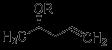
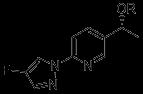
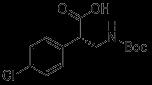
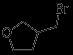
Accelerating Drug Discovery: Integrating In Vivo and In Vitro Testing with C.elegans
The Data Revolution in Drug Discovery: How Lab Automation & AI are Reshaping the Future of Medicine Moving Closer to Affordable Advanced Therapies: A Review of Current Trends in Downstream Processing
Delivering the Future: Overcoming Shipping Challenges in Cell & Gene Therapy
Sponsor Company:
DIRECTOR: Mark A. Barker
INTERNATIONAL MEDIA DIRECTOR: Anthony Stewart anthony@senglobalcoms.com
All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IBI will be published in Spring 2025.
ISSN No.International Biopharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
2024 Senglobal ltd.
Volume 7 Issue 4 – Winter 2024
04 Foreword
TALKING POINT
06 Polypure Celebrates and Reflects on 25 Years in the Pharmaceutical Industry
Polypure have been at the forefront of developments in the pharmaceutical industry for 25 years, being a driving force for high-quality PEG’s and PEG-derivatives. Chloe Euripides of the International Biopharmaceutical Industry Journal sits down with CEO Erik Agner to hear about all things Polypure, from successes and key milestones to what we should expect to see from Polypure in the coming years.
REGULATORY AND COMPLIANCE
08 Emerging Pharma vs. Big Pharma: Rethinking CRO Partnerships
Outsourcing has become essential to the development and commercialisation of drugs in the pharmaceutical landscape, especially for new and emerging pharma companies. Aashritha Marepalli of Covalent Bonds explains the importance of CROs expanding beyond the big pharma, and how targeting the needs of the start-ups and small companies can help us adapt the modern pharmaceutical world.
PRECLINICAL
12 Breath Analysis and Cancer: The Future of Early Detection and Personalised Medicine
Cancer management can be very challenging due to the nature of the delays in diagnostics, as well as the heterogeneity of the disease. Dr. Hsuan Chou and Lucy Godbeer of Owlstone Medical stress the urgent need for low cost and accessible diagnostic techniques and explain just how this can help improve earlier detection of cancer, assessment of treatment efficacy and overall personalised treatment.
18 In vitro Models Guiding Strategic Decision-making in Biotherapeutic Development
Evolving regulatory requirements are continually reshaping the drug development process, and navigating the intricacies and financial challenges of bringing new therapies to market, demands innovative approaches and strategic decision-making. Dr. Agapitos Patakas of RoukenBio examines one specific area of development in how the in vitro pre-clinical models have become indispensable in the way of creating life-saving treatments.
22 Accelerating Drug Discovery: Integrating In Vivo and In Vitro Testing with C. elegans
In Vivo and In Vitro models are at the forefront of drug discovery and development but, with in vitro often failing to capture the complexity of living organisms, and in vivo being too costly and time-consuming, David Weinkove of Magnitude Biosciences offers a new approach to adopting the models. He suggests that by integrating Caenorhabditis elegans (C. elegans) into in vitro testing, can allow for reduced failures and improved accuracy.
RESEARCH/INNOVATION/DEVELOPMENT
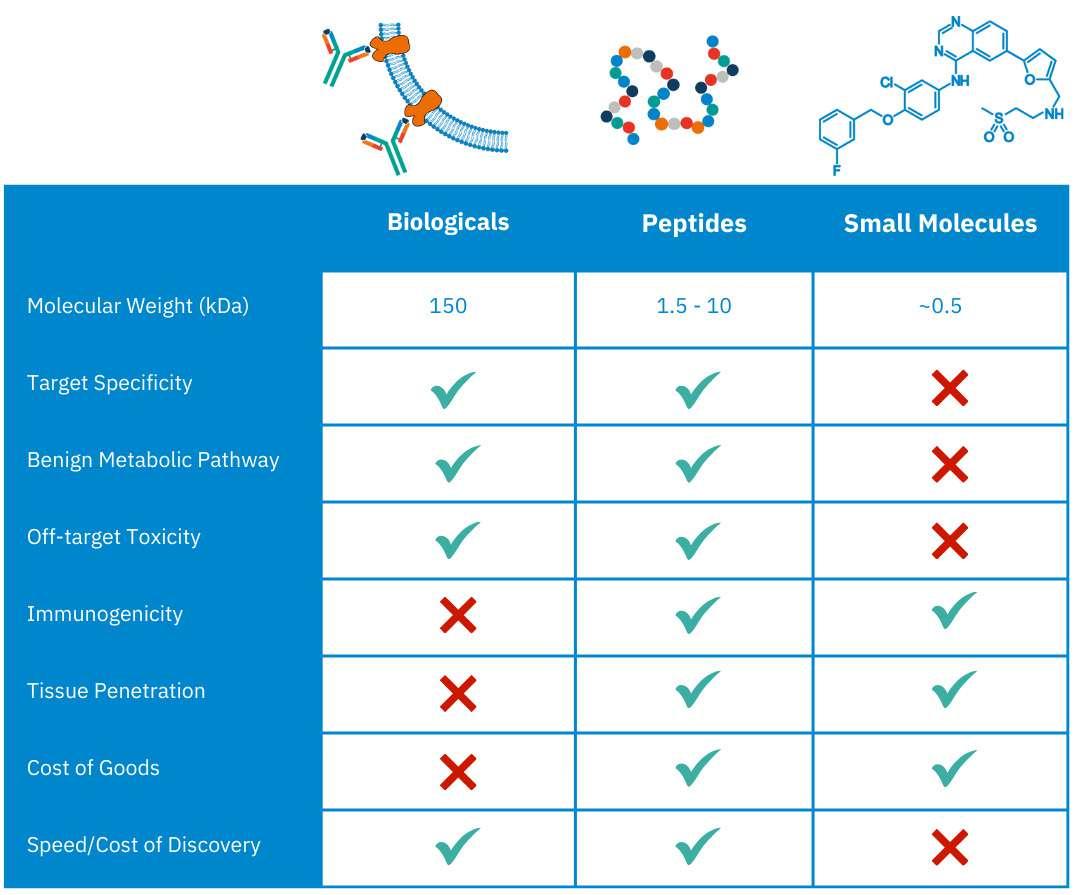
24 Peptide Therapeutics: Unleashing the Power of Precision Medicine
Innovation is a concept that is at the heart of drug development and as result of this has seen peptide therapies emerge into the
industry. Peter Timmerman of Biosynth delves into the unique niche between small molecule drugs and complex biologics peptides, and how this can improve precision and reduce small molecule side effects.
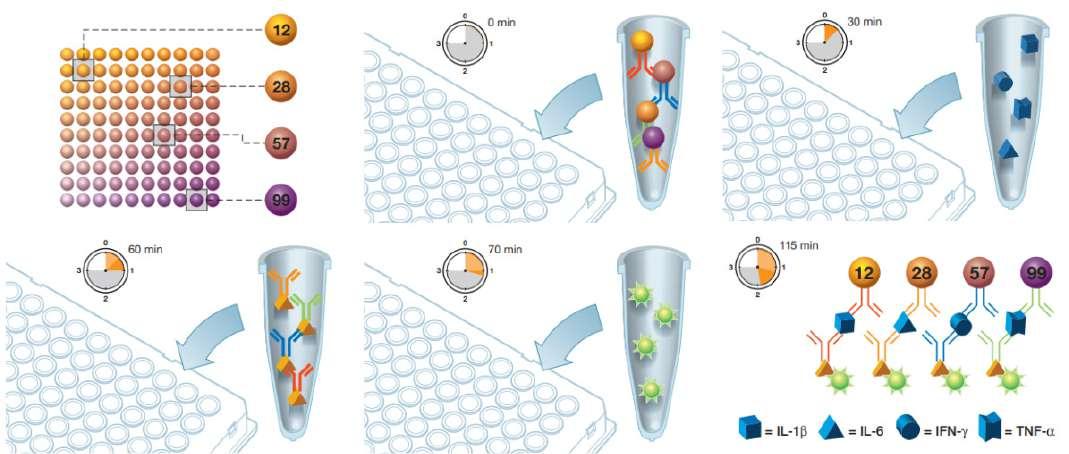
28 Advancing Cancer Immunotherapy Research: The Role of Multiplex Immunoassays
Immunotherapy has revolutionised cancer treatments. However, while it shows high efficacy against certain tumours, immunotherapy is not always effective. Thus, Vanitha Margan of Bio-Rad, makes a case for the adoption of biomarkers and how they can enable a better understanding of the immune landscape; ultimately leading to better predictions, personalised medicine and therefore, improved patient outcomes.
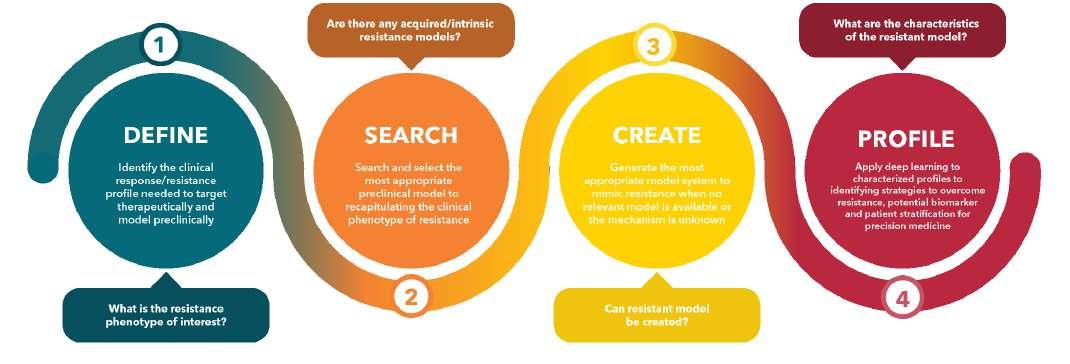
32 Overcoming Oncology Drug Resistance: Models and Strategies
Cancer is inherently difficult to detect and in addition to this sees some patients develop drug resistance to treatments. To best improve this Bindi Doshi of Crown Biosciences highlights the importance of taking time to choose the right preclinical models and once carried out what the significance of sharing our data collection can have.
TECHNOLOGY
36 The Data Revolution in Drug Discovery: How Lab Automation and AI are Reshaping the Future of Medicine
Evolving technologies are reshaping scientific progression, changing research methodologies and redefining drug discovery as we know it. Tom Fleming of Arctoris discusses how the adoption of Artificial Intelligence (AI) can be a means of improving the efficiency of fundamental drug processing and ultimately pave the way for further successful treatments available.
40 CRDMOs Redefining Drug Substance Development with Platform Technologies
The development of biopharmaceuticals is expanding at an unprecedented rate, however, the path from discovery-to-clinic poses quite a few challenges, seeing that there are immense complexities to finding robust enough bioprocesses. Dr. Rita Cruz of Ingenza suggests that to best tackle these complexities CRDMOs must integrate AI and Machine Learning (ML), to provide more seamless processes as well as increase the scalability of CRDMOs specialised expertise.
MANUFACTURING AND PROCESSING
46 Moving Closer to Affordable Advanced Therapies: A Review of Current Trends in Downstream Processing
Viral Vectors are an essential tool used withing the development of vaccines and gene therapies. However, adoption at a clinical grade does not come without its challenges and thus, Abdullah Sufan of Tozaro, reviews the best ways to address and tackle these in order to transform the access to advanced therapies.
49 Innovation for the Few: Key Development to Commercialisation Considerations for Drug-Device Combination Products in Rare Diseases
Rare disease drug-device combination products present both unique challenges and great opportunities for the biopharma industry, specifically in the way of offering novel solutions. Bill Welch of PCI explores and examines the key development considerations to
commercialisation for drug-device combination products, focalising specifically on that of rare diseases.
LOGISTICS AND SUPPLY CHAIN
54 Stability Secured: Leveraging Technology and Expertise to Protect Clinical Trial Supply
Complex biologics and next-generation cell and gene therapies demand precision handling to maintain stability and promote optimised and patient centric clinical trials. Sarah McAliskey of Almac exemplifies the importance of maintaining the appropriate environmental supply chain conditions, and just how detrimental it could be if this is not maintained from production to patient assignment.
CELL AND GENE THERAPY SUBSECTION
58 Particulates in Cell and Gene Therapies
Particulates in cell and gene therapies are greatly significant, but not identifying them correctly can be cause for complications. Alistair Michel of RSSL tells all about the importance of identifying the difference between organic and inorganic particulates and the implications of misidentifying when it comes to therapy quality and safety.
60 Exploring Cell & Gene Therapies Through the Ages: How Past Cell and Gene Advancements are Shaping Tomorrow’s Medicines
Cell and gene therapies are revolutionising medical landscape, offering cures for the once considered incurable and rewriting the future of development. Bill Vincent of Genezen delves into the Cell and Gene through time, pinpointing the challenges, the breakthroughs and the milestones that have fuelled scientific progression and rapid development across cell and gene as a whole.
64 Delivering the Future: Overcoming Shipping Challenges in Cell and Gene Therapy
The rise of cell and gene therapy is undeniably reason for key scientific advancements, however, this does not come without its obstacles. Alex Guillen of Tive addresses key logistical challenges, and considerations that must be taken to ensure treatments reach patients in the most efficient and safest way possible.
66 Overcoming Portal Fatigue in Cell and Gene Therapies: Optimising Orchestration
Technologies have pioneered scientific developments in ways beyond human capabilities, but with this comes the complexities of operating and managing these digital systems. Dr. Akshay Peer of Trakcel discusses the causes and consequences surrounding technologies, specifically addressing “portal fatigue” and the need for standardisations and collaborations.
APPLICATION NOTES
43 The Benefits of 5,000L Single-use Bioreactors for Biologics Manufacturing
Thermo Fisher give insight into the benefits of using 5,000L singleuse bioreactors for manufacturing, helping to bridge the gap between small and large-scale productions. They explain just how flexible this new technology is and how the fluctuations in demand can be addressed more efficiently and with greater ease.
Versatile design intuitive delivery
Your fill volume may change, with Aidaptus® auto-adjust plunger technology your auto-injector doesn’t need to
Commercially available
Accommodates both 1mL and 2.25mL glass syringes in the same device
See our auto-adjust plunger technology in action
Find out more by scanning the QR code or visiting ompharmaservices.com/ibi-sept2024
Looking back on 2024 we must recognise that the globe and our societies are in a worse situation than previous years. Globally we are faced with more conflicts, severe environmental catastrophes caused by climatic change, increasingly isolationist governments and a challenging global economy. Our industry is also still facing challenges of its own with the ever-growing costs of both energy and goods, as well as the increased strain on supply chains. But against these odds we have still seen significant progression, with 44 new drugs approved by the FDA in 2024 and the trend suggesting a similar trajectory of advancement to follow into 2025.
This issue of IBI is a testament to the innovative power of the biopharmaceutical industry, featuring a subsection dedicated to cell and gene therapy. The field of cell and gene therapy has come a long way from its inception, with a few early setbacks, but a great promise for many chronic conditions either caused by a gene defect and injured or fatigued tissues. Bill Vincent of Genezen gives a comprehensive overview of the challenges and success stories in cell and gene therapy through time.
Particulate contaminations in parenteral drug products are a quality issue and can have severe implications on patient safety. In any drug product in which the active itself is either formulated as a particle, e.g. lipid nanoparticles, or is a particulate in nature, can be used viral vectors in gene therapy. In these products the challenge is to distinguish between the active particulate and undesirable particulate impurities born from degradation or contamination. Alistair Michel of RSSL explains how to identify particulates in cell and gene therapies. As promising as these new developments are, for them to make a real impact in patients in need of these new drugs, they need to be made widely available and affordable.
Viral vectors, the main tool for most gene therapies, are challenging to be manufactured at scale and is one of the main reasons for the current high price tag of these drugs. Abdullah Sufan of Tozaro reviews the best ways to address and tackle these challenges to transform the access to these advanced therapies.
Finally, once cell and gene therapies have been produced the next step is ensuring they are delivered to the patient without
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Cellia K. Habita, President & CEO, Arianne Corporation
• Deborah A. Komlos, Senior Medical & Regulatory Writer, Clarivate Analytics
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in Ethics
• Hermann Schulz, MD, Founder, PresseKontext
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
impacting their quality or potency. For viral vectors this may require a supply chain enabling ultra-low temperatures as we have seen for the first generation of COVID vaccines. For cell-therapies this may mean that cells or engineered tissues need to be shipped at physiological temperature, minimising mechanical stress or reflecting the short stability of the product. Alex Guillen of Tive addresses key logistical challenges, and the considerations that must be taken to ensure treatments reach the patients in the most efficient and safest ways possible.
Before closing this foreword and this year, I want to thank the great editorial team and everybody at IBI for carefully curating the content of each issue, making sure that this publication offers added value to the pharmaceutical industry. It is with great optimism that I look forward to 2025, welcoming new developments and continuing to target biopharma’s goal of ultimately bettering patients in need.
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rafael Antunes, Vice President Business Development, Aurisco Pharmaceutical Europe
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steven A. Watt, CBDO (Chief Business Development Officer) at A&M STABTEST GmbH
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
Dr. Steven A. Watt, CBDO (Chief Business Development Officer) at A&M STABTEST GmbH
CONTRACT DEVELOPMENT AND MANUFACTURING OF BIOPHARMACEUTICALS
Richter BioLogics is a Germany-based GMP manufacturer specialized in products derived from bacteria and yeasts, with a proven 35-year track record.
Count on us to flexibly provide a comprehensive range of services and customized solutions. Clients worldwide have already benefited from our commitment to good manufacturing practice and total transparency. Our work focuses on recombinant proteins, plasmid DNA, antibody-like sca olds (VHH/Nanobody), cell-free expression and vaccines.
Your product – our competence and dedication. Contact us +49 40 55290-801 www.richterbiologics.eu
LEARN MORE ABOUT OUR SERVICES AND CAPABILITIES
Polypure Celebrates and Reflects on 25 Years in the Pharmaceutical Industry
Polypure have been at the forefront of developments in the pharmaceutical industry for 25 years, being a driving force for high-quality PEG’s and PEG-derivatives. Chloe Euripides of the International Biopharmaceutical Industry Journal sits down with CEO Erik Agner to hear about all things Polypure, from successes and key milestones to what we should expect to see from Polypure in the coming years.
The Pharmaceutical Industry is Undergoing Rapid Changes in Drug Delivery Systems, Biotechnology Developments, and Regulatory Frameworks. Where Do You See Polypure in this Landscape and How Can Polypure Capitalise on These Changes?
For 25 years Polypure has focused on high-quality monodisperse PEG’s and PEG-derivatives. It is well known that PEG can increase the solubility and stability of drugs before and after administration. Monodisperse and well-characterised PEG’s lead to higher reproducibility in production and dramatically reduce batch-to-batch variations. It also leads to more predictable and better understanding of behavior in therapeutic applications, and ultimately more efficient drugs and drug delivery systems. At the same time, there are many new developments in drug formulation and delivery systems where highly characterised and purified monodisperse compounds based on PEG have distinct advantages. It is a win-win situation for Polypure as
manufacturer and for pharma companies, therefore, I regard Polypure as well positioned to take part in future value creation in pharmaceutical development.
What Kind of Legacy Do You Hope to Leave as the CEO of Polypure? How Do You Envision the Company’s Role in Improving Global Healthcare in the Long Term?
Our main focus is quality and purity; we will never compromise on that. Research is the backbone of Polypure and we are committed to exploration and innovation. There are almost endless possibilities when it comes to making new molecules, and we enjoy working on the research frontier. The real challenge is how to identify and focus on the most relevant compounds; those that can be of real interest and useful to the pharmaceutical industry because of their innate properties. Polypure is not a drug development company, so we need to interact with pharmaceutical companies and CDMO’s etc. to learn and understand needs and trends in the industry. A successful strategy so far has been to develop a library of compounds that we believe can be useful, and to test their interest in the marketplace by offering them small-scale through our catalogue.
If You Were to Summarise Your Leadership Philosophy, What Would it Be?
Curiosity and scientific interest have always been my main driving force. I realised a long time ago that it was possible to do things and make compounds that no one had made before, and that
has been a huge inspiration. It made me want to start Polypure 25 years ago. I would say that my leadership philosophy is still based upon pride in doing good science. If you do good science, you can push boundaries and you can realise new products. I want the people who work here to like what they do and to be proud of what they do – they should really want to come to work! And this is to be combined with high ethical standards, ensuring a mutually open and honest relationship with our employees, customers and collaborators.
As Polypure Celebrates its 25th Anniversary, What Do You See as the Key Milestones that Have Defined the Company's Success so Far? How Would You Build on These Achievements as You Lead the Company into its Next 25 Years?
I think most important is that we have succeeded in keeping the original idea, building upon it and ensuring a continued process of development and commercialisation of new products. We have grown organically because we deliver unique high-quality products, therefore, we have satisfied recurring customers. At the same time, we have been able to spread the word about the benefits of monodisperse products and to find new customers. Through our commitment to exploration and innovation we have been able to develop projects that push the boundaries of possibilities and adapt to the ever-changing dynamics of the market. Our mission is to be the preferred global partner for delivery of single oligomer PEG derivatives for the biotech and pharmaceutical industries, and this applies also for the next 25 years!
Polypure has Built a Strong Reputation Over the Last 25 Years. As CEO, How Would You Balance Respecting the Company’s Legacy While Leading Cultural and Operational Changes that are Necessary to Keep Pace with Industry Evolution?
As mentioned before, research is the backbone of Polypure and our major focus has always been on delivering products of the highest quality and of uncompromised purity. This will not change as we move forward - we will continue to innovate, and we will not compromise when it comes to the quality or purity of new products. Combining chromatography with organic synthesis to make high-purity individual PEG oligomers and derivatives have always been our strength. Looking beyond the science; The fast-paced and continuously evolving pharmaceutical industry is also challenged with new regulations that struggle to keep up with the innovation taking place. Polypure now puts more effort into quality assurance and good documentation practice to further support the inherent quality of our products, making them attractive for the pharmaceutical industry. This is a key element in our professionalisation process as we continue to grow.
Tell us a Little About the Projects (such as PAVE, NOVA-MRI and PIANO) You Have Been Working on and What New R&D Ventures we Can Expect to See from You?
Polypure has been invited into several EU projects because we are an SME and an attractive collaboration partner due to our competence and capabilities to tailor-make useful compounds. Through the PIANO project, we have developed a new class of products with unique properties, monodisperse polypropylene glycol (PPG) and its derivatives. Since PPG is closely related
to PEG, it can be used as a linker or for improving solubility. But the slightly higher hydrophobicity and thermosensitivity of PPG, makes it also a promising candidate for amphiphilic copolymers and self-assembling formulations, like micelles and hydrogels. In NOVA-MRI we are attaching monodisperse PEG-derivatives onto fluorine labelled nanoparticles. We are investigating the impact of PEG length and choice of end-group on the binding of nanoparticles to various types of immune cells for improved biocompatibility. The PAVE project seeks to establish immunotherapy-driven strategies for treatment of pancreatic cancer. Polypure has developed homogenous PEG-based products; PEG-lipids and PEG-peptides (10.1021/ acsomega.4c02604) that can offer a better alternative to vaccine delivery systems based on lipid nanoparticles (LNPs) and peptide-based vaccines. Thanks to these EU projects, we have spent the last 5 years working in synergy with academia. We have gained an extensive network of universities and got to know experienced investigators working on the edge of peptide chemistry, nanotechnology, and biology. And thanks to this close cooperation, the projects were given access to custommade compounds which would not be available otherwise. Through these projects we have reached new audiences and spread awareness on the importance of use of well-defined and high-purity polymers in biotech and pharma applications.
Erik Agner
Erik Agner, CEO and founder of Polypure, has an academic background in chemistry from the University of Linköping in Sweden. Subsequently, he pursued his career by joining pharmaceutical companies in both Sweden and Norway where he engaged in peptide related projects. Through early work experiences, Erik Agner realized the challenges in generating high-quality materials for advancing pharmaceuticals and biotechnology. With interest in purification technology, he initiated a startup and filed patents in displacement chromatography. From that novel technology he founded Polypure in 1999.
Regulatory and Compliance
Emerging Pharma vs. Big Pharma: Rethinking CRO Partnerships
In today’s competitive pharmaceutical landscape, outsourcing has become essential to the development and commercialisation of drugs. This is especially true for emerging pharma companies, whose needs differ significantly from the more established, resource-rich big pharma firms. However, many contract research organisations (CROs) are still structured to cater primarily to big pharma, often overlooking the distinct needs of start-ups and smaller players.
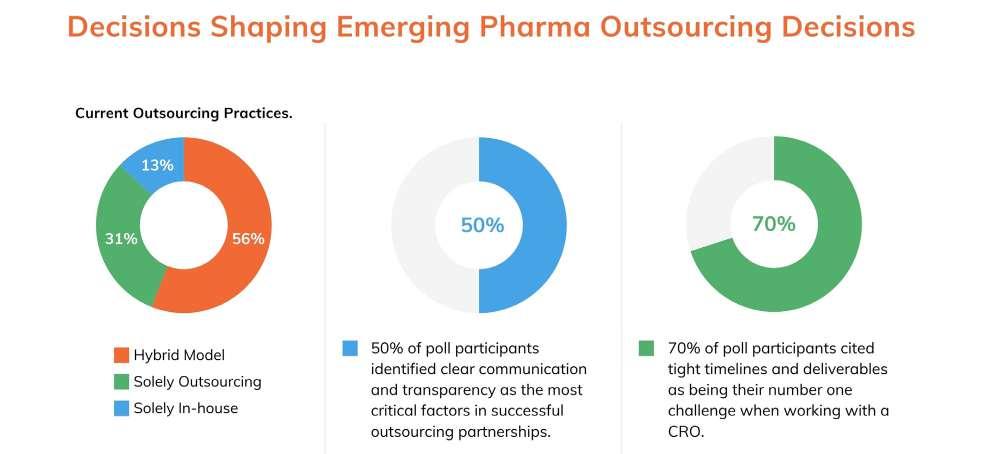
Emerging pharma companies face unique challenges in their path to market. While scientific capabilities and state-of-the-art facilities are important, polling during a recent webinar revealed that other, more human factors –such as clear communication and transparency – play a much more critical role in successful outsourcing relationships over time.1 Emerging pharma companies, unlike their larger counterparts, are looking for partnerships grounded in trust and collaboration. This deeper level of connection fosters research-minded problem-solving and allows for a more efficient and successful route to market.
The Growing Divergence Between Emerging and Big Pharma Emerging pharmaceutical companies are increasingly challenging the dominance of big pharma, particularly in innovative fields like gene therapy. As a result, nearly 54% of potential blockbuster drugs are now expected to come from smaller and emerging pharma companies.2 Despite this rapid growth, emerging companies continue to face challenges such as funding and access to trained personnel.
Strategic Decision-Making:
In-House Capabilities vs. Outsourcing
A critical decision for emerging pharma companies is whether to invest in building in-house capabilities or to rely on outsourcing. Both options have their advantages, but for many small and mid-sized companies, outsourcing offers a more strategic approach, particularly when working with bioanalytical testing or other specialised services.
Building in-house capabilities provides direct control over processes and decision-making. However, the costs associated with building and maintaining these capabilities, such as equipment, personnel, and compliance, can be prohibitive. In addition, the need for continuous training and upkeep can stretch the resources of smaller companies.
On the other hand, outsourcing to a specialised CRO can provide immediate access to state-of-the-art technology, expert teams, and the infrastructure needed for complex testing. For emerging pharma companies with limited resources, outsourcing allows them to focus on their core competencies, such as drug discovery and clinical strategy, while letting a
specialised partner handle the technical aspects of bioanalytical testing.
The Case for Specialised CROs
One of the biggest shifts in the emerging pharma landscape has been the move toward niche, specialised CROs. These CROs focus on specific areas of drug development, providing deeper expertise and more customised services than larger, generalist CROs.
• Agility: Specialised CROs excel at providing agility. They can quickly adapt to changes in project scope, timelines, or direction, making them ideal partners for companies that need to stay nimble. This is especially true for bioanalytical testing, where niche CROs can prioritise specific projects and provide faster turnaround times, helping smaller biopharma companies meet critical milestones.
• Deep Expertise: While large CROs offer a wide range of services, they may not have the specialised focus that niche CROs provide. Specialised CROs concentrate their resources on a few key areas, allowing them to stay at the cutting edge of their field. In bioanalytical testing, this means greater accuracy, more reliable data, and ultimately better decision-making for the client.
• Collaborative Communication: Specialised CROs are more likely to maintain close, collaborative relationships with their clients. Instead of simply delivering results, they work alongside emerging pharma companies to provide strategic insights and help troubleshoot challenges. This collaboration ensures that clients are always informed and involved, making the entire process more transparent and dynamic.
GREAT FLUIDIC SOLUTIONS
BIOTECH LIQUID µFLOWMETER
Continuous Flow Measurement 10 nl – 80 µl/min
Software compatibility with leading CDS
Regulatory and Compliance
Case Study: How to Get Ahead of Potential Delays
Recently, we collaborated with a biotherapeutic sponsor facing tight timelines to obtain the necessary data for an important grant deadline. Initially, the company engaged a large CRO to manage its preclinical study; however, a significant five-month delay between the dosing and bioanalysis timelines the CRO proposed posed a risk to the project’s timeline.
Recognising the urgency of the situation, the company engaged a consultant who understood the strategic importance of working with a specialised bioanalytical CRO. They chose to allow the large CRO to conduct the preclinical trial while sending the toxicology samples to our specialised bioanalytical CRO for analysis. Because this is our specialty, we were able to develop methods and conduct the necessary analysis in a relatively short time. By adopting this strategy, the sponsors successfully met their grant deadline.
This case study highlights the importance of two key factors:
1. Forward planning: The large CRO’s timeline included a significant five-month gap between dosing and bioanalysis. Had the sponsor engaged a specialised bioanalytical CRO from the beginning, they could have synchronised dosing and analysis schedules for a quicker data turnaround.
2. Engaging expert partners: Partnering with a specialised CRO enabled this sponsor to develop methods and conduct analyses more rapidly, allowing them to meet their tight deadlines.
This example underscores how partnering with specialised CROs can effectively address the unique needs of small to mid-sized pharmaceutical companies. Collaborating with niche CROs provides access to tailored expertise and dedicated support, empowering drug developers to meet tight deadlines efficiently. Furthermore, this approach highlights the value of strategic decoupling, which allows companies to focus on their core competencies while leveraging specialised services for critical project components.
Strategic Decoupling of Clinical and Bioanalytical Workflows
For smaller companies, efficiency is critical. Delays in drug
development can significantly impact their ability to secure investment and move their therapies forward. Unlike big pharma, which can sometimes absorb the financial impact of setbacks, emerging pharma companies depend on precise timelines and streamlined processes.
During a recent webinar, 70% of poll participants cited tight timelines and deliverables as being their number one challenge when working with a CRO. Often, delays stem from gaps in preclinical data, along with practical challenges like site selection and patient enrollment.
One solution is to decouple bioanalytical method development from clinical trials, allowing for parallel workflows that boost efficiency. For many emerging pharma companies, decoupling clinical and bioanalytical workflows offers significant advantages. By running these processes in parallel rather than sequentially, companies can save valuable time and gain access to critical data earlier in the development process.
Initiating the bioanalytical method development process prior to clinical trial start-up and recruitment, ensures no delays when the clinical samples are ready and allows for real-time data access. This approach can help identify potential issues early in the process, reducing the risk of costly delays later on. Specialised CROs, with their focus on bioanalytical testing, are well-suited to support this approach, offering the flexibility needed to adjust strategies as new data becomes available, while still meeting tight timelines.
Further, as drug development progresses from preclinical to in-human trials, having the bioanalytical method housed with a specialised CRO avoids the need for revalidation or loss of knowledge in optimising the method between trials. In an industry where speed is critical, this efficiency can provide a significant competitive advantage. By decoupling workflows and working with specialised CROs, these companies can accelerate their development timelines, gain early insights into drug efficacy, and avoid any costly delays.
The Benefits of a Partner Over a Vendor
A key to overcoming common outsourcing pain points lies in fostering open lines of communication with CRO partners, ensuring
Regulatory and Compliance
flexibility and agility in project management. In our polling, we were surprised to note the significance of communication in the decision of an outsourcing partner; 50% of poll participants identified clear communication and transparency as the most critical factors in successful outsourcing partnerships. These statistics demonstrate that for emerging pharma companies, choosing the right CRO is about more than just outsourcing services, it’s about building a strategic partnership.
Smaller boutique CROs that offer strong communication along with a scientific expertise, flexibility, and the ability to think outside the box are ideal partners for emerging pharma teams. In contrast to the traditional vendor model, this kind of engaged partnership brings added value by collaborating closely with the company, providing dynamic insights, and ensuring the project remains aligned with the company’s goals.
It All Comes Down to Partnership
Emerging pharma has very different needs from traditional large pharma companies. While some CROs are still trying to meet the needs of large pharma, emerging pharma is fast overtaking the market. Therefore, there is a growing space in the outsourcing landscape for a new kind of CRO, one that caters to the unique needs of emerging pharma. Strategic decoupling is an essential element in this strategy, overcoming many of the issues of delays that thwart start-ups all too often. Specialised CROs that are set up to facilitate this kind of strategic decoupling offer the benefits of deep expertise, cutting-edge facilities and efficiency. Embracing this kind of partnership over the old-style vendor relationship is essential for companies looking to compete in the modern pharmaceutical landscape.
People are at the heart of scientific research, and the relationships you build can make or break your project. Ask yourself, do you want to simply hand things over and wait weeks to find out if your experiment worked? Or would you
rather collaborate with someone who keeps you informed, helps you solve problems in real time, and genuinely wants to see your project succeed?
Working with a specialised CRO offers not only technical expertise but also the kind of collaborative communication that helps ensure success. And in the long run, it’s far more rewarding to work with a partner who genuinely understands your goals and wants to see your project succeed. In today’s competitive market, the right partner can make all the difference.
REFERENCES
1. Rethinking Emerging Biopharma’s Relationships with CROs. Xtalks. (2024). https://xtalks.com/webinars/rethinking-emergingbiopharmas-relationships-with-cros/, visited 22 Oct 2024.
2. Pangasa, A., Hohn, B., Rewari, P., Kumar, G. N. & Kumar, P. Emerging and Smaller Pharmaceutical are launching more $500M+ Products than Big Pharmaceutical. Zs. Emerging Biopharma. (2022). https:// www.zs.com/insights/emerging-and-smaller-pharma-to-launchmore-dollar-500-million-plus-products, visited on 22 Oct2024.
Aashritha Marepalli
Spearheading Sannova's commercial operations & marketing strategy, Aashritha brings a customer-centric and efficiency-driven approach to the team. She focuses on bridging study execution between the lab and clients with an emphasis on turn-around times and flexible study support. Aashritha began her career in investment management, and discovering a passion for growing companies, transitioned to fintech, where she worked with series A level startups in developing innovative client solutions. Aashritha holds an MBA from UVA's Darden School of Business, and a B.S. from NYU's Stern School of Business.
Breath Analysis and Cancer: The Future of Early Detection and Personalised Medicine
VOCs and Breath
There are many challenges associated with cancer management, including delayed diagnosis, low efficacy of treatment, and heterogeneity of disease. CT, MRI, and PET scans cannot identify small tumours, meaning it is difficult to achieve early diagnosis for patients. Histological biopsy is considered the gold standard for cancer diagnosis; however, it is invasive, time-consuming, and expensive. There is an urgent need for low cost and accessible diagnostic techniques that would allow for the earlier detection of cancer, personalised therapy, and assessment of treatment efficacy.
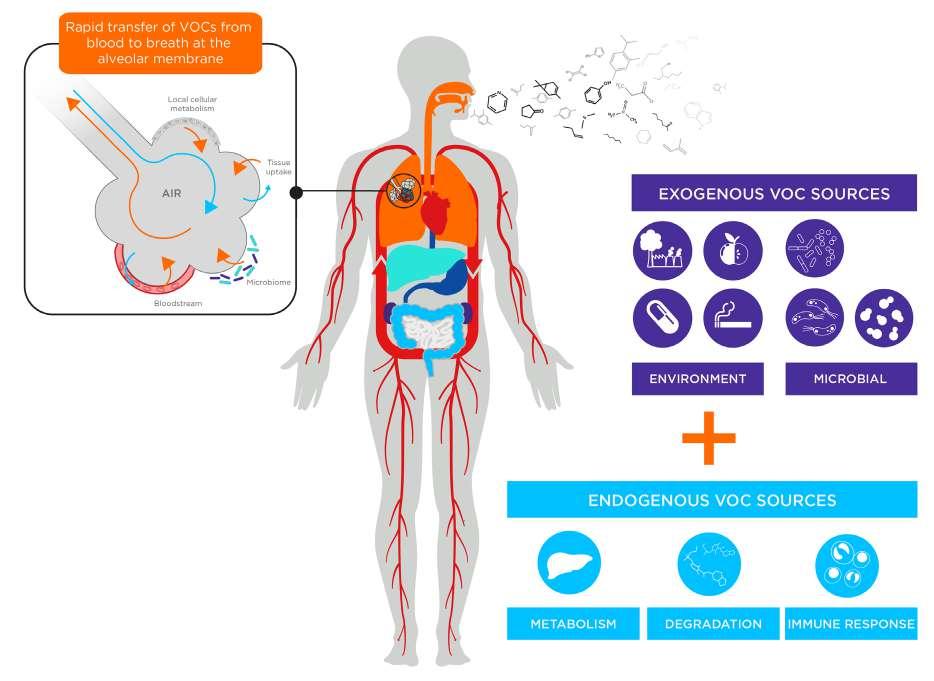
Volatile organic compounds (VOCs) are gaseous compounds produced throughout the body by numerous biological processes and released in biological samples such as faeces, urine, blood, and breath, with many advantageous attributes that position them as candidate novel biomarkers for cancer. The VOCs detectable in the breath can originate either from within the body (endogenous VOCs) or from external sources such as diet, medication, and environmental exposure (exogenous VOCs). Endogenous VOCs are produced throughout the body,
picked up, and distributed via the bloodstream, before being exhaled in breath (Figure 1). Exogenous VOCs interact with biological systems and provide valuable health and disease information. The collection, identification, and quantification of these VOCs can provide a window into what is happening inside the body, offering great potential as non-invasive biomarkers to indicate disease onset and progression.
Exhaled breath, enriched with VOCs, offers a promising sample matrix for clinical analysis and diagnosis of disease. By testing breath samples against established reference ranges, clinicians in the future could detect abnormal VOC levels that may signal the presence of disease. Compared to traditional sampling methods such as blood and faeces, breath analysis provides several key benefits; primarily its non-invasiveness, enhancing patient comfort and simplifying the approval process for clinical trials. Breath is an abundant and renewable resource, allowing for preconcentration of its compounds before analysis, which can improve test accuracy. Another important advantage is the flexibility of breath sampling, which can be conducted virtually anywhere. This opens the door to decentralised trial designs and at-home testing, making disease monitoring and diagnosis more accessible and convenient for patients.
Figure 1. Pathways of volatile organic compounds (VOCs) from exogenous (environmental and microbial) and endogenous (metabolic, degradation, and immune response) sources to exhaled breath
Measuring VOCs in Breath
Breath testing presents significant challenges due to the complex nature of breath itself, as well as the technical difficulties in the collection and analysis of exhaled breath. The VOCs are often found in very low concentrations in breath, frequently at trace levels, making them hard to detect without highly sensitive equipment. Researchers must also ensure that breath samples are uncontaminated with VOCs from background sources. Without proper collection and storage protocols, contaminants such as ambient air, bacteria, or particles from the collection apparatus can alter results. There are then often challenges associated with analysing the breath samples due to the complexity of breath and the thousands of compounds that are found in breath, many of which may not be characterised, as they require advanced analytical tools which can be time-consuming and costly. The entire breath collection process needs to be rigorously controlled to ensure reliable and reproducible results.1
To overcome these challenges, instruments such as the ReCIVA ® Breath Sampler have been developed by multi-disciplinary leaders in the breath research field as an optimised tool for robust and reliable breath sample collection whilst ensuring safety and comfort during use.2 This device enables the collection of replicate breath samples by directly capturing and pre-concentrating VOCs from breath onto sorbent tubes. The ReCIVA is connected to a source of clean air for users to breathe into, which minimises the contribution of contaminating VOCs entering a breath sample from the outside air, and therefore VOCs present in the breath can be distinguished more easily. Studies often utilise gas chromatography-mass spectrometry (GC-MS) due to its heightened sensitivity and capacity to handle a wide range of VOCs at low concentrations to detect and analyse VOCs accurately in the breath.
Metabolic Processes
Understanding the mechanistic origin of VOCs detectable in the
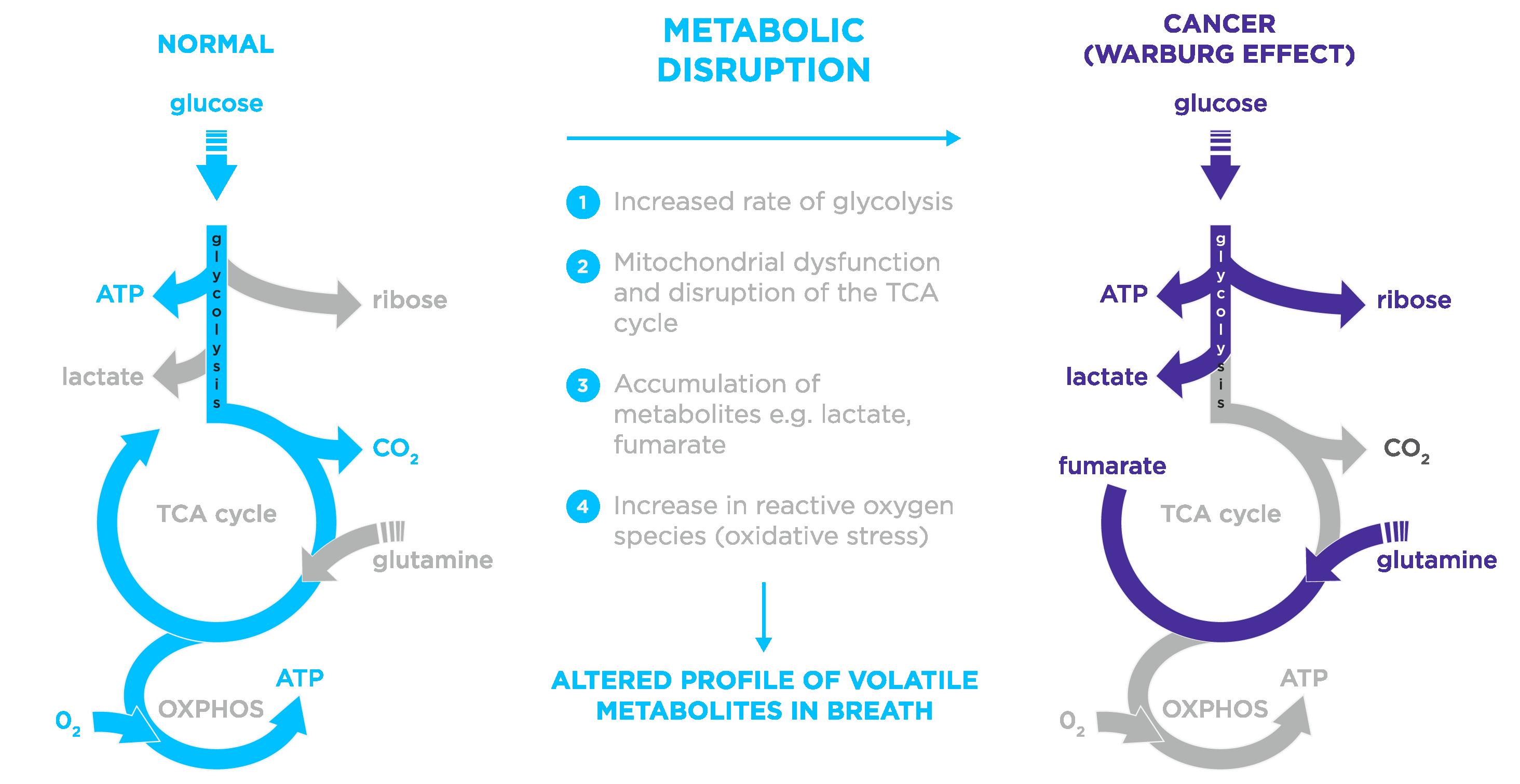
breath and identifying those compounds involved in cancerrelated pathophysiology and metabolic pathways is important in assessing whether they could serve as biomarkers for cancer. In normal cells, energy is produced through aerobic respiration, which includes glycolysis, the tricarboxylic acid cycle (TCA) cycle, and oxidative phosphorylation. When oxygen is limited, pyruvate, the end-product of glycolysis, no longer enters the TCA cycle but is converted into lactate. However, cancer cells require vast amounts of energy to support their rapid growth. The Warburg effect refers to the phenomenon of increased aerobic glycolysis and lactate production, along with the disruption of energy production in the TCA cycle, which cancer cells require for rapid proliferation. This altered metabolism results in increased production of acetyl-CoA, which in turn elevates the levels of ketones that can be detected in exhaled breath (Figure 2).
With the progress made in detecting and quantifying breath VOCs over the past decades, a recently published systemic review performed a meta-analysis of breath (and urinary) VOCs to assess their diagnostic potential in cancer detection.3 Based on the analysis of 85 publications, the authors reported that VOCs have a sensitivity of 0.89 and a specificity of 0.88 for cancer screening. The authors then focused on metabolic pathways, identifying several microbiota-related VOCs in lung, colorectal, breast, and liver cancers that enriched various metabolic pathways.3 Notably, butanoate metabolism was enriched across all four cancer types and was the most enriched pathway in lung, breast, and liver cancers.
These findings highlight the importance of one specific metabolite, butyrate, which is generated by gut microbial fermentation. Butyrate serves as a transport substrate in butanoate metabolism and plays a critical role in regulating host energy homeostasis. Since altered levels of butyrate have been associated with several diseases, including cancer, it holds potential as a biomarker.4,5,6 While butyrate can be detected
Figure 2. Comparison of normal metabolism and cancer metabolism (Warburg effect)
from blood samples, butyrate is volatile and detectable in exhaled breath, and so could support future clinical applications, including disease management and exploring therapeutic targets.
Cancer and Inflammation-Associated VOCs.
Chronic inflammation in the body can have many negative impacts on health. Specifically, chronic inflammation has been associated with the development and progression of various types of cancer.7 The mechanisms that link cancer and inflammation are complex and include interactions between immune cells and signalling molecules that can promote tumour growth and metastasis. Being able to detect signs of chronic inflammation in the body would, therefore, be useful for diagnosing and monitoring patients as well as in clinical research. As certain VOCs are byproducts of the metabolic processes altered by inflammation and cancer, breath analysis can offer a non-invasive way of detecting cancer earlier, as well as monitoring disease progression through the detection of inflammatory markers.
Studies have found that nearly all neoplastic legions (abnormal masses of tissue that form when cells divide and grow more than they should) contain immune cells at densities ranging from subtle infiltrations to gross inflammation.8 Research on the link between inflammation and cancer pathogenesis continues to produce results demonstrating the tumour-promoting effects that immune cells (mainly from the innate immune system) have on cancer progression. For example, inflammation can supply the tumour microenvironment with bioactive molecules. These molecules can include growth factors that increase proliferative signalling, factors that limit cell death, and enzymes that facilitate angiogenesis and metastasis.9 Angiogenesis plays a role in the growth of cancer as tumours need a consistent blood supply if they are to grow and metastasize. Tumours can cause this blood supply to occur by giving off chemical signals that stimulate angiogenesis and resulting blood vessels feed tumours with nutrients and oxygen, allowing tumours to metastasize.10
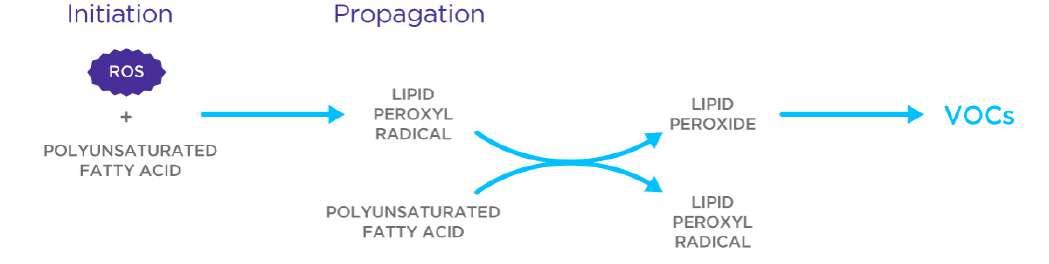
When inflammation occurs, the increased production of reactive oxygen species (ROS) can lead to oxidative stress, including lipid peroxidation. Lipid peroxidation is the oxidative degradation of lipids in cell membranes which generates VOCs such as alkanes and aldehydes. Oxidative stress and lipid peroxidation have been associated with cancer progression, specifically in lung carcinogenesis, as oxidative stress in lung tissue lipids can cause the presence of ROS in the lungs (Figure 3).11 Studies have been conducted to compare the
VOCs produced by lung cancer cells in vitro to the ones found in
breath from lung cancer patients.
A study by Buszewki et al. quantitatively measured VOCs in the headspace of healthy and lung cancer tissues and compared these results to VOCs obtained from breath samples of healthy individuals and lung cancer patients.12 A total of 22 compounds were found in both the headspace of cancerous tissues and the breath of lung cancer patients, including alcohols, aldehydes, ketones, and hydrocarbons. Compounds such as acetone, ethanol, 1-propanol, and carbon disulfide were found in higher levels in the headspace of cancerous tissues compared to healthy tissues. These same compounds were also found in increased concentrations in the breath samples of patients with lung cancer and could, therefore, be used as biomarkers of lung cancer. Aldehydes and alkanes are products of lipid peroxidation from inflammation, highlighting that the compounds found in exhaled breath in this study could be markers of inflammation caused by cancer and be used to identify and diagnose cancer at an earlier stage through exhaled breath analysis.
EVOC Probes and Lung Cancer
Understanding cancer-related metabolic pathways opens the door to developing powerful dynamic breath tests with boosted diagnostic accuracy, such as those that use an EVOC (exogenous VOCs) probe-based approach. EVOC probes are designed to target specific enzymes that are part of the affected metabolic processes in a disease state. Ingestion of an EVOC probe and measuring the probe and its metabolic products through a breath test has shown safety and great success in liver disease, where a longitudinal study showed that levels of the administered EVOC probe returned to baseline more slowly in cirrhotic patients than in controls after ingestion, with the best-performing timepoint observed at 60 minutes post-administration (AUC = 0.91).13
With the current understanding of cancer-related metabolic pathways, EVOC probes targeting the Warburg effect or lipid peroxidation pathways with high sensitivity and specificity could greatly benefit early detection in a non-invasive manner. In many cancer types, increased oxidative stress can lead to the overproduction of aldehydes, and the enzyme aldo-keto reductase (AKR) is suggested to effectively remove aldehydes by converting them into alcohols.14 Also, in healthy cells, certain enzymes are present inside the cells, whereas in cancer cell types these enzymes are found to leak out the cell and be present in the tumour microenvironment.15 Leveraging this concept, researchers have strategically designed an EVOC probe to amplify altered metabolic signals and measure
Figure 3. Lipid peroxidation pathway leading to the formation of volatile organic compounds (VOCs)
exhaled
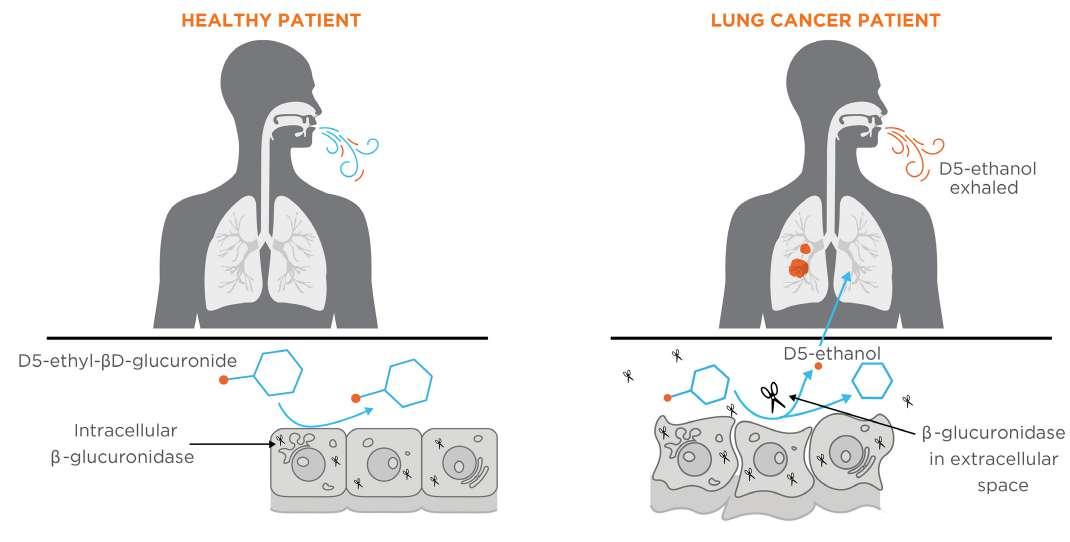
Figure 4. A schematic indicating how β-glucuronidase in the extracellular space around a tumor in the lungs can metabolise the D5-ethyl-βD-glucuronide probe into D5-ethanol
VOC production in exhaled breath for lung cancer detection. The probe uses D5-ethyl-βD-glucuronide as a substrate and targets β-glucuronidase , an enzyme with elevated levels in the extracellular space of cancer cells. The enzymatic reaction cleaves D5-ethanol, which can be measured in exhaled breath as a reporter for lung cancer screening (Figure 4).
The D5-ethyl-βD-glucuronide EVOC probe has demonstrated its safety, mechanism of action, and dosing range in the Phase I study, and a Phase II study is currently underway to confirm the robustness and specificity of this EVOC probe for lung cancer detection.16 As an innovative approach to breath testing, the use of EVOC probes is expected to expand rapidly in breath science over the coming years, highlighting the rapid evolution of the breath research field, and promising a future where early cancer detection and monitoring can be conducted non-invasively through a simple breath integrated into clinical practice. Other future applications of breath tests for cancer include the development of at-home devices for real-time sample analysis which utilise sensor technologies to detect concentration changes of targeted compounds. When developing portable breath analysis devices, it is crucial to focus not only on diseaserelevant biomarker selection, but also on the concentration range of targeted compounds in healthy versus diseased individuals and the detection limits of the chosen sensors.
Artificial Intelligence in Breath-based Diagnostics
With the breakthroughs in artificial intelligence (AI) in recent years, it is anticipated that AI could facilitate the processing of large and complex breath datasets, while machine learning algorithms for breath pattern recognition could enhance diagnostic capabilities.17 The future of breath analysis technology holds great promise for revolutionising disease diagnosis, management, and treatment monitoring, with breath tests for early cancer screening potentially being integrated into routine physical check-ups at clinics. With the right biomarkers, home-based breath testing devices could offer a convenient
solution to monitor health status after treatment, providing longitudinal data that could help physicians notice potential recurrences early. The non-invasive nature of breath tests holds the potential to reduce healthcare costs while improving, and even saving, millions of lives.
REFERENCES
1. Chou, H., Godbeer, L., Allsworth, M. et al. Progress and challenges of developing volatile metabolites from exhaled breath as a biomarker platform. Metabolomics 20, 72 (2024)
3. Zhou M, Wang Q, Lu X, Zhang P, Yang R, Chen Y, Xia J, Chen D. Exhaled breath and urinary volatile organic compounds (VOCs) for cancer diagnoses, and microbial-related VOC metabolic pathway analysis: a systematic review and meta-analysis. Int J Surg. 110(3):1755-1769 (2024)
4. Zhang Y, Zhou L, Bao YL, Wu Y, Yu CL, Huang YX, Sun Y, Zheng LH, Li YX. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem Biol Interact. 185(3):174-81 (2010)
5. Salimi V, Shahsavari Z, Safizadeh B, Hosseini A, Khademian N, Tavakoli-Yaraki M. Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids Health Dis. 16(1):208. (2017)
6. Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R, Lapek JD Jr, Zhang L, Wang WB, Hao S, Flythe MD, Gonzalez DJ, Cani PD, Conejo-Garcia JR, Xiong N, Kennett MJ, Joe B, Patterson AD, Gewirtz AT, Vijay-Kumar M. Dysregulated Microbial Fermentation of Soluble Fiber Induces Cholestatic Liver Cancer. Cell. 175(3):679694.e22 (2018)
7. Singh N, Baby D, Rajguru J, Patil P, Thakkannavar S, Pujari V. Inflammation and Cancer. Annals of African Medicine. 2019 Jul-Sep; 18(3): 121-126
8. Pagès, F., Galon, J., Dieu-Nosjean, MC. et al. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29, 1093–1102 (2010).
9. Hanahan, D., Weinberg, A. Hallmarks of cancer: the next generation. Cell. 144(5):646-674 (2011)
10. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications
of angiogenesis. Nature. 473(7347):298-307. (2011)
11. Hakim M, Broza YY, Barash O, Peled N, Phillips M, Amann A, Haick H. Volatile organic compounds of lung cancer and possible biochemical pathways. Chem Rev. 112(11):5949-66. (2012)
12. Buszewski B, Ulanowska A, Kowalkowski T, Cieśliński K. Investigation of lung cancer biomarkers by hyphenated separation techniques and chemometrics. Clinical Chemistry and Laboratory Medicine. 50(3): 573-581 (2012)
13. Ferrandino G, Ricciardi F, Murgia A, Banda I, Manhota M, Ahmed Y, Sweeney K, Nicholson-Scott L, McConville L, Gandelman O, Allsworth M, Boyle B, Smolinska A, Ginesta Frings CA, Contreras J, Asenjo-Lobos C, Barrientos V, Clavo N, Novoa A, Riviotta A, Jerez M, Méndez L. Exogenous Volatile Organic Compound (EVOC®) Breath Testing Maximizes Classification Performance for Subjects with Cirrhosis and Reveals Signs of Portal Hypertension. Biomedicines. 11(11):2957 (2023)
14. Ma J, Cao D. Human aldo-keto reductases: structure, substrate specificity and roles in tumorigenesis. Biomol Concepts. 2(1-2):11526 (2011)
15. Peltrini R, Cordell RL, Wilde M, Abuhelal S, Quek E, ZounematKermani N, et al. Discovery and Validation of a Volatile Signature of Eosinophilic Airway Inflammation in Asthma. American Journal of Respiratory and Critical Care Medicine. 2024 May 31
16. Van der Schee, M., Mizen, J., et al. Proof-of-mechanism for a diagnostic probe generating D5-ethanol as an on-breath reporter molecule for lung cancer – Evolution phase 1: https://www. owlstonemedical.com/media/uploads/files/2023-08_Evolution_ Poster_for_IASLC_Compressed.pdf
17. Skarysz A, Salman D, Eddleston M, Sykora M, Hunsicker E, Nailon WH, Darnley K, McLaren DB, Thomas CLP, Soltoggio A. Fast and automated biomarker detection in breath samples with machine learning. PLoS One. 17(4):e0265399. (2022)
Dr. Hsuan Chou
Dr. Hsuan Chou is a senior biomarker scientist at Owlstone Medical, where she ensures the successful delivery of customer project results and contributes to manuscript writing. She also supports the biological aspects of internal product development and plays an active role in creating scientific content for the company’s technical sales and marketing efforts. Dr. Chou holds a PhD in Plant Science from the University of Connecticut and has several years of postdoctoral experience working with omics data in the broader biology field at North Carolina State University before joining Owlstone in late 2021.
Email: hsuan.chou@owlstone.co.uk
Lucy Godbeer
Lucy Godbeer is a scientific marketing associate at Owlstone Medical, assisting with scientific content writing for the company. Lucy graduated with a BSc in Biology and has an MSc in Biomedical Science.
Email: lucy.godbeer@owlstone.co.uk
Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.
www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA POD
‘Putting science into conversation, and conversation into science.’Join some of the most esteemed and integral members of the Drug Discovery & Development world as they give insights & introspect into the latest movements, discoveries and innovations within the industry.
senglobalcoms.com
In vitro Models Guiding Strategic Decision-making in Biotherapeutic Development
Scientific advancements and evolving regulatory requirements are continually reshaping the drug development process. Navigating the intricacies and financial challenges of bringing new therapies to market in this dynamic landscape, demands innovative approaches and strategic decision-making. In this context, in vitro pre-clinical models have emerged as indispensable tools for accelerating the translation of promising discoveries into life-saving treatments.
As in vitro technologies advance, pre-clinical models, particularly those utilising human cells and tissues, offer a powerful platform for evaluating drug efficacy, safety and mechanisms of action early in the development pipeline. They provide a controlled environment where researchers can study complex biological processes and interactions, gaining valuable insights into drug behaviour and potential therapeutic benefits. By strategically leveraging in vitro models, biopharmaceutical companies can make more informed decisions, streamline development timelines and reduce reliance on animal testing, a critical consideration in modern drug development.
In this article, Dr Agapitos Patakas, RoukenBio’s Chief Scientific Officer (CSO), explores the multifaceted role of in vitro models in advancing therapeutic innovation. He examines the key drivers behind their increasing adoption, delves into the diverse types and applications of cell-based models and discusses strategies for ensuring their robustness and reliability. He also addresses the challenges and future directions in the field, highlighting how ongoing advancements and innovative approaches will shape the future of drug development.
The Evolving Landscape of in vitro Models
Technological advances are enhancing the predictive power of in vitro models in assessing eventual drug success in a clinical setting. Drug developers are increasingly relying on in vitro models in pre-clinical drug development as they offer the potential to streamline the process and improve efficiency. This growing reliance is further fuelled by a number of factors:
• Regulatory Evolution
The introduction of the FDA Modernisation Act 2.0 has allowed developers to utilise diverse testing methods –including human-relevant in vitro models – for evaluating the safety and efficacy of new drugs and biosimilars before they progress to human trials. This change in legislation reflects a growing recognition of the limitations of traditional animal testing and the need for more predictive and human-relevant pre-clinical models.
• Increasing Availability of Advanced In Vitro Technologies
The development of sophisticated three-dimensional (3D) cell culture systems, organ-on-a-chip platforms and
microphysiological systems has enabled researchers to create more complex and physiologically relevant models that better mimic human biology. Advancements in materials science have also been instrumental in replicating the extracellular matrix (ECM) of tissue microenvironment to facilitate the application of these technologies. Such advancements allow intricate drug interactions, cellular responses and disease mechanisms to be studied in a more controlled and human-focused environment.
• The Rising Costs and Complexities of Drug Development
Drug development is becoming increasingly expensive, placing a greater emphasis on efficiency and informed decision-making.1 In vitro models offer a cost-effective and time-saving approach to pre-clinical research, allowing developers to rapidly screen drug candidates and identify potential safety or efficacy issues early in the development process. As a result, researchers can prioritise promising candidates and allocate resources more effectively, ultimately accelerating the translation of discoveries into therapies.
• A Growing Emphasis on Ethical and Sustainable Practices in Research
By providing human-relevant data earlier in the drug development process, in vitro models can potentially limit the need for animal studies. Adopting effective in vitro modelling contributes to a more humane and responsible approach to drug development, aligning with the ethical concerns of researchers and the public. This is particularly important in areas such as immunology and immunooncology, where the intricacies of the human immune system can be challenging to replicate in animal models.
Types and Applications of In vitro Models
A wide variety of in vitro systems are available, each with its own strengths and limitations. Drug developers must select the appropriate model for a specific research question or development stage to obtain meaningful and reliable results.
Traditionally, 2D cell culture systems, where cells are grown on flat surfaces, have been the standard for most in vitro research. They offer simplicity, cost-effectiveness and ease of manipulation, making them valuable tools for studying basic cellular processes, screening drug candidates and assessing cytotoxicity. However, 2D cultures fail to recreate the tissue architecture and cellular interactions of living systems, limiting their predictive capacity for in vitro responses. To address the limitations of 2D cultures, researchers have developed more sophisticated in vitro models that better mimic the in vivo environment:
• Primary human cell models
In vitro assays utilising primary human cells, such as immune cells and tumour cells, are essential for
understanding disease mechanisms and evaluating novel therapies. Although limited by the access constraints of primary tissue, humanised cell-based assays provide valuable insights into the complex interactions between drugs and the immune system, enabling the development of more targeted and effective treatments. While human immortalised cell lines are available, the tissue source and genetic alterations made to these cells limit their translational benefit to drug discovery.
• Co-culture Models
Co-culture systems, which incorporate multiple cell types, such as immune cells and tumour cells, can provide a more accurate representation of the complex interactions within the tumour microenvironment. This approach allows researchers to study cell-to-cell interactions, the impact of the microenvironment on drug response, and the efficacy of immunotherapies. When combined with advanced synthetic biology and genetic engineering approaches these platforms can be powerful tools that allow the assessment of important translational questions, such as the impact of target antigen density on the safety and efficacy of antibody-based therapeutics.
Target antigen density is a crucial factor in the development of antibody-based therapeutics, including immune cell engagers, antibody-drug conjugates (ADCs), and Chimeric Antigen Receptor (CAR) therapies. It significantly influences both therapeutic efficacy and on-target toxicities. Traditionally, assessing target antigen density required the use of cell line panels expressing varying levels of the antigen of interest. However, this approach is logistically challenging and scientifically flawed, as intrinsic differences between cell lines can render them resistant or susceptible to therapeutic candidates in ways unrelated to antigen density. The IndEx-2 platform addresses these limitations by utilising chemically inducible proximity promoters to achieve a high dynamic range of tuneable and independent expression for up to two target antigens within a single, uniform cell line background. This enables precise control of antigen density, allowing for more accurate evaluation of therapeutic candidates. Using this system, it was demonstrated that CLN-978, a novel CD19-targeting T-cell engager (TCE) under development for B-cell malignancies and autoimmune diseases, retains
its activity even at very low levels of CD19 expression.2 This approach demonstrates how carefully designed co-culture models can provide crucial insights into drug mechanisms and efficacy, guiding further development decisions.
• Advanced and Humanised In Vitro Models
3D cell cultures, such as spheroids, organoids and materials that emulate the extracellular matrix (ECM) (e.g. hydrogels), allow cells to grow and interact in a more physiologically relevant manner, forming structures that resemble tissues and organs. These models allow researchers to study intricate processes like cell-to-cell and cell-ECM communication, tissue organisation and drug penetration, providing a more holistic view of drug effects.
With a growing understanding of human biology and technological advances, increasingly sophisticated in vitro models are being developed to better predict drug efficacy and safety in humans. Organoids, in particular, have shown great promise in modelling human diseases and predicting the efficacy of new drugs. These 3D structures, derived from stem cells or primary tissues, can mimic the complex architecture and function of organs, allowing researchers to study disease progression and drug response in a more realistic setting.
Tissue explant-based assay systems, which utilise small pieces of tissue taken directly from patients, offer another valuable approach. Unlike spheroids or organoids, these assays contextually preserve the native tissue architecture, pathology and cellular content and interactions, providing
Preclinical
a more accurate representation of the in vivo environment. This is particularly critical for evaluating immune-modulating therapies, as the pathology-specific microenvironment can profoundly influence immune cell function – a complexity that is difficult to replicate using blood-derived immune cells.
• Organ-on-a-chip Models
Organ-on-a-chip systems represent another in vitro modelling advancement where microfluidic devices incorporate living cells within microchannels, mimicking the structure and function of human organs. These models allow for the study of drug effects on multiple cell types simultaneously, as well as the assessment of drug transport and metabolism. Introducing flow to cell-based systems additionally replicates the natural stress and strain that assists physiologically relevant tissue maturation while providing a more realistic method of drug application to tissues. As a result, organ-on-chip systems offer a promising platform for predicting drug responses in humans and reducing reliance on animal testing.
• Stem Cell Technology
An alternative approach to using human primary cells is utilising stem cell technology, in particular, the development of induced pluripotent stem cells (iPSCs), that can be produced from adult cells or tissues. Stem cell technology offers access to human tissue without the constraints of taking tissue samples from vital organs. These stem cells and similar classes can be differentiated into multiple cell types to investigate human physiology in both health and disease while retaining the genetic information of the person providing the tissue. Stem cells can be combined with genetic engineering technologies like CRISPR to further improve the effectiveness of pre-clinical models. This allows researchers to introduce specific disease-related mutations or create reporter cell lines for more targeted studies, ultimately increasing the effectiveness of these models before moving to live systems.
When selecting an in vitro model, it is important to also consider the specific research question, stage of drug development and the desired level of complexity needed. Simple 2D cultures may be sufficient for initial screening workflows when throughput and assay robustness is of consideration, while more intricate 3D models or organ-on-achip systems may be necessary for studying intricate biological processes or predicting clinical outcomes. Careful selection and implementation of appropriate models allow researchers to gain valuable insights into drug behaviour, identify promising candidates, and accelerate the development of safe and effective therapies.
Ensuring Robustness and Reliability
Generating meaningful and reproducible data from in vitro models requires careful consideration of several key factors. Appropriate assay systems and technologies must be applicable to the specific research question, the drug candidate being evaluated and the desired endpoints. Employing multiple orthogonal assays can provide a more comprehensive understanding of drug effects and increase confidence in the data generated. Access to well-characterised biological
materials ensures the data generated is of relevance to the research question, minimising variability and ensuring the reproducibility of results.
Maintaining rigorous quality control throughout the experimental process is also essential in contributing to the reliability of in vitro data. This includes implementing standardised protocols, using validated reagents and equipment, and ensuring proper training and expertise of personnel involved in the study. Part of ensuring an effective drug discovery output is incorporating appropriate controls and benchmark molecules in in vitro studies, aiding data interpretation and validation. Positive and negative controls help establish the dynamic range and sensitivity of the assay, while benchmark molecules provide a reference point for comparing the activity of novel drug candidates.
Finally, embracing technological advancements and data analysis tools can significantly enhance the robustness and reliability of in vitro studies. Automated liquid handling systems, high-throughput screening platforms and advanced imaging techniques can improve precision, reduce human error and increase the throughput of experiments. Robust data analysis methods, including statistical modelling and machine learning, can help extract meaningful insights from complex datasets and identify potential biases or confounding factors.
Limitations and Advancements in In vitro Modelling
While in vitro models offer a powerful approach to pre-clinical research, they are not without limitations. One inherent challenge lies in replicating the intricate complexity of the in vivo environment. The human body is a dynamic and interconnected system, with multiple organs, tissues and cell types interacting in a tightly regulated manner. Even the most advanced in vitro models can only partially capture this complexity.
However, ongoing advancements in technology and innovative approaches are constantly pushing the boundaries of in vitro modelling, addressing these challenges and paving the way for more predictive and human-relevant systems. For example, researchers are exploring the potential of "body-on-a-chip" systems, which aim to connect multiple organ-on-a-chip models to create a more holistic representation of human physiology. This approach could enable the study of drug interactions with multiple organs and provide a more comprehensive understanding of systemic effects.
Another area of active research is the development of personalised in vitro models. These models utilise cells or tissues derived from individual patients, either from tissue biopsies or stem cell methods, allowing for the study of drug effects in a patient-specific context. Developing stratified approaches to in vitro models holds great promise for personalised medicine, enabling the identification of the most effective treatments for individual patients based on their unique genetic and phenotypic characteristics.
In vitro Models Driving the Future of Drug Development
In vitro pre-clinical models have become essential in producing innovative and effective therapies. These cell-based models provide valuable insights into drug efficacy, safety and mechanisms of action, cell-based models empower
biopharmaceutical companies to make informed decisions, streamline development processes and reduce reliance on animal testing.
As technology continues to evolve, we can anticipate even more sophisticated and human-relevant in vitro models to emerge, further revolutionising drug development. The future of drug discovery hinges on embracing these advancements and fostering collaboration among stakeholders.
To maximise the value and impact of cell-based models, the industry must prioritise robust quality control, meticulous experimental design and access to well-characterised biological materials. The continued integration of advanced technologies will enable the creation of highly complex and personalised models that more accurately predict drug behaviour in humans.
Ultimately, these efforts in developing and refining in vitro models will accelerate the translation of scientific discoveries into life-saving treatments, leading to improved human health and more effective therapies.
Dr. Agapitos Patakas, CSO, RoukenBio. As CSO, Agapitos strategically leads all R&D and new service development. He is co-inventor in several patents in the field of T-cell therapies and cell line development. Agapitos joined RoukenBio in 2018 where he established the translational immunology business unit, leading the development of several bespoke immunology assays such as our T-cell exhaustion, T-cell mediated cytotoxicity assays etc., significantly expanding the company’s service portfolio. He originally trained as a pharmacist, followed by an MSc in Immuno-Pharmacology & MPhil in Pharmacology (Uni of Strathclyde) and a PhD in Immunology/Rheumatology (Uni of Glasgow).
Creating Independent and Resilient Supply-Chains for Key Raw-Materials, Reagents and Building Blocks for the Fine-Chemical and Pharmaceutical Industry in Europe and North America. For customized products, adjusted specifications and packaging, with safe and sustainable manufacturing solutions made in Europe, contact us at info@anceus.com
Accelerating Drug Discovery:
Integrating In Vivo and In Vitro Testing with C. elegans
The pharmaceutical industry is continually challenged by high attrition rates (> 90 %) as drug candidates progress from discovery, through clinical trials and to market. Neurodegenerative disease research, for example, is well known for late-stage failures due to the limitations of traditional drug development pipelines, which rely heavily on in vitro and in vivo animal testing. These bottlenecks can be attributed to the inherent limitations of each method, with in vitro models often failing to capture the complexity of living organisms, and in vivo models being too costly and time-consuming for early-stage testing of lead molecules. Integrating Caenorhabditis elegans (C. elegans) early into these pipelines, by running concurrently with in vitro testing, offers a solution by allowing earlier detection of potential failures, thereby reducing time and costs. This integration provides a more nuanced approach to drug discovery, where both the cellular and whole-organism level response to the compounds is considered in tandem, offering a clearer picture of the potential success or failure of a compound.
The Bottleneck in Drug Discovery:
Transitioning from In Vitro to In Vivo Testing
One of the most significant challenges in drug discovery is the transition from in vitro (cell-based) models to in vivo (animalbased) models. In vitro testing is invaluable for its ability to screen large numbers of compounds quickly and cost-effectively, providing detailed insights into the biochemical and cellular effects of potential drugs. However, these models lack the complexity of an entire living organism, where factors such as metabolism, intra-tissue interactions, and whole-organism physiological processes can be considered. As a result, many compounds that perform well in vitro fail in subsequent in vivo tests, leading to wasted time and resources.
The use ofC. elegans as a model organism offers a promising solution. With its simple anatomy, well-understood genome, and significant genetic similarity to humans, C. elegans provides an efficient and ethically acceptable model for early-stage in vivo testing. By incorporating C. elegans into the pipeline, researchers can observe whole-organism effects early on, allowing for better prioritisation of compounds before they advance to more complex and costly studies in mammalian animal models.
Integrating In Vivo and In Vitro Testing:
A Synergistic Approach
Data from C. elegans experiments provide different forms of information than data obtained only from in vitro experiments in human cell lines, adding to the predictive power of early-stage drug discovery. The data can be used to inform decision-making, or at a greater scale, to train machine-learning approaches. Compounds that show positive results in both in vitro assays and C. elegans models are more likely to succeed in subsequent
stages of testing. This synergy allows for refining selection criteria, ensuring that only the most promising candidates progress to mammalian studies. By doing so, the overall number of mammals used in testing is reduced, aligning with the 3Rs principle of minimising animal use while maximising research efficiency, ultimately accelerating the drug discovery process and reducing the likelihood of costly late-stage failures.
Neurodegenerative Disease Research
Neurodegenerative disease drug development is notorious for its high rate of late-stage failures, with many diseasemodifying therapies for conditions like Alzheimer’s failing in clinical trials.1 One of the critical challenges in this field is the lack of physiological and predictive animal models that accurately mimic human disease progression. Current mouse models often rely on artificially accelerated pathology in young animals. These models do not replicate the natural course of neurodegenerative diseases in humans, which mostly occurs in older individuals. Age is the strongest risk factor for developing Alzheimer’s disease. The nematode C. elegans ages fast when compared to mammalian animal models and has emerged as a valuable model in the field of the biology of ageing. For example, discoveries in C. elegans showed that the Insulin/Insulin-like growth factor signalling, influences ageing, a finding that has been shown to be true in flies and mice. A genetic change in the downstream protein of this pathway has been shown to be associated with long-lived humans. In neurodegenerative disease research, combining in vitro approaches with C. elegans models offers a more robust and comprehensive strategy. in vitro models, such as induced pluripotent stem cells (iPSCs) derived from patients with neurodegenerative diseases, allow for the detailed study of cellular mechanisms and the initial screening of drug candidates.2
Drug Discovery Pathway
Potential Drug Discovery Pathway with C. elegans
C. elegans complements these in vitro approaches by providing a whole-organism context, where the effects of drug candidates can be monitored throughout the organism’s lifespan. For example, continuous monitoring of C. elegans can reveal early signs of mobility decline during ageing, which are indicative of neurodegenerative progression. This capability allows researchers to evaluate not just the cellular impacts of a compound, but also its effects on overall organismal health and behaviour.3 The synergy between in vitro and in vivo testing can lead to earlier identification of potential failures, reducing the risk of late-stage setbacks and ensuring that only the most promising candidates advance to clinical trials.
This integrated approach allows researchers to adjust dosing regimens, explore combination therapies, and gain a more accurate understanding of the long-term effects of potential treatments. By observing these effects in a whole organism, rather than just on isolated cells, researchers can better predict how a drug will perform in humans, significantly de-risking the transition from pre-clinical studies to human trials.
Expanding the Role of C. elegans in Drug Development and Beyond Looking forward, there is significant potential to expand the role of C. elegans in drug development. Advances in genetic engineering, imaging technologies, and high-throughput screening are making C. elegans an even more powerful tool for drug discovery. For example, CRISPR-Cas9 technology allows for precise genome editing in C. elegans, enabling the creation of more accurate disease models.
Furthermore, the integration of automated imaging systems and automated worm handling enhances the ability to monitor C. elegans in high-throughput screens.4 These systems provide detailed phenotypic data that can be analysed in real-time, further accelerating the drug discovery process. By combining these technological advancements with the inherent advantages of C. elegans, biotech and pharmaceutical companies can develop more robust and translatable pre-clinical models.
Additionally, the vast amounts of data generated from C. elegans experiments are increasingly being used in machine learning algorithms. For instance, recent studies have demonstrated how machine learning can predict lifespanextending chemical compounds in C. elegans by analysing large datasets of chemical interactions, thereby refining drug candidates earlier in the development process.5 This synergy between C. elegans models and machine learning is revolutionising preclinical testing, making the transition to clinical trials faster and more reliable.
Another promising area is the exploration of C. elegans for microbiome studies, which is gaining traction as we better understand the role of gut microbiota in health and disease. Given that C. elegans can be maintained germ-free, researchers can manipulate its microbiome with various bacterial strains to study their effects in a controlled environment.6 This capability is invaluable for testing the impact of probiotics and other microbiome-targeting therapies, allowing for a better understanding of how these treatments might work in humans.
Conclusion
The integration of in vivo and in vitro testing, particularly through
the use of a 3R-compliant animal model, C. elegans, represents a significant advancement in the field of drug discovery. By bridging the gap between these testing stages, pharmaceutical companies can more effectively validate drug candidates, reduce the time and cost of development, and adhere to ethical standards. This approach not only streamlines the drug discovery process but also aligns with the growing emphasis on ethical research practices, reducing the need for vertebrate animals in early-stage testing.
As the pharmaceutical industry continues to evolve, the adoption of innovative models like C. elegans will be essential for maintaining efficiency and ensuring the development of safe and effective therapies. By embracing these approaches, the industry can accelerate the transition from the lab bench to the bedside, ultimately benefiting patients and advancing public health. The ongoing development of technologies such as CRISPR-Cas9 and automated imaging systems, and machine learning promises to further enhance the capabilities of C. elegans as a model organism, paving the way for more personalised and precise therapeutic interventions. In this rapidly changing landscape, C. elegans stands out as a versatile and powerful tool that, when used in conjunction with in vitro methods and advanced data analysis techniques, can significantly enhance the robustness and translatability of pre-clinical drug development.
REFERENCES
1. Cummings et al., 2019. Alzheimer’s Research and Therapy. The ‘’rights’’ of precision drug development for Alzheimer’s disease.
2. Caldwell K. A., Willicott C. W., & Caldwell G. A. "Modeling neurodegeneration in Caenorhabditis elegans." Dis Model Mech., 13(10) (2020).
3. Roussos A., Kitopoulou K., Borbolis F., & Palikaras K. "Caenorhabditis elegans as a model system to study human neurodegenerative disorders." Biomolecules, 13(3) (2023).
4. Petrascheck M., Ye X., & Buck L. B. "A High-Throughput Screen for Chemicals that Increase the Lifespan of Caenorhabditis elegans." Annals of the New York Academy of Sciences, 1171(1) (2009): 95-106.
5. Ribeiro C, Farmer CK, de Magalhães JP, Freitas AA. Predicting lifespanextending chemical compounds for C. elegans with machine learning and biologically interpretable features. Aging (Alban y NY). 2023 Jul 13;15(13):6073-6099. doi: 10.18632/aging.204866. Epub 2023 Jul 13. PMID: 37450404; PMCID: PMC10373959.
6. Matty, M. A. & Chalasani, S. H., 2020. Microbial Mind Control. Cell Host & Microbe, 28(2), pp. 147-149.
David Weinkove
David is well respected in the academic fields of C. elegans and ageing biology. As a postdoc, he worked with David Gems, Ronald Plasterk and Erik Jorgensen. He is now Associate Professor at the Department of Biosciences, Durham University and Chair of the British Society for Research on Ageing. David is passionate about applying the strength of C. elegans research to industrial application and he co-founded Magnitude Biosciences to bring reliable automated technology together with experienced C. elegans scientists to bring increases in productivity to the whole field.
Email: david@magnitudebiosciences.com
Peptide Therapeutics: Unleashing the Power of Precision Medicine
In the dynamic world of drug discovery and development, where the quest for innovative and effective therapies is relentless, peptide therapeutics have emerged as a beacon of hope. Occupying a unique niche between small molecule drugs and complex biologics, peptides offer a compelling combination of advantages that position them at the forefront of modern medicine. Their inherent specificity, arising from their precise amino acid sequences, allows them to target disease-related proteins with laser-like precision, minimising the risk of unintended side effects often associated with small molecules. This specificity also empowers peptides to interact with challenging protein targets, including those involved in protein-to-protein interactions, that have historically been deemed "undruggable" by conventional drug modalities.
Moreover, peptides boast the advantage of being chemically synthesised. This synthetic nature translates to cost-effective and scalable production processes, contrasting with the complex and often expensive manufacturing of biologics. The inherent flexibility of peptide synthesis further enables scientists to fine-tune their physicochemical properties, such as solubility and stability, to optimise drug delivery and enhance therapeutic efficacy. The confluence of these advantages has positioned peptides as a powerful tool in the fight against a wide array of diseases, from cancer and metabolic disorders to infectious diseases and beyond. As the global peptide therapeutics market surges towards an estimated $75 billion by 2028, it is clear that the biopharmaceutical industry is on the brink of a transformative era in drug development.
Overcoming Challenges in Peptide Drug Development
Despite these compelling advantages, the path to peptide drug development is not without its hurdles. The design of potent and selective peptides requires a deep understanding of protein structure and function, coupled with sophisticated computational and experimental techniques. For instance, researchers often employ techniques like X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and computational modelling to gain insights into the intricate interactions between peptides and their target proteins. This knowledge is crucial for designing peptides that can bind with high affinity and specificity, maximising their therapeutic potential while minimising off-target effects. Furthermore, the synthesis and manufacturing of peptides, particularly those with complex structures or modifications, can be challenging and costly. Scaling up production from small-scale research to large-scale clinical trials and commercialisation demands specialised expertise and infrastructure. This is where Contract Development and Manufacturing Organisations (CDMOs) emerge as indispensable partners in the peptide drug
development journey. CDMOs possess the technical know-how, state-of-the-art facilities, and regulatory experience to navigate the complexities of peptide manufacturing, ensuring the seamless transition from bench to bedside. By leveraging their capabilities, biopharmaceutical companies can overcome the challenges associated with peptide development, accelerate timelines, and ultimately bring innovative therapies to patients in need.
Advances in Peptide Synthesis and Diversification
The field of peptide therapeutics has witnessed remarkable advancements in synthesis and diversification techniques, propelling the development of novel and effective treatments. The introduction of solid-phase peptide synthesis (SPPS) in the 1960s revolutionised the field, enabling the efficient and controlled synthesis of peptides with diverse sequences. This breakthrough paved the way for the development of numerous peptide-based drugs that are now used to treat a variety of conditions. Further innovations, such as microwave-assisted synthesis, ligation chemistry, and recombinant synthesis, have expanded the repertoire of accessible peptide structures and functionalities. The incorporation of unnatural amino acids, late-stage functionalisation, and chemo-selective ligation techniques has enabled the creation of peptides with enhanced stability, bioavailability, and target specificity. For example, the use of unnatural amino acids can introduce novel chemical properties into peptides, allowing them to interact with their targets in unique ways or resist degradation by enzymes. These advancements have significantly expanded the chemical space of peptide therapeutics, paving the way for the development of novel drugs targeting a wider range of diseases.
Peptide Discovery: Emerging Technologies
In addition to advancements in synthesis, the discovery of
Figure 1: Peptide technology in action
A dual-wavelength expansion module enables recombinant cascade reagents and chromogenic LALbased assays to be run on your αBET™ system, alongside the existing kinetic turbidimetric assays. First shipments will drop in March 2025 – scan the QR code to receive further information and we’ll be in touch!
The CMD αBETTM system is a fully integrated endotoxin testing system providing users with a rapid and sustainable solution to endotoxin testing without compromising on sensitivity or performance.
Research / Innovation / Development
peptide therapeutics has also benefited from the emergence of powerful new technologies. DNA-encoded libraries (DELs) and display techniques, such as phage display and yeast display, have revolutionised the screening and identification of peptides with high affinity and specificity for their targets. These high-throughput approaches allow for the rapid exploration of vast chemical libraries, significantly accelerating the discovery process. By combining these cutting-edge technologies with traditional screening methods, researchers can now efficiently identify and optimise peptide leads, expediting the development of novel therapeutics.
Case Study: Tirzepatide – A Dual-Action Peptide Targeting Diabetes and Obesity Care
Tirzepatide, a novel dual GIP and GLP-1 receptor agonist, epitomises the precision medicine approach within peptide therapeutics. By engaging both GIP and GLP-1 receptors, tirzepatide achieves superior glycemic control and weight reduction compared to selective GLP-1 agonists, demonstrating the potential of synergistic mechanisms. This targeted approach, coupled with flexible dosing options, allows for personalised treatment strategies tailored to individual patient needs. Furthermore, emerging evidence suggests potential disease-modifying effects, including preservation of beta-cell function and improvement in cardiovascular risk factors. While long-term safety data is still being gathered and cost remains a consideration, tirzepatide represents a significant advancement in peptide therapeutics, poised to reshape the management of type 2 diabetes and obesity.
Case Study: BT1718 – Targeting Metastasis in Cancer
Another remarkable success story in peptide drug development involves the application of CLIPSTM (Chemical Linkage of Peptides onto Scaffolds) technology. Researchers utilised CLIPSTM to identify a bicyclic peptide that selectively binds to membrane-type 1 matrix metalloproteinase (MT1-MMP) present on tumour cells. MT1-MMP is a key player in cancer metastasis, and by linking a toxin payload to this high-affinity bicyclic peptide, scientists created a 'Bicycle Drug Conjugate (BDC)' known as BT1718. This compound has demonstrated rapid tumour penetration and significant anti-tumour activity in preclinical studies, offering hope for a new weapon in the fight against metastatic cancers.
The Clinical Trials Landscape and Market Potential
The growing number of FDA-approved peptide drugs and the expanding pipeline of peptide-based therapeutics in clinical trials highlight the increasing recognition of their therapeutic potential. Peptides are being investigated for a wide range of applications, including cancer, metabolic disorders, infectious diseases, and more. The clinical trial landscape reveals a diverse range of drug targets and therapeutic areas, showcasing the versatility and adaptability of peptide-based therapies.
The global peptide therapeutics market is witnessing remarkable growth, fuelled by the rising prevalence of chronic diseases and the need for targeted and effective treatments. With a current market value of US$39.3 billion, peptide-based drugs represent a substantial and rapidly growing segment of the pharmaceutical sector. The continued advancements in peptide discovery, design, and manufacturing, coupled with
Figure 2: Positive Papanicolaou test the support of CDMOs, are expected to further drive the growth of this market, bringing forth a new generation of innovative therapies that will transform patient care.
Conclusion:
A Promising Future for Peptide Therapeutics
Peptide therapeutics stand at the forefront of a new era in drug discovery and development. Their unique advantages, coupled with ongoing advancements in synthesis, diversification, and discovery technologies, position them as a powerful tool in the fight against a wide range of diseases. The expanding clinical trials landscape and the robust growth of the peptide therapeutics market further underscore their potential to address unmet medical needs and improve patient outcomes. As the field continues to evolve, we can anticipate a future where peptide-based therapies play an increasingly vital role in the treatment of various diseases, ultimately shaping the future of healthcare.
REFERENCES
1. Bertoldo D et al (2016), ‘Phage Selection of Peptide Macrocycles against β-Catenin To Interfere with Wnt Signaling’, ChemMedChem, 11(8), 834–839.
2. Bozovičar K et al (2021), ‘Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties’, International Journal of Molecular Sciences, 22(4), Article 4.
3. Chen S et al (2012), ‘Structurally Diverse Cyclisation Linkers Impose Different Backbone Conformations in Bicyclic Peptides’, ChemBioChem, 13(7), 1032–1038.
4. Cook N et al (2019), ’Pharmacokinetic (PK) assessment of BT1718: A phase I/II a study of BT1718, a first in class bicycle toxin conjugate (BTC), in patients (pts) with advanced solid tumours’, Annals of Oncology, 30, v174.
5. DiMasi J et al (2018), ‘Assessing the Financial Benefits of Faster Development Times: The Case of Single-source Versus Multi-vendor Outsourced Biopharmaceutical Manufacturing’, Clinical Therapeutics, 40(6), 963–972.
6. Duengo S et al (2023), ‘Epimerisation in Peptide Synthesis’, Molecules, 28(24), 8017.
7. Fisher E et al (2019), ‘Peptide-Based Therapeutics for Oncology’, Pharmaceutical Medicine, 33(1), 9–20.
8. Grieco P et al (2019), ‘Natural and synthetic peptides in the cardiovascular diseases: An update on diagnostic and therapeutic potentials,’ Archives of Biochemistry and Biophysics, 662, 15–32.
Research / Innovation / Development
9. Heinis C et al (2009), ‘Phage-encoded combinatorial chemical libraries based on bicyclic peptides’, Nature Chemical Biology, 5(7), 502–507.
10. Imai K et al (2006), ‘Comparing antibody and small-molecule therapies for cancer’, Nature Reviews, 6(9), 714–727.
11. Itoh Y (2006), ‘MT1-MMP: A key regulator of cell migration in tissue’, IUBMB Life, 58(10), 589–596.
12. Iyengar S et al (2017), ‘The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine’, Pain, 158(4), 543–559.
13. Lee AC et al (2019), ‘A Comprehensive Review on Current Advances in Peptide Drug Development and Design,’ International Journal of Molecular Sciences, 20(10), 2383.
14. Li S et al (2023), ‘Therapeutic Peptides for Treatment of Lung Diseases: Infection, Fibrosis, and Cancer’, International Journal of Molecular Sciences, 24(10), 8642.
15. Robinson JA (2019), ‘Folded Synthetic Peptides and Other Molecules Targeting Outer Membrane Protein Complexes in Gram-Negative Bacteria’, Frontiers in Chemistry, 7.
16. Sharma K et al (2023), ‘Peptide-based drug discovery: Current status and recent advances’, Drug Discovery Today, 28(2), 103464.
17. Timmerman P et al (2009), ‘Functional Reconstruction of Structurally Complex Epitopes using CLIPS™ Technology’, The Open Vaccine Journal, 2(1).
18. Torres MDT et al (2019), ‘Peptide Design Principles for Antimicrobial Applications’, Journal of Molecular Biology, 431(18), 3547–3567.
19. Tufts Centre for the Study of Drug Development. (2017). Assessing
the Economics of Single-Source vs. Multi-Vendor Manufacturing [White paper].
20. Venneti NM et al. (2023), ‘Stretching Peptides Potential to Target Protein-Protein Interactions’, ACS Central Science, 9(4), 590–592.
21. Wang L et al (2022), ‘Therapeutic peptides: Current applications and future directions’, Signal Transduction and Targeted Therapy, 7(1), Article 1.
22. Wang X et al (2021), ‘Rational Design of Peptide-Based Inhibitors Disrupting Protein-Protein Interactions’, Frontiers in Chemistry, 9.
Peter Timmerman
Peter Timmerman, Head of Peptide Science at Biosynth, joined the group following the acquisition of Pepscan in 2022, where he had served as chief scientific officer since 2001. He drives advancements in protein mimicry for peptide drug discovery programs. Timmerman is the inventor of CLIPSTM technology and previously held a chair as a Professor at the University of Amsterdam. He obtained his Chemistry degree from Vrije Universiteit (Amsterdam) and earned his PhD cum laude from the University of Twente. Timmerman has co-authored over 80 scientific papers and holds more than 10 patents.
Advancing Cancer Immunotherapy Research:
The Role of Multiplex Immunoassays
Immunotherapy has revolutionised the treatment of cancer. However, while it shows high efficacy against certain tumours, immunotherapy is not effective against all types of cancers, or in all patients. For the development of more effective immunotherapies against a greater variety of cancers, a better understanding of the interaction between tumours and the immune system is needed. Biomarkers serve as useful indicators of the immune landscape, enabling scientists to gain deeper insight into the changes that occur during tumour progression. Profiling the immune landscape assists researchers to predict patient response to immunotherapy, monitor the tumour microenvironment, and the identification of biomarkers of adverse immune reactions. These insights can be used to guide the development of personalised therapies to improve patient outcomes.
The Complexities of the Tumour-immune Landscape
Alongside the transformation of cancer treatment, with both improved therapeutic efficacy and promise for future application, immunotherapy has also led to a deeper understanding of tumours and their surrounding landscape, including the tumour microenvironment (TME) and the immune system. The TME is a complex and evolving ecosystem, constituting changes to the extracellular matrix, environmental factors, and the presence of different cell types alongside the tumour cells. These changes help cancer cells to survive, which is partly achieved by influencing the interaction between tumours and the immune system. Understanding this complexity is important to provide insights into the mechanisms by which cancer cells adapt to survive. Cancer immunology focuses on the study of this interaction between tumours and the immune system and has revealed crucial insights leading to the development of cell therapies, immune checkpoint inhibitors (ICIs), monoclonal antibodies, and cancer vaccines.1
One of the many strategies cancers use to survive and progress involves evading detection by the immune system, which is achieved through a variety of mechanisms. It is well known that the TME can suppress immune activity by modifying the expression of immune checkpoint receptors and their ligands. The first ICI approved by the FDA for treatment of cancer was ipilimumab, an antibody targeting cytotoxic T lymphocyteassociated protein 4 (CTLA-4).2 Since then, ICIs targeting programmed cell death 1 (PD-1), and its ligand PD-L1, have also been approved, demonstrating promising efficacy against metastatic melanoma, urothelial carcinoma, non-small cell lung cancer, and Merkel cell carcinoma.3–4 Whilst efficacious in some patients, others either do not respond to ICIs or experience tumour regression but develop resistance to ICIs over time.
Chimeric antigen receptor (CAR)-T cell therapy represents another breakthrough in the field, with several already
approved by the FDA to treat a range of cancers.5 The generation of CAR-T cells involves the synthetic engineering of receptors, enabling them to recognise and target tumour cells expressing the corresponding antigen. Under normal T cell activation, the activated T cells release signaling molecules to attract other immune cells that transmit their own chemical signals, potentiating the response. As part of a negative feedback loop, regulatory T cells dampen the immune response to avoid damage to the surrounding cells. However, one major issue sometimes seen with CAR-T cell therapy is caused by strong T cell activation, induced due to the target antigen-receptor binding being independent from the major histocompatibility complex receptor, which can lead to cytokine release syndrome (CRS), otherwise known as a cytokine storm. The strength of this signaling overpowers the negative feedback, resulting in severe and potentially life-threatening toxicities.6 In addition, CAR-T cell therapy has limited anti-tumour activity against some types of tumours, particularly solid tumours, where it can show poor persistence and resistance can occur, for example, through antigen escape and poor trafficking and tumour infiltration. Consequently, despite its promising clinical value, the therapeutic efficacy and safety of CAR-T cell therapy is limited. To enable the development of new and more effective immunotherapy strategies using CAR-T cells as well as ICIs, a deeper understanding of the complex interplay between tumours and the immune system is required.
Characterising the Tumour-immune Landscape Through Biomarkers
Biomarkers can help to build a clearer picture of the interactions of tumours with the TME and immune system. As discussed, these can range from cytokines and chemokines to other immune- and tumour-related proteins such as growth factors, and immune checkpoint ligands and receptors, which affect the proteins that tumour cells express. However, this area of research is hindered by the need for precise, high-throughput assays that can measure multiple immune biomarkers simultaneously. A need for methods to investigate large numbers of proteins at the same time became apparent during the early research carried out by Dr. Steven Kornblau at the MD Anderson Cancer Center.7 Kornblau was characterising the abnormal expression and phosphorylation of retinol blastoma protein in leukemia and noticed a correlation with patient outcomes. After using reverse phase protein arrays to simultaneously evaluate multiple proteins, it became clear that the leukemic cells were interacting with their environment within the bone marrow. This led Kornblau and his scientists to investigate the chemokines and cytokines that the tumour cells were being exposed to, and the discovery that they themselves were prognostic. In search of ways to conduct these cytokine and chemokine studies, Kornblau’s lab determined that using ELISA kits would require significant amounts of samples and would not produce consistent results. Around the same time, large cytokine kits were being developed, which would enable numerous chemokines and cytokines to be analysed
Research / Innovation / Development
1. Example multiplex immunoassay workflow using bead-based xMAP technology. Colour-coded beads are incubated with the sample followed by incubations with detection antibodies and a reporter dye. Beads can then be classified by colour and the level of target present is measured through quantification of the associated reporter signal intensity.
simultaneously, significantly reducing the volume of sample required.
Studying the Immune Response Through Multiplex Immunoassays
In preclinical studies using small animal models, a typical 3-weekold mouse has a circulating blood volume of around 1.5–2.5 mL, with the volume of available plasma being less than half of this volume. This small volume limits the number of cytokines that can be studied per sample using traditional ELISA kits, thereby restricting the level of insights possible into the complexities of the tumour-immune landscape. Multiplex immunoassays offer a solution, enabling simultaneous measurement of multiple biomarkers in a single sample, overcoming issues relating to both sample volume and batch-to-batch variability (Figure 1).
Measuring multiple biomarkers using large cytokine panels allows scientists to obtain a comprehensive view of the tumourimmune landscape and cytokine network. This can provide critical insights to aid the prediction of patient responses to immunotherapy, monitor changes in the TME, and identify biomarkers associated with adverse immune events, to aid the development of more effective and safer immunotherapies.
I.Predicting Patient Response to Immunotherapy
A current challenge in the delivery of effective treatment for patients using immunotherapies such as ICIs and CAR-T cell therapy is the identification of patients who are most likely to respond, as some immunotherapies fail to induce a response in certain patients.8 Biomarkers identified through cytokine profiling research can support the identification and stratification of patients into subgroups based on their immune status, which can help scientists to predict therapeutic responses in translational and pre-clinical studies.
Findings from previous studies have already demonstrated the value of cytokine profiling in predicting therapeutic outcomes. For example, increased levels of the pro-inflammatory cytokines IL-6 and TNF-α have been linked by investigators to poor prognosis and increased immunosuppression in cancer,9–10
whilst elevated levels of IFN-γ and IL-12 have been linked by researchers to improved prognosis as these cytokines are associated with stronger immune responses, which indicates improved responses to ICIs.11 Not only can these insights be useful to scientists for predicting responses to immunotherapy, but they can also direct immunotherapeutic strategies to help improve efficacy.12
II. Monitoring the TME
As the TME hinders the efficacy of immunotherapy through immunosuppressive signaling, cytokine profiling provides insight into the mechanisms by which it promotes tumour cell growth and inhibits anti-tumour immune cell activity. By profiling the cytokines and chemokines present within the TME simultaneously through multiplex immunoassays, researchers can build a better picture of the immunosuppressive signaling within the TME. This includes chemokines that attract or repel specific cell types, which can be harnessed to develop novel immunotherapies. For example, IL-12 nanostimulant-engineered CAR T cell biohybrids have been developed to boost antitumour immunity of CAR T cells via immunofeedback. The release of IL-12 can promote the secretion of CCL5, CCL2 and CXCL10 to recruit and expand CD8+ CAR-T cells in tumours.13
III. Monitoring Immune-related Adverse Events
CRS greatly impacts the clinical safety and widespread adoption of CAR-T cell therapy. Other immunotherapies can also cause immune-related adverse events (irAEs), including autoimmunity and inflammation. Multiplex cytokine immunoassays can be used to assess complete cytokine profiles associated with CRS in research models to understand the correlation between irAEs and cytokines as these molecules work collectively to regulate immune responses. Similarly, cytokine dysregulation has been linked in literature to irAEs in patients receiving ICIs, particularly elevated IL-6, IFN-γ, CXCL2, and CCL17.14
Using the knowledge obtained through the identification of cytokine biomarkers, novel strategies are being developed to improve the safety of CAR-T cells. For example, the engineered T cells can be loaded with transgenic cytokine release cassettes.
Figure
Research / Innovation / Development
The encouraging outcome from this approach was demonstrated by Chmielewski and Abken using a CEA+ pancreatic tumour model system in immunocompetent mice.15 Through multiplex screening of pro-inflammatory cytokines, IL-18 and IL-21 were shown to favor acute pro-inflammatory responsiveness and resistance to immune exhaustion. Treatment of mice with CAR-T cells including an inducible IL-18 expression cassette strengthened the acute inflammatory response to increase the anti-tumour immune reaction. By augmenting the initial immune reaction, a stronger and more durable acute reaction was achieved, with a smaller systemic reaction. This outcome is likely to be protective against systemic side effects like CRS when applied clinically. In addition, this example demonstrates the potential of shaping the cytokine milieu to open avenues for the treatment of solid tumour lesions using CAR-T cells.
Conclusion
While immunotherapy holds great promise for the treatment of cancer, its potential is currently limited by the proportion of the patient population responding to treatment, associated irAEs, and resistance. To improve the efficacy and safety of immunotherapies, a deeper understanding of the complex interactions between tumours and the immune system is needed. Multiplex immunoassays allow the simultaneous measurement of large number of cytokines, chemokines, and other immune- and tumour-related proteins, overcoming issues relating to sample volumes and batch-to-batch variation, and enable more comprehensive analysis for preclinical and translational research, drug discovery, and monitoring of therapeutic interventions. These capabilities allow scientists to gain deeper insights into cancer immunology, to aid the development of novel immunotherapeutic strategies, which in turn improve therapeutic outcomes for patients.
REFERENCES
1. Waldman AD et al (2020). A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nature Reviews Immunolology, 20(11), 651–668. https://doi.org/10.1038/s41577-020-0306-5
2. Lipson EJ and Drake CG (2011). Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clinical Cancer Research, 17(22), 6958-6962, https://doi.org/10.1158/1078-0432.CCR-11-1595
3. Pardoll, DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer, 12(4), 252-264. https://doi. org/10.1038/nrc3239
4. Bagchi S, Yuan R and Engleman EG (2021). Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annual Review of Pathology, 6, 223–249. https://doi.org/10.1146/annurevpathol-042020-042741
5. Cappell KM and Kochenderfer JN (2023). Long-term outcomes following CAR T cell therapy: what we know so far. Nature Reviews. Clinical Oncology, 20(6), 359–371. https://doi.org/10.1038/ s41571-023-00754-1
6. Fajgenbaum DV and June CH (2020). Cytokine storm. The New England Journal of Medicine, 383(23), 2255–2273. https://doi. org/10.1056/NEJMra2026131
7. Kornblau SM et al (2010). Recurrent expression signatures of cytokines and chemokines are present and are independently prognostic in acute myelogenous leukemia and myelodysplasia. Blood, 116(20), 4251–4261. https://doi.org/10.1182/blood-201001-262071
8. Kim SK and Cho SW (2022). The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Frontiers in Pharmacology, 13, 868695. https://doi.org/10.3389/ fphar.2022.868695
9. Weber R et al (2021). IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cellular Immunology, 359, 104254. https://doi.org/10.1016/j.cellimm.2020.104254
10. Dixit A et al (2022). Targeting TNF-α-producing macrophages activates antitumor immunity in pancreatic cancer via IL-33 signaling. JCI Insight, 7(22), e153242. https://doi.org/10.1172/jci. insight.153242
11. Garris CS et al (2018). Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity, 49(6), 1148–1161.e7. https://doi. org/10.1016/j.immuni.2018.09.024
12. Liu J-Q et al (2022). Intratumoral delivery of IL-12 and IL-27 mRNA using lipid nanoparticles for cancer immunotherapy. Journal of controlled release, 345, 306–313. https://doi.org/10.1016/j. jconrel.2022.03.021
13. Luo Y et al (2022). IL-12 nanochaperone-engineered CAR T cell for robust tumor-immunotherapy. Biomaterials, 281, 121341. https:// doi.org/10.1016/j.biomaterials.2021.121341
14. Gonugunta AS et al (2021). Humoral and cellular correlates of a novel immune-related adverse event and its treatment. Journal for Immunotherapy of Cancer, 9(12), e003585. https://doi.org/10.1136/ jitc-2021-003585
15. Chmielewski M and Abken H (2017). CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell reports, 21(11), 3205–3219. https://doi.org/10.1016/j.celrep.2017.11.063
Vanitha Margan
Vanitha Margan, MBA, obtained her MBA from the University of Phoenix (USA) and a Bachelor of Applied Science in Biochemistry & Chemistry from Swinburne University of Technology (Australia). She has extensive experience in life sciences and clinical diagnostics fields, with her career spanning from clinical laboratory scientist, sales, marketing, and product management. She is a Global Product Manager at Bio-Rad Laboratories, leading the immunoassays product portfolio.
Overcoming Oncology Drug Resistance: Models
and Strategies
Cancer is inherently difficult to detect and treat. To add to this complication, some patients develop drug resistance to treatment, leading to tumour recurrence or metastasis. Much research is further needed to characterise and understand how tumours respond to drugs and therapy until the tumour is successfully eradicated completely.
To begin, it is important to have a better understanding of oncology drug resistance, how and why it occurs, and then how to avoid resistance. Oncology drug resistance is where the tumour initially responded to a treatment, but over time, the body develops mechanisms to bypass the drug’s effects. In some cases, the body did not respond to treatment from the outset. There are many mechanisms where resistance to cancer therapy is developed. Genetic mutations within the cancer cells can render treatment ineffective. Also, changes in the tumour microenvironment (TME) can have a role in drug resistance. Immune evasion is another situation where drug resistance can occur. In this case, there may be upregulation of immune checkpoints, downregulation of tumour antigens, or increased production of immunosuppressive cytokines. By understanding mechanisms used for resistance, better treatment and therapies can be developed for the patient.
Oncology Drug Resistance Models
Resistance is a multifaceted challenge that requires different approaches to find the right answer. As such, there's a variety of resistance models available that can provide different insights into the resistance mechanism of interest. On one hand, we have pre-existing models with intrinsic or acquired resistance. Within the acquired resistant models, pretreated models are a great tool to provide a more clinically relevant system. These are models (e.g., pretreated PDX models) that have been generated after the patient has received a certain treatment(s) and has relapsed. They can be used to investigate novel compounds or potential combination therapies to overcome the resistance being observed in the clinic. If a model of interest is not readily available, there are different approaches that can be used to generate resistance models. When investigating target resistance, engineering techniques such as CRISPR-Cas9 can be used to knock out the gene of interest and study the sensitivity of the compound compared to the wild-type version. On the other hand, if the interest lies in off-target resistance, it is possible to generate an in vitro or in vivo model by drug challenge, that is continuous dosing with the relevant SoC until the model acquires resistance. This approach can elucidate previously unknown mechanisms of resistance. Last, but not least, metastatic modeling is another category of resistant models, when the combination of in vivo models with imaging techniques can help track the progress of the disease in real time. Overall, a strategy needs to be developed to gain a full understanding of the potential resistance development and how to overcome it.
Patient-Derived Xenograft (PDX) Models
PDX models are where tumour tissue from a patient is implanted directly into immunocompromised mice. The models are created from patients who were treated with cancer therapy and either relapsed or developed treatment resistance. Tumour tissue from these patients is taken and implanted on a mouse. These models preserve the genetic and phenotypic characteristics of the human tumour, including heterogeneity, histology, and genetic mutations, making them an excellent platform for studying cancer progression and treatment resistance.
The major advantage of PDX models in drug resistance research is that it allows for the study of resistance mechanisms as they pertain to the patient's own tumour biology. When used in combination with serial treatments, PDX models can help identify changes in tumour genetics and phenotype that occur as the tumour adapts to therapy. By using drugs commonly administered in the clinic, such as chemotherapy agents or targeted therapies, researchers can monitor how tumours evolve in response to treatment.1,2
For example, PDX models have been used to study the resistance to commonly used chemotherapy drugs like cisplatin and doxorubicin, as well as, targeted therapies such as EGFR inhibitors in lung cancer and HER2-targeted therapies in breast cancer. Resistance can emerge due to genetic mutations in the drug target (e.g., mutations in EGFR or HER2), altered signaling pathways (e.g., activation of alternative growth pathways), or other mechanisms such as epithelial-mesenchymal transition (EMT).
PDX models offer a highly predictive platform for studying tumour biology and testing new therapies. Their ability to closely replicate human tumours makes them invaluable for preclinical drug development and personalised medicine approaches. Emerging trends in PDX models include humanised PDX models, organoid generation, single-cell sequencing, and multi-omics.13 As technology advances, PDX drug resistant models are becoming even more powerful, particularly with the integration of humanised systems and high-throughput capabilities.
Antibody-Drug Conjugate (ADC)
The use of monoclonal antibodies (mAbs) has changed cancer therapy due to its ability to target tumour surface antigens. However, treatment with mAbs is not as sufficient as conventional chemotherapy. Antibody-drug conjugate (ADC) is thought to bridge the specificity of mAbs and the efficiency of cytotoxic drugs.3 In this situation, mAbs target the tumour while carrying the cytotoxic drug to its target. ADC candidates are continuing to increase in clinical trials for various cancer treatments. However, similar to all cancer therapies, ADCs are not immune to resistance. Exploring resistance mechanisms to ADCs within pre-treated models is essential for understanding how tumours evade these advanced therapies and for developing strategies to overcome these challenges.
Research / Innovation / Development
Mechanisms of resistance to ADCs can be broadly classified into different categories: target-related resistance, drug-related resistance, linker-related resistance, and TME resistance. In tumour-related resistance, if the antigen epitope mutates, the ADC is unable to bind and is, therefore, ineffective. Tumours may also change the expression of the targeted antigen, resulting in an ineffective ADC. For drug-related resistance, the tumour develops resistance to the treatment drug. Linkerrelated resistance is where the linker is not cleaved or is cleaved prematurely, resulting in improper delivery of the drug to the tumour. Any changes in the TME can render the ADC unsuccessful.
PDX and GEMM models are significantly useful tools to understand ADC resistance because of their ability to mimic the most accurate in vivo conditions of the tumour environment in patients. For example, studies using PDX models have revealed that changes in antigen expression, such as HER2 downregulation, can lead to resistance to trastuzumab emtansine.2 Additionally, PDX models have been used to study how tumours acquire resistance to ADCs by upregulating ABC transporters or modifying the cellular machinery that processes the ADC payload.4
Model Selection
Numerous considerations are taken into account when selecting which PDX model to choose. First, the needs of the model need to be defined. Tumour type and molecular profiles are determined. The model should closely match the specific tumour being investigated. After determining the needs, search databases to see if the acquired/intrinsic model is available for use. If the model of interest has not been established, determine if the model can be created. If so, begin work to build and characterise the desired model. Next generation sequencing (NGS) can be used to confirm that the model closely resembles the genetic and transcriptome of the tumour. Since different models grow at different rates, select a model that has proven reliable and timely engraftment for the study timeline. The preclinical models then have deep learning applied to characterise and build strategies for drug development and treatment. Importantly for some studies, there are some PDX models that will express a desired biomarker relevant to the drug target. This biomarker expression helps understand drug response rates and therapeutic efficacy. PDX models that already have documentation, high-quality baseline data,
and are well characterised result in trustworthy data. In some situations, the PDX model may not be the appropriate model to use and, therefore, investigates the use of alternative models such as organoids, cell lines, etc.
Mouse Clinical Trials (MCT)
MCTs leverage PDX models to improve preclinical drug discovery by reflecting human clinical trials more closely. Unlike traditional approaches using few xenografts with large subject numbers, MCTs utilise many diverse PDX models with fewer subjects per model, enhancing clinical relevance.
MCTs simulate human clinical trials, with each PDX representing an individual "patient avatar" that collectively mirrors human patient diversity. This allows for predictive insights into responder and non-responder subgroups, guiding patient stratification and therapeutic strategies. Typically, MCTs involve randomisation, controls, and statistical rigor, making them a powerful tool for testing new therapies and combinations.4
Different MCT designs, such as indication-driven (targeting a specific cancer) or target-driven (targeting mutations across cancers), support specific study needs. For instance, an indication-driven MCT focuses on biomarker discovery within one cancer type, while target-driven MCTs validate therapeutic targets across cancers.
Designing effective MCTs requires expertise in model selection, statistical planning, and data interpretation. Crown Bioscience’s team offers comprehensive guidance for optimisng MCTs to improve drug development, ensuring translational insights and actionable data that enhance clinical trial success.
Summary
Cancer drug research is difficult. Each patient is unique, each cancer is unique, and the cancer itself can mutate, all factors complicating successful drug treatments and therapies. By utilising PDX or alternative models, many of these factors can be investigated before any drug reaches clinical trials, therefore, potentially identifying drugs that may not be effective from the beginning of treatment to final tumour eradication. Choosing a preclinical model is a balance between the tumour characteristics, the research goals, and practical considerations. By defining model needs, searching current established models,
Figure 1: 4 Step Approach for Resistance
Research / Innovation / Development
Figure 2: A Representative PDX Mouse Clinical Trial Design
or creating your model of interest, followed by profiling the model, successful studies can be achieved for understanding cancer resisting drug treatments. With each drug study and collection of data, knowledge in the field of cancer biology expands, expediting the progress to personalised, successful cancer treatments.
REFERENCES
1. Abdolahi, S., Ghazvinian, Z., Muhammadnejad, S., Saleh, M., Asadzadeh Aghdaei, H. and Baghaei, K. (2022). Patient-derived xenograft (PDX) models, applications and challenges in cancer research. Journal of Translational Medicine, 20(1). doi:https://doi. org/10.1186/s12967-022-03405-8.
2. Dobrolecki, L.E., Lewis, M.T. and et al. (2016). Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Reviews, [online] 35(4), pp.547–573. doi:https://doi.org/10.1007/s10555-016-9653-x.
3. Fu, Z., Li, S., Han, S., Shi, C. and Zhang, Y. (2022). Antibody drug conjugate: the ‘biological missile’ for targeted cancer therapy. Signal Transduction and Targeted Therapy, [online] 7(1). doi:https://doi. org/10.1038/s41392-022-00947-7.
4. Guo, S., Jiang, X., Mao, B. and Li, Q.-X. (2019). The design, analysis and application of mouse clinical trials in oncology drug development. BMC Cancer, 19(1). doi:https://doi.org/10.1186/s12885-019-5907-7.
5. Hidalgo, M., Villanueva, A. and et al. (2014). Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discovery, 4(9), pp.998–1013. doi:https://doi. org/10.1158/2159-8290.cd-14-0001.
6. Huang, L., Muthuswamy, S.K. and et, al. (2020). PDX-derived organoids model in vivo drug response and secrete biomarkers. JCI Insight, 5(21). doi:https://doi.org/10.1172/jci.insight.135544.
7. Liu, Y., Wu, W., Cai, C., Zhang, H., Shen, H. and Han, Y. (2023). Patientderived xenograft models in cancer therapy: technologies and applications. Signal Transduction and Targeted Therapy, [online] 8(1), pp.1–24. doi:https://doi.org/10.1038/s41392-023-01419-2.
8. Oser, M.G., MacPherson, D., Oliver, T.G., Sage, J. and Park, K.-S. (2024). Genetically-engineered mouse models of small cell lung cancer: the next generation. Oncogene, [online] 43, pp.457–469. doi:https://doi. org/10.1038/s41388-023-02929-7.
9. Rottenberg, S., Pajic, M. and Jos Jonkers (2009). Studying Drug Resistance Using Genetically Engineered Mouse Models for Breast Cancer. Methods in molecular biology, pp.33–45. doi:https://doi. org/10.1007/978-1-60761-416-6_3.
10. Samad Muhammadnejad, Seyed Mostafa Monzavi, Amir Arsalan Khorsand and Abdol Mohammad Kajbafzadeh (2021). PDX Clinical Trial Design in Anti-Cancer Research. Benthamdirect.com, [online] 10, pp.100–150. Available at: https://www.benthamdirect.com/ content/books/9789815039290.chapter-6 [Accessed 13 Nov. 2024].
11. Shi, J., Li, Y., Jia, R. and Fan, X. (2019). The fidelity of cancer cells in PDX models: Characteristics, mechanism and clinical significance. International Journal of Cancer, 146(8), pp.2078–2088. doi:https:// doi.org/10.1002/ijc.32662.
12. Yao, Y., Sun, S. and et al. (2022). Clinical utility of PDX cohorts to reveal biomarkers of intrinsic resistance and clonal architecture changes underlying acquired resistance to cetuximab in HNSCC. Signal Transduction and Targeted Therapy, [online] 7(1). doi:https:// doi.org/10.1038/s41392-022-00908-0.
13. Xu, X., Li, Q.-X. and et, al. (2023). A living biobank of matched pairs of patient-derived xenografts and organoids for cancer pharmacology. PLoS ONE, 18(1), pp.e0279821–e0279821. doi: https://doi. org/10.1371/journal.pone.0279821.
Bindi Doshi
Bindi Doshi, Product Marketing Manager at Crown Bioscience, has over 14 years of experience in the life sciences industry. With a PhD in Cell Biology from the University of Connecticut, where she focused on cell stress and movement, Bindi has greatly contributed to various research areas, particularly immunology and T cell detection using MHC Tetramers. Her deep scientific understanding and effective communication skills have enabled multiple successful collaborations, making her a crucial member of both technical support and marketing teams at Crown Bioscience.
TrueQuant Small RNA-Seq Kit
Key Benefits
• Ultra low sample Input: 10 pg total RNA – single cell
• Liquid biopsies / EVs from only 25 µl of plasma
• Fast, single tube protocol
• Gel free
• Diverse patented adapters for highest variety of small RNAs
• Sequencing on any Illumina® or Ion TorrentTM NGS instrument
Our TrueQuant library preparation methodology used in this kit enables accurate identification and quantification of miRNAs, piRNAs and other small RNAs.
Please contact us for more information.
GenXPro GmbH Altenhöferallee 3 60438 Frankfurt am Main Germany
Fon +49 69 95739710
Fax +49 69 95739706
info@genxpro.de www.genxpro.de
• Bioinformatics included
Technology
The Data Revolution in Drug Discovery: How Lab Automation and AI are Reshaping the Future of Medicine
In drug discovery, a technological shift is underway, promising to transform how we develop new medications and advance precision medicine. At the core of this transformation is the evolving nature of data – the foundation of scientific progress – being reshaped by the integration of laboratory automation and artificial intelligence (AI). This convergence of technologies is not only changing research methodologies but also redefining the fundamental processes of drug discovery, potentially leading to more efficient and successful development of treatments for a wide range of diseases.
Experimentation has always been central to scientific discovery, particularly in the pharmaceutical industry. The data generated from these experiments drive our understanding of diseases and potential treatments. High-quality, reliable data is crucial for forming accurate hypotheses and making informed drug efficacy and safety predictions. However, the traditional drug discovery landscape faces a significant challenge: the reproducibility crisis. Studies suggest that up to 85% of landmark scientific publications in the life sciences may be irreproducible, meaning independent scientists following the same protocols fail to reach the same conclusions. This crisis undermines establishing biological ground truths, impeding drug development efforts and contributing to high attrition rates in discovery and clinical trials.
The root of this crisis can be traced to limitations inherent in manual experimentation. Despite technological advancements in many areas, laboratory-based experimentation in drug discovery has largely remained a manual process. This reliance on manual methods introduces several critical issues that affect the drug discovery pipeline. Human error can lead to inconsistencies that skew results and potentially misdirect research efforts. The variability introduced by different researchers performing the same experiment differently further compounds this problem, leading to results that are difficult to replicate or build upon.
Moreover, the complexity of biological systems demands large-scale, systematic experiments that are challenging to conduct manually with the required precision and consistency. This scalability issue becomes particularly acute when dealing with the vast amount of reliable data needed to understand complex diseases and develop effective treatments. Data quality often suffers in manual processes, resulting in unstructured or inconsistent datasets that are difficult to analyse, compare, and interpret accurately. These limitations have significant consequences, not only slowing down the drug discovery process but also making it increasingly expensive and risky. As a result, the pharmaceutical industry faces substantial challenges, with drug development costs high and success rates remaining low.
In response to these challenges, innovative companies are advancing experimental processes through laboratory automation. While automation isn't new to drug discovery, its application has traditionally been limited to basic, repetitive tasks such as in high-throughput screening. The current wave of automation, however, is more comprehensive, sophisticated, and applied throughout the drug discovery journey. Advanced robotic systems can now automate a broad spectrum of processes across the entire drug discovery value chain from Target Identification and Validation, through Hit Identification, Hit-to-Lead, and Lead Optimisation. These systems aim to deliver precise, consistent, and scalable data, ultimately addressing the reproducibility issues that have long affected the industry.
The potential benefits of this approach are significant. Automated systems can perform experiments with high degrees of consistency, reducing variability between trials and enhancing reproducibility – a critical factor in establishing scientific validity. Automating processes requires digitisation of the protocol and, thereby, the elimination of ambiguity from its instructions down to the finest details, such as the amplitude, velocity, mix pattern, temperature, humidity, [CO2], and duration – all of those parameters that overlooked when a manual protocol simply states ‘mix’.
Whereas human scientists will easily make mistakes or become tired, the scalability of these robotic systems allows for continuous experimentation, potentially increasing throughput and accelerating the pace of discovery. Importantly, automation generates structured, systematic data that is well-suited for advanced analysis techniques, paving the way for a more sophisticated interpretation of results.
Parallel to these advancements in laboratory automation, artificial intelligence has emerged as a potentially transformative force in drug discovery. AI algorithms, particularly machine learning models, have shown promising capabilities in various aspects of the drug development process, from Target Identification to Lead Optimisation. However, the success of AI in drug discovery is intrinsically linked to the quality and quantity of data available for training and validation. This is where the potential synergy between laboratory automation and AI becomes evident.
The high-fidelity, structured data generated by automated platforms could provide the input AI models need to reach their full potential. This convergence is creating a relationship that may reshape the drug discovery landscape. Automated platforms can produce large amounts of consistent data to fuel AI models. In turn, AI algorithms can leverage these large datasets, assimilating patterns, trends and insights that might be challenging for human researchers to discern, enabling them to make predictions of new solutions that humans could not readily propose. This can lead to new hypotheses,
which can be tested using automated laboratory systems, creating an iterative cycle of experimentation, learning, and prediction that could accelerate the drug discovery process.
Several companies are working on integrating laboratory automation and artificial intelligence in drug discovery. Exscientia, a UK-based company, combines AI models with robotic lab automation in their drug discovery process. Their platform includes two main components: DesignStudio and AutomationStudio. DesignStudio uses AI to explore chemical space and generate drug candidates based on specified parameters. AutomationStudio handles the physical synthesis and testing of these candidates. This integration allows for a cycle where experimental results inform and refine subsequent AI-generated candidates. In 2020, Exscientia and Sumitomo Dainippon Pharma reported that a drug designed using this AI-driven approach had entered Phase I clinical trials for obsessive-compulsive disorder (OCD).
Recursion Pharmaceuticals uses a combination of AI and laboratory automation in their drug discovery efforts. The company focuses on phenomics, using AI to analyse high-dimensional assay data. Their approach involves conducting a large number of experiments weekly, including genome-wide knockouts, to explore gene functions and potential drug targets. Recursion has formed partnerships with pharmaceutical companies such as Bayer and Roche, focusing on areas like fibrosis and oncology. Recursion recently acquired Exscientia in a move that brings these two cuttingedge technology platforms together.
DeepCure employs an integrated platform that includes robotic automation software, robotic synthesis, and assay automation. Their platform uses deep reinforcement learning to design compounds, with a focus on generating molecules that are both potentially effective and synthetically accessible. This approach aims to streamline the design-build-test-learn cycle in drug discovery by closely linking computational design with experimental validation.
Insilico Medicine focuses on using AI across multiple stages of the drug discovery and development process. They have developed tools such as PandaOmics for target discovery and Chemistry42 for de novo small molecule design. In 2021, the company reported nominating a preclinical candidate for idiopathic pulmonary fibrosis using their AI platforms, a process they state took 18 months. While Insilico's primary focus is on AI rather than lab automation, their platforms are designed to work with automated experimental systems.
Arctoris operates as a tech-enabled Contract Research Organisation (CRO), combining their automation platform, Ulysses™, with scientific expertise to support drug discovery programmes. The Ulysses platform can perform a range of assays and experiments autonomously, from complex target validation to precision DMTA assays. Arctoris emphasises the generation of structured, machine-readable data that can be used directly in AI models. The company collaborates with various biotech and pharmaceutical corporations, often using their automated experimentation platform to conduct experiments designed by or for their partners' AI systems.
While these companies showcase the potential of integrating AI and lab automation, it's important to note that the field still faces significant challenges. AI models require large volumes of high-quality data for training, which is often lacking in drug discovery. Available data may be limited, noisy, or biased, which can affect the performance and reliability of AI models. There's also a lack of negative data (failed experiments), which is crucial for training robust machine learning models.
AI algorithms can inherit biases from the data they are trained on, potentially leading to skewed results that may not be generalisable. There's also a challenge in making AI models interpretable, which is critical for understanding the rationale behind predictions and gaining trust from researchers and regulatory bodies. The computational power required for training complex AI models, especially deep learning models, is substantial, which can be a barrier for smaller companies or those with limited resources.
While these approaches show promise, it's important to note that the integration of AI in drug discovery also faces significant challenges. AI models require large volumes of high-quality data for training, which is often lacking in drug discovery. Available data may be limited, noisy, or biased, which can affect the performance and reliability of AI models. There's also a lack of negative data (failed experiments), which is crucial for training robust and balanced machine learning models.
AI algorithms can inherit biases from the data they are trained on, potentially leading to skewed results that may not be generalisable. There's also a challenge in making AI models interpretable, which is critical for understanding the rationale behind predictions and gaining trust from researchers and regulatory bodies. The computational power required for training complex AI models, especially deep learning models, is substantial, which can be a barrier for smaller companies or those with limited resources.
While AI offers potential improvements, it must be effectively integrated with traditional experimental methods to validate predictions and ensure practical applicability. Combining AI with real-world experimental validation is necessary to overcome the limitations of purely computational approaches. Despite these challenges, many stakeholders believe that AI will drive significant improvements in the coming years, particularly in areas such as time and cost savings, increased probability of success, and the discovery of novel therapeutic agents.
Despite these challenges, many in the field believe that the combination of AI and laboratory automation will contribute to improvements in drug discovery, particularly in areas such as time and cost savings, increased probability of success, and the discovery of novel therapeutic agents.
As we look to the future, it's clear that the combination of laboratory automation and AI will likely play an increasingly important role in drug discovery. This technological alliance has the potential to accelerate discovery by speeding up experimentation and data analysis, allowing new drug candidates to be identified more quickly. The improved quality of data and more accurate predictions could reduce attrition
rates in clinical trials, potentially saving significant resources and development time.
Furthermore, the ability to process and analyse vast amounts of biological data could facilitate the development of personalised treatments, advancing precision medicine where therapies can be tailored to individual patients based on their unique genetic and molecular profiles. The increased efficiency and potentially higher success rates promised by this technological synergy could help lower the costs associated with drug development, potentially making new treatments more accessible to patients worldwide.
It's important to note that these technologies are not replacing human scientists but rather augmenting their capabilities. By automating routine tasks and providing powerful analytical and predictive tools, automation and AI can free researchers to focus on creative problem-solving and innovative thinking. This combination of human potential and advanced technologies creates an environment conducive to breakthrough discoveries. As we enter this new era in drug discovery, the potential for medical advancements is significant. The data revolution, driven by the synergy between laboratory automation and AI, represents an important moment in the history of medical science.
The future of drug discovery is likely to be increasingly data-driven, automated, and intelligent. As these technologies continue to evolve and integrate, we can anticipate a future
where drug discovery could become more efficient and potentially more successful. This technological alliance has the potential to impact many lives, bringing hope to patients worldwide who are waiting for new treatments and cures. As we embrace this data revolution, we move closer to a world where diseases might be treated more effectively, where personalised medicine becomes more common, and where human ingenuity is amplified by the precision of machines and the insights of artificial intelligence.
Tom Fleming
Tom Fleming is the CEO and Co-Founder of Arctoris, a CRO enabling next generation drug discovery with its fully automated laboratory platform, Ulysses™. He holds a Master's degree in Chemistry from the University of Southampton and was an Industrial Fellow at the University of Oxford and an SME Leadership Fellow at the Royal Academy of Engineering. Tom is recognised for his contributions to life sciences and is a Fellow of the Royal Societies of Chemistry, Biology, and Arts. He has received numerous accolades, including the ELRIG Impact Award and a spot in the Top 100 Most Influential People in Healthcare and Life Sciences. His work focuses on integrating robotics and data sciences into drug discovery to enhance efficiency and generate superior data.
Roald Dahl's Marvellous Children's Charity provides specialist nurses and support for seriously ill children.
Roald Dahl Nurses are a vital lifeline for children and their families, supporting them clinically and emotionally.
Roald Dahl's Marvellous Children's Charity
I don’t know where we’d be as a family without our Roald Dahl Nurse. She has been a beacon of light through such a difficult journey. Roald Dahl Nurses are invaluable!
There are thousands of other families who still don’t have access to this lifeline during the most difficult of times.
To donate or learn more about how your business could help, please contact partnerships@roalddahlcharity.org or scan the QR code.
Over 36,000
Over 150 children and young people supported Roald Dahl Nurses
Over 200,000 hours of care provided by Roald Dahl Nurses last year
Technology
CRDMOs Redefining Drug Substance Development with Platform Technologies
The development of biopharmaceuticals is expanding at an unprecedented rate, as the industry now comprises around 20 percent of the global pharmaceutical market.1 This momentum stems primarily from an escalating demand for advanced therapeutic treatments for both previously untreatable chronic illnesses and conditions resulting from an aging population.2 The appeal of biopharmaceuticals also reflects a shift toward precision medicine, as the unique molecular complexities of biotherapeutics tend to offer greater targeting capabilities and fewer side effects, aligning with growing consumer demand for better healthcare outcomes.1,3
This combination of factors means that biopharmaceuticals are opening doors to therapeutic approaches that were once out of reach with traditional small-molecule drugs. However, the impressive advances made in the sector often come with a range of critical challenges in process development. Even though each drug substance is unique, there is a common set of hurdles that each biomanufacturer faces during every process development journey. These can sometimes be related to the stability, solubility and purity of candidates, as well as bioprocessing hurdles, such as difficulty scaling up, low yields, the high cost of goods and even the need for complex characterisation methods. Taking just one of these factors as an example, scaling up production demands a high level of technical expertise and often imposes considerable constraints. These challenges can make the journey to the clinic a complex and resource-intensive process every time a new drug candidate is investigated, especially the complex ones. For instance, proteins are a highly diverse molecule class, defined by unique amino acid sequences, complex three-dimensional structures, specific post-translational modifications and, sometimes, specific cofactor requirements.4,5,6 This variability demands a highly customised approach to protein production, which often results in prolonged development timelines and elevated costs, and hinders the transition from discovery to larger-scale production.
Core Bioprocessing Challenges
As the biopharmaceutical sector grows, tackling the complexities of bioprocess development has become essential, and among the most pressing challenges for biomanufacturers are instability, low yields, and issues with purity and solubility.
Instability
Stability is a crucial factor in biopharmaceutical production, not least because instability of the final product frequently results in a shorter shelf life, limiting the timeframe within which biologics can be safely administered. Proteins can become biologically inactive or even provoke immune responses in patients when they lose their structural integrity, diminishing their therapeutic efficacy. 7 They are also susceptible to
aggregation, degradation, denaturation, or other structural alterations that can arise at various stages of the manufacturing process, from initial fermentation to final drug formulation. These issues can be mitigated using various approaches, including optimising cell lines, adjusting expression conditions, modifying amino acid sequences, and incorporating excipients or stabilising agents. However, this inevitably adds layers of complexity to the development process and ultimately increases manufacturing costs.
Low Yield
Maximising output from recombinant expression systems is vital for producing biotherapeutic proteins, but many proteins demonstrate low expression levels, reducing the yield in each production cycle. Low yield means that more resources, including raw materials, time, and labour, are required to produce adequate therapeutic quantities, raising production costs. This may also extend timelines, potentially delaying clinical readiness. Yield can be influenced by several factors, including the choice of host – such as bacterial, yeast, or mammalian cells – protein stability, folding efficiency, and post-translational modifications, so careful consideration needs to be given to each step in the process. Overall, enhancing yield can be a complex process that calls for sophisticated bioprocessing methods and expert handling.
Poor Purity and Solubility
Ensuring high purity and solubility is also essential to the safety and therapeutic effectiveness of biopharmaceuticals. Poor solubility can result from some proteins having a tendency to aggregate or precipitate, which can affect a drug’s bioavailability, restrict its therapeutic impact and complicate its formulation for clinical applications.8 Any drug substance must meet stringent purity standards to prevent contamination by common impurities, including host cell proteins, residual DNA, endotoxins, or other byproducts from the production process. Such contaminants can undermine the product’s safety and effectiveness, and may even trigger adverse immune responses in patients.
Addressing purity and solubility issues requires rigorous purification and filtration techniques, such as chromatography, depth filtration, and tangential flow filtration (TFF). However, these processes add considerable operational costs and often extend production timelines, posing additional challenges to large-scale biomanufacturing.
CRDMOs: Bridging Resource Gaps with Platform Technologies
Overcoming the complex challenges associated with bioprocessing requires considerable resources, and many biopharmaceutical companies find themselves constrained by limited time, facilities, and in-house expertise. This often makes it impractical to develop new cell lines or innovate manufacturing technologies internally and, as a result, companies are increasingly partnering with contract research, development,
and manufacturing organisations (CRDMOs) to bridge these gaps. CRDMOs provide deep expertise and advanced technological solutions across the entire biopharmaceutical production spectrum, offering support in areas from the selection of host cells to stringent quality assurance and control. CRDMOs are increasingly achieving this by establishing efficient, end-to-end platform technologies that quickly overcome the challenges faced during each stage of development, from host selection to analysis.
Advanced Technologies
Many CRDMOs incorporate state-of-the-art technologies like machine learning (ML) to precisely monitor and control numerous protein production stages. 9 For example, ML can be applied to refine and optimise gene sequences of recombinant proteins, thereby enhancing their expression and boosting overall yields. This approach addresses the inherent unpredictability in translating RNA to protein, resulting in more consistent production.
Host Selection and Screening
Selecting an appropriate host organism is the first pivotal step in biomanufacturing, as it directly impacts critical factors such as protein yield, solubility, and overall efficiency; the choice of host influences every subsequent stage of the process, including expression levels and scalability. Traditionally, biomanufacturers have favoured well-known hosts like mammalian cells and Escherichia coli. However, CRDMOs sometimes bring a wider variety of host options to the table, such as Gram-positive bacteria like Bacillus subtilis and yeasts like Pichia pastoris, which can be advantageous due to their rapid growth rates, simpler media requirements, protein secretion capabilities and genetic flexibility.
A skilled, platform-focussed CRDMO partner can use specialised strain and cell line engineering techniques to tailor the selected host for peak productivity. This includes offering an array of refined genetic components and expression vectors aimed at enhancing the solubility and expression of targeted proteins. Using ultra-high throughput systems also allows CRDMOs to swiftly identify high-performing ‘jackpot’ clones, greatly improving the efficiency of selecting ideal recombinant proteins for scaled-up manufacturing. Advanced capabilities in selecting and screening different hosts allow CRDMOs to help biomanufacturers overcome the challenges commonly experienced during bioprocess development, streamlining production with more efficient expression systems.
CRDMOs can refine the biomanufacturing process by optimising the strain or cell line. This iterative process enhances the target protein’s stability, proper folding and overall yield. For instance, codon optimisation can promote the production of molecular chaperones that support correct protein folding and stability, or prevent the expression of proteins – such as proteases – that could interfere with or degrade the therapeutic protein.
Process Development
CRDMOs also apply design of experiments (DOE) methodologies to precisely adjust key parameters in upstream processes, like fermentation, and downstream steps, such as purification.10 By meticulously adjusting these variables, they can create scalable,
consistent and efficient processes that can be smoothly transitioned to scale-up, ensuring that materials for clinical trials are manufactured with precision and efficiency.
Advanced Analytics and Quality Control
Collaborating with a CRDMO provides biomanufacturers with access to advanced analytical technologies that lead to high quality and consistent biologic products. One powerful tool in this arsenal is quadrupole time-of-flight liquid chromatographymass spectrometry (Q-TOF LC-MS). This technology separates, characterises and quantifies complex protein compounds, enabling precise analysis of their composition, structural integrity and biological activity. By evaluating critical quality attributes like purity, solubility and stability, Q-TOF LC-MS ensures that each batch meets stringent quality standards.
Case study of a Recombinant Subunit Vaccine11
Ingenza tackled the challenge of producing a recombinant pancoronavirus vaccine featuring eight distinct antigens displayed on a nanoparticle for cross reactive coronavirus immunity. Initially, the vaccine candidate required eight different mammalian cell lines for antigen production, while Escherichia coli was used for the nanoparticle, resulting in high production costs and endotoxin contamination. Ingenza used its knowledge of microbial host selection and process optimisation to move antigen production to Pichia pastoris, which grows in simple media, and nanoparticle production to Bacillus subtilis, which is naturally free from endotoxins. It also optimised upstream and downstream processes for the vaccine’s nine components and established a QTOF-MS characterisation method to show batch-to-batch reproducibility. This approach accelerated vaccine development, positioning it for IND submission by 2026, and supports global accessibility through cost-effective production.
Conclusion
Biopharmaceuticals are evolving rapidly, the path from discovery to clinic presents significant challenges – including issues with developing a scalable and robust bioprocess – that make the journey both resource-intensive and technically demanding. Partnering with a CRDMO that integrates platform technologies and ML into a seamless process enables biomanufacturers to address these challenges effectively at each stage from host selection to downstream processing. With capabilities across diverse host systems, innovative bioprocessing techniques and advanced analytical tools such as QTOF-MS, CRDMOs provide the specialised expertise and resources to optimise drug substance development. This collaborative model allows faster, more efficient and scalable production of biopharmaceuticals, better positioning the industry to meet rising healthcare demands and improve patient outcomes worldwide.
REFERENCES
1. Otto R, Santagostino A, Schrader U. Rapid growth in biopharma: Challenges and opportunities. In: From Science to Operations: Questions, Choices and Strategies for Success in Biopharma. McKinsey & Company.
2. Towards healthcare. Biopharmaceuticals Market Size to Hit USD 856.1 Billion by 2030. https://www.towardshealthcare.com/ insights/biopharmaceuticals-market-is-rising-rapidly
3. Within3. (2022). Why are biopharmaceuticals in high demand?
4. Sun PD, Foster CE, Boyington JC. Overview of protein structural and functional folds. Current Protocols in Protein Science. 2004;35(1).
5. Ramazi S, Zahiri J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database. 2021;2021.
6. Marchetti M, Puglisi R, Cellini B, Dindo M, Marchesani F. Editorial: The role of cofactors in protein stability and homeostasis: Focus on human metabolism. Frontiers in Molecular Biosciences. 2023;10.
7. Yasir M, Tripathi AS, Shukla P, Maurya RK. Immunogenicity of therapeutic proteins. Protein-based Therapeutics. 2023:251-273.
8. Stielow M, Witczyńska A, Kubryń N, Fijałkowski Ł, Nowaczyk J, Nowaczyk A. The bioavailability of drugs—the current state of knowledge. Molecules. 2023;28(24):8038.
9. Khuat TT, Bassett R, Otte E, Grevis-James A, Gabrys B. Applications of machine learning in antibody discovery, process development, manufacturing and formulation: Current trends, challenges, and opportunities. Computers & Chemical Engineering. 2024;182:108585.
10. Ingenza. Case study: Design-of-Experiment guided protein solubility optimisation. https://www.ingenza.com/design-of-experimentguided-protein-solubility-optimisation/
11. Ingenza. Nanoparticle vaccine technology – the key to pandemic preparedness. https://www.ingenza.com/blog/nanoparticlevaccine-technology-the-key-to-pandemic-preparedness/
Dr. Rita Cruz
Dr. Rita Cruz, Head of Molecular Biology at Ingenza, received her Ph.D. through the prestigious Marie Curie Industrial European Doctorate programme at the Centre for Bacterial Cell Biology at Newcastle University, in collaboration with DSM, a world leader in enzyme manufacturing. She joined Ingenza in 2016, where she leads strain development projects with both academic partners and high profile international corporations to deliver high quality research and steer programmes towards commercialisation. Successful applications include platform vaccine development, agrobiotechnology, and scalable production of enzymes for biocatalysis and home care products.
The Benefits of 5,000L Single-use Bioreactors for Biologics Manufacturing
Executive Summary
The entry of the 5,000L single-use bioreactor (SUB) into biologics manufacturing is providing an important bridge between small-scale and large-scale production. This new choice offers biotech and pharma companies, as well as CDMOs, more flexibility than ever before to respond to fluctuations in demand quickly and efficiently.
As the market for complex biologics continues to grow, effective capacity planning becomes more important – and more challenging.
• This report offers a guide for biopharmaceutical innovators on evaluating capacity needs and the advantages of using SUBs at each stage of development.
• Topics include:
• Features and benefits of 5,000L SUBs
• How improvements to turndown ratio, oxygen transfer rate, and more with SUBs provide a more efficient and flexible biologics development ecosystem and help accelerate scale-up and time to market
• Efficiency and sustainability advantages that SUBs offer over stainless-steel bioreactors, helping to accelerate time to market for biologics manufacturing and lowering carbon footprint
• What to look for in a CDMO partner for manufacturing with SUBs to ensure reliability, flexibility and collaboration
Introduction: Understanding the Importance of Capacity
The market for complex biologics including monoclonal antibodies, bispecific antibodies and fusion molecules is growing at a rapid pace. Recent estimates suggest the global market for monoclonal antibodies will grow 11% annually to reach nearly $500 billion in 2030.1 The global market for fusion proteins is expected to grow 4% a year to $33 billion in 2030, and the market for bispecific antibodies is projected to expand at a rapid 43% per year to $110 billion in that time period.2,3 This unprecedented growth is increasing the pressure on biopharma developers and their partners to improve efficiencies at every stage of the manufacturing process. Ensuring that the correct manufacturing capacity is in place for a new biologic is an essential part of that process.
Capacity planning, however, is challenging. Predicting future demand for a novel therapy that’s still in clinical development requires significant guesswork, as well as contingency planning in case projections are either too high or too low. Supply and
demand for marketed biologics can fluctuate, requiring that therapeutics manufacturers and CDMOs have the capacity to scale production up or down flexibly and efficiently.
The ability to quickly scale up capacity based on demand significantly impacts cost efficiency, potentially lowering costs per unit. What’s more, effective capacity planning can shorten the time to market for new biologic drugs. That gives developers a competitive advantage as they aim to speed patient access to new biologics.
Single-use bioreactors (SUBs) offer biologics manufacturers the flexibility to respond to rapid changes in demand for new therapeutics. Until recently, manufacturers typically started with 2,000L SUBs for biologics in clinical development, then jumped up to 10,000L or larger SUB or stainless steel bioreactors for commercialisation. But this limited their flexibility to respond to fluctuations in demand, and many traditional large-scale SUBs have suboptimal qualities, such as oxygen transfer rates that are not suitable for some cellular processes.
The recent introduction of 5,000L SUBs fills a significant gap in the industry. These new SUBs enhance scale-up and scale-down flexibility, as they can be used alone, at sub-5,000L volumes or in pairs at full capacity when more output is needed. Coupled with enhanced capabilities in singleuse technology, this new choice in capacity improves efficiencies and in turn helps accelerate biologics drug development and speed to market.
Thermo Fisher Scientific works with developers to project their capacity needs early in clinical development, and to put in place a flexible and scalable manufacturing process using SUBs. With its depth of biomanufacturing expertise and multiple sites around the world, Thermo Fisher can help ensure that realistic timelines are established and met, and that developers have a reliable supply at each stage of development.
Evaluating Capacity Needs and Single-use Bioreactors
To build the most efficient manufacturing ecosystem, manufacturers can partner with CDMOs to start capacity planning before a new therapeutic enters Phase I clinical trials. They can work together to forecast the quantity of drug needed at each stage of development based on the number of planned clinical trials, as well as the potential commercial requirements based on the target patient population.
Working with a CDMO allows biotech and pharma manufacturers to leverage the most advanced and up-to-date single-use platforms across a wide range of cell lines and processes. In situations where future demand may be hard to predict, the new generation of SUB technology offers the flexibility to scale up or down quickly. Thermo Fisher’s DynaDrive 5,000L SUBs, for example, have agitator shafts that extend the full length of the bag. This results in a turndown ratio of 20:1, offering more flexibility to adjust volumes than
Application Note
previous generations of SUBs, most of which have turndown ratios of 10:1.
Additionally, DynaDrive SUBs offer an optimal oxygen transfer rate (OTR) for advanced processes. An efficient drive train, coupled with multiple impellers, allows for lower RPM, and by extension less shear stress on cells. A combination of uniform mixing and a high oxygen transfer rate helps improve cell growth.
Other recent advances in SUBs are expanding their utility in biologics manufacturing. New features include improved container design optimised for advanced processes such as perfusion cell culture. Consumables are made with improved materials that are more pliable and durable than they were in the past, and that minimise leachables and extractables. And advanced sensor technology allows manufacturers to monitor
the environment inside each SUB, so they can ensure they are maintaining optimal conditions for cell growth.
Another valuable attribute of SUBs is that they help manufacturers advance their sustainability goals, while significantly reducing the cost associated with stainless steel bioreactors. Many biopharma developers are increasingly focused on reducing their carbon footprint, and as a result may be reluctant to deploy SUBs because of emissions concerns and the fact that the bioreactors generate waste from consumables such as bags, tubes, and filters. However, avoiding cleaning and sterilisation procedures that are necessary with stainless steel bioreactors significantly reduces emissions. Deploying SUBs also drives down the use of water and expensive chemicals, thereby lowering the amount of wastewater and chemical waste. As a result, SUBs reduce the overall size of the required manufacturing footprint. And biopharma companies can partner with third parties to recycle plastic consumables.
Benefits of 5,000L Single-use Bioreactors
By providing an alternative capacity option above 2,000L, the 5,000L SUB facilitates the logarithmic and efficient scale-up of a new biotherapeutic. Drug developers can work with CDMO partners to establish a roadmap for deploying SUBs that will be both flexible and cost efficient.
The ability to easily scale production up or down within a 5,000L SUB helps to increase efficiency and lower costs. For example, the DynaDrive 5,000L SUB can handle volumes starting as low as 250L, making it easy to deploy in early-stage clinical trials. As demand grows, volumes can be increased gradually. If demand surpasses the capability of the 5,000L, a second 5,000L SUB can be quickly deployed. Multiplexing with 5,000L SUBs is nimbler and more cost effective than scaling up with stainless steel bioreactors. In fact, the up-front investment required to deploy SUBs is significantly less than that of stainless steel.
SUBs reduce turnaround and cleaning costs, which can be substantially more per batch with stainless steel. The net result is lower operational costs across the board, including reduced spending on cleaning chemicals, water, energy and labour.
Another advantage of SUBs is that they reduce the risk of cross-contamination from other biologic products that are being manufactured in the same facility and from improper handling techniques. The DynaDrive 5,000L SUB’s high turndown ratio allows developers to consolidate several steps of the scale-up process in one bioreactor. The bioreactor also has semi-automated loading systems and fewer connections inside, lowering contamination risks by reducing the amount of manual labor needed.
Deploying 5,000L SUBs from clinical trials through early commercialisation eases the regulatory process for full commercial production, as well. That’s because the manufacturing process, equipment, and materials will have already been validated in clinical trials.
SUBs offer cost and efficiency benefits over stainless steel bioreactors. While the consumables costs are typically higher
than they are for stainless steel bioreactors, resulting in higher costs per run, savings come later in the process. SUBs eliminate turnaround and cleaning costs, which can be substantially more per batch with stainless steel. The net result is lower operational costs across the board, including reduced spending on cleaning chemicals, water, energy, and labour.
Choosing a CDMO with a Robust and Flexible Manufacturing Offering
When selecting a potential CDMO partner, there are several considerations to keep in mind. First, the CDMO should have extensive experience in the drug modality that’s being developed. That will ensure the partner has regulatory expertise and a track record of setting and meeting realistic timelines for customers.
Choosing a CDMO with multiple sites around the globe is also important, because it offers several advantages, including the opportunity to scale up quickly when demand is higher than expected. Thermo Fisher has four facilities in four countries that are equipped to manufacture complex biologics with SUBs. Thermo Fisher’s facility in Lengnau, Switzerland, was built to enable flex capacity, with multiple open manufacturing bays. This allows customers to quickly multiplex 5,000L SUBs – staggering harvesting and purifying steps – to efficiently fulfill growing demand. All facilities are equipped with similar, often identical, technology, making the tech-transfer process seamless.
Thermo Fisher also has expertise in developing predefined and qualified scale-down models for development with SUBs.
Additionally, Thermo Fisher offers end-to-end services, from cell-line development through fill-finish capabilities. This improves efficiency and minimises interruptions in the manufacturing timeline, with robust platforms that aim to provide reliable, on-time delivery from early development through commercialisation.
Conclusion
As the market for monoclonal antibodies, bispecific antibodies, fusion molecules, and other advanced biologics grows rapidly, so does the demand for manufacturing processes that are flexible, efficient, and environmentally sustainable.
SUBs with a capacity of 5,000L offer biopharmaceutical developers a cost-effective way to quickly scale up manufacturing in response to demand, from clinical development through commercial manufacturing. Thermo Fisher’s DynaDrive 5,000L SUBs have a turndown ratio of 20:1, a favourable oxygen transfer rate, and other features that improve the flexibility of scale-up and scale-down processes.
Thermo Fisher collaborates with drug developers to select, design, and implement an effective manufacturing strategy using SUBs. With its multiple, complementary sites around the world and end-to-end services, Thermo Fisher can help accelerate the path to market using flexible 5,000L SUBs.
Application Note
REFERENCES
1. Grand View Research. “Monoclonal Antibodies Market Size, Share & Trends Analysis Report By Source Type (Chimeric, Murine, Humanized, Human), By Production Type (In Vivo, In Vitro), By Application, By End-use, By Region, Segment Forecasts, 2023 –2030.”https://www.grandviewresearch.com/industryanalysis/ monoclonal-antibodies-market
2. MMR. “Fusion Protein Market: Global Industry Analysis by Market Share, Size, Competitive Landscape, Regional Outlook and Forecast (2024-2030).” https://www.maximizemarketresearch.com/marketreport/ fusion-protein-market/187945/
3. Grand View Research. “Bispecific Antibodies Market Size, Share & Trends Analysis Report By Indication (Cancer, Inflammatory & Autoimmune disorders), By Region (North America, Europe, Asia Pacific), And Segment Forecasts, 2023 – 2030.” https://www.grandviewresearch. com/industry-analysis/bispecificantibodies-market-report
Thermo Fisher Scientific Pharma Services
Thermo Fisher Scientific provides industry leading pharmaceutical services solutions for drug development, clinical trial logistics, and commercial manufacturing. With more than 60 locations worldwide, the company offers integrated, end-to-end capabilities across all phases of development. Pharmaceutical and biotech companies of all sizes gain instant access to a global network of facilities and technical experts. Thermo Fisher delivers integrated drug development and clinical services tailored to fit each drug development journey, ensuring high quality, reliability, and compliance as a leading pharmaceutical services provider.
Web: www.patheon.com
Manufacturing & Processing
Moving Closer to Affordable Advanced Therapies: A Review of Current Trends in Downstream Processing
Viral vectors are a crucial tool within the advanced therapy sector, providing an elegant solution for gene delivery in areas such as gene therapy and vaccine development. These viral particles, often uniquely engineered for a specific application, can effectively deliver genes into target cells, sometimes even enabling long term expression of the therapeutic gene of interest. However, producing clinical grade viral vectors comes with significant challenges and cost, partly due to comparatively low productivity of viral vector expression systems, in tandem with complex and onerous downstream processes.
In this article, we review viral vectors as a tool for advancing novel medicinal products and investigate the key steps and main challenges of viral vector purification. Finally, we will explore some of the emerging trends that hold the potential to transform access to advanced therapies.
Viral Vectors: The Essentials of Advanced Therapies In the decades since their discovery, many viral vectors have been developed for gene delivery in vitro, however, only a small subset of these have been established as safe for therapeutic use, including:
• Adeno-associated Viruses (AAVs): AAVs are non-pathogenic and can integrate into the host genome, offering long-term gene expression.
• Lentiviral Vectors (LVs): Derived from HIV, LVs can infect dividing and non-dividing cells, allowing stable gene integration and expression.
• Adenoviral Vectors (Ads): Derived from Adenoviruses, these vectors can carry large DNA inserts and are effective for transient expression of genes.
• Retroviral Vectors (RVs): These vectors integrate into the host genome, but primarily infect dividing cells.
There is significant variation between these vectors in terms of efficiency, stability of gene expression and versatility. The most appropriate vector, payload and dose is largely dictated by the target therapeutic application. For gene therapy applications, integrating non-pathogenic viruses will be favourable when delivering therapeutic genes to correct genetic disorders, such as cystic fibrosis or haemophilia.
On the other hand, when looking at cancer treatment, vectors with specific tropism and activation mechanisms will be preferred in order to induce apoptosis in malignant cells. Alternatively, vectors with high integration efficiency and long-term stable expression will be used to genetically modify cells ex-vivo, before re-administrating them to the patient to elicit an immune response.
However, use of viral vectors as a therapeutic agent, or as a raw material in the production of advanced therapies, also carries risk to the patient. This is mainly due to immunogenicity, where the viral vector itself triggers an immune response in the patient. Whilst in some cases an immune response may simply reduce the effectiveness of a therapy, in more serious cases it can lead to adverse responses and other safety concerns, including the risk of insertional mutagenesis or oncogenic potential.
Regulatory agencies, therefore, closely monitor the use of viral vectors in therapeutics, requiring extensive preclinical and clinical data to ensure safety and efficacy.
The Key Steps of Downstream Processing
Downstream processing for viral vectors typically begins with the bioreactor, where techniques used for harvesting vary depending on several factors, including the expression system and vector of choice. Where viral vectors are predominantly secreted by their expression system (i.e. the virus is mostly contained in the supernatant) it is preferable that the cells are not harvested and lysed, as this will increase the purification challenge further downstream. Conversely, where the vector is not secreted, a lysis step is usually required to extract the vectors from the expression system. Lysis may be carried out chemically, by addition of surfactants and detergents, but can also be done physically, using pressure or temperature – although the latter often proves more difficult to scale. Harvesting conditions must be selected carefully to achieve high product recovery, while maintaining particle stability and purity.
Upon harvesting of the vector, the feed stream must be clarified to remove cell debris and other larger contaminants. At small scale, this can be achieved using centrifugation or microfiltration, although clinical and commercial settings often utilise filtration with depth filters as the preferred method due to its high recovery and linear scalability. This technique uses inert filters with decreasing pore size, able to trap larger debris whilst allowing viral particles to pass through. Here, it is crucial to select a filter composition that ensures high product recovery, although certain compositions may also offer increased removal of process-related impurities such as host cell protein (HCP) and host cell DNA (hcDNA).
Following filtration, the final major step that remains is purification, often requiring several stages to achieve the final product. There are a wide array of purification methods currently in use, each with its own distinct benefits and drawbacks. Chromatography is highly effective for both capture and polishing of viral vectors, however, it is typically an onerous process step, due to raw material cost, and may require extensive development depending on the type of chromatography.
to an immobilised ligand within the column, allowing for targeted purification based on the surface properties of the virus. This is widely accepted as the ‘gold standard’ method for product capture, offering high recovery but generally relying on the availability of an antibody against the target product. Furthermore, traditional antibody-based affinity chromatography requires harsh elution conditions to effectively separate virus from ligand, which can have a detrimental impact on virus infectivity. This process may also result in leaching of the animal-derived ligand into the final product, a processrelated impurity that is highly scrutinised by regulators. Clearance of these impurities is a high priority during downstream processes, and must be demonstrated through rigorous quality control testing.
Added to this, affinity chromatography reagents currently on the market are expensive and offer much lower reusability compared to antibody-based reagents for other modalities. This method is also agnostic to product related impurities and will effectively co-purify non-target and non-intact viral vectors.
Ion Exchange Chromatography
Ion exchange chromatography separates viral vectors based on charge, which allows for additional purification and concentration. As the presence of a genetic payload modifies the overall charge of the viral particle, this method is widely utilised in large-scale production for separation of target from non-target viral vectors. While they are typically highly reusable and cost-effective, this method also often relies on harsh conditions to recover the virus from the column, which will have a detrimental impact on vector infectivity.
Alternative chromatography methods are available to purify the product based on other properties, (e.g., Size Exclusion Chromatography to remove smaller contaminants while concentrating the viral vector) but may be more challenging to develop or scale up.
To achieve the desired product profile, chromatography is usually used in tandem with filtration-based methods. Filtration is interesting operationally, as it is relatively simple to develop, scale and execute in a manufacturing environment relative to chromatography, and can often be less onerous, dependent on the choice of filter.
Dead-end Filtration
The most common, and perhaps simplest, type of filtration is dead-end filtration, where the complete feed flow is forced through the membrane, and filtered matter is accumulated on the surface of the membrane. Dead end filtration is most widely used to reduce bioburden or ensure sterility by passing the feed through filters with pore sizes between 0.22–0.45µm. Dead-end filters may also be functionalised using identical ligands to chromatography, particularly ion exchange ligands, which are smaller in size and easier to use. By functionalising a filter, filtrates can be retained based on other properties than size alone. This proves particularly useful when considering hcDNA, which can be easily removed from the feed material using a filter functionalised with ion exchange ligands.
Cross-flow Filtration
Cross-flow filtration, also known as tangential flow filtration
(TFF), gets its name because the majority of the feed flow travels tangentially across the membrane surfaces rather than through them. The main advantage here is that filtrate accumulation on the membrane surface is substantially reduced, increasing filter lifetime and capacity.
TFF can be deployed in ultrafiltration or diafiltration mode. In ultrafiltration, the viral vector is concentrated by using a membrane that will only allow particles below a certain size to filter through. This is predominantly used to bring the viral vector concentration up to the target dose concentration, but may also serve as an intermediate step to reduce sample volume and the overall downstream processing time. In diafiltration mode, often performed alongside ultrafiltration, the product is exchanged into a different buffer, removing salts and other small molecules.
Diafiltration is one of the final steps of a traditional downstream process, as it is the preferred method to exchange the product into its final formulation buffer. Certain stabilisers (e.g. sugars or surfactants) may decrease the diafiltration efficiency, and result in aggregation of product and contaminants, and are only added after completion of ultrafiltration and diafiltration.
Filtration is typically the last purification step applied to any viral vector product, as the material must be processed through a 0.22 µm sterilising filter to ensure that remaining contaminants such as bacteria are removed prior to long term storage. Once the virus is in its final storage condition, Quality Control on the final product may be performed, to ensure the vector meets purity standards.
By combining these different purification techniques, manufacturers can create a final viral vector product with a suitable product purity profile, however, there must be balance between purity, recovery and cost-effectiveness. Although increasing the number of purification steps may result in higher purity, each step comes with additional costs (e.g., raw materials, time, overhead), and usually reduces the total recovery of the viral vector product. Currently, recovery as low as 20% of the harvested amount is deemed acceptable, which raises questions as to the commercial viability of these products.
When low recoveries are looked at alongside particle infectivity, which is also reduced through standard purification techniques, the need for simpler, more cost-effective and gentler viral vector purification processes is evident.
New Technologies Revolutionising Downstream Processing
Many new technologies are emerging that promise to address some of the current challenges in viral vector purification. Synthetic affinity reagents, offering specificity and selective binding to viral vectors, have been demonstrated to offer gentle, high recovery purification. These ligands can be designed rationally, using molecular modelling tools that allow for targeting of specific surface epitopes, giving greater confidence that infectivity and tropism will not be affected through the bind and elute mechanism. This targeted approach also allows scientists to tailor for engineered viral vectors, which present a greater challenge due to their inherent uniqueness.
Manufacturing & Processing
Synthetic affinity ligands are synthesised from simple monomers, meaning they are chemically-defined, free of animal-derived components, and far more cost-effective than their antibody counterparts. The polymeric nature of these ligands also makes them highly resistant to extreme conditions (pH, temperature, pressure, shear), improving cleanability and reusability compared with traditional reagents.
Advanced solid supports for ligand immobilisation also offer an avenue for gentler, more cost-effective purification. Magnetic particles functionalised with affinity ligands can specifically and selectively bind viral particles, and then magnetically isolated and concentrated from the solution. When paired with gentle elution conditions, functionalised nanoparticles could be an excellent replacement for current column-based chromatography, alleviating the risk to viral integrity and infectivity posed by shear and pressure. The use of nanoparticles is also advantageous operationally, as material can be processed faster, and equilibrium conditions can be leveraged for higher binding capacity and recovery. Gold, silver and silica-based nanoparticles have also been used for small-scale purification, although they are typically not commercially viable at scale.
Alternative means of separating viruses from their specific ligands are also being investigated. Electrochemistry-based methods are a good example, where short electric impulses applied to the solid support can replace traditional methods of separation (pH, salt concentration). This offers several distinct advantages, including being gentler on the viral particle, and decreasing total buffer consumption, ultimately reducing downstream process costs.
With increasing reagent availability, high throughput testing methods are becoming increasingly important, particularly when looking at the increased throughput of upstream and analytical workflows. High-throughput testing methods in upstream process development is nothing new, and is essential for selecting optimal growth and transfection conditions to obtain the highest possible yield and purity. However, high-throughput methods for purification condition screening are less commonplace, meaning that downstream process development can only begin after the upstream conditions have been optimised and scaled. High-throughput downstream testing, for example microfiltration plates or miniature chromatography columns, are allowing us to bridge the throughput gap between upstream and analytics, and are increasingly deployed in industry for screening of large numbers of reagents and conditions. However, these methods typically require automated liquid handling and some machine learning to be performed at an appreciable pace, and the development of these automated screens may be resource intensive.
The Prospective Challenges in Downstream Processing
Despite a surge in the development of new and innovative technologies, some challenges remain unaddressed. While our understanding of the potentially deleterious effects of non-target viral vectors has increased greatly, our ability to produce and separate target vectors still has a long way to go. To date, much of the focus in this area has centred around increasing the yield of target vectors. Although this will undoubtedly be crucial to making advanced therapies widely available, development of more selective reagents
to effectively separate the target vector may hold the key to commercial viability.
This is particularly pertinent when considered alongside recent regulatory changes on the requirement for adventitious virus reduction/inactivation where possible. This requires two orthogonal methods, and is traditionally met by performing one inactivation step, consisting of a hold at low pH followed by a virus removal filtration using low nominal pore size filters, normally performed prior to the final formulation. Non-enveloped viruses such as AAVs may be amenable to such treatments given their higher pH tolerance and small size, however, enveloped viruses and those with lower pH tolerance are generally not amenable to either method. Although in these cases analytical methods are available for detection of adventitious virus contamination, their cell-based nature makes testing slow, and inadequate for in-process control. Consequently, there is significant demand for novel solutions to increase patient safety, de-risk manufacturing, and increase compliance.
Conclusions
Effective collaboration between regulatory bodies, technology developers, and therapeutics manufacturers will be essential in defining a new standard for production of viral vectors that maintains patient safety, without compromising commercial viability and accessibility to these cutting-edge advanced therapies.
While the challenges today remain great, the gene and cell therapy field must prepare itself for an increasing number of engineered and novel viral vectors currently in development, which hold great promise in increasing potency and improving tropism. Availability of specific and selective purification reagents alongside rapid, high-throughput screening techniques will be essential for development of high recovery downstream processes. Alongside increasing expression system productivity and developing a suite of robust analytical tools, overcoming these crucial hurdles will be essential to ensure viral vector-based therapies are widely available to the patients that need them.
Abdullah Sufan, Head of Cell and Gene Therapy Applications, joined Tozaro with over 6 years of experience in downstream purification of viral vectors and complex proteins. Abdullah has focused his career on the development and scale up of CGT processes for pre-clinical and clinical manufacturing. He is currently leading the Vector Production function at Tozaro, where he is responsible for production and purification of viral vector using novel synthetic affinity reagents in newly acquired laboratory space.
Abdullah Sufan
Innovation for the Few: Key Development to Commercialisation Considerations for Drug-Device Combination Products in Rare Diseases
Developing drug-device combination products for rare diseases presents unique challenges and opportunities for biopharmaceutical companies. These advanced drug delivery products which integrate a drug and a device into a single treatment, can be particularly beneficial for patients with rare diseases, offering novel solutions that may improve treatment efficacy, patient adherence, and quality of life. However, the development process for these products is complex, requiring careful consideration of regulatory requirements, technical feasibility, clinical efficacy, and patient-centred design.
Understanding the Unique Needs of Rare Disease Populations
Rare diseases, by definition, affect fewer than 200,000 people in the U.S, or less than one in 2,000 in Europe but globally, they impact some 300 million people.1 It is estimated there are more than 7,000 rare diseases. Yet, with a mere five to seven percent of these conditions having an FDA-approved drug, they remain largely untreated.
People with rare diseases face numerous challenges, including the pursuit of an accurate diagnosis, accessing suitable and sufficient medical care, and working toward better social inclusion and independence. Each condition has its own unique characteristics, progression, and symptoms, which often require tailored treatments. These variations necessitate a personalised approach in designing drug-device combination products.
Key considerations:
• Conduct thorough research on the rare disease to understand its pathology, symptoms, and treatment challenges.
• Engage with patient advocacy groups to gain insights into patient needs and preferences.
• Design drug-device combination products with flexibility to accommodate variations in disease presentation and severity.
Regulatory Pathways and Requirements
Navigating the regulatory landscape is arguably the most critical aspect of developing and commercialising drug-device combination products. Regulatory bodies such as the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have established specific guidelines to ensure the safety and efficacy of these complex products.
First, companies must determine the regulatory pathway applicable to their product. Understanding the regulatory classification of a drug-device combination product is essential and depends on its principal mode of action. This will dictate whether the product will be regulated as a medical device, a
drug, or a combination product. This determination influences the type of regulatory submission required and the specific requirements for demonstrating safety and efficacy.
In addition, rare diseases often qualify for expedited regulatory pathways, such as the EMA Accelerated Approval pathway or in the US Fast Track, Priority Review, Breakthrough Therapy or Accelerated Approval, which aim to speed up the approval process and provide incentives for developers.
Key considerations:
• Familiarise regulatory guidelines for combination products, which often differ from traditional drug or device approval.
• Understand the requirements for both drug and device components, including quality control, safety, and efficacy data.
• Leverage orphan drug incentives to offset costs and expedite the approval process, such as tax credits, fee waivers, and market exclusivity.
For those orphan drugs that are granted an expedited approval pathway, the challenge for the biopharmaceutical company is to leverage their own or their partnering CDMO’s drug development, manufacturing, drug-device design, final assembly and packaging knowledge. This expertise will help build a drug-device strategy that maintains the integrity, quality and timeliness of the manufacturing process, ensuring a complete regulatory submission package is generated to support filings. Aligning development, analytical, manufacturing, pack design and device packaging functions internally or via a CDMO providing integrated end-to-end advanced drug delivery solutions is vital to aid speed to patient and keep activities on the critical path for regulatory submission.
Formulation and Device Compatibility
The successful integration of a drug and a device requires meticulous design control to address the unique challenges posed by combination products. The compatibility of the drug product and the device, as well as the potential impact on the product's stability and performance, must be thoroughly assessed during the development phase.
For combination products, it is critical evaluate the chemical and physical compatibility of the drug with potential device materials and designs. Container closure studies are conducted during the development of drug products to compare the performance, stability, and compatibility of the product packaged in different container types.
Evaluating multiple container closure system configurations early in the development lifecycle, provides optimum flexibility and accelerates speed to market. For example, certain drugs may require specific types of delivery mechanisms, such as injectors, inhalers, or depending on viscosity – on-body
Manufacturing & Processing
injectors. If the device component is not properly designed to work with the drug product, it can lead to decreased efficacy, increased side effects, or device malfunctions.
Additional studies to conduct:
• Stability testing under various storage conditions (e.g., temperature, humidity) to evaluate the degradation kinetics of the drug product and ensure the drug product maintains its efficacy and safety when used in the device.
• Functional stability testing following ISO 11608-5 guidelines, testing cap removal force, activation force, extended needle length, dose accuracy, injection time, and lockout force to ensure the device can accurately and consistently deliver the required dose.
• Container Closure Integrity Testing (CCIT) is critical for maintaining the sterility and stability of the drug product. Both vials and prefilled syringes undergo testing to ensure that the closure system combination effectively prevents contamination and leakage. CCIT may be conducted in parallel with auto-injector functional stability testing.
Clinical Trial Design and Execution
The generation of robust clinical evidence is paramount for the successful commercialisation of drug-device combination products. Companies must design and conduct clinical trials that not only demonstrate the product's safety and efficacy but also provide meaningful real-world data on the interaction between the drug and the device.
The design of clinical studies should align with regulatory requirements and consider the unique challenges posed by drug-device combination products. Well-designed clinical trials should consider patient populations and clinical endpoints. Collecting data on both the drug and device components individually, as well as their combined effects, is essential for building a comprehensive body of evidence to support regulatory submissions.
Clinical trials for rare diseases however present unique challenges, such as small patient populations, heterogeneous disease presentations, and limited geographical distribution. For drug-device combination products, these challenges are compounded by the need to evaluate both the drug and the device in a single study.
Key clinical trial considerations:
• Design flexible and innovative trial protocols that accommodate small and diverse patient populations.
• Consider adaptive trial designs, which allow modifications based on interim results, to optimise limited patient numbers.
• Engage with rare disease networks and patient advocacy organisations to facilitate patient recruitment and retention.
• Real-World Evidence (RWE) can supplement clinical data, particularly post-market, to support ongoing effectiveness and safety. This is crucial for rare diseases, where patient numbers are limited.
Patient Centric Design and Usability
Human factors engineering (HFE) forms a critical aspect of drug-device combination product development, especially for
patients with rare diseases who often have specific needs and limitations, and may lack experience using medical devices. Therefore, creating a drug-device combination product that is easy to use and suited to their unique requirements is essential.
With a patient-centric focus, interaction between users and the product as-received is paramount. Understanding how patients and healthcare professionals interact with the packaging, IFU, and device itself is vital for optimising usability, minimising user errors, and enhancing overall safety, efficacy, and adherence to gain improved health outcomes.
Conducting usability studies and incorporating human factors considerations early in the design process can help identify potential issues and inform design modifications. Human factors and usability engineering is an integral component of regulatory submissions and is essential for demonstrating the product's usability and user comprehension.
Key design considerations:
• Involve patients and caregivers in the design process to ensure the device is user-friendly and addresses their needs.
• Implement human factors engineering (HFE) to enhance usability and adoption of self-administration devices.
• Design devices with intuitive controls, clear instructions, and support for various stages of disease progression.
One of the benefits of developing therapies for rare diseases is market exclusivity – seven years in the U.S or ten years in the EU. A key decision to be made regarding device strategy is whether there is a unique need for device innovation for specific patient populations or if traditional, readily available platforms would be suitable. Selecting established platforms that have received regulatory approval as part of a drug-device combination product previously, may be deemed lower risk for a rare disease therapy and may provide greater speed to market.
Manufacturing & Processing
For innovative platforms, as part of a company’s device strategy, securing intellectual property (IP) rights is crucial in the competitive landscape of drug-device combination products. Companies must carefully navigate the patent landscape to protect their device innovations and establish a strong IP position.
Advantages
Established platform
• Lower upfront costs
• Leverage of existing capital infrastructure
• Smoother regulatory path
• Robustness of device uses currently in the market
Proprietary platform
• Product differentiation –competitive advantage
• Custom design for specific applications
• Extend Intellectual Property (IP) life of the product
• Lower unit costs if scale is achieved
Key considerations
Disadvantages
• Limited product differentiation
• Higher unit costs
• Coemption of supply for popular devices
• Higher upfront time and costs, for example, design, IP, capital technology
• Complex regulatory path
• IP strategy should consider both the drug and device components individually and as a combination. This may require separate patents, as well as coordinated patent lifespans, to ensure competitive exclusivity.
• Determine if the high investment costs of device innovation are warranted for small patient populations and if the added cost can be recovered.
• Orphan drug exclusivity can be a major factor in recouping development costs and attracting investors.
Scalable Manufacturing and Securing Supply Chains
The small patient populations associated with rare diseases results in not only smaller clinical batch sizes but also commercial batch manufacturing which can pose challenges for relatively small campaigns and cost-efficiencies.
In sterile fill-finish often the use of disposable filling solutions provides complete manufacturing flexibility, maximising product yield, while minimising any potential risk
of cross-contamination at the manufacturing site. For the final assembly and packaging of drug-device combination products, device-agnostic scalable technologies with comprehensive tool sets can deliver optimised output, reducing the need to validate new processing lines, helping accelerate supply to patients and reduce costs.
Regardless of scale, ensuring the quality and consistency of drug-device combination products is paramount. Manufacturers, whether that be the biopharmaceutical company or their partnering CDMO, must adhere to good manufacturing practices (GMP) to guarantee the reproducibility and reliability of the product. This involves implementing rigorous quality control measures at every stage of production, from raw material sourcing, sterile filling of the drug product into the primary container to final drug-device combination product assembly, labelling and packaging.
Validating robust scalable manufacturing processes and controls is essential to produce combination products with consistent quality and performance throughout the drug-device combination product lifecycle, from clinical trials to commercial market supply. For rare diseases, scaling up should focus on flexibility without compromising quality, as increased patient reach may necessitate changes in manufacturing processes.
For the commercial success of rare disease drug-device combination products ensuring a robust and secure supply chain is vital. Companies should establish strong relationships with suppliers, partnering CDMOs, implement risk mitigation strategies, and have contingency plans in place to address potential disruptions. This is particularly important given the interconnected nature of the drug and device components, each with its own set of manufacturing and sourcing considerations.
Key considerations:
• Establish partnerships with specialised contract development and manufacturing organisations (CDMOs) experienced in SFF and final assembly, testing and packaging of combination products at various scales.
• Optimise manufacturing processes to reduce costs while maintaining high-quality standards.
Manufacturing & Processing
• Plan for scalability to meet demand as more patients are diagnosed and treatment adoption grows.
• Considerations such as dual sourcing for critical components and risk management for supply continuity are key.
Economic Considerations and Market Access
Rare disease therapies often come at high costs due to the limited patient base and the high cost of research and development. Small volume manufacturing, investment in device development, and final assembly and packaging in a drug-device combination format adds additional expense. Securing reimbursement from payers is essential for market success. However, payers may hesitate to cover high-cost products, especially for rare diseases with limited patient populations.
Due to a geographical disperse patient population best practice would be to consider multiple formats for regional presentations. Driven by cost and reimbursement, companies increasingly bring different injectable formats to different geographical territories. For example, some may commercialise vials with syringes for Eastern European markets but may look to more advanced prefilled syringes with safety devices or autoinjectors for the US or Western Europe.
Key considerations:
• Work with health economists to develop evidence of the product’s value and potential cost savings for healthcare systems by investing in a patient centric self-administered device.
• Gather real-world data to support reimbursement negotiations, demonstrating improved patient health outcomes and reduced hospitalisations.
• Develop drug-device combination product strategies with flexibility to accommodate geographical reimbursement variations.
Conclusion
Developing drug-device combination products for rare diseases requires a holistic approach that addresses regulatory, technical, patient-centred, and manufacturing aspects. From understanding patient needs to navigating complex regulatory pathways and designing a user-friendly product, each step is crucial to creating a solution that improves patient outcomes and quality of life.
While the development process is challenging, advances in technology, patient advocacy, and regulatory support provide an encouraging environment for innovation. By addressing these considerations, biopharmaceutical companies can create commercially viable, effective, safe, and accessible drug-device combination products that meet the needs of patients with rare diseases, ultimately improving lives.
Bill Welch is Executive Director of Market Development for PCI’s advanced drug delivery business segment, with a focus on injectable drug-device combination products. Bill has over 30 years contract development and manufacturing experience, with over 20 years in drug delivery devices and combination products. Prior to joining PCI, Bill served as Chief Technology Officer at Phillips-Medisize, leading a 900-person global innovation, development and new product introduction service segment. Bill holds a B.S in Industrial Engineering from the University of Minnesota, Duluth.
Logistics and Supply Chain
Stability Secured: Leveraging Technology and Expertise to Protect Clinical Trial Supply
Complex biologics and next-generation cell and gene therapies demand precision handling to maintain stability and promote optimised and patient centric clinical trials. As a result, biopharmaceutical organisations must adapt traditional supply chains to ensure that appropriate environmental conditions are maintained from production through to patient assignment. Get it wrong and the consequences can be catastrophic. Physical degradation, chemical instability and microbiological contamination can occur; compromising patient safety and commercial performance in one fell swoop.
While environmental factors, including light and humidity, play a key role in maintaining the stability of some investigational medicinal products, ‘the most important environmental parameter having significant potential to impact the quality of pharmaceutical products is temperature’.1
Maintaining strict temperature ranges for biopharmaceutical products through all handling, distribution, and storage steps in the supply chain isn’t a straightforward task. Multiple risk factors exist that can cause deviations and threaten drug stability. Considering the high cost of bringing new drugs to market, effectively addressing these risk factors isn’t just a matter of patient safety, but an opportunity to maximise return on investment potential.2
The trouble is when a fragmented approach to temperature management is employed, an approach that relies heavily on human intervention, error and inefficiency ensue. To secure a stable environment, a holistic, connected, and technologyenabled temperature surveillance and management strategy is required. One that is fuelled by accurate and complete data, and bolstered by expertise, to deliver end-to-end visibility and control, without creating additional workload for any one stakeholder group.
Understanding Temperature Excursions
When temperature-sensitive supplies leave the audited facilities of sponsors or CDMOs, and start the journey to sites and patients, the likelihood of temperature excursions increases, while detectability plummets. When drugs are in transit, it can be more difficult for sponsors to know if an excursion has occurred and whether it has been appropriately reported in line with defined processes. This reduced visibility and control extends to storage at clinical sites and, with the shift towards decentralised clinical trial models, within patients’ homes.
In an ideal world, if excursions occur, they should be reported in line with defined processes and appropriate action taken based on the outcome. The worst-case scenario with this is that drugs are rejected, potentially impacting the clinical site’s ability to dose the patient, resulting in perhaps losing the patient from the trial – increasing product resupply and recruitment costs, while delaying timelines. Indeed, temperature excursions are estimated to cost the pharmaceutical industry an average of $15 billion each year.3 The other side of the risk associated with excursions is that, if they go unreported, the compromised drug remains available and patient safety as well as corporate reputation, hangs in the balance.
Of course, keeping clinical supplies within specified ideal temperature ranges, as detailed on a product’s packaging label, is key to avoiding temperature excursions and securing stability. Yet sometimes, factors outside of a sponsor’s control, such as excessive time out of conditions while awaiting customs clearance in the destination country or a temperature excursion while the drug is stored at the clinical site, can make temperature excursions unavoidable.
When the Dots Don’t Connect
When these situations arise, fast identification, accurate adjudication, and clear communication are paramount. However, manual workflows can make it difficult for sponsors to detect and quickly and accurately adjudicate excursions. Manual
Logistics and Supply Chain
workflows can also limit a sponsor’s capacity to effectively and efficiently capture and centrally store all temperature data. This can pose serious problems when sponsors need to demonstrate that products, even those that haven’t been subject to a known temperature excursion, have maintained compliance with label claims throughout their lifecycle.
Let’s explore these challenges in more detail. When shipments arrive at sites, temperature monitors must be actioned correctly in line with pharmacy manual instructions. This typically requires site staff to plug monitor devices into a computer and forward a PDF by email to a designated contact. While it may sound straightforward, the process contains multiple points of failure. What if site staff are busy and forget? What if they send the email to an incorrect address? What if they send it to the correct email address but the inbox isn’t regularly reviewed and alarmed monitors go unnoticed for prolonged periods of time, and compromised supplies are incorrectly assigned to patients?
Similarly, without a streamlined computerised system to collate all required information for adjudication, sponsors have no choice but to painstakingly sift through multiple disjointed data sources to understand the impact. Even if alarmed monitor data is emailed the same day, sponsors must gather related data to support adjudication. This typically takes significant time, as information such as shipment numbers, kit numbers, monitor serial numbers and drug stability information must be pieced together from various, non-connected sources. The inevitable delay this causes can result in the need to reschedule patients, which in turn can delay key study milestones.
When data has finally been gathered to support adjudication, the next challenge with a manual process is effectively communicating outcomes to the relevant stakeholders. Relying on a person sending email notifications is problematic for the same reasons outlined above: there are too many points of failure. The person adjudicating the excursion, which very often is not their only study-related task, must remember who at the clinical site reported the excursion and send them an email notifying them of the adjudication outcome. A delay in sending this communication out can cause further delays down the line.
Audit readiness is another aspect that suffers under this manual approach, as there’s no complete and accessible audit trail to evidence adjudication decisions. Likewise, GDP regulations demand that clinical supplies maintain compliance with labelled storage claims during transportation. Evidencing this requires timely access to complete data sets for every drug shipment – not just those with alarmed monitors. This can represent vast quantities of information that can prove challenging and time-intensive for sponsors to stay on top of.
Leveraging Technology and Expertise
To truly secure the stability of biopharmaceutical supplies at each touchpoint in the supply chain, it’s necessary to leverage technology and expertise. Temperature management systems (TMS), designed and supported by temperature management experts, can transform the management of events that affect drug stability from fragmented and chaotic to streamlined and robust.
Next-generation TMSs achieve this by collating temperature data from any stakeholder, at every supply chain touchpoint – from transit to site storage – offering instant access to complete and trustworthy information. When these systems are designed and supported by experts, they serve to uphold the quality and integrity of products and support GDP and GCP compliance. By having an unbroken, cradle-to-grave temperature history of supplies, sponsors are empowered to make fast, informed decisions, achieve instant access to audit-ready data, and improve visibility of gaps with temperature data. This combines to deliver lower risk operations and improved patient safety and commercial performance. TMSs can also offer sponsors a flexible and failsafe mechanism for collating data to support the evolution of a drug’s stability profile as clinical trials progress.
Leveraging technology and expertise also enhances lifecycle tracking, which is essential for products that have a total allowable time outside of ideal conditions. Typically, it can be challenging for sponsors to calculate the effect of cumulative events that impact temperature-sensitive supplies – from in-transit and site-based excursions to time-out-of-conditions for production operations and freeze/thaw events. A connected, technology-enabled temperature management strategy empowers sponsors to obtain a drug’s complete temperature history instantly. With a single, centralised, and connected temperature management system in place, sponsors can easily ensure all time out of conditions is accurately recorded and automatically deducted and tracked at kit level for serialised material in a validated system. There are many advantages to this. Firstly, patient safety is improved by guaranteeing drugs with cumulative stability never exceed allowable limits. Full lifecycle reporting also facilitates kit-level visibility of a product’s remaining time out of condition and promotes audit readiness.
Visibility of missing data is another important benefit of using a TMS. If sites fail to upload monitor information, the monitor’s status will remain as ‘shipped’. In this event, the TMS will notify relevant stakeholders so they can easily pinpoint gaps in data and follow-up with sites to prompt compliance.
While this can drastically improve excursion management processes, auto-adjudication functionality contained within more advanced TMS applications can enhance the process of managing alarmed monitors that arrive at clinical sites. One of the key pitfalls of manual methods of temperature management is how time-consuming it can be for sponsors to adjudicate temperature excursions that occur in transit. Yet, when sponsors use a centralised TMS, with built-in process automation capability, adjudications of in-transit excursion events happen automatically. This provides site staff with an immediate, on-screen update on whether affected supplies are acceptable for use. Not only does this save time and ease site burden but, the likelihood of needing to reschedule patients is reduced. Armed with timely knowledge, site staff can be confident that drug stability has been upheld and dose patients quickly with an acceptable drug, even after an alarmed monitor has arrived at site. This boosts patient-centricity and operational performance. Furthermore, if auto-adjudication functionality rejects supplies that have been subject to excursion events, sponsors who partner with a dedicated
Logistics and Supply Chain
global team of temperature management experts benefit from specialist review of transit and site storage excursions within 24 hours. This belt and braces approach ensures high value, low yield clinical supply is only removed from the supply chain –and costly rework instructed – where necessary.
Harnessing automated adjudication functionality also removes the perceived need to build in time out of conditions when configuring temperature monitors. The premise of this common practice is to reduce site burden by lowering the likelihood of alarmed monitors arriving at sites. However, this can mask issues in the supply chain and mean sponsors are unable to get to the heart of the deviations they experience. This practice also doesn’t align well with GDP regulations that require drugs to remain within ideal storage per the defined limits on the outer packaging. When sponsors have access to auto-adjudication functionality, site burden is alleviated, regulatory compliance best practice is maintained, and sponsors are better able to identify issues in the supply chain that are causing deviations. This in turn can facilitate prevention of future excursion events.
Embracing Innovation, Lowering Risk
In summary, then, we know that if temperature excursions are not handled systematically, product quality can be adversely impacted and patient safety and commercial performance can suffer. We also know that fragmented processes for temperature management rely excessively on human intervention and can breed inefficiency and risk.4
While sponsors can face challenges maintaining product stability throughout the clinical supply chain, leveraging connected technology and expertise can support sponsors to obtain increased visibility and control. Use of advanced functionality, including lifecycle tracking and automated adjudication, can facilitate fast and informed decision-making and minimise the likelihood of needing to reschedule patients and conduct costly resupply activity.
Finally, prioritising complete and accurate data can support sponsors in managing drug stability more proactively and effectively, while facilitating audit readiness. Ultimately, by combining tried and tested technology and by working with experts, sponsors can better secure stability and ensure safe drug products reach the right patients, at the right time.
As Temperature Services Manager, Sarah McAliskey works closely with clients to understand the challenges they face with distributing temperature sensitive drug product and advising on the best solutions to implement for efficient management of temperature data on a global scale. Supply with Care is fundamental to Sarah's approach and evident in her unwavering commitment to supporting sponsors to deliver their temperature sensitive supply to patients compliantly and cost effectively. Sarah joined Almac in 2016 and has worked with a large number of Pharma and Biotech companies initially in developing proposals to fulfil packaging and distribution needs, aiding in the successful delivery of a range of clinical trials. After graduating from Queens University Belfast with a BEd in Business Studies, she gained a wide range of experience working in the clinical diagnostics industry, especially around solutions regarding shipping temperature sensitive product in challenging climates.
Subsection: Cell and Gene Therapy
Particulates in Cell and Gene Therapies
Over the past few months, I’ve had numerous conversations at work and conferences about particulates in cell and gene therapies. These discussions have highlighted to me two things. Firstly, the importance of identifying both inorganic and organic particulates in cell-based therapies. Secondly, many in the field may not be fully aware of the existing options for this identification, which has significant implications for the quality and safety of these advanced therapies.
Challenges in Cell-based Therapeutic Products
Building on these insights, it’s clear that manufacturing finished products for cell-based therapies presents unique challenges. These therapies often involve living cells, introducing complexities not found in traditional pharmaceuticals. The additional complexity arises from the presence of living cells and their surrounding secretome. For some therapeutics, this secretome is essential for their function and naturally contains organic particulates. Despite these complexities, ensuring the purity and integrity of these products is crucial, as any contaminants, including particulates, can significantly impact their efficacy and safety.
Another challenge caused by the presence of living cells and their surrounding secretome is that it is often not possible to test finished product for sub-visible particles. Therefore, it is essential that a baseline for sub-visible and visible particulate levels is established. To create a baseline, a comprehensive suite of testing of all materials and processes involved in the manufacture of finished product is typically performed. To ensure that the whole manufacturing process is tested, a mock run where the entire manufacturing process without growing cells allows the flushed fluid to be tested for particulates. At this stage, sub-visible, visible and even endotoxin testing may be performed. The data from this mock run helps in understanding the typical levels of particulates present, assessing the risk each component poses and implementing strategies to control this risk. It will also give the reassurance and confidence that baseline particulate levels are acceptable before the actual product is processed.
While the complexity of a cell-based therapeutic poses challenges when assessing particulates in the final product, in addition to establishing a baseline level generated by the manufacturing process, incoming raw materials should still be assessed for particulates. The materials used to produce the therapeutic will vary but often include common reagents such as DMSO, DPBS, and EDTA. These reagents should be assessed for particulates upon arrival and by screening the reagents before they are used in the manufacturing process means that additional particulates are not being introduce into the process.
When the likely particulate level produced during a manufacturing run has been determined, incoming raw materials analysed, and the risk is at a low and acceptable level. The finished product would still need to be assess for the presence of any visible foreign bodies. During this analysis, any extrinsic object in the samples should be investigated. Often these bodies are a fibrous material and could contain protein, silicone and non-silicone based plastics or a range of all three. Identifying these foreign bodies are crucial as they may be indicative of a fault or contamination occurring during the manufacturing process.
With foreign body analysis, the presence of living cells and their secretome, again adds complexity. Typically, analysis would involve running the sample through the particle counter and isolation of the foreign body would require passing the sample through various filters. In the case of cell-based samples, both the particle counter and filters with small pore sizes would clog. In addition, if the cell-based sample is run through a filter with a bigger pore size to avoid clogging, there would be an inherent risk that some particles are lost.
Once the foreign body has been isolated, work on identifying it can proceed. A plethora of analytical techniques can be implemented to determine the chemical or elemental composition of the extrinsic material. For elemental analysis, SEM and X-ray microanalysis are typically performed. For chemical composition, two complementary methods Fourier-transform infrared (FTIR) or RAMAN spectroscopy can be used. When single use plastic manufacturing consumables are used, identification
can be further enhanced by collecting and keeping a library of all components used in the manufacturing process. Once a foreign body has been isolated, both the foreign body and library database can then be spectroscopically analysed. If an exact match is generated, then this can help determine where the source of the particulate has come from.
Techniques and Approaches
After addressing the challenges, it is crucial to understand the advanced techniques and approaches used to identify particulates:
• Fourier-transform Infrared (FTIR) Spectroscopy: FTIR spectroscopy is a powerful analytical technique used to obtain the infrared spectrum of absorption or emission of a solid, liquid, or gas. It measures the intensity of infrared light absorbed by a sample at different wavelengths. This method is particularly effective for identifying inorganic particulates, as it can detect specific vibrational modes of chemical bonds. Each inorganic particulate has a unique spectral fingerprint, allowing for precise identification. By creating a library of all inorganic compounds found in the manufacturing process, the source of particulate contamination can be identified quickly and efficiently.
• Raman Spectroscopy: Complementary to FTIR, Raman spectroscopy is especially useful for materials rich in water, which can be challenging to analyse using FTIR. Raman spectroscopy provides detailed information about molecular vibrations, making it effective for identifying both organic and inorganic particulates.
• Scanning Electron Microscopy (SEM): SEM examines the surface morphology and ultrastructure. It provides high-resolution images and, when combined with energydispersive X-ray spectroscopy (EDS), determines the elemental composition of the particulates.
• Light Obscuration and Flow Imaging: These techniques count and size subvisible particulates in a sample. They are particularly useful for monitoring the presence of particulates in therapeutic proteins and other biopharmaceuticals.
• Confocal Microscopy: Confocal microscopy allows for high-resolution imaging of particulates and their distribution within a sample. It is often used in conjunction with fluorescent stains to differentiate between different types of particulates.
• HIAC Particle Counting: For therapeutic proteins, smaller volumes can be tested using HIAC particle counters to measure protein aggregation and particulate formation.
Practical Considerations
Implementing these techniques requires careful planning and execution. Here are some practical considerations to ensure effective particulate identification:
• Sample Collection and Preparation: Samples must be collected and prepared in a way that prevents additional contamination. This includes using clean containers and avoiding procedures that might introduce particulates.
• Method Validation and Standardisation: Ensuring that the methods used for particulate analysis are validated and standardised is crucial. This ensures consistency and reliability in the results.
• Comparing to Controls: Comparing particulate samples to controls is important to determine they are intrinsic to the product or extrinsic contaminants.
• Rapid Identification and Response: Once particulates are identified, it’s important to respond quickly to address the source of contamination. This might involve adjusting manufacturing processes or sourcing materials from different suppliers.
Future Directions
Looking ahead, the field of particulate identification in cell and gene therapies is continuously evolving. Advances in technology and methodologies are improving the accuracy and efficiency of particulate analysis. Some future directions include:
• Automation and High-throughput Analysis: Developing automation and high-throughput analysis techniques can speed up the process of particulate identification and characterisation, allowing for faster and more efficient quality control.
• Advanced Imaging Techniques: Emerging imaging techniques, such as super-resolution microscopy, provide even greater detail and accuracy in particulate analysis.
• Integration with Other Analytical Methods: Integrating particulate identification with other analytical methods, such as proteomics and genomics, offers a more comprehensive understanding of the sources and impacts of particulates in cell and gene therapies.
Conclusion
In summary, identifying particulates in cell and gene therapies is a critical aspect of ensuring the quality, safety, and efficacy of these advanced treatments. By combining advanced analytical techniques and practical approaches, manufacturers can effectively identify and address particulate contamination. This ultimately leads to delivering safer and more effective therapies to patients. Establishing a baseline through thorough testing of all materials and processes involved in production is essential. This proactive approach helps maintain the integrity of the final product and adhere to stringent regulatory standards.
Acknowledgements
I would like to thank my RSSL colleague who specialises in particulate analysis, Yohanes Tamene, for his valuable insights and contribution to this work.
Alistair Michel
Alistair Michel is an immunologist with a BSc (Hons) from the University of Edinburgh and an MSc from Imperial College London. He is a member of the British Society of Immunologists and the Royal Society of Biology. With over 20 years of experience as a bioanalytical scientist, he specialises in developing, validating, and optimising methods in GxP-compliant laboratories, focusing on ELISA and immunoassays. At Reading Scientific Services Ltd, Alistair serves as the technical lead for complex bioanalytical projects, delivering tailored solutions to address unique client challenges.
Exploring Cell & Gene Therapies Through
the Ages: How Past Cell and Gene Advancements are Shaping Tomorrow’s Medicines
Cell and gene therapies (C>s) are revolutionising medicine, offering the tantalising prospect of cures for diseases once considered incurable. This remarkable field has experienced rapid expansion, evolving from tentative experimental trials to groundbreaking U.S. Food and Drug Administration (FDA) approvals that have captured the world’s attention. But this journey hasn’t been without challenges. It’s a story of bold innovation and the unwavering determination to overcome significant hurdles.
In this article, Bill Vincent, a seasoned C> biotech founder and CEO, explores the key milestones that have shaped this dynamic field. He delves into the pivotal breakthroughs and setbacks fuelled by the relentless pursuit of scientific progress and the unyielding hope for a healthier tomorrow and examines the promising future that lies ahead.
Reimagining Modern Medicine with C>s
The C>s space is experiencing a surge of innovation and growth, poised to redefine the landscape of modern medicine. In 2023, C>s accounted for a remarkable 10% of all novel drug approvals by the FDA, up from 7% and 6% in 2022 and 2021, respectively.1 This trend underscores the accelerating pace of discovery and development in this field, with a robust approval pipeline promising even more transformative therapies in the years ahead.
Market projections are equally optimistic. The global C> market, valued at an impressive $18.13 billion in 2023, is expected to reach an estimated $97.33 billion by 2033.2 This represents a remarkable compound annual growth rate (CAGR) of 18.3% from 2024 to 2033, signaling a burgeoning industry brimming with potential.3
Groundbreaking advancements are driving this growth, offering the opportunity to transform the treatment landscape for previously intractable diseases. In recent years, we have witnessed the emergence of novel therapies offering newfound hope for patients battling conditions like spinal muscular atrophy (SMA), hemophilia and sickle cell disease.4–6
As researchers and clinicians continue to push the boundaries of scientific understanding, we can anticipate a future where these therapies play an increasingly pivotal role in transforming patient care and offering a brighter outlook to countless individuals worldwide.
Building on a Foundation of Innovation
The rapidly growing C> space is built upon decades of pioneering research and bold experimentation. Even before the full complexities of DNA were understood, scientists were exploring the therapeutic potential of cell-based interventions. Landmark events such as the first attempt to treat aplastic anemia
with bone marrow injections in 1939, laid the groundwork for the field’s evolution.7
In 1957, researchers attempted the first bone marrow stem cell transplant from a donor to a patient. Although there were complications due to immune system responses, this pioneering treatment highlighted the critical importance of ensuring compatibility between donor and recipient tissues. This spurred further research into the complex interactions between the immune systems of donors and recipients.8
However, progress in this field has not been without its challenges, particularly surrounding immunogenicity. The unfortunate 1999 death of Jesse Gelsinger, who experienced a severe immune reaction to the vector used in a gene therapy trial, served as a grave reminder of the potential risks associated with these treatments and emphasised the critical need for strict safety measures.9 Additionally, the unexpected development of leukemia in some patients taking part in several gene therapy trials in the early 2000s underscored the importance of refining gene delivery methods to minimise the risk of unintended genetic changes.10
These setbacks, while undeniably difficult to overcome, have driven crucial advancements in patient safety and the effectiveness of treatments.
Recent Advances Expanding the C> Space
Recent advancements have propelled the C> field into an era of unprecedented promise. Building upon the foundation laid by pioneering researchers, scientists and clinicians are leveraging cutting-edge technologies to address the limitations of traditional approaches and expand the therapeutic potential of C>s. From enhancing safety and efficacy to improving accessibility and affordability, these advancements are paving the way for a future where C>s are a cornerstone of modern medicine.
• Safer Gene Delivery
A pivotal step toward safer gene delivery was marked by the introduction of self-inactivating lentiviral vectors (LVVs) in 2010.11 These innovative vectors, designed to minimise the risk of unintended genetic changes, have significantly enhanced the safety profile of gene therapies.
The field’s unwavering commitment to progress is evident in the ongoing research focused on refining viral vector designs, optimising immune modulation strategies and developing even more precise gene editing tools. For example, researchers are utilising in silico approaches to predict novel capsid designs with properties that could lead to better targeting of specific tissues, improved transduction efficiency and reduced off-target effects.12,13
The development of refined separation methods is also enabling more efficient isolation of full capsids, minimising
Enable better vaccines with TrypsiNNex®
Introducing TrypsiNNex®, a trypsin product for bioprocessing built on our many years of experience in specialty enzymes manufacturing.
animal-free recombinant trypsin, it reduces costs by minimizing the risk of viral contamination, while improving process consistency through unmatched high trypsin purity for long-lasting performance.
In addition, TrypsiNNex® comprises a consistently high β-trypsin content of well above 70%, giving it the higher stability and consistency needed in advanced and highly regulated biopharma processing.
And once incorporated, its extensive documentation allows for easy qualification – with us here to help you enable better processes, too.
Learn more at novonordiskpharmatech.com
Application Note Cell and Therapy
the risk of adverse reactions associated with empty capsids. Concurrently, the engineering of specialised cell lines designed to package full capsids more effectively is further enhancing the quality, yield and potency of viral vectors.
• Enhancing Specificity
The pursuit of enhanced specificity in C> therapies is driving a wave of innovation across multiple fronts. Novel adeno-associated virus (AAV) serotypes and engineered capsids are being developed to improve targeting and efficiency, enabling more precise delivery of therapeutic payloads to specific tissues and cell types.14 The emergence of lipid nanoparticles (LNPs) as an alternative to viral vectors offers the potential for tissue-specific and targeted delivery while reducing the risk of adverse immune responses. For specialised applications like retinal gene therapy, alternative delivery methods are also being explored to overcome anatomical barriers and achieve targeted transduction.15
• Delivering ”Off-the-shelf” Potential
Allogeneic therapies, often referred to as “off-the-shelf” treatments, are set to transform the C> landscape. By utilising cells from healthy donors, these therapies offer the promise of broader patient access and reduced costs compared to personalised autologous approaches. The ability to manufacture and store allogeneic cell products in advance eliminates the need for lengthy and expensive patient-specific cell processing, making these life-saving treatments more readily available to a wider population.
However, allogeneic therapies present unique challenges, including the risk of graft-versus-host disease (GvHD) and
ensuring consistent cell quality across different batches. To address these hurdles, researchers are exploring innovative strategies such as developing universal donor cell lines engineered to circumvent GvHD, refining manufacturing processes to enhance scalability and implementing rigorous quality control measures to ensure product consistency.16–18
• Embracing the Power of Gene Editing
Over the past decade, the C> field has experienced a remarkable acceleration in innovation, with groundbreaking discoveries and technological advancements swiftly leading to approved therapies. A pivotal moment in this journey was the development of CRISPR/Cas9 in 2012, a revolutionary gene editing technology that has redefined genetic manipulation.19
With its ability to precisely target and modify genetic material, CRISPR/Cas9 holds immense promise for correcting disease-causing mutations and enhancing therapeutic outcomes. The transformative potential of CRISPR is exemplified by its rapid translation from the lab to the clinic. In just 11 years, the first FDA-approved therapy utilising CRISPR/Cas9 (marketed as CTX001™ and now Lovo-cel™), was approved for treating sickle cell disease.5 This remarkable achievement underscores the game-changing impact of CRISPR and its potential to reshape the future of medicine.
As the C> space rapidly evolves on the back of technical and scientific advancements, a collaborative approach will be paramount to realising the full potential of these therapies. Open dialogue and knowledge sharing between researchers, clinicians, manufacturers and regulators are essential to
navigate the complexities and address the formidable challenges in their production, including scalability, variability and regulatory compliance. By fostering collaboration and transparency, stakeholders across the C> ecosystem can leverage advancements, streamline development processes and ensure the safe and effective delivery of these transformative therapies to patients worldwide.
Ongoing Evolution and Unforeseen Possibilities
The current C> landscape is nothing short of remarkable, with over 30 gene therapies and more than 65 non-genetically modified cell therapies already approved globally.20 The field shows no signs of slowing down; new technologies and therapies are emerging at a breathtaking pace. Just this year, a novel gene editing technique derived from bacterial “jumping genes” has captured the imagination of researchers, offering the potential to overcome some of the limitations of CRISPR.21
The future of medicine is being rewritten with the advent of increasingly powerful C>s, but the road to widespread adoption will be paved with challenges. To navigate complexities surrounding scalability, variability and regulatory compliance, the C> industry must embrace collaboration, sharing insights and knowledge to help accelerate and broaden access to life-changing C>s.
7. George W. Santos, History of bone marrow transplantation, Clinics in Haematology, Volume 12, Issue 3, 1983, Pages 611-639, ISSN 0308-2261. https://doi.org/10.1016/S0308-2261(83)80003-4.
9. Sibbald B. Death but one unintended consequence of gene-therapy trial. CMAJ. 2001 May 29;164(11):1612. PMID: 11402803; PMCID: PMC81135.
10. Maetzig T, Galla M, Baum C, Schambach A. Gammaretroviral vectors: biology, technology and application. Viruses. 2011 Jun;3(6):677713. doi: 10.3390/v3060677. Epub 2011 Jun 3. PMID: 21994751; PMCID: PMC3185771.
11. Cavazzana-Calvo M, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010 Sep 16;467(7313):318-22. doi: 10.1038/nature09328. PMID: 20844535; PMCID: PMC3355472.
12. Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019 May;18(5):358378. doi: 10.1038/s41573-019-0012-9. PMID: 30710128; PMCID: PMC6927556.
13. Zinn E, Pacouret S, Khaychuk V, Turunen HT, Carvalho LS, Andres-Mateos E, Shah S, Shelke R, Maurer AC, Plovie E, Xiao R, Vandenberghe LH. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015 Aug 11;12(6):1056-68. doi: 10.1016/j.celrep.2015.07.019. Epub 2015 Jul 30. PMID: 26235624; PMCID: PMC4536165.
14. Issa SS, Shaimardanova AA, Solovyeva VV, Rizvanov AA. Various AAV Serotypes and Their Applications in Gene Therapy: An Overview. Cells. 2023 Mar 1;12(5):785. doi: 10.3390/cells12050785. PMID: 36899921; PMCID: PMC10000783.
Cell and Gene Therapy
15. Peters CW, Maguire CA, Hanlon KS. Delivering AAV to the Central Nervous and Sensory Systems. Trends Pharmacol Sci. 2021 Jun;42(6):461-474. doi: 10.1016/j.tips.2021.03.004. Epub 2021 Apr 13. PMID: 33863599; PMCID: PMC9302199.
16. Lv, Z., Luo, F., & Chu, Y. (2023). Strategies for overcoming bottlenecks in allogeneic CAR-T cell therapy. Frontiers in Immunology. https:// doi.org/10.3389/fimmu.2023.1199145
17. Lonez C, Breman E. Allogeneic CAR-T Therapy Technologies: Has the Promise Been Met? Cells. 2024 Jan 12;13(2):146. doi: 10.3390/ cells13020146. PMID: 38247837; PMCID: PMC10814647.
18. Jo, S., Das, S., Williams, A. et al. Endowing universal CAR T-cell with immune-evasive properties using TALEN-gene editing. Nat Commun 13, 3453 (2022).
Bill Vincent has over 35 years of experience working in the pharma and biotech sector. He is the board chair of Genezen, a biotech contract manufacturer of viral vectors, cell manufacturing, and testing services. He founded the company fourteen years ago and grew it from his one-person operation to its current 60 employees with an outside investment of $45 million. Bill has since become a consultant for early-stage biotech and medical device companies coming into the market from an academic background.
Cell and Gene Therapy
Delivering the Future: Overcoming Shipping Challenges in Cell and Gene Therapy
The rise of cell and gene therapy (CGT) is revolutionising how we treat complex diseases, offering life-changing treatments for patients in need. However, with these innovations come significant logistical challenges.
Ensuring these highly specialised therapies reach patients on time, without damage, is crucial. As the CGT market expands, logistics must keep pace to protect the integrity of these groundbreaking treatments.
High-Stakes Medicine Meets Outdated Logistics
The CGT industry is growing rapidly, but the logistics supporting it have struggled to keep pace. CGT treatments are personalised, highly sensitive, and time-critical, making their durability and availability incredibly limited. Simply put, these treatments are some of the most expensive and delicate shipments moving around the world right now – and that means the standard supply chain playbook no longer applies.
Reaching upwards of $4 million per dose, CGT offers life-altering potential at an incredibly high cost. Once manufactured, these therapies must be administered to patients within hours, and they often require ultra-low temperatures during transport. Should any of these treatments be lost, damaged, or delayed in transit, the manufacturing process would
have to start over. For patients waiting to receive these therapies, that’s time that they may not survive.
It’s no wonder the logistics behind this process are daunting. Not only do logistics teams face the possibility of losing millions of dollars in product, but they are responsible for delivering potentially life-saving treatments. The specific needs of these fragile therapies create a new set of issues that many companies don’t fully understand, and therefore, aren’t ready to support.
The CGT Cold Chain Challenge
One of the key issues in CGT shipping is outdated cold chain infrastructure. Traditional shipment tracking methods, such as barcode scanning, provide only a snapshot of the shipment’s location, leaving teams in the dark about the cargo's real-time condition. Considering how incredibly time-sensitive CGT shipments are, this lack of visibility can be catastrophic.
Additionally, the extreme temperature requirements of CGT products – often needing cryogenic conditions of -150°C or lower – further complicate shipments. If temperature control fails at any point, the entire shipment could be lost. Adding to the strain are stringent regulatory demands. CGT is still a relatively new area of medicine, and additional transport requirements like packaging, surcharges, and restrictions are constantly evolving. This web of intricate cold chain conditions,
Cell and Gene Therapy
regulatory compliance, and constant monitoring needs creates a perfect storm of risk.
The stakes are incredibly high. A single misstep could delay or prevent a patient from receiving treatment, underscoring the dire need for improving the CGT logistics market. As the industry expands, these challenges will only intensify unless there is a significant transformation in how CGT therapies are transported.
Innovative Solutions Transforming CGT Shipments
Fortunately, the logistics industry has begun to develop solutions tailored to the unique needs of CGT. New technologies and approaches are transforming how companies ship these sensitive therapies, providing the precision and reliability that the market demands.
The development of advanced cold chain technology is a critical innovation given the ultra-low temperatures CGT products require. To help maintain treatment integrity, many companies are now leveraging advanced cryogenic packaging that can maintain consistent temperatures over extended periods. When combined with real-time temperature monitoring, these containers can ensure a shipment's condition from origin to destination.
To that end, real-time visibility into shipment location and condition has become essential. GPS-enabled tracking devices offer a live feed of the shipment’s status, including temperature, humidity, and even shock events that might compromise the therapy. These devices enable real-time data collection and monitoring, so teams can not only react quickly if something goes wrong but pull from collected data to demonstrate compliance if need be. This visibility not only reduces risk for companies but also provides peace of mind to healthcare providers and patients.
Coupled with real-time visibility, predictive analytics can help not only prevent issues but improve overall operational efficiency. By analysing historical shipping data, weather patterns, and traffic conditions, companies can predict potential delays and proactively reroute shipments. On top of minimising disruptions, these insights can help guide future shipments, helping companies continually optimise their supply chains.
Of course, the importance of close collaboration with experienced logistics service providers (LSPs) cannot be overstated. Specialised LSPs that understand the complexities of CGT regulatory compliance, packaging, and temperature needs, are invaluable in navigating the intricacies of CGT logistics. Their expertise helps reduce the margin for error, ensuring that the product arrives intact and ready for patient administration.
Modernising CGT Supply Chains for Lifesaving Impact
While the challenges of CGT logistics are significant, they are not insurmountable. By embracing innovative cold chain technology, real-time monitoring, and predictive analytics, companies can navigate the complexity of CGT supply chains. These advancements are more than just technical upgrades –they’re lifesaving improvements.
The future of cell and gene therapy depends not only on scientific innovation but also on the logistics systems that bring these treatments to life. By adopting an integrated, real-time solutions, approach, companies can protect the integrity of their therapies and ensure they reach the patients who need them most.
Alex Guillen
Alex Guillen is an established executive with a proven record in global business and market development, with well-rounded experience in multicultural sales management and brand building. Guillen has extensive experience and expertise in cold chain; as Global SME, Life Science and Pharma at Tive, Guillen leads sales and business development within the company's rapid-growth Life Science division. Previously, Guillen served as a Board Member and leader of Corporate Strategy at SWITRACE S.A, a developer of temperature and humidity data loggers compliant to the Pharma and Biotech industries. Guillen’s extensive experience also includes serving as Global Cold Chain Director of Fisher Clinical Services, CEO of Escort Cold Chain Solutions SA and Director for Commercial Operations for Novartis Vaccines.
Cell and Gene Therapy
Overcoming Portal Fatigue in Cell and Gene Therapies: Optimising Orchestration
Frustration, exhaustion, and a sense of being overwhelmed – these are the hallmarks of “portal fatigue”, a growing problem for approved treatment center (ATC) staff in the cell and gene therapy (CGT) field. Juggling a multitude of digital platforms and systems, each with its own unique interface and login credentials is taking a toll on productivity, morale, and potentially, patient care.
This fatigue is particularly acute in CGT due to the intricate nature of the field, characterised by complex supply chain relationships between treatment providers, ATC staff, manufacturers, and other stakeholders. While technology platforms have emerged to streamline and simplify these processes, their rise has inadvertently introduced new challenges, such as communication barriers and process inconsistencies. These issues can lead to confusion, delays, and errors, further contributing to the burden of portal fatigue.
In this article, Dr Akshay Peer, Senior Vice President Product, TrakCel will explore the causes and consequences of portal fatigue in CGT, discuss the need for standardisation and collaboration, and offer solutions and recommendations for mitigating this growing challenge.
The Rise of Technology Platforms in CGT
The complexity of supply chains associated with personalised CGTs, combined with requirements such as ensuring the correct treatment reaches each patient, creates a challenging order-totreatment process. This complexity grows as therapies advance from clinical development to commercialisation, and the number of patients and other stakeholders in the supply chain increases. Manual approaches to supply chain co-ordination and management are difficult, if not impossible at scale. This has led to the development of supply chain orchestration software. Orchestration systems streamline and simplify the CGT process, offering benefits such as:
• Chain of custody and chain of identity: Ensuring the secure tracking of therapies throughout the supply chain.
• Improved efficiency: Coordinating and scheduling activities between stakeholders.
• Automated processes: Streamlining payer approval verification and order-to-cash processes.
• Real-time visibility: Providing updates on the progress of individual therapies.
Almost all commercial personalised CGT therapies are supported by their own custom orchestration platform. However, as an increasing number of CGTs are commercialised, the abundance of platforms has created new challenges, particularly for ATC staff who must navigate multiple systems, each with its own unique interface, login credentials, and processes.
The Pitfalls of Custom-built Solutions
Developing a custom-built orchestration platform can seem like the ideal solution, promising a perfect fit for the unique needs of cell and gene therapy. However, the allure of customisation often masks significant pitfalls. This approach frequently leads to unforeseen challenges such as spiralling development costs, integration complexities, and the burden of ongoing maintenance, which can strain resources and hinder the efficient delivery of therapies to patients.
One of the most significant pitfalls is the contribution to portal fatigue. Healthcare providers are increasingly burdened with a multitude of digital platforms, each with its own login credentials, user interface, and workflows. Custom-built solutions exacerbate this problem by adding yet another platform for ATC staff to learn and navigate. This lack of standardisation forces users to switch between different systems, leading to frustration, decreased efficiency, and the risk of errors that can compromise patient safety.
Custom-built solutions frequently overlook the importance of the user experience. By neglecting to incorporate feedback from ATC staff and other end-users, these systems can miss the mark in addressing their needs and streamlining workflows. This can lead to resistance to adoption and a less efficient process. In contrast, standardised platforms that offer a unified interface, streamlined workflows, and features like single sign-on (SSO) can significantly alleviate portal fatigue and enhance user experience.
Another significant challenge is the potential for scope creep and overcomplexity. As stakeholders identify new requirements or modifications, software can become increasingly complex. This can lead to delays, cost overruns, and a system that is difficult to use and maintain – such complexity can also hinder integration with other critical systems, creating data silos and workflow inefficiencies.
Maintaining and supporting a custom-built platform also poses its own set of challenges. Companies must invest in dedicated IT resources to manage the system, address bugs, and implement updates. This can divert resources from core competencies, such as research and development or patient care. Moreover, as technology and regulatory frameworks evolve, custom-built solutions can become obsolete, requiring further investment to remain functional and secure.
Furthermore, custom-built platforms may lack access to specialised but essential features readily available in commercial solutions. Features like SSO and advanced scheduling capabilities can significantly enhance security and efficiency but may require extensive development effort in a custom-built system.
The Consequences of Portal Fatigue
Portal fatigue, while a growing concern, has far-reaching consequences that extend beyond individual frustration. It
Cell and Gene Therapy
presents a significant challenge to healthcare organisations, impacting not only the well-being of ATC staff but also the efficiency, safety, and financial health of the institutions they serve.
For ATC staff, constantly juggling multiple platforms and logins increases workload and stress. This can lead to reduced efficiency and productivity, as valuable time is lost navigating complex systems and reconciling inconsistent information. The cognitive burden of managing disparate platforms can also increase the potential for medical errors, as attention is diverted from patient care. For example, treatment delays caused by navigating complex platforms with unfamiliar terminology and workflows can compromise the efficacy of time-sensitive CGT products. Similarly, difficulties accessing critical patient information due to a maze of different logins and interfaces can lead to frustration, wasted time, and potential errors in treatment decisions.
Beyond the individual level, portal fatigue places a significant financial and resource burden on healthcare organisations. The costs associated with training staff on multiple platforms, managing user access and permissions, and addressing IT issues related to system incompatibility with existing IT infrastructure can be substantial. Furthermore, the reduced efficiency and productivity resulting from portal fatigue can lead to increased operational costs and potentially impact the quality of care delivered.
The Need for Standardisation and Collaboration
Standardising processes and terminology across different platforms is crucial. This would reduce the cognitive burden on ATCs, who currently have to navigate a maze of different systems with varying requirements. Standardised data formats and workflows would improve efficiency, reduce the risk of errors, and facilitate better communication between stakeholders.
Collaboration between technology providers and ATC staff is equally important. Technology providers need to understand the needs and challenges faced by ATCs in their daily work, while ATCs need to be involved in the design and development of new technologies. This collaborative approach can ensure that platforms are user-friendly, intuitive, and tailored to the specific needs of the CGT workflow.
Several initiatives are underway to address portal fatigue and promote standardisation. For example, the TrakCel-led Industry Advancement Board (IAB) is bringing together ATCs, manufacturers, and technology providers to discuss challenges and develop solutions. Such collaborative efforts are essential for driving progress and creating a more unified and efficient CGT landscape.
By embracing standardisation and collaboration, the CGT industry can create a more user-friendly and efficient environment for healthcare providers, ultimately leading to better patient outcomes and a more sustainable future for this promising field.
Solutions and Recommendations
The consequences of portal fatigue highlight the urgent need for solutions that prioritise standardisation, interoperability, and user-centric design in CGT platforms. By streamlining workflows, reducing the cognitive burden on ATC staff, and improving efficiency, the industry can mitigate the negative impacts of portal fatigue and ensure that technology serves its intended purpose – enhancing patient care and accelerating the delivery of life-saving therapies.
Addressing the pervasive issue of portal fatigue requires a multi-pronged approach, encompassing technological solutions, collaborative initiatives, and a commitment to user-centric design.
1. Technology Solutions
Developing user-friendly and intuitive platforms is paramount. Complex, cumbersome interfaces exacerbate portal fatigue, while streamlined workflows and intuitive navigation can significantly reduce cognitive burden and improve user experience. Implementing SSO solutions can eliminate the need for ATCs to remember multiple login credentials, simplifying access and enhancing security. Additionally, minimising the number of clicks required to complete a task can significantly enhance usability for busy ATC staff.
Integrating with existing healthcare systems, such as electronic medical records (EMRs), can streamline data exchange and reduce the need for duplicate data entry. This not only saves time but also minimises the risk of errors. Prioritising interoperability between different platforms is also crucial, allowing for seamless data sharing and reducing the need for ATCs to switch between disparate systems.
2. Collaborative Initiatives
Collaboration between technology providers and ATC staff is essential. Technology providers need to actively seek feedback from ATCs to understand their needs and challenges, while ATCs need to be involved in the design and development of new technologies. This collaborative approach can ensure that platforms are tailored to the specific needs of the CGT workflow.
Industry-wide initiatives, such as the IAB can play a crucial role in driving standardisation and addressing portal fatigue. By bringing together stakeholders from across the CGT ecosystem, these initiatives can foster collaboration, knowledge sharing, and the development of best practices.
Cell and Gene Therapy
not only the well-being of ATC staff but also the financial and operational health of healthcare organisations.
• For Technology Providers: Prioritise user-centric design, interoperability, and integration with existing systems. Actively seek feedback from ATCs and involve them in the development process to ensure platforms truly meet their needs.
• For Healthcare Organisations: Invest in comprehensive training and support for ATCs, empowering them to effectively utilise technology platforms. Foster a culture of collaboration and knowledge sharing between departments and stakeholders to maximise efficiency and minimise confusion.
• For Policymakers: Promote policies that encourage standardisation and interoperability in CGT platforms. Support initiatives that foster collaboration between technology providers and technology users.
By embracing these solutions and recommendations, the CGT industry can mitigate the negative impacts of portal fatigue, empower ATC staff, and pave the way for a more efficient and patient-centric future.
Looking Ahead: The Future of CGT Platforms
Portal fatigue presents a significant challenge in the cell and gene therapy landscape, hindering efficiency, increasing the risk of errors, and potentially compromising patient safety. The consequences of this fatigue can be far-reaching, impacting
To address this challenge, the industry must prioritise standardisation and collaboration. By working together, technology providers and platform users can develop solutions that streamline workflows, reduce the cognitive burden on ATCs, and improve the overall efficiency and safety of CGT delivery. This includes initiatives like the IAB, which brings together stakeholders to identify challenges and develop solutions.
The future of CGT platforms lies in creating user-friendly, interoperable systems that prioritise the needs of the user. Let's move forward with a commitment to developing and adopting standardised practices, fostering open communication, and prioritising the needs of the ATC staff who are at the forefront of delivering these life-changing therapies.
Dr. Akshay Peer
Dr. Akshay Peer is the Senior Vice President of Product Development and a Co-founder of TrakCel. He has over 12 years of experience in creating technology-based solutions for cell and gene therapy industry. His long-standing tenure in this field reflects a dedicated commitment to advancing innovation and contributing to the industry's growth.
3. Recommendations
QC -Analysis of Inhaled Antisense Oligonuclotides and mRNA
Analytical support for ASO and mRNA based therapies from early development to batch release
Antisense oligonucleotides (ASOs) and full-length mRNA -based therapeutics promise diseasespecific treatment options . One of the challenges of these therapies is the tissue- or cellspecific delivery. Inhalation has been an effective means to deliver pharmaceutically active substances to the respiratory tract for centuries Today there is a growing interest in the industry to leverage on the experience with different inhalation technologies to deliver ASO and mRNAbased treatments directly to diseased respiratory tissues.
A&M STABTEST has over 15 years of experience in supporting drug development and QCanalysis of nasal and orally inhaled products Specially trained staff and dedicated laboratory facilities are the key to analyze inhaled products throughout their lifecycle with the same constantly high quality. In the past 10 years A&M STABTEST has gained in-depth experience in stability and release testing of nucleic acid -based therapies ranging from ASO to mRNA -LNP therapies Very recently we have embarked on a project to combine our know-how of inhalable drug and nucleic acid testing to support our clients with inhaled ASO and mRNA therapies
For QC-analysis of mRNAs formulated as lipid -nanoparticles
Figure 1: A) displays the different charge states of a 40 mer oligo in a negative mode full MS B) Deconvoluted spectrum resulting from the full MS spectrum The mass difference between the calculated mass and the observed mass is 8ppm C) Celibatarian curves from a parallel detection by UV (above) and MS (below)
A&M STABTEST has established the panel of methods described in the USP draft guidance on “Analytical Procedures for mRNA Vaccine Quality”. Especially the determination of polydispersity by DLS is a valuable indicator to evaluate how well different LNP formulations respond to aerosolization . Combined with our generic cell -based bioassay, we can help to predict the transfection efficiency of different LNP formulations A&M STABTEST has also developed a generic LC-MS/MS method to support early ASO development projects, offering data on quantity, purity and impurities in one analytical run.
A&M STABTEST can support your early development and more advanced ASO and mRNALNP projects whether inhaled or not With our experience spanning well over two decades of providing the pharmaceutical industry with high quality analytical services, we are well suited to find analytical solutions for the industries most innovative drug development programs
BC Aldevron LLC
Page 69
Page 21
Page 71
Page 9
IBC
Page 70
Page 25
Page 35
Page 73
IFC
Page 61
Page 3
Page 51
Page 29
Page 72
Page 5
Page 39
Page 17
Subscribe today at www.international-biopharma.com or email info@senglobalcoms.com
A&M STABTEST
Anceus LDA
Anglonordic Life Science Conference 2025
Biotech Fluidics
Biopharma Group Limited
BioTrinity 2025
FUJIFILM Wako Chemicals USA
GenXPro GmbH
London BioTechnology Show
Newcells Biotech
Novo Nordisk Pharmatech A/S
Owen Mumford Ltd.
PCI Pharma Services
Polypure AS
Pre-filled Syringes & Injectable Drug Devices (SAE Media Group)
Richter Biologics GmbH & Co. Kg
Roald Dahl’s Marvellous Children’s Charity
Senglobal Ltd
I hope this journal guides you progressively, through the maze of activities and changes taking place in the biopharmaceutical industry
IBI is also now active on social media. Follow us on:
The biotechnology industry is poised to deliver amazing products and solutions that will dramatically change millions of lives for the better. Aldevron is ready.
As a globally trusted CDMO, our years pioneering ground-breaking solutions to advance biologic manufacturing for research use, as well as cGMP for clinical and commercial applications, has enriched us with a body of knowledge that is critical to support these next series of breakthroughs.
Contact us to advance your program. www.aldevron.bio/contact-us