Cardiovascular News - Issue 75 - November 2024 (US)
5 RHEIA trial First all-women TAVI trial reports oneyear results
6 Chronic coronary syndromes New guidelines push imaging techniques
14 Profile Rafael Sádaba
The waiting is over: Trials shed new light on optimal timing for aortic stenosis intervention
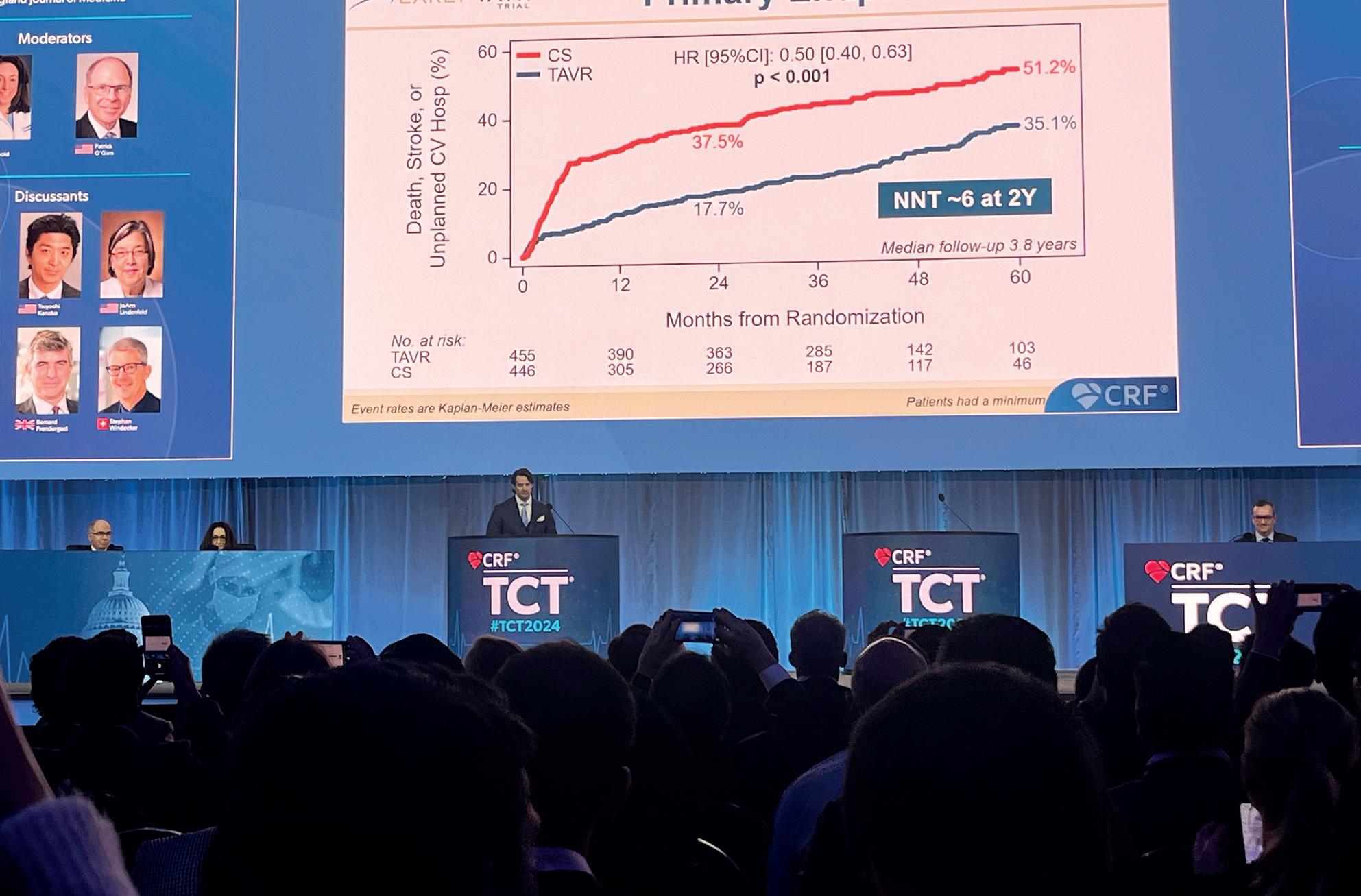
“The world has been waiting for the outcome of this trial,” Bernard Prendergast (St Thomas’ Hospital, London, UK) said in the opening of his commentary of the EARLY TAVR trial at TCT 2024 (27–30 October, Washington, DC, USA), in no way downplaying the significance of the study’s positive outcome favouring a strategy of early transcatheter aortic valve implantation (TAVI) in patients with asymptomatic, severe aortic stenosis.
EARLY TAVR was one of the most hotly anticipated trials at the TCT meeting, and the positive primary endpoint result was met with spontaneous applause within the main arena late-breaking trial session when presented by Philippe Généreux (Morristown Medical Center, Morristown, USA).
The results, published simultaneously in The New England Journal of Medicine, have led some to speculate that, instead of waiting for aortic stenosis symptoms to progress, physicians may now have the evidence they need to justify an early intervention in patients with severe, asymptomatic aortic stenosis. Indeed, Prendergast commented that the findings could warrant a “major reset in our approach” to treatment.
However, some have questioned the trial’s replicability in real-world practice, particularly given variable waiting times for procedures across the globe, and whether the lack of a significant benefit in mortality for patients who underwent an interventional procedure justifies moving to an earlier invasive strategy.
Conducted at 75 centres in the USA and Canada, EARLY TAVR looked at the safety and effectiveness of early intervention with TAVI using the Sapien 3 (Edwards Lifesciences) valve (n=455), compared to clinical surveillance in patients with asymptomatic severe aortic stenosis (n=446).
Between March 2017 and December 2021 investigators screened 1,578 patients for enrolment, ultimately randomising 901 patients to either TAVI (n=455) or surveillance (n=446).
Patients enrolled had an average age of 76 and average Kansas City Cardiomyopathy Questionnaire (KCCQ) overall summary score of 92.7, and were confirmed as asymptomatic through a protocol-mandated stress test and medical history evaluation.
Those randomised to TAVI were treated within a median time of 14 days.
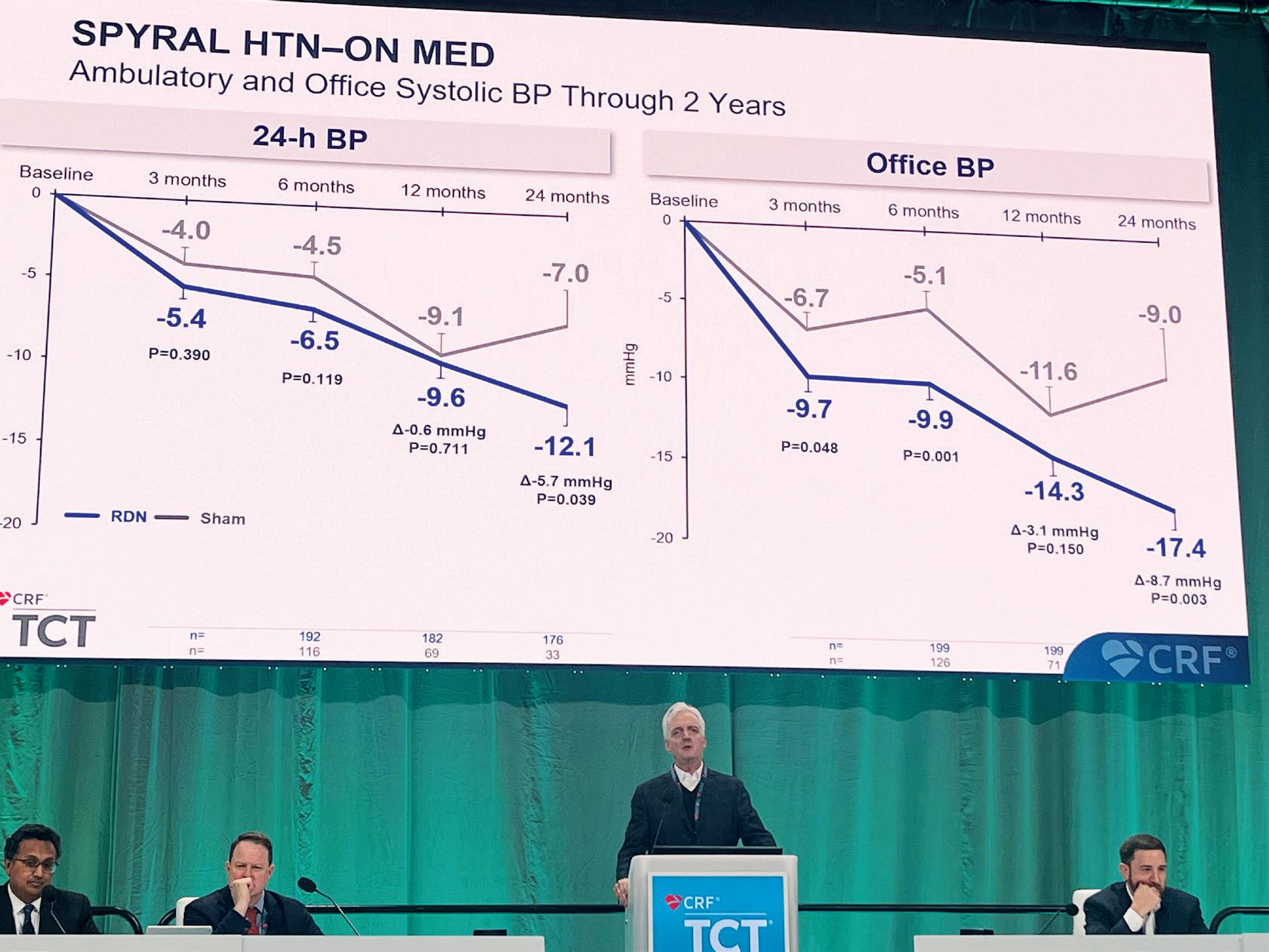
The trial’s primary endpoint, a composite of death, stroke, or unplanned cardiovascular hospitalisation, was evaluated for superiority in the intent-to-treat population after a minimum follow-up of two years. Genereux reported at TCT 2024 that early intervention with TAVI resulted in a significant reduction of the primary endpoint at two years as well as a median followup of 3.8 years, occurring at rate of 35.1% in the TAVI group compared with 51.2% in the surveillance group (p<0.001).
Breaking the result down further, Généreux showed that the difference between the two strategies at a median follow-up of 3.8 years was predominantly driven by a difference in rates of unplanned hospitalisation seen in the two arms (20.9% for TAVI vs. 41.7% for surveillance) whilst rates of all-cause death (8.4% vs. 9.2%) and stroke (4.2% vs. 6.7%) were relatively similar in the two arms, albeit favouring TAVI in both instances.
Additionally, Généreux reported that within the first six
Early TAVI may be preferred to clinical surveillance in patients with asymptomatic severe aortic stenosis”
18 TRISCEND II Tricuspid valve replacement data
New evidence could prompt rethink over use of colchicine following acute MI
THE LARGEST TRIAL TO DATE to study the impact of colchicine— an anti-inflammatory medicine commonly used to treat gout—in acute myocardial infarction (MI) has found that administering a low dose of the drug did not reduce cardiovascular death, MI, stroke or ischaemia-driven revascularisation compared to placebo.
Sanjit S Jolly (McMaster University and Hamilton Health Sciences, Hamilton, Canada), principal investigator in the trial— CLEAR SYNERGY (OASIS 9)— presented results at TCT 2024 (27–30 October, Washington, DC, USA), where he commented that the findings “should give clinicians pause” about whether to use colchicine therapy in their practice.
Colchicine has been touted as a route to improving cardiovascular outcomes in patients with MI by inhibiting inflammation. Two trials— CLEAR and COLCOT—have shown a significant benefit for colchicine for the treatment of coronary artery disease, and have driven its use in the setting of acute MI.
“We designed [CLEAR SYNERGY] prior to these trials and were running this as the largest trial of colchicine in coronary artery disease, with significantly more events and more power than the prior trial,” said Jolly. “We believe it is important to replicate the results particularly for them to change practice and to move to a class I indication in the guidelines.”
CLEAR SYNERGY included patients with ST-elevation myocardial infarction (STEMI) or non-ST elevation myocardial infarction (NSTEMI) who were randomised within 72 hours of percutaneous coronary intervention (PCI) to either colchicine or placebo.
Taking place at 104 sites in 14 countries between February
Continued on page 2
COLCHICINE
New evidence could prompt rethink over use of colchicine following acute MI
Continued from page 1
2018 and November 2022, investigators enrolled a total of 7,062 patients in the trial. Following the initial randomisation, subjects were also then randomised to either spironolactone or placebo. Results of the second phase of this analysis will be presented at the American Heart Association (AHA) 2024 scientific sessions (15–18 November, Chicago, USA), with findings due to be published in The New England Journal of Medicine (NEJM).
CLEAR SYNERGY was designed with 80% power to detect a 25% relative risk reduction in the primary outcome, a composite of cardiovascular death, MI, stroke or ischaemia driven revascularisation, as assessed using a Cox proportional hazards model, stratified by STEMI versus NSTEMI and spironolactone versus placebo.
At a median follow-up of three years, Jolly reported that the composite endpoint was not significantly different between the colchicine and placebo groups (p=0.93). Additionally, there were no significant differences in any of the individual components of the composite endpoint. Results were consistent between patients who discontinued the therapy, and those who carried on into the long-term, Jolly revealed.
One area where colchicine was observed to have a significant impact was in a reduction in C-reactive protein (CRP), a marker for inflammation, Jolly detailed.
“There has been a lot of interest in the C-reactive protein as a marker of prognosis, and really a marker of benefit on anti-inflammatory therapies. We found that CRP was high in the midst of the acute MI in both groups, but at three months CRP did go down in both groups, [and] was lower in the colchicine group, so colchicine was effective in reducing CRP.”
Adverse events were similar between both study cohorts except that diarrhoea was more common after colchicine than with placebo (10.2% vs. 6.6%, p<0.001).
“We believe, based on this large trial the role of colchicine post MI and long-term is uncertain,” Jolly commented at TCT 2024, noting that the results have already led him to halt use of the therapy among patients in his care, despite having been a “believer” in the therapy.
“On balance when you look at this trial, we didn’t see a reduction in cardiovascular outcomes and unfortunately there was a side-effect of the therapy, diarrhoea. As a patient you can decide for yourself, would you want to take this therapy?” he said.
Ajay Kirtane (Columbia University Irving Medical Center/NewYork-Presbyterian Hospital, New York,
Sanjit S Jolly
USA) went further, describing the results as “a big deal” and highlighting the lack of an association between the reduction of the inflammatory marker—CRP—and the clinical outcomes observed in the trial.
“When we talk about patients after an MI, you want to make sure they are taking their antiplatelet therapy, you want to make sure that their LV [left ventricle] is supported with all the guideline-directed medical therapy,” he said. “This medicine is not well tolerated, [and] the data previously were so-so. Now, with this type of trial, I fully agree that I would not want to start it [colchicine therapy] in a patient.”
Similarly, Wayne Batchelor (Inova Heart and Vascular Institute, Falls Church, USA) said that the trial’s results
We didn’t see a reduction in cardiovascular outcomes and unfortunately there was a side-effect of the therapy”
would limit his use of colchicine post-MI, but said it is important not to dismiss the potential role of reducing inflammation among these patients.
“We have to be careful that we don’t throw the baby out with the bathwater, so to speak,” Batchelor said. “Inflammation post-MI is still a very important thing to study, and to understand the science that underlies it. There is another trial, the ARTEMIS trial, that is using a very potent IL6 inhibitor, ziltivekimab, and that is going to be a very similar trial to this to study the effects of supressing inflammation at a much larger level than colchicine to see how that plays out. I think we have to be careful that we just assume that inflammation doesn’t play an important role.”
Editor-in-chief: Simon Redwood | Publisher: Stephen Greenhalgh
Content Director: Urmila Kerslake | Global Sales Director: Sean Langer
Senior editor: Will Date will@bibamedical.com | Editorial contribution: Jamie Bell, Jocelyn Hudson
Published by: BIBA News, which is a subsidiary of BIBA Medical Ltd BIBA Medical, Europe, 526 Fulham Road, Fulham, London, SW6 5NR, United Kingdom Tel: +44 (0) 20 7736 8788 BIBA Medical, North America, 155 North Wacker Drive, Suite 4250, Chicago, IL 60606, United States Tel: +1 708-770-7323
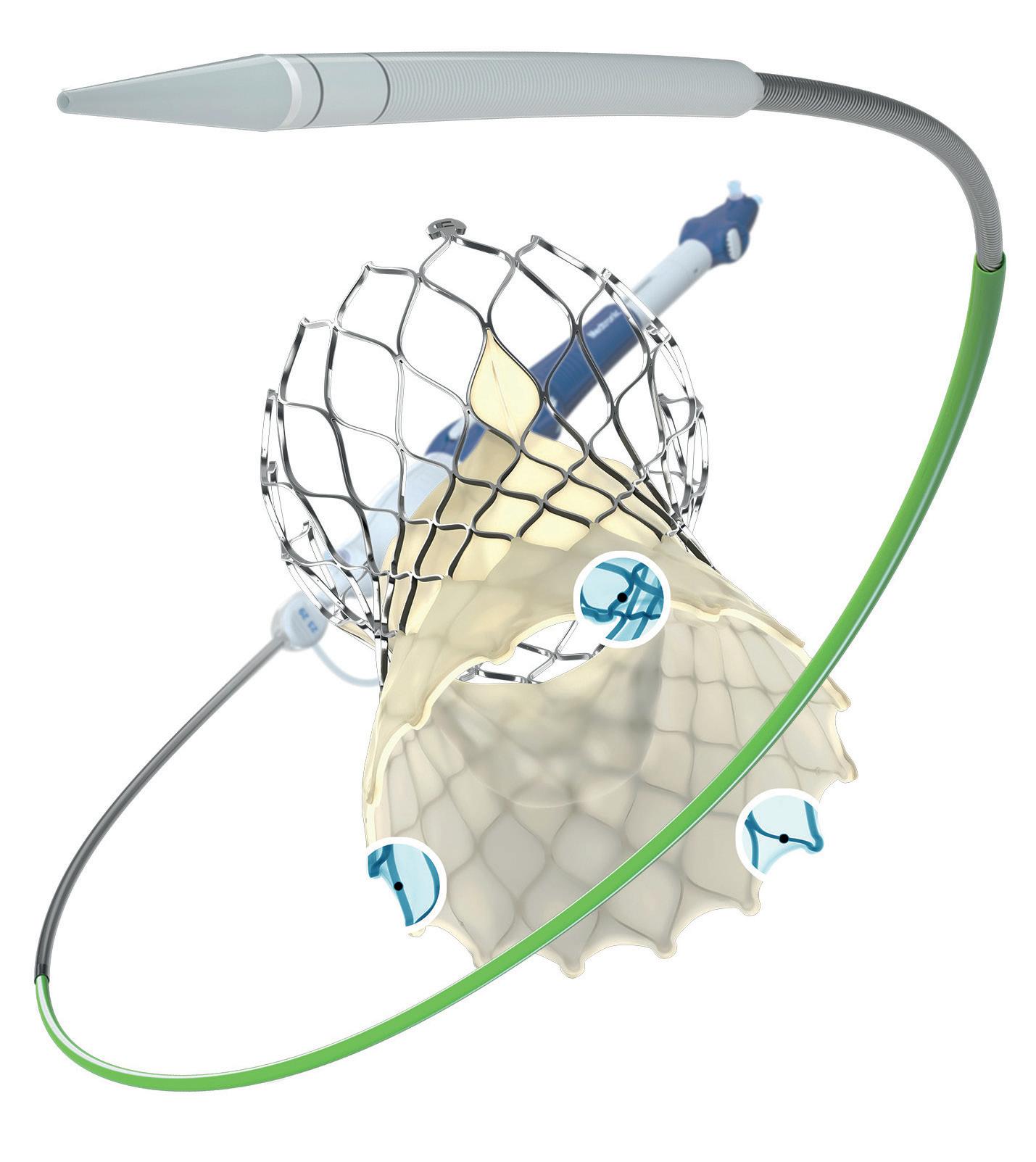
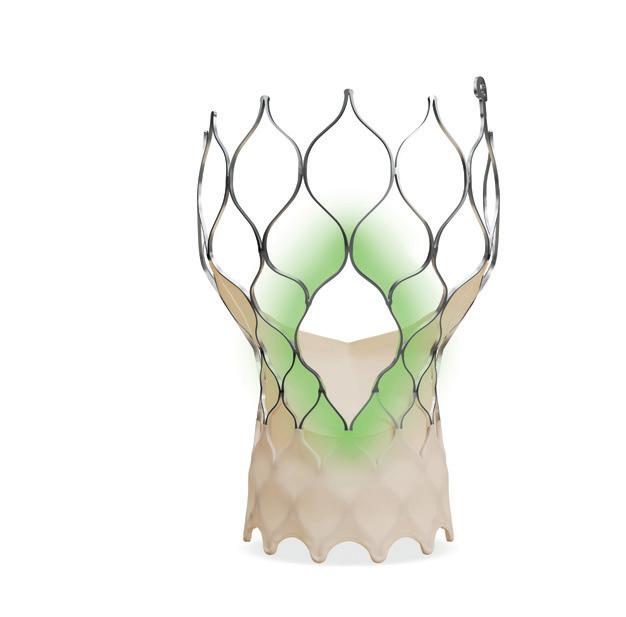
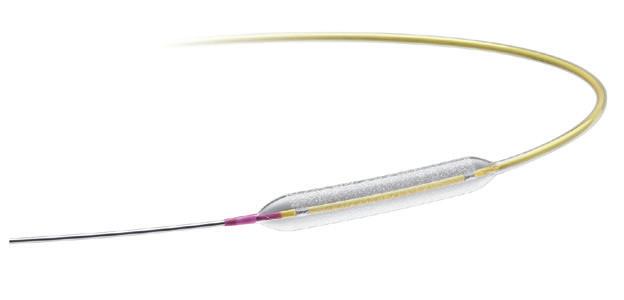
One-year data from the INFIINITYSWEDEHEART trial demonstrate the safety and efficacy of a coronary bioadaptor system for the treatment of coronary artery disaese. The novel device is implanted in a similar fashion to a drug-eluting stent (DES) but has an unlocking mechanism that begins after the device is encapsulated with tissue and its abosorbable polymer coating is resorbed and is intended to allow the vessel to grow and adapt to maintain established blood flow lumen.
For more on this story go to page 7.
n EACTS 2024:
Updates from the European Assocation of Cardio-Thoracic Surgery (EACTS) 2024 annual meeting (9–12 October, Lisbon, Portugal), which include new research on outcomes following the implementation of a digital prehabilitation programme prior to cardiac surgery, coronary artery bypass graft (CABG) among diabetic patients with multivessel disease, and long-term outcomes of a surgical aortic valve system.
For more on this story go to page 11.
n RENAL
DENERVATION:
Guideline changes from the European Society of Cardiology (ESC) are among a slew of updates concerning the use of renal denervation therapy, an interventional procedure that is intended to reduce blood pressure. As well as new long-term data, investigators have announced plans to widen the study of renal denervation to other vascular beds, with the aim of providing more consistent, sustainable blood pressure reductions.
For more on this story go to page 22.
Scan the QR code to subscribe
If you have comments on this issue or suggestions for upcoming editions write to will@bibamedical.com
COVER STORY continued
The waiting is over: Trials shed new light on optimal timing for aortic stenosis intervention
Continued from page 1
months, 26.2% of patients in the clinical surveillance arm converted to aortic valve replacement (AVR) with many presenting progressive or advanced symptoms. In the 12-month follow-up period after randomisation, the rate of conversion to AVR was 47.2%.
“Given the benefits observed and the lack of harm, early TAVI may be preferred to clinical surveillance in patients with asymptomatic severe aortic stenosis, especially when combined with the challenges of timely symptom recognition and prompt treatment in real-world settings,” Généreux said of the clinical implications of the trial.
Others have been less emphatic in their interpretation of the findings, believing the trial’s impact to be more muted in clinical practice, given the lack of a survival advantage seen between the two arms of the study.
“Quality of life is very important for patients,” David Kandzari (Piedmont Heart Institute, Atlanta, USA) tells Cardiovascular News, offering his analysis of the result. “It will open the
discussion for doctors to discuss with our patients about TAVI at a perhaps earlier stage, but I am not sure it will necessarily move the market so much for expanding this to a broader group of patients.”
The question of when to replace a stenotic aortic valve stood at the heart of a second trial, EVOLVED, which followed EARLY TAVR in the TCT main arena.
Guidelines have long recommended valve replacement in patients with severe aortic stenosis, study investigator Mark Dweck (University of Edinburgh, Edinburgh, UK) told TCT attendees, but only in those who have symptoms due to their valvular disease.
Dweck noted that assessing symptoms in elderly patients with multiple comorbidities can be challenging, prompting the question as to whether an earlier intervention—via either TAVI or surgical aortic valve replacement (SAVR)—could be warranted to improve outcomes among these patients.
EVOLVED, an international, multicentre, prospective randomised open-label blinded-endpoint trial, was conducted at 24 sites in the UK and Australia. The study involved patients with asymptomatic severe aortic stenosis and mid-wall myocardial fibrosis—a driver of left ventricular (LV) decompensation in aortic stenosis and a predictor of patient mortality— with cardiovascular magnetic resonance (CMR) used to determine the patients most likely to benefit from an early
intervention.
A total of 427 participants were screened for inclusion in the trial, with 278 deemed eligible for CMR assessment. Of these, 113 were randomised to undergo early intervention, with the use of TAVI or SAVR determined by a heart team, and 111 received guideline-directed conservative management.
The median time-to-intervention was starkly different with a gap of roughly 15 months between the two groups, Dweck reported at TCT, standing at
I think it will be a personalised decision for individual patients to make with their physicians”
five months in the early intervention arm and 20 months in the routine care arm. This difference in the timing of the procedure did not result in a meaningful difference in outcomes for patients, with the trial’s primary endpoint, a composite of all-cause death or unplanned aortic stenosis-related hospitalisation, occurring in 18% of patients in the early intervention group and 23% of patients in the conservative treatment group,
Adjustable valve replacement system wins TCT Shark Tank innovation prize
Symbiosis, the developer of an adjustable transcatheter mitral valve replacement (TMVR) system—Valsync—was chosen as the recipient of the Shark Tank innovation prize at the 2024 Transcatheter Cardiovascular Therapeutics (TCT) conference (27–30 October, Washington, DC, USA).
THE VALSYNC SYSTEM COMPRISES TWO highly compliant balloons, made of a flexible deformable material that can expand to fit the surrounding valve anatomy. The system is designed to be delivered transeptally, with an atrial balloon expanded and lowered onto supra-annular plane of the mitral valve. Afterwards a second balloon is secured in the ventricle, held in place by a series of barbs or arms.
The conformable design makes the device adaptable to unique valve shapes, Symbiosis says, and the balloons can be inflated or deflated based on real-time echo Doppler feedback to optimise the sealing effect and ensure proper alignment. Symbiosis founder and CEO Shira Burg, a qualified veterinary doctor with a PhD in cardiac electrophysiology, presented Valsync at TCT.
Presently Symbiosis is launching a chronic animal study using the device, and upon success anticipates a first-in-human trial to commence around late 2026. Though the valve is being tested in mitral applications, it could also be used to treat tricuspid valve disease. Symbiosis has recently been bolstered by the addition of Stanton Rowe, the former chief scientific officer at Edwards Lifesciences, who was instrumental in the commercialisation of the first transcatheter aortic valve implantation (TAVI) systems, to its
advisory board.
“We are currently in preclinical studies to test the implant and delivery system,” said Burg. “We are also filing a pre-submission to the US Food and Drug Administration (FDA) to approve the development plan and are aiming to enter first in human testing in the third quarter of 2026. Winning the TCT Shark Tank competition is an honour and validates the importance of the work we are doing.”
“We are thrilled to name Symbiosis this year’s TCT Shark Tank innovation competition winner,” said Juan F Granada, president and chief executive officer of the Cardiovascular Research Foundation (CRF), the organiser of TCT. “Their invention has the potential to overcome some of the limitations of current TMVR systems by adapting to more complex anatomies.”
Other finalists in the 2024 Shark Tank competition included AMX Technologies, which was named as the runner
Dweck revealed.
Unplanned aortic stenosis-related hospitalisation was less frequent in the early intervention group, however, whilst New York Heart Association (NYHA) symptom class at 12 months also favoured the early intervention group. “In patients who underwent early intervention, symptoms did not change at 12 months, but in the patients who were in the routine care group, the symptoms deteriorated with worsening NYHA status at 12 months,” Dweck said.
The finding prompted the EVOLVED investigators to conclude that the principal benefit of early intervention appears to be in the reduction of unplanned hospitalisation and in preventing the development of limiting symptoms among this patient population.
“I personally think this will be a fairly nuanced conversation with the patient,” said Dweck, reflecting on the results, which were also published simultaneously in the Journal of the American Medical Association. “The patients that I see fall broadly into two categories: elderly patients who are feeling well and don’t want to have an intervention; and other patients who want to be more proactive, they are healthy and well and want to stay healthy and well, and they are happy to undergo a procedure earlier to maintain their health status. So, I think it will be a personalised decision for individual patients to make with their physicians.”
up for its clip removal procedure for the removal of failed transcatheter edge-to-edge repair (TEER). Paul Sorajja (Minneapolis Heart Institute, Minneapolis, USA), who is the founder and chief medical officer of the company presented details of the system.
“A distinguished panel of multidisciplinary experts judged the entrants on the six criteria established for the competition at its inception: unmet clinical need, technology differentiation, intellectual property (IP) position/viability, biological proof of concept, regulatory pathway, and commercialisation potential,” said Greg L Kaluza, senior director of research, CRF Skirball Center for Innovation. “This year’s competition featured many groundbreaking advancements, and we are honoured to recognise a company like Symbiosis for their innovation and dedication to advancing the field.”
The TCT Shark Tank competition is delivered through a partnership with the Jon DeHaan Foundation, with the winner receiving a US$200,000 award. The competition aims to identify groundbreaking concepts in cardiovascular medicine.
“CRF is truly grateful to the Jon DeHaan Foundation for their generous continued support of pioneers in the field dedicated to developing novel technologies for the diagnosis and treatment of cardiovascular disease,”
(l-r) Shira Burg, Juan F Granada and Robert Schwartz
No advantage to continuing anticoagulation among patients undergoing TAVI
Patients undergoing transcatheter aortic valve implantation (TAVI) do not benefit from the continuation of oral anticoagulation compared to those whose anticoagulants were interrupted before the procedure, new research has shown.
THIS WAS AMONG THE FINDINGS OF the POPular PAUSE TAVI trial, an open-label, investigator-initiated, non-inferiority trial in patients receiving oral anticoagulation undergoing TAVI, comparing the two anticoagulation strategies. Results of the trial were presented at the 2024 European Society of Cardiology (ESC) congress (30 August–2 September, London, UK) and published in The New England Journal of Medicine
At ESC 2024, Dirk-Jan van Ginkel (St Antonius Hospital, Nieuwegein, The Netherlands), on behalf of the POPular PAUSE TAVI investigators, reported that the trial’s primary endpoint—a composite of cardiovascular mortality, stroke, myocardial infarction (MI), major vascular complications and major bleeding—occurred at 30 days in 16.5% of patients who continued oral anticoagulation during TAVI, compared to 14.8% of patients whose oral anticoagulation regime was interrupted.
The risk difference of 1.7% between the two strategies in favour of the interruption group meant that the trial’s non-inferiority margin was not met, hence POPular PAUSE TAVI investigators suggest that their data support the interruption of oral anticoagulation during TAVI.
“The current population undergoing TAVI is different than, for example, the PCI [percutaneous coronary intervention] population,” Van Ginkel said. “These patients are generally older, they have more comorbidities, for example, renal disease, peripheral arterial disease, and cerebrovascular disease. Also, larger catheters are needed to perform TAVI.”
“When such patients using oral anticoagulation undergo a high bleeding risk procedure, such as TAVI, general guidelines on perioperative anticoagulation management recommend interrupting oral anticoagulation for a couple of days,” he noted. Whilst this has been assessed in some observational studies, which have shown a potential decrease in the risk of stroke, without an increase in bleeding when oral anticoagulation was continued, there have been no randomised trials to compare the two strategies in patients undergoing TAVI.
Taking place in 22 European centres, POPular PAUSE TAVI saw 858 patients randomised 1:1 either to continue or stop oral anticoagulation at least 48 hours before their TAVI procedure. The mean age was 81 years and 34.5% were women, 81.9% were taking direct oral anticoagulants, with 18.1% taking vitamin k antagonists.
Meta-analysis supports DAPT de-escalation after PCI
De-escalation to ticagrelor monotherapy does not increase ischaemic risk and reduces the risk of major bleeding when compared to 12 months of dual antiplatelet therapy (DAPT) following percutaneous coronary intervention (PCI), particularly among patients with acute coronary syndromes (ACS).
THIS WAS THE FINDING OF A systematic review and individual patient data (IPD)-level meta-analysis of randomised trials comparing the safety and efficacy of ticagrelor monotherapy after short-term DAPT versus 12 months of DAPT in patients who have undergone PCI with a drug-eluting stent. Findings of the analysis were shared on behalf of the Single Versus Dual Antiplatelet Therapy (Sidney-4) collaborator group at the European Society of Cardiology (ESC) 2024 annual meeting (30 August–2 September, London, UK) and published simultaneously in The Lancet DAPT is currently recommended as the default antiplatelet regimen after coronary drug-eluting stent implantation, the authors, Marco Valgimigli (Cardiocentro Ticino
Foundation, Lugano, Switzerland) and colleagues, note in their Lancet paper, but some studies have shown that from a few weeks to a few months after initiation of DAPT, de-escalation of treatment to a strategy of P2Y12 inhibitor monotherapy does not increase ischaemic risk and is associated with less bleeding compared with continuation of standard DAPT.
The authors note, however, that these studies can be difficult to interpret due to differing study design, patient populations, and types of P2Y12 inhibitor used, and have insufficient power to estimate the treatment effect on
Secondary endpoints, including cardiovascular mortality, risk of thromboembolic events and ischaemic stroke were comparable between the groups, Van Ginkel noted, but bleeding events occurred in 31.1% of patients in the continuation group and 21.3% in the interrupted group.
BLEEDING
EVENTS
31.1%
OF PATIENTS WHO CONTINUED ORAL ANTICOAGULATION
“There was no advantage of continuing oral anticoagulation compared with interruption in patients undergoing TAVI with a need for anticoagulation and there was more bleeding in the continuation group,” he said. “Therefore, we think that this trial provides the first randomised data which support interruption of anticoagulation in patients undergoing TAVI.”
The findings of the trial should prompt a change in guidelines, discussant Gilles Montalescot (Pitié-Salpêtrière Hospital, Paris, France) said following the presentation of the results at ESC 2024.
“Patients anticoagulated for atrial fibrillation and scheduled for TAVI procedures should have their anticoagulants stopped without low molecular-weight heparin bridging,” Montalescot said. “This recommendation should be taken by the ESC guidelines, because so far we have no recommendations at the ESC level for this situation.”
14.5%
OF PATIENTS WHO PAUSED ANTICOAGULATION
relevant individual endpoints, such as mortality or stent thrombosis, or in sub-groups.
After analysing all available randomised evidence on DAPT deescalation to ticagrelor monotherapy, investigators selected data from six randomised trials with centrally adjudicated endpoints that assigned patients to ticagrelor monotherapy or DAPT, representing more than 24,000 patients in total.
For the three ranked coprimary endpoints—a composite of all-cause death, myocardial infarction, or stroke—the investigators were able to demonstrate that the DAPT de-escalation strategy met
The risks of Bleeding Academic Research Consortium (BARC) 3 or 5 bleeding and all-cause death were lower with ticagrelor monotherapy compared with DAPT. Trial sequential analysis showed robust evidence of noninferiority for
This trial provides the first randomised data which support interruption of anticoagulation in patients undergoing TAVI”
major adverse cardiovascular or cerebrovascular events (MACCE) and superiority for bleeding among the overall and ACS populations.
The treatment effects for MACCE were heterogeneous by sex, suggesting a benefit in women administered ticagrelor monotherapy, and by clinical presentation for BARC 3 or 5 bleeding, indicating a benefit in ACS with ticagrelor monotherapy, the investigators report.
“The present IPD meta-analysis provides evidence that de-escalating from DAPT to ticagrelor monotherapy from a few weeks to a few months after coronary drug-eluting stent implantation does not increase fatal and non-fatal ischaemic risk, and significantly reduces the risk of major bleeding compared with 12 months of DAPT in patients with ACS,” the study’s authors write. “There was a significant, yet inconclusive, mortality benefit with ticagrelor, particularly among women, which warrants further investigation. However, in patients with CCS [chronic coronary syndrome], the benefits and risks of DAPT de-escalation to ticagrelor monotherapy compared with continued DAPT remain unestablished.”
Marco Valgimigli
Dirk-Jan van Ginkel
First all-women TAVI trial sees transcatheter therapy outperform surgery
Randomised trial data point to transcatheter aortic valve implantation (TAVI) being superior to surgery for the treatment of severe aortic stenosis in women.
Aseries of positive trials comparing TAVI to surgical aortic valve replacement (SAVR) have fuelled the growth of transcatheter therapy across a broad range of risk profiles, but a predominantly male population in these trials has led to questions as to whether the results can be applied equally to both men and women.
That is why the results of the RHEIA trial—the first TAVI trial to enrol an exclusively female population— presented at this year’s European Society of Cardiology (ESC) congress (30 August–2 September; London, UK) have been eagerly anticipated, representing the first available comparable data of the two treatment strategies in what has, until now, been an underrepresented patient population.
“We have recent data that suggest the risk of mortality is higher after aortic valve replacement in women, but lower following TAVI in women versus men,” Hélène Eltchaninoff (University Hospital of Rouen, Rouen, France), the principal investigator in the RHEIA trial, told ESC delegates during her presentation of the results.
Differences in anatomy—in particular aortic annulus size— valve fibrosis, rates of frailty, and comorbidities are among the common differentiators seen in men and women that potentially impact outcomes.
The prospective RHEIA trial recruited an all-comers female population made up of patients with severe symptomatic aortic stenosis at any surgical risk status except for those deemed to be ‘prohibitive’.
A total of 48 sites in 12 European countries participated in the trial, randomising 443 patients 1:1 to undergo either TAVI using the Sapien 3 or Sapien 3 Ultra (Edwards Lifesciences) balloon-expandable valves or surgical valve replacement. Patients had a mean age of 73 years and a mean Society of Thoracic Surgeons (STS) risk score of 2.1–2.2%. They were followed out to one year.
At ESC, Eltchaninoff reported that TAVI resulted in a two-fold lower incidence of the composite of all-cause mortality, stroke or rehospitalisation for valve- or procedure-related symptoms or worsening of heart failure at one year, with the primary endpoint occurring in 8.9% of patients undergoing TAVI, compared to 15.6% for those receiving surgery (p=0.03).
This significant reduction was
predominantly driven by a reduction in rehospitalisation for valve- or procedure-related symptoms or worsening heart failure, which occurred in 4.8% in the TAVI group and 11.4% in the surgical group (p=0.02), though she reported that there was no significant difference in all-cause mortality or stroke.
“In women all comers with severe aortic stenosis, TAVI using Sapien 3 or Sapien 3 Ultra was superior to surgery for the primary composite endpoint of death, stroke or rehospitalisation at one year, and this superiority was essentially driven by the lower rate of rehospitalisation,” Eltchaninoff said. “TAVI, a less invasive technique, had a lower incidence of new onset atrial fibrillation, a quicker recovery, and a shorter length of hospital stay, but higher rates of mild paravalvular regurgitation, and new permanent pacemaker.”
“This is really a unique study; it is the first study only to recruit women patients and to compare TAVI versus surgery, so the results are very important, but we would have expected more, I would say,” Sabine Bleiziffer (Herz- und Diabeteszentrum NRW, Bad Oeynhausen, Germany), remarked following the RHEIA presentation, pointing to the fact that the endpoint was predominantly driven by a lower rate of rehospitalisation in the TAVI arm.
Pressed on this, Eltchaninoff explained that hospitalisations in the surgery arm were often more serious than those in the TAVI group, including complications related to the valve or procedure or worsening of heart failure, compared to local vascular complications or some cases of atrial fibrillation after TAVI.
“The trial certainly provides strong evidence in favour of TAVI in women with symptomatic severe aortic stenosis who are suitable for transfemoral replacement,” Mirvat Alasnag (King Fahd Armed Forces Hospital, Jeddah, Saudi Arabia) tells Cardiovascular News when asked whether the trial justifies TAVI as the preferred strategy for women with severe aortic stenosis.
“Not only did the trialists meet the noninferiority criteria but also superiority for the composite endpoint of all-cause death, stroke and rehospitalisation, with rehospitalisations primarily driving events.
“We know there are unique features
to women such as small annuli, fibrotic valves and concentric remodelling of the left ventricle with preserved systolic function, hence such a randomised trial was necessary. Of course, it doesn’t provide insights into those with high STS score, requiring alternate access or concomitant coronary revascularisation. In addition, these were the one-year outcomes of the RHEIA trial and we would need to see if the curves diverge further after a longer follow-up period.”
Balloon-expandable valves are the most common TAVI devices used in regions including France and the USA, Eltchaninoff explained when asked by Cardiovascular News on the choice of the Sapien platform as the basis for the trial instead of a self-expanding device, such as the Medtronic Evolut platform, which is also in wide use.
“We know that the results are quite different with other valves,
enrolled directly comparable populations, results of the SMART trial have led some to question whether a self-expanding valve should be the valve of choice when performing TAVI in women, as they often have small aortic annuli.
Eltchaninoff pressed back against some of the results seen in SMART, which she said paint a different picture of the performance of the balloon-expandable device than in the RHEIA trial, pointing in particular to differences in rates of patient-prosthesis mismatch and haemodynamic measures seen in the two trials.
“A sub-group analysis of the SMART trial in women, representing 87% of all those enrolled, demonstrated better haemodynamics in small annuli using self-expanding platforms at one year compared with the balloonexpandable platforms,” Alasnag tells
The trial certainly provides strong evidence in favour of TAVI in women with symptomatic severe aortic stenosis”
and we have a recent trial which was published with conclusions which were very different,” Eltchaninoff commented, referring to the SMART trial, a randomised trial comparing the self-expanding and balloon-expandable devices in the small aortic annulus population, which enrolled a majority female population.
The SMART trial, first reported in April this year, found that the supra-annular TAVI platform met the co-primary endpoint of clinical noninferiority against a composite of death, disabling stroke, or rehospitalisation for heart failure, tested for non-inferiority at one year, and a composite of bioprosthetic valve dysfunction, tested for superiority.
Though two studies have not
Cardiovascular News of the balance between the RHEIA and SMART trials. “From the PARTNER trial, however, we know this haemodynamic advantage in small annuli did not translate into better cardiovascular outcomes at five years.
“It would be difficult to draw across-trial conclusions about transcatheter heart valve durability or even in special subsets like bicuspid valves which were excluded. At this point, the type of transcatheter heart valve selection should be individualised.”
TAVI using balloon-expandable devices could be considered the preferred therapy in women with symptomatic severe aortic stenosis, Eltchaninoff concluded of the clinical implications of RHEIA.
She tells Cardiovascular News that the young mean age of the RHEIA trial population—at 73 years—should offer impetus to guideline writers in Europe, where transfemoral TAVI is seen as the gold standard for the treatment of severe, symptomatic aortic stenosis in patients over the age of 75, to lower the age threshold. “[RHEIA] gives some confirmation that maybe we could decrease the age for women, since we have the data for this population and we have excellent results,” she said.
RHEIA
Hélène Eltchaninoff
ESC chronic coronary syndrome guidelines clarify role of revascularisation and strengthen imaging recommendations
The European Society of Cardiology (ESC) has issued updated guidelines on the management of chronic coronary syndrome, with new recommendations covering diagnosis, timing of revascularisation, and a stronger emphasis on the use of intracoronary imaging to guide revascularisation procedures.
Released at ESC’s 2024 congress (30 August–2 September, London, UK), the guidelines should prompt cardiologists to rethink chronic coronary syndromes as caused “not only by blockages in large arteries but also by dysfunction of smaller vessels”, guideline committee co-chair Christiaan Vrints (Antwerp University Hospital, Antwerp, Belgium) comments.
At a session to mark the launch of the new guidelines at the ESC meeting, Francisco Javier Rossello (Son Espases University Hospital, Palma de Mallorca, Spain), a member of the writing committee, detailed that the guidelines advocate a ‘stepwise’ approach to managing individuals with suspected chronic coronary syndrome, incorporating four pillars that encompass general assessment, further assessment, confirmation of diagnosis, and treatment.
Updates in the latest version include a focus on the diagnosis and management of angina/ischaemia with non-obstructive coronary arteries (ANOCA/INOCA) caused by coronary artery spasm or microcirculatory dysfunction. Recommendations state that persistently symptomatic patients with suspected ANOCA/ INOCA who do not respond to guideline-derived medical therapy should undergo invasive coronary functional testing to determine underlying endotypes to guide appropriate medical therapy.
are non-operable or considered to be at high risk for surgery or with anticipated difficult rehabilitation.
Among patients with three-vessel disease without diabetes, CABG is recommended if the individual is not responding to medical therapy, but PCI is recommended in those with low-to-intermediate anatomic complexity.
Speaking at the ESC congress, Julinda Mehilli
on a multitude of randomised controlled trials showing consistent substantial clinical benefit,” said Lorenz Räber (Bern University Hospital, Bern, Switzerland), highlighting the importance of this new recommendation to Cardiovascular News. “Europe is lagging behind, as many believe angio is enough, so the upgrade is expected to fuel the use of imaging for complex lesions. Currently, there is no other technical refinement of PCI that reportedly improves clinical outcomes to the degree observed with intracoronary imaging.”
“Meta-analysis of randomised clinical trials had already shown that intracoronary image guidance of PCI improves patient outcomes and saves lives,” said Javier Escaned (Hospital Clinico San Carlos, Madrid, Spain). “But the IA recommendation for IVUS in the updated ESC guidelines is crucial, as it reflects expert consensus based on a definite body of evidence supporting the positive impact of IVUS, specifically for patients with anatomically complex lesions treated with PCI.”
Intracoronary pressure measurement, fractional flow reserve (FFR), instantaneous wave-free ratio (iFR), or quantitative flow ratio (QFR) are recommended to guide lesion selection in multivessel disease.
Europe is lagging behind as many believe angio is enough”
For individuals with symptoms suggestive of chronic coronary syndrome who have a low to moderate (>5%–50%) likelihood of obstructive coronary artery disease based on symptoms, age, sex and risk factors, the guidelines state that coronary computed tomography angiography (CCTA) is an effective means of ruling out coronary atherosclerosis or, at the other extreme, in estimating the risk of adverse events based on disease anatomy.
Revascularisation
Indications for coronary revascularisation in the 2024 guidelines remain similar to the previous iteration of the document, published in 2018, namely: symptoms related to ischaemia that are refractory to medical therapy alone, and/or significant disease of the left main stem, of the proximal left anterior descending artery, or of multiple large epicardial arteries.
Selection of the revascularisation modality should be based on the patient’s profile, coronary anatomy, procedural factors, patient preferences, and outcome expectations, the guidelines state. Coronary artery bypass graft (CABG) surgery is favoured over percutaneous coronary intervention (PCI) in patients with diabetes or reduced left ventricular ejection fraction (LVEF, <35%), though PCI may be considered as an alternative to CABG in those who
(Hospital Landshut-Achdorf, Landshut, Germany) stressed the importance of applying a shared decision-making process when considering these recommendations.
“We should apply a patient-centred decision, particularly for patients with LV dysfunction to choose between revascularisation or medical treatment,” said Mehilli, referring specifically to the recommendations relating to patients with reduced LVEF. “We need to have a careful evaluation, preferably by the heart team, of coronary anatomy, correlation between coronary anatomy and LV dysfunction, comorbidities, life expectancy, individual risk to benefit ratio, and what is the patient's perspective.”
Intracoronary imaging
Among the major changes to the latest version of the guidelines is a new emphasis intravascular imaging. When performing revascularisation via PCI, intracoronary imaging—in the form of intravascular ultrasound (IVUS) or optical coherence tomography (OCT) —is deemed as being helpful to guide interventions and enhance results, especially in complex scenarios including left main disease, bifurcations, or long lesions.
“The change to a IA recommendation is based
DCB or DES for in-stent restenosis?
Further recommendations apply when patients require a repeat revascularisation—representing around one in five of those undergoing CABG or PCI within five years, according to figures quoted within the guidelines. Significantly, recommendations favour the use of drug-eluting stents (DESs) over drug-coated balloons (DCBs) for the treatment of in-stent restenosis following PCI. This has elicited surprise from some corners, given that recent evidence has suggested that DCBs may be a favourable option in this scenario.
“I find it strange that after the positive results of the AGENT IDE trial, with consequently the possibility of using a DCB for in-stent restenosis in the USA (despite the lack of reimbursement so far), the ESC guidelines have decided to step back from a substantial equivalence in the indication in using a DCB or a DES, coming to favour a DES approach,” Bernardo Cortese (UH Harrington Heart & Vascular Institute, Cleveland, USA, and Fondazione RIC, Milan, Italy) tells Cardiovascular News
Cortese points out that the recently published 10year results of the ISAR DESIRE 3 trial—in which investigators compared PCI using paclitaxel-coated balloons and DESs for the treatment of in-stent restenosis—“showed substantial equivalence between DCB and DES from the clinical standpoint, with a signal of excess mortality in the DES group”, which, he says, makes it unclear why the guidelines now favour DES over DCB.
“I believe that we should run a modern study with an ad hoc protocol of lesion preparation before DCB or DES, with a stepwise implementation of intracoronary imaging during all the stages of ISR-PCI.”
Cortese also expressed surprise at the absence of an indication for DCB as a therapeutic alternative in the de novo setting, highlighting that several trials have shown good outcomes compared to stents, with similar or improved angiographic performance, along with improved long-term performance. A patient-level meta-analysis from the ANDROMEDA registry will offer yet further evidence in this area, he says.
Coronary bioadaptor noninferior to DES at one-year for TLF, with clinical benefit in complex subgroups
The DynamX (Elixir Medical) bioadaptor system met noninferiority at one year for target lesion failure (TLF) in a trial comparing the device against the Resolute Onyx (Medtronic) drug-eluting stent (DES) in a broad population of patients undergoing percutaneous coronary intervention (PCI).
THIS IS THE HEADLINE finding from the INFINITYSWEDEHEART trial, presented by David Erlinge (Lund University, Lund, Sweden) during a Hot Line trial session at the 2024 European Society of Cardiology (ESC) congress (30 August–2 September, London, UK).
The DynamX device is implanted in a similar fashion to a DES but begins “unlocking” after it is encapsulated with tissue and its absorbable polymer coating is resorbed. This is intended to allow the vessel to grow and adapt to maintain established blood flow lumen.
Erlinge later presented analysis at TCT 2024 (27–30 October, Washington, DC, USA) on the performance of the device in complex patient population subsets, including patients with acute coronary syndrome (ACS), small vessel lesions, and lesions within the left anterior descending (LAD) artery, demonstrating “significant benefit” of the bioadaptor compared to stent.
INFINITY- SWEDEHEART set out to evaluate the safety and efficacy of the bioadaptor compared to the DES in a population representative of every day clinical practice, including a large
proportion of ACS patients, from 20 sites across Sweden.
The trial enrolled 2,400 patients (aged between 18 and 85 years) requiring PCI who had previously untreated chronic coronary syndrome (CCS) or ACS, and had achieved a successful target vessel pre-dilation. Patients were randomised in a 1:1 ratio to either bioadaptor (1,201 patients; average age 68 years; 24% female) or DES (1,198 patients; average age 68 years; 24% female).
The primary endpoint was the TLF rate, defined as a composite of cardiovascular death, target vessel myocardial infarction (TV-MI) and
Orbital atherectomy fails to eclipse balloon angioplasty in randomised trial in severely calcified lesions
The routine use of orbital atherectomy prior to percutaneous coronary intervention (PCI) in severely calcified lesions does not improve outcomes compared to conventional balloon angioplasty, findings of the ECLIPSE clinical trial have shown.
THE LARGE-SCALE TRIAL, THAT enrolled more than 2,000 patients at over 100 sites throughout the USA, showed that the two strategies were similar for a primary clinical endpoint of target vessel failure (TVF), defined as the composite of cardiac death, target vessel-related myocardial infarction, or ischaemia-driven target vessel revascularisation at one year follow-up.
ischaemia-driven target lesion revascularisation (ID-TLR), at one year. At one year, Erlinge reported that there was an 18% reduction in the TLF rate of the bioadaptor compared to DES (2.35% vs 2.77%), demonstrating non-inferiority (p<0.001), driven by low rates of TV- MI and ischaemia-driven TLR with bioadaptor compared to DES.
The one-year results also demonstrated a 14% reduction in the target vessel failure (TVF) rate (3.03% vs. 3.52%) with the bioadaptor compared to DES.
Further prespecified powered landmark analyses showed a significant reduction and plateau in TLF (0.2% vs. 1.3%, p=0.003) and TVF (0.6% vs. 1.8%, p=0.008) events after six months, when the bioadaptor’s mechanism of action is enabled, driven by a
With sustained reduction and plateauing of TLF after six months as compared to stents, the data further validate the substantial clinical benefit of Bioadaptor”
favourable reduction in CV death, TVMI, and ischaemia-driven TLR with the bioadaptor compared to DES.
At TCT, Erlinge showed that after six
The results were presented by Ajay Kirtane (Columbia University Irving Medical Center, New York, USA) at TCT 2024 (27–30 October, Washington, DC, USA), and have led one investigator in the study to conclude that orbital atherectomy should be “reserved for the most extreme cases”, including those where the operator does not believe that balloon angioplasty would be likely to safely cross or predilate the calcified lesion.
From March 2017 to April 2023, a total of 2,005 patients (2,492 lesions) were enrolled, with a mean a age of 70 years. Among the patient cohort 27% were female, 44% had diabetes and 24% had chronic kidney disease.
By angiographic core laboratory analysis, mean reference vessel diameter was 3mm, mean lesion length was 28.7mm, and 97.1% of lesions met criteria for severe calcification. A large proportion (62%) of patients underwent intravascular imaging.
Patients were randomised after successful wire crossing to either the orbital atherectomy strategy (n=1,008) or conventional balloon angioplasty (n=997) prior to second generation drug-eluting stent implantation and optimisation. Procedural complications were largely similar between groups.
months, clinical results in patients with ACS demonstrated TLF of 0.3% versus 1.8% (p<0.018), translating into 83% reduction and significant benefit for the bioadaptor compared to DES.
Additional analysis of clinically complex lesions associated with adverse DES outcomes, such as the left anterior descending (LAD) artery and small vessels, demonstrated 73% reduction (0.2% vs. 2.2%) in TLF rate after six months with the bioadaptor compared to treatment with DES. There were no TLF events in patients with small vessels (less than or equal to 2.75mm) in the bioadaptor arm compared to 1.8% TLF with DES after six months.
“Historically, patients with ACS are at a higher risk for adverse events after PCI in part because of higher rates of comorbidities, reduced heart function, and more frail patient condition,” said Erlinge, the study’s principal investigator.
“Analysis of this critical population was key to understanding the effects of the bioadaptor’s unique mechanism of action on improving safety and effectiveness after PCI. With sustained reduction and plateauing of TLF after six months as compared to stents, the data further validate the substantial clinical benefit of Bioadaptor and its potential to impact the treatment and long-term success in high-risk patient populations.”
DynamX has been granted US Food and Drug Administration (FDA) breakthrough device designation for an indication to improve coronary luminal diameter, restore haemodynamic modulation, and reduce plaque progression in symptomatic ischaemic heart disease due to discrete de novo native coronary artery lesions.
The primary imaging endpoint consisted of the acute post-PCI minimal stent area at the site of maximum calcification as assessed by optical coherence tomography (OCT) in a pre-specified cohort of 555 subjects. Stent areas were not appreciably different between the two groups, the investigators reported.
The primary clinical endpoint of TVF at one year follow-up occurred in 11.5% in the orbital atherectomy group compared with 10% in the traditional balloon angioplasty group. The two secondary endpoints of procedural success and strategy success without the need for crossover were similar in both groups.
“Compared with conventional balloon angioplasty, the routine use of orbital atherectomy did not reduce minimal stent area or target vessel failure,” said Kirtane. “The high use of intravascular imaging within this trial was remarkable and was associated with improved outcomes in both treatment groups.
But the take home message for me is that we showed that adequate stent expansion and low rates of adverse outcomes are achievable with conventional balloon angioplasty if meticulous attention is paid to lesion preparation, further highlighting the importance of randomised trials to inform treatment strategies.”
David Erlinge
ORBITAL ATHERECTOMY
BALLOON ANGIOPLASTY
Invasive strategy does not reduce risk of cardiovascular death or non-fatal MI in older NSTEMI patients
A trial comparing an invasive and a conservative strategy to treat patients over the age of 75 years with a non-ST-elevation myocardial infarction (NSTEMI)—SENIOR-RITA—has shown that there was no significant reduction in the combined risk of cardiovascular death or non-fatal myocardial infarction (MI) with the invasive strategy.
PRESENTING THE RESULTS OF THE STUDY, the largest of its kind in this population, at the 2024 European Society of Cardiology (ESC) congress, study chair and chief investigator Vijay Kunadian (Translational and Clinical Research Institute, Newcastle University and Freeman Hospital, Newcastle-Upon-Tyne, UK) said that though the invasive strategy did not reduce the primary endpoint, it did appear to be safe overall in older patients.
“Among older adults with type 1 NSTEMI our study showed that an invasive strategy, doing an angiogram and revascularisation procedure,” Kunadian reported. “An invasive strategy did not actually risk the combined risk of cardiovascular death and nonfatal MI, as compared to a conservative strategy of medications alone in these patients.
“However, treatment with an invasive strategy did reduce the risk of non-fatal MI, and subsequent revascularisation. Our study, in a sense, provides a foundation for older heart attack patients and their clinicians to make informed decisions about whether they need to undergo invasive procedures or not.”
In higher-risk patients after NSTEMI, guidelines recommend an invasive strategy over medications alone. However, older patients with NSTEMI are less likely to receive guideline-recommended care including an invasive strategy, Kunadian detailed, citing a potential fear of the risk of procedural complications among the possible obstacles. Older patients have also been underrepresented in clinical trials of NSTEMI therapies.
In the open-label SENIOR-RITA trial, patients aged ≥75 years presenting with type 1 NSTEMI were randomly allocated (1:1) to one of two treatment groups. In the conservative strategy group, patients received ESC Guidelinerecommended secondary prevention therapy, including antiplatelet therapy, statins, angiotensin-converting enzyme inhibitors and beta-blockers.
Patients randomised to the invasive strategy group, in addition to these medications, patients had invasive coronary angiography and, if deemed necessary, coronary revascularisation— percutaneous coronary intervention (PCI) or coronary artery bypass graft (CABG) surgery.
48 NHS sites across England and Scotland. The mean overall age was 82.4 years and 72% were aged 80 years or older (the oldest being 103 years old). Almost half were female (45%). Overall, 80% of patients were classified as prefrail or frail, more than 60% had cognitive impairment and the majority had a comorbidity index of ≥5, indicating multiple concurrent long-term conditions. Medical therapy was balanced between the two groups.
In the invasive group, 90% had the intended angiography and 50% had revascularisation procedures during hospitalisation, Kunadian reported.
After median follow-up of 4.1 years, there was no difference in the primary endpoint of cardiovascular death or nonfatal MI between the invasive strategy group (25.6%) and the conservative strategy group (26.3%, p=0.53). This pattern was observed for the different prespecified subgroups (including those who were frail, cognitively impaired or had multiple comorbidities).
CARDIOVASCULAR DEATH OR NON-FATAL MI
No differences were observed for cardiovascular death (15.8% with invasive strategy vs. 14.2% with conservative strategy). There was a significant reduction in non-fatal MI, which occurred in 11.7% of patients in the invasive strategy group vs. 15% in the conservative strategy group.
All patients had formal assessment of frailty, cognition and co-morbidity at baseline and follow-up. The primary endpoint was time to cardiovascular death or non-fatal MI. Secondary endpoints included components of the primary endpoint, all-cause death, subsequent coronary revascularisation and bleeding complications.
Invasive strategy Conservative strategy
In total, 1,518 patients were recruited from around
New evidence suggests fasting not needed before cardiac catheterisation procedures
A randomised trial testing whether the removal of fasting requirements prior to cath lab procedures requiring conscious sedation has any impact on procedural safety and patient satisfaction has found no difference in complications in patients who fasted or did not fast before cardiac catheterisation procedures.
DAVID FERREIRA (JOHN Hunter Hospital, Newcastle, Australia) presented findings of the SCOFF trial during a Hot Line session at the 2024 European Society of Cardiology (ESC) Congress (30 August–2 September, London, UK), where he argued that these results, alongside other existing evidence from studies such as CHOWNOW, TONIC and Fast-CIED, should open the discussion about changes to guidelines recommending fasting as standard before cardiac catheterisation.
“This is an incredibly important question, because this is bread and butter cardiology,” said Ferreira. “Millions of patients undergo coronary-devicerelated procedures every year, and so this is an important clinical question.
“The main take home message from this trial is that, based on the data that we present, removing fasting requirements was safe, and improved patient satisfaction for those undergoing cardiac procedures that require conscious sedation.”
The investigator-initiated, randomised SCOFF trial, with a prospective openlabel, blinded endpoint design, assessed the non-inferiority of no fasting prior to cardiac cath lab procedures requiring conscious sedation. Patients who had been referred for coronary angiography, coronary intervention or cardiac implantable electronic device-related procedures were recruited.
They were randomised 1:1 to fasting before the procedure (no solid food for
Patients in the invasive strategy group also required fewer subsequent revascularisation procedures than those in the conservative strategy group (3.9% vs. 13.7%). There were no observed differences in the other secondary outcomes, including all-cause death, all MIs combined, stroke, hospitalisation for heart failure or any bleeding complications. The rate of procedural complications was less than 1%.
six hours and no clear liquids for two hours) or to no fasting where the patient was encouraged to eat as usual.
The primary composite endpoint was hypotension, aspiration pneumonia, hyperglycaemia and hypoglycaemia assessed with a Bayesian approach. Secondary endpoints included contrastinduced nephropathy, new intensive care admissions post-procedure, new ventilation requirements post procedure, new intensive care unit admissions, 30-day readmissions, 30-day mortality, 30-day pneumonia and pre-procedure patient satisfaction. In total, 716 patients were recruited from six sites in New South Wales, Australia. The mean age was 69 years and 35% were female.
The primary composite outcome occurred in 19.1% in the fasting group and 12% in the no-fasting group. In an intention to treat analysis, the estimate of the mean posterior difference was
−5.2% (95% confidence interval [CI] −9.6 to −0.9) favouring no fasting. This result confirmed the noninferiority of no fasting, based on a non-inferiority margin of 3% with a likelihood of greater than 99.5%. No fasting was also potentially superior to fasting for the primary outcome with a likelihood of 99.1%. There was an absolute risk difference between the groups of 7.1% in favour of no fasting, with a number needed to treat of 14.1 to prevent one primary outcome event. In analyses of secondary outcome events, no differences were observed without and with fasting. Patient satisfaction was significantly better without fasting versus with fasting when assessed via a questionnaire: 11 vs. 15 points where a lower score indicates greater satisfaction (posterior mean difference, 4.02 points; 95% CI 3.36 to 4.67; Bayes factor ≥100).
Removing fasting requirements was safe and improved patient satisfaction for those undergoing cardiac procedures that require conscious sedation”
Digital “prehabilitation”
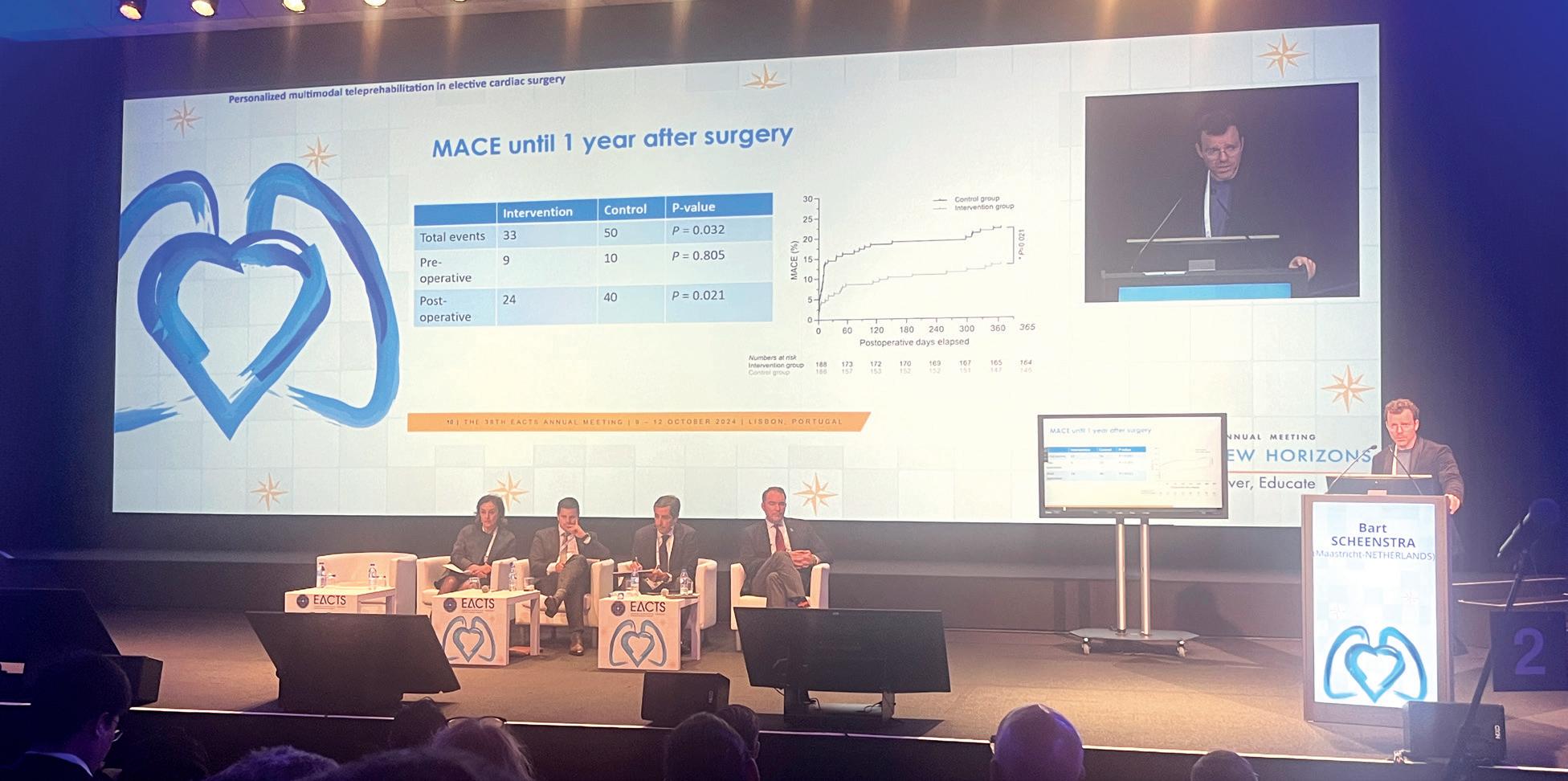
programme reduces adverse events after cardiac surgery
Patients awaiting cardiac surgery who underwent a digital “prehabilitation” programme benefited from an improvement in major adverse cardiovascular events (MACE) compared to patients who did not undergo the same intervention prior to their surgery.
This is the conclusion of the Digital Cardiac Counseling Trial, a randomised trial led by investigators in Maastricht, The Netherlands, who—during the COVID-19 pandemic—used a digital application to deliver patients a series of rehabilitation modules, tailored to their specific modifiable risk-factors, aimed at improving outcomes following their procedure.
Results of the trial were presented at the European Association of Cardio-Thoracic Society (EACTS) 2024 annual meeting (9–12 October, Lisbon, Portugal), and simultaneously published in the Journal of the American College of Cardiology (JACC), in what has been described as a first for research presented at a cardiothoracic surgery meeting.
“Cardiac surgery saves lives, there is no doubt, but what if we could improve patient outcomes by starting rehabilitation programmes weeks before the upcoming surgery?” posed study investigator Bart Scheenstra (Maastricht University Medical Center, Maastricht, The Netherlands).
“We already know from previous research in cardiac prehabilitation that it improves quality of life, it reduces length of hospital stay, and it reduces complications. We do not know what the effect is of these programmes on major adverse cardiovascular events,” he said.
To investigate this, the trial included 394 patients scheduled for elective cardiac surgery or transcatheter intervention who were referred at several centres throughout The Netherlands to the Maastricht University Medical Center. Patients enrolled were either randomised to the investigational group (n=197), where they were given access to an online multimodal teleprehabilitation programme, or assigned to a control arm
(n=197), where no additional intervention was given. Patients were screened prior to randomisation, and those in the intervention group were offered remote rehabilitation modules that included a smoking-cessation programme, nutritional counselling, psychological-education, inspiratory muscle training, and exercise training, based upon their individual risk factors, delivered over a period of around six to eight weeks.
The MACE endpoint consisted of a composite of cardiovascular death, myocardial infarction (MI), stroke, hospitalisation for heart failure or other life-threatening cardiac events, and earlier or repeated intervention. Outcomes were assessed by an independent, blinded, adjudication committee.
Results of the trial presented by Scheenstra and detailed in JACC showed that from randomisation until one year postoperatively, the primary endpoint occurred in 33 patients (16.8%) in the teleprehabilitation group and 50 patients (25.5%) in the control group.
The difference was primarily driven by a reduction in hospitalisations, with sensitivity analyses showed that treatment effect was mainly in the patients undergoing cardiac surgery rather than transcatheter procedure.
Teleprehabilitation also reduced the incidence of active smoking, elevated pulmonary risk scores, and elevated depression scores. There was no significant difference in postoperative length of hospital stay, occurrence of postoperative complications, physical fitness, incidence of obesity, or malnutrition the study shows.
“What is really important here is that this is a trial that empowered patients to take care of their care, and to do something about their risk factors at their own home,” the trial’s principal investigator Peyman Sardari Nia (Maastricht University Medical Center, Maastricht, The Netherlands) commented. “Even if they don’t participate in all those modules for which they actually have risk factors, this trial shows that you will still have an effect, because it is possible to integrate into the care that we already have, because it moves the care to the patients’ home.”
This is a trial that empowered patients to take care of their care, and to do something about their risk factors at their own home”
“No need” for potassium supplementation after CABG
SUPPLEMENTING POTASSIUM for patients who have undergone coronary artery bypass graft (CABG) surgery—a routine practice following the procedure—may not be necessary according to new evidence presented at the European Society of Cardiology (ESC) 2024 Congress (30 August–2 September, London, UK).
Investigator Benjamin O’Brien (Deutsches Herzzentrum der Charité, Berlin, Germany) reported that giving potassium supplements only when levels dropped below the lower limit of normal was non-inferior to routinely supplementing potassium to the upper limit of normal.
The trial enrolled patients with no history of atrial dysrhythmias and scheduled for isolated CABG surgery were recruited across 23 centres in the UK and Germany. Patients were randomised in a 1:1 ratio to a strategy of tight potassium control (potassium supplementation if serum levels fell below 4.5mEq/L) or relaxed potassium control (potassium supplementation only if serum levels fell below 3.6mEq/L).
The primary endpoint was the presence of new-onset atrial fibrillation (AF) after cardiac surgery (AFACS) in the 120 hours (5 days) after the operation, or until discharge, whichever was sooner. In total, 1,690 participants were randomised, with a mean age of 64.7 years and 15% were female. The mean European System for Cardiac Operative Risk Evaluation (EuroSCORE) II score was 1.5%.
There was no significant difference in the primary endpoint, which occurred in 27.8% of patients in the relaxed control group and 26.2% in the tight control group. The rate of AFACS detected by any means (clinically and/or by ambulatory heart rhythm monitoring) was 33% in both groups.
“We were able to show that routinely supplementing potassium for tight control offers no benefits compared with relaxed control but is more expensive. Unnecessary intervention can carry risks, such as drug errors, and can negatively impact the patient experience, for example, the unpleasant taste of oral potassium supplements,” said O’Brien. “So, the results from TIGHT-K are good news—we can safely stop the widespread practice of maintaining high-normal potassium levels after isolated CABG, improve the patient experience and also save money.”
EACTS
No-touch graft harvesting technique shows “no benefit, but significant harm” in SWEDEGRAFT study
A multicentre, registry-based, randomised trial of the ‘no-touch’ technique for graft harvesting for coronary artery bypass graft (CABG) surgery has shown that the technique is not superior to the conventional method for reducing vein graft failure or improving clinical outcomes.
The no touch technique, which was developed in Sweden, is an atraumatic approach to remove the saphenous vein complete with its cushion of surrounding tissue without touching the vessel at all, unlike traditional vein harvesting where all of the tissue is stripped from the vein.
Stefan James (Uppsala University Hospital, Uppsala, Sweden) presented findings of the
SWEDEGRAFT study comparing the two techniques at the European Society of Cardiology (ESC) 2024 Congress (30 August–2 September; London, UK) where he reported that as well as failing to show superiority over the traditional vein harvesting technique, the use of no-touch vein harvesting also raises severe safety concerns.
Several trials have shown improved graft patency using the no-touch technique, James said, and the evidence has led to no-touch vein harvesting holding a IIa B recommendation in 2018 ESC and European Association of Cardio-Thoracic Surgery (EACTS) guidelines on myocardial revascularisation. Many sites in Europe as well as the USA use this as a standard technique, he said.
SWEDEGRAFT set out to test whether this strategy was superior to conventional grafting in patients aged under 80 years old undergoing first-time isolated non-emergent CABG with at least one saphenous vein graft. Taking place at all eight surgical sites in Sweden and one in Denmark, investigators randomly assigned patients 1:1 to either of the two vein harvesting techniques.
The trial’s primary endpoint was the proportion
Robotic aortic valve replacement shows promise as a TAVI alternative
Robotic aortic valve replacement (RAVR) may offer an alternative to transcatheter aortic valve implantation (TAVI) as a minimally invasive option for the treatment of symptomatic aortic valve disease.
THIS WAS THE VIEWPOINT
offered by Vinay Badhwar (West Virginia University, Morgantown, USA) at the European Association of Cardio-Thoracic Surgery (EACTS) 2024 annual meeting (9–12 October, Lisbon, Portugal), where he reported longitudinal outcomes from the first 300 patients undergoing RAVR across 10 established robotic cardiac surgery programmes at centres in the USA, Europe, Saudi Arabia, Brazil and Australia covering 2020–2024.
“As we all know minimally invasive surgical and transcatheter options for the management of symptomatic aortic valve disease are increasingly sought by providers as well as patients, but the debate continues as the incursion of
transcatheter therapies enters into low to intermediate risk,” Badhwar said.
The RAVR procedure is performed in a similar fashion to robotic mitral valve surgery, with the patient positioned in the same way and accessed via a 3cm transaxillary right lateral mini thoracotomy. Importantly, according to Badhwar, the RAVR procedure facilitates the use of a traditional rather than a sutureless aortic valve prosthesis—for which there is a larger pool of available long-term outcome data—whilst also allowing concomitant mitral or tricuspid valve procedures, as well as arrhythmia therapies such as left atrial appendage closure.
At EACTS 2024, Badhwar reported 30-day and one-year echocardiographic
of patients with graft failure, defined as an occluded or stenosed graft >50% on coronary computed tomography (CT) angiography at least two years after the procedure, clinically driven coronary angiography demonstrating an occluded or stenosed >50% vein graft, or death within two years.
A total of 902 patients were enrolled in the trial, with a mean age of 67 years and 88% were male. James reported that there was no significant difference in the primary endpoint of graft failure within 3.5 years, which occurred in 19.8% of patients in the no-touch group and 24% of patients in the conventional group (p=0.15). There were no significant differences in the three individual components of the primary endpoint.
Our trial does not support the current guidelines in using this strategy for bypass surgery”
Regarding secondary endpoints, James reported that the incidence of major adverse cardiovascular events was similar 12.6% vs. 9.9% in the two groups at a mean follow-up of 4.4 years, but there were significantly more leg wound complications among patients randomised to the no-touch technique compared to conventional grafts at three months (24.7% vs. 13.8%) and more negative leg symptoms at 2 years (49.6% vs. 25.2%).
“It [the trial] did not show superiority for vein graft failure or clinical outcomes, but severely increased the risk of leg complications and residual symptoms in these patients, so our trial does not support the current guidelines in using this strategy for bypass surgery,” James said at ESC 2024.
and clinical outcomes for the first 300 patients to undergo the procedure up until July of 2024.
Patients had an average age of 67 years, with a mean predictive risk of mortality of 1.6% according to Society of Thoracic Surgeons (STS) criteria. All of the patients had severe aortic stenosis, 44.7% had bicuspid valves, and nearly 40% greater or equal to moderate aortic insufficiency. Over 10% had a concomitant aortic root enlargement, whilst 17% had concomitant procedures.
patients, whilst median length of stay stood at five days.
The mean aortic valve gradient stood at 9mmHg at 30 days, increasing to 10mmHg at one year, with prosthetic/ paravalvular leak (PVL) of 2+ reported in only two patients (<1%) across both timepoints. None had PVL of 3-4+.
2.3%
pacemaker implantation rate following robotic aortic valve replacement
Overall 30-day and one-year outcomes showed that the operative mortality rate stood at 0.7%, with a stroke rate of 1%, 1.7% experienced renal failure, 4.3% required prolonged ventilation, and 2.3% required pacemaker implantation, with Badwhar noting that this was seen to be “much higher” in trials of TAVI in lowrisk patient groups. Reoperation for bleeding was required in 8.3% of
“In centres with an established robotic programme, RAVR does appear to be reproducible and safe, with both 30-day and one-year outcomes that are at least equivalent to those of open surgery, with a lower trend, perhaps of stroke and pacemaker rates,” said Badhwar of the results. “A comment is that as transcatheter aortic procedures are being offered to lower and lower risk patients and perhaps even bicuspid disease, RAVR may provide an important alternative option for the management of symptomatic aortic valve disease and consideration in our global heart teams.”
Stefan James
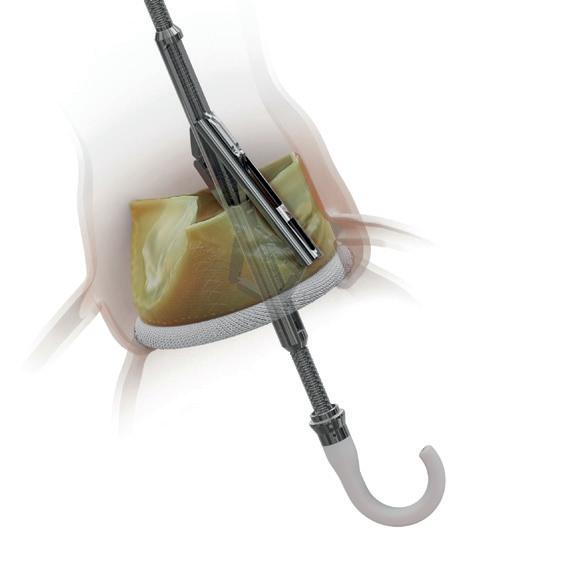
PERIGON trial demonstrates durability of Avalus aortic valve at seven years
Seven-year results of the PERIGON pivotal trial, assessing the clinical and haemodynamic performance of the Avalus (Medtronic) pericardial aortic bioprosthesis, demonstrate the sustained durability of the device, investigators say.
JOSEPH SABIK (CLEVELAND MEDICAL CENTER, Cleveland, USA), the trial’s North America principal investigator, presented the findings during a late-breaking trial session at the European Association of CardioThoracic Surgery (EACTS) 2024 annual meeting (9–12 October, Lisbon, Portugal). Sabik reported that the valve demonstrated low rates of mortality, reintervation, valve deterioration or dysfunction at the seven-year timepoint— describing this as a “critical time” to evaluate the durability and haemodynamic stability of new tissue valves.
“Bioprosthetic surgical valves are prone to deterioration over time,” he commented in his presentation of the results, which included data from 458 of the trial’s 1,132 patients who have reached the seven-year mark. A total of 39 North American and European centres participated in the study.
Patients had a mean age of 70 years, 75.4% were male and 84.3% were treated for aortic stenosis. Thirty percent of patients had a bicuspid aortic valve, Sabik noted. Multiple valve sizes were used, spanning 17–29mm. The majority of patients received 21mm (18.6%), 23mm (35.4%) or 25mm (30.9%) valves.
Sabik reported that at the seven-year timepoint, freedom from all-cause mortality stood at 83%, freedom from cardiac mortality at 91%, and freedom from valve-related mortality 97%. Additionally, freedom from all reintervention stood at 94.3%, while freedom from reintervention for structural valve deterioration or severe haemodynamic dysfunction stood at 98.8%.
PERIGON trial investigators defined structural valve deterioration as a change in the function of the valve, Sabik explained, resulting from an abnormality causing stenosis or regurgitation confirmed by examination of explanted valves. However, he detailed that was not always adequate information to definitively determine the cause of the valve failure in cases where this occurred.
A severe haemodynamic dysfunction endpoint was used to categorise potential safety events that were characterised by severe aortic stenosis, severe transvalvular regurgitation, or progressive severe dysfunction that did not have the evidence to meet the protocol definition of structural valve deterioration or non-structural valve deterioration.
“These cases were likely structural valve deterioration but were unable to be
adjudicated as structural valve deterioration due to a lack of explanted valves from transaortic valves and surgical aortic valves,” said Sabik.
Reinterventions were required in 5.7% of patients at seven years, he noted, adding that reintervention rates were similar to those aged 65 years and above or below 65. Haemodynamic results remained stable through seven years, with mean effective orifice areas (EOAs) of 2cm2 and mean gradients of 13mmHg.
“At seven years, 98.3% of patients had none or trace transvalvular regurgitation; 99% of patients had none or trace paravalvular leak, and at one and seven years nearly 80% of patients had no prosthesis-patient mismatch (PPM). Only 3.5% of patients had severe PPM at seven years,” revealed Sabik.
The PERIGON seven-year results demonstrate excellent durability of the Avalus valve”
“The PERIGON seven-year results demonstrate excellent durability of the Avalus valve, and continued low rates of mortality, reintervention and structural valve deterioration or severe haemodynamic dysfunction requiring reintervention,” he concluded. “Importantly, reinterventions were rare in both younger and older patients, and the haemodynamic performance was excellent and stable through seven years.”
Sabik’s presentation of the seven-year results comes as Medtronic launches the latest-generation of the Avalus device—Avalus Ultra—in Western Europe. Available in the USA since mid 2024, the valve is designed to facilitate ease of use at implant with a low valve profile, and a radiopaque coil to aid potential transcatheter aortic valve-in-surgical aortic valve (TAV-in-SAV) procedures.
The first implantation of the Avalus Ultra valve in Western Europe occurred at the Heart Center (Leipzig, Germany) where Michael Borger and his team successfully conducted two aortic valve replacements using the valve through minimally invasive access.
CABG shows benefit over PCI in patients with diabetes and multivessel disease in Swedish registry study
Coronary artery bypass graft (CABG) surgery is associated with significantly lower all-cause and cardiovascular mortality risk and longer median survival time compared to percutaneous coronary intervention (PCI) in patients with diabetes and multivessel coronary artery disease, a new analysis from the SWEDEHEART registry has shown.
THESE FINDINGS WERE presented by Emma Hansson (University of Gothenburg, Gothenburg, Sweden) as late-breaking science at the European Association of Cardio-Thoracic Surgery (EACTS) 2024 annual meeting (9–12 October, Lisbon, Portugal).
SWEDEHEART is the Swedish national registry recording outcomes of patients hospitalised for acute coronary syndrome or undergoing coronary or valvular intervention. Hansson and colleagues also used data from the country’s coronary angiography and angioplasty registry and other healthcare registries, with information on hospitalisation, drug prescription, socioeconomic status and mortality, to quantify the mean survival time after CABG or PCI.
Data were gathered for all revascularisations taking place between 2006–2020, excluding emergency cases, giving a total of 26,166 patients. Of these, 64% underwent PCI and 36% underwent CABG, with data available at a median follow-up of five years.
The investigators noted that there were differences in which treatment patients were likely to be offered based upon where in the country they lived, with patients in some regions eight times more likely to have PCI than in others.
CABG was associated with a lower rate of all-cause mortality, with cardiovascular mortality also lower among patients who underwent surgery. Surgical patients had a median survival benefit that was 0.9 years greater than those undergoing PCI, with this benefit seen to be larger among women (1.5 years).
Hansson reported a longer survival time in patients with left main stem stenosis, who gained a median of 4.3 years if they underwent CABG versus PCI. In patients with three-vessel disease, there was also a “pronounced” increase in median survival, she noted, though in those with a lower complexity of disease the same effect was not observed.
RAFAEL SÁDABA
Hailing from Pamplona, northern Spain, cardiac surgeon Rafael Sádaba was born and raised in a medical family. Following an education that took him across the world to Australia via East Yorkshire, Sádaba has returned to his native Spain where he is now professor of cardiac surgery at Complejo Hospitalario de Navarra. An active and prominent member of the cardiac surgery community, Sádaba tells Cardiovascular News about the importance of cross-border collaboration and where he sees the specialism heading in the future.
How did you arrive at a career in medicine, and then why did you gravitate to cardiac surgery?
When I was born in 1967 my father was working as a cardiologist at a lung hospital in northern Spain. Cardiac surgery was in its very beginnings there, with thoracic surgeons already starting to do some of these procedures. My father was a junior doctor at the time, and although he was a cardiologist, he was very much involved in the few cardiac surgery cases that were being done at the time.
One of my very early memories is of my mother telling me that my father was performing an “extracorporeal” operation that day, an operation with a heart-lung machine.
Though I didn’t know what it meant, I knew that he wouldn’t be home until very late into the evening. I spent my very first few years in that hospital as my parents had been living in hospital accommodation, so you could say that I was born into it.
You may ask why I didn’t follow my father’s footsteps into cardiology, but I always joke that sons should go further than their fathers! I like being more effective than prescribing drugs, that was what made me go into medicine and cardiac surgery.
As one of three brothers, my father was very supportive of us, and all three of us are doctors. One is an interventional cardiologist, one a radiologist, and myself a cardiac surgeon. Of all the subjects in medical school, the cardiovascular physiology and system was the one I always found the most interesting, dedicated more time to, and eventually that made me go into cardiac surgery.
Who have been the biggest influences on your career?
I went to medical school in Bilbao in the Basque country, but I always wanted to go elsewhere to see the world—I didn't care where, I just wanted to move. That took me to the UK, where I ended up in various different positions. I have a strong tie to Yorkshire, in northern England, having done my basic surgical training in Hull and then going on to do higher training in cardiothoracic surgery in Leeds. My eldest son was born in Beverley and my other two children, a son and a daughter, were both born in Leeds. After that I had a big opportunity to spend a year in Perth, Australia, to undertake cardiac surgery training.
Do I have a particular mentor? No. The surgeons with whom I worked in Leeds, Joe McGoldrick, Philip Kay and Chris Munsch, to name a few, were like my parents and I have great memories of all of them, as well as Mark Newman in Perth.
My experiences overseas have been crucial to my surgical career. I not only thoroughly enjoyed collaborating with diverse individuals and adapting to various work environments, but I also gained a broader perspective that allows me to respect and appreciate different methods for addressing similar challenges.
For me it was very enriching to go to these different places. Any doctor, whether they specialise in cardiac surgery, cardiology or any other specialty, should take the opportunity to go to new places to learn. You gain something just from being there and seeing how things are done differently. Not wrong, better or worse, but differently. You learn things from what you see, from ways of doing things that are different, and you learn a lot from the people you meet. That personal touch, knowing different personalities and how they face the same problem in different ways.
Perhaps the biggest influence, however, was that of my father. He basically taught me the craft of medicine, how to be a good doctor.
During your career what has been the biggest change in cardiac surgery?
Being able to offer less invasive treatment for patients has been a change in mentality. In the beginning, it was big surgeries with big wounds, but now there has been a shift to less invasive procedures. Why I think this is important goes back to what is valuable to a patient. If you operate on somebody and you tell them that they cannot go back home for another six or seven days, or back to work for another two or three months, that can be very difficult.
Minimally invasive surgery means we can get people back to their normal activities, quicker, with similar long-term results. That has been the important shift in mentality and what makes interventional cardiology very competitive. I have grown up in the age of maximally invasive surgery, so I don’t consider myself an expert in minimally invasive procedures, but I make sure that is how my team sees the future and what we are preparing to move towards.
What has been your main area of focus for research?
As a specialty, cardiac surgery has not been very research oriented and most of the research we do is based on retrospective analysis of patients. That is one of the things we need to improve upon and develop further, and something I have a special interest in. At my institution we have a basic science lab which concentrates on translational research in cardiology and my interest has been at the level of basic science in heart valve disease
FACT FILE
CURRENT APPOINTMENTS
Chief of cardiac surgery and associate professor of surgery, Navarra University Hospital, Pamplona, Spain
Area coordinator, cardiovascular and renal diseases, Health Research Institute Navarra
Chair, EACTS
Education Committee
International association representative, Latin American Association of Cardiac and Endovascular Surgery (LACES)
Associated editor, European Journal of Cardiothoracic Surgery
EDUCATION AND TRAINING
University of the Basque Country, Bilbao, Spain
Leeds General Infirmary, Leeds, UK
East Yorkshire Hospitals, Cottingham, UK
Sir Charles Gardner Hospital, Perth, Australia
MEMBERSHIPS
Spanish Society for Thoracic and Cardiovascular Surgery
Heart Valve Society Society for Thoracic Surgeons (STS)
mainly on aortic stenosis, but also in other types of aortic and mitral valve disease. It also helps to escape the stress of everyday surgery and complex cases and is something that I enjoy. Cardiac surgery is quite intense and the part of your day that is spent in the operating theatre and looking after patients is so time and attention consuming. It is so important to keep yourself up to date with new developments that are coming out. Techniques have developed so much that we now have sub-specialisation, so there is little time for surgeons to dedicate time to go to the lab and do basic science research. There also isn’t much profit in return for the time you invest in it.
In the past, cardiac surgeons were always at the forefront of science and developments, whereas now we don't have such an innovative mentality, and this is a problem for us. We have to think outside of the knife, needle and stitches, and look into important aspects of our daily activities.
Are cardiac surgery and interventional cardiology competing or complementary specialties?
My view is that they should be complementary, but the fact is that they are competing, and both sides have responsibility. Surgeons see interventional cardiology as competition because they feel that they are taking patients away. From the other side, interventional cardiologists want to get more and more patients, and they want to do more things that they weren’t doing before, so there is competition. Sometimes interventional cardiologists may want to go a bit too far too fast, but we will all be able to go much further if we work in a way that is complementary to each other.
In my environment, I am very lucky in that we have that view. Both sides are going to do far more if we help each other fully and truly. Once both sides understand that, life is so much easier.
What is your advice to young cardiac surgeons who are just starting out in the field?
My view is that the specialty is at a crossroads and in danger of lagging behind other alternative treatments if we don’t wake up and move forward. We must concentrate on adding value to the care we give to our patients, making sure that we put them, their interests and preferences, before anything else. Processes must be streamlined to offer safe procedures, early recovery, and long-lasting results free from complications. Demands on surgical abilities will increase, as surgical
procedures become more complex and the population ages.
How has your involvement in the European Association of Cardio-Thoracic Surgery (EACTS) contributed to your professional life?
One of the things I have found most interesting from being a part of EACTS is that you are always in contact with first-line research and scientific development. It is a scientific society which means that, not only do you have access to the research and science, but to the people who lead that research, and for me that is what has helped me the most. These scientific organisations give you the opportunity to attend courses and other educational activities that show you what you need to know and the skills you have to develop.
A big part is networking. It is an overused word, but it is true. You may get to know people who you admire, who are leaders in the field, and learn from these people. You learn a
lot from top surgeons, but also from peers who you may be able to talk more openly with.
EACTS is evolving, and I think this is what distinguishes us from our American colleagues, for instance, who acknowledge this when they come to our annual meeting. I have been a part of the programme committee for many years, and there is a willingness to constantly evolve and do better each year. We have to try new things and recognise that members and non-members attending the meeting want to feel a part of it. They want to see that there is more interaction and that they can play a bigger role, instead of just seeing the usual suspects who are always on the stage. That is why, in most of our presentations, we have six or seven minutes for the presentation and another six or seven minutes of discussion to give attendees an opportunity to speak.
Having more hands-on opportunities in the meetings, giving surgeons more opportunities to come and try things, giving industry the
“We must concentrate on adding value to the care we give our patients, making sure we put them, their interests and preferences before anything else”
opportunity to showcase their devices and giving them the opportunity to interact with surgeons, are also important.
We are the best attended meeting in the world for cardiac and thoracic surgery and I think that is a reflection of the quality that we offer. We are a European association but we try to open up globally and attract people from all parts of the world. We have interest from Latin America and we are working more in Africa to try to help surgeons and their patients in those parts of the world.
What does your life outside of medicine look like?
I try to play golf as much as I can. I try to go out to walk or run every day, even if it is just for a short time. I also like seeing different parts of the world, travelling and meeting new people—although outside of cardiac surgery I don't have many opportunities. Now that my children have grown up, I grasp every opportunity to have the five of us together.
Newest-generation TAVI platforms designed meet the demands of the modern cath lab
As transcatheter aortic valve implantation (TAVI) continues its evolution interventionalists need the latest generation of devices to offer features that respond to the needs of a diverse and changing population of patients. The EvolutTM (Medtronic) family of TAVI devices is among the most widely used TAVI systems in the world, and new iterations of the platform have sought to match the developing needs of users whilst maintaining the durability and reliability that physicians and patients expect from previous generations.
INTERVENTIONAL CARDIOLOGIST
Angela McInerney (Galway University Hospital, Galway, Ireland) spoke to Cardiovascular News about the features she sees as being important in the Evolut system and how newer iterations of the device potentially broaden the scope of the patients who may be offered treatment.
Evolut FX was launched in late 2023, bringing with it a series of features intended to enhance ease-of-use and control for operators. “In terms of redesigned features, Evolut FX includes a longer nose cone and a single spine delivery shaft, which both improve flexibility,” McInerney says. These enhancements help to increase the crossability of the device improving its ability to navigate tortuous and calcified anatomies or horizontal aortas, for example.
“The flexibility of the device gives us confidence to use it in these commonly encountered tortuous anatomies, knowing that the device will safely navigate the iliofemoral and aortic angles without the need for a large sheath,” McInerney comments. “This is a major change in comparison to previous Evolut iterations.”
A further addition in Evolut FX is that of radiopaque markers—gold markers located close to the inflow edge of the valve and under the commissures that are visible on fluoroscopy to help operators align the valve with the native annular plane—with the intention of achieving commissural alignment to improve future access to the coronary arteries.
“Commissural alignment has become so much more important for TAVI implanters, particularly when you are thinking about patients who have concomitant coronary artery disease, but also younger patients who could easily come back in the future with myocardial infarction (MI) or angina, where you not only want to image the coronary arteries, but also treat them,” says McInerney, commenting that the addition of radiopaque markers takes any concern off the table about future coronary access.
“I find achieving commissural alignment to be much less challenging and this has resulted in a change in our practice whereby we now perform percutaneous coronary intervention (PCI) in those with concomitant coronary artery disease (CAD) after the TAVI has been implanted,” she explains, adding that the likely ability to achieve commissural alignment also has a bearing on valve choice when considering treatment strategies in younger patients.
The Galway team has been using the Evolut FX platform since its launch over one year ago and McInerney says that she has found the deliverability of the device to be a positive change. The ability to navigate tortuous and calcified anatomies has also been impactful on her practice given the proportion of patients that come into the cath lab with comorbidities
that may predispose them to heavily calcified vasculature, such as advanced age, diabetes or chronic kidney disease.
Whether these new features translate into improved patient outcomes remains to be borne out in practice, but McInerney comments that being able to treat more patients with the self-expanding Evolut platform will likely be a plus given that the new device builds upon the legacy Evolut and CoreValve technologies, underpinned by a solid body of data. “The clinical data for the use of Evolut is continuing to grow and be reassuring,” she says. “Many of the recent studies have answered research questions that are very important for TAVI operators.”
TAVI (CoreValveTM, Evolut R, or Evolut PRO) devices to SAVR in low-risk patients with severe aortic stenosis—shared in late 2023, have shown that TAVI patients had a 26% reduction in hazard for death or disabling stroke compared to surgery, with the difference between the two groups continuing to increase over time.
Data on durability and valve performance also appear favourable. A five-year pooled analysis taken from the CoreValve US High Risk and SURTAVI randomised trials has demonstrated a significantly lower cumulative rate of bioprosthetic valve deterioration compared to surgery, as well as five-year rates of structural valve deterioration and nonstructural valve dysfunction that were significantly lower after TAVI compared to surgery.
McInerney says that the evidence of a divergence of the curves for all-cause mortality and stroke seen in the EVOLUT Low Risk trial can be considered “good news” for patients, whilst the haemodynamic results seen with the device also provide reassurance.
More recently the interventional community has digested findings from the SMART trial, the randomised trial in which Evolut PRO, PRO+ and FX systems were compared against the balloonexpandable Sapien 3 and Sapien 3 Ultra (Edwards Lifesciences) devices in patients with small aortic annuli. Though demonstrating a similar rate of death, disabling stroke or rehospitalisation for heart failure in both groups, Evolut was shown to be superior in bioprosthetic valve dysfunction through 12 months, with a rate of 9.4% compared to 41.6% with the balloon-expandable valve.
“Comparison between TAVI devices is always of interest to us as TAVI operators. We want to be confident in our choice of valve to ensure the safety of our patients as well as providing long-term durability,” says McInerney of the results. Patients with small annuli—a group that is disproportionately made up of women— often presents a challenge for TAVI operators, where optimising haemodynamic performance and avoiding patient prosthesis mismatch (PPM) is important. Crucially, SMART demonstrates a positive impact for Evolut in both.
Recent data releases support the performance of the Evolut family of devices across a broad range of patient groups, in particular low-risk patients and
The flexibility of the device gives us confidence to use it in these commonly encountered tortuous anatomies”
those with small aortic annuli, as well as highlighting the durability of the prosthesis when compared to surgical aortic valve replacement (SAVR).
Four-year findings from the EVOLUT Low Risk trial—a randomised, non-inferiority trial, comparing
Whilst the Evolut devices have shown a positive performance to date, development does not stand still. Already launched in the USA and now reaching European shores following its recent CE mark approval is the Evolut FX+, the newest addition to the Evolut family. Evolut FX+ offers three larger coronary access windows through a modified diamond-shaped cell design, which are four times larger than regular cells of the Evolut TAVI system. This provides increased space for catheter manoeuvrability to facilitate access to coronary arteries of varying patient anatomies, whilst also designed to maintain its structural strength and radial force.
“In the context of the shift in the ‘typical’ TAVI patient to those that are younger and lower risk, coronary re-access is essential, and these design features are very welcome and useful in that respect,” says McInerney of this latest innovation.
EvolutTM FX TAVI platform
EvolutTM FX+ TAVI platform
Angela McInerney
Structural Heart Interventions
RESHAPE-HF2 results move conversation forward on effectiveness of TEER in functional MR
A strategy of mitral transcatheter edge-to-edge repair (TEER) plus medical therapy in patients with heart failure and moderate to severe mitral regurgitation (MR), who were not eligible for surgery, was found to be more effective than optimal medical therapy alone, investigators in the RESHAPE-HF2 trial have reported at the European Society of Cardiology (ESC) 2024 Congress (30 August–2 September, London, UK).
The trial’s principal investigator, Stefan Anker (Charite University Hospital, Berlin, Germany) said the findings should provide reassurance about the benefits of TEER using the MitraClip device (Abbott) in patients with moderate to severe functional MR and heart failure and could strengthen and broaden recommendations for its future use. Critics, however, are less enthusiastic about the conclusions.
RESHAPE-HF2 comes against the backdrop of two previous trials having delivered diverging results of transcatheter therapy in this patient population. COAPT, which included patients with less advanced left ventricular (LV) dysfunction and more severe functional MR, demonstrated significant benefits of the treatment in reducing heart failure hospitalisations and mortality. However, MITRA-FR included patients with more advanced LV dysfunction, did not enforce as rigorous a standard for medical therapy optimisation, and did not show a significant benefit from TEER.
This latest trial, an investigatorinitiated randomised study, recruited patients with symptoms of heart failure despite optimal guideline-directed medical therapy, who had left ventricular ejection fraction (LVEF) ≥20% and ≤50%, MR grade 3+ or 4+—meaning that they had slightly more severe LV dysfunction than COAPT, but less than MITRA-FR—and had a recent hospitalisation for heart failure or elevated plasma natriuretic peptide concentrations within the last 90 days. Patients for whom mitral valve surgery was recommended were not eligible. Patients were randomised 1:1 to TEER using the MitraClip device plus optimal medical therapy or medical therapy.
The three primary endpoints were a composite rate of total (first and recurrent) heart failure hospitalisations and cardiovascular death within 24 months; the rate of total heart failure hospitalisations within 24 months; and the change from baseline to 12 months in the Kansas City Cardiomyopathy Questionnaire (KCCQ) overall score, which assesses the impact of heart failure on quality of life.
In total, 505 patients were randomised
from 30 centres in nine countries. The mean age was 70 years and 20% were female. The mean LVEF was 31% and HF was New York Heart Association (NYHA) class III or IV in around threequarters of patients. The mean KCCQ overall summary score was 46, which indicates significant symptoms and limitations due to heart failure.
At 24 months, Anker reported that the rate of total hospitalisations for heart failure and cardiovascular death was significantly lower in the TEER group compared to the control group (37 per 100 patient-years vs. 58.9 per 100 patient-years; rate ratio 0.64; 95% confidence interval [CI] 0.48–0.85; p=0.002).
The rate of total hospitalisations for heart failure within 24 months was significantly reduced by 41% in the TEER group vs. the control group (26.9 per 100 patient-years vs. 57.8 per 100 patient-years; rate ratio 0.59; 95% CI 0.42–0.82; p=0.002).
In addition, the mean (standard deviation [SD]) change from baseline to 12 months in the KCCQ overall summary score was greater (improved) in the TEER group (+21.6 [26.9]) than in the control group (+8.0 [24.5]), he reported, resulting in a mean difference of +10.9 points (95% CI 6.8–15.0; p<0.001).
During the total follow-up of 38.1 (18.1) months, there was no difference between the groups for all-cause mortality (17% in the M-TEER group vs. 18.5% in the control group; hazard ratio 0.90; 95% CI 0.71–1.13). Periprocedural adverse events were reported in four patients (1.6%) in the TEER group.
Speaking to Cardiovascular News, Anker said that the results should conclusively demonstrate the impact of transcatheter therapy in patients with severe
functional MR, and potentially point to a benefit in patients with less severe disease.
“I would hope that many colleagues in the power to make such recommendations will start to see that even patients with less than severe functional MR should at least be considered for this treatment approach, it may be a soft recommendation,” he said. “It is certainly not a negative [result]—I am positive about the results. They indicate to me that there is benefit to be had for these patients but we need confirmation of this to make this as valid a standpoint as we can. For severe functional MR, I think we have now, taking the results here together a good indication that this should be done.”
Questions as to whether the results of RESHAPE-HF2 close down the debate caused by the divergent results of the preceding trials still remain, however.
“I would be less enthusiastic than your conclusion,” Jean-Francois Obadia (Civils Hospices of Lyon, Lyon, France), the principal investigator in the MITRA-FR trial, said of the results of RESHAPE-HF2 in discussion that followed the presentation at ESC 2024, pointing in particular to the fact that there was no difference between the groups for all-cause mortality.
“The positive [result] relies mainly on criteria that have some weaknesses. If we stick on unquestionable criteria, like mortality—because death is death—or all hospitalisation, because an hospitalisation is a hospitalisation, [there is] no discussion, whatever the quality of the adjudication committee.
“Therefore, in RESHAPE, you have no impact on mortality [and] no impact on hospitalisation, which is not far from the result we have with MITRA-FR. So, what I mean is that RESHAPE does not conclude the discussion [of] the discrepancy observed between MITRAFR and COAPT, and my feeling today is that we should merge the three databases and try to perform a meta-analysis of the three studies.”
Anker agreed that analysing data from the three datasets in the form of an individual patient data meta-analysis would be a positive step in answering some of these points.
“What can I say—we all agree on the need for an individual patient metaanalysis to sort out, not only the overall effect size, because that is a question that people might estimate already and form their opinion about, but getting an idea for the reasons for the variability in results between trials and look at sub-groups to see who can particularly benefit.”
Results of the trial were published simultaneously in The New England Journal of Medicine
Trilogy valve shows favourable results at two years in aortic regurgitation patients
Latest data from the ALIGN AR trial, investigating the use of the Trilogy (JenaValve) transcatheter aortic valve implantation (TAVI) system in high-risk patients with symptomatic, severe aortic regurgitation, support the safety and efficacy of the device out to two years..
Thorsten Vahl (NewYorkPresbyterian/Columbia University Medical Center, New York, USA) presented two-year outcomes from the trial—a landmark prospective, single-arm investigational device exemption (IDE) study, intended to support US Food and Drug Administration (FDA) approval of the device—at TCT 2024 (27–30 October, Washington, DC, USA).
Design features of the Trilogy valve, including locators that clip onto native leaflets and enable secure anchoring in the absence of calcium, make the device well suited to the treatment of aortic regurgitation, investigators say, as well as helping to facilitate commissural alignment, which can be important when treating aortic stenosis. Trilogy also features large-open cells that are designed to support future coronary access.
ALIGN AR, taking place at 20 sites throughout the USA, follows the clinical, echocardiographic, functional and quality of life outcomes annually out to five years among 180 patients with symptomatic, severe aortic regurgitation, deemed high-risk for surgery. One-year findings from the trial, reported at TCT in 2023, showed that Trilogy met its non-inferiority safety and efficacy endpoints.
Vahl reported that at two years, all-cause mortality stood at 15.4%, continuing to fall within the threshold for non-inferiority. The rate of death attributed to cardiovascular causes stood at 6.2%. “This tells us that between year one and year two, the vast majority of patients, 11 out of 13, experienced non-cardiovascular death, that we attribute to the comorbidities of the patients,” said Vahl.
The haemodynamic performance of the valve also appears favourable according to Vahl, with mean effective orifice area (EOA) >2.5cm2 and mean transvalvular gradient <5mmHg, as well as no cases of haemodynamic valve deterioration.
Further study of the Trilogy valve is set to commence with the initiation of the ARTIST trial, which is due to begin enrolment shortly. The ARTIST trial is a randomised study comparing TAVI with Trilogy to surgical aortic valve replacement (SAVR) in non-high-risk patients with severe and moderate-to-severe aortic regurgitation.
Stefan Anker
TRISCEND II reports positive outcomes for tricuspid replacement system
Reductions in tricuspid regurgitation (TR) following transcatheter tricuspid valve replacement with the Evoque (Edwards Lifesciences) system were associated with significant improvements in symptoms, function and quality of life at one year, data from the TRISCEND II pivotal trial indicate.
FINDINGS FROM THE 400patient randomised trial, comparing tricuspid replacement to optimal medical therapy among patients with severe TR, were presented at the 2024 TCT meeting (27–30 October, Washington, DC, USA) by Susheel Kodali (Columbia University Irving Medical Center, New York, USA) and Suzanne Arnold (University of Missouri-Kansas City, Kansas City, USA). The investigators reported that tricuspid valve replacement using Evoque compared favourably to medical therapy in mortality and heart failure hospitalisation—with numerically fewer incidences of each outcome in the valve replacement arm.
Of the 400 patients enrolled at 45 sites in the USA and Germany, 267 were randomised to undergo tricuspid valve replacement, and 133 to the study’s control arm. One-year follow-up data were available in 224 patients in the device arm and 104 patients in the control arm. Kodali and Arnold detailed that the patient population was elderly, with an average age of 79 years, and it was a majority female cohort, with more
than 70% women in each arm.
Patients were described as “very symptomatic” with Kansas City Cardiomyopathy (KCCQ) scores of 52.8 among patients undergoing tricuspid replacement and 50.6 for those receiving only medical therapy at baseline. Most patients were treated for secondary TR, and more than half had massive or severe TR at baseline.
The Evoque valve was successfully implanted in 95.4% of patients, and of those who received the valve, nearly all (95.3%) achieved almost complete TR elimination with mild or less TR at one year, compared to 2.3% of patients receiving medical therapy alone, Kodali and Arnold revealed.
Cardiovascular mortality stood at 3.1% at 30 days in the device group and 0% in the control group, with late events occurring in 5.7% of patients in the Evoque arm compared to 7.8% in the control group. Severe bleeding occurred in 10.4% of tricuspid replacement patients. Overall, the investigators noted that between 30 days and one year there was no difference in safety signals
between the two groups.
Quality of life, as measured by KCCQ score, demonstrated an initial improvement in favour of transcatheter tricuspid replacement, with a difference of 11.8 points between the two arms at one month, which grew out to 20.8 points at six, and stood at 17.8 points at 12 months. As many as 58% of patients in the Evoque arm saw a large or moderate change in health status at one year, compared to 31% in the control group, whilst 24% in the interventional arm had worse health status or had died at the end of 12 months, compared to 45% receiving medical therapy.
“Given the large improvements in
TRISCEND II will change practice and supports transcatheter tricuspid valve replacement as the preferred management strategy for eligible patients who have symptomatic severe TR”
Latest TRILUMINATE and TRI.Fr data add to growing body of evidence for tricuspid repair
Data continue to emerge supporting the safety and efficacy of transcatheter-edge-toedge repair (TEER) for the treatment of tricuspid regurgitation (TR).
THREE-YEAR DATA FROM THE TRILUMINATE study, investigating TEER using the TriClip (Abbott) tricuspid repair system, published in September 2024 in JACC: Cardiovascular Interventions, follow shortly after the release of oneyear results of the TRI.Fr trial at the 2024 European Society of Cardiology (ESC) congress (30 August–2 September, London, UK).
“Tricuspid regurgitation is a marker of bad prognosis,” Erwan Donal (CHU Rennes, Rennes, France) commented at ESC Congress, where he reported the results of the TRI.Fr trial, a multicentre randomised evaluation of the use of the TriClip device in the treatment of severe TR. “The question is do we have to consider a treatment of this tricuspid regurgitation or not,” said Donal.
TRI.Fr assessed the safety and the efficacy of TEER, comparing TEER plus guideline-directed medical therapy to guideline directed medical therapy alone in 300 patients with symptomatic severe TR, who were ineligible for surgery. In total, 148 patients were randomised to undergo guideline-directed medical therapy and 152 to undergo TEER.
Investigators used a composite clinical score at 12 months to assess the efficacy of the two strategies,
comprised of the occurrence of major cardiovascular events, changes in New York Heart Association (NYHA) class, or patient-reported outcomes.
Secondary endpoints included the change in the degree of TR at one year, quality of life assessed using the Kansas City Cardiomyopathy Questionnaire (KCCQ) score, patient global assessment, and a hierarchical composite endpoint and including all-cause mortality, tricuspid valve surgery, time to heart failure hospitalisation and improvement of more than 15 points of the KCCQ score at 12 months, as well as major cardiovascular events, and deaths.
“We can observe that at one year follow up, 74% of the patients treated with transcatheter edge-to-edge repair improved, as opposed to 40% in the guideline directed medical therapy group. This is highly significant,” said Donal. “If we look in detail at all the components of the primary endpoint, and if we look at all the secondary endpoints, we observe that all the parameters are in favour of the treatment by TEER.” Donal concluded that much of the success of the treatment was driven by a significant reduction in the
quality of life and overall favourable clinical outcomes, TRISCEND II will change practice and supports transcatheter tricuspid valve replacement as the preferred management strategy for eligible patients who have symptomatic severe TR,” said Neil Fam (University of Toronto, Toronto, Canada) discussing the results following Kodali and Arnold’s presentation at TCT.
One of the important limitations of the trial raised during the discussion that followed the presentation, was the possibility of a placebo effect given that those undergoing intervention were not blinded to treatment, with a potential for this to skew quality-of-life outcomes.
“Patient-reported outcomes in an open-label study are always somewhat prone to bias, I think there are a number of factors here that support that at least some of this is biologically mediated,” said Arnold on this point. “I think one is the magnitude of effect that is much larger than what we would expect with a placebo, the duration of effect is important, but also interestingly here is that the change was delayed. The change from one month to six months to me supports the fact that this is not all placebo.”
One-year primary endpoint outcomes have been simultaneously published in The New England Journal of Medicine, and one-year quality-of-life (QoL) outcomes in the Journal of the American College of Cardiology.
Evoque received CE mark approval in October 2023 and US Food and Drug Administration (FDA) approval in February 2024
degree of TR. “We were also able to see that we have much less death and hospitalisation in our study than in previous registries and even the TRILUMINATE trial,” he said.
“TRI.Fr is a positive prospective study, confirming a clear benefit for patients suffering from severe tricuspid regurgitation,” Donal told Cardiovascular News. “It is important to note that medical treatment and strict monitoring of these patients, who are often in advanced heart failure, are equally crucial.
“What seems to be confirmed is that tricuspid regurgitation treatment with clips effectively complements medical therapy and is a therapeutic option that must be recognised, as overlooking tricuspid regurgitation in our patients should be avoided at all costs.”
TRILUMINATE: Three-year results
Representing the longest follow-up data to date of any tricuspid TEER therapy, the three-year results of the international, prospective, single-arm, multicentre TRILUMINATE trial include outcomes from 98 patients with symptomatic moderate or greater TR receiving the TriClip system.
Investigators report that at three years, TR was reduced to moderate or less in 79% of patients, with a reduction of at least one grade achieved in 92%. The TR reduction achieved at one year was sustained through to the three-year timepoint, the investigators report. The site-reported heart failure hospitalisation rate decreased from 0.43 events/ patient-year one year before device implantation to 0.06 events/patient-year one year after device implantation, representing a reduction of 86% .
Erwin Donal
TEER non-inferior to surgery in MATTERHORN trial but dividing lines remain
Investigators of a trial comparing transcatheter edge-to-edge repair (TEER) with surgery in patients with heart failure and secondary mitral regurgitation (MR) say that the results of their study—MATTERHORN—should prompt a reappraisal over guideline recommendations on the use of the transcatheter therapy in these patients.
Current European guidelines “somehow infer that surgery would be the procedure of choice”, said Volker Rudolph (Heart and Diabetes Center NRW, Bad Oeynhausen), presenting the MATTERHORN results at the 2024 European Society of Cardiology (ESC) congress (30 August–2 September, London, UK) where he acknowledged that, at present, TEER is only recommended for those deemed ineligible for surgery.
MATTERHORN, an investigator-initiated trial conducted at 16 centres in Germany, randomised 210 patients to undergo either TEER using the MitraClip (Abbott) device, compared to surgical mitral vale repair or replacement.
Rudolph reported that there was no significant
difference in the primary composite endpoint—death, heart failure hospitalisation, mitral reintervention, assist device implantation and stroke—at one year, which occurred in 16.7% of patients in the TEER group and 22.5% in the surgical group (p<0.01 for non-inferiority).
The secondary endpoint, recurrence of MR grade ≥3 at one year also demonstrated non-inferiority of the transcatheter treatment, occurring in 8.9% of patients in the TEER group compared to 1.5% in the surgical group (p=0.091), whilst 73.2% of patients in the TEER group and 87.3% of patients in the surgical group had MR grade ≤1 after one year.
Finally, Rudolph reported that the primary safety endpoint occurred in significantly more patients in
Analysis of EXPANDed registry finds clinical benefit to atrial secondary MR patients receiving MitraClip
Atrial secondary mitral regurgitation (MR) patients treated with the MitraClip (Abbott) transcatheter edge-to-edge repair (TEER) system experienced clinical benefit, new evidence from the EXPANDed registry has shown.
MARK RICCIARDI
(NorthShore Cardiovascular Institute, Chicago, USA) presented this and other findings as late-breaking data as part of the Heart Failure Society of America (HFSA) annual scientific meeting. The meeting had been due to take place 27–30 September in Atlanta, USA, but was cancelled due to Hurricane Helene. Late-breaking studies that had been due to be presented in-person at HFSA 2024 were live-streamed online.
Atrial secondary MR is a sub-group of the secondary MR patient population
that is broadly defined as having annular dilatation and/or left atrial margin in the face of preserved left ventricular (LV) systolic function, Ricciardi explained during his presentation. “Little is known about atrial secondary MR pathology, even less about the prognosis following edge-to-edge repair,” he added, noting that the point of the study was to evaluate the atrial secondary MR population and their outcomes following a MitrClip procedure.
Investigators used data from the EXPANDed registry, a patient-level
the surgical group (54.8%) than in the TEER group (14.9%; p<0.001), which was largely driven by more major bleeding (29% vs. 3%, respectively), all reinterventions (19% vs. 8%) and new-onset AF (33% vs. 9%).
“We know that this condition is associated with adverse outcomes in patients with heart failure—it more or less doubles the mortality of these patients. We have also seen now that treatment of this condition with transcatheter edge-to-edge repair can improve the prognosis of these patients,” said Rudolph.
“In patients with heart failure and secondary mitral regurgitation, intervention with edge-to-edge repair is non-inferior to surgery regarding clinical efficacy and has a much better safety profile,” he added of the overarching message of the trial. “We think that these results may extend the indication for interventional mitral repair to patients with secondary mitral regurgitation who are actually eligible for surgery.”
Despite the trialists’ enthusiasm over the results, critics of MATTERHORN have urged caution when interpreting the findings. Cardiac surgeon Joanna Chikwe (Smidt Heart Institute at Cedars-Sinai, Los Angeles, USA) acted as the discussant for the study at ESC 2024, and raised a number of points that she felt could limit the applicability of the research.
“The questions I would want to ask myself if I had a patient sitting in front of me in the office [are]: is this trial unbiased? Are the patients representative of my practice? Was the control treatment—surgery— standard of care? And, what can we decide based on 12-month data?” Chikwe said.
Of the short-term nature of the trial’s endpoints, Chikwe commented that surgery has a “hazard phase” and that the treatment’s effects are often seen late, potentially altering the perception of the trial at 12 months. Rudolph responded that longer term follow-up from the trial is being conducted.
Chikwe also argued that the trial included a wide margin for non-inferiority, which was explained by the trialists as being necessary due to the study’s sample size. “I would say that the study design is unfortunately not unbiased, it is a non-inferiority design with short follow-up, variation in the control arm, crossover and endpoints that favour TEER,” Chikwe commented, adding: “We need to do more to treat this disease of the ventricle, but the secret is not treating the leaflets.”
Findings of the study were published in the New England Journal of Medicine
pooled cohort study combining both the EXPAND and later EXPAND G4 post-market studies, that have gathered real-world evidence of the safety and efficacy of the MitraClip system from use in the USA, Canada, Europe, the Middle East and Japan, taking into account 160 atrial secondary MR patients in total.
Key outcomes assessed included procedural, echocardiographic, functional, and quality of life parameters, as well as all-cause mortality, heart failure hospitalisation, and safety outcomes, with follow-up out to one year.
The baseline characteristics of the patients showed that typically they were older than the general EXPANDed population, with an average age of 78% years, 50% were female, all had atrial fibrillation (AF) and 7.5% had a permanent pacemaker.
“Compared to overall secondary MR population within EXPANDed, in general the atrial secondary MR subjects were similarly symptomatic with fewer
cardiac rhythm interventions, smaller left ventricles, larger atria than the general population of secondary MR,” Ricciardi said.
Acute procedural success was achieved in 87.5% of patients, Ricciardi detailed, with an average procedural time of 92 minutes, and an average of 1.4 MitraClips implanted per patient. No procedural deaths or conversions to mitral valve surgery were reported.
Ricciardi reported that all-cause mortality stood at 8.4%, with two deaths occurring within 30 days and rates of myocardial infarction, stroke, mitral valve replacement, single leaflet device attachment, or embolization were low.
Investigators also saw a significant reduction in MR, with around 95% of patients having mild or less MR up to 12 months after the procedure.
“One-year outcomes suggest that atrial secondary MR patients in the EXPANDed study experienced clinical benefit when treated with the MitraClip system,” Ricciardi said of the study’s take-home messages.
Volker Rudolph
AI virtual voice assistant identifies complications after TAVI
Clinical follow-up using virtual voice technology helped identify complications after transcatheter aortic valve implantation (TAVI) with a high degree of patient satisfaction, according to research presented at the European Society of Cardiology (ESC) 2024 congress (30 August–2 September, London, UK).
EXPLAINING THE RATIONALE for the development of the virtual voice assistant for TAVI patients, study author Marta Herrero Brocal (Dr Balmis General University Hospital, Alicante, Spain), said: “Aortic valve stenosis is common, especially in the ageing population. It can be treated with surgery or with TAVI. Complications may occur after TAVI, especially within the first month, but due to a lack of resources, many hospitals are not able to provide the intense follow-up needed after patient discharge.
“Based on artificial intelligence and natural language processing, a new application was developed for the virtual voice assistant, ‘LOLA’, which is able to make more than 40 phone calls in two hours, allowing us to gather follow-up information and act accordingly. Results from the TeleTAVI study indicate that we can provide excellent care virtually, without substantially
increasing resources.”
TeleTAVI was a prospective, observational, single-centre study conducted at the Dr Balmis General University Hospital. All patients undergoing TAVI via the femoral artery in 2023, without language barriers, were offered the option of follow-up with the virtual voice assistant.
LOLA called the patients in week one, week two, month one, month three and month 12 after patient discharge. In these calls, a series of questions were asked, mainly related to vascular access and the patient’s cardiovascular situation. After finishing the call, all the information
New analysis shows difference in rates of stroke among US and nonUS cohorts of PROTECTED TAVR trial
New analysis of the PROTECTED TAVR trial has shown a reduction in stroke within the US portion of the trial.
DATA PRESENTED DURING A LATEbreaking trial session at TCT 2024 by Samir Kapadia (Cleveland Clinic, Cleveland, USA), simultaneously published in JAMA Cardiology, outline the regional differences in stroke outcomes among patients taking part in the trial.
PROTECTED TAVR was among the headlinegrabbing trials at TCT 2022, where Kapadia presented findings from the randomised, multicentre trial, conducted in 51 centres in North America, Europe and Australia. The trial was conducted among 3,000 aortic stenosis patients undergoing transcatheter aortic valve implantation (TAVI) with or without cerebral embolic protection using the Sentinel (Boston Scientific) device. Devices like Sentinel are intended to mitigate the risk of stroke during TAVI by capturing and removing debris dislodged during the procedure before it reaches the brain, but their benefit remains unproven. PROTECTED TAVR, however, failed to show a significant effect on the incidence of periprocedural stroke in favour of the device, even though a difference
collected was uploaded to a web platform where the data were monitored by healthcare professionals who acted where necessary.
A total of 274 patients were included. The mean age was 81 years and 49.3% were women. Only six patients refused the follow-up option. A total of 1,039 calls were made, involving 385 hours of autonomous conversation, with an average duration of four minutes and three seconds per call. The calls were completed in 94% of cases, with the degree of adherence above 85% throughout the follow-up period. The patient answered in 89% of calls, with 11% answered by family members or caregivers.
No alerts were detected in 44% of calls, eliminating the need for review. Among the remaining calls, there were 926 alerts resulting in at least one intervention in 57% of the calls. The number of alerts decreased as follow-up progressed, reflecting the need for closer monitoring in the early stages after
The virtual voice assistant also facilitated rapid patient discharge. Knowing that automated close follow-up was available, 40.1% of patients were able to be discharged within 24 hours of the procedure and 32.9% between 24 and 48 hours.
Patients generally
in rates of disabling stroke did favour Sentinel. More recently, analysis of data from the TVT registry has demonstrated a “small, borderline significant” reduction in in-hospital disabling stroke when using embolic protection. Of note, the research showed a steady growth in embolic protection device usage throughout the duration of the study followed by a decline and plateau coinciding with the release of the PROTECTED TAVR trial results.
Looking in more detail at the data from PROTECTED TAVR, Kapadia and colleagues divided the trial’s population into a US and non-US cohort, comprising 1,833 and 1,167 patients respectively. Among the US cohort, 919 underwent TAVI alone, and 914 underwent TAVI with Sentinel. In the non-US cohort, this number stood at 580 for TAVI alone and 587 for TAVI with Sentinel.
Kapadia and colleagues identified some key differences and baseline and procedural characteristics between the US and non-US cohorts of the trial, highlighting in particular a difference in the rates of medically-treated diabetes (29.7% in the US population and 24.9% non-US), history of peripheral vascular disease (13.2% vs. 7.6%), coronary artery disease (60.3% vs. 54.2%) and atrial fibrillation (29.9% vs. 37.3%), whilst more patients in the US cohort (77.7%) were treated using balloon expandable valves than among OUS patients (42.4%). Additionally 27.5% of patients underwent predilatation prior to TAVI in the US cohort, compared to 60.1% in the non-US cohort.
had a favourable response to the virtual system. The satisfaction score was 4.68/5 and 89% of patients reported good or very good satisfaction. In total, 86% of patients said that they would recommend the use of LOLA.
Herrero Brocal concluded: “The TeleTAVI study found that follow-up with a virtual voice assistant enabled safe and early discharge after TAVI, with a low complication rate and without increasing the burden on healthcare resources. Patients know that behind LOLA is a doctor or a nurse so they are very happy to speak to it, as reflected in our high patient satisfaction rates.”
A virtual voice assistant enabled safe and early discharge after TAVI, with a low complication rate and without increasing the burden on healthcare resources”
1.3% for patients in the TAVI + Sentinel arm, as well as disabling stroke, which occurred in 1.5% of TAVI only patients and 0.4% of Sentinel patients. In the non-US patients, these figures stood at 3.3% and 3.7% for stroke in non-Sentinel and Sentinel patients, and 1% vs. 0.7% for disabling stroke in Sentinel vs. nonSentinel patients.
Additionally, Kapadia reported that after a stroke, 36% of patients in the US cohort and 22% of patients non-US were discharged within 72 hours of the procedure, whilst US patients were more likely to be released home rather than to another hospital. In both regions, use of Sentinel was associated with a greater likelihood of being discharged home with no services. Furthermore, 58% of patients in the US and 68% of patients outside of the US required a hospital stay longer than 72 hours after stroke.
Kapadia described the findings as hypothesis generating, given that the sub-group analyses were not powered to detect treatment differences in clinical outcomes, but concluded that regional differences in patient characteristics and procedural practices may impact the effectiveness of Sentinel in reducing TAVI-related stroke.
Results showed that patients treated in the USA had a 50% relative reduction for both stroke, which stood at 2.6% for patients in the TAVI only arm and
“I am very careful about interpreting the data,” Kapadia commented. “The reason for doing this analysis is to say that there a lot of new devices being investigated in this field, and a lot of people are looking at it to see if it is worthwhile to do this type of investigation or not. The purpose of this analysis is to say that there is some debate and there is some light at the end of the tunnel, so it is worthwhile to do the trials.”
Samir
Kapadia
BECOME CRT 2025 FACULTY! BECOME CRT 2025 FACULTY!
CRT EVIDENCE
ABSTRACTS
Accepted abstracts published in JACC Interventions
Top 3 abstracts will be eligible to receive a cash prize.
DEADLINE: November 22, 2024
CRT PRACTICE
INTERESTING CASES
CRMIC Publication Opportunity
LATE-BREAKING TRIALS
DEADLINE: January 15, 2025
Top 3 cases will be eligible to receive a cash prize.
DEADLINE: December 10, 2024
Simulatenous Publication Opportunities
CRT INNOVATION
BEST INNOVATION COMPETITION
DEADLINE: January 10, 2025
The Top Innovation will be the recipient of our CRT 2025 Innovations Award
CRT 2025 faculty will receive the following benefits: SCAN FOR SUBMISSION PORTAL
• Waived registration fee
• Complimentary subscription to the CRM Journal and entry to the faculty lounge
• Upgrade to CRT Community Premium including all CRTmeeting Videos and PowerPoint archives
CRT SCHOLARSHIPS
Early Career Leaders
DEADLINE: December 12, 2024
Fellows Scholarship
DEADLINE: December 12, 2024
Women in Cardiology
DEADLINE: December 31, 2024
Renal denervation gains class IIb recommendation in ESC guidelines
Latest guidelines from the European Society of Cardiology (ESC) on the management of blood pressure and hypertension include a recommendation for the use of renal denervation as a tool to reduce blood pressure.
The treatment—in which energy is targeted through a catheter to the renal nerves to modulate the sympathetic signalling between the kidneys and brain to reduce blood pressure—has been included with a class IIb level of recommendation for treating resistant hypertension in patients with uncontrolled blood pressure, despite the use of three or more blood pressure-lowering drugs.
Additionally, clinicians can consider the interventional treatment in patients with increased cardiovascular risk who have uncontrolled hypertension on fewer than three drugs.
In both instances the guidelines stipulate that the treatment is contingent on patients expressing a preference for renal denervation after a shared riskbenefit discussion and multidisciplinary assessment, and that the procedure is performed at a medium-to-high volume centre. Furthermore, the therapy is not recommended for patients with highly impaired renal function (eGFR <40mL/ min/1.73m2) or secondary causes of hypertension.
During a question and answer session following the presentation of the new guidelines at the 2024 ESC congress (30 August–2 September, London, UK), co-chair of the writing committee that drafted the document, John William McEvoy (University of Galway School of Medicine, Galway, Ireland), explained why there is greater emphasis on additional pharmacological interventions— such as the diuretic medication spironolactone or beta blockers— ahead of the device-based treatment in the algorithm for treating resistant hypertension.
“We followed a policy that to meet a class I recommendation or a higher level, we would like to see evidence from trials not just showing a blood pressure-lowering effect but also an impact on outcomes,” McEvoy said. “When it comes to spironolactone and beta blockers, we felt there was sufficient evidence—it wasn’t always direct evidence in the context of resistant hypertension, but in other areas where these agents have shown benefit on cardiovascular disease outcomes.”
McEvoy said that evidence of the efficacy of renal denervation in improving cardiovascular outcomes, on top of its efficacy in reducing blood
pressure, would dictate any future changes to future versions of the guidelines.
Demonstrating that reductions in blood pressure resulting from the use of renal denervation do lead to improvements in cardiovascular events would likely elevate the therapy within guidelines, Felix Mahfoud (Saarland University Hospital, Homburg, Germany)—who has researched the technique extensively— tells Cardiovascular News, but the practicalities of conducting such a trial make it unlikely.
“Both randomised controlled trials and real-world registries have demonstrated the blood pressurelowering efficacy of renal denervation. It is highly likely that these reductions in blood pressure will translate into improved outcomes, including lower rates of stroke, myocardial infarction, heart failure, and other cardiovascular events,” he comments.
“However, from a purist research perspective, proving this association in a randomised controlled trial would be ideal, as it could eventually lead to a class I recommendation in guidelines. Conducting such an outcomes trial in renal denervation, however, would be challenging due to the need for a large sample size (>10,000 patients), longterm follow-up (five years), and the associated high costs.”
Elsewhere, the 2024 guidelines maintain an existing definition for ‘Hypertension’ as a blood pressure measuring ≥140/90mmHg, but introduce a new category of ‘Elevated blood pressure’ which is defined as 120–139/70–89mmHg. This new category is intended to facilitate consideration of more intensive blood pressure treatment targets among persons at increased risk for cardiovascular disease.
Another change sees the introduction of a new systolic blood pressure treatment target range of 120–129mmHg for most patients receiving blood pressure-lowering medication, with the proviso that the new target requires that treatment is well tolerated.
The addition of renal denervation— having previously been confined to use solely in the setting of clinical trials— has been described by advocates of the technique as a big step forward for patients requiring additional options to control hypertension.
“Renal denervation has returned to the guidelines and is now recommended for patients with uncontrolled resistant hypertension, as well as for those who are unable or unwilling to tolerate antihypertensive medications”, comments Mahfoud. “This is reassuring news for both physicians and patients. The guidelines
This is reassuring news for both physicians and patients”
also suggest that renal denervation should be considered as an alternative or additive approach to lowering blood pressure, used in combination with lifestyle modifications and antihypertensive drugs.”
ESC’s latest guidelines come shortly after the American Heart Association (AHA) acknowledged renal denervation as a “promising therapeutic approach” for some patients with uncontrolled hypertension in a scientific statement following a review of evidence into the therapy.
The statement was published in the journal Hypertension and recognises in particular that patients with uncontrolled hypertension, those with resistant hypertension or who have multiple medication intolerances, may be the most likely to benefit from the procedure, albeit with the caveat that further research is needed in areas such as patient selection and long-term efficacy of the technique.
“The AHA scientific statement presents an intensive review of trial evidence, followed by clinical considerations and practical recommendations for incorporating
Renal denervation therapy has been approved in several regions, including now in the USA
renal denervation into hypertension treatment programmes,” said Naomi Fisher (Harvard Medical School and Brigham and Women’s Hospital, Boston, USA), a co-author of the statement and consultant for Recor Medical, one of the two companies that has a renal denervation device approved for use by the US Food and Drug Administration (FDA).
“While more research is needed, most, but not all, of the new-generation randomised renal denervation control trials reached their primary endpoint, and the procedure carries a favourable safety profile. The AHA statement serves an important role by illuminating renal denervation as a new treatment option for many patients with uncontrolled hypertension, particularly those with resistant hypertension or who are intolerant to multiple medications.”
In November 2023, the field of renal denervation was buoyed by the approval of two devices by the US Food and Drug Administration (FDA)—the Paradise (Recor Medical) and Symplicity Spyral (Medtronic) systems—following a review of evidence from trials assessing both technologies.
ESC’s guideline recommendations add to recent releases from the Society for Cardiovascular Angiography & Interventions (SCAI)—which heralded the “tremendous potential” of renal denervation for the treatment of hypertension—as well as updated arterial hypertension guidelines from the European Society of Hypertension (ESH), stating that renal denervation can be proposed as an adjunctive therapy in select patients with resistant hypertension.
Attention turns to multi-organ denervation as new evidence supports “durability” of blood pressure reductions
New research will investigate the safety and efficacy of multi-organ, hepatic artery and renal artery denervation using the Symplicty Spyral (Medtronic) catheter in uncontrolled hypertension patients who are both on and off medications.
A PLANNED GLOBAL PILOT STUDY, SPYRAL GEMINI, represents the latest frontier in sympathetic denervation research, following the US Food and Drug Administration (FDA) approval of the Symplicity Spyral and Paradise (Recor Medical) renal denervation systems in late 2023. SPYRAL GEMINI will involve only the use of the Symplicity Spyral catheter.
“It is a nice way of saying that the story doesn’t stop here with renal denervation. We are exploring other vascular beds for renal denervation therapy, and it so happens that the hepatic artery is highly innervated with sympathetic nerves and would provide a rich target for sympathetic denervation,” David Kandzari (Piedmont Heart Institute, Atlanta, USA), who has investigated renal denervation using the Medtronic system, tells Cardiovascular News
“The pre-clinical data would suggest that hepatic denervation combined with renal denervation may not only achieve a potentially greater reduction in blood pressure, but an even more consistent reduction in blood pressure as well.”
The study programme is anticipated to begin in late 2024/early 2025 and will be “exciting with regard to seeing what the reductions in blood pressure are beyond renal denervation only”, according to Kandzari, and whether this may increase the number of individuals with a response from the therapy.
Kandzari recently presented two-year data from the SPYRAL HTN-ON MED clinical trial at TCT 2024 (27–30 October, Washington, DC, USA).
The trial investigated the safety and efficacy of renal denervation using the Symplicity Spyral radiofrequency catheter in 337 patients with uncontrolled hypertension prescribed one to three hypertensive drugs, who were randomised 2:1 to either renal denervation or a sham procedure.
The findings showed that, compared to patients treated with a sham procedure, those who underwent renal denervation had significantly greater reductions in 24-hour ambulatory systolic blood pressure and office-based blood pressure at two years.
The data showed significant group differences in 24-hour ambulatory systolic blood pressure and office-based systolic blood pressure in favour of renal denervation, despite significantly more medications detected in the sham group.
Ambulatory systolic blood pressure reduced by -12.1mmHg in the renal denervation group vs. -7mmHg in the sham group (treatment difference: -5.7 mmHg; p=0.039), and office-based systolic blood pressure: -17.4mmHg in the renal denervation group vs. -9mmHg in the sham group (treatment difference: -8.7mmHg; p=0.0034). Long-term safety was very favourable with no confirmed renal artery stenosis greater than 70% in the Spyral group at two years.
“Now that renal denervation has been approved with FDA regulation and made commercially available in the USA, broadening its use in more clinical applications worldwide, for many practitioners there remain outstanding issues with regard to the longterm durability of this therapy,” says Kandzari of the significance of the study’s findings. “Equally important is the long-term safety as well as other issues such as identifying which patients might benefit
most from the therapy.”
“At least in this regard, the two-year outcomes for the SPYRAL ON-MED expansion study, representing the largest contemporary comparative on-med clinical trial, highlights the sustainability of blood pressure lowering, if not [the] amplified blood pressure reductions over a long-term follow-up, which is quite a consistent finding during follow-up in the SPYRAL programme.”
This is consistent with findings from other clinical
clinical outcome for patients.”
With regard to the uptake of the therapy, Kandzari says that, in the USA at least, reimbursement remains the biggest sticking point. This situation shows signs of shifting, with the Centers for Medicare and Medicaid Services (CMS) having this month (November 2024) granted transitional pass-through (TPT) payment for both Medtronic’s Symplicity Spyral and Recor Medical’s Paradise catheters. TPT payment, which will be effective for up to three years beginning 1 January 2025, aims to support patient access to new and innovative technology.
“Receiving TPT approval for our renal denervation catheter is an important milestone for the Symplicity blood pressure procedure, as it will enable greater patient access to a breakthrough treatment by reducing cost barriers for healthcare systems,” said Jason Weidman, senior vice president and president of the coronary and renal denervation business within the cardiovascular portfolio at Medtronic, in a
Kandzari presents at TCT 2024
We are exploring other vascular beds for renal denervation therapy, and it so happens that the hepatic artery is highly ennervated with sympathetic nerves”
trials, Kandzari says, as well as from the nearly 3,000-patient Global SYMPLICITY registry. The twoyear SPYRAL ON-MED data also confirm the safety of renal denervation, he adds, showing that there are no late-term consequences of the therapy regarding renal artery stenosis or declining kidney function.
“If anything, we would hope that kidney function might be more preserved compared with those individuals with uncontrolled hypertension that poses a continued risk for them. And then ideally too, spending a greater time with blood pressure control or what we call time in target range, or time in therapeutic range, that this may translate to improved
company press release.
CMS has created a distinct category and code for ultrasound renal denervation in recognition of the differentiated technology and procedure with Paradise.
“TPT for ultrasound renal denervation increases access to a proven device-based hypertension treatment option to patients who have been unable to achieve blood pressure control with lifestyle changes and medications alone,” said Lara Barghout, president and chief executive officer of Recor Medical.
“The granting of TPT highlights the safety and efficacy of this breakthrough device, which together demonstrated that the Paradise ultrasound renal denervation system met the newness and significant clinical improvement criteria. By creating a distinct device category, CMS have also recognised that the Paradise ultrasound renal denervation system is a highly differentiated technology and that there are significant differences in comparison to other technologies available in the marketplace. This is a major step forward in the reimbursement available for the Paradise ultrasound renal denervation system, creating additional financial support for hospitals and physicians to provide this novel and effective therapy to their uncontrolled hypertension patients.”
David
Product News
ShortCut leaflet splitting device gains US FDA market clearance
The US Food and Drug Administration (FDA) has provided market clearance for the ShortCut (Pi-Cardia) dedicated leaflet modification device, which is designed to enable valve-in-valve transcatheter aortic valve implantation (TAVI) procedures in patients at risk of coronary obstruction.
“Lifetime management of aortic stenosis calls for leaflet modification solutions like ShortCut to ensure that we are carefully addressing the risk of coronary obstruction before implanting a valve,” said Martin B Leon (New York-Presbyterian/Columbia University Medical Center and Columbia University College of Physicians and Surgeons, New York, USA) who chairs the global steering committee for ShortCut studies.
“The rigorous pivotal study leading to this important market clearance by the FDA demonstrates that ShortCut was both safe and effective in achieving the intended leaflet split in all patients. Importantly—it also shows that mechanical splitting with ShortCut, in both single- and dual-leaflet cases, was a controlled and teachable procedure, making it adoptable by TAVI centres as a critical step pre-implantation, so that patients at risk of coronary obstruction may be safely treated, without disruption of TAVI workflow.”
As bioprosthetic valves degenerate over time, patients will at some point likely need a valve-in-valve procedure to be performed, and a significant portion of them who are at risk for coronary obstruction will require leaflet splitting with ShortCut, Pi-Cardia says in a press release.
Medtronic’s Evolut FX+ TAVI system gains CE mark
Medtronic has received a CE mark for the Evolut FX+ transcatheter aortic valve implantation (TAVI) system for the treatment of symptomatic severe aortic stenosis. This follows the recent US Food and Drug Administration (FDA) approval of the system in March 2024.
The latest Evolut FX+ TAVI system offers three larger coronary access windows through a modified diamondshaped frame design, which are four times larger than regular cells of the Evolut TAVI system.
Evolut FX+ provides increased space for catheter maneuverability to
facilitate access to coronary arteries of varying patient anatomies. The design builds on the leading valve performance, excellent durability, and excellent outcomes that clinicians expect from the Evolut platform.
“The Medtronic Evolut FX+ system represents a significant step forward in the evolution of heart valve disease care,” said Didier Tchétché, interventional cardiologist and director of the Structural Heart Disease Department at Clinique Pasteur in Toulouse, France. “This advanced technology enables coronary access across multiple patient anatomies and offers clinicians a contemporary tool to improve outcomes without compromising an established valve performance. We are excited to see this innovative solution approved in European regions and look forward to using it for the benefit of patients.”
The Evolut FX+ TAVI system is indicated for symptomatic severe aortic stenosis adult patients across all risk categories (extreme, high, intermediate, and low) in the European Union and is also indicated for symptomatic severe aortic stenosis patients across all risk categories in the USA. The Evolut FX+ system is expected to be commercially available across Europe in the coming weeks as teams and physicians are trained on this new technology.
AtriCure launches EnCompass clamp in Europe
AtriCure has received regulatory approval to sell the EnCompass clamp in CE-marked countries in the European Union, and European surgeons have recently performed the first series of cases with the device.
The EnCompass clamp received US Food and Drug Administration (FDA) 510(K) clearance and was launched in the USA in 2022.
“Launching our EnCompass clamp in Europe represents a significant expansion of our product line internationally,” said Michael Carrel, president and CEO of AtriCure.
“We have seen this product have a positive impact in the USA over the last two years by advancing treatment concomitant to cardiac surgery. We are excited to offer this safe, innovative, and effective therapy to patients and our physician partners in Europe.”
The EnCompass clamp provides a simpler and faster approach to ablating the heart in open-chest procedures, allowing physicians to perform a comprehensive epicardial ablation of the left atrium in just a few minutes. The EnCompass clamp includes the features of AtriCure’s existing Synergy clamp family, such as parallel closure, uniform pressure, and custom power using Synergy radiofrequency (RF).
The EnCompass clamp also allows for easier placement using a magnetic guide, which enables more efficient procedures by minimising tissue
dissection. Further, the EnCompass clamp is designed to fit cardiac anatomy, supporting surgical ablation in procedures where the atrium would normally not be opened such as CABG [coronary artery bypass grafting] and AVR [aortic valve repair]. AtriCure estimates approximately 400,000 cardiac surgeries occur annually in the European Union.
Adona interatrial shunt used in first human cases
Adona Medical has announced the successful first-in-human use of its novel interatrial shunt in patients with heart failure.
The initial cases were performed by George Khabeishvili (Tbilisi Heart and Vascular Clinic, Tbilisi, Georgia) and supported by US heart failure experts Gagan Singh and Edris Aman.
“I am incredibly excited to be involved in the initial procedures for this groundbreaking technology. The Adona implant represents the next-generation of shunt technology, designed for dynamic interatrial flow that can be modified based on patientspecific haemodynamic needs,” said Singh, clinical cardiovascular research unit director, University of California at Davis (Davis, USA). “Heart failure is not a static condition but requires lifetime management. The combination of adjustable shunt size and atrial pressure measurement represents a huge leap forward in device therapy solutions.”
The Adona device includes a shunt that features a flow channel with an adjustable geometry that can be made larger or smaller postimplantation via the use of a proprietary induction catheter.
This configurability holds the potential for more individualized treatments compared to current shunt technologies which feature a flow passage with one fixed geometry. In addition, the Adona device features integrated sensors designed to capture pressure readings from both the left and right atria multiple times per day without requiring patient interaction.
These daily readings hold promise to provide physicians with a more complete understanding of a patient’s hemodynamic status and could augment shunt therapy by enabling more informed medical management.
“Success with the initial clinical cases represents a significant milestone in our mission to offer improved solutions for the management of heart failure. We’re now one step closer to
validating the potential benefits that the Adona technologies may offer to a vulnerable and suffering patient population,” said Brian Fahey, cofounder and chief executive officer of Adona Medical. “This was an enormous team effort, and I’d like to thank all of the Adona employees for their many contributions and our clinical collaborators for providing the valuable insights that have guided our incredible journey.”
Adona Medical is a part of the Shifamed portfolio of companies.
AI-based aortic stenosis screening software gains US FDA breakthrough designation AccurKardia has announced that its aortic valve stenosis electrocardiogram (ECG)-based artificial intelligence (AI) screening software has been granted breakthrough device designation by the US Food and Drug Administration (FDA).
The screening software aims to leverage the ubiquity of the ECG to identify potential cases of aortic stenosis within millions of ECGs already present in healthcare system electronic health records in order to help identify and prioritise which patients should receive echocardiograms for definitive diagnosis, AccurKardia says in a press release.
“AccurKardia’s recent advancement has the potential to create a paradigm shift in the detection of aortic valve stenosis, where earlier detection and treatment may mean the difference between life or death,” said Eduardo Hernandez, president, The Texas Heart Institute Center for Cardiovascular Care (Houston, USA). “Once FDAcleared and successfully deployed, this technology could become established as a standard-of-care screening tool for aortic valve stenosis in elderly patients.”
AccurKardia’s novel AI technology is designed to address challenges in the detection and diagnosis of aortic stenosis by offering a practical solution that complements a readily available diagnostic test, the ECG, for identifying at-risk patients earlier and more efficiently, the company’s press release adds.
US FDA approves IDE application for Katana thrombectomy system The US Food and Drug Administration (FDA) has approved Akura Medical’s investigational device exemption (IDE) application to initiate the QUADRAPE study evaluating the Katana thrombectomy system in patients with acute pulmonary embolism (PE).
QUADRA-PE is a multicentre, international trial designed to enrol up to 118 patients with clinically significant acute PE at up to 26 sites globally. The co-principal investigators of the pivotal study are Sanjum Sethi (Columbia University Medical Center, New York, USA) and Ann Gage (Tristar Centennial Medical Center, Nashville, USA).
Shockwave Medical has announced the completion of enrolment in EMPOWER CAD, the first prospective all-female study of percutaneous coronary intervention (PCI) in complex calcific disease.
The study is seeking to confirm the benefits of coronary intravascular lithotripsy (IVL) in female patients with calcified lesions in a post-market, real-world, all-comers population. Primary endpoint results will be presented in 2025, and patients will subsequently be followed out to three years.
“The completion of enrolment in EMPOWER CAD is a major milestone in our desire to improve cardiovascular outcomes for women with challenging calcified lesions,” said Margaret McEntegart (Columbia University Medical Center/NewYork-Presbyterian Hospital, New York, USA), coprincipal investigator of EMPOWER CAD. “Not only will this study yield valuable insights on the performance of coronary IVL in female patients, but as the first prospective all-comers study of coronary IVL, we also hope to gain additional insights about the utility of the technology in more complex patients.”
“We are eager to analyse the data and share results next year, with a goal of helping to close the gap in treatment outcomes between men and women,” said Alexandra Lansky (Yale University School of Medicine, New Haven, USA), co-principal investigator of EMPOWER CAD. “EMPOWER represents a significant step forward in women’s heart health. We also look forward to seeing the participating female interventional cardiologists and clinical researchers in the study continue to take on clinical trial leadership roles in the years to come.”
The EMPOWER CAD study enrolled 400 participants across 48 sites, spanning five countries.
First US cases performed with Vertex PE system in SPIRARE II pivotal study
Jupiter Endovascular has announced that the first US patient has been treated in the SPIRARE II US pivotal study of the Vertex pulmonary embolectomy system featuring the company’s Endoportal control platform.
The Vertex system is designed to treat acute pulmonary embolism (PE) in an endovascular procedure offering an “unprecedented level of control and precision”, according to the company.
The first US case was performed at Staten Island University Hospital, Northwell Health (New York, USA) by Mitchell Weinberg and Vincent Gallo.
“The unique manoeuvrability of the large-bore Vertex endoportal device enabled us to easily navigate across multiple bends through the right
heart and into the pulmonary arteries. Once in the pulmonary arteries, the technology allowed us to stabilise the endoportal device, creating a secure base for us to safely advance the aspiration catheter deep within the pulmonary vasculature and capture hard-to-reach thrombi. With Endoportal control, we achieved an excellent procedural result with remarkable speed and ease, especially given that it was our first time using this technology,” said Weinberg.
SPIRARE II is a prospective, singlearm, multicentre pivotal trial that will enrol up to 145 patients with acute, intermediate-risk PE treated with the Vertex pulmonary embolectomy system at up to 25 US sites. Trial endpoints will characterise the procedural and clinical benefits of PE treatment with Endoportal control using the Vertex system, across measures of safety, right heart function, and clinical improvement from the time of the procedure to 30 days post-procedure.
Prevail DCB to be studied in US IDE trial
Medtronic has received approval of a US Food and Drug Administration (FDA) investigational device exemption (IDE) to initiate its pivotal clinical trial of the Prevail coronary paclitaxel drug-coated balloon (DCB) for in-stent restenosis (ISR) and de novo small vessel disease.
Data from the Prevail Global Clinical Program will support approval of the Prevail DCB in Japan and the USA.
The multicentre, dual-cohort clinical trial will enrol up to 1,205 patients with coronary artery disease from approximately 65 global centres across the USA, Europe and Asia Pacific.
The trial will include a randomised controlled evaluation of ISR patients and a single-arm evaluation of de novo small vessel disease patients to assess the safety and efficacy of the DCB.
“As physicians treat more patients with complex lesions, it is important to have a device that helps to maintain durable patency while preserving future treatment options,” said David Kandzari (Piedmont Heart Institute and Cardiovascular Services, Atlanta, USA), co-principal investigator of the Prevail Global Study. “Drug-coated balloons provide clinicians with an anti-restenosis solution, without
the need of a permanent stent. This groundbreaking trial will include the first head-to-head randomised trial of two drug-coated balloons in the USA and will provide important additional evidence for this growing therapy.”
The ISR Cohort will be led by Kandzari and Bruno Scheller (Saarland University, Homburg, Germany) and will randomise patients 1:1 with Prevail and Boston Scientific Agent DCB to assess non-inferiority.
The de novo small vessel (DNSV) cohort will be led by Azeem Latib (Montefiore Health System, New York, USA) and Darren Mylotte (Galway University Hospital, Galway, Ireland) and will compare Prevail DCB against drug-eluting stents, the standard of care for small vessel treatment, using a historical control from the extensive body of evidence from the Resolute Onyx Clinical Program. The primary endpoint for both cohorts will be target lesion failure (TLF) at 12 months. Patients will be followed out to five years.
The Prevail Global Study will build upon the experience with the use of Prevail DCB globally. Prevail DCB was launched in Europe in 2021 with indications for the treatment of de novo lesions, in-stent restenosis, and small vessel disease in the coronary arteries.
The Prevail DCB is commercially available in more than 79 countries globally. Within the Prevail Global Study, Prevail DCB is investigational. Prevail DCB is not approved or sold in the USA or Japan.
First patients treated with Duo tricuspid coaptation system in TANDEM II study
CroíValve has announced the first patient treated with the Duo system as part of the TANDEM II early feasibility study in the USA.
The Duo system is a transcatheter valve that works in tandem with the native tricuspid valve to restore valve function while preserving the native valve apparatus.
TANDEM II is a prospective, multicentre study in the USA evaluating the safety and performance of Duo in severe symptomatic tricuspid regurgitation (TR).The first implant was performed by interventional cardiologists Pradeep Yadav and James Stewart, and cardiac surgeon Vinod Thourani, principal investigator for TANDEM II, at Piedmont Heart Institute in Atlanta, USA.
“The Duo system offers a unique benefit for patients suffering from TR as the device is designed to treat a broad patient population. The Duo procedure is straightforward, leveraging standard interventional techniques and has the potential to
accelerate transcatheter tricuspid therapy adoption by more physicians due to the system’s simplicity and scalability. This is a limitation with many other devices. On behalf of the TANDEM II investigators, we look forward to advancing treatment options for patients suffering from tricuspid regurgitation,” said Thourani.
“Every patient has a unique tricuspid valve anatomy and disease aetiology which can make device procedures complex. The Duo system is an important option in my treatment toolbox,” said Yadav, director of structural heart interventions at Piedmont Heart Institute.
The Duo system includes a coaptation valve, an adjustable catheter system and a stent for anchoring. The coaptation valve is positioned between the native tricuspid leaflets to fill the leaky area and prevent regurgitation.
A novel anchor system leaves the right heart and native valve apparatus untouched. The Duo implant procedure uses standard imaging and is less reliant on expert intraprocedural imaging, enabling a scalable procedure for broad therapy adoption.
Six-month data reflect safety and efficacy of Lithix HC-IVL system
Six-month data from the PINNACLE I study, evaluating the safety and performance of the LithiX Hertz Contact (HC, Elixir Medical) intravascular lithotripsy (IVL) system for the treatment of moderate to severely calcified coronary artery lesions have been reported at TCT 2024 (27–30 October, Washington, DC, USA).
LithiX data results demonstrate 98.3% clinical success (primary effectiveness and safety endpoint) with 100% angiographic success across a range of moderate to severe calcium morphologies, alongside <30% residual diameter stenosis was achieved in 100% of the lesions. No procedural angiographic complications, including no severe dissections, perforation, or abrupt closure post-stenting.patients with severe symptomatic tricuspid regurgitation (TR).
The first implant was performed by interventional cardiologists Pradeep Yadav and James Stewart, and cardiac surgeon Vinod Thourani, the principal investigator for the TANDEM II study.
Duo system
Prevail
Vertex
Industry News
J&J completes acquisition of V-Wave
Johnson & Johnson has announced the completion of its acquisition of V-Wave, a developer of treatment options for patients with heart failure. V-Wave will operate as part of Johnson & Johnson MedTech.
V-Wave’s cardiovascular implant technology specifically targets heart failure with reduced ejection fraction (HFrEF). The Ventura interatrial shunt (IAS) is a novel implantable device designed to decrease elevated left atrial pressure seen in congestive heart failure by creating a shunt between the left and right atrium, thereby reducing cardiovascular events and heart failure hospitalisations.
The device received US Food and Drug Administration (FDA) breakthrough device designation in 2019 and CE mark in 2020.
Tim Schmid, executive vice president and worldwide chairman of Johnson & Johnson MedTech, said: “We’re excited to officially welcome V-Wave to Johnson & Johnson MedTech. V-Wave’s novel implantable device, the Ventura interatrial shunt, offers tremendous promise for patients experiencing heart failure with reduced ejection fraction. This technology has the potential to be the first device of its kind to market. We look forward to working with the talented V-Wave team to bring this transformative innovation to patients.”
Novostia announces new funding and leadership changes
Novostia has announced a leadership transition alongside the successful completion of a ₣5.6 million fund raise to advance the company’s development efforts and clinical evidence base.
Novostia is developing the Triflo artificial heart valve, a mechanical,
Conference calendar
25–27 January
Society of Thoracic Surgeons (STS) 2025 annual meeting Los Angeles, USA sts.org/education/
29–31 January
British Cardiovascular Intervention Society (BCIS) ACI London, UK bcis.org.uk/event/bcis-aci-25/
three-leaflet valve prosthesis which is currently undergoing human clinical trials. Triflo is intended to minimise the risks associated with prolonged anticoagulation, as well as addressing patient concerns including noiseinduced discomfort and enhancing quality of life.
Recent changes at the company include the appointment of Soad El Ghazouani as the chief executive officer, effective immediately, succeeding Alain Barbal who will join the board of directors.
“I am honoured to join Novostia at such a pivotal time for our company. The company’s groundbreaking Triflo heart valve has already demonstrated immense potential to transform patient care, and I am eager to guide our talented team as we continue to push the boundaries of innovation and deliver exceptional value to patients, employees and shareholders.”
as well as his medical and clinical expertise in support of the success of the next-generation Point-Guard and the company’s future clinical plans and strategy,” said Eric Goslau, president and chief executive officer of Transverse Medical. “I’ve admired and followed Dr Meredith throughout his career, and it gives me great pleasure to welcome him to the board.”
CorFlow completes series B funding round to evaluate therapeutic treatments for MVO using CoFl system
Ian Meredith joins board of directors at Transverse Medical Transverse Medical has announced the appointment of Ian Meredith to the board of directors.
Meredith brings over 35 years of experience and global leadership in cardiology coupled with successful clinical trials and device commercialisation of multiple interventional devices.
Transverse Medical is developing the Point-Guard cerebral embolic protection device, designed to protect patients from embolic debris that is dislodged and released into the cerebral blood flow and could lead to periprocedural stroke as a result of the transcatheter aortic valve implantation (TAVI) procedure. Meredith has agreed to join the company’s board of directors as an independent member. He retired from Boston Scientific in April 2023 following six years serving as its executive vice president and global chief medical officer.
“I’m looking forward to Dr Meredith joining the board of directors for his corporate experience and board role,
19–22 February JimGISE 2025 Rome, Italy jimgise2025.it/ 11–13 February Technology and Heart Failure Therapeutics (THT) 2025 Boston, USA tht2025.crfconferences.com
CorFlow Therapeutics AG (CorFlow) has announced that it has raised €44 million in series B funding, which will contribute to the MOCA II pivotal study intended to validate CorFlow’s CoFl system to diagnose microvascular obstruction (MVO) in heart attack patients immediately following stent implantation. The trial will run in the USA and Europe and aims to facilitate US market clearance for the CoFl system. Additionally, it will fund an adaptive platform therapy study evaluating treatment effects of therapeutic agents delivered locally through the CoFl system on heart attack patients diagnosed with MVO.
MVO affects more than half of all patients who suffer an acute heart attack and is an independent predictor for heart failure and mortality. Currently not routinely diagnosed, MVO remains largely untreated, leading to poor patient outcomes, and contributing to high healthcare costs associated with cardiovascular disease.
CoFl is being developed to provide timely, accurate and consistent detection of MVO while patients are still in the cath lab immediately following the reopening of the larger epicardial arteries with a stent. The technology has been designed to also enable localised delivery of therapeutics to the microvasculature upon MVO diagnosis.
The MOCA II investigational device exemption (IDE) trial is designed to confirm the CoFl system’s accuracy in diagnosing MVO in high-risk heart attack patients. Led by principal investigator Tim Henry (The Christ Hospital, Cincinnati, USA), the trial
28 February–1 March CTO PLUS 2025 New York, USA cto2025.crfconferences.com
8–11 March Cardiovascular Research Technologies (CRT) 2025 Washington, DC, USA crtmeeting.org/attend-crt/
will recruit several hundred patients undergoing stent implantation due to ST-elevation myocardial infarction (STEMI) and will compare CoFl’s proprietary dynamic diagnostic measurement of MVO to postprocedure contrast-enhanced cardiac magnetic resonance imaging (CMRI), the current gold standard for detecting MVO. The MOCA II trial leverages the learnings from the company’s firstin-human MOCA I trial conducted in Switzerland, Latvia and the UK.
Jeffrey Popma joins CRF as chief scientific and strategic officer
The Cardiovascular Research Foundation (CRF) has announced the appointment of Jeffrey Popma as chief scientific and strategic officer. Popma will drive forward pivotal programmes and initiatives that will shape CRF’s future and fuel innovation at the CRF Clinical Trials Center (CTC), the organisation said in a press release.
His leadership will be instrumental in the development of the recently launched Real-World Data and Outcomes Center, advancing CRF’s commitment to impactful research, the organisation adds. As programme director for New York Valves and TCT, CRF’s flagship scientific meeting, Popma will play a key role in guiding the organisation’s contributions to the field of cardiovascular medicine.
“Jeff was an early leader at CRF and TCT and has had an illustrious career as a practicing interventionalist, academician, and thought leader, “said Martin Leon, founder and chairman emeritus of CRF. “I am thrilled that Jeff is returning to CRF as the chief scientific and strategic officer, a leadership role that will strengthen our depth and focus in achieving future creative goals in academic achievement, advanced data science, and medical education.”
“Jeff’s extensive expertise and visionary leadership will be invaluable as we continue to drive innovation and excellence in cardiovascular research and education,” said Juan F Granada, president and chief executive officer of CRF. “We are excited about the contributions he will make in furthering our mission, advancing the understanding of cardiovascular disease, and improving patient outcomes.”
13 March Global Cardiovascular Awards 2025 London, UK globalcardiovascularawards.com
29–31 March American College of Cardiology (ACC) 2025 Scientific Session Chicago, USA expo.acc.org/acc25/public
23–25 April Charing Cross (CX) Symposium 2025 London, UK cxsymposium.com/
Ventura interatial shunt
Ian Meredith
Third-party brands are trademarks of their respective owners.
1 Based on bench test data on file at Medtronic. May not be indicative of clinical performance. N = 7 DES of each tested.
2 Based on bench test data on file at Medtronic. May not be indicative of clinical performance. N = 5 DES of each tested:
Onyx Frontier Zotarolimus-Eluting Coronary Stent System Brief Statement
Indications
The Onyx Frontier zotarolimus-eluting coronary stent system is indicated for improving coronary luminal diameters in patients, including those with diabetes mellitus or high bleeding risk, with symptomatic ischemic heart disease due to de novo lesions of length ≤ 35 mm in native coronary arteries with reference vessel diameters of 2.0 mm to 5.0 mm. In addition, the Onyx Frontier zotarolimus-eluting coronary stent system is indicated for treating de novo chronic total occlusions and non-left main bifurcation lesions utilizing the provisional bifurcation stenting technique.
Contraindications
The Onyx Frontier system is contraindicated for use in: • Patients with a known hypersensitivity or allergies to aspirin, heparin, bivalirudin, clopidogrel, prasugrel, ticagrelor, ticlopidine, drugs such as zotarolimus, tacrolimus, sirolimus, everolimus, or similar drugs or any other analogue or derivative • Patients with a known hypersensitivity to the cobalt-based alloy (cobalt, nickel, chromium, and molybdenum) or platinum-iridium alloy • Patients with a known hypersensitivity to the BioLinx™ polymer or its individual components.
Coronary artery stenting is contraindicated for use in: • Patients in whom antiplatelet and/or anticoagulation therapy is contraindicated • Patients who are judged to have a lesion that prevents complete inflation of an angioplasty balloon or proper placement of the stent or stent delivery system.
Warnings
• Ensure that the inner package has not been opened or damaged as this would indicate the sterile barrier has been breached. • The use of this product carries the same risks associated with coronary artery stent implantation procedures, which include subacute and late vessel thrombosis, vascular complications, and bleeding events. • This product should not be used in patients who are not likely to comply with the recommended antiplatelet therapy.
Precautions
• Only physicians who have received adequate training should perform implantation of the stent. • Subsequent stent restenosis or occlusion may require repeat catheter-based treatments (including balloon dilatation) of the arterial segment containing the stent. The long-term outcome following repeat catheter-based treatments of previously implanted stents is not well characterized. • The risks and benefits of the stent implantation should be assessed for patients with a history of severe reaction to contrast agents. • Do not expose or wipe the product with organic solvents such as alcohol. • The use of a drug-eluting stent (DES) outside of the labeled indications, including use in patients with more tortuous anatomy, may have an increased risk of adverse events, including stent thrombosis, stent embolization, myocardial infarction (MI), or death. • Care should be taken to control the position of the guide catheter tip during stent delivery, stent deployment, and balloon withdrawal. Before withdrawing the stent delivery system, confirm complete balloon deflation using fluoroscopy to avoid arterial damage caused by guiding catheter movement into the vessel.
• Stent thrombosis is a low-frequency event that is frequently associated with MI or death. Data from the RESOLUTE clinical trials have been prospectively evaluated and adjudicated using the definition developed by the Academic Research Consortium (ARC).
The safety and effectiveness of the stent have not yet been established in the following patient populations:
vessel diameters of < 2.0 mm or > 5.0 mm • Patients with evidence of an acute ST-elevation MI within 72 hours of intended stent implantation
• Patients with vessel thrombus at the lesion site • Patients with lesions located in a saphenous vein graft, in the left main coronary artery, or ostial lesions • Patients with diffuse disease or poor flow distal to identified lesions
• Patients with 3 vessel disease
The safety and effectiveness of the stent have not been established in the cerebral, carotid, or peripheral vasculature. Additionally, the safety and effectiveness of using atherectomy devices with the stent have not been established. The effect of potential drug interactions on the safety or effectiveness of the Onyx Frontier™ stent has not been investigated. Potential interactions of the stent with other drug-eluting or coated stents have not been evaluated and should be avoided whenever possible.
Clinical studies of the Resolute stent did not suggest any significant differences in safety and effectiveness for male and female patients and did not include sufficient numbers of patients to assess for differences in safety and effectiveness due to ethnicity.
Decisions about duration of DAPT are best made on an individual basis and should integrate clinical judgment, assessment of the benefit/risk ratio, and patient preference. Premature discontinuation or interruption of prescribed antiplatelet medication could result in a higher risk of stent thrombosis, MI, or death. Before PCI, if premature discontinuation of antiplatelet therapy is anticipated, physicians should carefully evaluate with the patient whether a DES and its associated recommended DAPT regimen is the appropriate PCI choice.
Following PCI, if elective noncardiac surgery requiring suspension of antiplatelet therapy is considered, the risks and benefits of the procedure should be weighed against the possible risk associated with interruption of antiplatelet therapy. Patients who require premature DAPT discontinuation should be carefully monitored for cardiac events. At the discretion of the patient’s treating physician(s), the antiplatelet therapy should be restarted as soon as possible.
Instructions for Stenting of Bifurcation Lesions
The provisional technique of bifurcation stenting recommends a single stent placement in the Main Vessel (MV), finalized with proximal optimization technique (POT). POT includes performing post-dilatation to achieve full apposition of the stent proximal to the bifurcation and reduce the risk of side branch (SB) compromise. If inadequate results are found in the SB such as: threatened SB closure, TIMI flow < 3, dissection type B or worse, or residual stenosis > 80%, the provisional bifurcation stenting technique recommends placing a second stent in the SB as a bailout. As per cardiology societal recommendations, two-stent techniques following single stent provisional bifurcation stenting including T, TAP, and Culotte stenting may be utilized as needed. However, the RESOLUTE ONYX PAS Bifurcation Cohort did not evaluate the safety and effectiveness of two-stent bifurcation techniques, including planned (upfront) two-stent bifurcation techniques (such as DK-crush). Additionally, two-stent bifurcation techniques may introduce additional forces and/or failure modes to the stents, and the performance of the Resolute Onyx stent has not been evaluated under these conditions in nonclinical testing.
Potential Adverse Events
• Coronary artery occlusion, perforation, rupture, or dissection • Coronary artery spasm • Death • Embolism (air, tissue, device, or thrombus)
• Emergency surgery: peripheral vascular or coronary bypass • Failure to deliver the stent • Hemorrhage requiring transfusion • Hypotension/ hypertension • Incomplete stent apposition • Infection or fever • MI
• Restenosis of the stented artery • Shock/pulmonary edema • Stable or unstable angina • Stent deformation, collapse, or fracture • Stent migration or embolization • Stent misplacement • Stroke/transient ischemic attack
• Thrombosis (acute, subacute, or late)
Adverse Events Related to Zotarolimus
Patients’ exposure to zotarolimus is directly related to the total amount of stent length implanted. The actual side effects/complications that may be associated with the use of zotarolimus are not fully known. The adverse events that have been associated with the intravenous injection of zotarolimus in humans include but are not limited to:
The potential adverse reactions in nursing infants from zotarolimus have not been determined. The pharmacokinetic and safety profiles of zotarolimus in infants are not known.
Adverse Events Related to BioLinx™ polymer
Although the type of risks of the BioLinx polymer coating are expected to be no different than those of other stent coatings, the potential for these risks are currently unknown as the coating has limited previous use in humans. These risks may include but are not limited to the following:
• Allergic reaction • Focal inflammation at the site of stent implantation
• Restenosis of the stented artery
Please reference appropriate product Instructions for Use for more information regarding indications, contraindications, warnings, precautions, and potential adverse events.
CAUTION: Federal (USA) law restricts this device to sale by or on the order of a physician.
For further information, please call and/or consult Medtronic at the toll-free numbers or websites listed.
• Patients with coronary artery reference
• Patients with target lesions that were treated with prior brachytherapy or the use of brachytherapy to treat in-stent restenosis of the stent • Women who are pregnant or lactating • Men intending to father children • Pediatric patients below the age of 18 years
Other risks associated with using this device are those associated with percutaneous coronary diagnostic (including angiography and IVUS) and treatment procedures. These risks (in alphabetical order) may include but are not limited to: • Abrupt vessel closure • Access site pain, hematoma, or hemorrhage • Allergic reaction (to contrast, antiplatelet therapy, stent material, or drug and polymer coating) • Aneurysm, pseudoaneurysm, or arteriovenous fistula (AVF) • Arrhythmias, including ventricular fibrillation • Balloon rupture • Bleeding • Cardiac tamponade