Figure 3 shows the FTIR obtained from the adsorption of the ions in the chitosan/glutaraldehyde hydrogel. In the range of 3900 and 3700 cm-1 the stretching of the OH bonds increased, although the small displacements that were appreciated in the adsorptions were due to the fact that the excited atoms of the cations caused greater vibration and therefore a change in their dipole moment[25], that is, the adsorbates are interacting with the O atoms of the hydroxyl groups of chitosan[35]. The FTIR indicated a balancing of the NH2 groups at 1690 and 1660 cm-1. Additionally, slight increases in the intensity of the peaks and displacements were observed in comparison with the hydrogel, indicating that the cations are also interacting with the N atoms through the lone pair electrons shared from such atoms to the empty orbitals of heavy metals[36]
Specifically, Figure 3a) shows the FTIR of hydrogelHg. The band assigned at 1667-1659 cm-1 indicates the swinging (scissoring) of the H-N-H bonds of the amino groups where Hg is bind. The peak at wave number 1492 cm-1 was attributed to the in-plane rocking of the O-H bonds of the hydroxyl groups. The bands assigned at 1375 and 592 cm-1 were attributed to the out-of-plane and in-plane rocking of the hydroxyl group where Hg binds, respectively. On the other hand, the bands located at 1259, and 1150-1089 cm-1 were assigned to C-O and C-C bonds of the chitosan monomers, as well as to the out-of-plane rocking of the H-C-H bonds respectively. The spectrum within the wavenumber of 345-294 cm-1 indicated the out-of-plane equilibrium of the O-H bonds of the hydroxyl groups. Figure 3b) shows the hydrogel-Ni system where, the band assigned at 1690 cm-1 indicated the rocking (scissoring) of the H-N-H bonds of the amino groups where the Ni ion binds. On the other hand, the peak at located at 514 cm-1 was attributed to the in-plane rocking of the hydroxyl group where Ni is bind.
The system hydrogel-Cu is shown in Figure 3c) where, the band located at 3647 cm-1 was attributed to the stretching of the hydroxyl group where the Cu ion is bind. Besides, the band located at wave number 3472 cm-1 was assigned to the symmetric stretching of the amino group where the same metal is bind.
On the other hand, in the band 1684-1655 cm-1 the rocking (scissor) vibrations of the amino groups were assigned, including the group where the Cu cation binds. The binary system of hydrogel-Hg-Ni is shown in Figure 3d) where, the band located at 3689 cm-1 was attributed to the stretching of the O-H bond where the Ni cation binds. The region between 3516 to 3351 cm-1 was assigned to the symmetric (3516-3491 cm-1) and asymmetric (3402 cm-1) stretching vibrations of the amino groups, including those where the Ni and Hg cations were bind. Figure 3e) shows the hydrogel-Hg-Cu where, the band located at 3646 cm-1 was attributed to the stretching vibration of the hydroxyl group where the Cu cation is bind. The bands between 3517 to

3342 cm-1 were assigned to the symmetric stretching vibrations of the amino groups, including those where the binding of heavy metals Hg and Cu occurs, instead, the band located at 1682 cm-1 was appreciated to the balancing (scissors) of the free amino groups and those occupied by the heavy metals Hg and Cu. Figure 3f) shows the hydrogel-Ni-Cu system where the peak located between 3695 and 3654 cm-1 was attributed to the stretching of the hydroxyl groups where the Ni and Cu cations are bind respectively.
Finally, Figure 3g) shows the multicomponent system hydrogel-Hg-Ni-Cu where, the characteristic bands located between 3911 and 3816 cm-1 were assigned to the stretching of the hydroxyl groups, where the Hg and Cu cations are bind, while the band located at 3686 cm-1 was localized to the stretching of the same functional group but where Ni binds. The first two low intensity bands within wave numbers 3531 and 3344 cm-1 indicated the stretching vibrations of the free amino acid groups and occupied by heavy metals, specifically in 3531, 3491 and 3474 cm-1 was assigned to the symmetric stretching of NH2 where Cu, Hg and Ni bind respectively, while at 3402, 3367 and 3344 cm-1 were
attributed to the asymmetric stretching of the amino groups where Ni, Hg and Cu bind respectively.
Figure 4 shows the electronic distribution of the different adsorptions where it is observed that heavy metal ions were adsorbed on the OH and NH2 functional groups simultaneously (red color) by forming covalent bonds forming an association ring[36]. The oxygen atoms (red spheres) and nitrogen atoms (blue spheres) presented the negative potential because they have a high electronegativity, that is, they have a greater capacity to attract electrons towards themselves. The carbon atoms (green spheres), mainly the C-C bonds of chitosan, generated neutral electrostatic potentials, that is, there are the electrons that carry out the union of these bonds.
Finally, the positive electrostatic potential was observed mainly around the hydrogen atoms (white sphere), because these atoms have the lowest value of electronegativity compared to the other atoms. In the adsorption systems, a change in the color scale (green-yellow) was observed, manly in the N and O atoms of the amino and hydroxyl functional groups, respectively, generating a less negative electrostatic
Analysis of the adsorption of Hg2+, Ni2+ and Cu2+ on chitosan hydrogels

4. Electrostatic potential maps of optimized structures where, (a) CG-Hg, (b) CG-Ni, (c) CG-Cu, (d) CG-Hg-Ni, (e) CG-Hg-Cu, (f) CG-Ni-Cu and (g) CG-Hg-Ni-Cu.
potential in both groups, which is due to the formation of metal complexes thanks to the donation of lone pairs electrons from N and O to the cations, thus reducing the density of the electron cloud of both functional groups, generating a negative electrostatic potential in the heavy metals[37]
Using computational chemistry methods, adsorption mechanisms can be predicted, as well as the viability
of the processes. In this case, adsorptions occur through the formation of multiple coordination bonds between chitosan functional groups and heavy metal ions, because chitosan-based adsorbent is a strongly chelating agent, therefore adsorption It will depend on several factors, such as electronegativity and polarity. It was appreciated that the adsorptions were spontaneous, stable, and exothermic. In addition, an increase in polarizability was observed specifically in the glutaraldehyde-crosslinked chitosan without the bound ions. The chitosan/glutaraldehyde hydrogel
Ávila-Camacho, B. A., & Rangel-Vázquez, N. A.
contains double bonds, which means that these atoms have delocalized electrons, so they are more polarizable, that is, they contribute to a temporary breaking of the dipole moments, causing complementary dipoles in neighboring molecules or ions, so that the adsorbents are attracted to the adsorbates. These studies can be applied to different adsorption systems to determine the viability of the adsorbent and the adsorption system itself.
5. Author’s Contribution
• Conceptualization – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Data curation – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Formal analysis – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Funding acquisition – NA.
• Investigation – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Methodology – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Project administration – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Resources – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Software – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Supervision – Norma Aurea Rangel-Vázquez.
• Validation – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Visualization – Billy Alberto Ávila-Camacho; Norma Aurea Rangel-Vázquez.
• Writing – original draft – Billy Alberto ÁvilaCamacho; Norma Aurea Rangel-Vázquez.
• Writing – review & editing – Billy Alberto ÁvilaCamacho; Norma Aurea Rangel-Vázquez.
6. Acknowledgements
Billy Alberto Ávila-Camacho thanks to National Council of Humanities, Sciences and Technologies (CONAHCYT) for the Doctorate in Engineering Science 834799 scholarship.
7. References
1 Villarín, M. C., & Merel, S. (2020). Assessment of current challenges and paradigm shifts in wastewater management. Journal of Hazardous Materials, 390, 122139. http://doi. org/10.1016/j.jhazmat.2020.122139 PMid:32007860.
2 Briffa, J., Sinagra, E., & Blundell, R. (2020). Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon, 6(9), e04691 http://doi.org/10.1016/j. heliyon.2020.e04691 PMid:32964150.
3 Zamora-Ledezma, C., Negrete-Bolagay, D., Figueroa, F., Zamora-Ledezma, E., Ni, M., Alexis, F., & Guerrero, V. H. (2021). Heavy metal water pollution: a fresh look about hazards, novel and conventional remediation methods. Environmental
Technology & Innovation, 22, 101504 http://doi.org/10.1016/j. eti.2021.101504
4 Ando, S., & Koide, K. (2011). Development and applications of fluorogenic probes for mercury (II) based on vinyl ether oxymercuration. Journal of the American Chemical Society, 133 (8 ), 2556 -2566 . http://doi.org/10.1021/ja108028m. PMid:21294513.
5 Taylor, A. A., Tsuji, J. S., Garry, M. R., McArdle, M. E., Goodfellow, W. L., Jr., Adams, W. J., & Menzie, C. A. (2019). Critical review of exposure and effects: implications for setting regulatory health criteria for ingested copper. Environmental Management, 65(1), 131-159 http://doi.org/10.1007/s00267019-01234-y PMid:31832729.
6 Zeng, X., Zhang, G., Zhu, J., & Wu, Z. (2022). Adsorption of heavy metal ions in water by surface functionalized magnetic composites: a review. Environmental Science. Water Research & Technology, 8(5), 907-925 http://doi.org/10.1039/D1EW00868D
7 Arora, R. (2019). Adsorption of heavy metals–a review. Materials Today: Proceedings, 18(Pt 7), 4745-4750 http:// doi.org/10.1016/j.matpr.2019.07.462
8 Li, Q., Dunn, E. T., Grandmaison, E. W., & Goosen, M. F. (1992). Applications and properties of chitosan. Journal of Bioactive and Compatible Polymers, 7(4), 370-397 http:// doi.org/10.1177/088391159200700406
9. Cheng, B., Pei, B., Wang, Z., & Hu, Q. (2017). Advances in chitosan-based superabsorbent hydrogels. RSC Advances, 7(67), 42036-42046 http://doi.org/10.1039/C7RA07104C
10. Ahmed, E. M. (2015). Hydrogel: Preparation, characterization, and applications: A review. Journal of Advanced Research, 6(2), 105-121 http://doi.org/10.1016/j.jare.2013.07.006 PMid:25750745.
11 Gulrez, S. K. H., Al-Assaf, S., & Phillips, G. O. (2011). Hydrogels: methods of preparation, characterization and applications. In A. Carpi (Ed.), Progress in molecular and environmental bioengineering-from analysis and modeling to technology applications (pp. 117150). Italy: InTech http:// doi.org/10.5772/24553
12. Silos-Llamas, A. K., Durán-Jiménez, G., Hernández-Montoya, V., Montes-Morán, M. A., & Rangel-Vázquez, N. A. (2020). Understanding the adsorption of heavy metals on oxygenrich biochars by using molecular simulation. Journal of Molecular Liquids, 298, 112069 http://doi.org/10.1016/j. molliq.2019.112069
13 Ngah, W. S. W., Ab Ghani, S., & Kamari, A. (2005). Adsorption behaviour of Fe (II) and Fe (III) ions in aqueous solution on chitosan and cross-linked chitosan beads. Bioresource Technology, 96(4), 443-450 http://doi.org/10.1016/j.biortech.2004.05.022 PMid:15491825.
14 Medina, R. P., Nadres, E. T., Ballesteros, F. C., Jr., & Rodrigues, D. F. (2016). Incorporation of graphene oxide into a chitosan–poly (acrylic acid) porous polymer nanocomposite for enhanced lead adsorption. Environmental Science. Nano, 3(3), 638-646 http://doi.org/10.1039/C6EN00021E
15. Kim, M. K., Sundaram, K. S., Iyengar, G. A., & Lee, K.-P. (2015). A novel chitosan functional gel included with multiwall carbon nanotube and substituted polyaniline as adsorbent for efficient removal of chromium ion. Chemical Engineering Journal, 267, 51-64. http://doi.org/10.1016/j.cej.2014.12.091.
16 Atangana, E., & Oberholster, P. J. (2020). Mathematical modeling and stimulation of thermodynamic parameters for the removal for Cr6+ from wastewater using chitosan cross-linked glutaraldehyde adsorbent. Alexandria Engineering Journal, 59(4), 1931-1939 http://doi.org/10.1016/j.aej.2019.12.012
17. Costa-Júnior, E. S., Barbosa-Stancioli, E. F., Mansur, A. A. P., Vasconcelos, W. L., & Mansur, H. S. (2009). Preparation and characterization of chitosan/poly (vinyl alcohol)
Analysis of the adsorption of Hg2+, Ni2+ and Cu2+ on chitosan hydrogels
chemically crosslinked blends for biomedical applications. Carbohydrate Polymers, 76(3), 472-481 http://doi.org/10.1016/j. carbpol.2008.11.015
18 Mirzaei, E. B., Ramazani, A. S. A., Shafiee, M., & Danaei, M. (2013). Studies on glutaraldehyde crosslinked chitosan hydrogel properties for drug delivery systems. International Journal of Polymeric Materials and Polymeric Biomaterials, 62(11), 605-611. http://doi.org/10.1080/00914037.2013.769165.
19 Li, B., Shan, C.-L., Zhou, Q., Fang, Y., Wang, Y.-L., Xu, F., Han, L.-R., Ibrahim, M., Guo, L.-B., Xie, G.-L., & Sun, G.-C. (2013). Synthesis, characterization, and antibacterial activity of cross-linked chitosan-glutaraldehyde. Marine Drugs, 11(5), 1534-1552 http://doi.org/10.3390/md11051534 PMid:23670533.
20 Galan, J., Trilleras, J., Zapata, P. A., Arana, V. A., & GrandeTovar, C. D. (2021). Optimization of chitosan glutaraldehydecrosslinked beads for reactive blue 4 anionic dye removal using a surface response methodology. Life, 11(2), 85 http:// doi.org/10.3390/life11020085 PMid:33504022.
21. Gamage, A., & Shahidi, F. (2007). Use of chitosan for the removal of metal ion contaminants and proteins from water. Food Chemistry, 104(3), 989-996 http://doi.org/10.1016/j. foodchem.2007.01.004.
22 Guibal, E. (2004). Interactions of metal ions with chitosan-based sorbents: a review. Separation and Purification Technology, 38(1), 43-74 http://doi.org/10.1016/j.seppur.2003.10.004
23 Guibal, E., Vincent, T., & Navarro, R. (2014). Metal ion biosorption on chitosan for the synthesis of advanced materials. Journal of Materials Science, 49(16), 5505-5518 http://doi. org/10.1007/s10853-014-8301-5
24. Alsamman, M. T., & Sanchez, J. (2021). Recent advances on hydrogels based on chitosan and alginate for the adsorption of dyes and metal ions from water. Arabian Journal of Chemistry, 14(12), 103455. http://doi.org/10.1016/j.arabjc.2021.103455.
25 González, A. J., & Vázquez, N. A. R. (2023). PM3 semiempirical method and Monte Carlo simulation application on pesticides adsorption on SWCNT. Colloid and Interface Science Communications, 53, 100699 http://doi.org/10.1016/j. colcom.2023.100699
26 Bader, R. F., Carroll, M. T., Cheeseman, J. R., & Chang, C. (1987). Properties of atoms in molecules: atomic volumes. Journal of the American Chemical Society, 109(26), 79687979 http://doi.org/10.1021/ja00260a006
27 Allred, A. L. (1961). Electronegativity values from thermochemical data. Journal of Inorganic and Nuclear Chemistry, 17(3-4), 215-221 http://doi.org/10.1016/0022-1902(61)80142-5
28 Zhu, H., Chen, S., & Luo, Y. (2023). Adsorption mechanisms of hydrogels for heavy metal and organic dyes removal: A
short review. Journal of Agriculture and Food Research, 12, 100552 http://doi.org/10.1016/j.jafr.2023.100552
29 Li, K., & Xue, D. (2006). Estimation of electronegativity values of elements in different valence states. The Journal of Physical Chemistry A, 110(39), 11332-11337. http://doi. org/10.1021/jp062886k PMid:17004743.
30 Basolo, F., & Pearson, R. G. (1967) Mechanisms of inorganic reactions a study of metal complexes in solution New York: John Wiley and Sons, Inc.
31. Pearson, R. G. (1963). Hard and soft acids and bases. Journal of the American Chemical Society, 85(22), 3533-3539 http:// doi.org/10.1021/ja00905a001
32 Boddu, V. M., Abburi, K., Randolph, A. J., & Smith, E. D. (2008). Removal of copper (II) and nickel (II) ions from aqueous solutions by a composite chitosan biosorbent. Separation Science and Technology, 43(6), 1365-1381 http:// doi.org/10.1080/01496390801940762
33 Kalyani, S., Priya, J. A., Rao, P. S., & Krishnaiah, A. J. S. S. (2005). Removal of copper and nickel from aqueous solutions using chitosan coated on perlite as biosorbent. Separation Science and Technology, 40(7), 1483-1495 http://doi.org/10.1081/ SS-200055940
34. Vieira, R. S., Oliveira, M. L. M., Guibal, E., RodríguezCastellón, E., & Beppu, M. M. (2011). Copper, mercury and chromium adsorption on natural and crosslinked chitosan films: an XPS investigation of mechanism. Colloids and Surfaces. A, Physicochemical and Engineering Aspects, 374(1-3), 108-114 http://doi.org/10.1016/j.colsurfa.2010.11.022
35 Jiang, C., Wang, X., Wang, G., Hao, C., Li, X., & Li, T. (2019). Adsorption performance of a polysaccharide composite hydrogel based on crosslinked glucan/chitosan for heavy metal ions. Composites. Part B, Engineering, 169, 45-54 http://doi. org/10.1016/j.compositesb.2019.03.082
36 Yu, K., Ho, J., Mccandlish, E., Buckley, B., Patel, R., Li, Z., & Shapley, N. C. (2013). Copper ion adsorption by chitosan nanoparticles and alginate microparticles for water purification applications. Colloids and Surfaces. A, Physicochemical and Engineering Aspects, 425, 31-41 http://doi.org/10.1016/j. colsurfa.2012.12.043
37 Li, N., & Bai, R. (2005). Copper adsorption on chitosan–cellulose hydrogel beads: behaviors and mechanisms. Separation and Purification Technology, 42(3), 237-247. http://doi.org/10.1016/j. seppur.2004.08.002
Received: Jun. 12, 2024 Revised: Aug. 08, 2024 Accepted: Aug. 14, 2024
Ávila-Camacho, B. A., & Rangel-Vázquez, N. A.
Supplementary Material
Supplementary material accompanies this paper.
Suplementary Information
This material is available as part of the online article from https://doi.org/10.1590/0104-1428.20240053
Polímeros, 34(3), e20240035, 2024
Ingrid Amélia dos Santos Matusinho1 , Ítalo Rocha Coura1 , Patterson Patricio de Souza1 and Patrícia Santiago de Oliveira Patrício1*
1Departamento de Química, Centro Federal de Educação Tecnológica de Minas Gerais - CEFET-MG, Belo Horizonte, MG, Brasil *patricia@cefetmg.br
Obstract
Banknotes often have a short lifespan and are frequently removed from circulation due to wear and tear, leading to a significant disposal challenge given the millions of banknotes discarded annually. This study evaluated the utilization of these waste banknotes, primarily composed of cotton fibers, in producing polymer composites. The mechanical and thermal properties, hydrophilicity, and degradation through accelerated aging of composites made from recycled polypropylene (PPR) and banknote residues were evaluated. The results indicated that including banknote waste increased the hydrophilicity of the composites relative to pure PPR and improved mechanical properties against weathering. Notably, composites with 10 wt% banknote content enhanced Young’s modulus and tensile strength, both in weathered and non-weathered conditions. This approach not only offers a potential solution for the disposal of waste banknotes but also aligns with the broader goal of reducing ecological impact and promoting the development of sustainable composite materials.
Keywords: composite, polypropylene, banknote.
How to cite: Matusinho, I. A. S., Coura, Í. R., Souza, P. P., & Patrício, P. S. O. (2024). Evaluation of recycled polypropylene properties with banknote-reinforced. Polímeros: Ciência e Tecnologia, 34(3), e20240036. https://doi. org/10.1590/0104-1428.20240044
The banknotes have a short useful life, with a period of use until they become unsuitable for circulation due to their dire conditions. In addition to issuing and monitoring, the issuing institutions are responsible for the activity of sanitation of the circulating medium, aiming to keep the money in circulation with good conditions of use, thus assuming also responsibility for solid waste management. This activity, in general, is regulated by law, providing principles and guidelines related to solid waste management[1]
In Brazil, the banking network collects millions of banknotes yearly, and those that are not in good circulation conditions are destroyed. Incineration is used to destroy banknotes, but this technique has become increasingly inadequate as it allows for the release of toxic waste into the environment[2] In this context, due to the growing concern with solid waste management and others, the enormous environmental impact they generate, studies, and improvements of processes that are efficient in recovering or reusing waste[1]
Some studies have been carried out to explore different destinations for waste banknotes, such as their use as a raw material in the production of biofuels and recycling for papermaking, seeking solutions for reusing this waste[2,3] However, it is essential to note that these alternatives usually involve multiple processing steps, often including the use of chemical reagents. Since banknotes are mainly composed of
cotton and have a high cellulose content, a promising option for its destination is to use it as a filler or reinforcement in polymer composites[4]. This utilization offers a sustainable approach to managing waste and provides an opportunity to enhance the properties of the polymer[5-7]. Fillers can be added to maintain or improve the properties of functional polymers when it comes to polymer recycling[6,7]. Recycling has been used as an alternative to reduce the amount of plastic waste discarded in the environment. However, some properties of polymers are compromised when subjected to processing conditions, high temperatures, and shear mechanical recycling[8]
Given this perspective, to reduce the accumulation of waste banknotes and polypropylene in the environment, the present work aims to develop composites using recycled polypropylene as a polymer matrix and waste banknotes as a filler. The composites were characterized using Infrared Spectroscopy (FTIR), Thermogravimetric Analysis (TG), Tensile Test, Differential Scanning Calorimetry (DSC), Wetting Test, and Accelerated Weathering Test.
The PP-banknote composites were prepared with recycled polypropylene (PPR) supplied in granular form
by RCA Plásticos Company. The banknotes (BK) were provided by the Central Bank of Brazil, Department of the Circulation, with particle sizes ranging from 2.0-3.0 mm.
The PPR and the residues of BK were placed in the mixing chamber of a Homogenizer (Drais), model MH-100 of the brand MH Equipment. The banknote contents were 10, 20, and 30 wt% of the mixture, and the identification is shown in Table 1. After the polymer melting, homogenization was maintained for approximately 5 seconds, then the equipment was turned off, and the mixture obtained was removed. The same procedure was performed with the pure matrix (PPR). The materials obtained by homogenization were ground in a knife mill, Marconi, model MA 580.
Infrared spectroscopy was performed using the Shimadzu IRPrestige-21 Spectrophotometer with an ATR accessory. Thirty scans were performed at room temperature in the region between 4000 and 400 cm-1
The materials’ thermogravimetric analyses (TGA) were carried out using DTG-60H thermal analyzer equipment, Shimadzu. The protocol used was heating from 25 °C to 800 °C, with a heating rate of 10 °C.min-1 and a synthetic air atmosphere. The materials were submitted to differential scanning calorimetry (DSC) using PerkinElmer DSC6000 equipment. The samples were heated from 25 °C to 250 °C and kept at a temperature of 250 °C for 3 min, then cooled to 25 °C and heated again to 250 °C, with a heating rate of 10 °C min-1 and nitrogen flow of 20.00 mL.min-1
The measurements of the sample contact angle (θ) were carried out in triplicate on different regions of the samples. It used a wettability test apparatus at an ambient temperature of 24 °C. The samples were placed on the support, and then deionized water of standardized volume (10.00 µL) was dripped onto the sample’s surface. The images were obtained by filming the test using the AMCap program, and the contact angle (θ) was calculated using the ImageJ program. The contact angle (θ) was given by measuring three drops deposited on each film at least twice. Later, the arithmetic mean of the measurements and the standard deviation were calculated.
The exposure test to accelerated weathering was performed in the Accelerated Weather Simulator, brand BASS, model UVV-STD-4400. Seven samples were exposed to weathering in 12-hour cycles divided into 6 hours of exposure to UV-A light at a maximum temperature of 60 ºC, without moisture, followed by 6 hours of lamps off, at a minimum temperature of 30 ºC, with forced humidity. A total of 124 cycles, equivalent to 1488 hours of material exposure to artificial weathering, was performed.
The tensile tests were carried out in the Instron universal testing machine, model EMIC 23-20, operated at room temperature (∼ 23°C), using material bodies exposed to accelerated weathering and pure matrix specimens, not weathering. The materials were submitted to the test using clamps for tensile testing at a test speed of 50 mm.min-1 The results were obtained from an average of 7 specimens tested for each composition of exposed materials and five pure matrixes not exposed to weathering. The tensile tests of the PPR-BK composites and PPR were carried out to
evaluate the influence of fillers on the mechanical properties of materials before and after exposure to weathering.
In the FTIR spectrum of the recycled polypropylene and the PPR-BK composites (Figure 1), it was possible to identify the typical bands of the polymer groups at 2950, 2866, and 2840 cm-1 refer to the asymmetric stretching of methylene (νassimC-H3), the symmetrical stretches (νsimCH3) and (νsimCH2), respectively. It can be observed two bands are associated with the deformations of the methyl group (-CH3) in the region of 1376 cm1 (δsimCH3) and 1453 cm -1 (δassimCH3). Low-intensity bands at 842 and 725 cm-1 correspond to the angular deformations of the C-H bonds (δC-H) and 1170 cm-1 carbon-carbon stretching (νC-C) of the polymer chain[9,10]. The 500-400 cm-1 bands can be attributed to stretching the iron-oxygen bond (νFe-O), corresponding to the pigments in recycled polypropylene. Black inorganic pigments applied to plastics are made of iron oxides (Fe3O4, (FeCO)Fe2O4)[11]. Furthermore, the contamination of recycled polypropylene by low-density polyethylene (LDPE) was identified in the FTIR spectrum of the PPR due to the appearance of the characteristic LDPE bands. The main bands that characterize polyethylene appear in the range of 2918 and 2840 cm-1 and 1463 cm-1, corresponding to the C-H stretching vibrations and the vibration deformation band of the CH2 group, respectively. Bands associated with polyethylene amorphous/crystal form are also noted at 720-730 cm-1[12,13]. The analyzed material (commercial recycled PP) may contain other polymers in addition to recycled polypropylene, such as low-density polyethylene
Table 1. Identification of the composites and correlation with the banknote.
Identification banknote (wt%)
PPR-10BK 10
PPR-20BK 20
PPRC-30BK 30

Figure 1. FTIR spectra of (a) PPR, (b) PPR-10BK, (c) PPR-20BK, (d) PPRC-30BK, and (e) banknotes.
Evaluation of recycled polypropylene properties with banknote-reinforced
and polystyrene, due to potential contamination during the recycling process.
The banknotes spectrum (Figure 1) shows typical cellulose bands associated with the cellulosic O-H stretching and asymmetric stretching C-H in the range of 3500 and 3200 cm-1 and at wavenumber at 2900 cm-1, respect. The band at 1312 cm-1 is associated with the C-O-H (δ C-O-H) bending vibrations, and at 1052 and 1000 cm-1 corresponds to the stretching of the C-O-C bonds[14,15]. It was also possible to detect the presence of bands attributed to the Metal-Oxygen bonds, corresponding to the pigments in the banknote, pearl effect pigments. Among the numerous pigments used, these have a metallic appearance resulting from different thicknesses and combinations of titanium and iron oxides on mica, providing an iridescence effect[14]. The bands in the region between 600 and 420 cm-1 correspond mainly to the stretching of the Metal-Oxygen bonds (νTi-O, ν Fe–O) of the titanium (TiO2) and iron (Fe2O3) oxides present in the pigment.
Figure 2 shows the contact angle measured between the drops of water on the surface materials at the top and the average values of the contact angles. It was observed that all samples presented a mean contact angle characteristic of hydrophilic materials. In the case of recycled polypropylene, the hydrophilicity is attributed to the presence of pores on the surface of the polymeric films and metallic oxides, as indicated by the FTIR spectra. For composites, the hydrophilicity is higher than pure PPR, which is justified by adding banknotes composed of cotton fibers. The hydroxyl groups (–OH) present in the chemical structure of the fibers promote hydrogen bonds between the cellulose of the cotton fiber and the water molecules[15]. The contact angle value of the composite with 30%wt cellulose is indeed close to that of pure PPR. This can be explained by the possible uniform dispersion of cellulose fibers within the PPR matrix, which reduces the exposure of hydrophilic cellulose on the composite’s surface
TGA and DTG curves obtained for the banknotes and composites are shown in Figure 3. The banknote TGA exhibits a mass loss stage around 25-120 ºC relative to the
adsorbed moisture. The thermal degradation of the banknote occurs in a temperature range between 240 and 442 ºC, presenting two stages of decomposition. According to the banknote curve, peaks of maximum loss at temperatures of 323 and 395 ºC were identified in the DTG curve. Other authors have found similar results, attributing the stages of grade degradation to the degradation of cellulose-based fibers[4]. The TGA curve of the PPR10BK composite is like the pure polymer. It can be seen through the DTG of PPR that its thermal degradation occurs in two consecutive stages of decomposition. The first stage proceeds slowly and can be evidenced by forming a broad peak in the curve, followed by another immediate step that presents a maximum peak of around 427 °C. The composites PPR20BK and PPR-30BK present two stages of decomposition that can be better visualized in the DTG curve (Figure 3b). The incorporation of residues decreased the temperature at which the first decomposition stage occurs. In addition, it is observed that the maximum mass loss peak temperature of the composites decreases as the banknotes content increases. The residual mass visualized in the TGA curves is related to PPR contaminants and pigments in the polymer and the banknotes.
In the DSC curves, Figure 4 shows the crystallization and melting temperatures for PPR, PPR-10BK, PPR-20BK, and PPR-30BK. The PPR curve shows two peaks associated with melting (peak 1 and peak 2). and two crystallization peaks (peak 3 and peak 4). The data obtained from the DSC curves (Figure S1) of the PPR and PPRbanknote materials are shown in Table 2. The peak at the highest temperature, close to 160 ºC, refers to the melting of polypropylene. The peak at temperatures ~120 ºC was associated with the melting of LDPE, present in the commercial sample of PPR, as suggested by its FTIR spectrum. In the work of McCaffrey et al.[13], similar behaviors were observed in the polypropylene-polyethylene blend. The composites PPR-10BK, PPR-20BK, and PPR-30BK showed similar T m, around 150 ºC. The presence of the banknote reduced the melting temperature of the PPR.

Table 2. Differential Scanning Calorimetry curves data: PPR, PPR-C10%, PPR-C20%, and PPR-C30%.
Heating
Material
Cooling

3. TGA (A) and DTG (B) curves of the (a) PPR, (b) PPR-10BK, (c) PPR-20BK, (d) PPR30BK, and (e) Banknotes.
In the cooling curve, the first crystallization peak was identified at 122 °C, which refers to polypropylene crystallization. A shift in the crystallization peak is observed with the addition of banknote with a decrease in T c of 6 °C for PPR-30BK. The second exothermic peak occurs at a temperature of 113 °C and is probably related to the crystallization of LDPE. From the analysis of the enthalpy variation during the LDPE melting, it was possible to determine the amount of this polymer in the mixture. The variation of the melting enthalpy obtained by the peak area was 4.31 J.g-1. Considering that for pure LDPE, the enthalpy change during melting (ΔHm°) is 140 J.g-1, the ratio of values (ΔHpeak/LDPE) was 0.0307, corresponding to approximately 3.1%wt of mixed polyethylene to the PPR.

4. DSC curves of the PPR and Composites PPR/Banknote.
Furthermore, an addition of the banknote in the polymer raises Young’s modulus of the exposure weathering PPR, reaching a 55% increase for the PPR-10BK, following the 27% and 9% for PPR-20BK and PPR-30BK respect. This result indicates that the degradation of the PPR is superficial, probably caused by the breakage of a few polymeric chains, disrupting the entanglements and allowing an additional crystallization, increasing the stiffness and resistance of the polymer. The loss of mechanical properties of polymers usually results in more
The results of the tensile tests of the materials PPR-BK composites, pure PPR exposed to accelerated weathering (PPR), and recycled polypropylene not exposed to accelerated weathering (PPRS) are presented in Figure 5 It is observed from the average maximum tensile strength values that exposure to weathering provided the pure polymer (PPR) with a decrease of approximately 53% of the tensile strength compared to recycled polypropylene not exposed to weathering (PPRS). In the work of Tian et al.[16] , which investigated the accelerated weathering properties of polypropylene-wood composites, the tensile strength of virgin PP showed the most significant decrease in tensile strength values for all composites were higher than those of virgin PP. The degradative processes of the polymer exposed to accelerated weathering provide the scission of the chains and, consequently, the drop in the material’s mechanical properties, such as the reduction of its tensile strength.
Evaluation of recycled polypropylene properties with banknote-reinforced

intensive degradations in the polymer matrix. Incorporating banknotes proved efficient in delaying the degradation of PPR when subjected to weathering. One of the hypotheses to explain this behavior is that the cellulose in the banknotes influences the decomposition reactions of polypropylene, deactivating free radicals. The higher filler content can compromise its dispersion in the matrix and, therefore, have a lower influence on the mechanical properties. Consequently, the dispersed phase starts as a defect, leading to early matrix rupture and compromising mechanical properties.
The difference between the mechanical properties found after the analysis of the materials was statistically evaluated by the F (variance) and T (mean) tests with a confidence level of 95%. The results obtained for the tensile strength and Young’s modulus values are presented in the work’s Table S1 (Supplementary Material). It can be seen in Figure 5 that the composites with 20% and 30 wt% of banknotes showed slightly higher values of tensile strength when compared to the PPR. However, according to the average (T-test) analysis, the PPR-20BK and PPR-30BK composites appear statistically equivalent to the PPR. Therefore, it is considered that these composites, in terms of tensile strength, are similar.
Finally, the mechanical properties of the PPR-10BK composite were compared with those of the pure polymer before weathering. Statistical analysis revealed that they are statistically equivalent with 95% confidence. This result indicates that the composite, even after exposure to weathering, was similar to the unexposed pure polymer (PPR) in terms of its tensile strength. Regarding statistical analysis, the T-test confirmed that the means of Young’s modulus values are statistically similar between PPR and PPR10BK and distinct from PPR and PPR-10BK with 95% confidence. This outcome suggests that the composite, regarding Young’s modulus, remained similar to the unexposed polymer. All variances were equivalent, indicating that the variation in the results of the analyses performed was identical between the materials.
In this work, the batch homogenization technique effectively incorporated waste banknotes into recycled
polypropylene for composite production. This method facilitated the development of materials without needing prior treatment of the banknote waste or additional reagents. Using polymeric composites presents an effective strategy to reduce the accumulation of discarded banknotes and polymers, addressing sustainability requirements and reducing environmental waste.
The FTIR analysis revealed the presence of distinct functional groups in the PPR and the waste banknotes. In the spectra of the BK, bands associated with cellulose at 1312 cm1 (δC-O-H) and at 1052 and 1000 cm-1 (νC-O-C bonds), as well as to additives such as metallic pigments, at 600 and 420 cm-1, correspond mainly to the stretching of the MetalOxygen bonds. In the spectrum of recycled polypropylene, typical polypropylene bands were observed, along with signs of contamination by low-density polyethylene (LDPE). These observations were confirmed by an exothermic transition related to the crystallization of LDPE in the DSC curves. Moreover, the results from the DSC analysis demonstrated that the inclusion of waste banknotes led to a reduction in the melting temperature as the banknote content increased. Thermogravimetric analysis revealed a decrease in the thermal stability of the composites with an increased concentration of banknote waste compared to pure recycled polypropylene. Nevertheless, incorporating these wastes conferred stability against degradation from weathering, resulting in materials with enhanced mechanical resistance, particularly in terms of Young’s modulus and tensile strength. The addition of 10 wt% of banknote waste significantly increased Young’s modulus of the composite compared to the pure matrix, suggesting the potential for application of these materials in outdoor environments and the manufacturing of durable items.
• Conceptualization – Patrícia Santiago de Oliveira Patrício; Patterson Patricio de Souza
• Data curation – Ingrid Amélia dos Santos Matusinho; Ítalo Rocha Coura.
• Formal analysis – Ingrid Amélia dos Santos Matusinho; Ítalo Rocha Coura.
Matusinho, I. A. S., Coura, Í. R., Souza, P. P., & Patrício, P. S. O.
• Funding acquisition – Patrícia Santiago de Oliveira Patrício.
• Investigation – Patrícia Santiago de Oliveira Patrício; Ingrid Amélia dos Santos Matusinho; Ítalo Rocha Coura.
• Methodology – Ingrid Amélia dos Santos Matusinho; Ítalo Rocha Coura.
• Project administration – Patrícia Santiago de Oliveira Patrício.
• Resources – Patrícia Santiago de Oliveira Patrício; Patterson Patricio de Souza
• Software – NA.
• Supervision – Patrícia Santiago de Oliveira Patrício.
• Validation – NA.
• Visualization – NA.
• Writing – original draft – Patrícia Santiago de Oliveira Patrício; Ingrid Amélia dos Santos Matusinho; Ítalo Rocha Coura.
• Writing – review & editing – Patrícia Santiago de Oliveira Patrício, Patterson Patricio de Souza.
6 Acknowledgements
This study was financially supported by the Financiadora de Estudos e Projetos (FINEP) 324 – Reference 1636/22 Contract 01.22.0546.00, CNPq (INCT-MIDAS), FAPEMIG.
7 References
1 Awino, F. B., & Apitz, S. E. (2023). Solid waste management in the context of the waste hierarchy and circular economy frameworks: an international critical review. Integrated Environmental Assessment and Management, 20(1), 9-35 http://doi.org/10.1002/ieam.4774. PMid:37039089.
2 Aghmashhadi, O. Y., Rocha-Meneses, L., Bonturi, N., Orupõld, K., Asadpour, G., Garmaroody, E. R., Zabihzadeh, M., & Kikas, T. (2021). Effect of ink and pretreatment conditions on bioethanol and biomethane yields from waste banknote paper. Polymers, 13(2), 239 http://doi.org/10.3390/polym13020239 PMid:33445706.
3 Yehia, A., Yassin, K. E., & Eid, A. (2017). Recycling of shredded currency waste of Egyptian Central Bank for making goodquality papers. Separation Science and Technology, 53(3), 544-550 http://doi.org/10.1080/01496395.2017.1395887
4 Gil, C. S. B., Boaventura, T. P., Paula, F. G. F., Castro, M. C. M., & Oréfice, R. L. (2019). Direct use of Brazilian banknotes residue for the production of reinforced composites based on low-density polyethylene. Journal of Applied Polymer Science, 136(47), 48232 http://doi.org/10.1002/app.48232
5 Gu, F., Hall, P., & Miles, N. J. (2016). Performance evaluation for composites based on recycled polypropylene using principal component analysis and cluster analysis. Journal of Cleaner Production, 115, 343-353 http://doi.org/10.1016/j. jclepro.2015.12.062
6 Sommerhuber, P. F., Wang, T., & Krause, A. (2016). Wood–plastic composites as potential applications of recycled plastics of electronic waste and recycled particleboard. Journal of Cleaner Production, 121, 176-185 http://doi.org/10.1016/j. jclepro.2016.02.036
7 Singh, N., Hui, D., Singh, R., Ahuja, I. P. S., Feo, L., & Fraternali, F. (2017). Recycling of plastic solid waste: a state of the art review and future applications. Composites. Part B, Engineering, 115, 409-422. http://doi.org/10.1016/j. compositesb.2016.09.013
8 Schyns, Z. O. G., & Shaver, M. P. (2020). Mechanical recycling of packaging plastics: a review. Macromolecular Rapid Communications, 42(3), e2000415 http://doi.org/10.1002/ marc.202000415 PMid:33000883.
9. Chibani, N., Djidjelli, H., Dufresne, A., Boukerrou, A., & Nedjma, S. (2014). Study of effect of old corrugated cardboard in properties of polypropylene composites: study of mechanical properties, thermal behavior, and morphological properties. Journal of Vinyl and Additive Technology, 22(3), 231-238 http://doi.org/10.1002/vnl.21437
10 Yao, Z. T., Chen, T., Li, H. Y., Xia, M. S., Ye, Y., & Zheng, H. (2013). Mechanical and thermal properties of polypropylene (PP) composites filled with modified shell waste. Journal of Hazardous Materials, 262, 212-217 http://doi.org/10.1016/j. jhazmat.2013.08.062. PMid:24036146.
11 Butylina, S., Martikka, O., & Kärki, T. (2016). Effect of inorganic pigments on the properties of coextruded polypropylene-based composites. Journal of Thermoplastic Composite Materials, 31(1), 23-33 http://doi.org/10.1177/0892705716646416
12 Borovanska, I., Dobreva, T., Benavente, R., Djoumaliisky, S., & Kotzev, G. (2012). Quality assessment of recycled and modified LDPE/PP blends. Journal of Elastomers and Plastics, 44(6), 479-497 http://doi.org/10.1177/0095244312441731
13. McCaffrey, Z., Torres, L., Flynn, S., Cao, T., Chiou, B.-S., Klamczynski, A., Glenn, G., & Orts, W. (2018). Recycled polypropylene-polyethylene torrefied almond shell biocomposites. Industrial Crops and Products, 125, 425-432 http://doi. org/10.1016/j.indcrop.2018.09.012
14 Rodrigues, A. R. N., Melquiades, F. L., Appoloni, C. R., & Marques, E. N. (2019). Characterization of Brazilian banknotes using portable X-ray fluorescence and Raman spectroscopy. Forensic Science International, 302, 109872 http://doi. org/10.1016/j.forsciint.2019.06.030 PMid:31310941.
15 Imperio, E., Calò, E., Valli, L., & Giancane, G. (2015). Spectral investigations on 1000 £ banknotes throughout Italian Republic. Vibrational Spectroscopy, 79, 52-58 http://doi.org/10.1016/j. vibspec.2015.05.004
16 Tian, S., Luo, Y., Chen, J., He, H., Chen, Y., & Ling, Z. (2019). A comprehensive study on the accelerated weathering properties of polypropylene: wood composites with non-metallic materials of waste-printed circuit board powders. Materials (Basel), 12(6), 876 http://doi.org/10.3390/ma12060876 PMid:30875982.
Received: Apr. 30, 2024
Revised: Aug. 12, 2024
Accepted: Aug. 19, 2024
Evaluation of recycled polypropylene properties with banknote-reinforced
Supplementary material accompanies this paper.
Table S1. Statistical Comparison of Mechanical Properties in PPR/BK Composites
Figure S1. DSC curves (a) PPR-30BK, (b) PPR-20BK, (c) PPR-10BK, (d) PPR-30BK.
This material is available as part of the online article from https://doi.org/10.1590/0104-1428.20240044

São Paulo 994 St. São Carlos, SP, Brazil, 13560-340
Phone: +55 16 3374-3949
Email: abpol@abpol.org.br
2021