PRIMAVERA BORELLI


Amanda Costa
Dra. Ana Lucia Borges Shimada
Profo. Dr. Antonio Altair Magalh„es Oliveira
Cadiele Oliana Reichert
Prof. Dr. Celso Spada
Dra. Dalila C. Oliveira
Prof™. Dra. Dulce Marta Schimieguel Mascarenhas Lima
Profo. Dr. Ed Wilson Santos
Profa. Dra. Graziela Batista da Silva
Profo. Dr. JosÈ Guilherme Xavier
Prof™. Dra. Ligia Ferreira Gomes
Profo. Dr. Marcelo Macedo Rogero
Mayara Caldas Ramos Cunha
Prof™. Dra. Primavera Borelli
Profo. Dr .Ricardo AmbrÛsio Fock
Prof™. Dra. Rita Font„o-Wendel
Prof™. Dra. Sandra Helena Poliselli Farsky
Prof™. Dra. Solange L˙cia Blatt
ValÈria Pereira Salgado
Fundamentos de Hematologia: teoria & prática
© 2023 Primavera Borelli (org.)
Editora Edgard Blücher Ltda.
Publisher Edgard Blücher
Editor Eduardo Blücher
Coordenação editorial Jonatas Eliakim
Produção editorial Aline Fernandes
Diagramação Joyce Rosa
Revisão de texto Flávia Carrara
Capa Laércio Flenic
Imagem da capa: iStockphoto
Desenhos Reynaldo Uezima
Esquemas Primavera Borelli

Rua Pedroso Alvarenga, 1245, 4o andar 04531-934 – São Paulo – SP – Brasil
Tel.: 55 11 3078-5366 contato@blucher.com.br www.blucher.com.br
Segundo o Novo Acordo Ortográfico, conforme 5. ed. do Vocabulário Ortográfico da Língua Portuguesa, Academia Brasileira de Letras, junho 2021.
É proibida a reprodução total ou parcial por quaisquer meios sem autorização escrita da editora.
Todos os direitos reservados pela Editora Edgard Blücher Ltda.
Dados Internacionais de Catalogação na Publicação (CIP) Angélica Ilacqua CRB8/7057
Fundamentos de hematologia: teoria e prática / organizado por Primavera Borelli. - São Paulo : Blucher, 2023. 894 p. : il., color.
Bibliografia
ISBN 978-65-5506-544-2 (eletrônico)
1. Hematologia I. Borelli, Primavera
22-1242
CDD 616.15
Índices para catálogo sistemático: 1. Hematologia
Este livro, com a importante colaboração dos coautores, aborda temas basilares da hematologia. Seus fundamentos teóricos e práticos destinam-se tanto aos profissionais, quanto aos estudantes e professores que atuam nessa área, assim como para pesquisadores das áreas da saúde e da biologia. Os temas são abordados de forma clara, objetiva e agradável, com uma linguagem acessível, mas não menos rigorosa, técnica e científ ica, trazendo uma visão integrada do sistema e os fundamentos para a compreensão da fisiologia e da patologia do sistema hemopoético.
A obra é resultado de nossas atividades acadêmicas e de pesquisa na hematologia clínica e experimental, nas disciplinas de hematologia da Faculdade de Ciência Farmacêuticas da Universidade de São Paulo, bem como da atividade profissional na referida Faculdade e no Laboratório Clínico do Hospital Universitário da USP.
O termo hemopoese, ou hematopoese, deriva do latim haemus, ou do grego haematus (sangue), e poiesis (formação), e envolve os processos relacionados a origem, proliferação, diferenciação, maturação e distribuição das células sanguíneas. É um fenômeno complexo e altamente regulado, sendo influenciado por vários estímulos que atuam em diferentes níveis do processo – central (órgãos hemopoéticos) e periférico (sangue circulante) – caracterizando-se pela contínua produção e liberação de células maduras para a circulação sanguínea.
A área hematológica apresentou, nos últimos quarenta anos, significativos avanços em relação à compreensão da fisiopatologia do sistema hemopoético como, também, nas técnicas diagnósticas e terapêuticas, especialmente na área da onco-hematologia, merecendo destaque os avanços obtidos na biologia da célula-tronco hemopoética.
Desde o século XVIII até meados do século XX, diversas teorias coexistiram, tentando explicar a origem das células sanguíneas bem como o local de sua produção.
Dentre as várias teorias, três se destacaram. Em primeiro lugar, a teoria polifilética, das quais destacamos a teoria dualista de Ehrlich e Naegli, que postulava que as células derivavam de um precursor mieloide e de um precursor linfoide. Em segundo, a teoria trialista de Schilling-Aschoff, que defendia a derivação de todas as células sanguíneas a partir de três precursores: o hemocitoblasto mieloide, a célula retículo-histiocitária e a célula linfocitopoética. Por fim, em contraponto a elas, Adolfo Ferrata, hematologista italiano (1880-1946), formulou, entre 1910 e 1918, a teoria monofilética ou unicista, preconizando que as células derivavam de uma célula pluripotente, que ele denominou de hemo-histioblasto, uma vez que essa população celular originaria as células sanguíneas e células do tecido conjuntivo e a existência do hemocitoblasto, célula que originaria as diferentes linhagens sanguíneas. Esta teoria, a partir dos dados experimentais e clínicos, se consolidou a partir da década de 1950, e desde então vem sendo adotada. Atualmente, essas células são denominadas, respectivamente, de célula-tronco embrionária e de célula-tronco hemopoética.
O desenvolvimento teórico e tecnológico nas áreas da genética, biologia celular, molecular, da física, bioquímica e farmacogenômica proporcionaram sensível avanço na elucidação da fisiologia do sistema hemopoético e das patologias que o acometem. Estas técnicas possibilitaram um refinamento no diagnóstico, contribuindo para prognósticos mais precisos e terapêuticas mais eficazes.
Devemos lembrar que os genes, sua transcrição e transdução, necessitam de um ambiente ou um microambiente específico e adequado para sua expressão, sendo necessário além do meio intracelular também o extracelular, permitindo as interações célula-célula e célula-matriz extracelular, e ainda, as interações matriz-matriz. Nas últimas décadas, a matriz extracelular, representada por células, proteínas adesivas e não adesivas e por substâncias solúveis (citocinas, fatores de crescimento, hormônios e nutrientes) deixou de ser vista apenas como um sistema de sustentação mecânico, mas como parte fundamental do processo regulatório. Esta estrutura extracelular proporciona não só a estrutura mecânica, mas possibilita a fixação de células e moléculas, regulando a osmolaridade, concentração de oxigênio, e demais substâncias e o pH local, o que possibilita a interação celular, sua comunicação e sinalização. Uma vez que as mensagens químicas que podem se traduzir na formação de segundo mensageiros e na sinalização ao genoma, depende da estrutura conformacional dessas moléculas e na forma das células, sem dúvida importante, é um dos aspectos da sinalização celular, pois o controle da transmissão da informação e sinal químico ou físico-químico dependem, também, da sua conexão com a rede externa e interna da célula.
A identificação de substâ ncias solúveis com ação sinalizadora, como os hormônios, citocinas e fatores de crescimento, e a descoberta dos receptores celulares para essas e para outras diversas moléculas, foram decisivas para a elaboração das vias de sinalização celular, possibilitando desvendar os fenômenos que envolvem a hemopoese: a autorrenovação da célula-tronco, os processos de proliferação, diferenciação das distintas linhagens sanguíneas e a maturação celular e, posteriormente, sua funcionalidade, proporcionando a descoberta de novos alvos-terapêuticos.
Assim, este livro pretende ser um manual importante para aqueles que trabalham com o diagnóstico hematológico e/ou com aspectos funcionais das células sanguíneas. A obra contempla desde a fisiologia do sangue até os procedimentos práticos mais rotineiros utilizados em um laboratório clínico de hematologia.
O livro é organizado em três grandes partes contendo sete seções e trinta capítulos. Os capítulos são ilustrados com esquemas de vias metabólicas e de regulação, desenhos e fotomicrografias dos tecidos e das células (desde precursores até células maturas) e da morfologia celular nos quadros de anemias, leucemias e demais patologias do sistema hemopoético, além de links que permitem a verticalização dos temas abordados. A descrição morfológica dos tipos celulares, suas etapas de maturação e as principais alterações morfológicas possibilitam entender as delicadas e complexas interações celulares com sua matriz.
Nesta edição, a Parte I – fisiologia do sistema hemopoético, capítulos de 1 a 14, aborda a fisiologia do sistema. O Capítulo 1 é uma breve introdução aos aspectos físico-químicos e funcionais do sangue e tem como objetivo apresentar, sucintamente, o panorama do mesmo para o organismo, aspectos que serão aprofundados nos capítulos posteriores.
A partir do Capítulo 2, descreve-se, detalhadamente, a ontogenia e fisiologia da produção das células sanguíneas, abordando-se a biologia da célula-tronco e dos órgãos hemopoéticos, com ênfase no microambiente da medula óssea, baço e linfonodos e os processos celulares e moleculares da hemopoese.
Além desses, há capítulos específicos sobre a eritropoese, plaquetopoese, granulopoese e sobre as séries monocítica e linfoide. Também são abordados os sistemas da coagulação e da fibrinólise, e há ainda um capítulo, com teoria e prática, sobre antígenos eritrocitários, plaquetários e leucocitários (imuno-hematologia).
Completam a primeira parte do livro os temas sobre citocinas, moléculas de adesão, cinética e mobilização de células e sua repercussão no hemograma, assuntos que normalmente não são encontrados nos textos de hematologia, e que ao nosso entender, são fundamentais para a compreensão da fisiologia hemopoética e sua regulação.
Na segunda parte do livro, capítulos 15 a 28, aborda-se a fisiopatologia das doenças que acometem o sistema: anemias (Capítulo 15), leucocitoses e leucopenias não malignas (capítulos 16 e 17), plaquetopenias, plaquetoses e hemorragias (capítulos 18 a 20), doenças onco-hematológicas (capítulos 21 a 28): mieloproliferações, leucemias agudas, mielodisplasias e linfomas, nos quais são tratadas a fisiopatologia e etiopatogenia dos processos, sinais e sintomas, classificações e diagnóstico laboratorial.
Na terceira parte (capítulos 29 e 30) são apresentadas as principais técnicas utilizadas na rotina hematológica, inclusive os testes citoquímicos. Cada técnica aborda: princípio, materiais e reagentes, protocolo de execução e interpretação dos resultados. Ênfase especial é dada no Capítulo 30, que trata da automação na área da hematologia.
Expressamos nossa gratidão aos demais autores que dedicaram seu tempo e expertise para a elaboração desta obra. Agradecimento especial às Dras. Amanda Krisma Rabelo e Karina Nakashima pelo aux í lio na organização do Capítulo 29 – Técnicas rotineiras em hematologia.
Agradecemos ao Reynaldo Uezima pela execução das ilustrações que compõe este livro e ainda à equipe editorial da Editora Blucher pelo apoio na edição da obra.
Este livro é dedicado aos professores Dr. Domênico H.G.P. Barbieri (in memorian) e Dra. Lucia Helena Meloni Bruneri (in memorian), docentes da disciplina de Hematologia Clinica da Faculdade de Ciências Farmacêuticas-USP, e meus mestres.
Às minhas filhas Priscilla e Melissa e aos meus netos, Gabriela, Ana Clara, Vitória, Luan e Teo.
Primavera Borelli
São Paulo, 2 de agosto de 2023
Primavera Borelli
Desde os primórdios da humanidade, o sangue (do latim, sangui) sempre foi considerado um elemento vital, essencial à vida. Descrições rupestres datadas da Pré-História mostram seres humanos ingerindo sangue. A Bíblia descreve que “a alma da carne é o sangue” (Lev. 17.11) e que “o sangue é a vida” (Deut. 12.23). Desenhos e pinturas da Idade Média evidenciam as primeiras tentativas de transfusões sanguíneas, e várias culturas, ainda hoje, mantêm o hábito de ingerir sangue como uma forma de obter ou manter a saúde. Por exemplo, membros da tribo africana dos Masai ingerem sangue de seu rebanho bovino; vários países europeus, asiáticos e latino-americanos utilizam sangue em diversos pratos da culinária, como o chouriço, o molho pardo e a sopa de osso buco ou “tutano”.
O sangue, na circulação, é um tecido líquido, móvel e disseminado por todo o organismo, sendo constituído por uma parte celular e por uma fração líquida denominada plasma (Figura 1.1).

Figura 1.1 - Tubo com sangue colhido com EDTA, evidenciando a camada de eritrocitos, a camada com leucócitos e plaquetas e o plasma. Fotografia gentilmente cedida por Alice Maria Serpentino.
Características biológicas e funções do sangue: considerações gerais
O plasma representa, em média, 55% da volemia total e contém cerca de 90% de água, encontrando-se dissolvidos nele proteínas e peptídeos com diferentes funções biológicas, aminoácidos, lipídeos, carboidratos, minerais e vitaminas.
O componente celular (Figura 1.2) representa, aproximadamente, 45% do volume plasmático e contém os eritrocitos, leucócitos (neutrófilos, eosinófilos, basófilos, linfocitos e monócitos) e as plaquetas, Tais células são produzidas nos órgãos hemopoéticos (ver Capítulo 2), que, após o nascimento, em seres humanos, são essencialmente medula óssea, timo, baço e linfonodos. A medula óssea é considerada o principal sítio anatômico da hemopoese extrauterina. Em condições fisiológicas apropriadas somente as células sanguíneas maduras atingem a circulação sanguínea.
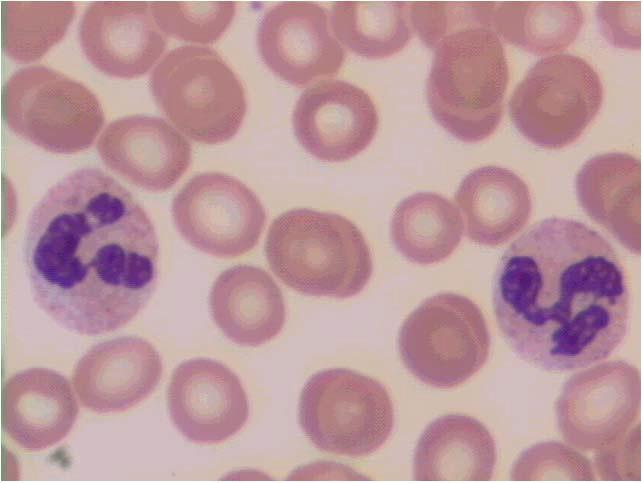
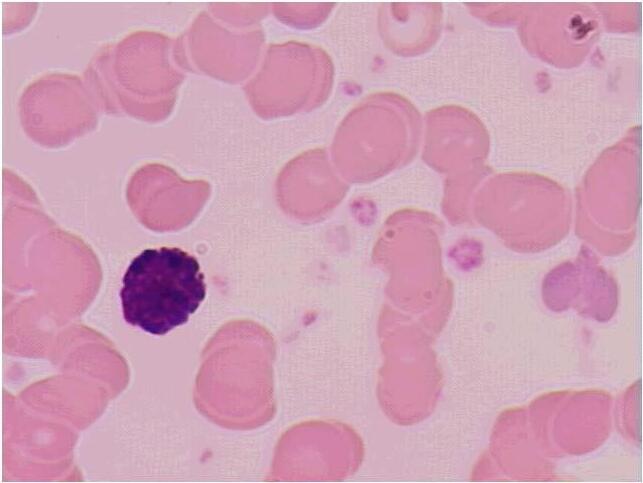
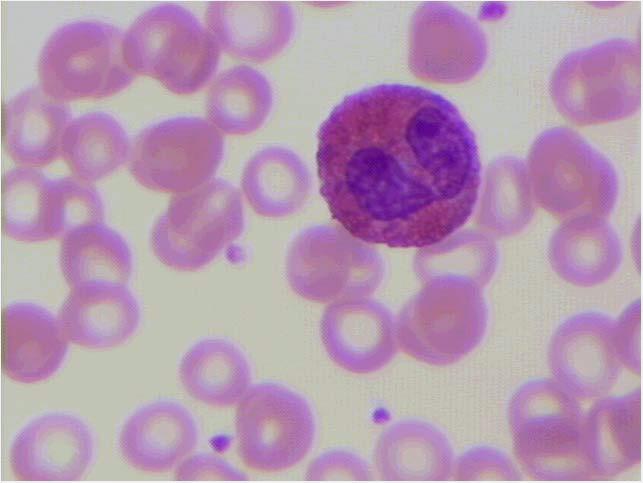
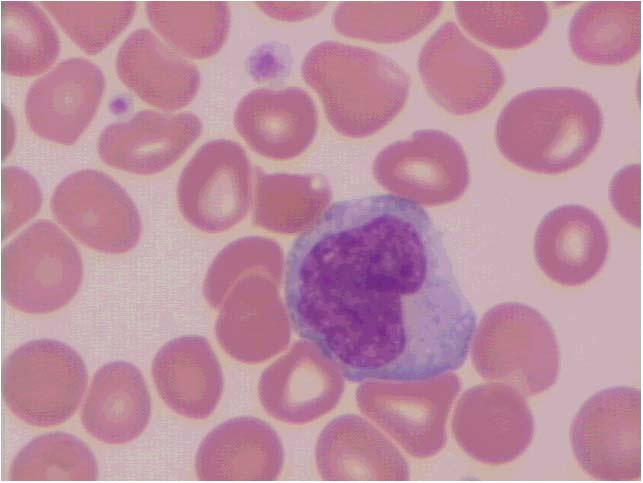
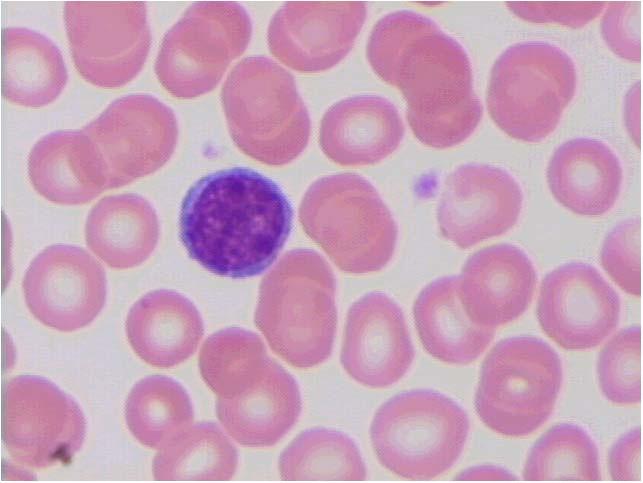
Figura 1.2 – Vista panorâmica de extensão de sangue periférico mostrando eritrocitos normais (1), neutrófilo segmentado (2); eosinófilo segmentado (3); plaqueta (4); basófilo (5); monócito (6) e linfocito (7). Todas as células apresentam morfologia típica. Coloração: May-Grunwald-Giemsa, modificado por Rosenfeld. Aumento 10x100. Fotomicrografia: Profa. Dra. Primavera Borelli, laboratório de Hematologia Clínica e Experimental, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo.
O sangue, diferentemente de outros tecidos, caracteriza-se por ter uma produção localizada em diferentes tecidos e órgãos, dependendo da fase de desenvolvimento do indivíduo (embrião, feto ou após o nascimento), e ainda pelo fato de, em condiçôes normais, somente as células maduras, dotadas de função efetora, migrarem para a circulação periférica. Nesse sentido, podemos dizer que o sistema é móvel e disseminado, ou seja, há células sanguíneas distribuídas por toda a rede vascular ao longo de todo o organismo.
O sangue é um tecido renovável. As células sanguíneas são continuamente formadas, uma vez que apresentam vida média limitada e relativamente curta na circulação (por exemplo, os eritrocitos permanecem, fisiologicamente, na circulação pelo período de 110-120 dias – vide Capitulo 8). Assim, as células precisam ser continuamente produzidas, e o sistema hemopoético apresenta elevada capacidade de replicação. Em média, um indivíduo adulto normal, de cerca de 70 kg, produz por volta de 2,5 × 109 eritrocitos/dia/kg; 1 × 109 plaquetas/dia/kg e 1 × 1010 leucócitos/dia/kg. Essa renovação celular é possível porque o sangue possui uma população de células-tronco denominadas células-tronco hemopoéticas (ou stem cell ) com alta capacidade de autorrenovação e diferenciação celular (vide Capítulo 5).
AGM: Aorta-gonada-mesonefro
BFU E: Unidade formadora de colonia da linhagem eritrocítica (Unit forming burst erythrocytic series)
CFU-G: Unidade formadora de colonia da linhagem granulocítica (Unity colony formating of granulocytic series)
CFU-GEMM: Unidade formadora de colonia das linhagens granulocítica, eritrocítica, megacariocítica e monocítica ou mono-macrofágica (Colony formating units of granulocytic, erythrocytic, megakaryocytic and monocytic series)
CFU-GM: Unidade formadora de colonia das linhagens granulocítica e monocítica ou mono-macrofágica (Unity colony formating of granulocytic and monocytic series)
CFU-M: Unidade formadora de colonia da linhagem monocítica ou mono-macrofágica (Unity colony formating of monocytic series)
CFU-Meg: Unidade formadora de colonia da linhagem megacariocítica (Unity colony formating of megakaryocytic series)
CFU-MegE: Unidade formadora de colonia das linhagens megacariocítica e eritrocítica (Unity colony formating of megakaryocytic and erythrocytic series)
CFU-S: Unidade formadora de colonia do baço (Colony formating unit-spleen)
CTH: Célula-tronco hemopoética
EPO: Eritropoietina
GM- CSF: Fator estimulador de colonias granulo-monocíticas (Granulocitic-monocitic stimulating factor)
IL-3: Interleucina 3
IL-6: Interleucina 6
IL-9: Interleucina 9
IL-11: Interleucina 11
MEC: Matriz extra celular
SCF: Fator estimulador de célula-tronco (Stem cell fator)
2.1 ONTOGENIA E FILOGENIA
Os estudos sobre o sangue e a circulação sanguínea remontam há cerca de 2000 anos. Galeno (129-199 d.C.) foi um dos pioneiros a descrever, com base em estudos em macacos, a pequena circulação, revelando que no interior dos vasos não existia água, e sim sangue. Miguel Servet descreveu, em 1551, a circulação pulmonar e o retorno do sangue ao ventrículo esquerdo. William Harvey publica em 1628 seus estudos descrevendo a circulação sanguínea e a distribuição de sangue pelo corpo em fluxo contínuo, concluindo que o “coração era uma bomba” . Diante desses conhecimentos, Descartes propôs que as “artérias e veias eram canos que levavam os nutrientes para o corpo”.
O aperfeiçoamento do microscópio realizado pelos holandeses Zacharias Janssen e Cornelius Drebbel e, posteriormente, por Christiaan Huygens (1629-1695) e Johannes Havelke (1611-1687) tornou possível a descoberta das células
Marcello Malpighi (1628-1694), biólogo e médico italiano, demonstrou, em 1661, que o sangue não se formava nos vasos periféricos, e, em 1666, foi o primeiro a observar os glóbulos vermelhos (eritrocitos) e a atribuir-lhes a cor do sangue. Foi Antonie van Leuuwenhoek (1632-1723), também holandês, quem descreveu, em 1668, a rede de capilares e como as células nela circulavam; em 1674, fez a primeira descrição de uma célula sanguínea: o eritrocito.
Durante os séculos XVIII, XIX e até meados do século XX, diversas teorias coexistiram tentando explicar a origem das células sanguíneas bem como o local de sua produção. Dentre as várias teorias duas se destacaram: a teoria polifilética e a monofilética.
A teoria polifilética, defendida por cientistas notáveis como Erik Undritz, postulava que as diferentes células sanguíneas derivavam de precursores distintos, independentes entre si (Figura 2.1).
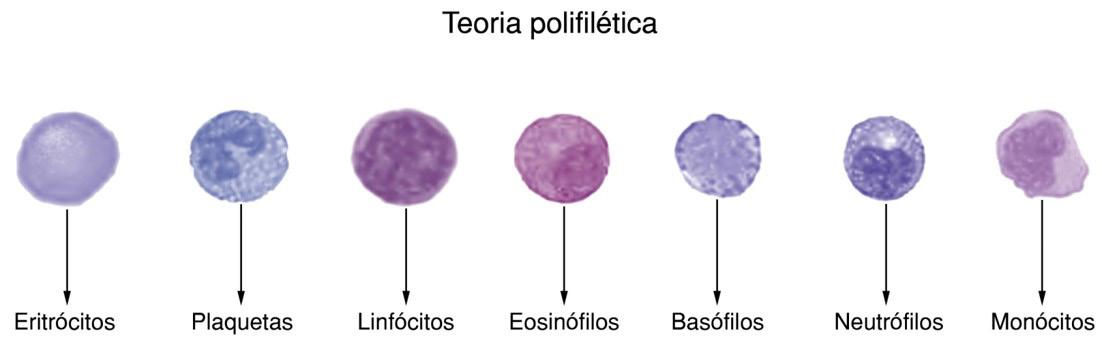
Figura 2.1 – Representação gráfica da teoria polifilética. Essa teoria preconiza que cada tipo celular sanguíneo deriva de um tipo específico de precursor, os quais são independentes entre si.
Variantes da teoria polifilética foram a teoria dualista de Ehrlich e Naegli, que postulava que as células derivavam de um precursor mieloide e de um precursor linfoide (Figura 2.2), e a teoria trialista de Schilling-Aschoff (Figura 2.3), que defendia a derivação de todas as células sanguíneas a partir de três precursores: o hemocitoblasto mieloide, a célula retículo-histiocitária e a célula linfocitopoética.
BFU-E: Unidades (primitivas) formadoras de células eritroides (Burst-Forming Unit-Erythroid )
CAR: Células adventícias reticulares. (Cells adventitial reticular)
CFU-GEMM: Unidade formadora de células granulocíticas-eritroides-macrofágicas-megacariocíticas. (Colony forming unit–Granulocyte-Erythroid-Makrophage-Megakaryocyte)
FCH: Fatores de crescimento hemopoéticos
FGF: Fator de crescimento de fibroblasto. (Fibroblast growth factor)
IGF1: Fator de crescimento similar a insulina. (Insulin(simile) growth factor-1)
BMP: Proteínas morfogenéticas do osso. (Bone morphogenetic proteins)
GM- CSF: Fator de crescimento de células granulo-macrofágicas. (Granulocyte-macrophage colony-stimulating factor)
IL-1: Interleucina 1
MEC: Matriz extracelular
PPARγ: Receptor ativado por proliferadores de peroxissoma gama; c/EBPS. (Enhancer binding proteins)
TGFβ: Fator de crescimento de tranformador beta. (Transforming growth factor beta)
TNF α : Fator de necrose tumoral alfa. (Tumor Necrosis Factor Alfa)
VLA4: Antígeno de aparecimento tardio 4 ou integrina α4β1 ou CD49d/CD2. (Very late antigen 4, or α4β1-integrin or CD49d/CD2)
VLA5: Antígeno de ativação de aparecimento ou integrina α 5β1. (Very late activation antigen 5 or α5β1 integrin)
3.1.1 MICROARQUITETURA MEDULAR
A medula óssea é encontrada no canal medular (Figuras 3.1 e 3.2) de ossos longos, até cerca dos 18 anos de idade, e nas cavidades de ossos esponjosos (ossos chatos) a partir dessa idade.
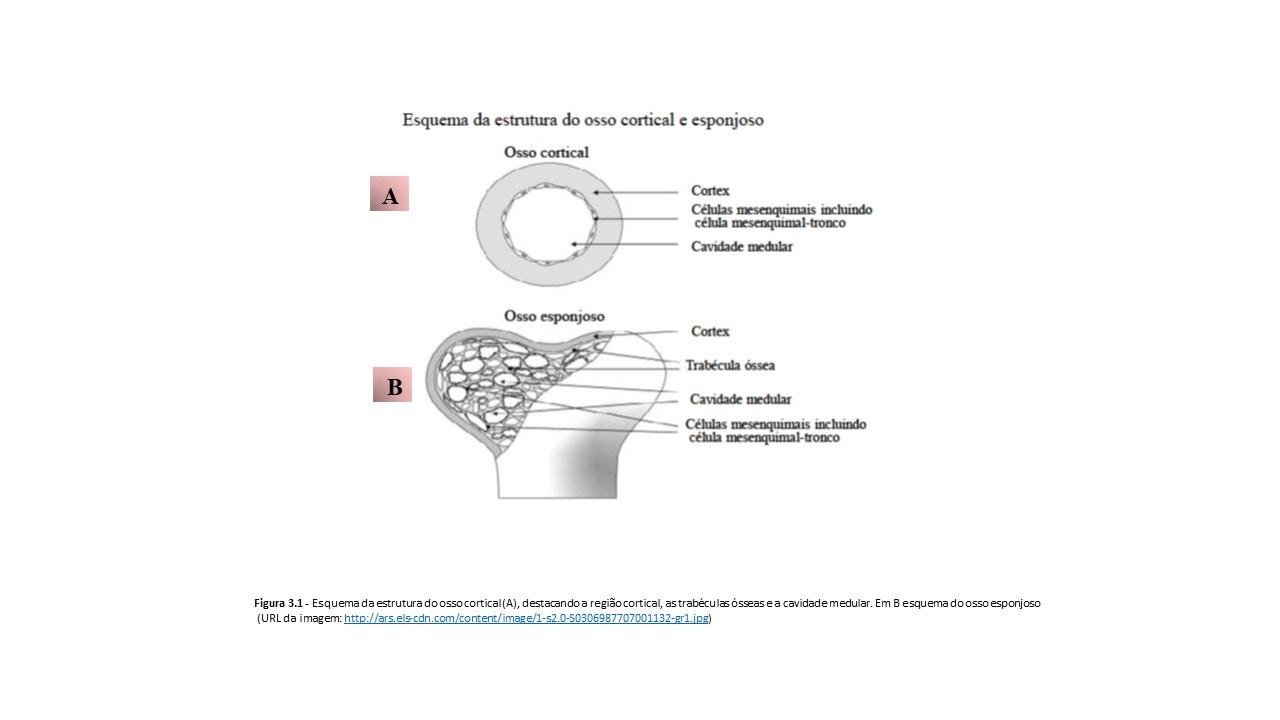
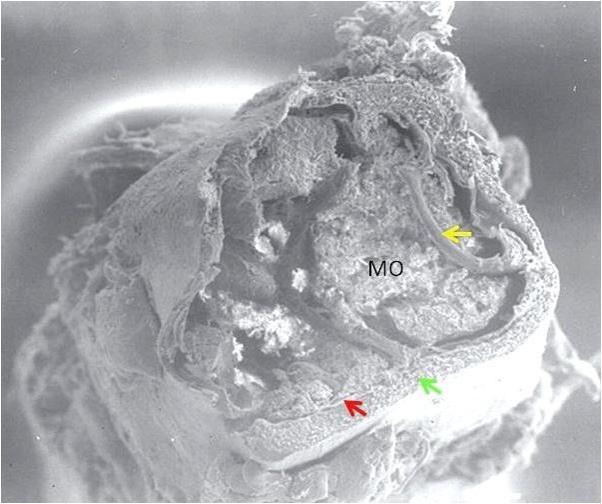
Figura 3.2 – Fotomicrografia de corte longitudinal de fêmur de camundongo, em microscopia eletrônica de varredura (×70). Observa-se o canal medular preenchido com a medula óssea (MO), a região cortical do osso (seta verde), a região subendosteal (seta vermelha) e a trabécula óssea (seta amarela). Fotomicrografia: Profa. Dra. Primavera Borelli, Laboratório de Hematologia Clínica e Experimental, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo.
À primeira vista, a medula óssea apresenta-se histologicamente como um tecido altamente celular pobre em estroma, de aspecto aparentemente anárquico, destacando-se traves ósseas, células sanguíneas em diferentes estágios de maturação e estruturas vasculares e nervosas. A superfície interna do córtex e a superfície externa das trabéculas ósseas são revestidas pelo endósteo (Figuras 3.1, 3.2 e 3.3), constituído por uma camada simples de células pavimentosas ou poliédricas associadas a um delicado estroma reticular (Figuras 3.3 e 3.4). Procedendo-
PALS: Bainha linfocitária periarteriolar. (Periarteriolar Lymphoid Sheath) interdigitantes (IDCs)
MHC-II+: Antígenos maior de histocompatibilidade II (Major Major Histocompatibility)
CAP: Cápsula esplênica
TNF: Fator de necrose tumora (Tumor necrosis factor)
IDC: Célula interdigitante
DAB: Diaminobenzidina
FDCs: Células foliculares dendríticas
SIRPα: Proteína alfa reguladora de sinal
O baço exibe algumas características que o tornam único dentro do organismo como um todo, pois apresenta-se como uma estrutura híbrida, relacionando-se aos sistemas linfo-hemopoético e circulatório. Como um órgão linfoide secundário, grupo do qual fazem parte também os linfonodos e os tecidos linfoides associados às mucosas, compartilha a capacidade de construir localmente uma resposta imunitária, apresentando, para tanto, uma população linfoide representativa, de componentes da imunidade inata e adaptativa, dispostos estrategicamente ao longo de sua extensão. No que tange ao sistema circulatório, ocupa uma posição central na circulação esplâncnica, funcionando como uma espécie de filtro, comprometido com a retirada de eritrocitos senescentes e com a reciclagem de ferro, exibindo em paralelo uma função específica na defesa imune contra antígenos carreados por via hematógena. A complexidade estrutural aliada à diversidade filogênica contribuíram para o relativo desconhecimento da histofisiologia do tecido até os dias de hoje. Em sequência, abordaremos a evolução histórica da compreensão do baço e suas principais características histofisiológicas, com especial referência à sua participação na biologia hemopoética.
Em 1706, sir Richard Blackmore publicou uma dissertação crítica acerca da natureza e cura da cólica e da melancolia, questionando, entre outras coisas, a utilidade do baço. Na verdade, essa discussão já atravessara séculos. O próprio termo “spleen” linguisticamente relaciona-se a melancolia em francês. Erisistrato médico alexandrino, fundador da anatomia, atraído pela simetria, sugerira que o baço contrabalançava, fisicamente, o fígado. Essa seria uma explicação para a sua existência, já que, ao contrário de outras vísceras, como os rins, o estômago, o fígado, o coração e os pulmões, vísceras historicamente relacionadas à descarga de urina, quimo, bile, sangue e ar,
respectivamente, o baço parecia não ter nenhuma comunicação com outras estruturas, constituindo-se em um órgão cego. Até mesmo o cérebro era compreendido, pois drenava pelo nariz. Tal perplexidade perdurou até os estudos de Galeno, que identificou uma pequena comunicação gastrolienal, relacionando o baço ao aparelho digestivo, visão que perdurou por mais de 1400 anos. Essa evidência só foi refutada por Vesalius, que demonstrou a inexistência de tal comunicação, reconduzindo o baço à condição de órgão enigmático. Apenas no século XVI o estudo de anatomia esplênica apresentou novidades, graças a Malpighi, que descreveu a presença da polpa vermelha e da polpa branca, esta última caracterizada pelos folículos, que lhe pareciam ácinos de glândulas endócrinas. Porém, na ausência de ductos excretores, tais estruturas foram tidas como secretórias para a circulação sanguínea. Já no século XIX, Billroth dividiu a polpa vermelha em seios venosos e cordões linfocitários, sendo posteriormente caracterizada sua atividade de reservatório sanguíneo por um cirurgião estadunidense, Benjamin Rush. Há 140 anos, von Koliker, autor do primeiro livro de Histologia, caracterizou a hemocaterese esplênica, considerando-a, apesar da forte oposição de cientistas, dentre eles Rudolph Virchow, como um evento fisiológico. A última das funções centrais esplênicas a ser identificada foi a imunitária. Em 1975, Veerman & van Ewijk realizaram uma descrição detalhada da polpa branca do baço de ratos, dividindo-a em três compartimentos: a bainha linfocitária periarteriolar (PALS), intimamente associada a ramificações da artéria esplênica, os folículos linfoides, localizados na periferia da PALS, e a zona marginal, circundando ambas as estruturas.
Em termos filogenéticos, o baço é uma estrutura exclusiva de vertebrados, sendo o primeiro tecido análogo a uma condensação linfomieloide encontrada no intestino de vertebrados primitivos, os ciclóstomos, como a lampreia. Também nesses animais surgem as primeiras formas de eritrocitos, sugerindo, desde há muito, uma relação entre esses elementos. A primeira aparição do baço como órgão isolado ocorre em peixes ósseos. Estruturalmente o compartimento esplênico primordial é a polpa vermelha, presente desde os peixes. A colonização linfoide esplênica é um evento posterior, estabelecendo-se inicialmente no entorno arterial, seguindo-se evolutivamente a organização de folículos linfoides, bainha linfocitária periarteriolar e centros germinativos. Os mecanismos moleculares envolvidos nessa organização ainda são desconhecidos. Apesar das diferenças no sistema imune entre os vertebrados, a organização histológica esplênica e sua participação na filtração sanguínea mantiveram-se preservadas, desde peixes, passando por anfíbios, répteis, aves e mamíferos. Nesse sentido, o baço pode ser percebido como uma espécie de filtro discriminatório constituído por estruturas vasculares especializadas e um estroma reticular. Ambos funcionam como sítios de passagem de sangue, sobre o qual exercem sua atividade, com a contribuição, em especial, de diferentes populações macrofágicas, distinguindo-se macrófagos associados aos elipsoides, macrófagos da zona marginal e macrófagos metalofílicos, e de componentes linfoides, distribuídos em compartimentos de células B e T e uma zona mista de interação.
Em mamíferos, com base em referências morfológicas, de acordo com a espécie animal, o baço pode ser identificado com duas configurações, descritas como primitiva e arquetípica.
O baço primitivo é o predecessor do baço típico dos mamíferos, sendo encontrado em monotrêmatos, insetívoros e sirênios. Neste identificam-se a cápsula, dotada de colágeno, fibras elásticas e musculatura lisa, seguindo-se três camadas: a polpa branca, a polpa vermelha e, entre elas, a zona intermediária, rica em sinusoides. A polpa branca já apresenta a compartimentalização clássica, composta de bainha linfocitária periarteriolar (PALS) e folículos linfoides. As arteríolas originárias da polpa branca comunicam-se diretamente com as vênulas, tanto na zona intermediária quanto na polpa vermelha, constituindo uma circulação fechada. Na polpa vermelha são observados os sinusoides venosos e os cordões esplênicos, nos quais o fluxo sanguíneo é lento. A atividade eritropoética é mantida e, em ornitorrincos, predominante sobre a medular.
Nos demais mamíferos evidencia-se o baço arquetípico, com a presença da zona marginal na polpa branca, topograficamente equivalente à zona intermediária do baço primitivo. Nessas espécies identifica-se um espaço vascular distinto, o seio marginal. Várias arteríolas correm externamente a partir das artérias centrais na polpa branca, formando capilares terminais que se encerram na zona marginal ou no seio marginal, enquanto outras adentram a polpa vermelha liberando o sangue na trama reticular dos cordões esplênicos, drenando para as vênulas da polpa.
Alguns autores preferem categorizar o baço de acordo com as características funcionais predominantes, destacando as atividades de "armazenamento sanguíneo" e de imunidade. Assim, referem o " baço de armazenamento", dotado de maiores dimensões e importante musculatura lisa, prestando-se particularmente ao armazenamento de sangue, presente em equídeos, cervídeos, felídeos e pinípedes. Em contrapartida, em algumas espécies o elemento de
Primavera Borelli, Maristela Tsujita, Karina Nakajima, Ricardo Ambrósio Fock e José Guilherme
Ang1: Ang iopoietina
BMI1: Repressor polycomb
BMP: Proteína morfogenética óssea (Bone morphogenetic protein)
BMPR1: Receptor da proteína proteína morfogenética óssea, tipo 1 (Bone morphogenetic protein, type I receptor)
C/EBPα: C/Potencializador da ligação a proteínas alfa l
CARs: Células adventícias reticulares (Cells adventitial reticular)
CBF1: Proteína recombinante de ligação 1b (Recombining binding protein suppressor of hairless)
CD: Determinantes de cluster (Cluster determinants)
CFU-GEMM: Unidade formadora de células granulocíticas, eritroides, megacariocíticas e monocíticas (Colony formating units of granulocytic, erythrocytic, megakaryocytic and monocytic series)
c-Kit: Receptor do fator de crescimento de célula-tronco (Receptor de stem cell factor)
CTH: Célula-tronco hemopoética
CTMs: Células-tronco mesenquimais
CXCL12: Fator 1 derivado de célula estromal (chemokine, Cxc Motif, Ligand 12 ou pre-B cell Growth-stimulating factor ou SDF-1:Stromal cell-derived factor 1)
CXCR4: C-x-c receptor de quimiocina tipo 4 (C-X- C chemokine receptor type 4)
DNA: Ácido desoxinucleico (Deoxynucleic acid )
EPO: Eritropoietina (Erythropoietin)
FAK: Quinase de adesão focal (Focal adhesion kinase)
LIF: Fator inibidor da leucemia (Leucemia inhibitory factor)
FCH: Fator de crescimento hemopoético
FGF: Fator de crescimento de fibroblasto (Factor growth fibroblast)
FLT3L: Fms ligante relacionado à tirosina quinase 3
PI3K: Fosfatidilinositol-3-quinase
GATA 1: Fator de transcrição da globina-tipo 1 (Globin transcription factor 1)
GATA 2: Fator de transcrição da globina-tipo 2 (Globin transcription factor 2)
Ho: Hoechst 33342
HOXB4: Proteína homebox HOx-4 (Homeobox protein Hox-B4)
ICAM-1: Molécula de adesão intercelular-1 (Intercellular Adhesion Molecule 1)
IGF-1: Fator de crescimento semelhante à insulina (Insulin-like growth fator)
Ikappa β kinase: I kappa B quinase beta (I kappa B kinase beta)
IL-1: Interleucina 1
IL-3: Interleucina 3
IL-7: Interleucina 7
LFA-1: A ntígeno associado à função de linfocitos (Antigen associated with lymphocyte function)
LTRC: Células com capacidade de reconstituição por tempo indefinido (Long-term reconstituting cells)
MEC: Matriz extra celular
MHC-I: Complexo principal de histocompatibilidade classe I (Major histocompatibility complex class I )
MSCs: Células-tronco mesenquimais (Mesenchymal stem cells)
MYB: Proteína proto-oncogênica (Myb proto-oncogene protein)
MYC: Proteína proto-oncogênica c-Myc (Myc proto-oncogene protein, também denominado de c-MYC)
NF-E2: Fator de transcrição nuclear E2 (Nuclear transcription factor NF-E2)
NFκB: Fator de transcrição nuclear kapa beta (Nuclear transcription factor NF kappa beta)
NK: Células matadoras naturais (Natural killer)
OPN: Osteopondina
OSX: Osterix
p18: Também denominada de Inibidor 2C de ciclina dependente de quinase (cyclin-dependent kinase inhibitor 2C)
p21 ou CDKN1A: Inibidor 1A ciclina independente de quinase (Cyclin-Dependent Kinase Inhibitor 1A)
p27: Também denominada de Inibidor de ciclina dependente de quinase (cyclin-dependent kinase inhibitor)
PHM: Progenitores hemopoético multipotentes
PKC: Proteína quinase C (Protein kinase C)
PL: Progenitor de linhagem linfóide
PLC: Fosfolipase C (Phospholipase C)
PPARγ: Receptor ativado por proliferadores de peroxissoma gama (Peroxisome proliferator-activated receptor gamma)
PTH: Hormônio paratiroidiano (Parathyroid hormone)
Pu-1 ou SPI1: Spi-1 proto-oncogene
RANKL: Ligante do receptor do fator nuclear kapa beta
RANTES: Regulador da ativação de células T normais expressas e secretadas ou CCL5 (Regulated on activation normal T-cell expressed and secreted ou CCL5)
RANK: Ligante do ativador do receptor do fator nuclear kapa beta (Receptor activator of nuclear factor kappa-B ligand )
PTHr: Receptor de hormônio paratireoidiano (Receptor of parathyroid hormone)
RUNX2: Fator de transcrição relacionado à Runx 2 (Runx-2: runt-related transcription factor 2)
Sca-1: Antígeno de célula-tronco 1 (Stem cell antigen-1)
SCF: Fator estimulante de célula-tronco (Stem cell factor)
SDF-1: Fator 1 derivado de célula estromal ou CXCL12 ou Pre -B fator de crescimento de célula pre-B (Pre-B cell growth-stimulating fator)
TGFβ: Fator de crescimento de transformação (tecidual) beta (Transforminng growth factor B)
APC: Células apresentadoras de antígenos
Ba-CFU: Unidades formadoras de células basofílicas (Unity colony formating of basophilic serie)
BCR: Receptor de células B
BFU-E: Unidades (primitivas) formadoras de células (ou colonias) eritroides (Burst-forming unit-erythroid )
CFU-Eo: Unidades formadoras de células (ou colonias) eosinofílicas (Unity colony formating of eosinophilic serie)
CFU-G: Unidades formadoras de células (ou colonias) granulocíticas. (Unity colony formating of granulocytic serie)
CFU-GEMM: Unidade formadora de células (ou colonias) granulocíticas-eritroides-macrofágicas-megacariocíticas (colony forming Granulocyte-erythroid-macrophage-megakaryocyte)
CFU-GM: Unidades formadoras de células (ou colonias) granulo-monocíticas (Unity colony formating of granulocytic and monocytic series)
CNTF: Fator neurotrofico ciliar (Ciliary neurotrophic factor)
CSF: Fatores estimuladores de colonias (Colony stimulating factors)
CT-1: Cardiotrofina-1
CTL: Linfocitos T citotóxicos (Cytotoxic T lymphocytes)
EPO: Eritropoietina
ERK: Proteína relacionada com sinais extracelulares da quinase (Extracellular signal-regulated kinases)
G- CSF: Fator de crescimento de células granulocíticas (Granulocyte colony-stimulating factor)
GM-CSF: Fator de crescimento de células granulo-monocíticas (Granulocyte-macrophage colony-stimulating factor)
IFN: Interferon
IFNR: Receptor do interferon I
IFN α : Interferon alfa
IFNβ: Interferon beta
IFN γ : Interferon gama
IkB: Proteína citoplasmática ligada ao inibidor IkB (I kappa B alpha)
IKK1: I kappa B Kinase 1 (I kappa kinase beta 1)
IKK2: I kappa B Kinase 2 (I kappa kinase beta 2)
IL: Interleucina
IL-1: Interleucina 1
IL-3: Interleucina 3
IL-5: Interleucina 5
IL-6: Interleucina 6
IL-11: interleucina 11
IL-6R: Receptor de interleucina 6
INFα/βIFNγ: Interferon alfa e gama
IL-22R: receptor de interleucina 22
IRAK: Serina-treonina proteína quinase do receptor de IL-1 (IL-1 receptor serine-threonine protein kinase)
IRF9: Fator regulador 9 de interferon (Interferon 1 Regulating Factor)
JNK: Quinase Jun N - terminal (Kinase Jun N – terminal )
LIF: Fator inibitório de leucemia (Leucemia inhibitory factor)
LIFR: Receptor do fator inibitório de leucemia (Leucemia inhibitory factor receptor)
LPS: Lipopolissacarídeo
MAPK: Proteínas quinases ativada por mitógenos (Mitogen activated protein kinases)
M- CSF: Fator de crescimento de célula (ou colonias) monocíticas (Monocytic colony-stimulating factor)
MEC: Matriz extracelular
Meg-CFU: Unidades formadoras de células (ou colonias) megacariocíticas (Unity colony formating of megacariocytic series)
NFkβ: Fator nuclear kappa β (Nuclear factor-κB)
NK: Celulas naturais matadoras (natural killer)
NNT-1/BSF-3/CLC: Nova neutrotrofina1/fator 3 estimulador de celulaB/citocina like cardiotrofina (novel neurotrophin-1/B cell-stimulating factor-3/cardiotrophin-like cytokine)
OSM: Oncostatina M
SCF: Fator de crescimento de célula-tronco (Stem cell factor)
STAT1: Transdutor e ativador de fator de transcrição 1 (Transducer and activator of transcription factor 1)
TGFβ: Fator transformador de crescimento beta (Transforming growth factor β)
Th2: Linfocitos T auxiliadores 2 (T helper lymphocytes 2)
TLR: Receptor tipo Toll (Toll-like receptors)
TNF-α: Fator de necrose tumoral alfa (Tumor necrosis factor alpha)
TPO: Trombopoietina
TRAF6: Fator de necrose tumoral associado ao fator 6 (Fator de necrose tumoral associado ao fator 6)
TSLP: Linfopoietina estromal tímica (Thymic stromal lymphopoietin)
TSLPR: Receptor da linfopoietina estromal tímica
TSLPR: Thymic stromal lymphopoietin receptor (TSLP receptor)
X-SCID: Imunodeficiência combinada grave ligada ao X (X-linked severe combined immunodeficiency)
Ana Lúcia Borges Shimada e Sandra Farsky
α4 β1: Very late antigen-4 ou CD49d/CD29
α4 β7: ou CD49/beta7
ADAM-17: ADAM metallopeptidase domain 17
AP-1: Antigen protein 1
CAR: Coxsackie and adenovirus receptor
ELAM-1: Endothelial-leukocyte adhesion molecule-1 ou E-selectina ou CD62E
ESAM: Endothelial cell-selective adhesion molecule
Gp150,95: ou CD11c/CD18
ICAM-1: Intercellular cell adhesion molecule-1 ou CD54
ICAM-2: Intercellular cell adhesion molecule-2 ou CD102
JAM-4: Junctional adhesion molecule-4
JAM-A: Junctional adhesion molecule-A
JAM-B: Junctional adhesion molecule-B
JAM- C: Junctional adhesion molecule- C
JAMs: Junctional adhesion molecules
LECAM-1: Lectin-like cell adhesion molecule-1 ou CD62L Mel-14
LFA-1: Lymphocyte function associated 1 ou CD11b/CD18 ou Mac-1 macrophage antigen-1
Mac-1: Macrophage antigen-1
NF-kappa β: Nuclear factor kappa B
NO: Óxido nítrico
PECAM-1: Platelet endothelial cell adhesion molecule-1 ou CD31
P-selectina: Granule membrane protein 14 ou selectina CD62P ou GMP140
PSGL-1: Selectina ligante-1
VAP-1: Vascular adhesion protein-1
VCAM-1: Vascular cell adhesion molecule-1 ou CD106
Desde o início até sua resolução, a resposta inflamatória é caracterizada pela infiltração sucessiva dos diferentes subtipos de células brancas. A mobilização dos leucócitos dos compartimentos de reserva e seus recrutamentos sequenciais no curso da resposta inflamatória – neutrófilo seguido pelas células mononucleares e eosinófilos – são processos complexos e vitais para eficiência da imunidade inata e adaptativa. Interferências nestes eventos acarretam prejuízos sérios à instalação, desenvolvimento e resolução de processos inflamatórios de diferentes origens.
A presença de leucócitos no tecido extravascular em resposta a uma lesão foi associada pela primeira vez no final do século XIX por Dutrochet (1824), Addison (1843) e Waller (1846), e este último também correlacionou o evento à formação de exsudato purulento. Subsequentemente, Cohnheim (1867) descreveu que as bases fundamentais para a resposta tecidual à injúria dependiam da presença de leucócitos e da formação do exsudato inflamatório, sendo que alterações na parede dos vasos da microcirculação próximos à área de lesão promoviam as interações celulares necessárias para a formação do exsudato purulento e a resposta inflamatória. Desta forma, foi descrita a primeira evidência da importância da interação dos leucócitos com a parede vascular, ou seja, a interação leucócito-endotélio, uma vez que a célula endotelial reveste a parte luminal dos vasos sanguíneos. Vinte e seis anos mais tarde, o brilhante Metchnikoff e seus discípulos postularam que o processo inflamatório é dependente da capacidade de leucócitos migrarem no tecido extravascular em direção ao foco de lesão, em resposta à ação de agentes químicos secretados na área inflamada. Assim, os primeiros conceitos da diapedese e da quimiotaxia começaram a ser descritos. Desde então, os mecanismos que regem as interações dos leucócitos em cada etapa do processo de mobilização do sangue para o tecido inflamado têm sido alvos de investigações intensas para a compreensão dos mecanismos da inflamação e para a identificação de alvos específicos para a terapêutica.
A mobilização dos leucócitos circulantes inicia-se com a mudança na localização das células na corrente sanguínea. Em condições normais de fluxo, os leucócitos circulam no centro da corrente sanguínea e suas interações com a parede vascular são muito pequenas, com exceções do pool periférico de leucócitos aderidos à parede dos vasos da microcirculação, principalmente pulmonar, e de linfocitos no processo de recirculação, os quais interagem com células endoteliais especializadas de tecidos linfoides. A posição central dos leucócitos na corrente sanguínea é dependente de seu maior tamanho em relação aos demais componentes sanguíneos. No entanto, na vigência de um processo inflamatório, a posição das células nos vasos da microcirculação próximos a área inflamada se modifica: os leucócitos passam a circular próximo às paredes dos vasos da microcirculação, enquanto os eritrocitos migram para parte central dos leucócitos. Este fenômeno é dependente, ao menos em parte, de alterações na hemodinâmica microvascular, desencadeadas pela vasodilatação e aumento de permeabilidade vascular, que levam a redução da velocidade do fluxo sanguíneo em capilares e vênulas pós-capilares. Em conjunto, as alterações microvasculares citadas provocam agregação de eritrocitos e redução na sua velocidade na circulação. Os agregados de células vermelhas, denominados de “roleaux”, passam, então, a circular no centro da corrente sanguínea e os leucócitos, na porção mais externa. O comportamento rolling foi a denominação conferida ao contato inicial dos leucócitos ao endotélio, pela habilidade dos leucócitos deslizarem sobre a parede vascular com uma velocidade menor que o fluxo sangüíneo e aderirem, momentaneamente, à célula endotelial. Com o progredir do processo inflamatório, o tempo e a extensão das interações dos leucócitos à parede vascular aumentam, desencadeando a firme adesão dos leucócitos ao endotélio. Estes eventos, denominados, em conjunto de interação leucócito-endotélio, são os iniciais e fundamentais para a subsequente diapedese e migração direcionada (quimiotaxia e haptotaxia) dos leucócitos no interstício extravascular adjacente.
As bases físicas descritas que influenciam a interação leucócito-endotélio não são suficientes para explicar o fenômeno. A complexidade do processo está centrada na habilidade dos leucócitos e da célula endotelial responderem a uma variedade de mediadores químicos, secretados ou gerados em decorrência da resposta inflamatória. A ligação dos mediadores químicos a receptores de membrana ou intracelulares, ou a ativação direta de segundos mensageiros, nos leucócitos, nas células da parede vascular e do interstício, promove alterações funcionais, bioquímicas e morfológicas necessárias para a interação leucócito-endotélio e para a diapedese. Neste contexto, alguns mediadores químicos induzem a expressão e/ou ativação de receptores glicoprotéicos nas superfícies celulares, que interagem entre si ou com componentes das membranas celulares, mediando a interação celular, evento este central no processo (Quadro 7.1). Pela habilidade de promoverem adesão celular, estes receptores foram inicialmente denominados moléculas de adesão, nomenclatura que permanece até o momento.
Δ-ALA: Ácido delta-levulínico
ABC: Transportadora mitocondrial (ATP-binding cassete)
AngII: Angiotensina II
ATP: Adenosina trifosfato ou trifosfato de adenosina (Adenosine triphosphate)
BFU-E: Unidade formadora de colonia da linhagem eritrocítica (Unit forming burst erythroid series)
BLC: Proteína do linfoma de células B (B-cell lymphoma protein)
CFU-E: Unidade formadora de colonia da linhagem eritrocítica (Unity colony formating of erythroid series)
CD: Determinantes de cluster (Cluster determinants)
CO: Monóxido de carbono
CO2: Dióxido de carbono
Dcyt b: Citocromo b duodenal
DMT1: Transportador de metal divalente (Divalent metal carrier 1)
DPG: Difosfoglicerato
EPO: Eritropoetina
FOG-1: Amigo da proteína GATA 1 (Friend of GATA protein 1)
GATA 1: Fator de transcrição da globina-tipo 1 (Globin transcription factor 1)
GPA: Glicorofina A
GSH: Glutationa
GSSG: Glutationa oxidada
Hb: Hemoglobina
IGF1: Fator-1 de crescimento semelhante à insulina (Insulin-Like Growth Factor-1)
IL: Interleucina
IRE: Elementos responsivos ao ferro (Iron responsive elements)
IRP: Proteínas regulatórias de ferro (Iron regulatory proteins)
NAD+: Dinucleótido de nicotinamida e adenina (Nicotinamide adenine dinucleotide)
NADP+: Fosfato de dinucleótido de adenina nicotinamida ( Nicotinamide adenine dinucleotide phosphate)
NADPH oxidase: Fosfato de dinucleótido de adenina nicotinamida oxidase (Adenine dinucleotide phosphate nicotinamide oxidase)
PKF: Fosfofrutoquinase (Phosphofructokinase)
PU.1: Spi-1 Proto-Oncogene
RNAm: RNA mensageiro
SCF: Fator de crescimento de célula-tronco (Stem cell factor)
T3: Tri-iodotiroxina
T4: Tiroxina
A estrutura básica do sistema eritropoetico é constituída por quatro compartimentos fundamentais: (i) o de células pluripotentes, (ii) o de células indiferenciadas comissionadas , (iii) o de precursores eritroides diferenciados (iv) e dos eritrocitos em circulação. O primeiro compartimento é comum a toda hematopoese, como já comentado, tanto o primeiro compartimento como os das células indiferenciadas comissionadas não possuem uma caracterização morfológica própria. (http://www.bloodjournal.org/content/bloodjournal/124/16/2479/F1.large.jpg)
O termo eritron é empregado para designar a população combinada dos eritrocitos e de seus precursores, no sangue, na medula óssea e em sítios intravasculares. O termo enfatiza a unidade funcional dos eritrocitos e de seus precursores. Estima-se que, para um volume sanguíneo aproximado de 5.000 mL de sangue de um indivíduo adulto e normal, cerca de 3.000 mL são de plasma e 2.000 mL são constituídos pelos glóbulos vermelhos que formam o volume globular normal dos eritrocitos em circulação. Sabendo-se que a hemácia apresenta em sua constituição 60% de água e 40% de parte sólida, sendo esta constituída principalmente pela hemoglobina (90%), teremos em um volume globular de 2.000 mL e 800 g somente de hemoglobina.
8.1.2
Eritropoese é o processo fisiológico de produção de hemácias na medula óssea. Esse processo compreende o comprometimento de células-tronco pluripotente com a diferenciação eritroide, sendo que esse processo pode ser dividido basicamente em duas fases: fase inicial do processo independente de eritropoetina (EPO) e fase final dependente de EPO (http://www.ncbi.nlm.nih.gov/pubmed/25887776).
Fisiologicamente o processo de produção de hemácias é constante, dessa forma, devem existir mecanismos de controle pelo qual essa produção seja regulada. O processo da eritropoese envolve o comprometimento da célula-tronco pela linhagem eritroide, e um mecanismo de proliferação e maturação eritroide até a formação dos eritrocitos.
Atualmente grande parte dos mecanismos envolvidos nesse processo são conhecidos, principalmente os mecanismos de controle das fases finais. Assim, sob ação de elevadas concentrações de EPO e outros fatores de crescimento como a interleucina 3 (IL-3) e o fator de células-tronco (SCF), células comprometidas com a linhagem eritroide são formadas.
As células mais primitivas comprometidas com a linhagem eritroide foram denominadas unidade formadora eritroide-explosiva (burst-forming unit erythroid, BFU-E). Sob o aspecto morfológico essa célula com característica blástica não pode ser identificada, mas em testes in vitro essas células são funcionalmente detectáveis por sua capacidade de formar colonias de eritroblastos (Figura 8.1).
BFU-E: é a célula progenitora, mais imatura, da linhagem eritroide, estando mais intimamente relacionada com a célula-tronco hematopoética multipotente, indicado pelo seu tamanho celular, densidade flutuante e porcentagem relativamente baixa de síntese ativa de DNA. Morfologicamente, a BFU-E possui característica de uma célula blástica, com citoplasma basofílico, cromatina nuclear delicada, alta relação núcleo/citoplasma e nucléolos visíveis. As BFU-E são células que apresentam alta densidade de receptores para IL-3 e pouca densidade para receptores de EPO Testes in vitro mostram que colonias de BFU-E conseguem sobreviver na ausência de EPO, mas não na ausência de IL-3. Mostrando, dessa maneira, que o estágio inicial de diferenciação e proliferação da BFU-E não é dependente de EPO, assim a BFU-E é considerada a célula mais jovem da linhagem eritroide e a progenitora da colony-forming unit erythroid, CFU-E. (https://link.springer.com/protocol/10.1007/978-1-4939-7428)()
Primavera Borelli
ANP: Peptídeo natriurético atrial (Atrial natriuretic peptide)
APC: Células apresentadoras de antígenos (Antigen-presenting cells)
ATP: Adenosina trifosfato ou trifosfato de adenosina (Adenosine triphosphate)
C/EBPα : C/ Potencializador da ligação a proteínas alfa l (Ccaat Enhancer Binding Proteins alpha ou cytosine-cytosine-adenosine-adenosine-thymidine Enhancer Binding Proteins alpha)
C/EBPε: C/ Potencializador da ligação a proteínas épsilon (Enhancer binding proteins epsilon)
C3a: Fração C3a do complemento
C3b: Fração C3b do complemento
C3bi: Fração C3bi do complemento
C3e: Fração do complemento C3e
CCL2: Proteína quimioatraente de monócitos-1.[(Chemokine (C- C motif) ligand 2 ou monocyte chemoatractant protein-1 MIP-1 ou MCP-1)]
CCL5: Ligante 5 de quimiocina (motivo C-C) ou RANTES [(Chemokine (C- C motif) ligand 5 or RANTES)]
CCL7: Proteína quimioatraente de monócitos-8 [(Chemokine (C- C motif) ligand 7 ou monocyte chemoatractant protein-8 or MCP-8)]
CCL11: Proteína quimiotática de eosinófilos ou Eotaxina (C- C motif chemokine 11)
CCL13: Proteína quimioatraente de monócitos-4 [(Chemokine (C- C motif) ligand 13 ou monocyte chemoatractant protein-4 MIP-4 ou MCP-4 )]
CCR1: Chemokine (C- C motif) receptor 1 ou CD191
CCR2: Receptor 1 de quimiocina [(motivo C-C) (Chemokine (C- C motif) receptor 2)]
CCR3: Receptor de eotaxina ou CCL11 [(Chemokine (C- C motif) receptor 3)]
CD14: Cluster de diferenciação 14. Receptor de LPS (lipopolissacarídeo)
CD45RA: Receptor do cluster de diferenciação 45
CFL1: Cofilina 1 não muscular (n-cofilin-1 non-muscle)
CFU-G: Unidade formadora de células (ou colonias) granulocíticas (Colony formating units of granulocitic serie)
CFU-GEMM: Unidade formadora de células (ou colonias) granulocíticas-eritroides-megacariocíticas-monocíticas (Colony formating units of granulocytic-erythrocytic-megakaryocytic and monocytic series)
CFU-GM: Unidade formadora de células (ou colonias) granulo-monocíticas (Unity colony formating of granulocytic and monocytic series)
CFU-M: Unidade formadora de células (ou colonias) monocíticas-macrofágicas (Unity colony formatting of monocytic serie)
CXCL12: Fator 1 derivado de célula estromal (Chemokine, Cxc Motif, Ligand 12 ou Pre-B cell growth-stimulating factor or SDF-1: stromal cell-derived factor 1)
CXCR1: C-x-C receptor de quimiocina tipo 1 (C-X- C chemokine receptor type 1)
CXCR2: C-x-C receptor de quimiocina tipo 2 (C-X- C chemokine receptor type 2)
CXCR3: C-x-C receptor de quimiocina tipo 3 (C-X- C chemokine receptor type 3)
CXCR4: C-x-C receptor de quimiocina tipo 4 (C-X- C chemokine receptor type 4)
CXCR-7: C-x-C receptor de quimiocina tipo 7 (C-X- C chemokine receptor type 7 )
DF32 P: Di-isofluorfosfato
ECP: Proteína catiônica eosinofílica (Eosinophil cationic protein ou RNase 3)
EDN: Neurotoxina derivada do eosinófilo (Eosinophil derived neuortoxin ou RNase-z)
EPX: Peroxidase derivada de eosinófilo (Eosinophil derived peroxidase)
FcRII: Receptor tipo RII de Fc de imunoglobulina (Immunoglobulin Fc-type RII receptor)
FcRIII: Receptor tipo RIII de Fc de imunoglobulina (Immunoglobulin Fc-type RIII receptor)
Fc γ R: Receptor de Fc de imunoglobulina gama (Gamma immunoglobulin Fc receptor)
Fc ε R: Receptor de Fc de imunoglobulina E (Epsolon immunoglobulin Fc receptor)
fMLP: Formil-metionil-leucilfenilalaninal peptídeo (N-Formylmethionyl-leucyl-phenylalanine or N-formyl-metleu-phe)
FOG-1: Amigo da proteína GATA 1 (Friend of GATA protein 1)
GATA 1: Fator de transcrição da globina-tipo 1 (Globin transcription factor 1)
GATA 2: Fator de transcrição da globina-tipo 2 (Globin transcription factor 2)
GATA 3: Fator de transcrição da globina-tipo 3 (Globin transcription factor 3)
G- CSF: Fator de crescimento de células granulocíticas (Granulocyte colony-stimulating factor)
GM-CSF: Fator de crescimento de células granulo-monocíticas (Granulocyte-macrophage colony-stimulating factor)
GSH: Glutationa reduzida
GSH-Px: Glutationa peroxidase
GSH-R: Glutationa redutase
3H-TdR: Timidina triciada
5-HPETE: Ácido 5-hidroperoxi-6,8,11,14-eicosatetraenóico (5-hydroperoxy-6,8,11,14-eicosatetraenoic acid)
HOXB4: Proteína homebox HOx-4 (Homeobox protein Hox-B4)
HRF: Fator liberador de histamina (Histamine release factor)
ICAM-1: Molécula de adesão intercelular-1 (Intercellular adhesion molecule 1)
ICSBP: Proteína de ligação de sequência de consenso de interferon (Interferon consensus sequence-binding protein)
IFN α : Interferon alfa
IFNβ: Interferon beta
IFN γ : Interferon gama
IgD: Imunoglobulina D
IgG: Imunoglobulina G
IGF-1: Fator de crescimento semelhante a insulina do tipo 1 (Insulin-like growth factor-1)
Ikaros ou IKZF1: Dedo de zinco da família IKAROS 1 (IKAROS family zinc finger 1)
AGM: Aorta-gônada-mesonefro
ANP: Peptídeo natriurético atrial (Atrial natriuretic peptide)
APC: Células apresentadoras de antígenos (Antigen-presenting cells)
ATP: Adenosina trifosfato ou trifosfato de adenosina (Adenosine triphosphate)
BFU E: Unidade formadora de colonia da linhagem eritrocítica (Unit forming burst erythrocytic series)
CFU-G: Unidade formadora de colonia da linhagem granulocítica (Unity colony formating of granulocytic series)
CFU-GEMM: Unidade formadora de colonia das linhagens granulocítica, eritrocítica, megacariocítica e monocítica ou mono-macrofágica (Colony formating units of granulocytic, erythrocytic, megakaryocytic and monocytic series)
CFU-GM: Unidade formadora de colonia das linhagens granulocítica e monocítica ou mono-macrofágica (Unity colony formating of granulocytic and monocytic series)
CFU-M: Unidade formadora de colonia da linhagem monocítica ou mono-macrofágica (Unity colony formating of monocytic series)
CFU-Meg: Unidade formadora de colonia da linhagem megacariocítica (Unity colony formating of megakaryocytic series)
CFU-S: Unidade formadora de colonia do baço (Colony formating unit-spleen)
CTH: Célula-tronco hemopoética
C5a: Fração do Complemento C5a
CD: Células Dendríticas (Dendritic cells)
CD: Determinantes de cluster (Cluster determinants)
CD14: Cluster de diferenciação 14. Receptor de LPS (Lipopolissacarídeo)
DC: Células Dendríticas (Dendritic cells)
FcγRIIIa: Receptor III da fração Fc da imunoglobulina gama
Fc ε R: Receptor de Fc de imunoglobulina E (Epsolon immunoglobulin Fc receptor)
FGF: Fator de crescimento de fibroblasto (Factor growth fibroblast)
GATA 1: Fator de transcrição da globina-tipo 1 (Globin transcription factor 1)
GATA 3: Fator de transcrição da globina-tipo 3 (Globin transcription factor 3)
G- CSF: Fator de crescimento de células granulocíticas (Granulocyte colony-stimulating factor)
GM- CSF: Fator estimulador de colonia granulo-monocíticas (Granulocitic-monocitic stimulating factor)
GM- CSF: Fator estimulador de colonias granulo-monocíticas (Granulocitic-monocitic stimulating factor)
H2O2: Peróxido de hidrogênio
ICAM-1: Molécula de adesão intercelular-1 (Intercellular Adhesion Molecule 1)
ICSBP: Proteína de ligação de sequência de consenso de interferon (Interferon consensus sequence-binding protein)
IFN γ : Interferon gama
IGF -1: Fator de crescimento semelhante à insulina (Insulin-like growth fator)
IL-1: Interleucina 1
IL-2: Interleucina 2
IL-3: Interleucina 3
IL-4: Interleucina 4
IL-5: Interleucina 5
IL-6: Interleucina 6
IL-8: Interleucina 8 ou CXCL8
IL-9: Interleucina 9
IL-11: Interleucina 11
LFA-1: A ntígeno associado à função de linfocitos tipo 1 (Lymphocyte function-associated antigen 1)
LPS: Lipopolissacarídeo
LTB 4: Leucotrieno B4
LTC 4: Leucotrieno C 4
MCP-4: Proteína quimioatraente de monócitos-4 ou CCL13 [Chemokine (C-C motif) ligand 13 (Monocyte chemoatractant protein-4 ou MIP-4 OU MCP-4)]
MCP-8: MCP-8 ou Chemokine (C-C motif) ligand 7 –CCL 7. Proteína quimioatraente de monócitos-8 (Monocyte chemoatractant protein-8)
M- CSF: Fator de crescimento de célula (ou colonia) monocíticas (Monocytic colony-stimulating factor)
MEC: Matriz extra celular
MHC I: Complexo principal de histocompatibilidade classe I (Major histocompatibility complex class I)
MHC II: Complexo principal de histocompatibilidade classe II (Major histocompatibility complex class II)
NAD+: Dinucleótido de nicotinamida e adenina (Nicotinamide adenine dinucleotide)
NADP+: Fosfato de dinucleotídeo de adenina nicotinamida (Nicotinamide adenine dinucleotide phosphate)
NADPH Oxidase: Fosfato de dinucleotídeo de adenina nicotinamida oxidase (Nicotinamide adenine dinucleotide phosphate-oxidase)
NK: Células naturais matadoras (Natural killer cells)
NO: Óxido nítrico
- O2: Ânion superóxido
PAR-1: Receptor 1 ativado por proteases-1 (Protein activated receptor-1)
PAS: Ácido periódico de Schiff (Periodic Schiff acid)
PECAM: Molécula de adesão de células endoteliais de plaquetas (Platelet endotelial cell adhesion molecule)
PGD2: Prostaglandina D2
PGG2: Prostaglandina G2
PGH2: Prostaglandina H2
BCR: Receptor de célula B (B cell receptor)
CCL19: Ligante de quimiocina 19 (C- C Motif Chemokine Ligand 19)
CCL21: Ligante de quimiocina 21 ou CCR7 ligante (C- C Motif Chemokine Ligand 21)
CD10: Cluster de diferenciação 10 (Differentiation cluster 10)
CD19: Cluster de diferenciação 19 (Differentiation cluster 19)
CD154 ou CD40L: Cluster de diferenciação ligante 154 (Differentiation cluster 154 ligand )
CD40L ou CD154: Cluster de diferenciação ligante 40 (Differentiation cluster ligand 40)
CTL: Linfocitos T citotóxicos (Cytotoxic T lymphocytes)
CTMs: Células-tronco mesenquimais
CXCL13: C-x-C receptor de quimiocina tipo 13 (C-X- C f Chemokine Ligand 13 ou CXCR5)
CXCR5: C-x-C receptor de quimiocina tipo 5 (C-X- C chemokine receptor type 5)
CXCR-7: C-x-C receptor de quimiocina tipo 7 (C-X- C chemokine receptor type 7 )
FoxP3: Forkhead box P3 ou scurfin
G- CSF: Fator de crescimento de células granulocíticas (Granulocyte colony-stimulating factor)
IFN: Interferon
IL: Interleucina
IL-1: Interleucina 1
IL-5: Interleucina 5
IL-6: Interleucina 6
IL-10: Interleucina 10
IL-13: Interleucina 13
MHC I: Complexo principal de histocompatibilidade classe I (Major histocompatibility complex class I)
MHC II: Complexo principal de histocompatibilidade classe II (Major histocompatibility complex class II )
RAGs (RAG 1 e 2): Genes ativadores de recombinação (Recombination-activating genes)
SCF: Fator de crescimento de célula-tronco (Stem cell factor)
SDF-1: Fator 1 derivado de célula estromal ou CXCL12 ou PRE-B fator de crescimento de Célula pre-B (Pre-B Cell growth-stimulating fator)
TCR: Receptor de células T
TdT: Enzima transferase deoxinucleotidil terminal (Terminal deoxynucleotide transferase enzyme)
TGFβ: Fator de crescimento (tecidual) transformador β (Transforming growth factor β)
Th1: Linfocito T auxiliar (Type 1 T helper linfocyte)
Th2: Linfocitos T auxiliadores 2 (T helper lymphocytes 2)
TNF a : Fator de necrose tumoral alfa (Tumor necrosis factor alpha)
Evolutivamente, os linfocitos surgiram juntamente com os primeiros vertebrados. O aparecimento dessas células com capacidade de resposta específica a uma diversidade quase infindável de antígenos e, mais que isso, de manter memória imunológica e responder mais vigorosamente em uma próxima exposição, foi essencial por superar as restrições do sistema imune inato e permitir que os organismos respondessem a um número mais amplo de patógenos e à pressão de seu contínuo processo evolutivo e o desenvolvimento de mecanismos de evasão.
Os linfocitos constituem de 20 a 30% dos leucócitos do sangue em humanos adultos. As diferentes classes de linfocitos T, linfocitos B e células NK (Natural Killer ou assassinas naturais) são formadas a partir de um precursor comum presente na medula óssea, a célula-tronco hemopoetica, porém diferem fenotípica e funcionalmente. Os linfocitos B e T possuem receptores específicos para antígenos, respectivamente, o BCR (receptor de células B) e o TCR (receptor de células T). Esses receptores possuem um repertório de reconhecimento de moléculas muito amplo, o qual decorre, dentre outros mecanismos, do processo combinatório de vários segmentos gênicos que ocorre durante o processo de maturação dessas células. Os linfocitos B são importantes, principalmente, na resposta a patógenos extracelulares. Essas células produzem anticorpos, proteínas com capacidade de ligação e neutralização de toxinas e microrganismos. Além disso, essas moléculas opsonizam microrganismos para a posterior fagocitose e destruição por fagócitos e células NK e ativam a via clássica do Sistema Complemento. Os linfocitos T são particularmente importantes nas respostas a patógenos intracelulares. Além disso, atuam no processo de ativação de linfocitos B no interior dos órgãos linfoides secundários para a produção de anticorpos específicos. As células NK, diferentemente dos linfocitos T e B, não apresentam receptores com alta diversidade responsáveis pelo reconhecimento de antígenos e estão envolvidas na resposta a patógenos intracelulares, particularmente, vírus e na destruição de células tumorais. Neste capítulo, inicialmente, serão abordados o processo de produção e as características e funções das principais classes de linfocitos. Posteriormente, serão discutidos os mecanismos regulatórios da linfopoese e, por fim, a caracterização morfológica e imunofenotípica dessas células. Populações linfoides atípicas, como os linfocitos TNK, células B1, linfocitos intraepiteliais e linfocitos B da zona marginal, as quais, juntamente com as células NK, possuem características intermediárias entre células da imunidade inata e adaptativa, serão descritas brevemente ao final do presente capítulo.
Os precursores linfoides são produzidos na medula óssea (Figura 11.1) a partir de um precursor comum a todas as células do sangue, a célula-tronco hemopoetica. Essa célula, que tem capacidade de autorrenovação e geração das diferentes linhagens celulares hemopoeticas, recebe a ação de fatores solúveis (ex., citocinas, fatores de crescimento e nutrientes) e insolúveis (ex., contato célula-célula e célula-matriz extracelular), prolifera-se e se diferencia dando origem aos precursores comuns mieloides (os quais dão origem a eritrocitos, plaquetas, granulócitos, monócitos e grande parte das células dendríticas) e linfoides. A diferenciação celular, conforme já ressaltado em capítulos anteriores, consiste no processo durante no qual as células se tornam especializadas e aptas a desempenharem as suas funções de maneira eficaz. Nesse processo ocorrem mudanças na expressão gênica decorrentes da ativação/inativação de conjuntos de fatores de transcrição e/ou modificações epigenéticas, que resultam em alterações morfológicas e funcionais nas células. Pode-se identificar, com base na expressão de proteínas de membrana, conjunto de fatores de transcrição ativados/inativados, capacidade funcional e características morfológicas, diversas etapas maturativas entre a célula-tronco hemopoetica e o linfocitos maduro, conforme discutido ao longo do capítulo.
A hemopoese no período embrionário se desenvolve em diferentes locais, conforme destacado em capítulos anteriores, incluindo saco embrionário, fígado, baço e, finalmente, medula óssea. Em particular, a linfopoese tem início no saco embrionário e depois se localiza no fígado, fase esta que se estende da 9ª à 24ª semana de gestação.
Primavera Borelli
AMP: Monofosfato de adenina (Adenosine monophosphate)
AMPc: Monofosfato de adenina cíclico (Cyclic adenosine monophosphate)
BFU-E: Unidades (primitivas) formadoras de células eritroides (Burst-Forming Unit-Erythroid )
CFU-G: Unidades formadoras de células granulocíticas (Unity colony formating of granulocytic series)
CFU-GEMM: Unidade formadora de células granulocíticas-eritroides-macrofágicas-megacariocítica (Colony forming unit – Granulocyte-Erythroid-Makrophage-Megakaryocyte)
CFU-GM: Unidades formadoras de células granulo-monocíticas (Unity colony formating of granulocytic and monocytic series)
GM-CSF: Fator de crescimento de células granulo-monocíticas (Granulocyte-macrophage colony-stimulating factor)
HETE: Ácido hidroxieicosatetranoico (Hydroxyeicosatetraenoic acid )
HPA: Antígenos plaquetários humanos (Human Platelet Antigens)
IL-3: Interleucina 3
IL-5: Interleucina 5
IL-6: Interleucina 6
IL-11: Interleucina 11
IMP: Monofosfato de inosina (IMP) (Inosine monophosphate)
Meg-CFU: Unidades formadoras de células megacariocíticas (Unity colony formating of megacariocytic series)
OCS: Sistema canalicular aberto ligado à superfície (Open canalicular system)
PGD: Endoperóxido de prostaglandina série D (Prostaglandin endoperoxides)
PGE2: Endoperóxido de prostaglandina série E2 (Prostaglandin endoperoxides)
PGF: Endoperóxido de prostaglandina série F (Prostaglandin endoperoxides)
SDF-1: Fator Derivado de Célula Estromal (Stromal derived fator -1)
SDM: Sistema tubular denso
SDS: Sistema de Demarcação de Membrana
TPO: Trombopoietina (Thrombopoietin)
TXA 2: Tromboxana A 2
TXB2: Tromboxana B2
Talvez devido ao seu pequeno tamanho (cerca de 2µ) e à sua morfologia, as plaquetas foram as últimas células sanguíneas a ser descritas. Provavelmente William Hewson (1739-1774) tenha sido o primeiro a observar as plaquetas, mas não as descreveu. No século XIX, diversos cientistas relatam a presença de corpúsculos menores e diferentes das hemácias e dos leucócitos no sangue. O médico francês Alfred Donné (1801-1878) é considerado o primeiro cientista a mencionar a presença das plaquetas no sangue, embora outros autores como o médico inglês George Gulliver (1804-1882) e Willian Addison também a tenham descrito em 1841. Em 1845, o anatomista Friederich Arnold (1803-1890) foi o primeiro a desenhar as plaquetas. Quanto à função plaquetária, os primeiros relatos são de Gustav Zimmermann, médico alemão, que descreveu a presença de “bilhões de certos corpúsculos incolores que tendem a se agrupar [...]” e que ele denominou “corpos elementares”. Em 1873, Edme Felix Alfred Vulpian (1826-1887) relatou que esses “corpos incolores” tinham a capacidade de aderir ao vidro e formar agregados. George Hayem (1841-1935) relatou que “no sangue existem pequenos elementos que têm uma tendência a agregar-se e a mudar de forma”, e em 1883 utilizou o termo “plaqueta” para descrever esses elementos. Hayem também descreveu a interação das plaquetas com a fibrina e a sua participação no controle de hemorragias, função essa também descrita por Giulio Bizzozero (1841-1901). Em 1890, Howell utilizou o termo “megacariócito” para descrever as células de grande tamanho observadas na medula óssea. A origem das plaquetas a partir do citoplasma dos megacariócitos foi descrita em 1906 por James Homer Wright (1869-1928). Durante todo o século XX as funções das plaquetas foram continuamente ampliadas, indo muito além da concepção original de que as plaquetas atuavam no controle das hemorragias por meio dos processos de adesão ao endotélio lesado, formando agregados estáveis. Atualmente sabe-se dos processos moleculares que controlam esses mecanismos, esclarecendo-se muito sobre a sua participação no controle das hemorragias e tromboses e no processo inflamatório (para saber mais, acesse http://www.bloodjournal.org/content/111/3/981.full-text.pdf+html).
Em uma extensão sanguínea, as plaquetas aparecem como estruturas azuladas (basofílicas), pequenas e de aspecto irregular. Elas circulam como células anucleadas e de formato discoide cujo diâmetro pode variar de 1,5 a 4 µm. O número normal de plaquetas no sangue periférico varia de 150.000 a 400.000/mm3 na dependência do método utilizado, com vida média na circulação de cerca de 10 dias. Em condições basais, a produção diária em indivíduos adultos é estimada em 1 × 109 plaquetas. Contudo, este número pode aumentar em até 10 vezes ou mais em casos de aumento de demanda (ver Figura 12.1).
As plaquetas são originadas de megacariócitos que no estado final de maturação têm a sua membrana citoplasmática rompida, liberando as plaquetas. Cada megacariócito pode produzir (valores estimados) entre 1.000 e 3.000 plaquetas. O número de megacariócitos na medula óssea é de 0,5-1% das células nucleadas e o número de plaquetas formadas é de 1 × 109/mL.
A linhagem megacariocítica é constituída de células em diversas etapas que seguem um continuum maturativo. Simplificadamente, podemos agrupá-las em quatro etapas morfologicamente reconhecidas. As principais características que distinguem essas etapas são o tamanho celular (em decorrência da endomitose, veja adiante), a relação núcleo/citoplasma, a basofilia ou acidofilia citoplasmática , o número de núcleos e o padrão de condensação da cromatina.
Megacarioblasto: também chamado de megacariócito de primeiro estágio e constitui cerca de 0,5% das células nucleadas da medula óssea. Apresenta-se de tamanho variável, de 8 a 24 µm, de aspecto arredondado e às vezes com certa irregularidade. Geralmente apresenta núcleo único que ocupa praticamente todo o citoplasma. O núcleo, com cromatina frouxa e vários nucléolos, pode ser redondo ou ligeiramente invaginado; o citoplasma é intensamente basófilo, sem organelas visíveis à microscopia óptica. Na microscopia eletrônica podem ser vistos ribossomos, mitocôndrias, granulos e o Complexo de Golgi. Neste estágio, também começa a se desenvolver um sistema citoplasmático de membranas denominado Sistema de Demarcação de Membrana (SDM), que é formado pela invaginação da membrana celular do megacarioblasto e que se constitui em um sistema aberto de comunicação com o meio extravascular. À medida que esse sistema se forma, pequenas áreas do citoplasma do megacarioblasto são separadas e parecem compartimentalizar o citoplasma que, posteriormente, originará pró-plaquetas e que, na etapa de megacariócito IV, originarão as plaquetas (ver Figura 12.2).
ADP: Trifosfato de adenosina
AAT: α1 antitripsina (α1-proteínase inhibitor-PI)
APC: Proteína C ativada
ATIII: Antitrombina III
ATP: Adenosina Trifosfato (Trifosfato de adenosina)
DDAVP: Desmopressina (1-desamino-8-D-arginina vasopressina), VIIIc: Fator VIII coagulante ou Fator anti-hemofílico)
Fator III: Fator tissular ou tecidual da coagulação
FGF: Fator de crescimento de fibroblasto (Factor growth fibroblast)
FT: Fator tissular ou tecidual da coagulação
FWv: Fator de von Willebrand
GM-CSF: Fator de crescimento de células granulo-monocíticas (Granulocitic-Macrophage Colony Stimulate Factor)
GPIa: Glicoproteína Ia da membrana plaquetária
GPIb: Glicoproteína Ib da membrana plaquetária
GPIIa: Glicoproteína IIa da membrana plaquetária
GPIIb: Glicoproteína IIb da membrana plaquetária
IL-6: Interleucina 6
IL-8: Interleucina 8 ou CXCL8
LPS: Lipopolissacarídeo
NO: Óxido nítrico
PAF: Fator de ativação plaquetária
PAI-1: Inibidor do ativador do plasminogênio-1 (Plaminogen acitvator inhibitor-1)
PAI-3: Inibidor do ativador do plasminogênio-3 (Plaminogen acitvator inhibitor-3)
PAR-1: Receptor 1 ativado por proteases-1 (Protein activated receptor-1)
PGH2: Prostaglandina H2
PGI2: Prostaciclina PGI2
PKC: Proteína quinase C (Protein kinase C)
TFPI: Inibidor da via do fator tecidual (Tissue factor pathway inibhitor)
TFPI α : Inibidor da fibrinólise ativado pela trombina alfa (Thrombin activatable fibrinolysis inhibitor alfa)
TFPIβ: Inibidor da fibrinólise ativado pela trombina beta (Thrombin activatable fibrinolysis inhibitor beta)
TNF α : Fator de necrose tumoral alfa (Tumor necrosis factor alfa)
t-PA: Fator tecidual ativador do plasminogênio (Plasminogen activator tecidual )
u-PA: Ativador do plasminogênio tipo uroquinase (Plasminogen activator type-1)
u-PAR: Receptor do ativador do plasminogênio tipo uroquinase (Plasminogen activator receptor urokinese type)
ZPI: inibidor de proteína Z (Protein Z dependent protease inhibitor)
Com o desenvolvimento de seres multicelulares, houve a necessidade de formação de um sistema de transporte de nutrientes, gases, células e remoção de metabólitos ou seja, do sistema circulatório. Por causa da crescente complexidade dos organismos, esse sistema evoluiu de aberto para fechado, desenvolvendo um sistema de bombeamento e de manutenção da integridade vascular que impedisse o extravasamento. Nesse contexto é que o sangue flui sob condições de pressão, através dos vasos, em um sistema fechado. Nos mamíferos, duas condições fisiológicas permitem o extravasamento de sangue para o meio extravascular: na menstruação e no parto; qualquer outra situação é considerada patológica. A hemostasia é responsável por manter a integridade vascular, impedindo o sangramento, restaurando o vaso lesado ou impedindo a ativação inadequada da cascata de coagulação que possa resultar em trombose. Para isso, envolve uma série de processos biológicos que englobam o sistema vascular, as plaquetas, o sistema da coagulação e fibrinolítico e, ainda, os mediadores da resposta inflamatória. A hemostasia não é um processo passivo, mas, sim, um conjunto de processos que permite a circulação do sangue no interior do vaso, impedindo o seu extravasamento (hemorragias) ou a formação de coágulos e trombos e reestabelecendo o sistema quando ele é lesado. Quando o sistema hemostático é alterado, uma série de interações é desencadeada, envolvendo fenômenos pró-coagulantes e, simultaneamente, a ativação do sistema fibrinolítico e de reparo vascular, processos esses que são regulados por sistemas de feedback positivos e negativos. A hemostasia envolve uma série de processos que se sobrepõem: vasoconstrição e vasodilatação, adesão das plaquetas, formando o “tampão” plaquetário (ver Capítulo 12), formação de fibrina pela ativação do sistema da coagulação e a remoção da mesma pelo sistema fibrinolítico. Esses fenômenos, associados também aos mediadores da resposta inflamatória, são interligados e interdependentes (Figura 13.1), mas, para fins didáticos, iremos, inicialmente, abordá-los individualmente.
AGH: Antiglobulina humana
DHRN: Doença hemolítica do recém-nascido
GIFT: Teste para anticorpos contra antígenos granulocíticos por imunofluorescência ( granulocyte immunofluorescence test)
HNA: Antígenos neutrofílicos humanos (Human neutrophil antigens)
HPA: Antígenos Plaquetários Humanos (Human platelet antigens)
IgA: Imunoglobulina humana classe A
IgG: Imunoglobulina humana classe G
IgM: Imunoglobulina humana classe M
ISBT: Sociedade Internacional de Transfusão Sanguínea
LISS: Soluções de baixa força iônica
MAIGA: Imobilização de antígenos granulocíticos por anticorpos monoclonais
MAIPA: Imobilização de antígenos plaquetários por anticorpos monoclonais
MHC: Complexo maior de histocompatibilidade (Major Histocompatibility Complex)
PBS: Solução salina tamponada com fosfato
PCR-SSP: Reação em cadeia da polimerase com sequências alelo-específicas
PIFT: Técnica de imunofluorescência plaquetária
SST: Solução salina tamponada
TNAI: Trombocitopenia neonatal aloimune
Imuno-hematologia é a ciência que estuda os antígenos e anticorpos ligados às células sanguíneas (hemácias, plaquetas e leucócitos) e está diretamente relacionada com transfusão de sangue e seus hemocomponentes
Os antígenos presentes nas células sanguíneas podem ser de origem proteica, glicoproteicas ou carboidratos. Estes antígenos são codificados por genes autossômicos levando à produção direta da proteína, no caso de antí-
Noções básicas em imuno-hematologia: antígenos eritrocitários, leucocitários e plaquetários
genos proteicos ou, na produção de enzimas que adicionam carboidratos a substratos específicos, caso de antígenos carboidratos. Estes antígenos, de forma geral, localizam-se na membrana celular das hemácias, plaquetas ou leucócitos. Para que um polimorfismo proteico seja considerado um antígeno eritrocitário, plaquetário ou leucocitário é necessário que o mesmo seja capaz de induzir uma resposta imune, produzindo anticorpos específicos contra o mesmo. A resposta imune provocada por estes antígenos é caracterizada por dois mecanismos distintos:
a) Reconhecimento direto, no qual os linfocitos T auxiliadores do receptor, ao entrarem em contato com as células do doador (como acontece quando um paciente recebe uma transfusão de sangue), interagem diretamente com as células apresentadoras de antígenos do doador, estimulando os linfocitos B do paciente a produzir anticorpos contra estes antígenos. Este tipo de reconhecimento está mais relacionado com a produção de anticorpos leucocitários (linfocitários)
b) Reconhecimento indireto, no qual as células do doador são fagocitadas e processadas pelas células apresentadoras de antígenos do paciente, que, por sua vez, irão apresentar estes antígenos para as suas próprias células T auxiliadoras, para então induzir a produção de anticorpos. Este mecanismo necessita da colaboração das células apresentadoras de antígenos do receptor, diferentemente do método direto, quando a apresentação é feita diretamente pelas células do doador. Esse tipo de sensibilização está mais relacionado com antígenos eritrocitários, plaquetários e neutrofílicos.
Os anticorpos formados inicialmente (após 2 a 12 semanas, em geral) são na grande maioria da classe IgM, sendo gradualmente substituídos por anticorpos da classe IgG (IgG 1 a 4) com fixação (subclasses IgG1 e IgG3) ou não de complemento (subclasses IgG2 e IgG4). Em alguns poucos casos podem surgir anticorpos do tipo IgA. Os anticorpos IgG atravessam a placenta e podem causar doença hemolítica do recém-nascido ou doença hemolítica perinatal. Em alguns casos, como o sistema ABO (discutido com mais detalhes na próxima seção), os anticorpos são considerados “naturais”, pois são produzidos por todos os indivíduos imunocompetentes a partir da idade aproximada de 3 meses. Atualmente acredita-se que estes anticorpos são produzidos devido a estímulos recebidos de carboidratos presentes em algumas bactérias do trato gastrointestinal.
A Sociedade Internacional de Transfusão Sanguínea (ISBT) reconhece, atualmente, a existência de 345 antígenos eritrocitários alocados em 38 sistemas (grupos de antígenos com a mesma localização cromossômica e/ou proteica). No Quadro 14.1 encontram-se todos os sistemas reconhecidos com seus respectivos antígenos e sua localização cromossômica (para maiores detalhes ver: https://www.isbtweb.org/working-parties/red-cell-immunogenetics-and-blood-group-terminology)
AHAI: Anemias hemolíticas autoimunes
ALAS: Enzima α-aminolevulinato sintetase
ARSA: Anemia Refratária com Sideroblastos em Anel
ATP: Adenosina trifosfato
BFU-E: Burst-forming unit erythroid
BRCA1: Breast cancer 1, early onset
BRCA2: Breast cancer 2, early onset
CD: Cluster of differentiation
CFU- C: Célula pluripotente medular
CHCM: Concentração de hemoglobina corpuscular média
CTLF: Capacidade total de ligação ao ferro
EDRF: Fator de relaxamento do endotélio
EH: Esferocitose hereditária
EPO: Eritropoetina
G/E: Relação granulocítica/eritroide
G6PD: Glicose 6 fosfato desidrogenase
G- CSF: Fator de crescimento de colonia granulocítica
GM- CSF: Fator de crescimento de colonia granulocítica e monocítica
GPI: Glicosil-fosfatidil-inositol
GSH: Glutationa
GSSG: Glutationa oxidada
Hb: Hemoglobina
HCM: Hemoglobina corpuscular média
HPN: Hemoglobinúria paroxística noturna
HRI: Inibidor regulado pelo heme
Ht: Hematócrito ou volume hematócrito
Ig: Imunoglobulina
IL: Interleucina
IMC: Índice de massa corporal
INF-γ: Interferon gama
IPR: Índice de produção de reticulócitos
LDL: Proteínas de baixa densidade
LES: Lúpus eritematoso sistêmico.
NAD+: Nicotinamida adenina dinucleótido
NADP+: Nicotinamida adenina dinucleótido fosfato
NADPH: Forma reduzida de NADP+
NO: Óxido nítrico
PK: Piruvato quinase
RDW: Red cell distribution width
SMD: Síndrome mielodisplásica
TNF-α: Fator de necrose tumoral alfa
VCM: Volume corpuscular médio
VHS: Velocidade de hemossedimentação
VLDL: Proteínas de densidade muito baixa
A anemia consiste na deficiência de hemoglobina circulante que pode ser originada de uma deficiência na massa eritrocitária ou por quantidade insuficiente de hemoglobina ou ainda por alteração na molécula da hemoglobina, nos eritrocitos, ocorrendo, assim, prejuízo no transporte de oxigênio para os tecidos e na oxigenação tecidual.
Geralmente, em clínica, a anemia é definida como a redução abaixo dos valores considerados de referência para idade, sexo e altitude do local de residência (espécie animal) da concentração de hemoglobina, com diminuição ou não do número de eritrocitos no sangue periférico.
A anemia, na verdade, é uma síndrome que pode acompanhar várias doenças subjacentes. O processo de oxigenação depende da hemoglobina no eritrocito, do próprio eritrocito além da respiração e da circulação, podendo haver algum tipo de compensação entre esses componentes em situações de lesão discreta. Uma quantidade de hemácias menor do que o normal reduz a capacidade do sangue em carrear oxigênio, ativando uma série de mecanismos compensatórios. As manifestações clínicas da anemia refletem justamente esses ajustamentos associados aos efeitos da hipóxia celular: taquicardia, hiperpneia, aumento do débito cardíaco e aceleração do fluxo sanguíneo. São secundários, principalmente, a redução da viscosidade sanguínea e da resistência vascular periférica.
A quantidade de oxigênio liberada em um tecido por um determinado volume de sangue depende da concentração de hemoglobina, do grau de saturação de oxigênio da hemoglobina, da afinidade da molécula da hemoglobina pelo oxigênio e da tensão de oxigênio no tecido. O número de eritrocitos presente na circulação decorre de um equilíbrio dinâmico entre a produção, distribuição na circulação e a remoção da circulação. A anemia pode ser decorrente de alteração na produção ou na destruição/perda ou de ambas.
A resposta do organismo à anemia depende da rapidez da sua instalação, da sua magnitude, da eficiência dos mecanismos compensatórios e das necessidades de oxigênio do indivíduo. Se a instalação da anemia é gradual, a volemia é mantida e os sinais predominantes são os de hipóxia. Esse indivíduo tolera mais de 50% de redução das hemácias sem grandes consequências entretanto, perdas agudas de cerca de 30% produzem profundas reações, levando à rápida diminuição da volemia e, consequentemente, ao choque.
Quando os valores de hemoglobina são menores do que 7,5 g%, geralmente aparece dispneia de esforço; abaixo de 3 g%, dispneia de repouso; e abaixo de 2,5 g% instala-se insuficiência cardíaca. Aumento do fluxo circulatório é devido à redução da resistência capilar. Como consequência, o fluxo renal é reduzido, conduzindo à retenção de sais e água. A redução do tônus vascular parece ser devida ao fator de relaxamento do endotélio (EDRF) ou ao
Primavera Borelli
CGD: Doença granulomatosa crônica (cronic granulomatous disease)
GCSFr: Receptor do fator de crescimento granulocítico ( granulocytic growth factor receptor)
gp91fox (Nox2): Glicoproteína 91 fox
HOCl: Ácido hipocloroso
LAD: Deficiência de adesão leucocitária (leukocyte adhesion deficiency –)
MPO: Mieloperoxidase
NADPH oxidase: Fosfato de dinucleotídeo de adenina nicotinamida oxidase (adenine dinucleotide phosphate nicotinamide oxidase)
Os leucócitos, ou glóbulos brancos, são células nucleadas cuja produção ocorre, após o nascimento, em seres humanos, na medula óssea. Como já vimos em capítulos anteriores, os leucócitos, assim como os eritrocitos e as plaquetas, derivam da célula-tronco hematológica, também denominada hemocitoblasto. Na medula óssea essa célula primitiva origina precursores morfologicamente indiferenciados da linhagem mieloide (Capítulos 8, 9, 10 e 12) e da linhagem linfoide (Capítulo 11). Sob condições do microambiente medular, tais precursores passam por estágios contínuos de proliferação, diferenciação e maturação, de modo a permitir a formação de células funcionalmente maturas. Sendo assim, na medula óssea são produzidos eritrocitos (Capítulo 8), polimorfonucleares (eosinófilos, basófilos e neutrófilos (Capítulo 9), monócitos (Capítulo 10) e plaquetas (Capítulo 12).
Fisiologicamente, as células maduras ganham o compartimento do sangue circulante, lembrando que a vida média biológica das células sanguíneas na circulação é relativamente curta: 6-7 horas para polimorfonucleares, cerca de 70 h para monócitos, 10 dias para plaquetas, 110-120 dias para eritrocitos e um período que pode ir de semanas a meses para as células linfoides. No curso de sua produção ou do período de tempo em que permanecem na circulação ou durante o processo de recirculação dos linfocitos, os leucócitos podem ter alterações em seu metabolismo em função de processos fisiológicos, mediadores químicos, toxinas ou fármacos com os quais entram em contato. Essas modificações podem alterar tanto o número e distribuição dos leucócitos (veja Capítulo 17) como sua função e morfologia. Algumas delas, embora sejam fisiológicas (outras não), são coletivamente agrupadas como alterações patológicas e, ao longo do texto e dos capítulos posteriores, comentaremos a natureza dessas modificações. Dessa maneira, as alterações que acometem os leucócitos podem ser classificadas como quantitativas, ou seja, leucocitose e leucopenias, e alterações qualitativas e/ou morfológicas. Tanto uma quanto a outra podem ter caráter benigno/reacional ou caráter maligno. Neste capítulo trataremos das alterações morfológicas de natureza não maligna, deixando para o Capítulo 17 as alterações quantitativas de caráter reacional, sendo que as patologias de natureza malignas que compõem as doenças onco-hematológicas serão abordadas na Parte VI deste livro.
As alterações qualitativas e/ou morfológicas podem estar presentes em mais de um tipo celular e uma mesma linhagem pode apresentar mais que uma alteração.
A cariorrexis ou necrobiose (Figura 16.1) é decorrente da morte celular, podendo afetar qualquer um dos leucócitos. Na cariorrexis ocorre a fragmentação do núcleo.
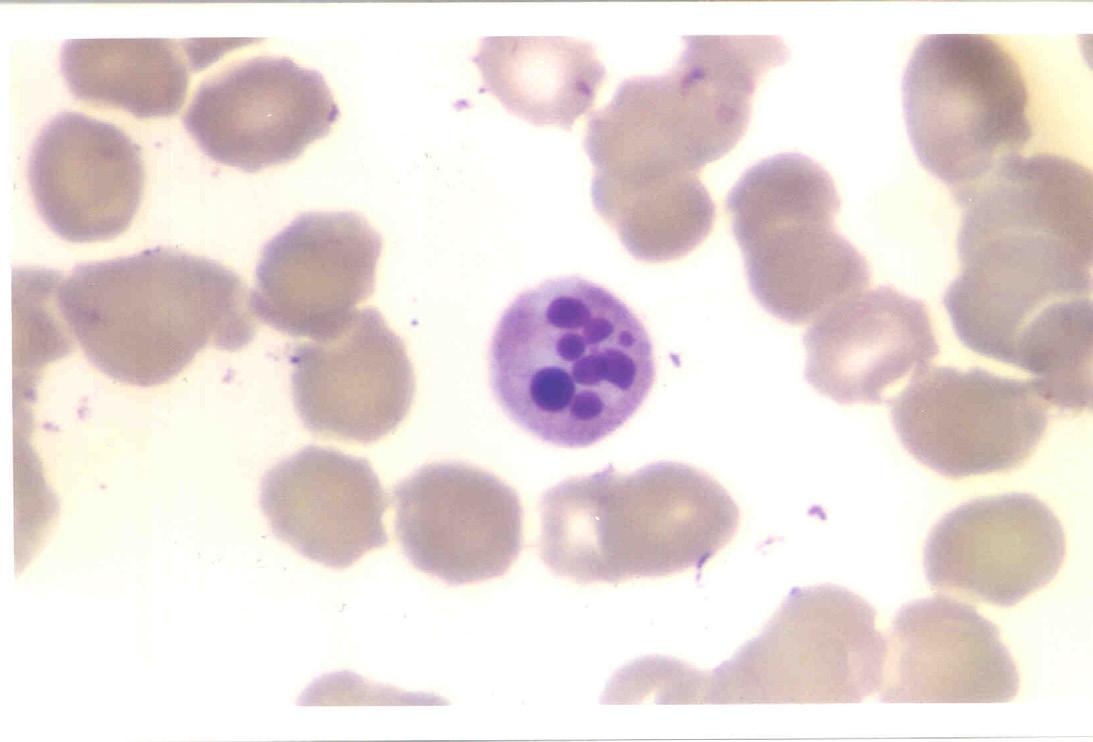
Figura 16.1 – Leucócito granulocítico em cariorrexis em extensão sanguínea feita após sete horas da coleta do material. Observar o núcleo fragmentado. Material: sangue periférico. Coloração: May-Grunwald-Giemsa, modificada por Rosenfeld (10×100). Fotomicrografia: Profª. Dra. Primavera Borelli, Laboratório de Hematologia Clínica e Experimental, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo.
A picnose é uma alteração nuclear na qual a cromatina é extremamente condensada, perdendo a sua estrutura e função (Figuras 16.2 e 16.3) e pode preceder à cariorrexis. De um modo geral, é raro encontrar essas formas in vivo, uma vez que elas são rapidamente retiradas da circulação pelos macrófagos esplênicos. Geralmente, esses achados estão relacionados a problemas técnicos da coleta, tempo e/ou armazenamento da amostra, portanto amostras que apresentem essas alterações devem ser descartadas, solicitando-se uma nova coleta, uma vez que outras alterações quali e quantitativas de qualquer das linhagens sanguíneas podem ocorrer, comprometendo, assim, o resultado e a análise do material.
Figura 16.2 - Leucócito granulocítico em cariorrexis em extensão sanguínea feita após sete horas da coleta do material. Observar o núcleo fragmentado. Material: sangue periférico. Coloração: May-Grunwald-Giemsa, modificado por Rosenfeld (10x100). Fotomicrografia: Profa. Dra. Primavera Borelli, Laboratório de Hematologia Clínica e Experimental, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo.
Primavera Borelli
Além das alterações qualitativas (morfológicas e funcionais), os leucócitos podem apresentar, concomitantemente ou não, alterações em seu número, sendo classificadas como alterações quantitativas, ou seja, leucocitoses e leucopenias. Tanto uma como a outra podem ser de caráter benigno, reacional ou de caráter maligno. Neste capítulo, trataremos das alterações de natureza não maligna, deixando para a Parte VI deste livro as patologias de natureza malignas que compõem as doenças onco-hematológicas (doenças mieloproliferativas, leucemias, linfomas e mielodisplasias) e com as quais se deverá fazer o diagnóstico diferencial.
A produção de células pela medula óssea é contínua e de grande intensidade, sendo produzida, para um indivíduo adulto, uma média de 1011 células/dia/kg de peso. Uma vez na circulação, os leucócitos se distribuem em dois compartimentos dinâmicos: cerca de 50% da população circulante encontram-se distribuídos mais centralmente no vaso (pool circulante) e os outros 50% encontram-se mais próximos à margem dos vasos (pool marginal). Fisiologicamente, os leucócitos saem dos vasos, exercendo, na maioria das vezes, suas funções em sítios extravasculares. Dessa maneira, o número de leucócitos na circulação depende (i) da produção medular e, no caso de linfocitos, também dos órgãos linfoides secundários; (ii) da sua distribuição no sangue circulante; (iii) da sua saída para sítios extravasculares (Figura 17.1), e (iv) da idade, gênero, ciclo circadiano, condição fisiológica, presença de patologias e/ou fármacos (veja item Regulação, Cinética e Mobilização dos Granulócitos no Capítulo 9).
Define-se como leucocitose a situação em que o número de leucócitos ultrapassa o valor de referência para a etnia e a idade do indivíduo (ver Quadro 9.3). A leucocitose pode ser devida ao aumento de uma ou mais linhagens leucocitárias: neutrófilos, eosinófilos, basófilos, linfocitos e monócitos, e essa condição deve ser avaliada e descrita. As leucocitoses podem decorrer de processos agudos ou crônicos, benignos ou reacionais e malignos. No caso das leucocitoses reacionais, o aumento do número de leucócitos é transiente, ou seja, assim que o processo for resolvido, o número de leucócitos retorna aos valores normais. Os mecanismos envolvidos nas leucocitoses são (Figura 17.1) (i) aumento de produção na medula óssea ou, no caso dos linfocitos, dos órgãos linfoides secundários; (ii) por aumento da mobilização do compartimento de células maturas e de células imaturas (desvio à esquerda, Quadro 17.1) para o sangue periférico; (iii) por deslocamento dos leucócitos do pool marginal para o pool circulante e, nesse caso, não há aumento de proliferação, da maturação, nem de mobilização da medula óssea para o sangue periférico, ou, ainda, (iv) por aumento de proliferação e maturação das células sanguíneas por doenças mielo ou linfoproliferativas. Devemos recordar que, na vigência de processos inflamatórios e/ou infecciosos, citocinas (TNFα , IL-1) e fatores de crescimento como G-CSF, GM-CSF, M-CSF, IL-3, IL-6 etc. podem ser produzidos, modificando a cinética de produção e migração dos diferentes tipos de leucócitos.
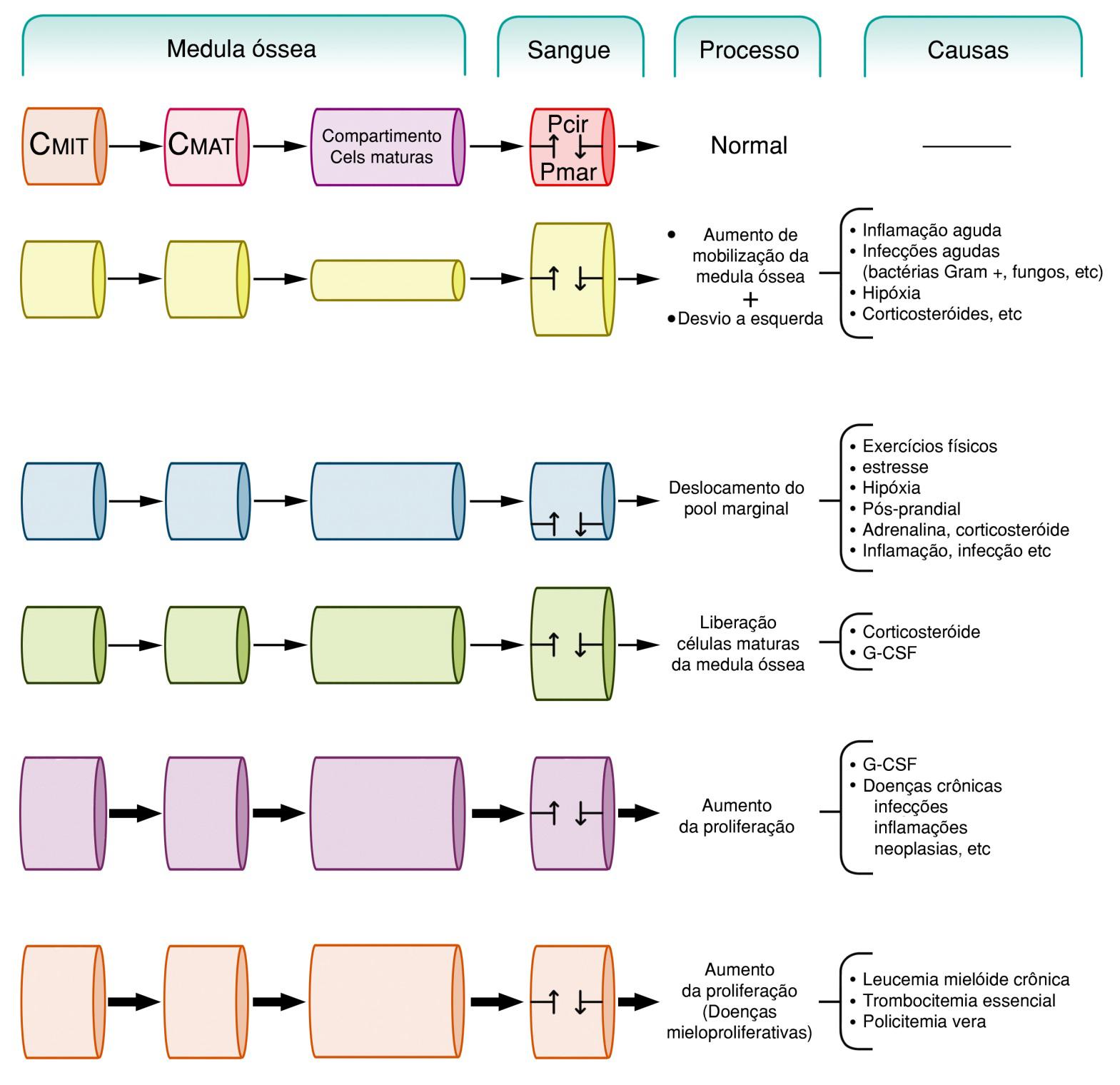
Figura 17.1 - Esquema mostrando os processos que podem levar a uma leucocitose sanguínea especialmente com neutrofilia. Observar que diferentes causas podem induzir a leucocitose por distintos ou não processos. O tamanho dos cilindros indica o tamanho da respectiva população celular. Cmit: compartimento mitótico-células primitivas Cmat: compartimento de proliferação e maturação de células já comprometidas Pcirc: pool circulante de leucócitos Pmar: pool marginal de leucócitos Adaptado de Bagby,C.J. Leukocytosis and leukemoid reactions.Cap 140.2, p.916, IN Cecil Textbook of Medicine, Bennett,J & Plum, F., W.B. Saunders Company, Philadelphia,2223 p,1996
Promielócito neutrófilo: 3%
Mielócito neutrófilo: 7%
Metamielócito neutrófilo: 10%
Bastonete neutrófilo: 14%
Segmentado neutrófilo: 42%
Eosinófilos:0%
Basófilos:0%
Linfocitos:15%
Monocitos:9%
Quadro 17.1 – Exemplo de desvio à esquerda da série neutrofílica com escalonamento maturativo preservado. Os estágios mais imaturos das células estão presentes em menor número que os estágios mais maturas.
Na avaliação do número de leucócitos, é importante para a interpretação do exame ter também a contagem diferencial dos diferentes tipos de leucócitos presentes. Devemos observar que, em algumas condições, pode haver aumento de uma ou mais das linhagens leucocitárias sem que o número total de leucócitos esteja aumentado. Em função dos diferentes tipos celulares, poderemos, então, ter: neutrofilias, eosinofilias, basofilias, linfocitoses e monocitoses.
17.2.1 NEUTROFILIAS
Define-se como neutrofilia a condição em que o número de neutrófilos está acima do valor de referência considerando a idade e a etnia. Recém-nascidos apresentam, fisiologicamente, valores mais elevados de neutrófilos (ver Quadro 9.3).
Primavera Borelli
ADAMTS-13: Desintegrina e metaloprotease com colonias tipo 1 repetida (Disintegrin and metalloprotease with thrombospondi type 1 repeated )
Apaf1: Fator de ativação da protease apoptótica 1 (Apoptotic protease activating factor 1)
Apo-3L: Apoliproteina 3 ligante
Apo-4L: Apoliproteina 4 ligante
Apo-5L: Apoliproteina 5 ligante
CAM: Trombocitopenia amegacariocítica congênita (Congenital amegakaryocytic thrombocytopenia)
CAR: Receptor de adenovírus coxsackie (Cosackie-adenovirus receptor)
CCL: Quimiocina (Chemokine (C- C motify) ligand )
CLEC-2: Família de domínio de lectina tipo C (C-type lectin domain family)
CMV: Citomegalovírus (Cytomegalovirus)
CR: Receptor do complemento (Complement receptor)
CTRUS: Trombocitopenia amegacariocítica congênita com sinostose radio-ulnar (Congenital amegakaryocytic thrombocytopenia whit radio-ulnar synostosis)
CXCR4: C-x-c receptor de quimiocina tipo 4 (C-X- C chemokine receptor type 4)
cyRII: Receptor Fc y RII; GP: glicoproteína (Fc receptor y RII; GP: Glycoprotei)
DC-SIGN: Não integrina de adesão intercelular específica para células dendríticas (Dendritic cell-specific intercelular adhesion molecule-3-grapping non-integrin)
DD: Domínios de morte citoplasmáticos
DEB: Diepoxibutano
DV: Virus da dengue (Dengue virus)
EBV: Virus de Epstein-Barr (Epstein-Barr virus)
EMCV: Virus da encefalomiocardite (Encephalomyocarditis virus)
ERO: Espécies reativas do oxigênio
Fab: Fragmento de ligação ao antígeno (Fragment antigen-binding)
FADD: Dinucleótido de flavina e adenina (Flavin adenine dinucleotide)
Alterações quantitativas, não malignas, das plaquetas: plaquetopenias
FasL: Fas ligante
GATA 1: Fator de transcrição da globina-tipo 1 (Globin transcription factor 1)
G- CSF: Fator de crescimento de células granulocíticas (Granulocyte colony-stimulating factor)
GM-CSF: Fator de crescimento de células granulo-monocíticas (Granulocyte-macrophage colony-stimulating factor)
GPIbα Glicopoteina Ib alfa
HCV: Vírus da hepatite C (Hepatites virus C)
HETE: Hydroxyeicosatetraenoic acid (Ácido hidroxieicosatetranoico)
HGF: Fator de crescimento de hepatócitos
IgA: Imunoglobulina A
IgG: Imunoglobulina G
IgM: Imunoglobulina M
IL-6: Interleucina 6
IL-8: Interleucina 8
MCP-1: Proteína quimioatraente de macrófago (Macrophage chemoattractant protein)
MPTP: Poro de transição de permeabilidade mitocondrial (Mitochondrial permeability transition pore)
OMI/HTRLA2: Proteína A2 requer alta temperatura (High-temperature requirement protein A2)
PAMPS: Padrões moleculares associados a patógenos (Molecular patterns associated with pathogens)
PAR1: Receptor-1 ativado por protease (Protease-activate receptor-1)
PAR2: Receptor-2 ativado por protease (Protease-activate receptor-2)
PAR3: Receptor-3 ativado por protease (Protease-activate receptor-3)
PAR4: Receptor-4 ativado por protease (Protease-activate receptor-4)
Smac: Segundo ativador derivado de mitocôndria de caspases ou diablo ou survivin (Second mitochondria-derived activator of caspases ou diablo ou survivin)
TC: Tempo de coagulação
TPPA: Tempo de protrombina parcial ativada
Como já mencionamos nos capítulos 12 e 13, as plaquetas exercem papel multifuncional, estando envolvidas em diversos processos fisiológicos: na hemostasia, na reparação tecidual e na resposta inflamatória, bem como na fisiopatologia de vários processos como trombose, púrpuras, aterosclerose e metástase tumoral. Na coagulação, as plaquetas têm papel crucial aderem ao endotélio lesado, formando uma barreira física (tampão primário) e, ao agregarem-se, formam uma estrutura mais estável, denominada tampão secundário. Nesse processo, além de estancarem o sangramento em vasos de pequeno calibre (especialmente da microcirculação), participam da ativação da cascata da coagulação via ativação de fator XII pelo fator plaquetário 3, contribuindo para a formação de fibrina sobre as plaquetas e, assim, estabilizando o tampão plaquetário. Alterações no número e/ou na função das plaquetas podem ocasionar doenças hemorrágicas, denominadas púrpuras e processos trombóticos. Neste capítulo, trataremos das plaquetopenias ou trombocitopenias (neste livro usamos indistintamente os dois termos. O consenso do III US National Health and Nutrition Examination Survey (NHANES III) (http://www.cdc.gov/ nchs/nhanes/about_nhanes.htm) considera como plaquetopenias as situações em que o número delas se encontra abaixo de 150.000/mm3 de sangue. Na dependência do número de plaquetas, podemos ter: (i) plaquetopenias discretas entre 100.000 e 150.000 plaquetas/mm3; (ii) plaquetopenias moderadas entre 50.000 e 100.000/mm3; (iii) plaquetopenias severas quando o número de plaquetas está abaixo de 50.000/mm3. A intensidade dos sintomas depende do grau da plaquetopenia que o paciente apresenta, ou seja, quanto menor o número de plaquetas, mais evidentes e mais severos serão os sinais e sintomas, entretanto pacientes com valores de plaquetas entre 100.000 e 150.000/mm3 podem não apresentar sintomatologia. Abaixo de 10.000 plaquetas/mm3 é significativa-
c-kit ligante: Fator de crescimento de célula-tronco (também chamado de fator steel) (Stem cell growth factor)
Fator Steel: Fator de crescimento de célula-tronco (também chamado de c-kit ligante) (Stem cell growth factor)
FL-3: Ligante
FLT3L: Fms ligante relacionado à tirosina quinase 3 (Fms-related tyrosine kinase 3 ligand)
G- CSF: Fator de crescimento de células da linhagem granulocítica (Granulocyte colony-stimulating factor)
GM- CSF: Fator de crescimento de células da linhagem granulo-monocíticas (Granulocitic-macrophage colony stimulate factor)
IFN α : Interferon alfa
IL-1: Interleucina 1
IL-3: Interleucina 3
IL-6: Interleucina 6
IL-11: Interleucina 11
JAK2: Janus quinase 2
MAPK: Proteína quinase ativada por mitógeno (Mitogen activator protein kinase)
MPL: Homólogo celular dos oncogenes do vírus da leucemia mieloproliferativa (Myeloproliferative leucemia virus oncogenes)
PI3K: Quinase do 4,5- fosfatidilinositol 3-fosfato (Phosphatidylinositol-4,5-bisphosphate3-kinase)
PI3K: 3-Fosfatidil inositol quinase (3-Phosphatidyl inositol kinase)
PV: Policitemia vera
RAS: Vírus do sarcoma de rato (Rat sarcoma Vírus)
SCF: Fator estimulante de célula-tronco (Stem cell factor)
SDF-1: Fator 1 derivado de célula estromal (Factor 1 derived from stromal cell)
STAT 5: Transdutor de sinal e ativador de transcrição 5 (Signal transducer and activator of transcription 5)
TE: Trombocitemia essencial
TGFβ: Fator de crescimento transformador de tecido beta (Transforming growth factor beta)
TNF α : Fator de necrose tumoral alfa (Tumor necrosis factor alfa)
TPO: Trombopoietina
não malignas, das plaquetas: plaquetoses
A plaquetose ou trombocitose (usamos indistintamente os dois termos neste livro) é definida quando, em adultos, o número das plaquetas encontra-se acima de 450.000/mm3
No transcorrer do século XX, as funções das plaquetas foram continuamente ampliadas, indo muito além do conceito inicial de que tinham como única função o controle das hemorragias através dos processos de adesão ao endotélio lesado, formando agregados estáveis. Atualmente, sabe-se que as plaquetas participam de diversos outros processos, como a manutenção da integridade capilar, cascata da coagulação, trombose, arteriosclerose, resposta inflamatória e modulação da resposta imune (para mais detalhes, acesse http://www.nature.com/nri/journal/v11/n4/full/nri2956.html) e promoção de reparo tecidual subsequente à injúria. O número normal de plaquetas no sangue periférico varia de 150.000 a 400.000/mm3 na dependência do método utilizado e apresentam vida média na circulação de cerca de 8 a 10 dias. A produção diária, em condições basais, em indivíduos adultos, é estimada em 1×1011 plaquetas, número este que pode se elevar em até 10 vezes ou mais em casos de aumento de demanda. Semelhantemente aos eritrocitos, as plaquetas estão restritas aos vasos sanguíneos, não fazendo recirculação linfática e, fisiologicamente, a interação delas com os leucócitos ocorre primariamente no baço, o que permite a modulação da resposta imune.
As plaquetas originam-se de megacariócitos que, no estado final de maturação, têm a sua membrana citoplasmática rompida, liberando as plaquetas. Como já mencionado no Capítulo 12, em seres humanos, após o nascimento, os precursores megacariocíticos derivam, na medula óssea, de um precursor mieloide comum que, por sua vez, deriva da célula-tronco hemopoética. A diferenciação, a proliferação, a maturação de megacariócitos e, portanto, do número de plaquetas, são mantidas por um sistema de regulação do qual participam hormônios, citocinas, fatores de crescimento e fatores de transcrição. Dentre os fatores solúveis que regulam a plaquetopoese temos IL-3, GM-CSF, FL-3 ligante, Fator Derivado de Célula Estromal (SDF-1), Fator de Crescimento de Célula-Tronco (também chamado de Fator Steel ou de c-kit ligante), IL-11, IL-6 e Trombopoietina (TPO). A trombopoietina atua sinergicamente com esses fatores e ainda é responsável pela maturação dos megacariócitos e pela produção de plaquetas.
Duas citocinas estão claramente associadas ao controle negativo da megacariopoese: o fator crescimento TGFβ e o interferon α (IFNα). O TGFβ é um modulador negativo da hemopoese e é encontrado em vários tecidos linfo-hemopoeticos e nos granulos alfa das plaquetas, particularmente ricos nessa citocina. As evidências experimentais mostram que o TGFβ bloqueia a progressão do ciclo celular via pRb. Por sua vez, o IFNα inibe diretamente a megacariopoese por diversas vias que controlam o ciclo celular, fato esse que é a base do uso terapêutico de IFNα em casos de plaquetoses (para mais detalhes do controle da produção de plaquetas, veja Capítulo 12).
Enquanto em adultos o número de plaquetas é mantido entre 150.000 e 400.000 plaquetas/mm3, podendo flutuar fisiologicamente nesse intervalo, em crianças os valores dependem da idade. Em recém-nascidos pode atingir 500.000/mm3, valor que pode permanecer durante os primeiros 12 meses de vida, quando começa a diminuir.
As plaquetoses são classificadas como primárias e secundárias ou plaquetoses reacionais (Quadro 19.1).
As plaquetoses primárias estão relacionadas às alterações que afetam a células hemopoéticas primitivas e geralmente são de:
ADAMTS-13: Desintegrina e metaloprotease com tromboSpondi tipo 1 repetida (Disintegrin and metalloprotease with thrombospondi type 1 repeated)
ADP: Difosfato de adenosina (Adenosine diphosphate)
AMPc: Monofosfato de adenina cíclico (Cyclic adenosine monophosphat)
PC: Proteína C ativada (Protein C activated)
ATIII: Antitrombina III
CAM: Trombocitopenia amegacariocítica congênita (Congenital amegakaryocytic thrombocytopenia)
CIVD: Coagulação intravascular disseminada
DDAVP: Desmopressina (1-desamino-8-D-arginina vasopressina)
DEB: Diepoxibutano
DV: Vírus da dengue (Dengue virus)
EBV: Vírus de epstein-barr (Epstein-barr virus)
EMCV: Vírus da encefalomiocardite (Encephalomyocarditis virus)
F13A1: Cadeia A – alfa- do fator de coagulação XIII (Coagulation factor XIII A chain)
F13B: Cadeia B- beta do fator de coagulação XIII) (Coagulation factor XIII B chain)
Fator I: Fibrinogênio
Fator II: Protrombina
Fator IX: Fator Christmas
Fator IX: C: Atividade coagulante do fator IX
Fator VIII: Fator anti-hemofílico
Fator VIII: C: Atividade coagulante do fator VIII
Fator X: Fator de Stuart-Prower
Fator XII: Fator de Hageman
FGα: Cadeia alfa do fibrinogênio (Fibrinogen alpha chain)
FGβ: Cadeia beta do fibrinogênio (Fibrinogen beta chain)
FGγ: Cadeia gama do fibrinogênio (Fibrinogen gamma chain)
FT: Fator tissular ou tecidual da coagulação
FV: Fator V
FVII: Ag: Antígeno de fator VII
FVII: C: Atividade coagulante do fator VII
FVIIa-r: FVIIa recombinante
FVL: Fator de Leiden
FVW: Fator de von Willebrand
FVW: Ag: Antígeno do fator de von Willebrand
FVW: CB: Ensaio de ligação entre o FVW e o colágeno
FVW: RCo: Atividade cofatora da ristocetina
GpIIb/IIIa: Glicoproteína IIb/IIIa
GpPIbα: Glicoproteína Ibα
GpIIa: Glicoproteína IIa da membrana plaquetária
HBPM: Heparinas de baixo peso molecular
HIT: Trombocitopenia induzida por heparina (Heparin induced thrombocytopenia)
HITT: Trombose trombocitopenica induzida por heparina (Heparin induced thrombocytopenia thrombosis)
HNP: Heparina não fracionada
INR: Razão normalizada internacional (International normalized ratio)
ISTH: Sociedade Internacional de Trombose e Hemostasia
LES: Lúpus eritematoso sistêmico
LMAN1: Lectina manose ligação 1 (Lectin, mannose binding 1)
MCFD2: Deficiência múltipla de fator de coagulação 2 (Multiple coagulation factor deficiency 2)
MTHFR: Metilhidrofolato redutase (Methyltetrahydrofolate reductase)
NOACs: Anticoagulantes orais não antagonistas da vitamina K (Non- vitamina K antagonist oral anticoagulants)
PAI-1: Fator inibidor do ativador do plasminogênio PAI-1 (Plasminogen activator inhibitor factor PAI-1)
PAI-3: Inibidor do ativador do plasminogênio-3 (Plaminogen acitvator inhibitor-3)
PAR-1: Receptor-1 ativado por proteases (Protease activated receptor-1)
PA-t: Ativador tecidual do plasminogênio (Tissue plasminogen activator)
PDF: Produtos de degradação de fibrina-fibrinogênio
PGE1: Endoperóxido de prostaglandina série E1 (Prostaglandin endoperoxides E1)
PGE2: Endoperóxido de prostaglandina série E2 (Prostaglandin endoperoxides E2)
PROC: Proteína C (Protein C)
PROS1: Proteína S1 (Protein S)
PIVKA: Proteínas induzidas pela ausência de vitamina K (Proteins induced by vitamin K absence)
RC: Retração do coágulo
PTI: Púrpura trombocitopênica idiopática
RIPA: Agregação de plaquetas induzida por ristocetina (Ristocetin induced platelet agregation)
SAF: Síndrome do anticorpo antifosfolipídeo
SERPINE1: Membro 1 da família Serpin E (Serpin family E member 1)
SERPINF2: Membro 2 da família Serpin E (Serpin family F member 2)
SIRS: Síndrome da resposta inflamatória sistemica (Systemic inflammatory response syndrome)
ABL1: Abelson murine leucemia
Akt: Serine/threonine kinase 1 human
ARSA/T: Anemia refratária com sideroblastos em anel associada à trombocitose
BCR: Creakpoint cluster region
BCR-ABL1-/+: BCR-ABL negativo/positivo
CBL: Cbl proto-oncogene human
CDKN2A: Cyclin dependent kinase inhibitor 2a
CDKN2B: Cyclin dependent kinase inhibitor 2b
C-MYC: Myc proto-oncogene protein
CSF3R: Colony stimulating factor 3 receptor
DDX47: DEAD-box helicase human
DNA: Ácido desoxirribonucleico
DRM: Doença residual mínima
ELN: European leucemia net
ERK: Extracelular regulates MAP kinase
ETNK1: Ethanolamine kinase 1
FDA: Food and Drug Administration
FGFR1: Fibroblast growth factor receptor 1
FISH: Hibridização fluorescente in situ (Fluorescence in situ hybridization)
GAP: GTPase activating protein
G- CSF: Granulocyte-macrophage colony-stimulating factor
G-CSFR: Granulocyte-macrophage colony-stimulating factor receptor
GDT: Guanosina difosfato
GEF: Guanine nucleotide exchange factor
GM- CSF: Granulocyte-macrophage colony-stimulating factor
GRB2: Growth factor receptor bound protein 2 human
GTP: Trifosfato de guanosina
HLA: Human leukocyte antigen
IDH2: Isocitrate dehydrogenase (NADP(+)) 2
IGSF2 ou CD101: Membro 2 da superfamilia das imunogobulinas (humano)
IKZF1: IKAROS family zinc finger 1
IKZF3: IKAROS family zinc finger 3
IL: Interleucina
IL-10: Interleucina 10
IL-1β: Interleucina 1 beta
IL-3: Interleucina 3
IL-6: Interleucina 6
INF-γ: Interferon gama
IRIS: Estudo internacional aleatorizado de interferon e STI571
IS: Padrão internacional
JAK: Janus kinase human
JAK2: Janus kinase 2 human
JUN: Jun proto-oncogene
Kb: Kilobase
Kit: Mastocitose
LA: Leucemia Aguda
LDH: Lactato desidrogenase
LEC: Leucemia eosinofílica crônica
LLA: Leucemia linfoblástica aguda
LMA: Leucemia mieloide aguda
LMC: Leucemia mieloide crônica
LMCa: LMC atípica
LMC-FA: Fase acelerada na LMC
LMC-FB: Fase blástica na LMC
LMC-FC: Fase crônica na LMC
LMC-N: Leucemia mieloide neutrofílica-crônica
LNC: Leucemia neutrofílica crônica
LTB4R: Leukotriene B4 receptor human
MAPK: Mitogen-activated protein kinase
M-BCR: Major cluster region
m-BCR-ABL: Minor cluster region
MEK: ERK kinase bombyx mori domestic silkworm
MFP: Mielofibrose primária
mTOR: Mechanistic target of rapamycin human
MYC: MYC proto-oncogene
NCCN: National Comprehensive Cancer Network
NMD: Neoplasias mielodisplásicas
NMP: Neoplasias mieloproliferativas
CALR: Calreticulina
CFU-GEMM: Unidade formadora de colonias granulocíticas, eritroides, megacariocíticas e monocíticas (Colony formating units of granulocytic-erythrocytic-megakaryocytic and monocytic series)
EPO: Eritropoetina
G- CSF: Fator de crescimento de granulócitos (Granulocyte colony-stimulating factor)
GM- CSF: Fator estimulador de colonias de granulócitos e macrófagos (Granulocyte-macrophage colony-stimulating factor)
IL-3: Interleucina 3
JAK2: Janus kinase 2
MAPK: Proteína quinases ativadas por mitógenos (Mitogen activated protein kinases)
MPL: Gene do receptor da trombopoetina
PI3K: Fosfatidil inositol 3-quinase (Phosphatidylinositol-4,5-bisphosphate 3-kinase)
PV: Policitemia Vera
PVSG: Grupo de estudos da policitemia vera
STAT:Transdutor e ativador de fatores de transcrição (Transducer and activator of transcription factors)
As neoplasias mieloproliferativas BCR-ABL negativas (Philadelphia negativo) são doenças mieloproliferativas crônicas e classicamente englobam a policitemia vera (PV), a trombocitemia essencial (TE) e a mielofibrose primária; têm em comum a proliferação excessiva de células progenitoras terminalmente diferenciadas que tiveram uma mutação somática, mas que são negativas para o cromossomo BCR-ABL. Os mecanismos que levam a essa mutação ainda não estão totalmente esclarecidos. Nessas neoplasias mieloproliferativas ocorre uma expansão de uma única linhagem ou então de múltiplas linhagens, assim, na policitemia vera há o aumento predominante da linhagem eritroide, acompanhado de hiperplasia granulocítica e megacariocítica; na trombocitemia essencial a expansão é prioritariamente no setor megacariocítico e em menor escala no granulocítico, sendo que o compartimento eritroide não é afetado. Já na mielofibrose primária, a mieloproliferação ocorre inicialmente na medula óssea para logo se expandir para sítios extramedulares. Frequentemente, a clínica dessas patologias é distinta, e não raro o paciente é assintomático, mas exige-se o diagnóstico diferencial entre elas.
A policitemia vera (PV) é uma doença mieloproliferativa ocasionada, na maioria das vezes, por uma mutação clonal e maligna da célula-tronco hemopoetica que leva à proliferação anormal de precursores eritroblásticos na medula óssea, originando o aumento anormal de eritrocitos. Em grande parte dos pacientes também há superprodução de granulócitos e plaquetas.
Classificação das policitemias
Policitemia Vera
Policitemia secundária ou poliglobulia
Mediada por EPO
Hipóxia dependente
Hipóxia central
Doença pulmonar crônica
Habitar em altitude elevada
Envenenamento por monóxido de carbono
Tabagismo (exposição a longo prazo por monóxido de carbono)
Síndromes de hipoventilação, incluindo apneia
do sono
Hipóxia periférica
Estenose da artéria renal
Hemoglobinopatia por alta afinidade ao oxigênio
Deficiência de 2,3-Difosfoglicerato mutase
Hipóxia independente (produção de EPO patológica)
Tumores malignos
Carcinoma hepatocelular
Câncer de células renais
Hemangioblastoma cerebelar
Carcinoma da paratireoide
Condições não malignas
Miomas
Cistos renais (doença renal policística)
Feocromocitoma
Meningioma
Mediada por EPO-R
Mutação do EPO -R
Droga associada
Doping por EPO
Tratamento com andrógenos
Mecanismos desconhecidos
A maioria dos casos de policitemia congênita autossômica dominante
Algumas formas de policitemia congênita autossômica recessiva
Eritrocitose pós-transplante renal
- Classificaç ão etiológica das Policitemias.
EPO: Eritropoietina; R-EPO: Rceptor de EP
Adap o de: Tefferi A. Polycythemia Vera: A Comprehen
Re Clin Proc. 2003;78:174 -194.
Quadro 22.1 – Classificação das policitemias.* EPO - eritropoietina; R-EPO - receptor de EPO. Adaptado de: Tefferi A. Polycythemia Vera: A Comprehensive Review and Clinical Recommendations.Mayo Clin Proc. 2003;78:174-194.
A policitemia vera deve ser diferenciada de outras condições em que também há eritrocitose e comumente são denominadas poliglobulias. Policitemia ou poliglobulia é a condição em que há o aumento das diversas linhagens sanguíneas, no entanto, o termo é mais frequentemente usado para designar aumento de eritrocitos, do volume hematócrito ou mesmo da concentração de hemoglobina. Também por isso, na maioria dos casos, é chamada de eritrocitose.
A poliglobulia pode ocorrer de forma fisiológica, por exemplo, quando viajamos para regiões de altitudes elevadas, onde o ar é rarefeito. Nesses casos a concentração de oxigênio diminui e o organismo produz mais eritropoetina (EPO), aumentando o número de eritrocitos e, consequentemente, estabilizando os níveis de oxigênio. Alguns autores consideram essa poliglobulia fisiológica como sendo secundária à hipóxia e mediada por EPO, visto que a baixa saturação de oxigênio leva ao aumento da EPO.
A poliglobulia pode ser secundária a outros distúrbios, como doença pulmonar, cardiopatia ou tabagismo elevado. O quadro ocorre pela oxigenação inadequada, levando a uma saturação de oxigênio inadequada. Pode ser em menor proporção, decorrente de nefropatias, alterando a síntese e o metabolismo da eritropoetina e levando a alterações na volemia eritroide.
A policitemia relativa, policitemia aparente, poliglobulia ou pseudopoliglobulia decorre da redução do volume plasmático por perda acentuada de líquidos ou ingestão insuficiente de líquidos, no entanto, a massa eritrocitária mantém seus níveis normais, levando a um aumento relativo dos eritrocitos. Apesar de a causa ser incerta, pode ocorrer em pacientes sob tratamento com diuréticos, tabagistas, alcoólatras, hipertensos ou com problemas cardiovasculares (Quadro 22.1). Costuma ser mais frequente que a policitemia vera.
Podemos, ainda, encontrar raros casos de policitemia congênita (familiar) onde há uma mutação no gene da proteína Hippel-Lindau, aumentando a afinidade ao oxigênio, com o consequente aumento da produção de EPO.
Também conhecida como policitemia rubra, a policitemia vera foi primeiramente descrita em 1892 por Vaquez e associada à hiperplasia mieloide da medula óssea em 1910. Em 1935, a mielofribrose e a leucemia mieloide aguda
CALR: Calreticulina
c-MPL: Gene do receptor da trombopoetina
LMC: Leucemia mieloide crônica
Ph1: Cromossomo Philadelphia 1
PV: Policitemia vera
STAT3: Transdutor e ativador de fator de transcrição 3 (Signal transducer and activator of transcription 3)
STAT5: Transdutor e ativador de fator de transcrição 3 (Signal transducer and activator of transcription 5)
TE: Trombocitemia essencial
A trombocitemia essencial (TE), também conhecida como trombocitemia idiopática, trombocitose essencial ou, ainda, trombofilia essencial, é uma síndrome mieloproliferativa crônica, Ph1 negativa, de origem clonal da célula-tronco hemopoetica, em que ocorre a hiperproliferação de megacariócitos funcionais, originando plaquetose periférica. O termo “trombocitemia” significa excesso de plaquetas no sangue. O termo “essencial” indica que o aumento das plaquetas é um problema inato da produção de células sanguíneas na medula óssea. A TE é uma doença clonal, maligna, mas que pode apresentar um curso indolente “benigno” quando devidamente controlada e, nesse caso, a sobrevida dos pacientes pode exceder quinze anos.
Apesar de afetar as linhagens eritroide, megacariocítica e granulocítica, a expressão fenotípica predominante é a megacariocítica, que é responsável pela produção e diferenciação de plaquetas.
Na TE ocorre uma hiperplasia de megacariócitos na medula óssea, ocasionando um número elevado de plaquetas na circulação. Além disso, a TE também pode promover esplenomegalia e quadros trombóticos e hemorrágicos.
A maioria dos casos de TE ocorre em indiv íduos acima de 50 anos de idade. A TE pode afetar igualmente mulheres e homens adultos, apesar de alguns estudos mostrarem tendência levemente maior em mulheres mais jovens. Raramente ocorre em crianças. A incidência média de TE é de aproximadamente 2,2 casos novos por 100.000 habitantes a cada ano.
Os mecanismos fisiopatológicos que levam à plaquetose na TE eram pouco conhecidos até 2005, quando foram descritas as mutações em JAK2 e, mais recentemente, as mutações em c-MPL (c-MPL: gene do receptor da trombopoetina) (W515, W515K) e em CALR (calreticulina).
As neoplasias mieloproliferativas Philadelphia negativas (policitemia vera, trombocitemia essencial e mielofibrose primária) têm como características a tendência à evolução para leucemias mieloides agudas e para o aparecimento de fenômenos tromboembólicos. Para saber mais, acesse “Somatic mutations of calreticulin in myeloproliferative neoplasms” e “CALR, JAK2, and MPL Mutation Profiles in Patients With Four Different Subtypes of Myeloproliferative Neoplasms”
A mutação JAK2V617F é encontrada em cerca de 95% dos pacientes com PV e em cerca de 60% dos pacientes com TE ou com mielofibrose primária. A proteína JAK2 é uma tirosina quinase que atua como segundo mensageiro fosforilando fatores de transcrição STAT3 e STAT5, fatores esses que controlam o ciclo celular. Essa mutação altera os mecanismos inibitórios fisiológicos da tirosina quinase e, dessa forma, a proteína JAK2 V617F adquire um aumento funcional na sua sinalização.
Outra importante mutação que tem sido descrita ocorre no gene que codifica o receptor da trombopoietina –MPL, sendo que a mais frequente são as mutações em triptofano no códon W515 na região citosólica do receptor MPL (troca por leucina – W515L; troca por lisina – W515K e troca por alanina – W515A). Essas mutações são encontradas entre 3% e 5% dos pacientes com TE e fazem com que o receptor MPL permaneça ativado mesmo na ausência do ligante (trombopoetina), induzindo, portanto, à proliferação de megacariócitos.
Em 2013 foram descritas mutações somáticas no gene da calreticulina (CALR), que codifica proteína ligante de cálcio associada ao ret ículo endoplasmático. A CALR possui diversas funções, inclusive de controle da apoptose e da proliferação celular, e modula a homeostasia do cálcio. A proteína não mutada CALR apresenta a região C-terminal carregada negativamente, enquanto a proteína mutada apresenta essa região carregada positivamente, o que ocasiona alteração na ligação com outras proteínas e, consequentemente, alteração na sinalização celular. Para saber mais, acesse “A novel assay to detect calreticulin mutations in myeloproliferative neoplasms”
Atualmente, são descritas mais de cinquenta mutações no gene da calreticulina, todas localizadas no éxon 9. Essas mutações somáticas são detectadas em 50-88% dos pacientes com ET ou mielofibrose primá ria JAK2/MPL negativos.
As duas mutações mais frequentes de CALR correspondem a uma deleção de 52 bp (p.L367fs*46), também chamada de tipo 1 e à inserção 5bp (p.K385fs*47), também chamada de tipo 2. Na TE as mutações tipo 1 estão presentes em 55% dos pacientes, enquanto as do tipo 2 estão presentes em 33%, frequências essas distintas daquelas observadas na mielofibrose primária, em que o tipo 1 é o predominante (75% dos casos).
Clinicamente, os pacientes portadores da mutação em CALR apresentam valores mais baixos para hemoglobina e para os leucócitos. O número de plaquetas encontra-se elevado, mas o risco de fenômenos trombóticos é relativamente baixo.
Outras mutações, como TET2, 10,11 CBL, 12,13 EZH2, 14,15 DNMT3A, 16, podem ser encontradas nessas neoplasias mieloproliferativas, mas, em geral, encontram-se concomitantemente com as mutações JAK2 e MPL. Veja no Quadro 23.1 as principais mutações associadas à TE.
Akt: Família de proteínas quinases contendo resíduos de serina/treonina também denominadas de proteína quinase B
G– CSF: Fator de crescimento de granulócitos (Granulocyte colony–stimulating fator)
GM– CSF: Fator estimulador de colonias de granulócitos e macrófagos (Granulocyte–macrophage colony–stimulating factor)
IL–3: Interleucina 3
JAK2: Janus kinase 2
LMC: Leucemia mieloide crônica
MAPK: Proteína quinases ativadas por mitógenos (Mitogen activated protein kinases)
MF: Mielofibrose
MPL: Vírus de leucemias mieloproliferativas (Myeloproliferative leukaemia virus)
PDGF: Fator de crescimento derivado de plaquetas (Platelet derived growth factor)
PI3K: Fosfatidil inositol 3–quinase (Phosphoinositide 3–kinase)
PV: Policitemia vera
Ras: Sarcoma vírus ou vírus do sarcoma de rato: proteína que controla a migração, proliferação, diferenciação, adesão e apoptose celular
STAT: Transdutor de sinal e ativador de transcrição (Signal transducer and activator of transcription)
TE: Trombocitemia essencial
VEGF: Fator de crescimento de endotélio vascular (Vascular endotelial growth fator)
A mielofibrose primária (MF), também denominada metaplasia mieloide agnogênica, mielofibrose idiopática ou essencial, mieloesclerose, osteomielofibrose e mielofibrose com metaplasia mieloide, se caracteriza pela mieloproliferação acompanhada da fibrose generalizada e progressiva da medula óssea e hemopoese extramedular.
As neoplasias mieloproliferativas BCR–ABL negativas (Philadelphia negativo), onde a mielofibrose é classificada, são doenças mieloproliferativas crônicas e englobam a policitemia vera (PV) a trombocitemia essencial (TE) e a mielofibrose primária e têm em comum a proliferação excessiva de células progenitoras terminalmente diferenciadas que tiveram uma mutação somática, mas que são negativas para o cromossomo BCR–ABL . Os mecanismos que levam a essa mutação ainda não estão totalmente esclarecidos. Nessas neoplasias mieloproliferativas
ocorre a expansão de uma única linhagem ou então de múltiplas linhagens, assim, na policitemia vera há o aumento predominante da linhagem eritroide acompanhada de hiperplasia granulocítica e megacariocítica; na trombocitemia essencial a expansão é prioritariamente no setor megacariocítico e em menor escala no granulocítico, sendo que o compartimento eritroide não é afetado. Já na mielofibrose primária, a mieloproliferação ocorre inicialmente na medula óssea para logo se expandir para sítios extramedulares e a medula óssea passa a ser fibrótica. Os quadros clínicos típicos dessas doenças apresentam características muitas vezes similares com prognósticos distintos e exigem diagnóstico diferencial. Essas patologias possuem complicações comuns: fenômenos trombo–hemorrágicos e a transformação leucêmica.
A MF é uma doença mieloproliferativa caracterizada pela fibrose generalizada e progressiva da medula óssea devido à proliferação anômala de megacariócitos associada à proliferação granulocítica. A proliferação anômala e clonal da célula–tronco hemopoética leva a alterações na produção de células sanguíneas, hemopoese extracelular, esplenomegalia e fibrose da medula óssea. A hiperplasia megacariocítica é acompanhada de diferenciação aberrante (mielodisplasia), causando a liberação de citocinas e de fator de crescimento de fibroblastos no microambiente medular, o que ocasiona a fibrose da medula óssea.
Dentre as mieloproliferações, a MF é o distúrbio mieloproliferativo crônico menos comum. Trata–se de uma enfermidade rara (cerca de cinco casos por milhão de habitantes por ano), ocorrendo predominantemente em indivíduos idosos (idade superior a 65 anos).
Para saber mais, acesse http://www.bloodjournal.org/content/bloodjournal/129/6/667.full.pdf e http://ac.els–cdn.com/S0303720717300722/1–s2.0–S0303720717300722–main.pdf?_tid=19b79bfe–2923–11e7–8be3–00000aab0f6c&acdnat=1493061819_b7d521e230cb05b79516fffc1513c50a.
A MF é uma síndrome mieloproliferativa crônica muitas vezes decorrente da evolução da policitemia vera ou da trombocitemia essencial. A MF está associada à mieloproliferação e diferentes graus de mielodisplasia. Apesar de o mecanismo não estar plenamente elucidado, entende–se que as células hemopoéticas anômalas da medula óssea podem estimular a proliferação de fibroblastos. Associado a isso, há o desenvolvimento de uma hemopoese extramedular, no baço e no fígado (chamada metaplasia mieloide).
A partir de 2005, os estudos citogenéticos foram fundamentais para decifrar o papel das mutações nas síndromes mieloproliferativas BCL–ABL negativas estando envolvidas principalmente as mutações em JAK2, o receptor de trombopoetina denominado cMPL e em CALR (calreticulina). Descobriu–se que cerca de 70% dessas doenças estão associadas a mutações somáticas adquiridas de JAK2V617: 95% dos pacientes com Policitemia Vera, cerca de 50% dos pacientes com TE e 60% dos pacientes com mielofibrose apresentam essa mutação do nucleotídeo 1849 no éxon 14 do gene para a JAK2 (9p24), o que resulta na substituição da valina para fenilalanina no códon 617 (JAK2V617F ou V617F), gerando uma proteína quinase anômala. A proteína JAK2 ( Janus kinase 2) é uma tirosina quinase ativada pela ligação dos receptores de IL–3, G– CSF, GM– CSF e trombopoetina aos seus respectivos ligantes. A substituição de uma valina por uma fenilalanina (mutação V617F) promove a ativação constitutiva levando à autofosforilação de JAK2 sem a ligação do fator de crescimento ou da citocina, promovendo, portanto, a ativação de vias envolvidas na proliferação, diferenciação e apoptose, tais como a via de sinalização STAT, Ras/MAPK e PI3K/Akt (Figura 22.1). Essa mutação pode se apresentar heterozigótica ou homozigótica. Em cerca de 4% dos pacientes pode ocorrer uma mutação em JAK2 no éxon 12 do gene 9p24. Veja no Quadro 24.1 a prevalência de mutações na mielofibrose primária.
Menos frequente que a mutação JAK2V617F são as mutações em MPL (gene do receptor da trombopoietina) e em CALR (calreticulina) que atinge cerca de 25% dos pacientes com MF. A mutação em MPL ocorre no códon 515 do MPL (W515L, W515K, S505N), alterando as vias de sinalização das moléculas STAT3, STAT5, MAPK e na fosfatidil–3 quinase ATK (PI3K/AKT). Para saber mais sobre as bases moleculares, acesse: https://www.ncbi.nlm. nih.gov/pmc/articles/PMC4841197/pdf/f1000research–5–8692.pdf; https://www.nature.com/bcj/journal/v7/n2/ pdf/bcj20176a.pdf; http://www.bloodjournal.org/content/bloodjournal/129/6/667.full.pdf?sso–checked=true.
A mutação JAK2V617F é encontrada em cerca de 50–60% dos casos de MF, e os indivíduos portadores dessa mutação apresentam valores de leucócitos no sangue circulante mais elevados em relação aos indivíduos JAK2V617F negativos. A necessidade de transfusões sanguíneas é menor em JAK2 V617F positivos. Alguns estudos mostram que a mutação possivelmente protege de uma anemia mais severa, no entanto o quadro clínico é mais agressivo e com sobrevida menor.
ATM: Gene mutado da Ataxia-telangiectasia (Ataxia-telangiectasia mutaded gene)
DHL: Desidrogenase lática sérica
FISH: Hibridização fluorescente in situ (Fluorescence in situ hybridization)
HLA: Antígenos de histocompatibilidade (Histocompatiblity leukocytes antigens)
HTLV: Vírus da leucemia humana de células T (Human T-cell lymphotropic virus)
HTLV-1: Vírus da leucemia humana de células T tipo 1 (Human T-cell lymphotropic virus type 1)
IgD: Imunoglobulina D
IGHV: Gene da região variável da cadeia pesada da imunoglobulina (Immunoglobulin heavy-chain variable region gene)
IgM: Imunoglobulina M
Inca: Instituto Nacional do Câncer
L/LTA: Linfoma/leucemia de células T do Adulto
LDGC: Linfoma não Hodgkin difuso de grandes células
LLC: Leucemia linfoide crônica
LPL-B: Leucemia pró-linfocítica de células B
LPL-L: Leucemia pró-linfocítica de células T
SmIg: Imunoglobulina ligada à superfície da membrana (Surface membrane bound immunoglobulins)
miRNA: Micro RNA
PD-1: Ligante 1 de morte programada (PD-1 programmed cell death-1 ligand )
PD-2: Ligante 2 de morte programada (PD-2 programmed cell death-2 ligand )
SR: Síndrome de Richter
SS: Síndrome de Sézary
TdT: Transferase terminal (Terminal deoxynucleotidyl transferase)
TRAP: Fosfatase ácida tartarato-resistente
As Leucemias linfoides crônicas (LLC) (também são tratadas no Capítulo 28) caracterizam-se pelo acúmulo de linfocitos pequenos, maduros e neoplásicos, principalmente linfocitos B no sangue periférico, medula óssea, baço
linfoide crônica e linfonodos. São desordens neoplásicas malignas, crônicas e heterogêneas, em que há a proliferação clonal de células B – e raramente de T – e que apresentam alterações na função imune.
As LLC compreendem um grupo heterogêneo de patologias com maior prevalência (3,5 a 5/100.000 ano) em indivíduos com idade acima de 50 anos e cuja incidência varia conforme a população, idade e sexo: aumenta com o passar dos anos e incide mais em homens. Caracterizam-se pelo acúmulo progressivo de células B CD5+ (9095% dos casos) ou, mais raramente, de células T no sangue periférico, medula óssea e órgãos linfoides ( baço e linfonodos). Em geral, a doença é assintomática, sendo, muitas vezes, detectada em exames de rotina.
Existe uma variabilidade na apresentação dos subtipos de LLC e respectivas evoluções clínicas. Estão descritos diversos mecanismos moleculares associados à LLC. Até poucos anos, acreditava-se que alterações genéticas induziam a um bloqueio no processo de morte celular (apoptose), com consequente inibição do mecanismo apoptótico, provocando acúmulo progressivo das células linfoides tanto na medula óssea quanto no sangue periférico. Atualmente, sabe-se que os mecanismos que levam ao acúmulo dessas células estão relacionados a sinais de sobrevivência (autofagia) induzidos nos subgrupos das células leucêmicas. Apesar de os avanços científicos progredirem no entendimento da doença, as causas que levam à LLC e aos seus subtipos ainda não estão totalmente compreendidas.
As características imunofenotípicas e os dados hematológicos apresentados pelos pacientes, bem como a evolução no conhecimento das bases moleculares e melhor entendimento da patologia, proporcionam informações que possibilitam a classificação clínica da patologia, permitindo um melhor direcionamento no tratamento do paciente.
A LLC é uma patologia que se manifesta com maior frequência nos países ocidentais (é relativamente rara na China e praticamente ausente no Japão, com exceção da tricoleucemia – veja adiante), representando 22% a 30% de todas as leucemias em adultos acima de 50 anos, sendo mais frequente em homens que em mulheres (1,5-2:1) e mais em brancos que em negros.
A idade média ao diagnóstico fica na faixa dos 64-70 anos de idade. Existe uma correlação entre casos familiares de LLC – verifica-se que um em cada dez pacientes acometidos possui algum outro caso de LLC ou de doença linfoproliferativa ou de imunodeficiência na família, existindo discreta associação (em 5 a 10% dos casos) com suscetibilidade familiar, embora o modo de herança ainda não esteja estabelecido. Não existem dados consistentes da correlação entre a LLC e a exposição ocupacional à radiação ou a agentes químicos, exceto nos casos de exposição a herbicidas, contudo acredita-se que a incidência dessa patologia esteja subestimada devido às características inerentes da doença, principalmente por ser uma patologia, na maioria das vezes, assintomática no momento do diagnóstico, dificultando o desenvolvimento de estudos epidemiológicos.
Existem poucos dados relatando a incidência da LLC na população brasileira. O Instituto Nacional do Câncer (Inca) não específica os subtipos de leucemias em seu Registro de Câncer em Base Populacional (RCBP): “[...] os maiores valores das taxas médias anuais de incidência, ajustadas por idade por 100 mil homens, foram constatadas em São Paulo (1997-1998: 10,2); Distrito Federal (1996-1998: 8,9) e Natal (1998-1999: 8,4). Na população feminina, as maiores taxas foram observadas em São Paulo (1997-1998: 7,4); Porto Alegre (1993-1997: 5,3) e Natal (1998-1999: 4,6). As menores taxas foram observadas nas cidades de Recife (1995-1998) em homens (3,0) e Salvador (1997-2001) em mulheres (2,3)”. http://www1.inca.gov.br/regpop/2003/index.asp?link=comentarios.asp&ID=1. Para saber mais, acesse http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2354.2004.00489.x/epdf e http://ac.els-cdn.com/S1040842807000844/1-s2.0-S1040842807000844-main.pdf?_tid=a14efc7e-4f9d-11e7-a9bd00000aab0f26&acdnat=1497292589_ad039d3d254b880e56f5c6ca8d282732.
No passado, acreditava-se de que a LLC se tratava de uma doença com origem celular homogênea derivada de linfocitos B naïve. Avanços na compreensão da patologia evidenciaram que as células leucêmicas presentes nos casos de LLC diferem no estado de ativação, maturação e subgrupo celular. Estudos apontam a ocorrência de um acúmulo de células devido à proliferação celular alterada associada a sinais de sobrevivência induzidos por estimulação de receptores nas células B, bem como por quimiocinas e citocinas.
A LLC é uma doença linfoproliferativa, comumente originada em 90-95% dos casos por linfocitos B e mais raramente originada por linfocitos T, sendo considerada uma doença clinicamente heterogênea, uma vez que os
ABL: Abelson Leucemia
ABL1: Abelson murine leucemia viral oncogene homolog 1
AF4: AF4/FMR2 family member 1
AIDS: Síndrome da imunodeficiência adquirida
ANAE: Alfa-Naftil-Acetato-Esterase
BCR: Breakpoint cluster region
CBFA2: Core-binding factor subunit alpha-2
CBFB: Core-binding factor beta subunit
CD: Grupo de diferenciação (Cluster of differentiation)
CDKN2A: Cyclin dependent kinase inhibitor 2a
CDKN2B: Cyclin dependent kinase inhibitor 2b
CEBPA: CCAAT/enhancer-binding protein alpha
C-FOS: Fos proto-oncogene
CFTR2: Cystic fibrosis transmembrane conductance regulator
cIgM: Imunoglobulina M de citoplasma
C-MYC: Myc proto-oncogene protein
DEK: DEK proto-oncogene
DNA: Ácido desoxirribonucleico
DNMT3A: DNA methyltransferase 3 alpha
E2A: Transcription factor 3
EBF1: Early B-cell factor 1
EBF1: Transcription factors EBF1
EBNA-1: Epstein-Barr nuclear antigen 1
EBV: Vírus Epstein-Barr
EGFR1: Epidermal growth factor receptor (EGFR) family
EGIL: European Group for the Immunological Characterization of Leucemia
ENV: Envelope protein
EPOr: Receptor de Eritropoietina
ERG: ETS transcription factor
ETP: Low molecular weight protein-tyrosine-phosphatase
ETP-LLA: Early T-cell Precursor Acute Lymphoblastic Leucemia
ETS: Transcription factors
ETV6: ETS variant 6
EVI1: Zinc finger protein Evi1
FAB: Franco-Americano-Britânica
FGFR1: Fibroblast growth factor receptor 1
FISH: Hibridização fluorescente in situ (Fluorescence in situ hybridization)
FLT3: Protein called fms-like tyrosine kinase 3
FSC: Forward Scatter
GAG: Group-specific antigen
GATA2: GATA binding protein 2
GM- CSF: Granulocyte-macrophage colony-stimulating factor
HHV: Herpes virus
HLA: Human Leukocyte Antigen
HOX11: Homeobox
HTLV: Human T Lymphotropic Virus
iAMP21: Intrachromosomal amplification of chromosome 21
IDH1: Isocitrate dehydrogenase (NADP(+) 1
IDH2: Isocitrate dehydrogenase (NADP(+) 2
IGH: Immunoglobulin heavy locus
IKZF1: IKAROS family zinc finger 1
IKZF3: IKAROS family zinc finger 3
IL: Interleucina
IL-3R: Receptor de interleucina 3 (Interleukin 3 receptor)
INCA: Instituto Nacional de Câncer José Alencar Gomes da Silva
KMT2A: Histone-lysine N-methyltransferase 2A
KMT2A: Lysine methyltransferase 2A
KRAS: KRAS proto-oncogene
LA: Leucemia Aguda
LCR: Líquido cefalorraquidiano
LDH: Lactato desidrogenase
LLA: Leucemia Linfoblástica Aguda
LLC: Leucemia Linfocítica Crônica
LLTA: Leucemia-linfoma de células T do Adulto
LMA: Leucemia Mieloide Aguda
LMP-1: Latent membrane protein 1
LPA: Leucemia Promielocítica
APC: Aloficocianina
AR: Anemia refratária
AREB: Anemia refratária com excesso de blastos
AREB-T: Anemia refratária com excesso de blastos em t ransformação
ARSA: Anemia refratária com sideroblastos em anel.
ASXL1: Regulador transcricional suplementar ao sexo tipo 1 (Additional sex combs like 1, transcriptional regulator)
ATG: Globulina antitimocítica
BMO: Biópsia de medula óssea
CCUS: Citopenia clonal de significado indeterminado
CD: Grupo de designação (Cluster designations)
CFM: Citometria de fluxo multiparamétrica
CHIP: Hemopoese clones de potencial indeterminado
CR: Citopenia refratária
CR/DML: Citopenia refratária com displasia de múltiplas linhagens
CRP: Citopenia refratária pediátrica
CTH: Célula-tronco hemopoetica
DNA: Ácido desoxirribonucleico.
EZH2: Potenciador de zeste homólogo 2 (Enhancer of zeste homolog 2)
ETV6: Gene variante 6 da translocação e vinte e seis (E Twenty six variant 6)
FAB: Franco-Americano-Britânico
FAS: Antígeno Faz
FITC: Isotiocianato de fluoresceína
GATA2: Proteína ligante 2 GATA (guanina, adenina e timina) (GATA binding protein 2)
G- CSF: Fator estimulante de colonias de granulócitos
HB: Hemoglobina
HIV: Vírus da imunodeficiência humana
ICUS: Citopenia idiopática de significado indeterminado
IDUS: Displasia idiopática de significado indeterminado
IPSS: Sistema de prognóstico internacional (International Prognostic Scoring System)
LMA: Leucemia mieloide aguda
LMMC: Leucemia mielomonocítica crônica
MO: Medula óssea
OMS: Organização Mundial da Saúde
PE: Ficoeritrina
PERC-P: Peridinina
RUNX1: Fator de transcrição relacionado com Runt 1 (Runt-related transcription factor 1)
SMD: Síndrome mielodisplásica
SMD/AREB: Síndrome mielodisplásica/Anemia refratária com excesso de blastos
SMD/ARS: Síndrome mielodisplásica/Anemia refratária com sideroblastos em anel
SMD/CR-AR: Síndrome mielodisplásica/Citopenia refratária – anemia refratária
SMD/CR-DML: Síndrome mielodisplásica/Citopenia refratária com displasia de múltiplas linhagens ou multilinear
SMD/CR-NR: Síndrome mielodisplásica/Citopenia refratária – neutropenia refratária
SMD/CR-TR: Síndrome mielodisplásica/Citopenia refratária – trombocitopenia refratária
SMD/I: Síndrome mielodisplásica/Inclassificável
SMD-P: Síndrome mielodisplásica pediátrica
SMD-P/CR: Síndrome mielodisplásica/Citopenia refratária pediátrica
SMD-SA: Síndrome mielodisplásica com sideroblastos em anel
SMD/5q-: Síndrome mielodisplásica associada a del (5q) isolada
SMDs: Síndromes mielodisplásicas
SP: Sangue periférico
TCTH-Alo: Transplante de células-tronco hemopoeticas alogênico
TNF: Fator de necrose tumoral
TP53: Proteína tumoral 53 (Tumor protein P53)
VCM: Volume corpuscular médio
WPSS: Sistema de prognóstico da Organização Mundial da Saúde (World Health Organization Classification–Based Prognostic Scoring System)
As Síndromes Mielodisplásicas (SMDs) ou mielodisplasias são neoplasias hematológicas caracterizadas pelo desenvolvimento anormal (displasia) da medula óssea (MO). Esta nomenclatura foi sugerida em 1975, em Paris, em uma conferência sobre leucemias inclassificáveis, sendo inicialmente denominada como displasia hemopoética, mais tarde abreviada para mielodisplasia
São formadas por um grupo de desordens hemopoeticas originadas de um defeito clonal na célula-tronco hemopoetica, possivelmente originado de aberrações epigenéticas e que tem como característica marcante a proliferação e a apoptose aumentadas das células hemopoéticas (hemopoese ineficaz). A consequência dessas alterações na produção e destruição celular é a presença paradoxal de citopenias periféricas variadas com hipercelularidade na medula óssea e alterações morfológicas displásicas (Figura 27.1).
São também característicos a presença de marcadores genéticos clonais e aumento da propensão para evolução em leucemia mieloide aguda (LMA).
AIDS: Síndrome da imunodeficiência adquirida
CD: Cluster designation
EBV: Vírus Epstein:Barr
FAB: French – American:British
HIV: Vírus da imunodeficiência humana
HLA: Antígenos leucocitários humanos
HRS: Células Hodgkin:Reed Sternberg
IPI: Indice de prognóstico internacional
LAGC: Linfoma anaplásico de grandes células
LCM: Linfoma de células do manto
LDGCB: Linfoma difuso de grandes células B
LF: Linfoma não Hodgkin folicular
LHC: Linfoma de Hodgkin clássico
LHC:CM: Linfoma de Hodgkin clássico subtipo celularidade mista
LHC:DL: Linfoma de Hodgkin Clássico subtipo depleção linfocitária
LHC-EN : Linfoma de Hodgkin Clássico subtipo esclerose nodular
LHC:RL: Linfoma de Hodgkin Clássico subtipo rico em linfocitos
LH: Linfoma de Hodgkin
LHPLN: Linfoma de Hodgkin com predomínio linfocitário nodular
LLC/LL: Leucemia linfoide crônica/linfoma linfocítico
LNH: Linfoma Não Hodgkin
LP: Predomínio linfocitário
LT:helper: linfocitos T auxiliares
LZM: Linfoma da zona marginal
MALT: Tecido linfoide associado à mucosas
NFκB: Fator Nuclear:kB
NK: Natural killer
OMS: Organização Mundial da Saúde
RS: Reed:Sternberg
SNC: Sistema nervoso central
TC: Tomografia computadorizada
TCR: Receptor de células T;
WF: Working Formulation
Os linfomas constituem um grupo heterogêneo de doenças malignas do tecido linfoide, de origem e evolução variáveis, que se localizam primariamente nos linfonodos, podendo também ocorrer na medula óssea e outros órgãos não hemopoéticos. São neoplasias clonais de células B e T/NK maturas ou imaturas em vários estágios de diferenciação
Histologicamente os linfomas podem ser classificados em linfomas de Hodgkin e linfomas não Hodgkin, com base na presença ou ausência das células de Reed-Sternberg e suas variantes, respectivamente.
Em um artigo histórico denominado “On Some Morbid Appearances of the Absorbent Glands and Spleen” (1832), Thomas Hodgkin (Figura 28.1), fez a primeira descrição dos linfomas, que posteriormente levariam seu nome, relatando alterações encontradas nos linfonodos e baço em sete casos clínicos, como sendo característicos de uma doença primária distinta dos processos inflamatórios ou neoplásicos até então descritos.
Quase 25 anos depois, Samuel Wilks (1856) nomeou os casos descritos em 1832, como doença de Hodgkin em homenagem ao médico inglês. Calr Sternberg (1898) e Dorothy Reed (1902), realizando estudos independentes, fizeram as primeiras descrições histopatológicas do linfoma de Hodgkin. Com base nos resultados observados nos casos clínicos estudados, os pesquisadores caracterizaram as células gigantes multinucleadas, como sinal morfológico característico desta doença, nomeadas como células de Reed-Sternberg (RS).

de Thomas Hodgkin (1832)”. Fonte: https://archive.org/details/onsomemorbidappe00hodg
Dorothy Reed concluiu em seu artigo que “[...] acreditamos que a partir das descrições na literatura e os achados em oito casos analisados, que a doença de Hodgkin tem um quadro histológico peculiar e típico e poderia, assim, justamente ser considerada uma nova entidade histopatológica”. Alguns anos mais tarde (1926), amostras restantes dos casos descritos por Dr. Hodgkin armazenadas no Museu do Hospital Guy foram revistas, utilizando os critérios histológicos descritos por Reed (1902). Notavelmente, quase um século depois da primeira análise histopatológica, a microanatomia estava preservada e foi possível confirmar a doença de Hodgkin em três dos sete casos originais de Thomas Hodgkin, um foi classificado como linfoma não Hodgkin, e os casos restantes como tuberculose e sífilis.
As células de Reed-Sternberg (Figura 28.2) derivam das células B do centro germinativo por meio de um desarranjo na transcrição da célula B, devido ao silenciamento epigenético e outras lesões no gene. Estas alterações
Nesta seção colocamos as principais técnicas hematológicas, manuais (Capítulo 29) e automatizadas (Capítulo 30), utilizadas rotineiramente em laboratórios. Cada técnica é apresentada individualmente com o Sumário, sua Lista de Abreviatura e Bibliografia. O capítulo sobre Automação em Hematologia finaliza esta seção.
LISTA DE ABREVIATURAS
EDTA: Ácido etilenodiaminotetracético (Ethylenediaminetetraacetic acid )
mm: Milímetro
ºC: Graus Celsius
q.s p: Quantidade suficiente para
29.1.1 INTRODUÇÃO
O sangue é um tecido conjuntivo que consiste em uma suspensão de células vermelhas (eritrocitos), brancas (leucócitos) e plaquetas em um meio líquido rico em proteínas. Fisiologicamente, o sangue circula por todos os tecidos do organismo por meio de de uma intrincada rede vascular, apresentando funções importantes na oxigenação de tecidos, no sistema imunológico, na hemostasia e no transporte de hormônios, citocinas, nutrientes e metabólitos. Quando coletado sem anticoagulantes, o sangue coagula em aproximadamente 5 a 8 minutos, ocorrendo separação entre a fração celular e a fração líquida (denominada soro); quando coletado com anticoagulantes, a suspensão celular é mantida e a separação entre a fração celular e a fração líquida (plasma) pode ser obtida por meio de de centrifugação. As análises laboratoriais são realizadas tanto com amostras de soro como com amostras de plasma, sendo que as análises bioquímicas são realizadas preferencialmente com amostras de soro. Já para a grande maioria das análises hematológicas é utilizado o sangue “colhido com anticoagulante ”, seja com EDTA (ácido etilenodiaminotetracético), citrato de sódio, oxalato de potássio ou heparina.
29.1.2.1 Sangue capilar
29.1.2.1.1 Metodologia
O sangue capilar é obtido das pontas do dedo ou do lóbulo auricular em adultos, e do calcanhar de crianças. Para tanto, a área deve ser esterilizada com álcool 70% e puncionada com uma lanceta a uma profundidade de 2 mm. A primeira gota de sangue deve ser descartada, limpando-se a área com gaze estéril, procedendo-se em seguida com a coleta das demais gotas. Deve-se evitar comprimir a região para obter mais sangue, pois isso pode alterar a composição do mesmo. Se a obtenção do sangue for difícil, a região pode ser aquecida ou deve-se permitir que o sangue circule para a extremidade dos dedos, mantendo as mãos abaixadas antes da coleta.
Vantagens
1. O sangue capilar pode ser obtido facilmente (por isso é muito utilizado para preparar extensões sanguíneas).
Desvantagens
1. A quantidade de sangue obtida é pequena e a amostra frequentemente sofre hemólise;
2. De modo geral, os resultados obtidos com sangue capilar não podem ser comparados com resultados obtidos de sangue venoso.
29.1.2.2 Sangue venoso
29.1.2.2.1 Metodologia
O sangue venoso é o mais comumente utilizado devido à facilidade de coleta empregada. Embora o sangue seja geralmente obtido das veias anticubitais, outras veias também podem ser escolhidas. Antes da coleta, a região superior do braço é garroteada com o auxílio de um torniquete para evitar que haja retorno venoso. É feita a assepsia do local com álcool 70% e, após a veia ser puncionada com uma agulha estéril de 25 × 0,7 mm ou 25 × 0,8mm, ou butterfly, deve-se afrouxar o torniquete. Caso o sangue seja coletado com seringa a vácuo, deve-se esperar até o preenchimento total do tubo; no caso de se coletar o sangue com anticoagulante, deve-se respeitar a relação entre anticoagulante/sangue e o tubo deve ser homogeneizado imediatamente.
Vantagens
1. A partir de uma única amostra, é possível realizar diversos exames;
2. Alíquotas da amostra (soro ou plasma) podem ser congeladas para possíveis contraprovas.
Desvantagens
1. A coleta de sangue venoso necessita de pessoas treinadas;
2. Quando a estase provocada pelo torniquete é muito prolongada, pode causar hemoconcentração, prejudicando algumas análises;
3. Podem-se formar hematomas e coágulos na região puncionada.
29.1.2.2.2 Cuidados gerais
1. A coleta de sangue capilar deve ser feita com lancetas afiadas e a região não deve ser pressionada;
2. Para coleta de sangue venoso, deve-se utilizar agulhas de grande calibre para evitar a hemólise;
3. O torniquete não pode estar muito apertado e deve ser afrouxado após a punção da veia;
4. Sangue coletado com anticoagulante deve ser homogeneizado imediatamente por inversão;
5. Se for necessária a obtenção do soro, deve-se esperar a formação do coágulo (2 horas a 37 ºC) antes da separação do soro por centrifugação.
29.1.3 ANTICOAGULANTES
A coagulação do sangue pode ser evitada por três mecanismos: remoção (reversível ou irreversível) de cálcio utilizando-se quelantes; inativação da trombina a partir da adição de heparina; e remoção da fibrina por defibrinação. Para cada exame deve-se levar em consideração a quantidade e o tipo de anticoagulante a ser utilizado.
29.1.3.1 Oxalatos
Isoladamente, o oxalato de potássio e o oxalato de amônio não podem ser utilizados para se analisar a morfologia das células sanguíneas, uma vez que o primeiro tende a enrugar as hemácias enquanto o segundo promove edema celular. Esse tipo de anticoagulante não é recomendado para a contagem de plaquetas e avaliação da sua
Em Hematologia, os principais exames automatizados são os de hemograma, reticulócitos, coagulação, velocidade de hemossedimentação (VHS) e agregação plaquetária. Laboratórios de pequeno, médio e grande porte geralmente utilizam aparelhos automatizados capazes de aumentar sua produtividade e eficiência operacional, fornecendo resultados precisos e exatos, podendo evitar riscos de contaminação, acidentes de trabalho e troca de amostras pelos operadores do instrumento.
Os instrumentos automatizados, em geral, têm elevado nível de reprodutibilidade e exatidão que é incomparável à das técnicas manuais. Quando calibrados e operados dentro das especificações da qualidade e do fornecedor, fornecem resultados muito acurados e reprodut íveis, porém, em alguns casos não conseguem identificar todas as anormalidades significativas identificadas pelo observador humano. Desta forma, alguns instrumentos liberam alarmes ( flags) qualitativos ou quantitativos, por exemplo: atípicos, bastonetes ou eritrocitos nucleados, para que a revisão do resultado bruto e a decisão final caiba ao profissional.
Para a instalação dos analisadores é importante que:
- o espaço físico seja adequado, de forma a acomodar todos os constituintes;
- o fornecimento de energia elétrica seja estável;
- as condições de temperatura e umidade estejam conforme indicação do fabricante.
Para a manutenção dos analisadores são realizadas as manutenções preventivas internas ou externas. A manutenção diária (por exemplo, a autolavagem das agulhas) é importante para a garantia da qualidade das análises. As manutenções preventivas externas devem ser realizadas periodicamente pela assistência técnica do fabricante ou fornecedor visando à limpeza de partes internas e à reposição de peças.
A comunicação bidirecional do computador do laboratório com o computador do equipamento é conhecida como interface e permite a identificação dos tubos de coleta por leitura de código de barras, identificação dos exames solicitados, transmissão de resultados brutos e liberação automática ou manual dos resultados.
Os equipamentos de hemograma e reticulócitos são conhecidos como contadores hematológicos. Os principais fabricantes são Abbott Diagnostics, Beckman Coulter, Horiba Medical, Siemens Healthcare Diagnostics e Sysmex America, que produzem equipamentos de pequeno até grande porte (Quadro 30.1).

Quadro 30.1 – Principais fabricantes e equipamentos em hemograma e reticulócitos.
30.2.1.1 Impedância Elétrica
Este método foi descrito por Coulter em 1956 e se baseia na detecção e mensuração da oscilação de corrente elétrica gerada pela célula quando esta passa por um campo elétrico estático (Figura 30.1).
Figura 30.1 – Método de impedância elétrica descrito por Coulter.
As células são suspensas em uma solução eletrolítica e, quando passam por um orif ício entre os dois eletrodos de platina, promovem uma resistência elétrica, ou seja, um impedimento da passagem da corrente elétrica gerando pulsos. A quantidade de pulsos gerada é proporcional ao número de células avaliadas, enquanto a amplitude dos pulsos é proporcional ao volume celular. Os sinais elétricos gerados são plotados em gráficos de histograma segundo sua frequência.
Devido à diferença de tamanho celular e o estabelecimento do limiar de separação entre as populações celulares, as células são divididas em grupos como leucócitos (WBC, White Blood Cell ), hemácias (RBC, Red Blood Cell ) e plaquetas (PLT, Platelet) ou em subgrupos como na contagem diferencial de leucócitos. Quando mais de uma célula passa pelo orif ício ao mesmo tempo, ou se esta não passar pelo centro da abertura, pode ocorrer a distorção de impulsos elétricos. Os dados são ajustados para excluir os picos eletronicamente distorcidos, assim como o limite superior e inferior do tamanho da partícula para excluir aglomerados ou restos celulares.
