9 minute read
Trapped Ion Mobility Mass Spectrometry (TIMS) Drives High-throughput Phosphoproteomics Research

from IPI Autumn 2020
by Senglobal

Unlike genomic and transcriptomic research, the measurement technologies for proteomics are still evolving, and the complete analysis of a proteome has not yet been achieved. The end goal of proteomics studies is not just to identify all the proteins that can be expressed, but also to uncover cellular protein events such as protein expression/ degradation, protein localisation, protein interaction, protein posttranslational modifications (PTM), and protein processing/splicing.
Protein phosphorylation is an abundant form of reversible PTM and plays a vital role in various cellular processes, such as protein synthesis, cell division, signal transduction, cell growth, development, and ageing1. As such, it regulates important metabolic, hormonal, developmental, and stress responses. Atypical phosphorylation can contribute to a range of disease states, including cancer and diabetes. Phosphoproteomics – the characterisation of proteins with phosphorylated PTMs – is therefore an important tool for obtaining insights into health and disease.
In cellular signal transduction networks, reversible phosphorylation is one of the key events in transducing a signal into the nucleus to control gene expression. Approximately 30% of human proteins were previously estimated to be phosphorylated, but researchers at the Laboratory of Molecular and Cellular BioAnalysis at Kyoto University developed a highly selective enrichment method for phosphopeptides and, when applied to proteome-wide acquisition of cellular phosphorylation status, revealed that at least 70% of human proteins are phosphorylated2,3 .
Recent technological advances have enabled the use of mass spectrometry (MS) in phosphoproteomic approaches to address the scope of phosphorylation. We have developed phosphoproteomics methods to carry out in vivo phosphoproteome profiling of kinase-targeting drugs, which would facilitate drug discovery and development for cancer therapy, as well as enable the exploration of the functional analysis of newly discovered phosphorylated molecules.
MS-based Phosphoproteomics Deep characterisation and quantitative analysis of proteins and PTMs are critical to understanding signalling pathways and abnormal disease states. Developments in high-resolution mass spectrometers and specific enrichment of phosphorylated peptides tailored for the global analysis of protein phosphorylation represent powerful tools for molecular and cellular biologists studying signal transduction pathways. Despite these advances, identifying PTMs remains significantly more challenging compared with unmodified peptides, as they often occur at low abundances and the differences in protein phosphorylation span several orders of magnitude, driving the need for instruments with higher sensitivity and increased peak capacities.
Protein expression varies depending on the genetic background of an individual, but also on time, localisation, and as a physiological response to external stimuli (stress, disease, ageing, effort, etc.). Moreover, because of the combined effects of alternative splicing, point mutation, PTM and endogenous proteolysis, a given protein (gene expression product) can be expressed as many different proteoforms, each having a dedicated biological activity. The sophistication of modern instrumentation enables the identification of tens of thousands of phosphopeptides in a singleshot liquid chromatography coupled with mass spectrometry (LC-MS) run, but the percentage occurring as positional isomers is unknown.
The combination of ion mobility spectrometry (IMS) with MS is a wellestablished technique that has shown considerable potential for improving peptide identification, providing structural information that is complementary to LC and MS. IMS-MS separates ions based on differences in their shape (IMS) and mass (MS), delivering information on the threedimensional (3D) structure of an ion. This added ability to separate ions by differences in conformation makes it possible to separate isobaric and isomeric species, such as phosphopeptide positional isomers (i.e., peptides that differ only by the residue that is phosphorylated), which are not easily distinguished by MS techniques alone4 .
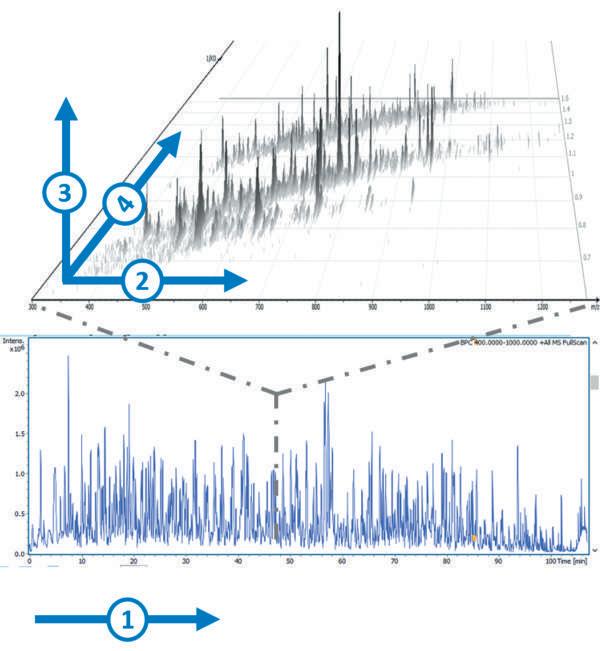
The commercialisation of trapped ion mobility spectrometry (TIMS) in 2016 built on the advances in IMS technology made over previous years. TIMS enables the interrogation and manipulation of mobility separated ion populations in the gas-phase, with high efficiency, duty cycle and high resolving power in millisecond-second timescales, and with the possibility to measure collisional cross section (CCS) using first principles that can be further utilised for structural assignments5, in CCS-aware workflows. The addition of TIMS provides a fourth dimension that is complementary to the previously used mass, intensity and retention time dimensions, resulting in 4D-Protoemics methods (Figure 1). TIMS is most often coupled with a time-of-flight (TOF) mass analyser to capitalise on its highspeed capabilities.
A fractionation approach is often used to obtain deeper proteome coverage, but this method is time-consuming, and if many clinical samples are analysed, the timeframe is not realistic. Modern TIMS-QTOF MS systems utilising parallel accumulation – serial fragmentation (PASEF) can provide the necessary high speed and increased sensitivity. The novel design allows for ions to be accumulated in the front section, while ions in the rear section are sequentially released depending on their ion mobility. Modern TIMS-QTOF MS with PASEF can provide sequencing speed > 100 Hz without losing sensitivity or resolution by synchronising the quadrupole isolation mass window with the elution time of the specific peptide packages from the TIMS funnel.
Our research found that PASEF can effectively increase MS/MS acquisition rates to reach new depths in phosphoproteomics, improving both identification and confidence in results. The high speed and sensitivity of the PASEF acquisition mode also facilitates high throughput on a TIMS-QTOF MS system.
Figure 1: Dealing with proteomics samples complexity can be reduced by spreading it into a fourth dimension: 1 – Retention time, 2 – Mass to charge ratio, 3 – Intensity, 4 –Ion mobility.
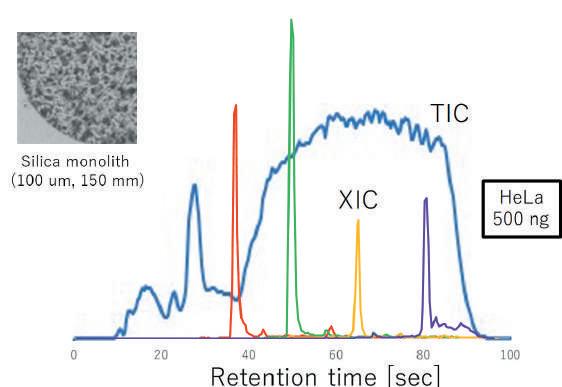
That capability is particularly important for processing the high quantity of samples required for metaproteomics studies. They now plan to take advantage of the PASEF acquisition mode in current research to investigate the interaction between humans and bacteria at the proteome level in order to uncover potential biomarkers of disease6 . one-minute gradient with the timsTOF Pro, a laboratory can investigate the mode of action of kinase inhibitors (Figure 2). Kinase networks are important for cellular signal transduction, by their catalysis of reversible protein phosphorylation, and kinase-mediated phosphorylation signals are known to cause or drive the progression of diseases such as cancer.
Protein Kinase Profiling TIMS-QTOF MS has implications in drug discovery, particularly for phosphorylations related to cancer. By implementing the Many drugs that inhibit a specific kinase or kinases have been developed for kinasetargeting therapy and, to date, more than 19,000 kinase inhibitors targeting ~260 protein kinases have been reported7, and ~30 small-molecular kinase inhibitors have been approved for clinical use by the United States Food and Drug Administration (US FDA)8. Our work aims to connect the kinases with their substrates to reveal the entire picture of the signalling network, using experimental and computational approaches.
For the past 20 years, identification of phosphorylated proteins based on MS has been used in many studies aimed at large-scale analysis of cellular signalling. Building on our work on the kinome with LC-MS/MS9,10, researchers are now achieving throughput levels only possible with TIMSQTOF MS.
Future Developments The increased versatility of computational proteomics, in combination with software and hardware developments, is a significant future direction for proteomics. An important advancement in MS-based phosphoproteomics is data-independent acquisition (DIA). DIA is a relatively recently developed MS acquisition technique where, unlike data-dependent acquisition (DDA), MS2 scans are acquired in a continuous and unbiased manner for all precursor ions falling within a specific mass range. The combination of next generation sequencing (NGS) with proteomics is becoming more widely used in multi-omics fields such as proteogenomics, which is finding application in clinical research, particularly precision oncology11 .
The combination of DIA with PASEF (Figure 3) allows researchers to compensate for the traditional DIA pitfalls by using a pattern of m/z isolation windows within consecutive TIMS events. The percentage of ions used in the dia-PASEF can be greatly increased (reaching up to 100% for low complexity samples, and still 5X higher than with traditional DIA methods using comparable isolation windows size and m/z range for high complexity samples). The diaPASEF cycle time can be reduced to make it compatible with short gradient separation while preserving a high selectivity. It benefits from the TIMS space concentration effect that allows for increased sensitivity and takes DIA selectivity to the next level with full 4D-proteomics support.
Over the last two decades, significant advances seen in technology and new methodologies have made proteomics
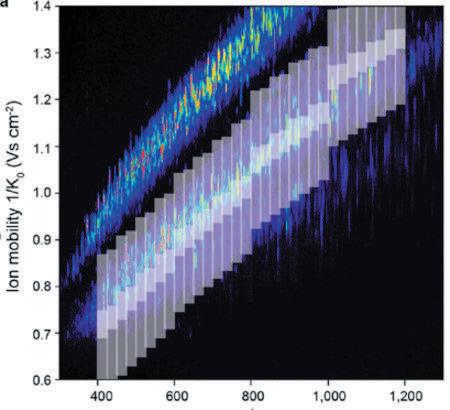
Figure 3: Example of diaPASEF windows distribution. Display of the 64 25 width m/z used with the 1,7 sec cycle time diaPASEF method. This window scheme uses 6.25% of the ions; an equivalent 3D DIA scheme would use only 1.25%.
an extremely powerful tool for protein scientists, biologists and clinical researchers. The current coverage of the proteome is far from 100%, and researchers are continually looking for improvements. It is clear that developments in MS-based proteomics, such as TIMS enabled 4D-Proteomics, have allowed researchers to gain deeper insights into the molecular and cellular functionality of the human body through the improvements in throughput and sensitivity. In particular, the PASEF acquisition method can provide extreme high speed and sensitivity to reach new depths in shotgun proteomics and phosphoproteomics, using low sample amounts. These 4D-Proteomics approaches will allow scientists to routinely explore parts of the proteome not previously accessible. REFERENCES
1. Ardito F, Giuliani M, Perrone D, Troiano G and Lo Muzio L (2017). The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int J Mol Med. 40(2):271-280. 2. Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S and Mann M (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis, Sci Signal, 3(104):ra3. 3. Sharma K, D’Souza RCJ, Tyanova S, Schaab C, Wiśniewski JR, Cox J and Mann M (2014). Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/ThrBased Signaling, Cell Reports, 8(5):1583-1594. 4. Glover MS, Dilger JM, Acton MD, Arnold RJ, Radivojac P and Clemmer DE (2016). Examining the Influence of Phosphorylation on Peptide Ion Structure by Ion Mobility SpectrometryMass Spectrometry, J Am Soc Mass Spectrom, 27(5): 786-794. 5. Fernandez-Lima F (2016). Trapped Ion Mobility Spectrometry: past, present and future trends, Int. J. Ion Mobil. Spec, 19:65–67. 6. Lin M-H, Sugiyama N, Ishihama Y. Systematic profiling of the bacterial phosphoproteome reveals bacterium-specific features of phosphorylation. Sci Signal, 8(394):rs10. 7. Hu Y, Furtmann N and Bajorath J (2015). Current compound coverage of the kinome, J Med Chem, 58(1):30-40. 8. Wu P, Nielsen TE and Clausen MH (2016). Small-molecule kinase inhibitors: an analysis of FDA-approved drugs, Drug Discovery Today, 21(1):5-10. 9. Sugiyama N and Ishihama Y (2016). Largescale profiling of protein-kinases for cellular signaling studies by mass spectrometry and other techniques, Journal of Pharmaceutical and Biomedical Analysis, 130: 264-272. 10. Sugiyama N, Imamura H and Ishihama Y (2019). Large-scale Discovery of Substrates of the Human Kinome, Scientific Reports, 9:10503. 11. Ang MY, Low TY, Lee PY, Nazarie WFWM, Guryev V and Jamal R (2019). Proteogenomics: From next-generation sequencing (NGS) and mass spectrometry-based proteomics to precision medicine, Clinica Chimica Acta, 498:38-46.

Yasushi Ishihama
Yasushi Ishihama is a professor of the Graduate School of Pharma-ceutical Sciences, Kyoto University. He has been working in the field of proteomics for 20 years. He has made contributions to this field especially by developing the basic technologies as well as by applying these tools to biology. His group also established ‘one-shot’ proteomics by ultrahigh resolution monolithic silica columns to uncover proteome on a microarray scale. He is also interested in comprehensive analysis of cellular signalling networks using experimental and computational approaches. He has published over 180 papers, cited over 20,100 times. He received the Research Award from the Mass Spectrometry Society of Japan (2011), the Award of the Japanese Proteomics Society (2013) and the Award of the Society for Chromatographic Sciences (2018). He is currently a president of the Japanese Proteomics Society, a vice president of the Japan Society for Analytical Chemistry and an executive council member of AOHUPO.








