Journal for Clinical Studies – ISSN 1758-5678 is published quarterly by Senglobal Ltd.
4 FOREWORD
WATCH PAGES
6 Site Selection in Clinical Trials: Current Challenges and Future Opportunities
In clinical research, site selection is an essential component as it determines whether the trial is successful or not. However, the current process of site selection has many challenges which affect the cost, efficiency and quality of the trial. Cristin MacDonald of WCG explains how there is a requirement for a more flexible and innovative approach to leverage the potential of artificial intelligence (AI) and machine learning (ML).
8 Settling the Score with Scleroderma While Waiting for a Cure
Scleroderma, also known as systemic sclerosis (SS) is a rare and progressive autoimmune rheumatic disease which currently has no cure. The disorder causes inflammation of the skin as well as in the body. Jaime Gavazzi at Clarivate, discusses the needs of addressing disease progression in clinical trials, and explores the novel therapies that are under evaluation for the treatment of scleroderma.
10 The Expanding Role of Functional Service Providers in Clinical Research
Functional Service Providers (FSPs) offer adaptable resources and specialised skills, and are currently shaping clinical research. As a result, biopharmaceutical and biotechnology organisations are now using FSPs in lieu of full-service outsourcing. Lisa Stetler and Ershlena McDaniel of Resourcing Operations, explore the importance of utilising FSPs in clinical research, and list the ways in which companies can choose the right model.
REGULATORY
14 Striking the Right Balance Between Transparency and Privacy in Clinical Data Sharing
Clinical trial researchers must be able to maintain transparency needs whilst protecting participants’ privacy and looking after the company’s intellectual property. Regulatory requirements ensure that best practices govern the information that is released. Michael Healey and others at ICON discuss how regulators and sponsors must find a fine balance between providing data and protecting the privacy of trial participants.
www.journalforclinicalstudies.com
MARKET REPORT
16 Generating Entire Regulatory Dossiers –The Logical Next Step for GenAI in Life Sciences?
Generative AI (GenAI) can analyse and summarise key findings across extensive and varied bodies of existing research and data. This technology is ideal for high-volume tasks that are carried out daily by regulatory affairs teams. Renato Rjavec of ArisGlobal, highlights how these have been placed into an easily accessible knowledge base which helps to accelerate the process and serves as a productivity tool.
RESEARCH AND DEVELOPMENT
18 Making AI Work for Drug Discovery: A Joined-up Approach
Data-driven AI-enabled drug discovery is becoming a reality. It is beginning to fulfil technology’s promise and is demonstrating its potential, as many pharmaceutical companies have now started to implement AI in their operations. Dr. Ben Sidders at Biorelate, explains how scientists need to determine how to effectively integrate data science into their workflows.
THERAPEUTICS
20 Enhancing Oncology Trials: How Generalised Pairwise Comparisons Drive Multi-faceted, Patient-focused Research
Traditional clinical trials typically focus on efficacy endpoints and can overlook the safety and quality of the trial. Therefore, there is a need for a new and improved statistical approach where the health and perspectives of patients’ takes precedence. Sebastien Coppe and Samuel Salvaggio at One2Treat, explore how the Generalised Pairwise Comparisons (GPC) method addresses gaps in statistical methodologies.
24 Challenges of Measuring Clinical Meaningful Changes in Alzheimer’s Disease
Clinical trials in Alzheimer’s Disease experience numerous difficulties and complexities. For example, there is insufficient evidence to
show how the outcome of a trial correlates with changes in disease progression and treatment response. The author, Catarina Cunha of WCG improves our understanding of how trial endpoints relate to clinical assessments could benefit patients’ and their healthcare providers.
TECHNOLOGY
28 Forecasting and Kit Design in Clinical Trials for the Pharmaceutical and Biopharmaceutical Industry.
The success of clinical trials is reliant on studious planning, precise forecasting and the consideration of factors of the kit design. These two components can significantly improve the timeline, cost and implementation of a clinical trial. Slava Shulov at PCI delves into the multiple factors that influence forecasting and designing kits, and how biopharmaceutical and pharmaceutical organisations can better patient outcomes and enhance business success.
LOGISTICS & SUPPLY CHAIN
30 New Developments in Clinical Trial Logistics
Technological advancements and operational changes such as decentralised clinical trials (DCTs), have forced clinical trials to adapt to an ever-changing environment. The pandemic, regional challenges and turbulence in the air freight industry have also had an influence on the life sciences sector. Steve Healy of COREX Logistics highlights what factors, amid new developments, are crucial to ensure a streamlined and effective clinical trial process.
34 Transforming Clinical Trials: A Decade of Change and a Future of Innovation
Technological innovations, regulatory changes and an increasing focus on the experiences of participants’ have been the main influences in the transformation of clinical trials. Dr. Daniel Arkwell at Envirotainer touches on how significant changes to clinical trials have enabled them to be more structured, accessible and universally connected. The author adds how the future of clinical trials looks even more promising due to advancements in multifaceted areas.
A diverse issue this Autumn, seeing discussions of new opportunities created by technological developments and the implementation of artificial intelligence, the concerns around patient-data protection as a result of this and, the reframing of approaches in tackling disease and rare disease so to best improve treatment and reduce early death.
In this issue we revisit the importance of early detection and preventing disease progression in rare disease. The FDA has been increasing its focus on rare disease research, trying to maximise the treatment options by recognising and improving patient input. Jamie Gavazzi of Clarivate shines a light on Scleroderma, an autoimmune disorder which causes fibrosis and, like many other rare diseases, has seen its symptoms addressed and managed but little done to target the direct cause of the disease itself. And thus, stresses that when moving forward with rare disease trials, that through placing focus more toward the prevention/halting of disease progression we could drastically and rapidly reduce deaths.
In a similar fashion our therapeutics asks us to reframe the ‘typical’ approach when delving into the best ways of enhancing oncological trials. When it comes to the trial process, the end and main goal is typically aligned with the efficacy endpoints and finding the most successful means of achieving our desired end goals. However, as Sebastien Coppe and Samuel Salvaggio of One2Treat argue, this usually results in trial safety and quality being overlooked, highlighting the limitations with single-to-endpoint approaches and the importance of simultaneously considering all the relevant outcomes. In reframing in this way it could not only improve our patient-focused analyses but the precision of the evolvement in future oncology clinical studies.
This section also explores the obstacles in Alzheimer studies, explaining the difficulty in capturing data for the disease, as there can be limitations in those considered in the trial. It asks us to reevaluate the ways we can considered factors and components to our trials and how innovation of the preexisting formulations and foundations can be just effective in progressing clinical trials.
Much like other areas of the life science industry currently, AI and Machine Learning is at the forefront of development. And across
JCS – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Principal Content Editor, Clarivate
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
• Georg Mathis, Founder and Managing Director, Appletree AG
the journal we hear of how, for clinical studies, this seems to be a driving force for the means of improved efficiency and reduced costs. Its ability to manage and retrieve extensive complex data logs has been capitalised, and has notably improved the ease, circulation and accessibility of research. While on the contrary, although furthering development, our regulatory section examines the risks of potential data breaches and how moving forward with technology calls for readdressing the protection of patient data.
An interesting selection of content for you this issue that ask us, as members of the industry, to be open to ideas of reworking and readjusting our approaches and offering new ways of thinking and enhancing our methodologies to keep the world of clinical trials ever evolving.
I do hope you enjoy this edition of Journal for Clinical Studies, and I look forward to meeting many you over the coming events!
Chloe Euripides – Managing Editor
• Hermann Schulz, MD, Founder, PresseKontext
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, AsiaPacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
Ramus Medical
is a part of Ramus Corporate Group. The company is managed under a centralised quality management and has developed an integrated QMS as well as specific standard operating procedures tailored for the clinical trials department that are fully harmonised with the GCP guidelines, and the local and European legislation.
Ramus Medical EOOD is a full-service contract research organisation (CRO) in Sofia, Bulgaria.
The company was created in 2009 as a natural development of the Medical Laboratory Ramus Ltd., the largest privately-owned medical laboratory in Bulgaria.
The company independently manages clinical research projects in Bulgaria and provides partnerships in multinational clinical projects providing a comprehensive range of clinical research services:
Core Services include:
• Medical writing
Our staff has extensive expertise in the preparation, adaptation and translation of a wide range of clinical trial documents that are fully compliant with the Good Clinical Practice (GCP) standards, the client’s specifications and the regulatory requirements.
• Study start-up
We offer full or partial study start-up assistance for different types of studies throughout Bulgaria.
• Regulatory submission
• Project management
• Monitoring
• Data Management
• Pharmacokinetic evaluation
• Biostatistics
• Regulatory advice and services
• Readability User Testing
• Registration of medicinal products on the territory of Bulgaria
• Pharmacovigilance services
• Logistic department
• Destruction of IMPs/IMDs & clinical samples – agreement with PUDOOS
• Archiving services
• DDD activities
Ramus Medical has gained its expertise during the completion of numerous clinical projects carried out over the past decade:
• Phases I to IV drug trials
• Non-interventional studies
• Pilot and Pivotal Medical Device investigations
The clinical trials we conducted facilitated the MA/CE mark granted by various European Agencies/Notified Bodies and Third Country Agencies.
Ramus Medical offers flexible clinical research services in various domains, with extensive experience in fields.
Our team comprises qualified, appropriately trained, experienced, motivated and collaborative professionals and is competent to
www.journalforclinicalstudies.com
communicate effectively across geographical and cultural boundaries to resolve any arising issues. We adhere strictly to the agreed timelines during the clinical investigations and strive to complete the tasks on time.
Why are we the solution for your projects? Ramus has its own:
Medical and Bioanalytical Laboratory
In 2018 the Medical Centre Ramus was established, located in Sofia, Bulgaria. Up to date, it has three separate locations, one of which is developed as an independent clinical research centre in compliance with the requirements for the phase I unit.
The Medical Centre Ramus allows the conduct of clinical trials in all phases in many therapeutic areas.
The Medical Centre meets all requirements for performing highquality clinical research and is designed to maximise the delivery of high-quality research data and was GCP-inspected.
Ramus Medical retains an extensive database of investigators and sites compiled through years of mutually beneficial collaboration.
Our bioanalytical laboratory is equipped with leveraging state-ofthe-art instrumentation (LC-MS/MS), techniques, and facilities, our team of experts has experience in a broad range of small molecules. Our Analytical laboratories provide method development, transfer, validation, and analysis of preclinical and clinical biological samples. We have extensive expertise in developing sensitive methods for LCMS/MS-qualifying multiple analytes and metabolites.
• Logistical company, certified for hazardous and biological samples transportation
• Clinical site facility and own catering company for hospitalised patients
Site Selection in Clinical Trials: Current Challenges and Future Opportunities
Site selection is a crucial step in clinical research as it affects the quality, efficiency, and cost of the clinical trial. However, many challenges exist in the current site selection process, such as low investigator participation, poor recruitment performance, and lack of data-driven decision making. This article aims to highlight some of the key factors that need to be considered when selecting sites, and to provide suggestions on how to improve the process and leverage the potential of artificial intelligence (AI) and machine learning (ML) in the future.
According to a recent WCG survey, only 28% of investigators participate in more than one clinical trial, and 40% of trials fail to meet their recruitment targets. Some of the reasons for these low numbers are the lack of adequate training, resources, and incentives for investigators, as well as the mismatch between the trial protocol and site characteristics. Therefore, it is important to evaluate a site's capabilities, experience, patient population, and infrastructure before selecting it for a trial. The decreasing number of investigators and increase in site burden, lead to the challenge of meeting recruitment targets and make site identification and selection an imperative part of clinical trial execution that should be continuously evaluated. It is essential to establish clear and transparent communication and collaboration between sponsors, sites, and patients at the start of the site identification/selection process and throughout the trial. This is to ensure that modifications, such as adding and/or removing sites or amending protocols, are included in all feedback loops.
Current Practises and Challenges in Site Selection
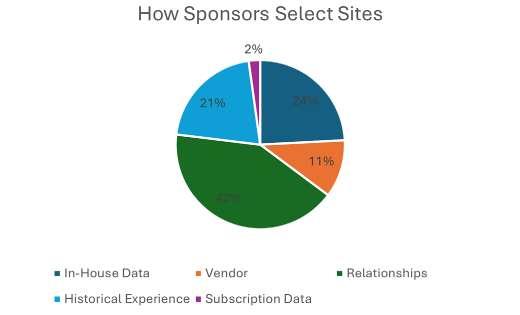
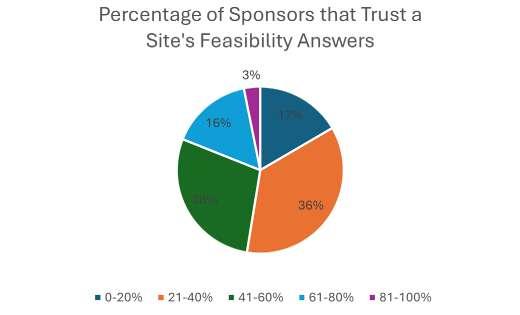
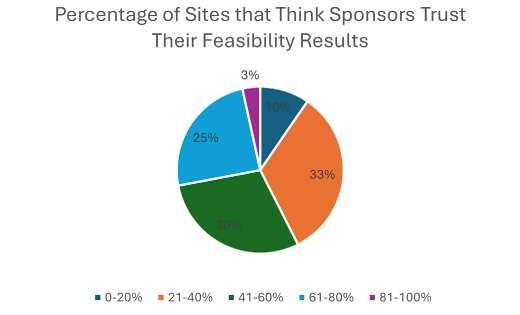
The traditional site selection process consists of two main stages: site identification and site feasibility. Site identification involves using various data sources, such as historical performance, claims data, regulatory compliance, and quality indicators, to generate a list of potential sites that match the study criteria. Despite the number of data sources available, a recent poll conducted by WCG revealed that more than half of sponsors are still making their site identification and selection decisions exclusively based on relationships and historical experiences, as opposed to databased selection (see Figure 1). The second step in the site selection process is site feasibility, which involves collecting data from the sites themselves such as, patient population, demographics, referral sources, and facilities to assess their suitability and interest in participating in the trial. However, in the same poll conducted by WCG, most respondents believe that sponsors have little trust in the feasibility responses they receive from sites (see Figure 2). Sites agree that sponsors are not likely to believe their responses (see Figure 3), particularly surrounding recruitment numbers.
It is not a surprise that the data collected at the time of site identification and feasibility may not always align with reality. This is because this process is often fraught with challenges, such as:
• Data Quality and Availability: The data used for site identification may be outdated, incomplete, or inaccurate, leading to suboptimal site selection. Moreover, the data collected from the sites during feasibility may not reflect the actual enrollment potential or performance of the sites, as they may overestimate or underestimate their capabilities or interest. This can result in wasted resources, delays, and low retention rates.
• Data Integration and Analysis: The data used for site identification may come from different sources, such as internal databases, subscription services, vendors, or CROs, and may not be integrated or analysed in a consistent or comprehensive way. Moreover, the data collected from the sites during feasibility may not be weighted or prioritised according to the study objectives or recruitment strategies, leading to biased or inefficient site selection. This can result in missed opportunities, misalignment of expectations, and poor performance.
• Data Utilisation and Feedback: The data used for site identification and feasibility may not be utilised or communicated effectively throughout the site selection process, leading to missed opportunities or misaligned expectations. Moreover, the data collected from the sites during feasibility may not be updated or validated regularly, leading to inaccurate or outdated site selection. This can result in loss of trust, dissatisfaction, and attrition among the sites and sponsors.
To address these issues, some of the best practices and recommendations for improving the site selection process are:
• Use reliable and relevant data sources that are updated and verified regularly, and which cover a wide range of site characteristics and performance indicators.
• Use standardised and transparent criteria and methods for integrating and analysing the data, that align with the study objectives and recruitment strategies.
• Use effective and timely communication and feedback mechanisms that involve the sites and sponsors throughout the site selection process, and allow for adjustments and corrections based on feedback during the site identification process, and throughout trial execution to allow the necessary mitigation strategies to be implemented.
Future Trends and Opportunities in Site Selection
As the complexity and competitiveness of clinical trials increase, there is a need for more innovative and adaptive approaches to site selection that can overcome the current challenges, and optimise the trial outcomes whilst continuing to use some of the tried and true qualitative and quantitative methods. Some of the emerging trends and opportunities in site selection are:
AI and ML: AI and ML can be used to enhance the data quality, integration, analysis, utilisation, and feedback in site selection, by applying advanced algorithms and models that can learn from the data
and provide insights and recommendations. For example, AI and ML can be used to:
• Predict the enrollment rate and performance of sites based on historical and real-time data, and adjust the site selection accordingly. This can help to optimise the site allocation, reduce the risk of under- or over-enrollment, and improve the trial efficiency and cost.
• Identify the best combination of site attributes and factors that align with the study objectives and recruitment strategies, and rank the sites accordingly. This can help to select the most suitable and interested sites, increase the site diversity and representation, and improve the trial quality and validity.
• Provide dynamic and interactive feedback to the sites and sponsors throughout the site selection process, and update the site selection based on the feedback. This can help to enhance site engagement and empowerment, foster a collaborative
and respectful relationship, and improve site retention and satisfaction.
Site Engagement and Empowerment: Site engagement and empowerment can be used to enhance site participation, recruitment, and retention in clinical trials, by providing incentives, support, and recognition for the sites. For example, site engagement and empowerment can be used to:
• Offer financial support to the sites to support mutually beneficial initiatives to recruit patients to the trials, and align with their diversity, equity and inclusion goals, and health literacy activities. This can help to motivate sites, increase their loyalty and commitment, and reduce site turnover and dropout as these activities help support the sponsor, patient, and site relationship.
• Provide training, education, and resources to sites to help them improve their clinical research capabilities and competencies. This can help to increase the site’s professionalism and quality, reduce its errors and deviations, and enhance its compliance and adherence.
• Listen to site feedback. Leverage qualitative and quantitative data to show their successes and shortcomings. It is worth having continuous feedback loops, like client satisfaction surveys, to obtain qualitative feedback from sites on what is working and what is not working so that you can have a real-time understanding of the current circumstances at the site in order to help provide mitigation support.
• Recognise and acknowledge sites for their achievements and challenges in clinical research and foster a collaborative and respectful relationship with them. This can help to increase site trust and confidence, reduce frustration and dissatisfaction, and improve communication and feedback.
Conclusion
Site selection is a critical component of clinical research, as it can determine the success or failure of the trial. However, the current site selection process is often inefficient, ineffective, and inconsistent, leading to poor site performance, recruitment, and retention. To address these issues, there is a need for more innovative and adaptive approaches to site selection that can leverage the power of AI and ML, and enhance site engagement and empowerment. By doing so, the site selection process can be improved and optimised, resulting in better trial quality, efficiency, and cost.
Cristin MacDonald
Cristin MacDonald, PhD, is Vice President, Client Delivery at WCG. As the leader of WCG Avoca’s integrated consulting and research solutions, Cristin provides consulting services to top pharmaceutical, biotech, and contract research organisations, and oversees client deliverables, systems, and processes.
Figure 1
Figure 2
Figure 3
Settling the Score with Scleroderma While Waiting for a Cure:
Addressing Disease Progression in New Clinical Trials
Scleroderma, also called systemic sclerosis (SSc), is a rare, progressive autoimmune connective tissue disorder with no cure. It causes inflammation in the skin and other parts of the body and triggers the immune system to produce excess collagen, which leads to hardening and tightening of skin and tissue (i.e., fibrosis). It is a heterogeneous disease that affects all patients differently, manifesting as limited (i.e., progressing more slowly) and diffuse (i.e., more advanced). The average age of disease onset is 30–50 years, and four out of five patients with scleroderma are women, according to the Scleroderma Research Foundation (SRF).1 The Johns Hopkins University, host to one of several designated scleroderma research and treatment centers in the United States (US), notes that approximately (~) 300,000 people in the US have been diagnosed with scleroderma, and ~10,000 die from the most serious forms of the disease each year.2
The US Food and Drug Administration (FDA) has increased its focus on specific disease areas, including rare diseases, and has been seeking input on what patients are hoping for when considering treatment options. Recognising the value of gathering patient input, the agency hosted several disease-specific patientfocused drug development (PFDD) public workshops after the passage of the fifth reauthorisation of the Prescription Drug User Fee Act (PDUFA V). In October 2020, the FDA held one such workshop to obtain patients’ perspectives on scleroderma, including effects on their health and well-being that most impact daily life and their experiences using prescription medical treatments and other treatments or therapies.2 Presentations at the meeting provided an overview of scleroderma, including the pathogenesis of the disease, which is not fully understood. However, over the past few decades, progress has been made in understanding its pathogenesis, which includes vascular involvement or vasculopathy (e.g. Raynaud’s phenomenon), dysregulation of the immune system, and fibrosis in the skin, musculoskeletal system, and internal organs (e.g., lungs, heart, kidneys).
Treatments generally address the symptoms of scleroderma and do not target the underlying cause of the disease. Typical therapies include proton pump inhibitors for digestive symptoms, medications to prevent organ rejection and/or treat arthritis (e.g., immunosuppressants), corticosteroids for skin and arthritis symptoms, blood pressure medications, and pain relievers. None of these treatments reverse the disease or halt its progression.
Historically, drug development for scleroderma has focused on reducing the severity of symptoms and managing or preventing challenges associated with disease progression. Interstitial lung disease (ILD) – a complication caused by scleroderma in ~40–75% of all patients with the disease – is the leading cause of death in this population.4 Scleroderma-associated ILD (SSc-ILD) has been and continues to be treated off label with immunosuppressive agents (e.g., mycophenolate mofetil, mycophenolic acid). However, in 2019, the FDA approved the first treatment for SSc-ILD, Ofev (nintedanib), from Boehringer Ingelheim Pharmaceuticals, Inc. The product
is indicated to slow the rate of decline in pulmonary function in patients with SSc-ILD. A second therapeutic for that same indication came to market in 2021 when the agency approved tocilizumab (Actemra), from Genentech, Inc. Despite these breakthroughs for scleroderma patients with lung involvement, a significant unmet need exists for treatments for all scleroderma patients, especially to treat the overall disease and halt its progression through achieving remission.
Novel Treatments on the Horizon
Several treatments are in development for scleroderma with various mechanisms of action. In addition to monoclonal antibodies (mAbs), antifibrotic agents and CAR T-cell therapies are under evaluation in several clinical studies across the globe.
CABA-201.
Perhaps the most promising novel approach to treating scleroderma is chimeric antigen receptor (CAR) T-cell therapy, which has traditionally been studied for oncology indications. Multiple CAR T-cell products are in development for scleroderma and other autoimmune diseases. Cabaletta Bio, Inc (Cabaletta), is recruiting ~12 adult participants for an open-label phase I/II study (RESET-SSc) to evaluate the safety and efficacy of CABA-201, a 4-1BB–containing fully human CD19-targeted CAR T-cell investigational therapy, for the treatment of SSc. This trial, which is part of the sponsor’s Chimeric Antigen Receptor T cells for Autoimmunity (CARTA) strategy, is evaluating the potential of CABA-201 to “transiently, but fully, eliminate B cells” to potentially enable durable remissions through a “reset” of the individual’s immune system, Cabaletta announced in a press release.5
In the study, subjects receive a single intravenous infusion of CABA-201 1 x 106 cells/kg following preconditioning with fludarabine and cyclophosphamide. The primary outcome measure is the incidence of adverse events (AEs), and efficacy is one of several secondary endpoints. In January 2024, the sponsor announced that it received fast track designation from the FDA for CABA-201 for the treatment of scleroderma. Then, in March 2024, the agency granted orphan drug designation to the CAR T-cell therapy for the treatment of SSc. The study began in June 2024 and is estimated to complete by July 2029.
FT011.
A novel first-in-class oral therapy, FT011 (asengeprast) from Certa Therapeutics (Certa) is in development for the treatment of chronic fibrosis in multiple organs. The sponsor completed a multinational, double-blind phase II trial that randomly assigned 30 participants to three treatment arms: oral FT011 400 mg, FT011 200 mg, or placebo once daily in addition to the standard of care for 12 weeks. Positive results reported in November 2023 showed a clinically meaningful improvement in 60% of participants treated with FT011 400 mg (p-value = 0.019) and 20% of participants in the FT011 200 mg group compared with 10% in the placebo group.6 Overall, significant improvements were observed in American College of Rheumatology Composite Response Index in Systemic Sclerosis (ACR-CRISS) score, skin thickness, lung function, physician-reported assessment, and quality-of-life evaluations. FT011 was well tolerated, and no
differences in AE rates were noted between the treatment arms. No serious AEs or AEs resulting in study drug interruption, withdrawal, or discontinuation were reported.
FT011 targets the G protein–coupled receptor GPR68, and transcriptomic research has shown that treatment with FT011 leads to reversal in the activation of genetic markers associated with fibrosis. This provides the potential for a precision therapy, Certa stated.7 In February 2024, the firm announced that FT011 was granted fast track designation by the FDA for the treatment of SSc after receiving orphan drug designation in October 2023 for the same indication. Following positive results from the phase II study, the sponsor has announced plans to begin a pivotal phase III study in late 2024. The fast-track status of the agent could lead to expedited review of an application through more frequent communication with the FDA, eligibility for accelerated approval and priority review, and a rolling review of the application.
Anifrolumab.
A multicentre, randomised, double-blind, placebo-controlled phase III study (DAISY) is recruiting adult participants with SSc who may be taking one or a combination of protocol-specified standard therapies to evaluate the efficacy and safety of anifrolumab (Saphnelo, from AstraZeneca), a fully human mAb that targets interferon alfa receptor subunit 1. Approximately 306 participants are randomised to receive anifrolumab or placebo subcutaneously once weekly for 52 weeks. The primary outcome measure is the number of participants responding to treatment based on the Revised CRISS. Begun in November 2023, the study is estimated to complete in December 2027.
These treatments are among many other novel therapies under evaluation for the treatment of scleroderma. While early detection and symptom management are paramount for these patients, preventing disease progression and halting the disease altogether have the potential to drastically reduce the number of deaths attributed to scleroderma each year.
REFERENCES
1. Scleroderma Research Foundation. https://srfcure.org/
2. Public Meeting on Patient-Focused Drug Development for Systemic
Sclerosis. Food and Drug Administration. https://www.fda.gov/drugs/ news-events-human-drugs/public-meeting-patient-focused-drugdevelopment-systemic-sclerosis-10132020-10132020#:~:text=On%20 October%2013th%2C%202020%2C%20FDA,patient%20views%20on%20 treatment%20approaches
3. Diagnoses & Tests. National Scleroderma Foundation. https:// scleroderma.org/diagnoses-tests/
4. Bernstein EJ, Huggins JT, Hummers LK, Owens GM. Systemic sclerosis with associated interstitial lung disease: management considerations and future directions. Am J Manag Care. 2021. https://www.ajmc.com/ view/systemic-sclerosis-with-associated-interstitial-lung-diseasemanagement-considerations-and-future-directions
5. Cabaletta Bio Announces FDA Granted Orphan Drug Designation to CABA-201 for Treatment of Systemic Sclerosis. Cabaletta Bio Website. https://www.cabalettabio.com/news-media/press-releases/ detail/107/cabaletta-bio-announces-fda-granted-orphan-drugdesignation
6. Certa Therapeutics presents positive data from a Phase 2 clinical study highlighting the potential benefit of FT011 as a novel treatment for scleroderma. Certa Therapeutics Website. https://certatherapeutics. com/certa-therapeutics-presents-positive-data-from-a-phase-2clinical-study-highlighting-the-potential-benefit-of-ft011-as-a-noveltreatment-for-scleroderma/
7. Certa Therapeutics’ FT011 Granted US FDA Fast Track for the Treatment of Systemic Sclerosis. Certa Therapeutics Website. https:// certatherapeutics.com/certa-therapeutics-ft011-granted-us-fda-fasttrack-for-the-treatment-of-systemic-sclerosis/
Jaime Gavazzi
Jaime Gavazzi is a Principal Content Editor for the Cortellis suite of life science intelligence solutions at Clarivate. Her previous roles include writing and editing for books, online magazines, educational coursework, government proposals, and government regulatory publications. Her primary assignments at Clarivate include reporting on FDA drug/device advisory committee meetings and drug approvals. She is also a scleroderma patient living with interstitial lung disease.
Email: jaime.gavazzi@clarivate.com
The Expanding Role of Functional Service Providers in Clinical Research
Functional service providers (FSPs) are reshaping clinical research by offering specialised skills and adaptable resources. As trials become more complex, many biotech and biopharma companies are opting for FSPs over full-service outsourcing. FSPs provide numerous advantages such as globalised strategies, expert domain knowledge, and efficient management, balancing quality, innovation, and cost. These partnerships speed up market introduction of new therapies and strengthen long-term collaborations, enhancing trial management. This article explores the strategic importance of FSPs in clinical research, emphasising their contribution to cost-efficiency, the acceleration of drug development, and technological innovation in trial management.
The Growing Role of FSPs in Biopharmaceutical Research
As both large and small biotech and pharmaceutical companies streamline their operations, outsourcing has become essential to drug development and clinical trials. The role of FSPs in outsourcing has expanded significantly, with their involvement in monitoring and data management tasks rising from 28 percent in 2018 to over 40 percent by 2021.1,2 As of 2023, the FSP market was valued at $15 billion and is projected to reach $29 billion by 2031, growing at a compound annual growth rate of more than 8 percent from 2024 to 2031.3 This projected growth highlights the industry's increasing reliance on FSPs to enhance operational efficiencies and indicates a strategic shift in resource allocation within pharmaceutical companies to optimise and accelerate the drug development process. To fully appreciate this trend, it is crucial to examine the specific advantages FSPs provide to sponsors of clinical research.
FSPs Offer Flexible and Comprehensive Solutions
FSPs offer versatile services such as biometrics, clinical operations, safety and medical writing. Unlike traditional full-service outsourcing, FSPs adopt a flexible, customisable approach, enabling sponsors to selectively outsource functions as required. This strategic flexibility boosts clinical research efficiency and adapts seamlessly to evolving industry needs. Common services include:
• Biostatistics: FSPs deliver biostatisticians to design study protocols, perform statistical analyses, and ensure accurate data interpretation, which is crucial for validating trial results and regulatory submissions.
• Clinical Monitoring: FSPs provide both on-site and remote monitoring to ensure that clinical trials comply with protocols, SOPs, Good Clinical Practice, and regulations.
• Data Management: This includes managing data collection, processing, and validation to ensure data integrity. Services cover database design, maintenance, and archiving.
• Regulatory Affairs: FSPs assist in navigating the complex regulatory landscape, preparing documentation and submissions, ensuring compliance with global standards, and maintaining communication with regulatory bodies.
• Patient Recruitment: Developing and implementing strategies to identify, screen, and enroll suitable trial participants for clinical trials.
• Pharmacovigilance: Monitoring, evaluating, and reporting adverse events and other drug-related risks to ensure patient safety.
• Project Management: Overseeing the entire lifecycle of a clinical trial, ensuring it progresses on schedule and budget while meeting all scientific benchmarks.
• Medical Writing: Creating essential clinical documents, such as study protocols, final study reports, consent forms, and scientific papers for publication.
• Quality Assurance: Ensuring all trial aspects are conducted and documented following applicable standards and regulations.
• IT and Technology Support: Offering and supporting key technologies such as electronic data capture systems and clinical trial management systems.
Strategic Advantages of Functional Service Providers
As previously noted, the strategic use of FSPs in clinical research provides tailored, efficient, and cost-effective solutions. The primary benefits of using an FSP partner include:
1. Cost Efficiency: FSPs enable sponsors to boost productivity while maintaining high proficiency at reduced costs. Unlike traditional full-service outsourcing, which often involves packaging entire deliverables to a contract research organisation, FSPs offer expertise for distinct functions. This tailored approach can minimise unnecessary expenditures and maximise cost-effectiveness.
2. Improved Quality and Governance: Collaborating with FSPs allows clinical research teams to access specialised expertise. By focusing on specific functions such as site selection, patient recruitment, and trial monitoring, FSPs enhance the seamless execution of trials, efficient resource allocation, and adherence to quality standards.
3. Flexibility and Scalability: FSPs provide unmatched flexibility, permitting organisations to outsource the essential functions of a trial. This modularity means resources can be scaled up or down as needed without the complexities of hiring and training staff.
4. Time-to-Market Reduction: Utilising the FSP model accelerates trial timelines. By leveraging specialised resources for dedicated tasks, sponsors can accelerate study execution, leading to faster product development and earlier market entry.
5. Tailored Solutions: FSPs create bespoke solutions that cater to the unique needs of each sponsor. This approach allows sponsors to combine different services from the same FSP, ensuring a perfect fit for their specific requirements.
6. Communication and Transparency: FSPs facilitate open communication channels with drug developers, ensuring that every stage of the trial is aligned with their goals.
7. Reduced Operational Burden: Outsourcing specific functions to FSPs alleviates the operational load of managing extensive in-house teams for every aspect of clinical research, allowing internal teams to focus on their core competencies.
8. Risk Mitigation: With their ability to deliver specialised expertise, FSPs reduce the risk of errors, delays, and compliance issues.
The cumulative advantages of these benefits are noteworthy. According to a report from the Tufts Center for the Study of Drug Development, companies employing FSPs have reported achieving cost savings of 20 to 30 percent compared to traditional outsourcing models.4 Similarly, research conducted by the Association of Clinical Research Organizations indicates that FSPs can reduce costs by 15 to 30 percent, underscoring their efficiency and economic benefits in the industry.5
Emerging Technological Trends
Technology also is reshaping clinical trial research, with innovations such as artificial intelligence (AI), blockchain, the Internet of Things, and real-time data analytics driving revolutionary changes. These technologies streamline trial processes, improve data quality, and accelerate timelines, enhancing operational efficiency and altering the clinical trial landscape.
AI, for example, accelerates data analysis, facilitating more informed decision-making, and fostering innovative trial designs. AI and machine learning are essential for interrogating large datasets, recognising patterns, and proposing tailored treatment strategies based on detailed data analysis.
Blockchain technology is crucial for maintaining data integrity and security in clinical trials. By providing a secure, unalterable record of clinical data, blockchain builds trust among stakeholders and simplifies data-sharing processes. The adoption of these technologies requires strict data security and compliance with regulations.
As FSPs integrate these new advanced technologies, they are advancing their role as strategic partners in the clinical enterprise. This infusion of new technologies allows FSPs to further enhance data quality and speed up trial timelines, positioning them at the forefront of a new era of innovation and efficiency in clinical trial management. For clinical sponsors, leveraging these technologies through FSPs is key to managing the complexities of clinical trials and securing long-term success in meeting unmet medical needs.
Selecting the Right Functional Service Provider
Selecting the right FSP is crucial for developers aiming to optimise clinical trial operations and achieve successful outcomes.
Below is a checklist of tips for choosing the right FSP:
• Align on Expertise and Specialisation: Ensure the FSP specialises in the specific services you need, such as biostatistics, clinical monitoring, or regulatory affairs. Assess their track record and expertise to confirm they can meet your needs.
• Evaluate Cultural Fit: The integration of the FSP team with your company's culture is vital. The FSP should align with your operational practices and values and demonstrate a commitment to seamless collaboration and transparency.
• Consider Scalability and Nimbleness: Choose an FSP that can scale services according to the evolving needs of your clinical trials.
• Assess Technological Capabilities: In today's data-driven trial environment, having an FSP with strong IT and data management capabilities is essential. Review their use of technology, such as electronic data capture and clinical trial management systems, and their ability to integrate with your technologies.
• Check for Quality and Compliance: The FSP should have a robust quality assurance system and a proven track record of compliance with regulatory standards.
• Review the Financial Health and Stability of the FSP: Ensuring that the FSP is financially stable and can sustain long-term operations is crucial for multi-year trials.
• Ask for Client References and Case Studies: References from other sponsors or case studies can provide insights into the FSP’s operational effectiveness and reliability.
• Discuss and Define Clear Expectations: Before finalising a partnership, define the scope of work, expectations, and deliverables. Transparent discussions about timelines, budgets, communication protocols, and escalation paths ensure smooth operations.
• Plan for Long-term Engagement: Look for an FSP that can meet current needs and has the potential to partner with you for future projects. A long-term partnership can yield significant benefits in terms of operational continuity and accumulated knowledge.
REFERENCES
1. PPD. (2021). FSP Outsourcing Trends in Clinical Research.
2. Shaw, E. (2023, November 13). Why are FSP models gaining traction within clinical research? Fierce Pharma
5. Association of Clinical Research Organizations (ACRO).
Decentralizing Clinical Trials: A New Quality-by-Design, Risk-Based Framework.
Lisa Stetler
Lisa Stetler, Senior Director at Biometrics Resourcing Operations, is a seasoned leader with over 25 years as a statistical programmer in the clinical research industry. During her career, she has built and led programming teams and currently oversees a large, high-performing FSP. Her extensive experience spans Phases 1 to 4 across various therapeutic areas, with expertise in DSMB, DMC, CSR, ISS/ISE reporting, and regulatory submissions to agencies like the FDA, EMA, and PMDA.
Ershlena McDaniel
Ershlena McDaniel, Senior Director at Resourcing Operations, is an experienced data manager by trade who has been in the industry since 2004, serving in various positions. She has domain experience through the lifecycle of clinical trials and has worked in multiple therapeutic areas. Ershlena joined Triangle Biostatistics (now Catalyst) in June 2013 as a contractor Sr. Clinical Data Manager and was promoted to Manager in 2019. In 2020, she began assisting with recruitment of biometrics resources as Catalyst worked to expand their FSP presence in the industry. In her current position, Ershlena leads our biometrics resourcing team. She and her team have been highly successful in placing qualified biostatisticians, statistical programmers, clinical programmers, EDC programmers, and data managers.
4. DiMasi, J. A., Grabowski, H. G., & Hansen, R. A. (2016). Innovation in the pharmaceutical industry: new estimates of R&D costs. Journal of Health Economics, 47, 20-33
(2020).
40 years of building powerful industry partnerships.
Broadest portfolio of self-injection products built on platforms
Serving pharma globally for all their originator and biosimilar needs
Over 70 products launched in 15 different therapeutic areas
Global manufacturing footprint spanning Switzerland, Germany, China and North America
Fully integrated strategic partner network
For more information visit www.ypsomed.com/yds
Ypsomed AG // Brunnmattstrasse 6 // 3401 Burgdorf // Switzerland T +41 34 424 41 11 // info@ypsomed.com
www.journalforclinicalstudies.com
Striking the Right Balance Between Transparency and Privacy in Clinical Trial Data Sharing
Clinical trial researchers must balance the at times contradictory needs for transparency, with protecting participants’ privacy and looking after companies' intellectual property. Transparency and information sharing can be voluntary and motivated by the desire to progress science through collaboration. However, increasingly sharing of trial documents and data is driven by regulatory requirements. The different global regulations ensure that the information released is governed by best practices which reduce the risk of re-identifying individuals represented in various data sets. In what may seem to be a conflicting end result, sponsors and regulators must find the delicate balance between protecting participant privacy, through redaction or anonymisation strategies, and providing data utility. This can be defined as the degree to which a reader can analyse and make meaningful interpretations from the information.
So how can pharmaceutical or academic researchers perform this delicate balancing act? And when operating in multiple geographic regions how can they meet the expectations of different regulatory bodies? In this article we outline the requirements of different regulators. We suggest ways that researchers can assess risk, plan and take steps that maximise data utility while meeting the expectations of regulators and patients.
Redaction vs Transformation
Redaction is a method of masking information by applying a box over direct or indirect identifiers. It can be done manually or semiautomatically using most common software tools. Because of this it can be perceived as attractive and cost-effective. For short documents with little personal information (PI) or Protected Personal Data (PPD) it may be the logical choice. However, it has little to no data utility since all of the information is fully hidden. In addition, deciding what to redact can be subjective.
Transformation is the process of pseudonymising, offsetting or generalising direct or indirect information relating to participants. Direct identifiers can be the full name, subject numbers, phone number, email address or a government ID number. Indirect identifiers alone might not lead to reidentification, but combined with other information could be used to identify an individual. They include city, state, demographics and sensitive medical information. In transformation, instead of including a participant’s age, participants’ ages would instead be banded into groups. Validated software tools can assess the risk of reidentification, establish transformation strategies and implement anonymisation techniques. This can save time by automatically applying anonymisation to multiple clinical datasets and documents. This automated quantitative approach, when combined with robust quality control steps, can ensure confidence in the outputs.
The Challenges of Voluntary Data Sharing
Many sponsors already share data beyond the regulatory requirements with several voluntary data sharing programmes established in recent years. The objectives are varied but include contributing to open research to benefit future research studies and improving patients’ access to information. Because the drivers are voluntary, decisions about subjectivity and other factors are varied and can be inconsistent. What gets shared, with whom, and for what purpose may be open to interpretation by the different sponsors and stakeholders. From an organisational perspective, it calls for a change in corporate planning, resourcing and putting in place processes to deal with information requests.
Regulatory Requirements
Regional regulatory bodies also have similarities and differences and different timelines for reporting information. These can be as detailed as which colour to use for overlays of PPD and confidential business information (CBI) or company confidential information (CCI). The Food and Drug Administration (FDA) and National Institute of Health allow redaction of information that the sponsor deems necessary to safeguard PPD and CCI. In Japan, the Pharmaceuticals and Medical Devices Agency (PMDA) requests justification for PPD and CCI redactions. These can be rejected by PMDA if the rationale for redaction is not acceptable. The European Medicines Agency (EMA) allows CCI redaction on a very limited basis and each proposed redaction must be accompanied by a justification which may be rejected. During the first year of the implementation of Policy 0070, of 1.3 million pages submitted just 134 pages, or 0.01% of the total pages published were accepted. A separate EU regulation includes an initiative to post plain language summaries alongside the final results summaries at the time of marketing authorisation. Health Canada’s Public Release of Clinical Information (PRCI) policy provides public access to clinical information that allows independent analysis of data and supports new scientific research directions. The guidance is similar to EMA’s Policy 0070 in overall scope and processes.
Reidentification Risk
If data are insufficiently anonymised there is a significant risk of reidentification. In a court case between the Southern Illinoisan and The Department of Public Health, expert witness Dr Latanya Sweeney successfully reidentified 18 out of 20 individuals in a neuroblastoma data set from the Illinois cancer registry. For the remaining two individuals, the witness was able to suggest one of two alternative names. While a graduate student at MIT, Dr Sweeney, identified the Governor of Massachusetts’s medical information using birth date, gender and ZIP code information and a publicly available database. In Canada, a national broadcaster aired a report of the death of a 26 year old student taking a particular drug. The student was reidentified using information from the adverse drugs reaction database released by Health Canada. Aside from the loss of privacy and distress to individuals or their families, these reidentifications can lead to lawsuits and pay outs.
Regulatory
Strategies to Minimise Risk and Maximise Research Success
Managing both voluntary and regulatory requirements is challenging for any research organisation. Advance planning, agreed standardised processes and clearly defined roles will help to ensure the correct measures are established and upheld. A small team of well-informed
cross-functional members is important to respond to data sharing initiatives. Proactive planning for the required disclosure activities should be built in from start up. Regulators look for justification for anonymising both PPD and CCI, and planning report writing in advance can save time later on. In addition, many of the tasks related to anonymisation and document preparation for release can be outsourced to relieve pressure on clinical teams. Having validated software tools and knowledgeable people to implement them can help you to avoid the potential pitfalls and ensure that your findings will contribute to future research.
REFERENCES
1. SOUTHERN ILLINOISAN v. ILLINOIS DEPARTMENT OF PUBLIC HEALTH (2006) | FindLaw
2. L. Sweeney. k-anonymity: a model for protecting privacy. International Journal on Uncertainty, Fuzziness and Knowledge-based Systems, 10 (5), 2002; 557-570
3. Khaled El Emam, Fida Kamal Dankar, Protecting Privacy Using k-Anonymity, Journal of the American Medical Informatics Association, Volume 15, Issue 5, September 2008, Pages 627– 637, https://doi.org/10.1197/jamia.M2716
Sarah Johnston
Sarah Johnston is Director of Clinical Trial Transparency at ICON, with over 20 years of industry experience spanning diagnostic assay development, pre-clinical and clinical project management, and technical writing. Over the past 7 years, Sarah has focused on Clinical Trial Transparency within Clinical Research Organisations. Since joining ICON in 2020, she has led diverse, global teams in all aspects of trial transparency, while managing high-profile client relationships and large-scale programs across both pharmaceutical and biotech sectors.
Maureen Kennedy
Maureen Kennedy holds a Bachelor's degree in Interdisciplinary Health Sciences from Saint Joseph's University in Philadelphia and brings over two decades of experience in pharmaceutical research. She joined ICON in 2009 and, for the past 14 years, has specialised in Clinical Trial Transparency. In her current role as Clinical Trial Transparency Manager, Maureen leads a dedicated team focused on the anonymisation and redaction of clinical trial documents for public disclosure.
Michael Healey
Michael Healey is an Associate Director of Clinical Programming at ICON, bringing over 20 years of industry experience. Since joining ICON's Statistical Consulting group in 2021, Michael has specialised in data anonymisation services, submission support, and providing expert guidance on CDISC SDTM and ADaM standards. His career spans a range of technical roles, including database development and analysis programming for regulatory submissions. Additionally, Michael has held managerial positions within ICON's Clinical Programming team, contributing to both technical leadership and team development.
Generating Entire Regulatory Dossiers – The Logical Next Step for GenAI in Life Sciences?
Generating Entire Regulatory Dossiers – The Logical Next Step for GenAI in Life Sciences?
Generative AI (GenAI)’s ability to digest, assess and summarise key insights and findings from across vast and diverse bodies of existing content, and data - even as this is being continuously refreshed –make the technology ideal for high-volume everyday tasks completed by regulatory affairs teams. As it is, GenAI is already making its mark with some impressive early pilot solutions.
For instance, initial GenAI applications have demonstrated the ability to pre-empt agency queries and build stronger marketing authorisation applications, by applying insights gleaned from historical health authority (HA) interactions, where these have been put into an accessible knowledge base – serving as a significant process accelerator and productivity tool. Across 23 different languages, pilot applications have seen more than a dozen fields of data extracted with 90% accuracy – with up to 80% faster processing and three times fewer handovers than if teams were trawling through agency correspondence themselves.
The technology is also demonstrating powerful potential in monitoring and proactively using the latest global regulatory intelligence, for instance as part of impact assessment/change management. Early pilot projects here too have yielded 50- 80% faster processing, and in this case half the handovers compared to manual lookup and intervention.
But the truly transformational potential is still ahead – on course to be realised within the next two years. This will be the point at which regulatory teams are able to lean on the technology to generate and cross-check entire regulatory submissions automatically, with a quality review from RA professionals requiring just a fraction of the effort expended today. This capability will be particularly powerful in transforming regulatory submission lifecycle management, which today consumes significant time and budget.
Despite the increasing trend of data-oriented submissions, the reality of content-based submissions is here to stay for the foreseeable future. At a conservative estimate, large pharma organisations typically generate around 600–800 submissions per month. Even a very modest time saving, of just 1–2 hours per submission, would make a substantial difference to associated resource allocation, and that is the minimum saving expected once GenAI is harnessed in earnest to automate the collation and assembly of content, extrapolating from initial regulatory use cases of the technology.
The Best is Yet to Come
Among the enablers of this automation leap within regulatory affairs,
to the creation of complete submissions, are GenAI’s accelerating pace of advancement, its steady maturation, and the technology’s rapid acceptance and perceived reliability.
Already, the technology is being used widely and with confidence to analyse and infer meaning from data and content in a wide range and formats, and distil what is needed into whatever the desired new format for the target context. This is true in most enterprise settings today – even, as we’ve seen, within the strictly-regulated life sciences industry, where GenAI is already trusted to transform not only the cost-efficiency and impact of marketing authorisation and licence maintenance, but also the affordability, speed and precision of realworld product safety monitoring.
The next wave of developments will build on all of this important progress, to enable end-to-end process transformation. Next use cases will include the provision of inline regulatory guidance to help users in submission compilation; generation of new draft submission content based on existing content; and cross-validation of final content against regulatory guidance and data (each of these may be delivered at the different times with different scope).
These targeted GenAI applications will be able to identify and draw from the latest correct sources, to collate and repurpose the relevant information and fill the respective submission outline. This will automatically involve cross-checking with the company’s regulatory information management (RIM) system, assess what has previously been submitted, ensuring that the new submission is accurate and consistent.
Better, Faster Submissions – in Any Market
Through all of this lightning-fast cross referencing (which will ensure that the correct excipient/ingredient information has been used, for instance), GenAI will expedite submissions compilation. It will also improve the quality, accuracy, and success rate of submission updates, reducing the ‘return’ rate, and boosting the company’s track record and associated standing with regulatory agencies.
In other words, on top of substantial time and cost savings, as GenAI does all the heavy-lifting and content cross-checking, significant additional benefits will include a significant uptick in quality as accuracy, consistency and submission success rates go up.
In the meantime, skilled teams will be free to focus more of their attention on scientific work – activities that add more value for the organisation.
In addition to fulfilling the demands of agencies in mature regulatory markets such as the EU and North America, advanced automation in regulatory submissions generation could transform the efficiency of dealing with less developed markets.
Emerging markets together account for a sizeable proportion of the global life sciences opportunity . Growth in pharma sales in emerging markets is set to accelerate over the next decade, with medicine use in Latin America and Asia expected to rise faster than other regions over the next five years.
As more mature markets lean toward well-defined electronic submissions, it is a stark reality that the rest of the world continues to rely heavily on non-electronic files; for submission to authorities whose requirements are less standardised. The ability to streamline associated submissions with advanced end-to-end automation promises to be very powerful in this context, to help companies navigate the differing requirements, deduce “what good looks like”, and swiftly collate and format what’s needed.
Building Knowledge Bases, Enriching Data and Experimenting with what is Possible
Additional opportunities for GenAI in a regulatory affairs context include automated cross-checks to identify discrepancies and anomalies in data and its formatting, as part of companies’ efforts to get their IDMP data standardisation in order, by honing and formalising associated data governance. Further possibilities include more efficient and effective maintenance of labelling compliance internationally across the product lifecycle, again boosted by automated, GenAI-enabled cross-referencing.
With all of this potential on the horizon, it is important that organisations across life sciences start to get to grips with GenAI technology now. Testing out the possibilities will give companies a feel for how far GenAI can go, how quickly results can be reliably honed, and how much time and budget this could buy back for hardpressed regulatory teams.
Simply adding a GenAI capability alone is no magic bullet, of course. The more robust the assets GenAI can draw from, the more reliable and transformational associated process automation initiatives will be. The more diverse the available checkpoints, meanwhile, the more confidence there will be in the newly-generated output.
In parallel, then, companies will need to do some work to proactively bolster their regulatory intelligence knowledge bases (comprising non-public information and soft intelligence that has accumulated within companies based on their experience and direct
HA relations). They should also continue or recommit to existing initiatives to clean up, standardise, and unify their product data. All of this is crucial groundwork that is needed anyway, and will optimise the success and impact of GenAI-based process automation.
REFERENCES
1. This is ArisGlobal’s own data from early customer pilots. Separately, McKinsey estimates that deploying next-generation AI to improve HA responses and their impact can reduce Agency follow-up by 50%.
2. Emerging Markets Offer Pharma Its Next Growth Opportunity - National governments and global non-governmental organizations are trying to expand access to essential medicines and treatments in developing countries: Pharmacy Times, February 2024 https://www.pharmacytimes. com/view/emerging-markets-offer-pharma-its-next-growth-opportunity
3. Latin America is one of the fastest-growing pharmaceutical markets in the world. With an increasing and aging population of 660 million people, it is forecast to grow at a compound annual growth rate (CAGR) of seven to ten percent between 2023 and 2027 (Statista, July 2024: https://www. statista.com/topics/12539/pharmaceutical-industry-in-latin-america/) In 2024, the projected revenue for the Pharmaceuticals market in Asia is expected to reach a staggering US$238.10bn. The largest market within this industry is Oncology Drugs, with a projected market volume of US$40.67bn in 2024. (Statista, June 2024: https://www.statista.com/outlook/hmo/ pharmaceuticals/asia)
4. Navigating Global Regulatory Requirements for Generic Drugs: A Comparative Study of MIST, BRICS, and ICH Countries, International Journal of Pharmaceutical Investigation, December 2023/updated February 2024: https://jpionline.org/article/32579
Renato Rjavec
Renato Rjavec is the Senior Director of Product Management at ArisGlobal, where he is shaping the future of regulatory information management as well as quality management for life sciences, with a keen focus on AI as a means for targeted automation of critical but labour-intensive processes where accuracy and precision are paramount. Renato has almost two decades of experience in ideation, development and implementation of regulatory and quality solutions for the life sciences industry.
Email: rrjavec@arisglobal.com; www.arisglobal.com
Making AI Work for Drug Discovery: A Joined-up Approach
AI/data-driven drug discovery is starting to evolve at an encouraging rate now. But the technology’s positive impact will depend on how well the technology, and the insights it elicits, are embedded into R&D, says Biorelate’s Dr. Ben Sidders.
Gradually, data-driven, AI-enabled drug discovery is becoming a reality, beginning to fulfil the technology’s promise and demonstrating its potential more tangibly. The broader signs are encouraging, too –such as the growing profile of ‘AI-first’ companies such as Recursion and Insilico Medicine, and the observation that many traditional pharmaceutical companies are now embracing AI across their businesses.
Where previously the perceived value of AI in drug discovery and development had failed to live up to the technology’s hype, targeted solutions are now emerging which are making a positive impact on aspects of R&D. In turn, these applications are providing some valuable lessons and feedback about how to successfully embed AI within R&D operations.
In target discovery, knowledge graphs are now proving adept at integrating a vast number of data sources into a query-able structure, forming the basis for informed and relatively unbiased target prioritisation decisions and chemistry, where transformers are accelerating small molecule design and synthesis.
Challenges remain, however, predicting synergistic drug combinations has been the topic of extensive research, with only limited success and almost no translational relevance. Nor are we any nearer to being able to predict the effect of a drug on a given patient without first running a clinical trial.
The overriding realisation is that AI’s role in life sciences R&D is directly dependent on how decisively, and how well, they integrate the technology – and the insights it surfaces – within the wider R&D operation. Achieving this, in turn, will require a structured approach to AI-enabled R&D transformation, spanning four parallel priority areas: data, model, culture, and validation. Here’s how that breaks down.
Data
AI has found most success where the data set is large, complete and in many cases has been generated specifically to solve the problem at hand. The UNI foundation model for computational pathology, for instance, was trained on >100 million images from 20 tissue types.
In contrast one of the largest datasets available to train models for drug combination synergy prediction has 910 combinations of 118 drugs – many orders of magnitude smaller.
Significantly, much of our biomedical knowledge is locked away in unstructured data sources such as the literature. This problem is further exacerbated when we look at data from clinical trial cohorts, which is often sparse, and inconsistent in what is measured. For example, one trial might collect demographics and data for a specific blood-based biomarker; another might also collect genomic data. Then there are differences in the analysis pipelines applied to all these data. Re-processing and harmonising all of these data types is highly labour intensive, and often only the start of the process.
The underlying issue, is that Pharma’s data, particularly that from clinical trials, was not generated for AI. To exploit data in a meaningful way using AI, companies must develop a data strategy, be willing to fund and generate data on clinical cohorts if possible, and adopt approaches to maximise the value of unstructured data.
Model
While AI models excel at classification and predictive problems, if AI is to revolutionise drug discovery it must incorporate causality. Predicting that a drug might work in a new indication is valuable, but it is not the same as explaining why the drug will work in that indication. To support internal and regulatory decision-making it is essential to have explainable biology that supports a mechanistic understanding of the particular drug or biology.
The integration of prior knowledge and data-driven insights offers a promising solution. AI combined with highly accurate causal relationships can distil both a broader array of targets with strong promise, and a mechanistic understanding of their biological role in disease.
Causal relationships can be mined from the literature and created from experimental data. These relationships, defining the regulatory interactions between two biological entities, can be combined into structural causal models – a framework to represent and analyse the causal relationships between variables. Such models provide a systematic way to model how changes in one variable can lead to changes in another. These could be used during the training process of more expansive foundation models, but also to build specific mechanistic models that further describe the output from an upstream finding.
Validation
The output from all AI solutions should be validated, experimentally if appropriate, with two provisos. First, the R&D function should be set up so that all data feeds back to the AI model. This helps to mitigate some of the challenges described above, while ensuring that the model can be continually improved.
Second, there needs to be a triage-based validation model. While an AI system is able to identify hundreds of targets, the challenge is to stay open to ‘left-field’ opportunities that AI might highlight.
Research and Development
Orthogonal in silico approaches might be used to go from 1000 to 100 targets, but to go from 100 to 10 the team should adopt the quickest, most high-throughput experiment to yield the next rung of supporting evidence.
Culture
Underlying many of the data, model and validation issues up to now has been the culture of the organisation and its failure to fully adapt to an AI driven way of thinking or working.
While there are increasing efforts to bridge this gap, upskilling or recruiting talent with AI expertise is essential. At the same time data scientists must be educated in the decision-making process of R&D, and understand/develop methods that directly support that. More could also be done to build the understanding that AI will raise the productivity level of all R&D researchers, and is therefore an opportunity and not a threat.
Building the Future Today
Very soon – within a decade, certainly – we can expect every major decision taken along the drug R&D pipeline to be accelerated by unprecedented access to knowledge. But that relies on companies having done the groundwork, to put the right measures in place.
Data scientists, for their part, will need to develop actionable models with causality at their heart. Biologists must determine how to effectively integrate data science into their workflows. And heads of R&D will need to orchestrate more seamless integration and symbiosis between the two sciences.
Only then, will step changes in R&D success be possible.
Dr. Ben Sidders
Dr. Ben Sidders, Chief Scientific Officer at Biorelate, has been working at the forefront of pharma data science for the last two decades. Formerly Executive Director and Head of Early Data Science within Oncology R&D at AstraZeneca, Ben also previously spent eight years at Pfizer, and has extensive experience of many aspects of drug discovery for major pharma.
Enhancing Oncology Trials: How Generalised Pairwise Comparisons Drive
Multi-faceted, Patient-focused Research
The need for innovative statistical methodologies that prioritise patients' perspectives in cancer treatment decisions is becoming increasingly recognised. Traditional clinical trials, which focus predominantly on efficacy endpoints, often overlook safety and quality of life considerations. This narrow focus can misalign with patient experiences, limiting a comprehensive understanding of treatment benefits and risks. The Generalised Pairwise Comparisons (GPC) method addresses these gaps by enabling a holistic analysis of treatment effects across multiple clinically relevant outcomes, facilitating patient-focused analyses. Additionally, GPC can significantly reduce sample size requirements in clinical trials. The SHAPERS trial exemplifies GPC's potential by transforming its design from non-inferiority to superiority, incorporating both efficacy and safety outcomes into a single and unified benefit-risk assessment. Ultimately, the broader adoption of GPC can enhance clinical research, enabling patients and clinicians to express their treatment preferences before any treatment decision, ensuring that patients remain at the forefront of medical advancements.
The need for innovative statistical methodologies that ensure patients' perspectives are central to treatment decisions is increasingly evident. Clinical trials are the cornerstone of medical advancements in cancer care, offering critical insights before the approval of new therapies. These trials typically focus on efficacy endpoints, which are designed to reflect the intended effects of a treatment and include a broad range of assessments, from survival time to tumour response, often relegating safety and quality of life outcomes to secondary considerations.
Treatments are often approved based on the results associated to a single dimension of the treatment effect. Although this primary endpoint is chosen for its clinical relevance by medical professionals, it may not always align with the daily experiences and preferences of patients, potentially overlooking broader effects critical to patient wellbeing. This narrow focus can limit the understanding of a treatment's benefits and risks, potentially overlooking important factors that might impact patient quality of life and overall satisfaction with the therapy.
Patient needs often include multiple dimensions, especially when evaluating treatments for diseases with different symptomatic expressions or functional impacts. This problem is exacerbated by the intrinsic limitations of traditional statistical tests frequently employed in clinical trials. These traditional methods are limited to the analysis of a single variable at a time, preventing a comprehensive understanding of how different outcomes of interest collectively influence the efficacy and safety of a treatment.
The need for a more inclusive and patient-focused trial design is underscored by real-world examples. For instance, the addition
of oxaliplatin to 5-fluorouracil for metastatic colorectal cancer initially failed to gain Food and Drug Administration (FDA) approval because its primary endpoint, overall survival (OS), lacked statistical significance.1 However, the treatment exhibited substantive benefits in progression-free survival, highlighting the complexity of evaluating treatments solely on OS. Oxaliplatin was ultimately approved and remains a standard of care today, reflecting its proven efficacy. Another example is the addition of erlotinib to gemcitabine for metastatic pancreatic cancer, which was approved based on a mild yet statistically significant improvement in progression-free survival.2 This evaluation did not consider important factors such as toxicities and a decrease in quality of life, failing to capture the drug's overall benefit-risk profile, which led to erlotinib's limited adoption by the medical community despite regulatory approval.
These examples emphasise the necessity of a paradigm shift which places patient insights and experiences at the heart of clinical research. To bridge these gaps, the GPC statistical method was developed.3 This innovative statistical method enables the multidimensional analysis of treatment effects across the most clinically relevant criteria, offering a broader view of a treatment's potential benefits.
GPC: A Powerful Enabler for Multi-dimensional Treatment Assessment
GPC is a powerful and flexible statistical methodology that extends the traditional non-parametric Wilcoxon-Mann-Whitney test. Unlike conventional statistical methods that typically focus on a single primary endpoint, GPC allows several outcomes to be analysed simultaneously within a single assessment, enabling a comprehensive comparison of treatments grounded in considerations of clinical relevance. For instance, outcomes can be ranked from highest to lowest priority based on expert consensus or tailored to reflect each patient’s preferences. This flexibility spurs unprecedented opportunities for listening to patients and clinicians’ needs, integrating these needs in trial designs’ primary endpoint and then conducting more patient-centric analyses of trials. This aligns with the increased emphasis on patient-centricity in clinical research, promoted by regulatory agencies and pharmaceutical companies.4
GPC works by comparing every possible pair of patients from different treatment groups within a trial. It assesses the probability that one treatment is more effective than another across various outcomes. Each patient in the experimental group is compared to each patient in the control group, and each pair is categorised as either “favourable” to experimental treatment, “unfavourable” to experimental treatment or “neutral.” If a pair is deemed neutral for the primary outcome, the comparison is then carried over to the next outcome. Envision a clinical trial in oncology where you want to compare a patient who received the experimental therapy, with a patient who received the typical standard of care. The objective of
the pairwise comparison is to determine which patient benefited the most from their treatment. You want to focus on the most important outcome first, e.g. OS. If a patient survived much longer than the other patient, it is easier to classify the pair. However, if both patients were alive at the end of the trial or survived to a similar number of months, you can consider the second outcome (e.g. the most severe adverse event) to determine which patient experienced the most significant benefit from treatment.
After the evaluation of all pairwise comparisons, the scores (i.e. favourable, unfavourable or neutral) are aggregated to provide an overall measure of treatment effect called the “Net Treatment Benefit” (NTB) that can be interpreted as the net difference in probability that a random patient in the experimental arm is doing better than a random patient in the control arm. Positive values of NTB indicate a beneficial treatment effect, while negative values favour the control arm. This measure can be understood by both clinicians and patients, and used to facilitate informed decision-making. GPC also allows for an easy, straightforward understanding of the outcome of each contribution to the NTB. This means that if one wants to evaluate the impact of four different outcomes on the treatment effects, the individual contribution of each outcome will be displayed. This will help patients and clinicians to rapidly understand the benefits and/or risks of the experimental treatment compared to the standard of care, leading to an easy computation of the NTB (the sum of the individual contributions).
A key feature of GPC is its flexibility in handling various types of outcomes, including continuous, binary, categorical, and time-toevent data. This flexibility allows researchers to integrate multiple outcomes into a single analysis, addressing the complex interplay between different treatment effects. For example, in oncology trials, researchers can simultaneously evaluate survival rates, tumour response, patient-reported outcomes, side effects, and quality of life leading to a more nuanced understanding of a treatment's holistic impact.
Moreover, GPC can lead to significant reductions in sample size requirements. By evaluating multiple outcomes simultaneously, GPC increases statistical power (i.e. the ability to detect an effect if it exists) with fewer patients. This reduction in sample size is particularly important in areas where patient recruitment is challenging. Achieving a well-powered clinical trial with fewer participants can expedite the trial recruitment and reduce costs and timelines, making it more feasible to bring new treatments to the market faster.
Case Study Example: The SHAPERS Trial
SHAPERS is a Belgian clinical trial that addresses the efficacy and safety of short-course radiotherapy versus total neoadjuvant therapy in patients aged 70 years and older with locally advanced rectal cancer (LARC).5 Rectal cancer, particularly in older patients, presents unique challenges due to the patients' age-related comorbidities and reduced physiological reserves. Traditionally, treatment approaches for LARC
Therapeutics
have involved a combination of chemotherapy, radiotherapy and surgery. Total neoadjuvant therapy has become a new standard of care for LARC, primarily based on results from phase III trials such as RAPIDO and PRODIGE-23, which showed improved disease-related outcomes.6,7 However, these trials often exclude or underrepresent older patients, who may experience different efficacy and toxicity profiles compared to younger populations.
Initially, a non-inferiority trial was considered, which aims to demonstrate that a new treatment is not significantly worse than an established one. However, this approach often requires large sample sizes (e.g., thousands of patients) and fails to consider the full spectrum of treatment effects, particularly those important to patients, such as quality of life and side effects.8
GPC was successfully proposed for this trial, allowing for a more holistic evaluation by prioritising outcomes based on their clinical relevance and patient preference. It allowed the transformation of the study design from a non-inferiority to a superiority trial. The new endpoint enabled by GPC incorporated four prioritised composite outcomes: OS at 3 years, progression-free survival (PFS) at 3 years, increased-grade peripheral sensory neuropathy at 3 years, and grade ≥ 3 toxicities during treatment. This approach included both efficacy (OS and PFS) and safety (neuropathies and other toxicities) outcomes in a single benefit-risk assessment. Consequently, allowing to reduce the trial sample size and making the trial feasible. Patient enrollment started in June 2024.
Conclusion
In conclusion, GPC represents a transformative advancement in oncology clinical trials by addressing the limitations of traditional single-endpoint approaches. By considering multiple clinically relevant outcomes simultaneously, GPC enables a comprehensive assessment of treatment effects, ensuring that patient priorities are at the forefront of decision-making. This comprehensive, multi-faceted assessment enhances the precision of clinical studies, reduces sample sizes, and facilitates faster approval of new treatments. The SHAPERS trial demonstrates GPC's ability to shift from non-inferiority to superiority designs, integrating both efficacy and safety outcomes into a holistic benefit-risk analysis. As oncology continues to evolve, we believe GPC will play a crucial role in advancing patient-centered care and improving treatment outcomes.
REFERENCES
1. André, T., Boni, C., Mounedji-Boudiaf, L., Navarro, M., Tabernero, J., Hickish, T., ... & de Gramont, A. (2004). Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. New England Journal of Medicine, 350(23), 2343-2351.
2. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, Clark G, Voskoglou-Nomikos T, Ptasynski M, Parulekar W. National Cancer
Institute of Canada Clinical Trials Group (2007) Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25: 1960–1966.
3. Buyse, M. (2010). Generalized pairwise comparisons of prioritized outcomes in the two-sample problem. Statistics in medicine, 29(30), 3245-3257.
4. Food and Drug Administration. FDA Patient-Focused Drug Development Guidance Series for Enhancing the Incorporation of the Patient’s Voice in Medical Product Development and Regulatory Decision Making. Consulted July 2024. https://www.fda.gov/drugs/development-approvalprocess-drugs/fda-patient-focused-drug-development-guidance-seriesenhancing-incorporation-patients-voice-medical
5. Saúde-Conde, R., Vandamme, T., De Backer, M., Martinive, P., Covas, A., Deleporte, A., ... & Sclafani, F. (2024). Efficacy and safety of short-course radiotherapy versus total neoadjuvant therapy in older rectal cancer patients: a randomised pragmatic trial (SHAPERS). ESMO Gastrointestinal Oncology, 4, 100067.
6. Bahadoer, R. R., Dijkstra, E. A., van Etten, B., Marijnen, C. A., Putter, H., Kranenbarg, E. M. K., ... & Silviera, M. L. (2021). Short-course radiotherapy followed by chemotherapy before total mesorectal excision (TME) versus preoperative chemoradiotherapy, TME, and optional adjuvant chemotherapy in locally advanced rectal cancer (RAPIDO): a randomised, open-label, phase 3 trial. The Lancet Oncology, 22(1), 29-42.
7. Conroy, T., Bosset, J. F., Etienne, P. L., Rio, E., François, É., MesgouezNebout, N., ... & Marquis, I. (2021). Neoadjuvant chemotherapy with FOLFIRINOX and preoperative chemoradiotherapy for patients with locally advanced rectal cancer (UNICANCER-PRODIGE 23): a multicentre, randomised, open-label, phase 3 trial. The Lancet Oncology, 22(5), 702-715.
8. Fleming, T. R. (2008). Current issues in non-inferiority trials. Statistics in medicine, 27(3), 317-332.
Sebastien Coppe
Sebastien Coppe, CEO of One2Treat, brings extensive experience in the field of clinical trials and artificial intelligence. Over the course of more than a decade at N-SIDE, he played a pivotal role in leading the company's life sciences activities, contributing significantly to its growth from a small start-up to a purpose-driven organisation with over 200 employees. With a deep understanding of the biopharmaceutical industry's challenges and opportunities, Sebastien made the bold decision to join Marc Buyse in his latest venture, aiming to industrialise his paradigm-shifting solution that enhances treatment decisions. Sebastien is driven by a strong conviction in the potential of innovative digital solutions to meet the overall patients’ needs and accelerate clinical research timelines. He is committed to pioneering approaches that prioritise patientcentricity and enhance biopharmaceutical productivity.
Samuel Salvaggio
Samuel Salvaggio, senior trial design lead of One2Treat, translates clinical outcomes into statistical insights, utilising his expertise in data analytics and applied biostatistics. His dedication to advancing patient-focused research is evident through his work, which includes scientific publications and educational efforts. At One2Treat, Samuel's passion fuels innovative research initiatives, while his teaching role at the Université libre de Bruxelles allows him to share his applied biostatistics expertise with medical professionals. This blend of professional activities underscores his commitment to enhancing patient-centric solutions through education and innovative research.
Challenges of Measuring Clinical Meaningful Changes in Alzheimer’s Disease
Alzheimer's disease (AD) is a progressive neurodegenerative disorder that profoundly impacts patients and public health. It manifests through cognitive decline such as memory loss and functional impairment, leading to a loss of independence and the need for full-time care.1 The number of patients with dementia worldwide is expected to exceed 150 million by 2050.2 The disease not only affects individuals but also significantly impacts families, caregivers, and society. Family members often become primary caregivers leading to emotional, physical, and financial strains.
Clinical trials in AD represent a critical frontier in the search for effective treatments and interventions, however, the field faces several challenges and complexities.1 As an example, with increased intervention targeting early-stage AD, the choice of specific and sensitive clinical endpoints to capture subtle cognitive and functional changes, and understanding their applicability in clinical practice, are becoming critical.3 Currently, there is a lack of definitive evidence of how trial outcome measures correlate with changes in disease progression and treatment response which creates ambiguity around their clinical relevance.3,4 Addressing this uncertainty would benefit patients, caregivers, primary care providers, and regulators, by improving our comprehension of how trial endpoints relate to everyday clinical assessments.
Presently, clinical trials in AD employ a variety of assessment tools, with considerable variability in the endpoints selected. This review will cover what constitutes clinically meaningful changes in early-stage AD, the most frequently used assessment tools, and discuss those measures in relation to disease progression and treatment efficacy.
Common Outcome Measures
AD clinical trial endpoints have been primarily tailored to track symptom changes in patients with moderate to severe AD dementia. However, as the focus has shifted towards developing therapeutics for mild cognitive impairment (MCI) due to AD and early-stage AD, there is a growing need to develop and validate clinical endpoints that can accurately detect changes during the earlier stages of the disease.4
Several outcome measures are commonly used to assess treatment efficacy and disease progression. These measures include cognitive assessments, such as standardised cognitive tests, the Mini-Mental State Examination (MMSE), the Montreal Cognitive Assessment (MoCA) and the Alzheimer's Disease Assessment Scale-Cognitive subscale (ADAS-Cog) that are used to evaluate changes in cognition over time.4,5,6 To capture the sensitivity of cognitive change in MCI due to AD, additional tools are used in clinical trials such as the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS), the Neuropsychological Test Battery (NTB) and the Preclinical Alzheimer’s Cognitive Composite (PACC).4,7 Assuming that functional changes in AD are due to cognitive decline, functional scales to assess activities of daily living (ADLs) and instrumental activities of daily living (IADLs) are crucial
to provide insight into the impact of treatment and cognitive decline on patients’ ability to perform everyday tasks independently. Both the Amsterdam Instrumental Activities of Daily Living (A-IADL) and the Alzheimer’s Disease Cooperative Study – Activities of Daily Living Scale for use in Mild Cognitive Impairment (ADCSADL-MCI) have regularly been used in early symptomatic AD trials to assess instrumental activities of daily living known to decline in early stages of AD due to the high level of cognitive ability associated with these tasks.4,8
The US Food and Drug Administration (FDA)9 recognises the value of composite assessments (both cognitive and function in a single scale) for the evaluation of patients with MCI due to AD. The Clinical Dementia Rating (CDR) is a semi-structured interview that was identified in the 2013 FDA guidance10 as a suitable tool and has served as a primary or secondary endpoint in multiple AD clinical trials. Recent studies3,4,6,11 have demonstrated the value of the CDR as an anchor measure to determine minimal clinically important differences (MCID). MCID provide a way of measuring changes in disease symptoms that go beyond just statistical significance. An MCID is defined as the smallest change in an outcome measure that results in a noticeable change in a patient’s life.12
The CDR global score (GS) is used to quantify stage severity of AD and MCI and ranges from 0 to 3 (0= none; 0.5 = questionable; 1 = mild; 2= moderate and 3 = severe). The CDR sum of boxes (CDR-SB) is a continuous measure of AD and MCI stage severity and ranges from 0 to 18.13 The CDR GS has been used as an anchor to estimate meaningful changes for other scales, but the CDR SB seems more efficient at capturing what is considered minimally important change.3,4
Andrews et al.,14 used the data from the National Alzheimer’s Coordinating Center Uniform Data Set (UDS) to estimate MCID for commonly used cognitive and functional assessment tools in AD, such as the MMSE, CDR-SB and the Functional Activities Questionnaire (FAQ). Their findings showed that as AD progressed, the MCID estimate increased. Specifically, a 1-3-point decrease in the MMSE, a 1-2-point increase in the CDR-SB, and a 3-5-point increase in FAQ were associated with meaningful clinical decline, depending on the severity of the disease.14 Other composite scales that have shown sensitivity to change in AD are the Alzheimer’s Disease Composite Score (ADCOMS) and the integrated Alzheimer’s Disease Rating Scale (iADRS)4,15,16
Assessing Clinically Meaningful Changes
Assessing clinically meaningful changes in AD trials requires robust methodologies that capture changes considered relevant to patients, caregivers, and clinicians. Several approaches have been proposed, including distribution-based and anchor-based methods. The distribution-based methods rely on statistical properties of the data distribution to determine what constitutes a meaningful change in a clinical outcome measure.4,17 These methods provide a quantitative approach to interpreting the clinical significance of changes observed in clinical trials or studies.
Common approaches include effect size calculations to measure the magnitude of change in a clinical outcome relative to the
variability of the measurements within the study population. It is typically calculated as the mean change divided by the standard deviation of the baseline scores.17 An MCID based on effect size indicates how many standard deviations of change are considered clinically meaningful.17,18 The anchor-based approaches rely on external criteria or anchors that are considered clinically meaningful by patients, caregivers or clinicians.4,19 These approaches establish a connection between changes observed in a specific outcome measure and their perceived clinical significance. They offer a patient-centered perspective and enhance the interpretability of changes observed in a clinical trial. By aligning outcome measures with the patient’s and clinician’s perspectives of meaningful change, these approaches provide valuable insights into treatment efficacy and the impact of interventions on patients’ lives. They complement distribution-based methods and contribute to a comprehensive understanding of what constitutes a clinically important change in AD.18,19
The FDA recommends using these anchor-based methods, along with empirical cumulative distribution methods, to determine a threshold or range of thresholds that signify a meaningful within-patient change score for the target outcome or the derived endpoint for the patient population.9,18 Anchoring measures serve as external benchmarks to identify patients who have experienced a meaningful change in their condition, with the outcome score change assessed in these patient groups.
Current State of Affairs
There has been a shift towards developing treatments that target the underlying pathology of AD, such as amyloid-beta and tau protein aggregation and AD clinical trials are increasingly focusing on disease-modifying treatments aimed at slowing or halting disease progression.4,11,12 Identification of these treatment effects on validated outcome measures is essential for regulatory approval. However, defining and measuring what constitutes a clinically meaningful benefit from these trials to various stakeholders, including clinicians and patients, needs further clarification.
While reviewing the definitions of clinically meaningful changes it is crucial to discuss misinterpretation regarding thresholds for meaningful within-patient progression and thresholds for determining meaningful group-level differences. In fact, a critical aspect of understanding clinical trial outcomes is distinguishing “between-group differences” from “within-individual change.”11,12
Measuring group-level differences is considered an appropriate statistical approach in parallel-group AD trials, but these estimates depend not only on changes observed within individuals but on several trial design factors, including sample size among others.20 Thus, group-level differences do not directly indicate likely treatment effects at the individual level. This is mostly relevant for the early stages of AD trials, where disease progression is slow and even highly effective treatments may yield relatively small effects.20,21 Consequently, large sample sizes and longer duration trials are often required to achieve sufficient statistical power to detect significant group-level differences during these stages.3,4,12
Utilising validated thresholds that represent the MCID in clinical outcome measures would help to objectively evaluate the clinical significance of trial results. However, MCID thresholds for most AD trial outcomes have not been established. For instance, when evaluating treatment efficacy based on the absolute point difference in change from baseline on a scale with a large range, small differences may be perceived as modest effects.20 Conversely, the same magnitude of difference on a scale with a narrower range may be seen as large effects. Moreover, researchers interpreting
these differences should consider the scale's range, reflecting the expected change over time for a specific cohort, which is often distinct from the full-score range of the scale. Without these reference points, it is challenging to assess the clinical relevance of small, yet statistically significant, differences between groups.3,20,21
Anti-amyloid monoclonal antibody therapies, which have recently garnered significant attention due to the FDA approval of lecanemab and donanemab, have demonstrated slowing of cognitive decline as measured by clinical endpoints such as the CDR-SB among others. In the Clarity AD phase 3 trial,22 the change from baseline at 18 months in the CDR-SB (primary endpoint) was less with lecanemab than with placebo and the other secondary endpoints (ADAS-Cog 14, ADCS-MCI-ADLI) were in the same direction, favouring lecanemab. However, the drugplacebo difference did not meet the boundary of definitive MCID and exceeded the boundary of no MCID established by Andrews et al.14,22
In the TRAILBLAZER-ALZ 2 phase 3 trial, the results showed that donanemab slowed the rates of cognitive and functional decline in participants with early AD.23 Additionally, a point change of -5 on the iADRS and +1 on the CDR-SB for those with MCI, or -9 on the iADRS and +2 on the CDR-SB for those with mild AD at consecutive visits from baseline, were also considered meaningful.
Furthermore, the CDR-SB is a pivotal endpoint in an Alzheimer’s disease trial providing critical insights into the efficacy of treatments like lecanemab, donanemab and aducanumab.22,23,24 Clinically meaningful changes in the CDR-SB scores might reflect real-world benefits emphasising the potential of these therapies to slow disease progression.
Future Directions
Despite significant advances and recent approvals of treatments aimed at modifying the course of AD, the clinical trials in the field remains challenging. AD encompasses a spectrum of clinical presentations, including early-onset and late-onset forms, as well as notable variations in disease progression and underlying pathology. Therefore, designing effective clinical trials for AD requires taking into consideration several key factors, including selecting appropriate outcome measures, defining clinically meaningful endpoints, and optimising trial duration and sample size.
The modest decrease in cognitive decline observed following treatment with lecanemab has reignited discussions on what constitutes “clinically meaningful” change. Recent literature3,4,12,20 has explored this topic, involving scientists from various fields who agreed that meaningfulness should be evaluated individually rather than based solely on group differences. There is an emphasis on the need to consider the effects of treatments over time and the importance of outcome measures that reflect patients’ priorities.
While clinical trials report statistical differences, they do not determine the minimum change noticeable to patients, caregivers, or physicians, which makes determining clinical meaningfulness at the participant or patient level challenging. Previous attempts to quantify meaningful change suggested a 1- to 2-point worsening on the CDR-SB scale,14 but applying this threshold to consider small benefits seen with emerging therapies seems insufficient. Different studies have reported similar conclusions, with the minimal meaningful difference varying by disease stage and suggesting the need for longer trials to assess disease-modifying therapies adequately.
Researchers also highlight the limitations of current measurement scales, advocating for scales that capture a broader range of relevant changes based on patients’ and caregivers’ perspectives.3,4,6,9,12 Therefore, multiple anchors, such as selfreported and partner-reported measures should be explored to gather comprehensive evidence to interpret a clinically meaningful within-patient score change, and these anchors should be used at comparable time points as the target outcome.9,8,25,26
It is also important to consider that patient-reported symptoms may become less predictive of objective cognitive impairment as the disease progresses due to loss of the patient’s insight into their symptoms (i.e., anosognosia), therefore the use of study-partner symptoms may become more predictive.26 Additional research is required to comprehend the impact of anchor agreement on MCID estimation in the context of AD severity.
RESOURCES
1. Kumar A, Sidhu J, Lui F, et al. Alzheimer Disease. [Updated 2024 Feb 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan
2. GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022; 7: e105–25
3. Borland E, Edgar C., Stomrud E, et al. Clinically relevant changes for cognitive outcomes in preclinical and prodromal cognitive stages. Implications for Clinical Alzheimer Trials. Neurology. 2022; 99(11) e1142-e1153. https://doi.org/10.1212/WNL.0000000000200817
4. Cohen S, Cummings J, Knox S, Potashman M, Harrison J. Clinical trial endpoints and their clinical meaningfulness in early stages of Alzheimer’s disease. J Prev Alzheimer’s Disease. 2022; 9 (3): 507-522. Doi:10.14283/ jpad.2022.41
5. Podhorna J, Krahnke T, Shear M, Harrison JE. Alzheimer's Disease Assessment Scale-Cognitive subscale variants in mild cognitive impairment and mild Alzheimer's disease: change over time and the effect of enrichment strategies. Alzheimer’s Res Ther 2016; 8:8; doi:10.1186/ s13195-016-0170-5
6. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer's disease drug development pipeline: 2020. Alzheimer’s Dement.2020;6(1):12050; doi:10.1002/trc2.12050
7. Karantzoulis S, Novitski J, Gold M, Randolph C. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): utility in detection and characterization of mild cognitive impairment due to Alzheimer’s disease. Arch Clin Neuropsychol. 2013; Dec; 28 (8): 837-44.doi 10.1093/ arclin/act057
8. Pedrosa H, De Sa A, Guerreiro M, et al. Functional evaluation distinguishes MCI patients from healthy elderly people—the ADCS/MCI/ADL scale. J Nutr Health Aging 2010;14(8):703-9; doi:10.1007/s12603-010-0102-1
9. Early Alzheimer’s Disease: Developing Drugs for Treatment, Guidance for Industry. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2018.
10. Early Alzheimer’s Disease: Developing Drugs for Treatment, Guidance for Industry. U.S. Department of Health and Human Services, Food and Drug Administration, CDER, CBER; 2013.
11. Stojanovic M, Mikula, C, John S, Kiselica A. Clinical importance in Alzheimer’s disease: effects of anchor agreement and disease severity. Aging Clinical and Exp Research. 2024; 36: 5.doi.org/10.1007/s40520-02302643-0
12. Petersen RC, Aisen PS, Andrews JS, et al. Expectations and clinical meaningfulness of randomized controlled trials. Alzheimer Dement. 2023; 19:2730-2736
13. McDougall F, Edgar C, Mertes M, et al. Psychometric properties of the clinical dementia rating - Sum of boxes and other cognitive and functional outcomes in a prodromal Alzheimer’s disease population. J Prev Alzheimer’s Dis. 2021;8(2):151-160.
14. Andrews JS, Desai U, Kirson NY, et al. Disease severity and minimal clinically important differences in clinical outcome assessments for Alzheimer’s disease clinical trials. Alzheimer’s Dement.2019;5:354-363.
15. Wessels AM, Siemers ER, Yu P, et al. A combined measure of cognition and function for clinical trials: the integrated Alzheimer's Disease Rating Scale (iADRS). J Prev Alzheimer’s Dis 2015;2(4):227-41; doi:10.14283/jpad.2015.82
16. Wang J, Logovinsky V, Hendrix SB, et al. ADCOMS: a composite clinical outcome for prodromal Alzheimer's disease trials. J Neurol Neurosurg Psychiatry 2016;87(9):993-9; doi:10.1136/jnnp-2015-312383
17. McLeod LD, Cappelleri JC, Hays RD. Best (but oft-forgotten) practices: expressing and interpreting associations and effect sizes in clinical outcome assessments. American Journ Clin Nutrition. 2016; 103(3). DOI:10.3945/ajcn.115.120378
18. Edgar CJ, Vradenburg G, Hassenstab J. The 2018 revised FDA guidance for early Alzheimer’s disease: establishing the Meaningfulness of treatment effects. J Prev Alzheimer’s Dis. 2019;6(4):223-227.
19. Zhang Yu, Xiaoyu X, Huang Y. The anchor design of anchor-based method to determine the minimal clinically important difference: a systematic review. Health and Quality of Life Outcomes. 2023; 21:74 https://doi. org/10.1186/s12955-023-02157-3
20. Tarawneh R, Pankratz V. The search for clarity regarding “clinically meaningful outcome” in Alzheimer disease clinical trials: CLARITY-AD and beyond. Alzheimer’s Research & Therapy. 2024; 16:37. https://doi. org/10.1186/s13195-024-01412-z
21. Rentz DM, Wessels AM, Annapragada AV, et al. Building clinically relevant outcomes across the Alzheimer’s disease spectrum. Alzheimer’s Dement. 2021;7e12181. https://doi.org/10.1002/trc2.12181.
22. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023; 388:9-21. https://doi.org/ 10.1056/ NEJMoa2212948
23. Sims JR, Zimmer JA, Evans C, et al. Donanemab in Early Symptomatic Alzheimer Disease. The TRAILBLAZER-ALZ2 Randomized Clinical Trial. JAMA. 2023;330(6):512-527. doi:10.1001/jama.2023.13239
24. Day G, Scarmeas, N, et al. Aducanumab use in symptomatic Alzheimer disease evidence in focus. Neurology. 2022; 98:619-631. Doi:10.1212/ WNL.0000000000200176
25. Kiselica AM, Johnson E, Benge JF. How impaired is too impaired? Exploring futile neuropsychological test patterns as a function of dementia severity and cognitive screening scores. J Neuropsychol. 2021; 15:410–427
26. Nosheny RL, Jin C, Neuhaus J, et al. Study partner-reported decline identifies cognitive decline and dementia risk. Ann Clin Transl Neurol. 2019; 6:2448–2459. https:// doi. org/ 10. 1002/ acn3. 50938
Catarina Cunha
Catarina Cunha is a Senior Clinical Scientist at WCG Clinical Research Solutions, overseeing clinical trials in neurology, focusing on conditions such as Alzheimer's disease, Parkinson's disease, Huntington's disease, progressive supranuclear palsy, and frontotemporal dementia. She has an MS in Clinical Psychology and is currently pursuing a PhD in Clinical Neuropsychology.
Vials with Exceptional Inner Surface Durability
For Biotech
Less interactions of drug molecules and formulations with the inner glass surface
• Optimized lyophilization process (less fogging)
• Reduced risk of glass delamination
For Diluents
Water for injection | Aqueous NaCl solution Lower pH shift
• Reduced risk of glass delamination
Forecasting and Kit Design in Clinical Trials for the Pharmaceutical and Biopharmaceutical Industry
Clinical trials are the cornerstone of drug development, providing the critical data needed to evaluate the safety and efficacy of new therapeutic agents. The success of these trials depends not only on meticulous planning, and accurate forecasting, but also considering the many factors of kit design. These elements are not just logistical concerns; they are strategic imperatives that can significantly influence the timeline, cost, and overall execution of a clinical trial.
Upstream Planning Activities that Define the Strategy Accurately predicting the demand for clinical supplies begins with a comprehensive understanding of the upstream planning activities that shape the overall strategy for a clinical trial. These activities include considerations related to risk tolerance, cost pressure, and time pressure – each of which plays a critical role in the decisionmaking process.
Different stakeholders within a clinical trial, such as clinical operations teams and supply chain managers, often have varying perspectives on material demand. For example, if there is a delay in opening a country for enrollment, the clinical operations team may prioritise kit availability to start new sites in an existing country or open a new country to increase enrollment. On the other hand, the supply chain team may focus on adjusting to enrollment rates that differ from initial predictions to ensure that supplies are available on time, avoiding study disruptions. Risk tolerance varies among these job functions and often these teams will not be aligned. Balancing these differing priorities requires open and honest communication among all stakeholders.
In the pharmaceutical industry, every decision has a cost implication. The clinical supply team must balance the cost implications of various scenarios, such as accelerating or delaying the start of a country’s trial phase against the need to procure additional drugs outside the planned budget. For example, if patient recruitment is slower than expected it may be necessary to delay the procurement of supplies to avoid overproduction and wastage. Conversely, if recruitment is ahead of schedule, additional supplies may need to be procured quickly, potentially at a higher cost. Time pressure is another critical factor that influences decision-making in clinical trials. The clinical operations team is driven by the need to meet study timelines, where the supply chain team must ensure kit availability. Effective forecasting helps to manage these time pressures by ensuring that supplies are produced and distributed according to the trial’s demand and timeline. Once each team has independently assessed these factors within their areas of expertise, they must come together to align on a strategy that addresses their collective priorities and constraints. This collaborative approach helps to ensure that the forecasting process is comprehensive and that all potential risks and challenges are considered.
Collaboratively Defining the Strategy
A well-defined forecasting strategy should include clear assumptions, identified risks, planned mitigation strategies, and estimated initial quantities to ensure that the clinical trial proceeds according to plan. Misunderstandings or inaccuracies in assumptions – particularly around timelines for procurement, packaging, and labelling – can lead to significant disruptions in the trial. For example, if the assumed lead time for drug manufacturing is inaccurate the trial may experience delays due to a lack of available supplies. Identifying potential risks is another critical component of the forecasting process and the strategy should also include detailed plans for mitigating these risks. For instance, if an anticipated retest date extension becomes impossible because it is not supported by stability data, then adjustments need to be made. Mitigations such as redirecting existing inventory may be required to maintain supplies until rapid changes to the production plan can be completed. Establishing initial quantities involves calculating the expected number of kits that will be dispensed and any other required supplies per treatment cycle. This provides a baseline of the minimum quantities required for dosing, facilitating further planning and budgeting discussions. Accurately determining initial quantities is essential for avoiding both overproduction, which leads to wastage, and underproduction, which can cause delays in the trial. There are many sources of material loss in a clinical trial. Some are anticipated such as inventory sent to sites that never enroll a subject and others are unexpected such as material lost due to a temperature excursion caused by a power outage. Sufficient overage and safety stock must be added to the initial quantities to account for all sources of loss. Communication between the stakeholders is required to create a forecast that balances oversupply against out of stock events. This will lead to effective risk management. A final, often overlooked, factor to consider is site stocking. If materials are to be shipped to sites prior to subject need that material must be accounted for as a separate portion of the material calculations. Failure to do so risks depleting the inventory as soon as the trial open.
The Role of Kit Design in Clinical Trials
The design of clinical trial kits can have a significant impact on the overall efficiency and cost-effectiveness of a clinical trial. A number of considerations must be taken into account when designing clinical trial kits including the dosing plan, flexibility, patient compliance, cost efficiency, and the impact on the supply chain. Clinical trial kits must be designed with flexibility in mind to accommodate changes in study protocols, use by patients, and regulatory requirements across different countries. For example, in global trials, countryspecific regulations may dictate different packaging and labelling requirements which must be anticipated during the design phase. Flexibility in kit design also allows for adjustments to be made in response to changes in patient dosing designs or study protocol changes. Proper kit design will help to avoid delays and ensure that the trial proceeds smoothly. By optimising the contents and packaging of kits, companies can reduce material costs and minimise waste. Additionally, well-designed kits can streamline the supply chain, reducing the need for last-minute changes and expensive rush shipments. For example, modular kit designs that allow for the easy addition or removal of components can reduce the need for multiple kit versions and simplify the manufacturing process. The design of the kit can affect the entire supply chain from manufacturing to distribution to site management. Kits that are too complex or contain unnecessary items can complicate the manufacturing process, leading to delays and increased costs. Conversely, streamlined kit designs that focus on essential components can simplify production and distribution ensuring that supplies reach sites on time and in the correct quantities. Additionally, well-designed kits can facilitate inventory management at clinical sites, reducing the risk of stockouts or overstocking.
Impact on Upstream and Downstream Planning
The importance of kit design extends beyond the physical kit itself. Decisions made during the design phase can have far-reaching effects on both upstream and downstream planning, influencing the overall success of the clinical trial.
During upstream planning the focus is on aligning kit design with the overall strategy of the clinical trial. This includes ensuring that the design meets the needs of the study while also being flexible enough to adapt to unforeseen changes, such as patient recruitment rates, study timelines, manufacturing lead times and distribution logistics. Effective communication is crucial during this phase to ensure that all perspectives are considered.
Once the kits are designed and produced, they enter the downstream phase of the supply chain. This includes distribution to clinical sites, inventory management, and real-time adjustments based on the progress of the trial. A well-designed kit can make this process smoother by reducing the likelihood of supply shortages or excess inventory. Additionally, well-designed kits can improve site management by reducing the complexity of kit handling and administration, leading to more efficient and accurate trial execution.
Conclusion
Forecasting and kit design are two critical components of clinical trial management in the pharmaceutical and biopharmaceutical industries. Accurate forecasting ensures that the right supplies are available at the right time, while thoughtful kit design can lead to cost savings, streamlined logistics, and a more efficient supply chain. By considering the various factors that influence forecasting and designing kits that are both flexible and cost-effective, companies can improve the overall success of their clinical trials.
In an industry where delays can be costly and time is of the essence, the ability to forecast accurately and design efficient kits is not just a competitive advantage – it is a necessity. As the complexity of clinical trials continues to grow, the importance of these elements will only increase making them key areas of focus for any company looking to succeed in the fast-paced world of drug development. By investing in advanced forecasting tools and innovative kit design strategies, pharmaceutical and biopharmaceutical companies can enhance their ability to bring new therapies to market more quickly and efficiently, ultimately improving patient outcomes and driving long-term business success.
Slava Shulov
Slava Shulov is a Senior Clinical Supply Manager for the EMEA/APAC regions and has been with PCI for five years. Slava has twelve years of experience in clinical manufacturing and supply chain for both sponsors and CDMOs, giving her valuable insight and experience in supporting clinical trials. In her current role, she provides clinical supply management expertise and oversight throughout the lifecycle of global clinical studies from the study concept phase to trial completion and product launch.
New Developments in Clinical Trial Logistics
Clinical trial logistics have seen major changes in recent years. Technological innovations, as well as operational changes such as the increasing popularity of decentralised clinical trials (DCTs) have seen the field have to adapt to this changing environment. The pandemic and other geopolitical factors have also had a large impact, amid changing regional challenges and turbulence in the air freight market.
Pandemic Impacts
To understand clinical logistics in recent years, it is best to start with the pandemic. Due to the global nature of clinical trials, many shipments are international and require shipments by air. Air freight was hugely affected, with staff having to stay home and flights grounded. However, items such as personal protective equipment (PPE) and vaccinations were moving, so clinical logistics businesses continued to operate albeit limited due to patients not being able to travel and stay at home.
However, the sector-wide lull created by the pandemic allowed air freight businesses to review their operations and find ways of increasing efficiency. As the pandemic began to subside, many airlines changed their schedules and reduced the number of flights, meaning logistic operators have had to adapt to a decrease in the availability of flights.
Post-pandemic, there has also been a period of high inflation driven by energy and fuel prices, increasing rates across the freight industry. Twinned with the decrease in capacity, prices have risen across the industry.
The clinical logistics industry has adapted to this in a number of ways. Clinical trial shipments are usually shipped to order on demand, but increasingly, clinical logistics is moving to a mixed model that requires establishing depots or warehouses within countries where medicines are stored, for final mile distribution as well as shipping on demand from the global central depot storage site. This is done to increase the resilience of the supply chain and reduce costs. However, new modes of clinical trials require a move in the other direction, with increased flexibility in where medicines are to be delivered. Getting the right balance is very important, with collaboration between CROs and clinical trial logistics companies essential. Clinical trial logistics companies need to understand the requirements and goals of sponsors and CROs so they can adapt their infrastructure and operations to fit their needs.
Medicines requiring specific conditions during transit, such as temperature-controlled medicines which must often remain at low temperatures throughout the supply chain, require effective partnerships with all stakeholders. The requirements need to be fully understood by the freight handler so specialists can be in the right
place at the right time to properly handle the product, so effective communication, collaboration and relationship building is paramount to build a high-performing supply chain. This can also include information and data sharing, so the supply chain can continue to improve over time.
Decentralised Clinical Trials
While traditional clinical trials will not be going anywhere any time soon, there has been a noticeable increase in popularity of DCTs in the year during and since the pandemic. This direct-to-patient model is ideal for accessing populations who are remote or for whom it would be difficult to travel to a trial site, such as those with debilitating illnesses. In this model patients are sent the drugs to administer themselves or with a local health professional, supported by members of the clinical trial team via video call or other digital communications.
This model is especially useful for sponsors researching rare diseases or vulnerable populations, which limits their potential patient recruitment pool for a trial. Rather than having to search for patients fulfilling their requirements within a geographical area, DCTs mean that they are no longer limited by geography and can recruit patients within much wider areas.
The model can also help with patient retention. With high levels of patient attrition throughout a trial common across the sector, DCTs allow patients to report from their own home, rather than reporting to a trial site. Removing the requirement for travel and reporting to a site has been shown to reduce attrition.
Decentralised trials require an efficient and effective last-mile delivery operation, with medicines often being delivered to remote and hard-to-reach locations where patients live. The coordination required is immense and utilises a mix of modes of transport. Last mile logistics providers share a duty to keep the medicine in good condition and fulfilling any temperature or environmental storage requirements.
Timing is also imperative. If a local health professional needs to be present to administer the drug, then further coordination will be required to ensure the drug arrives while the health professional is also present. As a result, specialist driver training is imperative, including training for reading temperature monitors, standards when dealing with patients and CROs, and best last-mile logistics practice in general.
This new approach requires a complete rethink of the usual clinical trial supply chains. Clinical trial medicines and materials would traditionally be sent to one location and stored in bulk, with enough provided for the length or phase of trial including contingencies. With DCTs shipping smaller amounts directly to patients, processes like the return of containers, destruction of excess supplies become much more complicated.
While there are challenges, DCTs offer opportunities for sponsors. Participants are less burdened by travel requirements and the resulting time and effort commitments; and trial organisers are able to take advantage of these efficiency savings, as well as increased ease of data collection and correction via digital channels. Collaboration between pharmaceutical logistics providers and CROs will be essential to fully harness the opportunities presented.
Regional Challenges
The increased popularity of decentralised clinical trials has allowed for increased access to naive patient populations that are sometimes lacking in other healthcare infrastructure, which can be advantageous to sponsors. Eastern Europe and countries from the former Soviet Union are fertile ground for finding these populations, but a lack of logistical infrastructure often requires complex, multi-mode logistics to transport medicines to patients, in turn requiring expert knowledge of the region and contacts with the appropriate expertise and training.
The war between Russia and Ukraine has further complicated operations in these areas. Accessing Ukraine for example now requires shipping to a neighbouring country, like Poland, and then using road freight to enter the country rather than direct flights into the country. These operations are further complicated by on-the-ground military movements and risk assessments are essential. Conversely, accessing Russia is complicated by a lack of direct flights and increased sanction regimes, as some technologies employed for clinical trial logistics, such as temperature monitors or communication devices, transmit real-time data on a product’s condition. These also have military uses and thus require special permits to export to Russia which complicates the process and can add costs.
Latin America is a key emerging market for clinical trials. Markets like Brazil are seeing an expansion of the middle class, increasing demand for Western medicines. However, this presents difficulties when shipping temperature-controlled containers into the country as containers may require a formal import procedure, but the lack of medicine production for export in the country means that there is then nothing to go into the container for the return trip, creating great expense to get the container back to a location where it can be used again.
Africa is a complex market. South Africa is a self-contained market of its own, and North Africa is well served by European businesses, whereas sub-Saharan Africa suffers from underdeveloped clinical trial and logistical infrastructure as well as underdeveloped regulatory knowledge and experience. Furthermore, developing countries with less developed infrastructure also often have lengthy distances between locations within them with unreliable routes. This makes them unpredictable in terms of delivery time, or ability for refuelling both for a vehicle and with materials necessary to keep medicines cool if a cold chain is required. This makes DCTs in particular very difficult, even if the populations are attractive to sponsors due to their lack of exposure to other medicines.
Across all regions, last-mile logistics and relationships with local stakeholders are key. Increased efficiency in this area will be essential to continue the development of DCTs and evolve the clinical trial process.
Types of Clinical Trial Logistics
Different types of clinical trial will have different logistical requirements, depending on the type and size of the trial and types of medicines involved. Here are some of the considerations and types of clinical trial logistics:
Clinical Trial Supply Basics
Clinical trial supply requires synergy between lots of different aspects of logistics. Drugs need to reach their destinations, whether that be a clinical trial site or direct-to-patient, but prior to that crucial step there are many requirements to fulfil.
Drugs will need to be produced and then stored, which can require specialist equipment to create the ideal conditions, warehouses will need to be properly equipped, and best monitoring practices put in place. Labelling requirements can differ by country or region also regulatory requirements will need to be fulfilled during export and import, with appropriate documentation provided to customs authority.
After the trial has been completed, any additional equipment will need to be returned, and any excess medicine will need to
Logistics & Supply Chain
be reconciled and destruction certificates provided in line with regulations.
Cold Chain Logistics
New innovations in medicine have required drugs to be kept at low temperatures during shipment to ensure they retain efficacy. Various degrees of refrigeration technology are employed to achieve this, along with temperature monitoring to ensure the medicine has remained within a ‘cold chain’ over the full journey.
This is a growing area of pharmaceutical logistics that has been fuelled by new drugs with this requirement entering the market, not least a significant proportion of COVID-19 vaccines that continue to be used as booster doses today. As a growing space, this also means that clinical trial logistics often require cold chains too.
Many of these medicines are sensitive to even the slightest variations in temperature, so training and expertise in this area is required for anyone involved in the handling of these payloads as they travel through the supply chain, including drivers, staff at airports and healthcare professionals.
Early Access Programmes
Drugs that have concluded a clinical trial and have not achieved regulatory approval are sometimes approved for use in specific circumstances. These drugs might have been approved in other countries or been shown to have some efficacy for patients with no other alternatives available in these cases, a drug may be approved for use in an early access programme.
These situations can provide vital data for sponsors, as it allows drugs to be used in ‘real-life’ situations as if the drug has been approved. However, getting the drug to a patient in a country where the drug has not been fully approved can mean additional considerations for logistics. Knowing and navigating that country’s regulatory environment is key. Achieving the correct documentation and permits can be difficult to do so in a timely manner without the requisite knowledge, presenting additional challenges.
There may also be additional regulations surrounding the logistic handling, labelling and delivery of such medicines, on top of drugspecific requirements such as a cold chain. Pharmaceutical logistic companies should have the on-the-ground expertise in the countries in which they operate to achieve this.
Technological Innovations
New technologies have allowed for more data about shipments,
shipping routes and conditions to be recorded, analysed and shared, creating exciting new possibilities for supply chain visibility.
Increasing visibility is all about making more information available to more parties involved in a shipment. This could include location data, inventory information and, for cold chain purposes, real-time temperature monitoring data.
In turn, this information creates opportunities for logistics providers to be reactive to changing conditions and ensure that more shipments reach their destinations in the proper condition. Any issues with temperature integrity can be spotted and dealt with, as well as providing data for more effective risk management and mitigation practices in the future. Even situation in which a payload is irrecoverable can be identified and the product reshipped in a timelier manner.
The Internet of Things (IoT) technologies and software systems that record each of these data points can be connected, allowing all stakeholders to remain informed, but this interconnectedness can also allow for technologies to work together automatically. IoT hardware can react to certain temperatures and increase or decrease environmental temperature controls accordingly to ensure the payload stays in range.
The applications of these technology are numerous and practical. Malfunctioning containers can be identified by their more extreme fluctuations, allowing maintenance teams to be placed at a future touch point to fix the issue. A package being unexpectedly opened could imply a security issue, so inventory checks can be carried out at the next opportunity. Other touch points like airports and depots can add their own data points, adding further interconnectivity.
For clinical trials, and DCTs in particular, this technology will allow for quick adaptations in last-mile logistics and lane mapping, while also enabling more detailed knowledge acquisition when working in new markets. CROs will be more informed of the status of their payloads and will be able to react effectively. However, interpreting all of the data, gaining insights and implementing efficiencies will require expertise on the part of all stakeholders, including CROs and pharmaceutical logistics companies.
Conclusion
These new developments present new challenges and opportunities for sponsors. Across all of these innovations, relationship building and knowledge sharing between pharmaceutical logistics businesses will be essential to ensure that clinical trials can operate efficiently and effectively.
Steve Healy
Steve Healy, CEO of COREX Logistics, has over twenty years’ experience in the pharmaceutical logistics sector, with focuses on commercial pharmaceutical distribution and clinical trial supply chains. With extensive knowledge of Good Distribution Practise and complex temperaturecontrolled supply chain solutions, including airfreight, road freight and specialist courier services. He has worked with pharmaceutical companies and central laboratories, and CROs to improve their logistics operations and supply chains. Steve has also worked with public sector institutions in the rollout of COVID-19 vaccines, and other public health initiatives.
AVAILABLE WORLDWIDE AVAILABLE WORLDWIDE AT YOUR FREIGHT FORWARDER AT YOUR FREIGHT FORWARDER
Temperature protection of pharmaceutical and healthcare products in airfreight (+15°C + 25°C°) and (+2°C + 30°C) (+15°C + 25°C°) and (+2°C + 30°C)
Multilayer thermal blanket for PMC-ULD - Euro and Block pallets
Stress-tested in summer (+46°C) and winter (-15°C) profiles
Airfield Tarmac tested on solar power and greenhouse effects
Tel. (Belgium): +32-11.26.24.20
E-mail: info@krautz.org Website: www.krautz.org
Logistics & Supply Chain
Transforming Clinical Trials: A Decade of Change and a Future of Innovation
Clinical trials play a pivotal role in advancing medical research and improving patient care. They are essential for new drug development, which is why they are constantly evolving.
In recent years, technological advancements, regulatory shifts and a growing focus on the needs and experiences of patients have been the main driving forces behind the significant changes in clinical trials. The Covid-19 pandemic has also significantly accelerated these trends, demonstrating the remarkable potential for rapid market deployment. While these changes have reshaped the conduct, regulation, and perception of clinical trials, the industry must remain aware of the possibilities and leverage the lessons learned to build on the successes achieved during the Covid-19 pandemic.
These changes have set the stage for potential significant advancements in the future. But first, let’s take a closer look at the current shifts shaping clinical trials today before diving into predictions for what lies ahead.
Current Trends in Clinical Trials
One of the most prominent recent developments in clinical trials is the increasing use of real-world evidence (RWE). RWE uses data from various sources, such as electronic health records, insurance claims and wearable devices. For example, data from wearable devices can provide continuous health monitoring, offering valuable insights into how patients respond to treatments in their daily lives. This method allows researchers to gather data beyond traditional clinical trial settings, providing a more comprehensive view of a treatment’s effectiveness and safety, with the additional advantages of optimising approval timelines and maximising cost efficiencies.
The pandemic has also driven the adoption of remote and decentralised clinical trials, known as virtual, at-home or site-less trials. These make use of telemedicine and home-based monitoring. Patients can participate from the comfort of their homes, eliminating the need to travel to specific sites. This approach has shown increased patient participation and study effectiveness, ultimately speeding up the time it takes to bring new drugs to market.
In addition, there is a growing emphasis on making clinical trials more patient-centric. Regulatory agencies and sponsors now actively involve patients in the trial design process to align research with their needs and preferences. This enhances the relevance of trials and improves patient recruitment and retention rates.
Regulatory Updates Around the World
Countries around the world are adapting their clinical trial regulations to the evolving landscape. In India, the Ministry of Health and Family Welfare updated the New Drugs and Clinical Trials Rules back in 2019. These updated rules set new time limits for responses to clinical trial applications, making the regulatory process simpler and more efficient.
More recently, in the United States, the FDA released a draft guidance in June 2023 titled “E6(R3) Good Clinical Practice (GCP)”. This guidance encourages innovation and quality in clinical trials and supports the use of digital health technologies like wearable sensors to improve the efficiency of data collection.
Meanwhile, the European Union implemented the Clinical Trials Regulation in early 2023. This new regulation simplifies approval processes, harmonises trial designs and establishes a single portal for trial submissions across member states. These changes aim to reduce administrative burdens and increase the competitiveness of EU clinical research.
The United Kingdom has also introduced new measures. In October 2023, the Medicines and Healthcare products Regulatory Agency announced a more streamlined and flexible framework for clinical trial approvals. This new framework supports diverse trial designs, including decentralised trials, and aims to speed up approvals without compromising safety.
These regulatory changes across different regions reflect a global effort to make clinical trials more efficient and patient-focused. Going forward, we can expect regulatory agencies to collaborate with the pharmaceutical industry even more closely and adopt more flexible approaches.
This will improve the clinical trial landscape and enable faster responses to emerging health crises. For example, during a sudden outbreak of a new disease, regulatory bodies might expedite the trial approval process for promising treatments. This will allow faster deployment of critical medications while maintaining safety standards.
What the Future Holds
As the landscape of clinical trials continues to evolve, several key trends, driven by technological advancements, will shape the industry in the coming years.
One major trend is the integration of advanced data analytics. Artificial intelligence (AI) and machine learning is already being used to transform data analysis in clinical trials, however future developments in this technology will advance this even further. Predictive modelling, data mining and real-time monitoring is enabling more efficient trial designs, better patient selection and early identification of safety concerns.
For example, AI has the potential to analyse patient data to predict which individuals are most likely to benefit from a specific treatment. This can make trials more precise. It can also reduce costs and speed up the time to market for new drugs.
Transport/Logistics Outlook
The trend of decentralised and remote trials will expand and make trial participation more convenient for patients. The cold chain logistics industry plays a crucial role in this expansion by safely transporting temperature-sensitive medications to patients. This maintains the integrity and efficacy of treatments during virtual
trials. With the support of reliable logistics, virtual check-ins with healthcare providers and clear regulations, more people will be able to join these trials, leading to better overall outcomes.
At the same time, clinical trials will adapt to consider individual genetic profiles and treatment responses. This shift is driven by advances in genomics and biomarker discovery, which are paving the way for targeted therapies. We will see the rise of innovative trial designs and methodologies that meet the unique needs of personalised medicine. For instance, a clinical trial for a cancer treatment might stratify participants based on their genetic mutations and test the new therapy on the most relevant patient groups. Similarly, single patient samples and specimens will increasingly be transported for clinical trials, highlighting the critical role of effective, safe and rapid cold chain logistics.
A New Era for Clinical Trials
The world of clinical trials is hardly recognisable from what it was a decade ago. There is now a strong focus on real-world evidence, patient-centric approaches and streamlined regulatory processes. These changes have made clinical trials more efficient, user-friendly and more globally connected. The future looks even more promising with advancements in data analytics, decentralised trials, global harmonisation and the rise of personalised medicine.
With this advancement comes increasing pressure on the cold chain industry to respond to the demand for safe, secure and swift transport of medicines and clinical samples. For example, as remote and decentralised trials become more common, there will be increased demand for innovative packaging solutions. Packaging suppliers will need to develop materials and containers that maintain the integrity and efficacy of medications during transport to patient homes and remote locations. This includes meeting stricter temperature requirements and regulations for new drugs and ensuring secure transportation systems.
This progress will result in safer and more effective treatments, reducing the time spent in the trial phase and significantly benefiting those in need. The evolution of clinical trials will not only impact pharmaceutical companies and patients but also drive innovation across the supply chain, including packaging and logistics providers who will be continually challenged to handle increased volumes more rapidly and with reduced risk.
Dr. Danial Arkwell, Head of Global Key Accounts, Pharma, at Envirotainer
Diane Onken
Diane Onken, Head of Sales, America at Environtainer.
IPI
Media and Communications
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.
www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA POD
‘Putting science into conversation, and conversation into science.’Join some of the most esteemed and integral members of the Drug Discovery & Development world as they give insights & introspect into the latest movements, discoveries and innovations within the industry.
senglobalcoms.com
Page 23 Autoscribe Informatics UK
IFC Catalyst Clinical Research
Page 36 DDL2024
Page 27 Nipro
Page 33 Krautz Temax
Page 3 PCI Pharma Services
Page 5 Ramus Medical Limited
OBC Trilogy Writing & Consulting Gmbh
Page 37 Senglobal Ltd
IBC UPC Cambridge
Page 13 Ypsomed AG
Subscribe today at Email: info@senglobalcoms.com www.journalforclinicalstudies.com
Journal for Clinical Studies
I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry JCS is also now active on social media. Follow us on: