Navigating the 2024 Life Science Landscape Trends, Challenges, and Opportunities
The Delivery of Pharmaceutical Drugs and How it Will Change the Future
Generative AI and its Impact on Speed to Market for Pharmaceuticals
Fighting Counterfeits in the Medical Devices Sector A Step Beyond the EU MDR

Sponsor Company:



www.international-pharma.com Volume 16 Issue 1
Peer Reviewed

PRINT QUALTY CONTROL FOR YOUR ENTIRE PROCESS www.EyeC.com Inspection, Security & Quality 100 % for your packaging materialwhether labels, folding cartons or leaflets • Easy to Use & Fast • Braille Inspection • 1D & 2D Code Inspection • Pharma Conformity • 21 CFR part 11 ready • Validation Support Package Visit us: drupa 2024 May 28th - June 7th Düsseldorf Hall 3 / A101
DIRECTOR: Mark A. Barker
BUSINESS DEVELOPMENT: Eliza Sarfaraz eliza@senglobalcoms.com
EDITORIAL: Virginia Toteva virginia@senglobalcoms.com
DESIGN DIRECTOR: Jana Sukenikova www.fanahshapeless.com
FINANCE DEPARTMENT: Akash Sharma accounts@senglobal.com
RESEARCH & CIRCULATION: Jessica Chapman info@senglobalcoms.com
COVER IMAGE: iStockphoto ©
PUBLISHED BY: Senglobal Ltd.
Unit 5.02, E1 Studios, 7 Whitechapel Road, E1 1DU, United Kingdom
Tel: +44 (0) 2045417569
Email: info@senglobalcoms.com www.international-pharma.com All
Craig Grant
Persistent challenges in poor solubility and low bioavailability have long impeded the pharmaceutical development pipeline, presenting ongoing hurdles for drug companies. IPI spoke with Craig Grant of Veranova to uncover best practices for formulating drugs with solubility issues.
REGULATORY & MARKETPLACE
10 Navigating The 2024 Life Science Landscape: Trends, Challenges, and Opportunities
The pharmaceutical and contract outsourcing space has shifted, with new trends emerging that will shape the future of this rapidly changing industry. Emma Bank at Ramarketing predicts the trends that will dominate the sector in 2024.
12 Proposed March-in Guidance Signals Funding Agencies to Actively Evaluate Government Rights Under the Bayh-dole Act
The Bayh-Dole Act governs the rights to inventions made with federal assistance. Rob Sahr and Curtis at Wolf Greenfield's Biotechnology Practice explains that, to shield against a potential loss of title to a funding agency, those that utilise federal funding to develop inventions should place a high emphasis on Bayh-Dole compliance.
16 Updating Your Regulatory Information Management Capability?
It Could Pay to Get Hands-on Letting business-savvy techies play around with a new RIM system ahead of proposed implementation could save Life Sciences companies a fortune. Romuald Braun at MAIN5, warns against hasty vendor selection and contracting.
DRUG DISCOVERY, DEVELOPMENT & DELIVERY
18 The Delivery of Pharmaceutical Drugs and How it Will Change the Future
Pharmaceutical drugs have seen a seismic shift over the past ten years, moving away from chemically synthesised drugs to focus more on a new class of biological drugs (biologics). Afzal R. Mohammed, Professor of Pharmaceutics, show how biologics have the potential to revolutionise the treatment of many common conditions that significantly affect millions of lives.
24 Big Data, Personalised Medicine and Support for Healthcare Professionals: What Will Drive Pharma in 2024?
In 2023, precision targeting for personalised medicines, improving the information flow between reps and healthcare professionals, and advancements in clinical trials have driven the adoption of new technologies. Chris Moore at Veeva Systems explains that pharma and healthcare providers who can successfully embrace data, analytics, and digital platforms will be the best positioned to succeed in 2024 and beyond.
26 Novel Drugs: Challenging Entrenched Prescriber/Investment Behaviour isn’t Just About Education
Behavioural science’s time has come in the pharma industry as medical communications adapt to a more ambitious and diverse treatment landscape. William Hind at Alpharmaxim brings to life the science of behavioural change.
28 Transforming the Biologics Product Lifecycle with Inhalation Innovation
In recent years, the biopharmaceuticals landscape has witnessed a revolutionary shift with the emergence of new nasal inhalation technologies. Nicolas Buchmann, CTO, and Frank Verhoeven, Business
INTERNATIONAL PHARMACEUTICAL INDUSTRY 1 www.international-pharma.com Contents
rights reserved. No part of this
may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
next issue of IPI will be published in Summer 2024. ISSN No.International Pharmaceutical Industry ISSN 1755-4578. The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright. 2024 Senglobal Ltd./Volume 16 Issue 1 – Spring – 2024
Editor’s Letter TALKING POINTS
publication
The
04
06 Harnessing the Power of Solid Form and Particle Engineering to Overcome Solubility and Bioavailability Challenges: An Interview with Veranova’s
Developer at Resyca, delve into the potential transformative impact of inhalation innovations on the biopharmaceutical product lifecycle.
CLINICAL & MEDICAL RESEARCH
38 Why is Real World Data the Key to Rare Disease Success?
The quest to understand and treat rare diseases is among the most challenging and vital missions in healthcare today. Karen Ooms at Quanticate shows how RWD can provide insight that you simply would not get in the traditional sense and opens patient populations that may not have been looked at previously.
40 Advancing Therapeutic Solutions with Antibody-drug Conjugates (ADCs)
As promising therapeutic agents for treating oncology indications, antibody-drug conjugates (ADCs) have become prominent in the biopharmaceutical market in recent years. Louise Duffy and Cambell Bunce at Abzena explores how innovation in the biopharma sector is driving the development of novel ADCs to treat indications beyond cancer.
TECHNOLOGY
42 Generative AI and its Impact on Speed to Market For Pharmaceuticals
Developing and bringing a new drug to market takes approximately seven years. However, this time can be significantly reduced if life sciences companies leverage generative AI to accelerate insight and content generation. Bryan Hill at Cognizant debates that this timesaving is crucial in clinical development.
44 The End to Painfully Slow Cloud Migration?
Despite the benefits of moving to the cloud, migration can still be prohibitively time-consuming and costly, especially in a highly regulated industry like pharma. Arjun Khanna at Kallik explains how these tools could accelerate digital transformation strategies in pharma.
MANUFACTURING
48 Reaching the Marketplace – Understanding the Complexities of Paediatric Drug Development and Manufacture
Bringing a paediatric drug product to the marketplace can be very complex. Tom Hegarty at Almac Pharma Services explains that adhering to strict guidelines and practices in the manufacturing process enables pharmaceutical companies to have surety that their paediatric patient is receiving the medication exactly as intended.
52 The Future of Sterility: Advancements and Innovations in Sterile Drug Product Manufacturing
Sterile drug product manufacturing is complex and plays a crucial role in the pharmaceutical industry, ensuring the production of safe and effective medications for patient use. Shawn Cain at PCI Pharma Services explores the key developments in sterile drug product manufacturing.
58 The Key to Formulation Development is in the Details
During the tablet manufacturing process, tooling and press manufacturers are often faced with ongoing challenges. If there is a change in formulation, a process following Scale-Up and Post Approval Changes (SUPAC) guidelines must occur before moving forward. Robert Sedlock at Natoli Engineering reviews the importance of the formulation development process and how a thorough approach can reduce manufacturing issues, costs, and downtime.
PACKAGING
62 Biocompatible, Pre-coloured and Sustainable ABS Optimised for Laser Marking to Support Medical UDI Identification Along with Sustainability Targets
EU’s Medical Device Regulation (MDR) and US Code of Federal Regulations
Title 21 referring to labelling for medical devices (21 CFR 801 Subpart B)
require a Unique Device Identification system (UDI) on each medical device. Luca Chiochia at ELIX Polymers reveals more information about the valid alternative of a laser marking.
66 Safety, Compliance and Quality Just a Click Away
Producing safe and compliant medicines, harmless cosmetics, or good food is sometimes like a puzzle with an infinite components number. That is only possible if all the individual parts are used and manufactured with the necessary care and precision. Dr. André Schwarz at EyeC GmbH explains why, quality and precision, reproducibility, and exact documentation are essential.
HEALTH OUTCOMES
72 Electronics and Data, How Effective Are Personal Electronics in Helping Patients to Maintain Good Health?
What Does the Future Hold for Patients?
There is only really a subtle difference between compliance and adherence systems. The former is doctor-led, and so the intention is for patient treatment data to be available remotely. Bill Treneman of UPC Cambridge Limited discuss Adherence e-aids which allows monitoring of the patient’s adherence to daily medicines by the parents or carers of that patient, or the patient themselves.
LOGISTICS & SUPPLY CHAIN MANAGEMENT
76 Fighting Counterfeits in the Medical Devices Sector –A Step Beyond the EU MDR
The counterfeiting of health products is a serious and growing concern, and it has come to the forefront of the public mind in recent years. Bart Vansteenkiste at Domino explains why all prescription pharmaceuticals sold in the EU and US are subject to complete end-to-end traceability.
APPLICATION NOTES
22 Exploring Device Interchangeability for Drug Delivery (OM)
Choosing the right injectable drug delivery device can be a crucial differentiator for pharmaceutical manufacturers. Alex Fong at Owen Mumford explains that the switching of drug delivery devices needs to maintain and ideally improve the patient experience and have a limited effect on patient behaviour.
32 Functional Testing of Prefillable Glass Syringes Aligned with ISO 11040 Guidelines
Having a primary packaging container that serves as both a sterile storage compartment and delivery device is the major driving force for injectable combination products using prefillable syringes. The authors at SCHOTT Pharma discuss that these benefits come along with more challenges than the traditionally used two-unit delivery systems vial and disposable syringe.
54 Automating Biotherapy Production at the Speed of Market Expansion
The effort to scale biotherapy production and meet the demand for personalised medicine is well underway, and robotic automation is taking on an expanded role. Rudolf M. Weiss at Stäubli Robotics explains that there are numerous possibilities for robotic automation in biotherapeutics, and by extension many other pharmaceuticals.
68 Automation of Manual Packaging for Small Batches in The Medical Device Industry
The pressure to automate the packaging process is increasing. Packing by hand? This is still the case in many areas of the medical device industry. Jürgen Sikora at Christ Packing Systems explains that if you want to produce more flexibly and be less dependent on staff availability, you need an automation solution.
2 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Contents
The future of synthetic endotoxin detection.

PYROSTAR ™
Recombinant Endotoxin Detection Reagent
Limulus Amebocyte Lysate
PYROSTAR™ Neo is a new endotoxin detection reagent that mirrors nature but is developed by recombinant technology.
FUJIFILM Wako Chemicals U.S.A. Corp.
© FUJIFILM Wako Chemicals U.S.A. Corp. - 2023
www.wakopyrostar.com ~ wkuspyrostarinfo@fujifilm.com

As we step into 2024, the global life science industry finds itself at a crossroads, facing plenty of challenges alongside an abundance of exciting opportunities.
The pharmaceutical and contract outsourcing space has shifted, with new trends emerging that will shape the future of this rapidly changing industry. The global stage has witnessed significant upheavals throughout 2023, with wars impacting everything from oil prices to work locations. Inflation and rising material costs are cutting into the profits of many life science companies, and a continuation of the COVID comedown has left vaccine producers facing challenges and emphasising the need for diversification of capabilities.
Pharmaceutical companies facing a range of challenges need to bring new products to market fast. To do it, they’re looking to stimulate innovation, attract the best people and drive seamless, efficient production.
Disruptions in concentrated markets, along with the need for comprehensive due diligence and third-party risk management, can significantly impact the supply chain. Additionally, ensuring drug safety in the cold chain and complying with traceability requirements pose further challenges for pharmaceutical companies.
ESG considerations are becoming a significant focus for the pharmaceutical industry in 2024, as public awareness of the industry’s environmental impact and social responsibilities increases, requiring companies
Editorial Advisory Board
Bakhyt Sarymsakova, Head of Department of International Cooperation, National Research, Center of MCH, Astana, Kazakhstan
Catherine Lund, Vice Chairman, OnQ Consulting
Deborah A. Komlos, Principal Content Writer, Clarivate
Diana L. Anderson, Ph.D president and CEO of D. Anderson & Company
Franz Buchholzer, Director Regulatory Operations worldwide, PharmaNet development Group
Francis Crawley. Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
to strengthen reporting, meet environmental benchmarks, scrutinise third parties, and enhance governance frameworks to ensure compliance with regulations and mitigate risks.
The pharmaceutical industry has plenty to celebrate. In the last decade, major therapeutic advances, such as immunotherapy and cell and gene therapy, have given new hope to patients.
But during that same period of groundbreaking innovation, pharmaceutical companies failed to keep pace with the capital markets. In fact, returns from pharmaceutical companies lagged the S&P 500 by about onethird, and biotech fared even worse.
Looking at the stock performance of the top 50 pharmaceutical companies, the divide between the leaders and laggards has been widening. In 2021, the five-year total shareholder return (TSR) for drugmakers in the top quintile was up by 29%, compared with a decline of 11% in the bottom quintile, according to a PwC analysis. As performance pressures mount, investors are taking a closer look at which pharma companies are positioned to win and allocating their investments accordingly. As leaders set the path ahead, they should incorporate a sharper lens on shareholder value creation into everyday decisionmaking. Connecting product-market decisions (e.g., portfolio choices, launch investments, production expansions) to shareholder value creation across the enterprise is essential to help translate great science into great returns.
In this 1st edition of IPI of 2024, we have a whole series of exciting articles, which I believe will be worthwhile reading for you.
We start off with an interview with Craig Grant of Veranova, discussing how to harness the power of solid form and article engineering to overcome solubility and bioavailability challenges.
Emma Banks, CEO of Ramarketing, navigates the 2024 Life Science Landscape, discussing trends, challenges, and opportunities.
Afzal R Mohammed, Professor of Pharmaceutics discusses the delivery of pharmaceutical drugs and how It will change the future. Pharmaceutical drugs have seen a seismic shift over the past ten years, moving away from chemically synthesised drugs to focus more on a new class of biological drugs (biologics). Biologics have the potential to revolutionise the treatment of many common conditions that significantly affect millions of lives.
Chris Moore at Veeva Systems discusses what will drive pharma in 2024, whether it will be big data, personalised medicine and support for healthcare professionals.
I hope you all enjoy this edition of IPI, and I look forward to meeting many of you at ACHEMA and other exhibitions coming up.
Virginia Toteva, Editorial Manager – IPI
Georg Mathis Founder and Managing Director, Appletree AG
Jagdish Unni, Vice President – Beroe Risk and Industry Delivery Lead – Healthcare, Beroe Inc.
Jeffrey Litwin, M.D., F.A.C.C. Executive Vice President and Chief Medical Officer of ERT
Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma
Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
Stanley Tam, General Manager, Eurofins MEDINET
(Singapore, Shanghai)
Steve Heath, Head of EMEA – Medidata Solutions, Inc
Patrice Hugo, Chief Scientific Officer, Clearstone Central Laboratories
Heinrich Klech, Professor of Medicine, CEO and Executive Vice President, Vienna School of Clinical Research
Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
Sanjiv Kanwar, Managing Director, Polaris BioPharma Consulting
Stefan Astrom, Founder and CEO of Astrom Research International HB
T S Jaishankar, Managing Director, QUEST Life Sciences
4 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Editor's Letter

Minimize your risks and get peace of mind
You can ensure optimal processes and even better medicines by prioritizing high quality, consistency and reliable GMP manufactured raw materials.
Our Benzalkonium Chloride is manufactured following cGMP guide ICH Q7 for APIs, the highest available quality standard in the industry.
Get peace of mind with:
• Full traceability
• High product purity
• Full pharmacopoeia compliance (PhEur, USP/NF, JP, ChP)
• Audit access
• Full regulatory documentation package
• Manufacturing under fully validated processes
• 30+ years of experience in cGMP manufacturing
Learn more about Benzalkonium Chloride at novonordiskpharmatech.com



INTERNATIONAL PHARMACEUTICAL INDUSTRY 5 www.international-pharma.com
Pharmaceutical grade Benzalkonium Chloride
Talking Point
Harnessing The Power of Solid Form and Particle Engineering to Overcome Solubility and Bioavailability Challenges
An Interview with Veranova’s Craig Grant
Persistent challenges in poor solubility and low bioavailability have long impeded the pharmaceutical development pipeline, presenting ongoing hurdles for drug companies. As the industry grapples with increasingly complex new chemical entities, the need for innovative solutions to overcome these challenges becomes more urgent.
To delve into this issue, explore current strategies for advancing poorly soluble drugs through the clinical pipeline, and uncover best practices for formulating drugs with solubility issues, IPI Journal spoke with Veranova’s Craig Grant, VP and General Manager, Cambridge.
With over two decades of solid form expertise, Craig is a founding figure behind Pharmorphix®, Veranova’s dedicated brand for solid form and particle engineering.
Q: It’s estimated that up to 40% of marketed drugs, and between 70 and 90% of drug candidates in the development stage, exhibit poor solubility.1 Could you explain why solubility and bioavailability continue to be such big issues in bio/pharmaceutical drug formulation?
A: In the pharma and biotech industries, the need for effective therapeutics is continually driving the discovery and development of novel active pharmaceutical ingredients (APIs) and new chemical entities (NCEs). This has resulted in the development of increasingly complex drug scaffolds, often with one or more chiral centres and which routinely possess high molecular weight. Though these drugs offer improved stereoselectivity, target specificity and activity, their complexity often causes them to be poorly soluble, which can cause a wide variety of knock-on issues, most notably poor bioavailability with low drug absorption in the body, resulting in such molecules being assigned to BCS Class II (high permeability, low solubility).2
Moreover, at the candidate selection stage, developers are more likely to focus on potency or efficacy, meaning the downstream developability of NCEs is often overlooked during early-stage development. This can lead to significant challenges further down the development pipeline, leading to costly delays and resource wastages.
Q: As increasingly complex drug molecules enter the development pipeline, how do you see solubility challenges evolving in future?
A: With small molecules becoming increasingly more complex, solubility challenges are here to stay. This is even more relevant when considering current pharmaceutical trends, with increased prevalence in existing modalities such as peptides, which, depending on size and/ or complexity, straddle the boundary between small and large molecules. The emergence of new “small molecule” modalities such as PROTACs (PROteolysis TArgeting Chimeras) also bring substantial solubility and developability challenges. PROTACs are a subset of TPD (targeted protein degraders), an emerging therapeutic modality used to treat previously difficult or undruggable targets. Unlike traditional protein inhibition methods, PROTACs are two-pronged molecular entities designed to seek out and degrade disease-causing proteins within the cell. However, due to their size and flexibility, PROTACs typically pose crystallisation and solubility challenges during solid form studies. PROTACs are just one example of how molecular complexity is likely to continue to present difficulties going forward.
Q: What are the most common approaches to overcoming solubility/ bioavailability issues during development? Which do you think are the most effective?
A: Obtaining the optimal solid form is pivotal in developing a drug product with good solubility and bioavailability as well
as many other required or desirable physical properties. For crystalline materials, optimal usually refers to the thermodynamically stable polymorph of the parent API, salt or cocrystal thereof. Metastable forms have their place too but the key to developing these is a rigorous understanding of the solid form landscape and interconversions that may take place to progress stable, developable forms.
Salts make up the majority of all marketed drugs and are typically the first port of call when attempting to improve aqueous solubility. Crucially, by conducting salt screening and identifying the optimal salt, it is possible to tune the physical properties of the drug in development. If there are no accessible ionisable centres and producing a salt is not possible, cocrystal screening and selection is an increasingly popular alternative.
Arguably less obvious to make, but just as attractive in their ability to modify physical properties as salts, cocrystals allow formulators to preserve the therapeutic benefits of an API but offer the potential to improve solubility and bioavailability. Unlike salts where acid-base chemistry is at play between the API and corresponding acid or base, interactions between an API and coformer are weaker and typically hydrogen bonding-driven. Despite an increase in regulatory approvals over the years, cocrystals still appear somewhat underutilised within the pharma industry, yet our understanding of how to effectively screen, select and scale developable cocrystals offers a significant opportunity.
Then, once the optimal salt, cocrystal or parent version of the API has been selected, it is necessary to carry out full polymorph screening. In fact, it is prudent to perform a preliminary polymorph assessment as part of salt or cocrystal screening, as this is likely to guide selection. Understanding the polymorphism behavior of an API is important for multiple reasons. It not only develops an understanding of the solid form landscape of the molecule in question, but solid forms are also patentable and, crucially, understanding polymorphism for an API is a regulatory requirement.
6 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Beyond modification of the crystal lattice as per methods already outlined, particle-size reduction methods including micronisation are commonplace in the industry, although typically in a “top down” approach. Methods for achieving ever smaller “nano” sized particles are becoming increasingly popular.
Exploring a variety of excipients and surfactants can also provide valuable uplifts in solubility during the early stages of formulation development. It may even be prudent to develop such an “enabling formulation” for toxicology studies, facilitating API progression whilst other crystal or particle engineering methods are pursued for the longer term. Either way, the method of choice needs to consider both the route of administration and the final dosage form to ensure effective formulations are designed with the end in mind.
Amorphous materials, being the most metastable of forms, also offer attractive solubility and bioavailability benefits, though this usually comes at a price. Such materials, where “unprotected”, are more unstable chemically and from a solid form perspective than their crystalline counterparts. Stabilisation via incorporation
into an appropriate polymer matrix to give a developable Amorphous Solid Dispersion (ASD) is one solution, however. In summary, there is no ‘one-size-fits-all’ approach, and the process will often be different for each drug molecule.
Q: Why is it so important to address solubility issues early in the drug development pipeline?
A: The further an API progresses down the development pipeline, the larger the ramifications will be if a problem is discovered. The main aim of performing solid form studies early is therefore to ‘derisk’ future development. Scientists must gain a deep understanding of the physical properties of the drug candidate as early as possible. Typically, solid form studies are performed in preclinical development, but you could argue that understanding the physical characteristics of molecules should be applied in late-discovery/lead-candidate selection. Issues uncovered at this early stage may even promote the selection of an alternate lead candidate but ultimately one that is developable! Solid form and particle engineering studies are vital in ensuring
speed to market – by taking all possible precautionary steps, the development process will be streamlined and its efficiency maximised.
Q: Have there been any recent technological advances that you think will have a significant impact on aiding solubility and bioavailability in the development pipeline?
A: Looking to the future, it is likely that predictive AI technologies will see increasing use in parallel with experimental work to help scientists conduct more pertinent experiments earlier on. Modelling tools are already used increasingly in the work we do spanning physicochemical and solubility predictions to crystallisation development. These along with other developments in polymorph prediction will all play their part in helping scientists get to the answer quicker.
Due to convenience and ease of manufacture, oral dosage forms continue to be one of the most preferable delivery routes, and improvements in the methods used to monitor their dissolution in the

INTERNATIONAL PHARMACEUTICAL INDUSTRY 7 www.international-pharma.com
Talking Point

gastrointestinal (GI) tract have been invaluable. Small-scale in situ dissolution studies have helped improve our understanding of the solubility and bioavailability of BCS Class II compounds, which are highly permeable in the GI tract but exhibit poor solubility.
There have also been some advancements in the use of porous excipients, such as mesoporous silica, often formulated as lipid nanoparticles to boost the solubility and bioavailability of known compounds. There have been no FDA-approved drugs utilising these technologies yet, but research in this area will likely advance in the coming years.
Q: Could you provide some best practices for formulators when dealing with poor solubility/bioavailability?
A: When dealing with poorly soluble APIs, the aim is always to get it right the first time. It is therefore vital for formulators to obtain as much information about the end goals of a novel drug product as early as possible. Factors such as the route of administration, drug target, and so on can impact the type of experimental design and yield the best value-added data.
Successfully overcoming solubility and bioavailability issues also requires the implementation of robust scale-up processes – for any solid form to have value, it must be scalable. Modelling tools are routinely employed as we design and build
robust crystallisation processes, enabling the development of one that will require minimal intervention as it is scaled up.
Should a company decide to collaborate with an outsourcing partner, it is important to carefully consider the requirements and complexity of the project in question. This ensures they select the right expertise for their drug candidate. At Veranova, we adopt a hierarchical approach, following a drug candidate at all stages of development and collecting as much data as possible. Nevertheless, a developer will likely need to draw on a wide range of expertise when managing difficult issues such as solubility, so it is important to fully consider the demands of their drug moiety.
Q: In what ways can a CDMO partner help pharmaceutical companies overcome solubility and bioavailability challenges during formulation?
A: CDMOs can provide pharmaceutical companies struggling with solubility and bioavailability with invaluable guidance. Using specialised technology and expertise, they can develop a tailored approach to each drug molecule, helping companies overcome development hurdles at every stage. A reliable and experienced CDMO company will have the capacity to react to challenges in both a proactive and reactive manner, ensuring that all bases are covered and that the project is brought to completion as efficiently as possible.
Though solubility and bioavailability challenges are central to drug development today, CDMOs can help pharmaceutical companies identify and overcome the full scope of problems associated with a particular drug moiety, from stability to hygroscopicity. By fully understanding the chemistry and selecting the optimal solid form of a drug candidate, CDMOs can help develop robust scalable processes, establish a robust IP position, and ultimately accelerate the therapeutic to market.
REFERENCES
1. Khan, KU et al. 2022 , Overview of nanoparticulate strategies for solubility enhancement of poorly soluble drugs, Life Sciences, 291, 120301. Available at https://doi. org/10.1016/j.lfs.2022.120301 [Accessed March 2024]
2. Samineni, R. et al. 2022, Emerging Role of Biopharmaceutical Classification and Biopharmaceutical Drug Disposition System in Dosage form Development: A Systematic Review, Turk J Pharm Sci, 19(6), 706-713. Available at https://doi.org/10.4274%2Ftjps. galenos.2021.73554 [Accessed March 2024]

Craig Grant
Craig Grant is Veranova’s Vice President and General Manager in Cambridge, UK. He heads up the solid form and particle engineering team. Craig has a Ph.D. in structural inorganic chemistry from the University of Edinburgh and has over 20 years’ experience in the health and pharmaceutical industry. He has worked across a range of servicebased companies, including Cambridge Combinatorial, Exova and Solid Form Solutions, as well as pharma companies, such as Millennium. In 2003, Craig cofounded Pharmorphix® Ltd which was acquired by Sigma-Aldrich in 2006, later by Johnson Matthey in 2015 and is now part of Veranova, a standalone CDMO with a global footprint and particular expertise in the development and manufacture of highly potent and controlled substances. Under the Pharmorphix® brand, Veranova offers one of the most comprehensive arrays of integrated solid form, preformulation, particle engineering and chemical development capabilities available to the pharmaceutical and biotechnology industries.
8 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Talking Point

Your quest for perfection ends here. Let’s revolutionize your manufacturing process, redefine precision, and gear up for excellence together. When you partner with KAHLE perfection isn’t a destination—it’s the journey we craft, gear by gear.


Kahle® is dedicated to providing custom automation machinery solutions for the Medical Device, Pharmaceutical, and Healthcare Industries around the world.



INTERNATIONAL PHARMACEUTICAL INDUSTRY www.international-pharma.com
U.S.A. | ITALY | CHINA
Visit www.KahleAutomation.com Contact: Kahle@KahleAutomation.com
Precision Revolution
Navigating the 2024 Life Science Landscape: Trends, Challenges, and Opportunities
As we step into 2024, the global life science industry finds itself at a crossroads, facing plenty of challenges alongside an abundance of exciting opportunities.
The pharmaceutical and contract outsourcing space has shifted, with new trends emerging that will shape the future of this rapidly changing industry. The global stage has witnessed significant upheavals throughout 2023, with wars impacting everything from oil prices to work locations. Inflation and rising material costs cutting into the profits of many life science companies, and a continuation of the COVID comedown, has left vaccine producers facing challenges and emphasising the need for the diversification of capabilities.
Here, Emma Banks, our CEO, predicts the trends that will dominate the sector in 2024.
Biotech Investment Slowdown: A Cautionary Post-COVID Tale
The roaring success of biotech investments during the pandemic has hit a speed bump. As other sectors gain traction postCOVID, biotech face a slowdown due to rapidly increasing interest rates and the diversification of investments. This shift poses risks and a reduction in the flow of money with less capital available for biotech companies.
Faced with this challenge, biotech firms are now reevaluating and being more cautious with their investment strategies, focusing on specific assets within their pipelines.
“...the biggest trend we've seen is the number of customers that are not necessarily well-funded. So many of them have far less cash than they've had in the past. As a result of that, they're much more cautious as to how they're spending their capital. They are very selective on their number of assets. We've seen a lot of deprioritisation of assets. They're looking for more creative funding, where they
give us stock. We can only take so many bets, and I think that's the challenge.” Outsourced vendor
Increasing Focus on Supply Chains
The vulnerabilities exposed by COVID have led to a renewed emphasis on securing and optimising supply chains.
Onshoring is gaining prominence due to geopolitical tensions, with more business flowing from East to West. That said, there are a growing number of Asian CDMOs coming to the fore of the industry, particularly in markets such as Japan and South Korea.
The concept of "domesticating" gains momentum, as countries like Canada invest
heavily to be better prepared for future pandemics.
“Canada has a nice R&D rebate program, which factored into our decision to go with our vendor. If it's qualified as research, you can get a good percentage of that back from the Canadian government.”
Biotech
Continued Explosion of Advanced Novel Modalities
Cell and gene therapies, CAR-Ts, and oligonucleotides continue to grow within the clinical pipelines, promising groundbreaking treatments. Despite regulatory challenges and complexities in commercialisation, the focus on these novel modalities remains
10 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Regulatory & Marketplace
Regulatory & Marketplace

strong. Companies specialising in platforms supporting these new products play a critical role in the success of these therapies.
Real Focus on Partnerships
The life science outsourcing market remains highly fragmented; with over 400 players in the biopharma CDMO space alone, businesses and investors face almost limitless options.
This trend is coupled with a shift towards innovative partnerships; biotech companies are now seeking risk-sharing models and flexible collaborations with CROs and CDMOs. It’s time for companies to explore models beyond traditional fee-for-service arrangements, ensuring maximum value and conservation of cash.
New breed of Biotech
Despite challenges, long-term funding trends for biotech companies look positive. A leaner and more capital-efficient approach
is emerging, with biotech relying more on outsourcing. Mastering the vendor ecosystem and investing in innovative partnership models are becoming key strategies for success.
“So, when I got to BMS, our process for developing a protocol took 12 months from the idea of a clinical trial to implementation. When I was at Rockefeller, the timeline from an idea to implementation was less than a month. Because we were an academic, we didn't have the process that a Big Pharma has and in the environment we’re in, we can't afford to delay 12 months to develop and launch a new clinical trial.”
Ian Walters, Portage Bio
Hear more about this trend from Molecule to Market’s interview with Ian Walters: Meet the lean, keen biotech.
AI – A Disruptive Force
AI and digitisation are transforming the
industry and we expect to see this evolve further in 2024. From drug discovery to clinical trials, AI is already beginning to save time, reduce manual efforts, and improve overall efficiency. Companies with AI-enabled platforms are set to become sought-after partners.
Marketing practices and processes are also being heavily influenced by AI. Both agencies and in-house teams are utilising tools to make efficiencies. There is still more to consider and refine as AI continues to evolve and the jury is still out on some of the ethical considerations regarding this technology.
“How does AI seep into this world? And does that put pressure on everybody to just get things done more quickly? Can I do some of that myself or hire those people to do that?”
Biotech VC
“Something may not be a big deal now but in five years, it may be a very big deal as you think about AI. If you can electronically collect, organise, and analyse your datasets faster, you will be well ahead with speed and quality compliance will be much easier.”
Outsourced vendor
Outsourcing is King R&D spending and clinical pipelines continue to grow, driving the need for outsourcing to access specialist capabilities, expertise, and capacity. The total addressable market for outsourcing in the life science sector is vast and continues to expand. Despite challenges, outsourcing remains a key driver of success for companies ranging from big pharma to virtual start-ups and biotech.

Emma Banks
Emma Banks is the CEO of Ramarketing with a Ph.D. in Immunology and has over 15 years of experience in life sciences business leadership. Emma oversees the entirety of Ramarketing's business operations across the UK, Europe, and North America and has led Ramarketing's 400% growth and a major equity partnership. An advocate for ESG, mental health, and female health issues, she's an industry influencer, driving the company's operations across multiple continents.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 11 www.international-pharma.com
Proposed March-in Guidance Signals Funding Agencies to Actively Evaluate Government Rights Under the Bayh-dole Act
The Bayh-Dole Act (“Bayh-Dole”) governs the rights to inventions made with federal assistance. It offers ownership rights to federal award recipients (“Contractors”) that “conceive or first actually reduce to practice” inventions utilising federal funding (“Subject Inventions”), but Bayh-Dole also comes with certain obligations that carry forward to licensees of governmentfunded technology. Bayh-Dole also provides rights to the U.S. government. Among these rights are two distinct but often conflated rights: the march-in and the request of title. Historically, the U.S. government has rarely exercised those rights. Indeed, in the nearly 45 years since the enactment of Bayh-Dole in 1980, no federal agency has exercised march-in rights (and the government has routinely declined to use march-in authority on request), and agencies have requested title only a handful of times.
On 8 December 2023, the Department of Commerce and the National Institute of Standards and Technology (NIST) published a Federal Register Notice titled “Draft Interagency Guidance Framework for Considering the Exercise of March-In Rights” (“Draft Framework”) detailing a new paradigm for the assessment of march-in rights. The Draft Framework reflects the current executive administration’s effort to more aggressively monitor compliance by Contractors and encourage the exercise of government rights under Bayh-Dole.
March-In Threat Level Increases
The “March-in right” refers to a federal funding agency’s right to require a Contractor, an assignee, or an exclusive licensee of a Subject Invention to grant a license to “a responsible applicant” (or applicants), or to grant a license itself, if certain conditions are met.1
The Draft Framework requests public comments on a proposed framework for the exercise of march-in. In view of President Biden’s Executive Order of 28 July 2023 (“Executive Order 14014”), which invoked changes to utilisation reporting,
the Draft Framework strongly signals that federal agencies will be more proactive in searching for effective opportunities to exercise march-in rights. Per the Executive Order and recently promulgated regulations by NIST, as of 1 October 2023, all agencies are required to collect annual utilisation reports for Subject Inventions, and NIST “strongly encourages” agencies not currently participating in iEdison to do the same. NIST has also provided standard utilisation questions, and answers to these utilisation questions will provide agencies with information to help assess whether exercising march-in is warranted and can be done effectively, according to the Draft Framework.
Executive Order 14014 places a clear emphasis on Bayh-Dole’s domestic manufacturing requirement for exclusive licensees of Subject Inventions. This domestic manufacturing requirement is a statutory requirement and a major component of the Draft Framework. NIST’s standard utilisation questions will help agencies assess whether this domestic manufacturing requirement is being met. In this light, it would not be surprising if the first product subject to march-in rights is a product that lacks compliance with Bayh-Dole’s domestic manufacturing requirement.
Product Pricing in the Spotlight
In addition to the domestic manufacturing requirement, much attention has been directed at the Draft Framework’s inclusion of the “reasonableness of price” of a product as a consideration for march-in. However, agencies were not previously precluded from reviewing product price as a consideration for marchin.2 As such, the inclusion of “reasonableness of price” in the Draft Framework as a march-in consideration may not be surprising to some, but it is noteworthy that the Draft Framework places a clear emphasis on instances in which product price is increased in response to increased demand and/or a health or other disaster. Indeed, each exemplary scenario provided in the Draft Framework that discusses the “reasonableness of price” relates specifically to this context (Scenario 5 describes a 10,000% price increase for a product following a spike in demand; and
Scenario 6 describes a 400% price increase for a product following a viral outbreak). It remains to be seen, but considerations of “reasonableness of price” may not weigh as heavily as feared outside of the context of a sudden product price increase.
While the Draft Framework reminds federal agencies that “march-in is an important tool for agencies,” it also highlights various hurdles to the effective use of march-in. For example, many products are protected by multiple patents. If only a subset of the patents is subject to Bayh-Dole, the exercise of march-in rights alone would not provide a clear path for “a responsible applicant or applicants” to make and/or sell the product. Similarly, some products (e.g. drug products) are subject to regulatory approval, and the exercise of march-in rights does not negate the requirement that a similar product produced via march-in would also need regulatory approval. In light of these (and other) hurdles, it seems unlikely that march-in rights will be exercised against the vast majority of subject inventions.
Request for Title Remains a Major Danger for Bayh-Dole Noncompliance
In addition to the march in, Bayh-Dole describes circumstances when a federal agency can take title to a patent/patent application claiming a Subject Invention (i.e. to become the owner). Specifically, if a Contractor fails to timely disclose a subject invention to the funding agency or to timely elect title to a subject invention, the agency may exercise its right to take title. Typically, one must disclose the subject invention to the funding agency within two months after the inventor discloses it in writing to the Contractor’s patent personnel, and one must elect title to the subject invention within two years of disclosure. Prior to 14 May 2018, Bayh-Dole regulations provided a 60-day window for a federal agency to take title if disclosure or election was not timely; in the absence of action by the federal agency title would remain with the Contractor or assignee. However, on 14 May 2018, this 60-day window was eliminated from the regulations, and as such, a funding federal agency may, in some instances, take title at any time, if disclosure or election is not timely.
12 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Regulatory & Marketplace
AVAILABLE WORLDWIDE AVAILABLE WORLDWIDE AT YOUR FREIGHT FORWARDER AT YOUR FREIGHT FORWARDER

TEMPERATURE TEMPERATURE QUALIFICATIONS & VALIDATIONS QUALIFICATIONS & VALIDATIONS
Multilayer thermal blanket for PMC-ULD - Euro and Block pallets


Stress-tested in summer (+46°C) and winter (-15°C) profiles
Airfield Tarmac tested on solar power and greenhouse effects

Tel. (Belgium): +32-11.26.24.20
E-mail: info@krautz.org
Website:

INTERNATIONAL PHARMACEUTICAL INDUSTRY 13 www.international-pharma.com
Sustainability program CO2-neutral
Manufacturing Recycling Material reuse
Customer use
TEMAX TEMAX RECYCLABLE THERMAL BLANKETS for healthcare airfreight
(+15°C + 25°C°) and (+2°C + 30°C) (+15°C + 25°C°) and (+2°C + 30°C)
www.krautz.org Temperature protection of pharmaceutical and healthcare products in airfreight
Regulatory & Marketplace
For Contractors, assignees, and licensees of patents claiming Subject Inventions, the threat of loss of title is significantly greater than the threat of march-in. Indeed, and in contrast to march-in, if title is requested by the government for failure to timely disclose an invention, a Contractor, prior assignee or licensee may be sued for patent infringement for practicing the Subject Invention (absent a license from the federal agency). As such, it is very important that Contractors comply with Bayh-Dole requirements to avoid this risk.
Although not explicit, the Draft Framework also suggests that federal agencies may be more proactive in searching for opportunities to request title moving forward. Specifically, in the first step of the Draft Framework, an agency is asked to consider whether BayhDole applies. In addition to considering whether the inventions were previously reported as Subject Inventions, the Draft Framework instructs agencies to actively review patents for “unreported subject invention[s]” (i.e. ones that were not properly disclosed). For example, the Draft Framework instructs funding agencies: to search for a publication(s) that relates to a patent to assess whether the publication acknowledges government funding, and to search for a funding agreement(s) that relates to a patent to assess whether the specific aims under the funding agreement are related to the claimed subject matter. Funding agencies would also be likely to review progress reports provided by the Contractor that correspond to any funding agreements identified as having potential relevance. With this information in hand, the Draft Framework instructs agencies to consider whether there is sufficient evidence
to confirm whether a patent includes an invention that was “conceived or first actually reduced to practice under the performance of work under a funding agreement” (i.e. includes a Subject Invention).
While this discussion in the Draft Framework is within the context of marchin rights, it is important to remember that if a funding agency identifies “an unreported subject invention,” its rights under BayhDole are not limited to march-in rights. Instead, the funding agency has the option to take title to the patent claiming the Subject Invention, which would be extremely problematic for the Contractor, current assignee, or licensee.
Thus, the December 8th Draft Framework suggests that federal agencies may be more proactive in searching for opportunities to exercise their rights under Bayh-Dole moving forward, including march-in rights and the right to take title. To shield against the exercise of march-in rights, those that utilise federal funding to develop inventions should develop a strategy that would result in a fact pattern that would – according to the Draft Framework – weigh against the exercise of march-in. To shield against a potential loss of title to a funding agency, those that utilise federal funding to develop inventions should place a high emphasis on Bayh-Dole compliance, in particular on the timeliness of Subject Invention disclosure. Establishing strong internal protocols to formalise the disclosure of inventions will aid in these endeavors.
REFERENCES
1. 35 U.S.C. § 203

2. A proposed rule in 2021 (Federal Register vol. 86 No. 1, January 4), never adopted, stated that “March-in rights shall not be exercised exclusively on the business decisions of the contractor regarding the pricing of commercial goods and services arising from the practical application of the invention.” It did not preclude pricing from being considered as a factor in combination with other factors.

Rob Sahr is a shareholder in Wolf Greenfield's Biotechnology Practice. Rob develops strategies for life science companies to maximise exclusivity for therapeutic and diagnostic products. He assists clients with building global patent portfolios aligned with product life cycle, and advises clients on regulatory exclusivities, patent term extensions, Orange Book listings, and biosimilars. In addition to pharma and biotech companies, Rob works with universities, research institutions, and venture capital investors, providing patentability and freedom-to-operate assessments, noninfringement and in-validity opinions, performing IP diligence, and preparing agreements. Rob also assists federal grant recipients and contractors with Bayh-Dole compliance, including use of iEdison.

Curtis Powell is an associate in Wolf Greenfield's Biotechnology Practice. Curtis focuses his practice on patent prosecution and counseling clients in the biotechnology and life sciences industries. In addition to advising clients on patent portfolio development and management, Curtis works with clients on: patentability, freedom-to-operate, infringement, and validity opinions; patent landscape studies; Bayh-Dole compliance; and due diligence. He has also worked with clients on contested matters before the USPTO and various foreign jurisdictions. Curtis's clients include start-ups, mid-size companies, large companies, and universities.
14 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Rob Sahr
Curtis Powell


INTERNATIONAL PHARMACEUTICAL INDUSTRY 15 www.pci.com talkfuture@pci.com Your sterile drug product, our world. OUR END-TO-END BIOLOGIC SOLUTIONS INCLUDE: • Sterile Formulation & Lyophilization Cycle Development • Lyophilization and Sterile Fill-Finish Manufacturing • Aseptic Robotic Technologies
Analytical Support
Clinical & Commercial Labeling & Packaging
Refrigerated/Frozen Storage & Distribution Driving development and
of
and lyophilized drug
we are dedicated to your
in bringing
therapies to patients. Lyophilization and Sterile Manufacturing
•
•
•
connecting commercialization
sterile
products,
success
life-changing
Updating Your Regulatory Information Management Capability? It Could Pay to Get Hands-on
Letting business-savvy techies play around with a new RIM system ahead of the proposed implementation could save Life Sciences companies a fortune, says Romuald Braun, Managing Partner at MAIN5, warning against hasty vendor selection and contracting.
Everyone is time-poor now. So, when Life Sciences companies are looking to formalise or refresh their regulatory information management (RIM) capability, it is tempting to default to the leading brand. After all, if peer companies have already done the research to arrive at this choice, why reinvent the wheel?
Yet, a lack of rigour in the requirements definition and vendor selection process could invite considerable risk and additional cost, if something important has been overlooked. As the EU IDMP grace period gives way to a hard mandate for data standards compliance, and as digital transformation ambitions expand beyond the Regulatory remit to encompass Quality, Safety & Clinical processes, it’s more important than ever that companies do their research when approaching RIM vendor selection and contracting. An overly generic request for proposal (RFP), or ‘safe’ shortlist made up of what everyone else seems to be using, is a risky starting point.
The Growing Expectations for RIM
Deploying a formal, optimised system or platform for regulatory information management is a given now for all Life Sciences companies, irrespective of their size and focus. Regulators expect this and, as data rather than static documents evolves to become the default means of submitting, exchanging and maintaining regulated product and process information, it follows that the systems for managing and keeping track of everything must be sophisticated and reliable.
In the 2020s, a strong, modern RIM platform should equip a company to:
• Fulfil all of the differing and continuously evolving health authority requirements internationally.
• Effortlessly and reliably track the status of products, their licences and current marketing authorisation applications.
• Maintain a single, authoritative version of regulated product/process/licence truth that is accessible centrally and locally by the people who need it.
• Support future ambitions for process transformation, for instance beyond the scope of Regulatory Affairs – potentially encompassing adjacent functions such as Quality, Safety and Clinical operations – through integrated system capabilities and readily exchangeable data.
Although individual events such as a company merger or acquisition, or EU IDMP compliance, may trigger the decision to invest in a new RIM capability, it’s important not to progress this decision without considering the broader associated opportunity – e.g. to address internal data control challenges; transform internal visibility and decisionmaking; and ultimately re-imagine processes so that they are more efficient and support the business strategy more directly.
All these considerations should feed into the RIM selection process, which requires that all of the various business (as well as technical) stakeholders are consulted early on for feedback about their requirements and current process pain points. Thought should be given not only to what the company and its functional teams want and need to be able to do, but also to scenarios they wish to avoid in future. These might include:
• Incurring delays/business interruption and new cost as new features and capabilities have to be added to the system later, e.g. in response to a change or update to regulatory requirements in one or more markets.
• Future problems with system or data incompatibility if the company later tries to improve the connection and information flow with other departments or part of the global organisation; or following a business merger resulting in system consolidation.
• Issues arising from a change to the software vendor’s circumstances, ownership or strategic focus (e.g. what will
happen to your data, and how you’ll extract it/get it back).
Assessing the Available Options, Using a Formal Structure
A structured, holistic approach to vendor selection is the best way to ensure that nothing is left to chance in the choice of a new system – from its long-term strategic fit, and lasting deliverability for all target users, to the fulfilment of procurement requirements around cost/value for money, sustainability and so on.
Taking a structured approach (e.g. applying an agreed vendor selection methodology) will make it possible to score each option/each long-listed vendor across the full range of criteria, in a meaningful and comparable way.
This should span:
• Expressed user requirements and priorities.
• The strategic/wider digital transformation roadmap perspective (e.g. a move away from isolated best-of-breed solutions towards a unified platform approach to application rollout and data sharing); and
• Any technical/IT considerations, such as system architecture specifications (e.g. cloud-hosted or cloud-ready, and compatibility/integration potential with adjacent legacy systems).
Raising Awareness, Fostering Inclusion
While no single solution will tick every box, following this formalised vendor selection approach will ensure that each RIM proposition and supplier is considered from every angle. This means that any tradeoffs (e.g. in specific user features that may be sacrificed for a more holistic platform serving multiple departments) can be duly considered, communicated properly and agreed pragmatically – an essential pillar of effective change management.
Starting with a standardised approach (to establishing user requirements, for instance) is a great way to get teams thinking about what is most important. That could be across a series of common or desired use
16 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Regulatory & Marketplace
Regulatory & Marketplace

cases. In a Regulatory context, these might include variations management in marketing authorisation; IDMP-specific processes; and investigation of new drugs in key target markets.
Thinking through each scenario will help focus teams on what they need from a new system – before they make their choice.
Tailored Demos & Hands-on Experimentation
There’s no substitute for seeing a new system in action. Ideally, this should happen via a bespoke demo applied to routine use cases and familiar scenarios – better still, using the company’s own data (e.g. via a demo ‘sandbox’ environment).
It is in the vendor’s interest for target users to understand the system’s potential in their own setting and routine context. Requesting and securing this will enable a more confident and better-informed decision. If the demo can also reflect processes as they might evolve in the future (e.g. as Regulatory is blended more seamlessly with Quality and Safety, if this is a strategic aim), so much the better.
A good pain-saving tip is to let businesssavvy technical enthusiasts within the company play around with capabilities to see what they can do, something that is easily provided for today via the cloud. This active user involvement is much more illuminating than watching a pre-set vendor demo and will drive home an understanding of what’s really important when specifying and choosing a new platform.
Payback: The Benefits of Early Scrutiny Investing the time in a proper needs
assessment is a powerful way to get everyone on board and manage expectations, which in turn will cement user acceptance and ease change management. The risk otherwise is being saddled with a solution that doesn’t fit the bill and which no one uses (a failed rollout), or which incurs a six/seven-figure cost and lengthy additional timescale to put right.
Or it could be that striving to fulfil all the immediate user requirements results in a fully featured, single-application system, yet curbs the potential for cross-organisational process transformation. From ordering regulatory information to catering for broader Quality, Safety and Clinical processes in the future, it’s important to look five-ten years ahead when assessing a RIM vendor and solution. Otherwise, it could take two years to implement the initial solution, only to find it needs replacing again a further two years down the line.
A further aspect of a solid vendor selection process is to perform supplier due diligence/background checks – looking into their financial stability, their existing client base and customer satisfaction ratings, and their longer-term product roadmap, for instance.
Performing all of this work up front is not only a good risk management strategy; it will also provide solid justification for the vendor/system decision if unforeseen issues arise later.
Contracting for Success
Using a formal methodology in vendor scoring and selection can help in the negotiation and contracting process too,
by drawing attention to what is essential versus less important. It’s also a means of ensuring that additional considerations (e.g. data preparations/migration, and issue resolution, and potential future data transition requirements) are factored into the contract and pricing.
Remember that data-related work is rarely a one-time event, so it will be important to specify, scope and assign ongoing processes – and relative accountability – around data (data governance).
Indeed, the success of any project requires that internal teams also allocate sufficient time and resources to its effective delivery. A good contract should protect the interest of both parties.
Be prepared – and pragmatic.
A final, but important, point in assessing potential suppliers is to consider what they will be like to work with on a day-to-day basis. Every company has its own culture, set of beliefs, and way of working, and a good match will help ensure a harmonious relationship and optimum outcome.
Not everything can be controlled, but being precise about what’s needed, and well prepared for every eventuality, can go a long way in ensuring a project’s successful delivery.

Romuald Braun
Romuald Braun is Managing Partner at MAIN5, a European consulting firm specialising in organisational and digitally enabled change in Life Sciences. MAIN5’s consultants are deeply experienced in vetting and implementing RIM and other critical Life Sciences platforms and systems for biopharma and medical device companies around the world. Its BPMN 2.0-based methodology for vendor selection fulfils 85–95% of most companies’ requirements when assessing a RIM or other platform vendor. MAIN5 also provides a range of services from supplier contract development to change management.
Email: romuald.braun@main5.de
INTERNATIONAL PHARMACEUTICAL INDUSTRY 17 www.international-pharma.com
The Delivery of Pharmaceutical Drugs and How it Will Change the Future
Pharmaceutical drugs have seen a seismic shift over the past ten years, moving away from chemically synthesised drugs to focus more on a new class of biological drugs (biologics).
Biologics have the potential to revolutionise the treatment of many common conditions that significantly affect millions of lives, such as rheumatoid arthritis, diabetes, autoimmune diseases and certain cancers. Nearly 50% of the new FDAapproved drugs have been biologics, showing the size of the shift.
Yet, biologics are, by nature, more fragile. In order to facilitate the mass adoption of these new biological drugs, the industry needs to find safe, effective and cost-efficient ways to deliver them.
Challenges with Delivering Biological Drugs
The previous generation of chemically synthesised drugs is pretty much one-sizefits-all. Drugs like paracetamol work for the majority of people in safe doses, they come in stable pill form, and it is difficult to accidentally overdose. Overall, they are safe to self-administer, relatively effective and cheap to produce, transport and sell.
Biological drugs, on the other hand, are very different in terms of properties and formulations. They are much more complicated, specific and targeted, meaning greater efficacy and fewer side effects. Yet, this also means they tend to be more vulnerable and more potent. A large set of biological drugs are delivered through the parenteral route in the form of injections. However, this presents issues such as a risk of infection, needle stick injuries and adherence. Major adversity includes vascular damage, injury to the nerve, tissue necrosis and muscle fibrosis.
Most biological drugs need to be stored in refrigerated units at exact temperature ranges to keep them stable. They also need to be administered in very specific doses, tailored to the individual and usually injected intravenously. As such,
they tend to be stored, handled and administered in hospitals, health centres and GP surgeries, complicating the delivery of these medications. Not only does this make it much more expensive to deliver these biological drugs but it also makes it harder to transport and store, especially in countries without reliable refrigeration or power, making the global adoption of these drugs much more difficult.
The complex nature of biologics structure presents the formulation scientist with a unique set of challenges. Biologics can be easily altered during the formulation process, storage, environmental conditions, and administration causing denaturation and degradation, resulting in the loss of some or all of the therapeutic efficacy. As such, these challenges lead to complex manufacturing processes and any deviation can significantly impact the potential adverse effects associated with the use of these biologics.
Delivery of biological drugs via the nonparenteral route will circumvent some of the major challenges associated with synthesis, storage and delivery. Any non-parenteral route is likely to address a significant subset of current challenges – it could improve patient compliance through selfadministration, reduce the cost of treatment if delivered in a non-specialist facility and/
or at home, improve storage conditions and eliminate the need to freeze or refrigerate the formulation. However, delivery of biologicals through a non-parenteral route requires careful consideration of drug properties, manufacturing methods and route of delivery.
For instance, when biologics are administered non-invasively, the absorption of biologics through the gastrointestinal tract mucosal membranes is hindered by their size, hydrophilicity, and susceptibility to enzymatic degradation, therefore presenting further challenges to effective drug delivery.
Additionally, the physiological barriers, such as the requirement to cross multicellular barriers to reach blood circulation, harsh pH environment and enzymatic activity, present significant formulation challenges for nonparenteral routes of delivery. It should be kept in mind that the choice of excipients in the formulation development of biologics requires careful selection and optimisation such that there is no interaction. For instance, the use of surfactants may impact the structure of the biological drug which, in turn, will compromise the stability and activity. Similarly, the process and method of manufacture should avoid/minimise the use of solvents, heat and multistep processing, to prevent any deleterious impact on the integrity and functionality of the biological molecule.
18 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Drug Discovery, Development & Delivery
Drug Discovery, Development & Delivery

These key challenges are holding the pharmaceutical industry back. Biological drugs are often far more effective, have fewer unwanted side effects and can be used to manage or cure a wide range of otherwise life-threatening illnesses.
Therefore, developing effective noninvasive formulation strategies is critical for overcoming biologic-related challenges, ensuring safe, patient-friendly, and efficient manufacturing processes. If we can solve the challenge of formulation, storage, transporting and delivering new biological drugs in a safe, effective and cost-effective way, we will unlock a new and exciting future for the medical and pharmaceutical world.
Exciting Advancements in Drug Delivery
There are two particularly promising

advancements currently being explored that could revolutionise biological drug delivery: liquid-based delivery and inhalation.
Liquid-based Delivery Systems
One common way to deliver synthetic drugs is via the gut (the buccal system). Over the years, the pharmaceutical industry has developed reliable ways to transport drugs to the gut without being damaged by stomach acid. The main difficulty with biological drugs, however, is that they tend to be much larger compounds, which are harder to absorb in the gut.
Drug delivery via the buccal route refers to a drug formulation that is placed in the mouth/oral cavity for local or systemic effects. The buccal route offers many advantages such as high compliance, painless administration, easily removable in case of irritation, bypassing the degradation in the liver (firstpass metabolism) and significant changes in pH and composition of the digestive enzymes in the gut.
One approach (being developed by Max Bio+) is to use a unique combination of polymers and lipids to create a liquidbased delivery system for biological drugs. This composite system creates nanostructures that hold and disperse biologics in an aqueous solution, such as water. The result is the creation of nanoparticulates that can permeate across multiple cell layers into the bloodstream via the buccal route (gut).
What’s more, this unique combination of polymers and lipids has been found to create a synergistic effect with biological drugs, increasing their potency.
The application of this approach could be incredibly broad, offering a stable oral liquid form of important drugs, such as insulin. So, rather than diabetics needing to inject insulin several times a day, they could simply drink a shot of insulin when required.
Other applications could include drugs that are otherwise insoluble in water, such as CBD which binds to fats and is often found as oils. Instead of requiring often unpalatable emulsifiers to mix these compounds into other water-based liquids, they can be added at higher concentrations without sacrificing flavour. The result could be more potent CBD-based drinks and foods, including alcoholic beverages.
Inhalation Delivery Systems
Another particularly interesting area of

INTERNATIONAL PHARMACEUTICAL INDUSTRY 19 www.international-pharma.com
Drug Discovery, Development & Delivery
biological drug delivery is inhalation systems. Inhalers for conditions like asthma have been around for years. However, formulating drugs for inhalers typically requires a lot of energy and, therefore, heat as well as solvents. While this isn’t a problem for most synthetic drugs, biological drugs are much more sensitive to heat and solvents. As a result, traditional inhalation manufacturing methods don’t lend themselves to formulating biological drugs.
Additionally, the majority of inhalation drugs are low-dose compounds, making it easier to reliably measure and deliver the correct dose into the lungs. For more potent or high-dose drugs, it can be a challenge to ensure that the right dose is delivered and, importantly, absorbed in the lungs. If the molecule is too light, it will simply be exhaled, if it’s too heavy it might not be absorbed.
An interesting approach being developed by Aston Particle Technologies uses isothermal dry particle coating technology to blend potent and high-dose biologics into inhalation formulas at ambient temperatures. No additional heat and no solvents are required, making it the ideal candidate for delivering biologics via inhalers.
The approach is also simpler than other methods of formulating high-dose drugs as it doesn’t require a complex multistage process, helping accelerate drug development and route to market timelines

while reducing costs. The fact that it doesn’t use excessive energy or solvents means it is also more environmentally friendly.
The Future of Drug Delivery
We are on the cusp of a transformative wave of biological drugs that will improve lives throughout the world. Some of these drugs, such as insulin, have already made huge differences in the quality and length of life for millions of people. The advancement of these types of drugs could lead to the easy and effective management of a wide range of illnesses.
By advancing the delivery of these biological drugs, we can make it safer, easier and cheaper to transport, handle

and store these drugs. Rather than going to a hospital or requiring daily injections, patients will be able to drink, eat, or inhale their medication.
In the wake of the COVID pandemic, we have understood how important it is to find quicker and easier ways to vaccinate large groups of people. As biologics, vaccines can benefit from these new delivery systems, allowing rapid distribution of vaccines as liquids or inhalers, for example.
It is exciting to think of how biologics will transform people’s lives over the next decade and beyond, with the potential to cure or manage debilitating and lifethreatening illnesses. The key to this transformation, however, lies in delivery. Safe, effective, cost-efficient delivery will allow everyone to participate in this medical revolution, helping end suffering across the world. That is the future we should all be working towards.

Afzal R. Mohammed
Professor Afzal R. Mohammed is Professor of Pharmaceutics and advisor to Max Bio+. Max Bio+ was founded in 2020 by Professor Sunil Shah, Consultant Ophthalmologist, Professor at Aston University, and philanthropist Sean Ngu. Max Bio+ has created revolutionary patented technology to solve the perennial problem of how to mix oil and water, specifically for drug delivery.
Web: www.maxbiology.com
20 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1

Application Note

Exploring Device Interchangeability for Drug Delivery
Alex Fong, Head of Insight, Owen Mumford Pharmaceutical Services
Devices as Differentiators
Choosing the right injectable drug delivery device can be a crucial differentiator for pharmaceutical manufacturers – particularly as the market becomes increasingly crowded with biosimilars. But any decision on switching to a different device needs to be underpinned by firm evidence on the likely reaction from patients. After all, any drug is only as good as the patient’s adherence to their treatment regimen.
If a patient struggles to make the switch from a familiar auto-injector to an innovative new one, there is a danger they will be less inclined to use the device and therefore miss out on vital doses of medication. This issue is particularly important now that many treatments for chronic conditions are self-administered by patients at home, often without the support of a healthcare professional. Ease of use encourages patient adherence and therefore benefits the patient, the healthcare system – and pharmaceutical companies.
Device Interchangeability
Alongside the trend for home selfadministration is the growth of biosimilars, as an increasing number of patents for biological medicines are now expiring. In the UK, the NHS expects to save up to £300 million a year by making more biosimilar alternatives available.1 Meanwhile, in the US, savings of $38.4 billion have been predicted for 2021–2025, compared with 2020, as the wider availability of biosimilar products creates a significantly more competitive market.2
Of course, as well as the cost savings of switching from originators to biosimilars, there also needs to be consideration of the most suitable device for administering the new drugs. To support the choice of this device, pharmaceutical companies should have access to data from human factors studies and any other user testing. This data can then support regulatory applications for determining device interchangeability.
The FDA requires a regulatory application to provide evidence that the impact of switching between delivery devices has been assessed, stating: “Data and information supporting the appropriate use and performance testing of the delivery device constituent part of the proposed interchangeable product should be submitted.”3 In Europe, the European Medicines Agency allows differences in the administration device, as long as there is no impact on safety and efficacy.
INTERCHANGEABILITY STUDY
Study Objectives
To provide pharmaceutical companies with evidence for making delivery device decisions for their drugs, there is a need for more studies on the ease of switching devices. This prompted Owen Mumford Pharmaceutical Services (OMPS) to commission an inde-pendent study to evaluate the ease with which patients were able to switch between two different autoinjectors.
Auto-injectors are routinely used to deliver biologics suitable for subcutaneous administration. They are convenient for patients and allow them to administer medication in their own homes without requiring the presence of a healthcare professional. The OMPS study aimed to discover whether regular users of a marketleading three-step auto-injector were able to switch to a new two-step auto-injector and successfully perform injections.
Study Design
The three-step auto-injector used in the study was SHL Medical’s button-activated DAI® device – one of the first auto-injectors to be commercialised for home injection (4). The two-step auto-injector was the springpowered Aidaptus® device from OMPS (5), which uses pressure to activate the injection – rather than having to push a button, the patient simply presses the device onto the injection site.
The study involved a total of 52 tests, with 26 participants in the UK and a further 26 in the US, all of whom had been using the DAI auto-injector for at least three months. The 34 women and 18 men were
aged between 16 and 75, with an average age of 51. The study followed the FDA guidance document: “Considerations in demonstrating interchangeability with a reference product.”6
No training, demonstration or coaching was provided to help the participants use the Aidaptus device – they were simply given unopened original boxes containing the device and its instructions for use (IFU). They were tasked with administering injections into an injection pad on a table, with facilitators briefed to intervene only if there was a risk to a participant. The participants were asked to carry out four injections in total – starting with the DAI auto-injector and then alternating with the Aidaptus.
The study was not designed to compare the two devices, as all the participants were already familiar with the DAI auto-injector, but to assess the risk of switching between the two. The aim was to discover how easily the participants could learn to use the new Aidaptus auto-injector and test their ability to switch from a familiar device to a new one – and successfully complete injections with the new auto-injector.
The Aidaptus device was filled with the same volume of injection solution as DAI, and the injection time was similar, replicating a comparable injection experience. Injection time was calculated from when a participant placed the auto-injector on the injection pad and started the injection process through to when the device was removed from the pad.
Measures Used
The study’s primary measure was injection success. An injection was classed as successful if the participant correctly delivered the injection into the pad, as described by the IFU, and allowed the contents of the autoinjector to be fully delivered into the injection pad before removing it.
Secondary measures included injection time (measured as described above), whether participants examined or read the IFU and their confidence in performing the injections.
22 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Application Note
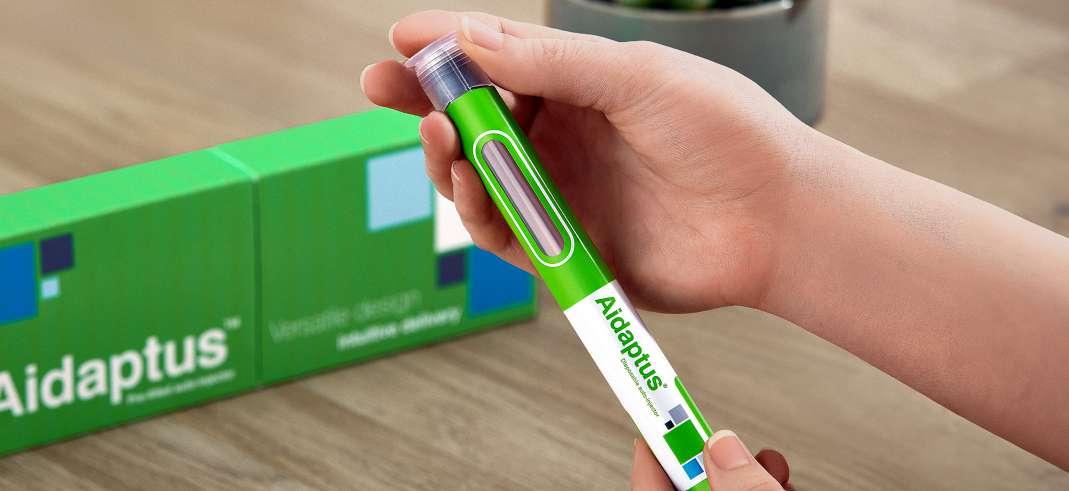
Videos of participants performing the tests were analysed to calculate the study’s secondary measures. As well as injection time, watching how participants handled the new device also gave clues as to ease of use.
STUDY RESULTS
Injection time
Each injection in the alternating sequence was completed successfully by all participants – giving a 100% success rate for both devices in the user tests. Even though they were all familiar with the DAI device, participants took longer to carry out the initial injection with the three-step DAI auto-injector than the first of the two-step Aidaptus auto-injector injections – 12.6 seconds compared to 11.4 seconds for Aidaptus. But for the second series of injections, the mean times were much closer to the expected times and were similar for both devices – at 8.8 seconds for DAI and 7.9 seconds for Aidaptus.
Demographic Differences
US study participants were, on average, 4.6 seconds faster than UK participants when it came to delivering the first Aidaptus injection, although the times were similar for the second injection. The female participants took, on average, 30% longer than the men to carry out the Aidaptus injections. The difference between the sexes for the DAI injections, however, was just one second for both injections. Injection times for left-handed participants were roughly the same for both auto-injectors. The over-40 age group took marginally longer to deliver the injections with both devices but still completed them successfully.
Device & IFU Examination
Overall, the study found that most participants only needed to examine a device once to deliver injections successfully. A higher percentage of US participants (92%) than UK participants (85%) examined the device and IFU before their first Aidaptus injection. When it came to the second Aidaptus injection, 73% of UK participants did not examine the device of the IFU beforehand, compared with 54% of US participants.
More men than women examined the device and/or IFU before the first injections using both devices – but more women than men examined them before the second injections. However, the success of the injections – and the ability to switch between the two devices – was not affected by gender. The percentage of participants who looked at the device and/or IFU beforehand across all four injections increased with age.
De-risking Device Selection
The switching of drug delivery devices needs to maintain – and ideally improve – the patient experience and have a limited effect on patient behaviour. Multiple factors are likely to be involved in the switching process, so more user studies are required for a thorough understanding of the issue. But the results of the OMPS user tests demonstrated that all the study participants were able to switch devices successfully – effectively derisking the choice of a two-step auto-injector rather than a three-step device.
REFERENCES
1. NHS England Website. Medicines: improving outcomes and value; Biosimilar medicines.
Available from:
2. Mulcahy A, Buttorff C, Finegold K, et al. Projected US savings from biosimilars, 2021 –2025. Am J Manag Care. 2022;28:329–335.
3. FDA Guidance Document: Considerations in demonstrating interchangeability with a reference product. Published May 2019. Available from: https://www.fda.gov/regulatoryinformation/search-fda-guidance-documents/ considerations-demonstrating-interchange ability-reference-product-guidance-industry
4. SHL Medical Website. DAI® product information. Available from: https://www.shl-medical.com/ products-and-services/dai-auto-injector/
5. Owen Mumford Website. Aidaptus®® product information. Available from: https://www. ompharmaservices.com/products/Aidaptus®auto-injector/
6. FDA Guidance Document: Considerations in demonstrating interchangeability with a reference product. Published May 2019. Available from: https://www.fda.gov/regulatoryinformation/search-fda-guidance-documents/ considerations-demonstrating-interchange ability-reference-product-guidance-industry

Alex Fong MBA is an experienced senior manager in the Insight, Analytics and Strategy fields. He has applied these skills in a broad range of Industries including the FMCG/CPG, retail, telecoms and mana-gement consulting sectors. Alex has worked and lived in several international markets throughout his career, including Hong Kong, USA, South Africa and France.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 23 www.international-pharma.com
Alex Fong
Big Data, Personalised Medicine and Support for Healthcare Professionals: What Will Drive Pharma in 2024?
In 2023, precision targeting for personalised medicines, improving the information flow between reps and healthcare professionals, and advancements in clinical trials have driven the adoption of new technologies, but we are only at the beginning. With these advancements come more valuable data, and the potential to make better-informed decisions. Using better connected data and processes will help reduce friction to speed up the delivery of treatments to the right patients, at the right time, as well as reducing cost.
As we progress through 2024, we are set to see all these areas continue to mature and evolve. Pharma and healthcare providers who can successfully embrace data, analytics, and digital platforms will be the best positioned to succeed in 2024 and beyond.
Development Teams will Rethink Big Data to Support Personalised Medicine
The life sciences industry has been waiting a long time for big data1 to transform the commercial viability of personalised medicine. With automation now coming of age, R&D teams can finally seize the opportunity – as long as their data is also clean, standardised, interoperable, and secure.
In 2024, companies will focus on making big data (which could range from raw trial and site-specific data to IT data points, such as cycle times) more usable, by resolving common pain points around cleaning, ownership, and standards. As a result, the volume and frequency of access to study data will increase exponentially. This will require a transparent data model with stringent user access controls to address data privacy and cyber-security concerns.
Leading companies will use automation to make hundreds of marginal and incremental efficiency gains across the development lifecycle, whether deep querying protocols, detecting patterns during medical imaging analysis, or verifying the origin of
chemical components. A growing industry momentum will lead to more direct data APIs between sponsors, health institutes, and regulatory authorities2 so that “big (clean) data” becomes a reality, creating the right conditions for commercially viable, personalised medicines to reach patients in need.
As Digital Therapeutics Evolve, Top Device Manufacturers Will Stake Their Claim in The Space
When Pear Therapeutics became the first company to receive payer funding for a mobile app to treat substance use disorder in 2021, it seemed like the industry was gearing up for digital therapeutics to go mainstream.3
However, a lot has changed in two years. The digital health market has suffered some losses that have cast a long shadow on the budding industry, with some companies filing for bankruptcy and other leaving the payer market to go direct-to-consumer.
Despite these setbacks, digital therapeutics will continue to gain traction in 2024 – especially for larger companies that have the resources to learn lessons from these early trailblazers in the space.
In the coming years, we expect to see top companies testing out varied commercial strategies for digital therapeutics, including going direct-to-consumer or developing companion apps to previously established therapies. Heavy “hitters” like Pfizer, AstraZeneca, Boehringer Ingelheim, and Roche have already joined the Digital Therapeutics Alliance,4 and more will likely join the FDA’s5 newly formed Digital Health Advisory Committee.
Specialised Medicines Will Push Biopharmaceutical Companies to Respond to Healthcare Professionals in Time of Need
In-person interactions with healthcare professionals (HCPs) are returning to pre-pandemic levels, but access is more selective. HCPs are prioritising access based on the value and timeliness of interaction. This is driving the inevitable new question: is the traditional push model supporting HCPs in their time of need?6
The complexity and precision of specialised medicines are putting increased pressure on biopharmaceutical companies to shift to a service-focused model. HCPs are having to keep track of a bewildering array of treatments and inconsistent levels of access to those based on treatment and reimbursement guidelines.
A deeper collaboration between commercial and medical teams will be critical to provide HCPs the on-demand education and resources in their time of need – even at the point of care. As the focus changes from promotional to servicecentric, field teams will augment in-person touchpoints with inbound capabilities that connect HCPs and reps in real time and prioritise responsiveness and customer value over quantitative sales and frequency measures.
Early adopters of this model are creating more than double the amount of digital interactions7 with HCPs without reducing in-person visits. Success requires executive sponsorship, investments in field training, compliant technology, and realignment of incentives to focus on the strength of relationships.
Most importantly, it requires that every HCP interaction builds off the last and adds value to the HCP in a timely manner that entails a fusion of in-person, digital, and ondemand interactions. In fact, the use of video meetings in conjunction with in-person meetings increases efficiency and impact. When engaging HCPs through a blend of in-person and virtual channels, the video engagement has three times the promotional response compared to in-person meetings alone, offering a significant advantage. We also see that when leveraging an additional inbound channel like compliant chat, HCPs reach out to the industry — initiating 30% of conversations8 with reps looking for information.
Advanced Analytics for Clinical and Medical Data Will Become a Key Differentiator of Speed to Market
Commercial teams have been using advanced analytics for years to measure commercial impact. In R&D, sponsors employ
24 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Drug Discovery, Development & Delivery
armies of data scientists to analyse data, but it is still early days for clinical and medical teams when it comes to using analytics to select suitable trial sites or to drive patient enrolment. In 2024, things are more likely to change as competition and external pressures, like the Inflation Reduction Act,9 will drive an increased need for speed and agility in R&D.
Clinical and medical teams will leverage advanced analytics to help drive site selection, support patient enrolment, and identify gaps in the standards of care. In doing so, they will have a greater impact on how companies go to market, rather than commercial.
Companies that can define the marketplace for their products, identify impacted patients, associate healthcare providers, and move faster than the competition will win. Clean, connected data with a common architecture across customers, patients, KOLs, and business operations will become a fundamental necessity to not only prove products efficiently and effectively, but also to lay the foundations for their future utilisation.

As we venture into 2024, the healthcare landscape is poised for transformative shifts. The convergence of technology, personalised medicine, and data analytics will redefine patient care, making it more tailored, efficient, and responsive than ever. By embracing these advancements, healthcare providers and pharmaceutical companies will be standing at the forefront of innovation, ready to address evolving challenges and improve outcomes. The key to success lies in not only acknowledging these trends, but actively participating in their evolution – nurturing a future when healthcare is no longer a mere service, but a dynamic and beneficial experience for every individual.
REFERENCES
1. How Big Data Will Lower Costs and Advance
Personalised Medicine (genengnews.com)
2. EU CTR challenges sponsors and CROs to get their house in order
3. New Insights, New Efficiencies: Veeva’s Top Life Sciences Predictions for 2022
4. https://dtxalliance.org/engage/dta-members/
5. FDA Digital Health Advisory Committee
6. 3 Ways Biopharmas Are Using Field Insights to Better Engage HCPs
7. 3 Ways Biopharmas Are Using Field Insights to Better Engage HCPs
8. 3 Ways Biopharmas Are Using Field Insights to Better Engage HCPs
9. Navigating the Inflation Reduction Act’s Impact on Drug Pricing and Innovation

Chris Moore
Chris Moore has been European President at Veeva Systems for the past six years, and he is responsible for developing the organisation's European business. Chris is an experienced life science professional and is leading the delivery of Veeva's Industry Cloud.

HIGH PURITY. HIGH DEMANDS.















Spring 2024 Volume 16 Issue 1 Drug
Development
Delivery
Discovery,
&
Pressure regulators Control valves Pipeline ancillaries Special equipment Steam traps Safety valves
purity
for
steam geral@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467, Guia PBL PORTUGAL PRODUCTS MANUFACTURED IN PORTUGAL AV 001 AP EN 02.20
High
equipment
clean
Novel Drugs: Challenging Entrenched Prescriber/ Investment Behaviour is not Just About Education
Behavioural science’s time has come in the pharma industry, as medical communications adapt to a more ambitious and diverse treatment landscape. Drawing on examples from across public health and life sciences, Alpharmaxim’s founder, William Hind, brings to life the science of behavioural change and how its strategic, structured application within medical communications could help deliver important new drugs into the hands of the patients who need them.
Behavioural science, long proven in influencing consumer behaviour (e.g. nudging the public to make greener or healthier choices), is rising swiftly up the pharma agenda – and for good reason.
Novel therapies, often with expensive price tags and more targeted patient populations, require strategic new positioning if they are to have a maximum positive impact on patients. Whether to challenge established prescribing practices, or to effectively get across a therapy’s value to health authorities and investors, drug developers and licence holders must adapt to the increased sophistication now needed in their communications strategies.
This isn’t simply a case of educating the market about the new product’s benefits, however. There are other factors that keep decision-makers coming back to habitual choices. It is here that behavioural science comes in, and in particular, the need for methodology when identifying how to use omnichannel communications to maximum impact.
What is Behavioural Science?
Behavioural science draws on psychological theory and the social sciences to understand why individuals follow or resist certain behaviours.
In a public health or ‘responsible citizen’ context, behavioural science has been used successfully across a range of highprofile cases. These include encouraging people to follow COVID-19 guidelines,1 or
adapt behaviour in line with climate change recommendations,2 for example by reducing air travel or meat intake. Behavioural science has also been used to encourage vaccine uptake,3 and motivate people to make healthier lifestyle choices, such as increasing physical exercise, reducing obesity and driving up the number of people who stop smoking.4
The Opportunity in Pharma
In Life Sciences, behavioural science has a powerful role to play in medical communications – specifically in overcoming barriers to changing prescribing behaviour.
This is important so that healthcare providers (HCPs) don’t automatically default to their habitual choices of medical or treatments but become more open to emerging options which may improve patient outcomes. So much so that the UK’s Medical Research Council (MRC) is now advocating the use of behavioural science in the design of interventions such as marketing campaigns, to ensure that important new biopharma innovation fulfils its potential for patients.
Evidence of Inertia & Other Barriers to New Treatment Pathways
While it’s possible to make an educated guess about HCPs’ reasons for falling back on tried and tested treatment choices (including officially recommended first-line treatments, budget restrictions, and/or a lack of knowledge of the emerging options), the reality is usually more complex.
The established COM-B model for behavioural science sets out 93 different techniques and how they can be successfully combined to address barriers to change, based on the relative roles of Capability, Opportunity, and Motivation (M) as determinants of current behaviour. (Examples follow below.)
As HCPs strive towards their claimed goals of increased patient centricity, and place more emphasis on the patient experience as well as quality of life, understanding and proactively overcoming a given combination of barriers to behavioural change in a very targeted way among prescribers, or drug
funders, will be important if patients are to gain access to the best options now available to them.
Indeed, detailed research is currently being carried out into why the latest ostensibly transformational treatments for Parkinson’s disease aren’t making headway, despite early promise. Once published, later this spring, the results are likely to further support the case for more sophisticated medical communications campaigns designed to challenge HCP preconceptions.
A Knowledge Gap is Rarely the Only Issue
Pharma companies tend to excel at educating funders and HCPs about new assets and their mechanism of action (MOA); the specifics around their efficacy and the improvements they will bring; as well as the target disease and the unmet medical need. Yet this activity only represents one-third (the Capability element) of the combined factors that may affect a change in behaviour.
It may be that the HCPs involved don’t have access to the right resources to see through a new treatment plan. Or perhaps prescribers are not being encouraged to try something new by their peers, or by the system they are in, are hampered by a lack of time or a failure to see the value of the new path.
When it comes to rare diseases, by their nature these are seldom seen by physicians’ day to day. Treatments might be perceived to have some symptomatic relief but nothing to impact on the disease progression; or patients or their HCPs may fail to act on the diagnosis out of a lack of belief that there is anything positive that can be done. Lack of motivation or opportunity can be as significant a barrier as lack of capability (e.g. awareness or education), so campaigns should address those factors too, rather than simply inform relevant stakeholders about the new drug.
In other cases, HCPs and patients may have accepted as inevitable what many people would consider a gruelling regime if it is keeping the individual alive. A new therapy might be seen to disrupt the equilibrium
26 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Drug Discovery, Development & Delivery
Drug Discovery, Development & Delivery
that had been achieved, even where there is a promise of an improved quality of life. In at least one case, further probing revealed that patients’ parents were the main source of resistance.
Confidence Issues: Negative Perceptions & Neurodegenerative Disease Treatments
In common with other neurodegenerative diseases such as Alzheimer’s, Parkinson’s has been the subject of important novel treatments which have been heralded with great fanfare but have not delivered for patients. After a series of failed launches, it would seem that no amount of new education can substantially tackle the sense of poor motivation, where confidence has been eroded. A more involved approach will be needed.
Distilling the particular combination of ‘blockers’ to behaviour change is the first step in designing an effective medical communications campaign. Ideally this needs to be guided by a structure, framework or methodology, to ensure that campaigns are evidence rather than assumption based and have maximum impact across the target blend of channels.
Knowing How and When to ‘Nudge’ Your Target Behaviour change frameworks contain a set of instructions to guide the development of
a behaviour change strategy, in this case a medical communications campaign.
Drawing on the COM-B model – which differentiates Capability, Opportunity and Motivation as drivers of Behaviour –medical communications teams can start to determine what kind of campaigns might work best to drive change.
In a public health context, COVID-19 required a series of new behaviours, starting with fastidious handwashing, face-covering and keeping a distance from others. Getting the public on board required improving their knowledge of what to do and how (Capability); ensuring they had access to soap/hand gel or face coverings (Opportunity); developing a clear plan of when they should do these things and align this with why they needed to do it or what they would get from it (Motivation - e.g. reduce the risk of negative outcomes/protect themselves and others).
At Amsterdam’s Schiphol Airport more than two decades ago, repeat issues with the cleanliness of the men’s toilets prompted alternative thinking. Putting up signs did nothing to drive ‘a better aim’, but when the creative team designed a realistic ‘fly’ into the ceramic of the urinals, suddenly customers’ ‘targeting’ became more accurate. The change was possible; it was a case of finding the right motivation. The impact was
striking, too, reportedly an 80% reduction in urinal spillage and an 8% reduction in total WC cleaning costs at the airport.
How a Framework Can Help
Behaviour change frameworks encourage those developing communication strategies to be specific about the changes in behaviour that are needed (e.g. losing weight is not a specific behaviour, whereas reducing highcalorie food intake or walking X number of steps is). With strong, evidence-based insights into the changes that are needed, and the specific barriers that need to be overcome, it becomes easier to identify the right type of message and content, as well as the most effective combination of modes of delivery – and when these should be targeted at a population vs more individual level, to maximise the omnichannel opportunity.
REFERENCES
1. Engagement with protective behaviours in the UK during the COVID-19 pandemic: a series of cross-sectional surveys (the COVID-19 rapid survey of adherence to interventions and responses [CORSAIR] study), National Library of Medicine, March 2022 https://pubmed.ncbi. nlm.nih.gov/35272652/
2. The implications of behavioural science for effective climate policy (CAST), Climate Change Committee, September 2023: https://www. theccc.org.uk/publication/the-implicationsof-behavioural-science-for-effective-climatepolicy-cast/
3. The London Borough of Havering: Using the COM-B framework to develop a vaccine take up strategy, Local Government Association, April 2021: https://www.local.gov.uk/case-studies/ london-borough-havering-using-com-bframework-develop-vaccine-take-strategy
4. Tackling inequalities in Coventry by supporting smokers to quit, Local Government Association, May 2023: https://www.local.gov.uk/casestudies/tackling-inequalities-coventrysupporting-smokers-quit
 William Hind
William Hind
William Hind founded Alpharmaxim in 2001. Alpharmaxim helps cross-functional biopharma teams communicate effectively with clinicians about new medicines or alternative treatment regimens and their potential to improve the patient experience, supported by relevant scientific evidence.
Email: william.hind@alpharmaxim.com
INTERNATIONAL PHARMACEUTICAL INDUSTRY 27 www.international-pharma.com

Transforming the Biologics Product Lifecycle with Inhalation Innovation
In recent years, the biopharmaceuticals landscape has witnessed a revolutionary shift with the emergence of new nasal inhalation technologies. Among these, soft mist nasal sprays have emerged as a promising avenue, particularly for the delivery of biologic drug products. Such nasal drug delivery innovations hold promise in transforming the way biologic drug products are administered and managed throughout their lifecycle.
In this article, Nicolas Buchmann, CTO, and Frank Verhoeven, Business Developer at Resyca (a joint venture between Bespak and Medspray), delve into the potential transformative impact of inhalation innovations on the biopharmaceutical product lifecycle.
A Dynamic and Growing Inhalation Market
The global market for inhalation drug delivery devices is experiencing rapid expansion, poised to reach a value of $20.7 billion by 2031, growing at a compound annual growth rate (CAGR) of 4.4%.1 This growth trajectory is propelled by several key drivers, notably the escalating prevalence of chronic lung conditions worldwide and the need for more efficient and patient-friendly drug delivery methods.
Chronic respiratory ailments such as asthma, cystic fibrosis, pulmonary arterial hypertension (PAH) and chronic obstructive pulmonary disease (COPD) have increasingly been diagnosed globally. In 2019, 262 million individuals were living with asthma worldwide, with approximately 1,000 asthmarelated fatalities occurring daily, many of which are preventable.2 Additionally, PAH affects up to 70 million individuals globally, accounting for nearly 1% of the global population, a figure expected to rise as the population grows older and larger.3 Cystic fibrosis, affecting 162,428 people globally, further underscores the demand for effective treatment modalities.4
Given the nature of these conditions, inhalation emerges as the preferred route for drug delivery, enabling targeted therapy
directly to the affected sites within the respiratory system.
The burgeoning biologics market is exerting an unexpected influence on the inhalation landscape. Projected to be valued at approximately $854.86 billion by 2032, with a remarkable CAGR of 7.9% from 2023 to 2032,5 the global biologics market is driving innovation in drug delivery methodologies.
Traditional approaches to drug delivery often present obstacles in achieving optimal therapeutic outcomes and patient compliance, particularly with biologic drugs. Given the sensitive nature of biopharmaceutical formulations, parenteral delivery routes are often required to bypass the gastrointestinal tract. However, this approach causes discomfort for patients and puts constraints on drug administration confined to clinical settings, creating inconveniences for patients and strain on healthcare provider (HCP) resources.
Innovations in inhalation delivery technologies, particularly in nasal inhalation such as soft mist nasal sprays, have opened avenues for novel drug delivery strategies. Nasal delivery now presents a viable alternative for administering a wide array of biologic formulations, offering improved patient experiences and expanding treatment accessibility beyond clinical settings.6
Large Molecule Innovation: Opportunities and Challenges for Developers
These developments in biopharmaceutical drug delivery present both opportunities and challenges for developers of large molecule drugs. As the biologics segment grows, competition within the biopharma industry will also intensify. Notably, major industry drugs, such as Humira, Keytruda, Revlimid and Eliquis, are set to lose their exclusivity in the coming years.7 The resultant industry changes, partly fueled by patent expiries, are catalysing the expansion of the biosimilars market, projected to expand at a CAGR of 18.32% until 2029, reaching an estimated $82.27 billion within the next decade.8
In this fiercely competitive landscape, biologics manufacturers must strategise effectively to maximise the lifecycle
value of their products. This includes exploring opportunities for reformulation to ensure continued relevance and market competitiveness. Soft mist nasal sprays and other innovative nasal inhalation devices have the potential to play a pivotal role in extending product lifecycles while enhancing patient outcomes.
The nascent rise of the nasal route for biologics delivery is underpinned by compelling advantages demonstrated in recent studies. Nasal inhalation emerges as a promising avenue for systemic delivery of protein and peptide drugs, leveraging the nasal mucosa’s extensive surface area (150 cm2) and high vascularity, similar to the small intestinal mucosa. This route offers multiple benefits, including ease of administration, non-invasiveness, rapid onset of action, and circumvention of gastrointestinal degradation and hepatic first-pass effects. Notably, nasal delivery holds immense potential for enhancing the delivery of insulin to distal brain regions, marking a significant advancement in diabetes management.9
For vaccines, mucosal delivery offers superior and longer-lasting efficacy compared to traditional injection routes.10 Mucosal vaccine delivery elicits robust protective immune responses at the sites of pathogen entry, strengthening the body’s defences against infections. This approach, characterised by the induction of adaptive immunity at mucosal sites, can prevent the establishment of infections, particularly in the case of viruses such as influenza and coronaviruses.
These promising findings are translating to enhanced market projections, with the nasal vaccine segment expecting exponential growth, forecast to rise from $416.8 million in 2023 to $742.6 million by 2030.11 This underscores the growing recognition of nasal delivery as a potent strategy for enhancing vaccine efficacy and combatting infectious diseases.
Reviving Product Lifecycles for Existing Biologics
To prolong the lifecycle of existing biologics, novel approaches need to be explored,
28 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Drug Discovery, Development & Delivery
Drug Discovery, Development & Delivery

including the adoption of new indications and formulation enhancements aimed at promoting patient convenience.
Nasal delivery presents an opportunity for developers to rejuvenate their products, harnessing a fresh format for their intellectual property while enhancing patient adherence and comfort.
The regulatory advantages inherent in nasal biologics further amplify their appeal. The non-invasive nature of nasal delivery and its potential for targeted drug delivery with reduced side effects streamline the regulatory approval process, facilitating product lifecycle optimisation for developers.
Improving Delivery and Efficacy of Sensitive Biologics
However, the adoption of nasal delivery for biologic formulations poses some development challenges, such as the susceptibility of biologic formulations to damage from standard administration devices during spraying or nebulising. For example, a study by the University of Amsterdam has shown that lipid nanoparticles of the kind
used in mRNA vaccines can be damaged by traditional nebulisers for oral inhalation.12,13 Additionally, preservatives used in nasal delivery devices are often incompatible with biopharmaceutical formulations, complicating formulation development.
Further to these issues, achieving optimal drug efficacy requires meticulous consideration of dosing parameters and nasal cavity dynamics. Conventional nasal spray devices may exhibit limitations in dose uniformity and drug deposition, necessitating innovative solutions to overcome these obstacles. For instance, a traditional nasal spray often features a swirl nozzle with a spray cone angle of between 60 and 90°. This can result in most of the formulation being deposited on the walls of the nose, rather than penetrating further into the nasal cavity, with ramifications for dose uniformity.
The Role of Soft Mist Nasal Sprays in Overcoming Product Lifecycle Challenges
Soft mist nasal sprays represent a cuttingedge innovation in the biologics field, offering distinct advantages for both new product initiatives and lifecycle extension
projects. The unique design features of these sprays streamline device customisation, significantly reducing development time and costs for biologic formulations.
Evolved from the soft mist inhaler (SMI) for oral delivery, soft mist nasal sprays deploy a liquid sprayer mechanism that generates a slow-moving aerosol cloud, facilitating efficient drug delivery to the posterior nasal cavity with minimal inspiratory effort from patients.
Advancements in soft mist nasal spray design have tailored these devices specifically for biologic formulations, with innovations such as more targeted nozzle designs. Narrow spray cone angles address dosing challenges inherent in traditional wide-cone nozzles, ensuring optimal drug deposition in the nasal cavity. Next-generation spray nozzles, enable customisable spray cone angles (from 0 to 30°), droplet sizes and plume velocities. When used in a soft mist nasal spray, such a nozzle can evenly distribute a soft mist into the nasal cavity, resulting in uniform dosing and enhancing drug distribution to critical nasal regions.14
INTERNATIONAL PHARMACEUTICAL INDUSTRY 29 www.international-pharma.com
Drug Discovery, Development & Delivery
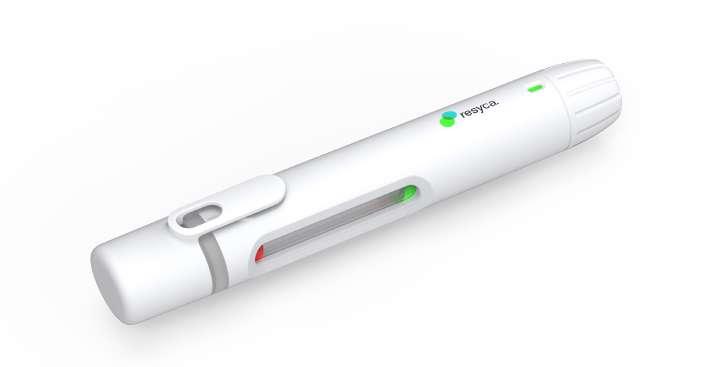
These nozzle designs not only optimise drug delivery but also mitigate formulation damage, particularly crucial for sensitive biologic products. By minimising shear forces on formulation particles during aerosolisation, soft mist nasal sprays ensure the integrity and effectiveness of biologic formulations.
Regulatory Approval in Nasal Delivery: Streamlining Development
In addition to enhancing drug delivery performance, recent soft mist nasal spray innovations promise to streamline development and regulatory processes, making nasal delivery an attractive option for biologic product lifecycle extension. Emerging device platforms, including multiuse devices combined with disposable, prefilled syringes, reduce the need for complex device customisation and high-volume manufacturing.
Moreover, customisable spray cone features simplify formulation-specific adjustments without necessitating extensive device redesign, further expediting development timelines and regulatory approvals.
These advancements not only make nasal delivery a viable option for new biologics but also present an opportunity for reformulating existing biopharmaceutical products to extend their lifecycle. By minimising development and regulatory hurdles while optimising manufacturing efficiency, soft mist nasal sprays pave the way for more convenient and user-friendly solutions for healthcare providers and patients alike.
Harnessing Nasal Innovation for Biologic Lifecycle Management
As the patents for numerous biologic products approach expiration, the demand for lifecycle extension solutions is growing. With increasing competition in the bio-
similar market, drug developers are increasingly turning to innovative strategies to differentiate their products and meet evolving patient needs.
By leveraging nasal delivery, drug developers can breathe new life into blockbuster biologic formulations, ensuring sustained market relevance and improved patient outcomes.
REFERENCES
1. HealthcareAnalyst, “Global Inhalation Drug Delivery Devices Market $17.6 Billion by 2027” (Jan 23, 2024) https://www.ihealthcareanalyst. com/global-inhalation-drug-delivery-devicesmarket/
2. “The Global Asthma Report 2022” (Jan 2024) http://globalasthmareport.org/
3. P.A. Corris, et al., “Call it by the correct name – pulmonary hypertension not pulmonary arterial hypertension: growing recognition of the global health impact for a well-recognised condition and the role of the Pulmonary Vascular Research Institute”, American Journal of Physiology-Lung Cellular and Molecular Physiology, (2020), 318(5), L992-L994.
4. J. Guo, et al., “Worldwide rates of diagnosis and effective treatment for cystic fibrosis” Journal of Cystic Fibrosis., (2022), 21(3), 456–462.
5. Novaoneadvisor, “Biologics Market Size, Share & Analysis Report, 2023-2032” (Jan 2024) https://www.novaoneadvisor.com/report/ biologics-market
6. J.O. Morales, et al., “Challenges and Future Prospects for the Delivery of Biologics: Oral Mucosal, Pulmonary, and Transdermal Routes” AAPS J., (2017), 19, 652–668. [and references therein].
7. Fierce Pharma, “The top 15 blockbuster patent expirations coming this decade” (July 2021) https://www.fiercepharma.com/specialreport/top-15-blockbuster-patent-expirationscoming-decade
8. Mordor Intelligence, “Biosimilars Market Size, Share, Trends & Industry Report” (Jan 2024) https://www.mordorintelligence.com/ industry-reports/global-biosimilars-marketindustry#:~:text=Biosimilars%20Market%20 Analysis,period%20(2024%2D2029).
9. D. Shah, J. Shao, “Nasal Delivery of Proteins and Peptides”. Glob J Pharmaceu Sci, (2017), 1(4), 555569.
10. E.C. Lavelle, R. W. Ward, “Mucosal vaccines – fortifying the frontiers”, Nat Rev Immunol, (2022), 22(4), 236-250.
11. Coherent Marketing Insights, “Nasal Vaccines Market Size, Trends and Forecast to 2030” (Jan 2024) https://www.coherentmarketinsights. com/market-insight/nasal-vaccines-market5818
12. D. M. Klein et al., “Degradation of lipid based drug delivery formulations during nebulisation”, Chemical Physics, (2021), 547, 11192.
13. C.J.M. van Rijn et al., ”Low energy nebulisation preserves integrity of SARS-CoV-2 mRNA vaccines for respiratory delivery”, Sci Rep, (2023), 13(1), 8851.

Nicolas Buchmann
Nicolas Buchmann, Chief Technology Officer, Resyca® has experience in developing oral and nasal inhalation drug delivery devices and has worked in this industry for most of his career. He has extensive knowledge in medical device development and in managing complex drug delivery programmes and portfolios for inhalation drug-device combination products. Before joining Resyca®, Nicolas held roles at Vectura (Chippenham, UK) as programme manager and at Pari GmbH (Starnberg, Germany) as technology manager. He holds a Ph.D. in biomedical engineering and is a certified senior project manager (IPMA).

Frank Verhoeven
Frank Verhoeven, Business Developer, Resyca® is an experienced product and business development manager with a degree in chemistry and nanotechnology from Saxion University. Before Resyca®, Frank worked at Medspray as an aerosol laboratory manager and product manager for Medspray’s soft mist inhaler technology. During this time, he managed the development of soft mist nasal delivery products with a focus on translating customer needs into customised products. In 2024, Frank joined Resyca, where he continues to develop Resyca’s unique soft mist nasal and oral inhalation platforms.
30 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1







INTERNATIONAL PHARMACEUTICAL INDUSTRY 31 www.international-pharma.com ––is
• • •
UPC’s
Application Note

Functional Testing of Prefillable Glass Syringes
Aligned with ISO 11040 Guidelines
Dr. Peer Schrader, Inge Burger, Dr. Florian Maurer, Dr. Alexander Traxl, Jens Herrmann, Dr. Uwe Rothhaar
Having a primary packaging container that serves both as sterile storage compartment and delivery device to administer drugs is the major driving force for the injectable combination products using prefillable syringes in a variety of applications. However, when it comes to the functional compatibility and delivery reliability between the syringe components (e.g., plunger, barrel, or closure cap) and the drug formulation, these benefits present more challenges than the traditionally used two-unit delivery systems vial and disposable syringe. Especially important properties like the gliding forces of the plunger or the flange strength need to be ensured over the entire shelf life. ISO 11040-4 defines a range of standardised test methods for prefillable glass syringes for the harmonisation of reliability within the industry. In this study, functional and strength tests according to the standards of ISO 11040-4 are performed, namely tests on flange breakage resistance, Luer-cone breakage resistance, needle pull-out force, gliding force, and pull-off force of closure caps. The results are statistically evaluated, thoroughly discussed, and the implications of the results for quality control are highlighted.
The percentage of prefillable syringes used for drug delivery on a yearly basis is steadily increasing in the pharmaceutical industry. Beginning with prefillable syringes, the pharmaceutical company/contract filling company is spared the responsibility for the washing, siliconisation, and sterilisation process. In addition, the packaging configurations for prefillable syringes (tubs and nests) are also suitable and relevant for smaller filling batches during small production campaigns. The subsequent versatility makes prefillable syringes suited for many pharmaceutical applications and encourages their increasing use.
To ensure patient safety, having a clear understanding of the behaviour of prefillable syringes in practical use is imperative. Therefore, ISO 11040-41 offers a wide range
of test methods meant to give the interested party (i.e. primary packaging engineer tasked with selecting between syringes from different companies; quality control tasked with troubleshooting production and/or field issues) insight into the most essential properties of prefillable syringes. Most of the described tests serve as reference methods to provide a measure for functionality, strength, and quality control in general during production. The same applies for ISO 11040-6,2 which stipulates test methods for the testing of prefillable polymer syringes that are based on methods as per ISO 11040-4.1 Intended for the manufacturers of prefillable syringes, it is making it mandatory to investigate whether the prefillable syringes will withstand the loads exhibited during filling, transportation, or the use in medical practice by medical staff or patients.
The present study gives an overview of the different tests applied on 1 ml long glass syringes. This includes the procedures for flange breakage, Luer-cone breakage, needle pull-out force, gliding force measurements, and the pull-off force of closure caps. For each of the aforementioned methods, 50 specimens are tested, gaining sufficient data to capture their statistical distributions. The experimental methods and the statistical analyses are also discussed in regard to their implications for quality control. As an overview, the test methods are briefly introduced in the following:
Test on flange breakage resistance: The test on flange breakage resistance is conducted to determine the strength of the fingerflange by applying axial load on a syringe barrel. As the flange is the most crucial for supporting the syringe barrel when a load is applied on the plunger, its structural integrity is of high importance during an injection process. Flange breakage during an injection will at least cause pain and discomfort for the patient and at worst have fatal consequences, for example, when a life-saving dose of a drug cannot be administered as quickly as it should. Therefore, having good knowledge of the flange breakage resistance is obligatory.
Test on Luer-cone breakage resistance: As the Luer-cone is the part of the prefillable
syringe subject to most critical loads due to its length and slenderness, it is necessary to thoroughly investigate its strength. Especially during transportation or when used as a subassembly with a vial adapter and lyophilised vial, the cone tip can happen to be loaded laterally, resulting in complex stress conditions at the base of the cone.
Test on needle pull-out force: A sufficient needle pull-out force is probably essential when it comes to the patient`s comfort, because a pulled-out needle during an injection would be the worst-case scenario, causing a lot of pain and discomfort to the patient. Therefore, it is mandatory to regularly check the quality of the bond between the needle and cone, to ensure a narrow distribution of the fracture forces at a comparatively large force magnitude.
Gliding Force between plunger and inner wall of the syringe: In practice, syringes must deliver and eject the filling solution in a smooth and controlled way. The gliding force test serves as an indicator for the siliconisation quality of the inner walls of the syringe barrel, as the gliding force has to be as low as possible whilst also guaranteeing tightness and plunger positioning accuracy.
Pull-off force of closure caps: Generally, closure caps help maintain sterility and protection of the cone or cannula. Additionally, Rigid Needle Shields (RNS) must protect the needle during production or during any handling, keeping the needle intact over the storage period. The pull-off force tests of the closure systems are meant to test whether the caps can be removed with an appropriate force by the user, along with a high enough resistance to being pulled off, as the integrity of the closure prior to injection should be absolute for the safety of the patient and handler.
SCHOTT Pharma Services provides the aforementioned tests put forward by ISO 11040-4 and ISO 11040-6 for glass and polymer syringes together with proven statistical evaluation procedures that support the pharmaceutical industry during design selection, verification, or quality control. A selection of data gathered for 1 ml
32 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
glass syringes is presented in the following sections.
Materials, Methods, and Equipment
All experiments were performed with a universal testing machine (INSTRON 5569, Instron GmbH, Darmstadt, Germany).
Prefillable SCHOTT 1 ml long glass syringes with staked 27G needles and RNS were tested. The typical components of such a prefillable syringe are presented in Figure 1. For each test series, 50 sample pieces were tested.
Application Note
agreement between syringe manufacture and pharmaceutical customer is required, which allows the option of different test parameters. The standard is designed for the testing to be conducted on empty syringes obtained prior to filling.
Test on flange breakage resistance: During the test on flange breakage resistance, an axial force is applied on the syringe barrel that has been placed in a cylindrical holder under the flange with a constant displacement
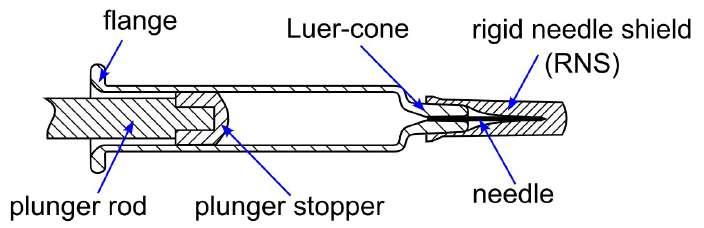
Experimental Investigations and Test Evaluations
All tests were performed in accordance with the methods described in ISO 11040-4.1 Schematic drawings of the test setups are shown in Figure 2. It has to be noted that while the test conditions described below fulfil the requirements of the standard, the standard clearly states that for most of the tests an
rate of 100 mm/min. The flange strength is determined as the peak force during the experiment, i.e. the force when the flange broke.
Test on Luer-cone breakage resistance: During the tests on Luer-cone breakage resistance, the cone of the syringe is mechanically loaded laterally at its tip, with
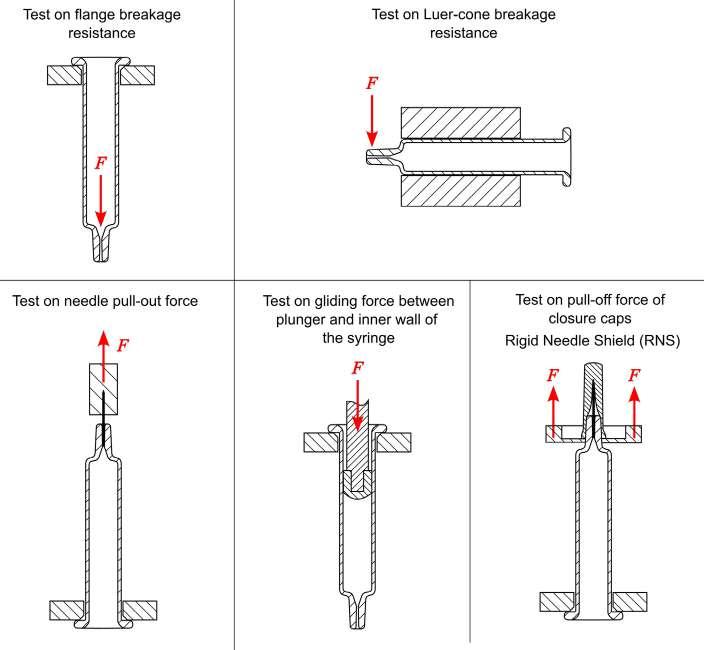
a constant displacement rate of 25 mm/min to simulate a side load on the cone that could arise during handling by the user or during transport. The cone’s strength is then determined as the measured peak force during the experiment, i.e. the force needed to break off the cone.
Test on needle pull-out force: During the test on needle pull-out force, the needle is gripped with clamps and pulled out at a constant loading rate of 50 mm/min. The syringe is held at the flange with a cylindrical holder. The needle pull-out force is determined as the peak force during the experiment.
Gliding force between plunger and inner wall of the syringe: During the test, the plunger is pushed along the syringe cylinder at a constant displacement rate of 100 mm/min and the occurring resistive forces are recorded. The syringe is held at the flange with a cylindrical holder. The mean and maximum gliding forces during testing are evaluated according to ISO 11040-41 and ISO 11040-6.2
Pull-off force of closure caps: During the tests, the caps (e.g. RNS) are gripped from below and are pulled off at a constant displacement rate of 280 mm/min. Here, the syringe is also held at the flange with a cylindrical holder. The pull-off force is determined as the peak force during the experiment.
Statistical Evaluations
For the flange breakage resistance, Luercone breakage resistance, and needle pullout force, statistical analyses are performed using the two-parameter Weibull distribution3 to investigate the fracture behaviour and give indications for quality control purposes. The Weibull distribution is preferably used to describe the fracture behaviour of glass and other brittle materials.4 For the pull-off force of closure caps, the normal distribution is used for statistical evaluation.
To develop a better understanding of the presentation of such statistical results, a brief digression on probability distributions should be made: in Figure 3, the number of specimens that failed in a given force range is plotted over the force at failure in a histogram (gray bars, left diagram). This can be then converted into the probability density, which shows the likelihood of individual results of the random experiment (blue curve, left diagram) and allows the determination of the bell-shaped probability density function. To compare the obtained probabilities against a specified
INTERNATIONAL PHARMACEUTICAL INDUSTRY 33 www.international-pharma.com
Figure 1: Principle illustration of a prefillable syringe with designation of single components
Figure 2: Schematic drawings of the test setups
Application Note
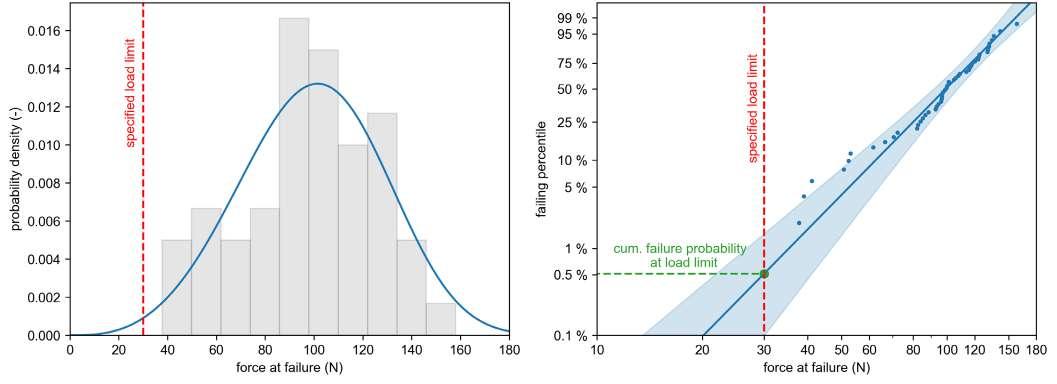
3:
evaluation and determination
limit (red line), the cumulative sum of these probabilities, the so-called failing precentile, is used. The resulting cumulative distribution is shown in the right diagram of Figure 3. This diagram clearly visualises the proportion of the sample that will already have failed at a given load, allowing a good comparison with a specified load limit (for the example given in Figure 3 a load limit of 30 N corresponds with a failure probability of approx 0.5%). Typically, the coordinate axes are scaled to achieve a linear appearance of the displayed dataset, which further facilitates the reading-off of corresponding values. This linearised display of the dataset and distribution also allows a visual control of the goodness-of-fit, as a linear alignment of the data can be observed if the data follows the predefined distribution.
Results
Flange Breakage Resistance
The Weibull distribution for the flange breakage experiments is shown in Figure 4. Typically, for the tested glass syringes,
breakage occurred at the radius between the flange and the syringe barrel, as the mechanical stress in this region is greatest for the given load configuration. Unfortunately, contact damages are frequently introduced during processing and production in the same region, lowering the strength of the flange.
Using the Weibull distribution for the statistical investigation of the data has a significant advantage: with a sufficiently large sample size, the resulting distributions can be extrapolated to estimate the failure rate at a specified load limit. For example, given an arbitrary flange breakage resistance limit of 20 N, a failing percentile of 0.003 % for the tested lot of glass syringes would be obtained. This, although it can only be seen as a rough estimate, can already be considered as a valuable insight into the durability of the lot of prefillable syringes in the later use case, constituting a suitable method for risk management.
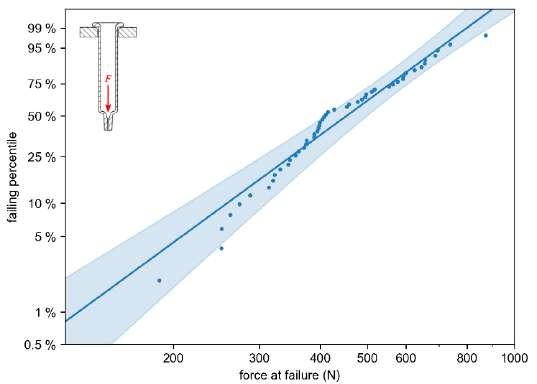
Although the glass syringes always broke in the region of the flange, it should be highlighted that, according to the standard ISO 11040-4,1 every breakage event is considered as flange breakage regardless of the position of the fracture origin, and no distinction is made as to whether the syringe is fractured at the cone, syringe barrel, or the flange during the test. Therefore, it is important for quality control purposes to define what is to be considered a flange breakage and that, depending on the aim of the experimental investigation, it could be beneficial to also record the position of the fracture origin in addition to the force values that must be documented according to the standards.
Furthermore, it must be stated that the force at flange breakage depends heavily on the diameter of the syringe holder, as the distance between the support points and the syringe barrel will influence the bending stress applied to the flange. As ISO 11040-41
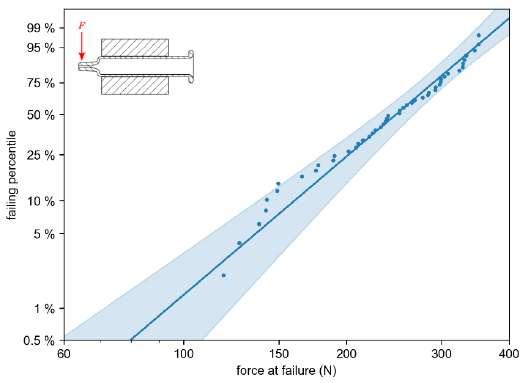
34 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Figure
Statistical
of the cumulative distribution function (exemplary data); histogram of total amount of failures (grey bars, left), probability density diagram (blue curve, left), and cumulative distribution (right) with individual data points (dots), obtained Weibull distribution (solid line) and 95% confidence interval (shaded area)
Figure 4: Obtained Weibull distribution for flange breakage resistance: individual data points (dots), obtained Weibull distribution (solid line) and 95% confidence interval (shaded area)
Figure 5: Obtained Weibull distributions for Luer-cone breakage resistance: individual data points (dots), obtained Weibull distribution (solid lines) and 95% confidence interval (shaded area)
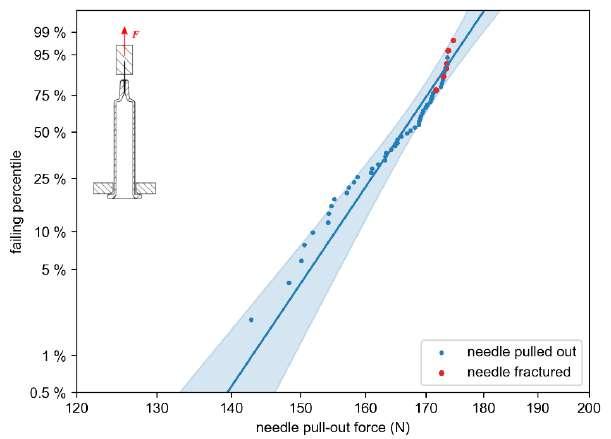
states, the dimensions of the syringe holder and loading pin must be, therefore, also agreed on between the test lab and the customer. Unfortunately, this means that comparing different syringe geometries in regard to their flange breakage strength is not easily done via the measured forces at fracture.
Luer-cone Breakage Resistance
The Weibull distribution for the Luer-cone breakage resistance is shown in Figure 5. When considering the fracture distribution, it should be noted that the test on the Luer-cone breakage resistance according to ISO 11040-41 is challenging because of the complex mechanical stress state induced in the cone: the applied force induces a bending load on the cone of the syringe. This means that the upper half of the cone is loaded by tensile stress, whereas the lower half of the cone is loaded by compressive stress, cf. Figure 6. As brittle materials tend to favorably fracture in tension rather than under compression, it is possible that, depending on the rotatory (azimuthal) position of the syringe, critical defects that would lead to failure under tension at a lower load could be located in the region of compressive stress, therefore yielding an artificially higher, overestimated Luercone breakage resistance for the tested syringe. This could, potentially, also yield a wider distribution of the forces at fracture, as critical flaws are randomly distributed along the circumference of the cone.
This issue can only be resolved with a very large number of samples, as it can be assumed that the errors due to the rotatory position compensate each other with increasing samples size. The investigator should be
aware of this, especially if the statistical distributions are to be used (or extrapolated) as a basis for batch comparisons or to evaluate changes in production conditions. However, it should also be mentioned that the setup proposed by the standards represents the application case, in which critical flaws will also not lie in the region of tensile stress, necessarily.
It should also be noted that ISO 11040-41 does not specify the exact position at which the syringe should be clamped in the test setup. This may also influence the complex stress state along with the Luer-cone slightly, leading to changes in the observed forces at failure. Therefore, it is imperative for quality control purposes that care must be taken to ensure that the distance between the point of load introduction and the beginning of the specimen clamping is similar when comparing different test series.
Needle Pull-out Force
For the tested 1 ml long glass syringes with 27G needles, two types of failure mechanisms were observed during the experiments on needle pull-out force: typically, the
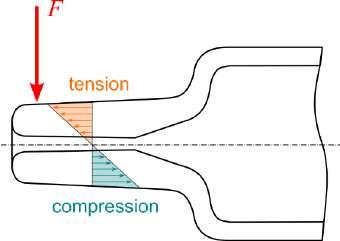
needle is pulled-out of the cone, resulting from a failure of the bond between Luercone and needle. However, in six of the 50 experiments performed, the needles could not be pulled out as they fractured before the bond between glass cone and needle could fail, meaning that the bond could have withstood larger loads if the needle would not have fractured prematurely. The Weibull distribution for the observed forces at fracture is shown in Figure 7, with the two failure mechanisms indicated by the color of the markers. As it can be deduced from the figure, at loads above 170 N, the needle tends to fail instead of the bond between cone and needle. Because the standard1 does not differentiate between the failure modes during the test on needle pull-out force, the values from the dataset at which the needle ruptured are not treated as rightcensored data, although, strictly speaking, this would be meaningful from a statistical point of view.
As stated in ISO 11040-4,1 the test should prove whether the method of bonding the needle to the cone is sufficient to comply with the regulations of ISO 7864,5 which states a minimum needle pull-out strength of 22 N for the tested 27G cannula. Given the results displayed in Figure 7, it can be observed that the distribution of fracture forces for the tested syringe type is well above the specified limit. When extrapolating the obtained distribution to the specification limit of 22 N, the estimated failure rate is negligible.
Gliding Force
A representative curve for the gliding force experiments is shown in Figure 8. Within this study, the length of the break-loose region was defined as the length of the used plunger, as settling processes, interactions between the siliconisation of the syringe’s inner wall and the elastomeric plunger stopper, and a possible tilting of the plunger in this region, could potentially influence the measured gliding force. This can also be observed in Figure 8, as strong oscillations are visible in the obtained force, which may be ascribed to setting processes between the lips of the elastomeric plunger stopper and the inner wall of the syringe barrel. The end of the gliding force region is taken as the maximum machine displacement minus the distance between the end of the thread for screwing in the plunger rod and the tip of the plunger. Within the gliding force region, the measured forces stay very much constant for the
INTERNATIONAL PHARMACEUTICAL INDUSTRY 35 www.international-pharma.com
Application Note
Figure 7: Obtained Weibull distribution for needle pull-out force: individual data points (dots), obtained Weibull distribution (solid lines) and 95% confidence interval (scatter bands)
Figure 6: Schematic representation of the load distribution in the Luer-cone during the test on Luer-cone breakage resistance
Application Note
tested glass syringes, indicating a uniform siliconisation of the syringe barrel.
In Figure 9 the results of the gliding force tests are displayed in a boxplot. In addition to the mean and maximum force in the gliding force region, i.e. the results demanded by the standard,1 the breakloose force (BLF, maximum force in the break-loose region), and the minimum force in the region of the gliding force are also considered. It must be stated, however, that the definition of gliding force region within the standards is ambiguous, as only a load-displacement diagram is shown in the standard to illustrate the definition of the break-loose region and gliding force region.1 Especially considering the quality control, this ambiguous definition requires prior agreement and good communication between customer and test lab. It should also be noted that strictly speaking, breakloose force determination is outside the scope of this standard.
For quality control, it is of special interest that the break-loose and gliding forces during applying a drug are generally low, without compromising on the tightness and positioning accuracy of the plunger. Therefore, the aim should be obtaining comparatively small differences between mean, minimum, and maximum forces in the gliding force region, as this indicates a uniform siliconisation of the syringe’s inner wall. Furthermore, the gliding forces should be similar within one lot, which implies a high processing accuracy during siliconisation. As it can be observed in Figure 9, the tests do not show a large variation between the
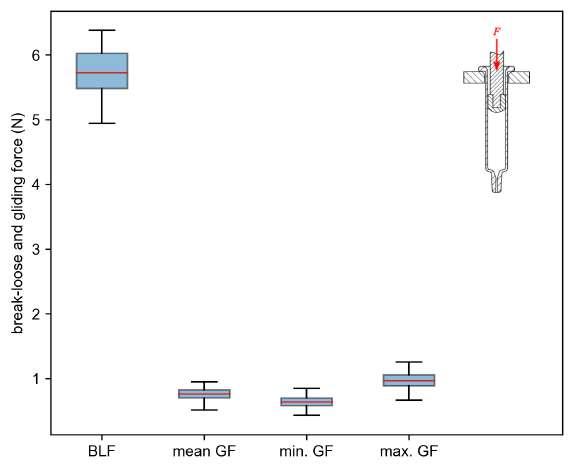
different syringes and the minimum, mean, and maximum gliding forces lie very close together,
a very good quality of the siliconisation process.
Pull-off Force of Closure Systems
The normal distribution for the pull-off force tests of the rigid needle shields (RNS) is shown in Figure 10. As can be observed, the pull-off forces of the RNS range between approx. 13 N and 23 N. Again, the statistical investigation of the data provides an advantage: the resulting distribution can, again, be extrapolated to different specified load limits. As the pull-off forces should neither be so large as to complicate handling nor so small that there could be a risk of the cap falling off, it could be advantageous
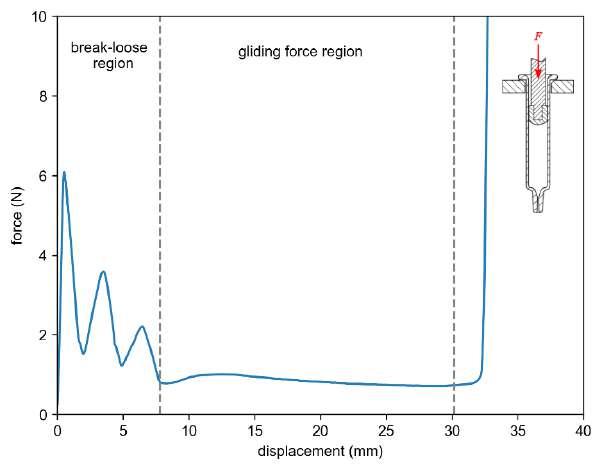
to specify minimum and maximum load limits for the pull-off force. If, for example, a minimum pull-off force of 10 N and a maximum pull-off force of 25 N was to be specified for the given lot, one would find from the extrapolation of the distribution that only 0.0002 % of caps would be pulled off at a force below 10 N, whilst 99.9995 % of caps would have come off until a force of 25 N was reached. This, again, could be a valuable insight into the homogeneity of the lot of prefillable syringes in the later use case, constituting a suitable method for risk management.
Discussion
As presented, the statistical evaluation of the data obtained from functional and strength tests according to ISO 11040 is especially useful for quality control purposes, and the understanding of underlying statistical distributions helps in comparing the data gathered from different lots of prefillable syringes and allows an estimation of the failing percentile in the later application.6 If the forces that are applied during usage are known, the investigator can simply estimate the value of the failing percentile of the lot from the statistical distributions at this given load, already providing a valuable insight on the number of syringes that could possibly malfunction during use. It must be noted, however, that a sufficient number of experiments has to be performed per lot to allow such extrapolations with sufficient certainty. It is therefore recommended that, for smaller sample sizes, the value of the upper limit of the confidence boundary to be used as a conservative approach.
36 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Figure 8: Representative gliding force measurement for the tested syringes and definition of gliding force region used for evaluation
Figure 9: Boxplots for break-loose force (BLF) and gliding force (GF): median (red line), boxes (standard deviation), whiskers (min. and max. values of the dataset)
thus indicating
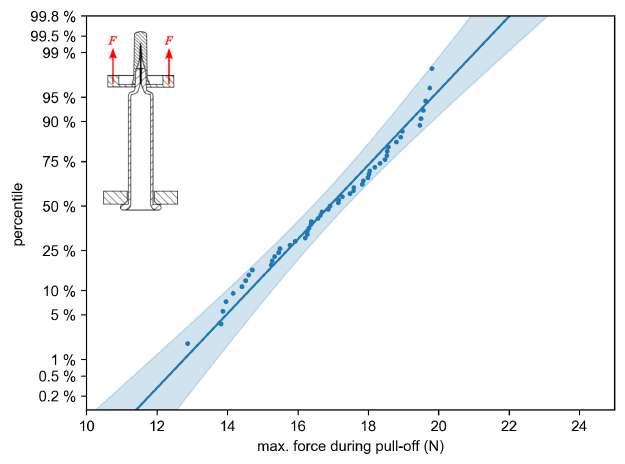
The standard ISO 11040-41 does not give any information on the number of samples to be tested. Because all the methods discussed in this study are of “destructive” nature, one should bear in mind that it is only possible to test a small amount of the complete lot. Hence, statistical investigations can only be an estimate for the behaviour of the underlying basic population. However, it should be emphasised that, if one wishes to investigate the strengths of the syringes statistically, a minimum sample size of 30 representative specimens is needed for a reasonably small confidence interval of the Weibull modulus.7 For normally distributed data, similar minimum sample sizes are needed to achieve an accurate estimation of the mean value.8 Therefore, if the data from the inspection is to be used for quality control purposes, e.g., when batches are to be compared with each other accompanying production, for design verification, or for specification testing, it is recommended to select a sample size of 50 specimens or more to decrease the uncertainty. Testing smaller sample sets may save costs in the short term, but in the long term a potential recall of a batch of prefillable syringes due to non-compliance with the specifications is certainly more expensive.
It must also be noted that the tests discussed in this study can be performed as well with polymer syringes according to ISO 11040-6.2 Because the test methods do not differ between standards, the above tests and findings can be also largely transferred to the testing of prefillable polymer syringes in accordance with ISO 11040-6.2
The strength test methods per ISO 11040 provide valuable insight in regard to the behaviour of the tested lots via fracture forces and allow an easy comparison between different lots of the same syringe format. It should be noted that it is not easily achievable to obtain the mechanical stress at fracture with these given methods. These mechanical stress values, however, are necessary for a comparison between different geometries or formats, which is not possible purely based on forces. To determine the mechanical stress, complex finite element analyses are necessary. Furthermore, combining the tests with fractography allows gaining further insight into the failure inducing flaws and the failure mode.9,10 It is therefore advised to also follow up the mechanical tests with a fractographic analysis.6
Conclusions
In this work, we covered the relevant functionality and strength tests for prefillable glass syringes and discussed the details per ISO 11040-4.1 The described methods are deemed indispensable for the design verification and quality control purposes of prefillable syringes. Combined with statistical evaluations, these tests provide a powerful tool for complying with specifications or finding the root cause of an unexpected behaviour. For adequate implementation, however, both experimental expertise and close consultation between the test laboratory and the customer are of the essence when it comes to determining relevant and representative parameters for the application case.
REFERENCES
1. International Organisation for Standardisation. (2015). Prefilled syringes – Part 4: Glass barrels for injectables and sterilised subassembled syringes ready for filling (ISO 11040-4:2015).
2. International Organisation for Standardisation. (2019). Prefilled syringes – Part 6: Plastic barrels for injectables and sterilised subassembled syringes ready for filling (ISO 11040-6:2019).
3. Weibull, W. (1951). A Statistical Distribution Function of Wide Applicability. Journal of Applied Mechanics, 293-297.
4. Berlinger, M. (2021). A Methodology to model the statistical fracture behaviour of acrylic glasses for stochastic simulation.
5. International Organisation for Standardisation. (2016). Sterile hypodermic needles for single use – Requirements and test methods (ISO 7864:2016).
6. Haines, Daniel & Maurer, Florian & Rothhaar, Uwe. (2016). Why Do Pharmaceutical Glass Containers Break: The Underestimated Power of Strength Testing and Fractography. International Pharmaceutical Industry, 8: 8892.
7. Danzer, R., Lube, T., & Supancic, P. (2001). Monte Carlo simulations of strength distributions of brittle materials - Type of distribution, specimen and sample size. Zeitschrift für Metallkunde: International Journal of Materials Research and Advanced Techniques, 92(7), 773783.
8. Berenson, M., Levine, D., Szabat, K. A., & Krehbiel, T. C. (2012). Basic business statistics: Concepts and applications.
9. Quinn, G. (2020). NIST Recommended Practice Guide: Fractography of Ceramics and Glasses, 3rd edition, Special Publication (NIST SP), National Institute of Standards and Technology, Gaithersburg, MD, [online], https://doi.org/10.6028/NIST.SP.960-16e3 (Accessed November 2, 2023)
10. Maurer, F. Tracking the cracking – How to optimise pharma fill-and-finish processes by use of strength testing and fractography [White Paper]. SCHOTT AG, 2021.
All authors are working in different roles providing analytical services focused on the compatibility between drug product and containment.
Inge Burger, Dr. Florian Maurer, Dr. Peer Schrader, and Jens Herrmann belong to the "Reliability & Lifetime" group of SCHOTT AG. A very experienced team in the areas of strength, fractography and functional testing. Dr. Alexander Traxl (SCHOTT Pharma) provides with his laboratory team application near tests of drug containments and delivery systems. Dr. Uwe Rothhaar is responsible for SCHOTT Pharma Services supporting pharmaceuticals companies with compatibility and compendial data.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 37 www.international-pharma.com Application Note
Figure 10: Obtained normal distribution for RNS pull-off force: individual data points (dots), obtained normal distribution (solid lines) and 95% confidence interval (scatter bands)
Why is Real World Data the Key to Rare Disease Success? Clinical and Medical Research
The quest to understand and treat rare diseases is among the most challenging and vital missions in healthcare today. As the industry seeks innovative solutions, Real World Evidence (RWE) studies utilising Real World Data (RWD) have grown in acceptance. It’s been argued by many that RWD provides valuable insight into how an Investigational Medicinal Product (IMP) performs in the real world, whereas a Randomised Clinical Trial (RCT) setting is heavily regulated and robustly follows the patient inclusion and exclusion criteria defined in a protocol and trial settings. RWD can provide insight that you simply would not get in the traditional sense, and this insight is worth reviewing and not ignoring, as well as opening you up to other patient populations that may not have been looked at previously. This paradigm shift from traditional clinical data to real-world insights marks a new era for researchers, physicians, and patients alike. As the industry adapts, the implications of RWD are revealed, shaping the future of diagnosis, treatment, and patient care.
The Emergence of Real-World Data
Rare diseases, characterised by their low prevalence and complex nature, have long presented unique challenges to researchers. With approximately 7,000 rare diseases affecting over 350 million people worldwide, the research community faces significant hurdles. Traditional clinical trials often fall short due to small patient populations, heterogeneous symptoms, and a lack of historical data, often leading to significant delays in drug development. In this context, Real World Data (RWD) emerges as a critical tool, by tapping into a diverse array of sources like electronic health records (EHRs), insurance claims, patient registries, and direct patient inputs, offering a more comprehensive understanding of rare diseases.
Unlike the structured environment of traditional clinical trials, RWD reflects the complexities of everyday clinical practice and offers a broader, more diverse patient population. It provides insights into disease
progression, treatment outcomes, and patient quality of life in a real-world context, thus enabling more informed decision-making and innovative trial designs. It can also give you more insight into how an IMP would react with other co-medications and treatments because you will get more situations where the trial drug is being taken alongside other medications.
Key Benefits of RWD in RWE Rare Disease Research
Access to a Global Pool of Patients: One of the most significant advantages of RWE studies is the ability to access a much larger and more diverse patient population than what is typically available in centralised clinical trials. For rare diseases, this is particularly crucial as patients are often scattered across the globe. RWD allows researchers to tap into international databases, patient registries, and health records, thus overcoming the geographical and logistical barriers that often hamper rare disease research. This globalisation can lead to more robust data sets, offering deeper insights and more generalisable findings.
Understanding Natural Disease Progression: Understanding natural disease progression through Real-World Data (RWD) provides researchers with critical insights into rare diseases. By analysing a variety of patient data over extended periods, researchers can discern patterns, variations, and outcomes of diseases, offering a comprehensive view of their progression and impact. This deep understanding, drawn from real-life contexts, is instrumental in designing effective treatments and care strategies, thereby enhancing both treatment effectiveness and patient outcomes.
Enhancing Drug Development and Approval Processes: RWE studies are becoming increasingly recognised by regulatory agencies for their role in supporting and complementing clinical trial data, including providing additional evidence on drug safety, efficacy, and identifying potential patient populations. This is especially beneficial for rare diseases as it streamlines the development, evaluation, and approval of new treatments, potentially accelerating
access to new therapies and improving postmarketing surveillance.
Facilitating Personalised and Adaptive Treatments: Every rare disease patient is unique, and so are their treatment needs. RWD enables a more personalised approach to treatment by allowing researchers and clinicians to understand individual variations in disease presentation and treatment response. This leads to more adaptive and patient-centred care models.
Empowering Patient-Centric Care: RWD enables a more patient-centric approach to treatment and care. By understanding the real-world experiences of patients, healthcare providers can offer more personalised and effective treatment strategies. Additionally, patients and caregivers can make informed decisions about their care, knowing it's based on a wide range of similar patient experiences. Patients also no longer have the burden of travelling to site and wearable devices that capture RWD improve patient adherence.
Cost-Effective Research and Treatment Models: Conducting traditional clinical trials for rare diseases is often prohibitively expensive due to the high cost of recruitment and the long duration of studies. RWE studies offer a more cost-effective alternative by leveraging existing data. This not only accelerates the research process but also reduces the financial burden on healthcare systems and patients.
Transformative Case Studies: The Power of RWD
Gene Therapy Trials: The integration of RWD is particularly impactful in the field of gene therapy, where understanding the long-term effects and variability of treatments is crucial. RWD facilitates this by offering longitudinal data, helping researchers track outcomes and adapt strategies accordingly. For rare genetic disorders, this means a more tailored and effective approach to treatment, informed by a wealth of real-world patient experiences.
Responding to Global Challenges: The COVID-19 pandemic underscored the versatility of RWD. As the world grappled with the virus, RWD provided timely insights into its
38 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Clinical and Medical Research

behaviour, treatment responses, and vaccine efficacy. This demonstrated the potential of RWD in responding to global health crises, offering lessons that are particularly relevant in the context of rare diseases.
Navigating the Challenges
The implementation of RWE studies and RWD offers undeniable benefits, but also presents significant challenges. There have been some reasons for hesitancy with RWD. Some argue that the data from the real world cannot be easily trusted because there are too many variables that cannot be controlled, unlike a traditional clinical trial with patient screening and recruitment processes and easily controlled dosage etc. It is sometimes difficult to unravel the data to determine whether the IMP was the cause in a patient’s improvement or not. For example, if a clinician can choose which treatment to give, they may decide to give the new unproven drug to a really poorly patient, and the standard drug to a less poorly patient, because there is less risk in both situations. But in these situations, it will be more difficult to detect that the new drug performs better than the standard one due to the differences in the underlying health of the patients. There is also the issue of potentially ‘dirty data’ as there is no team
behind the data checking for inconsistencies or errors.
Some statisticians argue that with almost a limitless amount of data, you can get lost in doing over analysis as you hope to find answers you want to see but may not see, so it has to be careful of defining how much RWD is being used and being strict in the analysis you do perform.
Ensuring data quality, privacy, standardisation, and interoperability are critical issues that need to be addressed to maintain the reliability and validity of RWD. As we leverage these vast data sources, we must develop robust analytical techniques and methodologies for the collection, analysis, and application of RWD. Navigating a complex and evolving regulatory environment is also essential to ensure that RWD is used ethically and effectively. The industry must continue to innovate and invest in technologies that enhance RWD's capabilities, grappling with these challenges to realise the full potential of RWD in advancing rare disease research and treatment.
A Collective Journey Towards a Brighter Future
RWD is revolutionising the rare disease
research, promising a future of more effective, efficient, and patient-centred approaches. This paradigm shift is not merely about adopting new data sources; it's about transforming the entire approach to understanding and treating these complex conditions, making every patient count, and every data point a step towards a cure. This leads to the potential of accelerated drug development and improved patient care. As the healthcare industry evolves, the integration of RWE studies utilising RWD is critical, offering comprehensive insights, better treatments, and improved outcomes. The data-driven approach taken by researchers, clinicians, and organisations is essential in advancing the treatment of rare diseases. By leveraging existing data from hospitals and patient registries, along with other key data sources, they are independently forging paths of innovation and creating new opportunities. This work is instrumental in bringing hope and improved care to patients with rare diseases, and to the stakeholders dedicated to positively impacting their lives.
However, there are still those who may be concerned by including the use of RWD compared to conducting the gold standard approach to clinical research, namely a randomised, placebo, controlled trial, which is often advocated as the most appropriate choice irrespective of the circumstances. We can’t deny the importance and value of the RCT, but not in all circumstances, including for some, or even most trials involved with rare diseases can follow RCT, and more trials will find themselves being a hybrid as they use both RWD and site data in their submission package.
The collaborative efforts of researchers, clinicians, and organisations are pivotal in this journey. Together, they are driving innovation, hope and opportunities for patients with rare diseases and the stakeholders committed to making a difference in their lives.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 39 www.international-pharma.com
Karen Ooms
Karen Ooms, Joint Chief Operating Officer, Quanticate, is a Chartered Fellow of the Royal Statistical Society and has a background in biostatistics spanning over 25 years. Prior to joining Quanticate in 1999 (Statwood), Karen was a Senior Statistician at Unilever.
Clinical and Medical Research
Advancing Therapeutic Solutions with Antibody-drug Conjugates (ADCs)
As promising therapeutic agents for treating oncology indications, antibodydrug conjugates (ADCs) have become prominent in the biopharmaceutical market in recent years. The approval of the first ADC in 2000 by the Food and Drug Administration (FDA) to treat acute myeloid leukaemia prompted a move in the oncology space toward a targeted cancer therapy.1 As of June 2023, 11 ADCs are available on the market, having gained the FDA approval for treating a range of cancers, including leukaemia, lymphoma, and cervical and ovarian cancer.2 With the global ADC market valued at $8.6 billion in 2022, and forecast to reach $23.9 billion by 2032, research into this promising drug method will continue to transform the therapeutic landscape.3 However, the complexity of ADCs means their design, development, and manufacturing are multifaceted, with many challenges that must be overcome to successfully deliver these therapies to the patients who need them.
In this article, Louise Duffy, Chief Technical Officer (CTO), and Campbell Bunce, Chief Scientific Officer (CSO) at Abzena, explore how innovation in the biopharma sector is driving the development of novel ADCs to treat indications beyond cancer. They also examine the difficulties that drug developers and manufacturers must overcome to ensure the ADC development and manufacturing success.
Constructing an ADC ADCs contain three separate components, including:1
• The antibody: Traditionally a monoclonal antibody (mAb), this part of the ADC is key to specifically binding the target antigen with high binding affinity.
• The linker: This component connects the toxic payload to the antibody. Ensuring that it remains bound during circulation, the linker is specifically designed to maintain the stability of ADC in circulation and release the payload at the target site. A cleavable linker will take advantage of the environmental differences between the
systemic circulation and the tumour cells to accurately release the drug. Alternatively, a non-cleavable linker will rely on the catabolic digestion of the whole ADC by proteases to release the payload.
• The toxic payload: These highly potent materials (HPAPI) exert cytotoxicity onto the cells targeted.
The design of these innovative therapies demands extensive teamwork and innovation across various disciplines and areas of expertise, to ensure the ADC is designed for optimal function and stability while enabling scalability, manufacturability, intellectual property, and funding.
Navigating the ADC Development ADCs represent a promising option when developing drugs for oncology. As a result of the rising interest in this drug method within the biopharmaceutical sector, there are currently over 100 ADCs in clinical trial studies.4
The design and development of ADCs begins following the identification of a suitable molecular target. Typically, the drug target is chosen based on its differential expression on the cells of interest, e.g. tumour cells that will allow them to be pinpointed by the ADC. Drug developers will then conduct a comprehensive evaluation of the safety implications of aiming at that particular target or pathway before initiating ADC design.
In addition to the careful design of each element, the assembly of all components is a key part of the ADC development and will significantly impact the drug performance. The antibody, the linker and the payload, all three elements need to be brought together in a way that produces an optimal form of the ADC, one that is stable, can be manufactured at scale, and is capable of delivering the required therapeutic effect safely. The key to achieving this is considering the target product profile (TPP) of the final ADC product when designing each element. As a result, the developers with a holistic overview of the end objective from the earliest development stages are best positioned for success.
Developing and optimising an ADC will rely on the testing of various linker designs,
payloads, antibody-to-drug ratios, and conjugation modalities using biopsy, and both in vitro and ex vivo systems. From this matrix design approach, the developers can identify a promising lead candidate and progress it through the development pipeline.
Overcoming Challenges in the ADC Development
With many factors to consider, navigating the ADC development is often highly complex, presenting many challenges that must be overcome to successfully deliver these innovative therapies to patients, and these include:
• Handling Toxic Payloads
The payload of the ADC is often inherently toxic, designed to induce a therapeutic effect such as the killing of cancer cells upon release at the target site. The development and manufacture of these materials can therefore pose a significant risk to the operator, which increases as the process is scaled.
• To mitigate these risks and remain compliant with good manufacturing practice (GMP), the ADC developers and manufacturers must utilise specialised facilities. These facilities often use containment strategies and equipment to form a barrier between the individual and the compound, maintaining the material within the defined operational exposure limits. In addition, the ADC producers will also need to introduce and continuously re-evaluate rigorous, well-defined safety processes, to ensure the safety measures remain compliant.
• Eliminating Analytical Bottlenecks
Analytical expertise and specialist equipment is essential to generate vital information to drive the development forward, allowing ADC producers to gain insight into component characteristics, stability, toxicity, and activity throughout development. With this information, developers can identify the best-suited elements to progress through to the conjugation stage to form the final ADC. However, inefficient method development can result in process bottlenecks, where waiting for the data regarding one
40 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
component can stall the development of the next.
Effective ADC development therefore requires effective coordination between all the development teams. With access to varied expertise and specialised equipment, the teams involved can better coordinate their efforts to maintain timelines and ensure the objectives are met.
• Determining Conjugation Accuracy
Selecting the best conjugation technology is essential in ADC development, determining the mode and extent of the release of the payload. When administered to a patient, a poor selection of a conjugation technology could result in the release of the payload in the periphery, generating off-target effects. Therefore, careful design and selection instils precision, enabling the accurate delivery of the drug to the target site. This not only increases therapeutic efficiency but also reduces systemic toxicity.
Designed with Patients in Mind
Meticulously reviewing the TPP at the earliest point of development means that the patient is considered at every step of the ADC production. This provides the opportunity to apply a holistic approach to the ADC design and development, with developers considering the patient population and the context of the disease throughout the process. It also provides an opportunity to revise the TPP as data is generated to ensure that patients benefit throughout.
In addition to the therapeutic benefits, having a holistic understanding of the ADC development and manufacturing process ensures the production speed is taken into consideration. The biopharmaceutical industry is under significant time pressure to deliver essential medicines to patients. While considering the best development strategy, having a well thought through TPP ensures that objectives are seamlessly met, and the treatments are delivered to patients swiftly.
Looking to the Future of ADCs
ADCs are proven oncology treatments, having shown their ability to treat various cancer types. However, innovation is needed to drive the next generation of therapies and improve available cancer treatments for patients. The biotech industry is increasingly harnessing biand tri-specific antibodies in ADCs to increase precision for more accurate drug delivery.5,6
Clinical and Medical Research
With ADCs having gained recognition for treating cancer alone, the potential of these therapeutics for treating disease indications beyond oncology is an area of significant potential.
Recent innovations in delivering peptides to a target using ADCs are providing developers with the opportunity to tackle a broader range of clinical indications in need of more effective therapies. The use of non-toxic, novel payloads provide an alternative way to influence particular disease pathways.
Immunological developments are also enhancing ADC capabilities. By enabling the delivery of toxic immune stimulators to specific targets, innovative new ADCs can produce a localised immune response avoiding widespread adverse reactions.
ADCs as Advanced Therapeutic Solutions
Since their approval, ADCs have been used as treatments for various cancer types. However, navigating the ADC development is a complex process and developers need to overcome several challenges to successfully design and manufacture all three elements required – the antibody, the linker and the payload.
With an effective strategy in place that considers the toxicity and the operator safety, harnesses seamless and integrated technical expertise and working practices, and selects the best technology and approach to use, ADCs can be effectively and efficiently developed.
Only with continuous innovation, ADCs will proceed with transforming the biopharmaceutical sector, providing patients with broader treatment options. As the industry continues to advance, the ADC pipeline will evolve to encompass more diverse, hard to treat disease indications, both within and beyond oncology.
REFERENCES
1. Antibody-drug conjugate: the “biological missile” for targeted cancer therapy, Accessed Jan 9 2024, from: https://www.nature.com/articles/s41392022-00947-7
2. Gogia, P., Ashraf, H., Bhasin, S., & Xu, Y. (2023). Antibody–Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers, 15(15). https://doi. org/10.3390/cancers15153886
3. Global Antibody Drug Conjugates Market Size To Grow USD, Accessed Jan 9 2024, from: https://www.globenewswire.com/en/newsrelease/2023/05/30/2678771/0/en/GlobalAntibody-Drug-Conjugates-Market-Size-To-
Grow-USD-23-9-Billion-By-2032-CAGR-of-10-7. html
4. Rise of Antibody-Drug Conjugates: The Present and Future | American Society of Clinical Oncology Educational Book, Accessed Jan 9 2024, from: https://ascopubs.org/doi/full/10.1200/ EDBK_390094
5. Beishenaliev A, Loke YL, Goh SJ, Geo HN, Mugila M, Misran M, Chung LY, Kiew LV, Roffler S, Teo YY. Bispecific antibodies for targeted delivery of anti-cancer therapeutic agents: A review. J Control Release. 2023 Jul;359:268-286. doi: 10.1016/j.jconrel.2023.05.032. Epub 2023 Jun 13. PMID: 37244297.
6. Jin, S., Sun, Y., Liang, X. et al. Emerging new therapeutic antibody derivatives for cancer treatment. Sig Transduct Target Ther 7, 39 (2022). https://doi.org/10.1038/s41392-021-00868-x

Louise Duffy
Louise Duffy, Ph.D., has a very extensive background in the global Bio/pharmaceutical industry with more than 30 years of experience in R&D and Commercial Supply having held senior roles in GlaxoSmithKline as VP & Global Supply Chain Head, Biopharmaceuticals, where she also served as VP and Corporate Officer of Human Genome Sciences, Inc,. and Janssen R&D (J&J) as VP & Global Head, Strategic Sciences. She has wide-ranging experience in developing, licensing, and supplying Biopharmaceuticals, Vaccines, Cell and Gene Therapy products for global markets. Louise has broad CMC and Regulatory experience including the development of strategic CMC plans to support regulatory filings such as INDs, IMPDs, BLA, MAAs.

Campbell Bunce, Ph.D., is Chief Scientific Officer (CSO) and Cambridge Site Head at Abzena. He leads a talented team of scientists across a diverse range of expertise and capabilities to support drug discovery, design, and developability; and cell line development. With over 25 years’ of experience working in the biotech and diagnostics sectors, he ensures Abzena’s strong innovation focus and depth of scientific expertise is maintained through technological developments.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 41 www.international-pharma.com
Campbell Bunce
Generative AI and its Impact on Speed to Market for Pharmaceuticals
Did you know that it takes approximately seven years to develop and bring a new drug to market? However, this time can be significantly reduced by months or even years if life sciences companies leverage generative AI to accelerate insight and content generation. This timesaving is crucial in clinical development, as it can expedite the availability of treatments, improving or even saving lives. It also presents a substantial revenue opportunity, with some industry sources suggesting that bringing new treatments to market ahead of schedule can translate to a daily value ranging from £500,000 to £6.5 million.
Nevertheless, the pharmaceutical industry faces an uncertain regulatory environment, despite the application of generative AI rapidly advancing. In the face of this, some companies are adopting a more cautious approach to adopting generative AI tools, postponing investments until the path forward becomes clearer. Although this may seem like a prudent approach, it could become a source of regret in the long term for these organisations. By delaying adoption, they risk missing out on the numerous opportunities presented by generative AI, including advancements in drug discovery and an accelerated speed to market that
their more forward-thinking competitors may benefit from.
For life sciences enterprises who want to stay ahead of the game and hasten their time to market, they should prioritise digitally transforming specific areas of the clinical development lifecycle.
Streamlining the Research Pipeline
Research and development (R&D) is often the most time-consuming part of the drug development process, but AI can accelerate this process by up to 50% as the technology has a multiplier effect wherever it is applied.
Life sciences can implement generative AI at the very beginning of the R&D cycle, to aid in searching and synthesising available literature on a specific potential drug. Instead of beginning with a manual keyword search and sifting through hundreds of articles across various sources, teams could prompt a generative AI-enabled tool to rapidly search, gather and distil relevant articles – or even suggest unanticipated information pathways to explore.
Generative AI also has the potential to change how researchers find existing literature. Usually, researchers simply type keywords into the search box. But with a generative AI tool, they could state their goal into the prompt, providing context and
intent, for the technology to find reference materials to support that specific ask, saving significant time while broadening the research horizon.
Speeding Up Clinical Trial Protocol Creation
Compiling a clinical trial protocol document is a lengthy process that can take anywhere from a few months to over a year. Generative AI technology’s capabilities can automate a substantial proportion of the protocol writing process, bringing it down to days or even mere hours.
Generative AI can be trained on thousands of existing protocols in industry databases and each company’s own research data so that it can identify the patterns relevant to investigational products, certain conditions, specific patient populations, or other factors. As the generative AI tool identifies relevant patterns, it can combine all the insights to design a baseline study, with a defined narrative that determines eligibility, drafts exclusionary criteria, and provides other necessary details. It can generate a number of draft options that would later be evaluated and refined by a human.
Facilitating Quicker Secondary Market Launches
Once a new therapy has been approved to launch in one market, many companies will be looking to expand the launch into others. This

42 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Technology
process takes a tremendous amount of time and resources, from strategy development and market research to agency engagement, content creation and material development. Much like in the research and protocol writing processes, a lot of the steps in this part of the process could be automated with generative AI.
For instance, when the drug is close to gaining approval, generative AI could support commercial teams’ research and compile strategy documents for secondary markets, taking into account specific regulations the therapy will need to adhere to in the new country. Similarly, generative AI can be used to adapt existing content – including website copy, brochures and other promotional materials – to the language and culture of the secondary market. This could shave up to a year off the go-to-market timeline in new countries and massively reduce marketing and design costs.
Setting the Groundwork
Introducing generative AI into a business should be done one step at a time. It starts with fostering a culture of AI literacy, where every employee understands how the technology can
be used to reshape and empower their role. It is also important to build a solid ecosystem of partners, which includes relationships with academic institutions, data providers, and specialty generative AI vendors that will support the business’ knowledge growth and internal capabilities.
Once generative AI is introduced, it is a good idea to establish a body within the business to supervise how the organisation uses the technology and manages the upskilling and development of employees engaging with the tech. This body should also establish best practices and develop frameworks that guide the deployment of generative AI across the business.
Transforming Patient Outcomes
Beginning to use generative AI in a life sciences company is a significant undertaking and understandably not something to rush. Nevertheless, it is a must-have for companies aspiring to stay ahead of their competitors and a changing market. Equally vital is the commitment to providing comprehensive training to employees so they feel comfortable with and can maximise the benefits of using this technology. Establishing an internal
Technology
governing body to oversee responsible deployment of generative AI is also imperative to prevent any potential misuse.
Companies are already gradually establishing the groundwork required to harness the full potential of these technologies. Through ongoing experimentation, companies can accelerate the discovery, testing, and market release of their drugs. This advancement improves patient outcomes through safer, more effective and affordable drug development, whilst amplifying revenue opportunities in a fiercely competitive market.

Bryan Hill


















INTERNATIONAL PHARMACEUTICAL INDUSTRY 43
Bryan Hill is Chief Technology Officer for Cognizant Life Sciences, responsible for digital solutions and technology innovation. His focus is on how emerging tech can help clients increase innovation to bring new therapies to market faster.
Pressure regulators Control valves Pipeline ancillaries Steam traps Special equipment STEAMING SOLUTIONS FOR ALL INDUSTRIES geral@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467, Guia PBL PORTUGAL PRODUCTS MANUFACTURED IN PORTUGAL AV 001 IN EN 02.20
The End to Painfully Slow Cloud Migration?
Despite the benefits of moving to the cloud, migration can still be prohibitively time-consuming and costly, especially in a highly regulated industry like pharma. But that's all starting to change, thanks to tools designed to automate and speed up the process. Here, Arjun Khanna, CTO at labelling and artwork management specialist Kallik, explains how these tools could accelerate digital transformation strategies in pharma –enabling companies to get products to market faster, improve compliance and reduce recalls.
The idea that the pharma industry is a digital foot-dragger compared to other industries has been dissipating for some time now. Of course, caution is needed in this highly regulated industry – but the past few years, in particular, have brought significant advances in the application of cloud technology.
From academia and research to manufacturing and distribution, the cloud has paved the way for the development and delivery of innovative new treatments.
Without the cloud, it's likely we wouldn't have seen the Covid-19 vaccine rolled out at the speed, and on the scale that we did. Far from being behind the curve when it comes to technology, the industry giants had embarked on their cloud strategies a decade
or more ago – so they already had proven solutions to support the vaccine programme.
Over the years, they've introduced a mix of off-the-shelf 'software as a service' (SaaS) solutions, and ones they've built in-house using the leading cloud platforms, Google Cloud, AWS (Amazon Web Services), and Microsoft Azure.
These platforms, and the solutions built on them, are continually improving their security, scalability and performance, enabling companies to drive innovation and tackle both long-standing inefficiencies and new challenges like the rise in counterfeit medicines.
Investment in cloud infrastructure means that pharma manufacturers can process and maintain data within a secure environment at scale and in near real-time, using Artificial Intelligence (AI), machine learning (ML), the Internet of Things (IoT), and blockchain. Every aspect of the drug lifecycle can be, or is being, transformed by smart technologies, which in turn promotes better health outcomes for patients, and improved performance for life sciences companies.
Now, we're in a position to quantify the benefits of a cloud compared to on-prem which, according to one report1 can be seen in four key areas: R&D, clinical development, commercialisation and manufacturing, and safety and compliance. They include a 31% uplift in revenue from new products; an 83%

increase in AI/ML usage; and a 27% drop in unexpected manufacturing-related downtime.
Because of benefits like these, we're seeing more companies press ahead with cloud migration, with the appointment2 of chief digital and technology officers (CDTOs) to drive their strategy forward.
While it might feel less risky, even wise, to take a piecemeal approach to cloud migration, companies that have lacked the direction of a CDTO, until now, may find that key processes have been overlooked. As such, they remain largely manual and generally unfit for purpose in the modern world.
Labelling – A Prime Candidate for Cloud Transformation
Labelling and artwork management is one of those areas that is often overlooked when it comes to cloud transformation, yet it is a prime candidate for change. Manually producing compliant labels can take weeks; now it can be achieved in seconds, using cloud-based software. By using pre-approved templates and automation, manufacturers are able to minimise human intervention and standardise processes to save time and reduce the risk of error.
Not only is it time-consuming to produce these labels in the first instance, reformulations, regulatory change, and labelling mistakes often require urgent label updates. But it can typically take many man-months, sometimes many man years depending on the size of the migration, to change all impacted product labels manually, and can be a costly investment as design teams are often required to make creative changes.
It's also common for labels to require changes five times per year, making it a resource-intensive and unsustainable process for companies whose priority is to get innovative products to market and remain both competitive and compliant.
Again, without cloud migration, this wouldn't be possible. AI depends on the scalability and computational power of the cloud because of the large amount
44 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Technology
Versatile design intuitive delivery
Your fill volume may change, with Aidaptus® auto-adjust plunger technology your auto-injector doesn’t need to




Now in production
Accommodates both 1mL and 2.25mL glass syringes in the same device
See our auto-adjust plunger technology in action
Find out more by scanning the QR code or visiting ompharmaservices.com/ipi-april2024
*In addition to an air bubble and overfill Aidaptus® is a registered trademark of Owen Mumford Ltd, ©️2024 OMPS/ipi/ad/ob/0324/7





Now in collaboration with
INTERNATIONAL PHARMACEUTICAL INDUSTRY 45 www.international-pharma.com
0.3mL - 1mL* 0.5mL - 2.0mL*
Auto-adjust plunger technology 2.25mL 1mL Auto-adjust plunger technology
of data required in model development, management and deployment. So, in the simplest terms, companies that don't migrate to the cloud cannot leverage AI/ ML, IoT, or blockchain.
Time is Still a Barrier
While the direction of travel across the industry, cloud migration isn't a straightforward task – especially for pharma companies with large and complex infrastructure, and data stored in multiple locations.
Any hesitancy, particularly around security, privacy and good data management, is entirely understandable and prudent in a highly regulated sector where patient information and intellectual property (IP) are sacrosanct. Indeed, we would be more concerned if companies were adopting new technologies too rapidly without due diligence.
The cloud can be more secure than onprem servers and software because it's regularly monitored for threats and uses sophisticated encryption to protect data from cyberattacks. In fact, public cloud platforms (like AWS, Google Cloud, Azure) tend to be more secure than the private cloud because providers are constantly addressing vulnerabilities.
Beyond security and regulatory compliance (like HIPAA and GDPR), one of the biggest concerns for pharma companies
is the migration process itself – specifically, the time it takes to onboard applications, move data, and get staff up-to-speed.
Even if they want to update their labelling processes, the sheer amount of content, in multiple formats, needing to be moved from on-prem labelling systems to cloud-based ones still represent a major barrier.
We've seen this for ourselves on numerous occasions. In one company we worked with, it took between 70 and 80 people a year to manually migrate the data from their previous system to our cloudbased one. It meant that the project took between six and nine months longer than expected, leaving them unable to use their new software for two years. Poor time-tovalue like this is unacceptable, from their perspective and ours, and is only going to increase cloud hesitancy.
Automating Cloud Migration
The obvious solution to labour intensive tasks is automation, which led us to develop an Assistive Technology for Migration (AToM) solution, specifically to aid migration of labelling and artwork management systems to the cloud – while also improving time-tomarket by 70 percent, migration accuracy over 80 percent and increasing operational efficiency by 70 percent.
Recently, we've been working with Aston University3 as part of a Knowledge Transfer Partnership (KTP) to see how we could use
Gen AI and ML to automate and improve the artwork creation stage of the labelling.
By leveraging AI/ML, it can extract data from labels and artwork, in any format, and produce branded and compliant label templates that enable labels to be created and updated quickly. It achieves this by establishing a single, reusable data repository for accurate artwork management and labelling – and, crucially, learning to optimise the way it manages data and minimise errors. Getting over 90 percent right the first time.
Using AToM, it can take just a few minutes or no more than an hour to migrate all assets which is a significant time saving compared to typical cloud migration projects which can take months or, in the case of pharma, years. Because the process is automated, it doesn't divert skilled technical staff away from their day-to-day work in the same way a traditional cloud migration project might.
It's already been trialled by a global consumer company, as part of its AI journey. Since then, it has seen improvements in both compliance and the time it takes to get products onto the shelves.
AToM, using AI and Machine Learning (ML), establishes a single, reusable data repository for accurate artwork management and labelling, learning to optimise the way it manages data and minimise errors. The cloud makes it possible to manage a vast

46 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Technology
Technology
and growing digital repository of data on a global scale.
Looking Ahead
Once companies have migrated to the cloud, the opportunities to improve performance are near limitless, especially as technologies like AI/ML, blockchain and predictive analytics become more sophisticated. Along with improved data processing, management and automation, we also envisage the rise of VR (virtual reality) and AR (augmented reality) labelling, where users scan products to find out more information. The ability to manage this data on the cloud is essential.
These technologies are both supporting and shaping wider trends in the pharma industry.
For example, the combination of AI and blockchain could be a key weapon in the fight against counterfeit medicine, enabling transparency and authenticity throughout the supply chain. Our ability to use AI to automate and predict with greater precision opens the possibility for transformative and personalised treatments, decentralised clinical trials, predictive machine maintenance, and highly efficient labelling and artwork management processes that ensure compliance with current and upcoming regulations.
The challenge for technology providers, is to ease the transition to the cloud as much as possible, talking to the industry and recognising where the biggest roadblocks are. As we've seen first-hand, a focus on

automating the resource-intensive elements of migration can result in significant timesavings, enabling companies to maximise the value of their cloud investments as soon as possible.
REFERENCES
1. Hackett_Business-Impact-Cloud-Adoption-inLife-Sciences.pdf (awscloud.com)
2. Rewired pharma companies will win | McKinsey
3. Kallik Partners With Aston University To Develop World-First Technology For Label Artwork Management System | Kallik

Arjun Khanna
Arjun Khanna is the Chief Technology Officer at labelling software specialists, Kallik. Arjun has over 20 years of experience in the technology sector, and considers himself a cloud architect, an AI/ML engineer, a full-stack developer and a UX and VFX designer. While at Kallik, Arjun has been spearheading the creation of the company's AToM solution; a tool developed to specifically aid the migration of labelling and artwork management systems to the cloud.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 47 www.international-pharma.com
Reaching the Marketplace
Understanding
the
Complexities of Paediatric Drug Development and Manufacturing
Bringing a paediatric drug product to the marketplace can be very complex and also incredibly important as it increases the limited treatment options available for children, especially for niche and novel medications. The formulation development and manufacturing of paediatric medications poses unique challenges compared to that of adult medications. Developing safe, effective, and age-appropriate drug formulations for children requires a careful consideration of factors such as taste, dosage form, dosing accuracy, and palatability. Adhering to strict guidelines and best practices in the manufacturing process enables pharmaceutical companies to ensure that their paediatric patient is receiving the medication exactly as intended. Exploring the key challenges encountered in the formulation development and manufacturing of paediatric medications enables us to address and continually improve the healthcare outcomes of children.
Paediatric medications prompt a very specific set of challenges. They are required to reach across a broad age and developmental range, must be easy to swallow correctly, while being palatable to the patient, as well as being administered with ease at the point of care. Hence, these medications have very stringent guidelines that regulate their development, manufacturing, and packaging. Having the knowledge and expertise to navigate the regulatory complexities it is therefore essential to making sure that when the drug product reaches the marketplace it can be held to the highest quality, integrity, and safety standards.
One of the significant challenges in paediatric formulation development is ensuring palatability and taste-masking. As children are often sensitive to taste, this can create difficulties in administering medications due to their aversion to bitter and unpleasant flavours. Ensuring that medications are palatable is also crucial in supporting adherence and compliance and
foster treatment plan effectiveness. Making use of various techniques such as flavouring, sweeteners, or encapsulation to mask the bitterness and enhance palatability is therefore essential. However, the tastemasking techniques can be complex and may affect the stability and bioavailability of the medication.
To establish the suitability and effectiveness of a medication such as minitablets, formulation scientists must conduct food-drug interaction studies. Food exposure assessments covering the dose range are carried out for multiple exposure times, mimicking in-use practices performed by parents and guardians. Carrying out this research is vital in establishing the effect that the food has had on the physical structure, assay, impurity and dissolution profile of a medicine. These studies should be carried out on new and aged mini tablets, demonstrating consistent characteristics throughout the shelf life of the drug to ensure effectiveness. In addition, the formulations must consider factors such as age-appropriate flavours, allergies, and other potential interactions. Finding the right balance between effectiveness, safety, and palatability is essential in paediatric medication development
The paediatric patients span a wide age range, from newborn to adolescents, each with unique anatomical and physiological characteristics. Ensuring the safety and effectiveness of medications across various age groups requires extensive testing to account for all the developmental differences and side effects that can occur. As children’s bodies metabolise drugs differently at various stages of their development, formulation scientists need to have a thorough understanding of age-specific pharmacokinetics and pharmacodynamics.
It is also essential to consider ageappropriate dosage forms that are easy to administer, safe, and suitable for any given developmental stage of a child. Infants and young children may require liquid formulations, while older children may have the ability to swallow tablets or capsules. Developing child-friendly formulations, such as mini-tablets, powders and granules,
liquids, and orodispersible tablets calls for extensive research and formulation optimisation.
Recent clinical studies have shown that for children aged between six months and six years, mini-tablets provide equal acceptance rates when compared with sweet-tasting syrup formulations, similar to those that have been historically used for paediatric patient populations.1 The industry is also experiencing an increase in demand for the more traditional powder/ granule formulations, which are filled into sachets or bottles for re-constitution at the point of care. However, there are additional technical considerations associated with the development of these specialised formulations. These can range from developing formulations with the right flow characteristics, to analytical studies designed to demonstrate the in-use stability for re-constituted formulations, or the compatibility of the formulations with various foods and other methods or routes of administration to the patient such as the nasogastric tubes.
Accurate dosing is critical for paediatric medications, as under-dosing can result in therapeutic failure, and over-dosing can lead to adverse and sometimes perilous effects. However, dosing accuracy is equally challenging due to the variations in body weight, age, and developmental stages of children. Formulations must be continually tested during their manufacturing process, to ensure uniform blend consistency throughout the batch. The weight of formulations should also be checked, and in some cases counted, throughout the manufacturing and packaging process, to warrant accuracy in dosage forms.
There should be a focus on simplifying dosing calculations, providing appropriate measuring devices, and offering a range of dosing strengths to facilitate accurate administration at the point of care. A range of varied packaging solutions enables a pharmaceutical company to select the most suitable delivery method for their treatment. Child-safe, tamper-evident packaging solutions such as sachets, bottles, and blister packs are ways of safeguarding young
48 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Manufacturing
Vials with Exceptional Inner Surface Durability
For Biotech
Less interactions of drug molecules and formulations with the inner glass surface
• Optimized lyophilization process (less fogging)
• Reduced risk of glass delamination

For Diluents
Water for injection | Aqueous NaCl solution
Lower pH shift
• Reduced risk of glass delamination
NIPRO PHARMAPACKAGING INTERNATIONAL Blokhuisstraat 42, 2800 Mechelen, Belgium | pharmapackaging@nipro-group.com | www.nipro-group.com
children and at the same time complying with all the regulatory guidelines, while still remaining user-friendly for adults. All packaging should be checked and weighed, such as weighing bottles prior to, and after filling for example, to maintain accuracy throughout the process. Solid oral dose medications delivered in specific weighed solutions such as stick packs and sachets allow pharmaceutical companies to have a higher level of control that the correct dosage amount will be administered to the patient without under or over-dosing occurring.
As is the case with adult formulations, paediatric formulations, also need to maintain stability and potency over their intended shelf life. Various factors such as temperature variations, light exposure, and moisture can affect the stability and effectiveness of medications.
Developing stable formulations that retain efficacy and palatability throughout their shelf life is especially challenging for
liquid dosage forms. Extensive stability studies should be carried out to assess the physical, chemical, and microbiological stability of paediatric formulations under different storage conditions.
Selecting the correct packaging for a drug product is also key in ensuring the intended shelf life is upheld. Moisture-resistant and dosage specific packaging aids maintaining the desired therapeutic properties of the medication by protecting the drug product from external factors. Paediatric medications often require an extended shelf life to accommodate their storage and administration over prolonged periods of time, therefore their packaging must also be able to complement this purpose. For instance, an orodispersible tablet will require moisture-resistant packaging, making it unaffected by where it's stored and able to retain its intended efficacy.
The fact that paediatric patients have different physiological characteristics from adult patients has an impact on drug
safety and pharmacokinetics. Children’s physiological and developmental differences from adult’s present unique challenges in drug development. Factors such as metabolism, maturity of organs, body composition, and cognitive development, all have an influence on drug response and safety. Therefore, understanding these variations is crucial for developing safe and effective paediatric medications.
Preclinical and clinical studies enable the development of safety profiles to ensure that the medication does not pose potential risks to paediatric patients. They aim to provide data that helps to guide the dosing recommendations and inform many formulation decisions. However, there is a limited availability of paediatric clinical trial data which poses a challenge in development. This is partly due to ethical concerns regarding clinical trials in children. Regulatory agencies have strict requirements for paediatric-specific studies, which leads to a lack of comprehensive information on dosing, safety, and effectiveness in
50 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Manufacturing
paediatric populations. Hence, formulation scientists must rely on the extrapolation of data from adult patient studies and pharmacokinetic modelling to bridge the knowledge gap and inform decisions.
The development of paediatric medications involves navigating various complex regulatory requirements. Regulatory agencies have specific guidelines for paediatric drug development to ensure patient safety and efficacy. For example, EMA Paediatric regulation (26 January 2007) sets out “to ensure that medicines for use in children are of high quality, ethically researched and authorised appropriately and improving the availability of information on the use of medicines for children. It aims to achieve this without subjecting children to unnecessary trials or delaying the authorisation of medicines for use in adults”.2 While being extremely important, compliance with these guidelines can be time-consuming and costly, and require obtaining informed consent from parents and guardians. Balancing the potential benefits of new drugs against the risks involved entails careful assessment and stringent ethical guidelines. These factors, in turn, result in a limited number of approved paediatric formulations. Familiarisation with the current guidelines from the early phases of development leads to a clearer and definite adherence and is crucial to improving access to safe and effective medications for children.
Many pharmaceutical companies choose to engage with a contract development and manufacturing organisation due to the high level of knowledge required and the restrictions surrounding paediatric treatments. Outsourcing to a provider who has a track record in paediatric medication production means that the knowledge base, technical experience, and equipment trains are already in place. The benefit of using an end-to-end single source provider also enables a seamless transfer of knowledge from early phase development through to manufacturing, and into commercialisation; along with providing access to dedicated teams of scientists, project managers, quality, regulatory, artwork, and packaging experts that are focused on the product and the end-user. This helps to manage costs and ensure that regulatory requirements are adhered to the highest standard throughout the process. Engaging with an established and experienced provider can be key to marketplace success during the development and launch of paediatric medications, especially when it is the first
Manufacturing
paediatric treatment that a pharmaceutical company is looking to produce.
Formulation development and manufacturing of paediatric medications is a complex task that requires careful consideration of a multitude of factors that are highly specific to the demographic. The challenges related to taste and palatability, dosage form selection, dosing accuracy and safety, drug delivery, and regulatory hurdles, all need to be met and overcome to ensure optimum outcomes for the patient. Developing a comprehensive understanding of paediatric pharmacokinetics, physiology, and patient needs can contribute to improving the development of paediatric specific medications. Making sure the correct dosage form is selected for the appropriate route of administration to the patient, and stringent testing as well as check-weighing are performed throughout the manufacturing process, guarantee that safe and effective medications are delivered at the point of care. The main responsibility lies with the pharmaceutical company (and CDMO if used) to ascertain that all testing has been rigorously carried out, the regulatory standards are met, and the medication is provided in child-safe packaging that clearly defines dosage by age and/or weight, dosage method, and usage instructions.
Most importantly, by working through the complexities with the patient and care-
giver in mind, pharmaceutical companies are able to develop effective and patientfriendly formulations that provide a wider availability of dosage forms and improve the treatment options available for children.
REFERENCES
1. Journal of Paediatrics, Volume 167, Issue 4, Pages 893-896. Klingmann et al.
2. European Medicines Agency, Paediatric Regulation, https://www.ema.europa.eu/en/ human-regulatory/overview/paediatricmedicines/paediatric-regulation

Tom Hegarty, Director – Operations, Manufacturing for Almac Pharma Services, has worked in the pharmaceutical industry for over 25 years fulfilling various technical, operational and managerial roles. During this time, he has worked for large multinational and smaller regional based companies. His current responsibilities include pharmaceutical formulation development, technical support and strategic capital investment projects.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 51 www.international-pharma.com
Tom Hegarty
The Future of Sterility: Advancements and Innovations in Sterile Drug Product Manufacturing
Sterile drug product manufacturing is complex and plays a crucial role in the pharmaceutical industry, ensuring the production of safe and effective medications for patient use. Over the years, the advancements and innovations in this field have transformed the manufacturing processes, enhancing product quality, efficiency, and safety. This article explores the key developments in sterile drug product manufacturing, from traditional methods to cuttingedge robotic technologies, addressing regulatory challenges, ensuring patient safety and highlighting the industry's commitment to continuous improvement.
Traditional Sterile Drug Product Manufacturing
Aseptic processing involves the sterilisation of components, equipment, and the control of the environment to prevent microbial contamination during drug product formulation and filling. Historically, sterile drug manufacturing relied on aseptic processing techniques such as filtration and terminal sterilisation to eliminate or control potential microbial contamination.
a. Sterile Filtration: Sterile manufacturing of monoclonal antibodies (mAbs) and other biologic modalities relies on effective and efficient filtration processes to remove micro-organisms and particles that compromise drug product purity. Sterilising-grade filters with extremely small pores, typically ranging from 0.1 to 0.2 micrometres in size are used in the manufacturing of sterile drug products and play a pivotal role in assuring final product sterility.
b. Terminal Sterilisation: Terminal sterilisation refers to the process of sterilising a drug product, typically in its final container, to eliminate any microorganisms that may be present and ensure its safety, efficacy and stability. This process is typically performed using methods such as steam, radiation, or chemical sterilisation. The method used depends on the drug product's sensitivity, for example, gamma radiation may be used for drug products that
cannot withstand heat. It is important to highlight that if a product can be terminally sterilised, it should not be filled aseptically alone. It can be processed aseptically if it is terminally sterilised after. Buffers, placebos and some small molecules would be representative of such products that require terminal sterilisation.
Novel Aseptic Techniques
From isolator technology to restricted access barrier systems (RABS), these innovations contribute to maintaining product integrity, safety and compliance with global regulatory standards. Inline monitoring and control systems further improve the aseptic processing environment, by separating operators from the product at all times.
1. Single-Use Technologies
One of the significant innovations in sterile drug manufacturing has been the adoption of single-use technologies (SUTs) to replace conventional reusable stainless-steel vessels and processing lines. Single-use systems, including disposable bags, filters, tubing, and connectors, have also gained popularity due to:
• Little or no cleaning required – replacing at the end of each batch, saves time and money involved with cleaning and validation requirements.
• Flexible and adaptable – single-use products can be quickly and easily modified to fit each process and scale requirements. They also allow for a safe and quick transition of changes, minimising validation costs.
• Time saving – Removing cleaning and validation or verification periods, helps reduce time to market and improves batch turnaround times.
• Reduced risk of product crosscontamination – single-use products are replaced after each batch, which eliminates the risk of cross-contamination between products.
• Creates a sealed barrier that separates the product from the operators, de-risking the process and enhancing sterility.
Although SUTs are gaining momentum, which technology is best for sterile manu-
facturing is dependent on various considerations such as batch sizes. In many cases, biopharmaceutical companies and CDMOs alike are reaping the benefits of implementing hybrid approaches across their global sterile fill-finish manufacturing networks. For instance, SUTs are possible up to 2,000 litres, however, bags at that size are expensive and do have a known failure rate. SUTs are valuable up to 500 to 600 litres but less so above this size. Another point to note is that SUTs do add some recurring batch costs that are not necessary if you are manufacturing commercial batches. Then it is just processing and energy costs.
2. Advanced Aseptic Processing
In recent years, advancements in aseptic processing techniques have enhanced the sterility assurance of drug products. New technologies focus on minimising human interventions, automating processes, and optimising cleanroom designs.
a. Closed Systems: Closed systems aim to minimise the exposure of drug products to the environment and humans by reducing the risk of contamination. Isolators and RABS provide physical barriers, ensuring a sterile environment during drug manufacturing processes. Isolators utilise glove ports to support necessary interventions. These gloves need to be tested regularly and do represent a potential point of failure/ risk.
b. Robotics and Automation: Integrating robotics and automation into aseptic processing reduces human interventions, lowering the risk of microbial contamination. Automated systems for vial filling, syringe filling, and loading/ unloading lyophilisation systems enhance precision and efficiency.
c. Gloveless Robotic Isolator Filling Technology: With no glove ports, validated robots perform all operations within a closed isolator system. Using ready-to-use (RTU) components and integrating filling and handling robotics within gloveless isolator technology platforms, further reduces the risk of microbial contamination and particulate generation, providing an increased quality and sterility assured drug
52 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Manufacturing
product. Having a robust and validated robotic system is critical, as if there is an intervention required that the robot cannot perform, then the batch will be aborted.
Advancements in automation and robotics are revolutionising sterile fill-finish processing. From vial loading to stoppering, advanced robotics are streamlining and enhancing the efficiency of sterile fillfinish operations with reduced human interventions, minimising contamination risks, while optimising production speed and time to provide life-changing therapies to patients.
Regulatory Landscape and Compliance
The regulatory landscape for sterile drug product manufacturing is stringent, reflecting the critical importance of ensuring product safety and efficacy. Regulatory authorities, such as the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), continue to evolve guidelines and standards to keep pace with the technological advancements.
a. Annex 1 Revision: EudraLex Volume 4 Annex 1 of the EU Good Manufacturing Practice (GMP) guidelines outlines requirements for sterile medicinal products. The revision of Annex 1, which became effective in August 2023, focuses on addressing new technologies and concepts, including Pre-Use Post Sterilisation Integrity Testing (PUPSIT), the use of closed systems (RABS and isolators), automation, and reinforces quality risk management principles and the requirement for contamination control strategies.
b. FDA Guidance on Sterile Drug Products
Produced by Aseptic Processing: The FDA provides comprehensive guidance on aseptic processing, covering various aspects, including facility design, environmental monitoring, and process validation.
While both the FDA guidance and EU Annex 1 address sterile drug production, they may differ in specific details and approaches. Manufacturers must carefully consider and align their practices with the relevant guidelines to ensure the safety and quality of sterile products comply with the geographies they intend to market their products. While there is some mutual recognition, there are still some differences in expectations and regulatory requirements.
Quality by Design (QbD) and Process Analytical Technology (PAT)
Quality by Design (QbD) and Process Analytical Technology (PAT) are regulatory initiatives that have influenced sterile drug product development and manufacturing. These approaches emphasise a systematic understanding of the processes and use realtime monitoring to ensure product quality.
a. QbD Principles: QbD involves designing and controlling manufacturing processes to ensure the desired product quality. By identifying critical process parameters (CPPs) and critical quality attributes (CQAs), manufacturers can optimise processes and enhance the overall quality of sterile drug products.
b. PAT Implementation: PAT incorporates real-time monitoring and control of critical process parameters during manufacturing. Techniques such as near-infrared spectroscopy, Raman spectroscopy, and mass spectrometry provide in-process analysis, allowing for immediate adjustments to maintain the target product quality.
Environmental Monitoring and Control
Maintaining a controlled and clean environment is paramount in sterile drug product manufacturing. Advances in environmental monitoring and control systems contribute to the prevention of microbial contamination and the assurance of product quality.
a. Real-Time Environmental Monitoring: Continuous, real-time monitoring of critical parameters such as non-viable and viable particulate counts and microbial levels allows for immediate corrective actions, reducing the risk of contamination. Advanced monitoring systems provide a more comprehensive understanding of cleanroom conditions.
b. Barrier Technologies: Innovations in barrier technologies, including isolators and RABS, contribute to the creation of controlled environments that minimise the risk of microbial contamination from manufacturing operators. These technologies offer enhanced protection for both personnel and drug products.
While manufacturing a safe sterile drug product for patient use is the primary objective, the impact of operations on the environment is becoming a greater area of focus within the industry. Considering environmental monitoring and impact, biopharmaceutical manufacturers and their partnering CDMOs are working to
Manufacturing
incorporate green initiatives into their sterile fill-finish processes, from using ecofriendly packaging materials to energyefficient manufacturing facilities.
Conclusion
Advancements and innovations in sterile drug product manufacturing have transformed the pharmaceutical industry, enabling the production of safer and more efficient lifechanging therapies. As the industry moves forward, aseptic liquid filling, automation, and environmental considerations will continue to shape the landscape of sterile fill-finish, with a focus on adherence to evolving regulatory standards. It is imperative that sterile fill-finish equipment and manufacturing environments advance to meet the “current” in cGMP.
The commitment to patient safety and product efficacy remains at the forefront of these advancements, driving continuous improvement in sterile drug product manufacturing. As technologies mature and regulatory frameworks adapt, the industry will undoubtedly witness further breakthroughs, ensuring the delivery of high-quality biologics and sterile injectables to patients around the world.

Shawn Cain
Shawn Cain is the SVP of Sterile Development and Manufacturing at PCI Pharma Services, a leading global CDMO. With over thirty years of experience, Shawn combines process engineering and project management to direct the development and manufacture of sterile pharmaceuticals, cell-based biologics, and medical devices. Most recently, he served as the COO of LSNE, which PCI Pharma Services acquired in 2021. Shawn has also worked at Organogenesis and served as the Director of Operations for another pharmaceutical CDMO, Formatech, Inc. Prior to these roles, he held positions as Interim President and CEO at Arbios Systems, Inc., and was employed at Becton Dickinson's Biologics Business. Additionally, he served as Vice President of Operations at Circe Biomedical, Inc., leading bioartificial liver technology development. Shawn holds an M.S. in Biological Sciences from the University of Massachusetts and a B.S. in Biological Sciences from Northeastern University.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 53 www.international-pharma.com
Application Note
Automating Biotherapy Production at the Speed of Market Expansion
The effort to scale biotherapy production and meet the demand for personalised medicine is well underway, and robotic automation is taking on an everexpanding role. There are numerous possibilities for robotic automation in biotherapeutics and by extension many other pharmaceuticals. Suppliers and system integrators are finding new ways to transform production, from Grade A/B environments to inspection, final packaging and palletising.
The term “biotherapies” generally refers to medicinal products, including biopharmaceuticals, that are derived or produced from biological sources such as bacteria or animal cells. Biotherapies can also be the product of gene therapy, involving the insertion of genetic material into cells; cell therapy, in which healthy cells are transplanted; or tissue therapy. Recombinant proteins, vaccines, and monoclonal antibodies are among the most significant products.
Biotherapies are particularly useful for treating chronic diseases such as cancer, diabetes, and rheumatoid arthritis. The techniques and technologies of biotherapy also play a central role in personalised medicine, where demand continues to surge. These and other drivers make biotherapeutics one of the pharmaceutical sector’s fastest growing segments. Producing biotherapeutics at scale, however, poses significant challenges. Minimising risk, maintaining consistent product quality, and optimising process control are all important factors.
The Bioproduction Challenge
The field of bioproduction encompasses all the complex manufacturing processes and biotechnologies that go into producing biotherapies and biopharmaceuticals. Often this includes modifying the genetic heritage of cells and cultivating them in large-scale bioreactors during upstream production. Risk management and quality control measures are critical at every stage to maintain optimal sterile conditions. Even though market forces exert pressure on manufacturers to minimise production


times and increase yields, safety and efficacy cannot be compromised.
Automation is turning out to be a major ally in the effort to meet such demands. An early forerunner, the PUMA robot was used in the Cellmate cell culturing system for the production of biotech drugs in 1993. Today, innovative robotic devices and systems are poised to shape the future of bioproduction, making it more flexible, reliable, and safer.
Case Study: Scaling CGT Manufacturing
Cell and gene therapy (CGT) manufacturing is a complex process, often involving hundreds of steps. Quite a few variables can affect the quality, safety, therapeutic efficacy and commercial viability of these sophisticated biologics. With the market growing rapidly, the race is on to scale manufacturing and meet rigorous GMP and cleanroom requirements. Technology offers ways to increase production and ensure quality
54 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
For scaling up your production, robots are placed in a row or the same robot operates on a seventh axis as part of a linear transfer system, thus maximising its capabilities.
CGT scalability, solved. A Stäubli TX2-60 Stericlean robot picks an IV bag from a conveyor for transfer to filling and sealing stations within BATG’s Vers-A-Tech™ aseptic fill-finish platform, yielding up to 300 IV bags per hour.

while decreasing risks and costs as volumes increase.
BAUSCH Advanced Technology Group (BATG) engineers and manufactures controlled and aseptic fill-finish machines for the life sciences sector, specialising in processing solutions for parenteral medications. The company developed its Vers-A-Tech™ (Versatile Aseptic Technology) platform specifically for CGTs, enabling the aseptic filling and closing of multiple dosing formats including nested vials and syringes as well as IV bags on a single machine.
The process includes loading, de-bagging and de-lidding, surface decontamination, filling and stoppering, lyophilisation (freeze drying), crimping and discharge, all with 100% IPC through integrated weighing cells. The system can be semi-automatic or fully automatic, with all sensitive processes taking place in isolators. Restricted access barrier systems (RABS) with glove ports ensure aseptic conditions during manual tasks.
Crucial to the Vers-A-Tech platform is the Stäubli TX2-60 Stericlean robot. Specialising in industrial automation, Stäubli works extensively with customers in the life science sector and has had numerous successes in pharmaceutical production automation. Its six-axis Stericlean robot proved to be the perfect solution for automating the highprecision aseptic pick and place operations involved in small- and medium-batch CGT processing.
An ISO Class 4 cleanroom-compliant robot designed to operate in a GMP
Grade A environment, the Stericlean can automatically process different types of containers in isolators with no risk of contamination. A fully enclosed structure with special seals prevents the ingress of airborne particles. The surface is completely smooth, eliminating retention areas, and protected by a high-resistance coating. All connections run through the base of the robot, safely outside the isolators.
The process begins with an operator loading the nested containers into the machine, where they are de-bagged and delidded either automatically by a robot or manually via RABS. The robot then transfers the containers to a fully enclosed sterile transfer chamber for decontamination. A second robot picks up the nested containers and places them on a transport conveyor for de-nesting, either by BATG’s custom nest holder or by another robot. Next, a robot holds the containers steady as they are filled by BATG’s peristaltic pump with filling accuracy of less than +/- 0.5%.
The robot stoppers the filled containers, and when complete, the batch is re-nested and passed through another isolated transfer chamber to the freeze dryer. After lyophilisation, they are crimped and discharged. When processing IV bags, the robots readily adapt. As the empty bags enter the first isolated processing station, a robot picks them up individually, cuts the tube (if needed), and inserts the open tube into the filling needle. Once the filling is complete the robot moves the IV bag to a heat-sealing station, and finally onto a transport conveyor for discharge.
Enabled by Stäubli Stericlean robots, specifically designed for aseptic processing, Vers-A-Tech can fill and finish up to 1,200 nested vials or syringes per hour and up to 300 IV bags per hour. A seamless changeover to different formats eliminates the need for separate fill-finish lines. As companies add new CGTs to their product portfolios, the system can continue aseptically filling orders at scale, thanks to its flexible modular design and the adaptability of the robots.
Confidential Upstream Processes, New Downstream Trends
Automating pharmaceutical applications like this is challenging due not only to the complexity of the production processes involved, but also to the confidential nature of these processes. While the process of producing generic drugs is commonly known, for example, the process of producing personalised medicine, notably ATMPs (advanced therapy medicinal products), remains closely guarded by pharmaceutical companies. This adds another hurdle to industrialisation, as suppliers are left to generalise robotic applications as “management of the process of cell & gene therapy.”
And yet, there are numerous opportunities for robotic automation in personalised medicine and biotherapy production. By nature, personalised medicine is small batch production, or even unique batch production. The need for machine builders to enable efficient, commercially sustainable small batch production lines, or standalone machines, has given rise to the need for a robotic arm that is more flexible and able to work safely within a small isolator.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 55 www.international-pharma.com Application Note
TX2-90 Stericlean robots. Robots are used in the production of biomedicines validated for pharmaceutical environments.
Application Note
The robot’s role is also expanding. As seen in BATG’s fill-finish system, it is no longer used for a single task, and while its workspace remains limited, it is now placed at the center of production. New developments in small batch equipment are expected to follow the rest of the process down to the logistics area, with small batch inspection and packaging machines.
Robots are proving their ability to adapt to these trends, provided they have a small footprint, the ability to operate safely in Grade A/B environments, and the flexibility to handle any product type. The use of robot tool changers in biopharmaceutical applications offers significant advantages, as this technology greatly expands the number of tasks that can be performed by a single robot.
Solutions for Scaling Up: Moving Toward Mobility
Producing a drug for a single patient has a high cost, and pharma is entering a new era of industrialisation, with the objective of scaling up by producing batches in parallel. One approach is to reduce the footprint of robotic cells and increase the number of cells in the line. Another is to increase the size of the robots and limit scaled-up production only to the area around the robot, where the number of stations dedicated to “management of cell & gene therapy” would multiply.
In yet another scenario, robots are placed in a row, as in conventional industrial production, or the same robot operates on a seventh axis as part of a linear transfer system. This solution is still under development, since currently there is no mobile robotic solution on the market for pharma cleanroom areas. A cleanable, Grade A-compatible AGV or mobile robot would provide a vital link between upstream and downstream processes –opening up the possibilities for scaling production in Grade A/B environments.
The impact of the revised GMP Annex 1 currently seen in generic drug manufacturing will push the industry overall to adopt more stringent contamination control strategies in terms of hygienic design and equipment cleanability. Manufacturers of personalised medicine will invariably follow the same rules. But thus far, the need for Grade A compatibility, and more recently GMP Annex 1 compliance, have posed a barrier to the use of mobile robotics in biotherapeutics.
The most promising solution lies in using robots that align with GMP standards and use

FDA-compliant materials in the production of biotherapies and biopharmaceuticals.
Stäubli
Stäubli is a global industrial and mechatronic solution provider with four dedicated Divisions: Electrical Connectors, Fluid Connectors, Robotics and Textile, serving customers who aim to increase their productivity in many industrial sectors. Stäubli currently operates in 28 countries, with agents in 50 countries on four continents. Its global workforce of 6,000 shares a commitment to partnering with customers in nearly every industry to provide comprehensive solutions with long-term support. Originally founded in 1892 as a small workshop in Horgen/ Zurich, Switzerland, today Stäubli is an international Group headquartered in Pfäffikon, Switzerland.
www.staubli.com
Stäubli Robotics
Stäubli Robotics’ unique product portfolio contains 4 and 6 axis industrial robots, cobots, mobile robotics and Automated Guided Vehicles. The powerful, high precision solutions allow clients in many demanding industries to tackle the challenges of Industry 4.0 under specific manufacturing conditions.
www.staubli.com/de/en/robotics.html
While the use of robotics in bioproduction is relatively still in its infancy, it is apparent that the parallel production method will bring even more robots into the bioproduction space. As this new bioproduction paradigm takes form, the future looks increasingly mobile.

Rudolf M. Weiss
Rudolf M. Weiss started in the senior management at the Stäubli Robotics Division of the STAUBLI Group in October 2020. He took over the global responsibility for robotic/automation business in pharma and medical devices as Global Head of Pharma and as a Member of the Global Management Team. After his studies in Stuttgart Rudolf M. Weiss started working at Servotech in 1999. From 2001 on, he was working at Groninger where he was responsible as Marketing Director, Senior Project Manager and Sales Director. After experiences at FIMA, he decided to go back to pharma and joining Bausch+Ströbel the global leading company for fill/finish equipment 2018 he was announced to be Authorized Officer and Director Sales & Marketing. He can draw on 20 years of experience in the field of Pharma Fill / Finish.
Email: rm.weiss@staubli.com
56 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
At BATG the Stäubli robot precisely stoppers a tray of nested vials after filling. Changing over seamlessly to different formats, Vers-A-Tech™ can process up to 1,200 nested vials or syringes per hour.
ROBOTICS
Unrivalled expertise for every pharmaceutical environment

Next-level pharmaceutical robots for optimal health
Highly aseptic, sterile or Grade C environment, demanding or routine tasks, Stäubli robots meet the highest hygiene and cleanliness standards, while bringing greater efficiency and reliability to your pharma applications.
Achema
10 – 14 June 2024
Hall 3.1, Booth J72
Stäubli – Experts in Man and Machine
www.staubli.com
INTERNATIONAL PHARMACEUTICAL INDUSTRY 57 www.international-pharma.com Stäubli Faverges SCA, Tél. +33 (0)4 50 65 62 87, robot.sales@staubli.com
The Key to Formulation Development is in the Details
During the tablet manufacturing process, tooling and press manufacturers are often faced with ongoing challenges. As a tooling and press manufacturer, you should be able to expect support from your partnering vendor during all aspects of the compression process. These services should include press operator training, maintenance /calibration services, quick delivery parts, tooling/ tablet design, and powder formulation support.
If any compression issues occur, these services act as a support tool to help you overcome manufacturing challenges. However, if there is a change in formulation, a process following Scale-Up and Post Approval Changes (SUPAC) guidelines must occur before moving forward. Time consuming and often frustrating, the SUPAC process is guaranteed to halt manufacturing and should be avoided.
This article will review the importance of the formulation development process and how a thorough approach can reduce manufacturing issues, costs, and downtime. The case studies provided in this article were performed at the Natoli Institute for Industrial Pharmacy Research & Development — Arnold and Marie Schwartz College of Pharmacy and Health Sciences on the Brooklyn, NY campus of Long Island University.
For both formulators and research professionals, developing a tablet formulation that is both robust and able to be scaled into manufacturing without issue can be a challenge. Tasked with this, formulators and research professionals use their understanding of the science side of powder compaction to effectively communicate with scale-up and manufacturing groups – increasing return on investment and reducing time to achieve a marketed product.
Single-station tablet presses offer many advantages during early development, such as requiring a limited amount of material
to characterise potential formulations. With only a few milligrams of material, the die filling process can be performed manually. Characterising just the API can also be performed on these machines allowing the scientist to select the appropriate excipients based off the results of the compacted API.
Figure 1 (below) is a tabletability profile performed on the NP-RD10A singlestation tablet press. This is an example of evaluating a high drug load directly compressible APAP formulation (DB) versus a wet granulation APAP formulation (WG).
Instead of collecting the data using compression force (kN), there is an advantage to normalising the punch tip face area and utilising compaction pressure. With any new formulation the required compression force is based on the material properties and tablet size. Performing a compaction study from 50MPa–300MPa covers the normal ranges for compressing pharmaceutical tablets of all sizes.
Furthermore, the tablet tensile strength is a normalisation of the tablet geometry. Target hardness or a breaking force value (kilopond or Newtons) cannot be determined until further testing is performed. Target tensile strength of 1–2MPa are representative values for a robust tablet that will withstand handling, friability, and coating operation.
The profile clearly shows that the wet granulation blend yields stronger tablets than the directly compressible blend throughout the compaction pressure ranges. Although


58 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Manufacturing
Formulation Composition Formulation Proportion (%) Acataminophen 74.40% Croscarmellose Sodium 9.00% Lactose Anhydrous 4.30% Polyvinylpyroolidine K 30 9.60% Talc 1.06% Colloidal Silicon Dioxide 0.53% Magnesium Stearate 1.06%
Figure 1
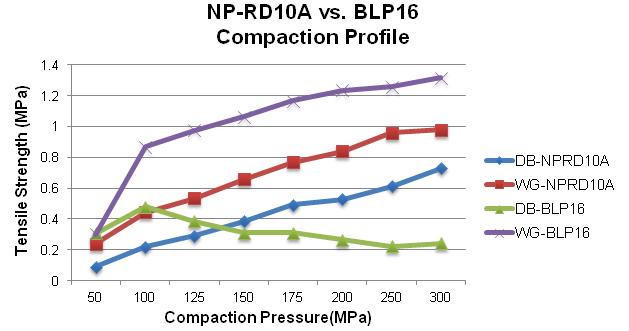
is a cost advantage with directly compressible blends as the equipment investment costs and process times are much lower. The next step in the process is to perform additional studies on a rotary tablet press.
Figure 2 depicts the tabletability profile performed on the BLP 16 rotary tablet press at 20 RPM turret speed for the DB and WG formulations. The BLP16 rotary tablet press is a 16-station B tooled machine that is capable of running at variable turret speeds.
The profile shows different results as compared to the RD10A single-station tablet press. This data is representative of what can be expected at the manufacturing level since the manufacturing machines utilise the rotary press design with upper and lower compression rollers.
In this case, the upper and lower punches are moving horizontally across the rollers and vertically to compress the powder whereas the single station machine only moves vertically for non-eccentric designs.
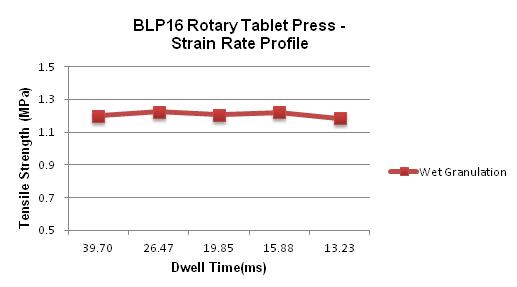
This difference changes the dynamics of the compression event and has an impact to the final tablet attributes as shown in this example.
Figure 3 Natoli – Single Station vs. Rotary Press Tabletability Profile depicts the compaction profiles for the two different machines.
The WG formulation run on the BLP16 yields robust tablets above 1MPa tensile strength at a reasonable compaction pressure where the DB formulation exhibits capping above 100MPa of compaction pressure. It is clear that the WG blend is the choice formulation, and that the BD blend will fail on a manufacturing rotary press.
In Figure 4 are examples of different tabletability profiles.
Profile “A” is an example of good tabletability, whereas Profile “B” is an example of when tabletability is acceptable at lower compaction pressures but deteriorates at higher pressures and Profile “C” is an example of poor tabletability.
Many factors can impact the tabletability curve including the powder deformation characteristics, particle size, shape, moisture content, amount of powder fines. Every product behaves differently on a tablet press, even varying when run on a different day. Variation is due to changes in the properties of the raw materials, API, and excipients from batch to batch.
As shown by the data in figure 4, it is crucial that your product has a robust tabletability profile. Profile “B” is an example of where your tablet attributes might be acceptable – at research or scale up levels – but fails at the manufacturing level. Operating too close to the peak of the curve or on the descending side offers no flexibility
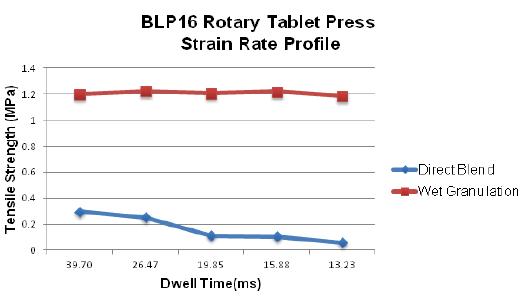
INTERNATIONAL PHARMACEUTICAL INDUSTRY 59 www.international-pharma.com
Manufacturing
Figure 2
Figure 3: Natoli – Single Station vs. Rotary Press
Figure 4 there

to the press operator when issues are found and with the many variables as described above this is crucial.
The use of pre-compression can improve your tabletability profile as it de-aerates and consolidates the material before the main compression event; but it is wise to save this tool as a backup when issues arise at the manufacturing level. Developing a robust formulation without pre-compression will allow the manufacturing press operators to make press adjustments to solve these issues.
Another study that is valuable in the development process is a strain rate study; where the compaction pressure is held constant and, in this case, we chose 150MPa since tablets were robust at this level, the turret speed was incrementally increased, and tablet attributes were evaluated.
Instead of evaluating tablet strengths at different turret speeds it is a more scalable approach to normalise for the turret pitch circle diameter, punch head flat diameter
and evaluate the tablet strengths as a function of dwell time which is the time under maximum compression force or when the punches are no longer moving vertically. Another important parameter to consider for press scalability is the punch vertical velocity or loading rate and the decompression event.
In Figure 4 we can observe that this WG formulation is not strain rate sensitive at dwell times as low as 13 milliseconds.
Ultimately the product will be produced on a high-speed manufacturing press and the dwell times can be calculated and further studies can be performed at the research level. As most research presses are designed with smaller diameter turrets, reaching similar velocities as the manufacturing machines cannot be achieved so your tooling head profile can be designed to simulate similar dwell times.
The methods above are ways to evaluate your potential formulation's tabletability and scalability; now let's discuss how the
ingredients that make up the formulation impact the final tablet quality. Apart from the tablet’s active ingredient, other essential components include diluents, binders, disintegrants, lubricants, glidants, coloring agents, and more. This next section will describe the importance of powdered lubrication (i.e. Magnesium Stearate) and the negative effects of excessive amounts.
Lubricants are essential for successful tablet manufacture. They provide a means to reduce the friction at the interface between a tablet’s surface and the die wall during the tablet ejection event. High ejection forces and excessive friction can cause premature wear to the lower punch heads and press cam tracks, visual striations on the tablet belly band surface, and lamination. Lubricants can also improve flowability as it reduces the inter-particle friction.
With the many benefits of adding lubricant, there are negative effects when the amounts are excessive and not optimised. An excessive amount of lubricant can cause tablet strength issues, and increased disintegration and dissolution times, and with the tablet press paddle feeder this issue can be exacerbated when feeder speeds are too high. The feeders could potentially overblend the formulation causing the lubricant to coat more particles inhibiting the bonding process during compression.
As lubricants are hydrophobic, it will prevent the penetration of fluids, hence the disintegration and dissolution issues. Furthermore, when the tablet press is not set up properly and the selected fill cam is not appropriate for the dosing cam setting, the excessive scrape-off material is recycled back into the feeder for further mixing.
Performing a lubricant study in the development process with varying amounts of lubricant will identify the optimum level. During a compaction profile study as described earlier, the ejection force can be recorded as the applied compression force is incrementally increased. Since the ejection force is the product of the residual radial die force and the coefficient of friction between the tablet belly band and die wall the ejection force should increase with increased compression. If the ejection force does not increase with increased compression, the friction is reduced, indicating the lubricant level can be decreased.
Furthermore, a properly designed ejection transducer is required to accurately measure
60 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Manufacturing
Manufacturing

the ejection event. As most tablet presses adjust the lower roller position to change compression force and tablet thickness the lower punch head will contact the ejection cam at a different location depending on the setting.
A full travel transducer design (Figure 5) is insensitive to punch position whereas a
button load cell design is only accurate at one contact point. Figure 5 is an illustration of the ejection event. This example adds another variable where the die has an excessive wear ring resulting in higher ejection forces and tablet quality issues.
Conclusion
With the many challenges in the pharma-
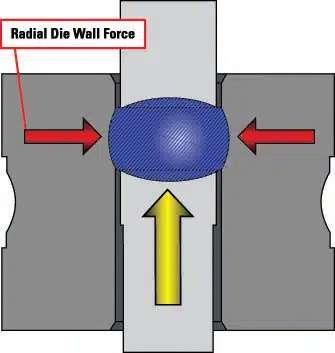
ceutical manufacturing world today, tooling and press manufacturers are your key partners in the support of delivering a quality product. Tablet manufacturing issues arise from many variables including operator training, calibration and maintenance of equipment, and the formulation that is compressed.
It is crucial in the development process to optimise the formulation and evaluate tablet robustness from Tabletability and scalability studies. This will help minimise the challenges faced at the manufacturing level and will allow the press operators to make press adjustments when many variables arise unexpectedly.

Robert Sedlock
Robert Sedlock is the Director of Natoli Scientific and brings over 25 years of industry experience. Robert's earlier career spans strain gage technology and data acquisition systems. When joining Natoli Engineering Company in 2015, Robert worked with the industrial pharmacy graduate students at Natoli Institute / Long Island University, AMS school of Pharmacy in Brooklyn, NY. This initiative brought in industry contract laboratory projects and developed the solid dosage manufacturing process training program. In 2018 Robert opened the Natoli Scientific, Telford PA facility which is now a 14,000 square foot facility that offers solid dosage training programs, contract research and development projects from preformulation to formulation development and start-up manufacturing GMP services. Robert is an invited speaker at many universities worldwide and has published many technical articles and peer-reviewed research papers.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 61 www.international-pharma.com
Figure 6
Biocompatible, Pre-coloured and Sustainable ABS Optimised for Laser-marking to Support
Medical UDI Identification along with Sustainability Targets
The EU’s Medical Device Regulation (MDR) and US’ Code of Federal Regulations Title 21, with regard to the labelling for medical devices (21 CFR 801 Subpart B), require a Unique Device Identification system (UDI) on each medical device, in addition to the creation of a central UDI database.
Several key public health targets are meant to be covered through UDI, such as anti-counterfeiting, safety communication promotion, reduction of inappropriate use of the device as well as the management of adverse events, incident reporting and efficient device recalls.
Laser-marking is a valid alternative to get the unique identifier permanently attached to the medical device and offers a series of additional advantages compared to alternative technologies like labelling, pad printing or hot stamping. Digital information can be created and stored in a digital database, facilitating traceability tasks and regulatory requirements, while digital data manipulation enables high flexibility to fit laser-marking according to the required size of the device, the available surface, and geometry. Adhesives or glues used in labelling or dyes /solvents needed in conventional printing processes are not required, avoiding contamination issues and the environmental impact related to the use of such substances.
There are some materials that can be more readily laser-marked than others, while some will give very poor results; indeed, that`s because it is very important to select the correct material to be laser-marked, besides choosing the correct laser-marking machine settings. In addition to the material, special attention should be given to its colour, in particular to the formulation of the colour that gets combined with the material and it’ s needed when creating certain lasermarking effects or contrasting colours.
In the case of reusable medical devices, frequent cleaning, disinfection, or sterilisation procedures need to be also
carried out to allow their safe reuse. The longterm readability of HRI (Human Readable Interpretation of the data characters) and AIDC (Automatic Identification and Data Capture through bar codes, smart cards, biometrics, and RFID) falling within the expected device lifetime is a challenging task. For this reason, it becomes even more important to maximise the contrast and resolution of the laser-marked text and make it indelible. Therefore, specific medical materials optimised for laser-marking have been developed.
Considering that ABS polymers are widely used in the production of external enclosures, covers, or shells for reusable medical devices to achieve a high level of aesthetic, functionality, and medical regulatory compliance, it’s important to bear in mind that ABS is in itself a lasermarkable material. Correct laser settings allow good laser-marking results to be achieved, nevertheless, there are special ABS formulations that have been optimised for laser-marking and can be used for boosting the contrast and resolution and attain the desired colour and contrasting effect on the surface of a medical device.
Special ABS formulations are particularly valuable when other relevant properties of the material come into play and must be maintained alongside the laser-
marking optimisation, like for example the biocompatibility requirement according to ISO 10993.
Biocompatible medical ABS-optimised formulations for laser-marking that capture the laser energy more efficiently have been developed, enhancing the lasermarking effect whilst guaranteeing medical compliance.
Since ABS and its colour are both important factors in obtaining the required laser-marking effect, creating a selection in the database of available pigments with the highest performance assessed under different laser systems is one of the critical elements to look at with regard to this type of development. Several combinations of material-colour samples must be prepared and pass the scrutiny of a laser processing qualification process. Previously, another essential aspect is obtaining the optimal compound out of the selected colour pigments in the colour formulation of the ABS material formulation. The mix of colour masterbatches with ABS material in natural colour during the injection moulding process cannot guarantee the same level of colour homogeneity as pre-coloured ABS. The reason resides in a better dispersion of pigment powders in the ABS matrix during the compounding extrusion process required to produce pre-coloured ABS. Instead, in

62 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Packaging
CONTRACT MANUFACTURING AND PRIVATE LABEL PRODUCTS





CONTRACT MANUFACTURING OF LIQUID DRUGS AND MEDICAL DEVICES
•BAG-ON-VALVE
•NASAL SPRAY PUMPS
•DERMAL SPRAY PUMPS
•AIRLESS SPRAY PACKAGING
•PureHale®
CE-MARKED PRIVATE LABEL PRODUCTS FOR YOUR BRAND
•SPARKLING SALINE NASAL SPRAY
•SALINE/SEAWATER NASAL SPRAY
•EAR CLEANSING SPRAY
•WOUND WASH SPRAY
•EYE WASH SPRAY
•ADHESIVE REMOVER SPRAY
•BURN GEL

www.aurenalabs.com • contact@aurenalabs.com
INTERNATIONAL PHARMACEUTICAL INDUSTRY 63 www.international-pharma.com
TODAY.
LET’S COLLABORATE TO CREATE YOUR NEXT PRODUCT. CONTACT AURENA

a masterbatch, the colour pigments have been previously dispersed in a carrier, an additional material in granules 100% compatible with the material to be coloured (e.g. SAN or ABS in the case of ABS), being only afterwards dispersed into the ABS material through an injection moulding machine, which has, in addition, less mixing power efficiency than a specialised compounding extrusion machine. For the same reasons, colour stability and consistency show higher quality levels in pre-coloured ABS than in natural ABS previously combined with a colour masterbatch.
In the case of medical devices requiring biocompatibility, among the available pigments that optimise laser-marking contrast only the ones with biocompatibility properties and staying within the legal admissible concentrations can be selected, to guarantee the required medical regulation compliance. A key support is provided for this task by the ELIX Product Stewardship department, which adds value through the constant surveillance of admitted substances and concentrations and by guaranteeing that the complete formulation, including the colour formulation, stays within legal limits. The re-combinations of the validated pigments result in new colour formulations for medical ABS, in which the laser-marking contrast is enhanced while guaranteeing the same material colour target.
In addition to regulatory compliance, biocompatibility, long-term UDI conformity and optimised colours and marking contrast, the medical industry is also looking for more sustainable ABS materials for medical devices.
High concern is given to CO2 emissions, which are strictly related to their
environmental impact such as global warming, but also other sustainability priorities are considered, for example the introduction of circular products that can avoid incineration, landfills (or dispersion into the environment in the worst-case scenario) at the end of life of the product.
Due to biocompatibility risks associated with recycled medical devices, it is not possible to directly apply circularity and reintroduce in the same medical application recycled ABS materials coming from medical devices. At least not a mechanically recycled ABS, that may include contaminants and does not imply a significant change in the chemical structure of the material. Instead, such a type of circularity would be possible if a chemical transformation takes place. The resulting pyrolysis oil obtained from waste can be used to feed the petrochemical crackers, as an alternative to NAFTA oil, and useful basic molecules can be obtained by chemical reactions and be reintroduced into the supply chain of ABS plastics production.
In this sense, ISCC+ certified chemical recycling and also bio-attributed certified feedstocks have such a great potential to create circularity, complementing and overcoming mechanical recycling limitations when it comes to the need for materials free of contaminants for medical applications.
Chemically recycled and bio-attributed materials have the same chemical composition and properties of a virgin medical resin. As a consequence, they fulfil the same medical applications and meet the stringent medical regulation requirements. All the colours and optimised medical formulations that are available as virgin medical ABS can also be used in their circular and bio-circular versions, guaranteeing not
only regulatory compliance but also the availability of bright and intense colours and optimised colour formulations for laser-marking in chemically recycled and/ or bio-attributed ABS formulations. These types of ABS medical formulations cannot be achieved with mechanical recycled content. In the near future we are likely to witness a redefinition of waste as a raw material even for stringent medical applications. The mission is to offer top-quality, sustainable solutions, pushing the value chain towards a circular economic model.
Conclusions
The medical device industry is looking for compliant materials that fulfil several strict regulatory requirements, like biocompatibility and Unique Device Identification, but are also oriented towards a lower environmental impact and CO2 emissions reductions. ABS materials are widely used to produce external enclosure components of medical devices due to their mechanical properties, colour appearance and post-processing versatility such as laser markability. Laser-marking represents a valuable technology for the Unique Device Identification of medical devices, supporting the traceability implementation and other related benefits. Further to recent ABS material developments of leading manufacturers, special pre-coloured circular and bio-circular ABS formulations with reduced CO2 emissions have been made available, in conformity with the stringent requirements of the medical industry. By doing so, regulatory compliance, biocompatibility, material properties, colour stability, waste or bio-based certified content and even laser-marking optimisation can be ensured at the same time.

64 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Packaging
Luca Chiochia
Luca Chiochia graduated at the Politecnico di Milano University, Italy, in Management Engineering and covers the position of Business Development Manager at ELIX Polymers for the healthcare/medical business since 2017. He is fluent in six languages and has over 15 years of experience in industrial applications from multiple sectors' perspectives including thermoplastics, thermoset plastics, composites, electrical insulation, and electronics.

Safety, Compliance and Quality Just a Click Away
The Crucial Role that Automated Print Inspection Systems Can and Should Play in Pharmaceutical Packaging.
Producing safe and compliant medicines – and harmless cosmetics, or good food for that matter – sometimes resemble a puzzle with countless number of components. It is only possible if all the individual parts are used and manufactured with the necessary care and precision. Quality and precision, reproducibility and exact documentation are therefore essential.
Every aspect of the manufacturing process must be closely monitored. Among the many critical procedures in this system, the inspection of the various packaging materials is an often overlooked but essential part. After all, compliant content reasonably requires compliant packaging.
This is the first point of contact between the product and the end user, where the latter receives information about the product, contents, dosage, or area of application, alongside the medicinal products. Incorrect or even false information can have fatal consequences, especially in the pharmaceutical sector.
In what follows, will be describing the importance of print inspection in the pharmaceutical industry – and it also applies, with only minor differences, to other regulated sectors such as the cosmetics and food industries – highlighting the advantages of automated inspection and discussing the safety aspects.
Why Printed Packaging Should Be Inspected
Compliance is the main keyword here. The pharmaceutical industry is subject to strict legal regulations issued by the relevant regulatory authorities such as the FDA (Food and Drug Administration) in the United States, the EMA (European Medicines Agency) in the European Union and similar authorities worldwide. The relevant regulations, such as 21 CFR Part 210 or in Germany the German Medicines Act, require clear and accurate labelling of

pharmaceutical products, including essential information such as dosage, expiry date and active ingredients. Any deviation or error in such labelling can lead to non-compliance. The consequences of such non-conformity can be manifold, including product recalls, fines, damage to the company reputation, or worst-case scenario, a health risk to the user. So how can you ensure that pharmaceutical packaging complies with regulations? A print inspection system plays a crucial role here by meticulously checking packaging materials for inaccuracies or defects. In such a system, the approved artwork is compared pixel by pixel with a sample of the packaging, and a scan-to-scan comparison is also possible. The system records all deviations between the two variants, which are displayed and documented for the user, much more accurately and precisely than the human eye could ever do.
This recording of deviations also serves to maintain the consistent quality of
packaging. Maintaining the highest quality standards is non-negotiable in this area. Print inspection helps maintain these standards by detecting defects on packaging materials that compromise product integrity. These would include printing errors, illegible text, stains or material inclusions, missing information (such as an artwork element obscuring the text). Any of these technical errors can influence the perceived quality of the product and affect consumer confidence.
The consumer confidence in the reliability and quality of a medicine is also a significant economic factor, as brand reputation is a priceless commodity in the highly competitive pharmaceutical market. Any association with inferior or defective products can irreparably damage a brand's reputation. The use of a print inspection system serves as a proactive measure to protect brand reputation by ensuring that the packaging meets the highest standards of accuracy and quality. And the growing use of highly refined or complex packaging in particular increases the demand for the quality-related appearance of the product. By preventing defective packaging from reaching the market, pharmaceutical companies can protect their brand value and maintain consumer confidence.
As mentioned above, automated print inspection is far superior to any visual inspection done by a human. Even if the four-eyes principle is applied, it can never be completely ruled out that errors will be overlooked. Computer-aided inspections,

66 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Packaging
Packaging
which analyse pixel by pixel, know no pause, no fatigue, no overlooking. Manual inspection of packaging materials is not only fragile, but also immensely time-consuming. In contrast, automated print inspection systems offer unrivaled speed, accuracy and efficiency. The scan of the packaging is completed within few seconds and the analysis is hardly any slower. With suitable systems, the accuracy and quality of codes or height and correct positioning of embossed Braille dots can be checked alongside the artwork in a single work step. In such a manner, production processes can be optimised and operating costs reduced by minimising the need for manual intervention and re-work. Such a system enables pharmaceutical manufacturers to significantly streamline their processes and maximise productivity while simultaneously reducing resource expenditure.
This efficiency extends to supply chains. These are complex and cover several stages from production to distribution. Any interruption or deviation in the packaging process can have far-reaching consequences that affect product availability. Packaging inspection ensures that packaging materials meet the required standards at every stage of the process.
What are the Benefits of Automated Print Inspection?
Automatic print inspection systems use advanced image processing technologies, high-precision camera systems, powerful computers and special software to analyse printed packaging with exceptional speed and efficiency. Even the smallest defects or deviations can be detected at 600 dpi resolution or in line with 8k cameras. The modern hardware and software also bring enormous efficiency. Manual inspection is labor-intensive and immensely timeconsuming, often leading to bottlenecks and production delays. The inspection system eliminates these inefficiencies by enabling fast and continuous monitoring

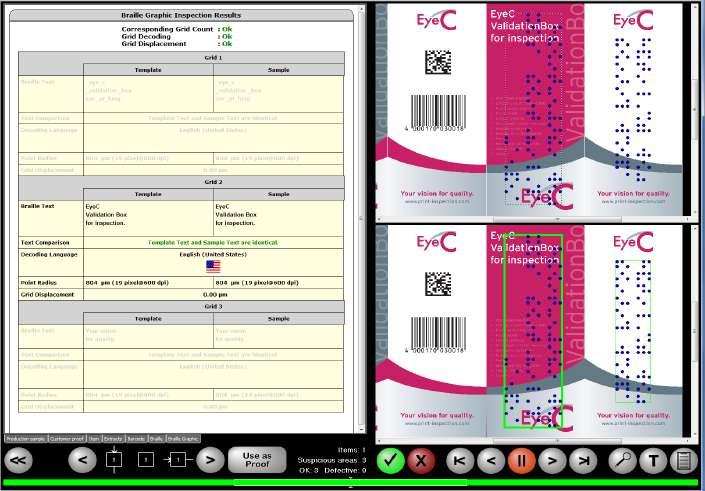
of the packaging material, speeding up the inspection process and increasing throughput. With large format scanners, multiple samples can even be inspected simultaneously, allowing demanding production schedules to be met without compromising on quality.
Another advantage is the direct feedback on the quality of the packaging material; corrective measures can be taken quickly in the event of deviations or defects. This not only minimises the risk of faulty products reaching the market but also facilitates continuous process improvement, as potential problems can be identified early in the production cycle. Modern print inspection systems are equipped with sophisticated analysis functions that enable users to gain valuable insights into their production processes. By analysing inspection data, companies can identify trends, patterns and causes of defects and thus implement targeted preventative measures to improve overall quality. The systems are highly scalable, allowing users to seamlessly adapt to changing production requirements and volumes. Whether small batches or large quantities are involved, these systems can adapt flexibly to different throughput volumes while maintaining uniform inspection standards. This is an invaluable advantage – especially for globally active companies, as it means that sites worldwide can be equipped with identical systems –that also considerably reduces the validation effort. Once a pilot system and the underlying process have been validated, the effort required for all other locations is significantly lower. All systems can be qualified with the
same measuring equipment, and URS, IQ and OQ documents or SOPs can be used and rolled out across all sites.
In summary, the print inspection of packaging is an indispensable part of pharmaceutical production and plays a crucial role in ensuring safety, quality and compliance. By using advanced technology and automation, pharmaceutical companies can improve the accuracy, efficiency, scalability and validation of their processes while minimising the risks associated with dosing errors, regulatory non-compliance or product recalls. Error-free and safe packaging is essential, and this can only be achieved with the use of advanced technology such as automated print image inspection.

Dr. André Schwarz is Head of Marketing, Documentation & Validation Support at EyeC GmbH. He holds a doctorate in German Language and Literature and has been working as a technical editor, marketing and computer validation expert at the Hamburg-based print inspection company since 2016. EyeC is a global supplier to the pharmaceutical industry, with 20 of the 25 largest pharmaceutical manufacturers relying on inspection systems from Hamburg.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 67 www.international-pharma.com
Dr. André Schwarz
Application Note

Automation of Manual Packaging for Small Batches in The Medical Device Industry Using the Example of Bone Screws and Surgical Implants
From Manual Packaging to Automated Solutions
Packing by hand? It remains the case in many areas of the medical device industry and there are many reasons why this is still happening: the batches are small; the products are sensitive and the packaging process is either specialised or varies greatly. But companies are increasingly looking for more flexibility and less dependence on staff availability, and there is a significant shift towards the automation of the packaging process in the medical device industry – even when dealing with small batches of medical devices.
One company that specialises in automating packaging processes for medical devices is Christ Packing Systems, based in Ottobeuren, Germany. Thanks to its modular and easily customisable BoxTeq cartoning machine, even the most complex manual packaging processes can be automated. This enables companies to produce more efficiently and with less reliance on human resources – a key competitive advantage in the medical device sector.
When it comes to high-value medical products, such as surgical implants or bone screws, they are often still produced in small batches of 50–100 units and manually packaged in folding cartons. The packaging process is complex due to varying product sizes and numerous packaging configurations for different target markets. In addition, the speed of the packaging process and the output is highly dependent on the staff availability, which becomes a problem especially during holidays and sick leave periods.

In this case, an automation solution is the answer. However, a standard cartoner cannot perform all the complex tasks of a manual packaging process, and developing a completely new packaging machine is often not feasible due to the time and cost involved.
As Standardised as Possible, as Customised as Necessary
Medical technology companies need a tailormade solution that meets their high-quality
standards and at the same time fits in with their internal cost and time schedules. This is where the modular concept of the BoxTeq from Christ Packing Systems comes into play. The cartoner can be easily customised and equipped with additional components.
"When customers visit our production facilities, they quickly realise that we have the experience and all the resources on site to successfully carry out such a project," explains Jürgen Sikora, Key Account Manager
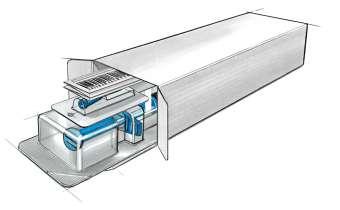
68 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Figure 1: Hard blister with sensitive medical products: A complex manual packaging process needs to be automated
Application Note

at Christ Packing Systems. With more than 70 years of experience, a production area of 12,500 m² and a high degree of in-house production depth/vertical integration, Christ can respond quickly and flexibly to customer-specific adaptations, making customisation easier and more predictable.
"Our vertical integration of around 87% gives us the flexibility to implement customerspecific adaptations quickly and easily".
Jürgen Sikora, Key Account Manager at Christ
Overview: Customised Packaging Solutions for Hard Blisters
This case study describes a specific customer project in which bone screws in hard blister packs with various inserts are being automatically packed into folding cartons and individually labelled. Christ has developed a comprehensive packaging solution consisting of three parts: a feeding solution, a BoxTeq cartoner and a LabelTeq labelling solution.
A special container was developed that enables the customer to feed the hard blisters into the cartoner, which acts as a reservoir for the blisters to be packaged. This was important as secondary packaging takes place in a separate room. The packaging process starts with a blister being automatically fed from the container onto the product transport.
A patient label, which is automatically printed and folded, is then added to the blister. Both are transported together to the BoxTeq cartoner where the folding cartons are fed, erected, filled and sealed.
The sealed folding carton is then transported to the LabelTeq labelling station, where multiple labels are printed and applied to different sides of the carton. The entire packaging process is monitored by sensors and cameras. All printed labels are checked for accuracy before application – only correctly printed labels are fed into the process. Electronic digital position indicators provide additional reliability to the process during format changeover.
Maintaining Process Standards
When it comes to medical devices, it is often easier to adapt the machinery to the existing packaging process than to change proven processes. This happens because changes to validated processes and packaging materials always involve risks and costs: if every move in the operating theatre is to be perfect, the surgeon must be able to rely upon the product coming out of the carton
in the correct position and the relevant information being readable at first glance.
In this customer project, for example, a special folding of the patient label specified by the customer had to be adhered to. Christ developed a special folding unit that uses three folds to bring the patient label into the required shape. "However, the special fold had posed a few challenges," recalls Karin Schmalholz, Project Manager at Christ. "The accordion-folded patient label sometimes bounced like a tight spring. But our engineering team quickly found the right solution for this too.”
Christ developed an additional fixture to hold the patient label in place. This allowed the products to be packaged according to the customer's requirements.
„Even for customer-specific requirements such as special label folding, our engineering team can quickly provide the right automation solution.”
Karin Schmalholz, Project Manager at Christ
Fast and Secure Changeover to Other Formats
The desire to switch between formats quickly and easily also leads to customer-specific adaptations: "For this customer project, we had the idea of adapting the drive of the product carriers in the LabelTeq," explains Jürgen Sikora. "Instead of the product carriers being coupled with only one drive, they were equipped with two independent drives, which enables automatic longitudinal adjustment of the bucket chain feed.
To ensure that the machine is always set correctly, Christ has equipped the cartoner's format changeover points with electronic digital position indicators, which automatically receive their target value from

INTERNATIONAL PHARMACEUTICAL INDUSTRY 69 www.international-pharma.com
Figure 2: Vertical integration: State-of-the-art production facilities and decades of experience enable customised and predictable automation solutions within the specified time and budget frame
Figure 3: The BoxTeq cartoner, the centrepiece of the automated packaging line, is modular and customisable
Application Note
the control system. In addition, a control mechanism has been integrated to ensure that the machine can only be switched on when everything is set to match the selected format. This provides greater process reliability.
Reliable Process Control with Sensors and Cameras
In the field of medical devices, it is particularly important that the packaging quality is monitored reliably and automatically.
In this specific example, extensive sensor technology ensures that all the required components such as blister packs and patient labels are included in the folding carton. The customer’s camera system, which is integrated into the LabelTeq, checks that the correct labels are applied in the correct places and around the specified sides.
If necessary, the packaging line can also be switched to manual feeding before the LabelTeq. This means that individual items can be labelled subsequently if required.
A Convincing Result
With packaging solutions from Christ, medical device industry customers can design their own packaging processes more efficiently.
Even for complex packaging tasks, including frequent format changeovers, Christ develops tailor-made solutions that guarantee high process reliability and consistent packaging quality.
Automating the packaging process is therefore also worthwhile for small product batches. As a result, customers are less dependent on staff availability and can scale their output more flexibly.
At a Glance
The automation of complex manual packaging in the medical device industry, using implants and bone screws as examples.
Situation
Many medical device companies are producing small batches that are packaged into folding cartons and labelled by hand. Manufacturers are dependent on reliable employees. Despite frequent format changeovers and monotonous work, quality must be ensured at all times.
Challenge
Due to the large number of manual activities, the process is difficult to scale. Staff shortages lead to fluctuations in output. The packaging process therefore needs to be automated. A standard cartoner is not suitable for this, as it cannot map all the complex processes involved.
Solution
Christ specialises in the automation of complex packaging processes. In this customer project, Christ adapts its modular cartoner to the specific requirements of the

70 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Figure 4: The fanfold label is automatically folded and inserted into the folding carton together with the hard blister –for greater scalability and consistently high packaging quality with less waste
Figure 5: Automatic longitudinal adjustment of the bucket chain feed and electronic digital position indicators ensure reliable format changeovers
Application Note
packaging and labelling process. A camera and sensor solution are integrated to ensure reliable quality control.
Result
The customer can flexibly adapt production to demand and switch between formats


quickly and reliably. This way, the company becomes less dependent on staff availability. Automation allows for consistently high packaging quality with less waste.
Requirements
• Patient label to be folded automatically according to customer specification.
• Hard blister and patient label to be packaged together in a folding carton.
• U and L labels to be printed and applied to the folding carton on a productspecific basis.
• Primary and secondary packaging are physically separated.
• Space for secondary packaging is extremely limited.
• Reliable process control.
Implemented Automation Solution
• Compact BoxTeq cartoner with customised infeed including individual magazine.
• Product transport with automatic longitudinal adjustment in the LabelTeq.
• Comprehensive quality control with sensor and camera systems.
• Customised patient label folding unit.
• Fast and reliable format changeover due to electronic digital position indicators with target value specification.

Jürgen Sikora
Jürgen Sikora started his career as a Service and Development Engineer, installing packaging machines worldwide, and then moved to the organisational side as Service Director for Christ Packing Systems. Since 2009 he has been deeply involved in technical sales, which culminated in his role as Key Account Manager in 2018. He is an expert in secondary packaging automation for pharmaceuticals and medical devices, and excels in technical consultancy. Working closely with pharmaceutical companies around the world has given him a deep insight into their unique packaging challenges and specific requirements.
Email: jsikora@christ-ps.com
INTERNATIONAL PHARMACEUTICAL INDUSTRY 71 www.international-pharma.com
Figure 7: BoxTeq in perspective
Figure 6: Integrated camera and sensor systems ensure reliable process control and guarantee consistently high packaging quality
Health Outcomes
Electronics and Data, How Effective are Personal Electronics in Helping Patients to Maintain Good Health?
What does the Future Hold for Patients?
In Oct 2017 the Adherium stock was trading around AUS $22 with the Rapihaler device FDA approved. Today 2024 – 16 different Hailie smart inhaler products are FDA approved for use. Yet the parent company Adherium stock has tanked from 2016 AUS$ 155.21 down to $0.04c.
Inhaler Compliance Technology
Not a Patient Success
One of the joys of leading an R&D healthcare company are the disparate opinions to be found amongst the employees on any subject. In 2017 when e-aids for both compliance and adherence monitoring of patients were a very hot ticket – there was strong and divisive debate about us engaging in this R&D area or not. My lone older voice could not quite understand what the fuss was all about with e-aids on medical devices?
• Why not just use an app on a smartphone to remind at treatment times and perhaps press a button to confirm taking the tablet? But a beeping inhaler is way cooler!
• Throwing away electronics every 30 days did not seem like a great idea for the planet, and safe battery disposal is not easy either for built-in devices.
• The multi-use additive e-aids, such as the Hailie system above on the Symbicort® Rapihaler® seemed like the only practical way forward, but they were a bit clumsy in-use and battery stocking and changing was ignored in advertising…
I am delighted to be looking at this area with fresh eyes. What does the future hold in the next few years for e-aids for compliance enhancement and adherence monitoring or just patient centric technology?
What Do Electronic Compliance/Adherence Devices Do For Patients?
There is only really a subtle difference between compliance and adherence systems, and that is the monitor data recipient. The former is doctorled, and so the intention is for patient treatment data to be available remotely for review and advisory purposes. For example, compliance devices would be very useful in Mental Health in-patients who are now recovering at home.
Adherence e-aids are to allow monitoring of the patient’s adherence to daily medicines by the parents or carers of that patient, or the patient themselves.
So, in summary compliance e-aids are for the doctor, and adherence e-aids are for the patients and/or carers. The devices log when you take medicine, and some nag to tell you to take it. However, the add-on devices are not linked to the drug container, so do not function as dose remaining counters.
Clinical Study Compliance Devices
A massive unintentional win in monitoring with add-on e-aids for any treatment, is in clinical studies for New Drug Applications (NDA). In this application the subjects (sometimes a patient) are monitored by spy-ware in the e-aid to ensure they take the correct dose of medication as prescribed and at the right time of day.
The simpler systems allow the data to be downloaded from the device at the end of the trial period and checked for compliance. A Yes /No data-driven instruction can then be given for subject’s inclusion in the clinical study report, or removal.
A more comprehensive approach for clinical study work is to use short-range Bluetooth® wireless to link through the internet for more frequent remote monitoring. In this way a home subject who is not complying can be contacted by
telephone or email and advised to ‘please’ comply with the study requirements.
2019, five years ago those promoting the technology for built-in compliance/ adherence e-aids for marketed drug delivery devices were touting the following as the benefits:
1. Remote monitoring of compliance data by healthcare professionals, improving patient outcomes.
2. Local device alerts, and technique aids (e.g. Metered Dose Inhalers) for chronic disease.
Just look at any mobile phone application store, personal telemedicine is here today via the many, many smart phone apps for e.g., blood pressure, diabetes, tablet taking, sleep monitoring, snore control, blood glucose monitoring, cardiac disease, etc.
But the technology push described in 1. for a doctor to remotely monitor patients via worn sensors, has not come into being. In the UK with its cost-conscious National Health Service, with doctors under great time pressure, the author believes it is unrealistic to dream of such a system to be used for the many conditions possible in the near or even far future.
Private healthcare systems for the relatively rich patient with a chronic disease is possibly another story, but not one that is of great commercial interest to medical device
72 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Health Outcomes
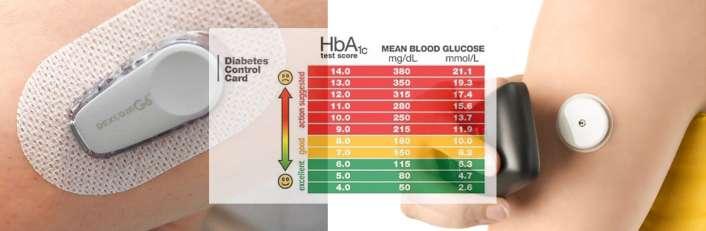
manufacturers. So, the author cannot see that being a win-win either.
The big next step in Type 1 diabetes control is full-feedback systems where insulin pumps can be direct controlled by ISF patches like the ones above. Low blood sugar reading = reducing insulin can be done today, but the regulatory authorities have put high hurdles for high blood sugar reading = increased insulin.
This is a safety-first situation, as a false reading for high blood sugar could put the patient into a coma, with the chance of death to themselves and/or others if they were driving a vehicle.
In the control of asthma Forced Expiratory Volume in one second (FEV1) is a common clinical measure of a patient’s disease condition. In 2019 a new company was promoting the gadget above with the ‘Artificial Intelligence’ or AI tag, which perhaps is pushing the bounds of UK advertising standards. However, the claim was that the pocket electronic liquid aerosol system could vary the drug dose, depending on the patient’s condition as measured by FEV1. Low FEV1 would trigger increased dose and vice versa.
For a bronchodilator (opens the airways) this could be useful, but experienced patients know that they can take more than one puff if they are wheezy. Unfortunately,
not enough asthma patients monitor FEV1. A personal electronic gadget retailing around £100 can measure FEV1 (search spirometers on the web) but are still not in common use, despite years of availability. When you are having difficulty breathing, you do not need a machine to tell you!
The purpose for adding electronics to a patient’s life, is that it must improve health outcomes, and compliance monitoring of asthma or COPD inhaler use, just does not achieve that goal.
Remote Doctor’s Appointments –Telemedicine or Improving Doctor’s Efficiency?
As we all know, Covid-2019 has pushed the global population into remote working (or hybrid working) using the PC-based internet technologies, or mobile phone 3G/Wi-Fi


technologies which allow video conferencing without the show-and-tell slide capabilities of the PC-based tech.
Doctors have adopted PC tech in medicine, but for security reasons they use less common software systems than we all use. I recently was asked by an NHS doctor to use one brand of video conference software, but it was Mac IOS incompatible. So, we ended up using mobile phone end to end encrypted commercial technology – with my prior consent. Unfortunately, the doctor’s phone screen side camera was broken, so she could see me, but I had a view of a coffee cup! My story illustrates how much tech must work for remote video conferences to be more successful than a telephone.
Taking a more Global view, countries with low population densities can now access doctors via the internet or mobile video conferences, and it helps improve the populations health through faster diagnosis. There remains the problem of access to medicine in low population density countries, as fuel and running vehicles is expensive for the long run to a pharmacy.
False Claims?
Below is a watch retailing at £35 claiming to monitor your blood glucose all day. How it works was not mentioned, nor whether it had FDA or other regulatory approvals. Is

this a fake, or the real thing – amazing value if the latter? The £25 retail micro-CPAP is intended to blow air up your nose all night and give snore-free nights. A friend bought one and it did not fit his nose and so was did not achieve purpose. The fancy $75 health watch does everything you could want, but in the small print states not for medical use.
These products highlight the problem for a patient hoping to improve their lifestyle
INTERNATIONAL PHARMACEUTICAL INDUSTRY 73 www.international-pharma.com
Local Device Alerts; Condition Improving and Technique Aids, The G6 Revolution® and Libre® interstitial fluid ‘blood glucose’ level monitoring systems photos from company marketing in 2019. These systems have revolutionised T1 diabetics control of their condition. The removal of frequent skin prick, blood droplet testing has meant insulin compliance is improved
Post Covid-19 remote doctor’s conference, not sure how the doctor’s stethoscope can be used... Home setting to Surgery in the same country or in another even. Better communication is achieved than by audio alone. However, PC/Laptop and reliable fast internet is required, many countries today rely on wireless mobile mast technology, rather than fibre or copper wire systems
The purpose for adding electronics to a patient’s life, is that it must improve health outcomes, and compliance monitoring of asthma or COPD inhaler use, just does not achieve that goal
Health Outcomes
and condition with medical devices. What is useful to buy and from what source? The internet market is full of choice, but also confusion.
This table is a possible prediction of patient-centric solutions for current disease groupings. As always forward-looking predictions can be inaccurate… Accurate sensing is more important than data mining, in the first instance triage and advice is helpful to reducing costs in the NHS. The Dr. Google approach for selfdiagnosis or condition advice is as poor as a medical dictionary was to former generations, so the NHS 111 service is an improvement.
What is Next in Patient-centric Electronics?
By 2030 I think there will be a natural cull of today’s health gadgets and the advent of much more focussed market of medical devices that can claim ‘for medical purposes.
Remote doctor’s consultations will be less frequent than in 2024, when seeing a GP face to face is a challenge. The jury appears out as to whether 111 phone systems for medical
I believe the practical approach of reliable health monitoring which is data logged for a doctor’s use will increase beyond blood pressure monitoring. The key R&D innovation activity is sensor development to monitor disease directly or find reliable analogues to the condition.
Data analysis based on a smart phone or tablet computer (portable to the doctor)

seems a clear winner, but again it must not give false warnings of disease, nor miss signs of serious disease. Early systems will merely warn of possible outcomes, but by 2030 they should have become more accurate and thus helpful by providing early warnings.

Bill Treneman Bsc AM I Mech E, Managing Director, UPC Cambridge Limited, has personally developed electronic, powder and gas-powered inhalation devices, as well as parenteral delivery systems such as auto-injectors starting in 1991. Prior to that he worked developing live vaccine making clean-room robot cells. Other successful projects were ultrasonic sensor systems for threads and then for sucrose particles. He has dreamed innovations all his long career, with numerous patents granted. Bill currently runs UPC, a successful forensic engineering science business, combining Medicine, Science and Engineering under one roof.
74 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Bill Treneman
Data-logging Personal Medical Devices – Take to Your Doctor’s Appointment
Blood Pressure monitors like this one which record and store for up to two people, can be used to reliably record BP for those moving towards hypertension. For less than £100 you can buy one and then take it to your doctor for your face-to-face appointment. The doctor can scroll through your data. A patient centric approach that is simple to use and for the doctor to access. Personal spirometers measuring lung function (FEV1) offer much the same data logging and recall as this device
2024 to 2030 R&D Predictions Constant Sensor Systems Wrist worn device With Bluetooth® link to Wi-fi> Web Smart Phone Apps FDA approved Asthma/COPD Pneumonia Throat sound Chest sound No Possibly Data analysis, to become useful crisis preventers Diabetes Type 1 Interstitial Fluid Insulin pumps/pens Smaller pump controllers Cable for firm-ware upgrades Not yet FDA approwed Diabetes Type 2 Arm or Body No No Data log & take to Doctors Appoitments Mental Health Sleeping Safely Brain Activity Yes – fixed band Yes, for alarm Data log & take to Doctors Appoitments Heart Disease Market Saturated with wrist and smart-phone add-ons? Data analysis, to become useful crisis preventers Cancer Prostate – marker? Breast – tissue density? No No Data log & take to Doctors Appoitments Arthritis Joint sounds Pain level sensor No No Data log & take to Doctors Appoitments






























INTERNATIONAL PHARMACEUTICAL INDUSTRY 75 www.international-pharma.com
Fighting Counterfeits in the Medical Devices Sector –A Step Beyond the EU MDR
The counterfeiting of health products is a serious and growing concern, and it has come at the forefront of the public mind in recent years with several strict regulations aimed at improving traceability and protecting patients and consumers from harm. Today, all prescription pharmaceuticals sold in the EU and US are subject to complete end-to-end traceability from the point of manufacturing to the point of dispensing. However, compliance for medical devices is not as strict.
Medical devices are an extremely broad category, covering everything from bandages and personal protective equipment (PPE) to pacemakers and ventilators. The implications of counterfeit medical devices can also vary greatly, from not performing quite as intended to putting patients and healthcare staff at serious risk of harm. Even seemingly ‘lowrisk’ devices such as PPE and face masks can have potentially life-threatening implications if they are not fit for purpose.
Counterfeiting is a serious problem in the global medical device industry. Vulnerabilities in global supply chains were brought to the fore at the very start of the COVID-19 pandemic, when counterfeit test kits, face masks, and PPE appeared seemingly overnight.
During the pandemic, many companies not previously manufacturing medical devices entered the marketplace with disposable masks and PPE to satisfy a dramatic increase in demand – and not all companies have manufactured products to the high standards required for use in medical settings. According to the National Institute for Occupational Safety and Health, 60% of KN95 face masks evaluated during the COVID-19 pandemic in 2020 and 2021 did not meet the requirements approved for healthcare use in the US.1
While we have come a long way since the start of the pandemic, this is still a significant issue, with COVID-related medical device fraud regularly topping the list of Interpol’s Operation Pangea. In March 2020,
Interpol seized some 37,000 counterfeit medical devices, most of which were surgical masks.2 The most recent operation, which ran from 3–10 October 2023, led to the seize of approximately 11,000 counterfeit COVID-19 test kits in Australia alone.3
The global rise in e-commerce has played a significant role in promoting counterfeit sales of medical devices, with online marketplaces and e-commerce platforms offering a convenient place for consumers to shop and compare prices to find the best deals while providing a breeding ground for counterfeit products. Findings from the US Government Accountability Office have suggested that as many as two of every five consumer products available online through third-party retailers could be counterfeit.4
The sale of counterfeit medical devices is, first and foremost, a safety concern but can also have significant economic repercussions. Counterfeit medical device sales distort competition and cause damage to legitimate producers who may experience direct loss of sales or see their reputation and brand name tarnished by counterfeit products using legitimate company trademarks.
What more can be done to protect against counterfeit medical devices?
Current Regulatory Requirements for Medical Devices
The upcoming EU Medical Device Regulation mandates that manufacturers of medical devices for sale within the EU must adhere to guidelines to ensure their products are safe to use. The regulations cover all medical devices sold in the EU regardless of where the manufacturer is located.
The regulations are strict but currently contain only minimal requirements for individual device identification. Under the regulations, all medical devices must be assigned a unique device identification (UDI) code and have their UDI recorded, indexed, and registered in EUDAMED, the Central European Database for Medical Devices.
Publication and full functionality of EUDAMED are expected to be in place by the end of 2024, and full use will be mandatory
for all medical device manufacturers with staggered deadlines from 2026 to 2028.
The UDI contains:
• Device identifier – a unique text code for each specific model of a particular device. The information is static, meaning it is the same for all instances of the product model.
• Production identifier – a variable text code comprising one or more variable characteristics, such as the date of manufacture, expiration date, lot number, or serial number.
Initially, serialisation and identification of products down to the individual item level will only be a requirement for active implantable devices, such as pacemakers, ventilators, and internal glucose monitors. However, companies whose products do not fall under the category would still benefit from equipping their production lines with technology to enable both UDI compliance and serialisation.
Taking such an approach will not only help combat the rise and risk of counterfeit medical devices but will also help manufacturers future-proof their lines should the requirement for serialisation be
76 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Logistics & Supply Chain Management

Hundred years of air cargo services

Finnair’s first commercially operated flight carried mail from Helsinki to Tallinn. Today Finnair Cargo’s extensive network offers frequent connections also for temperature controlled cargo between Asia, Europe and North America.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 77 www.international-pharma.com
more: cargo.finnair.com
Read
Logistics & Supply Chain Management
extended to include more product groups. In addition, those who implement serialisation can also benefit substantially from tracking a product’s progression from creation to the end user, with a wealth of supply chain data that can be used to enhance operational and commercial performance.
Learning from Experience, the EU FMD and DSCSA
The pharmaceutical industry has already deployed serialisation as an anticounterfeit measure globally for prescription pharmaceutical products. The EU FMD (Falsified Medicines Directive) and US DSCSA (Drug Supply Chain Security Act) are notable developments in the battle against counterfeiting and product diversion to protect patient health and safety.
The EU Falsified Medicines Directive (EU FMD), introduced in 2011 and updated most recently in 2019, requires that manufacturers mark prescription pharmaceuticals with a GS1 2D DataMatrix code, including four data elements – an individual product identifier, serial number, batch or lot number, and expiry date. Upon scanning, the DataMatrix code provides access to data transmitted to the European Medicines Verification System (EMVS) portal and corresponding national databases.
Any inconsistencies that arise when products are scanned are automatically
flagged, and the medicine in question is rejected. This information is then passed on to the Medicines and Healthcare Products Regulatory Agency for further investigation.
Similar serialisation requirements are required under the US Drug Supply Chain Security Act, which has been in force for pharmaceutical products sold in the US since 2017. All key players in the US pharma supply chain, including packagers, manufacturers, distributors, and dispensers, must generate, authenticate, and verify serial numbers for all packages travelling through the chain.
Under both regulations, the serialisation process allows all pharmaceutical products to be tracked and traced throughout their journey in the supply chain, from manufacturer to end user. Adopting protocols for medical devices based on these wellknown and trusted serialisation procedures is an essential step towards a secure system that ensures transparency and accountability in the medical device sector.
The Hidden Benefits of Serialised Track-and-trace
The benefits of pharmaceutical serialisation in terms of boosting patient safety and the security of legitimate medicines are well documented. What should also be considered is the wealth of data that serialisation presents for manufacturers and how this can be used to build
efficiencies in the broader pharmaceutical supply chain.
Legislative requirements such as the EU FMD and US DSCSA have helped manufacturers improve production line efficiency and provide access to supply chain data to enhance operational performance. In other industries serialisation has opened multiple commercial opportunities, as the foundation for digital capabilities in physical products, including personalised consumer engagement and improved brand positioning.
Via serialisation, manufacturers can gain holistic insights into a product’s lifespan and distribution trends to facilitate strategic planning. Such data could, regulations permitting, present an opportunity for sales teams to engage with pharmacy chains to improve sales performance and enable forward-thinking manufacturers to consider a whole new level of consumer-focused marketing or engagement.
Applying product serialisation can also help protect patient and consumer health and safety and make product information more dynamic and consumer-friendly. By containing serialised data within scannable 2D codes, medical device companies can help make product-specific information, such as user guides, easier for users to
78 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
ANYONE CAN DO COLD CHAIN PACKAGING.
But there’s only one who can do sustainable, zero-waste, modular and payload-optimized pharmaceutical packaging!


Get ready for retecma!
It’s the one and only AI-supported reverse logistics system that fights to keep all resources in the lifecycle as long as possible. Cold
INTERNATIONAL PHARMACEUTICAL INDUSTRY 79 www.international-pharma.com
Chain Excellence eutecma.com/en/reuse-rethought
access while also ensuring that the guidance is up-to-date and accurate.
Paper inserts and instructions for use present several practical challenges – not least the cost and environmental impact of supplying printed instructions with each pack, which may be inadvertently thrown away or damaged, and poor accessibility due to tiny print or language barriers. Today, with scannable 2D codes and serialisation, companies can provide more capabilities for users to interact digitally with products to find the information they need in a format that is accessible to them.
Of course, the standard 2D codes used in the pharmaceutical and medical device sectors today are GS1 DataMatrix codes, which unfortunately cannot be scanned by native camera apps on most smartphones –though there is currently lobbying to change this. However, consumers can still access the information using specialist 2D code scanner applications, which are widely available.
Managing the Cost of Implementation
A few years ago, when the pharmaceutical industry began preparing for the EU FMD and US DSCSA, the total cost of implementing serialisation was at the front of everyone’s mind. Compliance came at a significant expense for manufacturers, not just in terms of the capital cost – which can stretch from €100K for smaller manufacturers to €250K for mid to large-sized firms – but the time and resources taken to make the upgrade, and the overall cost of related downtime.
Indeed, before now, the high cost and perceived complexity of implementing serialised solutions have stood as a barrier to widespread implementation,
and examples of organisations adopting serialisation have mainly been limited to those with a compliance obligation and highvalue luxury goods manufacturers seeking to protect their brand.
But today, the cost of serialisation through 2D codes is no longer a barrier –the time, effort, and resources needed to upgrade lines have significantly reduced as the knowledge and know-how in this area have grown, making it much more efficient and cheaper to install across a range of markets. Today, serialised technology is being used, to great advantage, in many low-cost sectors, including fresh fruit and vegetables and even postage stamps.
Looking to the Future
The addition of serialisation via 2D codes for medical devices is only a small step beyond what is already required under existing EU MDR compliance – but one that can help to reduce the risk of counterfeit products further and bring many untold benefits to both end-users and manufacturers.
Beyond the current benefits, such advanced traceability metrics could allow further supply chain analytics using artificial intelligence and machine learning tools. With a thorough data set enabled via product serialisation, such tools could be applied to instantaneously examine vast quantities of data to identify trends and inconsistencies in the supply chain.
Globally, there are an increasing number of manufacturers beyond the pharmaceutical sector realising the opportunities of 2D code-enabled serialisation. For those not yet exploring the possibilities, the time to act is now.


REFERENCES
1. Types of Mask and Respirator https://www.cdc. gov/coronavirus/2019-ncov/prevent-gettingsick/types-of-masks.html, visited 14th March 2024.
2. Global operation sees a rise in fake medical products related to COVID-19 https://www. interpol.int/en/News-and-Events/News/2020/ Global-operation-sees-a-rise-in-fake-medicalproducts-related-to-COVID-19, visited 14th March 2024.
3. INTERPOL operation targets illicit medicines around the world https://www.interpol.int/fr/ Actualites-et-evenements/Actualites/2023/ Global-illicit-medicines-targeted-by-INTERPOLoperation, visited 14th March 2024.
4. Agencies Can Improve Efforts to Address Risks Posed by Changing Counterfeits Market https:// www.gao.gov/assets/gao-18-216.pdf, visited 14th March 2024.
Images are only for illustrative purposes and do not represent real codes or applications

Domino’s Global Life Sciences Sector Manager Bart Vansteenkiste has a 20year history with the company. He works with Domino’s European OEM partners and trade associations, including the European Federation of Pharmaceutical Industries and Associations (EFPIA), and Medicines for Europe. A Dutch, English, French, and Spanish speaker, Bart presents at conferences worldwide.
Email: bart.vansteenkiste@domino-uk.com
80 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1 Logistics & Supply Chain
Management
Bart Vansteenkiste

Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS

Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.

www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA’S DNA
Listen to industry experts on the latest in drug discovery, development, research, industry regulations and much more at Pharma,s DNA, the podcast channel by Senglobal Ltd., available on Sound Cloud, Spotify, iTunes and YouTube.
senglobalcoms.com



INTERNATIONAL PHARMACEUTICAL INDUSTRY 81 www.international-pharma.com



londonbiotechshow.com info@londonbiotechshow.com +44 (0)208 242 6566 Unlocking The Potential of Biotechnology For Revolutionising Medical & Healthcare Sectors Globally Orginised by Business Media
- 9 May 2024 Olympia West
8
INSPIRING SUSTAINABLE CONNECTIONS

10 - 14 June 2024
Frankfurt am Main, Germany
World Forum and Leading Show for the Process Industries
ACHEMA is the hotspot for industry experts, decision-makers and solution providers. Experience unseen technology, collaborate cross-industry and connect yourself worldwide to make an impact.
Are you ready? Join now!
#ACHEMA24
Page 83
IBC
Page 63
Page 21
Page 75
Page 79
IFC
Page 77
Page 3
Page 9
Page 13
Page 82
BC
Page 49
Page 5
Page 45
Page 15
Page 81
Page 57
Page 31
Page 25 & 43
Page 65
Subscribe today at www.international-pharma.com or email info@senglobalcoms.com
ACHEMA
Air France KLM Martinair Cargo
Aurena Laboratories
Biopharma Process Systems Ltd.
EMBALL’ISO
eutecma GmbH
EyeC GmbH
Finnair Cargo
FUJIFILM Wako Chemicals U.S.A. Corporation
Kahle Automation
Krautz Temax
London Biotechnology Show
Natoli Engineering Company
Nipro
Novo Nordisk A/S
Owen Mumford
PCI Pharma Services
Senglobal Ltd
Stäubli International AG
UPC Cambridge Ltd
Valsteam ADCA
Wool Packaging Company Limited

I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry
IPI is also now active on social media. Follow us on:
www.twitter.com/ipimediaworld
www.facebook.com/ipimediaworld
www.plus.google.com/+ipimediaworldmagazine
www.ipimediaworld.tumblr.com
84 INTERNATIONAL PHARMACEUTICAL INDUSTRY Spring 2024 Volume 16 Issue 1
Index
Advertisers
KEEPING YOU AND YOUR PHARMA COOL

With our premium pharma service and its advanced end-to-end cold chain solution, you can rest assured that your pharmaceutical shipment is at the right temperature from departure to arrival, and at every step in between. Visit afklcargo.com for more information.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 85 www.international-pharma.com

