Journal for Clinical Studies – ISSN 1758-5678 is published quarterly by Senglobal Ltd.
4 FOREWORD
WATCH PAGES
6 Ensuring Flexibility when Approaching CTIS
Here, we explore the regulation's impact on the submission process, highlighting the need for dynamic and flexible solutions tailored to specific scenarios. Inês Vale De Gato and Blaine Maloney, from Catalyst Oncology, discuss the implementation of the European Union Clinical Trial Regulation (EU CTR) across studies. Through three case studies, they illustrate Catalyst Oncology's approach to overcoming challenges in the Clinical Trials Information System (CTIS) process, emphasising the importance of close collaboration, creative problem-solving, and timely responses to ensure efficient regulatory approval timelines.
8 Pulse Oximeters and Addressing the Potential for Bias
The FDA discusses the increased use of pulse oximeters during COVID-19 and addresses potential racial bias in their readings. They explore the limitations of pulse oximetry, regulatory measures, and efforts to improve accuracy across different skin pigmentations. Jennifer Nguyen at Clarivate dives into the FDA's ongoing initiatives including seeking feedback from the Anaesthesiology and Respiratory Therapy Devices Panel, updating guidance, and funding real-world clinical trials to assess pulse oximeter performance in diverse patient populations.
REGULATORY
10 Changing the Narrative of First in Human Oral Drug Development: The SMART Advantage
PCI Pharma Services and Worldwide Clinical Trials discuss their SMART FHD initiative to expedite First Human Dose (FHD) studies. This partnership streamlines drug formulation and trial execution with a drug-in-capsule (DiC) process, reducing time and cost. Ed Groleau of PCI explores how their collaboration enhances pharmacokinetic/pharmacodynamic (PK/PD) studies, site selection, and participant recruitment. The initiative aims for faster market entry and plans to expand into sterile injectable forms.
12 Rethinking Big Data Will Lead to New Advances in Personalised Medicine
The life sciences industry is set to advance personalised medicine with AI and machine learning, provided data is clean and secure. Stephan Ohnmacht from Veeva notes how integrating diverse data sources, from patient records to genetic information, is crucial for evaluating treatments.
MARKET REPORT
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
Volume 16 Issue 2 Summer 2024 Senglobal Ltd.
www.journalforclinicalstudies.com
14 Reviewing Research Participant Payments Through a Diversity Lens
This in-depth analysis of five years of data from an independent IRB, which reviewed over 90% of FDA-approved drugs in 2023, offers valuable insights. Drawing from over 7,500 records, Kelly FitzGerald and Donna Libretti Cooke of WCG uncover how participant payments are structured and described across various study phases and therapeutic areas. The findings aim to assist sponsors and sites in designing more equitable and inclusive trials, thereby enhancing access and diversity for underserved populations.
RESEARCH AND DEVELOPMENT
18 Designing and Executing a Robust Feasibility in Rare Disease Clinical trials
Rare disease studies, especially in paediatric populations, face unique challenges due to complexity and stakeholder involvement. Thorough feasibility assessments are crucial for understanding study viability, site selection, support needs, recruitment timelines, and costs. Experts at The Emmes Company LLC explore how robust feasibility data helps mitigate risks and predicts patient enrolment speed, while industry practices often emphasise quick, superficial assessments.
THERAPEUTICS
22 Liver Disease, Obesity and the Value of Treatment
Obesity, affecting nearly one billion people, leads to serious liver complications like MASLD, MASH, and cirrhosis, and worsens the prognosis of cirrhosis while increasing the risk of hepatocellular carcinoma (HCC). Dr. Alan Baldridge and Jolanta M. Wichary at ICON dive into the advances made in medical, endoscopic, and surgical care – that offer new hope for managing obesity's impact on the liver.
TECHNOLOGY
24 Could GenAI be Pharma’s Silver Bullet for Medical Writing? Only if Aimed Carefully
With the rise in potential of Generative AI transforming medical
writing for regulatory and safety reports, many companies are struggling to develop these capabilities in-house. A recent survey by the Regulatory Affairs Professionals Society highlights a significant gap in these efforts, as noted by Punya Abbhi of Celegence, who points out the challenges companies face in addressing their regulatory content needs effectively.
28 Using RBQM and New Technologies to Enhance Data Analysis and Enable Faster Review and Decision Making
With the rise in clinical trial data, there’s a growing need for faster and more efficient data analysis. Sponsors require quick, insightful data reviews to make informed decisions swiftly. Francois Torche from CluePoints explores risk-based quality management (RBQM), coupled with new technologies, presents a solution to enhance data quality and integrity while boosting efficiency in this high-demand environment.
LOGISTICS & SUPPLY CHAIN
32 Ensuring Integrity in Clinical Trials: The Role of Small-Box Technology
The global clinical trials market is projected to hit $100 billion by 2030, driven by research advancements and regulatory changes. Ensuring the integrity of clinical trial materials during transit is crucial, and temperature-controlled packaging solutions are key to managing these logistics challenges effectively. Niall Balfour at Tower Cold Chain covers investing in these tailored solutions is essential for maintaining product quality and navigating complex supply chains.
Lyophilization and Sterile Manufacturing
Driving development and connecting commercialization of sterile and lyophilized drug products, we are dedicated to your success in bringing life-changing therapies to patients.
talkfuture@pci.com Your
www.pci.com
OUR END-TO-END BIOLOGIC SOLUTIONS INCLUDE:
• Sterile Formulation & Lyophilization Cycle Development
• Lyophilization and Sterile Fill-Finish Manufacturing
• Aseptic Robotic Technologies
• Analytical Support
• Clinical & Commercial Labeling & Packaging
• Refrigerated/Frozen Storage & Distribution
This summer edition of our journal for clinical studies is a page turner! Our world is changing dramatically, with competition sparking other competition – we’re refining ourselves at a brilliant pace, and the advancements we’re making are astonishing. From covering the importance of treatment and the impact of our lifestyles on our bodies, to diving into the care we must maintain to keep our end of bargains in clinical research, we’re doing our best to talk about the best of the best – what’s new!
Under our Regulatory heading, we take a leap into the impact of the European Union Clinical Trial Regulation (EU CTR) on submission processes. Inês Vale De Gato and Blaine Maloney from Catalyst Oncology provide insightful case studies on navigating the Clinical Trials Information System (CTIS), emphasising the necessity of flexible, collaborative approaches to regulatory approval – how to make things more efficient, how to be a little more creative with the ways we get things done.
Our Market Report section highlights the true importance of equitable participant compensation in clinical trials. Amazingly, Kelly FitzGerald and Donna Libretti Cooke of WCG analyse data from over 7,500 records to reveal how payment structures can be optimised to enhance access and diversity, promoting fairer trial participation. This is an engaging and eye opening read, and ideally holds us all to a better standard for how we go about participation moving forward.
In Therapeutics, we boast a feature on an in-depth look at the interplay between obesity and liver disease. Dr. Alan Baldridge and Jolanta M. Wichary from ICON explore recent advancements in treatment strategies for conditions, such as MASLD and cirrhosis, offering new hope for improved management and outcomes. With this lens on our lifestyles and the effect they have on our bodies, followed by the connections between strategy and management, this article offers a cohesive view, taking the context into account.
We wouldn’t be cutting edge without a segment on technology and AI, and we cover the potential of Generative AI in medical writing. Punya Abbhi from Celegence examines the challenges and opportunities of integrating AI into regulatory and safety documentation, highlighting the need for careful implementation to
JCS – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Principal Content Editor, Clarivate
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
• Georg Mathis, Founder and Managing Director, Appletree AG
maximise its benefits. Of course, the realm of AI is a heavily debated field, and has a plethora of uses; we’re interested in these changes and creative ways to make it work for us comfortably.
As you explore this edition, you'll find a wealth of insights into the ways our industry is advancing, from enhancing data analysis with risk-based quality management to addressing potential biases in pulse oximetry. We hope you find this issue both informative and inspiring, reflecting our commitment to innovation, equality, and progress.
Enjoy the read!
Sara Shikooh, Editorial Manager – IPI
• Hermann Schulz, MD, Founder, PresseKontext
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, AsiaPacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
Ramus Medical
is a part of Ramus Corporate Group. The company is managed under a centralised quality management and has developed an integrated QMS as well as specific standard operating procedures tailored for the clinical trials department that are fully harmonised with the GCP guidelines, and the local and European legislation.
Ramus Medical EOOD is a full-service contract research organisation (CRO) in Sofia, Bulgaria.
The company was created in 2009 as a natural development of the Medical Laboratory Ramus Ltd., the largest privately-owned medical laboratory in Bulgaria.
The company independently manages clinical research projects in Bulgaria and provides partnerships in multinational clinical projects providing a comprehensive range of clinical research services:
Core Services include:
• Medical writing
Our staff has extensive expertise in the preparation, adaptation and translation of a wide range of clinical trial documents that are fully compliant with the Good Clinical Practice (GCP) standards, the client’s specifications and the regulatory requirements.
• Study start-up
We offer full or partial study start-up assistance for different types of studies throughout Bulgaria.
• Regulatory submission
• Project management
• Monitoring
• Data Management
• Pharmacokinetic evaluation
• Biostatistics
• Regulatory advice and services
• Readability User Testing
• Registration of medicinal products on the territory of Bulgaria
• Pharmacovigilance services
• Logistic department
• Destruction of IMPs/IMDs & clinical samples – agreement with PUDOOS
• Archiving services
• DDD activities
Ramus Medical has gained its expertise during the completion of numerous clinical projects carried out over the past decade:
• Phases I to IV drug trials
• Non-interventional studies
• Pilot and Pivotal Medical Device investigations
The clinical trials we conducted facilitated the MA/CE mark granted by various European Agencies/Notified Bodies and Third Country Agencies.
Ramus Medical offers flexible clinical research services in various domains, with extensive experience in fields.
Our team comprises qualified, appropriately trained, experienced, motivated and collaborative professionals and is competent to
www.journalforclinicalstudies.com
communicate effectively across geographical and cultural boundaries to resolve any arising issues. We adhere strictly to the agreed timelines during the clinical investigations and strive to complete the tasks on time.
Why are we the solution for your projects? Ramus has its own:
Medical and Bioanalytical Laboratory
In 2018 the Medical Centre Ramus was established, located in Sofia, Bulgaria. Up to date, it has three separate locations, one of which is developed as an independent clinical research centre in compliance with the requirements for the phase I unit.
The Medical Centre Ramus allows the conduct of clinical trials in all phases in many therapeutic areas.
The Medical Centre meets all requirements for performing highquality clinical research and is designed to maximise the delivery of high-quality research data and was GCP-inspected.
Ramus Medical retains an extensive database of investigators and sites compiled through years of mutually beneficial collaboration.
Our bioanalytical laboratory is equipped with leveraging state-ofthe-art instrumentation (LC-MS/MS), techniques, and facilities, our team of experts has experience in a broad range of small molecules. Our Analytical laboratories provide method development, transfer, validation, and analysis of preclinical and clinical biological samples. We have extensive expertise in developing sensitive methods for LCMS/MS-qualifying multiple analytes and metabolites.
• Logistical company, certified for hazardous and biological samples transportation
• Clinical site facility and own catering company for hospitalised patients
When Catalyst Oncology implements the European Union Clinical Trial Regulation (EU CTR) across Sponsor studies the Site Activation Management (SAM) team is often asked what impact certain elements will have on the submission process. We frequently find ourselves responding with “it depends.” There is no one-size-fits-all approach to clinical research, and the EU CTR is no exception. As clinical trials become more complex and regulations evolve, Sponsors’ goals remain the same: a need for dynamic solutions requiring flexibility and creativity tailor–made to their specific scenarios.
Below are three case studies illustrating several challenges the SAM team has mitigated, the opportunities the team has explored with our Sponsors based on their strategies, and our approaches with EU CTR and the Clinical Trials Information System (CTIS) process. In these examples, we supported Sponsors throughout the process to ensure efficient regulatory approval timelines.
Close Collaboration for Overcoming Hurdles
Sponsors must understand that making changes to the protocol or other clinical trial documents after CTIS submission can create challenges. More important than the changes themselves, how the changes are responded to affects the activation timelines either positively or negatively; therefore, responses and experience are important in understanding the impact of critical decisions that need to be tailored to each project.
Case Study 1. During the part I assessment, the Sponsor received several queries related to the investigational medicinal product dossier (IMPD), investigator’s brochure (IB), and protocol resulting in a protocol amendment, and in other requested changes to the documents. Our SAM team instructed the Sponsor on the steps to take to understand the impact of the document changes, including the deadlines to hit the required response timeline and the effects on their strategy. Three days after the timeline was set, the Sponsor released a protocol amendment, updated IB, IMPD, and synopsis to our team. Some of the protocol amendment updates impacted the main informed consent form (ICF) and the country ICFs in Spain, France, and their translations. At this time, the part II assessment for Spain was concluded and closed with an approved country ICF version for Spain linked to the original protocol version submitted in part I.
Following consultation with the Sponsor, the SAM team contacted the Spanish ethics committee and requested re-opening the part II assessment to raise an additional query for the part II assessment in the system for the country ICF for Spain. This allowed Catalyst Oncology to make changes within the CTIS system part II and resubmit a revised country ICF for Spain linked with the new revised protocol version as a result of the queries received in the part I assessment review.
This complex strategy and thinking outside of the box enabled Catalyst Oncology to provide a new main ICF version and country ICF updates (Spain and France) with completed translations within 24 hours along with a submission to the committee’s RFI in part II. All other queries in the protocol, IB, and IMPD were addressed in parallel and on time with the new documents during the RFIs in part I.
As illustrated, it is vital to address queries and receive documents within the provided due dates to manage any hurdles. Otherwise, delays or amended documents can impact and derail timings. When such events occur, diligence, flexibility, creative thinking, and attention to the EU CTR guidance helps to mitigate challenges.
Flexibility in Addressing Delays
Case study 2. Formal selection of several sites in Spain occurred only two days ahead of a planned submission date due to a delay in site identification. In this scenario two standard valid options are advisable: first, delay the submission until all site-specific documents for the recent sites selected are collected; or second, submit the application without those sites in the initial application and add them as new sites at a later date. With the latter option, the Sponsor would need to accept a two-month delay in the site activation timeline for those sites.
Instead, the SAM team enabled a different successful approach. We proceeded with the application within the planned timeline and submitted it for all sites. While the application proceeded as planned, the part II package remained incomplete due to the missing sitespecific documents for the recent sites selected. Anticipating receiving validation queries for the missing site documents 10 business days after the submission, we worked with the sites’ principal investigators (PIs) and collected the required signed documents during the validation assessment timeline. We then answered the validation requests for information with the missing documents on time. Our strong relationship with these sites was critical to the success of this approach.
Our actions resulted in no impact on the submission or approval timelines. It also did not negatively impact the site activation projection plan for the sites in Spain as they were submitted and approved within the initial application.
Approaching Shortened Timelines
Case Study 3. In one of our studies, while collecting the required core regulatory documents for the CTIS submission part I deadline, we had not received all required core regulatory documents from the Sponsor. The SAM team remained flexible with a strategy in place that incorporated multiple supporting resources to review the lastminute documents when received from the Sponsor. We performed a final quality check, revised any necessary inconsistencies, and applied necessary document redactions within 24 hours.
While a new regulation and process can create difficulties for the Sponsor, they do not have to. Flexibility, creativity, risk mitigation, and dependability are all contributing factors for the continual success of the delivery of clinical trial submissions. In summary, here are the actions Catalyst Oncology took to ensure that there were no consequences on the submission timelines:
1. In the first case mentioned above, all queries were answered on time and within 12 days with a new protocol amendment, IB, and ICF in Spain and France with the synopsis translated.
2. In the second case, all sites in Spain were submitted in the initial application without impact on the approval timeline or the site activation plan.
3. In the third case, there were no negative effects on the approval timeline.
Each of these studies illustrate a high-level of expertise and close collaboration between the Sponsor and Catalyst Oncology. Throughout each study, Catalyst communicated with the Sponsor and kept in close contact to ensure any issues were anticipated and resolved quickly.
As previously mentioned, there is no “one-size-fits-all” approach to conducting clinical research. Maintaining thoughtful and simple processes that are built to allow customisation for study-specific needs are critical for the overall success of the EU CTR submission process. At Catalyst Oncology, we split the submission package preparation process into three main steps, and run them in parallel to allow an efficient and streamlined approach:
1. Pre-preparation – Catalyst Oncology supports the Sponsor throughout the critical steps with the European Medicines Agency (EMA) account, CTIS access, CTIS permissions assignments, creation of the new trial and getting the EU clinical trial number, and the investigational product (IP) registration in Eudravigilance.
2. Preparation – The SAM team prepares the part I and part II submission packages from the collection of the core regulatory submission documents from the Sponsor until the collection of signed site-specific documents from sites are available. The team prepares all country and site-specific documents, including any relevant translations and redactions, and the manual entry of the CTIS application form. During this step, in our experience, it is critical to set up a document delivery tracker to monitor the status of the required submission documents and deadlines per country and site. It is important to start filling out the CTIS application form on an ongoing basis, saving the form as information is added until its final completion, paying attention when uploading documents between the redacted versus nonredacted documents.
3. Review – SAM team completes an internal and external quality review process and Sponsor approval before the final application of part I and part II submissions packages. Catalyst Oncology uses quality checklists during the review process. The higher the quality of the documents being prepared, the faster the review process, which helps with meeting any submission target dates. To ensure the quality and the timeline of the submission, it is vital to conduct daily reviews of the submission inclusive of a completion status of the documents, and to closely communicate with the Sponsors on the status of the documents, translations, and redactions.
Catalyst Oncology knows that when it comes to the CTIS process, much rests on us and our expertise. The processes and actions above illustrate flexibility, creativity, and the dependable delivery for clinical trials; however, any solutions connected to CTIS will always depend on the specific needs of the clinical trial with a tailor–made response to achieve ultimate goals and objectives.
Inês Vale de Gato, MSc, Associate Director, Catalyst Oncology, brings 14 years clinical development research experience. Ines expertly supervises and manages operational study start-up and regulatory strategies for multiple studies and regions. Ines manages multiple site activation staff across multiple countries. She is an EU regulatory and CTIS certified expert and earned B.S. and M.S. degrees in microbiology from the Medicine and Sciences Faculty, the University of Lisbon, Portugal.
Blaine Maloney
Blaine Maloney, Associate Director, Catalyst Oncology, began his career in oncology, becoming passionate about helping to provide access to cutting-edge compounds in over 60 countries. In 2021, Blaine joined Catalyst Clinical Research to help create and develop global regulatory and site activations teams with the goal of inspiring people to design and deliver better clinical trials. Blaine earned a master’s degree in regulatory affairs from Northeastern University.
Inês Vale de Gato
Pulse Oximeters and Addressing the Potential for Bias
As a result of the coronavirus disease 2019 (COVID-19) public health emergency, pulse oximeter usage has increased in both hospital and home settings. The US Food and Drug Administration (FDA) describes a pulse oximeter as a device that is typically placed on a fingertip and uses light beams to provide an estimate of the oxygen saturation of the blood (SpO2) and the pulse rate. These non-invasive devices are widely used to obtain an indirect measure of arterial blood oxygen saturation (SaO2), the gold standard for evaluating blood oxygen saturation levels.
Pulse oximetry is built on two physical principles: 1) arterial blood creates a pulsatile signal and 2) oxygenated haemoglobin (HbO2) and reduced haemoglobin (HHb) have different absorption spectra.1 Pulse oximeters typically emit light in the red and infrared wavelength regions (660 nm and 940 nm, respectively). More infrared light is absorbed by HbO2, allowing red light to pass through, while HHb absorbs more red light and permits infrared light to pass through. The ratio of the red-light measurement to the infrared-light measurement is calculated for the systolic and diastolic phases. Afterward, the ratio of these ratios is calculated and converted to SpO2 2
The FDA published the guidance for industry Pulse Oximeters – Premarket Notification Submissions [510(k)s] in March 2013 to assist sponsors in preparing 510(k) submissions for pulse oximeters.3 It covers class II devices regulated under 21 Code of Regulations (CFR) 870.2700 for oximeters and 21 CFR 870.2710 for ear oximeters. Of note, these devices are categorised as prescription-use pulse oximeters that undergo clinical testing and FDA review before being granted 510(k) clearance. The guidance includes recommendations on topics such as how to evaluate the accuracy of pulse oximeters and what to include in labelling.
Limitations of Pulse Oximetry
In December 2020, results were published in the New England Journal of Medicine about the potential for racial bias in pulse oximetry measurement.4 The findings described in the article suggested that pulse oximeter readings may not be as accurate in people with darker skin pigmentation. Relying on pulse oximetry to adjust supplemental oxygen levels may increase the risk of hypoxemia for this population, the authors noted.
This article was referenced in an FDA safety communication issued in February 2021.5 The agency notified patients and healthcare providers that pulse oximeters “have limitations and a risk of inaccuracy under certain circumstances.” They were advised to not rely solely on pulse oximeters to assess oxygen levels and to be aware that factors such as poor circulation, skin pigmentation, skin thickness, and skin temperature can affect pulse oximeter readings. Additionally, the FDA specified that over-the-counter (OTC) pulse oximeters, which saw increased use due to COVID-19, do not undergo FDA review and are not intended for medical use.
Re-examining
Current Regulations
Among its efforts to address the limitations of pulse oximetry, the FDA
sought feedback from the Anesthesiology and Respiratory Therapy Devices Panel (ARTDP) of the Medical Devices Advisory Committee (MDAC). A meeting was held on November 1, 2022, focusing on the concern that pulse oximeters may be less accurate in individuals with darker skin pigmentation and the potential factors that affect pulse oximeter accuracy and performance. The ARTDP members acknowledged the disparate performance of pulse oximeters in this patient population. They recommended that future studies on these devices assess a full spectrum of skin pigmentation, that the labelling for pulse oximeters include a statement on their possible inaccuracy in relation to skin pigmentation, and that OTC pulse oximeters should indicate that they are not intended for medical use or clinical decision-making.
After this meeting, the FDA published the Approach for Improving the Performance Evaluation of Pulse Oximeter Devices Taking Into Consideration Skin Pigmentation, Race and Ethnicity to share a potential clinical study design that can incorporate a larger range of skin pigmentation.6 As noted in the discussion paper, the 2013 FDA guidance recommends that a study should involve participants with a range of skin pigmentation but does not explain how or at which site on the body to assess skin pigmentation. Elements of the proposed clinical trial include enrolling participants that span the entire Monk Skin Tone Scale (MST) and evaluating MST values at locations with a wide range of pigmentation levels (e.g., the forehead).
On February 2, 2024, the ARTDP reconvened to provide feedback on the discussion paper and consider other data that should be provided by manufacturers to the FDA. The panel members supported the use of the MST and the approach of evaluating it as an initial assessment of skin pigmentation, followed by an objective pigmentation assessment such as individual typology angle (ITA). However, they also had some critiques of the proposed clinical study design. For example, several panellists noted that the minimum sample size suggested by the FDA (24 participants) may not be sufficient. Because premarket studies generally include healthy participants and are conducted in controlled settings, the ARTDP emphasised the need for real-world data (RWD) to ensure that the devices perform as intended for patients.
Ongoing Efforts to Assess the Performance of Pulse Oximeters
Regarding RWD, the FDA has funded grants to the University of California San Francisco (UCSF)–Stanford University Center of Excellence in Regulatory Science and Innovation (CERSI) for two real-world, prospective clinical trials that are assessing pulse oximeter errors related to skin pigmentation in hospitalised adult and paediatric patients.7–8 Both studies are designed to measure several parameters, including SpO2, SaO2, and the site of pulse oximeter probe placement, across different patient groups. Additionally, skin pigmentation is being measured via colorimetry tools such as the MST, the Fitzpatrick Skin Type Scale, and the von Luschan's Chromatic Scale.
At the February 2024 ARTDP meeting, updates for each trial were provided by the study coordinators. Among their findings thus far, they have observed that the probe location varies in the real-world setting, and some pigment scales correlate moderately with ITA values. The FDA has yet to update its 2013 guidance
on pulse oximeters but noted that it intends to reassess current recommendations based on stakeholder feedback, results from the published literature, and outcomes from the UCSF-Stanford realworld studies.
REFERENCES
1. Jubran A. Pulse oximetry. Crit Care. 1999;3(2):R11-R17. https://pubmed.ncbi. nlm.nih.gov/11094477/
2. Nitzan M, Romem A, Koppel R. Pulse oximetry: fundamentals and technology update. Med Devices (Auckl). 2014;7:231-239. https://pubmed. ncbi.nlm.nih.gov/25031547/
3. Pulse Oximeters – Premarket Notification Submissions [510(k)s]: Guidance for Industry and Food and Drug Administration Staff. Food and Drug Administration webpage. https://www.fda.gov/regulatory-information/ search-fda-guidance-documents/pulse-oximeters-premarketnotification-submissions-510ks-guidance-industry-and-food-and-drug
4. Sjoding MW, Dickson RP, Iwashyna TJ, Gay SE, Valley TS. Racial bias in pulse oximetry measurement. N Engl J Med. 2020;383(25):2477-2478. https://pubmed.ncbi.nlm.nih.gov/33326721/
5. Pulse Oximeter Accuracy and Limitations: FDA Safety Communication. Food and Drug Administration web page. https://www.fda.gov/ medical-devices/products-and-medical-procedures/pulseoximeters#safetycommunication
6. Approach for Improving the Performance Evaluation of Pulse Oximeter Devices Taking Into Consideration Skin Pigmentation, Race and
www.journalforclinicalstudies.com
Ethnicity. Food and Drug Administration web page. https://www.fda.gov/ media/173905/download
7. Prospective Clinical Study of Pulse Oximeter Errors in Adult Hospitalized Patients with Varying Skin Pigmentation. Food and Drug Administration webpage. https://www.fda.gov/science-research/advancing-regulatoryscience/prospective-clinical-study-pulse-oximeter-errors-adulthospitalized-patients-varying-skin
8. Prospective Clinical Study to Evaluate the Accuracy of Pulse Oximeters in Children. Food and Drug Administration webpage. https://www.fda.gov/ science-research/advancing-regulatory-science/prospective-clinicalstudy-evaluate-accuracy-pulse-oximeters-children
Jennifer Nguyen
Jennifer Nguyen, PhD, is a Senior Content Editor for the Cortellis suite of life science intelligence solutions at Clarivate. She previously worked as a medical writer, which involved writing and editing scientific journal articles and materials for science conferences. Her current role includes reporting on FDA advisory committee meetings, drug approvals, and workshops.
Email: jennifer.nguyen@clarivate.com
Changing the Narrative of First in Human Oral Drug Development: The SMART Advantage
In the intricate realm of pharmaceutical development, First Human Dose (FHD) studies represent a pivotal juncture, replete with complexities yet essential for advancing patient safety and drug efficacy. These early-stage clinical trials aim to establish safety and refine the dose to minimise adverse events (AEs) with potentially some indication of efficacy. Traditional drug development pathways for solid oral formulations are often characterised by their transactional nature, contributing to prolonged timelines. Recognising the critical balance between expediency and safety, PCI Pharma Services (PCI), a world-leading CDMO, and CRO, Worldwide Clinical Trials provide an innovative end-to-end partnership designed to navigate the FHD process with unmatched efficiency from drug formulation, and clinical supply through to trial execution and study management.
In FHD development, formulating a drug is just the beginning. Other essential elements include executing Phase 1 trials with precise bioanalytical assessments, effective participant recruitment, and meticulously planned trial designs. These studies rely on healthy volunteers and expert teams to conduct detailed pharmacokinetic/ pharmacodynamic (PK/PD) analyses, which are crucial for positioning the novel drug for success in subsequent phases. This article will cover the importance of the partnership between CROs and CDMOs and how their individual capabilities have the power to expedite clinical trials and increase efficiency.
Transforming FHD with Drug in Capsule Technology and Pristine Supply Management
CDMOs have the unique opportunity to proactively recognise areas of improvement in the development and manufacturing process and implement innovative solutions to combat hurdles. PCI identified a way to transform their client’s studies and expedite the pathway into the FHD study phase with their Supply Management and Readiness Team (SMART) of development, manufacturing, and supply management experts. SMART is not only an acronym but a reflection of its mission to guarantee the timely provision of solid oral medications for Phase 1 clinical studies. SMART FHD transcends conventional oral drug development and stability study delays by eliminating early-stage formulation development to deliver strategic operational advantages, including a managed operational package and pivotal distribution options. This approach can propel drugs into the early clinical phase months or even a year ahead of traditional methods.
With SMART FHD, all that is needed from the client is the delivery of a sufficient volume of drug substance, and PCI can manage the rest. The critical difference is the streamlined drug-in-capsule (DiC) process that requires less back-and-forth on developing and optimising the drug formulation, moving a molecule to the clinical phase more quickly. In addition, there’s no need to add the potential for delays and higher costs from managing multiple vendors.
Comprehensive Solution Providing Time and Financial Savings
The SMART FHD plan offers a seamless package from the availability of the drug substance to delivery at the clinical site. The package provides a ready-made bundle of DiC development and flexible manufacturing, stability program management, packaging and labelling for clinical demand, regulatory dossier compilation, distribution, and clinical supply management.
The SMART approach is optimal for FHD studies as it provides the flexibility needed for dose-ranging studies. Eliminating timeconsuming processes and delays over formulation options and having the drug product supply available before any regulatory and clinical site approvals enables an FHD trial to start months faster.
SMART FHD distinguishes itself by providing a white-glove partnership instead of standard transactional manufacturing services. The holistic oversight capabilities encompass material management and inventory planning enhanced by strategic distribution capabilities through Canada for North American studies. Access to this facility circumvents typical IND filing delays but also facilitates an expedited supply chain, significantly reducing time to trial. The predetermined package and unique supply chain remove delays that would otherwise add unnecessary time and increase costs.
The Power of Partnership – The CRO & CDMO Relationship
Traditionally, the relationship between CDMO's and CRO's is very transactional or even non-existent if the client doesn't make that initial introduction. Once the drug is shipped by the CDMO there’s often little to no communication between the two parties. Collaborating with a CDMO and CRO in tandem presents a strategic opportunity to move through the lifecycle of a novel drug investigation efficiently. By having in-house Clinical Supply Managers, the CDMO can maintain that connection and prepare clients for the next phase of their clinical trial. This partnership offers a more complete package that goes from drug substance all the way to clinical data for phase I trials and potentially beyond.
PCI and Worldwide together provide a comprehensive service beginning with drug formulation and dosing through PK/PD and healthy volunteer recruitment and testing, allowing for reduced costs and increased speed to market. Overall, a shared vision and aligned leadership between PCI and Worldwide function to streamline the FHD process across the board.
Excellence in Pharmacokinetic/Pharmacodynamic Studies, Site Selection and Patient Recruitment
Worldwide's PK/PD capabilities enhances the drug development process from start to finish. At our state-of-the-art manufacturing facility, specialised teams analyse PK/PD data in a bioanalytical lab and review it thoroughly to ensure a deep understanding of the relationship between kinetic and dynamic components. Having an experienced team behind a study provides rigorous and accurate data
on drug interactions with target engagement and efficacy, saving time and money when moving forward in trials. This preemptive analysis also helps avoid delays later in the research, particularly concerning dosage adjustments.
Engaging with Worldwide’s PK/PD services for drug trials streamlines the development process, from initial dose planning to dose escalation in later trials. This efficiency is partly due to automated bioanalytic capabilities that reduce the risk of inaccuracies that traditionally arise during manual testing and partially from the expertise of the scientific staff. Worldwide has an extensive automation suite with various liquid handling workstations and microplate management systems. Beyond, Worldwide’s resources and capacity allow for state-of-the-art instrumentation to cover all drug discovery needs, spanning high-throughput mass spectrometry, Spectramax, MSD, Microlab Star, and liquid scintillation counter and oxidiser, which supports radiolabeled absorption metabolism and excretion (AME) studies. When combining the automated lab capabilities with staff that average more than 15 years of bioanalytical experience, the opportunities for research increase exponentially. The team prioritises data integrity, traceability, quality control, and on-time records, touting a 100% sponsor audit pass rate and a clean record for regulatory inspections. When working with Worldwide, you can expect direct and individualised attention from the in-house PK and biostatistics staff with the added assurance of knowing that their lab space is GLP and Part 11 compliant.
Whilst managing a novel drug investigation can at times be stressful, leveraging Worldwide's expertise in drug discovery minimises common challenges, particularly in solid dose formulation. where expertise extends beyond the lab to include site selection and patient
recruitment, adding value to clinical projects and helping to sidestep potential setbacks and unnecessary stress.
Worldwide delivers continued excellence in early-phase drug development through expertise in precision targeting of nuanced patient populations and maintaining the highest quality standards of scientific and operational acumen. Worldwide collectively provides a wide range of support throughout Phase 1 drug studies beyond PK/ PD in their expansive network of sites and participant recruitment, leveraging their highly differentiated contributions to the drug development industry. The innovation expands beyond participant recruitment; the intentionally located 200-bed facility close to their cutting-edge bioanalytical lab facilitates rapid data output that you can trust and position your drug to lead the industry.
Unique Innovation with an Opportunity for Expansion: The Future of Drug Development
PCI is looking forward with the SMART FHD initiative, which promises to set a new standard in the market for solid oral medications. With PCI's innovative SMART FHD offering and Worldwide's expertise in Phase 1 trial execution, together we provide a comprehensive solution that encompasses every aspect of development to get you from a molecule to clinical data available in the fastest time possible.
Looking forward, PCI also plans to revolutionise the pharmaceutical landscape by increasing the scale of this offering enabling a commercialisation strategy with a DiC formulation. By avoiding the delay and expense of formulating, there is the potential to reach the market years ahead of the traditional model with significant cost savings. Additional enhancements to the program are investigating the expansion of the SMART concept to include sterile injectable dosage forms.
Edward Groleau
Ed Groleau has over 30 years of experience in pharmaceutical drug development on both the pharma and vendor sides. The first half of his career was spent in various laboratories from analytical method development, to solid state characterization and polymorph screening, to stress degradation and chemical characterization. He left the labs and moved into clinical supplies in 2003 providing all aspects of CSM support to maintain clinical programs. He joined PCI in 2018 and became Sr. Director of PCI’s Supply Management And Readiness Team (SMART).
Rethinking Big Data Will Lead to New Advances in Personalised Medicine
Accelerating Time to Patient
The life sciences industry has waited a long time for big data to transform the potential of personalised medicine. With AI and machine learning now coming of age, R&D teams can finally seize the opportunity – as long as their data is also clean, standardised, interoperable, and secure.
To understand a treatment’s potential for a specific patient, biopharma companies have to layer together data from multiple disparate sources. Some of these data sources will be common to all disease areas: for example, patient demographics, electronic medical records, and quality-of-life scores. However, the majority (including genetic information, imaging, and activity data from wearable devices) will be unique to each individual. Since the clinical effectiveness and safety profile of a personalised treatment will be different from patient to patient, all relevant stakeholders must be able to trust the data to make medical and business decisions confidently.
Reappraising the approach to quality, ownership, and interoperability will bring usable data to the core of their strategy, even when working with potentially millions of relevant data points. Leading biopharma companies are also rethinking their existing ways of working and systems to get to first-time-right submissions. With access to a clean data foundation, they can identify which functional areas are most critical to speed time to market so that patients aren’t left waiting for innovative new treatments.
Historically, data collection initiatives have been broad in ambition and scope. These ranged from sequencing, imaging, and electronic health record data to text-based information such as interactions with health authorities and conference abstracts. The main objective was data completeness, and the scale of data points collected made it challenging to spot patterns or identify the most effective uses.
Today, the go-to-market and approval requirements of personalised treatments are more complex than anything seen before. As a result, biopharma companies seek to make appropriate use of their study data far sooner, which shifts the focus from data collection to governance and ownership. Gaining more control and oversight will change the dynamics in their relationships and contracts with third parties. Connected systems are becoming critical so relevant stakeholders can view the data at any time rather than waiting for data to be sent back in meta-data formats, or final text-based documents.
It is also becoming easier for sponsors to pinpoint the most impactful inefficiencies during the clinical development phase – vital to compressing time to market so that personalised medicines remain commercially viable. Analysing data on the cycle times between two critical clinical milestones could indicate whether inefficiencies and operational challenges typically arise during protocol design, site selection, initiation, or elsewhere. These insights can support the whole organisation to become more productive. A single source of accurate data can create competitive advantages by driving better decision-
making on patent filings or patient recruitment, or efficiency gains in outsourcing, procurement, or portfolio rationalisation.
Analytical and data science capabilities have improved but limitations remain. Raw data is not standardised and limited industry (or even intra-company) reference models exist. If common pain points around cleaning, ownership, and standards can be resolved, the volume and frequency of access to study data will increase. This will require a transparent data model with stringent user access controls to address privacy and cyber-security concerns.
Useful Big Data Requires Clean Data
Clarity of purpose is key when prioritising data initiatives. If the objective is to significantly reduce the time from ‘first patient, first visit’ to database lock, it's best to choose a group of experts before data is collected and cleaned to decide the approach and exact use cases. Data scientists, subject matter experts, and even external experts (e.g., HCPs, KOLs), could all help to make decisions and test hypotheses for improvements for this key clinical development milestone.
Many biopharma organisations have the right people and technology in place but struggle with effective governance. This may require collaboration between functions that have not worked
together much, such as research and basic science, business, and IT/ digital. Leadership commitment is a prerequisite for companies to start thinking this way. Management teams must test, learn, and further experiment with different working models before deciding which one best suits the company’s culture.
Once cross-functional roles and responsibilities have been defined, people, processes, and technology must align to wider corporate goals, the agreed problem statements, and hypotheses. Agile resourcing is essential. An urgent drug safety issue, for example, could have immediate clinical (and downstream commercial) repercussions for a company unless the right experts come together to tackle the challenge. Clean data sets from a single source of truth are critical for statisticians, molecular biologists, chemists, medical experts/ geneticists, and data scientists to work through to get the drug back on track.
Once the use cases for big data have been well defined and executed, we will see closer collaboration between teams within organisations all working toward a common goal. The result will be higher-quality documentation, reduced cycle times, and more rightfirst-time submissions. The growing impetus for direct data APIs with regulatory and health authorities (HAs), and potentially contract research organisations (CROs) and other third parties, could lead to more cooperation. The benefits of faster regulatory decisions will be felt directly by patients.
Smarter Data Use Throughout Drug Development
The costs and risks of developing personalised medicines challenge even the most efficient R&D functions. Now, we need a mindset shift to: “There are no big problems, there are just a lot of little problems.” Pivoting to smarter data use will help companies break down the long and complex drug development journey, and pinpoint which ‘little problem’ to solve first.
When big data is clean, standardised, and interoperable, other exciting possibilities can be explored, such as finding novel biological targets or net new patient populations. Eventually, a centralised approach to data management could support the long-held ambition of connecting real-world data (such as patient data, electronic medical records, digital therapeutics, etc.,) to clinical development, so we can improve the patient experience.
These advances move us toward the shared goal of providing lifeenhancing medicines to patients who need them.
Register for Veeva R&D and Quality Summit, Europe, and learn how to advance patient innovation
Stephan Ohnmacht
Stephan Ohnmacht, is the Vice President, R&D Business Consulting, Veeva Systems
With over twenty years of experience in the healthcare and life sciences industry, Stephan began his career as a researcher and scientist, and holds a Ph.D. in Organic Chemistry from the University of Edinburgh. For the last twelve years, he has brought his in-depth industry knowledge and expertise to the consulting sector. In his current role as Vice President, R&D Business Consulting at Veeva Systems, Stephan heads up Veeva's R&D consulting offerings in Europe, which is based around Veeva’s products and software, combined with Veeva's unique Vault industry data.
Reviewing Research Participant Payments Through a Diversity Lens
Whether and how a participant is compensated for taking part in a clinical study are two of the decisions an institutional review board (IRB) makes when approving research. The IRB must ensure participants are not unduly influenced to join the research and that the research is just, beneficial, and the participants are respected. Participant payments can include reimbursement of reasonable expenses for travel to and from the research site, fair compensation for the participant’s time, effort, burden, and inconvenience, token payments of appreciation, etc. A detailed description and analysis of participant payments was carried out by the Secretary’s Advisory Council on Human Research Protections (SACHRP),1 and this article uses the terms described by SACHRP.
Protecting Clinical Trial Participants IRBs are tasked with protecting the rights and welfare of research participants. IRBs came into being as a result of the Belmont Report.2 The Belmont Report established three ethical principles: justice, beneficence, and respect for persons. These principles form the basis of the U.S. Food and Drug Administration (FDA) and Office of Human Research Protections (OHRP) regulations governing research with human participants. Both agencies publish guidance to help IRBs interpret the regulations. In 2022, the DEPICT Act was passed, requiring applications to the FDA for an investigational use exemption for new drugs and devices “to include information about the demographic diversity of the clinical trial population and addresses related issues.”3
The three pillars of the Belmont Report are broad concepts. IRBs have further refined respect for persons to focus on the idea of autonomy. This concept undergirds ideas around informed consent and freedom from undue influences. Participant payments have historically been viewed through the lens of undue influence and that has led IRBs to minimise payments in order to allay those concerns.
More recently, scholars4 and SACHRP have published work supporting the idea that lowering payments does not decrease undue influence. In fact, they report that this approach can negatively impact respect for persons by failing to acknowledge the full context of participants’ lives and the sacrifices in time and effort they must make to participate in a research study.
Research should also be just. The burdens and benefits of research should be accrued by all. As outlined in the DEPICT Act, the industry has fallen short of ensuring that research participant populations reflect the general population, and this is a failure of justice as well as science. As IRBs evaluate diversity plans, one area warranting particular attention is participant payment or compensation.
Appropriate payment to research participants is critical for diversifying clinical research participation. When payments and reimbursements to research participants are limited, those who have less free time, less available income, or more burdensome lives are less likely to participate, resulting in a study with a participant population that does not reflect society. Sponsors, sites, and IRBs all play a role in approving participant payment plans, but payment structures are often not transparent, and decisions may be made without a deep understanding of how differences in payments affect participant enrollment and retention.5
Data Analysis of Participant Payment Plans
To explore how participant payment plans have been structured and described to potential participants, we conducted a systematic review of research payment plans during the past five years. We analysed the participant payment language from more than 7,500 site-level consent forms reviewed by an IRB between 2019 and 2024 in a wide variety of therapeutic areas across the U.S. The analysis was carried out using artificial intelligence (AI) to efficiently summarise this large data set. Trends and patterns in compensation are described, creating a baseline data set.
Methods
SACHRP published recommendations on participant payments, including commentary on FDA and OHRP guidance documents, in 2019. Its analysis provides a framework for thinking about undue influence and how different types of payments to participants can be categorised.
Common sense business terms are used in site budgets to describe participant payments. But there is no consistency in how the industry applies terms such as stipend, compensation, and participant payment. The following summary in Table 1 combines the SACHRP recommendations and a sponsor’s experience in the space and reflects the concepts featured in the data analysis.6 This underscores the need for standardisation and consistency in terminology.7
We started this study with 14,000 documents, which included extracts of payment information from each site for a given clinical trial that had been reviewed by the IRB. The documents were developed as part of the normal IRB operations to provide the board with a summary of the previously approved payments for a given study. The documents included unique site identifiers and investigator surnames in addition to the specific payment language from the informed consent document. This identifying information has been removed from this analysis, and only aggregate results are reported.
Once this data set was produced, we extracted payment language, terms, and components including payment amounts from the data using ChatGPT4. Average payments per visit were extracted, and they were allocated to various categories such as meals, travel, stipends, etc.
Payment
Term used to show that research participants may be paid for time, effort.
Stipend A fixed sum of money paid periodically for services or to defray expenses.
Reimbursement
Appreciation
for out-of-pocket expenses.
Compensation addresses the participant’s contribution of time and acceptance of research-related burdens and inconvenience, as distinguished from the out-of-pocket costs addressed through reimbursement.
Small payments or gifts that are not intended to meaningfully reimburse or compensate study participants.
Incentive Payment Payments beyond compensation for time and effort intended to encourage study recruitment and retention.
Completion Bonus Payments beyond compensation for time and effort intended to encourage study recruitment and retention.
Table 1: “X” represents the inclusion of this payment term in a given analysis.
Originally, we intended to calculate the total amount of compensation over the course of the trial. After a first refinement of the data, we realised that most payment language was structured as “per visit.” To calculate total compensation across the trial, the total number of visits would be needed. Given the wide variety of ways in which study visits were described in other sections of the protocol and informed consent document, we abandoned this effort, and concentrated on per visit compensation. The output from seven training cases was reviewed in detail to confirm that the system was correctly identifying payment terms. It took just over one hour of researcher time to analyse the payment document and accompanying data for the seven training cases using traditional methods.
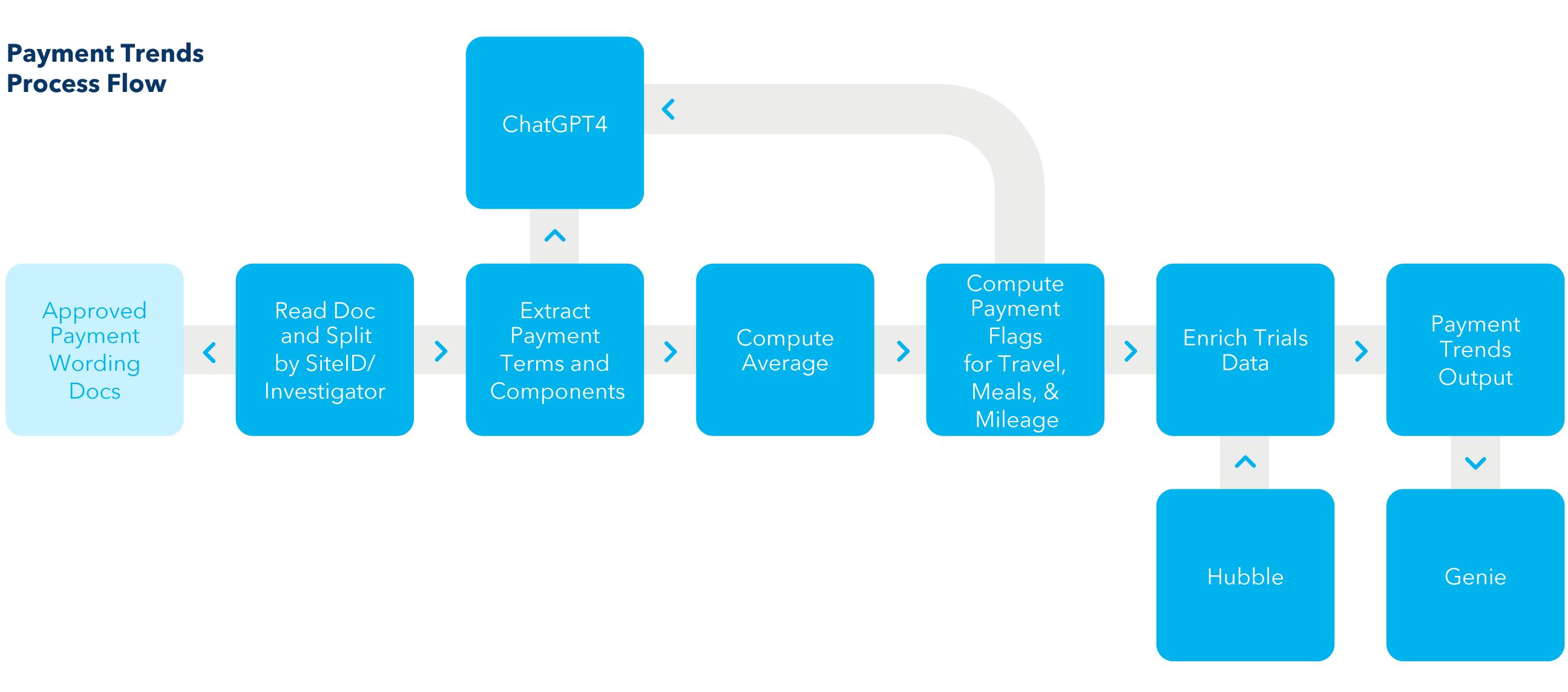
The documents were then matched to IRB site meta data to create the full data set. Because investigators change over time, we were unable to match all the studies to facility information, and we reduced our list of analysed documents to about 7,500 for analyses involving site location data, i.e., facility type and zip code. Figure 1 is a representation of the process.
Of significant note, before delving into the data analysis, is the uncertainty in terminology usage when describing certain payments. It cannot be discerned how certain sponsors are using the term stipend, while others opt to provide payment for time or effort as compensation. While stipend is commonly used across
1. Hubble is a central internal data repository used to match the results to clinical trial level meta data. Genie8 is a conversational natural language interface employed to facilitate analysis of the results.
Figure
the industry and appears in the data set, it is not a recommended term because it combines expenses and payment for services. Meanwhile, with the term participant payment it likewise cannot be discerned whether this is a separate payment for time and effort or if it includes reimbursement for expenses.
As such, the amounts allocated as stipend, compensation, and participant payment may include reimbursement amounts if that is how the language was used in the consent documents. More importantly, separating expense reimbursements from payments for time, effort, burden, and inconvenience (compensation) helps
distinguish true income from expenses for tax reporting purposes. This is crucial for participants who may decline to participate for fear of losing SNAP (Supplemental Nutrition Assistance Program) benefits because they may exceed the income threshold.
In summary, this is a call to action for the industry to agree on standards with respect to participant payments by (a) aligning on terminology for payment categories, and (b) separating reimbursements for expenses from compensation for time, effort, burden, or inconvenience. This will help alleviate some impact of the financial burden on research participation.
Figure 2
Figure 3
Figure 4
Market Report
Results
Prompt A:
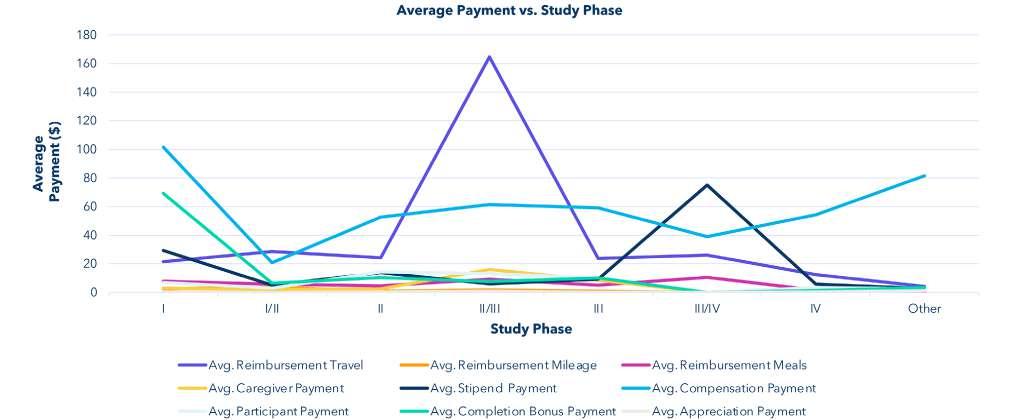
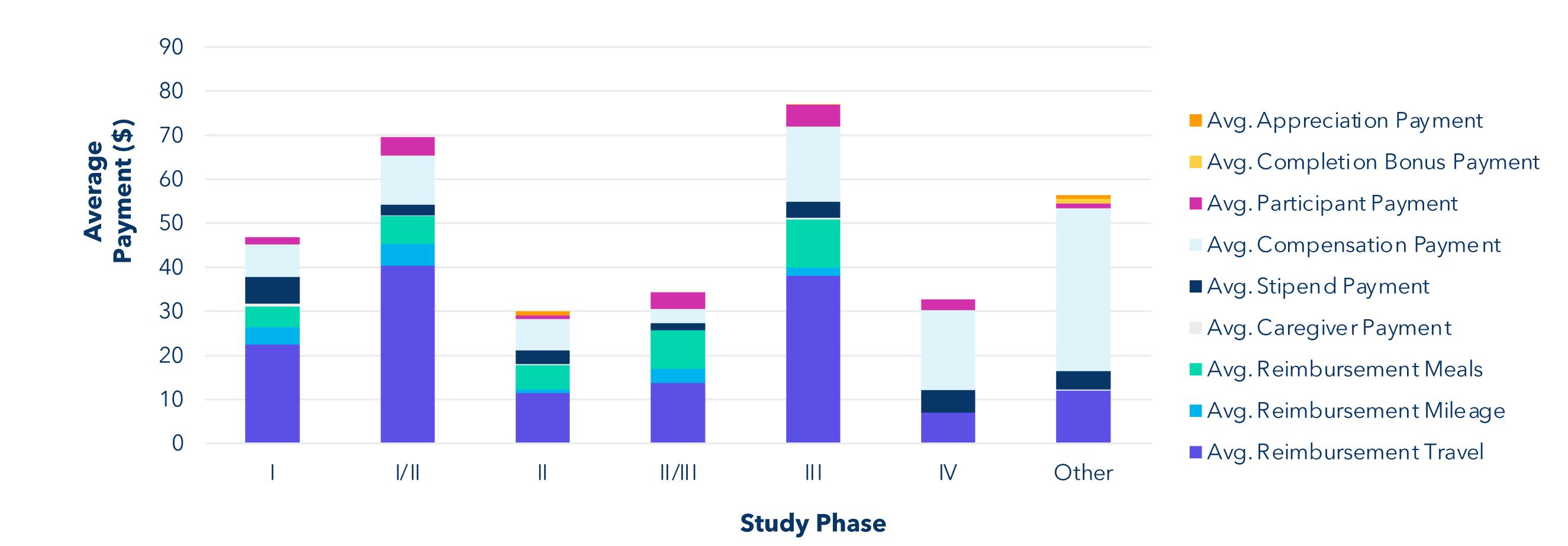
Find the average payment of each payment type based on the phase of the study. Ignore the rows where phase field contains the string "null." Add a new column to the table showing how many rows have that phase. The phase values in the table should follow the order: I, I/II, II, II/III, III, III/IV, IV, Other.
Key Insights
The highest total compensation is clustered around phase I and phase III/IV studies. “Other” studies are primarily composed of a mix of medical device trials, and any other research study that is not a clinical trial involving a drug, and which does not have a phase. The highest total average payment based on aggregate data is for travel reimbursement.
Prompt B:
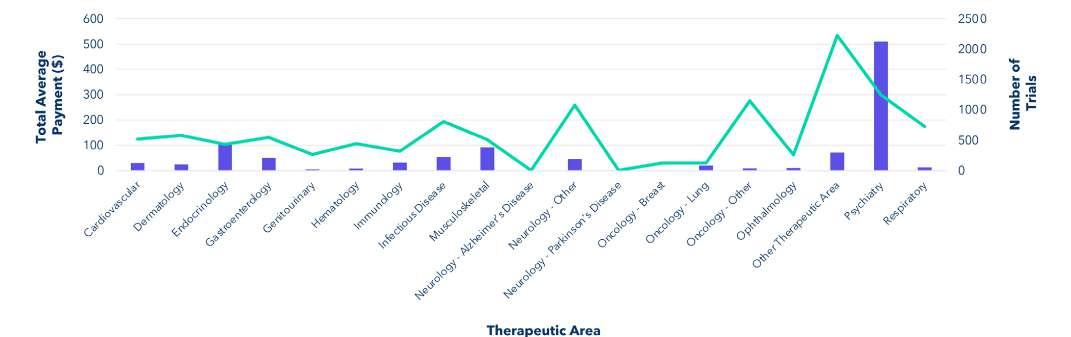
Find the total average payment across different therapeutic areas. Ignore the rows where the therapeutic area field contains the string "null" or "Not Specified." Add a new column to the table showing how many rows have that therapeutic area.
Key Insights
There is a wide range of compensation paid per visit for different therapeutic areas. Psychiatry trials represent the high end of this range and breast cancer trials represent the low end. The psychiatry category includes several in-patient, healthy-participant trials with larger than average payments and larger completion bonuses due to the nature of those studies. In future work, we intend to examine trends within therapeutic areas, but we did focus on oncology trials which make up more than 12% of all trials in the data set. Additionally, we have received anecdotal information from both sites and sponsors that they believe IRBs do not approve payments for oncology trials.
Prompt C:
Provide average payment for each payment term for various phases of studies where the therapeutic area contains the string "Oncology." Ignore the rows where the phase field contains the string "null." Add a new column to the table showing how many rows have that phase. The phase values in the table should follow the order: I, I/II, II, II/ III, III, III/IV, IV, Other.
Key Insights
While average payments for oncology trials are on the lower end, we do see several payment categories for oncology participants across all phases of studies. We hope this data serves to dispel the myth that IRBs do not approve payments for oncology trials.
Conclusions
Using ChatGPT4, we were able to analyse five years of data from an independent IRB. In 2023, this IRB participated in reviews of more than 90% of drugs approved by the FDA. The full data set involved more than 7,500 records. It allowed us to determine both the types of payments being offered to participants and how they were described in informed consent documents. We also learned how payments varied across study phases and therapeutic areas. We hope to provide access to this baseline data set to help sponsors and sites design trials with just and inclusive payments for participants to better reach underserved populations, increase access, and achieve more diversity in clinical trials.
REFERENCES
1. Attachment A – Addressing Ethical Concerns Offers of Payment to Research Participants. https://www.hhs.gov/ohrp/sachrp-committee/ recommendations/attachment-a-september-30-2019/index.html
2. The Belmont Report. https://www.hhs.gov/ohrp/regulations-andpolicy/belmont-report/index.html
4. Largent EA, Lynch HF. 2017. Paying Research Participants: The Outsized Influence of “Undue Influence.” IRB 39(4):1–9. PMID: 29038611; PMCID: PMC5640154
5. Anderson, E. E., & Brown, B. (2021). A Call for Radical Transparency Regarding Research Payments. The American Journal of Bioethics, 21(3), 45–47. https://doi.org/10.1080/15265161.2020.1870763
Kelly FitzGerald, PhD, CIP is Executive IRB Chair and Vice President IBC Affairs at WCG. She oversees the review teams for the IRB and several hundred IBCs and is responsible for their compliant and efficient operation.
Donna Libretti Cooke
Donna Libretti Cooke, JD is a Clinical Operations – Specialised Consultant. In her prior role, she was Director of the Global Contracts & Budgets team for Phase II–III clinical trials and Phase I oncology studies at Bayer.
Research and Development
Designing and Executing a Robust Feasibility in Rare Disease Clinical Trials
Global Feasibilities
A well designed and executed feasibility which builds on validated data is crucial for a successful clinical trial and the design of a solid product development plan. Ideally the feasibility should be impartial and results should solely be data driven.
Introduction
Rare disease studies, especially those conducted in paediatric populations, present considerable challenges at a country, site and patient level due to their complexity and the fact that many stakeholders need to be involved. A thorough and well-planned feasibility will allow sponsors to gain valuable insights enabling important decisions to be made, most importantly if the study is feasible to be conducted at all and if yes, where it will be conducted, under which conditions, how much support and intervention will be required and how long recruitment will take. All these factors will also affect the cost of the study. Only when a robust feasibility is conducted can these important questions be answered and an appropriate study execution strategy be developed. The more accurate the feasibility data the more likely any risks can be mitigated. Clinical trial feasibility is the best means for predicting the speed at which Investigators will enrol patients. The outcome may also be useful for pre-identification of patient pools for future studies, registries or for marketed products. In the industry it is common practice that feasibility is performed quickly and usually are superficial in nature, where the focus is on immediate study start and quick site set-up. Investors for example prefer to see more near-term KPIs which allows them to validate their investment progress as early as possible. Consequently, instead of focusing on when the last patient will enter the study, more attention is paid to when the first patient will be recruited which in the worst case scenario can lead to slipping timelines for drug approval. According to recent studies, identifying sites in rare disease studies is one of the most difficult tasks. A well designed and executed feasibility which builds on validated data is crucial for a successful clinical trial and the design of a solid product development plan. Ideally the feasibility should be impartial and results should solely be data driven. This white paper explores critical factors that should be taken into consideration when conducting feasibility studies.
Data Review, Epidemiology and Patient Identification
A high-level strategy would be to determine where patients with a particular rare disease are located around the world and to find where the highest concentrations of the patients exist, keeping in mind that practicalities like patient’s access to join the clinical trial are highly significant. The importance of data searches to pre- identify possible countries and sites is paramount, especially when no established database of Investigators for many of the rare disease indications exists and when information is scattered. In case very few patients are scattered globally, which may be the case in ultra-rare diseases, one strategy can be to first target countries with a high population
and density, relying on probability of finding more affected patients. Considering all aspects, it may still be useful to conduct feasibility in many countries and not be too restrictive. In some cases, pockets of a disease are found in certain regions, which may be due to factors such as consanguinity.
There may be countries that are not considered for various reasons, e.g. political unrest, but it would be prudent not to eliminate countries for reasons of perceived challenges too quickly. Taking a balanced approach between selecting high risk countries promising higher patient numbers and lower risk countries with lower recruitment potential should be considered especially in ultra-rare diseases. Once a thorough feasibility is performed and all relevant facts are known, an informed decision can be made. Extensive data research needs to be performed to consolidate all information that is already available about the disease under investigation. Availability of data will depend on how “common” a rare disease is, e.g. for more common diseases like Cystic Fibrosis a lot of data is available whereas in cases of ultra-rare diseases data is most likely to be scarce. Data for emerging regions such as Asia Pacific, South America and Africa may also be limited. As a starting point and where possible, epidemiology data should be established, however incidence and prevalence rates alone will not provide enough information to include in which countries the feasibility should be conducted. Prevalence in conjunction with the defined target population as per the protocol however will give a much better guidance which countries to choose.
Other information that will support the decision-making process with respect to narrowing the country selection and tailoring the feasibility according to the protocol requirements include:
• Genetic variability from country to country or regional variability
• Start-up timelines and regulatory environment
• Patient pathways and standard of care
• Competing trials
• Patient advocacy, support groups and social media
• Patient participation and travel support
• Availability and reimbursement of supporting concomitant medications or therapies
• Set-up of health system, referral networks and patient access
• Sites with clinical trial experience but lower population vs. clinical trial naive sites with higher patient population
Genetic Variability from Country to Country or Regional Variability
Depending on the degree of variability and the protocol requirements, this factor may not play such an important role. However, data should be collected and taken into consideration. If required, the sending of samples to genetic screening labs should be explored.
Start-up Timelines and Regulatory Environment
Assessing study start-up timelines in each potential country and comparing them to Sponsor projected timelines for study completion
(if available) is of significant interest when determining where to conduct feasibility. Feasibility teams should have knowledge of country specific challenges from experience and be able to provide valuable information such as overall start-up timelines from preparing regulatory submission to SIV, including specific Regulatory, Ethical and site contracting timelines with import and export licences as required. Country-specific nuances need to be taken into consideration too. For example, India does not usually allow Phase I studies, however under special circumstances approval may be granted for rare disease studies where there is no available treatment and the risk of the disease burden outweighs that of receiving treatment.
Patient Pathways and Standard of Care
When planning a clinical trial, it is essential to know the standard of care on a global, country and even site level. Sometimes guidelines on treatment exist which may provide some uniform guidance within a country or region such as the EU. Other countries in more emerging regions will most likely have a site-specific standard of care. It will be necessary to obtain further information for the specific indication by approaching each potential site. The patient pathway is of extreme importance for recruiting in rare diseases. As many rare diseases are affecting many organ systems, these patients are seen by many specialists. It will be important to determine how and by whom the patient is treated. This will enable your feasibility efforts to be directed at the appropriate personnel, where you may need to approach different disciplines for the same trial. Thus, enabling you to identify the most appropriate investigators for the clinical study. Some patients will only be eligible once the disease is active or severe and may first be seen in the emergency department. Knowing the patient flow at site and where these patients are coming from will ensure a maximum of eligible patients are directed towards the clinical trial and patients are not unnecessarily lost.
Competing Trials
Rare disease trials competing for the same or similar patient population can be a key concern, as there is a limited patient pool and limited number of key specialists working in the field. When selecting countries and sites it is prudent to take into consideration the competitive landscape, such as: number, location, phase and size of trials being performed in a country and at each site. The knowledge of existing marketed products or products close to market, in your specific indication in any particular country, may influence the decision on country selection. Information is readily available in databases such asclinicaltrials.gov and in the EU Clinical Trials Register. In addition, there are also country specific databases where all trials being performed in the country must be registered. However, the feasibility team should appreciate that from these databases a complete list may not be available and it will be best to ascertain the current status individually at each site. It may also be possible to get an indication of future planned trials at the site. If after careful
Research and Development
consideration it is not possible to avoid placing the trial at a particular site with other ongoing trials in a similar or the same indication, a detailed dialogue needs to take place with the site personnel outlining the recruitment strategy and impact on resources.
Patient Advocacy, Support Groups and Social Media
Each country and disease indication has patient advocacy and support groups that are established at different levels. However, it is less likely that groups are formed in emerging countries where disease specific advocacy is not set-up and patients may connect to umbrella organisations that cover the therapeutic area.
Identifying and connecting with these groups and engaging with them can support the trial in the following ways:
• Financing early development of clinical trials
• Providing early feedback/ input into the protocol design, to determine if the study is acceptable from a patient’s perspective
• Helping drive disease awareness
• Enhancing recruitment through connecting patients and running forums
In many developed countries social media is widely used to connect patients and during the feasibility social media channels can be identified that are disease specific and connect the community.
Patient Participation and Travel Support
Whilst conducting the feasibility it should be investigated if country and regional factors will influence patient participation. In some regions such as the Middle East it is part of the culture to involve the larger family and relatives in the decisionmaking process, before giving consent. The intensity of patient involvement in a trial may affect the consent process. Factors such as the extra burden to the carer, need for hospitalisation, missing school, frequent visits, repeated blood sampling and large travel distances may deter patients. For countries and sites where patient travel and additional support can be provided, a higher recruitment may be seen. Patient travel support can be considered for patients that are referred to a central location within a country where a key specialist is located.
Availability and Reimbursement of Supporting Concomitant Medications or Therapies
Lack of reimbursement or unavailability of concomitant medications / therapies that are required as specified in the protocol may result in the inability of the study to proceed in a particular country. Availability of already marketed products for the indication can also influence recruitment. Set-up of health systems, referral networks and patient access Patient access to specialists will be a requirement to conduct the trial and in four countries where there is access to electronic health records, patients can be more
Research and Development
easily identified. A key component to take into consideration is to validate the recruitment projections at any one site. This activity can be performed by reviewing historical information regarding patient numbers and reviewing anonymized patient data. Referral networks are set up for some indications especially in Europe and the US, however referring a patient to another site may also have its challenges, if patients have to travel long distances or are treated by a different specialist in an unfamiliar environment, where they are less likely to give consent.
Site and Investigator Selection
Selecting the right sites is fundamental to every clinical study, but the impact is greater if the wrong sites are recruited in rare disease trials, where misjudgements in the site selection can potentially lead to complete trial failure. The first step should begin with designing a detailed feasibility questionnaire, tailored to the specific study. Assessing Sites in a systematic way will give an indication of the sites’ competency to conduct the trial in terms of experience, resources, medical knowledge and patient access. Site infrastructure the type of site and infrastructure can vary considerably. It should be determined what kind of equipment, calibration and tests are available and required as per the protocol. Alternative sites may also be used to conduct some of the tests and additional equipment may need to be supplied.
Evaluating Investigators
Site specific experience in the disease under investigation is a clear advantage and investigators are more likely to be able to input into protocol development and design and be able to tell if study specific procedures are in line with existing medical practices. However, when finding sites on a global scale it is not always the case that all sites will have worked in the defined indication. Varying levels of experience is to be expected and occasionally it will be possible to transfer experience from a similar study indication. Sites with no experience should not be immediately excluded, especially if they have a good recruitment potential. Instead, it should be determined which kind of training is required, such as indication, protocol, assessments and GCP specific training. In some cases, site support is recommended, especially for sites in emerging countries where both experience and/ or site resources are scarce, but the investigator remains motivated and is willing to participate.
Providing a site coordinator can be beneficial and this strategy tends to work well, however most likely extra associated costs will be generated and this has to be weighed up with the number of patients the site will contribute to the overall study.
Patient Recruitment and Retention
Subject enrolment is particularly more complex when conducting rare disease studies, especially in paediatrics and can be considered the most important part of the feasibility. The recruitment potential of each individual site, indicated by subjects per month or over the entire duration of the study when low patient numbers are expected, should be used to track predicted vs actual recruitment. There are many factors that the investigator must take into consideration when providing an estimation of the recruitment. It is not sufficient to only consider the inclusion and exclusion criteria when other factors such as patient motivation and concomitant medications will also drive recruitment and eligibility. Often, patient recruitment is overestimated by the site which is seen in indications where there is diagnostic uncertainty as in complex rare diseases. Equally important is for the investigator to give a good prediction of screening failure, reasons for failure, drop-out rates and possible retention figures. Thus, once a completed feasibility questionnaire is received the information must be discussed in detail with the investigator by senior personnel within the feasibility team. Data should be confirmed and where
possible historical and current data should be validated and checked regarding patient recruitment. This may involve checking patient medical records or site databases, whilst adhering to data protection regulations in the specific country.
Data Mining and Undiagnosed Patients
Several data mining companies are now established that have access to patient data covering specific regions of the world. They usually have access to large data and patient records which include data on prescriptions and concomitant medications. These are then used in combination with refined algorithms to find both diagnosed and undiagnosed patients. With some accuracy it can be predicted if an undiagnosed patient is likely to have the disease under consideration. However, these services are usually engaged as a second avenue to find patients when recruitment is not on target.
Conclusion
Site selection as one of the most important steps in any trial can be a daunting task especially with the choice of trial countries expanding in recent years and the absence of an organised global network of trial centres in the rare disease arena. Rare disease clinical trial feasibilities should be approached in a holistic way moving away from a one size fits all approach as only a tailored feasibility will lead to a successful and efficient patient recruitment strategy. Listening to the client needs, their objectives and experiences should be the starting point to facilitate the design of a robust feasibility. It is important to work with the experts within the sponsor organisation to review the important criteria. Feasibilities should not be hurried as when they are performed within short timelines many aspects may be overlooked including the patient’s perspective. With appropriate resources and taking into consideration the above points, a comprehensive feasibility can be performed in six to twelve weeks. Aspects that can affect the delivery time can be the size of the feasibility, holidays, need of CDAs to be signed, review and approval of questionnaires etc. which all need to be factored in when setting up timelines.
At the conclusion of the feasibility process and when the data has been scrutinised and confirmed, a customised report should be produced that includes recommended sites to be further explored and identifies potential high enrolling sites. A comprehensive report highlighting challenges when selecting certain countries and sites will be of great value. Analysis of the feasibility data may show that it is necessary to conduct the trial in countries where there are certain underlying risks. Sometimes mitigation strategies can be put in place to address or reduce these risks. The sponsor should be made aware of the challenges as part of the feasibility, so an informed decision can be made when selecting the final sites for the clinical trial. Feasibilities when conducted with diligence and accuracy can lead to the successful trial completion within the planned timelines.
Emmes
For over 45 years, Emmes' team of 1,600+ experienced professionals have provided the full range of Contract Research Organization expertise necessary to conduct clinical research with a firm scientific basis that is fully compliant with national and international regulatory guidelines. With offices throughout the US, Europe, Canada, Latin America, and India, Emmes supports the advancement of global public health and biopharmaceutical innovation through disciplined science, rigorous research, fact-based decision-making, and operational excellence.
Liver Disease, Obesity and the Value of Treatment
Since 1990, the prevalence of obesity has more than doubled. In 2022, nearly one billion people were living with obesity.1 The health risks of being obese or overweight are well understood and include higher mortality and morbidity due to cardiovascular disease, diabetes, cancer, neurological disorders, chronic respiratory disease, and gastrointestinal disease. Here we discuss the hepatic complications of obesity and the value of treatment for obesity for those liver complications.
Metabolic-dysfunction associated steatotic liver disease (‘MASLD’) is found in the majority of obese patients and the prevalence correlates with the degree of obesity (i.e., the higher the grade of obesity the greater the likelihood of finding MASLD).2 Of patients with MASLD, 20–30% will have an inflammatory, fibrotic form of steatotic liver disease called Metabolic-dysfunction Associated SteatoHepatitis (‘MASH’) and 10–15% of MASH patients will progress to cirrhosis.3 Among patients with co-existing type 2 diabetes and obesity of all degrees, the prevalence of MASLD, MASH, and cirrhosis has been found to be 55.5%, 37.3%, and 17%.4 As well, obesity worsens the prognosis of patients with compensated cirrhosis due to other causes.5 Obesity is an independent risk factor for the development of hepatocellular carcinoma (‘HCC’) and is associated with a 2–3 fold increased risk for the development of HCC, a 4-fold increase in HCC-related mortality, and a 2-fold increase in serious surgical complications following HCC resection.6
Obesity results in multiple metabolic derangements that contribute to the development of steatotic liver disease and HCC. Insulin resistance and resulting hyperinsulinemia lead to diminished insulin responsiveness and failure of target organs to dispose of blood glucose. Subsequently, there is hyperglycemia, loss of inhibition of lipolysis, increased circulating free fatty acids and increased intra cellular free fatty acid intermediaries (lipotoxicity), decreased glycogen synthesis, and liver production of glucose.7 Lipid accumulation and lipotoxicity results from excessive lipid influx and impaired lipid export, and lipid accumulation in the liver is directly related to hepatic toxicity, oxidative stress, and endoplasmic reticulum stress. These stressors activate an inflammatory cascade that can recruit and activate inflammatory cells and trigger hepatic stellate cells ushering the transition from bland steatosis (MASLD) to MASH and increasing risk of HCC.8
Weight loss is the cornerstone of effective management of hepatic complications of obesity and is associated with improvement in all-cause mortality, MASLD, MASH, complications of cirrhosis, and HCC. A sustained weight loss of 5–7% of total body weight is required to achieve improvements in hepatic steatosis and hepatic steatohepatitis. Greater weight loss is required to show improvement in liver disease activity and fibrosis. Dietary modification and lifestyle interventions have been shown to achieve weight loss and
improvement in MASLD, but sustained weight loss is rarely achieved with lifestyle interventions alone.9
Bariatric surgery (BS) is an effective method to achieve substantial and sustained weight loss for patients with obesity. Depending on the procedure, performed weight reduction between 20–35% are anticipated after BS.10 Studies have shown marked improvement in hepatic disease after five years following BS with MASH resolution in 84%, fibrosis stage decrease in 70%, fibrosis resolution in 56%, and an 88% risk reduction in progression to major liver outcome. BS is also associated with a decrease in major adverse cardiovascular events and risk of obesity-related cancer.11 A BS patient cohort has been observed to have reduced progression to MASH or HCC as compared to a propensity-matched control group.12 Some have reported improvement and even resolution of cirrhosis after BS.13 Bariatric surgery is recommended for patients with a comorbidity with a BMI ≤35. It is recommended for patients without a comorbidity who have been unable to maintain weight loss with lifestyle changes with a BMI > 35. Unfortunately, BS is unavailable to or not feasible for many patients due to expense, healthcare resources, or patient preference.
Endoscopic bariatric procedures have emerged as a less invasive alternative to BS to facilitate similar weight loss less invasively. The anticipated total body weight loss for endoscopic procedures ranges from 10–20%. Each procedure (e.g., gastric balloon, transpyloric shuttle, endoscopic sleeve gastroplasty) works by reducing gastric volume and/or delaying gastric emptying. Some of these procedures are temporary and some patients experience weight gain after reversal.14 Similar to BS, endoscopic therapies have been found to improve metabolic parameters, reduce liver steatosis, hepatic inflammation and fibrosis in MASH.15 Long-term outcomes studies are ongoing to evaluate the efficacy of these procedures on liverrelated, cardiovascular, and cancer-related events.
Pharmacologic therapy for obesity has been available for more than two decades and is currently recommended in adults with a BMI ≥30 kg/m2, or ≥27 kg/m2 with one or more comorbidities. These medications include orlistat, phentermine/topiramate combination, naltrexone, bupropion/naltrexone combination, GLP-1 agonist, and GLP-1/GIP receptor dual agonist. Sustained weight loss with orlistat, phentermine/topiramate and naltrexone/bupropion ranged from approximately 6–10% in clinical studies. With the introduction of GLP-1 agonists and dual agonist therapy, otherwise known as integrin therapy, sustained weight loss has been observed in 8–20% of patients. The dual receptor agonists showed higher sustained weight loss efficacy. Patients treated with orlistat have been found to reduce liver fat on MRI content; serum markers of fibrosis have not been shown to decrease with treatment. No long-term data is available to clarify the hepatic effects of orlistat therapy.16 GLP-1 agonist therapy has been shown to improve liver steatosis on MRI, with a 2-fold increase in participants achieving a 30% reduction in liver fat over placebo. As well, histologic reduction in disease activity was observed
with GLP-1 administration. Long-term studies are ongoing to assess GLP-1 therapy in obesity and its impact on major liver outcomes and liver fibrosis.17 Dual GLP-1/GIP receptor agonist has shown the greatest benefit thus far in percentage of weight loss, up to 20%. Dual agonist therapy is associated with a 50% reduction in hepatic steatosis on MRI proton density fat fraction (PDFF), reduction of biomarkers of hepatocyte apoptosis and fibrosis. There are currently ongoing studies to evaluate histologic endpoints in MASH.18 A variety of triple agonists are in early clinical development. These triple agonists may be able to achieve weight reduction that rival BS. Unfortunately, the GLP-1 agonist and multiple agonist therapies share an adverse even profile that includes delayed gastric emptying, nausea, vomiting, diarrhoea and constipation. Many patients are unable to continue therapy to achieve the desired weight loss. Overall, up to 25% of patients discontinue GLP-1 therapy by three months and nearly one-third by six months before the benefit of therapy is achieved. Patients impacted by early discontinuation were disproportionately African American, Hispanic, lived in areas of high economic need, or were on public insurance programs.19 Further, the expense of these medications limit their long-term availability for many patients.
In conclusion, obesity is a growing health problem that impacts multiple organ systems. The liver is significantly impacted by the metabolic derangements associated with obesity. Weight loss, if achieved and sustained, can forestall and may even correct the hepatic complications of obesity. Recent advances in medical, endoscopic and surgical care have provided insight and hope for patients living with obesity.
REFERENCES
1. World Health Organization, https://www.who.int/news-room/factsheets/detail/obesity-and-overweight (last visited 30 May 2024).
2. Divella R, et. al., Obesity, Nonalcoholic Fatty Liver Disease and adipocytokines Network in Promotion of Cancer. Int. J. Biol. Sci 2019; 15(3): 610-616.
3. Armandi A and Bugianesi, Natural history of NASH. Liver Intl; 2021;41 (Suppl.1): 78-82.
4. Younossi Z, et. al., The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hep. July 4, 2019; 71(4): 793-801.
5. Berzigotti A, et.al., Effects of an intensive lifestyle intervention program on portal hypertension in patients with cirrhosis and obesity: The Sport Diet study. Hepatology April 2017; 65(4) 1293-1305.
6. Chen Y, et. al., Obesity, non-alcoholic fatty liver disease and hepatocellular carcinoma: current status and therapeutic targets. Front. Endocrinol. June 7, 2023; 14
7. Bansal S and Bansal M, Pathogenesis of MASLD and MASH – Role of insulin resistance and lipotoxicity. Alimentary Pharmacology and Therapeutics. 2024; 59(Suppl. 1): S10-S22.
8. Bansal S and Chen Y.
9. Storm A and Dayyeh B, Metabolic function and weight loss after
endoscopic sleeve gastroplasty: resistance is futile. Gastro. Endo; May 2021; 93(5) 1119-1120.
10. Alfadda A, et. al., Long-term weight outcomes after Bariatric Surgery: A single Center Arabian Cohort Experience. J. Clin. Med. Nov 2021; 10(21) 4922-4936
11. Aminian A, et. al., Association of Bariatric Surgery with Major Adverse Liver and Cardiovascular Outcomes in Patients with Biopsy -Proven Nonalcoholic Steatohepatitis. JAMA. November 11, 2021; 326(20): 2031-2042
12. Kwak M, et.al., Bariatric Surgery is associated with reduction in nonalcoholic steatohepatitis and hepatocellular carcinoma: a propensity matched analysis. Am. J. Surg. Mar. 2020; 219(3): 504-507.
13. Kral J, et.al., Effects of surgical treatment of the metabolic syndrome on liver fibrosis and cirrhosis. Surgery. Jan 2004; 135(1): 48-58.
14. Mauro A, et. al. A comprehensive review on bariatric endoscopy: where we are now and where we are going. Medicina. 2023; 59(3): 636-
15. Jirapinyo P, et.al. Effect of endoscopic bariatric and metabolic therapies on nonalcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. Mar 2022; 20(3): 511-524
16. Xiongcai F, et.al., Treatment of obesity and metabolic-associated fatty liver disease with a diet or orlistat: A randomised controlled trial. The American Journal of Clinical Nutrition Volume 117, Issue 4, April 2023: 691-700 https://doi.org/10.1016/j.ajcnut.2023.02.008
17. Chakhtoura M, et. al., Pharmacotherapy of obesity: an update on available medications and drugs under investigation. eClinical Medicine. Apr 2023; 58: DOI:https://doi.org/10.1016/j.eclinm.2023.101882
18. Machado M, MASLD treatment—a shift in the paradigm is imminent. Front Med. Dec 2023; doi: 10.3389/fmed.2023.1316284
19. Do D, et. al., GLP-1 receptor Agonist Discontinuation Among Patients with Obesity and/or Type 2 Diabetes. JAMA network open. May 2024; 7(5): e2413172. doi:10.1001/jamanetworkopen.2024.13172
Dr. Alan Baldridge
Dr. Baldridge is a Medical Doctor and Juris Doctor. He received his M.D. from Virginia Commonwealth University – Medical College of Virginia and his Juris Doctor from the University of Michigan Law School. He has 24 years of clinical experience in Pediatric Gastroenterology. He has been involved in clinical pediatric care and subspecialty resident training as a subspecialty training director in pediatric gastroenterology. He is a senior director in ICON Drug Development Services and provides advice regarding protocol and regulatory consideration, health authority submissions, and clinical trial design. He was one of the first pediatric investigators to describe MASH in pediatric patients. During his tenure at ICON, he has acted as a medical monitor for MASH phase 3 registration trials, post-marketing observational studies in rare disease patients, a phase 3 studies in rare pediatric disease. He has contributed to multiple protocols as a scientific advisor and regulatory body consultant in multiple indications. He has participated in assisting clients responding to regulatory agencies with responsive letters, submissions and meetings.
Jolanta M. Wichary
Jolanta M. Wichary, MD is VP, Therapy Area Lead for Gastroentetology & Hepatology in ICON Drug Development Services. She has contributed to multiple clinical development plans, protocols as a scientific advisor and regulatory body consultant in multiple indications including: IBD, IBS, EoE, Gastroparesis, MASH, PBC and PSC. Dr. Wichary supported several NDAs and MAAs in gastroenterology and hepatology.
Could GenAI be Pharma’s Silver Bullet for Medical Writing? Only if Aimed Carefully
As an acutely ambitious and overstretched Pharma industry increasingly identifies the vast potential of Generative AI to transform medical writing, across a range of use cases linked to regulatory documentation and safety report summaries, several companies are attempting to build capabilities in-house – and are coming unstuck. With reference to a new survey conducted with the Regulatory Affairs Professionals Society, Punya Abbhi, Chief Operating Officer and Co-Founder of Celegence, highlights a critical gap in capability as companies look for effective shortcuts to their most burdensome regulatory content needs.
Generative AI (GenAI) is seen as a panacea for transforming the way companies work with vast quantities of knowledge and content – sifting it and summarising it, and deriving new insights from the data it contains. And the technology certainly offers that potential, as long as it is harnessed appropriately and with the right controls. And this is where some life sciences companies are potentially opening themselves up to new risk, as they rush to embrace GenAI capabilities to make lighter work of their substantial medical writing requirements.
The Pharma industry is an ideal candidate for GenAI-based process transformation, because of its highly-regulated and painstakingly-templated repeat activities including licence application and maintenance and safety report writing. Even if GenAI-based ‘bots’ could distil the right content and craft a first draft of these typically hefty documents, for attentive checking and amendment by skilled humans, this would considerably reduce the strain on overstretched teams of Regulatory, Quality, Safety and Clinical professionals.
The trouble is that GenAI technology needs a lot of moulding and crafting to be of reliable use, even with the benefit of large language models (vast data sets) to build system ‘knowledge’. However advanced and intuitive the natural language processing and deep learning capabilities, AI models will still need to be guided in what to look for, how to repurpose information and data correctly, and what ‘good’ looks like. This requires a unique combination of AI proficiency and industry insider knowledge (hands-on experience).
Effective Shortcuts Start with Effective Instruments
In the context of Pharmaceutical medical writing, AI specialists, already in high demand in the war on talent,1 need to be proficient not just in the latest combinations and applications of natural language processing and deep learning, but also need access to experts in the life science industry to understand specialist language and vocabularies, required templates, and nuanced demands of each market. They need an intricate appreciation for what will be accepted by regulatory agencies, and how that differs from region to region, and
country to country, with a strong feel for specific medical writing best practices linked to each use case.
That’s because to maximise any return on investment and be trusted as a credible generator of first-round content, GenAI must be better and faster than humans at the initial collation and interpretation of what’s critical and required in technical documentation.
It also takes vast reams of successful example content to ‘teach’ a GenAI model what’s needed, and the ideal output to aim for. There needs to be a robust understanding of formal terminology (and its abbreviations and variants); of how to decipher tables, listings and figures; and of meaningful correlations between data and a drug or treatment. Only in this honed, specialist context can AI tools be trusted to interpret complex data and point to a logical outcome.
For Pharma companies to bring their own capabilities up to speed, and stay ahead, seems an impossible feat.
The Inhibitor to Advancement: Pharma’s Capability Gap
A new survey conducted with the Regulatory Affairs Professionals Society (RAPS)2 found that, in Pharma, medical writing remains a critical area for support. The main driver of this is the pressure on regulatory professionals’ time.
The appetite to intelligently automate some facet of the medical writing process is significant and growing stronger. Some 57 percent of surveyed companies specified planned investment in technology to improve medical writing over the year ahead, almost on a par with eCTD v4.0 spending (specified by 58 percent for the coming 12 months), the two categories dominating immediate Regulatory IT spending plans.
The primary medical writing needs identified by Regulatory professionals, in terms of requiring additional support, were clinical study protocol/report writing, and drafting of regulatory documents.
In the survey, more than half of respondents in the 2024 survey identified a need to harness AI in data extraction (56 percent) and information summarisation (53 percent), where just 9–10 percent are using AI for those purposes today – specifically within the context of medical writing. Twelve percent said they were actively in the process of incorporating AI into automated report generation from multiple sources, which is the ultimate opportunity on offer.
Despite these ambitions, more than half (53 percent) of survey respondents in Pharma conceded that their organisations did not possess sufficient knowledge internally to implement AI technologies themselves. This was further confirmed at May’s RAPS Euro Convergence 2024 event in Berlin, where Regulatory professionals also revealed some trepidation linked to what the use of AI might mean for their roles. (In reality, the technology offers to alleviate the
more burdensome and repetitive aspects of content drafting, allowing teams to focus more intently on final document quality.)
Confidentiality Concerns, Transparency and Compliance
Other potential barriers to AI uptake revolve around confidentiality and the scope for inadvertent breaches, linked in part to how the technology is ‘prompted’ to call up information. Because the technology is so complex ‘under the hood’, there are concerns that applying public cloud-based GenAI models could compromise internal data security.
All of these concerns are readily addressable in an appropriate ‘closed’ processing environment, which has been purposefully tailored to life sciences use cases. The closed environment ensures that all data interactions remain within a secure and controlled ‘space’, for data confidentiality and integrity purposes.
Other considerations include traceability and auditability: the ability to see where extracted data has come from or from which source content summarisation has been achieved (for instance, clear links in the final report to original documents). This is essential to build confidence and trust in solutions, so that they are seen to add significant value and save time; that they are not a ‘lazy’ option that requires painstaking checks (compromising the return on investment).
Knowing that there are solutions that can be validated for priority Pharma medical writing use cases will be an important facilitator for companies with a growing need for smarter support, as medical writing workloads continue to soar rather than diminish over time.
Transformed Process Efficiency, Plus Smarter, Leaner and Cleaner Output
In looking for the value from an effective (and ideally fully-managed) GenAI medical writing automation capability, Pharma companies need to look not just at the efficiency gains associated with smart data extraction, information summarisation, and narrative authoring – albeit that these are promising areas to start focusing on.
Further gains will come from improvements to consistency, and to leaner, tighter output once specified regulatory documents are being drafted according to training (from extensive exposure to approved documents) on what ‘good’ looks like.
Investing in R&D or partnership with external technology solution providers on a variety of use cases promises to make better use of scientific experts’ time, as use of their time shifts from initial drafting to the strategic thinking in collaboration with clinical development professionals. In a more strategic context, in early clinical evaluation, Gen-AI based tools could help summarise the vast wealth of existing
information on the internet to hone the focus of planned research in order to avoid wasted time investment. AI-assisted search across multiple sources can also summarise headline findings, while also highlighting what emerges as the best path to follow.
The Only Way to Advance is to Try the Technology First Hand
While Pharma companies may not have sufficient R&D resources internally to play around with possibilities, experimentation is a powerful way forward in determining where GenAI offers maximum value in transforming medical writing. It will be important, then, to find a way to at least co-design a pilot solution (for example with an appropriate technology or service partner), that can be tested with example documents – to determine how powerful and accurate the technology can be and how quickly it learns and adapts, to hone its output.
Only by trying out the technology will companies get a true feel for what’s possible, and the scale of difference it could make to their everyday Regulatory workflow. The risk otherwise is one of falling behind as industry trailblazers, the likes of Moderna (which has already deployed more than 750 GPTs across the Company that help drive automation and productivity3) continue to push the envelope.
REFERENCES
1. The AI talent shortage — can companies close the skills gap? Computerworld, 10 Apr 2024: https://www.computerworld.com/article/2086920/the-aitalent-shortage-can-companies-close-the-skills-gap.html
2. Pharmaceutical Industry Regulatory Readiness & Resources 2024 Survey Report, RAPS-Celegence – a survey of 139 Pharma companies headquartered chiefly in the US, Europe or Asia-Pac regions
Punya Abbhi is Chief Operating Officer & CoFounder at Celegence. She was previously Life Sciences Consultant at Capgemini Consulting and before that spent time at Kates Kesler Organisation Consulting and Gartner, and working for a specialist life sciences service provider. Punya holds an MBA from INSEAD and a BA in Politics, Philosophy, Economics (PPE) with a minor in Cognitive Science from the University of Pennsylvania.
Single-metered dose for systemic-acting drug administration
Using RBQM and New Technologies to Enhance Data Analysis and Enable Faster Review and Decision Making
An increase in clinical trial data is driving demand for enhanced clinical trial processes. Sponsors want to be able to review data which has been analysed quickly and efficiently so they can make informed decisions faster.
In this high-volume, high-demand landscape risk-based quality management (RBQM), supported by innovative new technologies, offers an opportunity to improve data quality and integrity, while still creating much-needed efficiencies.
Growth in RBQM Adoption
Evolving industry guidance has combined with growing recognition of the benefits of risk-based approaches to drive an uptake in RBQM adoption.
ICH E6 (R3) tied together the concepts of quality-by-design (QBD) and RBQM, bringing together risk assessment, risk mitigation strategy planning and risk detection. In 2023, the FDA issued additional guidance to facilitate sponsors’ implementation of riskbased monitoring.1
There is growing evidence to show how RBQM can help improve data quality. Traditional manual processes are labour-intensive, prone to
error and may not be effective for spotting critical issues. Source data verification (SDV) drives just 2.4% of queries in critical data suggesting it has a negligible effect on data quality2 despite being a drain on time and resources.
Analysis of key risk indicators (KRIs) from more than 200 studies conducted via RBQM software found quality improvement in 82.9% of KRIs as measured by statistical score and 81.1% for observed KRI value.3
A comprehensive assessment of RBQM adoption in clinical trials published by the Tufts Center for the Study of Drug Development found 78% of respondents trust RBQM will improve the overall quality of research and 63% trust it will enable efficiency and cost savings.4
Combining RBQM with Technology
Integrating new technologies with RBQM increases its potential to improve data quality and integrity from start to submission. Artificial intelligence (AI) in particular offers opportunities to streamline data management.
It can enable faster data analysis and identification of anomalies and help sponsors and contract research organisations (CROs) make sure errors are understood and prevented or spotted earlier in the trial.
For example, a deep learning (DL) module can be used to detect duplicate patients who might have enrolled in a study more than once at different sites by using a statistical-based comparison to determine the relative likelihood that any given pair of patients might be the same person. Intelligent patient profiles can be used to score patients statistically and identify those with unusual data patterns compared to others in the study.
Understanding Nuances in AI
While AI offers opportunities to further enhance RBQM, it has thousands, if not millions of potential applications, not all of which are relevant or useful for clinical data insights. To truly seize the opportunities on offer, we need to focus on the areas of machine learning (ML) and DL which can enhance our analysis, review, and decision-making processes.
Data anomaly detection is an example of where unsupervised ML can drive efficiencies. While there will always be a place for manual edit checks on critical data which needs to be 100% accurate, ML can help reduce the noise. Traditionally, around 30% of line listings
require a query but 70% of those are noise. This leaves data managers reviewing hundreds of pages of noise. Intelligent query detection using unsupervised machine learning can offer up to 98% accuracy, meaning, according to CluePoints’ customers, only 2% of noise is being reviewed. This frees up data managers to focus on critical queries and allows earlier and easier identification of any potential medical or patient safety issues.
Another example of combining new technologies with RBQM approaches is the use of DL for medical coding. A traditional medical coding workflow uses a rules-based automated coding tool, followed by two lines of manual review. Automated-coding tools typically successfully code 50–60% of simple input terms. This can be increased with the addition of a synonym library. However, building and maintaining such a library adds another labour-intensive task to the process.
A DL model removes the need for first-line medical coders and synonym list maintenance, automatically suggesting MedDRA and WHODrug dictionary codes for medical and drug terms reported either as part of a patient’s adverse events (AEs) or concomitant medications (CMs). It provides researchers with the correct corresponding dictionary term with more than 90% accuracy in seconds.5
Conclusion
The industry has dedicated a lot of time and attention to improving methods of data collection in recent years. However, sticking to traditional, manual processes risks creating a new bottleneck around data analysis, review and decision making.
RBQM allows efficient, effective analysis of data which highlights what really matters and empowers sponsors and CROs to make informed decisions. Combined with innovative new technologies it can truly enhance data analysis to create industry efficiencies and, ultimately, improve treatment efficacy and patient safety.
François holds a Master's in Business Administration from the ICHEC School of Management, Brussels, Belgium. Throughout his career in the pharmaceutical industry, he has held positions as a statistical programmer, SAS and JAVA developer, and IT project leader for companies such as GSK, UCB, and IDDI. During his ten-year tenure with IDDI as an IT Specialist, Mr. Torche assisted in the development of the SMART™ engine, a patent-pending software solution, and the underpinning of CluePoints. Francois served as CluePoints’ Chief Executive Officer since the company’s inception in 2012 until December 2022. More recently, he now holds the position of Chief Product & Technology Officer.
Ensuring Integrity in Clinical Trials: The Role of Small-Box Technology
The global clinical trials market is rapidly expanding, with recent projections estimating a value of nearly $100 billion by 2030, according to Fortune Business Insights. This growth is spurred by advancements in medical research and development, alongside evolving regulatory landscapes aimed at expediting trial approvals, like those introduced by the MHRA.
In this dynamic environment, the transportation of clinical trial materials – ranging from biological samples to personalised drugs – demands meticulous attention to ensure they maintain their integrity and efficacy until the point of use. Given the high value, perishable nature, and vulnerability of these products to temperature fluctuations and microbial contamination, maintaining a stable and secure environment during transit is paramount.
The implementation of small box, temperature-controlled solutions has emerged as a key strategy in addressing this logistics challenge. Investing in packaging solutions specifically tailored for smaller shipments, with an optimal packaging-topayload ratio, is vital for navigating the complexities of modern clinical trial supply chains.
Complexities in Clinical Trial Shipping
As clinical trials expand globally, maintaining temperature control during transportation over vast distances and diverse climates poses significant challenges. This issue is particularly pronounced in remote locations where local infrastructure and road conditions may be far from ideal – for example, if access to electricity is unreliable or nonexistent. The safe delivery of clinical trials in such areas requires robust cold chain solutions that can maintain internal temperatures and prevent damage during transit yet remain portable enough to ensure delivery anywhere in the world. Therefore, containers must strike the optimal balance between high performance, durability, and optimised size and weight.
And yet, pharmaceutical manufacturers face the additional challenge of ensuring all container systems pass rigorous qualification stages to meet regulatory requirements, industry standards, and performance expectations critical for the safety and quality of drug products. These qualification processes involve extensive documentation and testing, often consuming significant time and internal resources. Thus, to minimise time and expense, pharmaceutical companies frequently validate only a limited selection of containers, resulting in situations where the containers used are not optimised for the specific product being shipped. For instance, a half-empty box may be shipped because smaller, more appropriate containers were not validated despite being available. Additionally, manufacturers often choose single-use solutions for convenience, which raises sustainability concerns due to the waste generated.
Addressing the complexities of clinical trial shipping requires careful consideration of container selection to ensure alignment with the specific needs of the product being transported. Fortunately, pharmaceutical cold chain solution providers are continuously working to bridge this gap, helping manufacturers match the right solution to their products seamlessly.
Staying informed about the latest innovations in product design and material choice is essential for moving towards the optimal solution for clinical trial samples.
Matching the Product to the Container
Every container on the market fundamentally serves the primary purpose of facilitating the delivery of pharmaceutical shipments, while protecting the product from physical damage and temperature excursions. However, notable distinctions among container suppliers present pharmaceutical manufacturers with a range of decisions regarding thermal technology, materials, container design, and IoT capability.
While each option has its own set of pros and cons, evaluating containers based solely on the packaging-to-payload ratio can provide a clear perspective. Which solutions optimise volume and minimise environmental impact without compromising the container's essential function?
Understanding Passive Technology
When selecting a container for clinical trial shipping, understanding the underlying technology is crucial. Small box, passive technology systems utilise specialised materials such as lightweight vacuum insulation panels and phase-change materials (PCM) or dry ice to maintain stable internal temperatures from -60°C to +25°C without the need for manual assistance or electricity.
Recent advancements in PCM technology have significantly driven innovations in the cold chain market, leading to new configurations like pyramid-shaped designs that enhance thermal performance and efficiency. These innovations are particularly advantageous in clinical trials, where the integrity of sensitive materials like vaccines, biologics, and gene therapies must be preserved across global transport, spanning diverse climates and remote locations with limited infrastructure.
Tailored Small Box Solutions
One of the primary advantages of smaller temperature-controlled solutions is their ability to be customised to the specific needs of the shipment. Unlike larger, more generalised shipping containers, small boxes can be designed with an optimal packaging-to-payload ratio, maximising the use of space while minimising environmental impact. This efficiency is crucial in clinical trial logistics, where every cubic inch counts, and the cost of shipping can be substantial.
Logistics & Supply Chain
Tailored solutions also mean that the packaging can be designed to meet the precise thermal protection requirements of the materials being transported. This level of customisation ensures that the products remain stable and effective, reducing the risk of contamination or temperature excursions that could compromise the trial's validity.
Robust Design
In addition, the design of small box temperature-controlled solutions emphasises both robustness and reliability. These containers are engineered to withstand the rigours of transportation, including handling by multiple agents and exposure to various environmental conditions.
Moreover, the sealing mechanisms of these containers are designed to be weatherproof and washable, reducing the risk of microbial contamination. This is particularly important for clinical trial materials, which must remain uncontaminated to ensure patient safety and the accuracy of trial results.
Portable Containers
Another significant benefit of small box solutions is their portability. Unlike larger containers that require specialised handling equipment and infrastructure, small boxes can often be managed manually by one or two people. This portability is invaluable when delivering clinical trial materials to remote or last-mile locations, where access to electricity or other resources may be limited. The compact size and lightweight nature of such passive containers also allow for greater flexibility in transportation methods. Whether by air, sea, or land, small box solutions can be easily integrated into various logistics chains, ensuring timely and efficient delivery of critical materials.
Sustainability
With a growing focus on sustainability in supply chain decisions, opting for fully reusable, passive containers can significantly reduce a manufacturer's carbon footprint by keeping them in circulation for an extended period, thereby minimising the environmental impact of manufacturing.
It's crucial for reusable solutions to be robust, as discussed in the design considerations above, to endure years of transportation. The evident advantage lies in the substantial reduction of packaging waste compared to single-use alternatives that are discarded after a single journey, or semi-reusable containers that are prone to repairs or modifications. As such, some cold chain providers are offering collection initiatives, which facilitates the return of containers to the customer for reuse. This circular approach not only enhances sustainability by extending the lifecycle of the containers but also reduces the need for new packaging materials, further contributing to a greener supply chain.
In addition, maximising payload volume while considering the total cost of ownership (TCO) is critical for enhancing sustainability in clinical trial logistics. TCO encompasses all expenses associated with a container throughout its life cycle, including acquisition, maintenance and disposal costs. By optimising the payload volume with small box containers, pharmaceutical companies can reduce the number of shipments required, thereby decreasing fuel consumption and carbon emissions. Efficiently designed containers that maximise internal space and minimise unused volume contribute to significant cost savings and environmental benefits. Moreover, investing in reusable containers with a longer lifespan further reduces waste and resource usage, ultimately leading to a more sustainable and costeffective supply chain.
Logistics & Supply Chain
Effective Monitoring
With so much at stake for the clinical trials industry – financially and reputationally – it’s imperative that sample deliveries can be effectively monitored and tracked. The main source of reassurance at the end of a sample’s journey is its temperature data logger, which provides ongoing and real-time insight into whether there has been any temperature excursion during transit.
Handily, as more journeys are made and their data is recorded, providers of pharmaceutical cold chain solutions can advise potential customers of their typical excursion rates – the lower, the better. Shipment data confirms that, with modern pharmaceutical cold chain solutions, you can expect fewer than 0.1% temperature excursions per 25,000 shipments.
This means you can overlay a statistically modelled macro level of reassurance, onto the micro level from each individual transit.
Future Directions and Trends
Looking ahead, the future of clinical trial shipping will likely see continued advancements in small box temperature-controlled solutions. Emerging trends include the development of even more sustainable materials and designs, further enhancing the environmental benefits of these containers. Additionally, advancements in battery technology and renewable energy sources could lead to improved temperature control capabilities, even in the most challenging environments.
The integration of blockchain technology is another potential development, offering enhanced security and traceability for clinical trial shipments. By providing a centralised record of each shipment's journey, blockchain could further ensure the integrity and compliance of clinical trial logistics.
Addressing Market Demands
As the clinical trial market continues to expand, so too does the demand for efficient and reliable logistics solutions. Small box temperaturecontrolled containers are at the forefront of meeting these demands, offering a scalable and adaptable approach to clinical trial shipping. Their ability to provide precise temperature control, combined with their portability and sustainability, makes them an ideal choice for pharmaceutical companies navigating the complexities of global trials. As the industry continues to grow and innovate, these solutions will remain essential in maintaining the quality and efficacy of clinical trial products, ultimately contributing to the advancement of medical research and the development of new therapies.
Niall Balfour
Niall Balfour, Chief Executive Officer at Tower Cold Chain, explores the role of small box, temperature-controlled containers in the clinical trial market.
AVAILABLE WORLDWIDE AVAILABLE WORLDWIDE AT YOUR FREIGHT FORWARDER AT YOUR FREIGHT FORWARDER