Journal for Clinical Studies

Defining Success in Rare Disease Paediatric Trials
Advancing Research and Development of Multi-indication and Combination Therapies for Obesity
Conducting Clinical Trials in the Parallel Virtual Universe
Generative AI Is Revolutionising Life Sciences and We’re Just Scratching the Surface
www.journalforclinicalstudies.com
PEER REVIEWED Volume 16 Issue 1
Industry-leading global oncology CRO
Advancing hope, transforming lives: Inspiring biotech sponsors to develop breakthrough cancer therapies

Inspiring people to design and deliver better clinical trials
catalystcr.com

An innovative approach to functional outsourcing
Individually tailored and adaptable to meet the changing needs of clinical research
MANAGING DIRECTOR
Mark A. Barker
BUSINESS DEVELOPMENT info@senglobalcoms.com
EDITORIAL MANAGER
Beatriz Romao beatriz@senglobalcoms.com
DESIGNER
Jana Sukenikova www.fanahshapeless.com
RESEARCH & CIRCULATION MANAGER
Jessica Chapman info@senglobalcoms.com
ADMINISTRATOR
Barbara Lasco
barbara@senglobalcoms.com
FRONT COVER istockphoto
PUBLISHED BY Senglobal Ltd.
Unite 5.02, E1 Studios, 7 Whitechapel Road, E1 1DU, United Kingdom
Tel: +44 (0) 2045417569
Email: info@senglobalcoms.com
www.journalforclinicalstudies.com
Journal for Clinical Studies – ISSN 1758-5678 is published quarterly by Senglobal Ltd.
4 FOREWORD
WATCH PAGES
6 Quality-of-Life Improvements for Heart Health in Scarce Treatment Settings
When medical circumstances are severe and treatment options are limited in number or effectiveness, being able to help patients improve their quality of life (QOL) takes on an elevated importance. To effect changes in QOL, clinical studies should address outcomes that are meaningful to patients, especially for “insidious” diseases that slowly wear them down. Deborah Komlos at Clarivate explains the quality of life improvements for heart health in scarce treatment settings.
8 A Reflection on the European Regulatory Framework
Historically gaining regulatory approval across multiple countries within the European Union (EU) was segmented, excessively bureaucratic, and challenging to navigate. Sponsors had to submit clinical trial applications separately to national competent authorities (NCA) and ethics committees (ECs) in each country to gain regulatory approval to run a clinical trial. Individual countries dictated their own set of national submission requirements and gave local opinions on the trial design. Inês Vale De Gato and Louise Scott at Catalyst Oncology analyse the European regulatory framework.
10 Feasibility Assessments: What Investigators Need to Know
The feasibility of a clinical trial involves a thorough assessment of the potential and practicality of conducting a particular trial within a specified geographic area to ensure a project’s success in terms of timeline, target achievement, cost management, and various other essential factors. Nur Ain, Shu Hui, Liew Eu and Nur Aziemah outline key insights to investigators, underscoring the importance of feasibility assessments, the impact of feasibility on successful clinical trials, and recommendations for improving feasibility responses.
REGULATORY
12 Five Trends Shaping the Trial Master File
The trial master file is integral to demonstrating that a clinical trial has been conducted in accordance with good clinical practice (GCP). The sponsor must therefore ensure their TMF is complete, timely, and accurate at all stages of a trial and across the document lifecycle. Aaron Grant at Phlexglobal analyses five trends that are shaping trial master file.

Journal for Clinical Studies 1 www.journalforclinicalstudies.com
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright. Volume 16 Issue 1 Spring 2024 Senglobal Ltd. Contents Journal for Clinical Studies
14 Generative AI Drives efficient Regulatory Processes
As regulatory Agencies around the world continue to refine and update their requirements, artificial intelligence (AI) can help Life Sciences companies increase their chances of new products being accepted promptly. ArisGlobal’s Agnes Cwienczek & Renato Rjavec describe how AI, and Generative AI in particular, are distilling insights from regulatory exchanges and harnessing the latest global Regulatory intelligence to anticipate and respond to Agency requirements.
MARKET REPORT
16 Green Miracles: Cannabis and Kratom's Potential for Healing and The Industry in Malaysia
Global Cannabis market size was valued at USD 14154.75 million in 2023 and is expected to expand at a CAGR of 17.17% during the forecast period, reaching USD 36619.44 million by 2031. While Kratom on the other hand valued at USD 1.87 Billion in 2023 and is expected to grow at a CAGR of 17.2 percent from 2024–2030 to reach USD 5.69 Billion based on MMR. Nur Ain at Clinical Research Malaysia, aims to delve into the industrial strategical way and potential of cannabis and kratom, and their role in advancing treatments within Malaysia.
RESEARCH & DEVELOPMENT
18 Conducting Clinical Trials in the Parallel Virtual Universe
Quantitative systems pharmacology (QSP) and physiologically based pharmacokinetic (PBPK) modelling can be employed to create a parallel virtual workflow throughout drug development, beginning with preclinical research and continuing through phase IV clinical studies. Many clinical trials are conducted with virtual patients prior to selecting some trials to be carried out with real patients in real life. Amin Rostami-Hodjegan at Certara explains how to conduct clinical trials in the virtual world.
THERAPEUTICS
22 Advancing Research and Development of Multi-indication and Combination Therapies for Obesity
Obesity is commonly defined as excess fat accumulation that puts someone at a higher risk for adverse health outcomes. The simplicity of this definition can be misleading, because obesity’s pathophysiology is highly complex, and characterised by a multitude of interrelated factors, including metabolic and immune changes in response to energy surfeit; mechanisms such as oxidative stress, and local and systemic inflammatory responses; alterations in blood flow and perfusion; and the mechanical burden of weight. Dr. Jack L. Martin at ICON shows the advancing research and development of multi-indication and combination therapies for obesity.
24 Defining Success in Rare Disease Paediatric Trials
The effects of rare disease change everything from direct consequences of having the condition to impact on the child’s growth patterns and development, behaviour, psychology, self-worth, and self-belief. This is why paediatric trials in rare disease are so important; they help to develop the treatments of the future and by targeting treatment of paediatric patients, they also provide the chance to live a positive childhood with as much normal development as possible. Christine McSherry from The Emmes Company discuss the complexities of agreeing the definitions and approaches used to measure trial success that are meaningful for all involved.
30 Cell and gene Therapies are set to Revolutionise Healthcare
The first chemotherapy drugs were discovered by mistake – during World War II – when humans were accidentally exposed to mustard gas. At that time, the only other treatments for cancer were radiotherapy and surgery. But these weren’t necessarily promising for the treatment of all forms of cancer, especially cancers that had progressed. Richard Rossi at CRYOPDP outlines how some cell and gene therapies are set to revolutionise healthcare.
TECHNOLOGY
32 Generative AI Is Revolutionising Life Sciences, And We’re Just Scratching the Surface
On average, it takes about seven years to develop a new drug and bring it to market. For ambitious life science businesses, generative AI’s ability to generate insights and content in a fraction of the time of a human means wiping months, or even years, off that average. Bryan Hill at Cognizant Life Sciences outlines the impact of AI in life sciences.
34 Modern by (Trial) Design: Shedding Legacy EDC Systems to Gain Clinical Capacity
In today's context of complex trials, traditional electronic data capture (EDC) systems are not flexible enough to address frequent protocol amendments. They hinder study teams by creating hidden data management issues that can affect sponsors' and contract research organisations (CROs) clinical capacity. Richard Young at Veeva Vault CDMS shows how shedding legacy EDC systems help to gain clinical capacity.

2 Journal for Clinical Studies Volume 16 Issue 1
Contents



Lyophilization and Sterile Manufacturing
Driving development and connecting commercialization of sterile and lyophilized drug products, we are dedicated to your success in bringing life-changing therapies to patients.
www.pci.com
OUR END-TO-END BIOLOGIC SOLUTIONS INCLUDE:
• Sterile Formulation & Lyophilization Cycle Development
• Lyophilization and Sterile Fill-Finish Manufacturing
• Aseptic Robotic Technologies
• Analytical Support
• Clinical & Commercial Labeling & Packaging
• Refrigerated/Frozen Storage & Distribution
talkfuture@pci.com Your sterile drug product, our world.

There has been considerable international activity about rare diseases in the last few months. First, there are many 1000’s of rare disorders and diseases each of which is very different from the other, and whilst every person with a rare disease is a unique person, there are things that people with rare diseases may share, as may their families, due to the rareness of the syndrome. These include delays in diagnosis, the absence of treatments, and a lack of knowledge among health and other professionals. These problems associated with being ‘a rare disorder’ were universal across all the rare diseases and disorders represented. Secondly, there is very extensive national, regional and international activity going on to raise the profile of rare diseases led by the various national, regional, and international organisations.
The clinical presentation, natural history, pathophysiology, and often mysterious nature of rare diseases have fascinated physicians for centuries. Rare diseases provide opportunities to study human physiology and biomedical science from unique perspectives. Major scientific breakthroughs resulting from investigation of rare diseases have often provided insight into more common disorders. The satisfaction of diagnosing a patient with a rare disorder successfully is often rapidly countered by the realization that the ability to understand and treat the patient’s condition is limited by ignorance and the difficulties of studying the disease. Moreover, for the “interesting” patient with a rare disease, being a “fascinoma” to physicians may intensify suffering. Patients may feel that their physicians are in league with the “interesting” disease. Furthermore, for patients with a rare disorder, the disease is no longer rare – it is a constant part their lives and the life of their families.
The perspective of human rights is important. This goes beyond the rights of access to health care and to treatments that are approved and available to include the rights to family life, to opportunities, to respect – to a quality of life. The United Nations Convention on the Rights of Persons with Disabilities (UN CRPD) is seen as key to this.
There is still much to do to ensure that not only are health needs met, but that educational, emotional, and social needs are also met for all people with a rare disorder.
JCS – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Principal Content Editor, Clarivate
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
• Georg Mathis, Founder and Managing Director, Appletree AG
This year sees four new research projects to help children with rare diseases now underway. Co-funded with LifeArc, this amounts to almost £1 million of vital new research, giving hope to children facing some of the toughest fights.
Molecular genetics have characterized the cause of many rare diseases and provide unprecedented opportunities for identifying patients, determining phenotypes, and devising treatments to prevent, stabilise, or improve each disease. Rare disease research poses challenges to investigators requiring specific approaches to, the design of clinical studies, the funding of research programs, the discovery, testing, and approval of new treatments, and the training of clinical scientists. Rigorous, statistically valid, natural historycontrolled, cross-over, and n-of-1 trials can establish efficacy and support regulatory approval of new treatments for rare diseases.
Keeping this in mind, the cover story of JCS is a fascinating explanation by Christine McSherry from The Emmes Company in his article titled “Defining Success in Rare Disease Paediatric Trials” discuss the complexities of agreeing the definitions and approaches used to measure trial success that are meaningful for all involved.
In other articles, Amin Rostami-Hodjegan at Certara explains how to conduct clinical trials in the virtual world. Dr. Jack L. Martin at ICON shows the advancing research and development of multiindication and combination therapies for obesity and Bryan Hill at Cognizant Life Sciences outlines the impact of AI in life sciences.
I hope you all enjoy the first issue of JCS of 2024. My team and I look forward to bringing more exciting articles to you in the future issues.
Beatriz Romao, Editorial Manager, Journal for Clinical Studies

• Hermann Schulz, MD, Founder, PresseKontext
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, AsiaPacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
4 Journal for Clinical Studies Volume 16 Issue 1 Foreword

Ramus Medical
is a part of Ramus Corporate Group. The company is managed under a centralised quality management and has developed an integrated QMS as well as specific standard operating procedures tailored for the clinical trials department that are fully harmonised with the GCP guidelines, and the local and European legislation.
Ramus Medical EOOD is a full-service contract research organisation (CRO) in Sofia, Bulgaria.
The company was created in 2009 as a natural development of the Medical Laboratory Ramus Ltd., the largest privately-owned medical laboratory in Bulgaria.
The company independently manages clinical research projects in Bulgaria and provides partnerships in multinational clinical projects providing a comprehensive range of clinical research services:
Core Services include:
• Medical writing
Our staff has extensive expertise in the preparation, adaptation and translation of a wide range of clinical trial documents that are fully compliant with the Good Clinical Practice (GCP) standards, the client’s specifications and the regulatory requirements.
• Study start-up
We offer full or partial study start-up assistance for different types of studies throughout Bulgaria.
• Regulatory submission
• Project management
• Monitoring
• Data Management
• Pharmacokinetic evaluation
• Biostatistics
• Regulatory advice and services
• Readability User Testing
• Registration of medicinal products on the territory of Bulgaria
• Pharmacovigilance services
• Logistic department
• Destruction of IMPs/IMDs & clinical samples – agreement with PUDOOS
• Archiving services
• DDD activities
Ramus Medical has gained its expertise during the completion of numerous clinical projects carried out over the past decade:
• Phases I to IV drug trials
• Non-interventional studies
• Pilot and Pivotal Medical Device investigations
The clinical trials we conducted facilitated the MA/CE mark granted by various European Agencies/Notified Bodies and Third Country Agencies.
Ramus Medical offers flexible clinical research services in various domains, with extensive experience in fields.
Our team comprises qualified, appropriately trained, experienced, motivated and collaborative professionals and is competent to
communicate effectively across geographical and cultural boundaries to resolve any arising issues. We adhere strictly to the agreed timelines during the clinical investigations and strive to complete the tasks on time.
Why are we the solution for your projects? Ramus has its own:
Medical and Bioanalytical Laboratory
In 2018 the Medical Centre Ramus was established, located in Sofia, Bulgaria. Up to date, it has three separate locations, one of which is developed as an independent clinical research centre in compliance with the requirements for the phase I unit.
The Medical Centre Ramus allows the conduct of clinical trials in all phases in many therapeutic areas.
The Medical Centre meets all requirements for performing highquality clinical research and is designed to maximise the delivery of high-quality research data and was GCP-inspected.
Ramus Medical retains an extensive database of investigators and sites compiled through years of mutually beneficial collaboration.
Our bioanalytical laboratory is equipped with leveraging state-ofthe-art instrumentation (LC-MS/MS), techniques, and facilities, our team of experts has experience in a broad range of small molecules. Our Analytical laboratories provide method development, transfer, validation, and analysis of preclinical and clinical biological samples. We have extensive expertise in developing sensitive methods for LCMS/MS-qualifying multiple analytes and metabolites.
• Logistical company, certified for hazardous and biological samples transportation
• Clinical site facility and own catering company for hospitalised patients
• Integrated QMS


Ramus Medical Ltd
26 Kapitan Dimitar Spisarevski Street, 1592 Sofia, Bulgaria
Tel./Fax: +359 2 841 23 69
www.ramusmedical.com, www.ramuslab.com
email: office@ramusmedical.com

Journal for Clinical Studies 5 www.journalforclinicalstudies.com
Dimitar Mihaylov Marketing Director
Corporate Profile
! S ct a e f r e r , o f c ast, to V i e C r o e Tut

Quality-of-life Improvements for Heart Health in Scarce Treatment Settings
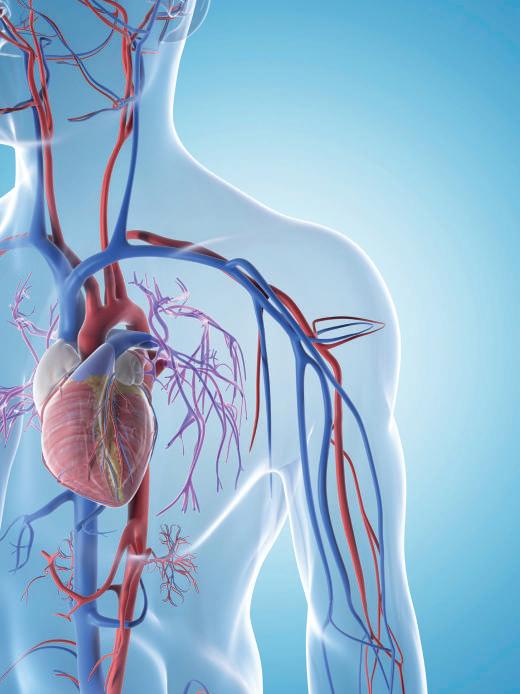
When medical circumstances are severe and treatment options are limited in number or effectiveness, being able to help patients improve their quality of life (QOL) takes on an elevated importance. To effect changes in QOL, clinical studies should address outcomes that are meaningful to patients, especially for “insidious” diseases that slowly wear them down.
This view recurred among public speakers and expert panelists at a February meeting of the Circulatory System Devices Panel (CSDP) of the Medical Devices Advisory Committee (MDAC), US Food and Drug Administration (FDA). The meeting concerned a premarket approval application (PMA) submission by Abbott Medical (Abbott) for the use of its TriClip G4 System (TriClip), which was proposed to improve the health status in a particular population of patients with symptomatic severe tricuspid regurgitation (TR). These patients were being treated with optimal medical therapy, were at intermediate or greater risk for surgery, and were determined by a heart team to be suitable candidates for tricuspid valve (TV) transcatheter edge-toedge repair (TEER).
TR occurs when the TV leaflets do not close completely during systole, the part of the cardiac cycle when the heart muscle contracts and pumps blood from the chambers into the arteries. Consequently, there is leakage or “regurgitation” of blood from the right ventricle into the right atrium. TR may lack symptoms, or the symptoms are vague (e.g., weakness, fatigue), the American Heart Association (AHA) states.
In the US, the treatment options for patients with severe TR are limited. As presented at the February meeting, these individuals experience debilitating symptoms that impact QOL. Medical therapy is largely driven by diuretic use to manage volume overload; however, it is often ineffective, particularly if diuretic resistance develops. While isolated TV surgery is an option, it is not commonly performed due to the high rate of operative mortality. In a study that evaluated patients aged >18 years who underwent TV repair or replacement from 2004 to 2013, operations per year increased from 290 in 2004 to 780 in 2013.1 Despite this increase in surgical volume, in-hospital mortality was 8.8% and remained unchanged over the study period. The FDA highlighted that most patients with moderate or severe TR are not offered surgery.
Classification of Health Status
To evaluate the impact on QOL, Abbott’s pivotal study of TriClip incorporated a patient-reported outcome (PRO) measure, the Kansas City Cardiomyopathy Questionnaire (KCCQ), in the primary endpoint of its pivotal trial, TRILUMINATE. For the randomised cohort, the endpoint was a hierarchical composite comprising time to all-cause mortality or TV surgery, number of heart failure (HF)
hospitalisations, and a ≥15-point improvement in KCCQ overall summary score from baseline at 12 months. A self-administered PRO, the KCCQ evaluates the health status of patients with HF, relying on several domains (e.g., QOL, social limitation, physical function, symptom stability) and two summary scores.
The KCCQ was qualified in 2020 by the FDA’s Center for Devices and Radiological Health in the Medical Device Development Tools (MDDT) program as a clinical outcome assessment PRO instrument for adults aged ≥18 years with symptomatic HF.2,3 The instrument can be used in feasibility and pivotal studies to evaluate treatment benefits for patients with symptomatic HF. According to an AHA/ American College of Cardiology (ACC)/Heart Failure Society of America (HFSA) guideline, symptomatic HF is categorized as stages C and D or classes II–IV per the New York Heart Association (NYHA) classification.4
From the FDA’s perspective, information obtained from “welldefined and reliable” PRO instruments can provide valuable knowledge for its benefit-risk determinations. In relation to medical device labelling, the agency explains in its Guidance for Industry and Food and Drug Administration Staff, and Other Stakeholders: Principles for Selecting, Developing, Modifying, and Adapting Patient-Reported Outcome Instruments for Use in Medical Device Evaluation5 from January 2022 that PRO data can be used to communicate the effect of a treatment on patient symptoms, functioning, or health-related QOL, provided the use is consistent with the documented and supported measurement properties of the PRO tool.
Incapacitating Symptoms
At the February MDAC meeting, the panel heard several testimonials during the open public hearing session regarding the poor QOL experienced by patients with TR, including challenges with shortness of breath, fatigue, weight gain due to swelling and its associated mobility issues, and the inability to sleep at night. The disease “slowly disables then kills patients,” said Patrick McCarthy, MD, a cardiac heart surgeon who was the inventor of the Edwards MC3 Tricuspid annuloplasty ring, from Edwards Lifesciences Corporation.6
Although the MC3 ring “works well” and is “the most commonly used tricuspid repair ring in the world,” a significant number of patients who received it “faced a difficult and slow recovery,” McCarthy noted. When the TV leak becomes severe, there is no good medical therapy, he explained, adding that the risk of openheart surgery to repair or replace the TV to stop leaking “is one of the highest risk elective operations that we perform in all of heart surgery.” Because the procedure is high risk, patients are rarely referred for it, and nearly all are maintained on ineffective drug and medical therapy, McCarthy said.
Matthew Price, MD, an interventional cardiologist who enrolled patients into TRILUMINATE, noted that he has observed patients
6 Journal for Clinical Studies Volume 16 Issue 1
Watch Pages
with severe TR with “really horrific symptoms,” including “legs like tree trunks” and “just feeling miserable.” Through his experience with the trial, Price said he learned “what TR means to patients and what resolving their TR means.” Looking at the “big picture,” the “day-today lives” of patients should be the primary focus, he said.
Measurements That Matter
The primary endpoint of TRILUMINATE was met, driven by KCCQ score improvement in the device group. Mortality or TV surgery rates were similar between treatment groups, and the HF hospitalization rate was numerically higher in the TriClip group versus the control group. In its summary comments about the PMA submission for TriClip, the FDA stated that TRILUMINATE was an unblinded (i.e., open label) randomised controlled trial and that PROs such as the KCCQ score could be subject to the placebo effect in an unblinded trial.
MDAC panelists at the February meeting expressed support for the use of KCCQ as a primary endpoint measure that reflected clinical significance. To the FDA’s point regarding the potential placebo effect, panelists commented that this concern was “mollified” by the fact that the treatment benefit continued through one year. Nonetheless, panel members urged the collection of longer term data to gain improved understanding about the device, including a better definition of which patient populations would benefit from it and to acquire more findings on “hard” endpoints such as mortality.
During the meeting, panelists and presenters occasionally referenced Abbott Vascular’s MitraClip Clip Delivery System (MitraClip), a TEER technology that was approved in 2013 to treat mitral regurgitation. TriClip involves the identical clip-based technology as the MitraClip but with a modified delivery system and steerable guide exclusively designed to access the TV. The MitraClip has been used off label for the treatment of TR since 2015,
highlighting the unmet need for the safe treatment of patients with symptomatic severe TR, Abbott noted.
A recent arrival to the treatment landscape for severe TR occurred just ahead of this MDAC meeting, when the FDA approved the Edwards EVOQUE Tricuspid Valve Replacement System from Edwards Lifesciences on February 1.7 As the first transcatheter replacement device indicated for use in the tricuspid position, the EVOQUE device is intended to treat patients with severe TR who continue to experience symptoms despite being on HF medications. One MDAC panelist expressed interest in learning how structural clinicians will make decisions on whether to use the EVOQUE system or TriClip, if approved by the FDA, and in which patients.
TRILUMINATE “was a success for both safety and efficacy,” McCarthy summarised at the conclusion of his comments. “I’m here,” he said, “because I hope that this treatment will be approved so we can help these patients who are suffering with little hope.” For their review of the trial data, Price urged the MDAC panel members “to remember the patients that are behind the numbers, who can now put on socks, who can go gardening, who can shop, and who can play with their grandchildren.” He added, “I think that’s what’s really important.”
REFERENCES
1. Zack CJ, Fender EA, Chandrashekar P, et al. National trends and outcomes in isolated tricuspid valve surgery. J Am Coll Cardiol. 2017;70(24):2953-2960. http://doi.org/10.1016/j.jacc.2017.10.039
2. Medical Device Development Tools (MDDT). Food and Drug Administration Website. https://www.fda.gov/medical-devices/ medical-device-development-tools-mddt#mddts
3. Medical Device Development Tool (MDDT) Qualification Decision Summary for Kansas City Cardiomyopathy Questionnaire (KCCQ). Food and Drug Administration Website. https://www.fda.gov/media/108301/ download?attachment
4. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A report of the ACC/ AHA Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145(18):e895-e1032. https://doi.org/10.1161/CIR.0000000000001063
5. Guidance for Industry and Food and Drug Administration Staff, and Other Stakeholders: Principles for Selecting, Developing, Modifying, and Adapting Patient-Reported Outcome Instruments for Use in Medical Device Evaluation. Food and Drug Administration Website. https://www.fda.gov/media/141565/download
6. Edwards MC3 Tricuspid Annuloplasty Ring with Template/Lanyard for Valvuloplasty. Edwards Lifesciences Corporation Website. https://eifu. edwards.com/eifu/5d4dd5ad46e0fb0001c61ac7/DOC-0198262A.pdf
7. Edwards EVOQUE Tricuspid Valve Replacement System – P230013. Food and Drug Administration Website. https://www.fda.gov/medicaldevices/recently-approved-devices/edwards-evoque-tricuspid-valvereplacement-system-p230013
Deborah Komlos

Deborah Komlos, MS, is a Principal Content Writer for the Cortellis suite of life science intelligence solutions at Clarivate. In this role, her coverage centres on FDA advisory committee meetings, workshops, and product approvals. Her previous positions have included writing and editing for magazines, newspapers, online venues, and scientific journals, as well as publication layout and graphic design work.
Email: deborah.komlos@clarivate.com
Journal for Clinical Studies 7 www.journalforclinicalstudies.com
Watch Pages
A Reflection on the European Regulatory Framework
Historically gaining regulatory approval across multiple countries within the European Union (EU) was segmented, excessively bureaucratic, and challenging to navigate. Sponsors had to submit clinical trial applications separately to national competent authorities (NCA) and ethics committees (ECs) in each country to gain regulatory approval to run a clinical trial. Individual countries dictated their own set of national submission requirements and gave local opinions on the trial design. The result of inconsistent feedback left Sponsors with the challenge of unifying recommendations within a single global protocol often leading to multiple protocol amendments or country-specific versions of the master protocol. These challenges subsequently resulted in inflated costs and long start-up timelines and the perception that the EU was a difficult place to run clinical research.
On 31 January 2022 the EU Clinical Trial Regulation (EU CTR) No 536/2014 came into effect and repealed the Clinical Trials Directive (CTD) 2001/20/EC and national implementing legislation in the EU Member States.1 The EU CTR is focused on ensuring high patient safety standards, collective decision-making on clinical trials and greater public transparency of clinical research, and aims to make the EU a more attractive place for conducting high-quality large-scale trials. There is a requirement to transition all clinical trials to the EU CTR by 31 January 2025 if they are still ongoing.
Streamlining the Regulatory Process
The EU CTR enables Sponsors to submit one multi-country application via a single online platform through the Clinical Trials Information System (CTIS) for approval to run a clinical trial in the EU and European Economic Area (EEA), making it more efficient to gain regulatory approval for multinational trials. The CTIS provides a single point of entry for clinical trial application procedures and publication of trial results over the life cycle of clinical trials run across the EU/EEA.
For multi-country clinical trial applications, a single, harmonised submission dossier is presented. The dossier is reviewed in two parts with the scientific assessment of the core documentation harmonised across all member states being led by one reporting Member State (part I) and ethical assessment of the country- and site-specific documentation (part II) completed by each country where the trial is to be conducted, creating a combined review and assessment process.
One of the principles of the EU CTR is that all submitted documents will be publicly available and the CTIS allows for searchable clinical trial information. CTIS enables redaction of documents as the method to protect personal data (PD) and commercial confidential information (CCI), if those are included in the documents. Having a single portal where the whole regulatory and ethical dossier of a trial is available has led to greater transparency of information.
Although the new submission process may have increased or decreased the overall timelines in some countries, it brings an increased predictability for clinical trial start-up timelines in the EU countries.
As with any new regulation, Sponsors, regulators, and ECs have faced challenges navigating the new requirements and process. Approximately one year on, Catalyst Oncology reflects on the impact this regulation has had on attaining approvals to run trials in the EU Member States and EEA countries and our experience to-date.
Reforming Operational Considerations
Data retrieved from CTIS, as of 19 February 2024, showed 3,872 clinical trial applications have been submitted since its launch and 2,473 have received a decision.4 The therapeutic areas mostly investigated are neoplasms (tumours).4
The EU CTR establishes an overall timeline of 60 days for the member states to evaluate an initial application. This deadline may be extended if requests for information (RFIs) are raised by a Member State Concerned throughout the evaluation process. Timelines can be extended up to 15 days for RFIs raised in the validation phase and up to 31 days for RFIs raised in the assessment phases. In total, the initial application type assessment takes up to 106 days.
Harnessing CTIS Submissions
Technical problems with CTIS have led to delays in study approvals, unnecessarily raised RFIs, and duplication of work. A significant challenge is the requirement for manual data entry and the duplication of efforts to upload two versions of documents: redacted version for publication and non-redacted version for authorities’ review, which results in a significant resource burden.
Another challenge is that the CTIS does not send alerts to users’ emails, forcing users to daily monitor incoming RFIs and notifications within the portal. Considering the 12-day response requirement for RFIs, it is critical to become aware of an RFI immediately.
Another issue is certain information is not received in a document format, such as query letters or acknowledgments of application, which prevents such documentation from being included in the electronic trial master file.
Meeting Deadlines
It is important to understand the implication of not meeting RFI timelines. If a Sponsor fails to meet a deadline for a response to an RFI for a country, the RFI lapses, the whole application withdraws automatically in the system for all countries, and part I and part II must be resubmitted.
As Catalyst Oncology, we dedicate significant time with the preparation and review of the submission documents to ensure quality and minimize potential RFIs. We implement a risk mitigation strategy with our Sponsors to anticipate potential RFIs and preprepare responses where possible while the assessment is underway. Close collaboration between Sponsor and the dedicated regulatory or site activation team is critical to success.
Documentation Needs
While approval timelines should be shorter with EU CTR compared to CTD, timelines and the number of documents required for
8 Journal for Clinical Studies Volume 16 Issue 1 Watch Pages
submission have increased (mainly for part II). Examples of new documents previously not required in many countries are:
• individual site feasibility forms
• CVs
• declarations of interest for all investigators
• recruitment arrangements
• financial arrangements
• data protection statements
• description of use of biological samples
Their inclusion in the submission dossier increases the number of required documents to be prepared, reviewed, and potentially queried, increasing preparation and review timelines.
Modifications of Submissions
It is possible to make substantial and non-substantial modifications to a trial after study approval, including changes to the protocol, other study documentation, and to add new sites in a country (part II substantial modification). Adding a new country is considered a new application for both part I and part II or only part II, if there was a partial application previously and can take up to 83 days. New sites can be added via a substantial modification (SM) and must be approved by both NCA and ECs before the site can start enrolling patients (EU CTR, Chapter III, Article 15). Where modifications are made, each SM can take from 60 to 95 days and this should be factored into site and country strategies.
Transparency
One of the principles of the EU CTR is that all submitted documents will be publicly available unless their confidentiality are justified. Deferral rules for varying amounts of time depending on the category of the clinical trial and document type were in place for Sponsors submitting in CTIS. The European Medicines Agency (EMA) also notes that PD and CCI should not be included in the structured data fields or uploaded documents within CTIS, as structured fields are meant for publication.
However, during May and June 2023, a public consultation was held with stakeholders to gather insights from the previous year. One key finding was that the transparency and associated deferral rules were too complex. The goal of these rules, which aims to strike a harmonious balance between informing patients and protecting their data, is inherently complex and ambitious. The European Medicines Agency has subsequently decided to eliminate deferrals for clinical trial information publication.
Because of the ongoing changes, contract research organisations must have dedicated teams staying updated on changes in guidelines and regulations. Sponsors should consider how this change might impact their strategy, especially when identifying in-scope documents.
Additional Considerations with the CTIS Application Process
Although the submission process is a two-part package, clinical trial applications can be submitted as a full application with both parts together for all countries. Or a partial application with a part I submission to the reporting Member State and part II, the country packages, submitted later for the national assessment.
A mixed application where a part I package and an initial country part II package is submitted, followed by a second part II package to add additional countries later is also a possible strategy with risks and benefits to consider.
It is important and critical for success of a planned CTIS submission to have the sites in a country pre-selected as part II

packages (country and site level) require the collection and review of site-specific documents signed by the principal investigator or institutional board (CVs, Declaration of Interest, site suitability forms, GCP certificates, etc.). We recommend expediting and streamlining the site feasibility and selection process to get all sites selected four to six weeks before the planned CTIS submission timeline.
Conclusion
The changes brought about by EU CTR have shifted the clinical trial landscape in the EU. While challenging regulatory segmentation has receded, the EU CTR and CTIS have yet to completely smooth out all the challenges. As Sponsors and contract research organisations continue to embrace the changes the EU CTR is working towards, the EU regulatory landscape will become an even more welcoming place for conducting high-quality large-scale trials.
REFERENCES
1. Clinical Trials Regulation | European Medicines Agency (europa.eu), visited 4 March 2024.
2. Guidance for the Transition of clinical trials from the Clinical Trials Directive to the Clinical Trials Regulation, 10c83e6b-2587-420d-9204d49c2f75f476_en (europa.eu), visited 4 March 2024.
3. EMA CTIS newsflash - 23 February 2024 (europa.eu), visited 4 March 2024.
4. Monitoring the European clinical trials environment A deliverable of the ACT EU Priority Action 2 November 2023, ACT EU KPI Report_November 2023 (europa.eu), visited 19 February 2024.
5. EMA Revised CTIS Transparency Rules Oct 2023, Revised CTIS transparency rules (europa.eu), visited 4 March 2024.
Inês Vale de Gato

Inês Vale de Gato, MSc, Associate Director, Catalyst Oncology, brings 14 years clinical development research experience. Ines expertly supervises and manages operational study startup and regulatory strategies for multiple studies and regions. Ines manages multiple site activation staff across multiple countries. She is an EU regulatory and CTIS certified expert and earned B.S. and M.S. degrees in microbiology from the Medicine and Sciences Faculty, the University of Lisbon, Portugal.
Louise Scott

Louise Scott, PhD, Director Oncology Drug Development, Catalyst Oncology, has over 25 years of oncology scientific and clinical development research experience. With an extensive background in clinical operations, Louise supports study optimisation and new initiatives across the commercial and operational teams at Catalyst. Louise started her industry experience as a cell biologist and has a PhD in molecular medicine from Keele University, UK and a BSc (hons) in applied biochemistry.
Journal for Clinical Studies 9 www.journalforclinicalstudies.com
Pages
Watch
Feasibility Assessments: What Investigators Need to Know
The feasibility of clinical trial involves a thorough assessment of the potential and practicality of conducting a particular trial within a specified geographic area to ensure a project’s success in terms of timeline, target achievement, cost management, and various other essential factors. The extent of feasibility outreach can constitute an initial assessment phase that provides a preliminary understanding of the disease prevalence and standard of care. In contrast, an extensive feasibility analysis encompasses patient criteria, site capabilities, regulatory requirements, and potential execution challenges, thus enabling informed decision-making while paving the way for a successful clinical trial execution and delivery.1 Clinical Research Malaysia (CRM) provides a centralised feasibility assessment to sponsors and contract research organisations (CROs) to match clinical trials with the right investigators and sites in Malaysia. CRM provides reliable insights and up-todate information that covers the hospitals in the public, private, and academia to ensure that potential centres and institutions are tapped into the study.2
This article offers key insights to investigators, underscoring the importance of feasibility assessments, the impact of feasibility on successful clinical trials, and recommendations for improving feasibility responses.
Feasibility Growth and Its Importance
Over the period spanning from 2018 to 2023, the growth of feasibilities conducted by CRM has been significant (Figure 1). In line with this, there has also been an increasing number of sponsors and Contract Research Organisations (CROs) conducting feasibility studies through CRM. Based on the annual customer satisfaction survey reported in the CRM Annual Report 2022, 94% of the respondents rated “good” and “very good” for CRM’s complimentary feasibility service.
There are a few components that should be considered by clinical investigators when responding to feasibility questionnaires, such as population profile and access,3 facilities and equipment accessibility, and investigators’ experience.1 Usually, each of the components has a weight assigned based on the sponsors’ evaluation of the site’s strengths and weaknesses and allows for objective comparison. Hence, due consideration should be given to each of the components in completing the questionnaires to meet the feasibility objective and facilitate site selection.
Estimating Potential Patient Population
A vital aspect of feasibility assessment is identifying the availability of patients with a certain disease condition specific to a particular trial. Investigators’ considerations encompass a thorough review of the study design, inclusion/exclusion criteria for patient recruitment, the current standard of care, frequency of visits, invasiveness level of the trial, and the provision of reimbursement for study-related procedures.1 Furthermore, correctly identifying the available patient pool, while exercising caution on underestimation, and ensuring realistic projections of patient numbers are crucial aspects to consider. References from Clinical Practice Guidelines (CPG), up-












to-date patient registries,4 and the availability of databases such as the Malaysian Health Data Warehouse (MyHDW) platform, could provide vital data during feasibility assessments. The integration of Electronic Medical Records (EMR) within hospitals supports investigators in providing real-time patient pool for a certain disease condition, further translating to a more reliable recruitment projection that ultimately meets the recruitment targets set.
Resources and Infrastructure
Assessing infrastructure in healthcare institutions is crucial in determining the site’s ability to conduct clinical trials.5 A thorough evaluation enables sponsors and investigators to anticipate and proactively address challenges during the feasibility stage. Having basic and adequate facilities such as freezer and fridge for investigational product storage and centrifuge with regular calibration taking place, will increase the chances of sites being selected during the feasibility assessment. Considering Malaysia’s resource allocation and population distribution, certain sites may lack essential diagnostic capabilities or long waiting times at diagnostic units. The sites’ readiness to utilize outsourcing services offers a valuable alternative for obtaining timely results. In addition, it also helps in mitigating the prolonged turnaround time of test results and alleviates overcrowding issues at public hospital laboratories. CRM provides the support to connect sponsors and CROs for the outsourcing services to ensure a successful trial.
Investigators’ Experience and Competing Studies
Investigators’ experience and their familiarity with the study protocols are vital for successful studies.6 In addition, identifying competing studies involving similar patient populations is crucial for patient recruitment. Investigators are encouraged to share their trial experience and disclose any ongoing or planned trials within the same patient pool at a particular locality. This could prevent challenges in recruiting patients for different trials which taps into the same patient pool especially if both trials are taking place at the same time. CRM continuously provides training to the study teams to uphold the quality of trial conduct and adherence to the protocol. The trainings include Nurturing New Talents in Sponsored Research, GCP refresher courses, Protocol Compliance Workshop, and Patient Recruitment and Retention Workshops.
10 Journal for Clinical Studies Volume 16 Issue 1 Watch Pages
Graph Pre Feasibility – Country outreach level Full feasibility – Feasibility study with protocol and survey
115 137 151 214 199 223 315 215 234 179 276 289 0 50 100 150 200 250 300 350 2018 2019 2020 2021 2022 2023 Total Full Feasibility (FF) Total Full Pre-Feasibility (PF) Numbers of Feasibility
Figure 1: Total full feasibility and pre-feasibility from 2018-2023
Year
2018–2023
Figure 1: Total full feasibility and pre-feasibility from
Challenges and Strategies
One of the challenges in meeting Sponsors’ requirements includes short timelines given to investigators to complete feasibility questionnaires, especially if the study is under the bidding stage by a CRO or if it’s a rescue study for an ongoing trial that is already happening in other parts of the world. Sponsors may also set short timelines to meet specific business objectives and address seasonal considerations, such as disease outbreaks, which require rapid trial initiation and completion. With digitalisation playing a larger role in clinical research, sponsors and CROs are introducing new digital platforms to implement feasibility assessments. Unfamiliarity with the use of these platforms among investigators also poses challenges in responding to feasibility in a timely manner. To address this, CRM feasibility team and onsite study coordinators support the investigator in completing the feasibility questionnaire, with input from investigators. Guidelines on navigating the feasibility platform and support in gathering information from other departments, such as the laboratory and pharmacy are provided. This collaborative effort expedites the feasibility process, leading to quicker site start-up if the sites are selected.
Other strategies aimed at improving feasibility timelines include the use of e-signatures in Confidential Disclosure Agreements (CDA)8 and utilising online questionnaires/surveys from sponsors. E-signature is legally recognised under the Electronic Commerce Act 2006 (ECA) in Malaysia. E-signature offers flexibility that can aid in recordkeeping, improve efficiency and deadlines, and reduce the overall timeline for feasibility assessment. The incorporation of e-signatures and online surveys aligns with CRM and the industry’s focus on operational efficiency in clinical trials.
Conclusion
Details and information shared by investigators in feasibility assessments carry a huge weight in sponsors’ decisions in site selection. Significant aspects including the availability of the patient pool, basic facilities, relevant investigator experience, and upcoming competing trials will also ensure that they can deliver the trial and meet the expectations of sponsors. Feasibility assessments enable investigators to anticipate and address challenges in clinical trials, which could facilitate resource utilisation and the development of effective strategies in trial initiation. At the end of the day, a good feasibility assessment and response translates into successful clinical trials. CRM continuously supports investigators in feasibility assessments and endeavours to ease communication between the investigator and sponsor throughout the process.
REFERENCES
1. Rajadhyaksha V. Conducting feasibilities in clinical trials: an investment to ensure a good study. Perspect Clin Res. 2010 Jul;1(3):106-9. PMID: 21814631; PMCID: PMC3146075.
2. Ooi, A.J.A & Khairul, F.K. A Unique Model to Accelerate Industry-Sponsored Research in Malaysia. Journal for Clinical Studies. 11(1), 24–27 (2019).

3. Identifying and selecting a clinical trial for your practice. J Oncol Pract. 2008 Jan;4(1):27-8. doi: 10.1200/JOP.0814602. PMID: 20859441; PMCID: PMC2793943.
4. Christian J, Dasgupta N, Jordan M, et al. Digital Health and Patient Registries: Today, Tomorrow, and the Future. In: Gliklich RE, Dreyer NA, Leavy MB, et al., editors. 21st Century Patient Registries: Registries for Evaluating Patient Outcomes: A User’s Guide: 3rd Edition, Addendum [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2018 Mar. 3. Available from:https://www.ncbi.nlm.nih.gov/books/NBK493822/
5. Buse JB, Austin CP, Johnston SC, Lewis-Hall F, March AN, Shore CK, Tenaerts P, Rutter JL. A framework for assessing clinical trial site readiness. J Clin Transl Sci. 2023 May 8;7(1):e151. doi: 10.1017/cts.2023.541. PMID: 37456265; PMCID: PMC10346039.
6. Cimino J, Braun C. Design a Clinical Research Protocol: Influence of RealWorld Setting. Healthcare (Basel). 2023 Aug 10;11(16):2254. doi: 10.3390/ healthcare11162254. PMID: 37628452; PMCID: PMC10454664.
7. M.Murad I.M, Johari, N.H, Kuppusamy V., and Yeoh J. CRM Study Coordinators –Making a Difference in Clinical TrialConduct in Malaysia, Journal for Clinical Studies Volume 15 Issue 1.
8. Roslan H.N.S, A. Muis N.S, and A. Rahman N.A Impact of Electronic Signature Towards Clinical Trial Agreements, Journal for Clinical Studies, Volume 14 Issue 6.
Nur Ain binti Amir

Nur Ain binti Amir graduated from Universiti Putra Malaysia with a Bachelor of Science in Cell and Molecular Biology. She is currently pursuing a master’s degree in Neuroscience from the same institute. She holds the position of Feasibility Specialist at Clinical Research Malaysia. In this role, she collaborates with doctors, pharmaceutical companies, and Clinical Research Organisations to conduct feasibility studies for clinical trials.
Email: nur.ain@clinicalresearch.my
Shu Hui, Cheng

Shu Hui is currently a Senior Feasibility Specialist at Clinical Research Malaysia. She has her bachelor’s degree with honours in Medical Biotechnology from both Sunway University Malaysia and Lancaster University UK. She has the experience in medical diagnostic as well as research and development field.
Liew Eu, Koh

Liew Eu graduated from Universiti Sultan Zainal Abidin with a Bachelor of Animal Production and Health (Hons) and later went on to obtain her master’s degree in Biomedical Science from Universiti Sains Malaysia. She had experience in the diagnostic lab before joining Clinical Research Malaysia and now she works as a feasibility specialist at CRM.
Nur Aziemah, Ab.Ghani

Nur Aziemah is currently working as a feasibility specialist at Clinic Research Malaysia. She has experience working in molecular microbiology wet lab research, before working as a study coordinator in clinical trials. She earned her bachelor's degree with honours in microbial biotechnology from The University of Queensland.
Journal for Clinical Studies 11 www.journalforclinicalstudies.com
Pages
Watch
Five Trends Shaping the Trial Master File
The trial master file is integral to demonstrating that a clinical trial has been conducted in accordance with good clinical practice (GCP). The sponsor must therefore ensure their TMF is complete, timely, and accurate at all stages of a trial and across the document lifecycle.
Managing the TMF, however, has often been a challenge for organizations due to resource shortages and document owners not having enough experience with the TMF. Increasingly, artificial intelligence is playing an integral role in improving management and oversight of the TMF, which will have hugely beneficial implications for industry.
Indeed, AI is already shaking up TMF inspection readiness and TMF health in many different ways. As adoption of AI grows and as the sophistication of AI models expands, several key trends will gain traction as companies seek to ensure they meet compliance requirements with the TMF.
Trend One: TMF Health is Becoming a Greater Priority Regulatory agencies are becoming more interested in the both the TMF itself and the processes around it, and industry is starting to see more guidelines around how their documents and data are stored.1 They are not only looking at what is in the TMF but also how it gets there, how it's managed on an ongoing basis, and its oversight.
In the past, inspectors focused mostly on ensuring the documentation was in the TMF. However, that led to organizations acting reactively, getting their documentation uploaded and doing a huge push on quality control only when they found out they were having an inspection. That undoubtedly became more and more apparent to the regulators during their inspections and audits, and increasingly they are emphasizing the importance of having the TMF in a good state at all times.
As a result, inspections now look more at the processes and workflows, requiring companies to demonstrate that they are uploading their documents in a timely fashion and that they are keeping track of what needs to be in the TMF.2
AI can help to get companies inspection ready, speed up the preparation process and improve their TMF health, particularly if they are resource constrained.
Trend two: Digital disruption, including AI, is shaking up the TMF Having AI solutions focused on the TMF is new to the industry, but there are growing options for using automation and AI to solve different problems. One example is document classification. Using AI to help classify your documents saves time for everyone involved in the TMF and helps those document owners that are less familiar with the filing structure.
From a TMF health perspective, therefore, AI will help to reduce misfiles, which is key when inspectors conduct an audit of the TMF, because misfiles will raise the risk of inspection findings. In fact, our internally generated data shows that between 9% and 12% of quality issues are due to documents being filed in the wrong place.
Another area where AI can come to the fore is with ongoing periodic review to ensure documents are organised and complete. While this is not something that is in use in production environments yet, many of the existing AI capabilities will be able to be utilized to enable this. With metadata extraction being combined with identifying document relationships, an AI solution will be able to proactively check the completeness of the TMF and identify gaps. AI also can shake up risk-based QC by doing the additional check on documents that are easier to classify, reducing the need to do that QC manually.
Trend Three: AI Lets Smaller Organizations Ensure they have a Healthy, Inspection-ready TMF Smaller and mid-size companies that we work with typically struggle to determine which documents to QC, because they don’t have the human resources to manage all the TMF documents. The option open to those companies has, until more recently, simply been to focus their limited resources on catching the biggest problems, knowing that other problems will slip through the cracks.
But when you have documents that will not have any oversight, it does raise concerns. For example, the document owners who filed them might not be very familiar with the TMF and might have misfiled those documents. If that occurs, the company may face an inspection finding.
AI can help to prevent some of these issues from occurring by providing document owners with proper classification suggestions when they are uploading their documents, and therefore this is one area they won’t have to QC later on.
Next, having automated QC checking for non-key documents means that at least those documents will get some oversight, given there just aren’t the human resources for those companies to review everything. Automation or AI can look for quality issues such as missing pages and carry out classification and other metadata checks.
If you can automate some processes, it will contribute to improving your TMF health without having to increase your staffing levels.
Trend Four: AI Places a Different Emphasis on Personnel Roles with the TMF
Having AI manage many of the less strategic processes could potentially revolutionize the role of the TMF expert. For example, rather than focusing their time on conducting quality control on
12 Journal for Clinical Studies Volume 16 Issue 1 Regulatory
documents, they could potentially use the time to engage with their clinical research organizations and improve the CRO process.
Another area where the TMF team could redirect their time and effort is in improving site engagement, understanding what worked, what didn't, what sites they would want to work with again and which principal investigators they would want to work with again.
Having AI manage mundane tasks would allow TMF experts to leverage their skills in different areas, expand their roles and potentially enjoy a positive career path within the TMF team, rather than spending all their time conducting document quality control.
While the issue of staff cuts is contentious and creates concerns about AI taking over jobs, there is no indication that AI would take over strategic roles. However, it does offer companies an opportunity to either cut some costs on less strategic roles or at least not to have to invest in more staffing to manage QC activities.
As an example, we've relatively recently introduced AI to enable our services team to better support clients. That has resulted in some job cuts, but, more specifically, it has allowed the services team to focus on more strategic support.
Ultimately, AI can help to make TMF teams more efficient while continuing to ensure the quality of the TMF. For those smaller companies that have had to balance where to focus their QC, AI helps to improve the overall quality and allows the team to focus on more strategic activities that will improve the TMF health.
Trend Five: AI will Become Your TMF Early-warning System
One important aspect of the TMF is what is referred to as periodic QC or completeness quality checking. This process is something that all TMF teams should be doing and it’s something regulatory agencies look at. By way of example, in its guidance on good clinical practice for clinical trials the UK’s Medicines and Healthcare products Regulatory Agency (MHRA) notes: “The complete TMF is the basis for inspection and all the documents in it must be made available to the inspectors.”3
The problem is that you have documents coming from different sources. Often those documents either indicate that there's another document necessary or it indicates that there's data that needs to be added into the system as a result of content of that document.
This might be site documents such as the site delegation log or very often the Form FDA 1572, which includes the list of investigatorsiv. Often that 1572 will look fine when it’s put into the TMF. However, what is often overlooked is ensuring all information about investigators in the trial was captured, including their CV, their professional license, a financial disclosure form, and training documentation. Part of the periodic completeness QC involves going through that document to ensure that information on each investigator listed in the document is captured in the TMF.
This is very manual, labor-intensive work since it requires having people look through all those documents. When you're talking about big studies, or companies that are running lots of studies, typically these completeness checks are only carried out on each site once a year. That means if there is an issue, it will be some time before that problem is noticed.
Having AI review these documents to check what is missing would offer companies a real-time early-warning system that information about an investigator was missing from the TMF, or, by way of another example, that documents from a protocol amendment are missing.

Eventually having AI conduct these types of checks and provide the TMF team with a to-do list to complete the TMF would allow companies to do away with periodic quality control checks.
While AI is not yet able to manage these more sophisticated QC checks, it is where the technology is heading and ultimately will give companies far better oversight of their TMF.
An Essential TMF companion
AI is becoming an important tool for good TMF health. As AI algorithms become more accurate and sophisticated, there is greater potential for AI to take over many of the mundane tasks that currently require TMF teams to manage, allowing those teams to focus on strategic activities and at the same time making sure the TMF is complete and inspection ready.
The contents of this article are solely the opinion of the author and do not represent the opinions of PharmaLex GmbH or its parent Cencora. PharmaLex and Cencora strongly encourage readers to review the references provided with this article and all available information related to the topics mentioned herein and to rely on their own experience and expertise in making decisions related thereto.
REFERENCES
1. Guideline on the content, management and archiving of the clinical trial master file (paper and/or electronic), EMA, 2018. https://www.ema.europa. eu/en/documents/scientific-guideline/guideline-content-managementarchiving-clinical-trial-master-file-paper/electronic_en.pdf
2. GCP Inspections: Expectations and the dos and don’ts for hosting, MHRA Inspectorate, March 2020. https://mhrainspectorate.blog.gov. uk/2020/03/10/gcp-inspections-expectations-and-the-dos-and-dontsfor-hosting/
3. Guidance: Good clinical practice for clinical trials, MHRA, updated April 2023. https://www.gov.uk/guidance/good-clinical-practice-for-clinicaltrials
4. Information Sheet Guidance for Sponsors, Clinical Investigators, and IRBs: Frequently Asked Questions – Statement of Investigator (Form FDA 1572), FDA, May 2010. https://www.fda.gov/media/78830/download
Aaron Grant

Aaron Grant is VP of Solutions Consulting at Phlexglobal, a PharmaLex company, where he is focused on the development of new solutions and helping clients to solve challenges through a mix of people, process and technology. Aaron has been innovating in the life science software for over 15 years and prior to joining Phlexglobal, he held various positions at PAREXEL/Liquent and Siemens Medical.
Journal for Clinical Studies 13 www.journalforclinicalstudies.com Regulatory

Generative AI Drives Efficient Regulatory Processes
As regulatory Agencies around the world continue to refine and update their requirements, artificial intelligence (AI) can help Life Sciences companies increase their chances of new products being accepted promptly. ArisGlobal’s Agnes Cwienczek & Renato Rjavec describe how AI, and Generative AI in particular, are distilling insights from regulatory exchanges and harnessing the latest global Regulatory intelligence to anticipate and respond to Agency requirements.
As life sciences companies’ investments in Regulatory data capture, enrichment and exchange continue to rise in line with Agency mandates, the potential scope for artificial intelligence (AI) and Generative AI in particular seems limitless in its promise to transform labour-intensive processes while maintaining high accuracy. But where could this technology deliver tangible everyday benefits?
One emerging example is in capturing, searching and distilling insights from across companies’ global regulatory exchanges, going back several years. Here, Regulatory Affairs teams are able to preempt Agency queries in their initial submissions and/or when trying to provide consistent replies to questions globally – thereby increasing their chances of new products or clinical trials, or changes thereto, being accepted quickly, and first time. Another is in making prompter and more value-added use of Regulatory intelligence. That’s as Agencies around the world continue to refine and update their requirements, with a bearing on development, marketing approval application and license maintenance processes. These highly tangible use cases, which are characterised by intense work volumes, are already being transformed by AI today.
In the case of Agency queries and information exchanges, all Life Sciences companies must capture and process high volume of interactions around the world, as follow-ups to initial authorisation applications or as part of registration maintenance. Additional or updated information may be required, concerns might be raised, or the query might seek specific clarification, for instance.
Tracking Agency Interactions
Keeping track of these interactions, wherever they might take place across the global organisation, can be a logistical challenge. Correspondence could come in different formats and languages, and be channelled via email, web form, letter, or phone call. If local affiliates are involved, those stakeholders may be juggling query resolution alongside a host of other regulatory tasks, so that manual intake and processing of each Agency enquiry presents a significant burden for them. Given that there is only a short window to respond to Agency queries, potential administrative backlogs can present a risk to authorisation.
On top of this, there is a further risk of inefficiency and inconsistency, as well as omission, if queries and their resolution are not recorded centrally. Ideally, all Regulatory exchanges should be routinely and systematically recorded and logged. There should be proof of their handling and any associated outcomes and, crucially,
the knowledge (of the additional input that was needed, and how that query was answered) can be fed into Regulatory submissions –ideally heading off similar queries in the future, and accelerating the approvals process.
Unless Agency interactions are stored as a searchable record within the central company regulatory information management (RIM) system, and unless everyone (including multi-tasking teams within local affiliates) has access to this, authorisations could be significantly delayed or their long-term maintenance at risk.
Intelligent automation in this use case involves scanning incoming queries and associated responses for metadata, and making that information searchable and reusable so that the responses can be pre-emptively added to future applications.
New Drug Application Case Study
Take the example of a response from the US FDA to a certain New Drug Application (NDA) for the registration of several new products. That letter is likely to contain a number of data points that could be captured as metadata within the RIM record. An optimised AI tool, overseen by a professional, could process all of this, to import, process, verify, and record the relevant knowledge – first classifying the correspondence (e.g. that it signals a deficiency in the application), who it’s from, the relevant application number, and how soon a response is needed, then generating a summary of all of this (using Generative AI) and relating this to other applicable registration processes.
Associated alerts are triggered automatically, meanwhile trend analysis can be drawn from across all Regulatory exchanges, to drive better Regulatory applications next time around. As a final performance-boosting stage, Generative AI can be used to collate and re-use accepted content from positive responses. Although a human review layer remains vital, AI acts as a significant process accelerator and productivity tool. Real applications of the technology in this context to date, across a sample of 23 different languages, have extracted up to 12 fields of data, with 90% accuracy, 80% faster processing, and three times fewer handovers - and with no need for AI ‘training’ thanks to the way generative AI ‘learns’ from vast amounts of data1. In short, it’s making a huge and tangible difference to busy teams’ working lives, while boosting overall output and efficiency.
Keeping Abreast of International Regulatory Intelligence
Linked to the transformation of health authority exchanges is another use case with similarly significant potential. This involves the tracking and proactive harnessing of the latest global Regulatory intelligence to improve the first-time success, and speed of approval of, development or marketing authorization applications.
Still, today, health authorities distribute news about updates to regulatory requirements in hugely diverse ways, ranging from web site posts to postal or email circulars. Again, these are likely to appear in local languages. Keeping track of all of the changes, internationally, can feel impossible – again, especially if this relies on overstretched local affiliates updating a central repository with the latest developments in their country. China’s eCTD implementation
14 Journal for Clinical Studies Volume 16 Issue 1
Regulatory

guide, for instance, contains important information on how to compile, publish and submit eCTD sequences to the National Medical Products Administration (NMPA), the Chinese Agency for regulating drugs and medical devices. The guidance is provided in simplified Chinese.
Optimised AI tools offer an important solution here. That’s not only in monitoring for emerging updates, in any language and format, and via any channel, or capturing this intelligence centrally; but also in proactively flagging the impact of those considerations and exposing it for further consumption – via the global organisation’s central RIM system, and alerts to the relevant stakeholders.
Meanwhile, as Regulatory teams compile new content for health authorities, they can ensure that the proposed new requirements are factored in from the outset, by simply asking their RIM system’s GenAI-enabled chatbot a direct question about the latest relevant requirements. To avoid any concerns about AI getting it wrong, human team members are encouraged to validate any Regulatory updates, while the GenAI chatbot provides complete links to its information sources (even down to the specific document, and page number), so that the findings can be verified.
Initial pilots have again yielded 90% accuracy, and 80% faster processing, with half the handovers of manual Regulatory intelligence lookup and implementation. Improved efficiency in capturing, processing and distribution of regulatory intelligence. Direct benefits include reduced risk of non-compliance with new or updated regulations, and improved clarity and faster decision-making thanks to automated summarization, translation and analytics.
The Tip of The Iceberg
These Regulatory use cases represent merely a glimpse of the vast possibilities within a discipline that is meticulously detail-oriented and constantly evolving. The logical progression involves Regulatory AI tools recommending enhancements to submissions while they are still in development. This will be based on automated lookups of past Agency correspondence and the latest Regulatory intelligence.
The intersection of artificial intelligence (AI) with the field of Life Sciences has massive potential. True transformation will emerge from the implementation of specific, well-defined use cases that directly tackle the everyday hurdles encountered in Regulatory
REFERENCES
1. This is ArisGlobal’s own data from early customer pilots. Separately, McKinsey estimates that deploying next-generation AI to improve HA responses and their impact can reduce Agency follow-up by 50%.
Agnes Cwienczek

Agnes Cwienczek is Director of Product Management at ArisGlobal, with a remit including the provision of business process and data management expertise in the areas of Regulatory Information Management, Document Management, Submission Management and Labelling Management. Agnes has previously worked at Merck in its Global Regulatory and Quality Assurance department, a milestone in a career spanning nearly two decades at the frontline of regulatory information management.
Email: acwienczek@arisglobal.com
Renato Rjavec

Renato Rjavec is Senior Director of Product Management at ArisGlobal, where he is shaping the future of regulatory information management as well as quality management for Life Sciences, with a keen focus on AI as a means for targeted automation of critical but labour-intensive processes where accuracy and precision are paramount. Renato has almost two decades of experience in ideation, development and implementation of regulatory and quality solutions for the Life Sciences industry.
Email: rrjavec@arisglobal.com
Journal for Clinical Studies 15 www.journalforclinicalstudies.com
Regulatory
Affairs. These targeted applications have the potential to catalyse significant advancements, revolutionising how regulatory processes are managed and enhancing overall efficiency and effectiveness in the Life Sciences sector.
Green Miracles: Cannabis and Kratom's Potential for Healing and The Industry in Malaysia
Global Cannabis market size was valued at USD 14154.75 million in 2023 and is expected to expand at a CAGR of 17.17% during the forecast period, reaching USD 36619.44 million by 2031.1 While Kratom on the other hand valued at USD 1.87 Billion in 2023 and is expected to grow at a CAGR of 17.2 percent from 2024–2030 to reach USD 5.69 Billion based on MMR.2 Cannabis and kratom have emerged as subjects of increasing interest due to their potential industrial applications and therapeutic properties. As the global perception of these substances evolves, exploring their significance in Malaysia's context is vital. Furthermore, as Malaysia continues to diversify its economy and explore new avenues for growth, tapping into the expanding global cannabis and kratom markets presents an opportunity for economic expansion and job creation. This writing aims to delve into the industrial strategical way and potential of cannabis and kratom, and their role in advancing treatments within Malaysia.
Kratom, an herb native to Southeast Asia, including Malaysia, presents promising industrial prospects. Its alkaloids, particularly mitragynine, have drawn attention to their potential pharmaceutical applications. The herb’s properties show promise in herbal supplements and cosmetics, potentially expanding Malaysia’s industrial landscape. Cultivating kratom could not only contribute to economic growth but also foster a thriving domestic industry. The therapeutic potential of cannabis and kratom extends beyond their industrial uses. Cannabis derivatives, notably CBD (cannabidiol), exhibit promise in managing various medical conditions. From alleviating chronic pain to assisting in anxiety and epilepsy treatment,3 ongoing research showcases its potential as a therapeutic agent. Similarly, kratom's alkaloids have been studied for their potential in managing pain and aiding in opioid addiction treatment.4,5 This is supported by a clinical trial conducted back in 2020 which reported significant paint tolerance increased 1 hour after kratom ingestion.6 Exploring these treatments could revolutionise healthcare practices in Malaysia.
According to data from clinicaltrial.gov website, in 2023 as per portrayed in Figure 1, America led in conducting trials on cannabis and kratom, followed by Europe, Oceania and Asia, which showed the least activity in this area. The clinical trial data from the platform indicates that the most prevalent condition being studied in relation to cannabis and kratom is pain, underscoring the significance of research in addressing this area of medical need. This emphasizes the importance for Malaysia to actively engage in research initiatives exploring the potential therapeutic effects of cannabis and kratom, particularly in addressing pain management within the local population. In addition to its potential in pain management, research into the therapeutic effects of cannabis and kratom has also shown promising results in addressing other medical conditions such as anxiety, depression and neurological disorders.
In order for Malaysia to effectively utilise the industrial potential and therapeutic advancements associated with cannabis and kratom, it is crucial to adopt an approach that is both informed and regulated. Establishing a comprehensive regulatory framework to govern cultivation, processing, and use is crucial. Furthermore, dedicated research initiatives, collaborations, and funding are necessary to explore these substances' potential fully. In 2021, the contentious use of certain drug ingredients was brought to the forefront by the former Health Minister, Mr Khairy Jamaludin. He emphasized the potential of cannabis and kratom, highlighting that these plants are locally accessible yet not fully utilised. Consequently, NPRA approved cannabis, kratom, and related ingredients for clinical trial use, as reported in the news in 2021. However, this approval was limited solely to end products. Table 1 shows a list of clinical studies in Malaysia on cannabis and kratom, indicating that the number of these trials should be increased in the future to align with market demand. Moreover, Malaysia’s diverse population and rich cultural heritage present unique opportunities for conducting ethnopharmacological studies on cannabis and kratom. Ny collaborating with local communities and traditional healers, Malaysia can leverage indigenous knowledge to inform scientific research and develop culturally sensitive healthcare
16 Journal for Clinical Studies Volume 16 Issue 1 Market Report
Figure 1: Numbers of studies in regions that conducted trial on cannabis and kratom in 2023 based on clinicaltrial.gov
Figure 2: Numbers of condition in clinical trial conduct on cannabis and kratom in 2023 based on clinicaltrial.gov
behavior syndrome abuse hiv mental adolescent anxiety drug use cannabis disorder disorders drinking health alcohol chronic substance pain post driving physical depressive substance-related sexual cancer dependence coronary musculoskeletal disease tobacco related healthy parenting neuropathic depression addiction brain risk opioid smoking stress injury back diseases thc peripheral
interventions. This approach not only enhances the relevance and applicability of research findings but also fosters inclusivity and equity in healthcare delivery.
including players in the clinical trial and pharmaceutical industries, along with healthcare authorities, should collaborate to turn this vision into reality.
REFERENCES
1. Global “Cannabis Market” Insight 2024
2. Kratom Market – Industry Analysis and Forecast (2024-2030)
3. Burke DJ, Mahonski SG, Van Cott AC. Breakthrough Seizure Associated With Kratom Use in Patients With Epilepsy. Neurol Clin Pract. 2021 Feb;11(1):78-84. doi: 10.1212/CPJ.0000000000000846. PMID: 33968476; PMCID: PMC8101317.
To initiate awareness in Malaysia, similar strategic avenues as Thailand could be pursued; Firstly, the exploration of legislative amendments to permit controlled medical research and supervised usage of cannabis and kratom under stringent regulations.7 Subsequently, the establishment of a consumption ensures strict adherence to quality and safety benchmarks. Encouraging collaborative research among institutions, universities, and medical entities to conduct comprehensive studies on the therapeutic potentials of cannabis and kratom. In addition, invest in developing a supportive healthcare infrastructure equipped to handle the med policy. Lastly, the launch of comprehensive public education campaigns aimed at emphasizing both the advantages and risks associated with these substances, engaging healthcare professionals, policymakers, and the wider public to facilitate wellinformed discussions.
Conclusion, cannabis and kratom hold immense potential in Malaysia, not only industrially but also in healthcare advancements. Exploring their significance requires a balanced approach that leverages their industrial utility while ensuring responsible usage in treatment. Malaysia stands poised to benefit from these resources by fostering innovation, economic growth, and advancements in healthcare. Therefore, stakeholders and authorities must materialise this concept to ensure the regulation and exploration of the potential ‘gems’ within Malaysia, particularly the underutilised valuable local herbs. Malaysia has recently explored collaboration with Thailand on this matter, and now various stakeholders,
4. Hossain R, Sultana A, Nuinoon M, Noonong K, Tangpong J, Hossain KH, Rahman MA. A Critical Review of the Neuropharmacological Effects of Kratom: An Insight from the Functional Array of Identified Natural Compounds. Molecules. 2023 Oct 31;28(21):7372. doi: 10.3390/ molecules28217372. PMID: 37959790; PMCID: PMC10648626.
5. Ismail I, Wahab S, Sidi H, Das S, Lin LJ, Razali R. Kratom and Future Treatment for the Opioid Addiction and Chronic Pain: Periculo Beneficium? Curr Drug Targets. 2019;20(2):166-172. doi: 10.2174/13894501 18666170425154120. PMID: 28443503.
6. Vicknasingam B, Chooi WT, Rahim AA, Ramachandram D, Singh D, Ramanathan S, Yusof NSM, Zainal H, Murugaiyah V, Gueorguieva R, Mansor SM, Chawarski MC. Kratom and Pain Tolerance: A Randomized, Placebo-Controlled, Double-Blind Study. Yale J Biol Med. 2020 Jun 29;93(2):229-238. PMID: 32607084; PMCID: PMC7309661.
7. Baratta F, Pignata I, Ravetto Enri L and Brusa P (2022) Cannabis for Medical Use: Analysis of Recent Clinical Trials in View of Current Legislation. Front. Pharmacol. 13:888903. doi: 10.3389/fphar.2022.888903
Nur Ain binti Amir
Nur Ain is a postgraduate with master’s degree in Neuroscience from Universiti Putra Malaysia. Her project of studies in headache and genetic studies related to migraine. She is experienced with clinical trial and wide in molecular study.
Email: ainamir136@gmail.com
Journal for Clinical Studies 17 www.journalforclinicalstudies.com Market Report
Table 1: List of Clinical Studies on Cannabis and Kratom conducted in Malaysia based on NMRR website and Clinical Trial.Gov
Number of Studies Type of Studies 3 Review 4 Observational 1 Case Report 1 Clinical Trial
Research and Development
Conducting Clinical Trials in the Parallel Virtual Universe
Quantitative systems pharmacology (QSP) and physiologically based pharmacokinetic (PBPK) modelling can be employed to create a parallel virtual workflow throughout drug development, beginning with preclinical research and continuing through phase IV clinical studies. Many clinical trials are conducted with virtual patients prior to selecting some trials to be carried out with real patients in real life. This is done in a reiterative fashion such that insights gleaned from the virtual studies are used to inform the actual clinical trial and vice versa. Thus, the results from the real clinical trial inform the next set of virtual trials and the iterations continue throughout the drug development process until the drug enters the market. At each stage, the workflows in virtual and real space have similar study objectives and endpoints. However, the virtual trials provide an economical alternative to conducting a wide range of studies with the aim of informing fewer optimal real trials. By taking advantage of events that have already happened in the virtual world, the clinical research can progress faster, and more predictably and reliably, which has huge economic implications.
When model-informed drug development (MIDD) was first introduced, some pharmaceutical executives thought that modelling clinical trials using virtual, computer-generated patients was higher risk than conducting real-life clinical trials because the results might not be accepted by the regulatory agencies. But now that the approach has been used for 15-20 years and shown to work well, precedent has been set, and that risk is greatly diminished.
MIDD is employed throughout the drug discovery and development process. It is used to help choose the best candidate molecules, select optimal drug doses, design clinical trials, inform
Virtual World
go/no-go decisions, and support regulatory approvals. In fact, global regulatory agencies now expect to see modelling and simulation included with new drug applications.
Drug development typically starts in the virtual world a few months or one year ahead of the real world. The arrows in Figure 1 show how information gleaned from the virtual clinical trials is used to design the next real trial. Then, when the real trial produces clinical results, they are used to optimise the next virtual trials. This feedback loop facilitates fine-tuning the clinical trial design, improving accuracy and reliability, and increasing confidence in the results throughout the drug development process.
Virtual clinical trials are based on mechanistic, physiologically based pharmacokinetic (PBPK) models, which focus on how the human body handles drugs, and quantitative systems pharmacology (QSP) models, which define the way drugs affect the body.1 A typical QSP model consists of a pharmacokinetic module, describing absorption, distribution, and elimination of the drug, connected to a systems biology model quantitatively describing the biology of the disease and mechanisms of drug action.2 All the available preclinical and clinical data are incorporated into these models in a very methodical way.
Each virtual clinical trial is also populated with a diverse participant cohort which closely resembles a real trial. Virtual participants of all ages, weights, genders, and ethnicities are included with different genetics, liver variations, and transporter and enzyme levels.
In Figure 1, the capsule in the virtual world is larger because there are many more studies being conducted there than in the real world. Virtual studies are run with many different doses and dosing schedules to determine the optimal approach to use in the real trial.


Actual World





18 Journal for Clinical Studies Volume 16 Issue 1
Figure 1: Exploring the Connections Between the Virtual and Real World
Investigating Drug Combinations
For example, if a patient has cancer and needs to receive a combination of drugs: What dose should they be given of each drug, in what order, and with what timing? Those decisions are complicated enough to make with two drugs but what if they need three? It is impossible to run all those clinical trials in real life, but all 20 or 30 permutations can be tested virtually. If researchers know the mechanism by which drug A works, and how drug B works, and the body clock regarding the circulation of the targets, how they come and go and resynthesize, they can determine by conducting virtual trials which drug to use first and second. Then, based on those results, the best options can be selected for investigation in clinical trials.
That was the approach we took with AstraZeneca’s COVID-19 vaccine to determine the right timing for their second booster shot. Scenarios were run using a four-, six- and eight-week delay in a large, virtual study. The optimal results of seven to eight weeks were confirmed six or seven months later in a two-arm clinical study. But if AstraZeneca had waited to receive the clinical trial results before deciding on the right timing, many more people would have died, or received unnecessary extra boosts when the cycle of production for the vaccine was not high enough to supply all patients even with their first injection.
Building a Virtual Clinical Trial
To be able to build a realistic virtual clinical trial, researchers need to have a model of the disease – an animal model or a microphysiological system (commonly known as an Organ-0n-aChip) – to use as a foundation on which to build. There also needs to be data available about the relevant enzymes, receptors, and targets in humans.
This is also an important approach to consider for orphan diseases where it may not be viable to conduct a clinical trial because there is little information available about the condition and few patients to enroll. While modelling without detailed disease knowledge is not an ideal scenario, it is often the best option to progress drug development for patients with rare diseases.
Making Virtual Trials Realistic
Consider a virtual clinical trial investigating the concentration time profile or blood pressure time profile of a drug. In a virtual trial, it is possible to obtain a sample every minute at a nominal cost. But in the real world, sampling might be scheduled to occur every hour for the first three hours, and then every four hours to provide 10 samples. But in the hospital, the nurse might be a few minutes late returning from getting a sandwich or coffee, resulting in the sample being taken at one hour and 10 minutes, rather than at one hour. Activities may not be as precise in the real world and those factors need to be incorporated as design elements into virtual clinical trials to make them more realistic. The simulation needs to incorporate some variability because the sample may be taken five minutes earlier or later than prescribed. The virtual clinical trials also need to include a small margin of error to account for an occasional mistake being made when recording a measurement or analysing an assay.
When MIDD first began, these adjustments were not made, but now a reality variation and a margin of error are incorporated into models to ensure that the virtual clinical trial results more closely resemble those gotten from a real clinical study.
Determining Statistical Power
As the cohorts included in virtual clinical trials are diverse, the study will not always produce a clear yes or no answer. Just like
a real clinical study, it will depend on how many participants are recruited.
But the advantage with a virtual trial is that the study can be run initially with 2,000 people to see what the result is. Then, that cohort can split into smaller groups – perhaps 10 trials of 100 people. If at the end of that study, eight of the trials show statistical advancement of the effect, but two of them are failing, the 100-person cohort gave 80% power. But if the response from five of those 100-person cohorts is yes, and five of them it is no, the chance is now 50%. Therefore, this approach can also predict the likelihood of success based on the size of the study. It can deliver insights such as, “Don’t run the study with 50 people, because the likelihood of success will only be 10%, and then you are just wasting everybody’s time.”
Conversely, the results may demonstrate that it is not necessary to run a study with 1,000 people at 97% confidence if it is possible to achieve the same result with 95% confidence using just 300 people.
Investigating Potential DDIs
Virtual trials can also help to determine the likelihood of drugdrug interactions (DDIs) occurring when patients are taking comedications. This is very important because it would not be practical or ethical to test all the potential drug combinations in real clinical trials.
To underscore the challenge involved: for a real clinical trial with 500 participants, at least 5% of them would need to be taking the specific co-medications to be able to detect a signal, even for the strongest DDI.7 Furthermore, there have been cases where a pharmaceutical company has decided not to take a drug forward because the risk of DDIs was considered too high to manage.
Reverse Translation
Virtual PBPK modelling also allows researchers to investigate scenarios that would be impossible to test in clinical studies, such as measuring toxicological or pharmacological responses to a drug in a human brain, kidney or fetus.
In the past, researchers used to “humanize” pre-clinical data by extrapolating directly from animals to humans, just using allometry to account for their difference in weight. But mice are not the same as humans! We now use an approach called reverse translation where we start with the animal data, then go back to determine which mechanisms created those responses, and forward to reproduce those effects in human form in a model by adding the relevant blood flows, receptors, and enzyme turnover.
For example, a PBPK model can be developed which can predict a drug’s nephrotoxicity risk in humans using in vivo study results from pre-clinical species and adding species-specific differences in renal physiology such as glomerular filtrate rate, blood flow and renal drug transporter expression/substrate affinity.4 Reverse translation is used to determine what drug doses and concentrations may cause some nephron-toxicity and must never be used in humans.
A similar approach can be taken to investigate fetal exposure to drugs travelling across the placenta. In this case, a feto-maternal PBPK model is developed using toxicity data from pre-clinical species combined with knowledge of the rapid changes in anatomy, biochemistry and physiology that occur as a fetus grows during pregnancy.5
Journal for Clinical Studies 19 www.journalforclinicalstudies.com Research and Development
Research and Development

Animal neurotoxicity/efficacy

Animal brain / CSF exposure
Pharmacodynamic/ toxicodynamic model
Predicted human neurotoxicity/efficacy

Animal systemic exposure
Brain 4-compartmental model within PBPK model
Estimated human brain / CSF exposure
In Figure 2, PBPK modelling is employed to estimate human brain tissue/cerebrospinal fluid (CSF) exposure to a drug and predict its pharmacological efficacy and neurotoxicity.6 Initially, the effectiveness of different drug doses is assessed in pre-clinical species and a pharmacodynamic/toxicodynamic model is used to determine the response-exposure relationship. Then a PBPK model of the human brain is used to factor in species differences such as CSF volume and flow rate and brain transporters.
The top right-side graph shows concentration versus time, and the top middle graph indicates temporal changes of effect. One reflects the physiological elements, which are determined by the enzymes, etc., and the other shows the pathways for the effect and the number of targets that are engaged. The effect of the drug dose over time can be seen, but instead of just looking at the end result, the focus is on linking it to the concentration at the site effect, resulting from engagement with the targets.
In the bottom row, the arrows are reversed because the model is being rebuilt based on knowledge of what happens in humans. In many cases, the mechanism is shared between humans and animals, hence it is assumed that they have the same engagement to targets. However, QSP models can incorporate the knowledge of variations in pathway to effects and the differences in the level of targets between animals and humans, which are obtained by analysis of relevant tissues and organs. Hence, the picture is just adjusted when humans have different numbers of those receptors.
Schizophrenia Case Study
But the conduct of successful virtual clinical trials, just like real clinical trials, requires medical researchers and modelers with extensive expertise. For example, when creating a virtual clinical trial to investigate smoking, the modelers needed to know not only how many cigarettes the participants were smoking, but also what type, and how deeply they were inhaling. That latter detail helped to determine whether the participants were keeping smoke in their lungs, which could cause enzyme induction.
While the association is unclear, people with schizophrenia tend to smoke a lot. A sponsor had a drug X to treat schizophrenia, which was metabolized by the hepatic enzyme cytochrome P450 1A2
Human systemic exposure
(CYP1A2), which is induced by smoking.9 But when those patients were hospitalised, they were not allowed to smoke, so the enzyme was de-induced and became less active. Therefore, if the patients were given their normal drug dose, it would intoxicate them, because the enzyme was not present to turn it over.
The researchers applied their knowledge of the pertinent metabolic pathway, and how much of it was going to be affected, to the model to determine the effect of smoking reduction on the drug. Then they determined how much each patient’s drug dose should be reduced, and on what day of their hospitalization that new dose should be applied. While a confirmatory clinical study was still required, their modelling identified the best clinical path, and removed the need for an exploratory clinical study. Their research also illustrated how, going forward, many smoking studies can be conducted as virtual studies.12
Valuable Resource
Underscoring the value of MIDD, Allerheiligen reported in a 2014 paper in Clinical Pharmacology and Therapeutics that during the previous three years the Modeling and Simulation group at Merck had “delivered more than half a billion dollars in cost avoidance [for studies not conducted], and it continues to enable approximately 10 critical decisions per year.”3
Conclusion
Virtual clinical trials save time because they reduce the number of drug combinations and various conditions in which the drugs must be studied in actual clinical trials. They obviate the need for conducting many large studies (which take longer to recruit participants) by making the likelihood of success clear with smaller studies. They also help to ensure that a study is not initiated which is too small to answer the question posed and will need to be redone. While it is still necessary to conduct clinical studies, there are many instances in which a virtual clinical trial will produce the same answer, faster, and at a much lower cost.
REFERENCES
1. Rostami-Hodjegan A. Reverse Translation in PBPK and QSP: Going Backwards in Order to Go Forward With Confidence. Clin Pharmacol
20 Journal for Clinical Studies Volume 16 Issue 1
Figure 2: Prediction of Pharmacological Efficacy and Neurotoxicity Using Brain PBPK Modeling. Reprinted from the Journal of Neurochemistry.6
Research and Development

2. Giorgi M, Desikan R, van der Graaf PH, Kierzek AM. Application of quantitative systems pharmacology to guide the optimal dosing of COVID-19 vaccines. CPT Pharmacometrics Syst Pharmacol. 2021 Oct;10(10):1130-1133. doi: 10.1002/psp4.12700. Epub 2021 Aug 12. https://www.ncbi.nlm.nih.gov/ pmc/articles/PMC8420530/
3. Allerheiligen SR. Impact of Modeling and Simulation: Myth or Fact? Clin Pharmacol Ther. 2014 Oct;96(4):413-5. doi: 10.1038/clpt.2014.122. PMID: 25236663.
4. Scotcher D, Jones C, Posada M, Galetin A, Rostami-Hodjegan A. Key to Opening Kidney for In Vitro-In Vivo Extrapolation Entrance in Health and Disease: Part II: Mechanistic Models and In Vitro-In Vivo Extrapolation. The AAPS Journal, Vol. 18, No. 5, September 2016 (# 2016) DOI: 10.1208/s12248016-9959-1. https://link.springer.com/article/10.1208/s12248-016-9959-1
5. Abduljalil K, Johnson TN, Rostami-Hodjegan A. Fetal PhysiologicallyBased Pharmacokinetic Models: Systems Information on Fetal Biometry and Gross Composition. Clin Pharmacokinet. https://doi.org/10.1007/ s40262-017-0618-1.
6. Al Feteisi H, Al-Majdoub ZM, Achour B, Couto N, Rostami-Hodjegan A, Barber J. Identification and quantification of blood-brain barrier transporters in isolated rat brain microvessels. J Neurochem. 2018 Sep;146(6):670-685. doi: 10.1111/jnc.14446. Epub 2018 Aug 1. PMID: 29675872.
7. Johnson TN, Kerbusch T, Jones B, Tucker GT, Rostami-Hodjegan A, Milligan PA. Assessing the efficiency of mixed effects modelling in quantifying metabolism-based drug-drug interactions: using in vitro data as an aid to assess study power. Pharm Stat. 2009 Jul-Sep;8(3):186-202. doi: 10.1002/pst.373. PMID: 19291743.
8. Milligan PA, Brown MJ, Marchant B, Martin SW, van der Graaf PH, Benson N, Nucci G, Nichols DJ, Boyd RA, Mandema JW, Krishnaswami S, Zwillich S, Gruben D, Anziano RJ, Stock TC, Lalonde RL. Model-Based Drug Development: A Rational Approach to Efficiently Accelerate Drug Development. Clin Pharmacol Ther. 2013 Jun;93(6):502-14. doi: 10.1038/ clpt.2013.54. Epub 2013 Mar 14. PMID: 23588322.
9. Plowchalk, DR, Rowland Yeo K. Prediction of drug clearance in a smoking
population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Clin Pharmacol (2012) 68:951–960. DOI 10.1007/ s00228-011-1189-y
10. PricewaterhouseCoopers, “Pharma 2020: Virtual R&D Which path will you take?” https://www.pwc.com/gx/en/pharma-life-sciences/pdf/pharma 2020_virtualrd_final2.pdf
11. PricewaterhouseCoopers, “Pharma 2020 Series Executive Summary.” https://www.pwc.com/gx/en/pharma-life-sciences/pdf/pharma-execsummary.pdf
12. Hermann R, Rostami-Hodjegan A, Zhao P, Ragueneau-Majlessi I. Seeing what is behind the smokescreen: A systematic review of methodological aspects of smoking interaction studies over the last three decades and implications for future clinical trials. Clin Transl Sci. 2023 May;16(5):742758. doi: 10.1111/cts.13494. Epub 2023 Feb 22. PMID: 36752279;
13. Mostafa S, Rafizadeh R, Polasek TM, Bousman CA, Rostami-Hodjegan A, Stowe R, Carrion P, Sheffield LJ, Kirkpatrick CMJ. Virtual twins for modelinformed precision dosing of clozapine in patients with treatment-resistant schizophrenia. CPT: Pharmacometrics and Systems Pharmacology, 19 January 2024. https://doi.org/10.1002/psp4.13093
Amin Rostami-Hodjegan
Professor Amin Rostami-Hodjegan, PharmD, PhD, FCP, FAAPS, FJSSX, FBPhS, is the Senior Vice President of Research & Development and Chief Scientific Officer at Certara. Previously, he was co-founder of Simcyp Limited, a University of Sheffield spin-off which was acquired by Certara. Amin is also a Professor of Systems Pharmacology and the Director of the Centre for Applied Pharmacokinetic Research at the University of Manchester. Ther. 2018 Feb;103(2):224-232. doi: 10.1002/cpt.904. Epub 2017 Nov 9. PMID: 29023678; PMCID: PMC5813098.

Journal for Clinical Studies 21 www.journalforclinicalstudies.com

Advancing Research and Development of Multi-indication and Combination Therapies for Obesity
Obesity is commonly defined as excess fat accumulation that puts someone at a higher risk for adverse health outcomes.1 The simplicity of this definition can be misleading, because obesity’s pathophysiology is highly complex, and characterised by a multitude of interrelated factors, including metabolic and immune changes in response to energy surfeit; mechanisms such as oxidative stress, and local and systemic inflammatory responses; alterations in blood flow and perfusion; and the mechanical burden of weight. Together, these factors contribute to the morbidity and mortality of the many obesity-related conditions.
The interdependent pathophysiology of obesity and comorbid conditions are relevant to treatment efficacy and have informed an interdisciplinary approach to obesity treatment in recent years, including the development of single treatments for multiple indications and combination interventions. Indeed, in a 2023 ICON survey of more than 100 professionals engaged in obesity-related clinical research, only 37% reported studying obesity alone. The indications most commonly included in obesity-related research were reported to be diabetes (55%), metabolic disease (48%), mental health (39%) and cardiovascular disease (38%).
Multi-indication Trials
Historically, drug development proceeded one indication at a time, from diabetes to weight loss and MASH indications. But, encouraged by the recent approval of GLP-1 receptor agonists for obesity – in addition to type 2 diabetes, and an improved understanding of the shared pathophysiology of obesity and other comorbid conditions – that has begun to shift. In the ICON survey, 50% of respondents reported employing multi-indication studies in their obesity-related trials.
Innovative approaches to trial design are often needed to demonstrate a treatment's efficacy for multiple indications. Here, sponsors may benefit from the use of master protocols, which are constructed to include multiple sub-studies, which have an overarching set of procedures to improve efficiency, such as inclusion of a common screening protocol or the sharing of control subjects. Master protocols with a basket trial design are well suited for multiindication treatments across the obesity related spectrum of diseases or for subtypes of a single disease, which have a common molecular characteristic.2
The Need for Long-term Follow-up
In the ICON survey, 44% of respondents reported that long-term follow-up was the biggest challenge in obesity-related clinical trials, Longer-term studies are needed to determine treatment effects on clinical events linked to obesity-related comorbidities, including major adverse cardiovascular events (MACE), chronic kidney disease and the onset of diabetes. This information is crucial for assessing the
cost effectiveness of new treatments and will be required to compete with products that have proven outcome benefits.
Novo Nordisk’s SELECT trial provides an excellent example. The primary endpoint of SELECT – a double-blind, parallel-group study evaluating weekly injections of the GLP-1 receptor agonist Wegovy® (semaglutide) in non-diabetic patients with a history of cardiovascular disease – was the reduction of MACE. In August 2023, Novo Nordisk reported a 20% reduction in MACE compared to placebo, outpacing the reduction analysts had been expecting, and boosting Wegovy’s prospects clinically and financially.3
Long term outcomes trials are very costly and there is an imperative for creating efficiencies in their execution. High levels of retention and compliance contribute to efficiency. This can be driven by easing patient burden with decentralised and hybrid approaches to follow-up as ICON successfully demonstrated in a recent heart failure registrational trial.4 Furthermore, we have employed our tokenisation capabilities to protect against loss of endpoint data and also to allow for continued data collection after the trial completes. Of note in the ICON survey only 22% of respondents were following patients beyond five years in their obesity-related clinical studies.
Traditional time-to-first-event analysis may lack efficiency compared with alternative approaches that have the potential to reduce sample size.5 Many cardiovascular outcomes trials have been underpowered for confirmatory endpoints. The use of historic electronic medical record (EMR) data can be leveraged to inform sample size estimates to assure that studies are adequately powered including for the key secondary endpoints.
Combination Approaches to Obesity Treatment
New obesity therapies have to be developed in the context of adequate background lifestyle counselling.6 As an example of this in practice, ICON is incorporating digital platforms to assure consistent counselling across study sites. The value of enhanced behavioural therapies was reflected by survey respondents, the majority of whom felt that this was a promising pathway for future treatment. Evolving digital therapeutic devices have the potential to advance the efficacy of long-term lifestyle interventions.
Even with recent advancements in obesity therapies only 17% of survey respondents felt that single pharmaceutical targets – such as GLP-1 receptor agonists – would dominate future research efforts. Instead, most respondents (64%) felt that there would likely be a focus on combination treatments.
Drug combinations have the potential to optimise a more personalized treatment approach. For example, GLP-1 receptor agonists result in loss of both fat and lean mass. Loss of lean mass is a particular concern for older patients with sarcopenia.7 Here the addition of medications that preserve muscle mass may not only
22 Journal for Clinical Studies Volume 16 Issue 1 Therapeutics
improve the quality of weight loss but also enhance fat loss given the role of skeletal muscle in energy expenditure. Medications targeting new molecular pathways will provide additional opportunities to explore multidrug synergies.
Development Considerations for Combination Interventions
Sponsors wishing to develop combination therapies for obesity will face added complexity during treatment development. For one thing, they need to consider potential drug-drug interactions. As with multiindication treatments, sponsors looking to validate combination treatments will need to employ innovative clinical trial designs. Here, master protocols using a platform trial design – which can evaluate and compare multiple interventions for one indication through multiple trial arms – may prove invaluable. Platform trials are adaptable, allowing researchers to begin additional arms or evaluate the results of an arm while the trial is ongoing. Moreover, a platform trial allows a greater percentage of participants to receive treatment, since only one of several arms receives a placebo.
Obesity Treatments Continue to Evolve
In recent decades, obesity has become a global epidemic, and there is a growing need for more effective treatments of this condition. Already, an increased understanding of obesity’s complex pathophysiology has led to better treatments. However, continued efforts are needed, particularly in approaches to treatments that account for obesity as a multi-condition risk factor. These must be supported by strategic and innovative approaches to therapeutic development and clinical trials design.
REFERENCES
1. Purnell, Jonathan Q. “Definitions, Classification, and Epidemiology of Obesity.” Endotext, edited by Kenneth R. Feingold et al., MDText.com, Inc., 2000, http://www.ncbi.nlm.nih.gov/books/NBK279167/.
2. Rhythm Pharmaceuticals. “Rhythm Pharmaceuticals Presents New Data from Phase 2 Basket Study Showing Continued Weight Loss at Up to Nine Months in Patients with HET Obesity on Setmelanotide at ENDO 2021.” Press Release. March 2021. https://ir.rhythmtx.com/news-releases/news-releasedetails/rhythm-pharmaceuticals-presents-new-data-phase-2-basket-study
3. “SELECT: Semaglutide Reduces Risk of MACE in Adults With Overweight or Obesity.” American College of Cardiology, https://www.acc.org/Latestin-Cardiology/Articles/2023/08/10/14/29/http%3a%2f%2fwww.acc.
org%2fLatest-in-Cardiology%2fArticles%2f2023%2f08%2f10%2f14%2f29%2fS ELECT-Semaglutide-Reduces-Risk-of-MACE-in-Adults-With-Overweightor-Obesity. Accessed 22 Sept. 2023
4. Claggett BL. Et al. Quantifying Treatment Effects in Trials with Multiple Event-Time Outcomes. NEJM Evid 2022; 1 (10) https://evidence.nejm.org/ doi/full/10.1056/EVIDoa2200047
5. Spertus JA, et al. The SGLT2 inhibitor canagliflozin in heart failure: the CHIEF-HF remote, patient-centered randomized trial. Nat Med 2022 Apr;28(4):809-813. doi: 10.1038/s41591-022-01703-8. Epub 2022 Feb 28
6. Hall, Kevin D., and Scott Kahan. “Maintenance of Lost Weight and LongTerm Management of Obesity.” The Medical Clinics of North America, vol. 102, no. 1, Jan. 2018, pp. 183–97, https://doi.org/10.1016/j.mcna.2017.08.012.
7. Ji T, Li Y, Ma L. Sarcopenic Obesity: An Emerging Public Health Problem. Aging Dis. 2022;13(2):379-388. Published 2022 Apr 1. doi:10.14336/ AD.2021.1006
Dr. Jack L. Martin

Dr. Martin, MD, FACC, is board certified in Cardiovascular Diseases and Interventional Cardiology. He has over 35 years of clinical practice and investigational experience. Jack is an experienced consultant for pharmaceutical and medical device companies. This includes all phases of product development including device design, trial design, FDA pre-sub and panel meetings. Dr. Martin has served as study chairman or the coordinating investigator for multiple multicenter international pharmaceutical and device trials. His previous roles included Assistant Professor of Medicine, University of Pennsylvania School of Medicine, Philadelphia, Chief, Division of Cardiovascular Diseases and Chief of Interventional Cardiology, Health System. He has served as President and a Board Member of several research foundations and is a respected educator having served as an Interventional Cardiology Fellowship Program Director. He has numerous peer-reviewed publications, is an active journal reviewer and has been a frequent invited speaker at national and international professional conferences. While at ICON, Jack has provided medical oversight for numerous cardiometabolic studies and has focused on cross functional team building to provide novel solutions for the effective delivery of drug and device trials.
Journal for Clinical Studies 23 www.journalforclinicalstudies.com Therapeutics
Therapeutics
Defining Success in Rare Disease Paediatric Trials
Rare diseases are often chronic conditions that begin to manifest during childhood.1 The effects of disease change everything from direct consequences of having the condition to impact on the child’s growth patterns and development, behaviour, psychology, self-worth, and self-belief. All these alongside the untold impact on the families who support them. At the current time there are very few treatments available for rare conditions, but even with supportive therapies, the earlier that medical and supportive intervention takes place, the bigger the impact because it not only helps to slow the disease, but also gives the child the opportunity to mature and integrate into diverse friendship groups for longer. This is why paediatric trials in rare disease are so important; they help to develop the treatments of the future and by targeting treatment of pediatric patients, they also provide the chance to live a positive childhood with as much normal development as possible.
Every individual is affected differently by the condition and therefore mainstream trial design, analysis, and real-world data methods are suboptimal. Collaboration between all stakeholders are essential for studies that are feasible to run and treatments that matter most to those who take them.
In this whitepaper we discuss the complexities of agreeing the definitions and approaches used to measure trial success, that are meaningful for all involved. Experts might imply we are missing out the family/patient voice.
Success Means Different Things to Different People
Clinical trial results need to give people enough information to guide treatment decisions. Two key trial terms that are routinely discussed are:


When combined, these terms help to define what to look for and how to record change, as well as the degree of change that is important.
Trials must characterise both the benefits and risks of a new treatment. Working out what potential benefits are possible and then which are most important to measure is a complicated task. There are many people who have opinions on what should be chosen to study, and what level of change in this is meaningful. These include but are not limited to: (See Figure 1)
Traditionally two main factors have influenced what is chosen. Firstly, giving regulators what they expect to see when they review the drug at the end of development. This is important because if they don’t see something that they believe is valid then the drug will not be approved, and people will not be able to access it. Secondly, what clinical experts state is required to best tackle the disease. Again, this is logical. If the drug is not changing the disease, then it’s not working.
Both are essential considerations for any medicine – it needs to provide predictable results and the evidence showing this needs to be reliable and understandable. Trials need to go beyond this though. Children and their families live with the condition 24/7 full spectrum of the disease's consequences. They are in the perfect position to offer advice on which disease consequences matter most for improved quality of life, the amount of change that will make a meaningful difference and suggest ways to gather data to measure that change. Their insight goes beyond the clinical mechanism of action and its patients and their families, working with clinicians, that provide the essential linkage with real-world life.
The Traditional Approach
For years patient and caregiver quality of life was seen as ‘extra’ data to capture, and the ‘core’ trial results continued to be gathered unchanged.2 A comparison of clinical trials for rare disease with those for common diseases in the EU clinical trials register, showed that although many adaptations were made to trial design and delivery to make them more practical, things like primary endpoints and trial durations tended to stay the same.3 Information about patient or caregiver experiences and quality of life was instead gathered using add-on questionnaires called either PREMS (Patient Reported Experience Measures) or PROMS (Patient Reported Outcome Measures). These have now become a normal and essential component of nearly all trials and are used to capture patient perspectives and experiences within the trial. The focus of PREMS/PROMS is usually quality of life, daily functioning and symptoms.4
Traditional PREMS and PROMS have been created on the patients’ behalf by researchers and are very important because they add context and understanding around endpoints and can be used as part of the evidence given to regulators and payers when they make decisions about whether to approve and reimburse the drug.5
24 Journal for Clinical Studies Volume 16 Issue 1





Journal for Clinical Studies 25 www.journalforclinicalstudies.com
A few years ago, the International Rare Diseases Research Consortium (IRDiRC) taskforce focused on trying to make these measures more patient-centered. They concluded that multi-stakeholder input is required to develop and validate true Patient-Centered Outcome Measures (PCOMs). Their suggestion was to use mixed methods psychometric research to create PCOMs in rare diseases because it integrates different types of information and is tailored for small numbers of respondents.6 However, even after this initiative proposed a method for developing optimal PCOMs, a recent review found that such bespoke assessments tended to be used for understanding the realworld cost of illness for those involved, and quality of life assessments remained traditional.7 This has created a problem as over reliance on traditional quality of life assessments can overlook aspects of the condition meaningful to patients themselves. In addition, it’s trial endpoints that define success and these were not found to be changed or influenced by PREM or PROM approaches.
We Need to Do More
In 2020 a research group searched for evidence that patients were involved in defining success in primary and secondary clinical trial endpoints and concluded that, at most, there was very limited action.8
As described above, the definition of a trial’s success rests on whether the primary and secondary endpoints meet pre-defined targets. If this clinical target is not seen as relevant by patients, is unrealistic, or is bigger than that needed to give benefit to patients and families, then a drug can be rejected without patients ever even knowing that there was something with the potential to make their lives better. This has happened in the past for Duchenne Muscular Dystrophy (DMD) where, undertaking a six-minute walk test is an accepted and standard endpoint for trials. This test uses the distance that a person can walk in six minutes as a measure of performance capacity. This is less relevant for young men with Duchenne who have no desire to walk fast. A race for distance-covered is simply not meaningful when compared to loss
of independence. For a young man who has lost ambulation and relies on a powerchair, the ability to reach down the side of his chair and place his arm back on the arm rest may equate to selfsufficiency.
Regulatory authorities around the world have started to acknowledge this problem, with the FDA releasing guidance that is leading the charge. A few years ago, the FDA created a group called Patient-Focused Drug Development (PFDD) who have authored 4 guidance documents to date.9 These build up into a series and describe how to involve patients better in study design and the creation of more relevant, blended endpoints for trials. The latest guidance that has been released for comment, shows researchers how to begin to analyse results, define thresholds of success, and gather evidence that this change is meaningful to patients themselves.10,11 This outlines the expected approach for future trials.
As with every other step of this evolution – there isn’t one simple answer that fits perfectly. It’s a thought process, and every situation is different requiring a balance of the strengths and limitations of each possible way to measure change,12 but it can be achieved through collaboration between experts and solid research being gathered to underpin decisions and build the evidence base. Reflecting on her attendance at a recent World Orphan Drug Congress (WODC) conference to discuss rare disease endpoint advancement, Managing Director, Strategic Collaborations at Emmes' Rare Disease and Paediatric Center, Christine McSherry stated: “Research is developing personalised medicine through novel approaches, however we aren't using the same approach in our outcomes and endpoints. For research to evolve we need to reassess this for the good of patients”.
Action, Not Talk. Early-stage Development of New Measures
There are several examples of patient and research organisations working together to develop tools that can profile how a rare disease is affecting children as they grow. This is an essential starting point for any future endpoint creation as a robust understanding of the variation in how the disease affects each individual informs how measures can be created. One of these early assessment tools can be found in the rare condition Tuberous Sclerosis Complex (TSC).
26 Journal for Clinical Studies Volume 16 Issue 1
Figure 1
Therapeutics
TSC is a disease in which non-cancerous tumours begin to grow in the brain and elsewhere in the body. It is an incurable condition that is often first recognised in early childhood.
An individual’s development, functioning, and interaction with others is highly variable and very dependent on the symptoms of the disease that they experience. These symptoms often include seizures, delays in cognitive development, and behavioural issues such as aggression, attention deficit hyperactivity disorder, obsessivecompulsive disorder, and repetitive, destructive, or self-harming actions that can be difficult to manage. There is also a strong link with autism.
Behavioural problems associated with TSC often have a profound impact on both the person exhibiting the behaviour and those around them. In some cases, behaviour causes more daily concern and has a greater impact on ability for independent daily activities than other aspects of TSC.13 Yet very few trials of new medicines measure these effects or consider them when defining success.
questionnaire associated.16,17 The results are used within healthcare by consultants and families to create a mental health treatment plan for those with TSC and integrate this with their overall clinical care.
This is currently not a trial endpoint, but it is an example of one of the many initiatives to give medical communities a way of engaging in a structured conversation for the benefit of the patient, a method of collecting a consistent dataset of individualised profiles, and an agreed scientific wording for publishing and comparing results. This may also be of utility to families who may be able to recognise and understand characteristics of the condition as they arise during developmental milestones.
TAND is now being modified into a validated longitudinal measure for patients that has the potential to be even more useful for future study consideration. If used, this will mean that clinical trials will finally be able to measure the effects of future treatments across the TAND areas that are so important for children’s ability to communicate, interact, and be socially accepted as they grow.
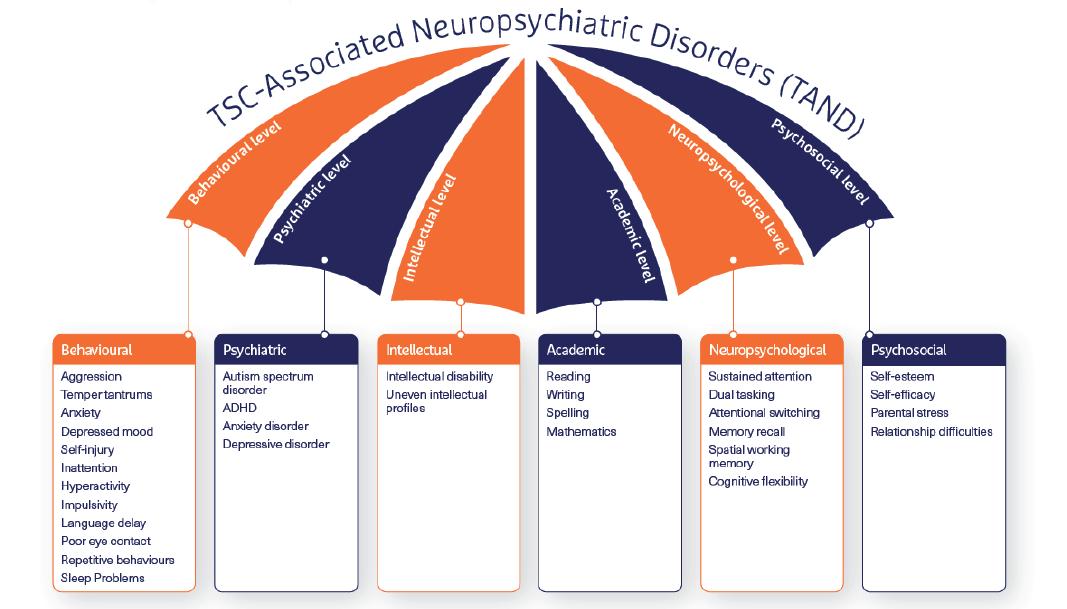
Recent pilot data supported the discussions that medical and patient family communities been having for a while. The earlier on a child receives the correct treatment the bigger the benefit on the way that young children learn to communicate as they grow.14 Further research is needed, but for those experiencing this, there is no time to wait.
An interesting initiative was started to produce a recognized classification chart, ‘TAND’ (TSC-Associated Neuropsychiatric Disorders). The inspiration behind the idea came from the HIV community where they produced a similar tool for increasing awareness and consistency in language around a poorly understood aspect of the disease.15
This TAND checklist is a screening tool represented by an umbrella covering several key domains, each of which lists out the main characteristics of disease impact and has a validated
Designing Endpoints that Matter
Emmes Endpoint Solutions (previously Casimir) is an example of a service validating such patient-centered tools to clinical trial standards. The company was founded by parents of children with Duchenne Muscular Dystrophy (DMD). This is a progressive condition caused by changes in some of the genes responsible for the structure and functioning of a person's muscles. As the disease progresses, the muscles' ability to function reduces, causing increasing disability and dependence on others.
The founders of Casimir were parents with children who took part in clinical studies for DMD. These trials were considered to have ‘failed’ because they didn’t meet their primary clinical endpoints. However, parents and children taking part saw life-altering improvements whilst on the trial drug. Something had to change and parents began to work with the FDA’s Center for Drug Evaluation Research (CDER) and clinicians to innovate.
Journal for Clinical Studies 27 www.journalforclinicalstudies.com
Figure 2
Therapeutics
Therapeutics
They used a video-based approach to characterise outcomes of importance to patients and families, then blended these with activities of daily living (ADLs) and assessments of functional ability to measure and quantify change.18
Casimir was born, and they started their own trial that subsequently demonstrated real and quantified improvements that transformed the lives of the boys taking part. Even more importantly, these measured changes were focused on areas of greatest relevance to the people affected and the endpoint measures accounted for differences in the way that disease presented during the growth of each child. Eteplirsen, a drug that had previously ‘failed’ in a trial for DMD, was then approved by the FDA as a supportive care therapy for use in patients with DMD. This is now available in the USA for others living with DMD to help them maintain meaningful activities for independence and socialisation.

Conclusion
As an industry we need to do more. In rare disease we can often improve lives most by intervening and developing therapies for children. This brings with it all the added complexities relating to paediatric studies as well as the need to adapt to the highly variable way that rare diseases may present during childhood.
As researchers we need to accept this challenge and change the way we do research to ensure that therapies address the underlying clinical condition and what will make the most meaningful difference for those affected. We are honoured to have Emmes Endpoint Solutions as part of our team. Together we are leading the way in patient-centered rare disease trials and embracing the urgent requirement for novel, robust, trial endpoints that measure the success that transforms lives.
REFERENCES
1. Gunne E, McGarvey C, Hamilton K, Treacy E, Lambert DM, Lynch SA. A retrospective review of the contribution of rare diseases to paediatric mortality in Ireland. Orphanet Journal of Rare Diseases. 2020 Dec;15(1):1-8.
2. Deal LS, Goldsmith JC, Martin S, Barbier AJ, Roberds SL, Schubert DH. Patient voice in rare disease drug development and endpoints. Therapeutic Innovation & Regulatory Science. 2017 Mar;51:257-63.
3. Logviss K, Krievins D, Purvina S. Characteristics of clinical trials in rare vs. common diseases: a register-based Latvian study. PLoS One. 2018 Apr 3;13(4):e0194494.
4. Slade A, Isa F, Kyte D, Pankhurst T, Kerecuk L, Ferguson J, Lipkin G, Calvert M. Patient reported outcome measures in rare diseases: a narrative review. Orphanet Journal of Rare Diseases. 2018 Dec;13:1-9.
5. Contesse MG, Valentine JE, Wall TE, Leffler MG. The case for the use of patient and caregiver perception of change assessments in rare disease clinical trials: a methodologic overview. Advances in Therapy. 2019 May 1;36:997-1010.
6. Morel T, Cano SJ. Measuring what matters to rare disease patients–reflections on the work by the IRDiRC taskforce on patient-centered outcome measures. Orphanet journal of rare diseases. 2017 Dec;12(1):1-3.
7. Delaye J, Cacciatore P, Kole A. Valuing the “burden” and impact of rare diseases: a scoping review. Frontiers in Pharmacology. 2022;13.
8. Lanar S, Acquadro C, Seaton J, Savre I, Arnould B. To what degree are orphan drugs patient-centered? A review of the current state of clinical research in rare diseases. Orphanet Journal of Rare Diseases. 2020 Dec;15(1):1-8.
9. Perfetto EM, Burke L, Oehrlein EM, Epstein RS. Patient-focused drug development: a new direction for collaboration. Medical care. 2015 Jan 1;53(1):9-17
10. Food and Drug Association. FDA Patient-Focused Drug Development Guidance Series for Enhancing the Incorporation of the Patient’s Voice in Medical Product Development and Regulatory Decision; April, 2023. Available from: https://www.fda.gov/drugs/development-approval-processdrugs/fda- patient-focused-drug-development-guidance-series-enhancingincorporation-patients-voice-medical
11. Food and Drug Association. Rare Disease Endpoint Advancement Pilot Program [Internet]; June, 2023. Available from: https://www.fda.gov/drugs/ development-resources/rare-disease-endpoint- advancement-pilot-program
12. Busner J, Pandina G, Domingo S, Berger AK, Acosta MT, Fisseha N, Horrigan J, Ivkovic J, Jacobson W, Revicki D, Villalta-Gil V. Clinician-and Patientreported Endpoints in CNS Orphan Drug Clinical Trials: ISCTM Position Paper on Best Practices for Endpoint
13. Massachusetts General Hospital. How TSC Affects Metal Health; Accessed June, 2023. Available from: https://www.massgeneral.org/neurology/tsc/ patient-education/how-tsc-affects-mental-health
14. McDonald NM, Hyde C, Choi AB, Gulsrud AC, Kasari C, Nelson III CA, Jeste SS. Improving developmental abilities in infants with tuberous sclerosis complex: a pilot behavioral intervention study. Infants and young children. 2020 Apr;33(2):108.
15. TANDem Empowering Families Through Technology. About TSC, About TAND; Accessed June, 2023. Available from: https://tandconsortium.org/ about/
16. Cervi F, Saletti V, Turner K, Peron A, Bulgheroni S, Taddei M, La Briola F, Canevini MP, Vignoli A. The TAND checklist: a useful screening tool in children with tuberous sclerosis and neurofibromatosis type 1. Orphanet journal of rare diseases. 2020 Dec;1
17. De Vries PJ, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, Dahlin M, D’Amato L, d’Augères GB, Ferreira JC, Feucht M. TSC-associated neuropsychiatric disorders (TAND): findings from the TOSCA natural history study. Orphanet journal of rare disease
18. Pandina G, Busner J, Horrigan JP, McSherry C, Bateman-House A, Pani L, Kando J. Implications of Paediatric Initiatives on CNS Drug Development for All Ages–2020 and Beyond: Second of Three Sets of Expanded Proceedings from the 2020 ISCTM Autumn Conference
Christine McSherry

Christine McSherry founded Jett Foundation in 2001 after her son was diagnosed with Duchenne Muscular Dystrophy. As Executive Director, she co-founded the International Duchenne Alliance, which has funded nearly $15 million in research. As a Patient Advocate, Christine advises on measuring more meaningful outcomes for clinical trial participants and caregivers in rare disease for rare disease organizationsand bio-pharmaceutical companies. In April 2016, Christine successfully presented the Jett Foundation's Patient Centered Outcomes report to the FDA's Peripheral and Central Nervous System Drugs Advisory Committee for the review of Exondys 51, marking a historic moment in FDA history. Christine’s experience and expertise led her to co-founded Casimir Trials in 2016 to utilize her experience with the first approved drug for Duchenne, Exondys 51, for the benefit of drug developers, patient organizations, and regulators. Casimir Trials specializes in rare disease studies, including pre-clinical and real-world evidence studies in nearly 20 indications, using remote applications. Acquired by Emmes Group in March 2022, Casimir now brings additional rare disease expertise and advocacy in partnership with Emmes' commitment to excellent clinical trial management.
28 Journal for Clinical Studies Volume 16 Issue 1


Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS

Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.

www.international-biopharma.com
PNP
Pharma
Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA’S DNA
Listen to industry experts on the latest in drug discovery, development, research, industry regulations and much more at Pharma,s DNA, the podcast channel by Senglobal Ltd., available on Sound Cloud, Spotify, iTunes and YouTube.
senglobalcoms.com



Therapeutics
Cell and Gene Therapies are set to Revolutionise Healthcare: Here’s How
The first chemotherapy drugs were discovered by mistake –during World War II – when humans were accidentally exposed to mustard gas. At that time, the only other treatments for cancer were radiotherapy and surgery. But these weren’t necessarily promising for the treatment of all forms of cancer, especially cancers that had progressed.
Scientists were recruited to study the symptoms of people that were exposed to copious amounts of mustard gas. Based on their research, mustard gas agents were transformed into chemotherapy drugs used for the treatment of cancers. Chemotherapy is still the standard first line of cancer therapy for many people with cancer. Because chemotherapy doesn’t only target cancer cells, they are associated with many side effects including diarrhoea and hair loss.
But with a focus on patient centricity, the life science industry as we know it is changing.
Over the last couple of years, we have seen personalized medicine take the centre stage. One particular class of products, called Advanced Therapeutic Medicinal Products (or ATMPs, for short), is gaining a lot of interest from scientists, doctors, researchers, investors, and patients, alike. Put simply, these therapies utilize cells, tissues, and/or gene approaches to develop products that can repair, generate, or replace faulty cells that cause diseases.
ATMPs, including cell and gene therapies (CGTs), have emerged as a groundbreaking solution, offering long-term symptom relief and even complete cures for patients who were previously considered incurable. These therapies have the potential to shift the paradigm in chronic and rare diseases, transitioning from mere disease management to achieving full recoveries. What was once a distant aspiration for scientists is now becoming a tangible reality, as various CGTs receive approvals based on promising results from clinical trials.
But with a focus on patientcentricity, the life science industry as we know it is changing.
A Case for CGTs: Diffuse Large B-Cell Lymphoma
What is Diffuse Large B-Cell Lymphoma?
Diffuse Large B-Cell Lymphoma (DLBCL) is a type of cancer affecting the lymphatic system. It is characterized by its aggressive nature and rapid growth in the lymph nodes, liver, bone marrow, spleen, and other organs. DLBCL arises from the production of abnormal B lymphocytes, which normally play a crucial role in combating infections. However, in DLBCL, these abnormal cells fail to fully mature and accumulate in the lymph nodes and other organs, compromising the patient’s ability to fight infections effectively.
Regardless of the chosen medical approach, DLBCL treatment is often intense and requires frequent hospital visits over a short period. Coping with the diagnosis and treatment of this cancer can be challenging for patients, leaving them feeling fatigued and drained, particularly during and after treatment.
Standard Treatment for Diffuse Large B-Cell Lymphoma
Standard treatment for Diffuse Large B-Cell Lymphoma (DLBCL) is determined by various factors, such as the location and stage of the cancer, as well as the patient’s symptoms. The treatment goal can range from aiming for a cure to controlling the symptoms and the progression of the cancer for as long as possible.
The standard therapy typically involves a combination of chemotherapy and immunotherapy. Chemotherapy is used to destroy cancer cells, while immunotherapy involves the use of antibodies that attach to cancer cells, marking them for destruction by the immune system. However, a drawback of these standard treatments is that they are not targeted and can also affect healthy cells, resulting in side effects like hair loss.
How CGTs are Changing the Game?
Cell and gene therapies (CGTs) are revolutionising the approach to DLBCL treatment. In some cases, patients may undergo high dose chemotherapy followed by a stem cell transplant. Autologous transplants involve using the patient’s own stem cells collected prior to the procedure, while allogeneic transplants use stem cells from a donor. Stem cell transplantation can potentially address the underlying issue by replacing problematic cells and offering a potential cure.
Another innovative therapy gaining prominence is CAR-T therapy, which is a personalized form of immune-cellbased treatment. CAR-T therapy involves modifying a patient’s own immune cells to recognize and target cancer cells more effectively. This tailored approach has shown promising results in DLBCL and other types of cancer, providing new hope for patients.
CGTs are transforming the treatment landscape for DLBCL, offering targeted and potentially curative approaches that hold great promise for patients.
Gene therapy, specifically CAR-T therapy, has become available to patients with certain types of lymphoma. One notable example is Breyanzi, a CAR-T therapy that received approval from the U.S. FDA and the European Medicines Agency (EMA) in 2022 for the treatment of large B-cell lymphoma, including DLBCL.
To create each dose of Breyanzi, the patient’s blood is extracted, and T-cells, a type of white blood cell crucial for the immune system, are collected. These T-cells are then modified in a laboratory to express chimeric antigen receptors (CARs) that allow them to recognize and bind to proteins on cancerous B-cells. This modification flags the cancer cells for elimination.
Clinical studies evaluating the effectiveness of Breyanzi have shown significant clinical benefits. Patients treated with Breyanzi demonstrated meaningful disease control, event-free survival, complete responses to therapy, and progression-free survival, especially in comparison to patients who experienced relapse within 12 months after standard first-line therapy. A high percentage of patients (86%) achieved either a complete or partial response, with 66% achieving a complete response.
30 Journal for Clinical Studies Volume 16 Issue 1

While there are potential risks of side effects associated with Breyanzi therapy, the therapy has received approval because the benefits outweigh the risks. A single treatment with Breyanzi offers patients a more favourable prognosis compared to standard combination therapy with chemotherapy and immunotherapy.
In summary, Breyanzi has shown improved survival rates and, in some cases, the potential for curing patients. Its success has rapidly established CAR-T therapies as a valuable treatment option for patients with certain types of lymphoma.
Cell and gene therapies (CGTs) are shaping the future of oncology, bringing about a significant transformation in the field. The emergence of personalized medicine has challenged the traditional one-size-fits-all approach in healthcare, emphasizing the need for better health outcomes and patient-centric strategies. Consequently, CGTs have garnered substantial funding and support, revolutionizing the treatment landscape.
The potential of CGTs is evident in the FDA’s approval of over 20 cell and gene therapy products. These innovative therapies have been authorized for the treatment of various chronic conditions, including cancers, mucogingival conditions, retinal dystrophy, and spinal muscular atrophy. This remarkable progress highlights the shift from disease management to seeking genuine, long-term cures facilitated by CGTs.
However, despite the promising prospects, several challenges lie ahead. The implementation of cell and gene therapies faces obstacles related to ethics, regulations, costs, and logistics. These factors could
impede the translation of life-saving therapeutics from the research bench to the patient’s bedside. Overcoming these hurdles is crucial to unlock the transformative potential of CGTs and extend their benefits to millions of individuals suffering from incurable chronic and rare diseases, thereby enhancing their quality of life.
In conclusion, CGTs represent the future of medicine, offering hope and transformative potential in treating conditions that previously had no effective treatments. While there are obstacles to overcome, the continued development and integration of cell and gene therapies have the potential to revolutionize the life science and pharmaceutical industries, paving the way for better outcomes and improved quality of life for patients. life-saving therapeutics from the bench to the bedside
Richard Rossi

Richard is the Global Cold Chain & Strategic Projects Director at CRYOPDP. With over 20 years of experience in both diagnostic pathology, clinical trial pharmaceutical storage and logistics management, my industry knowledge allows me to apply best practices in everything that I do. From the creation of temperature-controlled warehouses for clinical trial product storage across Australia, Singapore and Japan, to launching 3PL pharma storage solutions in Australia, my history of leading and delivering state of the art pharma storage and supply lends itself to operational excellence.
Journal for Clinical Studies 31 www.journalforclinicalstudies.com Therapeutics
Generative AI is Revolutionising Life Sciences, And We’re Just Scratching the Surface
On average, it takes about seven years to develop a new drug and bring it to market. For ambitious life science businesses, generative AI’s ability to generate insights and content in a fraction of the time of a human means wiping months, or even years, off that average. When it comes to clinical development, saving time translates to saving lives, or at least improving them, through faster availability of treatments. It also translates into revenue opportunity – and a significant one at that. Some industry sources estimate that bringing new treatments to market ahead of schedule can be worth between £500,000 to £6.5 million per day. However, due to uncertain regulatory landscapes, coupled with the rapidly evolving nature of the technology, some companies are taking a wait-and-see approach to adopting generative AI, delaying investments until the course forward is clearer. While this may seem a prudent approach, it will be one organisation’s regret in the long run as they miss out on the opportunities generative AI holds, such as drug discovery and speed to market, and as competitors jump ahead and take the lead.
For life sciences companies looking to seize a competitive edge and supercharge their speed to market, there are a few key areas of the clinical development lifecycle they should focus on first.
Simplifying the Research Process
Research and development (R&D) is often the most time-consuming part of the drug development process, but AI can accelerate this process by up to 50% as the technology has a multiplier effect wherever it is applied.
Life sciences can implement generative AI at the very beginning of the R&D cycle, to aid in searching and synthesising available literature on a specific potential drug. Instead of beginning with a manual keyword search and sifting through hundreds of articles across various sources, teams could prompt a generative AI-enabled tool to rapidly search, gather and distil relevant articles – or even suggest unanticipated information pathways to explore.
Generative AI also has the potential to change how researchers find existing literature. Usually, researchers simply type keywords into the search box. But with a generative AI tool, they could state their goal into the prompt, providing context and intent, for the technology to find reference materials to support that specific ask, saving significant time while broadening the research horizon.
Automating Clinical Trial Protocol Writing
Compiling a clinical trial protocol document is a lengthy process that can take anywhere from a few months to over a year. Generative AI technology’s capabilities can automate a substantial proportion of the protocol writing process, bringing it down to days or even mere hours.
Generative AI can be trained on thousands of existing protocols in industry databases and each company’s own research data in order to identify the patterns relevant to investigational products, certain conditions, specific patient populations, or other factors. As the generative AI tool identifies relevant patterns, it can combine all the insights to design a baseline study, with a defined narrative that determines eligibility, drafts exclusionary criteria, and provides other necessary details. It can generate a number of draft options that would later be evaluated and refined by a human.
Expediting Launch Processes in Secondary Markets
Once a new therapy has been approved to launch in one market, many companies will be looking to expand the launch into others. This process takes a tremendous amount of time and resources, from strategy development and market research to agency engagement, content creation and material development. Much like in the research and protocol writing processes, a lot of the steps in this part of the process could be automated with generative AI.
For instance, when the drug is close to gaining approval, generative AI could support commercial teams’ research and compile strategy documents for secondary markets, taking into account specific regulations the therapy will need to adhere to in the new country. Similarly, generative AI can be used to adapt existing content –including website copy, brochures and other promotional materials – to the language and culture of the secondary market. This could shave up to a year off the go-to-market timeline in new countries and massively reduce marketing and design costs.
Taking the First Steps
Introducing generative AI into a business should be done one step at a time. It starts with fostering a culture of AI literacy, where every employee understands how the technology can be used to reshape and empower their role. It is also important to build a solid ecosystem of partners, which includes relationships with academic institutions, data providers, and speciality generative AI vendors that will support the business’ knowledge growth and internal capabilities.
Once generative AI is introduced, it is a good idea to establish a body within the business to supervise how the organisation uses the technology and manages the upskilling and development of employees engaging with the tech. This body should also establish best practices and develop frameworks that guide the deployment of generative AI across the business.
A Life-saving Revolution
Introducing generative AI into a pharmaceutical business is no mean feat and is understandably very daunting. It is, however, essential for companies that want to stay ahead of their competitors and the market to invest in generative AI. Likewise, it is crucial to ensure employees are provided training on how to best optimise the
32 Journal for Clinical Studies Volume 16 Issue 1 Technology

technology and create a body that supervises how the technology is being deployed across the business to avoid any misuse. As companies continue to experiment with generative AI across its various use cases, they will begin to lay the foundation needed to harness the full potential of this transformative technology, discovering, testing and bringing their drugs to market sooner. This improves patient outcomes due to safer, more effective and affordable drug development and increases revenue opportunities in a highly competitive market, driving value and improving patient outcomes at the same time.
Bryan Hill is Chief Technology Officer for Cognizant Life Sciences, responsible for digital solutions and technology innovation. His focus is on how emerging tech can help clients increase innovation to bring new therapies to market faster.

Journal for Clinical Studies 33 www.journalforclinicalstudies.com Technology
Bryan Hill
Technology
Modern by (Trial) Design: Shedding Legacy EDC Systems to Gain Clinical Capacity
As trials become increasingly complex, companies share how leaving behind legacy EDC systems can save thousands of hours during study build and testing.
In today's context of complex trials, traditional electronic data capture (EDC) systems are not flexible enough to address frequent protocol amendments. They hinder study teams by creating hidden data management issues that can affect sponsors' and contract research organisations (CROs) clinical capacity. Data managers have to navigate the manual workarounds and custom programming required by legacy systems, which cause bottlenecks. Organisations already grappling with the exponentially increasing volume of data (through new sources like wearables, ePRO, genetic biomarkers, etc.,) lack a single source of truth now that most critical trial data can come from outside EDC. Delayed clinical insights and the increased cost of executing studies invariably feed through to trial outcomes.
Leading sponsors and CROs can avoid common pitfalls by ending their reliance on custom programming and embedding rules and dynamics to reduce study build times. Others have introduced risk-based user acceptance testing (UAT) rather than attempting to validate hundreds of possibilities before launching a study. Given that protocol changes are inevitable (and sometimes even preferable) in complex studies, it is better to eliminate downtime and/or migrations so that amendments become a positive change agent rather than something to be avoided at all costs.
Since companies started to adopt EDC decades ago, the lag period between data being recorded during a patient's visit and appearing in the data management workstream has decreased only marginally, from eight to six weeks. The burden of data entry shifted from biopharma sponsors to sites, which fragmented the data management model without addressing core challenges. With new systems, we are getting closer to complete and concurrent data review.
Enhancing Clinical Capacity
Most traditional EDC systems need custom programming to achieve desired study designs, partly because the study architecture isn't flexible to nuanced trial requirements. The more custom programming is needed, the greater the investment to build, test, and maintain these programs.
Custom functions are particularly challenging for biotech companies because skilled resources are at a premium and needed elsewhere in the trial. As a result, these small and midsize companies often partner with CROs: Deepak Mahadevaiah, senior director for clinical data sciences at Agenus, points out, "We don't have the skillset in-house and are dependent on a technology partner." The costs can rack up quickly. In a recent study (not undertaken for Agenus), one CRO spent over 1,100 hours building and testing over 200 custom functions.
Meanwhile, sponsors and CROs that minimise custom programming are accelerating study design and build. Lotus Clinical Research, a CRO specialising in supporting novel drug development,
not only reduced study startup time by 67%1 with Veeva Vault CDMS but can now implement changes to the EDC nine times faster than before.
Manual workarounds in legacy EDC cost valuable time during study updates. When studies change, teams spend their time trying to prevent custom functions from breaking down. For example, as Platform Life Sciences (PLS) prepared to conduct an outpatient COVID-19 trial with 14,000 patients and 12 interventions, it realised the volume of manual workarounds required2 would make it impossible to secure clinical insights in a timely manner. Michael Zimmerman, AI infrastructure executive at PLS, recalls: "We encountered multiple inefficiencies and had an inability to move fast."
A more effective approach uses pre-defined variables and functionality within the EDC system to replace custom functions. Study teams can then create rules to automate tasks, complete edit checks, and generate treatment cycles, visits, and forms while avoiding custom programming entirely. This makes the data manager's job easier and boosts productivity. Once PLS adopted a rules engine to replace custom functions, it could support studies with multiple amendments and drugs across geographies with less manual work and reduced testing (and re-testing) of study elements.
Early protocol changes can delay database builds and the study going live; post-production changes are equally stressful. Although amendments are inevitable, eliminating downtime mitigates their impact. This way, companies can continue to make protocol changes mid-study without risking the trial's overall timeframe. For example, biotech company Cara Therapeutics, which advances therapies for those experiencing chronic pruritus, made 11 post-production changes with zero downtime on migrations.3
Testing is another key driver of delays in study builds. This is most evident in complex branching trials when teams must validate hundreds of possibilities before the launch. However, sponsors can get studies up and running faster by taking a risk-based approach that identifies pre-approved trial elements and removes them from testing (for example, Vertex reduced release times through riskbased testing4). Similarly, if teams can complete live, dynamic design reviews of the study in its proper environment, they will have richer conversations and feedback — and make better decisions than when they collaborate via email or spreadsheets.
Business Benefits of Modern EDC
Agile design can support a rapid evolution in trial capacity and embed new ways of working.
Global CRO Fortrea needed to streamline decision-making, eliminate downtime, and speed deployment of amendments. The company improved responsiveness and eliminated pain points by unifying its clinical foundations, spanning randomisation trial supply and management (RTSM), quality management software (QMS), clinical trial management software (CTMS), and electronic Trial Master File (eTMF). It drove the new approach by seeking feedback from staff and sponsors and dedicating enough training time for study team members and experts in global offices.
34 Journal for Clinical Studies Volume 16 Issue 1

Successful change management also requires close alignment between technology partners and study teams, who can provide input to future technology development. Fortrea achieved this by clearly communicating where issues might arise to build trust and understanding up front. The company acknowledged that there would be some hurdles to implementation while ensuring user input so that the first studies to go live had the best chance of success.
Now that multiple teams have been trained and use the same platform, Fortrea has minimised programming and time spent on manual-intensive activities, including reducing its use of unique forms by 20–30%. Jerry Yarem, vice president of data management at Fortrea, observes: "That increased efficiency is because the system design allows us to focus on quality."
Data Foundation for Decision Confidence
There is work ahead to bring the full benefits of modern EDC to trial design. First, by introducing standards for more complex studies, including those involving electronic health and medical records. We need to increase automation in study design – and are making advances in this area5 – to get trials up and running faster. Most importantly, we can tackle inefficiencies in the non-EDC dataflow, where integrated data reconciliation and cleaning are critical.
A modern approach to clinical data will be a catalyst for more productive trials by helping sponsors and CROs to accelerate their study builds and make clinical decisions with confidence. With a robust data management foundation, teams can focus on science,
and move faster to bring new innovations to patients – even for the most complex studies.
REFERENCES
1. https://www.veeva.com/customer-stories/lotus-clinical-research-speedsstudy-startup-by-67-with-veeva-clinical-suite/
2. https://www.veeva.com/resources/why-3-biotechs-upgraded-their-edcto-veeva-vault/
3. https://www.veeva.com/customer-stories/from-early-adopter-tooptimised-trials-cara-therapeutics-and-veeva-vault-cdms/
4. https://www.veeva.com/resources/how-a-risk-based-approach-to-uatand-real-time-updates-are-shaving-weeks-off-edc-study-builds/
5. https://www.veeva.com/resources/new-veeva-link-applications-give-clinicalteams-real-time-intelligence-to-optimise-site-selection-and-trial-design/
Richard Young

Richard Young is Vice President, Vault CDMS, at Veeva Systems. With over 25 years of experience in life sciences, Richard is known for his executive vision and proven operational experience in data management, eClinical solutions, and advanced clinical strategies. His role at Veeva is helping to transform the way we approach clinical data, unifying every contributor and consumer in a single platform.
Journal for Clinical Studies 35 www.journalforclinicalstudies.com
Technology
36 Journal for Clinical Studies Volume 16 Issue 1



londonbiotechshow.com info@londonbiotechshow.com +44 (0)208 242 6566 Unlocking The Potential of Biotechnology For Revolutionising Medical & Healthcare Sectors Globally Orginised by Business Media 8 - 9 May 2024 Olympia West
Page 36 Advances in Cell-Based Screening in Drug Discovery 2024
Page 36 AI in Drug Discovery
Page 25 Autoscribe Informatics UK
Page IFC Catalyst Clinical Research
Page 37 London Biotechnology Show
Page 3 PCI Pharma Services
Page 5 Ramus Medical Limited


38 Journal for Clinical Studies Volume 16 Issue 1 Ad Index
for
Page 29 Senglobal Ltd OBC Trilogy Writing & Consulting Gmbh IBC UPC Cambridge Subscribe today at Email: info@senglobalcoms.com www.journalforclinicalstudies.com I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry JCS is also now active on social media. Follow us on: www.twitter.com/jforcsjournal www.facebook.com/Journal-for-Clinical-Studies www.plus.google.com/+Jforcsjournal www.journalforclinicalstudies.com Journal
Clinical Studies






––is
• • •
UPC’s


A podcast to discuss topics important for the profession of medical writing
Available on Apple Podcasts, Google Podcasts, Spotify, YouTube Podcasts, and TrilogyWriting.com/TriloTalk

