14 minute read
Interview: A conversation on cancer research with Dr Anscombe


One faulty cell
An interview with Dr Anscombe
Advertisement
Research is undoubtedly one of the most arduous yet rewarding careers in science. From trials and errors to meticulously analysing your findings, science research has the potential to shape the future of the world. Vanessa Y, Lily and Emma Tse sat down with Dr Anscombe to discuss her journey through a career in cancer therapy research. We’d like to thank Dr Anscombe for providing such great insight into the world of biochemical research - we hope you enjoy learning about her experiences as much as we did!
Background knowledge
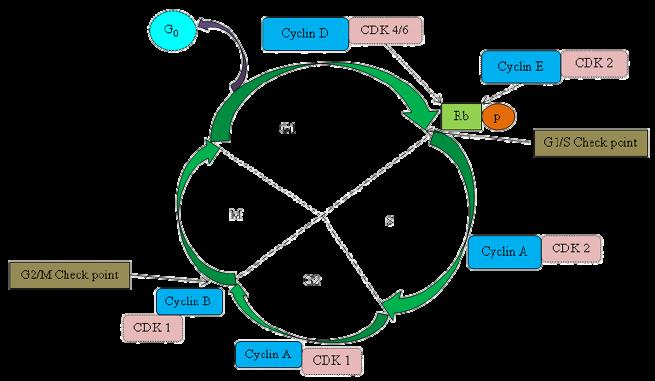
The cell cycle is regulated by cyclin-dependent kinases (CDKs). Kinases are enzymes that add phosphate groups to other molecules, a process called phosphorylation. Throughout the cell cycle, proteins called cyclins bind to CDKs, which allows CDKs to phosphorylate proteins such as pRb. pRb, short for retinoblastoma protein, is a tumour suppressor protein, which means it acts to inhibit cell proliferation through slowing mitosis or inducing apoptosis. Once pRb is phosphorylated, the cell is given the green light to continue to the next phase of the cell cycle; if not, the cell cannot progress further. When it comes to proteins themselves, understanding their 3D structure is imperative, because structure equates to function. X-ray crystallography is a widespread technique that can be used to find a protein's structure, which involves bouncing Xrays off a protein crystal and measuring the diffraction angles (in simple terms). Once the X-ray crystallography data is obtained, it's processed through a computer programme that executes a Fourier Transform, which is an image-processing tool that breaks down an image into cycles more specifically, their sine and cosine components. A simple analogy is to think of being given a smoothie and finding its recipe!
Could you summarise the research that you did for your PhD?
There are four phases in a normal cell cycle, G1, S, G2 and M (mitosis), and within M you ’ ve got subphases. The protein I looked at was CDK2 (cyclin-dependent kinase 2) and that regulates the transition between G1, where the cell is growing fairly normally, and S, where it starts to replicate its DNA. Between each stage of the cell cycle, there are checkpoints that check, for instance, that there’s no damage to the DNA before replication, whether you ’ ve got the right number of copies of chromosomes, or that the cell is generally ready to enter the next phase of the cell cycle. We were looking at one particular cell cycle transition and the kinase involved in regulating it. We were essentially trying to halt that process because in cancer cells, the processes at these checkpoints become disrupted, so cancer cells escape the normal checks and balances a cell goes through. These checks keep cells functioning properly, avoiding things like DNA damage by UV or reactive oxygen species being passed down to the next generation of cells. Cancer cells usually have mutations that allow them to escape this G1-S checkpoint, so they don’t respond to pRb, for instance. So you want to switch off the CDK2 and upregulate the pRb activity. The idea we were looking at was whether we could reactivate those checkpoints and therefore stop cancer cells from dividing further. If we could, that would imply it could be used as cancer therapy! Essentially, the CDK2 phosphorylates another protein, and in order to do that, it has to bind ATP (the energy currency of the cell). In the binding site where the ATP binds to the kinase, we looked at developing inhibitors that bound there to stop ATP from working, and therefore stop the kinase from working. My PhD involved my team and I getting sent molecules, which were the potential candidates we would test. I was based at Oxford, and a lab in Newcastle would send us possible drug molecules that might potentially bind to this protein. We would see if they did bind and if we could determine the structure of the protein, in addition to looking at the structure with the drug molecule it bound to, how closely it bound and whether it was a stable interaction or not.
So you’re basically trying to inhibit the CDK2, which is involved in the G1-S checkpoint, in order to reactivate that checkpoint. How exactly does that work?
Here is a simplified diagram of the cell cycle. This protein, pRb, which stands for the retinoblastoma protein, is a tumour suppressor. The function of this tumour suppressor protein, which determines whether DNA synthesis can start, is determined by CDK2. There are lots of cyclins in the cell cycle - CDK2 phosphorylates cyclin E, ATP has to bind to CDK2 in order to phosphorylate the protein, and if we can stop the ATP from binding to the CDK2, phosphorylation can’t take place.
In the process of your research, what did you do?
The day-to-day process was that we would get sent a new set of chemicals to test - they ’ ve done all the basic chemistry and replicated all these potential drug molecules. They would send us a big batch to test, and we firstly did various assays to see if they-
In the process of your research, what did you do?
-would bind to the proteins. I used radioactive phosphorus to follow the process of phosphorylation. We put the radioactivity under the photographic film and it makes a dark spot on the film which can be used to see if the kinase is still working. If the kinase has not been working, this suggests that the drug molecule bound to it has had some effect because the kinase is not phosphorylating things anymore. So this was a functional test. If the drug molecule was working, we would look at a picture of what the protein looked like bound to the drug molecule by crystallising the CDK2 and conducting X-ray crystallography, which is a way of taking a photo of a protein. We use X-rays because proteins are way smaller than the wavelength of light so they are too small to take a light photo. Usually, proteins are too small to get a good picture even with electron microscopy. X-ray crystallography is quite a complicated process - you make a crystal so you have a regular arrangement of proteins which amplifies the signal. You then shine X-rays at it and look at the scattering pattern. If you use some complicated maths called the Fourier transform, you can work backwards to the original structure. Lots of computer programs were involved, and you need an X-ray source and to do lots of maths afterwards. We used to go to Diamond Light source, which was a big synchrotron. The first time I went there I was amazed - you feel like you ’re in a Bond movie! Inside, there are lots of beam lines and there is a smaller ring in the middle. They basically send electrons really, really fast around this ring structure, and when you bend an electron round a corner, it releases certain types of radiation depending on how much it bends. So, if you control how fast an electron is going and how much you ’re bending them, you can get different types of radiation. We used X-rays, and we used to take our crystals here and we would get a few hours here to irradiate them and get a pattern at the end.

Image credits: Copyright © 2020 Diamond Light Source

So a famous X-ray diffraction pattern you might have seen is that of DNA, and that’s because DNA has a regular structure with a double helix so you get a cross shape.
X-ray diffraction pattern of crystallized 3Clpro, a SARS protease Jeff Dahl, X-ray diffraction pattern 3clpro, CC BY-SA 3.0
But proteins are more irregular, so you get something that looks more like this (see diagram below) - lots of dots. In a nutshell you feed this into a computer program which does a Fourier transform and you have to do some interpretation of that yourself, so it’s a mathematical procedure that turns it back into an electron density map. And then you have to interpret that and try to drop amino acids that make up the protein into the right places and understand the protein structure. Once I got that protein, I looked at whether the drug molecule was bound and where it was bound - if it was in the binding site where the ATP should be, was it fully occupying it? Which bits of the protein were in contact with it? So we did that technique crystallography - and we did NMR as well as mass spectrometry which enables you to find out the Mr (relative molecular mass) of the molecule to find out whether the drug molecule was bound.
So what did you find in your results?
CDK2 was one of the protein targets I had. The other target I looked at was p53, another tumour suppressor also known as the “ master switch” of the cell involved in processes like apoptosis (programmed cell death). In that regard, I was looking at p53 binding to another protein called MDM2, and I was looking at trying to disrupt that interaction between the two because that turns the tumour suppressor back on. I was looking for a drug molecule that bound in the interface between the two molecules by “ getting in the way ” . We managed to find some good candidates and structures, some of which are currently going through clinical trials. We got some patents on the drug molecules that we tested and they are currently being tested to see if they can be used. There’s a long process in drug
development after you ’ ve done the basic research in cells and you ’ ve got to test it in organisms, animals and eventually humans, and most tend to fail in that stage due to safety or functional issues.
It’s a difficult one with things like patents because there’s this argument that drugs should be cheaper. You get generic drugs, which is where a drug has gone out of patent and another drug company makes something with exactly the same structure and marks it with a different name. There’s a limitation for how many years a drug stays in patent for, kind of like copyright. The reason why the patent is there is to protect the investment of the companies that are going to spend lots of money developing it. Obviously, you have to also try to get the prices to come down because it’s not fair if prices are very high - you want as many people as possible to benefit from it, so I think it is right for drugs to come out of patent so that others can produce it. Since they haven’t put in as much money into the research, they can produce the drugs more cheaply as they can essentially copy what the other drug companies have done.
If drug development wasn’t so confidential and more open to researchers, would developments happen quicker?
Since we’ve opened the ethical can of worms now, do you think patenting drugs is ethical if research is primarily directed in a way for companies to make profits? This means a lot of people go into cancer research because it’s very well-funded, and other diseases like Alzheimer’s get less funding as a result. Do you think that’s ethical?
With patents, for me, I think they ’ ve got the balance probably about right at the moment, in terms of protecting the companies enough that you ’re still able to fund things. If you don’t incentivise new developments, people will stop doing that research and stick with drugs they have, and research will slow down and stop. For example, if you look at the COVID-19 vaccines, there’s enough incentive for drug companies to want to produce this to make money so that we get vaccines quickly. The main issue is that drug companies will fund what they know will make them money. So that’s why things like cancer get funded a lot more, and things like malaria and orphan diseases (rare diseases) tend to get little funding. And there are other things like Alzheimer’s, which is becoming so much more of a problem as the population ages, that are still massively underfunded compared to cancer in terms of research. Some of that is to do with what we value in countries that are wealthier. In Western countries, rates of cancer are much higher, and Western countries tend to do most of the funding, therefore cancer is funded more. But malaria is more prevalent in other countries, so some of that has to change in terms of research to strike a better balance.
I agree - I remember reading a statistic that Alzheimer’s affects even more people than cancer worldwide, but Alzheimer’s gets much less funding than cancer shockingly.
Yeah, and I think cancer is such an emotive thing because most people know someone - a relative or a friend - that has experienced it. When I was growing up, the reason why I went into cancer research was because my granddad died of lung cancer when I was 14, I didn’t really understand the concept that there wasn’t a single cure for cancer, but I wanted to help with the effort. But I think, whatever your best intentions are, especially as an individual researcher like a PhD candidate, you can’t really change what’s being funded. You don’t really have much say over what projects you do, you just choose between projects that are being funded and go for it. If you stayed and worked in science you could have an impact on what kind of research is funded, but it’s difficult because deciding what gets funded is more of a political issue. There’s a body called the National Institute for Clinical Excellence (NICE) and they make a lot of decisions about what gets funded - BBSRC (Biotechnology and Biological Sciences Research Council) decides a lot of the funding. But there are charities that fund things independently, so that’s how you change things being funded. But it definitely is a difficult one.
How did you find research in general? I read a very interesting Chemistry World article that said 2/3 of people in academia would consider leaving in the next 5 years or so?
I left research - I did enjoy it, and I found a lot of things I did interesting. I loved going to Diamond Light Source and I did projects in different places around the world as well and they were really interesting! But the thing that I missed in doing research was I spent a lot of time doing crystallography...
By yourself?
Well, with this big robot thing. Occasionally some people would come in to check their crystals, say “Hi” and go, and I’d be stuck in this basement for quite a long time. Or I’d be pipetting stuff for hours on end. It is interesting, but the day-to-day bit that I missed was the social aspect. I’ ve always enjoyed science communication, and one of the things I considered going into at university was science communication and science writing, like you ’re obviously interested in.
(Lily and Vanessa laugh)
So I considered doing a Master’s in science writing! I really missed the human aspect while working in research, and the nice thing about working here is being able to work with talented colleagues and interested pupils, and obviously have an impact on what they go on to study and what they do with their futures. I feel like I enjoy that more, having an overview of science. When you work in science, it can get really specific when you focus on only one project which is really, really interesting, but quite narrow. On the other hand, when you ’ ve involved in things like science writing or teaching, you get a much broader overview of everything. But it is fascinating to be at the cutting-edge of science - I really enjoyed it. I also think another issue with losing people from science is that women tend to struggle to move up the academic pathway. The proportion of women decreases from undergraduate, to master’s and PhDs and postgraduate students and professors. I think that should be addressed as well - why is that? I think they should look into it! Where have all the women gone? I was really lucky - when I was an undergraduate, I had a really, really inspirational female professor, Elspeth Garman, who did X-ray crystallography and I did my undergraduate project with her. She inspired me to carry on with research!








