11 minute read
Returning to Basics of siRNA Design to Fulfil Therapeutic Potential

from IPI Autumn 2020
by Senglobal

The recent FDA approval of siRNA therapeutics has re-energised the RNA interference (RNAi) field. The RNAi mechanism was first proposed with the discovery of microRNA in 19931,2 and soon after it was demonstrated that microRNAs regulate gene expression in normal cellular biology and disease. In a Nobel prize-winning discovery in 1998, RNAi was described in C. elegans, where small interfering RNA (siRNA) caused sequence-specific gene knockdown3. The discovery of RNAi-mediated silencing in mammalian cells4 and parallel availability of the whole human genome sequence allowed for creation of research tools to study gene function using siRNA. Manufacturing siRNAs using chemical synthesis was straightforward and the prevailing thought was that siRNA could be used to silence any target gene in any cell. In fact, siRNAs targeting the whole human genome were synthesised in 2006 and whole genome screens have shown them to be a powerful genomics research tool5. However, due to challenges in siRNA delivery and pharmacokinetics, its potential as a therapeutic was not realised until the FDA approval of the siRNA therapeutics ONPATTRO® (patisiran) and GIVLAARI® (givosiran) in 2018 and 2019, respectively. The success of these therapeutics builds on almost two decades of siRNA design and encourages the field to pursue additional gene targets for therapeutic intervention.
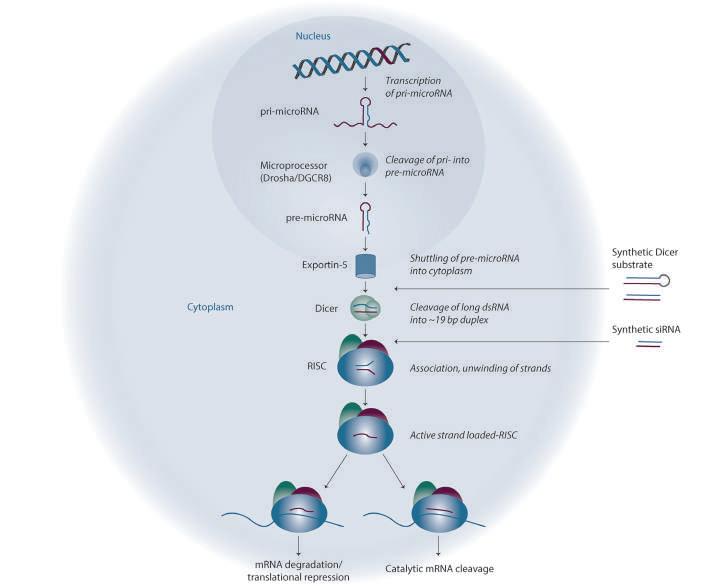
Mechanism To design a functional and specific synthetic siRNA therapeutic, the RNAi mechanism must be considered. The endogenous RNAi mechanism uses transcribed microRNA to regulate gene expression (Figure 1). Synthetic RNA can enter the RNAi mechanism at different steps to effect gene knockdown. Longer double-stranded RNAs [>19 base pairs (bp)] can enter the pathway as Dicer substrates which are cleaved into siRNAs by Dicer, or synthetic siRNAs (<19 bp) can enter the cell and be loaded directly
Figure 1: Synthetic siRNA can enter the endogenous RNAi mechanism to specifically downregulate gene expression. Primary microRNAs are transcribed in the nucleus and processed by the Drosha-containing complex (microprocessor) to yield the precursor microRNA, which is then shuttled out of the nucleus into the cytoplasm by Exportin-5. The Dicer complex cleaves the pre-microRNA hairpin into a double-stranded microRNA duplex. The double-stranded microRNA is unwound, and one strand is selected for incorporation into the RISC. RNA-programmed RISC binds to target mRNA with imperfect (seedmediated) or perfect complementarity, respectively. Seed-mediated complementarity causes transcript destabilisation and subsequent downregulation of protein expression while perfect complementarity causes catalytic mRNA cleavage and robust gene silencing.
into the RNA-induced silencing complex (RISC). These synthetic siRNAs must be able to enter the cell and interact with RNAi machinery including: Dicer to cleave the molecule into siRNA, RISC components for unwinding, loading, and then finally WatsonCrick base pairing one strand to the mRNA target to result in gene silencing.
RNA Synthesis and Chemical Modifications This discovery that siRNAs can cause robust gene knockdown excited the field because now it is possible to easily knock down the expression of a gene by delivering a complementary RNA sequence. siRNAs are comprised of two strands (active and passenger) that form a helical duplex (Figure 2). Compared to traditional organic molecule therapeutics, siRNAs do not have drug characteristics. They are relatively large in size, the backbone contains many negative charges, and the molecule is hydrophilic. Fortunately, the short length of siRNA allows almost any sequence to easily be synthesised by various chemistries: TBDMS (tert-butyldimethylsilyl), ACE (5'-silyl-2'-acetoxyethylorthoester), and TOM (2'-O-[(triisopropylsilyl)oxy]methyl) chemistries have all been used6. In addition, chemical modifications can be introduced to the siRNAs at multiple positions (nucleoside base, phosphate backbone, 5’ or 3’ ends, demonstrated by the coloured ovals in Figure 2) to alter the therapeutically relevant properties of the molecule. The advantage of siRNA is that its pharmacokinetic properties are primarily defined by the molecular structure, while the targeting properties are defined by the RNA sequence. This means that a successful molecular structure can be applied to a different target sequence and the development time to therapeutic can be substantially shortened. In theory, it should be possible to target any gene, making previously undruggable proteins amenable to therapeutic approaches.
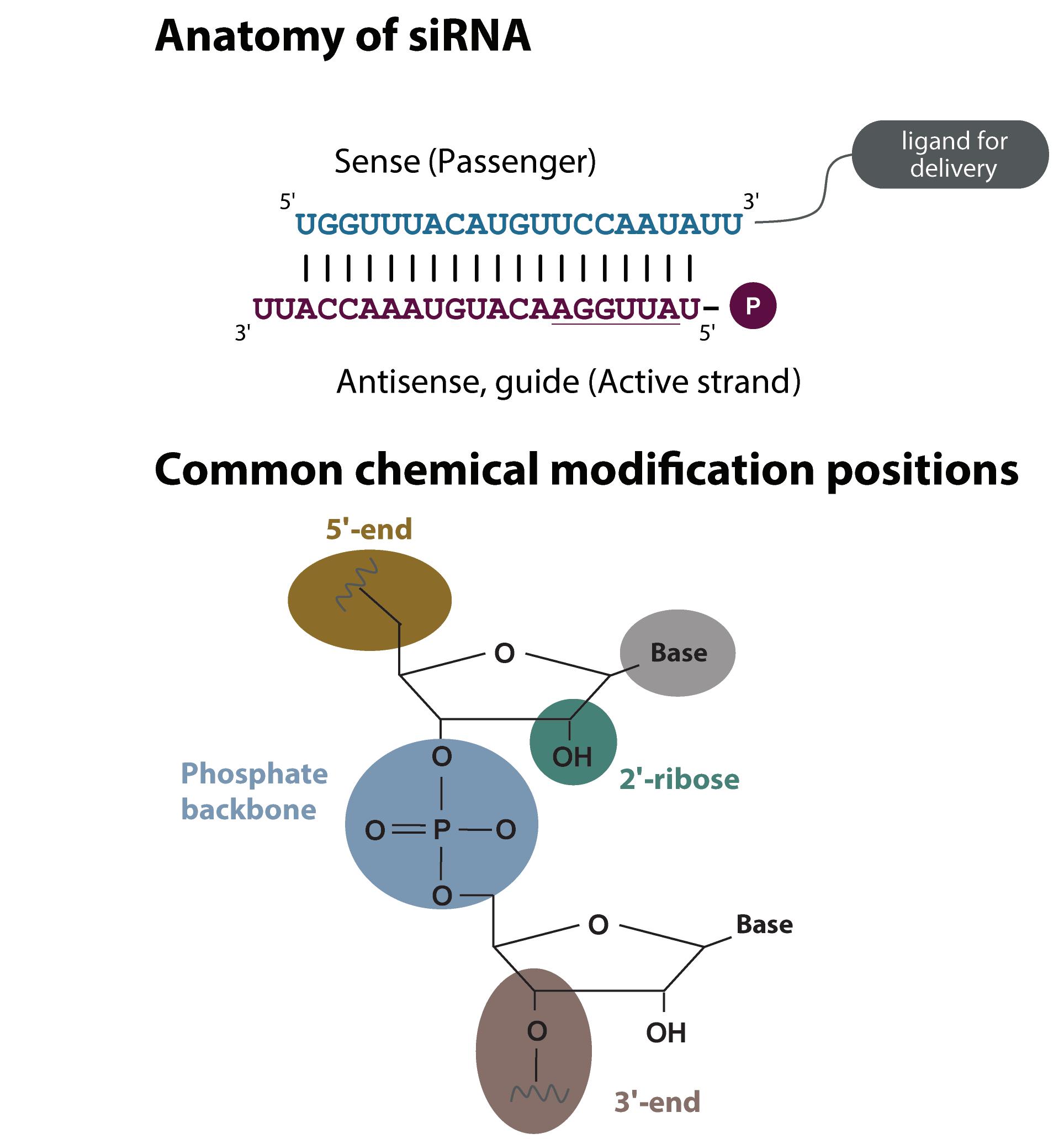
Figure 2: Composition of siRNA and placement of chemical modifications. Top: Synthetic siRNA is comprised of an active (red) and a complementary passenger (blue) strand to make a duplex of approximately 20 base pairs. Phosphorylation on the 5’ end of active strand is required for RISC loading and function. The seed region (underlined in figure) is also involved in downregulation. Conjugation of a ligand such as cholesterol, cell penetrating peptide or GalNAc can facilitate delivery of siRNA into the cell. Bottom: Chemical modifications can be applied to any position on the RNA (common positions are indicated by colours). These modifications improve characteristics of siRNA including function, off-targeting, nuclease stability and tolerance by the innate immune system.
Improving Nuclease Stability When unmodified siRNAs are injected into the bloodstream, they are degraded within minutes, due to the 2’-hydroxyl on the sugar making RNA susceptible to degradation by RNAses. Modifications on the 2’ position of the sugar can improve stability against RNAses, and 2’- fluoro (2’-F) and 2’-O-methyl (2’-OMe) modifications are commonly applied to both 5’ and 3’ terminal nucleotides of the siRNA to protect against exonucleolytic degradation. Backbone modifications, such as phosphorothioates (PS), can also improve nuclease resistance against endonucleolytic attack.
Innate Immune System Unmodified siRNA can cause toxicity in cells as it may stimulate the innate immune system. The innate immune system is activated by longer double-stranded RNA structures and consequently minimising duplex lengths reduces immune systemassociated toxicity.
Both sequence motifs and chemical modifications, or the combination of these, can cause cellular toxicity. For example, PS modifications on the backbone can cause toxicity when used in certain sequence contexts. Also, there are known toxic motifs associated with locked nucleic acid modifications. Avoiding combinations of these motifs and modifications is key to designing an effective and non-toxic siRNA. In addition to increasing cellular stability, modifications to the 2’-positions of the sugar such as 2’-F and 2’-OMe help the siRNA evade the innate immune system.
Unwinding and Strand Selection Unwinding of the duplex is an essential step of loading the guide strand (and not the passenger strand) for effective silencing and preventing off-targeting by the passenger strand.
Chemical modifications can make base pairing between guide and passenger strands so strong that unwinding is impaired and siRNA functionality is impacted. Data have shown that thermodynamic stability of the duplex requires the 5’ end of the guide RNA to be paired more weakly, followed by a higher affinity region through the seed sequence and finally a lower affinity region toward the 3’ end to promote strand release during unwinding. The thermodynamic stability of the duplex can easily be tuned through application of chemical modifications. For example, 2’-F and 2’-OMe modifications increase the local melting temperature, while PS modifications reduce melting temperature.
A 5’-phosphate on the guide strand is required for RISC recognition and loading. Chemical modifications can be used to block the phosphorylation and loading of the passenger strand and promote guidestrand-only loading. In addition, chemical stabilisation of the phosphate can improve loading and the resulting activity of the siRNA in vivo.
Delivery and Bioconjugation Since siRNA is negatively charged and hydrophilic, crossing the cell membrane is a major obstacle. A variety of delivery approaches have been, and are continuing to be, explored. siRNA can be encapsulated in polymeric and lipid nanoparticles for in vivo delivery. In addition, conjugation to delivery moieties have also been researched. Several ligands have shown promise, including conjugation to cholesterol, lipids, antibodies, aptamers, N-acetylgalactosamine (GalNAc), among others. The position of the conjugated ligand is important for maintaining siRNA function with the 3’ end of the passenger strand being a preferred location. Also, the length and composition of the linker between the siRNA and ligand can affect delivery and functionality.
Many of these bioconjugation strategies have shown promise, but finally after decades of research investment, two RNAi therapeutics have been approved. One aspect of their siRNA design that contributed to success is the use of GalNAcconjugated siRNA for specific delivery to liver hepatocytes. GalNAc binds to the asialoglycoprotein receptors which are expressed at a high level in hepatocyte cells; once bound, the GalNAc and siRNA are brought into the cell by endocytosis. GalNAc-siRNA conjugates provided an exciting advancement in delivery of siRNA
to hepatocyte cells and paved a path for RNAi therapeutics moving forward.7
Modification Strategy An effective synthetic siRNA must be recognised by the RNAi mechanism components for processing, unwinding, RISC loading and mRNA targeting. While some modifications can improve the molecular properties of the siRNA, others can compromise gene knockdown. Chemical modifications affect many molecular properties including the sugar conformation, winding of the duplex, and helix conformation. The chemical modification pattern that is applied must leave the RNA looking like an A-form helix. In general, 2’-F more closely mimics the 2’-hydroxyl in size and charge, so this modification is often applied to the guide RNA strand, which is more sensitive to modification. 2’-OMe modifications are often applied to the passenger strand that is not loaded into RISC. It has also been found that these types of modifications are tolerated well when applied in an alternating pattern to maintain the A-form helical structure.
Overall, the goal is to modify siRNA to improve nuclease stability, evade innate immune system response and improve delivery while maintaining ability of siRNA to interact with the RNAi machinery. (Reviewed in8,9)
Rational siRNA Design Many algorithms have been developed to design sequences that are both functional and specific. A workflow for efficient siRNA design and selection will include multiple steps to ensure functional knockdown of the intended target while avoiding offtargeting.10 First, target gene sequence space is defined and used to build a comprehensive siRNA candidate list. This is followed by removal of candidate sequences outside the open reading frame and 3’ UTR, not present in all of the transcript variants, or containing characterised SNPs. Additional filtering of sequences that are likely to decrease function is required. Filtered candidates include sequences with degenerate base symbols, toxic motifs, repetitive bases, excessively high or low GC content, microRNA seed matches, and sequences that cause secondary structure in siRNA. The remaining siRNA candidates are scored according to a functionality algorithm to select highly functional siRNAs; criteria may include thermodynamic asymmetry, possible fold-over structures and positionspecific nucleotide preferences. Finally, candidate sequences are aligned to all targets in the transcriptome to eliminate sequences that have perfect or near-perfect complementarity to unintended targets.
Additional design parameters may be required depending on the specific therapeutic siRNA programmes. For example, it may be necessary to design siRNA sequences that are identical or similar enough to be functional in mouse, rat and human ortholog gene to facilitate animal studies, or the disease application may require targeting all the protein-coding transcripts while avoiding a non-coding transcript.
A large body of work has defined how sequences are selected and how chemical modification can affect silencing efficiency. However, each unique application requires empirical testing. Success of an siRNA programme depends on testing multiple sequence candidates with the desired chemical modifications to find the most potent siRNA.
REFERENCES
1. Lee, R.C., R.L. Feinbaum, and V. Ambros, The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 1993. 75(5): p. 843-54. 2. Wightman, B., I. Ha, and G. Ruvkun, Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell, 1993. 75(5): p. 855-62. 3. Fire, A., et al., Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 1998. 391(6669): p. 806-11. 4. Elbashir, S.M., et al., Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 2001. 411(6836): p. 494-8. 5. Mohr, S.E., et al., RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol, 2014. 15(9): p. 591-600. 6. Caruthers, M.H., A brief review of DNA and RNA chemical synthesis. Biochem Soc Trans, 2011. 39(2): p. 575-80. 7. Springer, A.D. and S.F. Dowdy, GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther, 2018. 28(3): p. 109-118. 8. Khvorova, A. and J.K. Watts, The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol, 2017. 35(3): p. 238-248. 9. Takahashi, M., et al., Current Progress and Future Prospects in Nucleic Acid Based Therapeutics. 2017. 10. Birmingham, A., et al., A protocol for designing siRNAs with high functionality and specificity. Nat Protoc, 2007. 2(9): p. 2068-78.
Annaleen Vermeulen
Annaleen Vermeulen is a Principal Scientist at Horizon Discovery. She studied nucleic acid structure by NMR at the University of Colorado at Boulder where she received her doctoral degree in chemistry. Annaleen joined the Research and Development Team at Dharmacon (now part of Horizon Discovery) in 2004 and contributed to development of several products in the gene editing and RNAi fields.
Email: annaleen.vermeulen@ horizondiscovery.com
Amanda Haas
Amanda Haas is a Product Manager at Horizon Discovery. Amanda joined in 2005 and worked in multiple departments including Manufacturing, Chemistry R&D, Biology R&D and Product Management. She is responsible for custom synthetic manufacturing and also contributed to new product development in multiple areas across RNA interference and CRISPR-Cas9 genome engineering. She received her degree in chemistry at The University of Miami.
Email: amanda.haas@horizondiscovery.com








