3 minute read
10. ANEMIA DREPANOCÍTICA


Generalidades
Es una enfermedad hemolítica del grupo de las hemoglobinopatías de tipo cualitativo (ESSALUD 2018). Es la hemoglobinopatía más frecuente. Esta es una anemia hemolítica congénita e intrínseca. Es más frecuente en la raza afroamericana.
Etiología
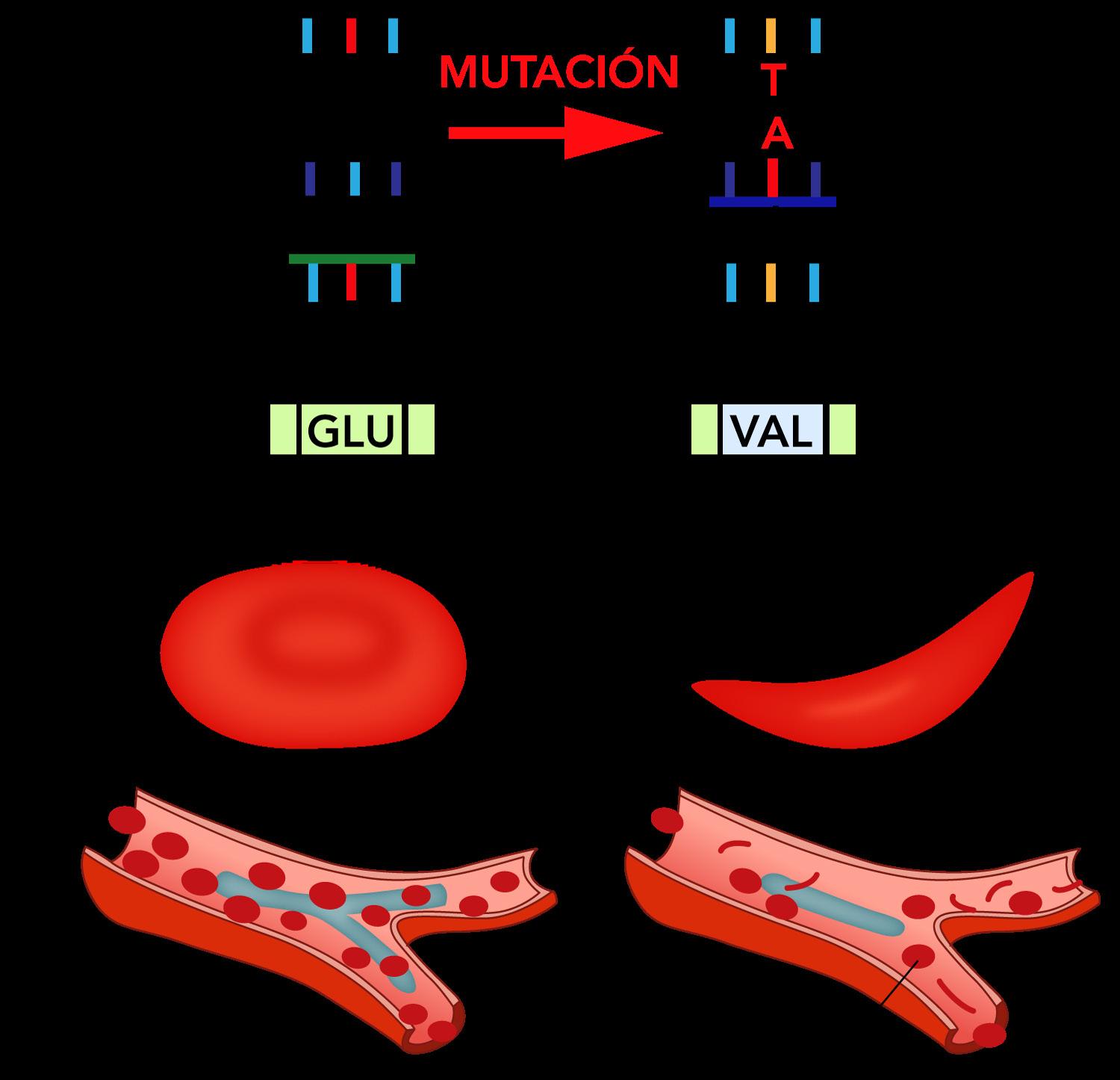
Mutación del gen de la beta globina. Tiene herencia autosómica recesiva.
Fisiopatología
En la sexta posición de la globina beta el ácido glutámico es sustituido por valina, luego dos globinas beta “erróneas” se unen a dos globinas alfa normales para formar una hemoglobina A “errónea” llamada hemoglobina S (HbS). Cuando la HbS está totalmente oxigenada permanece soluble en el citoplasma del eritrocito, pero al desoxigenarse conlleva a que estas HbS hagan un “apareamiento” entre ellas dentro del eritrocito, volviéndose menos soluble y formando cristales líquidos de polímeros de Hb S que crecen en longitud más allá del diámetro del eritrocito, y esto determina la forma de drepanocitos o células falciformes. Con forme se van formando los eritrocitos falciformes, la sangre se vuelve más viscosa y lenta, para que al final genere la oclusión de las arteriolas y capilares por los drepanocitos y la presencia de infarto de los tejidos circundantes.
Diagnóstico
Clínica
No presentan síntomas hasta la segunda mitad del primer año de vida debido al efecto protector de la hemoglobina fetal (HbF), ya al final de los primeros 6 meses de vida se empieza a producir cadenas beta mutadas y por ende la formación de HbS, los eritrocitos ahora son susceptibles a la hemolisis y puede manifestarse anemia hemolítica progresiva y esplenomegalia. La característica distintiva de la drepanocitosis es la oclusión vascular.
1. Oclusión vascular
A. Causas
Acidosis, hipoxemia, deshidratación, infección, fiebre, frio extremo
B. Manifestaciones clínicas
Recuerda
La anemia drepanocítica ejerce un efecto protector para malaria.
Recuerda
El paciente con drepanocitosis es susceptible a la infección por Salmonella.
(ESSALUD 2008)
– Huesos: dolor, dactilitis de manos y pies, infección (osteomielitis)
Pulmones: neumonía, síndrome torácico agudo
– Hígado: hepatomegalia, ictericia
Bazo: secuestro esplénico y esplenomegalia, autoesplenectomia (ESSALUD 2002)
– Pene: priapismo
Ojos: hemorragia retiniana
– Sistema nervioso central
Aparato urinario: necrosis papilar renal
– Úlceras de las piernas
2. Infecciones bacterianas
A. Sepsis
B. Neumonía
C. Osteomielitis
3. Defectos hematológicos
A. Anemia hemolítica crónica
B. Crisis megaloblástica
C. Crisis aplásicas
4. Defectos cardiacos
A. Cardiomegalia
B. Soplos cardiacos
5. Otras características clínicas
A. Detención del crecimiento
B. Embarazo de alto riesgo
Exámenes
Hemoglobina disminuida
VCM normal
HCM normal
Reticulocitos elevados
DRW aumentado
Lámina periférica se verá poiquilocitosis y anisocitosis, con eritrocitos en forma semilunar o falciforme, eritrocitos normales, dianocitos, punteado basófilo, cuerpos de Pappenheimer y cuerpos de Howell-Jolly.
Trombocitosis suele estar presente
Médula ósea con hiperplasia eritroide
Inmunoglobulina A elevada
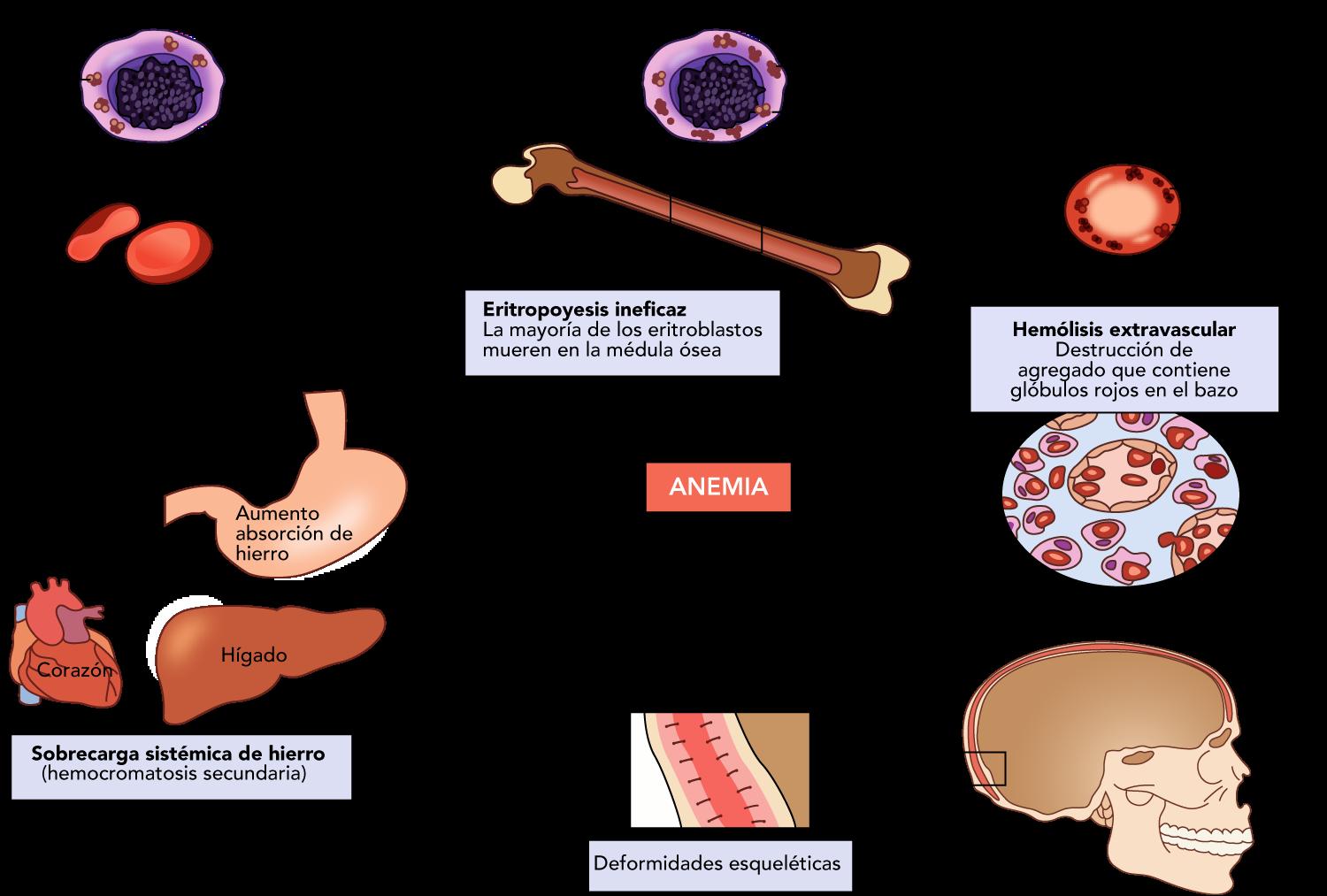
Marcadores de hemolisis crónica extravascular
Para niños y adultos, la combinación de HPLC (cromatografía líquida de alta resolución) e IEF (enforque isoeléctrico de capa fina) permite un diagnóstico definitivo.
Recuerda
La presencia de drepanocitos y dianocitos es la característica distintiva de la drepanocitosis.
Tratamiento
El manejo de sostén es crucial e incluye: hidratación adecuada, vitaminoterapia profiláctica, evitar ambientes con poco oxígeno (evitar ejercicio vigoroso, ir a grandes alturas y los viajes con aire no presurizado), uso de analgésicos para el dolor y antibióticos ante los primeros signos de infección.
La exanguinotransfusión es una opción en algunos casos.
Debido al riesgo de infección por bacterias capsuladas, la vacunación contra estas es crucial en los primeros años de vida.
Uso de transfusiones periódicas con uso de quelantes de hierro.
Hidroxiurea se usaría para aumentar la proporción de HbF
Trasplante de médula ósea resulta exitoso en algunas personas.
Generalidades
Las talasemias son un grupo diverso de trastornos hereditarios. Es considerada un tipo de hemoglobinopatía cuantitativa. Es una hemolisis congénita e intrínseca.
Etiología
Se debe a una alteración genética que reduce o impide la síntesis de una o más de las cadenas de globina del tetrámero de la hemoglobina. Es de herencia autosómica recesiva.
Clasificación
Se dividen en betatalasemias y alfa-talasemias. Aquí revisaremos las beta-talasemias y dentro de ellas principalmente la beta-talasemia mayor.
La beta-talasemia se puede dividir en cuatro síndromes clínicos:
Beta-talasemia mayor
Beta-talasemia intermedia
Beta-talasemia mayor
Beta-talasemia menor
Portador asintomático de beta-talasemia
Se caracteriza por una anemia grave detectada por primera vez en la infancia cuando ocurre en cambio de gamma globina por beta globina, ósea entre los 4 a 6 meses de vida. Generalmente se les diagnóstica entre los 6 meses y 2 años de vida.
Diagnóstico Recuerda
Tanto la drepanocitosis y la beta-talasemia suelen empezar entre los 4 a 6 meses de vida, cuando se requiere beta-globina para la formación de hemoglobinas normales.
Clínica
Hepatoesplenomegalia marcada
Ictericia
Cambios óseos marcados
Facies típica de “cara de ardilla” por prominencia de la frente, pómulos y maxilar superior.
Crecimiento físico y desarrollo alterados
Exámenes
Anemia muy severa
VCM muy bajo
HCM muy bajo
Reticulocitos elevados, aunque no tanto a lo esperado.
Lámina periférica con anisocitosis y poiquilocitosis, con dianocitos, formas de lágrimas y eliptocitos.
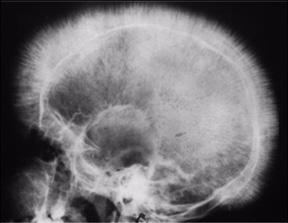
Radiografía de cráneo con aspecto típico de “cráneo en cepillo”
Los estudios de electroforesis o HPLC muestran que la mayor parte de hemoglobina es HbF, con un aumento leve de la concentración de HbA2
Médula ósea muestra marcada hiperplasia eritroide
Marcadores de hemolisis extravascular
Tratamiento
Transfusiones sanguíneas, se suelen iniciar en el primer año de vida. Es la principal opción terapéutica en estos pacientes.
Debido a la hipertransfusión es necesario agregar quelantes de hierro. Esplenectomía es una opción
El trasplante de médula ósea es el tratamiento curativo de la beta-talasemia mayor. Hidroxiurea podría ser una opción, aunque no todos los pacientes parecen responder.





