



Sponsor Company: www.international-pharma.com Volume 15 Issue 3 Peer Reviewed The Great Why Questions About Dry Powder Inhalers High Efficiency Safety Profile for Recurrent Vaginitis and Biofilm Treatment Promising Advances in Plasma Proteomics for Early Disease Detection and Personalised Treatments Taking Healthcare Out of the Hospital How Device Design Can Empower Users

DIRECTOR: Mark A. Barker
BUSINESS DEVELOPMENT: Eliza Sarfaraz eliza@senglobalcoms.com
EDITORIAL: Virginia Toteva virginia@senglobalcoms.com
DESIGN DIRECTOR: Jana Sukenikova www.fanahshapeless.com
FINANCE DEPARTMENT: Akash Sharma accounts@senglobal.com
RESEARCH & CIRCULATION: Jessica Chapman jessica@senglobalcoms.com
COVER IMAGE: iStockphoto ©
PUBLISHED BY: Senglobal Ltd.
Unit 5.02, E1 Studios, 7 Whitechapel Road, E1 1DU, United Kingdom
Tel: +44 (0) 2045417569
Email: info@senglobalcoms.com www.international-pharma.com
All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IPI will be published in Winter 2023. ISSN No.International Pharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
2023 Senglobal Ltd./Volume 15 Issue 3 – Autumn– 2023
04 Editor’s Letter
TALKING POINTS
06 Rethink, Redesign, Recycle – Pharma's 100% PET Blister
IPI Speaks with Alberto Negra, Technical Manager at Rotor Print about creating a pharmaceutical blister which can be recycled, but without losing the properties of the conventional blisters.
09 A New Age of Pharmaceutical Innovation
IPI Speaks with PharmaLex’s Senior Director Dr. Patrick Lancier, Strategy Product Development Solutions, EU & US, on Next-generation Small Molecules.
12 Enhancing Clinical Succes During Early Development
IPI Speaks with Dr. Victor Diaz, Operations Director at Solitek on a new concept in solid state development services for the pharmaceutical, agrochemical and fine chemical industries.
16 Integrated CDMO Partner for Parenteral Drug Products
IPI Speaks with Mr. Anil Busimi, VP, Strategy & Marketing of Terumo Medical Care Solutions (TMCS) on the recent changes in the CDMO industry and what the market is looking for now.
REGULATORY & MARKETPLACE
20 5 Steps to Improving Pharmacovigilance System Master File (PSMF) Management
Large pharmaceutical companies are often restricted by a lack of ability to exchange information and access data from other departments. Christian Schmitz-Moormann at Generis considers the role of this essential master resource in maximising opportunities to improve R&D processes.
24 Why Pharma Dealmaking is a Sector that’s Ripe for Growth
The global pharmaceutical sector is booming thanks to continued growth in R&D spending in new technologies and therapeutic areas. Michael Jewell at finnCap Cavendish analyses the market and explains what we look forward to seeing what’s next.
DRUG DISCOVERY, DEVELOPMENT & DELIVERY
26 Antibody Drug Conjugates and the Link to Fighting Cancer Antibody drug conjugates are a big deal in oncology. Campbell Bunce at Abzena shows how, as we strive forward, pushing the science and generating more and better drugs, the future of ADCs looks set to live up to the lofty claim of revolutionizing cancer therapy.
30 Promising Advances in Plasma Proteomics for Early Disease Detection and Personalised Treatments
The early detection of disease poses a substantial challenge in the field of human health, necessitating the development of more accurate biomarkers. Shourjo Ghose and Andreas Schmidt at Bruker Daltonics explains the recent breakthroughs in plasma proteomics.
34 High Efficiency Safety Profile for Recurrent Vaginitis and Biofilm Treatment
There are over 200 million annual cases worldwide of vaginitis. Emeritus Giora Kritzman, a renowned scientist in the field of microbiology & Amnon Kadron, entrepreneur, and inventor, details a laboratory study, to address the state of the vaginal biofilm.
CLINICAL & MEDICAL RESEARCH
42 Keeping eCOAs off the Critical Path – Key Considerations
The prevalence of decentralised clinical trials has led to a compound annual growth in the use of connected digital devices. Brian Lillis, PhD at ICON and Janick Michel at Mapi Research Trust discuss that it cannot only improve the efficiency of clinical trials but also contribute towards long-term cost savings.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 1 www.international-pharma.com
Contents
TECHNOLOGY
44 Can a Modular Approach to Cleanroom Technology Deliver Flexible Sterile Manufacturing Capacity?
The sterile pharmaceutical manufacturing segment has been expanding rapidly in recent years. Trista Hager at AES Clean Technology explores the challenges facing pharmaceutical manufacturers in expanding their cleanroom capacity to meet burgeoning market demand.
MANUFACTURING
48 At Last! The Advent of Better Software Application UX to Manage Critical Manufacturing Processes in Highly Regulated Industries
For too long, professionals in the manufacturing industry have had to cope with software applications that are designed more for function than usability. Gurdip Singh at Kallik discuss why prioritising user experience (UX) and the use of intuitive software applications will be crucial in highly regulated manufacturing industries.
PACKAGING
60 Addressing the Challenges of Serialisation
Drug counterfeiting poses a significant threat to the pharmaceutical industry, risking patient safety and undermining the integrity of the global supply chain. Patrick Ferguson, of Tjoapack, explores the evolving landscape of serialisation and its implications for the pharmaceutical industry.
64 What does the Future Hold for Delayed Release Empty Hard Capsules?
The term “delayed-release” formulation refers to finished dosage form tablets or capsules, which are acid-resistant and protect the release of a medicament until the capsule has passed through the stomach. Subhashis Chakraborty of ACG Capsules explores, Enteric Release Capsules in the Nutraceutical and Pharmaceutical Markets.
68 Chemical Recycled ABS Materials for the Transition to a Circular Economy Model
The demand for new, sustainable ABS materials for drug delivery device applications is growing in the healthcare sector. Luca Chiochia of ELIX Polymers analyses an easier transition towards the use of more sustainable ABS medical materials in drug delivery and other medical devices in the coming years.
70 Navigating the Shift in Pharmaceutical Packaging Beyond Traditional Structures towards Healthier and more Sustainable Future
In the past the choice of packaging strategy in pharmaceutical industry was often purely based on functional criteria that defined which packaging material delivers the most stable performance for a safe, secure, and efficient drug delivery. Agnieszka Van Batavia at Constantia Flexibles, shows how the right packaging partner can help navigate the complex landscape of pharma packaging.
HEALTH OUTCOMES
72 Taking Healthcare Out of the Hospital
How
Device Design Can Empower Users
From point of care diagnostics to auto-injectors, medical devices are increasingly enabling more procedures and healthcare activities to be conducted outside of the clinic. Alex Driver of Team Consulting discuss how device manufacturers are utilising a range of tools and approaches, from digital support solutions to packaging innovations and more.
80 Through the Lens of Human Factors Developing Connected Drug Delivery Devices
The market for drug delivery devices continues to grow at a rapid rate. Finola Austin of Owen Mumford explains that as a growing number of biosimilars come onto the market, it is likely the market for subcutaneous
drug delivery devices will develop further.
LOGISTICS & SUPPLY CHAIN MANAGEMENT
82 Pharmaceutical Cold Chain Risks and How to Mitigate Them
Shipping temperature sensitive pharmaceuticals involves a range of risks that can impact the quality, safety, and availability of live saving treatments for patients. Nick Gilmore of Tower Cold Chain explores how risk can be mitigated through strategic partnerships with specialist temperaturecontrolled packaging suppliers.
86 Putting Safety at the Heart of Supplier Controls
Responsibility for ensuring the continuous monitoring of the safety of a medicinal product across the marketing authorisation lifecycle includes accountabilities for all third-party suppliers. Anna Lukyanova at Arriello, provides practical tips for maintaining a robust safety profile.
88 What does the Future of Temperature-controlled Pharma Supply Chain Look Like and How Do We Shape It?
Airfreight plays a vital role in the pharmaceutical industry, especially for economically export-oriented countries. Susanne Wellauer at Swiss WorldCargo discuss the future of the temperature controlled pharmaceutical supply chain.
INHALATION
92 The Great Why Questions About Dry Powder Inhalers
The research pertains to the economic burden of asthma and chronic obstructive pulmonary disease and the impact of poor inhalation techniques with commonly prescribed dry powder inhalers. Amnon Kritzman Kadron of Can Dapi shows how the patented DPI based on VibraMeshTM, solved major issues of the existing DPI model.
96 Clinical Gamma Scintigraphy for the Evaluation of Inhaled Medicine
IPI speaks with Simon Warren, Research Director at Cardiff Scintigraphics Ltd. on the use of Scintigraphy in inhalation drug development.
99 Vaping – Success or Disaster?
To use a vape as a medical product the technical hurdle would include large scale in vivo safety studies firstly in animals and then a human study to prove safety and efficacy. Bill Treneman of UPC Cambridge analyses that with proven safety of the inhaled vapour, then the scope to use this technology for small molecule delivery.
APPLICATION NOTES
50 Tablet Coating: Optimal Uniformity and Flexibility Tablet Coater Saves Resources
Tobias Borgers of L.B. Bohle explains the improvements of the BFC series through an intensive exchange with customers and continuous crossdepartmental research.
52 How to Make the Grade in Pharma Manufacturing
Rudolf-Michael Weiss of Stäubli discuss SKAN’s collaboration, which involved putting Stäubli’s then-current offering of Stericlean robots through intensive tests for cleanability, resistance, and movement.
56 High Potent Manufacture
David O'Connell at PCI Pharma Services discuss the role of CDMOs that can develop, manufacture and package drug products containing HPAPIs is more vital than ever.
76 Nanoformulation for Enhanced Drug Delivery and Better Patient Compliance
Dr. Maj-Britt Cepok of Beneo explains how Losan Pharma and BENEO, pool their collective resources and develop a convenient-to-take oncea-day oral solid dosage form prototype of Dexamethasone, an API that’s practically insoluble in water.
2 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Contents
+




INTERNATIONAL PHARMACEUTICAL INDUSTRY 3 www.international-pharma.com
our reputation We are Scientific Protein Laboratories. With over 40 years of expertise in development and cGMPcompliant manufacturing, we have become a trusted global source for innovation, customization, and the manufacturing of high quality API’s and naturally derived pharmaceutical products. • Custom process development and formulations • Traceable supply chain of natural ingredients • Scale up and cGMP production • Worldwide regulatory support • Decades of experience manufacturing naturally derived materials including heparin and pancreatic enzymes Put our quality team to work on your product solution. Visit us at CPhl Worldwide 2023 in October in Barcelona. Booth #7J60 splpharma.com 700 E. Main Street Waunakee, WI 53597 USA +1 (608) 849-5944 Scientific Protein Laboratories LLC part of Shenzhen Hepalink Pharmaceutical Group Co.,Ltd.
nature innovation
Two reasons for outsourcing, cost and focus on the core competencies of the enterprise. Surveying U.S., U.K., and Continental Europe companies concerning current and future outsourcing trends positions outsourcing as a prominent strategic lever. Achieving “best-ofbreed” practice is predicted to occur through new technology. Also, human resources and IT outsourcing will become more prominent. The results strongly indicate that partnership alliances and performance-driven contracts will become as important as the current preferred, trusted supplier relationship.
The pharmaceutical industry is under mounting scrutiny because of rapidly increasing expenditures for drugs in most countries. Drug expenditures are now the fastest-growing component of health care costs, increasing at the rate of about 15 percent per year. The increase is due both to a greater use of drugs and to higher prices for individual drugs.
Our Regulatory Section starts with, Christian Schmitz-Moormann at Generis
considering the role of this essential master resource in maximising opportunities to improve R&D processes and wider product lifecycle management.
Biopharma M&A is back, and IPOs are possible, but most biotech’s aren’t out of the woods. Most deals focus on late-stage or marketed assets, as pharma buyers attempt to fill gaps left by blockbusters set to lose patent exclusivity. There’s resurgent interest in treatments for chronic, widespread conditions across immunology and cardio-metabolic diseases, although rare diseases remain popular among some buyers. Initial public offerings (IPOs) and follow-on public financings are also reappearing, as investors take confidence from pharma’s renewed M&A activity. Michael Jewell at finnCap Cavendish in his article, “Why Pharma Dealmaking is a Sector that’s Ripe for Growth” analyses the market and explains what we look forward to seeing what’s next.
Our Drug Discovery section features an exciting study “High Efficiency Safety Profile for Recurrent Vaginitis and Biofilm Treatment” by Emeritus Giora Kritzman, a renowned
Page 73 Aurena Laboratories
Page 23 Austria Wirtschaftsservice Gesellschaft mbH
Page 91 Cardiff Scintigraphics Ltd
Page 41 FUJIFILM Wako Chemicals U.S.A. Corporation
Page 61 Gerresheimer AG
Page 93 H&T Presspart
Page 31 Kahle Automation
AdPage 85 Klinge Corporation
Page 89 Krautz Temax
Page 67 MM Packaging GmbH
BC Natoli Engineering Company
Page 45 Nemera
Editorial Advisory Board
Bakhyt Sarymsakova, Head of Department of International Cooperation, National Research, Center of MCH, Astana, Kazakhstan
Catherine Lund, Vice Chairman, OnQ Consulting
Deborah A. Komlos, Principal Content Writer, Clarivate

Diana L. Anderson, Ph.D president and CEO of D. Anderson & Company
Franz Buchholzer, Director Regulatory Operations worldwide, PharmaNet development Group
Francis Crawley. Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organization (WHO) Expert in ethics
Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
scientist in the field of microbiology & Amnon Kadron, entrepreneur, and inventor, detailing a laboratory study, to address the state of the vaginal biofilm using natural substances, enzymes, and bacteria to stabilise the system.
You must read our Health Outcomes section, where we feature a variety of features related to patient adherence and compliance. Alex Driver of Team Consulting discuss how device manufacturers are utilising a range of tools and approaches, from digital support solutions to packaging innovations and more to enable patients to adhere to their treatment regime and Dr. Maj-Britt Cepok of Beneo explains, Nanoformulation for Enhanced Drug Delivery and Better Patient Compliance.
This issue of IPI will be at a variety of exhibitions and conferences, namely CPHI in Barcelona, Bio Europe in Munich & DDL in Edinburgh. We hope to see most of you at these meetings. Enjoy this edition and my team and I look forward to bringing you more interesting articles and features in the December issue.
Virginia Toteva, Editorial Manager – IPI
IBC Nipro
Page 5 Novo Nordisk A/S
Page 27 Owen Mumford
Page 57 PCI Pharma Services
Page 7 Rotoprint
Page 65 Securikett®
Page 3 SPL Scientific Protein Laboratories
Page 53 Stäubli International AG
IFC Stoelzle Glass Group
Page 19 Terumo
Page 21 UPC Cambridge Ltd
Page 39 & 47 Valsteam ADCA
Georg Mathis Founder and Managing Director, Appletree AG
Jagdish Unni, Vice President – Beroe Risk and Industry Delivery Lead – Healthcare, Beroe Inc.
Jeffrey Litwin, M.D., F.A.C.C. Executive Vice President and Chief Medical Officer of ERT
Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma
Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
Stanley Tam, General Manager, Eurofins MEDINET
(Singapore, Shanghai)
Steve Heath, Head of EMEA – Medidata Solutions, Inc
Patrice Hugo, Chief Scientific Officer, Clearstone Central Laboratories
Heinrich Klech, Professor of Medicine, CEO and Executive Vice President, Vienna School of Clinical Research
Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
Sanjiv Kanwar, Managing Director, Polaris BioPharma Consulting
Stefan Astrom, Founder and CEO of Astrom Research International HB
T S Jaishankar, Managing Director, QUEST Life Sciences
4 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Editor's Letter
Index
Pharmaceutical grade
Benzalkonium Chloride
Reduce the risk for your raw material

If you want to reduce the risk for your raw materials, pharmaceutical grade Benzalkonium Chloride is the way to go –both for APIs (Active Pharmaceutical Ingredients) and excipients.
Our Benzalkonium Chloride is manufactured following cGMP guide ICH Q7 for APIs, the highest available quality standard in the industry. And we have 30+ years of experience in cGMP manufacturing.

This means you will get:
• Full traceability
• High product purity
• Full pharmacopoeia compliance (PhEur, USP/NF, JP, ChP)

• Audit access
• Full regulatory documentation package
• Manufactured under fully validated processes
Learn more about Benzalkonium Chloride at novonordiskpharmatech.com


Learn more at booth 7K20
October 24-26
INTERNATIONAL PHARMACEUTICAL INDUSTRY 5 www.international-pharma.com
Rethink, Redesign, Recycle – Pharma's 100% PET Blister
IPI Speaks with Alberto Negra, Technical Manager at Rotor Print about creating a pharmaceutical blister which can be recycled, but without losing the properties of the conventional blisters.

Q: Rotor Print provides all kinds of solutions for flexible packaging to the pharma industry. Can you tell our readers a brief history of the company, how you started and your growth so far?
A: The original idea for the project of Rotor Print was released during 2010. At that time, we detected some significant needs in the pharma primary packaging market. The ISO 15378 was just released in its first version in 2006, and only a few converters were able to adapt their existing facilities to those exigent requirements. Building a new plant according to these requirements was easier than adapting an existing one. Additionally, just the previous year, the merger between the two main players in the Spanish market (Amcor & Alcan) was completed and that opened an opportunity for other converters in those customers that were concentrated in the big new player.
Q: The manufacture of packaging materials for medicines requires compliance with standards of hygiene to ensure that the product is not contaminated with external elements that may alter the product. Can you tell us what these regulations are and how does your operations adhere to these guidelines?
A: The Standard required for primary packaging materials production is the ISO 15378 (current version is from 2017). This is equivalent to the traditional ISO 9001 Quality Standard plus the Good Manufacturing Practice (GMP) Standards. In this Standard of course there are a significate number of procedures to be followed, but I would like to emphasize that all the production processes are done inside a Certified Clean Room. This is very important, as there are not too many converters who have Rotogravure printers, Flexographic printers and Laminators inside a Clean Room. Most companies do the final process, slitting, in a Clean Room, while Rotor Print is doing 100% of their activity.
A Certified Clean Room is a facility built following the recommendations of ISO 16444
and are validated afterwards, achieving a minimum qualification of ISO-8. This means that you have a maximum number of particles per square meter of air inside the room, and this air is changed at least 20 times per hour.
Q: Have you noticed any recent changes in the industry? What are customers looking for now? How are you addressing these changes?
A: During the last 5 years, we have been introducing new structures using more sustainable materials for the flexible packaging in the food industry. The pharma market was initially reluctant to make these changes, but recently a bigger number of customers are asking how we can help them to make their blister packs, sachets or stick packs more sustainable.
Q: As a, industrial giant, I am sure you are governed by the vision of sustainability, emission control and circular economy. What steps are you taking to lead in this category, and what commitments have you made and gained from your customers and suppliers?
A: The use of flexible packaging by itself helps the companies to reduce their carbon
footprint if we compare them to the use of other kinds of packaging. For instance, now we are in a project to calculate the carbon footprint reduction of a company who wants to switch from rigid PP pots to stand-up pouches.
In terms of circular economy, we are following the recommendations of different EU organizations such as Ceflex, Citteo or Recyclass, to produce flexible packaging laminates fully compatible with a further mechanical recycling process.
One of our principal aim is to substitute those standard flexible packaging structures, combining different molecules of plastic compounds and aluminium foil, for another structure using in all layers of the same family of molecules, to be fully compatible with the existing recycling requirements. In some cases, we must help the customers to adapt their existing packaging machinery. In other cases, the material can run in their existing machines with minimum adjustments.
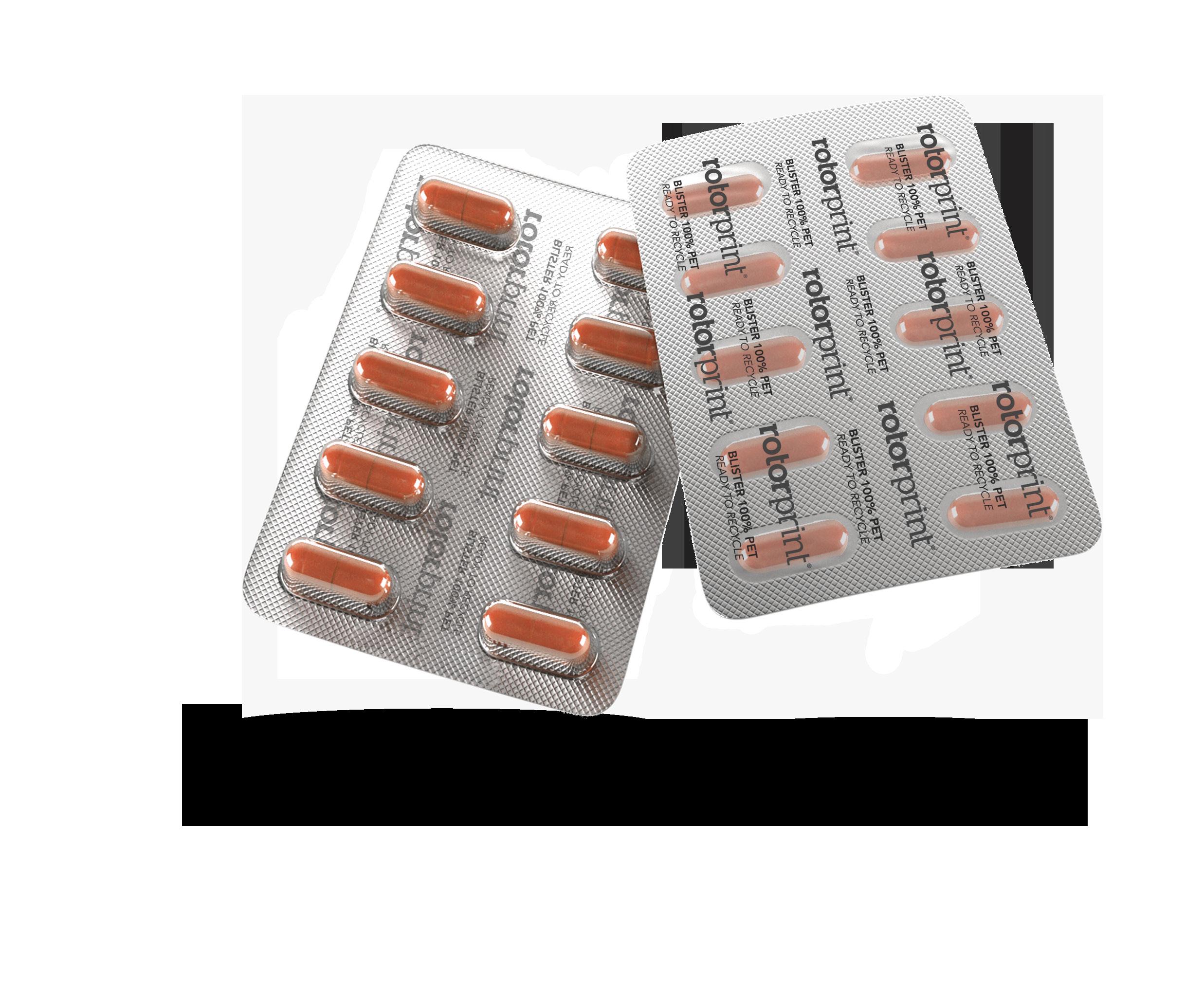
Q: You have an interesting strapline –Rethink, Redesign, Recycle, to refer to your – 100% PET Blister. Can you tell us about it. How will this impact the industry?
A: The blister packaging as we know it, is using a rigid film of PVC for the bottom and a lacquered aluminium foil for the lid.
6 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Talking Point






INTERNATIONAL PHARMACEUTICAL INDUSTRY 7 www.international-pharma.com
In this current situation, we are using two materials completely incompatible for the recycling process, which cannot be separated from each other after disposal.
At Rotor Print we are committed to sustainability, and so, with the recyclability of the packaging concept. Our goal was to create a pharmaceutical blister which can be recycled, but without losing the properties of the conventional blisters.
In our 100% PET blister we use a PET foil for the bottom and a PET foil for the lidding film. It is mono material and it’s not opaque, so fits the recycling plants requirements to be recycled. It has a pushthrough system that allows the user to extract the pill with the same convenience as with the aluminium. And the barrier properties are the same as the conventional blisters.
Q: The rotogravure printing system is the origin of your company. Can you explain the technology further & why is this unique process beneficial to your clients?
A: The origin of Rotor Print was only with rotogravure printing, as it was the technology offering a premium quality at that time.
During the last 10 years, the flexographic printing has achieved a significant improvement in its quality, using machinery equipped with the latest technologies in this field.


By the end of 2018 we introduced new machinery with the state-of-the-art technology in flexographic printing, and now we are glad to offer both technologies to the market, offering the same level of high quality.
Q: The pharma industry faces challenges from global competition, shorter innovation cycles, legal regulations for safety and environment, and individualized product demand. How does your company help ramp up production faster and accelerate faster products to market to combat new diseases?
A: In a few years, due to the UE regulations, the recyclability of pharmaceutical packaging will be mandatory to commercialize it. That’s why at Rotor Print we keep on investing on
I+D+I, to be able to create recyclable solutions of the conventional structures.
This regulation poses a huge challenge for the industry regarding the environment. Historically, pharmaceutical packaging was created focused on preserving the product, without considering the environmental impact of the packaging.
Nowadays, the industry might start to move into a more sustainable packaging concept. This move will pose other challenges, to move to a recyclable packaging meanwhile it keeps preserving the product.
In Rotor Print we help the laboratories to move to a recyclable packaging with the correct barrier properties to preserve the product without any additional investment in new machineries.

Q: What is Rotor Print’s vision for the future? What projects are you most looking forward to?
A: Rotor Print vision is recyclability and reducing the environmental impact. That’s
why we keep developing recyclable solutions for all kind of packaging (sachets, sticks, blisters, etc). Talking about the reduction of the environmental impact, we are creating life cycle assessment, being able to provide to our customers the environmental impact of the production of the packaging.
Chemical Engineer specialised in plastics and packaging materials. Since 1992 he has worked in the manufacture of flexible packaging for the food and pharmaceutical industries. In 2010 he was part of the group of professionals who founded Rotor Print SL. During the last 6 years he has been especially involved in the development of new structures and combinations of materials that replace those existing on the market to have the most sustainable solution possible.

8 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Alberto Negra
Talking Point
A New Age of Pharmaceutical Innovation
IPI Speaks with Dr Patrick Larcier, Senior Director, Strategy Product Development Solutions, EU & US, of PharmaLex, on Next-generation Small Molecules
Q: Can we start with a bit of an overview of what Small Molecule Excellence from PharmaLex involves, including the types of experts involved and the overarching objective?
A: Despite the high-profile advances in the field of biotechnology, small molecules continue to be the mainstay in disease treatment. Nevertheless, these new small molecule products require expertise in order to move seamlessly through the product development process. That’s where the concept of excellence comes from – it’s about having that depth of knowledge in the field and across the development life cycle. With this in mind, we involve experts focused on operational excellence in every different part of the process, meaning CMC experts, non-clinical experts, as well as clinical experts, in order to build the appropriate steps, paths and studies needed in each module of the regulatory dossier, while considering time constraints and using the most appropriate regulatory pathways at all development stages. Through this expertise and extensive experience in developing small molecules, we’re able to help companies save time and effort in the development of these products.
Q: Let’s break down some of the areas a bit more, specific to the type of support offered to small molecule innovators (for example, non-clinical/quality, strategy, marketing authorization, scope, regulatory support)
A: It's really on a case-by-case basis, based on the experience and expertise we have. This allows us to adapt the classic development steps or recommendations to each program and provide very specific support for the development of these molecules at the CMC, non-clinical, and even clinical stages. We combine this expertise with recommendations on interactions with the health authorities. The idea is to propose smart development plans to enable successful outcomes for our customers.
Q:
What is unique, or different, about the new generation of small molecules?
A: First of all, these products are typically more diverse and complex than traditional chemical entities and they target pathologies that previous generations of small molecules did not. By way of example, now we find small molecules under development in neurological conditions, such as Alzheimer’s disease and multiple sclerosis, as well as in the field of oncology.1 That’s different to what we have seen previously, with companies largely focusing on the development of what we would call “classical biologics” (or large molecules) for these diseases – such as monoclonal antibodies, fusion proteins, and so on. So now some companies are, in some ways, reversing, by turning to small molecules for these pathologies. We’re seeing that at companies like Bayer, which is targeting at the RNA level through small molecule innovation,2 and Pfizer, which has had success with breakthrough small molecule treatments for arthritis and ulcerative colitis.3 We’re also observing this trend with smaller companies, such as Agios Pharma with Pyrukynd for adults with pyruvate kinase (PK) deficiency which results in chronic anemia,4 as well as with Accent Therapeutics, Epics Therapeutics, Skyhawk Therapeutics among others.
Q: Why is a high level of expertise important when working with companies developing next-generation small molecules?
A: Some of these small molecules have come out of RNA research and are targeting RNA functions, which is why they are diverse and complex from both a technical and scientific standpoint. This requires specific knowledge and experience with working on the development of these small molecules. This is what we’re seeing with new products approved for cancer, in neurological conditions, and even in some inflammatory bowel diseases, as is the case with Pfizer’s tofacitinib (Xeljanz®) for ulcerative colitis and arthritis or Amgen’s RNA degraders in different therapeutic areas.
What’s really exciting about these breakthroughs is they are targeting difficultto-treat pathologies and adding to the therapeutic armamentarium. It’s important that these innovative therapies have the best chance of success because the need is great. Even if companies can get new biologics on the market, it seems many patients are not getting the long-term benefit that one would hope. They may respond to treatment for a period between 12 months and two years and then their condition deteriorates. Often then, the options for treatment are very limited. There might be another biologic that can be used, but these treatments may have quite similar efficacy with potentially fewer severe side effects.5 So, the hope with innovative small molecules is not only from the efficacy side, but also from the safety perspective to enable a more patient-centric approach. Again, having that expert insight into potential safety issues is important in the development process.
Q: What are the biggest changes influencing innovation with small molecules?
A: In addition to breakthroughs in RNA research, innovation is also coming from new approaches from both the pharmaceutical and biotech industry. Traditional biotech companies like Amgen and Biogen are now developing small molecules because they see a potentially smoother path on two fronts: manufacturing and regulatory.
On the manufacturing side, biological products, including both “complex biologics” (such as cell and gene therapies) but also “classical biologics”, typically face manufacturing challenges that can require more time and resources to overcome. Our regulatory expertise leads us to conclude that it’s likely that small molecules will face fewer questions from the health authorities, meaning those interactions might be shorter and questions regarding the future dossier might be easier to address. Fewer questions and a more seamless regulatory pathway inevitably mean shorter timelines for approval and faster availability of these treatments to patients. And that’s
INTERNATIONAL PHARMACEUTICAL INDUSTRY 9 www.international-pharma.com
Talking Point PHARMACEUTICAL INDUSTRY www.international-pharma.com
particularly important when a company is targeting conditions with huge unmet medical need.
Q: Can you describe any regulatory incentives that can help to create opportunities for the developers of novel small molecules?
A: Regulatory authorities offer a number of incentives to support innovation, particularly when tackling unmet medical need. In the EU, the PRIME scheme provides enhanced support for innovative therapies,6 as does the UK’s Innovative Licensing and Access Pathway (ILAP)7 and Fast Track Designation (FTD) or Breakthrough Designations (BTD) in the US.8 There are also Orphan Drug designations (ODDs) in many jurisdictions that companies should consider.9 All these kinds of programs may help small molecule innovators with their next steps.
There are also collaborative efforts such as the FDA-run Project Orbis,10 which developers of innovative oncology projects can take advantage of to gain access to several markets, and therefore propose new treatments to patients and generate revenue earlier, and in more jurisdictions.
These and other types of incentives or programs need to be considered in a global light, which is why working with an experienced service provider is important. Companies need that global perspective and the expertise required to solve CMC quality challenges that might be encountered, the right insight to define the non-clinical and clinical strategy for developing a small molecule, and the ability to put all of that into perspective in order to leverage appropriate regulatory tools, like Project Orbis or PRIME, particularly for the products in the field of oncology.
Q: What are the biggest challenges companies face when navigating the development and regulatory landscape with innovative new molecules?
A: While the challenges on the CMC side are less pressing than with biologics, there are still issues to overcome because these small molecules are structurally diverse and complex. For example, when working on RNA functions, there will be complexities that need to be addressed. That’s not just on the CMC quality side, but also pharmacological considerations, since companies must be able to demonstrate the effects and benefits their product is expected to have
on a specific disease, especially when we talk about neurological disorders such as Alzheimer's and multiple sclerosis, which are difficult to treat diseases. As I mentioned earlier, there are a number of regulatory tools such as PRIME, ILAP and Project Orbis (the latter for oncology developers, when appropriate), among others, that might be helpful, so getting the right advice and recommendations from an experienced service provider can be extremely helpful.
Q: What role does technology, including AI, play in supporting small molecule innovation, and what technologies is PharmaLex focused on for this purpose?

A: Through our Global Statistics and Data Science team of experts, PharmaLex supports artificial intelligence/machine learning from discovery to manufacturing of small molecules. Areas of expertise span:
• Target identification and related processes using real-world “omics” data
• Leveraging knowledge in pharmacology and pharmacometrics for the distribution, metabolization, elimination, dose, mechanism of action of new structures
10 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Talking Point
• Using modeling and ML for the “manufacturability” of new compounds.
In summary, AI plays an important role in supporting small molecule innovation.
Q: What is the vision of the PharmaLex small molecule team in terms of supporting the industry and raising the profile of small molecule innovation?

A: The vision is to support companies developing these small molecules to address treatment of patients suffering from diseases with high unmet medical need. One area, in particular, where small molecules could be particularly helpful is in the fight against antimicrobial resistance (AMR). This is an issue we are already facing and one that could be even more difficult to manage in the coming 25 years. We have an initiative at PharmaLex to support companies developing small molecules (in particular that target these pathogens) as we are committed to being part of the fight against AMR. By this, I’m not just talking about antibiotics, but also antifungals, anti-malarials, antiprotozoals –all these pathologies are emerging and could potentially result in more deaths than cancers by 2050.11
Unfortunately, despite this threat, there has been very limited research into fighting these pathogens and currently there are only around 30 new compounds under development in this area, which is tiny when compared with the field of oncology or autoimmune disorders, with about 4,000 agents. By being part of this fight and playing a key role in product development to address AMR, we believe we can really help to make a difference.
The contents of this article are solely the opinion of the author and do not represent the opinions of PharmaLex GmbH or its parent Cencora Inc. PharmaLex and Cencora strongly encourage readers to review the references provided with this article and all available information related to the topics mentioned herein and to rely on their own experience and expertise in making decisions related thereto.
REFERENCES
1. Application of Small Molecules in the Central Nervous System Direct Neuronal Reprogramming, Frontiers in Bioengineering and Biotechnology, July 2022. https://www. frontiersin.org/articles/10.3389/fbioe.2022. 799152/full
2. RNA-Targeting Small Molecules, Bayer. https://
www.bayer.com/en/pharma/rna-targetingsmall-molecules

3. JAK inhibitors: A new dawn for oral therapies in inflammatory bowel diseases, Frontiers in Medicine, March 2023. https://www.frontiersin. org/articles/10.3389/fmed.2023.1089099/full
4. PYRUKYND® (mitapivat) Approved in the EU for the Treatment of Pyruvate Kinase (PK) Deficiency in Adult Patients, Nov 2022. https://investor.agios.com/news-releases/ news-release-details/pyrukyndr-mitapivatapproved-eu-treatment-pyruvate-kinase-pk
5. Comparing Small Molecule and Biologics Drug Development Challenges, Pharma News Intelligence, May 2023. https://pharmanewsintel. com/news/key-differences-in-small-moleculebiologics-drug-development
6. PRIME: priority medicines, European Medicines Agency. https://www.ema.europa.eu/en/ human-regulatory/research-development/ prime-priority-medicines
7. Innovative Licensing and Access Pathway, Medicines & Healthcare products Regulatory Agency. https://www.gov.uk/guidance/ innovative-licensing-and-access-pathway
8. Fast Track, Breakthrough Therapy, Accelerated Approval, Priority Review, U.S. Food and Drug Administration. https://www.fda.gov/patients/ learn-about-drug-and-device-approvals/fasttrack-breakthrough-therapy-acceleratedapproval-priority-review
9. Orphan Drug Designation, PharmaLex. https:// blue-reg.com/glossary/odd/
10. Project Orbis, FDA. https://www.fda.gov/about-
fda/oncology-center-excellence/project-orbis
11. Antimicrobial resistance could kill more people than cancer by 2050, experts say, The Straits Times, Feb 2023. https://www.straitstimes.com/ singapore/new-drugs-needed-or-antimicrobialresistance-could-kill-more-people-thancancer-by-2050#:~:text=Antimicrobial%20 resistance%20could%20kill%20more%20people%20than%20cancer%20by%20 2050%2C%20experts%20say,-A%20new%20 network&text=SINGAPORE%20%2D%20 Antimicrobial%20resistance%20(AMR),2050%20 than%20cancer%2C%20say%20experts.
Dr. Patrick Larcier is Senior Director at PharmaLex and has worked in drug development and regulatory affairs for 30 years at biotech companies, at CROs and in consulting. Until April 2022, he led drug development and pharmacovigilance activities for PharmaLex France and Benelux; he now provides support for PharmaLex’s growing EU and US activities in these areas.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 11 www.international-pharma.com
Dr. Patrick Larcier
Talking
www.international-pharma.com
Point
Enhancing Clinical Succes During Early Development
IPI Speaks with Dr. Victor Diaz, Operations Director at Solitek on a New Concept in Solid State Development Services for the Pharmaceutical, Agrochemical and Fine Chemical Industries.
Q: Solitek brings a new concept in solid state development services for the pharmaceutical, agrochemical and fine chemical industries. Can you tell our readers a brief history of the company, how you started and your growth so far?
A: I met Steve in Cambridge, UK, in 2000, through some common acquaintances from the Chemistry Department at Lensfield Road. At the time, we both were working for discovery companies. We became good friends and have been in touch over the years, even after moving to different countries.
Both Steve and I did our PhDs and started our professional careers doing synthetic organic chemistry. But, by pure serendipity, we both ended up in the world of solid state development. Steve specialised in the development of crystallisation processes, while I spent most of the time providing solid state services for small and medium size biotech and pharma companies mainly, initially working hands on in the lab, and later leading large teams in some wellknown CDMOs in UK.
During this time, we got to understand solid state profoundly, and realised that to achieve its maximum potential, we needed to move away from the constraints we had in our jobs at the time. For instance, to work on different industries optimising the physical properties of the different compounds, which would ultimately lead to an improvement on the performance for the application for which these compounds have been designed, was not an option in labs and manufacturing plants that were designed to produce materials that were going to be administered to patients. It was also an opportunity to develop areas that were the normal next step in the work that we had been doing to date, something that we could not do in our previous jobs, since it escaped from our core roles at the time.
So, we got together, and we set up what was initially a consultancy company, but then very soon it became evident that we needed to bring some niche services in. We made our
business plan, put our savings together and got a couple of loans from banks to bring some analytical instrumentation specific for solid state research. When we started in July 2021, it was just Steve and me. Now we have €400K worth of instruments, labs in the Parc Cientific de Barcelona, and another three people on board who are really the ones responsible for the growth of the company. And we are envisaging additional growth in the coming months.
Q: I understand that you have set up a new advisory board. Who are they, and what value will they add to your services?
A: The formation of the Advisory Board marks a significant milestone for Solitek, signaling the company's dedication to enhancing its capabilities, staying at the forefront of the pharmaceutical sector, and ensuring excellence in its service offerings.
Dr. Sudhakar Garad is currently Global Head of Chemical and Pharmaceutical Profiling at Novartis Institutes for BioMedical Research (NIBR), and he is a recognised figure in the pharmaceutical industry. He has held key leadership positions in various global pharmaceutical companies, contributing to the successful development of numerous drugs. Dr. Garad's expertise will be invaluable in guiding Solitek's strategic decisions.
Dr. Michael J. Wilkins, currently a pharmaceutical consultant, after retiring from his position as Head of Pharmaceutical Formulation Development at Almac Pharma Services, has a wealth of experience in preclinical, clinical and commercial formulation development, and has played an instrumental role in advancing cutting-edge drug delivery systems. His profound knowledge of drug formulation technologies and industry trends will empower Solitek to create innovative and efficient pharmaceutical solutions.
We are delighted to welcome Sudhakar and Michael to our Advisory Board. Their exceptional expertise and accomplishments in the pharmaceutical sector will undoubtedly
strengthen our position in the industry and accelerate our efforts in developing groundbreaking solutions for the benefit of patients worldwide. The combined experience of the newly appointed advisors will complement the existing strengths of Solitek's dedicated team of scientists, researchers, and professionals. With a shared vision for pushing the boundaries of pharmaceutical innovation, the Advisory Board will foster an environment of collaboration and excellence.
Q: Solid-state characterisation allows scientists to understand the properties of formulation and formulation components, the first step in rational formulation development. Can you explain in detail the services you offer, and the value you add within the pharmaceutical development process?
A: When developing a new drug, we first need to understand the application for which it has been designed. Depending on the intended route of administration, dosage form, desired onset of the effects, and duration of the treatment, a different solid form with different physical properties may be recommended. For instance, non-prescription ibuprofen is available as a tablet, chewable tablet, capsule, gel capsule, suspension (liquid), and drops (concentrated liquid). Each one of those formulations would have required the active ingredient to exhibit different properties, and in fact, ibuprofen is marketed depending on the specific application as a sodium salt, lysine salt of free acid parent compound.
What we intend is to provide the necessary tools for our clients to make informed decisions that will benefit the progression of their development programs. Unlike many other companies which focus on the larger, more profitable studies, we thrive in problem solving. Being able to solve problems is a skill that we have acquired over many years working in this field, we have seen lots of different situations, and each one teaches you something new. If we can help our clients to solve the pressing problems they are facing now, we trust they will come back to us in the future with larger studies.
12 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Talking Point
The management team at Solitek is also very close to the science. Both Steve and I are, first and foremost, scientists. We get heavily involved in the discussions with our clients and we prepare the proposals that are designed to address the issues concerning them right now. So, although I very rarely work in the lab these days, I still do a lot of literature research and data analysis to provide direction to our team working in the lab. And because we get so involved in the projects, it is so easy to change direction when and if the findings suggest that the initial proposal is no longer appropriate.
Our services revolve around solid state characterisation, solid state screening and selection, development of crystallisation processes, development of early enabling preclinical formulations and training and consultancy. However, we don’t like to think of our services as independent entities with no connection between them. In fact, we think of these services together as a toolbox, to be deployed as required depending on the problem in question. Sometimes the clients do understand what they need, but others, the clients only know what their problem is. For instance, we have had clients telling us that they needed to reach a particular concentration in solution. Whether this requires a change on solid form, a new formulation, or changes in the particle size distribution, may not be clear at the beginning. Working alongside the clients, we can deliver the most efficient solutions for their problems.
Finally, we really are trying to push the boundaries of science. For some of our screening projects, particularly for cocrystal selection projects, we have partnered with specialist companies who have excellent computational tools to apply AI in the selection of the potential coformers, thus increasing the chances of successfully

finding new cocrystal species. Obviously, the use of these AI tools comes at a cost, but it really reduces the scope of the experimental part of the study, thus saving material and reducing the cost of the experimental part. But introducing changes takes time and we need to work with our clients for them to see the benefits of this approach, as opposed to the more traditional one, using large batteries of compounds to be able to identify new cocrystal species.
Q: Solid-state transformations may occur during any stage of pharmaceutical processing and upon storage of a solid dosage form. Early detection and quantification of these transformations during the manufacture of solid dosage forms is important since the physical form of an active pharmaceutical ingredient can significantly influence its processing behaviour, including powder flow and compressibility, and biopharmaceutical properties such as solubility, dissolution rate and bioavailability. How would you analyse solid-state transformations of pharmaceutical compounds using vibrational spectroscopy?
A: It is true that solid state transformations may occur during processing and storage of solid dosage forms. This is typically addressed by selecting the most stable form in the conditions likely to be encountered. However, the most stable form is not always the one that will provide the best performance, and in those cases, it becomes critical to be able to monitor and quantify the transformation of the crystalline solids present in the dosage form.
Another common case is when we get a client that has come up with a new form of an existing drug and they want to confirm
that their form does not transform on one of the previously known forms, potentially infringing a patent. Or the opposite situation, when an innovator suspects that a generic company is infringing their patent and wants to demonstrate precisely that this transformation does occur.
Our preferred way to detect and quantify forms on a tablet is typically by XRPD. Of course, it is dependent on identifying a window in the diffractogram in which there are no interfering peaks from the excipients, and we can focus only on the peaks of the API. Also, things like the loading of the sample are critical. We have just completed a study in which we investigated a new form of a known API. In this case the API was only present in the tablet on a 2.5% w/w ratio, which is probably in the low end of the limit of detection for this technique, but we successfully developed an XRPD method, and we managed to compare with 11 previously described forms of this compound and determined what transformation was taking place.
We have used vibrational spectroscopy in some cases, but it is not typically our preferred approach to monitor and quantify solid-sate transformations.
Q: There has been a significant discussion on Solid-state study of polymorphic drugs: carbamazepine. Can you shed some light on it. How do you analyse such materials?
A: Carbamazepine is a great model compound. It is a very small molecule (MW 236), very rigid, with hydrogen bond donors and acceptors, and it possesses the most common synthon when making cocrystals, which is the amide (urea) group. On top of that, it also contains aromatic groups that could potentially contribute to intermolecular π-π stack interactions. All these properties make carbamazepine an extraordinary target for new cocrystals.
To analyse these new species, the first thing we would do is to establish whether they display a new XRPD pattern. Even before solving the 3D structure, we can determine the unit cell parameters from the XRPD pattern, and often we may be able to establish the stoichiometry of the new form based upon the volume of the unit cell. After this, we would normally try to grow single crystals for structure collection by X-ray or
INTERNATIONAL PHARMACEUTICAL INDUSTRY 13 www.international-pharma.com Talking Point www.international-pharma.com
electron diffraction, and we would establish the purity of the sample by HPLC and /or 1H-NMR followed by a battery of analyses to fully characterise the new species, like DSC, TGA or DVS.
Once the new species has been characterised, we would need to establish whether it offers an advantage with regards to the starting parent compound, which could be an improvement in some of their physical properties (i.e. stability or solubility) or a lower or melting point which may have utility for a new formulation, for instance or advantages derived from the processing of the material (i.e. increased in purity or different morphology with better bulk properties.
Since you mention carbamazepine, it is important to determine the objective of the exercise. In this case is not likely to lead to a new, marketable form of the drug, but it can open the door to new strategies to identify new cocrystals and may be useful to establish strategies for the development of new, more valuable drugs in the future.
Q: Have you noticed any recent changes in the industry? What are customers looking for now? How are you addressing these changes?
A: A few years ago, there was some reluctancy to perform solid state studies too early. Since the success rate of new drug approvals was of only ~10%, many felt that spending money here was not the best use of their budget, and these studies were reserved for compounds that had reached the clinical candidate status. Plus, let’s not forget, many new drugs were being discovered by small companies intending to out-license their programs to larger pharmaceutical companies who would make the decisions on the development of the early candidates.
More recently, many companies have implemented a more holistic approach, in which a strong cross-functional team made of medicinal chemists, biochemists, pharmacologists, formulation scientists and clinicians collaborate to build the appropriate physicochemical attributes into the design of the NCEs (e.g. pKa, logP/D, solubility, stability, etc.), select the solid forms susceptible of being developed for the intended application for which these were designed (e.g. salts, cocrystals, polymorph, etc.) or to choose the optimal delivery

strategy (e.g. route of administration and formulation principles), thus increasing the chances of success.
If these strategies lead to unsatisfactory results, it is probably wise to consider bringing the project to a halt. This is a great mechanism to mitigate risks and prioritise the development of candidate drugs with greater chances of success, before incurring in much higher expenditure.
For some organisations in which some of these functions are not present, there are well positioned contract service companies like Solitek that can take on the lacking functions and act as strategic partners. This is often an efficient way to incorporate these functions into your own organisation, without the need to increase overheads or the additional costs of infrastructure and headcount.
Q: As you are involved in formulation development and chemical compounds, I am sure you are governed by the vision of sustainability, emission control and circular economy. What steps are you taking to lead in this category, and what commitments have you made and gained from your customers and suppliers?
A: It would be naïve on my part to think that a company as small as like Solitek can have any significant impact in terms of sustainability, emission control and circular economy. However, we try to convey those values in the work we do, always recommend our clients to go with the parent compound, if possible, since a salt or cocrystal would extend the preparation and require additional reagents and solvents. Also, whenever possible, we advocate to use formulation approaches that are more environmentally friendly. For instance, if you need to prepare amorphous
dispersions, instead of using spray-drying technologies, which often requires large volume of solvents, we recommend investigating technologies like hot melt extrusion, which has no waste. But in these cases, we can only make suggestions, since the impact of the work we do on a lab scale, in terms of sustainability, is negligible in comparison with the manufacturing efforts that will come later.
Having said this, we try to establish a no waste philosophy in our labs and offices. We are virtually a paper free company, we use electronic LNBs and never print proposals, updates, or reports. And the data is typically collected, stored, and reviewed electronically. Equally, CDAs, MSAs, invoices, etc., are all of them handled electronically, as it is our quality system and internal and external audits. And we are big advocates for electronic signatures, so there is no need to ever print a paper to get it signed. But this is just a drop in the ocean, a lot more should be done by everybody.
Q: The pharma industry faces challenges from global competition, shorter innovation cycles, legal regulations for safety and environment, and individualised product demand. How does your company help ramp up production faster and accelerate faster products to market to combat new diseases?
A: I believe that the biggest waste of time and money in our industry comes from the development of suboptimal candidate drugs, with lack of control on their properties or with physical properties that are not appropriate for its intended use. Having to repeat preclinical studies because the form has changed during the initial tests is an absolute waste of time and money that today should
14 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Talking
Point
not happen. And even worse if we get into the really expensive clinical trials.
By selecting a candidate with the right biopharmaceutical attributes, the right solid form, and the right balance of physical and physicochemical properties, we will be en route for a much more successful development program, thus saving money and time along the way.

If we must recommend bringing a program to a halt, which is not something we will be doing lightly, but when we have to do it, the purpose will always be to redirect resources towards programs that are
more likely to bring benefits to the patients in the long term.
Q: What is Solitek’s vision for the future? What projects are you most looking forward to?
A: For the future, as well as continuing to provide solutions to our clients, we want to fully embrace the age of AI. It is early days for AI in certain sectors of science, and all the new AI development news has been received with caution. However, we do believe that all the new computer
power that was not available that long ago can be used to improve the outcome of our research. For instance, we discussed above how to use AI tools to aid the selection of coformers for cocrystal selection studies. But this is not the only one. What if we could use AI to establish what parameters are going to be important to control particle attributes during crystallisation processes?
Finally, this is something I have been thinking about for a while. Small molecule organic compounds will behave similarly, no matter for which industry they have been designed. So, applying what we know from the pharmaceutical industry into other industries (food, flavors and fragrances, dyes and pigments, cosmetics, etc.) to modify the physical properties of these compounds, will likely lead to an enhanced performance of these active ingredients.
In my (very little) spare time, I like to get in the lab and try a few experiments, and every now and again, something interesting comes out of those. If, as well as being interesting, these results turn out to be commercially viable, we may be looking at a new business line in the future. Quoting Michael Ende, “But that is another story and shall be told another time.”
Victor Diaz trained as a synthetic, organic chemist. He completed his PhD on the synthesis of pseudo-oligosaccharides with carbamide-type bridges and glycomimetics related to polyhydroxyindolizidines and on studies on enzymatic inhibition from the University of Seville, Spain. Victor is a passionate leader in the world of solid state and preclinical development of small molecule active pharmaceutical ingredients. With almost three decades of experience in the pharmaceutical industry and nearly 20 years in preclinical development, Victor has established himself as a driving force behind some of the larger European teams providing solid state services for the biotech and pharmaceutical industries while at Sigma-Aldrich, Johnson Matthey and Almac.
Email: victor.diaz@solitekpharma.com

INTERNATIONAL PHARMACEUTICAL INDUSTRY 15 www.international-pharma.com Talking
www.international-pharma.com
Point
Victor Diaz
Talking Point
Integrated CDMO Partner for Parenteral Drug Products
IPI Speaks with Mr Anil Busimi, VP, Strategy & Marketing of Terumo Medical Care Solutions (TMCS) on the Recent Changes in the CDMO Industry and what the Market is Looking for Now.
Q: Terumo is a core player in the healthcare market. Can you tell our readers a brief history of the company, how you started and your growth so far?
A: Terumo Corporation is a global leader in medical technology and has been committed to "Contributing to Society through Healthcare" for over 100 years. Based in Tokyo and operating globally, Terumo employs more than 25,000 associates worldwide to provide innovative medical solutions in more than 160 countries and regions. The company started as a Japanese thermometer manufacturer and has been supporting healthcare ever since. Now, its extensive business portfolio ranges from vascular intervention and cardio-surgical solutions, blood transfusion and cell therapy technology, to medical products essential for daily clinical practice such as transfusion systems, diabetes care, and peritoneal dialysis treatments. Terumo will further strive to be of value to patients, medical professionals, and society at large.
Terumo has three companies encompassing global businesses.

• Cardiac and Vascular Company
• Medical Care Solutions Company
• Blood and Cell Technologies Company
As Part of Terumo Medical Care Solutions, the Pharmaceutical Solutions Division develops patient-oriented parenteral delivery solutions for therapeutic performance and safety.
We offer pharmaceutical and medical device manufacturers around the world comprehensive product design and development services through our decades of experience collaborating with pharmaceutical companies from the earliest phases of drug development to product commercialisation to optimise critical aspects of parenteral drug delivery.
Innovation and creativity are central to our value proposition. Our expert teams lead the industry in developing and manufacturing advanced, high-
performing infusion and injection technologies, including pre-fillable syringes, CDMO services for all parenteral applications.
Q: Terumo Corporation has recently renamed the “General Hospital Company” to its new name “Medical Care Solutions Company”. What prompted this move, and how will this impact your standing in the industry?
A: The new Brand Medical Care Solutions Company was launched as part of our continued growth and Terumo’s 5-Year Growth Strategy (GS26).
The landscape of healthcare today is facing a paradigm shift. The point of care is not limited to in-hospital treatments but has expanded to in-home care. There are increased needs for a more personalised treatment, as well as improving hospital management efficiency and safety. The brand promise of Terumo Medical Care Solutions, "Quality time for better care," embodies the values and the wide scope of solutions the Medical Care Solutions Company provides today and carries the promise to provide this quality time for better care to everyone involved in healthcare.
Q: Can you give a brief overview of Pharmaceutical Solutions Division and CDMO platform?
A: Pharmaceutical Solutions Division develops patient-oriented parenteral delivery solutions for therapeutic performance and safety. Globally trusted for quality and precision, we offer pharmaceutical and medical device manufacturers around the world comprehensive product design and development services.
We have decades of experience collaborating with pharmaceutical companies from the earliest phases of drug development to product commercialisation to optimise critical aspects of parenteral drug delivery.
Innovation and creativity are central to our value proposition. As a partner for global pharma customers to offer parenteral drug solutions with five business platforms:
1. Injection Platform includes our K-Pack™ II Hypodermic needles.
2. Infusion Platform features our Surflo™ Winged Infusion Sets.
3. Primary Packaging Platform with our Prefillable Syringes PLAJEX™ range.
4. Drug Delivery Devices with our intradermal injection device, Immucise™.
5. CDMO – Our parenteral Contract Development and Manufacturing Organisation (CDMO) brings customers a uniquely integrated service offering. We bring together our high-quality polymer prefillable syringe (PFS) manufacturing and our complete CDMO service offering that includes formulation development, fill-finish services, device assembly and packaging all in one trusted partner. For our customers, integrated innovation means end to end services that deliver injectable products of the highest quality, compliance, and with reduced time to market. Our Japanese manufacturing sites are cGMP compliant and are certified by the Japanese Pharmaceuticals and Medical Devices Agency (PMDA). Both of our aseptic fill finish sites have been successfully inspected by the U.S. FDA and the EMA has inspected one site for a commercial product inspection. This track record of success with leading international regulatory bodies demonstrates Terumo’s continuing commitment to achieving the highest quality standards for its pre-filled syringe CDMO customers.
Q: Have you noticed any recent changes in the CDMO industry? What are customers looking for now? How are you addressing these changes?
A: During COVID-19 pandemic many pharma companies struggled to find manufacturing capacity in a short time, CDMOs filled the gap and will continue to play this role in future.
16 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
We see the following major changes in the global CDMO landscape..
• Increasing capacity and modernization: Increasing number of biologics in the pipeline demands specialised aseptic fill & finish operations and support. Globally, we see significant investments by CDMOs to increase the capacity across the value chain to meet this demand. Some CDMOs are updating their facilities with latest technologies and fill & finish equipment.
• Quality & Compliance: Continuous improvements to enhance the quality and compliance means latest technology, equipment, procedures.
• Flexibility: in operations due to drugs being very specific to a patient population or therapeutic area. CDMOs needs to adapt to provide smaller batch sizes, multiple packaging formats, just in time (JIT) production models.
• Integrated services: Many biosimilars or bio-betters in late stage and most of these companies rely on CDMOs for their manufacturing, supporting these pharma companies from pre-clinical to commercial stage is critical to avoid changes along the value chain.
• Collaboration: Industry wide collaboration to have a robust supply chain and offer best solutions to the pharma companies and ultimately to patients.
Q: How do you support companies for different stages of drug development (clinical to commercial)?
A: Terumo’s CDMO services span the full range of services required to take a customer molecule from drug formulation to fully packaged commercial product, all in one company.
We can help the pharma companies simplify their supply chain as One single source for device technology, and drug product development/manufacturing services – from clinical, small batches to commercial scale production.
Whether if it is a small, medium, and large organisation, we can customise our services to meet the customer requirements. For example, a small company may not have resources and capabilities for drug compatibility, device testing or regulatory experience, whereas the big pharma may have those capabilities in-house.
Depending on the customer’s needs and capabilities we either offer the full package service or collaborate closely to split the responsibilities without compromising the quality and time to market for final product.
Q: Terumo Medical Care Solutions is a leading manufacturer of primary containers, you have launched a readyto-fill polymer syringe for challenging biotech drugs with larger volume. Can you explain to our readers the latest trends in this market and how they are evolving?
A: There are more than 3000 injectable drugs in the pipeline – many of which are sensitive biologic drugs with potential stability challenges. Thus, primary packaging solutions must reduce drug-container interactions throughout the shelf life. In addition, many biologic drugs in the pipeline are targeted towards smaller patient populations, which requires the manufacture of smaller batches even in pre-filled syringes – without compromising on quality. Flex filling lines in combination with ready-to fill components like PLAJEX™ is the answer to address this trend.
Prefilled syringes kickstarted a trend that moved injectable drug delivery from hospital to home environments. Today's pharma companies want to make the self-administration of injectables as safe, convenient, and painless as possible. Combining pre-filled syringes with autoinjectors is a popular approach. And it is also possible to combine drug delivery devices with electronics to collect data at the time of administration. For example, we work with external partners to ensure compatibility of PLAJEX™ with autoinjectors and safety devices so that our customers have an integrated solution.
Q: The pharma industry faces challenges from global competition, shorter innovation cycles, legal regulations for safety and environment, and individualised product demand. How will integrated CDMO help ramp up production faster and accelerate faster products to market to combat new diseases?
A: Combining the global standard in polymer pre-fillable syringe supply with
unrivalled sterile formulation development services by a uniquely coordinated approach, provides significant advantages to Terumo’s CDMO customers. Working with one supplier that has an unmatched understanding of the capabilities of their own syringes, allows Terumo to select best device for each individual project. It also allows Terumo’s CDMO teams to coordinate the formulation and device development steps, and to quickly arrive at an optimal formulation, that considers the challenges of potential interaction with the device. Terumo’s highly efficient sterile filling operations utilise modern, flexible, and highly automated sterile filling lines suitable for both clinical and commercial manufacturing volumes. The result is that Terumo’s integrated innovation approach can result in a significantly derisked project with accelerated timelines.
Q: As an industrial giant, I am sure you are governed by the vision of sustainability, emission control and circular economy. What steps are you taking to lead in this category, and what commitments have you made and gained from your customers and suppliers?
A: Based on the Group Mission of Contributing to Society through healthcare, Terumo believes that its social mission (corporate purpose) is to lead the advancement of healthcare and the enhancement of patients’ QOL. To achieve this, Terumo will strive for the utmost quality in all activities, create solutions of value by utilising new technologies, and spread those solutions globally.
Amid drastic changes in social and global environments, Terumo will also take leadership toward solving a variety of social issues, and to meet the expectations of its broad range of stakeholders.
We position the reduction of environmental burden as one of the most important sustainability priorities. By committing to the 1.5°C pathway and working with various stakeholders to achieve the goal, we will further accelerate our efforts to address the global issue of climate change. Terumo Group’s targets for Greenhouse Gas Emissions reduction include:
• Achieve carbon neutrality by 2040.
• Increase a ratio of renewable electricity use up to 50% by 2030.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 17 www.international-pharma.com Talking Point www.international-pharma.com
• Reduce absolute GHG emissions 50.4% by 2030 from a 2018 base year.
• Reduce GHG emissions 60% per unit of revenue by 2030 from a 2018 base year.
• On our rooftop in Belgium, we will install 3000 solar panels by early next year, sufficient to meet the annual electricity consumption of some 385 households. This investment is a fitting example of sustainability in action, contributing to Terumo Corporation’s goal to be carbon neutral by 2040.
The Terumo Group has established key sustainability activity themes to resolve issues related to healthcare, society, and the global environment, and is working to realise a sustainable society and sustainable growth for the Terumo Group while also listening to the voices of various stakeholders. Through these sustainability efforts, Terumo will contribute to the realisation of a sustainable society, create new Terumo strengths, and culture, and bring about sustainable corporate growth.
Q: What is Terumo’s / PSDs vision for the future? What projects are you most looking forward to?
A: With the Terumo Mission Statement of ‘Contributing to Society through Healthcare’, and the Medical Care Solutions Company promise of ‘Quality time for better care’, the Pharmaceutical Solutions Division support this through their strategic vision of aspiring to be the preferred partner of pharma customers for innovative medical devices and injectable drug delivery solutions, providing safe and effective products that improve people’s lives. We recently announced our global expansion of our more than 20 years established integrated CDMO in Japan offering end-toend services supporting customers with pre-filled syringe (PFS) design, moulding, drug preparation, filling, assembly, and final packaging for challenging biotech drugs and small molecules.
Our CDMO capabilities with three CDMO sites in Japan, will involve engaging on projects that cover the early development stage to large scale commercial production for global pharmaceutical customers including assembly of PFS with devices like autoinjectors, needle safety devices, and supporting customers for regulatory submissions globally. Terumo has invested to expand the CDMO production facilities for pre-
filled syringes at its wholly owned subsidiary Terumo Yamaguchi corporation Ltd. The total investment, including peripheral equipment, will be 15 billion yen (100m USD). As a result, Terumo Yamaguchi D&D's production capacity will increase by 3.5 times compared to when the plant started operation in 2016. This global expansion is just one example of an exciting outlook for us and aligns with our growth strategy. Moreover, the Kofu Factory site will be extended with an entire new building dedicated to the Medical Care Solutions Company. The total investment will be 52.2 billion JPY (350m USD), and the construction will be completed in fiscal year 2025. Once operation starts, the new manufacturing facility will be utilised for the contract development and manufacturing organisation (CDMO) business, as well as products related to peritoneal dialysis. Terumo is a global leader for injection devices like needles, we have capabilities to customise the needles for a specific therapeutic area. We are looking forward to expanding our portfolio. Terumo will continue to contribute to better patient care and transformation by further enhancing our solutions that leverage the strengths it has cultivated over the years,
thereby providing “yasashii*” medical care for everyone involved in healthcare.
* “Yasashii” is a Japanese term that suggests a combination of kind, caring, friendly, and thoughtful.
Anil Busimi almost 20 years experience built profound understanding of the global injectable drugs industry. He is passionate about providing solutions to pharma customers and end users in the areas of primary packaging, drug delivery devices, and fill and finish aspects for injectable drugs. Anil is currently working at Pharmaceutical solutions Division of Terumo Medical Care Solutions (TMCS) as VP, Strategy & Marketing and be part of Terumo's corporate mission: "Contributing to Society though Healthcare."

www.terumopharmaceuticalsolutions.com

18 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Talking Point
Anil Busimi

Integrated CDMO partner for parenteral drugs Let’s find your best solution We provide sterile injectable contract development and manufacturing (CDMO) services from initial combination device design and formulation development to commercial manufacturing, supporting better patient outcomes around the world. Published by Terumo Europe 2023PM-06511 www.terumopharmaceuticalsolutions.com
5 Steps to Improving Pharmacovigilance System Master File (PSMF) Management
Christian Schmitz-Moormann, a senior consultant and pharmacovigilance (PV) expert, and partner of Generis, considers the role of this essential master resource in maximising opportunities to improve R&D processes and wider product lifecycle management.
Large pharmaceutical companies are often restricted by a lack of ability to exchange information and access data from other departments. Prompted by regulatory changes, they have largely transformed marketing authorisation (eCTD) submissions and pharmacovigilance (PV) reporting, so now the attention is turning to the management of the Pharmacovigilance System Master File (PSMF). That’s because of its bearing on so many other aspects of research and development, wider product lifecycle management, and associated information management and reporting.
The PSMF is a core document describing the pharmacovigilance system used by the marketing authorisation holder (MAH) with respect to one or more authorised medicinal products. Bringing its management and data exchange capabilities up to scratch digitally makes sense now, given that its contents are so comprehensive and wide-reaching.
A Vital Master Resource
The PSMF’s numerous sections and appendix require information and input from areas across the Quality, Safety, and Regulatory domains. Despite this, many organisations still use manual processes and tools such as Microsoft Word or Excel to maintain and track the master file. As well as exposing companies to non-compliance, inefficiency and delays, a manually-maintained PSMF limits visibility and transparency: characteristics which, if addressed, could be invaluable right across the business.
Part of the challenge around PSMF management is that so many different people are involved in providing contributory information. The file needs to be handy at all times, too – in a single, definitive location, and suppliable to an authority in the defined structure (also printable, or in an acceptable
electronic format) within one week of a request being made, in the EU at least.
Although companies do have the option to lean on an external service provider to maintain and update the PSMF, the price tag for this can be hefty – running into the low six-digit range in the case of a very large pharma organisation with a great number of products, for each fresh version that is needed (not including the annual maintenance cost).
The alternative approach is to ‘divide and conquer’, allowing approved, live information to be pulled from individual, relevant departmental systems, and extracted and put together to form a comprehensive and accurate PSMF. This can be achieved with by integrating a PSMF management capability with existing functional systems across the pharma organisation, or by implementing a single open platform where all of the master information resides, already supporting any necessary integration and formatting standards.
Transforming Manufacturing Change Control
The impact of more fluid data interchange between systems, drawing on a common information core, would be widely felt.
Take the example of manufacturing and packaging, and the need to ensure that patient information leaflets reflect the current Summary of Product Characteristic (SmPC) – which outlines important information about medicines such as form, clinical parameters and pharmacological properties – plus the correct latest version of artwork. Ensuring that all of this up-to-date detail is reflected in the PSMF is crucial for all manufacturers. If teams have to trawl through 20 different SAP systems to find the correct batch information for products made across the world, this process could take up to 18 months and still be at risk of error or omission. Improved integration and data exchange between systems, supported by a common platform, can alleviate much of that time and effort.
Processes such as change control and deviation management also become much easier to manage once variations
and deviations can simply be tagged for automatic inclusion in PSMF, as appropriate.
Enabling Seamless Data Sharing
Beyond compliance, the benefits of more dynamic PSMF creation and management, via shared and connected data, are to do with improved collaboration and communication across a national or international business group, keeping all teams singing from the same hymn sheet.
The better the state of the PSMF, the more powerful the lever to improve business processes. Strategically, that could include identification of PV risks or using the PSMF as a complete planning and tracking tool for the company’s approach to PV.
This consistent rigour can help smooth PV compliance in other regions, too – even if the authorities concerned don’t (yet) have a specific requirement for a PSMF. In the US, for instance, although the FDA doesn’t demand a detailed PV master file, the authority can ask for evidence of the robustness of a company’s PV provisions. Having standardised documentation on tap globally can be very reassuring here.
A standardised approach to PSMF management could also support new cooperation between companies to enhance pharmacovigilance and associated learning for the benefit of all.
Lessons from the Leading Edge:
5 Tips to Maximising PSMF Management
With all of this in mind, here are five specific ways a unified platform for Life Sciences processes can ease the management of the PSMF, with knock-on benefits across and beyond the enterprise:
1. Version Certainty
Using manual processes to collaborate and contribute to PSMF makes it difficult to maintain a single source of truth and know which version is current. With real-time collaboration and version control, you can be certain that you are working on the most upto-date version, and any changes made are tracked on the master copy. Version control not only allows you to see what the correct version was at any point in the document
20 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Regulatory & Marketplace





INTERNATIONAL PHARMACEUTICAL INDUSTRY 21 www.international-pharma.com ––is UPC’s • • •
lifecycle, but also revert to previous versions when needed, and provides the ability to quickly pull up the most up-to-date version for audits, inspections, and regulatory submissions.
2. Granularity in Documentation Presentation & Access
The PSMF as a process does not operate in a vacuum, and often the supporting information is needed elsewhere in the company – for periodic safety update reports (PSURs), or a PSMF for another product for example.
With a data-first platform, the content of the PSMF can be separated into compartments, allowing for the flexibility of multiple data owners. Users can extract individual compartments from the PSMF for use as standalone documents, or for use in other items in the product development lifecycle, the compartments can either be presented as a "PSMF document view" or any other view, individually or in groups, giving the flexibility to use the same single data elements in multiple document views.
Simple tagging, searching, and filtering make it easy to retrieve content from the PSMF as a whole without wasting time going through folders file by file.
3. Risk Tracking
Risk management plans and associated documents need to be created and distributed to Affiliates, Distributors and Authorities. Traditionally those tools involve external systems for sharing content and a separate system for registering and reporting on implementation status. This can leave companies exposed to non-compliance if the process is not managed properly.
A platform designed specifically for pharma companies is likely to have security and compliance at its core, allowing for controlled access and granular user permissions. This means that users can only access the information or key parts of the PSMF that they need to do their work.
4. ‘Submit & Go’
A single, centralised system can also help streamline the organisation and submission of the eCTD, during marketing authorisation applications.
Metadata makes it easy to create and pull together all the required components of the eCTD, such as the PSMF summary, with the ability to add cover pages and other finishing touches ready to submit to regulatory
authorities. Storing the information as components means that dossiers and submissions can be easily assembled and re-assembled as per the requirements of different regulatory authorities. Automations can ensure that submissions are not open to human error and reduce the stress of the risk of non-compliance.
5. 360-degree Oversight
Last but not least, a data-driven platform should provide a clear, 360-degree view of an entire organisation’s content and information, and the relationships between them. With a single, structured information ‘lake’ that all linked apps can draw on, features such as ‘where used’ will make traceability and impact analysis simple.
With metadata relationships between different objects, it becomes easy to see where a change to one content item would impact items like the PSMF further down the line, for instance – all at the click of a button. Users should be able to easily access reporting and dashboards so that they can clearly visualise their processes.
By overcoming the technological, operational and financial drawbacks of a network of individual systems, via an open platform, the different functions of the pharma organisation can consolidate processes, data and content and harness their choice of apps for Regulatory, Quality, Safety and Clinical operations.
Meanwhile respective users can create, locate, and re-use information instantly in any process across the organisation drawing on a single, structured master data/ information/content resource, enabling accelerated work with greater accuracy of information.
It’s no coincidence that Life Sciences companies are moving towards PSMF transformation now. Taking what they’ve learned from eCTD and PV system optimisation, many see this as the next logical step in their digital process transformation efforts.
Christian Schmitz-Moormann is a senior consultant and pharmacovigilance (PV) expert with over 30 years of experience in services, training and change consultancy, including an impressive history of senior roles in Safety and Quality at Life Sciences companies such as Merck and Boehringer Ingelheim. He is a strategic partner of Generis, providing specialist expertise around Life Sciences PV case management and other aspects of safety management. Generis is the creator of CARA™, a data and content management platform that helps companies in regulated industries transform their complex business processes. Generis is dedicated to helping organisations in Life Sciences and other safety-critical sectors move forward with modern, innovative solutions to make day-to-day work easier. Its CARA Life Sciences Platform streamlines the management of business processes, data, and content across the entire Life Sciences product development lifecycle.
Email: christian.schmitz-moormann@ generiscorp.com

22 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Regulatory & Marketplace
Christian SchmitzMoormann

INTERNATIONAL PHARMACEUTICAL INDUSTRY 23 www.international-pharma.com www.lifescienceaustria.at Advancing Austrian life science at the heart of Europe www.lifesciencesdirectory.at Find your right partner in Austria Meet us at CPhI, October 24 – 26, 2023 BIO-Europe, November 6 – 8, 2023 Medica, November 13 – 16, 2023
Why Pharma Dealmaking is a Sector that’s Ripe for Growth
The global pharmaceutical sector is booming thanks to continued growth in R&D spend in new technologies and therapeutic areas, putting the industry back on a more normal track following a tumultuous period during the pandemic. According to a report by the IQVIA Institute, R&D expenditure by big pharma companies reached a record $138 billion in 2022, marking a huge 43% increase since 2017.
It’s been encouraging to see pipeline activity innovation regain its pre-pandemic momentum, and we expect this to drive a continued increase in M&A, as cash-rich, mature companies buy access to developing product areas and expertise to drive forward future growth.
According to a report from Goldman Sachs, the global pharmaceutical sector has a war chest of approximately $700 billion to acquire other companies and invest in R&D, a significant amount of potential M&A firepower. This is only growing, as combined total revenue for the largest global pharmaceutical companies reached $737 billion in 2022, representing a 4.7% rise in net sales compared to the $704 billion recorded in 2021, further increasing deployable cash flows.
As ever a major driver remains the need for pharma companies to continue to invest due to the “patent cliff” – the potential sharp decline in revenues upon patent expiry of one or more leading products which then become generics.
A Deeper Insight into Big Pharma
Record levels of drugs in the pipeline, improved speed productivity drivers in clinical trials and the emergence of increasing numbers of smaller biopharma companies are all factors widening big pharma’s interest in M&A. With accumulation of substantial cash reserves during the coronavirus pandemic, coupled with investor worries regarding future growth prospects we believe there will be a further upsurge in activity.
Recent acquisitions in the pharmaceutical and biotech industries have completely
bucked the overall trend in the M&A market which has been relatively lacklustre. Reuters Breakingviews echoed this sentiment in a recent article, stating that “drugmakers are primed for a shopping spree in 2023” with chief executives having a “potential half-trillion-dollar war chest to use on dealmaking”. Therefore, we could be well on track to reach exceptional levels of deals this year, far higher than last year’s total value of biopharma deals reaching $127bn.
If a company possesses promising late-stage R&D assets, it becomes that much more appealing as a potential acquisition target for the cash rich major pharmaceutical companies. If the ongoing product development pipeline activity remains at a high, there’s no doubt we’ll see continued positive sentiment across the sector and M&A, both bolstering share prices and overall capital markets activity.
Earlier this year, Novartis AG, Switzerlandheadquartered pharma company, announced its acquisition of United States-based clinical-stage biopharmaceutical company, Chinook Therapeutics Inc. The deal, valued at $3.45 billion, involved Novartis acquiring Chinook at a price of $40 per share, with an additional contingent payment of $4 per share. This transaction exemplifies the ongoing trend of major pharma companies seeking growth opportunities by acquiring smaller counterparts in the industry.
It’s likely that established early-stage treatments being acquired by big pharma companies will continue to be a recurring trend if we see this ongoing increase in investment levels.
A Greater Look at the R&D Pipeline
It's promising to see the marked increase in the number of companies contributing to the growing pharma R&D pipeline – over 2,800 companies were reported at the last count, a 49% growth in numbers reported since 2017, according to the IQVIA Institute. This growth lies in part down to a focus on multiple therapeutic areas ranging from oncology development (having seen 5% growth over the past year), rare diseases, as well as areas such as neurology research, centred on Alzheimer’s and Parkinson’s, alongside
the growing significance of depression and mental health conditions.
One of the biggest trends we are seeing in R&D is the greater presence and sizeable investment of China. Products from companies with headquarters in China now make up 15% of the R&D pipeline, up from 2% in 2007 and 6% five years ago. Indeed, this active pipeline from China has more than tripled in the last five years, showing clear scope for growth thanks to the significant investment made into the country’s life sciences sector. However, there are clear geopolitical risks associated with China. As reported in the Financial Times, Astra Zeneca may opt to carve out its China business with a separate listing as tensions heighten between the US and China and businesses look to insulate themselves from the region’s geopolitics.
Industry experts are also projecting a key area of growth in the next-generation biotherapeutic pipeline, which are focused on gene editing, CAR T-cell and other cell therapies. With a 20% CAGR since 2017, the next-generation biotherapeutic pipeline has expanded dramatically in recent years, according to research from the IQVIA Institute. Given that 40% of these are being researched for a variety of cancers, primarily non-rare solid tumour malignancies, cell treatments make up the greatest portion of the next-generation biotherapeutic pipeline, with nearly all of them being developed for cancer.
Small, Emerging Biopharma Companies are Having Their Moment
Smaller and emerging biopharma companies are making a massive dent in the R&D landscape. Significantly, these smaller companies were responsible for only onethird of innovation in 2002, which has since converted to over two-thirds of the R&D pipeline. Not only that, IQVIA research notes that in 2022, emerging biopharma companies were responsible for 67% of all new drugs, and successfully launched 69% of them, highlighting their increasing self-reliance in bringing innovative products to market.
Interestingly, this trend speaks to the broader growth from China. Indeed, China-
24 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Regulatory & Marketplace
based companies now make up 20% of the global emerging biopharma pipeline, a significant increase from 9% in 2017, surpassing China's share of the overall pipeline. Notably, China experienced the strongest growth in the emerging biopharma pipeline compared to other regions, with a 19% increase in the past year, although there is significant pricing pressure in China. And while the United States remains the largest contributor to the emerging biopharma pipeline, its share has slightly declined in recent years, from a peak of 50% in 2016 to 46% in 2022.
The Acceleration of Clinical Trials Processes
As well as the growing spend and number of companies developing novel products, increased speed productivity drivers in clinical trials, which are essential for driving innovation and treatment approvals, are also on the rise.

Trial success rates and trial durations have remained essentially flat over the last 5 years – due largely to the pandemic but also now the difficulties trials face in recruiting patients to take part in rare disease and longer trials. However, last year, we saw an increase in clinical development productivity for the first time in 10 years, driven by AI, virtual clinical trials and the flexibility of regulators on real world data.
Large pharma has typically not had much expertise in AI and machine learning. It’s therefore a gap they are determined to plug. It has exploded in recent times in terms of its application in drug discovery, clinical trials, healthcare analytics, diagnostics and personalised care. These new AI tools are having a significant impact on areas such as speeding up clinical trials, as well as influencing spend on R&D. Many companies are looking aggressively at ways to integrate AI technology into their businesses and M&A is likely to follow this path.
Additionally, there has been a consistent increase in the number of remote, virtual, or decentralised (RVD) trials aligned with industry trial starts. Although RVD trials involve a greater number of subjects, sites, countries, and endpoints compared to traditional trials, they have shorter durations, indicating their potential for accelerated progress. There is also an increased focus on RVD trials whereby data is collected in the context of routine delivery as opposed to clinical trials and where trial controls are not representative of real-world outcomes.
Continued improvements in these areas, meaning faster results from clinical trials which are essential for driving new product innovation, will lead to more and quicker new product releases across the pharmaceutical industry. This in turn will drive the tide of M&A within the pharma industry.
Yet, Big Pharma Acquisitions Can Pose a Risk
The Federal Trade Commission (FTC) created a stir recently by filing a lawsuit to halt Amgen's $28 billion acquisition of Horizon Therapeutics. This was the FTC’s first attempt in over a decade to block a merger in the pharmaceutical industry, driven by concerns over rising prices for patients resulting from excessive consolidation within the sector. The FTC employed a unique argument, suggesting that the transaction would grant Amgen the ability to use rebates on its established blockbuster drugs as leverage to pressure insurance companies and pharmacy benefit managers into favouring Horizon's two dominant products. This increasing scrutiny from US antitrust regulators poses a threat to the recovery of big pharma deal-making.
In addition, the US gene-sequencing firm Illumina was fined €423 million for acquiring Grail, the cancer test developer, without its approval. The maximum penalty
was intended as a signal to deter others from side-stepping European merger rules.
The tougher stance on pharma/biotech deals risks stifling innovation, as smaller entities engaged in high-risk research, typically acquired by larger companies to complete trials and commercialise drugs, may be deterred. It’s also likely to prolong the deal-making process and potentially alter companies’ risk appetite when considering M&A activities.

Conclusion
Large pharmaceutical companies remain compelled to persist in their pursuit of M&A despite the introduction of new regulations. This drive stems from the necessity to address the gaps in their drug pipelines and ensure a continuous flow of innovation that will be essential to the health of the sector overall.
Clearly, the need to address the drug pipeline is propelling M&A activity, driving increased investment in R&D healthcare and the introduction of innovation medications. As record-breaking budgets are being allocated to R&D, there is huge scope for expansion of next-generation treatments and medications, that puts both smaller biopharma and big pharma on the same table. So, despite the increased regulatory scrutiny challenges to deal-making, it’s an exciting time of innovation for the sector, and we look forward to seeing what’s next.
Michael joined Cavendish in January 2006 and has advised on many successful sale mandates across a range of sectors with a particular focus on business services and healthcare. Michael’s transaction experience includes arranging sales to some of the largest global corporates such as the sale of Rapidscan Pharma Solutions to GE Healthcare, Polar Speed Thermologistics to UPS and the sale of Finsbury Orthopaedics to Johnson and Johnson, as well to private equity purchasers such as the sale of Zenith Hygiene to Bain Capital backed Johnson Diversey.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 25 www.international-pharma.com Regulatory & Marketplace
Michael Jewell
Drug Discovery, Development & Delivery
Antibody Drug Conjugates and the Link to Fighting Cancer
Antibody drug conjugates (ADCs) are a big deal in oncology. And rightly so: they combine highly specific targeting with a potent cytotoxic payload to deliver tremendous results. Since the approval of the first ADC in 2000, Mylotarg® for adults with acute myeloid leukemia (AML), ADCs have continued to make waves – something we can honestly say has revolutionised the way we approach oncology. Right now there are 15 approved ADC drugs for cancer indications, with reported hundreds more ADC candidates in pipelines or clinical trials,1 testament to the extensive interest and promise of this therapeutic approach.
But we have to ask, why have they been so successful where small molecules haven’t? What drives their success and potentially limits their efficacy? And what challenges still have to be overcome?
A Quick ADC Track Record
Despite their current success, ADCs’ adoption was a little slow to start. Interestingly, the complexity of ADC design, the combination of a cytotoxic drug, an antibody, a linker, and of course the conjugation technology, was both the core of its success and the main reason it took so long for the field to initially take off. ADC developers had to wrestle with early issues such as the lack of stable conjugation methods, limited therapeutic profiles, and poorly designed linkers. However, advances in our understanding of selectivity, site-specific conjugation, and improved stability have led to this area of R&D thriving like it is.
Over the past two years, the ADC industry has witnessed significant milestones. A few of the most notable achievements include the approval of ADCs by the US Food and Drugs Administration (FDA), such as Elahere® (Mirvetuximab soravtansine) for ovarian cancer and Tivdak® (Tisotumab vedotin) for cervical cancer. We’ve also seen investment from big pharma into smaller biotech ADC companies that has brought not only a hefty dose of confidence but also a host of promising prospects to the field.
If we look at the R&D side of things, we’ve seen marked improvements in linker and conjugation chemistries that translate into clinical successes – you can see this in action when you take a look at Enhertu® (Trastuzumab deruxtecan), which is the first HER2-directed therapy for patients with HER2-low metastatic breast cancer to be approved in the EU.2 Enhertu® was developed with a novel linker–payload combination (a tetrapeptide linker and an exatecan derivative as a payload), which makes it so effective in the treatment of many HER2-positive cancers.3
ADCs have a great track record, and with next-generation ADCs coming onto the scene, they look to be going from strength to strength. But what makes them so successful?
Of mAbs and Linkers
To get a better insight into why ADCs have been so successful we need to look at what they are and how they work. Central to this is their targeted approach to deliver potent cytotoxic drugs directly to cancer cells, and this is thanks to the inclusion of monoclonal antibodies (mAbs), which even on their own have transformed the way we approach cancer therapy.
But of course, while mAbs provide more targeted treatment compared to traditional chemotherapy, their efficacy when used in killing cancer cells alone is limited. This limitation birthed the concept of ADCs: “What if we link mAbs to cytotoxic drugs?”
And so an ADC combines the accuracy of a tumor-targeting mAb with a cytotoxic payload through an intricately designed chemical linker. While they may not be considered “magic bullets,” (although some have called them “biological missiles”4) ADCs deliver highly specific and targeted chemotherapy while also aiming to minimise – though sadly not always eliminating – offtarget effects.
Though it’s easy to get caught up in mAb specificity and payload potency, it’s this link between mAb and payload that plays a crucial role in how well the ADC functions. This is because that linker influences the ADC stability, payload release profiles, and,
ultimately, the therapeutic index (TI; the ratio between the toxic dose and the dose at which the drug becomes effective).
Ideally, a linker needs to avoid causing ADC aggregation and ensure a controlled release of the payload at the targeted sites while simultaneously minimising premature release in plasma. You can generally divide linkers into cleavable and non-cleavable. Cleavable linkers respond to external cues or enzymes, which enables the targeted release of the cytotoxic payload, whereas noncleavable linkers provide plasma stability and controlled release through proteasemediated degradation. Both linker types offer unique advantages for optimising ADCs – but we’ll come back to these in a bit. Let’s clear something up first about ADCs and effective dosing.
They Work, But Not Like That
Contrary to one particular and widely quoted notion, the clinical data from human studies just don’t support the idea that ADCs increase the therapeutic window by lowering the minimum effective dose (MED) or increasing the maximum tolerated dose (MTD) compared to small molecules. 5 However, when administered at the MTD, ADCs do seem to show improved efficacy over small molecules in oncology trials.
While there’s a distinct lack of direct trials comparing small molecules with ADCs, we can take a look at data from ADC trials and contrast them with trials involving analogous small molecules. Let’s look at the 2021 DESTINY gastric cancer trial where the ADC Enhertu® showed an objective response rate (ORR) of 42%, while the ORR for the small molecule Irinotecan was 12.5%.5 ADC treatments frequently achieve ORRs above 40% in breast, gastric, urothelial, and lung cancer trials, whereas small molecules rarely exceed 25%. There’s a degree of interpretation here, but these results would suggest that ADCs could offer greater beneficial outcomes for patients.
A recent discussion from Gerber et al. put forward a revised model where anti-tumour activities of ADCs, and by extension their TIs, aren’t solely associated with changes in just their MTDs, but also in their MEDs.6 This ties
26 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
UniSafe® Platform

Delivering the future with safety and simplicity
Developed around UniSafe®, our platform meets the needs of you, our partner, and your varying patients’ needs both now and in the future.
A spring-free, passive safety device for 1mL pre-filled syringes, designed for simple assembly and use.
A spring-free, passive safety device for 2.25mL pre-filled syringes, designed for simple assembly and use.


Proven, on market, in patient use Coming soon Coming soon
Fully industrialised and ready to supply
A reusable companion auto-injector for UniSafe® 1mL.
A reusable connected companion auto-injector for UniSafe® 1mL.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 27 www.international-pharma.com
UniSafe® is a registered trademark of Owen Mumford Ltd. © 2023 OMPS/ipi/ad/ob/0923/7
Want to know more? Visit ompharmaservices.com/ipi-sept2023 or email pharmaservices@owenmumford.com
in with the way using their exposure-based TI calculation approach lets us more easily explain the improved anti-tumour activities of ADCs and highlight the success of new ADCs in the clinic.
If we want to optimise the TI ADCs, we have to explore dose optimization. Dosing regimens, including body weight capping, treatment duration, dosing frequency, and response-guided dosing are already being used to increase the therapeutic index of approved ADC treatments.7 Comparisons of the MTDs between approved ADCs and small molecule drugs with equivalent cytotoxic payloads indicate that ADCs do not increase the MTD. The reasons behind this are not fully understood, but factors such as payload-linker stability and deconjugation in plasma are believed to play a significant role.
So while ADCs do not improve the MED or significantly differ in MTD compared to small molecules, they do seem to show better efficacy in oncology trials. Dose optimisation and understanding the factors influencing ADC TI will help unlock their full potential in targeted cancer therapy.
The Link to Success
With all that in mind, let’s return to linkers. Currently, most of the ADCs approved for
use or in clinical trials are produced by coupling a maleimide moiety on the linker payload to the antibody. The problem with this bioconjugation method is that it can lead to instability in the ADC, resulting in deconjugation via a retro-Michael reaction.8 Consequently, you can end up with a lot of variability in your drug-to-antibody ratio (DAR).
But like everything in biology, things are rarely simple. So, on one hand, deconjugation before engaging with target cancer cells can have a positive impact on reducing tumour cells by allowing the cytotoxic moiety to interact with cancer cells. On the other hand, this deconjugation can lead to a reduced concentration of the cytotoxic agent reaching the cancer cells, causing the unintended destruction of healthy cells. For example, deconjugation in plasma can result in albumin binding, leading to non-specific payload disposition and direct tumour albumin uptake.
As you can see, the linker is a major player in how successful your ADC is. And we’re not suggesting that maleimide is a poor bioconjugation – not at all – it’s just that there are better options. This is why we’ve seen a noticeable shift toward more site-selective conjugation technologies in pre-clinical and clinical-stage ADCs. These
advancements include ThioBridge™ cysteine re-bridging technology, peptide-directed lysine conjugations such as AJICAP™, and enzyme-directed approaches like microbial transglutaminase and GlycoConnect™ enzyme-mediated glycan remodelling technology.
If we take a deeper look at ThioBridge™, for example, we’ll see how it overcomes some of the shortcomings of firstgeneration conjugation technologies. Unlike some of the first-generation conjugation technologies, ThioBridge™ delivers a more uniform profile compared to maleimide conjugation and enables site-specific conjugation to antibody interchain disulfides without requiring antibody engineering. Additionally, ThioBridge™ allows for stable attachment of the linker to the antibody through a 3-carbon bridge, resulting in optimised pharmacokinetic (PK) properties and architectural design flexibility. This conjugation technology lets scientists rapidly produce and profile ADCs in complex in vivo evaluations. Overall, ThioBridge™ gives people the power and control to generate homogeneous constructs with precise drug loading and predetermined, controlled sites of attachment.
It’s these collective advancements in linker technology and bioconjugation that
28 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
&
Drug Discovery, Development
Delivery
will help deliver ADCs with improved safety, efficacy, and pharmacokinetic properties –which only bolsters their already incredible success in targeted cancer therapy.
Future Challenges and Opportunities
Despite rocketing to success, ADCs have faced challenges in their development – particularly in optimising the aspect of the monoclonal antibody, linker, and payload – but the ingenuity of scientists has so far overcome them.

But we’re not done yet: there are more hurdles to clear and opportunities to seize. Bispecific antibody technology and dual-payload ADCs, for example, show tremendous potential in improving antibody internalisation and tumour specificity and overcoming the ever-present menace of drug resistance. Other groups are exploring alternative structures for ADCs, such as smaller molecular weight polypeptide fragments, which can overcome penetration efficiency issues and improve payload delivery. And then there’s payload selection, something that’s now expanded beyond cytotoxic drugs to include targeted and immune drugs, opening even more new possibilities for ADC therapies.
As long as there’s room for improvement and challenges to address, the outlook for ADCs remains promising. A
better understanding of payload release mechanisms, exploration of unique linker strategies, and targeting strategies will continue to drive enhanced efficacy coupled with fewer off-target effects.
Decades of research and development have led to the success we now see with ADC therapies, and this benefits even more cancer patients. And as we strive forward, pushing the science and generating more and better drugs, the future of ADCs looks set to live up to the lofty claim of revolutionizing cancer therapy.
REFERENCES
1. In Vivo [Internet]. 2023 [cited 2023 Jul 11]. ADCs Coming Of Age: Deals, Targets And Catalysts. Available from: https://invivo. pharmaintelligence.informa.com/IV147692/ ADCs-Coming-Of-Age-Deals-Targets-AndCatalysts
2. Enhertu approved in the EU as the first HER2directed therapy for patients with HER2-low metastatic breast cancer [Internet]. 2023 [cited 2023 Jul 11]. Available from: https:// www.astrazeneca.com/media-centre/pressreleases/2023/enhertu-approved-in-the-euas-the-first-her2-directed-therapy-for-patientswith-her2-low-metastatic-breast-cancer.html
3. Shin SH, Park Y, Park SS, Ju EJ, Park J, Ko EJ, et al. An Elaborate New Linker System Significantly Enhances the Efficacy of an HER2‐Antibody‐Drug Conjugate against Refractory HER2‐Positive Cancers. Adv Sci. 2021 Oct 18;8(23):2102414.
4. Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug
conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022 Mar 22;7(1):1–25.
5. Colombo R, Rich JR. The therapeutic window of antibody drug conjugates: A dogma in need of revision. Cancer Cell. 2022 Nov 14;40(11):1255–63.
6. Gerber HP, Gangwar S, Betts A. Therapeutic index improvement of antibody-drug conjugates. mAbs. 15(1):2230618.
7. Liao MZ, Lu D, Kågedal M, Miles D, Samineni D, Liu SN, et al. Model-Informed Therapeutic Dose Optimization Strategies for AntibodyDrug Conjugates in Oncology: What Can We Learn From US Food and Drug AdministrationApproved Antibody-Drug Conjugates? Clin Pharmacol Ther. 2021 Nov;110(5):1216–30.
8. Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ, et al. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug Chem. 2008 Mar;19(3):759–65.
Campbell Bunce is Chief Scientific Officer (CSO) and Cambridge Site Head at Abzena. He leads a talented team of scientists across a diverse range of expertise and capabilities to support drug discovery, design, and developability; and cell line development. He ensures that Abzena’s strong innovation focus and depth of scientific expertise is maintained through technological developments and works in partnership with clients to design and deliver solutions that support their program needs. Campbell has over 25 years of experience working in the biotech and diagnostics sectors. Before joining Abzena in 2015, he held multiple positions of increasing responsibility in Biotech including Head of Cellular Immunology at Cantab Pharmaceuticals, Director of Programs at Piramed Pharma, and R&D Director at Immune Targeting systems. Throughout his career he has applied innovative solutions for the design, manufacture and clinical evaluation of novel products including vaccines, biologics, and small molecules in multiple therapeutic areas. These include inflammation, cancer, infectious disease, and addiction. Campbell has a PhD in Immunology from the University of Manchester, an Executive MBA from Judge Business School, Cambridge University and has published a number of papers on cell-mediated immunity, immunotherapy and vaccines.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 29 www.international-pharma.com Drug Discovery, Development & Delivery
Campbell Bunce
Promising Advances in Plasma Proteomics for Early Disease Detection and Personalised Treatments
The early detection of disease poses a substantial challenge in the field of human health, necessitating the development of more accurate biomarkers, enhanced patient stratification techniques, and predictive methods for treatment response. Proteins present in the circulatory system reflect an individual's physiological state, and protein levels are typically measured using singleprotein immunoassays. However, recent breakthroughs in plasma proteomics show great potential in improving the comprehension of disease in clinical practice. The use of high-throughput, quantitative analysis through mass spectrometry (MS)-based proteomics of blood, plasma, and serum has been shown to be highly effective in improving the detection and quantification of low-abundance proteins and posttranslational modifications (PTMs).1
Comprehensive analysis of the plasma proteome provides valuable insights into
an individual's health status. Research has demonstrated the efficacy of a rapid and highly reproducible proteomic workflow that allows for the quantitative analysis of hundreds of plasma proteomes obtained from just one µL of blood from a single finger prick. This innovative three hour workflow utilises a single-run shotgun proteomic method that eliminates the need for protein depletion and produces protein data that reflects allele differences, metabolic risk, and inflammatory status.2
Nevertheless, this approach presents analytical challenges due to the wide range of protein abundances found in these samples. Protein expression is influenced not only by an individual's genetic background but also by various factors such as time, location, and physiological responses to external stimuli like stress, disease, aging, and physical activity. Furthermore, a single protein or gene expression product can manifest in multiple proteoforms due to alternative splicing, point mutations, PTMs, and endogenous proteolysis — each with distinct biological activities.3-4
To address this complexity, the implementation of orthogonal approaches in plasma proteomics has demonstrated the ability to facilitate more accurate and comprehensive data analysis. Three recent technological advancements open new possibilities for enhancing early disease detection and personalised treatment strategies with plasma proteomics.
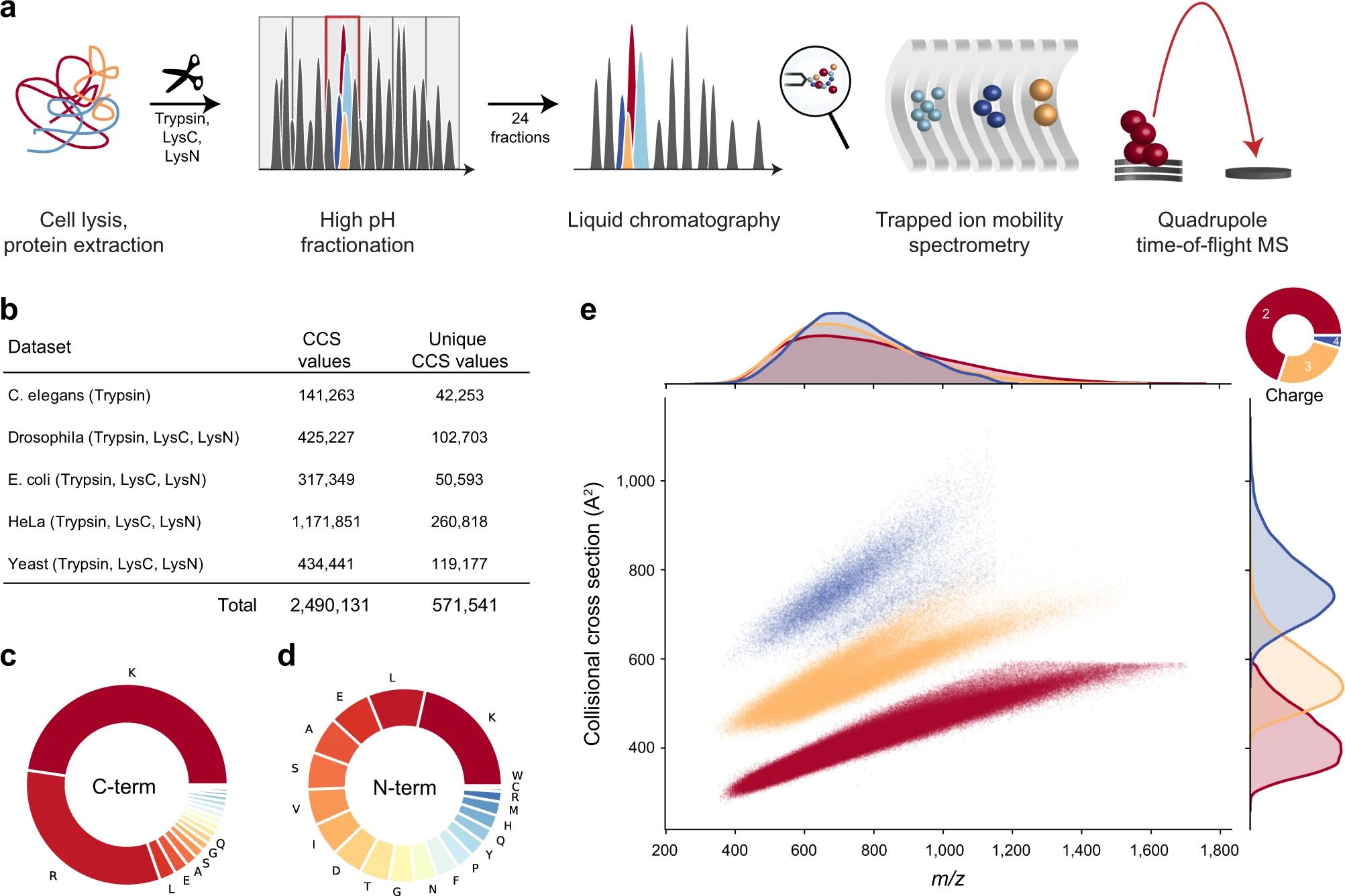
Advanced Proteomics Analysis with prm-PASEF
Parallel accumulation serial fragmentation (PASEF) is an MS-based proteomics method employed for peptide sequencing and identification. A novel advancement called parallel reaction monitoring-PASEF (prmPASEF) integrates ion mobility as an additional dimension to liquid chromatography-MS (LCMS) proteomics. This innovative approach enhances proteome coverage while reducing analysis time. By combining trapped ion mobility spectrometry (TIMS) with a highspeed time-of-flight (TOF) mass spectrometer, the sensitivity for proteomics is significantly improved, enabling the detection of minute protein quantities present in single cells.
30 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Drug Discovery, Development & Delivery
Figure 1: Large-scale peptide CCS measurement with TIMS and PASEF.

































INTERNATIONAL PHARMACEUTICAL INDUSTRY 31 www.international-pharma.com KahleAutomation.com We’ve merged with BBS Automation Our ideas just got a whole lot brighter Announcing a partnership in automation equipment that brings a whole new light to medical and pharmaceutical device manufacturing. Visit www.KahleAutomation.com or contact Kahle@KahleAutomation.com U.S.A. | ITALY | CHINA Kahle® is dedicated to providing custom automation machinery solutions for the Medical Device, Pharmaceutical, and Healthcare Industries around the world.
Using a dual ion mobility trap, prm-PASEF facilitates highly multiplexed targeted acquisition. This technology enables a shorter chromatographic separation while maintaining the number of targeted peptides.5 Furthermore, the design of TIMS allows for the consistent measurement of collisional cross section (CCS) values for all detected ions. The enhanced selectivity of the system enables researchers to obtain more reliable relative quantitation information from complex samples and short gradient analyses (Figure 1).6
Analysis of Proteins with 4D Proteomics
The field of four-dimensional (4D) proteomics aims to enhance protein analysis by integrating the traditional three dimensions of proteomics (protein identity, abundance, and post-translational modifications) with a fourth dimension: the spatial localisation of proteins within their cellular and tissue environments. Through the analysis of proteins in all four dimensions, 4D proteomics strives to offer a more comprehensive and intricate comprehension of protein function and

regulation. This approach holds significant potential in the advancement of personalised medicine by facilitating the identification of disease-specific biomarkers and potential targets for drug development.
Using a TIMS mass spectrometer with PASEF and high-throughput LC separation, research has shown 4D proteomics using a match between runs (MBR) approach can extract intensity information for all peptides to be quantified using narrow mass-tocharge ration (m/z) and retention time
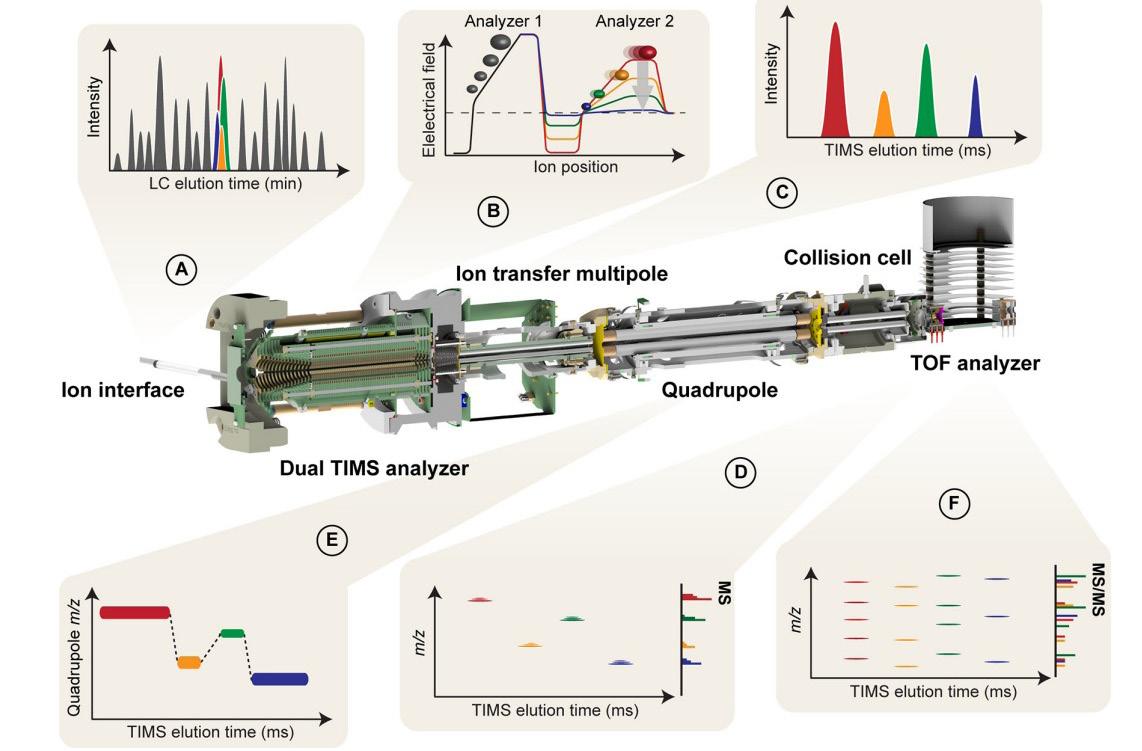
32 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Drug Discovery, Development & Delivery
Figure 3
Figure 2: 4D-MBR applied on high-throughput plasma proteomics
windows for biomarker discovery in large sample cohorts of human blood plasma. 4D-MBR adds an extra CCS filter to the first m/z and retention time analysis, allowing for much greater specificity (Figure 2).7
Data-independent Acquisition Approach with PASEF


Unlike traditional PASEF, which uses a datadependent acquisition (DDA) approach where precursor ions are selected based on their intensity, dia-PASEF uses a dataindependent acquisition (DIA) approach where all precursor ions are fragmented, allowing for the detection and quantification of a larger number of peptides and proteins in a sample. Dia-PASEF combines DIA with the high-resolution ion mobility separation provided by TIMS, enabling the rapid and sensitive identification and quantification of peptides in complex mixtures (Figure 3). Compared to traditional DIA methods, this technique has the potential to offer greater coverage and accuracy in the identification and quantification of peptides.
Dia-PASEF has been used in a variety of proteomics applications, including the identification and quantification of proteins in clinical samples, the analysis of PTMs, and the characterisation of proteinprotein interactions. Research has shown dia-PASEF delivers extremely reproducible identification and quantification information over a concentration of five orders of magnitude making the approach perfectly suited for the challenges of quantitative proteomics.8
Figure 3. Overview of a dia-PASEF workflow. A) Peptides eluting from the chromatographic column are ionized and enter the mass spectrometer through a glass capillary. B) In the dual TIMS analyser, the first TIMS section traps and stores ion packets, and the second resolves them by ion mobility. C) Ion mobility separated ions are released sequentially from the second TIMS analyser as a function of decreasing electrical field strength. D) and E) For dia-PASEF couple DIA isolation windows to the precise ion mobility elution of the corresponding ions. Within a single TIMS separation, multiple precursor windows are set.
Conclusion
Plasma and serum are rich sources of proteins that are commonly used for clinical proteome profiling and biomarker discovery. Comprehensive analysis of the plasma proteome can provide valuable insights into
an individual’s health status. The progress made in unbiased and comprehensive 4D plasma proteomics offers distinct advantages by virtue of its exceptional sensitivity and selectivity. Notably, the combination of TIMS and PASEF facilitates high-throughput analysis of intricate protein samples, resulting in superior sensitivity, dynamic range, and throughput when compared to conventional proteomics methods.
These technological advancements hold immense potential for unveiling novel insights into disease mechanisms and identifying potential therapeutic targets. High-dimensional profiles could indicate current disease risk as well as efficacy of lifestyle changes or pharmacological interventions, thereby contributing to individual and public health. While further research is necessary, the implementation of orthogonal approaches in plasma proteomics shows great potential for widespread utilisation in the field of biomedicine.
REFERENCES
1. Geyer, P. E., Holdt, L. M., Teupser, D., & Mann, M. (2017). Revisiting biomarker discovery by plasma proteomics. Molecular systems biology, 13(9), 942. https://doi.org/10.15252/msb.20156297
2. Geyer, P. E., Kulak, N. A., Pichler, G., Holdt, L. M., Teupser, D., & Mann, M. (2016). Plasma Proteome Profiling to Assess Human Health and Disease. Cell systems, 2(3), 185–195. https://doi. org/10.1016/j.cels.2016.02.015
3. Ignjatovic, V., Geyer, P. E., Palaniappan, K. K., Chaaban, J. E., Omenn, G. S., Baker, M. S., Deutsch, E. W., & Schwenk, J. M. (2019). Mass SpectrometryBased Plasma Proteomics: Considerations from Sample Collection to Achieving Translational Data. Journal of proteome research, 18(12), 4085–4097. https://doi.org/10.1021/acs. jproteome.9b00503
4. Gegner, H. M., Naake, T., Dugourd, A., Müller, T., Czernilofsky, F., Kliewer, G., Jäger, E., Helm, B., Kunze-Rohrbach, N., Klingmüller, U., Hopf, C., Müller-Tidow, C., Dietrich, S., Saez-Rodriguez, J., Huber, W., Hell, R., Poschet, G., & Krijgsveld, J. (2022). Pre-analytical processing of plasma and serum samples for combined proteome and metabolome analysis. Frontiers in molecular biosciences, 9, 961448. https://doi.org/10.3389/ fmolb.2022.961448
5. Brzhozovskiy, Alexander & Kononikhin, Alexey & Bugrova, Anna & Kovalev, Grigoriy & Schmit, Pierre-Olivier & Kruppa, Gary & Nikolaev, Evgeny & Borchers, Christoph. (2022). The Parallel Reaction Monitoring-Parallel Accumulation–Serial Fragmentation (prm-PASEF) Approach for Multiplexed Absolute Quantitation of Proteins in Human Plasma. Analytical Chemistry. https:// doi.org/10.1021/acs.analchem.1c03782
6. Meier, F., Köhler, N.D., Brunner, AD. et al. Deep learning the collisional cross sections of the peptide universe from a million experimental
values. Nat Commun 12, 1185 (2021). https://doi. org/10.1038/s41467-021-21352-8
7. Kosinski, T., Heilig, R., Bensaddek, D., Bache N., Hørning, O.B., Fischer, R., Koch, H. Plasma proteomics goes high throughput – timsTOF Pro with PASEF and 4D feature alignment to quantify 500 plasma proteins in 11.5 min. Bruker Daltonics 03-2019, LCMS-151, 1867805-lcms-151plasmaproteomics-ebook-rev-01.pdf
8. Kaspar-Schoenefeld, S., Marx, K., Gandhi, T., Reiter, L., Meier, F., Brunner, A-D., Frank, M., Ha, A., Lubeck, M., Raether, O., Distler, U., Tenzer, S., Aebersold, R., Collins, B., Rost, H., Mann, M. diaPASEF: label-free quantification of highly complex proteomes. Bruker Daltonics 09-2019, LCMS 160, https://www.bruker.com/ en/applications/academia-life-science/ proteomics/4d-proteomics/_jcr_content/ root/sections/section_1201413713/sectionpar/ twocolumns/contentpar-1/text_1512135182. download-asset.pdf/e45214d3-a5e0-4cb2-9f1e6ecf1029778e/1871458-lcms-160-diapasef-labelfree-quantification-09-2019-ebook-rev-01.pdf
Shourjo Ghose
Shourjo Ghose got his Ph.D. in Biochemistry from Montana State University and went on to do his postdoctoral work at the Scripps Research Institute followed by a business certification from Harvard Business School. Dr. Ghose joined Bruker as an application scientist in 2017 working on proteomics workflows on the timsTOF line of systems. Dr. Ghose is presently responsible for managing the Proteomics business in North America.
Andreas Schmidt
Andreas Schmidt did his undergraduate studies in chemistry in Leipzig, Germany and continued study protein posttranslational modifications with mass spectrometry to earn his PhD in natural sciences from the University of Vienna, Austria. After post-graduate stays at the LMU Munich and University of Cologne (Germany), studying the epigenetic code and composition, development, and differentiation of the muscle, he joined Bruker Daltonics in 2022. Dr. Schmidt has more than 16 years of experience in the field of proteomics.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 33 www.international-pharma.com Drug Discovery, Development & Delivery
High Efficiency Safety Profile for Recurrent Vaginitis and Biofilm Treatment

Sustainable system for the treatment of vaginal diseases, part I – pre-clinical laboratory study A holistic approach, to address the state of the vaginal biofilm using natural substances, enzymes and bacteria to stabilise the system. In the first set of experiments, we showed that yeast (Candida albicans) growth inhibition promoted by unique combination of Lactobacilli species and yeast-specific degradative enzymes. Part I of the research, based on sample received from the clinic taken from women of different ethnic groups who suffered from one or more symptoms as well as, healthy women. We understand that the main problem of the low successes of the recommended treatment is the biofilms. Vaginal swabbing to collect vaginal samples from women was carried out with the approval of the Helsinki Committee at an HMO clinic in Haifa and with informed consent obtained for each subject. Our first step in the research was to create a condition for the development of biofilm that will be similar to the source. We chose candida as a representative pathogen. In this research we developed methods that enables us to prove under laboratory condition the following steps: A) to grow biofilms from vaginal biofilms samples of women from different ethnic groups. B) decomposed any kind of vagina biofilm of all the ethnic groups that we tested. C). eradication of pathogens originating from the biofilm that has been dismantled. D) construction of a new vaginal biofilm that will be sustainable and has a suppressive feature (inhibiting) integration of vaginal pathogens.
There are over 200 million annual cases worldwide of vaginitis.1,2 FemLife is targeting the multi-billion-dollar worldwide women's health market for treating vaginal infections, while finding an innovative solution for recurrent vaginal infections.6,7,8 FemLife's advanced local treatment aims to heal and improve vaginal health by creating healthy flora, with the aim of replacing other
drugs,9 and can be used as prevention for the development of recurrent infections. The average prevalence in the world for vaginal infections in women for vaginal yeast (Candida) is about 27% and for Bacterial Vaginosis over 40%. The total number of recurrent and chronic vulvovaginal diseases in the world is over 50 million women every year. It is well known phenomena that each ethnic group is characterised by a different microbiome, different biofilm naturally developed in the vagina.3,4,5,8,9
Materials and Methods
In vitro Yeast and Lactobacilli Culture and Co-Culture and Viability Effects: Candida albicans strains used in this study are listed in Table 1. Wild-type (WT-1) Candida
albicans was isolated from an infected woman in a Health Clinic,6 in accordance with Helsinki protocols, and as described in12 and confirmed using the Chromogenic medium – CHROMagar (CHROMagar Candida, France). Pure cultures were stored frozen in small aliquots (1ml) in glycerol (50% V/V). Strains were sub-cultured from freezer stocks on YPD plates containing yeast extract, peptone, dextrose and incubated at 37 °C overnight. Cultures were washed in saline (0.85g NaCl/1Li), counted, and diluted in saline for inoculation at a final concentration of 0.4 OD at 540 nm. L. crispatus, [as freeze-dried powder, 3.0x1010 viable cells], L. rhamnoses [as freeze-dried powder 4.0x1010 viable cells] L. acidophilus [as freeze-dried powder 6.0x1010 viable cells] and L. gasserii [as
34 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Drug Discovery, Development & Delivery
Symbol Treatment Yeast CFU/ml Yeast SE (+) Lactobacilli CFU/ml Lactobacilli SE (+0) A Yeast substrate + buffer 0 0 0 0 A Yeast substrate + buffer 0 0 0 0 B Lactobacilli medium + buffer 0 0 0 0 B Lactobacilli medium + buffer 0 0 0 0 C Yeast in a yeast + buffer medium 1.2x105 177 0 0 C Yeast in a yeast + buffer medium 1.1x105 181 0 0 D Lactobacilli in Lactobacilli medium + buffer 0 0 1.7x106 5.6x103 D Lactobacilli in Lactobacilli medium + buffer 0 0 1.5x106 4.8x103 E Yeast + Lactobacilli in substrate * + buffer 1.2x104 3.9x102 1.6x105 1.5x103 E Yeast + Lactobacilli in substrate * + buffer 1.0x104 4.1x102 1.6x105 1.3x103 F Yeast + Lactobacilli in substrate * + buffer + enzyme 7.6x103 1.6x102 1.0x105 1.8x103 F Yeast + Lactobacilli in substrate * + buffer + enzyme 7.4x103 1.7x102 1.1x105 1.6x103 G Yeast + Lactobacilli in substrate * + water + enzyme 7.55x103 1.8x102 1.11x105 1.7x103 G Yeast + Lactobacilli in substrate * + water + enzyme 7.4x103 1.6x102 1.2x105 1.6x103 Table
1 describes the strains and conditions evaluated, T=0.
Drug Discovery, Development & Delivery
freeze-dried powder 1.0x1010 viable cells] were purchased from Wuhan Health-dream Biological Technology Co. Ltd. as also listed in Table 1 (together referred to herein as “lactobacilli”). Freeze-dried lactobacilli powders containing 109 CFU/gr were stored at 4 0C. A mixture of the bacteria were used, in equal ratio/concentrations per strain, and cultured on MRS plates (BD Difco™ Lactobacilli MRS Broth). Growth media was prepared in phosphate buffered solution (pH 5.8) or in water. Co-cultures were prepared with yeast and lactobacilli strains, as indicated in Table 1, where media for each organism (YPD and MRS) were included in a 1:1 ratio. Co-cultures were also further subjected to contact with a degradative enzyme mixture of B-(1-3) D glucanase, (Sigma G67138) at a final concentration containing 0.5 units/ml and Chitinase (Sigma C8241), was included at a final concentration of 0.6 units/ml, both in 0.05M Acetate, at pH 5.0. Data presented is the time zero mean value of 6 samples, assessed twice per condition tested, and Standard error of the mean was calculated (SE) in Table 1 and after a 17-hour incubation period in Table 2, presented hereinbelow. Table 1 describes the strains and conditions evaluated, at time “0” (T=0).
Yeast substrate = YDP liquid media, prepared in water or in buffer. Lactobacillus was grown in MRS liquid media, prepared in water or in buffer. CFUs were determined by plating on agar containing the respective media (1.8 % W/V). As is clear in Table 1, the strain designations (A–G), describe different culture conditions, where samples A and B are negative controls of freshly thawed seed lots which were not grown in culture; Samples C and D represent individual growth in appropriate buffered media conditions
at the indicated CFU values, and Samples E, F and G describe co-culture conditions with the indicated strains in buffered
media alone (E), or in the presence of the degradative enzymes (F) or in the presence of the degradative enzymes in water. Table 2 describes the results of the culture conditions after 17 hours of incubation.
Student’s T test value of <0.05 were achieved in pairwise comparisons as indicated, with respect to the indicated sample. In addition to assessing CFU changes, culture aliquots were taken for microscopic analysis of the yeast for viability effects. Comparing the data in Table 1 versus Table 2 shows that the culture conditions for growth of yeast alone or lactobacilli alone promoted a two-log increase in CFU within the 17hours incubation period. When Yeast and lactobacilli were co-cultured in media alone, while the yeast CFU increased substantially, there was a clear growth inhibition of the lactobacilli, reducing the CFU by 1-log, compared to the T=0 culture
INTERNATIONAL PHARMACEUTICAL INDUSTRY 35 www.international-pharma.com
Infection status in each woman, at sample collection Ethnic origin #Sample Healthy European 1 BV+CAN European 2 BV+CAN European 3 CAN European 4 HEALTHY Ethiopian 5 BV African/Asian 6 BV +GBS European 10 CAN European 11 CAN + GBS European 12 CAN + MYCOPLASME HOMANIS European 13 CAN European 14 CAN European 15 CAN African/Asian 16 CAN European 20 CAN African/Asian 21 CAN African/Asian 22 CAN + BV European 30 CAN European 31 CAN + BV Asian 32 CAN Asian 33 Symbol Treatment Yeast CFU/ml Yeast SE (+) Lactobacilli CFU/ml Lactobacilli SE (+0) A Yeast substrate + buffer 0 0 0 0 B Lactobacilli medium + buffer 0 0 0 0 C Yeast in a yeast + buffer medium 4x107 A 8.4x103 0 0 D Lactobacilli in Lactobacilli medium + buffer 0 0 1.2x108 2.1x104 A E Yeast + Lactobacilli in substrate * + buffer 2.1x106 B 5x103 1.6x105 1.3x103 B F Yeast + Lactobacilli in substrate * + buffer + enzyme 1.0x103 C 1.6x102 1.2x108 3.6x104 A G Yeast + Lactobacilli in substrate * + water + enzyme 2.4x103 C 1.6x102 2.5x108 3.7x104 A
Table 2 describes the results of the culture conditions after 17 hours of incubation.
Table 3 describes the ethnicities of the women from which each sample number was derived, as follows
alone sample. Adding the degradative enzymes promoted lactobacilli growth with CFU levels comparable to lactobacilli alone culture values, while the Yeast CFU show a reduction, as compared to the same time point, when cultured in buffer alone. Comparable results were obtained whether the enzymes and strains were co-cultured in a buffered solution or in water. Samples were collected from the following women with different ethnicities, as indicated in Table 3 described further hereinbelow. Samples were taken from women of each ethnicity, some of whom were healthy, having no history of vaginal diseases in the last two years, some suffering from a vaginal fungal infection with Candida, some suffering from Bacterial Vaginosis and some suffering simultaneously from Candida infection and Bacterial Vaginosis.
Following sample collection swabs were placed in saline, mixed well, and stored under refrigeration at a temperature of 4–7°C, with aliquots in glycerol (10% V/V) also maintained at -20°C. 200 microliter aliquots of each sample were added to different wells in a 96-well ELISA plate, to assess different media/growth conditions and their impact on biofilm formation. Ten different media were tested for the creation of biofilm in the wells of Elisa plates. MRS broth (Sigma) 25 grams/l, pH 7.1 was found to support biofilm of all the samples of the different ethnic groups. Samples were incubated at 37 0C for 17 hours, in a humidified chamber using buffer Potassium – Phosphate 0.1M pH 7.1 in a total volume of 1 ml. Following incubation, wells were inspected for biofilm formation. Briefly, wells were washed and stained with Gentian Violet (1.5% solution in ethanol W/V), which was taken up by the biofilm. Dye is then washed in water, which removed unbound dye. An acetic acid solution (33%) was applied, and dye is then quantified by ELISA reader at 590 nm. Table 4 describes the OD values obtained in treated versus untreated samples 10–16 and provides the Student’s T-test values as obtained, as well. Statistical significance was achieved for each of samples 10–33, as well.
To further extend these studies, where using the 1X enzymatic mixture, robust biofilm degradation was readily seen that was statistically significant, it was of interest to discern if better results could be obtained, using a more concentrated enzymatic solution. Thus, biofilms were prepared from samples 20–23, as indicated, and treated with 1X dose or 2X doses of
test, pairwise comparison, Treated vs Average OD following incubation, following enzymatic treatment
36 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Drug Discovery, Development & Delivery
Average OD following incubation, no enzymatic treatment Sample # 1.43E-20 0.101 0.309 10 1.34E-07 0.023 0.099 11 1.34E-08 0.09 0.136 12 4.13E-12 0.021 0.145 13 5.24E-07 0.06 0.112 14 6.75E-19 0.092 0.211 15 1.62E-07 0.095 0.162 16 8.53E-25 0.118 0.329 30 1.63E-34 0.074 0.303 31 1.58E-34 0.105 0.459 32 1.96E-20 0.107 0.307 33 Sample # OD Following incubation, no enzymatic treatment OD Following incubation, 1x enzymatic treatment OD Following incubation, 2x enzymatic treatment T- test, 1X Treatment vs Untreated T- test, 2X Treatment vs Untreated T- test, 2X Treatment vs 1X Treatment 20 0.941 0.371 0.341 6.08E-24 2.15E-25 0.023431 21 0.258 0.182 0.111 0.003219 1.14E-09 0.004109 22 0.658 0.563 0.318 7.6939E-08 3.97E-08 0.099759 23 0.237 0.084 0.046 1.32E-09 4.75E-15 0.000834 Sample # Original CFU/ml Sample # After dilution 1:100 CFU/ml Samples # Original CFU/ml following enzymatic treatment Diluted CFU/ml following enzymatic treatment Original 10 106x9.88 2.4x105 10 1.4x104 2.3x102 11 106x3.1 105x8.3 11 1.2x104 7.5x102 12 107x1.6 105x8.8 12 1.1x105 8.2x102 13 107x1.7 105x9.7 13 6/3x105 6.7x102 14 106x1.56 104x5.1 14 1.2x104 3.3x102 15 106x3.12 105x5.7 15 1x104 6.2x102 16 106x7.55 105x5.8 16 5x104 4.1x102
T-
Table
4: OD at 590 nm results and statistical significance for enzymatic degradation of biofilms formed from samples 10-33.
Table 5: Biofilm degradation – Comparative results between 1X dose or 2X doses of enzymatic mixture (two times concentration of the enzymes) to the untreated control.
Table
6: Yeast CFUs following second enzymatic treatment: original population and after dilution of the original population 1:100
The results shown in Table 5 demonstrate that in these samples, as well, the enzymatic treatment produced a statistically significant destruction of the biofilms formed, in 1X and 2X concentration. Comparing the more concentrated mixture showed an improved effect over the original enzymatic mixture concentration, which achieved statistical significance in three out of the four samples assessed here.
When the candida population was high, around 107/CFU ml, 10 percent of the population survived the treatment. But if the population before the treatment was around 105CFU/ml, only 0.1 percent of the population survived the treatment reduction of 3 logs units after one treatment.
These results were further extended to show the effects of lactobacilli co-culture in the above system. Biofilms were subjected to enzymatic treatment to liberate the yeast component, then further cultivated and exposed to the second yeast-specific degradative enzyme treatment. Parallel samples were then cultivated with and without lactobacilli addition to the culture media containing the degradative enzymes. Table 7 presents the results obtained.
As can be seen from the table, enzymatic treatment reduced yeast CFU by almost 3 logs, whereas having lactobacilli present resulted in a surprising almost 6-log reduction in CFUs in native and diluted samples.
Yeast growth inhibition promoted by unique combination of Lactobacilli species and
yeast-specific degradative enzymes in newly developed culture methods.
Materials and Methods
Mimicking Human Vaginal Organ Culture in an Infection Model:
Human vaginal flora samples were isolated and prepared as described in Example 2. In order to re-capitulate formation of biofilms in vivo, a culture method was developed. Toward this end, dialysis bags (Sigma- Dialysis tubing cellulose membrane, Typical molecular weight
cut-off = 14,000. Supplied in rolls, dry average flat width 33 mm (1.3 in.) were purchased, filled with media and flora from samples 6, 20 and 30 were independently inoculated into multiple dialysis bags.
Biofilm formation was allowed to proceed by cultivating the dialysis bags under low oxygen conditions at 37°C. Recovery of the biofilm was via saline wash of sonicated bags and CFUs assessed.
Biofilms of parallel sample inoculated dialysis bags were subjected to the biofilm degradative enzyme mixture containing Lipase, Deoxyribonuclease I, and B-(1-3) glucanase (hereinafter referred to as “treatment 1”), as described in Example 2, to assess the breakdown of the biofilm to liberate the biofilm components. CFUs were determined from these samples to demonstrate the efficacy of the enzymatic treatment on biofilm degradation.
To further assess the impact of the second Candida eradicating enzymatic treatment specifically targeting yeast growth the second enzymatic treatment containing B-(1-3) D glucanase, and Chitinase (hereinafter, referred to as “treatment 2”) was applied as listed in Example 2 to
INTERNATIONAL PHARMACEUTICAL INDUSTRY 37 www.international-pharma.com Drug Discovery, Development & Delivery
Yeast
Sample # 5x103 +/- 64 9x106 +/-2.1x103 1x109 +/- 3.6x105 32 22 +/- 1 1.2x102 +/- 7.2 8x105+/- 4.1x102 Diluted 32 Candida CFU/ml recovered Lactobacilli CFU/ml recovered Description of Treatment 0.0 6.3x108 Following simple incubation of sample 1 0.0 1.5x103 Sample 1 post enzymatic treatment I 0.0 8.6x109 Sample 1 post enzymatic treatment II
Yeast CFU/ml obtained, second enzymatic treatment + lactobacilli combination
Yeast
CFU/ml obtained, second
enzymatic
treatment only
CFU/ml obtained in untreated samples
Table 7: Yeast growth following enzymatic treatment +/- lactobacilli co-culture. The results in the table are the mean of 5 replications for each sample.
Table 8: CFU recovery in samples exposed to both enzymatic treatments: enzymatic mixture and compared to untreated samples. The results are provided in Table 5.
Candida CFU/ml recovered Lactobacilli CFU/ml recovered Description of Treatment 2.6x108 3x102 Following simple incubation of sample 30 3x104 1.3x103 Sample 30 post enzymatic treatment I 0 7.9x109 Sample 30 post enzymatic treatment II 1.8x108 3.1x103 Following simple incubation of sample 6 1x109 3.6x103 Duplicate simple incubation of sample 6 1.3x104 5x103 Sample 6 post enzymatic treatment I 1.6x104 4.6x103 Duplicate, Sample 6 post enzymatic treatment I 0 4x109 Sample 6 post enzymatic treatment II 22 5.8x109 Duplicate, Sample 6 post enzymatic treatment II 2x107 2x106 Following simple incubation of sample 20 4.9x104 3.1x104 Duplicate simple incubation of sample 20 1.4x103 3x106 Sample 20 post enzymatic treatment I 10 4.5x105 Duplicate, Sample 20 post enzymatic treatment I 16 7.5x108 Sample 20 post enzymatic treatment II 0 1x109 Duplicate, Sample 20 post enzymatic treatment II Table 9: CFU recovery in additional samples exposed to both enzymatic treatments:
another parallel sample inoculated dialysis bag, with concomitant introduction of the four lactobacilli species, delivered as dry powder, as also described in Example 2 (Acidophilus, Crispatus, Rhamnosus. Gasserii) further supplemented with media, where following incubation, samples were then washed and plated for CFUs, on parallel agar plates containing supportive media for yeast and lactobacilli growth, respectively.
The enzymatic treatments 1 and 2 were applied to the dialysis bags delivered in a Pullulan capsule (LFA USA, Empty Pullulan Capsules – Size #0 Clear). The capsule was loaded in the dialysis bag via syringe, containing a cut end to expand the bore. Pullulan serves as a carbon source for the lactobacillus, which liberates its contents upon degradation.
Results
Following the positive results obtained in table 7, it was of interest to create an in vitro culture system that more closely mimics in situ conditions. Toward that end a system was developed to prepare biofilms isolated from patients using dialysis bags and modified culture conditions, to assess independent growth of Candida and lactobacilli and to assess the effect of the enzymatic treatments on as well.
Table 8 describes the results obtained in assessing Sample 1, sample taken from healthy women. In the table, CFUs of lactobacilli and candida as a function of the treatment applied to the model system.
Table 8 demonstrates that the biofilm of healthy women is typically rich in lactobacilli, without appreciable candida.
Samples 30, 6 and 20 were similarly probed and Table 9 describes results obtained in assessing Sample 30, Sample 6 and Sample 20, CFUs of lactobacilli and candida as a function of the treatment applied to the model system.
It is clear that while at the outset, significant candida was recovered from the biofilms in each sample, and far fewer cultivable lactobacilli species were cultivatable, following treatment to disrupt the biofilm, candida CFUs precipitously dropped, while lactobacilli CFUs rose.
Completing the exposure to the second degradative enzymatic treatment almost abrogated candida presence, while
supporting robust lactobacilli CFUs in this model system.
To further extend these studies, it was of interest to determine if additional modifications to the assay system could further support positive lactobacilli growth.
Toward this end, 3 additional samples were assessed as above, including establishing the biofilms from Samples 4, 5 and 22 in the dialysis bags containing MRS and YDP broth at a 1:1 V/V ratio. Following addition of the first enzymatic biofilm degradative mixture containing Lipase, Deoxyribonuclease I, and B-(1-3) glucanase, the second candida enzymatic degradative mixture containing B-(1-3) D glucanase, and Chitinase mixture was applied, in the presence of the lactobacilli mixture, with the media provided being supplemented with inulin and Pullulan to serve as lactobacilli carbon sources.
Table 10 provides the results of these studies assessing candida and lactobacilli CFUs, respectively, recovered as described hereinabove, with media enriched lactobacilli growth.
Candida growth declined substantially after the first enzymatic treatment and was almost undetectable following the second enzymatic
treatment, as is evident in columns 2 and 4 of the table. A concomitant increase in lactobacilli reconstitution is seen following the second enzymatic treatment with lactobacilli spp.
In table 10, candida growth declined substantially after the first enzymatic treatment and was almost undetectable following the second enzymatic treatment, as is evident in columns 2 and 4 of the table. A concomitant increase in lactobacilli reconstitution is seen following the second enzymatic treatment with lactobacilli spp.
To further extend the above studies, biofilms were prepared and assessed in the in vitro model system probing effects on Sample 3, assayed as 4 replicates for each treatment. Average CFUs for lactobacilli and Candida are provided in Table 11 following each treatment, as follows:
Treatment C as listed in the table refers to samples where a biofilm was prepared, with no further treatment applied to same (i.e. no enzymatic degradation exposure is provided). Treatment I comprised the first enzymatic biofilm degradative mixture containing Lipase, Deoxyribonuclease I, and B 1-3 glucanase, as described hereinabove. Treatment II comprised the second candida enzymatic degradative mixture containing
38 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Drug Discovery, Development & Delivery
test T test T test T test T Candida Lactobacilli Treatment IY-IIY CY-IY IB-IIB CB-IB 3.2x106 3.5x104 C 2.5x102 2.5x102 I 17 5.3x108 II 2.18E-07 6.47E-06 6.71E-06 0.005 Sample & treatment Description Lactobacilli CFU/ml Candida CFU/ml % Of the Control Lactobacilli % Of the Control Candida Following simple incubation of sample 4 3.5x103 1.4x108 100.0 100.0 Sample 4 post enzymatic treatment I 4.8x103 1.5x104 137.1 0.1 Sample 4 post enzymatic treatment II 4.9x109 11 1.4x107 0 Following simple incubation of sample 5 1.2x103 3x109 100.0 100.0 Sample 5 post enzymatic treatment I 1.5x103 6.3x104 125.0 0.021 Sample 5 post enzymatic treatment II 5x108 6x102 4.2x107 0.0 Following simple incubation of sample 22 8x103 5x106 100.0 100.0 Sample 22 post enzymatic treatment I – – – –Sample 22 post enzymatic treatment II 1.6x109 30 1.9x106 0.0
Table 11: Statistical significance for all treatments
Table 10: Influence of additional lactobacilli carbon sources on lactobacilli growth in the model system
B-(1-3) D glucanase, and Chitinase mixture, applied in the presence of the lactobacilli mixture of species, wherein the media provided is supplemented with the carbon sources inulin and Pullulan for use by the lactobacilli.
Statistical significance was assessed using the Student’s T-test, in pairwise comparisons between CB–IB, representing lactobacilli CFUs compared in samples treated per C conditions versus those subjected to Treatment I, IB–IIB, compares lactobacilli CFUs post Treatment II versus Treatment I, and CY–IY relates to pairwise comparisons of candida CFUs compared in samples treated per C conditions versus those subjected to Treatment I, and IY–IIY relates to pairwise comparisons of candida CFUs compared in samples post Treatment II versus Treatment I.
It is noted that each treatment showed statistical significance in terms of the impact of the treatment on both Candida and lactobacilli growth.
Discussion
In this research we achieved our six goals:
I. Grown any kind of vaginal biofilm from natural source of different women of different ethnic groups.
II. We were able to decompose those different biofilms.
III. Eradication of pathogens originating from the biofilm that had been dismantled.
IV. We developed a method based on two capsules for two-step treatments of vaginal infections of any type based on natural substances (enzymes and additives). and focusing on the acute area of recurrent vaginal infections, according to the steps described.


V. Construction of a new vaginal biofilm composed of mixture of lactobacilli spp. applying with two special carbon sources that can be utilized by the lactobacilli and not by the pathogens. The new biofilm, will be sustainable and has a suppressive feature (inhibiting) integration of vaginal pathogens.
VI. This treatment will be easy to use by friendly applicator.
















Scientifically, we developed a new method for growing biofilms from common clinic vagina samples that are sent to the diagnostic laboratories. Quantitative method was developed for estimations biofilms using our stained with Gentian Violet as described earlier followed by using Eliza reader to quantify the intensity of the colour that represent the amount of the biofilm. We found the suitable enzymes to break the biofilms as well as to controlling the candida – a common pathogen in the vagina`s system. We invented a mimic tool made of dialysis tub, that served as a vagina model for the laboratory experiment with the uses of Sonicator that enabled us to estimate microflora that creates biofilm including pathogens.
The most important innovation is the creation of the two capsules for treatment I and treatment II. – the enzymes, the carbon sources and the mixture of the lactobacilli spp.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 39 Drug Discovery, Development & Delivery
Pressure regulators
Control valves
Pipeline ancillaries
Steam traps
SOLUTIONS FOR ALL INDUSTRIES adca@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467, Guia PBL PORTUGAL PRODUCTS MANUFACTURED IN PORTUGAL AV 001 IN EN 01.20
Special equipment STEAMING
As demonstrated herein, an inexpensive treatment regimen using specific combinations of readily available enzymes and lactobacilli strains can be used as an effective therapy to limit growth of yeast and other vaginal flora components to favor lactobacilli growth. Some unique aspects to our invention include the ability to assess BV and/or candida treatment regimens in female subjects, and offer the promise of reconstitution of vaginal flora without need for antibiotic therapy, which in turn helps reduce the known global burden of antibiotic resistance.
We developed a model system to specifically assess vaginal flora from affected patients having the potential to form biofilms, in terms of the sensitivity of the flora to our unique combination therapy.
Our newly developed system provided a means to qualitatively and quantitatively assess growth of pathogenic flora, the impact of our treatment regimen on the pathogenic components and concomitant positive impact on the growth of protective lactobacilli species.
Some unique aspects to our invention include the ability to assess BV and/or candida treatment regimens in female subjects, and offer the promise of reconstitution of vaginal flora without need for antibiotic therapy, which in turn helps reduce the known global burden of antibiotic resistance.
Furthermore, repeat treatments, as needed would not be expected to be reduced in efficacy, since the enzymatic treatment specifically targets components critical for the survival and niche creation of the pathogenic organisms.
Summary
The two treatments effect on vagina pathogens:
Before the treatment: present of biofilm composed of bacteria mixture and fungi including pathogens covered by extracellular matrix: polysaccharides, lipids, DNA and proteins which protect them from the common medicines.
After the first treatment (capsule I): the biofilm matrix is broken down by the enzymes, the microflora that was a part of the biofilm separates to free cells.
After the second treatment (capsule II): the pathogens were attacked by enzymes that broke their cell walls, as a result, their population dramatically decreased. The lactobacilli complex that was inserted with the enzymes and with two carbon sources (that can’t be utilised by the pathogens). Under this enrichment conditions, the lactobacilli spp. Created within a short period time new sustainable suppressive biofilm towards the pathogens.
REFERENCE
1. Bignoumba, M., Moghoa, M. K. H., MuandzeNzambe, J.U., Kassa Kassa, R. F., Ndzime, Y.M., Gafou, A., Pendy. N. M. L., Onanga, R., Kumulungui. B.S., Vaginal Infections’ Etiologies in SouthEastern Gabon – An Overview International Journal of Women’s Health 14, 505-515, (2022).
2. Cornforth, T. The 3 Most Common Vaginal Problems Recognizing Yeast Infections, Trichomoniasis, and Bacterial Vaginosis Very Well Health, https://www.verywellhealth. com/vaginal-infections-and-other-concerns3522673 (2023).
3. Hardy, L. Cerca ,N. Jespers ,V. Vaneechoutte ,M., Crucitti, T. Bacterial biofilms in the vagina Research in Microbiology,168, 865-874 (2017).
4. Karygianni, L., Ren, Z., Koo, H., Thurnheer, T. Biofilm Matrixome: Extracellular Components in Structured Microbial Communities Trends in Microbiology, 28, 668 -681 (2020).
5. Lene, K. V., Torstein G., Roger S., Live L. N. Bacterial Biofilm and its Role in the Pathogenesis of Disease Antibiotics, 59, 1-29. (2020).
6. Montes, K., Ortiz B, Galindo, C.C., Figueroa I., Braham, S., Fontecha, G. Identification of Candida Species from Clinical Samples in a Honduran Tertiary Hospital Pathogens, 8, 237-247 (2019) doi:10.3390/pathogens8040237
7. Morales-brown, P. About candida albicans:
natural yeast and problematic infection. Medical News Today, article 322727. (2023).
8. Reid, G. Therapeutic opportunities in the vaginal microbiome. Microbiol Spectrum, 5, 1-9 (2017).
9. Roachford O.S.E., Alleyne A.T, Nelson K.E. Insights into the vaginal microbiome in a diverse group of women of African, Asian and European ancestries. PeerJ, 10: e14449 (2020), https:// doi.org/10.7717/peerj.14449
10. Schalkwyk, J. & Yudin, M. H. Infectious Disease Committee. Vulvovaginitis screening for and management of trichomoniasis, vulvovaginal candidiasis, and bacterial vaginosis. J Obstet Gynecology Can, 37, 266-274, (2015).
11. Xiaodi, C. , Yune, L.,Tao, C., Rongguo, L. The Female Vaginal Microbiome in Health and Bacterial Vaginosis. Front Cell Infect Microbiol. 11, 1-15 (2021)
12. Yazdanpanah, A., & Khaithir T.M.N. Issues in Identifying Germ Tube Positive Yeasts by Conventional Methods. Journal of Clinical Laboratory Analysis 28: 1–9 (2014)
Scientist in the field of microbiology. Expert in the fields of bacterial diseases, bacteria and fungi interaction and biological control. Has more than 120 publications. Was the President of the Israeli Phyto Pathological Society and was active in 78 Meeting and Congresses around the world. Last possession – The head of the Institute of Plant Protection, ARO, The Volcani Center Israel.

40 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Giora Kritzman
Drug Discovery, Development & Delivery


www.international-pharma.com Neo PYROSTAR ™ Recombinant Endotoxin Detection Reagent www.wakopyrostar.com ~ wkuspyrostarinfo@fujifilm.com FUJIFILM Wako Chemicals U.S.A. Corp. © FUJIFILM Wako Chemicals U.S.A. Corp. - 2023 PYROSTAR ™ Neo is a new endotoxin detection reagent developed by recombinant technology. A new synthetic colorimetric method that can be run using a standard absorbance microplate reader. Convenient and Easy to Use.
Clinical and Medical Research
Keeping eCOAs Off the Critical Path – Key Considerations
The prevalence of decentralised clinical trials, rising demand in preventive healthcare solutions, a market driven by the need for more and better-quality data and the importance of patientcentricity in medical practices have led to a compound annual growth in the use of connected digital devices. The Covid-19 pandemic has been instrumental in proposing innovations to existing sitebased trial designs. Remote patient monitoring and reporting services provided by digital health technologies can expand and diversify the study cohort for clinical trials, benefitting a much larger cross-section of the global population. It can not only improve efficiency of clinical trials but also contribute towards longterm cost savings.
These developments have also led to an increase in the use of electronic Clinical Outcome Assessments (eCOAs). Digital devices like smartphones, tablets, wearables and web-based platforms are increasingly being used to measure the impact of a disease and/or treatment on patients' quality of life and are an important way we can capture the patient’s voice in a clinical trial. These assessments monitor the patient’s health by analysing several parameters such as any symptoms of diseases, adverse events, quality of life or other health-related outcomes. The eCOA system consists of the devices used for data collection, specialised eCOA software installed in these devices for collecting the data or access to web-based eCOA platforms and a centralised database or server to store, manage and analyse the data securely.
eCOA solutions come with several benefits in a study trial set-up such as:
• Interface: The digital devices offer interactive, user-friendly and engaging interfaces as opposed to paperbased questionnaires, leading to increased patient engagement and data completion rates. eCOAs support delivery of automatic notifications, reminders and alerts that can improve patient compliance.
• Analytics: eCOAs are ideal for realtime data capture, which can help in monitoring and reviewing data in realtime and on a continuous basis. This allows researchers and healthcare professionals to access real-time patient-reported data for analysis and decision-making. They also support collection of patient-reported outcomes remotely and enforcing a time limit for filling up electronic questionnaires that helps in avoiding the “parking lot effect,” allowing timely collection of patient data. Electronic assessment methods reduce the possibility of data entry and transcription errors ensuring accuracy and reliability of collected data.
• Profitability: eCOAs reduce administrative burden, data entry and paperrelated costs. They eliminate the need for data transcription from paper to electronic and streamline data collection, management and analysis processes, improving trial efficiency and reducing overall study timelines.
Ideally, critical path analysis is used in clinical trial study set-up to forecast and manage critical milestones. Mitigating potential risks and delays and understanding dependencies between project milestones and activities is an important part of critical path management. Perforce, sponsors want to ensure electronic data is collected and managed in a way that there are no delays or risks that can affect the strict timelines followed in clinical trials. Against this backdrop, it’s important to understand that eCOA implementation is a complex process that comes with its own set of challenges such as:
• Technical compatibility: Since eCOAs require specific devices, software or operating systems for data collection, the software’s compatibility across devices and platforms could pose as a challenge, especially if study spans multiple locations or countries. Additionally, there could be system related issues with software that powers the eCOA instrument.
• Participant acceptance: Introducing new technologies to a study cohort could be challenging, particularly older adults
or people with limited technological literacy. This can lead to recruitment issues in a study startup. Ensuring eCOA services are easy-to-use, intuitive and accessible to diverse populations is important to increase participation in trials.
• Training and support: Lack of availability of training and support for study participants, site staff and investigators can also impact a trial. Providing comprehensive training materials, user guides and responsive helpdesks can mitigate challenges related to user adoption.
• Infrastructural assistance: Since eCOAs rely on fast and stable internet connectivity for data collection, areas with limited or unstable connectivity can cause infrastructural hindrances. It is important to have alternate data collection methods in place to avoid such situations.
• Integration with existing systems: Integrating eCOAs with existing systems such as Electronic Data Capture (EDC) systems or clinical trial management systems can also result in a delay in implementation. Seamless integration of eCOAs with other supporting platforms is important for efficient data management.
The timely delivery of eCOAs entails seamless co-ordination between several service providers such as licensing service providers and translation service providers or vendors, to keep eCOAs off the critical path of a clinical trial startup. eCOAs are based on electronic versions of paper questionnaires and assessments that are copyright protected. Licensing teams help sponsors liaise with the copyright holders to obtain permission to use and implement the electronic versions of the original COAs. Assessments for global trials require translation to regional languages and sponsors often need help from translation providers for this task. Successful implementation of eCOAs is also dependent on appropriate vendor selection. The vendor is responsible for the software used for collecting and analysing electronic data.
42 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
The vendor will develop a design document that includes the requirements for building the system, generating screenshots in US English, overseeing User Acceptance Testing (UAT), training of sites and shipping of devices.
There are two aspects to a clinical trial study setup: the patient-facing materials and readiness of the system that will use this material for the trial study to be completed within a stipulated period. A thoroughly planned schedule can ensure eCOA timelines are in keeping with study target dates. Since there are so many service providers working together, it is important to have a study development plan in place that takes into consideration the sequence of events involved in this kind of a setup. Therefore, lack of co-ordination between all the functional units that put together an eCOA system can lead to delays and contingencies in adopting eCOAs for trials. A delay in acquiring licensing or unavailability of screenshots for eMigration of translations can lead to workflow delays.

eCOA Implementation Best Practices

A trial plan should follow a comprehensive and realistic understanding of the critical path and mitigate any contingencies that can affect the trial timelines. An optimised eCOA implementation strategy can minimise several risks and challenges associated with clinical trials. For a successful eCOA implementation, multiple service providers need to collaborate efficiently and effectively. The purpose of an
Clinical and Medical Research
implementation plan can be adapted as needed.
Sponsors can find co-ordinating with multiple vendors for eCOA development a daunting project. A best-practice approach to viewing and managing eCOA implementations as a single project can mitigate risks, thereby keeping eCOAs off the critical path.
Brian Lillis
implementation strategy is to ensure there are no coordination failures in the process of setting up a study startup.
eCOA implementation best practices should include:
• Early eCOA assessment: The eCOA assessment should ideally happen at the design phase before the system build and UAT testing of the eCOA software. It can help identify potential risks in licensing, translations and electronic implementation plans by experts in the eCOA field.
• Create eCOA implementation plan: A stable eCOA implementation plan will depend on a stable eCOA strategy. A dedicated project manager should be assigned for end-to end eCOA implementation and creation of a single eCOA plan defining all the deliverables for each stakeholder involved in the implementation design, confirming target dates for these deliverables are in tandem with study startup timelines. A thorough risk assessment and an accompanying mitigation strategy should be developed in parallel with the implementation plan.
• Form an eCOA team: The project manager should ensure all stakeholders involved in the different stages of the plan, from licensing, system design to UAT and electronic implementation of translations, appoint a representative for the eCOA team and conduct regular meetings for better co-ordination and faster eCOA implementation.
• Engage study startup: It is important that the eCOA project manager stays closely connected to the study startup targets. Any information on any changes to the study startup plan can be shared with the eCOA team so that the
Brian Lillis, Director, eCOA Technology at ICON has over 13 years' experience in the development of site and patient facing eClinical Technology solutions. He has a broad range of experience across product development, operations, and solution architecture. In this current role, he oversees the implementation of eCOAs on ICON trials and is currently focused on how to streamline the eCOA implementation process across all stakeholders to ensure it does not become the critical path in clinical trial start up. Brian holds a PhD from University College Cork, Ireland.
Janick Michel
Janick Michel, Director, Client Services at Mapi Research Trust has broad experience in clinical and post-marketing studies spanning over 20 years. She has dedicated the past 15 years to patient centred services and direct-to-patient contact aiming to facilitate patients' journey and experience in clinical research. This includes strategies for patient recruitment, retention and multilingual/multimedia PROs data collection for global Phase II to postmarketing programs across multiple therapeutic areas. In her current role within Mapi Research Trust, she is responsible for the global client services team including successful delivery to clients for COA distribution and licenses, business development and strategic initiatives in collaboration with other Patient Centred Sciences, and Mapi Trust experts.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 43 www.international-pharma.com
Paper Translations (Linguistic Validations) Award FAP EC/IRB Submission (Master) Go Live (FSIV) Go Live* (Other) EC/IRB Submission (Other) Licensing Team eCOA Vendor UAT Team (or Sponsor) Translations SME Licensing System Go Live* (Other Countries) System UAT & Go Live (US English) System Design Screenshots (Master) System Build Electronic Implementation of Translations
Figure 1: eCOA timeline in the context of study startup
Can a Modular Approach to Cleanroom Technology Deliver Flexible Sterile Manufacturing Capacity?
Trista Hager, Vice President of Sales at AES Clean Technology, explores the challenges facing pharmaceutical manufacturers in expanding their cleanroom capacity to meet burgeoning market demand. She discusses how a modular approach to new cleanroom installation can help deliver the flexible sterile manufacturing infrastructure to stay ahead of industry needs.
The sterile pharmaceutical manufacturing segment has been expanding rapidly in recent years and is showing no signs of slowing down. Worth $899 billion in 2021, the market is expected to grow by more than 50% by 2028, reaching $1,358 billion by the end of the forecast period.1
Several drivers are responsible for this strong market growth:
• An increase in vaccine manufacturing needs: The successful development of several effective COVID-19 vaccines and the need to vaccinate an unprecedented number of people worldwide has led to increased demand for limited sterile manufacturing capacity. A significant investment in expanding this cleanroom infrastructure by national governments and pharmaceutical companies enabled the rapid roll-out of these vaccines. Since then, companies have begun exploring the potential of the messenger (m)RNA technology platform used in several COVID-19 vaccines to develop new products to tackle other viruses, such as influenza.
• A rise in the number of biopharmaceuticals in development: The biopharmaceuticals market is projected to surpass $566 billion by 2032, growing at a compound annual growth rate (CAGR) of 8.2% throughout the decade.2 This is a result of the rapidly rising number of new biopharmaceutical projects in the development pipeline, with 8,000 medicines currently in development, 74% of which are potentially first-in-class.3
• Advanced therapy medicinal products (ATMPs) on the rise: Alongside other biopharmaceuticals, ATMPs based on
genes, tissues and cells are making up a growing share of the development pipeline. The market for contract development and manufacturing organisations (CDMOs) specialising in these therapies is expanding rapidly and is expected to reach $18.8 billion by 2030, growing at a CAGR of 18.8% over the next seven years.4
The reason all these developments are driving demand for sterile manufacturing is simple. Due to their sensitivity to degradation in the gastrointestinal tract combined with bioavailability challenges, the majority of these treatments must be administered by injection, rather than any other route of administration. Injection bypasses the body’s principal defenses against infection. As such, these therapies require sterile processing capacity when they are approved for the market to ensure they are safe for patients to use.
To meet the future processing needs of all of these therapies, CDMOs and pharmaceutical manufacturers are under increasing pressure to invest in their cleanroom infrastructure. Novel ways to build cleanrooms, such as a modular approach to design and construction, are becoming increasingly important to deliver this new capacity quickly and efficiently.
The Challenges of Increasing Sterile Manufacturing Capacity
Efforts to meet the pharmaceutical industry’s growing sterile manufacturing needs are complicated by several issues:
• The requirements of the products that need to be processed: Ensuring the cleanroom caters to the target product characteristics is essential to maintain high quality and stability. For example, temperature and humidity control may need to be tightly controlled throughout manufacture and downstream processing for a highly sensitive biologic. Where the production line is being built for a specific large-volume project, this is relatively straightforward. However, as small-volume projects, such as ATMPs, begin to dominate the market, it will be important to take the needs of multiple products into account when designing new lines.
• The need for flexibility: Equipment and production line features designed to simplify product changeovers are a key requirement in this new age of smallervolume runs. Many ATMPs in particular are designed to treat rare or orphan diseases, meaning they have smaller batch sizes and runs. Sterile processing lines need to be flexible to allow them to be easily changed over to other treatments at the end of each campaign. Facilities must be flexible enough to accommodate a change to process equipment or to the room conditions required to maintain ISO classification.
• Limited factory floor space: The square footage available for a cleanroom installation may be limited based on various building constraints, so the design must be optimised to accommodate all required functionality within the given area.
• Integration into existing processes: New cleanroom infrastructure may need to be easily incorporated into lines and processes previously in place. As such, the equipment must be compatible with the machinery already installed on the line. The isolator technology must also be able to seamlessly connect with surrounding technology. In addition, the new cleanroom may need to be easily incorporated into existing operator working preferences to minimise the need to rewrite standard operating procedures (SOPs) or to retrain team members. Designing a cleanroom that meets these needs can empower operators to carry out their day-to-day activities efficiently, with no disruption.
• The need for speed: To meet the industry’s present high demand for sterile capacity, many manufacturers are under pressure to ensure that their new lines are designed, installed and validated as quickly as possible. Alternative approaches are needed to achieve this goal within a short turnaround time.
• Regulatory compliance: In addition to all of the above, cleanroom environments must adhere to good manufacturing practice (GMP) guidelines for sterile products. In the US, the Food & Drug Administration (FDA)
44 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Technology



Your partner for drug delivery device solutions and combination product services
• classifies cleanrooms from ISO8 (the least strict) to ISO5 (the most stringent) to remain compliant with ISO14644. In the EU, the cleanrooms are categorized as Grade A, B, C or D. Manufacturers producing treatments destined for the EU market must also ensure that their cleanrooms comply with the amendments to Annex 1 of the GMP, in force from 2023.5
Drawbacks of Traditional Cleanroom Design
The traditional approach to designing and building sterile capacity is to custom build a new line, planning the layout and features from the ground up to meet the needs of a particular treatment. The line is designed either to fit into an existing space or a new facility is planned to house the equipment. The technology is selected both to integrate with existing SOPs and worker requirements, according to the product characteristics of the project, and each machine is sourced from the manufacturer. Then, the cleanrooms are built on-site using stick-built drywall construction methods.
This approach means that the final cleanroom meets the needs of the existing project and can ensure the line offers flexibility for future product changeovers. However, it comes with some drawbacks:
• Unpredictable costs: A custom build, combined with the sourcing of individual pieces of machinery according to the specific needs of the line can mean that the costs are unpredictable until the entire line has been planned and built. Unexpected delays in the delivery of equipment can also increase costs, as the completion of the entire project will need to be pushed back.
• Long lead times: The traditional approach to cleanroom design and build can take a considerable amount of time, due to sequential design and construction activities. As such, it can be difficult for CDMOs and manufacturers to be responsive to surges in demand for sterile capacity.
“Cleanroom by committee” makes accountability a challenge: In the traditional design approach, no single entity is responsible for the entire design and build process, making it hard to establish accountability or to address issues with the potential to cause delays. In addition, sourcing equipment from many individual
suppliers complicates the management of the entire project. A large number of vendor relationships need to be handled simultaneously, which can take time and human resources.
Pre-integration of Technologies Can Help
These drawbacks can be mitigated by taking a modular approach to cleanroom design and pre-integrating required technologies before installation in the field.
Modular cleanroom infrastructure is preengineered, pre-manufactured in individual modules that are built off-site and designed to be simply installed and locked together on-site for ease and accuracy of cleanroom construction. These modules can come as ready-designed turnkey units for speed and ease of build or can be customised to meet a sterile manufacturer’s specific needs.
They feature integral heating, ventilation and air conditioning (HVAC) systems to control temperature, humidity, cleanliness, pressure cascade and containment. As they are fully integrated into the unit, they can control these parameters more effectively, enhancing energy efficiency. Integrated linear LED lighting and controls also reduce energy consumption.
Pre-populated modular electrical raceways and standardised utility connections within each unit simplify installation. Integrated control and monitoring technologies also allow plug-and-play integration.
Modular units come with pre-engineered architecture, featuring antimicrobial materials and finishes applied in a controlled factory setting, ensuring optimum and consistent sterility once installed and streamlining washdown regimes during use. Walkable ceilings can be easily incorporated into the units, allowing easy operational access to critical systems that serve the cleanroom without interrupting operations within it.
Glass walls and prefabricated curtaining can be designed off-site to maximise visibility within the cleanroom environment post-installation compared with the traditional design approach. This can simplify monitoring for workers, making it easier for them to ensure the line continues to operate effectively.
The Value of Modular Design
Thanks to all of these features, modular cleanrooms accelerate project schedules and lend themselves to fundamental and superior
capital investment benefits that traditional methods can’t accommodate.
The modular cleanroom systems available today are engineered to accommodate virtually any commercially viable floor plan and a wide range of manufacturing needs. They offer many benefits, including:
• Greater flexibility: It is possible to source modular units with special features designed to streamline product changeovers and modifications while in use.
• Cost-effectiveness: Manufacturers only need to work with a single supplier that will have access to equipment that offers not only high performance but also reduced cost, cutting the investment required for the entire project.
• Superior quality: The nature of the modular approach assures enhanced sterile integrity to other build methods, supporting companies to maintain compliance with increasingly strict regulatory requirements.
• Expedited validation timelines: Modular cleanrooms can be designed, installed, validated and put into operation faster than those built using traditional approaches.
• Clear accountability for design: There is a single source of responsibility to design, fabricate, install and commission the cleanroom facility. Accountability is clearer, speeding up response should any unexpected events occur during construction to ensure they don’t delay completion. Advanced modular cleanroom manufacturers also provide support from dedicated experts post-validation and launch to ensure that the technology continues to offer the highest performance throughout its life.
With these advantages, modular cleanrooms can be an ideal solution to help CDMOs and other pharmaceutical manufacturers equip themselves with the high-performance sterile processing capacity they need to meet present and future aseptic demand rapidly and effectively.
Time to Act to Meet Changing Industry Needs
Given the rapid growth of the biologics sector in particular, sterile manufacturing will
46 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Technology
account for an ever greater share of CDMOs’ revenue in the future. Taking into account the number of biopharmaceutical projects in the development pipeline, it is unlikely that there will be a slowdown in demand for specialist capacity.
Companies cannot afford to wait to invest in their cleanroom capacity. Working with experts that specialise in modular approaches to design, CDMOs and biopharmaceutical manufacturers can be confident they will very quickly have the future-proofed cleanroom capacity they need to thrive in the future.

REFERENCES
1. https://www.mckinsey.com/industries/lifesciences/our-insights/how-sterile-pharmamanufacturers-can-grow-capacity-withoutcapital-investment
2. https://www.globenewswire.com/newsrelease/2023/05/12/2667540/0/en/GlobalBiopharmaceuticals-Market-is-Expected-toHave-a-Value-of-USD-566-Billion-in-2032CAGR-of-8-2.html#:~:text=New%20York%2C%20 May%2012%2C%202023,USD%20263%20 Billion%20in%202022.















3. https://phrma.org/en/Scientific-Innovation/ In-The-Pipeline

4. https://www.prnewswire.com/news-releases/ advanced-therapy-medicinal-products-cdmomarket-size-share--trends-analysis-report-byproduct-by-phase-by-indication-by-region-andsegment-forecasts-2023---2030-301757859.html
5. https://health.ec.europa.eu/system/files/ 2022-08/20220825_gmp-an1_en_0.pdf
Trista Hager
With over 20 years of cleanroom experience, Trista Hager, brings her extended company knowledge and sales understanding for continued company success. Being able to help clients build facilities that will support treatments of novel lifesaving therapies and pharmaceuticals is the main reason for her continued dedication to the company and why she believes strongly in AES' core values. Trista has been part of the AES team for over 20 years and has held numerous roles including Project Coordinator, Project Manager and for the last 15 years, Project Development Engineer. She has played a key role within the sales team promoting the AES brand, developing regional presence along with being a part of many important projects, including (6) ISPE Facility of the Year Awarded winners.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 47
Technology HIGH PURITY. HIGH DEMANDS. Pressure regulators Control valves Pipeline ancillaries Special equipment Steam traps Safety valves High purity equipment for clean steam adca@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467, Guia PBL PORTUGAL PRODUCTS MANUFACTURED IN PORTUGAL AV 001 AP EN 01.20
At last! The Advent of Better Software Application UX to Manage Critical Manufacturing Processes in Highly Regulated Industries
For too long, professionals in the manufacturing industry have had to cope with software applications that are designed more for function than usability despite the fact that it is people who are the ones using these solutions in their everyday work lives. From the shop floor and production, to packaging and distribution, the easier and more accessible the solution, the more likely business benefits will accrue. This is particularly relevant for manufacturing businesses in highly regulated industries where mistakes can result in huge fines and brand damage.
At last, the development of engaging user interfaces (UIs) commonplace in consumer applications is coming to manufacturing with the development of a new generation of intuitive UIdesigned software applications, from shop floor through to packaging and labelling. Gurdip Singh, CEO at Kallik, argues that prioritising user experience (UX) and the use of intuitive software applications will be crucial in highly regulated manufacturing industries to ensure compliance with the ever growing rules and regulations, all the way through to the label.
Software companies have severely underestimated the needs of label and artwork professionals for sophisticated ways to use solutions, particularly in regulated industries where mistakes can mean fines and damaging recalls. We have all become accustomed to high quality UX in our everyday lives, with intuitive and engaging UI enabling consumers to perform tasks from booking a car service to high-level transactions such as autonomous online banking. This expectation has now reached the manufacturing industry, where label and artwork professionals need the same level of ease of use and convenience from their industrial software as their personal applications.
The Label has the Power to Make or Break a Business
UX is no small matter. The IEEE reports three
of the main reasons for software failure are directly related to UX. The usability of an industrial software application that manufacturing and labelling and artwork professionals have to use every day to do their jobs, has a deep impact that stretches across entire operations from design and production through to distribution and can even influence overall business performance.
First and foremost, poor UX has a significant impact on productivity. If software applications are too complex and are difficult to navigate, this will significantly slow down operations. The lack of intuitive design can even heighten the risk of errors, an issue that can be detrimental in highly regulated industries. With far greater legal, financial and safety implications than most other industries, even the smallest error could result in costly delays to market or damaging product recalls, which could lead to medical device manufacturers, for example, seeing a 10% drop in share price or food and beverage manufacturers seeing up to 55% of customers leave their brand, even if only temporarily.
With nearly 15% of pharmaceutical recalls between 2017–2019 a result of labelling issues, getting it right the first time and keeping it right in label and artwork management is vital but poor software usability can cause unnecessary roadblocks to get the product to market.
Poor UX Impacts More than just Operations
Not only are there financial ramifications for poor UX, but there are much deeper business issues too. A lack of software usability leads to lowered employee satisfaction where software applications make professionals’ jobs harder or wastes their valuable time on fixing problems. This impact can even go as far as raising levels of staff attrition.
Label and artwork professionals have to manage thousands of labels across multiple product ranges and languages. For example, one leading multinational consumer goods company alone has over 300,000 current pieces of artwork, which require up to 60,000 minor changes per year. Label and artwork professionals already have a time and labour-intensive job with a single artwork
taking up to one hour to change manually. They need a software solution that will relieve cognitive overload to make mental space for higher level operations such as design optimisation and branding.
There are three essentials to high-quality UI and UX in label and artwork management software that allow professionals to flourish at their jobs without the burden of dealing with process changes and incurring possible errors.
1. Designed by manufacturing professionals for manufacturing professionals
Often those who design industrial software are doing so from a desk in an office, completely separate from the actual environment where the application will be used. This can immediately limit the usability of the software. The environments where industrial software is used, whether it is a plant floor, a print room, or a warehouse, can be challenging or even hazardous. So, attention to the UI and cognitive load is paramount. A software UI that takes up a large amount of cognitive load is not practical to use in such high-intensity environments, therefore, in the case of label and artwork, a UI design that allows label and artwork professionals to seamlessly work within different industrial spaces is essential.
The UI for label and artwork management (LAM) solutions should be developed through close collaboration with existing customers who are key industry players. It should then be rigorously tested in real manufacturing scenarios to allow the creation of a label and artwork management solution that is perfectly adapted to the environments that label and artwork professionals work in. This level of understanding of label and artwork management processes can only be achieved through the close collaboration and expertise of relevant industry players.
2. Intuitive use by design
An intuitive design helps industrial professionals do their job not just efficiently, but more accurately, reducing errors and decreasing unexpected costs and delays. Software applications should leverage symbols and UI features common in personal
48 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Manufacturing
applications to enhance the usability of the application. This creates the ultimate software application UX that reduces the need for extensive prior training as its systems for use reflect those of popular personal applications – allowing quick adoption of new software.
The software application interface also enables the mobility for plant operators to move between sites with ease while inspecting operations and allows designers and managers to access the software database on the move, from different work devices. Industry professionals need solutions that are designed with the end-user in mind and with the deep domain knowledge of manufacturing systems and operations
to develop a UI that truly optimises critical manufacturing processes.

3. Streamlines complex functions into one manageable system Industrial software solutions should make the lives of label and artwork professionals easier. That means they should be designed to simplify processes and streamline different functions into one, easy to navigate system. Quality UX design will collect all the tools and functionalities needed to complete tasks in one place for easy access and maximum efficiency. One UI, one view of all the processes involved.
The software design should also allow new integrations and capabilities to be
added with ease over time to continuously improve UX and keep up with changes to the industrial environment. When designing label and artwork management applications, software providers should seamlessly integrate more advanced userfacing features, that can quickly be made available to users. This could include the potential to build more mobile-enabled functionality into the software application in the future, such as graphical packages, 3D rendering technology or augmented reality views of labelling and artwork on the factory floor. Automatic updates allow for label and artwork professionals to receive consistently high standards of UX even as the industry changes.
The Future of Label and Artwork Management Software is UX Optimised UX optimised label and artwork management (LAM) software is the next step to optimising manufacturing processes. It is an investment worth making. For every dollar a company invests in improved UX, that dollar returns $100 in revenue. High-quality UX improves productivity for everyone involved in the labelling and artwork process, from designers and label creators to reviewers and auditors, and operations teams, all the way up to upper-level management.
Ensuring LAM software applications have a good UX benefits the entire value chain. The optimised operations boost speed to market and customer satisfaction, both crucial parts in an ever-changing industrial landscape that demands time critical, accurate, and value-added service and delivery.
Gurdip Singh
Gurdip Singh is a deeply experienced leader and practitioner in life sciences industry transformation. Before joining Kallik as CEO, he held senior life sciences and healthcare industry leadership roles at CSC, then DXC Technology. He enjoys being an executive hands-on leader, driving organizational transformation in heavily-regulated industries, where he has been actively delivering change for the last decade as a visionary, an advisor and practitioner in leadership, growth, transformation and complex program delivery.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 49 www.international-pharma.com
Manufacturing
Tablet Coating: Optimal Uniformity and Flexibility Tablet Coater Saves Resources
Tablets are the most important oral dosage form in the pharmaceutical industry. The advantages of tablets are numerous. First and foremost is the ability to precisely control dosage and the high stability of active ingredients. In addition, tablets can be produced quickly and in large quantities and offer flexible design freedom in terms of size, shape, and color.
Tablets can be effervescent, dissolving, or dispersible, dissolving tablets, lozenges, or enteric-coated and retarding tablets, as well as uncoated and coated tablets.
Tablets are often coated. This involves applying a thin film of one or more polymers and other functional excipients (colorants or humectants) to the tablet, which can perform a variety of functions.
Tablets are coated to modify the release of the active ingredient, such as in entericcoated or extended-release dosage forms, to protect the active ingredient from light or moisture, or to mask a bitter taste in the tablet formulation. In addition, tablets are coated to improve swallowability or to mask with colour (for unique identification or marketing purposes).
Coating of active ingredients is becoming increasingly important. This includes combination products as well as the combination of two incompatible active ingredients in one dosage form. In addition, different release profiles of the same active ingredient can be combined. In this case, the core contains the slow-release component, and the tablet coating contains the fastrelease initial dose. Formulation approaches sometimes consist of up to four different film types. This results in long process times. In order to successfully develop and produce such formulations, coating uniformity is a mandatory requirement.
Coating uniformity is indicated by the Relative Standard Deviation (RSD) and has a very high analytical significance. The smaller the RSD, the more uniform the coating on the tablet. However, the exact
determination of the RSD requires a high analytical effort, since the same individual tablets have to be analysed before and after coating, or components of the coating have to be quantified by content determination methods.
Not to
be confused with RSD uniformity
is the determination of color difference (ΔE) for coloured coatings. High ΔE values describe a noticeable color difference. The dimensionless quantity ΔE is often mistakenly equated with coating uniformity in % RSD, but this is a gross error and can completely distort the validity of a process.
Mixing, Spraying and Drying
The interaction of mixing, spraying, and drying is critical in tablet coating. Mixing, spraying and drying must be performed simultaneously and with the correct settings to achieve optimum coating uniformity.
Mixing
Smooth and gentle movement of the tablet cores under the spray cones is essential. The tablet cores must not be subjected to excessive mechanical stress to avoid damage.
For more than 20 years, L.B. Bohle (Ennigerloh, Germany) has successfully used an enlarged coating drum (length/diameter (L/D) > 1) with welded-in mixing spirals. The mixing spirals ensure continuous and gentle mixing of the tablet bed. Homogeneous mixing is achieved within minutes and is maintained throughout the process. The flat tablet bed reduces the melt pressure in the tablet bed. Due to the continuous guidance of the mixing spirals, the tablets are not strongly accelerated. Tablet breakage and twinning do not occur.
Drum Geometry, Spray arm Design and Reduced Coating Time
The drum geometry of the L.B. Bohle BFC tablet coater creates a large spray area in the moving tablet bed. This allows more spray nozzles to be used compared to shorter drums, resulting in a larger total spray area and higher spray rate. In addition to the coating suspension, the nozzle type, number of nozzles and nozzle to nozzle distance are of particular importance.
L.B. Bohle offers various solutions for adjusting the nozzle bed distance, the spray angle of the nozzles and the pressure parameters for atomisation.
Typically, the amount of suspension mass in film-coated tablets is 5–15% of the core mass. Of particular importance is the film thickness, which is not only important for active ingredient coatings, but must also be uniform for thin color (protective) coatings. Uneven film application within a batch, for example, results in color variations that degrade product quality or poor compliance.
Compared to conventional tablet coaters with L/D ratios <1, systems with enlarged drums allow up to 40% shorter process times due to higher spray rates.
Drying
It is critical to ensure optimal energy and mass transfer. This means that the energy must be applied directly to the tablet bed. The air flows directly and quietly into the tablet bed and effectively ensures rapid drying of the sprayed suspension. There is no heating of the coater periphery or housing.
Optimal airflow creates a smooth spray pattern that reduces spray drying. Spray nozzles are not hit by the supply air stream and remain cool during the spraying process. This minimises spray drying effects and achieves coating uniformity of >97% and better.
World premiere at Interpack 2023 – Coater optimised in terms of process, technology, and machine execution.
L.B. Bohle tablet coaters have been firmly established in the market for more than two decades and are recognised as technologically advanced.
L.B. Bohle has been able to further improvements of the BFC series through an intensive exchange with customers and continuous cross-departmental research and process optimisation. This has resulted in improvements in the areas of technology / machine equipment, cleaning, userfriend-liness and safety, hygienic design,

50 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Application Note
and sustainability. The goal is to optimise the daily production process by making production not only more flexible and faster, but also more resource-efficient with the best possible uniformity.
In the area of machine equipment, the focus is on a new machine control system. The iFix application makes it possible to monitor and control the processes. As an HMI (Human Machine Interface), iFix provides the necessary elements for operation. In addition, important data such as long-term data storage of measured values, alarms and messages, as well as data interfaces to external systems and a recipe management system are recorded.
The importance of coating small batches is constantly increasing. Despite existing laboratory coaters, there is also a need for high flexibility in production machines. The BFC tablet coater offers maximum flexibility, allowing batch sizes with a fill level between 10 and 100%. With a BFC 400, this corresponds to batch sizes of 65–650 l.

Application Note
In the BFC, L.B. Bohle has redesigned the nozzle block. Nozzle angle and distance adjustment is motorised and automatic. In addition, the nozzle block is now slimmer and rounded. This makes it easier to clean, and an enlarged sight glass allows better process monitoring.
Easier to Operate and Safer
The coating process is already highly automated. Nevertheless, operators are present during the process to perform inprocess controls, visual assessments or to take material samples.

In the new tablet coater, L.B. Bohle has implemented the operators' instructions in particular. The opening and closing of the side door is now assisted by pneumatic cylinders, reducing the amount of force required. The air supply shoes are separated, which means a significant reduction in weight and easier handling.
Safety aspects have also been taken into account. Inflatable seals are now standard. Fully automatic sampling and loading of tablets with the door closed increases operator safety.
The control panel has been redesigned in hygienic design and a status light embedded in the front of the machine supports process monitoring.
Fast Discharge and Effective Cleaning
At the end of the coating process, the coater is quickly, gently, and completely discharged by reversing the direction of rotation and adjusting the inclination. This is followed by fully automatic cleaning. An
additional cleaning lance at the bottom of the unit is now integrated as standard. Further improvements reduce the cleaning time and ensure that the cleaning medium drains off quickly and without residue.
Focus on Sustainability
The pharmaceutical industry emits large amounts of greenhouse gases and generally has high energy consumption. L.B. Bohle has addressed the issue of energy consumption in the new design of the coater. For the first time, L.B. Bohle uses optional heat recovery in the ventilation system, which significantly minimises energy consumption.

A new energy monitoring system for the energy supply of the coater gives the manufacturing companies a complete overview of their own process and consumption.
Tobias Borgers is a Marketing Professional, with a huge experience of multi -channel marketing initiatives. Proven abilities in creating successful exhibitions, integrated digital and traditional marketing campaigns, social media marketing, content management, lead generation, event and project management. Tobias holds a B.A. from University of DuisburgEssen and joined L.B. Bohle in 2012.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 51 www.international-pharma.com
Tobias Borgers
How to Make the Grade in Pharma Manufacturing
The current era in pharmaceutical manufacturing is defined by challenging market demands and stricter regulations, particularly with new, more comprehensive GMP standards codified in the EU’s revised Annex 1 and updated CGMP standards from the FDA. Industrial automation pioneer Stäubli Robotics, in a show of foresight, has entered it armed with solutions to bring the efficiency of robotics to more applications while assuring compliance – and delivering the documentation to prove it.
Regulations vary around the globe and are often a source of confusion. But their goals are both straightforward and universal: to avoid contamination, chiefly by minimising human intervention, and thus ensure the safety of products and patients. Separating operators from processes is the overriding theme of Annex 1, and any contamination control strategy (CCS) must make this its focus. From this unifying standpoint, Stäubli Robotics has developed a robust portfolio of pharma robots designed to reliably handle processes in aseptic areas and withstand rigorous cleaning procedures.
This mission started long before the EU or FDA set out to enact stricter requirements for aseptic processing. Stäubli Robotics is a pioneer in the field, having served the pharmaceutical industry for decades. In 2009, the company launched its groundbreaking Stericlean series, the world’s first robot specifically designed to operate inside isolators and RABS. Today these robots are used in pharmaceutical and biotech labs and production facilities worldwide for a range of tasks in material handling, filling and closing, as well as troubleshooting, inspections and packaging.
Joining Forces to Meet New Challenges
In the 15 years since Stericlean was launched, Stäubli has worked with its installed customer base to learn from their experiences and refine the robot’s design. The company has also cultivated partnerships with OEMs and integrators specialising in pharma to expand and adapt its portfolio of robots to handle more applications and meet the industry’s
changing compliance and commercial needs. The pharma community witnessed the next leap forward at CPHI Frankfurt in 2022, when Stäubli launched its next-generation Stericlean+ series, designed exclusively for use in isolators, with the flagship TX2-60.
In many ways, this is a classic story of evolution through adaptation, specifically to GMP grades and ISO classes. It is also a story of partnership. To fine-tune its aseptic robot design and help end users meet the standards of Grades A/B through D/E and ISO Classes 1 through 8, Stäubli partnered with SKAN, the market leader in isolator systems. Stäubli also sought to proactively provide customers with the validation and documentation packages they would need to prove that their equipment is compliant. SKAN proved an ideal partner for this purpose as well. Through its SKANalytix service, the company conducts comprehensive thirdparty testing and analysis of isolator and cleanroom technologies.
Putting Aseptic Robots to the Test Stäubli and SKAN’s collaboration involved putting Stäubli’s then-current offering of Stericlean robots through intensive tests for cleanability, resistance, and movement. The object was to determine what improvements could be made to make the high-performance robots even more fit for use in cleanrooms and
isolators. These tests, including actual cleaning processes, helped define the R&D roadmap for the new design, which also benefited from testing and validation by a neutral third party.

Ultimately this rigorous process would result in the development of new features for the Stericlean+ robot Stäubli was developing. But first, there was painstaking work to be done. Ensuring beyond a doubt that Stäubli’s next generation of Stericlean robots would be optimally suitable for aseptic manufacturing in highly aseptic Grade A (ISO 5) areas was not to be taken lightly. The robots were evaluated inside and out against the highest hygienic standards in terms of design, materials and components.
The robots would need to operate in highly monitored fill-finish lines in the production
52 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Application Note
Stäubli Robot with fluorescent tracer substance (riboflavin) and cleaning. (Source: Stäubli/Skan)
Engineer proceeding cleanroom test. (Source: Stäubli/Skan)
ROBOTICS
Discovering new pharma automation solutions





Robots for Life
Sterile or standard environments, high-end or routine tasks, Stäubli robots deliver clean, consistent performance ensuring the highest levels of product hygiene, safety, flexibility and productivity. Benefit from our know-how and discover the new automation possibilities of intelligent and safe robot technology.




CPHI Barcelona
October 24 – 26, 2023
Hall H3.0, Booth 3T61
Stäubli – Experts in Man and Machine
www.staubli.com

INTERNATIONAL PHARMACEUTICAL INDUSTRY 53 www.international-pharma.com Discover new pharma website
Stäubli Faverges SCA, Tél. +33 (0)4 50 65 62 87, robot.sales@staubli.com
of drugs that are not only highly potent and/ or toxic, but also very costly. The safety of people and products had to be absolute. And crucially, the robot’s entire outer surface would need to withstand regular cleaning and decontamination with VHP (H2O2) in isolators. All areas of the machine had to be easy to clean and free of any gaps, surface roughness, or other areas where substances, particles or microorganisms could adhere and accumulate.
Individually, each test provided information about one aspect of the robot’s design, specifically equipment cleanability, chemical and microbiological resistance, adsorption and desorption (H2O2), D-value, particle emissions, surface roughness, and seal tightness. Together, the tests painted a complete picture of the robots and how well adapted they were for use in isolators.
Applying Test Results to Improvements in Hygienic Design
SKAN’s testing, combined with Stäubli’s own, resulted in major benefits. The new data propelled and informed many aspects of R&D. Since, for example, cleanability depends both on the materials used and ease of access, Stäubli’s engineers focused on simplicity of form. The robot’s joints demanded especially close attention, since moving parts represent the greatest risk of particle generation. The robots would need to interact harmoniously with laminar airflow, generating no turbulence despite their rapid movements during operation to keep particles at a low threshold in accordance with ISO 5 standards.
Crucially, Stäubli obtained a much more detailed understanding of the shortcomings in its existing robots in the context of new standards and requirements for aseptic equipment. In the 15 years since the release of the original Stericlean, much has changed commercially as well. The steep increase in demand for personalised medicine and small batch production runs demands utmost flexibility. The new robots must not only handle an expanded repertoire of tasks, but also switch from one to the next with no errors or significant time loss.
The new Stericlean+ series, designed specifically for use in isolators, pushes the frontier of aseptic process automation and helps future-proof production. The
range includes four six-axis models with an improved hygienic design on the covers and new pharma industry-approved dynamic sealing on each joint, as well as a new FDAcompliant coating on the entire surface, which is VHP/H2O2 compatible for higher cleaning performance and chemical resistance. Stäubli also offers a hollow wrist option with all cables running inside the robot to the flange.

A Validated and Documented Solution
The scientific data based on SKAN’s test results is now part of the validation and documentation package delivered along with every Stäubli robot, to the benefit of machine builders and end users alike. For machine builders, it provides support for the qualification documentation of the whole

54 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Application Note
TX2-40 Stericlean+ is designed exclusively for use in isolators with an improved hygienic design on the covers and new FDA-compliant dynamic sealing on each joint
Unique hygienic robots for pharma aseptic processes
Stericlean+ series of TX2-40, TX2-60 and TX2-90.
machine, helps with global contamination risk assessment, and increases acceptance amongst end users. It gives end users greater transparency as to how the robot was developed and validated, while confirming its mechanical and aseptic capabilities and compatibility with highly aseptic ISO 5 / Grade A environments.

“Providing test results isn’t something we have to do,” says Rudolf M. Weiss, Global Head of Pharma at Stäubli Robotics. “It’s a commitment that we’re making to strengthen trust among our customers and partners.” Customers now have neutral, documented results that Stäubli robots fulfill their essential role in isolators. A comprehensive and well-documented testing package adds value for the pharmaceutical industry and ultimately for patient safety.
Together, Stäubli’s original Stericlean and Stericlean+ robot series have every area of the pharmaceutical production environment covered, from the most restrictive isolators in Grade A to logistics, including RABs, freeze dryers, autoclaves, various levels of inspection, and secondary packaging. They are also used in cell & gene therapy (CGT), biotherapy, API research and production, lab automation, auxiliary processes, and other areas within pharmaceuticals.
Extending the Advantages to Diagnostics

Boosted by the pandemic, as well as the boom in personalised medicine, diagnostic tests have come to play a vital role in global healthcare. In personalised medicine, they help curb high costs by enabling pharmaceutical companies to predict the effects of personalised formulations and refine them for patients. Stäubli robots are increasingly being deployed in the diagnostics segment to support the growing footprint of automation and enable efficient high-throughput screening (HTS).
In laboratory settings, robots cooperate safely with humans and take over repetitive
tasks. Mobile robots are proving especially valuable here. While no mobile solution exists that is compatible with Grade A/B or even C environments in drug production, laboratories are less restrictive, opening the door to the integration of modular Stäubli robots designed for general industry. This includes autonomous mobile robots (AMRs), which help with the handling and storage of blood bags, for instance. They not only increase productivity, but also collect data that can be used to optimise workflows and traceability.
Reagent production is another key area of diagnostics where robotics is having an impact. The processes are similar to those in drug manufacturing, consisting of pick and place, filling and closing tasks. While an aseptic environment is essential in drug manufacturing, reagent production requires a DNA-free environment. The same technical features that make Stäubli robots safe and effective in drug production apply.
Bench-top cells remain the primary domain of robots in diagnostic testing, delivering safer, more reliable results. Combined with a tool changer, Stäubli robots can perform multiple tasks within a small cell, such as handling a PCR plate for a moment and then moving into vial handling. The Stericlean version is often preferred to control contamination. Both stationary and mobile robots perform these and other tasks such as pipetting and
centrifuge loading. Automation is making it possible to meet demand amidst a lack of resources to manage routine tasks while relieving operators of neuromuscular problems resulting from repetitive actions. The safety of both operators and patients is assured.
Groudbreaking Stericlean evolves to Stericlean+
The flexibility of Stäubli’s standard, Stericlean and Stericlean+ robots is also a key factor: Whether in a small isolator or in a complete production line equipped with multiple robots, within highly controlled areas including freeze dryers impacted by the new GMP Annex 1, non-classified rooms, or intermediate levels where cleanability remains essential, they can manage a variety of different RTU containers, making changeovers to different formats and sizes quick and easy, thanks to the Stäubli Connectors division and its variety of tool changers. This is also becoming important as personalized medicine enters the industrialization phase with production scaling up.
CPHI worldwide attendees will have the opportunity to see Stäubli’s robots in action at Stand 3T61, Hall H3.0 in the Pharma Machinery and Equipment Zone.

Rudolf-Michael Weiss
Rudolf-Michael Weiss started in the senior management at the Stäubli Robotics Division of the STAUBLI Group in October 2020. He took over the global responsibility for robotic/automation business in pharma and medical devices as Global Head of Pharma and as a Member of the Global Management Team. After his studies in Stuttgart Rudolf M. Weiss started working at Servotech in 1999. From 2001 on, he was working at Groninger where he was responsible as Marketing Director, Senior Project Manager and Sales Director. After experiences at FIMA, he decided to go back to pharma and joining Bausch+Ströbel the global leading company for fill/finish equipment 2018 he was announced to be Authorized Officer and Director Sales & Marketing.He can draw on 20 years of experience in the field of Pharma Fill / Finish.
Email: rm.weiss@staubli.com
INTERNATIONAL PHARMACEUTICAL INDUSTRY 55 www.international-pharma.com Application
Note
TX2-90 Stericlean+
Application Note
High Potent Manufacture
The global market for drug products containing highly potent active pharmaceutical ingredients (HPAPIs) is currently on a growth fast track. Data indicates a Compound Annual Growth Rate (CAGR) of 8.7% to 2032, with oncology a major driver, capturing a market share of 76% in 2022.1 Additionally, even though biologic dosage forms are experiencing significant growth, the shift from intravenous (IV) dosing towards solid oral dosing is likely to gain momentum; the main driver for this is patient experience, as taking solid oral dosage forms is far less stressful than spending days or weeks in hospital receiving chemotherapy infusions.

Therefore, the role of CDMOs that are able to develop, manufacture and package drug products containing HPAPIs is more vital than ever. There should be a robust approach to all stages of the process, including containment strategies, formulation development, scalable manufacture and potent packaging operations.
Containment Strategies
Throughout the pharmaceutical industry, there is a lack of consistency regarding the assignment of Occupational Exposure Limits (OELs) to specific bands. It is therefore critical to have a unified understanding of all relevant definitions, as described below.
• PDE (Permitted Daily Exposure), also known as ADE (Acceptable Daily Exposure), represents a substancespecific dose that is unlikely to cause an adverse effect if an individual is exposed at or below this dose every day for a lifetime.
• NOEL (No Observed Effect Level) is the highest tested dose at which no “critical” effect is observed. If the critical effect is observed in several animal studies, the associated NOEL should be used to calculate the PDE value.
• OEL (Occupational Exposure Limit) is the average concentration load of an airborne active pharmaceutical
ingredient (API) in μg/m3 that’s acceptable during an 8-hour timeweighted average with no negative impact on workers/environment.
• OEB (Occupational Exposure Band) is the banding of OEL into ranges for which appropriate measures to protect workers and facilities/equipment can be defined in common for APIs with different OELs within this band.
• COSHH (Control of Substances Hazardous to Health) assessment.
Table 1 below illustrates the difference in banding applied by two pharmaceutical companies, demonstrating that there is a significant lack of common terminology, and therefore true understanding, of potent classification.
contamination controls are essential, with a risk assessment performed in accordance with four principle modes:
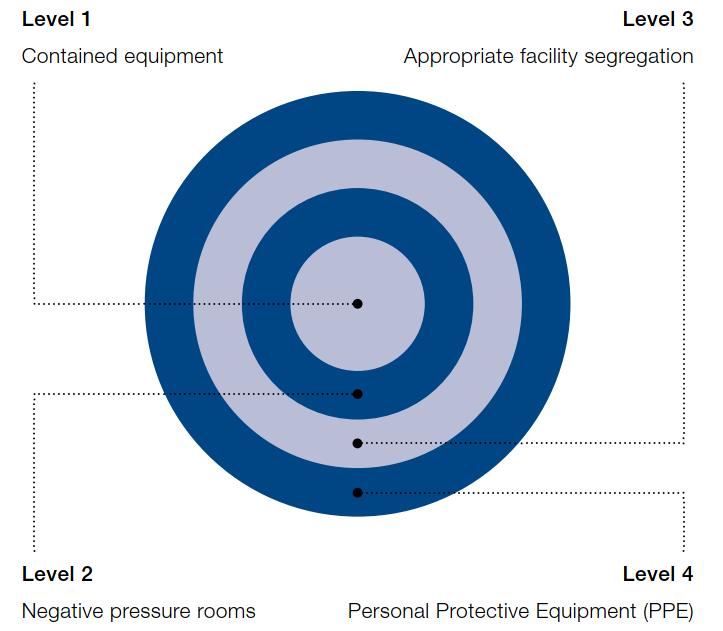
• Airborne;
• Mechanical;
• Personnel transfer;
• Retention of product on contact surfaces.
With traditional, non-potent drug products, avoiding contamination from personnel involved in the production process is, of course, critical. However, when manufacturing drug products containing HPAPIs, processes carried out under negative pressure process rooms isn’t enough. High-grade specialised containment equipment is also required in order to protect employees from the API
When developing and manufacturing highly potent drug products, there should always be an OEL monitoring program at each stage of the process. Strict cross-
itself, and to ensure drug product integrity in a multi-product facility.
Understanding the levels of containment is important when determining the
56 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Band Company A OEL Range Company B OEL Range 1 > 100μg/m3 > 1–10mg/m3 2 10–100μg/m3 > 0.1–1mg/m3 3 or 3A 1– 10μg/m3 > 0.01–0.1mg/m3 3B or 3+ 0.05–1μg/m3 > 0.001–0.01mg/m3 4 < 0.05μg/m3 ≤ 0.001mg/m3 5 N/A ≤ 0.001mg/m3 (Robotics only) Table 1
Figure 1
Your high potent manufacturing destination. Our world.
Rely on us for global, integrated CDMO services.
Building capacity today, based on demand predictions of the future.
Significant investment at our world-class high potent facilities doubles manufacturing and packaging capacity, addressing the growing need for expert integrated oncology outsourcing solutions.

HIGH POTENT SOLUTIONS INCLUDE:
• High potent pharmaceutical development, clinical trial and commercial manufacture
• Containment down to an OEL of 0.01µg/m3
• Xcelodose® microdosing technology and roller compaction
• Tablets, capsules, powders, liquids, suspensions, semi-solids and drug-in-capsule/vial
• Analytical laboratory and QP services
• Clinical and commercial packaging
Your bridge between life-changing therapies and patients talkfuture@pci.com

INTERNATIONAL PHARMACEUTICAL INDUSTRY 57
www.pci.com
appropriate handling of highly potent compounds (Figure 1).
Designing a safe and compliant high potent containment facility is a demanding process. Primary containment requires the use of suitable equipment that runs under negative pressure and effectively serves as a “cleanroom” in its own right, with secondary containment being the facility itself. The appropriate solution depends on the product potency and batch size being handled, as different solutions may need to be considered for laboratory, small- and commercial-scale use. Containment must therefore address the potency of the product with careful consideration being given to dust extraction (heating, ventilation and airconditioning systems [HVAC] and central dust collection) and breach control procedures. HVAC systems should be controlled using a building management system (BMS), which must be suitably designed with terminal high efficiency particulate air (HEPA) filters (H14 grade).
Additional safeguarding measures such as single-pass air ensures a full room evaluation of any potential loss of containment, suitable automatic equipment cleaning and waste disposal systems. In addition to the room pressure differentials, airlocks adjacent to the manufacturing area are necessary for high risk activities such as API dispensing, along with utilities such as purified water and effluent treatment capabilities. Restricted access is also a requirement, ensuring that only essential staff can access the HPAPI processing areas, with cleaning and decontamination areas provided for employees.
It is therefore clear that containment is a technology in its own right, and for CDMOs working with HPAPIs, it is just as important as granulation, compression, or roller compaction technologies. A sound understanding of what containment really mean is therefore essential for CDMOs operating in the high potency development and manufacturing space.
High Potent Development and Manufacture
A noticeable trend in the high potent formulation development space is the use of a Design of Experiment (DoE)/Quality by Design (QbD) approach at earlier stages of the product lifecycle. DoE is a systematic, statistical approach with the aim of optimising the product and the process by understanding the relationship between various factors (input variables) and responses (output
variables). This method helps identify the most influential factors, determine their optimal levels, and establish robust and efficient processes while minimising the number of experimental runs.
During formulation development, multiple factors can influence the quality, safety, and efficacy of the final drug product, such as the choice of excipients, API concentration, processing conditions, and manufacturing equipment. Traditional trialand-error methods can be time-consuming, resource-intensive, and may not identify the best combination of factors to produce a high-quality drug product.
DoE provides a deeper understanding of the interactions between factors and their impact on the final product, enabling the identification of critical process parameters (CPPs) and critical quality attributes (CQAs). By using a structured approach to experiment design, DoE allows for the simultaneous assessment of multiple factors and their interactions, reducing the total number of experiments required, which saves time and resources—an ideal scenario when handling often expensive high potency APIs.
Regulatory agencies, such as the FDA, encourage the use of Quality by Design (QbD) principles in pharmaceutical development, which includes the application of DoE to ensure a science-based and risk-based approach.
By considering the scale-up process during the development stage of the project, formulation development teams can design processes that are more easily scaled, reducing potential challenges
and delays during the transition from development to commercial production. Scalable manufacture within a single CDMO is becoming increasingly desirable. Moving from a smaller more niche CDMO to a larger CDMO later in the drug development journey involves additional technical transfer, introducing complexity and risk to the supply chain and delaying timelines. In-house scalability using geometrically similar fluid bed granulation equipment, for example, ensures the end-product quality is reproducible from small to large-scale.
Cleaning Verification and Validation
When developing and manufacturing drug products containing HPAPIs, having a robust approach to cleaning is essential. Each time a new HPAPI is introduced into a multiproduct facility, the following considerations must be assessed:

• Can the new drug be effectively cleaned?
• Can the residue be detected at low enough levels to confirm cleaning?
• Does the mode of action present specific risks to existing drugs?
• Do existing drugs present a risk to the new one?
Starting with the right equipment is vital to supporting effective cleaning. The most basic consideration is that the machinery has an appropriate surface finish, lacks crevices and can be easily cleaned. However effective a cleaning system is, the most critical thing from a cleaning validation perspective is that all the parameters affecting the cleaning process are consistent. Periodic checks alone are not sufficient; as such, alarms for flow rate or pump pressure and temperature are essential.
Some equipment parts represent more of a cleaning challenge than others. Hard, flat 316 L stainless-steel surfaces, for example, are easy to clean and sample reliably. Materials such as EPDM, however, present difficulties. At the low levels required to clean highly potent molecules, surface interaction becomes more relevant, as does the potential for chemicals being absorbed and leaching gradually with time. In such cases, the preference is to dedicate parts to specific projects. That said, not every part that presents a challenge can be dedicated. For example, multilayer stainlesssteel gauze filters used in certain fluid bed drying technologies are not easy to clean or swab; but, it’s simply not practical to dedicate these because of the amount of time required to fabricate them. One method
58 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Application Note
for such parts is to apply a second cycle in the parts washer, following the initial wash-in-place. Additionally, as swabs can’t identify what’s inside the layers of the gauze filters, rinse sampling may be performed as a further measure.

It’s important to have a range of detergents to provide different cleaning modes of action for different types of molecule. Appropriate detergents are selected by running lab-scale cleaning trials. This is something that’s traditionally done by suppliers of cleaning additives; but, as most suppliers aren’t able to handle potent drugs, this often needs to be performed in-house by the CDMO.
Suppliers of cleaning agents often recommend the degradation of molecules, but this presents its own risks. A highly potent molecule is less likely to be degraded to something more harmful – as would be the case with less potent molecules – but, unless there is sufficient evidence to suggest otherwise, degradation products must be treated as a patient risk and therefore addressed accordingly.
Some materials can limit the choice of appropriate cleaning agents. For example, soft metals will be more prone to corrosion by caustic cleaning solutions, and plastics may deform when washed at high temperatures. Such limitations cause restrictions on the TACT (temperature, agitation, concentration and time) cleaning model. As time is not a major requirement to achieve effective cleaning, this can be addressed during the method development stage, along with factors such as the concentration of the cleaning agent, the chemicals added using detergent pumps and/or the temperature of the cleaning medium. Attrition is more relevant, as increasing pressure or changing spray balls/jets is required to achieve more attrition on equipment. In this instance,
requalification of the system is required to ensure that spray coverage and consistency are maintained.
High Potent Packaging
When packaging drug products containing highly potent APIs, certain considerations have to be made in terms of facility design.
Many clients today are interested in small-volume products, such as orphan drugs or highly targeted therapies for very small patient populations, many of which contain highly potent APIs. Building flexibility into facilities and equipment trains ensures that a CDMO can cope with such requests. For example, blister lines with HAPA digital printers means that with just one reel of lidding foil, artwork can be printed for different market requirements on one line, all within an envelope of very high grade control of airborne particulates. This, in turn, grants clients considerable flexibility and reduces waste of pre-printed materials.
In high risk areas, HVAC systems with a minimum of 20 air changes per hour ensure additional safety when packaging highly potent drug products. With air constantly being circulated through the facility, any loss of highly potent material would be captured in the HVAC and drawn through very high classification filters.
As with development and manufacture, multiproduct packaging facilities require robust cleaning procedures. Equipment with a low Ra (surface roughness) value of 0.8 or less essentially gives a mirror finish to equipment and prevents product from being ingrained. Along with walls made of an easily cleanable surface, this makes cleaning easier and prevents the risk of contamination and carryover.

Strategic Outsourcing
Ensuing that you partner with a CDMO
with the relevant experience, expertise, equipment and facilities will significantly reduce the amount of risk in your supply chain, saving time, money and drug product along the way. It’s important to remember that the right CDMO is there to help you achieve your clinical and commercial goals. They know their processes and equipment trains, and have a vast amount of experience in their areas of expertise. By identifying the right CDMO and establishing a strong collaborative relationship during the development stage, sponsors can be rest assured that their drug product will achieve speed to patient, study, approval, and commercial launch.
REFERENCES
1. https://www.precedenceresearch.com/highpotency-active-pharmaceutical-ingredientsmarket#:~:text=ingredients%20market%20 size%3F-,The%20global%20high%20potency%20 active%20pharmaceutical%20ingredients%20 market%20size%20was,USD%2055.66%20 billion%20by%202032
David O'Connell is the Director of Scientific Affairs at PCI Pharma Services. After graduating with a BSc. in Applied Bioscience, David spent seven years as a Supervisory Scientist working for Aptuit before moving to Penn Pharma in 2009. He played a vital role in the design of the potent Contained Manufacturing Facility (CMF), which won the ISPE Facility of the Year award for Facility Integration (2014). In 2013 David took on the role of Director, Pharmaceutical Development and in 2017 became Director of Scientific Affairs.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 59 www.international-pharma.com
Application Note
David O'Connell
Addressing the Challenges of Serialisation
Drug counterfeiting poses a significant threat to the pharmaceutical industry, risking patient safety and undermining the integrity of the global supply chain. As illicit activities continue to evolve, with counterfeit drug seizures in 2021 increasing 101% over the previous year,1 pharmaceutical companies face a pressing need for robust solutions to combat counterfeit drugs.
Serialisation has been a powerful weapon in the battle against counterfeiting for over a decade, and regulations offer a systematic approach to product tracking and authentication. By assigning unique identifiers to each unit of a product as a regulatory requirement, serialisation has been a comprehensive means of ensuring the authenticity and traceability of pharmaceuticals throughout the supply chain, from factory to patient.
Serialisation frameworks, such as the EU Falsified Medicines Directive (FMD) and the US Drug Supply Chain Security Act (DSCSA) – with its impending phase II changes – act as safeguards against counterfeit drugs for their respective pharmaceutical markets.
Additionally, flexibility in the supply chain, cost-effective manufacturing and labelling processes, collaboration with external experts, and the benefits of partnering with contract packaging organisations (CPOs) in the implementation of serialisation measures are vital to pharmaceutical integrity. Non-compliance with these serialisation requirements may lead to penalties, market restrictions and compromised patient safety.
In this article, Patrick Ferguson, Managing Director TPU, explores the evolving landscape of serialisation and its implications for the pharmaceutical industry.
Worldwide Serialisation Legislation and Directives
Regulations regarding serialisation vary from region to region and from market to market, making compliance a complex affair for pharma companies looking to operate
internationally. The key facts that all pharma companies need to be aware of include:
• International requirements
• Language variations
• Country specific regulations and serialisation guidelines
• Unique data elements
• Formatting requirements
• Submission processes
• EU legislation complexity
The EU FMD was established to ensure a consistent level of harmonisation across member states and address the growing threat of counterfeit drugs, placing the onus on pharmaceutical manufacturers to integrate serialisation into their operations. These directives were first adopted by the European Parliament and Council in 2011 and they outline the requirements and timelines for serialisation compliance. The final part of the directive came into effect in 2019 and concerned the following safety features:
• Unique identifiers: A 2D data matrix code and legible information placed on medical products that can be scanned at fixed points along the supply chain
• Tamper evident features: Anti-tampering devices on the packaging
Manufacturers operating in the EU are required to comply with these directives and regulations, ensuring that their products are serialised and traceable at every stage of the supply chain.
Changing US requirements
The DSCSA serves as the foundation for serialisation regulations in the US, aiming to enhance traceability and combat the infiltration of counterfeit drugs into the supply chain. Phase II updates to DSCSA 2023 requirements come into effect in November 2023, and are comprised of three specific yet highly interrelated components:
• Interoperable verification: Trading partners must be able to verify the product identifier on a package or sealed homogeneous case in a secure, electronic, interoperable manner.
• Interoperable exchange: Trading partners must exchange required transaction information (TI) and transaction statements (TS) in a secure, electronic, interoperable manner and the TI must include the product identifier at the package level.
• Interoperable tracing: Trading partners must maintain secure, electronic, interoperable systems and processes to provide TI and TS in response to regulator requests and to promptly gather the information necessary to produce the TI for each transaction going back to the manufacturer.
Although the Phase II 2023 DSCSA and EU FMD do mandate aggregation, many organisations are already implementing it to support harmonisation and increased end-to-end visibility of the supply chain. Pharma companies seeking to supply the US market must stay updated on evolving US serialisation requirements, engage with solution providers and industry experts, and adopt agile strategies to navigate the complexities of compliance effectively.
Challenges posed by incoming US requirements
The implementation of these upcoming US requirements presents several challenges for pharmaceutical manufacturers:
• Compliance timeline: Meeting the deadlines set by regulatory bodies, such as the DSCSA November 2023 deadline, requires careful planning, investment and industry coordination.
• Technical integration: Implementing serialisation systems and integrating them with existing manufacturing processes involves complex technical considerations, including technology selection, infrastructure setup and data management systems.
• Data management and standardisation: Effective management, standardisation and interoperability of serialisation data across systems and stakeholders are crucial to ensure accurate and secure data exchange.
60 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Packaging
Solving the challenges for CGT

— Suitable for deep cold storage


High break resistance
Treated to prevent delamination
Available gamma irradiated
— Meets ISO standards
INTERNATIONAL PHARMACEUTICAL INDUSTRY 61 www.international-pharma.com CPHI Barcelona | October 24 – 26, 2023 | Barcelona, Spain | Hall 2, Booth 2H50 gerresheimer.com
solutions
COP parenteral vials for biological
Packaging
• Supply chain collaboration: Close collaboration and data sharing among multiple supply chain stakeholders is needed to ensure successful implementation. Establishing communication channels and data governance frameworks facilitate effective collaboration.
• Cost considerations: Serialisation implementation involves significant financial investment, and manufacturers must carefully balance compliance requirements with cost-efficiency without compromising patient safety.
• Operational impact: Serialisation brings operational changes that require manufacturers to assess and optimise production lines, packaging operations and inventory management to minimise disruptions and maximise efficiency.
Addressing these challenges requires proactive planning, collaboration and a commitment to leveraging technology and industry best practices.
Pharmaceutical manufacturers need to stay informed about the evolving serialisation legislation and directives within their respective regions. By adhering to these regulations, manufacturers can demonstrate their commitment to patient safety, regulatory compliance and the integrity of the pharmaceutical supply chain.
How Serialisation Requirements Exacerbate Existing Challenges in the Industry
The implementation of upcoming serialisation not only introduces challenges but also exacerbates existing ones, adding complexity to the pharmaceutical industry. These challenges include:
• Integration complexity: Integrating serialisation systems with existing manufacturing processes becomes challenging, considering the need to align with current and upcoming requirements.
• Data harmonisation: Harmonising data formats and communication protocols across supply chain stakeholders is crucial for interoperability and reducing the risk of errors or delays.
• Supply chain collaboration: Effective collaboration among partners is crucial for serialisation success, but new requirements may strain existing efforts and communication channels.
• Infrastructure and resource constraints: Implementing serialisation requires significant investments in technology infrastructure and resources, which may be constrained by existing limitations.
• Time and cost considerations: Meeting US serialisation deadlines adds urgency and cost pressures, potentially leading to rushed decisions and increased expenses.
• Regulatory complexity: The introduction of new US requirements adds to the existing regulatory complexity, requiring alignment with multiple bodies and staying updated on evolving guidelines.
By exacerbating existing challenges, serialisation necessitates a comprehensive approach to compliance. Manufacturers must be proactive with careful planning, resource allocation, collaboration and leveraging technology solutions to successfully achieve compliance, and strengthen supply chain integrity and security. Ultimately, overcoming these challenges will result in a more secure and transparent pharmaceutical supply chain, ensuring patient safety and regulatory compliance in worldwide markets.
Supply Chain Flexibility and Meeting Demand
Meeting existing and future demands is of paramount importance in the dynamic pharmaceutical industry. By effectively addressing and managing the challenges associated with serialisation, manufacturers can proactively prevent disruptions in the supply chain and maintain continuous and reliable availability of pharmaceutical products to patients and healthcare providers alike.
With evolving serialisation requirements and potential expansions to serialisation scopes, such as aggregation and additional data elements, a flexible supply chain allows manufacturers to easily incorporate these changes without major disruptions or significant reconfigurations.
Efficient tracking through serialisation provides opportunities for improved demand forecasting and inventory management, enabling manufacturers to proactively optimise production schedules, reduce wastage and avoid shortages. Introducing such sophisticated track and trace methods to gain the added value of serialisation data requires the integration of technology.
Technological Innovations in Serialisation
Achieving future demands of the pharmaceutical industry will require technological innovation to enhance supply chain flexibility. Leveraging advanced technologies such as blockchain, data digitisation and artificial intelligence (AI) offers transformative solutions that streamline operations and enable efficient serialisation implementation.
• Blockchain technology: This is a method of recording information that makes it impossible (or at least difficult) for the serialisation system to be altered, hacked or otherwise manipulated. Blockchain makes product tracking more secure, with fewer errors, and has the potential to fix global supply chain vulnerabilities, accelerate collaboration among companies, reduce fraud and assure product authenticity.
• Data digitisation: The digitisation of supply chain data is a crucial enabler of serialisation implementation. Capturing and storing data electronically improves efficiencies across other operational areas. Digitisation processes will also play a role in safeguarding the integrity of primary and secondary packaging with technologies that allow for more rigid authentication and better quality control.
• AI: AI technologies offer powerful capabilities for optimising supply chain processes. They are used in drug development to analyse large amounts of data and identify potential drug targets, but can potentially transform drug manufacturing and packaging. One area where enhancement can be expected through AI is predictive maintenance to reduce downtime by implementing preventative measures before equipment begins to fail. Process optimisation and inventory management are other areas where AI can more effectively identify potential improvements and ensure that necessary stock levels can be forecast.
Serialisation technology has revolutionised the pharmaceutical industry by enhancing supply chain security and regulatory compliance. However, implementing serialisation requires addressing various manufacturing and labelling processes to ensure seamless integration and effective execution.
62 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Addressing Manufacturing and Labelling Processes
Serialisation implementation significantly impacts manufacturing and labelling processes in the pharmaceutical industry. Manufacturers face issues in production efficiency and cost considerations when optimising these processes to ensure cost-efficiency while complying with serialisation requirements.
Collaborative Efforts to Implement Effective Solutions
Collaborative efforts between manufacturers and external suppliers have become key to overcoming the challenges associated with serialisation and labelling processes. Working together, they can implement effective solutions and leverage expertise to optimise cost-efficiency and ensure compliance.
• Solution providers: Collaborating with specialised solution providers grants manufacturers access to serialisation expertise, technology platforms and integration support. These providers offer guidance on equipment selection, system integration, data management and workflow optimisation, ensuring efficient serialisation implementation.
• Regulatory authorities and industry networks: Engaging with regulatory authorities and participating in industry networks and associations facilitate knowledge sharing, regulatory compliance and adoption of best practices. Manufacturers benefit from staying updated on changing requirements, gaining insights into serialisation trends and accessing collective experiences and guidance.
• CPOs: Partnering with CPOs can assist manufacturers in managing serialisation activities, particularly in packaging and labelling processes. Manufacturers can outsource specific serialisation tasks or rely on CPOs for end-to-end serialisation requirements. This collaborative approach optimises cost-efficiency, scalability and utilisation of specialised resources.
Benefiting from CPOs
CPOs play a crucial role in the pharmaceutical industry, particularly in the context of serialisation implementation. These specialised service providers offer expertise, resources and infrastructure to support pharmaceutical manufacturers in meeting their packaging needs, including serialisation requirements.
CPOs work closely with manufacturers to understand their specific serialisation requirements and develop tailored solutions to meet those needs. Manufacturers can leverage their specialised capabilities by collaborating with CPOs and ensuring efficient serialisation implementation while focusing on their core competencies.
The Specialised Capabilities of CPOs
It is important to recognise that although CPOs provide valuable support, there are specific complexities inherent to complex packaging, like pre-filled syringes (PFS) that demand careful attention. Understanding these unique considerations is crucial for pharmaceutical manufacturers seeking to optimise the serialisation process for PFS and ensure the highest standards of quality and safety.
The vital role filled by CPOs addresses the unique challenges associated with packaging and labelling during serialisation implementation. By partnering with CPOs, manufacturers can navigate the complexities of serialisation and ensure the integrity, safety and regulatory compliance of their products.
Collaboration, Innovation and Adaptability in Serialisation
Serialisation has emerged as a crucial solution to combat counterfeiting and ensure compliance in the pharmaceutical industry. Implementing serialisation measures allows manufacturers to track and trace products throughout the supply chain, provide a robust framework for product authentication, improve patient outcomes and enhance regulatory compliance.

However, the successful implementation of serialisation requires collaboration, innovation and adaptability. Pharmaceutical
companies must work closely with regulatory bodies, technology providers and other stakeholders to navigate the complexities and challenges that arise. Flexibility in the supply chain, adoption of advanced technologies and proactive strategies are essential for meeting the evolving demands of serialisation.
As the industry continues to evolve, embracing innovations and leveraging the expertise of external partners such as CPOs can streamline serialisation efforts and ensure compliance with legislation across any border. The expertise provided by experienced CPOs becomes increasingly important as the complexity of packaging develops from increased demand for PFS and other personalised and unique modalities.
REFERENCES
1. https://www.psi-inc.org/incident-trends
Seasoned operations and compliance professional with 20+ years of experience in Pharmaceutical Manufacturing and Packaging Operations, Compliance, and Regulatory Affairs. I thrive on managing and leading challenging operations and compliance units and contributing individually where needed. As Managing Director, I have overall responsibility for all operational activities for the Tjoapack US location. I have a BS in Chemistry and a BS in Biology from Missouri State University and a Master's in Business Administration from Park University.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 63 www.international-pharma.com
Patrick Ferguson
Packaging
What Does the Future Hold for Delayed Release Empty Hard Capsules? An Exploration of Enteric Release Capsules in the Nutraceutical and Pharmaceutical Markets
The term “delayed-release” formulation refers to finished dosage form tablets or capsules, which are acid-resistant and protect the release of a medicament until the capsule has passed through the stomach into the small intestine.
Generally, in the case of capsules, either the final formulation is coated, or it is filled with pellets coated with acid-resistant polymers. Alternatively, the contents (powder, granules, or pellets), are filled into an empty hard capsule shell specially designed to demonstrate delayed release behavior. The industry is equipped with –and has already developed products using – these two approaches. However, there remains an expectation on the healthcare industry to receive “ready-to-fill” delayed release empty hard capsule shells to help support operational convenience and reduce the processing time required for coating.
As a result of this demand, the capsule manufacturing fraternity has been proactive when it comes to developing such functional capsules to support the industry’s development pipeline. In recent years, the discussion around these functional acidresistant capsules has also moved across into the pharmaceutical and nutraceutical industries. This has been illustrated with the launch of these capsules with further evolving technologies which provide better acid protection and have also been made available for pharmaceutical applications. As a result of this evolution, a variety of market intelligence predictions indicate a strong growth potential for these enteric release capsules from 2023–2030.
Unfortunately, despite the need for one, there is no pharmacopeial test method available to qualify a “ready- to-fill” delayed release empty hard capsule. This is undoubtedly due to the absence of any marketed pharmaceutical product using such capsules. However, given the industry demand for these capsules it is worth creating all possible control parameters to support their initial development. This should include all test methods, to ensure a current quality product, which is capable
of being commercialised in the future. In the meantime, as an interim arrangement, the industry has adopted the pharmacopeial method of delayed release finished formulation.
Accordingly, the delayed release empty hard capsules should withstand a one-hour disintegration test in 0.1N HCL while the end formulation (filled capsules) must protect the inner content for at least two hours in 0.1N HCL (<10% drug release), followed by >75% release in 6.8 pH buffer within 45 mins unless otherwise specified in a dissolution study.
For further clarity, the delayed release capsules could be classified into “enteric capsules” and “modified release capsules”. Enteric capsules, also called gastro-resistant capsules, are capsule shells coated with a special polymeric film that acts as a barrier to the acidic environment of the stomach. Thus, allowing the intact capsules to be carried forward, released, and absorbed in the alkaline environment of the small intestine.
There are a couple of methods by which enteric empty hard capsules can be manufactured. The first is by spraying a fine film of the polymeric solution on the prelocked capsules using conventional pan coating technology. The second is to use the traditional process of dipping the cap and the body separately in an acid-resistant polymer solution, followed by drying and finally joining them into a pre-lock state, thus resulting in a bi-layer capsule shell. These technologies ensure the formation of a continuous and uniform film on the shell surface capable of providing acid
protection of the fill for at least two hours. Usually, the polymers used are methacrylic acid co-polymers, cellulose acetyl succinate, phthalates, along with other additives to aid the coating process and stabilize the film properties. On the other hand, anionic polysaccharides like pectin, gellan gum, etc. – either alone or in combination along with HPMC as the base film forming polymer –have been used to produce a conventional monolayered delayed release capsule shell. A system like this is effective in protecting the release of the fill in acidic environment for shorter durations of less than an hour, depending on the concentration and type of the polysaccharides used. These may be termed as “modified release capsules”.
Modified release capsules have demonstrated sufficient protection to deliver acid sensitive nutraceutical ingredients like probiotics, enzymes, etc. They have effectively been able to address critical consumer complaints such as aftertastes relating to the ingestion of fish oil and other omega-3 fatty acids. Moreover, these capsules also keep the lining of stomach protected from exposure to potentially harmful or irritating substances that can lead to nausea, vomiting, and ulcers. They also protect the enzymes so that they can be absorbed at their highest activity levels in the small intestine.
In pharmaceutical applications, enteric coating is a useful strategy for the oral delivery of labile entities such as peptides, nucleotides, live biopharmaceutical products, and vaccines which rapidly degrade in the stomach. While they are still relatively nascent in their development, enteric coated
64 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Packaging
Contact
YOUR SECURE SUPPLIER FOR TAMPER EVIDENT SEALING TO ACHIEVE SUSTAINABLE PACKAGING




The PAPER-based security seal with VOID technology is now available!
• Works on almost all cardboard surfaces
• Underlying text and printed codes remain legible

• Compliant with FMD safety requirement

• Serialisation of each individual product

• 'One package - one material' - this way you improve the recycling process and make a positive contribution to the recycling management


• Paper seals are produced from renewable raw materials

Our seals are easy to understand multifunctional security locks against tampering and counterfeiting. They are used and tested on pharmaceutical packaging worldwide.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 65
us for free samples Visit us at AIPIA World Congress Amsterdam November 14-15, 2023 Booth 31 Applied Peel-Off Tampered
capsules have demonstrated the protection of insulin from the acidic environment of the stomach, thereby enhancing the intestinal absorption of insulin and providing a prolonged hypoglycemic effect. “Readyto-fill” capsules with integrated enteric protection functionality enable possibilities for rapid formulation screening, prototype development, and allow for the rapid testing of in vivo performance. Use of these capsules can enable oral delivery of heat sensitive actives which otherwise would require exposure to high temperatures during the coating process.
While all the above pharma applications indicate new drug developments, it would be interesting to delve into the lucrative space of generics as well. It is important to recognize that the level of pharma expertise already existing in the field of enteric coated tablets or enteric coated pellets filled in capsules is impeccable. The infrastructure, expertise, and technology that enables the development of such products is available to almost every company. The pharmaceutical market is flooded with such products at affordable costs. Moreover, as compared to larger dosage forms like tablets, enteric-coated multi-particulate (pellets or minitablets) capsule dosage forms are known to reduce variability in bioavailability and minimize the likelihood of a therapeutic failure when coating defects occur during manufacturing.
Before responding to the question: ‘is there still a scope left for such products to be redeveloped into a formulation filled into a “ready-to-fill” enteric hard capsule?’, we need to appreciate the repercussions of undertaking such a developmental approach from a bioequivalence standpoint. It is obvious that the in vivo behavior of enteric
coated pellets filled in capsule vs uncoated pellets filled in enteric capsules would be significantly different. The dissolution rate of a dosage form is a function of its surface area, and the total surface area of multiple tiny pellets together is exponentially higher than a single capsule. This results in a completely different release profile. Therefore, although there could be a possibility to improve the bioavailability, it would be extremely challenging to establish a bioequivalency as per the acceptance criteria defined by the authorities. Moreover, there are currently no enteric empty hard capsules-based innovator products unless enteric coating is done on a filled capsule, thus leaving limited room for generic drug product development. In this context, the next question would be, ‘what would motivate researchers to venture into such a product development strategy?’.
Based on the facts above, it appears that enteric “ready-to-fill” empty hard capsules hold great promise for future pharma developments. However, the journey to commercial success may extend beyond a decade. The generic space, often considered as a quick launchpad in the pharmaceutical market seems to be limited for these capsules due to the complexities involved in replicating the delivery mechanism with similar therapeutic efficacy.
However, the nutraceutical space and demand for natural products have grown significantly since the pandemic, presenting an attractive opportunity to explore the potential of enteric capsules.
Active players in the nutra industry –have been successful in partnering with customers and supporting them with the right capsules for product development of
acid sensitive molecules. With their ability to deliver substances to the intestines, these capsules could play a significant role in the nutraceutical sector's growth and profitability.
With such ongoing developments, the regulatory bodies need to consider framing the required analytical methods for accessing the quality, efficacy, and safety of these delayed release “ready-to-fill” empty hard capsules.
Subhashis Chakraborty

Dr. Subhashis Chakraborty holds a PhD in Pharmaceutical Sciences. He has more than 15 years of experience with different MNCs in senior management roles, working across R&D, business development and product management. Presently, Dr. Chakraborty holds the position of General Manager, Global Product Management, for ACG Capsules. In this role, he is responsible for spearheading the new capsule technologies for pharmaceutical and nutraceutical applications, right from development to commercialization.
Shincy Jacob
Sr. Manager, Global Product Management, ACG Capsules
Ganesh Adasul
Sr. Manager, Global Product Management, ACG Capsules
66 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Packaging
Unlocking Smarter Pharma Packaging Production
 Laura Lopez Marketing Manager MM Packaging, Pharma & Healthcare
Laura Lopez Marketing Manager MM Packaging, Pharma & Healthcare

It’s time to think smarter. For pharma businesses of every category or size, disruptions and bottlenecks come with significant consequences – but can the packaging supply chain work harder to avoid this?
Today’s pharma supply chains are complex and interconnected, as globalisation becomes the standard rather than the exception.
Whether caused by demand outstripping capacity, inefficiencies or malfunctioning equipment, supply chain bottlenecks are prompting pharmaceutical companies worldwide to streamline their packaging production. Left unattended, repeated
equipped assembly lines to simple, taskspecific process enhancements like sensors or gating mechanisms.
Bringing powerful efficiency benefits, automated packaging solutions can also be used to enhance areas such as labelling, carton production, inspection and more, to boost throughput and reduce the potential for errors.
By replacing time and labour-intensive manual processes with digital technology, pharma businesses can expand capabilities and meet tighter lead times. Production teams can spend more time on value-adding tasks, and less time on troubleshooting and maintenance. This, in turn, better equips businesses to meet spiking demand levels through greater agility.
Automation can also contribute to businesswide sustainability in a number of ways, including more effective use of energy, and reducing the potential for human error during
logy to produce high-contrast precise patterns or codes tailored for Optical Character Recognition (OCR) Vision Systems.
This next-generation coding system produces codes that are completely scuff and smudge-resistant and are not susceptible to environmental degradation, providing companies with track and trace capability as well as the ability to remain agile in the face of market changes.
Finding the Right Solution
Although the benefits of smart packaging manufacturing are clear, for businesses looking to drive competitive advantage, the most effective option is to work closely with a partner that understands the intricacies of the pharmaceutical supply chain, such as the team at MM Packaging.

Our team knows that there is no ‘one size fits all’ template for supply chain automation – workflows are simply too varied from one business to another, and we strive to stay ahead of the curve with our manufacturing techniques, which enable us to lead the market on many fronts.
disruptions can bring increased costs for business, harm customer satisfaction and damage a brand’s reputation.
Instead, smart packaging manufacturing systems can provide a suite of benefits that help pharma brands maintain a crucial competitive edge. At MM Packaging, we know that an automated workflow can be a very powerful production tool, easing the pressure on manufacturing for pharma businesses and ensuring treatments reach patients as quickly as possible.
What Does Smart Manufacturing Look Like?
The word ‘automation’ still tends to conjure images of science fiction, but as technology scales and advances, automation through the manufacturing supply chain has never been more accessible. Examples in the pharma supply chain can include anything from robot-
the packing process, resulting in less waste, which has an ultimate cost to the environment.
To further enhance pharma workflows, automation can be combined with digitalisation tools to enhance data collection and monitor inventory as it moves throughout the supply chain. This data can help to identify further bottlenecks and areas for improvement, as well as help businesses forecast demand and adapt to accommodate fluctuations.

Furthermore, supply chain transparency and efficiency can be boosted when combined with cutting-edge serialisation technology such Clear Code®. The solution uses a CO2, fibre or UV laser alongside proprietary colour-changing coating techno-
MM operates 28 dedicated pharma packaging manufacturing sites that are fully equipped to offer services such as co-packing, meaning our customers can take advantage of our state-of-the-art equipment and fully automated logistics. Combined with our expertise in sustainable packaging design, operational bottlenecks and delays could soon become a thing of the past.
To find out more about how MM Packaging can support your business in overcoming disruptions, get in touch by emailing pharmahcpackaging@mm.group.
To learn more about MM Packaging and its activities, please visit www.mm-packaging.com
Advertorial
Chemical Recycled ABS Materials for the Transition to a Circular Economy Model

The demand for new, sustainable ABS materials for drug delivery device applications is growing in the healthcare sector. However, because of the risk of cross-contamination, medical regulatory compliance requirements cannot be fully met with mechanically recycled ABS materials. Fortunately, new chemically recycled and bio-based ABS materials are already available, which possess the same chemical composition and properties of virgin medical ABS. This means that they fulfil the same medical applications and meet medical regulation requirements. All the colours that are available as virgin medical ABS can also be used in the bio-circular version, guaranteeing not only regulatory compliance, but also the availability of bright and intense colours in chemically recycled ABS formulations. These types of colours cannot be achieved with mechanical recycled content.
Some clarifications are needed to understand how it is possible for chemical recycled medical ABS to fully comply with medical applications and which are their main sustainability advantages versus medical ABS with entire fossil origin.
There are several types of chemical recycling technologies and different kinds of waste that can be chemically recycled. In this case we are talking about conversion chemical recycling, which is breaking down the target waste through pyrolysis (a thermal decomposition process without the presence oxygen) to obtain an oil like feedstock. Very important is the entrance point of this pyrolysis oil into the ABS polymer supply chain, which is at its very start (upstream). This means, not only before the production of ABS starting raw materials (Acrylonitrile, Butadiene and Styrene), but also before all the steps that are needed to produce such raw materials, without introducing any process change in them. The ISCC+ certification with a mass balance approach guarantees the sustainable nonfossil content, and the whole supply chain can benefit of this with the already existing
production processes, without making first huge investments which would make this impossible from an economical perspective. Already the chemical recycling processes themselves, the transformations from waste into pyrolysis oil, are represented by different types of expensive alternative technologies that depend on the type of considered waste and that need to be optimised and scaled up to make chemical recycling economically feasible.
An example is the incorporation of pyrolysis oil obtained from used tires in the supply chain production of Styrene. The only change is the partial substitution of Nafta oil with Pyrolysis oil to feed the steam cracking process, that is needed to convert large hydrocarbons contained in the oil into smaller ones. As it happens in the case of 100% fossil oil, the same molecules such as Ethylene and Benzene can be extracted, as it occurs for several other ones. These can be used as reagents in the production of Ethylbenzene and use the same exact processes that is used since many years for the fossil version of these input substances. The next step in the supply chain is the production of Styrene, which is obtained starting from Ethylbenzene with its standard production process which is also the same and has been also optimised since many years. The only difference is that, if chemical recycled content from used tires is present instead of fossil content, each one of these supply chain steps must be certified by ISCC+
with a mass balance approach. The same occurs in the following production passage at the ABS manufacturer, where Styrene is used as raw material to be polymerised with Butadiene and Acrylonitrile to obtain ABS.
It has been possible to obtain the ISCC+ certification with a mass balance approach and to offer ABS grades with chemical recycled content to the market. Medical grades are already used since many years in medical applications such as drug delivery devices and medical device housings, and now are available with up to 70% ISCC+ certified raw materials content (chemical recycled + bio-based content), ISCC+ certified with a mass balance approach. The same special GMP procedures implemented for medical ABS with fossil content are used, and there is no difference in the chemical composition, process, or properties between the fossil and chemical recycled version.
Medical devices need approval from authorities before market introduction. Specific and confidential information about material formulation must be included in the requested documentation to support the approval process. Medical ABS FC composition is registered in a Drug Master File (DMF) by the material manufacturer at the FDA and can be accessed by regulatory authorities to verify medical device compliance for the specific medical device application. A Letter of Authorization is released by the material manufacturer to grant the medical OEM the right to incorporate the information contained within the DMF into their medical device application. This also grants FDA reviewers permission to review the proprietary information within the DMF in reference to that specific medical device application. Each DMF contains all formulation detailed information regarding not only the medical ABS FC composition, but also all the biocompatible colour formulations that are available for that ABS FC grade. Periodically, when a new medical ABS FC colour is developed on customer demand, the FDA is contacted to get permission to include that specific colour formulation in the corresponding ABS FC Drug Master File. Only authorised colour pigments within the maximum allowed concentrations can be
68 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Packaging
used, pigments which have been previously biocompatibility tested at recognised laboratories according to ISO 10993.
The formulations with chemical recycled content (ISCC+ certified) go through the same process and are finally registered at the FDA in the same DMF M203FC or M205FC, in a similar way as it happens for new medical FC colours. In this way, all information regarding the specific ABS material composition with exact chemical recycled content and in the requested colour is included in DMF #25288 (or #25284 in the case of ELIX M205FC).

Once discussed how it is possible for a medical ABS ISCC+ certified with chemical recycled content to fully comply with medical regulations and healthcare applications, it is important to consider which are the sustainability advantages in comparison with a full fossil-based medical ABS. The key variables to take into account are the type of waste used, the level CO2 emissions related with the waste recycling process, the efficiency of the process (which % of the waste can be exploited) and the possible presence of biogenic content in the waste itself. In addition, from an end-of-life perspective, also the CO2 emission reduction due to the avoided waste incineration process can be also considered, since this step is eluded when creating circularity.
When the waste is methodically separated and sorted by type, the related recycling process can be fine-tuned on that specific type of waste, with a consistent reduction of energy inputs required and consequent CO2 emissions outputs. If we compare for example used tire waste with undefined mixed plastic waste, the first one has recently implemented
a much more efficient chemical recycling process, with much lower CO2 emissions and no solid/liquid residues generated. In addition, the controlled source used (only tires) improves the traceability of the process, which would solve a big problem in the European tire industry.
The average tire composition from this source of used waste includes synthetic rubber but also natural rubber, carbon black, textile fiber and steel. With the identified recycling process, also carbon black can be recovered, representing an additional positive environmental impact. The volatiles generated during the process can be reincorporated to feed energy into the same recycling process, reducing the related CO2 process emissions. Furthermore, the natural rubber content in the used tires has great importance. Natural rubber was
extracted from plants in the past, and those plants where absorbing CO2 emissions during their previous life instead of emitting CO2. This biogenic content in tires provides therefore a relevant contribution in CO2 emissions reduction.
The percentage content of sustainable certified raw materials in the medical ABS composition can be adapted to the OEMs sustainability and economic targets. For example, ABS grades with 25%, 50% or up to 70% ISCC+ certified raw material contents (chemical recycled and/or bio-based) are possible but also any specific customer percentage configuration is possible. Even 100% is in theory possible option, and it depends on available chemical recycled raw material sources, incremental environmental contribution, recycling technology state of the art, scale synergies and related cost.
Most important is to start adopting this type of solution including recycled waste as raw materials, even with low percentages. This would be the correct attitude that will support an easier transition towards the use of more sustainable ABS medical materials in drug delivery and other medical devices in the coming years.

Luca joined ELIX Polymers in February 2017 and works in the position of Business Development Manager for the Healthcare and Consumer sectors. He is responsible for the identification, development and validation of new market/segments, products & applications opportunities and for translating customer needs into practice. Luca graduated in management engineering at the "Politecnico di Milano" University (Milan, Italy) and has at present over 15 years’ experience in the fields of thermoplastics, thermoset plastics, composites, electrical insulation, and electronics. He worked in the past in different positions as Business Development Manager, Head of Sales Energy & Composite Materials, Export Manager Europe & Asia and Sales Application Engineer Iberia & South France.
Email: luca.chiochia@elix-polymers.com

INTERNATIONAL PHARMACEUTICAL INDUSTRY 69 www.international-pharma.com
Luca Chiochia
Packaging
Navigating the Shift in Pharmaceutical Packaging: Beyond Traditional Structures towards Healthier and more Sustainable Future
In the past the choice of packaging strategy in pharmaceutical industry was often purely based on functional criteria that defined which packaging material delivers the most stable performance for a safe, secure, and efficient drug delivery.
Predictable interaction between active ingredients and the packaging material can only be guaranteed through extensive testing of a specific combination of a drug formulation and dedicated packaging material. It is a time consuming and rather expensive process. Once the winning match has been confirmed, it becomes a part of an official pharma dossier that is submitted for a legal approval and registration by regulatory bodies. In this context it needs to be understood that most changes to primary packaging specification would directly require new stability testing and new submission of a dossier with all the associated costs and effort. Which is why such changes are unlikely to be implemented unless public safety is at stake, great financial risk or gain has been identified or a new legal requirement has been introduced.
For years, the pharmaceutical industry has relied on combinations of PVC and aluminum for medication packaging. However, as sustainability climbs up the global agenda, this traditional approach is coming under scrutiny. With products like mono-polymer and PVC-free aluminium coldform blisters leading the change, the industry is adopting innovative and more environmentally sound packaging solutions. Read how a transition is both timely and challenging and how to maneuver through this journey.
The Changing Landscape of Pharmaceutical Packaging
Driven by the quest for more sustainable solutions, the material choices for blister packaging will shift towards less complex substrates. Halogenated polymers like PVC, PVDC, and PTFE for mid to high barrier applications will most probably be less acceptable as the need for alternative
solutions, that are more compatible with circular systems requirements will grow.
The Rise of Coldform Blisters
Coldform blisters without PVC sealing layers are increasingly garnering attention for their ability to substitute other less preferential options for high barrier applications. While not a new concept, it's quickly becoming a low-hanging fruit in the mission for sustainable packaging transition due to several other benefits that those structures have to offer.
In addition to being PVC-free, the new generation of coldform blisters contains less plastic in weight and therefore the relative share of aluminium is increased without adding more metal. Higher aluminium content contributes to a better sorting and higher material recovery rate in waste processing in all life cycle stages of the material, including the end-of-life of the final blister pack.
Replacing PVC with polyolefins also results in coldform blister material that is flatter and less prone to wrinkling, which then leads to a better machinability with a potential for a more efficient blister forming performance.
Exploring Alternative Polymers
Not all pharmaceutical products require the level of protection the coldform blisters deliver. In cases where an absolute barrier isn't a top priority, and the packaging mainly functions as a protective dust cover, companies’ main priority tends to be moving away from PVC-based bottom webs. They're opting instead for alternative polymers such as PET, PP, and PE because these materials are better suited for recycling, aligning closely with sustainable design principles. It's becoming an increasingly common choice for corporations focused on environmental responsibility.
The Quest for Mono-material Blister
Packaging
Medicine blister is a two-component packaging structure, so the challenge doesn't stop at replacing the bottom web. Pharmaceutical companies must also embark on the search for new lidding materials that can be paired with non-PVC bottom webs to create a mono-
material blister that complies with designfor-recycling guidelines and can be therefore considered recyclable.
While it seems like a straightforward and most logical solution, is it the most effective one for a pharma packaging?
Recyclability is a System Challenge
The Complexity of Recyclable Pharma Packaging
Just because a material is technically recyclable doesn't mean the finish packaging will be. Even if a package adheres to designfor-recycling guidelines, it's not automatically guaranteed to be recycled. The recyclability of packaging depends on several factors such as local collection systems and the sorting and recycling infrastructure that is in place for specific materials. Moreover, the type of product being packaged further complicates matters. While the material itself may be recyclable, pharmaceutical packaging often can't be included in general recycling streams due to the risk of contamination from medication residues.
Study of EPR systems of 68 countries worldwide shows that merely 17 countries explicitly mention pharmaceutical packaging in recyclability and sorting guidelines. Only 5 of those countries allow certain medicine packaging in their recycling streams, one of it being medicine cardboard boxes in South Africa, which is not representative for the case, as this is not the immediate packaging of medicinal products associated with the contamination risk.
Most countries, work with a positive list for the recyclables collection. If a packaging type is not mentioned, it is not included in the collection, as only items that are on the list are allowed. Those are typically beverage containers, glass, and metal packaging. Therefore, it is rather unlikely that plastic pharmaceutical packaging will be considered recyclable in any of the remaining 51 countries where the blister packs are not mentioned.
But that’s not all. Even when the blister material complies with the design for recycling guidelines and contamination risk is mitigated, the small size of most
70 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Packaging
pharmaceutical packaging makes it difficult to sort and recycle within the existing waste management infrastructure.
The only flexible pharma format that can currently be considered recyclable in practice is PVC free aluminium coldform blister. Although they will not be collected for recycling, unlike plastic, when aluminium coldform foil blisters end up in general waste the resources are not entirely lost. Polymers and potential medicine residues are incinerated but a large percentage of aluminium is recovered from bottom ashes of the furnaces, allowing that part of the material to be returned to the loop.
The Dilemma of Polymeric Film Lidding
Replacing aluminum lidding with polymeric film is a trend that comes with its own set of complications. Aluminium provides a robust barrier against migration from inks used for printing the necessary medicine information on the lidding, something that might not be entirely replicable with polymer films. In addition, the sealing of polymeric lidding film comes with challenges that don’t apply to traditional aluminum blister lidding foils. Last but not least, aluminum lidding offers a well-established, child proof and senior friendly opening mechanism for the blister: pushing the tablet through the lidding allowing for convenient and safe access to the medicine. This straightforward, push-through experience is difficult to replicate with polymeric options. Plasticbased materials often lack the same functional ease, making it less convenient for the patient to access their medication. Therefore, before making the transition to polymeric film liddings, it's crucial to weigh the benefits and drawbacks carefully. The material you choose could have a lasting impact on user satisfaction and convenience, while the potential environmental benefit
of enhanced recyclability might not be available in practice.
Advanced Recovery Technologies
As the industry struggles with recycling of multi-layer, multi-material structures, new technologies are emerging to tip the balance. Advanced, solvent-based technologies are being developed to allow for the full material recovery of both aluminum and polymeric films. These technologies are not only being tested but are also being scaled up to fit into existing waste management and recovery ecosystems.
Case Studies: German and EU Initiatives
• Saperatec in Germany: This technology specializes in post-patient/consumer resource recovery by separating multilayer, multi-material packaging into mono substrates without damaging the structure of the material. It produces clean flakes that are ready for mechanical recycling process. A first industrial scale plant is currently under construction in East Germany.
• AR in Germany: Another German program targets advanced recovery methods for packaging materials generating sorted recyclates that are suitable for processing in a wide range of applications. Pilot plant is operating in the industrial Park in Frankfurt.
• Solrec: Part of the Horizon2020, this EU-wide project is pioneering scalable solutions for sorting and recycling of multilayer, flexible packaging materials.
The Path Forward in Pharmaceutical Packaging
While the most apparent solutions may seem logical, they may not always offer the best
long-term benefits. The primary function of pharmaceutical packaging—ensuring product protection – must be balanced against its environmental impact. Packaging that does not fulfil its necessary function is not sustainable.

Although sustainable packaging is no longer a consumer goods domain, it is yet to be seen if applying the same criteria to consumer goods packaging and pharma packaging will leads us towards the future proof, circular packaging system that the industry and society are looking for. There's no room for compromise when public health is concerned, and a one-size-fits-all solution is illusory, especially with forthcoming legislation likely to raise the bar even higher.
Business as usual scenario is no longer viable. Being agile and proactive in adopting new technologies and legal requirements has never been more crucial. It's equally important to choose a partner who understands these complex system dynamics. With introduction of innovative more sustainable products and the development of advanced recovery technologies, the industry is taking steps in the right direction, but the journey toward sustainable pharmaceutical packaging is far from over.
The right packaging partner can help navigate this complex landscape, ensuring compliance, sustainability, and – most importantly –patient safety.
Agnieszka Van Batavia
Agnieszka Van Batavia is the Sustainability Manager Pharma at Constantia Flexibles, where she spearheads eco-friendly initiatives. Previously, she served as a Strategic Sustainability and Regulatory Advisor at The LCA Centre Netherlands, guiding stakeholders in sustainable packaging. A pioneer in circular packaging systems, Agnieszka is also an expert in packaging legislation. She is a frequent lecturer and keynote speaker, advocating for a science-based approach to sustainability. Additionally, she acts as a strategic policy advisor to companies, institutions, and NGOs.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 71 www.international-pharma.com
Packaging
Taking Healthcare Out of the Hospital: How Device Design Can Empower Users

From point of care diagnostics to auto-injectors, medical devices are increasingly enabling more procedures and healthcare activities to be conducted outside of the clinic. What is paramount to the success of these devices is making them simple and intuitive to use, so that they can be operated by non-clinically trained individuals. To achieve this, device manufacturers are utilising a range of tools and approaches, from digital support solutions to packaging innovations and more.
The Medical Devices Democratising Healthcare
Creating medical technologies to conduct healthcare outside of clinical settings often involves a certain degree of up-skilling nonspecialist users. The idea is that by putting the right technology and support in the hands of more healthcare professionals (HCPs) and providers, patient access to care can be increased, a phenomenon known as the democratisation of healthcare.
There are many examples of devices being used to help democratise healthcare.
Wearable devices are empowering users to track and better understand their health, providing a wealth of digital biomarkers that can be used to better inform health decisions. Meanwhile, point of care diagnostics are providing lab comparable results in minutes, enabling faster diagnosis and greater access for patients.
Healthcare at Home –The Challenge of Training Patients
In addition to medical devices designed for use by clinicians and healthcare professionals, there are also occasions when patients are required to self-train and administer therapy which would otherwise be carried out by an HCP in a clinical setting.
Take for example home auto-injection systems for a condition like rheumatoid arthritis. Traditionally, a patient would be instructed in first use of an auto-injector by an HCP during a face-to-face meeting, who would be able to support the patient with challenges such as injection anxiety and how to rotate the injection site. Increasingly, patients are now expected to self-train in the use of auto-injectors, sometimes having to rely solely on instructions for use (IFUs) to learn how to use their device.
In reality, many people either don’t read instructions fully or don’t follow the use steps correctly. This has been a significant driving factor in the simplification of use steps in recent years, with many modern auto-injectors now following a simple twostep process of ‘remove cap’ and ‘hold against skin’. Whilst this has reduced the opportunity for user error, it has also come at the expense of device complexity.
Even something as simple as an asthma inhaler poses significant challenges in terms of self-training and adherence for patients, as delivery technique is critical. With pressurised metred dose inhalers (pMDIs), users often struggle with coordinating the button press with inhalation, the result being that most of the drug is deposited on the back of the throat, which can lead to further complications. Inhaler spacers, which are plastic tubes that are attached to the mouthpiece, go some way to addressing the coordination challenge, but they are bulky and require cleaning. This has driven innovations in inhaler design such as breath actuation, where the plume of drug is not released until the user starts inhaling. The challenge here again is that this comes at the price of increased device complexity.
72 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Health
Outcomes
Figure 1
CONTRACT MANUFACTURING AND PRIVATE LABEL PRODUCTS
CONTRACT MANUFACTURING OF LIQUID DRUGS AND MEDICAL DEVICES

•BAG-ON-VALVE
•NASAL SPRAY PUMPS
•DERMAL SPRAY PUMPS
•AIRLESS SPRAY PACKAGING

•PureHale®
CE-MARKED PRIVATE LABEL PRODUCTS FOR YOUR BRAND
•SPARKLING SALINE NASAL SPRAY




•SALINE/SEAWATER NASAL SPRAY


•EAR CLEANSING SPRAY
•WOUND WASH SPRAY
•EYE WASH SPRAY
•EMOLLIENT SPRAY
•ADHESIVE REMOVER SPRAY
•BURN GEL
INTERNATIONAL PHARMACEUTICAL INDUSTRY 73 www.international-pharma.com
• contact@aurenalabs.com
COLLABORATE TO CREATE YOUR NEXT PRODUCT. CONTACT AURENA TODAY.
www.aurenalabs.com
LET’S
Health Outcomes
When you factor in all the additional support previously provided to a patient by an HCP, device manufacturers are therefore asking much more of their users and requiring more effective user-centred design. This has pushed companies to look beyond the device itself, to other elements of the design ecosystem such as the packaging and quick reference guides.
Figure 1 shows a flip-book packaging solution that walks the user through the features of an auto-injector pen before they’ve even removed it from the packaging. This draws upon a principle from digital User Interface (UI) design called 'progressive disclosure', in which features and information are presented to the user in bite-sized chunks, gradually building up a picture rather than bombarding them with all the information at once. The images on the packaging prime the user for how the device should be held and can include a QR code and ‘call to action’ to prompt the user to engage with digital training content on a companion app. Using digital content such as instructional videos has the potential to be more dynamic than traditional paper IFUs and perhaps even tailored to the needs of an individual.
Digital Tools to Support Users
The benefits of using smart devices go beyond
training. Sensors enable the recording of usage data, which can be shared with HCPs to ensure patients are adhering to their treatment, or with device manufacturers to make improvements to their product offering. Patients can also access more general information and support around managing their condition.
Another area where smart devices are proving to be useful is in the decentralisation of clinical trials. As the name suggests these would typically be conducted in a clinical setting which may therefore limit the number and type of participants that pharma companies can engage with. Additionally, a clinical setting can influence the behaviour of a participant.
Smart devices are now enabling clinical trials to be conducted over longer periods of time, in the participant’s home environment. This gives pharma companies much greater access to patients – especially more vulnerable cohorts who would previously have been excluded from attending clinical settings, such as cystic fibrosis patients. It also provides more confidence that the data collected is representative of reality. In short, smart devices in clinical trials allow for higher quantities of more targeted data, which can help to promote safer and more effective treatments.
While these digital solutions may seem like an obvious benefit for both users and pharma companies, the system design needs to work hard to engage and motivate patients, particularly if additional user steps are required such as downloading an app or sharing personal data. Designing solutions to prompt and motivate users to adopt a digital solution requires a deep understand of their desires and un-met needs. This comes down to performing good contextual research from the outset and testing ideas often with the target user group.
Ensuring User-centred Design in a Development
Taking healthcare out of the hospital and clinical settings has the potential to improve access, enable faster, more accurate diagnosis, reduce burden on the healthcare system and ultimately create better health outcomes. However, doing so places greater burden on both the devices enabling this and the users themselves. It is therefore important to think carefully about what is being lost by taking a healthcare application out of the hands of an experienced HCP. Achieving the right balance involves developing devices which people can and want to use, which are simple, intuitive and safe. This is easier said than done. The following are some of the key steps that can be employed in a device’s development to achieve successful, user-centred design.
74 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Contextual Research
The key to effective design is understanding the underlying behaviours, beliefs and unmet needs of your users, followed by a cycle of developing and prototyping solutions and testing them frequently with stakeholders along the way. This process should ideally begin with conducting contextual research. When developing a diagnostics device for example, it can be useful to visit labs, GP surgeries, hospitals and pharmacies, ideally in different locations. This ensures that regional differences are captured and insights gathered from a sample of relevant stakeholders, which may include patients, carers, HCPs, buyers and regulators.
User Journey Mapping
Once a contextual understanding of the user and environment of use has been developed, a useful next step is creating a user journey map. The aim is to map the different use steps and highlight pain points in a user’s journey – the areas of friction where improvements can be made. If there are multiple stakeholders involved, this is also where device manufacturers can identify conflicting requirements for the device.
From here, key design challenges can be identified and prioritised, which can then be expressed as “how might we?” statements, such as “how might we reduce the number of use steps required to set up a test?”. These insights can then be used to inform the concept generation phase.
Concept Generation
Getting something into the hands of the target users early and often is essential to allow effective testing and iteration of device
Health Outcomes
expectations and their comprehension of the system. This allows design decisions to be based on first-hand evidence.
The Design Ecosystem
Sometimes a complicated challenge can’t be solved through device design alone and requires looking at the packaging and information design, digital design and even service design to achieve a desired outcome.

designs. This can range from simple foam handling models to functional demonstrators. Creating something physical is a valuable way of identifying potential usability issues and design flaws that are often difficult to foresee. This may involve simple ergonomic issues such as how a user naturally holds the device, or functional problems involving the position of different features. By creating quick and dirty prototypes, device manufacturers can work through a cycle of prototyping, testing and iterating the design, until converging on a few suitable options to take forward.
Human Factors Studies
As the development team begins to converge on a suitable design, it is a good idea to start conducting more formal human factors observational studies. This provides the opportunity to explore specific aspects of the device’s use in more detail, to understand the root causes of any issues. It is again important to consider these usability challenges early on in the project, before beginning the physical development of the device. This can be particularly useful for understanding how a user interacts with a digital user interface (UI). For example, there are a number of UI prototyping tools available for smartphones and tablets that allow development teams to quickly and cheaply prototype a workflow for a user study. Recording these interactions allows a review of what the user focuses on at different moments of time, if they are experiencing an issue.
In medical device development, the process of learning from target users is often continuous and should occur throughout the development. The above steps should come together to provide a rich picture of user
One of the common challenges with designing medical devices is how to deal with the existing mental models of a user. This might range from perceived ways to administer an injection dose, to applying a sample off-instrument for a diagnostic device. To tackle any preconceived mental models, designers can utilise the various parts of the design ecosystem to nudge and guide the user towards a desired behaviour, from the packaging and instructions for use, to the design of the device itself.
Conclusion
As the industry continues to move towards providing more healthcare outside of the hospital, it is essential to ensure that the medical devices needed to enable this are safe, effective and easy to use. Achieving this requires device manufacturers to build a strong understanding of their users, including their challenges, motivations and unmet needs. In doing so, they can continue to create medical devices that empower users to take on tasks previously limited to trained HCPs.
Alex Driver, Head of Industrial Design, Team Consulting, has over 15 years' experience in consultancy and design research. His consultancy work has covered a range of consumer products and medical devices, whilst his spell as a researcher involved collaboration with scientists at the University of Cambridge on the development of early-stage science and technology. In his role at Team Consulting, he works across a variety of medical device projects, from immersion and concept generation through to detailed design and development.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 75 www.international-pharma.com
Alex Driver
Nanoformulation for Enhanced Drug Delivery and Better Patient Compliance
The increasing occurrence of highly potent or frequent-administration therapies in the drug development pipeline means that the low solubility of drugs is becoming a potential bottleneck for pharmaceutical formulators. This not only makes getting new medicines to market more time-consuming, it also hampers the lives of patients in need. In this case study, Losan Pharma – a contract development and manufacturing organization (CDMO) focusing on improved active pharmaceutical ingredient (API) performance –and BENEO, a multifunctional excipient supplier, pool their collective resources. In doing so, they managed to develop a convenient-to-take once-a-day oral solid dosage form prototype of Dexamethasone, an API that’s practically insoluble in water.
One of the most challenging issues to overcome in terms of getting a new chemical entity (NCE) to market, or reaching its full potential, is solubility. Approximately 60% of potential new products exhibit greater or lesser solubility problems, with some industry experts suggesting that the proportion of poorly soluble drugs in the current pipeline is as high as 90%.1,2 Not only do APIs with solubility issues require more complex preclinical studies and clinical trials, they also present formulators with considerable technical challenges.3 The search for appropriate technologies and formulation methods to enhance aqueous solubility, permeability and dissolution rate can be problematic; plus, poorly watersoluble drugs often need high doses to reach therapeutic plasma concentrations after oral administration.4 It perhaps comes as no surprise, therefore, that improving drug solubility and, consequently, bioavailability, remains crucial for the entire pharma sector.

Wanted: An Easy-to-use Glucocorticoid

The demand for pharmaceutical substances with improved properties continues to drive progress in the field of outsourced product development. Today, many pharma companies are turning to external specialists that offer formulation know-how, appropriate
technologies and available capacity – key requirements to reduce both research and development (R&D) costs and time to market. To cite a recent example, two Europebased experts in formulation development – Losan Pharma and BENEO – joined forces to design a novel administration format for the poorly soluble glucocorticoid medication: dexamethasone (with Dexamethasonratiopharm® 4 mg as a reference listed drug). To reach this goal, Losan Pharma applied its long-term expertise in the development of nanoformulations with improved API performance. BENEO supplied the carrier, which needed to both convert the API suspension into a dry format and bring the chosen administration format to fruition; this was a convenient and easy-to-use stickpack containing biphasic release pellets (Figure 1).
oxygen support or mechanical ventilation.7 “In this project, dexamethasone served as a model drug because it is practically insoluble in water,” said Oliver Luhn, Head of Pharmaceutical Technology at BENEO. Generally, the solubility of an API is a critical factor for drug formulators as, first and foremost, it determines the overall efficacy of the drug. If the API is only partly or not fully dissolved in the gastrointestinal fluids at the site of absorption, the drug can’t properly reach the systemic circulatory pathway and evoke the required pharmacological response.4
Oliver Luhn continues: “When administered in tablets, dexamethasone needs to be taken at a high frequency during the day for most indications. This means
From API to administration: optimizing drug delivery Dexamethasone is a synthetic corticosteroid with antiinflammatory and immunosuppressive properties. It is primarily used to treat various inflammatory and autoimmune conditions, such as allergies, asthma, rheumatoid arthritis and inflammatory bowel disease.5 According to the National Cancer Institute (USA), dexamethasone is also approved in combination with other drugs to treat certain types of cancer, including leukemia, lymphoma and/or multiple myeloma.(6) In recent times, dexamethasone has gained attention for its potential to reduce mortality in hospitalized COVID-19 patients requiring
that patients need to adhere to a special dosage regime, which can pose a considerable challeng to them. Thus, we were tasked by Losan Pharma to deliver a carrier system that facilitated the development of an alternative oral dosage form with increased API bioavailability and minimized the need for repeated administration throughout the day.”
The developers opted to use stickpacks with dual-release pellets: the overall formulation contained both immediate release (IR) and delayed release (DR) pellets. Although the API released from the IR component reaches the bloodstream
76 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Application Note
Figure 1: A stickpack with biphasic release pellets
quickly after administration, the drug in the DR portion maintains the effective therapeutic dose for a prolonged period of time. This biphasic drug delivery system allows the frequency of dosing to be reduced and contributes to the long-term therapy of chronic disease conditions by maintaining the patient’s drug plasma concentration within its therapeutic range.
(8) Matthias Rischer, Director Drug Delivery & Innovation Projects at Losan Pharma, comments: “We see stickpack technology as a way to facilitate oral intake and increase both application convenience and patient compliance, especially for elderly people and for frequently dosed formulations. Last but not least, drugs in a stickpack provide an easy-to-use administration format as they can be swallowed without water.”
Fast-track Development of an API Nanoformulation
The drug formulation process centered on establishing a stable API nanosuspension. Pharmaceutical nanosuspensions are defined as submicron colloidal dispersions of nanosized drug particles stabilized by surfactants and polymers.9 Generally, creating nanosuspensions offers a way to enhance the solubility of “brick dust” APIs. Reducing the particle size to nanoscale significantly increases their surface area, which results in a much higher dissolution rate.3 This may subsequently contribute to improved bioavailability.
To develop the dexamethasone nanosuspension, the researchers turned to rapid API wet ball milling in a dual centrifuge (ZentriMix 380R) (Figure 2). Unlike common


separation techniques, dual centrifugation (DC) induces additional rotation of the samples during the spinning process. This causes the strong centrifugal forces inside the vials to change direction continuously; combined with the in situ milling beads, this powerful movement results in very effective milling of the samples and excellent homogenization.10 Additionally, developing nanosuspensions of poorly soluble APIs with DC can be very time saving. Of note, the development of a suitable nanosuspension of a poorly soluble drug is often time consuming as a high number of different combinations/ratios of polymers and surfactants have to be tested for every drug compound by milling; in addition, the particle size distribution and the stability of the agglomerates of each combination have to be measured.10
However, by being able to test 40 small-batch (1 g) samples per DC run, the developers were able to rapidly screen for suitable formulation variables (copolymers and surfactants). Incorporating suitable stabilizers into the nanosuspension helps to prevent particle aggregation and Ostwald ripening (redeposition of small particles onto larger crystals) as they create a protective layer around the particles. This reduces their tendency to grow or coalesce. The formulation screening process also included the particle size distribution assessment of the samples by laser diffraction. Afterwards, trial formulations were fine-tuned and scaled-up (2 to 10 g). With the Zeta potential (a measure of the magnitude of the electrostatic or charge repulsion/attraction between particles) in
mind, the appearance and bulk stability of selected nanosuspensions were tested after 2 weeks of storage at 2–8 °C.

Moving Towards a Marketable Product
The next step was transferring the resulting nanosuspensions into a dry state to prevent Ostwald ripening.3 This was done by fluid bed layering the nanosuspension onto a carrier system. During this process, carrier particles are fluidized, applied (sprayed on) with a coating fluid (active layer of nanoparticles) and then dried. Small droplets and a low viscosity spray medium are crucial to ensure an even product coating. These “dry” suspensions can then be used to produce solid dosage forms such as tablets, capsules or stickpacks with granulates or pellets.
At this drug development stage, special attention was paid to choosing the most suitable base material. It was crucial for the starter particles to experience as little abrasion in the fluid bed as possible – but to benefit from excellent flowability and ensure smooth processing. The developers tested various different carriers during the trials and finally opted for a sieved grade of BENEO’s isomalt with a bulk density of 820 g/L and a flowability of 27 s/100 g (orifice d = 6 mm) (Figure 3).
Critical to this decision was isomalt’s ability to enhance the nanoformulation process. Oliver Luhn from BENEO explains: “As well as good abrasion resistance, the carrier had to be almost instantly soluble in aqueous media. This is key to ensure rapid dissolution of the drug and fast release of the nanoparticles. Also, a highly soluble drug core facilitates particle size measurement as there is no interference with non-soluble carrier particles. Not only is the chosen grade of sieved isomalt suitable for fast formulation screening according to the particle size distribution, it also minimizes electrostatic charging, which can lead to segregation or demixing of the components.”
INTERNATIONAL PHARMACEUTICAL INDUSTRY 77 www.international-pharma.com Application Note
Figure 2: Dual centrifugation was used to expedite the development of an API nanoformulation
Figure 3: Scanning electron microscope image of BENEO’s carrier particles (magnification x 50)
Table 1: IR pellet formulation
The nanosuspension preparation (batch size 40 g, nanobeads 0.3 mm, 2000 rpm, 90 min, 0°C) was coated onto isomalt particles using the MiniGlatt Micro-Kit with topspray (batch size = 32 g) to prepare the immediate release pellets (Table I). To form the delayed release (DR) components, pellets were covered with a preparation
of Eudragit L100 and S100 (3:1) (Table II). This enteric coating allows the pellets to pass through the stomach and reach the intestine before dissolution begins, thus delaying API release. Finally, IR and DR granules were filled in a 1:1 ratio into a stickpack using a volumetric slider system with two dosing chambers.
Conclusion
During the subsequent in vitro dissolution tests, the IR granules demonstrated complete release of the active ingredient within 10 min (dissolution USP II, 50 rpm, 37 °C, 0.1 M HCI). The modeled dissolution of DR granules showed that release of the second fraction would start about an hour postintake with complete dissolution within the next 2 hours (USP buffer pH 6.8) (Figure 4). Therefore, the developers concluded that the 1:1 filling ratio delivered the desired release profile — immediate release directly after administration and delayed release of the second fraction. Additionally, the obtained results showed no leaking of the modified release pellets. Particle size measurements revealed that the API was maintained in nanoscale during in vitro dissolution and was confirmed by a short-term stability study.
Wolfgang Mohr, Head of R&D at Losan Pharma, says: “We are pleased to note that we were able to achieve a stable nanosuspension of dexamethasone with improved bioavailability using a streamlined production process. The applied nanotechnology offers the advantage of fast set-up and easy integration into a conventional manufacturing line. In addition, using multiparticulate formulations in stickpacks makes a wide range of applications available owing to the different target release profiles (modified, sustained, dual release) that can be achieved by simply changing the filling ratio in the stickpack.”
Oliver Luhn from BENEO adds: “With a carrier system that facilitates the development of dosage formats with both immediate and delayed release of the API, it was possible to design a medicinal product that offers extended delivery of the active ingredient in a once-a-day format. Granules in stickpacks, which can be taken without water, lead to improved compliance, better treatment outcomes and higher patient satisfaction. By diversifying their product portfolio with innovative delivery formats, pharma manufacturers can better cater to different patient needs and gain a competitive edge in the market. For more information about dualrelease pellets and the R&D insights, you can watch our webinar, to be broadcasted on 12 October 2023 (registration via QR Code).
Table
DR
formulation

78 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Application Note Winter
2:
pellet
Dexamethasone [mg/SD] 10.00 Kollidon VA64 fine [mg/SD] 3.00 SDS [mg/SD] 0.50 Purified water [mg/SD] 86.50 Total [mg/SD] 13.50 Nano suspension [mg/SD] 13.50 Isomalt starter pellet galenIQ 960 [mg/SD] 197.00 Kollidon VA64 fine [mg/SD] 3.0 Purified water nano suspension [mg/SD] 86.50 Purified water [mg/SD] 28.00 Total [mg/SD] 213.50 Dexamethasone [mg/SD] 10.00 Kollidon VA64 fine [mg/SD] 3.00 SDS [mg/SD] 0.50 Purified water [mg/SD] 86.50 Total [mg/SD] 13.50 Nano suspension [mg/SD] 13.50 Isomalt starter pellet galenIQ 960 [mg/SD] 197.00 Kollidon VA64 fine [mg/SD] 3.0 Purified water nano suspension [mg/SD] 86.50 Purified water [mg/SD] 28.00 Total [mg/SD] 213.50 API Pellets [mg/SD] 213.50 Eudragit L100 [mg/SD] 58.83 Eudragit S 100 [mg/SD] 19.61 Triethyl citrate [mg/SD] 7.79 Talc [mg/SD] 20.52 Purified water [mg/SD] 45.56 Isopropanol [mg/SD] 549.59 Total [mg/SD] 320.25 Weight gain [%] 50
REFERENCES
1. Swati, S., George, M. & Lincy, J., Improvement in solubility of poor water-soluble drugs by solid dispersion, International Journal of Pharmaceutical Investigation, 2(1):7-12, (2012)
2. https://www.pharmaceutical-technology.com/ comment/cphi-experts-90-current-pipelineapis-poorly-soluble, visited on 14 Apr 2023

3. Hagedorn, M. et al, Rapid development of API nano-formulations from screening to production combining dual centrifugation and wet agitator bead milling, International Journal of Pharmaceutics, Vol. 565,187-198 (2019)
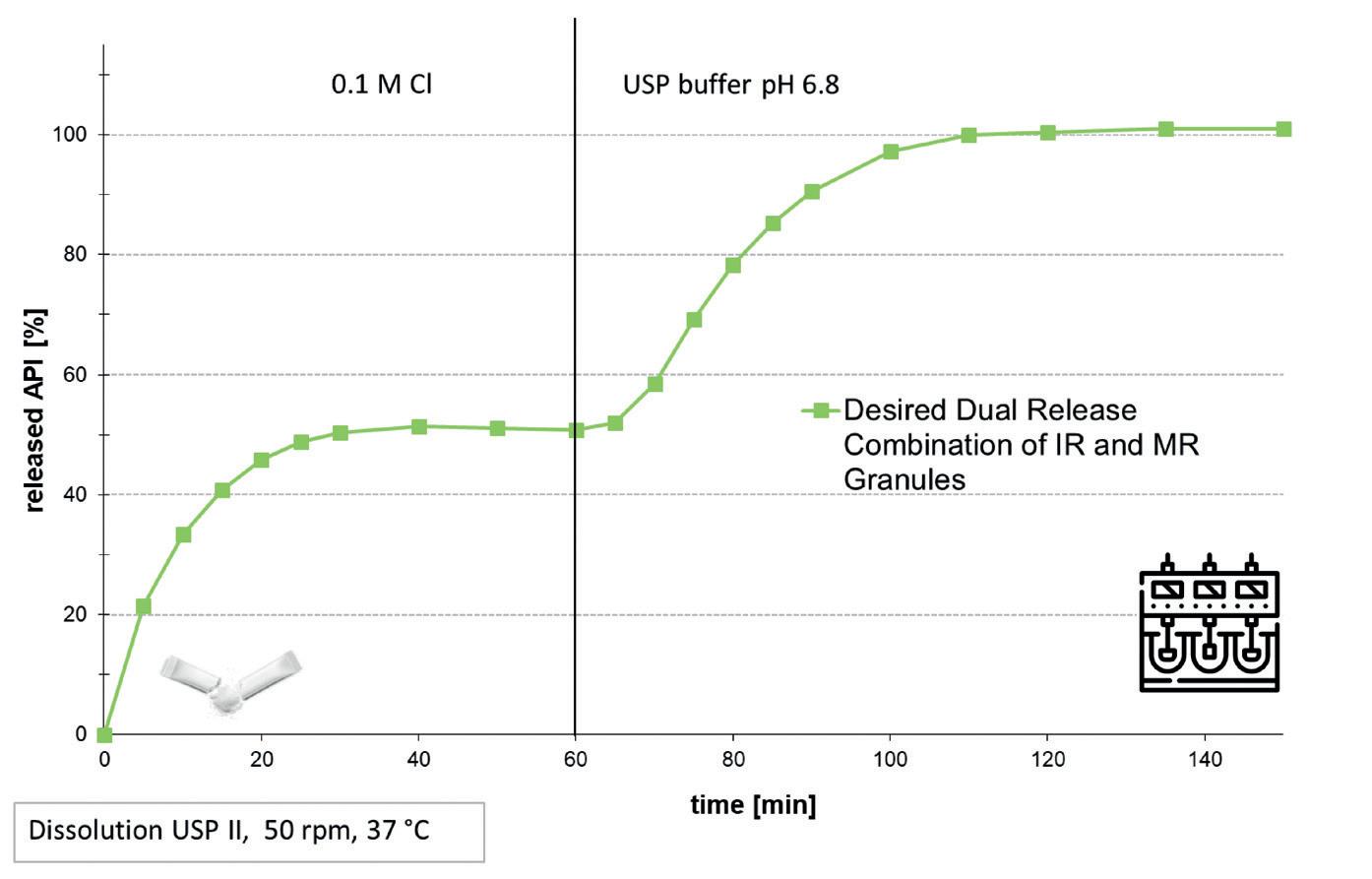
Application
Note
4. Savjani, K.T., Gajjar, A.K. & Savjani J.K., Drug Solubility: Importance and Enhancement Techniques, ISRN Pharmaceutics, Vol. 2012, Art. ID 195727
5. Madamsetty, V.S. et al, Dexamethasone: Insights into Pharmacological Aspects, Therapeutic Mechanisms, and Delivery Systems, ACS Biomater Sci Eng, 8(5):1763-1790, (2022)
6. https://www.cancer.gov/about-cancer/treatment/ drugs/dexamethasone, visited on 14 Apr 2023
7. Sobia, N., Irsah, M. & Asadullah, M., Dexamethasone: Therapeutic potential, risks, and future projection during COVID-19 pandemic, Eur J Pharmacol. 2021 Mar 5; 894, (2021)
8. Hamed, R. & Omran, H., Development of dual–release pellets of the non-steroidal anti–inflammatory drug celecoxib, Journal of Drug Delivery Science and Technology, Vol. 55, February 2020, 101419, (2020)
9. Aher, S.S., Malsane, S.T. & Saudagar, R.B., Nanosuspension: an overview, International Journal of Current Pharmaceutical Research, Vol 9, Issue 3, 19-23, (2017)
10. Hagedorn, M. et al, Dual centrifugation – A new technique for nanomilling of poorly soluble drugs and formulation screening by an DoE-approach, International Journal of Pharmaceutics, Vol. 530, Issues 1–2,79-88 (2017)
Dr. Maj-Britt Cepok
Dr. Maj-Britt Cepok is a Food Chemistry graduate and holds a PhD in Analytical Chemistry. She has experience in both the food and pharmaceutical industries, and her particular specialties are the pharmaceutical development of solid dosage forms and taste profiling. Dr. Maj-Britt Cepok joined BENEO in 2005 and has held a variety of roles in global sales, technical services, product management and business development for the Pharma business unit of the company.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 79 www.international-pharma.com
-
Figure 4: Dissolution of the IR and DR pellets
Through the Lens of Human Factors: Developing
Connected Drug Delivery Devices
Designing Devices for Self-management
The market for drug delivery devices continues to grow at a rapid rate,1 largely due to an expanding elderly population with an increasing frequency of chronic diseases. Biotherapeutics have played a significant part in this growth, with biologics frequently administered via subcutaneous injection. As a growing number of biosimilars come onto the market, it is likely the market for subcutaneous drug delivery devices will develop further.
More than ever before, chronic conditions are being treated outside of a hospital setting, with drugs administered by carers and patients themselves, without advanced medical training. As greater agency is placed on drug delivery devices, reliable and intuitive design is essential alongside ensuring safe and effective treatments, while maximising patient comfort and convenience.
Increasing Device Functionality with Connectivity
The growing trend of adding connected functionality to these devices allows the medical profession to simplify the capture, transfer and display of injection data; automatic upload of key treatment data can be crucial in tracking and improving patient adherence to treatments. Meanwhile, introducing additional electronic features to the device itself provides an opportunity to offer real time step-by-step feedback to the patient during the injection procedure. In addition, educational tools and instructions provided by digital content in the associated app can help to simplify the injection process.
As with any system, the temptation to increase the functionality of a medical device comes with potential to increase the complexity of the user interface and impact ease of use. A user-centred design approach is desirable to optimise the user experience and associated benefits, and essential to ensure that the potential for use error is managed.
This article will look at the importance of applying human factors engineering (HFE)
during the development of new connected drug delivery devices and how we employed these principles when creating our first connected auto-injector.
Human Factors and Usability Engineering Requirements
In the EU, manufacturers must demonstrate compliance with requirements set out in the Medical Device Regulation (MDR). These requirements align with the international standard for IEC-62366-1:2007 (note it is recommended that HFE is conducted according to the 2015 update), with IEC-623662 (2016) as a guide. Meanwhile, in the US, the Food Drug Administration (FDA) provides recommended human factors engineering guidance for both medical devices and combination products, and more recently has provided further draft guidance on the inclusion of human factors and usability engineering as part of the market approval process. Therefore, most developers will incorporate usability engineering as a matter of protocol.
Although some differences exist between the various sources above as well as guidance drafted in other territories, the general principles are the same: understanding users and use, and use environments, generating user interface requirements, conducting a use-related risk analysis, and evaluating the product through formative and human factors validation tests.
Human Factors Engineering in Practice
In the US, the FDA identifies three key components that underpins human factors engineering and usability engineering: intended users, intended use environment and device user interface.2 Typical profiles of intended users are built up by examining characteristics such as physical strength, dexterity, sensory abilities, literacy and the ability to learn how to operate a new device. Use environments consider factors such as ambient lighting and noise levels and possible distractions to the user that could impact the injection process. Lastly the device user interface considers all elements of the device with which the user interacts – those parts that the user can see, hear and touch. A good integrated human factors process ensures a focus on these three elements is
sustained throughout medical device design and development.
Preliminary Analysis
Although formative evaluations are fundamental to many human factors initiatives, there are some key preliminary activities that should precede any user testing in order to optimise the interface and lay the foundation for successful user testing plans and outcomes. Preliminary activities are geared towards getting the best understanding of the intended users and their strengths and limitations, the intended context of use, and intended use scenarios. Early user research around these topics also seeks to understand the known use issues with similar on market devices. As early concepts and ideation start to formulate, these activities help to generate a picture of use related tasks, and inform the first iteration of a use related risk analysis (URRA).
Use Related Risk Analysis
A central pillar of any human factors programme is the identification of use related risk and its mitigation. The relationship between the human factors process and Risk Management Standard 14971 is perhaps best illustrated in IEC 62366; Human Factors activities are designed to systematically flush out potential use error, by examining the interface from different viewpoints (users), under different conditions (e.g., emergency, maintenance, low battery), in different environments (e.g., military, roadside, clinical).
Formative User Testing
The human factors testing process requires both formative and summative studies to evaluate the intended use of a product, test the device in anticipated use environments and demonstrate its effectiveness for different user groups. The group of test subjects must therefore represent the full range of potential device users, incorporating people of relevant ages, gender, training levels, reading ages, physical and sensory impairments, and previous device experiences.
In formative tests, products are placed into the hands of users early in the development cycle, deliberately testing
80 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Logistics & Supply Chain
Management
how patients and carers utilise devices in real-world situations. The results of that testing enable exploration of issues from needle phobia to dexterity challenges, to identify potential problems with the size of visual displays, dosage settings and alerts and to address these issues, before releasing the product to market. Finally, summative tests will evaluate the device that is going to be launched to market – validating its effectiveness, usability and safety.
When developing our own connected product, we prepared for the worst-case injection scenario. In practice, patients introduced to a drug delivery device would most likely initially be instructed directly by healthcare workers on how to use it. However, our study placed our auto-injector in front of patients with no training and without directions to read the instructions provided before use.

Human Factors for Connected Devices
In many ways the human factors programme for a connected device is no different to any other product. We began early with a preliminary analysis, including a contextual enquiry to establish an understanding of users interacting with existing on market products with and without electronic components and or connectivity.
The platform device interface was intended for a wide audience, so a broad use specification was established and used
to inform and shape the ergonomics to ensure ease of use across multiple patient demographics. The psychology of the user interface design had a big impact on key decisions around the Bluetooth® connection process, placement of Bluetooth button, and power management. But designers were also clear that any added functionality should not in any way hinder safe and effective injection procedures.
The human factors team also conducted studies on the digital interface of devices once an acceptable level of reliability had been reached. A landscape-style generic IFU was also generated to clearly demonstrate intended user steps, and the team also experimented with different colours on the key touch points to guide loading and unloading of the syringe into the device. The smartphone application to go alongside the device was quickly generated and simulated in Adobe XD before writing the software, whereas the device components and electronics were developed at a slower pace, necessitating their testing at different points. With differing speeds of development resulting from various device components, our team was compelled to mimic connectivity in the app during studies to utilise testing time effectively.
Learnings from testing reinforced our initial commitment to creating a demonstrator app, a supporting element to promote safe and effective injection practices.
Importance of Human Factors in Future Developments
The integration of a human factors approach into the development and testing of connected drug devices is critical to create devices with a user-centric design. When users are prioritised from the very beginning of development, it helps to ensure that devices are intuitive and have considered the needs of different potential user groups.
Manufacturers wishing to seize a share of the expanding drug delivery device market must employ human factors approach to ensure products are suitable for patients in a new era for healthcare.
REFERENCES
1. Precedence Research. (2023). Drug Delivery Devices Market (By Route of Administration: Oral, Ocular, Inhalation, Nasal, Injectable, , Topical, and Others; By Application: Cardiovascular, Diabetes, Cancer, Infectious Diseases, and Others; By End Users: Hospitals, Ambulatory Services, and Home Healthcare) - Global Industry Analysis, Size, Share, Growth, Trends, Regional Outlook, and Forecast 2023 – 2032. https://www.precedenceresearch.com/drugdelivery-devices-market
2. FDA. (2017). Human Factors Considerations. https://www.fda.gov/medical-devices/humanfactors-and-medical-devices/human-factorsconsiderations
Finola Austin
A highly experienced Human Factors Engineering Manager, Finola Austin boasts 20 years’ experience in mentorship and management of human factors services in safety-critical industries. Her career began in Occupational Therapy within acute, long term and community settings, and her training in accessibility has given her special insight into the needs of impaired users. Since then, Finola has successfully planned and delivered Human Factors activities for hundreds of handheld medical devices, including auto injectors, emergency use devices, inhalers, injection pens, and lancets, and is proficient in the generation and review of documentation. She has executed numerous user evaluation studies in the UK and USA – including studies on safety engineered devices, injection pens, and colour differentiation.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 81 www.international-pharma.com Logistics & Supply Chain
Management
Pharmaceutical Cold Chain Risks and How to Mitigate Them
Shipping temperature sensitive pharmaceuticals involves a range of risks that can impact the quality, safety, and availability of live saving treatments for patients. Nick Gilmore, Global Head of Sales and Marketing of Tower Cold Chain, explores how risk can be mitigated through strategic partnerships with specialist temperature-controlled packaging suppliers.
The pharmaceutical industry is growing at a rapid rate, and as the most current data from Statistica has demonstrated, it has now surpassed the $1.48 trillion mark in revenue. With such growth comes an increased level of complexity in the pharmaceutical supply chain, and thus, the reason why implementing best practices for managing risk is crucial.
Securing the integrity and quality of medical and biological products during transportation stands as a pivotal challenge for both pharmaceutical manufacturers and logistics providers. With many biopharmaceuticals requiring strict temperature control from manufacturer to patient, precise handling and storage is essential. The potential loss of cargo poses a significant risk to patient health and wellness. Coupled with increasing regulatory requirements whilst serving an international market, players need to implement robust quality assurance practices to mitigate these risks and ensure products are transported reliably across the supply chain.
Reliable Shipping
Timely delivery, intact condition, security and a stable temperature – these are the fundamentals required for successful pharmaceutical shipping, and it’s here that the choice of temperature-controlled solution and supplier makes all the difference.
Manufacturers require a container robust enough to safeguard its contents, and eliminate breakage, throughout the various pressures faced during international freight. Such container needs to effectively regulate temperature, remove manual intervention, and prevent excursions that
might compromise the efficacy of the cargo. And finally, an optimal packaging-to-payload ratio is required, adhering to transport safety regulations.
Yet fundamentally, every solution available on the market does the same thing. The use of temperature-controlled containers ensures palletised delivery of pharmaceutical shipments, in a way that keeps the product safe from physical damage and temperature excursions. Yet the distinction lies between the suppliers, in terms of how well-equipped they are to mitigating risk.

Increasingly, pharmaceutical manufacturers are partnering with cold chain shipping providers with risk-first mindsets. Knowledge of the nature of the supply chain’s risks is the first step in establishing supply-chain resilience.
Cold Chain Weak Spots
Transfer points where multiple stakeholders are involved in the handover and stopover elevates the chance of damage and/or temperature excursion. This often occurs during the unloading and loading process, through either improper handling or through delayed shipments sitting on tarmac or storage depots.
Further to this, the distribution of vaccines during the COVID-19 pandemic
brought forth a number of issues with the global cold chain, not least the fact that many developing nations are not as prepared to receive and transport temperature-sensitive products. Airports often have less capacity or lack of on-site temperature-controlled facilities. There is also often limited knowledge on the ground, which can lead to serious errors leading to spoiled shipments, pilferage, or mishandled packaging.
Beyond major transport hubs, the issue of delivering vaccines, medicines and other temperature-sensitive payloads to rural areas in developing nations is a challenge. Road infrastructure is often poorly maintained, and in many places not passable for many delivery vehicles.
And, as we see more extreme weather conditions due to climate change, with natural disasters and power cuts becoming more prominent, the focus on how packaging performs will intensify.
Delays in customs clearance can be another reason for deviations and the damage caused to temperature sensitive goods. The import of pharmaceutical products often requires various permits and extensive documentation, the incompleteness of which can quickly lead to queries and delays, which in turn pose a risk to the goods.
82 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Logistics & Supply Chain
Management
However, the right combination of suitable packaging and a specialist cold chain shipping partner can significantly reduce the risk of weak points in the cold chain. Passive containers offering both internal temperature control and robust external protection can provide up to 5 days’ worth of contingency and are built to be fully reusable, withstanding 10+ years of freighting. Providing peace of mind in cases of disruption, whether this be diversions, breakdown or even labour strikes, passive containers consist of materials intended to keep the internal contents of the package within a specific temperature range for a defined period of transport without any means of manual intervention or power source.
In comparison, active containers use mechanical and electric systems powered by an energy source, combined with thermostatic control in order to maintain proper product temperatures.

From the pharmaceutical brand’s viewpoint, the critical issue is reliability, and some understandably take comfort in having a powered solution. Yet, as advances in passive technology have progressed, and we continue to face supply chain challenges, passive containers are quickly becoming a sensible choice to mitigate risk in the industry.
Regulatory Compliance
Pharmaceuticals are a highly regulated
industry for a good reason: safety, effectiveness, and the wider impact medicines can have on patient’s lives. The ramifications of not complying with regulatory affairs can profoundly impact a company’s reputation, finances, and customers, and can cause significant legal implications and criminal consequences. Thus, compliance is a top priority for manufacturers.
Due to the risk of non-compliance, players in the pharmaceutical cold chain should have a sound understanding of the varying regulatory bodies that govern them and be aware of new guidance and how it applies to them.
To build up a successful and sustainable pharma-distribution operation, the nuances and key characteristics per country and region need to be handled bi-laterally. Logistic providers must provide the agility and flexibility demanded of them, offering a global network for localised deliveries – all whilst complying with each country’s regulations. Investment in Good Distribution Practice (GDP) training for local teams, enables organisations to tailor to the needs of specific regional markets guaranteeing compliance with all pharmaceutical regulations within all market geographies.
And, as sustainable supply chain management comes to the fore, the pharmaceutical industry is under pressure to
optimise environmental performance, whilst still meeting stringent safety standards. The implementation of environmental, social and corporate governance (ESG), combined with increasing consumer struggles with single-use plastics, has led pharmaceutical companies to favour sustainability focused packaging and transport partners, which can not only provide robust and reliable solutions, but ones that are fully reusable too.
Quality Control
As more and more pressure is put on global cold chains, the higher the stakes become for those responsible for delivering safe and secure logistics networks and cold chain processes.
The risks of cross contamination and mishandling can lead to physical damage, contamination, or tampering of pharmaceutical products which ultimately compromises product quality and safety. Cold chain container providers safeguard against temperature fluctuations, assuring the stability of temperature-sensitive drug, with tracking and tamper-evident features to deter any form of unauthorised access or damage.
And whilst training and investment in infrastructure are key, a move to more automated processes and the deployment of technology will ultimately reduce risk from human error in cold chain logistics. Implementation of tracking tools such as internal data loggers provide peace of mind
INTERNATIONAL PHARMACEUTICAL INDUSTRY 83 www.international-pharma.com Logistics & Supply Chain
Management
Logistics & Supply Chain Management
by monitoring the internal temperature of the shipment and can notify users in the rare case of a temperature excursion, as well as providing automatic data downloads throughout the transit. By using Bluetooth Low Energy Technology, each logger communicates wirelessly, sending accurate data to the cloud. This digital innovation allows for accurate compliance checks and on-delivery sign-off, empowering operations with clear, transparent information no matter the mode of transport used.
Companies who invest in digitalisation are likely to see the clear benefits of time, cost and resource savings in addition to customer satisfaction and most importantly, patient safety.
Experts in Risk Mitigation
Evidentially, the potential pitfalls in the pharmaceutical cold chain are multifarious. Containers and their contents can suffer damage during cargo handling. Pharmaceuticals may endure temperature excursions due to extreme weather, improper storage or training, that undermine their efficacy. The involvement of numerous stakeholders further complicates the management of these factors, intensifying the associated risks. For instance, research by the Institute for Human Data Science found that approximately $35 billion is lost annually to failures in temperature-controlled logistics.
Certainly, this is a scenario nobody desires and so it is vital pharmaceutical companies
implement robust quality assurance practices. Partnering with experts in cold chain shipping is a sure way to mitigate risk, helping businesses adhere to regulatory guidelines, use validated packaging and shipping methods and ultimately, ensure that pharmaceutical products are delivered to patients, safely and securely.
In short, by looking carefully at the options available on the market, you can find the container and dedicated supplier that meets all of your needs and simultaneously helps mitigate risk in the pharmaceutical cold chain.


Nick Gilmore has 24 years sales and marketing experience in international public and private equity owned companies. He is an energetic business leader working cross-functionally and with boards to deliver winning commercial strategies delivering long term, sustainable growth for multi-site manufacturing facilities supplying global customers.

84 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Nick Gilmore
PROTECT YOUR PHARMA PRODUCT WITH OUR QUALIFIED REFRIGERATED AND DEEP FREEZER CONTAINERS.
REDUNDANT REFRIGERATED & -70°C DEEP FREEZER CONTAINERS FOR PHARMA PRODUCTS
Klinge Corporation has been safely transporting and storing pharmaceuticals for nearly 40 years.
Avoid Temperature Excursions with Redundant Reefer/ Freezer Containers


Keep pharma products at the required temperature from +25°C down to -70°C
Meet international transport regulations
Safeguard pharma products with temperature recorder & online temperature tracking

inquiry@klingecorp.com


www.klingecorp.com
USA: + 1-717-840-4500
Putting Safety at the Heart of Supplier Controls
Marketing authorisation holders’ responsibility for ensuring the continuous monitoring of the safety of a medicinal product across the marketing authorisation lifecycle includes accountability for all third party suppliers with a potential impact on that safety profile. Arriello’s COO, Anna Lukyanova, provides practical tips for maintaining a robust safety profile through tighter controls right along the supply chain.
The global Life Sciences supply chain has experienced unprecedented turmoil recently, creating a challenge when it comes to ensuring end-to-end quality and safety. Suppliers could include anyone from local distributors or qualified persons to IT system partners, security providers and even auditors themselves. Increasing changes to suppliers and service providers affect the vendor records and controls companies must maintain to ensure end-to-end quality and safety.
If control issues are exposed during inspections, that could lead to fines, or even product withdrawal from affected markets. Scenarios potentially triggering an inspection could include a distributor failing to flag an effective product recall in a particular market; a local partner failing to implement additional risk minimisation measures (aRMMs); or a local qualified person being unreachable by the relevant authority due to out-of-date contact details or the vendor going into liquidation.
Regulators are expected to issue firmer guidance on vendor management controls sooner rather than later. In the meantime, robust vendor management is an expectation under EU GxP requirements. So, if drug developers or licence holders fall short, they could risk their reputations as well as significant fines and ultimately product withdrawals.
Start by Creating a Definitive List
To address this issue, it’s important to start by identifying ALL suppliers with a potential bearing on a products’ safety profile, however
tenuous. Once a definitive list has been compiled, each supplier can be reviewed for potential risk/safety impact and appropriate due diligence. (Ideally, such factors should form part of the risk-based evaluation conducted before entering into a contract with a new supplier or service provider.)
Suppliers range from sales and marketing companies which capture data and could come across adverse events (AEs); outsourced service providers – e.g. of data management; biostatistics; medical writing; pharmacovigilance (PV) service providers; IT providers that host the company’s safety database or ensure the safety data being stored; and independent PV or Clinical Safety contract auditors. Then each third party can be assessed for potential safety profile impact, the current supply/contract status and any monitoring, and required next steps.
Vendor and Quality Management Go Hand in Hand
A robust vendor management system (VMS) is every bit as important as a robust quality management system (QMS). Ultimately it should form an integral part of the QMS, and awareness, training and buy-in to vendor management practices should be formalised with appropriate communication and training.
Governance and the sharing of business decisions with the Safety/Pharmacovigilance Quality Assurance (PVQA) department prior to contract emerge as important, positive practices, too. Additionally, all contractual arrangements with PV agreements, clauses or a PV section should be reviewed by the Safety/PVQA department. MAHs should have all responsibilities clearly documented in an appropriate agreement, to avoid misunderstandings around relative responsibilities.
Tailor risk-based Questionnaires
It is key to assess the potential impact of any proposed new vendor on the safety profile based on the services it is going to provide. For instance, a third party that is providing services that are critical to the delivery of the safety profile, maintenance, evaluation of the product (e.g. CRO, PV service provider, medical information provider, outsourced clinical services) will require greater vigilance than
one that does not have a direct impact on the safety profile, maintenance or delivery of product and whose failure would not disturb clinical or PV operations.
Suppliers posing a 'significant' if not critical risk, in that their failure could disrupt the business, must be included too. Performing an audit or risk-based questionnaire on all critical and significant third parties is suggested best practice. A templated questionnaire for each service type means this can be sent swiftly to proposed new partners with a timeframe for delivery that minimises any impact on business timelines.
Although the level of initial due diligence required may be determined to an extent by per third- party type, each respective relationship will need to be reviewed in its own right to allow for other variances and influencing factors. A critical or significant lone contractor that uses the company’s internal quality system might be made exempt from this type of questionnaire where proof of training, CV, and certification may be deemed adequate, for instance.
Due diligence could take one or more forms, including: risk-based questionnaires based on the contractual obligations of the third party; Pre-qualifications audits; proof of qualification documentation; proof of training and proof of required registrations and relevant certification; and/or references, CVs and interviews.
Once the new partner has been classified, the associated risk level must be assessed through a process of due diligence. The objective here is to gather and review all relevant information to facilitate an informed, risk-based decision as to whether your organisation should enter into a contractual relationship. Performing an audit or risk-based questionnaire on all critical and significant third parties is suggested best practice.
Finally, a third-party score (TPS) is given. The final TPS derived from the due diligence process may identify the need for a prequalification audit where not otherwise conducted. While a risk-based questionnaire
86 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Logistics & Supply Chain Management
should not supersede the need for audit with critical and significant vendors, it may provide justification for the timeline of the scheduled audit.
Understand the Cut-off Point if Breaches are Untenable
Other potential risk factors may also need to be mitigated through evidential proof of compliance, or potentially via an audit. Only when a potential risk becomes a confirmed risk would a ‘for-cause’ audit be required, or if deemed appropriate, a review of the vendor themselves to consider the continuation of the contractual arrangement.
All contractual arrangements affecting the safety profile should contain a clause to ensure that, in the event of confirmed noncompliance with evidential proof, the contract can be ended. This is important. Although contract termination is a last resort, the option to end the relationship is paramount to ensure ongoing compliance if so needed.
It is a good idea to keep an eye on the audit programme and ensure it is updated to identify potential and emerging risks. Audit program updates should ensure that changes to the audit cycle are based on potential risk or confirmed risk (for-cause audits), so that all open, potential risks are reviewed during the audit – if not mitigated through other means prior to audit.
Risk-based questionnaires, previous audit findings, and logged potential risks can lead to a better more focused thirdparty audit and therefore more relevant/ detailed findings and better corrective and preventative actions (CAPAs).
Maintaining open, transparent communication with third-party vendors throughout the audit and due diligence program can help pinpoint potential inspection findings internally so that these can be avoided, averting regulatory-led penalties for noncompliance.

Eight Steps to Safety Along the Supply Chain
1. Advise everyone concerned about the impact of all kinds of third-party suppliers and service partners on safety.
2. Get the Safety team involved in vendor selection due diligence before the contract is signed.
3. Practise ongoing communication and open sharing of information – all
paramount to a good third-party relationship.
4. Review potential risk factors continuously, to ensure ongoing compliance.
5. Set out respective responsibilities clearly in contracts to ensure there is no misunderstanding of responsibilities around sharxing changes to information.
6. Make sure your chosen vendor management system, like a good QMS, reflects the company’s organisational complexity.
7. Audits are not the definitive solution to effective vendor monitoring and management. Don’t rely on audits to flag changes and issues. Foster good relationships with relevant third parties, and keep re-evaluating them.
8. Support buy-in to supply chain safety management from everyone – from business development and contract management, to marketing and beyond.
Ongoing Due Diligence
Identify and review any contractual changes made throughout the contract to ensure continued safety. Regular testing of contact details to ensure they are still valid and correct offers one simple way to uncover changes that may not have been identified otherwise through routine process.
From a post-marketing perspective, under GVP Module IV.B.1. (Pharmacovigilance audit and its objective) there must be ongoing due diligence and assessment, alongside an audit program based on risk. This should reflect evolving facxtors such as changes to legislation and guidance, or a major reorganisation/restructuring of the PV system (e.g. following a merger/ acquisition).
Ongoing due diligence should ideally include the maintenance of the initial due diligence documentation (e.g. request a
CV every two years, if certification requires renewal, request renewal); the reporting/ gathering of any changes that could impact the third- party risk profile score (TPS); sufficient tracking of potential risks and review of any potential or confirmed risks that change the TPS, and identify mitigations to ensure the TPS is kept within acceptable boundaries. Everyone in the organisation should be aware of the vendors in their ecosystem and should be clear on how to report any changes to safety/PVQA to investigate the potential risk of those changes.
Rigour and consistency are essential at a worldwide level, given the global nature of today’s businesses and their supply chains. As authorities globally are closing in on vendor management and working towards more formal requirements, all drug licence holders must take back control of their endto-end safety profile through robust vendor management.
Find the full Guide 'Vendor Selection & Management: Maintaining a robust safety profile through tighter supplier controls' here – https://arriello.com/guides-ebooks/ vendor-selection-management-maintaininga-robust-safety-profile-through-tightersupplier-controls/

INTERNATIONAL PHARMACEUTICAL INDUSTRY 87 www.international-pharma.com Logistics & Supply Chain Management
and solutions
of risk management and compliance services to the pharmaceutical industry. Email: anna.lukyanova@arriello.com
Anna Lukyanova Anna Lukyanova is COO of Arriello, a consultancy
provider
What Does the Future of Temperature-controlled Pharma Supply Chain Look Like and How Do We Shape It?
Importance of Airfreight for the Pharma Industry
Airfreight plays a vital role in the pharmaceutical industry, especially for economically export-oriented countries like Switzerland. Throughout the pandemic, air cargo actors specialized in the efficient transportation of highvalue and care-intensive shipments played a crucial part in ensuring the distribution of vital vaccines and medical supplies, including masks. This highlights the essential role of airfreight in times of necessity.
The transportation of these shipments requires careful consideration of several crucial factors, including service excellence, dependability, swiftness, security, and meticulous temperature regulation. Also, transporting these highly sensitive shipments with limited lifespans demands reliable services and clearly defined processes. Due to the products’ lifespan, it is very important to offer a stable and reliable network. This should be complemented by quick turnaround times, short acceptance times, and rapid execution of transfers and deliveries.
The Future of the Pharma Logistics Industry
What can we expect in terms of the future development of such shipments?
The pharma and healthcare transport business grows continuously, offering many opportunities as well as challenges. In addition, customer demands will continue to grow and get more complex in the future; as a result, all actors involved will need to ensure their preparedness to offer solutions that correspond to these changing dynamics.
For example, to address the present and expanding trends within the industry, all actors involved would need to actively engage in industry forums, conferences, and collaborations. This will ensure they remain informed about the most recent developments and play a role in shaping forthcoming standards and practices.
In the area of the temperaturecontrolled pharmaceutical supply chain we can expect significant advancements and transformations in the future, driven by (1) technological innovations, (2) regulatory changes, and industry demands regarding (3) sustainability, (4) resilience, (5) education & collaboration.
Below we will dive into these trends and factors that could shape the future of the pharma logistics sector, along with strategies to proactively shape this state.
1. Technological Advances
First of all, technological advances such as digitalisation are bringing changes with them:
A. Advanced Temperature Monitoring and Control
The supply chain landscape has evolved through the integration of IoT sensors for real-time monitoring of temperature and environmental conditions during storage and transportation. This technology, coupled with data analytics and AI, ensures continuous oversight and prompt intervention in case of temperature deviations, enhancing the reliability and visibility of the entire supply chain. This synergy of IoT and AI marks a significant advancement in supply chain management, minimising risks and optimising processes.
B. Blockchain and Digitalisation
Utilising blockchain technology ensures the establishment of a transparent supply chain record, maintaining data accuracy and regulatory compliance. The digitalisation of these supply chain processes further facilitates seamless tracking, traceability, and accountability throughout the entire journey.
C. Cold Chain Logistics Innovations
The pandemic acted as a catalyst for digitalising the complete supply chain process, including everything from demand management to shipment flows.
For example, the development of innovative packaging materials and insulation technologies to ensure prolonged temperature stability will also impact the industry. Additionally, in the future, autonomous vehicles and drones might be
employed for temperature-sensitive lastmile deliveries, reducing transit time, and increasing efficiency.
A future strategy in the pharma logistics industry should therefore also focus on technological integration. In fact, digital transformation is fundamental for global supply chain progress. The challenge is that, considering various external interfaces, certain freight processes, e.g. clearance documentation, still rely on non-digital methods. Nonetheless, collaborative initiatives are on their way with the aim of minimising paper-centric freight handling. Furthermore, advancements in IoT, AI, blockchain, and data analytics are vital because they will bolster supply chain precision, visibility, and efficiency. For example, by placing emphasis on electronic and web-based technologies, the goal is to enhance transportation processes, resulting in improved visibility of shipments and heightened excellence in the pharmaceutical logistics supply chain.
2. Global Regulatory Logistics Standards
In addition to technological transformations, the airfreight industry is also confronted with regulatory changes.
Maintaining regulatory compliance remains paramount as there will be increased enforcement for product quality

88 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Logistics & Supply Chain Management
© Swiss WorldCargo



INTERNATIONAL PHARMACEUTICAL INDUSTRY 89 www.international-pharma.com RECYCLABLE THERMAL BLANKETS For Healthcare Airfreight Temperature Qualifications & Validations - Multilayer thermal blanket for PMC-ULD – Euro and Block pallets - Stress tested in summer (+46°C) and winter ( -15°C) profiles - Airfield Tarmac tested on solar power and greenhouse effects Recycling Qualification - NOT laminated = Dismantlable = Recyclable = Remanufacturing Available worldwide at your freight forwarder KRAUTZ - TEMAX Group Tel: (Belgium) +32-11.26.24.20 / Website www.krautz.org / Email info@krautz.org Temperature protection of pharmaceuticals and healthcare products in airfreight (+15°C +25°C) and (+2°C +30°C)
Logistics & Supply Chain Management
and patient safety. Ensuring product integrity by adhering to temperature requirements throughout transportation is critical also due to stringent global regulations in handling pharmaceutical and healthcare items. Industry collaboration aims to create worldwide strategies to ensure that standards for pharmaceutical logistics supply chains are respected at all times, optimising supply chain processes.
In response to these regulatory shifts, an appropriate strategy for the near future involves staying informed about the evolving regulatory requirements and maintaining strict adherence to quality and safety standards. This proactive approach helps ensure compliance while upholding the highest levels of quality and safety in operations.
3. Sustainability and Environmental Consideration
Furthermore, sustainability demands are also increasing and, as a result, the industry is moving towards adopting environmentally friendly packaging materials, refrigerants, and containers to minimise the ecological impact of temperature-controlled logistics operations. This initiative goes hand in hand with the focus on reducing energy consumption and carbon emissions throughout the entire supply chain.
In the airfreight industry, the journey towards greener cold chain logistics relies on data, innovation, and collaboration. Data empowers us with tools like transparent carbon emissions tracking and AIoptimised routes. Innovation introduces new solutions like lightweight packaging and recycling initiatives across the value chain. Collaboration is vital, uniting stakeholders for a collective impact on this important journey.
4.
Supply Chain Flexibility and Resilience
Additionally, in order to navigate the present and shape the future of pharmaceutical logistics, a resilient supply chain and solid risk management is required.
In fact, in response to unforeseen disruptions like natural disasters or pandemics, there is a growing trend towards the establishment of more flexible supply chain models that allow companies to respond swiftly. Additionally, the diversification of sourcing locations and suppliers is being pursued as a means to mitigate risks effectively.
For these reasons, in anticipating the future, a focused approach that includes risk management is essential: In the airfreight industry, we aim to craft comprehensive strategies that address potential disruptions to ensure business continuity. This proactive stance empowers us to navigate complexities and maintain a steady course even in the face of challenges.
5. Education & Collaboration
Finally, increased education and collaboration are factors that will bring about various changes, shaping the future of the temperaturecontrolled pharmaceutical supply chain sector.
A.
Education and Workforce Development
To navigate the evolving temperaturecontrolled supply chain effectively, there's a need to invest in educating and training a skilled workforce. This preparation will enable experts to competently and effectively manage the complex technologies and processes required in this dynamic industry.
B. Collaboration
Here, in recognising the significance of collaboration among stakeholders in cold chain logistics, a shared vision has emerged. For this joint purpose, active interaction with regulatory bodies, industry associations, and advocacy groups remains a priority.
In fact, air cargo experts, manufacturers, distributors, logistics providers, and regulators, are increasingly collaborating to share best practices, which will then result in an enhanced synergy. These partnerships are being formed to optimise supply chain operations, reduce waste, and thereby improve overall efficiency levels. This effort involves for example eliminating data duplications, reducing paperwork, and strengthening air cargo security while offering better shipment visibility.


Conclusion
In conclusion, the future of the temperaturecontrolled pharmaceutical supply chain is marked by technological innovation, regulatory compliance, sustainability efforts, supply chain flexibility, and collaborative
partnerships. Airfreight's vital role during the pandemic underscores its importance in this industry. Advanced technologies like IoT, AI, and blockchain will enhance monitoring and transparency. Conforming to changing regulations, adopting sustainable practices, and building resilient supply chains are additional essential strategies. Education and collaboration will drive workforce development and industry-wide improvements. By embracing these trends, the pharmaceutical supply chain is set to guarantee efficient and secure delivery of crucial medical supplies in the years to come.
Susanne Wellauer, Vertical Industry Pharma & Healthcare representative at Swiss WorldCargo, began her career at Swissair in 1996 as Cargo Expert in Basel. Since then, she has held several Sales and Marketing positions at Swisscargo and Swiss WorldCargo based in Switzerland. Before being appointed Vertical Industry Pharma & Healthcare representative in 2014, she was the Cargo Manager for the Basel region. In her current role, she monitors the industry needs and trends on a global scale and liaises closely with cold chain experts at freight forwarding companies, as well as with major pharmaceutical and healthcare companies.

90 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Susanne Wellauer
© Swiss WorldCargo
© Swiss WorldCargo
An independent company providing innovative R&D through to clinical evaluation of inhalation products for global pharma and medical technology companies since 1992.




SERVICES
• pMDI, DPI, Nebuliser & Device Evaluation

• Formulation R&D
• Product Development
• Accelerated Stability Studies
• Pilot Batch Production
• Technology Transfer
• Scintigraphy Clinical Trials
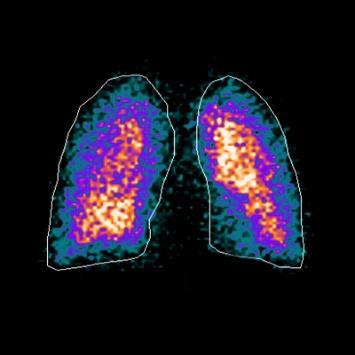
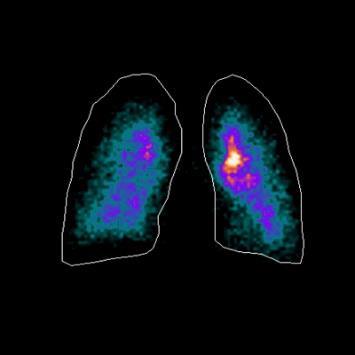
• Respitab™ pMDI platform technology for NCE’s & generics (WO2015/121653)
• Optimised for low global warming potential propellants
• Flexible batch sizes, simple scaling from pilot to commercial batches
• No pressure vessel, propellant top-up or homogenisation challenges
• Simplified cleaning, reduced waste & cost, & improved productivity
• Reduced operator input minimising hazards
• Minimal production facility/equipment refits
INTERNATIONAL PHARMACEUTICAL INDUSTRY 91 www.international-pharma.com
i2c Pharmaceutical Services: www.i2cpharm.co.uk +44 29 2075 7865 info@i2cpharm.co.uk
LICENSING OPPORTUNITY
0 10 20 30 40 50 Inlet / S1 S2 S3 S4 S5 S6 S7 MOC % Salbutamol Recovery Next Generation Impactor Recovery Profiles for Salbutamol from Respitab ® & Marketed Comparator Respitab HFA 152a Marketed HFA 134a Product
The Great Why Questions About Dry Powder Inhalers
The research pertains to the economic burden of asthma and chronic obstructive pulmonary disease and the impact of poor inhalation techniques with commonly prescribed dry powder inhalers as illustrated in three European countries. Currently, the direct cost burden of managing asthma and COPD for people using DPIs is €813 million, €560 million, and €774 million in Spain, Sweden, and the UK, respectively. Poor inhalation techniques comprised 2.2–7.7% of direct costs, totalling €105 million in these three countries alone. When lost productivity costs were included, total expenditure increased to €1.4 billion, €1.7 billion, and €3.3 billion in Spain, Sweden, and the UK, respectively, with €782 million attributable to poor inhalation technique across the three countries.1,2
Airflow resistance
A major issue is current airflow resistance: “Current guidance is in the line with these observations, suggesting that passive DPIs are all flow-rate dependent, and that young children and elderly patients are at risk of not being able to achieve the flow rates necessary to effectively disperse the powder. The underlying factors controlling inspiratory pressures, flow rates and dispensing, and dispersion characteristics of the various DPIs explain why this is the case. While it is also clear then that some patients at the extremes of the population, with poor muscle strength, may not be able to achieve the inspiratory flow rates to utilise a given DPI”.3 “In particular, the inspiratory airflow generated by the patient represents the only active force (a passive force for the device) able to produce the micro-dispersion (even if differently sized for each device) of the powdered drug to inhale. On the other hand, the extent of the patient’s inspiratory airflow depends on the patient’s airway and lung conditions, and, partially, on the intrinsic resistive regimen of the device”. “While low resistance DPIs are still regarded as the easiest and the most comfortable devices for the patient, they instead require a high inhalation airflow rate to the patient, not always
achievable. The reason is that the role of the other possible force involved in drug deagglomeration”.4
De-agglomeration
De-agglomeration is a major parameter in DPI design. “Dry powders designed for inhalation are very fine and can easily form agglomeration due to cohesion between individual particles and are hard to aerosolize. Despite the inhaler and formulation designs, patients are required to generate a forceful and deep inhalation through the DPI to de-agglomerate the powder formulation into respirable particles (with an aerodynamic size ≤ 5 µm) for efficient delivery to the lungs”.5
“In the inhaler, de-agglomeration is one of the most important particle behaviours since it determines the final particle size distribution and hence the FPF. For CapsuleBased DPI, the presence, dimension and movement of the capsule could greatly influence the flow field in the inhaler, as well as the de-agglomeration and dispersion of particles. The capsule is initially placed in the capsule chamber of the DPI and pierced in a certain way before inhalation to allow particles to be fluidised and move out of the capsule. During the inhalation, the high-speed air flow (normally around 60 L/min) from the inlet of the inhaler forces the capsule to rotate and collide frequently with the inhaler wall. This would, for a certain degree, help the de-agglomeration process of the particle clusters and enhance the performance of the DPI. The drug emptying process normally takes about several seconds. It is a rather complex process, involving the interaction of solid capsule, particles and air flow. At an even higher air flow, for example, over 100 L/ min, the shattering of the capsule may occur, which will further increase the complexity of the problem”.6 Although Capsule DPIs are relatively simple and low-cost device it is absolutely limited their use for patient with high inhaling force over 60L/min which is insufficient for babies, young children, elder people and many lung diseases patients. The result of shaking (rotating) the capsule inside the Capsule DPI still does not guarantee, according to studies, the solution to the agglomeration problem when inhaling into the lungs.
The Great Why Questions
• Why after decades of producing and using DPI's, still the fine particle lung deposition is so low?
• Why is the use of DPI's required complicated, especially for children, babies, the elderly, and those with disabilities?
• Why is the airflow resistance so high in capsule inhalers and is it not suitable for many populations that suffer from breathing problems?
• Why is the high price of inhalers that does not contribute to health in the third world where 3.2 million people die from lung diseases every year?
• Why is it forbidden to exhale into the inhaler and why is it impossible to breathe through the inhaler?
• Why do you need expensive digital inhalers just to cover up the inefficiency of delivering the drug to the lungs?
• Why practically the existing inhalers are not suitable for use for new drugs or biological drugs aiming as lifesaving drugs suffering from the combination of the DPI's inefficiency with the high price of the drug?
• Why is the use of hazardous medical cannabis cigarettes and VAPs still in use?
The answer to all questions is: Engineers, companies, and entrepreneurs are captive to old concepts and they only try to improve them and complicate them in more complex systems to cover the basic performances which do not contribute to users, medical insurance companies, and global health.
As Mr. Jacek Olczak, CEO, of Philip Morris International speech regarding stopping smoking, it's very true for DPI: "It's time to try something else, to try including an innovative approach"
Like years of using polluting vehicles, a new generation of hybrid and electric vehicles is starting!
Hence the revolutionary solution, the result of joint research and development by the author of the article, who has many inventions in the field of medical devices
92 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Inhalation


INTERNATIONAL PHARMACEUTICAL INDUSTRY 93 www.international-pharma.com Accelerate your route to market with us Let’s work together Contact us: www.presspart.com Visit us at stand 2F21 at CPHI 2023 Manufacturing & Assembly Program Management Regulatory Support Device Development Analytical Services Small Series & Pilot Phase Scale-up & Industrialisation Impacting Tomorrow. Together.
Inhalation
with Prof. Dan Adler from the Jet Engine Department at the Faculty of Aeronautics in the Israeli Technion.
And here is the solution:
Revolutionary low cost, instructions free VibraMeshTM Dry Powder Inhaler (VDPI)–having >90% Lung Particles Deposition.
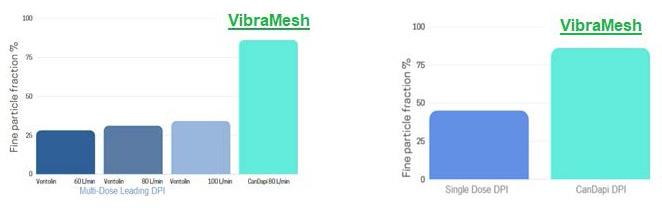

The Solution
CanDapi revolutionary DPI is based on a VibraMeshTM technology activated only when the patient inhales; creating a highfrequency vibration of an internal leaf. The vibrations shake the drug chamber, which is covered by a sieve. The vibrations break powder agglomeration and release particles at a specific size that is streamed to the lungs until all powder is consumed. The device achieved the highest score of lung particle deposition scores vs. competitors during the development of the prototypes (~80% efficiency, Kiel University, Germany)7,8 and 91.8% with cannabinoids dry powder as tested by Copley lung simulator. The patient can breathe through the device with the lowest known airflow resistance without special warnings. The cost of the inhaler without the drug is less than 2€.
Key Message
The most important finding of The VibraMeshTM DPI:
• The most important finding of The VibraMeshTM DPI
• >90% efficiency
• Works only during inhaling (breath through DPI).
• Lowest air-flow resistance (0.01 KPa1/2/ l/min)
• Simple and easy-to-use DPI
• Suits everyone – Multi-age and disabled patients
• Customisable
• delivery of soluble cannabinoid powder
• Very low cost
• Very long shelf life
Grandma sifting flour

Clarinet single reed diffuser

Efficacy Test Results
Dose DPI: Air-flow Resistance Test
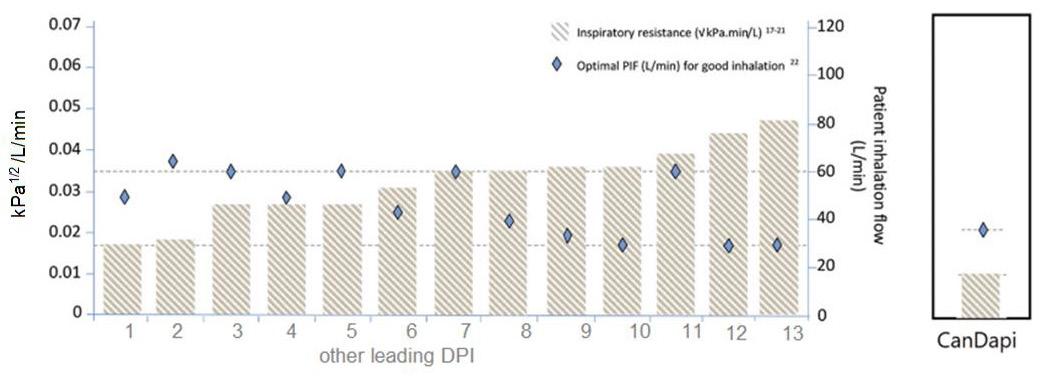
De-agglomeration
According to “The Investigation of the “New Single-Use Dry Powder Inhaler” by Wagenseil, L., Menge, and Steckel, H. Department of Pharmaceutics and Biopharmaceutics, Christian Albrecht University, Grasweg 9a, 24118 Kiel, Germany,
Minimum airflow resistance and creating a "jet" air stream based on aeronautical principles

Low-cost
the VibraMeshTM Dry powder inhalers are well-established in the treatment of various lung diseases and are also useful for systemic drug delivery. They can simply be adapted to the requirements of a single use device, which provides several advantages such as application in acute therapy, economic integration into treatment regimen in hospitals and onceonly use. Therefore, preferred features of a disposable device are, amongst others, a low resistance, an effective powder deagglomeration and a constant powder release at clinically relevant air flow-rates. Intuitive and simple use of the device is essential as well. The de-agglomeration efficiency of the device was calculated in percentage based on the obtained primary particle size distribution, which was assumed to be 100% de-agglomerated.
94 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
materials and manufacturing The scientific principles
Summary of the scientific technology solution and testing results and achievements
Comparison of Fine particle others to CanDapi DPI
Analyzing VibraMeshTM air-flow resistance by Copley DFM4 – Flow Meter vs. Others9
Cannabinoids VibraMeshTM Test Results
Note: The cannabinoids formulation is a water-soluble dry powder.
• > 90% of the cannabinoid-lactose formulations material was released from the ESD
• No increase in degradation, photodegradation or oxidation products in comparison to control (d8-THC, CBL or CBN).
• Cannabinoid profile remain with no change.
• Cannabinoids powder DPI test results shows excellent performance
Conclusion
The patented DPI based on VibraMeshTM, solved four major issues of the existing DPI:
1. Efficiency
2. Solving high airflow resistance issues
3. Solving DPI powder agglomeration issues
4. Simplicity of use for all ages and medical condition
5. Price for the end user, the healthcare provider and a solution for developing countries
6. Replacing harmful cannabis cigarettes and cannabis VAPEs with safer DPI solution
REFERENCES
1. chronic obstructive pulmonary disease and the impact of poor inhalation technique with commonly prescribed dry powder inhalers in three European countries A. Lewis1, S. Torvinen2, P. N. R. Dekhuijzen3, H. Chrystyn4, A. T. Watson1, M. Blackney1* and A. Plich2, Lewis et al. BMC Health Services Research (2016) 16:251 DOI 10.1186/s12913-016-1482-7
2. The Confusing World of Dry Powder Inhalers: It Is All About Inspiratory Pressures, Not Inspiratory Flow Rates, Andrew R. Clark, PhD,1 Jeffry G. Weers, PhD,2 and Rajiv Dhand, MD3, JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY, Volume 33, Number 1, 2020, Mary Ann Liebert, Inc. 1–11, DOI: 10.1089/jamp.2019.1556
3. The Confusing World of Dry Powder Inhalers: It Is


All About Inspiratory Pressures, Not Inspiratory Flow Rates Andrew R. Clark, PhD, Jeffry G. Weers, PhD, and Rajiv Dhand, MD. JOURNAL OF AEROSOL MEDICINE AND PULMONARY DRUG DELIVERY Volume 33, Number 1, 2020 Mary Ann Liebert, Inc. Pp. 1–11, DOI: 10.1089/jamp.2019.1556

4. Dry powder inhalers and the right things to remember: a concept review, Roberto W Dal Negro, Dal Negro Multidisciplinary Respiratory Medicine (2015) 10:13, DOI 10.1186/s40248-0150012-5
5. Dry Powder for Pulmonary Delivery: A Comprehensive Review Birendra Chaurasiya and You-Yang Zhao Pharmaceutics 2021, 13, 31. https:// dx.doi.org/10.3390/pharmaceutics13010031
6. Flow and Particle Modelling of Dry Powder Inhalers: Methodologies, Recent Development and Emerging Applications by Zhanying Zheng, ORCID, Sharon Shui Yee Leung 2ORCID and Raghvendra Gupta, Pharmaceutics 2021, 13(2), 189; https://doi.org/10.3390/ pharmaceutics13020189
7. Wagenseil, L., Menge, A.‐K., Solomon, I., Steckel, H. "Optimization and performance of the ResQhaler‐a single‐use disposable dry powder inhaler" DDL22, Edinburgh, Scotland (2011)
8. Investigation of Single-use disposable ResQhaler New Single-Use Dry Powder Inhaler, Wagenseil, L.1, Menge, A.-K., and Steckel, Department of Pharmaceutics and Biopharmaceutics, Christian Albrecht University, Grasweg 9a, 24118 Kiel, Germany, Kiel university publications
9. (EXPERT OPINION ON DRUG DELIVERY, 2017 VOL. 14, NO. 4, 499–512)
Amnon Kritzman Kadron, Founder and Inventor, Engineer (M.Sc.) From the Technion, Israel. Amnon is a serial entrepreneur and inventor with over 20 patents. CEO and CTO of several medical devices and biotechnology engineering companies based on his inventions. Amnon is currently the CEO and founder of CanDapi in Israel based on his invention of the innovative new Dry Powder Inhaler (DPI).
INTERNATIONAL PHARMACEUTICAL INDUSTRY 95 www.international-pharma.com
Amnon Kritzman Kadron
Test Material Powder type Amount Loaded (mg) Total amount released (mg) Total amount released (%) THC 1 High THC 5.52 5.07 91.8
Inhalation
Clinical Gamma Scintigraphy for the Evaluation of Inhaled Medic

IPI Speaks with Dr. Simon Warren, Research Director at Cardiff Scintigraphics on the benefits of scintigraphic evaluation in respiratory drug delivery.
Q: Cardiff Scintigraphics Limited (CSL) is an Established Provider of Clinical Gamma Scintigraphy Services for the Evaluation of Pharmaceutical Formulations & Devices. Can you tell our readers a brief history of the company, how you started and your growth so far?

A: Cardiff Scintigraphics Limited (CSL, https://www.scintigraphics.co.uk) was formed in 1992, by three distinguished research academics from Cardiff University who had pioneered the application of gamma scintigraphy to the evaluation of pharmaceutical products. One of those founding directors, Prof Glyn Taylor, remains closely involved with the company.
CSL was established as an independent company to provide scintigraphy services to the pharma industry with a particular emphasis on respiratory drug delivery. CSL has for over 25 years performed clinical scintigraphy studies in collaboration with a local full-service contract research organisation (CRO), Simbec Research Limited. Together, in the early 2000’s, we established a purpose-built scintigraphy suite and GMP formulation laboratory at Simbec’s site and we continue to offer this unique combination of scientific, clinical, analytical, regulatory and project management expertise.
To promote the full range of research and development expertise available from CSL we started trading as i2c Pharmaceutical Services (https://www.i2cpharm.co.uk) to advance research and testing capabilities in pharmaceutical formulation and device development in addition to clinical scintigraphy services.
i2c’s laboratory key offerings include small scale manufacture (100’s–1000’s) of pressurised metered dose inhalers (pMDI’s) and dry powder inhalers (DPI’s), accelerated stability testing, particle size analysis by inertial impaction, chemical analysis by HPLC, and moisture analysis (Karl Fischer). i2c also performs analysis of oral dosage forms including tablet disintegration and dissolution and has a small-scale single punch tablet machine. Accelerated stability
assessments of all types of dosage forms are performed using four environmental cabinets at standard ICH and custom conditions.
In addition to the bespoke service offerings, CSL has developed a novel pMDI formulation & production approach and licencing opportunities are available for this Respitab™ platform technology (www. Respitab.co.uk) which offers affordable & accelerated development for NCE and generic products.
The experienced team of scientists at CSL / i2c provide independent services to global pharma, medical technology companies and emerging clients for the development of pharmaceutical formulations and inhaler devices and has performed studies for more than 30 different pharma companies, including half of the ‘Global Top 10’.
Q: You serve as the Research Director at Cardiff Scintigraphics. What does your day-to-day role look like and what are your daily goals and objectives?
A: As Research Director in a thriving R&D company, every day is different and that is what makes my role both challenging and exciting. For scintigraphy studies my day-to-day role involves close liaison with Simbec Research, in particular with the IMP management services group i.e. Research
Pharmacist & QP, functions essential in the successful production of small scale batches of radiolabelled product formulated to GMP quality standards.
For i2c studies my day-to-day role is defined by close interaction with the R&D team to achieve our goal of providing excellent customer service by ensuring the integrity and interpretation of laboratory data and ensuring that detailed updates are maintained and provided to the sponsors of external research and development projects.
For all types of study, I interact with sponsor companies to develop and evaluate research proposals and assure the smooth running of projects and allocate internal resources to ensure their timely conduct and reporting.
Q: What is the role of scintigraphic evaluation in respiratory drug delivery.
A: Gamma scintigraphy is a safe non-invasive method for determining the biodistribution of drug delivery systems under physiological conditions, providing critical data early in the clinical development process. This provides information on the efficiency of deposition and the distribution in the lung. It also gives a crucial insight into the inter- and intra-subject variability in the performance of the drug / device in vivo and
96 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
Inhalation
using this methodology early in the clinical development process gives valuable insights for the design of larger clinical trials.
The radiolabelling processes used by CSL are fully validated. For pulmonary delivery systems this involves standard Pharmacopoeial inertial impaction techniques combined with chemical assay (HPLC) of API and imaging of radiolabel, these processes are performed to demonstrate that the radiolabel is an accurate surrogate for the drug.
Scintigraphy can be used to evaluate all types of respiratory delivery systems, e.g., pMDI, DPI nebulisers, air-jet, electronic and soft mist inhalers, in patient populations and healthy volunteers. Studies can be designed to evaluate device variables e.g., mouthpiece configurations, formulation factors e.g., excipient levels or patient interactions e.g., inhalation flow rate and inhaled volume. Since the clinical end points i.e., efficiency of lung deposition and distribution pattern can be accurately measured relatively small populations can be evaluated to provide critical data in terms of product performance and variability. Study protocols may also incorporate simultaneous PK evaluations.
Q: Understanding the relationship between in vitro and in vivo data for inhaled drug products is an important goal. In vitro aerodynamic particle size distributions (APSDs) are expected to have some predictive power not only for drug deposition, but also for clinical effects. Can you tell our readers about the current needs and trends in inhalation drug delivery field?
A: In vitro data can form a critical part of regulatory submission, EMA guidance indicates that in theory, if not practice, in vitro equivalence of test and reference products is sufficient for product registration, in the US in vitro data forms a part of the weight of evidence approach.
Key in vitro data for inhalation products are generated by inertial impaction assessments whereby aerosolised particles are fractionated according to their aerodynamic properties. The Next Generation Impactor (NGI) is the most widely used instrument for this purpose coupled with a sensitive and accurate analytical technique such as HPLC-UV. The data are evaluated to
determine key aerosol characteristics e.g., fine particle fraction (FPF), fine particle dose (FPD) mass median aerodynamic diameter (MMAD).
These methods have great utility for QC purposes e.g. determining batch-tobatch consistency. In terms of product development, the impact of formulation variables e.g., ethanol levels in pMDI formulations, on the resultant aerosol properties can be effectively measured thus enabling optimisation of aerosol performance in idealised laboratory testing conditions.
In terms of predicting in vivo deposition, however, in vitro tests are often poorly predictive tending to overestimate lung deposition. Patient variables such as upper airway geometry, and inspiratory flow profiles contribute to the deviation from in vitro data typically generated by impactors using standard induction ports i.e., non-anatomical throats and operating square wave airflows. Methods have been introduced to improve the predictive power of in vitro tests, including the use of more anatomically representative throats (inlets) and air flow profiles/volumes derived from patient data.
The industry is attempting to develop in vitro models to be more predictive of in vivo deposition. Successful models may allow product development to be streamlined reducing clinical data requirements for registration of products. Gamma scintigraphy is a critical tool for validating the predictive power of in vitro models used to estimate in vivo lung dose and distribution.
We see increased interest in the treatment of diseases such as idiopathic pulmonary fibrosis and pulmonary arterial hypertension and formulations/devices have been refined to maximise the therapeutic efficacy in these disease states.
We have recently been part of a consortium to develop of a novel proof of concept aerosol formulation for the treatment of antimicrobial resistant chronic lung infections. The concept, known as Light4Lungs (https://light4lungs. eu) utilises novel breathable light emitting nanoparticles that kill pathogenic bacteria by photodynamic effects irrespective of multidrug resistance properties. Uniquely, the formulation is being developed so that the nanoparticles are inhaled, deliver the light energy and are then exhaled. We are
currently working on optimising the aerosol properties of these nanoparticles.
Q: Reformulation of pMDI’s using low global warming potential propellants is underway, how may γ-Scintigraphy help provide data to support the regulatory process? What kind of work are you doing currently, and what are clients wanting from you?
A: Several pharma companies e.g., AstraZeneca, Chiesi and GSK have publicly stated their intention to reformulate pMDI products using low GWP (Global Warming Potential) propellants.
Recent EMA guidance states that when transitioning to the new propellants, whether that be HFA152a or HFO1234ze, an assessment of possible effects on ciliary function, i.e., a mucociliary clearance (MCC) study should be performed. The guidance reports that gamma scintigraphy would be an acceptable option for performing this assessment.
In addition to MCC studies, gamma scintigraphy can also help clients in understanding the performance of their low GWP MDI formulation in the clinical setting. For these types of studies, the active ingredient is surface labelled with Technetium-99m in the case of suspension systems, or a soluble form of Tc-99m is used to radiolabel solution formulations. In vitro characterisation of the radiolabelled product is performed to demonstrate the validity of the radiolabel i.e., that the radiolabelled is an accurate surrogate for the active ingredient and that the radiolabelling process has not changed the aerosol characteristics relative to control samples.
Following validation of the process the radiolabelled products are tested in the clinic. The results from the test formulations can be compared with those from radiolabelled reference products to enable assessments of in vivo equivalence. As a consequence of accurate end points i.e. dose to lung, distribution within the lung, such studies can be conducted in relatively small populations i.e. 10’s of subjects. To ensure accuracy of the procedures Krypton-81m gas ventilation imaging is used to define the lung margins, and Cobalt-57 transmission imaging enables regional tissue attenuation of the radiotracer signal to be accurately determined and appropriate corrections made.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 97 www.international-pharma.com Inhalation
We have recently performed some radiolabelling development work on model formulations prepared in HFA 152a and HFO 1234ze and found we were able to accurately match radiolabel and drug deposition in vitro. These data were published at DDL last year (https://ddl-conference.com/ ddl2022/conference-papers/acceleratedstability-studies-with-pmdis-of-respitabhfa-152a-propellant-dispersible-salbutamoltablets/).
Results from scintigraphy studies we have performed have been included in product registration documents for locally acting respiratory products and other products intended for sustained delivery to the lung.
Q: As a key player in the pharmaceutical industry, I am sure you are governed by the vision of sustainability and circular economy. What steps are you taking to lead in this category, and what commitments have you made and gained from your customers and suppliers?
A: i2c has an established track record in supporting companies in transitioning to sustainable products and reducing environmental impact. In the early 2010’s, i2c was part of a consortium that worked with the United Nations Industrial Development Organisation (UNIDO) to help pharma companies in Egypt and Mexico reformulate CFC based inhalers to HFA 134a products thus reducing the use of ozone depleting propellants. Successful technical transfer procedures were implemented at the benefactor companies that resulted in the successful registration and marketing of five products.
Current F-gas regulations mean that there will be a phase down in the use of HFA 134a and HFA 227ea due to their global warming potential. The industry is now focussed on reformulation activities using alternative
low GWP propellants such as HFA 152a and HFO 1234ze.
i2c is using its formulation experience and capabilities for small scale laboratory batch preparation using low GWP propellants to assist companies in their research. We have equipment for single- and two-stage filling of small batches of pMDI and environmental cabinets for accelerated stability storage. These facilities are complemented by analytical methods for drug and moisture analysis so that aerosol properties can be fully characterised during stability studies.
Of the new low GWP propellants HFA 152a presents challenges for scale up to commercial manufacturing due to its classification as a flammable gas. Traditional single stage pressure filling necessitates the use of large pressure vessels which have been identified as a hazard. Eliminating pressure vessels and associated operator interventions during propellant filling of the vessel and drug addition and subsequent cleaning of vessels/ filling lines will significantly reduce risks.
i2c has a unique solution to these problems in the form of a propellant dispersible tablet i.e., Respitab™ a patented (WO2015121653A1) formulation /manufacturing approach the eliminates the higher-risk steps in production of pMDI using flammable gases. The process also reduces waste and is thereby more sustainable and cost effective. The API in the form of standard micronised material is compressed into a tablet with inhalation grade lactose and an approved dispersant i.e., menthol, both excipients have been used previously in marketed pMDI’s. The inclusion of lactose in the formulation enhances product stability and generates high performance aerosol properties.

The Respitab platform technology can be applied to a range of API’s to produce single and combination products. Propellant dispersible tablets may be dispensed into canisters and crimped prior to the addition of the propellant. Filling can be performed
at any time after crimping and may even be performed at a different site meaning facilities without experience or capability for complex inhaler manufacturing processes could quickly set-up a finishing service for pMDI production.
Q: What is Cardiff Scintigraphic’s vision for the future? What projects are you looking forward to?
A: CSL’s vision is to enhance its capabilities as a contract research centre of excellence for inhaler development and testing. Capitalising on CSL’s unique combination of knowledge, skills and experience of early-stage pharmaceutical formulation/ device development and clinical gamma scintigraphy, it is intended that services will be extended into a GMP environment to provide clients with a world-class facility enabling a swift and cost-effective route to first-in-human trials.
We are particularly looking forward to facilitating projects with the development and clinical trials of Next Generation Inhalers with lower carbon footprints and lower GWP. Included in this objective is finding commercial partners for our Respitab™ technology which can provide excellent aerosol performance, stability and will enable rapid and effective transition to Next Generation Inhalers with associated environmental benefits.
Dr. Simon Warren is the Research Director at Cardiff Scintigraphics. He joined Scintigraphics in 1994 with responsibility for radiolabelling development and validation programs for oral and inhalation delivery systems. He has extensive experience in small scale GMP manufacture of radiolabelled pMDIs, dry powder inhalers and nebulizer systems for use in clinical trials.
Dr. Warren is also the co-inventor of several patents describing novel formulation and manufacturing methods for pMDIs using propellants with low global warming potential.
Email: simon.warren@scintigraphics.co.uk www.scintigraphics.co.uk

98 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Inhalation
Simon Warren
Vaping – Success or Disaster?
This month disposable vapes (also known as e-cigarettes) have hit the headlines – this time we have finally woken up to the fact that millions of disposable vapes are being thrown away every week in the UK, and of course in other countries.
Q1: Are vapes being targeted at children, by using candy colours and flavours?
Q2: Are vapes just intended to help smoking cessation?
Q3: How safe is it to inhale vapourised PG and VG, the e-liquids, regularly?
Q4: Can this technology be used to deliver life-saving drugs via the lung?
These £5 sales price products are now significantly cheaper than a pack of twenty cigarettes, and come in many attractive sweet flavours masking the nicotine addiction, some say promoting under-age nicotine addiction. Published US government statistics show that an average of 20% of school children claim to have vaped.
I find it difficult to understand why the vaping market in the UK is unregulated, aside from CE/CA UK marking which is a DIY activity requiring the manufacturer to keep a technical file on their product design and manufacture. Such marking does not prove that a product is safe to use as an inhaler. Indeed the number of cases of lipid (fat deposit) pneumonia related to vaping use are rising, and the FDA in 2019 (four years ago) proposed to put the

– EC’s addicts than Tobacco products…
e-liquid main ingredients on the ‘potentially hazardous to inhale’ register.
To use a vape as a medical product the technical hurdle would include large scale in vivo safety studies firstly in animals and then a human study to prove safety and efficacy. It is my understanding that no vaping product has been registered voluntarily with any regulatory authority. The timescale to do so,


99 www.international-pharma.com Inhalation
Inhalation
and cost would make that product many times more costly to buy. It just does not make economic sense to the manufacturers, unless it is mandatory for all to do so. Governments must enforce safety in vaping.
Lithium Salt is a Well-known Poison
Aside from the safety issue of inhalation of e-liquid vapour, I was shocked to find out today that all disposable vaping products contain lithium batteries. To my mind using Lithium-ion batteries only once, in these disposable products, is environmental piracy. Vapes get thrown away in general rubbish and so end up in landfill, with both underground fire risk due to battery crushing at the site, as well as long-term rainwater leachate of the poisonous Lithium salts.
The numbers of disposable vapes, as reported by the BBC and other sources suggest that in the UK alone we are putting up to 35 tonnes of Lithium Carbonate Equivalent into landfill – every year. It will take some years for the battery casing to break down and release the lithium load into rainwater, thus penetrating the soil. So the Lithium experiment is only at the beginning.

25gm of Lithium Carbonate ingested is the toxic limit for a human. A few gm ingested repeatedly will induce nausea and migraine headaches in most people. So every year we are burying enough Lithium from disposable vapes alone to potentially kill 1.4m people, or
give 14m (20% of the UK population) lithium induced illnesses.
2015 UK (8 years ago) ‘Vapes are 95% safer than cigarette smoking’ Public Health England reported in 2015 (8 years ago) that E-cigarettes (EC’s) are ‘95% safer’ for addicts than tobacco products. The report’s authors wanted to encourage the use of EC’s as nicotine replacement therapies.
In 2019 (four years ago) two of the authors of the 2015 PHE report are named in a clinical study of circa 900 smokers at three smoking cessation centres, demonstrating that 18% of EC users did not revert to tobacco, whereas only 10% of those using replacement therapies were successful in stopping smoking. It was not a safety study for vaping.


Deep-dive tear down of a single typical UK sourced CE and CA UK marked product, bought from a large chain supermarket for £5.
The core technology now includes an inhaled air flow pressure sensor at the distal end to the user’s mouth. This e-switch is only Ø6mm by 2.8 mm thick and includes a miniature diaphragm and a packaged control chip for the heating coil. This flow switch could easily be re-purposed for use in an e-haler of any type, or for providing user compliance monitoring by detecting inhalation, or for breathing apparatus as a demand system.
There is also a very small LED (blue in this case) that mimics the first e-cigarette of Ho Lin, a Chinese pharmacist and smoker
100 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3
–
(2003). His invention had a large red LED on this distal end to encourage a tobacco smoker by mimicking the combustion product.
It is reported that Schenzen in China is the global centre for making vapes, so Ho Lin has helped generate considerable profit for his country over the last twenty years.
Inside the mouthpiece of this used vape can be seen a white foam oil collector than has been discoloured by the flavoured orange oil that has leeched from the reservoir next to the oil catcher. My understanding is that the oil, which contains nicotine, is toxic if ingested, so this oil catcher is a safety feature of some sort.

This vape has 21 individual components, much like a state-of-of-the art complicated inhaler used to treat asthma or COPD*. Single-use Lithium batteries are not common in any products, but their ability to generate high currents is helpful when using a hot element to vapourise the PG and VG.
Lithium battery fires are fairly well known, following crushing or just shock damage. When combusting acutely poisonous gases are released by the batteries, so I was a little surprised to see that in this case the lithium battery was directly in the air stream leading to the user’s mouth. Although the likely hood of battery combustion during inhalation is low – the outcome could be death of the user from hydrogen fluoride gas and hydrofluoric acid vapour. For this reason in a medical device the use of a lithium battery in the drug path or air path would not be advisable, and not approvable by most regulatory authorities.
The majority of the product weight is in the aluminium tube and battery (62%) with the majority of remaining weight in hard and soft plastics and foam. The only unusual pieces are the steel chimney found in the centre of the product, and the very small steel structure wire mesh used to vapourise the polypropylene glycol (PG) ‘oil’ and vegetable glycerine (VG).


These products vary in weight by is we use 25gm as a typical weight when used, then the UK landfill sites will receive 6,500 tonnes of them per year – assuming no recycling occurs. Perhaps some 250 bin lorries full of vapes, every year.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 101 www.international-pharma.com Inhalation

102 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3 Inhalation Description of component Manufacturing Process Most likely bulk Material Weight gm % overall weight 1 Outer coloured & printed body tube Aluminium, anodised & tampo printed Aluminium 8.052 32.4% 2 Lithium-ion battery- re-chargeable 356mAh Complex - poisonous chemical Lithium Salt 7.327 29.5% 3 Proximal reservoir plug Injection moulding Elastomer 1.727 7.0% 4 Distal reservoir plug Injection moulding Elastomer 1.363 5.5% 5 Mouthpiece Injection moulding ABS 1.327 5.3% 6 Reservoir foam tube - some liquid Foamed tube Polyester 0.994 4.0% 7 Reservoir tube Extruded Polycarbonate 0.939 3.8% 8 Metal chimney - coil to mouth Stainless tube laser cut Stainless 302 0.852 3.4% 9 Flow sensor holding plug Injection Moulding Elastomer 0.759 3.1% 10 Air inlet cap Injection moulding ABS 0.68 2.7% 11 Flow sensor, coil switch & LED Multiple station automated Plastic & Aluminium 0.258 1.0% 12 Mouthpiece back catcher Foamed tube Polyester 0.165 0.7% 13 Printed circuit board - coil Glass fibre board - metal flashing Glass fibre board 0.16 0.6% 14 Hot wick in contact with coil Folded Fibre glass sheet Glass fire weave 0.113 0.5% 15 Green insulating tab Punch sheet Card & foam 0.063 0.3% 16 Expanded mesh heating coil Nickel Chromed steel Steel 0.034 0.1% 17 Battery anti-rattle wrap Punch sheet Foam & Adhesive 0.023 0.1% 18 Brown wire Aluminium & plastic 0.0% 19 Black wire Aluminium & plastic 0.0% 20 Purple wire Aluminium & plastic 0.0% 21 Blue wire Aluminium & plastic 0.0% Overall 'empty' weight typical disposable £5 retail vape - grammes 24.836
This pie chart shows that the aluminium, battery and plastics have roughly equal shares of product. Manual disassembly is needed to recover the internal battery and aluminium tube body. The rest could be perhaps used as part of the fuel of a combined heat and power plant for a town – a rare item in the UK, but more common in Scandinavia.
It would seem that the economic return of such re-cycling work is low, unless there is a use for millions of 400 to 500mWh lithium batteries to power other products? LED head torches perhaps, made in developing countries, re-purposing vape batteries?
The pie chart shows 39% of a Lithium battery is Cobalt, which is undesirable in rainwater leachate. The data was sourced from a ‘Green’ site, and 16% seems slightly high for LCE.

*Chronic Obstructive Pulmonary Disease (COPD) is generally considered a tobacco related disease, when lung function is impaired after longer-term exposure to smoke and the other tobacco contents.
There is not common medical name yet for those adversely affected by vape inhalation – but the chronic symptoms are of lipid pneumonia due to a sticky substance blocking the lung. By 2019 there had been 42 deaths reported in the US considered related to lung disease due to vaping – but reports suggest that most individuals had used THC in their vape, so vape issues are not proven beyond doubt, at the time of writing.
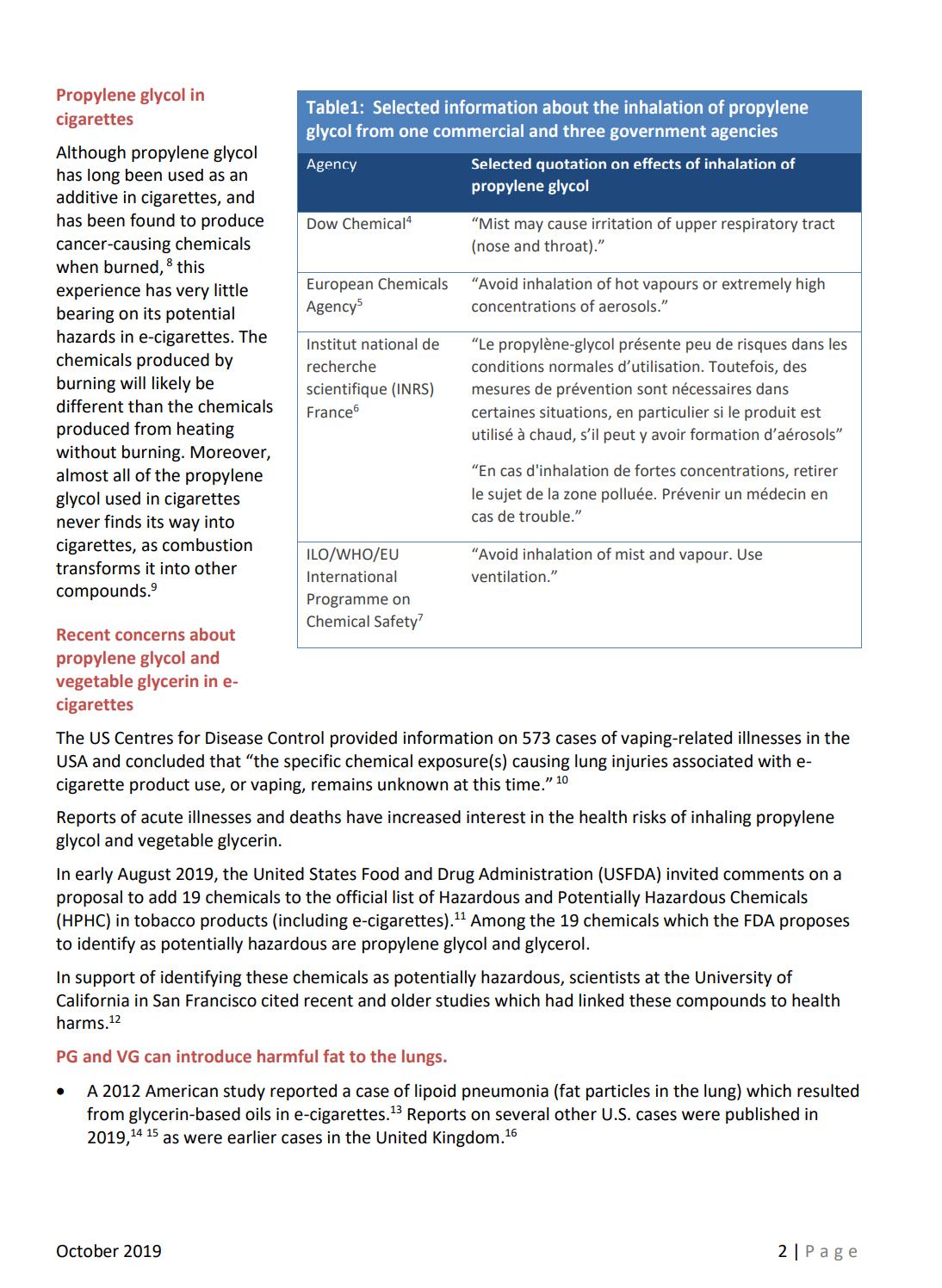
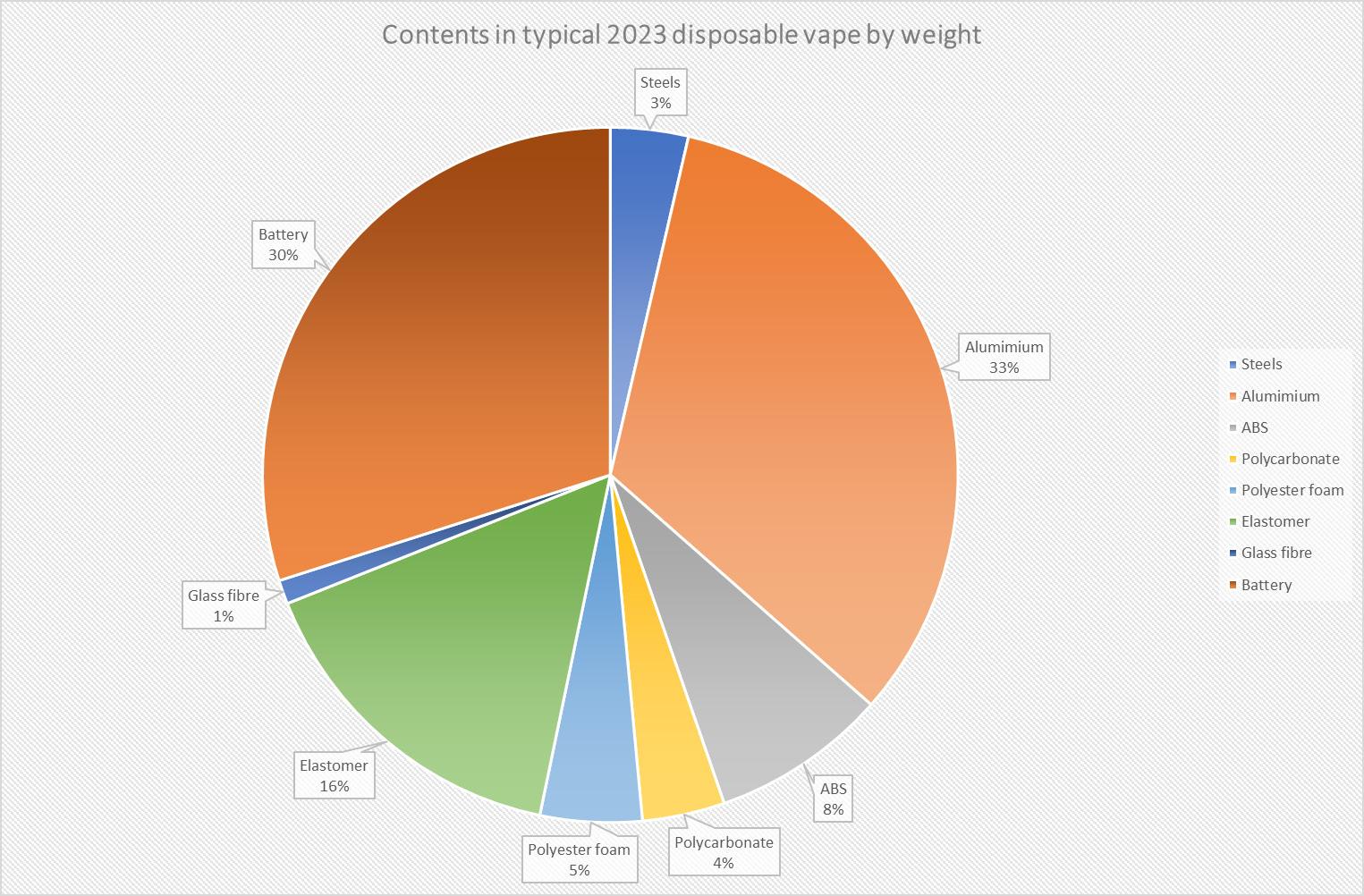
INTERNATIONAL PHARMACEUTICAL INDUSTRY 103 www.international-pharma.com Inhalation
––
Inhalation
US President Trump in his last moments of office in late 2019 was considering a ban on flavoured vapes. Why not just follow Canada and Australia who have always banned all vaping products from the outset, using their poisons legislation?

Conclusions
• Inhalation of primarily PG and VG vapour by smokers was considered ‘safe’ by Public Health England in 2015, and ‘95% safer than smoking’. The ‘E-cigarettes –evidence update’ PHE report continued to promote the use of vaping for smoking cessation. {A search of the 113-page document using ‘safety study’ yields no results; ‘safety’ yields results, but never expressing or agreeing with the concerns or data raised.}
• In 2019 a 3 UK centre ~900 person study was completed that showed e-cigarettes (vapes) helped 18% of smokers to stop using tobacco products, when compared with 10% success with
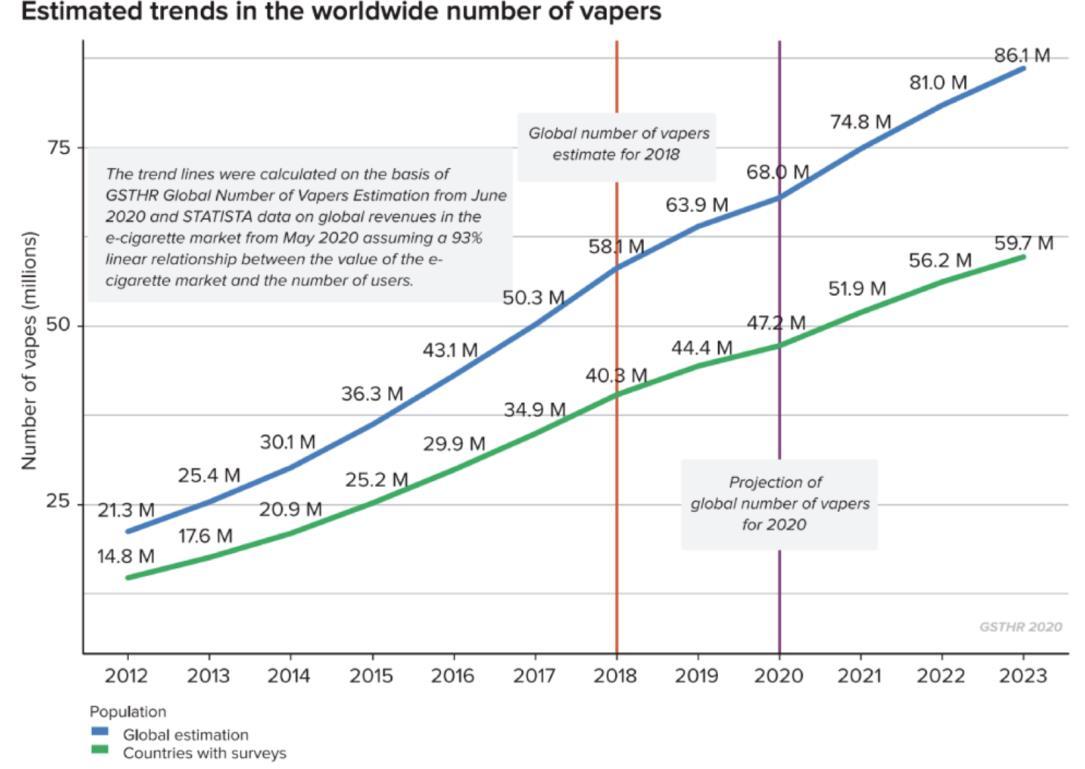
other products. {This was not a safety study, but a promotion/efficacy study of e-cigarettes as NRT’s}
• Also in 2019 concerns were raised about 42 US deaths from lung disease in vapers. These concerns have not yet been converted into government led actions to regulate vapes as medicinal products, or even treat the vape inhalers as potential sources of toxins and so require mandatory safety testing before sale.
• In 2023 the free-to-the-vape manufacturers human ‘Beagle’ experiment continues, but with disposable vape devices under threat of a ban mostly due to the Lithium Batteries
underground fire risk in land fill, as well as rainwater leachate of poisons into the soil in the next decade.
• The technology developed to trigger the e-cigarette or vape by inhalation sensing should have many uses in the inhalation field. A very positive result from the experiment.
• With proven safety of the inhaled vapour, then the scope to use this technology for small molecule delivery is potentially disruptive in pharmaceutical use. It seems however than liquids other than PG and VG might well prove more optimal to safe delivery to the human lung.
 Bill Treneman
Bill Treneman
Bill is the Founder and Engineer from UPC Cambridge Limited, UK. Bill has been working continuously for thirty years in the field of inhalation to the lung, as well as parenteral, nasal and diabetes fields. Prior to inhalation Bill specialised in fuel two phase aerosols and engine cooling at LandRover. Bill currently is the Managing Director at UPC Cambridge, a medical system consultancy.

104 INTERNATIONAL PHARMACEUTICAL INDUSTRY Autumn 2023 Volume 15 Issue 3

www.international-pharma.com NIPRO PHARMAPACKAGING INTERNATIONAL Blokhuisstraat 42, 2800 Mechelen, Belgium | pharmapackaging@nipro-group.com | www.nipro-group.com
to by-pack with drug products!
needle is sterile packed individually
Packaging design supports compatibility with automated pick-and-place systems/feeders
Ideal
Each
•
labels and barcode on label
in-line product identification
Needles in Hard Plastic Unit Packaging
• Color-coded
enable
Hypodermic





scan the QR to receive your free digital copy. Request your Multi-tip Tooling Guide Today, Dramatically increase your production without adding a new tablet press or extending press run time. With innovative tooling designs, superior customer service, and incomparable construction, Natoli is your number one choice. Natoli Multi-Tip Tooling natoli.com
