Journal for Clinical Studies – ISSN 1758-5678 is published quarterly by Senglobal Ltd.
4 FOREWORD
WATCH PAGES
6 FDA Regulatory Perspectives on Barth Syndrome and Rare Diseases
Barth syndrome (BTHS) is a rare paediatric heart disorder with no cure and is typically managed through medications that alleviate symptoms. Stealth BioTherapeutics Inc. (Stealth) has developed a novel molecular entity and mitochondrial protective agent that has shown promising potential in treating the condition. Asher Madan at Clarivate highlights how despite the need for further data, particularly in infants, the drug could become a first-in-class treatment.
8 Developing Robust Submission Strategies for Combined Studies
Navigating the complex regulatory landscape for clinical trials involving medical devices and combination products requires careful planning. Vesta Marciulioniene, Laura Van Vaeck, and Ewa Gawlik-Perek at ICON explain how a well-defined EU submission strategy with clear timelines and attention to country-specific requirements can improve the process and ensure compliance. With a robust approach, sponsors can avoid unnecessary setbacks.
10 The Strategic Imperative of Quality Management in Pharmaceuticals and Biotechnology
Quality management is essential in pharmaceutical and biotechnology sectors to ensure safety, compliance, and competitiveness. Trends like digitalisation and continuous improvement boost efficiency and preparedness. Alok Mehrotra of Syngene stresses how proactive strategies, including strong Quality Management Systems (QMS) and thorough training, maintain high standards. Emphasising a qualityfocused culture helps organisations handle regulatory challenges effectively and achieve sustained success.
REGULATORY
12 Good Review Process – The New Panacea
Optimising the review process for regulatory documents can greatly minimise inefficiencies and accelerate timelines. By concentrating on content-driven reviews and prioritising actionable feedback, teams can sidestep common challenges. Julia Forjanic Klapproth of Trilogy Writing & Consulting, an Indegene Company, emphasises that with the right training and clear guidelines, reviewers can enhance the process, ensuring high-quality submissions.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
Volume 16 Issue 4 Winter 2024, Senglobal Ltd.
www.journalforclinicalstudies.com
18 Next-generation AI Process Automation Essential to Cope with Soaring Regulatory Workloads, Survey Finds
Generative AI holds significant potential to transform regulatory processes in the pharmaceutical industry by enhancing efficiency, accuracy, and compliance. Renato Rjavec at ArisGlobal adds how despite hesitancy around data quality and technology maturity, adoption is expected to accelerate as AI solutions address regulatory challenges.
MARKET REPORT
20 Study Designs and Digital Technologies for Sustainable Clinical Trials
Adopting sustainable solutions such as patient-centric designs, decentralised trials and digital technologies can significantly reduce the environmental impact of clinical trials while improving efficiency and patient access. Dr. Bipin Patel of ElectronRx explains how barriers like data security and accessibility will be essential for realising these benefits.
22 The Role of Company Culture in Functional Service Provider (FSP) Partnerships
A strong and focused emphasis on culture is essential for the success of FSP partnerships in clinical research. By aligning values, fostering open communication, and empowering resources, FSPs can create long-term, mutually beneficial relationships with clients. Allison Crumpler and Svetlana Kolchinsky at Catalyst Clinical Research, demonstrate that cultural alignment not only drives operational success but also builds innovation in clinical research collaborations.
RESEARCH
AND DEVELOPMENT
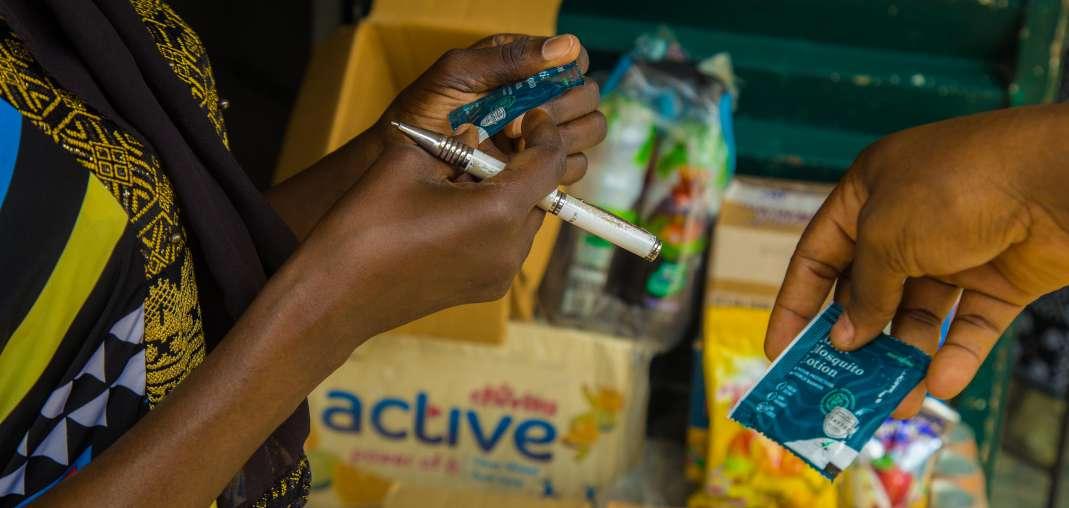
24 Targeting the End of Malaria by 2050
LivFul has successfully developed an innovative mosquito repellent that addresses adoption and adherence challenges, offering longlasting protection without the need for reapplication. Livful Inc’s
Hogan Bassey, demonstrates that rigorous testing, including trials conducted in Ghana, revealed a substantial reduction in malaria cases and strong user compliance within affected communities.
THERAPEUTICS
28 An Interview with Dr. John Stone, Steritas
Steroid toxicity is a growing concern in autoimmune disease treatment, with long-term use leading to significant patient and healthcare system impacts. Dr. John Stone of Steritas discusses an initiative which enables better monitoring of steroid-related harm, and aims to drive safer, more effective prescribing practices globally, improving patient outcomes and minimising healthcare burdens.
TECHNOLOGY
32 Digital Health Technologies are Inherently Remote –Let’s not Burden Sites with Them
Managing digital health technologies (DHTs) through clinical sites creates inefficiencies and unnecessary burdens on vendors, sites, and participants. Geoffrey Gill at Verisense Health explains how a more effective approach involves treating DHTs like tech products, with remote management and direct vendor-patient interactions. This approach could streamline operations and improve participant experience.
LOGISTICS & SUPPLY CHAIN
34 System Integration’s Role in Reducing the Likelihood and Increasing Detectability of Temperature Excursions
Implementing system integration in temperature management processes can greatly mitigate risks and enhance compliance in clinical trials. Sarah McAliskey from Almac Clinical Services highlights how streamlining workflows and enabling smooth data flow between systems helps sponsors safeguard patient safety, eliminate inefficiencies, and accelerate trial timelines.
Ramus Medical
is a part of Ramus Corporate Group. The company is managed under a centralised quality management and has developed an integrated QMS as well as specific standard operating procedures tailored for the clinical trials department that are fully harmonised with the GCP guidelines, and the local and European legislation.
Ramus Medical EOOD is a full-service contract research organisation (CRO) in Sofia, Bulgaria.
The company was created in 2009 as a natural development of the Medical Laboratory Ramus Ltd., the largest privately-owned medical laboratory in Bulgaria.
The company independently manages clinical research projects in Bulgaria and provides partnerships in multinational clinical projects providing a comprehensive range of clinical research services:
Core Services include:
• Medical writing
Our staff has extensive expertise in the preparation, adaptation and translation of a wide range of clinical trial documents that are fully compliant with the Good Clinical Practice (GCP) standards, the client’s specifications and the regulatory requirements.
• Study start-up
We offer full or partial study start-up assistance for different types of studies throughout Bulgaria.
• Regulatory submission
• Project management
• Monitoring
• Data Management
• Pharmacokinetic evaluation
• Biostatistics
• Regulatory advice and services
• Readability User Testing
• Registration of medicinal products on the territory of Bulgaria
• Pharmacovigilance services
• Logistic department
• Destruction of IMPs/IMDs & clinical samples – agreement with PUDOOS
• Archiving services
• DDD activities
Ramus Medical has gained its expertise during the completion of numerous clinical projects carried out over the past decade:
• Phases I to IV drug trials
• Non-interventional studies
• Pilot and Pivotal Medical Device investigations
The clinical trials we conducted facilitated the MA/CE mark granted by various European Agencies/Notified Bodies and Third Country Agencies.
Ramus Medical offers flexible clinical research services in various domains, with extensive experience in fields.
Our team comprises qualified, appropriately trained, experienced, motivated and collaborative professionals and is competent to
Corporate Profile
communicate effectively across geographical and cultural boundaries to resolve any arising issues. We adhere strictly to the agreed timelines during the clinical investigations and strive to complete the tasks on time.
Why are we the solution for your projects? Ramus has its own:
Medical and Bioanalytical Laboratory
In 2018 the Medical Centre Ramus was established, located in Sofia, Bulgaria. Up to date, it has three separate locations, one of which is developed as an independent clinical research centre in compliance with the requirements for the phase I unit.
The Medical Centre Ramus allows the conduct of clinical trials in all phases in many therapeutic areas.
The Medical Centre meets all requirements for performing highquality clinical research and is designed to maximise the delivery of high-quality research data and was GCP-inspected.
Ramus Medical retains an extensive database of investigators and sites compiled through years of mutually beneficial collaboration.
Our bioanalytical laboratory is equipped with leveraging state-ofthe-art instrumentation (LC-MS/MS), techniques, and facilities, our team of experts has experience in a broad range of small molecules. Our Analytical laboratories provide method development, transfer, validation, and analysis of preclinical and clinical biological samples. We have extensive expertise in developing sensitive methods for LCMS/MS-qualifying multiple analytes and metabolites.
• Logistical company, certified for hazardous and biological samples transportation
• Clinical site facility and own catering company for hospitalised patients
Welcome to the Winter edition of JCS. This journal is packed full of features on a diverse range of topics such as the benefits of artificial intelligence, how to improve the regulatory document review process, and how innovative solutions offer hope for more effective prevention and treatment of infectious disease.
As we enter 2025, the world of clinical trials is experiencing notable developments. While the industry continues to evolve, it faces both unprecedented challenges and exciting opportunities.
Artificial Intelligence (AI) is emerging as a game-changer in improving efficiency and outcomes. AI-driven technologies optimise patient recruitment and predict trial success while streamlining operations and reducing costs. With its ability to analyse vast datasets, AI enhances decision-making and speeds up drug development. As clinical trials grow increasingly complex, embracing AI is crucial for staying ahead, driving innovation, and ultimately bringing life-saving therapeutics to market faster.
We begin this issue with a feature on the FDA regulatory perspectives on Barth syndrome and rare diseases. Barth syndrome (BTHS) represents a profound challenge for the medical community, exemplifying the complexity of ultra-rare paediatric-onset diseases. This article highlights the critical unmet needs of individuals living with BTHS, including the absence of curative treatments. Asher Madan of Clarivate details the groundbreaking development of a promising mitochondrial protective agent that garnered significant attention during its FDA review.
A personal favourite of mine is an interview with Dr. John Stone of Steritas, in which he sheds light on a public initiative aimed at accelerating a shift in steroid prescribing patterns. This is crucial for minimising steroid toxicity and promoting steroid-sparing treatments. This feature was enlightening for me, as I, along with millions of others globally, have used steroid medications to manage inflammatory and autoimmune diseases.
Another article that captures my attention is a piece by Renato Rjavec at ArisGlobal, on how next-generation AI process automation is essential to cope with soaring regulatory workloads. This article sheds light on the growing regulatory workloads faced by the pharma
JCS – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Principal STEM Content Analyst, Clarivate
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
• Georg Mathis, Founder and Managing Director, Appletree AG
and biopharma sectors, underscoring the urgency for innovative solutions like Generative AI (GenAI). Through insights from a recent survey, it explores the industry's readiness to adopt AI and the barriers hindering its widespread application.
We conclude this journal with a write-up on how ensuring product integrity is pivotal to safeguarding patient safety and achieving successful clinical trial outcomes. In today’s complex landscape, maintaining drug stability is increasingly challenging, especially with biologics requiring precise temperature controls throughout the supply chain. This contribution by Sarah McAliskey from Almac Clinical Services explores the challenges, solutions, and transformative potential of system integration in clinical trials.
This journal offers valuable insights, and as we continue to share knowledge with one another, we make progress in improving the realm of clinical trials. I hope you enjoy this remarkable collection of work, and I am excited to meet some of you at the forthcoming events!
Kelly Woods, Editorial Manager
• Hermann Schulz, MD, Founder, PresseKontext
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, AsiaPacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
40 years of building powerful industry partnerships.
Broadest portfolio of self-injection products built on platforms
Serving pharma globally for all their originator and biosimilar needs
Over 70 products launched in 15 different therapeutic areas
Global manufacturing footprint spanning Switzerland, Germany, China and North America
Fully integrated strategic partner network
For more information visit www.ypsomed.com/yds
Ypsomed AG // Brunnmattstrasse 6 // 3401 Burgdorf // Switzerland
T +41 34 424 41 11 // info@ypsomed.com
www.journalforclinicalstudies.com
FDA Regulatory Perspectives on Barth Syndrome and Rare Diseases
Barth syndrome (BTHS) is a paediatric-onset, ultra-rare disease characterised by cardiac abnormalities that can result in various health challenges, including an enlarged and weakened heart, heart failure, exercise intolerance, fatigue, and short stature.1
Currently, BTHS has no cure and can only be treated with medications to help manage symptoms and prevent complications. With an eye to fill this unmet medical need, Stealth BioTherapeutics Inc (Stealth) developed elamipretide hydrochloride injection, a new molecular entity and mitochondrial protective agent. The firm’s resubmitted new drug application (NDA) was recently reviewed at a meeting of the US Food and Drug Administration’s (FDA’s) Cardiovascular and Renal Drugs Advisory Committee (CRDAC) and garnered broad support by the panel to introduce a new treatment option for BTHS.
Approximately (~) 250 individuals worldwide and ~130 people in the US live with BTHS, and cardiomyopathy is considered the leading cause of death. The X-linked condition is caused by mutations in the TAFAZZIN gene and is inherited as a recessive allele.2 The National Library of Medicine of the National Institutes of Health (NIH) notes in MedlinePlus that the tafazzin protein is located in mitochondria and is responsible for altering a lipid called cardiolipin. Cardiolipin maintains mitochondrial shape, energy production, and protein transport. TAFAZZIN mutations can lead to decreased levels of adenosine triphosphate, a molecule that stores and provides energy for cells.3 Elamipretide functions by distributing to and improving the function of cardiolipin-deficient mitochondria in patients with BTHS.
Clinical Evidence for Rare Diseases
The US Orphan Drug Act defines a rare disease as any disease or condition that either affects <200,000 people in the US or affects >200,000 people and “there is no reasonable expectation that the cost of developing and making available in the United States a drug for such disease or condition will be recovered from sales.”4 Section 115(a) of the Food and Drug Administration Modernization Act allows the FDA to determine if data from a single adequate and well-controlled clinical trial plus confirmatory evidence are sufficient to establish effectiveness.5 However, particular clinical circumstances (e.g., unmet medical need) can impact the degree of certainty supporting the conclusion that substantial evidence of effectiveness has been demonstrated.6
At the CRDAC meeting in October 2024,2 the committee reviewed evidence provided by Stealth for its NDA resubmission in the form of clinical study results and nonclinical findings. The sponsor originally submitted its NDA for elamipretide in August 2021 and subsequently received a refusal-to-file letter from the FDA. Stealth proceeded to develop the needed data to suitably support the efficacy of the drug to treat BTHS, leading it to resubmit the NDA in early 2024.
While the majority of the CRDAC found data from the animal models and echocardiography to be “disappointing and unclear,” several panelists supported the efficacy assessment after listening to testimonies shared during the open public hearing (OPH) of positive experiences with elamipretide. The OPH is a staple of FDA advisory committee meetings that gives the panelists a chance to receive input from members of the public on the topic at hand. Given the rarity of BTHS, many CRDAC members commented on the importance of the OPH.
The majority of patients, caregivers, and healthcare providers who spoke during the OPH stated that they witnessed a marked improvement in quality of life after they, their loved ones, or their patients began treatment with elamipretide. Before the OPH, several CRDAC panelists emphasised how the data were insufficient to support efficacy and stated they looked forward to hearing from patients directly. Afterward, most of the committee members noted that the OPH made “a huge difference” to their viewpoints and said the patient experiences with elamipretide were “hard to ignore” and “compelling.” As a result, the panel majority agreed that the drug was shown to be effective and even went as far as to recommend its approval.
Despite the CRDAC’s recommendation, panelists acknowledged the need to collect more data on the use of elamipretide, particularly its effect on infants since results suggested a clearer efficacy signal in that population. A few committee members hypothesised that treating infants with elamipretide may “halt” the disease before it leads to irreversible heart and skeletal muscle damage. Some CRDAC members also expressed concern about the methods used by the FDA when “dealing with rare diseases.” The agency should establish “a different structure for rare disease treatment approvals,” especially for those as rare as BTHS, one individual suggested.
If elamipretide is approved by the FDA, it would be a first-in-class treatment for BTHS.1 As noted, the current treatment approaches manage the symptoms of BTHS without addressing the underlying disease. The FDA is set to make a decision about Stealth’s NDA by the end of January 2025.
Pipeline Candidates, Future Treatments
Another treatment avenue under investigation for BTHS is gene therapy, and a considerable amount of the research is being conducted by the NIH. According to the Barth Syndrome Foundation, the goal for gene therapy is to deliver a working copy of the TAFAZZIN gene to prevent the impact of BTHS on the heart and skeletal muscle in paediatric patients. Currently, gene therapies for BTHS have only been tested in animal models and human cells. No human subject testing has been conducted.7
The Harvard Stem Cell Institute (HSCI) has successfully tested gene therapy in mice with BTHS.8 William Pu, MD, and colleagues
were able to use gene therapy to replace defective TAFAZZIN in mice by injecting an engineered virus to deliver a working copy of the gene. The mice that received gene therapy were able to survive into adulthood. The HSCI noted that treatment prevented cardiac dysfunction when given to newborn mice and reversed cardiac dysfunction in older mice. Until gene therapy becomes a standard of care in medicine, the FDA should continue to accelerate the development of medications for rare diseases such as BTHS by offering incentives and support to drug manufacturers.
REFERENCES
1. Advisory Committee Meeting: Stealth Briefing Document NDA#215244. Food and Drug Administration Website. https://www.fda.gov/media/ 182554/download
2. Advisory Committee Meeting: FDA Briefing Document NDA#215244. Food and Drug Administration Website. https://www.fda.gov/media/182553/ download
3. Tafazzin Gene: MedlinePlus Genetics. MedlinePlus, National Library of Medicine. https://medlineplus.gov/genetics/gene/tafazzin/
4. H.R.5238 - 97th Congress (1981-1982): Orphan Drug Act. Library of Congress. http://www.congress.gov/bill/97th-congress/house-bill/5238
5. S.830 – 105th Congress (1997–1998): Food and Drug Administration Modernization Act of 1997. Library of Congress. https://www.congress.
www.journalforclinicalstudies.com
gov/bill/105th-congress/senate-bill/830
6. Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products: Guidance for Industry. Food and Drug Administration Website. https://www.fda.gov/media/133660/download
7. Gene Replacement Therapy for Barth Syndrome. Barth Syndrome Foundation Website. https://www.barthsyndrome.org/file_download/ inline/7b31bd51-ddb6-4ddb-9b37-8b5c4b930219
8. Gene Therapy for Barth Syndrome. Harvard Stem Cell Institute Website. https://www.hsci.harvard.edu/news/gene-therapy-barth-syndrome/
Asher Madan
Asher Madan, MBBS, is a Senior STEM Content Analyst for the Cortellis suite of life science intelligence solutions at Clarivate. After medical school, he worked on tuberculosis research and contributed to a number of technology publications. His current role includes reporting on FDA advisory committee meetings and workshops.
Email: asher.madan@clarivate.com
Developing Robust Submission Strategies for Combined Studies
Medical interventions increasingly involve the use of technology and devices. New treatments may involve a medical device, combination product or in vitro diagnostic device. For sponsors this means their studies have additional regulatory requirements to meet, each on very distinct submission pathways. The assessment of the safety or performance of the device is not in scope, but the device is necessary to conduct the study. Clinical trials with human subjects where such devices are deployed must comply with applicable regulations EU CTR (Regulation (EU) 536/2014), MDR (Regulation (EU) 2017/745) and/or IVDR (Regulation (EU) 2017/746).
Identifying and managing the additional regulatory requirements for these studies can be confusing. Multiple factors must be considered, such as whether the device is CE-marked, is being used outside of its intended purpose or is custom-made. The submissions process may be further complicated by additional national regulatory and ethical requirements.
Even where there is some overlap with the study documentation, the additional device-specific regulations require extra time, effort and expertise. Missing information or a misfiled application can negatively impact a study timeline. Planning a route through the labyrinth of regulations can avoid these delays. Time dedicated to building a robust EU submission strategy in advance means time saved later on in the submissions process.
Conformity Assessment of Medical Devices and in Vitro Diagnostic Devices
In the EU, the device manufacturer is responsible for complying with and demonstrating safety standards. Medical devices do not require official authorisation from a public agency in the EU, unlike medicinal products. However, they need a conformity assessment procedure. Devices that meet EU requirements bear the CE mark upon market entry. Conformity assessment procedures vary according to the devices’ risk classes. High risk devices have greater regulatory oversight than low risk devices. The rules for risk classification of devices are listed in Annex VIII of MDR and IVDR and in the corresponding Medical Device Coordination Group (MDCG) guidance documents including MDCG 2021–24 and 2020–16.
For low-risk devices, with some exceptions, the manufacturer can conduct the conformity assessment procedure themselves and issue the EU Declaration of Conformity. Higher risk devices require an independent conformity assessment procedure by a notified body (NB). These conformity assessment procedures can consist of one or more modules, depending on the risk characteristics. The modules concern the manufacturer’s quality management system, the product’s technical documentation, the individual type-examination, production quality assurance or conformity to an EU type-examination. Typically, the process includes an audit of the manufacturer’s quality system. NBs must consult specific expert panels before granting a Certificate of Conformity for some high-risk devices. Certificates of Conformity are valid for the period indicated and shall not exceed five years. Manufacturers can request an extension of validity for further periods
(less than five years). A re-assessment of conformity must be conducted as part of the extension request.
Medical devices and IVDs certified under Medical Device Directive (MDD), Active Implantable Medical Device Directive (AIMDD) or In Vitro Device Directive (IVDD) should undergo a conformity assessment procedure under the new regulations. These assessments must be carried out before the end of the transitional periods stipulated by MDR and IVDR. The large number of MDs and IVDs that must undergo new conformity assessments has exceeded the capacity of NBs. This has led to several extensions of the transitional periods.
Drug-device Combination Products (DDC)
The multipurpose nature of drug-device combination products has blurred their definitions depending on the applicable regulations. In July 2021 the EMA published the Guideline on Quality Documentation of Medicinal Products when Used with a Medical Device. The guidelines define three types of combination products as medicinal products.
1. Integral DDCs: the medical device and/or device part and the medicinal product form an integral product that is not reusable and where the action of the medicinal product is principal. Example: a single-use dry powder inhaler preassembled with the medicinal product. It is ready for use but cannot be refilled when all doses are exhausted.
2. Co-packed DDCs: the medical device is packed (or packaged) together with the medicinal product. Example: Reusable dry powder inhaler packed with dry powder capsules.
3. Referenced DDCs: the product information refers to a specific medical device to be used with the medicinal product, and the medical device is obtained separately by the user of the medicinal product. Example: Reusable dry powder inhaler and dry powder capsules packed separately but the patient information leaflet indicates that the capsules can only be used with a specific inhaler.
Article 117 of the MDR requires a NB to provide an opinion on the conformity of the DDC against MDR Annex I. Devices already CEmarked do not require an opinion on conformity.
Tips for Creating Robust Submission Strategies:
1. Seek Submission Route Clarity: Clarify the regulatory requirements of the various regulatory authorities (RAs) before mapping your study’s submission route. There may be country-specific requirements in addition to EU regulatory requirements.
2. Develop a Plan for Updating Documents: Where additional country-specific requirements apply, find a way to add these considerations to the overlapping documentation for other RAs.
3. Map a Regulatory Pathway with Clear Timelines for each Requirement:
Agree internal deadlines for drafting and reviewing documentation. Define the roles and responsibilities for document preparation and the processes to follow. The device manufacturer may need to be involved in the regulatory process as well as the sponsor.
4. Anticipate Potential Questions and Provide Relevant Information Upfront: It may be prudent to create an accompanying cover letter with additional supplementary details. This could include descriptions of the roles of investigation sites and central testing sites and other relevant information about patient recruitment and enrolment. By providing these supporting materials sponsors can minimise the number of potential validation questions from each RA.
Conclusion
While the regulatory route is convoluted, it is not impassable. Having a robust strategy in place from the outset ensures that sponsors take the most direct route, avoiding wrong turns, dead ends and delays.
Ewa Gawlik-Perek
Ewa Gawlik-Perek, Senior Regulatory Affairs Manager at Global Regulatory Clinical Service Operational Delivery, ICON. Ewa has been working in Clinical Research for the last two decades building extensive experience of drug delivery at the Clinical Trial Site and at the CRO. From 2022, she focused on “combined trials” delivery, a simultaneous investigation of a medicinal product and an IVD.
Laura Van Vaeck
Laura Van Vaeck, Site Activation Manager at ICON, oversees the success and timeliness of global investigative site activation and maintenance in medical device and in vitro diagnostic medical device studies. With broad expertise in EU MDR and IVDR, Laura acts as one of the experts in this complex space for regulatory, start up and clinical safety reporting processes.
Vesta Marciulioniene
Vesta Marciulioniene, Director Regulatory Affairs at ICON, has spent more than a decade at Pfizer and a decade at Covance & Labcorp. She has extensive clinical research expertise with a strong focus in start-up, having formerly taken global roles of Head of Start Up Centre of Excellence and Head of Site ID in previous organisations. Vesta has been leading multifunctional teams regionally and globally, aiming to connect the start-up and regulatory world with clinical project delivery.
The Strategic Imperative of Quality Management in Pharmaceuticals and Biotechnology
In the highly regulated pharmaceutical and biotechnology industries, quality management serves as the bedrock for ensuring product safety, efficacy, and regulatory compliance. These sectors operate within stringent frameworks established by global regulatory bodies such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), necessitating robust Quality Management Systems (QMS). Effective quality management not only safeguards patients but also underpins organisational reputation, operational efficiency, and market competitiveness.
Recent trends in quality management are reshaping how organisations approach compliance and operational excellence. Key trends include the integration of digital technologies, the decentralisation of quality responsibilities, the emphasis on continuous improvement, and the prioritisation of employee training and supplier quality standards. Understanding these trends is crucial as we explore their impact on the effectiveness of quality management practices in the pharmaceutical and biotechnology sectors.
A QMS encompasses a comprehensive set of processes, from supplier qualification and process validation to employee training and continuous improvement initiatives. By embedding these systems, organisations can standardise procedures, minimise risks, and meet high-quality benchmarks consistently. The importance of fostering a culture of quality, where every employee takes responsibility for maintaining standards, cannot be overstated. Such an environment ensures preparedness for regulatory changes, product recalls, or unexpected disruptions, all of which can significantly impact operations.
Proactive Quality Management Approaches
Contract Development and Manufacturing Organisations (CDMOs) play a pivotal role in the pharmaceutical supply chain, offering specialised services that enable pharmaceutical and biotechnology companies to bring products to market efficiently. For CDMOs, quality management is not merely about regulatory compliance but also about creating a competitive advantage. By maintaining exceptional quality standards, CDMOs build trust with clients and differentiate themselves in a crowded marketplace.
To achieve these objectives, CDMOs must integrate robust quality management practices into their operations. A "state of readiness" philosophy, exemplified by Syngene International’s "Anytime Audit Ready" approach, ensures organisations are consistently prepared for audits, moving beyond the reactive approach of preparing only for scheduled inspections. This proactive strategy reflects a deep-seated commitment to quality, enabling companies to maintain compliance amidst evolving regulatory landscapes.
Central to successful quality management is a dynamic QMS, which supports continuous adaptation and improvement. This system provides a foundation for meeting international regulatory requirements while integrating operational feedback to refine
processes. A critical component of the QMS is the development and maintenance of Standard Operating Procedures (SOPs). These documents guide essential operations, ensuring consistency and adherence to regulatory standards across all functions. Regular updates to SOPs incorporate new regulatory requirements, technological advancements, and industry best practices, ensuring the organisation remains at the forefront of quality management.
However, even the most comprehensive QMS and SOPs are only as effective as the people executing them. Recognising this, organisations invest heavily in ongoing employee training to ensure staff competence. By providing education and development opportunities, companies equip their workforce to adhere to SOPs meticulously, maintaining operational integrity and minimising errors.
Importance of Internal Audits
Internal audits form a cornerstone of effective quality management frameworks. Designed to mimic external regulatory inspections, these audits ensure organisations consistently meet or exceed required standards. They serve multiple purposes, including verifying compliance, identifying improvement areas, and maintaining a continuous state of readiness. By addressing potential issues proactively, internal audits mitigate risks, uphold quality standards, and strengthen client trust.
Integrating Digital Technology in Quality Management Strategies
The pharmaceutical industry’s shift towards digitalisation has significantly impacted quality management practices. The adoption of digital tools such as automated quality management systems and electronic document management systems exemplifies how technology can enhance operational efficiency. These tools enable real-time oversight, reduce human error, and improve traceability. Additionally, transitioning to a paperless environment reflects a broader industry trend towards leveraging digital solutions to streamline processes and boost accuracy.
Learning Management Systems (LMS) further underscore the role of digital innovation. These platforms facilitate continuous employee education, ensuring staff remain up-to-date with evolving protocols and regulatory expectations. In an industry where non-compliance can have severe consequences, such systematic training is invaluable for maintaining high standards.
Continuous improvement is a central tenet of quality management. Quality Improvement Plans (QIPs) focus on enhancing operational efficiency, reducing errors, and maintaining quality at every production stage. A "right first time" approach exemplifies this philosophy by minimising mistakes during laboratory work. Precision and accuracy are critical in pharmaceutical operations, where even minor errors can have far-reaching consequences for product safety and efficacy.
Decentralisation of Quality Responsibility
Another distinguishing feature of effective quality management strategies is the decentralisation of responsibility. Empowering line leaders to take ownership of quality within their areas ensures
that accountability is embedded at every organisational level. This decentralised approach fosters a culture where quality is a shared responsibility, integral to all processes and decisions.
Infrastructure and Supplier Quality Standards
Investment in state-of-the-art infrastructure underpins the ability to meet modern pharmaceutical and biotechnology demands. Advanced technology and specialised facilities enable organisations to handle complex projects while maintaining high standards. Additionally, rigorous supplier qualification programmes ensure that all materials and components meet stringent quality benchmarks. By prioritising supplier quality, companies strengthen their supply chain’s integrity, directly impacting the safety and efficacy of their products.
Adherence to global quality standards is validated by regulatory approvals from leading bodies such as the FDA, EMA, and PMDA. These certifications reflect a company’s compliance capabilities and ability to navigate complex international regulatory landscapes. Maintaining such approvals requires continuous vigilance and adaptation, underscoring the importance of a robust QMS.
The Future of Quality Management
As the pharmaceutical and biotechnology industries evolve, quality management remains a critical focus. The increasing complexity of operations and rising regulatory expectations demand a proactive approach to compliance and quality assurance. Insights from industry leaders highlight the importance of prioritising continuous improvement, embracing digital transformation, and fostering a culture of shared accountability to enhance quality management practices and maintain competitiveness.
Quality management extends beyond regulatory compliance; it is a comprehensive strategy that drives efficiency, innovation, and competitive advantage. A proactive approach — anchored in robust
systems, advanced technology, and a culture of excellence — serves as a model for these sectors. By investing in infrastructure, empowering employees, and upholding stringent supplier standards, organisations can meet current regulatory demands and position themselves for future success.
As global regulatory frameworks become more rigorous, the emphasis on quality will only increase. Companies that adopt advanced technologies, decentralise accountability, and cultivate a culture of continuous improvement will be better equipped to thrive. This journey underscores the transformative potential of quality management in safeguarding compliance while driving operational excellence in an increasingly complex industry landscape.
Alok Mehrotra
Syngene's CQO, Alok Mehrotra leads the Quality and Compliance function and is tasked with further strengthening the company's exemplary track record. He holds an M. Tech in Chemical Technology (Food Technology) from Harcourt Butler Technological Institute. He has more than 25 years of experience spread across Manufacturing Operations, Quality Assurance, Sustainability/ EHS, Production, and Supplier Technical Assurance across varied industries. Over the years, Alok has worked with leading corporates, including Dr. Reddy's, Reckitt Benckiser, Pepsi Foods Ltd, Godrej Foods, and Beverages Ltd. In his recent assignment as Head of Global Quality Management at Dr. Reddy's, he harmonised and integrated Quality management systems Globally and was also responsible for quality from all external suppliers and vendors.
Good Review Process – The New Panacea
Over the last 25 years, the way we write regulatory documents has evolved – from lean writing to technology that aids in populating the reports we write, things have gotten more streamlined. Which is a good thing, considering the goal of this endeavour is to get new drugs out to patients who need them as quickly as possible. Yet there is one area that has seen very little progress during that time: the review process by which teams of co-authors review and comment on drafts of a document.
The industry as a whole is striving to reduce the time needed to complete the regulatory documents we prepare. Statistics show that, on average, it takes about 8 weeks to prepare a clinical study report, ranging up to 33 weeks.1 Many people are looking to technology to make a dent in these times. However, technology can only really help reduce time on the first step of drafting an initial draft of any document. What it cannot do is replace the cycles of review and crafting of the text that we humans do to develop the storyline and ensure the focus is on the right data. Unfortunately, that is where most of the blood, sweat, and tears are spent on preparing these reports. A survey of members of the European Medical Writers Association found that the average time spent on review cycles is about 60% of the total time required to complete a clinical study report and can be as high as 73%.1 Although crafting a document depends on team discussion and it is an iterative process to refine the thoughts of interpreting the data, much of that 73% is due to inefficiencies.
So why is this process so inefficient? The review process has many different areas that need to work for it to run efficiently and many of these are often not done properly. There are 5 key problem areas
with the review process: a lack of focus and strategic input, poor prioritisation, revisiting previously agreed sections, comments that are late or unfocused, and too many reviewers. The good news is that these are all problems of poor process and/or a lack of understanding on the part of the reviewers about how to provide meaningful feedback. Through some best practices, reviews can be streamlined considerably by helping the reviewers understand how to avoid these problems.
Like with most things in life, doing something well begins with understanding what you are meant to do. How many people who are reviewing these regulatory documents ever have any training on what a good review looks like? Very few. In the absence of a clear concept of how to do something, most humans will fall back on how they have seen something similar done elsewhere and try to mimic that. So, what kind of role models do we have for providing feedback on documents we write? Generally, these are our school teachers and our parents who corrected things we wrote while at school. What did they do when they checked our documents? They got a big red pen and circled all the spelling mistakes and corrected all the commas. So that seems like the obvious thing to do when we provide feedback on these documents.
Yet, this could not be further from the mark of what is needed. The reason we have documents reviewed by teams of people is because different people see different things and bring a different background and perspective to what we are reporting. We also want to get input from each functional area (clinical, statistics, regulatory, safety, etc) on their area of expertise. What we need from these subject matter experts is their specialised input on content related to a particular topic. Their time is precious and their insight is valuable. When we
already have a mechanism in place to ensure grammar and spelling are corrected, these specialists should not be spending their time correcting those things, but rather the area for which only they can provide input.
Another way of looking at this is that we have compiled a team of people who all have a particular role to play. Like on any team, if someone is meant to be doing one thing but does something else, it makes the whole system unravel. A pitcher on a baseball team shouldn’t be running to the outfield to catch the balls. A violinist shouldn’t be grabbing the trumpet in an orchestra. A statistician shouldn’t be changing the text to reflect their personal preferences on English style issues (that have been agreed in a style guide). If the functional area experts spend their time correcting 100 commas, that is time they are not spending focused on the content. Teamwork depends on trust: each reviewer needs to trust their colleagues to do the piece they are responsible for.
So what should they be doing? Every study is done for a regulatory reason – to answer a specific question about how the drug works or what its safety profile looks like. Our job when reporting on that study is to explain what that question is and what answer we got to the question. This means reviews of any document need to be strategic and focused (Figure 1).
• A strategic review asks whether the text and data presented accurately and appropriately tell the story and answer the specific questions. If not, the reviewer should suggest how to do this. If yes, the reviewer can confirm it does it well.
• A focused review means the reviewer only reads specific parts of the document: those areas the reviewer provides content expertise (see examples in Figure 2) and those that need to be read in given round of review (i.e. if a section has been agreed previously, don’t read it again).
There is something called reviewer fatigue, which is the loss of concentration that sets in over the course of reviewing a document in a single sitting. Generally, a flurry of comments is made within
the first 10-15 pages, after which the number of comments drops off as the reviewer begins to lose focus. Sequential review of a document (i.e. reading the document from page 1 like a book) leads to reviewer fatigue in later sections of the document, which can result in the need for additional review cycles because reviewers did not effectively read those sections the first time through and then need to review them again. By applying focused reviews, where the reviewer does not start on page 1 each time but goes directly to the sections for which they need to provide content expertise, reviewer fatigue is avoided and the quality of the review of those sections is better.
From a process perspective, there are a few key rules that make for a good review practice (Figure 3). The overarching goal of these rules is to help reviewers think about how to make sure their feedback aids rather than hinders the process. Will a medical writer and the rest of the team be able to do something with their comments? There is no point in making the effort to comment on something if nobody knows what you are trying to communicate.
First, all comments should provide clear directions on what the reviewer is asking or thinking. Comments must be actionable and make clear what the reviewer wants done. Unactionable comments are those that do not inform or instruct, such as the following:
• “?”
• “Bad” or “Unclear”
• “Can’t you make it more convincing?”
• “Can we reword this?”
• “I think we said this better someplace else”
• “Perhaps we should be applying for a different dose”
By providing a comment that is unactionable, it will never be possible for someone to just take the comment and do something with it. They will have to come back to the reviewer to clarify exactly what should be changed. Medical writers and the rest of the authoring team should not be guessing about what the reviewer thinks the problem is. That is not an efficient means of communicating and it wastes everyone’s time.
Similarly, efficient reviews provide comments in full sentences. Many people have the odd idea that when writing something in a comment bubble it should be in shorthand. It happens frequently that for comments made in shorthand and incomplete sentences, not only does the medical writer not know what is meant, when the reviewer is asked what they wanted to say, they no longer understand what they wrote. So, although they took the time to read the text and write a comment about it, their idea is now lost because they did not write the comment as a full thought, and they do not remember what it was about. Not only was their time wasted, and the processing of their review slowed by the need to clarify the comment, we might have lost a really important idea! So, teach reviewers to give comments that will be clear to everyone who reads them (themselves included).
Proactive revision is more effective than posing a question or just making a comment about what should be changed. If a reviewer thinks a sentence can be improved, it is much more constructive to show the team what they are thinking by rewriting it with revision marks. They should not, however, spend time changing stylistic issues or proof reading. They should stay focused on the content they are responsible for. If needed, the medical writer will top and tail the text to ensure it complies with language and style requirements.
Reviewers should help the team to triage their feedback by prioritising their comments. There is never enough time to deal with every reflection that each reviewer has. Comments should be categorised into major (critical change that needs discussion with the team – you would not sign-off if this issue is not resolved or changed) or minor (content-related but not critical), where minor edits can be ignored if needed. When time is tight, it can speed up processing the review feedback by telling the writer and rest of team which points must be dealt with and which are just nice to have.
Remember that every review cycle counts. Reviewers should not think that because it is an early stage (e.g. Shell) they don’t need to pay as much attention. The whole point of having a detailed review of early drafts is to avoid needing to make big changes once the document is fully populated with lots of content. They need to think about each section carefully, even if there is no data yet (or dummy data) and imagine how the planned order of information will flow.
Giving good input early avoids the need for rewrites later that result in extra rounds of review and discussion.
Respect locked-down sections. It is essential for the process that reviewers do not revisit texts the team has already agreed on and now consider final. Their time and focus are now needed elsewhere in the document. By reviewing locked-down sections, they are inevitably going to rehash conversations that the rest of the team considers closed and this is not a good use of resources. In addition, if they spend the time allocated for a particular review reading sections they are not meant to be reading, they will need more time to complete the review. Which may delay completion of the review cycle. At a very minimum, they will be spending time on something that is not needed or effective for the process.
Although different functions perform different reviews that focus on their content expertise, there should be just ONE set of comments per functional area. Each functional area should work together to develop a consistent, cohesive position on a given topic. Reviewers should discuss things thoroughly within their function and find proper agreement before giving their comments back to the team. Not doing this results in inconsistent and often conflicting opinions on a particular topic. It is also not an efficient use of the team’s time during comment resolution meetings if people from the other functions have to sit and listen to 3 people from the same function debate their stance on something. A unified functional comment saves everyone time because the writer will have a clear instruction on what needs to be done, reducing the amount of follow-up needed.
Finally, for a review process to run smoothly, all reviewers must buy into and comply with agreed timelines for providing their feedback in each round of review. Each document is a wheel in the bigger machine of everything project teams are working on. They are rarely being written in isolation and project plans are juggling the activities of many people working on multiple documents and projects. Not providing comments on time slows down the whole process, throwing out timelines of other documents and teams.
In addition, it is disrespectful to one’s teammates if someone does not deliver on time. Every person on a team is working on several
Figure 3
projects and is up against multiple deadlines. Everyone has to plan their workload to make all their activities happen on time. Barring extenuating circumstances, if one person decides they can just take more time, they are implying that their time is more valuable than their teammates because they are, by definition, making the other people wait for them.
If comments come in late, it makes dealing with all comments less efficient. This is because the medical writer and the rest of the team need to know everyone’s thoughts before making any decisions on major rewrites or changes. Concerns about timelines that seem unfeasible should be raised right at the start of a project so that all interconnected activities can be adjusted. Or, at a minimum, if someone is not going to make a deadline, they should discuss it with the whole review team and agree together on an extension. This gives everyone the opportunity to juggle their priorities to accommodate the change.
In summary, there are several aspects of reviews that could be done more efficiently in most companies. The good news is that these are frequently the result of people not clearly understanding what they should be focussing on and the impact of their actions. By providing training to reviewers, they can learn these concepts and develop better, more effective review habits. If reviewers follow a few key rules for good review practice (Figure 3), the whole process can
and will be a more streamlined activity. Who knows, it might even be fun!
REFERENCES
1. Hamilton S. Effective authoring of clinical study reports: a companion guide. Medical Writing. 2014;23(2).
Julia Forjanic Klapproth
Senior Partner at Trilogy Writing & Consulting, an Indegene Company. After receiving her PhD in Developmental Neurobiology, Julia started her career as a medical writer in the pharmaceutical industry at Hoechst Marion Roussel (later Sanofi) in 1997. In 2002, Julia co-founded Trilogy Writing & Consulting, a company specialised in providing medical writing of regulatory documentation. She has also been President of the European Medical Writers Association (EMWA) twice and is an experienced speaker and trainer of medical writers, regularly running workshops for EMWA, American Medical Writers Association (AMWA), and pharmaceutical companies.
Next-generation AI Process Automation Essential to Cope with Soaring Regulatory Workloads, Survey Finds
A new survey of senior US regulatory professionals has confirmed a direct correlation between unsustainable regulatory workloads and planned AI investment. That’s as long as the function can accept advances in the technology and its demonstrated applicability in a pharma regulatory context, says ArisGlobal’s Renato Rjavec.
For the pharma/biopharma industry to ensure affordable access for patients as well as commercial viability as products grow ever more sophisticated, companies must become smarter in how they allocate resources to routine late-stage R&D processes, including regulatory workloads.
It is in this context that organisations are turning towards artificial intelligence (AI), and in particular next-generation technologies such as Generative AI (GenAI) powered by large language models (LLMs), even though confidence in the technology is taking time to catch up.
To understand the evolving balance between AI appetite and barriers to adoption, ArisGlobal recently commissioned a Censuswide survey, with 100 senior regulatory professionals in US pharma and biopharma organisations.
Regulatory Inefficiency is Intensifying
The poll, conducted in September, found that almost all (97% of) respondents had seen their regulatory obligations swell over the last five years, with three in five (60%) citing the increase as beyond what might be expected as the result of company growth. The trend is almost unanimously expected to continue over the next five years, with 41% saying next increases will be significant.
Particular process challenges include excessive time spent producing submissions/dossiers; maintaining labelling compliance; inputting data/documents into IT systems; verifying submission correctness/completeness; performing regulatory impact assessments; and locating data or documents in existing IT systems. Further
barriers to efficiency include responding to agency queries; inadequacy of current IT systems; and time lost to data quality checks, assessing submission readiness, and other administrativelydemanding preoccupations.
A lack of qualified people was not identified to be a great concern; preferred strategies do not involve allocating more people to processing regulatory workloads. Rather, pharma and biopharma regulatory functions are looking to smarter use of technology to ease the impact of their rising workloads.
Determining AI’s Value in a Regulatory Context
There is general acceptance of AI’s potential usefulness in solving information or process bottlenecks in a regulatory context, with 96% of survey respondents citing its current or potential value here, and almost half (45%) describing AI as “very useful”.
Almost all respondents could see direct potential for AI in transforming labelling compliance and deviations maintenance; capturing, searching, filtering the latest regulatory requirements; automating the intake of Health Authority interactions; automating regulated content translations for different markets; automating the authoring of responses to Health Authority queries; suggesting improvements to submissions/dossiers; performing regulatory impact assessments; authoring submission documents; automating document summarisation; and generating entire regulatory submissions.
Over a third (35%) of respondents claimed to be using AI for regulatory purposes in some form already, while 42% plan to invest in the next 18 months. A further 15% are looking at a timeframe beyond that, but do also have plans to roll out AI within the regulatory function.
Bases for AI Hesitancy
When asked what might be holding back initial or further investment in AI for Regulatory purposes, respondents most commonly cited outdated existing IT landscapes (45%); a belief that risks currently
outweigh the benefits (44%); and inadequate availability/quality/ consistency of data or content resources to derive the value from AI (42%).
Additionally, 39% of respondents felt the technology remained too immature/unproven; similarly, that the tools do not exist today to address their particular regulatory pain points. Sixteen per cent blamed a lack of trust in AI currently. This was ahead of budget challenges: only 15% named a lack of budget as a barrier to AI investment.
Drivers of Adoption
The research also identified the factors most likely to convert interest and inertia into active projects. Here, respondents most commonly cited the discovery that their competitors are using the technology (41%); soaring workloads/continued resource pressures (40%); advances with the technology (36%); the availability of specific tools geared to the tasks regulatory teams find most challenging or expensive (35%); and relevant IT systems becoming easier and more affordable to deploy (33%).
Beyond those drivers, 31% said updating their upgrades to existing IT set-ups (making it possible to use AI reliably) would prompt investment. Endorsement or recommendation of AI by regulators would inspire investment also for just under a third of respondents.
Barriers are Surmountable
Budget constraints did not appear to be a particular barrier to investment plans: just 18% indicated that the availability of new budget would unlock AI investment. That budget constraints are not a major barrier is encouraging, because hesitancy linked to “a lack of confidence to deploy” is surmountable and readily addressable now.
AI technology, including Generative AI (GenAI) is maturing and advancing at an accelerating pace, and specialist applications for target use cases in a life sciences regulatory context are being actively developed and piloted today, showcasing what is possible. This is in keeping with Gartner’s prediction that, by 2027, more than 50% of the GenAI models used by enterprises will be specific to either an industry or business function, up from just 1% in 2023.
Regulatory AI Has a Strong Trajectory
Finally, almost half (48%) of respondents agreed that, in time, AI
would transform a lot of routine regulatory work and considerably streamline processes, pointing to a strong future for the technology in a regulatory context.
Over 2 in 5 (43%) felt AI would drive up accuracy and quality in the information they produce for regulators and patients. Almost 2 in 5 (39%) respondents believed AI would be critical to the regulatory function’s ability to keep pace with market demands. And over a third (35%) of respondents agreed that AI would save a lot of time and money.
As the technology continues to mature, and as specialist GenAI solutions that target specific regulatory pain points with demonstrable benefits become available, the practical challenge becomes a question of “how”. How can companies take advantage of AI? How can they deploy it within their existing IT estates? How will they know how to use it, and that they can trust it?
Identifying a targeted use case to test what the technology can do offers a practical way forward here. Taking an agile, incremental, use-case-by-use-case approach to GenAI deployment will be faster, and represent lower cost and lower risk, than a big-bang “AI project”. It is also more likely to build engagement, as specific incremental wins are demonstrated.
REFERENCES
1. 3 Bold and Actionable Predictions for the Future of GenAI, Gartner, April 2024: https://www.gartner.com/en/articles/3-bold-and-actionable-predictionsfor-the-future-of-genai
Renato Rjavec
Renato Rjavec is the Senior Director of Product Management at ArisGlobal. He is shaping the future of regulatory information management, as well as quality management for life sciences. Renato has a keen focus on AI as a means for targeted automation.
Study Designs and Digital Technologies for Sustainable Clinical Trials
The clinical trial industry is essential for ensuring the safety and efficacy of new medicines; however, it is also a significant contributor of greenhouse gas emissions. The Sustainable Healthcare Coalition estimates clinical trials may be responsible for up to 100 megatons of CO2 emissions annually, about the same emissions produced by a country the size of Belgium.1 The main sources of these emissions are thought to include trialrelated travel by study participants and site staff, deliveries of trial equipment and energy usage by clinical study sites.2 Given the clear role of CO 2 emissions in climate change, the clinical trial industry must adopt new solutions to improve the sustainability, while enhancing patient access and retention as well as the patient experience. Those solutions that are showing promise include patient-centric study designs, virtual clinical studies, adaptive study designs, electronic patient-reported outcomes (ePRO) and the use of digital technologies.
Patient-centric Study Designs
Patient-centric study designs consider a patient's needs and preferences with the aim of reducing the burden on study participants. Those patient-centric approaches that may impact trial sustainability include the use of shorter trial durations and reductions in the frequency and number of required clinic visits e.g., using remote monitoring technologies or telemedicine. An example of a patientcentric study design can be seen in the CHIEF-HF trial, which aimed to evaluate whether the SGLT2 inhibitor, canagliflozin significantly reduces symptom burden in patients with heart failure.3 This trial was designed to reduce the need for in-person visits with direct engagement of patients through a study website, electronic informed consent, direct home delivery of study medication, reporting of the primary endpoint by a mobile application, and use of a Fitbit to monitor activity.3 Patient-centric approaches can make trials more convenient for patients through fewer patient journeys, which in turn can lower fuel consumption and help reduce CO2 emissions. The involvement of patients in the design of clinical trials can also benefit patient recruitment and retention. It is estimated that more than 80% of clinical studies face problems with study recruitment resulting in delays and the need for additional study sites.4 Furthermore, almost a quarter of participants involved in cardiovascular clinical studies drop out before completion.5 Through improved recruitment and retention, patient-centric approaches can help reduce the number of participants required to meet the study outcomes and the resources needed to complete the study.
Decentralised Clinical Trials
Decentralised clinical trials (DCTs) describe trials where some of the trial activities take place at sites other than the clinical investigation site, such as the patient's home or a local clinic. DCTs often involve the use of digital tools and platforms to facilitate communication between research teams and study participants. A notable example includes
the DeTAP study – a trial, involving the physiologic monitoring of patients with atrial fibrillation receiving oral anticoagulation therapy.6 The DeTAP study utilised telemedicine, eConsent, remote monitoring devices, and ePROs collected via a mobile application to fully decentralise research activities.6 DCTs can improve trial sustainability by reducing the distance or frequency participants travel to clinical study sites and CO2 emissions. The improved accessibility associated with DCTs can help speed up study recruitment, reducing timelines and the need for additional study sites. DCTs can also lead to the more efficient use of energy resources through reduced reliance on physical study sites and the generation and handling of study-related paperwork. However, the manufacture, distribution and use of digital tools and data storage platforms can also contribute to energy usage and CO 2 emissions. Nonetheless, one study estimated that the full digitalisation of a traditional clinical study could lead to a more than 90% reduction in energy usage.7
Adaptive Study Designs
Adaptive study designs allow for the modification of an ongoing clinical trial design based on evidence accumulated during the study. This is in contrast with traditional clinical studies, where the study design is fixed at the outset and does not change during the course of the study. These modifications may include refinement of sample sizes, targeting specific patient subpopulations, treatment or dosage regimens, study endpoints and trial duration. Adaptive trial designs have been utilised in cardiovascular studies such as the ANTHEMHFrEF, a study evaluating the impact of autonomic regulation therapy in heart failure patients, which employed a Bayesian approach for sample size selection.8 Adaptive study designs can help improve trial sustainability by reducing participant numbers and study timelines, potentially reducing the resources needed to complete the study. The optimisation of clinical trial designs can also help increase the likelihood that the study will meet its endpoints, saving the time and resources spent on failed studies.
Electronic Patient-reported Outcomes
ePROs are health outcomes directly reported by patients by means of electronic devices such as smartphones, tablets or web applications. ePROs are commonly used in clinical trials to provide information about a participant's symptoms, daily functioning, quality of life and response to therapy. Notably, more than a quarter of the clinical trials currently being conducted are thought to involve ePROs.9 The CHIEF-HF study is one such study, where the outcomes were assessed using an app version of the Kansas City Cardiomyopathy Questionnaire for quantification of symptom frequency and severity, and Fitbit monitoring of daily step counts.3 ePROs can enhance study data quality by enabling more frequent and real-time reporting of a study participant's health status. ePROs can contribute specifically to trial sustainability by reducing the number of appointments participants need to attend, thereby reducing required clinic time and trial-related travel and its environmental impacts. Additionally, the incorporation of ePROs
into study designs can conserve resources by reducing study-related paperwork and the associated administrative burdens.
Digital Technologies
Digital technologies are increasingly being used to enhance data quality within the clinical trial setting by enabling the continuous and remote collection of health data from study participants during their everyday activities. These digital technologies range widely and may include software (e.g., smartphone apps), and hardware (e.g., wearables) solutions. The number of clinical trials incorporating digital technology was estimated at 11% in 2020 and is projected to reach 70% by 2025.10 One example is the LINK-HF study, which utilised a multi-sensor disposable patch to continuously monitor a range of physiological parameters and detect oncoming heart failure exacerbations.11 Another recent example, the eBRAVE study, utilised a smartphone app to screen for pulse wave irregularities and detect atrial fibrillation in individuals at risk of stroke.12 Digital technologies can contribute to trial sustainability by reducing the need for participants to travel to study sites to have measurements performed and conserving energy associated with study site operations. Additionally, digital tools can provide more accurate and real-world data as well as additional data points, helping to reduce the likelihood that the study will need to be extended or repeated.
Sustainable Solutions Driving Clinical Trials
New solutions are crucial for improving the sustainability of the clinical trial industry and reducing its contribution to climate change. The main contributing factor is CO2 emissions, resulting from trialrelated travel and energy usage by clinical study sites. Those solutions showing promise in this regard revolve around adjustments in study designs or the incorporation of digital technologies. These solutions can also improve recruitment and retention by enhancing the clinical trial experience for study participants. However, several barriers exist to their adoption, including concerns over data security and privacy, data quality, participant safety in the absence of physical visits, and accessibility for those with poor computer literacy. Addressing these concerns will not only help minimise the environmental impact of the industry but also help ensure the continued development of new medicines.
REFERENCE
1. https://shcoalition.org/clinical-trials/, visited on 9 October 2024.
2. Sustainable Trials Study Group. Towards sustainable clinical trials. BMJ.
334(7595), 671–673 (2007).
3. Spertus, J.A., Birmingham, M.C., Nassif, M., et al. The SGLT2 inhibitor canagliflozin in heart failure: the CHIEF-HF remote, patient-centered randomized trial. Nat. Med. 28(4), 809–813 (2022).
4. Desai, M. Recruitment and retention of participants in clinical studies: Critical issues and challenges. Perspect. Clin. Res. 11(2), 51 (2020).
5. Hutchison, E., Zhang, Y., Nampally, S., et al. Modeling Clinical Trial Attrition Using Machine Intelligence: A driver analytics case study using 1,325 trials representing one million patients. medRxiv. 2021.11.12.21266277 (2021).
6. Sarraju, A., Seninger, C., Parameswaran, V., et al. Pandemic-proof recruitment and engagement in a fully decentralized trial in atrial fibrillation patients (DeTAP). NPJ Digit. Med. 5(1), 80 (2022).
7. Kohl, S.H., Schmidt-Lucke, C. Clinical trials to go green–A sustainable argument for decentralised digital clinical trials. PLOS Digit. Health. 2(10), e0000366 (2023).
8. Konstam, M.A., Udelson, J.E., Butler, J., et al. Impact of Autonomic Regulation Therapy in Patients with Heart Failure. Circ. Heart Fail. 12(11) (2019).
9. Vodicka, E., Kim, K., Devine, E.B., et al. Inclusion of patient-reported outcome measures in registered clinical trials: Evidence from ClinicalTrials. gov (2007–2013). Contemp. Clin. Trials. 43, 1–9 (2015).
10. Chandrasekaran, R., Katthula, V., Moustakas, E. Patterns of Use and Key Predictors for the Use of Wearable Health Care Devices by US Adults: Insights from a National Survey. J. Med. Internet Res. 22(10), e22443 (2020).
12. Rizas, K.D., Freyer, L., Sappler, N., et al. Smartphone-based screening for atrial fibrillation: a pragmatic randomized clinical trial. Nat. Med. 28(9), 1823–1830 (2022).
Dr. Bipin Patel
Bipin Patel Ph.D. is the CEO and Founder of electronRx, a deep-tech startup developing novel chronic disease and hospital patient management solutions. He is a key digital health thought-leader with over 20 years’ experience in medical engineering, drug development and commercialisation and holds a PhD in Medical Engineering from UCL, UK.
Email: enquiries@electronRx.com
The Role of Company Culture in Functional Service Provider (FSP)
Partnerships
Pharmaceutical and clinical research sponsors (clients) have long outsourced clinical development work, utilising both fullservice contract research organisations (CROs) and functional service provider (FSP) models. FSPs play a critical role in clinical research by offering tailored solutions to clients with diverse needs. In recent years, many clients have realised the benefits that narrower, functionally specific FSP models provide. These include increased operational and resourcing flexibility along with different pricing plans in comparison to traditional full-service CRO offerings. However, domain knowledge, technical strengths and technology aspects of these partnerships are not always enough to guarantee success.
Culture as a Cornerstone of FSP Partnerships
Company culture can and does serve as a critical foundational element for effective collaboration, addressing challenges, and driving sustained achievements that allow clients to succeed at their research. A deliberate focus on culture – both within the FSP engagement and in alignment within sponsor organisations – can elevate partnerships from transactional relationships to transformative alliances. This article explores the interplay of CRO, client, and FSP cultures, the role of intentionality in creating successful collaborations, and how these cultural dynamics result in long-term research and operational successes.
The Client’s Culture: The Roadmap to Success Catalyst Flex emphasises that understanding a client’s culture is a non-negotiable first step in forming a successful partnership. This cultural understanding serves as a roadmap that informs how FSPs interact with clients, communicate expectations, and align service delivery. For FSPs, culture influences every facet of its operations, from initial conversations to ongoing project management.
The Importance of Understanding a Client’s Corporate Culture
Clients’ cultures vary widely, shaped by their size, operational maturity, and therapeutic focus. For instance, startups with minimal infrastructure may demand resource professionals capable of thriving in dynamic, less-structured environments. In contrast, larger, established organisations often prefer staff who excel in independent, process-oriented roles. By actively engaging with clients to assess these factors, an FSP should ensure a seamless cultural fit, setting the stage for efficient and productive collaborations.
Listening as a Key to Cultural Alignment
A service provider should prioritise active listening during the early client engagement as well as during ongoing interactions throughout the engagement phases. Active listening goes beyond passively hearing a client’s needs – it involves internalising their goals, reflecting insights back to them, and adjusting approaches based on their feedback. This practice demonstrates commitment to building trust and understanding client priorities at a deeper level. It also sets a precedent for open communication throughout the partnership.
For instance, during intake discussions where clinical monitoring or project management roles are being considered, Catalyst enquires
about aspects such as communication preferences, decision-making styles, and site-level operations. As an illustration, some clients require professional research resources adept at maintaining close relationships with research sites, while others seek professionals who can manage broader project oversight. Tapping professionals with in-depth skills and ensuring they align with corporate culture is a successful combination. These tailored interactions illustrate deliberate approaches to client engagement.
Internal Culture: A Model of Intentionality
An organisation’s internal culture should be defined by its core values. At Catalyst Flex, ours are listening, learning, flexibility, commitment, and collaboration, which help us provide unwavering support to our full-time staff, contract research professionals and clients. Such values form the bedrock of the organisation and are deliberately reinforced at every level.
Core Values Driving Organisational Behaviour
Corporate cultures do not just drift toward greatness. This is also true of FSP relationships with clients. In these engagements, there is a joint culture that must be established between the FSP and the client, which must be cultivated with precision and sustained through intentional practices. A core aspect of an FSP culture should be around creating a safe space for employees and resources. Professionals will be encouraged to express their concerns, provide candid feedback, and acknowledge areas where they may require additional support. This approach fosters trust and accountability, allowing team members to operate with confidence in their roles.
Empowering Learning and Growth
An FSP model that invests heavily in the professional and personal growth of its employees and resources helps enrich professional and personal lives. As we have found, a dedicated learning and training team ensures that team members have access to the resources and support they need to excel in their roles. These programmes not only enhance technical skills but also reinforce cultural values like adaptability and resilience, ensuring that research professionals are well-equipped to navigate diverse client environments.
Bridging Cultures: Aligning FSP and Client Values
An FSP should excel in aligning its culture with the unique dynamics of its clients. This alignment begins with a detailed intake process, such as the one Catalyst Flex uses, which assesses both technical requirements and cultural nuances. Key considerations include the client’s size, infrastructure maturity, and management style. For example, smaller clients often require more frequent communication and hands-on engagement, while larger organisations may prefer a more independent approach.
Tailored Matching Processes
This cultural alignment extends to resource allocation. The FSP needs to carefully match resources with clients based not only on technical expertise but also on compat ibility with the client’s cultural environment. In our experience, an FSP’s deliberate matching process can result in long-term relationships, with some resources remaining with the same client for over five years. This model of an FSP can
prove its value through team retention numbers and repeat business, which reinforces the intentional approach.
Creating
Feedback Loops for Continuous Improvement
While Catalyst Flex views feedback as a gift that enhances relationships and drives operational improvements, not every FSP organisation or client does. By maintaining open communication channels, an FSP organisation can encourage both clients and resources to share their experiences and insights. This feedback informs the FSP’s approach to future engagements, ensuring that all parties feel heard and valued.
Building
Mutual Trust and Respect
A defining strength for an FSP is its ability to create environments where clients and resources feel mutually respected. Resources are treated as integral members of the client’s team, fostering a sense of ownership and collaboration. This approach not only enhances project outcomes but also builds loyalty among both clients and professionals. For instance, some resources have transitioned overtime from resource roles to managerial positions within Catalyst Flex, reflecting their long-term commitment to the organisation.
Measurable Outcomes of Cultural Alignment
High Retention Rates
Catalyst Flex has found an intentional approach to culture has resulted in exceptional retention rates. The organisation has a 99% project retention rate, a figure that underscores its success in fostering a supportive and empowering environment. A high retention rate benefits clients, resources, and an FSP by ensuring continuity and stability in long-term engagements.
Sustained Client Relationships
Emphasis on cultural alignment also translates into strong, enduring client relationships and long-term partnerships. Many clients return to an FSP for subsequent projects, often requesting the same resources who have proven to be a good fit. Other clients request Catalyst FSP services once they land in other organisations. We have seen high levels of repeat business from clients – nearly 80% – and from resources who have enjoyed their collaborations with the FSP group and come back for new engagements and recommend new resources into our network. This repeat business highlights the trust and satisfaction that clients associate with the intentional cultural approach.
Conflict Resolution as a Byproduct of Cultural Understanding
A well-aligned culture also facilitates conflict resolution. When challenges arise, the shared understanding between the FSP and its clients allows for open, constructive conversations. This proactive approach to problem-solving strengthens partnerships and ensures that issues are addressed collaboratively, minimising disruptions to project timelines or resulting in finding a replacement FSP.
Creating a Culture of Empowerment and Innovation
An intentional culture at an FSP helps to empower resource professionals to excel by providing them with the tools and support they need to succeed. This includes tailored onboarding, ongoing mentoring, and opportunities for professional growth. Resources are encouraged to be proactive, take ownership of their roles, and contribute meaningfully to their teams. All of which differentiates the FSP.
Fostering Innovation Through Collaboration
The synergy between an FSP’s culture and its clients’ environments creates fertile ground for innovation. By aligning values and expectations, the FSP can facilitate seamless collaboration, enabling teams to develop creative solutions to complex challenges. This
innovative spirit drives research success and reinforces the FSP’s reputation as a trusted partner.
The Human Element in Clinical Research
At its core, an FSP model should prioritise the ubiquitous human element of clinical research. By valuing individuals and fostering connections, an FSP organisation can create partnerships that are as meaningful as they are effective. This focus on people – whether they are clients, resources, or employees – distinguishes an FSP organisation in a competitive industry, making it a model for other FSPs.
Culture as a Strategic Asset in FSP Partnerships
Catalyst Flex’s approach to culture exemplifies the transformative potential of intentionality in clinical research partnerships. By prioritising cultural alignment, empowering professionals, and fostering open communication, we have achieved remarkable outcomes for our clients, our team members, and our resources. The organisation’s success serves as a compelling case study for the importance of culture as a strategic asset in FSP operations.
Our experiences underscore how deliberate corporate cultural practices drive trust, collaboration, and long-term success in FSP partnerships.
Allison Crumpler
Allison Crumpler, Vice President of People & Culture, Catalyst Clinical Research, has worked in clinical research for over 15 years. She is a Professional Certified Coach, at International Coaching Federation.
Allison has served as an internal coach providing executive, leadership and team coaching to senior executives. She designs and delivers leadership training and onboarding programmes and oversees clinical technology trainers. Allison focuses on creating an organisational culture with competitive recruitment and retention advantages.
Svetlana Kolchinsky
Svetlana Kolchinsky brings over 20 years of experience in clinical operations and leadership. She joined clinical research overseeing operational aspects of investigator-initiated clinical trials.
Svetlana is passionate about building strong operational teams and supporting them in project delivery and client governance oversight roles. In 2020, Svetlana joined Catalyst Clinical Research. She leads the Catalyst Flex clinical operations team and works closely with her counterparts to build FSP solutions for Catalyst clients.
Research and Development
Targeting the End of Malaria by 2050
Malaria continues to have a significant impact on half of the world’s population who live in tropical regions. It claims 600,000 lives each year, the majority of whom are children under 5-years of age. Over the last century, scientific advances have managed to bring the death toll down from its historic annual peak of around five million fatalities, but even with today’s vaccines, mosquito nets, and indoor residual pesticide spraying programmes, we have not yet eradicated the disease’s threat.
Some would go further and express concerns that we are losing momentum. As Peter Sands, Executive Director of the Global Fund puts it: “More than ever, we are at risk of losing our fight against this disease. Progress has ground to a halt, and in some places is reversing. Unless we act now, malaria could resurge dramatically.”1
Malaria is caused by a parasite, transmitted to the human bloodstream through the bites of infected mosquitoes. Once the parasite is inside the body, it multiplies, attacking red blood cells. This causes symptoms like chills, flu-like symptoms, extreme exhaustion, and more. Without treatment, the effects of malaria can become severe and even fatal, especially for children and pregnant women. Severe malaria can even cause permanent brain damage to unborn children.
The Relationship Between Malaria and Economic Development
It is undeniable that the prevalence of malaria in modern day is linked to inequities in access to health innovation. There is a direct correlation between health access and economic development. While the efforts to prevent and manage malaria have significantly lowered the disease's impact in numerous nations in recent years, such as India, Sri Lanka, and Vietnam, more detailed evaluations of health outcomes reveal that poor and vulnerable populations miss out on the benefits of these malaria control initiatives.
An estimated 58% of malaria deaths occur among the poorest 20% of the world’s population.2 This staggering figure results from numerous contributing factors: inadequate housing and overcrowding without use of mosquito preventatives, poorly paid farming occupations, nearby mosquito-filled forests, malnutrition that limits the body’s ability to fight malaria, and lack of access to preventative agents. Note that in many of these contexts, infrastructure related to economic development impacts the lifestyle of communities which increases the risk of contracting malaria and vector borne diseases in general.
Malaria control relies heavily on prevention, with prompt treatment being the most crucial method to avoiding malaria-related deaths. However, when the poorest, most vulnerable in society lack access to preventative measures or quality treatment, the disease is free to spread.
As reported by WHO in 2022, four African countries accounted for almost 50% of all malaria deaths worldwide: Nigeria (26.8%), the Democratic Republic of the Congo (12.3%), Uganda (5.1%) and Mozambique (4.2%).3
This data showcases the inequity in treatment of the disease in countries where large populations are living in poverty, extreme poverty, or beginning to recover from poverty. In these nations, poverty is heavily linked to lack of healthcare access, and therefore, lack of access to preventative measures and treatments of malaria.
Several high-GDP countries with subtropical and tropical regions once struggled with the prevalence of malaria but saw the incidence of malaria cases decline rapidly in line with the development of their economies. The United States of America is a clear example of this often-untold history.
Malaria was brought to the Americas during the transatlantic slave trade, primarily through infected African slaves and European colonisers. In particular, the strain of malaria (P. falciparum) carried by enslaved Africans was deadly. It led to the establishment of malaria as a major health problem in the southern region of the USA.
In hot climates such as South Carolina, the disease was rampant in sugar, cotton, and rice plantations. The US was plagued by malaria until the mid-20th century, infecting presidents from Washington to Lincoln, weakening Civil War soldiers by the hundreds of thousands, and draining the country’s physical and economic health in the process.
The nation saw a burst of hope in the 1930s when the Tennessee Valley Authority modernised the rural south with hydroelectric power. But just as the United States was eradicating its last pockets of infection, malaria wormed back into the USA’s spotlight during World War II. During the early days of the Pacific campaign, more soldiers were killed by malaria than by enemy forces.
The leading public health agency in the United States, the Centers for Disease Control and Prevention (CDC) was founded in 1946 to combat malaria. Through heavily funded mosquito control programmes, surveillance, and publicity campaigns denouncing the insects, in combination with a robust private market, malaria was officially eradicated from the USA in 1951. Today, the CDC remains very involved with malaria research around the globe.
This history of malaria in the USA is a prime example of the power of economic development when combatting the mosquitotransmitted disease. As the USA transformed into a developed country, the nation had the national resources and the private sector market to eradicate malaria, even back in 1951. Economic development also brought changes to how people lived. Better sewage and waste management systems, transition away from unclean
tight living quarters, and changes to animal rearing and agricultural practices all changed proximity to breeding grounds for mosquitoes.
In contrast, nations with high levels of poverty remain disproportionally plagued by the disease today.
Vaccines – The Latest Breakthrough in Malaria Prevention
The high-profile research breakthroughs we have seen in the last decade include the development and rollout of the RTS,S vaccine, commonly known as Mosquirix.4 In Phase 3 clinical trials conducted between 2009 and 2014, the RTS,S vaccine that was given in 4 doses over 18 months reduced severe malaria cases by 22% and deaths by 13%4 in children aged 5–17 months. With repeated booster vaccines over a 4-year period, efficacy can reach up to 36%.5
Since then, we have seen pilot programmes start in 2019 in Ghana, Kenya, and Malawi which vaccinated over 1.7 million children. The World Health Organization’s (WHO) modelling study estimates the vaccine could prevent 5.4 million cases of malaria and 23,000 deaths annually in the vaccinated population. Global estimates of malaria cases are 249 million.6
Based on the success of pilot programmes, in October 2021 WHO recommended the widespread use of the RTS,S vaccine for children
Research and Development
in areas of moderate to high malaria transmission. The decision represented a landmark in global malaria control efforts, but we do not yet have the panacea the world is waiting for.
RTS,S is seen as a complementary tool, rather than a standalone solution. Mosquirix offers approximately 30% protection against malaria in children during the first year after vaccination,7 with its efficacy dropping over time which requires the need for booster doses. These efficacy numbers are only attainable in areas where insecticide-treated nets are widely used and there is good access to diagnosis and treatment.
Plus, malaria vaccines on the market do not provide complete immunity, meaning that vaccinated individuals can still contract the disease, though they are likely to experience less severe symptoms. Protection provided by vaccines is also strain-specific, primarily targeting Plasmodium Falciparum, the deadliest species of malaria parasite, and offering limited defence against other species such as P. vivax
Vaccines are part of the solution, but beyond the scientific challenge of discovery, safety, and efficacy, there are also logistical challenges such as cold storage of the vaccine and the practicalities of multi-dose programmes, and high costs of widespread administering of vaccinations, especially in resource-limited areas.
Other Interventions
When used properly, mosquito nets are one of the most effective tools for preventing mosquito-borne diseases. However, as a preventative measure it does suffer from several drawbacks. It is common for untreated nets to have holes in them, trapping mosquitos inside and attracting more bites as a result. In hot, humid conditions, such as the subtropical regions where malaria is rife, the netting can also be too thick and uncomfortable to sleep underneath, limiting air circulation and discouraging people from using them altogether.
Other options include residual sprays that can help reduce the mosquito population in specific areas by coating surfaces where mosquitoes are likely to land with insecticides. While effective to deter mosquitos, these sprays can also affect other species, including beneficial insects like bees and butterflies, and can even pose health risks to humans when stringent safety guidelines aren’t followed.
Research and Development
Symptoms of exposure can include respiratory issues, skin irritation, or more severe health effects with long-term exposure – again, deterring individuals from using the sprays. This preventative method is also less effective in very high humidity or when washed away by rain, limiting its convenience in high malaria-risk areas.
The reality is there is no silver bullet. We need an integrated vector control system that gives people and public health agencies more options to design the best system to suit their context.
Battling a Mutating Species
Fighting malaria is like fighting several diseases at once. There are 156 different species of Plasmodium, the parasite that carries malaria. Five of these species infect humans. This doesn’t count other vector borne disease such as dengue, zika, and others.
Each species goes through six different lifecycles and each lifecycle is like dealing with a completely different creature, similar to the difference between a pupa and a butterfly. This is why creating a vaccine is extremely difficult. This is further complicated by the speed the parasite mutates. Mosquito mutation also compounds this challenge. This is why insecticides become increasingly less effective over time for insecticide treated bed nets and indoor residual sprays, the primary tools currently used to eliminate the spread of malaria.8
Recently, it’s been reported that the species of mosquitos which carry malaria, typically night biters, are biting earlier in the day. This limits the effectiveness of pesticides and indoor residual sprays which focus on protection in the home.
This complex context is why there is a need to expand the pool of integrative tools. This is already common practice in many parts of the United States where a combination of local government control measures combined with private sector activities play an important role in mosquito population control. These government programmes, such as on-going seasonal surveillance, larvicide, and aerial fumigation work best in combination with private sector practices like mosquito repellent use and home pest control services. Therefore, developing countries must find the right integrative solutions that fit their circumstances.
Mosquito repellents have significant potential as part of an integrative set of tools to combat mosquito borne diseases. Unlike pesticides, because repellents do not kill, insects do not become resistant over time.
The Case for a New Generation of Mosquito Repellents
Historically, mosquito repellents have proven inconclusive as an effective prevention tool in community-wide studies for vector control. Discussions with vector control experts indicate this has primarily been due to problems with adoption and adherence during studies. In short, people don’t like using the current mosquito repellent options on a daily basis.
A new type of repellent is needed, one that solves the adoption and adherence challenges in current repellents. A generation of mosquito repellents that are safe for daily use, do not require reapplication, and can fit into the daily skincare routine of communities most at risk of vector borne diseases.
The UK has many public health challenges, but malaria is not one of them. Nevertheless, the country has some of the world’s leading scientists and institutions focused on infectious disease. These include the major research-led universities and other
specialist research centres such as Liverpool School of Tropical Medicine.
Based around a famous deep-water port with a long history of global trade, Liverpool City Region is also the UK location of LivFul. Our organisation joined the conversation in 2015 from a strategic base on the Sci-Tech Daresbury campus, a national science and innovation campus. The organisation set out to develop an improved repellent designed to prevent mosquito-borne infections.
As a Co-CEO and Co-Founder of LivFul, I grew up in Nigeria and suffered several malaria infections by the age of 10. I saw first-hand its impacts in my community and family. But motivated to change my situation, I created my own repellent in my bathroom from simple household products. Only later did I learn that repellents already existed – I just didn’t have access to them. The experiences I had as a child all helped me to realise that lack of access is a systemic issue, so I co-founded LivFul to find a holistic way to solve health access.
After moving to the United States as a teenager, I devoted my career to forming a company to work on a more effective insect repellent. Our small team at LivFul began research into tangibility by speaking with individuals in developing countries who attend to the malaria epidemic every day – including health professionals and locals in malaria-afflicted communities.
We sought to create a product that avoided some of the common complaints about existing mosquito repellents – that they were often sticky, uncomfortable, not long lasting, and carried an unpleasant odour.
LivFul went on to produce an enhanced insect repellent (EIR) featuring IR3535®, a well-known active ingredient recognised in the scientific community for effectively repelling mosquitoes without posing health or environmental risks. To enhance its effectiveness, LivFul also developed a patented technology, STAYTEC™, which works alongside IR3535® to ensure the repellent lasts on human skin for over 14 hours.
LivFul has set out to make long-lasting, affordable repellents desirable to fit into daily skincare routines, comparable to the use of moisturiser.
In partnership with the pharmaceutical and chemical manufacturers Merck KGaA, LivFul tested the repellent (consisting of the active ingredient IR3535® and LivFul’s patented STAYTEC™ technology).
The 16-hour study involved placing the arms of volunteers into boxes filled with 200 female mosquitos to test the effectiveness of the repellent over long time periods. Prior to the study, mosquitos were starved for 24 hours to ensure they would be most aggressive and hungry during this time. Upon fully passing this study, the same test was carried out in Ghana in partnership with the University of Ghana’s Noguchi Memorial Institute for Medical Research.
The next study involved testing against the Anopheles Gambie species of mosquito, the deadliest in the world. LivFul partnered with Noguchi in Ghana to test their repellent’s success against this species, as many repellents fail to achieve consistent success against the Anopheles Gambia species due to their erratic, aggressive nature. However, LivFul’s repellent passed with flying colours, where no volunteers were bitten or therefore infected during the study.
During their continued success throughout Africa, LivFul began gathering data in Congo and Uganda, showcasing their repellent’s
Research and Development
more than 90% adoption and adherence rate. Everyone was surprised to see that when using LivFul’s repellent, people weren’t getting bitten at all – despite concerns over human error and the possibility of ‘missing a spot’. LivFul measured the levels of malaria in the blood of users, and after 60 days, users were clearer of the disease than ever.
Earlier this year, LivFul received the final report of a study conducted with the Noguchi Medical Institute in Ghana. The significant results of the study were shared by Merck KGaA at the American Mosquito Control Association annual meeting, the National Malaria Eradication Program meeting in Ghana, and by LivFul to the NMEP in Nigeria.
Not only did its EIR demonstrate maintained repellency (reaching 100%–96.2% over 9 hours), the test had a significant impact on the Entomological Inoculation Rate, while also showing high efficacy against mutating mosquitoes; more than 98% fewer landings over a 16-day period.
To assess the repellent’s economic viability in impoverished regions, LivFul conducted a trial with Ghanian security guards who earn $3 a day. At the end of the trial, security guards talked about the quality-of-life improvement as a result of using the repellent, which not only protects them from a deadly disease but acts as a luxury item in everyday life. While security guards couldn’t justify the purchase of items like lotion or deodorant, LivFul’s repellent sachets offer an array of benefits such as a pleasant odour, moisturising properties, as well as 14 hours of mosquito protection, rationalising the purchase.
LivFul Founder, Hogan Bassey, was a typical child growing up in the suburbs of Lagos, Nigeria. He went to school, played sports with the neighbourhood kids, and contracted malaria multiple times. As he grew up, he realised a lack of access to basic health solutions an endemic and structural problem in global healthcare. His "success" in disease prevention motivated him first to pursue a solution to the problem of insect-borne diseases. Hogan's determination resulted in the creation of a new generation of repellent. LivFul Inc. is a company that supports better health with ground-breaking science and oneof-a-kind products, beyond vector-borne disease control.
An Interview with Dr. John Stone, Steritas
John Stone, MD MPH, Chair, Scientific
Advisory Board, Steritas
There is increasing, and necessary, attention on an open secret: the epidemic of steroid-toxicity. We sat down with Dr. John Stone, Professor of Medicine at Harvard Medical School, rheumatologist, the Edward A. Fox Chair in Medicine at the Massachusetts General Hospital, and Chair of the Scientific Advisory Board at Steritas. To learn more about the impact of steroid exposure on patients and healthcare systems, we posed a series of questions including about The Great TaperTM initiative, a public health movement to alter steroid-prescribing patterns. After graduating from Harvard Medical School, Dr. Stone completed his internal medicine training at Johns Hopkins University School of Medicine and a rheumatology fellowship at the University of California-San Francisco. Before being recruited to Mass General in Boston, he co-founded and directed the Vasculitis Center of Excellence at Johns Hopkins in Baltimore. His expertise is in vasculitis, a group of inflammatory diseases that target blood vessels. Dr. Stone is also a thought leader on a newly-described multi-organ condition known as IgG4-related disease (IgG4-RD). He has led pivotal trials in ANCA-associated vasculitis, giant cell arteritis, and IgG4-RD, all leading to the worldwide approval of new treatments for those diseases.
Since 2015, drawing on 25 years of experience with glucocorticoid treatments for his own patients, Dr. Stone has focused on the measurement and prevention of glucocorticoid toxicity to help transition the treatment of patients with inflammatory diseases into an era of safe, effective, steroid-toxicity sparing medications. Recognising the need to research, monitor, and reduce the use of steroids in patients and populations, he convened an international group of experts to lay the foundations for development of the Glucocorticoid Toxicity Index (GTI), now part of the STOX Suite, a collection of validated clinical outcome assessment (COA) to measure steroid-toxicity.
Q. How are steroids used in the treatment of autoimmune and autoinflammatory diseases?
A. The first clinical use of steroids was in 1948 for the treatment of a young woman with rheumatoid arthritis, a medical breakthrough that earned the researchers the 1950 Nobel Prize in Medicine. Since then, steroids have been the first-line treatment for dozens of diseases caused by inflammation. Steroids are a class of medications that includes cortisone, hydrocortisone, prednisone, prednisolone, and others. Together, these medications are referred to as glucocorticoids, or corticosteroids. Glucocorticoids are useful in treating inflammatory diseases such as arthritis, asthma, Crohn’s disease, lupus, vasculitis, autoimmune blistering skin conditions, and sarcoidosis.
Steroids suppress the immune system quickly and are employed in the interest of exerting fast-acting control over inflammation by
inhibiting the initial events of an inflammatory response, including reducing vasodilation and decreasing white blood cell emigration to the site of injury or inflammation. Glucocorticoid receptors are expressed throughout the body, making steroids effective against almost all immune cells, but also leading to multiple potential offtarget effects.
Within 23 days of that first steroid dose administered in 1948, the first steroid toxicities were reported and thus began the first steroid taper to a lower therapeutic dose. Since then, leading experts have been looking to research, monitor, and reduce the use of steroids in patients and populations. But progress in tapering glucocorticoids has been slow, due largely to the lack of effective alternatives, consensus among clinicians on which health domains to measure and how to measure them, and the technology to make such measurement practical. Now we have the tools to quantify steroid-toxicity, we’re in a position to accelerate a shift in treatment options. This includes recent developments in our understanding of the glucocorticoid receptor itself, opening new avenues for novel steroid-sparing treatments with fewer damaging side effects.
Q. What are the potential impacts of long-term steroid use on both patients and healthcare systems?
A. Steroids play a role in treating many diseases, and yet, they cure essentially none of them. While steroids may provide relief in the short-term, the side effects that accompany both short and longterm usage can be severe, including diabetes, neuropsychiatric manifestations, osteoporosis, bone fractures, cataracts, and cardiovascular events.
Over 80 steroid-toxicities have been documented, ranging from inconvenient to life-threatening. In paediatric illnesses, the detrimental impact of steroids can be especially cruel and persist into adulthood – affecting growth and disrupting the psychosocial development of children. Not only can the impact on quality of life be devastating, the cost implications and risk to healthcare systems can be significant, with over 50 million people worldwide taking long term oral glucocorticoids.1 At a glance, steroids appear to be lower cost than other medications, but research continues to reveal the true economic impact of steroid use.
Studies have revealed that the adverse effects of glucocorticoids accounted for 40% of medical costs2 for patients taking glucocorticoids in high doses (>15 mg/day) or for longer than 60 days, for a range of autoimmune and inflammatory diseases. Strikingly, only 25% of the medical costs for these patients were related to the underlying disease. For every 1 mg/day increase in steroid average dosage, it is estimated that a patient’s average annual healthcare cost increases 1.07 times.3 Thus, a 5 mg increase in dose can increase the cost of caring for the patient by 40%. This is unsurprising, given that studies have indicated daily doses of prednisone even lower than 5 mg are
associated with an increase in the risk of infections severe enough to cause hospitalisation.
Q. What is the difference between ‘steroid-sparing treatments’ and ‘steroid-toxicity sparing treatments’, and how are the two concepts influencing the development of new therapies for autoimmune disorders?
A. “Steroid-sparing” is a term of use that many researchers have settled upon, referring to the reduction in reliance on steroids which was previously considered an advancement. There is little evidence that conventional “steroid-sparing” agents actually achieve the purpose for which they are used, as these drugs have not been studied rigorously in this context despite decades of use. Moreover, reducing dosage does not necessarily equate to a reduction in toxicity. While striving to reduce cumulative steroid exposure through the employment of conventional steroid-sparing agents is a step in the right direction, it may not reduce long-term harm. The only way to confirm that a medication actually achieves a reduction in steroidtoxicity is to measure toxicity over time.
Accomplishing this is only possible via a systematic approach such as the GTI. The GTI offers a validated tool to quantify the heavy burdens of steroid-toxicity on patients and has an added benefit of cost savings to healthcare systems as a by-product of the effort. Steroid-toxicity reduction leads to both improved patient outcomes and minimises healthcare burden.
Q. How are existing steroid-sparing treatments helping to alleviate the clinical burden of long-term steroid usage?
A. The introduction of steroid-sparing treatments and an increased awareness of steroid-toxicity are leading to a shift in mindset around ‘traditional’ steroid-prescribing patterns as doctors and patients recognise the associated risks and the new capacity to measure outcomes.
Novel steroid-sparing treatments are facilitating a reduction in steroid prescribing for conditions such as asthma, where the anti-interleukin monoclonal antibody, benralizumab, offers an alternative. While the treatment has already shown to be effective in severe eosinophilic asthma, a study published in November 2024 demonstrated the drug’s potential to routinely treat acute exacerbations most commonly treated with oral corticosteroids. 4
Another study, published in January 2022, evaluated the effectiveness and safety of a rapid steroid-reduction plan after benralizumab initiation, with almost all enrolled patients eliminating oral glucocorticoid use or reducing the daily dose to 5 mg or less (prednisone equivalent).5 This shift in steroid-prescribing can reduce healthcare burden associated with emergency room visits alone. The study may be template for the development of other steroid-reduction protocols.
Q. What clinical tools are available to monitor and quantify steroid-toxicity, and to evidence the need for new treatments?
A. Within days of the first cortisone treatment, doctors recognised the need to find the lowest therapeutic dose to manage disease and eliminate steroid toxicity. Over the 75 years following the first cortisone dose, progress in managing associated toxicity has been slow and steroids have become a keystone treatment for autoimmune and inflammatory diseases. The ability to effectively minimise symptoms from the outset makes steroids indispensable. However,
the persistence of inflammatory conditions frequently requires longterm use, increasing the risk of steroid-toxicity.
We developed the GTI for use in research to demonstrate the steroid-sparing benefits of new drugs and treatment protocols. The GTI provides weighted outcome scores of steroid-toxicity to demonstrate the safety of new treatments in comparison to steroids, especially important when the new therapy is shown to be noninferior to standard of care. The first use of the GTI in a clinical trial in ANCA-associated vasculitis was published in 2021 in the New England Journal of Medicine, in which the measure was identified as the most important secondary outcome measure of efficacy.6
A recent study in the journal Rheumatology investigated the use of the GTI in a clinical practice setting for individual patients. The cohort exhibited a range of GTI scores, which researchers used to differentiate among toxicities associated with the current dose of steroids and those that correlated with cumulative dose, demonstrating that the GTI is sensitive enough to determine prominent patient-specific differences in steroid-toxicities.7 The recent development of the GTI-MD, an abridged outcome assessment of steroid toxicity, which correlates highly to the GTI, makes the same scientific rigor possible in routine care without excessive burden to the practice.
Q. How do these tools support Health Economics Outcomes Research (HEOR)?
A. Health economics outcomes research (HEOR) is an approach that seeks to illustrate the value of a new therapeutic by quantifying the link between treatments and outcomes in large datasets to provide evidence-based information that guides drug development. HEOR can also be used to better understand drug prescribing patterns by unmasking steroid-toxicity in populations.
In the context of glucocorticoids, HEOR informs drug developers and healthcare decision makers on how steroids and their toxicities influence the value, cost, and cost-effectiveness of available treatments.
Incorporating the GTI-MD into HEOR studies provides scientifically validated assessments that reveal steroid-toxicity in retrospective studies of large patient populations. GTI-MD scores can inform treatment guidelines, payer coverage, identify where the greatest medical needs lie, and where to focus prospective research into novel steroid-toxicity sparing treatments.
Q. In which diseases do you anticipate these tools and steroidtoxicity sparing treatments having the most benefit?
A. Whatever the disease for which they are being used, if steroids are the standard of care to reduce inflammation, then patients suffer from the same condition: steroid-toxicity. The GTI and other STOX assessments can be used to optimise research in virtually any autoimmune disease. The pGTI was developed by another team of paediatric experts for children aged 2–18 years.
Steroids can severely impact growth and development in children that can lead to adverse events in puberty and well into adulthood. The pGTI differs from the GTI in its weighting and the addition of paediatric-specific parameters, including the growth domain, which allows for age-related references, gender-specific reference ranges, and paediatric neurological side effects.
A recent paper in Seminars in Arthritis and Rheumatism demonstrated the pressing need for management of steroid-toxicity
Therapeutics
in paediatric patients with systemic lupus erythematosus (SLE), an autoimmune condition with a variety of symptoms that can cause organ damage in the heart, lungs, brain, kidneys, and skin.8 The study revealed the significant burden of steroid-toxicity, including increased blood pressure, mood disturbances, BMI increases, and decreased growth velocity, highlighting the urgent need for further evidence-based research into alternative treatment strategies.
Outside of paediatrics, the GTI is also being deployed to change prescriber behaviour in indications where steroid-sparing therapies are already available, such as ANCA-associated vasculitis and asthma, and to support the development of novel steroid-toxicity sparing therapeutics for indications including sarcoidosis, myosotis, and other chronic inflammatory diseases.
Q. How can we continue to expand clinical tools to support the tapering of steroid-prescribing practices?
A. An increased understanding of the impact of emerging clinical tools and the value they deliver is leading to their global deployment; the GTI has now been used at 2,100 sites across 80 countries, in more than 30 diseases, evidencing the need for a shift in prescribing patterns.
The Great TaperTM initiative focuses on optimising steroid use. Using the GTI and its sister COAs in research in practice is part of the global effort. An online portal to educate and equip patients for shared decision making with their providers will drive awareness about steroid-toxicity and the need for a taper to the lowest therapeutic dose, either alone or with steroid-toxicity sparing therapies.
Adding new COAs to the STOX Suite will extend the scientific applications of the GTI; the GTI MD is an abridged COA which provides a systematic approach to assessing steroid-toxicity in the clinic and existing population datasets using data captured in routine clinical practice It is fast, accurate, and easily integrated into point-of-care programmes, as well as being compatible with electronic medical records, limiting the manual burden of use in a clinical setting. Increasing the accessibility of such clinical tools will continue to facilitate a global reduction in steroid-prescribing patterns, evidencing the urgent need for novel steroid-toxicity sparing treatments.
REFERENCES
1. Fardet, L., Petersen, I., et al. (2011). Prevalence of long-term oral glucocorticoid prescriptions in the UK over the past 20 years.
2. Rice, J. B., White, A. G., Johnson, M., et al. (2018). Healthcare resource use and cost associated with varying dosages of extended corticosteroid exposure in a US population. Journal of medical economics, 21(9), 846–852. https://doi.org/10.1080/13696998.20 18.1474750
3. Kabadi, S., Yeaw, J., Bacani, A. K., et al. (2018). Healthcare resource utilization and costs associated with long-term corticosteroid exposure in patients with systemic lupus erythematosus. Lupus, 27(11), 1799–1809. https://doi.org/10.1177/0961203318790675
4. Ramakrishnan, S., Russell, R. E. K., et al. (2024). Treating eosinophilic exacerbations of asthma and COPD with benralizumab (ABRA): a double-blind, double-dummy, active placebo-controlled randomised trial. The Lancet. Respiratory medicine, S2213-2600(24)00299-6. Advance online publication. https://doi.org/10.1016/S2213-2600(24)00299-6
5. Menzies-Gow, A., Gurnell, M., et al. (2022). Oral corticosteroid elimination via a personalised reduction algorithm in adults with severe, eosinophilic asthma treated with benralizumab (PONENTE): a multicentre, open-label, single-arm study. The Lancet. Respiratory medicine, 10(1), 47–58. https://doi.org/10.1016/ S2213-2600(21)00352-0
6. Jayne, D. R. W., Merkel, P. A., et al. (2021). Avacopan for the Treatment of ANCA-Associated Vasculitis. The New England journal of medicine, 384(7), 599–609. https://doi.org/10.1056/ NEJMoa2023386
7. Bahap-Kara, M., Sariyildiz, E., et al. (2024). Prospective Assessment of Glucocorticoid Toxicity in Rheumatology Practice: A Focus on the Glucocorticoid Toxicity Index. Rheumatology (Oxford, England), keae288. Advance online publication. https://doi. org/10.1093/rheumatology/keae288
8. Zhang, E., Capponi, S., et al (2024). Real-world application of the pediatric Glucocorticoid Toxicity Index in childhood-onset lupus. Seminars in arthritis and rheumatism, 68, 152516. https://doi. org/10.1016/j.semarthrit.2024.152516
Dr. John Stone
Dr. John Stone, Professor of Medicine at Harvard Medical School and Rheumatologist. He is also the Edward A. Fox Chair in Medicine at the Massachusetts General Hospital, and Chair of the Scientific Advisory Board at Steritas.
Vials with Exceptional Inner Surface Durability
For Biotech
Less interactions of drug molecules and formulations with the inner glass surface
• Optimized lyophilization process (less fogging)
• Reduced risk of glass delamination
For Diluents
Water for injection | Aqueous NaCl solution Lower pH shift
• Reduced risk of glass delamination
Digital Health Technologies are Inherently Remote – Let’s not Burden Sites with Them
There is ongoing and increasing interest in using digital health technologies (DHTs) in clinical trials for clinical endpoints. By changing the yardstick by which outcomes are measured, DHTs have the potential to improve the accuracy of results and/ or reduce the sample size necessary to achieve the required significance level. They can also be used to add label claims for secondary effects.
However, the desire to add DHTs to clinical trials may be hampered by the growing site capacity problem. The number, size, and complexity of clinical trials keeps increasing. The number of new drug candidates has doubled in the past 10 years.1 All must be tested in clinical trials. Furthermore, the average cost of a single trial has increased by 76% over roughly the same period.2 If one assumes that cost is roughly equivalent to effort, the demand for clinical site capacity is roughly 3.5 times higher than 10 years ago. In addition, clinical sites are not immune to the ongoing cost and productivity pressures that affect the entire healthcare system. As a result, site capacity has become a significant challenge when conducting clinical trials.
Impact of Decentralised Trials
Decentralised clinical trials (DCT), where a remote organisation manages the complete trial, were employed extensively during the COVID pandemic when there was little choice. It quickly became clear that a full DCT approach was difficult, if not impossible, for most trials. There are almost always tests and assessments that must be performed at a clinical site. Furthermore, principal investigators must provide medical supervision. The emphasis has now swung to “hybrid” trials, where certain functions that need to be performed in a central location (e.g., MRI scans) are done at the site. Activities that can be performed remotely (e.g., recording electronic patient reported outcomes, ePROs) are done remotely. Hybrid trials have helped somewhat, but some of the benefits of reducing site visits have been mitigated by increased coordination efforts with the remote functions. Thus, the overall impact on site capacity has been minimal.
This is not to say that hybrid trials provide little value. Reducing the number of site visits for participants is incredibly important for reducing the burden of participating in clinical trials. It also makes it feasible to enroll participants from a wider geographic area. Enrolling participants is almost always the chief roadblock in executing clinical trials quickly, so anything that can be done to improve that is vital.
The Site Burden
Although it may seem like DHTs shouldn’t be much of a burden to sites, the reality is that they are. Sites need to manage the inventory of DHT devices for patients to take home, but they are generally not set
up to do that. They don’t have the space or the processes to become a warehouse. Beyond the hardware for patients, there is typically another platform that sites need to keep track of and use to monitor compliance on a regular basis, often daily. Clinical sites often have upwards of 15 different logins for each trial. Given that sites often have only a few participants at a time in a particular trial, logging on to yet another system to check on them creates a significant additional burden on a per participant basis.
Furthermore, every tech vendor has their own platform, and it is not reasonable to expect site staff to master all of them. Even routine questions require site staff to involve the vendor, putting an additional burden on the vendor and clinical research organisation (CRO). For example, even the most routine of events, like a participant not wearing a device, becomes a significant project. To avoid potential confusion, many trials require that there be a single point of contact for each site. What usually happens is that the DHT vendor notices the non-compliance issue first. The vendor must notify the CRO, and copy the sponsor. The CRO then contacts the site involved (while copying the sponsor and tech vendor). The site then calls the participant (often only making contact after several tries). If the participant has any technical questions, the communication often goes back up the chain to the supplier. In a recent non-wear example, getting a participant to start wearing their watch again took six emails with about 20 people on each, plus several calls to the participant from the site.
Challenges in Site Management
Clinical sites do not add value to the deployment of DCTs. In fact, involving clinical sites adds a whole series of costs. In terms of logistics, deploying DHTs at all the sites requires additional inventory, and 30% of sites typically never enroll a single participant. Furthermore, all the site staff who will be working on the trial need to be trained, and there is often a time lag between the training and actual enrollment of participants. As clinical personnel are sometimes not familiar with the DHT, any delay between training and implementation can require refresher training. Personnel turnover is also a problem that requires additional training.
The biggest issue, however, is communication. Having multiple extra layers of communication creates a situation like the old game of telephone, where a message gets completely distorted after going through multiple people. Coordinating a time where all the parties can engage in a real-time conversation can be difficult though and different time zones and languages can make it virtually impossible.
These challenges are not just cost and capacity issues. They create bad experiences for the participants, too. Trying to work through an issue with multiple layers of communication takes more time for the participants. It can also be frustrating for them to deal with support personnel that may not understand the technology any better than
they do. In the end, this approach is more expensive and burdensome for everyone, the vendor, site staff, and participants.
Managing DHTs Remotely
The question is, why do the sites need to be involved at all, especially when the tech vendor is most often the first to notice the problems? Most DHTs are meant to be monitored remotely. The data is generally sent to a central server from where it is analysed and distributed as appropriate. Tech companies deploy similar digital technologies remotely all the time.
It is possible to approach this situation more like a tech company. One can think of a robust process whereby the vendor sends the device directly to the participant. The vendor supports the device installation remotely or, when onsite installation is needed, sends a trained person to the participant. The vendor does the remote monitoring and compliance management, contacting the participant directly if needed. In many cases, remote monitoring can be completely automated. For example, a participant not wearing a device when required could trigger an automated text message, asking them to put it on (replacing the chain of six emails with 20 people copied on each). This process could be much more efficient and effective than the current one.
This approach is possible because most DHTs are used as observational, not interventional, tools. As a result, they are safer than most interventions. A failure in this process results in a loss of data, not injury to a person. The involvement of a principal investigator to monitor the safety of the DHT is generally unnecessary. Furthermore, since the data from DHTs can be monitored remotely, quality checks on compliance and data quality can be implemented and often automated. The risks associated with managing DHTs remotely are generally less than relying on a clinical site to monitor them.
Challenges
Of course, there are challenges to this approach. The typical clinical trial participant is probably less technically savvy than the average population. We want the participants to feel well supported, or compliance may decrease and dropouts increase. The traditional tech vendor support approach needs to be significantly upgraded; 24-hour call centres are a start, but if there is a need for in-person service for installation or trouble shooting, having a local support solution will often be necessary.
Additionally, a clinical trial needs to be run with more care than the typical tech product. The vendor needs to be qualified in, and employ, Good Clinical Practices (GCPs), especially those related to privacy and adverse event reporting. As part of the participant onboarding process, it needs to be made clear that any participant issue relating to the DHT should be directed to the vendor and all other issues should go to the clinical site. Regardless of what they are told, participants may call whomever they think will support them better, irrespective of whom they should call. There needs to be a seamless way to manage those calls/inquiries.
Summary
Managing DHTs through clinical sites is inefficient, ineffective, and creates unnecessary burden on the participants, clinical sites, and DHT vendor. There is an opportunity to dramatically improve the process by working more like a typical tech vendor and managing everything remotely. However, these projects are still clinical trials and need to be managed more carefully than typical consumer electronics.
REFERENCES
1. Pharmaprojects, January 2024
2. Measuring the Return on Pharmaceutical Innovation, April 2024, Deloitte, US
Geoffrey Gill
Geoffrey Gill, MS, is CEO of Verisense Health, a digital health technology and data company, and Co-founder of the Open Wearables Initiative (OWEAR). Verisense Health is dedicated to producing clinicalgrade digital health data and solving digital health data access and reuse problems. Geoffrey joined Verisense Health from Shimmer Research, the global wearable technology provider, where he served as President of Shimmer Americas. He received his MS in Management of Technology from the MIT Sloan School of Management.
System Integration’s Role in Reducing the Likelihood and Increasing Detectability of Temperature Excursions
Upholding product integrity is mission critical to protecting patients and achieving safe and compliant clinical trial outcomes. It also has a pivotal role in controlling study costs and promoting timely trial completion; both essential components for making breakthrough therapeutics available to patients sooner and maximising a sponsor’s return on investment. However, upholding product integrity is becoming increasingly challenging for sponsors dealing with complex biologics that demand stringent temperature control to maintain drug stability through each stage of the supply chain.
Mitigating the risks requires sponsors to implement preventative measures to both reduce the likelihood of excursions and maximise detectability. Increasingly, it also requires sponsors to embrace digital transformation: to replace existing manual approaches to temperature management with automation that has the potential to lower the likelihood of an excursion occurring, while increasing detectability and promoting fast and accurate adjudication. Integration of core supply chain technology – including forecasting, ERP, IRT and temperature management systems (TMS) – can promote end-to-end drug temperature data visibility and help mitigate risk.
Mitigating the Risk of Temperature Excursions 101
Before exploring the opportunities that system integration can present, it’s important to understand the fundamentals of mitigating the risk of temperature excursions. This starts with identifying the risk and outcomes if those risks are realised. If a drug is exposed to adverse temperatures, one of two scenarios will unfold. The excursion event will either be reported, or it won’t. Each presents its own ultimate risk.
If excursions occur and are reported, drugs are rejected, and the patient cannot be dosed and is possibly lost from the trial – increasing trial costs in both product rework and recruitment. Meanwhile, if excursions go unreported, the compromised drug remains available and patient safety hangs in the balance. Both scenarios can have a significant impact on the patient (their experience and their safety) and the trial; delaying timelines and introducing further cost.
A Preventative Approach
Key to mitigating the risks associated with each scenario is taking preventative action. It’s necessary to consider how the level of control sponsors have over their supply differs throughout the supply chain. Risk assessing each stage, from production line to patient administration, to understand what mitigations can be used and where (prior to study start-up) will facilitate fewer excursions and enhance detectability. The audited facilities of sponsors and CMOs represent a relatively low risk for temperature excursions. However, once temperature-sensitive supply begins its journey to sites and patients, the likelihood of temperature excursions increases, while
detectability plumets. Drugs in transit are at the mercy of third parties, making it harder for sponsors to know if an excursion has occurred and, if it has, whether it has been appropriately reported in line with defined processes. This typical lack of visibility and control extends through global distribution to storage at clinical sites and within patients’ homes.
The question sponsors need to ask at this point is how visible are excursions that happen and what facilities will be responsible for reporting, and how? Whereas CMOs operate audited facilities with validated processes to identify and report excursions, processes are typically less robust further along the supply chain. The further from the sponsor products travel, the more difficult detectability becomes.
Decreasing the Likelihood, Increasing Detectability
Prevention is better than cure and it pays to take the time to consider the factors at play that will impact the likelihood of excursions occurring. Reviewing key factors and investing in appropriate mitigations will help save drug product and protect patients.
The type of container the drug is being shipped in should be a prime consideration. Phase change shippers for example are qualified against recognised thermal conditions, with the technology validated to maintain defined temperature for a set period to reduce the potential for excursions occurring in transit. Another consideration is who will be responsible for shipping the drug and what facilities, technology, and processes are in place to safeguard temperature through transit.
Working with experienced distribution staff, who know the pitfalls to avoid and have defined processes in place to support sponsors on developing best-fit solutions – from monitor profiles to appropriate shipping containers and premium courier services - will help ensure drugs make it to sites safely. Access to a ‘control tower’ of global shipping lane data is also essential – if drugs are likely to be exposed to longer lead times, risks can be identified early, and appropriate interventions put in place. Drug stability is another key area to scrutinise. Reviewing stability data and time out of condition criteria will help to establish the best route forward and reduce opportunity for excursions to occur. Sponsors should also analyse the process for shipping product and work with a qualified vendor to establish effective and efficient processes that promote smooth custom clearance, appropriate handling of products and optimised distribution strategy to minimise time in transit.
A final factor that needs to be considered in a bid to reduce the likelihood of excursions is the process for receiving drug shipments at site. Establishing standardised and robust processes with sites for handling drug receipt is dependent on clear communication and instruction to site personnel. Helping sites to understand and comply with the process will limit false excursion recordings, due to devices
not being stopped, and also improve site compliance with associated processes, such as handling reusable phase change shippers.
Taking a preventative approach and risk assessing the supply chain to identify areas where excursions are more likely to occur provides the best chance of spotting and mitigating issues early. However, even with the best planning and preventative measures in place, excursions will still happen. When they do, it’s vital that they are detected quickly. There are several techniques available to increase detectability. For example, using temperature monitors in shipments or remote monitoring devices, although the cost and hardware implications of the latter option will need to be weighed up. Establishing robust processes for returning temperature data and reporting excursions, and effectively managing sites to ensure compliance with both processes, will also support enhanced detectability.
The Pitfalls of a Traditional Approach
The reason tradition excursions management processes at site are unfit for purpose is because they are built around several manual steps, with few (typically no) electronic systems is use. This leaves the door wide open for human error and inefficiency to enter.
The traditional process starts when a shipment is received and an alarm is raised, prompting the site to consult the pharmacy manual before notifying the relevant contact. When the contact receives the email, material is quarantined in the IRT so it won’t be available for patient assignment. The contact will then review the temperature data vs. stability information before updating the drug status and notifying the site. The drug status can then be updated again in the IRT and re-supply arranged, if drug product has been rejected.
Theoretically, this process works. Practically, it falls incredibly short. This is because every stage is relying on a person to do everything right and everything quickly, which simply isn’t realistic. Sites are busy. It isn’t uncommon for site personnel to forget to stop temperature monitors or notice that they’re alarmed upon shipment receipt. Dealing with multiple protocols for the same sponsor, there’s also a significant risk that site staff notify the incorrect contact, or that the contact might not see the email. Both cause delays with adjudications.
Similarly, the contact or site may forget to quarantine drugs in the IRT, which increases the risk of it being assigned to patients, whilst pending adjudication. The sponsor contact may also have several excursions to manage for different sites, making the process difficult to keep track of. This increases the risk of delay and miscommunication when responsible for notifying sites regarding the outcome of the excursion and (if drug has been rejected) organising timely resupply. Operating a traditional excursions management process at site can mean it takes several days to know if resupply needs to be arranged. In today’s competitive drug development landscape, few sponsors can afford to operate with such high levels of risk and inefficiency.
Adopting System Integration
Integrating core supply chain systems can help to close the gaps left by manual processes and reduce risk and inefficiency. Instead of viewing TMS, and the data they capture and store, in isolation, it’s important to look at the bigger picture. TMS data is critically connected to data stored in other systems, including ERP, IRT and even forecasting. TMS need information about shipments, which it gets from the ERP. If an excursion occurs, the IRT needs to know to quarantine kits, until disposition is available. Depending on the outcome, the IRT may also need to order replacement inventory,
Logistics & Supply Chain
which is where the connection to forecasting comes into play. It might not be an immediate integration, but it is important.
Accurate forecasting is dependent on knowledge of material available in the field, along with its status and patient demand. Excursions impact the drug status, while replacement shipments impact what is available. With core supply chain systems already operating with interconnected data, there is opportunity to develop further integration that ensures data flows between systems in an accurate and timely manner. In doing so, it is possible to reduce the likelihood of excursions and boost detectability.
Better Connected Data in Practice
In contrast to the traditional approach, an integrated model can tackle the problem of reduced detectability at site. When temperature sensitive supply is needed at site, the IRT creates an order, which is transmitted to the ERP, prompting two actions. Physically, the material handed to a courier and delivered to site. This stage in the process represents the first opportunity to introduce system integration by connecting the ERP to the TMS so it has all shipment information.
Once shipments arrive, site personnel acknowledge receipt transaction in the IRT. Unlike site-based temperature monitor upload transactions, which vary per protocol, introduce inefficiency and have poor compliance rates, this is a standardised process with high levels of compliance. Through system integration, it is possible to couple the acknowledgement of receipt transaction with the monitor information upload to create a single, seamless, and simple workflow. And because the IRT now has temperature information from the monitor, it can share it with the TMS. If there are no alarmed monitors or excursions, the process is complete: sponsors have full data and do not need to wait for it to be emailed.
However, if an alarmed monitor is recorded, the TMS will have immediate visibility; allowing it to automatically quarantine inventory in the IRT to prevent potentially compromised material being assigned to patients. In this integrated approach, the IRT understands the status of the material, as well as upcoming patient demand, so it can determine if a replacement order is needed and generate it automatically if required. In turn, through integrating multiple systems, forecasting platforms have data about site, patient, and medication events from the IRT, along with inventory from the ERP if replacement orders have been generated, helping sponsors optimise supply and demand.
The Benefits of Adopting System Integration
GDP guidelines state that it is ‘necessary to demonstrate that drug has not been exposed to conditions that could compromise quality and integrity’. By coupling the monitor data upload and the acknowledgement of receipt transaction into a single workflow, upload rates increase, which is vital for audit readiness.
Logistics & Supply Chain
Reviewing site upload performance and finding 6 unreported alarms, with the potential to cause significant patient and regulatory impact, one sponsor recently opted to introduce system integration. By coupling the two workflows the sponsor boosted its site-based temperature monitor upload rate from 65% to 99%. Detectability of excursions also increases, as sponsors are receiving a complete data set because of the established workflow that forces site upload by quarantining material until data is inputted.
A recent comparison of the performance of 25 non-integrated and 25 integrated studies revealed a significant difference in upload rate – 65% for non-integrated, 97% for integrated; demonstrating how effective integration can be in boosting site compliance with temperature monitor data upload. However, it also highlighted a notably lower alarm rate for non-integrated studies, 3% compared with 4.5%. The implication being that these non-integrated studies were under-reporting alarms, risking patient safety and leaving sponsors vulnerable to regulatory action. It may seem counterintuitive but more alarms are a good thing when the objective is to increase detectability. If sponsors know about alarms, they can deal with them
appropriately and ensure the rejected drug is taken out of the supply chain.
The integrated approach also increases accuracy and speed, helping to deliver more efficient and cost-effective processes. Site personnel only need to complete one task in one system, which streamlines the process. Meanwhile, automated rejected drug notifications mean resupply occurs quickly, helping to minimise patient impact. Critically, system integration keeps patients front and centre – preserving their safety and experience by closing all gaps associated with the manual process. It achieves this by boosting detectability and through the automatic quarantining of alarmed kits. The full data set integration offers serves to protect patients too. Afterall, there’s no chance for unreported alarms if sponsors know the outcome of all monitors shipped in their protocol.
Striving for Continuous Process Improvement
Coupling the monitor upload and acknowledgment of receipt transactions to improve upload compliance and enhance detectability is one example of process improvement delivered via system integration. However, it is important to always strive for continuous process improvement in our mission to mitigate risk and uphold patient safety.
Another example of integration driving temperature management improvements can be found in the adjudication process. If an alarmed monitor is recorded upon site receipt, instead of the TMS simply sending a message to quarantine the material in the IRT, the system can be harnessed to conduct an automated adjudication. If the system deems the shipment acceptable, based on the data it has, it can autorelease the product. If the data is not deemed acceptable, the system can quarantine material and request a manual adjudication. This model brings together TMS and IRT further and offers a reduction in human effort and potential error, as well as increasing the speed of disposition.
Boosting temperature monitor upload compliance at site, automating quarantine of at-risk materials, enhancing audit readiness, better protecting patients, and delivering expedited (and more cost-effective) clinical trial operations are key benefits of moving away from manual, error-prone processes. Ultimately, sponsors need to be confident that their temperature management processes are working hard to keep patients safe and can demonstrate a robust supply chain to auditors. If sponsors can’t be certain this is the case with existing approaches, it may be time to consider system integration.
Sarah McAliskey
Sarah McAliskey is Temperature Services Manager at Almac Clinical Services. She works closely with clients to understand the challenges they face with distributing temperature sensitive drug products, and advising on the best solutions to implement for efficient management of temperature data on a global scale. Sarah joined Almac in 2016 and has worked with a large number of Pharma and Biotech companies initially in developing proposals to fulfil packaging and distribution needs, aiding in the successful delivery of a range of clinical trials. Prior to joining Almac, she worked in the clinical diagnostics industry, gaining in solutions for shipping temperature sensitive product in challenging climates. Sarah holds a BEd in Business Studies from Queens University Belfast.
Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.
www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA POD
‘Putting science into conversation, and conversation into science.’Join some of the most esteemed and integral members of the Drug Discovery & Development world as they give insights & introspect into the latest movements, discoveries and innovations within the industry.
senglobalcoms.com
AVAILABLE WORLDWIDE AVAILABLE WORLDWIDE AT YOUR FREIGHT FORWARDER AT YOUR FREIGHT FORWARDER
TEMPERATURE TEMPERATURE QUALIFICATIONS & VALIDATIONS QUALIFICATIONS & VALIDATIONS
Temperature protection of pharmaceutical and healthcare products in airfreight (+15°C + 25°C°) and (+2°C + 30°C) (+15°C + 25°C°) and (+2°C + 30°C)
Multilayer thermal blanket for PMC-ULD - Euro and Block pallets
Stress-tested in summer (+46°C) and winter (-15°C) profiles
Airfield Tarmac tested on solar power and greenhouse effects