Subsection: Nasal & Pulmonary Drug Development & Delivery










Q&A
with Will Ganley, Senior
Specialist, Nanopharm
Can you Explain ‘in Silico Modeling’ and What a PBPK Model is?
In silico modeling involves using computer simulations to predict how drugs behave in the human body. An example is physiologically based pharmacokinetic (PBPK) modelling, which uses drug property, physiology, and biochemical data to predict absorption, distribution and excretion (ADME) processes using pharmacokinetic models.
The outcome is a system of equations employing a 'bottom-up' strategy that begins with modeling each specific process that moves the drug throughout the body. Starting from these foundational elements and progressing to a comprehensive model that integrates data from various sources, such as in vitro tests and clinical trials, we can replicate observed clinical outcomes. By developing a model that assigns significance to input data based on physiological relevance, we create solutions that accurately predict clinical outcomes, resulting in a trustworthy PBPK model.
How is the PBPK Model Applied Specifically to Inhaled Drug Products?
A typical whole-body PBPK model can be regarded as a large flow diagram, which maps mass transport processes across different types of tissue and blood vessels. If all the drug mass starts in the lung after a subject has inhaled, the model shows that the drug mass moves through the rest of the body and is exposed to the different tissues and blood compartments before it is ultimately metabolised and eliminated. For orally inhaled products, we are particularly interested instead in the details of what happens in the lungs.
The lung can be divided into two broad regions: the central region, which encompasses the upper airways where the blood flow goes from the arterial blood through to the venous blood, and the lower peripheral airways where the blood flow
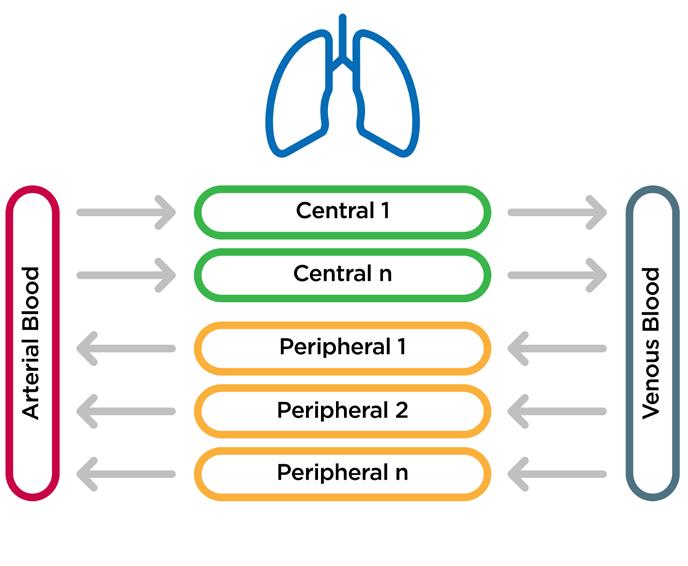
is reversed. We can further segment both regions into more granular compartments.
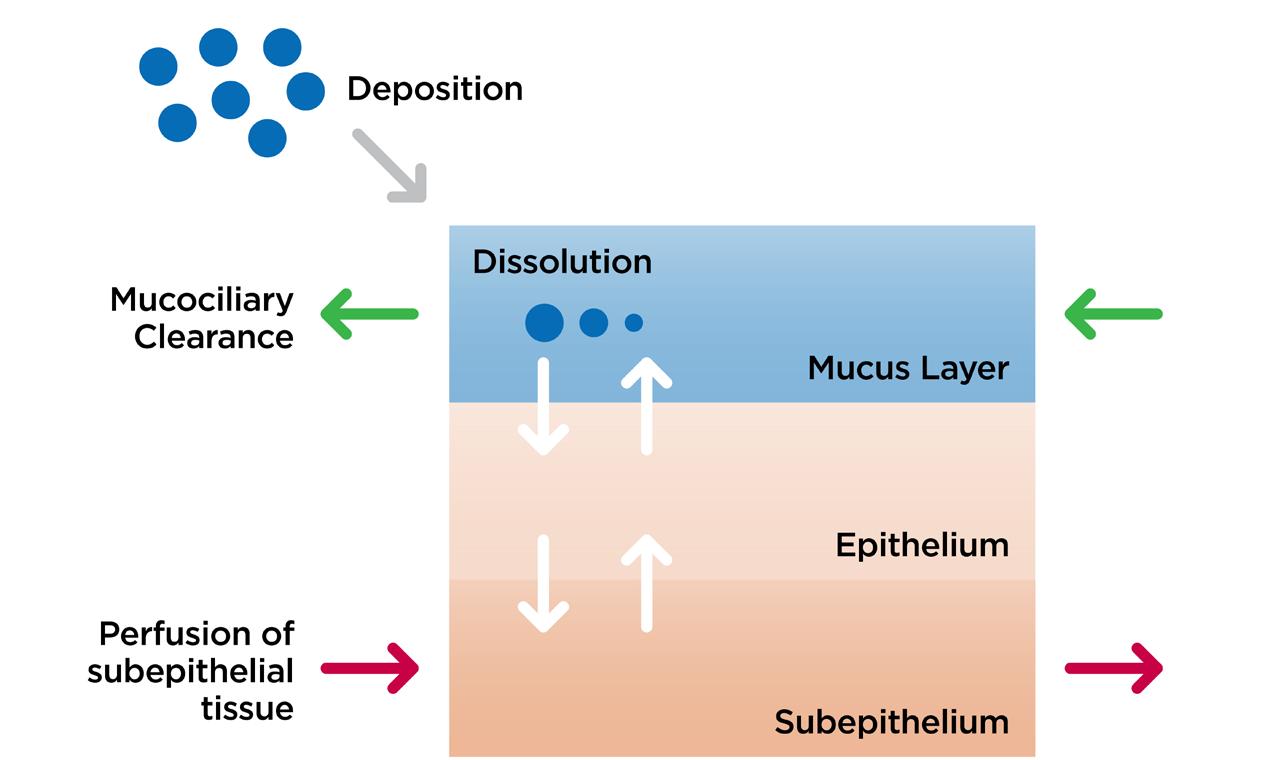
Within each compartment, the mucus layer is where the drug will typically start its journey as either a completely dissolved system, for example from a nebuliser, or as undissolved particles. For the latter, we directly simulate the particles’ dissolution in the mucus layer before they permeate down into the lung tissue.
In the mucus layer, dissolved drug will passively diffuse into the epithelium, then subepithelium, before entering the blood vessels and being carried away to the rest of the body. Knowing this, it is important that we consider the inputs to the model when developing and designing studies that allow us to generate the model’s input parameters.
There are two key types of input parameters for orally inhaled PBPK models. Deposition data that is obtained through deposition models or imaging data. Commonly used
deposition models include computational fluid dynamics (CFD) or semi-empirical methods (such as the National Council on Radiation Protection and Measurements model). The advantage of CFD models is that computed tomography (CT) scans of healthy volunteers and patient airways can be used in the simulations to generate realistic and subject specific deposition predictions in the lungs.
The other form of input data is dissolution data, typically through in vitro studies. At Nanopharm, we have developed a dose collection system called Dissohale™ that allows us to deposit the lung dose uniformly and measure its dissolution. We then use a modelling approach that allows us to determine the dissolution of deposited particles that are irregular in shape and agglomerated. We take the fitted equivalent spherical representation of these particles and scale down the dissolution medium volume, matching the mucus volume in each region of the lung, which can be input into our PBPK model.
How Do You Use This Information to Generate Data?
Once we have the input parameters for the PBPK model, the next step is to address the questions posed by clinical product development.
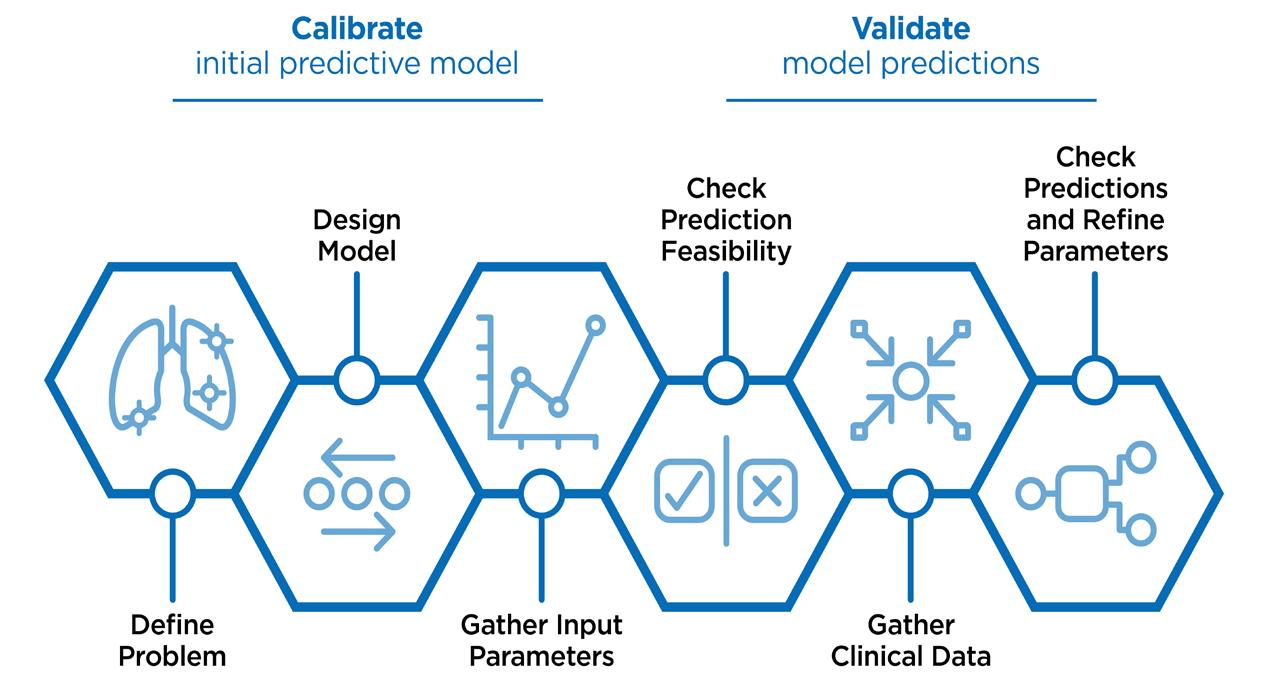
To start, we define the problem – whether it be as simple as observing the interplay between two competing processes, through
to a complex scenario of simulating a clinical trial. This information allows us to determine the number of lung compartments required, the dissolution model, and whether we will obtain the input parameters from pre-clinical data or literature.
Following on from that, we verify the prediction feasibility to check whether the predictions follow the right time course, or the right curve shape and ensure they are not producing wildly unexpected results. To do this, we recommend gathering clinical data either generated as part of the drug development programme or from literature. For any uncertainty with initial data, we recommend that parameters be refined until the model is producing the data that is expected within the model.
Can You Introduce Simhalation™, and Discuss How it is Being Used in Drug Development?
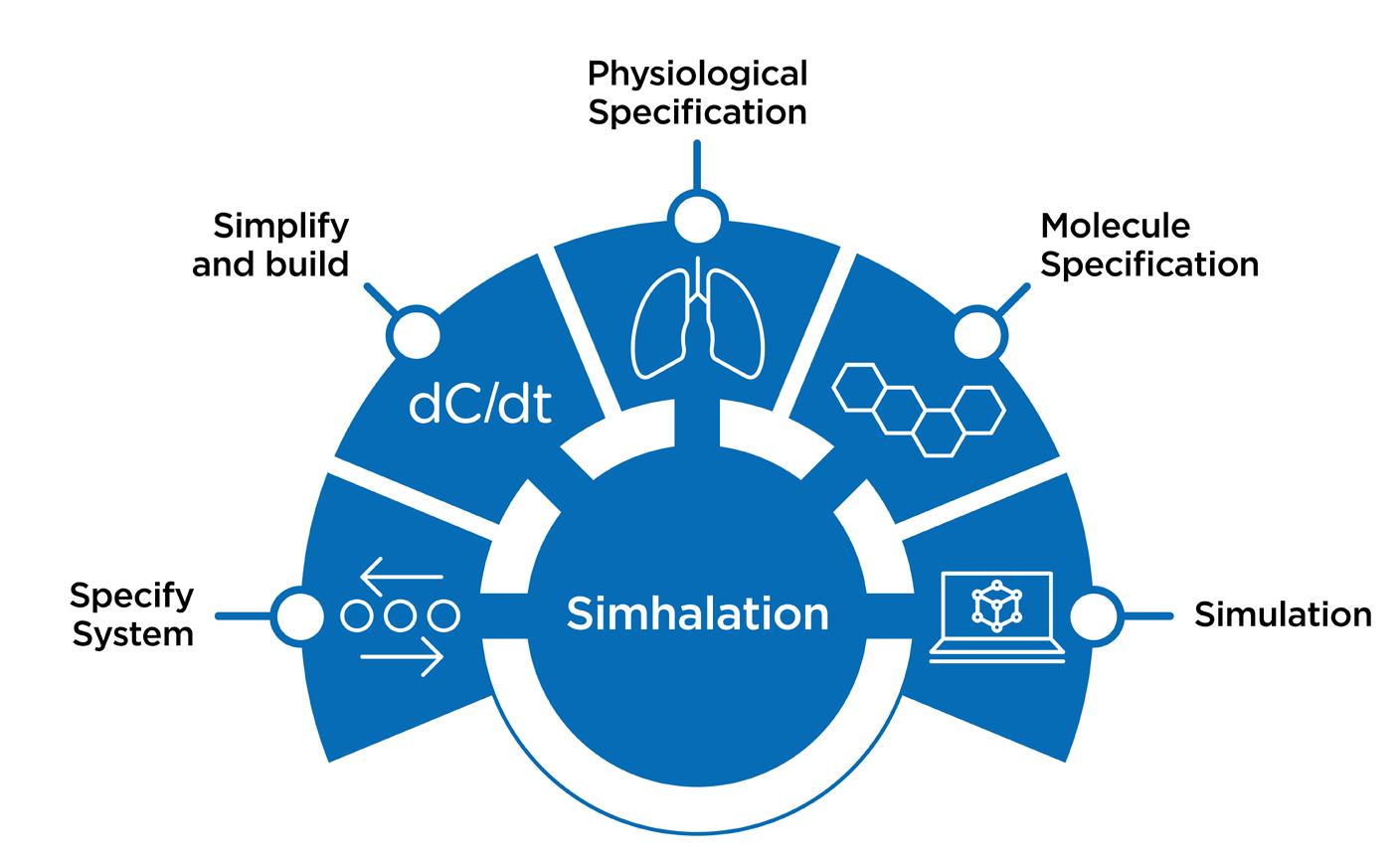
Simhalation™ is an in-house PBPK model, developed at Nanopharm. Our software will generate a set of equations representing the system and build executable computer code.
The model will ask us for two things: physiological specifications (for example, the lung surface areas of the subjects we are interested in) and the molecular specification (such as log pKa and the rate of hepatic clearance). This allows us to input parameters for a range of patients and a range of formulations or molecules, run our simulations and finally generate the output we are looking for.
The Simhalation PBPK model can be used across the entire product development life cycle for orally inhaled drug products, but first
it is important to understand how different ‘levers’ in the model will affect the exposure of a drug at the site of action.
Factors such as how the angle of insertion from different device variants impacts the deposition within the nose, dissolution rates and mucociliary clearance (MCC) which can be propagated through to the local or systemic exposure in the PBPK model to understand the effect on the exposure of your target drug at the site of exposure. Using this information, we can run multiple simulations for repeated dosing and assess things like inter- and intrasubject variability as well.
How Can You Ensure The PBPK Model is Credible Enough to Provide the Answers You Need?
There are two key concerns I see with PBPK models:
The first concern is that PBPK models are so highly parameterised that users think it is possible to achieve any answer by tuning the parameters accordingly, and secondly, are we
able to justify using the model if we are going to have to perform a clinical study anyway?
Before starting any work, it makes sense to address both concerns by revisiting how the model is going to be used. Are we using it to answer some simple drug product development questions, or are we trying to replace a clinical study or use the predicted data to aid regulatory decision making?
Because these two things require different levels of credibility to be established, it can be useful to understand the model constraints, and the data available.
Even if we do not know the exact value of parameters for a given model – such as the lung epithelial permeability – for the types of molecules we are looking at, we might know their values to within a few orders of magnitude. Applying all the known or estimated model parameter ranges constrains the prediction space, meaning that the scope of possible predictions is quite limited.
Secondly, look at the data available or consider how it could be generated. Can we access data from available literature, or do we need to generate this for ourselves? Once a model is verified for a particular type of molecule, say a lowly soluble and highly soluble molecule, the model validation data should be applicable to other molecules and products of the same type. The regulatory landscape surrounding this is evolving rapidly now and should allow the use of validated modelling “platforms” in submissions soon using frameworks such as the Model Master File, which is current being developed by the United States Food and Drug Agency.
What is useful about this framework is that we can do it as a paper-based exercise before modelling commences to understand what
we can and can’t use the model for, whether we need to perform any lab work or if we can already answer our questions of interest.
Can You Share Some Examples Where You Have Used PBPK Modeling For OINDPs at Nanopharm?
There are a few examples of how we’ve used PBPK modeling for orally inhaled drug product development here at Nanopharm.
We wanted to look at the dissolution of dry powder inhalers (DPIs) to understand what the influence of the dissolution of fluticasone propionate is on the systemic exposure of these drug products.
We were able to parameterise the model for fluticasone propionate using data from a literature study to reproduce three different doses by tweaking the dose in the model but keeping all other parameters the same.
Within the study data, three different dry powder formulations of fluticasone propionate had been developed with different aerodynamic and dissolution rates. Using this, we refined our model parameters against one of them and then using the in vitro data that was generated in that study, we extracted the dissolution rates and input that in the model for the other two formulations.
We found that with the PBPK model, we were able to reproduce the shapes of the systemic pharmacokinetic curves by tuning the dissolution parameters and show that dissolution was the driving factor and make credible predictions by using the data. Also, we predicted the systemic exposure of different parameters as if we were developing a product.
The model ended up concluding that you can deliver different doses fluticasone propionate to the lung with different dissolution rates, and if the total dose is fixed, then the total exposure that the subject received will be the same across different formulations but the maximum concentration will be different. This is an important consideration for drug product safety and would be useful information at the start of a new development program.
In another example, we were looking specifically at the delivery of spray dried amikacin sulphate powder to cystic fibrosis patients using a spray dried powder characterised using the anatomical mouth-throat models and realistic breathing profiles. We were very interested in the difference in exposure for both cystic fibrosis patients and healthy volunteers.
We parameterised the model using literature data and then worked to generate CFD deposition data using two lung geometries from our database. When we ran these simulations comparing a cystic fibrosis patient and a healthy volunteer, we found that when the same dose was delivered to these two subjects, the healthy volunteer would receive a lot more of it to their peripheral lung whereas the cystic fibrosis patient would receive a lot more to their central lung. Inputting this data into our PBPK model and running a sensitivity analysis similar, we were interested in disease state differences rather than differences in performance parameters.
When we input some other aspects of cystic fibrosis into the model – such as mucus thickness – we're able to see what effect that might have on systemic exposure. In this case, it effectively dilutes the mucus layer and means less of the drug can get through to the tissue as quickly. But due to the thick and sticky mucus of cystic fibrosis patients, the cilia beating is inhibited resulting in mucus clearance that isn't particularly effective. So, in the model, we can represent this by effectively turning mucus clearance off.
What we found is that this gave us a negligible change compared to having mucus clearance on, and our hypothesis is to this might be the case is that the spray dried amikacin sulphate actually dissolves very quickly and permeates through the lung, before the mucociliary clearance would have been able to have an effect.
It depends on the regulators that we work with, but in our experience at Nanopharm, we've had a lot of interaction with the US
FDA at various points over the past couple of years.
They have been interested in how simulation approaches might be used to form part of alternative bioequivalence approaches for orally inhaled and nasal drug products. The feedback we have gotten during our interactions has been very positive because they are really interested to see how this technology could be used to reduce the burden on clinical testing and complexity required to get drugs to market.
The FDA are currently working on the concept of a model master file which would allow technology providers like ourselves to keep validation data on file at the FDA for drug developers to reference. This has the potential to reduce duplication of effort and broaden access to modeling and simulation for drug submissions.
I think we're getting there, and over the next few years I expect to see more drug products approved using simulations like CFD and PBPK as a key part of the data package.

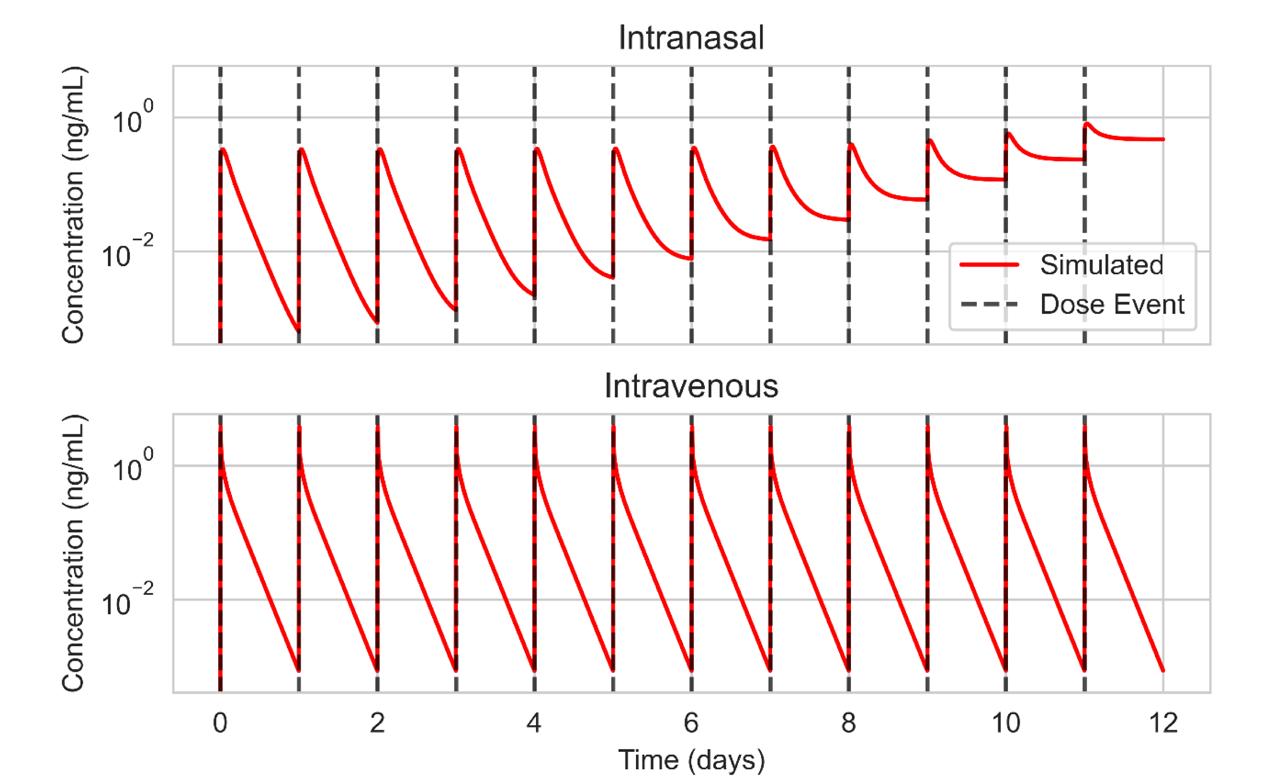
Repeated dosing can refer to multiple doses during a day, or multiple doses over a predetermined time. But with the PBPK model, we can inject doses at any time point during the simulation (Figure 5). If a drug is to be delivered once daily, we can simulate how it might build up in the body over time.
This is particularly useful when considering repurposing a drug that is taken via a different route – intramuscular injection or intravenous infusion, for example – and how that builds up in the body over time. Using the PBPK model, we can try to replicate this with our orally inhaled product before proceeding with clinical development.
How Do You Validate Your Model? Do You Have to Validate Every Molecule?
We need to make sure that the data we produce using these models is robust and valid. The US FDA are quite public about their expectations, and this is making the process of validation easier.
At Nanopharm, we perform validation typically through a risk-based framework called the ASME V&V 40, that has been provided to us by the US FDA. We identify how a model is going to be used and the credibility level is required, so that we can design a validation package based on that.
The validation framework is used to verify that your model solves the numerical problem to an appropriate degree of accuracy, and that your model outputs can be compared to clinical data. Currently we perform this to validate each individual molecule that we investigate.
Recently, the term ‘platform validation’ has been used a lot – this is where you take a simulation methodology, such as PBPK or CFD, and demonstrate that it is valid for typical use cases. This could be where the model works for varying solubilities and permeabilities,
and a combination of the two. Provided that we can show that the model predicts well for the extremes of these cases, we can claim validity for the cases in between too.
Could Simulation Replace Certain Clinical Studies for Orally Inhaled Products?
We can say that PBPK model structures for orally inhaled and nasal drug products are very well established. The main challenge in making them work is generating the required parameters and input data from your preclinical and formulation development work.
There are many different applications for PBPK models in the product development for OINDPs – spanning from early development where you're interested in identifying your critical parameters, all the way through to clinical validation where you're trying to either derisk or replace clinical studies.
Understanding what it is that you are using your model for means that you can establish what validation data are required and what the possibilities for using that model are. It is worth noting that most of the applications that we have seen fall somewhere in between those requiring that robust clinical validation, such as the granting of biowaivers, and those requiring only inputs literature studies, such as addressing low risk product development decisions.


Will Ganley, Senior Specialist at Nanopharm, an Aptar Pharma company, is a Physical Chemist with a PhD from the University of Bristol, UK. He started his career as a postdoc in Pharmaceutical Surface Science Lab at the University of Bath, UK. His focus was on advancing physical characterisation and simulation techniques for dry powder inhaler formulations, aiming to better understand the connection between physical attributes and delivery to patients. In 2019 Will joined Nanopharm as Head of Computational Pharmaceutics where he led the development of a number of statistical and mechanistic modelling methodologies, notably Nanopharm’s Simhalation PBPK platform. Will is now a senior member of the Science & Technology department at Nanopharm where he supports Nanopharm’s customers in product development and regulatory strategy and manages a portfolio of internal research and development projects aimed at advancing Nanopharm and Aptar Pharma’s scientific excellence in the use of advanced physical characterisation and digital technologies in inhaled and nasal drug product development. Will has authored a number of peer reviewed publications on pharmaceutics, statistics and physical chemistry, has presented his work at a range of international conferences and is a Scientific Advisor for the Drug Delivery to the Lungs conference.




Takeaway Message
MRX004 SMI delivers single breath doses 3 to 5 times higher than nebulisers.
We contend that nebulisers are serial SMIs and only achieve mg delivered doses because of repeat dosing.
In short, Keep Breathing! SMIs deliver more – Q.E.D.
For many years, nebulisers have been the preferred choice for respiratory drug developers needing an inhaler to deliver high doses to the lungs. This preference is largely due to nebulisers' ability to deliver a continuous stream of aerosol that can be inhaled via natural, tidal breathing over several minutes, thereby seemingly facilitating the delivery of high doses. However, a shift in perspective reveals a superior alternative: the soft mist inhaler (SMI), particularly the MRX004. SMIs deliver medication as a fine mist, which can be deeply inhaled in a single breath, offering greater efficiency and convenience (www.mrx004.com).
Delivering High Doses To The Lungs –Nebulisers Are A Legacy Technology
To understand why an alternative to nebulisers is desirable, it is important to recognise their limitations. Nebulisers are bulky devices that require an external electric power source to aerosolise a drug solution. The patient sits and inhales for several minutes, making the use of a nebuliser a time-consuming process. This is compounded by the high level of manual handling required through the assembly of the device, dispensing of the dose, and cleaning and maintenance of the nebuliser. Nebulisers are not portable inhalers, unlike pMDIs, DPIs and SMIs.
Nebulisers are comparatively wasteful. The need for electricity creates a demand for power that is unnecessary for purely mechanical devices like SMIs. In addition to spare parts requiring changing (metering chambers, tubing, masks), the aerosol is continuous, meaning that the patient is only breathing in half of the emitted dose. The other half is exhaled, wasting valuable
drug product and contaminating the air around the patients. Nebulisers rely on tidal breathing to deliver the drug to the lungs; this is characterised by shallow breathing with a breathing cycle of about 15 breaths (inhale/exhale cycle) per minute, each characterised by an inhalation/exhalation cycle, or 6 effective inhalations per minute. This translates into a 4 seconds breathing cycle and an inhalation duration of 2 seconds. A single dose from a nebuliser therefore takes 2 seconds to be delivered; bear that number in mind.
The efficiency of a nebuliser is characterised by the fine particle fraction delivered to the deep lung, i.e. the proportion of droplets below 5 mm. Typically this is about 30%, in some exceptional cases 50%, once the losses from exhalations are discounted.
Are Nebulisers Serial SMIs? – The Challenge Of Comparing SMIs And Nebulisers SMIs, such as Merxin Ltd's MRX004, deliver drugs to the lung by producing a slow-moving mist that patients inhale via a bolus or deep inhalation. Unlike nebulisers, SMIs deliver a discrete metered dose upon activation rather than a continual stream. In the case of MRX004, each dose is delivered in 1.5 seconds. No drug is wasted during exhalation, as an entire unit dose is delivered in a single, deep inhalation, rather than requiring the patient to sit and breathe with the device for an extended period as with a nebuliser.
The precise dosing capability of SMIs allows developers to accurately determine the amount of drug delivered with each activation. This precision has sometimes led to the perception of SMIs as a low-dose format. However, this is only the case if we limit the SMI to single doses. By administering multiple activations in sequence – similar to how nebulisers deliver medication over multiple continuous breaths – SMIs can effectively deliver high drug doses to the lungs. In this context, it is possible to see a nebuliser as a serial SMI, delivering drug over multiple continuous breaths where an SMI delivers doses one breath at a time.
What matters therefore is to compare the single dose delivered by an SMI with

the single dose delivered in one inhalation by a nebuliser.
Thinking in this way, a fair comparison of SMIs and nebulisers requires comparing them breath-for-breath, single dose for single dose, metered dose for metered dose. However, doing so from existing literature is difficult as there is a wide variety of performance characterisation across nebulisers, droplet size, formulation concentration, physical properties, and drug wastage during exhalation, as well as a paucity of single breath/dose measurements. To make the task of accurate comparison even more difficult, the in vitro performance testing requirements for nebulisers and SMIs in the EU and US Pharmacopoeias are not aligned; nebulisers are averaged over the therapy’s duration (a timescale of minutes), while SMIs are assessed on a single spray (a matter of seconds). Traditionally the output of nebulisers is given in mL/min, not corrected for fine particle fraction and breathing cycles, while the output of portable inhalers (pMDIs, DPIs and SMIs) is quoted in weight of the fine particle dose (droplet/particles below 5 μm) of API per metered dose ex-device delivered at a fixed flow rate.
Taking these limitations into account, the best way to make a direct comparison of a nebuliser with an SMI is to calculate the fine particle dose (FPD) of a single breath for each. For an SMI, this value can be calculated from a single activation and is already standardised. For nebulisers, a new calculation is required, and this is what we are going to use in this article.
Let us assume that the entire content of a nebule is delivered in a single sitting, so we can use the following formula to calculate the FPD per breath:
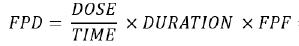
Here, TIME is the time taken for the entire dose to be delivered during nebulisation (usually of the order of minutes). DOSE is the dose contained in the nebuliser (typically 1 mg in a 1 mL nebule). The DOSE/TIME ratio is in fact the output of the nebuliser and is a physical limitation of the nebuliser. FPF is the fine particle fraction of the delivered dose (typically 30% for nebuliser due to
inefficiencies and tidal breathing that do not promote deep lung deposition). DURATION is the length of one breath, which as mentioned above is defined as 2 seconds by the US Pharmacopeia and ISO standards.2
Using this formula in conjunction with available literature, one can calculate a theoretical FPD per breath emitted from a nebuliser. Assuming a nebule concentration of 1 mg/mL and a nebule volume of 1 mL to normalise the calculations, the DURATION of the inhalation is fixed at 2 seconds as per the pharmacopoeia. One can calculate the FPD by varying the TIME and FPF. The theoretical range of nebuliser FPDs are listed in Table 1.
Assuming a 50% FPF and a TIME of 8 minutes, the FPD per breath of an average nebuliser is 1.67 μg.
Where CONCENTRATION is the API bulk concentration in the cartridge and MC is the volume of the metering chamber (15 µL).
MRX004 has a FPF of 70%. If we assume an API bulk concentration of 1 mg/mL, the unit dose per breath is 10.5 µg, or about 5 times the single breath FPD of a nebuliser (cf 1.67 μg).
Theoretical FPDs of MRX004 for a range of FPFs are listed in Table 3.
Recent publications at DDL 2023 on the FPD of MRX004 can be compared with theoretical values. The experimental FPD per inhalation of MRX004 SMI for Dornase Dornase Alfa (2.5 mg/ 2.5 mL) was 6.28 μg4 and Salbutamol 5 mg/mL was 35.1 μg, or 7 μg
inhaler MRX004 SMI is and that SMIs can achieve much higher doses than nebulisers.
We therefore contend that nebulisers are serial SMIs and only achieve mg delivered doses because of the repeat dosing. In short, Keep Breathing! SMIs deliver more.
More work is required to establish a direct experimental comparison between SMIs and nebulisers, but this initial review demonstrates that by viewing nebulisers as serial SMIs and comparing them on a breath-for-breath basis, most available nebulisers struggle to compete with SMIs. If patients just keep breathing and take multiple doses from an SMI, they can benefit from all the advantages of SMIs as a patientfriendly delivery device,1 along with excellent efficiency for delivering high doses to the lungs.
1. Aqrawe Z, Rogueda P, Murnane D, Eng B, “Soft Mist Inhalers Versus Nebulisers: Delivery Equivalence & Future Therapies”. ONdrugDelivery, Issue 158 (Apr 2024), pp 24–28.
2. US Pharmacopeia. (1601) Products for Nebulization – Characterization Tests.
Table 1: Theoretical FPD per inhalation range of nebulisers as a function of the required nebulisation TIME (fixed by the nebuliser hardware) and a range of FPFs. The nebule concentration is assumed to be 1 mg/mL in a 1 mL nebula.
Nebuliser
MicroBase µSMI 44.3 ± 1.7 0.26 ± 0.01
Aerogen Solo 47. ± 5 0.30 ± 0.04
Philips Innospire Go
PARI eRapid
± 1 0.21 ± 0.02
± 2 0.10 ± 0.01
Most nebulisers are not so efficient; assuming an FPF of 30% and 12 minutes required nebulisation time, the FPD becomes 0.83 μg per breath. This can be compared with the data from Table 2 with effective experimental FPDs for a small selection of commercial nebulisers. The experimental FPDs of these commercial nebulisers is much lower than the theoretical ones calculated in Table 1.
Let us now turn to the MRX004 SMI. The FPD per breath for an SMI is calculated thus:
when normalised to 1 mg/mL (assuming no effect on the aerosolisation process from the dilution). This is 3 to 4 times higher than nebuliser single breath FPDs.
This demonstrates that MRX004 SMI outperforms nebulisers on a single inhalation basis by delivering a unit dose up to 5 times higher. Add to this that the plume duration of MRX004 is 1.5 seconds but a nebuliser single breath is 2 seconds, and that SMIs deliver their payload to the deep lung when nebuliser aerosols deposit mostly in the upper respiratory track, it is then obvious what a superior
3. Hsiao S et al, “A Novel Contact-triggered Vibrating Mesh Nebulizer: Aerodynamic Performance and Drug Distribution of Suspension Drug Delivered with MicroBase μSMI”. RDD 2018, 2018, Vol 2, pp 459–462.
4. Antoniak D et al, “Inhaled biologics: from nebules to SMIs – Dornase alfa in MRX004”. Poster Presentation at DDL 2023.

Philippe Rogueda is the Chief Business officer and a co-founder of Merxin Ltd. Merxin Ltd makes inhalers. He is an inhaled formulation expert with a global reputation and an enviable track record of developing pMDIs, DPIs, Nebules, Nasal and soft mist inhalers for both generic and originator companies. Philipe trained in Physical Chemistry at the Universities of Bordeaux (France) and Bristol (U.K.), where he obtained his PhD. He subsequently worked for AstraZeneca, Novartis, Actavis before founding Inhalation Asia (Hong Kong) and Merxin Ltd (U.K.).
www.linkedin.com/in/philipperogueda
Medical device bright and intense colours are relevant for patients because they stimulate a positive attitude to take the drug medication, they help the distinction of different device part and facilitate the way how to use the device itself. In addition, they help to distinguish different types of drugs that are targeting to different types of diseases. Despite colour in medical device applications is such a key property, there are strict regulatory limitations in the type of pigments admitted in the colour formulations, and in their maximum allowed concentrations. When the biocompatibility requirement according to ISO 10993 must be met, not only the base ABS material must be biocompatible but also all the additives compounded with the material, including the colour formulation with all its different pigments. No biocompatible pigments must be directly excluded, and maximum allowed pigment concentrations must not be exceeded. Furthermore, special attention must be given to possible mutual interactions among different pigments, ABS material and other additives. ELIX Polymers eliminates those risks to OEMs, processors and developers providing a medical precoloured ABS material formulation that includes the complete colour recipe and does not need any further material modification. The complete formulation has been fully reviewed and pre-tested to meet biocompatibility according to ISO 10993 and other regulatory requirements. This is a much safer approach for medical OEMs and moulders instead of choosing a medical ABS in natural colour and compound it themselves with a masterbatch colour, during the injection moulding process.
The ABS market offers mostly natural ABS, forcing processors, OEMs and moulders to buy natural ABS and assume most responsibilities and risks, additional quality control costs and regulatory compliance verifications at different development and production stages.
As mentioned, in the case of pre-tested precoloured medical ABS, the material

formulation must not be modified by the customer, reducing responsibilities, supporting the medical device approval process, and avoiding the risk of compounding mistakes during the injection moulding production process. All required medical compliance certifications are already provided by ELIX and are referring to the complete material formulation, including all included colour pigments, additives, and related concentrations.
Regulatory compliance is a pre-requisite for inhalation medical device, but there are also other important quality properties that are strictly related with colour: its homogeneity along the complete device part, its consistency from lot to lot productions, or required colour target contrast in case of surface laser marking (typically for traceability reasons to comply with UDI EU MDR and US CFR regulations). In all these cases colour deviations are not admitted, and a pre-coloured ABS can bring again relevant advantages when compared with natural material post coloured (with masterbatch during the injection moulding process).
Pre-coloured ABS is obtained during a compounding extrusion process, mixing the ABS intermediate materials directly with colour pigments in powder form. Three important elements to consider come into
play at this point to optimise dispersion homogeneity in the material compound: the type of technology used (extrusion compounding), the fact that the colour pigments are in powder form, and the mixing step that happens when the base ABS material is not already a compound but still a “set of ingredients” made of different ABS intermediates raw materials, like for example the ABS rubber phase, which also comes in powder form, and the ABS matrix phase (SAN). This combination of factors is not possible in the case of an injection moulding process, as injection moulding machines are not specifically designed to mix optimally different ingredients together but to melt, feed and inject specific types of materials into a mould in a reasonable cycle time. On the other hand, compounding extrusion machines can handle powder recipes and have a specific double screw design that optimises dispersion homogeneity. Twin screw extruders have the right length, L/D relation and helical elements shape to provide adequate compound mixing and better interaction of all ABS intermediate raw materials, but also better interaction with pigments and additives employed. In this way pre-coloured ABS offers better colour pigment dispersion and homogeneous distribution, which is consistent from lot to lot, and also laser marking enhancers benefit of this optimal compounding ability.

In the case of natural ABS post coloured during the injection process, there is also an additional product needed which does not exist in the situation of pre-coloured ABS: a colour masterbatch. This includes a carrier (an additional material to the ones mentioned until now) and a concentration of colour pigments. The carrier is needed to incapsulate the colour pigments and help the pigment distribution with the natural ABS during the injection moulding process. Due to the mentioned compounding limitations during the injection moulding process, the targets of colour dispersion and lot to lot consistence are more difficult to achieve compared to pre-coloured ABS. Masterbatch carrier compatibility with base material and other additives must be assured, and production personnel needs additional training and competences for colouring with masterbatch and managing possible unexpected situations (e.g. colour differences in different injection cycles, production stop, colour trouble shooting, specific interactions between ABS with

MB carrier or colour formulation). Even when colour targets may be achieved with a masterbatch, there is still the doubt of biocompatibility compliance of the final compound ABS + Masterbatch for the easy reason that no biocompatibility test according ISO 10993 will be conducted on the resulting compound. Such test must be conducted on the final devices for medical approval but not before. Instead, in the case of ELIX pre-coloured medical ABS, the biocompatibility tests are already conducted and passed on the complete compound ABS + colour formulation + additives.
When it comes to sustainable design for medical devices, colour quality turns into an even more critical and sensitive property that needs to be preserved. The demand for new sustainable ABS materials for drug delivery devices applications is growing in the healthcare sector. Due to the risk of cross contamination, medical regulatory compliance cannot be fully fulfilled with mechanically recycled ABS materials. On the other hand, the new scenarios of chemically recycled and bio-based ABS materials are already available and offer the same exact chemical composition and properties of virgin medical ABS, fulfilling the same drug delivery applications along with medical regulations requirements. All the colours that are available in the virgin medical ABS version can be also used in the bio-circular version, guaranteeing not only regulatory compliance, but also the availability of bright and intense colours in chemically recycled ABS formulations. These types of
colours cannot be achieved in any case with mechanical recycled content.
ELIX vision is to be a driving force of the new plastics economy in the next years, participating in the redefinition of plastic waste as raw material. The mission is offering top-of-the-line sustainable solutions in our markets, promoting the transformation of the value chain towards a circular economy model.
The company was the first ABS manufacturer to get the International Sustainability and Carbon Certification (ISCC+ certification) for sustainable materials. The certified raw materials content of ELIX M203FC and M205FC medical grades can be adapted according to the customer OEMs’ sustainability targets.
ELIX medical ABS formulations with chemically recycled and/or bio-based content have been approved by the FDA for the inclusion in the same Drug Master Files (DMF) of standard virgin ELIX medical ABS formulations M203FC and M205FC. This will support an easier transition towards the use of more sustainable ABS medical materials in drug delivery devices in the coming years.

Graduated in management engineering at the "Politecnico di Milano" University (Milan, Italy), Luca has 20 years’ experience in the fields of plastics, composites and OEMs devices. Luca joined ELIX Polymers in 2017 in the position of Business Development Manager for the healthcare strategic sector. Since 2020 he is actively involved in the development of ELIX E-LOOP sustainable solutions and circular innovations, that include a new growing sustainable ABS and blends material portfolio, with chemically recycled, bioattributed, bio-based and mechanically recycled content. Luca wrote several technical articles on behalf of ELIX about specialties and sustainable ABS for medical applications that were published on several renowned medical and pharmaceutical magazines. He lived in different European countries and speaks fluently 6 languages (Italian, English, German, Spanish, French and Catalan).
Switching from current pressurised metered dose inhaler (pMDI) propellants to new propellants with lower global warming potential (GWP) could help to reduce the carbon footprint of pMDIdelivered medicines significantly. When looking at the reformulation of an existing product, there is also a need to review the analytical methodology and overall chemistry, manufacturing and controls approach.
With changes being made to the formulation, a suitable validation gap analysis of methods will be required as these methods will underpin the generation of critical data supporting product characterisation, stability and in vitro bioequivalence work. This presents opportunities to address key analytical issues associated with contaminants such as nitrosamines or leachables which represent significant risks to patient safety.
Nitrosamine Formation in pMDIs
Since the EMA and FDA introduced guidelines in 2020 on controlling nitrosamine impurities in medicinal products, industry stakeholders have undertaken extensive risk assessments and testing. These efforts are time-consuming and costly, requiring the development of sensitive detection methods and comprehensive root cause analysis, potentially leading to significant reformulation to comply with new intake limits.
The transition to low GWP propellants in pMDIs provides a chance to proactively address nitrosamine formation early in formulation development. Utilising advanced analytical techniques, such as Liquid Chromatography Mass Spectrometry (LC-MS) and Gas Chromatography Mass Spectrometry (GC-MS) can help streamline efforts to meet regulatory requirements for nitrosamine levels. Tandem LC-MS/MS provides high sensitivity and specificity for detecting APIspecific nitrosamines, even in complex pMDI matrices. GC-MS is effective for analysing volatile, non-API-specific nitrosamines and potential degradation products from
propellants. Utilising both LC-MS and GCMS in early trials enables comprehensive screening, identifies nitrosamine sources and ensures regulatory compliance.
Nitrosamines primarily form through reactions between secondary or tertiary amines and nitrosating agents, like nitrous oxides or nitrites, under acidic conditions. In pMDIs, nitrosamine formation can result from interactions among the propellant system, active pharmaceutical ingredients (APIs), excipients, and device materials. The introduction of low GWP propellants to pMDIs necessitates thorough analytical evaluation to minimise the risk of nitrosamine formation.
Next generation low GWP propellants, such as HFO-1234ze and HFA-152a, differ chemically from traditional propellants. As unsaturated compounds, HFOs can potentially react with other formulation components or degrade over time, forming reactive species that may contribute to nitrosamine formation. The interactions between these new propellants and other formulation components, especially in the presence of moisture, heat, and light, are not yet fully understood.
Each component of the pMDI formulation, including APIs, excipients, and device materials, must be evaluated for its potential to contribute to nitrosamine formation. Therefore, reformulation efforts should include rigorous testing with robust and validated methods at all stages of development, to assess any increased risk when using these new propellants.
The conditions under which pMDIs are stored and used (temperature, humidity, and exposure to light) can significantly influence nitrosamine formation and should be carefully controlled. Accelerated stability studies can help predict how formulations might behave over time and under different environmental conditions, providing essential data for formulation decisions.
The new low- GWP propellant systems could mean changes in recommended long
term storage conditions or packaging to provide the re-formulated product with the necessary stability to remain a viable commercial product.
Reformulating pMDIs with low GWP propellants allows addressing nitrosamine concerns early in the drug development process (something that wasn’t considered when many of the current marketed pMDI products were being developed in the 1990s and 2000s). However, analytical strategies must align with regulatory guidelines and include robust risk assessments that consider all potential sources of nitrosamine contamination, including the supply chain, raw materials, and manufacturing processes.
The different chemical and physical properties of the new low GWP propellants will mean that adjustments to existing formulations, different drug and excipient combinations and inhaler designs will be necessary to account for the different characteristics of the low GWPs.
Due to the new production processes of the low GWP propellants, a unique impurity profile is to be expected. These impurities must be tested for their concentration and toxicity before the new propellants are used in clinical or commercial products.
The physical and structural properties such as density, vapor pressure, polarity and viscosity (Figures 1–3 and Table 1)1 undoubtedly affect the extraction behaviour and interaction with contact materials or storage containers, resulting in different profiles of extractables and leachables after long-term storage in pMDIs.

In a recent publication from Faucard et al., three fluorinated propellants (HFO-1234ze, HFA-152a and HFA-134a) have been compared regarding their leaching behaviour.


In the study by Faucard et al, a typical pMDI valve, consisting of the following materials: PBT (polybutylene terephthalate), EPDM (ethylene propylene diene monomer) and COC (cyclic olefin copolymer), was exposed to these propellants and tested after intervals of T=0, T=1, T=3 and T=6 months’ storage time with different analytical screening techniques.2 The leaching of target compounds was investigated with multiple analytical techniques, such as GC-FID and LC-UV.
The PBT dimer and trimer was investigated as a representative compound for a nonvolatile leachable. HFA-152a showed the largest increase in leaching of the dimer after 6 months, and the leaching of the trimer was also higher compared to the other blowing agents, but generally at a much lower level.
As representatives for the semi-volatile leachables, the “antioxidants” and “other semivolatile leachables” have been investigated and reported as individual compound-groups. The extracted concentration of each group was investigated per time point. THF was chosen as representative example of a highly volatile leachable.
The total concentration of leached species was below the toxicological limits, but there were differences reported in the leaching behaviour between the individual propellants.
Both the extractable concentration of the individual substance groups and the speed of extraction appear to depend strongly on the structures and physical properties of the gas molecules. If we look at the properties in Table 1, the different structure is also associated with differences in the physical characteristics.
HFA 152a shows a higher extractability of the non-volatiles and THF as highly volatile. While for the antioxidants and other volatiles, the HFA 152a and HFO 1234ze showed a slightly stronger and partially faster extraction behaviour which indicates that more parameters than just the volatility of the leachable compounds play a role for the leaching behaviour. Unfortunately, only sum parameters or specific target compounds were investigated in this study, which prevents a correct correlation of the leaching behaviour with different molecular structures of the leachable compounds. There could be an increased selectivity of the new propellants, which leads to a pronounced leaching of compounds with special structures.
In a pre-study performed by Intertek in Switzerland, a rubber material component used as an inhaler valve gasket was analysed by Thermodesorption Gas Chromatography – Mass Spectrometry (TDS-GC/-MS). TDSGC/-MS is a powerful technique often used for extractables profiling.3 As the thermal desorption approach does not involve a solvent and trapping of compounds, it covers multiple compound classes which could be thermally desorbed including volatiles and many semi-volatile species.
Due to the trapping of desorbed compounds, even species with very low concentration can be detected. Table 2 shows a list of the compounds detected and identified in the rubber seal. Many different compounds from various substance classes were detected, such as hexane, a residual solvent, butylated hydroxytoluene, a stabiliser, partially halogenated rubber oligomers, a by-product of rubber production, and diethyl phthalate, a plasticiser. It is obvious that the rubber gasket contains many small molecules of varying structure and polarity, all of which could be selectively leached out by formulations including the new low GWPs.
As already shown in the previous systematic studies, the extraction behaviour indeed depends on the physical properties and structures of the new low GWPs.2 Since pMDI systems, and especially the valves, are typically composed of many different materials, all with unique additive sets and other low molecular weight impurities, there is some risk of leaching of new compounds or higher levels from the traditional materials used in pMDIs if low GWPs are introduced.
The unique extraction selectivity of the new fluorinated propellants must be investigated in more detail to understand the differences and to rule out a toxicity issue for dedicated compounds. In the so far published literature, only a few targets or sum parameters have been investigated.2 The next step will be a dedicated generic
Adapted from Buttini, F., Glieca, S., Sonvico, F., & Lewis, D. A. (2023). Expert Opinion on Drug Delivery, 20(8), 1131–1143. https://doi.org/10.1080/17425247 .2023.22641841
n-Hexane
Hexanal
1-Bromohexane
2,6-Dimethyloctane
2,2,4,6,6-Pentamethylheptane
2-Octanone
Octanal
Limonene
p-(1-Propenyl)-toluene
Nonanal
1,3,3-Trimethyl-2-(2-methylcyclopropyl)-1cyclohexene
4a-Methyldecahydro-2H-benzo[a]cyclohepten2-one
Rubber oligomer with sum formula C13H24
Decanal
1,2-Dibutylcyclopentane
Undecanal
Rubber oligomer with sum formula C14H26
Brominated Rubber oligomer
2,6-Di-tert-butyl-4-hydroxy-4methylcyclohexa-2,5-dien-1-one
Rubber oligomer with sum formula C14H28
2,6-Di-tert-butylbenzoquinone
Rubber oligomer with sum formula C13H23Br
Butylated Hydroxytoluene
Diethyl Phthalate
Rubber oligomer with sum formula C13H23Br
2-Bromo-4,6-di-tert-butylphenol
3,5-di-tert-Butyl-4-hydroxybenzaldehyde
Rubber oligomer with sum formula C25H48
Rubber oligomer with sum formula C21H39Br
Dibutyl phthalate
Rubber oligomer with sum formula C21H39Br
1-Hexadecanol
Bromo alkane
Rubber oligomer with sum formula C25H48
Table 2: Compounds in a bromobutyl rubber material detected and identified by TDS-GC/MS, sorted according to increasing retention time
extractables study (performed by Intertek in Switzerland) which compares the extraction behaviour of traditional propellants (including also HFA 227) against the new low GWP candidates in further detail. In particular, the effects on a wide range of extractable compounds with different
structures and polarities should and will be investigated in order to assess the risk of pronounced leaching for certain compounds and the potential toxicity problem associated with this.
According to USP <1663>, extractables screening of individual construction materials is helpful to assess complex container closure systems, such as pMDI products.4 Techniques such as thermal desorption are cited as the method of choice to detect a wide range of compounds with little effort, as direct analysis is possible without prior extraction.
This material-specific extractables screening study is important for tracing the extractables detected after incubation of the combined pMDI device. Quantitative screening techniques, such as GC-MS and LCMS, need to be optimised to characterise the extract and capture a wide range of different compounds.
As the extractables content in most plastics is typically very low, even a small variation in the additive, as would be expected in different batches of material, could distort the results and make it difficult to adequately compare the different propellants.
For the planned exemplary comparative study on leachables, it is important to compare the same type of inhaler, ideally from the same batches of material incubated with the different types of propellants.
The pure propellant gas should be used as the incubation medium to also exclude variations in the drug formulation. Alternatively, a suitable control sample or blank sample of the formulation must be stored in an inert container under the same conditions and analysed using the same techniques to exclude effects not caused by the inhalation system.

After the quantitative evaluation of the results, the differences in extraction behaviour and extraction selectivity of the new low GWPs compared to the traditionally used propellants will be investigated peak by peak to prove whether there are major differences for specific compounds, compound classes or whether the extraction behaviour is similar for a wider range of compounds and has a similar selectivity to the old solvents.
After the results of this more substancespecific extractables study are known, a general indication could be given if big differences in extraction behaviour should be expected for the new low GWPs. As a “best case”, there is only a small difference over all classes of extractables and no selective effect in specific plastic ingredients. In such a case, the traditional construction materials could be further used without concern.
The transition to next-generation low GWP propellants in pMDIs is a crucial step toward reducing the environmental impact of respiratory treatments. However, this introduces new analytical challenges in ensuring product safety, particularly concerning nitrosamine formation and leachables. A comprehensive analytical approach is necessary to understand and mitigate these risks. By optimising formulation components and employing advanced analytical techniques, the pharmaceutical industry can successfully reformulate pMDIs with low GWP propellants while ensuring
patient safety and compliance with regulatory standards.
1. Buttini F, Glieca S, Sonvico F, Lewis DA. Metered dose inhalers in the transition to low GWP propellants: what we know and what is missing to make it happen. Expert Opin Drug Deliv. 2023 Jul-Dec;20(8):1131-1143. doi: 10.1080/17425247.2023.2264184. Epub 2023 Oct 16. PMID: 37767756.
2. Faucard P, Fontaine I, Rives S, Le Corre B, Cannette C, Ferrao J. Leachables Assessment from a New Generation of pMDI Using Low Global Warming Potential Propellants.

Dr. Tino Otte, Managing Director at Intertek Switzerland, is an expert for extractables and leachables studies. He holds a degree in polymer chemistry from the University of Halle/Saale and a Ph.D. from the Darmstadt Technical University. Prior to joining Intertek, he worked with different research, development and manufacturing companies where he served in several functions in product management and development of analytical services. He has over 20 years of experience in GMP regulated environment within multiple areas of product analysis, including method development, validation, and QC.
Respiratory Drug Delivery 2023: 163-168.
3. Scherer, Nicole Marion Doris (2019): Leachable and extractable studies on single-use system technologies in commercial scale drug filling lines. Dissertation, LMU München: Faculty of Chemistry and Pharmacy. 10.5282/edoc.24707
4. United States Pharmacopeia (2024). General Chapter, (1663) Assessment of Extractables Associated with Pharmaceutical Packaging/ Delivery Systems. USP-NF. Rockville, MD: United States Pharmacopeia. DOI: https://doi. org/10.31003/USPNF_M7126_03_01

John McLaughlin is a Principal Scientist at Intertek Melbourn, where he specialises in LC-MS analysis for the Biologics team. He manages LC-MS work, covering a wide range of samples with expertise in small molecule impurities in pharmaceuticals and large molecule proteins and oligonucleotides in inhaled formulation products. His focus is on method development and validation in GxP regulated environments. John earned his master's degree in Chemistry with Medicinal Chemistry from the University of Warwick in 2018. Prior to joining Intertek, he has worked in various research organisations, focusing on bioanalysis of small and large molecules.

Chris Vernall is the Commercial Director at Intertek Pharmaceutical Services, leading the business development team at the inhaled and nasal drug product center near Cambridge, UK. With a master's degree in Medicinal and Pharmaceutical Chemistry from Loughborough University, he started his career as an Inhalation Scientist, specialising in formulation development and analytical testing, before moving into commercial roles. Passionate about advancing respiratory drug product development, Chris fosters partnerships and collaborations across the industry. He advises companies on technical and commercial strategies and chairs the Inhaled and Nasal Biologics and DNA Forum, an annual conference in Cambridge, now in its third year.
Nasal sprays have become an important route of delivery for a variety of indications and as their use becomes more widespread, regulatory bodies across the globe have issued various guidelines and recommendations to ensure their safety and efficacy. However, the landscape of these regulations, particularly concerning preservatives, remains complex, leading to confusion and inconsistency of application. This article delves into the current guidance on the use of preservatives in nasal sprays, explores the market trends, and discusses the manufacturing requirements essential for producing safe and effective nasal spray products.
In Europe, the regulatory landscape for nasal sprays is influenced significantly by the recommendations of the European Medicines Agency (EMA) and the German Federal Institute for Drugs and Medical Devices (BfArM). Both agencies have recommended the removal of one preservative – benzalkonium chloride (BAK or BAC) – in nasal sprays, citing potential long-term adverse effects on patients. However, it is crucial to note that these are recommendations rather than mandates. The EMA has also updated its requirements for patient labeling on formulations containing BAK, reflecting a growing awareness of preservative-related concerns.1,2
Despite these recommendations, there has been no definitive guidance mandating the development of preservative-free nasal formulations. This regulatory ambiguity has led to a perception that the trend towards preservative-free formulations is more of a regulatory compulsion, rather than an industry-driven tendency.
Similarly, the BfArM has echoed the EMA's stance on BAK, recommending its removal but stopping short of enforcing a mandate. This cautious approach highlights the ongoing debate within regulatory circles about the necessity and feasibility of eliminating preservatives from nasal formulations.
Across the Atlantic, the U.S. Food and Drug Administration (FDA) has provided more detailed guidance on various aspects of nasal spray development. According to the latest FDA guidelines, numerous factors need to be considered during the development program of nasal sprays. These include formulation, container closure systems, manufacturing processes, stability, and controls of critical steps and intermediates. The FDA emphasises that any changes in these aspects can significantly impact the product's ability to deliver reproducible doses to patients throughout its shelf life but stops short of recommending or issuing guidance on removing preservatives in formulations.3,4
The FDA acknowledges that if a formulation is preservative-free, it requires the use of a preservative-free device designed to prevent microbial ingress. This can be achieved using .22 sterile filters and other closure mechanisms in the nasal devices.5
The global regulatory landscape for nasal sprays reflects a cautious and evolving approach towards preservative-free formulations. Although there is a tendency towards such formulations, there is still no clear mandate from regulatory authorities. This ongoing debate underscores the need for continuous review and adaptation of manufacturing practices to meet both regulatory expectations and market demands.
Until recently, nasal sprays have been primarily used for treating allergies, rhinitis and sinusitis, with products ranging from over the counter (OTC) products to prescription (Rx) decongestants, antihistamine and antiinflammatory products. The largest class of commercially available medicines are the nasal corticosteroids, or nasal steroid sprays, with drugs such as fluticasone, mometasone, and triamcinolone.
Since the late 1990s, intranasal drug delivery for systemic indications, especially for rescue therapies, have overshadowed all commercial approvals. This is due to a rapid onset of action of the drug, ease of administration and convenience as opposed to injections, oral delivery or other routes of administration. Indications range from
treatment of opioid overdose, migraine, seizure, anaphylaxis and a variety of other indications. Many of today’s leading Rx nasal sprays, some of which are now OTC, utilise preservatives like benzalkonium chloride and sodium phosphate, to ensure their stability and efficacy over time (Table 1).
In the United States, there are only five products that are preservative-free: Migranal®, Noctiva®, Nayzilam®, Rivive® and Zavpret®. Of these five only one appears to be manufactured in sterile conditions –Noctiva. Specifically, the approved labeling states “nasal sprays are not typically required to be sterile. Noctiva is manufactured under aseptic conditions but becomes non-sterile once in use. The device adequately prevents ingress of bacteria, which is important because the product does not contain a preservative and bacterial contamination could degrade desmopressin”.7 The other products specifically state they are “non-sterile” and/or submitted to the appropriate microbial growth controls during the manufacturing process.8,9,10,11
While it may appear that there is a shift towards preservative-free nasal sprays, it is evident that this trend is more driven by industry innovation and consumer preference rather than stringent regulatory requirements. The perception of a strong movement towards preservative-free formulations needs to be balanced with the reality of regulatory guidance and market reality.
Manufacturing nasal sprays involves adhering to several key requirements to ensure product safety, efficacy, and quality. The FDA's guidance on nasal spray manufacturing emphasises the critical need to ensure that the product can deliver consistent and reliable doses with the highest quality throughout its shelf life. This involves rigorous attention to formulation, container closure systems, manufacturing processes, stability, and process controls at every stage of production. Each of these aspects must be tailored and maintained to the specific product requirements to sustain its efficacy and safety.
Developing preservative-free nasal sprays introduces additional complexities, which
Reference Drug Molecule Indication Preservative Year Approved
Flonase® and generics fluticasone propionate perennial allergic or nonallergic rhinitis benzalkonium chloride 1994 OTC in 2003
Nasarel® flunisolide seasonal or perennial rhinitis benzalkonium chloride 1995 (discontinued)
Afrin® and generics oxymetazoline hydrochloride relief of nasal congestion
chloride 1966 OTC in 1975
Nasacort® and generics triamcinolone seasonal and perennial allergic rhinitis benzalkonium chloride 1996 OTC in 2013
Nasonex® and generics acetonide seasonal allergic or perennial rhinitis benzalkonium chloride 1997 OTC in 2022
Migranal® and generics mometasone furoate acute treatment of migraine none 1997
Imitrex® and generics sumatriptan acute treatment of migraine Sodium phosphate 1997
Rhinocort® and generics budesonide seasonal or perennial allergic rhinitis
Zomig® and generics zolmitriptan acute treatment of migraine
sorbate 1999 OTC in 2015
phosphate 2003
Dymista® and generics azelastine hydrochloride and fluticasone propionate seasonal allergic rhinitis benzalkonium
Narcan® and generics naloxone opioid overdose
Xhance® fluticasone propionate chronic rhinosinusitis without nasal polyps.
Noctiva® desmopressin nocturia due to nocturnal polyuria
Tosmyra® sumatriptan acute treatment of migraine
Spravato® esketamine hydrochloride treatment resistant depression and depressive symptoms in adults with MDD with acute suicidal ideation or behavior
midazolam
diazepam
2015
nalmefene
Neffy® epinephrine emergency treatment of allergies/ Anaphylaxis
Table
require adherence to stringent guidelines to ensure patient safety. These formulations have a unique set of requirements that pose significant challenges. These challenges include the following: advanced formulations techniques, sterile environmental handling, processing, controls, and the use of delivery devices capable of maintaining sterility. Additionally, these formulations require thorough risk assessments and validation studies to demonstrate that the product can maintain sterility and efficacy throughout its shelf life.
Nasal sprays can be confidently manufactured utilising a preserved formulation or a preservative free formulation with a sensible approach to risk. The following techniques minimise bioburden to ensure one can produce a product of the highest quality.
• Aseptic Techniques: Mimicking conditions before and during the transfer of formulations into nasal delivery devices is critical. This includes the use of single-use disposable vessels and tubing during compounding and filling processes.
• Low Bioburden Grade C Environment: Manufacturing in a controlled Grade C environment helps minimise microbial and particulate contamination. This baseline allows for limited to no environmental impact to the formulation.
• Dedicated Single-Use Product Contact Pieces: Using dedicated single-use contact pieces wherever possible further reduces contamination risks.
• Closed System and Sterilisation Filtration:
The manufactured solution poststerilisation filtration remains within a sanitised system (hold vessel, single use tubing, filling needles) until moment of fill. Ultimately, limiting exposure to the open Grade C environment to seconds prior to stoppering/capping. between filling and stoppering/capping of finished product.
• Personnel Practices: Personnel must adhere to strict aseptic techniques, including the use of personal protective equipment (PPE) and rigorous cleaning practices.
• Terminal Sterilisation: Techniques such as radiation can
be used to inactivate any potential microbial contamination post-filling.
Drug development companies need to evaluate how the chosen production pathway of their drug candidate and the related costs of such will impact its commercialisation success. For example, the cost implications of manufacturing nasal sprays, particularly preservative-free formulations in sterile conditions, are substantially higher than those of preserved formulations and will lead to a higher cost of goods (COGS) and can directly impact the profitability of the product for commercialisation. Sterile manufacturing of nasal products is also not as widely available, thus limiting third-party manufacturing options. Low bioburden manufacturing, as before, is generally less expensive than sterile manufacturing, offering a cost-effective alternative while still ensuring high levels of safety, quality, and efficacy.
Adopting low bioburden manufacturing techniques can provide several benefits for drug companies. It allows them to meet regulatory and market demands for preservative-free products without incurring the higher costs associated with sterile manufacturing. Also, it supports producing high-quality nasal sprays safe for acute and chronic use by patients.
In the evolving landscape of nasal spray manufacturing, regulatory recommendations and market trends are shaping the development of preservative-free formulations. While the debate on preserved or preservative-free continues, the industry must assess what is best for the drug candidate considering current recommendations and consumer preferences. As a contract development and manufacturing organisation (CDMO), our role is to support innovators in navigating these complexities, offering expertise in both traditional and preservative-free nasal spray manufacturing. By leveraging advanced aseptic techniques and ensuring low bioburden levels, CDMOs help clients achieve their formulation goals while maintaining the highest standards of safety and efficacy.
The future of nasal sprays will see continued innovation and adaptation as regulatory bodies refine their guidelines and the market evolves. In the meantime, a comprehensive understanding of current requirements and best practices remains

essential for successful product development and patient care.
1. Riechelmann H et al, “Nasal toxicity of benzalkonium chloride”. Am J Rhinol, 2004, Vol 18(5), pp 291–299.
2. European Medicines Agency. Questions and answers on benzalkonium chloride used as an excipient in medicinal products for human use. Accessed online March 14, 2023 at: ema. europa.eu/en/documents/scientific-guideline/ questions-answers-benzalkonium-chlorideused-excipient-medicinal-products-humanuse_en.pdf
3. Guidance for Industry Nasal Spray and Inhalation Solution, Suspension, and Spray Drug Products – Chemistry, Manufacturing, and Controls Documentation. Center for Drug Evaluation and Research (CDER) July 2002.
4. Pharmacopeial Convention; 2019. Accessed online March 14, 2023: https://www.usp.org/ compounding/general-chapter-797
5. Ehrick JD, Shah SA, Shaw C, Kulkarni VS, Coowanitwong I, De S, Suman JD. Considerations for the Development of Nasal Dosage Forms. Sterile Product Development. 2013 Jun 22;6:99–144. doi: 10.1007/978-1-4614-7978-9_5. PMCID: PMC7120012. article
6. The above information was compiled using the information from each respective product on the Drugs@FDA database: Drugs@FDA: U.S. Food & Drug Administration: Drugs@FDA [Internet]. Silver Spring, Maryland. 1938-present [accessed August 7, 2024]. Available at: www.fda.gov/drugsatfda
7. Acerus Pharmaceuticals. Noctiva (desmopressin acetate) [package insert]. U.S. Food and Drug Administration website. https://www.accessdata. fda.gov/drugsatfda_docs/labe/2027/201656lbl. pdf. Revised March 20, 1027. Accessed August 07, 2024.
8. Center for Evaluation of Drugs and Research. Product Quality Review, Application Number: 211321 [Memorandum, May 14, 2019]. https:// www.accessdata.fda.gov/drugsatfda_docs/ nda/2019/211321Orig1s000ChemR.pdf.
9. Center for Evaluation of Drugs and Research. Chemistry Review, Application Number: 20148 [Memorandum, October 6, 1992]. https:// www.accessdata.fda.gov/drugsatfda_docs/ NDA/97/20148_Migranal_chemr_EA.pdf
10. Center for Evaluation of Drugs and Research. Product Quality Review, Application Number: 217722 [Memorandum, June 13, 2023]. https:// www.accessdata.fda.gov/drugsatfda_docs/ nda/2023/217722Orig1s000ChemR.pdf
11. Center for Evaluation of Drugs and Research. Product Quality Review, Application Number: 216386 [Memorandum, May 12, 2022]. https:// www.accessdata.fda.gov/drugsatfda_docs/ nda/2023/217722Orig1s000ChemR.pdf

Carolyn Berg has over 25 years of experience in pharmaceutical sales, marketing and business development. She is currently Vice President, Business Development for Catalent's inhaled drug delivery solutions where she is responsible for growing the Inhalation business in North America and Europe. Carolyn holds a Bachelor of Journalism in Public Relations and a Bachelor of Arts in French from the University of Texas at Austin, and an MBA from the University of South Carolina.

David Wilcox, Director of Inhalation Product Development, has over 25 years of pharmaceutical industry experience, with substantial expertise and knowledge in formulation, development, and manufacturing of orally inhaled and nasal drug products. In his role, he is responsible for providing operational, scientific, and technical leadership for inhalation product development activities. David holds a Bachelor of Science degree in Chemistry from Wofford College in Spartanburg, South Carolina.

Mark Ignaczak, Director of Innovation and Partnerships for Nasal Delivery at Catalent, has over 18 years of pharmaceutical experience and over 15 years of direct experience with nasal spray products. He has worked on more than 75 nasal programs at varying stages of the project lifecycle and held various roles within bioprocessing engineering, product development, supply chain and nasal delivery program management and strategy. He holds a Bachelor's degree in biochemical engineering from Rutgers University, New Brunswick, US.


spray drying
encapsulation & blistering
Catalent is your preferred CDMO for pulmonary and nasal delivery, with clinical to commercial-scale development and manufacturing capabilities for dry powder inhalers, unit-dose, bi-dose and multi-dose nasal sprays.
With over 30 years of inhalation product experience and state-of-theart facilities in Boston, Massachusetts and Morrisville, North Carolina, Catalent can handle the most complex projects and help bring your inhaled therapy to market, faster.
characterization
development & manufacture of spray dried & carrierbased powders
spray drying expertise for small & large molecules
scale up from lab to psd-1, psd-4 & psd-7
nasal liquid spray & powder
unit-dose, bi-dose & multidose filling & assembly
The pulmonary route is gaining increasing attraction not only for low-dose locally acting therapies, e.g. Asthma and COPD, but also for systemic applications often require higher doses or new formulation technologies.
High-performance devices need to be developed to accommodate the requirements to deliver these new drugs/ formulations efficiently. To ensure the best performance, the development of the formulation and the device should go hand in hand.
Dry Powder inhaler devices are used to deliver the medication in powder form to the lungs via oral inhalation. Medication in powder form can be filled in either a capsule, blister, reservoir or cartridge based on the drug product configuration. The device can be either an active or a passive device. In many cases, capsule-based inhalers are preferred solutions for new applications because they offer several interesting features, e.g. possibility to use a wide range of different APIs and doses generally ranging from 5 mg to 50 mg. Further advantages include ease of use and a good feedback mechanism for the patient.
Due to the complexity and cost of manufacturing multidose DPI devices, pharma companies want to minimise cost and risks in the new applications by using a simpler and more affordable device.
For some new applications, e.g. pain relievers or antibiotics, a reusable dose is beneficial; for others, e.g. vaccines, a disposable single-use is required. In both cases, capsule-based inhalers can be a good solution.
Covid 19 has made it imperative that an easy-to-use vaccine is available worldwide and remains stable at ambient conditions. Research continues to happen to deliver the vaccine by the inhalation route.1
However, there are some drawbacks associated with current capsule-based DPI devices, e.g. Low performance is an inherent
feature of many capsule-based DPI devices on the market. Even newly developed integrated solutions for antibiotics, e. g, Tobramycin, do not reach a higher FPF than 35%. In order to achieve the therapeutic dose of the antibiotic, the patient has to inhale 4 capsules a day.2
Many of the strategies for overcoming the inherent challenges of most existing devices are done by focusing on improved dispersion properties of the formulations. Not a lot of attention was paid to improving the devices either by technological advancements of the device performance (higher deagglomeration and less retention of powder in the device/ capsule) or on increasing patient adherence for better handling and thus, higher deposition in the deep lungs.3
The novel Presspart DPI device addresses the above problems and challenges of capsule-based inhalers. The most important features of an ideal capsule-based DPI device are, the ease of use with a good feedback mechanism to the patient coupled with a cost-effective design. In addition, dose delivery with high reliability and consistency, and high-performance efficiencies for a wide range of applications are desired.
While developing the novel capsule-based inhaler, the main focus was to create a high-efficiency engine to de-aggregate and aerosolise the powder formulation: a medium resistance and relatively flowrateindependent device. Several prototypes have been tested and studied throughout the development journey.
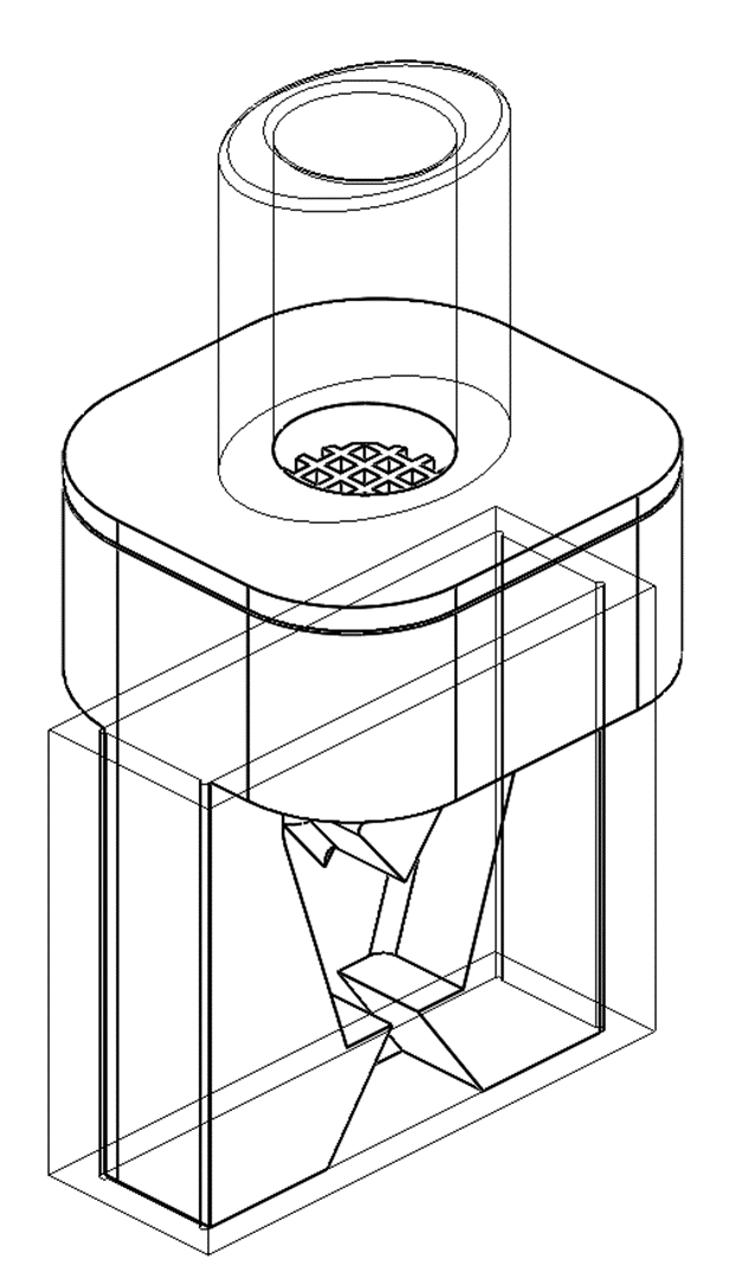
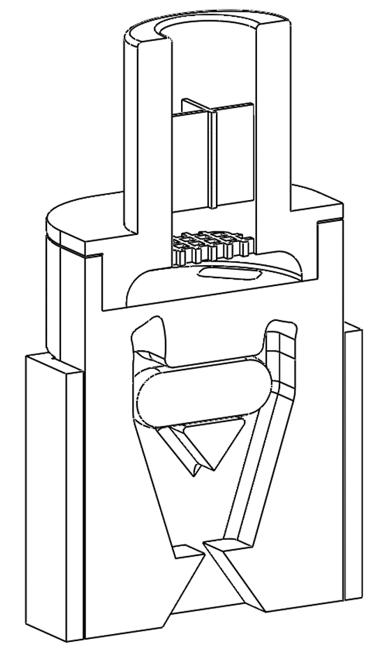
The airflow through the capsule chamber was designed so that the capsule oscillates. This causes impaction of the capsule within the capsule chamber leading to the breakdown of big powder aggregates inside the capsule and, consequently, an efficient release of the powder from the capsule.
After the initial de-aggregation of the powder in the capsule, the powder evacuates from the capsule and reaches the swirl chamber. Further de-aggregation takes place due to the shear forces caused by the turbulent airflow created in the swirl chamber and the impaction of the powder on the walls
of the swirl chamber. The combination of the capsule movement and powder flow in the swirl chamber leads to a highly dispersed powder. This powder exits into the tubular mouthpiece through the mesh.
The powder de-aggregation and dispersion potential in a device are crucial parameters to achieve high efficiencies of the device. Various techniques can be used to study airflow and powder de-aggregation behaviour within a device. Here, a combination of a simulation with CFD (computational fluid dynamics) modelling in a steady state and an experimental approach was used to develop the new device.
In one experimental design CFD was used to study the airflow structures within the device. Two prototypes were tested prototype 1 (Figure 1a) without a flow straightener and prototype 2 (Figure 1b) with an integrated flow straightener in the mouthpiece. The test of fine particle assessment by NGI was conducted on the prototypes. As seen in Figure 1c, the CFD data of Prototype 1 exhibits a swirling flow that proceeds out of the mouthpiece, reflecting a higher deposition and a swirl pattern observed in the induction port of the NGI. However, with the introduction of the flow straightener in Prototype 2, the CFD data shows a high reduction of the swirl exiting the mouthpiece (Figure 1d). This is also in correlation when comparing CFD data with our NGI data (Figure 2). There was a higher deposition in the induction port at both the flow rates for prototype 1, and it was significantly higher @ 30 LPM(p<0.05). The statistical analysis performed using an independent Student t-test gave probability values of less than 0.05 which was considered as a significant difference. The flow straightener reduced the swirl of the airflow exiting the device, thereby reducing the deposition in the induction port. There was no significant difference in Fine Particle Fraction (FPF) between the two devices indicating that major deaggregation occurs within the device. CFD proved to be a valuable tool for studying the air flow dynamics of the dry powder inhaler. NGI testing provided the supporting data and visual observation of the drug deposition in the induction port, which indicates/simulated probable oropharyngeal deposition.
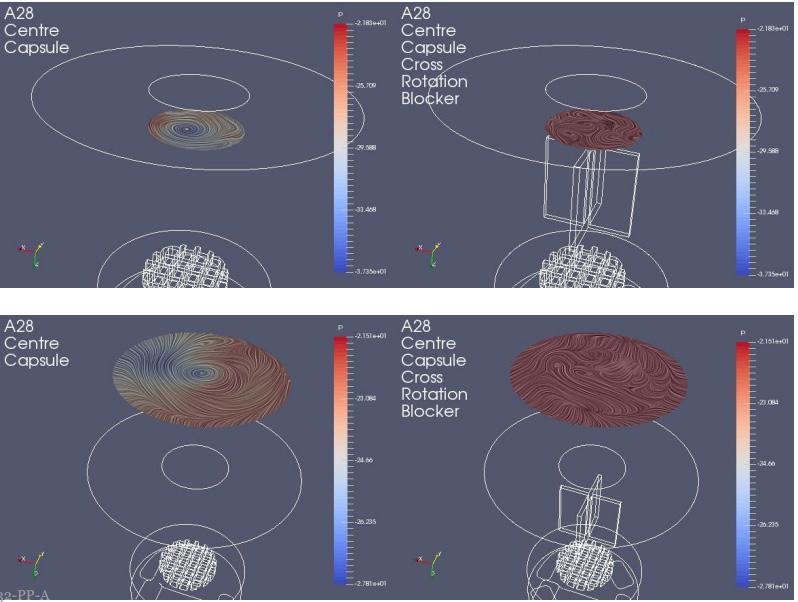
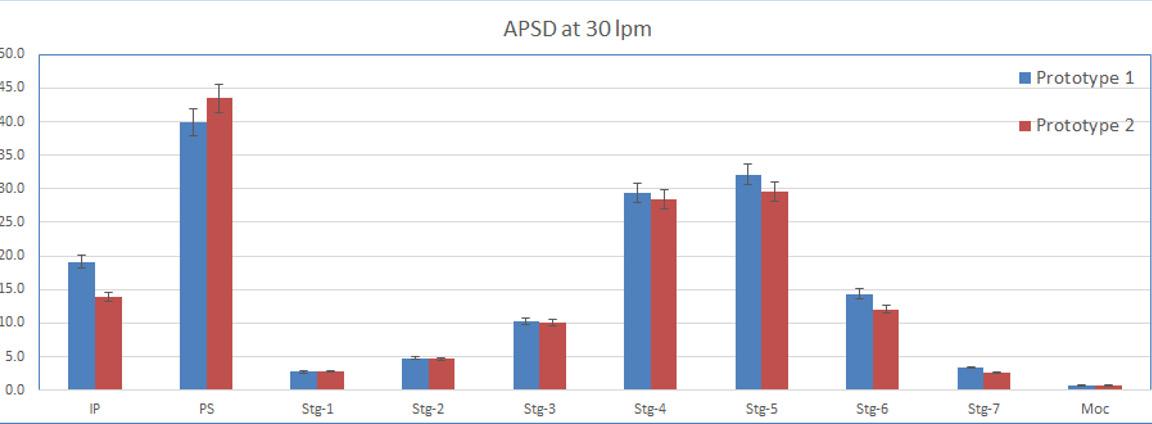
A modular engine set-up during development allowed for adapting small changes in the prototypes. Significant improvements could be achieved by changing different engine parameters. Figure 3 demonstrates the improvement in performance of the test data from Prototype 1 to 4. The NGI testing demonstrated the reduction in deposition in the Induction Port and pre-separator stage and increase in the lower stages of the impactor thereby increasing Fine Particle Fraction as we moved along our design development.
The intrinsic resistance of a device is often discussed controversially in the literature. For Asthma/COPD applications, often lowresistance devices seem to be more beneficial for the patient struggling to achieve high flow rates. However, low resistances of a device often come along with a lower deposition of the fine particles of the API in the deep lung. Medium resistances have the big advantage of creating a deeper lung deposition.4 The inspiratory resistance of the new Presspart device was determined to be designed as a medium resistance device.
Another development target was to achieve a relatively flow rate-independent device. The airflow path was designed and optimised such that there was no significant difference in the fine particle fraction for flow rates ranging from 30ltr/min to 90ltr/ min (Figure 4).
Two types of formulations were studied a binary mixture and spray-dried engineered particles.
Budesonide a glucocorticoid is known for its property of its sticky nature, and its difficulty to de-aggregate was chosen as the candidate formulation. A binary mixture of a marketed formulation of lactose and budesonide 200 mcg per dose with a capsule fill weight of 25mg was selected to test the prototype. In addition, the performance was compared to a marketed Plastiape RS01 equivalent device. The novel Presspart device exhibited a high fine particle fraction compared to the marketed Plastiape RS01 device confirming the engine's efficiency (Figure 5).
In a study in a corporation with the University of Bonn, the performance of the novel DPI Device was assessed by testing a spray-dried Rifampicin formulation. More detailed information on that can be
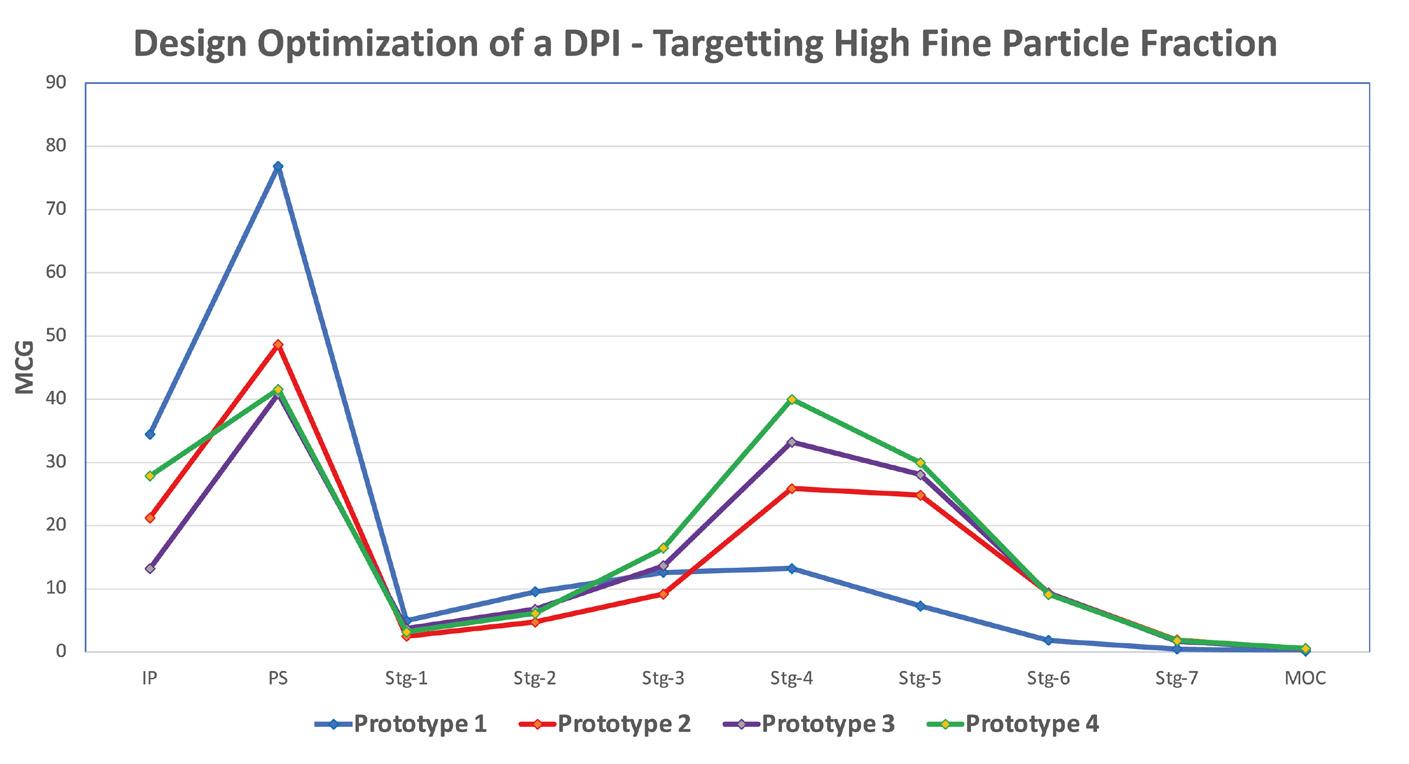
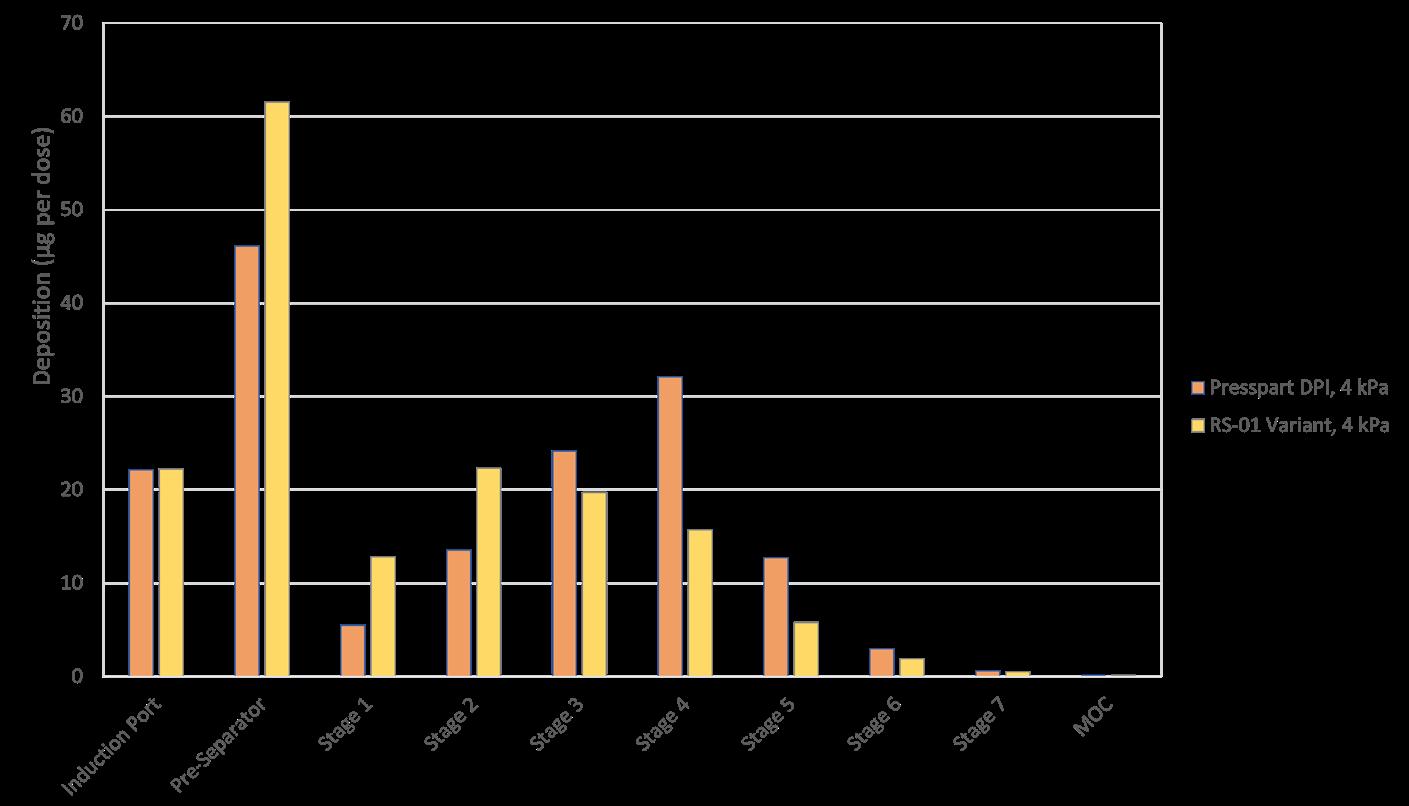
found in the recently published article.5 The formulation was used to benchmark the Presspart device performance against devices already well introduced in the market. As shown in Table 1, the FPFs generated are highest for the PP device when compared to the commercially available standard devices Handihaler and RS01 equivalent.
Conclusion
Several key factors influence the performance of the dry powder inhaler device. The most important ones are the patient's inhalation technique, device handling and the device engine i.e. the efficiency of de-aggregation mechanism of the device. Targeting a medium resistance and a relatively flow rate independency were critical factors within the device development approach.
Various complementary techniques were used to study airflow and powder

de-aggregation behaviour within a device. Combining different development approaches of fast-paced prototyping, CFD technique and laboratory data for verification can effectively develop cost-effective high-performance DPIs for the inhalation market. These techniques enabled H&T Presspart to develop a higperformance prototype independent of the formulation type tested (carrier-based as well as an engineered formulation).
1. MIT technology review September 8, 2022
2. Geller DE, Weers J, Heuerding S, “Development of an inhaled dry-powder formulation of Tobramycin using PulmoSphere™ technology”. J Aerosol Med Pulm Drug Deliv, 2011,24, pp 175–82.
3. Hoppentocht M, Hagedoorn P, Frijlink HW, de Boer AH, “Technological and practical challenges of dry powder inhalers and formulations”. Advanced Drug Delivery Reviews, 2014, 75, pp 18–31.

4. Dal Negro RW, “Dry powder inhaler and the right things to remember: a concept view”. Multidisciplinary Respiratory Medicine,2015, pp 10-13
5. Groß R, Berkenfeld K, Schulte C, Ebert A, Sule S, Sule A, Lamprecht A, „State of the Art in CapsuleBased Dry Powder Inhalers: Deagglomeration Techniques and the Consequences for Formulation Aerosolization”. Pharmaceutics, 2022, 14, pp 1185

Ameet Sule, Director, Inhalation Product Technology Centre, H&T Presspart, is a pharmaceutical professional having worked in the industry for more than 20 years, specialising in the development of inhalation products and devices. Mr Sule works closely with H&T Presspart’s customers around the globe, understanding and mitigating the frontend development challenges of new and generic products for inhalation drug delivery.

Sunita Sule, Inhalation Product Consultant, is a pharmaceutical professional who has over 25 years of experience in the pharmaceutical industry, with core expertise in formulation and device development of inhalation products.

Mirjam Kobler, PhD, is currently Global Business Development Manager for H&T Presspart. Dr. Kobler oversees the management and business development activities for H&T Presspart’s DPI device portfolio. Before joining H&T Presspart Dr. Kobler worked at Meggle, where she headed the R&D Department of Meggle’s Excipients and Technology business group. Dr. Kobler’s background includes seven years of experience in various areas of lactose excipients, especially for DPIs.


