18 minute read
Ethan Liu '25


Potential Culprit of Alzheimer’s Disease - Hyperphosphorylated Tau
BY ETHAN LIU '25
Advertisement
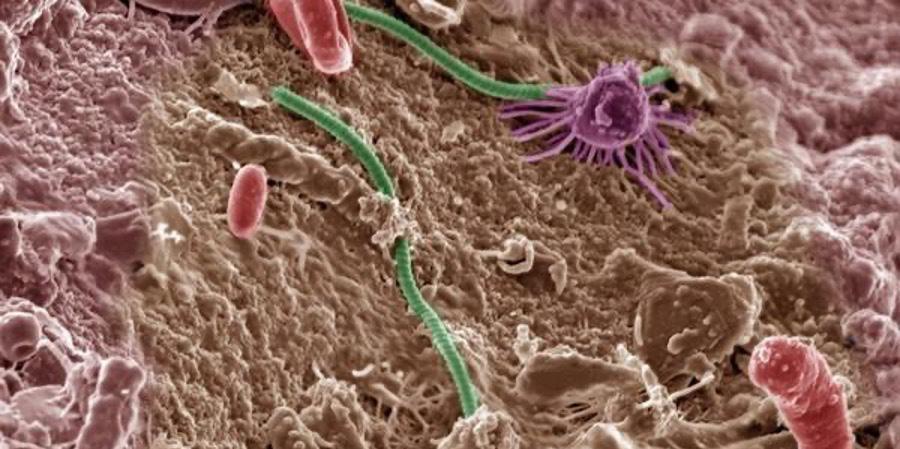
Cover Image: Neuro brillary tangles in the Hippocampus of an old person with Alzheimer-related pathology. Image Source: Wikimedia Commons Abstract
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder and the leading cause of dementia. Since the rst report of this disease by Dr. Alois Alzheimer in 1906, extensive studies have revealed several pathological features, including extracellular neuritic plaques containing Aβ and intracellular neuro brillary tangles mainly composed of hyperphosphorylated tau. According to the Aβ hypothesis, which has dominated the eld in the last two decades, the primary cause of neurodegeneration in AD is the abnormal accumulation or disposal of Aβ. However, the successive failure of several clinical trials targeting Aβ-associated pathology have raised doubts in its role in neurodegeneration. It is bene cial to revisit the importance of another essential protein, tau, in the pathogenesis of AD to explore more strategies for treating this devastating disorder. is report will review the physiological and pathological functions of tau and its relation to neurodegeneration in AD. Additionally, recent evidence supporting the tau hypothesis will be addressed.
Introduction
neurodegenerative disorder associated with aging. It is clinically characterized by dementia followed by other cognitive impairments, such as aphasia (inability to understand/express speech), agnosia (inability to interpret sensations), apraxia (inability to speak), inability to interpret sensations, and behavioral disturbance. e disease a ects approximately 10% of individuals older than 65 and about 33% of individuals 85 and older. Progressive neuronal loss caused by the disease is associated with the accumulation of insoluble brous materials within the brain, both intracellularly and extracellularly. e extracellular deposits consist of aggregated betaamyloid protein (Aβ), which is derived from a precursor protein called β-amyloid precursor protein (APP) that undergoes sequential cleavages by proteinases (Hebert et al., 2003). Aβ is a short peptide, and, although it is initially non brillar, it progressively transforms into brils called neuritic plaques. Intracellularly, cell bodies and apical dendrites of neurons are occupied by intraneuronal lamentous inclusions, which are called neuro brillary tangles (NFTs). NFTs mainly consist of hyperphosphorylated forms of microtubule-associated protein (MAP) and tau (Serrano-Pozo et al., 2011). Although Alzheimer’s Disease is pathologically characterized by the
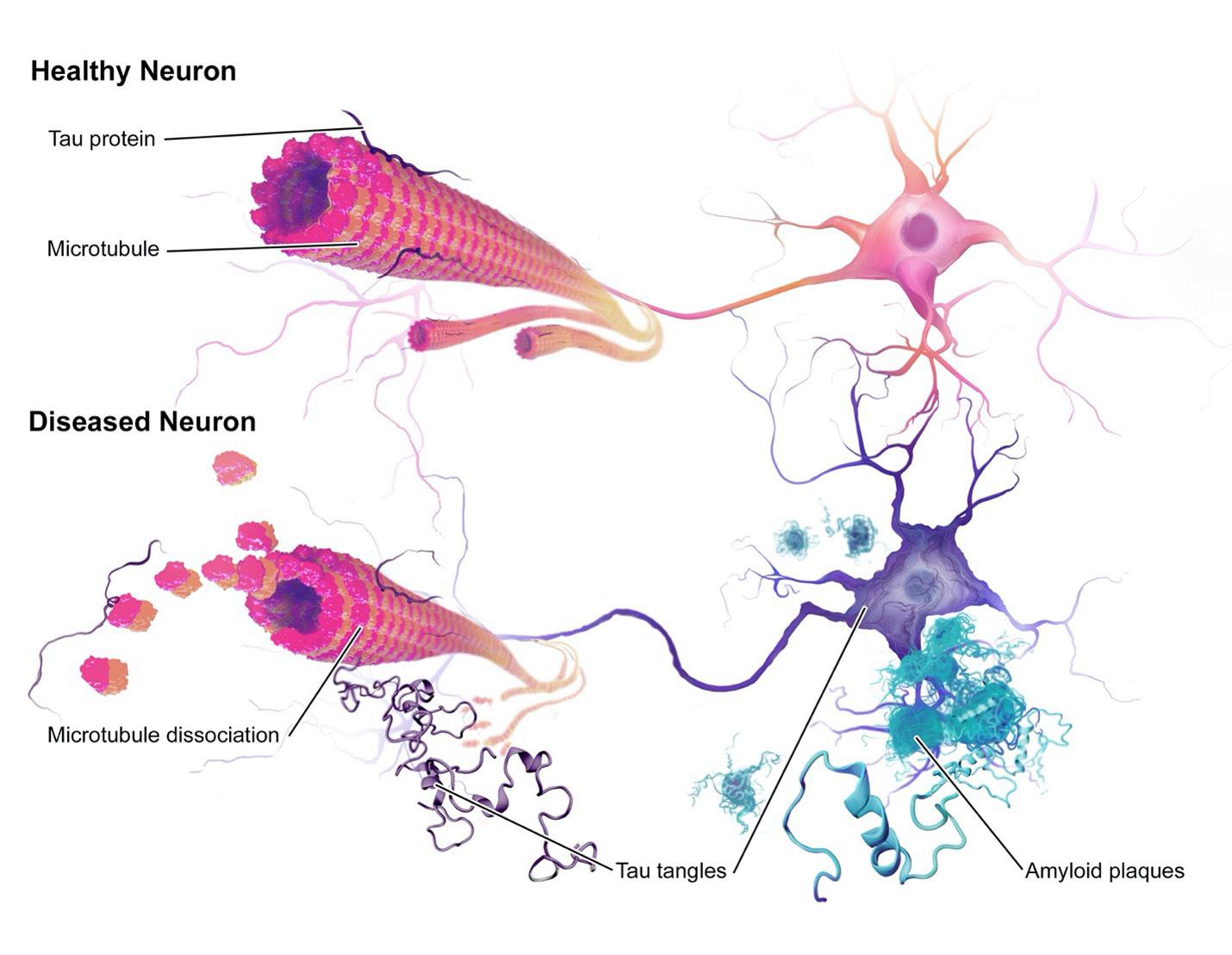
Image Source: Wikimedia Commons
presence of both neuritic plaques and NFTs, recent evidence indicates that neuro brillary tangles could have a more signi cant impact on the progression of the disease.
First, the extent and topographical distribution of neuro brillary lesions strongly correlates with the degree of dementia. e progression of NFTs in AD follows a typical spatiotemporal progression, which can be characterized in six neuropathological stages. During stages I and II, the super cial cell layers of the trans-entorhinal region and entorhinal regions are a ected. Patients are still clinically unimpaired at this stage. In stages III and IV, severe NFTs develop extensively in the hippocampus and the amygdala with limited growth in the association cortex. In stages V and VI, signi cant neuro brillary lesions appear in the isocortical association areas. e patient meets the criteria for the neuropathological diagnosis of AD during these two stages (Braak et al., 2014). e development of neuro brillary lesions contrasts with the development of Aβ deposits. Aβ deposits show a density and distribution pattern that varies signi cantly amongst individuals and is not a reliable indicator of Alzheimer’s disease. In contrast, the gradual and patterned development of neuro brillary lesions allows scientists to gauge the progression of the disease (Serrano-Pozo et al., 2011). exclusive to AD patients, Aβ deposition can be observed in healthy individuals. Neuro brillary lesions are only observed in neurodegenerative disorders such as AD, amyotrophic lateral sclerosis (ALS), Down syndrome, and postencephalitic parkinsonism (Rodrigue et al., 2009). erefore, it has been theorized that neuro brillary lesions have a more signi cant impact on Alzheimer’s patients than neuritic plaques. is paper provides a summary of the physiological and pathological roles of tau in AD and discusses the advancement of the tau hypothesis in understanding AD disease progression.
Tau Protein and its Physiological Function
Tau is a phosphoprotein and belongs to the microtubule associated protein (MAP) family. Microtubules are structures in the cell cytoskeleton which supports neurons with structural integrity and cellular transport Tau t is an intrinsically disordered protein (lacking a xed threedimensional shape) and is naturally unfolded and soluble under normal conditions. Although it is primarily found in neurons in the central nervous system, a relatively high concentration of tau is present in the heart, kidneys, lungs, skeletal muscles, pancreas, and testis. Trace amounts can also be found in the adrenal gland, stomach, and liver (Gu et al., 1996). In the adult brain, there
are six tau isoforms: products of alternative mRNA-splicing from a single gene, MAPT. e six isoforms, each ranging from 352 to 441 amino acid residues, di er in the number of amino acid N-terminal inserts (0N, 1N, or 2N) and the presence of three or four microtubule binding repeats (3R or 4R) near the C-terminal end. Each isoform has a di erent function on microtubules, and they are not equally expressed in cells. Tau is developmentally regulated since the abundance of di erent isoforms change as people age.
Tau plays an essential role in regulating microtubule dynamics and serves as a potent promoter of tubulin polymerization in vivo. e protein increases the rate of association and decreases the rate of dissociation of tubulin proteins at the growing ends of microtubules. It also reduces the dynamic instability of microtubules (the tendency for microtubules to change lengths rapidly) by preventing catastrophe, a period where microtubules rapidly shorten in response to stimuli (Barbier et al., 2019). ese phenomena have been observed in vivo in neurons when tau is injected into broblasts. An increase in microtubule mass and increased microtubule resistance to depolymerizing agents were observed in those cells (Drubin & Kirschner, 1986). Other than directly binding to microtubules, tau proteins also bind to other cytoskeletal components, such as spectrin and actin laments. ese interactions may allow microtubules to interconnect with other cytoskeletal components, such as neuro laments, to change cell shape and exibility (MietelskaPorowska et al., 2014). Additionally, tau serves as a postsynaptic sca old protein that modulates actin binding to substrates by regulating kinases which phosphorylates actin (Sharma et al., 2007). Overall, under normal physiological conditions, tau is involved in the regulation of microtubule dynamics, spatial arrangement, and cell stability, which a ects a variety of cellular functions.
Tau Phosphorylation and Aggregation
e phosphorylation of tau is regulated by kinase and phosphatase activity, although the speci c kinases (which phosphorylate proteins) and phosphatases (which dephosphorylate proteins) involved are still under investigation. Phosphoryalted forms of tau are high in fetuses and decrease with age due to phosphatase activation. Glycogen synthase kinase 3 (GSK3), cyclindependent kinase 5 (CDK5), and microtubulea nity-regulating kinase (MARK) are promising candidates currently under study for regulating the phosphorylation of tau. Previous studies showed that the inhibition of GSK3 by addition of lithium chloride decreased tau phosphorylation and lowered levels of aggregated and insoluble tau in vivo, suggesting that GSK3 could be a drug target for treating AD (Noble et al., 2005). In comparison to kinases, phosphatases decrease tau phosphorylated forms. One study found that when PP2A protein phosphatase-2A (PP2A) activity is increased following downregulation of its endogenous inhibitors, I1pp2Aand I2pp2A, there is an increased level of phosphorylated tau (Chen et al., 2008). erefore, disturbances
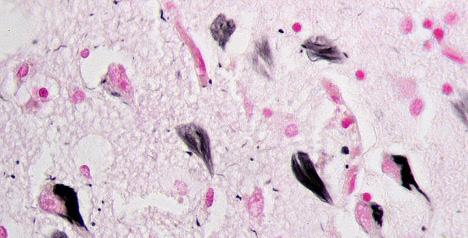
Image 2: Healthy Map2-tau in neurons. Map2 is stained in green. Tau is stained in red. DNA is stained blue. Tau is also in the yellow regions, where its red stain superimposes with the green stain in MAP to create the yellow colour. Image Source: Wikimedia Commons
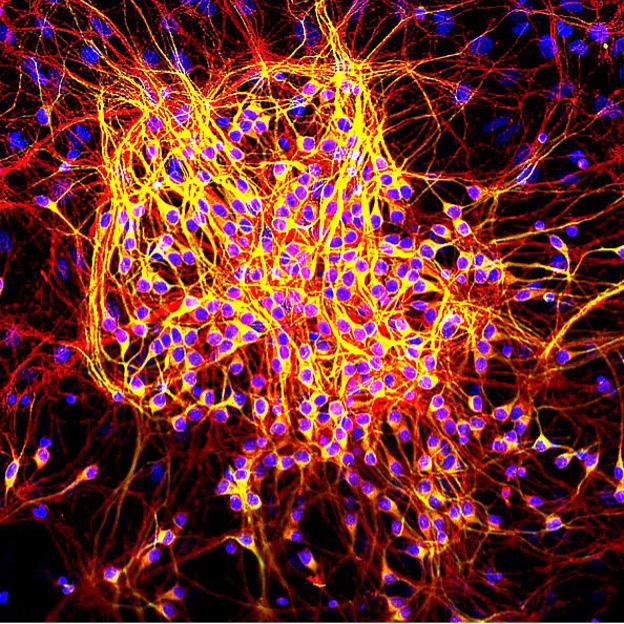
in the balance between kinase and phosphatase activities is suspected to contribute to the hyperphosphorylated status of tau protein and toxicity to neurons.
In terms of pathological status, hyperphosphorylated tau that are dissociated from microtubules become unbound tau in the cytoplasm. It has been speculated that this increased hyperphosphorylated cytosolic tau promotes the conformational change in favor of tau misfolding and aggregation. NFTs are mainly made up of paired helical laments (PHF), which are composed of tau in an abnormally phosphorylated state. All six isoforms have been detected in neuro brillary lesions in varying levels. With the help of cryo-electron microscopy, a precise microscopy technique which involves cooling the sample to cryogenic temperatures, it was discovered that the 3R tau isoforms assembled into twisted, paired, helical-like laments, while the 4R tau isoforms assembled into straight laments (Fitzpatrick et al., 2017). Although the stepwise process for generating tau aggregation is not well-understood, one hypothesis for a nucleation-dependent assembly mechanism suggests that tau is dissociated from the microtubules and undergoes a conformational change more prone to misfolding. Early deposits of tau which promote the formation of pretangles are suggested to lack β-sheets structures, since they cannot be detected by histochemistry, which involves staining the protein with dyes that detect speci c structures. However, later deposits of tau are detected to have β-sheets which can be detected by histochemistry, suggesting a structural transition. Recent evidence suggests that this structure change may be caused by tubulin dimers and oligomers, conglomerations of microtubule subunits. A er e cient lament addition and elongation, the mature PHFs are eventually formed (Meraz-Rios et al.). More work is still required to understand the factors promoting tau hyperphosphorylation and the nucleating steps.
Tau Hypothesis in the Pathogenesis of AD and Its Recent Updates
e tau hypothesis is one of the two major hypotheses to explain the pathogenesis and disease progression in AD (the other being the Aβ hypothesis). e tau hypothesis states that tau phosphorylation and aggregation are the primary cause of neurodegeneration in AD. Firstly, the topographical distribution of NFTs can characterize the progression of AD. Clinicopathological studies have demonstrated that the amount and distribution of NFTs correlates with the severity and the duration of dementia, while the distribution of Aβ does not (Kametani & Hasegawa, 2018). Second, it has been demonstrated that tau-related pathology occurs before the onset of Aβ accumulation. According to recent ndings from Dr. Braak and Dr. Del Tredici, who outlined the original six stages of AD (previously described in this paper), the system has been revised to include the progression of pre tangles formed in subcortical and transentorhinal regions of the brain prior to the development of NFT stage I. During these stages, Aβ deposits are not accompanied by tau lesions (Braak & Del Tredici, 2014). ird, tau has been involved in mediating Aβinduced neurodegeneration from animal studies. Knocking out the tau genes in the APP/PS1 (Amyloid Precursor Protein) mice conferred protection not only against memory impairment,
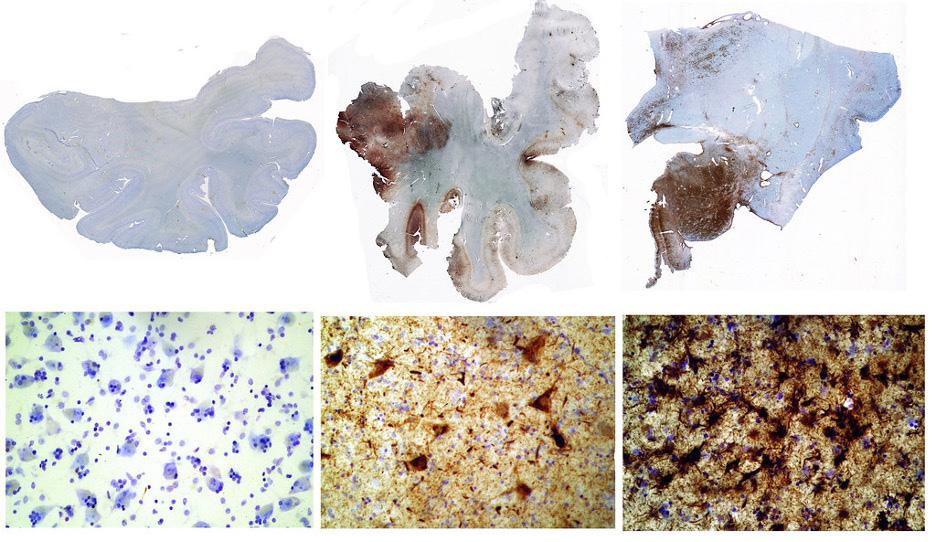
Image 3: Brain sections showing tau protein (brown spots). From le to right, 1) the brain of a healthy 65-year-old, 2) the brain of a former NFL linebacker who su ered eight concussions and died at age 45, John Grimsley, 3) the brain of a 73-year-old boxer who su ered from extreme dementia pugilistica. Image Source: Flickr
but also against synaptic loss and premature death. APP/PS1 mice that lacked tau had lesser plaque burdens than age-matched APP/PS1 mice that expressed tau (Leroy et al., 2012). Fourth, tau can elicit neuronal toxicity independent of Aβ. e pathological accumulation of tau within di erent cell types is responsible for a heterogeneous class of neurodegenerative diseases, which are termed tauopathies. Taumediated toxicity is extensively supported by ndings from familial frontotemporal dementia (a condition formerly known as frontotemporal dementia with parkinsonism-17) (Goedert et al., 2017).
With the advancement of AD studies through genetic animal models, the Tau P301S mouse model demonstrated synaptic de cits in the hippocampus before the formation of NFT. Another tau mouse model (rTg4510), which conditionally expresses P301L mutant tau, showed cell loss without the presence of NFTs, suggesting that early pathological species other than NFTs mediate cellular toxicity. e puzzle of how NFTs are formed is being solved. One step to solving this puzzle is identifying an intermediate species between tau monomers and NFTs, called tau oligomers. With the help of atomic force microscopy, granular oligomers were identi ed in Braak stage 0 brain samples. ese oligomeric species were suggested to increase with the advancement of pre-symptomatic stages, possibly explaining the synaptic degeneration that precedes NFTs. Oligomers were also detected in the P301L transgenic mice and rTg4510 mice (Meraz-Rios et al., 2010). While the results are still inconclusive, these oligomeric species could be the potential cause of NFT formation.
Another recent update of the tau hypothesis has emphasized the propagation of tau lesions through a prion-like mechanism or “seeding” concept. Prions are misfolded proteins that convert normal proteins into their misfolded shapes. Although there is no evidence supporting tau as an infectious entity, its capacity to transform healthy tau species to aggregation-prone species resembles the cell-to-cell transmission of prions in vivo. Guo and Lee (2011) demonstrated that minute quantities of misfolded preformed brils could rapidly yield large amounts of lamentous inclusions resembling NFTs when the misfolded preformed brils were introduced into tau expressing cells. e aggregates they produced resemble the NFTs found in AD patients; they have highly ordered β-pleated sheet structures and resemble NFTs in AD patients based on several analyses, including immuno uorescence, amyloid dye binding, immune-EM, and biochemical analysis. ese results indicate that preformed brils can potentially act as a prion to transform healthy tau species into NFTs (Guo & Lee, 2011). e hypothesis of tau-induced seeding was supported by numerous studies conducted in both cell and animal models. In these studies, tau seeds – derived from brain
Image 4: Model mouse brain with Neuro brillary Tangles (blue). Neurons are stained green, while blood vessels are red.
Image Source: ickr
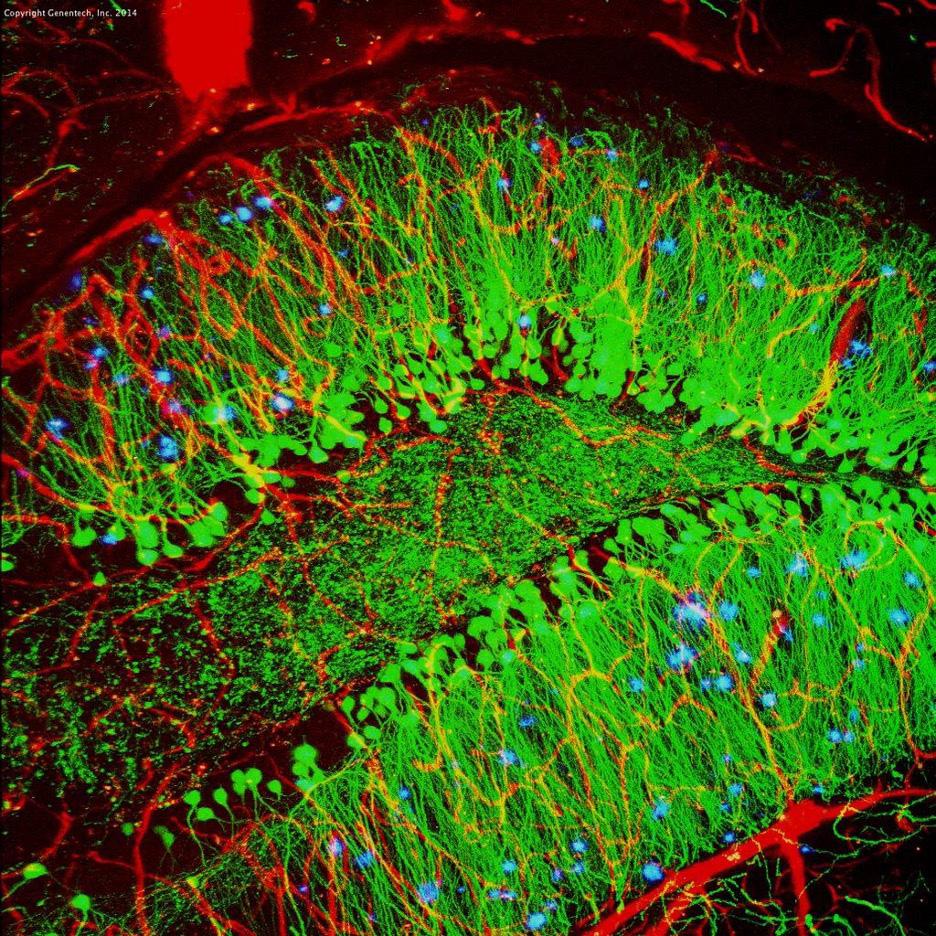
homogenates of tauopathy patients, symptomatic tau transgenic mice, or from recombinant tau in vitro – generated tau aggregate-bearing transfected cells. In all instances, tau aggregates were competent for seeding, though to di erent degrees (Goedert et al., 1989). To demonstrate that tau aggregates engage in a prion-like behaviour, more evidence is required to elucidate the mechanisms of cellular uptake, templated seeding, and subsequent intercellular transfer to induce similar aggregation in recipient cells.
Conclusion
Tau is a natively unfolded protein, and its ability to oligomerize and aggregate is regulated by post-translational modi cations, with hyperphosphorylation being the most detrimental change. e pathological tau species confer their neurotoxicity through both gain-offunction and loss-of function mechanisms, which have been the foundations of the tau hypothesis in AD. A growing body of data further identi es that tau oligomers are the conformers mediating neurodegeneration. Anti-amyloid (Aß) therapy has been the focus of the eld for the past 25 years, but due to a lack of clinical e cacy, scientists have recently been focused on anti-tau treatments (Vaz & Silvestre, 2020). Currently, there are studies targeting kinases involved in tau phosphorylation, such as glycogen synthase kinase 30beta (GSK-3ß) (Lauretti et. al., 2020). Several drugs, including lithium, a treatment for bipolar disorder, have been considered for their ability to inhibit GSK-3ß (Carmassi et al., 2016). e prevention of tau aggregation is also being researched using Methylthioninium Blue (also called Methylene Blue) or its derivatives, which have been shown to prevent aggregation in vitro (Wischik et al., 2015). ere are also studies on tau clearance, which removes abnormal tau through immunotherapy. is approach is still in its early clinical process and requires more time to be tested (Bittar et al., 2020). However, all these therapies are still in phase II or III of clinical trial and are still in development. Hopefully, some of them will be successful and bring us closer to understanding and defeating this mysterious disease.
References
Barbier, P., Zejneli, O., Martinho, M., Lasorsa, A., Belle, V., Smet-Nocca, C., Tsvetkov, P. O., Devred, F., & Landrieu, I. (2019). Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Frontiers in aging neuroscience, 11, 204. https://doi.org/10.3389/ fnagi.2019.00204 Bittar, A., Bhatt, N., & Kayed, R. (2020). Advances and considerations in AD tautargeted immunotherapy. Neurobiology of Disease, 134, 104707. https://doi.org/10.1016/j. nbd.2019.104707
Braak, H., & Braak, E. (1995). Staging of Alzheimer's disease-related neuro brillary changes. Neurobiol Aging, 16(3), 271-278; discussion 278-284.
Braak, H., & Del Tredici, K. (2014). Are cases with tau pathology occurring in the absence of Abeta deposits part of the AD-related pathological process? Acta Neuropathol, 128(6), 767-772. doi: 10.1007/s00401-014-1356-1
Carmassi, Claudia & Del Grande, Claudia & Gesi, Camilla & Musetti, Laura & Dell'Osso, Liliana. (2016). A new look at an old drug: Neuroprotective e ects and therapeutic potentials of lithium salts. Neuropsychiatric Disease and Treatment. Volume 12. 1687-1703. 10.2147/NDT.S106479.
Chen, S., Li, B., Grundke-Iqbal, I., & Iqbal, K. (2008). I1PP2A a ects tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J Biol Chem, 283(16), 1051310521. doi: 10.1074/jbc.M709852200
Drubin, D. G., & Kirschner, M. W. (1986). Tau protein function in living cells. J Cell Biol, 103(6 Pt 2), 2739-2746. doi: 10.1083/jcb.103.6.2739
Fitzpatrick, A., Falcon, B., He, S., Murzin, A. G., Murshudov, G., Garringer, H. J., Crowther, R. A., Ghetti, B., Goedert, M., & Scheres, S. (2017). Cryo-EM structures of tau laments from Alzheimer's disease. Nature, 547(7662), 185–190. https://doi.org/10.1038/nature23002
Goedert, M., Eisenberg, D. S., & Crowther, R. A. (2017). Propagation of Tau Aggregates and Neurodegeneration. Annu Rev Neurosci, 40, 189210. doi: 10.1146/annurev-neuro-072116-031153
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., & Crowther, R. A. (1989). Multiple isoforms of human microtubuleassociated protein tau: sequences and localization in neuro brillary tangles of Alzheimer's disease. Neuron, 3(4), 519-526. doi: 10.1016/08966273(89)90210-9
Goedert, M., Spillantini, M. G., Potier, M. C., Ulrich, J., & Crowther, R. A. (1989). Cloning and sequencing of the cDNA encoding an isoform of "Tau-mediated toxicity is extensively supported by ndings from familial frontotemporal dementia ..."
microtubule-associated protein tau containing four tandem repeats: di erential expression of tau protein mRNAs in human brain. EMBO J, 8(2), 393-399.
Gu, Y., Oyama, F., & Ihara, Y. (1996). Tau is widely expressed in rat tissues. J Neurochem, 67(3), 12351244. doi: 10.1046/j.1471-4159.1996.67031235.
Guo, J. L., & Lee, V. M. (2011). Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem, 286(17), 15317-15331. doi: 10.1074/jbc. M110.209296
Harada, A., Oguchi, K., Okabe, S. et al. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 369, 488–491 (1994). https://doi.org/10.1038/369488a0
Hebert, L. E., Scherr, P. A., Bienias, J. L., Bennett, D. A., & Evans, D. A. (2003). Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol, 60(8), 1119-1122. doi: 10.1001/archneur.60.8.1119
Hutton, M., Lendon, C. L., Rizzu, P., Baker, M., Froelich, S., Houlden, H., Pickering-Brown, S., Chakraverty, S., Isaacs, A., Grover, A., Hackett, J., Adamson, J., Lincoln, S., Dickson, D., Davies, P., Petersen, R. C., Stevens, M., de Graa , E., Wauters, E., van Baren, J., … Heutink, P. (1998). Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature, 393(6686), 702–705. https:// doi.org/10.1038/31508
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., . . . Montine, T. J. (2012). National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement, 8(1), 1-13. doi: 10.1016/j.jalz.2011.10.007
Kametani, F., & Hasegawa, M. (2018). Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. Front Neurosci, 12, 25. doi: 10.3389/fnins.2018.00025
Lauretti, E., Dincer, O., & Praticò, D. (2020). Glycogen synthase kinase-3 signaling in Alzheimer's disease. Biochimica et biophysica acta. Molecular cell research, 1867(5), 118664. https://doi.org/10.1016/j.bbamcr.2020.118664
Leroy, K., Ando, K., Laporte, V., Dedecker, R., Suain, V., Authelet, M., Héraud, C., Pierrot, N., Yilmaz, Z., Octave, J. N., & Brion, J. P. (2012). Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. e American journal of pathology, 181(6), 1928–1940. https://doi. org/10.1016/j.ajpath.2012.08.012
Meraz-Rios, M. A., Lira-De Leon, K. I., CamposPena, V., De Anda-Hernandez, M. A., & MenaLopez, R. (2010). Tau oligomers and aggregation in Alzheimer's disease. J Neurochem, 112(6), 1353-1367. doi: 10.1111/j.1471-4159.2009.06511.
Mietelska-Porowska, A., Wasik, U., Goras, M., Filipek, A., & Niewiadomska, G. (2014). Tau protein modi cations and interactions: their role in function and dysfunction. Int J Mol Sci, 15(3), 4671-4713. doi: 10.3390/ijms15034671
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., Gaynor, K., Wang, L., LaFrancois, J., Feinstein, B., Burns, M., Krishnamurthy, P., Wen, Y., Bhat, R., Lewis, J., Dickson, D., & Du , K. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proceedings of the National Academy of Sciences of the United States of America, 102(19), 6990–6995. https:// doi.org/10.1073/pnas.0500466102
Rodrigue, K. M., Kennedy, K. M., & Park, D. C. (2009). Beta-amyloid deposition and the aging brain. Neuropsychol Rev, 19(4), 436-450. doi: 10.1007/s11065-009-9118-x
Serrano-Pozo, A., Frosch, M. P., Masliah, E., & Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med, 1(1), a006189. doi: 10.1101/ cshperspect.a006189
Sharma, V. M., Litersky, J. M., Bhaskar, K., & Lee, G. (2007). Tau impacts on growth-factorstimulated actin remodeling. J Cell Sci, 120(Pt 5), 748-757. doi: 10.1242/jcs.03378
Vaz, M., & Silvestre, S. (2020). Alzheimer's disease: Recent treatment strategies. European journal of pharmacology, 887, 173554. https:// doi.org/10.1016/j.ejphar.2020.173554
Wischik, C. M., Sta , R. T., Wischik, D. J., Bentham, P., Murray, A. D., Storey, J. M., Kook, K. A., & Harrington, C. R. (2015). Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. Journal of Alzheimer's disease : JAD, 44(2), 705–720. https://doi.org/10.3233/JAD-142874