6 minute read
Distrofia areolar central


Autores: Borghi, Santiago; Usuna, María Paz; Medina, Débora; Fangio, Rocío; López, Martín; Núñez Sánchez, Carlos; Aghetoni, Fernando y Julieta Mitchelena, Clínica Privada de Ojos Santa Lucía (Mar del Plata).
La Distrofia Areolar Central o CACD (Central Areolar Choroidal Distrophy) es una enfermedad hereditaria de la retina poco frecuente, de carácter autosómico dominante, ligada al cromosoma 17p13, aunque se han descrito pedigríes recesivos en algunos casos. Se caracteriza por la pérdida de la coriocapilar y atrofia del epitelio pigmentario de la retina en un área circunscripta que afecta la zona foveal y parafoveal, lo que resulta en una pérdida visual progresiva y profunda.
Caso clínico
PALABRAS CLAVES
DISTROFIA RETINAL
CADC
ATROFIA RETINAL

DISMINUCIÓN DE AV
ENFERMEDAD HEREDITARIA
POCO FRECUENTE
DMAE
Se presenta a la consulta un paciente masculino, de 80 años de edad manifestando disminución de agudeza visual bilateral lentamente progresiva y metamorfopsias. No informa antecedentes personales de relevancia.
Examen oftalmológico
AV sin corrección 5/10 ao, no mejora con estenopeico.
PIO 12 mmHg ao.
BMC pseudofaquia ao.
Test de Amsler positivo ao.
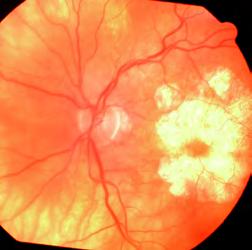
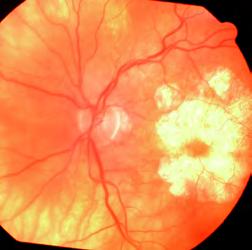
Se realiza fondo de ojos bajo dilatación donde se distingue atrofia retinal geográfica bien delimitada, con compromiso macular en ambos ojos.
Ateneo Mo
Se realiza tomografía de coherencia óptica (OCT) y angiofluoresceinografía (AFG).
En la OCT se observa disminución de las capas plexiforme y nuclear externas, con conservación del área foveal.
En la AFG aneritra se observan lesiones redondeadas con centro oscuro. Mientras que en el tiempo arterio-venoso se notan hipofluorescencias.
Diagnóstico
Teniendo en cuenta el cuadro clínico con el que se presentó el paciente, el análisis oftalmológico y los exámenes complementarios, se descarta la degeneración macular asociada a la edad planteada en un principio y se determina el diagnóstico de atrofia areolar central. Se explica al paciente y su familiar acompañante la evolución de esta enfermedad y el pronóstico visual reservado que implica. Se indican controles cada 6 a 12 meses.

Ateneo Mo
Discusión
La distrofia coroidea areolar central fue descrita por primera vez en 1884 por Nettleship que por entonces la llamó distrofia areolar central senil.
En 1939, Sorsby la renombró como Angio-esclerosis Coroidea, enfermedad que cuenta con 5 pedigríes de los que solo 1 o 2 fueron autosómicos dominantes y algunos de ellos presentaban drusas y flecks (por lo que podría confundirse con Stargart o Degeneración Macular Asociada con la Edad).
Se trata de una patología retinal poco frecuente que afecta por igual a hombres y a mujeres.
En la mayoría de los casos, la CACD se encuentra relacionada con una herencia autosómica dominante heterogénea, ligada al cromosoma 17p13 sin la presencia de drusas. Asimismo, pueden describirse 5 mutaciones del gen periferina/degeneración lenta de la retina (RDS): p.Arg142Trp, p.Arg172Trp, p.Arg172Gln, p.Arg195Leu y p.Leu307fsX83. De estas, la p.Arg142Trp es la mutación más fuertemente asociada a drusas y flecks, sin embargo, no son comunes de ver en esta patología.
La CACD es un desorden hereditario que afecta principalmente la mácula, que resulta en un área bien definida de atrofia del epitelio pigmentario de la retina (EPR) y coriocapilar. En un principio no se asocia a la presencia de drusas o flecks, aunque puede haber excepciones.
En etapas tempranas presenta una sutil despigmentación moteada en polo posterior. Ya en últimas instancias, aparece el reflejo tapetorretiniano y, finalmente, la despigmentación se agranda en un área redonda u ovalada de atrofia del EPR y la coriocapilar en el centro de la mácula.
En muchos casos presenta un borde marcado que separa la retina sana de la enferma. Es muy típico que persista una pequeña isla central de 1° o 2° rodeada por una zona de atrofia que produce un escotoma en anillo de los 20° centrales. Los grandes vasos coroideos subyacentes se hacen visibles.
Los pacientes usualmente presentan manifestaciones clínicas entre los treinta y cuarenta años de edad, con agudezas visuales (AV) de 0.1, por lo cual suele llevar a un mal diagnóstico de DMAE seca.
Pero, entre los cuarenta y setenta años, la atrofia progresiva lleva a una pérdida dramática de AV y severa discapacidad visual.
Presenta 4 estadios clínicos descriptos por Deutman:
Estadio I: despigmentación moteada fina y leve parafoveal, de comienzo en la edad adulta y asintomática.
Estadio II: moteado rodeando la fóvea.
Estadio III: aparecen gradualmente áreas redondas u ovales de atrofia geográfica (ausencia de coriocapilar) simétricas, bien delimitadas, inferiores a 3-4 diámetros papilares y sin afectación foveal.
Estadio IV: afectación central.
Debido a las alteraciones inespecíficas vistas en CACD, los diagnósticos diferenciales pueden ser un desafío. Además de la degeneración macular asociada con la edad, muchas de las distrofias maculares pueden presentarse con moteado simétrico del EPR, maculopatía en ojo de buey y atrofia central geográfica, imitando aquellos cambios tempranos vistos en la distrofia coroidea areolar central. Entre ellos se pueden considerar:
1. Drusas maculares dominantes (DD) que evolucionan a atrofia, presentan aparición precoz (20/30 años) con un patrón de distribución diferente, predominando en la mitad nasal del polo posterior, provocan disminución AV alrededor de los 50 años
2. Enfermedad de Stargardt dado que es la segunda patología hereditaria de retina más frecuente tras la retinosis pigmentaria, en la mayoría de los casos aparece entre los 8 y 14 años. La presencia de flecks amarillos que dejan una zona de retina sana alrededor del NO, la elevada fluorescencia, junto con la presencia de silencio coroideo permiten sospechar esta enfermedad
3. Distrofias en Patrón (DP) se caracterizan por la acumulación de lipofucsina en el EPR.
Conclusión
La CACD es una enfermedad poco frecuente que suele ser sub-diagnosticada por su parecido a la Degeneración Macular Asociada a la Edad y otras enfermedades retinales. Precisamente, ante cuadros incompletos o confusos, además del examen oftalmológico completo, OCT y AFG como estudios complementarios, es importante considerar realizarle al paciente un mapeo genético para arribar a un diagnóstico de certeza.









