haematologica


VOL. 108 JUNE 2023 Journal of the Ferrata Storti Foundation ISSN 0390 - 6078 haematologica.org
J/"#,-#%3,0%&'()*#5*-K%*-%%
4'+5%+*$/1%6,'0-")%
78&"+$%!"+$,0%9:9;<%;;=:>%%% %?*$/@+,0/%9:9;<%;;=A%%%
!"#$%0/B*/C%&0,+/##%
@'(8*##*,-%→%;#$%1/+*#*,-<%;D%1"E#%

!"#$%&'()*+"$*,-%

."&/0#%*##'/1%2'#$%"3$/0%"++/&$"-+/
F,C%&'()*+"$*,-%+,#$%

G5/%&'()*#5/0%*#%"%-,-H&0,3*$% %!,'-1"$*,-%$5"$%I//&#%$5/%% %+,#$%3,0%"'$5,0#%"#%),C%"#%&,##*()/%
6,'0-")%,3%$5/%!/00"$"H%@$,0$*%!,'-1"$*,-%
h aematologica
haematologica
Editor-in-Chief
Jacob M. Rowe (Jerusalem)
Deputy Editors
Carlo Balduini (Pavia), Jerry Radich (Seattle)
Associate Editors
Shai Izraeli (Tel Aviv), Steve Lane (Brisbane), Pier Mannuccio Mannucci (Milan), Pavan Reddy (Houston), David C. Rees (London), Paul G. Richardson (Boston), Francesco Rodeghiero (Vicenza), Gilles Salles (New York), Kerry Savage (Vancouver), Aaron Schimmer (Toronto), Richard F. Schlenk (Heidelberg), Sonali Smith (Chicago)
Statistical Consultant
Catherine Klersy (Pavia)
Editorial Board
Walter Ageno (Varese), Sarit Assouline (Montreal), Andrea Bacigalupo (Roma), Taman Bakchoul (Tübingen), Pablo Bartolucci (Créteil), Katherine Borden (Montreal), Marco Cattaneo (Milan), Corey Cutler (Boston), Kate Cwynarski (London), Ahmet Dogan (New York), Mary Eapen (Milwaukee), Francesca Gay (Torino), Ajay Gopal (Seattle), Alex Herrera (Duarte), Martin Kaiser (London), Marina Konopleva (Houston), Johanna A. Kremer Hovinga (Bern), Nicolaus Kröger (Hamburg), Austin Kulasekararaj (London), Shaji Kumar (Rochester), Ann LaCasce (Boston), Anthony R. Mato (New York), Matthew J. Mauer (Rochester) Neha Mehta-Shah (St. Louis), Moshe Mittelman (Tel Aviv), Alison Moskowitz (New York), Yishai Ofran (Haifa), Farhad Ravandi (Houston), John W. Semple (Lund), Liran Shlush (Toronto), Sarah K. Tasian (Philadelphia), Pieter van Vlieberghe (Ghent), Ofir Wolach (Haifa), Loic Ysebaert (Toulouse)
Managing Director
Antonio Majocchi (Pavia)
Editorial Office
Lorella Ripari (Office & Peer Review Manager), Simona Giri (Production & Marketing Manager), Paola Cariati (Graphic Designer), Giulia Carlini (Graphic Designer), Debora Moscatelli (Graphic Designer), Igor Poletti (Graphic Designer), Marta Fossati (Peer Review), Diana Serena Ravera (Peer Review), Laura Sterza (Account Administrator)
Assistant Editors
Britta Dost (English Editor), Rachel Stenner (English Editor), Anne Freckleton (English Editor), Rosangela Invernizzi (Scientific Consultant), Marianna Rossi (Scientific Consultant), Massimo Senna (Information Technology), Luk Cox (Graphic Artist)
Haematologica | 108 - June 2023
Brief information on Haematologica
Haematologica (print edition, pISSN 0390-6078, eISSN 1592-8721) publishes peer-reviewed papers on all areas of experimental and clinical hematology. The journal is owned by a non-profit organization, the Ferrata Storti Foundation, and serves the scientific community following the recommendations of the World Association of Medical Editors (www.wame.org) and the International Committee of Medical Journal Editors (www.icmje.org).
Haematologica publishes Editorials, Original articles, Review articles, Perspective articles, Editorials, Guideline articles, Letters to the Editor, Case reports & Case series and Comments. Manuscripts should be prepared according to our guidelines (www.haematologica.org/information-for-authors), and the Uniform Requirements for Manuscripts Submitted to Biomedical Journals, prepared by the International Committee of Medical Journal Editors (www.icmje.org).
Manuscripts should be submitted online at http://www.haematologica.org/.
Conflict of interests. According to the International Committee of Medical Journal Editors (http://www.icmje.org/#conflicts), “Public trust in the peer review process and the credibility of published articles depend in part on how well conflict of interest is handled during writing, peer review, and editorial decision making”. The ad hoc journal’s policy is reported in detail at www.haematologica.org/content/policies.
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articles to the Ferrata Storti Foundation. No formal permission will be required to reproduce parts (tables or illustrations) of published papers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with commercial intent will require written permission and payment of royalties.
Subscription. Detailed information about subscriptions is available at www.haematologica.org. Haematologica is an open access journal and access to the online journal is free. For subscriptions to the printed issue of the journal, please contact: Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, E-mail: info@haematologica.org).
Rates of the printed edition for the year 2022 are as following:
Institutional: Euro 700
Personal: Euro 170
Advertisements. Contact the Advertising Manager, Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, e-mail: marketing@haematologica.org).
Disclaimer. Whilst every effort is made by the publishers and the editorial board to see that no inaccurate or misleading data, opinion or statement appears in this journal, they wish to make it clear that the data and opinions appearing in the articles or advertisements herein are the responsibility of the contributor or advisor concerned. Accordingly, the publisher, the editorial board and their respective employees, officers and agents accept no liability whatsoever for the consequences of any inaccurate or misleading data, opinion or statement. Whilst all due care is taken to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this journal, should only be followed in conjunction with the drug manufacturer’s own published literature.
Direttore responsabile: Prof. Carlo Balduini; Autorizzazione del Tribunale di Pavia n. 63 del 5 marzo 1955. Printing: Press Up, zona Via Cassia Km 36, 300 Zona Ind.le Settevene - 01036 Nepi (VT)

Associated with USPI, Unione Stampa Periodica Italiana. Premiato per l’alto valore culturale dal Ministero dei Beni Culturali ed Ambientali
Haematologica | 108 - June 2023
Table of Contents
Volume 108, Issue 6: June 2023
About the Cover
Image taken from the Editorial by de Gaetano in this issue.
Landmark Paper in Hematology
1453 The International Prognostic Index: still relevant 30 years later
Ann S. LaCasce
https://doi.org/10.3324/haematol.2023.283097
Editorials
1455
What about (MG)US? Towards tailored testing in monoclonal gammopathies
Friederike Bachmann and Stefan Knop
https://doi.org/10.3324/haematol.2022.282271
1458
Disrupting autophagy in FLT3-mutant acute myeloid leukemia
Steven Grant
https://doi.org/10.3324/haematol.2022.282054
1461
Cytomegalovirus-specific T cells following haploidentical transplants: reshaping a repertoire by half Richard J. O’Reilly and Zaki Molvi
https://doi.org/10.3324/haematol.2022.282132
1463 DUSP22-rearranged ALK-negative anaplastic large cell lymphoma is a pathogenetically distinct disease but can have variable clinical outcome
Kerry J. Savage and Graham W. Slack
https://doi.org/10.3324/haematol.2022.282025
1468
Targeting glutaminase to starve lymphoma cells
Charles Dumontet
https://doi.org/10.3324/haematol.2022.282348
1470 Do older patients truly benefit from advances in myeloma care?
Moshe E. Gatt and Eyal Lebel
https://doi.org/10.3324/haematol.2022.281897
1473 How different are blood platelets from women or men, and young or elderly people?
Giovanni de Gaetano et al.
https://doi.org/10.3324/haematol.2022.282131
Perspective Article
1476 Monoclonal gammopathy of increasing significance: time to screen?
Lucia Y. Chen et al.
https://doi.org/10.3324/haematol.2022.281802
Haematologica | 108 - June 2023 I
Review Articles
1487 Aspirin in essential thrombocythemia. For whom? What formulation? What regimen?
Marco Cattaneo
https://doi.org/10.3324/haematol.2022.281388
Articles
1500 Acute Myeloid Leukemia
Concomitant targeting of FLT3 and BTK overcomes FLT3 inhibitor resistance in acute myeloid leukemia through the inhibition of autophagy
Weiguo Zhang et al.
https://doi.org/10.3324/haematol.2022.280884
1515 Bone Marrow Failure
Somatic genetic alterations predict hematological progression in GATA2 deficiency
Laetitia Largeaud et al.
https://doi.org/10.3324/haematol.2022.282250
1530 Cell Therapy & Immunotherapy
Cytomegalovirus-specific T cells restricted for shared and donor human leukocyte antigens differentially impact on cytomegalovirus reactivation risk after allogeneic hematopoietic stem cell transplantation
Elena Tassi et al.
https://doi.org/10.3324/haematol.2022.280685
1544 Cell Therapy & Immunotherapy
Therapeutic potential of fetal liver cell transplantation in hemophilia A mice
Simone Merlin et al.
https://doi.org/10.3324/haematol.2022.282001
1555 Chronic Myeloid Leukemia
Differential inhibition of T-cell receptor and STAT5 signaling pathways determines the immunomodulatory effects of dasatinib in chronic phase chronic myeloid leukemia
Patrick Harrington et al.
https://doi.org/10.3324/haematol.2022.282005
1567 Chronic Myeloid Leukemia
Molecular response in newly diagnosed chronic-phase chronic myeloid leukemia: prediction modeling and pathway analysis
Jerald P. Radich et al.
https://doi.org/10.3324/haematol.2022.281878
1579 Coagulation & its Disorders
Complement protein C3a enhances adaptive immune responses towards FVIII products
Eva Ringler et al.
https://doi.org/10.3324/haematol.2022.281762
1590 Non-Hodgkin Lymphoma
ALK-negative anaplastic large cell lymphoma with DUSP22 rearrangement has distinctive disease characteristics with better progression-free survival: a LYSA study
David Sibon et al.
https://doi.org/10.3324/haematol.2022.281442
1604 Non-Hodgkin Lymphoma
DUSP22 rearrangement is associated with a distinctive immunophenotype but not outcome in patients with systemic ALK-negative anaplastic large cell lymphoma
Lianqun Qiu et al.
https://doi.org/10.3324/haematol.2022.281222
Haematologica | 108 - June 2023 II
1616 Non-Hodgkin Lymphoma
Targeting glutaminase is therapeutically effective in ibrutinib-resistant mantle cell lymphoma
Lingzhi Li et al.
https://doi.org/10.3324/haematol.2022.281538
1628 Plasma Cell Disorders
High-dose carfilzomib achieves superior anti-tumor activity over low-dose and recaptures response in relapsed/refractory multiple myeloma resistant to low-dose carfilzomib by co-inhibiting the β2 and β1 subunits of the proteasome complex
Xiang Zhou et al.
https://doi.org/10.3324/haematol.2022.282225
1640 Plasma Cell Disorders
Improved survival in myeloma patients–a nationwide registry study of 4,647 patients ≥75 years treated in Denmark and Sweden
Kari Lenita Falck Moore et al.
https://doi.org/10.3324/haematol.2021.280424
1652 Red Cell Biology & its Disorders
Comprehensive in silico and functional studies for classification of EPAS1/HIF2A genetic variants identified in patients with erythrocytosis
Valéna Karaghiannis et al.
https://doi.org/10.3324/haematol.2022.281698
Letters
1667 Age- and gender-matched controls needed for platelet-based biomarker studies
Siamack Sabrkhany et al.
https://doi.org/10.3324/haematol.2022.281726
1671 IELSG40/CLEO phase II trial of clarithromycin and lenalidomide in relapsed/refractory extranodal marginal zone lymphoma
Maria Cristina Pirosa et al.
https://doi.org/10.3324/haematol.2022.281963
1676 Allogeneic hematopoietic cell transplant for hairy cell leukemia: EBMT experience
Dai Chihara et al.
https://doi.org/10.3324/haematol.2022.281754
1680 Light chain amyloidosis associated with Waldenström macroglobulinemia: treatment and survival outcomes
Joshua N. Gustine et al.
https://doi.org/10.3324/haematol.2022.282264
1685 Patient-reported fatigue and pain in Erdheim-Chester disease: a registry-based, mixed methods study
Anne S. Reiner et al.
https://doi.org/10.3324/haematol.2022.282287
1691 Retrospective analysis of a cohort of 41 de novo B-cell prolymphocytic leukemia patients: impact of genetics and targeted therapies (a FILO study)
Caroline Algrin et al.
https://doi.org/10.3324/haematol.2022.282162
1697 End-of-treatment PET in early-stage Hodgkin lymphoma: valuable in addition to interim PET
Karan L. Chohan et al.
https://doi.org/10.3324/haematol.2022.282115
Haematologica | 108 - June 2023 III
Case Reports
1702 Sirolimus as frontline therapy for PTEN-mutated histiocytic sarcoma
Karan L. Chohan et al.
https://doi.org/10.3324/haematol.2022.282207
1707 Clinical response to dabrafenib and chemotherapy in clonally-related histiocytosis and acute lymphoblastic leukemia
Gervaise Hubert et al.
https://doi.org/10.3324/haematol.2022.281926
Haematologica | 108 - June 2023 IV
The International Prognostic Index: still relevant 30 years later
Ann S. LaCasce
 Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA
E-mail: Ann_LaCasce@dfci.harvard.edu

https://doi.org/10.3324/haematol.2023.283097
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
AUTHORS Shipp MA, Harrington DP, Anderson JR, et al.
JOURNAL
The New England Journal of Medicine. 1993;329(14):987-94. PMID: 8141877.
Thirty years ago, the International Prognostic Index (IPI) for aggressive lymphomas was published by Shipp and colleagues in the New England Journal of Medicine.1 Although the pathologic classifi cation schema for lymphoma has evolved dramatically with the introduction of immunohistochemical and molecular testing, the IPI remains relevant today. At the time of publication, the paper was groundbreaking in many ways. The work was an international collaboration (16 institutions and co-operative groups across the US, Canada and Europe) with a very large data set used for discovery and validation. The five risk factors of age (> 60 years), stage (3/4), more than one extranodal site, performance status (> or = to 2), and elevated lactate dehydrogenase, are simple and accessible across all regions, including resource-poor settings.
The IPI has held the test of time. It is used in most prospective, randomized trials to stratify risk and ensure balance between groups. In nearly all retrospective analyses, the IPI is an independent prognostic factor for progression-free and overall survival on multivariate analysis. The IPI also sheds light on why the outcomes of clinical trials are uniformly more favorable than in the “real world”. Patients with high-risk disease are under-represented in clinical trials. This reflects, in part, the difficulty of enrolling patients on a study who have high-risk disease and may be hospitalized and/or in urgent need of therapy. In addition, it facilitates the investigation of the independent impact of other prognostic biomarkers through adjusted analyses.
The IPI inspired other clinical prognostic scales across
 TITLE
A predictive model for aggressive non-Hodgkin's lymphoma: the International NHL Prognostic Factors Project.
TITLE
A predictive model for aggressive non-Hodgkin's lymphoma: the International NHL Prognostic Factors Project.
Haematologica | 108 June 2023 1453 LANDMARK PAPER IN HEMATOLOGY A.S. LaCasce
Figure 1. Overall survival according to International Prognostic Index risk group. L: low; LI: low-intermediate; HI: highintermediate; H: high. Figure adapted with permission from Shipp et al. N Engl J Med 1993.
many different types of lymphoma, including follicular lymphoma, T-cell lymphoma and Burkitt lymphoma. A number of groups have designed alternative versions of the IPI, incorporating additional risk factors or refining ranges of risk factors. The National Comprehensive Cancer Network (NCCN)-IPI is one such example.2 Although the NCCN-IPI improves the distinction between risk groups in terms of survival, the IPI remains the predominant index
References
1. International Non-Hodgkin's Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin’s lymphoma. New Engl J Med. 1993:329(14):987-94.
used in clinical practice and clinical trials given it is easy to use and its widely accepted status. Furthermore, it forms the basis of the central nervous system (CNS)-IPI which also includes renal/adrenal involvement, to predict the risk of CNS relapse.
Disclosure
No conflicts of interest to disclose.
2. Zhou Z, Sehn LH, Rademaker AW, et al. An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood. 2014;123(6):837-842.
Haematologica | 108 June 2023 1454 LANDMARK PAPER IN HEMATOLOGY A.S. LaCasce
What about (MG)US? Towards tailored testing in monoclonal gammopathies
Friederike Bachmann1 and Stefan Knop2
In this issue of Haematologica , Chen and co-workers examine the multitude of challenges around monoclonal gammopathies with their numerous clinical facets.1 These gammopathies include malignant diseases such as multiple myeloma (MM) as well as exceedingly rare conditions, for instance POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal protein, Skin changes) syndrome, glomerulopathies, and skin disorders.2 Most likely, the fate of a given individual diagnosed with a monoclonal gammopathy of undetermined significance (MGUS) (note the “undetermined“) is, in fact, actually determined by an array of variables. The challenge for a physician when encountering an M spike on an electropherogram is to anticipate whether the underlying clone will remain stable (“true” or “benign” MGUS), develop towards a disease with a malignant phenotype or evolve into one of the rare disorders with devastating end-organ damage. Genetic alterations as well as differential usage of immunoglobulin variable genes have a significant impact on the phenotype and the longitudinal behavior of plasma cell diseases.3-5 Much work has gone into the analysis of genomic and transcriptional changes occurring during the evolution from early to advanced and symptomatic stages of plasma cell diseases.3,4,6 Very recently, a group from the USA published their insights into how different genetic subtypes of smoldering multiple myeloma (SMM) predispose to specific progression dynamics and clinical outcomes.6 The authors identified six subgroups that rely on different gene enrichments. They were able to identify three SMM groups at high risk of progression to active MM. These findings may guide future attempts at early interception (particularly in the three high-risk categories) with differential approaches depending on dysregulated molecular and oncogenic networks.6
Given the enormous expense required for sequencing technologies and bioinformatics, such comprehensive analyses are not yet ready for clinical-scale use. This is why, for the time being, clinical stratification models are important. These cover two areas of interest: first, the
Correspondence: S. Knop stefan.knop@klinikum-nuernberg.de
Received: November 14, 2022.
Accepted: November 22, 2022.
Early view: December 1, 2022.
https://doi.org/10.3324/haematol.2022.282271
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
identification of populations who are at high risk of having MGUS or SMM and, second, the characterization of subjects with evolving SMM in whom the initiation of systemic therapy prior to the development of overt MM will be beneficial. While a first true population-based screening study is underway but still far from its readout,7 there is clear evidence to support targeted screening in known high-risk groups. The incidence of MGUS is as high as 25% in black people aged 50 years or older who have at least one family member with MM. This incidence was also found in people with a different ethnic background aged 50 and older if at least two family members were diagnosed with MM.8 Such a screening approach could result in placing subjects with high-risk SMM (still defined by clinical and laboratory parameters) on systemic therapy to prevent end-organ damage due to symptomatic MM. With many studies examining novel combinations of therapy still underway, there is some evidence that treatment with lenalidomide and dexamethasone in high-risk MM prolongs progression-free and overall survival when compared to observation only.9
A further layer of complexity in monoclonal gammopathies is the existence of very rare, non-malignant, albeit severely disabling entities, such as light-chain amyloidosis or renal, neurological or myopathic disease for which the abbreviation “MGCS” (monoclonal gammopathy of clinical significance) was coined.2 These entities are typically characterized by their underlying lowburden plasma cell dyscrasias and by a broad spectrum of clinical symptoms. This highlights the need for more sensitive laboratory tests with respect to both confirming the presence of a monoclonal gammopathy and monitoring treatment response. Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry has the potential to identify even small amounts of monoclonal proteins that go unrecognized by serum protein electrophoresis and serum immunofixation. A large study has proven its superiority in detecting monoclonal gammopathies in a defined screening
1Department of Nephrology and Medical Intensive Care, Charité University Medicine, Berlin and 2Nuremberg General Hospital, Department of Hematology and Oncology and Paracelsus Medical School, Nuremberg, Germany
Haematologica | 108 June 2023 1455 EDITORIAL F. Bachmann and S. Knop
cohort.10 Mass spectrometry could in the future replace serum immunofixation, resulting in greater accuracy in detecting gammopathies and excluding false-positive cases on immunofixation. The incremental benefit of this advanced technology is most likely to occur in low-level conditions. From a diagnostic perspective, certain “red flags” may serve as initial clues to the underlying clonal B-/plasma cell proliferation. Awareness is a prerequisite to allow for a timely diagnosis.
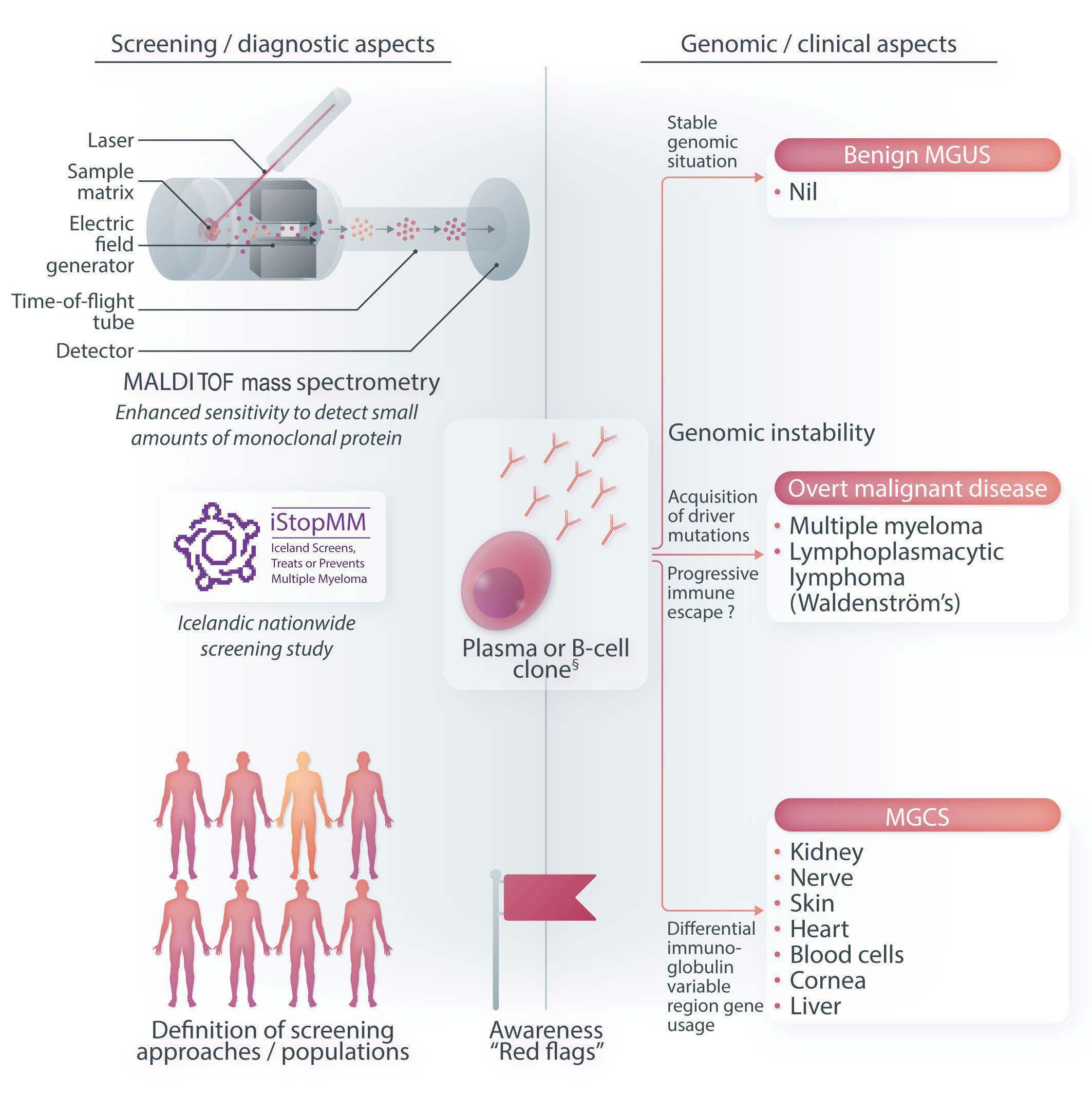
In conclusion, a complex and clinically significant spectrum of disease takes center stage in the review by Chen et al.1 The authors touch on all topics that are currently under debate: the question of whether screening for the “pre-symptomatic” condition is justified; the different
mechanisms contributing to organ damage; the importance of early recognition of a monoclonal gammopathy; and the dilemma over how to establish the best screening algorithm. The review is an important contribution to the field and will certainly attract the interest of readers.
Disclosures
SK has received honoraria from BMS, Celgene, Janssen, Sanofi, and Pfizer, and a travel grant from Sobi. FB has no conflicts of interest to disclose.
Contributions
FB and SK wrote the manuscript and both authors approved its final version.
Haematologica | 108 June 2023 1456 EDITORIAL F. Bachmann and S. Knop
Figure 1. Considerations regarding diagnosis and surveillance of clonal B-cell/plasma-cell diseases. MALDI-TOF: matrix-assisted laser desorption/ionization-time of flight; MGUS: monoclonal gammopathy of undetermined significance; MGCS: monoclonal gammopathy of clinical significance.
References
1. Chen LY, Drayson M, Bunce C, Ramasamy K. Monoclonal gammopathy of increasing significance: time to screen? Haematologica. 2023;108(6):1476-1486.
2. Moreno DF, Rosinol L, Cibeira MT, Bladé J, Fernández de Larrea C. Treatment of patients with monoclonal gammopathy of clinical significance. Cancers (Basel). 2021;13(20):5131.
3. Farswan A, Gupta A, Jena L, Ruhela V, Kaur G, Gupta R. Characterizing the mutational landscape of MM and its precursor MGUS. Am J Cancer Res. 2022;12(4):1919-1933.
4. Dutta AK, Alberge JB, Sklavanitis-Pistofidis R, Lightbody ED, Getz G, Ghobrial IM. Single-cell profiling of tumour evolution in multiple myeloma - opportunities for precision medicine. Nat Rev Clin Oncol. 2022;19(4):223-236.
5. Kourelis TV, Dasari S, Theis JD, et al. Clarifying immunoglobulin gene usage in systemic and localized immunoglobulin lightchain amyloidosis by mass spectrometry. Blood. 2017;129(3):299-306.
6. Bustoros M, Anand S, Sklavenitis-Pistofidis R, et al. Genetic
subtypes of smoldering multiple myeloma are associated with distinct pathogenic phenotypes and clinical outcomes. Nat Commun. 2022;13(1):3449.
7. Rögnvaldsson S, Love TJ, Thorsteinsdottir S, et al. Iceland screens, treats, or prevents multiple myeloma (iStopMM): a population-based screening study for monoclonal gammopathy of undetermined significance and randomized controlled trial of follow-up strategies. Blood Cancer J. 2021;11(5):94.
8. Rajkumar S. The screening imperative. Nature. 2020;587(7835):S63.
9. Mateos MV Hernández MT, Giraldo P, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369(5):438-447.
10. Murray D, Kumar SK, Kyle RA, et al. Detection and prevalence of monoclonal gammopathy of undetermined significance: a study utilizing mass spectrometry-based monoclonal immunoglobulin rapid accurate mass measurement. Blood Cancer J. 2019;9(12):102.
Haematologica | 108 June 2023 1457 EDITORIAL F. Bachmann and S. Knop
Disrupting autophagy in FLT3-mutant acute myeloid leukemia
Steven Grant
Division of Hematology/Oncology and Palliative Care, Virginia Commonwealth University Health Sciences Center, Richmond, VA, USA
In the study by Zhang et al., published in this issue of Haematologica, 1 the authors examine the mechanisms by which the multi-kinase inhibitor CG-806 kills FLT3-mutant acute myeloid leukemia (AML) cells, with an emphasis on modulation of autophagy and related endoplasmic reticulum stress-associated pathways. They report that in FLT3-mutant cells, exposure to FLT3 inhibitors e.g., sorafenib and quizartinib, elicits an autophagic response, operating, at least in part, through microenvironmental factor-mediated induction of Bruton tyrosine kinase (BTK). Using both pharmacological (e.g., BTK inhibitors) and genetic strategies (e.g., Atg7 knockdown), they demonstrate that disruption of BTK-induced autophagic responses increases the lethal effects of FLT3 inhibitors. Building on this foundation, they investigated the antileukemic activity of a novel multi-kinase inhibitor, CG-086, which inhibits FLT3, BTK, and in addition, aurora kinase A (AURA). Notably, they found that CG-806 induced cell death in association with inhibition of these kinases, as well as by disrupting autophagic events. Interestingly, the authors observed that interference with autophagy was primarily effective in FLT3-mutant AML models, but less so in wildtype FLT3 cells, raising the possibility that cytoprotective autophagy is selectively relevant for the former cells. Nevertheless, the ability of CG-806 to kill FLT wildtype cells could be attributed to the ability of this agent to interfere with AURA function, causing cells to die from a form of mitotic catastrophe. Importantly, the authors demonstrated that CG-086 was active in primary AML cells obtained from patients resistant to a FLT3 inhibitor (e.g., sorafenib), and was quite tolerable and effective in a FLT3-mutant patient-derived xenograft model. Collectively, these findings argue that CG-086 represents a potentially important addition to the therapeutic armamentarium for FLT3-mutant AML, and potentially other AML subtypes.
The involvement of autophagy in FLT3 inhibitor-associated resistance in AML is an interesting concept, and one with obvious translational potential. The authors have pre-
Correspondence: S. Grant steven.grant@vcuhealth.org
Received: October 27, 2022. Accepted: November 9, 2022. Early view: November 17, 2022.
https://doi.org/10.3324/haematol.2022.282054
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
viously described this phenomenon,2 focusing on genetic strategies (e.g., Atg7 knockdown) to validate its role in cell death. Here, they emphasize the potential for circumventing resistance by disabling this process pharmacologically. It should be noted that autophagy is a highly complex process, and its effects on cell death may vary extensively with cell context. For example, while autophagy can be cytoprotective under some circumstances,3 under other circumstances it can also be cytotoxic or cytostatic.4 Targeting cytoprotective autophagy, for example by agents such as chloroquine, has been the subject of great interest,5 and the authors demonstrated that chloroquine did indeed enhance the effectiveness of CG-806. The concept of disabling autophagy by dual strategies, such as targeting signaling pathways implicated in autophagy induction (e.g., FLT3 and BTK) as well as the autophagic apparatus directly (e.g., via chloroquine), warrants future consideration. It should be kept in mind that preventing the induction of autophagy, for example, by inhibiting signaling pathways may differ fundamentally from interfering with lysosome function, for example, by agents such as chloroquine.
The role of BTK, implicated in lymphomagenesis, in myeloid malignancies has previously been described,6 including in FLT-mutant AML.7 However, the mechanism by which inhibition of this signaling molecule induces AML cell death has not been identified. The results of the present study suggest that the actions of CG-806 may involve interruption of both BTK as well as microenvironmental factors, resulting in the prevention of cytoprotective autophagy. If validated in lymphoid malignancies, such findings could provide a foundation for employing this agent in the setting of non-Hodgkin lymphoma and potentially chronic lymphocytic leukemia.
It is interesting, but at the same time puzzling, that mechanisms responsible for the antileukemic effects of CG-086 appeared to be operative primarily in FLT3-mutant AML, but not in wildtype cells. Such findings raise the possibility that the ability of the former cells to mount a cytopro-
Haematologica | 108 June 2023 1458 EDITORIAL S. Grant
Figure 1. Mechanisms of CG-806 lethality in mutant and wildtype acute myeloid leukemia. In mutant FLT3 disease, the lethal effects of inhibition of FLT3 are opposed by microenvironmental factor-mediated induction of cytoprotective autophagy operating through a BTK-dependent mechanism. In FLT3 mutant cells, CG-806 inhibits both FLT3 as well as BTK, resulting in a marked increase in cell death. This process is not operative in wildtype FLT3 acute myeloid leukemia, but such cells are killed by CG-806 through inhibition of FLT3 as well as disruption of aurora kinases (AURK), leading to death via G2M arrest and polyploidy. Thus, CG-806 is effective against both FLT3-mutant and wildtype disease.
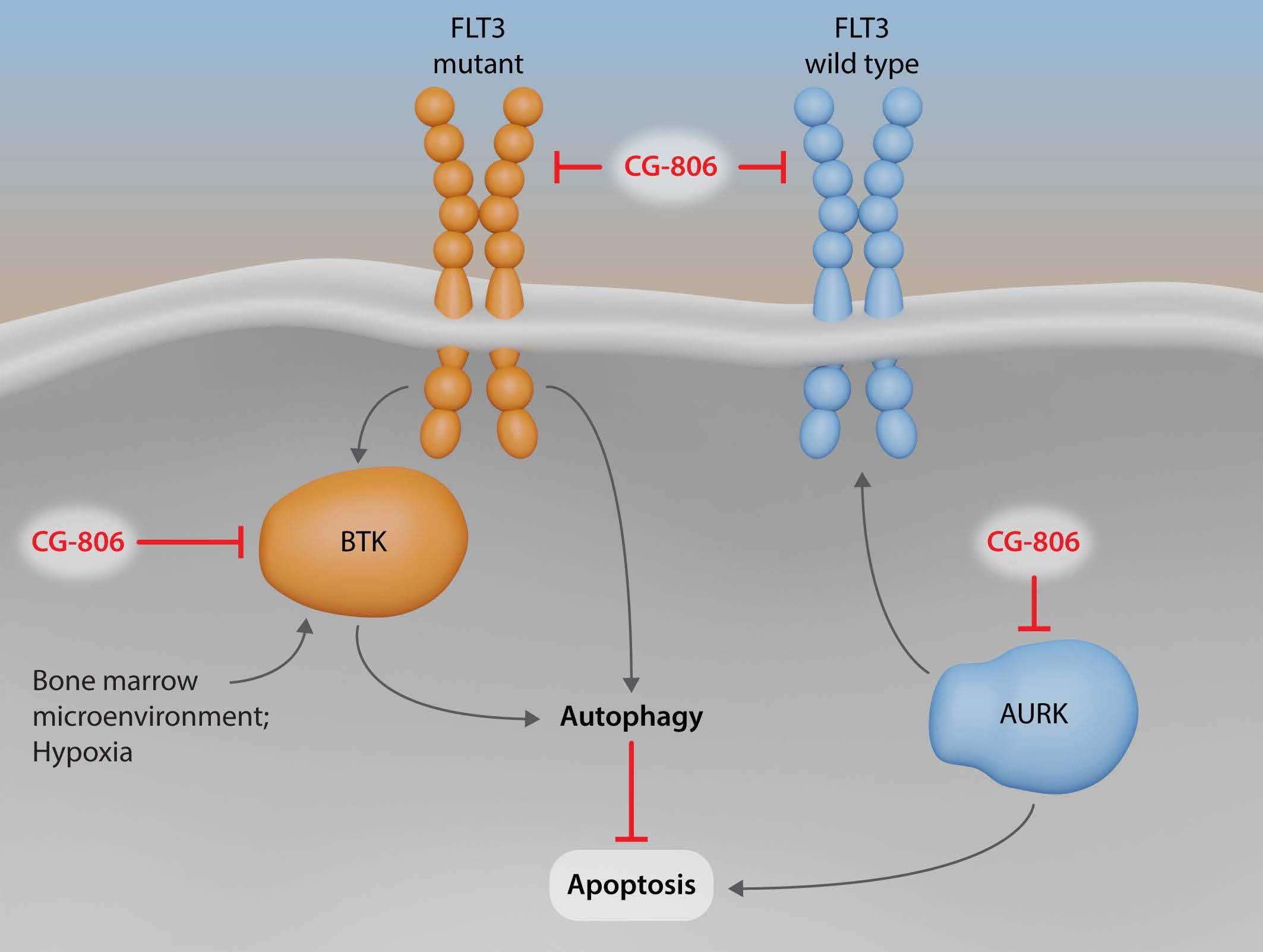
tective autophagic response may contribute to the poor prognosis of this particular subtype of AML. Nevertheless, wildtype cells remained susceptible to CG-086, a phenomenon attributed to the ability of this agent to inhibit AURA, resulting in inappropriate G2M phase transition and induction of a form of mitotic catastrophe.8 This observation argues that while highly selective kinase inhibitors offer the promise of diminished off-target effects, multikinase inhibitors, such as CG-086, which disrupt multiple survival pathways may provide countervailing advantages in certain settings.
The ultimate role that CG-086 will play in the treatment of FLT3-mutant AML remains to be determined, but early clinical results appear to be promising, and at the very least suggest that this agent is tolerable in humans. Whether it will prove superior to other FLT3 inhibitors remains to be established, as does its role in the treatment of wildtype disease. It should be kept in mind that while the contribution of FLT3 inhibitors in AML is now firmly established, it is unclear whether such approaches will eradicate leukemia stem cell-like cells, given that FLT3 mutations can be relatively late-appearing genetic aberrations, and that elimination of FLT3-positive cells is not by itself a curative strategy. However, the multiple mechanisms of action of CG-086 may address this issue, and
preclinical studies examining the effects of CG-086 on more primitive AML progenitors, such as stem cell-like cells, are likely to be informative. Another question to be addressed is which of the multiple mechanisms of action of CG-086, for example, inhibition of FLT3, BTK, AURA, microenvironmental factors, and/or autophagy, is/are primarily responsible for antileukemic activity. It would also be interesting to determine whether and to what extent common survival pathways downstream of FLT3 and BTK, for example, MAPK and AKT pathways, contribute to the actions of this agent. For example, FLT3 interruption may sub-optimally inhibit these pathways9 whereas concomitant BTK disruption may enhance signaling blockade. Finally, the possibility that these actions may cooperate to trigger leukemic cell death is quite likely, and adds to the complexity. In any event, the present observations, along with early clinical findings, indicate that CG-086 represents an interesting new FLT3 and multi-kinase inhibitor that warrants further scrutiny. The results of ongoing clinical trials are eagerly awaited and should help to determine whether this agent deserves a place in the therapeutic armamentarium for FLT3-mutant AML.
Disclosures No conflicts of interest to disclose.
Haematologica | 108 June 2023 1459 EDITORIAL S. Grant
References
1. Zhang W, Yu G, Zhang H, et al. Concomitant targeting of FLT3 and BTK overcomes FLT3 inhibitor resistance in acute myeloid leukemia through inhibition of autophagy. Haematologica. 2023;108(6):1500-1514.
2. Piya S, Kornblau SM, Ruvolo VR, et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood. 2016;128(9):1260-1269.
3. Moreau K, Luo S, Rubinsztein DC. Cytoprotective roles for autophagy. Curr Opin Cell Biol. 2010;22(2):206-211.
4. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. 2014;74(3):647-651.
5. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528-542.
6. Elgamal OA, Mehmood A, Jeon JY, et al. Preclinical efficacy for a
novel tyrosine kinase inhibitor, ArQule 531 against acute myeloid leukemia. J Hematol Oncol. 2020;13(1):8.
7. Pillinger G, Abdul-Aziz A, Zaitseva L, et al. Targeting BTK for the treatment of FLT3-ITD mutated acute myeloid leukemia. Sci Rep. 2015;5:12949.
8. Mathison A, Salmonson A, Missfeldt M, et al. Combined AURKA and H3K9 methyltransferase targeting Inhibits cell growth by inducing mitotic catastrophe. Mol Cancer Res. 2017;15(8):984-997.
9. Siendones E, Barbarroja N, Torres LA, et al. Inhibition of Flt3activating mutations does not prevent constitutive activation of ERK/Akt/STAT pathways in some AML cells: a possible cause for the limited effectiveness of monotherapy with small-molecule inhibitors. Hematol Oncol. 2007;25(1):30-37.
Haematologica | 108 June 2023 1460 EDITORIAL S. Grant
Cytomegalovirus-specific T cells following haploidentical transplants: reshaping a repertoire by half
Richard J. O’Reilly1,2 and Zaki Molvi2
1Bone Marrow Transplant Service, Department of Pediatrics and 2Immunology Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA
Studies addressing the impact of human leukocyte antigen (HLA) disparities on the reconstitution of antigen-specific immunity following allogeneic HLA haploidentical hematopoietic cell transplant (HCT) are very limited. The paper by Tassi et al.1 in this issue of Haematologica is the first to analyze the pattern of HLA restrictions exhibited by antigen-specific T cells emerging in a cohort of adult patients following HLA haploidentical transplants for hematologic malignancies. This report provides important new evidence documenting limitations to the repertoire of cytomegalovirus (CMV) epitope-specific HLA-restricted T cells achievable following HLA-haploidentical HCT.
In 1974, Zinkernagel and Doherty2 first demonstrated that T cells recognize viral antigens only when presented by self major histocompatibility complex (MHC) alleles. Subsequent studies demonstrated that T cells bearing such MHC-restricted antigen-specific receptors (TCR) are positively selected during their development through interactions with cortical epithelial cells in the thymus.3 Thereafter, they are negatively selected to deplete potentially autoreactive T cells specific for self-peptides presented by MHC alleles expressed by thymic dendritic cells and other antigen-presenting cells. After selection, the surviving, mature but naïve T cells are exported to the periphery and maintained in homeostasis until activated in response to antigenic challenge.4
While HLA restriction is now well recognized as a hallmark characteristic of TCRα/b antigen-specific T cells, information regarding the contributions of T cells restricted by shared versus donor or host unique alleles to reconstitution of pathogen-specific immunity following HLA partially matched or haploidentical HCT is minimal. Prior studies were almost exclusively focused on reconstitution of T-cell immunity in children with severe combined immunodeficiency diseases (SCID) who had received either T-cell-depleted grafts from an HLAhaploidentical parent or a fully allogeneic fetal liver, usually administered after no or minimal conditioning. Because the thymic epithelium is of host origin and, in
Correspondence: R.J. O’Reilly oreillyr@mskcc.org
Received: November 10, 2022. Accepted: November 22, 2022. Early view: December 1, 2022.
https://doi.org/10.3324/haematol.2022.282132
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
murine models, positive selection depends on the interaction of the TCR of T-cell precursors with MHC alleles of thymic cortical epithelial cells,4 it would be expected that donor-type tetanus toxoid antigen-specific T cells developing in the host thymus would be selectively restricted by HLA alleles shared by donor and host and that T cells restricted by host-unique alleles would not be detected. In fact, T-cell responses to tetanus presented by host-unique HLA alleles were equal to responses to epitopes presented by HLA alleles shared by donor and host.5,6 However, prior to transplantation, the thymus in infants with SCID is embryonal, without evidence of lymphoid or other hematologic elements. Its development, including corticomedullary differentiation, is only initiated after transplantation. It is thus possible that concomitant differentiation of a functional thymic cortical epithelium, which is induced by prothymocytes,7 could also be influenced by cells in the donor graft to permit positive selection of developing T cells recognizing host-unique HLA alleles.
In contrast, the CMV-specific T cells analyzed by Tassi et al.1 were from adult recipients of HLA-haploidentical HCT whose thymuses would be fully differentiated from birth to the time of transplant. In these patients, CMV-specific T cells isolated by dextramers selectively recognized epitopes presented by shared or donor-unique HLA alleles. As observed in murine models, no CMV-specific T cells restricted by host-unique HLA alleles were detected, either early or up to 3 years after transplantation, likely reflecting the failure of T-cell progenitors in the graft bearing TCR specific for epitopes presented by donor-unique HLA alleles to be engaged and positively selected by cortical epithelial cells of the host thymus. Furthermore, while CMV-specific T cells restricted by donor-unique HLA alleles (D-restricted CMV-specific T cells) were detected in most patients who received transplants from CMV-seropositive donors, they were detected in only a minority of recipients of transplants from seronegative donors, consistent with D-
Haematologica | 108 June 2023 1461 EDITORIAL R.J. O’Reilly and Z. Molvi
restricted CMV-specific T cells being derived from mature T cells in the graft. Furthermore, these T cells expanded later than CMV-specific T cells restricted by shared alleles, persisted for only 6 months and maintained a less differentiated phenotype. In contrast, CMV-specific T cells restricted by shared alleles expanded rapidly in most (but not all) patients, and to a greater degree. They also persisted at a high frequency for ≥1 year and contained more TEM and TEMRA cells. These findings support the authors’ hypothesis that the donor CMV-specific T cells restricted by shared HLA alleles are responding to CMV epitopes presented by infected host-type nonhematopoietic cells or residual antigen-presenting cells. They also indicate a primary role for CMV-specific T cells restricted by shared HLA alleles in the initial and sustained control of CMV reactivation after transplantation. However, the proportion of T cells restricted by shared alleles that are derived from T-cell precursors maturing in the host thymus and their repertoire remains to be determined.
Transplants from HLA non-identical unrelated and HLA haploidentical related donors are increasingly invoked for the treatment of patients lacking a matched related or unrelated donor. Such transplants have been associated with higher incidences of CMV infection and associated non-relapse mortality.8 In part, this increased incidence can be ascribed to graft-versus-host disease or the use of more intensive treatment with immunosuppressive drugs to control it, or to impairments to early reconstitution associated with T-cell-depleted grafts, in vivo T-cell
References
1. Tassi E, Noviello M, De Simone P, et al. Cytomegalovirusspecific T cells restricted for shared and donor human leukocyte antigens differentially impact on cytomegalovirus reactivation risk after allogeneic hematopoietic stem cell transplantation. Haematologica. 2023;108(6):1530-1543.
2. Zinkernagel RM, Doherty PC. Restriction of in vitro T cellmediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature. 1974;248(5450):701-702.
3. Lo D, Sprent J. Identity of cells that imprint H-2-restricted Tcell specificity in the thymus. Nature. 1986;319(6055):672-675.
4. Viret C, Janeway CA Jr. MHC and T cell development. Rev Immunogenet. 1999;1(1):91-104.
5. Geha RS, Rosen FS. The evolution of MHC restrictions in antigen recognition by T cells in a haploidentical bone marrow transplant recipient. J Immunol. 1989;143(1):84-88.
6. Roberts JL, Volkman DJ, Buckley RH. Modified MHC restriction of donor-origin T cells in humans with severe combined
depletion by pre-transplant antithymocyte globulin or post-transplant cyclophosphamide. However, the findings of Tassi et al.1 raise the possibility that the inability of virus-specific donor T cells to recognize or be primed to epitopes presented by host-unique alleles could also retard or impair development of an effective immune response. We previously described a recipient of a haploidentical HCT with an Epstein-Barr virus (EBV)positive lymphoma of host origin who failed treatment with donor EBV cytotoxic T lymphocytes that were restricted only by an HLA allele not shared by the patient but responded to third-party EBV cytotoxic T lymphocytes restricted by a shared allele.9 To what degree such limitations contribute to the heightened risk of CMV or other latent viral infections in haploidentical transplant recipients warrants examination since the repertoire of CMV-specific T cells contracts over time and, in the graft, can be limited to responses to immunodominant viral epitopes presented by a single HLA allele. The repertoire of donor T cells developing in the host thymus may also be constricted by graft-versus-host disease and older age.10 Thus, if epitopes presented by shared alleles are less immunogenic, early recovery of functional CMV-specific T cells may be delayed or inadequate to control disease.
Disclosures
No conflicts of interest to disclose.
Contributions
RJO and ZM co-wrote the editorial.
immunodeficiency transplanted with haploidentical bone marrow stem cells. J Immunol. 1989;143(5):1575-1579.
7. Hollander GA, Wang B, Nichogiannopoulou A, et al. Developmental control point in induction of thymic cortex regulated by a subpopulation of prothymocytes. Nature. 1995;373(6512):350-353.
8. Oltolini C, Greco R, Galli L, et al. Infections after allogenic transplant with post-transplant cyclophosphamide: impact of donor HLA matching. Biol Blood Marrow Transplant. 2020;26(6):1179-1188.
9. Doubrovina E, Oflaz-Sozmen B, Prockop SE, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119(11):2644-2656.
10. Trofimov A, Brouillard P, Larouche JD, et al. Two types of human TCR differentially regulate reactivity to self and nonself antigens. iScience. 2022;25(9):104968.
Haematologica | 108 June 2023 1462 EDITORIAL R.J. O’Reilly and Z. Molvi
DUSP22-rearranged ALK-negative anaplastic large cell lymphoma is a pathogenetically distinct disease but can have variable clinical outcome
Kerry J. Savage1,2 and Graham W. Slack1,3
1Centre for Lymphoid Cancer; 2University of British Columbia and Division of Medical Oncology and 3University of British Columbia and Department of Pathology, BC Cancer, Vancouver, British Columbia, Canada
In comparison to ALK-positive anaplastic large cell lymphoma (ALCL), ALK-negative ALCL has been more difficult to define. It was first characterized by morphological similarity to ALK-positive ALCL, including the presence of pathognomonic ‘hallmark cells’, but lacking expression of the ALK protein. Very early reports suggested a similar prognosis to peripheral T-cell lymphoma – not otherwise specified1 but larger studies indicated that the prognosis, although still unsatisfactory, was better than that of peripheral T-cell lymphoma – not otherwise specified.2 However, outcomes are notably variable across studies, as described by Hapgood and Savage.3 This is in part due to clinical risk factors as captured by the International Prognostic Index score.2,4 Firm recognition of ALK-negative ALCL as a distinct entity came in the World Health Organization’s (WHO) 4th edition update following the description of unique molecular features.5,6
The identification of two recurrent rearrangements represented key milestones in deciphering ALK-negative ALCL. The first rearrangement involves the DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement [DUSP22-R]) and the other involves TP63 on 3p28 (TP63 rearrangement [TP63-R]). The clinical significance of the DUSP22-R was first evaluated in a series of 73 patients with ALKnegative ALCL in which 22 cases were found to harbor the rearrangement, representing 30% of all ALK-negative ALCL. The TP63-R was identified in six cases (8%) and the remainder were deemed to have ‘triple-negative’ ALKnegative ALCL meaning they lacked any rearrangement and are also referred to as DUSP22-NR (nonrearranged)/TP63-NR. Cases with a DUSP22-R had a 5year overall survival (OS) of 90%, which was similar to that of a comparison group of cases of ALK-positive ALCL. In contrast, those with a TP63-R had a dismal 5year OS of only 17%. The majority of cases in the series were triple-negative and had an intermediate prognosis
Correspondence: K.J. Savage
ksavage@bccancer.bc.ca
Received: October 20, 2022.
Accepted: November 18, 2022.
Early view: December 1, 2022.
https://doi.org/10.3324/haematol.2022.282025
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
(5-year OS of 42%). Pathological evaluation of DUSP22-R tumors revealed sheet-like growth of classic hallmark cells, fewer pleomorphic cells and, as assessed by immunohistochemistry (IHC), reduced expression of epithelial membrane antigen (EMA) and cytotoxic markers (TIA1, granzyme B, perforin) (Table 2). Subsequent studies of these cases described characteristic ‘doughnut’ cells’,7 and a molecular profile characterized by overexpression of immunogenic cancer testis antigen (CTA) genes, a signature of marked DNA hypomethylation and diminished expression of STAT3 and programmed death ligand (PDL), with consequential lack of pSTAT3 and PDL as determined by IHC.8 Exome sequencing identified a recurrent mutation in MSCE116K in almost all cases.9
Two subsequent small series of four and five patients each with DUSP22-R ALK-negative ALCL also demonstrated a favorable prognosis10,11 (Table 1). In contrast, in our BC Cancer study of 12 cases, a less favorable prognosis was observed with a 5-year progression-free survival (PFS) and OS of only 40%.12 A range of clinical courses were noted including one patient with central nervous system relapse and another patient managed palliatively had a relapsing/remitting disease course over 4 years, which was reminiscent of cutaneous ALCL. In contrast, IHC features were as expected: EMAnegative, infrequent expression of cytotoxic markers and all cases were pSTAT3- and PDL1-negative (Table 2). In this issue of Haematologica, two studies have further evaluated the pathological characteristics and prognostic significance of DUSP22-R ALK-negative ALCL.13,14 The study by Sibon and colleagues represents the largest series to date of 47 cases of DUSP22-R ALK-negative ALCL derived from the TENOMIC database, a translational lymphoma research consortium of the LYSA group. In total, 47/104 (45%) cases harbored a DUSP22-R which is a significantly higher proportion than in other studies, and
Haematologica | 108 June 2023 1463 EDITORIAL K.J. Savage and G.W. Slack
aReported as skin involvement during disease course. bRemaining patient received radiotherapy alone. cIncludes CHOP (N=22); CHOEP (N=11); Ro-CHOP (N=1); CH(E)P-BV (N=3); mini-CHOP (N=5); ACVBP (N=2). d5-year point estimate not reported in the paper but estimated from its Figure 6B. DUSP22-R: DUSP22-rearranged; nr: not reported; PS: Performance Status; IPI: International Prognostic Index: CNS: central nervous system; CHOP: cyclophosphamide, doxorubicin, vincristine; prednisone; auto-SCT: autologous stem cell transplantation; PFS: progressionfree survival; OS: overall survival.
the authors acknowledge a selection bias as cases are submitted to the LYSA TENOMIC database with an aim of compiling those of clinical interest.13 In the subset enrolled on clinical trials, estimates are more in keeping with other studies (23-35%). Regardless, this is an extensively curated database with detailed clinical information which has been lacking in most prior studies (Table 1). Frequent bone involvement was observed, which was also found in the BC Cancer series. Pathologically, cases had the expected morphology and immunophenotype (Table 2) although, curiously, 27% of cases expressed at least one cytotoxic marker (‘cytotoxic profile’), which appears to be higher incidence than in other reports although most other studies only reported on individual marker frequency (Table 2). With a median follow-up of 5 years, the PFS was superior in DUSP22-R cases than in non-rearranged cases (5-year PFS: 48% vs. 25%, respectively; P=0.025); however, the point estimates are far lower than in the original series, and what would be expected for ALK-positive ALCL. Furthermore, OS was not statistically different (5-year OS: 58% vs. 44%,
respectively; P=0.20). Confining the analysis to the 39 DUSP22-R cases treated with curative intent, anthracycline chemotherapy (all but 3 cases), and confirmed to have TP63-NR status, demonstrated a more favorable PFS (5-year PFS: 57% vs. 26%, respectively; P=0.001) but not OS (5-year OS: 65% vs. 41%, respectively; P=0.07) although it must be acknowledged that there was limited power to detect a smaller difference. This larger dataset enabled exploration of factors associated with survival. Those cases with a poor performance status (PS) (≥2) and elevated b2-microglobulin had an inferior PFS and OS. Cytotoxic marker expression was also associated with an inferior PFS and the individual factors of granzyme B or perforin expression, but not TIA1, were associated with inferior PFS and OS. However, only PS and DUSP22-NR status were included in the final model because of missing information, and both were associated with PFS, but only PS was also associated with OS. Using these two factors, DUSP22-R patients with a poor PS, a group representing 29% of all cases (11/38, with one patient not included due to missing PS), had a 5-year PFS and OS of only 27% and
Feature First author (Study location/group) Parilla-Castellar (Mayo)6 Pedersen (Denmark)10 Hapgood (BC Cancer)12 Onaindia (Spain)11 Sibon (LYSA)13 Qiu (MDACC)14 DUSP22-R cases, N 22 5 12 4 47 22 DUSP22-R among 30 19 19 18 45 28 ALK-negative cases, % Age in years, median 53.5 49 61.5 57.5 60 52 Range 36-76 35-85 50-86 39-71 40-86 33-79 Stage 3 or 4, % 85 80 75 100 64 71 Missing data, % 68 25 2 37 PS ≥2, % nr 0 25 40 30 nr Missing data, % 50 IPI ≥3, % 42 40 42 0 48 33 Missing data, % 36 50 2 32 Extranodal sites, % Bone nr nr 33 nr 32 nr Bone marrow nr nr 17 25 13 6 Skin 28a 60 25 25 15 nr Liver nr nr 8 nr 19 nr CNS relapse % nr nr 8 nr nr nr Treatment, % CHOP(like) 90 100 92 50b 94c 90 Consolidative auto-SCT 5 50d 8 0 19 27 Missing treatment data 36 25 5-year PFS, % nr nr 40 nr 57 40d 5-year OS, % 90 80 40 100 65 40
Table 1. Summary of studies to date evaluating the prognosis in DUSP22-rearranged ALK-negative anaplastic large cell lymphoma.
Haematologica | 108 June 2023 1464 EDITORIAL K.J. Savage and G.W. Slack
Table 2. Immunophenotypic features of DUSP22 ALK-negative anaplastic large cell lymphoma cases across studies.
DUSP22-R: DUSP22-rearranged; nr: not reported; TIA-1: T-cell intracellular antigen; EMA: epithelial membrane antigen: PD-L1: programmed death ligand 1. *P<0.05 vs DUSP22-not rearranged. #Not compared. ^Reported as % mean of lymphoma cells (2% of lymphoma cells in DUSP22-R cases are pSTAT3-positive vs. 36% in non-rearranged cases; 3% of lymphoma cells in DUSP22-R cases are PDL1-positive vs. 26% in non-rearranged cases).
29%, respectively, which was indistinguishable from the survival outcomes of triple-negative cases (see Figure 6E, F in the accompanying article).13
In the second paper, Qiu and colleagues from the MD Anderson Cancer Center evaluated 22 cases of DUSP22-R ALK-negative ALCL, representing 28% of all ALK-negative ALCL cases with pathological features, also consistent with previous reports (Table 2).14 Treatment information was available for 16 patients with DUSP22-R, 13 of whom received anthracycline-based chemotherapy; follow-up information was available for 18 patients and nine (50%) had died. With a median follow-up of 19 months, the projected 5-year OS was only 40%, which was similar to that of DUSP22-NR cases (P=0.275) and inferior to that of ALK-positive ALCL cases (5-year OS 82%; P=0.005). Similarly, 5-year PFS was only 40% (P=0.275 vs. TP63-NR). The treatment landscape of ALCL has changed over the last decade with the approval of brentuximab vedotin (BV) for the treatment of relapsed/refractory ALCL15 and more recently, approval of CHP (cyclophosphamide, doxorubicin, prednisone)-BV for newly diagnosed systemic ALCL based on superior PFS and OS over CHOP (cyclophosphamide, doxorubicin, vincristine; prednisone), as shown in the ECHELON-2 study in CD30+ peripheral T-cell lymphomas.16
In the LYSA study, survival was notably poor from first relapse/progression, regardless of DUSP22-R status, suggesting that any prognostic relevance may diminish in this high-risk setting (4-year OS 21% [DUSP22-R] vs. 34% [triple-negative]; P=0.62) (Figure 5A in the accompanying
article).13 The use of BV in relapsed/refractory ALCL improved outcomes across genetic subgroups but similarly, no outcome difference was noted (Figure 5E, F in the accompanying article).13 Of note, the prognostic impact of DUSP22-R in patients with treatment-naïve ALK-negative ALCL who were treated with CHP-BV remains unknown. Collectively, unique morphological, immunophenotypic and molecular features support the designation of DUSP22-R ALK-negative ALCL as a distinct entity as proposed by the International Consensus Classification17 although the WHO 5th edition update (WHO-HAEM5) applied only a provisional designation due to uncertainty around prognosis18 (Figure 1). In contrast, cases with a TP63-R are important to recognize given the poor prognosis, but further genetic studies are still required to fully characterize them. Furthermore, although these rearrangements are usually mutually exclusive, rare ‘double-hit’ cases have been reported.19 Considering all studies to date, the prognosis of DUSP22-R ALK-negative ALCL is more variable than that of the typically favorable ALK-positive ALCL, but even that entity can have a poor outcome.2 The LYSA study highlights that clinical information, such as PS, must also be taken into consideration when making management decisions. Of note, as a composite risk score, the International Prognostic Index did not reach statistical significance and, with limited numbers, it was not specifically applied to DUSP22-R cases to judge its utility.
There may also still be unknown pathobiological and
Immunohistochemical Feature First author (study location/group) ParillaCastellar (Mayo)6 Pedersen (Denmark)10 Hapgood (BC Cancer)12 Onaindia (Spain)11 Sibon (LYSA)13 Qiu (MDACC)14 DUSP22-R, N 22 5 12 4 47 22 CD2+, % (N/N) 83 (15/18)* nr 83 (10/12)* nr 87 (33/38)* 77 (13/17) CD3+, % (N/N) 81 (17/21)* nr 83 (10/12)* 100 (4/4) 62 (29/47)* 76 (16/21) CD5+, % (N/N) 12 (2/17)# nr nr nr 43 (19/44) 35 (14/19) CD4+, % (N/N) 53 (9/17)# nr nr nr 72 (34/47) 74 (14/19) CD8+, % (N/N) 12 (2/17)# nr nr nr 11 (6/44) 28 (5/18)* TIA1+, % (N/N) 10 (2/21)* nr 8 (1/12)* 0 (4/4) 13 (5/38)* 30 (3/10) Granzyme B+, % (N/N) 5 (1/21)* nr 0 (0/12)* 0 (4/4) 11 (5/44)* 0 (0/12)* Perforin+, % (N/N) 0 (0/10)# nr 8 (1/12)* 0 (4/4) 12 (4/33)* 25 (1/4) Positive for any cytotoxic marker, % (N/N) nr nr 8 (1/12)* 0 (4/4) 27 (8/30)* nr EMA+, % (N/N) 0 (0/20)* nr 0 (0/12)* nr 13 (5/38)* 18 (2/11)* pSTAT3+, % (N/N) nr nr 0 (0/12)* 0 (3/3) 10 (2/20)* 2*^ PD-L1+, % (N/N) nr nr 0 (0/12)* nr nr 3*^
Haematologica | 108 June 2023 1465 EDITORIAL K.J. Savage and G.W. Slack
Figure 1. Current classification of systemic anaplastic large cell lymphoma. The most common t(2;5) (ALK-NPM) is shown. Rare variant rearrangements involving the ALK gene on 2p23 and different partner genes on other chromosomes can occur in 15-25% of cases of ALK-positive anaplastic large cell lymphoma. ALCL: anaplastic large cell lymphoma; WHO: World Health Organization; ALK: anaplastic lymphoma kinase.

genetic factors contributing to outcome in DUSP22-R ALKnegative ALCL. Cases are typically negative for cytotoxic markers but rare cases may be positive, and previous studies have shown an association with inferior outcomes across ALK-negative ALCL.6 DUSP22-R in ALCL was originally shown to occur as a result of a balanced translocation involving the DUSP22 phosphatase gene on 6p25.3 and the FRA7H fragile site on 7q32.3, resulting in downregulation of the DUSP22 gene.20 Subsequent studies assessing DUSP22-R in ALCL have used break-apart fluorescence in situ hybridization and the translocation partner has not been determined. Could alternate translocation partners occur in DUSP22R ALCL and might these account for the more aggressive clinical behavior seen in some cases? Further investigations are required to
References
1. ten Berge RL, de Bruin PC, Oudejans JJ, Ossenkoppele GJ, van der Valk P, Meijer CJ. ALK-negative anaplastic large-cell lymphoma demonstrates similar poor prognosis to peripheral T-cell lymphoma, unspecified. Histopathology.
extend our understanding of the underlying molecular mechanisms that result in these phenotypic and behavioral differences.
Disclosures
KJS has received honoraria from and acted as a consultant for BMS, Merck, Seagen, Kyowa, Novartis, Janssen and Abbvie; has participated in a steering committee for Beigene; and has sat on a Data and Safety Monitoring Committee for Regeneron. GWS has received honoraria from and acted as a consultant for Seattle Genetics.
Contributions
KJS and GWS wrote and approved the manuscript.
2003;43(5):462-469.
2. Savage KJ, Harris NL, Vose JM, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not
Haematologica | 108 June 2023 1466 EDITORIAL K.J. Savage and G.W. Slack
otherwise specified: report from the International Peripheral TCell Lymphoma Project. Blood. 2008;111(12):5496-5504.
3. Hapgood G, Savage KJ. The biology and management of systemic anaplastic large cell lymphoma. Blood. 2015;126(1):17-25.
4. Shustov A, Cabrera ME, Civallero M, et al. ALK-negative anaplastic large cell lymphoma: features and outcomes of 235 patients from the International T-Cell Project. Blood Adv. 2021;5(3):640-648.
5. Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123(19):2915-2923.
6. Parrilla Castellar ER, Jaffe ES, Said JW, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473-1480.
7. King RL, Dao LN, McPhail ED, et al. Morphologic features of ALK-negative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.
8. Luchtel RA, Dasari S, Oishi N, et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood. 2018;132(13):1386-1398.
9. Luchtel RA, Zimmermann MT, Hu G, et al. Recurrent MSC (E116K) mutations in ALK-negative anaplastic large cell lymphoma. Blood. 2019;133(26):2776-2789.
10. Pedersen MB, Hamilton-Dutoit SJ, Bendix K, et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: a Danish cohort study. Blood. 2017;130(4):554-557.
11. Onaindia A, de Villambrosia SG, Prieto-Torres L, et al. DUSP22-rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica. 2019;104(4):e158-e162.
12. Hapgood G, Ben-Neriah S, Mottok A, et al. Identification of high-risk DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. Br J Haematol. 2019;186(3):e28-e31.
13. Sibon D, Bisig B, Bonnet C, et al. ALK-negative anaplastic large cell lymphoma with DUSP22 rearrangement has distinctive disease characteristics with better progression-free survival: a LYSA study. Haematologica. 2023;108(6):1590-1603.
14. Qiu L, Tang G, Li S, et al. DUSP22 rearrangement is associated with distinctive immunophenotype but not outcome in patients with systemic ALK-negative anaplastic large cell lymphoma. Haematologica. 2023;108(6)1604-1615.
15. Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic largecell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190-2196.
16. Horwitz S, O'Connor OA, Pro B, et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet. 2019;393(10168):229-240.
17. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140(11):1229-1253.
18. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
19. Karube K, Feldman AL. "Double-hit" of DUSP22 and TP63 rearrangements in anaplastic large cell lymphoma, ALKnegative. Blood. 2020;135(9):700.
20. Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117(3):915-919.
Haematologica | 108 June 2023 1467 EDITORIAL K.J. Savage and G.W. Slack
Targeting glutaminase to starve lymphoma cells
Charles Dumontet
Correspondence: C. Dumontet charles.dumontet@chu-lyon.fr
Received: November 16, 2022.
Accepted: November 23, 2022.
Early view: December 1, 2022.
https://doi.org/10.3324/haematol.2022.282348
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The discovery of Bruton tyrosine kinase (BTK) inhibitors such as ibrutinib has had a significant impact on the outcome of patients with mantle cell lymphoma. However, most of these patients will relapse under BTK inhibitor therapy, with a poor prognosis since overall survival after failure of BTK inhibitor therapy is less than 12 months.1 In their article, published in this issue of Haematologica , Li et al. provide promising preclinical evidence suggesting that tumor cell glutaminase (GLS) could constitute a potential therapeutic target in this difficult-to-treat population of patients.2
Glutamine addiction has been reported in various subtypes of hematologic malignancies, including acute lymphoblastic leukemia and NK-cell lymphoma, allowing neoplastic cells to thrive in glucose-low or hypoxic environments. Le et al. showed that Myc induction enhanced glucose consumption and lactate production in a model non-Hodgkin lymphoma line and that glutamine contributed significantly to citrate carbons under hypoxic conditions.3 This work demonstrated the existence of an alternative energy-generating glutaminolysis pathway involving a glucose-independent tricarboxylic acid cycle. Glutamine metabolism thus appears to be essential for cell survival and proliferation under conditions of hypoxia and glucose deprivation. Gao et al. reported that c-Myc induces increased expression of mitochondrial GLS, upregulating glutamine conversion to glutamate, which is further catabolized in the tricarboxylic acid cycle to generate ATP.4 Using cell lines containing GLS variants as well as in vivo modulation of murine and human GLS, Xiang et al. showed that targeted inhibition of tumor-specific GLS reduced tumorigenesis in a human non-Hodgkin lymphoma xenograft model. 5 There does, therefore, seem to be a well-established correlation between Myc, tumor cell GLS and the use of glutamine as a key ATP-generating energy substrate in lymphomas.
Targeting glutamine addiction in cancer has been explored in various preclinical settings and more recently in early phase clinical trials using teglenastat. Targeting mitochondrial GLS has been shown to inhibit oncogenic transformation in preclinical models of fibroblasts and
breast cancer.6 Matre et al. reported that inhibition of GLS by various inhibitors blocked the growth of acute myeloid leukemia cell lines as well as a subset of primary acute myeloid leukemia samples.7 Interestingly, the antitumor effect of recombinant L-asparaginase, which is widely used to treat various lymphoid malignancies, is believed to rely at least in part on its GLS activity which results in extracellular glutamine depletion.8
Telaglenastat (CB-839) has been evaluated in early phase clinical trials, mainly in combination regimens in patients with solid tumors. In a combination study with cabozantinib or everolimus, telaglenastat displayed promising activity in patients with advanced or metastatic renal cell carcinoma, with mostly grade 1 to 2 treatment-related adverse events.9 A single-agent phase I study has been conducted in patients with hematologic malignancies (NCT02071888) but the results have not yet been reported.
Targeting GLS appears to be particularly relevant in the context of ibrutinib resistance. Lee et al. analyzed the impact of ibrutinib in various mantle cell lymphoma lines and found that inhibition of BTK had a profound effect on several metabolic pathways, including glutaminolysis.10 Importantly, glutaminolysis was found to contribute to over 50% of mitochondrial ATP production. By showing that GLS expression and glutamine addiction are enhanced in ibrutinib-resistant mantle cell lymphoma models, Li et al. provide compelling evidence suggesting that targeted inhibition of GLS could benefit patients with mantle cell lymphoma whose disease has progressed under BTK inhibitor therapy.
More generally these results support the tantalizing possibility that tumor-associated metabolic specificities may represent an Achilles heel allowing the selective destruction of neoplastic cells. Exploiting these characteristics, either using single agent therapies or in the context of synthetic lethality approaches, has proven to be challenging. To date attempts to target the Warburg effect, i.e. preferential cytosolic fermentation of glucose to lactic acid rather than mitochondrial oxidative fermentation even in the presence of abundant oxygen, has not led to major breakthroughs in cancer therapy. These
INSERM UMR1052/CNRS5286, Hospices Civils de Lyon and University of Lyon, Lyon, France
Haematologica | 108 June 2023 1468 EDITORIAL C. Dumontet
attempts highlight the difficulty of inducing systemic alterations in key metabolic processes to specifically target tumors while preserving healthy tissues. For this reason it is possible that the use of metabolic inhibitors, such as telaglenastat, in combination with BTK in -
References
1. Jain P, Kanagal-Shamanna R, Zhang S, et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br J Haematol. 2018;183(4):578-587.
2. Li L, Nie L, Jordan A, et al. Targeting glutaminase is therapeutically effective in ibrutinib-resistant mantle cell lymphoma. Haematologica. 2023;108(6):1616-1627.
3. Le A, Lane AN, Hamaker M, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110-121.
4. Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762-765.
5. Xiang Y, Stine ZE, Xia J, et al. Targeted inhibition of tumorspecific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest. 2015;125(6):2293-2306.
hibitors, rather than after failure of such therapies, will reduce or defer the emergence of resistant phenotypes.
Disclosures
CD has received research funding from Roche.
6. Wang JB, Erickson JW, Fuji R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207-219.
7. Matre P, Velez J, Jacamo R, et al. Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget. 2016;7(48):79722-79735.
8. Sugimoto K, Suzuki HI, Fujimura T, et al. A clinically attainable dose of L-asparaginase targets glutamine addiction in lymphoid cell lines. Cancer Sci. 2015;106(11):1534-1543.
9. Meric-Bernstam F, Tannir NM, Iliopoulos O, et al. Telaglenastat plus cabozantinib or everolimus for advanced or metastatic renal cell carcinoma: an open-label phase I trial. Clin Cancer Res. 2022;28(8):1540-1548.
10. Lee SC, Shestov AA, Guo L, et al. Metabolic detection of Bruton's tyrosine kinase inhibition in mantle cell lymphoma cells. Mol Cancer Res. 2019;17(6):1365-1377.
Haematologica | 108 June 2023 1469 EDITORIAL C. Dumontet
Do older patients truly benefit from advances in myeloma care?
Moshe E. Gatt1,2 and Eyal Lebel1,2
The survival of patients with multiple myeloma (MM) has increased dramatically during the last two decades, alongside the advent of novel anti-myeloma treatments and their incorporation into front-line therapy. Recent studies document a significant increase in the prevalence of MM, especially in the elderly,1 highlighting the importance and relevance of real-world reports of this population. As the elderly population is regularly underrepresented in clinical trials, questions arise about whether the improved outcomes reported truly reflect the real-world situation, and whether the elderly population does indeed benefit from the advances seen in the field.
In their study, published in this issue of Haematologica, Moore et al.2 not only present data from a very large Nordic registry of over 4,600 elderly patients ( ≥ 75 years) with MM, thus capturing close to 100% of the elderly, affected population of the two countries, but further compare their outcomes with those of younger patients, and with parallel data from key clinical trials.
The authors highlighted differences in baseline parameters between younger and older patients, with higher International Staging System (ISS) stage and higher frequency of anemia and kidney dysfunction in the elderly. Overall, and not surprisingly, older age was confirmed as a predictor of worse outcome. However, the analysis confirmed that survival has improved significantly over time also among older patients. The population-based design of this study enabled the assessment of relative survival rates as opposed to overall survival, taking into account competing causes of mortality. The improved relative survival over time among the older patients points to better myeloma care as a key contributing factor, and rules out the general improved survival of the whole population as the sole explanation. This improvement in survival coincided with a dramatic increase in the use of novel agents, and with improved response rates achieved with these agents. Moreover, the observed benefit in relative survival was greater for the older patients than for the younger ones. Even in the octogenarians, although mortality rates remained relatively high, a net survival benefit was observed, highlighting that age itself is not a reason to withhold treatment with
novel agents.
Correspondence: M. Gatt
rmoshg@hadassah.org.il
Received: October 5, 2022.
Accepted: October 18, 2022.
Prepublished: October 27, 2022.
https://doi.org/10.3324/haematol.2022.281897
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Compared with key clinical trials in transplant-ineligible patients (i.e., a relatively older population), the population-based study by Moore et al. included a larger proportion of patients ≥75 years, and a higher frequency of those with advanced ISS stage. These differences limit the external validity of key clinical trials, and myeloma physicians as well as health care authorities should be aware of them. An analysis of the relative importance of different outcome predictors in different ages confirmed notable differences between younger and older patients.3 In younger patients cytogenetic risk had more influence on survival, while in older patients the effect of cytogenetics on outcome was considerably weaker.
Specifically, while 17p deletion was associated with adverse prognosis in patients of all ages, t(4:14) and 1q gain were associated with adverse outcomes only in younger patients. On the other hand, ISS stage was a stronger predictor in older patients than in younger ones. Performance status commonly predicted survival at all ages, suggesting that physical frailty rather than numerical age is more predictive of outcome. In view of this observation, it is less surprising that age ≥70 years was not associated with worse outcomes in MM patients who underwent autologous transplant, as long as melphalan 200 mg/m2 was given, as shown in a large report from the Center for International Blood and Marrow Transplant Research (CIBMTR).4 This observation probably reflects the fitness of older patients who were judged to be eligible for this therapy, and highlights the importance of comprehensive assessments of function and frailty. Recent studies confirmed that frailty status is predictive of outcomes, 5 can be objectively assessed using validated scoring systems, and can influence treatment decision-making.6 Puyade et al. described major age-related disparities in adherence to guidelines,7 and concluded that older patients are less likely to undergo all necessary diagnostic procedures and to receive adequate therapy in accordance with guidelines. These conclusions are complex to interpret considering, as discussed above, that the validity of guidelines for the older population is uncertain, given the underrepresentation of this group in clinical trials. Taken together, it is poss-
1Faculty of Medicine, The Hebrew University of Jerusalem and 2Hematology Department, Hadassah-Hebrew University Medical Center, Jerusalem, Israel
Haematologica | 108 June 2023 1470 EDITORIAL M.E. Gatt and E. Lebel
ible that the excess mortality in older patients may be partially explained by underutilization of novel agents, as well as limited assessments of frailty, leading to suboptimal selection of older patients for front-line regimens of various intensities.
Last, but not least, in this Nordic population study, Moore et al. found that despite more effective treatment, and the decline if not disappearance of conventional chemotherapy from usage, early mortality has not decreased in older patients, and remains strikingly high.2 As expected, age is an established predictor of early mortality in many cancers, including MM. However, the incorporation of novel agents was consistently associated with a lower risk of early mortality.8 This finding should highlight the importance of close monitoring of older patients, as well
References
1. Turesson I, Bjorkholm M, Blimark CH, Kristinsson S, Velez R, Landgren O. Rapidly changing myeloma epidemiology in the general population: increased incidence, older patients, and longer survival. Eur J Haematol. 2018;101(2):237-244.
2. Moore KLF, Turesson I, Genell A, et al. Improved survival in myeloma patients–a nationwide registry study of 4,647 patients ≥75 years treated in Denmark and Sweden. Haematologica. 2023;108(6):1640-1651.
as consideration of dose reduction (particularly of steroids), in order to reduce infection rates and mortality.9
In conclusion, older patients ≥ 75 years, accounting for about 40% of all patients with MM, do indeed benefit from the recent advances in the field, as they more often nowadays receive novel agents, achieve deeper responses and survive longer. However, there are still gaps (highlighted in Figure 1) in the incorporation of better assessments of fitness and frailty, adherence to guidelines, understanding of the relative importance of different risk factors and, most importantly, in the critical need for early reduction in the mortality rate.
Disclosures
No conflicts of interest to disclose.
3. Pawlyn C, Cairns D, Kaiser M, et al. The relative importance of factors predicting outcome for myeloma patients at different ages: results from 3894 patients in the Myeloma XI trial. Leukemia. 2020;34(2):604-612.
4. Munshi PN, Vesole D, Jurczyszyn A, et al. Age no bar: a CIBMTR analysis of elderly patients undergoing autologous hematopoietic cell transplantation for multiple myeloma. Cancer. 2020;126(23):5077-5087.
Figure 1. Suggested steps to improve survival of older patients with multiple myeloma. ISS: International Staging System.
Haematologica | 108 June 2023 1471 EDITORIAL M.E. Gatt and E. Lebel
5. Patel BG, Luo S, Wildes TM, Sanfilippo KM. Frailty in older adults with multiple myeloma: a study of vs veterans. JCO Clin Cancer Inform. 2020;4:117-127.
6. Grant SJ, Freeman CL, Rosko AE. Treatment of older adult or frail patients with multiple myeloma. Hematology Am Soc Hematol Educ Program. 2021;2021(1):46-54.
7. Puyade M, Defossez G, Guilhot F, Leleu X, Ingrand P. Agerelated health care disparities in multiple myeloma. Hematol
Oncol. 2018;36(1):224-231.
8. Galli M, Paris L, Pavoni C, Delaini F, Rambaldi A. Effect of novel agents on the risk of early death of newly diagnosed symptomatic multiple myeloma patients: a single centre retrospective analysis. Am J Hematol. 2019;94(1):E11-E13.
9. Diamond E, Lahoud OB, Landau H. Managing multiple myeloma in elderly patients. Leuk Lymphoma. 2018;59(6):1300-1311.
Haematologica | 108 June 2023 1472 EDITORIAL M.E. Gatt and E. Lebel
How different are blood platelets from women or men, and young or elderly people?
Giovanni de Gaetano, Marialaura Bonaccio and Chiara Cerletti
Department of Epidemiology and Prevention, IRCCS Neuromed, Pozzilli, Italy
Correspondence: G. de Gaetano
giovanni.degaetano@moli-sani.org
Received: October 18, 2022.
Accepted: November 9, 2022.
Prepublished: November 17, 2022.
https://doi.org/10.3324/haematol.2022.282131
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Gender-specific medicine is the study of how (sex-based) biological and (gender-based) socioeconomic and cultural differences influence people’s health. Significant differences are currently described in the development, progression and clinical signs of conditions common to men and women, such as the response to treatments and nutrients, and in lifestyles. Several determinants (genetic, epigenetic, hormonal and environmental) reportedly account for differences between cells from women and men and/or from young or elderly people. To be truly effective, prevention, diagnosis and management of a given condition should be considered in relation to the biological sex of the individual/patient, as well as to other parameters, such as gender identity, age, ethnicity, level of education, religious beliefs, sexual orientation, social and economic conditions.
A Letter to the Editor published in this issue of Haematologica1 gives a small but significant contribution to the progress of gender-specific medicine.
It is well known that platelet count variability is dependent upon genetic factors and is highly heritable. Several genes have been identified to concomitantly influence platelet count, mean platelet volume and platelet activation/function. Many of the variants are in non-coding regions of the genome, suggesting that they may play a role in the expression/modulation of these regions, possibly through epigenetic mechanisms.2
Studies trying to identify the non-genetic factors associated with platelet count identified sex, age and ethnicity as major variables. Studies in the general population and in different Italian geographical isolates showed no difference in platelet count in men and women until the age of 15, but subsequently women constantly had more platelets than men, with a slow, progressive, parallel decline with aging in both sexes.3 These Italian data have been confirmed and extended in other geographical settings, as now also reported in the Letter to the Editor by Sabrkhany et al.1
The results presented in that Letter suggest that some platelet function parameters may also differ between women and men and change with progression of age. Integrin
αIIbb3 activation increased with age, a finding possibly in agreement with previous observations of increased platelet response to ADP with aging, while P-selectin expression decreased with age. Age was found to be an independent predictor of platelet growth factor (PGF) content and was negatively correlated with intra-platelet concentrations of platelet factor 4 (PF4), connective tissue activating peptide III (CTAP-III) and platelet-derived growth factor (PDGF). In contrast, the relationship with thrombospondin-1 (TSP-1) was not significant. In isolated platelets, the concentration of PDGF, but not of other platelet-derived biomarkers, was significantly higher in women than in men. However, taking into account the higher number of platelets in women, the total circulating concentrations of all biomarkers were significantly higher in women, even when adjusted for age. The translational aspects of these data remain to be defined, but the authors underline the importance of ageand sex-matched controls for future platelet-based biomarker studies.
A previous proposal by Bijno et al.3 to use different normal ranges of platelet count that take into account sex and age has not apparently become a diffuse practice, but has occasionally been adopted. The use of personalized reference intervals, instead of the traditional ones, resulted in relevant differences in the number of patients classified as thrombocytopenic or affected by thrombocytosis; the proportion of subjects with unexplained thrombocytopenia was also smaller.4 A sex-stratified approach also revealed peculiar relationships between platelet distribution width – an index of platelet size variability – and the intensity of depressive symptoms.5 Within the Moli-sani Study cohort, using personalized (sex- and age-specific) reference intervals of platelet count, the reduction of the number/proportion of subjects with thrombocytopenia was confirmed; interestingly, the group of possibly true thrombocytopenic subjects (identified by personalized range intervals) had a higher risk of total mortality compared with subjects classified as thrombocytopenic by traditional range intervals. In the same Moli-sani cohort, better adherence to a Mediterranean diet, rich in fibers and antioxidants, was associ-
Haematologica | 108 June 2023 1473 EDITORIAL G. de Gaetano et al.
ated with reduced platelet (and leukocyte) counts as compared to personalized (sex- and age-specific) reference intervals of platelet count.6
The reasons why platelet count and/or some platelet function parameters (and perhaps the response to antiplatelet drugs) vary by age and sex (and possibly by other factors, such as different lifestyles) remain speculative at present.2 The differences between platelet counts of individuals adhering or not to a Mediterranean diet,6 as well as those reported in the Letter by Sabrkhany et al., 1 are statistically significant, but quite small in absolute terms. Although it is obvious that statistical significance does not necessarily imply biological or clinical relevance,7 it has been observed that a difference of only 10,000 platelets/mL corresponds to an average of 50 billion circulating platelets.2 On the other hand, very small variations in platelet count were reported in men at different 10-year risk of developing cardiovascular disease. Only a few thousand circulating platelets are reportedly able to prevent serious hemorrhage in adults with acute myeloid leukemia. Thus, even minor changes in platelet number and/or function could be associated with different phenotypes and lead to different health outcomes, such as neuropsychiatric and neurodegenerative disorders.5,8
The following statement made by Giulio Bizzozero in 1882 “it is hardly permitted to assume that elements represented in blood in a constant fashion and at great number, as is the case for blood platelets, are active only under abnormal or pathological conditions. Their physiological significance therefore remains to be investigated…” is still worth consideration. Adopting personalized, sex- and agematched controls of platelet number and/or platelet-de-
rived biomarkers will help the studies of platelet (micro)variability in health and disease. This may open a new interesting window on platelet physiology and will also contribute to better targeted and successful antiplatelet therapy. For example, in a meta-analysis of 53 different studies (and 6,450 individuals) on aspirin-treated normal subjects or cardiovascular patients, the response variability to aspirin, as assessed by the point-of-care platelet function test PFA-100, was similar between men and women, but populations with higher mean age had a significantly higher prevalence of non-responders to aspirin than those with a lower mean age.9
Aging is the time-dependent functional decline of an organism at all levels and lies at the intersection of genetics, biology, and the environment; it exhibits marked disparity among individuals. The concept of “biological age” has, therefore, recently attracted interest, as chronological age fails to account for the heterogeneity with which individuals age; indeed, concepts of “biological age” have emerged from the need to account for this variability better and are currently a major focus of research.10 Whether the data presented by Sabrkhany et al.1 and those discussed in this editorial will contribute to introducing the concepts of biological age into platelet research is not known at the moment, but is a reasonable, exciting perspective.
Disclosures
No conflicts of interest to disclose.
Contributions
GdG wrote the draft; MB and CC contributed information and reviewed the final manuscript.
Haematologica | 108 June 2023 1474 EDITORIAL G. de Gaetano et al.
Figure 1. Age, sex and platelets. Some characteristics of human platelets vary according to age and sex. The changes in platelet count are emblematic. The number of platelets decreases with age and after 15 years of age, women have more platelets than men.
References
1. Sabrkhany S, Kuijers MJE, van Kuijk SMJ, Griffioen AW, Oude Egbring MGA. Age- and gender-matched controls needed for platelet-based biomarker studies. Haematologica. 2023;108(6):1667-1670.
2. Izzi B, Bonaccio M, de Gaetano G, Cerletti C. Learning by counting blood platelets in population studies: survey and perspective a long way after Bizzozero. J Thromb Haemost. 2018;16(9):1711-1721.
3. Biino G, Santimone I, Minelli C, et al. Age- and sex-related variations in platelet count in Italy: a proposal of reference ranges based on 40987 subjects' data. PLoS One. 2013;8(1):e54289.
4. Zaninetti C, Biino G, Noris P, Melazzini F, Civaschi E, Balduini CL. Personalized reference intervals for platelet count reduce the number of subjects with unexplained thrombocytopenia. Haematologica. 2015;100(9):e338-e340.
5. Izzi B, Tirozzi A, Cerletti C, et al. Beyond haemostasis and thrombosis: platelets in depression and its co-morbidities. Int J
Mol Sci. 2020;21(22):8817.
6. Bonaccio M, Di Castelnuovo A, De Curtis A, et al; Molisani Project Investigators. Adherence to the Mediterranean diet is associated with lower platelet and leukocyte counts: results from the Molisani study. Blood. 2014;123(19):3037-3044.
7. Di Castelnuovo A, Iacoviello L. Moving beyond p-value. Bleeding, Thrombosis, and Vascular Biology 2022;1:30.
8. Tirozzi A, Parisi R, Cerletti C, et al. Genomic overlap between platelet parameters variability and age at onset of Parkinson disease. Appl Sci. 2021;11:6927.
9. Crescente M, Di Castelnuovo A, Iacoviello L, Vermylen J, Cerletti C, de Gaetano G. Response variability to aspirin as assessed by the platelet function analyzer (PFA)-100. A systematic review. Thromb Haemost. 2008;99(1):14-26.
10. Gialluisi A, Di Castelnuovo A, Donati MB, de Gaetano G, Iacoviello L; Moli-sani Study Investigators. Machine learning approaches for the estimation of biological aging: the road ahead for population studies. Front Med (Lausanne). 2019;6:146.
Haematologica | 108 June 2023 1475 EDITORIAL G. de Gaetano et al.
Monoclonal gammopathy of increasing significance: time to screen?
Lucia Y. Chen,1 Mark Drayson,2 Christopher Bunce2 and Karthik Ramasamy1
1Oxford University Hospitals NHS Foundation Trust, Oxford and 2University of Birmingham, Birmingham, UK
Abstract
Correspondence: K. Ramasamy karthik.ramasamy@ndcls.ox.ac.uk
Received: July 19, 2022.
Accepted: October 28, 2022.
Prepublished: November 10, 2022.

https://doi.org/10.3324/haematol.2022.281802
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Monoclonal gammopathy (MG) is a frequently detected clonal B-cell or plasma-cell disorder. Importantly, every multiple myeloma (MM) case is preceded by MG. Although clinical algorithms now allow earlier treatment of patients with biomarkers of malignancy before MM-induced tissue damage (CRAB) occurs, most patients are still diagnosed late. It is important to revisit how MG should be managed in clinical practice and whether screening is required. As the prevalence of MG and other medical co-morbidities both rise with increasing age, the degree of contribution of MG to disease states other than malignant progression is often unclear. This can lead to monitoring lapses and under recognition of the organ dysfunction that can occur with monoclonal gammopathy of clinical significance (MGCS). Therefore, models of progression to MM and/or MGCS require further refinement. While MG is currently detected incidentally, a case for screening has been made with ongoing studies in this area. Screening has the potential benefit of earlier detection and prevention of both MGCS and delayed MM presentations, but important drawbacks include the psychosocial impact on individuals and resource burden on healthcare services. MG terminology should transition alongside our increasing understanding of the condition and genomic characterization that have already begun to revise the MG nomenclature. The biology of MG has been poorly understood and is often inferred from the biology of MM, which is unhelpful. We review the literature and case for MG screening in this paper. In particular, we highlight areas that require focus to establish screening for MG.
Introduction
The incurable plasma-cell malignancy multiple myeloma (MM) accounts for 2% of all cancer diagnoses and cancer deaths in the UK1 and the USA.2 MM is consistently preceded by well-defined earlier states termed monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM).3,4 The recognition that a period of MGUS universally heralds MM, alongside the advent of less toxic MM therapies, have strengthened the argument for earlier intervention. As a result, therapeutic algorithms for MM have transitioned over the last decade to treating earlier stages of disease, in patients with biomarkers of malignancy and no end-organ damage, using standard anti-myeloma therapy.5 However, the increasing focus on early intervention in MM highlights the need to re-assess methods of early detection, including screening.
Monoclonal gammopathy (MG) describes a clonal B-cell or plasma-cell dyscrasia leading to the production of a
monoclonal protein discernible against a background of polyclonal immunoglobulins. Traditionally, MG is considered a benign premalignant condition and therefore research on MG has thus far concentrated on drivers of malignant transformation. However, there is growing evidence that MG can cause organ damage via mechanisms independent of tumor growth. For example, there has been increasing attention on the ability of small but ‘dangerous’ B-cell clones to cause paraprotein-mediated tissue damage, a phenomenon termed monoclonal gammopathy of clinical significance (MGCS). Furthermore, several large epidemiological studies have reported excess morbidity and mortality associated with a diagnosis of MG, with uncertain biological mechanisms.6 Thus, accumulating evidence suggests that MG is of increasing significance.
Here, we outline current understanding, describing both the malignant and non-malignant mechanisms by which MG can cause tissue damage. Speci fically, we question whether active systematic identification of MG cases
Haematologica | 108 June 2023 1476 PERSPECTIVE ARTICLE
through population or targeted screening should be considered in the context of growing evidence of the clinical importance of this condition.
Monoclonal gammopathy: is it harmful enough to warrant screening?
The mechanisms by which different types of MG (Table 1) cause organ dysfunction and morbidity are incompletely understood. Although MG can lead to malignant transformation and paraprotein-mediated tissue damage, it has also been repeatedly associated with increased occurrence of other diagnoses (MGCS). The biological explanation for MGCS is even less well understood; it is possible that the mechanisms leading to cancerous and non-cancerous consequences overlap.
Malignant transformation
The prevalence of MG is 3.2% in those over 50 years and increases with age.7,8 Non-IgM MG typically progresses to MM at a rate of 1% per year;9 light-chain MG progresses to
MM less frequently at a rate of around 0.3% per year,10 while IgM MG progresses to B-cell malignancies such as Waldenström macroglobulinemia (WM) at a rate of 1.5% per year.11 Rare cases of IgE and IgD MGUS/MM have also been described.12,13
Progression from MG to plasma-cell or B-cell malignancies is the principal cause of MG-related morbidity and mortality, and the risk of malignant progression is not uniform.9 At present, there are two major risk predictors for progression to MM: (i) genomic and (ii) secreted protein profiles. Genomic myeloma-defining events, including MYC activation, driver gene mutations and mutant apolipoprotein B mRNA-editing enzyme, catalytic polypeptide (APOBEC) activity, help to distinguish indolent MG from MG with malignant potential.14 In addition, secreted protein profiles are established risk factors for malignant transformation and include abnormal serum free light chain (SFLC) ratio, paraproteinemia >15 g/L, and non-IgG subtype.15,16 Patients with no risk factors and those with all three risk factors (highrisk) have a 5% and 58% absolute risk, respectively, of MM progression at 20 years.15 Further studies have identified baseline SFLC >100 mg/L,16 immunoparesis17 and pathologi-
1. Abnormal free light chain ratio (<0.26 or >1.65) with increased level of the appropriate involved light
2. Increased concentration of involved light chain
3. Complete loss of heavy chain immunoglobulin expression
1. Hematologic clonal disorder producing a monoclonal paraprotein that causes renal injury
2. Absence of: light chain cast nephropathy, or monoclonal plasma-cell infiltration in kidney biopsy
Monoclonal gammopathy of neurological significance (MGNS)
Monoclonal gammopathy of cutaneous significance
Smoldering multiple myeloma (SMM)
Peripheral neuropathy associated with a monoclonal paraprotein, without other obvious cause
Varied group of MG-associated cutaneous presentations, some of which demonstrate a strong pathological link
23, 28
37
1. Serum paraprotein (IgG or IgA) ≥30 g/L or urinary M-protein >500 mg/24 h and/or clonal bone marrow plasma cells 10-59% 5
2. Absence of myeloma-defining events* or amyloidosis
*Myeloma-defining events (SLiM-CRAB criteria): S: ≥60% plasma cells in bone marrow; Li: involved:uninvolved light chain ratio ≥100 (provided the involved light chain is >100 mg/L); M: two or more focal lesions on magnetic resonance imaging (>5 mm in size); C: hypercalcemia (>2.75 mmol/L or >0.25 mmol/L higher than upper limit of normal); R: renal insufficiency (serum creatinine >177 mmol/L or creatinine clearance <40 mL/min); A: anemia: hemoglobin <100 g/L or 20 g/L below lower limit of normal; B: one or more lytic bone lesion on X-ray, computed tomography or positron emission tomography/computed tomography (>5 mm in size).
Haematologica | 108 June 2023 1477
Monoclonal gammopathy (MG) disorder Definition References Non-IgM monoclonal gammopathy of undetermined significance (MGUS)
Serum monoclonal immunoglobulin ≤3 g/dL
Plasma cells in the bone marrow ≤10% 3. Absence of: lytic bone lesions, anemia, hypercalcemia, and renal impairment 5 IgM MGUS 1. Serum monoclonal immunoglobulin ≤3 g/dL 5
Lymphoplasmacytic
in
bone marrow ≤10%
Absence
Light chain MGUS
1.
2.
2.
cells
the
3.
of: constitutional symptoms or symptoms and signs of hyper-viscosity, anemia or lymphadenopathy
5, 23 Monoclonal gammopathy of
significance
Organ dysfunction or damage caused by a MG-related clonal disorder via
mechanisms 23 Monoclonal
clinical
(MGCS)
different
gammopathy of renal significance (MGRS)
27,
28
Table 1. Definitions of conditions relating to monoclonal gammopathy.
PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
cal SFLC N-glycosylation18 to be additional risk factors for progression. Risk stratification using select parameters has since led to a distinct management pathway for high-risk MGUS involving additional investigations and more frequent follow-up in secondary care.19
Several risk stratification models of progression from IgM MG and smoldering WM to WM have also been proposed20,21 and include measures of disease burden, such as bone marrow infiltration and IgM level, as well as immunoparesis, albumin and b2-microglobulin levels. Wildtype MYD88 status has also been shown to be an independent risk factor for progression21 and mortality22 despite correlating with lower tumor burden at diagnosis.
Paraprotein-mediated tissue damage
MGCS has become a well-recognized entity that includes a wide range of non-cancerous MG-associated clinical presentations.23,24 The mechanisms reported thus far include deposition of monoclonal immunoglobulin or amyloid fibrils (for example, in type I cryoglobulinemia and light chain amyloidosis; AL amyloidosis), autoantibody activity of the immunoglobulin (for example, anti-MAG antibodies in IgMrelated neuropathy) and aberrant complement-activation (for example, in C3-glomerulonephritis and atypical hemolytic uremic syndrome).23 The most recognized forms of MGCS are AL amyloidosis, monoclonal gammopathy of renal significance (MGRS), monoclonal gammopathy of neurological significance (MGNS) and monoclonal gammopathy of cutaneous significance.
The incidence of AL amyloidosis is around 12 cases per million person-years and the prevalence is around 30,000 to 45,000 cases in Europe and the USA.25 As with many forms of MGCS, AL amyloidosis remains underdiagnosed and earlier detection is key to improving survival.26 Presenting symptoms are often non-specific; therefore, a high index of clinical suspicion alongside screening tests for organ damage, such as albuminuria and cardiac biomarkers, can be key to an early diagnosis. Despite advances in treatments, the reported mortality rate is 25% within 6 months of diagnosis.25
MGRS represents a spectrum of MG-induced renal conditions diagnosed via renal biopsy and is defined as a hematologic clonal disorder producing a nephrotoxic monoclonal protein.27,28 In an Austrian cohort of nearly 3,000 MGUS patients, the rate of MGRS (around 80% biopsyproven) was 1.5%,29,30 and the estimated prevalence of MGRS is 0.5% among people aged 70 or older in the general population.31 However, accurate case detection is impacted by the rising prevalence of chronic kidney disease with age32 and difficulty in obtaining histological diagnoses in an older cohort with multiple co-morbidities.
MGNS is defined as neuropathy caused by a monoclonal protein, and often requires input from a neurology specialist for diagnosis.28 Peripheral neuropathy is a frequent find-
ing in MG patients, with the prevalence being up to 30-50% in IgM MG patients, 5% in IgG MG, and 15% in IgA MG.33 Furthermore, large population studies have demonstrated that MG patients have a 2.7-fold higher risk of peripheral neuropathy compared to matched controls34 and a 5.9-fold higher risk of chronic inflammatory demyelinating polyradiculoneuropathy.35 Despite diagnostic challenges, up to 50% of cases of demyelinating neuropathies are likely linked to a causal IgM MG,36 with anti-myelin-associated glycoprotein (MAG) neuropathy accounting for a large proportion of cases.
Cutaneous manifestations of MG are classified into several subgroups. Group I conditions are pathologically caused by malignant or clonal plasma cells (for example, polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin disease; POEMS), group II conditions are strongly associated with a MG, group III conditions are anecdotally linked to MG and group IV conditions are related to immunoglobulins or M-proteins that may or may not be clonal.37 Treatments are generally specific to the dermatological condition, aside from group I/II conditions for which clonally directed treatment may be employed. The true incidence and prevalence of MGCS are unknown due to suboptimal monitoring, lack of established diagnostic criteria and the reliance on multiple specialties to identify MGCS through a high index of suspicion. The main challenge in MGCS is distinguishing symptoms caused directly by the MG clone and its resultant monoclonal protein, and those that are merely coincidental; for example, only half of patients with clinically suspected MGRS have the condition on biopsy.38 Evidence suggests that treatment of the underlying plasma-cell or B-cell clonal disorder can ameliorate symptoms and prevent irreversible organ damage in MGCS.28 Further studies, both large-scale epidemiological and biological, are required to fully understand the etiology of MG-related disorders. In order to achieve this, thought needs to be given to how cohorts of MG patients can be established, and we hypothesize that improving detection and monitoring of MG will enhance recognition of MGCS cases to this end.
Other monoclonal gammopathy-related morbidity
Higher mortality rates in MG unrelated to malignant progression have been observed. A UK cohort study of 2,193 newly diagnosed MG patients demonstrated excess morbidity and mortality associated with the diagnosis.39 MG patients had a 5-year overall survival of 71.9% compared to 80.1% in age-matched controls and were significantly more likely to have a higher comorbidity index score.39 MG patients also had significantly higher rates of hospital attendance, particularly for renal and rheumatological issues, both prior to and after diagnosis.39 Several large populationbased studies show similar findings of increased morbidity and mortality associated with MGUS;6,40,41 for example, in a
Haematologica | 108 June 2023 1478 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
Swedish population study, MG patients had a survival ratio of 0.70 compared to matched population controls.6 In keeping with these findings, results from the PROMISE study and Mass General Brigham Biobank, investigating patients at high risk of developing MG, demonstrated an increased all-cause mortality associated with patients who had screen-detected MG.42
The reason for increased morbidity and mortality in nonmalignant MG patients is unclear but may be related to the increased rate of other medical conditions. The link between MG and bone fractures as well as osteoporosis is well established,43-45 and a recent study demonstrated a detection rate of MGUS of one in 13 patients with osteoporotic fractures in the fracture clinic screened for MM.46 Further studies have also confirmed a higher risk of thrombophlebitis44 and a two-fold increased risk of developing viral and bacterial infections.47 MG patients in the Swedish cohort had a significantly increased risk of co-existing medical conditions such as ischemic heart disease, and renal disorders.6 Furthermore, a recent Korean study demonstrated a concurrent diagnosis of hypertension, hyperlipidemia, diabetes, and osteoarthritis in 80% of patients with MG followed up for 10 years.48
The challenge with these MG disease-associations is demonstrating causality, given that most patients being tested for MG are older and more likely to have pre-existing medical diagnoses, leading to inherent bias in these studies. However, as epidemiological evidence continues to accumulate, it becomes increasingly important to initiate thorough investigation into the possible biological causes of MGCS-associated morbidity and define an ICD-10 code to capture data more reliably. Clonal hematopoiesis of indeterminate potential (CHIP), an analogous but more genetically defined precursor state, has in recent years been associated with increased atherosclerosis,49 with loss of TET2 function in hematopoiesis proven to accelerate atherogenesis in murine models. This demonstrates the potential of clonal hematopoietic disorders to cause organ pathology and supports the need to further investigate the relationship between MG and other disease states. An etiological role of chronic inflammation and immune stimulation in MG is plausible50 and needs to be explored.
Monoclonal gammopathy: the importance of early detection and intervention
That MM is preceded by MG creates the opportunity for early intervention and possible prevention. The argument for early detection of MG includes improving quality of life for MM patients through reduced end-organ complications and potentially improving survival of MM patients. In ad-
dition, early identification of MG could, in theory, lead to earlier intervention and better outcomes for non-malignant MG-related morbidities and MGCS.
Less than 10% of MM patients are diagnosed at the MG stage51 and it is estimated that by the time patients are formally diagnosed with MG, the clonal disorder has been present for at least 10 years.52 Currently, a diagnosis of MM requires significant burden of disease meaning that, de facto, most MM diagnoses occur in a late stage of disease. Real-world data from Europe have demonstrated that around 85% of patients present with International Staging System stage II/III disease and over 50% present with at least two bone lesions.51 Further cohort studies have shown that the median time from symptom onset to MM diagnosis is around 4 to 6 months, and while this diagnostic delay was not associated with adverse survival, it likely contributed to the significant burden of MM-related complications seen in a large proportion of patients at diagnosis.53 Therefore, by the time most patients are diagnosed with MM, the time for early intervention has been missed. Screening would lead to earlier detection of MG, including MGCS, MM and WM; however, current guidelines do not recommend this due to the lack of clinically proven low-toxicity interventions at the precursor stage.19,43,54,55 There is some evidence that knowledge of prior MG before MM diagnosis can improve survival, although whether this is solely due to early detection remains unclear.56 MG patients under regular monitoring have been shown to suffer significantly fewer major complications (such as dialysis use, cord compression and fracture) at MM diagnosis, and significantly improved disease-specific and overall survival when compared to patients with MG who were not actively managed.57 However, these studies may suffer from lead-time bias. Importantly, the first screening study for MG, iStopMM,58 has shown higher detection rates of Band plasma-cell malignancies through screening; however, whether this enhanced detection will lead to clinical benefit is unknown. Further refinement of risk prediction in MG, using novel genomic59 and biochemical18 biomarkers, may help to define a groups of high-risk MG patients who would benefit from high-intensity follow-up and early intervention, as well as a low-risk MG group who may need less frequent or no monitoring.
Studies investigating low-toxicity treatments at the precursor stage are ongoing. Treatment of SMM with lenalidomide-dexamethasone and single-agent lenalidomide has been shown to improve progression-free survival in two randomized controlled trials60,61 and to delay organ damage.62 However international consensus on the treatment of SMM has not been reached. While the National Comprehensive Cancer Network recommends lenalidomide treatment for SMM, the European Myeloma Network does not advocate treatment of SMM outside of a clinical trial setting.63 A phase II study (CENTAURUS) demonstrated
Haematologica | 108 June 2023 1479 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
the safety and activity of an anti-CD38 monoclonal antibody, daratumumab, as a single agent in intermediate- and high-risk SMM patients.64 A small phase II study of carfilzomib, lenalidomide, and dexamethasone including highrisk SMM patients demonstrated minimal residual disease-negative responses in 11 of 12 patients,65 which is significant given minimal residual disease negativity correlates with improved survival.66 There is an ongoing debate surrounding the goal of treating SMM – to delay progression versus cure – and studies are currently addressing this. Studies investigating treatment at the MG stage are also underway. For example, phase II studies investigating treatment of high-risk MG patients with both daratumumab (NCT03236428) and isatuximab (NCT02960555) are ongoing and a phase I trial is evaluating the role of rifaximin in patients with MG (NCT03820817).67 Potential opportunities exist for early intervention studies in MG targeting the microenvironment. This strategy is supported by single-cell RNA sequencing studies that have identified early changes in the bone marrow immune microenvironment in MG.68,69 A study on patients with bi-clonal gammopathy highlighted that MG clones can be more difficult to eliminate with standard myeloma treatment because of having a very low proliferative fraction.70 A combination approach with simultaneous targeting of the clone and its resident microenvironment may be required and warrants further investigation.
The value of early detection in asymptomatic WM (including IgM MGUS and smoldering WM) is less clear. There is evidence that the progression rate of smoldering WM to WM decreases after the first 5 years71 and prior studies have also shown that the overall survival of patients with smoldering WM and the general population is similar.72 Accordingly, early treatment before the symptomatic stage in WM has not thus far been recommended, and therefore the benefits of early detection of IgM MG to prevent malignant progression may be limited. However, the role of early detection in improving outcomes for IgM MGCS patients requires further study. Enhanced pick-up of IgM MG may lead to earlier diagnosis of IgM-related neuropathy as well as other IgM-related disorders, in which clonally targeted treatments have been effective.73
Monoclonal gammopathy: is screening warranted?
Does monoclonal gammopathy meet screening criteria?
The purpose of screening is to identify asymptomatic individuals at higher risk of developing a particular disease so that they may benefit from early intervention that can lead to improved survival or quality of life. The benefits to those who screen positive must also outweigh any poten-
tial harm to those who screen negative. International screening principles have been widely used to guide the development of screening programs,74 such as the breast, cervical and colorectal cancer screening initiatives in the UK,75–77 which have been shown to reduce mortality. MG fulfils many of the criteria for screening (Table 2). However, two main contentions exist: firstly, whether the collective health risk of MG on a population level is great enough to warrant screening, and secondly, whether an effective intervention for MG exists to reduce mortality and morbidity. These contentions to MG screening require re-appraisal in the context of new emerging evidence. It is also important to consider population versus targeted screening, which may have implications for the risk-benefit ratio of testing.
Who, if anyone, should we screen for monoclonal gammopathy?
As early interventions for MG continue to develop, it is also important to consider which population group would benefit the most from screening. A recent review of National Health Service screening programs highlighted targeted screening as a means of improving cost-effectiveness and reducing the risk-to-benefit ratio by focusing on individuals at a higher risk of developing the condition.78 Several wellestablished risk factors for MG provide a strong basis for defining a population for targeted screening, including increasing age,7 male gender,7 black ethnicity79 and having a first-degree relative with MG.80 Other potential risk factors for MG, such as high body mass index81 and immune-related conditions,50 may further contribute to delineating a high-prevalence group suitable for screening. The PROMISE study is an example of targeted screening of those within a higher-prevalence group, and includes adults aged over 40 years old, identified as Black/African American or with a family history of myeloma or a precursor state.42 Interim 3-year data on the first 2,960 participants screened demonstrated a 10% prevalence of MG,42 a higher rate than previous estimates in the Minnesota cohort,7 which therefore helps to corroborate this approach. An alternative strategy would be opportunistic screening, for example combining serum protein electrophoresis with other primary care screening blood tests, such as cholesterol. A recent study demonstrated an increased prevalence of MG (5.3%) in unselected emergency medical admissions,82 which also highlights medical inpatients as a possible group for opportunistic screening.83 However, further prospective evidence is required to assess the long-term implications of opportunistic screening as an approach. Population screening carries the highest resource burden and risk of psychosocial impact on otherwise healthy individuals. A population-based MG screening study ongoing in Iceland, iStopMM, has screened 75,422 individuals over the age of 40 and identified 3,725 individuals with MGUS.58 Patients were randomized to three arms: no follow-up,
Haematologica | 108 June 2023 1480 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
standard follow-up according to current practice, or an intensive diagnostic and follow-up pathway. After 3 years of follow-up, MG patients in the intensive follow-up arm of the study had significantly higher detection rates of lymphoproliferative disorders, specifically smoldering WM,
Wilson & Junger principles of early disease detection74
The condition sought should be an important health problem
There should be an accepted treatment for patients with recognized disease
Case-finding should be a continuing process and not a “once and for all” project
SMM and MM,84 demonstrating that early detection of these malignancies through screening is possible. Results from longer-term follow-up are required to determine whether this enhanced detection translates into better outcomes for patients.
Criteria met? Explanation
Contentious MM is an incurable life-limiting hematologic malignancy and accounts for 2% of all cancer deaths in the UK
MGUS is not infrequent; the age-standardized prevalence in UK is estimated at 8.7/100,000 and prevalence increases with age
However, absolute risk of progression to MM remains low at 0.5-1% per year
MG can lead to morbidity through MGCS independently of progression to MM
Contentious Identification and routine monitoring of MGUS may improve outcomes and survival upon progression to MM
Risk stratification helps to identify high-risk MGUS patients who have higher rates of progression to MM and in whom early intervention may be more valuable
There are no proven low toxicity treatments to eliminate MGUS clones
Treatment of early MM at the asymptomatic SMM stage improves survival
MGUS patients have excess morbidity and mortality independently of progression to MM; screening may help early identification of MGCS such as MGRS and prevent irreversible end-organ damage
References
1,6,8,9,39,45
15,56,57,60,62
Unknown Future prospective studies may help to determine whether screening for MG can be cost-effective The blood test required for diagnosis is inexpensive
Yes If MG screening is justified and of proven benefit in a particular population, continual screening could need to be organized
MM: multiple myeloma; MGUS: monoclonal gammopathy of undetermined significance; MG: monoclonal gammopathy; MGCS: monoclonal gammopathy of clinical significance; SMM: smoldering multiple myeloma; MGRS: monoclonal gammopathy of renal significance. IMWG: International Myeloma Working Group.
Facilities for diagnosis and treatment should be available Yes Phlebotomy and laboratory services are available and widely accessible There should be a recognizable latent or early symptomatic stage Yes It is well established that MGUS constantly precedes MM as a precursor state 3 There should be a suitable test or examination Yes Diagnosis of MG via peripheral blood serum protein electrophoresis and immunofixation has a high sensitivity and specificity 94 The
the population Yes The blood test diagnosis for MG is non-invasive and convenient The natural history of the condition
be adequately understood Yes Large longitudinal studies have helped our understanding of the natural history of MGUS Further studies are required to understand MGCS 9 There should be an agreed policy
whom to treat as patients Yes IMWG guidelines for MGUS 19
test should be acceptable to
should
on
The cost of case-finding should be economically balanced in relation to possible expenditure on medical care as a whole
Table 2. Interrogation of suitability of asymptomatic monoclonal gammopathy for screening using Wilson and Junger’s principles of early disease detection.
Haematologica | 108 June 2023 1481 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
Limitations to screening for monoclonal gammopathy
Despite significant advances in early intervention and risk identification, the potential adverse effects of screening for MG need to considered carefully. A consensus evidence-based treatment for MG that improves morbidity and mortality by preventing malignant progression or allowing for earlier treatment of MGCS is required. The diagnosis of a pre-malignant condition through positive MG screening in otherwise “well individuals” would inevitably create health anxiety. Both the iSTOPMM and PROMISE studies have incorporated patient questionnaires to measure the psychosocial impact of screening for MG. Thus far, the PROMISE study has shown no significant difference in cancer-related anxiety or health-related quality of life in participants who screened positive for MG,42 however much longer follow-up is required to determine the true psychological impact of screening in these patients. As with all screening programs, there is the potential of over-diagnosis,85 which leads to the possibility of overtreatment. Furthermore, MG screening would likely create significant time and cost burdens on primary care clinicians requesting and interpreting the test results, as well as specialist teams monitoring high-risk MG patients. Uncovering unexpected MG cases would increase referrals to myeloma and cancer specialist services, at a great cost to already strained health services. The resource burden to specialist teams could be offset if screening were shown to be successful in preventing cases of advanced malignant disease. Furthermore, a recent study identified that using a modified monoclonal antibody threshold of 10 g/L and an extended range of SFLC ratios (0.15-3.36) excluded 89% of MGUS but importantly still identified 99% of MM patients.86 Thus, a strategy for screening for MG that does not overload hematology referrals querying MM appears possible.
Monoclonal gammopathy: unanswered questions and future steps
Genomic studies have begun to provide explanations for the heterogeneity of MG and SMM.87 Recent advances in low-input whole-genome sequencing in MG and MM has led to the delineation of two distinct entities within asymptomatic MG: those with a low burden of myelomadefining genomic events and indolent phenotype, and those with sufficient myeloma-defining genomic events to cause malignant transformation.88 Increasing availability and use of these technologies could provide enhanced molecular inspection of the plasma- and B-cell clones in MG patients. The mechanisms that trigger an indolent phenotype MG to become a malignant phenotype and whether there are genetic drivers associated with MGCS are yet to be understood.
Fluorescence in situ hybridization panels frequently fail to identify genetic abnormalities in MG patients. Use of targeted gene panels such as the Myeloma Genome Project next-generation sequencing panel, which comprises 228 genes/exons for mutations, six regions for 40 translocations, and 56 regions for copy number abnormalities, could overcome this limitation.89 This panel can be employed in a routine diagnostic laboratory and detailed genomic characterization could serve as a potential predictor of disease progression. Recent observation of higher rates of pathological N-glycosylation noted in cold hemagglutinin disease as well as AL amyloidosis provides vital routes to develop proteomic research to better understand causality of these post-translational modifications.90
It is inescapable that a major stumbling block to introducing MG screening approaches is the lack of early intervention options that could be applied in MG to prevent progression to either MGCS or MM. If such safe and affordable interventions were available, the arguments for screening would be significantly changed. Making this a reality will require novel research focused on the biological features of MG, which are likely to be distinct from MM and which represent a potential Achilles’ heel for an MG clone. For example, one biological question yet to be addressed is: what is the repopulating cell responsible for maintaining early-stage MG? In MM, it is widely accepted that plasma cells are the propagating cells.91 However, the involvement of the B-cell hierarchy in earlier stage plasma cell dyscrasias has been under-investigated and may provide potential interventional strategies in earlier stage disease.
Monoclonal gammopathy terminology
As the genomic and biological understanding of MG progresses it is important that the field also updates the MG terminology to reflect this transition. There is increasing recognition of the need to move from “cancer burden to cancer genomics”.92 With the recent characterization of myeloma-defining genomic events using whole-genome sequencing,14 three simplified genetically defined (rather than clinically defined) myeloma categories have been proposed: MG, early MM and MM.88 Using in-depth genomic and biological characterization of monoclonal gammopathies, MGCS-defining events and features of ‘benign’ MG should be identified (Figure 1).
Conclusion
Recent research has started to unpick the ‘undetermined’ aspect of MGUS, with accumulating evidence that MG is
Haematologica | 108 June 2023 1482 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
Figure 1. Proposed schema for classification of monoclonal gammopathies. §The cell intrinsic/extrinsic factors that lead to the persistence and/or progression of plasma/B-cell clones are still unknown. ❋The events, e.g. genomic/proteomic changes, defining monoclonal gammopathy of clinical significance have not yet been defined and require further research. ✢Myelomadefining events have recently been described.88 △The interplay and relationship between benign monoclonal gammopathy, monoclonal gammopathy of clinical significance and multiple myeloma/Waldenström macroglobulinemia is not fully understood; it is not clear whether the genomic and biological drivers of these conditions are shared or distinct. MG: monoclonal gammopathy; MDE: myeloma-defining event; MGCS: monoclonal gammopathy of clinical significance; MM: multiple myeloma; WM: Waldenström macroglobulinemia.

heterogeneous and more clinically significant than initially thought.88,93 MG is perfectly poised as a condition in which early detection and intervention could make a significant impact on the morbidity and mortality of patients by preventing irreversible organ damage. Despite recent advances, such as improved risk stratification of MG patients, effective treatment of asymptomatic SMM and enhanced awareness of MGCS, further prospective data are needed before widespread screening of MG can be recommended. Results from a single randomized trial of screening for MG (iStopMM) are eagerly awaited and a large prospective observational MG study in the UK is underway (SECURE study; NCT05539079). We believe that more trials of MG screening and monitoring are warranted, as enhanced risk stratification of both malignant progression and identification of MGCS is likely to provide benefit to patients. Continual re-appraisal of the balance between risk and benefit of a targeted screening program for MG is required as the field of early intervention continues to evolve. Further research into the biology of MG as an independent entity is important to understanding MGCSdefining genomic and molecular events and will help to inform methods of effective early therapeutic intervention.
Disclosures
MD owns shares in Abingdon Health. LYC, CB, KR have no conflicts of interest to disclose.
Contributions
LYC wrote the manuscript. LYC, CB, KR, and MD developed the concept of the article, edited several revisions of the paper and approved the final manuscript.
Acknowledgments
We acknowledge Professor Rafael Fonseca and the Myeloma UK Monoclonal Gammopathy Working Group, including Dr Jenny Bird, Dr Stella Bowcock, Dr Aris Chaidos, Dr Kassim Javaid, Tom Jennis, Dr Martin Kaiser, Dr Constantinos Koshiaris, Dr Ira Laketic-Ljubojevic, Dr Charlene McShane, Dr Brian Nicholson, Dr Jennifer Pinney, Prof Guy Pratt, Dr Suzanne Renwick, Dr Judith Richardson, Prof Alex Richter, Dr Simon Stern, Mairi Whiston, and Dr Fenella Willis, for their comments on the manuscript.
Data-sharing statement
For data requests, please contact the corresponding author via email.
Haematologica | 108 June 2023 1483 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
References
1. Cancer Research UK. Myeloma incidence statistics. https://www.cancerresearchuk.org/health-professional/cancerstatistics/statistics-by-cancer-type/myeloma/incidence#ref [accessed April 30, 2022]
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7-33.
3. Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412-5417.
4. Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113(22):5418-5422.
5. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-548.
6. Kristinsson SY, Björkholm M, Andersson TM-L, et al. Patterns of survival and causes of death following a diagnosis of monoclonal gammopathy of undetermined significance: a population-based study. Haematologica. 2009;94(12):1714-1720.
7. Kyle RA, Therneau TM, Rajkumar SV, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362-1369.
8. Smith A, Howell D, Crouch S, et al. Cohort Profile: The Haematological Malignancy Research Network (HMRN): a UK population-based patient cohort. Int J Epidemiol. 2018;47(3):700-700g.
9. Kyle RA, Larson DR, Therneau TM, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018;378(3):241-249.
10. Pelzer BW, Arendt M, Moebus S, et al. Light chain monoclonal gammopathy of undetermined significance is characterized by a high disappearance rate and low risk of progression on longitudinal analysis. Ann Hematol. 2018;97(8):1463-1469.
11. Kyle RA, Dispenzieri A, Kumar S, Larson D, Therneau T, Rajkumar SV. IgM monoclonal gammopathy of undetermined significance (MGUS) and smoldering Waldenström’s macroglobulinemia (SWM). Clin Lymphoma Myeloma Leuk. 2011;11(1):74-76.
12. Hejl C, Mestiri R, Carmoi T, et al. IgE monoclonal gammopathy: a case report and literature review. Clin Biochem. 2018;51:103-109.
13. Bladé J, Kyle RA. IgD monoclonal gammopathy with long-term follow-up. Br J Haematol. 1994;88(2):395-396.
14. Oben B, Froyen G, Maclachlan KH, et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat Commun. 2021;12(1):1861.
15. Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood. 2005;106(3):812-817.
16. Gran C, Liwing J, Wagner AK, et al. Comparative evaluation of involved free light chain and monoclonal spike as markers for progression from monoclonal gammopathy of undetermined significance to multiple myeloma. Am J Hematol. 2021;96(1):23-30.
17. Turesson I, Kovalchik SA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance and risk of lymphoid and myeloid malignancies: 728 cases followed up to 30 years in Sweden. Blood. 2014;123(3):338-345.
18. Dispenzieri A, Larson DR, Rajkumar SV, et al. N-glycosylation of monoclonal light chains on routine MASS-FIX testing is a risk
factor for MGUS progression. Leukemia. 2020;34(10):2749-2753.
19. Kyle RA, Durie BGM, Rajkumar SV, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24(6):1121-1127.
20. Moreno DF, Pereira A, Tovar N, et al. Defining an ultra-low risk group in asymptomatic IgM monoclonal gammopathy. Cancers (Basel). 2021;13(9):2055.
21. Bustoros M, Sklavenitis-Pistofidis R, Kapoor P, et al. Progression risk stratification of asymptomatic Waldenström macroglobulinemia. J Clin Oncol. 2019;37(16):1403-1411.
22. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791-2796.
23. Fermand J-P, Bridoux F, Dispenzieri A, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood 2018;132(14):1478-1485.
24. Merlini G, Stone MJ. Dangerous small B-cell clones. Blood. 2006;108(8):2520-2530.
25. Gertz MA, Dispenzieri A. Systemic amyloidosis recognition, prognosis, and therapy: a systematic review. JAMA. 2020;324(1):79-89.
26. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-2654.
27. Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15(1):45-59.
28. Castillo JJ, Callander NS, Baljevic M, Sborov DW, Kumar S. The evaluation and management of monoclonal gammopathy of renal significance and monoclonal gammopathy of neurological significance. Am J Hematol. 2021;96(7):846-853.
29. Jain A, Haynes R, Kothari J, Khera A, Soares M, Ramasamy K. Pathophysiology and management of monoclonal gammopathy of renal significance. Blood Adv. 2019;3(15):2409-2423.
30. Steiner N, Göbel G, Suchecki P, Prokop W, Neuwirt H, Gunsilius E. Monoclonal gammopathy of renal significance (MGRS) increases the risk for progression to multiple myeloma: an observational study of 2935 MGUS patients. Oncotarget. 2018;9(2):2344-2356.
31. Ciocchini M, Arbelbide J, Musso CG. Monoclonal gammopathy of renal significance (MGRS): the characteristics and significance of a new meta-entity. Int Urol Nephrol. 2017;49(12):2171-2175.
32. Hounkpatin HO, Harris S, Fraser SDS, et al. Prevalence of chronic kidney disease in adults in England: comparison of nationally representative cross-sectional surveys from 2003 to 2016. BMJ Open. 2020;10(8):e038423.
33. D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol. 2017;176(5):728-742.
34. Rögnvaldsson S, Steingrímsson V, Turesson I, Björkholm M, Landgren O, Yngvi Kristinsson S. Peripheral neuropathy and monoclonal gammopathy of undetermined significance: a population-based study including 15,351 cases and 58,619 matched controls. Haematologica. 2020;105(11):2679-2681.
35. Chaudhry HM, Mauermann ML, Rajkumar SV. Monoclonal gammopathy-associated peripheral neuropathy: diagnosis and management. Mayo Clin Proc. 2017;92(5):838-850.
Haematologica | 108 June 2023 1484 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
36. Tatum AH. Experimental paraprotein neuropathy, demyelination by passive transfer of human IgM anti-myelin-associated glycoprotein. Ann Neurol. 1993;33(5):502-506.
37. Claveau J-S, Wetter DA, Kumar S. Cutaneous manifestations of monoclonal gammopathy. Blood Cancer J. 2022;12(4):58.
38. Paueksakon P, Revelo MP, Horn RG, Shappell S, Fogo AB. Monoclonal gammopathy: significance and possible causality in renal disease. Am J Kidney Dis. 2003;42(1):87-95.
39. Lamb MJ, Smith A, Painter D, et al. Health impact of monoclonal gammopathy of undetermined significance (MGUS) and monoclonal B-cell lymphocytosis (MBL): findings from a UK population-based cohort. BMJ Open. 2021;11(2):e041296.
40. Schaar CG, le Cessie S, Snijder S, et al. Long-term follow-up of a population based cohort with monoclonal proteinaemia. Br J Haematol. 2009;144(2):176-184.
41. Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564-569.
42. El-Khoury H, Lee DJ, Alberge J-B, et al. Prevalence of monoclonal gammopathies and clinical outcomes in a high-risk US population screened by mass spectrometry: a multicentre cohort study. Lancet Haematol. 2022;9(5):e340-e349.
43. Berenson JR, Anderson KC, Audell RA, et al. Monoclonal gammopathy of undetermined significance: a consensus statement. Br J Haematol. 2010;150(1):28-38.
44. Bida JP, Kyle RA, Therneau TM, et al. Disease associations with monoclonal gammopathy of undetermined significance: a population-based study of 17,398 patients. Mayo Clin Proc. 2009;84(8):685-693.
45. Melton LJ, Rajkumar SV, Khosla S, Achenbach SJ, Oberg AL, Kyle RA. Fracture risk in monoclonal gammopathy of undetermined significance. J Bone Miner Res. 2004;19(1):25-30.
46. Agarwal G, Milan C, Mohsin Z, et al. Multiple myeloma screening within a fracture liaison service (FLS). Osteoporos Int. 2022;33(4):937-941.
47. Kristinsson SY, Tang M, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance and risk of infections: a population-based study. Haematologica. 2012;97(6):854-858.
48. Kang K-W, Song JE, Lee B-H, et al. A nationwide study of patients with monoclonal gammopathy of undetermined significance with a 10-year follow-up in South Korea. Sci Rep. 2021;11(1):18449.
49. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111-121.
50. Pang L, Rajkumar SV, Kapoor P, et al. Prognosis of young patients with monoclonal gammopathy of undetermined significance (MGUS). Blood Cancer J. 2021;11(2):26.
51. Yong K, Delforge M, Driessen C, et al. Multiple myeloma: patient outcomes in real-world practice. Br J Haematol. 2016;175(2):252-264.
52. Therneau TM, Kyle RA, Melton LJ, et al. Incidence of monoclonal gammopathy of undetermined significance and estimation of duration before first clinical recognition. Mayo Clin Proc. 2012;87(11):1071-1079.
53. Graziani G, Herget GW, Ihorst G, et al. Time from first symptom onset to the final diagnosis of multiple myeloma (MM) –possible risks and future solutions: retrospective and prospective ‘Deutsche Studiengruppe MM’ (DSMM) and ‘European Myeloma Network’ (EMN) analysis. Leuk Lymphoma. 2020;61(4):875-886.
54. van de Donk NWCJ, Palumbo A, Johnsen HE, et al. The clinical relevance and management of monoclonal gammopathy of
undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica. 2014;99(6):984-996.
55. Bird J, Behrens J, Westin J, et al. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol. 2009;147(1):22-42.
56. Sigurdardottir EE, Turesson I, Lund SH, et al. The role of diagnosis and clinical follow-up of monoclonal gammopathy of undetermined significance on survival in multiple myeloma. JAMA Oncol. 2015;1(2):168-174.
57. Go RS, Gundrum JD, Neuner JM. Determining the clinical significance of monoclonal gammopathy of undetermined significance: a SEER-Medicare population analysis. Clin Lymphoma Myeloma Leuk. 2015;15(3):177-186.e4.
58. Rögnvaldsson S, Love TJ, Thorsteinsdottir S, et al. Iceland screens, treats, or prevents multiple myeloma (iStopMM): a population-based screening study for monoclonal gammopathy of undetermined significance and randomized controlled trial of follow-up strategies. Blood Cancer J. 2021;11(5):94.
59. Zhao S, Choi M, Heuck C, et al. Serial exome analysis of disease progression in premalignant gammopathies. Leukemia. 2014;28(7):1548-15452.
60. Mateos M-V, Hernández M-T, Giraldo P, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med. 2013;369(5):438-447.
61. Lonial S, Jacobus S, Fonseca R, et al. Randomized trial of lenalidomide versus observation in smoldering multiple myeloma. J Clin Oncol. 2020;38(11):1126-1137.
62. Gourd E. Early intervention effective in smouldering multiple myeloma. Lancet Oncol. 2019;20(12):e666.
63. Musto P, Engelhardt M, Caers J, et al. 2021 European Myeloma Network review and consensus statement on smoldering multiple myeloma: how to distinguish (and manage) Dr. Jekyll and Mr. Hyde. Haematologica. 2021;106(11):2799-2812.
64. Landgren CO, Chari A, Cohen YC, et al. Daratumumab monotherapy for patients with intermediate-risk or high-risk smoldering multiple myeloma: a randomized, open-label, multicenter, phase 2 study (CENTAURUS). Leukemia. 2020;34(7):1840-1852.
65. Korde N, Roschewski M, Zingone A, et al. Treatment with carfilzomib-lenalidomide-dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. 2015;1(6):746-754.
66. Perrot A, Lauwers-Cances V, Corre J, et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood. 2018;132(23):2456-2464.
67. Ho M, Patel A, Goh CY, Moscvin M, Zhang L, Bianchi G. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Leukemia. 2020;34(12):3111-3125.
68. Zavidij O, Haradhvala NJ, Mouhieddine TH, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer. 2020;1(5):493-506.
69. Bailur JK, McCachren SS, Doxie DB, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight. 2019;5(11):e127807.
70. Campbell JP, Heaney JLJ, Pandya S, et al. Response comparison of multiple myeloma and monoclonal gammopathy of undetermined significance to the same anti-myeloma therapy: a
Haematologica | 108 June 2023 1485 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
retrospective cohort study. Lancet Haematol. 2017;4(12):e584-e594.
71. Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood. 2012;119(19):4462-4466.
72. Gobbi PG, Baldini L, Broglia C, et al. Prognostic validation of the international classification of immunoglobulin M gammopathies: a survival advantage for patients with immunoglobulin M monoclonal gammopathy of undetermined significance? Clin Cancer Res. 2005;11(5):1786-1790.
73. Lunn MPT, Nobile-Orazio E. Immunotherapy for IgM anti-myelinassociated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst Rev. 2012;(5):CD002827.
74. Wilson JMG, Jungner G. Principles and practice of screening for disease. World Health Organization. Geneva. 1968.
75. Broeders M, Moss S, Nyström L, et al. The impact of mammographic screening on breast cancer mortality in Europe: a review of observational studies. J Med Screen. 2012;19(Suppl 1):14-25.
76. Logan RFA, Patnick J, Nickerson C, et al. Outcomes of the Bowel Cancer Screening Programme (BCSP) in England after the first 1 million tests. Gut. 2012;61(10):1439-1446.
77. Peto J, Gilham C, Fletcher O, Matthews FE. The cervical cancer epidemic that screening has prevented in the UK. Lancet. 2004;364(9430):249-256.
78. Richards M. The independent review of adult screening programmes in England. 2019. NHS England.
Publication reference 01089
79. Landgren O, Graubard BI, Katzmann JA, et al. Racial disparities in the prevalence of monoclonal gammopathies: a populationbased study of 12,482 persons from the National Health and Nutritional Examination Survey. Leukemia. 2014;28(7):1537-1542.
80. Vachon CM, Kyle RA, Therneau TM, et al. Increased risk of monoclonal gammopathy in first-degree relatives of patients with multiple myeloma or monoclonal gammopathy of undetermined significance. Blood. 2009;114(4):785-790.
81. Georgakopoulou R, Andrikopoulou A, Sergentanis TN, et al. Overweight/obesity and monoclonal gammopathy of undetermined significance. Clin Lymphoma Myeloma Leuk. 2021;21(6):361-367.
82. Atkin C, Reddy-Kolanu V, Drayson MT, Sapey E, Richter AG. The prevalence and significance of monoclonal gammopathy of
undetermined significance in acute medical admissions. Br J Haematol. 2020;189(6):1127-1135.
83. Turesson I. Monoclonal gammopathy of undetermined significance in medical hospital admissions - a new strategy for screening? Br J Haematol. 2020;189(6):1010-1011.
84. Kristinsson SY, Rögnvaldsson S, Thorsteinsdottir S, et al. Screening for monoclonal gammopathy of undetermined significance: a population-based randomized clinical trial. First results from the Iceland Screens, Treats, or Prevents Multiple Myeloma (iStopMM) study. Blood. 2021;138(Suppl 1):156.
85. Brodersen J, Schwartz LM, Woloshin S. Overdiagnosis: how cancer screening can turn indolent pathology into illness. APMIS. 2014;122(8):683-689.
86. Heaney JLJ, Richter A, Bowcock S, et al. Excluding myeloma diagnosis using revised thresholds for serum free light chain ratios and M-protein levels. Haematologica. 2020;105(4):e169-e171.
87. Lionetti M, da Vià MC, Albano F, Neri A, Bolli N, Musto P. Genomics of smoldering multiple myeloma: time for clinical translation of findings? Cancers (Basel) 2021;13(13):3319.
88. Landgren O. Advances in MGUS diagnosis, risk stratification, and management: introducing myeloma-defining genomic events. Hematology Am Soc Hematol Educ Program. 2021;2021(1):662-672.
89. Truger M, Hutter S, Meggendorfer M, et al. FISH and WGS in newly diagnosed and relapsed/refractory multiple myelomaWGS will affect future treatment decisions. Blood. 2021;138(Suppl 1):397.
90. Kumar S, Murray D, Dasari S, et al. Assay to rapidly screen for immunoglobulin light chain glycosylation: a potential path to earlier AL diagnosis for a subset of patients. Leukemia. 2019;33(1):254-257.
91. Johnsen HE, Bøgsted M, Schmitz A, et al. The myeloma stem cell concept, revisited: from phenomenology to operational terms. Haematologica. 2016;101(12):1451-1459.
92. Maura F, Bolli N, Rustad EH, Hultcrantz M, Munshi N, Landgren O. Moving from cancer burden to cancer genomics for smoldering myeloma: a review. JAMA Oncol. 2020;6(3):425-432.
93. Munshi NC, Jagannath S, Avet-Loiseau H. Monoclonal gammopathy may be of unpredictable significance. JAMA Oncol. 2019;5(9):1302-1303.
94. Katzmann JA. Screening panels for monoclonal gammopathies: time to change. Clin Biochem Rev. 2009;30(3):105-111.
Haematologica | 108 June 2023 1486 PERSPECTIVE ARTICLE - MG-related conditions: role of screening L.Y. Chen et al.
Aspirin in essential thrombocythemia. For whom? What formulation? What regimen?
Marco Cattaneo
Fondazione Arianna Anticoagulazione, Bologna, Italy
Abstract
Correspondence: M. Cattaneo marco.natale.cattaneo@gmail.com
Received: October 6, 2022.
Accepted: December 23. 2022.
Early view: January 12. 2023.
https://doi.org/10.3324/haematol.2022.281388
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Essential thrombocythemia (ET) is a BCR-ABL1-negative myeloproliferative neoplasm, the most common clinical manifestations of which include arterial and venous thrombosis, bleeding and vasomotor/microvascular disturbances. Low-dose (81-100 mg) aspirin once daily, which irreversibly inhibits platelet thromboxane A2 (TxA2) production by acetylating cyclo-oxygenase-1, is the recommended treatment for the control of vascular events in all ET risk categories, except patients at very low risk, who need aspirin for treatment of vasomotor/microvascular disturbances only. Simple observation should be preferred over aspirin prophylaxis in low-risk patients with platelet counts >1,000x109/L or harboring CALR mutations. Plain aspirin should be preferred over enteric coated aspirin because some ET patients display poor responsiveness (“resistance”) to the latter. When treated with a once daily aspirin regimen, adequate inhibition of platelet TxA2 production (measured as serum thromboxane B2 level) does not persist for 24 h in most patients. This phenomenon is associated with the patients’ platelet count and the number (but not the fraction) of circulating immature reticulated platelets with non-acetylated cyclo-oxygenase-1 and is therefore consequent to high platelet production (the hallmark of ET), rather than increased platelet turnover (which is normal in ET). Twice daily aspirin administration overcame this problem and proved safe in small studies. Although additional data on gastrointestinal tolerability will be useful, the twice daily regimen could already be implemented in clinical practice, considering its favorable risk/benefit profile. However, patients whose platelet count has been normalized could still be treated with the once daily regimen, because they would otherwise be unnecessarily exposed to a potential small risk of gastrointestinal discomfort.
Essential thrombocythemia in the realm of myeloproliferative neoplasms
Essential thrombocythemia (ET) belongs to the group of BCR-ABL1-negative myeloproliferative neoplasms, which also includes polycythemia vera and primary myelofibrosis. Myeloproliferative neoplasms are hematopoietic stem-cell disorders that are characterized by the presence of mutually exclusive driver mutations of the downstream kinase JAK2 (Janus kinase 2), the endoplasmic reticulum chaperone CALR (calreticulin) or the thrombopoietin receptor MPL (myeloproliferative leukemia virus oncogene), which are associated with constitutive activation of hematopoietic pathways. In ET, the most frequent mutation is JAK2V617F (present in about 55% of cases), followed by CALR mutations (about 20%) and MPL mutations (about 4%); no
known mutation is detected in about 20% of ET patients.1 Isolated thrombocytosis may be the first manifestation of polycythemia vera or primary myelofibrosis. It is indispensable to distinguish ET from pre-fibrotic primary myelofibrosis or other myeloid neoplasms, mostly by means of morphological examination of the bone marrow.1,2 Rare cases have been described of hereditary, familial thrombocythemia associated with germline mutations of JAK2, MPL or the thrombopoietin gene (THPO).3 Major criteria for a diagnosis of ET are: (i) platelet count >450x109/L, (ii) bone marrow megakaryocyte proliferation and loose clusters, (iii) not meeting World Health Organization (WHO) criteria for other myeloid neoplasms, and (iv) mutated JAK2/ CALR/MPL. Minor criteria include the presence of other clonal markers and no evidence of reactive thrombocytosis. All four major or three major plus one minor criteria are required to make the diagnosis.4
The annual incidence of ET is estimated to be between
Haematologica | 108 June 2023 1487 REVIEW ARTICLE
1.2 and 3.0 cases per 100,000,5,6 while the estimated prevalence in Western countries is 24-30 cases per 100,000.7,8 The clinical picture is highly variable, with most patients being asymptomatic at presentation. Signs and symptoms include microcirculatory and vasomotor manifestations, such as vascular headaches, dizziness, vertigo, visual disturbances, acral dysesthesia, acrocyanosis and erythromelalgia.5,9 The most common severe clinical manifestations include arterial and, more rarely, venous thromboembolic events and bleeding manifestations.5,9,10 The clinical course is usually mild, with a median survival that is not very different from normal.5,7 The 10-year incidence of transition to acute myeloid leukemia or post-ET myelofibrosis is rare (<1%).10 The main goal of treatment of ET is to alleviate the microcirculatory and vasomotor signs and symptoms and reduce the risk of thrombotic and hemorrhagic complications. Aspirin is the recommended treatment for the control of vascular events in most ET patients, although its use is still a matter of debate.11,12
Thrombosis and bleeding in essential thrombocythemia
Incidence and prevalence
Thrombosis and bleeding are among the initial manifestations of ET: a meta-analysis of published studies up to August 2018 revealed that, among 6,610 patients, the prevalence of thrombotic and bleeding events at diagnosis was 20.7% (95% confidence interval [95% CI]: 16.6-25.5) and 7.3% (95% CI: 5.3-10.0), respectively.13 In an international collaborative study of 891 patients, 109 (12%) experienced arterial (n=79) or venous (n=37) thrombosis after a median follow-up of 6.2 years.14 Venous thrombotic events may involve atypical vascular districts, such as cerebral sinuses and splanchnic veins.10 An analysis of studies published in the previous 15 years showed that the median incidences of bleeding and major bleeding events were 2.2% and 0.79% patient-years, respectively.15 Most bleeds were gastrointestinal, while the most frequent fatal bleeding was intracerebral hemorrhage.15
The occurrence of both thrombotic and bleeding events in ET highlights an apparently paradoxical coexistence of opposite clinical manifestations of abnormalities of hemostasis, which stimulated several investigations of hemostasis parameters in these patients.
Hemostasis parameters in essential thrombocythemia Global tests of primary hemostasis
As in congenital hemostatic defects,16 global tests of primary hemostasis display different sensitivities to the acquired defects of ET patients: prolongation of the bleeding time is observed in a minority of patients,17-21 while pro-
longed closure times of both PFA-100 cartridges are more frequent.22 Prolongation of the bleeding time after the oral administration of aspirin is more pronounced in ET patients than in healthy subjects,23,24 suggesting that aspirin can unmask underlying defects of primary hemostasis.
Platelet thromboxane A2 production
Some studies have found high total serum levels of thromboxane B2 (TxB2), a stable metabolite of thromboxane A2 (TxA2) in ET patients;25-27 however, these high levels were most likely a reflection of thrombocytosis, because when results were expressed relative to the platelet count, they were comparable to normal.25,27
Platelet aggregation in vitro
Some studies showed that high percentages of ET patients display spontaneous platelet aggregation,19,20,28,29 which is associated with increased risk of vascular events in the general population.30,31 Studies of in vitro agonistinduced platelet aggregation revealed a high degree of inter-individual variability and contrasting results.32 In general, results obtained by the traditional light transmission aggregometry (LTA) technique in citrate-anticoagulated platelet-rich plasma whose platelet count had been normalized to pre-defined standardized values by the addition of autologous plasma usually documented defects of agonist-induced platelet aggregation.18,19,21-23,32-34 However, the normalization of platelet count in platelet-rich plasma by autologous plasma is now contraindicated,35 because LTA results are not affected by the sample platelet count36,3 and dilution of platelet-rich plasma by autologous plasma inhibits platelet aggregation.33,36 In contrast to LTA studies, experiments performed in whole blood by multiple electrode aggregometry showed normal or even increased platelet aggregation,28,29,32,34,38,39 consistent with the demonstration that there is a strong positive correlation between the sample platelet count and platelet aggregation measured by multiple electrode aggregometry.37 A study in which platelet aggregation was tested in parallel in the same patients under five different experimental conditions confirmed that agonist-induced aggregation of ET platelets is normal when confounders do not influence the results.34 The only agonist that consistently did not induce normal aggregation of ET platelets was epinephrine,1719,25,34,40 due to decreased expression of α 2-adrenergic receptors and/or abnormalities of the transduction pathway.17,40 JAK2V617F did not affect the in vitro aggregation response of ET platelets to agonists.34,41
Platelet granule content
ET platelets often display acquired storage pool deficiency, characterized by deficiency of the constituents of platelet granules.17,23,25,34,42 Although the presence of an abnormal clone with defective d-granules has been hypo-
Haematologica | 108 June 2023 1488 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
thesized,25 “exhaustion” of platelet granules by in vivo platelet activation and participation in microthrombi is the prevalent pathogenic mechanism.
Markers of in vivo activation of hemostasis
In vivo activation of hemostasis in ET is documented by the presence in peripheral blood of platelets expressing activation markers, platelet/leukocytes hetero-aggregates, high plasma levels of platelet granule constituents and, less frequently, markers of activation of coagulation.32,34,43-45 The levels of urinary 11-dehydro-TxB2 were higher in 40 ET patients than in 26 healthy sex- and age-matched controls and were decreased by aspirin, suggesting that this urinary metabolite is largely derived from in vivo platelet activation.46 In vivo platelet activation causes the release of procoagulant platelet-derived microparticles47 and contributes to dysfunction of von Willebrand factor (VWF).48
Plasma von Willebrand factor
Acquired VWF deficiency with loss of VWF high molecular weight multimers was first described in seven patients,49 associated with increased proteolysis50 and high platelet count51 and improved after reduction of platelet counts by chemotherapy.49 These findings were later confirmed by other investigators.10,52 ET patients who showed an excessive prolongation of bleeding time by aspirin had significantly decreased levels of large VWF multimers in plasma.24
Platelet survival and turnover
Platelet turnover increases under conditions in which decreased platelet survival caused by heightened peripheral platelet consumption is associated with increased compensatory platelet production. The most commonly used method to evaluate platelet turnover is based on the measurement of reticulated platelets, i.e., newly formed platelets retaining some RNA. An increased percentage of circulating reticulated platelets is suggestive of increased
turnover, while an increased number but normal percentage is simply a reflection of high platelet count. The number of reticulated platelets was increased, but its percentage was normal in ET patients without vascular disorders (Table 1).39,53,54 In contrast, ET patients with vascular disorders display decreased platelet survival10,55 and an increased percentage of circulating reticulated platelets.56,57
Aspirin treatment not only cured the signs and symptoms of erythromelalgia,10 but also normalized platelet survival and the percentage of reticulated platelets.10,56 Therefore, platelet turnover is normal in ET patients without vascular disorders, while an increased count with a normal percentage of reticulated platelets is simply a reflection of increased platelet production.
Potential mechanisms of bleeding and thrombosis in essential thrombocythemia
Platelets of ET patients interact with activated leukocytes,42,45,47,58-60 to form thrombi, especially in the microcirculation. The release of procoagulant platelet-derived microparticles contributes to the dissemination of the thrombogenic stimuli. Platelets that deaggregate from these thrombi and return to the circulation are “exhausted”, displaying acquired storage pool deficiency17,23,25,34,42 and contributing to increase the bleeding risk together with acquired von Willebrand disease in patients with extremely high platelet counts. The thrombotic risk is increased by the presence of JAK2V617F , 62,63 which plays a role in neutrophil extracellular trap formation.64 In contrast, the risk of thrombosis is low in patients with CALR-mutated ET, which affects relatively young individuals and is characterized by markedly elevated platelet count.65
Risk factors for bleeding and thrombosis in patients with essential thrombocythemia
Clinical risk factors for bleeding are not particularly useful to define the bleeding risk in ET patients. Duration of dis-
Results are expressed as mean±standard deviation or median (interquartile range), as in the original publications. The data refer to patients with essential thrombocythemia (ET) without thrombotic events. In the study by Rinder et al., seven additional ET patients with thrombotic events had increased reticulated platelet count (94±33x109/L) and reticulated platelet fraction (13.8±5.1%),56 suggesting that increased platelet turnover is associated with thrombosis and not with ET (see text).
Study Reticulated platelet count, x109/L Reticulated platelet fraction, % Healthy subjects ET patients Healthy subjects ET patients Kienast et al.53 21.3±7.9 75.5 8.6±2.8 8.2 N=50 N=2 N=50 N=2 Rinder et al.56 6±6 36±14 3.4±1.3 4.2±1.9 N=83 N=5 N=83 N=5 Pedersen et al.39 6.9 (5.5-10.3) 12.3 (9.8-18.7) 2.6 (2.1-3.9) 2.8 (2.3-3.4) N=24 N=24 N=24 N=24 Scavone et al.54 19.4 (17.1-25.2) 34.3 (26.9-50.7) 10.6 (7.7-12.8) 9.1 (7.5-12.3) N=8 N=15 N=8 N=15
Table 1. Reticulated platelet count and reticulated platelet fraction in healthy subjects and patients with essential thrombocythemia.
Haematologica | 108 June 2023 1489 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
ease, hypertension, bleeding history, splenomegaly and male sex were identified as bleeding risk factors in some studies.15 In a retrospective analysis of 891 ET patients, previous hemorrhage and aspirin use were independently associated with bleeding risk.66 The bleeding risk associated with aspirin use was found to be particularly relevant for patients harboring CALR mutations67 or with platelet counts >1,000x109/L.66,68 The combination of platelet count >1,000x109/L, leukocytosis and acquired von Willebrand disease was identified as a biological risk factor for bleeding.15
Risk stratification for thrombosis in ET patients has been mostly based on the presence of conventional risk factors, such as age >60 years and a positive history of thrombosis.5 The revised International Prognostic Score for Thrombosis in ET (IPSET-thrombosis) model also considered the presence of a JAK2 mutation and of additional cardiovascular risk factors to stratify ET patients into four categories of thrombosis risk:69,70 (i) very low risk: age ≤60 years, absent JAK2 mutation, no prior history of thrombosis; (ii) low risk: age ≤60 years, JAK2 mutation, no prior history of thrombosis; (iii) intermediate risk: age >60 years, absent JAK2 mutation, no prior history of thrombosis; and (iv) high risk: history of thrombosis at any age or age >60 years with JAK2 mutation (Table 2). The revised IPSETthrombosis model was validated in 1,381 patients and found to be a better fit than the earlier IPSET-thrombosis score.71 A recent retrospective real-world analysis of Medicare patients (aged ≥65 years) newly diagnosed with intermediate/high-risk ET revealed that the mortality risk
during a 25.5-month follow-up was significantly higher among patients who experienced a thrombotic event.72
Prevention of thrombosis in essential thrombocythemia patients
Prevention of recurrent thrombosis in ET patients must follow the general guidelines for secondary prevention in the general population, including antiplatelet agents and anticoagulant drugs, depending on the clinical setting. Primary prevention of thrombosis is based on the prophylactic use of low-dose aspirin (81-100 mg daily) and cytoreduction. Aspirin is recommended for all risk categories, with the exception of patients at very low risk, who should only be given aspirin for treatment of vasomotor/microvascular disturbances;70 cytoreduction with hydroxyurea or, alternatively, with peginterferon α 2a or anagrelide is usually restricted to high-risk patients (Table 2).70 Cytoreductive treatment should, however, also be considered in other risk categories, depending on the presence of additional conditions, such as von Willebrand disease, extreme thrombocytosis and/or leukocytosis, splenomegaly, vasomotor/microvascular disturbances or other disease-related symptoms not responsive to aspirin. The efficacy of aspirin in controlling vasomotor/microvascular disturbances is well recognized, despite the lack of controlled trials, thanks to the dramatic improvement of patients’ signs and symptoms in response to aspirin treatment.10,52,73,74 One study elegantly documented the tem-
α2a
Risk category Patients’ characteristics Rate of thrombosis Management Without CV risk factors With CV risk factors Very low Age ≤60 years No JAK2 mutation No history of thrombosis 0.44%/year 1.05%/year Manage CV risk factors
disturbances) Low Age ≤60 years JAK2 mutation No history of thrombosis 1.59%/year 2.57%/year Manage CV risk factors Aspirin (81-100 mg) Intermediate Age >60 years 1.44%/year 1.64%/year Manage CV risk factors No JAK2 mutation Aspirin (81-100 mg) No history of thrombosis High Any age + History of thrombosis Age >60 years + JAK2 mutation 2.36%/year 4.17%/year Manage CV risk factors Aspirin (81-100 mg)
therapy:
Hydroxyurea
Anagrelide
No aspirin (aspirin 81-100 mg for patients with vasomotor/microvascular
Cytoreductive
Preferred:
Other recommended regimens: Peginterferon
CV: cardiovascular. Haematologica | 108 June 2023 1490 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
Table 2. National Comprehensive Cancer Network (NCCN) Guidelines (version 3.2022) for risk stratification and management of patients with essential thrombocythemia.
poral association between increased urinary levels of TxA2 metabolites and the development of erythromelalgia, which were both dramatically inhibited by the administration of low-dose aspirin.75
The efficacy of aspirin in primary prevention of major thrombotic events in ET has not been documented by randomized controlled trials. The potential benefit of aspirin is inferred from analogies with its use in polycythemia vera, in which low-dose aspirin has been shown to cause an approximately 60% reduction in the risk of the combined endpoint of nonfatal myocardial infarction, nonfatal stroke or death from cardiovascular causes and of venous thromboembolism, without increasing the risk of bleeding complications.76 The indication for the use of aspirin also comes from observational, retrospective studies. Alvarez-Larrán et al. showed that, among ET patients <60 years of age and without a positive history of thrombosis, aspirin reduced the incidence of venous thromboembolism in carriers of JAK2V617F and of arterial thrombosis in patients with cardiovascular risk factors, while it was ineffective in the remaining patients and increased the incidence of bleeding in those with a platelet count >1,000x109/L.77 Another study on the same type of ET patients confirmed the safety and protective effect of aspirin against thrombosis in JAK2V617F carriers, but not in CALR mutation carriers, in whom aspirin was not protective and increased the incidence of bleeding.67 A recent consensus of experts on the management of CALR-mutated ET recommends a pure observational approach over aspirin prophylaxis in asymptomatic low-risk ET patients without cardiovascular risk factors, while cytoreduction should be preferred over aspirin for low-risk symptomatic patients with a platelet count of 1,000-1,500x109/L.78 An observational study including high-risk ET patients >60 years old showed that the combination of aspirin plus cytoreductive therapy was superior to cytoreductive therapy alone in the primary prevention of thrombosis.79
Overall, protection from thrombosis by aspirin in ET appeared modest in a systematic review of 24 observational studies including 6,153 ET patients, which showed an estimated 26% reduction of thrombotic events from antiplatelet therapy (aspirin in 80% of patients),80 lower than that observed in polycythemia vera.76 Although this observation may also have alternative interpretations, it raised the question of whether the inhibition of TxA2 biosynthesis by aspirin is inadequate in all ET patients, because inadequate inhibition of TxA2 biosynthesis by aspirin is associated with insufficient antithrombotic efficacy.81-83
Aspirin as an antiplatelet and antithrombotic drug
Aspirin acetylates a serine residue at position 529 of cyclo-oxygenase-1 (COX-1), thus irreversibly inhibiting its
metabolic pathway, which is responsible for the production of the platelet agonist and vasoconstrictive molecule TxA2.83 Virtually complete inhibition of TxA2 synthesis throughout the 24-hour interval between doses is necessary to prevent thrombotic events.83 Aspirin is widely used as an antithrombotic drug for the treatment of acute coronary syndromes and cerebrovascular accidents and for their secondary prevention.83 Although the net clinical benefit of aspirin in primary prevention of coronary and cerebrovascular disorders in the general population is unclear,83,84 it is well established in patients with polycythemia vera.76 The very good antithrombotic efficacy of aspirin, despite its very selective pharmacodynamics, is explained by the fact that TxA2 contributes to the amplification of platelet activation by almost any platelet agonist and is essential for the full aggregation response of platelets.85
Inter-individual variability of pharmacological response to aspirin (“aspirin resistance”)
At the beginning of the 21st century, several studies documented a high prevalence of poor inhibition of platelet function by aspirin (defined in some reports as “aspirin resistance”) in treated patients.86 However, a careful analysis of the published studies revealed flaws in the evaluation of the pharmacological response to aspirin, which was studied by LTA or other non-specific tests of platelet function, such as the PFA-100.86,87 LTA is sensitive to several variables and should be performed only in specialized laboratories by dedicated personnel, following standardized procedures.35,85 Although TxA2 contributes to the final extent of platelet aggregation,85 aspirin cannot inhibit the initial response to the platelet agonists that are used in LTA studies which, therefore, lack the necessary specificity for the inhibitory effects of the drug.85,86 Even when arachidonic acid, the direct precursor of TxA2, is used as a platelet agonist, the results obtained with this technique may overestimate the prevalence of poor responders to aspirin.85 Methods that measure the capacity of platelets to synthesize TxA2 directly are preferable. Of these, measurement of the urinary levels of the TxB2 metabolite is not highly specific for platelet COX-1, because about 30% of it (or more in pathological conditions) derives from extra-platelet sources.81 In contrast, serum TxB2 reflects the total capacity of platelets to synthesize TxA2 and is highly specific, because the contribution of other blood cells to its synthesis is marginal.83,85 When the response to aspirin was correctly tested by measuring serum TxB2 levels, the frequency of poor responders was very low, suggesting that monitoring aspirin treatment with laboratory tests should not be implemented in the clinical setting, but limited to research studies.87 Some studies showed that poor responsiveness to aspirin was relatively more frequent in users of enteric coated (EC) aspirin.
Haematologica | 108 June 2023 1491 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
Plain aspirin versus enteric coated aspirin
EC aspirin was developed with the aim of reducing the incidence of gastrointestinal discomfort, mucosal erosions/ulcerations and bleeding, which are common complications of chronic treatment with aspirin.83 While plain aspirin is absorbed in the stomach, EC aspirin is absorbed in the small intestine. Compared with plain aspirin, the pharmacokinetics of EC aspirin are less favorable: the time to peak maximal concentration (Tmax) is higher, while maximal concentration (Cmax) and area under the curve (AUC) are lower.52,84 In addition, the ability of EC aspirin to inhibit platelet TxA2 production is lower,54,85-87 especially in subjects with high body weight88-91 or with type 2 diabetes mellitus.92 Such inferiority in the pharmacological efficacy of EC aspirin seems to have a clinical impact, as a meta-analysis of seven randomized controlled trials of low-dose aspirin in primary cardiovascular prevention showed that the clinical efficacy of aspirin decreased with increasing body weight, particularly in subjects treated with EC aspirin.93
Several studies showed that EC aspirin is not safer than plain aspirin, thus disappointing the expectations that fostered its development. Endoscopic studies in asymptomatic aspirin-treated subjects showed that, compared with plain aspirin, the administration of EC aspirin is associated with fewer gastric mucosal lesions94 but with more frequent lesions of the small bowel mucosa,95 thus suggesting that asymptomatic gastrointestinal mucosal lesions are caused by topical effects of aspirin in the region of its absorption. Most importantly, several studies failed to provide data supporting the clinical benefit of EC aspirin in terms of prevention of gastrointestinal bleeding and ulcers,96 which are likely caused by the systemic effects of the drug. Therefore, plain aspirin has a more favorable pharmacological profile than EC aspirin and, in my opinion, should be preferred over EC aspirin for cardiovascular prevention in all patients at risk.
Inadequate inhibition of TxA2 biosynthesis by aspirin (“aspirin resistance”) in essential thrombocythemia
In the last decade, some well-conducted studies evaluated the ability of aspirin to inhibit TxA2 biosynthesis in ET patients. The published studies can be divided into two categories: the first category evaluated the pharmacological response to aspirin by measuring TxB2 production serially at 0.5-8.0 hours after aspirin ingestion, while the second category measured TxB2 production the day after the last ingestion of aspirin, thus really exploring the recovery of the ability of platelets to synthesize TxA2.
Evaluation of the pharmacological response to aspirin (“aspirin resistance”)
A study of aspirin pharmacokinetics and pharmacodynamics and of the potential mechanisms causing poor
pharmacological responsiveness to aspirin in ET patients was published by Scavone et al. in 2020.54 Seventeen ET patients on chronic treatment with 100 mg EC aspirin once daily and ten healthy subjects on 100 mg EC aspirin once daily for 7 days were enrolled. Blood samples were collected before the morning administration of aspirin (exactly 24 hours after the last dose) and between 2 and 8 hours after the morning dose. Based on their high serum TxB2 levels 6 hours after dosing (when the nadir values in healthy subjects were observed), six patients were identified as poor responders (Figure 1). All of them had plasma levels of aspirin and salicylic acid that were much lower than those of controls and ET good responders (Figure 1). Their plasma and whole blood activity of esterases were normal, thus ruling out the possibility that poor aspirin response was attributable to accelerated de-acetylation of the drug. When experiments were repeated in the same subjects using 100 mg plain aspirin instead of EC aspirin, all studied parameters were normal in all ET patients, thus suggesting that some ET patients are “resistant” to EC aspirin, but not to plain aspirin. Similar results had previously been obtained in type 2 diabetic patients92 and in subjects with high body weight.89-91
Evaluation of the recovery of the ability of platelets to synthesize TxB2 after aspirin administration
In a study of 60 healthy subjects, it was shown that serum TxB2 levels measured 24 hours after the ingestion of one 325 mg aspirin tablet were highest in subjects displaying the highest percentage of reticulated platelets.97 The ability of reticulated platelets to form TxB2 was inhibited in vitro by a COX-1 antagonist and, albeit less efficiently, by a COX-2 inhibitor, suggesting that newly formed platelets synthesize TxB2 through the action of uninhibited COX-1 and, partially, of COX-2,97 which is present in immature, but not in mature platelets.27
A similar study in 41 ET patients and 24 healthy controls treated with EC aspirin (100 mg once daily) revealed that urinary 11-dehydro-TxB2 and total serum TxB2 levels, measured the day after the last ingestion of aspirin, were higher in ET patients than in controls.98 Both metabolites were partially (about 25%) reduced by the in vivo administration of the COX-2 inhibitor etoricoxib on top of aspirin for 7 days. Similarly, in vitro addition of the COX-2 inhibitor NS-398 (1 m mol/L) reduced serum TxB2 levels by about 30%, which, in contrast, were completely inhibited by 5 0 mmol/L aspirin. Platelet COX-2 expression was increased in ET patients and correlated with circulating reticulated platelets. Therefore, this study suggested that COX-1 and, partly, COX-2 in newly formed, non-acetylated platelets are responsible for the observed high levels of TxA2 metabolites the day after aspirin ingestion by ET patients.98
The same group of investigators later showed in the same
Haematologica | 108 June 2023 1492 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
Figure 1. Variations in serum thromboxane B2 levels and plasma concentrations of acetylsalicylic acid following the oral administration of 100 mg enteric coated aspirin or plain aspirin to healthy subjects and patients with essential thrombocythemia. Study subjects were on treatment with once daily 100 mg enteric coated (EC) aspirin tablets (left graphs) or plain aspirin tablets (right graphs) for ≥7 days. Measurements were performed on serum or plasma samples obtained before (time 0, which corresponds to 24 hours after the previous aspirin ingestion) and at the indicated time points after the witnessed oral administration of the drug in the morning of the experiment. Open squares, healthy subjects; open circles, ET patients who were poor responders to EC aspirin (ET-PR); closed circles, ET patients who were good responders to EC aspirin (ET-R). Each point in the graphs refers to the median value obtained in ten healthy subjects, six ET-PR and ten ET-R. Interquartile ranges [IQR] are not indicated for the sake of better legibility of the graphs. The tables under the graphs indicate the median [IQR] of maximal plasma aspirin concentration (Cmax) and of the area under the curve (AUC). TxB2: thromboxane B2; ASA: acetylsalicylic acid (aspirin); HS: healthy subjects. Data were analyzed by Kruskal Wallis analysis of variance. Adapted from Scavone et al. 52
41 EC aspirin-treated ET patients that there was a good correlation between the total serum TxB2 levels measured 24 hours after the last 100 mg dose of EC aspirin and the reticulated platelet count, while the correlation with reticulated platelet fraction was much weaker (Table 3).99 In addition, in a randomized cross-over study, the authors evaluated the effects of different aspirin regimens in 21 ET patients with high total serum TxB2 levels (≥4 ng/mL) 24 hours after the administration of aspirin: EC aspirin 100 mg twice daily, EC aspirin 200 mg once daily, or plain aspirin 100 mg once daily. Compared with EC aspirin 100 mg once daily, EC aspirin 100 mg twice daily reduced 24-hour total serum TxB2 by 88%, EC aspirin 200 mg once daily by 39%, while plain aspirin 100 mg once daily did not have a statistically significant effect. The authors concluded that
abnormal megakaryopoiesis accounts for the shorter-lasting inhibitory effect of aspirin in ET patients, which can be rescued by increasing the frequency of aspirin doses.99 The already mentioned study by Scavone et al. confirmed that total serum TxB2 levels 24 hours after EC aspirin (100 mg once daily) were higher in ET patients than in controls.54 A good correlation was found between the number of reticulated platelets and the 24-hour total serum TxB2 levels. However, there was no correlation between the percentage of reticulated platelets and total serum TxB2 levels, indicating that high reticulated platelet count in ET is a reflection of increased platelet production (the hallmark of ET), rather than increased platelet turnover as has been suggested (Table 3).10,100-103 Consistent with this interpretation was the finding that the platelet count cor-

Haematologica | 108 June 2023 1493 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
Table 3. Coefficients of correlation between platelet count, reticulated platelet count and fraction with serum thromboxane B2 levels 24 hours after the oral administration of aspirin to patients with essential thrombocythemia.
*The mild statistically significant correlation with reticulated platelet fraction suggests that some patients had increased platelet turnover, in addition to the predominant increase in platelet production. Indeed, 12 (29.3%) of the 41 patients enrolled in the study by Pascale et al. had had previous thrombotic events, which are associated with increased platelet turnover in ET patients.56,57 na: not available.
related with the 24-hour serum TxB2 levels to exactly the same extent as the reticulated platelet count (Table 3) and that no statistically significant correlation was observed between reticulated platelet count and serum TxB2 levels expressed relative to the sample platelet count (ng/108 platelets). The 24-hour total serum TxB2 levels were also high after treatment of the same patients with plain aspirin (albeit lower than 24 hours after EC aspirin), in contrast with the low levels (comparable to those in healthy controls) measured shortly after drug administration. This finding, which replicates the results by Pascale et al., 99 is not surprising, because the initial response to aspirin cannot influence the rate of platelet production. Finally, twice daily administration of 100 mg aspirin maintained the serum TxB2 levels low also in the pre-dose (after 12 hours) samples, as already shown by Pascale et al. 99
Aspirin Regimens in Essential Thrombocythemia (ARES) is an ongoing parallel-arm, placebo-controlled, randomized phase II trial in 300 ET patients. It is testing the effects of 100 mg EC aspirin twice or three times daily, compared to the usual once daily dose, on the inhibition of platelet TxA2 and vascular prostacyclin production.102 The first phase of the trial, which was completed in 245 ET patients, showed that the twice daily regimen was superior in inhibiting TxA2 production, while the vascular production of prostacyclin was reduced by 35% in both arms. Administering aspirin three times daily did not further reduce TxA2 production and was associated with more gastrointestinal discomfort.104
Two mechanisms of inadequate aspirin efficacy in patients with essential thrombocythemia
To summarize, it appears that two independent mechanisms are responsible for inadequate pharmacological effects of aspirin in ET (Figure 2): poor drug absorption and increased recovery of the capacity to synthesize high levels of TxA2. Poor drug absorption was observed when EC aspirin was used and would be easily overcome by using plain aspirin, which is readily absorbed with negligible inter-individual variability.54 Increased recovery of the
capacity to synthesize high levels of TxA2 is caused by increased platelet production, rather than increased turnover, and may be overcome by administering aspirin twice daily instead of once daily.54,99-101
Conclusions and suggestions
Low-dose aspirin is the recommended treatment for the control of vascular events in all risk categories of ET patients (Table 2), with the exception of patients at very low risk, who should be given aspirin for treatment of vasomotor/microvascular disturbances only. A simple observational approach should be preferred over aspirin prophylaxis in low-risk patients with platelet counts >1,000x109/L or harboring CALR mutations. Cytoreduction should be preferred over aspirin prophylaxis for high-risk or symptomatic patients with platelet counts >1,000/109/L. Plain aspirin should be preferred over EC aspirin because some ET patients display poor responsiveness to the latter,54 similarly to patients with type 2 diabetes mellitus and subjects weighing more than 70 Kg.89-92 Although the clinical efficacy of the two aspirin formulations has not been compared in randomized controlled trials, the more efficient inhibition of TxA2 production by plain aspirin would predict a more efficient protection from thrombosis.81-83 On the other hand, comparisons of the gastrointestinal toxicity of the two formulations have been performed in many studies, which indicated their equivalence, thus crippling the rationale for the use of EC aspirin in clinical practice.
When treated with the once daily aspirin regimen, most ET patients do not display virtually complete inhibition of platelet TxA2 production persisting for 24 hours, which is necessary for the prevention of thrombosis.83,104 This phenomenon is attributable to the increased daily platelet production, which causes the presence of a high number of immature platelets with non-acetylated COX-1 in the circulation. Several studies showed that twice daily aspirin administration overcomes this problem.54,99-103,104 The results of these studies are certainly very important and,
Independent
Pascale
al.99 Scavone
54 Coefficient of correlation (R) P Coefficient of correlation (R) P Reticulated platelet count 0.61 <0.001 0.61 0.0018 Platelet count na na 0.62 0.0018 Reticulated platelet fraction 0.34* 0.03 0.03 0.88
variable
et
et al.
Haematologica | 108 June 2023 1494 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
although additional long-term safety data may be necessary to evaluate the incidence of dose-related gastrointestinal side effects,total serum TxB2 levels could already change our clinical practice. Safety data on bleeding complications are not strictly necessary, as they would hardly be different from results of randomized controlled trials and real-world data obtained in millions of non-ET subjects treated with once daily aspirin causing virtually com-
plete inhibition of TxA2 synthesis throughout the 24-hour dosing interval.83,104 In addition, phase III randomized controlled trials comparing the safety and efficacy of the two aspirin dose regimens in ET would be practically impossible to organize, because of the very high number of these relatively rare patients who should be enrolled. Concerns about acquired VWF defects should influence the decision of whether to treat or not treat ET patients with aspirin:

Figure 2.
representation of the mean extent of inhibition of platelet thromboxane A2 production over time in healthy subjects and patients with essential thrombocythemia treated with different regimens of enteric coated aspirin or plain aspirin. Oval green symbols represent resting, non-acetylated platelets; oval red symbols inside the green ovals represent the mean percent inhibition of platelet thromboxane A2 production by aspirin. Mean percent inhibition after enteric coated (EC) aspirin is lower than that after plain aspirin in patients with essential thrombocythemia (ET) at each time point because some ET patients are not responsive to EC aspirin. The mean percent inhibition of platelets from ET patients at 24 hours after once daily (od) administration of aspirin is significantly decreased compared to that at earlier time points, because the recovery of the ability to produce thromboxane A2 by newly formed non-acetylated platelets at 24 hours is significantly increased compared to normal, due to the increased daily production of platelets in ET. The mean percent inhibition of platelets from healthy controls at 24 hours after once daily administration of aspirin is negligibly decreased compared to that at earlier time points, because some newly formed platelets are formed by megakaryocytes that have been acetylated by aspirin in the bone marrow. Twice daily (bid) administration of any formulation of aspirin to ET patients increases the percent platelet inhibition at the 24-hour time-point, compared to once daily aspirin, which is comparable to that at the 12-hour time-point because the number of newly produced platelets by megakaryocytes at 12 hours is 50% lower than after 24 hours. The indicated percentages of thromboxane A2 inhibition do not correspond exactly to actual measurements because available data from corresponding studies are not sufficient to allow reporting of accurate numbers; differences have been amplified for better clarity of this visual representation.
Haematologica | 108 June 2023 1495 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
Schematic
when the decision to use aspirin is taken, the most efficient dose regimen must be selected, because undertreatment would still carry the risk of side effects, while likely failing to be clinically effective. On the other hand, it is reasonable to predict that not all ET patients should be treated with twice daily aspirin.54 The demonstration that recovery of the ability to produce high levels of TxA2 after aspirin ingestion in ET is a function of the patient’s platelet count54 provided evidence that, for instance, ET patients whose platelet count has been normalized by treatment do not need the twice daily regimen, which would expose them unnecessarily to a potential increased risk of gastrointestinal side effects. In contrast, the suggestion to use aspirin twice daily
References
1. Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95(12):1599-1613.
2. Barosi G. Essential thrombocythemia vs. early/prefibrotic myelofibrosis: why does it matter. Best Pract Res Clin Haematol. 2014;27(2):129-140.
3. McMullin MF. Diagnostic workflow for hereditary erythrocytosis and thrombocytosis. Hematology Am Soc Hematol Educ Program. 2019;2019(1):391-396.
4. Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200-1228.
5. Tefferi A, Pardanani A. Essential thrombocythemia. N Engl J Med. 2019;381(22):2135-2144.
6. Verstovsek S, Yu J, Scherber RM, et al. Changes in the incidence and overall survival of patients with myeloproliferative neoplasms between 2002 and 2016 in the United States. Leuk Lymphoma. 2022;63(3):694-702.
7. Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32(3):171-173.
8. Ma X, Vanasse G, Cartmel B, Wang Y, Selinger HA. Prevalence of polycythemia vera and essential thrombocythemia. Am J Hematol. 2008;83(5):359-362.
9. Michiels JJ. Aspirin cures erythromelalgia and cerebrovascular disturbances in JAK2-thrombocythemia through platelet cyclooxygenase inhibition. World J Hematol. 2017;6(3):32-54.
10. Barbui T, Finazzi G, Tefferi A. Thrombocytosis and essential Thrombocythemia. In: Michelson AD, Cattaneo M, Frelinger AL, Newman PJ, editors. Platelets, 4th ed. Cambridge (MA): Academic Press. 2019. p. 863-876.
11. Alberio L. Do we need antiplatelet therapy in thrombocytosis? Pro. Diagnostic and pathophysiologic considerations for a treatment choice. Hamostaseologie. 2016;36(4):227-240.
12. Scharf RE. Do we need antiplatelet therapy in thrombocytosis? Contra. Proposal for an individualized risk-adapted treatment. Hamostaseologie. 2016;36(4):241-260.
13. Rungjirajittranon T, Owattanapanich W, Ungprasert P, Siritanaratkul N, Ruchutrakool T. A systematic review and metaanalysis of the prevalence of thrombosis and bleeding at diagnosis of Philadelphia-negative myeloproliferative neoplasms. BMC Cancer. 2019;19(1):184.
only in ET patients with a particularly high risk of thrombosis due to the coexistence of cardiovascular risk factors or evidence of treatment failure10,103 is, in my opinion, questionable, because all patients with an indication for drug treatment should be given an effective drug regimen, independently of the severity of their condition.
Disclosures
No conflicts of interest to disclose.
Data-sharing statement
Data and protocols can be requested by sending an email to the author.
14. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857-5859.
15. Nicol C, Lacut K, Pan-Petesch B, Lippert E, Ianotto JC. Hemorrhage in essential thrombocythemia or polycythemia vera: epidemiology, location, risk factors, and lessons learned from the literature. Thromb Haemost. 2021;121(5):553–564.
16. Podda GM, Bucciarelli P, Lussana F, Lecchi A, Cattaneo M. Usefulness of PFA-100 testing in the diagnostic screening of patients with suspected abnormalities of hemostasis: comparison with the bleeding time. J Thromb Haemost. 2007;5(12):2393-2398.
17. Swart SS, Pearson D, Wood JK, Barnett DB. Functional significance of the platelet alpha2-adrenoceptor: studies in patients with myeloproliferative disorders. Thromb Res. 1984;33(5):531-541.
18. Tobelem G. Essential thrombocythaemia. Baillieres Clin Haematol. 1989;2(3):719-728.
19. Hehlmann R, Jahn M, Baumann B, Köpcke W. Essential thrombocythemia. Clinical characteristics and course of 61 cases. Cancer. 1988;61(12):2487-2496.
20. Hattori A, Nagayama R, Kishi K, et al. Primary thrombocythemia in Japan: a survey of 225 patients. Leuk Lymphoma. 1991;4(3):177-186.
21. Finazzi G, Budde U, Michiels JJ. Bleeding time and platelet function in essential thrombocythemia and other myeloproliferative syndromes. Leuk Lymphoma. 1996;22(Suppl 1):71-78.
22. Cesar JM, de Miguel D, García Avello A, Burgaleta C. Platelet dysfunction in primary thrombocythemia using the platelet function analyzer, PFA-100. Am J Clin Pathol. 2005;123(5):772-777.
23. Barbui T, Buelli M, Cortelazzo S, Viero P, De Gaetano G. Aspirin and risk of bleeding in patients with thrombocythemia. Am J Med. 1987;83(2):265-268.
24. van Genderen PJ, van Vliet HH, Prins FJ, et al. Excessive prolongation of the bleeding time by aspirin in essential thrombocythemia is related to a decrease of large von Willebrand factor multimers in plasma. Ann Hematol. 1997;75(5-6):215-220.
25. Pareti FI, Gugliotta L, Mannucci L, Guarini A, Mannucci PM. Biochemical and metabolic aspects of platelet dysfunction in
Haematologica | 108 June 2023 1496 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
chronic myeloproliferative disorders. Thromb Haemost. 1982;47(2):84-89.
26. Zahavi J, Zahavi M, Firsteter E, Frish B, Turleanu R, Rachmani R. An abnormal pattern of multiple platelet function abnormalities and increased thromboxane generation in patients with primary thrombocytosis and thrombotic complications. Eur J Haematol. 1991;47(5):326-332.
27. Rocca B, Secchiero P, Ciabattoni G, et al. Cyclooxygenase-2 expression is induced during human megakaryopoiesis and characterizes newly formed platelets. Proc Natl Acad Sci U S A. 2002;99(11):7634-7639.
28. Balduini CL, Bertolino G, Noris P, Piletta GC. Platelet aggregation in platelet-rich plasma and whole blood in 120 patients with myeloproliferative disorders. Am J Clin Pathol. 1991;95(1):82-86.
29. Manoharan A, Gemmell R, Brighton T, Dunkley S, Lopez K, Kyle P. Thrombosis and bleeding in myeloproliferative disorders: identification of at-risk patients with whole blood platelet aggregation studies. Br J Haematol. 1999;105(3):618-625.
30. Trip MD, Cats VM, van Capelle FJ, Vreeken J. Platelet hyperreactivity and prognosis in survivors of myocardial infarction. N Engl J Med. 1990;322(22):1549-1554.
31. Breddin HK, Lippold R, Bittner M, Kirchmaier CM, Krzywanek HJ, Michaelis J. Spontaneous platelet aggregation as a predictive risk factor for vascular occlusions in healthy volunteers? Results of the HAPARG study. Haemostatic parameters as risk factors in healthy volunteers. Atherosclerosis. 1999;144(1):211-219
32. Kvernberg J, Grove EL, Ommen HB, Hvas AM. Platelet function and turnover in essential thrombocythemia: a systematic review. Semin Thromb Hemost. 2021;47(1):90-101.
33. Grignani C, Noris P, Tinelli C, Barosi G, Balduini CL. In vitro platelet aggregation defects in patients with myeloproliferative disorders and high platelet counts: are they laboratory artefacts? Platelets. 2009;20(2):131-134.
34. Lussana F, Femia EA, Pugliano M, et al. Evaluation of platelet function in essential thrombocythemia under different analytical conditions. Platelets. 2020;31(2):179-186.
35. Cattaneo M, Cerletti C, Harrison P, et al. Recommendations for the standardization of light transmission aggregometry: a Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J Thromb Haemost. 2013;11(6):1183-1189.
36. Cattaneo M, Lecchi A, Zighetti ML, Lussana F. Platelet aggregation studies: autologous platelet-poor plasma inhibits platelet aggregation when added to platelet-rich plasma to normalize platelet count. Haematologica. 2007;92(5):694-697.
37. Femia EA, Scavone M, Lecchi A, Cattaneo M. Effect of platelet count on platelet aggregation measured with impedance aggregometry (Multiplate™ analyzer) and with light transmission aggregometry. J Thromb Haemost. 2013;11(12):2193-2196.
38. Panova-Noeva M, Marchetti M, Russo L, et al. ADP-induced platelet aggregation and thrombin generation are increased in essential thrombocythemia and polycythemia vera. Thromb Res. 2013;132(1):88-93.
39. Pedersen OH, Larsen ML, Grove EL, et al. Platelet characteristics in patients with essential thrombocytosis. Cytometry B Clin Cytom. 2018;94(6):918-927.
40. Kaywin P, McDonough M, Insel PA, Shattil SJ. Platelet function in essential thrombocythemia. Decreased epinephrine responsiveness associated with a deficiency of platelet alphaadrenergic receptors. N Engl J Med. 1978;299(10):505-509.
41. Hattori N, Fukuchi K, Nakashima H, et al. Megakaryopoiesis and platelet function in polycythemia vera and essential thrombocythemia patients with JAK2 V617F mutation. Int J Hematol. 2008;88(2):181-188.
42. Scharf RE. Acquired disorders of platelet function. In: Michelson AD, Cattaneo M, Frelinger AL, Newman PJ, editors. Platelets, 4th ed. Cambridge (MA): Academic Press. 2019. p. 905-20.
43. Falanga A, Marchetti M, Evangelista V, et al. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood. 2000;96(13):4261-4266.
44. Blann A, Caine G, Bareford D. Abnormal vascular, platelet and coagulation markers in primary thrombocythaemia are not reversed by treatments that reduce the platelet count. Platelets. 2004;15(7):447-449.
45. Kaplar M, Kappelmayer J, Kiss A, Szabo K, Udvardy M. Increased leukocyte-platelet adhesion in chronic myeloproliferative disorders with high platelet counts. Platelets. 2000;11(3):183-184.
46. Rocca B, Ciabattoni G, Tartaglione R, et al. Increased thromboxane biosynthesis in essential thrombocythemia. Thromb Haemost. 1995;74(5):1225-1230.
47. Feng Y, Zhang Y, Shi J. Thrombosis and hemorrhage in myeloproliferative neoplasms: the platelet perspective. Platelets. 2022;33(7):955-963.
48. Lancellotti S, Dragani A, Ranalli P, et al. Qualitative and quantitative modifications of von Willebrand factor in patients with essential thrombocythemia and controlled platelet count. J Thromb Haemost. 2015;13(7):1226-1237.
49. Budde U, Schaefer G, Mueller N, et al. Acquired von Willebrand's disease in the myeloproliferative syndrome. Blood. 1984;64(5):981-985.
50. Budde U, Dent JA, Berkowitz SD, Ruggeri ZM, Zimmerman TS. Subunit composition of plasma von Willebrand factor in patients with the myeloproliferative syndrome. Blood. 1986;68(6):1213-1217.
51. Budde U, Scharf RE, Franke P, Hartmann-Budde K, Dent J, Ruggeri ZM. Elevated platelet count as a cause of abnormal von Willebrand factor multimer distribution in plasma. Blood. 1993;82(6):1749-1757.
52. Michiels JJ, Berneman Z, Schroyens W, et al. Platelet-mediated erythromelalgic, cerebral, ocular and coronary microvascular ischemic and thrombotic manifestations in patients with essential thrombocythemia and polycythemia vera: a distinct aspirin-responsive and coumadin-resistant arterial thrombophilia. Platelets. 2006;17(8):528-544.
53. Kienast J, Schmitz G. Flow cytometric analysis of thiazole orange uptake by platelets: a diagnostic aid in the evaluation of thrombocytopenic disorders. Blood. 1990;75(1):116-121.
54. Scavone M, Rizzo J, Femia EA, et al. Patients with essential thrombocythemia may be poor responders to enteric-coated aspirin, but not to plain aspirin. Thromb Haemost. 2020;120(10):1442-1453.
55. van Genderen PJ, Michiels JJ, van Strik R, Lindemans J, van Vliet HH. Platelet consumption in thrombocythemia complicated by erythromelalgia: reversal by aspirin. Thromb Haemost. 1995;73(2):210-214.
56. Rinder HM, Schuster JE, Rinder CS, Wang C, Schweidler HJ, Smith BR. Correlation of thrombosis with increased platelet turnover in thrombocytosis. Blood. 1998;91(4):1288-1294.
57. Kissova J, Bulikova A, Ovesna P, Bourkova L, Penka M. Increased mean platelet volume and immature platelet fraction as potential predictors of thrombotic complications in BCR/ABLnegative myeloproliferative neoplasms. Int J Hematol. 2014;100(5):429-436.
58. Carobbio A, Finazzi G, Guerini V, et al. Leukocytosis is a risk factor for thrombosis in essential thrombocythemia: interaction with treatment, standard risk factors, and Jak2 mutation status.
Haematologica | 108 June 2023 1497 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
Blood. 2007;109(6):2310-2313.
59. Carobbio A, Antonioli E, Guglielmelli P, et al. Leukocytosis and risk stratification assessment in essential thrombocythemia. J Clin Oncol. 2008;26(16):2732-2736.
60. Carobbio A, Finazzi G, Antonioli E, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood. 2008;112(8):3135-3137.
61. Barbui T, Carobbio A, Rambaldi A, Finazzi G. Perspectives on thrombosis in essential thrombocythemia and polycythemia vera: is leukocytosis a causative factor? Blood. 2009;114(4):759-763.
62. Lussana F, Caberlon S, Pagani C, Kamphuisen PW, Büller HR, Cattaneo M. Association of V617F Jak2 mutation with the risk of thrombosis among patients with essential thrombocythaemia or idiopathic myelofibrosis: a systematic review. Thromb Res. 2009;124(4):409-417.
63. Michiels JJ, Berneman Z, Schroyens W, Finazzi G, Budde U, van Vliet H. The paradox of platelet activation and impaired function: platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost. 2006;32(6):589-604.
64. Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436):eaan8292.
65. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544-1551.
66. Finazzi G, Carobbio A, Thiele J, et al. Incidence and risk factors for bleeding in 1104 patients with essential thrombocythemia or prefibrotic myelofibrosis diagnosed according to the 2008 WHO criteria. Leukemia. 2012;26(4):716-719.
67. Alvarez-Larrán A, Pereira A, Guglielmelli P, et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica. 2016;101(8):926-931.
68. Campbell PJ, MacLean C, Beer PA, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood. 2012;120(7):1409-1411.
69. Barbui T. Refining prognostication of thrombosis in ET. Am J Hematol. 2016;91(4):361-363.
70. NCCN Guidelines Version 3.2022 Myeloproliferative Neoplasms (https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf) Accessed on September 4, 2022.
71. Stuckey R, Ianotto JC, Santoro M, et al. Validation of thrombotic risk factors in 1381 patients with essential thrombocythaemia: a multicentre retrospective real-life study. Br J Haematol. 2022;199(1):86-94.
72. Pemmaraju N, Gerds AT, Yu J, et al. Thrombotic events and mortality risk in patients with newly diagnosed polycythemia vera or essential thrombocythemia. Leuk Res. 2022;115:106809.
73. Griesshammer M, Bangerter M, van Vliet HH, Michiels JJ. Aspirin in essential thrombocythemia: status quo and quo vadis. Semin Thromb Hemost. 1997;23(4):371-377.
74. Preston FE. Aspirin, prostaglandins, and peripheral gangrene. Am J Med. 1983;74(6A):55-60.
75. van Genderen PJ, Prins FJ, Michiels JJ, Schrör K. Thromboxanedependent platelet activation in vivo precedes arterial thrombosis in thrombocythaemia: a rationale for the use of low-dose aspirin as an antithrombotic agent. Br J Haematol. 1999;104(3):438-441
76. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of lowdose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114-124
77. Alvarez-Larrán A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood. 2010;116(8):1205-1210.
78. Alvarez-Larrán A, Sant'Antonio E, Harrison C, et al. Unmet clinical needs in the management of CALR-mutated essential thrombocythaemia: a consensus-based proposal from the European LeukemiaNet. Lancet Haematol. 2021;8(9):e658-e665.
79. Alvarez-Larrán A, Pereira A, Arellano-Rodrigo E, HernándezBoluda JC, Cervantes F, Besses C. Cytoreduction plus low-dose aspirin versus cytoreduction alone as primary prophylaxis of thrombosis in patients with high-risk essential thrombocythaemia: an observational study. Br J Haematol. 2013;161(6):865-871.
80. Chu DK, Hillis CM, Leong DP, Anand SS, Siegal DM. Benefits and risks of antithrombotic therapy in essential thrombocythemia: a systematic review. Ann Intern Med. 2017;167(3):170-180.
81. Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105(14):1650-1655.
82. Frelinger AL 3rd, Li Y, Linden MD, et al. Association of cyclooxygenase-1-dependent and -independent platelet function assays with adverse clinical outcomes in aspirintreated patients presenting for cardiac catheterization. Circulation. 2009;120(25):2586-2596.
83. Patrono C, García Rodríguez LA, Landolfi R, Baigent C. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353(22):2373-2383.
84. Zheng SL, Roddick AJ. Association of aspirin use for primary prevention with cardiovascular events and bleeding events: a systematic eview and meta-analysis. JAMA. 2019;321(3):277-287. Erratum in: JAMA. 2019;321(22):2245
85. Cattaneo M. Resistance to antiplatelet drugs: molecular mechanisms and laboratory detection. J Thromb Haemost. 2007;5(Suppl 1):230-237.
86. Cattaneo M. Aspirin and clopidogrel: efficacy, safety, and the issue of drug resistance. Arterioscler Thromb Vasc Biol. 2004;24(11):1980-1987.
87. Cattaneo M. Laboratory detection of 'aspirin resistance': what test should we use (if any)? Eur Heart J. 2007;28(14):1673-1675.
88. Bochner F, Somogyi AA, Wilson KM. Bioinequivalence of four 100 mg oral aspirin formulations in healthy volunteers. Clin Pharmacokinet. 1991;21(5):394-399.
89. Maree AO, Curtin RJ, Dooley M, et al. Platelet response to lowdose enteric-coated aspirin in patients with stable cardiovascular disease. J Am Coll Cardiol 2005;46(7):1258-1263.
90. Cox D, Maree AO, Dooley M, Conroy R, Byrne MF, Fitzgerald DJ. Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke 2006;37(8):2153-2158.
91. Peace A, McCall M, Tedesco T, et al. The role of weight and enteric coating on aspirin response in cardiovascular patients. J Thromb Haemost 2010;8(10):2323-2325
92. Bhatt DL, Grosser T, Dong JF, et al. Enteric coating and aspirin nonresponsiveness in patients with type 2 diabetes mellitus. J Am Coll Cardiol. 2017;69(6):603-612.
93. Rothwell PM, Cook NR, Gaziano JM, et al. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: analysis of individual patient data from randomised trials. Lancet. 2018;392(10145):387-399.
Haematologica | 108 June 2023 1498 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
94. Dammann HG, Burkhardt F, Wolf N. Enteric coating of aspirin significantly decreases gastroduodenal mucosal lesions. Aliment Pharmacol Ther. 1999;13(8):1109-1114.
95. Endo H, Sakai E, Kato T, et al. Small bowel injury in low-dose aspirin users. J Gastroenterol. 2015;50(4):378-386.
96. García Rodríguez LA, Hernández-Díaz S, de Abajo FJ. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiologic studies. Br J Clin Pharmacol. 2001;52(5):563-571.
97. Guthikonda S, Lev EI, Patel R, et al. Reticulated platelets and uninhibited COX-1 and COX-2 decrease the antiplatelet effects of aspirin. J Thromb Haemost. 2007;5(3):490-496.
98. Dragani A, Pascale S, Recchiuti A, et al. The contribution of cyclooxygenase-1 and -2 to persistent thromboxane biosynthesis in aspirin-treated essential thrombocythemia: implications for antiplatelet therapy. Blood. 2010;115(5):1054-1061.
99. Pascale S, Petrucci G, Dragani A, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood.
2012;119(15):3595-3603.
100. Dillinger JG, Sideris G, Henry P, Bal dit Sollier C, Ronez E, Drouet L. Twice daily aspirin to improve biological aspirin efficacy in patients with essential thrombocythemia. Thromb Res. 2012;129(1):91-94.
101.Perrier-Cornet A, Ianotto JC, Mingant F, Perrot M, Lippert E, Galinat H. Decreased turnover aspirin resistance by bidaily aspirin intake and efficient cytoreduction in myeloproliferative neoplasms. Platelets. 2018;29(7):723-728.
102. De Stefano V, Rocca B, Tosetto A, et al. The Aspirin Regimens in Essential Thrombocythemia (ARES) phase II randomized trial design: implementation of the serum thromboxane B2 assay as an evaluation tool of different aspirin dosing regimens in the clinical setting. Blood Cancer J. 2018;8(6):49.
103. Tefferi A. Overcoming "aspirin resistance" in MPN. Blood. 2012;119(15):3377-3378.
104. Rocca B, Tosetto A, Betti S, et al. A randomized double-blind trial of 3 aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood. 2020;136(2):171-182.
Haematologica | 108 June 2023 1499 REVIEW ARTICLE - Aspirin in essential thrombocythemia M. Cattaneo
Concomitant targeting of FLT3 and BTK overcomes FLT3 inhibitor resistance in acute myeloid leukemia through the inhibition of autophagy
Correspondence: M. Andreeff mandreef@mdanderson.org
Received: February 18, 2022.
Accepted: September 28, 2022.
1Section of Molecular Hematology and Therapy, Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Department of Hematology, Nanfang Hospital, Southern Medical University, Guangzhou, Guangdong, China; 3Aptose Biosciences, San Diego, CA, USA and 4Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
*WZ and GY contributed equally as co-first authors.
Abstract
Prepublished: October 13, 2022.
https://doi.org/10.3324/haematol.2022.280884
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Strategies to overcome resistance to FMS-like tyrosine kinase 3 (FLT3)-targeted therapy in acute myeloid leukemia (AML) are urgently needed. We identified autophagy as one of the resistance mechanisms, induced by hypoxia and the bone marrow microenvironment via activation of Bruton tyrosine kinase (BTK). Suppressing autophagy/BTK sensitized FLT3mutated AML to FLT3 inhibitor-induced apoptosis. Furthermore, co-targeting FLT3/BTK/aurora kinases with a novel multikinase inhibitor CG-806 (luxeptinib) induced profound apoptosis in FLT3-mutated AML by co-suppressing FLT3/BTK, antagonizing autophagy, and causing leukemia cell death in FLT3-wildtype AML by aurora kinase-mediated G2/M arrest and polyploidy, in addition to FLT3 inhibition. Thus, CG-806 exerted profound anti-leukemia activity against AML regardless of FLT3 mutation status. CG-806 also significantly reduced AML burden and extended survival in an in vivo patient-derived xenograft leukemia murine model of FLT3 inhibitor-resistant FLT3-ITD/TKD double-mutant primary AML. Taken together, these findings indicate that CG-806 has a unique mechanistic action and pre-clinical activity, which is presently undergoing clinical evaluation in both FLT3 wildtype and mutant AML.
Introduction
Acute myeloid leukemia (AML) is a diverse group of hematologic malignancies characterized by clonal evolution and genetic heterogeneity.1,2 Mutations in the FMS-like tyrosine kinase 3 (FLT3) gene are detected in approximately onethird of patients with newly diagnosed AML. These mutations include the common FLT3 internal tandem duplication (FLT3-ITD) in approximately 20-25% of AML and point mutations in the tyrosine kinase domain (FLT3TKD) in approximately 5-10% of AML cases. FLT3-targeted therapy represents an important paradigm in the management of patients with highly aggressive, FLT3-mutated AML. A number of FLT3 inhibitors have been developed in recent years, including the small molecular inhibitors sorafenib3 and quizartinib.4 While midostaurin and gilteritinib are approved by the Food and Drug Administration, all inhibitors show only limited efficacy in clearing leukemic blasts from the bone marrow (BM) microenvironment and inducing sustained remissions, resulting in relapse and/or
resistance.4,5 Thus, it is of paramount importance to understand the underlying mechanisms of this resistance. We and others have reported that acquired secondary FLT3-TKD mutations, including mutations of residues D835, Y842, and F691, which have been identified in relapsed patients receiving FLT3-targeted therapy, can contribute to resistance.6-8 It has also been reported that sorafenib-induced macroautophagy, hereafter referred to as autophagy, through induction of endoplasmic reticulum stress and 5’ AMP-activated protein kinase (AMPK)-dependent mammalian target of rapamycin complex 1 (mTORC1) inhibition in liver cancer cells,9 and in human myeloid dendritic cells.10 Thus, AMPK-mTORC1 is potentially a key player in sorafenib-induced autophagy,11 which could be associated with resistance to FLT3 inhibitors. Autophagy is a process of intracellular degradation of proteins, organelles, etc. in response to various stressors, including chemotherapy in leukemia.12 In the cancer context, autophagy has a dual role both as a tumor initiator, by inducing DNA damage and genetic instability, and as a
Weiguo Zhang,1* Guopan Yu,1,2* Hongying Zhang,3 Mahesh Basyal,1 Charlie Ly,1 Bin Yuan,1 Vivian Ruvolo,1 Sujan Piya,1 Seemana Bhattacharya,1 Qi Zhang,4 Gautam Borthakur,1,4 Venkata L. Battula,1 Marina Konopleva,4 William G. Rice3 and Michael Andreeff1,4
Haematologica | 108 June 2023 1500 ARTICLE - Acute Myeloid Leukemia
tumor promoter, by providing cancer cells with the necessary nutrients for survival.13 As autophagy has an adaptive tumorigenic function, it may also provide leukemia cells with a mechanism of resistance to FLT3 inhibitor-mediated cytotoxicity. Evidence has shown that autophagy sustains the FLT3-ITD-dependent proliferation of leukemic cells through the activation of transcription factor 4 (ATF4). Targeting either autophagy or ATF4 reduces AML tumor burden in mice.14 In addition, we reported that autophagy targeting sensitized AML to chemotherapy,15 implying an association between autophagy and chemoresistance. Thus, inhibition of autophagy may be a potentially effective therapeutic strategy in AML by overcoming resistance to FLT3-targeted therapy. In the present study, we observed upregulation of phospho-Bruton tyrosine kinase (BTK) levels and an increase of ATF4 accompanying autophagy in FLT3 inhibitor-resistant leukemia cell lines and in primary AML samples from patients who had received sorafenib monotherapy and developed resistance during a clinical trial of FLT3-targeted therapy. In addition, conditions mimicking the BM microenvironment (e.g., hypoxia and the presence of mesenchymal stem cells [MSC]) also triggered an increase of autophagy in FLT3-mutated AML cells. By repressing autophagosome-lysosome fusion or BTK activation, we enhanced quizartinib-induced apoptosis in FLT3 inhibitor-resistant leukemia cells. Of note, blockade of BTK/FLT3 with a small molecule multi-kinase inhibitor, CG-806, exerted impressive anti-leukemia activity against FLT3 inhibitor-resistant leukemias in vitro and in vivo in a patient-derived xenograft (PDX) leukemia murine model engrafted with FLT3-inhibitor-resistant primary AML, suggesting that co-targeting BTK and FLT3 may provide a novel strategy for preventing or overcoming FLT3 inhibitor resistance.
Methods
Cell lines and patients’ samples
The human AML cell lines MOLM14, MV4-11 (harboring FLT3-ITD mutations), OCI-AML3 and THP-1 (harboring FLT3 wildtype [WT]); the murine leukemia cell lines Ba/F3 (harboring different FLT3 mutations including ITD, TKD or ITD+TKD double mutations or FLT3 WT); and MSC were used for this study. Details of the cell lines and culture conditions are provided in the Online Supplementary Methods. All cell lines were validated by STR DNA fingerprinting using the AmpFISTR Identifier kit according to the manufacturer's instructions (Applied Biosystems cat. 4322288).
AML patients’ samples were obtained after written informed consent following institutional guidelines of the University of Texas MD Anderson Cancer Center and in ac-
cordance with the principles of the Declaration of Helsinki. Mononuclear cells were purified from primary samples by Ficoll-Hypaque (Sigma-Aldrich) density-gradient centrifugation and were cultured in RPMI 1640 culture medium supplemented with 10% fetal bovine serum before treatment.
Compounds
Quizartinib, ibrutinib and SNS-314 were purchased from Selleckchem (Houston, TX, USA). CG-806 (luxeptinib) was provided by Aptose Biosciences (San Diego, CA, USA). The molecular structures of the kinase inhibitors are shown in Online Supplementary Figure S1. Chloroquine was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Apoptosis assays
Cells were treated with drugs or an equivalent amount of dimethylsulfoxide for 48-72 h. Cells were harvested and stained with annexin V-fluorescein isothiocyanate/propidium iodide according to the manufacturer’s instructions. Apoptosis induction was analyzed by measuring annexin V positivity and propidium iodide positivity with fluorescence activated cell sorting as described previously.16
Cell cycle and polyploidy analysis
Cell cycle progression was measured using flow cytometric analysis of DNA content and BrdU incorporation. A DNA histogram was plotted to show the cell cycle, and diploid and polyploid distributions were measured with a FACScalibur (Becton Dickinson, Franklin Lakes, NJ, USA). The details are provided in the Online Supplementary Methods.
BTK and ATG7 knockdown
BTK protein was knocked down by transfecting BTK short interfering (si)RNA into MOLM14 cells. ATG7 protein, an essential effector enzyme for canonical autophagy, was knocked down either by electroporation transfection of ATG7 siRNA into MV4-11 cells or lentiviral transduction of ATG7 short hairpin (sh)RNA into OCI-AML3 cells. Details are provided in the Online Supplementary Methods.
Patient-derived xenograft murine leukemia model
A primary BM specimen was collected with informed consent from an AML patient who had been treated with sorafenib and developed resistance (harboring FLT3-ITD/D835 mutations) and was xenografted using the University of Texas MD Anderson Cancer Center Institutional Review Board protocol Lab02-395. Nod.CgPrkdcscidIL2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Laboratories and handled according to Institutional Animal Care and Use Committee-approved protocol #00001303-RN01. The leukemia cells were administered intravenously into the NSG mice at a dose
Haematologica | 108 June 2023 1501 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
of 3.5x106 cells/mouse. The mice were randomly separated into two groups, one given the vehicle (15% Transcutol plus 85% PEG400, n=13 mice) and the other given the treatment (CG-806 at a dose of 100 mg/kg, n=13 mice). The vehicle or CG-806 was given orally every day for 5 days on, followed by 2 days off/week starting from when the leukemia cells reached about 1% engraftment in the blood (day 27 in this case). Leukemia cell engraftment was monitored by measuring hCD45+/mCD45– cells in mouse peripheral blood by flow cytometry and the mice’s body weight and vital signs were monitored simultaneously. Three mice from each group were sacrificed 72 days after injection of the leukemia cells. Peripheral blood, BM and spleen were collected and leukemia cell engraftment was assessed by determining the hCD45+/mCD45– cell population by flow cytometry. The survival curve was plotted and analyzed by GraphPad Prism 7 using the Kaplan-Meier method.17
Statistical analyses
The Student t test or two-way analysis of variance was used to analyze immunoblot and cell apoptosis data. A P value ≤0.05 was considered statistically significant. All statistical tests were two-sided and the results are expressed as the mean of triplicate samples/experiments ± standard deviation or 95% confidence intervals (error bars). The efficacy of CG-806 with respect to survival was estimated by the Kaplan-Meier method,17 and log-rank analysis was used to test for differences in survival.
Results
Upregulation of autophagy is associated with resistance to FLT3 inhibitors and is further increased by hypoxia and co-culture with mesenchymal stem cells
Since leukemia cells bearing TKD or ITD/TKD double mutations are resistant to certain FLT3 inhibitors, in comparison to cells with only ITD mutations,6-8 we sought to determine whether autophagy was associated with the resistance. Since LC3-II, but not LC3-I, is bound to phosphatidylethanolamine (PE) in the autophagosome membrane and closely correlates to the number of autophagosomes, therefore serving as a good indicator of autophagosome formation,18 we determined autophagy levels by measuring LC3-II (or LC3-II/I ratios) based on semi-quantitative analyses of immunoblotting data of murine leukemia cell lines harboring FLT3 WT or FLT3 mutations (ITD or ITD+TKD dual mutations). The results demonstrated that the autophagy levels were positively associated with upregulation of ATF4 and Beclin1, which is another inducer of autophagy (Figure 1A). High autophagy levels were associated with high values of IC50 (50th percentile of the maximal inhibitory concentration) to FLT3 inhibition as well, especially in cells with FLT3-TKD
mutations (Online Supplementary Table S1). The cells with higher autophagy levels also had higher levels of phosphoFLT3 or phospho–ERK, which are associated with resistance to FLT3 inhibitors.14,19 Interestingly, high phospho-BTK levels were observed in cells with high autophagy levels, implying a hitherto unknown correlation between the two proteins in these cell lines (Figure 1A).
We further compared autophagy levels in paired primary AML samples before and after the administration of sorafenib in a clinical trial. The patients were given sorafenib and either did not respond to the therapy or died during treatment (Online Supplementary Table S2). The resistant samples (showing upregulation of phospho-FLT3 after sorafenib treatment in comparison to pre-treatment levels in these FLT3 mutated samples) had higher LC3-II levels than their pre-treatment counterparts, a phenomenon accompanied by upregulation of phosphorylated Unc-51-like autophagy-activating kinase 1 (ULK1) in addition to upregulation of ATF4 and phospho-BTK (Figure 1B). The phosphorylation of ULK1 at Ser556 is AMPK-dependent and is required for the activation of downstream autophagy.20,21 The results imply that sorafenib resistance may be associated with upregulation of autophagy, which is likely related to upregulation of ATF4 and phospho-ULK1 as well as phospho-BTK. We further assessed whether the BM microenvironment modulates autophagy levels in leukemia cells. The BM niche provides a sanctuary for AML cells and protects them from targeted therapies.22 FLT3-mutant MOLM14 cells were cultured in BM niche-mimicking conditions (i.e. hypoxia [1% oxygen tension] or in the presence of MSC) in vitro for 48 h. The BM niche-mimicking conditions modestly upregulated LC3-II/I and Beclin-1 levels and these effects were accompanied by an increase of phospho-BTK and hypoxia inducible factor-1α (HIF-1α) (Figure 1C). Coculture of the MOLM14 cells with MSC also led to a significant increase in autophagosomes as compared to the number in MOLM14 cells alone in normoxia without MSC (Figure 1D, E). These observations suggest a possible association between autophagy and hypoxia or MSC co-culture and the protection (resistance) afforded by the BM microenvironment against AML therapies.
Chloroquine enhances quizartinib-induced apoptosis and partially abrogates the protection of acute myeloid leukemia cells mediated by mesenchymal stem cells To validate if autophagy levels were associated with FLT3 inhibitor resistance, we sought to sensitize AML cells to FLT3 inhibitor-induced killing by reducing autophagy. We first tested the sensitivity to apoptosis induction by using the second-generation FLT3 inhibitor quizartinib and impairing autophagy with chloroquine, a lysosomotropic agent that inhibits lysosomal degradation of the autophagosome.23 The presence of chloroquine sensitized cells to quizartinib-induced apoptosis, as determined by annexin
Haematologica | 108 June 2023 1502 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
Figure 1. Upregulation of autophagy is associated with resistance to FLT3-targeted therapy, which further increases in hypoxia and mesenchymal stem cell co-culture of FLT3-mutated acute myeloid leukemia. (A) Basal expression levels of autophagyrelated proteins was assessed by immunoblotting in a panel of murine leukemia cell lines with different FLT3 mutational status. (B) Autophagy-related proteins were investigated in samples from patients with acute myeloid leukemia, which were collected before and after administration of sorafenib in a clinical trial of sorafenib. (C) FLT3-ITD-mutated MOLM14 cells were cultured together with mesenchymal stem cells (MSC) or in hypoxic conditions for 48 h and autophagy-related proteins were analyzed by immunoblotting. (D) MOLM14 cells were co-cultured with or without (w/o) MSC for 48 h and the ultrastructure of autophagosomes was observed with transmission electron microscopy (TEM). Nuclei, endoplasmic reticulum, and mitochondria are marked. Red arrows indicate autophagosomes. Left panel: scale bar represents 2 mm. Right panel: scale bar represents 0.5 mm. (E) A statistical analysis of autophagosome numbers based on TEM observation of at least 20 cells per sample. The numbers shown in the western blot data are from semi-quantitative analyses of protein levels, comparing them to the control samples of each group. ***P<0.001. WT: wildtype; ITD: internal tandem duplication; Tx: treatment with sorafenib; N: nuclei; ER: endoplasmic reticulum; m: mitochondria; MSC: mesenchymal stem cells; w/o: without.

A B C D E Haematologica | 108 June 2023 1503 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
V staining in Ba/F3-FLT3-ITD and Ba/F3-ITD/D835Y mutant cell lines (Figure 2A). Immunoblot analysis confirmed that chloroquine enhanced quizartinib-induced apoptosis (shown as an increase of cleaved caspase 3) in FLT3-ITD mutant MOLM14 cells and in FLT3 inhibitor-resistant Ba/F3-ITD+D835Y cells, an effect that was accompanied by increased suppression of FLT3 and its downstream signaling pathways (i.e. p-ERK, p-mTOR and p-S6K) (Figure 2B). As expected, LC3-II was not decreased in the presence of chloroquine. In fact, chloroquine acts on autolysosome degradation to impair autophagic flux by decreasing autophagosome-lysosome fusion instead of reducing autophagosome formation, which was demonstrated in a previous study.18 Further investigations showed that chloroquine partially abrogated MSC-mediated protection and resensitized leukemia cells to quizartinib-induced apoptosis in co-culture of leukemia cells with MSC (Figure 2C), suggesting that pharmacological disruption of autophagosome-lysosome fusion resensitizes FLT3-mutated leukemia cells to FLT3-targeted therapy.
BTK inhibition sensitizes FLT3-mutated leukemia cells to quizartinib-induced
killing
Since we observed an association between phospho-BTK and the autophagy-related proteins Beclin-1 and LC3-II in cells resistant to FLT3 inhibitors, in both relapsed primary AML patients’ samples and in FLT3-ITD-mutant MOLM14 cells under hypoxic culture or in the presence of MSC (Figure 1B, C), we sought to investigate whether suppression of BTK sensitized cells to quizartinib-induced apoptosis. BTK inhibition, achieved using the BTK-specific inhibitor ibrutinib,24 significantly enhanced quizartinib-induced apoptosis in both FLT3-ITD-mutant and ITD/D835Y mutant cells in the presence or absence of MSC (Figure 3A, B, Online Supplementary Figure S2). Immunoblot analysis further demonstrated that ibrutinib suppressed autophagy and abrogated MSC-mediated protection through suppression of phospho-BTK and its downstream phosphoERK, -mTOR, and -S6K signaling pathways (Figure 3C, Online Supplementary Figure S3). Furthermore, knockdown of BTK by siRNA markedly reduced levels of the autophagy
Figure 2. Autophagy inhibition with chloroquine enhances quizartinibinduced apoptosis and partially abrogates mesenchymal stem cell-mediated protection. (A) Murine leukemia cell lines harboring either FLT3-ITD or FLT3-ITD/D835Y mutations were exposed to increasing doses of quizartinib for 48 h in the presence or absence of chloroquine in vitro. Apoptosis induction was then assessed by measuring annexin V positivity by flow cytometry. (B) FLT3-ITD-mutated MOLM14 cells and Baf3-ITD/D835Y cells were exposed to increasing doses of quizartinib for 24 h in the presence (red box) or absence (green box) of chloroquine and then subjected to immunoblot analysis. (C) MOLM14 cells were exposed to the indicated doses of quizartinib for 48 h in co-culture with (yellow box) or without mesenchymal stem cells (blue box) in the presence or absence of chloroquine (50 µM). Apoptosis induction was assessed by measuring annexin V positivity by flow cytometry. ***P<0.001; ****P<0.0001 (two-way analysis of variance). Error bars represent the standard deviation from three independent experiments. w/o: without; CQ: chloroquine.

A C B Haematologica | 108 June 2023 1504 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
Figure 3. BTK inhibition sensitizes leukemic cells to quizartinib-induced killing and abrogates mesenchymal stem cell-mediated protection in FLT3-mutated leukemia cells. (A) MOLM14 cells were exposed to ibrutinib, quizartinib or their combination for 48 h and apoptosis induction was assessed by measuring annexin V positivity by flow cytometry. (B) MOLM14 cells were treated with quizartinib, ibrutinib or their combination for 48 h in the presence or absence of mesenchymal stem cells (MSC), and annexin V positivity was measured by flow cytometry. (C) MOLM14 cells were exposed to ibrutinib for 24 h in the presence or absence of MSC, and protein levels were analyzed by immunoblotting. The numbers are from semi-quantitative analyses of protein levels in comparison to the control samples of each group. (D) MOLM14 cells were transfected with BTK siRNA for 48 h, and protein expression was determined by immunoblotting. (E) BTK knockdown (BTK-KD) MOLM14 cells and cells transfected with scrambled RNA were exposed to quizartinib in the presence or absence of MSC for 48 h. Apoptosis induction was assessed by measuring annexin V positivity by flow cytometry. The numbers over the individual bars indicate exact levels of apoptosis induction. (F) BTK knockdown and MOLM14 cells transfected with scrambled RNA were co-cultured with or without MSC for 24 h, followed by immunoblot analysis. *P<0.05; **P<0.01. Error bars of the flow cytometry data represent the standard deviation from three independent experiments. Quiz: quizartinib; Ibru: ibrutinib; w/o: without; KD: knockdown; NC: normal control (transfected with scrambled RNA); DMSO: dimethylsulfoxide.

A B C D E F Haematologica | 108 June 2023 1505 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
inducers ATF4 and Beclin-1 (Figure 3D), triggered similar levels of quizartinib-induced apoptosis, abrogated MSCmediated protection, increased the pro-apoptotic protein Bim and cleaved caspase-3, although it showed only marginal modulation of Beclin-1 and LC3-II levels (Figure 3E, F). However, BTK knockdown by siRNA was marked by decreased ATF4 levels (Figure 3D), suggesting a correlation between BTK levels and autophagy modulation. At least, these findings suggest that high BTK activity is associated with resistance to FLT3 inhibitors, and that this effect could be overcome by suppressing BTK activation, implying that BTK is also involved in the modulation of autophagy.
Co-targeting FLT3 and BTK with the multi-kinase inhibitor CG-806 abolishes mesenchymal stem cell- and hypoxia-mediated protection and induces apoptosis in FLT3-mutated leukemia cells
We further tested the anti-leukemia efficacy of a novel small molecule kinase inhibitor CG-806, which has multikinase inhibitory activity against FLT3, BTK, and aurora kinases (AURK) at low IC50 values (0.82, 5.0 and 0.38 nM against FLT3 ITD, BTK and aurora A, respectively) in a cellfree system (Online Supplementary Table S3). CG-806 has much lower IC50 values than most commercially available FLT3 inhibitors, including the Food and Drug Administration-approved small molecule FLT3 inhibitors midostaurin and gilteritinib, especially in AML cells with FLT3-ITD/TKD double mutations (Online Supplementary Table S4). CG806 also demonstrated marked pro-apoptotic efficacy in AML cell lines and patients’ samples harboring these mutations and in FLT3 WT cells as well (Online Supplementary Figures S3 and S4). Further investigations demonstrated that CG-806 completely abrogated MSC-mediated chemoprotection and triggered the induction of apoptosis in FLT3-ITD-mutant leukemias, accompanied by suppression of autophagy (Figure 4A-C, Online Supplementary Figure S5). ATG7 knockdown enhanced CG-806-induced apoptosis in FLT3-ITD mutant MV4-11 cells (Figure 4D, Online Supplementary Figure S6), suggesting an association between autophagy impairment and sensitivity to CG-806. Mechanistically, CG-806 profoundly suppressed FLT3, BTK, c-Myc, and ATF4 (Figure 4E, Online Supplementary Figure S7). Of note, CG-806 alone had stronger pro-apoptotic activity than could be induced by co-targeting FLT3 and autophagy, or BTK, with quizartinib and chloroquine or ibrutinib, respectively, in primary AML patients’ samples (Figure 4F, Online Supplementary Figure S8).
CG-806 induces G2/M arrest and promotes polyploidy through aurora kinase inhibition in FLT3 WT leukemia cells
Since CG-806 exerted profound anti-leukemia effects in Baf3-FLT3 cells and primary AML samples with mutant FLT3 (Online Supplementary Figures S3 and S4), we further
investigated whether the mechanism of triggering apoptosis in FLT3-mutated cells also applied to FLT3 WT cells (i.e., targeting FLT3/BTK to suppress autophagy). Unexpectedly, CG-806 did not suppress autophagy in FLT3 WT cells. Instead, it increased the LC3-II:I ratio (P<0.001) and this was accompanied by upregulation of the pro-autophagic proteins Beclin-1, ATG7, and phospho-ULK1 after exposure to CG-806 for 48 h (Figure 5A, Online Supplementary Figure S9). CG-806 predominantly triggered suppression of phospho-aurora kinases, especially aurora B and C, but had less effect on the modulation of phosphoFLT3 and -BTK signaling (Online Supplementary Figure S10). In addition, CG-806 showed enhanced activity against proliferating cells. The IC50 values were 3.88, 11.81, and 21.99 nM in the FLT3 WT cells THP-1, OCI/AML3, and Kasumi-1, respectively. Interestingly, CG-806 had much weaker apoptogenic effects in FLT3 WT cell lines such as THP-1 and Kasumi-1, and the EC50 (concentration producing half-maximal response) could not be reached even at micromolar concentrations (data not shown).
Cell cycle analysis by BrdU incorporation and propidium iodide staining revealed a marked increase in G2/M cells and polyploidization, with FLT3 WT leukemia cells having a tetraploid DNA content even after very low doses of CG806 (Figure 5B-D). Immunoblot analysis demonstrated that CG-806 induced upregulation of the anti-proliferative proteins p53, p2125 and the DNA repair-related protein γH2AX in addition to decreasing PLK1 and phosphorylatedhistone H3 -CDC25c, and -CDK2, which are closely associated with induction of polyploidization26 (Figure 5E). To determine whether the effects of CG-806 on cell cycle progression and autophagy regulation were off-target or specific to aurora kinase inhibition, a specific aurora kinase inhibitor SNS-31427 was used for reference in the FLT3 WT cells. The results revealed induction of G2/M arrest, polyploidy and a slight increase in autophagy levels (Figure 5F, Online Supplementary Figure S11), implying an association between aurora kinase inhibition and CG-806induced cell cycle perturbation and polyploidization in FLT3 WT cells.
Impairment of autophagy re-sensitizes FLT3 WT cells to CG-806-induced pro-apoptotic effects
To better understand the roles of autophagy and polyploidy in CG-806-induced killing of FLT3 WT leukemia cells, we suppressed autophagy by chloroquine or ATG7 knockdown using shRNA in FLT3 WT leukemia cells (Online Supplementary Figure S12). Inhibition of autophagy by chloroquine or ATG7 knockdown profoundly re-sensitized cells to CG-806-induced apoptosis (Figure 6A-C), suggesting an association between autophagy levels and sensitivity to inhibition of FLT3/BTK/aurora kinases. Meanwhile, inhibition of autophagy also reduced CG-806induced polyploidy, but only modestly modulated G2/M
Haematologica | 108 June 2023 1506 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
Figure 4. The FLT3/BTK inhibitor CG-806 abolishes mesenchymal stem cell/hypoxia-mediated protection and induces apoptosis in FLT3-mutated leukemia cells. (A) MOLM14 cells were exposed to either CG-806 or quizartinib for 48 h in the presence or absence of mesenchymal stem cells (MSC), and apoptosis induction was assessed by measuring annexin V positivity by flow cytometry. Data are from three independent experiments. (B) MOLM14 cells were exposed to either CG-806 or quizartinib for 24 h in the presence or absence of MSC and then cellular proteins were assessed by immunoblot analysis. (C) MOLM14 cells were exposed to CG-806 for 24 h and then cellular ultrastructure was observed using transmission electron microscopy. Nuclei, endoplasmic reticulum, and mitochondria are marked; the red arrows indicate autophagosomes. Left panel: the scale bar represents 2 µm. Right panel: the scale bar represents 0.5 mm. (D) FLT3-ITD-mutated MV4-11 cells were transfected with ATG7 siRNA or scrambled siRNA for 48 h and then exposed to CG-806 for an additional 48 h. Apoptosis induction was assessed by measuring annexin V positivity by flow cytometry.
(E) MOLM14 cells were exposed to CG-806 in either normoxic or hypoxic conditions in the presence or absence of MSC for 24 h, and then subjected to immunoblot analysis. (F) FLT3-ITD-mutated primary AML blasts were exposed to either CG-806 or quizartinib ex vivo for 48 h. Apoptosis induction was assessed by measuring annexin V positivity by flow cytometry. w/o: without; DMSO: dimethylsulfoxide; N: nuclei; ER: endoplasmic reticulum; m: mitochondria; KD: knockdown.

A B C D E F Haematologica | 108 June 2023 1507 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
Figure 5. CG-806 induces G2/M arrest and polyploidy and is accompanied by suppression of pro-autophagic proteins through aurora kinase inhibition in FLT3-WT leukemia cells. (A) The FLT3 WT leukemia cell line OCI/AML3 was exposed to CG-806 for 48 h and autophagy-related proteins were analyzed by immunoblotting. (B) The graphs show data representative of three independent experiments in which cells were stained with propidium iodide and anti-BrdU antibodies and analyzed by flow cytometry. Polyploidy is indicated by red arrows. (C) Percentage of cells in G2/M and polyploidy distribution, expressed as the mean of three independent experiments. Error bars represent the standard error of mean. Statistical analysis was carried out using the t test based on three independent experiments. (D) FLT3 WT cells were treated with the indicated concentrations of CG-806 for 24 h and analyzed by immunofluorescence using γ-tubulin (green) and α-tubulin (red) antibodies. 4’,6-Diamidino-2-phenylindole staining (blue) indicates a cell’s nucleus (scale bar represents 10 mm). (E) The FLT3 WT cells were exposed to CG-806 for 24 h and then the protein levels were assessed by immunoblotting. (F) FLT3 WT cells were exposed to the BTK inhibitor SNS-314 for 24 h and DNA content was measured by flow cytometry. Red arrows indicate polyploidy. The data are from three independent experiments. DMSO: dimethylsulfoxide; PI: propidium iodide.

A B C D E F Haematologica | 108 June 2023 1508 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
cells (Figure 6D, Online Supplementary Figure S13). Nevertheless, these results suggest that inhibition of autophagy enhances CG-806-triggered, pro-apoptotic effects and partially abolishes CG-806-induced polyploidy in FLT3 WT cells.
CG-806 induces marked in vivo anti-leukemia effects in acute myeloid leukemia resistant to FLT3 inhibitors
We tested the pro-apoptotic effects of CG-806 and quizartinib in an FLT3-ITD/D835-mutated primary AML sample ex vivo by isolating hCD45+/mCD45– leukemia cells from the peripheral blood of a mouse xenografted with leukemia cells from a patient resistant to FLT3 inhibitors and exposed the cells to either agent for 48 h. The resistant leukemia cells were indeed resistant to quizartinib, but were sensitive to CG-806 (Online Supplementary Figure S14). We next tested the anti-leukemia activity of CG-806 in a PDX murine model by injecting FLT3-ITD/D835-mutated primary AML blasts via the tail vein. CG-806 markedly reduced leukemia cell burden in the peripheral blood, spleen, and BM after several weeks of drug administration (Figure

7A-C, Online Supplementary Figure S15). We observed a statistically significant prolongation of survival from a median of 72 days for controls to 113 days for the CG-806treated (100 mg/kg) group (Figure 7D). Of note, the engrafted leukemia cells also exhibited a decrease in autophagy, as evidenced by LC3-II-PE fluorescence after CG-806 treatment (Figure 7E), suggesting that autophagy suppression through BTK inhibition has a benefit in overcoming FLT3 inhibitor resistance in AML.
Discussion
The mechanisms associated with autophagy and drug resistance during FLT3-targeted therapy of AML have not been extensively explored. In this study, we confirmed that leukemia cells harboring resistance-related FLT3 mutations have higher basal levels of autophagy as well as higher phospho-FLT3, implying an association between autophagy and drug resistance during FLT3-targeted therapy. Mechanistically, there was a notable association between auto-
Figure 6. Inhibition of autophagy with chloroquine or with ATG7 knockdown re-sensitizes FLT3 WT cells to CG-806-triggered proapoptotic effects. (A, B) Apoptosis induction was assessed in the FLT3 WT leukemia cell lines THP-1 and OCI-AML3 by exposing the cells to the indicated concentrations of CG-806 for 24-48 h in the presence or absence of chloroquine and then measuring annexin V positivity by flow cytometry (A) and cleaved caspase-3 by immunoblotting (B). (C) ATG7 knockdown and control FLT3 WT OCI-AML3 leukemia cells were exposed to the indicated concentrations of CG-806 for 48 h, and apoptosis induction was assessed by flow cytometric by measuring annexin V positivity. (D) THP-1 cells were exposed to different concentrations of CG-806 for 24 h in the presence (+) or absence (-) of chloroquine and the cell cycle phase distributions were determined by staining the cells with propidium iodide and assessing their DNA content using flow cytometry. Data in panels (A), (C) and (D) are presented as the means of three independent experiments ± standard deviations. w/o: without; CQ: chloroquine; casp: caspase; NC: normal control; KD: knockdown; PI: propidium iodide. *P<0.05.
A B C D Haematologica | 108 June 2023 1509 W. Zhang et al. ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML
Figure 7. CG-806 has efficient anti-leukemia activity in FLT3 inhibitor-resistant acute myeloid leukemia, represses autophagy induction, and prolongs mouse survival in an FLT3-ITD/D835 bearing patient-derived xenograft leukemia model. (A) Leukemia cell engraftment in mouse peripheral blood was assessed by measuring the percentage of the hCD45+/mCD45– population in vehicle- and CG-806-treated mice in a patient-derived xenograft model. CG-806 treatment started from day 27 after the injection of leukemia cells. (B) The percentage of leukemia cell engraftment was assessed by measuring the hCD45+/mCD45– population in bone marrow and spleen 25 days after CG-806 treatment. (C) Leukemia cell engraftment was assessed by hematoxylin and eosin staining and anti-hCD45 (green) immunostaining of mouse organs. The scale bar represents 200 mm. (D) Mouse survival was estimated by the Kaplan-Meier method and log-rank statistics were used to test for differences in survival. The arrow indicates the duration of treatment. (E) Autophagy levels were assessed by flow cytometry after staining with anti-LC3II-PE and gated on the hCD45+ population. BM: bone marrow; Ctrl: control; H&E: hematoxylin and eosin; Tx: treatment; MFI: mean fluorescence intensity; PE: phosphatidylethanolamine.

A C B D E Haematologica | 108 June 2023 1510 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
phagy and the increase in phospho-FLT3, phospho-BTK, and ATF4 in the resistant AML cells/blasts (Figure 1A, B, Online Supplementary Figure S16). A recent study also indicated that FLT3 activation drove autophagy in FLT3-ITD AML cells through upregulation of ATF4 protein in a manner independent of eukaryotic initiation factor 2α (eIF2α), an upstream protein of ATF4.14 An increase of ATF4 was also associated with sorafenib resistance in hepatocellular carcinoma.19 Of note, the increase of autophagy was also observed in FLT3-ITD-mutated AML cells in a BM-mimicking, co-culture system (Figure 1C), which implies that BM-mediated resistance may result from the upregulation of autophagy in addition to CXCL12-CXCR4 axis-mediated leukemia cell homing and HIF1α-induced modulation of cell survival signaling.28,29 HIF1α may also be involved in the upregulation of autophagy, as shown in Figure 1C. Indeed, HIF1α or HIF2α/BNIP3 triggered induction of autophagy during hypoxia in prostate carcinoma cells,30 and recently the HIF 1α/miR 224 3p/ATG5 axis was reported to be involved in autophagy regulation and induced drug resistance in glioblastoma and astrocytoma cells.31 Hypoxia can activate autophagy through the endoplasmic reticulum stress-related PERK/eIF2α/ATF4 signaling pathway as well.32 Interestingly, we observed a positive association between the upregulation of phospho-BTK and autophagy in the resistant cells, especially in the presence of BM-mimicking hypoxia or MSC, implying a pivotal role for BTK level in the modulation of autophagy. In addition, BTK could be activated by FLT3 activation and sensitizes AML to BTK inhibition,33 supporting the putative association of BTK and FLT3 activation, which may also result in the upregulation of BTK in FLT3-mutated AML cells along with resistance to FLT3 inhibitors (sorafenib in this case). In terms of BMmediated resistance, BTK could also be upregulated by CXCL12 in chronic lymphocytic leukemia,34 and its high level is associated with the upregulation of CXCR4 in myeloma as well.35 AML cells with FLT3 mutations demonstrate a high level of CXCR4,36 which could be further upregulated with FLT3-targeted treatment (Online Supplementary Figure S17). A clinical trial with a combinatorial regime co-targeting FLT3 and CXCR4 with sorafenib and plerixafor led to substantial response rates in relapsed/refractory FLT3-mutated AML.37 Therefore, suppression of BTK might provide additional benefit for overcoming FLT3 inhibitor resistance of the FLT3/CXCR4 combinatorial regime in a BM microenvironment scenario by repressing BTK-mediated upregulation of autophagy. It has been observed that suppression of BTK with ibrutinib, BTK knockdown or CG-806 profoundly abrogated the resistance and sensitized AML cells to FLT3-targeted therapy, accompanied by a decrease of phospho-BTK, ATF4 (or beclin-1) and autophagy in our in vitro and in vivo experiments, suggesting a potential of targeting BTK in overcoming FLT3inhibitor resistance in FLT3-mutated AML.
Unexpectedly, targeting BTK with CG-806 did not inhibit autophagy in FLT3 WT AML cells. Conversely, we observed upregulation of the pro-autophagic proteins Beclin-1, ATG7 and phospho-ULK1 (Figure 5A). Our data documented the profound suppression of aurora kinases B and C, rather than BTK inhibition, in these cells (Online Supplementary Figure S10). Several groups have reported that targeting aurora kinase triggers pro-autophagic effects in an AMPKULK1-dependent manner in cancer cells.38-40 The multi-kinase inhibitor CG-806 exerts potent inhibition of FLT3, BTK, and aurora kinases at relatively low concentrations (e.g., IC50 values less than 5 nM) ( Online Supplementary Table S3). However, it mainly triggers aurora kinase inhibition in FLT3 WT cells. In fact, FLT3 WT cells demonstrated higher activation of aurora kinase (i.e., a high basal level of phospho-aurora kinase) compared to phosphoFLT3 and phospho-BTK, implying that aurora kinase, but not FLT3 or BTK, might be a driver of survival signaling in FLT3 WT AML cells (data not shown). Aurora kinase signaling has recently been identified by Druker’s group as an important mechanism of early resistance to the FLT3 inhibitor gilteritinib, and pharmacological inhibition of aurora kinase re-sensitized gilteritinib-induced anti-leukemia effects in FLT3-mutant AML.41 Of note, CG-806 led to impressive suppression of aurora kinase not only in FLT3 WT cells but also in FLT3-mutant AML cells (data not shown), implying that targeting aurora kinase in addition to FLT3 can overcome resistance to FLT3 inhibitors in treating both FLT3 WT and FLT3-mutant AML patients.
Our data confirm that pharmacological inhibition of autophagy using chloroquine, or knockdown of ATG7, notably enhanced sensitivity of the FLT3 WT AML cells to CG-806 (Figure 6A-C), suggesting a protective effect of autophagy in FLT3 WT AML cells as well. Of note, CG-806 showed much more pronounced cytostatic effects than cytotoxic effects in FLT3 WT leukemia, even at extremely low concentrations (the IC50 values were 3.88 and 11.81 nM in THP1 and OCI/AML3 cells, respectively), but no apoptogenic effect was observed even at micromolar concentrations (data not shown) . Thus, CG-806 predominantly inhibits aurora kinase activity in FLT3 WT cells and further downregulates the phospho-histone-H3 and CDC25c-CDK2cyclin B1 axis, which triggers G2/M arrest, but not apoptosis, resulting in preferential inhibition of proliferation, which may be one of main anti-leukemia mechanisms of CG-806 in FLT3 WT AML.
Interestingly, we observed marked G2/M arrest in the FLT3 WT cells OCI/AML3 and THP-1 (Figure 5B), but only G1 arrest in the FLT3-mutated cells MOLM14 and MV4;11 (Online Supplementary Figure S18), after being exposed to low IC50 doses (5 to 20 nM) of CG-806. The increase in DNA content of more than 4N within a single nucleus of the FLT3 WT cells may have resulted from abrogation of mitosis as well as S-phase perturbation.
Haematologica | 108 June 2023 1511 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
The formation of polyploidy is frequently observed when cancer cells are exposed to DNA-damaging agents at doses that are insufficient to induce apoptosis, such as those used in chemotherapy or radiotherapy. DNA-damaged cancer cells stop mitotic procession while undergoing repeated cycles of DNA synthesis, leading first to an increase in cells in the G2 phase of the cell cycle, which can then lead to polyploidy. Although the impact of polyploidy on leukemia cell fate is still largely unclear, inhibition of aurora kinase has been reported to induce a mitotic block resulting in apoptosis-resistant polyploidy, which can be enhanced by cyclin G1 in breast cancer cells.42 Activation of aurora kinase is important for mitotic procession.43 Selective inhibition of aurora kinase B triggered inhibition of phosphorylated-histone H3, inducing polyploidy formation. Further inhibition of proliferation of a variety of leukemia, lymphoma, and solid tumor cell lines was eventually followed by cell death after 48 h.44-46 Although we have no direct evidence to address whether polyploidization plays a role in leukemia cell killing in FLT3 WT cells after exposure to CG-806, polyploid AML cells that transition through mitosis would be expected to trigger lethal multipolar cell division in the next interphase.47 Our data have indeed shown a marked inhibition of proliferation
in FLT3 WT cells exposed to low doses of CG-806, which was closely associated with G2/M arrest, polyploidy formation, and DNA damage. The last effect was shown with immunoblotting by an increase of γH2AX (Figure 5E). γH2AX is a central player in DNA damage repair.48 In fact, it has been reported that polyploid cancer cells enter aberrant mitosis leading to cell death.49 PLK1 is a key intermediate in triggering mitotic exit through activation of cyclin B1 and cdc25c,50 which have been observed following exposure of FLT3 WT cells to low doses of CG-806. It has been reported that inhibition of PLK1 resulted in G2/M cell cycle arrest and polyploidy, which mediated anti-tumor activity in a PDX model of colorectal cancer.51
Taken together, our results suggest that autophagy is accompanied by an increase in BTK activity as one of the potential mechanisms of resistance, which can be induced by culture conditions that mimic the BM microenvironment. Pharmacological inhibition of autophagy re-sensitized resistant leukemia cells to cell death and co-targeting BTK/FLT3 with CG-806 exerted a marked anti-leukemia effect in FLT3ITD and FLT3-ITD/TKD double-mutated AML cells by suppressing survival signaling and autophagy. In addition, CG-806 had a robust anti-leukemia effect in FLT3 WT AML by arresting cell cycle progression and suppressing DNA re-
Figure 8. Summary of proposed effects of CG-806 in acute myeloid leukemia. FLT3-ITD mutations constitutively activate downstream proliferative signaling pathways, including the MEK/ERK, PI3K/Akt/mTOR and STAT5 pathways, and lead to aberrant growth of leukemia cells. Leukemia cells harboring TKD mutations (with or without internal tandem duplications) demonstrate higher basal levels of autophagy, which can be upregulated under FLT3-inhibitor stress. Phospho-FLT3, phospho-BTK and bone marrow microenvironment-mediated HIF1α also upregulate ATF4 and result in further upregulation of autophagy. The autophagy level is strongly associated with resistance to FLT3 inhibitors. Suppression of autophagy by knocking down BTK or impairing autophagic flux by chloroquine (CQ) through decreasing autophagosome-lysosome fusion re-sensitizes FLT3-mutated acute myeloid leukemia (AML) cells to FLT3 inhibition or CG-806induced pro-apoptotic effects and G1 arrest, leading to the killing of leukemia cells. On the other hand, aurora kinase plays a critical role in driving survival signaling in FLT3 WT AML cells. Targeting aurora kinase with either its specific inhibitor SNS314 or CG-806 upregulates pro-autophagic proteins phospho-AMPK, -ULK1, -ATG7, and -Beclin-1. CG806 exerts dominant inhibition of aurora kinase activity in FLT3 WT cells and further downregulates PLK-1, phospho-Histone-H3 and the proteins of the CDC25c-CDK2-cyclin B1 axis, which trigger G2/M arrest, but not apoptosis. Inhibition of aurora kinase also impairs the procession through mitosis and results in polyploidization. Targeting FLT3 WT AML cells with CG-806 triggers cell growth inhibition through G2/M arrest, polyploidy formation, and DNA damage by dominant suppression of aurora kinase.

Haematologica | 108 June 2023 1512 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
plication upon aurora kinase inhibition. Mechanistically, low doses of CG-806 triggered the inhibition of proliferation in FLT3 WT AML through induction of G2/M phase arrest and polyploidization, while high doses triggered apoptosis (Figure 8). This is the first report that the novel multi-kinase inhibitor CG-806 counteracts FLT3-inhibitor resistance in AML by suppressing autophagy through co-targeting BTK and FLT3. It was recently reported that CG-806 demonstrated greater potency against primary AML samples from 364 patients with relapsed or transformed AML.52 Our work provides additional evidence to suggest that CG-806 could serve as a clinical drug for treating relapsed/refractory AML irrespective of FLT3 mutational status. A clinical trial in AML (https://clinicaltrials.gov/ct2/show/NCT04477291) is ongoing with recently observed complete remission in a FLT3-inhibitor-resistant patient.53 More detailed data will be reported at the 2022 ASH Annual Meeting (Goldberg AD, et al. ASH Abstract #2676).
Disclosures
HZ and WGR are employees of Aptose Biosciences; MA serves on the Aptose Biosciences Scientific Advisory Board.
Contributions
WZ contributed to the design and conduct of the experiments, animal studies, data analysis, and preparation of the manuscript; GY performed most of the in vitro experiments including transmission electron microscopy and analyzed the
References
1. Medinger M, Passweg JR. Acute myeloid leukaemia genomics. Br J Haematol. 2017;179(4):530-542.
2. Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510.
3. Zhang W, Konopleva M, Shi YX, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100(3):184-198.
4. Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681-3687.
5. Borthakur G, Kantarjian H, Ravandi F, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011;96(1):62-68.
6. Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia. 2015;29(12):2390-2392.
7. Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260-263.
8. Zhang W, Gao C, Konopleva M, et al. Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clin Cancer Res. 2014;20(9):2363-2374.
data; HZ and WGR provided the CG-806 and were involved in analyzing the data; MB and CL performed some of the in vitro experiments; BY and QZ assisted with conducting in vivo studies; VR contributed to vector construction and gene knockdown experiments; SP, SB, GB, VLB and MK contributed to discussions about the manuscript; and MA contributed to the experimental design, data analysis and interpretation, and reviewed and edited the manuscript.
Acknowledgments
The authors would like to thank Dr. Neil Shah for providing FLT3-ITD/TKD double mutant cells.
Funding
This work was supported in part by a grant from Aptose Biosciences, the Paul and Mary Haas Chair in Genetics, a National Institutes of Health Cancer Center support grant (P30CA016672), and CPRIT grant RP130397 (to MA). This work used MD Anderson Cancer Center Flow Cytometry and Cell Imaging, Research Animal Support, and Characterized Cell Line Core Facilities, which were all supported by a National Institutes of Health Cancer Center Support Grant (P30CA016672).
Data-sharing statement
The data that support the findings of this study are available from the corresponding author (MA), upon reasonable request.
9. Rodriguez-Hernandez MA, Gonzalez R, de la Rosa AJ, et al. Molecular characterization of autophagic and apoptotic signaling induced by sorafenib in liver cancer cells. J Cell Physiol. 2018;234(1):692-708.
10. Lin JC, Huang WP, Liu CL, et al. Sorafenib induces autophagy in human myeloid dendritic cells and prolongs survival of skin allografts. Transplantation. 2013;95(6):791-800.
11. Prieto-Dominguez N, Ordonez R, Fernandez A, et al. Modulation of autophagy by sorafenib: effects on treatment response. Front Pharmacol. 2016;7:151.
12. Takahashi H, Inoue J, Sakaguchi K, Takagi M, Mizutani S, Inazawa J. Autophagy is required for cell survival under L-asparaginaseinduced metabolic stress in acute lymphoblastic leukemia cells. Oncogene. 2017;36(30):4267-4276.
13. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856-880.
14. Heydt Q, Larrue C, Saland E, et al. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene. 2018;37(6):787-797.
15. Piya S, Kornblau SM, Ruvolo VR, et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood. 2016;128(9):1260-1269.
16. Clodi K, Kliche K-O, Zhao S, et al. Cell-surface exposure of phosphatidylserine correlates with the stage of fludarabine-
Haematologica | 108 June 2023 1513 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
induced apoptosis in chronic lymphocytic leukemia (CLL) and expression of apoptosis-regulating genes. Cytometry. 2000;40(1):19-25.
17. Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457-481.
18. Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720-5728.
19. Adjibade P, St-Sauveur VG, Quevillon Huberdeau M, et al. Sorafenib, a multikinase inhibitor, induces formation of stress granules in hepatocarcinoma cells. Oncotarget. 2015;6(41):43927-43943.
20. Popelka H, Klionsky DJ. Post-translationally-modified structures in the autophagy machinery: an integrative perspective. FEBS J. 2015;282(18):3474-3488.
21. Zachari M, Ganley IG. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017;61(6):585-596.
22. Tabe Y, Konopleva M. Role of microenvironment in resistance to therapy in AML. Curr Hematol Malig Rep. 2015;10(2):96-103.
23. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435-1455.
24. Ishizawa J, Kojima K, Chachad D, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53independent apoptosis in hematological malignancies. Sci Signal. 2016;9(415):ra17.
25. Wawryk-Gawda E, Chylinska-Wrzos P, Lis-Sochocka M, et al. P53 protein in proliferation, repair and apoptosis of cells. Protoplasma. 2014;251(3):525-533.
26. Bagheri-Yarmand R, Nanos-Webb A, Biernacka A, Bui T, Keyomarsi K. Cyclin E deregulation impairs mitotic progression through premature activation of Cdc25C. Cancer Res. 2010;70(12):5085-5095.
27. Arbitrario JP, Belmont BJ, Evanchik MJ, et al. SNS-314, a panaurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2010;65(4):707-717.
28. Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10(8):858-864.
29. Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood. 2009;113(7):1504-1512.
30. Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570-2581.
31. Huang S, Qi P, Zhang T, Li F, He X. The HIF1alpha/miR2243p/ATG5 axis affects cell mobility and chemosensitivity by regulating hypoxia-induced protective autophagy in glioblastoma and astrocytoma. Oncol Rep. 2019;41(3):1759-1768.
32. Song S, Tan J, Miao Y, Sun Z, Zhang Q. Intermittent-hypoxiainduced autophagy activation through the ER-stress-related PERK/eIF2alpha/ATF4 pathway is a protective response to pancreatic beta-cell apoptosis. Cell Physiol Biochem. 2018;51(6):2955-2971.
33. Oellerich T, Mohr S, Corso J, et al. FLT3-ITD and TLR9 use Bruton tyrosine kinase to activate distinct transcriptional programs mediating AML cell survival and proliferation. Blood. 2015;125(12):1936-1947.
34. Montresor A, Toffali L, Rigo A, Ferrarini I, Vinante F, Laudanna C. CXCR4- and BCR-triggered integrin activation in B-cell chronic lymphocytic leukemia cells depends on JAK2-activated Bruton's
tyrosine kinase. Oncotarget. 2018;9(80):35123-35140.
35. Bam R, Ling W, Khan S, et al. Role of Bruton's tyrosine kinase in myeloma cell migration and induction of bone disease. Am J Hematol. 2013;88(6):463-471.
36. Fukuda S, Broxmeyer HE, Pelus LM. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood. 2005;105(8):3117-3126.
37. Borthakur G, Zeng Z, Cortes JE, et al. Phase 1 study of combinatorial sorafenib, G-CSF, and plerixafor treatment in relapsed/refractory, FLT3-ITD-mutated acute myelogenous leukemia patients. Am J Hematol. 2020;95(11):1296-1303.
38. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287-1295.
39. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-141.
40. Xie X, Lin W, Zheng W, et al. Downregulation of G2/mitoticspecific cyclinB1 triggers autophagy via AMPK-ULK1-dependent signal pathway in nasopharyngeal carcinoma cells. Cell Death Dis. 2019;10(2):94.
41. Joshi SK, Nechiporuk T, Bottomly D, et al. The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell. 2021;39(7):999-1014.e8.
42. Zhang W, Xu J, Ji D, et al. CyclinG1 amplification enhances aurora kinase inhibitor-induced polyploid resistance and inhibition of Bcl-2 pathway reverses the resistance. Cell Physiol Biochem. 2017;43(1):94-107.
43. Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4(11):842-854.
44. Payton M, Cheung HK, Ninniri MSS, et al. Dual targeting of aurora kinases with AMG 900 exhibits potent preclinical activity against acute myeloid leukemia with distinct post-mitotic outcomes. Mol Cancer Ther. 2018;17(12):2575-2585.
45. Glaser KB, Li J, Marcotte PA, et al. Preclinical characterization of ABT-348, a kinase inhibitor targeting the aurora, vascular endothelial growth factor receptor/platelet-derived growth factor receptor, and Src kinase families. J Pharmacol Exp Ther. 2012;343(3):617-627.
46. Carpinelli P, Moll J. Aurora kinase inhibitors: identification and preclinical validation of their biomarkers. Expert Opin Ther Targets. 2008;12(1):69-80.
47. Nakayama Y, Inoue T. Antiproliferative fate of the tetraploid formed after mitotic slippage and its promotion; a novel target for cancer therapy based on microtubule poisons. Molecules. 2016;21(5):663.
48. Nakamura AJ, Rao VA, Pommier Y, Bonner WM. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle. 2010;9(2):389-397.
49. Erenpreisa J, Kalejs M, Cragg MS. Mitotic catastrophe and endomitosis in tumour cells: an evolutionary key to a molecular solution. Cell Biol Int. 2005;29(12):1012-1018.
50. Craig SN, Wyatt MD, McInnes C. Current assessment of polo-like kinases as anti-tumor drug targets. Expert Opin Drug Discov. 2014;9(7):773-789.
51. Klauck PJ, Bagby SM, Capasso A, et al. Antitumor activity of the polo-like kinase inhibitor, TAK-960, against preclinical models of colorectal cancer. BMC Cancer. 2018;18(1):136.
52. Rice WG, Howell SB, Zhang H, et al. Luxeptinib (CG-806) targets FLT3 and clusters of kinases operative in acute myeloid leukemia. Mol Cancer Ther. 2022;21(7):1125-1135.
53. Goldberg AD, Ohanian M, Koller P, et al. A phase 1a/b dose escalation study of the mutation agnostic FLT3/BTK inhibitor luxeptinib (CG-806) in patients with relapsed or refractory acute myeloid leukemia. Blood. 2021;138(Suppl 1):1272.
Haematologica | 108 June 2023 1514 ARTICLE - Co-targeting FLT3/BTK eliminates FLT3i-resistant AML W. Zhang et al.
Somatic genetic alterations predict hematological progression in GATA2 deficiency
Laetitia Largeaud,1,2 Matthew Collin,3 Nils Monselet,4 François Vergez,1 Vincent Fregona,2 Lise Larcher,5 Pierre Hirsch,6 Nicolas Duployez,7 Audrey Bidet,8 Isabelle Luquet,1 Jacinta Bustamante,9,10,11 Stéphanie Dufrechou,1 Naïs Prade,1 Marie Nolla,12 Camille Hamelle,12 Suzanne Tavitian,13 Christophe Habib,14 Mateo Meynier,14 Christine Bellanné-Chantelot,15 Jean Donadieu,16 Flore Sicre de Fontbrune,17 Claire Fieschi,18 Alina Ferster,19 François Delhommeau,6 Eric Delabesse,1,2# and Marlène Pasquet2,12# for the French GATA2 study group
1Laboratory of Hematology, Institut Universitaire du Cancer de Toulouse, Toulouse, France; 2Université de Toulouse, Inserm, CNRS, Université Toulouse III-Paul Sabatier, Centre de Recherches en Cancérologie de Toulouse, Toulouse, France; 3Human Dendritic Cell Laboratory, Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, UK; 4Department of Bioinformatics, Institut Claudius Rigaud, Toulouse, France; 5Laboratory of Hematology, Hôpital Saint-Louis, APHP, Paris, France; 6Sorbonne Université, INSERM, Centre de Recherche Saint-Antoine, CRSA, AP-HP, SIRIC CURAMUS, Hôpital Saint-Antoine, Service d’Hématologie Biologique, Paris, France; 7Laboratory of Hematology, CHU Lille, Lille, France; 8Laboratory of Hematology, CHU Bordeaux, Bordeaux, France; 9Center for the Study of Primary Immunodeficiencies, Paris Cité University, Necker Hospital for Sick Children, APHP, Paris, France; 10Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Imaging Institute, Paris, France; 11St Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, Rockefeller University, New York, NY, USA;
12Department of Pediatric Hematology and Immunology, CHU Toulouse, Toulouse, France; 13Adult Hematology Department, CHU Toulouse, Toulouse, France; 14Bioinformatics Department, CHU Toulouse, Toulouse, France; 15Laboratory of Medical Genetics, Hôpital Pitié Salpêtrière, APHP, Paris, France; 16Pediatric Hematology Department, Hôpital Trousseau, APHP, Paris, France; 17Hematology Department, Hôpital Saint-Louis, APHP, Paris, France; 18Department of Clinical immunology, Hôpital Saint-Louis, APHP, Université Paris Cité, Paris, France and 19Pediatric Hematology, Hôpital Reine Fabiola, Bruxelles, Belgium
#ED and BP contributed equally as co-senior authors.
Abstract
Correspondence: M. Pasquet pasquet.m@chu-toulouse.fr
Received: October 13, 2022.
Accepted: January 19, 2023.
Early view: February 2, 2023.
https://doi.org/10.3324/haematol.2022.282250
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Germline GATA2 mutations predispose to myeloid malignancies resulting from the progressive acquisition of additional somatic mutations. Here we describe clinical and biological features of 78 GATA2-deficient patients. Hematopoietic stem and progenitor cell phenotypic characterization revealed an exhaustion of myeloid progenitors. Somatic mutations in STAG2, ASXL1 and SETBP1 genes along with cytogenetic abnormalities (monosomy 7, trisomy 8, der(1;7)) occurred frequently in patients with GATA2 germline mutations. Patients were classified into three hematopoietic spectra based on bone marrow cytomorphology. No somatic additional mutations were detected in patients with normal bone marrow (spectrum 0), whereas clonal hematopoiesis mediated by STAG2 mutations was frequent in those with a hypocellular and/or myelodysplastic bone marrow without excess blasts (spectrum 1). Finally, SETBP1, RAS pathway and RUNX1 mutations were predominantly associated with leukemic transformation stage (spectrum 2), highlighting their implications in the transformation process. Specific somatic alterations, potentially providing distinct selective advantages to affected cells, are therefore associated with the clinical/hematological evolution of GATA2 syndrome. Our study not only suggests that somatic genetic profiling will help clinicians in their management of patients, but will also clarify the mechanism of leukemogenesis in the context of germline GATA2 mutations.
Introduction
During the last 15 years, with the development of next-generation sequencing (NGS), familial predisposition has
emerged as an important issue in hematology, with the identification of recurrent mutated genes leading to myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML). These germline mutations frequently encode master
Haematologica | 108 June 2023 1515 ARTICLE - Bone Marrow Failure
regulatory transcription factors such as RUNX1, 1 CEBPA2 or GATA2. 3-7 Myeloid neoplasms with germline predisposition became a separate entity of the World Health Organization (WHO) hematopoietic neoplasm classification in 2016.8 Germline heterozygous mutations of GATA2 account for heterogeneous clinical and hematological manifestations encompassing immunodeficiency (monocyte, B-cell, dendritic cell and natural killer [NK]-cell deficiencies) responsible for recurrent atypical mycobacterial, fungal, bacterial and viral infections,9 vascular disorders such as lymphedema (Emberger syndrome)4,6 or defects of alveolar macrophages leading to pulmonary alveolar proteinosis (PAP).10 Eighty percent of GATA2-deficient patients develop hematological disorders before the age of 40.11 Most patients display hypocellular bone marrows with or without myelodysplastic related changes.12,13
In hematological germline predisposition syndromes, additional somatic alterations could promote leukemic transformation,14-17 but few studies detailed the molecular landscape at different stages in patients with germline GATA2 mutations. Thanks to a series of 78 patients with a long-term follow-up, we now specify a correlation between the GATA2 genotype and the clinical phenotype. Furthermore, bone marrow cytological examination of each patient led to a stratification of hematological spectra, which correlates with somatic alterations.
Methods
Primary samples and diagnostic procedures
Sixty-two patients with heterozygous germline GATA2 mutations included in the GATA2 French-Belgium registry were enrolled in this survey, from 2011 to 2022. All participants gave written informed consent to participate in the study. This registry was labeled by the French health authorities in 2008 with a clinical database approved by the French national data protection agency (CNIL certificate 97.0). Sixteen patients were enrolled in the study from the UK with material obtained with informed consent from the Newcastle Biobank (Newcastle and North Tyneside 1 research Ethics Committee Reference 17/NE/0361.75). Thirty patients were previously reported.11,18-20 Data from 500 sporadic adult AML samples (2018-2022) were extracted from the AML database of Toulouse University Hospital registered at the Commission Nationale de l’Informatique et des Libertés (CNIL, #1778920), and sequenced with the same panel, coverage and sensibility of the technique. Demographics, biological parameters and infectious status were recorded. Age at first symptom was defined as the age at the first pathological manifestation including cytopenia, MDS, chronic myelomonocytic leukemia (CMML) or AML, any infection (including recurrent bacterial infections, mycobacterial, fungal, human papillomavirus [HPV]
and Eppstein Barr virus [EBV] infections), PAP, lymphedema and deafness. Peripheral blood counts were reported at the time of bone marrow evaluation. Bone marrow smears and karyotypes were evaluated by each center (n=76). Spectrum 0 was defined as a bone marrow with normal density without myelodysplastic related changes or normal blood counts, spectrum 1 as a hypocellular bone marrow and/or low-grade MDS (without excess blasts, <5%) and spectrum 2 includes patients with MDS with excess blasts (≥5%) or AML or CMML.
GATA2 germline analysis
GATA2 germline analyses were performed by Sanger or targeted NGS of exons 2 to 6 and the regulatory region in intron 4 using as reference sequence NM_032631.4. Large genomic deletions were investigated by quantitative polymerase chain reaction (PCR) and/or multiplex ligationdependent probe amplification (MLPA) (MRC-Holland, SALSA MLPA Probemix P437 Familial MDS-AML). GATA2 germline mutation status was confirmed by analysis of non-hematopoietic tissue (cultured skin fibroblasts, hair follicles or nails). Mutations were classified as probably null (nonsense, frameshift, essential-splicing site mutations or large deletion), missense, intronic or synonymous, according to the potential consequence on protein function.
Somatic variant analysis
Mononuclear cells from bone marrow or blood samples were centrally collected from 76 patients and isolated by Ficoll centrifugation. Genomic DNA was extracted using standard procedures and sequenced using an Illumina NextSeq500 sequencer and Sureselect capture in-house panel (Agilent, Santa Clara, CA, USA) targeted on the complete coding regions (i.e., all exons were covered) and -5 to +5 splicing sites of 91 genes recurrently mutated in myeloid neoplasms (Online Supplementary Table S1). Deep sequencing was estimated with an average coverage of 4,859X, and specifically 3,379X for STAG2 exons (sensitivity 1%). Raw NGS data were analyzed using MuTect2, HaplotypeCaller (both from the GATK suite developed by the Broad Institute) and SureCall (Agilent) algorithms for variant calling aggregated in the in-house remote pipeline (Institut Universitaire de Toulouse-Oncopôle) for data visualization, elimination of sequencing/mapping errors and retention of variants with high quality metrics. Variant interpretation was performed considering minor allele frequencies (MAF) in the public GnomAD database of polymorphisms (variants with MAF >0.02 in overall population/global ancestry or sub-continental ancestry are excluded), variant allele frequencies (VAF), prevalence and clinical interpretation (COSMIC, protein impact). All variants were checked manually on IGV and compared with other samples to check for possible sequencing artifacts, then named according to the Human Genome Variation Society, and compared with se-
Haematologica | 108 June 2023 1516 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
quencing results generated by each local center. Cancer cell fractions (CCF) were calculated from VAF taking into account the chromosomal location of the genes and karyotype. All data are available on ENA_PRJEB55350.
Hematopoietic stem and progenitor cell phenotyping
Hemotopoietic stem and progenitor cell (HSPC) phenotyping was performed on fresh bone marrow samples using an antibody combination targeting CD34, CD38, CD133, CD135, CD45 and CD45RA (Online Supplementary Table S1) and analyzed on Navios instruments (Beckman-Coulter, Miami, FL) and compared to patients without hematological diseases (n=22) or aplasia (n=8) or AML (n=155). HSPC subpopulations were classified as multipotent progenitors (MPP), erythroid/myeloid progenitors (EMP), lymphoid/yeloid primed progenitors (LMPP), common myeloid progenitors (CMP), megakaryocyte erythroid progenitors (MEP), granulocyte macrophage progenitors (GMP) as described in the Online Supplementary Table S1
Statistics
Data were summarized by frequency and percentage for categorical variables and median and range for continuous variables. Associations between variables were evaluated using Chi-square or Fisher’s exact test for qualitative variables and Kruskall-Wallis test for continuous variables. All survival times were calculated from the biological sampling date and survival are described using the graphical representation of Kaplan-Meier with death from any cause for overall survival (OS) as event. Patients who survived were censored at their last follow-up or for transplanted patients at their allograft day. Univariable analyses were performed using log-rank test. Tests were two-sided and P values <0.05 were considered significant. Statistical analyses were conducted using Stata, version 16. Statistical significance of differences between CBC counts, age or CCF data was determined using multiple unpaired t-test (GraphPad Prism 7.0) and P values ****P<0.001, ***P<0.005, **P<0.01, *P<0.05 were considered significant.
Results
GATA2-mutated patients display heterogeneous clinical disorders, depending of their genotype
We investigated a series of 78 patients with heterozygous germline mutations of GATA2 (62 from the French-Belgian series and 16 British patients) from 61 kindreds bearing 46 distinct GATA2 germline mutations (Figure 1A, B; Online Supplementary Table S2). The mutations were either missense, located on or close to the second C-terminal zinc finger (44 patients; 56%) or predicted to be a null allele (32 patients; 41%) due to nonsense (11), frameshift (18) or splice defect mutations (1) or large deletions (2). In addition, one
patient has an enhancer mutation in intron 4 and one a synonymous mutation (Thr117Thr) as described recently21 (Figure 1A, B).
Age at the time of analysis ranged from 0 to 62 years with a median age of 21 years, a male/female ratio of 1.2 (Figure 1C). Seventy-three of 78 patients were symptomatic. The first symptoms occurred at a median age of 16 years, ranging from 7 months to 61 years. First symptoms were predominantly infections (27; 38%), hematological malignancies (17; 24%), cytopenias (11; 15%) or congenital abnormalities such as deafness or lymphedema (9; 13%). Eight patients combined infections with cytopenia (3; 4%), hematological malignancies (4; 6%) or lymphedema (1; 1%). There was a trend for first clinical symptoms occurring at a younger age in patients harboring null mutations, with a median age of detection of 13 years, compared to 17 years for patients with missense mutations (P=0.077). Chronic infectious complications reported before bone marrow transplantation (i.e., bacterial pneumonia, otitis, cellulitis, enteritis, arthritis) were more frequent in patients with null mutations (23 vs. 11; P<0.001). In contrast, hematological disorders were more frequent at the time of diagnosis in patients with missense mutations (17 vs. 4; P=0.005). Frequency of other infections (mycobacterial, EBV), PAP or lymphedema were similar regardless of the mutation type (Table 1). Half of the patients (41; 53%) underwent bone marrow transplantation as curative treatment with no difference regarding the type of GATA2 mutations (missense vs. null; P=0.46). OS censored at allograft is 89% and 81% with a follow up of 1 and 2 years, respectively (Online Supplementary Figure S1). A total of 61 patients are still alive (78%).
GATA2-deficient patients acquire somatic mutations in a different pattern than those with sporadic acute myeloid leukemia
Disease progression in sporadic MDS/AML is associated with karyotypic abnormalities and somatic genetic mutations.22 We compared GATA2-deficient patients to a series of 500 AML without germline GATA2 mutation (sporadic adult AML, 467 molecular samples sequenced with the same panel, coverage and threshold and 431 karyotypes). Karyotype was normal in 38 GATA2-deficient patients (50%) and 237 sporadic AML (55%; P=0.68). When compared to sporadic AML, GATA2-deficient patients more frequently have monosomy 7 (29% vs. 8%; P<0.0001), and der(1;7) (9% vs. 1%; P<0.0001), while rate of trisomy 8 was not different in the two groups (16% vs. 10%; P=0.48). Few other cytogenetic abnormalities were detected in GATA2-deficient patients (12% vs. 28% in sporadic AML; P=0.03) (Online Supplementary Figure S2A). Karyotypic abnormalities were more frequent in men (27 men vs. 11 women; P=0.006) es-
pecially chromosome 7 abnormalities (20 vs. 6; P=0.006) and trisomy 8 (10 vs. 2; P=0.033) despite a male/female ratio in the whole cohort of 1.2.
Haematologica | 108 June 2023 1517 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
Molecular analysis was performed in 76 patients with germline GATA2 mutations at a median age of 21 years (range, 0-62). One hundred and forty-one somatic mutations were identified with an average of two mutations per patient (range, 1-13; Figure 2A, B; Online Supplementary Table S4). Fifty patients (66%) had at least one mutated gene with no significant difference between missense and null GATA2 mutations (P=0.13; Online Supplementary Table S1). The most frequently mutated genes were STAG2 (53 mutations in 25 patients, 33% vs. 4% in sporadic AML; P<0.0001), ASXL1 (18 mutations in 17 patients, 22% vs. 8%; P=0.0015), SETBP1 (11 patients, 15% vs. 1%; P<0.0001), EZH2 (6 patients, 8% vs. 3%; P=0.31), the RAS pathway (11 mutations combining PTPN11, NRAS, KRAS and CBL in 5 patients, 7% vs. 26%; P=0.031) and RUNX1 (5 patients, 7% vs. 11%; P=0.75) (Online Supplementary Figure S2A). Notably, somatic GATA2 mutations were identified in four patients including two missense mutations located in the first zinc finger domain and two in-frame mutations in the second zinc finger domain (Online Supplementary Table S3). The male/female ratio was not different across mutational identities.

In contrast, frequent mutations in sporadic AML were completely or almost absent in patients with germline GATA2 mutations. DNMT3A (3% vs. 28%; P<0.0001) and TET2 (3% vs. 17%; P=0.015) mutations were found in only two GATA2deficient patients each. No mutations of NPM1 (37% in sporadic AML; P<0.0001), FLT3 (37% in sporadic AML; P<0.0001), IDH2 (10% in sporadic AML; P=0.03), IDH1 (9% in sporadic AML; P=0.06) or SRSF2 (6% in sporadic AML; P=0.16) were detected in GATA2-deficient patients. The mutation profile was mainly C>T transitions (33%). A higher rate of deletions (23% vs. 8%; P<0.0001) and a lower rate of insertions (18% vs. 31%; P=0.012) compared to sporadic AML were a consequence of the absence of FLT3 and NPM1 mutations23, 24 (Online Supplementary Figure S2B).
GATA2-deficient patients have an exhaustion of common myeloid and granulocyte macrophage progenitor
hematopoietic stem cells
In order to further depict GATA2 syndrome at a cellular level, we analyzed the HSPC compartment of patients with germline GATA2 mutations (n=11 including 5 in spectrum 0 and 6 in spectrum 1). The majority had an EMP bias (63%
A B C Haematologica | 108 June 2023 1518 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
Figure 1. Characterization of germline GATA2 mutations. (A) Distribution of coding germline GATA2 mutations across GATA2 protein. Predicted protein domains are indicated inside each bar (dark: link zinc finger domain, grey: enlarged zinc finger domain); each dot represents 1 single patient. Each color indicates a type of mutations (frameshift: purple; nonsense: magenta; missense: teal; synonymous: orange). (B) Proportion of germline GATA2 mutation types. (C) Distribution of patients (males, females) according to their age in years at the time of analysis.
Hematological type: acute myeloid leukemia, myelodysplatic syndromes (spectrum 1 and 2); univariable analyses were performed using logrank test. Tests were two-sided and P values <0.05 were considered significant. HPV: human papillomavirus; EBV: Epstein Barr virus.
vs. 13% in normal bone marrows; P<0.0001; Figure 3A, B; Online Supplementary Figure S3) with a loss of heterogeneity in contrast to normal bone marrow where MPP was the major population in the HSC compartment (79%; Figure 3A, B). Specifically, at the progenitor level, the MEP subpopulation was overrepresented compared to normal bone marrow and AML patients (66% vs. 27% and 10%, respectively; P<0.0001; Figure 3A, B; Online Supplementary Figure S3) characterizing a GATA2-specific profile. This pattern is close to aplastic patients with a decrease in MPP and CMP
proportions. However, GATA2-deficient patients harbor a higher proportion of MEP and a lower proportion of GMP than aplastic anaemia patients (Figure 3B; Online Supplementary Figure S3). Moreover, it has been reported that the number of HSPC in GATA2-deficient patients is decreased in the same way as aplastic patients.25 Altogether, the differentiation bias in GATA2-deficient patients appears to be related rather to an exhaustion of CMP and GMP populations than to a proliferation of MEP cells. We note that part of the MEP population in GATA2 condition strongly
ex-
Type of GATA2 mutation, N (%) P value Missense (N=44) Null (N=32) Type of first event (N=70) Congenital Cytopenia Hematological Infectious Infectious + congenital Infectious + cytopenia Infectious +hematological Asymptomatic Missing data 3 (7.7) 6 (15.4) 14 (35.9) 10 (25.6) 0 (0.0) 3 (7.7) 3 (7.7) 5 0 6 (19.4) 4 (12.9) 3 (9.7) 16 (51.6) 1 (3.2) 0 (0.0) 1 (3.2) 0 1 0.017 Infection as first event (N=70) No Yes Missing data 23 (59.0) 16 (41.0) 5 13 (41.9) 18 (58.1) 1 0.157 Hematological sign as first event (N=70) No Yes Missing data 22 (56.4) 17 (43.6) 5 27 (87.1) 4 (12.9) 1 0.005 Age in years at first event (N=70) Median Range Missing data 17.0 0.6-61.0 4 13.0 0.9-30.0 2 0.077 Chronic infection during follow-up (N=76) No Yes 33 (75.0) 11 (25.0) 9 (28.1) 23 (71.9) <0.001 HPV (warts + cancers) (N=76) No Yes 28 (63.6) 16 (36.4) 19 (59.4) 13 (40.6) 0.706 Chronic EBV (N=76) No Yes 41 (93.2) 3 (6.8) 30 (93.8) 2 (6.3) 1.00 Mycobacteria (N=76) No Yes 39 (88.6) 5 (11.4) 27 (84.4) 5 (15.6) 0.734 Pulmonary alveolar proteinosis (N=76) No Yes 41 (93.2) 3 (6.8) 29 (90.6) 3 (9.4) 0.692 Lymphoedema (N=76) No Yes 39 (88.6) 5 (11.4) 25 (78.1) 7 (21.9) 0.215
Table 1. Genotype/phenotype correlation.
Haematologica | 108 June 2023 1519 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
press the CD133 marker which is usually little or not expressed on MEP surface26 (Figure 3A; Online Supplementary Figure S3). This observation may suggest that germline
GATA2 mutations also lead to a phenotypic expression bias in the remaining majority population.
Figure 2. GATA2 de
ciency syndrome defines a distinct entity regarding molecular profiles. (A) Molecular and cytogenetic abnormalities in the cohort of 78 GATA2-mutated patients. (B) Somatic mutation occurrence. Summary of patients with GATA2 deficiency (n=78) organized by spectra (spectrum 0: grey, spectrum 1: blue and spectrum 2: red), germline GATA2 mutation type (missense: teal, null: fuchsia, other including synonymous and intronic mutations: turquoise), survival and bone marrow transplantation status and somatic mutation and cytogenetic status. Each vertical row represents 1 patient. Grey boxes indicate no data for that parameter. Cytogenetic abnormalities are grouped together in the same manner as the main molecular abnormalities (STAG2, ASXL1 and SETBP1). The number of abnormalities is indicated in each square. The other mutations are listed below including genes encoding transcription factors, splicing factors, chromatin modifiers, cohesin members, signaling pathway genes.

A B Haematologica | 108 June 2023 1520 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
fi

Continued on following page. A B Haematologica | 108 June 2023 1521 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
Figure 3. Phenotypical characterization of hematopoietic stem and progenitor cells revealed loss of heterogeneity associated with common myeloid and granulocyte macrophage progenitors exhaustion. (A) Comparison of hematopoietic stem and progenitor cell (HSPC) phenotypic profiles visualized by a non-linear dimensionality reduction technique (t-SNE) between GATA2 deficiency patients (GATA2, blue, n=11) and control patients without hematological diseases (normal bone marrow [NBM] CD34+, grey, n=22) or acute myeloid leukemia (AML) (red, n=155) or aplasia (purple, n=6) patients without germline GATA2 mutations. Merged samples are localized at the left plots. The first plot line represents density of each condition, the second line the distribution of the different HSC and hematopoietic progenitor cells (HPC) subpopulations, the lines below show normalized mean fluorescent intensity (MFI) according the color scale of markers (CD38, CD45RA, CD135 and CD133). The black arrow on the CD133 plot of GATA2 condition identifies of megakaryocyte erythroid progenitors (MEP)-expressing CD133 marker. (B) Proportion of HSPC populations in CD34+ CD38- compartment (erythroid/myeloid progenitors [EMP]: CD135- CD45RA-; multipotent progenitors [MPP]: CD135+ CD45RA-; lymphoid/myeloid primed progenitors [LMPP]: CD135+ CD45RA+) and CD34+ CD38+ compartment (MEP: CD135CD45RA-; common myeloid progenitors [CMP]: CD135+ CD45RA-; granulocyte macrophage progenitors [GMP]: CD135+ CD45RA+) of GATA2-deficient patients (blue) compared with NBM CD34+ (grey), AML (red) and aplastic (purple) patients. Spectrum 0 (diamond), spectrum 1 (round); median and P values are calculated using non-parametric unpaired Mann-Whitney test ****P<0.001,***P<0.005 **, P<0.01, *P<0.05.
Hypocellular bone marrow - low-grade myelodysplastic syndromes represents the main hematological spectrum in GATA2-deficient patients
In order to further investigate the impact on hematopoiesis, we reviewed bone marrow smears of 76 patients and defined three distinct morphological categories: normal bone marrow (spectrum 0), hypoplastic and/or low-grade MDS (spectrum 1) or overt transformation (spectrum 2) (Figure 4A). The majority of GATA2-mutated patients had features of spectrum 1 (47; 62%), 19 had evidence of hematopoietic transformation (19; 25%) and only ten are in spectrum 0 (13%) (Figure 4B). Despite a different bone marrow pattern, there was no difference on survival, probably because most patients received an HSC transplantation before progression. Age had no impact (Figure 4C; P=0.83) but patients in spectrum 2 are at high risk of progression to MDS or AML in the year after diagnosis of the first event (P=0.036). As expected, null alleles are associated with spectrum 1 in contrast to missense mutations enriched in spectrum 2 (P=0.023; Figure 4D). Infections (mycobacterial, EBV), PAP or lymphoedema were not correlated with the spectra, with the exception of chronic HPV infections which were enriched in spectrum 1 (52 vs. 16; P=0.008).
Regarding the parameters of the peripheral blood count, absolute lymphocyte count showed no significant differences between spectra while hemoglobin concentration, platelet, absolute neutrophil and monocyte counts decreased progressively with advanced spectra. Six patients with spectrum 2 have an increased monocyte count (Figure 4E; Online Supplementary Table S5).
Somatic molecular and karyotypic abnormalities drive hematopoietic evolution
None of the patients at spectrum 0 have somatic mutations, or karyotypic abnormalities (Figure 5A). Mutation numbers increase at spectrum 2 (median of 3 vs. 1 at spectrum 1; P=0.022; Figure 2B) with an enrichment in SETBP1, RAS pathway and RUNX1 mutations (47% vs. 6% at spectrum 1; P<0.001; 29% vs. 0%; P<0.001; 23% vs. 2%; P=0.015, respectively). Karyotypic abnormalities increased from
spectrum 0 to spectrum 2 (0% at spectrum 0, 47% at spectrum 1 and 84% at spectrum 2) and with an enrichment of the other cytogenetic abnormalities (42% vs. 2%; P<0.001). Notably, monosomy 7 increased through spectra (none at spectrum 0, 28% at spectrum 1 and 47% at spectrum 2; P=0.12; Figure 5B). Leukemic transformation is associated with a high clone size at spectrum 2 as demonstrated by patients #62, #63 and #70 (Figure 5C; Online Supplementary Tables S2 and S4). In patients #62 and #63, karyotypic abnormalities were the first event, followed by SETBP1 and PTPN11 mutations respectively. Patient #70 had a major driver clone with monosomy 7 and several mutated genes, with acquisition of monosomy 21. In patient #22, clonaldynamics analysis showed a selective advantage of the major clone with monosomy 7 and SETBP1 mutation over STAG2-mutated clones detected at diagnosis (Figure 5D). These results suggest that the selective advantages of the clones were shaped by the acquisition of new abnormalities in distinct genes. According to these results, SETBP1 mutations may be critical for leukemic transformation in GATA2-deficient patients. Nine of 11 SETBP1-mutated patients also harbored a monosomy 7 (P<0.001), and their association was significantly enriched at spectrum 2 (31% vs. 6%; P=0.008). This co-occurrence was associated with a higher monocyte count (Figure 5E; P<0.001). Interestingly, SETBP1 mutations and monosomy 7 were the earliest oncogenic events to occur in patients (Figure 5F; median age 9.3 years). Patients with RUNX1 mutations had the highest median age (46 years) while being also enriched in spectrum 2. Patients with ASXL1 or EZH2 mutations or trisomy 8 were also older with a median age of 30 years, 26 and 27 years, respectively, without significant differences according to spectra. The only mutated gene significantly enriched in spectrum 1 was STAG2 (47% vs. 18%; P=0.035; Figure 5B) with a median age of 28 years (Figure 5F), and there was no difference across blood parameters, compared to patients in spectrum 1 without STAG2 mutation (Online Supplementary Figure S3). Our results strongly suggest that somatic acquired mutations in mutated GATA2 patients have a different impact on the leukemogenesis process.
Haematologica | 108 June 2023 1522 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
Figure 4. Classification in three hematological spectra based on cytological evaluation. (A) Representative pictures of normal density bone marrow defining spectrum 0 (left, objective 10x), hypocellular bone marrow (objective 10x) and an example of myelodysplastic-related changes: multinuclear megakaryocyte (objective 63x) reported in patients in spectrum 1 (middle), blast cells defining overt transformation for spectrum 2 including myelodysplastic syndromes (MDS) with excess of blasts, acute myeloid leukemia (AML) and chronic myelomonocytic leukemia (CMML) (objective 63x, right). (B) Proportion of patients in each spectrum (0, 1 and 2), P=not significant. (C) Distribution of patient age in each spectrum. (D) Percentage of patients with missense or null mutations in each spectrum. (E) Blood count parameters (hemoglobin level, platelet, granulocyte, monocyte and lymphocytes counts) of 78 patients in each spectrum. All data points represent each patient values according to spectra with median (wide dots) ± quartiles (small dots). P values are calculated using unpaired t-tests ***P<0.001, **P<0.01, *P<0.05.
Somatic mutations of STAG2 do not drive leukemic transformation in contrast to SETBP1 mutations

In order to further characterize the impact of somatic mutations in hematopoietic transformation, cancer cell fraction (CCF) was calculated for the most frequently mutated genes. STAG2 mutations exhibited a considerably lower CCF than those of ASXL1 and SETBP1 (median of 6% vs. 24% and 78%, respectively; Figure 6A). Some patients in spectrum 1 harbored a single mutation with low CCF (patient #65 for ASXL1 or #73 for STAG2), while others exhibited several mutations occurring in unique or probably multiple clones (Figure 6B, patients #13, #17, #5 and #43). Interestingly, despite a high proportion of mutated cells, patients #17 and #5 had no excess of blasts suggesting
that not all abnormalities lead to spectrum 2. STAG2 mutations were identified in 25 patients (10 males and 15 females) with up to eight different STAG2 mutations in the same individual (mean 2.1). STAG2 mutations are loss of function, mainly due to the introduction of premature stop codons leading to a destabilization of the cohesin complex27,28 (Figure 6C). Low CCF (≤20%) for STAG2 mutations were rarely associated with other molecular abnormalities in spectrum 1 patients (Figure 6D). Among the STAG2-mutated patients, only three were classified at spectrum 2. STAG2 mutations in patients #39 and #46 had low CCF suggesting that other mutations drove the leukemic transformation. Patient #43 exhibited a higher STAG2 CCF but in a leukemic clone probably driven
A B C D E Haematologica | 108 June 2023 1523 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
by the primary SETBP1 mutation (Figure 6D). In order to gain insight in the potential of clonal growth linked to STAG2 mutations, we monitored longitudinal follow-up of two patients. After detection, STAG2-mutated clones could be little selected (patient #51) or with little CCF variations upon time (patient #48). Importantly, these two patients remained at spectrum 1 after several years (Figure 6E). Overall, our analysis demonstrate that STAG2 somatic mutations were recurrent in GATA2-deficient patients, indicating that they may provide a selective advantage to GATA2-deficient hematopoietic cells although insufficient for their transformation.

Discussion
While the majority of MDS and AML are sporadic, rare germline predisposition syndromes have been delineated.29 Myeloid transformations in the context of germline alterations have variable latency but usually occur in younger patients than sporadic malignancies, with the exception of germline DDX41 mutations.17 GATA2 deficiency syndrome, recognized as a major MDS/AML predisposition in the WHO classification, has a high although variable penetrance.11,30 This series highlights that patients with missense mutations may have a higher risk of transformation, and are thus
Figure 5. Stratification of genetic abnormalities according hematological spectra. (A) Distribution of genomic alterations including molecular and cytogenetic abnormalities in each spectrum. (B) Proportion of patients in each spectrum according to their mutational and cytogenetic abnormalities (blue: spectrum 1; red: spectrum 2). (C) Clonal hierarchy of 3 patients at spectrum 2, evaluated thanks to cancer cell fraction (CCF). (D) Clonal dynamics evaluated by molecular and cytogenetic follow-up of one patient with two STAG2 mutations at diagnosis (spectrum 1) which disappeared 1 year later (spectrum 2), concomitantly to the appearance of a clone with monosomy 7 and a SETBP1 mutation. CCF (%) was evaluated using variant allelic frequency for mutations and polymorphisms located on chromosome 7 for monosomy 7. (E) Monocyte count in patients with the association of SETBP1 mutation and monosomy 7 (n=9) or the monosomy 7 only (n=11) or patients without SETBP1 mutations (n=45). (F) Median age in years of patients according their genetic profiles. Patients harboring the association monosomy 7 and SETBP1 mutation were compared to STAG2-mutated patients, patients with trisomy 8 and with ASXL1 mutation. All data points represent each patient values according to hematological spectra with median ± quartiles and P values are calculated using unpaired t-tests ***P<0.001, **P<0.01, *P<0.05.
A B C D E F Haematologica | 108 June 2023 1524 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
over-represented in the spectrum 2 group, as opposed to patients with null mutations. The latter seems to correlate with an earlier age of diagnosis and chronic infections. Most patients have hypocellular and/or myelodysplastic bone marrows, including four patients with no somatic mutations, raising the interesting possibility that germline GATA2 mutations may intrinsically induce this condition. In line with this hypothesis, McReynolds and colleagues described in 2019 a GATA2 deficiency-related bone marrow and immunodeficiency disorder (G2BMID),13 with bone marrow hypocellularity, atypical megakaryocytes and minimal dysmyelopoiesis and dyserythropoiesis. As dysplastic threshold in GATA2 deficiency remains hard to define, we chose to group hypoplastic bone marrow and low-grade MDS in the same spectrum. In the recent report by West and colleagues,31 13 GATA2-deficient patients were asymptomatic with a normal bone marrow. Only three had somatic mutations including DNMT3A, that can be explained by the older median age of patients in link with age-related clonal hematopoiesis.32
The molecular and cytogenetic profiles of GATA2-deficient patients differ from those of sporadic AML and adult MDS,33
and get closer to pediatric MDS.16,34 Indeed, no or very few mutations of NPM1, FLT3 or epigenetic-related genes such as IDH1/2, TET2 or DNMT3A or splicing genes (SRSF2, SF3B1, U2AF1 and SRSF2) have been identified in GATA2-deficient patients at spectrum 1 or 2, suggesting differences in the mechanism of clonal selection.33 In GATA2-deficient patients, we confirm in our study that the most frequent cytogenetic abnormalities involved chromosome 7 and somatic mutations target STAG2, ASXL1 and SETBP1. This profile more closely reflects pediatric hypocellular MDS also harbors monosomy 7 and SETBP1, ASXL1, RAS pathway and RUNX1 mutations.34,35 Sahoo et al. observed similar features in SAMD9/SAMD9L germline predisposition.16 However, GATA2-mutated patients harbored numerous STAG2 mutations, which are also reported in Down syndrome involving another GATA factor36 and in MDS with a poor OS,33 but not specifically in pediatric MDS, suggesting a specific mutational profile in GATA2-deficient patients.31,37 The analysis of HPC compartment38 reveals a loss of heterogeneity with exhaustion of CMP and GMP populations suggesting that hematopoiesis is less efficient in GATA2deficient patients. Interestingly, some residual MEP showed
Figure 6. Clonal hematopoiesis mediated by STAG2 mutations and clonal selection. (A) Comparison of cancer cell fraction (CCF) of the 3 main molecular abnormalities: SETBP1 (n=11), ASXL1 (n=17) and STAG2 (n=53) mutations. Blue and red dots correspond to spectra 1 and 2, respectively. (B) Representative examples of clonal hierarchy evaluated by CCF. (C) Proportion of the different STAG2 mutation types (n=53). (D) Percentage of STAG2 (purple) and the other (dark) mutation CCF from the 25 STAG2-mutated patients associated with the hematological spectrum (blue: spectrum 1; red: spectrum 2), sex (female: burgundy; male: green) and the cytogenetic profile (normal karyotype, monosomy 7, trisomy 8, der(1;7) and other cytogenetic abnormalities). The dotted line allows to visualize mutations with CCF less than or equal to 20%. (E) Clonal dynamic evaluated by longitudinal follow-up of 2 patients mutated only for STAG2

A B C D E Haematologica | 108 June 2023 1525 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
aberrant strong expression of CD133 marker suggesting that GATA2 deficiency also alters the remaining progenitor compartment. Functional and quantitative defects in the Gata2 +/- mouse model also suggest that germline GATA2 mutations could impair HSC fitness.39 Recently, the loss of fi tness in HSC has been described in some inherited bone marrow failures also associated with a higher risk of myeloid transformation by acquisition of genetic alterations.40 Recent studies have highlighted the ability of some abnormalities to drive a leukemic clone while others preferentially define a state of clonal hematopoiesis.16,41 We found that SETBP1 , RAS pathway, and RUNX1 mutations were enriched in spectrum 2, data which could be confirmed in a larger cohort. Monosomy 7 was present at spectrum 1 and 2, but the association with SETBP1 mutation was clearly enriched at spectrum 2. This co-occurrence suggests that the monosomy 7 clones can accumulate other genetic abnormalities and lead to leukemic transformation, corroborating recent findings in the context of other germline predisposition such as SAMD9 syndromes.16 Interestingly, patients with co-occurring monosomy 7 and SETBP1 mutations had a higher monocyte counts suggesting that it could improve
monocytic differentiation in GATA2-deficient patients. As previously reported, STAG2 mutations are recurrent in GATA2 defi ciency,31 we found that patients harboring STAG2 mutations are classified in spectrum 1, and were older than patients with SETBP1 mutations and monosomy 7. To go further, we showed that CCF of STAG2 mutations were lower than those of SETBP1 or ASXL1 mutations with up to eight STAG2 mutations per patient. Altogether, these observations suggest a different impact of STAG2 mutations, without obvious oncogenic potential but prone to induce a non-aggressive clonal hematopoiesis. Although our molecular analysis does not allow us to demonstrate that different STAG2 mutations were found in different clones, the presence of several low CCF mutations in distinct clones is a mechanism already reported in other IBMFS.41 Single- cell DNA sequencing analysis is required to confi rm it in GATA2 defi ciency. Moreover, mutations that modify the fitness of cells without association with transformation have been described in Shwachman-Diamond syndrome (SBDS) patients.41,42 The authors proposed that EIF6 mutations can counterbalance the initial deficiency induced by germline SBDS mutation and act as a somatic genetic rescue mechan-
Figure 7. Distinct pathways of somatic clonal progression in GATA2 deficient patients. Summary of the different possibilities of evolution according to the spectra. Proposal of a personalized follow-up of patients from each spectrum.
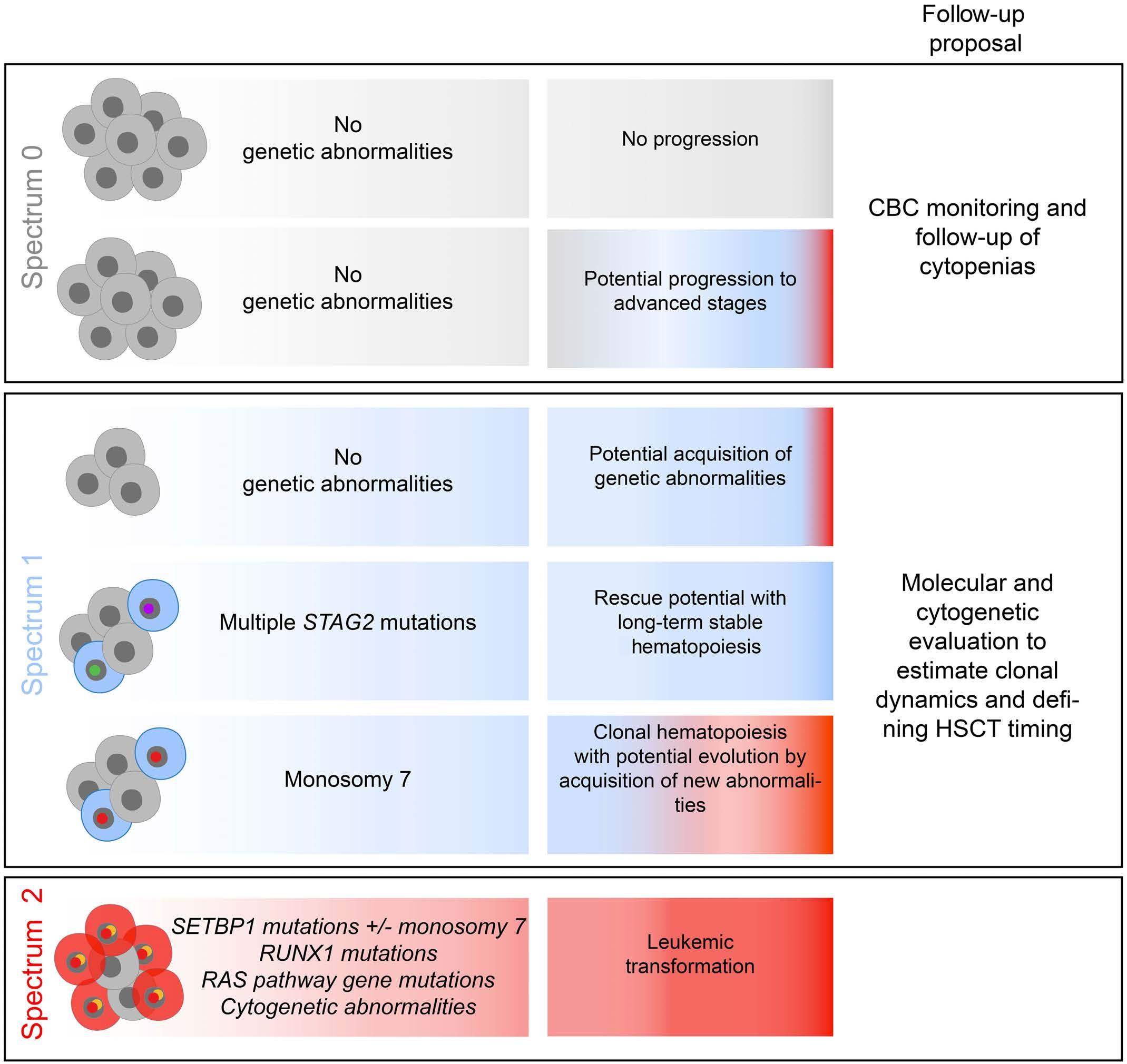
Haematologica | 108 June 2023 1526 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
ism. These mutations exhibit low VAF and up to eight different EIF6 mutations were present in the same individual, similar to what we found for STAG2 mutations. These mutations were found in multiple clones defining clonal hematopoiesis.42 STAG2 is a member of the cohesin complex, implicated in the three-dimensional folding of DNA and, thus in the regulation of numerous transcription factors28 such as GATA2 in HSPC. Indeed, its alteration can open chromatin at GATA and RUNX sites.27,43 This suggest that STAG2 mutations may increase the expression of GATA2 target genes by increasing GATA site opening. STAG2 mutations could act as a potential compensatory pathway, improving the fitness of clones with limited leukemic potential.
Overall, monitoring these genetic abnormalities is consequently of importance in the context of GATA2 deficiency (Figure 7).
Disclosures
No conflicts of interest to disclose.
Contributions
LL, NP, SD, CH, MM, ED and MP performed the experiments. LL, NP, SD, MN, ED and MP analyzed the data. LL, ED and MP wrote the manuscript. NM performed the statistical analysis. FV performed the flow cytometry experiment and analyzed the data. PH, FD, LL, CH, ND, ST, AB, IL, JB, JD, CBC, FSF, CF and AF shared the DNA samples and clinical data. FSF, CF, FD, VF and MC revised the manuscript. LL and MP designed the research. All authors have read and approved the final submitted version of the manuscript.
Acknowledgments
The team is also supported by the associations “Enfants Cancers et Santé”, “Capucine”, “Constance la Petite Guerrière Astronaute”. We acknowledge the contribution of the tumor bank HIMIP. We wish to thank all clinicians and biologists who participated in this study: Wadih Aboudchala, Pediatric Hematology, CHU Lille, Lille; Oana Balasanu, Department of Hematology, CHU Nancy, Nancy; Vincent Barlogis, Department of Pediatric Oncology and Hematology, CHU Marseille La Timone, Marseille; Sarah Beaussant Cohen, Pediatry Unit, CHU Besançon, Besançon; Nicolas Blein, Department of Hematology, CHU Nantes, Nantes; Anne Sophie Brunel, Department of Infectious Disease, CHU Montpellier, Montpellier; Benedicte Bruno, Pediatric Hematology, CHU Lille, Lille; Helene Cavé, APHP RobertDebré, Paris; François Chabot, Department of Hematology, CHU Nancy, Nancy; Sophie Ducastelle Leprete, Department of Hematology, CHU Lyon, Hôpital Lyon SUD, Lyon; Bernard Drenou, Department of Hematology, CH Mulhouse, Mulhouse; Viviana Marin Esteban, UMR 996, University Paris 11, Clamart; Jean-Hugues Dalle Department of Pediatric
Hematology and HSCT, APHP Robert Debré, Paris; PierreYves Dumas, Department of Hematology, CHU Bordeaux, Bordeaux; Laurence Faivre, Department of Genetics, CHU Dijon, Dijon; Pascale Flandrin, Laboratory of Hematology, CHU Saint Etienne, Saint Etienne; Lionel Galicier, Department of Immunology, APHP St Louis, Paris; Virginie Gandemer, Pediatric Hematology, Rennes; Federica Giannotti, Department of Hematology, APHP Saint-Antoine, Paris; Jean Gutnecht, Department of Internal Medicine, CH Frejus Saint-Raphael, Fejus; Sébastien Héritier, Department of Pediatric Oncology and Hematology, Registre National des Neutropénies Chroniques, APHP Trousseau, Paris; Eric Jeziorski, Department of Pediatrics, CHU Montpellier, Montpellier; Thierry Lamy de la Chapelle, Department of Hematology, CHU Nantes, Nantes; Hélène Lapillone, Hematology Laboratory, APHP Trousseau, Paris; Yannick le Bris, Hematology Laboratory, Nantes; Guy Leverger, Department of Pediatric Oncology and Hematology, Registre National des Neutropénies Chroniques, APHP Trousseau, Paris; Ludovic Martin, Department of Dermatology, CHU Angers, Angers; Gérard Michel, Department of Pediatric Oncology and Hematology, CHU Marseille La Timone, Marseille; Fabrice Monpoux, Department of Pediatric Oncology and Hematology, CHU Nice, Nice; Despina Moushous, Department of Pediatric Immunology, APHP Necker-Enfants
Malades, Paris; Benedicte Neven, Department of Pediatric Immunology, APHP Necker-Enfants Malades, Paris; Raphaele Nove Josserand, Department of Internal Medicine, CHU Lyon, Hôpital Lyon Sud, Lyon; Catherine Paillard, Department of Pediatric Oncology and Hematology, CHU Strasbourg, Strasbourg; Régis Peffault de la Tour, Department of Hematology, HSCT Unit, APHP Saint-Louis, Paris; Isabelle Pellier, Pediatric Hematology and Immunology, Angers; Arnaud Petit, Department of Pediatric Oncology and Hematology, Registre National des Neutropénies Chroniques, APHP Trousseau, Paris; Gaetan Sauvetre, Department of Internal Medicine, CHU Rouen, Rouen; Nicolas Schleinitz, Immunology and Internal Medicine, CHU Marseille La Timone, Marseille; Jean Soulier, APHP Saint Louis, Paris; Suzanne Tavitian, Department of Hematology, CHU Toulouse, Toulouse; Louis Terriou, Internal Medicine, CHU Lille, Lille; Pierre Van de Perre, ICR, Toulouse; Norbert Vey, Department of Hematology, HSCT Unit, Institut PaoliCalmette, Marseille and Stéphane Vignes, Department of Internal Medicine, Fondation Cognacq Jay, Paris, France.
Funding
This project was supported by associations “111 des arts”, “Toulouse Recherche Enfant Cancer”, INCa and CONECTAML project.
Data-sharing statement
Data will be shared by email request directly to the corresponding author.
Haematologica | 108 June 2023 1527 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
References
1. Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166-175.
2. Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351(23):2403-2407.
3. Dickinson RE, Griffin H, Bigley V, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656-2658.
4. Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012-1017.
5. Hsu AP, Sampaio EP, Khan J, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653-2655.
6. Ostergaard P, Simpson MA, Connell FC, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43(10):929-931.
7. Pasquet M, Bellanne-Chantelot C, Tavitian S, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. 2013;121(5):822-829.
8. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
9. Vinh DC, Patel SY, Uzel G, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115(8):1519-1529.
10. Spinner MA, Sanchez LA, Hsu AP, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809-821.
11. Donadieu J, Lamant M, Fieschi C, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. 2018;103(8):1278-1287.
12. Wlodarski MW, Hirabayashi S, Pastor V, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387-1397.
13. McReynolds LJ, Yang Y, Yuen Wong H, et al. MDS-associated mutations in germline GATA2 mutated patients with hematologic manifestations. Leuk Res. 2019;76:70-75.
14. Preudhomme C, Renneville A, Bourdon V, et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood. 2009;113(22):5583-5587.
15. Duployez N, Jamrog LA, Fregona V, et al. Germline PAX5 mutation predisposes to familial B-cell precursor acute lymphoblastic leukemia. Blood. 2021;137(10):1424-1428.
16. Sahoo SS, Pastor VB, Goodings C, et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat Med. 2021;27(10):1806-1817.
17. Duployez N, Largeaud L, Duchmann M, et al. Prognostic impact of DDX41 germline mutations in intensively treated acute myeloid leukemia patients: an ALFA-FILO study. Blood. 2022;140(7):756-768.
18. Ciullini Mannurita S, Vignoli M, Colarusso G, et al. Timely followup of a GATA2 deficiency patient allows successful treatment. J Allergy Clin Immunol. 2016;138(5):1480-1483.
19. Tholouli E, Sturgess K, Dickinson RE, et al. In vivo T-depleted reduced-intensity transplantation for GATA2-related immune dysfunction. Blood. 2018;131(12):1383-1387.
20. Sekhar M, Pocock R, Lowe D, et al. Can somatic GATA2 mutation mimic germ line GATA2 mutation? Blood Adv. 2018;2(8):904-908.
21. Kozyra EJ, Pastor VB, Lefkopoulos S, et al. Synonymous GATA2 mutations result in selective loss of mutated RNA and are common in patients with GATA2 deficiency. Leukemia. 2020;34(10):2673-2687.
22. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221.
23. Osorio FG, Rosendahl Huber A, Oka R, et al. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. 2018;25(9):2308-2316.
24. Lee-Six H, Obro NF, Shepherd MS, et al. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018;561(7724):473-478.
25. Ganapathi KA, Townsley DM, Hsu AP, et al. GATA2 deficiencyassociated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125(1):56-70.
26. Gorgens A, Radtke S, Mollmann M, et al. Revision of the human hematopoietic tree: granulocyte subtypes derive from distinct hematopoietic lineages. Cell Rep. 2013;3(5):1539-1552.
27. Ochi Y, Kon A, Sakata T, et al. Combined cohesin-RUNX1 deficiency synergistically perturbs chromatin looping and causes myelodysplastic syndromes. Cancer Discov. 2020;10(6):836-853.
28. Viny AD, Bowman RL, Liu Y, et al. Cohesin members Stag1 and Stag2 display distinct roles in chromatin accessibility and topological control of HSC self-renewal and differentiation. Cell Stem Cell. 2019;25(5):682-696.
29. Fenwarth L, Duployez N, Marceau-Renaut A, et al. Germline pathogenic variants in transcription factors predisposing to pediatric acute myeloid leukemia: results from the French ELAM02 trial. Haematologica. 2021;106(3):908-912.
30. Wlodarski MW, Collin M, Horwitz MS. GATA2 deficiency and related myeloid neoplasms. Semin Hematol. 2017;54(2):81-86.
31. West RR, Calvo KR, Embree LJ, et al. ASXL1 and STAG2 are common mutations in GATA2 deficiency patients with bone marrow disease and myelodysplastic syndrome. Blood Adv. 2022;6(3):793-807.
32. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111-121.
33. Bernard E, Tuechler H, Greenberg PL, et al. Molecular International Prognostic Scoring System for myelodysplastic syndromes. NEJM Evid. 2022;1(7).
34. Schwartz JR, Ma J, Lamprecht T, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017;8(1):1557.
35. Pastor V, Hirabayashi S, Karow A, et al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia. 2017;31(3):759-762.
36. Yoshida K, Toki T, Okuno Y, et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet. 2013;45(11):1293-1299.
Haematologica | 108 June 2023 1528 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
37. Brown AL, Hahn CN, Scott HS. Secondary leukemia in patients with germline transcription factor mutations (RUNX1, GATA2, CEBPA). Blood. 2020;136(1):24-35.
38. Vergez F, Green AS, Tamburini J, et al. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica. 2011;96(12):1792-1798.
39. Rodrigues NP, Janzen V, Forkert R, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106(2):477-484.
40. Tsai FD, Lindsley RC. Clonal hematopoiesis in the inherited
bone marrow failure syndromes. Blood. 2020;136(14):1615-1622.
41. Kennedy AL, Myers KC, Bowman J, et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat Commun. 2021;12(1):1334.
42. Tan S, Kermasson L, Hilcenko C, et al. Somatic genetic rescue of a germline ribosome assembly defect. Nat Commun. 2021;12(1):5044.
43. Mullenders J, Aranda-Orgilles B, Lhoumaud P, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med. 2015;212(11):1833-1850.
Haematologica | 108 June 2023 1529 ARTICLE - Somatic alterations in germline GATA2 deficiency L. Largeaud et al.
after allogeneic hematopoietic stem cell transplantation
Elena Tassi,1,2* Maddalena Noviello,1,2* Pantaleo De Simone,1,2 Maria T. Lupo-Stanghellini,3 Matteo Doglio,1 Francesca Serio,3 Danilo Abbati,1 Valeria Beretta,1,2 Veronica Valtolina,1,2
Giacomo Oliveira,1° Sara Racca,4 Edoardo Campodonico,3 Eliana Ruggiero,1 Daniela Clerici,3 Fabio Giglio,3 Francesca Lorentino,3 Roee Dvir,4 Elisabetta Xue,3 Francesca Farina,3 Chiara
Oltolini,5 Francesco Manfredi,1 Luca Vago,6,7 Consuelo Corti,3 Massimo Bernardi,3 Massimo
Clementi,4 Liselotte Brix,8 Fabio Ciceri,3,7 Jacopo Peccatori,3 Raffaella Greco3# and Chiara Bonini1,2,7#
1Experimental Hematology Unit, Division of Immunology, Transplantation and Infectious Diseases, Ospedale San Raffaele Scientific Institute, Milan, Italy; 2Cell Therapy Immunomonitoring Laboratory (MITiCi), Division of Immunology, Transplantation and Infectious Diseases, Ospedale San Raffaele Scientific Institute, Milan, Italy; 3Unit of Hematology and Bone Marrow Transplantation, Ospedale San Raffaele Scientific Institute, Milan, Italy; 4Laboratory of Clinical Microbiology and Virology, Ospedale San Raffaele Scientific Institute, Milan, Italy; 5Infectious Disease Unit, Ospedale San Raffaele Scientific Institute, Milan, Italy; 6Unit of Immunogenetics, Leukemia Genomics and Immunobiology, Division of Immunology, Transplantation and Infectious Diseases, Ospedale San Raffaele Scientific Institute, Milan, Italy; 7Università Vita-Salute San Raffaele, Milan, Italy and 8Immudex ApS, Virum, Denmark.
*ET and MN contributed equally as co-first authors.
#RG and CB contributed equally as co-last authors.
°Current address: Dana-Farber Cancer Institute, Boston, MA, USA.
Abstract
Correspondence: C. Bonini bonini.chiara@hsr.it
Received: January 18, 2022.
Accepted: September 27, 2022.
Early view: October 6, 2022.
https://doi.org/10.3324/haematol.2022.280685
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
After allogeneic hematopoietic stem cell transplantation (HSCT), the emergence of circulating cytomegalovirus (CMV)specific T cells correlates with protection from CMV reactivation, an important risk factor for non-relapse mortality. However, functional assays measuring CMV-specific cells are time-consuming and often inaccurate at early time-points. We report the results of a prospective single-center, non-interventional study that identified the enumeration of Dextramerpositive CMV-specific lymphocytes as a reliable and early predictor of viral reactivation. We longitudinally monitored 75 consecutive patients for 1 year after allogeneic HSCT (n=630 samples). The presence of ≥0.5 CMV-specific CD8+ cells/mL at day +45 was an independent protective factor from subsequent clinically relevant reactivation in univariate (P<0.01) and multivariate (P<0.05) analyses. Dextramer quantification correlated with functional assays measuring interferon-γ production, and allowed earlier identification of high-risk patients. In mismatched transplants, the comparative analysis of lymphocytes restricted by shared, donor- and host-specific HLA revealed the dominant role of thymic-independent CMV-specific reconstitution. Shared and donor-restricted CMV-specific T cells reconstituted with similar kinetics in recipients of CMV-seropositive donors, while donor-restricted T-cell reconstitution from CMV-seronegative grafts was impaired, indicating that in primary immunological responses the emergence of viral-specific T cells is largely sustained by antigen encounter on host infected cells rather than by cross-priming/presentation by non-infected donor-derived antigen-presenting cells. Multiparametric flow cytometry and high-dimensional analysis showed that shared-restricted CMV-specific lymphocytes display a more differentiated phenotype and increased persistence than donor-restricted counterparts. In this study, monitoring CMV-specific cells by Dextramer assay after allogeneic HSCT shed light on mechanisms of immune reconstitution and enabled risk stratification of patients, which could improve the clinical management of post-transplant CMV reactivations.
Cytomegalovirus-specific T cells restricted for shared and donor human leukocyte antigens differentially impact on cytomegalovirus reactivation risk
Haematologica | 108 June 2023 1530 ARTICLE - Cell Therapy & Immunotherapy
Introduction
The outcome of allogeneic hematopoietic stem cell transplantation (HSCT), a therapeutic option for several hematologic diseases,1 has improved dramatically in recent years.2 However, cytomegalovirus (CMV) reactivation is still one of the most important complications of allogeneic HSCT.
CMV serological status of the donor and recipient represents a well-recognized risk factor for CMV disease after allogeneic HSCT.3 Its impact on survival remains controversial, dampened by the introduction of new detection methods, highly effective prophylactic (e.g., letermovir) and/or preemptive strategies, but amplified by the growing use of new transplant platforms such as post-transplant cyclophosphamide,4,5 associated with a high incidence of viral infections, including CMV.6,7 Post-transplant immune reconstitution profoundly influences the risk of CMV reactivation and survival.8,9 Immune reconstruction occurs through different mechanisms, including thymus-independent expansion of naïve and antigen-experienced T cells present in the graft, and differentiation of donor-derived hematopoietic precursors educated in the host thymus.10 The relative contribution of these mechanisms in protecting patients from CMV still needs to be fully elucidated. Functional CMV-specific immune reconstruction, evaluated by either by interferon (IFN)-γ enzymelinked immunospot (ELISpot),11,12 flow cytometry13,14 or QuantiFERON-CMV,15,16 correlates with a lower rate of CMV reactivation. Nonetheless, IFN-γ ELISpot and cytokine detection by flow cytometry are cumbersome and time-consuming techniques. QuantiFERON-CMV on whole blood can be more easily implemented by routine diagnostic laboratories but a large percentage (38%) of results are “indeterminate”.17 Circulating CMV-specific T cells can also be enumerated directly by using fluorescent-labeled multimers of MHC-peptide complexes.18-20 Gratama et al. rigorously demonstrated that the recovery of CMV-specific T cells, evaluated by multimer staining, within 65 days after allogeneic HSCT is associated with a reduced incidence of CMV reactivation.19 Recently, more powerful tools have been developed, consisting of multiple MHC-peptide complexes and fluorochromes covalently bound to a dextran polymer backbone (i.e., Dextramer reagents). The higher number of MHC-complexes increases the binding avidity of these reagents compared to tetramers, allowing more specific and sensitive monitoring of CMV-specific CD8+ T cells early after allogeneic HSCT.21 The availability of a large repertoire of Dextramer reagents allows the evaluation of patients with different human leukocyte antigen (HLA) haplotypes.22
Recent European Guidelines for CMV management suggest monitoring IFN-γ-producing CMV-specific T lymphocytes to better identify patients at highest risk of
developing viral reactivation,23 but no consensus has yet been found on the best setting for this monitoring.24 The inclusion of CMV-specific T-cell responses in the risk stratification and clinical decision-making of patients undergoing allogeneic HSCT needs further validation in prospective clinical trials.
We report herein the results of a prospective, singlecenter, non-interventional study to ascertain the potential of CMV-specific T-cell reconstitution assessed by Dextramer assay in conferring protection against CMV-related clinically relevant events (CRE) after allogeneic HSCT with post-transplant cyclophosphamide. Furthermore, taking advantage of the possibility afforded by flow cytometry to quantify and characterize CMV-specific T cells restricted for different HLA alleles, we describe the differential contribution of CD8+ T lymphocytes restricted for either shared or donor-specific HLA to the viral-specific immune reconstitution after HLA-mismatched HSCT.
Methods
Allogeneic hematopoietic stem cell transplantation procedures
The majority of patients received a treosulfan-based conditioning regimen with post-transplant cyclophosphamide (50 mg/kg/day), on days +3 and +4, and sirolimus.25,26 Patients received antiviral prophylaxis with acyclovir, according to institutional guidelines. Until 100 days after HSCT, CMV was monitored weekly in whole blood by realtime quantitative polymerase chain reaction assay. CRE were defined as viral disease or viremia requiring preemptive treatment,27 and CMV end-organ disease was classified according to reported criteria.28 Anti-viral therapy was started after the first detection of CMV DNAemia above the cutoff (10,000 IU/mL). Furthermore, in the case of detectable CMV DNAemia below the cutoff value, even at low levels (<100 IU/mL), the analysis was repeated every 2-3 days until negativity or positivity above the threshold. All patients were treated according to institutional programs upon written informed consent for transplant procedures, use of medical records and immunological studies, within the non-interventional “CMVMON study”, approved by Ospedale San Raffaele Institutional Ethical Committee on 09/03/2017.
Dextramer staining on fresh blood
CMV-specific CD8+ T cells were quantified using a Dextramer CMV kit (Immudex) according to the manufacturer’s instructions. High HLA resolution was required to identify the correct Dextramer reagent to be used. More details, including the limit of detection and the analytical sensitivity of the assay, are provided in the Online Supplementary Materials.
Haematologica | 108 June 2023 1531 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
Flow cytometry analysis on frozen peripheral blood mononuclear cells
Peripheral blood mononuclear cells were obtained from whole blood by density gradient centrifugation (Lymphoprep, Sentinel diagnostics) and frozen. After thawing, cells were stained with a 20-color panel. High-dimensional analysis was performed using the application cytoChain.29
Interferon-γ ELISpot
Frequencies of CMV-specific T cells were assessed on frozen peripheral blood mononuclear cells by IFN-γ ELISpot (Lophius). Specific spot-forming cells (sfc) were counted by the ImmunoCapture 7.0 software (TLC ELISpot Reader). Results were expressed as CMV-specific sfc/µL.30
QuantiFERON-CMV
Peripheral blood (3 mL) was collected into QuantiFERONCMV (Qiagen) tubes and analyzed according to the manufacturer’s instruction. Data and results were analyzed using REVELATION DSX® software.
Statistical analysis
Statistical analyses were performed by GraphPad Prism 9 and Jmp 13 software. Comparisons between two groups were carried out by the Mann-Whitney test. For the comparison of more than two groups, in the presence of one variable, a Kruskal-Wallis test was used followed by the Dunn multiple comparison test. In the presence of two variables, the comparison between groups was carried out by two-way analysis of variance followed by the Sidak (for related factors) or uncorrected Fisher LSD (for not related factors) multiple comparison tests. The Shapiro-Wilk test was employed to check for normality. Multivariate analysis was performed using logistic regression. For the comparison of cumulative incidences, the Gray test was used. The time-event analysis was performed using a logrank test and Wilcoxon survival test.
More details on the Methods are included in the Online Supplementary Materials.
Results
Characteristics of the patients and allogeneic transplants
Between December 13, 2017 and February 23, 2019, 84 consecutive adult patients who underwent allogeneic HSCT in our center were screened for inclusion in the study (Figure 1A). HLA class-I alleles were not covered by the Dextramer CMV Kit in only nine patients (10.7%), who were therefore excluded.
The characteristics of the patients and their transplants are shown in Table 1. Most patients were affected by myeloid malignancies (acute myeloid leukemia, 51%; myelo-
dysplastic syndrome, 9%). At HSCT, 38% of patients were not in complete remission. According to the Disease Risk Index, patients were stratified in low-intermediate (61%), high (23%), and very high (12%) risk; three patients affected by benign disorders were not classifiable. Conditioning was myeloablative in most of cases (76%). The sources of the stem cells for transplantation were matched-unrelated donors (n=33), mismatched-related donors (n= 26), matched-related donors (n=10) and cord blood units (n=6).
Engraftment was obtained in 72 patients within a median of 24 and 22 days after HSCT for neutrophils and platelets, respectively. There were two deaths within the first 30 days, before engraftment, and one graft failure.
Eighteen patients developed grades II-IV acute graft-versus-host disease (GvHD; 24%), and 11 patients reported grades III-IV acute GvHD (15%). Relapse occurred in 18 patients (24%). At last follow-up 50 patients were alive. Causes of death were disease (n=11) or non-relapse events (n=14).
Management of clinically relevant events and immunomonitoring
CRE occurred in 39 patients (52%) at a median time of 68 days (range, 0-208) (Table 2). These patients received preemptive treatment, mainly consisting of foscarnet, ganciclovir or valganciclovir. Subsequent viral reactivations occurred in 7% of patients. Moreover, 11 patients developed end-organ CMV disease (3 cases of pneumonia and 8 of colitis/gastroenteritis), and were treated with foscarnet (n=3) or ganciclovir (n=8).
A total of 630 samples, comprising the donor apheresis and peripheral blood harvested before lymphodepletion and at eight time-points after HSCT for up to 1 year, were analyzed using the Dextramer CMV Kit (Figure 1B, Online Supplementary Figure S1), which allows the evaluation of seven HLA alleles covering ~95% of the European population (Online Supplementary Table S1). In these samples we evaluated CMV-specific CD8+ T cells restricted for HLA molecules shared (S) between the donor and the host, and for donor-specific (D) and host-specific (H) HLA. In about half the patients (47.9%) CMV-specific T cells restricted for only one HLA allele could be evaluated with the Dextramer CMV Kit, whereas for the remaining patients we could evaluate two (37.0%), three (13.7%) or four (1.4%) HLA. No differences were observed in CMV-specific T lymphocyte counts detected in patients with different numbers of evaluable restrictions (Online Supplementary Figure S2A), whether D-specific or S (Online Supplementary Figure S2B). CMV-specific T cells from 21 patients who received their transplant from HLA-mismatched donors and were evaluable for either D- or H-specific HLA restrictions were deeply characterized by polychromatic flow cytometry after thawing at +30, +60, +90, +180 and
Haematologica | 108 June 2023 1532 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
+365 days (n=84 samples). CMV-specific T-cell responses were also quantified by quantiFERON-CMV on fresh peripheral blood samples (n=47, based on the availability of the diagnostic kit) and by IFN-γ ELISpot on frozen peripheral blood mononuclear cells (n=113 samples) at +30, +60, +90 and +180 days after HSCT (Figure 1A).
Early reconstitution of cytomegalovirus-specific CD8+ T cells is
associated with reduced subsequent clinically relevant events
The majority of CRE occurred in the first 120 days after HSCT. Within the same timeframe CMV-specific T cells increased in patients, reaching the highest level at 150 days (mean=45.0 CMV-specific T cells/mL) and then remaining stable up to day 365 (Figure 2A). Notably, donor CMV-seronegative status was associated with an increased incidence of CRE (P=0.0414) (Online Supplementary Figure S3A). Despite this, counts of CMV-specific CD8+ T cells infused with the graft were comparable in patients who did or did not experience post-transplant CRE (Online Supplementary Figure S3B). This finding was confirmed even
Figure 1. Study design and enrollment flow chart. (A) Diagram showing the numbers of patients and samples included in each analysis. Eighty-four consecutive patients undergoing allogeneic hematopoietic stem cell transplantation (HSCT) were screened for inclusion in the study. Nine patients were excluded because no HLA class-I allele of either the patient or the donor was evaluable for assessment by the Dextramer CMV Kit. For the remaining 75 patients, in 630 longitudinal fresh samples CMV-specific CD8+ T cells were enumerated by the Dextramer CMV Kit and 47 fresh samples from 16 patients were analyzed by QuantiFERON-CMV. Eighty-four frozen samples from the 21 patients in whom donor- or host-specific restrictions could be evaluated were characterized by an extensive flow cytometry panel. IFN-γ ELISpot was also used to detect CMV-specific T cells in all the samples analyzed by QuantiFERON-CMV and/or in-depth flow cytometry. (B) Design of the study. Peripheral blood samples were taken before conditioning and at the indicated timepoints (solid blue arrows) after HSCT and used for the Dextramer CMV Kit and QuantiFERON-CMV assays. Donors’ apheresis samples were analyzed on the day of infusion. At selected time-points (dotted blue arrows) peripheral blood mononuclear cells were also frozen for subsequent flow cytometry analysis and IFN-γ ELISpot assay. HLA: human leukocyte antigen; CMV: cytomegalovirus; FACS: fluorescence activated cell sorting; IFN: interferon; ELISpot: enzyme-linked immunospot; qPCR: quantitative polymerase chain reaction; PET: pre-emptive antiviral therapy; PT-Cy: post-transplant cyclophosphamide; allo-HSCT: allogeneic hematopoietic stem cell transplantation; PBMC: peripheral blood mononuclear cells.
considering only patients receiving transplants from seropositive donors, the only grafts with detectable CMVspecific cells (Online Supplementary Figure S3B). These data indicate that the infusion of CMV-specific primed T cells is necessary but not sufficient for early protection from CRE, and that early post-HSCT factors (e.g., prophylaxis and treatment for GvHD) may impinge on in vivo CMV-specific T-cell expansion. Indeed, among post-transplant variables, the occurrence of acute GvHD (P=0.0116) (Online Supplementary Figure S3C), particularly those cases requiring administration of systemic steroids (grade II-IV, P=0.0019) (Online Supplementary Figure S3D), was associated with an increased incidence of CRE.

After HSCT, patients showed fast reconstitution of CD3+, CD8+ T lymphocytes and NK cells, and delayed reconstitution of B cells, CD4+ and regulatory T lymphocytes (Online Supplementary Figure S4A), as previously described in allogeneic HSCT with post-transplant cyclophosphamide and sirolimus.25
Interestingly, at 30 days ( P=0.0088) and 45 days (P=0.0084) after HSCT, CMV-specific CD8+ T-cell counts
A B Haematologica | 108 June 2023 1533 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
Table 1. Characteristics of the patients (N=75) and their transplants.
were higher in patients who did not experience CRE after these time-points than in patients with subsequent CRE, indicating a protective role for early CMV-specific immunity against succeeding viral reactivations (Figure 2B, Online Supplementary Table S2). This correlation was not found at day +30 for any other immune subset evaluated (Online Supplementary Figure S4B). High amounts of total CD8+ T cells were associated with reduced subsequent CRE only at 90 days (Figure 2C). No association between CMV-specific CD8+ T cell counts and subsequent CRE was found from 60 to 180 days after HSCT (Figure 2B, Online Supplementary Figure S5A). Receiver operating characteristic (ROC) analysis identified the threshold of 0.5 CMV-specific CD8+ T-cells/mL at day 45 after HSCT as the best predictor of subsequent CRE (P=0.0094, sensitivity=88.24%, specificity=67.74%) (Figure 3A, Online Supplementary Table S3). Reaching this threshold was confirmed as an independent protective factor from CRE in both competitive risk analysis with non-relapse mortality (P=0.02) (Figure 3B) and in a multivariate analysis including acute GvHD and host/donor serostatus (P=0.04) (Figure 3C).
CMV-specific CD8+ T-cell counts in patients’ peripheral blood prior to HSCT did not correlate with CRE risk (Online Supplementary Figure S5B). Interestingly, reduced risk of CMV disease was associated with the presence of CMVspecific T lymphocytes in pre-HSCT host peripheral blood but not at any post-HSCT timepoint (Online Supplementary Figure S5B, C and data not shown), suggesting a role in the protection against tissue CMV-mediated damage for the recently described peripheral host T cells remaining after HSCT.31
Overall, these data highlight the importance of an early evaluation of CMV-specific T cells as a reliable predictor of CRE.
Quantification of cytomegalovirus-specific T cells by Dextramer staining correlates with functional assays
To further investigate the predictive value of CMV-specific T-cell quantification, QuantiFERON-CMV15,32 and IFN- γ ELISpot functional assays were also performed on a selected subset of patients (Figure 1A).
HSCT, hematopoietic stem cell transplantation; HCT-CI, hematopoietic cell transplant-comorbidity index; MAC: myeloablative conditioning; RTC: reduced-toxicity conditioning; GvHD: graft-versus-host disease; PTCy: post-transplant cyclophosphamide; MMF: mycophenolate mofetil. *Values may not add up to 100% due to rounding.
IFN-γ ELISpot at +30 days failed to predict subsequent CRE (Online Supplementary Figure S6A-F). At later timepoints absolute counts of CMV-specific sfc/mL were lower in patients who experienced subsequent CRE than in patients able to control the virus (P=0.0249 for 60 days; P=0.0402 for 90 days after HSCT) (Online Supplementary Figure S6B). ROC analysis performed on data collected at +60 and +90 days identified 1.75 CMV-specific sfc/mL as the threshold to discriminate patients at higher risk of viral reactivations (P=0.0018, sensitivity=92.86%, specificity=67.67%) (Online Supplementary Figure S6G).
Dextramer staining showed 81.94% agreement with IFN-γ ELISpot (n=59/72 concordant results) (Figure 4A, Online
Characteristic Patients’ age in years, median (range) 56 (22-75) Diagnosis, N (%)* Myeloid diseases Acute myeloid leukemia Myelodysplastic syndrome Myeloproliferative neoplasm Lymphoid diseases Non-Hodgkin and Hodgkin lymphomas Acute lymphoblastic leukemia Chronic lymphocytic leukemia Multiple myeloma Other diseases 38 (51) 7 (9) 5 (7) 8 (11) 9 (12) 2 (3) 2 (3) 4 (5) Disease status at HSCT, N (%) First complete remission Beyond first complete remission Partial remission Active disease 26 (35) 20 (27) 7 (9) 22 (29) Refined Disease Risk Index at HSCT, N (%) Low Intermediate High Very high Not applicable 7 (9) 39 (52) 17 (23) 9 (12) 3 (4) HCT-CI score, median (range) 2 (0-9) Type of donor, N (%)* Matched related Matched unrelated (9/10) Matched unrelated (10/10) Mismatched related Cord blood 10 (13) 16 (22) 17 (23) 26 (35) 6 (8) Stem cell source, N (%) Bone marrow Peripheral blood Cord blood unit 1 (1) 68 (91) 6 (8) Conditioning regimen, N (%) Treosulfan-based MAC regimens Treosulfan-based RTC regimens Thiotepa, busulfan, fludarabine 55 (73) 18 (24) 2 (3) N. of cells infused x106/kg, median (range) CD34+ CD3+ 6.52 (0.06-10) 247 (2-775) GvHD prophylaxis, N (%) PTCy-Rapamycin-MMF PTCy-Rapamycin Others 59 (79) 9 (12) 7 (9) Acute GvHD, N (%) Grade I Grades II-IV Grades III-IV 5 (7) 18 (24) 11 (15) Chronic GvHD, N (%) Overall Severe 35 (47) 8 (11) Disease relapse N (%) Time in days until relapse, median (range) 18 (24) 176 (33-686) Alive at last follow-up, N (%) 50 (67) Follow-up in days, median (range) 737 (28-1,246)
Haematologica | 108 June 2023 1534 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
Supplementary Table S4) and 88.89% agreement with QuantiFERON-CMV (n=24/27) (Figure 4B, Online Supplementary Table S4). The two functional assays showed a concordance of 81.82% (n=27/33) (Online Supplementary Table S4). Linear regression analysis confirmed the agreement between CMV-specific T-cell counts evaluated by
Dextramer assay and those obtained by either IFN-γ ELISpot (P<0.0001) (Figure 4C) or QuantiFERON-CMV (P=0.0007) (Figure 4D). Collectively, these correlations indicate that quantification of CMV-specific T lymphocytes by Dextramer staining represents a strong surrogate biomarker of functional viral-specific T-cell responses, able to predict protection from viral reactivations earlier and more rapidly than functional assays.
Cytomegalovirus-specific T cells restricted by shared, donor, and host HLA molecules reconstitute with different kinetics and have different impacts on the incidence of clinically relevant events

After HSCT, the expansion of memory T cells restricted for S HLA might be sustained by either host cells or donorderived hematopoietic cells, whereas only the latter express D-specific HLA and therefore activate D-restricted CMV-specific T lymphocytes. Being absent in the donor Tcell repertoire, H-restricted T cells require maturation in the host thymus and priming by host antigen-presenting cells (APC) (Figure 5A). To characterize these different dynamics, we separately analyzed CMV-specific CD8+ T cells restricted for S (n=64 patients), D (n=19 patients) or H (n=13 patients) HLA molecules.
The kinetics of immune reconstruction were evaluated in
Figure 2. Early reconstitution of cytomegalovirus-specific immunity is associated with reduced subsequent clinically relevant events. (A) Longitudinal evaluation of cytomegalovirus (CMV)-specific immunity by Dextramer staining (blue line, mean value of CMV-specific CD8+ T cells/mL in all patients) and of the percentage of evaluable patients experiencing clinically relevant events (CRE; gray bars) at each time-point after hematopoietic stem cell transplantation (HSCT). (B) Absolute numbers of CMV-specific CD8+ T lymphocytes at the indicated time-points after HSCT in patients experiencing or not subsequent CRE. Continuous lines, mean ± standard deviation. The dotted lines indicate the level of 0.5 CMV-specific T cell/mL. (C) Absolute numbers of total CD8+ T lymphocytes at the indicated time-points after HSCT in patients experiencing or not subsequent CRE. Continuous lines, mean ± standard deviation. The analyses in (B) and (C) were performed with the MannWhitney test: **P<0.01; ns: not significant.
Characteristic CMV serostatus (H/D), N (%) Positive/positive Positive/negative Negative/positive 49 (66) 25 (33) 1 (1) CMV reactivation (any viremia) N (%) Time of onset in days, median (range) 42 (56) 69 (0-879) Clinically relevant CMV infection N (%) Time of onset in days, median (range) 39 (52) 68 (0-208) CMV disease N (%) Time of onset in days, median (range) 11 (15) 69 (19-210)
Table 2. Cytomegalovirus reactivation and disease after hematopoietic stem cell transplantation.
CMV:
cytomegalovirus; H/D: host/donor.
A B C Haematologica | 108 June 2023 1535 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
CMV-seropositive patients who received their grafts from either seropositive (Figure 5B-D) or seronegative (Figure 5E-G) donors. When the donor is seronegative, D-restricted CMV-specific T cells can be primed and stimulated in the host only by donor-derived hematopoietic cells, whereas S-restricted lymphocytes can be primed by either D-derived or residual host APC and can be further stimulated by any infected cell. We found that Drestricted reconstitution was impaired in the case of seronegative donors, differently from S-restricted immune reconstruction (P <0.001 for time, P=0.04 for restriction type) (Figure 5E, F, H), indicating that in primary immunological responses the emergence of viral-specific T cells is largely sustained by the antigen encounter on host infected non-hematopoietic or residual tissue-resident myeloid cells rather than by cross-priming/presentation by non-infected donor-derived APC. Accordingly, S- and D-restricted T lymphocytes reconstituted with similar magnitudes and kinetics when the donor was seropositive (Figure 5B, C, H), indicating an equal ability of infected donor hematopoietic cells and host cells in presenting viral antigens, thus sustaining secondary immune re-

sponses. H-restricted T cells were not detected in postHSCT samples for up to 1 year (Figure 5D, G). H-restricted T lymphocytes were absent also at 2 or 3 years after HSCT in the patients evaluable at those time-points (n=4 and n=2, respectively) or in three other younger patients (aged 20-29) evaluated for up to 4 years after haploidentical HSCT (data not shown).
High-dimensional analysis of cytomegalovirus-specific CD8+ T lymphocytes reveals earlier maturation in T cells restricted by donor-specific HLA molecules
Divergent differentiation, activation or exhaustion of CMVspecific T cells might account for the different dynamics we observed in T lymphocytes restricted for S or D HLA. To shed light on these potential mechanisms, the phenotype of CMV-specific CD8+ T lymphocytes was investigated by multiparametric flow cytometry with additional Dextramer reagents in 21 patients for whom either a S or a D-specific HLA restriction could be separately evaluated by Dextramer analysis. Overall, 68 CMV-specific T-cell subpopulations (n=15 patients) were detected at different time-points. A representative gating strategy is shown in Online Supplementary Figure S7. CMV-specific CD8+ T-cell counts in frozen samples correlated with those evaluated in fresh samples (P<0.001) (Online Supplementary Figure S8A). By manual gating we found that CMV-specific CD8+ T cells contained more TEM (P=0.002) and fewer TN (P=0.003), TCM (P=0.002) and early-differentiated CD28+CD27+ cells (P<0.001) compared to bulk CD8+ T lymphocytes (Online Supplementary Figure S8B). CMV-specific CD8+ T cells restricted by D HLA molecules displayed a less differentiated phenotype compared to S-restricted ones (P<0.001 for CD28–CD27–; P=0.002 for CD28–CD27+) (Figure 6A).
Figure 3. The presence of ≥0.5 cytomegalovirus-specific CD8+ T cells/ mL at 45 days after hematopoietic stem cell transplantation protects against subsequent clinically relevant events. (A) Receiver operating characteristic analysis for the identification of a protective amount of cytomegalovirus (CMV)-specific CD8+ T cells/mL at 45 days after hematopoietic stem cell transplantation (HSCT). (B) Competitive risk analysis for the risks of clinically relevant events and non-relapse mortality in patients having or not at least 0.5 CMV-specific CD8+ T cells/mL at 45 days after HSCT. (C) Left panel: forest plot showing CRE risk according to the occurrence of acute graft-versus-host disease, host/donor CMV serostatus or the presence of at least 0.5 CMV-specific CD8+ T cells/mL at 45 days after HSCT. Right panel: results of the statistical analysis. The analysis in (B) was performed with the Gray test and that in (C) by logistic regression: *P<0.05. AUC: area under the curve; DEX: Dextramer; aGvHD: acute graft- versus-host disease; OR: odds ratio; CI: confidence interval.
To identify S and D-specific signatures, we performed high-dimensional analysis and clustering by cytoChain29 on CMV-specific CD8+ T-cell populations with at least 65 events (n=55). The results revealed a clear segregation of the metaclusters depicting S- and D-restricted CMV-specific CD8+ T cells (Figure 6B). Interestingly, this segregation was also maintained in T cells derived from the same samples but differentially restricted for D or S alleles (Online Supplementary Figure S8C). In detail, metaclusters n. 9 (P<0.001), 15 (P=0.002), 20 (P=0.02) and 21 (P=0.01) were enriched in D-restricted T lymphocytes (Figure 6C), and were characterized by enhanced expression of early-differentiation markers (CD62L, P=0.046; CD27, P=0.014) and by lower KLRG1 expression (P=0.026) compared to those enriched in S-restricted T cells (metacluster n.1, P=0.01; 3, P<0.001; 8, P=0.009; and 11, P=0.002) (Figure 6C-E, Online Supplementary Figure S8D). Furthermore, HLA-DR expression was high in metaclusters n. 20 and 21, indicating recent activation of D-restricted lymphocytes. S-restricted metaclusters showed a trend to reduced CD127
A B C Haematologica | 108 June 2023 1536 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
expression, confirming a late differentiated phenotype. No differences were detected in the expression of inhibitory receptors, such as PD-1 and TIM-3, nor in 4-1BB or CD28 between D- and S-specific metaclusters (Figure 6D). Longitudinal analysis revealed variation across time of two metaclusters. Metacluster n. 14 was enriched at +30 days compared to other time-points (P<0.001), and was composed of activated TCM cells. Metacluster n. 24, which comprises early differentiated activated TSCM/TCM lymphocytes, peaked at +90 days after HSCT compared to later time-points (P=0.03 for day +180, P=0.007 for day +365) (Figure 6E, F), in line with an early triggering of CMV-specific cells by CMV reactivation events. Consistently, we also found early peaks of metaclusters expressing high CD28 and low PD-1 (n. 6 and n. 12), features that have been associated with higher anti-CMV activity33,34 (Online Supplementary Figure S8E). Interestingly the majority of metaclusters that accumulated in time (n. 1, 3, 8 [all Sspecific], and 9 [D-specific]) displayed a TEM/TEMRA phenotype, in part characterized by the expression of CD27 (metaclusters n. 1 and n.9) but not CD28, a phenotype typical of memory inflation, often observed in chronic infections (Figure 6E, F).35

Kinetics of D-restricted metaclusters showed the rise of metacluster n. 9, depicting CD27+ TEM/TEMRA, and a stable curve for metacluster n. 21, composed of activated TCM cells, while metaclusters n. 15 and n. 20 contracted rapidly (Figure 6E, F).
Overall, these results suggest that although D- and S-restricted CMV-specific T lymphocytes are triggered simultaneously, early after transplantation, the quality of this triggering is different, leading to the brisk accumulation of a pool of early memory D-restricted cells, while S-restricted cells expand more robustly and persist longer.
Figure 4. Quantification of cytomegalovirus-specific T cells by Dextramer staining correlates with functional assays. (A, B) Concordance between cytomegalovirus (CMV)-specific T-cell quantification by the Dextramer assay (positivity threshold 0.5 cells/mL) and either IFN-γ ELISpot for CMV (A, positivity threshold 1.75 sport-forming cells/ m L after CMV antigen stimulation) or QuantiFERON-CMV (B, positivity threshold 0.2 IU/mL in the CMV antigen tube). (C, D) Linear regression analysis between CMVspecific CD8+ T cells quantified by the Dextramer assay and the presence of functional IFN- γ producing CMV-specific T lymphocytes detected by either IFN-γ ELISpot (C) or QuantiFERON-CMV (D). CMVag: cytomegalovirus antigen; sfc: spot-forming cells; IFN: interferon.
Discussion
CMV-specific T cells protect patients not only from CMV reactivations but also from severe infections overall,30 thus representing a good surrogate for general immune competence after allogeneic HSCT. There is, therefore, a growing interest in the use of immunomonitoring to adjust treatment according to individual risk. In this prospective, observational study, we performed a detailed longitudinal analysis of CMV-specific immune reconstruction and its dynamics.
Looking for a highly sensitive assay we relied on MHCmultimer staining with Dextramer reagents to enumerate CMV-specific T cells in whole blood by flow cytometry. Allogeneic HSCT is the ideal context for MHC multimerbased immunomonitoring, since the HLA type of donor-host couples is known. We observed an inverse correlation between CMV-specific T-cell counts at +30/+45 days after allogeneic HSCT and the risk of experiencing subsequent CRE. Most of the previously tested biomarkers of CMV-specific immunity predicted relevant clinical events at later time-points (tetramer staining at day +65;19 QuantiFERON-CMV at day +90;15 and IFN-γ ELISpot at day +10012), thus reflecting the advantage of Dextramer staining in the management of allogeneic HSCT patients. One of the main concerns related to MHC multimer-based immunomonitoring is the possibility of evaluating limited HLA restrictions. Strikingly, the large repertoire of Dextramer reagents allowed us to evaluate nearly 90% of enrolled patients. The majority of patients in our cohort were Caucasian (76 out of 84; 90.5%), followed by six Hispanics (7.1%) and two Asians (2.4%). Interestingly, five out of six Hispanic patients and one out of two Asian patients could be enrolled in our study.
A B C D Haematologica | 108 June 2023 1537 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
Figure 5. Cytomegalovirus-specific T cells restricted by shared, donor or host HLA molecules reconstitute with different kinetics and differentially affect the incidence of clinically relevant events. (A) Immune reconstruction after hematopoietic stem cell transplantation (HSCT) with T lymphocytes restricted for shared, donor- or host-specific HLA molecules require priming/activation by different antigen-presenting cells or infected cells. (B-G) Longitudinal reconstitution of cytomegalovirus (CMV)-specific CD8+ T lymphocytes evaluated by Dextramer assay at the indicated time-points before and after HSCT is shown separately for CMVseropositive recipients infused with cells from either seropositive (panels B-D) or seronegative (panels E-G) donors. Absolute counts of T cells restricted for shared (B and E), donor-specific (C and F) or host-specific (D and G) HLA alleles are reported. Lines, mean ± standard deviation. (H) Means of longitudinal reconstitution of CMV-specific CD8+ T lymphocytes restricted for either shared or donor-specific HLA alleles in CMV-seropositive patients receiving grafts from either seropositive or seronegative donors, as indicated in the graph legend. The analysis in (H) was by two-way analysis of variance. *P<0.05. DC: dendritic cell; HLA: human leukocyte antigen; H; host; D: donor

A B
D E F G H Haematologica | 108 June 2023 1538 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
C
These data highlight the feasibility of this technique in the Caucasian population, and possibly in other ethnicities.
It is largely recognized that both CD4+ and CD8+ CMV-specific T cells contribute to anti-viral immune surveillance.36,37 Thus, Dextramer reagents evaluating only CD8+ T cells can theoretically underestimate CMV-specific Tcell immunity. In addition, MHC-multimer staining does not measure T-cell function. To verify the impact of these potential limitations, we compared different techniques for the evaluation of CMV-specific immunity. The enumeration of CMV-specific T cells by Dextramer staining showed strong concordance with functional assays, proving a good surrogate biomarker of functional immunity. Despite evaluating both CD4+ and CD8+ T-cell responses, IFN- γ ELISpot predicted the clinical outcome at later time-points (day +60/+90) than did the Dextramer-binding assay (day +30/45), underlining the higher sensitivity of the latter and confirming previous results obtained in different allogeneic HSCT transplant platforms.30,38 The Dextramer CMV kit is already available as an in vitro diagnostic test, is characterized by reliable and reproducible performances, requires a smaller amount (1 mL) of peripheral blood and should be preferred when studying individuals with evaluable HLA. As an alternative assay for patients lacking evaluable HLA types, we propose IFN-γ ELISpot, which is not dependent on pre-defined HLA alleles and which predicted a lower risk of developing CMV reactivations when the threshold of 1.75 specific sfc/ m L was reached at 60/90 days after HSCT.
Flow cytometry performed after Dextramer staining also allows single-cell multiparametric characterization of CMV-specific lymphocytes. We exploited this feature to shed light on the kinetics and weight of viral-specific responses that emerge during post-transplant immune reconstruction through different mechanisms. Our data suggest that up to 1 year after HSCT the CMV-specific immune reconstruction mainly relies on mature T cells infused within the graft, which could be antigen-experien ced or naïve depending on the donor’s CMV serostatus and can recognize CMV epitopes restricted by S or D-specific HLA. In the same time-frame, thymic instruction of donor-derived progenitors is impaired. H-restricted subpopulations of CMV- and flu-specific T cells have been detected in adult patients 6-14 years after HSCT;39 the absence of the thymic contribution to CMV-specific immune reconstruction in our cohort (median age, 56 years) could be due to age-related involution of the host thymus or to thymic damage caused by conditioning regimens or by subclinical GvHD.40 For this reason, in adult patients, we suggest that Dextramers restricted by D or S HLA alleles should be preferentially used, rather than H HLA alleles. In recipients of grafts from CMV-seropositive donors, we observed similar kinetics in S- and D-restricted CMV-specific T cells, pointing to an equal ability of donor hemato-
poietic cells and host cells in presenting viral antigens thus sustaining secondary immune responses to CMV. Interestingly, in recipients of CMV-seronegative donors, Drestricted CMV-specific cells appeared with delayed kinetics and at a reduced frequency compared to S-restricted cells. This observation suggests that the emergence of viral-specific T cells in primary responses is largely sustained by infected non-hematopoietic cells, presenting only epitopes on shared HLA alleles. Considering the wide tropism of CMV,41 we may assume that the amount and duration of antigenic stimulation may favor S-restricted T cells. An alternative explanation for this observation could be early priming by residual infected host APC,42 able to prime S-restricted T cells from seronegative donors, thus providing them an initial advantage and impairing the contribution of the latecoming D-restricted cells.43 The more extensive evaluation of S- and D-restricted CMV-specific T cells in patients receiving transplants from CMV-seronegative donors and that experience delayed CMV reactivation in the absence of host APC, such as in the context of prophylaxis with letermovir,27 could allow these hypotheses to be discriminated. The memory phenotype of CMV-specific T lymphocytes, a critical parameter for protective CMV-specific immunity after HSCT,44,45 indicates that CMV-specific cells restricted for D or S HLA alleles contribute differentially to memory subpopulations. D-restricted T lymphocytes display an earlier memory phenotype and lower persistence than Srestricted cells. In contrast, S-restricted cells expand progressively, peaking at 150 days, when CMV reactivations contract, and remain stable thereafter.
Our study indicates the benefits of evaluating CMV-specific immunity by Dextramer assay and allowed us to identify a threshold of CMV-specific T cells which stratifies patient’s risk of CRE in the context of allogeneic HSCT with post-transplant cyclophosphamide. Letermovir has recently been approved for the prophylaxis of CMV infection in seropositive transplant recipients27 and is associated with reduced non-relapse mortality by preventing or delaying CRE.46 However, late CMV reactivations occur,27,47 and in the few patients experiencing CMV DNAemia during letermovir prophylaxis the currently used assays cannot discriminate between non-infective DNA from abortively infected cells and infective virions.48 Impaired polyclonal T-cell reconstitution and CMV-specific immunity have recently been reported in patients receiving prophylaxis with letermovir.49,50 In this context, enumeration of CMV-specific T cells might allow identification of the patients who need prophylaxis to be prolonged beyond day 100.
In conclusion, monitoring CMV-specific T-cell counts in peripheral blood by Dextramer assay may become an important biomarker-driven strategy to facilitate risk stratification and to optimize anti-viral therapy, minimizing
Haematologica | 108 June 2023 1539 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.

Continued on following page. A B C D E F Haematologica | 108 June 2023 1540 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
Figure 6. Unsupervised analysis of CD8+Dextramer+ T lymphocytes reveals earlier maturation in donor-restricted T cells. (A) Manual gating analysis for the distribution of differentiation T-cell subsets in CD8+Dextramer+ T lymphocytes restricted for shared or donor-specific HLA. Differentiation T-cell subsets were defined according to the expression profile of CD45RA, CD62L and CD95 markers (left panel) or to the combination of CD28 and CD27 (right panel). TN (T-naïve): CD45RA+CD62L+CD95–; TSCM (T stem cell memory): CD45RA+CD62L+CD95+; TCM (T central memory): CD45RA–CD62L+; TEM (T effector memory): CD45RA–CD62L–; TEMRA (T effector memory RA): CD45RA+CD62L–. (B) tSNE map (left panel) and Flow-SOM metacluster overlay (middle panel) of CD8+Dextramer+ T cells from all the concatenated samples. The right panel shows the different distribution of Dextramer+ T lymphocytes restricted for donor-specific (blue) or shared (red) HLA molecules. (C) Frequency of metaclusters expressed as the percentage of cells in each metacluster for each type of HLA restriction among total CD8+Dextramer+ T lymphocytes. Shared subpopulations (n=29), donor-specific subpopulations (n=26). (D) Percentage of events positive for the indicated markers in the metaclusters enriched in CD8+Dextramer+ T cells restricted for donor-specific (blue, metaclusters n. 9, 15, 20 and 21) or shared (red, metaclusters n. 1, 3, 8, and 11) HLA alleles. (E) Heatmap for the 25 Flow-SOM metaclusters; the ratio of fluorescence intensity of each marker with respect to the maximum is indicated in the color-legend. Blue and red rectangles highlight the clusters significantly enriched in CD8+Dextramer+ T cells restricted for donor-specific or shared alleles, respectively. (F) The frequency of each metacluster over time expressed as the percentage of events in each metacluster among total CD8+Dextramer+ T lymphocytes. Among the cytomegalovirus (CMV)-specific T-cell populations evaluated, three were obtained at 30 days after hematopoietic stem cell transplantation, 11 at day +60, 16 at day +90, 12 at day +180 and 13 at day +365. Red, blue and black contours highlight metaclusters enriched in shared or donor-restricted CMV-specific T cells or common metaclusters, respectively. The analyses in (A), (C) and (D) were conducted using two-way analysis of variance. *P<0.05; **P<0.01; ***P<0.001. DEX: Dextramer; tSNE: t-distributed stochastic neighbor embedding; HSCT: hematopoietic stem cell transplantation.
drug exposure, thus improving the clinical management of patients undergoing allogeneic HSCT.
Disclosures
CB has received research support from Intellia Therapeutics and is a member of advisory boards or a consultant or speaker for Molmed, Intellia, TxCell, Novartis, GSK, Allogene, Kite/Gilead, Miltenyi, Kiadis, QuellTX, and Janssen. RG discloses honoraria for speaking at educational events supported by Biotest, Medac, Pfizer and Magenta.
Contributions
ET and MN designed the study, conducted laboratory experiments, analyzed and interpreted data and wrote the paper; PDS designed the study, conducted laboratory experiments, and analyzed and interpreted data; MTLS, FS, EC, DC, FG, FL, EX, FF, CO, CC and MB provided clinical data and samples and participated in the data interpretation; MD performed statistical analyses; DA and FM participated in the high dimensional analysis of flow cytometry data; VB and VV participated in the laboratory experiments; GO participated in the design of the study; SR, RD and MC performed the QuantiFERON-CMV, and analyzed and interpreted the results; ER and LV participated in the discussion and interpretation of data; LB
References
1. Passweg JR, Baldomero H, Chabannon C, et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: monitoring of activities and trends over 30 years. Bone Marrow Transplant. 2021;56(7):1651-1664.
2. Penack O, Peczynski C, Mohty M, et al. How much has allogeneic stem cell transplant-related mortality improved since the 1980s? A retrospective analysis from the EBMT. Blood Adv. 2020;4(24):6283-6290.
3. Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in
participated in the study design and reviewed the paper; FC and JP designed and supervised the study; RG and CB designed and supervised the study and wrote the paper.
Acknowledgments
The authors would like to thank San Raffaele URC (Clinical Trial Office), the participating patients and their families, as well as all nurses and data managers who contributed to this study.
Funding
This work was partially supported by Immudex. Funding was also provided by the Associazione Italiana per la Ricerca sul Cancro (AIRC-IG 18458 and AIRC 5 per Mille 22737), the Italian Ministry of Education, University and Research (PRIN 2017WC8499), Alliance Against Cancer (Ricerca Corrente CAR T project: RCR-2019-23669115), EHA, Cellnex (ACT4Covid) to CB; the Italian Ministry of Health (RF-COVID-19) to CB and FC; and the Italian Ministry of Health (GR-2016-02364847) to ER.
Data-sharing statement
The datasets generated for this study are available on request to the corresponding author.
hematopoietic stem cell transplant recipients. Hematol Oncol Clin North Am. 2011;25(1):151-169.
4. Luznik L, Fuchs EJ. High-dose, post-transplantation cyclophosphamide to promote graft-host tolerance after allogeneic hematopoietic stem cell transplantation. Immunol Res. 2010;47(1-3):65-77.
5. Mussetti A, Greco R, Peccatori J, Corradini P. Post-transplant cyclophosphamide, a promising anti-graft versus host disease prophylaxis: where do we stand? Expert Rev Hematol.
Haematologica | 108 June 2023 1541 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
2017;10(5):479-492.
6. Goldsmith SR, Abid MB, Auletta JJ, et al. Posttransplant cyclophosphamide is associated with increased cytomegalovirus infection: a CIBMTR analysis. Blood. 2021;137(23):3291-3305.
7. Oltolini C, Greco R, Galli L, et al. Infections after allogenic transplant with post-transplant cyclophosphamide: impact of donor HLA matching. Biol Blood Marrow Transplant. 2020;26(6):1179-1188.
8. Li CR, Greenberg PD, Gilbert MJ, Goodrich JM, Riddell SR. Recovery of HLA-restricted cytomegalovirus (CMV)-specific Tcell responses after allogeneic bone marrow transplant: correlation with CMV disease and effect of ganciclovir prophylaxis. Blood. 1994;83(7):1971-1979.
9. Seggewiss R, Einsele H. Immune reconstitution after allogeneic transplantation and expanding options for immunomodulation: an update. Blood. 2010;115(19):3861-3868.
10. Bosch M, Khan FM, Storek J. Immune reconstitution after hematopoietic cell transplantation. Curr Opin Hematol. 2012;19(4):324-335.
11. El Haddad L, Ariza-Heredia E, Shah DP, et al. The ability of a cytomegalovirus ELISPOT assay to predict outcome of low-level CMV reactivation in hematopoietic cell transplant recipients. J Infect Dis. 2019;219(6):898-907.
12. Wagner-Drouet E, Teschner D, Wolschke C, et al. Standardized monitoring of cytomegalovirus-specific immunity can improve risk stratification of recurrent cytomegalovirus reactivation after hematopoietic stem cell transplantation. Haematologica. 2021;106(2):363-374.
13. Avetisyan G, Aschan J, Hagglund H, Ringden O, Ljungman P. Evaluation of intervention strategy based on CMV-specific immune responses after allogeneic SCT. Bone Marrow Transplant. 2007;40(9):865-869.
14. Navarro D, Amat P, de la Camara R, et al. Efficacy and safety of a preemptive antiviral therapy strategy based on combined virological and immunological monitoring for active cytomegalovirus infection in allogeneic stem cell transplant recipients. Open Forum Infect Dis. 2016;3(2):ofw107.
15. Yong MK, Cameron PU, Slavin M, et al. Identifying cytomegalovirus complications using the Quantiferon-CMV assay after allogeneic hematopoietic stem cell transplantation. J Infect Dis. 2017;215(11):1684-1694.
16. Krawczyk A, Ackermann J, Goitowski B, et al. Assessing the risk of CMV reactivation and reconstitution of antiviral immune response post bone marrow transplantation by the QuantiFERON-CMV-assay and real time PCR. J Clin Virol. 2018;99-100:61-66.
17. Tey SK, Kennedy GA, Cromer D, et al. Clinical assessment of anti-viral CD8+ T cell immune monitoring using QuantiFERONCMV(R) assay to identify high risk allogeneic hematopoietic stem cell transplant patients with CMV infection complications. PLoS One. 2013;8(10):e74744.
18. Cwynarski K, Ainsworth J, Cobbold M, et al. Direct visualization of cytomegalovirus-specific T-cell reconstitution after allogeneic stem cell transplantation. Blood. 2001;97(5):1232-1240.
19. Gratama JW, Boeckh M, Nakamura R, et al. Immune monitoring with iTAg MHC tetramers for prediction of recurrent or persistent cytomegalovirus infection or disease in allogeneic hematopoietic stem cell transplant recipients: a prospective multicenter study. Blood. 2010;116(10):1655-1662.
20. Varanasi PR, Ogonek J, Luther S, et al. Cytomegalovirus-specific CD8+ T-cells are associated with a reduced incidence of early
relapse after allogeneic stem cell transplantation. PLoS One. 2019;14(3):e0213739.
21. Kato R, Tamaki H, Ikegame K, et al. Early detection of cytomegalovirus-specific cytotoxic T lymphocytes against cytomegalovirus antigenemia in human leukocyte antigen haploidentical hematopoietic stem cell transplantation. Ann Hematol. 2015;94(10):1707-1715.
22. Tario JD Jr, Chen GL, Hahn TE, et al. Dextramer reagents are effective tools for quantifying CMV antigen-specific T cells from peripheral blood samples. Cytometry B Clin Cytom. 2015;88(1):6-20.
23. Ljungman P, de la Camara R, Robin C, et al. Guidelines for the management of cytomegalovirus infection in patients with haematological malignancies and after stem cell transplantation from the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect Dis. 2019;19(8):e260-e272.
24. Greco R, Ciceri F, Noviello M, et al. Immune monitoring in allogeneic hematopoietic stem cell transplant recipients: a survey from the EBMT-CTIWP. Bone Marrow Transplant. 2018;53(9):1201-1205.
25. Cieri N, Greco R, Crucitti L, et al. Post-transplantation cyclophosphamide and sirolimus after haploidentical hematopoietic stem cell transplantation using a treosulfanbased myeloablative conditioning and peripheral blood stem cells. Biol Blood Marrow Transplant. 2015;21(8):1506-1514.
26. Greco R, Lorentino F, Morelli M, et al. Posttransplantation cyclophosphamide and sirolimus for prevention of GVHD after HLA-matched PBSC transplantation. Blood. 2016;128(11):1528-1531.
27. Marty FM, Ljungman P, Chemaly RF, et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017;377(25):2433-2444.
28. Ljungman P, Boeckh M, Hirsch HH, et al. Definitions of cytomegalovirus infection and disease in transplant patients for use in clinical trials. Clin Infect Dis. 2017;64(1):87-91.
29. Manfredi F, Abbati D, Cianciotti BC, et al. Flow cytometry data mining by cytoChain identifies determinants of exhaustion and stemness in TCR-engineered T cells. Eur J Immunol. 2021;51(8):1992-2005.
30. Noviello M, Forcina A, Veronica V, et al. Early recovery of CMV immunity after HLA-haploidentical hematopoietic stem cell transplantation as a surrogate biomarker for a reduced risk of severe infections overall. Bone Marrow Transplant. 2015;50(9):1262-1264.
31. Divito SJ, Aasebo AT, Matos TR, et al. Peripheral host T cells survive hematopoietic stem cell transplantation and promote graft-versus-host disease. J Clin Invest. 2020;130(9):4624-4636.
32. Cantisan S, Lara R, Montejo M, et al. Pretransplant interferongamma secretion by CMV-specific CD8+ T cells informs the risk of CMV replication after transplantation. Am J Transplant. 2013;13(3):738-745.
33. Zielinski M, Tarasewicz A, Zielinska H, et al. CD28 positive, cytomegalovirus specific cytotoxic T lymphocytes as a novel biomarker associated with cytomegalovirus viremia in kidney allorecipients. J Clin Virol. 2016;83:17-25.
34. Pei XY, Zhao XY, Chang YJ, et al. Cytomegalovirus-specific T-cell transfer for refractory cytomegalovirus infection after haploidentical stem cell transplantation: the quantitative and qualitative immune recovery for cytomegalovirus. J Infect Dis. 2017;216(8):945-956.
35. Snyder CM, Cho KS, Bonnett EL, et al. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity. 2008;29(4):650-659.
Haematologica | 108 June 2023 1542 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
36. Gabanti E, Bruno F, Lilleri D, et al. Human cytomegalovirus (HCMV)-specific CD4+ and CD8+ T cells are both required for prevention of HCMV disease in seropositive solid-organ transplant recipients. PLoS One. 2014;9(8):e106044.
37. Jackson SE, Sedikides GX, Mason GM, Okecha G, Wills MR. Human cytomegalovirus (HCMV)-specific CD4(+) T cells are polyfunctional and can respond to HCMV-infected dendritic cells in vitro. J Virol. 2017;91(6):e02128-16.
38. Chemaly RF, El Haddad L, Winston DJ, et al. Cytomegalovirus (CMV) cell-mediated immunity and CMV infection after allogeneic hematopoietic cell transplantation: the REACT study. Clin Infect Dis. 2020;71(9):2365-2374.
39. Oliveira G, Ruggiero E, Stanghellini MT, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Sci Transl Med. 2015;7(317):317ra198.
40. Velardi E, Tsai JJ, van den Brink MRM. T cell regeneration after immunological injury. Nat Rev Immunol. 2021;21(5):277-291.
41. Sinzger C, Digel M, Jahn G. Cytomegalovirus cell tropism. Curr Top Microbiol Immunol. 2008;325:63-83.
42. Collin MP, Hart DN, Jackson GH, et al. The fate of human Langerhans cells in hematopoietic stem cell transplantation. J Exp Med. 2006;203(1):27-33.
43. D'Souza WN, Hedrick SM. Cutting edge: latecomer CD8 T cells are imprinted with a unique differentiation program. J Immunol. 2006;177(2):777-781.
44. Liu J, Chang YJ, Yan CH, et al. Poor CMV-specific CD8+ T central
memory subset recovery at early stage post-HSCT associates with refractory and recurrent CMV reactivation. J Infect. 2016;73(3):261-270.
45. La Rosa C, Longmate J, Lingaraju CR, et al. Rapid acquisition of cytomegalovirus-specific T cells with a differentiated phenotype, in nonviremic hematopoietic stem transplant recipients vaccinated with CMVPepVax. Biol Blood Marrow Transplant. 2019;25(4):771-784.
46. Ljungman P, Schmitt M, Marty FM, et al. A mortality analysis of letermovir prophylaxis for cytomegalovirus (CMV) in CMVseropositive recipients of allogeneic hematopoietic cell transplantation. Clin Infect Dis. 2020;70(8):1525-1533.
47. Hill JA, Zamora D, Xie H, et al. Delayed-onset cytomegalovirus infection is frequent after discontinuing letermovir in cord blood transplant recipients. Blood Adv. 2021;5(16):3113-3119.
48. Cassaniti I, Colombo AA, Bernasconi P, et al. Positive HCMV DNAemia in stem cell recipients undergoing letermovir prophylaxis is expression of abortive infection. Am J Transplant. 2021;21(4):1622-1628.
49. Sperotto A, Candoni A, Gottardi M, et al. Cytomegalovirus prophylaxis versus pre-emptive strategy: different CD4(+) and CD8(+) T cell reconstitution after allogeneic hematopoietic stem cell transplantation. Transplant Cell Ther. 2021;27(6):518.e1-518.e4.
50. Zamora D, Duke ER, Xie H, et al. Cytomegalovirus-specific T-cell reconstitution following letermovir prophylaxis after hematopoietic cell transplantation. Blood. 2021;138(1):34-43.
Haematologica | 108 June 2023 1543 ARTICLE - Dextramer-monitoring of CMV-specific IR after HSCT E. Tassi et al.
Therapeutic potential of fetal liver cell transplantation in hemophilia A mice
Correspondence: A. Follenzi antonia.follenzi@med.uniupo.it
M.J. Sanchez mjsansan@upo.es
1University of Piemonte Orientale, Department of Health Sciences, School of Medicine, Novara, Italy; 2Centro Andaluz de Biologia del Desarrollo (CABD), University Pablo de Olavide, CSIC, Seville, Spain and 3Department of Genetics, Physiology and Microbiology, School of Biology, Complutense University of Madrid, Madrid, Spain
+SM, SA and MJS contributed equally.
Abstract
S. Merlin simone.merlin@med.uniupo.it
Received: August 31, 2022.
Accepted: January 13, 2023.
Early view: January 26, 2023.
https://doi.org/10.3324/haematol.2022.282001
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Hemophilia A (HA) cell therapy approaches in pediatric individuals require suitable factor (F)VIII-producing cells for stable engraftment. Liver sinusoidal endothelial cells (LSEC) and hematopoietic stem cells (HSC) have been demonstrated to be suitable for the treatment of adult HA mice. However, after transplantation in busulfan (BU)-conditioned newborn mice, adult LSEC/HSC cannot efficiently engraft, while murine fetal liver (FL) hemato/vascular cells from embryonic day 11-13 of gestation (E11-E13), strongly engraft the hematopoietic and endothelial compartments while also secreting FVIII. Our aim was to investigate the engraftment of FL cells in newborn HA mice to obtain a suitable “proof of concept” for the development of a new HA treatment in neonates. Hence, we transplanted FL E11 or E13 cells and adult bone marrow (BM) cells into newborn HA mice with or without BU preconditioning. Engraftment levels and FVIII activity were assessed starting from 6 weeks after transplantation. FL E11-E13+ BU transplanted newborns reached up to 95% engraftment with stable FVIII activity levels observed for 16 months. FL E13 cells showed engraftment ability even in the absence of BU preconditioning, while FL E11 cells did not. BM BU transplanted newborn HA mice showed high levels of engraftment; nevertheless, in contrast to FL cells, BM cells cannot engraft HA newborns in BU non-conditioning regimen. Finally, none of the transplanted mice developed anti-FVIII antibodies. Overall, this study sheds some light on the therapeutic potential of healthy FL cells in the cure of HA neonatal/pediatric patients.
Introduction
Spontaneous hemorrhagic events occurring in hemophilia
A (HA) patients are caused by a reduced or absent coagulation factor (F)VIII activity.1 Presently, these bleeding events are managed by replacement therapy compelling the patients to frequent infusions of exogenous FVIII as prophylaxis,2–4 with a short FVIII half-life (~10-12 hours) and high treatment costs representing the major drawbacks. Moreover, approximately 30% of severe HA patients develop anti-FVIII neutralizing antibodies (inhibitors), thus reducing or nullifying the effectiveness of the replacement therapy.5 New therapeutic approaches rapidly evolved in the last decade as extended half-life FVIII concentrates, FVIII mimetics (bi-speci fi c antibodies), and molecules targeting natural anti-coagulant pathways
(e.g., Fitusiran).6,7 However, all these strategies exert only a temporary therapeutic effect, while cell and/or gene therapy approaches aim to “one-time treatment” able to induce long-term correction with sustained FVIII expression in HA patients.6,7 Indeed, ongoing phase I/II and phase III adeno-associated virus (AAV) liver-directed FVIII gene therapy clinical trials ( clinicaltrials gov. Identi fi er: NCT03734588, NCT03588299, NCT03003533, NCT04370054 and NCT03370913) have been showing promising results for the treatment of adult HA patients.8,9 More recently, the European Commission has granted the conditional marketing authorization to ROCTAVIANTM gene therapy for the treatment of adult patients affected by the severe form of HA (https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-16-1622, https://investors.biomarin.com/2022-08-24-First-Gene-Therapy-for-Adults-with-Se
Simone Merlin,1+ Saicharan Akula,1+ Alessia Cottonaro,1 Tamara Garcia-Leal,2 Luis Javier Serrano,3 Ester Borroni,1 Vakhtang Kalandadze,1 Rocio Galiano,2 Chiara Borsotti,1 Antonio Liras,3 María José Sanchez2+ and Antonia Follenzi1
Haematologica | 108 June 2023 1544 ARTICLE - Cell Therapy & Immunotherapy
vere-Hemophilia-A,-BioMarins-ROCTAVIAN-TM-valoctocogene-roxaparvovec-,-Approved-by-European-CommissionEC). However, several difficulties remain, including lack of viral vector infection specificity, pre-existing immunity to viral vectors, and the inability to insert the full-length F8 gene due to restrictive viral cargo sizes. Additionally, all the above-mentioned clinical trials have included only adult HA patients, while none have involved pediatric patients who are not optimal candidates for these approaches due to the non-integrating nature of AAV and consequent transgene dilution during physiological liver growth.10 For these reasons additional studies and possibly alternative approaches are required for the treatment of early age/neonate HA patients.
Several studies demonstrated that FVIII is produced by endothelial cells, mainly liver sinusoidal endothelial cells (LSEC)11–17 and to a lesser extent by bone marrow (BM)derived hematopoietic and mesenchymal cells,18–21 that showed stable cell engraftment and sustained production of therapeutic FVIII levels following transplantation in preclinical HA murine models.
Following transplantation, the efficiency of vascular and hematopoietic engraftment depends on the transplantation procedure and the preconditioning regimen of the recipient. Transplanted adult LSEC require preconditioning regimens using toxic drugs/compounds (e.g., monocrotaline [MCT]), partial hepatectomy or irradiation 12,22 which are not suitable for the treatment of HA patients and would be even more damaging for pediatric individuals. Thus, less harmful preconditioning regimens with possibly lower/minor and well-defined side effects might represent a valid and more acceptable alternative. One possible treatment is preconditioning with Busulfan (BU), used in murine models23–25 and in clinics with both pediatric and adult patients.26,27 BU induces myeloablation and additionally, as a side effect, damages the vascular endothelium,28 thus possibly promoting both hematopoietic and endothelial cell engraftment in experimental models. Efficiency of cell engraftment is also dependent on the donor and recipient age, as shown for different tissues,29 which represents a relevant issue in a pediatric context. In fact, milder engraftment potential of adult LSEC and adult (A)BM-derived HSC compared to FL LSEC when transplanted to MCT-conditioned adult recipient mice has been previously shown.30 In general, FL cells seem to possess stronger repopulation activity compared to adult HSC31–33 and LSEC,23 thus constituting a potential source for cell therapy approaches.
In order to harness the potential of novel cell types for HA therapeutic purposes in neonatal individuals, it is central that transplanted cells engraft efficiently and differentiate into long-term repopulating FVIII-producing cells. We previously reported that different percentages of HSC and endothelial progenitor cell populations from FL are
capable to reconstitute the hematopoietic and liver endothelial compartments when transplanted into BU-conditioned newborn mice or adult irradiated recipient mice.23,24 However, whether healthy mouse FL cells can engraft, proliferate and reconstitute the hemato/vascular compartment of newborn HA mice is still unknown. Furthermore, it remains to be established whether these cells will assure long-term FVIII production and secretion at therapeutic levels. Here, using healthy FL cells in a preclinical neonatal murine model of HA we determined the conditions and a rational method to establish a novel cell therapy approach for the treatment of HA pediatric patients, supporting the potential of FL cells as a source of FVIII production and establishing the “proof of concept” that cell therapy can be used in pediatric hemophilic patients.
Methods
Animals
Animal studies were approved by the Animal Care and Use Committee of the Università del Piemonte Orientale "A. Avogadro" (Novara, Italy) and the by the Italian Ministry of Health with the authorization no. 758/2021-PR, and the Ethical Review Board of the Universidad Pablo de Olavide (Seville, Spain) according to the Eurpoean Union regulations. In vivo experiments were performed on recipient newborn and adult HA mice in a C57BL/6 background (C57BL/6-HA).18 Donor FL and BM cells were isolated from green fl uorescent protein-positive (GFP+ ) mice in a C57BL/6 background (C57BL/6-Tg(ACTbEGFP)1Osb/J, strain #:003291).18 FL cells were obtained from embryonic day (E)11 or E13 of gestation.23,24 Timed breedings of GFP+ transgenic mice were established to obtain the fetuses. Vaginal plugs were checked daily and the day a plug was detected was considered as E0. In order to generate recipient conditioned newborn mice, HA pregnant females were treated with BU (15.5 mg/kg; Sigma-Aldrich) plus 1 international unit (IU) recombinant human (rh) FVIII (ReFacto®, Pfizer) for 48 hours (h) and 24 h (BU group) or 24 h (1/2 BU1x group) before delivery, while adult mice (8 weeks old) received BU injections (30 mg/kg/injection) 48 h and 24 h before transplantation.
Cell isolation and transplantation
Adult BM (ABM), FL E11 and E13 cell isolation and transplantation into newborns and adult HA mice were performed as previously described.18,24 Briefly, fetuses were harvested from GFP+ females from E11 to E13 of gestation. FL were dissected and transferred individually into icecold D-phosphate-buffered saline (D-PBS) with Ca2+ and Mg2+ (Sigma-Aldrich) supplemented with 5% fetal bovine serum (FBS) (Euroclone). FL cells were isolated by mech-
Haematologica | 108 June 2023 1545 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
anical disaggregation. Only FL samples presenting GFP+ cells by flow cytometry analysis (FACSCalibur, BD) were pooled and resuspended in D-PBS 1% FBS. Adult BM cells were flushed from tibias and femurs of 6-week-old GFP+ mice, treated with red blood lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA), washed and resuspended in D-PBS 1% FBS; 0.3-5x106 cells were resuspended in 50 mL of D-PBS 1% FBS containing 0.2 IU rh FVIII and injected in 30 seconds in the super fi cial temporal vein or facial vein of day 2 HA newborn mice, while cells were resuspended in 300 mL and injected into the tail vein of adult mice. The procedure or the injected volume did not cause adverse effects or severe harm to the recipient mice.
FVIII activity, tail clip assay and enzyme-linked immunosorbant assay
Plasma FVIII activity was measured using the activated partial thromboplastin time (aPTT) as previously described.34 Standard curves were generated by serially diluting plasma pooled from GFP + mice into HA pooled mouse plasma. The presence of anti-FVIII antibodies in plasma of treated mice was evaluated by indirect competitive enzyme-linked immunsorbant assay (ELISA) as previously described. 34,35 The tail clip assay was performed as previously described.34
Flow cytometry analysis
Cells from peripheral blood (PB) and organs were prepared as previously described.36 Liver non-parenchymal cells (NPC) were isolated after liver perfusion as previously published.12 Samples were stained with antibodies listed in the Online Supplementary Table S1. Samples were acquired on the Attune NxT Acoustic Focusing Cytometer (Thermofi sher Scienti fi c) and analysis was performed by FlowJo (Tree Star Inc.).
Immunofluorescence
The organs harvested from treated mice were processed as previously described.34,37 Cryostat sections of 4- m m thickness were blocked in blocking buffer (5% goat serum, 1% BSA, 0.1% Triton X-100 in PBS), incubated with primary antibodies at room temperature (RT), and then incubated in the dark at RT with the secondary antibody along with DAPI. Sections were finally mounted with Mowiol mounting media (Sigma-Aldrich) and observed under a fluorescence microscope (LEICA DM5500B) using Leica Application Suite X (LAS X) software.
RNA isolation and real-time polymerase chain reaction for F8
For quantitative real-time polymerase chain reaction (qRTPCR), total RNA was extracted and cDNA was obtained as previously described.38 Results were analyzed using the relative expression method (2- Δ Ct). The PCR
primers designed for mouse f8 and GAPDH are: mouse f8 E16 F 5’ TGGCACCCACAGAAGATGAG 3’ and mouse f8 E17 R 5’ GGCAAATCAGAAGGGGTCCA 3’ (amplicon size 108 bp); GAPDH F 5’ CATGGCCTTCCGTGTTCCTA 3’ and GAPDH R 5’ GCGGCACGTCAGATCCA 3’ (amplicon size 55 bp).
Statistical analysis
The statistical analysis was performed with GraphPad Prism 5.0 (GraphPad Software). Data were analyzed for normal distribution of population with D’Agostino-Pearson omnibus normality test followed by a one-way analysis of variance (one-way ANOVA). Two-way ANOVA followed by a post hoc Bonferroni’s test was run to compare engraftment between groups. Pearson’s correlation test was performed to correlate percentage of engraftment and FVIII activity in all mice. Statistical significance was assumed for P<0.05.
Results
Engraftment of fetal liver cells in hemophilia A newborn recipient mice contributes to long-term FVIII production at therapeutic levels
In order to determine whether engraftment of FL cells into HA neonates could ameliorate the bleeding phenotype, we injected FL cells from congenic GFP + mice into the facial vein of BU-treated newborn HA mice (Figure 1A). We transplanted different numbers of cells from FL cells E11 or E13 according to the developmental stage (Table 1).23,24 After transplantation, mice were periodically monitored for engraftment (GFP+ cells in PB) and plasma FVIII activity. Flow cytometry analysis showed GFP+ cells in PB of all mice receiving FL cells with BU preconditioning up to 16 months (Figure 1B). Moreover, the FL E13 group displayed significantly higher chimerism than the FL E11 mice, starting from 12 months after transplantation.
Along with the engraftment we evaluated mouse FVIII (mFVIII) activity: all transplanted mice showed FVIII activity >5% (Figure 1C; Table 1) without anti-FVIII antibody production (Figure 1D). Following transplantation, the percentage of PB GFP+ cells and FVIII activity showed a direct significant correlation (Pearson’s correlation test, P <0.0001) ( Online Supplementary Figure S1), suggesting that FVIII production correlated with the PB engraftment level. The correction of the bleeding phenotype in treated mice was evaluated by tail clip challenge at 16 months after transplantation, showing a significant reduction in blood loss in transplanted mice compared to control BUtreated HA mice (noFL+BU) (Figure 1E). This indicates that engraftment of FL cells in HA newborn recipient mice can contribute to a life-long correction of the HA bleeding phenotype.
Haematologica | 108 June 2023 1546 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
Fetal liver-derived hematopoietic cells are responsible for long-term FVIII production in transplanted mice

In order to characterize engrafted cells, GFP+ cells were analyzed in spleen, BM and liver. Flow cytometry and immunofluorescence analysis showed that GFP+ cells from the spleen and BM of recipient mice were mainly of hematopoietic origin (CD45+) in both FL E11 and FL E13 groups in accordance with the percentage of GFP+ cells in PB (Figure 2A-E) 16 months after transplantation.
As FL cells showed the ability to reconstitute liver endothelial cells and LSEC,13,23 we performed flow cytometry analysis on the hepatic non-parenchymal cell (NPC) fraction and immunofluorescence on liver sections from FL
Figure 1. Engraftment of fetal liver cells into newborn hemophilia A mice. (A) Schematic representation of the experimental procedure. Hemophila A (HA) pregnant mice received busulfan (BU) 2 days and 1 day prior to birth. At 2 days after birth, HA mice were transplanted with fetal liver (FL) embyonic day 11 (E11) and embryonic day 13 (E13) cells from green fluorescent protein-positive (GFP+) mice. (B) Engraftment of transplanted GFP+ cells, was evaluated by flow cytometry on peripheral blood cells at different time points up to 16 months after transplantation. GFP+ cells were detectable in peripheral blood of all transplanted mice (n=10-11). Number of cell transplanted per recipient: FL E11 0.3±0.1x106; FL E13 5x106. (C) FVIII activity in plasma of transplanted mice was evaluated by activated partial thromboplastin time (aPTT) assay. Murine FVIII activity was detectable up to 16 months in all transplanted mice. (D) Enzymelinked immunosorbant assay showing that none of the transplanted mice developed anti-FVIII antibodies. Plasma samples were tested 12-16 months after transplantation (plasma dilution: 1:200 and 1:2,00; control [Ctr+]: plasma from mice immunized with FVIII). (E) Tail clip assay performed at 16 months after transplantation showed correction in all recipient mice (n=5 each group). Graphs show single values and mean values ± standard deviation. Ctr+: GFP+ mice(n=3 each). *P<0.05; **P<0.01. OD: optical density; FACS: fluorescence-activated cell sorting; IgG: immunoglobulin G.
E11 and FL E13 transplanted mice. The data showed the presence of GFP+ cells in the liver of all treated mice, where hepatic NPC GFP+ cells were mainly hematopoietic cells (CD45+), while only a low percentage of liver CD31+CD45endothelial cells/LSEC showed GFP expression (Figure 2B, C).
Immunostaining on spleen and liver sections from these mice confirmed long-term contribution of FL cells to hematopoietic cells (CD45+) and low contribution to endothelial cells (CD31+) in the liver (Figure 2D, E). In accordance with flow cytometry and immunofluorescence data, f8 mRNA was mainly detected in the spleen, BM and liver NPC (Figure 2F). Altogether, these data confirm the po-
A B C D E Haematologica | 108 June 2023 1547 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
GFP eng.: mice with ≥4% green fluorescent protein-positive (GFP+) cells in the peripheral blood; FVIII act.: mice with ≥2% FVIII activity; control: not transplanted hemophila A mice; BM: bone marrow; BU: busulfan; BU2x: pretreated with the standard dose of BU; BU1x: pretreated with half of the standard dose of BU; FL: fetal liver; E11: embryonic day 11; E13: embryonic day 13; mth: months; eng: engraftment; act: activity.
tential of FL cells to engraft in newborn HA mice pretreated with BU.
Effect of busulfan dosage on fetal liver cell engraftment in newborn hemophilia A mice
Since we observed high engraftment (up to 95%) in mice with BU pretreatment, we evaluated the possibility of obtaining high engraftment levels while reducing the preconditioning regimen. We, thus repeated the transplantation studies in two groups of newborn HA mice, one pretreated with the standard dose of BU (BU2x) while the second group received half dosage (BU1x). Pretreated newborns were transplanted with FL E11 or FL E13 cells as described above, and engraftment level was regularly evaluated. Six months after transplantation both BU1x groups showed significantly lower engraftment compared to their BU2x counterparts with the FL E11 cell-injected mice displaying the lowest level (~2.5%) (Figure 3; Table 1; Online Supplementary Table S2). As described previously, FVIII activity correlated with engraftment levels. Therefore, higher levels of engraftment and therapeutic levels of FVIII were achieved following the full BU treatment regimen.
Bone marrow cell transplantation in newborn hemophilia A mice
We previously showed that the transplantation of ABM cells can correct the bleeding phenotype of adult HA mice.18 On this basis, we decided to evaluate the ability of ABM cells to engraft and correct the bleeding phenotype of newborn HA mice. Following transplantation in newborn HA mice, we evaluated the engraftment and the phenotypic correction at 6 weeks, 3 months and 6 months
and we compared these results with those obtained with mice receiving FL E13 cells. Mice transplanted with FL cells showed significantly higher engraftment than mice transplanted with BM at 6 weeks (FL 79.5±4.5% vs. BM 68.53±5.9%) and 6 months (FL 90.2±2.6% vs. BM 74.9±6.6%) after transplantation ( P<0.05) (Figure 4A); however, 6 weeks after transplantation mice receiving BM cells showed marginally higher mFVIII activity than mice transplanted with FL E13 (FL 11.8±3.6% vs. BM 14.7±4.9%; P<0.05), while FL E13-transplanted mice showed higher mFVIII activity compared to mice transplanted with BM cells after 6 months (FL 14.3±3.8% vs. BM 12.7±3%; P<0.05) (Figure 4B). Interestingly, in our control mice not treated with BU (noBU), we observed engraftment in mice receiving FL E13 cells but not in mice transplanted with ABM cells (FL E13 ~15% vs. BM <1%; P<0.01) (Figure 4C), and mFVIII activity levels correlating with engraftment levels, showing to be significantly higher when compared with ABM cells (FL E1 36.3% vs. BM <0.1%; P<0.01) (Figure 4D). Levels of mFVIII activity were additionally confirmed by bleeding assay (Online Supplementary Figure S2). These data confirm that the correction of the bleeding phenotype using ABM cell transplantation requires BU preconditioning in newborn HA mice, while FL E13 cells showed long-term engraftment potential and mFVIII production ability even in the absence of BU preconditioning.
Fetal liver cells engraftment in adult hemophilia A mice requires busulfan pretreatment
It has been previously shown that FL E9.5-10.5 cells showed preferential engraftment in neonatal mice, while HSC from a later embryonic developmental stage or from
Recipient mice Donor cells Transplanted cells x106 Positive mice/analyzed mice, N/N 4 mth 12 mth no BU BU2X BU1X no BU BU2X GFP eng. FVIII act. GFP eng. FVIII act. GFP eng. FVIII act. GFP eng. FVIII act. GFP eng. FVIII act. Newborn FL E11 0.3-0.4 11/11 11/11 2/8 0/8 9/9 9/9 FL E13 2-5 19/20 18/20 20/20 20/20 7/7 7/7 6/7 4/7 20/20 20/20 BM 2-5 0/9 0/9 7/7 7/7 Control 0 0/6 0/6 0/21 0/21 0/6 0/6 0/21 0/21 Adult FL E13 3-5 0/5 0/5 6/6 6/6 BM 5 0/5 0/5 5/5 5/5 Control 0 0/5 0/5 0/5 0/5
Table 1. Peripheral blood engraftment and FVIII activity in plasma of different groups of transplanted mice.
Haematologica | 108 June 2023 1548 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
adult BM showed higher engraftment activity in adult mice.31 Thus, we evaluated the engraftment ability of FL E13 in adult (8-week-old) HA mice with or without sublethal myeloablation. After transplantation, mice receiving FL cells without BU preconditioning showed no engraftment, and we observed engraftment only in BU pretreated
mice. Six weeks after transplantation, GFP+ cells in PB ranged from 50% to 70%, while after 6 months more than 90% of cells were GFP (Figure 5A), with a concomitant bleeding phenotype correction ranging from ~9% at 6 weeks to 20% after 6 months (Figure 5B); data was additionally confirmed by tail clip assay (Online Supplementary
Figure 2. Engrafted donor cells characterization. (A, B) Representative flow cytometry analysis for characterization of green fluorescent protein-positive (GFP+) cells in spleen, bone marrow (A) and liver non-parenchymal cell (NPC) fraction (B) 16 months after transplantation. (C) Characterization of GFP+ cells in hepatic NPC fraction. Virtually all GFP+ cells in the spleen, bone marrow and liver showed CD45 expression (A-C), while few GFP+ events (<1%) showed CD45- and CD31+ expression in NPC (B, C). (D, E) Flow cytometry data were confirmed by immunofluorescence in liver (D) and spleen (E) showing that GFP+ cells were virtually all CD45+. (F) mRNA expression analysis showed murine FVIII mRNA expression mainly in hemopoietic organs. Graphs are showing single values and mean values ± standard deviation.

A B C D E F
Haematologica | 108 June 2023 1549 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
Figure S3).
These data indicate that correction of the bleeding phenotype of adult HA mice requires preconditioning treatments to allow the engraftment of FL cells.
atric HA patients. We previously demonstrated that FL cells engrafted and repopulated the hemato/vascular compartment of wild-type (wt) newborn recipient mice,24 and additionally characterized in FL a unique cell population capable of a stable multi-organ endothelial reconstitution, mostly composed of endothelial committed cells.23 More recently, we also reported that FVIII mRNA progressively increases in FL and in other different embryonic locations from E9 to E12 of gestation, in parallel to the expansion of the vascular network.38
In this study, following transplantation in newborn HA mice, both FL E11 and FL E13 cells showed long-term engraftment potential,23,24 and were able to correct the bleeding phenotype of the recipient hemophilic mice. These results are in line with the ones obtained in lethally irradiated adults HA mice, in which we observed a longterm phenotypic correction following total BM cell transplantation.18
Figure 3. Effect of busulfan dosage on fetal liver cell engraftment in newborn hemophila A mice. Six months following transplantation of fetal liver embryonic day 11 (FL E11) (0.3x106 cells per mouse) or FL E13 (5x106 cells per mouse) engraftment was ≥70% in mice treated with 15.5 mg/kg busulfan (BU) 48 and 24 hours before transplantation (standard dose of BU [BU2x]). In mice treated with half BU dosage (BU1x) (15.5 mg/kg 24 hours before transplantation) engraftment significantly dropped but was higher in mice receiving FL E13 compared to FL E11 (FL E11+BU2x, n=11; FL E11+BU1x, n=8; FL E13+BU2x, n=18; FL E13+BU1x, n=7). Graph is showing single values and mean values ± standard deviation. ns: not significant; **** P<0.0001.
Discussion
Over the last two decades many attempts have been made to develop long-term treatment for HA by using cell and/or gene therapy strategies. Despite numerous studies on adults, few data are currently available on newborn HA mice following cell and gene therapy.6,8,39
New cell therapy approaches able to ensure stable longterm FVIII production at therapeutic levels are extremely attractive, especially for the treatment of early age/neonate patients. Selection of the right cell type for transplantation is the key element in a cell-based therapy approach. Endothelial cells, mainly LSEC are the principal source of FVIII within the body;12,16,40 additionally, hematopoietic cells are able to produce and secrete FVIII, although to a lesser extent compared to endothelial cells, thus correcting the bleeding phenotype of adult recipient mice.18,19
Neonatal recipients are more permissive for embryonic/early fetal hematopoietic progenitor cell engraftment31,41 and higher proliferative activity of fetal-derived hematopoietic progenitors also confers increased engraftment potential in newborns as well as in adult recipients.31,33 Taking these previous studies into consideration, we investigated in a murine preclinical model the potential of FL cells a source for the development of a cell therapy approach to treat pedi-
Since BM is a readily available clinical source for cell therapy,19,27 we transplanted BM cells into newborn HA mice. Previous studies showed that FL cells exhibited a higher capacity for long-term and multi-lineage hematopoietic reconstitution than equal numbers of BM cells transplanted into lethally irradiated adult mice31,33 or into newborn recipients.31 Similarly, in our settings FL E13 cell engraftment in neonate HA mice was higher compared to ABM cells, although the difference was not statistically significant at all time points. Despite lower engraftment, BM cell-transplanted mice showed higher mFVIII activity up to 4 months. Analysis of the lineage output of transplanted HSC through development has previously revealed a trend for reduced myeloid lineage output from FL E13 compared to adult BM HSC at 4 months post-transplant in irradiated adult recipient mice.42 Considering that myeloid cells are the main producers of FVIII among the hematopietic lineage,19 this data can support the notion that BM cells may better contribute to the myeloid compartment and hence to FVIII production in newborns. Further work is necessary to characterize any differences in engraftment and lineage output of BM and FL cells transplanted HSC into HA newborn mice.
In our study, FL cells, particularly FL E13 cells, were able to engraft and produce therapeutic levels of FVIII even in the absence of preconditioning in newborn HA mice. This ability was not displayed by ABM cells, whereas both FL and ABM cells did not engraft in adults without preconditioning. Increasing circulating levels to 2-3% of normal FVIII activity can significantly reduce the risks of spontaneous bleeding and represent a clinically relevant achievement from the patient management point of view,43 even though several studies showed that levels of 20-30% may be required to prevent joint bleeding events, while 3-5% can lower the risk to 1-2 joint bleeding episodes per year.4 Despite the fact that our results were obtained using a

Haematologica | 108 June 2023 1550 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
Figure 4. Bone marrow cell engraftment in newborn hemophilia A mice. (A) Following transplantation in bulsufan (BU)-conditioned newborn hemophilia A (HA) mice, embryonic day 13 (E13) cell engraftment was significantly higher than adult bone marrow (BM) cells. (B) Interestingly, murine FVIII (mFVIII) levels were higher in mice receiving BM cells, despite the lower engraftment level. (C, D) In mice receiving cells without prior treatment (noBU), FL E13 cells showed higher green fluorescent protein-positive (GFP+) cell engraftment (C) and mFVIII activity (D) compared to BM cells (n=8-10; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001). Graphs show single values and mean values ± standard deviation.
preclinical mouse model of HA, we hypothesized that our strategy of cell transplantation without preconditioning could represent an alternative therapeutic approach which can improve patients’ quality of life, while avoiding adverse effects of preconditioning chemotherapy. Additionally, despite the potential demonstrated in this study using a preclinical HA mouse model, it is unlikely that FL cells will be used in a clinic setting for several reasons, such as availability, allogenic immune responses, therapeutic efficacy and ethical issues.
We are still far from fully understanding stem/progenitor cell engraftment in HA neonates, particularly related to conditioning regimens for efficient and safe endothelial progenitor cell engraftment. Studies related to engraftment of HSC in WT mice are more advanced and alternative methods for HSC transplantation in adults have been described including transplantation of a high number of HSC in non-conditioned hosts44 and novel conditioning methods inducing partial host BM ablation.45,46 Moreover, it has been shown that active cell cycle enhances neonatal engraftment.31 Considering that most of FL hematopoietic stem and progenitor cells are actively cycling from E11 to E13-13.5 whereas ABM cells divide infrequently47 this could confer FL cell engraftment advantages under non-conditioning regimen.
We speculate that the difference in engraftment ability between FL E11 and FL E13 cells could be explained by HSC ontogeny and their commitment where FL E10.5 HSC migrate from the aorta-gonads-mesonephros region (AGM) to FL, whereas FL E13 HSC start to move towards BM.31 Additionally, previous transplantation studies using irradiated NOD/SCID mice have shown that the number and self-renewal activity of human lympho-myeloid stem cells within the CD34+CD38- population were similar in FL and cord blood (CB) and decrease during ontogeny in ABM. However, although FL cells presented more self-renewal capacity (of approximately 7-fold to CB and 300-fold to ABM)48 and high engraftment in an immunodeficient mouse model,49 CB and ABM cells showed higher output of mature myeloid cells (CD45/71+CD15/66b+).48 Also, CB cells can be an effective source of endothelial colonyforming cells, a subset of circulating endothelial progenitor cells, with recombinant FVIII production capacity and long-term endothelial engraftment potential in newborn and adult HA mice.50 Therefore, CB cells constitute promising fetal-like candidates for the use as cell-based therapy for efficient treatment of newborn HA individuals. More studies are necessary to determine the output of human CB cells starting from preclinical models such as transplantation into immunodeficient newborn HA mice.

A B C D Haematologica | 108 June 2023 1551 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
Figure 5. Fetal liver cell engraftment and bleeding correction in adult hemophilia A mice. Fetal liver (FL) E13 cells were able to engraft (A) and produce mouse factor VIII (mFVIII) (B) following transplantation in adult hemophilia A (HA) mice pretreated with 2x30 mg/kg busulfan (BU), while no cells engrafted following transplantation in untreated adult HA mice (no BU) (n=5-6; ***P<0.001, ****P<0.0001). Graphs show single values and mean values ± standard deviation.
Further studies on different cell sources, in combination with the use of alternative non- or less-damaging conditioning regimens in newborns will lay the foundation to delineate molecular mechanisms involved in transplanted cell engraftment that should lead to new possibilities for bleeding phenotype correction in pediatric HA individuals. Moreover, we envisage the incorporation of gene therapy approaches, thus potentially further increasing FVIII production from transplanted cells.
Overall, this study has increased our knowledge on healthy FL cells and their possible usage in cell therapy approaches for the treatment of newborn patients. Future studies aimed at determining whether FL-derived hematopoietic and/or endothelial precursors would support higher engraftment ability and therapeutic potential compared to total FL cells could be envisaged. This study provides useful information regarding the hemato/vascular compartment reconstitution/repair in a neonatal preclinical model of HA, thus paving the way for studies focused at obtaining long-term reconstituting progenitors from other sources, such as CB or induced pluripotent stem cells, for the treatment of HA.
Disclosures
No conflicts of interest to disclose.
Contributions
SM, SA and MJS designed and performed experiments and
References
1. Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388(10040):187-197.
2. Arcieri R, Calizzani G, Candura F, Mannucci PM. Recommendations for factor VIII product source to treat patients with haemophilia A. Blood Transfus. 2017;15(3):285.
3. Coppola A, Santagostino E, Hassan HJ, et al. The increased demand for plasma-derived factor VIII in Italy between 2011 and

analysed data. SM, SA and AC conducted the in vivo studies. LJS and AL performed the mRNA analysis. EB, VK, TGL, RG and CB set up immunofluorescence and flow cytometry analysis. AF and MJS conceiv ed the study. AF generated most funding, supervised the whole project and analysed data. SM, SA, MJS and AF drafted the paper that was completed by all authors who critically reviewed the manuscript and approved the final version.
Funding
AF was supported in part by Telethon grant no. GGP19201 and by Horizon 2020, HemAcure grant no. 667421, Vanguard grant no. 874700. SM was partially supported by the Università del Piemonte Orientale (FAR 2017) and by Bando Roche per la Ricerca 2019. The Junta de Andalucia Research Funding Program PAI-BIO295 supported the work of MJS. MJS also acknowledges financial support from the Maria de Maeztu-CABD MDM-2016-0687 and CEX-2020001088-M grants and the Empleo Juvenil-JA 2018 program that supported the work of RG. LJS and AL were supported by the Andalusian Association of Hemophilia ASANHEMO FV2016-20.
Data-sharing statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
2014 is attributable to treatment of adult patients rather than paediatric or previously unexposed patients with severe haemophilia A. Blood Transfus. 2017;15(3):281.
4. Marchesini E, Morfini M, Valentino L. Recent advances in the treatment of hemophilia: a review. Biol Targets Ther. 2021;15:221-15235.
5. van Velzen AS, Eckhardt CL, Peters M, et al. Intensity of factor VIII treatment and the development of inhibitors in non-severe
A B
Haematologica | 108 June 2023 1552 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
hemophilia A patients: results of the INSIGHT case-control study. J Thromb Haemost. 2017;15(7):1422-1429.
6. Batty P, Lillicrap D. Advances and challenges for hemophilia gene therapy. Hum Mol Genet. 2019;28(R1):R95-R101.
7. Weyand AC, Pipe SW. New therapies for hemophilia. Blood. 2019;133(5):389-398.
8. Batty P, Lillicrap D. Hemophilia gene therapy: approaching the first licensed product. Hemasphere. 2021;5(3):e540.
9. Tomeo F, Mariz S, Brunetta AL, Stoyanova-Beninska V, Penttila K, Magrelli A. Haemophilia, state of the art and new therapeutic opportunities, a regulatory perspective. Br J Clin Pharmacol. 2021;87(11):4183-4196.
10. Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAVmediated in vivo gene therapy. Mol Ther Methods Clin Dev. 2018;8:87-104.
11. Do H, Healey JF, Waller EK, Lollar P. Expression of factor VIII by murine liver sinusoidal endothelial cells. J Biol Chem. 1999;274(28):19587-19592.
12. Follenzi A, Benten D, Novikoff P, Faulkner L, Raut S, Gupta S. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118(3):935-945.
13. Fomin ME, Zhou Y, Beyer AI, Publicover J, Baron JL, Muench MO. Production of factor VIII by human liver sinusoidal endothelial cells transplanted in immunodeficient uPA mice. PLoS One. 2013;8(10):e77255.
14. Hellman L, Smedsröd B, Sandberg H, Pettersson U. Secretion of coagulant factor VIII activity and antigen by in vitro cultivated rat liver sinusoidal endothelial cells. Br J Haematol. 1989;73(3):348-355.
15. Kumaran V, Benten D, Follenzi A, Joseph B, Sarkar R, Gupta S. Transplantation of endothelial cells corrects the phenotype in hemophilia A mice. J Thromb Haemost. 2005;3(9):2022-2031.
16. Shahani T, Covens K, Lavend’homme R, et al. Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J Thromb Haemost. 2014;12(1):36-42.
17. van der Kwast T, Stel H, Cristen E, Bertina R, Veerman E. Localization of factor VIII-procoagulant antigen: an immunohistological survey of the human body using monoclonal antibodies. Blood. 1986;67(1):222-227.
18. Follenzi A, Raut S, Merlin S, Sarkar R, Gupta S. Role of bone marrow transplantation for correcting hemophilia A in mice. Blood. 2012;119(23):5532-5542.
19. Zanolini D, Merlin S, Feola M, et al. Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica. 2015;100(7):881-892.
20. Caselli D, Morfini M, Paolicchi O, Frenos S, Casini T, Aricò M. Cord blood hematopoietic stem cell transplantation in an adolescent with haemophilia. Haemophilia. 2012;18(2):e48-e49.
21. Ostronoff M, Ostronoff F, Campos G, et al. Allogeneic bone marrow transplantation in a child with severe aplastic anemia and hemophilia A. Bone Marrow Transplant. 2006;37(6):627-628.
22. Krause P, Rave-Fränk M, Wolff HA, Becker H, Christiansen H, Koenig S. Liver sinusoidal endothelial and biliary cell repopulation following irradiation and partial hepatectomy. World J Gastroenterol. 2010;16(31):3928-3935.
23. Cañete A, Comaills V, Prados I, et al. Characterization of a fetal liver cell population endowed with long-term multiorgan endothelial reconstitution otential. Stem Cells. 2017;35(2):507-521.
24. Garcia-Ortega AM, Cañete A, Quintero C, et al. Enhanced hemato-vascular contribution of SCL-3′Enh expressing fetal liver cells uncovers their potential to integrate in extra-
medullary adult niches. Stem Cells. 2010;28(1):100-112.
25. Peake K, Manning J, Lewis CA, Barr C, Rossi F, Krieger C. Busulfan as a myelosuppressive agent for generating stable high-level bone marrow chimerism in mice. J Vis Exp. 2015;1(98):e52553.
26. Ciurea SO, Andersson BS. Busulfan in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2009;15(5):523-536.
27. Zao JH, Schechter T, Liu WJ, et al. Performance of busulfan dosing guidelines for pediatric hematopoietic stem cell transplant conditioning. Biol Blood Marrow Transplant. 2015;21(8):1471-1478.
28. Zeng L, Jia L, Xu S, Yan Z, Ding S, Xu K. Vascular endothelium changes after conditioning in hematopoietic stem cell transplantation: role of cyclophosphamide and busulfan. Transplant Proc. 2010;42(7):2720-2724.
29. Lau A, Kennedy BK, Kirkland JL, Tullius SG. Mixing old and young: enhancing rejuvenation and accelerating aging. J Clin Invest. 2019;129(1):4-11.
30. Filali EE, Hiralall JK, Van Veen HA, Stolz DB, Seppen J. Human liver endothelial cells, but not macrovascular or microvascular endothelial cells, engraft in the mouse liver. Cell Transplant. 2013;22(10):1801-1811.
31. Arora N, Wenzel PL, McKinney-Freeman SL, et al. Effect of developmental stage of HSC and recipient on transplant outcomes. Dev Cell. 2014;29(5):621-628.
32. Bowie MB, Kent DG, Dykstra B, et al. Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc Natl Acad Sci U S A. 2007;104(14):5878-5882.
33. Szilvassy SJ, Meyerrose TE, Ragland PL, Grimes B. Differential homing and engraftment properties of hematopoietic progenitor cells from murine bone marrow, mobilized peripheral blood, and fetal liver. Blood. 2001;98(7):2108-2115.
34. Merlin S, Cannizzo ESES, Borroni E, et al. A novel platform for immune tolerance induction in hemophilia a mice. Mol Ther. 2017;25(8):1815-1830.
35. Sabatino DE, Freguia CF, Toso R, et al. Recombinant canine Bdomain–deleted FVIII exhibits high specific activity and is safe in the canine hemophilia A model. Blood. 2009;114(20):4562-4565.
36. Merlin S, Famà R, Borroni E, et al. FVIII expression by its native promoter sustains long-term correction avoiding immune response in hemophilic mice. Blood Adv. 2019;3(5):825-838.
37. Famà R, Borroni E, Merlin S, et al. Deciphering the Ets-1/2mediated transcriptional regulation of F8 gene identifies a minimal F8 promoter for hemophilia A gene therapy. Haematologica. 2020;106(6):1624-1635.
38. Serrano L, Cañete A, Garcia-Leal T, et al. Searching for a cellbased therapeutic tool for haemophilia A within the embryonic/foetal liver and the aorta-gonads-mesonephros region. Thromb Haemost. 2018;118(08):1370-1381.
39. Cantore A, Naldini L. WFH State-of-the-art paper 2020: in vivo lentiviral vector gene therapy for haemophilia. Haemophilia. 2021;27(S3):122-125.
40. Pan J, Dinh TT, Rajaraman A, et al. Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo. Blood. 2016;128(1):104-109.
41. Kieusseian A, de la Grange PB, Burlen-Defranoux O, Godin I, Cumano A. Immature hematopoietic stem cells undergo maturation in the fetal liver. Development. 2012;139(19):3521-3530.
42. Papathanasiou P, Attema JL, Karsunky H, Xu J, Smale ST,
Haematologica | 108 June 2023 1553 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
Weissman IL. Evaluation of the long-term reconstituting subset of hematopoietic stem cells with CD150. Stem Cells. 2009;27(10):2498-2508.
43. Sokal EM, Lombard C, Mazza G. Mesenchymal stem cell treatment for hemophilia: a review of current knowledge. J Thromb Haemost. 2015;13(Suppl 1):S161-S166.
44. Shimoto M, Sugiyama T, Nagasawa T. Numerous niches for hematopoietic stem cells remain empty during homeostasis. Blood. 2017;129(15):2124-2131.
45. Ganuza M, McKinney-Freeman S. Hematopoietic stem cells under pressure. Curr Opin Hematol. 2017;24(4):314-321.
46. Taya Y, Ota Y, Wilkinson AC, et al. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science. 2016;354(6316):1152-1155.
47. Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell
heterogeneity takes center stage. Cell Stem Cell. 2012;10(6):690-697.
48. Holyoake TL, Nicolini FE, Eaves CJ. Functional differences between transplantable human hematopoietic stem cells from fetal liver, cord blood, and adult marrow. Exp Hematol. 1999;27(9):1418-1427.
49. Vanuytsel K, Villacorta-Martin C, Lindstrom-Vautrin J, et al. Multi-modal profiling of human fetal liver hematopoietic stem cells reveals the molecular signature of engraftment. Nat Commun. 2022;13(1):1103.
50. Gao K, Kumar P, Cortez-Toledo E, et al. Potential long-term treatment of hemophilia A by neonatal co-transplantation of cord blood-derived endothelial colony-forming cells and placental mesenchymal stromal cells. Stem Cell Res Ther. 2019;10(1):34.
Haematologica | 108 June 2023 1554 ARTICLE - Fetal liver cell therapy for HA newborns S. Merlin et al.
Differential inhibition of T-cell receptor and STAT5 signaling pathways determines the immunomodulatory effects of dasatinib in chronic phase
chronic myeloid leukemia
Correspondence: H. De Lavallade
h.delavallade@nhs.net
hugues.2.de_lavallade@kcl.ac.uk
1Department of Hematology, Guy’s International Center of Excellence in Myeloid Disorders, Guy’s and St Thomas NHS Foundation Trust, London, UK; 2School of Cancer and Pharmaceutical Sciences, King’s College London, London, UK; 3Medical and Molecular Genetics, King’s College London, London, UK; 4Service d’Hematologie, Centre Hospitalier de Versailles, Université de Versailles Paris-Saclay, UMR1184, CEA, Paris, France; 5Department of Cellular Pathology, Guy’s and St Thomas’ NHS Foundation Trust, London, UK and 6Integrated Toxicology Laboratory, Synnovis, King’s College Hospital NHS Foundation Trust, London, UK
Abstract
Received: August 26, 2022.
Accepted: January 16, 2023.
Early view: January 26, 2023.
https://doi.org/10.3324/haematol.2022.282005
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Dasatinib is a multi-kinase inhibitor with activity against the SRC kinase LCK, which plays a critical role in T-cell receptor signaling. Dasatinib, initially developed as an immunosuppressive agent, is by contrast, also noted to result in enhanced tumor immunity in a subset of patients. We studied the impact of dasatinib in chronic myeloid leukemia patients and compared it with patients taking other tyrosine kinase inhibitors (TKI) and healthy controls. We found that patients on dasatinib showed inhibition of both T-cell receptor (TCR) and STAT5 signaling pathways, and reduced expression of Teffector pro-inflammatory cytokines. In addition, dasatinib induced selective depletion of regulatory T cells (Tregs) and effector Tregs, particularly in patients with clonal expansion of effector CD8+ T cells, who demonstrated greater and preferential inhibition of Treg TCR intracellular signaling. In addition, we show that dasatinib selectively reduces Treg STAT5 phosphorylation via reduction of IL-2, in relation with the marked reduction of plasma IL-2 levels in patients taking dasatinib. Finally, patients on other TKI had significantly increased TCR signaling in TIM3+ cells compared to patients taking dasatinib, suggesting that chronic SRC kinase inhibition by dasatinib may play a role in preventing TIM-3-mediated T-cell exhaustion and preserve anti-tumor immunity. These data provide further insight into the selective immunomodulatory effects of dasatinib and its potential use for pharmacologic control of immunotherapies..
Introduction
The second-generation tyrosine kinase inhibitor (TKI) dasatinib (Bristol Myers Squibb) is an effective treatment used in the management of patients with chronic myeloid leukemia (CML).1-3 It is utilized both as a first-line agent and in patients resistant to other TKI, with an estimated 325-times greater affinity for BCR-ABL1 than imatinib.4 Dasatinib is a multi-kinase inhibitor with activity against the SRC family kinase LCK, which is inhibited at nanomolar concentrations.5 LCK plays a critical role in signaling from the T-cell receptor (TCR) and expression is largely restricted to T cells making it a promising target for suppression of T-cell activity.6,7 Immediate downstream targets from LCK are the key signaling molecules ZAP70
and LAT, which have both been implicated in regulatory T-cell (Treg) development also. Several studies have previously reported on the inhibitory effect of dasatinib on the function of T effectors, with reduction in signaling, proliferation and expression of pro-inflammatory cytokine expression.8-10 T-cell immunoglobulin domain and mucin domain 3 (TIM-3) is an immune-checkpoint molecule that has been proposed to promote T-cell exhaustion through initial enhancement of TCR signaling pathways.11
Tregs are a suppressive subset of CD4+ T cells, identifiable by high expression of the interleukin 2α (IL-2α) receptor CD25 and the transcription factor FOXP3.12, 13 In vitro analyses of Tregs from healthy controls have shown that increasing concentrations of dasatinib result in reduced expression of FOXP3 and reduced Treg function.14 Tregs
Patrick Harrington,1,2 Richard Dillon,1,3 Deepti Radia,1 Philippe Rousselot,4 Donal P. McLornan,1,2 Mark Ong,5 Anna Green,5 Alessandro Verde,6 Farzana Hussain,6 Kavita Raj,1 Shahram Kordasti,1,2 Claire Harrison1,2 and Hugues de Lavallade1,2
Haematologica | 108 June 2023 1555 ARTICLE - Chronic Myeloid Leukemia
have been shown to be increased at diagnosis in CML patients, with subsequent reduction following effective TKI therapy.15,16 They have also previously been shown to be increased in patients with advanced-phase disease and high-risk prognostic scores.17,18 STAT5 is a critical signaling molecule that propagates cytokine responses and is the downstream target of IL-2. STAT5 signaling plays a critical role in Treg differentiation and the JAK2-STAT5 pathway plays a pivotal role in survival and proliferation of CML leukemic stem cells.
A subset of patients treated with dasatinib are observed to develop clonal large granular lymphocytosis (LGL) which is associated with immune-mediated toxicity and improved CML-related outcomes.19-21 It has previously been shown that patients who develop LGL have a reduced frequency of Tregs.22 We hypothesized that these patients have a reduction in both number and function of Tregs, resulting in increased toxicity but also allowing for enhancement of the anti-leukemic immune response. We hereby report findings from an ex vivo functional assessment of the effect of dasatinib on immune cell subsets including Tregs in CML patients.
Methods
Patients and controls
Twenty-five Patients with a World Health Organizationdefined diagnosis of chronic phase CML (dasatinib n=18, nilotinib n=5, imatinib n=2) and seven healthy controls (HC) (median age 46 years, range, 31-78; 57% male) were recruited in accordance with the Regional Research and Ethics Review Board. Most patients had achieved major molecular response (MMR/MR3) or deeper at the time of inclusion and patient characteristics are summarized in the Online Supplementary Table S1. Patients administered TKI therapy at a uniform time on the morning of blood sampling. Peripheral blood mononucelar cells (PBMC) were isolated from venous blood samples using standard Ficoll density centrifugation and were cryopreserved in 30% dimethyl sulfoxide (DMSO) and fetal calf serum (FCS).
Intracellular phospho-specific flow cytometric assay
Phosphoflow cytometry was performed in Tregs, T effectors and natural killer (NK) cells to assess the effect of dasatinib on signaling downstream from the TCR and activating NK-cell receptors as well as cytokine signaling pathways. Briefly, cells were stained with a viability dye prior to activation with H2O2 (50 mM), due to its activity as a potent phosphatase inhibitor. Cells were fixed and permeabilized (BD Phosphoflow Buffer) prior to staining with antibodies directed against surface and intracellular markers (Online Supplementary Appendix). Cells were ana-
lyzed for phosphorylation of the signaling proteins ZAP70, LAT and STAT5. Flow cytometry analysis was performed on BD Fortessa and analysis was performed using FlowJo v10.3.
Intracellular cytokine flow cytometric assay
Intracellular flow cytometry was also performed to assess the impact of dasatinib on T-effector cytokine production including tumor necrosis factor α (TNFα), interferon γ (IFNγ), IL-2, IL-4 and IL-10, after stimulation with OKT3. Cells were thawed, then rested prior to adding OKT3 (BioLegend) and subsequently Brefeldin-A (BFA). Unstimulated cells were used as a negative control and PMA and ionomycin (Millipore) were used as a positive control. Cells were stained with a viability dye, then stained with antibodies directed against surface markers, and fixed and permeabilized (BD, CytoFix/Cytoperm) prior to staining with antibodies directed against intracellular cytokines.
In vitro dasatinib culture
In vitro analysis was performed on PBMC from HC (n=1 per experiment) using the above-mentioned techniques. Fresh cells were cultured alongside increasing concentrations of dasatinib (Cambridge Bioscience) in RPMI and 10% FCS for 3 hours. For phosphoflow assays, cells were stimulated with either H2O2 or IL-2 (Merck) for 15 minutes prior to fixation, permeabilization and staining with surface and intracellular antibodies.
Plasma cytokine analysis
Baseline cytokine levels were evaluated using the Luminex Flexmap3D platform with analysis performed on thawed plasma samples using the Thermofisher 9-Plex Human ProcartaPlex Panel as per manufacturer’s instructions. Briefly, plasma samples were incubated with magnetic capture beads with specific spectral properties for 18 hours, prior to streptavidin-RPE being added followed by a biotinylated detection antibody. Data was acquired on the Luminex Flexmap3D.
Plasma dasatinib levels
Plasma or serum samples are prepared by methanol/acetonitrile protein precipitation followed by phospholipid depletion using phospholipid-removal plates (Phree, Phenomenex). Extracts were then analyzed directly by mass spectrometry using the Agilent 6460 mass spectrometer and Infinity II MultiSampler.
T-cell receptor clonality analysis
TCR clonality analysis was performed using the Biomed2 PCR protocol with primers analyzing both TCR-B and TCRG genes for evidence of clonality on the CEQ-8000 Genetic Analysis System sequencer (Beckman Coulter, Brea), as described previously.23
Haematologica | 108 June 2023 1556 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
Figure 1. Dasatinib reduces regulatory T cells and inhibits cell signaling pathways. (A, B) Regulatory T-cell (Treg) gating strategy including identification of Treg subsets. CD4+/CD25+/FOXP3+/CD127lo cells were used for identification of Tregs. (C) Treg subsets were: FOXP3hi/CD45RA- cells denoting effector Tregs, FOXP3lo/CD45RA+ cells denoting resting Tregs and FOXP3lo/CD45RA- cells denoting the nonTreg population. (D) Treg frequency as proportion of total CD4+ cells in total cohort of patients on dasatinib compared with both patients taking other tyrosine kinase inhibitors (TKI) and healthy controls (HC). (E) Effector Treg frequency as % of total Tregs in total cohort of patients on dasatinib compared with both patients taking other TKI and HC.
Statistical analysis
Data was reported as mean values and P values were from independent sample t-tests, with values less than 0.05 considered statistically significant. Distribution of data was assessed using Levene’s test for equality of variance. All reported P values are two-sided. Analyses were performed using SPSS version 24 (IBM, Armonk, NY) and Prism v8.
Results
Patients on dasatinib have reduced frequency of Tregs and effector Tregs
Patients on dasatinib had a lower proportion of Tregs compared with the patients taking other TKI and HC, with a mean proportion of Treg from total CD4+ cells of 2.6% in dasatinib-treated patients, 4.4% in CML patients on other TKI without significant SRC kinase inhibitory effects and 4.7% in HC (P=0.002, P=0.0006; Figure 1A-D). Dasatinibtreated patients also had lower ratio of effector Tregs/total Tregs than the other two groups at 11.5% ver-
sus 23% in patients on other TKI and 19.6% in HC (P=0.003 and P=0.014 respectively; Figure 1E).
Dasatinib inhibits T-cell receptor downstream signaling pathway compared to other tyrosine kinase inhibitors
In order to assess if TKI inhibit TCR downstream signaling, we examined their impact on phosphorylation of ZAP70 and LAT on gated T cells. We demonstrated a reduction in levels of pZAP70 in patients on dasatinib as assessed by relative fluorescence intensity (RFI, RFI=mean fluorescence intensity [MFI] of stimulated sample/MFI of unstimulated sample), compared with HC and patients taking non-SRC kinase inhibitor TKI in CD4+ cells, CD8+ cells and Tregs (Figure 2A, B). The mean RFI of pZAP70 was 2.1 in patients on dasatinib versus 8.8 in HC in CD4+ cells, 3.1 versus 11.4 in CD8+ cells and 2.1 versus 6.3 in Tregs (P=0.004, P=0.005, P=0.041) (Figure 2D). This was also lower in dasatinib patients compared with patients on other TKI, who had mean RFI of 5.4 in CD4+, 6.1 in CD8+ and 4.4 in Treg (P=0.035, P=0.093, P=0.176) (Figure 2D). Analysis of patients on 100 mg dosage of dasatinib
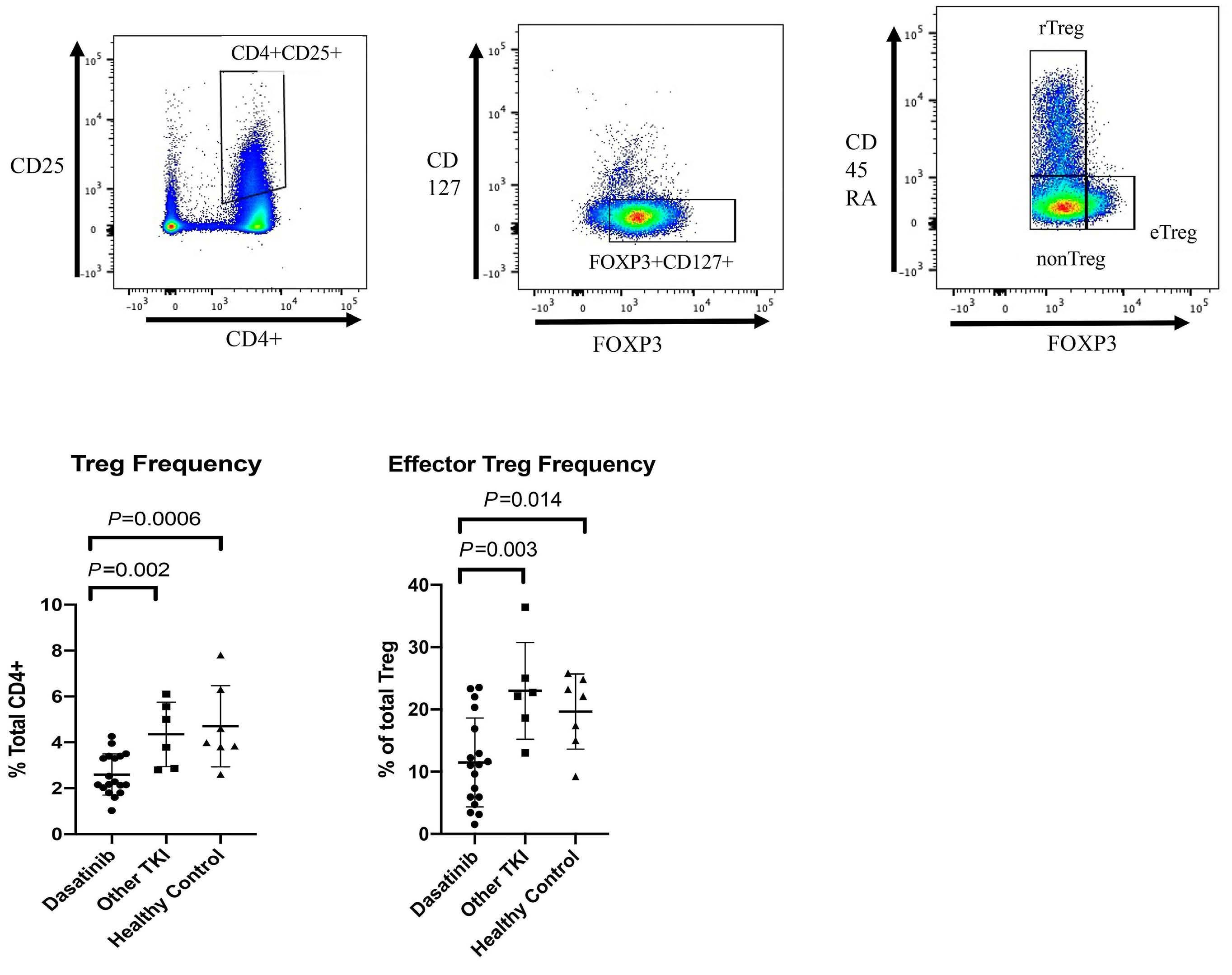
A B C D E Haematologica | 108 June 2023 1557 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
Figure 2. Dasatinib inhibits T-cell receptor and STAT5 signaling pathways with dose effect observed. (A) CD4+ pZAP70 expression in 2 patients with chronic myeloid leukemia (CML). Red events – unstimulated, blue events – H2O2 stimulated. Left image - nilotinib patient, right image – dasatinib patient. (B, C) Histogram showing (B) pZAP70 in CD4+ cells and (C) pSTAT5 in total CD3+ cells in 2 patients with CML, red peak – unstimulated, blue peak – H2O2. Left image - nilotinib patient, right image – dasatinib patient. (D) Relative fluorescence intensity (RFI) for total cohort of patients on dasatinib compared with patients on other tyrosine kinase inhibotors (TKI) and healthy controls (HC) in CD4+ cells for pZAP70. (E, F) RFI for patients on 100 mg dasatinib dosage compared with 50 mg dosage, patients on other TKI and HC in CD4+ cells for pLAT and pSTAT5.
showed greater inhibition of RFI compared with HC and patients on other TKI in CD4+ at 1.2, CD8+ 2.3 and Treg 1.2 (CD4+ HC P=0.004; other TKI P=0.001; CD8+ P=0.007, P=0.011; Treg P=0.01, P=0.005). Similarly, the dasatinib group had lower mean RFI in all Tcell subsets evaluated for pLAT (Figure 1Gii). In CD4+ cells mean RFI was 7.8 versus 22.9 in healthy controls and 19.5 in other TKI, (P=0.026, P=0.097), in CD8+ 6.9 versus 15 versus 19.5 (P=0.012, P=0.14) and Tregs mean RFI was 5.3 versus 14.6 in HC and 14.7 in other TKI (P=0.05, P=0.037). There was no difference in levels of pLAT in Tregs between patients on other TKI and HC. Comparison of patients on 100 mg dosage of dasatinib and other TKI showed greater difference in RFI, in the 100 mg group mean RFI was 3.1 in CD4+ cells, four in CD8+ cells, and 2.7 in Tregs (CD4+ HC P=0.001; other TKI P=0.006; CD8+ P=<0.001, P=0.013; Treg P=0.005, P=0.01) (Figure 2E).
Dasatinib inhibits STAT5 signaling pathway compared to other tyrosine kinase inhibitors
Patients on dasatinib showed significantly reduced pSTAT5 compared to HC and patients on other TKI. In CD4+ cells mean RFI for pSTAT5 was reduced at 7.7 in the dasatinib group versus 21.9 in HC and 20.8 in patients on other TKI (P=0.001 and P=0.004, respectively). Similarly, in CD8+ cells pSTAT5 RFI was 10.1 versus 31.1 and 28.1, respectively (P<0.001 and P=0.002) and in Tregs 7.3 versus
21.9 and 19.1, respectively (P=0.005 and P=0.011) (Figure 2C). In patients on 100 mg dasatinib there was greater difference in pSTAT5 RFI when compared with HC and other TKI groups with mean RFI of 3.8 in CD4+, 6.1 in CD8+ and 3.9 in Tregs (CD4+ HC P<0.001; other TKI P<0.001; HC P<0.001, other TKI P<0.001; Treg P<0.001; HC P<0.001) (Figure 2F). There was no difference in levels of pSTAT5 in Tregs between patients on other TKI and HC.
Dasatinib inhibits signaling pathways within natural killer cells
Signaling pathways within CD56+ NK cells were also inhibited in patients taking dasatinib when compared with HC. Patients on dasatinib had mean pZAP70 RFI within CD56+ cells of 4.6 compared with 17.8 in HC. Phosphorylated LAT and pSTAT5 were also reduced at 6.1 versus 33.5 and 15.2 versus 46.2 respectively (P =0.014, P=0.032 and P=0.008) (Online Supplementary Figure S1). Moreover, patients taking 100 mg dosage of dasatinib had a mean RFI for pZAP70 of 8.1 versus 3 in other TKI (P=0.008) and for pSTAT5 of 46.5 versus 8.2 (P<0.0001; data not shown).
Dasatinib reduces expression of pro-inflammatory cytokines in CD4+ and CD8+ cells, including interleukin 2 Patients on dasatinib had lower absolute increase in TNFα and IFN γ expression in CD4+ cells after activation with OKT3, when compared with patients on other TKI and HC.

A D E F B C Haematologica | 108 June 2023 1558 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
Figure 3. Dasatinib inhibits pro-inflammatory cytokines including interleukin 2. (A) Absolute increase in interleukin 2 (IL-2) expression in CD4+ cells (IL-2 expression in unstimulated sample subtracted from IL-2 expression in OKT3 stimulated sample). (B) Absolute increase in IL-2 expression in CD8+ cells. (C) IL-2 expression in CD4+ cells in 2 patients and a healthy control (HC). Left column – unstimulated, right column – OKT3. Top – HC, middle – intermediate dose (50 mg) dasatinib (DAS), bottom – full dose dasatinib (100 mg). TKI: tyrosine kinase inhibitor.
Mean absolute increase in the total dasatinib group was 9.5 for TNFα and 5.1 for IFNγ, compared with 25.7 and 16.3 for other TKI, and 26.8 and 16.9 for HC (P=0.027, P=0.028, P=0.001, P=0.007; Online Supplementary Figure S2).
The mean absolute increase in IL-2 expression was also lower in the dasatinib group in both CD4+ and CD8+ cells when compared with that of other TKI and HC. Mean absolute increase in CD4+ cells in dasatinib patients was 1.1, compared to 5 in other TKI and 9.1 in HC (P=0.0003, P<0.0001) (Figure 3A, C). Mean absolute increase in CD8+ cells in dasatinib patients was 0.3, compared to 2.4 in other TKI and 2.9 in HC (P=0.02, P=0.005) (Figure 3B).

Patients on reduced dasatinib dosage have preserved T-cell function
Six patients were managed with reduced dose dasatinib at a dose of 50 mg daily. These patients had significantly less inhibition of T-cell signaling within immune cell subsets when compared with those taking 100 mg daily. For pLAT RFI was higher at the 50 mg dosage in CD4+ cells at 17.2 versus 3.1, CD8+ T cells at 12.9 versus 4 and in Tregs at 10.4 versus 2.7 (P=0.025, P=0.083, P=0.049) (Figure 2E). Comparison of pSTAT5 between patients on different dosage also showed those on 50 mg dose to have significantly higher RFI than in those on the 100 mg dose within CD4+ cells at 15.4 versus
3.8 respectively, CD8+ cells at 18.1 versus 6.1 and within Tregs at 14 versus 3.9 (P=0.006, P=0.03, P=0.028) (Figure 2F). Similarly, patients at the 50 mg dose had significantly higher proportional increase in CD8+ cell TNF expression at 6 versus 3 (P=0.044). Proportional increase in IL-2 expression was also significantly higher at the 50 mg dose in both CD4+ cells, at 24.3 versus 2.5, and CD8+ cells at 4.8 versus 1.5 (P=0.035, P=0.006). No significant differences were observed in the frequency of Tregs between patients on different dasatinib dosage.
Dasatinib plasma levels were measured in ten patients (100 mg dosage n=6, 50 mg dosage n=4). Patients on 50 mg dosage had levels of <5 mg/mL whilst patients on 100 mg had a mean level of 14.7 mg/L (range, 5-26 mg/L). We observed a moderate inverse correlation between dasatinib level and RFI for pZAP70, pLAT and pSTAT5 across immune cell subsets (Online Supplementary Appendix; Online Supplementary Figure S3).
Effect of dasatinib on T-cell signaling is reversible
One patient was analyzed at three time points due to the development of pleural effusion and brief drug discontinuation. This patient demonstrated strong inhibition of cell signaling at both time points when taking dasatinib for each phosphoprotein evaluated. However, when ana-
A B C
Haematologica | 108 June 2023 1559 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
lyzed 1 week following treatment discontinuation a significant increase in phosphorylation was noted for all signaling proteins evaluated and across immune cell subsets, with strong inhibition again observed upon analysis 1 week after treatment re-initiation (Figure 4A-C).

Dasatinib enhances clonal expansion of CD8+ T cells
TCR gene rearrangement analysis was performed in 16 patients taking dasatinib. We identified seven patients as having clonal T-cell populations (Figure 5A), with nine having a polyclonal pattern. Of the patients with confirmed TCR clonality there was a reversal of the normal CD4:CD8 ratio in six of the seven in keeping with expansion of a clonal CD8+ T-cell population. Of note, two of the patients with confirmed clonality were taking a reduced dosage of dasatinib at 50 mg orally once daily (OD).
Patients with clonal CD8+ lymphocytosis have greater inhibition of Treg intracellular signaling
Patient with clonal T-cell populations had a lower Treg proportion of total CD3+ cells when compared with other patients on dasatinib with a mean value of 0.9 versus 1.7 (P=0.018). Moreover, these patients also had a lower RFI for pSTAT5 within isolated Tregs when compared with other patients on dasatinib with polyclonal T cells (mean RFI 1.7 vs. 11.8; P=0.04) (Figure 5B, C). Patients with clonal T-cell populations also had greater inhibition of TCR signaling within Tregs compared with other patients on da-
Figure 4. Patients on reduced dosage of dasatinib have preserved cell signaling. (A-C) Total CD3+ T cells show inhibition of pZAP70 expression when taking dasatinib (left and right plots), however, during treatment ‘holiday’ restoration of (D) ZAP70, (E) pLAT and (F) pSTAT5 phosphorylation is evident (middle plots).
satinib (mean RFI pZAP70 0.9 vs. 3.2; P=0.24, mean RFI pLAT 1.8 vs. 8.5; P=0.1).
Preferential inhibition of Tregs in patients on dasatinib Patients on 100 mg dasatinib had significantly reduced pZAP70 RFI in Tregs compared to effector cells, with mean of 1.2 in Tregs compared with 2.3 in CD8+ cells and 3.0 in NK cells (P=0.059, P=0.019) (Figure 6A, B). This effect was particularly noted in patients with clonal CD8+ lymphocytosis (Figure 6A). In addition, patients on the 50 mg dosage of dasatinib had greater difference in pSTAT5 RFI between Tregs and CD8+ T cells and NK cells, again suggesting a relative sparing of effector immune-cell inhibition. The mean difference in pSTAT5 RFI between Tregs and CD8+ T cells was 0.7 for those on 100 mg dasatinib compared with 5.4 in those on 50 mg dasatinib (P=0.005) (Figure 6B). Similarly, the mean difference in pSTAT5 RFI between Tregs and NK cells was 3 in those on 100 mg dose compared to 14.6 in those on 50 mg (P=0.08) (Figure 6D).
Dasatinib reduces phosphorylation of STAT5 via reduction of interleukin 2
We next investigated the mechanism through which dasatinib exerts an inhibitory effect on Treg STAT5 signaling, performing experiments on Tregs isolated from HC treated with dasatinib. We compared H2O2 and IL-2 as alternate methods of cell stimulation for STAT5 signaling. Using a uniform concentration of IL-2 (2U) to stimulate cells there was no in-
A B C Haematologica | 108 June 2023 1560 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
Figure 5. Patients on dasatinib with clonal lymphocytosis have increased inhibition of regulatory T-cell signaling. (A) Representative Tcell receptor (TCR) clonality analyses showing peaks in TCR B gene consistent with clonal T-cell population, in patient treated with dasatinib. (B) Histogram showing regulatory T cells (Treg) pSTAT5 mean fluorescence intensity/relative fluorescence intensity (MFI/RFI). Left – healthy control, middle – dasatinib patient with polyclonal T cells, right – dasatinib patient with clonal CD8+ lymphocytosis. (C) Comparison of RFI for Treg pSTAT5 between patients on dasatinib with clonal CD8+ T-cell populations and

hibitory effect on pSTAT5 from dasatinib, even at supratherapeutic concentrations (Figure 7B, C). In contrast, there was clear and dose-dependent inhibition of pSTAT5 by dasatinib when cells were activated with H2O2 (Figure 7A, C) suggesting that inhibition is dependent on reduction in IL-2. Supporting evidence for the central role of IL-2 inhibition came from additional experiments investigating effects of dasatinib on T-effector pro-inflammatory cytokine expression. Cells from HC were cultured with increasing concentrations of dasatinib, and then stimulated with OKT3. Levels of TNF, IFN and IL-2 were reduced in dosedependent manner in cells treated with both 100 nM and 500 nM concentrations of dasatinib, to levels approaching or less than the unstimulated control (Figure 7D-F).
Dasatinib reduces plasma levels of pro-inflammatory cytokines including interleukin 2
Plasma cytokine levels were evaluated in CML patients, including 11 taking dasatinib (100 mg dosage n=8, 50mg dosage n=3) and four taking nilotinib, and compared with six HC. In support of the above findings, most patients on dasatinib had undetectable levels of IL-2 (<0.571 pg/mL), significantly lower than both HC and patients taking other TKI (mean level dasatinib 0.9, HC 3.8, other TKI 7.5; P=0.0075, P=0.01). Moreover, levels of other pro-inflammatory cytokines including TNFα, IFNγ, IL-6 and IL-4 were lower in pa-
tients on dasatinib compared with HC and patients taking other TKI (mean levels TNFα – dasatinib 0.86, HC 20.4, other TKI 21.5, P=0.006/0.005; IFNγ - 7.3, 24, 18.5, P=0.2/0.3; IL-62.6, 16.1, 13.6, P=0.0004/0.004; IL-4 – 6, 36.3, 36.3, P =0.01/0.05) (Online Supplementary Figure S4B, C). No differences in plasma cytokine levels were observed between patients taking dasatinib 50 mg and 100 mg dosage.
Increased T-cell receptor signaling in TIM3+ T cells in chronic myeloid leukemia patients treated with other tyrosine kinase inhibitors is limited in those taking dasatinib
In keeping with previous studies that have shown a role for excessive TCR signaling in TIM-3-mediated T-cell exhaustion, we observed an increase in pZAP70 in TIM3+ cells in four patients analyzed on non-SRC kinase inhibitor therapy (3 nilotinib, 1 imatinib). The MFI was 2,313 in CD8+TIM3+ and 1,899 in CD4+TIM3+ cells, compared with 1,628 and 1,424 in CD4+TIM3- and CD8+TIM3- cells (P=not significant [ns]) (Figure 8Ai, ii). In contrast, this effect was not observed in four patients taking dasatinib at 100 mg dosage. The mean difference in MFI between CD4+TIM3+ cells was 475 in the nonSRC kinase inhibitor TKI group, compared with 66 in patients on full dosage dasatinib (P=0.0001) (Figure 8B). Similarly, in CD8+ cells the mean difference in MFI between TIM3+ and TIM3- cells was 95 in patients on 100 mg dasatinib compared
A B C Haematologica | 108 June 2023 1561 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
those with polyclonal T cells.
Figure 6. Preferential inhibition of regulatory T cells when compared with effector immune cell subsets. (A) Patient on dasatinib with clonal CD8+ lymphocytosis and showing preferential inhibition of pZAP70 in regulatory T cells (Tregs), (Ai) pZAP70 expression in Tregs, (Aii) pZAP70 in CD4+ cells, (Aiii) pZAP70 in CD8+ cells and (Aiv) pZAP70 in natural killer (NK) cells. (B) Comparison of mean florescence intensity (MFI) in immune cell subsets in patients on 100 mg dosage of dasatinib showing increased inhibition in Tregs compared with effector cell subsets. (C) Comparison of relative fluorescence intensity (RFI) pSTAT5 in CD8+ T cells – RFI pSTAT5 in Tregs, between patients on 50 mg and 100 mg doses of dasatinib. (D) Comparison of RFI pSTAT5 in CD56+ NK cells –RFI pSTAT5 in Tregs, between patients on 50 mg and 100 mg doses of dasatinib.

with 685 in those on other TKI (P=0.0016) (Figure 8C). Analysis was also performed in two patients on reduced dosage of dasatinib at 50 mg daily who had a mean difference in MFI in TIM3+ and non-TIM-3 expressing cells of 475 and 462 in CD4+ and CD8+ cells respectively (P=0.12/P=0.16).
Effector immune cells are activated during acute viral infection in patients treated with dasatinib
A patient taking 100 mg dasatinib OD was analyzed during acute viral infection with varicella zoster, prior to initiation of antiviral therapy. Baseline plasma IL-6 levels and IFNγ, were elevated at 76.081 pg/mL/14.6 pg/mL (average in HC 17.63 pg/mL/2.69 pg/mL) and C reactive protein levels were raised at 160 mg/L consistent with acute infection. There was significant expansion of CD56dim NK cells, accounting for >50% of the total live lymphocyte population (Online Supplementary Figure S5A). In addition, there was a notable reduction in the effector Treg (FOXP3hi/CD45-) subset of Tregs, with expansion of resting Tregs (FOXP3hi/ CD45+) (Online Supplementary Figure S5B). Of note, there was also pre-
served signaling within effector cells following stimulation with H2O2, however the signaling from the TCR and of pSTAT5 within Tregs remained largely inhibited (Online Supplementary Figure S5C, D). Repeat analysis performed 1 week after the resolution of infection showed a reduction in NK cells at 30% of total lymphocytes, although remaining Tregs increased (normal range in healthy controls, 5-20%).
Discussion
The positive effect of the development of lymphocytosis on CML response in patients treated with dasatinib was demonstrated from a review of 1,402 patients, in whom a third developed lymphocytosis.24 Patients with lymphocytosis were more likely to meet major response milestones including MMR/MR3 and deep molecular response (DMR/MR4). Iriyama and colleagues analyzed lymphocyte dynamics in patients treated with dasatinib and found that cytotoxic lymphocyte or NK-cell counts at 1 month were
Ai Aii Aiii Aiv B C D Haematologica | 108 June 2023 1562 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
Figure
STAT5 signaling within regulatory T cells via inhibition of interleukin 2. (A) Dose-dependent inhibition of pSTAT5 with increasing concentrations of dasatinib (DAS) in gated regulatory T cells (Tregs) from healthy control (HC, n=1), following stimulation with H2O2. Circles on graph represent positive events and squares represent mean florescence intensity (MFI). (B) Direct comparison between H2O2 and interleukin 2 (IL-2) stimulation techniques, showing dose-dependent inhibition of pSTAT5 by dasatinib in gated Tregs from HC (n=1), in H2O2-stimulated samples, with lack of inhibition in IL-2-stimulated samples, even at supra-therapeutic concentrations. (C) Histograms showing pSTAT5 in gated Tregs from HC (n=1) following treatment with increasing doses of dasatinib, left histogram shows IL-2-stimulated cells and right shows H2O2-stimulated cells. (D) IL-2 expression in CD4+ T cells from HC (n=1) treated with increasing concentrations of dasatinib, following OKT3 activation. (E) TNFα expression in CD8+ T cells from HC (n=1) treated with increasing concentrations of dasatinib, following OKT3 activation. (F) TNFα expression in CD8+ T cells from HC (n=1). Top left – unstimulated, top right – OKT3, bottom left – dasatinib 100 nM + OKT3, bottom right – dasatinib 500 nM + OKT3. +ve: positive; UNSTIM: unstimulated.
significantly higher in those patients who went on to achieve DMR.25 Further analysis from the same study showed that patients with Tregs below a determined threshold at 12 months, had significantly improved rates of DMR.16 It was also shown that Treg inhibition was inversely correlated with the NK-cell differentiation, with an increased proportion of mature CD57+ NK cells observed as Treg number reduced, suggesting that Tregs may impede the immune response against CML cells by restricting NK cell differentiation.
In our study treatment with dasatinib resulted in a reduction in the proportion of Tregs in CML patients when compared with HC and patients taking other TKI. We also demonstrated, for the first time, that dasatinib reduces the effector Treg cell subset, which are the subset with most suppressive activity and highest expression of key suppressor molecules such as CTLA-4.13 A recent study
suggests that imatinib causes depletion of effector Tregs, through inhibition of LCK, due to the relatively low expression of LCK in effector Tregs in comparison with other T-cell subsets, rendering them selectively susceptible to signal-deprived apoptosis.26 From our analysis dasatinib causes greater depletion of effector Tregs, as would be expected in view of its significantly greater inhibitory effect on LCK.
We describe the first report of the differential inhibitory effect of dasatinib on signaling pathways within immune cell subsets including Tregs and NK cells in an ex vivo analysis. We show that TCR signaling is inhibited most strongly in Tregs when compared with effector cell subsets in patients on dasatinib, again likely explained by low Treg expression of LCK. This may have potential implications given the welldemonstrated impact of NK cell subsets around TKI discontinuation outcomes.27 Our data also supports evaluating Treg

A B C D E Haematologica | 108 June 2023 1563 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
7. Dasatinib inhibits
function as an additional potential biomarker for TFR outcome in patients treated with dasatinib as suggested in a study on dasatinib discontinuation, which found reduced Tregs to correlate with improved TFR outcome.28
We found that patients with clonal CD8+ T cells have a lower proportion of Tregs when compared with patients on dasatinib without clonality, as well as reporting a novel finding of increased Treg STAT5 inhibition in patients with clonal T-cell populations.22 Dasatinib-treated patients with clonal lymphocytosis regularly develop immune-mediated adverse effects, with accumulation of clonal CD8+ T cells within affected organs.29,30 Reduction in number and function of Tregs may explain the enhanced tumor immunity effects seen in this group.
The roles of IL-2 and its downstream target STAT5 in the function of Tregs are well recognized. Tregs have abundant expression of the IL-2α receptor on the cell surface and binding results in STAT5 engaging with the promoter region of the FOXP3 gene, controlling Treg differentiation through expression of FOXP3. We have shown that dasatinib causes a significant reduction in pro-inflammatory cytokine expression within CD4+ cells, with greatest effect seen against IL-2, providing a mechanistic insight into the inhibition of Treg function by dasatinib through reduction of STAT5 phosphorylation. Our in vitro analysis of Tregs suggests pSTAT5 inhibition by dasatinib is primarily dependent on reduction in IL-2 levels. This was supported by the finding of undetectable IL-2 levels in patients receiving dasatinib, as well as confirmation of in vitro inhibition of IL-2 production in effector T cells following OKT3 activation, at nanomolar concentrations of dasatinib.
We also describe the effect dasatinib exerts on restricting the increased TCR signaling that is observed in TIM-3+ T cells in CML patients taking other TKI. TIM-3 contains no

recognized inhibitory motifs and as such is thought to result in T-cell exhaustion through sustained and uncontrolled increased TCR signaling under acute conditions, which is mediated by SRC kinases.31 In view of these observations, we suggest that chronic SRC kinase inhibition may play a role in preventing TIM-3-mediated T-cell exhaustion and enhance the immunostimulatory effects observed in certain patients on dasatinib.
Interestingly we demonstrate, for the first time in an ex vivo analysis, that patients on reduced dasatinib dosage have preserved T-cell function, suggestive of a dose-dependent effect of dasatinib. This may have important clinical implications when determining dasatinib dose reduction in certain cases. Moreover, we report ex vivo reversibility of the dasatinib effect on T-cell signaling in one patient. Importantly, we found expansion of NK cells and restoration of TCR downstream signaling in effector immune cells during acute viral infection in another patient taking dasatinib, suggesting the presence of mechanisms to overcome inhibitory effects in acute infection.
Our study is limited by relatively small sample size, and as such there was no identifiable association between presence of clonality and outcome in dasatinib-treated patients, although this has been demonstrated in other large clinical studies. In addition, patients in the dasatinib group had lower rates of DMR compared to those taking other TKI, however as most patients on dasatinib had achieved MMR or greater, we feel this is unlikely to cause significant differences in immune cell populations between groups. We also recognize that dasatinib has diffuse inhibitory effects across the kinome and that it has not been possible to evaluate all aspects in this ex vivo analysis.
In summary, dasatinib exerts selective depletion of effector Tregs and functional impairment of Tregs through in-
Ai Aii B C Haematologica | 108 June 2023 1564 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
Figure 8. Effect of dasatinib on TIM3+ T-cell receptor signaling and reversibility of dasatinib effect. (A) Increased mean florescence intensity (MFI) for pZAP70 in (Ai) CD4+ and (Aii) CD8+ cells expressing TIM3 compared with non-TIM-3+ cells. (B, C) Comparison of difference in MFI between TIM3+ and TIM3- (B) CD4+ and (C) CD8+ cells show significant increase in patients taking other tyrosine kinase inhibitors (TKI) compared to that observed in dasatinib (DAS)-treated patients.
hibition of TCR and STAT5 signaling leading to increase in effector CD8+ T cells. We show that Treg STAT5 inhibition is primarily mediated via reduction of IL-2 signaling and that SRC kinase inhibition may also prevent TIM-3-mediated CAR T-cell exhaustion. Our data are also of relevance to the potential use of dasatinib for pharmacologic control of CAR-T cell function as well as management of cytokine release syndrome and may help with future use of dasatinib alongside other immunotherapeutic approaches.32-34 The use of small molecule inhibitors that selectively target Tregs is an attractive option that might be beneficial across oncology practice and warrants further investigation.
Disclosures
PH and HdL have received research funding from Bristol
References
1. Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260-2270.
2. Shah NP, Guilhot F, Cortes JE, et al. Long-term outcome with dasatinib after imatinib failure in chronic-phase chronic myeloid leukemia: follow-up of a phase 3 study. Blood. 2014;123(15):2317-2324.
3. Hochhaus A, Muller MC, Radich J, et al. Dasatinib-associated major molecular responses in patients with chronic myeloid leukemia in chronic phase following imatinib failure: response dynamics and predictive value. Leukemia. 2009;23(9):1628-1633.
4. O'Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65(11):4500-4505.
5. Lombardo LJ, Lee FY, Chen P, et al. Discovery of N-(2-chloro-6methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658-6661.
6. Nyakeriga AM, Garg H, Joshi A. TCR-induced T cell activation leads to simultaneous phosphorylation at Y505 and Y394 of p56(lck) residues. Cytometry A. 2012;81(9):797-805.
7. de Lavallade H, Khoder A, Hart M, et al. Tyrosine kinase inhibitors impair B-cell immune responses in CML through offtarget inhibition of kinases important for cell signaling. Blood. 2013;122(2):227-238.
8. Weichsel R, Dix C, Wooldridge L, et al. Profound inhibition of antigen-specific T-cell effector functions by dasatinib. Clin Cancer Res. 2008;14(8):2484-2491.
9. Schade AE, Schieven GL, Townsend R, et al. Dasatinib, a smallmolecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008;111(3):1366-1377.
10. Blake S, Hughes TP, Mayrhofer G, et al. The Src/ABL kinase inhibitor dasatinib (BMS-354825) inhibits function of normal human T-lymphocytes in vitro. Clin Immunol. 2008;127(3):330-339.
11. Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol. 2014;193(4):1525-1530.
Myers Squibb. All other authors have no conflicts of interest to disclose.
Contributions
PH and HdL designed the research study, analyzed the data, and wrote the manuscript. PH, AV and FH performed the research. RD, DR, DM, PR, MO, AG, KR, SK and CNH assisted with patient recruitment and reviewed the paper.
Funding
This research was supported by a grant from Brystol-Myers Squibb CA180-642.
Data-sharing statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
12. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. 2019;50(2):302-316.
13. Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6):899-911.
14. Fei F, Yu Y, Schmitt A, et al. Dasatinib inhibits the proliferation and function of CD4+CD25+ regulatory T cells. Br J Haematol. 2009;144(2):195-205.
15. Hughes A, Clarson J, Tang C, et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood. 2017;129(9):1166-1176.
16. Najima Y, Yoshida C, Iriyama N, et al. Regulatory T cell inhibition by dasatinib is associated with natural killer cell differentiation and a favorable molecular response - the final results of the Dfirst study. Leuk Res. 2018;66:66-72.
17. Bachy E, Bernaud J, Roy P, et al. Quantitative and functional analyses of CD4(+) CD25(+) FoxP3(+) regulatory T cells in chronic phase chronic myeloid leukaemia patients at diagnosis and on imatinib mesylate. Br J Haematol. 2011;153(1):139-143.
18. Zahran AM, Badrawy H, Ibrahim A. Prognostic value of regulatory T cells in newly diagnosed chronic myeloid leukemia patients. Int J Clin Oncol. 2014;19(4):753-760.
19. Kim DH, Kamel-Reid S, Chang H, et al. Natural killer or natural killer/T cell lineage large granular lymphocytosis associated with dasatinib therapy for Philadelphia chromosome positive leukemia. Haematologica. 2009;94(1):135-139.
20. Lee SJ, Jung CW, Kim DY, et al. Retrospective multicenter study on the development of peripheral lymphocytosis following second-line dasatinib therapy for chronic myeloid leukemia. Am J Hematol. 2011;86(4):346-350.
21. Kreutzman A, Juvonen V, Kairisto V, et al. Mono/oligoclonal T and NK cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood. 2010;116(5):772-782.
22. Mustjoki S, Ekblom M, Arstila TP, et al. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia. 2009;23(8):1398-1405.
23. van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and
Haematologica | 108 June 2023 1565 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257-2317.
24. Schiffer CA, Cortes JE, Hochhaus A, et al. Lymphocytosis after treatment with dasatinib in chronic myeloid leukemia: effects on response and toxicity. Cancer. 2016;122(9):1398-1407.
25. Iriyama N, Fujisawa S, Yoshida C, et al. Early cytotoxic lymphocyte expansion contributes to a deep molecular response to dasatinib in patients with newly diagnosed chronic myeloid leukemia in the chronic phase: results of the D-first study. Am J Hematol. 2015;90(9):819-824.
26. Tanaka A, Nishikawa H, Noguchi S, et al. Tyrosine kinase inhibitor imatinib augments tumor immunity by depleting effector regulatory T cells. J Exp Med. 2020;217(2):e20191009.
27. Rea D, Henry G, Khaznadar Z, et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. 2017;102(8):1368-1377.
28. Okada M, Imagawa J, Tanaka H, et al. Final 3-year results of the
dasatinib discontinuation trial in patients with chronic myeloid leukemia who received dasatinib as a second-line treatment. Clin Lymphoma Myeloma Leuk. 2018;18(5):353-360.
29. de Lavallade H, Punnialingam S, Milojkovic D, et al. Pleural effusions in patients with chronic myeloid leukaemia treated with dasatinib may have an immune-mediated pathogenesis. Br J Haematol. 2008;141(5):745-747.
30. Bergeron A, Rea D, Levy V, et al. Lung abnormalities after dasatinib treatment for chronic myeloid leukemia: a case series. Am J Respir Crit Care Med. 2007;176(8):814-818.
31. Lee J, Su EW, Zhu C, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. 2011;31(19):3963-3974.
32. Weber EW, Lynn RC, Sotillo E. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019;3(5):711-717.
33. Mestermann K, Giavridis T, Weber J, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. 2019;11(499):eaau5907.
34. Leonard JT, Kosaka Y, Malla P, et al. Concomitant use of a dual Src/ABL kinase inhibitor eliminates the in vitro efficacy of blinatumomab against Ph+ ALL. Blood. 2021;137(7):939-944.
Haematologica | 108 June 2023 1566 ARTICLE - Dasatinib T-cell signaling P. Harrington et al.
Molecular response in newly diagnosed chronic-phase chronic myeloid leukemia: prediction modeling and pathway analysis
Jerald P. Radich,1* Matthew Wall,2* Susan Branford,3 Catarina D. Campbell,4 Shalini Chaturvedi,5 Daniel J. DeAngelo,6 Michael W. Deininger,7 Justin Guinney,2 Andreas Hochhaus,8 Timothy P. Hughes,9 Hagop M. Kantarjian,10 Richard A. Larson,11 Sai Li,12 Rodrigo Maegawa,5 Kaushal Mishra,5 Vanessa Obourn,4 Javier Pinilla-Ibarz,13 Das Purkayastha,12 Islam Sadek,5 Giuseppe Saglio,14 Alok Shrestha,5 Brian S. White2 and Brian J. Druker15
1Fred Hutchinson Cancer Research Center, Seattle, WA, USA; 2Sage Bionetworks, Seattle, WA, USA; 3SA Pathology, Centre for Cancer Biology, Adelaide, South Australia, Australia; 4Novartis Institutes for Biomedical Research, Cambridge, MA, USA; 5Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA; 6Dana-Farber Cancer Institute, Boston, MA, USA; 7Huntsman Cancer Institute, The University of Utah, Salt Lake City, UT, USA; 8Universitätsklinikum Jena, Jena, Germany; 9South Australian Health and Medical Research Institute and University of Adelaide, Adelaide, South Australia, Australia; 10MD Anderson Cancer Center, University of Texas, Houston, TX, USA; 11MedicineHematology/Oncology, University of Chicago, Chicago, IL, USA; 12Novartis Pharmaceuticals Corporation, Basel, Switzerland; 13Moffitt Cancer Center, University of South Florida, Tampa, FL, USA; 14University of Turin, Turin, Italy and 15Knight Cancer Institute, Oregon Health and Science University, Portland, OR, USA
*JPR and MW contributed equally as co-first authors.
Abstract
Correspondence: J.P. Radich jradich@fredhutch.org
Received: August 5, 2022.
Accepted: January 20, 2023.
Early view: February 2, 2023.
https://doi.org/10.3324/haematol.2022.281878
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Tyrosine kinase inhibitor therapy revolutionized chronic myeloid leukemia treatment and showed how targeted therapy and molecular monitoring could be used to substantially improve survival outcomes. We used chronic myeloid leukemia as a model to understand a critical question: why do some patients have an excellent response to therapy, while others have a poor response? We studied gene expression in whole blood samples from 112 patients from a large phase III randomized trial (clinicaltrials gov. Identifier: NCT00471497), dichotomizing cases into good responders (BCR::ABL1 ≤10% on the International Scale by 3 and 6 months and ≤0.1% by 12 months) and poor responders (failure to meet these criteria). Predictive models based on gene expression demonstrated the best performance (area under the curve =0.76, standard deviation =0.07). All of the top 20 pathways overexpressed in good responders involved immune regulation, a finding validated in an independent data set. This study emphasizes the importance of pretreatment adaptive immune response in treatment efficacy and suggests biological pathways that can be targeted to improve response.
Introduction
Tyrosine kinase inhibitor (TKI) therapy has revolutionized the treatment of chronic myeloid leukemia (CML), giving patients with chronic phase (CML-CP) a near-normal ageadjusted life span.1,2 In the ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials-Newly Diagnosed Patients, clinicaltrials gov. Identifier: NCT00471497 ) study, rates of complete cytogenetic response and major molecular response (MMR) for patients receiving either 300 mg nilotinib twice daily (BID), 400 mg nilotinib BID, or imatinib 400 mg once daily were 80% and 44%, 78% and
43%, and 65% and 22%, respectively, at 12 months.3 The second-generation TKI (bosutinib, dasatinib, and nilotinib) are more potent than imatinib, a first-generation TKI, and thus result in deeper molecular responses and lower rates of progression to advanced-phase disease, although overall survival rates are comparable to those of imatinib.3-8 The depth and kinetics of molecular response have clinical implications. Chronologically, the first milestone of molecular response is a BCR::ABL1 on the International Scale (IS) level of ≤10% after 3 to 6 months of therapy, denoted as early molecular response (EMR).9-13 EMR is associated with lower rates of progression to
Haematologica | 108 June 2023 1567 ARTICLE - Chronic Myeloid Leukemia
advanced-phase disease and superior long-term responses with all TKI.6,7,9,10,12,14-16 Lack of EMR (BCR::ABL1IS >10% after 3-6 months of therapy) occurs in approximately 30% of patients with CML-CP treated with imatinib and roughly 5% to 15% of patients treated with dasatinib or nilotinib and is associated with worse outcomes.6,7,10,12-17 For example, in an analysis of 282 patients with CML-CP treated with imatinib, the 8-year overall survival, progression-free survival, and event-free survival rates were 93%, 93%, and 65%, respectively, for patients with BCR::ABL1IS of ≤10% at 3 months compared with 57%, 57%, and 7%, respectively, for those with BCR::ABL1IS of >10% at 3 months.9
MMR is another important milestone because it is associated with a very low rate of progression and resistance (thus, MMR is referred to as a “safe haven”).12,13 Many patients who achieve MMR have a continued molecular response, reaching a BCR::ABL1IS level of ≤0.01%, referred to as a deep molecular response (DMR).12,13 DMR is clinically relevant because many studies have shown that, of patients who maintain DMR for several years, roughly 40% to 50% can successfully discontinue TKI therapy and achieve treatment-free remission (TFR).18-20 Thus, the two main clinical questions in CML therapy are when to change and when to stop therapy. The biological corollary of these clinical questions is: why does TKI therapy produce good responses in some patients and poor responses in others? If we understood the biology of good and poor responses, could we predict response before treatment, add new therapies for patients bound for a poor response, and change patients’ fate to that of a good responder?
A prior report by Branford et al. studied the transcriptome profiles of 46 patients with CML-CP who mostly received imatinib as their initial therapy. The study determined genetic alterations at baseline and blast crisis to determine which genetic alterations underlie disease transformation.21 We used pretreatment diagnostic samples from the ENESTnd study, which compared imatinib with nilotinib in patients with newly diagnosed CML-CP, to study the difference in RNA expression in the subsets of patients with good and poor responses, to define genetic predictors of response, and to infer the biological pathways and processes that drive response. We then validated these predictions and inferences using the previously established baseline transcriptome profiling data set from Branford et al.
Methods
Patients and samples
The randomized trial ENESTnd compared imatinib 400 mg once daily (n=283), nilotinib 300 mg twice daily (BID)
(n=282), and nilotinib 400 mg BID (n=281) (Online Supplementary Figure S1). This study was approved by an Ethics Committee or Review Board at Novartis and participating institutions. All patients provided informed consent. Study procedures were conducted in accordance with the ethical standards of the Declaration of Helsinki and local laws and regulations.
Sample processing, quality control, and genomic analysis
Detailed methods are available in the Online Supplementary Appendix. In brief, RNA was extracted from baseline, pretreatment whole blood samples and used for RNA sequencing (RNA-seq) library construction. RNA libraries were sequenced using HiSeq 2500 (Illumina) in six armand response group–matched batches to a depth of 50 million reads per sample. Reads were mapped to the reference human genome (build hg19), assembled into transcripts, normalized for abundance, and counted.
Bioinformatic analysis
All statistical analyses were performed in the R programming language. Statistical comparisons between responder groups were performed using the Wilcoxon rank-sum test unless otherwise stated.
Gene expression profile analyses
The edgeR package was used to normalize the RNA-seq counts via the trimmed mean of M values method and perform a log2(counts per million [cpm] +1) transformation. Genes with insufficient expression (i.e., log2[cpm +1] <1.1 in at least 50% of samples) were removed. The log2(cpm +1) data were standardized by a z-score transformation such that the mean expression of each gene across all samples was 0 and the standard deviation was 1.
Deconvolution of cell types
Inference of relative abundance of ten cell types in each sample was performed using the MCP-counter algorithm applied to the log2(cpm +1) gene expression data.22
Bootstrapped prediction of responder status
Detailed methods can be found in the Online Supplementary Appendix. In short, penalized logistic regression models were constructed from gene expression, clinical variables, normalized enrichment scores of biological pathways, and inferred cell type compositions. Each input data set was subject to 250 iterations in which bootstrapping (i.e., random sampling with replacement) of the input samples was performed to create random subsets of data on which the model was trained and evaluated via area under the curve (AUC). A final predictive model was trained on all the ENESTnd samples using logistic ridge regression.
Haematologica | 108 June 2023 1568 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
Prediction of responder status in validation cohort
A final logistic ridge regression model was trained on the gene expression data of all 112 ENESTnd samples and validated using the Branford et al. validation data by AUC as described on page 5 of the Online Supplementary Appendix (Online Supplementary Table S1).
Interpretation of predictive models
Gene set enrichment analysis using the fGSEA package was used to interpret the gene expression–based predictor by using the model coefficients assigned to each gene.23 The databases for canonical pathways and gene ontology biological processes were downloaded from MSigDb and used as references for these tests. For models not based on gene expression, interpretation centered on the relative importance of individual features as ranked by their model coefficients.
Results
Differences in patient response observed by 12 months persisted up to 10 years
Patients were dichotomized into good and poor responder groups. Good responders were defined as those who achieved BCR::ABL1IS ≤10% by 3 and 6 months and MMR by 12 months. Patients who did not meet both criteria were labeled as poor responders. One patient could not be labeled by these definitions after exhibiting BCR::ABL1IS <10% by 3 and 6 months but was lost to follow-up after 9 months without achieving BCR::ABL1IS ≤0.1%. As this patient exhibited a trajectory consistent with good responders, including early molecular response and BCR::ABL1IS=0.12% by 9 months, we labeled this patient as a good responder. In total, there were 40 good responders and 72 poor responders (Table 1). The BCR::ABL1IS levels of good and poor responders remained significantly different at 10 years (Figure 1; P<0.001). In our cohort, 95% of good responders achieved a DMR by 5 years, compared to 17% of poor responders (Online Supplementary Figure S1), consistent with published literature.12,13
Baseline gene expression predicted tyrosine kinase inhibitor response
We next studied how clinical features and gene expression signatures related to responder status. For clinical features, only female sex was associated with response in a multivariate analysis of clinical variables (P<0.001), although duration of treatment (P=0.05), age between 45 and 55 years (P=0.06), and treatment with either 300 mg (P=0.06) or 400 mg (P=0.06) of nilotinib trended with response (Figure 2).
We next examined the differential gene expression across the two responder groups. We developed a logistic ridge
regression model to predict responder status based on baseline gene expression (13,575 genes; see Methods). The principal component analysis (PCA) plots based on response criteria, which demonstrate the distribution of good and poor responders predicted in the logistic ridge model across the two variables most influencing their distribution, are shown in the Online Supplementary Figure S2A. The performance of the model (AUC=0.76; standard deviation [SD] =0.07) was significantly better than that of a null model (P=7.7×10-5) at a 95% confidence interval (CI) (Table 2; Online Supplementary Figure S3). Incorporating clinical variables into the model did not significantly improve prediction performance (AUC=0.75; SD=0.07; P=1.5×10-4).
We validated the model on an independent data set of 46 patients with CP-CML reported by Branford and colleagues.21 These 46 patients underwent transcriptome sequencing at diagnosis and mostly received imatinib as their initial therapy. In the study, 19 good responders achieved durable MMR, whereas the remaining 25 poor responders progressed to blast phase (n=24) or did not respond to four TKI (n=1). A total of 41 patients received

Treatment type Good responders, N Poor responders, N Imatinib 12 35 Nilotinib 28 37
Table 1. Distribution of responders versus poor responders split by treatment type.
Haematologica | 108 June 2023 1569 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
Figure 1. Early differences in patient tyrosine kinase inhibitor response persisted up to 10 years. BCR::ABL1IS (log10 scale) over time for good (blue) and poor (red) responders. IS: International Scale. Calculations of BCR::ABL1IS levels >10% are less accurate than those ≤10%.45
imatinib, including all the good responders, and three patients received nilotinib. The model developed on the ENESTnd data set predicted the responder status of the Branford validation data significantly better than a random model (AUC=0.67; P=0.02). Taken together, these results demonstrated that baseline gene expression was strongly associated with response to TKI therapy and that it provided more predictive power than clinical variables alone.
Tyrosine kinase inhibitor response was associated with increased immune response and cytotoxic lymphocyte activity at baseline

We next interrogated the differential gene expression set for genes and pathways associated with the two response groups. In order to identify correlates of response, we performed gene- and pathway-level analysis. A total of 458 genes were differentially expressed (false discovery rate
Figure 2. Multivariate analysis of clinical variables vs responder status. The most influential variable was sex, with women responding best. An odds ratio >1 indicated an association with good responders whereas those <1 indicated association with poor responders. BID: twice daily; CI: confidence interval. aIn the ENESTnd trial, prior tyrosine kinase inhibitor treatment was not allowed except for ≤2 weeks’ duration of imatinib.
[FDR] <5%) between good and poor responders (Online Supplementary Figure S3; Online Supplementary Table S2). The top differentially upregulated genes in the good responder group were enriched for genes associated with the immune system, whereas genes upregulated in poor responders were enriched for genes associated with cell cycle and metabolism (Figure 3). Among the genes upregulated in good responders were programmed death-ligand 1 (PD-L1) programmed cell death protein 1 (PD-1) death 1 ligand 1 (PD-L1) (Online Supplementary Figure S4).
In the Branford data set, PD-L1 was also upregulated in the good clinical response group. We then performed gene enrichment analyses to infer functional differences between good and poor responders. Good responders exhibited strong positive enrichment for expression of immune-related genes. Indeed, all 20 of the most significantly enriched gene sets were related to immunity (Fig-
Haematologica | 108 June 2023 1570 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
ure 4; Online Supplementary Table S3). Immune-related pathways were likewise positively associated with good response in the Branford data set (Table 3). Additionally, drug catabolism was associated with poor response in both data sets. As an alternative approach for a pathwaybased predictor, we next used the normalized enrichment scores of biological pathways instead of gene expression, and this different approach was also successful in predicting response (AUC=0.73; SD=0.09; P=3.9×10-3) and showed results that were consistent with the expressionbased predictor.
We next examined which specific immune-cell types were participating in the response, using the MCP-counter algorithm to infer cell types from the gene expression data. We deconvolved the bulk expression data into ten immune compartments. We found that a good response was associated with increased activity of natural killer (NK) (P=0.01) and CD8+ T cells (P=0.02) (Figure 5; Online Supplementary Table S4). Consistently, response was also associated with populations of cytotoxic lymphocytes (i.e., NK cells and CD8+ T cells; P=0.0037) and T cells (P=0.02). These associations also held true in the Branford data set
(NK cells, P=0.04; CD8+ T cells, P=0.04; cytotoxic lymphocytes, P=0.08; T cells, P=0.06) (Figure 5; Online Supplementary Table S4). The immune concordance across data sets was especially striking given that only 17 genes were differentially expressed (FDR <5%) between optimal and poor responders in the Branford data set, and only one of these (USP6) was differentially expressed in both the ENESTnd and Branford data sets (Online Supplementary Table S2).
We also explored if peripheral blood lymphocyte counts at baseline could influence the gene expression signature. Overall, there was no difference in the total white blood cell count between response groups. There were no significant differences between absolute and percentage lymphocytes across response groups (Online Supplementary Table S5). Taken together, the data suggest that good response, compared to poor response, is strongly influenced by immune processes governed largely through cytotoxic lymphocytes.
Chronic myeloid leukemia regulatory network analysis
We then asked how gene expression programs were being
Figure
in good and poor responders. Genes listed are those that were found in over 10% of the gene expression models (see Methods). The top upregulated genes in the good responder group were enriched in genes associated with immunity, in agreement with the immune pathway enrichment in good-responders (below, Figure 4).

Data type Number of model variables Model AUC (SD), mean Empirical P Significant at 95% CI Gene expression 13,201 0.76 (0.07) 7.7×10-5 Yes Clinical 24 0.59 (0.07) 0.12 No Gene expression and clinical 13,225 0.75 (0.07) 1.5×10-4 Yes AUC: area under the curve; SD: standard deviation; CI: confidence interval.
Table 2. Expression-based models predicted tyrosine kinase inhibitor response.
Haematologica | 108 June 2023 1571 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
3. Genes upregulated
controlled at the transcription factor level. We inferred a CML gene regulatory network (GRN) from the ENESTnd expression data using MINER.24 The CML GRN comprises 304 transcription factors regulating 8,827 genes partitioned into 2,479 regulons (Online Supplementary Table S6). Eighty-eight genetic programs were identified and their activity status in each sample was determined using MINER.

We evaluated the CML responder status with respect to the activity of the GRN programs using the Fisher exact test. There were several programs whose activity was significantly different (P<0.05) between good and poor responders (Online Supplementary Table S7). The program whose activity was most significantly associated with good response was program Pr-9 (P=9.2x10-4). The genes
of this program were regulated by interferon regulatory factors (IRF1-IRF5, IRF7 and IRF8), and were enriched for the hallmark pathways of interferon α response (adjusted P=4.9x10-34), interferon γ response (adjusted P=2.0x10-33), and immune system (Reactome; adjusted P=5.3x10-15). Moreover, these genes were significantly enriched for the experimental targets of IRF1 overexpression, consistent with the inferred regulation mechanism.
Several genetic programs were overactive in the poor responders of ENESTnd. A decision tree predictor trained on the genetic program activities of ENESTnd was predictive of poor response in both ENESTnd (AUC=0.75±0.06) and the Branford validation data set (AUC=0.70). The decision tree predictor could be further pruned to require only three genetic programs to achieve optimal performance
Figure 4. Immune-related pathways were enriched in good responders. (A) Normalized enrichment score (NES) and associated P value of 3,478 gene ontology gene sets. Immune-related gene sets are indicated in blue. Other gene ontology gene sets are in red. Positive NES indicates positive association with good response. (B) P values of the 20 most significantly dysregulated gene sets; all are immune-related.
A B
Haematologica | 108 June 2023 1572 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
Positive normalized enrichment score (NES) values indicated correlation to good response, whereas negative values were correlated with poor response. ATP: adenosine triphosphate; FDR: false discovery rate.
(Online Supplementary Figure S5). These three genetic programs tended to be active in distinct subsets of patients. Thus, they may represent three alternative transcriptional pathways to therapy resistance.24
Tyrosine kinase inhibitor response remained associated with cytotoxic lymphocyte activity when deep molecular response was used as a clinical endpoint In order to determine whether our results were sensitive to a later clinical endpoint, we repeated our analysis after redefining good responders as those patients who
reached DMR by 5 years and poor responders as those who did not. Not surprisingly, this definition partitioned patients somewhat differently than the original response definitions applied above (Online Supplementary Table S8). Nonetheless, immune-related genes (Online Supplementary Table S9; AUC=0.76; SD=0.07; P=9.5×10-5) and pathways (Online Supplementary Table S10; AUC=0.73; SD=0.07; P=4.6×10-4) remained the strongest predictors of good response (Online Supplementary Table S11), and drug catabolism (FDR <5%) was still significantly correlated with poor response. Increased activity of B cells (P=0.04),
Table 3. Gene ontology terms that were enriched (false discovery rate <10%) in both the ENESTnd and Branford data sets.
Gene ontology NES ENESTnd FDR ENESTnd NES Branford FDR Branford Adaptive immune response 3.00 1.49x10-71 1.47 5.67x10-2 Cell recognition 2.51 8.12x10-16 1.75 3.87x10-2 DNA replication -2.18 6.88x10-12 -1.50 6.99x10-2 T-cell activation 1.83 1.33x10-7 1.39 7.67x10-2 DNA conformation change -1.96 3.29x10-7 -1.62 2.04x10-2 Lymphocyte costimulation 2.28 8.71x10-7 2.14 6.09x10-3 T-cell differentiation 1.94 2.13x10-6 1.53 5.62x10-2 α-b T-cell activation 1.99 2.86x10-5 1.59 9.68x10-2 DNA packaging -1.92 6.03x10-5 -1.73 1.58x10-2 ATP synthesis-coupled electron transport -2.02 1.25x10-4 -1.71 5.63x10-2 α-b T-cell differentiation 1.98 1.83x10-4 1.69 7.62x10-2 Cellular response to toxic substance -1.68 1.24x10-3 -1.61 4.61x10-2 Drug catabolic process -1.91 1.88x10-3 -1.78 5.10x10-2 Protein-DNA complex subunit organization -1.60 2.35x10-3 -1.63 2.30x10-2 Antibiotic metabolic process -1.98 2.91x10-3 -1.95 1.58x10-2 T-cell selection 1.90 3.55x10-3 1.94 3.28x10-2 Detoxification -1.79 4.46x10-3 -1.76 3.87x10-2 Antibiotic catabolic process -1.94 5.41x10-3 -2.11 1.12x10-2 Response to toxic substance -1.46 5.71x10-3 -1.45 5.87x10-2 Cofactor catabolic process -1.93 7.55x10-3 -1.95 3.24x10-2 Positive regulation of cell cycle arrest -1.82 9.19x10-3 -1.74 9.68x10-2 Response to oxidative stress -1.42 1.04x10-2 -1.59 8.55x10-3 G protein–coupled receptor signaling pathway 1.42 1.05x10-2 1.47 1.51x10-2 Regulation of cytosolic calcium ion concentration 1.56 1.15x10-2 1.64 2.86x10-2 Nucleosome organization -1.62 1.90x10-2 -1.79 7.59x10-3 Erythrocyte homeostasis -1.56 2.15x10-2 -1.74 4.00x10-2 Hydrogen peroxide catabolic process -1.86 2.43x10-2 -2.13 7.59x10-3 Chromatin assembly or disassembly -1.58 2.54x10-2 -1.93 1.24x10-3 Chromatin assembly -1.57 3.56x10-2 -1.95 1.24x10-3 T-helper 17–type immune response 1.74 4.47x10-2 1.95 5.89x10-2 Haematologica | 108 June 2023 1573 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
NK cells (P=0.03), and aggregate cytotoxic lymphocytes (P=0.01) were also still significantly associated with achieving DMR (Online Supplementary Table S12). Thus, the finding of a strong immune influence on responder status is robust across at least two different definitions of clinical response.
Discussion
We found that pretreatment immunologic features, including upregulation of genes related to the immune system, pathways, and immune regulatory cells such as NK cells and cytotoxic lymphocytes, were associated with good response after initiation of TKI therapy. The adaptive immune response was the most influential biological process in predicting response of the ENESTnd patients,
as determined by gene-set enrichment analysis of the expression-based predictor. Further evaluation of gene networks controlled by the expression of transcription factors differentially expressed in good and poor responders also pointed to activation of interferon and immune regulatory networks in good responders. The predictive importance of immunologic features was validated in an independent data set, a remarkable finding given the small size of the validation study and the fact that patient features, treatments, and sample handling were undoubtedly not uniform across both studies. The pathway-based predictor offered valuable complementary information to the gene expression-based predictor. Specifically, if a pathway was predictive of response but the specific genes that are overexpressed in that pathway varied between patients, a pathwaybased predictor would capture the pathway’s impor-

Haematologica | 108 June 2023 1574 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
Figure 5. Cytotoxic lymphocytes were enriched in responders. Comparison of immune cell infiltration (inferred by MCP-counter) in the ENESTnd and Branford data sets. NK: natural killer.
tance, but an expression-based predictor might not. While upregulated pathways in good responders were predominantly involved in immunity, an important pathway in predicting good response by this approach was downregulation of transforming growth factor b (TGF- b ) receptor signaling. TGF- b signaling has been shown to promote leukemic stem cell survival in CML.25 Consistent with this finding, we observed that lower TGF- b activity correlated with better response ( Online Supplementary Table S2). Thus, it is intuitive that downregulation of TGFb receptor signaling correlates with good response. Among the pathways most predictive of poor response in the pathway-based predictor was disassembly of the b -catenin destruction complex and recruitment of axin to the membrane. This pathway is activated in response to Wnt signaling and results in b - catenin accumulation. 26,27 This finding agrees with the observation that WNT6 was among the genes most predictive of poor response in the gene expression models ( Online Supplementary Table S2) . Of note, Wnt and b - catenin upregulation has been shown to be associated with progression from CP to blast-phase CML and cell-intrinsic TKI resistance, strengthening the association of poor response in CP with future progression.28,29
CML is among the most immunogenic malignancies. Prior to the advent of TKI, the clinical evidence of the importance of the immune system in CML was as apparent by responses to interferon, allogeneic transplantation, and donor lymphocyte infusions. The use of TKI has further illuminated the importance of the immune system during disease pathogenesis and after the initiation of TKI therapy. A series of murine clinical and in vitro studies, largely based on sophisticated isolation of immune cells and accessing immune factor levels in blood, have demonstrated the role of immune exhaustion in CML pathogenesis, with an activation of the immune system after the initiation of TKI therapy30-33 and (reviewed34,35). Several lines of in vitro data suggest a fluid immunologic state in CML, whereby during disease progression, an expansion of myeloid-derived suppressor cells occurs that downmodulates NK and other T-cell activities, allowing the CML clone to expand.32,36-38 Additionally, it has previously been shown that PD-1 and PD-L1 are upregulated in CML at the time of diagnosis. 39 Under therapy, TKI stimulate immune function, certainly by direct killing of CML cells but also perhaps indirectly by other TKI effects on the immune system. 32,40 Our gene expression data support and complement these findings of the importance of the immune system in CML response (including the aforementioned PD-1 and PD-L1) and show that these differences in immune pathway use (and, by inference, biology in the actual patient) strongly segregate with good or poor response status. Moreover, using gene expression data has the added advantage of being unbi-
ased in discerning gene and pathway involvement, and may uncover targetable pathways that can potentially move a patient from poor to good response.
NK cell biology has been of particular interest in studying TKI response in CML.36 Some studies have reported that prior to therapy, patients with CML had decreased numbers of NK cells compared with healthy controls,41 and increased numbers of NK cells during DMR seemed to correlate with improved odds of remaining in TFR after TKI discontinuation.42-44 Our data, derived by inferring cell type by gene expression, support and complement these findings, as well as show the potential importance of other types of immune cells (e.g., cytotoxic T cells). The interpretation of patterns in bulk gene expression data from unfractionated blood samples is complicated by the possibility of significant differences in cell type distribution between samples. However, pretreatment cell counts showed little differences in cell type proportions as a function of responder status (i e , no significant differences at FDR <10%; Online Supplementary Table S5). Although we do not have data on the cell counts of cytotoxic lymphocytes, we applied a deconvolution algorithm (MCP-counter) to the gene expression data that was designed to estimate the abundance of different cell types, including cytotoxic lymphocytes, from complex mixtures. Thus, we expect that the significant differences in the inferred abundance of cytotoxic lymphocytes such as T cells and NK cells versus response status reflects a true difference in these populations, rather than an artifact of the varying composition of other cell types in the unfractionated samples.
Thus, predictive models based on multiple lines of analysis appear to converge on signatures of T-cell, NK-cell, and B-cell activation as predictive of good response, whereas Wnt signaling, TGF- b signaling, and cell-cycle progression predict poor response. The observation that patients with signatures of cytotoxic lymphocytes at baseline tend to respond best to TKI therapy suggests that the state of the immune system prior to therapy is predictive of response. These observations have two important implications. First, a panel of genes and quantification of cell types at diagnosis could be used to predict the likelihood of poor and good responses, and this prediction could be used to shape expectations, inform monitoring, and direct patients to clinical trials to improve response or to TFR strategies. Secondly, further understanding the role of the immune pathways involved in response might allow the pharmacological alteration of these pathways, potentially turning poor responders into good responders. The results of this work lead to several interesting and testable questions for the biology and treatment of CML, and we encourage our readers to explore the data sets provided and pursue these issues.
Haematologica | 108 June 2023 1575 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
Disclosures
All authors received non-financial support (assistance with manuscript preparation from ScientificPathways, Inc, which received funding from Novartis). JPR received research funding from TwinStrand Biosciences and Novartis. MW, JG, and BSW were supported by a contract with Novartis for this study. SB is a member of the advisory boards of Qiagen, Novartis, and Cepheid and has received hono- raria from Qiagen, Novartis, Bristol Myers Squibb, and Cepheid, as well as research support from Novartis. SB is also supported by the National Health and Medical Research Council of Australia APP1027531 and APP1104425, the Ray and Shirl Norman Cancer Research Trust, and the Royal Adelaide Hospital Research Foundation. RAL has acted as a consultant or advisor to Novartis, Amgen, Ariad/Takeda, Astellas, Celgene/Bristol Myers Squibb, CVS/Caremark, Epizyme, and MorphoSys and has received clinical research support from Novartis, Astellas, Celgene, Cellectis, Daiichi Sankyo, Forty Seven, and Rafael Pharmaceuticals and royalties from UpToDate. HMK received honoraria from AbbVie, Amgen, DaiichiSankyo, Novartis, Pfizer, Adaptive Biotechnologies, Aptitude Health, BioAscend, Delta Fly, Janssen Global, Oxford Biomedical, and Takeda, received grants from Ascentage, Bristol Myers Squibb, DaiichiSankyo, Immunogen, Jazz, Novartis, Pfizer, and Sanofi, and acted as a member of an advisory board for Actinium. MD is a paid consultant for Blueprint, Fusion Pharma, Takeda, Novartis, Incyte, Sangamo, SPARC, Pfizer, Medscape, and Dispersol, received research funding from Blueprint, Takeda, Novartis, Incyte, LLS, Pfizer, and SPARC, and participated as a member of advisory boards for Blueprint, Takeda, Incyte, and Sangamo. JP-I received personal fees from Takeda, AbbVie, Janssen, Novartis, Gilead, and TEVA. DJD received honoraria from Amgen, Autolus, Agios, Blueprint, Forty Seven, Incyte, Jazz, Kite, Novartis, Pfizer, Servier, and Takeda, and research support from AbbVie, GlycoMimetics, Novartis and Blueprint Pharmaceuticals. IS is employed by and has equity in Novartis. SC was employed by Novartis during the conduct of the study. CDC is employed by and is a shareholder of Novartis. RM, SL, KM, DP, AS, and VO are employed by Novartis. BJD Scientific Advisory Board of Aileron Therapeutics, Therapy Architects (ALLCRON), Cepheid, Vivid Biosciences, Celgene, RUNX1 Research Program, Novartis, Gilead Sciences (inactiv e) and Monojul (inactive); Scientific Advisory Board and is a stockholder of Aptose Biosciences, Blueprint Medicines, Enliven Therapeutics, Iterion Therapeutics, Third Coast Therapeutics and
References
1. Bower H, Bjorkholm M, Dickman PW, Hoglund M, Lambert PC, Andersson TM. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol. 2016;34(24):2851-2857.
GRAIL (Scientific Advisory Board inactive); is a scientific founder of MolecularMD (inactive, acquired by ICON); is on the board of directors and a stockholder of Amgen; is on the board of directors of Burroughs Wellcome Fund and CureOne; is a member of the joint steering committee of Beat AML LLS; is a founder of VB Therapeutics; has a sponsored research agreement with Enliven Therapeutics; and has received clinical trial funding from Novartis, Bristol Myers Squibb and Pfizer; has received royalties from patent 6958335 (Novartis exclusive license) and OHSU and DanaFarber Cancer Institute (one Merck exclusive license and one CytoImage, Inc. exclusive license). AH received research support from Novartis, Bristol Myers Squibb, Pfizer and Incyte. TPH is a consultant/has received research funding from Novartis, Bristol Myers Squibb, and Enliven. GS received personal fees from Novartis, Bristol Myers Squibb, ARIAD, and Pfizer.
Contributions
JPR conceived the experimental concept and design. CDC, MW, BSW, and JG performed the computational analysis. JPR, MW, and BSW wrote the manuscript with editorial support from Scienti ficPathways, Inc. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Acknowledgments
We thank Michelle Chadwick, PhD and Chris Hofmann, PhD (ScientificPathways, Inc) for medical editorial assistance with this manuscript. We thank Yumeng Wang for assistance with the mutation analysis.
Funding
This study was supported by funding from Novartis. Financial support for medical editorial assistance was provided by Novartis.
Data-sharing statement
Data files may be found in the Online Supplementary Appendix available with the online version of this article. This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the corrresponding author. The processed de-identified data used in this report can be found on the Synapse platform: https://www.synapse.org/#!Synapse:syn19551222/files/
2. Jabbour E. Chronic myeloid leukemia: first-line drug of choice. Am J Hematol. 2016;91(1):59-66.
3. Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med.
Haematologica | 108 June 2023 1576 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
2010;362(24):2251-2259.
4. Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260-2270.
5. Cortes JE, Saglio G, Kantarjian HM, et al. Final 5-year study results of DASISION: the dasatinib versus imatinib study in treatment-naive chronic myeloid leukemia patients trial. J Clin Oncol. 2016;34(20):2333-2340.
6. Hochhaus A, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30(5):1044-1054.
7. Kantarjian HM, Hughes TP, Larson RA, et al. Long-term outcomes with frontline nilotinib versus imatinib in newly diagnosed chronic myeloid leukemia in chronic phase: ENESTnd 10-year analysis. Leukemia. 2021;35(7):2142-2143.
8. Brummendorf TH, Cortes JE, Milojkovic D, et al. Bosutinib versus imatinib for newly diagnosed chronic phase chronic myeloid leukemia: final results from the BFORE trial. Leukemia. 2022;36(7):1825-1833.
9. Marin D, Ibrahim AR, Lucas C, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. 2012;30(3):232-238.
10. Hanfstein B, Muller MC, Hehlmann R, et al. Early molecular and cytogenetic response is predictive for long-term progressionfree and overall survival in chronic myeloid leukemia (CML). Leukemia. 2012;26(9):2096-2102.
11. Neelakantan P, Gerrard G, Lucas C, et al. Combining BCR-ABL1 transcript levels at 3 and 6 months in chronic myeloid leukemia: implications for early intervention strategies. Blood. 2013;121(14):2739-2742.
12. NCCN Clinical Practice Guidelines in Oncology. Chronic Myeloid Leukemia. V1.2021.
13. Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966-984.
14. Hughes TP, Saglio G, Kantarjian HM, et al. Early molecular response predicts outcomes in patients with chronic myeloid leukemia in chronic phase treated with frontline nilotinib or imatinib. Blood. 2014;123(9):1353-1360.
15. Jain P, Kantarjian H, Nazha A, et al. Early responses predict better outcomes in patients with newly diagnosed chronic myeloid leukemia: results with four tyrosine kinase inhibitor modalities. Blood. 2013;121(24):4867-4874.
16. Jabbour E, Kantarjian HM, Saglio G, et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2014;123(4):494-500.
17. Hochhaus A, Rosti G, Cross NC, et al. Frontline nilotinib in patients with chronic myeloid leukemia in chronic phase: results from the European ENEST1st study. Leukemia. 2016;30(1):57-64.
18. Mahon FX, Nicolini FE, Noël MP, et al. Preliminary report of the STIM2 study: a multicenter stop imatinib trial for chronic phase chronic myeloid leukemia de novo patients on imatinib. Blood. 2013;122(21):654.
19. Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029-1035.
20. Ross DM, Branford S, Seymour JF, et al. Safety and efficacy of
imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122(4):515-522.
21. Branford S, Wang P, Yeung DT, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132(9):948-961.
22. Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17(1):218.
23. Korotkevich G, Sukhov V, Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2019 Feb 1. https://doi.org/10.1101/060012 [Preprint, not peer-reviewed].
24. Wall MA, Turkarslan S, Wu WJ, et al. Genetic program activity delineates risk, relapse, and therapy responsiveness in multiple myeloma. NPJ Precis Oncol. 2021;5(1):60.
25. Naka K, Hoshii T, Muraguchi T, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676-680.
26. Song X, Wang S, Li L. New insights into the regulation of Axin function in canonical Wnt signaling pathway. Protein Cell. 2014;5(3):186-193.
27. Gerlach JP, Emmink BL, Nojima H, Kranenburg O, Maurice MM. Wnt signalling induces accumulation of phosphorylated betacatenin in two distinct cytosolic complexes. Open Biol. 2014;4(11):140120.
28. Radich JP, Dai H, Mao M, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103(8):2794-2799.
29. Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocytemacrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657-667.
30. Christiansson L, Soderlund S, Svensson E, et al. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS One. 2013;8(1):e55818.
31. Christiansson L, Soderlund S, Mangsbo S, et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol Cancer Ther. 2015;14(5):1181-1191.
32. Hughes A, Clarson J, Tang C, et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood. 2017;129(9):1166-1176.
33. Mumprecht S, Schurch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. 2009;114(8):1528-1536.
34. Hughes A, Yong ASM. Immune effector recovery in chronic myeloid leukemia and treatment-free remission. Front Immunol. 2017;8:469.
35. Hsieh YC, Kirschner K, Copland M. Improving outcomes in chronic myeloid leukemia through harnessing the immunological landscape. Leukemia. 2021;35(5):1229-1242.
36. Carlsten M, Järås M. Natural killer cells in myeloid malignancies: immune surveillance, NK cell dysfunction, and pharmacological opportunities to bolster the endogenous NK cells. Front Immunol. 2019;10:2357.
37. Giallongo C, Parrinello N, Brundo MV, et al. Myeloid derived suppressor cells in chronic myeloid leukemia. Front Oncol. 2015;5:107.
38. Bizymi N, Bjelica S, Kittang AO, et al. Myeloid-derived suppressor cells in hematologic diseases: promising biomarkers and treatment targets. Hemasphere. 2019;3(1):e168.
Haematologica | 108 June 2023 1577 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
39. Norde WJ, Maas F, Hobo W, et al. PD-1/PD-L1 interactions contribute to functional T-cell impairment in patients who relapse with cancer after allogeneic stem cell transplantation. Cancer Res. 2011;71(15):5111-5122.
40. Giallongo C, Parrinello N, Tibullo D, et al. Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with polymorphonuclear leukocytes (PMNs) in chronic myeloid leukemia patients. PLoS One. 2014;9(7):e101848.
41. Chen CI, Koschmieder S, Kerstiens L, et al. NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR-ABL-positive mice. Leukemia. 2012;26(3):465-474.
42. Irani YD, Hughes A, Clarson J, et al. Successful treatment-free
remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. Br J Haematol. 2020;191(3):433-441.
43. Ilander M, Olsson-Stromberg U, Schlums H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia. 2017;31(5):1108-1116.
44. Rea D, Henry G, Khaznadar Z, et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. 2017;102(8):1368-1377.
45. Arora R, Press RD. Measurement of BCR-ABL1 transcripts on the International Scale in the United States: current status and best practices. Leuk Lymphoma. 2017;58(1):8-16.
Haematologica | 108 June 2023 1578 ARTICLE - Molecular response in newly diagnosed CP-CML J. Radich et al.
Complement protein C3a enhances adaptive immune responses towards FVIII products
Eva Ringler,* Samira Ortega Iannazzo,* Jessica Herzig, Lisa M. Weiss, Martina Anzaghe, Lilija Miller and Zoe Waibler
3/1 ”Product Testing of Immunological Biomedicines”, Paul-Ehrlich-Institut, Langen, Germany
*ER and SOI contributed equally as co-first authors.
Abstract
Correspondence: Z. Waibler zoe.waibler@pei.de
Received: July 14, 2022.
Accepted: January 20, 2023.
Early view: February 2, 2023.
https://doi.org/10.3324/haematol.2022.281762
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The most serious complication in the treatment of hemophilia A (HA) is the development of factor (F)VIII inhibitors or antidrug antibodies (ADA) occurring in 25-35% of patients with severe HA. The immunological mechanisms underlying the development of ADA against FVIII products have not been completely understood yet. Immunological danger signals associated with events such as infection or surgery have been suggested to play a critical role. In previous studies, we demonstrated that plasma-derived (pd)FVIII but not recombinant (r)FVIII can activate human monocyte-derived dendritic cells (DC) in a danger signal-dependent manner, which subsequently mediate the proliferation of autologous CD4+ T cells. In this study, we investigated the ability of plasma components, naturally present in pdFVIII products, to mediate T-cell responses. In fact, we show that addition of plasma to rFVIII plus lipopolysaccharide (LPS)-stimulated DC induces proliferation of autologous CD4+ T cells. Interestingly, although DC pulsed with LPS plus plasma induce T-cell proliferation upon co-culture, the addition of FVIII significantly increases the number of proliferating as well as FVIII-specific CD4+ T cells. Total proliferating CD4+ T cells and FVIII-specific subsets were identified mainly as central memory T cells. Experiments using blocking antibodies and receptor antagonists revealed that the complement proteins C3a and, to a lesser extent, C5a are critically involved in these LPS-mediated T-cell responses. Collectively, our results indicate that complement proteins are potent drivers of T-cell responses to FVIII. Data presented provide a model how event-related substitution of FVIII in HA patients might contribute to inhibitor development.
Introduction
Hemophilia A (HA) is a hereditary bleeding disorder characterized by the complete absence or functional deficiency of the coagulation factor VIII (FVIII). In order to restore normal hemostasis, HA patients are treated with infusions of either plasma-derived (pd) or recombinant (r)FVIII products. A serious complication upon administration is the development of anti-FVIII antibodies, so called inhibitors, which can substantially limit treatment efficacy.1 The reported inhibitor incidence in treated patients with severe HA is approximately 25-35%.2 The risk for inhibitor development is higher in those HA patients completely lacking endogenous FVIII when compared to those suffering from mild or moderate HA with only a mutated or truncated FVIII protein.3 However, this genetic predisposition does not necessarily predict the risk of inhibitor development of an
individual patient. Patients with minor FVIII variations can develop inhibitors and patients with large deletions or a complete FVIII protein deficiency can remain free from inhibitor development. Consequently, it has been demonstrated that other factors such as ethnicity, HLA genotype, polymorphisms in immune regulatory genes, and/or the intensity and context of FVIII-treatment contribute to inhibitor development.4 An additional risk factor in the development of inhibitors is provided by the presence of immunological danger signals, in particular during intensive FVIII-treatment episodes. It has been observed that avoiding FVIII-treatment during acute infectious diseases or vaccinations, events associated with an enhanced presence of danger signals, limits the risk of inhibitor formation.5 In line with this, we have previously shown that in the presence of the danger signal lipopolysaccharide (LPS), human dendritic cells (DC) can be synergistically activated by pdFVIII
Section
Haematologica | 108 June 2023 1579 ARTICLE - Coagulation & its Disorders
but not rFVIII products in vitro 6 Furthermore, we have demonstrated that only pd but not rFVIII plus LPS-stimulated DC induce proliferation of co-cultured autologous T cells.7 As a medicinal product derived from human plasma, pdFVIII products co-deliver other blood-derived proteins. In fact, within pdFVIII products, a maximum of 2% is made up by FVIII while ~98% consist of (other) plasma proteins.8 Some of the plasma components such as proteins of the complement system, may promote immune responses. Of note, plasma components can either be delivered by pdFVIII products as in in vitro experimental settings or are ubiquitously present in pdFVIII- or rFVIII-treated patients per se.
The complement system is an ancient and evolutionary conserved arm of innate immunity. However, its diverse role in regulating adaptive immune responses has been underestimated for a long time. Complement serves as a natural adjuvant for B-cell activation and antibody production and has modulating effects on T-cell immunity.9 Particularly, the complement components C3a and C5a, which interact with their respective receptors C3aR and C5aR on antigen presenting cells (APC) and T cells, were shown to alter T-cell responses both directly and indirectly via APC.10,11 Furthermore, there is a body of evidence pinpointing towards a substantial crosstalk between complement and Toll-like receptor (TLR) signaling pathways resulting in synergistic interactions.12 For example, the TLR4 ligand LPS has been shown to activate complement which can lead to differential LPS-induced inflammatory processes.13 In line with this, excessive complement activation can contribute to destructive inflammatory conditions present in sepsis, allergy, transplant rejection, or autoimmunity.14 However, experimental data investigating the impact of plasma components and LPS on FVIII inhibitor development are largely missing. In our current study, we investigated whether plasma, and in particular the complement proteins C3a and C5a, contribute to FVIII plus LPS-mediated T-cell responses. In addition, we explore which cellular and molecular mechanisms are involved in this T-cell activation elicited by both pd and rFVIII, depending on the experimental setting. In order to further shed light on these mechanisms, we quantified and characterized proliferating and FVIII-specific CD4+ T cells and their subsets.
Methods
Ethics statement
The research was approved by the Ethics Committee of the Medical Faculty of the Goethe University Frankfurt, Germany (trade number 70/15) and is in accordance with the Declaration of Helsinki of 1975, revised in 2008. All volunteers have given written consent to the study.
FVIII products
One pdFVIII (corresponding to FVIII1 in Miller et al. 2015 and 20186,7) and one full-length rFVIII product produced in a baby hamster kidney cell line were used throughout the study. DC were incubated with 1 IU/mL of FVIII products. The generation of plasma is described in the Online Supplementary Appendix.
Quantification of C3a and C5a
In order to quantify C3a/C3adesArg and C5a/C5adesArg, OptEIA enzyme-linked immunsorbant assay (ELISA) kit from BD Biosciences (San Diego, USA) was used in accordance with the manufacturer's instructions.
T-cell proliferation assay
Proliferation assays were performed as previously described.7 In experiments performed using autologous, FVIII-deficient (Siemens Healthcare Diagnostics GmbH), and heat-treated plasma, cells seeded in different ratios (see the Online Supplementary Appendix for details) were pooled for analysis.
Complement blocking
Blocking of complement receptors was performed by incubating DC for 1 hour with the indicated concentrations of a specific C3aR antagonist (C3aRA) or C5aR antagonist (C5aRA; both from Calbiochem, now Sigma-Aldrich; dissolved in dimethyl sulfoxide [DMSO]) prior to treatment with pdFVIII plus LPS. As a control, DC were incubated with DMSO only (Sigma-Aldrich). Complement proteins were blocked by treating DC with pdFVIII plus LPS in the presence of the following neutralizing monoclonal antibodies: anti-C3/C3a/C3adesArg (clone K13/16), antiC5/C5a/C5adesArg (clone G25/2, both isotype IgG1, BioLegend, Fell, Germany), eculizumab (anti-C5, isotype IgG2/4, Alexion, Paris, France), or the C3-targeting peptide compstatin (Tocris Bioscience, Bristol, UK). An IgG1 isotype control antibody (clone MOPC-21, BioLegend) and ovalbumin (OVA) 323-339 (ISQAVHAAHAEINEAGR, AnaSpec, Fremont, USA) served as controls.
Flow cytometry
For cell proliferation analyses by flow cytometry, T cells were collected by up- and down-pipetting. Complement receptors on DC and T cells were stained for 20 minutes (min) at 4°C using anti-human C3aR-APC (clone hC3aRZ8, BioLegend) and anti-C5aR-PE (clone D53-1473, BD Pharmingen, San Diego, USA) at their optimal concentrations. As isotype control, IgG2b-APC (clone 27-35) and IgG1-PE (clone MOPC 21, both from BD Pharmingen) were used. Tcell surface markers were stained for 20 min at 4°C using the following antibodies: anti-CD3-AmCyan (clone SK7, BD Pharmingen), anti-CD4-PacBlue, anti-CD4-APC (both clone RPA-T4, BD Pharmingen), anti-CD45RA-PerCP (clone
Haematologica | 108 June 2023 1580 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
HI100, BioLegend), anti-CD45RO-AF700 (clone IV N31, BioLegend), and anti-CCR7-PE-Cy7 (clone 3C12, BD Pharmingen). For the detection of FVIII-specific CD4+ T cells, cells were harvested, incubated for 20 min with polyglobin (Bayer Vital, Leverkusen, Germany), and stained for 2 hours with HLA-DRB1*11:01 ProT2589-608 (ENIQRFLPNPAGVQLEDPEF) tetramer-PE (Proimmune, Oxford, UK). Subsequently, cells were stained with antibodies for 20 min at 4°C, washed, and fixed with 1% paraformaldehyde. All fluorescence-activated cell sorting (FACS) analyses were performed using LSR II and LSR Fortessa flow cytometers. Data were analyzed with the BD FACSDiva software version 8.0.1 (BD Biosciences) or FlowJo software version 7.6.5 or 10.0.8 (Tree Star, Ashland, USA).
Statistical analysis and calculations
Statistical analyses are further described in the Online Supplementary Appendix.
Results
Addition of human plasma to recombinant FVIII plus lipopolysaccharide-treated dendritic cells increases T-cell proliferation

We have previously shown that pd but not rFVIII-treated DC synergistically mediate T-cell proliferation, when applied along with LPS.7 This is reproduced in Figure 1 where DC were treated either with a pd or rFVIII product, LPS, or with FVIII plus LPS. Subsequently, PKH26-labeled autologous T cells were added for 9 days and T-cell proliferation was analyzed as a measure of PKH26 dilution. In order to investigate if plasma components contribute to the immunogenicity of pdFVIII products, plasma was added to rFVIII plus LPS-treated DC. If not stated otherwise, the plasma used throughout this study was collected and pooled from ten individual donors. With the exception of this exogenously added plasma, all experi-
Figure 1. Addition of human plasma to recombinant FVIII plus lipopolysaccharide-treated dendritic cells induces increased T-cell proliferation. Dendritic cells (DC) were treated for 24 hours with (A) plasma-derived (pd) FVIII or (B) rFVIII (each with 1 IU/mL), lipopolysaccharide (LPS) (0.1 mg/mL), or with FVIII plus LPS. In addition, DC were treated with rFVIII plus LPS in presence of plasma (2.5 mL/mL). Untreated DC served as control. PKH26-labeled autologous T cells were added and at day 9 of co-culture, cells were harvested and percentages of proliferated T cells were analyzed by fluorescence-activated cell sorting (FACS). Flow cytometry plots of 1 representative donor are shown in (A) and (B). Data of several donors are summarized from at least 3 independent experiments for (C) pdFVIII (n=14) and (D) rFVIII treatment (n=7-10). Statistical significance was determined using the Wilcoxon matched-pairs signed rank test. Error bars indicate the means ± standard error of the mean. *P<0.05; ***P<0.001.
A B C D
Haematologica | 108 June 2023 1581 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
ments throughout this study were performed in serumfree media. As shown in Figure 1B, D, addition of plasma to rFVIII plus LPS-treated DC indeed induced proliferation of co-cultivated autologous T cells. These data indicate that plasma components which are co-delivered by pd but not rFVIII products cause the synergistically enhanced induction of T-cell proliferation and thus the immunogenicity of pdFVIII products observed in vitro. This might also hold true for the in vivo situation, where plasma components are ubiquitously present within an FVIII-treated HA patient.
Proliferating T cells are FVIII-specific and predominantly central memory cells
In order to verify that co-stimulation with LPS plus plasma indeed amplifies T-cell responses towards FVIII, we determined FVIII-specific T cells within proliferating CD4+ T cells. For this purpose, DC were stimulated with LPS plus plasma in the presence or absence of rFVIII, cocultivated with autologous T cells, and proliferated CD4+ FVIII-specific T cells were analyzed using a FVIII-specific tetramer. As shown in Figure 2B, DC pulsed with rFVIII alone did not mediate expansion of FVIII-specific T cells, while LPS plus plasma-stimulated DC mediated some expansion of FVIII-specific T cells. Of note, stimulating DC with LPS plus plasma in the presence of rFVIII significantly increased FVIII-specific T-cell counts within the total number of proliferating CD4+ T cells (Figure 2B). These data strongly indicate that addition of an antigen such as FVIII to LPS plus plasma-pulsed DC directs the induced T-cell response towards this given antigen.

In order to further dissect T-cell subsets proliferating
upon co-culture with LPS plus plasma and rFVIII-stimulated DC, we classified proliferating T cells as published by Thome et al. 15 According to this classification, naïve T cells are CD3+CD4+CD45RO-CD45RA+CCR7+ while effector T cells are CD3+CD4+CD45RO-CD45RA+CCR7-. Furthermore, central memory (CM) T cells are characterized as being CD3+CD4+CD45RO+CD45RA-CCR7+ and effector memory (EM) T cells as being CD3+CD4+CD45RO+CD45RA-CCR7- 15 All four subsets were analyzed within the proliferating Tcell population. As shown in Figure 3B, C, both T-cell populations, total proliferating CD3+CD4+ T cells as well as proliferating CD3+CD4+ FVIII-specific T cells, consisted of approximately 72% or 67% CM and 17% or 19% EM T cells, respectively, while naïve or effector T cells were clearly underrepresented with a maximum of approximately 2%. Thus, in the presence of the antigen rFVIII, LPS plus plasma-stimulated DC predominantly induce the expansion of antigen-specific CM CD4+ T cells.
C3a and C5a are present in plasma-derived FVIII products
It has been shown before that C3a is present in various pdFVIII products.16 In order to investigate whether C3a and/or C5a are present in FVIII products used in our study, we determined C3a and C5a levels within these products by ELISA. As shown in Figure 4A, both C3a and C5a were detectable in the pdFVIII product, whereas both were absent in the solvent (water for injection [WFI]) and the rFVIII product. Heat treatment of plasma is a rather crude but commonly applied laboratory method to inactivate complement.17 As shown in Figure 4B, C, T-cell proliferation was strongly re-
Figure 2. Recombinant FVIII co-stimulation with lipopolysaccharide plus plasma induces the proliferation of FVIII-specific T cells. Dendritic cells (DC) from HLA-DRB1*11-positive donors were stimulated with recombinant (r)FVIII (1 IU/mL), lipopolysaccharide (LPS) (0.1 mg/mL), and plasma (2.5 mL/mL). Control DC were left untreated. After 24 hours, carboxyfluorescein succinimidyl ester (CFSE)-labeled autologous T cells were co-cultured with DC for 9 days. On day 9, T cells were harvested and stained with HLA-matched FVIII-specific tetramer and anti-CD3 and anti-CD4 antibodies for fluorescence-activated cell sorting (FACS) analyses. (A) Gating strategy for 1 representative donor is shown. In order to analyze only living T cells, gates were set in the forward/side scatter (FSC/SSC) and further on CD3+CD4+ T cells. T-cell proliferation was assessed by measuring the decrease of CFSE fluorescence intensity in the histogram. Then, CFSE- cells were defined in the dot plot as CD4+tetramer+ FVIII-specific T cells. (B) The proliferated FVIII-specific T cells are shown as absolute numbers in events. Data in (B) were obtained from 5 independent experiments (n=6-11). Statistical significance was determined using the Wilcoxon matched-pairs signed rank test. Error bars indicate the means ± standard error of the mean. *P<0.05.
A B Haematologica | 108 June 2023 1582 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
duced when DC were stimulated with rFVIII plus LPS in the presence of heat-treated plasma when compared to untreated plasma. Collectively, these data provide a first hint that complement components C3a and/or C5a might be involved in the induction of T-cell proliferation under the above-mentioned conditions.
C3a promotes pdFVIII plus lipopolysaccharide-mediated T-cell proliferation
The expression of C3aR and C5aR is not limited to cells of the myeloid lineage,18 but has been reported for lymphocytes as well.11,19 Expression of the respective receptors on cells used within our study is the prerequisite for the suggested (direct) action of C3a and/or C5a on these cells. As expected, FACS analyses confirmed the expression of C3aR and C5aR on the surface of in vitro-differentiated DC and freshly isolated T cells (Figure 5A). However, the expression levels of C3aR and C5aR were found to be donor-specific (data not shown) as reported before.20-22
In order to investigate the role of C3a and/or C5a in synergistically enhanced T-cell responses observed upon coculture with pdFVIII plus LPS-treated DC, we blocked these
complement components. For blocking, we used a monoclonal antibody (mAb) binding an epitope shared by both C3 and C3a, and C5 and C5a, respectively. As shown in Figure 5B, T-cell proliferation mediated by pdFVIII plus LPS-treated DC was significantly reduced upon blocking of C3/C3a. Blocking C5/C5a by a specific mAb also significantly reduced T-cell proliferation. However, also applying the isotype control mAb had an inhibitory effect. Using compstatin, a specific peptide inhibitor of C3, did not reduce T-cell proliferation indicating that T-cell proliferation mediated by pdFVIII plus LPS-treated DC indeed requires C3a but not its uncleaved precursor C3 (Figure 5B). Along this line, no reduction of T-cell proliferation was observed upon blocking C5 using the therapeutic mAb eculizumab (Figure 5B). Eculizumab is a chimeric IgG2/4 construct; hence, a corresponding isotype control was not available to us.

In order to confirm the role of C3a and/or C5a in T-cell proliferation mediated by pdFVIII plus LPS-stimulated DC, we made use of C3aR and C5aR antagonists (C3aRA and C5aRA).10,23 As shown in Figure 5C and D (upper panels), blocking C3aR significantly reduced T-cell proliferation when compared to the solvent control (DMSO). Similarly,
Figure 3. Recombinant FVIII co-stimulation with lipopolysaccharide plus plasma mainly induces proliferation of central memory T cells. Dendritic cells (DC) from HLA-DRB1*11-positive donors were treated with recombinant (r)FVIII (1 IU/mL), lipopolysaccharide (LPS) (0.1 mg/mL), and plasma (2.5 mL/mL). After 24 hours, carboxyfluorescein succinimidyl ester (CFSE)-labeled autologous CD4+ T cells were added. On day 9 of co-culture, T cells were harvested and stained with HLA-matched FVIII-specific tetramer and an antibody mix containing anti-CD3, anti-CD4, anti-CD45RA, anti-CD45RO, and anti-CCR7 antibodies. The CD4+ T cells were defined as 4 subsets: central memory (CM) T cells (CD45RO+CD45RA-CCR7+), effector memory (EM) T cells (CD45RO+CD45RA-CCR7), naïve T cells (CD45RO-CD45RA+CCR7+), and effector T cells (TE) (CD45RO-CD45RA+CCR7-). (A) Data from 1 representative donor are shown. (B) Percentage distribution of CD4+ T-cell subtypes from proliferated CD4+ T cells (n=17, 5 donors from 7 independent experiments) and (C) from proliferated CD4+ FVIII-specific T cells (n=7, 6 donors from 2 independent experiments) are summarized. Error bars indicate the mean ± standard error of the mean. SSC: side scatter; FSC: forward scatter.
A B C
Haematologica | 108 June 2023 1583 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
C5aR blocking resulted in significantly lower T-cell proliferation rates when compared to controls (Figure 5C, D lower panels). Of note, using the indicated concentrations of C3aRA and C5aRA had no effect on T-cell viability but using C5aRA at 10 mM reduced T-cell viability (data not shown) which is in line with Lalli et al. showing that some C5aR signaling is necessary for maintaining murine T-cell survival.24 For this reason, we omitted data for C5aRA treatment at 10 mM.
Collectively, data presented here indicate that pdFVIII products co-deliver C3a and C5a peptides of which particularly C3a is critically involved in the induction of synergistically enhanced T-cell responses upon co-cultivation with pdFVIII plus LPS-stimulated DC.
The antigen recombinant FVIII increases lipopolysaccharide- plus plasma-mediated T-cell proliferation

Adding plasma to rFVIII plus LPS-treated DC increases the proliferation of co-cultivated, autologous T cells (Figure 1C). However, enhanced T-cell proliferation was also observed upon co-culture with DC treated with LPS plus plasma in the absence of FVIII as an antigenic component (Figure 2B; Figure 6A). These observations raised two
questions: first, since the plasma used in our study is of allogenic origin, non-self determinants are present and might contribute to DC-mediated T-cell proliferation; second, whether FVIII as an antigenic component contributes to T-cell proliferation at all. In order to answer the first question, we collected plasma from ten individual donors which was used to stimulate cells from each corresponding donor. As a control, the pooled plasma was used throughout the study. As shown in Figure 6B, there was no difference in T-cell proliferation no matter whether the plasma pool or autologous plasma was used in the absence or presence of LPS, indicating that mechanisms independent of self/non-self discrimination are involved. In order to investigate, whether the presence of the antigen FVIII contributes to T-cell proliferation, we treated DC with LPS and FVIII-deficient plasma in the presence or absence of rFVIII, and analyzed proliferation of co-cultured T cells. As shown in Figure 6C, D, percentages of proliferated T cells were significantly increased when rFVIII was added. This increase in proliferating T cells in the presence of rFVIII was consistent for all individual donors tested (Figure 6E). These data show that rFVIII increases T-cell proliferative responses and suggests that if an antigen such as FVIII is present, plasma plus LPS-
Figure 4. C3a and C5a are present in plasma-derived FVIII products. (A) Plasma-derived (pd) or recombinant (r)FVIII products (5 IU each) were analyzed for C3a (black bars) and C5a (white bars) by enzymelinked immunosorabant assay (ELISA) with water for injection (WFI) as solvent control. Error bars indicate standard deviations from triplicate or quadruplicate ELISA measurements. (B) Dendritic cells (DC) were treated for 24 hours with rFVIII (1 IU/mL), lipopolysaccharide (LPS) (0.1 mg/mL), or with rFVIII plus LPS in the presence or absence of plasma or heat-treated plasma (both at 2.5 mL/mL). Control DC were left untreated. PKH26-labeled autologous T cells were added and 9 days after co-culture, cells were harvested and percentages of proliferated T cells were analyzed by fluorescence-activated cell sorting. Data of 1 representative donor are shown. (C) Summarized results of several donors from 2 independent experiments are given (n=4). Error bars indicate the means ± standard error of the mean. SSC: side scatter; FSC: forward scatter.
A C B Haematologica | 108 June 2023 1584 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
Figure 5. C3a promotes plasma-derived FVIII plus lipopolysaccharide-mediated T-cell responses. (A) Untreated dendritic cells (DC) and freshly isolated T cells were stained with antibodies directed against C3aR and C5aR (solid lines), respective isotype controls (dashed lines), or were left unstained (grey-shaded curves), and analyzed by fluorescence-activated cell sorting (FACS). Overlay histograms of 1 representative donor are shown. (B) DC were treated for 24 hours with plasma-derived (pd)FVIII (1 IU/mL) plus lipopolysaccharide (LPS) (0.1 m g/mL) in the presence or absence of the following complement inhibitors (inhib) or controls (ctrl): anti-C3/C3a/C3adesArg (αC3/C3a, n=16), anti-C5/C5a/C5adesArg (αC5/C5a, n=10), isotype control antibody (at 2 or 5 mg/mL), C3-targeting peptide inhibitor compstatin (n=7), control peptide (OVA323-339, both at 1 mg/mL), or anti-C5 (eculizumab, at 2 mg/mL, n=11). On day 9 of co-culture with PKH26-labeled autologous T cells, T-cell proliferation was assessed by FACS. Shown are the percentages of proliferated T cells from at least 3 independent experiments. (C) and (D) DC were pretreated for 1 hour with antagonists for C3aR (C3aRA) or C5aR (C5aRA) at the indicated concentrations followed by treatment with pdFVIII (1 IU/mL) plus LPS (0.1 mg/mL). Control DC were pretreated with dimethyl sulfoxide (DMSO) prior to stimulation with pdFVIII plus LPS. 24 hours later, PKH26-labeled autologous T cells were added and co-cultured for 9 days before the percentages of proliferated PKH26-labeled T cells were analyzed by flow cytometry. (C) FACS plots of 1 representative donor are given for C3aRA applied at 10 mM and C5aRA at 0.1 mM. (D) Summarized data of several donors (n=8) from 3 independent experiments are shown. Statistical significance was determined using the Wilcoxon matched-pairs signed rank test. Error bars indicate the means ± standard error of the mean.*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; ns: not significant. SSC: side scatter; FSC: forward scatter.

A B C D Haematologica | 108 June 2023 1585 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
treated DC mediate adaptive immune responses towards this given antigen.
Discussion
We have previously shown that pd but not rFVIII products
plus LPS synergistically activate DC and that these DC subsequently mediate enhanced proliferation of autologous T cells in vitro. 6,7 Here we show that DC pulsed with rFVIII plus LPS mediate synergistic T-cell activation when human plasma is added (Figure 1; Figure 6). Until now, it is still under debate whether pd or rFVIII products are more immunogenic. There are studies claiming higher in-
Figure 6. Addition of the antigen FVIII increases T-cell proliferation. (A) Dendritic cells (DC) were either left untreated or treated with lipopolysaccharide (LPS) (0.1 mg/mL), plasma (2.5 mg/mL), or plasma plus LPS. 24 hours later, PKH26-labeled autologous T cells were added and co-cultured for 9 days before fluorescence-activated cell sorting (FACS) analysis. T-cell proliferation is indicated as percentages of PKH26-labeled T cells from 7 independent experiments (n=18). (B) DC were left untreated or treated for 24 hours with LPS (0.1 mg/mL), plasma of either autologous (auto) or foreign origin (each at 25 µmL/mL), or with LPS plus the indicated plasma followed by co-cultivation with PKH26-labeled autologous T cells for 9 days. The percentages of proliferated T cells were analyzed for 10 donors by flow cytometry. (C) Treatment of DC for 24 hours with rFVIII (1 IU/mL), LPS (0.1 mg/mL), or FVIII-deficient plasma (plasmadef, 2.5 mL/mL), each alone or in combination as indicated. Untreated DC served as control. PKH26-labeled autologous T cells were added and after 9 days, T-cell proliferation was analyzed by FACS. Representative data for 8 donors analyzed are shown. (D) Summarized data from 8 donors and 3 experiments. (E) Comparison of the percentages of proliferated T cells obtained in the presence or absence of rFVIII. Statistical significance was determined using the Wilcoxon matched-pairs signed rank test. Error bars indicate the means ± standard error of the mean. **P<0.01; ***P<0.001; ns: not significant. SSC: side scatter; FSC: forward scatter.

A C D E B Haematologica | 108 June 2023 1586 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
hibitor rates for one or the other type of product as well as those showing similar risks for both.4 In a randomized trial (SIPPET), the incidence of FVIII inhibitors was assessed among patients treated with pd or rFVIII. Here, Peyvandi et al. 25 found that patients treated with pdFVIII had a lower incidence of inhibitors than those treated with rFVIII. Subsequently, they suggested that rFVIII products might be more immunogenic.
We hypothesize that plasma components, which are either co-delivered by pd but not rFVIII products within in vitro experiments, or which are ubiquitously present in FVIII-treated patients per se, contribute to the observed T-cell activation. This explains why we observe synergistic DC and T-cell activation in vitro only in the presence of pdFVIII or plasma (this study and6,7), while in HA patients both pd and rFVIII products can induce the generation of inhibitors. Of note, pdFVIII products consist of approximately 2% FVIII while 98% are made up by plasma proteins others than FVIII.26
Interestingly, it has been shown that plasmapheresis, the most common method to obtain plasma for fractionation of pdFVIII, can induce complement activation resulting in the formation of C3a and C5a27,28 and consequently, we and others detected C3a and C5a in various pdFVIII products (Figure 4, data not shown, and 16). Complement components are broadly recognized for their pro-inflammatory and immunomodulatory effects. Accordingly, they can even be involved in autoimmunity (summarized in29-31). Assembling this information, we suggested a role for the complement components C3a and C5a in FVIII-specific T-cell activation and indeed, we identified C3a and, to a lesser extent, C5a as mediators of enhanced proliferation of total CD3+CD4+ and FVIII-specific T cells (Figure 5).
In our experimental setting, C3a and C5a act in concert with LPS in order to mediate T-cell activation (Figure 5). Stimulating T cells with DC pulsed with LPS or plasma alone minimally induces T-cell proliferation while DC pulsed with both LPS plus plasma clearly induce T-cell proliferation (Figure 6A). Interestingly, it has been assumed for decades that within LPS-stimulated peripheral blood mononuclear cells, T-cell proliferative responses can be enhanced if human serum is added.32 In line with this, Morrison et al showed that LPS can activate the complement system via the classical or the alternative pathway13 and Zhang et al observed an enhancement of TLR-mediated inflammatory responses by complement components.33 Additionally, the expression of C3a and C5a receptors on human DC can be increased when cells are treated with LPS.34 Hence, it is most likely that the TLR and the complement pathway crosstalk in order to mediate T-cell proliferative responses when T cells are co-cultured with pdFVIII plus LPS-pulsed or rFVIII plus LPS plus plasma-pulsed DC. Whether these direct effects of complement are at the DC or T-cell level or whether C3a and C5a have an impact on
both cell types cannot be dissected within our experimental setting. However, we have previously shown a synergistic effect of pdFVIII plus LPS directly on human DC which upregulated co-stimulatory molecules and enhanced the expression of pro-inflammatory cytokines,6,7 suggesting an effect on the level of DC for our recent analyses as well. This is in line with data showing that stimulation of human DC with C3a and C5a increases the expression of HLA-DR and CD86 as well as the expression of pro-inflammatory cytokines.35 Of note, Gutzmer et al also observed the upregulation of co-stimulatory molecules CD83 and CD86 on human DC when stimulated with recombinant C3a. However, these C3a-stimulated DC did not mediate enhanced proliferative responses of autologous T cells towards the tetanus toxoid antigen.21 Thus, stimulation of DC with complement alone does not seem to be sufficient to mediate full-blown T-cell responses which is reflected also by our data showing that DC pulsed with pdFVIII or plasma alone do not mediate T-cell proliferation but addition of LPS is needed (Figure 1; Figure 6).
Of note, a direct effect of C3a and/or C5a on T cells might also contribute to pdFVIII- or plasma-mediated T-cell proliferation since also T cells express C3aR and C5aR (Figure 5 and 11,19) and expand upon co-culture with DCderived C3a or C5a.10
It has been reported before that complement component C3 can mediate antigen uptake by APC36 and that C3 and its cleavage product C3b can enhance FVIII endocytosis by DC in vitro. 37 Whether these mechanisms are also of importance in our experimental setting has not been elucidated yet.
Under the experimental conditions chosen here, a source of complement is the pdFVIII product or the exogenously added plasma. However, it has been repeatedly shown that also immune cells themselves including DC and T cells can produce and secrete complement components such as C3a and C5a.10 In particular, co-stimulatory interactions of cognate APC:T cell partners can generate C3a and C5a locally and moreover, upregulate C3aR and C5aR.38,39 In vivo, plasma, complement components, and danger signals are provided by the FVIII-treated patients themselves. Especially in episodes of massive bleeding, injuries, or surgeries, complement can be activated and danger signals can be present. Interestingly, it has been shown that event-related FVIII substitution, which is required in exactly these aforementioned situations, is associated with enhanced inhibitor development.4,40,41
As can be deduced from our experiments using plasma plus LPS to induce T-cell proliferation (Figure 6) and from our analyses detecting FVIII-specific T cells (Figure 2), a substantial number of T cells proliferate under these experimental conditions that have a specificity different from FVIII. It is a well-known in vitro phenomenon that bystander
Haematologica | 108 June 2023 1587 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
cells, in particular CD4+ memory T cells, can be activated independently of antigen by cytokines secreted by T cells that have been activated antigen-dependently.42-44 However, our data clearly indicate that adding FVIII as antigen significantly enhances total T-cell proliferation and expansion of FVIII-specific T cells (Figure 2; Figure 6). Whether this FVIII-specific T-cell activation/proliferation translates into antibody formation is still an open question. However, development of FVIII-specific antibodies is not restricted to HA patients. Natural FVIII-specific autoantibodies are found in 19% of healthy subjects45,46 and FVIII-specific CD4+ T cells are found in healthy donors and show a naïve and CM phenotype.47
Collectively, our data suggest a model in which danger signals such as LPS and the complement components C3a and/or C5a act in concert to synergistically enhance CD4+ T- cell responses towards a given antigen, present at this particular time point. In case of FVIII-substituted HA patients, specifically upon an event-related FVIII substitution, these danger signals plus complement-mediated T-cell responses are (largely or to a certain amount) directed towards the infused FVIII. Whether this mechanism of enhancing CD4+ T-cell responses is specific for FVIII or holds true for a number of (self)antigens will be a matter of future investigations.
References
1. Graw J, Brackmann H-H, Oldenburg J, Schneppenheim R, Spannagl M, Schwaab R. Haemophilia A: from mutation analysis to new therapies. Nat Rev Genet. 2005;6(6):488-501.
2. Male C, Andersson NG, Rafowicz A, et al. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica. 2021;106(1):123-129.
3. Oldenburg J, Schröder J, Brackmann HH, Müller-Reible C, Schwaab R, Tuddenham E. Environmental and genetic factors influencing inhibitor development. Semin Hematol. 2004;41(1 Suppl 1):S82-88.
4. Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046-4055.
5. Kurnik K, Bidlingmaier C, Engl W, Chehadeh H, Reipert B, Auerswald G. New early prophylaxis regimen that avoids immunological danger signals can reduce FVIII inhibitor development. Haemophilia. 2010;16(2):256-262.
6. Miller L, Weissmüller S, Ringler E, et al. Danger signaldependent activation of human dendritic cells by plasma-derived factor VIII products. Thromb Haemost. 2015;114(2):268-276.
7. Miller L, Ringler E, Kistner KM, Waibler Z. Human dendritic cells synergistically activated by FVIII plus LPS induce activation of autologous CD4+ T cells. Thromb Haemost. 2018;118(4):688-699.
8. Timperio AM, Gevi F, Grazzini G, Vaglio S, Zolla L. Comparison among plasma-derived clotting factor VIII by using monodimensional gel electrophoresis and mass spectrometry.
Disclosures
No conflicts of interest to disclose.
Contributions
ER, SOI, JH, LMW and LM performed experiments. ER, SOI, JH, MA, LM and ZW analyzed data. ER, SOI, JH, MA and ZW wrote the manuscript. ER, SOI, JH and ZW prepared the figures. MA, LM and ZW designed the study.
Acknowledgments
We thank Stefanie Kronhart and Dorothea Kreuz for excellent technical support, Peter Crauwels for providing an isotype control, Kay-Martin Hanschmann for statistical analyses, and Ger van Zandbergen and Andreas Hunfeld for fruitful discussion. We are thankful to Gerrit Praefcke for critically reading the manuscript.
Funding
This study was supported by a scholarship from the Stiftung Polytechnische Gesellschaft (Frankfurt, Germany) (to JH).
Data-sharing statement
For original data and protocols, please contact the corresponding author.
Blood Transfus. 2010;8(Suppl 3):S98-104.
9. Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5(10):981-986.
10. Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS. Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant. 2013;13(10):2530-2539.
11. Liszewski MK, Kolev M, Le Friec G, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):11431157.
12. Hajishengallis G, Lambris JD. Crosstalk pathways between Tolllike receptors and the complement system. Trends Immunol. 2010;31(4):154-163.
13. Morrison DC, Kline LF. Activation of the classical and properdin pathways of complement by bacterial lipopolysaccharides (LPS). J Immunol. 1977;118(1):362-368.
14. Klos A, Tenner AJ, Johswich K-O, Ager RR, Reis ES, Köhl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46(14):2753-2766.
15. Thome JJC, Yudanin N, Ohmura Y, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell. 2014;159(4):814-828.
16. Brodde MF, Kehrel BE. Markers of blood cell activation and complement activation in factor VIII and von Willebrand factor concentrates. Transfus Med Hemother. 2010;37(4):175-184.
17. Soltis RD, Hasz D, Morris MJ, Wilson ID. The effect of heat inactivation of serum on aggregation of immunoglobulins. Immunology. 1979;36(1):37-45.
18. Dustin ML. Complement receptors in myeloid cell adhesion and
Haematologica | 108 June 2023 1588 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
phagocytosis. Microbiol Spectr. 2016;4(6):10.
19. Arbore G, West EE, Spolski R, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016;352(6292):aad1210.
20. Nataf S, Davoust N, Ames RS, Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. J Immunol. 1999;162(7):4018-4023.
21. Gutzmer R, Lisewski M, Zwirner J, et al. Human monocytederived dendritic cells are chemoattracted to C3a after up-regulation of the C3a receptor with interferons. Immunology. 2004;111(4):435-443.
22. Werfel T, Kirchhoff K, Wittmann M, et al. Activated human T lymphocytes express a functional C3a receptor. J Immunol. 2000;165(11):6599-6605.
23. Camous L, Roumenina L, Bigot S, et al. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117(4):1340-1349.
24. Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112(5):1759-1766.
25. Peyvandi F, Mannucci PM, Garagiola I, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054-2064.
26. Basilico F, Nardini I, Mori F, et al. Characterization of factor VIII pharmaceutical preparations by means of MudPIT proteomic approach. J Pharm Biomed Anal. 2010;53(1):50-57.
27. Sonntag J, Emeis M, Vornwald A, Strauss E, Maier RF. Complement activation during plasma production depends on the apheresis technique. Transfus Med. 1998;8(3):205-208.
28. Burnouf T, Eber M, Kientz D, Cazenave J-P, Burkhardt T. Assessment of complement activation during membranebased plasmapheresis procedures. J Clin Apher. 2004;19(3):142-147.
29. Kemper C, Köhl J. Novel roles for complement receptors in T cell regulation and beyond. Mol Immunol. 2013;56(3):181-190.
30. Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol. 2015;194(8):3542-3548.
31. Karasu E, Demmelmaier J, Kellermann S, et al. Complement C5a induces pro-inflammatory microvesicle shedding in severely injured patients. Front Immunol. 2020;11:1789.
32. Miller RA, Gartner S, Kaplan HS. Stimulation of mitogenic responses in human peripheral blood lymphocytes by lipopolysaccharide: serum and T helper cell requirements. J Immunol. 1978;121(6):2160-2164.
33. Zhang X, Kimura Y, Fang C, et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110(1):228-236.
34. Li K, Fazekasova H, Wang N, et al. Expression of complement components, receptors and regulators by human dendritic cells. Mol Immunol. 2011;48(9-10):1121-1127.
35. Li K, Fazekasova H, Wang N, et al. Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology. 2012;217(1):65-73.
36. Jacquier-Sarlin MR, Gabert FM, Villiers MB, Colomb MG. Modulation of antigen processing and presentation by covalently linked complement C3b fragment. Immunology. 1995;84(1):164-170.
37. Rayes J, Ing M, Delignat S, et al. Complement C3 is a novel modulator of the anti-factor VIII immune response. Haematologica. 2018;103(2):351-360.
38. Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28(3):425-435.
39. Lubbers R, van Essen MF, van Kooten C, Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. 2017;188(2):183-194.
40. Gouw SC, van der Bom JG, van den Marijke Berg H. Treatmentrelated risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood. 2007;109(11):4648-4654.
41. Eckhardt CL, van der Bom JG, van der Naald M, Peters M, Kamphuisen PW, Fijnvandraat K. Surgery and inhibitor development in hemophilia A: a systematic review. J Thromb Haemost. 2011;9(10):1948-1958.
42. Bangs SC, Baban D, Cattan HJ, Li CK-F, McMichael AJ, Xu X-N. Human CD4+ memory T cells are preferential targets for bystander activation and apoptosis. J Immunol. 2009;182(4):1962-1971.
43. Di Genova G, Savelyeva N, Suchacki A, Thirdborough SM, Stevenson FK. Bystander stimulation of activated CD4+ T cells of unrelated specificity following a booster vaccination with tetanus toxoid. Eur J Immunol. 2010;40(4):976-985.
44. Boyman O. Bystander activation of CD4+ T cells. Eur J Immunol. 2010;40(4):936-939.
45. Whelan SFJ, Hofbauer CJ, Horling FM, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. 2013;121(6):1039-1048.
46. Lacroix-Desmazes S, Misra N, Bayry J, Mohanty D, Kaveri SV, Kazatchkine MD. Autoantibodies to factor VIII. Autoimmun Rev. 2002;1(1-2):105-110.
47. Meunier S, Menier C, Marcon E, Lacroix-Desmazes S, Maillère B. CD4 T cells specific for factor VIII are present at high frequency in healthy donors and comprise naïve and memory cells. Blood Adv. 2017;1(21):1842-1847.
Haematologica | 108 June 2023 1589 ARTICLE - C3a enhances immune responses towards FVIII E. Ringler al.
a LYSA study
anaplastic
cell lymphoma
David Sibon,1,2* Bettina Bisig,3* Christophe Bonnet,4 Elsa Poullot,2,5 Emmanuel Bachy,6 Doriane Cavalieri,7 Virginie Fataccioli,2,5 Cloé Bregnard,3 Fanny Drieux,8 Julie Bruneau,9 François Lemonnier,1,2 Aurélie Dupuy,2 Céline Bossard,10 Marie Parrens,11 Krimo Bouabdallah,12 Nicolas Ketterer,13 Grégoire Berthod,14 Anne Cairoli,15 Gandhi Damaj,16 Olivier Tournilhac,7 JeanPhilippe Jais,17 Philippe Gaulard2,5# and Laurence de Leval3#
1Lymphoid Malignancies Department, Henri-Mondor University Hospital, Assistance Publique-Hôpitaux de Paris (AP-HP), Créteil, France; 2Faculty of Medicine and Health, Campus Henri Mondor, Paris-Est Créteil University and INSERM U955, Créteil, France; 3Institute of Pathology, Department of Laboratory Medicine and Pathology, Lausanne University Hospital and Lausanne University, Lausanne, Switzerland; 4Hematology Department, Liège University Hospital, Liège, Belgium; 5Department of Pathology, Henri Mondor University Hospital, Créteil, France; 6Hematology Department, Lyon-Sud University Hospital, Lyon, France; 7Hematology Department, Clermont-Ferrand University Hospital, Clermont-Ferrand, France; 8Pathology Department, Henri Becquerel Cancer Center, Rouen, France; 9Pathology Department, Necker University Hospital, Paris, France; 10Pathology Department, Nantes University Hospital, Nantes, France; 11Pathology Department, Bordeaux University Hospital, Bordeaux, France; 12Hematology Department, Bordeaux University Hospital, Bordeaux, France; 13Clinique Bois-Cerf, Lausanne, Switzerland; 14Hospital Center for Valais Romand (CHVR), Martigny Hospital, Martigny, Switzerland; 15Service of Hematology, Department of Oncology, Lausanne University Hospital and Lausanne University, Lausanne, Switzerland; 16Institut d'Hématologie de Basse-Normandie, Caen University Hospital, Caen, France and 17Department of Biostatistics, Necker University Hospital, Paris, France
*DS and BB contributed equally as co-first authors #PG and LdL contributed equally as co-senior authors
Abstract
Correspondence: D. Sibon
david.sibon@aphp.fr
L. de Leval
Laurence.DeLeval@chuv.ch
Received: May 21, 2022.
Accepted: September 7, 2022.
Prepublished: December 1, 2022.
https://doi.org/10.3324/haematol.2022.281442
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
ALK-negative anaplastic large cell lymphoma (ALCL) comprises subgroups harboring rearrangements of DUSP22 (DUSP22R) or TP63 (TP63-R). Two studies reported 90% and 40% 5-year overall survival (OS) rates in 21 and 12 DUSP22-R/TP63not rearranged (NR) patients, respectively, making the prognostic impact of DUSP22-R unclear. Here, 104 newly diagnosed ALK-negative ALCL patients (including 37 from first-line clinical trials) from the LYSA TENOMIC database were analyzed by break-apart fluorescence in situ hybridization assays for DUSP22-R and TP63-R. There were 47/104 (45%) DUSP22-R and 2/93 (2%) TP63-R cases, including one DUSP22-R/TP63-R case. DUSP22-R tumors more frequently showed CD3 expression (62% vs. 35%, P=0.01), and less commonly a cytotoxic phenotype (27% vs. 82%; P<0.001). At diagnosis, DUSP22R ALCL patients more frequently had bone involvement (32% vs. 13%, P=0.03). The patient with DUSP22-R/TP63-R ALCL had a rapidly fatal outcome. After a median follow-up of 4.9 years, 5-year progression-free survival (PFS) and OS rates of 84 patients without TP63-R treated with curative-intent anthracycline-based chemotherapy were 41% and 53%, respectively. According to DUSP22 status, 5-year PFS was 57% for 39 DUSP22-R versus 26% for 45 triple-negative (DUSP22-NR/TP63-NR/ALK-negative) patients (P=0.001). The corresponding 5-year OS rates were 65% and 41%, respectively (P=0.07). In multivariate analysis, performance status and DUSP22 status significantly affected PFS, and distinguished four risk groups, with 4-year PFS and OS ranging from 17% to 73% and 21% to 77%, respectively. Performance status but not DUSP22 status influenced OS. The use of brentuximab vedotin in relapsed/refractory patients improved OS independently of DUSP22 status. Our findings support the biological and clinical distinctiveness of DUSP22R ALK-negative ALCL. Its relevance to outcome in patients receiving frontline brentuximab vedotin remains to be determined.
Haematologica | 108 June 2023 1590 ARTICLE - Non-Hodgkin Lymphoma
ALK-negative
large
with DUSP22 rearrangement has distinctive disease characteristics with better progression-free survival:
Introduction
Anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma (ALCL) is one of the four ALCL entities recognized in the current World Health Organization (WHO) classification of lymphoid neoplasms. It is a systemic disease entity defined as a CD30-positive T-cell neoplasm that is not reproducibly distinguishable on morphological grounds from ALK-positive ALCL but lacks ALK protein expression.1 Before 2017, ALK-negative ALCL was listed as a provisional entity, because of overlapping features with CD30-positive peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS), and the lack of established diagnostic criteria. Improved criteria for routine diagnostic practice, together with results from several studies suggesting distinguishing molecular features, led to the validation of ALKnegative ALCL as a definitive entity.1,2
Multiple studies over the past years have highlighted the heterogeneity of ALK-negative ALCL, and emphasized that this entity is not merely defined by the lack of ALK gene fusions, but comprises a heterogeneous genomic landscape including subgroups harboring DUSP22 or TP63 rearrangements (DUSP22-R or TP63-R) or lacking both (DUSP22NR/TP63-NR/ALK-negative, referred to as triple-negative ALCL). Other recurrent alterations consist of somatic mutations of JAK1, STAT3 or MSC, the expression of ERBB4aberrant transcripts, or a deregulated BATF3/IL-2R module.3-7 In particular, it has been shown that ALK-negative ALCL with DUSP22-R is characterized by a distinct gene expression signature, recurrent MSC mutations, lack of STAT3 activation and DNA hypomethylation.6,8 For these reasons, the recently released International Consensus Classification of lymphoid neoplasms, but not as yet the 5th Edition of the WHO-HAEM classification, considers DUSP22-R ALCL as a distinct genomic subtype.9,10
With conventional therapy, 5-year overall survival (OS) of ALK-negative ALCL patients is approximately 50%.11–15 It has been suggested that DUSP22-R could impact this survival rate. In the first clinical report from a multi-institution US study, the 5-year OS of 21 patients with DUSP22-R/TP63NR ALK-negative ALCL was 90%. Later on, a similar favorable outcome was reported in five patients in a Danish study (5-year OS, 80%) and in four patients from Spain (5year OS, 100%).16,17 However, in another recent work from the British Columbia Cancer Agency (BCCA) database, the 5year OS of 12 patients with DUSP22-R/TP63-NR ALKnegative ALCL was 40%.18 Thus, the prognostic impact of DUSP22-R in ALK-negative ALCL is currently unclear. The National Comprehensive Cancer Network guidelines suggest that treatment of the DUSP22-R subgroup according to the ALK-positive ALCL algorithm may be considered.19 However, this could lead to undertreating patients if the prognosis of DUSP22-R is not as favorable as expected.
In this retrospective study of 104 patients with ALK-negative
ALCL from the TENOMIC database of the Lymphoma Study Association (LYSA), we analyzed the pathological characteristics, clinical features, and outcomes of patients according to DUSP22 and TP63 status.
Methods
Patients and samples
Patients with ALK-negative ALCL diagnosed between January 2001 and January 2020 were retrieved from the TENOMIC database, the translational T-cell lymphoma research consortium of the LYSA. Thirty-seven patients had been enrolled in first-line clinical trials (26 Ro-CHOP, 8 AATT, 3 ECHELON-2 studies), and six in the TOTAL study for relapsed/refractory patients, the results of which have been reported,20-23 and nine patients were from a previous study.24 Other patients had been treated in routine care. Inclusion criteria required availability of diagnostic tissue (or existing documentation of a DUSP22 fluorescence in situ hybridization [FISH] result), and of clinical data including treatment and follow-up. Among the cases for which DUSP22 FISH was performed secondarily, we recorded a failure in five cases. These cases have not been included in the series. Special attention was paid in order to exclude patients with primary cutaneous ALCL. Diagnostic histological slides were reviewed by at least two expert pathologists and clinical data were collected (details are provided in the Online Supplementary Appendix). The study was approved by the ethics committee of the TENOMIC program (Comité de Protection des Personnes Ile-de-France IX 08-009).
Fluorescence in situ hybridization
Break-apart FISH assays to explore rearrangements of DUSP22/IRF4 and TP63 were performed on formalin-fixed paraffin-embedded tissue sections, using laboratory-developed probes,25 or commercial probes (ZytoLight SPEC IRF4, DUSP22 Dual Color Break Apart Probe [ZytoVision GmbH, Bremerharven, Germany]; and TP63 Split FISH Probe [Abnova, Taipei, Taiwan]), as previously described.26 At least 50 tumor nuclei were evaluated. The cutoff to consider a rearrangement was ≥10% of rearranged nuclei. Copy gains or losses of the explored loci were recorded qualitatively for rearranged and non-rearranged alleles.
Statistical analyses
The statistical analyses are described in the Online Supplementary Appendix.
Results
Patients’ and disease characteristics
In total, 104 ALK-negative ALCL patients newly diagnosed
Haematologica | 108 June 2023 1591 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
between January 2001 and January 2020 were analyzed, including 37 patients from first-line clinical trials and 67 patients treated in routine care. Baseline patients’ and disease characteristics did not differ significantly between patients included in first-line clinical trials and the others (Online Supplementary Table S1). At diagnosis, the median age of the 104 patients was 60 years (range, 39-86), 74% were male, 36% had a performance status (PS) ≥2, 72% had stage 3-4 disease, bone was the most frequently involved extranodal site, and the International Prognostic Index (IPI) score was equally distributed across the four risk groups (Table 1). Ten patients who had skin involvement had advanced stage disease and not just involvement of a draining lymph node. Most patients (97/104, 93%) were treated frontline with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP)/CHOP-like regimens, and seven patients received non-curative intent care.
The diagnostic samples were mostly lymph nodes (91/104 cases, 88%), and the majority were surgical biopsies. The other tissues examined were from the nasopharynx and tonsil (3/104), liver (3/104), mediastinum (1/104), and other extranodal organs (parotid, lung, intestine, maxillary sinus) (6/104). In all cases the tumor consisted of large cells strongly positive for CD30 and negative for ALK protein expression. Other immunophenotypic features are summarized in Table 2. Expression of pan-T-cell antigens was variably detected; the most commonly expressed was CD2 (66/87, 76%) followed by CD3 (49/104, 47%), CD5 (36/97, 37%) and CD7 (11/75, 15%). Expression of at least one cytotoxic molecule was demonstrated in 45/101 (45%) cases. Co-expression of EMA was common (41/87 cases, 47%). CD4 and CD8 were expressed in 72/97 (74%) and 11/89 (12%) cases, respectively. Phospho-STAT3 (pSTAT3) was positive in 21/44 (48%) samples.
Fluorescence in situ hybridization results
The DUSP22 locus was rearranged in 47/104 cases (45%), with several distinct hybridization patterns observed (Figure 1). Among DUSP22-R cases, 38/47 (81%) showed a classical break-apart pattern, i.e. one normal fusion signal and one red and one green separated (split) signals representing the rearranged allele (Figure 1C); or variant classical patterns, comprising several pairs of separated red and green signals. This group included three cases in which two rearranged alleles were present in the absence of any non-rearranged allele, reflecting biallelic rearrangements (Figure 1D). The remaining 9/47 (19%) DUSP22-R cases featured “atypical” hybridization patterns, consisting of at least one isolated green (3’) signal, in the absence of isolated red (5’) signals (Figure 1E); in one of these cases, tight clusters of more than ten green signals were detected, in addition to fusion signals (Figure 1F); in another case, only one or two isolated green signals could be seen, without any detectable fusion signal.
FISH assay for TP63 was contributive in 93/99 cases, indicating a failure rate of 6%, and could not be performed in five cases (no material available). The TP63 locus was rearranged in 2/93 cases (2%), including one case with dual DUSP22-R and TP63-R. Both TP63-R cases showed a “classical” break-apart pattern, with a relatively small distance between the separated red and green signals of the rearranged allele (Figure 2), consistent with an inv(3)(q26q28) resulting in the TBL1XR1::TP63 fusion, although dual fusion FISH probes were not tested to prove this. Among the samples lacking structural alterations of the explored loci, low-level (3 to 4) (Figure 1A) or highlevel (≥5) copy gains of DUSP22 were observed in the majority of cases (23/57 [40%] and 15/57 [26%], respectively), including three samples with tight clusters of up to 20 fusion signals, consistent with DUSP22 locus amplification (Figure 1B). Copy gains of TP63 were mostly of low level (47/91, 52%), with 4/91 samples (4%) showing up to five copies per nucleus.
Distinctive pathological and clinical features according to DUSP22 status
A morphological spectrum was observed irrespective of DUSP22 rearrangement, with marked overlap between the two genomic groups (Online Supplementary Figure S1). Although doughnut-type cells were essentially seen in the DUSP22-R subgroup, hallmark-type cells were otherwise seen as a prominent or more discrete component of the tumor cell population irrespective of the genomic status in most cases. Marked pleomorphism was seen in some cases of both DUSP22-R and DUSP22-NR.
Considering the immunophenotype of the neoplastic cells (Table 2), CD3 and CD2 were more often positive among DUSP22-R cases than in DUSP22-NR tumors (62% vs. 35%, P=0.01; and 87% vs. 67%, P=0.044 of the cases, respectively). The expression of other T-cell markers (CD4, CD5, CD7, CD8) was otherwise not significantly different between the two groups. Remarkably, the distribution of the tumors according to CD4 and CD8 expression was almost identical in the two subgroups, the usual profile being CD4+ CD8– (71% and 67% of the cases in DUSP22-R and DUSP22-NR cases, respectively), followed by CD4– CD8–(19% of the cases in both subgroups) and CD4– CD8+ (9% and 10% of the DUSP22-R and DUSP22-NR cases, respectively). Overall, there were only three CD4+ CD8+ cases. Conversely, the two genetic subgroups differed markedly in the frequency of expression of cytotoxic proteins, EMA and pSTAT3. Expression of TIA1, granzyme B or perforin was seen in 11-13% of the DUSP22-R group versus 40-63% of DUSP22-NR cases. Overall, considering the cases tested for all three cytotoxic markers, 8/30 (27%) of DUSP22-R cases versus 37/45 (82%) of DUSP22-NR cases (P<0.001) exhibited a cytotoxic profile, i.e. expressed at least one cytotoxic marker. Similarly, EMA was significantly less ex-
Haematologica | 108 June 2023 1592 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
ALK: anaplastic lymphoma kinase; ALCL: anaplastic large cell lymphoma; PET: positron emission tomography; CT: computed tomography; IPI: International Prognostic Index; CHOP: cyclophosphamide, doxorubicin, vincristine, and prednisone; CHOEP: CHOP + etoposide; BV: brentuximab vedotin; CH(E)P: cyclophosphamide, doxorubicin, (etoposide), prednisone; ACVBP: doxorubicin, cyclophosphamide, vindesine, bleomycin, prednisone; autoSCT: autologous stem-cell transplantation; alloSCT: allogeneic stem-cell transplantation. Statistically significant value shown in bold.
Clinical features at diagnosis All patients DUSP22-non rearranged ALK-negative ALCL DUSP22-rearranged ALK-negative ALCL P Number 104 57 47 Period of diagnosis 2001-2020 2001-2020 2004-2019 Age, years Median (range) 60 (39-86) 61 (39-85) 60 (40-86) >60, N (%) 53/104 (51) 29/57 (51) 24/47 (51) 1 Male, N (%) 77/104 (74) 39/57 (68) 38/47 (81) 0.225 Performance status ≥2, N (%) 37/103 (36) 23/57 (40) 14/46 (30) 0.403 Staging at diagnosis, N (%) 0.701 PET 84/100 (84) 45/55 (82) 39/45 (87) CT 16/100 (16) 10/55 (18) 6/45 (13) Ann Arbor stage, N (%) 0.862 (for 1-2 vs. 3-4) 1 8/104 (8) 3/57 (5) 5/47 (11) 2 21/104 (20) 12/57 (21) 9/47 (19) 3 20/104 (19) 16/57 (28) 4/47 (8) 4 55/104 (53) 26/57 (46) 29/47 (62) Involved site (any), N (%) Bone 22/103 (21) 7/56 (13) 15/47 (32) 0.031 Liver 17/103 (17) 8/56 (14) 9/47 (19) 0.692 Bone marrow 13/103 (13) 7/56 (13) 6/47 (13) 1 Lung 13/103 (13) 5/56 (9) 8/47 (17) 0.350 Spleen 12/103 (12) 5/56 (9) 7/47 (15) 0.528 Soft tissue 12/103 (12) 10/56 (18) 2/47 (4) 0.067 Skin 10/103 (10) 3/56 (5) 7/47 (15) 0.196 Gastrointestinal tract 7/103 (7) 4/56 (7) 3/47 (6) 1 Parotid 4/103 (4) 1/56 (2) 3/47 (6) 0.490 Nasopharynx 3/103 (3) 1/56 (2) 2/47 (4) 0.877 Tonsil 2/103 (2) 1/56 (2) 1/47 (2) 1 Sinus 2/103 (2) 1/56 (2) 1/47 (2) 1 Thyroid 1/103 (1) 0/56 (0) 1/47 (2) 0.930 Adrenal 1/103 (1) 0/56 (0) 1/47 (2) 0.930 Blood 1/103 (1) 1/56 (2) 0/47 (0) 1 Ascites 1/103 (1) 0/56 (0) 1/47 (2) 0.930 Pleura 0/103 (0) 0/56 (0) 0/47 (0) Extranodal site >1, N (%) 29/104 (28) 15/57 (26) 14/46 (30) 0.862 Elevated lactate dehydrogenase, N (%) 58/103 (56) 30/57 (53) 28/46 (61) 0.523 b2-microglobulin ≥3 mg/L, N (%) 24/55 (44) 17/34 (50) 7/21 (33) 0.352 IPI score, N (%) 0.358 0-1 29/103 (28) 13/57 (23) 16/46 (35) 2 24/103 (23) 16/57 (28) 8/46 (17) 3 26/103 (25) 16/57 (28) 10/46 (22) 4-5 24/103 (23) 12/57 (21) 12/46 (26) Patients in first-line clinical trials, N (%) 37/104 (36) 24/57 (42) 13/47 (28) 0.185 Primary therapy, N (%) 0.292 CHOP 45/104 (43) 23/57 (40) 22/47 (47) CHOEP 24/104 (23) 13/57 (23) 11/47 (23) Romidepsin-CHOP 10/104 (10) 9/57 (16) 1/47 (2) BV-CH(E)P 6/104 (6) 3/57 (5) 3/47 (6) Mini-CHOP 7/104 (7) 2/57 (4) 5/47 (11) ACVBP 5/104 (5) 3/57 (5) 2/47 (4) Non-curative care 7/104 (7) 4/57 (7) 3/47 (6) Consolidative transplantation 0.218 AutoSCT 14/104 (13) 5/57 (9) 9/47 (19) AlloSCT 5/104 (5) 2/57 (4) 3/47 (6) Auto-mini-alloSCT tandem 1/104 (1) 1/57 (2) 0/47 (0)
Haematologica | 108 June 2023 1593 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
Table 1. Patients’ and disease characteristics.
*Taking into consideration only fully conclusive cases, either negative for the three cytotoxic molecules analyzed, or positive for at least one of them. NR: not rearranged; R: rearranged; ALK: anaplastic lymphoma kinase; ALCL: anaplastic large cell lymphoma. Statistically significant values shown in bold. N+/N: number positive/number tested.
pressed in DUSP22-R cases, being positive in 13% versus 73% of DUSP22-NR cases (P<0.001). Phospho-STAT3 was positive in only 2/20 (10%) DUSP22-R samples versus 19/24 (79%) of DUSP22-NR cases (P<0.001).
Comparing the characteristics of DUSP22-R and DUSP22NR patients (Table 1), there was no significant difference in median age or sex, and IPI score was equally distributed. The only statistically significant difference was bone involvement, which was more frequent in DUSP22-R cases (32% vs. 13%, P=0.031). The two groups of patients did not differ regarding involvement of other extranodal sites. Of note, the frequency of DUSP22-R was 35% (13/37) for patients included in clinical trials and 51% (34/67) for patients treated routinely (P=0.185) (Online Supplementary Table S1).
After a median follow-up of 5 years, the 5-year PFS and OS of the 104 patients were 36% and 50%, respectively (Figure 3A, B). According to DUSP22 status, 5-year PFS was 48% versus 25% for 47 DUSP22-R and 57 DUSP22-NR patients, respectively (P=0.025) (Figure 3C), and 5-year OS was 58% versus 44% for DUSP22-R and DUSP22-NR patients, respectively (P=0.2) (Figure 3D).
Treatment response, survival, and prognostic factors
Analyses of treatment response, survival, and prognostic factors were restricted to patients for whom FISH in-
formation was complete, who had a confirmed TP63-NR status, and who were treated with curative intent with front-line anthracycline-based chemotherapy. This set consisted of 84 patients (39 DUSP22-R/TP63-NR and 45 triple-negative ALCL). The patients’ and disease characteristics are shown in Online Supplementary Table S2, and their immunophenotypic characteristics are presented in Online Supplementary Table S3
Four patients (1 DUSP22-R and 3 DUSP22-NR) were not evaluable for response because of early death (mainly due to infections). The overall response rate and the complete response rate were 75% and 67%, respectively, without significant difference between triple-negative and DUSP22-R/TP63-NR patients (Online Supplementary Table S4).
The median follow-up of the 84 patients was 4.9 years (range, 0.9 to 10 years). Their 2- and 5-year PFS rates were 45% (95% CI: 36%-57%) and 41% (95% CI: 31%-53%), respectively, and the 2- and 5-year OS rates were 67% (95% CI: 57%-78%) and 53% (95% CI: 42%-66%), respectively.
PFS rates were significantly higher in DUSP22-R/TP63-NR patients than in triple-negative patients (2-year PFS, 67% vs . 26%; 5-year PFS, 57% vs . 26%, P =0.001) (Figure 4A).
However, the OS rates were not significantly different between DUSP22 -R/ TP63 -NR and triple-negative patients (2-year
OS, 74% vs. 60%; 5-year OS, 65% vs. 41%,
All patients (N=104) DUSP22-NR ALK-negative ALCL (N=57) DUSP22-R ALK-negative ALCL (N=47) P CD30, N+/N 104/104 57/57 47/47 1 ALK, N+/N 0/104 0/57 0/47 1 T-cell antigens, N+/N (%) CD3 49/104 (47) 20/57 (35) 29/47 (62) 0.01 CD5 35/97 (36) 17/53 (32) 19/44 (43) 0.296 CD2 66/87 (76) 33/49 (67) 33/38 (87) 0.044 CD7 11/75 (15) 7/40 (18) 4/35 (11) 0.528 CD4 72/97 (72) 38/50 (76) 34/47 (72) 0.817 CD8 11/89 (12) 5/45 (11) 6/44 (11) 0.758 CD4+ CD8- 60/87 (69) 28/42 (64) 32/45 (69) 0.817 CD4- CD8- 16/87 (18) 8/42 (19) 8/45 (18) 1 CD4- CD8+ 8/87 (9) 4/42 (10) 4/45 (9) 1 CD4+ CD8+ 3/87 (3) 2/42 (5) 1/45 (2) 0.608 EMA, N+/N (%) 41/87 (47) 36/49 (73) 5/38 (13) <0.0001 Cytotoxic markers, N+/N (%) TIA1 21/78 (27) 16/40 (40) 5/38 (13) 0.01 Granzyme B 26/92 (28) 21/48 (44) 5/44 (11) 0.001 Perforin 31/76 (41) 27/43 (63) 4/33 (12) <0.0001 Cytotoxic profile* 45/75 (60) 37/45 (82) 8/30 (27) <0.0001 pSTAT3, N+/N (%) 21/44 (48) 19/24 (79) 2/20 (10) <0.001
P=0.07)
Haematologica | 108 June 2023 1594 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
Table 2. Immunophenotypic characteristics of the 104 tumors.
(Figure 4B). Importantly, PFS and OS were similar for patients included or not in fi rst-line clinical trials ( Online Supplementary Figure S2).
Clinical and laboratory features were subjected to univariate analyses to evaluate their impact on PFS and OS ( Online Supplementary Table S5). PS (Figure 4C, D), b 2microglobulin level, granzyme B and perforin expression signi fi cantly infl uenced PFS and OS, whereas DUSP22 status and cytotoxic profile affected only PFS. Only PS (01 vs ≥ 2) and DUSP22-R/DUSP22-NR status were retained for multivariate analysis because of missing data for the
other factors. Both PS and DUSP22 status significantly affected PFS, but only PS remained significant for OS (Table 3). These two variables delineated four risk groups (Figure 4E, F): DUSP22-R/TP63-NR and PS 0-1, with 4-year PFS and OS rates of 73% and 77%, respectively; DUSP22R/TP63-NR and PS ≥2, with 4-year PFS and OS rates of 27% and 29%, respectively; triple-negative and PS 0-1, with 4-year PFS and OS rates of 33% and 62%, respectively; and triple-negative and PS ≥2, with 4-year PFS and OS rates of 17% and 21%, respectively ( P <0.001 for PFS and P=0.001 for OS).
Figure 1. DUSP22 fluorescence in situ hybridization patterns. The range of fluorescence in situ hybridization (FISH) patterns observed for the DUSP22 locus (right column: ZytoLight SPEC IRF4, DUSP22 Dual Color Break Apart Probe, ZytoVision) is illustrated, with the corresponding hematoxylin & eosin (H&E) images (left column). DUSP22 non-rearranged cases (A, B) included a majority of samples showing copy gains (A: 3 to 4 fusion signals per nucleus), and a few characterized by an amplification of the DUSP22 locus (B: tight clusters of fusion signals). Among DUSP22rearranged cases (C-F), approximately 80% showed a classical break-apart pattern of the DUSP22 locus or variants thereof (C: separated red and green signals for the rearranged allele, with an additional fusion signal representing the non-rearranged allele; D: biallelic rearrangements), while 20% featured various atypical break-apart patterns (E: rearrangement with deletion of the red 5’ portion of the probe, resulting in an isolated green 3’ signal, in addition to the non-rearranged allele; F: variant of the pattern shown in E, presenting tight clusters of green 3’ signals, in addition to fusion signals representing the non-rearranged allele). All H&E images were taken at an original x400 magnification and the FISH images at x630.

Haematologica | 108 June 2023 1595 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al. A B C D E F
Post-progression survival
Of the 84 patients, 43 (14 DUSP22-R and 29 triplenegative) progressed or relapsed after frontline treatment. From this event, the 4-year OS (OS2) was 29% (21% in DUSP22-R/TP63-NR vs. 34% in triple-negative patients, P=0.62) (Figure 5A). Information on salvage treatment was retrieved for 40/43 patients. The 4-year OS2 was 44% for the 27 patients who received brentuximab vedotin (BV) at relapse (only one patient had previously received frontline BV) versus 0% for the 13 patients who received standard treatment, mainly cytarabine-based regimens or benda-
mustine (P<0.001) (Figure 5B). Figure 5C illustrates OS2 according to DUSP22 status and BV as salvage treatment. In multivariate analysis of these two parameters, only BV affected OS2 (P<0.001; HR=0.119, 95% CI: 0.041-0.343). Indeed, when restricting the OS2 analysis to the patients who received BV as salvage treatment, there was no significant difference according to DUSP22 status (Figure 5D).
Characteristics of the two patients with TP63rearranged ALK-negative anaplastic large cell lymphoma The patient with the dual TP63 and DUSP22 rearrangement
Figure 2. ALK-negative anaplastic large cell lymphoma with dual TP63 and DUSP22 rearrangement. (A, B) The tumor comprises cohesive sheets of atypical lymphoid cells including anaplastic-type “hallmark” cells (hematoxylin & eosin, original magnifications x400 and x800); (C-J) on immunohistochemical stains the neoplastic cells are strongly CD30+ (C), CD3+ (D), CD5+ (E), CD7– (F), CD4+ (G), CD8– (H), with a high Ki67 proliferation index (I) and negative for TIA-1 (J) (all immunoperoxidase; original magnification x400); (K-L) representative nuclei from the fluorescence in situ hybridization assays for DUSP22 (K) and TP63 rearrangement (L) showing a pattern indicative of a break for the two tested loci (original magnification x630).

Haematologica | 108 June 2023 1596 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al. A B C D E F G H K L I J
was a 43-year-old man presenting with cervical lymphadenopathy and an IPI score of 0. The tumor consisted of diffuse sheets of medium-sized to large atypical lymphoid cells with frequently reniform or horseshoe-shaped nuclei (Figure 2). In addition to being positive for CD30, the tumor cells were CD3+, CD4+, CD5+, CD7–, CD8–, EMA–, TIA-1–, granzyme B–, perforin–, pSTAT3– and p63+. Re-biopsy at relapse 1 year later showed identical features. The patient with an isolated TP63 gene rearrangement was a 52-year-old woman with an IPI score of 2 (Ann Arbor stage 3 and elevated lactate dehydrogenase). A lymph node biopsy showed cohesive sheets of large cells with
oval nuclei and prominent nucleoli, associated with diffuse interstitial fibrosis (Online Supplementary Figure S3).
The neoplastic cells were strongly positive for CD30, CD2+, CD3–, CD4+, CD5–, CD8–, TIA1+, granzyme B+, perforin+ with nuclear p63 protein expression.
Both patients reached CR after CHOP (the DUSP22R/TP63-R case) or CHOEP (CHOP with etoposide) (the TP63-R case) regimens and underwent consolidative autologous stem-cell transplantation. They both relapsed after transplantation: the patient with a dual rearrangement died from lymphoma 5 months after relapse, and the other remains in CR more than 2 years after salvage treat-

Haematologica | 108 June 2023 1597 ARTICLE
R
D. Sibon
al. A B C D
Figure 3. Survival of the 104 patients with ALK-negative anaplastic large cell lymphoma. (A) Progression-free survival and (B) overall survival of the whole cohort. (C) Progression-free survival and (D) overall survival according to DUSP22 status. R: rearranged; NR: not rearranged.
- Impact of DUSP22-
in ALK-negative ALCL: a LYSA study
et
Figure 4. Survival of the 84 TP63 -non-rearranged patients treated with anthracycline-based chemotherapy with curative intent. (A) Progression-free survival (PFS) and (B) overall survival (OS) according to DUSP22 status. (C) PFS and (D) OS according to performance status. (E) PFS and (F) OS according to both factors. R: rearranged; NR: not rearranged; PS: performance status.

Haematologica | 108 June 2023 1598 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al. A B C D E F
Table 3. Parameters influencing progression-free survival and overall survival in multivariate analyses in 83 patients.

PFS: progression-free survival; OS: overall survival; HR: hazard ratio; 95% CI: 95% confidence interval; PS: performance status; NR: not rearranged.
Figure 5. Post-progression overall survival (OS2). Overall survival following relapse/progression (A) according to DUSP22 status, (B) according to brentuximab vedotin (BV) use at relapse/progression, (C) according to both parameters, and (D) when restricting the analysis to the patients who received BV as salvage treatment. R: rearranged; NR: not rearranged.
Parameter PFS OS P HR 95% CI P HR 95% CI PS ≥2 0.005 2.259 1.271-4.013 <0.001 3.024 1.593-5.741 DUSP22-NR 0.008 2.256 1.233-4.127 0.194 1.556 0.799-3.031
Haematologica | 108 June 2023 1599 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al. A B C D
ment with BV + gemcitabine and allogeneic stem-cell transplantation.
Discussion
We report here the clinical and pathological findings of 104 patients with ALK-negative ALCL according to DUSP22 status (47 DUSP22-R and 57 DUSP22-NR) and TP63 status (2 TP63-R and 91 TP63-NR), including 39 DUSP22-R/TP63NR and 45 triple-negative cases. This represents the largest such series published so far. The main conclusions of our study are: (i) DUSP22-R ALCL encompasses a spectrum of FISH patterns, has distinctive immunophenotypic features and more frequently involves bone; (ii) the 65% 5-year OS of DUSP22-R patients is intermediate between those previously documented in an US study (90%) and by the BCCA investigators (40%); (iii) both DUSP22 status and PS have independent impacts on PFS; (iv) OS was mainly affected by PS; and (v) OS2 was markedly improved by the use of BV as salvage treatment, without DUSP22 status having a significant influence on this post-progression survival.
With the comparison group (DUSP22-NR ALK-negative ALCL) consisting of 57 individuals, the DUSP22-R cases constituted 45% of our study population. Strikingly, this proportion is higher than in other studies from North America and Europe, in which the frequency of DUSP22-R has been reported to be between 18% and 30%.3,16-18 However, the mode of recruitment of samples and patients precludes conclusions being drawn regarding the relative prevalence of ALK-negative ALCL genomic subgroups. Of note, the distribution of DUSP22-R/DUSP22-NR cases was different among the 37 patients enrolled in first-line clinical trials (13/37 [35%] DUSP22-R, including 6/26 [23%] in the Ro-CHOP study) versus the others collected through the TENOMIC network (34/67 [51%]). Since all cases of ALK-negative ALCL patients from the clinical trials were included in this study when possible, they represent an “unbiased” group of cases and their characteristics in terms of DUSP22 status are much consistent with the existing literature, confirming the 30% prevalence of DUSP22-R in the multi-institution US study.3
There are several explanations for the relatively numerous DUSP22-R cases among the non-clinical trial patients in our study. The collection of patients’ data and samples through TENOMIC primarily aims at collecting high-quality data and cases of medical and scientific interest, which may be influenced by specific topics of interest such as the current project on ALCL with DUSP22-R.27 Moreover, the most active participants are referral centers with expert pathologists being consulted for unusual or difficult cases, or for ancillary techniques such as FISH. In addition, it is worth mentioning the use of cases from a former pub-
lication, among which a majority (7/9) harbored a DUSP22R.24 In fact, five of these cases, all DUSP22-R that had been coded as CD30-positive PTCL-NOS in that study because they did not fulfill the stringent immunophenotypic criteria originally used for the diagnosis of ALK-negative ALCL (i.e., requiring the expression of at least one cytotoxic molecule or EMA), became consistent with ALK-negative ALCL in the light of updated criteria developed later. We found only 2/93 (2%) TP63-R cases in our series, which is at the lower end of previously reported frequencies (28%) in ALK-negative ALCL.3,16,18 It might be argued that the exclusive use of a break-apart FISH probe to explore the TP63 locus may have missed cases harboring a TBL1XR1::TP63 intrachromosomal inversion, due to the small distance between the split signals in this context. Nonetheless, being aware of the risk of false negative results, the slides were examined very carefully, and we believe that the low prevalence of TP63-R truly reflects the biology of our cohort. On the other hand, cryptic TP63 rearrangements cannot formally be excluded, as recently described.28 These latter would however not have been detected in previously published series based on FISH assays.
A spectrum of DUSP22 FISH patterns was observed (Figure 1). In addition to extra copies of the intact (non-rearranged) DUSP22 locus, which could represent either specific gains or polysomy of chromosome 6, three DUSP22-NR cases featured a FISH pattern consistent with DUSP22 locus amplification. This observation has not previously been reported, and its biological consequence is unclear. The DUSP22 gene encodes a dual specificity phosphatase that functions as a tumor-suppressor gene by exerting an inhibitory effect on various signaling pathways.29,30 While it has been shown that DUSP22 gene rearrangements lead to the downregulation of the enzyme, it is questionable how an amplification could result in its silencing, unless the amplified allele encodes an altered, non-functional isoform. Alternatively, the pathogenic effect in such cases could be mediated by the amplification of another neighboring gene with an oncogenic function (e.g., IRF4).
Among DUSP22-R cases, we observed both the most classical break-apart FISH pattern and variants of it, including cases with biallelic rearrangements or extra copies of both the rearranged and non-rearranged alleles. Although details regarding the FISH patterns encountered are frequently missing in the literature (the result being commonly limited to binary information: rearranged or not), the classical break-apart pattern is the most frequently described one in the series and case reports published so far on DUSP22. In our cohort, however, approximately 20% of DUSP22-R cases were characterized by atypical hybridization patterns, featuring one or several extra copies of isolated green signals, suggesting a re-
Haematologica | 108 June 2023 1600 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
arrangement with subsequent deletion of the 5’ side of the locus (telomeric red probe) and preservation of its 3’ side (centromeric green probe). This configuration, which reflects an unbalanced translocation, has been recurrently described in earlier series of cutaneous CD30+ T-cell lymphoproliferations, when the gene believed to be involved in 6p25.3 locus rearrangements was IRF4, but it has been reported once in systemic ALK-negative ALCL.31,32 Nonetheless, in a case of lymphomatoid papulosis characterized by a similar atypical DUSP22 FISH pattern, Karai and colleagues could demonstrate by FISH that the partner locus of the translocation was at 7q32.3, similar to what has been described for the classical break-apart pattern.29,33
The immunohistochemistry results on our series are overall consistent with the range described in previous reports.3,18,34 In addition, we documented CD4 and CD8 expression profiles which were evaluated in the majority of cases (87/104) and were remarkably similar irrespective of DUSP22 status, being most commonly CD4+ CD8– (67% of the cases) or CD4– CD8– (21% of the cases). In addition, our findings confirm significant differences between DUSP22-R and DUSP22-NR cases in terms of cytotoxic profile. Of note, while confirming the lack of cytotoxic phenotype as a characteristic feature of DUSP22-R cases, we also found that a significant minority of these (8/30, 27%) expressed one or several cytotoxic marker(s), which is a higher proportion than the approximately 10% in previously reported series.3,18 EMA and pSTAT3 expression were also much less common in DUSP22-R cases, and there was less frequent CD3 positivity in DUSP22-NR ALCL.3,8,18 The case with dual DUSP22 and TP63 rearrangements (Figure 2) was CD3+ CD4+ CD8– EMA– pSTAT3– and non-cytotoxic. Similar findings have been reported in the other ALK-negative ALCL cases with that rare genomic configuration, suggesting that the immunophenotype is likely driven by the DUSP22 rearrangement in those tumors.35,36
We found that among ALCL patients treated with chemotherapy with curative intent, DUSP22-R was a significant determinant of improved PFS in both univariate and multivariate analyses, with 57% 5-year PFS in DUSP22-R/TP63NR versus 26% in triple-negative patients. In comparison, in the BCCA study, the 5-year PFS of 11 DUSP22-R/TP63NR patients treated with curative-intent chemotherapy was 44%.18 PFS was not reported in the US study.3 Unlike previous reports, the advantage in OS for our DUSP22R/TP63-NR patients compared to triple-negative patients (5-year OS: 65% vs. 41%, respectively) did not reach statistical significance. We also found that PS affected PFS and was the prominent factor affecting OS in multivariate analysis in our series. Indeed, we identified a low-risk group characterized by DUSP22-R and PS of 0-1, with a 4year PFS of 73% and 4-year OS of 77%. Conversely, pa-
tients with DUSP22-R and PS ≥2 had 4-year PFS and OS rates of 27% and 29%, respectively, demonstrating the major impact of PS on outcome. In a recent report from the International T-Cell Project, PS ≥2 was the factor with the strongest impact on PFS and OS in multivariate analysis with hazard ratios of 3.69 and 4.04, respectively, but genomic subtyping of these ALK-negative ALCL was not studied.15
BV has previously been shown to improve OS2 after progression/relapse of ALK-negative ALCL patients compared to historical controls.37,38 Here, we also confirm that OS2 was markedly improved by salvage treatment with BV, which was the main prognostic factor in multivariate analysis. Interestingly, we found no significant difference in OS2 according to DUSP22 status and an overall similarly good outcome in patients who received BV at relapse/progression in DUSP22-R/TP63-NR and triple-negative patients, suggesting that response to BV in relapsed/refractory patients is not influenced by DUSP22 status.
PFS rather than OS may better capture the prognostic impact of DUSP22-R since it is not influenced by salvage treatment, while OS analysis is more complex to interpret and should take into account potential differences in salvage treatment. It turned out that, at relapse/progression, 21/26 (81%) triple-negative patients but only 6/14 (43%) DUSP22-R patients received BV. Therefore, this imbalance could contribute to the absence of a significant difference in OS between DUSP22-R and DUSP22-NR patients. Despite limitations inherent to a retrospective study with unbalanced distribution of DUSP22 -R/ DUSP22 -NR patients, incomplete TP63 FISH data, and heterogeneity in first-line treatments, our findings support the biological and clinical distinctiveness of DUSP22 -R ALK-negative ALCL. Moreover, our results confi rm a better PFS of DUSP22 -R/ TP63 -NR cases compared to triple-negative ALCL, but clearly inferior to that of a historical series of ALK-positive ALCL patients.39 Of note, with the limitation of low statistical power of small groups, outcome did not differ according to first-line treatment (CHOP, CHOEP or BV-CH(E)P; data not shown), but only a small fraction of our patients received frontline BV. Given the benefi t of BV-CHP over CHOP in ALK-negative ALCL in the ECHELON-2 trial with an improved 5-year PFS (but not OS), BVCHP has become the standard of care for fi rst-line treatment of ALK-negative ALCL. 22 However, since genomic subtyping was not reported, its potential impact on the PFS difference observed between the BV-CHP and CHOP arms is unknown. Future studies will be necessary to clarify this point and the impact of DUSP22 status in newly diagnosed patients with ALK-negative ALCL treated with frontline BV.
Disclosures No conflicts of interest to disclose. Haematologica | 108 June 2023 1601 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
Contributions
DS collected and reviewed clinical data, analyzed data, designed the research, and wrote the manuscript. BB performed morphological diagnoses and FISH studies, analyzed data and wrote the manuscript; ChrB, EB, DC, FL, KB, NK, GB, AC, GD, and OT reviewed and interpreted clinical data. EP performed morphological diagnoses and FISH analyses. VF supported material and data acquisition and collected data. ChlB and AD performed FISH analyses. FD, JB, CélB, and MP performed morphological diagnoses. JPJ analyzed data and supervised the statistical analyses; PG and LdL performed morphological diagnoses, designed and sustained the research, collected and analyzed data, and wrote the manuscript.
Acknowledgments
The authors would like to thank Mrs Nadine Vailhen and Jacqueline Polyte from the LYSA pathology platform, and Dr Nathalie Piazzon and Mr Jean-Daniel Roman from the Institute of Pathology (Lausanne) for their administrative and technical assistance and for management of digital
References
1. Swerdlow S, Campo E, Harris N, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). IARC: Lyon 2017.
2. Attygalle AD, Cabeçadas J, Gaulard P, et al. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward –report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology. 2014;64(2):171-199.
3. Parrilla Castellar ER, Jaffe ES, Said JW, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473-1480.
4. Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27(4):516-532.
5. Scarfò I, Pellegrino E, Mereu E, et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood. 2016;127(2):221-232.
6. Luchtel RA, Zimmermann MT, Hu G, et al. Recurrent MSCE116K mutations in ALK-negative anaplastic large cell lymphoma. Blood. 2019;133(26):2776-2789.
7. Liang H-C, Costanza M, Prutsch N, et al. Super-enhancer-based identification of a BATF3/IL-2R module reveals vulnerabilities in anaplastic large cell lymphoma. Nat Commun. 2021;12(1):5577.
8. Luchtel RA, Dasari S, Oishi N, et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood. 2018;132(13):1386-1398.
9. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood. 2022;140(11):1229-1253.
10. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720-1748.
11. Savage KJ, Harris NL, Vose JM, et al. ALK- anaplastic large-cell
slides. The work was supported by the histopathology, immunopathology and FISH laboratories and the digital pathology unit of the Institute of Pathology of Lausanne. The authors wish to thank Dr Stefano Caruso from the Department of Pathology in Creteil for performing the statistical analyses on the pathological data. The work was presented in part at the 16th International Conference on Malignant Lymphoma (Lugano, 2021).
Funding
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Swiss National Foundation (SNF).
Data-sharing statement
Anonymized data can be made av ailable on request to the corresponding authors by independent researchers, with a collaborative agreement, through a standard process which includes an internal feasibility assessment and scientific review process by the LYSA.
lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood. 2008;111(12):5496-5504.
12. Schmitz N, Trümper L, Ziepert M, et al. Treatment and prognosis of mature T-cell and NK-cell lymphoma: an analysis of patients with T-cell lymphoma treated in studies of the German HighGrade Non-Hodgkin Lymphoma Study Group. Blood. 2010;116(18):3418-3425.
13. Sibon D, Fournier M, Brière J, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d’Étude des Lymphomes de l’Adulte trials. J Clin Oncol. 2012;30(32):3939-3946.
14. Ellin F, Landström J, Jerkeman M, Relander T. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: a study from the Swedish Lymphoma Registry. Blood. 2014;124(10):1570-1577.
15. Shustov A, Cabrera ME, Civallero M, et al. ALK-negative anaplastic large cell lymphoma: features and outcomes of 235 patients from the International T-Cell Project. Blood Adv. 2021;5(3):640-648.
16. Pedersen MB, Hamilton-Dutoit SJ, Bendix K, et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: a Danish cohort study. Blood. 2017;130(4):554-557.
17. Onaindia A, Villambrosía SG de, Prieto-Torres L, et al. DUSP22rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica. 2019;104(4):e158-e162.
18. Hapgood G, Ben-Neriah S, Mottok A, et al. Identification of highrisk DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. Br J Haematol. 2019;186(3):e28-e31.
19. Horwitz SM, Ansell S, Ai WZ, et al. T-cell lymphomas, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2022;20(3):285-308.
Haematologica | 108 June 2023 1602 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
20. Bachy E, Camus V, Thieblemont C, et al. Romidepsin plus CHOP versus CHOP in patients with previously untreated peripheral Tcell lymphoma: results of the Ro-CHOP phase III study (conducted by LYSA). J Clin Oncol. 2022;40(3):242-251.
21. Schmitz N, Truemper L, Bouabdallah K, et al. A randomized phase 3 trial of autologous vs allogeneic transplantation as part of firstline therapy in poor-risk peripheral T-NHL. Blood. 2021;137(19):2646-2656.
22. Horwitz S, O’Connor OA, Pro B, et al. The ECHELON-2 trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol. 2022;33(3):288-298.
23. Tournilhac O, Hacini M, Bouabdallah K, et al. Addition of brentuximab vedotin to gemcitabine in relapsed or refractory Tcell lymphoma: results of a LYSA multicenter, phase II study. “the TOTAL trial.” Blood. 2020;136(Suppl 1):15-16.
24. Bisig B, Reyniès A de, Bonnet C, et al. CD30-positive peripheral Tcell lymphomas share molecular and phenotypic features. Haematologica. 2013;98(8):1250-1258.
25. Letourneau A, Maerevoet M, Milowich D, et al. Dual JAK1 and STAT3 mutations in a breast implant-associated anaplastic large cell lymphoma. Virchows Arch. 2018;473(4):505-511.
26. Bisig B, Cairoli A, Gaide O, et al. Cutaneous presentation of enteropathy-associated T-cell lymphoma masquerading as a DUSP22-rearranged CD30+ lymphoproliferation. Virchows Arch. Virchows Arch. 2022;481(4):653-657
27. Lemonnier F, Couronné L, Parrens M, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7):1466-1469.
28. Ahmed N, Ketterling RP, Nowakowski GS, Dasari S, Feldman AL. RNAseq identification of FISH-cryptic BCL6::TP63 rearrangement in ALK-negative anaplastic large-cell lymphoma. Histopathology. 2022;81(2):275-278.
29. Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood.
2011;117(3):915-919.
30. Mélard P, Idrissi Y, Andrique L, et al. Molecular alterations and tumor suppressive function of the DUSP22 (Dual Specificity Phosphatase 22) gene in peripheral T-cell lymphoma subtypes. Oncotarget. 2016;7(42):68734-68748.
31. Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130(3):816-825.
32. Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24(4):596-605.
33. Karai LJ, Kadin ME, Hsi ED, et al. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am J Surg Pathol. 2013;37(8):1173-1181.
34. Hsi ED, Said J, Macon WR, et al. Diagnostic accuracy of a defined immunophenotypic and molecular genetic approach for peripheral T/NK-cell lymphomas: a North American PTCL Study Group Project. Am J Surg Pathol. 2014;38(6):768-775.
35. Karube K, Feldman AL. “Double-hit” of DUSP22 and TP63 rearrangements in anaplastic large cell lymphoma, ALK-negative. Blood. 2020;135(9):700.
36. Klairmont MM, Ward N. Co-occurring rearrangements of DUSP22 and TP63 define a rare genetic subset of ALK-negative anaplastic large cell lymphoma with inferior survival outcomes. Leuk Lymphoma. 2022;63(2):506-508.
37. Pro B, Advani R, Brice P, et al. Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood. 2017;130(25):2709-2717.
38. Morel A, Brière J, Lamant L, et al. Long-term outcomes of adults with first-relapsed/refractory systemic anaplastic large-cell lymphoma in the pre-brentuximab vedotin era: a LYSA/SFGM-TC study. Eur J Cancer. 2017;83146-153.
39. Sibon D, Nguyen D-P, Schmitz N, et al. ALK-positive anaplastic large-cell lymphoma in adults: an individual patient data pooled analysis of 263 patients. Haematologica. 2019;104(12):e562-e565.
Haematologica | 108 June 2023 1603 ARTICLE - Impact of DUSP22-R in ALK-negative ALCL: a LYSA study D. Sibon et al.
DUSP22 rearrangement is associated with a distinctive immunophenotype but not outcome in patients with systemic ALK-negative anaplastic large cell lymphoma
Correspondence: J. Xu
jxu9@mdanderson.org
Received: April 11, 2022.
Accepted: September 7, 2022.
Prepublished: December 1, 2022.
https://doi.org/10.3324/haematol.2022.281222
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Abstract
DUSP22 rearrangement (R) has been associated with a favorable outcome in systemic ALK-negative anaplastic large cell lymphoma (ALCL). However, a recent study found that patients with DUSP22-R ALK-negative ALCL have a poorer prognosis than was reported initially. In this study, we compared the clinicopathological features and outcomes of patients with ALKnegative ALCL with DUSP22-R (n=22) versus those without DUSP22-R (DUSP22-NR; n=59). Patients with DUSP22-R ALCL were younger than those with DUSP22-NR neoplasms (P=0.049). DUSP22-R ALK-negative ALCL cases were more often positive for CD15, CD8, and less frequently expressed pSTAT3Tyr705, PD-L1, granzyme B and EMA (all P<0.05). TP63 rearrangement (TP63-R) was detected in three of the 66 (5%) ALK-negative ALCL cases tested and none of these cases carried the DUSP22-R. Overall survival of patients with DUSP22-R ALCL was similar to that of the patients with DUSP22-NR neoplasms regardless of International Prognostic Index score, stage, age, or stem cell transplantation status (all P>0.05), but was significantly shorter than that of the patients with ALK-positive ALCL (median overall survival 53 months vs undefined, P=0.005). Five-year overall survival rates were 40% for patients with DUSP22-R ALCL versus 82% for patients with ALK-positive ALCL. We conclude that DUSP22-R neoplasms represent a distinctive subset of ALK-negative ALCL. However, in this cohort DUSP22-R was not associated with a better clinical outcome. Therefore, we suggest that current treatment guidelines for this subset of ALK-negative ALCL patients should not be modified at present.
Introduction
Anaplastic large cell lymphoma (ALCL) is a mature T-cell neoplasm characterized by large pleomorphic neoplastic cells with kidney-shaped nuclei (so-called “hallmark” cells) and uniform, strong CD30 expression. Based on the presence or absence of an anaplastic lymphoma kinase gene (ALK) rearrangement and resultant ALK expression, ALCL is further classified into ALK-positive (+) and ALKnegative types.1 Although ALK-negative ALCL is morphologically indistinguishable from ALK+ ALCL, patients with systemic ALK-negative ALCL are usually older and have a more aggressive clinical course and poorer outcome with 5-year overall survival (OS) rates of <50% compared with 80-90% for patients with ALK+ ALCL.2-7
ALK-negative ALCL is a genetically heterogeneous entity, with 13-30% of cases harboring a DUSP22 rearrangement (R) and 2-8% of cases carrying a TP63-R.6,8-11 TP63-R and
DUSP22-R are nearly mutually exclusive. DUPS22, also known as c-Jun N-terminal kinase (JNK) pathway-associated phosphatase (JKAP), is a tumor suppressor gene located on chromosome 6p25.3. DUPS22 encodes the dual-specificity phosphatase-22 which plays a role in inhibiting T-cell receptor (TCR) signaling.12,13 DUSP22 knockout enhances T-cell activation and TCR signaling and produces enhanced T-cell-mediated immune responses in a mouse model.12 Restoring expression of DUSP22 in DUSP22-deficient malignant T cells inhibits cellular expansion by stimulating apoptosis and impairs clonogenicity and tumorigenicity.13 DUSP22-R often results from t(6;7)(p25.3;q32.3) and is associated with as much as a 50-fold reduction of DUSP22 expression.14 DUSP22-R ALKnegative ALCL cases appear to represent a distinctive subset of ALK-negative ALCL cases with unique morphology, immunophenotype, and molecular signature.9,11,15-17 TP63 is located at chromosome 3q28. TP63-R results in a
Lianqun Qiu,1 Guilin Tang,1 Shaoying Li,1 Francisco Vega,1 Pei Lin,1 Sa A. Wang,1 Wei Wang,1 Swaminathan P. Iyer,2 Luis Malpica,2 Roberto N. Miranda,1 Sergej Konoplev,1 Zhenya Tang,1 Hong Fang,1 L. Jeffrey Medeiros1 and Jie Xu1
1Department of Hematopathology, The University of Texas MD Anderson Cancer Center and 2Department of Lymphoma/Myeloma, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Haematologica | 108 June 2023 1604 ARTICLE - Non-Hodgkin Lymphoma
p63 fusion protein with structural homology to oncogenic ΔNp63. Among ALK-negative ALCL patients, the subgroup with TP63-R neoplasms has a poor prognosis.6
The prognostic significance of DUSP22-R in ALK-negative ALCL is currently controversial. A Mayo Clinic group initially reported that patients with DUSP22-R ALK-negative ALCL (n=22) have a favorable clinical outcome with a 5year OS rate of 90%, similar to that of ALK+ ALCL patients.6 Later on, two small studies of five and four patients, respectively, showed a similarly favorable prognosis in patients with DUPS22-R ALCL.9,10 Although the case numbers were limited in these studies, others have wondered if the treatment guidelines for patients with DUSP22-R ALK-negative ALCL should be modified. However, a recent study by Hapgood and colleagues reported a 5-year OS of 40% for their cohort of 12 patients with DUSP22-R ALK-negative ALCL.11
In this study, we used fluorescence in situ hybridization (FISH) to determine the status of DUSP22 and TP63 in cases of ALK-negative ALCL. We further focused on DUSP22-R ALK-negative ALCL cases to characterize their clinicopathological and immunophenotypic features and the patients’ outcomes.
Methods
Case selection
We searched the database of the Department of Hematopathology at the MD Anderson Cancer Center from January 1, 2007 through December 31, 2021 for cases of systemic ALK-negative ALCL. The diagnosis of ALKnegative ALCL was based on criteria specified in the 5th edition of the World Health Organization classi fication.1 The following cases were considered as primary cutaneous ALK-negative ALCL and were excluded from the study: (i) patients with cutaneous disease alone without extracutaneous involvement; and (ii) patients with concurrent cutaneous disease and involvement of regional lymph nodes but no other extracutaneous involvement.1,18 For comparison of survival, we compared this cohort with a group of patients with ALK+ ALCL seen at our institution during the same time interval. Some data for the group of ALK+ ALCL patients have been published previously.19 Clinical information was obtained by review of medical records. This study was approved by the Institutional Review Board.
Immunophenotypic analysis
Immunohistochemical studies were performed as described previously.20 The antibodies used were specific for: ALK1, BCL2, CD2, CD3, CD4, CD5, CD7, CD8, CD15, CD20, CD30, CD43, CD45, CD56, EMA, granzyme B, Ki-67, MUM1, MYC, PAX5, PD-L1, perforin, phospho-STAT3Tyr705, and TIA1.
The percentages of lymphoma cells positive for CD15, phospho-STAT3Tyr705 and PD-L1 were read by two hematopathologists (JX and SL) with estimation to the closest 5%, and then the average of the two reads was used for the final reading of each case.
Flow cytometry immunophenotypic analysis was performed using either a FACSCanto II or a FACSCalibur cytometer (Becton-Dickinson Biosciences, San Jose, CA, USA) as described previously.21 The panel of antibodies employed included CD2, CD3, CD4, CD5, CD7, CD8, CD10, CD25, CD30, CD45, CD52, CD56, TCR α/b and TCR γ/d (Becton-Dickinson Biosciences, San Jose, CA, USA).
Fluorescence in situ hybridization
FISH analysis was performed on formalin-fixed, paraffinembedded tissue sections using IRF4/DUSP22 dual-color break-apart probes (3’IRF4/DUSP22 – centromeric, labeled with red; 5’IRF4/DUSP22 – telomeric, labeled with green; CytoTest, Rockville, MD, USA) and TP63 dual-color breakapart probes (Cytocell, Cambridge, UK) according to the manufacturers’ instructions. Two hundred interphase nuclei were analyzed.22 The cutoff value was 10.7% for the IRF4/DUSP22-R and 11.1% for the TP63-R. IRF4/DUSP22-R and TP63-R were assessed in a blinded fashion without knowledge of the pathological diagnosis or results of other FISH analyses.
Statistical analysis
Statistical analyses were performed using Graph-Pad Prism 8 and SPSS 26.0 software (IBM Corporation, Armonk, NY, USA). The Fisher exact test was used to compare clinicopathological features between the DUSP22-R and DUSP22-NR groups in patients with ALK-negative ALCL. OS was calculated from the date of initial diagnosis to the date of death or last follow-up. Progression-free survival was calculated from the date of diagnosis to the date of progression/relapse or, if no progression/relapse, the date of death or last follow-up. Survival was analyzed using the Kaplan-Meier method and was compared using the log rank test. A P value of less than 0.05 was considered statistically significant.
Results
Clinical and fluorescence in situ hybridization findings
The results of FISH analysis showed that a DUSP22-R was present in 22 (28%) cases (DUSP22-R group), and absent in 59 (72%) cases (DUSP22-NR group). The clinical features of these patients are summarized in Table 1. The DUSP22R group comprised 15 men and seven women with a median age of 52 years (range, 33-79 years) at the time of diagnosis. Six (40%) patients with available data had B symptoms. Lymphadenopathy was identified in 16 of 17
Haematologica | 108 June 2023 1605 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al.
(94%) patients, and nine of 15 (60%) patients had extranodal involvement including skin (6 cases; 40%), bone (3 cases; 20%), soft tissue (2 cases; 13%), liver (1 case; 7%), lung (1 case; 7%), and muscle (1 case; 7%). Peripheral blood and bone marrow involvement were seen in one of eight (13%) and one of 18 (6%) patients, respectively. Fifteen patients were fully staged, and 13 (87%) had stage III or IV disease. Five of 15 (33%) patients had an International Prognostic Index (IPI) score of ≥3. One of 14 (7%) patients had leukocytosis and one of 13 (8%) patients had absolute lymphocytosis. Anemia was observed in seven of 14 (50%) patients and thrombocytopenia was present in two of 14 (14%) patients. An elevated serum lactate dehydrogenase level was detected in seven of 14 (50%) patients. The DUSP22-NR group consisted of 38 men and 21 women with a median age of 61 years (range, 21-95 years) at the time of diagnosis. Twenty-seven (63%) patients had B symptoms. Forty-one of 51 (80%) patients had lymphade-
nopathy and 34 of 45 (76%) patients had extranodal disease. The involved extranodal sites included lung (10 cases; 22%), skin (9 cases; 20%), bone (7 cases; 16%), liver (6 cases; 13%), soft tissue (5 cases; 11%), gingiva/oropharynx/nasphoarynx (3 cases; 7%), spleen (2 cases; 4%), muscle (2 cases; 4%), kidney (1 case; 2%), and stomach (1 case; 2%). Peripheral blood and bone marrow involvement were seen in four of 18 (22%) cases and eight of 46 (17%) patients, respectively. Forty-one patients were fully staged, and 27 (66%) had stage III or IV disease. Eighteen of 36 (50%) patients had an IPI scores of ≥3. Seven of 31 (23%) patients had leukocytosis and no patients (of 30) had absolute lymphocytosis. Anemia was observed in 20 of 31 (65%) patients and thrombocytopenia was present in 4 of 31 (13%) patients. An elevated serum lactate dehydrogenase level was detected in 16 of 26 (62%) patients. Compared to patients in the DUSP22-NR ALK-negative ALCL group, patients with DUSP22-R neoplasms were
DUSP22-R: with DUSP22 rearrangement; DUSP22-NR: without DUSP22 rearrangement; IPI: International Prognostic Index; WBC: white blood cell; *female <12.0 g/dL, male <14.0 g/dL; LDH: lactate dehydrogenase; FISH: fluorescence in situ hybridization; CHOP: cyclophosphamide, doxorubicin, vincristine, and prednisone; CR: complete response; SCT: stem cell transplant; OS: overall survival. Bold: statistically significant, i.e., P<0.05. P/T tested: positive/total cases tested.
Total (N=81) DUSP22-R (N=22) DUSP22-NR (N=59) P value Clinical features at diagnosis Male:female (N/N) 1.9:1 (53/28) 2:1 (15/7) 1.8:1 (38/21) 0.80 Age in years, median (range) 60 (21-95) 52 (33-79) 61 (21-95) 0.049 B symptoms, % (P/T tested) 57 (33/58) 40 (6/15) 63 (27/43) 0.14 Lymphadenopathy at diagnosis, % (P/T tested) 84 (57/68) 94 (16/17) 80 (41/51) 0.27 Extra-nodal involvement, % (P/T tested) 72 (43/60) 60 (9/15) 76 (34/45) 0.32 Peripheral blood involvement, % (P/T tested) 19 (5/26) 13 (1/8) 22 (4/18) 1.00 Bone marrow involvement, % (P/T tested) 14 (9/64) 6 (1/18) 17 (8/46) 0.43 Stage III or IV, % (P/T tested) 71 (40/56) 87 (13/15) 66 (27/41) 0.19 IPI ≥ 3, % (P/T tested) 45 (23/51) 33 (5/15) 50 (18/36) 0.36 Laboratory findings at diagnosis, % (P/T tested) Elevated WBC, >11x109/L 18 (8/45) 7 (1/14) 23 (7/31) 0.40 Absolute lymphocytosis, >4.8x109/L 2 (1/43) 8 (1/13) 0 (0/30) 0.30 Anemia* 60 (27/45) 50 (7/14) 65 (20/31) 0.51 Thrombocytopenia, <140x109/L 13 (6/45) 14 (2/14) 13 (4/31) 1.00 Elevated LDH 58 (23/40) 50 (7/14) 62 (16/26) 0.52 TP63 rearrangement by FISH, % (P/T tested) 5 (3/66) 0 (0/19) 6 (3/47) 0.55 Initial treatment, % (P/T tested) CHOP or modified CHOP 90 (55/61) 81 (13/16) 93 (42/45) 0.18 Others 10 (6/61) 19 (3/16) 7 (3/45) Initial CR, % (P/T tested) 54 (32/59) 75 (12/16) 47 (20/43) 0.08 Relapse, % (P/T tested) 52 (28/54) 43 (6/14) 55 (22/40) 0.54 SCT+, % (P/T tested) 29 (15/51) 36 (5/14) 27 (10/37) 0.73 Outcome Alive, % (P/T tested) 54 (36/67) 50 (9/18) 55 (27/49) 0.79 Dead, % (P/T tested) 46 (31/67) 50 (9/18) 45 (22/49) OS, months, median (range) 36 (1.1-94) 53 (8-85) 36 (1.1-94) 0.64
Table 1. Clinical features of patients with ALK-negative anaplastic large cell lymphoma with or without a DUSP22-rearrangement.
Haematologica | 108 June 2023 1606 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al.
younger (median age 52 vs. 61 years, P=0.049). There were no other significant differences in clinical features between patients in these two groups (all P>0.05) (Table 1).
TP63 rearrangement by fluorescence in situ hybridization analysis
FISH analysis was performed to evaluate the status of TP63-R in 66 cases (19 DUSP22-R and 47 DUSP22-NR) with material available. Three (~5%) cases were positive for TP63-R and all were in the DUSP22-NR group (Table 1). None (n=19) of the DUSP22-R ALK-negative ALCL cases tested carried a TP63-R. The difference in the frequency of TP63-R between the DUSP22-R and DUSP22-NR groups was not statistically significant (P=0.55).
Morphological and immunophenotypic findings
DUSP22-NR ALK-negative ALCL cases in tissue specimens have morphological features of the so-called “common pattern” described in ALK+ ALCL. In comparison, the lymphoma cells in DUSP22-R neoplasms were relatively more monotonous in appearance, smaller, and more often had central nuclear pseudoinclusions (“doughnut” cells). Hallmark cells were present in all cases of DUSP22-R ALCL, but large pleomorphic cells were seen only occasionally. A DUPS22-R ALK-negative ALCL case is shown in Figure 1
(lymph node) and Figure 2 (bone marrow and peripheral blood).
The lymphoma cells of DUSP22-R ALK-negative ALCL cases were positive for CD45 (12/13; 92%), BCL-2 (6/7; 86%), CD43 (5/6; 83%), CD2 (13/17; 77%), CD3 (16/21; 76%) and CD4 (14/19; 74%) (Table 2 and Figure 3). Expression of other T-cell-associated antigens was less frequent: TCR α / b (3/7; 43%), CD52 (2/5; 40%), CD5 (6/17; 35%), CD25 (2/6; 33%), CD7 (2/7; 29%) and CD8 (5/18; 28%). Cytotoxic markers were positive in only a few cases of DUSP22-R ALK-negative ALCL including TIA1 (3/10; 30%) and perforin (1/4; 25%) and granzyme B was consistently negative (n=12). Small subsets of cases were positive for EMA (2/11; 18%) and CD56 (1/12; 8%). All seven cases examined for TCR γ/d were negative. The proliferation index as assessed by Ki67 was high (~90%).
CD15 was positive in 12 of 15 (80%) DUSP22-R cases examined. A mean of 26% lymphoma cells in DUSP22-R cases were positive for CD15. Three types of CD15 staining pattern were observed: a Golgi-like (perinuclear dot) pattern, a membranous/cytoplasmic pattern, and a combination of the Golgi and membranous/cytoplasmic patterns (Figure 4).
The immunophenotype of DUSP22-NR ALK-negative ALCL cases was similar except for the following significant dif-
Figure 1. Lymph node biopsy from a case with DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. (A, B) The nodal architecture is effaced by sheets of monotonous, intermediate sized lymphoma cells. Some lymphoma cells show kidney-shaped nuclei (hallmark cells) (arrows, A) or central nuclear pseudoinclusions (“doughnut” cells) (arrows, B). (C-G) The lymphoma cells are strongly and diffusely positive for CD30 (C) and CD3 (D), and are negative for ALK1 (E), granzyme B (F) and EMA (G). (H) Fluorescence in situ hybridization analysis using IRF4/DUSP22 break-apart probes, ×600. The nuclei showing a DUSP22 rearrangement (with red and green split signals) are indicated by white arrow heads. (A, B) Hematoxylin-eosin stain, x600 (A) and x600 (B). (C-G) Immunohistochemistry, x400.
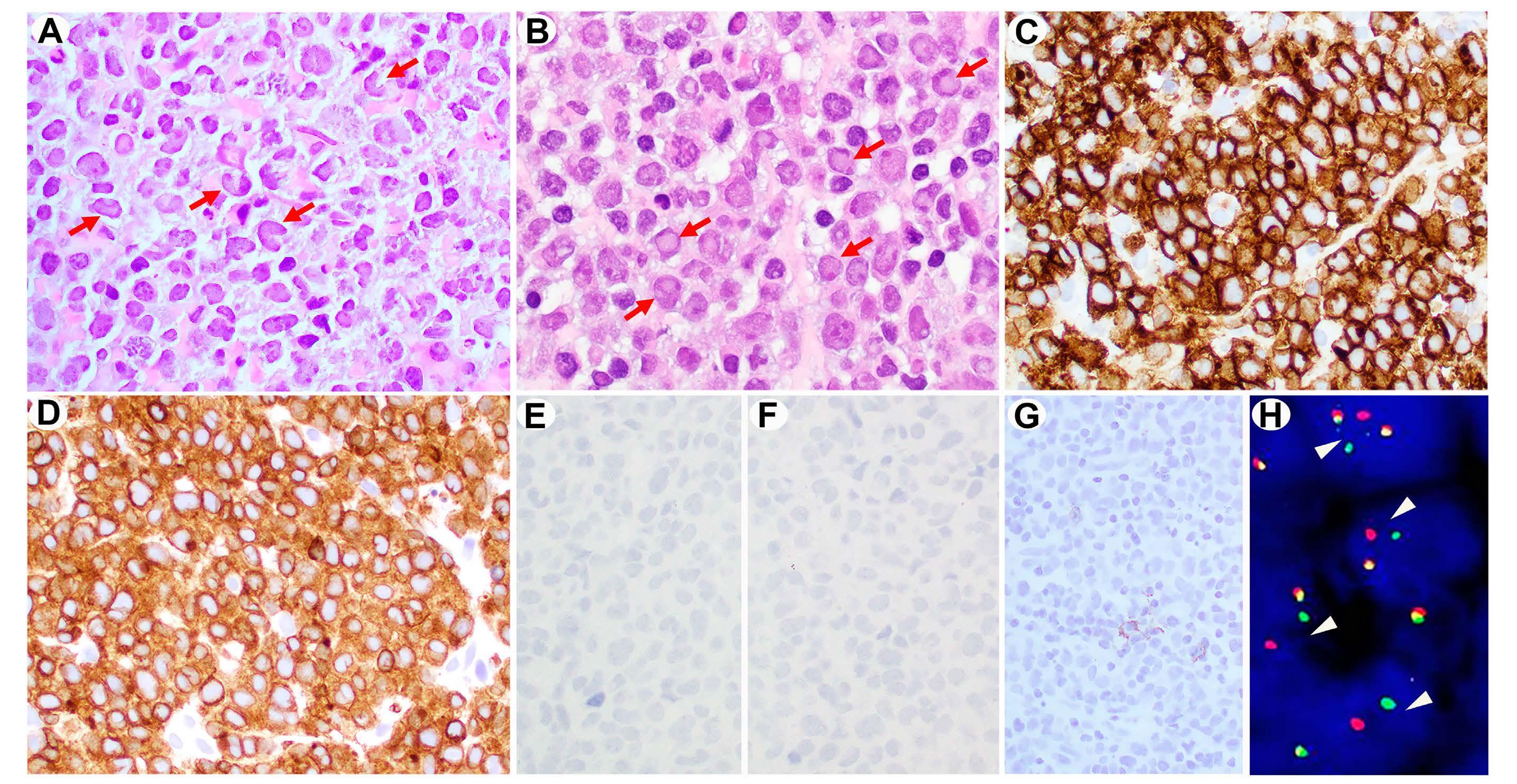
Haematologica | 108 June 2023 1607 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al. A B C
G
E
H
F
D
Figure 2. The DUSP22-rearranged ALK-negative anaplastic large cell lymphoma shown in Figure 1 also involves bone marrow and peripheral blood. (A) This bone marrow core biopsy specimen shows a hypercellular bone marrow infiltrated by lymphoma cells in an interstitial pattern. The lymphoma cells are morphologically similar to those in the lymph node described in Figure 1. (B) The lymphoma cells in the bone marrow are positive for CD30 by immunohistochemistry. (C) Bone marrow aspirate smear showing lymphoma cells, mostly small to intermediate size, with irregular nuclear contours and basophilic cytoplasm. A lymphoma cell shows kidney-shaped nuclei. (D-G) Flow cytometric immunophenotypic analysis of the peripheral blood shows a large population (75%) of lymphoma cells (red dots) which are positive for CD45 (D), CD30 (E), CD3 (partial/decreased, E), CD8 (dim, F), and CD7 (partial, G), and negative for CD4 and CD26, immunophenotypically similar to the lymphoma cells in the lymph node described in Figure 2. The purple and blue dots represent background benign CD4+ and CD8+ T cells, respectively. (A) Hematoxylin-eosin stain, x500. (B) Immunohistochemistry, x500. (C) Wright-Giemsa stain, x1000.

ferences that included CD15 being usually negative (9% vs 80%, P=0.0001), lower frequency of CD8 (7% vs. 28%, P=0.045), and more common expression of granzyme B (65% vs. 0%, P<0.0001) and EMA (57% vs. 18%, P=0.04).
STAT3 activation and PD-L1 expression
Activation of STAT3 was examined by assessing nuclear expression of phosphorylated STAT3 (pSTAT3Tyr705). A mean of 2% lymphoma cells in DUSP22-R ALK-negative ALCL cases showed nuclear staining for pSTAT3Tyr705, a value significantly lower than the mean of 36% observed in DUSP22-NR tumors (P=0.001) (Figure 5A-C). PD-L1, a downstream molecule regulated by the JAK/STAT3 signaling pathway, was positive in 3% lymphoma cells in the DUSP22-R ALCL group compared to 26% positive cells in DUSP22-NR tumors (P=0.01) (Figure 5D-F).
Treatment and response
Treatment information was available for 16 patients with DUSP22 -R ALK-negative ALCL and 45 patients with DUSP22 -NR neoplasms. All patients were treated with chemotherapy regimens over the time interval of this
study, with or without consolidation with stem cell transplant (SCT). Fifty-five of 61 (90%) patients were treated with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or modified CHOP: 13 of 16 (81%) patients in the DUSP22-R group and 42 of 45 cases (93%) in the DUSP22-NR group. Of the six patients treated with non-CHOP-based chemotherapy regimens, four received brentuximab vedotin-based therapy, one received ifosfamide, carboplatin and etoposide (ICE), and one received etoposide, methylprednisolone, high-dose cytarabine and cisplatin (ESHAP) in combination with gemcitabine and vinorelbine. After initial induction chemotherapy, 12 of 16 (75%) patients in the DUSP22-R group and 20 of 43 patients (47%) in the DUSP22 -NR group achieved complete remission. Patients with DUSP22-R ALCL tended to have a higher initial complete response rate than the patients with DUSP22-NR neoplasms, but this difference did not reach statistical significance ( P =0.08). Five of 14 (36%) patients in the DUSP22 -R group and ten of 37 (27%) patients in the DUSP22-NR group underwent SCT. There was no significant difference in initial treatment or SCT rates between
Haematologica | 108 June 2023 1608 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al. A B C D E F G
Table 2. Immunophenotypic features of ALK-negative anaplastic large cell lymphoma with or without a DUSP22-rearrangement
patients with DUSP22 -R or DUSP22- NR ALK-negative ALCL (all P>0.05) (Table 1).
Outcome
After a median follow-up of 19.2 months (range, 1.1-94 months), 31 of 67 (46%) patients with clinical follow-up data died, including nine of 18 (50%) patients in the DUSP22-R group and 22 of 49 (45%) patients in the DUSP22-NR group. In the DUSP22-NR group, two of three (67%) patients carrying a TP63-R died. Comparing the patients with DUSP22-R, DUSP22-NR/TP63-NR (so called “triple-negative”), and DUSP22-NR/TP63-R ALK-negative ALCL, no signifi cant differences were observed in OS (median 53 vs. 36 vs. 21 months, all P>0.05) or in progression-free survival (median 53 vs. 20 vs. 9 months, all P>0.05) between these groups (Figure 6A, B). Although the patients with TP63-R neoplasms tended to have shorter progression-free survival than those without TP63-R, this difference did not reach statistical significance. After excluding the TP63-R cases, the OS of the DUSP22-R and DUSP22-NR groups was further compared after stratifying patients by IPI score (≥3 or <3), clinical stage (stage III/IV or I/II), age (≥50 or <50 years [Figure 6C]; ≥40 or <40 years,
data not shown; ≥60 or <60 years, data not shown), and SCT status, there were still no significant differences in OS between the DUSP22-R and DUSP22-NR groups (all P>0.05) (Figure 6C-J).
We also compared the OS of patients with DUSP22-R ALKnegative ALCL to that of a group of patients with ALK+ ALCL. The OS of patients with DUSP22-R ALK-negative ALCL was significantly poorer than that of patients with ALK+ ALCL (median OS 53 months vs. undefined, P=0.005) (Figure 6A). The 5-year OS rates were 40% for patients with DUSP22-R ALK-negative ALCL as compared to 82% for patients with ALK+ ALCL.
Discussion
The DUSP22-R has been reported most often in cases of systemic ALK-negative ALCL and primary cutaneous ALCL, occasionally in lymphomatoid papulosis and rarely in peripheral T-cell lymphoma, not otherwise specified, but not in ALK+ ALCL.23,24 A DUSP22-R occurs in 13-30% cases of ALK-negative ALCL.6,8-11 Consistent with prior reports, 27% of systemic ALK-negative ALCL cases in the present study
Immunophenotype Total (N=81) DUSP22-R (N=22) DUSP22-NR (N=59) P value % (Positive/Total tested) CD2+ 72 (38/53) 77 (13/17) 69 (25/36) 0.75 CD3+ 60 (47/78) 76 (16/21) 55 (31/57) 0.12 CD4+ 82 (56/68) 74 (14/19) 86 (42/49) 0.29 CD5+ 45 (28/62) 35 (6/17) 49 (22/45) 0.40 CD7+ 30 (7/23) 29 (2/7) 31 (5/16) 1.00 CD8+ 13 (8/60) 28 (5/18) 7 (3/42) 0.045 CD15+ 31 (15/48) 80 (12/15) 9 (3/33) 0.0001 CD25+ 60 (9/15) 33 (2/6) 78 (7/9) 0.14 CD30+ 100 (81/81) 100 (22/22) 100 (59/59) 1.00 CD43+ 87 (26/30) 83 (5/6) 88 (21/24) 1.00 CD45+ 81 (39/48) 92 (12/13) 77 (27/35) 0.41 CD52+ 7 (2/29) 40 (2/5) 0 (0/24) 0.44 CD56+ 10 (3/31) 8 (1/12) 11 (2/19) 1.00 TCR αb+ 55 (18/33) 43 (3/7) 58 (15/26) 1.00 TCR γd+ 0 (0/17) 0 (0/7) 0 (0/10) 1.00 BCL-2+ 75 (15/20) 86 (6/7) 69 (9/13) 0.61 Granzyme B+ 41 (13/32) 0 (0/12) 65 (13/20) <0.0001 TIA1+ 50 (18/36) 30 (3/10) 58 (15/26) 0.26 Perforin+ 50 (5/10) 25 (1/4) 83 (5/6) 0.19 EMA+ 46 (18/39) 18 (2/11) 57 (16/28) 0.04 MUM1+ 91 (21/23) 100 (7/7) 88 (14/16) 1.00 MYC+ 43 (22/51) 41 (7/17) 44 (15/34) 0.54 Mean Ki-67, % (N) 88 (35) 91 (11) 85 (24) 0.16
DUSP22-R: with DUSP22 rearrangement; DUSP22-NR: without DUSP22 rearrangement.
for CD15 positivity was ≥5%. Bold: statistically significant, i.e., P<0.05 Haematologica | 108 June 2023 1609 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al.
The cutoff value
harbored a DUSP22-R. We also showed that patients with DUSP22-R ALCL were ~10 years younger than patients with DUSP22-NR neoplasms. Except for age, the present study showed no significant differences in other clinical features between patients with DUSP22-R versus DUSP22-NR ALKnegative ALCL.
Constitutive activation of the JAK/STAT3 signaling pathway is a central pathogenic feature of ALK+ as well as ALKnegative ALCL. DUSP22 has been shown to inhibit STAT3
signaling.25 Since DUSP22 expression was significantly decreased in the presence of a DUSP22-R, it seemed reasonable to expect enhanced STAT3 activation in DUSP22-R ALCL. Surprisingly, total STAT3 protein level and STAT3 activation (measured by pSTAT3Y705) in DUSP22-R ALCL was significantly decreased, at least partially due to significantly reduced expression of STAT3 and other genes in the JAK/STAT3 pathway.16 Lack of STAT3 activation in DUSP22R ALCL has also been shown by others.9,11 The JAK/STAT3

Figure
the lymph node biopsy specimen shown in Figure 1. (A-H) The lymphoma cells (red dots) are positive for CD45 (A), CD30 (B), CD3 (decreased, B), CD8 (partial, C), CD2 (D), CD7 (partial/decreased, F), and T-cell receptor (TCR) α/b (decreased, H), and negative for CD4 (C), CD5 (D), CD26 (F), CD25 (G), and TCR γ/d (H). The lymphoma cells are medium-sized by forward scatter (E). The purple and blue dots represent background benign CD4+ and CD8+ T cells, respectively.
Figure 4.
expression with three staining patterns in DUSP22-rearranged cases of ALK-negative anaplastic large cell lymphoma. (A) Golgi-like pattern. (B) Membranous/cytoplasmic pattern. (C) Combination of the Golgi-like and membranous/cytoplasmic patterns. (D) The percentage of CD15+ lymphoma cells in DUSP22-rearranged cases of anaplastic large cell lymphoma cases is significantly higher than that in the cases without a DUSP22 rearrangement. (A-C) Immunohistochemistry, x500. DUSP22-R: with DUSP22 rearrangement; DUSP22-NR: without DUSP22 rearrangement.
Haematologica | 108 June 2023 1610
 3. Flow cytometric immunophenotypic analysis of
CD15
3. Flow cytometric immunophenotypic analysis of
CD15
ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al. A B C D E F G H A B C D
pathway regulates expression of many downstream molecules including PD-L1.20,26,27 Therefore, one would predict that lack of pSTAT3 in DUSP22-R ALCL would result in absent PD-L1 expression, as shown here and elsewhere.11,16 Expression of other known STAT3 target genes, such as granzyme B and CD25, was also low in DUSP22-R ALCL in this cohort.16 DUSP22-R ALCL cases were nearly always negative or infrequently expressed cytotoxic markers and EMA.6,9,11,15 Based on gene expression profiling, ALK-negative ALCL cases included two distinct subgroups: cytotoxic and non-cytotoxic. The non-cytotoxic subgroup showed high expression of CD30 but not perforin or granzyme B, with half of cases harboring a DUSP22-R.28 In keeping with prior reports, the levels of expression of granzyme B and EMA in our DUSP22-R ALK-negative ALCL cases were significantly lower than those in the DUSP22-NR cases. DUSP22-R results in inhibition of the JAK/STAT3 pathway, but it may also lead to activation of other pathways, as supported by the increased frequency of CD15 observed in the current study. A high CD15 positivity rate in DUSP22R ALK-negative ALCL cases has not been previously reported. Three CD15 staining patterns were observed in
DUSP22-R ALCL cases: a Golgi-like pattern, a membranous/cytoplasmic pattern, and a combination of the Golgilike and membranous/cytoplasmic patterns. CD15 positivity with a Golgi-like staining pattern is unusual but has been reported in a case of DUSP22-R ALK-negative ALCL.29 Another case report of ALK-negative ALCL showed membranous and Golgi-like staining pattern of CD15, but DUSP22-R status was unknown.30
Although CD15 has received little attention in DUSP22-R ALK-negative ALCL, CD15 expression was observed in ALCL previously. Felgar et al. reported CD15 expression in three of 17 (18%) T- or null cell ALCL with unknown ALK status; these cases were all negative for TIA1, suggesting to the authors that the CD15+ ALCL cases may be different from other ALCL cases.31 Gorczyca et al. reported CD15 positivity in two of 26 (8%) ALK+ ALCL cases and seven of 30 (23%) ALK-negative CD30+ T-cell lymphomas.32 Four CD15+ ALKnegative ALCL cases were submitted to the 2005 Society of Hematopathology/European Association for Hematopathology Workshop: one of three (33%) cases was positive for EMA, and one of two (50%) cases was positive for TIA1.33 Retrospectively, we speculate that at least some of
Figure 5. Comparison of STAT3 activation (pSTAT3Tyr705) and PD-L1 expression in ALK-negative anaplastic large cell lymphoma with or without a DUSP22 rearrangement. (A, D) Negative pSTAT3Tyr705 (A) and PD-L1 (D) expression in a DUSP22-rearranged case. (B, E) Diffuse positivity of pSTAT3Tyr705 (B) and PD-L1 (E) in a case without a DUSP22-rearrangement. (C, F) The percentage of pSTAT3Tyr705-positive (C) or PD-L1-positive (F) lymphoma cells in DUSP22-rearranged cases of anaplastic large cell lymphoma is significantly lower than that in cases without a DUSP22 rearrangement. DUSP22-R: with DUSP22 rearrangement; DUSP22-NR: without DUSP22 rearrangement.

Haematologica | 108 June 2023 1611 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al. A B C F E D
previously reported CD15+ ALK-negative ALCL cases were likely neoplasms with DUSP22-R. The potential explanation for CD15 expression in DUSP22-R ALCL is unknown and needs to be investigated further.
CD15 has been a valuable marker for distinguishing classic Hodgkin lymphoma from other CD30+ lymphomas including ALK-negative ALCL. The differential diagnosis between ALK-negative ALCL and classic Hodgkin lymphoma is usually not difficult based on their morphological and immunophenotypic differences. However, a subset of tumors may occasionally show morphological and/or immunophenotypic overlap, posing a diagnostic challenge. For the CD30+ CD15+ cases with overlapping features of ALCL and classic Hodgkin lymphoma, FISH for DUSP22-R may be helpful in reaching the correct diagnosis because a positive result for DUSP22-R points to the diagnosis of ALKnegative ALCL.
The unique morphological, immunophenotypic, and mol-
ecular/genetic features of DUSP22 -R ALCL have been reported consistently, suggesting that DUSP22-R cases are a distinct subset of ALK-negative ALCL .9,11,15-17 However, the prognostic significance of DUSP22 -R in ALK-negative ALCL has been controversial. DUSP22 -R was initially reported to be associated with a favorable clinical outcome in systemic ALK-negative ALCL patients with a 5-year OS rate of 90%, similar to that of patients with ALK+ ALCL.6 However, in a recent study from Vancouver, the 5-year OS of DUSP22-R ALCL patients was only 40%.11 Unlike earlier studies with small cohorts, the present study of 81 ALK-negative ALCL patients shows that patients with DUSP22-R neoplasms have a relatively poorer outcome, similar to that of patients in the DUSP22 -NR group and substantially poorer than that of ALK+ ALCL patients. The discrepancies observed between studies on the outcome of DUSP22 -R cases could potentially be attributable to some missed breakpoints. Different FISH

Haematologica | 108 June 2023 1612 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al.
A B
Continued on following page.
Figure 6. DUSP22 rearrangement has no prognostic significance in patients with ALK-negative anaplastic large cell lymphoma regardless of International Prognostic Index score, stage, age, or transplantation status. (A, B) Comparison of overall survival (A) and progression-free survival (B) among the patients with ALK+ anaplastic large cell lymphoma (ALCL), DUSP22R (all were TP63-NR) ALK-negative ALCL, DUSP22-NR/TP63-NR (triplenegative) ALK-negative ALCL, and DUSP22-NR/TP63-R ALK-negative ALCL. *P<0.05, when comparing ALK+ ALCL versus DUSP22-R ALK-negative ALCL or comparing ALK+ ALCL versus DUSP22-NR/TP63-NR ALK-negative ALCL. (C, D) Comparison of overall survival in patients with International Prognostic Index score ≥3 (C) and <3 (D). (E, F) Comparison of overall survival in patients with stage III-IV (E) and stage I-II (F) disease. (G, H) Comparison of overall survival in patients aged ≥50 years (G) and <50 years (H). (I, J) Comparison of overall survival in patients with stem cell transplantation (I) and without (J). DUSP22-R: with DUSP22 rearrangement; DUSP22NR: without DUSP22 rearrangement; TP63-R: with TP63 rearrangement; TP63-NR: without TP63 rearrangement; IPI: International Prognostic Index; OS: overall survival; PFS: progression-free survival; SCT: stem cell transplant. (C-J) TP63-R cases were excluded.

probes were used in different studies: some were prepared in-house6,10,11 whereas others were purchased,8,9 as in the current study. It is possible that break-apart FISH probes may not detect all DUSP22 -R if the breakpoints are located outside of the probe coverage or if the rearrangement is through insertion. If true, differences in probes used could alter the results of survival analyses. We note that patients with DUSP22-R ALK-negative ALCL in this cohort tended to have a higher initial complete response rate than the patients with DUSP22 -NR neoplasms, although this difference did not reach statistical significance. This result might be due to the relatively small size of this cohort and larger-sized studies are needed for further investigation.
In this cohort, ~5% of patients with ALK-negative ALCL had TP63-R, consistent with the reported frequency (2-8%) of TP63-R cases. All TP63-R cases in this study were negative for DUSP22-R. TP63-R and DUSP22-R are nearly mutually exclusive,6 although rare cases harboring both rearrangements have been reported.34,35 In a past study TP63-R (n=6) was reported to be associated with poorer prognosis in patients with ALK-negative ALCL.6 The present study shows no association between TP63-R and OS, but the patients with TP63-R neoplasms tended to have shorter progression-free survival than those without TP63-R. Given the limited number of TP63-R cases in our cohort, the prognostic significance of TP63-R cannot be determined.
Haematologica | 108 June 2023 1613 L. Qiu et al. ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma C D E F G H I J
In summary, our data support the idea that DUSP22-R ALK-negative ALCL is a distinctive subset of ALCL. As suggested by others, our data showed that these neoplasms have minimal STAT3 activation and PD-L1 expression, and no/low expression of cytotoxic markers. We report a novel finding in this study that CD15 is commonly expressed in DUSP22-R ALK-negative ALCL cases. Our data also suggest that patients with DUSP22-R ALK-negative ALCL do not have an excellent prognosis as earlier studies suggested, but instead have a prognosis similar to that of patients with DUSP22-NR ALK-negative ALCL. We therefore suggest that further investigation is needed before modifications to the treatment of patients with DUSP22-R ALK-negative ALCL are proposed.
References
1. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022;36(7):1720-1748.
2. Vose J, Armitage J, Weisenburger D; International T-Cell Lymphoma Project. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124-4130.
3. Falini B, Pileri S, Zinzani PL, et al. ALK+ lymphoma: clinicopathological findings and outcome. Blood. 1999;93(8):2697-2706.
4. Sibon D, Fournier M, Briere J, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte trials. J Clin Oncol. 2012;30(32):3939-3946.
5. ten Berge RL, de Bruin PC, Oudejans JJ, et al. ALK-negative anaplastic large-cell lymphoma demonstrates similar poor prognosis to peripheral T-cell lymphoma, unspecified. Histopathology. 2003;43(5):462-469.
6. Parrilla Castellar ER, Jaffe ES, Said JW, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124(9):1473-1480.
7. Ellin F, Landstrom J, Jerkeman M, et al. Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: a study from the Swedish Lymphoma Registry. Blood. 2014;124(10):1570-1577.
8. Parkhi M, Bal A, Das A, et al. ALK-negative anaplastic large cell lymphoma (ALCL): prognostic implications of molecular subtyping and JAK-STAT pathway. Appl Immunohistochem Mol Morphol. 2021;29(9):648-656.
9. Onaindia A, de Villambrosia SG, Prieto-Torres L, et al. DUSP22rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica. 2019;104(4):e158-e162.
10. Pedersen MB, Hamilton-Dutoit SJ, Bendix K, et al. DUSP22 and TP63 rearrangements predict outcome of ALK-negative anaplastic large cell lymphoma: a Danish cohort study. Blood. 2017;130(4):554-557.
11. Hapgood G, Ben-Neriah S, Mottok A, et al. Identification of highrisk DUSP22-rearranged ALK-negative anaplastic large cell lymphoma. Br J Haematol. 2019;186(3):e28-e31.
12. Li JP, Yang CY, Chuang HC, et al. The phosphatase JKAP/DUSP22
Disclosures
No conflicts of interest to disclose
Contributions
LQ, LJM and JX collected and analyzed data and wrote the manuscript; GT and ZT analyzed FISH studies; SL, FV, PL, SW, WW, RNM, SK, and HF contributed to data collection, data analysis, and reviewed the manuscript; SPI, LM contributed to the patients’ care, and reviewed and edited the manuscript; JX designed and supervised the study.
Data-sharing statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat Commun. 2014;5:3618.
13. Melard P, Idrissi Y, Andrique L, et al. Molecular alterations and tumor suppressive function of the DUSP22 (Dual Specificity Phosphatase 22) gene in peripheral T-cell lymphoma subtypes. Oncotarget. 2016;7(42):68734-68748.
14. Feldman AL, Dogan A, Smith DI, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117(3):915-919.
15. King RL, Dao LN, McPhail ED, et al. Morphologic features of ALKnegative anaplastic large cell lymphomas with DUSP22 rearrangements. Am J Surg Pathol. 2016;40(1):36-43.
16. Luchtel RA, Dasari S, Oishi N, et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood. 2018;132(13):1386-1398.
17. Ravindran A, Feldman AL, Ketterling RP, et al. Striking association of lymphoid enhancing factor (LEF1) overexpression and DUSP22 rearrangements in anaplastic large cell lymphoma. Am J Surg Pathol. 2021;45(4):550-557.
18. Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000;95(12):3653-3661.
19. Khanlari M, Li S, Miranda RN, et al. Small cell/lymphohistiocytic morphology is associated with peripheral blood involvement, CD8 positivity and retained T-cell antigens, but not outcome in adults with ALK+ anaplastic large cell lymphoma. Mod Pathol. 2022;35(3):412-418.
20. Shen J, Li S, Medeiros LJ, et al. PD-L1 expression is associated with ALK positivity and STAT3 activation, but not outcome in patients with systemic anaplastic large cell lymphoma. Mod Pathol. 2020;33(3):324-333.
21. Shen J, Medeiros LJ, Li S, et al. CD8 expression in anaplastic large cell lymphoma correlates with noncommon morphologic variants and T-cell antigen expression suggesting biological differences with CD8-negative anaplastic large cell lymphoma. Hum Pathol. 2020;98:1-9.
22. Lyapichev KA, Tang G, Li S, et al. MYC expression is associated with older age, common morphology, increased MYC copy number, and poorer prognosis in patients with ALK+ anaplastic
Haematologica | 108 June 2023 1614 ARTICLE - DUSP22-R ALK-negative anaplastic large cell lymphoma L. Qiu et al.
large cell lymphoma. Hum Pathol. 2021;108:22-31.
23. Feldman AL, Law M, Remstein ED, et al. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009;23(3):574-580.
24. Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24(4):596-605.
25. Sekine Y, Tsuji S, Ikeda O, et al. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene. 2006;25(42):5801-5806.
26. Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105(52):20852-20857.
27. Atsaves V, Tsesmetzis N, Chioureas D, et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia. 2017;31(7):1633-1637.
28. Drieux F, Ruminy P, Abdel-Sater A, et al. Defining signatures of peripheral T-cell lymphoma with a targeted 20-marker gene expression profiling assay. Haematologica. 2020;105(6):1582-1592.
29. Chapman J, Vega F: Indolent ALK-negative anaplastic large-cell lymphoma, DUSP22 rearranged, with an unusual
immunophenotype in a human immunodeficiency virus patient. Histopathology. 2017;70(7):1173-1175.
30. Arun I, Roy P, Arora N, et al. PAX-5 positivity in anaplastic lymphoma kinase-negative anaplastic large cell lymphoma: a case report and review of literature. Int J Surg Pathol. 2017;25(4):333-338.
31. Felgar RE, Salhany KE, Macon WR, et al. The expression of TIA1+ cytolytic-type granules and other cytolytic lymphocyteassociated markers in CD30+ anaplastic large cell lymphomas (ALCL): correlation with morphology, immunophenotype, ultrastructure, and clinical features. Hum Pathol. 1999;30(2):228-236.
32. Gorczyca W, Tsang P, Liu Z, et al. CD30-positive T-cell lymphomas co-expressing CD15: an immunohistochemical analysis. Int J Oncol. 2003;22(2):319-324.
33. Medeiros LJ, Elenitoba-Johnson KS. Anaplastic large cell lymphoma. Am J Clin Pathol. 2007;127(5):707-722.
34. Karube K, Feldman AL. "Double-hit" of DUSP22 and TP63 rearrangements in anaplastic large cell lymphoma, ALKnegative. Blood. 2020;135(9):700.
35. Klairmont MM, Ward N. Co-occurring rearrangements of DUSP22 and TP63 define a rare genetic subset of ALK-negative anaplastic large cell lymphoma with inferior survival outcomes. Leuk Lymphoma. 2022;63(2):506-508.
Haematologica | 108 June 2023 1615 ARTICLE - DUSP22-R ALK-negative anaplastic large
L. Qiu et al.
cell lymphoma
Targeting glutaminase is therapeutically effective in ibrutinib-resistant mantle cell lymphoma
Correspondence: V.C. Jiang cjiang@mdanderson.org
Received: June 6, 2022.
1Department of Lymphoma and Myeloma and 2Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract
Accepted: November 10, 2022.
Early view: November 24, 2022.
https://doi.org/10.3324/haematol.2022.281538
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Mantle cell lymphoma (MCL) is an incurable B-cell non-Hodgkin lymphoma characterized by frequent relapses. The development of resistance to ibrutinib therapy remains a major challenge in MCL. We previously showed that glutaminolysis is associated with resistance to ibrutinib. In this study, we confirmed that glutaminase (GLS), the first enzyme in glutaminolysis, is overexpressed in ibrutinib-resistant MCL cells, and that its expression correlates well with elevated glutamine dependency and glutaminolysis. Furthermore, we discovered that GLS expression correlates with MYC expression and the functioning of the glutamine transporter ASCT2. Depletion of glutamine or GLS significantly reduced cell growth, while GLS overexpression enhanced glutamine dependency and ibrutinib resistance. Consistent with this, GLS inhibition by its specific inhibitor telaglenastat suppressed MCL cell growth both in vitro and in vivo . Moreover, telaglenastat showed anti-MCL synergy when combined with ibrutinib or venetoclax in vitro , which was confirmed using an MCL patient-derived xenograft model. Our study provides the first evidence that targeting GLS with telaglenastat, alone or in combination with ibrutinib or venetoclax, is a promising strategy to overcome ibrutinib resistance in MCL.
Introduction
Mantle cell lymphoma (MCL) is highly refractory to clinical treatments and very frequently relapses, which has created a great demand for more effective therapy.1 For example, ibrutinib is a potent inhibitor of Bruton tyrosine kinase (BTK) with proven efficacy in the treatment of MCL,2 but nearly all initially responsive patients eventually develop ibrutinib resistance. Our group has recently shown that ibrutinib-resistant MCL cells exhibit increased glutaminolysis, the process of glutamine metabolism contributing to the tricarboxylic acid cycle.3 This suggests that glutaminolysis could be targeted as a potential therapeutic strategy to overcome ibrutinib resistance. The rapid growth of cancer cells requires a fast metabolic rate and large energy supply, so cellular metabolic processes, especially glycolysis and glutaminolysis, are often dysregulated.4 Glycolysis is preferentially used for fuel in many types of cancer (the Warburg effect), generating a sufficient ATP supply to support cancer cell growth while
using less oxygen than the canonical Krebs cycle.5 However, there is increasing evidence that glutaminolysis is the main source of ATP supply in some other types of cancer, including MCL3 and multiple myeloma.6 Glutamine is first transported from the extracellular space to the cytosol and then to mitochondria for glutaminolysis.7 The first step of glutaminolysis is catalysis of glutamine to glutamate, which is performed by the glutaminase (GLS) enzymes coded by two paralogous genes: GLS on chromosome 2 and GLS2 on chromosome 12.8 We previously showed that overexpression of GLS, but not GLS2, is associated with ibrutinib resistance in MCL.3 This suggests that GLS is potentially vulnerable for targeting of glutaminolysis to overcome ibrutinib resistance in MCL.
In this study, we confirmed that GLS expression is increased in multiple ibrutinib-resistant cell lines and primary patients’ samples. We investigated the role of glutamine dependency and GLS overexpression in ibrutinib-resistant MCL cells. Furthermore, we evaluated the preclinical efficacy of targeting GLS either alone or in
Lingzhi Li,1 Lei Nie,1 Alexa Jordan,1 Qingsong Cai,1 Yang Liu,1 Yijing Li,1 Yuxuan Che,1 Jovanny Vargas,1 Zhihong Chen,1 Angela Leeming,1 Wei Wang,1 Yixin Yao,1 Michael Wang1,2 and Vivian Changying Jiang1
Haematologica | 108 June 2023 1616 ARTICLE - Non-Hodgkin Lymphoma
combination with BTK or BCL2 inhibitors to overcome therapeutic resistance in vivo and in vitro.
Methods
Patients and collection of their samples
Fresh surgical biopsies and peripheral blood specimens were collected from patients after provision of informed consent and approval by the Institutional Review Board at the MD Anderson Cancer Center. Ficoll-Hypaque density centrifugation was used to isolate mononuclear cells as previously reported.9 The patients’ characteristics are summarized in Online Supplementary Table S1.
Cell culture and reagents
The MCL cell lines JeKo-1, Rec-1, Mino, Maver-1, and Z138 were obtained from the American Type Culture Collection. JeKo-1, Rec-1, and Mino are ibrutinib-sensitive cell lines, while Maver-1 and Z138 cell lines show primarily resistance to ibrutinib. The ibrutinib-resistant JeKo-R and JeKo-BTK-KD cell lines were established as previously described.10 Briefly, JeKo-R cells were established from JeKo-1 cells by exposing these latter to stepwise dose increases of ibrutinib, while JeKo-BTK KD cells were genetically edited by CRISPR-Cas9 technology to have one allele of the BTK gene knocked out, resulting in dramatically reduced expression of BTK. The MCL cells were cultured in a 5% CO2 incubator at 37°C. Telaglenastat (A14396) was purchased from Adooq Biosciences; ibrutinib (S2680) and venetoclax (S8048) were purchased from Selleck Chemicals; V-9302 (HY-112683) was purchased from MCE/Fisher.
Cell viability, apoptosis and cell cycle assay
Cell viability, apoptosis and cell cycle assays were performed as described previously by Zhang et al.11
Western blot analysis
Western blots were performed as described previously.10 The antibody against GLS was purchased from Abcam (Cat. # AB156876) and the antibody against b - actin was purchased from Sigma (A1978-200UL); all the other antibodies were purchased from Cell Signaling Technology (GAPDH, Cat. # 5174s; MYC, Cat. # 9402s; ASCT2, Cat. # 8057s; cyclin D1, Cat. # 2922s; CDK4, Cat. # 12790s; cyclin B1, Cat. # 4138s; ERK, Cat. # 12950s; cleaved PARP, Cat. # 5625S; and cleaved caspase 3, Cat. # 9661s).
Mitochondrial membrane potential and ATP production assays
Assays of mitochondrial membrane potential and ATP production were performed as described previously. 3
Lentiviral packaging and lentiviral transduction for gene knockdown or overexpression
Lentiviral packaging and lentiviral transduction for gene knockdown or overexpression of genes were performed as described previously by Jiang et al. 12
Mantle cell lymphoma cell line-derived xenograft or patient-derived xenograft mouse models
All experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center. Drug evaluation, using cell line-derived xenograft (CDX) or patient-derived xenograft (PDX) mouse models, was performed as described previously.11 Briefly, CDX and PDX tumors were established in mice that were treated orally twice daily with 100 mg/kg telaglenastat or daily with 50 mg/kg ibrutinib, 10 mg/kg venetoclax, or vehicle, until the maximum diameter of the tumor reached 15 mm. Tumor size was measured weekly, and tumor volume (in mm3) was calculated as 0.5 × length × width2. Tail blood was collected biweekly, and serum b2-microglobulin level was measured according to the manufacturer’s protocol.11 The percentage of tumor cells (CD20+CD5+) in each tumor was determined by flow cytometry.
Gene set enrichment analysis and correlation with gene expression and patient survival. These analyses were performed as described previously.11 Briefly, alignment and read counting of the RNA-sequencing data were performed using STAR 2-pass alignment (v2.7.8a)13 with the parameter quantMode as GeneCounts. Reads overlapping with the exons of each gene in the STAR ReadsPerGene.out.tab were determined. The batch effect was corrected using R package sva and differentially expressed genes were identified by DESeq2 (v1.32.0).14 The enriched signaling pathways were determined by DESeq2 and gene set enrichment analysis (GSEA)15 against the curated gene sets C2 from MSigDB.15,16 Pathways related to glutamate metabolism were selected for further analysis. The GSEA pathway signature score was generated by the package TBSignatureProfiler with the gene list of interested pathways. Kaplan-Meier survival curves were analyzed with package survival.17
Statistical analysis
All statistical analyses were performed using GraphPad Prism. Data are represented as mean ± standard error of mean. An unpaired Student t test was used for comparisons between two groups. One-way or two-way analysis of variance was performed for comparisons of two or more groups. P values <0.05, 0.01, 0.001, and 0.0001 are flagged with * , ** , *** and ****, respectively. In vitro experiments were performed at least twice, and each sample was tested in technical replicates.
Haematologica | 108 June 2023 1617 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
Results
Ibrutinib-resistant mantle cell lymphoma cells overexpress glutaminase and are metabolically dependent on glutamine
Our previous study revealed that glutaminolysis is upregulated in ibrutinib-resistant MCL cells.3 To validate this, we compared glutaminolysis signaling pathways in ibrutinibresistant versus ibrubtinib-sensitive patients. Multiple signaling pathways involved in glutaminolysis were increased in ibrutinib-resistant patients (n=35) compared to ibrutinib-sensitive patients (n=35) (Figure 1A). Importantly, GLS expression correlated with glutaminolysis, including glu-
tathione synthesis and recycling pathways (R=0.27, P=0.025) (Figure 1B). These data demonstrate the clinical significance of glutaminolysis and GLS overexpression in contributing to ibrutinib resistance. To further validate GLS overexpression in ibrutinib-resistant cells, we checked GLS protein expression by re-visiting our previous reverse phase protein array dataset.3 GLS protein was confirmed to be overexpressed in ibrutinib-resistant cells (Z138 and Maver-1) compared to ibrutinib-sensitive cells (Rec-1 and Mino) (Figure 1C). This was further validated in additional MCL cell lines (n=5, P=0.0405) and primary patients’ samples (n=6, P=0.0047) by western blotting (Online Supplementary Figure S1A, B). Based on
Figure 1. Ibrutinib-resistant mantle cell lymphoma cells overexpress glutaminase and are dependent on glutaminolysis. (A) Hallmark pathways from gene set enrichment analysis of bulk RNA-sequencing datasets were analyzed in ibrutinib-resistant (IBNR) patients’ samples (n=35) compared with ibrutinib-sensitive (IBN-S) patients’ samples (n=35). (B) Pearson correlation of the glutamate and glutamine metabolic pathways with glutaminase (GLS) expression in patients’ samples. (C) Reverse phase protein array in IBN-S and IBN-R cell lines as indicated. (D, E) IBN-S cell lines (JeKo-1 and Mino) and IBN-R cell lines (Z138 and Maver-1) were cultured in medium with (Q-/glucose+) or without (Q+/glucose+) depletion of glutamine (Q) for 24 h. Cell viability (D) and ATP production (E) were determined. The results are represented as mean ± standard deviation. An unpaired Student t test was used to determine statistical significance. *P<0.05; **P<0.01; ***P<0.001; ****P< 0.0001.

A B C D E Haematologica | 108 June 2023 1618 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
these observations, we hypothesized that the survival and growth of ibrutinib-resistant MCL cells are dependent on glutaminolysis and glutamine supply. To test this, we depleted glutamine from the culture medium. Ibrutinib-resistant Z138 and Maver-1 cells, but not ibrutinib-sensitive Rec-1 and Mino cells, showed cell growth defects when glutamine was depleted (Figure 1D). Correspondingly, ATP production was greatly reduced by glutamine depletion (Figure 1E). Taken together, these data demonstrate that ibrutinib-resistant MCL cells overexpress GLS, the first enzyme in the process of glutaminolysis, and are metabolically dependent on the glutamine supply.
The glutamine transporter ASCT2 is required for ibrutinib resistance-associated glutamine dependency
To be used for glutamate synthesis in mitochondria, extracellular glutamine must be transported into the cytosol and then into the mitochondria. The transport into the cytosol relies on glutamine uptake mediated by the
glutamine transporter ASCT2. We therefore examined ASCT2 expression in patients’ samples. In the same cohort of patients as in Figure 1A-C, ASCT2 expression correlated well with glutathione synthesis and recycling pathways in ibrutinib-resistant MCL cells (R=0.61, P=1.4x10-8) (Figure 2A) and with poor outcomes (P=0.019) (Figure 2B). Therefore, ASCT2 expression correlates with glutamine dependency in ibrutinib-resistant MCL cells.

To address whether there is any correlation between ASCT2 and GLS, we treated MCL cells with V-9302, an ASCT2-specific inhibitor, to block glutamine uptake. ASCT2 inhibition reduced GLS expression in JeKo-R and Z138 cells that normally express high levels of GLS (Figure 2C). To further address this, we knocked down GLS in Z138 cells using two independent short hairpin (sh)RNA (Figure 2D). Again, ASCT2 inhibitor treatment dramatically reduced the expression of GLS in Z138 with shCtrl, but no further reduction was observed in Z138 with shGLS#1 or shGLS#4. These data demonstrate that GLS expression
Figure 2. Glutaminase expression is regulated by the MYC-ASCT2 axis. (A) Pearson correlation of the glutamate and glutamine metabolic pathways with ASCT2 expression in samples from patients with mantle cell lymphoma (MCL), using the same cohort of patients as that in Figure 1A, B. (B) Kaplan-Meier survival curve plotted for patients’ outcome based on ASCT2 expression using the same cohort of patients as that in Figure 1A, B. (C) JeKo-1 and Z138 cells were treated with 20 µM V-9302 (an ASCT2 inhibitor) for 48 h and harvested for immunoblotting with the indicated antibodies. (D) Z138 cells stably transduced with shCtrl, shGLS#1, or shGLS#4 were treated with 20 µM V-9302 for 8 h and harvested for immunoblotting with the indicated antibodies. (E) JeKo-1 cells stably transduced with shMYC or the corresponding control vector (shCtrl) were subjected to immunoblotting with the indicated antibodies. (F) Z138 cells were transduced with a control empty vector (EV) or MYC overexpression (MYC) were subjected to immunoblotting with the indicated antibodies. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
A B C D E F Haematologica | 108 June 2023 1619 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
was sensitive to the blockage of glutamine uptake. ASCT2 is a MYC target18 and MYC promotes GLS transcription through targeting of miR-23;19 therefore, MYC is likely to be the master regulator of ASCT2-GLS-mediated glutaminolysis. To test this hypothesis, we knocked down MYC expression by shRNA in JeKo-1 cells. When MYC expression was low, GLS expression was barely detectable (Figure 2E). Meanwhile, when MYC was overexpressed in Z138 cells, GLS expression was upregulated (Figure 2F). These data suggest a positive feedback loop of MYCASCT2-GLS-mediated glutaminolysis: MYC promotes the expression of ASCT2 and GLS, and ASCT2-GLS-mediated glutaminolysis in turn leads to upregulated MYC expression.
Glutaminase is crucial for glutamine dependency in ibrutinib-resistant mantle cell lymphoma cells
To understand the role of GLS in contributing to glutamine dependency and ibrutinib resistance, we generated MCL cells with stable GLS overexpression from ibrutinib-sensitive JeKo-1 and Rec-1 cells (Figure 3A, Online Supple-

mentary Figure S2A). We observed increased ibrutinib resistance in both cell lines engineered to overexpress GLS (Figure 3B, Online Supplementary Figure S2B). These GLSoverexpressing cells also showed enhanced dependency on glutamine for cell growth (Figure 3C, Online Supplementary Figure S2C). To investigate this further, we knocked down GLS expression in ibrutinib-resistant JeKoR and Z138 cells (Figure 3D, Online Supplementary Figure S2D) with two independent shRNA. When GLS was knocked down, these cells became more sensitive to ibrutinib treatment (Figure 3E, Online Supplementary Figure S2E) and more dependent on glucose (Figure 3F, Online Supplementary Figure S2F). Taken together, these data demonstrated that GLS was crucial for glutamine dependency in ibrutinib-resistant MCL cells, and its overexpression contributed to ibrutinib resistance.
Glutaminase-mediated glutamine dependency promotes cell proliferation and ATP production in ibrutinibresistant cells
To study the mechanism underlying cellular dependence
Figure 3. Glutaminase expression affects cell proliferation and ibrutinib sensitivity of mantle cell lymphoma cell lines. (A) JeKo-1 cells with or without stable glutaminase (GLS) overexpression (JeKo-1 EV and JeKo GLS, respectively) were harvested for immunoblotting with the indicated antibodies. (B) JeKo-1 EV and JeKo-1 GLS cells were treated with ibrutinib for 72 h at the indicated concentrations and cell viability was determined and plotted. (C) JeKo-1 EV and JeKo GLS cells were seeded for 24 h in culture medium with or without depletion of glutamine (Q) and cell viability was determined. (D) Z138 cells with or without stable GLS knockdown by shRNA (Z138-shCtrl, -shGLS#1, and -shGLS#4) were harvested for immunoblotting with the indicated antibodies. (E) Z138-shCtrl, -shGLS#1 and -shGLS#4 cells were treated with ibrutinib for 72 h at the indicated concentrations and cell viability was determined and plotted. (F) Z138-shCtrl, -shGLS#1 and -shGLS#4 cells were seeded for 24 h in culture medium with or without depletion of glucose and cell viability was determined. Two-way analysis of variance was used to determine statistical significance. ns: not significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; IBN: ibrutinib; Q: glutamine; EV: empty vector; GLS: glutaminase.
A B C D E F Haematologica | 108 June 2023 1620 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
on GLS-mediated glutamine metabolism, we examined GLS expression by depleting either glutamine or glucose in the culture medium. GLS expression was diminished by glutamine depletion but not glucose depletion in ibrutinib-resistant Z138 cells with control shRNA. Interestingly, cyclin D1 showed a similar expression pattern to GLS in these cells upon glutamine depletion (Figure 4A). Consistent with this, expression of cyclin D1 and GLS was higher in cells with acquired resistance to ibrutinib (JeKo-IBNR) or venetoclax (Rec-Ven-R and Mino-Ven-R) compared to parental cells (Online Supplementary Figure S3A). Furthermore, expression of Ki-67, as a proliferation marker, and MYC was also higher in these resistant cells. This sug-
gests that the MYC-GLS axis may promote cell cycle progression and cell proliferation. Indeed, cell proliferation was significantly reduced in ibrutinib-resistant Z138 and JeKo-R cells when GLS expression was depleted by shGLS#4 (Figure 4B, Online Supplementary Figure S3B). Z138-shGLS#4 consequently became more sensitive to glucose depletion (Figure 4C, D).

The maintenance of mitochondrial membrane potential is essential for cell proliferation.20 Glycolysis is important for maintaining mitochondrial membrane potential and glutaminolysis associates with increased mitochondrial potential.21 Both glutamine and glucose depletion in culture medium triggered a loss in mitochondrial potential in
Figure 4. Glutaminase-mediated glutamine dependency promotes cell growth in ibrutinib-resistant cells. (A) Z138-shCtrl or Z138-shGLS#4 cells were cultured in medium with or without depletion of glutamine or glucose for 24 h and harvested for immunoblotting with the indicated antibodies. The dotted line indicates that the data were from different parts of the same gels. (B) Z138-shCtrl or Z138-shGLS#4 cells were cultured for 7 days and the cell titers were monitored. (C-F) Z138-shCtrl or Z138shGLS#4 cells were seeded in complete medium containing glutamine and glucose (Q+/glucose+), or media deprived of glutamine (Q-/glucose+) or glucose (Q+/glucose-) and cultured for 24 h before proceeding with cell number counting (C), a cell viability assay (D), tetramethylrhodamine ethyl ester assay for mitochondrial membrane potential (E), or ATP generation assay (F). Twoway analysis of variance was used to determine statistical significance. ns: not significant; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. GLS: glutaminase; Q: glutamine; TMRE: tetramethylrhodamine ethyl ester; RFU: relative fluorescence units.
A B C D E F Haematologica | 108 June 2023 1621 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
Z138-shCtrl cells (Figure 4E), while Z138-shGLS#4 cells showed a further loss of mitochondrial potential in response to glucose depletion but not glutamine depletion. Moreover, glutamine-dependent ATP production was greatly decreased by GLS knockdown (Figure 4F). Together, these data demonstrate that GLS-mediated glutamine dependency correlates with cell growth and ATP production in ibrutinib-resistant MCL cells. The data support our hypothesis that targeting GLS has the thera-
peutic potential to overcome ibrutinib resistance in MCL.
Glutaminase inhibition suppresses glutaminolysis and cell growth in mantle cell lymphoma cells. Multiple GLS inhibitors are under clinical investigation for a broad range of solid tumors but not yet for MCL. These inhibitors include telaglenastat (CB-839), a specific, firstin-class GLS inhibitor that is in multiple clinical trials in
Figure 5. The glutaminase inhibitor telaglenastat suppresses growth of mantle cell lymphoma cells in vitro and in vivo. (A) Mantle cell lymphoma (MCL) cell lines were treated with telaglenastat at a 2-fold serial dilution from 20 µM and cell viability was determined at 72 h after treatment. The half maximal inhibitory concentration (IC50) values were calculated and are shown on the right. (B) A patient’s sample (Pt7) was treated with telaglenastat and cell viability was determined at the indicated time. The IC50 values were calculated as shown on the right. (C) JeKo-1 cells with or without stable glutaminase (GLS) overexpression (JeKo-1 EV and JeKo-1 GLS, respectively) were treated with telaglenastat at a 2-fold serial dilution from 20 µM and cell viability was determined at 72 h after treatment. (D) Z138-shCtrl and shGLS#4 cells were treated with telaglenastat at a 2-fold serial dilution from 20 µM and cell viability was determined at 72 h after treatment. (E) JeKo-1 or Z138 cells were treated with telaglenastat and cell cycle status was determined at 24 h after treatment. (F) JeKo-1 EV and JeKo-1 GLS and Z138 cells with or without GLS knockdown (Z138-shCtrl and Z138-shGLS#4) were treated with dimethylsulfoxide or 10 µM or 2 µM telaglenastat, respectively. The status of apoptosis was determined at 24 h after treatment. **P<0.01; ***P<0.001; ****P<0.0001. EV: empty vector; GLS: glutaminase; n/a: not available; DMSO: dimethylsulfoxide.

A B C D E F Haematologica | 108 June 2023 1622 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
combination with other regimens. We therefore chose telaglenastat to inhibit GLS in MCL cells in this study. Telaglenastat effectively inhibited the proliferation of MCL cell lines with an IC50 of 0.22-9.2 µM (Figure 5A) and patients’ MCL cells with an IC50 of 11.3 µM (Figure 5B) upon 72 hours of treatment. Telaglenastat showed greater efficacy in JeKo-1 cells with GLS overexpression than in the cells transduced with an empty vector control (Figure 5C). In line with this, Z138-shGLS#4 cells were more resistant than Z138-shCtrl cells to GLS inhibition (Figure 5D). Telaglenastat induced limited cell cycle arrest at the G0/G1 phase in JeKo-1 cells and more extensive arrest in Z138 cells (Figure 5E). Telaglenastat induced apoptosis in JeKo1 EV cells but less in JeKo-1 cells with GLS overexpression (Figure 5F, left panel). Telaglenastat also induced apoptosis in Z138 with GLS knockdown but only slightly in Z138 control cells (Figure 5F, right panel). These data suggest that telaglenastat inhibits tumor cell growth primarily by
blocking cell proliferation and not by inducing apoptosis in GLS-overexpressing cells. Indeed, telaglenastat treatment led to reduced expression of cell cycle-related proteins, including CDK4, cyclin D1, and cyclin B, in both JeKo-1 and Z138 cells (Online Supplementary Figure S3C). To assess the in vivo efficacy of GLS inhibition, we established a CDX model in immunodeficient NSG mice using Z138 cells. Telaglenastat treatment (200 mg/kg, orally, twice daily) significantly inhibited the growth of Z138 CDX tumors (P=0.004) and prolonged mouse survival (P=0.0017) (Figure 6A-C). As a systemic marker of tumor burden, serum b2-microglobulin levels were also reduced in telaglenastat-treated mice compared to vehicle-treated ones (Figure 6D). Flow cytometry analysis using the tumor-specific cell surface markers CD20 and CD5 confirmed the tumor cell growth in each CDX tumor (Online Supplementary Figure S4A, B). Expression of Ki67 was markedly suppressed by telaglenastat treatment in vivo,
Figure 6. The glutaminase inhibitor telaglenastat suppresses cell growth of Z138 cells in vivo. (A) In vivo scheme of Z138 subcutaneous xenografts treated with vehicle or telaglenastat (200 mg/kg, twice/day) orally. (B) Tumor volume was documented at the indicated time points, with statistical significance of differences calculated at the endpoint. (C) A Kaplan-Meier plot for mouse survival was generated based on mouse survival after tumor inoculation. (D) Serum b2-microglobulin levels were determined by enzyme-linked immunosorbent assay at the indicated time points. (E) Mouse body weight was monitored along with the treatments and percentage of body weight change relative to day 8 is shown. Two-way analysis of variance was used to determine statistical significance. ns: not significant; **P<0.01; ***P<0.001; ****P<0.0001. s.c.: subcutaneous; B2M: b2-microglobulin.

A B C D E Haematologica | 108 June 2023 1623 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
when compared to that of vehicle-treated controls (Online Supplementary Figure S4C). No gross abnormalities were observed in the telaglenastat-treated mice, and there was no change in body weight (Figure 6E). These data demonstrate that targeting GLS by telaglenastat can effectively and safely inhibit the growth of Z138 CDX tumors and overcome ibrutinib resistance in vivo

Telaglenastat improves the efficacy of ibrutinib or venetoclax in vitro and in vivo
Based on our data and previous studies,22 telaglenastat acts primarily by inhibiting cell proliferation and not by inducing apoptosis. The aim of combination therapy in cancer treatment is to prevent the occurrence of resistance by acting synergistically.23 We hoped that a com-
bination of the cell proliferation blocker telaglenastat with clinically proven inducers of apoptosis, such as ibrutinib (a BTK inhibitor) and venetoclax (a BCL-2 inhibitor), might be synergistic. Indeed, we observed anti-MCL synergy in all MCL cell lines exposed to either telaglenastat plus ibrutinib or telaglenastat plus venetoclax (Figure 7A, B, Online Supplementary Figure S5A, B and Online Supplementary Table S2). Consistent with this, anti-MCL synergy was observed in the primary cells from a patient with MCL and two MCL PDX models (Online Supplementary Figure S7C, D, Online Supplementary Table S2). No apparent toxicity was observed for telaglenastat in peripheral blood mononuclear cells from a healthy donor (Online Supplementary Figure S5C, D), indicating that the dose range used (0-30 µM) does not cause off-target effects in
Figure 7. Telaglenastat shows synergistic anti-mantle cell lymphoma activity in combination with ibrutinib or venetoclax in vitro. (A, B) Cell viability was determined in Z138 cells treated with telaglenastat alone or in combination with ibrutinib (A) or venetoclax (B) for 72 h. The 2-fold serial dilutions of ibrutinib (A) and venetoclax (B) are indicated on the x-axis. The 2-fold serial dilutions of telaglenastat (0-20 µM) were used alone or in combination with ibrutinib or venetoclax, but are not indicated on the x-axis. (C-F) JeKo-1 and Z138 cells were treated with telaglenastat alone or in combination with ibrutinib (C, D) or venetoclax (E, F) for 24 h. The cells were harvested for assessment of apoptosis status by flow cytometry (C, E) and for immunoblotting using the indicated antibodies (D, F). Two-way analysis of variance was used to determine statistical significance. ns: not significant; *P<0.05; **P<0.01; ****P<0.0001. Combo: combination treatment; DMSO: dimethylsulfoxide; GLS: glutaminase.
A B C D E F Haematologica | 108 June 2023 1624 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
healthy cells. The combinations of telaglenastat plus ibrutinib (Figure 7C), and telaglenastat plus venetoclax (Figure 7E) both showed potent synergy in inducing apoptosis, which was further validated by immunoblotting showing increased cleavage of caspase 3 and PARP (Figure 7D, F). Together, these data show the synergy between the clinically safe and potent drugs ibrutinib or venetoclax in combination with telaglenastat in MCL cells. To further establish the clinical relevance of our findings, we took advantage of the previously established PDX-2 mouse model to investigate the combined effects of the

drugs in vivo (Online Supplementary Figure 6A). The PDX2 cells were not completely resistant to either ibrutinib or venetoclax when tested in vitro for 24 hours, suggesting that there is a window for improving efficacy. We hypothesized that targeting GLS might augment the efficacy of ibrutinib or venetoclax in the PDX, which did indeed show partial sensitivity to ibrutinib or venetoclax (Figure 8A, B). Both telaglenastat plus ibrutinib (Figure 8A, C), and telaglenastat plus venetoclax (Figure 8B, D) showed improved efficacy beyond that of single agents. This was further confirmed by serum b2-microglobulin levels at day 86, an
Figure 8. Telaglenastat in combination with ibrutinib or venetoclax shows synergistic anti-mantle cell lymphoma activity in a patient-derived xenograft model . PDX-2 subcutaneous xenografts were established in NSG mice. The mice were randomly grouped and treated orally with vehicle (daily), telaglenastat (100 mg/kg, twice daily), ibrutinib (50 mg/kg, daily), venetoclax (10 mg/kg, daily) or combinations of drugs at day 56 after cell inoculation. (A, B) Tumor volume at the indicated time-points, with the statistical significance of differences calculated at the endpoint when the first mouse in the treatment group reached the limit of tumor size. (C, D) Kaplan-Meier plots were generated based on the survival time of each individual mouse. (E, F) Serum b2-microglobulin levels were determined by enzyme-linked immunosorbent assay at day 15 (pre-treatment), day 56 (start of treatment) and day 86 (after 30 days on treatment), before the tumors in all mice carrying the patient-derived xenograft model became measurable. Two-way analysis of variance was used to determine statistical significance. ns: not significant; *P<0.05; **P<0.01; ****P<0.0001. Combo: combination treatment; B2M: b2-microglobulin.
A B C D E F Haematologica | 108 June 2023 1625 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
early time-point of tumor growth before the tumors in all mice carrying the PDX model became measurable (Figure 8E, F). Again, we confirmed that the percentages of MCL cells present in the tumors were comparable across the treatment groups (Online Supplementary Figure S6B-D), and no gross abnormalities were observed in mice treated with any of the single agents or combinations (Online Supplementary Figure S6E, F). Taken together, these findings demonstrated improved efficacy of the two combinations tested in MCL cells, both in vitro and in vivo, paving the way for future clinical investigation of telaglenastat drug combination strategies in MCL.
Discussion
For the many types of cancer relying on glutaminolysis for energy metabolism to support rapid tumor growth,24 targeting glutaminolysis is a plausible therapeutic strategy. To examine this possibility, we determined the role of GLS in ibrutinib resistance and assessed the potential of targeting GLS alone or in combination with either ibrutinib or venetoclax in preclinical MCL models. We confirmed that GLS expression was upregulated in ibrutinib-resistant MCL cells and associated with a poor clinical outcome. The cancer hallmark MYC_TARGETs_v1 was the predominant signaling pathway associated with ibrutinibresistance in MCL patients.3 The proto-oncogene MYC promotes glutaminolysis and glutamine dependency by inducing expression of the major glutamine transporters ASCT2 and SN2 and the enzyme GLS, which is responsible for the first catalytic event of glutaminolysis. In this study, we confirmed that the expression of both GLS and ASCT2 was associated with glutaminolysis in MCL. Blocking glutamine uptake with the ASCT2-specific inhibitor V9302 led to marked reduction of both GLS expression and MYC expression. MYC knockdown resulted in reduced GLS and ASCT2, while MYC overexpression promoted their expression. Therefore, glutamine dependency in ibrutinibresistant MCL cells is likely due to a positive feedback loop of MYC-ASCT2-GLS-glutaminolysis (Online Supplementary Figure S7). It will be interesting to investigate whether this positive feedback loop in contributing to therapeutic resistance to BTK inhibitors also applies to other types of non-Hodgkin lymphoma with MYC overexpression, such as diffuse large B-cell lymphoma. Depleting GLS from formerly GLS-overexpressing cells led to release from glutamine dependency and a switch to greater dependency on glycolysis. Conversely, GLS ectopic overexpression resulted in enhanced glutamine dependency for tumor cell growth. Therefore, GLS overexpression promotes glutaminolysis. GLS depletion did not affect cell survival, but greatly affected mitochondrial membrane potential, ATP production, and cell growth. Consistent with
this, GLS inhibition by telaglenastat in cells with high GLS expression did not cause marked cell death, but significantly slowed cell growth. However, we observed strong synergy in inducing cell death when telaglenastat was combined with the potent apoptosis inducers ibrutinib or venetoclax. It is possible that targeting GLS to turn off glutaminolysis-dependent cellular energy metabolism may prime cells expressing high levels of GLS to be more sensitive to agents such as ibrutinib and venetoclax, which potently induce apoptosis. Therefore, GLS is a therapeutic target for MCL cells with glutamine dependency, and GLS inhibition-based combination treatment will act synergistically to induce tumor vulnerability. A synergistic anti-tumor activity of telaglenastat in combination with venetoclax has also been seen in acute myeloid leukemia.25 It is, therefore, possible that a combination of a GLS inhibitor with other agents used in the treatment of non-Hodgkin lymphomas, such as cytotoxic agents, could also augment the efficacy of therapy beyond that of the single agents, although this requires further investigation.
This is the first study dissecting the role of GLS in glutamine dependency associated with ibrutinib resistance in MCL and targeting glutaminolysis using the clinically investigated GLS inhibitor telaglenastat in MCL cells. Telaglenastat is currently under multiple clinical investigations in combination with other regimens to enhance the clinical efficacy over that of single agents. Our study provides preclinical evidence supporting the combination approach in targeting GLS and other vulnerabilities in treating MCL.
Disclosures
MW has received research support from Acerta Pharma, AstraZeneca, BeiGene, BioInvent, Celgene, Genmab, Genentech, Innocare, Janssen, Juno Therapeutics, Kite Pharma, Lilly, Loxo Oncology, Molecular Templates, Oncternal, Pharmacyclics, VelosBio, and Vincerx; has received speaker’s honoraria from Acerta Pharma, Anticancer Association, AstraZeneca, BeiGene, BGICS, BioInvent, CAHON, Clinical Care Options, Dava Oncology, Eastern Virginia Medical School, Epizyme, Hebei Cancer Prevention Federation, Imedex, Janssen, Kite Pharma, Leukemia and Lymphoma Society, LLC TS Oncology, Medscape, Meeting Minds Experts, Miltenyi Biomedicine GmbH, Moffit Cancer Center, Mumbai Hematology Group, OMI, OncLive, Pharmacyclics, Physicians Education Resources (PER), Practice Point Communications (PPC), and The First Afflicted Hospital of Zhejiang University; and is a consultant to AstraZeneca, BeiGene, CSTone, Deciphera, DTRM Biopharma (Cayman) Limited, Epizyme, Genentech, InnoCare, Janssen, Juno Therapeutics, Kite Pharma, Lilly, Loxo Oncology, Miltenyi Biomedicine GmbH, Oncternal, Pharmacyclics, and VelosBio. The other authors have no conflicts of interest to disclose.
Haematologica | 108 June 2023 1626 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
Contributions
MW, VCJ, YY, and LL conceived the study; LL, VCJ, and YY designed it; VCJ, LN, and YY supervised the study. LL, LN, QC, AJ, YL, YJL, YC, JV, ZC, AL, and WW acquired the data; LL, VCJ, LN, and YY analyzed and interpreted the data. LL wrote the original draft of the manuscript; VCJ and LN reviewed and edited the manuscript; MW acquired funding for the study.
Acknowledgments
The authors would like to thank the patients and their families who contributed to this research study and Paul
References
1. Jain P, Dreyling M, Seymour JF, Wang M. High-risk mantle cell lymphoma: definition, current challenges, and management. J Clin Oncol. 2020;38(36):4302-4316.
2. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507-516.
3. Zhang L, Yao Y, Zhang S, et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci Transl Med. 2019;11(491):eaau1167.
4. Duraj T, Carrion-Navarro J, Seyfried TN, Garcia-Romero N, Ayuso-Sacido A. Metabolic therapy and bioenergetic analysis: the missing piece of the puzzle. Mol Metab. 2021;54:101389.
5. Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009;69(6):2163-2166.
6. Bolzoni M, Chiu M, Accardi F, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood. 2016;128(5):667-679.
7. Patel D, Menon D, Bernfeld E, et al. Aspartate rescues S-phase arrest caused by suppression of glutamine utilization in KRasdriven cancer cells. J Biol Chem. 2016;291(17):9322-9329.
8. Márquez J, Campos-Sandoval JA, Peñalver A, et al. Glutamate and brain glutaminases in drug addiction. Neurochem Res. 2017;42(3):846-857.
9. Guo H, Zeng D, Zhang H, et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene. 2019;38(11):1802-1814.
10. Li CJ, Jiang C, Liu Y, et al. Pleiotropic action of novel Bruton's tyrosine kinase inhibitor BGB-3111 in mantle cell lymphoma. Mol Cancer Ther. 2019;18(2):267-277.
11. Zhang S, Jiang VC, Han G, et al. Longitudinal single-cell profiling reveals molecular heterogeneity and tumor-immune evolution in refractory mantle cell lymphoma. Nat Commun. 2021;12(1):2877.
12. Jiang C, Zhu Y, Zhou Z, et al. TMEM43/LUMA is a key signaling component mediating EGFR-induced NF-kappaB activation and tumor progression. Oncogene. 2017;36(20):2813-2823.
Dolber for critical editing of the manuscript.
Funding
This study was supported by various philanthropic donors.
Data-sharing statement
The RNA sequencing dataset reported in our previous study has been deposited in the European Genome-Phenome Archive (EGA) database and the access number is EGAS00001003418. Other original data are available from the corresponding author on reasonable request.
13. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21.
14. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
15. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-15550.
16. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417-425.
17. Li JCA. Modeling survival data: extending the Cox model. Sociol Method Res. 2003;32(1):117-120.
18. Ren P, Yue M, Xiao D, et al. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J Pathol. 2015;235(1):90-100.
19. Gao P, Tchernyshyov I, Chang TC, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762-765.
20. Vasan K, Clutter M, Dunne SF, et al. Genes involved in maintaining mitochondrial membrane potential upon electron transport chain disruption. Front Cell Dev Biol. 2022;10:781558.
21. Friday E, Oliver R, Welbourne T, Turturro F. Glutaminolysis and glycolysis regulation by troglitazone in breast cancer cells: relationship to mitochondrial membrane potential. J Cell Physiol. 2011;226(2):511-519.
22. Lee P, Malik D, Perkons N, et al. Targeting glutamine metabolism slows soft tissue sarcoma growth. Nat Commun. 2020;11(1):498.
23. Hanahan D, Bergers G, Bergsland E. Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105(8):1045-1047.
24. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. 2017;19:163-194.
25. Jacque N, Ronchetti AM, Larrue C, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015;126(11):1346-1356.
Haematologica | 108 June 2023 1627 ARTICLE - Targeting glutaminase in mantle cell lymphoma L. Li et al.
2 and b1 subunits of the proteasome complex
Xiang Zhou,1* Andrej Besse,2* Jessica Peter,1 Maximilian Johannes Steinhardt,1 Cornelia Vogt,1 Silvia Nerreter,1 Eva Teufel,1 Emilia Stanojkovska,1 Xianghui Xiao,1 Hannah Hornburger,1 Larissa Haertle,1 Max Mendez-Lopez,2 Umair Munawar,1 Angela Riedel,3 Seungbin Han,1 Elmer Maurits,4 Herman S. Overkleeft,4 Bogdan Florea,4 Hermann Einsele,1 K. Martin Kortüm,1 Christoph Driessen,2 Lenka Besse2# and Leo Rasche1,3#
1Department of Internal Medicine II, University Hospital of Würzburg, Würzburg, Germany; 2Experimental Oncology and Hematology, Department of Oncology and Hematology, Cantonal Hospital St. Gallen, St. Gallen, Switzerland; 3Mildred Scheel Early Career Center, University of Würzburg, Würzburg, Germany and 4Gorlaeus Laboratories, Leiden Institute of Chemistry and Netherlands Proteomics Center, Leiden, the Netherlands
*XZ and AB contributed equally as co-first authors. #LB and LR contributed equally as co-senior authors.
Abstract
Correspondence: C. Driessen christoph.driessen@kssg.ch
Received: October 6, 2022.
Accepted: January 20, 2023.
Early view: February 2, 2023.
https://doi.org/10.3324/haematol.2022.282225
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Optimal carfilzomib dosing is a matter of debate. We analyzed the inhibition profiles of proteolytic proteasome subunits b5, b2 and b1 after low-dose (20/27 mg/m2) versus high-dose (≥36 mg/m2) carfilzomib in 103 pairs of peripheral blood mononuclear cells from patients with relapsed/refractory (RR) multiple myeloma (MM). b5 activity was inhibited (median inhibition >50%) in vivo by 20 mg/m2, whereas b2 and b1 were co-inhibited only by 36 and 56 mg/m2, respectively. Coinhibition of b2 (P=0.0001) and b1 activity (P=0.0005) differed significantly between high-dose and low-dose carfilzomib. Subsequently, high-dose carfilzomib showed significantly more effective proteasome inhibition than low-dose carfilzomib in vivo (P=0.0003). We investigated the clinical data of 114 patients treated with carfilzomib combinations. High-dose carfilzomib demonstrated a higher overall response rate (P=0.03) and longer progression-free survival (PFS) (P=0.007) than low-dose carfilzomib. Therefore, we escalated the carfilzomib dose to ≥36 mg/m2 in 16 patients who progressed during low-dose carfilzomib-containing therapies. High-dose carfilzomib recaptured response (≥ partial remission) in nine (56%) patients with a median PFS of 4.4 months. Altogether, we provide the first in vivo evidence in RRMM patients that the molecular activity of high-dose carfilzomib differs from that of low-dose carfilzomib by coinhibition of b2 and b1 proteasome subunits and, consequently, high-dose carfilzomib achieves a superior anti-MM effect than low-dose carfilzomib and recaptures the response in RRMM resistant to low-dose carfilzomib. The optimal carfilzomib dose should be ≥36 mg/m2 to reach a sufficient anti-tumor activity, while the balance between efficacy and tolerability should be considered in each patient.
Introduction
The proteasome is a multi-subunit complex that is responsible for intracellular protein degradation. Only three proteasome subunits harbor proteolytic activity, b1 (caspase-like), b2 (trypsin-like), and b5 (chymotrypsin-like), which cleave peptide bonds C-terminally of acidic, basic,
and hydrophobic amino acid residues, respectively.1 Currently, proteasome inhibition is a major treatment strategy for multiple myeloma (MM).2 All currently approved proteasome inhibitors (PI) primarily target the rate-limiting b5 subunit.3 Carfilzomib (CFZ), a second-generation epoxyketone-based PI selectively targets the b5 subunit at low concentrations but co-inhibits the b2 and b1 subunits
High-dose carfilzomib achieves superior anti-tumor activity over low-dose and recaptures response in relapsed/refractory multiple myeloma resistant to lowdose carfilzomib by co-inhibiting the b
Haematologica | 108 June 2023 1628 ARTICLE - Plasma Cell Disorders
only at high concentrations, which subsequently enhances the cytotoxic activity against MM in vitro. 4
CFZ-containing therapies have shown outstanding anti-MM activity in patients with relapsed/refractory (RR) MM.5-10 As of August 2022, CFZ has been approved for RRMM in various combination regimens, such as Kd (CFZ and dexamethasone), KRD (CFZ, lenalidomide, and dexamethasone), and D-Kd (daratumumab, CFZ, and dexamethasone).11 However, with the approved dose ranging from low-dose (20-27 mg/m2) to high-dose (up to 70 mg/m2 once weekly or twice weekly), optimal CFZ dose is still a matter of debate. In clinical practice, the relationship between CFZ dose and inhibition profiles of proteasome subunits is largely unknown. Moreover, it remains to be explored whether CFZ dose escalation may recapture the clinical response in RRMM patients progressing under low-dose CFZ-containing treatments. Furthermore, real-world data on high-dose CFZ are still very limited for the currently approved Kd, KRD, and D-Kd combination regimens. Therefore, the aim of the current study was to address these issues by analyzing the inhibition profiles of proteolytic proteasome subunits b1, b2, and b5 in RRMM treated with different CFZ doses and to investigate the clinical efficacy and safety of high-dose CFZ in RRMM treated with Kd, KRD, and D-Kd combinations. In addition, we aimed to evaluate CFZ dose escalation treatment in patients with RRMM resistant to low-dose CFZ-containing treatments.
Methods Patients
First, 103 pairs of peripheral blood mononuclear cell (PBMC) samples were collected before and 3 hours after CFZ treatment from RRMM patients (defined by the current International Myeloma Working Group guidelines12) and were included in the study. The patient cohort characteristics are presented in Table 1. Second, the clinical data of 114 RRMM patients treated with CFZ combination regimens (Kd, KRD, and D-Kd) were investigated. The patient characteristics are summarized in Table 2. Third, the clinical data of 16 heavily pretreated RRMM patients, in whom we escalated CFZ dosing due to progression during low-dose CFZ-containing therapy, were analyzed. The characteristics of these patients and their CFZ-containing treatment regimens are shown in Table 3 and the Online Supplementary Table S1. Patient demographics, MM-related data, therapy responses, adverse events (AE), and survival outcomes were investigated. AE during treatment were classified according to the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. All procedures were performed in accordance with the Declaration of Helsinki and the national ethical standards. Informed consent was obtained from all patients included in the study.
Sampling and sample preparation
Proteasome inhibition in PBMC largely mirrors proteasome inhibition in plasma cells in vivo. 4,13 Therefore, we analyzed proteasome inhibition in PBMC of RRMM patients in our study. The detailed description of sampling and sample preparation is provided in the Online Supplementary Appendix
ADC: antibody drug conjugate; BCMA: B-cell maturation antigen; CAR T cell: chimeric antigen receptor modified T cell; CFZ: carfilzomib; EMD: extramedullary disease; ImiD: immunomodulatory drug; LC: light chain; PI: proteasome inhibitor; RRMM: relapsed/refractory multiple myeloma; SCT: stem cell transplant; #defined as the presence of at least one of the following: t(4;14), t(14;16), t(14;20), del17p and amp1q21.
Haematologica | 108 June 2023 1629 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al. Parameter Patients, N 103 Sex, N (%) Male Female 55 (53.4) 48 (46.6) Age in years at sampling (range) 64 (41-83) Subtype, N (%) IgG Non-IgG LC 61 (59.2) 30 (29.1) 12 (11.7) EMD, N (%) EMD adjacent to bone EMD without bone contact 47 (45.6) 13 (12.6) 34 (33.0) Cytogenetics, N (%) High risk# Standard-risk 59 (57.3) 44 (42.7) Prior lines of therapies at sampling, N(%) 1-3 4-6 >6 50 (48.6) 30 (29.1) 23 (22.3)
exposure at sampling, N (%) IMiDs Lenalidomide Pomalidomide PI Bortezomib Carfilzomib Monoclonal antibody Elotuzumab Daratumumab Prior SCT Autologous SCT Allogeneic SCT BCMA-directed therapy ADC CAR T cell Bispecific antibody 100 (97.1) 72 (69.9) 89 (86.4) 97 (94.2) 37 (35.9) 87 (84.5) 85 (82.5) 14 (13.6) 3 (2.9) 5 (4.9) 2 (1.9)
dosing, N (%) 20 mg/m2 27 mg/m2 36 mg/m2 56 mg/m2 23 (22.3) 27 (26.2) 38 (36.9) 15 (14.6)
Drug
CFZ
Table 1. Characteristics of 103 relapsed/refractory multiple myeloma patients treated with respective carfilzomib doses.
Parameter All Kd KRD D-Kd P Patients, N 114 33 71 10 Sex, N (%) Male Female 76 (66.7) 38 (33.3) 17 (51.5) 16 (48.5) 51 (71.8) 20 (28.2) 8 (80.0) 2 (20.0) 0.31 Age in years at diagnosis of MM, median (range) Age in years at Kd/KRD/D-Kd, median (range) 58 (34-81) 63 (39-83) 58 (45-73) 65 (54-76) 57 (34-81) 62 (39-83) 56 (44-71) 63 (52-80) 0.69 0.33 Subtype, N (%) IgG non-IgG LC 68 (59.6) 32 (28.1) 14 (12.3) 22 (66.7) 8 (24.2) 3 (9.1) 41 (57.8) 19 (26.8) 11 (15.5) 5 (50.0) 5 (50.0) 0 (0.0) 0.12 Cytogenetics, N (%) High-risk# Standard-risk 63 (55.2) 51 (44.8) 18 (54.5) 15 (45.5) 28 (39.4) 43 (60.6) 7 (70.0) 3 (30.0) 0.21 Prior lines of therapy, N (%) 1-3 4-6 >6 72 (63.1) 28 (24.6) 14 (12.3) 17 (51.5) 9 (27.3) 7 (21.2) 48 (67.6) 17 (23.9) 6 (8.5) 7 (70.0) 2 (20.0) 1 (10.0) 0.22 Pretreatment prior to the current line of therapy, N (%) IMiDs Lenalidomide Pomalidomide PI Bortezomib Carfilzomib Monoclonal antibodies Daratumumab Elotuzumab Exposed/Refractory 80 (70.2)/ 50 (43.9) 80 (70.2)/40 (35.1) 38 (33.3)/34 (29.9) 106 (92.9)/53 (46.5) 104 (91.2)/49 (42.9) 18 (15.8)/12 (10.5) 32 (28.1)/30 (26.3) 18 (15.8)/12 (10.5) Exposed/Refractory 28 (84.8)/19 (57.6) 28 (84.8)/13 (39.4) 16 (48.5)/14 (42.4) 28 (84.8)/17 (51.5) 27 (81.8)/16 (48.5) 6 (18.2)/3 (9.1) 9 (27.3)/9 (27.3) 7 (21.2)/4 (12.1) Exposed/Refractory 43 (60.6)/26 (36.6) 43 (60.6)/23 (32.4) 17 (23.9)/15 (21.1) 68 (95.8)/32 (45.1) 67 (94.4)/30 (42.3) 8 (11.3)/5 (7.0) 17 (23.9)/16 (22.5) 10 (14.1)/7 (9.9) Exposed/Refractory 9 (90.0)/5 (50.0) 9 (90.0)/4 (40.0) 5 (50.0)/5 (50.0) 10 (100.0)/4 (40.0) 10 (100.0)/3 (30.0) 4 (40.0)/4 (40.0) 6 (60.0)/5 (60.0) 1 (10.0)/1 (10.0) 0.15/0.25 0.15/0.24 0.38/0.39 0.48/0.08 0.47/0.11 0.46/0.49 0.36/0.36 0.40/0.43 Prior SCT, N(%) Autologous SCT Allogeneic SCT 100 (87.7) 6 (5.3) 29 (87.9) 0 (0.0) 62 (87.3) 6 (8.5) 9 (90.0) 0 (0.0) 0.40 0.43 Treatment response and toxicity Parameter All Kd KRD D-Kd P Number of cycles, median (range) 3 (1-10) 3 (1-20) 3 (1-5) 0.11 Maximum CFZ dose, N (%) 15 mg/m2 20 mg/m2 27 mg/m2 36 mg/m2 45 mg/m2 56 mg/m2 5 (4.4) 8 (7.0) 64 (56.1) 9 (7.9) 1 (0.9) 27 (23.7) 2 (6.1) 4 (12.1) 7 (21.2) 1 (3.0) 0 (0.0) 19 (57.6) 3 (4.2) 3 (4.2) 54 (76.1) 7 (9.9) 1 (1.4) 3 (4.2) 0 (0.0) 1 (10.0) 3 (30.0) 1 (10.0) 0 (0.0) 5 (50.0) 0.35 Maximum LEN dose (QD), N (%) 5 mg 10 mg 15 mg 20 mg 25 mg / / / / / 8 (11.3) 10 (14.1) 13 (18.3) 2 (2.8) 38 (53.5) / / / / / Best response, N (%) CR VGPR PR MR PD na 7 (6.1) 29 (25.4) 36 (31.6) 19 (16.7) 21 (18.4) 2 (1.8) 1 (3.0) 4 (12.1) 11 (33.3) 8 (24.3) 8 (24.3) 1 (3.0) 4 (5.6) 24 (33.8) 22 (31.0) 10 (14.1) 10 (14.1) 1 (1.4) 2 (20.0) 1 (10.0) 3 (30.0) 1 (10.0) 3 (30.0) 0 (0.0) 0.04
Basic characteristics
Continued on following page. Haematologica | 108 June 2023 1630 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
Table 2. Characteristics of 114 relapsed/refractory multiple myeloma patients treated with Kd, KRD or D-Kd.
CFZ: carfilzomib; CR: complete remission; D-Kd: daratumumab, carfilzomib, dexamethasone; ImiDs: immunomodulatory drugs; Kd: carfilzomib, dexamethasone; KRD: carfilzomib, lenalidomide, dexamethasone; LC: light chain; LEN: lenalidomide; MM: multiple myeloma; MR: minor response; na: not available; PD: progressive disease; PI: proteasome inhibitors; PR: partial remission; SCT: stem cell transplant; VGPR: very good partial remission; #defined as presence of at least one of the following: del(17p), amp1q21, t(4;14), t(14;16) and t(14;20). IgG: immunoglobulin G.
Proteasome b-subunits profiling with activity-based proteasome probes labeling
Proteasome subunit activity was assessed using protein lysate from PBMC. Briefly, PBMC pellets were lysed with a lysis buffer. The lysates were then labeled for 1 hour at 37°C with subunit-selective, fluorescent, activity-based proteasome probes (ABPP) that visualized b1, b2, and b5 activity of the constitutive proteasome and immune proteasomes as previously described by de Bruin et al. 14 (Online Supplementary Figure S1), and proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDSPAGE). In order to limit variability, samples from respective patients were always run on the same gel and to minimize differences in gel exposure, a sample prepared from a pool of PBMC obtained from healthy donors was run on each gel. After SDS-PAGE, gel images were acquired using Fusion Solo S Western Blot and Chemi Imaging System (Vilber Lourmat, Collégien, France). Active proteasome subunits were quantitatively assessed in each sample by densitometry using Fiji (an open-source image processing package based on ImageJ)15 and normalized to a fluorescence intensity obtained from the PBMC sample on each gel. For each sample, the activity of each proteolytically active b subunit was calculated by summarizing the normalized band fluorescence intensity of the respective constitutive (c) proteasome subunit and the corresponding subunit of the immuno (i) proteasomes (i.e., b1c+i, b2c+i, and b5c+i). The
inhibition of subunit activity after CFZ exposure in relation to a paired sample before CFZ exposure was calculated for each individual patient (Figure 1A, B). Total proteasome activity was defined as the average activity of the b1c+i, b2c+i, and b5c+i proteasome subunits. For further analysis, we dichotomized the patients into two groups: low-dose (≤27 mg/m2) and high-dose (≥36 mg/m2) CFZ.
Kd, KRD, and D-Kd regimens
A detailed description of the drug administrations and treatment regiments is provided in the Online Supplementary Appendix.
Statistical evaluation
Statistical analyses were performed using GraphPad Prism 9.0 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was set at 0.05 (P value <0.05). A more specific description of the statistical evaluation used in this study is provided in Online Supplementary Appendix.
Results
Inhibition profile of proteolytic proteasome subunits at different carfilzomib doses in relapsed/refractory multiple myeloma patients
CFZ has a very short half-life of <30 minutes (min) and
Treatment response and toxicity Parameter All Kd KRD D-Kd P Non-hematologic toxicities grade
N (%) Cardiotoxicity Pneumonia Fever of unknown origin Respiratory infection Fatigue Renal failure Thromboembolic events
associated infection Liver enzyme elevated Bacterial meningitis Delirium Infection of urinary tract Herpes zoster Soft tissue infection 11 (9.6) 5 (4.4) 3 (2.6) 11 (9.6) 2 (1.8) 4 (3.5) 1 (0.9) 1 (0.9) 2 (1.8) 1 (0.9) 1 (0.9) 2 (1.8) 1 (0.9) 1 (0.9) 5 (15.2) / 3 (9.1) 5 (15.1) 2 (6.1) 1 (3.0) 1 (3.0) 1 (3.0) / / / / / / 6 (8.5) 5 (7.0) / 6 (8.5) / 3 (4.2) / / 2 (2.8) 1 (1.4) 1 (1.4) 1 (1.4) 1 (1.4) / / / / / / / / / / / / 1 (10.0) / 1 (10.0) Hematologic toxicities grade ≥3, N (%) Anemia Leukopenia Thrombocytopenia Neutropenia 29 (25.4) 34 (29.8) 37 (32.4) 30 (26.3) 6 (18.2) 7 (21.2) 14 (42.4) 2 (6.1) 19 (26.8) 21 (29.6) 18 (25.4) 23 (32.4) 4 (40.0) 6 (60.0) 5 (50.0) 5 (50.0) 0.29 0.30 0.35 0.30
≥3,
Catheter
Haematologica | 108 June 2023 1631 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
reaches full proteasome inhibition within 60-90 min after CFZ treatment in peripheral tissues in vivo. 16 Additionally, in vitro, proteasome activity recovers to baseline after 24 hours from CFZ treatment and remains largely constant at later time points.4 Therefore, we investigated proteolytic proteasome subunit activity in PBMC before and 3 hours after CFZ treatment, and we combined the activity of constitutive (c) and immuno-proteasome (i), i.e., b1c+i, b2c+i, and b5c+i. In our cohort, 23, 27, 38, and 15 patients received 20, 27, 36, and 56 mg/m2 of CFZ, respectively (Table 1). All 103 patients were treated with twice-weekly CFZ regimen, and the PBMC samples were collected on day 1 before and 3 hours after CFZ infusion. Generally, proteasome subunit activity decreased with increasing CFZ dose, with the strongest inhibition of b5c+i, followed by b2c+i, and b1c+i at all dose levels (Figure 1B). Biologically meaningful inhibition (median residual activity <50% of baseline) of b5c+i was already achieved with 20 mg/m2, whereas b 2c+i and b 1c+i activity remained largely unchanged at this dosing level. Meaningful co-inhibition of b2c+i and b1c+i was observed only at higher doses of 36 and 56 mg/m2, respectively. Interestingly, the active b2c+i and b 1c+i subunits were moderately upregulated upon b5c+i inhibition by CFZ only at 20 mg/m2, possibly contributing to compensatory activity. In patients treated with 56 mg/m2, all b 1c+i, b 2c+i, and b 5c+i were inhibited by >50% compared to baseline before CFZ. We noticed a significant difference in b 2c+i and b 1c+i subunit inhibition between the groups treated with 20 mg/m2 versus 36 mg/m2 with a median residual activity of b2c+i of 120.1% (95% confidence interval [CI]: 76.8-174.6) versus 47.2% (95% CI: 31.2-53.8; P<0.0001); median residual activity of b1c+i of 132.8% (95% CI: 62.4-188.0) versus 60.7% (95% CI: 39.3-77.1; P=0.0009). The same held true for the groups CFZ treated with 27 mg/m2 versus 56 mg/m2 with a median residual activity of b2c+i of 65.9% (95% CI: 32.588.4) versus 39.5% (95% CI: 17.8-54.9; P=0.035); median residual activity of b1c+i of 81.2% (95% CI: 53.8-116.9) versus 42.1% (95% CI: 23.8-69.5; P=0.017). In contrast, b5c+i inhibition did not differ significantly between the groups at 20 mg/m2 versus 36 mg/m2 or 27 mg/m2 versus 56 mg/m2 (P>0.05). In terms of total proteasome inhibition (average of b1c+i, b2c+i, and b5c+i), we observed a significant difference between the groups treated with 20 mg/m2 versus 36 mg/m2 with a median residual total proteasome activity of 91.7% (95% CI: 56.1-133.1) versus 41.7% (95% CI: 28.2-52.7; P=0.0003). Similarly, CFZ treatment with 56 mg/m2 showed significantly superior total proteasome inhibition over 27 mg/m2 with a median residual total proteasome activity of 55.0% (95% CI: 37.8-86.7) versus 33.2% (95% CI: 23.8-35.7; P=0.019). We then dichotomized the CFZ dosing into two groups: low-dose (≤27 mg/m2) and high-dose (≥ 36 mg/m2) CFZ. Between both groups, b 2c+i and b 1c+i inhibition significantly differed
Table 3. Characteristics of patients with relapsed/refractory multiple myeloma progressing from low-dose carfilzomib and treated with carfilzomib dose escalation.
≥3 after CFZ
Hematologic toxicities grade ≥3 after CFZ dose escalation,
ADC: antibody drug conjugate; BCMA: B-cell maturation antigen; BITE: bispecific T-cell engager; CAR T cell: chimeric antigen modified T cell; CFZ: carfilzomib; EMD: extramedullary disease; ImiDs: immunomodulatory drugs; MM: multiple myeloma; MR: minor response; PD: progressive disease; PI: proteasome inhibitors; PR: partial remission; RR: relapsed/refractory; SCT: stem cell transplant; VGPR: very good partial remission; IgG: immunoglobulin G; #defined as presence of at least one of the following: del(17p), amp1q21, t(4;14), t(14;16) and t(14;20); ‡defined as refractory to bortezomib, carfilzomib, lenalidomide, pomalidomide and daratumumab.
Parameter Patients, N 16 Sex, N (%) Male Female 10 (62) 6 (38) Age in years at diagnosis of MM, median (range) 65 (41-79) Age in years at CFZ dose escalation, median (range) 71 (45-83) Time in months between diagnosis of MM and CFZ dose escalation, median (range) 61 (22-144) Subtype, N (%) IgG non-IgG 11 (69) 5 (31) Cytogenetics, N (%) High-risk# Standard-risk 11 (69) 5 (31) EMD, N (%) EMD without bone contact no EMD 6 (38) 10 (62) Prior lines of therapy, N (%) 2-4 5-7 ≥ 8 5 (31) 5 (31) 6 (38) Pretreatment prior to the current line of therapy, N (%) IMiDs Lenalidomide Pomalidomide PI Bortezomib Carfilzomib (low-dose) Monoclonal antibodies Daratumumab Elotuzumab Penta-refractoryǂ Prior SCT Autologous SCT BCMA-directed therapy ADC CAR T cell BITE Exposed/Refractory 15 (94)/13 (81) 15 (94)/11 (69) 14 (88)/13 (81) 16 (100)/15 (94) 16 (100)/14 (88) 8 (50)/8 (50) 15 (94)/15 (94) 3 (19)/3 (19) 6 (38) 12 (75) 2 (13) 2 (13) 1 (6) CFZ dose escalation, N(%) 15→36 mg/m2 20→56 mg/m2 27→36 mg/m2 27→56 mg/m2 1 (6) 1 (6) 6 (38) 8 (50) Best response to CFZ dose escalation, N (%) VGPR PR MR PD 5 (31) 4 (25) 6 (38) 1 (6) Non-hematologic toxicities grade
dose escalation,
Pneumonia Fever of unknown origin Fatigue Cardiotoxicity Corona virus infection HKU1 Renal failure 4 (25) 2 (13) 1 (6) 1 (6) 1 (6) 1 (6)
N(%)
N(%) Anemia Leukopenia Thrombocytopenia Neutropenia 10 (63) 10 (63) 8 (50) 4 (25)
Haematologica | 108 June 2023 1632 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
with a median residual activity of b2c+i of 81.9% (95% CI: 63.3-104.6) versus 45.5% (95% CI: 26.8-52.8; P=0.0001; median residual activity of b1c+i of 92.8% (95% CI: 65.7127.6) versus 51.0% (95% CI: 39.3-69.5; P=0.0005). However, high-dose CFZ did not show significantly superior b5c+i inhibition (P>0.05) compared to low-dose CFZ. Taken together, high-dose CFZ demonstrated superior total proteasome inhibition compared to low-dose CFZ through
the co-inhibition of b2c+i and b1c+i proteasome subunits activity with a median residual total proteasome activity of 65.8% (95% CI: 47.7-91.8) versus 35.7% (95% CI 28.2-43.7; P=0.0003) (Figure 2A-E).
High-dose carfilzomib showed more effective antimultiple myeloma activity than low-dose
In order to address the issue of whether high-dose CFZ
Figure 1. Experimental design and a representative gel image. In (A) the experimental design of our study is depicted. We collected 103 paired peripheral blood mononuclear cell (PBMC) samples (before and 3 hours [h] after carfilzomib [CFZ]) from patients with RRMM. CFZ was given at different doses. PBMC were lysed and then proteolytically active constitutive (c) proteasome and immuno (i) proteasome subunits, b1c+i, b2c+i, and b5c+i were labeled using activity-based proteasome probes. Subsequently, proteins and labeled proteasome subunits were separated with SDS-PAGE, and the b1c+i, b2c+i, and b5c+i activity was evaluated using densitometric analysis. Panel (B) shows shows a representative gel (SDS-PAGE) with proteasome subunits activity before and 3 h after CFZ.

A B Haematologica | 108 June 2023 1633 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
could achieve more effective anti-MM efficacy and superior progression-free survival (PFS) compared to lowdose CFZ in routine clinical practice, we investigated the real-world data of 114 RRMM patients who were treated with three currently approved CFZ-containing combinations Kd, KRD, and D-Kd. The median age at therapy initiation was 63 years (range, 39-83 years). In our study, 33, 71, and ten patients received Kd, KRD, and D-Kd combinations, respectively. In total, 20, 11 and five patients were treated with high-dose CFZ in subgroups, Kd, KRD, and D-Kd, respectively. The remaining 78 patients received low-dose CFZ in three different combinations. CFZ and/or LEN dosing was individually determined by the treating physician based on the expected tolerability of each pa-
tient. The median number of prior therapies was two (range, 1-12). Patients received a median of three (range, 1-20) cycles of CFZ combinations. Patient characteristics are summarized in Table 2. The median follow-up time was 15.6 months in this cohort. Overall, 72 patients achieved partial remission (PR) or better, yielding an overall response rate (ORR) of 64.3% in 112 patients with response data (Table 2). Notably, in patients treated with the Kd combination, high-dose CFZ showed a signi ficantly higher ORR compared to low-dose CFZ (ORR: 73.8% vs. 15.4%; P=0.003) (Online Supplementary Figure S2 ). We then analyzed survival outcome in the entire group and found that patients who had received highdose CFZ showed significantly superior PFS compared to
Figure 2. Proteasome subunit activity evaluated in peripheral blood mononuclear cells of multiple myeloma patients undergoing carfilzomib treatment and their outcome upon dose escalation. Panels (A-C) display proteasome subunit activity of b1c+i, b2c+i, and b5c+i pre-carfilzomib (CFZ) vs. 3 hours (h) post CFZ. Panel (D) demonstrates the whole proteasome activity at respective doses prior CFZ vs. 3 h post CFZ treatment. In (E), residual proteasome (subunit) activity 3 h after CFZ infusion is illustrated. ns: not significant; P value of two-tailed unpaired Student’s t-test. c: constitutive; i: immuno.

A D E B C Haematologica | 108 June 2023 1634 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
low-dose (median PFS: 11.7 vs. 4.5 months; P=0.007) (Online Supplementary Figure S3). In the subgroup analysis of Kd, high-dose CFZ likewise demonstrated improved PFS over low-dose (median PFS: 11.7 vs . 2.1 months; P=0.0006) (Figure 3A, B). In the D-Kd subgroup, we also observed superior PFS in the high-dose CFZ group than in the low-dose CFZ group. However, owing to the low number of cases in this subgroup, the difference in PFS was not statistically significant. (Figure 3C, D). In patients treated with KRD, PFS was significantly longer in patients who had received high-dose CFZ than low-dose (median PFS: 13.2 vs. 5.6 months; P=0.02), while LEN dose did not aff ect PFS in our cohort. (Figure 4). Overall, the most common non-hematologic AE grade ≥3 included cardiotoxicity (n=11, 9.6%) and respiratory infections (n=11, 9.6%). Importantly, among the 11 patients who suffered from cardiotoxicity ≥ 3, only two of them received high-dose CFZ, while the remaining nine patients were treated with low-dose CFZ. Regarding hematologic AE, 29 (25.4%), 34 (29.8%), 37 (32.4%) and 30 (26.3%) patients developed anemia, leukopenia, thrombocytopenia , and neutropenia grade ≥3, respectively. Of note, in the entire group, the frequencies of hematologic AE grade ≥3 were not significantly higher in patients who received high-dose compared to low-dose CFZ (Online Supplementary Figure S4).
Carfilzomib dose escalation recaptured clinical response in relapsed/refractory multiple myeloma patients who were resistant to low-dose carfilzomib

Considering the afore-mentioned findings, we treated 16 patients with RRMM who progressed during low-dose CFZ-containing therapy by escalating CFZ dosing as a personalized treatment approach. The median age of the patients was 71 years (range, 45-83 years), and high-risk cytogenetics was present in 11 (69%) patients. The patients were heavily pretreated with a median of six (range, 2-13) lines of therapies. In prior lines of therapies, all 16 patients received at least one PI, and the vast majority (n=15, 94%) was pretreated with at least one immunomodulatory drug (IMiD). Daratumumab was administered to 15 (94%) patients in prior treatments. All 16 patients were refractory to their last treatment line. Eleven (69%) patients were refractory to LEN, 13 (81%) to pomalidomide, 14 (88%) to bortezomib (BTZ), eight (50%) to lowdose CFZ, and 15 (94%) patients were refractory to daratumumab in prior lines of therapy. Six (38%) patients were penta-refractory (daratumumab, BTZ, low-dose CFZ, LEN, and pomalidomide). One patient had relapsed MM after treatment with a B-cell maturation antigen (BCMA)targeted bi-specific antibody, and two patients relapsed after chimeric antigen receptor-modi fied T-cell (CAR T)
Figure 3. Survival outcome in multiple myeloma patients that received immunomodulatory imide drug-free carfilzomib-containing regimens: Kd and D-Kd. Panels demonstrate progression-free survival (PFS) and overall survival (OS) of patients treated with high-dose carfilzomib (CFZ) vs. low-dose CFZ in the group of patients who (A, B) received Kd (carfilzomib, dexamethasone) and (C, D) recieved D-Kd (carfilzomib, daratumumab, dexamethasone).
Haematologica | 108 June 2023 1635
A C D B
ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
therapy. In the current line of therapy, all 16 patients showed progression during low-dose CFZ-containing combination regimens (range of CFZ dose, 15-27 mg/m2 twice weekly), and six patients presented true extramedullary disease (EMD) without bone contact. Therefore, we escalated the CFZ dose in these patients to high-dose (36 or 56 mg/m2), while the doses and schedules of all other anti-MM drugs remained the same (Table 3; Online Supplementary Table S1). After a median time to response of 0.7 (range, 0.3-1.1) months, high-dose CFZ recaptured response in nine (56%) patients, including five and four patients with very good partial remission (VGPR) and PR, respectively. Additionally, high-dose CFZ controlled disease progression (minor response) in six (38%) patients, yielding a clinical benefit rate of 94%. Importantly, four of six patients with true EMD achieved a PR (n=2) or VGPR (n=2), and one patient showed a minor response after CFZ dose escalation. This finding underlined that even highrisk RRMM patients with EMD might benefit from CFZ dose escalation. The only patient who progressed after CFZ dose escalation harbored multiple high-risk features, such as high-risk cytogenetics (amp1q21, t(4;14)) and EMD,17-19 which were potentially associated with aggressive disease and drug resistance, suggesting that CFZ resistance may be related to factors other than CFZ dosing.20
Serial PBMC samples before and 3 hours after CFZ administration at different dose levels were evaluated in a patient with CFZ dose escalation (patient #1 in the Online Supplementary Table S1). As expected, the b2c+i proteasome subunit was inhibited more effectively at 36 mg/m2 (residual β2c+i activity: 52.8%) than at 20 mg/m2 (residual b2c+i activity: 76.5%), whereas the b5c+i subunit was already meaningfully inhibited at 20 mg/m2 (residual b5c+i activity: 29.1%) (Online Supplementary Figure S5). After a median follow-up time of 13.0 months, high-dose CFZ achieved a median PFS of 4.4 (95% CI: 4.0-4.8) months and a median overall survival (OS) of 8.9 (95% CI: 6.0-11.7) months in our cohort of patients progressing under low dose CFZ therapy (Figure 5; Online Supplementary Figure S6). However, increased doses of CFZ may cause more severe side effects. Indeed, non-hematologic AE grade ≥3 were present in six (38%) patients after CFZ dose escalation to high-dose, while only four (25%) patients showed non-hematologic AE grade ≥3 during low-dose CFZ-containing treatments. Pneumonia (n=4, 25%) was the most common non-hematologic AE observed after CFZ dose escalation. Interestingly, cardiotoxicity grade ≥3 was observed in only one (6%) patient after CFZ dose escalation. However, two (12%) patients experienced cardiotoxicity grade ≥3 during the low-dose CFZ phase before dose es-
Figure 4. Survival outcome in multiple myeloma patients that received immunomodulatory imide drug-containing carfilzomibcontaining regimen. (A) Progression-free survival (PFS) and (B) overall survival (OS) of patients treated with high-dose carfilzomib (CFZ) vs. low-dose CFZ in patients who received KRD (carfilzomib, lenalidomie, dexamethasone). (C, D) PFS and OS of patients who received lenalidomide (LEN) 25 mg vs. < 25 mg.

A C D B Haematologica | 108 June 2023 1636 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
calation, and both patients tolerated high-dose CFZ without cardiotoxicity. Importantly, two patients who achieved PR with high-dose CFZ required CFZ dose reduction to 27 and 20 mg/m2 due to cardiotoxicity and fatigue, respectively. Notable, both patients showed prompt disease progression after the CFZ dose reduction ( Online Supplementary Table S1).

Discussion
In our study, high-dose CFZ showed significantly more effective proteasome inhibition than low-dose CFZ in vivo by co-inhibiting the b2 and b1 proteasome subunits, suggesting that patients resistant to low-dose CFZ-containing therapies could recapture the clinical response by escalating the CFZ dose. Moreover, high-dose CFZ resulted in longer PFS in RRMM patients treated with CFZcontaining combinations.
In the last few years, CFZ has been evaluated in different dosing regimens within clinical trials. In the phase III ENDEAVOR study evaluating the Kd combination (CFZ 56 mg/m2 twice weekly), CFZ showed a significantly longer PFS compared to BTZ (median: 18.7 months in the Kd group vs. 9.4 months in the BTZ group) in RRMM patients.6 Moreover, additional use of daratumumab to the Kd regimen (D-Kd) further improved PFS in RRMM (median: not reached in the D-Kd group vs. 15.8 months in the Kd group), as suggested by the phase III CANDOR trial.5 In terms of immunomodulatory imide drugs (IMiD)-containing combinations, the KRD regimen demonstrated superior PFS compared with the control group RD (lenalidomide, dexamethasone) (median: 26.3 months in the KRD group vs. 17.6 months in the RD group). However, only low-dose CFZ (27 mg/m2 twice weekly) was administered to avoid severe AE in the KRD combination.7 Indeed, at present, the optimal CFZ dose remains controversial in clinical practice.
High-dose once weekly CFZ is being developed as a mainstream regimen to improve patients’ compliance with a more convenient proteasome inhibition.21,22 In the Kd combination, Moreau et al. reported that CFZ 70 mg/m2 once weekly significantly improved PFS and ORR compared with 27 mg/m2 twice weekly in RRMM.21 Moreover, in real-world data, high-dose CFZ (56 mg/m2 twice weekly or 70 mg/m2 once weekly) likewise showed significantly superior patients survival outcomes when compared to low-dose CFZ (20-27 mg/m2 twice weekly) in patients treated with Kd combination.23 In contrast, Ailawadhi et al. did not observe a significant ORR or PFS benefit with twice-weekly CFZ 56 mg/m2 over 27 mg/m2 in the Kd regimen.17 Regarding the IMiD-containing CFZ combination, that is, the KRD regimen, our results demonstrate that high-dose CFZ can significantly improve PFS, while the LEN dose does not show any significant impact on patient outcome. Importantly, in different data sets, the safety profile of high-dose CFZ appears similar to that of low-dose CFZ.17,21,24,25 Here, we provide the first in vivo evidence in a clinical setting that the molecular activity of high-dose CFZ (≥36 mg/m2) differs from that of low-dose CFZ (≤27 mg/m2) by means of b2 and b1 proteasome subunit co-inhibition, which may be a potential mechanism to overcome low-dose drug resistance in RRMM patients. Although the patients in the current study were relatively heterogeneous, including patients treated with Kd, KRD and D-Kd regimens, high-dose CFZ showed improved survival outcome compared with low-dose in the entire cohort as well as in each subgroup Kd, KRD and D-Kd. This is in line with our previous in vitro findings4 and may explain the superior anti-MM activity of higher doses of CFZ compared to lower doses. PI-resistant cells change the level of the b2 and b1 proteasome subunits and become insensitive to sole b5 inhibition. At the same time, co-inhibition of other subunits, especially the b2 subunit, is able to overcome PI resistance.4,26,27 However, in our study, there were also patients who did not respond
A B Haematologica | 108 June 2023 1637 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
Figure 5. Survival outcome in multiple myeloma patients that received carfilzomib dose escalation. (A) Progression-free survival (PFS) and (B) overall survival (OS) of 16 relapsed/refractory multiple myeloma patients treated with carfilzomib (CFZ) dose escalation.
to CFZ dose-escalation, meaning that high-dose CFZ was not a “game changer” in all patients with heavily pretreated RRMM. In contrast, disease progression during CFZ-containing treatment may be related to mechanisms other than proteasome inhibition, such as high-risk cytogenetics, EMD, and epigenetic changes.18,19,28 The underlying resistance mechanisms should be further investigated. In addition, it could not be excluded that low-dose CFZ might potentially achieve a cumulative proteasome inhibition effect on the second day in twice-weekly regimens, and this issue should be addressed in future studies. In recent years, marizomib, a third generation blactone- γ -lactam PI, has been developed for the treatment of RRMM, and this novel agent is characterized by its irreversible inhibition of all three proteolytic subunits b5, b2, and b1 of the proteasome complex.29,30 Marizomib has shown high anti-MM activity in RRMM patients and may overcome BTZ and CFZ resistance.31,32 Therefore, marizomib-containing regimens might be a further option for patients resistant to low-dose CFZ.
Taken together, high-dose CFZ demonstrates superior anti-MM effect to low-dose CFZ by co-inhibiting b2 and b1 proteasome subunits, and resistance to low-dose CFZ does not exclude sensitivity to high-dose CFZ. The optimal CFZ dose should be ≥36 mg/m2 to achieve sufficient anti-MM activity, while the balance between efficacy and tolerability should be taken into account during treatment decision-making in each patient. In patients with RRMM refractory to low-dose CFZ, dose escalation to ≥36 mg/m2 may be worthwhile, as suggested by our data. Our findings provide a rationale for selecting high-dose CFZ to achieve
References
1. Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(1):12-36.
2. Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017;36(4):561-584.
3. Nunes AT, Annunziata CM. Proteasome inhibitors: structure and function. Semin Oncol. 2017;44(6):377-380.
4. Besse A, Besse L, Kraus M, et al. Proteasome inhibition in multiple myeloma: head-to-head comparison of currently available proteasome inhibitors. Cell Chem Biol. 2019;26(3):340351.
5. Dimopoulos M, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): results from a randomised, multicentre, open-label, phase 3 study. Lancet. 2020;396(10245):186-197.
6. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38.
maximum proteasome inhibition, an optimal anti-MM effect, and to avoid adaptive resistance. The mechanisms of toxicity, particularly cardiotoxicity, of high-dose CFZ remain to be addressed. In addition, future work is required to compare the effects of high-dose CFZ between onceweekly and twice-weekly schedules.
Disclosures
No conflicts of interest to disclose.
Contributions
XZ, AB, KMK, CD, LB and LR designed the research. XZ, AB, CV, SN, ET, ES, LH, MML, UM, SH and LB performed the experiments. XZ, JP, MJS, XX, HH, AR, HE, KMK and LR provided patient samples and clinical data. EM, BF and HSO provided the proteasome activity based probes. XZ, AB, CD, LB and LR wrote the manuscript which was approved by all authors and all authors analyzed and interpreted the data.
Funding
This work was supported by Swiss Cancer Research Foundation (KFS-4990-02-2020) and the German Cancer Aid via the Mildred Scheel Early Career Center Würzburg (MSNZ Würzburg), Interdisciplinary Center for Clinical Research Würzburg (IZKF Würzburg), German Research Foundation (DFG) (KFO5001), and Stifterverband und Verein Hilfe im Kampf gegen Krebs.
Data-sharing statement
The data generated in this study are available upon request from the corresponding author.
7. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372(2):142-152.
8. Moreau P, Dimopoulos MA, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet. 2021;397(10292):2361-2371.
9. Zhou X, Fluchter P, Nickel K, et al. Carfilzomib based treatment strategies in the management of relapsed/refractory multiple myeloma with extramedullary disease. Cancers (Basel). 2020;12(4):1035.
10. Zhou X, Ruckdeschel A, Peter J, et al. Salvage therapy with "Dara-KDT-P(A)CE" in heavily pretreated, high-risk, proliferative, relapsed/refractory multiple myeloma. Hematol Oncol. 2022;40(2):202-211.
11. Jayaweera SPE, Wanigasinghe Kanakanamge SP, Rajalingam D, Silva GN. Carfilzomib: a promising proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Front Oncol. 2021;11:740796.
12. Rajkumar SV, Harousseau JL, Durie B, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel
Haematologica | 108 June 2023 1638 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
1. Blood. 2011;117(18):4691-4695.
13. Kleiveland CR. Peripheral Blood Mononuclear Cells. In: Verhoeckx K, Cotter P, Lopez-Exposito I, Kleiveland C, Lea T, Mackie A, et al., editors. The Impact of Food Bioactives on Health: in vitro and ex vivo models. Cham (CH): Springer; 2015.
14. de Bruin G, Xin BT, Kraus M, et al. A set of activity-based probes to visualize human (immuno)proteasome activities. Angew Chem Int Ed Engl. 2016;55(13):4199-4203.
15. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an opensource platform for biological-image analysis. Nat Methods. 2012;9(7):676-682.
16. Alsina M, Trudel S, Furman RR, et al. A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clin Cancer Res. 2012;18(17):4830-4840.
17. Ailawadhi S, Sexton R, Lentzsch S, et al. Low-dose versus highdose carfilzomib with dexamethasone (S1304) in patients with relapsed-refractory multiple myeloma. Clin Cancer Res. 2020;26(15):3969-3978.
18. Sevcikova S, Minarik J, Stork M, Jelinek T, Pour L, Hajek R. Extramedullary disease in multiple myeloma - controversies and future directions. Blood Rev. 2019;36:32-39.
19. Corre J, Perrot A, Caillot D, et al. del(17p) without TP53 mutation confers a poor prognosis in intensively treated newly diagnosed patients with multiple myeloma. Blood. 2021;137(9):1192-1195.
20. Schwestermann J, Besse A, Driessen C, Besse L. Contribution of the tumor microenvironment to metabolic changes triggering resistance of multiple myeloma to proteasome inhibitors. Front Oncol. 2022;12:899272.
21. Moreau P, Mateos MV, Berenson JR, et al. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018;19(7):953-964.
22. Auner HW, Yong KL. More convenient proteasome inhibition for improved outcomes. Lancet Oncol. 2018;19(7):856-858.
23. Raje N, Medhekar R, Panjabi S, et al. Real-world evidence for
carfilzomib dosing intensity on overall survival and treatment progression in multiple myeloma patients. J Oncol Pharm Pract. 2021 Jun 10. doi: 10.1177/10781552211015283. [Epub ahead of print]
24. Dimopoulos MA, Goldschmidt H, Niesvizky R, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an openlabel, randomised, phase 3 trial. Lancet Oncol. 2017;18(10):1327-1337.
25. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood. 2016;127(26):3360-3368.
26. Ruckrich T, Kraus M, Gogel J, et al. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia. 2009;23(6):1098-1105.
27. Kraus M, Bader J, Geurink PP, et al. The novel beta2-selective proteasome inhibitor LU-102 synergizes with bortezomib and carfilzomib to overcome proteasome inhibitor resistance of myeloma cells. Haematologica. 2015;100(10):1350-1360.
28. Haertle L, Barrio S, Munawar U, et al. Cereblon enhancer methylation and IMiD resistance in multiple myeloma. Blood. 2021;138(18):1721-1726.
29. Rajan AM, Kumar S. New investigational drugs with single-agent activity in multiple myeloma. Blood Cancer J. 2016;6(7):e451.
30. Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem Int Ed Engl. 2003;42(3):355-357.
31. Levin N, Spencer A, Harrison SJ, et al. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br J Haematol. 2016;174(5):711-720.
32. Spencer A, Harrison S, Zonder J, et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): final study results. Br J Haematol. 2018;180(1):41-51.
Haematologica | 108 June 2023 1639 ARTICLE - High-dose carfilzomib in RRMM X. Zhou et al.
Improved survival in myeloma patients–a nationwide
registry study of 4,647 patients ≥75 years treated in Denmark and Sweden
Correspondence: K.L.F. Moore. k.l.f.moore@medisin.uio.no
Received: January 10, 2022.
Accepted: August 16, 2022.
1KG Jebsen Center for B-Cell Malignancies, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; 2Oslo Myeloma Center, Department of hematology, Oslo University Hospital, Oslo Norway; 3Department of Hematology and Oncology, Stavanger University Hospital, Stavanger, Norway; 4Department of Hematology, Skåne University Hospital Malmö/Lund, Malmö, Sweden; 5Regional Cancer Center of the Western Region, Sahlgrenska University Hospital, Gothenburg, Sweden; 6Department of Hematology, Herlev Hospital, Herlev, Denmark; 7Department of Hematology, Uddevalla Hospital, Uddevalla, Sweden; 8Department of Hematology, Odense University Hospital, Odense, Denmark; 9Department of Hematology, Sahlgrenska University Hospital, Gothenburg, Sweden; 10Faculty of Medicine, University of Iceland, Reykjavik, Iceland; 11Copenhagen Prospective Studies on Asthma in Childhood, Herlev and Gentofte Hospital, University of Copenhagen, Denmark; 12Department of Hematology, Rigshospitalet, Copenhagen, Denmark and 13Institution of Internal Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
#AJV and CHB contributed equally as co-senior authors.
Abstract
Prepublished: October 27, 2022.
https://doi.org/10.3324/haematol.2021.280424
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
The prevalence of multiple myeloma (MM) is increasing in Nordic countries and the rest of the western world. Patients aged ≥75 years at diagnosis constitute an increasing proportion of all MM patients, but are underrepresented in randomized clinical trials. There is an urgent need for studies of the characteristics, treatment and outcome in this cohort. We present data from two nationwide population-based registries of all MM patients diagnosed in Denmark from January 1, 2005 until February 18, 2020, and in Sweden from January 1, 2008 until December 31, 2019, including treatment data for patients diagnosed until 2018 (Denmark) and 2019 (Sweden). In total 4,647 patients were ≥75 years at diagnosis, compared to 7,378 younger patients. Patients ≥75 years, accounting for approximately 40% of all MM patients, are a distinct cohort with more advanced disease at diagnosis, reflected by higher International Staging System (ISS) stage, and a higher proportion have renal failure and anemia. We found a more gradual introduction of modern medications in the older cohort than in the younger, despite simultaneous changes in guidelines. Compared to the cohorts in randomized controlled trials that guide the treatment of non-transplant eligible patients, we found a higher proportion of patients ≥75 years and presenting with ISS III in the real-world populations. Nevertheless, response rates and survival are increasing, indicating that modern treatment regimens are effective and well tolerated also in elderly MM patients in real-world populations.
Introduction
The incidence of multiple myeloma (MM) increases with advancing age.1-3 In recent population-based studies with high case ascertainment in the western world, the median age at diagnosis was 71-72 years.1,4 In the Surveillance, Epidemiology and End Results (SEER) database in the USA, approximately one third of patients are ≥75 years at diagnosis.5 With an aging population in many countries and improved life expectancy for patients with MM,1,3 the
number of elderly patients living with MM is increasing rapidly
Turesson et al. have previously described the increasing crude incidence and prevalence of MM in Europe and the USA, and the expected increase in Asia and Africa.3 Data from the NORDCAN database of the Association of the Nordic Cancer Registries, show a striking increase in the prevalence of plasma cell neoplasms. This is particularly evident in the older cohort, in which the prevalence has doubled in Denmark for the population ≥75 years in 2005-2018, with
Kari Lenita Falck Moore1,2,3 Ingemar Turesson,4 Anna Genell,5 Tobias W. Klausen,6 Dorota KnutBojanowska,7 Louise Redder,8 Ingigerdur Sverrisdottir,9,10 Jonathan Thorsen,11 Annette J. Vangsted12# and Cecilie H. Blimark9,13# on behalf of the Nordic Myeloma Study Group
Haematologica | 108 June 2023 1640 ARTICLE - Plasma Cell Disorders
a similar increase in Sweden (Figure 1, Online Supplementary Table S1).6
Current treatment guidelines7-10 are based on results from randomized clinical trials,11-14 only some of which report results separately for patients aged ≥75 years. Patients in randomized clinical trials do not reflect the real-world population; in particular elderly and frail patients are underrepresented, and it has been shown that patients ineligible for inclusion in trials have a poorer outcome than patients who are eligible.15-18 This highlights the need for knowledge on disease characteristics, optimal treatment and outcome in patients aged ≥75 years.
The aim of this study was to examine the differences in clinical characteristics between patients aged ≥75 years
and <75 years, and how their characteristics and outcome compare to the patients in randomized clinical trials underpinning national and international treatment guidelines. In two nationwide real-world populations, we investigated how treatment patterns change over time and whether this translates into a better outcome in MM patients, particularly in the older cohort.
Methods
The Danish Multiple Myeloma Registry (DMMR) and the Swedish Myeloma Registry (SMR) are population-based nationwide registries, and have previously been described in
Figure 1. Prevalence (numbers per 100,000) of plasma cell neoplasms. (A) Patients <75 years at diagnosis in Denmark 1962-2018 and Sweden 1980-2018. (B) Patients ≥75 years at diagnosis in Denmark 1962-2018 and Sweden 1980-2018. Data from the NORDCAN database of the Association of the Nordic Cancer Registries May 3, 2021. The increasing prevalence is particularly evident in the older cohort. In Denmark the prevalence increased from 77.7 to 151.2 per 100,000 men aged ≥75 years, and from 53.33 to 112 per 100,000 women ≥75 years 2005-2018. In Sweden the prevalence increased from 92.7 to 134.2 per 100,000 in men aged ≥75 years, and from 99.3 to 151.2 per 100,000 women ≥75 years from 2008 to 2018.

Haematologica | 108 June 2023 1641 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al. A B
detail.19-21 They were established on January 1, 2005 and January 1, 2008, respectively. Both Denmark and Sweden have personal identification code systems which are unique for every citizen and enable close to 100% coverage and follow-up. In this study, we analyzed baseline characteristics and survival for patients diagnosed with MM reported to the registries from their establishment until February 18, 2020 (DMMR), and December 31, 2019 (SMR). Patients with smoldering MM reported to the registries were included only after progression to MM. Treatment and response data were reported with at least 1-year follow-up and included patients diagnosed until December 31, 2018 (DMMR) and December 31, 2019 (SMR) to allow adequate time for reporting. Patients were followed for survival until March 20, 2020 (DMMR) and April 30, 2021 (SMR).
We performed a retrospective analysis of baseline characteristics of two age cohorts, patients ≥75 years or <75 years at diagnosis in both nationwide registries, and compared treatment, response, and outcome among Danish and Swedish patients in the older cohort. Furthermore, we compared the characteristics of our older cohort with patients included in the randomized clinical trials that are the foundation of Danish, Swedish and international treatment guidelines: VISTA (bortezomib, melphalan and prednisolone [VMP] vs. melphalan and prednisolone [MP]), FIRST (lenalidomide and dexamethasone continuous [Rd] vs. lenalidomide and dexamethasone for 18 months [Rd18] vs melphalan, prednisolone and thalidomide [MPT]), ALCYONE (daratumumab, bortezomib, melphalan and prednisolone [D-VMP] vs. bortezomib, melphalan and prednisolone [VMP]) and MAIA (daratumumab, lenalidomide and dexamethasone [D-Rd] vs. lenalidomide and dexamethasone [Rd])
Statistical methods
For categorical variables we used the c2 test to examine statistical significance. P values <0.05 were considered statistically significant. Survival time was calculated from diagnosis until death or censoring. Patients were censored at the end of the study (April 30, 2021 [SMR] and March 20, 2020 [DMMR]) and at loss to follow-up. Overall survival and relative survival ratios are presented as graphs of 6-, 12and 36-month survival by year of diagnosis with 95% confidence intervals. When estimating relative survival we used the Ederer II method for expected survival, relative to the expected survival of each country’s population.22 We used a Cox proportional hazards model to compare the outcomes of the subgroups of patients 75-84 and ≥85 years compared to those <75 years, for the entire follow-up period, while adjusting for year of diagnosis. Early death was defined as death within 6 months of myeloma diagnosis. Patients who did not receive any treatment were excluded from analyses regarding treatment. We handled missing data by complete case analysis. R software was used for statistical analyses.23
Ethical approval
The study was approved by the Danish Data Protection Agency (18/22825) and the Danish Patient Safety Authority (3-3013-2047/2r). Ethical approval was also obtained in Sweden (Dnr 2020-01729) and from the Data Protection authorities (Datauttagsansökan SV-2079). The study was conducted in accordance with the Helsinki Declaration of 1975, revised in 2008.
Results
In total, we compared the characteristics of 4,647 Danish and Swedish MM patients aged ≥75 years or older at diagnosis with those of 7,378 Danish and Swedish MM patients <75 years. The proportion of all newly diagnosed MM patients ≥75 years was similar in both registries, 36% in DMMR and 40% in SMR (Table 1). Altogether, 3,904 patients ≥75 years were available for analysis of treatment data, and 3,490 patients were analyzed for response. There were no missing data for survival.
Baseline characteristics
Significant differences in International Staging System (ISS) stage and CRAB-criteria (hypercalcemia, renal failure, anemia, osteolytic skeletal lesions) were found between age groups. Patients ≥75 years had more advanced disease, as 46% of patients in this age group presented with ISS stage III in both registries, compared to 30% in the Swedish cohort <75 years, and 35% in the Danish cohort <75 years. This difference was consistent over time. The proportion of patients presenting with anemia was higher in the older group compared to the younger cohort, as was the proportion with renal failure (Table 1). There were more men than women diagnosed in both age groups, but the difference was less pronounced among patients aged ≥75 years due to the higher number of women in the population in higher age groups (Table 1). Overall, data describing patients aged ≥75 years from the two registries were consistent.
First-line treatment
The first-line treatment guidelines in patients not eligible for autologous stem cell transplantation were similar in Denmark and Sweden during the study period.19 We found that MP was replaced by bortezomib-based regimes from around 2012, while lenalidomide-based treatment increased in recent years (Figure 2, Online Supplementary Table S2). The proportion of patients receiving an immunomodulatory drug or proteasome inhibitor as part of firstline treatment increased dramatically in patients ≥75 years (from 18.1% in 2005 to 89.1% in 2018 in Denmark, and from 29.9% in 2008 to 95.5% in 2018 in Sweden) (Figure 3, Online Supplementary Table S3).
Haematologica | 108 June 2023 1642 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al.
Treatment response and survival
Parallel to the increased use of modern agents, the proportion of patients aged ≥75 years who achieved at least very good partial remission more than doubled in the studied time period to 40% in Denmark and 45% in Sweden (Figure 4, Online Supplementary Table S4).
Simultaneously, the median relative survival for the same patient group in Denmark increased from 25 months for patients diagnosed in 2005-2007 to 36 months for patients diagnosed in 2015-2016. In Sweden the median relative survival increased from 24 months for patients diagnosed in 2008-2009 to 42 months in patients diagnosed in 2016-
Population Danish Multiple Myeloma Registry N=4,691 P value Swedish Myeloma Registry N=7,334 P value Age group <75 years ≥75 years <75 years ≥75 years Number (%) 3,003 (64.0) 1,688 (36.0) 4,375 (59.7) 2,959 (40.3) Age in years, median (range) 65 (30-74) 80 (75-98) 66 (19-74) 81 (75-100) Gender 0.0009 <0.0001 Male, N (%) 1,736 (57.8) 891 (52.8) 2,649 (60.5) 1,567 (53.0) Female, N (%) 1,267 (42.2) 797 (47.2) 1,726 (39.5) 1,392 (47.0) Multiple myeloma type IgG, N (%) 1,597 (57.6) 937 (61.2) 2,195 (50.6) 1,571 (53.8) IgA, N (%) 595 (21.5) 345 (22.5) 821 (18.9) 588 (20.1) IgD, IgE, IgM, N (%) 49 (1.8) 17 (1.1) 44 (1.0) 19 (0.7) Light chain disease, N (%) 444 (16.0) 202 (13.2) 1,078 (24.8) 647 (22.2) Non-secretory, N (%) 76 (2.7) 26 (1.7) 151 (3.5) 60 (2.1) Mixed, N (%) 11 (0.4) 4 (0.3) 50 (1.2) 35 (1.2) Missing, N 231 (NA) 157 (NA) 36 (NA) 39 (NA) ISS stage < 0.0001 <0.0001 I, N (%) 724 (28.5) 201 (15.1) 860 (25.6) 191 (10.6) II, N (%) 936 (36.9) 511 (38.4) 1,482 (44.1) 774 (42.8) III, N (%) 877 (34.6) 617 (46.4) 1,019 (30.3) 845 (46.7) Missing, N 466 (NA) 359 (NA) 1,014 (NA) 1149 (NA) Hb <100 g/L < 0.0001 <0.0001 No, N (%) 1,834 (61.9) 903 (54.0) 3,035 (69.5) 1,809 (61.3) Yes, N (%) 1,131 (38.1) 770 (46.0) 1,332 (30.5) 1,143 (38.7) Missing, N 38 (NA) 15 (NA) 8 (NA) 7 (NA) Calcium Ionized calcium >1.35 (mmol/L) 0.4 Ionized calcium >1.35 (mmol/L) or total calcium >2.75 (mmol/L) 0.005 No, N (%) 2,119 (72.7) 1,205 (73.9) 3,790 (86.6) 2,630 (88.9) Yes, N (%) 797 (27.3) 426 (26.1) 585 (13.4) 329 (11.1) Missing, N 87 (NA) 57 (NA) 75 (NA) 52 (NA) Creatinine >177 mmol/L or CrCl <40 mL/min 0.04 0.01 No, N (%) 2,438 (82.4) 1,329 (80.0) 3,677 (84.3) 2,418 (82.0) Yes, N (%) 519 (17.6) 332 (20.0) 686 (15.7) 531 (18.0) Missing, N 46 (NA) 27 (NA) 12 (NA) 10 (NA) Skeletal disease <0.0001 <0.0001 ≥ 1 osteolytic lesions, N (%) 2,201 (77.4) 1,035 (66.7) 2,903 (68.0) 1,636 (59.2) Osteopenia and vertebral compression, N (%) 79 (2.8) 119 (7.7) 482 (11.3) 400 (14.5) None, N (%) 565 (19.9) 398 (25.6) 882 (20.7) 728 (26.3) Missing, N 158 (NA) 136 (NA) 107 (NA) 195 (NA)
Table 1. Baseline characteristics of patients in the Danish Multiple Myeloma Registry and Swedish Myeloma Registry.
Haematologica | 108 June 2023 1643 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al.
CrCl: creatinine clearance; DMMR: Danish Multiple Myeloma Registry; Hb: hemoglobin; ISS: International Staging System; NA: not applicable; SMR; Swedish Multiple Myeloma Registry.
2017 (Online Supplementary Table S5). The 3-year overall survival in Sweden was 30.9% for patients diagnosed in 2008-2009 and 44.3% for those diagnosed in 2018-2019 among patients ≥75 years. In Denmark, the 3-year overall survival for patients ≥75 years was 32.3% in 2005-2007 compared to 44.1% from 2017 (Figure 5B, Online Supplementary Table S6).
To further study this improvement in survival, we performed a post hoc subgroup analysis, splitting the age cohort ≥75 year into two age groups: 75-84 and ≥85 years at diagnosis, and compared them with the <75-year-old cohort (Online Supplementary Table S7). Using a Cox proportional hazards model, we found a hazard ratio for death for Danish patients ≥85 years of 4.24 (95% confidence interval [95% CI]: 3.75-4.79) while it was 2.43 (95% CI: 2.24-2.63) for Danish patients 75-84 years. For Swedish patients ≥85 years the hazard ratio for death was 5.78 (95% CI: 4.60-7.26)
Figure 2. Changes in first-line treatment over time for patients ≥75 years in (A) Denmark and (B) Sweden. Cd: cyclophosphamide, dexamethasone; CRd: cyclophosphamide, lenalidomide, dexamethasone; CTd: cyclophosphamide, thalidomide, dexamethasone; Mono: monotherapy; MP: melphalan, prednisolone; MPR: melphalan, prednisolone, lenalidomide; MPT: melphalan, prednisolone, thalidomide; MPV: melphalan, prednisolone, bortezomib; Rd: lenalidomide, dexamethasone; Td: thalidomide, dexamethasone; Td-ixa: thalidomide, dexamethasone, ixazomib; VCd: bortezomib, cyclophosphamide, dexamethasone; Vd: bortezomib, dexamethasone; VRd: bortezomib, lenalidomide, dexamethasone; VTd: bortezomib, thalidomide, dexamethasone.

while it was 2.72 (95% CI: 2.36-3.14) for those aged 75-84 years at diagnosis.
Finally, we analyzed possible interactions, and found that there was no association between the difference between the age groups and the year of diagnosis, and that the difference between the age groups appeared to be constant during the study period.
However, there was no improvement in relative survival at 6 months for either age group, which remained stable in the range of 78.2-83.9% (SMR) and 68.4-81.1% (DMMR) for patients ≥ 75 years (Figure 6, Online Supplementary Table S6).
Comparison with randomized clinical trials
In a comparison with important randomized clinical trials (VISTA, FIRST, ALCYONE and MAIA) supporting guidelines in MM patients not eligible for autologous stem cell trans-
A Haematologica | 108 June 2023 1644 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al. B
Figure 3. Use of immunomodulatory drugs and/or proteasome inhibitors in first-treatment line of multiple myeloma by country and age group. In 2009 the Danish treatment guidelines changed to recommending bortezomib and dexamethasone (VD) in first line for patients eligible for autologous stem cell transplant (ASCT) and melphalan, prednisolone and thalidomide (MPT) or melphalan, prednisolone and bortezomib (MPV) for those ineligible (marked by the red vertical line). In Sweden, the treatment guidelines changed in 2010 (blue vertical line). Bortezomib and thalidomide were recommended as part of standard induction treatment before ASCT. MPT was recommended as standard for patients not eligible for ASCT, while MPV and melphalan and prednisolone were treatment options.

plantation, we found a higher proportion of patients ≥75 years and more patients with advanced disease at diagnosis (Table 2).
The exception is the FIRST trial which reports the results of patients >75 years using International Myeloma Working Group criteria and overall survival. A similar proportion of patients in this age cohort on the trial presented with ISS stage III (48%), yet response rates were significantly higher than for patients receiving standard of care in Denmark and Sweden in the same time period (Table 2). In the FIRST trial 42-45% of patients treated with lenalidomide and 31% treated with MPT, achieved very good partial remission or better, compared to an average of 16.8% of Danish patients and 23.1% of Swedish patients achieving the same response rates during the trial period 2008-2011.12 In contrast, 69% of patients aged ≥75 years in more recent trials with CD38antibody combinations achieved very good partial remission or better.13
A similar difference was seen for survival with the median overall survival being 46-48 months for FIRST trial patients treated with lenalidomide, and 38 months for those receiving MPT, while contemporaneous Danish and Swedish patients had a median overall survival of approximately half of this (20.9 months and 20.3-21.5 months, respectively) (Table 2). This is as expected with selected populations in clinical trials. However, the overall survival for our realworld population is increasing in parallel to the increasing use of modern medications in more recent time periods.
Discussion
The prevalence of plasma cell neoplasms has increased in Nordic countries and the entire western world, particularly in the elderly. At least three important factors contribute to this; increasing crude incidence as the population ages, improved case ascertainment, and increased survival of patients diagnosed with MM. This makes the description of the characteristics, treatment and survival of the elderly MM population urgent.
In this study, we present data on 12,025 patients from two nationwide registries including all patients diagnosed with MM. We report real-world data on the characteristics, treatment and outcome of the largest, unselected population of patients aged ≥75 years to date. The results from the DMMR and SMR are consistent with those from other registry studies regarding the proportion of patients ≥75 years at diagnosis.29-31 We clearly show that the older MM cohort differs from the younger in clinical characteristics, with more advanced disease stage and higher rates of myeloma complications, such as anemia and renal failure, at diagnosis.
Even so, our comparison shows that randomized clinical trials supporting guidelines in elderly MM patients include a lower proportion of patients ≥75 years at diagnosis, and fewer patients with ISS stage III than our population, with the exception of the FIRST trial.26 An age-related decline in albumin and renal function, associated with increasing b2 -
Haematologica | 108 June 2023 1645 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al.
microglobulin levels, may account for some of the difference in ISS staging between the age cohorts in our study. However, age alone does not explain the difference in proportion of patients with ISS stage III disease between real-world populations and populations in randomized clinical trials. Our data show that more than 90% of patients diagnosed at ≥75 years received first-line treatment in Denmark and Sweden in 2008-2019. Other European registry and crosssectional studies have shown similar results.30,32 Data from the SEER database in the USA for the period 2007-2013 documented that only 51% of patients aged ≥80 years received treatment within the first 6 months of diagnosis, although this rate has increased in recent years.33
We found that the introduction of modern agents in the treatment of older patients has been much more gradual compared to that for younger patients in Denmark and Sweden, despite simultaneous changes in national guidelines (Figure 3, Online Supplementary Table S3). Mian et al. examined Canadian administrative health care data and found the same when comparing myeloma patients ≤65 years and >65 years from 2007 to 2017.34

Our data clearly show improved response rates and increased survival with modern treatment, also in MM patients ≥75 years. A subgroup analysis revealed that this improvement is also seen in the patients ≥85 years at MM diagnosis, although this age group has a significantly higher hazard ratio for death compared to patients 75-84 years and may constitute a possible new age-defined frail population. This is important information that requires data from an unselected real-world population and cannot be obtained from randomized clinical trials that commonly exclude a high proportion of elderly MM patients. Other possible contributions to increased survival are improved
supportive measures and better treatment of comorbidities.35 However, the improvement in relative survival over time supports that myeloma treatments and response are significant contributors to improved survival.
Despite more effective treatment, early mortality did not decrease for patients ≥75 years in our study. This matches findings in a study of Canadian patients diagnosed between 2007-2017.34 There remains an unmet clinical need to tailor treatment and supportive measures for elderly patients, particularly in the critical, early months after diagnosis when the risk of complications of both their MM and toxicity of treatment is high.36
We propose that future trials differentiate between frailty caused by myeloma tumor cell burden which may improve during treatment,37 and frailty related to age and comorbidity. It is hoped that ongoing and planned clinical studies, such as the Myeloma XIV study, which adjusts treatment regimens according to repeated assessments of patient frailty (clinicaltrials.gov NCT03720041), will contribute to finding tools to improve treatment strategies. Another possible strategy is to allow rescue treatment to correct myeloma-related complications such as renal failure before patients’ inclusion into randomized clinical trials. Our study is limited by lack of data on comorbidities, frailty status, quality of life, detailed information on medication dosing schedules, and later lines of treatment. However, providing the best possible first-line treatment is important, as two European studies have shown that between 20% and 30% of patients never receive any later myeloma treatment.38,39 As in other population-based registries, the reporting of fluorescence in situ hybridization data is limited in patients ≥75 years, but increasing rapidly and has been reported for more than 50% since 2014.
Figure 4. Response rate for very good partial remission or better by country and age group. From 2013, multiple myeloma patients ≥75 years are gradually approaching the high response rates on firstline treatment seen among younger patients. Simultaneously, the proportion of non-responders decreased significantly from 57.9% in 2005 to 32.2% in 2019 in Denmark, and from 38% to 16.8% from 2008 to 2019 in Sweden (Online Supplementary Table S4). VGPR: very good partial remission.
Haematologica | 108 June 2023 1646 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al.
The greatest strength of our study is the large number of unselected patients from two nationwide registries, as well as close to 100% case ascertainment. Both Denmark and Sweden have national publicly funded health care systems and national treatment guidelines, and our study shows that the patient populations are very similar. Another strength of our study is the population-based design that allows the use of relative survival rather than overall survival to measure outcome. In an elderly popu-
lation, comorbidities not related to MM contribute more to mortality than in younger patients and there is a risk that using overall survival could underestimate the beneficial effect of MM treatment in the elderly.
In conclusion, MM patients aged ≥75 years have more advanced disease at diagnosis. This is not reflected in the selected patient populations of the majority of randomized clinical trials guiding the treatment of elderly patients. Both healthcare policy makers and designers of

A Haematologica | 108 June 2023 1647 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al. B
Figure 5. Overall survival by country and age group. (A, B) Overall survival of patients with multiple myeloma in Denmark 20052018 (A) and Sweden 2008-2019 (B). mo: months.
clinical trials must consider this for the benefit of future MM patients. The introduction of modern treatment is more gradual in patients ≥75 years, but coincides with improved response rates and survival.
Disclosures
IT has received honoraria from Bristol-Myers Squibb. LR has received research funding from Janssen-Cilag. JT has received speaking fees from Astra Zeneca. CHB has received
to this work.
Contributions
AJV, CHB, and IT conceived and designed the study. KLFM, CHB, AJV, and IT wrote the manuscript. TK and JT (Denmark) and AG (Sweden) performed the statistical analysis. AJV, CHB, IT, KLFM, LR, DKB, IS, AG, TK, and JT revised and commented on the manuscript.
 honoraria from Amgen, BMS, Takeda and Janssen unrelated
honoraria from Amgen, BMS, Takeda and Janssen unrelated
A Haematologica | 108 June 2023 1648 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al. B
Figure 6. Relative survival by country and age group. (A, B) Relative survival of patients with multiple myeloma in Denmark 20052018 (A) and Sweden 2008-2019 (B). mo: months.
Table 2. Baseline characteristics, response rates and survival of patient populations in the Danish Multiple Myeloma Registry, Swedish Myeloma Registry and key randomized controlled trials.
Median OS ≥75 years: >75 years: VMP: 43 mos Rd cont: 48 mos
Rd18: 46 mos
MPT: 38 mos
$>75 years, not reported for ≥75 years. *Range. DMMR: Danish Multiple Myeloma Registry; SMR: Swedish Myeloma Registry; VISTA: bortezomib, melphalan and prednisolone (VMP) vs. melphalan and prednisolone (MP).11,24 FIRST: lenalidomide and dexamethasone continuous (Rd) vs. lenalidomide and dexamethasone for 18 months (Rd18) vs. melphalan, prednisolone and thalidomide (MPT).12,25,26 ALCYONE: daratumumab, bortezomib, melphalan and prednisolone (D-VMP) vs. VMP13,27. MAIA: daratumumab, lenalidomide and dexamethasone (D-Rd) vs. Rd.28 CR: complete response; CrCl: creatinine clearance; EBMT: European Society for Blood and Marrow Transplantation; EC: exclusion criteria; Hb: hemoglobin; IC: inclusion criteria; ISS: International Staging System; mos: months; NR: not reached; OS: overall survival; PR: partial response; VGPR: very good partial response.
DMMR ≥75 years SMR ≥75 years VISTA FIRST ALCYONE MAIA Time period 2005-2019 2008-2019 2004-2006 2008-2011 2015-2016 2015-2017 Patients ≥75 years, N (%) 1,688 (100) 2,959 (100) 208 (30) 567$ (35) 211 (30) 321 (44) Patients with ISS III N=617 (46%) N=845 (47%) VMP: 35% MP: 34% N=659 (41%) >75: N=273 (48%) N=271 (38%) N=227 (29%) Patients with anemia Hb <100 g/L: N=770 (46%) Hb <100 g/L: N=1,143 (39%) VMP: Median Hb: 104 g/L (range, 64-159)* MP: Median Hb: 106 g/L (range, 73-165)* Not reported. No IC/EC IC: Hb ≥75 g/L IC: Hb ≥75 g/L Patients with renal failure CrCl <40 mL/min or creatinine >177 mmol/L (>2 mg/dL): N=332 (20%) CrCl <40 mL/min or creatinine >177 mmol/L (>2 mg/dL): N=531 (18%) CrCl <60 mL/min: VMP: 54% MP: 55% 30-60 mL/min: VMP: 48% MP: 50% <30 mL/min: VMP: 6% MP: 5% EC:
<60
N=779
<30 mL/min: N=147 (9.1%) IC: CrCl
mL/min CrCl <60 mL/min: N=295 (42%) IC: CrCl
mL/min CrCl
60 mL/min: N=304
>75 years CrCl <30 mL/min: N=74 (13%) CrCl 30-49 mL/min: N=209 (37%) ≥VGPR, N(%) 2005: 9 (12) 2016: 49 (39) 2008: 27 (20) 2016: 74 (39) EBMT criteria CR or PR: Rd cont: 258 (48) Rd18: 255 (47) MPT: 166 (30) D-VMP: 255 (73) VMP: 177 (50) D-Rd: 298 (81) Rd: 210 (57) 2019: 12 (40) 2019: 62 (45) VMP: 238 (71) MP: 115 (35) Overall: Overall: >75 years: ≥75 years: 334 (24) 654 (31) CR: Rd cont: 84 (45) D-VMP: 72 (69) VMP: 102 (30) Rd 18: 81 (42) VMP: 52 (49) MP: 12 (4) MPT: 58 (31) Overall survival 3-year OS ≥75 years 2005-07: 32% 3-year OS ≥75 years: 2008-09: 31% 3-year OS: VMP: 69% MP: 54%. 3-year OS: Rd cont: 70% Rd18: 66% 3-year OS: D-VMP: 78% Estimated 60month OS: D-Rd: 66.3% Rd: 53.1% Median OS NR in either group at 40.1 mos 2017-18: 44% Median OS ≥75 years: 2018-19: 44% Median OS ≥75 years: Median OS: VMP: NR MP: 43 mos MPT: 62% Median OS: Rd cont: 59 mos Rd18: 62 mos Median OS NR in either group at 60 mos 2005-07: 20 mos 2008-09: 20 mos
30 mos 2018-19:
Median OS
dialysis
mL/min:
(48%)
≥40
≥30
≤
(41%)
2017-18:
34 mos
Haematologica | 108 June 2023 1649 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al.
Acknowledgments
The authors would like to thank the steering groups of the DMMR and SMR for maintaining high quality registries and all the patients, as well as their doctors and nurses who have reported to the registries . We would also like to thank Tom Børge Johannesen at NORDCAN and the Association of the Nordic Cancer Registries.
Funding
This investigation was supported by the Nordic Cancer Union, project grant number R241-A15003 to the NMSG Real-World-Evidence group, the Swedish State under the
References
1. Langseth ØO, Myklebust T, Johannesen TB, Hjertner Ø, Waage A. Incidence and survival of multiple myeloma: a population-based study of 10 524 patients diagnosed 1982-2017. Br J Haematol. 2020;191(3):418-425.
2. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046-1060.
3. Turesson I, Bjorkholm M, Blimark CH, Kristinsson S, Velez R, Landgren O. Rapidly changing myeloma epidemiology in the general population: increased incidence, older patients, and longer survival. Eur J Haematol. 2018;101:237-244.
4. Turesson I, Velez R, Kristinsson SY, Landgren O. Patterns of improved survival in patients with multiple myeloma in the twenty-first century: a population-based study. J Clin Oncol. 2010;28(5):830-834.
5. Howlader N NA, Krapcho M, Miller D, et al. (eds). National Cancer Institute, Bethesda, MD, USA. SEER Cancer Statistics Review, 1975-2018. https://seer.cancer.gov/csr/1975_2018/. Accessed 25 April, 2021.
6. Larønningen S FJ, Bray F, Engholm G, et al. Association of the Nordic Cancer Registries. Cancer Registry of Norway. NORDCAN: Cancer Incidence, Mortality, Prevalence and Survival in the Nordic Countries. https://nordcan.iarc.fr/. Accessed 3 May, 2021.
7. Dimopoulos MA, Moreau P, Terpos E, et al. Multiple myeloma: EHA-ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Hemasphere. 2021;5(2):e528:1-12.
8. Rajkumar SV (ed). mSMART: Stratification for Myeloma & RiskAdapted Therapy. Treatment of newly diagnosed myeloma Feb 2021.
https://static1.squarespace.com/static/5b44f08ac258b493a2509 8a3/t/601dd971c6429e2b6fa12fd5/1612568946139/Treatment-ofNewly-Diagnosed-MyelomaFeb2021_FINAL.pdf. Accessed 6 January, 2022.
9. Danish Myeloma Study Group. Sekretariatet for Kliniske Retningslinjer på Kræftområdet. Primær behandling af myelomatose hos patienter, som ikke er kandidater til højdosis kemoterapi med stamcellestøtte.
https://myeloma.hematology.dk/index.php/vejledninger-dmsg [Danish]. Accessed 22 February, 2022.
10. Regionala Cancercentrum i samverkan. Myelom Nationellt vårdprogram.
https://kunskapsbanken.cancercentrum.se/diagnoser/myelom/va rdprogram/ [Swedish]. Accessed 22 February 2022.
11. Mateos MV, Richardson PG, Schlag R, et al. Bortezomib plus melphalan and prednisone compared with melphalan and prednisone in previously untreated multiple myeloma: updated
Agreement between the Sw edish Government and the City Councils, and the ALF-agreement ALFGBG- 523261 to CHB.
Data-sharing statement
The data that support the findings of this study are available from the Danish and Swedish myeloma registries. Restrictions apply to the availability of these data, which were used under license for this study. Data are available at www.myeloma.dk and www.cancercentrum.se/myelom with the permission of the CPUA authorities in Sweden and The Danish Myeloma Study Group.
follow-up and impact of subsequent therapy in the phase III VISTA trial. J Clin Oncol. 2010;28(13):2259-2266.
12. Facon T, Dimopoulos MA, Dispenzieri A, et al. Final analysis of survival outcomes in the phase 3 FIRST trial of up-front treatment for multiple myeloma. Blood. 2018;131(3):301-310.
13. Mateos MV, Cavo M, Blade J, et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, openlabel, phase 3 trial. Lancet. 2020;395(10218):132‐141.
14. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115.
15. Chari A, Romanus D, Palumbo A, et al. Randomized clinical trial representativeness and outcomes in real-world patients: comparison of 6 hallmark randomized clinical trials of relapsed/refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2020;20(1):8-17.
16. Costa LJ, Hari PN, Kumar SK. Differences between unselected patients and participants in multiple myeloma clinical trials in US: a threat to external validity. Leuk Lymphoma. 2016;57(12):2827-2832.
17. Klausen TW, Gregersen H, Abildgaard N, et al. The majority of newly diagnosed myeloma patients do not fulfill the inclusion criteria in clinical phase III trials. Leukemia. 2019; 33(2):546-549.
18. Shah JJ, Abonour R, Gasparetto C, et al. Analysis of common eligibility criteria of randomized controlled trials in newly diagnosed multiple myeloma patients and extrapolating outcomes. Clin Lymphoma Myeloma Leuk. 2017;17(9):575-583.
19. Blimark CH, Vangsted AJ, Klausen TW, et al. Outcome data from >10 000 multiple myeloma patients in the Danish and Swedish national registries. Eur J Haematol. 2022;108(2):99-108.
20. Blimark CH, Turesson I, Genell A, et al. Outcome and survival of myeloma patients diagnosed 2008-2015. Real-world data on 4904 patients from the Swedish Myeloma Registry. Haematologica. 2018;103(3):506-513.
21. Gimsing P, Holmstrom MO, Klausen TW, et al. The Danish National Multiple Myeloma Registry. Clin Epidemiol. 2016;8:583-587.
22. Hakulinen T, Seppä K, Lambert PC. Choosing the relative survival method for cancer survival estimation. Eur J Cancer. 2011;47(14):2202-2210.
23. R Core Team. R Foundation for Statistical Computing. R: a language and environment for statistical computing. https://www.R-project.org/ Accessed 12 October, 2020.
24. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus
Haematologica | 108 June 2023 1650 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al.
melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359(9):906-917.
25. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371(10):906-917.
26. Hulin C, Belch A, Shustik C, et al. Updated outcomes and impact of age with lenalidomide and low-dose dexamethasone or melphalan, prednisone, and thalidomide in the randomized, phase III FIRST trial. J Clin Oncol. 2016;34(30):3609-3617.
27. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518-528.
28. Facon T, Kumar SK, Plesner T, et al. Daratumumab, lenalidomide, and dexamethasone versus lenalidomide and dexamethasone alone in newly diagnosed multiple myeloma (MAIA): overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2021;22(11):1582-1596.
29. Offidani M, Corvatta L, Polloni C, et al. Assessment of vulnerability measures and their effect on survival in a real-life population of multiple myeloma patients registered at Marche Region Multiple Myeloma Registry. Clin Lymphoma Myeloma Leuk. 2012;12(6):423-432.
30. Oortgiesen BE, van Roon EN, Joosten P, et al. The role of initial clinical presentation, comorbidity and treatment in multiple myeloma patients on survival: a detailed population-based cohort study. Eur J Clin Pharmacol. 2017;73(6):771-778.
31. Bergin K, Wellard C, Moore E, et al. The myeloma landscape in Australia and New Zealand: the first 8 years of the Myeloma and Related Diseases Registry (MRDR). Clin Lymphoma Myeloma Leuk. 2021;21(6):e510-e520.
32. Raab MS, Cavo M, Delforge M, et al. Multiple myeloma: practice patterns across Europe. Br J Haematol. 2016;175(1):66-76.
33. Fiala MA, Foley NC, Zweegman S, Vij R, Wildes TM. The characteristics, treatment patterns, and outcomes of older adults aged 80 and over with multiple myeloma. J Geriatr Oncol. 2020;11(8):1274-1278.
34. Mian HS, Seow H, Wildes TM, et al. Disparities in treatment patterns and outcomes among younger and older adults with newly diagnosed multiple myeloma: a population-based study. J Geriatr Oncol. 2020;12(4):508-514.
35. Sverrisdóttir IS, Rögnvaldsson S, Thorsteinsdottir S, et al. Comorbidities in multiple myeloma and implications on survival: a population-based study. Eur J Haematol. 2021;106(6):774-782.
36. Holmstrom MO, Gimsing P, Abildgaard N, et al. Causes of early death in multiple myeloma patients who are ineligible for highdose therapy with hematopoietic stem cell support: a study based on the nationwide Danish Myeloma Database. Am J Hematol. 2015;90(4):E73-74.
37. Uttervall K, Duru AD, Lund J, et al. The use of novel drugs can effectively improve response, delay relapse and enhance overall survival in multiple myeloma patients with renal impairment. PLoS One. 2014; 9(7):e101819:1-10.
38. Yong K, Delforge M, Driessen C, et al. Multiple myeloma: patient outcomes in real-world practice. Br J Haematol. 2016; 175(2):252-264.
39. Szabo AG, Iversen K. F, Møller S, Plesner T. The clinical course of multiple myeloma in the era of novel agents: a retrospective, single-center, real-world study. Clin Hematol Int. 2019;1(4):220-228.
Haematologica | 108 June 2023 1651 ARTICLE - Improved survival among elderly myeloma patients K.L.F. Moore et al.
Comprehensive in silico and functional studies for classification of EPAS1/HIF2A genetic variants identified in patients with erythrocytosis
Valéna Karaghiannis,1,2* Darko Maric,3,4* Céline Garrec,5 Nada Maaziz,6 Alexandre Buffet,7,8 Loïc Schmitt,2 Vincent Antunes,3,4 Fabrice Airaud,5 Bernard Aral,6 Amandine Le Roy,2 Sébastien Corbineau,2 Lamisse Mansour-Hendili,9,10 Valentine Lesieur,2 Antoine Rimbert,2 Fabien Laporte,2 Marine Delamare,2 Minke Rab,11,12 Stéphane Bézieau,2,5 Bruno Cassinat,13 Frédéric Galacteros,10,14 Anne-Paule Gimenez-Roqueplo,7,8 Nelly Burnichon,7,8 Holger Cario,15 Richard van Wijk,11 Celeste Bento,16 ECYT-4 consortium,° François Girodon,6,17,18# David Hoogewijs,3,4# and Betty Gardie1,2,18#
1Ecole Pratique des Hautes Etudes, EPHE, Université Paris Sciences et Lettres, Paris, France; 2Nantes Université, CNRS, INSERM, l’Institut du Thorax, Nantes, France; 3Section of Medicine, Department of Endocrinology, Metabolism and Cardiovascular System, University of Fribourg, Fribourg, Switzerland; 4National Center of Competence in Research “Kidney.CH”, Switzerland; 5Service de Génétique Médicale, CHU de Nantes, Nantes, France; 6Service d’Hématologie Biologique, Pôle Biologie, CHU de Dijon, Dijon, France; 7Université Paris Cité, INSERM, PARCC, Paris, France; 8Département de Médecine Génomique des Tumeurs et des Cancers, AP-HP, Hôpital Européen Georges Pompidou, Paris, France; 9Département de Biochimie-Biologie
Moléculaire, Pharmacologie, Génétique Médicale, AP-HP, Hôpitaux Universitaires Henri Mondor, Créteil, France; 10Université Paris-Est Créteil, IMRB Equipe Pirenne, Laboratoire d'Excellence
LABEX GRex, Créteil, France; 11Central Diagnostic Laboratory - Research, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands; 12Department of Hematology, University Medical Center Utrecht, Utrecht University, Utrecht, the Netherlands; 13Université
Paris Cité, APHP, Hôpital Saint-Louis, Laboratoire de Biologie Cellulaire, Paris, France; 14Red Cell Disease Referral Center-UMGGR, AP-HP, Hôpitaux Universitaires Henri Mondor, Créteil, France; 15Department of Pediatrics and Adolescent Medicine, University Medical Center, Ulm, Germany; 16Hematology Department, Centro Hospitalar e Universitário de Coimbra, CIAS, University of Coimbra, Coimbra, Portugal; 17Université de Bourgogne, INSERM U1231, Dijon, France and 18Laboratoire d’Excellence GR-Ex, Paris, France
*VK and DM contributed equally as co-first authors. #FG, DH and BG contributed equally as co-senior authors.
°An appendix with all ECYT-4 consortium members can be found at the end of the manuscript.
Abstract
Correspondence: B. Gardie betty.gardie@inserm.fr
Received: September 1, 2022.
Accepted: January 17, 2023.
Early view: January 26, 2023.
https://doi.org/10.3324/haematol.2022.281698
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Gain-of-function mutations in the EPAS1/HIF2A gene have been identified in patients with hereditary erythrocytosis that can be associated with the development of paraganglioma, pheochromocytoma and somatostatinoma. In the present study, we describe a unique European collection of 41 patients and 28 relatives diagnosed with an erythrocytosis associated with a germline genetic variant in EPAS1. In addition we identified two infants with severe erythrocytosis associated with a mosaic mutation present in less than 2% of the blood, one of whom later developed a paraganglioma. The aim of this study was to determine the causal role of these genetic variants, to establish pathogenicity, and to identify potential candidates eligible for the new hypoxia-inducible factor-2 α (HIF-2α) inhibitor treatment. Pathogenicity was predicted with in silico tools and the impact of 13 HIF-2b variants has been studied by using canonical and real-time reporter luciferase assays. These functional assays consisted of a novel edited vector containing an expanded region of the erythropoietin promoter combined with distal regulatory elements which substantially enhanced the HIF-2α-dependent induction. Altogether, our studies allowed the classification of 11 mutations as pathogenic in 17 patients and 23 relatives. We described four new mutations (D525G, L526F, G527K, A530S) close to the key proline P531, which broadens the spectrum of mutations involved in erythrocytosis. Notably, we identified patients with only erythrocytosis associated with germline mutations A530S and Y532C previously identified at somatic state in tumors, thereby raising the complexity of the genotype/phenotype correlations. Altogether, this study allows accurate clinical follow-up of patients and opens the possibility of benefiting from HIF-2α inhibitor treatment, so far the only targeted treatment in hypoxia-related erythrocytosis disease.
Haematologica | 108 June 2023 1652 ARTICLE - Red Cell Biology & its Disorders
Introduction
Erythrocytoses are characterized by an elevated red cell mass of more than 125% of the predicted value for the age and body mass of the subject, usually reflected by increased levels of hemoglobin (Hb) and/or hematocrit (Ht) values.1 Erythrocytoses can be acquired as in the primary polycythemia vera, a myeloproliferative neoplasm as a consequence of a gain of function mutation in the JAK2 gene (p.Val617Phe), or secondary to diverse pathological situations (pulmonary or heart disease, kidney cancer, carbon monoxide poisoning, etc.) due to an increased secretion of erythropoietin (EPO). On the other hand, erythrocytosis can be observed in a context of inherited disease, which can be primary when there is an intrinsic defect in the progenitor cells of the bone marrow (EPOR mutations), or secondary when the oxygen-sensing pathway is dysregulated and EPO is produced at a high level (VHL, EGLN1/PHD2, EPAS1/HIF2A mutations).
This study focuses on the EPAS1 gene that encodes the hypoxia-inducible factor-2 α (HIF-2 α ), a major player in the oxygen-sensing pathway, also known as the hypoxia pathway.
HIF is a transcription factor that is stabilized when the oxygen concentration is reduced.2 HIF is a α/b hetero-dimer consisting of a tightly regulated oxygen-labile α-subunit and a constitutive b-subunit. The HIF-α subunits (HIF-1α, 2α, 3α) contain an oxygen-dependent degradation (ODD) domain, and two independent transcriptional activation domains. In the presence of oxygen, the HIF-α subunits are hydroxylated by prolyl hydroxylases (PHD1-4)3 that regulate their stability and an asparaginyl hydroxylase factor inhibiting HIF (FIH) that regulates their transcriptional activity.4 The PHD hydroxylate proline residues are located within the HIF-α ODD (P402 and P564 for HIF-1α; P405 and P531 for HIF-2α). This hydroxylation allows the binding of the von Hippel-Lindau (VHL) protein, a recognition subunit of an E3 ubiquitin ligase multiprotein complex. Binding of VHL to HIF-α subunits induces ubiquitination, which targets them for degradation by the proteasome. Under hypoxic conditions, when the co-factor oxygen is limiting, hydroxylation of HIF-α subunits slows down and results in its stabilization. HIF-α then translocates to the nucleus, associates with the HIF-1α subunit and, upon recruiting appropriate co-activators, the HIF-α/b heterodimer binds to hypoxia response elements (HRE) within DNA and activates expression of HIF target genes.5 HIF regulate the transcription of more than 200 genes involved in many pathways, notably erythropoiesis via the synthesis of erythropoietin (EPO)6 and iron metabolism regulation (tranferrin, transferrin receptor, divalent metal transporter 1 [DMT1], ferroportin). HIF-2 is the main isoform that controls EPO expression which regulates the proliferation and differentiation of erythroid progenitors, thereby linking decreased tissue oxy-
genation to an adequate erythropoietic response.7
HIF-2α is an 870 amino acid protein encoded by the EPAS1 gene located in 2p21, that contains 16 exons. The first mutation in the EPAS1 gene has been described in 2008 associated with polycythemia, also called erythocytosis (ECYT) developed by patients over three generations.8 Since then, more than 40 other cases have been published (Online Supplementary Table S1). These are always germline missense mutations in the heterozygous state. Patients described with a mutation in the EPAS1 gene developed erythrocytosis frequently associated with high EPO levels and are at increased risk of pulmonary hypertension and thrombotic events (thrombosis, infarction, pulmonary embolism).8-11 This disease has been classified as ECYT4 (OMIM#603349, MIM#611783).
Importantly, mutations in EPAS1/HIF2A gene are also responsible for the development of pheochromocytomas, paragangliomas (PPGL), somatostatinomas and ocular lesions12 sometimes associated with erythrocytosis and rare cases of cyanotic congenital heart disease and hemangioblastomas (Online Supplementary Table S2).13,14,12,15 In these cases, the mutations have been found at the somatic level within the tumor, but further investigations sometimes revealed that the mutation was acquired during development and was actually present in a mosaic state.16
The majority of these missense mutations are located in exon 12 and cluster close to the proline residues Pro531, hydroxylation of which regulates HIF-2α stability. Combined biochemical and cellular assays showed that the majority of these mutations may reduce both hydroxylation of HIF-2α by the PHD, and subsequent recognition of HIF-2α by pVHL. Functional studies showed that mutations associated with tumor development have a more severe gain-of-function than mutations associated with erythrocytosis alone.17 These most deleterious mutations (amino acids 529-532) affect residues close to the key proline 531.
In the present collaborative study, we describe a unique series of 43 patients diagnosed with an erythrocytosis in whom we identified 33 different genetic variants in EPAS1. The aim of this study is to determine the causal role of these genetic variants in the pathogenesis of erythrocytosis and the potential risk for developing tumors.
In order to study the functional consequences of the different HIF-2 α mutations, we performed luciferase reporter assays. We used a vector encoding the firefly luciferase driven by multiple proximal and distal regulatory elements of the HIF-2 target gene EPO, the key target gene linked to the development of erythrocytosis. A recent publication suggested that the EPO gene may contain complex regulatory elements in its proximal promoter based on the identification of a mutation located in a region (c.-136, upstream the ATG codon18) not included in
Haematologica | 108 June 2023 1653 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
previously existing luciferase constructs.19,20 We therefore generated a luciferase vector driven by a substantially extended region of the EPO promoter and distal enhancer regions. In order to accurately quantify subtle changes in HIF-2 α activity real-time periodical luminescence measurements were performed.
Methods
Sequencing
Samples were obtained from laboratories specialized in diagnosis and research of idiopathic erythrocytosis after exclusion of classical causes of erythrocytosis (polycythemia vera or secondary erythrocytosis associated with particular renal, cardiac or pulmonary disorders). After receiving approval from the Ethics Committee and obtaining written informed consent from the proband and family members, blood samples were collected and DNA was extracted for genetic analysis. Molecular screening of genes associated with erythrocytosis was performed by high throughput sequencing with different technologies, depending on the sequencing center (see the Online Supplementary Appendix).
In silico analysis
Genetic variants located in the EPAS1 gene with a global frequency lower than 5.10-4 in the gnomAD v3 database were selected. The MetaDome21 analysis was performed on the EPAS1 gene by using the GENCODE: ENST00000263734.3, RefSeq: NM_001430.4 and UniProt: Q99814. The effect of each non-synonymous variant was assessed using Protein Variation Effect Analyzer (PROVEAN)21 Protein Batch v1.1.3 prediction integrative webtool. ENSEMBL ID HIF-2α wildtype amino acid sequence (ENSP00000263734) was used as a reference. Cutoff of -2.5 and -4.1 were applied to PROVEAN predictions. EPAS1 variants were studied by using MobiDetails,22 an annotation platform for DNA variants. Values obtained by single-and meta-predictor tools were normalized (0-1), 0 being the less damaging and 1 the most damaging for each predictor. For the final interpretation and classification of the variants, we used the ACMG (American College of Medical Genetics and Genomics) criteria and guidelines.23 The ACMG uses the following classification to describe variants identified in Mendelian disorders: class 1: benign; class 2: likely benign; class 3: variant of uncertain significance (VUS); class 4: likely pathogenic; class 5: pathogenic. Criteria used in our study are detailed in the Online Supplementary Appendix.
Full erythropoietin promoter plasmid generation
The full human core EPO promoter sequence was recovered from ENSEMBL (7 dna:chromosome:GRCh38:7: 100720400:100721001:1, ENSR00000833692). Briefly, the
pGL3-5’HRE290-FullProm-3’HRE126 EPO promoter-driven luciferase plasmid was generated after polymerase chain reaction (PCR) amplification of a 604 bp fragment of genomic DNA extracted from HeLa cells that was inserted into a pGL3-luciferase vector (details are provided in the Online Supplementary Appendix).
End point luciferase reporter assays
Cells were cultured as detailed in the Online Supplementary Appendix. Briefly, 4x105, 3.5x105 and 6.5x105 cells were transiently transfected with 500 ng or 100 ng reporter plasmid in a 6-well format using CaCl2 or JetOptimus (Polyplus), respectively for HEK293T and Hep3B or Kelly cells, and 1 mg of YFP-HIF-1α, YFP-HIF-2α or pcDNA3-HA-HIF-2α (Addgene) constructions. In order to control for differences in transfection efficiency and extract preparation, 50 ng or 75 ng pRL-SV40 Renilla luciferase reporter vector (Promega) was co-transfected, respectively for HEK293T and Hep3B or Kelly cells. The next day, cultures were evenly split onto 6-well plates, incubated for an additional 24 hours, under normoxic or hypoxic conditions (0.2% O2, 5% CO2 and 37°C). Cells were lysed with passive lysis buffer and luciferase activities of duplicated wells were determined using the Dual Luciferase Reporter Assay System (Promega) as described before.24 Reporter activities were expressed as relative Firefly/Renilla luciferase activities normalized to control under hypoxic conditions. All reporter gene assays were performed at least three times independently. Proteins were extracted and immunoblotted to quantify the HIF-2α proteins as described before25 (see the Online Supplementary Appendix).
Real-time luciferase reporter assays
Luciferase assays were performed on HEK293T cells seeded in 24-well Black Visiplate Perkin Elmer) (1×105 cells per well), 24 hours before transfection. Cells were transfected by using jetPRIME® (Ozyme Polyplus). The expression vectors pcDNA3-HA-HIF-2 α (25 ng) were co-transfected with the pGL3-5’HRE-FullProm-3’HRE-EPO promoter-driven luciferase plasmid (100 ng), and empty vector for a total amount of 500 ng transfected DNA. Luciferase activity was monitored over 48 hours using the bioluminometer WSL-1565 Kronos HT® (ATTO). Cells were harvested and lysed in extraction buffer (Macherey Nagel) for quantification of transfected plasmids by PCR (for details see the Online Supplementary Appendix).
Statistical analysis
Values in the figures of the end point luciferase assays are presented as mean ± standard error of the mean (SEM). For the real-time luciferase reporter assay, differences in means among multiple groups were analyzed by using one-way ANOVA of Kruskal-Wallis and Dunn's post hoc tests. All statistics were performed with GraphPad Prism
Haematologica | 108 June 2023 1654 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
software 7.05. Values of P≤0.05 were considered statistically significant.
Results
Diagnosis and genetic screening
Patients with erythrocytosis were selected after exclusion of classical causes of erythrocytosis (polycythemia vera or secondary erythrocytosis associated with particular renal, cardiac or pulmonary disorders). Genetic screening was performed by using next-generation sequencing (NGS) panels dedicated to erythrocytosis, and a total of 1,450 patients were sequenced at all centers. Results are presented in Table 1 and show 33 missense genetic variants identified in a total of 43 patients (41 patients with heterozygous germline variant and 2 patients with mosaicism) and 28 relatives from four different European countries. Ten variants have already been described.15,26-43 Twenty-four variants are located in exon 12 and extend from amino acids 525 to 658 surrounding the key proline in position 531. Family history of erythrocytosis is present in 14 families. The pedigrees are of variable size (Figure 1; Online Supplementary Figure S1) and can show a multigenerational history of erythrocytosis (family [F] 14, F22), but sometimes also display a family history

related to the consequences of undiagnosed erythrocytosis, such as ischemic stroke (F13). Clinical manifestations are in general variable, ranging from mild to severe erythrocytosis (Ht up to 77.5% in patient 19) (Table 1 for probands; Online Supplementary Table S3 for relatives). The serum EPO level is rarely elevated except in two patients (up to 7,500 UI/mL in patient 20). However, we were unable to obtain full information on the patient's phlebothomy time frame that may influence these factors.
Associated symptoms characteristic to EPAS1 mutations have been observed in a limited number of patients. Pulmonary arterial hypertension (PAH) was developed by two patients and ocular lesions have been detected in three patients as previously reported in association with EPAS1 mutations.12 A history of thrombosis was reported in six patients and six relatives which mainly were of the stroke type. Close examination of the medical records of family 13 (variant D525H identified in the 76-year-old proband and her son with no history of thrombosis) found a stroke in five sisters at an age of onset between 60 and 80 years, however, no genetic test has been done in these relatives to indicate that the variant is associated with thrombosis. We also found a stroke in a child (patient 20, variant Y532C) with a Moya-Moya type cerebral vascular malformation. Unfortunately, no information could be ob-
Haematologica | 108 June 2023 1655 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
Figure 1. Pedigree of patients carrying a mutation in EPAS1. Roman numerals indicate generations. Squares indicate men and circles women; black filling indicate the development of confirmed erythrocytosis; gray filling indicate a suspected erythrocytosis; arrows indicate probands; +, indicates a positive genetic screen.
ARTICLE - EPAS1 variants in patients with erythrocytosis
Table 1. Clinical data of probands with a genetic variant identified in EPAS1
ID Ex Pos. cDNA Pos. protein Age/age at diagnosis in years Sex Hb Ht RBC EPO Other symptoms Family history Ref Diagnostic center P#1 2 c.181A>G p.Ile61Val 17 M 17.1 49.3 5.89 6.9 na No / Coimbra P#2 6 c.587C>T p.Thr196Met 55 M 16.5 55 6 4.8 None No / Nantes P#3 6 c.734T>A p.Leu245Gln 57 M 16 48.5 5.94 5.8 na No / Dijon P#4 7 c.818T>G p.Leu273Arg 19 M 17.1 46 6.05 12.8 na ne / Nantes P#5 9 c.1046A>G p.Lys349Arg 57 M 17.8 53.6 na 5.5 na na / Dijon P#6 9 c.1057G>C p.Val353Leu 32 M 20 60 na 3 Pulmonary embolism at 30 yrs No / Nantes P#7 9 c.1121T>A p.Phe374Tyr 57 M 17.6 50 5.71 na None na G: 26-28 S: 15 Dijon P#8 9 c.1121T>A p.Phe374Tyr 57 M 17.5 50.5 5.88 5.6 None No G: 26-28 S: 15 Dijon P#9 9 c.1121T>A p.Phe374Tyr 57 M 18.5 55 6.2 5.7 Portal vein thrombosis at 1 yr No G: 26-28 S: 15 Paris P#10 11 c.1478A>G p.Asp493Gly 75/53 M na na na 7 na No / Nantes P#11 11 c.1510C>G p.Leu504Val 65 M 19 55.8 6.15 7.9 Tumors ne Yes (2) / Dijon P#12 11 c.1510C>G p.Leu504Val 48/63 M 17.2 51 5.6 4.4 None Yes (2) / Nantes P#13 12 c.1573G>C p.Asp525His 76 F 19.4 56 6.53 4.9 None; 5 deceased sisters after stroke Yes (2) G: 29 Nantes P#14 12 c.1574A>G p.Asp525Gly 44 M 20.5 56.9 6.54 16.8 ne Yes (6) / Nantes/ Dijon P#15 12 c.1578G>C p.Leu526Phe 62/49 M 19.8 57.1 6.46 13.5 Stroke, myocardial infarction, pulmonary embolism Yes (2) / Nantes P#16 12 c.1579G>A p.Glu527Lys 51 F 18.6 56.9 6.4 12.5 Congenital cataract Yes (3) / Dijon P#17 12 c.1588G>T p.Ala530Ser 33/28 M 23 68 na Norm. None Yes (2) / Nantes P#18 12 c.1589C>A p.Ala530Glu 1.5% reads 6/9 months M 19.5 65.6 10.04 na na No S: 27, 30 Nantes P#19 12 c.1591C>T p.Pro531Ser 1.9% reads 11/16 months F 24.2 77.5 9.4 573 Paragangliomas, cardiac symptomes, ocular lesions1 No S: 27, 31-36 Nantes P#20 12 c.1595A>G p.Tyr532Cys 9/7 M 14.8 53.7 8.33 >450-7,885 Stroke, PAH, ocular lesions,2 tumors ne, Moya-Moya disease No S: 27, 37 Dijon P#21 12 c.1597A>G p.Ile533Val 32 M 20.7 58.5 6.87 15.3 None, tumors ne Yes (3) G: 38 Nantes P#22 12 c.1604T>C p.Met535Thr 17/6 M 18.2 52 5.89 na None Yes (5) G: 39, 42 Nantes/ Créteil P#23 12 c.1604T>C p.Met535Thr 65/29 F 18.4 54 6.33 16.4 AHT Yes (5) G: 39, 42 Paris P#24 12 c.1609G>C p.Gly537Arg 26 M 20.2 59.8 6.7 13.6 ne No G: 9, 40, 41, 43 Nantes P#25 12 c.1609G>A p.Gly537Arg 11 M 18.2 53 6.13 10.1 None No G: 9, 40, 41, 43 Nantes P#26 12 c.1609G>A p.Gly537Arg 29 M 21.8 59 na 12.3 None No G: 9, 40, 41, 43 Nantes P#27 12 c.1609G>A p.Gly537Arg 23 F 19.0 55 6.4 13.1 PAH Yes (3) G: 9, 40, 41, 43 Nantes P#28 12 c.1609G>A p.Gly537Arg /32 M 14.5 50 na Norm. na No G: 9, 40, 41, 43 Nantes P#29 12 c.1609G>A p.Gly537Arg 39/23 F 20 58 na 41 Cerebral thrombophlebitis Yes (2) G: 9, 40, 41, 43 Nantes P#30 12 c.1612G>A p.Glu538Lys 54/33 M 18.2 54 na 24 None No / Dijon P#31 12 c.1620C>A p.Phe540Leu 45 M 20.3 60.5 6.7 8.3 None na G: 28, 42 Dijon P#32 12 c.1642G>A p.Glu548Lys /28 M 19.5 62 6.52 17 None, tumors ne Yes (3) / Utrecht P#33 12 c.1671G>C p.Gln557His 53 M 18.2 50.7 5.68 7.4 None, tumors ne Yes (2) / Nantes/ Dijon P#34 12 c.1679C>A p.Pro560His /32 M 17.9 51 6.23 na na na / Utrecht P#35 12 c.1685A>T p.His562Leu /49 M 15.0 50 6.28 9 Stroke at 35 yrs, died of sepsis na / Utrecht P#36 12 c.1700T>C p.Met567Thr 63 M 17.6 53 5.98 Norm. Cardiac symptomes No S: 32 Nantes P#37 12 c.1700T>C p.Met567Thr na F 20.4 59.7 6.39 3.7 na na S: 32 Mondor P#38 12 c.1705A>G p.Asn569Asp 34/27 F 16.5 49 na 8.5 AHT, eclampsia na / Paris P#39 12 c.1750C>T p.Leu584Phe /43 M 18.4 53 5.88 7 na No / Utrecht P#40 12 c.1805G>A p.Arg602Gln /30 M 17.3 52 5.9 1.7 na na / Nantes P#41 12 c.1960G>A p.Val654Ile 24 M na 52 na na None na / Nantes P#42 12 c.1973G>A p.Arg658His 73/68 M 18.6 56.5 6.55 14 Stroke, ocular lesions, tumors ne No / Nantes P#43 15 c.2474G>A p.Arg825Gln 66 M 16.9 50 5.75 5.9 na na / Dijon
Continued on following page Haematologica | 108 June 2023 1656
V. Karaghiannis et al.
Position of the genetic variants are indicated. They have all been identified in the heterozygous state (approximatively 50% of the reads obtained by next-generation sequencing), except 2 mosaic patients (percentage of reads indicated in bold). All patients presented with erythrocytosis and additional clinical manifestations are indicated. ID: identification; P#: patient number; Ex: exon; Pos: position; diagnosis: the age is indicated in years or the number of months is specified; M: male; F: female; Hb: hemoglobin in g/dL (normal range, 13-16.5 g/dL for men; 12-16 g/dL for women); Ht: hematocrit in % (normal range, 40-49% for men; 37-48% for women); RBC: red blood cells in x1012/L (normal range, 4.2-5.7x1012/L for men, 4.2-5.2x1012/L for women); EPO: erythropoietin in mU/mL (normal range, 5–25 mU/mL); PAH: pulmonary arterial hypertension; AHT: arterial hypertension; ne: not explored; Norm.: normal; yrs: years old; na: no data available. Ocular lesions1: bilateral major papilloedema with fibrotic aspect and vascular shunts; Ocular lesions2: monocular blindness due to retinal atrophy. Family history: the number of family members presenting erythrocytosis is indicated in brackets. Ref: references detailed in the bibliography of the main manuscript. G: germline mutation; S: somatic mutation identified in the tumor.
tained regarding the existence of any other cardiovascular risk factor in the relatives.
Of major importance, a paraganglioma has been detected in one mosaic patient (patient 19). Patient 19 presented at 16 months old with a strongly elevated Ht (77.5%), Hb (24.2 g/dL) and EPO (573 UI/mL). A first screening by NGS reads did not reveal germline mutations. In the presence of such a severe diagnosis, whole-exome and subsequently whole-genome sequencing were performed on the patient and parent’s DNA, but no significant genetic abnormality was identified. At the age of 10 years, due to severe hypertension highly suggestive of the presence of a catecholamine secreting tumor, a deeper re-analysis of NGS reads allowed the identification of the c.1591C>T, p.Pro531Ser variant at a maximum rate of 1.9% of the reads that was confirmed by using droplet PCR (Online Supplementary Figure S2). Indeed, two abdominal paragangliomas were identified and resected. Sequencing of the tumor using droplet PCR showed a variant allele frequency of 60%, confirming the role of EPAS1-mutated cells in oncogenesis (Online Supplementary Figure S2B). After surgical resection of the paragangliomas, disappearance of the hypertrophic cardiomyopathy and the noncompaction syndrome initially found was noted. A second patient presented at a very young age (patient 18, 9 months old) with severe erythrocytosis. The mutation c.1589C>A, p.Ala530Glu was detected at 1.5% of reads by NGS and confirmed by droplet PCR. The risk of tumor development in this patient is carefully monitored.
In silico analysis of the genetic variants
Subsequently, we employed the MetaDome web server to analyze the mutation tolerance at each position of the HIF-2α protein. The amino acids targeted by the missense genetic variants identified in our study were localized on the HIF-2α protein map (Figure 2). The resulted tolerance is reported in Table 2 for each variant, and the detailed score are indicated in the Online Supplementary Table S4 The majority of the variants target amino acids are located in the oxygen-dependent degradation (ODD) domain, an intolerant zone surrounding the key residue proline P531. The location of this hydroxylation site is shown in the 3D structure of HIF-2α (upper left panel of Figure 2).
We then used the Mobidetails online platform which
gathers many sources of data for the interpretation of DNA variants in the context of molecular diagnosis. For each variant, we analyzed the scores obtained with single- and meta-predictors and classified the variants as benign when both scores were <0.5, and as deleterious when both scores were >0.7 (Figure 3A).
These scores are illustrated with a Radar view presenting the prediction results obtained from the different in silico tools (Online Supplementary Figure S3).
The analysis was completed by using the PROVEAN in silico tool. We classified the variants depending on their score regarding the cutoff: eutral >-2.5, possibly deleterious <-2.5 and >-4.1, deleterious <-4.1 (Figure 3B).
The final classification of the variants was based on a global analysis of in silico and functional studies (see below) with the ACMG (American College of Medical Genetics and Genomics) criteria which previously developed guidance for the interpretation of sequence variants (see the last column of Table 2, and details of scores in the Online Supplementary Table S4)23
Generation of an erythropoietin promoter-driven reporter vector
As HIF-2α-dependent regulation of EPO expression represents the key pathophysiological mechanism involved in the occurrence of erythrocytosis, we focused our functional studies on reporter assays using a luciferase gene driven by EPO regulatory elements. Previously existing luciferase constructs under the control of the EPO promoter contain the proximal region located between position -194 and -341 upstream the ATG codon (termed minimal promoter, labeled in black; Online Supplementary Figure S4).19,20 A mutation located at position -136 in the EPO promoter has been recently described and linked to the development of erythrocytosis in two families.18 We therefore introduced a larger promoter region into the reporter constructs from position -17 to -564 of the EPO promoter (construct termed Full EPO promoter, labeled in gray; Online Supplementary Figure S4 ).
Functional studies by using end point and real-time reporter luciferase assay
We performed luciferase reporter assays using this novel EPO promoter-driven construct in the absence or presence of the distal 5’ and 3’ hypoxia-responsive elements (HRE)
Haematologica | 108 June 2023 1657 ARTICLE
EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
-
responsible for tissue-specific and hypoxia-inducible regulation of EPO gene expression in the kidney and the liver, respectively. Transient transfection of these constructs showed higher luciferase activity of the full promoter under hypoxic conditions compared to the minimal promoter. Furthermore, transient co-transfection of vectors encoding
wild-type HIF-1α or HIF-2α displayed an increased luciferase signal of both the minimal and enlarged promoter, but resulted in preferential response to HIF-2α only when the enlarged promoter was present under both normoxic and hypoxic conditions. A similar preferential HIF-2α-dependent increase in luciferase activity was obtained with reporter
Figure 2. HIF-2α three-dimensional structure and HIF-2α protein tolerance landscape. The main domains of the HIF-2α protein are presented in purple in the middle of the figure. The severity of the impact induced by sequence variation targeting each amino acid was determined by using the MetaDome in silico analysis tool. It is presented with a color code, from blue (tolerant) to red (intolerance) on the right. The variants identified in our series are listed. PAS (Per-ARNT-Sim) domain: dimerization domain, ODD: oxygen dependent degradation domain, c-TAD: c-terminal transactivation domain. In the upper left panel, the three-dimensional structure of HIF-2α is presented (from AlphaFold Protein Structure Database). The HIF-2α inhibitor (MK-6482/Belzutifan/Welireg) binds to a region that causes a conformational change of the amino acids H293 and M252 (red stars in the figure), while the amino acids found mutated in patients target the close vicinity of the hydroxylation proline P531 located in the ODD domain.
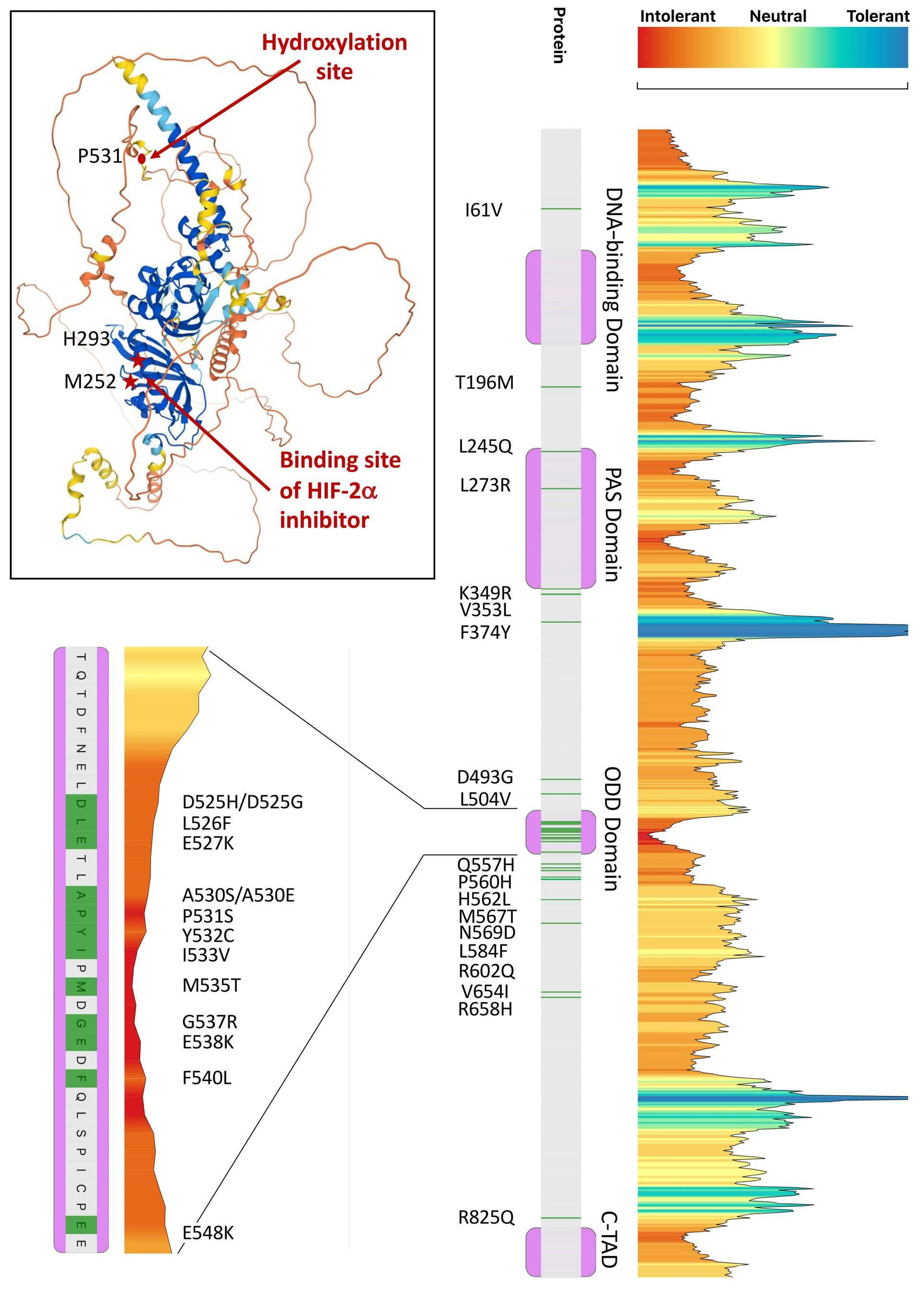
Haematologica | 108 June 2023 1658 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
Table 2. Results of in silico analysis and functional studies of a genetic variant identified in EPAS1
The prediction of each bioinformatic tool is illustrated by a color: red for a pathogenic/damaging classification, green for benign/neutral impact and orange for intermediate/unknown signification. The prediction obtained with the Mobidetails single predictors are shown and additional information is added (/) when the prediction obtained with the meta-predictors is divergent. The classification obtained with the functional tests (end point luciferase assay performed in different cell lines; activity and/or velocity of the reaction obtained by real-time luciferase assay) is illustrated by a color: red for a significant gain of function obtained in at least 2 assays, orange for a significant gain of function obtained in at least 1 experiment and green for no significant gain of function or a function close to the wild-type (WT) protein. Pos: position; Int.: intolerant; Tol.: tolerated; Neut.: neutral; Del.: deleterious; Prob.: probably; Dam.: damaging; Ben.: benign; Sign.: significant; GOF: gain of function; PROVEAN: protein variation effect analyzer; ACMG: American College of Medical Genetics and Genomics; VUS: variant of unknown signification. Percentage of reads indicated in bold in 2 mosaic patients.
ID Ex Pos. cDNA Pos. protein Pos. protein Global gnomAD v3 frequency Metadome prediction Prediction PROVEAN Radar Mobidetails prediction Compiled luciferase functional test ACMG class Conclusion/ classification P#1 2 c.181A>G p.Ile61Val I61V 2.094x10-5 Slighly Int. Neut. Prob. Dam./Tol. / 3 VUS P#2 6 c.587C>T p.Thr196Met T196M 7.678x10-5 Int. Delet. Prob. Dam./Tol. / 3 VUS P#3 6 c.734T>A p.Leu245Gln L245Q 0 Neut. Delet. Possibly Dam./Tol. / 3 VUS P#4 7 c.818T>G p.Leu273Arg L273R 0.0005 Int. Delet. Prob. Dam./Tol. / 3 VUS P#5 9 c.1046A>G p.Lys349Arg K349R 0 Int. Neut. Ben. / 3 VUS P#6 9 c.1057G>C p.Val353Leu V353L 0 Int. Neut. Possibly Dam./Tol. / 3 VUS P#7 9 c.1121T>A p.Phe374Tyr F374Y 0.0045 Tol. Neut. Ben. No GOF 2 Likely Ben. P#8 9 c.1121T>A p.Phe374Tyr F374Y 0.0045 Tol. Neut. Ben. No GOF 2 Likely Ben. P#9 9 c.1121T>A p.Phe374Tyr F374Y 0.0045 Tol. Neut. Ben. No GOF 2 Likely Ben. P#10 11 c.1478A>G p.Asp493Gly D493G 0 Int. Prob. Delet. Ben. No GOF 3 VUS P#11 11 c.1510C>G p.Leu504Val L504V 6.977x10-6 Slighly Int. Neut. Prob. Dam./Tol. No GOF 3 VUS P#12 11 c.1510C>G p.Leu504Val L504V 6.977x10-6 Slighly Int. Neut. Prob. Dam./Tol. No GOF 3 VUS P#13 12 c.1573G>C p.Asp525His D525H 0 Int. Delet. Prob. Dam. / 4 Likely Path. P#14 12 c.1574A>G p.Asp525Gly D525G 0 Int. Delet. Prob. Dam. Sign. GOF 5 Path. P#15 12 c.1578G>C p.Leu526Phe L526F 0 Int. Prob. Delet. Prob. Dam. Sign. GOF 4 Likely Path. P#16 12 c.1579G>A p.Glu527Lys E527K 0 Int. Prob. Delet. Prob. Dam. Sign. GOF 4 Likely Path. P#17 12 c.1588G>T p.Ala530Ser A530S 0 Int. Prob. Delet. Prob. Dam. Sign. GOF 5 Path. P#18 12 c.1589C>A p.Ala530Glu 1.5% reads A530E 0 Int. Delet. Prob. Dam. / 5 Path. P#19 12 c.1591C>T p.Pro531Ser 1.9% reads P531S 0 Highly Int. Delet. Prob. Dam. Sign. GOF 5 Path. P#20 12 c.1595A>G p.Tyr532Cys Y532C 0 Int. Delet. Prob. Dam. Sign. GOF 5 Path. P#21 12 c.1597A>G p.Ile533Val I533V 0 Highly Int. Neut. Prob. Dam. / 5 Path. P#22 12 c.1604T>C p.Met535Thr M535T 0 Highly Int. Delet. Prob. Dam. / 5 Path. P#23 12 c.1604T>C p.Met535Thr M535T 0 Highly Int. Delet. Prob. Dam. / 5 Path. P#24 12 c.1609G>C p.Gly537Arg G537R 0 Highly Int. Prob. Delet. Prob. Dam./Tol. Sign. GOF 5 Path. P#25 12 c.1609G>A p.Gly537Arg G537R 0 Highly Int. Prob. Delet. Prob. Dam./Tol. Sign. GOF 5 Path. P#26 12 c.1609G>A p.Gly537Arg G537R 0 Highly Int. Prob. Delet. Prob. Dam./Tol. Sign. GOF 5 Path. P#27 12 c.1609G>A p.Gly537Arg G537R 0 Highly Int. Prob. Delet. Prob. Dam./Tol. Sign. GOF 5 Path. P#28 12 c.1609G>A p.Gly537Arg G537R 0 Highly Int. Prob. Delet. Prob. Dam./Tol. Sign. GOF 5 Path. P#29 12 c.1609G>A p.Gly537Arg G537R 0 Highly Int. Prob. Delet. Prob. Dam./Tol. Sign. GOF 5 Path. P#30 12 c.1612G>A p.Glu538Lys E538K 0 Highly Int. Prob. Delet. Prob. Dam. Sign. GOF 3 VUS P#31 12 c.1620C>A p.Phe540Leu F540L 0 Int. Delet. Prob. Dam. Sign. GOF 3 VUS P#32 12 c.1642G>A p.Glu548Lys E548K 1.396x10-5 Int. Neut. Prob. Dam./Tol. / 3 VUS P#33 12 c.1671G>C p.Gln557His Q557H 6.983x10-5 Int. Neut. Possibly Dam./Tol. Close to WT 3 VUS P#34 12 c.1679C>A p.Pro560His P560H 4.187x10-5 Int. Neut. Possibly Dam./Tol. / 3 VUS P#35 12 c.1685A>T p.His562Leu H562L 0 Int. Neut. Ben. / 3 VUS P#36 12 c.1700T>C p.Met567Thr M567T 0.0002 Int. Neut. Ben. / 3 VUS P#37 12 c.1700T>C p.Met567Thr M567T 0.0002 Int. Neut. Ben. / 3 VUS P#38 12 c.1705A>G p.Asn569Asp N569D 4.192x10-5 Int. Neut. Ben. / 3 VUS P#39 12 c.1750C>T p.Leu584Phe L584F 6.978x10-6 Neut. Neut. Ben. / 3 VUS P#40 12 c.1805G>A p.Arg602Gln R602Q 1.397x10-5 Slighly Int. Neut. Ben. / 3 VUS P#41 12 c.1960G>A p.Val654Ile V654I 2.094x10-5 Slighly Int. Neut. Ben. / 3 VUS P#42 12 c.1973G>A p.Arg658His R658H 0.0002 Slighly Int. Neut. Ben. / 3 VUS P#43 15 c.2474G>A p.Arg825Gln R825Q 6.98x10-6 Neut. Neut. Ben. / 3 VUS
Haematologica | 108 June 2023 1659 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
genes in the presence or absence of distal HREs (Figure 4; Online Supplementary Figure S5). The same experiments were performed with co-transfection of plasmids encoding the mutated HIF-2α proteins, including P531A as positive control in HEK293, Hep3B and Kelly cells (1 clone for each variant in triplicates). We observed a significant
gain of function for the P531S mutant in all three cell lines and for A530S, G537R and F540L in Hep3B cells (Figure 5; Figure 6B; Online Supplementary Figure S7A). We also performed these experiments under hypoxic conditions confirming the results under normoxic conditions, with a significant gain of function for the P531S mutant in three
Figure 3. In silico prediction of the impact of an amino acid substitution on the HIF-2α protein. (A) Representation score obtained by the predictors analyzed by the Mobidetails annotation platform. Values are normalized (0-1), 0 being the less damaging and 1 the most damaging for each predictor. The graph indicates the mean normalized score obtained by single predictors (SIFT, Polyphen 2 HumDiv an0d HumVar) and meta-predictors (Fathmm, REVEL, ClinPred, Meta SVM, Meta LR, Mistic). (B) Representation of protein variation effect analyzer (PROVEAN) analysis performed on each non-synonymous HIF-2α variant. Variants with a score above -2.5 were predicted to be neutral. A threshold score below -2.5 was predicted to be deleterious and a more stringent threshold score of -4.1 is associated with increased specificity

A B Haematologica | 108 June 2023 1660 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
Figure 4. End point reporter gene assays demonstrate increased erythropoietin promoter-driven luciferase activity with a full erythropoietin promoter compared to minimal construct. HEK293 cells were co-transfected with the erythropoietin (EPO) minimal or full promoter-driven constructs containing the 5’HRE and 3’HRE and with HIF-1α or HIF-2α isoform overexpression plasmids, as indicated. The luciferase activity is reported as the induction compared to the control (Ctrl) under hypoxic condition and represents the ratio of firefly (FF) to Renilla (RL) relative light units (RLU). Each column represents the mean ± standard error of the mean of 4 to 14 different experiments performed in duplicate. One-way ANOVA (*P≤0.05; **P≤0.01; ****P≤0.0001).
cell lines and for the E538K in Hep3B (Online Supplementary Figures S6 and S7B).
In order to increase the sensitivity of the reporter assay we reduced the amount of transfected HIF-2α expression vectors. We used HEK293 cells in order to avoid additional induction of the reporter vector by endogenous HIF-2 α (expressed in Hep3B and Kelly cells). In order to determine the optimal time frame to quantify the luciferase activity after transfection, we followed it using a real-time luciferase measurement system for 48 h (Figure 6A). We set the 100% activity to the relative light unit (RLU) values obtained at the plateau of the wild-type protein after 30 h of expression from the beginning of the reaction (i.e., 34 h after transfection). We observed a large scale of activity from wild-type (around 100%) to elevated gain-of-function (150-300%). The mean of results derived from repeated experiments obtained at the plateau are shown in Figure 6B. We calculated the slope of the different curves to obtain an indicator of the velocity of the reaction (Figure 6C). The use of three independent clones for each variant shows consistent results with strong reliability of this test within an experiment, but some variants display variable behavior between experiments, demonstrating the importance to replicate assays. Altogether, a significant increase of the activity and/or the velocity of the reaction was found for the tested variants identified in patients targeting amino acids from D525 to E538. One variant presents an activity very close to the wild-type protein and was classified as benign (Q557H), the other variants have been classified as variant of unknown signification (VUS).

Discussion
We report here the largest collection of EPAS1/HIF2A gene variants from a large cohort of patients with idiopathic erythrocytosis. Variants were characterized using detailed in silico and new functional studies.
The use of a wide range of in silico prediction tools can be very useful for classifying variants in genetic diagnosis. However, the pathogenicity of a genetic variant can be difficult to classify as pathogenic when the disease is associated with a gain-of-function mutation. For this reason, functional studies need to be included in the diagnostic tools. In the case of erythrocytosis, the functional tests must be very sensitive because it is known that many mutations may be hypomorphic,44–46 especially because the genes of the hypoxia pathway play a major role in physiology and a germline alteration that is too severe and would likely be incompatible with life. The construction of an improved, more sensitive EPO promoter-driven vector in combination with the use of a real-time luciferase reporter assay measurements allowed us to accurately measure gain of function of new HIF-2α mutants. Altogether, combined in silico, segregation and functional studies performed in this study allowed the classification of 11 variants as likely pathogenic or pathogenic (including 4 never previously described) in 17 patients and 23 relatives. Our study further expanded the region of HIF-2α associated with erythrocytosis. Thus far, this region spanned from amino acids 529 to 537, but the classification of pathogenic variants of amino acids 525, 526 and 527 prompt an expansion of this
Haematologica | 108 June 2023 1661 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
ARTICLE - EPAS1 variants in patients with erythrocytosis
area. Additional investigations will be necessary to definitely classify the E538K and F540L variants. Our results will allow an appropriate follow-up of families at the clinical level, especially with regard to complications and associated diseases. It is indeed intriguing that so few patients carrying pathogenic variants present with additional symptoms associated with EPAS1 mutations:
PAH was only described in two patients (P#20 and 27), ocular lesions in two patients (P#19, 20) and thrombosis and ischemic accidents in five families (P#13, 15, 20, 22, 29). Interestingly, patient 20 carrying the variant p.Tyr532Cys also suffers from the Moya-Moya disease. This is a rare and chronic disease of mostly unknown causes that affects the blood vessels in the brain. No link has ever
Figure 5. End point reporter gene assays demonstrate a significant gain of function of the P531S HIF-2α mutant. HEK293 cells (A) and Hep3B cells (B) were transfected with 5’HRE and 3’HRE-dependent erythropoietin (EPO) full promoter-driven construct and different HIF2α mutants identified from patients, as well as with wild-type (WT) and positive control P531A HIF-2α constructs, as indicated. Luciferase activity is reported as the induction compared to the control (Ctrl) under normoxic conditions and represents the ratio of firefly (FF) to Renilla (RL) relative light units (RLU). Each column represents the mean ± standard error of the mean of 3 to 5 different experiments performed in duplicate. (C) Expression levels of the different HIF-2α proteins were assessed with a HIF-2α antibody. Actin and α-tubulin were used as loading controls. One-way ANOVA, compared to HIF-2α WT (*P≤0.05; ***P≤0.001; ****P≤0.0001).
Haematologica | 108 June 2023 1662

A B C
V.
Karaghiannis et al.
been found with the hypoxia pathway genes and additional investigations should be performed on the potential role of EPAS1 in the etiology of Moya-Moya disease. Some families are currently being closely monitored due to the high risk of evolution of tumor development. In the literature, somatic or mosaic pathogenic variants described in EPAS1 target the amino acid from position 529 to 532. In our study, four families carry mutations that target these amino acids. Among them, only a single patient (P#19), carrying the p.Pro531Ser pathogenic variant at a mosaic state, developed multiple paragangliomas at the

Figure 6. Real-time reporter gene assays demonstrate a significant gain of function for additional HIF-2α variants.
(A) HEK293 cells were transfected with 3’HRE erythropoietin (EPO) full promoter luciferase reporter construct and different HIF-2α mutants. Relative light unit (RLU) was measured every 30 minutes for 48 hours (h). A plateau was obtained 30 h after the beginning of reaction (i.e., 34 h after transfection). The 100% was set on the activity of the wild-type (WT) protein at this plateau. Each mutant curve represents the mean of 3 independent clones. (B) Representation of luciferase activity obtained with the real-time luciferase assay, at the plateau obtained 30 h after the beginning of the reaction (i.e., 34 h after transfection); The 100% was set on the activity of the wild-type protein at this plateau. Each column represents the mean ± standard error of the mean of experiments performed in triplicate and each chip represents 1 clone, 3 different clones was tested each time. The quantity of transfected HA-plasmid was verified by polymerase chain reaction targeting the HA tag. The results obtained on agarose gel are shown. One-way ANOVA, Kruskal–Wallis tests with Dunn’s post hoc analysis for multiple comparisons with HIF-2α WT was performed (*P≤0.05; **P≤0.01; ***P≤0.001). (C) Representation of curve slopes obtained from the realtime luciferase measurement between 10 h and 15 h after the beginning of the reaction (see grey triangle in Figure 6A), normalized to the WT protein. Each column represents the mean ± standard error of the mean of an experiment performed in quadruplicate, each color represents an experiment and each chip represents 1 clone, 3 different clones being were tested each time. One-way ANOVA, Kruskal–Wallis tests with Dunn’s post hoc analysis for multiple comparisons with HIF-2α WT was performed (*P≤0.05; **P≤0.01; ***P≤0.001).
age of 10 years. Two other young patients at high risk carry EPAS1 variants already described in tumors and will need close follow-up: patient 18 (mosaic p.Ala530Glu, 6 years old) and patient 20 (germline p.Tyr532Cys, 9 years old).
Noteworthy, it would be important to extend the followup of tumor occurrence to the family carrying the variant p.Leu526Phe. This variant indeed targets the consensus hydroxylation motif “LXXLAP” (L526XXL529A530P531) that plays a major role in tumor occurrence when mutated.
It is interesting to note the complexity of genotype/phenotype correlations in this disease17 with the example of
A C B Haematologica | 108 June 2023 1663 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
family 17 who carries the p.Ala530Ser variant (a substitution never described before this study) and do not present a tumor history despite a highly significant gain of function. A similar case has been described in a family who developed only erythrocytosis associated with the germline mutation variant p.Tyr532His.40 The precise substitution targeting key amino acids may therefore be of major importance for the severity of the disease. Of note, our study reintroduces the debate about the variant F374Y that was initially described as causal in patients with erythrocytosis and paraganglioma15,26-28 and found to be deleterious in one functional study.47 Here this variant, that could be considered as a polymorphism due to its high frequency in the population, has been clearly classified as benign. For patients in for whom variants have been classified as VUS with predictions in favor of a benign effect (i.e., patients 38 to 42), additional investigations should be conducted to identify the cause of the carrier's pathology. The search for associated variants in the same gene was carried out and only one was found: the c.1218C>G, p.Thr406Thr variant (P#41), whose high frequency provides little support for a gain of function. The association with other variants in the hypoxia pathway genes associated with erythrocytosis (EGLN1, VHL) should also be explored, including in intronic regions that may impact splicing.45 As a matter of fact, substantial research efforts remain to be made as approximately 70% of erythrocytosis cases are still of unknown genetic cause. The identification of two young cases carrying a variant at a mosaic state at a rate up to 1.9% of reads shows the potential failure to diagnose a number of patients with erythrocytosis. The development of a paraganglioma 8 years after the discovery of a major erythrocytosis, associated with very high EPO levels, is instructive: mosaicism may be present at a very low level that is not automatically detectable by usual NGS analysis. It is therefore necessary to lower the variant detection thresholds or to manually analyze the NGS results for the EPAS1 gene, notably in young children with severe erythrocytosis. Interestingly, our study opens up new research avenues related to HIF-2α-dependent EPO gene regulation. Indeed, expansion of the DNA sequence surrounding the minimal promoter previously described in the reporter vector not only resulted in substantially increased luciferase activity under both normoxic and hypoxic conditions, but also conferred more specificity towards HIF-2 α (vs. HIF-1 α ) under overexpression conditions. This additional sequence does not contain any consensus HRE (G/ACGTG)5 and strongly suggests the presence of additional regulatory sequences within the EPO promoter. It will be of interest to analyze which factors bind to this region and if these proximal elements further contribute to tissue-specific and conditional EPO gene regulation in co-operation with distal enhancers.
Overall, this study allowed the classification of EPAS1 variants classified as causal mutations in 40 individuals (17 patients and 23 relatives). This classification is of major importance giving the new therapeutics that specifically target and inhibit the HIF-2α protein. Indeed, a clinical trial with the HIF-2α inhibitor (MK-6482/Belzutifan/Welireg) on a single patient with a HIF-2α mutation has been recently published.48 This patient carried the p.Ala530Glu pathogenic variant at a mosaic state and the treatment led to a rapid resolution of the erythrocytosis, hypertension, headaches in addition to paraganglioma response.48 This remarkable effect opens up the possibility to treat more patients carrying mutations in EPAS1 which may potentially modify the drug binding capacity. Indeed, the chemical drug binds to a region that causes a conformational change of the amino acids H293 and M252 located in the HIF-2α-PAS-B domain. This drug-induced shift of the residue position, with a move of the side chains towards the binding surface, weakened the binding capacity of HIF-2α with HIF-b (ARNT-PASB) to form an active transcription factor.49 Importantly, none of the mutations associated with erythrocytosis are located in the drug-targeted region (Figure 2, upper left panel). Therefore, all the patients carrying a causal mutation in EPAS1, located between amino acids 525 to 537, are theoretically eligible for HIF2α inhibitor treatment.
Conclusions
Our collaborative study showed that enhanced functional assays in combination with in silico methods can improve the diagnosis in patients with erythrocytosis with an unclassified mutation in EPAS1. We also demonstrated the advantage of federating all diagnostic laboratories working on this rare pathology. This allows to increase the number of cases and the power of the analyses that can render genetic variants informative. It is also necessary, for a pathology linked to frequently hypomorphic mutations, to multiply the in silico analysis and to refine the functional approaches. A precise classification of mutations is indeed essential for a better diagnosis, clinical follow-up and access to a targeted treatment.
Disclosures
No conflicts of interest to disclose.
Contributions
VK, DM, AB, LS, VA, ALR, SC, VL, MD, DH and BG performed experiments. AR, FL, performed bioinformatics analyses.
CG, NM, FA, BA, LM, MR, SB, BC, FG, AP GR, NB, HC, RvW, CB, FG and the consortium ECYT4 conducted the medical and diagnostic studies. BG, DH and FG wrote the manuscripts. BG, FG and DH designed the study. BG directed the study. All authors contributed to the research and approved the final manuscript.
Haematologica | 108 June 2023 1664 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
Funding
This study was supported by grants from the Agence Nationale de la Recherche (ANR; PRTS 2015 “GenRED”; AAPG2020 "SplicHypoxia"), the Labex GR-Ex, reference ANR-11-LABX-0051, the Fondation Maladies Rares (FMR) and Kiwanis project FONDATION-GenOmics 2017, and the associations VHL Alliance USA, VHL France and Génavie. This work was also supported as a part of NCCR Kidney.CH, a National Center of Competence in Research, funded by the Swiss National Science Foundation (grant number 183774) and by Swiss National Science Foundation project grant 310030_207460.
Data-sharing statement
Data and detailed inform ation related to the study
References
1. Pearson TC, Guthrie DL, Simpson J, et al. Interpretation of measured red cell mass and plasma volume in adults: expert panel on radionuclides of the International Council for Standardization in Haematology. Br J Haematol. 1995;89(4):748-756.
2. Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268(29):21513-21518.
3. Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458-38465.
4. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858-861.
5. Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12.
6. Semenza GL, Koury ST, Nejfelt MK, Gearhart JD, Antonarakis SE. Cell-type-specific and hypoxia-inducible expression of the human erythropoietin gene in transgenic mice. Proc Natl Acad Sci U S A. 1991;88(19):8725-8729.
7. Scortegagna M, Ding K, Zhang Q, et al. HIF-2alpha regulates murine hematopoietic development in an erythropoietindependent manner. Blood. 2005;105(8):3133-3140.
8. Percy MJ, Furlow PW, Lucas GS, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(2):162-168.
9. Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112(3):919-921.
10. Martini M, Teofili L, Cenci T, et al. A novel heterozygous HIF2AM535I mutation reinforces the role of oxygen sensing pathway disturbances in the pathogenesis of familial erythrocytosis. Haematologica. 2008;93(7):1068-1071.
11. Gordeuk VR, Miasnikova GY, Sergueeva AI, et al. Thrombotic risk in congenital erythrocytosis due to up-regulated hypoxia sensing is not associated with elevated hematocrit. Haematologica. 2020;105(3):e87-e90.
12. Pacak K, Chew EY, Pappo AS, et al. Ocular manifestations of hypoxia-inducible factor-2α paraganglioma-somatostatinomapolycythemia syndrome. Ophthalmology. 2014;121(11):2291-2293.
are available from the corresponding author upon request.
Appendix: ECYT-4 members
Marc Bernard, Charles Bescond, Anaïse Blouet, Juliette Bouteloup, Françoise Boyer, Aisha Bruce, Emilie Cayssials, Nicole Casadevall, Brieuc Cherel, Marie Laure Couec, Guillaume Denis, Louis Devron, Cécile Dumesnil, Thierry Lamy de la Chapelle, Marion Gambart, Loïc Garçon, Brigitte Granel, Pierre Hirsch, Arnaud Hot, Agnès Lahary, Tabita Maia, Amira Mejri, Sandrine Meunier, Franck-Emmanuel Nicolini, Marie Nolla, Natalina Miguel, Nathalie Parquet, Emmanuel Raffoux, Dana Ranta, Laure Ricard, Nicolas Schleinitz, Jean-Baptiste Valentin and Mathieu Wemeau.
13. Vaidya A, Flores SK, Cheng Z-M, et al. EPAS1 mutations and paragangliomas in cyanotic congenital heart disease. N Engl J Med. 2018;378(13):1259-1261.
14. Zhuang Z, Yang C, Lorenzo F, et al. Somatic HIF2A gain-offunction mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367(10):922-930.
15. Taïeb D, Barlier A, Yang C, et al. Somatic gain-of-function HIF2A mutations in sporadic central nervous system hemangioblastomas. J Neurooncol. 2016;126(3):473-481.
16. Buffet A, Smati S, Mansuy L, et al. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab. 2014;99(2):E369-373.
17. Tarade D, Robinson CM, Lee JE, Ohh M. HIF-2α-pVHL complex reveals broad genotype-phenotype correlations in HIF-2αdriven disease. Nat Commun. 2018;9(1):3359.
18. Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47(7):717-726.
19. Orlando IMC, Lafleur VN, Storti F, et al. Distal and proximal hypoxia response elements cooperate to regulate organspecific erythropoietin gene expression. Haematologica. 2020;105(12):2774-2784.
20. Storti F, Santambrogio S, Crowther L, et al. A novel distal upstream hypoxia response element regulating oxygendependent erythropoietin gene expression. Haematologica. 2014;99(4):e45-e48.
21. Wiel L, Baakman C, Gilissen D, Veltman JA, Vriend G, Gilissen C. MetaDome: pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Human Mutation. 2019;40(8):1030-1038.
22. Baux D, Van Goethem C, Ardouin O, et al. MobiDetails: online DNA variants interpretation. Eur J Hum Genet. 2021;29(2):356-360.
23. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424.
24. Koay TW, Osterhof C, Orlando IMC, et al. Androglobin gene expression patterns and FOXJ1-dependent regulation indicate its functional association with ciliogenesis. J Biol Chem. 2021;296:100291.
Haematologica | 108 June 2023 1665 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
25. De Backer J, Maric D, Bosman M, Dewilde S, Hoogewijs D. A reliable set of reference genes to normalize oxygen-dependent cytoglobin gene expression levels in melanoma. Sci Rep. 2021;11(1):10879.
26. Lorenzo FR, Yang C, Ng Tang Fui M, et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl). 2013;91(4):507-512.
27. Welander J, Andreasson A, Brauckhoff M, et al. Frequent EPAS1/HIF2α exons 9 and 12 mutations in non-familial pheochromocytoma. Endocr Relat Cancer. 2014;21(3):495-504.
28. Oliveira JL, Coon LM, Frederick LA, et al. Genotype-phenotype correlation of hereditary erythrocytosis mutations, a single center experience. Am J Hematol. 2018;93:1029-1041.
29. Schelker RC, Herr W, Grassinger J. A new exon 12 mutation in the EPAS1 gene possibly associated with erythrocytosis. Eur J Haematol. 2019;103(1):64-66.
30. Toyoda H, Hirayama J, Sugimoto Y, et al. Polycythemia and paraganglioma with a novel somatic HIF2A mutation in a male. Pediatrics. 2014;133(6):e1787-1791.
31. Favier J, Buffet A, Gimenez-Roqueplo AP. HIF2A mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367(22):2161; author reply 2161-2.
32. Comino-Mendez I, de Cubas AA, Bernal C, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013;22(11):2169-2176.
33. Toledo RA, Qin Y, Srikantan S, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2013;20(3):349-359.
34. Därr R, Nambuba J, Del Rivero J, et al. Novel insights into the polycythemia-paraganglioma-somatostatinoma syndrome. Endocr Relat Cancer. 2016;23(12):899-908.
35. Vaidya A, Flores SK, Cheng Z-M, et al. EPAS1 mutations and paragangliomas in cyanotic congenital heart disease. N Engl J Med. 2018;378(13):1259-1261.
36. Abdallah A, Pappo A, Reiss U, et al. Clinical manifestations of Pacak-Zhuang syndrome in a male pediatric patient. Pediatr Blood Cancer. 2020;67(4):e28096.
37. Pacak K, Jochmanova I, Prodanov T, et al. New syndrome of paraganglioma and somatostatinoma associated with
polycythemia. J Clin Oncol. 2013;31(13):1690-1698.
38. Perrotta S, Stiehl DP, Punzo F, et al. Congenital erythrocytosis associated with gain-of-function HIF2A gene mutations and erythropoietin levels in the normal range. Haematologica. 2013;98(10):1624-1632.
39. Alaikov T, Ivanova M, Shivarov V. EPAS1 p.M535T mutation in a Bulgarian family with congenital erythrocytosis. Hematology. 2016;21(10):619-622.
40. Camps C, Petousi N, Bento C, et al. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica. 2016;101(11):1306-1318.
41. Liu Q, Tong D, Liu G, et al. HIF2A germline-mutation-induced polycythemia in a patient with VHL-associated renal-cell carcinoma. Cancer Biol Ther. 2017;18(12):944-947.
42. Percy MJ, Chung YJ, Harrison C, et al. Two new mutations in the HIF2A gene associated with erythrocytosis. Am J Hematol. 2012;87(4):439-442.
43. Percy MJ, Beer PA, Campbell G, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood. 2008;111(11):5400-5402.
44. Couvé S, Ladroue C, Laine E, et al. Genetic evidence of a precisely tuned dysregulation in the hypoxia signaling pathway during oncogenesis. Cancer Res. 2014;74(22):6554-6564.
45. Lenglet M, Robriquet F, Schwarz K, et al. Identification of a new VHL exon and complex splicing alterations in familial erythrocytosis or von Hippel-Lindau disease. Blood. 2018;132(5):469-483.
46. Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359(25):2685-2692.
47. Dwight T, Kim E, Bastard K, et al. Functional significance of germline EPAS1 variants. Endocr Relat Cancer. 2020;28(2):97109.
48. Kamihara J, Hamilton KV, Pollard JA, et al. Belzutifan, a Potent HIF2α Inhibitor, in the Pacak-Zhuang Syndrome. N Engl J Med. 2021;385(22):2059-2065.
49. Yu Y, Yu Q, Zhang X. Allosteric inhibition of HIF-2α as a novel therapy for clear cell renal cell carcinoma. Drug Discov Today. 2019;24(12):2332-2340.
Haematologica | 108 June 2023 1666 ARTICLE - EPAS1 variants in patients with erythrocytosis V. Karaghiannis et al.
Age- and gender-matched
controls needed for plateletbased biomarker studies
Thrombocytes, or blood platelets, regulate thrombosis and hemostasis,1 but also play a key role in inflammation, tumor angiogenesis, and cancer progression.2-4 Consequently, platelets are considered a valuable source of biomarkers of disease. It is known that platelet count and volume are dependent on both age and sex;5-8 however, it is not known whether this is also the case for other platelet characteristics. We recently reviewed the available literature on studies of platelet cancer biomarkers9 and observed that approximately 60% of the published studies on humans in which platelet content was investigated lacked control groups matched for age or sex. This suggests that the interpretation of the results of many platelet biomarker studies may be unreliable. Therefore, we
examined the relationship of both age and gender with phenotypic and functional platelet characteristics. We present the association of age and gender with platelet growth factor content, state of activation, and response to stimulation.
This study was performed in accordance with the principles of the Declaration of Helsinki and was approved by the local medical ethical committee (Maastricht University Medical Center+ [MUMC+], n. 114117). All study subjects provided written informed consent. Using a strict protocol,10 we collected blood from 94 healthy volunteers: 50 men (average age 65.9 years, range 43.6-85.3) and 44 women (average age 63.7 years, range 40.9-84.7). Platelet activation was quantified by whole blood flow cytometry
Pearson correlation was used to calculate the regression coefficient (r) between independent variables and age and gender. A multivariable linear regression analysis was used to calculate the combined effect of age and gender on platelet (plt) characteristics. MPV: mean platelet volume; ADP: adenosine biphosphate; PF4: platelet factor 4; CTAPIII: connective tissue activating peptide III; TSP-1: thrombospondin 1; PDGF: platelet-derived growth factor; VEGF: vascular endothelial growth factor.
Variables Age as independent predictor (r) P Gender as independent predictor (r) P Age and gender as predictors (r) P Platelet count -0.21 0.042 0.322 0.001 0.368 0.001 MPV fL 0.293 0.004 -0.201 0.026 0.339 0.004 % αIIbβ3 activation Thrombin 0.1 nM 0.012 0.09 -0.112 0.141 0.112 0.561 Thrombin 1 nM 0.164 0.104 -0.105 0.158 0.19 0.188 ADP 1 nM 0.431 <0.0001 0.079 0.226 0.433 <0.0001 ADP 10 nM 0.403 <0.0001 0.159 0.063 0.419 <0.0001 Convulxin 0.5 ng/mL -0.116 0.133 0.003 0.488 0.116 0.539 Convulxin 50 ng/mL 0.009 0.932 -0.029 0.392 0.029 0.962 % P-selectin expression Thrombin 0.1 nM -0.228 0.013 -0.089 0.197 0.225 0.047 Thrombin 1 nM -0.201 0.026 0.155 0.068 0.242 0.064 ADP 1 nM -0.03 0.387 -0.002 0.492 0.031 0.958 ADP 10 nM -0.036 0.366 0.182 0.04 0.182 0.215 Convulxin 0.5 ng/mL -0.289 0.002 0.047 0.325 0.29 0.018 Convulxin 50 ng/mL -0.217 0.018 0.023 0.413 0.217 0.111 Platelet content PF4 ng/106 plt -0.347 <0.0001 0.189 0.034 0.379 0.001 CTAPIII ng/106 plt -0.222 0.016 0.091 0.193 0.232 0.08 TSP-1 ng/106 plt -0.169 0.052 0.108 0.15 0.192 0.182 PDGF pg/106 plt -0.425 <0.0001 0.153 0.071 0.439 <0.0001 VEGF pg/106 plt 0.122 0.122 0.13 0.106 0.17 0.267
Table 1. Correlation of platelet characteristics with age and gender.
Haematologica | 108 June 2023 1667 LETTER TO THE EDITOR
of platelet membrane integrin α IIb b 3 conformational change and expression of P-selectin, before and after stimulation with 2Me-S-adenosine biphosphate (ADP), thrombin, or convulxin.10 Platelet-derived growth factor (PDGF), platelet factor 4 (PF4), connective tissue activating peptide III (CTAPIII), thrombospondin-1 (TSP-1), and vascular endothelial growth factor (VEGF) concentrations in platelets and platelet-free plasma (PFP) were measured with human Duo Set ELISA assays (R&D Systems, Abington, UK).
Age was found to be an independent predictor for platelet activation (Table 1). Integrin α IIb b 3 activation increased with age after stimulation with low and high concentrations of ADP (Table 1, Figure 1Ai). Hyper-reactivity at older age was also observed when platelets were stimulated
with a low dose of thrombin (Figure 1Ai), although correlation analysis showed a non-significant trend (Table 1). Our findings may explain the earlier observation of increased platelet aggregation in response to ADP with age.11 ADP is considered a weak platelet agonist with limited effect on platelet secretion, which explains the difference between integrin αIIbb3 activation (Figure 1Ai) and P-selectin expression (Figure 1Aii). These differences may be due to higher levels of platelet hydrogen peroxide in older individuals.12 Increased intraplatelet hydrogen peroxide, a critical mediator of the increased inside-out activation of integrin αIIbb3, leads to hyperactivation of platelets resulting in amplified αIIbb3 activation and fibrinogen binding, while it has no effect on α -granule secretion as measured by P-selectin expression.12 This may also explain
Figure 1. Platelet activation and growth factor content changes with age. (A) Effect of thrombin, adenosine biphosphate (ADP) and convulxin (CVX) on activation of αIIbb3 (i) and expression of P-selectin (ii) on platelets of healthy individuals. Whole blood flow cytometry was used to measure platelet activation upon stimulation with vehicle (hepes), thrombin (0.1 and 1 nM), ADP (1 and 10 nM) and convulxin (0.5 and 50 ng/ml). Individuals aged <65 years (n=46) are shown with light colored circles; individuals aged >65 years (n=48) are shown with dark colored circles. Scatter plots show the effect on binding of FITC-conjugated monoclonal antibody PAC-1 which binds to activated αIIbb3 (i) and expression of P-selectin (ii). (B) Platelet growth factor content changes with age. Concentrations of (i) platelet factor 4 (PF4), (ii) connective tissue activating peptide III (CTAPIII), (iii) thrombospondin 1 (TSP-1), (iv) platelet-derived growth factor (PDGF), and (v) vascular endothelial growth factor (VEGF) were determined by ELISA in platelets of healthy individuals. Individuals aged <65 years are shown with light colored circles; individuals aged >65 years are shown with dark colored circles. Data are presented as scatterplot with a horizontal line as median. *P<0.05; **P<0.01; ***P<0.0001.

ii Bi ii iii iv v Ai
Haematologica | 108 June 2023 1668 LETTER TO THE EDITOR
why we observed a small, but significant increase in integrin αIIbb3 activation in resting platelets of older individuals (Figure 1Ai). At the same time, P-selectin expression decreased with age after stimulation with both low and high concentrations of the strong agonists thrombin and convulxin (Table 1, Figure 1Aii). It is unclear whether this reduction is due to a decrease in P-selectin density in platelet α-granules, a decline in platelet secretory capability, or a reduction in the number of secretory granules in platelets of older individuals.
Age was also found to be an independent predictor for platelet growth factor content (Table 1). Age appeared to be strongly and negatively correlated with intraplatelet concentrations of PF4, CTAPIII and PDGF, whereas the relation with TSP-1 levels was not significant. In addition, no correlation between the VEGF content of platelets and age was found; this may be explained by the fact that the VEGF present in platelets is mainly derived from sequestration from plasma and to a lesser extent from synthesis by megakaryocytes.13 When we divided the groups into individuals aged <65 versus >65 years (this being approximately the average age of the study cohort) (Figure 1B), the concentrations of PF4, PDGF and VEGF in platelets were significantly lower in older subjects (Figure 1Bi, iv, v). Different results were obtained for CTAPIII and VEGF when analyzing the data either for the correlation between age and platelet growth factor content (Table 1) or the difference between individuals aged <65 or >65 years (Figure 1B). In these cases, we believe that the correlation results from Table 1 are the most conclusive, as these are independent of any age limit. No significant associations were found between plasma concentrations of any of these proteins and age (data not shown). The negative correlation between platelet α-granule protein content and age fits with the decline of P-selectin expression after stimulation with strong agonists in older individuals (Table 1, Figure 1Aii). This could be due to a reduced number of α-
granules in platelets in elderly subjects, a decline in protein synthesis by megakaryocytes, reduced uptake of proteins from plasma by megakaryocytes and platelets, or a combination of these effects. Little research has been done on the effects of aging on megakaryocytes and platelets, although one can hypothesize that age-related changes in bone marrow activity, such as a reduction in the amount and functional activity of the hematopoietic stem cells,14 may have direct or indirect effects on platelets. This hypothesis is supported by the present findings, and other studies,5,7,8 showing that platelet count decreases with age in both men and women (r=-0.210, P<0.05), while mean platelet volume (MPV) increases (r=0.293, P<0.01).
In isolated platelets, the concentration of PDGF (per 106 platelets) was significantly higher in women compared to men (Online Supplementary Figure S1D), while no substantial differences in platelet concentrations of PF4, CTAPIII, TSP-1, or VEGF were detected (Online Supplementary Figure S1A, B, C, E). This suggests that the concentration per platelet of some platelet-derived growth factors are gender-dependent. As platelets are the major circulating source of these factors in blood, the total circulating platelet concentrations of PF4, CTAPIII, TSP-1, PDGF and VEGF (platelet content multiplied by platelet count per milliliter) was also calculated, and was found to be signi ficantly higher in women (Figure 2); this difference remained statistically significant when adjusted for age (data not shown). This gender-related effect appeared to be primarily due to the higher platelet count in women compared to men (median 233x109/mL vs. 197x109/mL; P<0.001). This underlines the importance of matching for gender and correcting for platelet count in studies where potential biomarkers are platelet-derived. The study of Biino et al. on the Italian population suggested that reference intervals for platelet count should be age- and sex-specific to allow for better diagnosis of thrombo-

A B C D E
Figure 2. Total circulating platelet-derived growth factor concentrations are higher in women compared to men. Concentrations of (A) platelet factor 4 (PF4), (B) connective tissue activating peptide III (CTAPIII), (C) thrombospondin 1 (TSP-1), (D) plateletderived growth factor (PDGF), and (E) vascular endothelial growth factor (VEGF) were determined in platelets of healthy men (n=50) and women (n=44). The total circulating platelet concentrations of these proteins were calculated by multiplying the concentrations per platelet by the number of circulating platelets per milliliter of whole blood. Data are presented as scatterplot with a horizontal line as median. *P<0.05; **P<0.01; ***P<0.001.
Haematologica | 108 June 2023 1669 LETTER TO THE EDITOR
cytopenia and thrombocytosis.8 Furthermore, the mean platelet volume (MPV) was found to be smaller in women than in men (Table 1), confirming data from earlier studies.5,6 No significant gender-related differences in platelet reactivity were detected upon stimulation with thrombin, ADP or convulxin (Online Supplementary Figure S2). This was also the case for the concentrations of platelet-derived proteins in plasma (Online Supplementary Figure S3).
In conclusion, we confirm that platelet features differ between men and women, and change with increasing age. The hitherto underestimated association of platelet features with age and gender may mean the interpretation of data in earlier studies was unreliable and questions their conclusions. This emphasizes the importance of ageand gender-matched control groups in studies investigating platelet characteristics or platelet-derived biomarkers of disease.
Authors
1Department of Physiology, Cardiovascular Research Institute Maastricht, Maastricht University, Maastricht; 2Department of Biochemistry, Cardiovascular Research Institute Maastricht,
References
1. van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. 2019;16(3):166-179.
2. Koupenova M, Clancy L, Corkrey HA, et al. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122(2):337-351.
3. Sabrkhany S, Griffioen AW, oude Egbrink MG. The role of blood platelets in tumor angiogenesis. Biochim Biophys Acta. 2011;1815(2):189-196.
4. Haemmerle M, Stone RL, Menter DG, et al. The platelet lifeline to cancer: challenges and opportunities. Cancer Cell. 2018;33(6):965-983.
5. Segal JB, Moliterno AR. Platelet counts differ by sex, ethnicity, and age in the United States. Ann Epidemiol. 2006;16(2):123-130.
6. Panova-Noeva M, Schulz A, Hermanns MI, et al. Sex-specific differences in genetic and nongenetic determinants of mean platelet volume: results from the Gutenberg Health Study. Blood. 2016;127(2):251-259.
7. Lippi G, Meschi T, Borghi L. Mean platelet volume increases with aging in a large population study. Thromb Res. 2012;129(4):e159-160.
8. Biino G, Santimone I, Minelli C, et al. Age- and sex-related
Maastricht University, Maastricht; 3Department of Clinical Epidemiology and Medical Technology Assessment, Maastricht University Medical Center+, Maastricht; 4Angiogenesis Laboratory, Department of Medical Oncology, Cancer Center Amsterdam, Amsterdam UMC, Amsterdam, The Netherlands
Correspondence:
M.J.E. KUIJPERS - Marijke.Kuijpers@maastrichtuniversity.nl
https://doi.org/10.3324/haematol.2022.281726
Received: July 7, 2022.
Accepted: September 23, 2022.
Early view: October 6, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
SS, MK, AG and MoE were involved in study design, sample collection, and writing the manuscript. SvK performed statistical analysis and helped write the manuscript.
Data-sharing statement
The data that support the findings of this study are available upon reasonable request to the corresponding author.
variations in platelet count in Italy: a proposal of reference ranges based on 40987 subjects' data. PLoS One. 2013;8(1):e54289.
9. Sabrkhany S, Kuijpers MJE, oude Egbrink MGA, et al. Platelets as messengers of early-stage cancer. Cancer Metastasis Rev. 2021;40(2):563-573.
10. Sabrkhany S, Kuijpers MJE, van Kuijk SMJ, et al. A combination of platelet features allows detection of early-stage cancer. Eur J Cancer. 2017;80:5-13.
11. Meade TW, Vickers MV, Thompson SG, et al. Epidemiological characteristics of platelet aggregability. Br Med J. 1985;290(6466):428-432.
12. Dayal S, Wilson KM, Motto DG, et al. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation. 2013;127(12):1308-1316.
13. Klement GL, Yip TT, Cassiola F, et al. Platelets actively sequester angiogenesis regulators. Blood. 2009;113(12):2835-2842.
14. Flach J, Bakker ST, Mohrin M, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512(7513):198-202.
Siamack Sabrkhany,1 Marijke J.E. Kuijpers,2 Sander M.J. van Kuijk,3 Arjan W. Griffioen4 and Mirjam G.A. oude Egbrink1
Haematologica | 108 June 2023 1670 LETTER TO THE EDITOR
IELSG40/CLEO phase II trial of clarithromycin and lenalidomide in relapsed/refractory extranodal marginal zone lymphoma
Currently, there is not a single standard of care for patients with relapsed/refractory (R/R) marginal zone lymphoma (MZL), in whom avoiding overtreatment and superfluous toxicity is particularly important since they have an overall good prognosis.1
Lenalidomide as a single agent induced objective responses in roughly 60% of a series of 18 patients with either newly diagnosed or R/R extranodal MZL enrolled in a phase II trial at the University of Vienna.2 Clarithromycin has clinical antineoplastic activity in MZL;3,4 moreover, its addition to lenalidomide has been able to revert lenalidomide resistance in multiple myeloma patients.5 Based on these data, the International Extranodal Lymphoma Study Group (IELSG) designed the IELSG40/CLEO phase II trial (NCT03031483/EudraCT2015-003168-35) to investigate efficacy and safety of a full oral combination of clarithromycin plus lenalidomide in patients with R/R extranodal MZL. Patients received clarithromycin 500 mg b.i.d. on days 128 and lenalidomide 20 mg once daily on days 1-21 of repeated 28-day cycles. All patients but one received concomitant deep venous thrombosis prophylaxis. The dose of lenalidomide was reduced to 15 mg/day (and, if needed, to 10 mg/day in subsequent cycles) after any adverse event of grade >2 according to NCI-CTCAE version 4.03. No dose reduction was planned for clarithromycin. After the first three cycles, patients with stable disease or a better response received three further cycles. After the sixth, and again after the ninth cycle, treatment was discontinued in patients with progressive disease or complete response (CR), while patients with a partial response (PR) or stable disease received three further cycles, up to a maximum of 12.
The primary endpoint was the overall response rate, defined as the proportion of patients with a CR or PR according to the revised response criteria for malignant lymphoma.6 Patients with gastric lymphoma were evaluated endoscopically and histologically using the GELA scoring system.7 The hypothesis for sample size calculation was that clarithromycin can overcome resistance to lenalidomide leading to a clinically relevant 15% improvement of the overall response rate (from the 60% expected with lenalidomide alone).2 The trial was planned, with 5% significance and 80% power, according to the Simon minimax design,8 with the study treatment being considered of no interest if fewer than 19 of the first 30 patients achieved a PR or CR. Conversely, the treatment was con-
sidered active in the case of at least 44 responses in a total of 62 patients. Enrollment was not halted while waiting for response assessment in the first 30 patients. Secondary endpoints included safety, progression-free survival (calculated from the start of treatment to relapse/progression or death of any cause), overall survival (calculated from the start of treatment to death of any cause) and duration of response (calculated from achievement of partial or complete response to relapse/progression).6
Patients were enrolled in ten institutions, between March 2017 and October 2019. Patients older than 18 years with extranodal MZL refractory to or following ≥1 relapses after radiotherapy and/or chemotherapy and/or immunotherapy were eligible. Inclusion criteria comprised the presence of measurable disease or non-measurable lesions for which response was evaluable by non-imaging means (e.g., gastric or bone marrow infiltrations). Other eligibility criteria included Eastern Cooperative Oncology Group performance status ≤2, and adequate organ function. Exclusion criteria comprised a lymphoma histology other than extranodal MZL and clinically significant comorbidities. Ethics committees of the participating centers approved the study and all patients provided written informed consent. Forty-four patients were enrolled. One of them did not receive the study treatment after a revised diagnosis of diffuse large B-cell lymphoma. Enrollment was terminated after the first-step analysis in the first 30 patients showed an overall response rate of 44%, which did not meet the predefined threshold, while the patients already recruited continued the study treatment.
Table 1 summarizes the characteristics of the 43 eligible patients; one-third of them had a high-risk MALT-IPI score9 and 53% had stage IV disease. Forty-one patients (95%) had received one to six lines of previous systemic therapy and two had previously received only radiotherapy. The enrollment of patients with multiple relapses (47% had received ≥2 prior therapies) may explain the disease localizations at anatomic sites rarely involved at presentation.
At a median follow-up of 23.5 months (interquartile range, 9.4-37 months), the best response was a CR in six patients, PR in 13 and stable disease in 14 (Table 2). Five patients had progressive disease (3 with gastric, 1 with lung and 1 with salivary gland lymphoma). Response was not evaluable in five patients: three because of consent with-
Haematologica | 108 June 2023 1671 LETTER TO THE EDITOR
drawal and two because of early treatment discontinuation after serious adverse events (fever and pulmonary thromboembolism). In the intent-to-treat population (n=43), the overall response rate was 44% (95% confidence interval [95% CI]: 29-60%) with a CR rate of 14% (95% CI: 5-28%). The overall response rate and CR rate were 50% (95% CI: 33-67%) and 16% (95% CI: 6-31%), respectively, in the subset of 38 evaluable patients, of whom
29 were assessed by computed tomography scan, and nine (with localized gastric involvement) by repeat endoscopic biopsy only.
Twenty-one (49%) patients completed the entire protocol-planned treatment program; six of them achieved a CR, and eight had a PR (Table 2). Six of the eight patients previously exposed to the study drugs were assessable for response; two had progressive disease, two had stable disease and two had a PR. Notably, the response quality improved over time: of 11 patients with a PR at 3 months, three achieved a CR at 1 year; of 20 patients with stable disease at 3 months, one achieved a CR and three a PR at 1 year. Figure 1A depicts the individual patients’ best response in radiologically measurable lesions. The median duration of resposne was not reached (Figure 1B) and the 2-year continuous remission rate in the 19 patients achieving a CR or PR was 71% (95% CI: 44-87%). Response rates by anatomic site are summarized in Online Supplementary Table S1
In the intention-to-treat cohort, the median progressionfree survival was 40 months with a 2-year progressionfree survival rate of 53% (95% CI: 35-69%). Five deaths were reported. One patient in CR died from a second tumor (esophageal carcinoma, diagnosed 2 years after the completion of 12 cycles of treatment) and four due to lymphoma progression (with no biopsy performed to seek histological transformation). The median overall survival was not reached, with 86% (95% CI: 66-95%) of patients being alive at 2 years.
No toxic deaths were recorded. The most frequent adverse events of any grade were rash (35%), neutropenia (35%), asthenia (28%), and dysgeusia (28%). Diarrhea, vertigo, and arthralgia/myalgia were also frequently seen (21% each). Neutropenia was the commonest grade 3-4 adverse event, occurring in nine (21%) patients and lasting a median of 7 days; five patients were treated with granulocyte colony-stimulating factor. Thirteen serious adverse events were observed, with four reported as related to the study drug lenalidomide: basal cell carcinoma diagnosed 2 years after the study treatment completion, febrile neutropenia, fever, and pulmonary thromboembolism in the patient who was not given prophylaxis against deep vein thrombosis. No other severe thrombotic or hemorrhagic events were observed; only one case of superficial venous thrombosis (grade 1) and two episodes of bleeding (grade 2, gastric and retinal) were reported. The dose of lenalidomide was reduced to 15 mg/day in four patients, in one because of grade 3 hepatotoxicity and in three because of neutropenia.
*All with no evidence of H. pylori infection at study entry. **All either C. psittaci-negative at diagnosis or with successful Chlamydia eradication prior to study entry. ECOG: Eastern Cooperative Oncology Group; LDH: lactate dehydrogenase; MALT-IPI: Mucosa-associated lymphoma tissue-International Prognostic Index.
Although our study was formally negative for its primary endpoint, the observed overall response rate in the efficacy population is comparable to response rates reported by the studies leading to US Food and Drug Administration approval of targeted therapies for R/R MZL, namely ibruti-
Clinical features Age, years, median (range) 69 (43-87) Sex N (%) Male Female 24 (56) 19 (44) ECOG performance status, N (%) 0 1 39 (91) 4 (9) Anemia, N (%) 5 (12) Elevated serum LDH, N (%) 8 (19) Stage, N (%) I-II III-IV 19 (44) 24 (56) MALT-IPI prognostic index, N (%) Low risk Intermediate risk High risk 6 (14) 23 (53) 14 (33) N of prior systemic treatments, N (%) 1 2 3 6 23 (54) 10 (23) 7 (16) 1 (2) Type of previous systemic treatments, N (%) Anti-CD20 antibody in any previous line of therapy Anti-CD20 antibody in the last line before the present study Alkylating agents Lenalidomide Clarithromycin 39 (91) 28 (65) 18 (42) 5 (12) 7 (16) Prior radiotherapy only, N (%) 2 (5) Main site of relapsing/refractory disease, N (%) Stomach* Ocular adnexa** Lung Salivary glands Liver Skin Subcutaneous tissue Breast Kidneys Bone Muscle 11 (26) 8 (19) 6 (14) 4 (9) 4 (9) 3 (7) 2 (5) 2 (5) 1 (2) 1 (2) 1 (2) Table
1. Characteristics of the patients (N=43) at study entry.
Haematologica | 108 June 2023 1672 LETTER TO THE EDITOR
Figure 1. Response of patients with refractory/relapsed mantle zone lymphoma to treatment with the CLEO regimen. (A) Waterfall plot of percent change in radiologically measurable disease by primary anatomic site. The chart includes all the 28 evaluable patients with extra-gastric localizations and one patient who had gastric lymphoma with additional radiologically measurable disease (perigastric adenopathy). In the remaining nine evaluable patients, all with gastric lymphoma, response was assessed only by endoscopy and repeat biopsy, hence, they cannot be included in the graph. (B) Kaplan-Meier estimate of response duration in the 19 patients who achieved an objective response (complete or partial).

A B Response ITT patient population (total 43 patients) N (%; 95% CI) Evaluable patients (total 38 patients) N (%; 95% CI) Treatment entirely completed (total 21 patients) N (%; 95% CI) Overall response rate 19 (44; 29-60) 19 (50; 33-67) 14 (67; 43-85) Complete response 6 (14; 5-28) 6 (16; 6-31) 6 (29; 11-52) Partial response 13 (30; 17-46) 13 (34; 20-51) 8 (38; 18-62) Stable disease 14 (33; 19-49) 14 (37; 22-54) 7 (33; 15-57) Progressive disease 5 (12; 4-25) 5 (13, 4-28) 0 Not evaluable 5 (12; 4-25) na na
ITT: intent-to-treat; 95% CI: 95% confidence interval; na: not applicable. Haematologica | 108 June 2023 1673 LETTER TO THE EDITOR
Table 2. Best response.
nib,10 copanlisib,11 umbralisib12 and zanubrutinib.13 Our results also appear at least not inferior to those of a more recent phase II study evaluating the novel oral dual inhibitor of PI3K-d/γ, duvelisib.14 Moreover, the duration of response in our study (71% at 2 years, median not reached) appears very promising in comparison with the one reported with ibrutinib (median, 28 months)10 and copanlisib (median, 17 months).11
On the other hand, the efficacy of our clarithromycin-lenalidomide regimen appears similar to that observed with clarithromycin alone.3,4 Moreover, the overall response rate seems lower than the one reported in the study of lenalidomide alone, which we used for the sample size calculation.2 Since there are no reasonable grounds to assume that the combination is detrimental, the most likely explanation for these findings is the inclusion of a sizable number (15% to 60%) of untreated patients and the significantly higher proportion of stage I patients (over 70%) in the prior studies.2-4 This may also explain a higher lymphoma-related mortality (9%) in the present study than in the prior ones.2-4 However, it is worth noting that, despite a lower overall response rate, the duration of response and progression-free survival are not inferior to those observed in the trial that led to approval by the Food and Drug Administration of the lenalidomide-rituximab combination for previously treated patients with indolent lymphoma.15
Even if we cannot draw definitive conclusions, the explored lenalidomide-clarithromycin combination was feasible and moderately active, with a favorable safety profile and efficacy analogous to that reported for other approved oral drugs. Given the relatively limited therapeutic options for patients with extranodal MZL experiencing multiple relapses, the CLEO combination (clarithromycin and lenalidomide) remains a potential alternative, particularly for frail patients who, all the more in times of the pandemic of coronavirus disease 2019, may not be suitable for treatments that weaken the immune response.
Authors
Maria Cristina Pirosa,1,2* Marianna Sassone,3* Barbara Kiesewetter,4* Armando Lopez Guillermo,5 Liliana Devizzi,6 Eva Domingo
Domènech,7 Alessandra Tucci,8 Donato Mannina,9 Michele Merli,10 Antonio Salar,11 Carlo Visco,12 Fabiana Esposito,1 Luisella Bonomini,2
Emanuele Zucca,1,2,13# Andrés J. M. Ferreri3# and Markus Raderer4#
1Oncology Institute of Southern Switzerland, Ente Ospedaliero
Cantonale, Bellinzona, Switzerland; 2Institute of Oncology Research, Bellinzona, Switzerland; 3Lymphoma Unit, Department of OncoHematology, IRCCS San Raffaele Scientific Institute, Milan, Italy; 4Department of Internal Medicine I, Division of Oncology, Medical
University Vienna, Vienna, Austria; 5Department of Hematology, Hospital Clínic de Barcelona, Barcelona, Spain; 6Division of Hematology, Fondazione IRCCS Istituto Nazionale dei Tumori Milano, Milan, Italy; 7Department of Hematology, Institut Català d’Oncologia, Hospital Duran i Reynals, IDIBELL, Barcelona, Spain; 8Division of Hematology, AO Spedali Civili di Brescia, Brescia, Italy; 9Hematology Unit, AO Papardo, Messina, Italy, 10Department of Hematology Ospedale di Circolo di Varese, Varese, Italy;
11Department of Clinical Hematology Hospital del Mar, Barcelona, Spain; 12Department of Medicine, Section of Hematology, University of Verona, Verona, Italy and 13Faculty of Biomedical Sciences, Università della Svizzera Italiana, Bellinzona, Switzerland.
*MCP, MS and BK contributed equally as co-first authors. #EZ, AJMF and MR contributed equally as co-senior authors.
Correspondence:
M.C. PIROSA - maria.pirosa@eoc.ch
https://doi.org/10.3324/haematol.2022.281963
Received: August 18, 2022.
Accepted: December 7, 2022.
Early view: December 22, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
MCP, MS, LD, AT, MM, FE and LB have no conflicts of interest to disclose. BK has received honoraria from AAA, Boehringer Ingelheim, Ipsen, Novartis, MSD, and Eli Lilly and honoraria for advisory boards from Ipsen, Roche, and MSD. ALG has received honoraria from Roche, Celgene, Bristol-Myers Squibb, Janssen, Pfizer, Incyte, Takeda, Kern Pharma, and Gilead Kite. EDD has received honoraria from Takeda and Bristol-Myers Squibb. DM has received honoraria from GSK. AS has received honoraria from Roche (research funding, speakers bureau), Janssen (consultancy, speakers bureau), Gilead (research funding), and BMS/Celgene and BeiGene (consultancy). CV has received honoraria from Pfizer, AbbVie, Gilead, Janssen, Istituto Gentili, Novartis, Roche, BMS/Celgene, and Incyte. EZ has received honoraria from AstraZeneca, BeiGene, Celgene, Incyte, Janssen, Merck, Roche AbbVie, Miltenyi Biomedicine, Celltrion HealthCare, and Kite, a Gilead company. AJMF has received speaker fees from Gilead and Roche; was a member of advisory boards of Gilead, Juno, Novartis, PletixaPharm, AstraZeneca, BMS, and Roche; currently receives research grants from ADC Therapeutics, Bayer HealthCare Pharmaceuticals, Beigene, Bristol Myers Squibb, Genmab, Gilead, Hutchison Medipharma, Incyte, Janssen Research & Development, MEI Pharma, Novartis, PletixaPharm, Pharmacyclics, Protherics, Roche, and Takeda; and holds patents on NGR-hTNF in brain tumours and NGR-hTNF/R-CHOP in relapsed or refractory PCNSL and SNGRhTNF in brain tumors. MR has received honoraria from Celgene/BMS, Ipsen, Gilead, Novartis, Eisai, Eli Lilly, and Johnson & Johnson.
Haematologica | 108 June 2023 1674 LETTER TO THE EDITOR
Contributions
AJMF, MR, and EZ designed the trial and wrote the study protocol. MCP, MS, BK, AJMF, MR and EZ analyzed the data and wrote the manuscript. ALG, LD, EDD, AT, DM, MM, AS, and CV registered and treated patients, provided experimental data, and critically reviewed the manuscript draft. EZ, MCP, FE, and LB accessed and verified the trial data. LB coordinated regulatory activities and collection, assembly, and management of the data. All authors approved the definitive version of the manuscript and its submission.
Acknowledgments
We are indebted to our patients and their families for their commitment. We thank all the clinical investigators and research nurses. We appreciate the excellent assistance of the study coordinators at each study center as well as the administrative
References
1. Zucca E, Arcaini L, Buske C, et al. Marginal zone lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2020;31(1):17-29.
2. Kiesewetter B, Troch M, Dolak W, et al. A phase II study of lenalidomide in patients with extranodal marginal zone B-cell lymphoma of the mucosa associated lymphoid tissue (MALT lymphoma). Haematologica. 2013;98(3):353-356.
3. Ferreri AJM, Cecchetti C, Kiesewetter B, et al. Clarithromycin as a "repurposing drug" against MALT lymphoma. Br J Haematol. 2018;182(6):913-915.
4. Pokorny A, Kiesewetter B, Raderer M. Experience with clarithromycin as antineoplastic therapy for extranodal marginal zone B-cell lymphoma of the mucosa associated lymphoid tissue (MALT-lymphoma) outside of clinical trials: real-world data from the University of Vienna. Hematol Oncol. 2020;38(3):409-411.
5. Ghosh N, Tucker N, Zahurak M, Wozney J, Borrello I, Huff CA. Clarithromycin overcomes resistance to lenalidomide and dexamethasone in multiple myeloma. Am J Hematol. 2014;89(8):E116-120.
6. Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579-586.
7. Copie-Bergman C, Wotherspoon AC, Capella C, et al. GELA histological scoring system for post-treatment biopsies of patients with gastric MALT lymphoma is feasible and reliable in routine practice. Br J Haematol. 2013;160(1):47-52.
support with data collection and study conduction from the clinical project manager and the central study team at the IELSG Coordinating Center (Bellinzona, Switzerland). We also express gratitude to Irene Corradino for her valuable contribution to protocol writing and to Rita Gianascio Gianocca for her excellent secretarial assistance.
Funding
The IELSG is supported by the Swiss Cancer Research foundation and the Swiss Cancer League. The IELSG40/CLEO academic trial was sponsored by the IELSG and was funded in part by an unrestricted research grant from Celgene.
Data-sharing statement
The clinical trial (NCT03031483/ EudraCT 2015-003168-35) protocol will be made available upon request.
8. Shan G, Zhang H, Jiang T. Minimax and admissible adaptive two-stage designs in phase II clinical trials. BMC Med Res Methodol. 2016;16:90.
9. Thieblemont C, Cascione L, Conconi A, et al. A MALT lymphoma prognostic index. Blood. 2017;130(12):1409-1417.
10. Noy A, de Vos S, Coleman M, et al. Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: long-term follow-up and biomarker analysis. Blood Adv. 2020;4(22):5773-5784.
11. Panayiotidis P, Follows GA, Mollica L, et al. Efficacy and safety of copanlisib in patients with relapsed or refractory marginal zone lymphoma. Blood Adv. 2021;5(3):823-828.
12. Fowler NH, Samaniego F, Jurczak W, et al. Umbralisib, a dual PI3Kd/CK1ε inhibitor in patients with relapsed or refractory indolent lymphoma. J Clin Oncol. 2021;39(15):1609-1618.
13. Opat S, Tedeschi A, Linton K, et al. The MAGNOLIA trial: zanubrutinib, a next-generation Bruton tyrosine kinase inhibitor, demonstrates safety and efficacy in relapsed/refractory marginal zone lymphoma. Clin Cancer Res. 2021;27(23):6323-6332.
14. Flinn IW, Miller CB, Ardeshna KM, et al. DYNAMO: a phase II study of duvelisib (IPI-145) in patients with refractory indolent non-Hodgkin lymphoma. J Clin Oncol. 2019;37(11):912-922.
15. Leonard JP, Trneny M, Izutsu K, et al. AUGMENT: a phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol. 2019;37(14):1188-1199.
Haematologica | 108 June 2023 1675 LETTER TO THE EDITOR
Allogeneic hematopoietic cell transplant for hairy cell leukemia: EBMT experience
Hairy cell leukemia (HCL) is a rare indolent B-cell neoplasm accounting for 2% of leukemias, with an estimated incidence of <1 case per 100,000 population.1 Purine analogs such as cladribine and pentostatin are potent agents in HCL with complete remission (CR) rates approaching 75-90% with monotherapy.2,3 Despite these response rates, there is no evidence of cure and the majority of patients experience recurrence of the disease and require multiple treatments; repeated exposure to purine analogs yields lower response rates and shorter durations of remission.2 Moreover, the distinct, variant subtype of HCL (HCLv) is more resistant to standard purine analogs,4 so the median overall survival from diagnosis of patients with HCLv is 6-9 years compared to >25 years for those with classical HCL.4-6 First-line chemoimmunotherapy, adding rituximab concurrently with or after treatment with a purine analog, improved CR rates with longer remission periods in both HCL and HCLv.5,7,8 However, treatment for ‘high-risk’ patients and those with disease resistant to purine analogs remains challenging. Allogeneic hematopoietic cell transplant (allo-HCT) can potentially produce long-term remission in many diseases, including chronic leukemias; however, evidence to support the use of allo-HCT in refractory/multiple relapsed HCL has been limited to case reports only.9 Here we report an analysis of adult patients undergoing a first allo-HCT for HCL between 1996-2018, using either myeloablative or reduced-intensity conditioning defined by standard criteria. The Kaplan-Meier estimator was used to estimate overall survival and progression-free survival and the log-rank test was used to compare groups. The crude cumulative incidence estimator was used to estimate non-relapse mortality and relapse incidence within a competing risk setting. The crude cumulative incidence estimator was also used to estimate the cumulative incidence of acute graft- versus-host disease (GvHD) with death before acute GvHD as a competing event, as well as the cumulative incidence of chronic GvHD with death before chronic GvHD as a competing event. Events were artificially censored at 5 years except acute GvHD and death before acute GvHD, which were censored at 100 days after allo-HCT.
A total of 24 patients from 19 transplant centers were included in this study (Table 1). The median age at allo-HCT was 50 years (range, 32-68). The median number of lines of treatment and purine analogs prior to allo-HCT was six (range, 1-10) and two (range, 0-4), respectively. The median time from diagnosis to allo-HCT was 55.3 months (range,
1.6 months - 26.9 years). Details of prior lines of therapy were available for 11 patients (Online Supplementary Table S1). Six patients received rituximab, three in two lines of treatment. Three patients received BRAF inhibitors prior to allo-HCT. Disease status at allo-HCT (available for 20 patients) was reported as CR (n=7, 33%), partial remission (n=3, 14%), stable disease (n=5, 24%) and progressive/refractory disease (n=5, 24%). Among the 11 patients with details on prior treatment, 30% were refractory to the regimen preceding allo-HCT. Donor type was as follows: matched sibling (37%), syngeneic (4%), matched related (4%), mismatched related (4%), matched unrelated (29%), mismatched unrelated (17%) and unrelated, match unknown (4%). Myeloablative and reduced-intensity conditioning regimens were utilized in 59% and 41% of the patients, respectively. Eight patients (35%) received totalbody irradiation as part of conditioning. Regarding stem cell source, peripheral blood was used in 22 patients (92%), bone marrow in one (4%) and cord blood in one (4%).
No follow-up data were available for two patients and hence further results are reported on 22 patients only. The cumulative incidence of grade 2-4 acute GvHD at day 100 was 15% (95% confidence interval [95% CI]: 0-31%). The cumulative incidence of chronic GvHD at 2 years was 47% (95% CI: 25-70%). Non-relapse mortality at day 100 was 14% (95% CI: 0-28%). The best overall response rate was 64% (95% CI: 41-83%) and the CR rate was 59% (95% CI: 36-79).
With a median follow-up after allo-HCT of 54.5 months (interquartile range: 15.4 - not reached), 5-year estimated non-relapse mortality, progression-free survival and overall survival rates were 26% (95% CI: 6-46%), 33% (95% CI: 9-56%) and 46% (95% CI: 22-70%), respectively (Figure 1AC). The 5-year cumulative relapse incidence was 41% (95% CI: 17-66%). Causes of death were infection (in 36% of all deceased patients), multi-organ failure (27%), disease progression (18%), secondary malignancy (9%) and unknown (9%). The two patients with HCLv were alive and relapse free at the end of the reported follow-up (+12 and +20 months). The 2-year progression-free survival of patients transplanted in CR was 80% (95% CI: 45-100%) compared to 44% (95% CI: 16-72%) for those transplanted while not in CR (log-rank P=0.15). The 2-year progressionfree survival of patients undergoing allo-HCT during 20102018 was 67% (95% CI: 40-94%), as compared to 53% (95% CI: 19-87%) for those transplanted between 19962009 (log-rank P=0.43).
Haematologica | 108 June 2023 1676 LETTER TO THE EDITOR
Allo-HCT: allogeneic hematopoietic cell transplant; IQR: interquartile range; HCL: hairy cell leukemia; PA: purine analogs; TBI: total body irradiation.
This study represents the largest one to date describing outcomes following allo-HCT for patients with HCL. Overall, allo-HCT produced long-term survival in a significant percentage of heavily pre-treated patients for whom many likely had limited options at that time. However, it is clear that a significant number of patients continued to experience recurrence and there was no plateau in progressionfree survival in this cohort.
The treatment landscape for HCL has been revolutionized in the last decade with the availability of newer agents such as BRAF inhibitors and the anti-CD22 antibody-drug conjugate, moxetumomab pasudotox. Tiacci et al. reported response rates of 96-100% with vemurafenib monotherapy in relapsed/refractory HCL,10 although the only modest CR rates (35-42%) and persistent minimal residual disease (MRD) even in CR, are likely reflected by the short progression-free survival of 19 months. More recently, Tiacci et al. demonstrated the beneficial impact of adding rituximab to vemurafenib.11 A CR rate of 87% was reported, and 65% of patients achieved a negative MRD status with 78% of patients (85% among responders) remaining relapse-free at around 3 years. Another BRAF inhibitor, dabrafenib, in combination with a MEK inhibitor also produced a high CR rate of 65.5% and approximately half of these patients achieved MRD negativity.12 Moxetumomab produced durable responses in 36% of patients, and 61% of patients who achieved CR remained in CR at 5 years.13-15 Overall, 82% of patients who achieved CR were MRD negative, and the median duration of CR for those patients was >5 years. Supported by improvements in available therapeutics, the survival of patients with HCL diagnosed in the modern era has certainly improved.16 Although many patients treated with the new agents remain positive for MRD, which is concerning for future disease progression, these treatments will undoubtedly reduce the number of patients who need to be considered for allo-HCT. However, challenges may remain, particularly in high-risk patients such as those with HCLv, who can display high rates of chemotherapy resistance (particularly with TP53 mutation),5 and those who are negative for the BRAF-V600E mutation, who have fewer treatment options. In selected high-risk patients the indication for allo-HCT should be considered with the utmost caution, balancing risks and potential benefits.
Several limitations of our study should be noted. These are related to the study’s retrospective nature and cohort size which limit analysis exploring transplant-specific risk factors and improved allo-HCT strategies over time. Moreover, in this study we lacked an independent central review to confirm the diagnosis. The median time from diagnosis to allo-SCT was 55.3 months which is short considering the median progression-free survival of over 10 years following first-line purine analog monotherapy in HCL.2 This may be due to selection of very aggressive
Characteristics Total patients, N 24 Age at allo-HCT in years, median (IQR) 50.4 (46.6-55.6) Patients’ sex, Male, N (%) Female, N (%) 17 (70.8) 7 (29.2) Type of HCL Classical, N (%) Variant, N (%) 22 (91.7) 2 (8.3) Karnofsky status at allo-HCT ≥ 90, N (%) ≤ 80, N (%) Missing, N 15 (75.0) 5 (25.0) 4 N of lines of any treatment Median (IQR) Missing, N 6 (4.5-6) 13 N of lines of treatment including PA Median (IQR) Missing, N 2 (1-2.5) 13 Any rituximab prior to allo-HCT No, N (%) Yes, N (%) Missing, N 5 (45.5) 6 (54.5) 13 Any BRAF inhibitor prior to allo-HCT No, N (%) Yes, N (%) Missing, N 8 (72.7) 3 (23.7) 13 Disease status at allo-HCT Complete remission, N (%) Partial remission, N (%) Stable disease, N (%) Primary refractory, N (%) Relapse/progression, N (%) Untreated, N (%) Missing, N 7 (33.3) 3 (14.3) 5 (23.8) 1 (4.8) 4 (19.0) 1 (9.1) 3 Sensitivity at allo-HCT* Sensitive, N (%) Resistant, N (%) Missing, N 7 (70.0) 3 (30.0) 14 Months from diagnosis to allo-HCT, median (IQR) 55.3 (8.1-116.9) Year of allo-HCT 1996-1999, N (%) 2000-2009, N (%) 2010-2018, N (%) 3 (12.5) 6 (25.0) 15 (62.5) Conditioning regimen intensity Standard, N (%) Reduced, N (%) Missing, N 13 (59.1) 9 (40.9) 2 TBI at allo-HCT No, N (%) Yes, N (%) Missing, N 15 (65.2) 8 (34.8) 1
Table 1. Patient, disease and transplantation characteristics.
of disease to last regimen given before first allo-HCT. Haematologica | 108 June 2023 1677 LETTER TO THE EDITOR
*Sensitivity
cases such as IGHV4-34 unmutated HCL;6 however, it also raises the possibility of misclassification between HCL and HCLv. Nevertheless, patients in this study received a median of six lines of treatment, which suggests that they had very aggressive HCL. Allo-HCT provided meaningful progression-free survival considering the number of treatments patients had required in a short period before alloHCT, although it should be noted that the treatments patients received in this study do not reflect current standard treatment.
In conclusion, this study represents the largest reported evaluation of outcomes of allo-HCT for patients with HCL. In this heterogeneous cohort, allo-HCT produced meaningful response rates and durable remissions in some cases although, interestingly, there was no sign of a plateau in the progression-free survival, i.e. cure of HCL from allo-HCT. With the development of chimeric antigen receptor T-cell therapy and bispecific antibodies, these agents warrant evaluation for patients progressing following current standard treatments, including BRAF inhibitors and moxetumomab. The outcomes of allo-HCT in patients with HCL will provide references for future trials of novel agents.

Authors
Dai Chihara,1 Luuk Gras,2 Nienke Zinger,3 Nicolaus Kröger,4 Jiri Mayer,5 Jakob Passweg,6 Régis Peffault de Latour,7 Jenny Byrne,8 William Krüger,9 Jan-Paul Bohn,10 Uwe Platzbecker,11 Igor Wolfgang Blau,12 Francesca Bonifazi,13 Grzegorz Helbig,14 Andrew McDonald,15 Martin Mistrik,16 Mohamad Mohty,17 Ron Ram,18 Jaime Sanz,19 Carlos Vallejo
1Department of Lymphoma and Myeloma, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2EBMT Statistical Unit, Leiden, the Netherlands; 3EBMT Study Unit, Leiden, the Netherlands; 4University Hospital Eppendorf, Hamburg, Germany; 5University Hospital Brno, Brno, Czech Republic; 6University Hospital Basel, Basel, Switzerland; 7Hopital St. Louis 207, Paris, France; 8Nottingham University, Nottingham, UK; 9Klinik fuer Innere Medizin C, Greifswald, Germany; 10University Hospital Innsbruck, Innsbruck, Austria; 11Medical Clinic and Polycinic 1, Leipzig, Germany; 12Medizinische Klinik m. S. Hämatologie, Onkologie und Tumorimmunologie, Berlin, Germany; 13IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy; 14Silesian Medical University, Katowice, Poland; 15Alberts Cellular Therapy, Netcare Pretoria East Hospital, Pretoria, South Africa; 16University Hospital, Bratislava, Slovak Republic; 17Sorbonne University, Saint-Antoine Hospital, AP-HP, and INSERM UMRs 938, Paris, France; 18Tel Aviv Sourasky Medical Center and Soursky Faculy of Medicine, Tel Aviv, Israel; 19University Hospital La Fe, Valencia, Spain, 20Hospital Universitario Donostia, S Sebastian, Spain; 21Laboratory of Molecular Biology, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA; 22Department of Haematology, Trinity College Dublin, St. James’s Hospital, Dublin, Ireland, 23University College London Hospitals NHS Trust, London, UK; 24Department of Hematology and Cell Therapy, CHU Estaing, Université Clermont Auvergne, Clermont-Ferrand, France; 25University Hospital Maastricht, Maastricht, the Netherlands and 26CHU de Lille, Univ Lille, INSERM U1286, Lille, France
Correspondence:
D. CHIHARA - DChihara@mdanderson.org
O. TOURNILHAC - otournilhac@chu-clermontferrand.fr
Haematologica | 108 June 2023 1678
Figure 1. Outcomes after allogeneic hematopoietic cell transplantation in patients with hairy cell leukemia. (A) Cumulative incidence of non-relapse mortality. (B) Probability of progression-free survival. (C) Probability of overall survival. Numbers below the graph indicate the number of patients at risk. The shaded areas show the 95% confidence intervals.
Llamas,20 Robert J. Kreitman,21 Patrick J. Hayden,22 Donal McLornan,23 Olivier Tournilhac,24 Michel van Gelder25 and Ibrahim Yakoub-Agha26
LETTER TO THE EDITOR
https://doi.org/10.3324/haematol.2022.281754
Received: September 4, 2022.
Accepted: December 12, 2022.
Early view: December 22, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
DC, OT, MvG, RJK and IYA designed the study; NZ, JM, JP, RPL JB,
References
1. Teras LR, DeSantis CE, Cerhan JR, et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J Clin. 2016;66(6):443-459.
2. Else M, Dearden CE, Matutes E, et al. Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol. 2009;145(6):733-740.
3. Saven A, Burian C, Koziol JA, Piro LD. Long-term follow-up of patients with hairy cell leukemia after cladribine treatment. Blood. 1998;92(6):1918-1926.
4. Matutes E, Wotherspoon A, Brito-Babapulle V, Catovsky D. The natural history and clinico-pathological features of the variant form of hairy cell leukemia. Leukemia. 2001;15(1):184-186.
5. Chihara D, Arons E, Stetler-Stevenson M, et al. Long term follow-up of a phase II study of cladribine with concurrent rituximab with hairy cell leukemia variant. Blood Adv. 2021;5(23):4807-4816
6. Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood. 2009;114(21):4687-4695.
7. Chihara D, Arons E, Stetler-Stevenson M, et al. Randomized phase II study of first-line cladribine with concurrent or delayed rituximab in patients with hairy cell leukemia. J Clin Oncol. 2020;38(14):1527-1538.
8. Chihara D, Kantarjian H, O'Brien S, et al. Long-term durable remission by cladribine followed by rituximab in patients with hairy cell leukaemia: update of a phase II trial. Br J Haematol. 2016;174(5):760-766.
WK, DW, UP, IWB, FB, GH, AM, MM, MM, RR, JS, CVL, PJH and DM contributed data and reviewed the manuscript; LG analyzed the data; DC wrote the draft, and all the authors wrote and approved the manuscript.
Acknowledgments
The study was performed on behalf of the Chronic Malignancies
Working Party of the EBMT. The authors would like to thank the contribution of the EBMT and CMWP data managers to this work.
Data-sharing statement
The data are not available for sharing.
9. Zinzani PL, Bonifazi F, Pellegrini C, et al. Hairy cell leukemia: allogeneic transplantation could be an optimal option in selected patients. Clin Lymphoma Myeloma Leuk. 2012;12(4):287-289.
10. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med. 2015;373(18):1733-1747.
11. Tiacci E, De Carolis L, Simonetti E, et al. Vemurafenib plus rituximab in refractory or relapsed hairy-cell leukemia. N Engl J Med. 2021;384(19):1810-1823.
12. Kreitman RJ, Moreau P, Ravandi F, et al. Dabrafenib plus trametinib in patients with relapsed/refractory BRAF V600E mutation-positive hairy cell leukemia. Blood. 2023;14(9):996-1006.
13. Kreitman RJ, Dearden C, Zinzani PL, et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia. 2018;32(8):1768-1777.
14. Kreitman RJ, Dearden C, Zinzani PL, et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term followup from the pivotal trial. J Hematol Oncol. 2021;14(1):35.
15. Kreitman RJ, Tallman MS, Robak T, et al. Minimal residual hairy cell leukemia eradication with moxetumomab pasudotox: phase 1 results and long-term follow-up. Blood. 2018;131(21):2331-2334.
16. Bohn JP, Neururer S, Pirklbauer M, Pircher A, Wolf D. Hairy cell leukemia patients have a normal life expectancy - a 35-year single-center experience and comparison with the general population. Cancers (Basel). 2022;14(5):1242.
Haematologica | 108 June 2023 1679 LETTER TO THE EDITOR
Light chain amyloidosis associated with Waldenström macroglobulinemia: treatment and survival outcomes
Light chain (AL) amyloidosis is an uncommon clinical manifestation of Waldenström macroglobulinemia (WM), an IgM-secreting lymphoplasmacytic lymphoma characterized by recurrent mutations in MYD88 and CXCR4 WM-associated AL (WM-AL) amyloidosis is distinct from typical AL amyloidosis not only because of its underlying lymphoplasmacytic neoplastic clone, but also because of the absence of t(11;14) and higher rates of soft tissue, lymph node, lung, and peripheral nerve involvement.1,2 The occurrence of WM-AL amyloidosis confers a worse prognosis in WM patients,3 and the management approach is not standardized. Commonly used treatment regimens are derived from those used in WM without concurrent AL amyloidosis or in typical AL amyloidosis with a pure plasma cell neoplastic clone.4-6 Here we describe the treatment and survival outcomes of a cohort of patients with WM-AL amyloidosis.
We identified consecutive patients with WM-AL amyloidosis evaluated at the Boston University Amyloidosis Center between 2006 and 2022. All patients met consensus clinicopathological criteria for a diagnosis of WM (i.e., presence of a serum IgM paraprotein and bone marrow infiltration by lymphoplasmacytic lymphoma of any extent)7 and had positive Congo red staining of a biopsy specimen with typing confirming AL amyloidosis. The amyloidogenic protein was typed by immunohistochemistry, immunogold electron microscopy, or liquid chromatography and tandem mass spectrometry. Hematologic and organ responses to treatment for AL amyloidosis and WM were assessed using consensus definitions.8,9 Event-free survival was defined as the time between the diagnosis of WM-AL amyloidosis among treated patients and next line of treatment or death, whichever occurred first. Overall survival was defined as the time between the diagnosis of WM-AL amyloidosis and death from any cause or last follow-up. Logistic regression models were fitted to identify predictors of hematologic response. Time-to-event outcomes were calculated using the Kaplan-Meier method, and the log-rank test was used to compare estimates between groups. The Cox-proportional hazard regression method was used to fit models for event-free and overall survival. P values <0.05 were considered statistically significant. The study cohort consisted of 49 patients with WM-AL amyloidosis. Ten patients (20%) were diagnosed simultaneously with WM and AL amyloidosis. In the remaining 39 patients (80%) AL amyloidosis was diagnosed after WM, with a median time to diagnosis of 3 months (range, 0201); 12 patients (24%) were diagnosed with AL amyloido-
sis more than 5 years after the diagnosis of WM. Eight patients (16%) received a median of two WM-directed therapies (range, 1-4) before the diagnosis of AL amyloidosis. The patients’ baseline clinical characteristics at the time of WM-AL amyloidosis diagnosis are shown in Table 1. The presenting symptoms were heterogeneous and included peripheral edema (n=14; 29%), dyspnea (n=8; 17%), paresthesia (n=7; 14%), syncope (n=5; 10%), pleural effusion (n=5; 10%), diarrhea (n=4; 8%), foamy urine (n=4; 8%), carpal tunnel syndrome (n=3; 6%), atrial fibrillation (n=3; 6%), acute kidney injury (n=3; 6%), periorbital ecchymosis (n=2; 4%), macroglossia (n=2; 4%), lymphadenopathy (n=2; 4%), and subcutaneous mass (n=2; 4%).
Forty-four patients (90%) received at least one treatment after the diagnosis of WM-AL amyloidosis; five patients did not receive treatment due to poor performance status and/or patients’ preference (Table 2). Hematologic response assessments using serum free light chain (FLC) and IgM levels were available for 43 of 44 patients. Based on FLC criteria, the overall, complete, very good partial, and partial response rates were 77%, 26%, 26%, and 26%, respectively. Based on IgM criteria, the overall, complete, very good partial, partial and minor response rates were 86%, 26%, 26%, 27%, and 7%, respectively. There was discordance between the categories of FLC and IgM responses (partial response or better) in six of 43 patients (14%); three patients had deeper category responses by IgM criteria, while another three patients had deeper category responses by FLC criteria. No baseline clinical factors were associated with achieving a hematologic complete or very good partial response by either FLC or IgM criteria (P>0.05 for all comparisons). Cardiac, renal, and hepatic organ response rates were 67% (n=6/9), 52% (n=12/23), and 67% (n=2/3), respectively. Patients with a hematologic complete or very good partial response had significantly higher organ response rates by both FLC criteria (78% vs. 17%; P=0.002) and IgM criteria (83% vs 9%; P<0.001).
After a median follow-up of 2.6 years (95% confidence interval [95% CI]: 1.6-5.2), 21 patients (43%) had died. The median event-free survival was 4.9 years (95% CI: 2.3-not reached [NR]), and the estimated 5-year event-free survival rate was 48% (Figure 1A). The median overall survival was 7.3 years (95% CI: 5.4-NR), and the estimated 5-year OS rate was 70% (Figure 1B). A baseline serum creatinine >2.0 mg/dL was independently associated with both a shorter event-free survival (0.7 vs. 6.1 years; hazard ratio [HR]=4.20, 95% CI: 1.51-11.7; P=0.003) and a shorter overall
Haematologica | 108 June 2023 1680 LETTER TO THE EDITOR
Patients’ characteristics
All patients (N=49)
survival (2.5 vs. 10 years; HR=3.91, 95% CI: 1.29-11.8; P=0.02) (Online Supplementary Table S1, Online Supplementary Figure S1). There was also a trend to shorter overall survival in patients with B-natriuretic peptide levels >81 pg/mL (5.2 vs. 10 years; HR=2.31, 95% CI: 0.93-5.77; P=0.07) (Online Supplementary Table S1, Online Supplementary Figure S1). Using the Boston University cardiac staging system, patients with stage I, II, and III disease had estimated 5-year overall survival rates of 81%, 61%, and 25%, respectively (P=0.10) (Online Supplementary Figure S1). The depth of hematologic FLC and IgM responses was significantly associated with both event-free and overall survival (Figure 1C-F). The median overall survival from the time of WM diagnosis was 12.8 years (95% CI: 10.8-NR).
The response and survival outcomes for each frontline treatment regimen are summarized in Table 2. Maintenance rituximab was administered to seven of 33 patients (21%) who achieved a partial response or better to a rituximabcontaining frontline regimen. Among these patients, maintenance rituximab was associated with a significantly higher 5-year event-free survival rate (100% vs. 41%; P=0.02) and a trend to a higher overall survival rate (100% vs. 67%; P=0.05) (Online Supplementary Figure S1).
dFLC: difference between involved and uninvolved serum free light chain; BM: bone marrow; LPL: lymphoplasmacytic lymphoma; NTpro-BNP: N-terminal pro-brain natriuretic peptide; BU: Boston University; IPSSWM: International Prognostic Scoring System for Waldenström macroglobulinemia.
Eleven of 44 treated patients (25%) received salvage therapy, which most commonly was a bortezomib- and/or bendamustine-based regimen (Online Supplementary Table S2). Two patients received ibrutinib monotherapy without achieving either a hematologic or organ response. One patient was treated with venetoclax-obinutuzumab after being refractory to bortezomib, dexamethasone, and rituximab and bendamustine and rituximab, and achieved a hematologic partial response with stable proteinuria. The occurrence of WM-AL amyloidosis is an uncommon complication that alters the natural history of WM. We observed a median overall survival of 7.3 years from the diagnosis of WM-AL amyloidosis. This survival estimate compares favorably to the median overall survival of 2.5 years published by the Mayo Clinic,3 perhaps due to a lower frequency of cardiac involvement in our cohort of patients (35% vs. 57%). Both studies identified cardiac involvement as an adverse prognostic factor for overall survival, suggesting the potential relevance of the Boston University and Mayo cardiac staging systems in patients with WM-AL amyloidosis. We also identified renal dysfunction as an important prognostic factor for both eventfree and overall survival. In contrast to the study by the Mayo Clinic group,3 we included patients with <10% bone marrow involvement by lymphoplasmacytic lymphoma according to the consensus diagnostic criteria for WM.7 This difference in study design is unlikely to explain the observed survival discrepancy, as bone marrow involvement by lymphoplasmacytic lymphoma (<10% vs ≥10%) was not prognostic for survival. Importantly, we show that
Age, years Median (range) >65 years, N (%) 68 (56-86) 30/49 (61) Sex, N (%) Male Female 27/49 (55) 21/49 (45) Light chain isotype, N (%) Kappa Lambda 19/49 (39) 30/49 (61) Hemoglobin level, g/dL Median (range) ≤11.5 g/dL, N (%) 12.4 (9.2-18.1) 13/48 (27) Platelet count, x109/L Median (range) ≤100 x109/L, N (%) 263 (126-652) 0/48 (0) b2-microglobulin, mg/L Median (range) >3 mg/L, N (%) 3.2 (1.6-22.2) 26/48 (54) Serum IgM level, mg/dL Median (range) >4,000 mg/dL, N (%) 1418 (284-5,498) 6/49 (12) dFLC, mg/L Median (range) >180 mg/L, N (%) 73.7 (5.1-1,333.5) 10/49 (20) BM involvement by LPL, % Median (range) >10%, N (%) 20 (10-60) 41/48 (85) Tumor genotype, N (%) MYD88 mutation CXCR4 mutation t(11;14) 17/21 (81) 3/9 (33) 0/27 (0) Serum creatinine, mg/dL Median (range) >2.0 mg/dL, N (%) 0.9 (0.5-4.9) 7/48 (15) Urine protein excretion, mg/24 h Median (range) >5,000 mg/24 h, N (%) 655 (0-14,064) 13/48 (27) Alkaline phosphatase, IU/L Median (range) >150 IU/L, N (%) 91 (36-924) 8/47 (17) Brain natriuretic peptide, pg/mL Median (range) >81 pg/mL, N (%) 77 (3-2,163) 23/48 (48) NT-pro-BNP, pg/mL Median (range) >332 pg/mL, N (%) 554 (62-5,732) 13/22 (59) Troponin I, ng/mL Median (range) >0.1 ng/mL, N (%) 0.012 (0.006-0.599) 4/48 (8) BU cardiac stage, N (%) I II III 25/48 (52) 19/48 (40) 4/48 (8) IPSSWM stage, N (%) Low Intermediate High 12/48 (25) 31/48 (65) 5/48 (10) Organ involvement, N (%) Renal Cardiac Peripheral nervous system Autonomic nervous system Gastrointestinal Lymph node Pulmonary Skin/soft tissue Hepatic 25/49 (51) 17/49 (35) 16/49 (33) 10/49 (20) 8/49 (16) 8/49 (16) 7/49 (14) 7/49 (14) 3/49 (6)
Table 1. Baseline clinical characteristics at the time of diagnosis of AL amyloidosis in patients with Waldenström macroglobulinemia.
Haematologica | 108 June 2023 1681 LETTER TO THE EDITOR
the established response criteria for both AL amyloidosis and WM are prognostic for survival and predictive of organ response in patients with WM-AL amyloidosis.8,9

We also describe the timing of diagnosis of AL amyloidosis in WM patients. In most cases, AL amyloidosis was diagnosed within a few months of WM; however, 24% of patients were diagnosed more than 5 years later. This could be because of delayed recognition of the clinical syndrome of amyloidosis or because AL amyloidosis may be a late complication in some cases of WM. Neverthe-
less, our finding highlights the importance of monitoring for red flag symptoms of AL amyloidosis in WM patients throughout their entire disease course. In particular, cardiac AL amyloidosis should be considered in WM patients on BTK inhibitors who develop atrial fibrillation, a wellrecognized side effect. In one series, approximately 8% of WM patients who developed atrial fibrillation on ibrutinib had underlying cardiac AL amyloidosis.10 AL amyloidosis should also be considered in the differential diagnosis of IgM monoclonal gammopathy of unknown significance
Figure
(A, B) Kaplan-Meier curves for event-free survival (A) and overall survival (B) for the entire cohort. (C, D) Kaplan-Meier curves for event-free survival (C) and overall survival (D) stratified by depth of free light chain response. (E, F) Kaplan-Meier curves for event-free survival (E) and overall survival (F) stratified by depth of IgM response. WM-AL: Waldenström
AL
NR: no response; PR: partial response; VGPR: very good partial response; CR: complete response; MR: minor response.
A C E B D F Haematologica | 108 June 2023 1682 LETTER TO THE EDITOR
1. Survival in patients with AL amyloidosis associated with Waldenström macroglobulinemia.
macroglobulinemia associated with
amyloidosis;
Table
the frontline regimen used for patients with AL amyloidosis associated with Waldenström macroglobulinemia.
*All patients treated with high-dose melphalan and stem cell transplantation (HDM/SCT) received pre-transplant induction therapy (BDR: N=8; Benda-R: N=1). Patients treated with HDM/SCT had an estimated event-free survival of 88% at both 5 and 10 years, and there was no 100-day treatment-related mortality. #Total number of patients with involvement of the respective organ. FLC: free light chain; ORR: overall response rate; PR: partial response; VGPR: very good partial response; MR: minor response; EFS: event-free survival; OS: overall survival; NR: not reached; Benda-R: bendamustine and rituximab; BDR: bortezomib, dexamethasone, and rituximab; CPR: cyclophosphamide, prednisone, and rituximab; CyBorD±R: cyclophosphamide, bortezomib, dexamethasone, and rituximab; Flu-R: fludarabine and rituximab.
in the appropriate clinical scenario, particularly given the lower serum IgM levels we observed in patients with WMAL amyloidosis.
Prospective data to define the optimal treatment regimen for WM-AL amyloidosis are lacking. Our findings demonstrate that standard WM regimens (such as bortezomib, dexamethasone, and rituximab or bendamustine and rituximab) can also be effective in WM-AL amyloidosis. Previous studies in which bendamustine and rituximab were given to patients with IgM-AL amyloidosis did not delineate outcomes based on the underlying neoplastic clone.4,5 We report deep and durable responses with frontline use of high-dose melphalan and stem cell transplantation (HDM/SCT), which is typically reserved for the salvage setting in WM. HDM/SCT should be considered in selected patients with WM-AL amyloidosis, particularly since HDM/SCT can induce prolonged survival (>20 years) in typical AL amyloidosis.11 We also observed improved event-free survival in patients given maintenance rituximab. Based on the MAINTAIN trial,6 maintenance rituximab is not routinely used in WM but our data suggest it may have a role in WM-AL amyloidosis for patients who respond to induction therapy. Venetoclax represents a novel treatment option for WM,12 and we present the fi rst published case of its use in a patient with WM-AL amyloidosis. Unlike in WM, ibrutinib is associated with mixed effi cacy and tolerability in WM-AL amyloidosis and must be used with caution, particularly in patients with cardiac involvement, given its pro-arrhythmic properties.13,14 Second-generation BTK inhibitors, such as zanubrutinib, which have less cardiotoxicity than
ibrutinib, warrant further investigation in WM-AL amyloidosis. Finally, there are currently no data on daratumumab in patients with WM-AL amyloidosis, but a phase II trial in WM was stopped due to futility.15
The limitations of the current study include the inherent selection bias associated with a non-randomized, observational study from a tertiary referral center. However, this study is the largest to date describing treatment outcomes in patients with WM-AL amyloidosis. Prospective studies are needed to optimize the management of patients with this condition.
Authors
Amyloidosis Center and Section of Hematology and Medical Oncology, Boston University School of Medicine and Boston Medical Center, Boston, MA, USA
Correspondence:
V. SANCHORAWALA - vaishali.sanchorawala@bmc.org
https://doi.org/10.3324/haematol.2022.282264
Received: October 13, 2022.
Accepted: December 12, 2022.
Prepublished: December 22, 2022.
Joshua N. Gustine, Raphael E. Szalat, Andrew Staron, Tracy Joshi, Lisa Mendelson, J. Mark Sloan and Vaishali Sanchorawala
Treatment regimen N FLC response N (%) IgM response N (%) Organ response N (%) Survival years, median (5-year survival, %) ORR (≥PR) ≥VGPR ORR (≥PR) ≥VGPR Cardiac Renal EFS OS Benda-R 15 12/15 (80) 8/15 (53) 15/15 (100) 10/15 (67) 3/4# (75) 5/6# (83) 5.4 (65) 7.3 (86) BDR 9 6/9 (67) 6/9 (67) 7/9 (78) 3/9 (33) 1/2# (50) 2/6# (33) 4.4 (48) 7.3 (57) HDM/SCT* 9 9/9 (100) 8/9 (89) 9/9 (100) 8/9 (89) 2/2# (100) 5/6# (83) NR (88) NR (86) CPR 5 3/5 1/5 4/5 1/5 0/2# 1.7 12.0 CyBorD±R 2 2/2 0/2 2/2 0/2 0/2# 0.7 2.5 Melphalan 2 1/2 0/2 0/2 0/2 0/1# 2.8 7.7 Rituximab 1 0/1 0/1 0/1 0/1 1.3 1.6 Flu-R 1 0/1 0/1 0/1 0/1 0/1# 0.9 1.2
2. Clinical outcomes based on
Haematologica | 108 June 2023 1683 LETTER TO THE EDITOR
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
All authors contributed to the study design, data analysis, manuscript preparation, and patient care. All authors critically reviewed and approved the manuscript.
Acknowledgments
The authors thank the current and past members of the Amyloidosis
References
1. Sidana S, Larson DP, Greipp PT, et al. IgM AL amyloidosis: delineating disease biology and outcomes with clinical, genomic and bone marrow morphological features. Leukemia. 2020;34(5):1373-1382.
2. Sissoko M, Sanchorawala V, Seldin D, et al. Clinical presentation and treatment responses in IgM-related AL amyloidosis. Amyloid. 2015;22(4):229-235.
3. Zanwar S, Abeykoon JP, Ansell SM, et al. Primary systemic amyloidosis in patients with Waldenström macroglobulinemia. Leukemia. 2019;33(3):790-794.
4. Manwani R, Sachchithanantham S, Mahmood S, et al. Treatment of IgM-associated immunoglobulin light-chain amyloidosis with rituximab-bendamustine. Blood. 2018;132(7):761-764.
5. Milani P, Schönland S, Merlini G, et al. Treatment of AL amyloidosis with bendamustine: a study of 122 patients. Blood. 2018;132(18):1988-1991.
6. Rummel MJ, Lerchenmüller C, Hensel M, et al. Two years rituximab maintenance vs. observation after first line treatment with bendamustine plus rituximab (B-R) in patients with Waldenström's macroglobulinemia (MW): results of a prospective, randomized, multicenter phase 3 study (the StiL NHL7-2008 MAINTAIN trial). Blood. 2019;134(Suppl_1):343.
7. Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: Consensus Panel Recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003;30(2):110-115.
8. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain
Center, Cancer Clinical Trials Office, Stem Cell Transplant Program, and Center for Cancer and Blood Disorders at the Boston University School of Medicine and Boston Medical Center. JNG received a Young Investigator Award at the 11th International Workshop on Waldenström’s Macroglobulinemia in Madrid, Spain (October 2022), for his part in this research.
Funding
This research was supported by the Amyloid Research Fund.
Data-sharing statement
The dataset analyzed during the current study is available from the corresponding author on reasonable request.
amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541-4549.
9. Owen RG, Kyle RA, Stone MJ, et al. Response assessment in Waldenström macroglobulinaemia: update from the VIth International Workshop. Br J Haematol. 2013;160(2):171-176.
10. Gustine JN, Meid K, Dubeau TE, Treon SP, Castillo JJ. Atrial fibrillation associated with ibrutinib in Waldenström macroglobulinemia. Am J Hematol. 2016;91(6):E312-313.
11. Gustine JN, Staron A, Szalat RE, et al. Predictors of hematologic response and survival with stem cell transplantation in AL amyloidosis: a 25-year longitudinal study. Am J Hematol. 2022;97(9):1189-1199.
12. Castillo JJ, Allan JN, Siddiqi T, et al. Venetoclax in previously treated Waldenström macroglobulinemia. J Clin Oncol. 2022;40(1):63-71.
13. Pika T, Hegenbart U, Flodrova P, Maier B, Kimmich C, Schönland SO. First report of ibrutinib in IgM-related amyloidosis: few responses, poor tolerability, and short survival. Blood. 2018;131(3):368-371.
14. Bou Zerdan M, Valent J, Diacovo MJ, Theil K, Chaulagain CP. Utility of Bruton's tyrosine kinase inhibitors in light chain amyloidosis caused by lymphoplasmacytic lymphoma (Waldenström's macroglobulinemia). Adv Hematol. 2022;2022:1182384.
15. Castillo JJ, Libby EN, Ansell SM, et al. Multicenter phase 2 study of daratumumab monotherapy in patients with previously treated Waldenström macroglobulinemia. Blood Adv. 2020;4(20):5089-5092.
Haematologica | 108 June 2023 1684 LETTER TO THE EDITOR
Patient-reported fatigue and pain in Erdheim-Chester disease: a registry-based, mixed methods study
Erdheim-Chester disease (ECD) is a rare hematologic cancer with varied clinical manifestations. There have been 1,500 cases described since the first report.1 To date, there has been no investigation of ECD symptoms with validated patient-reported outcomes (PRO). There is increasing evidence that PRO can result in improved decisions about patientcentered care and improve the quality of care2 especially for patients with rare diseases who often endure heterogeneous and poorly understood symptoms.3 In patients with cancer, fatigue and pain have a markedly deleterious impact on health-related quality of life and are associated with greater financial stress, increased healthcare utilization, and less employment participation.4,5 We previously developed a symptom inventory evaluating patient-reported ECD-specific symptoms, their frequency, and severity. In 50 patients, we found that 72% reported fatigue and 58% reported pain,6 but did not investigate the severity and interference associated with fatigue and pain, nor whether clinical variables modified symptoms or their severity. In the current study, we implemented validated inventories for pain and fatigue to identify factors associated with these prevailing symptoms in a large cohort of ECD patients.
This is an Institutional Review Board-approved registrybased cohort study maintained at Memorial Sloan Kettering Cancer Center (NCT03329274). Participants with ECD who provided informed consent and were enrolled from 20182020 with completed PRO assessments were included. The Brief Pain Inventory (BPI)7 and Brief Fatigue Inventory (BFI)8 were used to assess pain and fatigue, respectively; these are two validated measures to assess the impact and severity of these symptoms in cancer patients. To elicit an indepth description of patients’ symptomatology in their own words, a subset of patients participated in a qualitative semi-structured interview. Participants were selected in a purposeful approach to represent a diversity of perspectives with respect to age, sex, race, ethnicity, and ECD treatment, with the goal of interviewing at least 12 to achieve thematic saturation.9
Clinically relevant fatigue and clinically relevant pain were each categorized a priori using a clinically meaningful cutoff of having at least one BFI or BPI item, respectively, scored ≥4 to reflect moderate to severe symptomatology.10,11 Univariable associations of numeric variables with clinically relevant fatigue or pain were performed with the Wilcoxon rank sum test. Univariable associations of categorical variables with clinically relevant fatigue or pain were identified with the Fisher exact test or c2 test as appropriate. Next, in a complementary analysis, recursive partitioning analysis
(RPA) was used across all variables to identify subgroups statistically more likely to experience clinically relevant fatigue or pain separately. An internal validation of 10-fold cross-classification was performed to prune each RPA tree to a more parsimonious model. The tree selected minimized both the complexity parameter and the cross-validated error. The Spearman correlation coefficient was used to correlate BFI and BPI scores. Association between clinically relevant fatigue and clinically relevant pain within patients was assessed with the McNemar test. In an exploratory analysis, the association between employment status and RPA-identified groups was investigated descriptively. All tests were two-sided with a statistical level of significance <0.05. Analyses were performed using SAS v9.4 and R v3.6.0.
Qualitative interviews were audio-recorded and transcribed verbatim. Transcripts were analyzed by two coders using a thematic content analysis approach. The codebook consisted of a priori codes derived from the domains of the interview guide as well as inductive codes based on recurring patterns in the data. Team members independently coded each transcript, meeting regularly to achieve consensus on emerging concepts. Once transcripts had been coded, the team grouped codes into conceptual categories and identified primary themes. Transcripts were coded using NVivo Pro version 12.0 (QSR International).
There were 127 ECD patients who enrolled in the parent registry protocol and completed PRO assessments. Cohort characteristic distributions are provided in Online Supplementary Table S1. Seventy-four percent reported clinically relevant fatigue and 53% reported clinically relevant pain. Twenty-six percent reported only clinically relevant fatigue, 5% reported only clinically relevant pain, and 48% experienced both (Online Supplementary Table S2). Among patients with clinically relevant pain, 91% also reported clinically relevant fatigue; and among patients who experienced clinically relevant fatigue, 65% reported clinically relevant pain.
The mean BFI total score was 3.8; 4.2 for BFI severity and 3.6 for BFI interference (with daily activities). The highest individual item mean for the fatigue severity construct was worst fatigue and that for fatigue interference was normal work (Table 1). The mean BPI total score was 2.6; 2.5 for BPI pain severity and 2.6 for BPI pain interference (with daily activities). The highest individual item mean for the pain severity construct was worst pain and that for pain interference was normal work.
The BPI pain severity construct score was moderately cor-
Haematologica | 108 June 2023 1685 LETTER TO THE EDITOR
Table 1. Distribution of patient-reported fatigue and pain at enrollment.
PRO: patient-reported outcome; BFI: Brief Fatigue Inventory; BPI: Brief Pain Inventory; N: number; SD: standard deviation. aThe BFI severity subscale comprises three items rated on a numerical scale of 0-10 concerning fatigue severity: right now, usual, and worst. bThe BFI interference subscale comprises six items rated on a numerical scale of 0-10 concerning interference of fatigue with daily living: general activity, mood, walking ability, normal work, relationships, and enjoyment of life. cThe BFI total comprises all nine items: three from the BFI severity subscale and six from the BFI interference subscale. dThe BPI severity subscale comprises four items rated on a numerical scale of 0-10 concerning pain severity: worst, least, average, and right now. eThe BPI interference subscale comprises seven items rated on a numerical scale of 0-10 concerning interference of pain with daily living: general activity, mood, walking ability, normal work, relationships, sleep, and enjoyment of life. fThe BPI total comprises all 11 items: four from the BPI severity subscale and seven from the BPI interference subscale.
related with BFI fatigue severity construct score (r=0.58) and BPI interference severity construct score was moderately correlated with BFI interference severity construct score (r=0.53). Finally, BPI total score was moderately correlated with BFI total score (r=0.56). Within patients, clinically relevant fatigue was statistically significantly
correlated with clinically relevant pain (P<0.0001) (Online Supplementary Table S2).
In univariable analysis, patients who were BRAFV600E-wildtype were more likely to report clinically relevant fatigue (P=0.04) (Table 2). RPA did not identify any subgroups more likely to report clinically relevant fatigue. The only individual
PRO Item/ Construct Description N (%) Mean SD None (0) N (%) Mild (1-3) N (%) Moderate (4-6) N (%) Severe (7-10) N (%) Unknown N (%) BFI Item 1 Fatigue right now 123 (97) 3.5 2.9 37 (29) 23 (18) 41 (32) 22 (17) 4 (3) BFI Item 2 Usual fatigue 123 (97) 3.9 2.7 30 (24) 20 (16) 49 (39) 24 (19) 4 (3) BFI Item 3 Worst fatigue 123 (97) 5.1 3.4 30 (24) 8 (6) 32 (25) 53 (42) 4 (3) BFI Item 4 General activity 123 (97) 4.1 3.3 34 (27) 20 (16) 35 (28) 34 (27) 4 (3) BFI Item 5 Mood 123 (97) 3.3 3.2 43 (34) 31 (24) 23 (18) 26 (20) 4 (3) BFI Item 6 Walking ability 122 (96) 3.5 3.6 50 (39) 17 (13) 26 (20) 29 (23) 5 (4) BFI Item 7 Normal work 123 (97) 4.2 3.6 38 (30) 18 (14) 28 (22) 39 (31) 4 (3) BFI Item 8 Relationships 122 (96) 3.1 3.2 48 (38) 26 (20) 22 (17) 26 (20) 5 (4) BFI Item 9 Life enjoyment 123 (97) 3.8 3.5 40 (32) 22 (17) 29 (23) 32 (25) 4 (3) BFI Severitya Subscale Items 1-3 123 (97) 4.2 2.9 30 (24) 21 (17) 48 (38) 24 (19) 4 (3) BFI Interferenceb Subscale Items 4-9 123 (97) 3.6 3.1 33 (26) 30 (24) 40 (32) 20 (16) 4 (3) BFI Totalc Items 1-9 123 (97) 3.8 2.9 30 (24) 31 (24) 43 (34) 19 (15) 4 (3) BPI Item 1 Worst pain 122 (96) 3.7 3.6 49 (39) 15 (12) 20 (16) 38 (30) 5 (4) BPI Item 2 Least pain 122 (96) 1.6 2.1 63 (50) 41 (32) 12 (9) 6 (5) 5 (4) BPI Item 3 Average pain 120 (94) 2.5 2.5 49 (39) 26 (20) 36 (28) 9 (7) 7 (6) BPI Item 4 Right now pain 122 (96) 2.1 2.6 60 (47) 28 (22) 24 (19) 10 (8) 5 (4) BPI Item 5 General activity 122 (96) 2.9 3.3 57 (45) 17 (13) 23 (18) 25 (20) 5 (4) BPI Item 6 Mood 122 (96) 2.3 3.0 63 (50) 21 (17) 22 (17) 16 (13) 5 (4) BPI Item 7 Walking ability 122 (96) 2.8 3.5 62 (49) 17 (13) 17 (13) 26 (20) 5 (4) BPI Item 8 Normal work 122 (96) 3.0 3.7 63 (50) 14 (11) 16 (13) 29 (23) 5 (4) BPI Item 9 Relations 122 (96) 1.9 2.9 71 (56) 22 (17) 16 (13) 13 (10) 5 (4) BPI Item 10 Sleep 122 (96) 2.6 3.3 63 (50) 19 (15) 19 (15) 21 (17) 5 (4) BPI Item 11 Life enjoyment 121 (95) 2.7 3.4 62 (49) 15 (12) 21 (17) 23 (18) 6 (5) BPI Severityd Subscale Items 1-4 120 (94) 2.5 2.5 49 (39) 35 (28) 27 (21) 9 (7) 7 (6) BPI Interferencee Subscale Items 5-11 122 (96) 2.6 3.0 53 (42) 28 (22) 26 (20) 15 (12) 5 (4) BPI Totalf Items 1-11 120 (94) 2.6 2.7 49 (39) 31 (24) 32 (25) 8 (6) 7 (6)
Haematologica | 108 June 2023 1686 LETTER TO THE EDITOR
variable associated with clinically relevant pain was lines of prior therapy (P=0.04) (Table 2). RPA identified subgroups associated with clinically relevant pain (Figure 1). Patients who were ≥70 years at ECD diagnosis were least likely to report clinically relevant pain whereas patients <70 years at ECD diagnosis with an ECD duration of 9.3 months or longer who were anemic (hemoglobin <13 g/dL) were most likely to report clinically relevant pain (P<0.0001).
We further explored the distribution of current employment status for the RPA-identified group most likely to report clinically relevant pain. Among these patients, 100% reporting unemployment also reported clinically relevant pain. Among patients in the other four RPA-identified groups who were less likely to report clinically relevant pain, only 47% reporting unemployment also reported clinically relevant pain.
Continued on following page.
Variable Level BPI Total: Any Item 4+ BFI Total: Any Item 4+ No Yes P value* No Yes P value* Median age, years 56.2 54.6 0.20 56.0 55.7 0.37 Median duration of diagnosed ECD illness, years 4.8 4.9 0.12 5.1 4.8 0.94 Brain parenchyma involvement, N (%) No 15 (52) 14 (48) 0.63 10 (32) 21 (68) 0.50 Yes 23 (58) 17 (43) 10 (25) 30 (75) Neurological involvement, N (%) No 8 (44) 10 (56) 0.29 8 (40) 12 (60) 0.17 Yes 30 (59) 21 (41) 12 (24) 39 (76) Median number of sites of disease 4 4 0.72 4 4 0.56 Sites of disease, N (%) 1 1 (25) 3 (75) 0.28 1 (25) 3 (75) 0.88 2 1 (25) 3 (75) 2 (40) 3 (60) 3 6 (50) 6 (50) 4 (31) 9 (69) > 3 30 (61) 19 (39) 13 (27) 36 (73) Lines of prior therapy, N (%) 0 or 1 29 (58) 21 (42) 0.04 12 (24) 38 (76) 0.67 2+ 28 (39) 43 (61) 20 (27) 53 (73) BRAFV600E status, N (%) BRAFV600E wildtype 17 (37) 29 (63) 0.07 7 (15) 40 (85) 0.04 BRAFV600E mutated 38 (54) 32 (46) 22 (31) 48 (69) Anemia, N (%) No 33 (52) 30 (48) 0.38 17 (27) 47 (73) 0.80 Yes 16 (43) 21 (57) 9 (24) 28 (76) Elevated CRP, N (%) No 9 (31) 20 (69) 0.19 9 (31) 20 (69) 0.19 Yes 5 (17) 25 (83) 5 (17) 25 (83) Treatment, N (%) None 19 (54) 16 (46) 0.31 7 (19) 29 (81) 0.29 Any 38 (44) 48 (56) 25 (29) 62 (71) None 19 (54) 16 (46) 0.51 7 (19) 29 (81) 0.41 Conventional 4 (36) 7 (64) 2 (17) 10 (83) Targeted 34 (45) 41 (55) 23 (31) 52 (69) Targeted middle, reduced, or intermittent 18 (50) 18 (50) 0.56 13 (36) 23 (64) 0.27 Targeted high or reduced high 16 (43) 21 (57) 9 (24) 28 (76)
Table 2. Clinically relevant fatigue and pain by variables of interest.
Haematologica | 108 June 2023 1687 LETTER TO THE EDITOR
BPI: Brief Pain Inventory; BFI: Brief Fatigue Inventory; ECD: Erdheim-Chester disease; CRP: C-reactive protein; PET: positron emission tomography; RPA: recursive partitioning analysis; Hb: hemoglobin. *Tests of association were performed using the c2 test, Fisher test, or Wilcoxon rank sum test, depending on the type and distribution of the variable. **The RPA identified subgroups of patients who were more likely to report clinically relevant pain but did not identify subgroups of patients who were more likely to report clinically relevant fatigue.
Thematic saturation was achieved after 13 patients had been interviewed. In the theme related to physical symptom burden, ten patients (77%) described pain and nine (69%) described fatigue. Online Supplementary Table S3 contains illustrative quotes on the nature of pain and fatigue. Pain was characterized as “diffuse” or “electrical” neuropathic pain. A few patients specifically mentioned joint pain as a treatment side effect. Patients generally described pain as worse at night and amplified by fatigue. Fatigue was described as a “constant” lack of energy and exacerbated by physical exertion.
This is the first study to identify specific subgroups of ECD patients who may benefit most from pain and fatigue interventions. While no other studies have detailed patientreported pain and fatigue, some have detailed general symptomatology in patients with ECD.6,12,13 Our current estimate of pain prevalence sits at the higher end of those previously reported for clinical presentation and our fatigue prevalence is three times that reported by Estrada-Veras et al. 12 We hypothesize that eliciting patient-reported symp-
toms, rather than gleaning symptoms documented by healthcare providers, may demonstrate a greater frequency of symptoms. We further hypothesize that these symptoms may become more severe over time, therefore more frequently reported in a patient population that spans time since ECD diagnosis, and this is supported by our RPA results.
The finding that BRAFV600E-wildtype patients reported more fatigue is intriguing, but difficult to interpret because few differences in disease phenotype or biology have been noted between BRAFV600E-mutated and wildtype patients. However, it is plausible that BRAF inhibitors have positively affected symptomatology of BRAFV600E-mutated patients. The observed association between fatigue and pain also raises the question of whether unmanaged pain interrupts sleep or rest and contributes to fatigue, underscoring the importance of optimal pain management.
We observed that pain was more frequent in younger patients, which has not been observed in a large meta-analysis of characteristics associated with cancer pain14 but has
Variable Level BPI Total: Any Item 4+ BFI Total: Any Item 4+ No Yes P value* No Yes P value* Diabetes mellitus, N (%) No 53 (48) 56 (51) 0.44 30 (27) 80 (73) 0.73 Yes 4 (36) 7 (64) 2 (17) 10 (83) Diabetes insipidus, N (%) No 20 (48) 22 (52) 0.74 12 (29) 30 (71) 0.73 Yes 16 (52) 15 (48) 8 (25) 24 (75) Hypertension, N (%) No 33 (49) 34 (51) 0.67 20 (30) 47 (70) 0.32 Yes 24 (45) 29 (55) 12 (22) 43 (78) Clinical response, N (%) Complete response 5 (83) 1 (17) 0.13 4 (67) 2 (33) 0.10 Partial response 26 (46) 31 (54) 14 (24) 44 (76) Stable disease 4 (33) 8 (67) 3 (25) 9 (75) Best response on PET, N (%) Complete response 6 (50) 6 (50) 0.49 5 (42) 7 (58) 0.34 Partial response 21 (49) 22 (51) 9 (20) 35 (80) Stable disease 4 (31) 9 (69) 3 (23) 10 (77) BPI RPA** Age ≥70 years 12 (92) 1 (8) <0.0001 Age <70 years, ECD duration <9.3 months 11 (85) 2 (15) Age <70 years, ECD duration ≥9.3 months, Hb ≥13 g/dL, Intermittent or high targeted treatment 8 (89) 1 (11) Age <70 years, ECD duration ≥9.3 months, Hb ≥13 g/dL, All other treatments 13 (37) 22 (63) Age <70 years, ECD duration ≥9.3 months, Hb <13 g/dL 6 (19) 25 (81)
Haematologica | 108 June 2023 1688 LETTER TO THE EDITOR
Final recursive partitioning analysis model for clinically relevant pain. Five subgroups and their associated predicted probabilities of having clinically relevant pain are shown. The following variables were entered into the recursive partitioning analysis model: age (continuous), sex (male/female), hypertension at enrollment (yes/no/unknown), presence of diabetes mellitus at enrollment (yes/no/unknown), BRAFV600E mutational status (mutated/wildtype/unknown), steroid treatment prior to enrollment (yes/no/unknown), individual organ system involvement (yes/no): bone, neurological, brain/parenchyma, cardiovascular, pulmonary, retroperitoneum, abdomen, lymph nodes, other, or unknown, presence of diabetes insipidus at enrollment (yes/no/unknown), number of prior lines of systemic treatment (continuous), undiagnosed Erdheim-Chester disease (ECD) duration from first symptom to official ECD diagnosis (continuous), diagnosed ECD duration from official ECD diagnosis until assessment of patient-reported outcomes (continuous), C-reactive protein (elevated/not elevated/unknown), hemoglobin (Hb; g/dL) at enrollment (continuous), clinical status at the time of survey completion (resolved/improved but not resolved/stable/not evaluable), ECD disease status as measured by positron emission tomography (resolved/improved but not resolved/stable/not evaluable), treatment at enrollment (none/conventional/targeted high dose/targeted reduced high dose/targeted middle dose/targeted reduced dose/targeted intermittent/targeted other), and anemia at enrollment (none/mild/moderate/severe/unknown).
been observed in one meta-analysis of breast cancer patients.15 We also observed that patients were more likely to report pain if they were anemic. Pain and anemia are most likely correlates of overall disease burden rather than mechanistically related. Altogether, our study suggests that ECD pain is highly prevalent, is likely persistent despite treatment, and should be discussed with all patients but particularly with individuals younger than 70. This is of great importance as pain in this group can be so debilitating as to prohibit gainful employment, with significant implications for disability program qualifications. In exploratory analysis, all patients who were most likely to report pain based on RPA were also unemployed, suggesting that pain is disruptive with respect to employment.
Our study has some limitations. This registry-based, crosssectional cohort is heterogeneous by design. We did not collect information on medications for pain or fatigue management. Furthermore, the rarity of this disease also precludes the external validation of the RPA results.
Using a registry-based study design of ECD patients with PRO-based methodology, we found that fatigue and pain were prevalent, severe, and interfered in the daily lives of

patients. Fatigue and pain were moderately associated, raising the notion that these may potentiate the impact of one another. Fatigue and pain were unrelated to treatment response emphasizing the importance of PRO assessments when evaluating the impact of therapies. Future investigations of the evolution of fatigue and pain over time may yield additional information about how best to evaluate and manage these symptoms.
Authors
Anne S. Reiner,1 Dana Bossert,2 Justin J. Buthorn,2 Allison M. Sigler,2 Selin Gonen,3 Deanna Fournier,4 Kathleen Brewer,5 Jessica Corkran,5 Gaurav Goyal,6 Carl E. Allen,7 Kenneth L. McClain,7 Thomas M. Atkinson,8 Kathleen A. Lynch,8 Jun J. Mao,9 Katherine S. Panageas1 and Eli L. Diamond2
1Department of Epidemiology and Biostatistics; Memorial Sloan Kettering Cancer Center, New York, NY; 2Department of Neurology; Memorial Sloan Kettering Cancer Center, New York, NY; 3Hunter
Haematologica | 108 June 2023 1689 LETTER TO THE EDITOR
College High School, New York, NY; 4Histiocytosis Association, Pitman, NJ; 5Erdheim-Chester Disease Global Alliance, DeRidder, LA; 6Division of Hematology-Oncology, University of Alabama at Birmingham, Birmingham, AL; 7Texas Children's Cancer Center, Department of Pediatrics, Baylor College of Medicine, Houston, TX; 8Department of Psychiatry and Behavioral Sciences, Memorial Sloan Kettering Cancer Center, New York, NY and 9Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
Correspondence:
E.L. DIAMOND - diamone1@mskcc.org
https://doi.org/10.3324/haematol.2022.282287
Received: October 21, 2022.
Accepted: December 14, 2022.
Early view: December 22, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
ELD discloses unpaid editorial support from Pfizer Inc and serves on an advisory board for Day One Therapeutics and Springworks Therapeutics, both outside the submitted work. CEA serves on an advisory board for Sobi and receives research funding from Genentech. KLM serves on advisory boards for Sobi and Atara Biotherapeutics, Inc. GG serves on an advisory board for Springworks Therapeutics. None of the other authors disclosed any conflicts of interest.
References
1. Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease. Blood. 2020;135(16):1311-1318.
2. Kotronoulas G, Kearny N, Maguire R, et al. What is the value of the routine use of patient-reported outcome measures toward improvement of patient outcomes, process of care, and health service outcomes in cancer care? A systematic review of controlled trials. J Clin Oncol. 2014;32(24):1480-1501.
3. Slade A, Isa F, Kyte D, et al. Patient reported outcome measures in rare diseases: a narrative review. Orphanet J Rare Dis. 2018;13(1):61.
4. Thong MSY, van Noorden CJF, Steindorf K, Arndt V. Cancer-related fatigue: causes and current treatment options. Curr Treat Options Oncol. 2020;21(2):17.
5. Neufeld NJ, Elnahal SM, Alvarez RH. Cancer pain: a review of epidemiology, clinical quality and value impact. Future Oncol. 2017;13(9):833-841.
6. Diamond EL, Reiner AS, Buthorn JB, et al. A scale for patientreported symptom assessment for patients with Erdheim-Chester disease. Blood Adv. 2019;3(7):934-938.
7. Cleeland CS, Ryan KM. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Sing. 1994;23(2):129-138.
8. Mendoza TR, Wang XS, Cleeland CS, et al. The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer. 1999;85(5):1186-1196.
9. Guest G, Bunce A, Johnson L. How many interviews are enough?
Contributions
ASR conceived the study, was responsible for the formal statistical analysis, interpreted the research , and drafted the manuscript. DB, JJB and AMS conceived the study, collected and interpreted data and drafted the manuscript. DF, KB, GG, JC, CEA, KLM, TMA, and JJM conceived the study, interpreted the research, and drafted the manuscript. SG conceived the study and drafted the manuscript. KAL and KSP conceived and supervised the study, interpreted the research and drafted the manuscript. ELD conceived and supervised the study, collected and interpreted data and drafted the manuscript. All authors reviewed and edited the final version of the manuscript and agreed with the submission.
Acknowledgments
The authors acknowledge the patients with Erdheim-Chester disease who are contributing to the ongoing registry.
Funding
This work was supported by the National Institutes of Health/National Cancer Institute (P30 CA008748) and Population Sciences Research Program award (ELD, KSP), as well as the National Cancer Institute (R37CA259260; ELD, KSP). This work was also supported by the Frame Family Fund (ELD), the Joy Family West Foundation (ELD), the Applebaum Foundation (ELD), and the Erdheim-Chester Disease Global Alliance (ELD).
Data-sharing statement
For original data, please contact diamone1@mskcc.org.
An experiment with data saturation and variability. Field Method. 2006;18(1):59-82.
10. Mao H, Bao T, Shen X, et al. Prevalence and risk factors for fatigue among breast cancer survivors on aromatase inhibitors. Eur J Cancer. 2018;101:47-54.
11. Romero SAD, Jones L, Bauml JM, Li QS, Cohen RB, Mao JJ. The association between fatigue and pain symptoms and decreased physical activity after cancer. Support Care Cancer. 2018;26(10):3423-3430.
12. Estrada-Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017;1(6):357-366.
13. Cavalli G, Guglielmi B, Berti A, Bampochiaro C, Sabbadini MG, Dagna L. The multifactorial clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis. 2013;72(10):1961-1965.
14. MH van den Beuken-van Everdingen, Hochstenbach LM, Joosten EAJ, Tjan-Heijnen VCG, Janssen DJA. Update on prevalence of pain in patients with cancer: systematic review and meta-analysis. J Pain Symptom Manage. 2016;51(6):1070-1090.
15. Wang L, Guyatt GH, Kennedy SA, et al. Predictors of persistent pain after breast cancer surgery: a systematic review and meta-analysis of observational studies. CMAJ. 2016;188(14):E352-E361.
Haematologica | 108 June 2023 1690 LETTER TO THE EDITOR
Retrospective analysis of a cohort of 41 de novo B-cell prolymphocytic leukemia patients: impact of genetics and targeted therapies (a FILO study)
B-cell prolymphocytic leukemia (B-PLL) is a very rare lymphoid neoplasm accounting for less than 0.5% of lowgrade mature B-cell lymphomas.1-3 B-PLL is a poor-prognosis disease with a historical median overall survival (OS) of 3 years.3 B symptoms, marked lymphocytosis, cytopenia and massive splenomegaly are hallmarks of B-PLL patients at diagnosis.1-3 A minority of patients present with an indolent phase, not requiring immediate therapy. The characterization of B-PLL genetic landscape has led to the identification of recurrent abnormalities including complex karyotype (CK), MYC translocations/gains (MYC aberration), 17p deletion (del17p) and TP53 mutations (TP53 abnormalities [TP53abn]).1,4-6 Due to the rarity of the disease, the absence of international guidelines and randomized clinical trial data, most therapeutic approaches used chemoimmunotherapy (CIT) or are executed according to chronic lymphocytic leukemia (CLL) guidelines. A watch and wait strategy is proposed to asymptomatic B-PLL patients while, for symptomatic B-PLL, rituximab-based CIT, alemtuzumab and more recently BCR inhibitors (BCRi) have been used for frontline therapy. Recent case reports and small series have indeed described the efficacy of Bruton tyrosine kinase inhibitor (BTKi)7-10 and phosphoinositide 3-kinase inhibitor (P13Ki),11 notably in frontline and relapse TP53abn B-PLL population. Allogeneic stem cell transplantation (allo-SCT) is still considered as the only curative therapy for eligible and responsive B-PLL patients. Our group has recently shown in a retrospective cohort of 34 patients with rigorous diagnostic criteria for B-PLL that three distinct cytogenetic risk groups could be identified: low (no MYC aberration), intermediate (MYC aberration but no del17p), and high-risk (MYC aberration and del17p), with profound impact on OS5.
Despite these advances, several questions remain unanswered in B-PLL. Factors associated with initial asymptomatic disease and those predicting time to first treatment (TFT) are still unknown as the impact of CIT or BCRi in different genetic subgroups. Based on our previous biological work,5 we describe here the clinical outcomes and associated prognostic factors of initially asymptomatic or symptomatic diseases, and the impact of different therapies in an extended and homogenously defined cohort of 41 de novo B-PLL patients.
We conducted a retrospective analysis of French adult patients with a diagnosis of de novo B-PLL, according to the
2016 World Health Organization classification criteria1 after thorough exclusion of potential differential diagnoses as described previously.5 While the 2022 World Health Organization classification included it in “splenic B-cell lymphoma/leukemia with prominent nucleoli”12 that also includes “hairy cell leukemia variant” (HCL-v), B-PLL is still recognized as a specific entity by the International Consensus Classification of mature lymphoid neoplasms.13 Forty-one patients diagnosed between 1992 and 2020 in 17 French centers were included after the reviewing of blood smears by three independent expert cytologists. In addition to the 2016 classification criteria (i.e., prolymphocytes accounting for at least 55% of the lymphoid cells in peripheral blood), inclusion cytological criteria included round nucleus and prominent central nucleolus while presence of hybrid features overlapping HCL, such as hairy projections was a strict exclusion criterion. Although we cannot exclude that some cases previously defined as HCL-v may be present in our cohort, it should be noted that only one case harbored usage of the IGHV4-34 gene family (n=1/25) and no MAP2K1 mutation was observed in the 20 cases explored by next-generation sequencing. Patients with a history of another B-cell malignancy (CLL or marginal zone lymphoma [MZL]) were excluded and diagnosis of MCL was ruled out according to karyotype and fluorescence in situ hybridization (FISH) assays looking for CCND1 rearrangements or translocations involving CCND2 or CCND3. Cytogenetic and molecular analyses for detection of MYC aberration and TP53abn were performed as described previously.5 All cases were analyzed for del17p by FISH and 23 of 41 had DNA available for evaluation of TP53 mutations by molecular analyses. CK and high CK (HCK) were defined by the presence of ≥3 and ≥5 chromosomal abnormalities respectively. The study was performed in accordance with the Declaration of Helsinki and was approved by the local Investigational Review Board (CPP Ile-de-France VI, Paris, France, 05/21/2014). The primary endpoints were overall survival (OS) defined as time from diagnosis to death from any cause or last follow-up, and progression-free survival (PFS) defined as time from first treatment initiation to progression, death, or last follow-up. TFT was defined as time from diagnosis to first therapy in asymptomatic B-PLL. Response to therapy was evaluated using modified criteria from those of iwCLL.9,14 Baseline characteristics are described as median and range for continuous variables and frequency and percen-
Haematologica | 108 June 2023 1691 LETTER TO THE EDITOR
tage for categorical variables. Comparisons between categorical variables were performed by c2 or Fisher’s exact test, as appropriate. Comparisons between continuous variables were performed by Wilcoxon-Mann-Whitney non-parametric test. Median follow-up was calculated using the reverse Kaplan-Meier method. Survival curves were calculated by using the Kaplan-Meier method, and the log-rank test was used for comparisons between groups. Univariate analyses were performed using the Cox proportional hazards model. All tests of statistical signifi-
cance were two-sided, and a P value <0.05 was considered statistically significant. All statistical analyses were performed using the R statistical package (version 4.1.0, R Core Team, 2021) and the RStudio software (versions 1.2.5033).
The main characteristics of the whole cohort at diagnosis are detailed in Table 1 and correspond to those previously reported in the literature.3 Median age at B-PLL diagnosis was 72 years old (range, 46-88 years) and most patients were male (25/41, 61%). Splenomegaly was present in 62%
Table 1. Characteristics at diagnosis and their prognostic impact on overall and progression-free survivals in the whole B-cell prolymphocytic leukemia cohort. Univariate analysis was performed using log-rank tests. Complex karyotype and high complex karyotype were defined by the presence of ≥3 and ≥5 chromosomal abnormalities respectively.
HR: hazard ratio; 95% CI: 95% confidence interval; NA: not assessed; PFS: progression-free survival; OS: overall survival; LDH: lactate dehydrogenase; del: deletion.
Parameter Whole cohort (N=41) OS PFS HR [95% CI] P value HR [95% CI] P value Age in years at diagnosis Median (range) 72 (46-88) 1.03 [0.99-1.07] 0.16 1.02 [0.98-1.06] 0.4 Sex, N (%) Male 25/41 (61) 0.92 [0.66-3.21] 0.92 1.36 [0.59-3.15] 0.47 Constitutional symptoms, N (%) Yes 4/29 (14) 2.05 [0.44-9.51] 0.36 na na Splenomegaly, N (%) Present 24/39 (62) 1.25 [0.56-2.81] 0.59 0.63 [0.28-1.45] 0.28 Lymphadenopathy, N (%) Present 11/39 (28) 2.31 [0.9-5.89] 0.08 1.11 [0.43-2.85] 0.83 Extranodal disease, N (%) Present 4/32 (12) 5.71 [1.34-24.4] 0.02 2.97 [0.79-11.1] 0.11 Lymphocytes, x109/L Median (range) 32 (5-227) 1 [0.99-1.01] 0.86 1 [0.99-1.01] 0.78 Prolymphocytes, % among lymphocytes Median (range) 83 (61-100) 0.96 [0.92-1.01] 0.1 0.97 [0.93-1.02] 0.2 Hemoglobin value, g/dL Median (range) 12 (8-17) 0.87 [0.74-1.03] 0.11 0.94 [0.79-1.11] 0.44 Platelets, x109/L Median (range) 135 (25-316) 0.99 [0.98-0.99] 0.02 0.99 [0.99-1] 0.41 LDH, N % Increased 15/24 (62) 1.44 [0.41-5.03] 0.57 1.45 [0.38-5.51] 0.58 b-2-microglobulin, N (%) Increased 11/17 (65) 1.71 [0.42-7.04] 0.46 1.07 [0.21-5.56] 0.94 Complex karyotype, N (%) Present 28/41 (68) 1.08 [0.45-2.6] 0.86 2.03 [0.75-5.53] 0.17 High complex karyotype, N (%) Present 19/41 (46) 1.39 [0.63-3.1] 0.41 2.39 [0.99-5.71] 0.05 del17p, N (%) Present 16/40 (40) 2.8 [1.19-6.59] 0.02 3.38 [1.42-8.07] 0.006 TP53 mutation, N (%) Present 9/23 (39) 2.41 [0.76-7.65] 0.14 1.72 [0.52-5.72] 0.38 del17p or TP53 mutation, N (%) Present 18/31 (58) 2.78 [0.99-7.78] 0.05 2.9 [1.05-7.97] 0.04 MYC gain or rearrangement, N (%) Present 30/41 (73) 1.4 [0.59-3.35] 0.45 0.5 [0.21-1.19] 0.12 MYC / del17p status, N (%) MYC WT / no del17p 3/40 (7.5) MYC WT / del17p 8/40 (20) 4.59 [0.79-26.6] 0.09 1.6 [0.38-6.65] 0.52 MYC aberration / no del17p 21/40 (52) 2.76 [0.56-13.7] 0.23 0.42 [0.11-1.68] 0.22 MYC aberration / del17p 8/40 (25) 11 [1.84-65.9] 0.009 1.89 [0.45-7.9] 0.38
Haematologica | 108 June 2023 1692 LETTER TO THE EDITOR
of patients (24/39) while lymphadenopathy (11/39, 28%) and extranodal disease (4/32, 12%) were rarer. The distribution of cytogenetic abnormalities was as follows: MYC aberrations (30/41, 73%), CK (28/41, 68%), high CK (HCK, 19/41, 46%) and del17p (16/40, 40%). TP53abn (mutation or del17p) was detected in 18 0f 31 patients (58%). Del17p was significantly associated with lymphocytosis, CK, HCK and TP53 mutations, while MYC aberration was enriched in patients lacking del17p (Online Supplementary Table S1). Median follow-up for the whole cohort was 102 months (range, 0.2-171 months). B-PLL was symptomatic at diagnosis in 22 of 39 (56%) patients. Main differences observed between asymptomatic and symptomatic B-PLL patients are summarized in Table 2. As expected, the proportion of patients with cytopenia and/or tumoral disease
was higher in the symptomatic B-PLL subgroup. Interestingly, significantly more symptomatic B-PLL patients harbored both MYC aberrations and del17p (7/22, 32%) than asymptomatic B-PLL patients (0/16, 0%; P=0.04). Among the 17 asymptomatic B-PLL patients, 13 (76%) progressed with a median TFT of 46.9 months. Of note, the four patients who did not progress harbored either MYC aberration (n=3) or del17p (n=1) but no HCK (0/4 compared to 8/13 asymptomatic B-PLL who progressed; P=0.08). During follow-up, 34 of 41 (83%) B-PLL patients required therapy. The median number of therapeutic lines was two (range, 1-4). Frontline therapies consisted of chemotherapy (CT) (9/34, 26%), CIT (15/34, 44%), BTKi (5/34, 15%), alemtuzumab (3/34, 9%) and rituximab monotherapy (1/34, 3%). Only two patients received allo-SCT. Overall-
Parameter Asymptomatic B-PLL (N=17) Symptomatic B-PLL (N=22) P value Age in years at diagnosis Median (range) 72 (51-87) 70 (46-88) 0.9 Sex, N (%) Female 5/17 (29) 9/22 (41%) 0.5 Constitutional symptoms, N (%) Yes 0/15 (0) 3/13 (23%) 0.09 Splenomegaly, N (%) Present 6/16 (38) 16/21 (76%) 0.02 Lymphadenopathy, N (%) Present 0/16 (0) 10/21 (48%) 0.002 Extranodal disease, N (%) Present 0/14 (0) 4/17 (24%) 0.11 Lymphocytes, x109/L Median (range) 20 (5-175) 41 (5-227) 0.2 Prolymphocytes, % among lymphocytes Median (range) 78 (66-94) 83 (61-100) 0.4 Hemoglobin value, g/dL Median (range) 14 (8-17) 11 (8-14) 0.02 Platelets, x109/L Median (range) 162 (91-316) 128 (25-206) 0.02 LDH, N % Increased 6/12 (50) 9/12 (75%) 0.4 b-2-microglobulin, N (%) Increased 4/10 (40) 7/7 (100%) 0.04 Complex karyotype, N (%) Present 14/17 (82) 13/22 (59%) 0.12 Highly complex karyotype, N (%) Present 8/17 (47) 10/22 (45%) 0.9 del17p, N (%) Present 5/16 (31) 10/22 (45%) 0.4 TP53 mutation, N (%) Present 3/8 (38) 5/14 (36%) 0.9 MYC gain or rearrangement, N (%) Present 10/17 (59) 18/22 (82%) 0.2 MYC / del17p status, N (%) 0.04 MYC WT/no del17p 2/16 (12) 1/22 (4.5%) MYC WT/del17p 5/16 (31) 3/22 (14%) MYC aberration/no del17p 9/16 (56) 11/22 (50%) MYC aberration/del17p 0/16 (0) 7/22 (32%)
Table 2. Characteristics at diagnosis according to the asymptomatic (asymptomatic B-cell prolymphocytic leukemia, N=17) or symptomatic (symptomatic B-cell prolymphocytic leukemia, N=22) presentation.
Haematologica | 108 June 2023 1693 LETTER TO THE EDITOR
B-PLL: B-cell prolymphocytic leukemia; LDH: lactate dehydrogenase; del: deletion.
response rates (ORR) for frontline CT (78%), CIT (60%), BTKi (100%) and alemtuzumab (100%) were not significantly different (P=0.3) (Online Supplementary Figure S1). In the whole cohort, median PFS of patients receiving frontline therapy was 30 months and median OS was 67 months (Figure 1A, B). Twenty-six of 41 patients (63%) have
died due to B-PLL progression (38%), therapy-related toxicity (23%), other causes (27%) and of unknown origin (12%). Main clinical and biological factors associated with PFS and OS in univariate analyses are summarized in Table 1. Median OS was significantly longer in asymptomatic B-PLL compared to symptomatic B-PLL (126 vs. 54
A B C D
Figure 1. Outcomes of B-cell prolymphocytic leukemia patients. Kaplan-Meier estimates of (A) overall survival (OS) (n=41) and (B) progression-free survival (PFS) (n=34) in the whole cohort. Kaplan-Meier estimates of (C) OS and (D) PFS according to the presence of del17p (no del17p, red; del17p, blue). Kaplan-Meier estimates of (E) OS and (F) PFS according to the treatment received (blue: Bruton tyrosine kinase inhibitor [BTKi]; red: chemotherapy or chemoimmunotherapy [CT/CIT]) among del17p patients.

E F Haematologica | 108 June 2023 1694 LETTER TO THE EDITOR
months; P=0.003; Online Supplementary Figure S2A). The presence of del17p pejoratively influenced both PFS (median 8 vs. 45 months; P=0.004) and OS (median 34 vs. 126 months; P=0.02) (Figure 1C, D) while low platelet count (hazard ratio [HR]=0.99, 95% confidence interval [CI]: 0.980.99) and extranodal disease (HR=5.71, 95% CI: 1.34-24.4) were associated with shorter OS (Table 1). The poorest median OS and PFS were observed in MYC aberration / del17p patients (20 and 6 months, respectively) (Table 1; Online Supplementary Figure S2B, C) while patients with either MYC aberration (101 and 58 months, respectively) or del17p (78 and 9 months, respectively) harbored comparable intermediate prognosis inferior to those without any of these abnormalities (145 and 36 months, respectively). The presence of TP53 mutation was highly correlated to the presence of del17p (Online Supplementary Table S1). Although the presence of TP53 mutation did not significantly affect OS (HR=2.41, 95% CI: 0.76-7.65; P=0.14) or PFS (HR=1.72, 95% CI: 0.52-5.72; P=0.38), the statistical analysis is limited by the reduced number of patients with available TP53 mutation status. The specific prognostic impact of TP53 mutation was not possible due to the very few cases (n=2) of TP53 mutation without del17p.
We eventually interrogated the impact of different frontline therapies on outcomes (Online Supplementary Figure S2D, E ). The type of frontline therapies did not significantly modify PFS or OS but was not randomly distributed among genetic subgroups as patients that received BTKi all harbored del17p. Looking specifically in the del17p subgroup, patients receiving BTKi (n=5) displayed better outcomes compared to those receiving CT/CIT (n=7) (respective median PFS and OS of 24 and 101 vs. 3 and 15 months; P=0.03 and P=0.2) (Figure 1E, F).
Although definitive conclusions are limited by its retrospective nature, the small numbers of patients in specific subgroups and lack of available DNA for TP53 mutation analysis in all samples, our study of the largest B-PLL cohort to date provides meaningful insights in this very rare disease. We identified that asymptomatic B-PLL represents around 40% of patients at diagnosis, do not harbor both MYC aberration and del17p, and displays a significant better OS. We highlight the pejorative impact of del17p and confirm that the subgroup presenting with both MYC aberration and del17p has the worst outcome with a median OS shorter than 2 years. Finally, albeit on a small number of patients, we confirm the efficacy of frontline BTKi in del17p B-PLL.7,8,9
Justine Siavellis,9 Alain Delmer,10 Anne-Sophie Michallet,11 Emmanuelle Ferrant,12 Pierre Feugier,13 Cécile Tomowiak,14 Annie Brion,15 David Ghez,16 Luc-Matthieu Fornecker,17 Sarah Ivanoff,9 Stéphanie Struski,18 Laurent Sutton,19 Isabelle Radford-Weiss,20 Virginie Eclache,21 Christine Lefebvre,22 Véronique Leblond,2 Florence Nguyen-Khac,3,4# and Damien Roos-Weil2,4# for the FILO (French Innovative Leukemia Organization) group, see Appendix below.
1Groupe Hospitalier Mutualiste de Grenoble, Service d’OncoHématologie, Grenoble; 2Sorbonne Université, Service d’Hématologie Clinique, Hôpital Pitié-Salpêtrière, AP-HP, Paris; 3Sorbonne Université, Unité de Cytogénétique Hématologique, Hôpital Pitié-Salpêtrière, AP-HP, Paris; 4Centre de Recherche des Cordeliers, INSERM, Drug Resistance in Hematological Malignancies (DRIHM) Team, Sorbonne Université, Université Sorbonne Paris Cité, Université Paris Descartes, Université Paris Diderot, Paris; 5Hôpital Lyon Sud, Laboratoire d’Hématologie, Hospices Civils de Lyon, Pierre-Bénite; 6Sorbonne Université, Service d’Hématologie Biologique, Hôpital Pitié-Salpêtrière, AP-HP, Paris; 7Hôpital Ambroise Paré, Laboratoire d’Hématologie, AP-HP, Boulogne-Billancourt; 8CHRU Nancy, Service d’Hématologie Biologique, INSERM 1256 NGERE, Nancy; 9Hôpital Avicenne, Service d’Hématologie Clinique, AP-HP, Bobigny; 10CHU Reims, Service d’Hématologie Clinique, Reims; 11Centre Léon Bérard, Service d’Hématologie Clinique, Lyon; 12Hôpital Lyon Sud, Service d’Hématologie Clinique, Hospices Civils de Lyon, Pierre-Bénite; 13CHU Nancy, Service d’Hématologie Clinique, Vandœuvre-lès-Nancy; 14CHU Poitiers, Service d’Hématologie Clinique, Poitiers; 15CHU J Minjoz, Service d’Hématologie Clinique, Besançon; 16Institut Gustave Roussy, Service d’Hématologie Clinique, Villejuif; 17Service d’Hématologie, Institut de Cancérologie Strasbourg Europe (ICANS), Strasbourg; 18IUCT Oncopole - Toulouse, Laboratoire d’hématologie/Plateau Technique Hématologie-Oncologie, Toulouse; 19CHU Tours, Service d’Hématologie Clinique, Tours; 20CHU Necker-Enfants Malades, Laboratoire de Cytogénétique, APHP, Paris; 21Hôpital Avicenne, Service d’Hématologie Biologique, AP-HP, Bobigny and 22CHU Grenoble Alpes, Laboratoire de Cytogénétique des Hémopathies, Grenoble, France
*CA, LP and EC contributed equally as co-first authors. #FNK and DRW contributed equally as co-senior authors.
Correspondence:
C. ALGRIN - caroline.algrin@avec.fr
D. ROOS-WEIL - damien.roosweil@aphp.fr
https://doi.org/10.3324/haematol.2022.282162
Received: September 26, 2022.
Accepted: December 14, 2022.
Early view: December 22, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Authors
Caroline Algrin,1* Louis Pérol,2* Elise Chapiro,3,4* Lucile Baseggio,5 Karim Maloum,6 Catherine Settegrana,7 Jean-François Lesesve,8
Haematologica | 108 June 2023 1695 LETTER TO THE EDITOR
Disclosures
No conflicts of interest to disclose.
Contributions
CA, EC, VL, FNK and DRW designed the study; CA, LP, EC, VL, FNK and DRW analyzed data; CA, LP and DRW wrote the manuscript; CA, LP, EC, LB, KM, CS, JFL, JS, AD, ASM, EM, PF, CT, AB, DG, LMF, SI, SS, IRW, VE, CL, LS, VL, FNK and DRW recruited patients. All authors critically reviewed and approved the manuscript.
Acknowlegments
We thank Mohamad Sabbah for his assistance in data collection.
Funding
This study was supported in part by grants from INCA-DGOSINSERM 12560 (SiRIC CURAMUS is financially supported by the French National Cancer Institute, the French Ministry of Solidarity and Health and INSERM with financial support from ITMO Cancer AVIESAN).
Data-sharing statement
The data that support the findings of this study are available on request from the corresponding author.
Appendix
FILO group members: Caroline Algrin (Grenoble), Thérèse Aurran (Marseille), Marie Christine Béné (Nantes), Fontanet Bijou
References
1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
2. Dearden C. Management of prolymphocytic leukemia. Hematol Am Soc Hematol Educ Program. 2015;2015:361-367.
3. Cross M, Dearden C. B and T cell prolymphocytic leukaemia. Best Pract Res Clin Haematol. 2019;32(3):217-228.
4. Hercher C, Robain M, Davi F, et al. A multicentric study of 41 cases of B-prolymphocytic leukemia: two evolutive forms. Leuk Lymphoma. 2001;42(5):981-987.
5. Chapiro E, Pramil E, Diop M, et al. Genetic characterization of Bcell prolymphocytic leukemia: a prognostic model involving MYC and TP53. Blood. 2019;134(21):1821-1831.
6. Lens D, De Schouwer PJ, Hamoudi RA, et al. p53 abnormalities in B-cell prolymphocytic leukemia. Blood. 1997;89(6):2015-2023.
7. Damlaj M, Al Balwi M, Al Mugairi AM. Ibrutinib therapy is effective in B-cell prolymphocytic leukemia exhibiting MYC aberrations. Leuk Lymphoma. 2018;59(3):739-742.
8. Gordon MJ, Raess PW, Young K, Spurgeon SEF, Danilov AV. Ibrutinib is an effective treatment for B-cell prolymphocytic leukaemia. Br J Haematol. 2017;179(3):501-503.
(Bordeaux), Annie Brion (Besançon), Julien Broséus (Nancy), Guillaume Cartron (Montpellier), Aline Clavert (Angers), Florence Cymbalista (Bobigny), Frédéric Davi (Paris), Caroline Dartigeas (Tours), Sophie De Guibert (Rennes), Alain Delmer (Reims), Marie Sarah Dilhuydy (Bordeaux), Bernard Drenou (Mulhouse), Jehan Dupuis (Créteil), Emmanuelle Ferrant (Lyon), Pierre Feugier (Nancy), Luc Matthieu Fornecker (Strasbourg), David Ghez (Villejuif), Romain Guièze (Clermont-Ferrand), Charles Herbaux (Lille), Bénédicte Hivert (Lille), Luca Inchiappa (Marseille), Kamel Laribi (Le Mans), Katel Le Dû (Le Mans), Magali Le Garff-Tavernier (Paris), Marielle Le Goff (Le Mans), Véronique Leblond (Paris), Stéphane Leprêtre (Rouen), Rémi Letestu (Bobigny), Vincent Lévy (Bobigny), Béatrice Mahé (Nantes), Fatiha Merabet (Versailles), Anne Sophie Michallet (Lyon), Lysiane Molina (Grenoble), Pierre Morel (Amiens), Florence Nguyen-Khac (Paris), Bertrand Pollet (Boulogne), Stéphanie Poulain (Lille), Anne Quinquenel (Reims), Daniel Ré (Antibes), Damien Roos-Weil (Paris), Laurence Simon (Corbeil Essonnes), Cécile Tomowiak (Poitiers), Olivier Tournilhac (Clermont-Ferrand), Xavier Troussard (Caen), Malgorzata Truchan-Graczyk (Saumur), Maud Voldoire (La Roche sur Yon), Lise Willems (Paris), Loïc Ysebaert (Toulouse), and Jean Marc Zini (Paris)
9. Moore J, Baran AM, Meacham PJ, Evans AG, Barr PM, Zent CS. Initial treatment of B-cell prolymphocytic leukemia with ibrutinib. Am J Hematol. 2020;95(5):E108-E110.
10. Xing L, He Q, Xie L, Wang H, Li Z. Zanubrutinib, rituximab and lenalidomide induces deep and durable remission in TP53mutated B-cell prolymphocytic leukemia: a case report and literature review. Haematologica. 2022;107(5):1226-1228.
11. Eyre TA, Fox CP, Shankara P, Went R, Schuh AH. Idelalisibrituximab induces clinical remissions in patients with TP53 disrupted B cell prolymphocytic leukaemia. Br J Haematol. 2017;177(3):486-491.
12. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720-1748.
13. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood. 2022;140(11):1229-1253.
14. Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745-2760.
FILO Recherche Clinique members: Laetitia Auvray, Alexandra Fayault, Lamya Haddaoui, Delphine Nollet and Valérie Rouille
Haematologica | 108 June 2023 1696 LETTER TO THE EDITOR
End-of-treatment PET in early-stage Hodgkin lymphoma: valuable in addition to interim PET
Classic Hodgkin lymphoma (HL) is a B-cell malignancy that is associated with high rates of cure with front-line therapy.1 Based on major clinical trials assessing treatment strategies adapted according to the findings of interim positron emission tomography (PET), the treatment paradigm for early-stage HL has evolved dramatically in the past decade.2-4 While clinicians commonly rely on interim-PET to make treatment escalation or de-escalation decisions, an area that remains less well investigated is the role of end-of-treatment (EOT)-PET. Literature has questioned the necessity of EOT imaging, especially in interim-PET-negative individuals, and clinical trials are lacking.5-7 There are limited real-world data assessing the prognostic value of EOT-PET and the importance of this scan in relation to interim-PET, especially in patients with early-stage disease. Thus, the primary objective of our study was to determine the utility of EOT-PET and its association with outcomes in early-stage HL. Our secondary objectives were to compare outcomes stratified by the interim-PET response and to assess treatment strategies used for EOT-positive disease. Consecutive adult patients ( ≥ 18 years old) with previously untreated early-stage (IA-IIB) HL evaluated at the Mayo Clinic in Rochester, Arizona, and Florida from January 1, 2010 to December 31, 2020 were retrospectively assessed. Patients with missing clinical data, or EOT-PET unavailable for radiological review were excluded. This study was approved by the Mayo Clinic Institutional Review Board. Patients’ data were collected through electronic chart review. Treatment modality was stratified into chemotherapy-alone or combined modality therapy. The number of treatment cycles was divided into four or fewer cycles, six cycles, and a novel consolidation group. EOT scans were identified as the first scan conducted after the completion of all frontline therapies (including consolidation therapies or radiotherapy). The standard at our institution is 6 weeks after chemotherapy completion, or 3 months after radiotherapy. Independent radiological review of PET2 scans (PET after 2 cycles of chemotherapy), end-of-chemotherapy (after the last chemotherapy cycle before radiotherapy), and EOT-PET scans was performed by a board-certified nuclear radiologist blinded to the patients’ treatment and outcomes. The Deauville score (DS) was calculated for all scans: a DS >3 was used to characterize EOT-positive disease. Primary study endpoints were progression-free survival (PFS) and overall survival (OS). Cox-proportional hazard models were used to determine hazard ratios (HR). Survival analyses were conducted using the Kaplan-Meier method. Time-to-event analyses were based on the date
of the PET scan to the date of the event. Progression events were determined from the date of a positive biopsy. A P value <0.05 was considered statistically significant. Analyses were conducted using SPSS 27, and BlueSky Ver 7.4.
Of 93 patients identified with early-stage HL (36 [39%] females, median age: 32 years [range, 18-78]), 83 (89%) patients were EOT-negative, and ten (11%) were EOTpositive. The patients’ baseline and treatment-related characteristics are displayed in Table 1. Ninety-two (99%) patients received ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) or AVD (doxorubicin, vinblastine, dacarbazine)-based regimens, and one received a front-line combination of brentuximab vedotin and nivolumab. Ten (11%) patients received novel agent consolidation with single-agent nivolumab or brentuximab vedotin after front-line chemotherapy as part of clinical trials. All patients receiving combined modality therapy (n=38) had a plan for radiotherapy determined at baseline.
Comparing PET2 results with EOT outcomes, patients who were EOT-positive had a greater degree of PET2positive disease (DS >3) compared to those who were EOT-negative (40% vs. 9%, P=0.006). Importantly, 60% of patients who were EOT-PET-positive were interim-PETnegative (DS ≤3). Among the patients receiving combined modality therapy (n=38), two (5%) were found to be EOTpositive. Prior to radiotherapy, 25 patients had an evaluable end-of-chemotherapy PET and three were found to be PET-positive, all of whom became negative on the EOT scan after radiotherapy. An end-of-chemotherapy scan was not available in the two patients who were positive at the EOT-PET after radiotherapy. No significant associations were observed between end-of-chemotherapy response (P=0.71) and EOT response in patients receiving combined modality therapy.
With a median follow-up of 48.1 months (95% confidence interval [95% CI]: 35.0-61.1), PFS was significantly reduced in patients who were EOT-positive compared to those who were EOT-negative (2-year PFS: 30% vs. 91%, respectively; P <0.001) (Figure 1). No significant association for OS was observed (P=0.34). Assessing hazard ratios, both PET2-positive (DS >3: HR=8.4 [95% CI: 2.9-24.3], P<0.001) and EOT-positive (DS >3: HR=15.1 [95% CI: 5.4-42.5], P<0.001) disease conferred an elevated risk of progression. Importantly, four (40%) of the ten patients with EOT-positive disease were found to have biopsy-proven progression despite a negative interim-PET. The inferior PFS associated with EOT-positive findings remained consistent regardless of the presence of disease bulk, or treatment modality. No PFS difference was
Haematologica | 108 June 2023 1697 LETTER TO THE EDITOR
found comparing patients who received novel consolidation (n=10) to those who did not (n=83) (P=0.17). Excluding patients who received novel consolidation, those with EOT-positive disease continued to have significantly reduced PFS (median 0.2 months) compared to those with EOT-negative disease (median not reached) (P<0.001). In the small number of patients with EOT-PET DS 3 (n=4), a 5-year OS of 100%, and 2-year PFS of 75% were observed (Figure 2).
Individual treatment outcomes for patients with EOTpositive disease are listed in Online Supplementary Table S1. Of the ten patients with EOT-positive imaging, eight (80%) underwent a biopsy either immediately or after follow-up scanning and all eight were found to have active disease. The remaining two (20%) were not biopsied; they
were monitored, and continued to remain in complete response at last follow-up. Biopsies were not performed in these patients because one had active diarrhea at the time of the scan as a possible explanation of new mesenteric lymph node uptake, and the other because of patient-provider preference with repeat scanning showing stability of lymph nodes. All of the eight patients with biopsy-proven disease had residual disease identified on EOT-PET in an initial site of HL involvement. All eight patients with active disease proceeded to salvage therapy, with two (25%) receiving salvage radiotherapy due to localized disease, and six (75%) receiving systemic salvage therapy with autologous stem cell transplant due to more diffuse involvement. After autologous stem cell transplantation, only one patient progressed to needing
CMR: combined modality therapy; PET2: positron emission tomography scan after two cycles of chemotherapy; DS: Deauville score; HL: Hodgkin lymphoma. Statistically significant differences are shown in bold.
Table 1. Patients’ baseline characteristics and differences between those positive and negative at the end-of-treatment positron emission tomography.
Characteristic Whole cohort (N=93) EOT-PET negative (N=83) EOT-PET positive (N=10) P Age, years, median (range) 32 (18-78) 32 (18-78) 32 (22-49) 1.00 Female sex, N (%) 36 (38.7) 33 (39.8) 3 (30.0) 0.55 Histology, N (%) Nodular sclerosis 59 (63.4) 53 (63.9) 6 (60.0) 0.71 Mixed cellularity 4 (4.3) 3 (3.6) 1 (10.0) Lymphocyte-rich 4 (4.3) 4 (4.8) 0 Classic 26 (28.0) 23 (27.7) 3 (30.0) Stage, N (%) IA 2 (2.2) 2 (2.4) 0 0.74 IB 0 0 0 IIA 62 (66.7) 56 (67.5) 6 (60.0) IIB 29 (31.2) 25 (30.1) 4 (40.0) Prognostic factor, N (%) Unfavorable disease 62 (66.7) 55 (69.6) 7 (87.5) 0.29 Favorable disease 25 (26.9) 24 (30.4) 1 (12.5) Bulky disease (≥7 cm diameter), N (%) 35 (37.6) 30 (36.1) 5 (50.0) 0.39 Treatment strategy, N (%) CMT 38 (40.9) 36 (43.4) 2 (20.0) 0.16 Chemotherapy alone 55 (59.1) 47 (56.6) 8 (80.0) Treatment cycles, N (%) ≤ 4 cycles 42 (45.2) 37 (44.6) 5 (50.0) 0.51 6 cycles 41 (44.1) 36 (43.4) 5 (50.0) Novel consolidation 10 (10.8) 10 (12.0) 0 PET2 response, N (%) Negative (DS ≤2) 70 (81.4) 65 (85.5) 5 (50.0) 0.007 Positive (DS ≥3) 16 (18.6) 11 (14.5) 5 (50.0) Negative (DS ≤3) 75 (87.2) 69 (90.8) 6 (60.0) 0.006 Positive (DS >3) 11 (12.8) 7 (9.2) 4 (40.0) Survival outcomes, N (%) Relapsed/refractory disease 15 (16.2) Death 4 (4.3) HL-related death 2 (2.2) Haematologica | 108 June 2023 1698 LETTER TO THE EDITOR
Figure 1. Association of end-of-treatment positron emission tomography findings with overall survival and progression-free survival for the whole cohort. (A) Overall survival. (B) Progression-free survival. N: number of patients; mOS: median overall survival; EOT: end of treatment; pos: positive; neg: negative; PFS progression-free survival; mPFS: median PFS; NR: not reached The 95% confidence interval is indicated in brackets after the median time.
Figure 2. Association of end-of-treatment positron emission tomography findings with overall survival and progression-free survival for the whole cohort and stratified by Deauville score. (A) Overall survival. (B) Progression-free survival. N: number of patients; mOS: median overall survival; DS: Deauville score; PFS progression-free survival; NR: not reached; mPFS: median PFS. The 95% confidence interval is indicated in brackets after the median time.


an allogeneic transplant. At last follow-up, among the eight patients found to have active disease, six patients remained in complete response, one had died due to graft- versus -host disease after allogeneic stem cell transplantation, and one continued to undergo treatment.
Most recent trials have not utilized EOT-PET as an endpoint, and observational literature largely predates the standardized evaluation using the DS.5,6,8,9 In the present study, both interim-positive and EOT-positive PET strongly predicted PFS, with significant associations between both results. Despite the strong prognostic value of the interim scan reported in previous trials and real-
world studies, a considerable fraction of patients who were EOT-positive with active disease had an interimnegative scan. 2-4,10 Previously, literature has pointed to EOT-PET having an improved sensitivity over interim-PET due to a potential tumor stunning effect early in treatment, and refractory clones showing initial response but late resurgence during the course of treatment.8 A previous assessment of 76 patients with HL of all stages found, similarly to our study, that both interim-PET-positive (HR 3.79 [95% CI: 1.37-10.49]) and EOT-PET-positive (≥3) (HR 24.02 [95% CI: 6.59-87.47]) disease were associated with reduced PFS.11 Certainly, important financial and resource implications need to be considered; how-
A B Haematologica | 108 June 2023 1699 LETTER TO THE EDITOR A B
ever, our data suggest that in clinical practice, EOT is useful. Suspicion of progression should remain high for those who are EOT-positive regardless of interim-PET results.
Non-specific findings are frequently found on PET scanning, and there are a host of other benign etiologies that can appear like active lymphoma.7,12 Similar to the findings in our study, a systematic review and meta-analysis found a false-positive rate of 23.1% (95% CI: 4.7%-64.5%) for EOT-PET in HL with the majority of these false-positive cases being due to inflammatory changes.12 In clinical practice, the decision to observe, biopsy, administer radiotherapy or begin salvage therapy can be exceedingly difficult. In our study, we found that most patients with EOT-positive disease ended up requiring a biopsy, even if initially observed. In those found to have progression, most patients were successfully salvaged with either high-dose chemotherapy and autologous stem cell transplantation or salvage radiotherapy due to localized disease in a small number of patients. Certainly, a riskbenefit relationship exists in obtaining a biopsy. Our cohort suggests that a tissue sample should be obtained soon after an EOT-positive scan, unless there is convincing evidence of another ongoing non-malignant process. The strengths of this study include the blinded uniform review of all PET scans, and granularity regarding outcomes and treatment. Limitations include the retrospective methodology and the associated biases of this design. Due to these, we had incomplete data and imaging on patients also followed outside our institution and were unable to determine exactly the underlying reasons for treatment approaches taken for EOT-positive disease. Additionally, even in assessing a 10-year cohort, as most patients with early-stage HL achieve a complete response in the front-line setting, conclusions were drawn using a small number of patients.
In conclusion, despite recent literature demonstrating the significant prognostic and treatment-related implications of the interim-PET scan, EOT-PET still adds value. With confirmatory biopsy and timely treatment initiation, most patients with EOT-positive disease and biopsyproven progression can be successively salvaged and have a comparable OS outcome to those with EOTnegative findings. Overall, in early-stage HL, EOT-PET is important for identifying patients with relapsed or refractory disease and is necessary even for those with interim-PET-negative responses.
References
1. Brockelmann PJ, Sasse S, Engert A. Balancing risk and benefit in early-stage classical Hodgkin lymphoma. Blood. 2018;131(15):1666-1678.
2. Johnson P, Federico M, Kirkwood A, et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin's
Authors
Karan L. Chohan,1 Jason R. Young,2 Scott Lester,3 Muhamad Alhaj Moustafa,4 Allison Rosenthal,5 Han W. Tun,4 Bradford S. Hoppe,6 Patrick B. Johnston,7 Ivana N. Micallef,7 Thomas M. Habermann7 and Stephen M. Ansell7
1Department of Medicine, Mayo Clinic, Rochester, MN; 2Department of Radiology, Mayo Clinic, Jacksonville, FL; 3Department of Radiation Oncology, Mayo Clinic, Rochester, MN; 4Division of Hematology and Medical Oncology, Mayo Clinic, Jacksonville, FL; 5Division of Hematology, Mayo Clinic, Phoenix, AZ; 6Department of Radiation Oncology, Mayo Clinic, Jacksonville, FL and 7Division of Hematology, Mayo Clinic, Rochester, MN, USA
Correspondence: STEPHEN M. ANSELL - ansell.stephen@mayo.edu
https://doi.org/10.3324/haematol.2022.282115
Received: September 16, 2022.
Accepted: December 14, 2022. Early view: December 22, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
HWT serves in a consulting/advisory role for Acrotech, Gossamerbio, and ADC Therapeutics, and receives research funding from Acrotech. TMH serves on scientific advisory boards for Eli Lilly & Co., Morphosys, Incyte, Biegene, and Loxo Oncology, has received research funding from Genentech, and serves on the data monitoring committee for Seagen, and Tess Therapeutics. SMA receives research funding from Bristol-Myers Squibb, Seattle Genetics, Affimed Therapeutics, Regeneron, Trillium Therapeutics, AI Therapeutics, and ADC Therapeutics. KLC, JRY, SL, MAM, AR, BSH, PBJ, and INM, have no conflicts of interest to disclose.
Contributions
KLC, JRY, and SMA designed this study, analyzed and interpreted the data, and wrote the manuscript. SL, MAM, AR, HWT, BSH, PBJ, INM, and TMH interpreted the data and assisted in writing the manuscript. All authors provided final approval of the manuscript and are accountable for all aspects of the work.
Acknowledgments
We would like to acknowledge the University of Iowa/Mayo Clinic Lymphoma SPORE CA97274-19 for support of this study.
Data-sharing statement:
The data presented in this study are not available for sharing.
lymphoma. N Engl J Med. 2016;374(25):2419-2429.
3. Andre MPE, Girinsky T, Federico M, et al. Early positron emission tomography response-adapted treatment in stage I and II Hodgkin lymphoma: final results of the randomized EORTC/LYSA/FIL H10 trial. J Clin Oncol. 2017;35(16):1786-1794.
Haematologica | 108 June 2023 1700 LETTER TO THE EDITOR
4. Straus DJ, Jung SH, Pitcher B, et al. CALGB 50604: risk-adapted treatment of nonbulky early-stage Hodgkin lymphoma based on interim PET. Blood. 2018;132(10):1013-1021.
5. Strobel K, Schaefer NG, Renner C, et al. Cost-effective therapy remission assessment in lymphoma patients using 2-[fluorine18]fluoro-2-deoxy-D-glucose-positron emission tomography/computed tomography: is an end of treatment exam necessary in all patients? Ann Oncol. 2007;18(4):658-664.
6. Adams HJA, Kwee TC. Systematic review on the value of endof-treatment FDG-PET in improving overall survival of lymphoma patients. Ann Hematol. 2020;99(1):1-5.
7. London J, Grados A, Ferme C, et al. Sarcoidosis occurring after lymphoma: report of 14 patients and review of the literature. Medicine (Baltimore). 2014;93(21):e121.
8. Hindié E, Mesguich C, Bouabdallah K, Milpied N. Advanced Hodgkin’s lymphoma: end-of-treatment FDG-PET should be
maintained. Eur J Nucl Med Mol Imaging. 2017;44(8):1254-1257.
9. Barnes JA, LaCasce AS, Zukotynski K, et al. End-of-treatment but not interim PET scan predicts outcome in nonbulky limited-stage Hodgkin's lymphoma. Ann Oncol. 2011;22(4):910-915.
10. Chohan KL, Young JR, Lester SC, et al. A real-world study of combined modality therapy for early-stage Hodgkin lymphoma: too little treatment impacts outcome. Blood Adv. 2022;6(14):4241-4250.
11. Mesguich C, Cazeau AL, Bouabdallah K, et al. Hodgkin lymphoma: a negative interim-PET cannot circumvent the need for end-of-treatment-PET evaluation. Br J Haematol. 2016;175(4):652-660.
12. Adams HJA, Kwee TC. Proportion of false-positive lesions at interim and end-of-treatment FDG-PET in lymphoma as determined by histology: systematic review and meta-analysis. Eur J Radiol. 2016;85(11):1963-1970.
Haematologica | 108 June 2023 1701 LETTER TO THE EDITOR
Sirolimus as frontline therapy for PTEN-mutated histiocytic sarcoma

Histiocytic sarcoma (HS) is an extremely rare non-Langerhans histiocytic neoplasm that can present as unifocal or multifocal extranodal disease.1 The limited reports available for this malignancy have estimated a median overall survival of 6 months, with an especially poor prognosis for multifocal disease.2 Given the rarity and lack of prospective trials, there are no established standard-of-care therapies. In multifocal disease, previous reports have utilized regimens typically administered for aggressive lymphomas, such as ICE (ifosfamide, carboplatin, and etoposide) and CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone).3 However, these combination chemotherapy regimens have limited efficacy and are associated with significant hematologic toxicity. Herein, we present a case of HS complicated by severe cytopenias achieving objective disease response and prolonged survival with frontline sirolimus.
A 63-year-old woman presented with bilateral leg edema, fatigue, and weight loss. Laboratory workup demonstrated macrocytic anemia (hemoglobin [Hb]: 7.7 g/dL [normal, 11.6-15.0 g/dL], mean corpuscular volume [MCV]: 120.4 fL [normal, 78.2-97.9 fL]), severe thrombocytopenia (platelet count: 11x109/L [normal, 157-371x109/L]), and borderline leukopenia (white blood cell [WBC] count: 3.5 x109/L [normal, 3.4-9.6x109/L]). The absolute neutrophil count (ANC) was 2.0x109/L (normal, 1.56-6.45 x109/L), with some elev-
ation in early myeloid progenitor cells (metamyelocytes 1% [normal, <1%]; myelocyte 2% [normal, <0.5%]). Folic acid: 9.7 mcg/L (normal, ≥4 mcg/L), vitamin B12: 218 ng/L (normal, 180-914 ng/L), and methylmalonic acid: 0.15 nmol/mL (normal, ≤0.40 nmol/mL) levels were determined to be within normal limits. Bone marrow (BM) biopsy initially revealed normal cytogenetics, with no features of myelodysplastic syndrome or neoplasia. Concurrently, during routine cardiac workup, the patient was noted to have lymphadenopathy on computed tomography (CT). This prompted positron emission tomography (PET)-CT which revealed multiple F-18 fluorodeoxyglucose (FDG) avid thoracic and abdominal lymph nodes, an enlarged spleen (largest diameter: 18.7 cm) with innumerable FDG-avid foci, and diffuse increased marrow FDG uptake (Figure 1A). In the weeks following presentation, the patient developed significant transfusion dependence requiring weekly packed red blood cells (pRBC) and platelets. Due to minimal improvement in the platelet count, pancytopenia of undetermined origin, and splenomegaly with hypersplenism, the patient underwent splenectomy with liver biopsy. Biopsies from the liver and spleen showed diffuse involvement by overtly malignant cells characterized by marked nuclear pleomorphism with occasional multinucleation, and abundant pale eosinophilic cytoplasm. Immunohistochemistry (IHC) showed the malignant cells had a histio-
A B
Figure 1. Maximum intensity projection images from F-18 fluorodeoxyglucose positron emission tomography. (A) Time of diagnosis and (B) 12 months after therapy demonstrating complete resolution of F-18 fluorodeoxyglucose avid lymph nodes (arrows) and splenic disease (bracket) post-splenectomy. Incidental biopsyproven benign right thyroid nodule noted (circle).
Haematologica | 108 June 2023 1702 CASE REPORT
cyte/macrophage phenotype with expression of CD4, CD11c (partial, i.e., 50% of tumor cells positive), CD14 (partial), CD68, CD163 (partial), cyclin D1, lysozyme (partial), and S100 (focal) (Figure 2). Additional markers were negative including CD1a and langerin. Thus, a diagnosis of multifocal HS involving the spleen, liver, and lymph nodes was made. The initial BM biopsy was re-reviewed and revealed 5-10% involvement by HS without features of an underlying myelodysplastic/myeloproliferative neoplasm. Subsequently, tissue-based multigene next-generation sequencing (NGS) (Tempus xT assay) performed on the splenic specimen demonstrated the following mutations: PTEN p.K223fs frameshift-loss of function (LOF), variant allele frequency (VAF) 13.9%; SETD2 p.V2108fs frameshiftLOF, VAF 14.7%; FGFR3 c.2131C>T p.H711Y missense variant, VAF 13.5%. Given the mutation involving the PTEN gene, additional immunostains performed for phosphor-AKT (pAKT) and phospho-ERK (p-ERK) revealed moderate (2+) p-AKT positivity and weak (1+) p-ERK expression in the tumor cells.

Due to underlying cardiac comorbidities and severe transfusion-dependent cytopenias, the patient was not a can-
didate for aggressive combination chemotherapy. Given the PTEN LOF and the role of this gene in the PI3k-AKTmTOR pathway, the patient was initiated on sirolimus 2 mg with a goal trough of 8-12 ng/mL and prednisone 1 mg/kg with a tapering dose over 3 months.4 During therapy, the patient had an objective clinical and radiological response. A repeat staging PET-CT demonstrated a favorable partial anatomic and metabolic response at 3 months with a complete anatomic and metabolic response after 12 months of therapy (Figure 1B). Interestingly, a repeat BM biopsy after 12 months of therapy demonstrated stable 5-10% involvement of HS. The patient experienced a drastic reduction in the requirement for both pRBC and platelet transfusions while on therapy (Figure 3). Unfortunately, despite the initial clinical improvement, she developed grade 3 anemia and thrombocytopenia after 13 months of therapy, requiring weekly transfusions (Figure 3). Out of concern for its potential myelosuppressive effect, sirolimus was stopped after 15 months of therapy. Shortly after, the patient presented with septic shock, anemia, and severe thrombocytopenia, which was transfusion refractory. Diagnostic imaging at the time revealed
Figure 2. Light microscopy images. Spleen showing involvement by large pleomorphic cells (hematoxylin and eosin stain - top left) with positive staining for CD11c (partial), CD163, and CD68 (magnification, X400).
Haematologica | 108 June 2023 1703 CASE REPORT
extensive fluid overload with diffuse anasarca. The patient’s condition rapidly deteriorated, and she ultimately chose to pursue comfort care measures and died 1 month after discontinuation of sirolimus, surviving 19 months from initial diagnosis. A final autopsy was not done per the patient’s preference.
The clinical course of multifocal HS is typically highly aggressive. Due to its rarity, there is a lack of established therapies, and multiple chemotherapeutic agents have been utilized in case reports with limited efficacy. Moreover, the administration of combination chemotherapy can be challenging when significant cytopenias and comorbidities are present. In patients with targetable mutations, BRAF and mitogen-activated protein kinase (MEK) inhibitor therapies with agents such as dabrafenib, vemurafenib and trametinib have also been previously utilized.5-7 Through genomic analysis, multiple genetic alterations have been implicated in HS. Previously, in a study of 21 patients with HS, whole-exome sequencing demonstrated a high prevalence of genetic alterations in the MAPK-ERK pathway.8 Additionally, the authors found that 19% of patients had mutations in the PI3k-AKTmTOR pathway. In another study of 28 patients with HS, targeted NGS revealed a MAPK-ERK pathway mutation in the majority of patients (57%), with a subset (21%) having mutations of the PI3k-AKT-mTOR signaling pathway. 9 These case series suggest possible distinct molecular subtypes of HS, allowing for potentially tailored targeted treatment strategies.

PTEN, the gene found to be implicated in our report, acts
as a tumor suppressor and negatively regulates the PI3kAKT-mTOR pathway. The positive p-AKT testing further indicated the PTEN LOF led to downstream activation of this pathway. The mammalian target of rapamycin (mTOR) pathway is involved in regulating cell growth, proliferation, apoptosis, and metabolic processes by integrating extracellular and intracellular signals.10 Previously, somatic PTEN alterations have been shown to be implicated in HS and other hematological malignancies, where they have been hypothesized to contribute to tumor initiation and progression.11,12 The PTEN LOF in our case indicated a potential benefit from mTOR inhibitor therapy.13 Sirolimus is a potent macrocyclic lactone immunosuppressant inhibitor of the mTOR signaling pathway, with antiproliferative and immunosuppressive properties.14 Previously, in a study of patients with Erdheim-Chester disease (ECD), another rare non-Langerhans histiocytic neoplasm, an open-label trial assessed sirolimus (target levels 8-12 ng/mL) in combination with prednisone.4 Among the ten patients enrolled, eight had an objective response or disease stabilization. Upon comprehensive literature review, we did not find any reports of sirolimus used to treat primary HS; however, mTOR-directed therapy (1 dose of temsirolimus and daily oral sirolimus) led to symptomatic and radiological improvement in an 18-month-old boy with recurrent mTOR-mutated secondary HS and history of T-cell acute lymphoblastic leukemia.6 We hypothesize that in our case, the objective response observed was likely secondary to targeted inhibition of the driver mutation with sirolimus, along with the immunosuppressive and anti-
Figure 3. Transfusion requirement of packed red blood cells and platelets relative to the duration of therapy.
Haematologica | 108 June 2023 1704 CASE REPORT
proliferative properties of mTOR inhibition. Despite the initial improvement in transfusion dependence in our case, the eventual worsening of cytopenias and transfusion burden led to the discontinuation of sirolimus due to concern for BM suppression. Sirolimus-related adverse events include thrombocytopenia, leukopenia, and hyperlipidemia, and previous cohorts have demonstrated these toxicities are generally self-limited.15 In the trial of patients with ECD, mild toxicity was observed, with several patients having cushingoid changes, hypercholesterolemia and hypertriglyceridemia; however, the authors did not report significant cytopenias.4 The cause of the worsening cytopenias in our case is unclear, as there was no significant improvement after sirolimus cessation. Given the initial improvement after sirolimus initiation, and then worsening of transfusion dependence, it raises the question of whether progressive malignancy contributed to BM suppression as opposed to sirolimus toxicity. It is possible that the disease ultimately progressed due to the contribution of other mutations like SETD2 through an escape mechanism.
To the best of our knowledge, this is the first report of treatment of primary HS with sirolimus. HS is an exceedingly rare histiocytic neoplasm with a paucity of literature on optimal therapeutic management. Our case highlights that a patient with PTEN-mutated HS achieved an over 1year objective response to the mTOR inhibitor sirolimus. With the PI3k-AKT-mTOR pathway being frequently implicated in patients with HS, further investigation is certainly needed to assess the role of these agents.
Authors
Karan L. Chohan,1 Jithma P. Abeykoon,2 Jason R. Young,3 W. Oliver Tobin,4 Mathew J. Koster,5 Mithun V. Shah,2 Jay H. Ryu,6 Robert Vassallo,6 Karen L. Rech,7 Aishwarya Ravindran,8 Gaurav Goyal,9 Ronald S. Go2 and N. Nora Bennani2
1Department of Medicine, Mayo Clinic, Rochester, MN; 2Division of
References
1. Skala SL, Lucas DR, Dewar R. Histiocytic sarcoma: review, discussion of transformation from B-cell lymphoma, and differential diagnosis. Arch Pathol Lab Med. 2018;142(11):1322-1329.
2. Kommalapati A, Tella SH, Durkin M, et al. Histiocytic sarcoma: a population-based analysis of incidence, demographic disparities, and long-term outcomes. Blood. 2018;131(2):265-268.
3. Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic
Hematology, Mayo Clinic, Rochester, MN; 3Department of Radiology, Mayo Clinic, Jacksonville, FL; 4Department of Neurology, Mayo Clinic, Rochester, MN; 5Division of Rheumatology, Mayo Clinic, Rochester, MN; 6Division of Pulmonary and Critical Care Medicine, Mayo Clinic, Rochester, MN; 7Division of Hematopathology, Mayo Clinic, Rochester, MN; 8Laboratory of Medicine-Hematopathology, Department of Pathology, University of Alabama, Birmingham, AL and 9Division of Hematology-Oncology, University of Alabama, Birmingham, AL, USA
Correspondence: N.N. BENNANI - bennani.nora@mayo.edu
https://doi.org/10.3324/haematol.2022.282207
Received: October 1, 2022.
Accepted: December 1, 2022.
Prepublished: December 7, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
KLC, GG and NNB conceived, wrote the first draft and modified the final draft; JPA, JRY, WOT, MJK, MVS, JHR, RV, KLR, AR and RSG critically appraised the manuscript and approved the final draft of the manuscript.
Funding
NNB gratefully recognizes support from the K12 Paul Calabresi Program in Clinical/Translational Research at Mayo Clinic (award N 2K12CA090628-21). NNB, JPA, and GG are supported in part by the University of Iowa/Mayo Clinic Lymphoma SPORE CA97422. KLR is supported by the Department of Laboratory Medicine and Pathology and GG is supported by the Walter B. Frommeyer, Jr., Fellowship Award in Investigative Medicine, University of Alabama at Birmingham.
Data-sharing statement
There is no relevant data to disclose.
sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol. 2004;28(9):1133-1144.
4. Gianfreda D, Nicastro M, Galetti M, et al. Sirolimus plus prednisone for Erdheim-Chester disease: an open-label trial. Blood. 2015;126(10):1163-1171.
5. Idbaih A, Mokhtari K, Emile JF, et al. Dramatic response of a BRAF V600E-mutated primary CNS histiocytic sarcoma to vemurafenib. Neurology. 2014;83(16):1478-1480.
6. Massoth LR, Hung YP, Ferry JA, et al. Histiocytic and dendritic
Haematologica | 108 June 2023 1705 CASE REPORT
cell sarcomas of hematopoietic origin share targetable genomic alterations distinct from follicular dendritic cell sarcoma. Oncologist. 2021;26(7):e1263-e1272.
7. Gounder MM, Solit DB, Tap WD. Trametinib in histiocytic sarcoma with an activating MAP2K1 (MEK1) mutation. N Engl J Med. 2018;378(20):1945-1947.
8. Egan C, Nicolae A, Lack J, et al. Genomic profiling of primary histiocytic sarcoma reveals two molecular subgroups. Haematologica. 2020;105(4):951-960.
9. Shanmugam V, Griffin GK, Jacobsen ED, et al. Identification of diverse activating mutations of the RAS-MAPK pathway in histiocytic sarcoma. Mod Pathol. 2019;32(6):830-843.
10. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361-371.
11. Carrasco DR, Fenton T, Sukhdeo K, et al. The PTEN and
INK4A/ARF tumor suppressors maintain myelolymphoid homeostasis and cooperate to constrain histiocytic sarcoma development in humans. Cancer Cell. 2006;9(5):379-390.
12. Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27(41):5443-5453.
13. Komiya T, Blumenthal GM, DeChowdhury R, et al. A pilot study of Sirolimus in subjects with Cowden syndrome or other syndromes characterized by germline mutations in PTEN. Oncologist. 2019;24(12):1510-e1265.
14. Kahan BD. Sirolimus: a comprehensive review. Expert Opin Pharmacother. 2001;2(11):1903-1917.
15. Hong JC, Kahan BD. Sirolimus-induced thrombocytopenia and leukopenia in renal transplant recipients: risk factors, incidence, progression, and management. Transplantation. 2000;69(10):2085-2090.
Haematologica | 108 June 2023 1706 CASE REPORT
Clinical response to dabrafenib and chemotherapy in clonally-related histiocytosis and acute lymphoblastic leukemia
Histiocytoses encompass a heterogeneous group of disorders characterized by tissue infiltration of cells with morphological and phenotypic features of macrophages or dendritic cells, which have been reclassified into five groups: i) L group - Langerhans cell histiocytosis (LCH)/Erdheim Chester disease (ECD); ii) C group - cutaneous histiocytoses; iii) M group - malignant histiocytoses; iv) R group - Rosai-Dorfman disease and v) H group - hemophagocytic lymphohistiocytosis (HLH).1 Histiocytoses rarely occur during acute lymphoblastic leukemia (ALL) treatment, potentially due to trans-differentiation2 or a common progenitor cell,3 and there is no standard treatment in this particular situation. We herein report a case of BRAF-mutated non-LCH arising during T-ALL therapy who responded to dabrafenib and chemotherapy combination. Our patient was a 7-year-old boy diagnosed in March 2020 with central nervous system (CNS)-positive T-ALL harboring the oncogenic STIL-TAL1 fusion. He initially presented with right facial nerve palsy and hyperleukocytosis with an initial white blood cell count at 205x109/L. He received fourdrug induction chemotherapy, achieved morphologic remission with positive end-induction minimal residual disease (MRD) by flow cytometry. He then received postinduction therapy according to Arm D of AALL0434 protocol,4 with a negative flow-based end-consolidation MRD. In October 2020, during delayed intensification (DI), he developed persistent thrombocytopenia refractory to corticosteroids and intravenous immunoglobulins. Extensive investigation for refractory thrombocytopenia came back negative. However, a positron emission tomography (PET) scan showed hypermetabolic focal lesions in the mediastinum, 5th right rib and right tibial tuberosity. In December 2020, 7 months from T-ALL diagnosis, biopsy of the rib lesion revealed proliferation of multinucleated giant cells with emperipolesis that were CD68+, CD163+, S100+, fascin+, lysozyme+ and BRAF+, suggestive of Rosai-Dorfman disease (RDD). Whole-transcriptome analysis of the rib lesion revealed a BRAF V600E mutation and the STIL-TAL1 fusion present at T-ALL diagnosis, suggesting a common clonal origin. Since RDD and T-ALL were clonally-related, leukemia treatment was prioritized and our patient pursued DI and maintenance therapy, including cranial irradiation. A follow-up PET scan in March 2021 showed histiocytosis progression despite ALL-based chemotherapy, which provided the rationale to introduce a BRAF inhibitor. Considering pre-existing transaminitis and thrombocytopenia, ALL maintenance chemotherapy was stopped and da-
brafenib monotherapy at 5.25 mg/kg/day was initially started in April 2021, with a rapid metabolic response 1 month post-dabrafenib. In order to pursue T-ALL therapy, low-dose ALL maintenance chemotherapy was combined with dabrafenib in June 2021 and titrated based on patient’s tolerance (monthly vincristine 1.5 mg/m2/dose, prednisone 20 mg/m2/dose twice a day for 5 days every month, daily 6-mercaptopurine 20 mg/m2/dose, weekly methotrexate was omitted because of thrombocytopenia). Combination of dabrafenib and chemotherapy was well-tolerated. Unfortunately, the patient experienced an isolated CNS relapse in September 2021, 17 months from T-ALL diagnosis and 9 months from onset of histiocytosis. Dabrafenib was stopped at the time of relapse to begin ALL reinduction chemotherapy. Of note, thrombocytopenia <50x109/L without clinically active bleeding persisted from October 2020 to September 2021. PET scans prior to relapse showed progressive hypermetabolic uptake in the liver. A liver biopsy was inconclusive for etiology. Relapse was treated with intrathecal chemotherapy and daratumumab, to provide systemic therapy and potentially address his refractory thrombocytopenia,5 followed by two cycles of the NECTAR regimen.6 After a conditioning regimen with VP-16, anti-thymocyte globulin and total body irradiation, he proceeded to a matched-sibling donor hematopoietic stem cell transplantation (HSCT) in December 2021. At the time of this report, the patient is 7 months post-HSCT without evidence of T-ALL and histiocytosis.
Histiocytoses arising during ALL therapy are exceedingly rare, although they can also occur at diagnosis or following treatment completion. Our case is unique in several ways and expands the paradigm of molecularly-targeted therapies in histiocytic neoplasms. First, we report a rapid metabolic response in BRAF-mutated histiocytic lesions refractory to conventional chemotherapy after only 1 month of dabrafenib monotherapy. Donadieu et al 7 previously reported rapid response within 2 months of vemurafenib in children with BRAF V600E-mutated refractory LCH. Given the co-existence of clonally-related BRAF-mutated RDD and T-ALL, ALL-directed therapy was prioritized prior to histiocytosis treatment. However, since RDD lesions were refractory to conventional chemotherapy, we report the feasibility of combining dabrafenib and lowdose maintenance ALL therapy to treat both diseases simultaneously. Combination of dabrafenib and chemotherapy was well-tolerated, without worsening pre-existing hematologic and hepatic toxicities. Although this
Haematologica | 108 June 2023 1707 CASE REPORT
Table 1. Clinical characteristics and outcome of children with co-occurrence of histiocytic
group disorders.
Continued on following page. Article Journal Age a (yr) Sex ALL immuno- phenotype Histiocytoses Organ involved Delay b (mth) ALL treatment phase BRAF mutation Proven clonal relationship c Treatment Outcome Alten 2015 Pediatric Blood & Cancer 6 M T HS (+HLH) na 12 Maintenance No Yes Treatment for secondary HLH (DEX, VP16, ATG, Basiliximab) DOH Aparicio 2008 Pediatric Dermatology 3 M B JXG Skin 6 na na na None DOL (relapse) Bleeke 2019 Pediatric Blood & Cancer 11 na B/myeloid MPAL HS Liver, spleen 5 na No Yes na DOH Cheon 2017 Pediatric and Developmental Pathology 16 M B JXG Skin, bone, bone marrow 4 Interim Maintenance na na ALL treatment continued Alive, RH and RL, 14 mth Chiles 2001 J Am Acad Dermatol 5 M T LCH Skin then bone marrow/pleura 7 Maintenance na na PRED,VBL,VP16/topical nitrogen mustard DOH Egeler 1998 Hematology/ Oncology Clinics of North America 3 M na LCH na 12 Maintenance* na na Chemotherapy NOSRadiotherapy Alive, RL but not RH, 2 yrs Egeler 1998 Hematology/ Oncology Clinics of North America 6 M na LCH na 6 na na na Chemotherapy NOS Alive, RH and RL, 2 yrs Egeler 1998 Hematology/ Oncology Clinics of North America 4 F na LCH na 12 Maintenance na na Chemotherapy NOS DOH Egeler 1998 Hematology/ Oncology Clinics of North America 10 M na LCH na 12 Maintenance na na Chemotherapy NOS DOH Egeler 1998 Hematology/ Oncology Clinics of North America 3 M na LCH na 12 Maintenance na na Chemotherapy NOS DOH Egeler 1998 Hematology/ Oncology Clinics of North America 9 M na LCH na 6 na na na Surgery DOL (relapse) Egeler 1998 Hematology/ Oncology Clinics of North America 13 M na LCH na 6 na na na Chemotherapy NOS Alive, RL but not RH, 6 mth Feldman 2004 Lancet Oncology 14 M B HS Spleen, kidney, bone 21 Maintenance na Yes VCR, CPM, DAUNO, MTX, VP16, CYTA, PRED then HSCT Alive, RL, 10 mth Ganapula 2014 Indian J Hematol Blood Transfus 4 M T HS Pleura, bone 18 Maintenance na na None Died NOS Jansen 2020 Pediatric Blood & Cancer 4 F T LCH Bone then pleura, digestive tract, pancreas, kidney 6 Maintenance No na ALL treatment continued then LCH-IV protocol (PRED, VBL) then Clofarabine DOH
neoplasms
leukemia therapy
H
Haematologica | 108 June 2023 1708 CASE REPORT
during acute lymphoblastic
excluding
anti-thymocyte globulin; CARBO: carboplatin; CPM: cyclophosphamide ;
and/or same mutation identi fi ed a t ALL diagnosis. ALL: acute lymphoblastic leukemia;
CSA: cyclosporin; CYTA: cytarabine; DAUNO: daunorubicin; DEXA: dexamethasone; DOH: died of histiocytosis; DOL: died of leukemia; E
CD: Erdheim-Chester disease; HLH: hemophagocytic lymphohistiocytosis; HS: histiocytic sarcoma; HSCT: hematopoietic stem cell transplantation; IFO: ifosfamide; JXG: juvenile xanthogr anuloma; LCH: Langerhans cell histiocytosis; LS: Langerhans sarcoma; MTX: methotrexate; na: not available; NOS: not otherwise speci fi ed; PRED: pr ednisone; RH: remission of histiocytosis; RL: remission of leukemia; VBL: vinblastine; VCR: vincristine; THL : true histiocytic lymphoma.
Article Journal Age a (yr) Sex ALL immuno- phenotype Histiocytoses Organ involved Delay b (mth) ALL treatment phase BRAF mutation Proven clonal relationship c Treatment Outcome Kanter 1976 Oral Surg 3 M na LCH Bone 4 na na na ALL treatment continued, surgery, CPM DOH (concomittant ALL relapse) Kato 2015 British Journal of Hematology 8 F T LCH Skin, lungs 8 Maintenance No Yes na Died NOS Kumar 2011 Pediatric Blood & Cancer 4 M B HS Bone 7 Maintenance na Yes DEXA, CPM, MTX, IFO, CYTA, VP16 then palliative radiation therapy DOH Onciu 2004 Am J Clin Pathol 13 M B HS Spleen 3 na na Yes Surgery na Pastor Jané 2011 Am J Dermatopathol 18 M B Indeterminate Skin, bone, bone marrow, liver, spleen 3 Consolidation na na ALL treatment continued then CYTA and Cladribine Died of infection, histiocytosis not in remission Pawi ń skaWa sikowska 2020 Frontiers in Oncology 15 M B JXG (+ HLH) Bone, skin 3 Consolidation na Yes HLH2004 (PRED, VP16, CSA) then Tocilizumab and HSCT Alive, RH and RL, 2 1/2 yrs Perez Becker 2010 Pediatric Blood & Cancer 5 F T JXG (atypical) Nodes then liver, kidney, lungs, digestive tract 5 na na Yes ALL treatment continued then LCH-III protocol (MTX, VBL, PRED) DOH Rodig 2008 American Journal of Hematology 3 F T LCH (then LS) Skin 18 Maintenance na Yes na Died of infection, histiocytosis not in remission Soslow 1996 Blood 8 M B THL Paraspinal mass 10 Maintenance na na VP16, PRED DOH Soslow 1996 Blood 6 M na THL Bone 20 Maintenance na na VP16, IFO, CARBO Alive, RL but not RH, 16 mth Vallonthaiel 2016 World Journal of Radiology 6 F B ECD Bone 24 Maintenance No na None Alive, RL but not RH Venkataraman 2020 Pediatric Blood & Cancer 0,5 M T HS Temporal mass 14 Maintenance Yes Yes Targeted therapy (dabrafenib, trametinib) Alive, RH and RL, 14 mth Wang 2021 European Journal of Nuclear Medicine and Molecular Imaging 12 M T LCH Bone marrow, nodes, liver, spleen 10 na na na na na Wongchan- chailert 2002 Med Pediatr Oncol 8 F na THL Extradural mass, bone 6 Maintenance na na CHOP regimen (CPM, DAUNO, VCR, PRED) Died of infection, histiocytosis and ALL not in remission Yokokawa 2015 Genes Chromosomes and Cancer 7 M T LCH Skin then lungs 22 Maintenance na Yes JLSG-02 protocol (CYTA, VCR, PRED) DOH a Age at ALL diagnosis (years [yr] old); b Delay between ALL diagnosis and onset of histiocytosis (months [mth]), c Proven clonal relationship between ALL and histiocytosis: same TCR gene rearrangement
ATG:
Haematologica | 108 June 2023 1709 CASE REPORT
combination resulted in a significant metabolic response of RDD lesions, leukemia remission was not durable as patient experienced CNS relapse 3 months following combination therapy. Of note, Gaspari et al. 8 also reported the safety and efficacity of vemurafenib combined with vinblastine/prednisone in a newborn with multisystem LCH. A literature review of children with co-diagnosis of histiocytosis (excluding H group disorders) during ALL treatment identified 30 cases. Median age at ALL diagnosis was 6 years old (range, 0.5-18 years) and median time between ALL diagnosis and histiocytosis onset was 7.5 months (range, 3-24 months). Most patients were boys. Leukemia immunophenotype included ten T-ALL. Histiocytic disorders arising during ALL treatment comprises LCH (47%), histiocytic sarcoma (23%), juvenile xanthogranuloma (13%), true histiocytic lymphoma (10%), Erdheim-
Chester disease (ECD) (3%) and indeterminate (3%). Prognosis was poor with 19 deaths (12 related to histiocytosis). Twenty-one patients did not achieve remission of histiocytosis. ALL treatment alone in this context appeared ineffective (remission in 1/5 cases). Four children experienced ALL relapse following diagnosis of histiocytosis, with two dying of ALL progression. The clinical course of our patient is consistent with key findings summarized above. Although the morphologic appearance of our patient’s histiocytic lesions is highly suggestive of RDD, the BRAF V600E mutation is not characteristic of RDD, but rather represents a molecular hallmark of LCH and/or ECD.9 This morphologic/molecular discrepancy is reminiscent of mixed histiocytosis arising as part of malignant hemopathy associated with clonal hematopoiesis previously reported in adults with ECD.10,11
Figure 1. Pathology of rib lesion and evolution of metabolic response over the course of therapy. (A) Rib lesion depicting predominant infiltration of large, multinucleated histiocytic cells with evidence of emperipolesis in the absence of necrosis or mitosis. The histiocytic cells are strongly positive for CD68 (not shown), (B) CD163, (C) Fascin, (D) weak and focal S100 staining and (E) negative for CD1a and CD207 (not shown). The following immunostains are negative: CD15, CD20, CD30, CD45, CD117, ALK, PAX5, EMA, HLA-DR, and MPO (not shown). Evolution of metabolic response by 18F-fluorodeoxyglucose positron emission tomography (PET) scans over the course of therapy. (F) At diagnosis of histiocytosis: mediastinal nodules (long arrow) SUVmax 4.2 and 8.2, right tibial diaphysis lesion (arrowhead) SUVmax 6.8 and 5th rib lesion (thick arrow) SUVmax 8.1; (G) post-rib biopsy and continuation of ALL therapy: progressive disease in the mediastinum SUVmax 10.1 and 12.4 and tibia SUVmax 11.5; (H) 1 month post-dabrafenib monotherapy: almost complete metabolic response in the mediastinum SUVmax 3.0 and right tibia SUVmax 1.5, new hepatic focus SUVmax 3.4 (curvilinear arrow) with remaining liver SUVmax 1.6 and liver size 14.5 cm CC; (I) 3 months post-dabrafenib and maintenance chemotherapy combination: complete metabolic response in the mediastinum and stable uptake in right tibia SUVmax 2.0. Progressive uptake of liver lesion SUVmax 4.3 with remaining liver SUVmax 2.0; (J) at isolated central nervous system relapse: no significant uptake in mediastinum or right tibia, progression in the uptake of liver lesion SUVmax 5.6 with diffuse hyperactivity of the remaining liver SUVmax 3.5; (K) after re-induction chemotherapy with nelarabine: no mediastinal lesion. Discrete uptake right tibia SUVmax 2.6. Persistent increased uptake in liver lesion SUVmax 4.0 and remaining liver SUVmax 3.2; (L) 100 days post-hematopoietic stem cell transplantation: no mediastinal or tibial lesions. Stable uptake in liver lesion SUVmax 4.0 and remaining liver SUVmax 3.0.

A F G H I J K L B C D E
It Haematologica | 108 June 2023 1710 CASE REPORT
Figure 2. Timeline illustrating different events and treatments of the case report. ALL: diagnosis of acute lymphoblastic leukemia; Conso: consolidation; CRT: cranial irradiation; DI: delayed intensification; FU: last follow-up; H: diagnosis of histiocytosis; HSCT: hematopoetic stem cell transplantation; Ind: induction; IM: interim maintenance; M: maintenance, R: relapse of acute lymphoblastic leukemia; Reind : reinduction.

remains unclear whether co-occurrence of histiocytosis confers a worse prognosis when associated with ALL or vice versa, although our literature review signals a high rate of histiocytosis-related mortality. Recent evidence of MAPK pathway activation in most histiocytic disorders paves the way for molecularly-targeted therapies in combination with conventional chemotherapy, as illustrated in this case, to treat leukemic and histiocytic entities concomitantly. This therapeutic combination strategy warrants further validation; however, prospective assessment of such strategy is unforeseeable due to the rarity of these pathologies. Therefore, our case provides a proofof-concept demonstrating safety of dabrafenib in combination with chemotherapy, and may represent an alternative therapeutic option for BRAF-mutated histiocytosis arising during ALL therapy, either as a definitive treatment or as a bridge to HSCT consolidation, given their poor outcome. Furthermore, since ALL maintenance chemotherapy is similar to LCH-based backbone, future prospective evaluation of BRAF inhibitor in combination with conventional chemotherapy for high-risk BRAF-mutated multisystemic LCH may be warranted.
Authors
Gervaise Hubert,1 Henrique Bittencourt,1,2 Caroline Laverdière,1,2 Pierre Teira,1,2 Sonia Cellot,1,2 Sylvie Langlois,2 Alexandre Rouette,3 Thomas Sontag,1 Daniel Sinnett,1,2 Dorothée Dal-Soglio,4 Sophie Turpin5 and Thai Hoa Tran1,2
1Division of Pediatric Hematology-Oncology, Charles-Bruneau
References
1. Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell
Cancer Center, CHU Sainte-Justine; 2Department of Pediatrics, Université de Montréal and CHU Sainte-Justine; 3Department of Laboratory Medicine, CHU Sainte-Justine; 4Department of Pathology, CHU Sainte-Justine and 5Department of Medical Imaging, Nuclear Medicine, CHU Sainte-Justine, Montréal, Québec, Canada
Correspondence:
T.H. TRAN - thai.hoa.tran@umontreal.ca
https://doi.org/10.3324/haematol.2022.281926
Received: August 10, 2022.
Accepted: November 10, 2022.
Prepublished: November 17, 2022.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
GH and THT designed the study, reviewed the literature, analyzed the data and wrote the manuscript; SL, AR, SC, DS and THT performed molecular analysis; DDS and ST provided pathology and radiology review; HB, CL, PT, SC and THT provided patient care and clinical information. All authors revised and approved the manuscript.
Data-sharing statement
Additional data can be requested via the corresponding author by email.
lineages. Blood. 2016;127(22):2672-2681.
2. Castro ECC, Blazquez C, Boyd J, et al. Clinicopathologic
Haematologica | 108 June 2023 1711
CASE REPORT
features of histiocytic lesions following ALL, with a review of the literature. Pediatr Dev Pathol. 2010;13(3):225-237.
3. Bleeke M, Johann P, Gröbner S, et al. Genome wide analysis of acute leukemia and clonally related histiocytic sarcoma in a series of three pediatric patients. Pediatr Blood Cancer. 2020;67(2):e28074.
4. Dunsmore KP, Winter SS, Devidas M, et al. Children’s Oncology Group AALL0434: a phase III randomized clinical trial testing nelarabine in newly diagnosed T-cell acute lymphoblastic leukemia. J Clin Oncol. 2020;38(28):3282-3293.
5. Migdady Y, Ediriwickrema A, Jackson RP, et al. Successful treatment of thrombocytopenia with daratumumab after allogeneic transplant: a case report and literature review. Blood Adv. 2020;4(5):815-818.
6. Whitlock J, dalla Pozza L, Goldberg JM, et al. Nelarabine in combination with etoposide and cyclophosphamide is active in first relapse of childhood T-acute lymphocytic leukemia (T-ALL) and T-lymphoblastic lymphoma (T-LL). Blood.
2014;124(21):795-795.
7. Donadieu J, Larabi IA, Tardieu M, et al. Vemurafenib for refractory multisystem Langerhans cell histiocytosis in children: an international observational study. J Clin Oncol. 2019;37(31):2857-2865.
8. Gaspari S, Di Ruscio V, Stocchi F, Carta R, Becilli M, De Ioris MA. Case report: early association of vemurafenib to standard chemotherapy in multisystem Langerhans cell histiocytosis in a newborn: taking a chance for a better outcome? Front Oncol. 2021;11:794498.
9. Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919-1923.
10. Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non–Langerhans cell histiocytosis. Blood. 2017;130(8):1007-1013.
11. Emile JF, Cohen-Aubart F, Collin M, et al. Histiocytosis. Lancet. 2021;398(10295):157-170.
Haematologica | 108 June 2023 1712 CASE REPORT

haematologica
Journal of the Ferrata Storti Foundation
haematologica.org
