Subsection: Cell and Gene Therapy


The evolution of disease modelling has seen a paradigm shift from the traditional use of animal models to more advanced in vitro human-based systems. This has been driven by the need for more accurate predictions of human disease mechanisms and drug responses in humans. Advanced in vitro models, including 2D cell cultures, organoids, and organ-on-chip systems, offer a closer simulation to human tissue environments. This article discusses the advantages these models can bring to drug discovery, emphasising their application in complex diseases like fibrosis, or development of ocular therapies. Despite the advancements, challenges such as donor variability, model validation, and the need for greater complexity and reproducibility remain. Continued innovation in combining existing models, refining culture conditions, and integrating modern technologies is crucial for enhancing the predictive power of these models and improving clinical outcomes. Industry has continued investing and there are currently commercially available retinal organoid models that allow for disease modelling and lung fibrosis models that overcome predictivity limitations to accelerate drug development.
Accurate and relevant disease models play a critical role in understanding the disease mechanism for developing targeted, effective therapies in humans. Traditionally, animal models have been crucial for understanding disease mechanisms and predicting treatment outcomes. They help assess the efficacy and safety of new drugs, providing critical insights for regulatory applications and the design of human clinical trials. However, due to significant genetic, anatomical, and physiological differences between animals and humans, animal models can sometimes inaccurately predict human responses to diseases and treatments. For example, thalidomide, an immunomodulatory agent, did not show side effects in animal models but caused birth defects in humans. Consequently, drug development is shifting towards human-based disease models to improve accuracy.
Two-dimensional (2D) cultivated patient-derived cells are an invaluable tool to study disease phenotypes and mechanisms, especially during the early phases of drug development. Donor-derived primary human cells are preferred for their higher genetic heterogeneity compared to immortalised cell lines. Induced pluripotent stem cell (iPSC)-derived lines are also used for disease modelling as they also preserve of donor genetics. Bioengineered tissue models involve cultured tissue pieces or 3D bio-printed mixtures of cells and biomaterials. The main advantage of these 3D models is their ability to better mimic the complex architecture and microenvironment of living tissues, providing more accurate physiological and cellular interactions compared to 2D models. This makes them more representative of in vivo conditions even if they tend to be more heterogenous.
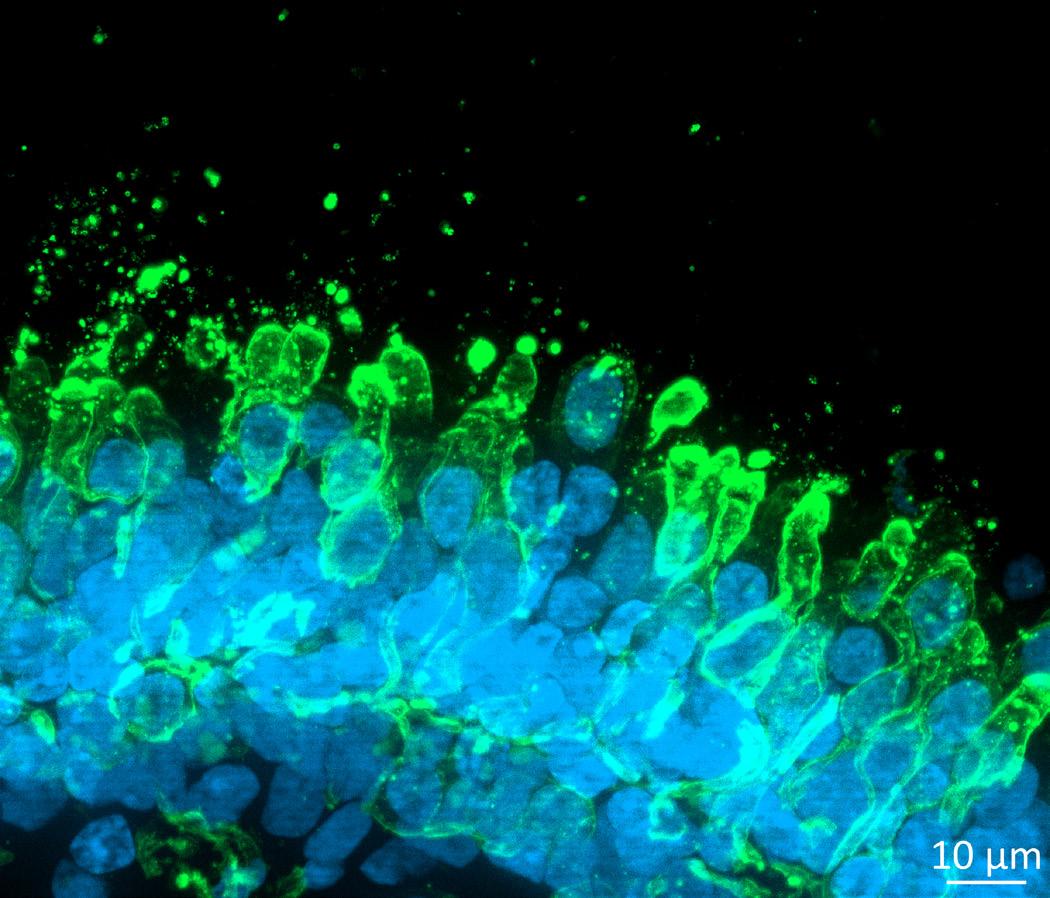
Organoids represent another sophisticated 3D model as they are self-organising structures derived from adult stem cells or induced pluripotent stem cells (iPSCs). In the appropriate culture conditions, these cells spontaneously forming organ-like structures ranging in size from 100 µm for lung organoids, 600 µm for retinal organoids to 2 mm for brain organoids. Organoids from patient-derived IPSCs or from gene-edited iPSCs for inherited disease, replicate the diseased genotype, thus allowing for complex cell pathways to be modelled and accurate drug screening. The industry has invested heavily into validation of iPSC-derived retinal organoids (Figure 1) in 96-well plates format to enable high-throughput drug screening of multiple candidates, making them an ideal platform to obtain predictive data in a physiologically relevant way. Well-validated retinal organoids present of all major retinal cell types and spatial cell arrangement that mimics the native tissue. The retinal organoids also show batch-to-batch consistency with expression of critical markers that define functionality and relevance for disease and drug efficacy studies. Another advantage of disease modelling in retinal organoids is the possibility of performing parallel studies in isogenic cell types like retinal pigment epithelium cells.
Organ-on-chip (OoC) systems are advanced microfluidic platforms that integrate bioengineered or miniaturised tissues or organs connected by 3D microchannels or undergoing fluidic flow. They mimic in vivo functions, biomechanics, and physiological responses, making them useful for disease modelling. However, OoC systems face challenges such as high technical complexity, cost, scalability issues, and difficulties in achieving precise cell placement and density. Additionally, they do not always replicate human
For more information scan the QR code or visit: newcellsbiotech.co.uk/retina

Wondering which retinal cell types are transduced by your viral vector or how safe it is?
Human iPSC Derived Retinal Organoids and RPE Models That Give You Confidence to Progress In Vivo
Gene therapy vector assessment
Disease model development
Drug testing
The only functionally validated human cell-based models of the retina to study efficacy and safety of new gene therapy vectors1 and licensed for commercial development of therapies.
A unique preclinical platform for ocular drug discovery, offering tailored solutions for retinal gene therapy viral vector evaluation and drug development for retinal disease.
We can design and deliver a customised study such as determining if new viral vectors mainly transduce photoreceptors or if they target a broader range of cells in the retinal organoids. The organoids are available for purchase live or frozen to conduct your studies internally.
organ complexity accurately and generate complex data difficult to interpret.
Overall, In vitro disease models are crucial for understanding diseases and developing new therapies, leading to better clinical outcomes. They are particularly popular for modelling prevalent and complex diseases like cancer, neurodegenerative diseases, cardiovascular diseases, diabetes, and fibrosis. Fibrosis is especially challenging to model due to its complex nature, involving tissue scarring and organ dysfunction.
Numerous in vitro models have been developed to simulate the condition of fibrosis disease in many organs including liver, lung and kidney. Each model replicates certain aspects of tissue fibrosis, so scientists need to select the one that fits their needs best.1,2 2D in vitro models are emerging as being particularly relevant by enabling predictive disease modelling and rapid screening of anti-fibrotic compounds. Mono or co-culture fibrosis models continue to be popular due to their simplicity and reliability, serving as platforms for understanding the basic disease mechanisms. They also enable rapid high-throughput screening of compounds that either promote or inhibit fibrosis. The advantages of these models include easy setup and handling, the ability to multiplex, high reproducibility, and the possibility to be imaged at single-cell level. They also allow for omics-based profiling (including genomics, transcriptomics, and proteomics) under various culture conditions,3 potentially leading to the discovery of several cellular markers associated with fibrosis. However, they lack in complexity as mono or co-cultured fibrosis 2D models are primarily based on the activation of primary fibroblasts arranged in a monolayer in tissue culture supplemented with TGF-β1 that differentiates them into myofibroblasts (MFs). These MFs are highly proliferative, contractile, produce extracellular matrix (ECM) and closely resemble their in vivo counterparts.4 Single-cell analysis has commonly been used to isolate and characterise these lineage-mapped MFs. Techniques such as single-cell RNA sequencing and transcriptome analysis carried out on these models help uncover the molecular reprogramming mechanisms and provide insights into the fibroblast to myofibroblast transition in fibrotic diseases.5,6
Liver In Vitro Fibrosis Models
Primary human hepatocytes (PHHs) and hepatocyte-like cells (HLCs) are the most common cell model for liver fibrosis using primary cells obtained from liver resections or tissues unfit for organ transplantation. These cells are highly functional i.e. they express critical hepatic markers and enzymes. The limited lifespan of such models in culture and donor variability, however, poses challenges.7–9 Hepatocyte-like cells (HLCs) are derived from induced pluripotent stem cells allowing the generation of patient-specific cells for drug screening. The main limitation of this model is the difficulty of scaling up production and the increased risk of teratoma formation.
Lung In Vitro 2D Fibrosis Models
Numerous important in vitro fibrosis studies have been conducted using lung fibroblasts derived from both healthy individuals 10,11 and patients suffering from Idiopathic
Pulmonary Fibrosis (IPF).12 These cells have been shown to model the effect of profibrotic molecules and cell pathways involved in fibroblast-to-myofibroblast transition.13–15 Other studies11,16,17 have utilised immortalised cell lines to understand pulmonary fibrosis progression and treatment. A lung fibrosis assay, based on TGF-β1 induction in primary lung fibroblasts from both healthy donors and IPF patients has been functionally validated and shown to be predictive. 18 The optimised conditions of the assay use reduced serum conditions to decrease cell proliferation.19 Furthermore, the incorporation of a macromolecular crowder promotes collagen type I deposition boosting assay sensitivity.20 Conducted in a 384-well format, it allows for high throughput and detailed phenotypic analysis (Figure 2), making it highly physiologically relevant and sensitive compared to other. Epithelial cells including primary human bronchial and alveolar epithelial cells, as well as lung carcinoma cells (A549 and BEAS2B) are also used as lung fibrosis models. These cell models can undergo Epithelial-Mesenchymal Transition (EMT)21,22 in the lab, making them useful for studying pulmonary fibrosis. When epithelial cells undergo EMT, they show a decrease in epithelial markers23 and an increase in mesenchymal markers leading to ECM deposition and increased alpha-smooth muscle actin (α-SMA) expression.
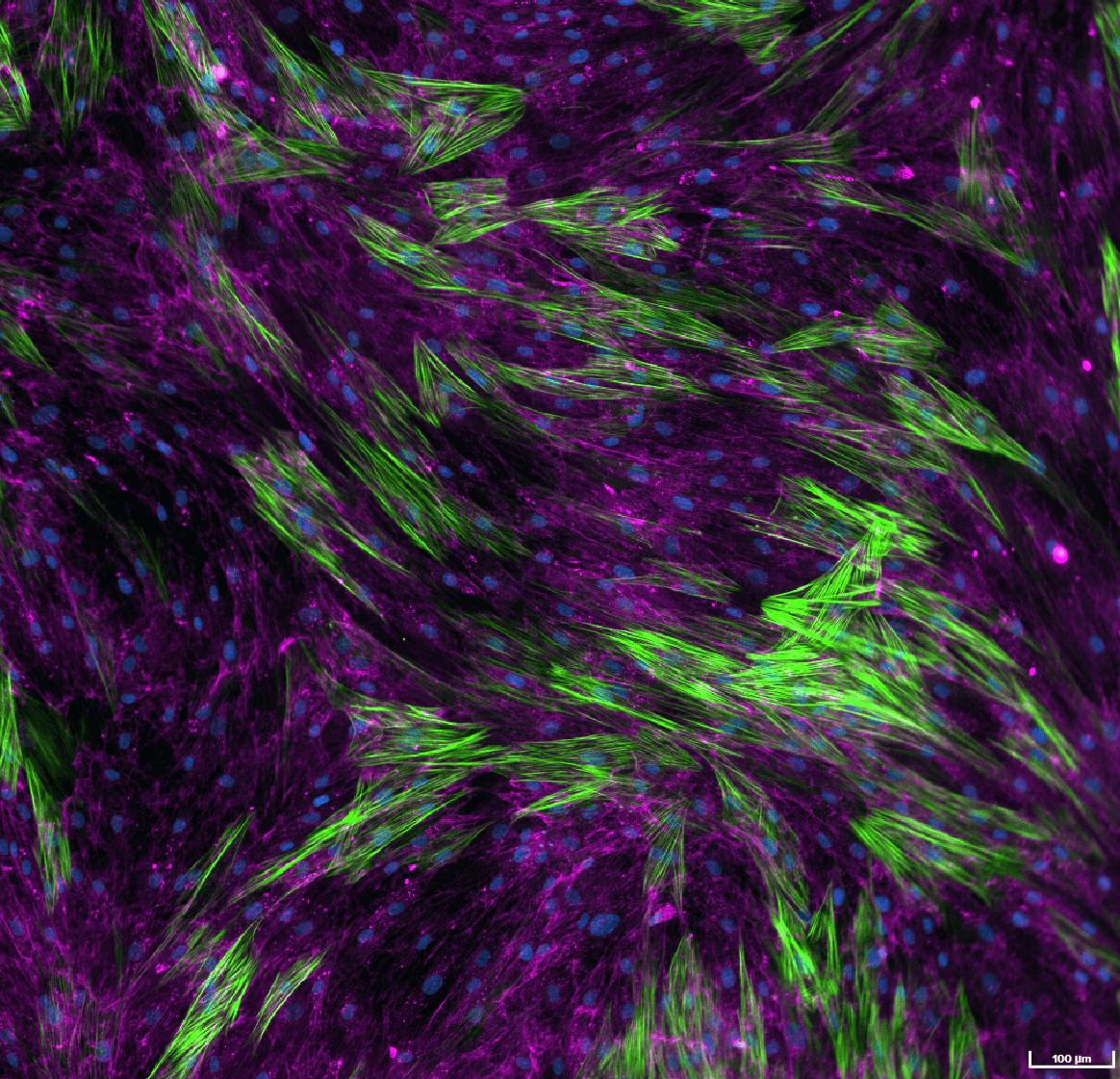
Mesangial cells, make up 30–40% of the cells in the glomerulus, playing a role in the development of kidney fibrosis. Under pathological conditions, they become activated leading to hyperproliferation, excess ECM accumulation, and secretion of inflammatory signals, all contributing to renal glomerular fibrosis.24,25 These models help identify anti-fibrotic drugs targeting TGF-β-induced pro-fibrotic signalling pathways. Renal fibroblasts are also used to create 3D fibrosis models, though more expensive and time-consuming to validate. Upon activation, they secrete ECM proteins, which accumulate and cause kidney scarring26 as part of the fibrosis process. Renal fibrosis can also be induced in renal epithelial cells and
When culturing any kind of cells in a laboratory environment, avoiding contamination is always a chief concern. Biological contaminants are often the focus of such efforts, and they also can be the most straight-forward to detect and avoid.
The most popular endotoxin testing option is the Limulus amebocyte lysate (LAL) assay. In the presence of endotoxins, an enzyme in the assay initiates a clotting cascade, which can be used to quantify endotoxin levels with high precision and sensitivity.
It is recommended to perform regular LAL testing on all cell culture reagents, particularly prior to any large-scale or crucial in vitro experiments. The ease and low cost required for the LAL assay makes it an ideal option for frequent endotoxin testing.
www.wakopyrostar.com

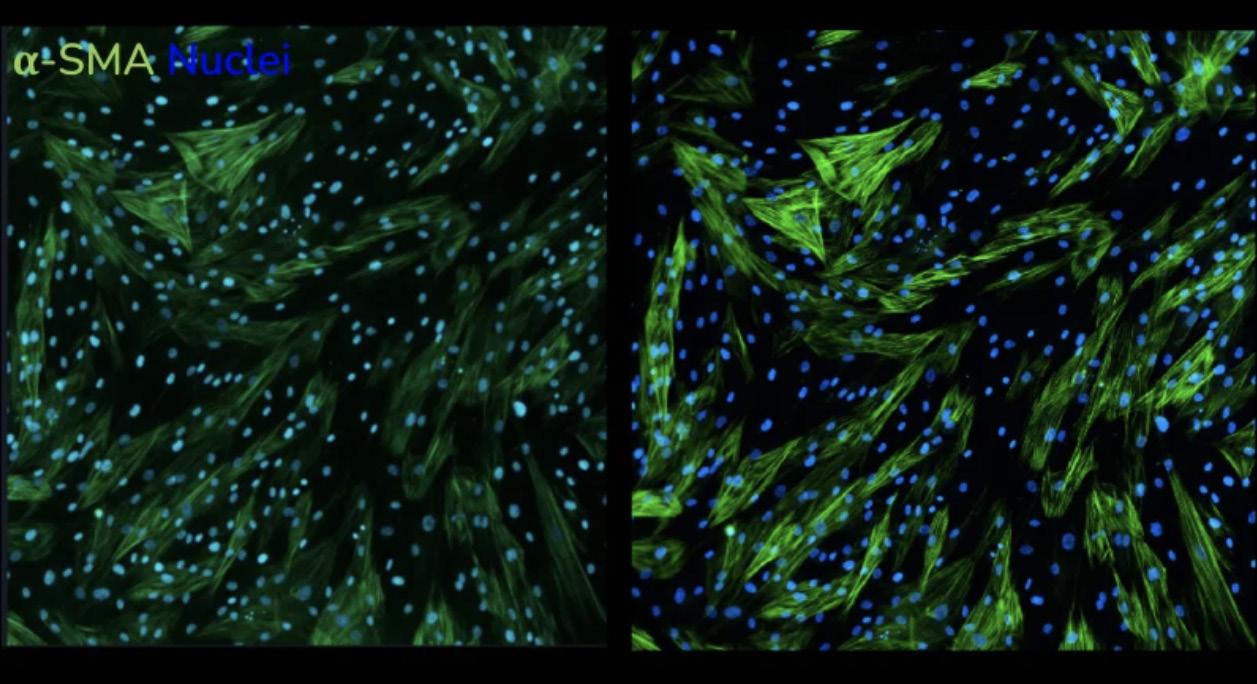
resolution imaging data obtained as part of the HTHS FMT assay service. α – SMA staining following stimulation of human lung fibroblasts with
captured using LED-based imaging (left) vs high resolution laser-based imaging (right).
podocytes, through the addition of TGF-β1 as shown in single culture of renal tubular epithelial cells27 and in a co-culture of epithelial cells and renal fibroblasts. These cells undergo a process known as EMT or endothelial-mesenchymal transition that contributes to the accumulation of fibroblasts and myofibroblasts seen in renal fibrosis.
Conclusion
Analysis of fibrosis is challenging due to the complexity of pathways and cell types involved. While 2D and 3D in vitro models have provided significant mechanistic insights including profibrotic cells, myofibroblast origins, immune cell involvement and mis-regulated signals, effective treatments remain very limited. Key challenges include donor variability, validation, reproducibility, and maintaining native tissue architecture in models. Advances are expected from combining models, adjusting culture conditions, and using perfusion systems, offering new hope for patients. The shift from traditional animal models to advance in vitro disease models marks a significant evolution in medical research and drug development. The emergence of 2D cell cultures, organoids, and organ-on-chip systems offers a closer representation of human tissue environments, enabling precise studies of disease mechanisms and drug responses. Examples include human iPSC-derived retinal organoids to model retinal diseases. Similarly, In vitro models of fibrosis in organs such as the liver, lungs, and kidneys show potential in replicating complex disease pathways and facilitating high-throughput screening of novel drug candidates. However, challenges such as donor variability, model validation, and the need for greater complexity and reproducibility must be addressed to achieve physiological relevance and predictive results.
[For more information about predictive, physiologically relevant in vitro models and disease modelling visit newcells. co.uk]
1. Lee YS, Seki E. In Vivo and In Vitro Models to Study Liver Fibrosis: Mechanisms and Limitations. Cell Mol Gastroenterol Hepatol 2023 16(3):355–67.
2. Kolanko E, Cargnoni A, Papait A, Silini AR, Czekaj P, Parolini O. The evolution of in vitro models of lung fibrosis: promising prospects for drug discovery. European Respiratory Review 2024 33(171).
3. De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, et al. Gene Expression Profiles During Hepatic Stellate Cell Activation in Culture and In Vivo. Gastroenterology. 2007;132(5):1937–46.
4. Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nature Medicine 2007;13(11):1324–32
5. Xie T, Wang Y, Deng N, Liang J, Noble PW, Correspondence J. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis Data and Software Availability GSE104154 Xie et al. CellReports. 2018; 22:3625–40.
6. Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. Journal of Clinical Investigation. 2014 Jun 2;124(6):2299–306.
7. Zeilinger K, Freyer N, Damm G, Seehofer D, Knö spel F. Cell sources for in vitro human liver cell culture models. Exp Biol Med. 2016; 241:1684–98.
8. Bell CC, Dankers ACA, Lauschke VM, Sison-Young R, Jenkins R, Rowe C, et al. Comparison of Hepatic 2D Sandwich Cultures and 3D Spheroids for Long-term Toxicity Applications: A Multicenter Study. Toxicol Sci 2018;162(2):655–66.
9. Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Vol. 87, Archives of Toxicology. 2013. p. 1315–530.
10. Weigle S, Martin E, Voegtle A, Wahl B, Schuler M. Primary cell-based phenotypic assays to pharmacologically and genetically study fibrotic diseases in vitro. J Biol Methods. 2019;6(2): e115.
11. Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast
transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 2003;29(5):537–44.
12. Hostettler KE, Zhong J, Papakonstantinou E, Karakiulakis G, Tamm M, Seidel P, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res 2014;15(1).
13. Lu Y, Azad N, Wang L, Iyer AKV, Castranova V, Jiang BH, et al Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am J Respir Cell Mol Biol. 2010;42(4):432–41.
14. Joannes A, Brayer S, Besnard V, Marchal-Sommé J, Jaillet M, Mordant P, et al. FGF9 and FGF18 in idiopathic pulmonary fibrosis promote survival and migration and inhibit myofibroblast differentiation of human lung fibroblasts in vitro. Am J Physiol Lung Cell Mol Physiol. 2016; 310:615–29.
15. Moodley YP, Misso NLA, Scaffidi AK, Fogel-Petrovic M, McAnulty RJ, Laurent GJ, et al. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol Biol. 2003;29(4):490–8.
16. Dunkern TR, Feurstein D, Rossi GA, Sabatini F, Hatzelmann A. Inhibition of TGF-beta induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur J Pharmacol. 2007;572(1):12–22.
17. Kohyama T, Liu X, Wen FQ, Yun KZ, Wang H, Hui JK, et al. PDE4 inhibitors attenuate fibroblast chemotaxis and contraction of native collagen gels. Am J Respir Cell Mol Biol. 2002;26(6):694–701.
18. Leslie F, Birch O, Whiting C, Webster M. High content imaging (HCI) assays enable the study of the fibrotic processes: fibroblastto-myofibroblast transition (FMT) and epithelial-to-mesenchymal transition (EMT) presented at Extracellular Matrix Pharmcology Congress poster; 2024 Jun 17-19; Copenhagen, Denmark
19. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, et al. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol. 2017;313(6): L1164–73.
20. Zeugolis DI. Bioinspired in vitro microenvironments to control cell fate: focus on macromolecular crowding. American Journal of Physiology-Cell Physiology 2021;320(5):C842–9.
21. Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res 2009;10(1).
22. Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky


Rhodopsin and Opsin staining of retinal organoids from Day 120 to 270 of differentiation in vitro
D V., Murray LA, et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med 2009;180(2):122–33.
23. N. Lekkerkerker A, Aarbiou J, van Es T, A.J. Janssen R. Cellular players in lung fibrosis. Curr Pharm Des 2012;18(27):4093–102.
24. Zhao JH. Mesangial Cells and Renal Fibrosis. Adv Exp Med Biol. 2019; 1165:165–94.
25. Xu Q, Norman JT, Shrivastav S, Lucio-Cazana J, Kopp JB. Innovative Methodology In vitro models of TGF-induced fibrosis suitable for high-throughput screening of antifibrotic agents. Am J Physiol Renal Physiol. 2007; 293:631–40.
26. Wang C, Li SW, Zhong X, Liu BC, Lv LL. An update on renal fibrosis: from mechanisms to therapeutic strategies with a focus on extracellular vesicles. Kidney Res Clin Pract. 2023;42(2):174.
27. Shen H, He Q, Dong Y, Shao L, Liu Y, Gong J. Upregulation of miRNA-1228-3p alleviates TGF-β-induced fibrosis in renal tubular epithelial cells. Histol Histopathol 2020;35(10):1125–33.

Dr. Tanmay Gharat works as Product and Marketing Manager at Newcells. He started his career with benchwork then carved a niche for himself at the intersection of business and technology. Dr. Tanmay has a solid grasp on product development bridging the gap between research and customer expectations. Dr. Tanmay holds a PhD. in Chemical & Biological Engineering.
Email: tanmay.gharat@newcellsbiotech.co.uk

Dr. Emanuela Costigliola is the Chief Marketing Officer at Newcells. With nearly 20 years in the life science and biotechnology industry, she has held senior roles at GE Healthcare, Qiagen, Life Technologies, and AstraZeneca. She was Head of Marketing at the Cell and Gene Therapy Catapult and has consulted on commercial capabilities in immuno-oncology. Dr. Emanuela holds a Ph.D. in Immunology and Molecular Pathology.
Email: emanuela.costigliola@newcellsbiotech.co.uk









• Ultra low sample Input: 10 pg total RNA – single cell
• Liquid biopsies / EVs from only 25 µl of plasma

• Fast, single tube protocol
• Gel free
• Diverse patented adapters for highest variety of small RNAs
• Sequencing on any Illumina® or Ion TorrentTM NGS instrument

Our TrueQuant library preparation methodology used in this kit enables accurate identification and quantification of miRNAs, piRNAs and other small RNAs.
Please contact us for more information.
Fon +49 69 95739710 Fax +49 69 95739706 info@genxpro.de www.genxpro.de
• Bioinformatics included

Advances in CAR-T cell therapy have carved a path for improved treatments in the field of oncology, in particular haematological cancers. In 2018, the European Medicine’s Agency approved the first two CAR-T cell therapies, Kymriah® and Yescarta®, for marketing authorisation for the treatment of certain cancer.1 Since then, CAR-T cell therapy has been rolled out across Europe, largely for the treatment of large B-cell lymphoma, mantle cell lymphoma, and acute lymphoblastic leukaemia. However, the same success has not been realised for solid tumours.2
To address the challenge of cell therapy for the treatment of solid tumours, many have looked towards the power of natural killer (NK) cells as a potential therapy. In particular, CAR-NK cell therapy is thought to have several advantages over CAR-T cell therapy, such as an improved safety profile with a reduced risk of cytokine release syndrome and lower incidence of graft versus host disease.3 CAR-NK cells may also provide the much-needed improved efficacy in treating solid tumours. It is believed that CAR-NK cells may be able to overcome tumour escape problems as seen with CAR-T cells due to their retention of antigen-independent activity even when there is loss of antigen dependent killing.4
As advancements in the NK cell therapy area are gaining speed, so is the complexity of the IP landscape. International and European patent filings mentioning NK cell therapy
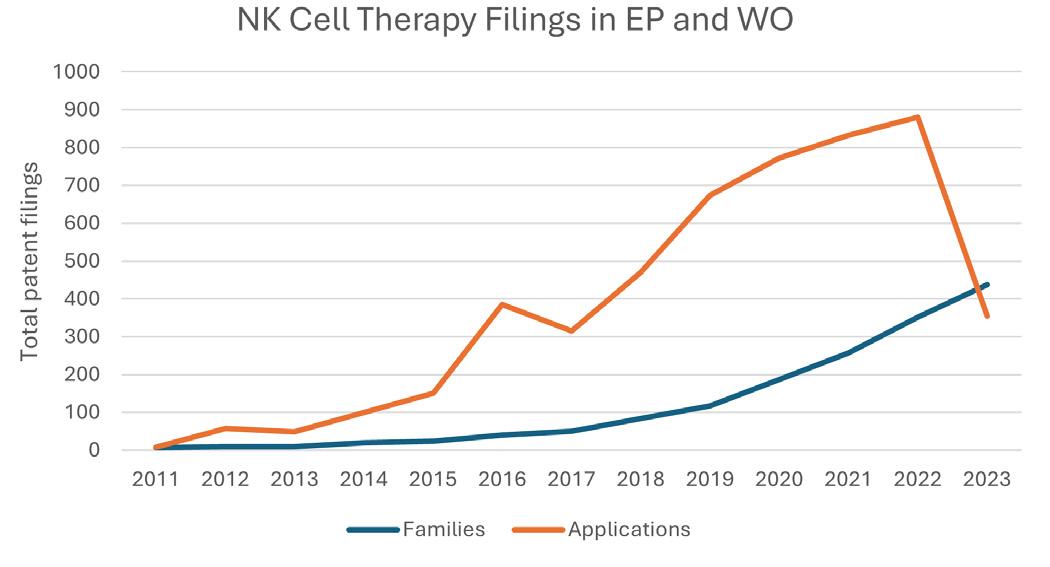
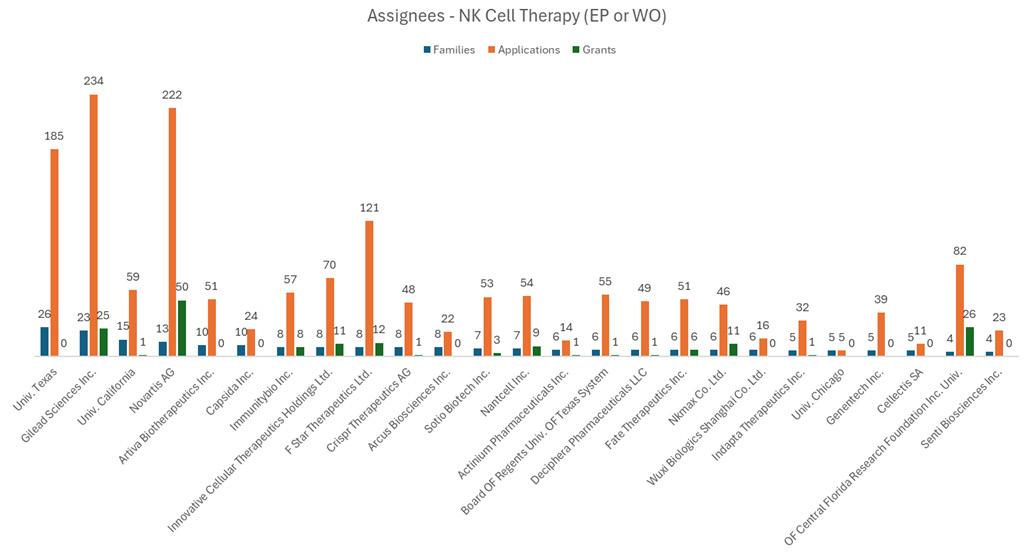
have been rising sharply over the last 5 years (Figure 1), with a number of large players in the game. Gilead Sciences Incorporated and Novartis AG have strong patent portfolios in this area (Figure 2), each with over 200 applications pending and 25–50 granted patents that at least mention NK cell therapy in the text of the applications. The University of Texas MD Anderson Cancer Center also holds a number of pending and granted patents directed towards methods of producing CAR-NK cells and claims towards therapeutic compositions of NK cells with engineered receptors.
To further strengthen their patent portfolios, a number of high-profile collaborations and deals have been struck. Gilead have partnered with Dragonfly Therapeutics to further strengthen their patent portfolio and to gain access to the
5T4-targeting investigational immunotherapy program and, upon completion of certain preclinical activities, a licence to use Dragonfly’s TriNKET™ technology to develop and commercialise NK cell engagers. Dragonfly Therapeutics have further capitalised on their IP position by licencing multiple NK cell therapy candidates to Merck and Bristol Myers Squibb, in the fields of oncology and neuroinflammation.
MD Anderson have partnered with Takeda to develop off the shelf NK cell products and, more recently, have granted exclusive licences to the Norwegian company, Zelluna Immunotherapy, to expand their offering of off the shelf TCR-NK cells for the treatment of cancer.5 Under the agreement with MD Anderson, Zelluna will have exclusive rights to develop and commercialise certain TCR-NK products. These TCR-NK products include allogeneic cellular therapies that combine the inherent killing mechanism and the allogeneic nature of NK cells with the antigen-specific targeting capabilities of TCRs.
As evidenced by Dragonfly’s IP position for their NK cell platform technology and MD Anderson’s position for off the shelf NK cell therapies, valuable gains can be made by having a solid and well thought out IP position. As this field grows, the already crowded IP landscape will continue to expand and become more challenging to navigate for companies wishing to expand in the field and to get their products to market.
However, despite the rapid rise in patent filings in Europe, relatively few patents have been granted. This adds to the complexity of the patent landscape, as those wishing to develop their products in this area will have to continually monitor the progress of these patent applications until grant or make licencing decisions before the scope of the patents and their jurisdictions are clear. One approach to strengthen one’s IP position, is to develop a robust patent portfolio for particular NK cell products or methods of manufacture in the European market and seek to cross licencing where possible.
One example of a European company making strides in this area is ONK Therapeutics. ONK, an Irish based company, offer engineered NK cell therapy candidates that express CAR for the treatment of AML, multiple myeloma and a number of solid tumour cancers such as prostate and triple negative breast cancer.6 ONK exclusively in-licence a recently granted
European patent from Australia’s Walter and Eliza Hall Institute of Medical Research (WEHI) covering CISH knockout NK use in cancer therapy. This licencing deal builds on ONK’s already growing patent portfolio on multiple different NK cell checkpoints.
There is clearly competition in this space, therefore a knowledgeable understanding of what competitors and academic institutions are doing is essential. As mentioned above, there is a backlog of pending patent applications in Europe that will proceed to grant over the coming years. This will likely result in a spike in opposition proceedings and litigation action. It is advisable that any organisation wishing to enter the NK cell therapy market has a freedom to operate assessment performed to avoid any contentious action.
Overall, the advancement of NK cell therapeutics is an exciting development to the cell therapy space, in particular as there is a clear need not being met by CAR-T cells. However, there is much to be considered with respect to patent filing strategy in Europe and beyond but also with respect to freedom to operate and licencing strategies to support NK cell therapies being brought to market. Those that will see success, are those organisations that not only develop the best technology but those who make the right decisions early on with developing their own patent portfolios and striking the best licencing deals.
1. Outcomes-based reimbursement for gene therapies in practice: the experience of recently launched CAR-T cell therapies in major European countries. Jørgensen, J., Hanna, E., & Kefalas, P. 1, s.l. : Journal of Market Access & Health Policy,, 2020, Vol. 8.
2. NHS England. [Online] https://www.england.nhs.uk/cancer/cdf/ car-t-therapy/.
3. Emerging roles of CAR-NK cell therapies in tumor immunotherapy: current status and future directions. Liu, Yan Zhong & Jingfeng. 218, s.l. : Cell Death Discovery , 2024, Vol. 10.
4. Charting a killer course to the solid tumor: strategies to recruit and activate NK cells in the tumor microenvironment. Ana L. Portillo, Jonathan K. Monteiro, Eduardo A. Rojas. 2023.
5. Zelluna Immunotherapy. [Online] [Cited: 23 07 2024.] https://www. zelluna.com/news/exclusive-license-agreements-to-develop-andcommercialize-mage-a4-and-vgll1-targeting-allogeneic-off-theshelf-tcr-based-natural-killer-tcr-nk-cell-therapies.
6. Labiotech. [Online] [Cited: 23 07 2024.] https://www.labiotech. eu/best-biotech/nk-cell-therapy-companies/.

Amy Dawson is a patent attorney in the Life Sciences group at HGF. She holds a first-class MSci in Immunology and a PhD in Cancer Studies from the University of Glasgow, where she studied the interaction of leukaemic stem cells with immune cells in the cancer microenvironment. Since joining HGF, she has experience with client management, IP landscaping, drafting and UK and European prosecution. In most recent years, Amy has experience in the cell and gene therapy field, with particular experience in programmable gene therapy technologies and synthetic promoters.
Cell and gene therapies are treatments that currently revolutionise medicine as they have the potential to cure serious and incurable diseases. In cell therapy, living cells from a patient or healthy donor are used to repair damaged tissue or boost the functioning of the immune system. Gene therapy aims to replace or repair defective genes with healthy ones in order to cure genetic diseases. Technologies for the development of cell and gene therapies center around gene transfer and genetic modification of cells. Today, there is enormous hope for these forms of advanced therapies, but major challenges in terms of safety, efficacy and production remain.
This article spotlights two emerging technologies for cell and gene therapy: mRNA technology and adeno-associated virus (AAV) technology.
I. mRNA-based Cell and Gene Therapies
mRNA-based therapies utilise the genetic information of messenger RNA (mRNA) to produce specific proteins within the patient's cells. These proteins can serve various therapeutic functions, such as stimulating the immune response against diseases (such as in the well-known COVID-19 vaccines), replacing missing or defective proteins, or inhibiting the function of disease-causing proteins. One example, is the application of mRNA as a non-viral vector for the genetic modification of immune cells to transform them into cell therapeutics by introducing chimeric antigen receptors (CAR) as new surface receptors.
mRNA-based CAR-T Cells
Immune cells modified with new surface receptors, known as CAR-T cells, are clinically well-established cell therapies that
are used to treat blood cancers that do not respond to any other treatment. All approved CAR-T cell therapies use lentiviral vectors for genetic transfer of the CAR into T cells. However, using lentiviral vectors the CAR gene is permanently integrated into the genome of the patient's own immune cells. Using mRNA for this genetic modification instead of viral vectors has various advantages: ensuring temporal restriction of the CAR expression, allowing better control over immune activation and reducing potential side effects. In addition, it minimises the risk of insertional mutagenesis, a problem associated with DNA-based gene transfer, since mRNA does not integrate into the host cell's DNA. This makes mRNA-based CAR-T cell therapy a promising and presumably safer approach for treating certain types of cancer and more recently also autoimmune diseases.
Here’s how it works (simplified):
1. Collection: T cells, a type of immune cell, are collected from the patient's blood. This is usually done by apheresis at the point of need, i.e., in the clinic. The apheresis is then frozen and transported to an appropriate production facility.
2. Engineering: At the manufacturing site, the isolated T cells are genetically modified using synthetically produced mRNA that carries the genetic instructions for the therapeutic relevant molecule. In this case, it instructs the T cells to produce chimeric antigen receptors (CARs) on their surface.
3. CAR Function: CARs are designed to recognise and bind to specific proteins on the surface of diseased cells. This binding signals the T cells to attack and destroy the diseased cells and triggers further elements of the immune response.
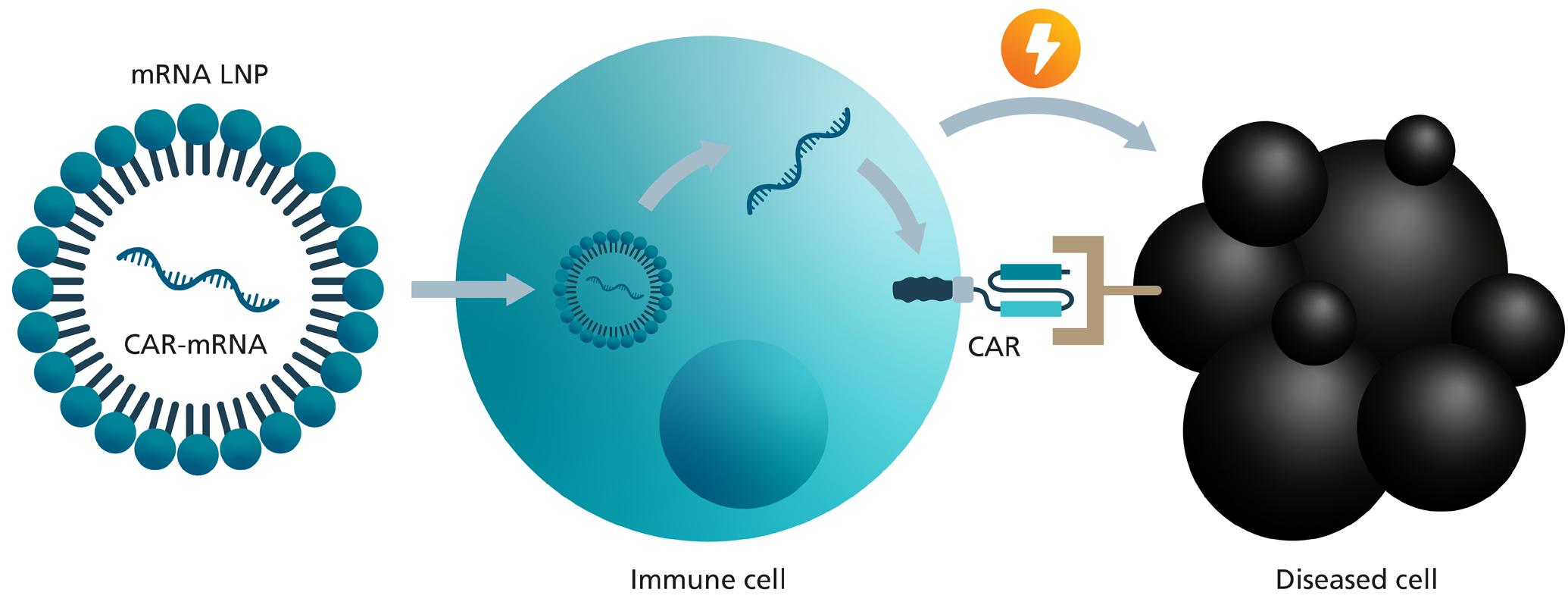

4. Reintroduction to Administration The engineered T cells, now equipped with CARs, are infused back into the patient’s bloodstream.
5. Targeting diseased cells (e.g., cancer cells): CAR-T cells circulate in the patient's body, search for diseased cells and eliminate them.
A critical point for mRNA-based CAR-T cell therapies is the efficient and safe transfer of the mRNA into the T cells. Lipid nanoparticles (LNP) are used to ensure this, which are tiny particles made of lipids that can encapsulate and protect mRNA from being broken down. LNP deliver mRNA into cells by merging with the cell membrane, allowing the mRNA to enter the cell's cytoplasm where it can be used to generate proteins. LNPs improve the efficiency of mRNA delivery compared to different other methods, ensuring more immune cells are successfully modified.
The LNP delivery of CAR-mRNA is a significant advancement in CAR-T cell therapy. It offers a less invasive and potentially safer alternative to existing CAR-T cell therapies. Beyond that, this method could make the production process simpler, reduce costs, and improve access, making personalised immunotherapy more available. Furthermore, by adding targeting moieties for uptake by specific cell types, LNPs have the potential for in vivo CAR-T cell generation in the future, which means making CAR-T cells directly within the patient’s own body.
The mRNA technology can help decrease costs for cell therapy manufacturing in several ways. The production process is simplified and less complex in comparison to viral vectors, since mRNA can be synthesised in vitro using cell-free systems, reducing the need for complex cell culture facilities. The cell-free production process minimises the risk of contamination, reducing the need for extensive safety testing and quality control measures. The production of mRNA is highly scalable, allowing for large quantities to be produced more efficiently. Cell therapies based on mRNA underlie a shorter development time reducing time to market and associated costs. These factors collectively contribute to reducing the overall costs associated with the manufacturing of cell therapies.
Pros Cons
Rapid development and production
Potential for immune reactions
High specificity and personalised medicine Stability and delivery challenges
Non-integrative and transient expression
Reduced risk of insertional mutagenesis
Ability to quickly adapt to new variants
Potential for in vivo application
Adeno-associated viruses (AAV) can be used as gene modification or transfer method. AAV vectors make it possible to transport genes into human cells and thereby supply a healthy version of the defective gene within the cells.
AAVs consist of a single-stranded DNA genome enveloped in a protein capsid. To use the wild-type AAV virus as a vector system for gene therapy, the viral genes of the DNA genome are replaced with the gene of interest. Thus, an AAV vector does not contain any viral genes. Some advantages of AAVs are their high transduction rates, broad tissue tropism, low immunogenicity, and long-lasting therapeutic effects. While other commonly used viral vectors, such as lentivirus and herpes-simplex virus 1, integrate their genetic payload into the host chromosomal DNA, AAVs in principle do not integrate in the host DNA. In contrast, their DNA typically remains episomal within non-dividing cells, in cells with high expansion the AAV vectors get lost over time. This greatly lowers the risk of genotoxicity and immunotoxicity for patients receiving such treatments. Due to their many advantages, AAV-based therapies have emerged as the preferred viral vector platform for gene therapies due to their unique safety and efficacy features.
Here’s how it works (simplified):
1. Identification: The defective target gene that causes the disease must be identified.
2. Engineering: A vector with the healthy target gene is generated using molecular biology.
3. Production: The viral vectors containing the healthy gene are first produced in a cell culture, then purified and formulated for use in humans.
4. Administration: A few milliliters of the finished medication are administered to the patient. This can be done intravenously or directly into the affected organ.
5. Transfection & Mode of action: In the patient's body, the capsid binds to the cell membrane of target cells, where it is internalised through endocytosis. Following release to the cytoplasm, the vector transits to the nucleus. Inside the cell nucleus, the capsid envelope is degraded and the DNA is released. Once the vector DNA transforms into episomal DNA, it is transcribed and the resultant mRNA is translocated to the cytoplasm where the protein of interest is produced by protein biosynthesis.
Compared to other methods of gene therapy, the use of AAV as viral vectors has the advantage that they transfer the
Name Company Disease treated FDA Approval Date
Luxturna® Spark Therapeutics Retinal dystrophy 2017
Requires cold storage and transport
Limited long-term data on efficacy and safety
Regulatory and approval hurdles
Zolgensma® Novartis Spinal muscular atrophy (SMA) 2019
Hemgenix® CSL Behring Hemophilia B 2022
Elevidys Serepta Therapeutics Duchenne muscular dystrophy 2023
Roctavian™ BioMarin Hemophilia A 2023
Durveqtix® Pfizer Hemophilia B 2024
Summary of Pros and Cons of mRNA-based Cell and Gene Therapies FDA-Approved AAV Gene Therapies

• Central Lab: Kitting, Logistics, and Biostorage With Virtual Sample Inventory Management
• Biospecimens: Curated Inventory and Analysis With Advanced Biobanking
• Preclinical: Advanced Cell-Based Assays and Target Validation
• Genomics: Single-Cell to Multiplex Studies
• Bioanalytics: Immunogenicity and PK Testing
• Immune Monitoring: Comprehensive Cell Phenotyping and Profiling
• Tissue and Liquid Biopsy: Rare Cell and CTC Isolation and Analysis
• Clinical Trials: Design, Strategy, and Full-Spectrum Support
• Diagnostics and CDx: Regulatory Consulting, Companion Dx, and NGS
• Data Sciences: Biometrics and Biostatistics via QuartzBio® precisionformedicine.com
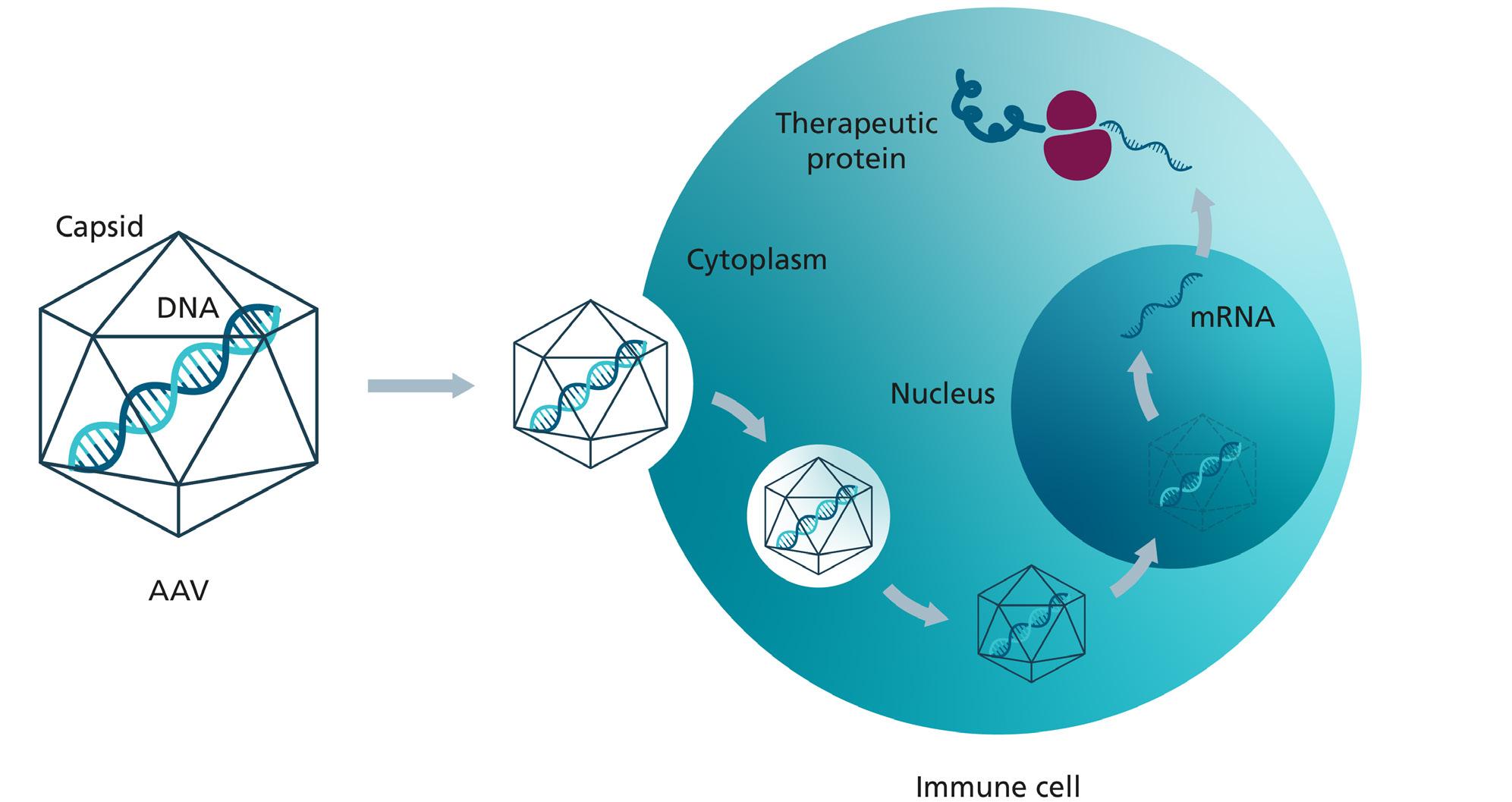
genes into the cells in a targeted manner with comparatively few side effects. AAVs are non-pathogenic and naturally occur in the human population. The immune reaction to AAVs in comparison to other viral vector systems is rather mild. Another advantage of AAVs is that they are not able to replicate without the presence of a helper virus such as Adenovirus, nonetheless they can maintain their therapeutic expression for years in non-dividing tissue.
Currently there are roughly 240 clinical studies in the AAV field targeting hematological, neurological, ocular and metabolic diseases. In addition, around 7000 monogenetic diseases are known to date that could potentially be cured with AAV-based gene therapies.
One of the biggest hurdles in the clinical application of AAVs is the pre-existing immune tolerance. As AAVs occur naturally, exposure to wild-type AAV virus results in the development of both humoral and T cell mediated immunity against the virus. AAVs are highly prevalent in the human population depending on geographics. AAV2 is the most prevalent serotype with roughly 30 to 60% of people carrying antibodies against
Pros Cons
Non-replicative viral vector Limited viral load capacity
High transduction rates High manufacturing costs
Direct in vivo application
Low unwanted immune responses (in comparison to other viral vectors)
Long-lasting efficacy
Capsid engineering broadens tissue tropism
One-shot therapies due to the development of immunity
Mutagenicity of delivered transgene over time reduces therapeutic efficacy
High prevalence of AAVs reduce applicability due to pre-existing immunity
Dilution of transgene due to non-integrative nature
Summary of Benefits and Limitations of AAV Gene Therapies
this specific serotype. As neutralising antibodies are known to have a negative impact on the therapeutic efficiency of the treatment, people with pre-existing immunity are currently exempt from clinical trials with AAV therapeutics. Additionally, pre-existing immunity carries the risk of dangerous acute immune responses. A work around strategy is to treat patients as early in life as possible to decrease the risk of previous AAV exposure. Additionally, immune inhibitors are given to patients during application of the therapy, aiming to prevent unwanted immune responses.
Due to the immune tolerance limitations, current AAV treatments can only be administered as single-shot therapies. The therapeutic efficiency can also be reduced by the development of mutations within the delivered genetic material over time. Naturally, AAV vectors are not able to replicate within the host cell and do not integrate their genetic payload within the host chromosome, thus reducing downstream genotoxic risks. On the downside AAVs DNA genome is constricted to an approximate size of 4.7 kb DNA. Although AAVs tolerate a certain degree of overlength of the genome, this goes hand in hand with a reduction of production efficiency of AAV vectors. As many genetic diseases would require DNA in the viral vector to greatly exceed 4.7 kb, AAV vectors can only be applied to a limited number of diseases. Creative approaches are necessary to increase viral payload, for example through virus capsid modification, using multiple AAV vectors for one therapy or combining AAV vectors with other therapies.
The bottleneck of mRNA and AAV vector development is the manufacturing of material for clinical trials. The development times for a clinical-grade GMP (Good Manufacturing Practice) products are long, require large facilities and have high labor costs, slowing down the application of new mRNA and AAV products to the clinic. The high costs of GMP process development arise primarily during upscaling, the part of process development in which the manufacturing volumes are gradually increased.

To facilitate the development of new AAV and mRNA-based cell and gene therapies, the Fraunhofer Institute for Cell Therapy and Immunology is investing in the optimisation of GMP process development through Quality by Design. As a non-profit organisation, Fraunhofer is thus making it easier for researchers, start-ups and small biotechnology companies to make the leap from laboratory scale to clinical trials.
The use and development of viral and non-viral vectors in cell and gene therapy will be discussed at the Leipzig Immune ONcology (LION) Conference on November 12 and 13, 2024. The conference will bring together clinicians, scientists, and industry to discuss latest developments and highlights on special issues of immune oncology. The annual meeting is organised by the Fraunhofer Institute for Cell Therapy and Immunology and the University Cancer Center of the University Hospital Leipzig.


Dr. Sandy Tretbar is a molecular biologist and RNA biochemist. She obtained a PhD in Molecular Biology at Leipzig University in Germany and has worked as Postdoc in the field of RNA biochemistry and biophysics and in immunology. Currently heading a research unit within the department Cell and Gene Therapy Development at the Fraunhofer Institute for Cell Therapy and Immunology, Sandy combines long-standing RNA research experience with cell therapy development in immuno-oncology to investigate mRNA-based cell and gene therapies for the treatment of diseases like cancer.

Dr. Jacqueline Breuer is a biotechnologist with expierience in viral vector production and purification, and quality management and control. She obtained a Doctor of Natural Sciences Molecular Medicine at Medical School Hanover and subsequently worked as a Research Associate in different Companies and Scientific Institutions. At the Fraunhofer Institute for Cell Therapy and Immunology, she is responsible for establishing the AAV manufacturing technology and various research projects in the same field.
A special mention to authors Dr. Thomas Schmid and Dr. Ulrich Blache for also collaborating on this piece.
Gene therapy represents a groundbreaking approach in the field of medicine, aiming to treat or prevent diseases by modifying the genetic material within a patient's cells. The concept involves introducing, removing, or altering genetic sequences to correct or mitigate the effects of diseasecausing mutations. Over the past few decades, advancements in molecular biology and genetic engineering have significantly propelled the development and application of gene therapy. Here, we provide our insights on progressing a novel gene therapy into clinical trials and various options that need to be considered along the way.
Gene therapy encompasses various strategies, including in vivo and ex vivo methods. In vivo gene therapy involves directly delivering genetic material into a patient's body, while ex vivo techniques involve modifying cells outside the body before reintroducing them to the patient. Viral vectors, such as adenoviruses and lentiviruses, are commonly used to deliver therapeutic genes due to their efficiency in transducing cells. Non-viral methods, including liposomes and nanoparticles, offer alternative delivery mechanisms with potentially fewer immunogenic issues.
Ensuring long-term efficacy, avoiding immune responses, and addressing ethical and regulatory concerns are some of the key challenges that still remain. However, ongoing research and clinical trials continue to refine these therapies, offering hope for curing previously untreatable diseases and revolutionising modern medicine.1,2
Virus-free Peptide-based Systems for Gene Therapies
Successful gene therapies depend on efficient methods to introduce DNA into target cells and ensure effective protein expression. Traditionally, viral vectors have been the preferred tool for DNA delivery due to their high efficiency. However, they come with significant drawbacks. Viral vectors can provoke strong immune responses, leading to inflammation and reduced therapy effectiveness. Moreover, viral vectors have a limited capacity for carrying genetic material, and their complex, expensive production processes further complicate their use. Additionally,
there is a risk of insertional mutagenesis, where viral DNA integrates into the host genome, potentially disrupting essential genes and posing serious safety concerns.
In contrast, peptides are emerging as a promising alternative to traditional viral vectors for DNA delivery due to their unique advantages. Unlike viruses, peptides offer a safer and more controlled delivery method by facilitating the condensation of DNA into nanoparticles, enhancing cellular uptake, and promoting subcellular release – key factors for effective gene expression. Peptides are also highly customisable, allowing for precise adjustments to improve biocompatibility, stability, and to overcome cellular barriers that often limit non-viral gene delivery efficiency. Additionally, they can be more easily scaled and produced at a lower cost. Crucially, unlike viral vectors, peptides bypass the issue of pre-existing immunity that can hinder the effectiveness of viral therapies and do not present safety concerns related to genome integration, thus eliminating potential long-term risks.3,4,5 Overall, peptide-based systems present a versatile, biocompatible, and cost-effective alternative to viral vectors, addressing many of the limitations associated with viral gene delivery systems and advancing the field of gene therapy.
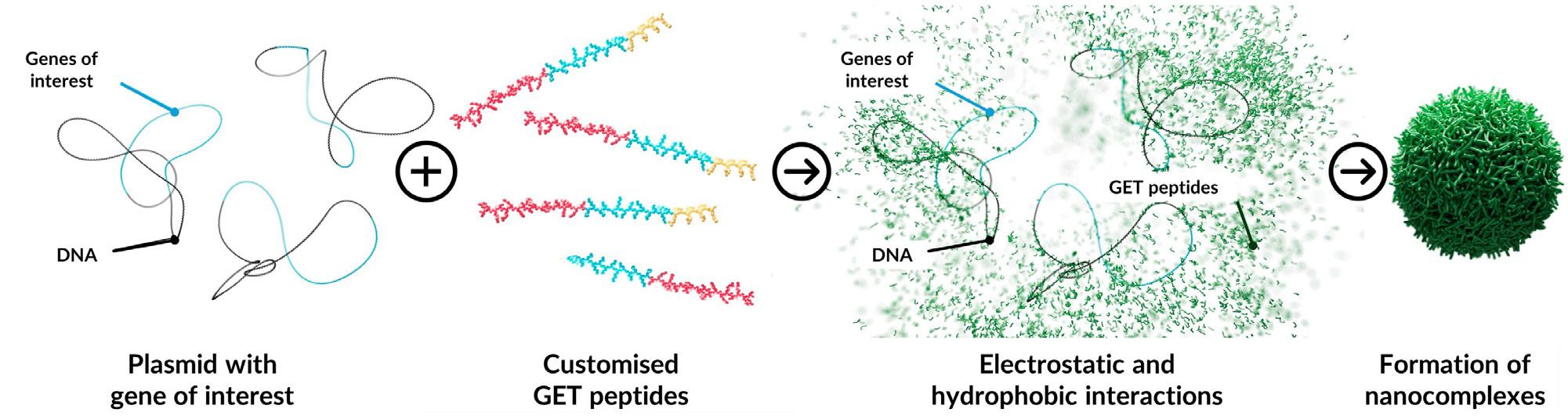
One such approach by Dr. James Dixon from the University of Nottingham has led to the development of a non-viral gene delivery system designed to deliver various types of therapeutic payloads, termed glycosaminoglycan-binding enhanced transduction (GET).6
The basic DNA delivery technology for gene therapy comprises two main components: DNA plasmids and engineered peptides. Due to their differing charges, DNA plasmids/other cargo (negatively charged) and peptides (positively charged and amphipathic) exhibit strong electrostatic attraction and hydrophobic interactions. This interaction between DNA and peptides suppresses the electrostatic repulsion of anionic nucleotides, leading to the formation of spherical nanocomplexes with hydrodynamic sizes ranging from 10 to several hundred nanometres, making them suitable for delivering multiple DNA molecules into target cells (Figure 1).7,8

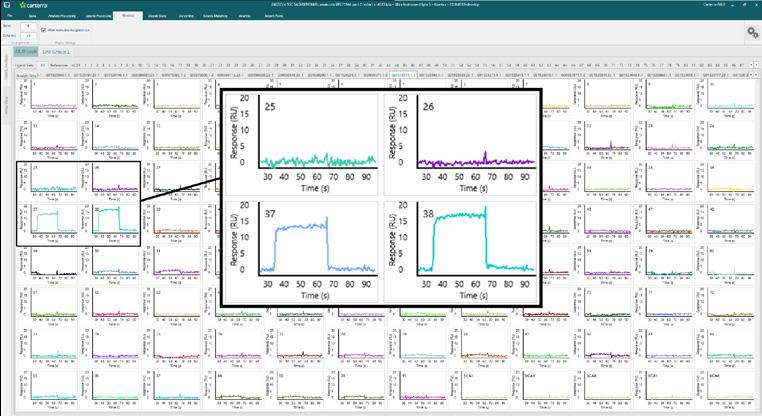
Fragment hit identi cation against 96 proteins using the Carterra Ultra platform.




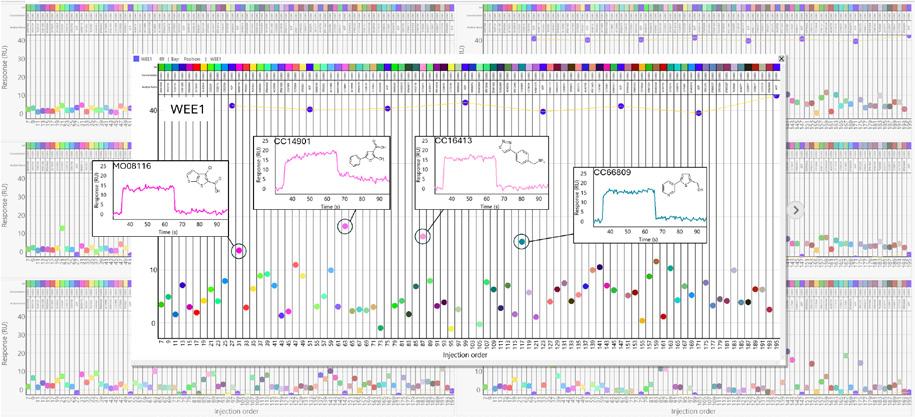
Maybridge fragment library screening against a kinase panel.





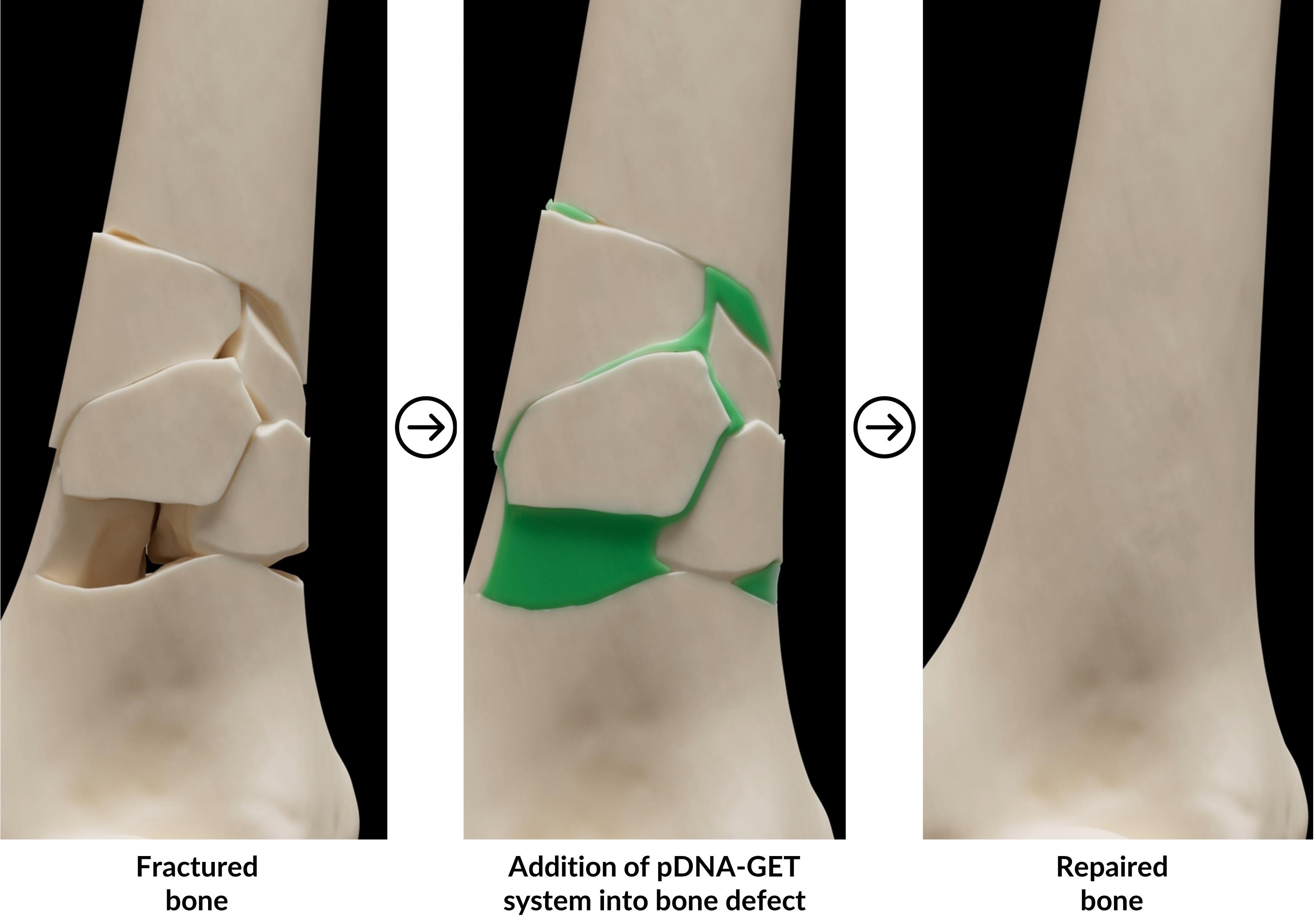
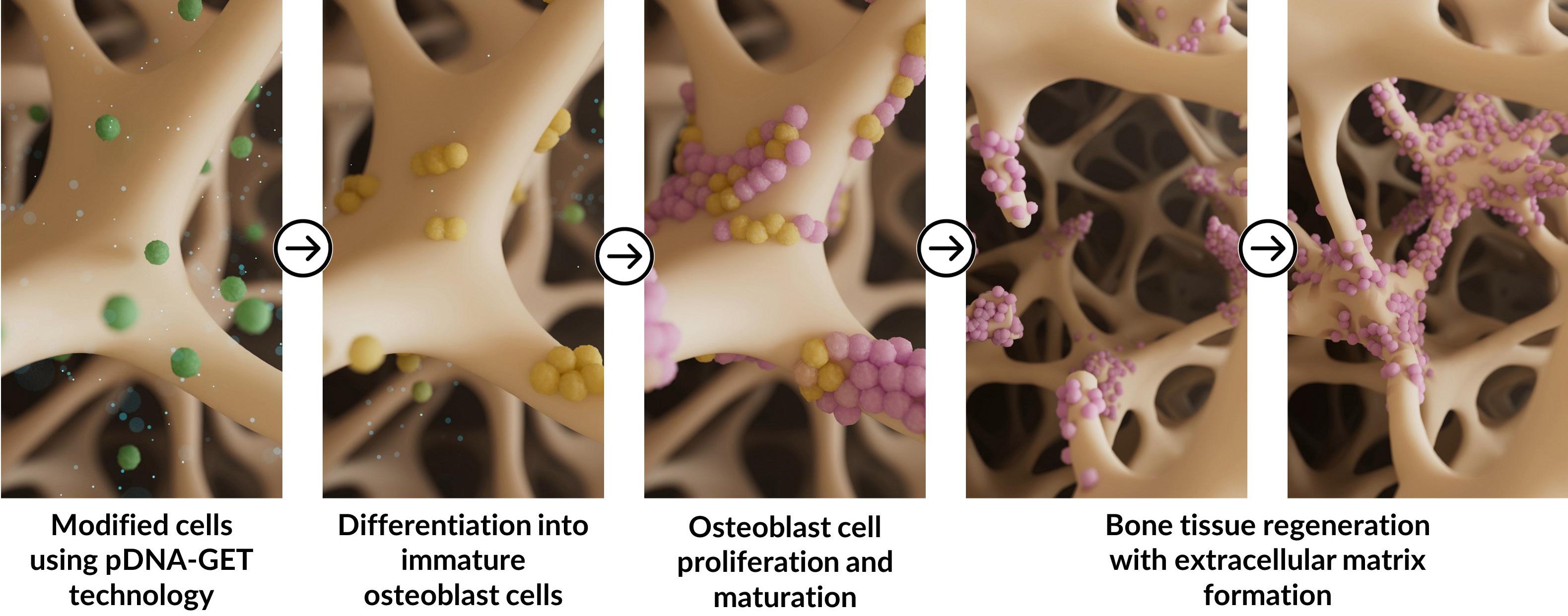
GET technology is very versatile and can be utilised to deliver therapeutic payloads to various cell types. It is currently being applied to improve bone graft augmentation for orthopaedic applications by researchers at TherageniX. This innovation aims to improve bone grafting outcomes by using autologous or allogeneic donor cells to enhance the expression of osteogenesis-related genes, like bone morphogenic proteins (BMPs). This approach aims to boost the regenerative capacity of skin, bone, muscle, and cartilage post-surgery.
On the other hand, this technology offers remarkable versatility through the straightforward substitution of genes inserted into plasmids or the introduction of alternative cargoes, enabling applications across various medical fields. By modifying the genetic payload, for instance, this approach can be tailored for use in dermatology, cardiology, ophthalmology, and immunology, offering precise, personalised treatments across multiple tissues and conditions.
To minimise risks and costs in drug development, particularly for smaller biotech companies, investing effectively in early-stage development is crucial. In the gene therapy field, this involves several key aspects. As an example, plasmid optimisation is critical for enhancing gene expression efficiency. This process involves carefully selecting and incorporating optimal and/or engineered promoters, enhancers, and regulatory elements to maximise transgene expression and stability, especially in the context of a specific tissue target. Moreover, effective plasmid design with minimal bacterial sequences and incorporation of features for efficient packaging and delivery into target cells can enhance robust and stable transgene expression, thereby maximising the therapeutic potential of the gene therapy. This includes optimising codon usage and avoiding unwanted immune responses.9
In the design of cutting-edge gene therapies, such as TherageniX’s virus-free peptide-based system, choosing the right peptide sequences for targeting and entry into specific cell types is another key factor to ensure that the therapeutic gene reaches the desired location and functions effectively within the target cells.10 In addition, conducting comprehensive in vitro screening to assess the efficiency of gene delivery, expression, and functionality of the therapeutic gene in various in vitro cellular models helps identify and select the most promising candidates and refine the delivery system.11,12
Pre-clinical early development also involves safety and efficacy testing, pre-formulation studies, and drug product evaluations in vitro and in vivo to support clinical trial designs. Key activities include safety pharmacology, pharmacodynamics, toxicology, and pharmacokinetic studies. Evaluating potential safety issues, such as immune responses or off-target effects, early in the development process helps to minimise risks and improve the therapeutic potential of the gene therapy.13
The selection of appropriate in vivo animal models is essential for the development and evaluation of gene therapy drug products as it directly impacts the accuracy of pre-clinical assessments, including safety, efficacy, and potential clinical outcomes.
Generally, animal models are broadly categorised into small and large species. In orthopaedic research, small species like rodents are commonly used in early research due to their genetic similarities to humans, cost-effectiveness, and ease of handling, though their size limits their ability to fully replicate human bone structure and function. Therefore, larger animals like sheep, pigs, dogs, and non-human primates are employed in the later stages for more accurate simulations of human conditions, such as joint mechanics and long-term healing. For example, sheep are useful for studying bone density, though their bone quality differs from humans; pigs closely resemble human bone structure, but their rapid growth complicates direct comparisons; dogs, with similar bone characteristics to humans, also present variability in bone turnover.15,16,17
In the context of in vivo models, ectopic bone formation models involve implanting osteogenic materials or cells into non-bone tissues such as subcutaneous, intramuscular, or renal sites. This approach facilitates controlled studies of bone development outside its natural environment, making it ideal for the preliminary evaluation of new materials and techniques. These models provide valuable insights into the processes of osteogenesis within a simplified and controlled setting. Conversely, orthotopic bone models replicate specific orthopaedic conditions within the natural bone environment. These models are essential for studying bone repair and regeneration in clinically relevant settings, providing more accurate insights into treatments. Both ectopic and orthotopic models are crucial for advancing orthopaedic care and improving human treatment outcomes.18
Overall, researchers must carefully select in vivo models, based on specific research questions, and confirm results across different animal types before clinical application. Additionally, advances in in vitro systems and bioresorbable implants aim to address ethical concerns and improve the translation of research findings to human medicine.
Cell and gene therapies began receiving regulatory approval in the mid-2010s. Since then, the number of annual approvals has gradually increased, with more than 30 approvals in the US and Europe reported as of early August 2024.19
Manufacturing cell and gene therapy products is crucial, with most therapies currently being autologous, focussing on process and method development due to scalability challenges. Higher throughput can be achieved by increasing manufacturing capacity or exploring decentralised, point-of-care production. Companies are also exploring allogeneic therapies, which facilitate classical scale-up, potentially reducing costs and speeding up treatment delivery. As demonstrated by Dr. Dixon’s group and his colleagues,6, 14 most recent advancements using cell-free peptide-based approaches enable gene therapy production through established plasmid manufacturing and synthetic peptide synthesis techniques, offering the possibility of scalable and cost-effective production.
Another key consideration for companies is whether to handle manufacturing in-house or outsource it. In-house manufacturing offers greater control and expertise but requires significant capital investment in facilities, personnel, and


Document, search, and collaborate
Scientific data management with new add-on functionality to make discoveries faster
Generate, QC, and analyze results
Register entities to secure your assets
Keep track of entities
Deep learning and AI
Connect instruments, data, and tools
Manage and analyze experimental data
Assay informatics
Plot and mine results

Application of GMP to manufacturing steps is shown in dark grey GMP Principles should be applied where shown in light grey starting material – active substance – finished product
In vivo gene therapy: mRNA Plasmid Manufacturing and linearisation In vitro transcription
In vivo gene therapy: non-viral vector (e.g. naked DNA)
In vivo gene therapy: viral vectors
Ex vivo: genetically modified cells3
Plasmid Manufacturing
Plasmid Manufacturing
Donation, procurement and testing of tissues / cells1
Establishment of bacterial bank (MCB, WCB)
Establishment of a cell bank (MCB, WCB) and virus seeds when applicable
Establishment of a cell bank (MCB, WCB) and virus seeds when applicable
Plasmid Manufacturing Vector Manufacturing
mRNA
Manufacturing and purification Formulation, filling
DNA
Manufacturing, fermentation and purification
Vector
Manufacturing and purification
Genetically modified cells
Manufacturing
filling
Formulation, filling
Formulation, filling
Examples of where full good manufacturing practice (GMP) or GMP principles apply in the manufacturing steps of in vivo gene therapy and ex vivo genetically modified cells (adapted from20). Abbreviations: MCB and WCB stand for master cell bank and working cell bank, respectively.
maintenance. Therefore, companies must conduct a “build vs buy” analysis to see what approach would suit them better. Currently, a hybrid model is emerging, where companies can rent pre-built, qualified facilities or train their staff to execute manufacturing steps. This approach allows companies to leverage their expertise without the need to invest heavily in their own manufacturing infrastructure.
Besides business considerations, chemistry, manufacturing, and controls (CMC) regulatory aspects also need to be considered. Companies need to have a good idea of possible countries they plan to conduct their first clinical trials in and the relevant regulations that govern them. In Europe, cell and gene therapies are considered an advanced therapy medicinal product (ATMP). It's necessary to clearly define which manufacturing steps and quality control tests apply to the ATMP's starting materials (SMs), active substance (AS), and drug product (DP) evaluated in accordance with the guidance “Questions and Answers on the Principles of GMP for the Manufacturing of Starting Materials of Biological Origin Used to Transfer Genetic Material for the Manufacturing of ATMPs” (EMA/246400/2021) released by the European Medicines Agency (EMA).20 This highlights (Figure 2) where good manufacturing practice (GMP) or GMP principles apply in the manufacturing steps of in vivo gene therapy and ex vivo genetically modified cells.
Other general regulatory considerations include identifying the country where the product is being developed and its intended use. Understanding the clinical application is vital for designing preclinical studies that are representative of the intended use. Knowing the country where clinical trials will be conducted enables manufacturers to address the specific quality, safety, and efficacy requirements, which may encompass aspects like sourcing SMs, developing QC testing
strategies, defining product control, release parameters, and manufacturing critical process parameters. For instance, in the United States, Phase I clinical trials do not require full GMP compliance for manufacturing, a requirement that is mandated in Europe. Furthermore, regulatory frameworks differ across countries, with variations in quality requirements for SMs, drug substances (DS), and DPs (e.g., United States Pharmacopeia vs European Pharmacopoeia). There are no standardised clinical and non-clinical development programmes and achieving full regulatory harmonisation is difficult due to the continuous evolution of scientific knowledge and technology. Therefore, regulatory authorities encourage sponsors to seek regulatory advice early when preparing first-in-human (FIH) data packages.
While developing a novel gene therapy product, it is vital to interact with key stakeholders from the beginning. Depending on the type of therapy, these stakeholders may vary. Key opinion leaders in the therapeutic area have extensive knowledge and experience and can contribute to clinical and non-clinical development. Understanding a clinician’s pain point and the improved treatment and therapies they need to help their patients is an important part in ensuring that the developed product meets the end-user requirements. While developing rare disease treatments, engagement with patient groups is also vital.
As the field of gene therapy is constantly evolving, it is extremely valuable to engage with relevant regulatory agencies early on during product development. This would serve the dual purpose of introducing your novel therapy to the regulatory agencies who would evaluate clinical trial applications while also providing the company with valuable feedback on their products. Various types of meetings and mechanisms exist for these depending on geographical areas.

Single-length polyethylene glycol derivatives for demanding applications in pharma
One of the vital building blocks for the success of early-stage companies developing novel therapies is to build a team that delivers. Start-ups, especially spin outs from universities, have a very strong understanding of the science and technology as the founder/co-founder might have helped develop it as part
of their research. While this understanding is important, there are various other aspects that are critical.
Industry experience helps in enhancing the understanding of how therapies are developed and results in acquisition of key transferable skills to the start-up setting. Early-stage
companies also need to develop strategic and long-term thinking and be comfortable dealing with uncertainties. This is extremely important as the company embarks on a new journey and will never have all the information or data available at the needed time to make decisions. A good risk management approach coupled with scenario planning would help navigate these unknown waters. Building a team that has the right mindset, capability and capacity is a big factor that drives success. Companies are often faced with the option of in house hiring or engaging consultants. A hybrid approach, where certain key roles are internal to the company while engaging external partners for certain services, could work favourably in the initial phases. Currently, fractional services are also offered for various roles which help minimise costs and obtain relevant capabilities within the organisation. Building a team is challenging but greatly rewarding.
Developing novel gene therapies is an exciting and extremely challenging endeavour. Strong fundamental research, a sound pre-clinical development plan, manufacturing strategy based on "build vs buy" and early interactions with various stakeholders are key components to progress gene therapy forward. A passionate and dedicated team will help manage the challenges ahead and successfully guide the product to its desired destination.
1. Bulaklak, K. & Gersbach, C.A. The once and future gene therapy. Nat Commun. 11(1), 5820 (2020).
2. Chancellor, D., Barrett, D., Nguyen-Jatkoe, L., et al. The state of cell and gene therapy in 2023. Mol Ther. 31(12), 3376–3388 (2023).
3. Urello, M., Hsu, W.H. & Christie, R.J. Peptides as a material platform for gene delivery: Emerging concepts and converging technologies. Acta Biomater. 117, 40–59 (2020).
4. Tarvirdipour. S., Skowicki, M., Schoenenberger, C.A, et al. PeptideAssisted Nucleic Acid Delivery Systems on the Rise. Int J Mol Sci. 22(16), 9092 (2021).
5. Hadianamrei, R. & Zhao, X. Current state of the art in peptide-based gene delivery. J Control Release. 343, 600–619 (2022).
6. Dixon, J.E., Osman, G., Morris, G.E., et al. Highly efficient delivery of functional cargoes by the synergistic effect of GAG binding motifs and cell-penetrating peptides. Proc Natl Acad Sci U S A. 113(3), E291–9 (2016).
7. Boisguérin, P., Konate, K., Josse, E., et al. Peptide-Based Nanoparticles for Therapeutic Nucleic Acid Delivery. Biomedicines. 9(5), 583 (2021).
8. https://theragenix.health/, visited on 12 Aug 2024.
9. Folarin, O., Nesbeth, D., Ward, J.M., et al. Application of Plasmid Engineering to Enhance Yield and Quality of Plasmid for Vaccine and Gene Therapy. Bioengineering (Basel). 6(2), 54 (2019).
10. Todaro, B., Ottalagana, E., Luin, S., et al. Targeting Peptides: The New Generation of Targeted Drug Delivery Systems. Pharmaceutics. 15(6), 1648 (2023).
11. Wei, F., Wang, S. & Gou, X. A review for cell-based screening methods in drug discovery. Biophys Rep. 7(6):504-516 (2021).
12. Astashkina, A., Mann, B. & Grainger, D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol Ther. 134(1), 82–106 (2012).
13. 13) https://www.pharm-int.com/2022/02/08/the-importance-ofearly-stage-development/, visited on 12 Aug 2024.
14. Raftery, R.M., Walsh, D.P., Blokpoel Ferreras, L., et al. Highly versatile cell-penetrating peptide loaded scaffold for efficient and localised gene delivery to multiple cell types: From development to application in tissue engineering. Biomaterials. 216, 119277 (2019).
15. https://www.intechopen.com/chapters/69075, visited on 12 Aug 2024.
16. Schindeler, A., Mills, R.J., Bobyn, J.D., et al. Preclinical models for orthopedic research and bone tissue engineering. J Orthop Res. 36(3), 832–840 (2018).
17. Hixon, K.R. & Miller, A.N. Animal models of impaired long bone healing and tissue engineering- and cell-based in vivo interventions. J Orthop Res. 40(4), 767–778 (2022).
18. Scott, M.A., Levi, B., Askarinam, A., et al. Brief review of models of ectopic bone formation. Stem Cells Dev. 1(5), 655–67 (2012).
19. https://www.fda.gov/vaccines-blood-biologics/cellular-genetherapy-products/approved-cellular-and-gene-therapy-products, visited on 12 Aug 2024.
20. https://www.ema.europa.eu/en/documents/other/questionsand-answers-principles-gmp-manufacturing-starting-materialsbiological-origin-used-transfer-genetic-material-manufacturingatmps_en.pdf, visited on 12 Aug 2024.

Anandkumar Nandakumar is the CEO and Co-founder of TherageniX. He has a PhD in Biomedical Engineering that focussed on bone tissue regeneration. Anand’s professional journey spans across various product types – medical devices, vaccines, orphan drugs, cell and gene therapy in start-ups, scale ups and established companies. He has played a key role in bringing various early-stage assets to early-stage clinical trials, building teams, setting up a manufacturing facility and launching drugs.
Email: anand@theragenix.health

Dr. Stefania Fedele
Stefania Fedele is a Scientific Project Manager at TherageniX. She holds a PhD in Structural and Functional Genomics and has completed postdoctoral research in Embryology and Stem Cell Biology. Stefania has driven the development of innovative drug therapies to enhance human health at several biotech companies, establishing transformational strategies in neurodegeneration, immuno-oncology, and rare diseases, utilising viral- and lipid-based particles for the delivery of DNAs, RNAs, proteins, functional enzymes, and antibodies.
Email: stefania@theragenix.health

Dr. James Dixon
James Dixon is an Associate Professor in the School of Pharmacy at the University of Nottingham, UK and developer and inventor of the GET peptide delivery system for gene therapy. His research group has funding from charity, governmental, venture capital and military sources to develop gene therapies for vaccinology, oncology, gene correction/ augmentation/editing and for regenerative medicine, covering wound healing and orthopaedics. He has a PhD in Biomedical Science from Imperial College London.
Email: james@theragenix.health
Join us at the Novotel London West from November 6–8 2024, for Cell 2024, where you'll explore the full potential of the CGT value chain – from discovery and development to successful commercialisation.
This three-day event features the Cell Culture Congress, Advanced Therapy Development Congress, and Cell & Gene Therapy Manufacturing Congress, with over 100 industryleading speakers, 600 delegates, 50+ partners, 150+ case study presentations, and 15 diverse content tracks, including a new innovation track.
Engage with key topics like Gene Therapy Discovery, CGT Commercialisation, and Stem Cell Therapy Development through dynamic presentations, interactive sessions, and exceptional networking opportunities.
Don't miss your chance to immerse yourself in the evolving CGT landscape.

07 - 08 November 2024 | Boston, MA

• 2-day Event
• In-Person Congress & Exhibition
• Track 1: Novel Targets Beyond The Rule Of Fives
• Track 2: Novel Models For ADME-Tox Research & Disease Modelling
• Track 3: Augmenting R&D With AI and Automation
any queries please contact:
• Track 1: Innovative Technologies For Hit Identification & Screening
• Track 2: Computational & Medicinal Chemistry
• Track 3: Genomics-Driven Drug Discovery & Bioinformatics

06 - 08 November 2024 | London, UK

• 3-day Event
• In-Person Congress & Exhibition
Join our premier event to unlock the potential of the CGT value chain, from discovery and development to successful commercialisation
100+ Industry-Leading Speakers Including...

DOLORES SCHENDEL, Chief Scientific Officer, Medigene AG

MARIA LUISA GIORELLO, Global Gene Therapy Platform Enablement Director, Pfizer

JOHN GILL, Senior Director Cell Line Development, Gilead Sciences

ELI GILSOHN, Vice President Intellectual Property, Resolution Therapeutics

JULIA MATILAINEN, Head Patient Operations CGT, Roche

• 600+ leading pharma, biotech & academic delegates
• 30+ hours of presentations, discussions & interactive content at both events
• 13 hours of networking breaks, including speed networking & refreshments
• 230 pre-arranged 1-2-1 meetings, facilitating business growth
For any queries please contact: marketing@oxfordglobal.com
Scan the QR code to visit the Oxford Global website

Animal Free
Bioactive
Easy to use
Simple Protocol
Innovative Technology
Characterised and Defined
Scalable Production
Multiple Bioactive Modules
Based on using engineered Caf1 proteins produced recombinantly in bacteria to mimic the action of bioactive proteins such as extracellular matrix (ECM) proteins and growth factors.
A leading supplier of serum and plasma in the United Kingdom and Europe.
Normal Serum and Plasma
Individual Donor Units, Mixed Pools, Specific Pools
Wide range of origins and pack sizes available, including bespoke filling
Disease State Serum and Plasma
Prospective collections, US and EU Origin
Fetal Bovine Serum
Donor Bovine Serum
Newborn Calf Serum
Adult Calf Serum
Donor Horse Serum Bovine Serum Albumin Porcine Serum
Medica 11-14 Nov 2024
EBF 20-22 Nov 2024
ESACT UK 8-9 Jan 2025
MedLab 3-6 Feb 2025


PYROSTARTM Neo+ is based on a genetically engineered approach to produce reactive factors required for endotoxin detection at pharmaceutical and medical equipment manufacturing sites. With greater stability of the negative control and better endotoxin recovery in heparin and heparin-based compounds, Neo+ facilitates the same sensitivity of endotoxin detection as our traditional limulus amebocyte lysate (LAL) reagent, through a more sustainable and environmentally friendly method.
• Colorimetric method, can be used with an absorbance plate reader
• 3-Factor system mimics the same cascade reaction as traditional LAL
• Endotoxin-specific reagent eliminates the risk of false positives from (1-->3) ß-D-Glucan
• 100% free of horseshoe crab blood
• Quantitative range: 0.001 to 50EU/mL
• High sensitivity with less lot-to-lot variation
• Stable storage after dissolution (4 hrs. at 2-8°C and 2 weeks at -30°C)



