haematologica

VOL. 108 OCTOBER 2023 Journal of the Ferrata Storti Foundation ISSN 0390 - 6078 haematologica.org


Editor-in-Chief
Jacob M. Rowe (Jerusalem)
Deputy Editors
Carlo Balduini (Pavia), Jerry Radich (Seattle)
Associate Editors
Shai Izraeli (Tel Aviv), Steve Lane (Brisbane), Pier Mannuccio Mannucci (Milan), Pavan Reddy (Houston), David C. Rees (London), Paul G. Richardson (Boston), Francesco Rodeghiero (Vicenza), Gilles Salles (New York), Kerry Savage (Vancouver), Aaron Schimmer (Toronto), Richard F. Schlenk (Heidelberg), Sonali Smith (Chicago)
Statistical Consultant
Catherine Klersy (Pavia)
Editorial Board
Walter Ageno (Varese), Sarit Assouline (Montreal), Andrea Bacigalupo (Roma), Taman Bakchoul (Tübingen), Pablo Bartolucci (Créteil), Katherine Borden (Montreal), Marco Cattaneo (Milan), Corey Cutler (Boston), Kate Cwynarski (London), Ahmet Dogan (New York), Mary Eapen (Milwaukee), Francesca Gay (Torino), Ajay Gopal (Seattle), Alex Herrera (Duarte), Martin Kaiser (London), Marina Konopleva (Houston), Johanna A. Kremer Hovinga (Bern), Nicolaus Kröger (Hamburg), Austin Kulasekararaj (London), Shaji Kumar (Rochester), Ann LaCasce (Boston), Anthony R. Mato (New York), Matthew J. Mauer (Rochester) Neha Mehta-Shah (St. Louis), Moshe Mittelman (Tel Aviv), Alison Moskowitz (New York), Yishai Ofran (Haifa), Farhad Ravandi (Houston), John W. Semple (Lund), Liran Shlush (Toronto), Sarah K. Tasian (Philadelphia), Pieter van Vlieberghe (Ghent), Ofir Wolach (Haifa), Loic Ysebaert (Toulouse)
Managing Director
Antonio Majocchi (Pavia)
Editorial Office
Lorella Ripari (Office & Peer Review Manager), Simona Giri (Production & Marketing Manager), Paola Cariati (Graphic Designer), Giulia Carlini (Graphic Designer), Debora Moscatelli (Graphic Designer), Igor Poletti (Graphic Designer), Marta Fossati (Peer Review), Diana Serena Ravera (Peer Review), Laura Sterza (Account Administrator)
Assistant Editors
Britta Dost (English Editor), Rachel Stenner (English Editor), Anne Freckleton (English Editor), Rosangela Invernizzi (Scientific Consultant), Marianna Rossi (Scientific Consultant), Massimo Senna (Information Technology), Luk Cox (Graphic Artist)
Haematologica (print edition, pISSN 0390-6078, eISSN 1592-8721) publishes peer-reviewed papers on all areas of experimental and clinical hematology. The journal is owned by a non-profit organization, the Ferrata Storti Foundation, and serves the scientific community following the recommendations of the World Association of Medical Editors (www.wame.org) and the International Committee of Medical Journal Editors (www.icmje.org).
Haematologica publishes Editorials, Original articles, Review articles, Perspective articles, Editorials, Guideline articles, Letters to the Editor, Case reports & Case series and Comments. Manuscripts should be prepared according to our guidelines (www.haematologica.org/information-for-authors), and the Uniform Requirements for Manuscripts Submitted to Biomedical Journals, prepared by the International Committee of Medical Journal Editors (www.icmje.org).
Manuscripts should be submitted online at http://www.haematologica.org/.
Conflict of interests. According to the International Committee of Medical Journal Editors (http://www.icmje.org/#conflicts), “Public trust in the peer review process and the credibility of published articles depend in part on how well conflict of interest is handled during writing, peer review, and editorial decision making”. The ad hoc journal’s policy is reported in detail at www.haematologica.org/content/policies.
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articles to the Ferrata Storti Foundation. No formal permission will be required to reproduce parts (tables or illustrations) of published papers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with commercial intent will require written permission and payment of royalties.
Subscription. Detailed information about subscriptions is available at www.haematologica.org. Haematologica is an open access journal and access to the online journal is free. For subscriptions to the printed issue of the journal, please contact: Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, E-mail: info@haematologica.org).
Rates of the printed edition for the year 2022 are as following:
Institutional: Euro 700
Personal: Euro 170
Advertisements. Contact the Advertising Manager, Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, e-mail: marketing@haematologica.org).
Disclaimer. Whilst every effort is made by the publishers and the editorial board to see that no inaccurate or misleading data, opinion or statement appears in this journal, they wish to make it clear that the data and opinions appearing in the articles or advertisements herein are the responsibility of the contributor or advisor concerned. Accordingly, the publisher, the editorial board and their respective employees, officers and agents accept no liability whatsoever for the consequences of any inaccurate or misleading data, opinion or statement. Whilst all due care is taken to ensure that drug doses and other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage, and described within this journal, should only be followed in conjunction with the drug manufacturer’s own published literature.
Direttore responsabile: Prof. Carlo Balduini; Autorizzazione del Tribunale di Pavia n. 63 del 5 marzo 1955. Printing: Press Up, zona Via Cassia Km 36, 300 Zona Ind.le Settevene - 01036 Nepi (VT)

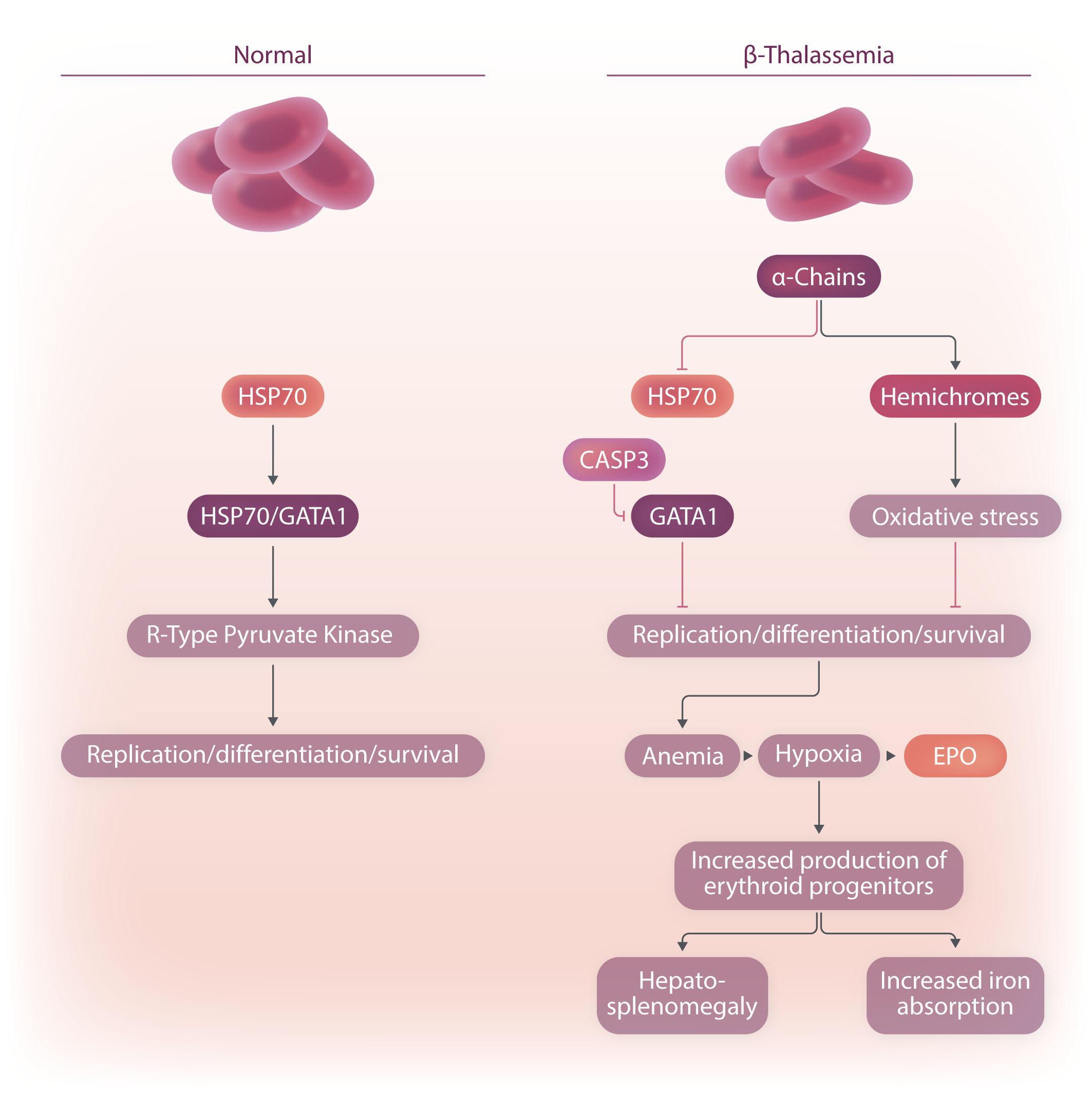
Image taken from the Review by Amaliris Guerra et al. in this issue.
2559 The French-American-British classification of myelodysplastic syndromes
Moshe Mittelman
https://doi.org/10.3324/haematol.2023.284054
2561 Can measurable residual disease assessment be reliably used to defer allogeneic stem cell transplant in patients with intermediate-risk acute myeloid leukemia?
Mahesh Swaminathan and Farhad Ravandi
https://doi.org/10.3324/haematol.2023.283120
2564 On either side of homeostasis: EPAS1 gain- and loss-of-function mutations
Josef T. Prchal and Gregg L. Semenza
https://doi.org/10.3324/haematol.2023.283285
2566 Correcting the aberrant Fanconi anemia transcriptional program by gene therapy
Yigal Dror
https://doi.org/10.3324/haematol.2023.283031
2568 CAR T-cell treatment of high-risk multiple myeloma: will there be a cure?
Gösta Gahrton
https://doi.org/10.3324/haematol.2023.282896
2570 Down syndrome and leukemia: from basic mechanisms to clinical advances
André Baruchel et al.
https://doi.org/10.3324/haematol.2023.283225
2582 Novel potential therapeutics to modify iron metabolism and red cell synthesis in diseases associated with defective erythropoiesis
Amaliris Guerra et al.
https://doi.org/10.3324/haematol.2023.283057
2594 Shwachman-Diamond syndromes: clinical, genetic, and biochemical insights from the rare variants
Nozomu Kawashima et al.
https://doi.org/10.3324/haematol.2023.282949
2606 Back to the future: the amazing journey of the therapeutic anti-leukemia enzyme asparaginase
Erwinia chrysanthemi
Wing H. Tong and Carmelo Rizzari
https://doi.org/10.3324/haematol.2022.282324
2616 Acute Myeloid Leukemia
Higher-dose venetoclax with measurable residual disease-guided azacitidine discontinuation in newly diagnosed acute myeloid leukemia
Jonathan A. Gutman et al.
https://doi.org/10.3324/haematol.2023.282681
2626 Acute Myeloid Leukemia
Co-targeting BCL-XL and BCL-2 by PROTAC 753B eliminates leukemia cells and enhances efficacy of chemotherapy by targeting senescent cells
Yannan Jia et al.
https://doi.org/10.3324/haematol.2022.281915
2639 Blood Transfusion
Reticulocytes in donor blood units enhance red blood cell alloimmunization
Tiffany A. Thomas et al.
https://doi.org/10.3324/haematol.2023.282815
2652
Bone Marrow Failure
Gene therapy restores the transcriptional program of hematopoietic stem cells in Fanconi anemia
Miren Lasaga et al.
https://doi.org/10.3324/haematol.2022.282418
2664 Chronic Lymphocytic Leukemia
Subgroup-specific gene expression profiles and mixed epistasis in chronic lymphocytic leukemia
Almut Lütge et al.
https://doi.org/10.3324/haematol.2022.281869
2677
Hematopoiesis
Aging-induced pseudouridine synthase 10 impairs hematopoietic stem cells
Yuqian Wang et al.
https://doi.org/10.3324/haematol.2022.282211
2690
Hematostasis
Platelet phosphatidylserine is the critical mediator of thrombosis in heparin-induced thrombocytopenia
Jan Zlamal et al.
https://doi.org/10.3324/haematol.2022.282275
2703
Iron Metabolism & its Disorders
Ferroportin inhibitor vamifeport ameliorates ineffective erythropoiesis in a mouse model of b-thalassemia with blood transfusions
Natarajaswamy Kalleda et al.
https://doi.org/10.3324/haematol.2022.282328
2715 Myelodysplastic Syndromes
Inactivation of p53 provides a competitive advantage to del(5q) myelodysplastic syndrome
hematopoietic stem cells during inflammation
Tomoya Muto et al.
https://doi.org/10.3324/haematol.2022.282349
2730 Myeloproliferative Disorders
Safety and efficacy of zinpentraxin alfa as monotherapy or in combination with ruxolitinib in myelofibrosis: stage I of a phase II trial
Srdan Verstovsek et al.
https://doi.org/10.3324/haematol.2022.282411
2743 Non-Hodgkin Lymphoma
Germline HAVCR2 mutations and their relation to the clinical spectrum of subcutaneous panniculitis-like T-cell lymphoma and hemophagocytic lymphohistiocytosis: results from a multicenter study and meta-analysis
Chatphatai Moonla et al.
https://doi.org/10.3324/haematol.2022.282419
2753
Plasma Cell Disorders
Randomized phase II study of weekly carfilzomib 70 mg/m2 and dexamethasone with or without cyclophosphamide in relapsed and/or refractory multiple myeloma patients
Borja Puertas et al.
https://doi.org/10.3324/haematol.2022.282490
2764 Plasma Cell Disorders
IgM monoclonal gammopathy of undetermined significance: clinicopathologic features with and without IgM-related disorders
Frido K. Bruehl et al.
https://doi.org/10.3324/haematol.2022.282389
2774 Plasma Cell Disorders
Real-world study of the efficacy and safety of belantamab mafodotin (GSK2857916) in relapsed or refractory multiple myeloma based on data from the nominative ATU in France: the IFM 2020-04 study
Alexis Talbot et al.
https://doi.org/10.3324/haematol.2022.281772
2783 Platelet Biology & its Disorders
Adolescents and young adults with newly diagnosed primary immune thrombocytopenia
Alexandra Schifferli et al.
https://doi.org/10.3324/haematol.2022.282524
2794
Measurable residual disease-guided therapy in intermediate-risk acute myeloid leukemia patients is a valuable strategy in reducing allogeneic transplantation without negatively affecting survival
Jesse M. Tettero et al.
https://doi.org/10.3324/haematol.2022.282639
2799
Impact of high-risk disease on the efficacy of chimeric antigen receptor T-cell therapy for multiple myeloma: a meta-analysis of 723 patients
Nico Gagelmann et al.
https://doi.org/10.3324/haematol.2022.282510
2803
Functional, biochemical, molecular and clinical characterization of antithrombin c.1157T>C (p.Ile386Thr), a recurrent Polish variant with a founder effect
Anna Weronska et al.
https://doi.org/10.3324/haematol.2022.282459
2808 Clinical and laboratory diversity of diffuse large B-cell lymphomas in children with Nijmegen breakage syndrome
Agata Pastorczak et al.
https://doi.org/10.3324/haematol.2022.282325
2814
Post-transplantation Burkit lymphoma: a retrospective study of 55 patients
Pierre Walczak et al.
https://doi.org/10.3324/haematol.2022.282297
2820 Survival outcomes with oral azacitidine maintenance in patients with acute myeloid leukemia in remission by receipt of initial chemotherapy: subgroup analyses from the phase III QUAZAR AML-001 trial
Andrew H. Wei et al.
https://doi.org/10.3324/haematol.2022.282296
2826
Gemtuzumab ozogamicin plus midostaurin in conjunction with standard intensive therapy for FLT3-mutated acute myeloid leukemia patients – Czech center experience
Barbora Weinbergerová et al.
https://doi.org/10.3324/haematol.2022.282263 Haematologica
2830 KIR3DL2 may represent a novel therapeutic target in aggressive systemic peripheral T-cell lymphoma
Amandine Decroos et al.
https://doi.org/10.3324/haematol.2022.282220
2837 Menin inhibitor zifomenib (KO-539) synergizes with drugs targeting chromatin regulation or apoptosis and sensitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax
Johanna Rausch et al.
https://doi.org/10.3324/haematol.2022.282160
2844 Impact of vincristine dose reduction on outcomes of patients with aggressive B-cell lymphoma treated with (R)-CHOP
Moritz Bewarder et al.
https://doi.org/10.3324/haematol.2022.282126
2850
Subcutaneous azacitidine maintenance in transplant-ineligible patients with acute myeloid leukemia: a single-center retrospective study
Natacha Johnson et al.
https://doi.org/10.3324/haematol.2022.282009
2855
The Glanzmann Thrombasthenia Registry: safety of platelet therapy in patients with Glanzmann thrombasthenia and changes in alloimmunization status
Man-Chiu Poon et al.
https://doi.org/10.3324/haematol.2022.281973
2859
Characterization of a novel MEF2D-BCL9 fusion-positive acute lymphoblastic leukemia cell line Inge van Outersterp et al.
https://doi.org/10.3324/haematol.2022.281712
2865 KMT2A partner genes in infant acute lymphoblastic leukemia have prognostic significance and correlate with age, white blood cell count, sex, and central nervous system involvement: a Children’s Oncology Group P9407 trial study
Blaine W. Robinson et al.
https://doi.org/10.3324/haematol.2022.281552
2872
One gene, two opposite phenotypes: a case report of hereditary anemia due to a loss-of-function variant in the EPAS1 gene
Barbara Eleni Rosato et al.
https://doi.org/10.3324/haematol.2022.282457
2877 ARID5B influences B-cell development and function in mouse Charnise Goodings et al.
https://doi.org/10.3324/haematol.2023.283687
Moshe Mittelman

Tel Aviv Sourasky Medical Center, Tel-Aviv University, Tel-Aviv, Israel
E-mail: moshemt@gmail.com
https://doi.org/10.3324/haematol.2023.284054
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license

TITLE Proposals for the classification of the myelodysplastic syndromes.
JOURNAL British Journal of Haematology 1982;51(2):189-199. PMID: 188440.
In 1976, Dr. John M. Bennett and colleagues, the FrenchAmerican-British (FAB) cooperative group (Figure 1), published a seminal paper, the first classification of acute myeloid leukemia (AML).1 Interestingly, for decades it had been recognized that many AML patients experience a preleukemic phase. Rhoads & Barker had reported on 100 patients with “refractory anemia” in 1938.2 Several terms were subsequently suggested, including: “preleukemic leukemia”, “smoldering leukemia”, “preleukemic syndrome (hemopoietic dysplasia)” and “dysmyelopoietic syndrome”. The 1976 paper, summarizing data from 150 patients, was the first to distinguish between common AML of recent onset, requiring immediate treatment, and less acute di-

sorders regarded as “preleukemia”, with no need for urgent treatment.1 The authors also noted that many patients with “preleukemia” never evolve to AML and succumb to the complications of bone marrow failure.
Later on, the FAB group realized that further clarifications of the preleukemic phase were required, especially characterization of blasts, and definition of conditions with excess blasts, monocytosis and leukemic progression. Reviewing slides from an additional 80 patients led to the landmark report, changing the field.
The paper introduced the term “myelodysplastic syndrome (MDS)” and proposed guidelines for its diagnosis and classification based on morphological findings, dysplasia and percentage of blasts in the peripheral blood and bone marrow. Five MDS groups were described (Table 1): (i) refractory anemia; (ii) refractory anemia with ring sideroblasts; (iii) refractory anemia with excess of blasts; (iv) chronic myelomonocytic leukemia (which was included among the MDS, despite controversy); and (v) refractory anemia with excess of blasts ‘in transformation’ (a new term). A main distinguishing feature of these conditions is the proportion of blast cells in the peripheral blood and/or bone
AUTHORS Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Figure 1. The French-American-British Leukemia Cooperative Group. Figure provided by Dr. John Bennett, the head of the group. Reproduced with permission.MDS: myelodysplastic syndrome; RBC: red blood cells; NRBC: nucleated red blood cells. Table with permission from Bennett et al. Br J Haematol 1982.

marrow. The morphological features of blasts were redefined and over 30% bone marrow blasts were proposed to diagnose AML.
Several aspects made this original report a landmark paper. First, the proposed term, MDS, was new and descriptive but also reflected the biology, i.e., syndromes (not a single disease) of the bone marrow (“myelo”), characterized by defective production (dysplasia). Second, clear guidelines, with some objectivity and quantitation, although arbitrary, for MDS diagnosis were described. The diagnostic criteria are based mainly on peripheral blood and bone marrow morphology, still the backbone of diagnosis even today, despite the developments of novel tools (cytogenetics, immunophenotyping, molecular analysis). Third, this allowed the use of a common language and terminology, providing a platform for research, communication, and permitting comparisons between patients, and led to collaborative clinical trials. Finally, this was an academic collaborative international effort, without external funding.
The MDS FAB guidelines soon became the worldwide gold-standard for the diagnosis and classification of MDS. Numerous publications validated it, leading to more research and clinical trials.3 Not surprisingly, despite new clinical, biological and genetic information that required updated classifications (the World Health Organization classification, Revised International Prognostic Scoring System, Molecular International Prognostic Scoring System), the FAB classi fication remained the backbone, and all these classifications maintained the structure and philosophy of the FAB proposal.
In summary, the MDS FAB classification manuscript proposed a new term, defined a new disease entity, suggested diagnostic tools and risk stratification and led to a diagnostic, prognostic and therapeutic paradigm change.
Disclosure
No conflicts of interest to disclose.
In writing the manuscript I was assisted by Dr. John Bennett, a colleague, friend, mentor, pioneer and the founder and “father” of MDS. I cannot imagine the field of MDS without the superb and unbelievable contribution of John.
hundred cases. JAMA. 1938;110(11):794-796.
1. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias: French-American-British Cooperative Group. Br J Haematol. 1976;33(4):451-458. 2. Rhoads CP, Barker WH. Refractory anemia: analysis of one 3. Vardiman J. The classification of MDS: from FAB to WHO and beyond. Leuk Res. 2012;36(12):1453-1458.Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Correspondence: F. Ravandi fravandi@mdanderson.org
Received: May 22, 2023.
Accepted: June 16, 2023.
Early view: June 22, 2023.
htps://doi.org/10.3324/haematol.2023.283120
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Efforts to detect minute numbers of leukemic blasts and differentiate them from normal cells date back to the 1980s.1 Immunofluorescence microscopy was the first method adopted to assess measurable residual disease (MRD), but it is no longer commonly used. More modern assays such as multicolor flow-cytometry (MFC), real-time quantitative polymerase chain reaction (RT-qPCR), nextgeneration sequencing (NGS), and PCR-NGS are signi ficantly more sensitive tools to detect residual leukemic cells that are conceptually the cause of future relapse.2 MRD is now routinely used in clinical practice as a guide to select post-remission therapies, including allogeneic stem cell transplant (allo-SCT), especially in pediatric patients with acute lymphoblastic leukemia (ALL).3,4 Similarly, MRD is shown to be highly prognostic in specific subsets of acute myeloblastic leukemia (AML), such as acute promyelocytic leukemia (APL), core-binding factor leukemia, and NPM1-mutated AML.5 This has led to significant recent interest in considering MRD detection as a potential endpoint in clinical trials. The European LeukemiaNet (ELN) has established guidelines for MRD assessment in specific subsets of AML.6 However, the role of MRD testing in ascertaining the type of post-remission therapies has not yet been fully characterized.
In this issue of the journal, Tettero et al. report the outcome of patients with intermediate-risk AML treated in the HOVON-SAKK132-trial (HO132, conducted from 20142017) who received MRD-guided post-remission therapy.7 The study included a comparator arm of patients with intermediate-risk AML treated in other HOVON-SAKK trials (conducted from 2006-2013) who received post-remission treatment without planned MRD guidance. Both groups of patients were compared using propensity score match analysis. MRD was assessed by MFC and/or RT-PCR for NPM1 mutations after cycle 2 (C2) of treatment in responders. One hundred and fifty-three patients with intermediate-risk AML received MRD-guided post-remission
therapy. Among them, 110 became MRD-negative, and 43 had persistent detectable MRD. Forty-four percent of MRD-negative patients received allo-SCT even though the HO132 trial recommended non-allo-SCT consolidation. Of note, half of these patients had a complex karyotype (46%), and a third achieved complete remission only after two courses of treatment. There was no difference in the event-free survival (EFS) of patients with MRD-positive and MRD-negative status (Hazard Ratio [HR]: 1.24; 95% Confidence Interval [CI]: 0.75-2; P=0.42), and the 3-year EFS was 47% and 54%, respectively. Similarly, there was no significant difference in overall survival (OS) (HR: 1.50; 95% CI: 0.85-2.64; P=0.16), with 5-year OS of 54% and 65%, respectively. The observations were similar in the comparator arm, which included patients who received non-MRD-adapted consolidation therapies. The subgroup analysis comparing the outcomes of MRD-negative patients in both groups showed that the MRD-adapted consolidation approach did not significantly influence the survival (3-year EFS [HR: 0.86; 95% CI: 0.56-1.33; P=0.50] and 5-year OS [HR: 0.84; 95% CI: 0.5-1.4; P=0.50]) suggesting that allo-SCT could be safely deferred in some patients without adversely affecting outcomes. This study has a few limitations that need to be addressed. Firstly, intermediate-risk AML is a diverse population. Patients with FLT3, IDH, and RAS pathway mutations are categorized as intermediate-risk AML by the ELN 2022 AML risk stratification if they do not have favorable or adverse-risk characteristics. The study had 42 patients and 50 patients with FLT3 mutations in the MRD-guided and MRD-unguided groups, respectively. These patients did not receive FLT3 inhibitors as midostaurin had not been approved at the time of treatment. (Currently, the standard therapy for these patients involves the combination of midostaurin with intensive chemotherapy.) The RATIFY trial demonstrated a longer median OS in patients who underwent allo-SCT at first
complete remission (CR1).8 However, no MRD information was available from the RATIFY trial. Nevertheless, the post hoc analysis of patients treated on the trial showed that the 3-year cumulative incidence of relapse (CIR) in responders (with non-AML death and transplant as competing risks) was approximately 32% in the midostaurin and 40% in the placebo arms. Similarly, in transplanted patients, the 3-year CIR (with non-AML death as a competing risk) was approximately 22% in the midostaurin and 32% in the placebo arms.9 This highlights the potential benefit of consolidation allo-SCT to improve outcomes in specific molecular subsets of AML. Recently, quizartinib in combination with intensive chemotherapy was evaluated in the phase III QuANTUM-First trial.10 This trial used a FLT3ITD-specific PCR-NGS technique, which has a sensitivity of 10-4-10-5, for MRD assessment.11 The investigators reported that the 3-year CIR was 34% (95% CI: 26-42 months) in the quizartinib arm as compared to 45% (95% CI: 37-53 months) in the placebo arm.10 Schlenk and colleagues reported that patients treated with quizartinib followed by allo-SCT at CR1 had a significantly longer OS (HR: 0.424; 95% CI: 0.301 - 0.597; P<0.0001) than their counterparts treated with placebo, regardless of pre-allo-SCT MRD status.12 This further signifies the importance of adding a FLT3 inhibitor in patients with FLT3-mutated AML receiving intensive chemotherapy. Other investigators have reported the outcomes of patients with FLT3-ITD-mutated AML based on the pre-allo-SCT MRD, assessed by FLT3ITD PCR-NGS. They showed that 81% of patients, who were FLT3-ITD MRD-negative by capillary electrophoresis, were MRD-positive by PCR-NGS. All these patients had inferior survival outcomes with a 3-year CIR of approximately 70%.13 Intriguingly, the 3-year CIR was approximately 20% when FLT3-ITD PCR-NGS variant allele frequency (VAF) was <0.001% compared to 70% in VAF <0.1%. The latter emphasizes the importance of a sensitive MRD assay and the prognostic significance of using such highly sensitive tests.14 These data further support the importance of assessing MRD by a sensitive assay, but do not yet support the decision to abandon allo-SCT in specific subsets. There is no precise optimal timing of MRD assessment in AML. In the HO132 trial, MRD assessment was performed
1. Bradstock KF, Papageorgiou ES, Janossy G, et al. Detection of leukaemic lymphoblasts in CSF by immunofluorescence for terminal transferase. Lancet. 1980;1(8178):1144.
2. Grimwade D, Freeman SD. Defining minimal residual disease in acute myeloid leukemia: which platforms are ready for "prime time"? Blood. 2014;124(23):3345-3355.
3. Pui CH, Pei D, Coustan-Smith E, et al. Clinical utility of sequential minimal residual disease measurements in the context of risk-based therapy in childhood acute lymphoblastic leukaemia: a prospective study. Lancet Oncol. 2015;16(4):465-474.
after C2, whereas in other trials, such as the QuANTUMFirst trial, MRD assessment was made after the induction cycle. In the retrospective study by Australian colleagues,13 MRD was assessed after C2 of chemotherapy. Clearly, standardization and harmonization of MRD assays, as well as the timing of testing, will be desirable in order to arrive at definitive recommendations for the use of MRD testing in selecting post-remission therapy.
The use of an MRD-adapted approach to select post-remission therapy is a compelling concept, especially in intermediate-risk AML, where alternative options for consolidation or maintenance may become available in the near future. While allo-SCT is preferred in some molecular subtypes of AML, the higher transplant-related mortality (TRM) (5-35%) makes it less favored in others.15 Therefore, it is essential to identify patients in whom allo-SCT is beneficial. However, several questions must be addressed before this becomes an accepted approach. First, the ideal timing of the MRD assessment still has to be defined; this may also be influenced by the intensity of the induction regimen. Second, with the development of small molecule inhibitors, such as venetoclax and other targeted inhibitors, it may be possible to improve outcomes in patients with MRD-negative disease with non-intensive maintenance strategies rather than with consolidation allo-SCT. Finally, highly sensitive MRD assays are required to identify patients with a very low risk of relapse who could then avoid a higher TRM. We have witnessed this in patients with Philadelphia chromosome-positive ALL, where historical reliance on allo-SCT for long-term remission has progressively diminished with the introduction of more effective agents.16,17 Furthermore, there are ongoing efforts to develop therapeutics that could eradicate MRD in AML, as has been seen with blinatumomab in patients with ALL. This is likely to see our approach towards consolidating remission in patients with AML evolve further.
MS and FR have no relevant conflicts of interest to disclose.
MS and FR wrote and revised the manuscript.
4. Vora A, Goulden N, Wade R, et al. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): a randomised controlled trial. Lancet Oncol. 2013;14(3):199-209.
5. Short NJ, Ravandi F. How close are we to incorporating measurable residual disease into clinical practice for acute myeloid leukemia? Haematologica. 2019;104(8):1532-1541.
6. Heuser M, Freeman SD, Ossenkoppele GJ, et al. 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138(26):2753-2767.
7. Tettero JM, Ngai LL, Bachas C, et al. Measurable residual disease-guided therapy in intermediate-risk acute myeloid leukemia patients is a valuable strategy in reducing allogeneic transplantation without negatively affecting survival. Haematologica. 2023;108(10):2794-2798.
8. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464.
9. Larson RA, Mandrekar SJ, Huebner LJ, et al. Midostaurin reduces relapse in FLT3-mutant acute myeloid leukemia: the Alliance CALGB 10603/RATIFY trial. Leukemia. 2021;35(9):2539-2551.
10. Erba HP, Montesinos P, Kim HJ, et al. Quizartinib plus chemotherapy in newly diagnosed patients with FLT3-internaltandem-duplication-positive acute myeloid leukaemia (QuANTUM-First): a randomised, double-blind, placebocontrolled, phase 3 trial. Lancet. 2023;401(10388):1571-1583.
11. Levis MJ, Erba HP, Montesinos P, et al. QuANTUM-First trial: FLT3-ITD-specific MRD clearance is associated with improved overall survival. Blood. 2022;140(Suppl 1):546-548.
12. Schlenk R, Montesinos P, Romero-Aguilar A, et al. Impact of allogeneic hematopoietic cell transplantation in first complete remission plus FLT3 inhibition with quizartinib in acute myeloid
leukemia with FLT3-ITD: results from QuANTUM-First. EHA: 2023 5/11/23. https://library.ehaweb.org/eha/2023/eha2023congress/387837/richard.schlenk.impact.of.allogeneic.hematopoi etic.cell.transplantation.in.html?f=listing%3D0%2Abrowseby%3D 8%2Asortby%3D1%2Asearch%3Dimpact+allogeneic+hematopoiet ic+cell+transplantation+complete+remission+plus+flt3+inhibitio n+quizartinib+acute+myeloid+leukemia+flt3itd%3A+results+quantum-first.
13. Loo S, Dillon R, Ivey A, et al. Pretransplant FLT3-ITD MRD assessed by high-sensitivity PCR-NGS determines posttransplant clinical outcome. Blood. 2022;140(22):2407-2411.
14. Dillon LW, Gui G, Page KM, et al. DNA sequencing to detect residual disease in adults with acute myeloid leukemia prior to hematopoietic cell transplant. JAMA. 2023;329(9):745-755.
15. Loke J, Malladi R, Moss P, Craddock C. The role of allogeneic stem cell transplantation in the management of acute myeloid leukaemia: a triumph of hope and experience. Br J Haematol. 2020;188(1):129-146.
16. Ghobadi A, Slade M, Kantarjian H, et al. The role of allogeneic transplant for adult Ph+ ALL in CR1 with complete molecular remission: a retrospective analysis. Blood. 2022;140(20):2101-2112.
1University of Utah, Salt Lake City, UT and 2Department of Genetic Medicine and Armstrong Oxygen Biology Research Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Correspondence: J.T. Prchal
josef.prchal@hsc.utah.edu
Received: May 17, 2023.
Accepted: May 29, 2023. Early view: June 8, 2023.
htps://doi.org/10.3324/haematol.2023.283285
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
In this issue of Haematologica, Rosato and colleagues from the laboratory of Dr. Roberta Russo, and other prominent collaborators, report their studies of a boy with anemia referred to them by clinical hematologists from Ancona, Italy, who noted that the child had moderate anemia but did not have appropriately elevated erythropoietin (EPO) expression.1 In fact, his EPO level was low, suggesting that it was the cause of his anemia. All the common causes of anemia were excluded. The presence of a similar phenotype in the father and his older sister suggested a genetic disorder with autosomal dominant inheritance. The delivery of O2 by red cells to all the other cells of the body is based on a beautiful homeostatic system in which hypoxia leads to increased EPO production, which binds to its receptor (EPOR) on erythroid progenitor cells and stimulates increased red blood cell formation, thereby ameliorating tissue hypoxia. The response to hypoxia is mediated by a short DNA sequence located downstream of the EPO gene that is known as the hypoxia response element, which contains a binding site for hypoxia-inducible factors (HIF).2 HIF are heterodimeric transcription factors that consist of an O2-regulated HIF-1α, HIF-2α or HIF-3α subunit and a constitutively expressed HIF-1β subunit.3 HIF-1 is expressed in virtually all nucleated cells, whereas HIF-2 and HIF-3 have limited tissue expression. In the presence of O2, HIF-α subunits are modified by a family of prolyl hydroxylase domain proteins (PHD1-3) and the hydroxylated proteins are bound by the von Hippel-Lindau (VHL) protein, which recruits a ubiquitin protein ligase, leading to the ubiquitination and proteasomal degradation of the HIF-α proteins. Under hypoxic conditions, the hydroxylation reaction is inhibited, and HIF-α subunits rapidly accumulate and regulate the expression of thousands of genes, including EPO, which mediate adaptive responses to hypoxia.
The key role of the HIF-PHD-VHL pathway in regulating erythropoiesis was underscored by the finding that familial erythrocytosis (i.e., abnormally increased red blood cell levels) is in some cases due to: a mutation in PHD2 that
decreases hydroxylase activity; a mutation in VHL that decreases its binding to hydroxylated HIF; or a mutation in HIF-2α that protects it from hydroxylation (Table 1 lists all loci encoding HIF pathway components at which mutations have been identified; the references are available in the paper by Semenza3). However, the report by Rosato et al. provides the first example of the converse: that loss of HIF-2α expression results in anemia.1 The authors of this paper found in the propositus and his anemic family members a novel EPAS1 mutation, c.(61del), consisting of deletion of the 61st nucleotide of the coding sequence. This single nucleotide deletion alters the reading frame, resulting in the generation of a premature stop codon, such that the protein translated from the mutant mRNA would only contain the first 20 amino acids of HIF-2α. This severely truncated protein is likely degraded, resulting in a null allele and haploinsufficiency for HIF-2α, which leads to deficient EPO synthesis and anemia.
HIF-2, like HIF-1, has other systemic regulatory functions. EPAS1 gain-of-function mutations have diverse pathophy-
Table 1. Identified causes of familial erythrocytosis.
ECYT* Gene Protein Mutation§ Effect Genetics
ECYT1 EPOR EPOR W439X GOF AD
ECYT2 VHL VHL R200W LOF AR
ECYT3 EGLN1 PHD2 P317R LOF AD
ECYT4 EPAS1 HIF-2α G537W GOF AD
ECYT5 EPO EPO c.32delG GOF AD
*ECYT6-8 are not shown: these are due to mutations in β-globin, αglobin, and bis-phosphoglycerate mutase, respectively, which affect hemoglobin-O2 affinity. §At each locus, multiple mutations have been identified in different affected individuals; only the first reported mutation is shown. All are missense mutations except ECYT1 (nonsense) and ECYT5 (single nucleotide deletion). ECYT: familial erythrocytosis; EPOR; erythropoietin receptor; GOF: gain-of-function; AD: autosomal dominant inheritance; VHL: von Hippel-Lindau; LOF: loss-of-function; AR: autosomal recessive inheritance; PHD2: prolyl hydroxylase domain protein 2; HIF-2α: hypoxia-inducible factor-2α; EPO: erythropoietin.
siological consequences in addition to erythrocytosis, including pulmonary hypertension4 and thrombosis.5 Whether there is impairment of any other HIF-2-regulated systemic function associated with the EPAS1c.(61del) genotype remains to be determined. One recent scientific advance may point to a potential therapy for affected individuals, which is the Food and Drug Administration’s approval, just several months ago, of daprodustat, a selective inhibitor of the HIF prolyl hydroxylases, which increases HIF activity and thereby increases EPO production.6 The drug has been approved for the treatment of anemia in patients with dialysis-dependent chronic kidney disease, which leads to a progressive loss of EPO production. It is likely that daprodustat would boost HIF-2α levels in EPAS1c.(61del) carriers,
1. Rosato BE, Marra R, Del Giudice F, et al. One gene, two opposite phenotypes: a case report of hereditary anemia due to a lossof-function variant in the EPAS1 gene. Haematologica. 2023;108(10)2872-2876.
2. Wang GL, Semenza GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447-5454.
3. Semenza GL. Regulation of erythropoiesis by the hypoxiainducible factor pathway: effects of genetic and pharmacological perturbations. Annu Rev Med. 2023;74:307-319.
4. Gale DP, Harten SK, Reid CDL, Tuddenham EGD, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial
leading to increased EPO mRNA and protein expression. The close coupling of this drug approval and the report by Rosato et al. highlight the remarkably rapid progress that has been made since the discovery of HIF-1 three decades ago,2 with the development of the HIF stabilizer daprodustat for the treatment of anemia6 and the HIF-2α inhibitor belzutifan for the treatment of renal cell carcinoma.7 This is just the beginning.
Disclosures
No conflicts of interest to disclose.
Contributions
Both authors contributed equally.
hypertension associated with an activating HIF2α mutation. Blood. 2008;112(3):919-921.
5. Gordeuk VR, Miasnikosa GY, Sergueeva AI, et al. Thrombotic risk in congenital erythrocytosis due to up-regulated hypoxia sensing is not associated with elevated hematocrit. Haematologica. 2020;105(3):e87-e90.
6. Singh AK, Carroll K, Perkovic V, et al. Daprodustat for the treatment of anemia in patients undergoing dialysis. N Engl J Med. 2021;385(25):2325-2335.
7. Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for renal cell carcinoma in von Hippel-Lindau disease. N Engl J Med. 2021;385(22):2036-2046.
Genetics and Genome Biology, Research Institute, The Hospital for Sick Children; Institute of Medical Science, Faculty of Medicine, University of Toronto; and Division of Haematology/Oncology, Department of Paediatrics, The Hospital for Sick Children and University of Toronto, Toronto, Ontario, Canada
Correspondence: Y. Dror
yigal.dror@sickkids.ca
Received: May 5, 2023.
Accepted: May 29, 2023.
Early view: June 8, 2023.
htps://doi.org/10.3324/haematol.2023.283031
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Fanconi anemia (FA) is a multisystem disorder, but hematopoietic derangements are the most common causes of morbidity and mortality, which include bone marrow failure and leukemic transformation. FA is a genetically diverse disorder with 22 associated genes.1 FANCA is the most commonly mutated gene, and about 60% of the patients have mutations in this gene.
All FA genes are involved in DNA repair.2 FANCA is part of nine FA genes that are recruited to fork-like DNA structures, and form a single large nuclear protein called “core complex”. The core complex functions as a ubiquitin ligase that monoubiquinates a downstream heterodimer composed of two FA proteins, FANCI and FANCD2, which bind to an arrested replication fork at an interstrand DNA crosslink. The monoubiquitinated FANCI and FANCD2 form a binding interface for single- and double-stranded DNA and downstream effector complexes with additional FA and other proteins to cleave DNA interstrand crosslinks, resulting in DNA adducts and double-stranded DNA breaks. The latter aberrant DNA fragments are resolved by exonucleases and by homologous recombination, both involving additional FA proteins.
Currently, the only curative treatment for the hematopoietic failure in FA is hematopoietic stem cell transplantation. However, short-term and long-term complications (e.g., serious infections, organ failure and graft-versus-host disease) are not uncommon.3 Furthermore, hematopoietic stem cell transplantation significantly increases the risk of solid tumors in this population of patients.4
Due to the limited treatment options and their associated risks, gene therapy provides an attractive alternative possibility. However, there are several challenges with gene therapy which are specific to inherited bone marrow failure syndromes such as FA. For example, although mobilization, collection and cryopreservation of sufficient peripheral blood CD34+ cells from FA patients are feasible with the administration of granulocyte colony-stimulating factor or plerixafor,5 this process is challenging for most such patients. The reason is that most FA patients are diagnosed
when they have substantial cytopenia and their hematopoietic stem cells are depleted.6
Several gene therapy trials have been conducted in FA and various others are currently being conducted in Europe and the USA. Preliminary data showed that FANCA gene therapy without conditioning therapy is safe in FA and corrected cells are engrafted and manifest a proliferative advantage.5,7,8 Furthermore, reasonable blood counts were sustained for several years.5 However, long-term follow-up studies are lacking. Therefore, we do not know whether gene therapy can abrogate the FA phenotype of a gradual depletion of hematopoietic stem cells and decline in blood cell counts. Furthermore, we do not know whether gene therapy can provide hematopoietic stem cells with the long-term ability to maintain normal or sufficient molecular and functional properties. We also do not know how long corrected hematopoietic stem cells can generate hematopoietic stem and progenitor cells with normal or sufficient molecular and functional properties.
A study by Lasaga and colleagues, published in the current issue of Haematologica, 9 aimed to address some of the above issues by investigating four previously described FA patients with FANCA mutations who had successfully undergone gene therapy using a PGK-FANCA-WPRE lentiviral vector.5 The vector contains a phosphoglycerate kinase (PGK) promoter, woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) sequence that enhances expression, and FANCA. Bone marrow CD34+ cells were harvested from the patients, transduced with the vector, and then infused back into the patients without conditioning. The analysis in the work by Lasaga et al. was performed 25 years after the gene therapy. The corrected cells were estimated to constitute 26-77% of bone marrow CD34+ cells. Using single-cell RNA sequencing the authors demonstrated that the lentiviral-mediated gene therapy resulted in substantial correction of the transcriptional program in hematopoietic stem and progenitor cells of patients with FA, bringing it closer to the transcriptional program of normal hematopoietic stem and progenitor cells.9 Importantly,
the expression of several genes that had been shown to be upregulated and mediate bone marrow failure in FA, such as p2110 and TGFβ, 11 were normalized after gene therapy. The authors also showed that gene therapy improved cellular properties, including reduced chromosomal breakage of peripheral blood T cells in response to diepoxybutane, and a slower rate of telomere length reduction in peripheral blood cells, as determined by quantitative polymerase chain reaction, compared to the cellular properties of a control group of patients who were not engrafted after cell transduction. Furthermore, gene therapy improved cellular function, specifically survival of hematopoietic progenitors plated in cultures with mitomycin C and their ability to form colonies, which likely facilitates a progressive increase in
1. Peake JD, Noguchi E. Fanconi anemia: current insights regarding epidemiology, cancer, and DNA repair. Hum Genet. 2022;141(12):1811-1836.
2. Badra Fajardo N, Taraviras S, Lygerou Z. Fanconi anemia proteins and genome fragility: unraveling replication defects for cancer therapy. Trends Cancer. 2022;8(6):467-481.
3. Bonfim C, Ribeiro L, Nichele S, et al. Long-term survival, organ function, and malignancy after hematopoietic stem cell transplantation for Fanconi anemia. Biol Blood Marrow Transplant. 2016;22(7):1257-1263.
4. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica. 2018;103(1):30-39.
5. Rio P, Navarro S, Wang W, et al. Successful engraftment of genecorrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat Med. 2019;25(9):1396-1401.
6. Heidemann S, Bursic B, Zandi S, et al. Cellular and molecular
the number of bone marrow and peripheral blood genecorrected cells.
The findings described in the paper by Lasaga et al. are important and document positive effects of gene therapy. Although the results are very promising, there are still questions that need to be addressed. For example, the ability of gene therapy to provide sufficient hematopoietic stem and progenitor cell numbers and blood cell counts for the full lifespan of a patient needs to be documented. The sustainability of the transcriptional program, DNA integrity and telomere length also still need to be shown.
Disclosures
No conflicts of interest to disclose.
architecture of hematopoietic stem cells and progenitors in genetic models of bone marrow failure. JCI Insight. 2020;5(4):e131018.
7. Kelly PF, Radtke S, von Kalle C, et al. Stem cell collection and gene transfer in Fanconi anemia. Mol Ther. 2007;15(1):211-219.
8. Adair JE, Becker PS, Chandrasekaran D, et al. Gene therapy for Fanconi anemia in Seattle: clinical experience and next steps. Blood. 2016;128(22):3510.
9. Lasaga M, Río P, Vilas-Zornoza A, et al. Gene therapy restores the transcriptional program of hematopoietic stem cells in Fanconi anemia. Haematologica. 2023;108(10):2652-2663.
10. Ceccaldi R, Parmar K, Mouly E, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11(1):36-49.
11. Zhang H, Kozono DE, O'Connor KW, et al. TGF-β inhibition rescues hematopoietic stem cell defects and bone marrow failure in Fanconi anemia. Cell Stem Cell. 2016;18(5):668-681.
Department of Medicine, Karolinska Institutet, Huddinge, Stockholm, Sweden
Correspondence: G. Gahrton gosta.gahrton@ki.se
Received: February 24, 2022.
Accepted: March 1, 2023. Early view: March 9, 2023.
htps://doi.org/10.3324/haematol.2023.282896
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Multiple myeloma (MM) is a cunning disease. Despite enormous progress in its treatment, cure is uncommon. Not long ago, some of the most important researchers in the field seemed to be happy if it could be turned into a chronic disease. Others went for cure.
The first attempts to cure the disease included autologous1 and allogeneic2 transplants after high-dose myeloablative treatment. Occasional patients survived 20 years or more, mainly following allogeneic transplantation. The idea that high-dose treatment was not the most important part of the allogeneic approach, but rather the graft-versus-myeloma effect exerted by the transplanted allogeneic lymphocytes, induced a modification of the procedure using a reduced intensity conditioning and a preceding auto-transplant.3 Results were encouraging, but relapses continued to occur, and cures were rare. Donor lymphocyte transfusion to treat relapses were successful in the short term4,5 and were proof of principle that cell therapy with allogeneic lymphocytes could induce responses lasting for several months or even years. However, the relatively short-term donor lymphocyte effect was unspecific and frequently caused graft-versushost disease. Thus, the real breakthrough was creating a lymphocyte, i.e., a T lymphocyte, that could target specific antigens on the tumor cells. An effective chimeric antigen receptor (CAR) T cell had already been produced in the early 1990s,6 and, after some delay, CAR T cells for clinical use were developed. From the beginning of the 21st century until now, the number of clinical cancer trials with such cells has exploded.
In MM, the most common target for CAR T cells has been B-cell maturation antigen (BCMA), but many other antigens on myeloma cells are currently being explored as targets. CAR T cells are T cells modified ex vivo to express a chimeric receptor. This receptor contains an antigen receptor, i.e., a single chain variable fragment (scFv), and an intracellular T-cell receptor (TCR) signaling domain. The scFv recognizes the target tumor cells, while the intracellular domain contains various components to
enhance efficacy and safety. In most of the ongoing studies, autologous cells are used for CAR T-cell production.
Two such CAR T-cell products have been approved for treatment of MM by both the US Food and Drug Administration and the European Medicines Agency, based on two phase II studies including 128 patients treated with idecabtagene vicleucel (ide-cel)-KarMMa,7 and 97 treated with ciltacabtagene autoleucel (cilta-cel)-CARTITUDE,8 respectively. Both trials have shown impressive results in triple-penta refractory patients with a median of six lines of previous treatment. A recent updated retrospective comparison has shown superior response (objective response rate 97.9%; stringent complete response 82.5%), progression-free survival (PFS) (62% reduction in risk), and overall survival (OS) (57% reduction in risk) with cilta-cel as compared to ide-cel.9 Other studies including fewer patients have also shown impressive results short term. Although these results are encouraging, the number of patients investigated in each of the studies is not high enough to provide reliable information of the impact of prognostic factors such as presence of extramedullary disease or high-risk cytogenetics.
In the present issue of Haematologica, Gagelmann et al.10 attempt to analyze these important prognostic factors retrospectively by compiling results from available CAR T-cell clinical trials. Out of 769 screened articles, they found 17 including 723 patients with heavily pre-treated relapsed or refractory MM patients with data including enough information about these prognostic factors. A comprehensive statistical analysis was able to conclude that extramedullary disease (EMD) did not hamper response, but PFS was decreased by 44% and OS was shorter due to EMD. Cytogenetic high-risk patients did even worse in comparison to the non-high-risk patients. The risk of no response was increased by 14%, the risk of minimal residual disease positivity by 23%, and the risk of progression/relapse was increased by 70%, affecting the OS that was significantly shortened.
Thus, it seems that CAR T-cell treatments currently in use do not overcome the poor prognostic impact of EMD or highrisk cytogenetics; new approaches are, therefore, warranted. In most of the included patients, BCMA was the target for the CAR T cells and autologous cells were used. Ongoing studies using CAR T cells directed against other antigens, such as the orphan G protein-coupled receptor, class C group 5 (GPRC5),11 bidirected CAR T cells, allogeneic CAR T cells, natural killer (NK) cells12 or CAR NK cells may show better response and outcome for high-risk relapsed and refractory myeloma patients. Future studies should move CAR T-cell treatment to an earlier stage of the disease. Myeloma
1. Barlogie B, Hall R, Zander A, Dicke K, Alexanian R. High-dose melphalan with autologous bone marrow transplantation for multiple myeloma. Blood. 1986;67(5):1298-1301.
2. Gahrton G, Ringden O, Lonnqvist B, Lindquist R, Ljungman P. Bone marrow transplantation in three patients with multiple myeloma. Acta Med Scand. 1986;219(5):523-527.
3. Gahrton G, Iacobelli S, Bjorkstrand B, et al. Autologous/reduced-intensity allogeneic stem cell transplantation vs autologous transplantation in multiple myeloma: long-term results of the EBMT-NMAM2000 study. Blood. 2013;121(25):5055-5063.
4. Tricot G, Vesole DH, Jagannath S, Hilton J, Munshi N, Barlogie B. Graft-versus-myeloma effect: proof of principle. Blood. 1996;87(3):1196-1198.
5. Aschan J, Lonnqvist B, Ringden O, Kumlien G, Gahrton G. Graftversus-myeloma effect. Lancet. 1996;348(9023):346.
6. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720-724.
7. Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J
with EMD and high-risk cytogenetics may well be treated up front with CAR T-cell therapy, and responses consolidated with other treatments, such as bispecific antibodies or allogeneic transplants. The goal must be cure.
Disclosures
GG is Advisor to the Fujimoto Pharmaceutical Corporation, Japan, and has received honorarium from them. In 2022 GG received honorarium from an interview with BMS. GG has shares in a small innovation Karolinska Institutet associated company called XNK (with no value on the market). GG has shares in Astra Pharmaceuticals.
Med. 2021;384(8):705-716.
8. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 openlabel study. Lancet. 2021;398(10297):314-324.
9. Martin T, Usmani SZ, Schecter JM, et al. Updated results from a matching-adjusted indirect comparison of efficacy outcomes for ciltacabtagene autoleucel in CARTITUDE-1 versus idecabtagene vicleucel in KarMMa for the treatment of patients with relapsed or refractory multiple myeloma. Curr Med Res Opin. 2023;39(1):81-89.
10. Gagelmann N, Ayuk FA, Klyuchnikov E, Wolschke C, Berger SC, Kröger N. Impact of high-risk disease on the efficacy of chimeric antigen receptor T-cell therapy for multiple myeloma: a metaanalysis of 723 patients. Haematologica. 2023;108(10):2799-2802.
11. Zhang M, Wei G, Zhou L, et al. GPRC5D CAR T cells (OriCAR-017) in patients with relapsed or refractory multiple myeloma (POLARIS): a first-in-human, single-centre, single-arm, phase 1 trial. Lancet Haematol. 2023;10(2):e107-e116.
12. Nahi H, Chrobok M, Meinke S, et al. Autologous NK cells as consolidation therapy following stem cell transplantation in multiple myeloma. Cell Rep Med. 2022;3(2):100508.
André Baruchel,1 Jean-Pierre Bourquin,2 John Crispino,3 Sergi Cuartero,4 Henrik Hasle,5 Johann Hitzler,6 Jan-Henning Klusmann,7 Shai Izraeli,8,9 Andrew A. Lane,10 Sébastien Malinge,11 Karen R. Rabin,12 Irene Roberts,13 Sandra Ryeom,14 Sarah K. Tasian15 and Elvin Wagenblast16
1Hôpital Universitaire Robert Debré (APHP and Université Paris Cité), Paris, France; 2University Children’s Hospital, Zurich, Switzerland; 3St. Jude Children’s Research Hospital, Memphis, TN, USA; 4Josep Carreras Leukemia Research Institute, Barcelona, Spain; 5Department of Pediatrics and Adolescent Medicine, Aarhus University Hospital, Aarhus, Denmark; 6The Hospital for Sick Children, Toronto, Ontario, Canada; 7Department of Pediatrics, Goethe University Frankfurt, Frankfurt, Germany; 8Schneider Children's Medical Center of Israel, Petah Tikva, Israel; 9Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel; 10Dana-Farber Cancer Institute, Boston, MA, USA; 11Telethon Kids Institute - Cancer Centre, Perth, Western Australia, Australia; 12Baylor College of Medicine, Texas Children's Cancer Center, Houston, TX, USA; 13University of Oxford, Oxford, UK; 14Department of Surgery, Herbert Irving Comprehensive Cancer Center, Columbia University Irving Medical Center, New York, NY, USA; 15Children’s Hospital of Philadelphia and University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA and 16Icahn School of Medicine at Mount Sinai, New York, NY, USA
Correspondence: S. Cuartero scuartero@carrerasresearch.org
S. Malinge sebastien.malinge@telethonkids.org.au
S. Ryeom swr2117@cumc.columbia.edu
Received: May 2, 2023.
Accepted: June 29, 2023.
Early view: July 13, 2023.
htps://doi.org/10.3324/haematol.2023.283225
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Children with Down syndrome (DS, trisomy 21) are at a significantly higher risk of developing acute leukemia compared to the overall population. Many studies investigating the link between trisomy 21 and leukemia initiation and progression have been conducted over the last two decades. Despite improved treatment regimens and significant progress in identifying genes on chromosome 21 and the mechanisms by which they drive leukemogenesis, there is still much that is unknown. A focused group of scientists and clinicians with expertise in leukemia and DS met in October 2022 at the Jérôme Lejeune Foundation in Paris, France for the 1st International Symposium on Down Syndrome and Leukemia. This meeting was held to discuss the most recent advances in treatment regimens and the biology underlying the initiation, progression, and relapse of acute lymphoblastic leukemia and acute myeloid leukemia in children with DS. This review provides a summary of what is known in the field, challenges in the management of DS patients with leukemia, and key questions in the field.
Children with Down syndrome (DS, trisomy 21) have a significantly increased risk of both myeloid and lymphoid leukemia compared to the general pediatric population.1 Understanding the mechanisms of leukemia predisposition related to constitutive trisomy 21 (T21) and characterizing the genetic landscape and multistep pathogenesis of DS-associated leukemias have led to major discoveries over the last two decades 2-11 (Figure 1). Notably, many of the genetic, cellular, and molecular mechanisms found in DS-associated leukemias are relevant in non-DS individuals, as gain of chromosome 21 is also frequently observed in hematologic malignancies as a somatic event.12
Significant progress has been made in the treatment of
children with myeloid leukemia of Down syndrome (MLDS) with 5-year survival now approaching 90%. In contrast, children with Down syndrome-associated Bcell acute lymphoblastic leukemia (DS-ALL) have worse outcomes than non-DS children with ALL, in part due to a high sensitivity to chemotherapy.13,14 Outcomes for relapsed/refractory leukemia in children with DS are extremely poor, highlighting the need to improve quality of care for these children who have other T21-associated health issues that complicate their chemotherapy treatment. This review summarizes the current knowledge in the field and the discussions of a panel of scientists and clinicians on topics that range from treatment regimens and complications, to leukemia predisposition and initiation, from mechanistic insights to key questions in the field.
Studies on the unique epidemiological patterns of cancer in DS include those that identified an increased risk of leukemia in children with DS ranging from a 3- to 100-fold
increase with the true risk increase estimated at 10- to 20-fold. The ratio of lymphoid to myeloid leukemias is higher in non-DS children, at 5:1, than it is in children with DS, in whom the ratio is closer to 1:1. One of the largest studies investigating the incidence of leukemia in DS was a Danish study1 examining 2,814 children with DS with long-term updates provided in 2016.15 Overall, the cumulative risk of leukemia in children with DS is 2% by 5 years
Figure 1. Overview of Down syndrome-associated leukemia. Trisomy 21 affects fetal blood formation, causing an increase in hematopoietic stem cells and megakaryocyte-erythroid progenitors, but a decrease in B-cell progenitors. Mutations in GATA1 cause transient abnormal myelopoiesis, which can lead to myeloid leukemia of Down syndrome (ML-DS) upon acquisition of additional somatic mutations. In the lymphoid branch, alterations in CRLF2 or JAK2 can lead to Down syndrome-associated acute lymphoblastic leukemia (DS-ALL). In both ML-DS and DS-ALL, the increased dosage of genes located on chromosome 21 cooperates with somatic mutations in disease onset and progression. Figure generated with BioRender.com. HSC: hematopoietic stem cells; MEP: megakaryocyte-erythroid progenitors; chr 21: chromosome 21.

of age and 2.5% by 30 years of age. In DS-ALL, about 50% of patients overexpress CRLF2 and 20% have JAK2 mutations. GATA1 mutations are seen in almost all patients with ML-DS but are effectively absent in non-DS children. MLDS is also associated with somatic mutations in other genes, with the next most common group of alterations found in genes encoding effectors of the cohesin complex as well as epigenetic regulators.3,4,6 The unique ML-DS disease may also occur in rare individuals with a germline GATA1 mutation and acquired T21.16
Another unusual aspect of the cancer spectrum in DS is the reduced frequency of solid tumors in this population,15,17,18 with the exception of testicular cancer. Possible explanations for this decreased cancer incidence were discussed in a report by Ossuna-Marco and colleagues.19 Based upon these observations, cancer screening recommendations for adults with DS20 include screening for colon cancer similar to the general public, no screening for breast cancer, and screening for cervical cancer only in sexually active women 25 years and older. Annual screening for testicular cancer is recommended between the ages of 15 and 45 years.
The key clinical subsets of DS-associated leukemias are: (i) transient abnormal myelopoiesis (TAM); (ii) ML-DS with GATA1s mutation; and (iii) DS-ALL. A recent study examining leukemia risk in a cohort of 3.9 million children, of whom 4,401 had DS, documented a statistically significant risk of acute myeloid leukemia (AML) before the age of 5 years with the risk being highest for ML-DS. This raises the question of whether all children with DS should be empirically screened for GATA1s. Early intervention approaches, such as the TMD 2007 prevention trial (TMD07) in which 102 infants with DS and clinical symptoms of TAM were treated at diagnosis or 8 weeks after positive minimal residual disease (MRD) detection with low-dose cytarabine chemotherapy, showed a reduction in TAM-related mortality, but did not prevent disease progression to future ML-DS.21 ML-DS can be associated with low blood counts and bone marrow blasts, which may be lower than 20% in the bone marrow and mostly show a megakaryocytic phenotype (previously classified as acute megakaryocytic leukemia in children with DS). Central nervous system involvement and chromosomal translocations are observed more frequently in non-DS acute megakaryocytic leukemia than in ML-DS. DS blasts are hypersensitive to chemotherapy, especially cytarabine, etoposide, and anthracyclines. Outcomes for children with ML-DS are favorable with event-free survival approaching 90%. The early treatment experience showed that high dose-intensive chemotherapy was not beneficial in children with ML-DS due to high treatment-related mortality.22 Subsequently, clinicians in-
troduced DS-specifi c myeloid-directed chemotherapy protocols that pursued a stepwise reduction of treatment intensity and achieved a reduction of both treatment-related mortality and cardiotoxicity. A recent study by the Children’s Oncology Group (COG) in children with ML-DS reduced treatment intensity through approaches including: (i) anthracycline chemotherapy reduction; (ii) etoposide reduction; and (iii) identification of patients on the basis of flow MRD or high-risk ML-DS for treatment with high-dose cytarabine-containing cycles. However, the trial was stopped due to the inability to define standard-risk patients with ML-DS for high-dose cytarabine omission by flow cytometry MRD assessment.23 While very low-dose cytarabine regimens can be curative,24 an appropriate subset of patients who can be cured with therapy reduction cannot currently be identified.
The most recent COG AAML1531 phase III clinical trial for children with ML-DS investigated whether it is necessary to use high-dose cytarabine as part of treatment in all patients with ML-DS given that the bulk of infectious adverse events is associated with this treatment element. Results of this study called into question whether stratification of treatment intensity for ML-DS can be based on MRD. MRD levels were measured in the bone marrow by flow cytometry after the first course of treatment. Patients with ML-DS who were MRD-negative were treated without high-dose cytarabine but turned out to have an inferior probability of survival compared to historical controls (treated with high-dose cytarabine). The conclusion was that the intensity of treatment could not be reduced by omitting high-dose cytarabine even for MRD-negative patients.23 While treatment-related mortality has been successfully reduced for children with ML-DS, management of relapsed/refractory ML-DS is emerging as a therapeutic challenge. Whereas patients with primary ML-DS have a very favorable prognosis, the outlook for those with relapsed/refractory disease is dire.
Overall, challenges of ML-DS treatment include how to perform risk stratification integrating new information about molecular subgroups of ML-DS, how to reduce relapse events, how to improve outcomes for refractory and relapsed ML-DS and how to get access to new agents for patients with DS, who historically were routinely excluded from early phase trials. There is a need for a leukemia prevention strategy in children with DS if it becomes feasible to identify the subset of patients with TAM at highest risk of progression to ML-DS and to develop an early, safe, and efficacious intervention.
Analysis of eight clinical trials across ten countries with matched DS-ALL and non-DS-ALL patient characteristics found lower event-free survival for patients with DS-ALL. Event-free survival for patients with ETV6-RUNX1 fusions
was 79% in DS-ALL and 96% in non-DS-ALL cases. Patients with DS-ALL harboring IKZF1 deletions were also noted to be at highest risk of MRD positivity and/or relapse. Dr. Baruchel discussed the ongoing European ALLTogether clinical trial (NCT03911128) that includes pediatric patients with DS-ALL treated with blinatumomab (a monoclonal antibody targeting CD19) replacing two chemotherapy consolidation blocks. In this trial, patients with standard-risk DS-ALL are not receiving blinatumomab, while those with intermediate-risk disease are being randomized to blinatumomab-containing therapy or standard-of-care chemotherapy. A recent study of 16 children with relapsed/refractory DS-ALL treated with CD19-directed chimeric antigen receptor (CAR) T-cell therapy found clinical outcomes and toxicities that were comparable to those of children with non-DS-ALL,25 further supporting the potential use of CD19-targeted immunotherapy for children with DS-ALL in first relapse in an attempt to avoid the toxicity of hematopoietic stem cell (HSC) transplantation. In an ongoing study of the French LEA cohort with more than 6,000 children cured of acute leukemia, 67 patients with DS-ALL are being compared to 201 matched patients with non-DS ALL and also to age-matched DS patients without leukemia followed at the Fondation Jerôme Lejeune (Paris, France). This study will help to dissect what complications are related to ALL treatment and which are predictably related to DS, including overweight, cataracts, bone mineral deficiency, and hypothyroidism (L. Nizery, C. Mircher, G. Michel, A. Baruchel; personal communication). Outcome data from cases of DS-ALL (n=743) compared to non-DS-ALL (n=21,703) treated on four COG trials conducted between 2003-2019 demonstrate the challenges in the clinical management of DS-ALL.26 Favorable cytogenetic alterations (ETV6::RUNX1 and trisomy of chromosomes 4 and 10) were significantly less frequent in DS-ALL than in non-DS-ALL (14.4% vs. 46.7%, P<0.0001, as were unfavorable cytogenetic alterations (BCR::ABL1, KMT2A-R, hypodiploid, and iAMP21) (0.7% vs. 7.2%, P<0.0001), whereas neutral cytogenetic alterations were more frequent (84.9% vs. 46.2%, P<0.0001). Early treatment-related mortality was observed among DS-ALL patients; the deaths were primarily due to overwhelming sepsis during periods of neutropenia. Treatment modi fi cations were then implemented, including reductions in anthracyclines and intravenous methotrexate, administration of leucovorin rescue after intrathecal methotrexate, shortened maintenance duration for boys, and reduction of vincristine/steroid pulses during maintenance. Enhanced supportive care measures were instituted, including inpatient observation during intensely myelosuppressive phases, antibiotic and antifungal prophylaxis, and IgG monitoring and replacement. The 5-year event free survival and overall survival were poorer for DS-ALL than for non-DS-ALL patients overall, and within the National Cancer Institute’s
standard-risk and high-risk subgroups. The cumulative incidence of relapse was higher (11.7% vs. 9.3%, P=0.0198), as were the risks of induction death (3.4% vs. 0.8%, P<0.0001) and death in remission (4.8% vs. 1.7%, P<0.0001). DS-ALL cases with CRLF2 overexpression had a better 5year event free survival and better overall survival than non-DS-ALL cases. In multivariable analysis, risk factors independently associated with inferior event-free survival in DS-ALL included age at diagnosis ≥10 years, initial white blood count ≥50x109/L, and end-induction MRD ≥0.01%. Patients with DS also exhibited increased rates of mucositis, infection, hyperglycemia, and seizures.
The most recent COG trial for children with DS incorporates blinatumomab to replace some elements of intensive chemotherapy. An increased risk of seizures was observed among DS-ALL patients over 10 years old, leading to an amendment requiring antiseizure prophylaxis during blinatumomab infusions for these patients. The trial has been temporarily suspended because it met a futility rule for inability to demonstrate a statistically significant decrease in treatment-related mortality in high-risk DS patients. The next COG trial for DS-ALL will likely employ a different approach for high-risk DS-ALL patients, extensively utilizing immune-targeted therapies such as blinatumomab and inotuzumab (a monoclonal antibody targeting CD22) to replace intensive chemotherapy blocks (K. Rabin; personal communication).
The frequency of CRLF2 rearrangements (either P2RY8::CRLF2 fusions or IGH::CRLF2 translocations) is high in DS-ALL and these rearrangements are also the most common genetic alteration in approximately 50% of Philadelphia chromosome-like (Ph-like) ALL.27,28 Preclinical and clinical studies demonstrated that constitutive JAK/STAT and other kinase signaling in CRLF2-rearranged Ph-like ALL may be targetable by tyrosine kinase inhibitors.28-31 Emerging data from Dr. Tasian’s laboratory suggest that preclinical CRLF2-rearranged DS-ALL and non-DS Ph-like ALL patientderived xenografts (PDX) are similarly sensitive to the selective JAK1/2 inhibitor ruxolitinib in vivo. Following demonstration of safety and identification of a recommended phase II dose of ruxolitinib in children with relapsed/refractory cancers in the COG ADVL1011 phase I trial,32 ruxolitinib in combination with multi-agent chemotherapy is under evaluation in children, adolescents, and young adults with newly-diagnosed CRLF2-rearranged or other JAK pathway-mutant Ph-like ALL via the recently completed COG AALL1521 phase II clinical trial that is currently awaiting outcome data to address its primary endpoint.33 However, patients with DS-ALL were excluded from participation in ADVL1011 and AALL1521, so the potential clinical activity of JAK inhibitor strategies in this population remains unknown.
Given the inferior clinical outcomes of children with DSALL and their increased risk of toxicity, particularly with
HSC transplantation, significant interest exists in development of immunotherapeutic strategies. In addition to the aforementioned CD19- and CD22-targeting antibodybased and cellular immunotherapies, recent preclinical studies have shown potent activity of CAR T cells targeting the thymic stromal lymphopoietin receptor (TSLPR) encoded by CRLF2 34 A first-in-human phase I clinical trial of TSLPR-CAR T-cell immunotherapy for adults and children with relapsed/refractory TSLPR/CRLF2-positive leukemia is planned to open in 2023. Additional preclinical studies in CRLF2-rearranged Ph-like and DS-ALL PDX models are also investigating the therapeutic potential of combining CAR T cells and kinase inhibitors to augment long-term remission. A recent report by Tasian and colleagues showed that co-treatment with ruxolitinib and TSLPR CAR T cells impaired T-cell functionality, although it was beneficial in reducing life-threatening inflammatory cytokine production. Conversely, delayed administration of ruxolitinib augmented anti-leukemia activity in both Ph-like ALL and DS-ALL and suggests future potential for a ‘maintenance’ inhibitor therapeutic strategy following CAR Tcell immunotherapy.35
The current European IntReALL clinical trial for children with relapsed ALL includes correlative biology studies of biospecimens from patients with very high-risk non-DS ALL and DS-ALL with the goal of drug response profiling. Machine learning and artificial intelligence can be used to perform such profiling in ALL cells with the ability to classify specific cell types (e.g., leukemia versus stromal) and perform cell segmentation analysis. An individual drug sensitivity profile can be developed for patient cohorts and will allow the development of new disease management strategies for patients with relapsed/refractory leukemia. Drug response profiling can also be utilized as a tool to identify appropriate precision therapies for interval stabilization of patients prior to CD19-CAR T-cell immunotherapy. Anecdotes of patients in whom drug response profiling was used as a ‘bridge’ until the next treatment include a highly chemorefractory ALL patient who underwent drug response profiling and was treated with cytarabine and the MEK inhibitor, trametinib due to the presence of a KRASG12D mutation. Leukemia burden was markedly reduced, and the patient was ultimately able to proceed to HSC transplantation in remission. Drug response profiling was conducted in another patient with a CNTRL::ABL fusion and identified sensitivity to ponatinib (a tyrosine kinase inhibitor), which was used for leukemia stabilization prior to CD19 CAR T-cell therapy. Drug response profiling complements existing technology platforms for precision oncology, and an International Leukemia Board for relapsed/refractory disease is now utilizing patients’ data from drug response profiling studies. A Pan-European precision tumor board is further prioritizing targets and therapy options by embedding drug
response profiling into early phase clinical trials to guide therapy or individualized therapies as well harmonizing the data dictionary and decentralizing data management.
The role of T21 in disrupting fetal hematopoiesis occurs through genome-wide transcriptional perturbation, including genes encoding transcription factors, pro-inflammatory cytokines, and various microRNA in fetal hematopoietic stem and progenitor cells and in stromal cells. Infants with DS display expanded prenatal HSC and myeloid progenitors, with fetal liver HSC being significantly biased toward erythro-megakaryopoiesis compared to disomic controls. The impact of T21 in early hematopoiesis has been confirmed using human induced pluripotent stem cells (iPSC).36-38 Indeed, reproducing hematopoiesis from trisomic iPSC has shown that T21 alone is sufficient to enhance erythropoiesis, and that DS fetal-like hematopoietic progenitors have an increased capacity to form myeloid and megakaryoblastic colonies.
The pre-leukemic step, TAM (which precedes ML-DS) is unique to DS, is almost exclusively due to acquired GATA1s in T21 cells and often undergoes spontaneous remission shortly after birth (before 90 days). However, in some cases the GATA1s mutation persists, and children with DS develop ML-DS. The transcription factor GATA1 is a master regulator of blood cell development, especially erythropoiesis and megakaryopoiesis, and mutations are found in virtually all TAM and ML-DS cases. This somatic mutation leads to the expression of a shorter isoform named GATA1s, lacking the N-terminal transactivation domain. Interestingly, these mutations have been recently identified in familial congenital anemia39 and in a subset of Diamond-Blackfan anemia that displays dyserythropoiesis and dysmegakaryopoiesis,40,41 as well as somatically in megakaryoblastic leukemias in children without DS who present a genetic landscape similar to that of ML-DS, including gain of chromosome 21.42-44 Roberts and colleagues at Oxford have identi fied GATA1s mutations present in ~25% of neonates with DS.45 Seven of these infants ultimately developed ML-DS (6%); one of these patients had a clinically ‘silent’ TAM, with blasts <10%, no clinical features and a low variant allele frequency of a GATA1s mutation. More than 90% of GATA1 mutations and blasts resolve spontaneously, disappearing before 90 days of life with no GATA1s mutations acquired postnatally. It has been found that GATA1s blasts are much less proliferative in the postnatal environment until additional muta-
tions are acquired and ML-DS develops. Applying CRISPR/Cas9-mediated genome engineering strategies in T21 livers from normal and DS fetuses, Wagenblast and colleagues showed that GATA1s induces a bias toward megakaryopoiesis and cooperates with T21 to lead to a TAM phenotype.10 In vitro analyses of human iPSC derived from TAM specimens or genetically engineered to express GATA1s, confirmed that DS hematopoietic progenitors expressing GATA1s are biased toward the myelo-megakaryocytic compartment.38,46,47
The existence of a pre-leukemic state in DS-ALL is currently unknown, but recent results suggest that T21 alone has an impact on fetal B lymphopoiesis, compatible with a prenatal origin. Jardine and colleagues described the effect of T21 on fetal bone marrow hematopoiesis with a predominance of erythroid cells, a significant loss of B progenitors, and a skewing of B and NK lymphoid output of fetal bone marrow HSC and multipotent progenitors.9 These cells are unable to make B cells even in vitro 48 T21 human iPSC also have a decreased ability to form B cells in vitro. 49 Fetal T21 cells are less efficient at making T cells and there is an activation of pro-inflammatory patterns of gene expression.9 T21 also perturbs gene expression in stromal cells9 and studies of stromal cell function are currently ongoing.
A genome-wide association study of inherited genetic variants associated with ALL susceptibility in children with DS7 included 542 cases (children with DS-ALL) and 1,192 controls (children with DS without ALL). The study identified four susceptibility loci at genome-wide significance, at IKZF1, CDKN2A, ARID5B, and GATA3. While these loci have also been associated with ALL susceptibility in children without DS, T21 appears to modify the penetrance of inherited ALL susceptibility with a greater magnitude of effect size for CDKN2A. A whole genome sequencing study is in progress, which will enable identification of rare, structural, and chromosome 21 variants associated with ALL susceptibility in DS. Another study in progress is evaluating the association between co-occurring birth defects and ALL risk in children with DS. Dr. Rabin presented results of a study of somatic genomic characterization of a cohort of 295 cases of DS-ALL characterized by whole genome and whole transcriptome sequencing.50 In accordance with prior reports, CRLF2 rearrangements were identified in 54% of DS-ALL, versus 6% of a non-DS-ALL comparator cohort. CRLF2-rearranged cases were significantly younger at diagnosis and more frequently involved P2RY8 versus IGH as the fusion partner in DS-ALL compared to non-DS-ALL.
Mouse models of DS have been utilized to ‘hunt’ for oncogene(s) on T21. Dr. Klusmann’s laboratory performed a CRISPR screen for potential new oncogenes on T21 using
CMK cells (ML-DS) and identified RUNX1 as a candidate.51 Deletion of RUNX1 in an ML-DS PDX model suppressed xenograft growth. RUNX1 is transcribed from two different promoters and has three isoforms in humans: RUNX1A, 1B, and 1C. ML-DS patients present a disequilibrium of these RUNX1 isoforms with a higher RUNX1A/RUNX1C ratio. RUNX1A blocks megakaryoblast differentiation while the 1C isoform blocks proliferation. RUNX1A expression cooperates with GATA1s to increase megakaryocytic phenotypes and induce an ML-DS-like phenotype in vivo. Mechanistically, RUNX1A binds to the MYC binding partner MAX allowing for upregulation of a MYC/E2F-induced proliferative program. Interestingly, GATA1s plus RUNX1A synergize with miR-125b expression to further enhance a malignant phenotype. Recent observations suggest that the therapeutic potential of MYC inhibitors, including MYCi361, to disrupt the RUNX1A/MAX interaction should be explored in ML-DS, as well as in other myeloid leukemias harboring T21.52
ERG is an ETS-family transcription factor very similar to FLI1, and both are involved in oncogenic fusions found in Ewing sarcoma. As ERG is located on chromosome 21, studies have addressed a possible role for ERG overexpression in fetal liver hematopoiesis, which could explain the aberrant hematopoietic differentiation of T21 and possible involvement in subsequent TAM or ML-DS. Dr. Izraeli and colleagues showed that ERG overexpression transforms mouse fetal liver megakaryocytic progenitors and increases the production of megakaryocyte-erythroid progenitors, especially in cooperation with GATA1s. High ERG expression correlates with poor prognosis in AML and is associated with an increased fraction of leukemic stem cells. Forced expression of ERG represses myeloid differentiation genes and the activation of HSC self-renewal genes. It was found that a single amino acid substitution (proline 199 to leucine: P199L) at the end of the ‘pointed’ (PNT) domain is involved in protein-protein interactions. In pre-leukemic conditions, the PNT domain has a role in preserving the stem cell phenotype associated with ERG, thereby preventing premature differentiation.53 Although it does not cause leukemia on its own or change the secondary structure of ERG, P199L is sufficient to disrupt ERG’s transforming ability in fetal liver transplantation assays and self-renewal in colony-forming assays. Interestingly, the myeloid differentiation block normally induced by ERG was partially reversed by the P199L mutant. Using Bio-ID proximity ligation mass spectrometry, Dr. Izraeli found that the P199L mutant was unable to interact with the HDAC3/NCoR repressive complex. While H3K27 acetylation at myeloid differentiation genes was lost in ERG-transduced cells, this did not happen in cells with the ERG P199L mutant. In AML cell lines that were dependent on ERG for growth, targeting HDAC3 blocked their growth in vitro and in vivo, but did not in AML
cells that were ERG-independent, suggesting that HDAC3 inhibition may be a therapeutic strategy for ERG-dependent AML (S. Izraeli, unpublished data). Future challenges include the investigation of the role of epigenetic interactors with ERG in leukemia and other malignancies. Dr. Klusmann and colleagues have also investigated three chromosome 21 microRNA (miRNA) for their contribution to ML-DS. Notably, these three miRNA are located in a region of chromosome 21 that is not included in the commonly used Ts65Dn mouse model of DS. The three candidate miRNA: miR-125b, miR-99a and let-7c were investigated in vitro using mouse fetal liver cells genetically engineered by CRISPR to generate a GATA1s mutation. Studies by Dr. Klusmann’s group investigating each of the miRNA in different combinations found that only miR-125b cooperates with GATA1s to increase the pool of megakaryocyte progenitors in vitro and led to aggressive acute megakaryocytic leukemia when transplanted into recipient mice.54 They identified the epigenetic regulator ARID3A as the main target of miR-125b and showed that ARID3A is highly expressed during normal megakaryopoiesis. This study also showed that ARID3A is post-transcriptionally repressed by mir-125b, which is highly expressed in TAM and ML-DS. Loss or knock-down of ARID3A leads to enhanced proliferation and prevents differentiation of megakaryocyte progenitors promoting an ML-DS-like signature, as well as enrichment of GATA1s-related E2F and MYC targets. Their data show that ARID3A is a megakaryocytic transcription factor cooperating with GATA1s and is a novel tumor suppressor gene in TAM/ML-DS.
Global chromatin alterations are associated with T21 in both myeloid and lymphoid leukemias. Lane and colleagues have shown transcriptional similarities between DS and other B-ALL with gain of chromosome 21. They identified HMGN1, located on 21q22, as an oncogene in DSALL and other B-ALL with somatic +21.8 HMGN1 is a chromatin structural protein that de-compacts chromatin and acts in opposition to histone H1. Modulation of HMGN1 expression levels induces a genome-wide increase in transcription55 and a specific de-repression of genes controlled by the PRC2 complex in B-ALL. This is due to an increase in H3K27 acetylation and an increase of B-cell factors. Trisomy of HMGN1 in a DS mouse model (Ts1Rhr) or its ectopic expression promotes pre-B-cell colony expansion and self-renewal, a phenotype that can be reversed with GSK-J4, which targets HMGN1 via inhibition of histone demethylases. Loss-of-function models demonstrated that HMGN1 is necessary for a pro-B phenotype. Interestingly, HMGN1 has been shown to upregulate the expression of CRLF2, a gene that is somatically altered in about 50% of DS-ALL.56
The role of HMGN1 in AML has also been investigated.57 HMGN1 is highly expressed in stem/progenitor cells and is necessary for differentiation of myeloid progenitors. Its
overexpression in myeloid progenitors increases chromatin accessibility and transcription in the HOXA cluster. The HMGN1 nucleosome binding domain is sufficient for the differentiation phenotype, and HMGN1 promotes hematopoietic and leukemia stem cell activity. This HMGN1 overexpression phenotype can be reversed by targeting the histone acetyl transferases CBP/p300. A newly developed mouse model of conditional Hmgn1 deletion shows a possible effect on Kmt2a-Mllt3 (formerly Mll-AF9) induced leukemogenesis. Global chromatin profiling studies during normal and malignant myeloid differentiation have revealed H3K23 acetylation as one of the most dramatic changes during normal versus AML differentiation. Since histone H3K23 acetylation is catalyzed by the lysine acetyltransferase KAT6A/6B and with recent reports showing that KAT6A and ENL form an epigenetic transcriptional control to drive leukemogenesis,58 studies should explore whether there is a common T21 epigenetic phenotype and whether DS, hematopoietic differentiation, and leukemia are connected in the epigenome. Therapeutic approaches targeting epigenetic regulators have been investigated in pre-clinical models and in patients. Klusmann and colleagues tested LSD1 inhibition in combination with JAK/STAT inhibition in a preclinical MLDS model and proposed this combination for future clinical trials.59 Other epigenetic therapies, such as histone deacetylase inhibitors or azacytidine, have been administered to individual patients with good responses,3,60-63 further supporting a role for epigenetic therapies in the treatment of DS-associated leukemias.
Interestingly, chromosome 21 encoded genes may also play a role in the treatment-related mortality observed in DS-ALL patients as well as the decreased incidence of solid tumors associated with DS. Four inhibitors of the calcineurin-NFAT signaling pathway, S100b, PCP4, DSCR1, and DYRK1A, are located on chromosome 21 and all function to negatively regulate the calcium-sensitive ser/thr phosphatase calcineurin. The calcineurin pathway is important for neutrophil and macrophage function. Dr. Ryeom has hypothesized that attenuation of this pathway in innate immune cells blocks their ability to effectively eliminate infections observed in patients with DS-ALL, leading to increased treatment-related mortality. Dr. Ryeom’s laboratory has developed a mouse model of DS with attenuation of calcineurin-NFAT signaling by the expression of a third copy of Dscr1 and can model treatmentrelated mortality in these mice after treatment with dexamethasone followed by challenge with the bacterial endotoxin, lipopolysaccharides. Ongoing studies in her laboratory are investigating neutrophil and macrophage dysfunction in this DS mouse model with chromosome 21 genes suppressing calcineurin signaling in these immune cells with the intent of identifying biomarkers of early treatment-related mortality in patients with DS-ALL. Pre-
vious studies by Ryeom and colleagues identifi ed suppression of calcineurin signaling in endothelial cells as a mechanism underlying the reduced incidence of solid tumors observed in individuals with DS.64 They and others found defects in tumor angiogenesis in DS mouse models and teratomas derived from T21 iPSC, suggesting that attenuation of calcineurin signaling blocks endothelial cell activation and suppresses tumor angiogenesis, thereby preventing the expansion of microscopic dormant tumors. Other studies investigating cancer protection in the DS population involve the contribution of chromosome 21 encoded tumor suppressor genes, such as ETS2. 64-67
Key somatic mutations in Down syndrome-associated leukemia
Dr. Crispino’s laboratory has been instrumental in determining the role of GATA1s mutations and T21 in driving DS leukemogenesis and has discovered key chromosome 21 genes (ERG, CHAF1B and DYRK1A) that are responsible for the increased risk of ML-DS in children with DS. Crispino and colleagues have also demonstrated that beyond the known expansion of megakaryopoiesis, GATA1s expression also impairs erythropoiesis.68 The erythroid phenotype can be seen during fetal life through at least 20 months, with the knock-in Gata1s mouse model displaying macrocytic anemia and bone marrow fibrosis.68,69 Mechanistically, GATA1s expression in erythroid cells is associated with major deficiencies in gene regulation which can be rescued by loss of one copy of GATA2, as shown in collaboration with Dr. Izraeli’s laboratory.70 These data re-emphasize the key role of GATA1 and of its alteration in the erythromegakaryocytic compartment.
Previous large-scale sequencing studies found that the most frequently mutated protein complex in ML-DS is cohesin, one of the main drivers of three-dimensional genome folding. Interestingly, the main partner of cohesin, the DNA-binding protein CTCF, is also recurrently mutated among patients with ML-DS.3,4 While both proteins are also frequently mutated in other cancers, it is unusual to find both mutated at such high frequencies in the same cancer type. Cohesin and CTCF are responsible for partitioning the genome into topologically associating domains, which are functional units of gene regulation that both facilitate and limit the range of action of enhancer-promoter interactions.71 However, only a minor fraction of genes are highly sensitive to perturbations in the levels of cohesin or CTCF. By depleting cohesin in mouse macrophages and hematopoietic progenitors, studies by Cuartero and colleagues showed a dramatic reduction in inflammatory gene expression, indicating a key role for cohesin in the activation of inducible genes.72 Interestingly, a similar phenotype was observed after depletion of CTCF.73 The inability to respond to inflammatory signals was linked to the incapacity of AML cells to differentiate, as normal HSPC
differentiate in response to inflammation.74 However, how the control of inducible gene expression plays a role in the transition from TAM to ML-DS remains an unresolved issue, as does the possibility of interplay between cohesin/CTCF and mutant GATA1 chromatin binding.
DS leukemogenesis can be modeled using primary human HSPC and xenotransplantation in mice. Utilizing fetal liver from normal and DS fetuses, Wagenblast and colleagues found that T21 HSC are immunophenotypically expanded.10 Applying CRISPR approaches to functionally interrogate the impact of GATA1s and STAG2, alone or in cooperation, they found a lower CD45+ engraftment for T21 fetal liver long-term HSC as compared to non-DS HSC except when GATA1s is expressed and STAG2 deleted. Interestingly in this model, T21 is required for pre-leukemia initiation but seems to be dispensable for leukemia progression upon STAG2 mutation. Blasts recapitulate the immunophenotype of TAM and ML-DS with STAG2 knockout cells in DS fetal liver HSC surviving long-term. Recently, a separate study showed that loss of the cohesin effector SMC3 and knock in of MPL-activating mutations gradually transformed T21/GATA1s human iPSC into ML-DS blasts by promoting a differentiation blockade and proliferation.47 Dr. Wagenblast showed that the predisposition/initiation step is at least partly controlled by chromosome 21 miRNA (miR-99, miR-125b-2 and miR-155a). Simultaneous overexpression of all three miRNA recapitulates a T21-like hematopoietic state based on differentiation potential, transcriptional and open chromatin accessibility profiles. Furthermore, knock-out of these three miRNA led to reduced blast accumulation in mice. Dr. Wagenblast also demonstrated that expression of CD117 in the CD34+ compartment marks pre-leukemia and leukemia-initiating cells, and that TAM can be therapeutically targeted with KIT inhibitors.
Wagenblast and colleagues are now using a similar CRISPR approach in which a 320 kb interstitial deletion is induced in cord blood and fetal liver HSPC to model P2RY8::CRLF2 fusions in DS-ALL, which leads to pre-leukemia development upon xenotransplantation in mice (E. Wagenblast, unpublished data).
PDX models from primary patients’ specimens (peripheral blood or bone marrow samples obtained from children with DS) that are propagated in immune-deficient NSG mice are being developed by Dr. Malinge and colleagues to facilitate preclinical studies of DS-ALL. These new models are undergoing comprehensive characterization at genetic, transcriptional, phenotypic and functional levels and are representative of several molecular subtypes of DS-ALL. Using these models, it was found that RAS/MAPK activation cooperates with T21 in DS-ALL. NRAS and KRAS mutations are frequently found in high hyperdiploid ALL,
a subtype of B-ALL that often harbors trisomy, tetrasomy, or even pentasomy of chromosome 21. Treatment of DSALL PDX models with MEK inhibitors (e.g., selumetinib, trametinib) decreased cell growth in vitro and leukemia burden in vivo, prolonging survival of DS-ALL PDX, as well as other models of B-ALL with somatic +21 (e.g., iAMP21, high hyperdiploid). In vitro and in vivo drug combinations show that RAS/MAPK inhibition in combination with vincristine and dexamethasone is effective in several DS-ALL PDX models with various genetic backgrounds (eg, KRASor CRLF2/JAK2-mutant). Malinge and colleagues work suggests that RAS/MAPK inhibition may be a promising therapeutic strategy in DS-ALL and other B-ALL with +21.12 Recent studies showed that the chromosome 21 kinase DYRK1A is not only overexpressed in DS-ALL, but also in other subtypes of B-ALL with +21.75 DYRK1A was inhibited by the small molecule EHT1610 in preclinical DS-ALL models and demonstrated that DYRK1A phosphorylates key targets in B-ALL cells (e.g., cyclin D3, FOXO1, STAT3). Inhibiting DYRK1A activity directly or one of its downstream targets prolongs survival of non-DS-ALL and nonDS B-ALL +21 models.
Despite the significant progress made over the past two decades in understanding the mechanisms driving leukemia in DS individuals, as well as improvements in treatment regimens, there is still much to be learned. Led by Dr. Crispino, the final session of the symposium was a thoughtprovoking discussion on key questions in the field of DS and leukemia. Somatic GATA1 mutations are probably the most distinctive trait of ML-DS, but why are these mutations so frequent in neonates with DS? A very strong selective pressure is undoubtedly in place, but the highly proliferative nature of fetal liver progenitors may also increase the rate of DNA lesions, which would help to explain this unusual frequency (~25%) of GATA1 mutations. This raises another important, yet not fully resolved question: what are the most important GATA1 target genes that are dysregulated in TAM/ML-DS? GATA1 is a transcription factor, and any selective advantage acquired by cells is most likely through dysregulated gene expression. Yet, full-length GATA1 and the shorter GATA1s share the same DNA-binding domain, and the mechanistic differences in how they control gene expression are still unclear. More broadly, identifying with precision the chromosome 21-encoded genes responsible for ML-DS and DS-ALL leukemogenesis would open up significant therapeutic opportunities. Despite numerous studies seeking such genes, the most likely explanation is a combined effect rather than a single gene. Finally, it was widely agreed that PDX models will be key in investigating these issues and identifying new treatments
for children with ML-DS and DS-ALL. Expanding and sharing the existing repertoire of DS leukemia-specific PDX resources for research is a high scientific priority.
Our understanding of the multi-step progression of leukemia in DS will undoubtedly improve in the upcoming years, and such an advancement will offer important insights into the development of all leukemias beyond those occurring in children with T21. One of the main goals of the meeting in France was the establishment of new collaborations with the intent of bringing together DS and leukemia experts in 2024 for a 2nd International Symposium for DS and Leukemia to discuss progress made and the most recent knowledge regarding the causes of DS-associated leukemias, the optimal treatment regimens and best practices for affected patients.
JHK has advisory roles for Bluebird Bio, Boehringer, Novartis, Roche and Jazz Pharmaceuticals . SKT receives or has received research funding for unrelated studies from Beam Therapeutics, Incyte Corporation, and Kura Oncology, has consulted for Bluebird Bio, has received travel support from Amgen, and serves on scientific advisory boards of Aleta Biotherapeutics, Kura Oncology, and Syndax Pharmaceuticals.
AB, JPB, JC, SC, HH, JH, JHK, SI, AL, SM, KR, IR, SR, SKT, and EW all contributed to writing the manuscript.
The authors would like to acknowledge Dr Maria Rujano and the Fondation Jérôme Lejeune for organizing the inaugural International Symposium for Down Syndrome and Leukemia.
The 1st International Symposium on Down Syndrome and Leukemia (ISDSL) was supported by a grant to SC, SM and SR from the Fondation Jérôme Lejeune, Paris France (grant #2081). JC is supported by the St. Jude Children’s Research Hospital/ALSAC. SC is supported by an ASH Global Research Award and by the Spanish Ministry of Science and Innovation (PID2020-117950RA-I00). JHK receives funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement #714226) and is a recipient of the St. Baldrick’s Robert Arceci Innovations Award. JHK is also supported by the German Research Foundation (DFG; KL-2374/1-3; KL2374/5-1), BMBF (01GM1911D myPred) and the DKH (#109251 and #110806). AAL is supported by Alex’s Lemonde Stand. SM is supported by a Fellowship from the Cancer Council Western Australia (CCWA) & Child Cancer Research Foundation (CCRF). SR was supported by the
NIH/NCI R01CA118374 and a grant from the Fondation Jérôme Lejeune (Project #2000). SKT is supported by NIH/NCI 1U01CA232486 and 1U01CA243072, Department of Defense Translational Team Science Award CA180683P1, V Foundation for Cancer Research, and philanthropy from the Croo-
1. Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355(9199):165-169.
2. Hitzler JK, Zipursky A. Origins of leukaemia in children with Down syndrome. Nat Rev Cancer. 2005;5(1):11-20.
3. Labuhn M, Perkins K, Matzk S, et al. Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with Down syndrome. Cancer Cell. 2019;36(3):340.
4. Yoshida K, Toki T, Okuno Y, et al. The landscape of somatic mutations in Down syndrome–related myeloid disorders. Nat Genet. 2013;45(11):1293-1299.
5. Malinge S, Bliss-Moreau M, Kirsammer G, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122(3):948-962.
6. Nikolaev SI, Garieri M, Santoni F, et al. Frequent cases of RASmutated Down syndrome acute lymphoblastic leukaemia lack JAK2 mutations. Nat Commun. 2014;5:4654.
7. Brown PA, Ji L, Xu X, et al. A randomized phase 3 trial of blinatumomab vs. chemotherapy as post-reinduction therapy in high and intermediate risk (HR/IR) first relapse of B-acute lymphoblastic leukemia (B-ALL) in children and adolescents/young adults (AYAs) demonstrates superior efficacy and tolerability of blinatumomab: a report from Children’s Oncology Group Study AALL1331. Blood. 2019;134(Suppl_2):LBA-1.
8. Lane AA, Chapuy B, Lin CY, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet. 2014;46(6):618-623.
9. Jardine L, Webb S, Goh I, et al. Blood and immune development in human fetal bone marrow and Down syndrome. Nature. 2021;598(7880):327-331.
10. Wagenblast E, Araújo J, Gan OI, et al. Mapping the cellular origin and early evolution of leukemia in Down syndrome. Science. 2021;373(6551):eabf6202.
11. Schwartzman O, Savino AM, Gombert M, et al. Suppressors and activators of JAK-STAT signaling at diagnosis and relapse of acute lymphoblastic leukemia in Down syndrome. Proc Natl Acad Sci U S A. 2017;114(20):E4030-E4039.
12. Laurent AP, Siret A, Ignacimouttou C, et al. Constitutive activation of RAS/MAPK pathway cooperates with trisomy 21 and is therapeutically exploitable in Down syndrome B-cell leukemia targeting RAS/MAPK pathway in DS-ALL. Clin Cancer Res. 2020;26(13):3307-3318.
13. Buitenkamp TD, Izraeli S, Zimmermann M, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014;123(1):70-77.
14. Ceppi F, Stephens D, den Hollander BS, et al. Clinical presentation and risk factors of serious infections in children with Down syndrome treated for acute lymphoblastic leukemia. Pediatr Blood Cancer. 2016;63(11):1949-1953.
kes/Burke Family to her laboratory in memory of Charlotte Clare Burke. SKT is a Scholar of the Leukemia & Lymphoma Society and holds the Joshua Kahan Endowed Chair in Pediatric Leukemia Research at the Children’s Hospital of Philadelphia.
15. Hasle H, Friedman JM, Olsen JH, Rasmussen SA. Low risk of solid tumors in persons with Down syndrome. Genet Med. 2016;18(11):1151-1157.
16. Hasle H, Kline RM, Kjeldsen E, et al. Germline GATA1s-generating mutations predispose to leukemia with acquired trisomy 21 and Down syndrome-like phenotype. Blood. 2022;139(21):3159-3165.
17. Satgé D, Sommelet D, Geneix A, Nishi M, Malet P, Vekemans M. A tumor profile in Down syndrome. Am J Med Genet. 1998;78(3):207-216.
18. Patja K, Pukkala E, Sund R, Iivanainen M, Kaski M. Cancer incidence of persons with Down syndrome in Finland: a population-based study. Int J Cancer. 2006;118(7):1769-1772.
19. Osuna-Marco MP, López-Barahona M, López-Ibor B, Tejera ÁM. Ten reasons why people with Down syndrome are protected from the development of most solid tumors - a review. Front Genet. 2021;12:749480.
20. Rethoré M-O, Rouëssé J, Satgé D. Cancer screening in adults with Down syndrome, a proposal. Eur J Med Genet. 2020;63(4):103783.
21. Flasinski M, Scheibke K, Zimmermann M, et al. Low-dose cytarabine to prevent myeloid leukemia in children with Down syndrome: TMD Prevention 2007 study. Blood Adv. 2018;2(13):1532-1540.
22. Uffmann M, Rasche M, Zimmermann M, et al. Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial. Blood. 2017;129(25):3314-3321.
23. Hitzler J, Alonzo T, Gerbing R, et al. High-dose AraC is essential for the treatment of ML-DS independent of postinduction MRD: results of the COG AAML1531 trial. Blood. 2021;138(23):2337-2346.
24. Tandonnet J, Clavel J, Baruchel A, Nacka F, Pérel Y. Myeloid leukaemia in children with Down syndrome: report of the registry-based French experience between 1990 and 2003. Pediatr Blood Cancer. 2010;54(7):927-933.
25. Laetsch TW, Maude SL, Rives S, et al. Three-year update of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia in the ELIANA trial. J Clin Oncol. 2023;41(9):1664-1669.
26. Rabin KR, Chen Z, Devidas M, et al. Outcomes in children with Down syndrome (DS) and B-lymphoblastic leukemia (B-ALL): a Children’s Oncology Group (COG) report. J Clin Oncol. 2020;38(15_suppl):10510.
27. Harvey RC, Tasian SK. Clinical diagnostics and treatment strategies for Philadelphia chromosome–like acute lymphoblastic leukemia. Blood Adv. 2020;4(1):218-228.
28. Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome–like acute lymphoblastic leukemia. Blood. 2017;130(19):2064-2072.
29. Tasian SK, Doral MY, Borowitz MJ, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120(4):833-842.
30. Maude SL, Tasian SK, Vincent T, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute
lymphoblastic leukemia. Blood. 2012;120(17):3510-3518.
31. Hurtz C, Wertheim GB, Loftus JP, et al. Oncogene-independent BCR-like signaling adaptation confers drug resistance in Ph-like ALL. J Clin Invest. 2020;130(7):3637-3653.
32. Loh ML, Tasian SK, Rabin KR, et al. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: a Children’s Oncology Group phase 1 consortium study (ADVL1011). Pediatr Blood Cancer. 2015;62(10):1717-1724.
33. Tasian SK, Assad A, Hunter DS, Du Y, Loh ML. A phase 2 study of ruxolitinib with chemotherapy in children with Philadelphia chromosome-like acute lymphoblastic leukemia (INCB18424269/AALL1521): dose-finding results from the part 1 safety phase. Blood. 2018;132(Suppl 1):555.
34. Chien CD, Nguyen SM, Qin H, Jacoby E, Fry TJ. CRLF2/Tslpr overexpressing acute lymphoblastic leukemia relapse is driven by chemotherapy-induced TSLP from bone marrow stromal cells. Blood. 2015;126(23):1432-1432.
35. Bagashev A, Loftus J, Niswander L, et al. P1421: bimodal targeting of cytokine receptor-like factor 2 (CRLF2) with JAK inhibition and chimeric antigen receptor T cell Immunotherapy in Down syndrome acute lymphoblastic leukemia. Hemasphere. 2022;6(Suppl):1305-1306.
36. Maclean GA, Menne TF, Guo G, et al. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc Natl Acad Sci U S A. 2012;109(43):17567-17572.
37. Chou ST, Opalinska JB, Yao Y, et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood 2008;112(12):4503-4506.
38. Banno K, Omori S, Hirata K, et al. Systematic cellular disease models reveal synergistic interaction of trisomy 21 and GATA1 mutations in hematopoietic abnormalities. Cell Rep. 2016;15(6):1228-1241.
39. Hollanda LM, Lima CSP, Cunha AF, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006;38(7):807-812.
40. Sankaran VG, Ghazvinian R, Do R, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122(7):2439-2443.
41. van Dooijeweert B, Kia SK, Dahl N, et al. GATA-1 defects in Diamond–Blackfan anemia: phenotypic characterization points to a specific subset of disease. Genes. 2022;13(3):447.
42. Labuhn M, Perkins K, Matzk S, et al. Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with Down syndrome. Cancer Cell. 2019;36(2):123-138.
43. Yoshida K, Toki T, Okuno Y, et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet. 2013;45(11):1293-1299.
44. de Rooij JDE, Branstetter C, Ma J, et al. Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49(3):451-456.
45. Roberts I, Alford K, Hall G, et al. GATA1-mutant clones are frequent and often unsuspected in babies with Down syndrome: identification of a population at risk of leukemia. Blood. 2013;122(24):3908-3917.
46. Byrska-Bishop M, VanDorn D, Campbell AE, et al. Pluripotent stem cells reveal erythroid-specific activities of the GATA1 Nterminus. J Clin Invest. 2015;125(3):993-1005.
47. Arkoun B, Robert E, Boudia F, et al. Stepwise GATA1 and SMC3 mutations alter megakaryocyte differentiation in a Down syndrome leukemia model. J Clin Invest. 2022;132(14):e156290.
48. Roy A, Cowan G, Mead AJ, et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci U S A. 2012;109(43):17579-17584.
49. MacLean GA, McEldoon J, Huang J, Allred J, Canver MC, Orkin SH. Downregulation of endothelin receptor B contributes to defective B cell lymphopoiesis in trisomy 21 pluripotent stem cells. Sci Rep. 2018;8(1):8001.
50. Li Z, Chang T-C, Junco JJ, et al. Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood. 2023;142(2):172-184.
51. Gialesaki S, Bräuer-Hartmann D, Issa H, et al. RUNX1 isoform disequilibrium promotes the development of trisomy 21 associated myeloid leukemia. Blood. 2023;141(10):1105-1118.
52. Klusmann J-H, Li Z, Böhmer K, et al. miR-125b-2 is a potential oncomiR on human chromosome 21 in megakaryoblastic leukemia. Genes Dev. 2010;24(5):478-490.
53. Kugler E, Birger Y, Yong D, et al. AML-079: the PNT domain of ERG regulates hematopoietic stemness and is required for the maintenance of ERG-induced leukemia. Clin Lymphoma Myeloma Leuk. 2020;20:S180-S181.
54. Alejo-Valle O, Weigert K, Bhayadia R, et al. The megakaryocytic transcription factor ARID3A suppresses leukemia pathogenesis. Blood. 2022;139(5):651-665.
55. Mowery CT, Reyes JM, Cabal-Hierro L, et al. Trisomy of a Down syndrome critical region globally amplifies transcription via HMGN1 overexpression. Cell Rep. 2018;25(7):1898-1911.
56. Page EC, Heatley SL, Eadie LN, et al. HMGN1 plays a significant role in CRLF2 driven Down syndrome leukemia and provides a potential therapeutic target in this high-risk cohort. Oncogene. 2022;41(6):797-808.
57. Cabal-Hierro L, van Galen P, Prado MA, et al. Chromatin accessibility promotes hematopoietic and leukemia stem cell activity. Nat Commun. 2020;11(1):1406.
58. Yan F, Li J, Milosevic J, et al. KAT6A and ENL form an epigenetic transcriptional control module to drive critical leukemogenic gene-expression programs. Cancer Discov. 2022;12(3):792-811.
59. Grimm J, Bhayadia R, Gack L, Heckl D, Klusmann J-H. Combining LSD1 and JAK-STAT inhibition targets Down syndrome-associated myeloid leukemia at its core. Leukemia. 2022;36(7):1926-1930.
60. Stankov MV, El Khatib M, Kumar Thakur B, et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia. 2014;28(3):577-588.
61. Scheer C, Kratz C, Witt O, Creutzig U, Reinhardt D, Klusmann JH. Hematologic response to vorinostat treatment in relapsed myeloid leukemia of Down syndrome. Pediatr Blood Cancer. 2016;63(9):1677-1679.
62. Tamura A, Ishida T, Saito A, et al. Low-dose azacitidine maintenance therapy after allogeneic stem cell transplantation for high-risk pediatric acute myeloid leukemia. Pediatr Blood Cancer. 2018;65(10):e27284.
63. Becktell K, Houser K, Burke MJ. Epigenetic therapy in a patient with Down sndrome and refractory acute myeloid leukemia. J Pediatr Hematol Oncol. 2019;41(1):e38-e40.
64. Baek K-H, Zaslavsky A, Lynch RC, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459(7250):1126-1130.
65. Sussan TE, Yang A, Li F, Ostrowski MC, Reeves RH. Trisomy represses Apc Min-mediated tumours in mouse models of Down’s syndrome. Nature. 2008;451(7174):73-75.
66. Zorick TS, Mustacchi Z, Bando SY, et al. High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Hum Genet. 2001;9(11):811-814.
67. Reynolds LE, Watson AR, Baker M, et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Down’s syndrome. Nature. 2010;465(7299):813-817.
68. Ling T, Zhang K, Yang J, Gurbuxani S, Crispino JD. Gata1s mutant mice display persistent defects in the erythroid lineage. Blood Adv. 2023;7(13):3253-3264.
69. Li Z, Godinho FJ, Klusmann J-H, Garriga-Canut M, Yu C, Orkin SH. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37(6):1619-1631.
70. Ling T, Birger Y, Stankiewicz MJ, et al. Chromatin occupancy and epigenetic analysis reveal new insights into the function of the GATA1 N terminus in erythropoiesis. Blood 2019;134(19):1619–1631.
71. Merkenschlager M, Nora EP. CTCF and cohesin in genome
folding and transcriptional gene regulation. Annu Rev Genomics Hum Genet. 2016;17:17-43.
72. Cuartero S, Weiss FD, Dharmalingam G, et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat Immunol. 2018;19(9):932-941.
73. Stik G, Vidal E, Barrero M, et al. CTCF is dispensable for immune cell transdifferentiation but facilitates an acute inflammatory response. Nat Genet. 2020;52(7):655-661.
74. Cuartero S, Innes AJ, Merkenschlager M. Towards a better understanding of cohesin mutations in AML. Front Oncol. 2019;9:867.
75. Bhansali RS, Rammohan M, Lee P, et al. DYRK1A regulates B cell acute lymphoblastic leukemia through phosphorylation of FOXO1 and STAT3. J Clin Invest. 2021;131(1):e135937.
Correspondence: S. Rivella rivellas@chop.edu
1Department of Pediatrics, Division of Hematology, The Children’s Hospital of Philadelphia (CHOP); 2University of Pennsylvania, Perelman School of Medicine; 3RNA Institute, University of Pennsylvania; 4Cell and Molecular Biology affinity group (CAMB), University of Pennsylvania; 5Raymond G. Perelman Center for Cellular and Molecular Therapeutics, CHOP; 6Penn Center for Musculoskeletal Disorders, CHOP and 7Institute for Regenerative Medicine, University of Pennsylvania, Philadelphia, PA, USA
Received: April 1, 2023.
Accepted: June 15, 2023.
Early view: June 22, 2023.
htps://doi.org/10.3324/haematol.2023.283057
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Under normal conditions, iron metabolism is carefully regulated to sustain normal cellular functions and the production of hemoglobin in erythroid cells. Perturbation to the erythropoiesis-iron metabolism axis can result in iron imbalances and cause anemia or organ toxicity. Various congenital and acquired diseases associated with abnormal red cell production are characterized by aberrant iron absorption. Several recent studies have shown that improvements in red blood cell production also ameliorate iron metabolism and vice versa. Many therapeutics are now under development with the potential to improve a variety of hematologic diseases, from β-thalassemia and iron-refractory iron deficiency anemia to anemia of inflammation and polycythemia vera. This review summarizes selected mechanisms related to red cell production and iron metabolism and describes potential therapeutics and their current uses. We also consider the potential application of the discussed therapeutics on various diseases, alone or in combination. The vast repertoire of drugs under development offers new opportunities to improve the clinical care of patients suffering from congenital or acquired red blood cell disorders with limited or no treatment options.
Erythropoiesis, the production of mature enucleated erythrocytes from early progenitor cells, is regulated by molecules that control proliferation and differentiation.1 For brevity, the descriptions of the molecules that regulate erythropoiesis and iron metabolism will be limited to those relevant to the diseases and potential therapies discussed in this review.
GATA1 is a key transcription factor essential for erythropoiesis (Figure 1).2 GATA1 promotes maturation and survival of erythroblasts, which develop into enucleated red cells.3
GATA1 stimulates several pathways, including those responsible for hemoglobin synthesis and stress erythropoiesis.4,5 The erythrocyte-specific isoform of pyruvate kinase, R-type pyruvate kinase (Figure 1), is regulated by GATA1 and is involved in the final steps of glycolysis, which leads to the formation of two adenosine triphosphate and two pyruvate molecules per glucose molecule (Figure 1).6,7 R-type pyruvate kinase is required for the survival of ma-
ture red cells and to sustain the tricarboxylic acid cycle in erythroid precursors.7-9
Erythropoietin (EPO) binds to the EPO receptor (EPOR) to regulate early-stage erythropoiesis, primarily by phosphorylation of the JAK2 kinase (Figure 2).10 Phosphorylated JAK2 kinase is responsible for the activation of downstream pathways that regulate expression of genes that control proliferation, survival, and iron metabolism, such as the hormone erythroferrone (ERFE) (Figure 2).11 A subset of transforming growth factor-β superfamily receptors, ligands and SMAD2/3 transcription factors may play a major role in late-stage erythropoiesis by modulation of differentiation and red cell production (Figure 2).12,13
Hepcidin (encoded by HAMP) is a liver-secreted hormone that plays a major role in iron homeostasis (Figure 1).14 It binds and inhibits the iron exporter ferroportin, preventing release of iron into the circulation from duodenal enterocytes, macrophages, and hepatocytes.15 Increased hepcidin expression causes anemia by limiting iron availability, whereas low hepcidin expression leads to iron overload.14,16
Amaliris Guerra,1 Hamideh Parhiz2,3 and Stefano Rivella1,2,3,4,5,6,7Activation of hepcidin transcription is mediated by the ironsensor complex on liver parenchymal cells, which includes bone morphogenetic protein (BMP) receptors from the TGF- β receptor superfamily as well as coreceptors, ligands, and modulators (Figure 2).16 Liver endothelial cells secrete the BMP ligands BMP2 and BMP6, which bind class I and II BMP receptors in hepatocytes and activate the SMAD1/5/8 complex and HAMP expression (Figure 2).16 Hemojuvelin (HJV) acts as a BMP coreceptor, whereas TMPRSS6 (which encodes for matriptase-2, a transmembrane serine protease) downregulates hepcidin by cleaving HJV (Figure 2).17
Additional proteins in the liver modulate the activity of the iron-sensor complex by sensing the level of iron bound to transferrin (TF), the main molecule responsible for the transportation of iron from the duodenum (Figure 2).16 Increasing amounts of diferric TF displace HFE from
TF receptor 1.16 This not only enables iron uptake in the liver but also stabilizes TF receptor 2 (TFR2), potentiating signaling of the iron-sensor complex (Figure 2).16 Although TFR2 was thought to only modulate hepcidin synthesis in the liver, it was recently shown that TFR2 colocalizes with EPOR on erythroid precursor cells to modulate EPO sensitivity (Figure 2).16,18 The role of TFR2 in this setting is thought to be that of finetuning erythropoiesis based on the amount of iron bound to TF.19
Erythropoiesis can be altered by genetic mutations that

impair red cell production, such as in congenital anemias or polycythemia vera (PV), or by conditions that perturb normal iron metabolism, such as iron-refractory iron deficiency anemia (IRIDA).20 Conditions associated with chronic inflammation, such as chronic kidney disease, are also characterized by reduced red cell production and survival.21 In this review, we focus on diseases that illustrate key pathophysiological mechanisms responsible for altered red cell production, the role of iron metabolism, and strategies targeting these mechanisms that may serve as potential novel treatment approaches. Anemia is a consequence of inadequate functional red cells in circulation, which can result from reduced red cell production, increased red cell destruction, or a combination of these two processes. Diseases associated with genetic mutations that directly alter red cell production are generally characterized by defects at the erythroid
cell-differentiation stage at which the mutated genes exert their functions. These mutations may also affect the quality and lifespan of surviving mature red cells.

β-thalassemia (BT) and α-thalassemia (AT) are two of the most widespread forms of congenital anemias.22 These diseases are triggered by mutations that reduce the synthesis of β-globin or α-globin, respectively.22 Heterozygosity for BT null mutations (BT trait) is usually clinically silent but becomes symptomatic with coinheritance of extra α-globin genes, as a consequence of a relatively increased amount of free α-globin protein.23,24 Hence, the globin-chain imbalance with an accumulation of free α-globin is a major determinant of BT pathophysiology and causes a variety of effects that
impair red cell maturation and survival (Figure 2).22 Excess α-globin chains precipitate as hemichromes, which are insoluble aggregates of α-chains and heme molecules that form large inclusion bodies and cause iron accumulation and free radical production (Figure 1).25,26 These events lead to premature destruction by apoptosis of a portion of red cell precursors in the bone marrow as well as the production of abnormal enucleated erythrocytes with reduced lifespan.27 In addition, excess α -globin chains also interfere with erythroid differentiation. In normal erythropoiesis, chaperone heat shock protein 70 (HSP70) protects GATA1 from caspase-3 cleavage (Figure 1).28 However, in BT, HSP70 interacts directly with free αglobin chains, and therefore HSP70 is sequestered and unable to protect GATA1, resulting in end-stage maturation arrest (Figure 1).28 Altogether, this mechanism limits the production of enucleated red cells and consequent oxygen delivery (Figure 1). The resulting hypoxia increases the production of EPO in the kidneys. EPO further stimulates the production and expansion of early erythroid progenitors without generating sufficient mature red cells (Figure 1).27 This leads to a net imbalance between nucleated and enucleated erythroid cells - the number of terminally differentiated red cells is disproportionally low compared with the number of progenitor red cells (Figure 1).27 This abnormal production of erythroid progenitors, combined with a hypoxic environment, profoundly affects iron metabolism. The production of ERFE, by the early progenitor red cells, increases as the number of erythroid precursor cells increases, leading to suppression of hepcidin and results in enhanced iron absorption (Figure 2).27 In addition, the hypoxic environment also triggers the synthesis of genes responsible for iron absorption in the duodenum, further exacerbating the process of iron overload in organs.27,29
Iron accumulation results in free radical damage to cells and organs and contributes to the morbidity and mortality of iron overload (Figure 1).30 Although the main source of extra iron in patients with severe BT comes from blood transfusions, the main cause of iron overload in patients who only sporadically receive transfused blood is from increased iron absorption.31 When total body iron is significantly elevated the TF iron-binding ability is exceeded and circulating non-TF-bound iron appears in the plasma. NonTF-bound iron rises markedly when the TF saturation reaches high levels, and a reactive Fe2+ subspecies of nonTF-bound iron called labile plasma iron increases concomitantly.32,33 It is important to underline that clinical observations suggested that non-TF-bound iron was absent in non-transfusion-dependent thalassemia patients without previous transfusions.32,33 Further, labile plasma iron was not detected in most non-transfusion-dependent thalassemia patients, whereas other iron storage and turnover markers were elevated.32,34 Nevertheless, in both
transfusion- and non-transfusion-dependent thalassemia patients, iron chelation is the only treatment to eliminate excess iron and prevent organ damage.31
Following extensive studies in mouse models and humans, many novel drugs and genetic approaches are now in development to improve the quality of life of or provide a cure for patients with BT. These drugs can be loosely classified based on their mechanism of action, such as improvement of red cell quality and production (e.g., luspatercept, mitapivat, and drugs that target excess αglobin chains), improvement of anemia, suppression of erythropoiesis and improvement of splenomegaly (e.g., JAK2 inhibitors), limitation of iron absorption and/or erythroid iron intake (e.g., hepcidin agonists, hepcidin inducers, ferroportin or ERFE inhibitors, and TF therapy), all which lead to improvements of iron overload, and, potentially, improvement of red cell production over time (Table 1).35,36 In addition, novel gene therapy approaches and strategies aimed to pharmacologically reactivate fetal hemoglobin have the potential to significantly improve symptoms of BT or to cure BT, but these topics are beyond the scope of this review.
Luspatercept (Reblozyl®) is a ligand trap made using the extracellular domain of the activin receptor type IIB (Table 1).37,38 This drug binds to a subset of TGF-β ligands to inhibit aberrant SMAD2/3 signaling and enhances late-stage erythropoiesis.37-39 Luspatercept has recently been approved in the USA and Europe for the treatment of adult patients with transfusion-dependent BT (TDBT).37 In the pivotal clinical trial, a significantly greater proportion of patients in the luspatercept group had a reduction in transfusion burden compared with the placebo group.37 Additional observations also suggest that patients with non-transfusion-dependent BT (NTDBT) may also benefit from this drug.38 Although luspatercept treatment reduced transfusion burden and iron loading in patients affected by TDBT, NTDBT, or myelodysplastic syndrome, likely caused by improved erythropoiesis and iron consumption, luspatercept treatment also increased ERFE levels and, consequently, reduced hepcidin levels.40 It is, therefore, possible that luspatercept may negatively modulate hepcidin transcription by binding TGF-β family ligands implicated in hepcidin synthesis.41 These observations suggest it could be beneficial to use luspatercept in combination with iron chelators or other therapeutics, as elaborated in the section “Anemias associated with impaired iron absorption.”
Mitapivat (AG-348; Pyrukynd®) is a small-molecule activator of R-type pyruvate kinase which enhances the affinity
Potential use of drugs already approved or under development for diseases discussed in this review. The plus sign (+) indicates drugs in use in preclinical studies or discussed in this review. The question mark (?) indicates the potential use of the drugs in other conditions, but these drugs have not been discussed in this review. A dash (–) indicates not applicable or not determined. The plus signs and question marks in this table are purely speculative. The use of these drugs under these conditions would need to be tested in animal models and preclinical trials to prove their ef fi cac y and safety. NTDBT: non-transfusion-dependent β -thalassemia; NTDAT: non-transfusion-dependent α -thalassemia; TDBT: transfusiondependent β -thalassemia; TDAT: transfusion-dependent α -thalassemia; PV: polycythemia vera; CDA-II: congenital dyserythropoietic anemia type II; DBA: Diamond-Blackfan anemia;
MDS: myelodysplastic syndrome; IRIDA: iron-refractory iron de fi cienc y anemia; Myel.: myelo fi brosis; CKD: chronic kidney disease; SMAD: small mothers against decapentaplegic; EPO: erythropoietin; TFR2: transferrin receptor 2; JAK2: Janus kinase 2; mTORC1: mammalian target of rapamycin complex 1; TMPRSS6: t r ansmembrane protease, serine 6; mRNA: messenger
RNA; ASO: antisense oligonucleotide; ERFE: erythroferrone; TF: transferrin; ALK2: activin-like receptor 2.
of R-type pyruvate kinase for its substrate, phosphoenolpyruvate (Table 1).42,43 Mitapivat was initially developed to treat patients with pyruvate kinase deficiency, as it can improve pyruvate kinase activity, increase red cell deformability, and reduce transfusion burden in some patients who receive regular blood transfusions.44 Additional observations found that mitapivat induces a rapid and sustained increase in hemoglobin levels in approximately 50% of patients with pyruvate kinase deficiency who did not receive regular blood transfusions.43 In mouse models of BT, oral mitapivat administration ameliorated ineffective erythropoiesis and anemia by improving red cell metabolism.9 Likely caused by improved erythropoiesis, mitapivat administration resulted in reduced levels of soluble ERFE, increased liver hepcidin expression, and diminished liver iron overload.9 Mitapivat also reduced duodenal iron absorption, suggesting that this drug could further reduce iron overload independently of its indirect effect on hepcidin expression.9,45 Ongoing observations in patients with non-transfusion-dependent AT (NTDAT) and NTDBT treated with mitapivat suggest that this drug could ameliorate anemia in these patients.45
Other studies are investigating the possibility of targeting pathways that enhance the removal of excess α-globin in BT erythroid precursor cells (Table 1).24 These types of detoxification strategies would allow BT to be treated similarly to other protein aggregation disorders.46 Pharmacological induction of protein-targeted quality control pathways and autophagy to eliminate relevant unstable proteins could be potentially beneficial in patients with BT.47,48 As mTORC1 inhibits autophagy, systemic treatment with the mTORC1 inhibitor rapamycin reduces α-globin precipitates and lessens pathologies in BT mice.49 These findings may allow investigation of additional pathways that can be regulated by drugs for amelioration of BT symptoms. However, we do not know if rapamycin or similar drugs also improve iron metabolism in patients with BT, although rapamycin has been shown to increase hepcidin expression in wild-type mice.50 Selective deletion of Tfr2 exclusively in cells of the bone marrow in both NTDBT and TDBT mice significantly ameliorated anemia and highlight a major role for TFR2 in modulating the relationship between erythroid delivery of iron by TF, EPO sensitivity, and ineffective erythropoiesis (Table 1).51,52
In BT, in response to anemia and hypoxia, high levels of EPO are associated with increased activation of the JAK2 pathway and increased numbers of erythroid progenitors in both the bone marrow and the spleen.53 This extramedullary hematopoiesis leads to hepatosplenomegaly,
which enhances the entrapment of red cells in the spleen and exacerbates the anemia, further worsening the hepatosplenomegaly.27,53 Additionally, erythropoiesis occurs in extramedullary sites, most commonly resulting in a paraspinal mass.54 Unfortunately, splenectomy considerably increases the rate of thromboembolic complications.31 Although administration of JAK2 inhibitors does not reduce transfusion burden, the combination of blood transfusions with JAK2 inhibitors could be effective in reversing splenomegaly and could, therefore, be used as an alternative to splenectomy (Table 1).55,56
Identification of hepcidin and elucidation of the pathways that control its synthesis have been crucial to the development of pharmacological compounds that mimic the activity of hepcidin, increase hepcidin expression, and decrease ferroportin or ERFE activity, which all lead to decreased iron absorption and TF saturation.36 In a similar fashion, administration of exogenous apo-TF can also decrease levels of TF saturation.57 These potential therapeutics have reduced iron overload in animal models of BT.36 Intriguingly, in BT mice, the same drugs also lead to reduction in anemia.35,58 These drugs decreased not only iron absorption but also erythroid iron intake.35,59 In thalassemic erythroid progenitor cells, reduced iron uptake reduces the detrimental effects of oxidative stress triggered by the excess iron and heme that are not included in functional hemoglobin molecules.35,58 Overall, the consequences of iron restriction to erythroid cells improve the quality and lifespan of mature red cells and increase circulating hemoglobin levels.35,58 An ERFE antibody is also a potential therapeutic that targets a component of the iron-regulation pathway, as it prevented hepcidin suppression and corrected the iron-loading phenotype in a mouse model of BT.60 These observations suggested that iron-restricted erythropoiesis could benefit BT patients in a similar fashion, and a series of clinical trials were designed using drugs that restrict iron absorption. The hepcidin mimetic rusfertide® (PTG-300) can reduce iron absorption by targeting ferroportin.61 Rusfertide treatment improved iron metabolism and erythropoiesis in BT mice and decreased serum iron levels in patients with TDBT (https://www.protagonist-inc.com/investors-media/press-releases/default.aspx) but failed to improve symptoms in patients who received regular blood transfusions.62 The ferroportin inhibitor vamifeport® (VIT2763) showed similar endpoints in BT mice. However, also in this case it failed to improve symptoms in NTDBT patients (https://thalassaemia.org.cy/fr/clinical-trial-updates/ferroportin-inhibitors/#:~:text=A%20randomized%2C%20controlled%2C%20multi-centred%20phase%202%20proof-of-co
ncept%20trial,serum%20iron%20for%20up%20to%2024%20 hours%20post-dose).63 Since, administration of these reagents in these clinical trials was associated with decreased levels of TF saturation, it is possible that the biology of human red cells may be less sensitive to the deleterious effect of hemichromes or that the effects of erythroid iron restriction on the viability of BT red cells may surpass the beneficial effects of reducing hemichrome formation. Alternatively, the use of these drugs may require some administration of EPO, as it has been shown to be very efficacious in BT mice.64 However, the ability of these reagents to modulate hemichrome formation in the red cells of BT patients has never been investigated. Although these drugs failed to improve anemia in BT patients, it is important to underline that they were efficacious in reducing dietary iron intake and TF saturation, which is a prerequisite to improving iron overload. This suggests that these drugs could be used to prevent iron overload or could be combined with other medications that improve erythropoiesis or reduce iron overload (e.g., luspatercept, mitapivat or iron chelators) in order to obtain a rapid reduction of iron overload, as shown in animal studies.35,64,65
Bitopertin is an oral inhibitor of glycine transporter type 1, a key membrane transporter required to supply red cells with sufficient glycine to support erythropoiesis.66 By limiting glycine uptake, bitopertin regulates downstream heme synthesis.66 In a mouse model of erythropoietic protoporphyria, bitopertin reduced the excessive production of protoporphyrin IX (a precursor of heme) and improved liver fibrosis.67 Based on these preclinical studies, bitopertin is currently being tested in patients with erythropoietic protoporphyria.67 Bitopertin also limited abnormal levels of heme and reduced anemia in BT mice.66 However, a clinical trial in patients with NTDBT failed to reverse the anemia, with decreased levels of mean corpuscular hemoglobin and hemoglobin.68
α-thalassemia
Patients with severe forms of AT who require chronic blood transfusion therapy for survival (i.e., who have TDAT) almost exclusively make hemoglobin H, which is a tetramer made of four β-globin chains that has extremely high oxygen affinity but provides poor oxygen delivery to tissues.69,70 These patients share several pathophysiological manifestations with patients with TDBT, such as iron overload and the requirement for iron chelation therapy.71 However, patients with TDAT require a more aggressive transfusion regimen
than patients with TDBT.72 If not, patients with TDAT continue to have a high proportion of circulating non-functional hemoglobin H and exhibit features of hypoxia and EPOdriven increased erythropoietic activity.71,73 These observations suggest that ineffective erythropoiesis in patients with TDAT may be qualitatively and quantitively different from that observed in patients with TDBT, with potentially more enucleated but non-functional cells in circulation and increased iron absorption. If these predictions are confirmed by future studies, this will indicate that drugs that alter red cell quality may not be efficacious in patients with TDAT, although therapeutics that limit iron absorption or splenomegaly (such as JAK2 inhibitors) may be beneficial (Table 1). In contrast, the less severe forms of AT (i.e., NTDAT), in which some level of functional hemoglobin is present, may benefit from the same therapeutics that are being tested for patients with NTDBT, such as luspatercept and mitapivat. In a preliminary study, patients with NTDAT seemed to benefit from administration of mitapivat.45 As is the case with severe forms of AT, other drugs that limit iron absorption or erythroid iron intake could also be effective in the less severe forms of AT associated with iron overload (Table 1).
There are conditions in which modulation of iron metabolism could be beneficial to reduce excessive production of red cells. Mutations in JAK2 can lead to PV, which is characterized by erythrocytosis, bone marrow erythroid and megakaryocytic hyperplasia, fatigue, and splenomegaly.74 Patients with PV are frequently iron deficient at the time of diagnosis, which is further exacerbated by therapeutic phlebotomies to maintain the hematocrit below 45% in order to decrease the risk of thrombosis.75,76
Studies in PV mice demonstrated that administration of exogenous hepcidin or induction of endogenous hepcidin using TMPRSS6-ASO, an antisense oligonucleotide to TMPRSS6 messenger RNA, was able to reverse erythrocytosis, decrease splenomegaly, and sequester iron in splenic macrophages, suggesting that approaches that limit erythroid iron absorption and/or heme synthesis could be beneficial in patients with PV (Table 1).35,77,78 Additionally, preliminary results from clinical trials that aimed to evaluate the safety and efficacy of the hepcidin mimetic rusfertide in phlebotomy-requiring patients with PV suggest that approaches that limit erythroid iron absorption and/or heme synthesis could eliminate phlebotomy requirements, increase systemic iron stores, and decrease systemic symptoms.79
In brief, some conditions, such as congenital dyserythropoietic anemia type II, myelodysplastic syndrome, and Diamond-Blackfan anemia, may benefit from therapeutics that
modulate erythropoiesis, erythroid iron intake, heme synthesis, and/or decreased dietary iron absorption. For instance, a subset of patients affected by myelodysplastic syndrome benefit from luspatercept treatment and may also benefit from iron restriction.80 Similar approaches may also benefit a subset of patients with congenital dyserythropoietic anemia type II (mutations in SEC23B) who have anemia associated with increased ERFE expression, decreased hepcidin levels, and iron overload.81,82 Finally, erythroid cells in patients with Diamond-Blackfan anemia synthesize excess heme, which could be reduced by limiting erythroid iron intake and/or heme synthesis by treatment with a drug such as bitopertin.83
Other forms of anemia can help us to further understand the relationship between ineffective erythropoiesis and iron metabolism. In this part of the review, we will discuss informative forms of anemia and speculate on novel potential drugs to treat them.
Anemia of inflammation (AI) and IRIDA are two of the clinical manifestations associated with inappropriately high hepcidin levels.21,84,85 AI is common in inflammatory diseases with complex pathophysiological features, such as chronic kidney disease, autoimmune diseases, and some forms of cancer and myelofibrosis.21,85,86 Here, we review conditions in which inflammatory cytokines together with hepcidin contribute to iron-restricted erythropoiesis.
The primary clinical goal for patients with AI is to treat the cause of inflammation; however, treatment for anemia is often managed by administration of intravenous iron and erythropoiesis-stimulating agents,85 which is ineffective for some patients and is associated with adverse effects.85 Myelofibrosis is a myeloproliferative neoplasm characterized by splenomegaly, debilitating constitutional symptoms, and bone marrow failure.86 Disease-related anemia is common and associated with increased expression of hepcidin.86,87
IRIDA develops when mutations in TMPRSS6 lead to high hepcidin levels.20 Patients with IRIDA have microcytic anemia and low plasma-iron levels and are refractory to oral iron therapy but are partially responsive to parenteral iron administration.20,84
In all these conditions, high levels of hepcidin not only limit iron absorption but also sequester the iron provided to the patients by oral or intravenous administration to parenchymal cells and macrophages.16 Therefore, drugs that target proteins that interfere with the iron-sensor complex and decrease hepcidin levels or that can redistribute iron to TF may make iron more available for erythropoiesis. In addition, drugs that increase EPO sensitivity could also improve red
cell production.
Activin-like receptor 2 (ALK2) belongs to the TGF-β receptor superfamily and is part of the iron-sensor complex.88,89 ALK2 is the target for the drugs momelotinib and LJ000328.90,91 These drugs can downregulate hepcidin expression and increase availability of iron for erythropoiesis.90,91 In clinical testing, momelotinib improved hemoglobin levels and reduced transfusion burden in patients with myelofibrosis and baseline anemia, while also reducing spleen size and symptom burden.91 In mice, LJ000328 repressed hepcidin activity and significantly reduced the symptoms of IRIDA.90
Mutations in HJV are associated with a severe form of hemochromatosis.20,92 Therefore, pharmacological targeting of HJV could lead to hepcidin suppression. Anti-HJV antibodies, such as DISC-0974, have been effective in suppressing hepcidin expression in normal individuals and in preclinical models of AI and IRIDA, in which the antibodies were shown to increase hemoglobin levels in both inflammatory and non-inflammatory states.93-96
It has recently been observed that luspatercept treatment in patients with BT or myelodysplastic syndrome resulted in the release of excess iron from stored organs into peripheral blood.40 This was associated with decreased hepcidin levels, suggesting that luspatercept (which targets a subset of TGF-β ligands) may also inhibit the activity of the iron-sensor complex, possibly by targeting BMP or BMP-like molecules involved in hepcidin synthesis.41 These observations provide a mechanistic rationale for evaluating luspatercept also in diseases of elevated hepcidin synthesis, such as AI and IRIDA.
Deferiprone (Ferriprox®) does not directly affect iron absorption, but it chelates iron and is used to treat iron overload in BT.97 Deferiprone readily reaches the major intracellular sites of iron accumulation, facilitating the extraction of iron deposited in organs.98 The iron affinity of deferiprone is lower than that of TF, so deferiprone could redistribute iron sequestered in organs to erythroid cells.99 Therefore, in patients with high hepcidin levels, deferiprone could shuttle the sequestered iron to unbound TF, paradoxically improving erythroid iron consumption and anemia in AI and IRIDA. This strategy may also require concurrent iron administration to provide the optimal amount of iron needed for efficient erythropoiesis.
Finally, it could be beneficial to patients with many of the above-mentioned conditions to target TFR2 on erythroid cells, as deletion of TFR2 in hematopoietic cells can improve EPO sensitivity and increase red cell production.100 Because deletion of TFR2 in the liver decreases hepcidin expression and leads to increased iron absorption, it is currently challenging to target TFR2 selectively to erythroid cells and improve anemic conditions associated with iron overload (e.g., AT and BT). However, it may be beneficial to target TFR2 on both erythroid cells and liver cells in conditions in which increases in both EPO sensitivity and iron absorption may be
beneficial (i.e., chronic kidney disease).101
Current nucleoside-modified mRNA-lipid nanoparticle technology has successfully paved the way for next-generation vaccinations against severe acute respiratory syndrome coronavirus-2 during the COVID-19 pandemic.102 This emerging technology is also being used for additional applications, such as the ability to deliver small interfering RNA, messenger RNA, or plasmid DNA to treat most genetic diseases by silencing pathological genes, expressing therapeutic proteins, or gene editing. Using unmodified or targeted lipid nanoparticles encapsulating mRNA, several studies are underway to treat a variety of non-liver and liver diseases.103,104 As the pathways that control iron metabolism are primarily localized to the liver, it is only a question of time before this technology will be applied to modify the expression of iron-related genes and to improve iron overload or to correct genetic defects directly, as in IRIDA or HFE-related hemochromatosis.
Future clinical trials should clarify which drugs benefit all or a subset of patients with diseases associated with defective erythropoiesis. Combination therapies may emerge as the best approach, not only to improve anemia and iron overload but also to enhance treatment efficacy in a larger number of patients. Drug combinations that prevent excess iron absorption and improve red cell production have al-
1. An X, Schulz VP, Mohandas N, Gallagher PG. Human and murine erythropoiesis. Curr Opin Hematol. 2015;22(3):206-211.
2. Fujiwara Y, Browne CP, Cunniff K, Goff SC, Orkin SH. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci U S A. 1996;93(22):12355-12358.
3. Weiss MJ, Orkin SH. Transcription factor GATA-1 permits survival and maturation of erythroid precursors by preventing apoptosis. Proc Natl Acad Sci U S A. 1995;92(21):9623-9627.
4. Gutierrez L, Tsukamoto S, Suzuki M, et al. Ablation of Gata1 in adult mice results in aplastic crisis, revealing its essential role in steady-state and stress erythropoiesis. Blood. 2008;111(8):4375-4385.
5. Liao R, Bresnick EH. Heme as a differentiation-regulatory transcriptional cofactor. Int J Hematol. 2022;116(2):174-181.
6. Max-Audit I, Eleouet JF, Romeo PH. Transcriptional regulation of the pyruvate kinase erythroid-specific promoter. J Biol Chem. 1993;268(8):5431-5437.
ready demonstrated clear additive benefits in animal models.64
In conclusion, a variety of promising novel drugs may expand the armory of therapeutics available to patients with diseases associated with defective erythropoiesis. If proven to be safe, selective, and effective, these drugs will increase the chance for treatment success, and competition between drug companies will likely diminish treatment costs.
Disclosures
SR is a member of the scientific advisory boards of Ionis Pharmaceuticals, Meira GTx, Vifor and Disc Medicine. He has been or is a consultant for Incyte, Cambridge Healthcare Res, Celgene Corporation, Catenion, First Manhattan Co., FORMA Therapeutics, Ghost Tree Capital, Keros Therapeutics, Spexis AG, Noble insight, Protagonist Therapeutics, Sanofi Aventis U.S., Slingshot Insight, Techspert.io, BVF Partners L.P., Rallybio, LLC, venBio Select LLC and GSK.
Contributions
AG, HP and SR wrote and revised the manuscript.
Funding
SR acknowledges the support of the Commonwealth Universal Research Enhancement (C.U.R.E.) Program Pennsylvania Department of Health and National Institute of Diabetes and Digestive and Kidney Diseases Institute of the National Institutes of Health (R01 DK090554, R01 DK095112), Institute for Translational Medicine and Therapeutics (ITMAT), Irish Health Research Board-Health Research Charities Ireland (HRCI-HRB) and The Sickle Cell and Red Cell Disorders Curative Therapy Center (CuRED)-Frontier Program. AG acknowledges the support of the Faculty CHOP Program.
7. Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007;21(4):217-231.
8. Max-Audit I, Kechemir D, Mitjavila MT, Vainchenker W, Rotten D, Rosa R. Pyruvate kinase synthesis and degradation by normal and pathologic cells during erythroid maturation. Blood. 1988;72(3):1039-1044.
9. Matte A, Federti E, Kung C, et al. The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a betathalassemia mouse model. J Clin Invest. 2021;131(10):e144206.
10. Witthuhn BA, Quelle FW, Silvennoinen O, et al. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74(2):227-236.
11. Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678-684.
12. Dussiot M, Maciel TT, Fricot A, et al. An activin receptor IIA
ligand trap corrects ineffective erythropoiesis in betathalassemia. Nat Med. 2014;20(4):398-407.
13. Guerra A, Oikonomidou PR, Sinha S, et al. Lack of Gdf11 does not improve anemia or prevent the activity of RAP-536 in a mouse model of beta-thalassemia. Blood. 2019;134(6):568-572.
14. Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98(15):8780-8785.
15. Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090-2093.
16. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. 2017;168(3):344-361.
17. Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8(6):502-511.
18. Nai A, Lidonnici MR, Rausa M, et al. The second transferrin receptor regulates red blood cell production in mice. Blood. 2015;125(7):1170-1179.
19. Parrow NL, Li Y, Feola M, et al. Lobe specificity of iron binding to transferrin modulates murine erythropoiesis and iron homeostasis. Blood. 2019;134(17):1373-1384.
20. Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet. 2008;40(5):569-571.
21. Nemeth E, Ganz T. Hepcidin and iron in health and disease. Annu Rev Med. 2023;74:261-277.
22. Steinberg MH. Thalassemia: molecular pathology and management. Am J Med Sci. 1988;296(5):308-321.
23. Higgs DR. The thalassaemia syndromes. Q J Med. 1993;86(9):559-564.
24. Khandros E, Weiss MJ. Protein quality control during erythropoiesis and hemoglobin synthesis. Hematol Oncol Clin North Am. 2010;24(6):1071-1088.
25. Mannu F, Arese P, Cappellini MD, et al. Role of hemichrome binding to erythrocyte membrane in the generation of band-3 alterations in beta-thalassemia intermedia erythrocytes. Blood. 1995;86(5):2014-2020.
26. Rachmilewitz EA, Thorell B. Hemichromes in single inclusion bodies in red cells of beta thalassemia. Blood. 1972;39(6):794-800.
27. Rivella S. Iron metabolism under conditions of ineffective erythropoiesis in beta-thalassemia. Blood. 2019;133(1):51-58.
28. Arlet JB, Ribeil JA, Guillem F, et al. HSP70 sequestration by free alpha-globin promotes ineffective erythropoiesis in betathalassaemia. Nature. 2014;514(7521):242-246.
29. Anderson ER, Taylor M, Xue X, et al. Intestinal HIF2alpha promotes tissue-iron accumulation in disorders of iron overload with anemia. Proc Natl Acad Sci U S A. 2013;110(50):E4922-4930.
30. Cabantchik ZI, Breuer W, Zanninelli G, Cianciulli P. LPI-labile plasma iron in iron overload. Best Pract Res Clin Haematol. 2005;18(2):277-287.
31. Musallam KM, Cappellini MD, Viprakasit V, Kattamis A, Rivella S, Taher AT. Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am J Hematol. 2021;96(2):E54-E56.
32. Porter JB, Cappellini MD, Kattamis A, et al. Iron overload across the spectrum of non-transfusion-dependent thalassaemias: role of erythropoiesis, splenectomy and transfusions. Br J Haematol. 2017;176(2):288-299.
33. Porter JB, Walter PB, Neumayr LD, et al. Mechanisms of plasma non-transferrin bound iron generation: insights from comparing
transfused Diamond Blackfan anaemia with sickle cell and thalassaemia patients. Br J Haematol. 2014;167(5):692-696.
34. Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583-588.
35. Casu C, Oikonomidou PR, Chen H, et al. Minihepcidin peptides as disease modifiers in mice affected by beta-thalassemia and polycythemia vera. Blood. 2016;128(2):265-276.
36. Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. 2018;131(16):1790-1794.
37. Cappellini MD, Viprakasit V, Taher AT, et al. A phase 3 trial of luspatercept in patients with transfusion-dependent betathalassemia. N Engl J Med. 2020;382(13):1219-1231.
38. Piga A, Longo F, Gamberini MR, et al. Long-term safety and erythroid response with luspatercept treatment in patients with β-thalassemia. Ther Adv Hematol. 2022;13:20406207221134404.
39. Attie KM, Allison MJ, McClure T, et al. A phase 1 study of ACE536, a regulator of erythroid differentiation, in healthy volunteers. Am J Hematol. 2014;89(7):766-770.
40. Ahsan A, Fang W, Ugidos M, et al. Investigations into the mechanisms and clinical implications of modulation of hepcidin levels by luspatercept in TD MDS and TD β-thalassemia. Blood. 2022;140:(Suppl 1): 8188-8189.
41. Arezes J, Foy N, McHugh K, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. 2018;132(14):1473-1477.
42. Kung C, Hixon J, Kosinski PA, et al. AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood. 2017;130(11):1347-1356.
43. Grace RF, Rose C, Layton DM, et al. Safety and efficacy of mitapivat in pyruvate kinase deficiency. N Engl J Med. 2019;381(10):933-944.
44. Rab MAE, Van Oirschot BA, Kosinski PA, et al. AG-348 (mitapivat), an allosteric activator of red blood cell pyruvate kinase, increases enzymatic activity, protein stability, and ATP levels over a broad range of PKLR genotypes. Haematologica. 2021;106(1):238-249.
45. Kuo KHM, Layton DM, Lal A, et al. Safety and efficacy of mitapivat, an oral pyruvate kinase activator, in adults with nontransfusion dependent alpha-thalassaemia or beta-thalassaemia: an open-label, multicentre, phase 2 study. Lancet. 2022;400(10351):493-501.
46. Khandros E, Thom CS, D'Souza J, Weiss MJ. Integrated protein quality-control pathways regulate free alpha-globin in murine beta-thalassemia. Blood. 2012;119(22):5265-5275.
47. Lithanatudom P, Wannatung T, Leecharoenkiat A, Svasti S, Fucharoen S, Smith DR. Enhanced activation of autophagy in beta-thalassemia/Hb E erythroblasts during erythropoiesis. Ann Hematol. 2011;90(7):747-758.
48. Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015;47(3):e147.
49. Lechauve C, Keith J, Khandros E, et al. The autophagy-activating kinase ULK1 mediates clearance of free alpha-globin in betathalassemia. Sci Transl Med. 2019;11(506):eaav4881.
50. Colucci S, Pagani A, Pettinato M, et al. The immunophilin FKBP12 inhibits hepcidin expression by binding the BMP type I receptor ALK2 in hepatocytes. Blood. 2017;130(19):2111-2120.
51. Artuso I, Lidonnici MR, Altamura S, et al. Transferrin receptor 2 is a potential novel therapeutic target for beta-thalassemia: evidence from a murine model. Blood. 2018;132(21):2286-2297.
52. Di Modica SM, Tanzi E, Olivari V, et al. Transferrin receptor 2 (Tfr2) genetic deletion makes transfusion-independent a murine model of transfusion-dependent beta-thalassemia. Am J Hematol. 2022;97(10):1324-1336.
53. Libani IV, Guy EC, Melchiori L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in betathalassemia. Blood. 2008;112(3):875-885.
54. Tyler PA, Madani G, Chaudhuri R, Wilson LF, Dick EA. The radiological appearances of thalassaemia. Clin Radiol. 2006;61(1):40-52.
55. Taher AT, Karakas Z, Cassinerio E, et al. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: results from a phase 2a study. Blood. 2018;131(2):263-265.
56. Casu C, Presti VL, Oikonomidou PR, et al. Short-term administration of JAK2 inhibitors reduces splenomegaly in mouse models of β-thalassemia intermedia and major. Haematologica. 2018;103(2):e46-e49.
57. Li H, Rybicki AC, Suzuka SM, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16(2):177-182.
58. Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109(11):5027-5035.
59. Guo S, Casu C, Gardenghi S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123(4):1531-1541.
60. Arezes J, Foy N, McHugh K, et al. Antibodies against the erythroferrone N-terminal domain prevent hepcidin suppression and ameliorate murine thalassemia. Blood. 2020;135(8):547-557.
61. Langer AL, Esrick EB. β-Thalassemia: evolving treatment options beyond transfusion and iron chelation. Hematology Am Soc Hematol Educ Program. 2021;2021(1):600-606.
62. Taranath R, Bourne G, Zhao L, Frederick B, King C, Liu D. Regulation of iron homeostasis by PTG-300 improves disease parameters in mouse models for beta-thalassemia and hereditary hemochromatosis. Blood. 2019;134:(Suppl_1):3540.
63. Manolova V, Nyffenegger N, Flace A, et al. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of beta-thalassemia. J Clin Invest. 2019;130(1):491-506.
64. Casu C, Pettinato M, Liu A, et al. Correcting beta-thalassemia by combined therapies that restrict iron and modulate erythropoietin activity. Blood. 2020;136(17):1968-1979.
65. Guerra A, Demsko P, McVeigh P, et al. Combination of a luspatercept-like drug (RAP-GRL) and Tmprss6-ASO is superior to either drug alone for correcting β-thalassemia. Blood. 2021;138(Suppl 1):2013.
66. Matte A, Federti E, Winter M, et al. Bitopertin, a selective oral GLYT1 inhibitor, improves anemia in a mouse model of betathalassemia. JCI Insight. 2019;4(22):e130111.
67. Wu M, Ducamp S, Xiang Y, et al. Bitopertin, a selective glycine transporter 1 inhibitor, reduced protoporphyrin IX (PPIX) level and improved liver fibrosis in a mouse model of erythropoietic protoporphyria (EPP). Blood. 2022;140:(Suppl 1):8192-8193.
68. Taher AT, Viprakasit V, Cappellini MD, et al. Haematological effects of oral administration of bitopertin, a glycine transport inhibitor, in patients with non-transfusion-dependent betathalassaemia. Br J Haematol. 2021;194(2):474-477.
69. Papassotiriou I, Kanavakis E, Stamoulakatou A, Kattamis C. Tissue oxygenation in patients with hemoglobinopathy H. Pediatr Hematol Oncol. 1997;14(4):323-334.
70. Mettananda S, Higgs DR. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am. 2018;32(2):177-191.
71. Amid A, Chen S, Athale U, et al. Iron overload in transfusiondependent survivors of hemoglobin Bart's hydrops fetalis. Haematologica. 2018;103(5):e184-e187.
72. Amid A, Barrowman N, Odame I, Kirby-Allen M. Optimizing transfusion therapy for survivors of haemoglobin Bart's hydrops fetalis syndrome: defining the targets for haemoglobin-H fraction and "functional" haemoglobin level. Br J Haematol. 2022;197(3):373-376.
73. Amid A, Chen S, Brien W, Kirby-Allen M, Odame I. Optimizing chronic transfusion therapy for survivors of hemoglobin Barts hydrops fetalis. Blood. 2016;127(9):1208-1211.
74. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755-761.
75. Ginzburg YZ, Feola M, Zimran E, Varkonyi J, Ganz T, Hoffman R. Dysregulated iron metabolism in polycythemia vera: etiology and consequences. Leukemia. 2018;32(10):2105-2116.
76. Barbui T, Passamonti F, Accorsi P, et al. Evidence- and consensus-based recommendations for phlebotomy in polycythemia vera. Leukemia. 2018;32(9):2077-2081.
77. Grisouard J, Li S, Kubovcakova L, et al. JAK2 exon 12 mutant mice display isolated erythrocytosis and changes in iron metabolism favoring increased erythropoiesis. Blood. 2016;128(6):839-851.
78. Casu C, Liu A, De Rosa G, et al. Tmprss6-ASO as a tool for the treatment of polycythemia vera mice. PLoS One. 2021;16(12):e0251995.
79. Pemmaraju N, Kuykendall A, Kremyanskaya M, et al. MPN-469 rusfertide (PTG-300) treatment interruption reverses hematological gains and upon reinitiation, restoration of clinical benefit observed in patients with polycythemia vera. Clin Lymphoma Myeloma Leuk. 2022;22(Suppl 2):S338-S339.
80. Zeidan AM, Platzbecker U, Garcia-Manero G, et al. Longer-term benefit of luspatercept in transfusion-dependent lower-risk myelodysplastic syndromes with ring sideroblasts. Blood. 2022;140(20):2170-2174.
81. Russo R, Andolfo I, Manna F, et al. Increased levels of ERFEencoding FAM132B in patients with congenital dyserythropoietic anemia type II. Blood. 2016;128(14):1899-1902.
82. Iolascon A, Andolfo I, Russo R. Congenital dyserythropoietic anemias. Blood. 2020;136(11):1274-1283.
83. Yang Z, Keel SB, Shimamura A, et al. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci Transl Med. 2016;8(338):338ra367.
84. Heeney MM, Finberg KE. Iron-refractory iron deficiency anemia (IRIDA). Hematol Oncol Clin North Am. 2014;28(4):637-652.
85. Batchelor EK, Kapitsinou P, Pergola PE, Kovesdy CP, Jalal DI. Iron deficiency in chronic kidney disease: updates on pathophysiology, diagnosis, and treatment. J Am Soc Nephrol. 2020;31(3):456-468.
86. Birgegard G, Samuelsson J, Ahlstrand E, et al. Inflammatory functional iron deficiency common in myelofibrosis, contributes to anaemia and impairs quality of life. From the Nordic MPN Study Group. Eur J Haematol. 2019;102(3):235-240.
87. Zhou A, Kong T, Fowles JS, et al. Hepcidin is elevated in primary and secondary myelofibrosis and remains elevated in patients treated with ruxolitinib. Br J Haematol. 2022;197(4):e49-e52.
88. Steinbicker AU, Bartnikas TB, Lohmeyer LK, et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood. 2011;118(15):4224-4230.
89. Traeger L, Gallitz I, Sekhri R, et al. ALK3 undergoes ligandindependent homodimerization and BMP-induced heterodimerization with ALK2. Free Radic Biol Med. 2018;129:127-137.
90. Belot A, Gourbeyre O, Fay A, et al. LJ000328, a novel ALK2/3
kinase inhibitor, represses hepcidin and significantly improves the phenotype of IRIDA. Haematologica. 2020;105(8):e385-e388.
91. Tremblay D, Mesa R. Momelotinib for the treatment of myelofibrosis with anemia. Future Oncol. 2022;18(20):2559-2571.
92. Nili M, Shinde U, Rotwein P. Soluble repulsive guidance molecule c/hemojuvelin is a broad spectrum bone morphogenetic protein (BMP) antagonist and inhibits both BMP2- and BMP6-mediated signaling and gene expression. J Biol Chem. 2010;285(32):24783-24792.
93. Boser P, Seemann D, Liguori MJ, et al. Anti-repulsive guidance molecule c (RGMc) antibodies increases serum iron in rats and Cynomolgus monkeys by hepcidin downregulation. AAPS J. 2015;17(4):930-938.
94. Kovac S, Boser P, Cui Y, et al. Anti-hemojuvelin antibody corrects anemia caused by inappropriately high hepcidin levels. Haematologica. 2016;101(5):e173-176.
95. Novikov N, Buch A, Yang H, et al. DISC-0974, an antihemojuvelin antibody, reduces hepcidin and mobilizes iron in healthy volunteers. Blood. 2022;140(Suppl 1):5339-5340.
96. Wu M, Wang K, MacDonald B. DISC-0974, an anti-hemojuvelin (HJV) monoclonal antibody, reduced hepcidin and improved anemia in a rat model of chronic kidney disease. Blood. 2022;140:(Suppl 1):8153-8154.
97. Kwiatkowski JL. Current recommendations for chelation for transfusion-dependent thalassemia. Ann N Y Acad Sci. 2016;1368(1):107-114.
98. Glickstein H, El RB, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106(9):3242-3250.
99. Tricta F, Fradette C, Temin N, Rozova A, Lee D, Cabantchik I. Efficacy and safety of early-start deferiprone in infants and young children with transfusion-dependent beta thalassemia: evidence for iron shuttling to transferrin in a randomized, double-blind, placebo-controlled clinical trial (START). Hemasphere. 2022;6(Suppl):1439-1440.
100. Silvestri L, Nai A, Pagani A, Camaschella C. The extrahepatic role of TFR2 in iron homeostasis. Front Pharmacol. 2014;5:93.
101. Olivari V, Di Modica SM, Lidonnici MR, et al. A single approach to targeting transferrin receptor 2 corrects iron and erythropoietic defects in murine models of anemia of inflammation and chronic kidney disease. Kidney Int. 2023;104(1):61-73.
102. Oliver SE, Gargano JW, Marin M, et al. The Advisory Committee on Immunization Practices' interim recommendation for use of Pfizer-BioNTech COVID-19 vaccine - United States, December 2020. MMWR Morb Mortal Wkly Rep. 2020;69(50):1922-1924.
103. Rizvi F, Everton E, Smith AR, et al. Murine liver repair via transient activation of regenerative pathways in hepatocytes using lipid nanoparticle-complexed nucleoside-modified mRNA Nat Commun. 2021;12(1):613.
104. Rurik JG, Tombacz I, Yadegari A, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375(6576):91-96.
1Department of Pediatrics, Nagoya University Graduate School of Medicine, Nagoya, Japan; 2Departments of Pediatrics and Cancer Biology, Cleveland Clinic, Cleveland, OH, USA and 3Cystic Fibrosis Center, Azienda Ospedaliera Universitaria Integrata, Verona, Italy
Correspondence: S.J. Corey coreys2@ccf.org
Received: February 28, 2023. Accepted: May 17, 2023.
Early view: May 25, 2023.
htps://doi.org/10.3324/haematol.2023.282949
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Shwachman-Diamond syndrome is a rare inherited bone marrow failure syndrome characterized by neutropenia, exocrine pancreatic insufficiency, and skeletal abnormalities. In 10-30% of cases, transformation to a myeloid neoplasm occurs.
Approximately 90% of patients have biallelic pathogenic variants in the SBDS gene located on human chromosome 7q11. Over the past several years, pathogenic variants in three other genes have been identified to cause similar phenotypes; these are DNAJC21, EFL1, and SRP54. Clinical manifestations involve multiple organ systems and those classically associated with the Shwachman-Diamond syndrome (bone, blood, and pancreas). Neurocognitive, dermatologic, and retinal changes may also be found. There are specific gene-phenotype differences. To date, SBDS, DNAJC21, and SRP54 variants have been associated with myeloid neoplasia. Common to SBDS, EFL1, DNAJC21, and SRP54 is their involvement in ribosome biogenesis or early protein synthesis. These four genes constitute a common biochemical pathway conserved from yeast to humans that involve early stages of protein synthesis and demonstrate the importance of this synthetic pathway in myelopoiesis.
Shwachman-Diamond syndrome (SDS) is a rare inherited bone marrow failure syndrome (IBMFS), which occurs in 1/75,000 live births.1 The syndrome is characterized by neutropenia, exocrine pancreatic insufficiency, and skeletal abnormalities (reviewed by Burroughs, Woolfrey, and Shimamura2). Other common manifestations include failure to thrive, transient hepatitis, attention deficit disorder, and eczema.3-5 In 10-30% of cases, transformation to a myeloid neoplasm, myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) occurs.6 Prognosis is poor for patients with SDS and MDS/AML due to therapy-resistant disease and treatment-related toxicities.7
Approximately 90% of patients with SDS have biallelic pathogenic variants in the Shwachman-Bodian-Diamond syndrome gene (SBDS) located on chromosome 7q11. Complicating the sequence analysis is the presence of a pseudogene, SBDSP1 with 97% homology to SBDS. The most common pathogenic variants are c.258+2T>C and c.183_184TA>CT. In a cohort of 158 unrelated individuals with SDS, 89% of patients had at least one allele mutated due to
gene conversion with the pseudogene, and 60% of patients had two of these converted alleles. (The cis/trans orientation of the alleles were not determined).8 Half were compound heterozygotes for c.258+2T>C and c.183_184TA>CT.
Over the past several years, pathogenic variants in three other genes have been identified in children with SDS-like disease: DnaJ heat shock protein family (Hsp40) member C21 (DNAJC21),9,10 signal recognition particle 54 (SRP54),11,12 and elongation factor-like GTPase 1 (EFL1).13 Common to these three genes and SBDS is their involvement in protein synthesis. SBDS, EFL1, and DNAJC21 are proteins that affect ribosome biosynthesis. SBDS co-operates with the GTPase EFL1 to catalyze the release of eukaryotic translation initiation factor 6 (eIF6) from the pre-60S subunit. Release of eIF6 is essential for the assembly of the 80S ribosomal subunit from the 40S SSU (small subunit) and the 60S LSU (large subunit).14,15 SRP54 facilitates the emergence of the nascent polypeptide in ribosome-associated signal recognition particle (SRP). Genotype-phenotype correlations among the variants of SBDS, DNAJC21, SRP54, and EFL1 have not been comprehensively discussed.
We used PubMed to identify published case reports or series with SDS caused by DNAJC21, SRP54, and EFL1 pathogenic variants. We searched for publications through to February 10, 2023, limited to human subjects, used the terms “DNAJC21”, “SRP54”, and “EFL1”, and extracted clinical information. We identified 17 publications that described 63 individual patients with Shwachman-Diamond-like syndromes. 9-13,16-27 We excluded those that were genetically diagnosed with asymptomatic carriers of SDS-associated variants and whose phenotypes were not described in the reports. Some phenotype data were not available, and the values below were calculated based on the number of cases with data for that specific phenotype. This review will use the terms ShwachmanDiamond-like syndrome or Shwachman-Diamond syndromes.
The affected genes among the 63 SDS-like patients were: SRP54 33 cases (52%), DNAJC21 17 cases (27%), and EFL1 13 cases (21%) (Table 1). Disease was inherited as either autosomal recessive (DNAJC21 and EFL1) or autosomal dominant (SRP54). Distribution plots of pathogenic/likely pathogenic variants are shown for DNAJC21, EFL1, and SRP54 (Figure 1). About half of the patients with DNAJC21 variants harbored biallelic missense mutations; one-third had biallelic null variants. In patients with EFL1 variants, compound heterozygous missense mutations were most frequent, with a few harboring compound missense and null variants. Three patients with EFL1 variants were found to have somatic uniparental disomy of the EFL1 locus in the hematopoietic cells. This generated homozygosity for the relatively milder variant, which conferred selective advantages.25 No patients harbored biallelic null variants. (This feature of having no biallelic null variants has also been observed in patients with SBDS.) Patients' characteristics are described in Table 2. The median age at diagnosis in the genetic groups was 1.2, 0.4, and 0.2 years in DNAJC21, SRP54, and EFL1, respectively. The male to female ratio was 1:1, 2:1, and 1:1.2 in DNAJC21, SRP54, and
EFL1, respectively.
The central hematologic hallmark of SDS is neutropenia with different degrees of severity. The bone marrow (BM) is generally hypocellular with a lower frequency of CD34+ cells as well as myeloid precursors through the metamyelocyte stage.28 Sbds deletion through downregulation of the gene in Cebpa-expressing murine cells also suggested that SBDS is critical for full myelocyte survival and differentiation.29 Almost all (93%) of the patients with mutated SRP54 showed maturation arrest of myeloid cells in the BM, a characteristic observed in patients with severe congenital neutropenia. The variable degree of neutropenia may be due to dosage effect of mutant SRP54, mitigated partially by transcription factor X-box binding protein.30 In contrast, this maturation arrest was not observed in patients with DNAJC21 and EFL1 variants. In addition, two patients (6%) with SRP54 showed cyclic neutropenia, which is typically observed in patients with congenital neutropenia harboring ELANE mutations.31
Pancytopenia was not observed in patients with the SRP54 variants, whereas 94% and 38% of patients with DNAJC21 and EFL1 variants, respectively, developed pancytopenia during their disease course. Since less than 40% of patients with SBDS variants develop pancytopenia by middle age (50 years old),32 the incidence of pancytopenia is higher in DNAJC21. The cumulative incidence of severe neutropenia, thrombocytopenia, and anemia in an Italian cohort of 88 patients with SDS secondary to SBDS variants was at 30 years of age, respectively, 60% (95% Confidence Interval [CI]: 46.9-76.5), 67% (95% CI: 52.4-85.1), and 20% (95% CI: 8.4-48.7). In the Italian study with complete information, 10 patients developed MDS/myeloid leukemia and 9 patients developed BM failure (BMF) / cytopenia. The 20-year cumulative incidence of myeloid neoplasia or BMF / severe cytopenia was 10% (95% CI: 3.719.5) and 10% (95% CI: 4.4-17.8), respectively.32
Once a patient with SBDS-associated SDS developed MDS/AML, the prognosis was fair (median survival: 7.7 years) for those with MDS and poor (median survival: 0.99 years) for AML, with persistent neoplastic disease and infectious complications being the predominant causes of death.7 Three-year survival for SDS patients with AML was
ficationin the cohort.
+: wild-type; -: null variant (nonsense and frameshift, excluding splice site variant); mis: missense variant; ss: splice altering variant. For individual variants, see Online Supplementary Table S1. *Genotype +/mis included 19 patients with a single amino acid deletion in the SRP54 gene. **The patient harbored a de novo recurrent (p.Gly274Asp) and a hotspot (p.Ile225Asp) variant in the SRP54 gene.
human DNAJC21,
and EFL1 showing position of disease-associated variants. Lollipop plot indicating the locations of variants in the DNAJC21 , SRP54 , and EFL1 genes. An unknown variant which resulted in loss of protein expression in a case with EFL1 was not included. Protein domains and structur es were obtained from Pfam and Uniprot databases.

yrs: years; AML: acute myeloid leukemia; MDS: myelodysplastic syndrome; IUGR: intrauterine growth restriction; CNS: central nervous system; NA: not available or applicable. Fisher's exact test was used to compare phenotypes in the gene groups. *Short telomeres were defined as below the 10th percentile of the age-matched controls. Table produced using previously published data.9-13,16-27
11% and 51% for those with MDS.6,7 The European Society for Blood and Marrow Transplantation - Severe Aplastic Anaemia Working Party recently performed a meta-analysis on 229 cases of allogeneic stem cell transplantation in SDS patients. They recommended regular and structured hematologic follow-up, reduction of transplant-related mortality through reduced-intensity conditioning regimens, the limitation of total body irradiation, and the early diagnosis of clonal malignant evolution and use of stem cell transplantation.33
Little is known about the risk of myeloid neoplasia among those with the rare genetic variants. This is likely due to the small number of patients and the relatively brief period of follow-up. Transformation to acute leukemia was reported in a patient with DNAJC21 variants and two with an SRP54 variant. The former, harboring a homozygous DNAJC21 p.Pro32Ala mutation, developed acute megakaryocytic leukemia (AML-M7) at 12 years of age, when genetic studies were carried out.9 A 15-year old boy with congenital neutropenia treated with granulocyte colony-stimulating factor presented with AML with myelodysplasia-related changes and was then diagnosed with the pathogenic variant p.Thr117del in SRP54 . Additional genetic lesions were found: del(5q31.2), CSF3R
p.Gln776* and RUNX1 p.Pro113Leu. For this one case, treatment with daunorubicin/cytarabine and stem cell transplant has been successful, albeit with a short follow-up.27 The other individual had acute lymphoblastic leukemia (ALL), even though ALL has rarely been reported in patients with SDS.34 This patient presented with neutropenia at the age of five weeks and was subsequently identified to harbor a de novo SRP54 p.Cys136Tyr variant. She was treated with granulocyte colony-stimulating factor from the age of four months until 10 years of age, when she was diagnosed with B-cell precursor ALL harboring aberrant expression of CD13 and CD33. Karyotype analysis showed del(5q) in the major clone with minor clones bearing del(7q). High-throughput sequencing revealed RUNX1 (NM_001754.5:c.958C>T;p.Arg320*) and CSF3R (NM_156039.3:c.2302C>T;p.Gln768*) variants, but no TP53 variants. She achieved complete remission after ALL-type induction chemotherapy and those mutations disappeared from BM cells. She received a cord blood transplant and has remained in remission for seven months.12,35
While predisposition to leukemia has been established in patients with SDS, there is less evidence for predisposition to non-hematologic cancers. Seven cases with solid
tumors have been reported, including 3 cases among 155 cases in the French Registry for Severe Chronic Neutropenia.36 This study reported two cases with breast adenocarcinoma, one ovarian cancer, one pancreatic adenocarcinoma, one esophageal squamous cell carcinoma, one peritoneal carcinoma, and one dermatofibrosarcoma. All had been diagnosed in their 30s / 40s, except one who was diagnosed with dermatofibrosarcoma at the age of 17 years. Currently, no patients with DNAJC21, SRP54, and EFL1 have been diagnosed with solid tumors. However, solid tumors in patients with SBDS mutations are predominantly diagnosed in adults, so patients with DNAJC21, SRP54, and EFL1 variants need continuous close monitoring starting once they enter the fourth decade of life. Another hallmark of SDS is exocrine pancreatic insufficiency, and approximately 95% of patients with SBDS are affected with pancreatic dysfunction associated with gastrointestinal symptoms. The developing pancreas is destined to become one of the most metabolically active secretory organs, producing approximately one liter of fluid rich in digestive enzymes every day.37 Produced by acinar cells, these digestive enzymes are critical for absorption of macronutrients (protein and lipids). Exocrine pancreatic insufficiency results in stunted growth and malnutrition. In SDS, exocrine pancreatic insufficiency results from pancreatic acinar cell atrophy, and adipocyte replacement without appreciable inflammation.38 Almost all patients harboring SBDS mutations present with pancreatic insufficiency in early life, whereas 40-60% became pancreatic sufficient over time.3 It has been reported that lipase output changes from low/absent at the time of diagnosis to normal range, whereas amylase, trypsin and chymotrypsin activity may increase but always remains below normal values.39 The incidence of exocrine pancreatic dysfunction was variable among patients harboring mutations in other SDS-like genes. All the patients with EFL1 variants manifested pancreatic dysfunction, while patients with DNAJC21 and SRP54 variants developed pancreatic insufficiency in fewer individuals (50% and 25%, respectively). In addition, endocrine pancreatic dysfunction has been reported in some SDS cases. Although at diagnosis the incidence of Type 1 diabetes is low (3.2%), it is almost 30-fold higher than the rate of Type 1 diabetes in the general population.40 No data are available for EFL1 and DNAJC21 and diabetes. Recently SRP54 levels were reported to be reduced in a murine model of diabetes,41 suggesting that SRP54 may contribute to the impairment of pre-proinsulin synthesis due to endoplasmic reticulum stress.
Growth abnormalities are commonly observed in patients with SDS.42 These may present as intrauterine growth restriction (IUGR) or short stature. IUGR or short stature were observed in 90% of patients with SBDS variants and the 50th percentile of the SDS population correspond to
the 3rd percentile of the healthy population.43 All patients with DNAJC21 and EFL1 variants had growth abnormalities, whereas only 28% of patients with SRP54 variants had a history of IUGR or developed short stature.
Neurocognitive disorders, such as delayed motor and language developmental skills and limited attention span were found in more than half the children / adolescents with SDS.44 Patients with DNAJC21 and EFL1 variants frequently had neurocognitive symptoms, whereas those with SRP54 variants were less affected. Microcephaly may occur in SDS patients due to SBDS, but this was not consistently reported. Cardiac abnormalities, such as cardiomegaly, have been reported in SDS, but most reports did not mention this.45,46 Other affected organs in SDS include the skin and the eye. Skin examinations showed eczema, café-au-lait spots, and hypo- or hyperpigmentation.47 Interestingly, skin findings were common in patients with DNAJC21, but they were absent in patients with SRP54 and EFL1. Retinal disease was observed in half of the patients with DNAJC21 disease, but in none of the patients with SRP54 and EFL1 variants. The extent of these and other non-classical manifestations due to any of the SDS-associated genes and their genotype-phenotype needs further clarification, although this will be difficult due to variations in evaluation and clinical reporting.
Chemotaxis defects in SDS patients48,49 along with neutropenia raise concern for recurrent and/or severe infection. Neutrophils isolated from SDS patients exhibit dysregulated chemotaxis due to altered F-actin polymerization capability. They may be responsive to common chemotactic stimuli; however, SDS cells cannot normally migrate towards the chemoattractant and generate random movements.48 Patients with SRP54 frequently had history of recurrent/severe infection. Attenuated chemotaxis in neutrophil was shown in a srp54 knockdown zebrafish model.11 Neutropenia was observed in all SRP54 patients that may have been the main cause of this high frequency. On the other hand, nearly half the patients with DNAJC21 variants had recurrent/severe infections, while patients with EFL1 variants were less likely to develop infection; even the frequency of neutropenia was similar. Loss of SBDS expression has been associated with shortened telomeres in a proportion of SDS patients.50,51 One study showed that most patients with DNAJC21 had short telomeres which were defined as below the 10th percentile of the age-matched controls,16 while patients with SRP54 and EFL1 were not evaluated for shortened telomeres. One of six patients with short telomeres was below the 1st percentile in granulocytes and lymphocytes, and the others were between the 1st and 10th percentiles. From a diagnostic perspective, telomere studies have high sensitivity with specificity for dyskeratosis congenita (or short telomere syndrome). For individuals with neutropenia, pancreatic insufficiency, short stature and/or skeletal
anomalies, and failure to thrive, next generation sequencing of BMF syndromes that includes SBDS, DNAJC21, EFL1, and SRP54 is the diagnostic test of choice. It is readily available, quick, and cost-beneficial.52 Having ruled out cystic fibrosis, and with no pathogenic or likely pathogenic variants identified in these four genes, the next step for diagnosis is whole exome sequencing.53 Measurement of telomere length is of limited benefit.
In mammalian cells, more than 200 proteins, ribosomal RNA species, and up to 75% of cellular energy are devoted to producing as many as 20,000 ribosomes per minute.54 This prodigious amount of synthetic activity is tightly regulated.55 Protein synthesis requires ribosomal biosynthesis and ribosomal translation of mRNA into nascent polypeptides that are processed via the SRP (Figure 2). Only a minority of the 200 proteins are structural components, while the majority serve as assembly factors or chaperones. Ribosomal biogenesis begins with the import of proteins and processing of ribosomal RNA in the nucleolus. The small (40S) and large (60S) subunits are constructed and exported out of the nucleus via nuclear pores into the cytoplasm, and especially in the endoplasmic
reticulum.56 DNAJC21 binds precursor 45S ribosomal RNA and may be involved in early nuclear ribosomal RNA biogenesis and maturation of the 60S ribosomal subunit.9 SBDS and EFL1 interact to promote the release of eIF6 to facilitate the assembly of the mature 80S ribosome. The 80S monosomes aggregate to form polysomes for more efficient translation and protein production. As the polypeptides are synthesized, they are processed and transported to organelles. For proteins directed to the endoplasmic reticulum, this process occurs co-translationally, which is mediated by the SRP complex. In eukaryotes, the SRP complex consists of six proteins arranged on a long, non-coding 7SL RNA, which help route the nascent polypeptides to the Golgi apparatus for further modification and shipping to specific subcellular compartments.57 Two other components of the SRP complex, SRP72 and SRP19, are associated with dysgranulopoiesis.58,59 How these fundamental and ubiquitous cellular processes that lead to protein synthesis result in specific, limited phenotypes remains poorly understood. Mammalian cells provide tissue-specific experimental models to study the pathophysiology of SDS. In patients with SBDS-mutated SDS, CD34+ hematopoietic cells were reduced in the BM.60 These hematopoietic cells had impaired colony formation and long-term colony formation. BM mesenchymal stem cells from SBDS-mutated SDS pa-
Figure 2. Components of the Shwachman-Diamond syndrome pathway participate in ribosomal biosynthesis and initial escort by the signal recognition particle. Ribosome maturation begins in the nucleus with the formation of the pre-60S and pre-40S subunits. These subunits traffic through the nuclear pores to the cytosol where the final steps of ribosome maturation occur, forming the 80S ready for translation of mRNA into a nascent polypeptide. The polypeptide emerges from the ribosome and is further processed via the signal recognition particle. See text for further details.

tients had reduced ability to support normal CD34+ hematopoietic cells. A role for mesenchymal cells in altering the BM microenvironment has been advanced and may contribute to dysmyelopoiesis or leukemogenesis.61-63 Increased apoptosis was observed in SDS-derived BM in combination with p53 overexpression.64,65 In studies of human cells, SDS patient cells were hypersensitive to low doses of actinomycin D, an inhibitor of rRNA transcription and its administration abolished nucleolar localization of SBDS.66 Downregulation of SBDS in HEK293 cells showed alterations in both the mRNA levels and mRNA polysome loading of genes implicated in nervous system development, bone morphogenesis, and hematopoiesis.67
SBDS co-localizes with the mitotic spindle by immunofluorescence. Recombinant SBDS bound to purifi ed microtubules in vitro resulting in microtubule stabilization both in vitro and in vivo 68,69 Cultured cells from SDS patients exhibited an increased incidence of mitotic aberrations, characterized by multipolar spindles and centrosomal amplification, compared with controls.68
Knockdown of SBDS expression with siRNA in human fibroblasts recapitulated this phenotype, but only after two weeks in culture, suggesting that the mitotic defects were a downstream result of SBDS loss. Loss of SBDS was associated with increased apoptosis when checkpoint pathways were intact but resulted in aneuploid cells when p53 was inactivated. The increased apoptosis observed in classical SDS hematopoietic progenitors has been associated with dysregulated FAS expression onto plasma membrane.70,71 Apoptosis induced by loss of SBDS expression in human breast cancer cells can be reverted by inhibitors of caspase 8, suggesting this protease contributes to this process.72 Loss of SRP54 expression has been associated with p53-dependent increased apoptosis instead.12 Little is known about the role of DNAJC21 and EFL1 in regulating apoptosis.
Patient-derived cells have limitations in that the numbers of hematopoietic cells are limited in this rare form of IBMFS. In addition, the genes more recently found to be associated with SDS (DNAJC21, EFL1, and SRP54) need to be further clarified, although all, including SBDS, play a role in ribosome biogenesis and function. Thus, there is a high demand for disease models to be established in SDS. The disease models caused by DNAJC21, EFL1, and SRP54 in relationship with human disease are summarized in Table 3.
DNAJC21 encodes a member of the DnaJ heat shock protein 40 family of proteins that contains two N-terminal tetratricopeptide repeat domains and a C-terminal DNAJ domain.73 DNAJC21 binds the precursor 45S ribosomal RNA, which is processed to form the 18S, 5.8S and 28S rRNA.74 DNAJC21 interacts with ZNF622 to stimulate the ATPase activity of HSPA8, promoting the release of the nuclear export receptor PA2G4 from the pre-60S ribosomal
subunit.9 Thus, DNAJC21 also participates in the maturation of the 60S subunit. In yeast, efficient removal of Tif6p (human eIF6) depends on the prior release of Arx1p (human PA2G4) by this pathway.9 This may link DNAJC21 and SBDS-EFL1-eIF6 in the late cytoplasmic 60S ribosomal subunit maturation. The missense variant p.P32A in DNAJC21 is thought to alter the fold of the critical J domain His, Pro, Asp (HPD) motif, disrupting the interaction with HSPA8 and stimulation of its ATPase activity. The p.K34E missense variant reverses the surface charge of a key amino acid adjacent to the HPD motif and likely disrupts the interaction with HSPA8.9
Knockdown of DNAJC21 in HeLa cells via small interfering RNA caused cytoplasmic accumulation of PA2G4, elongated cell morphology, and cell death. Reintroduction of DNAJC21 rescued cell viability and restored normal PA2G4 trafficking. Tummala et al. concluded that DNAJC21 is involved in nucleolar rRNA biogenesis and in cytoplasmic recycling of nuclear export factor PA2G4 for 60S ribosomal subunit maturation.9 In zebrafish using CRISPR / Cas9 knocking out dnajc21, reduced growth and abnormal yolk sac development were observed. This coincided with reduced lipid distribution in the vasculature and caudal hematopoietic tissue region at 48 hours post fertilization (hpf).75
SRP54 has basal GTPase activity and stimulates reciprocal GTPase activation of the SRP receptor subunit alpha (SRPRA).11,76 SRP54 is a component of co-translational targeting of secretory and membrane proteins to the endoplasmic reticulum by SRP complex.76 SR compaction and GTPase-mediated rearrangement of SRP receptor drive SRP-mediated co-translational protein translocation into the endoplasmic reticulum.76 SRP54 requires the presence of SRP9/SRP14 and/or SRP19 to stably interact with RNA. In patient-derived BM cells, GTPase activity was decreased,11 although another report showed that the same SRP54 variant (Gly226Glu) displayed basal GTPase activity and stimulates GTPase reactions with the receptor as efficiently as wild-type.76 Differentiation of patient-derived CD34+ hematopoietic cells resulted in decreased granulocytes.12 Patient-derived granulocytes showed increased endoplasmic reticulum stress and autophagy. In a zebrafish model using morpholino antisense oligonucleotides for srp54, decreased neutrophils and decreased mpx expression at 48 hpf was noted. Pancreas markers, trypsin and ptf1a expression at 72 hpf were also decreased.11
EFL1 is involved in the biogenesis of the 60S ribosomal subunit and translational activation of ribosomes.77 Together with SBDS, EFL1 triggers the GTP-dependent release of eIF6 from 60S pre-ribosomes in the cytoplasm, thereby activating ribosomes for translation competence by allowing 80S ribosome assembly and facilitating eIF6 recycling to the nucleus,78 where it is required for 60S rRNA processing and nuclear export. EFL1 also shows low
intrinsic GTPase activity.77 GTPase activity is increased by contact with 60S ribosome subunits.15
In patient-derived fibroblasts and lymphoblastoid cell lines, release of eIF6 was impaired. Using siRNA knock-
down or CRISPR/Cas9 edited HeLa cells and K562 cells, ribosome assembly was impaired.13 RNA-seq expression profile in K562 edited to harbor patient-derived variant in EFL1 showed different expression profiles than wild-type.
Gene
DNAJC21
SRP54
EFL1
Human cells
Mouse Zebrafish Yeast Patient-derived cells Cell lines C57BL/6J S. cerevisiae
Decreased proliferation by PHA/IL-2 (T cells)9
Morphological changes by shRNA (HeLa)9 NA
Reduced growth and abnormal yolk sac development, reduced lipid distribution in the vasculature and caudal hematopoietic tissue region at 48 hpf (CRISPR/Cas9 KO)75
NA
Reduced expression of rRNA (LCL)9 - - - -
Aberrant micronuclei in CBMN-cyt assay (lymphocyte) - - - -
Decreased GTPase activity (BM cells)11
Decreased granulocytic differentiation (CD34+ cells)12
Decreased GTPase activity by overexpressing the mutant protein (HEK293)11
Decreased proliferation, increased ER stress/autophagy using shRNA (HL-60)12
Increased ER stress/autophagy (granulocytes)12 -
Impaired eIF6 release (fibroblasts and LCL)13
Impaired ribosome assembly using siRNA and CRISPR/Cas9 (HeLa and K562)25
Decreased neutrophils; decreased mpx at 48 hpf, trypsin, and ptf1a expression at 72 hpf revealed using WISH and transgenic fish (MO)11
-
RNA-seq expression profiles after editing by CRISPR/Cas9 (K562)25
Loss of weight, decreased fat accumulation, reduced bone mass density, and decreased HSC/HSPC (N-ethyl-N-nitrosourea mutagenesis)13
Efl1-/- were embryonic lethal, while Efl1+/and Efl1mis/mis were healthy; Efl1mis/- were small, died earlier, and developed pancytopenia (CRISPR/Cas9 KO and KI)25
Smaller heads and eyes, slightly bent tails; decreased neutrophils and erythrocytes (MO)25
Rescuing slow growth of RIA1 (EFL1)Δ by expressing mutant ria123
-
Relocalization of Tif6p (eIF6) to the cytoplasm23
PHA: phytohemagglutinin; CBMN Cyt: cytokinesis-block micronucleus cytome; BM: bone marrow; ER: endoplasmic reticulum; LCL: EpsteinBarr-virus-transformed lymphoblastoid cell lines; HSC/HSPC: hematopoietic stem and progenitor cells; KO: knockout; KI: knock-in of variants; hpf: hours post fertilization; WISH: whole-mount in situ hybridization; MO: morpholino antisense oligomers; NA: not available.
In the yeast studies, expression of patient-corresponding variant ria1 (EFL1 ortholog) rescued slow growth of RIA1null yeast. This was associated with relocalization of Tif6p (eIF6) to the cytoplasm.23 In mice, introduction of variants close to patient-derived variants by N-ethyl-N-nitrosourea random mutagenesis resulted in loss of weight, decreased fat accumulation, reduced bone mass density, and decreased hematopoietic stem and progenitor cells.13 In other mice models, Efl1-/- were embryonic lethal, whereas Efl1+/and Efl1mis/mis were healthy. Efl1mis/- were small, died earlier, and developed pancytopenia.25 In a zebrafish model using morpholinos for efl1, smaller heads and eyes, and slightly bent tails were noted; neutrophils and erythrocytes were decreased in this model.25 Zebrafish lacking efl1 phenocopied some of the molecular and morphologic features of SDS. In addition, results from efl1-/- zebrafish were consistent with those from sbds-/- zebrafish strains, emphasizing a common molecular pathway induced by the dyad of eIF6 dissociating factors.79
The mechanistic target of rapamycin (mTOR) pathway is up-regulated in SBDS-deficient cells from patients. Our research group reported that mTOR phosphorylation of serine 2448 residue (activator) is signi ficantly elevated compared to healthy control cells, possibly as a compensatory mechanism in response to energy deficiency due to ribosome impairment.80-82 The mTOR pathway, triggered by the upstream activation of the phosphatidylinositol 3kinase (PI3K) and AKT kinase, can induce cell proliferation through mitochondria and ribosome biogenesis.83 Interestingly, Conn and Qian showed that constitutively activation of the complex 1 of mTOR can increase the speed of ribosomal elongation leading to decreased translation fidelity, therefore emphasizing a role of mTOR in maintaining protein homeostasis.84 No association between SRP54, EFL1, nor DNAJC21 and mTOR has been reported so far, and this should be clarified in future studies.
The initial description of SDS featured its hallmarks of exocrine pancreatic insufficiency and neutropenia.85,86 Cloning of the gene led to the characterization of its genotypes, demonstrating the complexity because of its pseudogene in humans and its limited number of variants.8 Since then, as many as 90% of individuals identified with the classical triad (neutropenia, exocrine pancreatic insufficiency, and skeletal anomalies) carry biallelic mutations. In 2011, consensus guidelines for the diagnosis of SDS defined the clinical diagnostic criteria with the presence of cytopenia of any given lineage and exocrine pancreas dysfunction.87 Bone abnormalities and behavioral problems were categorized as supportive evidence. Over the last several years, pathogenic variants have been at-
tributed to DNAJC21, EFL1, and SRP54. According to these guidelines, patients with DNAJC21 (BMF syndrome-3, OMIM #617052) and SRP54 (severe congenital neutropenia-8, OMIM #618752) may not be diagnosed as bona fide SDS. However, their clinical characteristics do not fit in other IBMFS categories such as dyskeratosis congenita or severe congenital neutropenia. Because of the small numbers involved, generalizations regarding phenotypes are to be made with caution.
Our review of genotype-phenotype correlations of patients with DNAJC21, EFL1, and SRP54 may refine the clinical diagnostic criteria of SDS. Such a classification conundrum has also arisen for other BMF syndromes. Dyskeratosis congenita encompasses a range of monogenic disorders involving telomere maintenance or stability. Involving blood, skin, lung, gut or genitourinary systems, these disorders are increasingly referred to as short telomere syndromes.88 Similar to dyskeratosis congenita in its restriction to a particular physiologic process is Fanconi anemia, which is due to one of 23 genes involved in different steps to detect and repair DNA interstrand crosslink damage. Defying thematic unity in its pathophysiology, severe congenital neutropenia is due to a number of genes that vary in their biochemical and cellular function.89
SBDS, EFL1, DNAJC21, and SRP54 encode proteins involved in ribosome assembly and nascent polypeptide synthesis. SDS has been viewed as a ribosomopathy.90 This term has been applied to diverse diseases with germline or somatic mutations, such as Treacher Collins syndrome, DiamondBlackfan anemia, cartilage hair hypoplasia, and del(5q) MDS.91 We suggest using the term Shwachman-Diamond syndromes or Shwachman-Diamond-like syndrome to denote disorders that may involve blood and/or pancreatic abnormalities, and which result from germline variants that encode proteins affecting ribosome biogenesis and early protein synthesis. The term Diamond-Blackfan anemia should be reserved for those with congenital hypoplastic anemia.
Limitations of this analysis for human phenotypes of SDS due to DNAJC21, EFL1, or SRP54 variants include missing data from patients in the literature and the nature of the descriptive research that did not provide an adequate sample size for statistical analyses to be performed. To date, there have been few organismal models to characterize phenotypes that copy human SDS. The molecular pathways underlying these entities have fallen short on identifying precise mechanisms for developing BMF and pancreatic insufficiency. In addition, even though embryonic lethality was avoided, neoplastic transformation (the major concern for SDS patients in late adolescence-early adulthood) has not been modeled in mice or zebrafish.
Comparison of phenotypes should promote a better understanding of the disease entities covered by the term SDS. This may contribute to early diagnoses, more effec-
tive treatment options, improved surveillance for neoplastic complications, design of chemopreventive strategies, and healthier outcomes. As children, adolescents, and young adults are being diagnosed at earlier ages, and with better monitoring and management, it is likely that new manifestations of SDS will be revealed as these patients live into middle age and beyond. Understanding the molecular pathophysiology of SDS will also likely provide major new insights into the fundamental conserved mechanisms of ribosome assembly and protein synthesis, their quality control, and neoplastic transformation.
The authors have no conflicts of interest to disclose.
1. Minelli A, Nicolis E, Cannioto Z, et al. Incidence of Shwachman-Diamond syndrome. Pediatr Blood Cancer. 2012;59(7):1334-1335.
2. Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009;23(2):233-248.
3. Cipolli M. Shwachman-Diamond syndrome: clinical phenotypes. Pancreatology. 2001;1(5):543-548.
4. Myers KC, Bolyard AA, Otto B, et al. Variable clinical presentation of Shwachman-Diamond syndrome: update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr. 2014;164(4):866-870.
5. Lange L, Simon T, Ibach B, Rietschel E. [Shwachman-diamond syndrome as cause of infantile eczema associated with failure to thrive]. Klin Padiatr. 2009;221(2):89-92.
6. Donadieu J, Fenneteau O, Beaupain B, et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012;97(9):1312-1319.
7. Myers KC, Furutani E, Weller E, et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: a multicentre, retrospective, cohort study. Lancet Haematol. 2020;7(3):e238-e246.
8. Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97-101.
9. Tummala H, Walne AJ, Williams M, et al. DNAJC21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60S ribosome subunit maturation. Am J Hum Genet. 2016;99(1):115-124.
10. Dhanraj S, Matveev A, Li H, et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood. 2017;129(11):1557-1562.
11. Carapito R, Konantz M, Paillard C, et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J Clin Invest. 2017;127(11):4090-4103.
12. Bellanne-Chantelot C, Schmaltz-Panneau B, Marty C, et al. Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome. Blood. 2018;132(12):1318-1331.
NK, VB and UO performed the research. NK analyzed the data. NK, UO, MC, VB and SJC wrote the paper.
This work is supported by the SENSHIN Medical Research Foundation and the Mochida Memorial Foundation (to NK), a VeloSano grant (to UO), and R01 HL128173, NIH R21 CA159203, DOD Idea Award, Hyundai Hope on Wheels, VeloSano and Lisa Dean Moseley Foundation Award (to SJC).
All the data analyzed in this paper are available in the publications referred to in the text.
13. Tan S, Kermasson L, Hoslin A, et al. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood. 2019;134(3):277-290.
14. Menne TF, Goyenechea B, Sanchez-Puig N, et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39(4):486-495.
15. Weis F, Giudice E, Churcher M, et al. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat Struct Mol Biol. 2015;22(11):914-919.
16. D'Amours G, Lopes F, Gauthier J, et al. Refining the phenotype associated with biallelic DNAJC21 mutations. Clin Genet. 2018;94(2):252-258.
17. Chirita-Emandi A, Petrescu C-A-M, Zimbru CG, et al. Case report: novel biallelic variants in DNAJC21 causing an inherited bone marrow failure spectrum phenotype: an odyssey to diagnosis. Front Genet. 2022;13:870233.
18. Alsavaf MB, Verboon JM, Dogan ME, et al. A novel missense mutation outside the DNAJ domain of DNAJC21 is associated with Shwachman-Diamond syndrome. Br J Haematol. 2022;197(6):e88-e93.
19. Carden MA, Connelly JA, Weinzierl EP, Kobrynski LJ, Chandrakasan S. Severe congenital neutropenia associated with SRP54 mutation in 22q11.2 deletion syndrome: hematopoietic stem cell transplantation results in correction of neutropenia with adequate immune reconstitution. J Clin Immunol. 2018;38(5):546-549.
20. Saettini F, Cattoni A, D’Angio’ M, et al. Intermittent granulocyte maturation arrest, hypocellular bone marrow, and episodic normal neutrophil count can be associated with SRP54 mutations causing Shwachman-Diamond-like syndrome. Br J Haematol. 2020;189(4):e171-e174.
21. Goldberg L, Simon AJ, Rechavi G, et al. Congenital neutropenia with variable clinical presentation in novel mutation of the SRP54 gene. Pediatr Blood Cancer. 2020;67(6):e28237.
22. Tamura T, Yagasaki H, Nakahara E, et al. A Filipino infant with severe neutropenia owing to SRP54 mutations was successfully treated with ethnically mismatched cord blood transplantation from a Japanese cord blood bank. Ann Hematol. 2021;100(11):2859-2860.
23. Stepensky P, Chacón-Flores M, Kim KH, et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in a
Shwachman-Diamond like syndrome. J Med Genet. 2017;54(8):558-566.
24. Tan QK-G, Cope H, Spillmann RC, et al. Further evidence for the involvement of EFL1 in a Shwachman--Diamond-like syndrome and expansion of the phenotypic features. Cold Spring Harb Mol Case Stud. 2018;4(5):a003046.
25. Lee S, Shin CH, Lee J, et al. Somatic uniparental disomy mitigates the most damaging EFL1 allele combination in Shwachman-Diamond syndrome. Blood. 2021;138(21):2117-2128.
26. Erdős M, Boyarchuk O, Maródi L. Case report: association between cyclic neutropenia and SRP54 deficiency. Front Immunol. 2022;13:975017.
27. Sabulski A, Grier DD, Myers KC, Davies SM, Rubinstein JD. Acute myeloid leukemia in SRP54-mutated congenital neutropenia. eJHaem. 2022;3(2):521-525.
28. Mercuri A, Cannata E, Perbellini O, et al. Immunophenotypic analysis of hematopoiesis in patients suffering from Shwachman-Bodian-Diamond syndrome. Eur J Haematol. 2015;95(4):308-315.
29. Zambetti NA, Bindels EM, Van Strien PM, et al. Deficiency of the ribosome biogenesis gene Sbds in hematopoietic stem and progenitor cells causes neutropenia in mice by attenuating lineage progression in myelocytes. Haematologica. 2015;100(10):1285-1293.
30. Schurch C, Schaefer T, Muller JS, et al. SRP54 mutations induce congenital neutropenia via dominant-negative effects on XBP1 splicing. Blood. 2021;137(10):1340-1352.
31. Horwitz MS, Corey SJ, Grimes HL, Tidwell T. ELANE mutations in cyclic and severe congenital neutropenia: genetics and pathophysiology. Hematol Oncol Clin North Am. 2013;27(1):19-41.
32. Cesaro S, Pegoraro A, Sainati L, et al. A prospective study of hematologic complications and long-term survival of Italian patients affected by Shwachman-Diamond syndrome. J Pediatr. 2020;219:196-201.
33. Cesaro S, Donadieu J, Cipolli M, et al. Stem cell transplantation in patients affected by Shwachman-Diamond syndrome: expert consensus and recommendations from the EBMT Severe Aplastic Anaemia Working Party. Transplant Cell Ther. 2022;28(10):637-649.
34. Furutani E, Liu S, Galvin A, et al. Hematologic complications with age in Shwachman-Diamond syndrome. Blood Adv. 2022;6(1):297-306.
35. Calvo C, Lainey E, Caye A, et al. Leukaemic transformation in a 10-year-old girl with SRP54 congenital neutropenia. Br J Haematol. 2022;198(6):1069-1072.
36. Bou Mitri F, Beaupain B, Flejou J-F, et al. Shwachman-Diamond syndrome and solid tumors: three new patients from the French Registry for Severe Chronic Neutropenia and literature review. Pediatr Blood Cancer. 2021;68(7):e29071.
37. Logsdon CD, Ji B. The role of protein synthesis and digestive enzymes in acinar cell injury. Nat Rev Gastroenterol Hepatol. 2013;10(6):362-370.
38. Tourlakis ME, Zhong J, Gandhi R, et al. Deficiency of Sbds in the mouse pancreas leads to features of Shwachman-Diamond syndrome, with loss of zymogen granules. Gastroenterology. 2012;143(2):481-492.
39. Cipolli M, D'Orazio C, Delmarco A, Marchesini C, Miano A, Mastella G. Shwachman's syndrome: pathomorphosis and longterm outcome. J Pediatr Gastroenterol Nutr. 1999;29(3):265-272.
40. Gana S, Sainati L, Frau MR, et al. Shwachman-Diamond syndrome and type 1 diabetes mellitus: more than a chance association? Exp Clin Endocrinol Diabetes. 2011;119(10):610-612.
41. Kobiita A, Godbersen S, Araldi E, et al. The diabetes gene JAZF1 is essential for the homeostatic control of ribosome biogenesis
and function in metabolic stress. Cell Rep. 2020;32(1):107846.
42. Bogusz-Wojcik A, Kolodziejczyk H, Moszczynska E, et al. Growth hormone improves short stature in children with ShwachmanDiamond syndrome. Pediatr Endocrinol Diabetes Metab. 2021;27(2):87-92.
43. Cipolli M, Tridello G, Micheletto A, et al. Normative growth charts for Shwachman-Diamond syndrome from Italian cohort of 0-8 years old. BMJ Open. 2019;9(1):e022617.
44. Kerr EN, Ellis L, Dupuis A, Rommens JM, Durie PR. The behavioral phenotype of school-age children with shwachman diamond syndrome indicates neurocognitive dysfunction with loss of Shwachman-Bodian-Diamond syndrome gene function. J Pediatr. 2010;156(3):433-438.
45. Lawal OS, Mathur N, Eapi S, Chowdhury R, Malik BH. Liver and cardiac involvement in Shwachman-Diamond syndrome: a literature review. Cureus. 2020;12(1):e6676.
46. Toiviainen-Salo S, Pitkanen O, Holmstrom M, et al. Myocardial function in patients with Shwachman-Diamond syndrome: aspects to consider before stem cell transplantation. Pediatr Blood Cancer. 2008;51(4):461-467.
47. Scalais E, Connerotte AC, Despontin K, et al. ShwachmanDiamond syndrome presenting with early ichthyosis, associated dermal and epidermal intracellular lipid droplets, hypoglycemia, and later distinctive clinical SDS phenotype. Am J Med Genet A. 2016;170(7):1799-1805.
48. Orelio C, Kuijpers TW. Shwachman-Diamond syndrome neutrophils have altered chemoattractant-induced F-actin polymerization and polarization characteristics. Haematologica. 2009;94(3):409-413.
49. Stepanovic V, Wessels D, Goldman FD, Geiger J, Soll DR. The chemotaxis defect of Shwachman-Diamond syndrome leukocytes. Cell Motil Cytoskeleton. 2004;57(3):158-174.
50. Thornley I, Sutherland R, Wynn R, et al. Early hematopoietic reconstitution after clinical stem cell transplantation: evidence for stochastic stem cell behavior and limited acceleration in telomere loss. Blood. 2002;99(7):2387-2396.
51. Calado RT, Graf SA, Wilkerson KL, et al. Mutations in the SBDS gene in acquired aplastic anemia. Blood. 2007;110(4):1141-1146.
52. Tsangaris E, Klaassen R, Fernandez CV, et al. Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population-based cohort and identification of novel mutations. J Med Genet. 2011;48(9):618-628.
53. Bluteau O, Sebert M, Leblanc T, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018;131(7):717-732.
54. Klinge S, Woolford JL Jr. Ribosome assembly coming into focus. Nat Rev Mol Cell Biol. 2019;20(2):116-131.
55. Elhamamsy AR, Metge BJ, Alsheikh HA, Shevde LA, Samant RS. Ribosome biogenesis: a central player in cancer metastasis and therapeutic resistance. Cancer Res. 2022;82(13):2344-2353.
56. Warren AJ. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv Biol Regul. 2018;67:109-127.
57. Kellogg MK, Miller SC, Tikhonova EB, Karamyshev AL. SRPassing co-translational targeting: the role of the signal recognition particle in protein targeting and mRNA protection. Int J Mol Sci. 2021;22(12):6284.
58. Kirwan M, Walne AJ, Plagnol V, et al. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am J Hum Genet. 2012;90(5):888-892.
59. Linder MI, Mizoguchi Y, Hesse S, et al. Human genetic defects in SRP19 and SRPRA cause severe congenital neutropenia with distinctive proteome changes. Blood. 2023;141(6):645-658.
60. Dror Y, Freedman MH. Shwachman-Diamond syndrome: an inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999;94(9):3048-3054.
61. Bardelli D, Dander E, Bugarin C, et al. Mesenchymal stromal cells from Shwachman-Diamond syndrome patients fail to recreate a bone marrow niche in vivo and exhibit impaired angiogenesis. Br J Haematol. 2018;182(1):114-124.
62. Zha J, Kunselman LK, Xie HM, et al. Inducible Sbds deletion impairs bone marrow niche capacity to engraft donor bone marrow after transplantation. Blood Adv. 2022;6(1):108-120.
63. Zambetti NA, Ping Z, Chen S, et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19(5):613-627.
64. Dror Y, Freedman MH. Shwachman-Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood. 2001;97(10):3011-3016.
65. Elghetany MT, Alter BP. p53 protein overexpression in bone marrow biopsies of patients with Shwachman-Diamond syndrome has a prevalence similar to that of patients with refractory anemia. Arch Pathol Lab Med. 2002;126(4):452-455.
66. Ganapathi KA, Austin KM, Lee C-S, et al. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110(5):1458-1465.
67. Hesling C, Oliveira CC, Castilho BA, Zanchin NIT. The Shwachman–Bodian–Diamond syndrome associated protein interacts with HsNip7 and its down-regulation affects gene expression at the transcriptional and translational levels. Exp Cell Res. 2007;313(20):4180-4195.
68. Austin KM, Gupta ML Jr, Coats SA, et al. Mitotic spindle destabilization and genomic instability in Shwachman-Diamond syndrome. J Clin Invest. 2008;118(4):1511-1518.
69. Orelio C, Verkuijlen P, Geissler J, van den Berg TK, Kuijpers TW. SBDS expression and localization at the mitotic spindle in human myeloid progenitors. PLoS One. 2009;4(9):e7084.
70. Rujkijyanont P, Watanabe K, Ambekar C, et al. SBDS-deficient cells undergo accelerated apoptosis through the Fas-pathway. Haematologica. 2008;93(3):363-371.
71. Sen S, Wang H, Nghiem CL, et al. The ribosome-related protein, SBDS, is critical for normal erythropoiesis. Blood. 2011;118(24):6407-6417.
72. Lee J, Ko P, You E, et al. Shwachman-Bodian-Diamond syndrome protein desensitizes breast cancer cells to apoptosis in stiff matrices by repressing the caspase 8-mediated pathway. Anim Cells Syst (Seoul). 2019;23(6):414-421.
73. Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63(22):2560-2570.
74. Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8(7):574-585.
75. Ketharnathan S, Prykhozhij S, Cordeiro A, Dror Y, Berman JN. Zebrafish models provide novel insights into the disease biology of Parn-mutant dyskeratosis congenita and DNAJC21-mutant Shwachman-Diamond syndrome. Blood. 2021;138(Suppl 1):1110-1111.
76. Lee JH, Jomaa A, Chung S, et al. Receptor compaction and GTPase rearrangement drive SRP-mediated cotranslational protein translocation into the ER. Sci Adv. 2021;7(21):eabg0942.
77. Finch AJ, Hilcenko C, Basse N, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25(9):917-929.
78. Jaako P, Faille A, Tan S, et al. eIF6 rebinding dynamically couples ribosome maturation and translation. Nat Commun. 2022;13(1):1562.
79. Kawashima N, Anderson R, Corey SJ, Oyarbide U. Deletion of efl1 in zebrafish recapitulates the spectrum of ShwachmanDiamond syndrome. Blood. 2022;140(s1):2597-2598.
80. Bezzerri V, Vella A, Calcaterra E, et al. New insights into the Shwachman-Diamond Syndrome-related haematological disorder: hyper-activation of mTOR and STAT3 in leukocytes. Sci Rep. 2016;6:33165.
81. Ravera S, Dufour C, Cesaro S, et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci Rep. 2016;6:25441.
82. Vella A, D'Aversa E, Api M, et al. mTOR and STAT3 pathway hyper-activation is associated with elevated interleukin-6 levels in patients with Shwachman-Diamond syndrome: further evidence of lymphoid lineage impairment. Cancers (Basel). 2020;12(3):597.
83. Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63-71.
84. Conn CS, Qian SB. Nutrient signaling in protein homeostasis: an increase in quantity at the expense of quality. Sci Signal. 2013;6(271):ra24.
85. Shwachman H, Diamond LK, Oski FA, Khaw KT. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964;65:645-663.
86. Nezelof C, Watchi M. [Lipomatous congenital hypoplasia of the exocrine pancreas in children. (2 cases and review of the literature)]. Arch Fr Pediatr. 1961;18:1135-1172.
87. Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011;1242:40-55.
88. Armanios M. The role of telomeres in human disease. Annu Rev Genomics Hum Genet. 2022;23:363-381.
89. Oyarbide U, Corey SJ. SRP54 and a need for a new neutropenia nosology. Blood. 2018;132(12):1220-1222.
90. Johnson AW, Ellis SR. Of blood, bones, and ribosomes: is Swachman-Diamond syndrome a ribosomopathy? Genes Dev. 2011;25(9):898-900.
91. Farley-Barnes KI, Ogawa LM, Baserga SJ. Ribosomopathies: old concepts, new controversies. Trends Genet. 2019;35(10):754-767.
1Department of Public Health and Primary Care (PHEG), Leiden University Medical Center, Leiden, The Netherlands; 2Argos Zorggroep “DrieMaasStede”, Center for Specialized Geriatric Care, Schiedam, The Netherlands; 3Department of Pediatrics, Foundation IRCCS San Gerardo dei Tintori, Monza, Italy and 4Department of Medicine and Surgery, University of Milano-Bicocca, Milano, Italy
°Current address: Evean "Oostergouw", Center for Specialized Geriatric Care, Zaandam, The Netherlands
Correspondence: W.H. Tong
w.h.tong@lumc.nl
Received: March 14, 2023.
Accepted: July 10, 2023.
Early view: July 20, 2023.
htps://doi.org/10.3324/haematol.2022.282324
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
For several decades, asparaginase has been considered world-wide as an essential component of combination chemotherapy for the treatment of childhood acute lymphoblastic leukemia (ALL). Discovered over 60 years ago, two main unmanipulated asparaginase products originated from primary bacteria sources, namely Escherichia coli and Erwinia chrysanthemi, have been available for clinical use. A pegylated product of the Escherichia coli asparaginase was subsequently developed and is now the main product used by several international co-operative groups. The various asparaginase products all display the same mechanism of action (hydrolysis of circulating asparagine) and are associated with similar efficacy and toxicity patterns. However, their different pharmacokinetics, pharmacodynamics and immunological properties require distinctive modalities of application and monitoring. Erwinia chrysanthemi asparaginase was initially used as a first-line product, but subsequently became a preferred second-line product for children who experienced immunological reactions to the Escherichia coli asparaginase products. An asparaginase product displaying the same characteristics of the Erwinia chrysanthemi asparaginase has recently been produced by use of recombinant technology, thus securing a preparation available for use as an alternative, or as a back-up in case of shortages, for the non-recombinant product. The long journey of the Erwinia chrysanthemi asparaginase product as it has developed throughout the last several decades has made it possible for almost every child and adult with ALL to complete the asparaginase-based protocol treatment when an immunological reaction has occurred to any Escherichia coli asparaginase product.
Asparaginase (ASNase) is a therapeutic enzyme used now for over 50 years as a component of multi-agent chemotherapy for children with ALL.1-8 ASNase preparations available in the market mainly derive from the Escherichia coli (E. coli) and from the Erwinia chrysanthemi (Erwinia c.) strains,9-14 display the same mechanism of action, have different pharmacokinetic (PK) and pharmacodynamic (PD) properties, and are available for clinical use in different forms: native E. coli and Erwinia c. products and the PEGylated E. coli product (PEG-ASNase) (Figure 1). ASNase mediates the breakdown of asparagine into aspartic acid and ammonia thus depleting the patient's
bloodstream of the amino acid asparagine, which is indispensable for the leukemic blast metabolism, thus exploiting the metabolic weakness of leukemic blasts to autonomously produce asparagine, an amino acid important for the synthesis of several proteins.15,16 In addition to asparagine (ASN) depletion, each ASNase product also results in a variable degree of glutaminase activity, whose significance remains partially unexplained and somewhat controversial. As an example, while Panosyan et al. found that deamination of glutamine (GLN) is a prerequisite for optimal ASN deamination by ASNases in vivo, 17 Chan et al suggested, in contrast, that glutaminase-negative variants of ASNase would provide larger therapeutic indices than wild-type ASNase for asparagine synthetase (ASNS)negative cancers.18 Since GLN is the most abundant amino acid in the blood, its depletion under ASNase treatment is limited; however, a substantial reduction of
Monday, Wednesday, Friday schedule of dosing. The original NCT trials are available at: https://clinicaltrials.gov/.

blood GLN levels leads to extensive ammonia production and has been associated with ASNase-induced liver dysfunction and neurotoxicity. Since the glutaminase activity of the Erwinia c. ASNase is approximately fi ve times higher than that of the E. coli ASNase products, it is important to consider such differences when investigating infusion-related and toxic side effects of the Erwinia c. ASNase products.19,20 Also, it has been suggested that, although GLN is broken down to glutamic acid by ASNase, this may not necessarily lead to a high GLN depletion, as this amino acid can be supplemented from other organs in vivo 21
Native E. coli ASNase has a half-life of 1.24 days following an initial dose of 25,000 IU/m2 (high dose ASNase, intramuscular [IM]), therefore multiple doses are necessary to attain prolonged and profound asparagine depletion.22 In 1978, this preparation was authorized by the US Food and Drug Administration (FDA), but is currently no longer available in the United States for use in children under 18 years of age. Another paper reported that the half-life of native E. coli ASNase after an intravenous (IV) dose of 6,000 IU/m2 is 17.3-19 hours.23 In the 1970s, Erwinia c. ASNase
was found to have PK characteristics similar to that of native E. coli ASNase, but with a shorter half-life of 0.65 days after IM administration.22 In 2011, Erwinia c. ASNase (dosed at 25,000 IU/m2) was approved by the FDA as a treatment option for ALL patients who experienced a hypersensitivity reaction (HSR) to E. coli-derived ASNase preparations.13
PEG-ASNase, a native E. coli ASNase product linked to polyethylene glycol (PEG), results in substantially prolonging the native E. coli ASNase half-life and reducing the associated hypersensitivity phenomena. PK and PD studies of PEG-ASNase began in the 1980s, and were usually conducted in patients who had HSR to native E. coli ASNase.24-26 The half-life of PEG-ASNase given at 2,000 and 2,500 IU/m2 was 357 +/- 243 hours (IV). 24 In 2006, based on a toxicity profile similar to that of the native E. coli ASNase and on a decreased incidence of antibody formation, the FDA approved PEG-ASNase for first-line use in patients with ALL.27,28
In 2008, the results of a study conducted in ALL children on a recombinant variant of native E. coli ASNase were reported by Pieters et al. They concluded that the recom-
binant and the native E. coli ASNase products were bioequivalent and had the same PD and toxicity profile.29 This recombinant E. coli ASNase product is currently not available in the United States.
In 2018, the FDA approved a long-acting product similar to PEG-ASNase, namely calaspargase pegol-mknl (Cal-PEG).30 This formulation used a different linker to E. coli ASNase, resulting in a longer half-life and shelf life. Patients treated with Cal-PEG every three weeks had a similar safety profile and event-free survival (EFS) to those treated with the traditional PEG-ASNase product.30
In 2021, based on the interim results of the COG AALL1931 trial study on the use of a recombinant Erwinia c. ASNase derived from a novel expression platform, the FDA approved its IM use at the dosage of 25 mg/m2. In 2023, Maese et al. reported that this recombinant Erwinia c. ASNase was efficacious at 25/25/50 mg/m2 when given IM on a MondayWednesday-Friday (MWF) schedule.31 The use of this preparation resulted in a safety profile consistent with other ASNases that had earlier been approved by the FDA. An interesting paper by Brigitha et al. recently evaluated how much ASNase is needed for optimal outcome in childhood ALL.32 The authors concluded that: i) the level and duration of exposure have usually been based on the PK profile of the drug and on the assumption that a trough ASNase activity level of 100 U/L or greater is necessary to obtain a complete ASN depletion; ii) the level of exposure has not yet been associated with the outcome as long as the therapeutic level was reached. Authors also concluded that no clear cut-off for optimal exposure duration could be determined and that the achievement of this goal can also depend on immunophenotype, (cyto)genetic subgroups, risk group stratification, and backbone therapy.32 Clearly, the various ASNase products are not readily interchangeable and need to be used at specific schedules and doses. This is especially true for the Erwinia c. ASNase when it is used as second-line agent to replace the E. coli ASNase products when a clinically overt HSR or silent inactivation (SI) phenomena occurs.2 Since several clinical studies have established the efficacy of Erwinia c. ASNase as a first- or second-line product, this specific aspect will be addressed later on in this Spotlight Review. Given that the incidence of E. coli ASNase and PEG-ASNase-associated HSR and SI are overall reported to be up to 60-70%, the importance of the availability of Erwinia c. ASNase is readily apparent.33-39
The benefits derived from an intensive use of any ASNase products, including the Erwinia c. ASNase, have been extensively reported. Table 1 shows all relevant clinical trials (including their main characteristics and relative results)
where the Erwinia c. ASNase has been used as first-line product. Beginning in the 1990s, the UK Medical Research Council ALL consortium conducted a non-randomized trial comparing the toxicity of the E. coli and of the Erwinia c. ASNases given IM to a large cohort of ALL patients as firstline therapy.40 Patients treated with E. coli ASNase had a significantly higher incidence of neurotoxicity, pancreatitis, and life-threatening sepsis than those receiving Erwinia c. ASNase. With a minimum follow-up of 4.5 years, the cohort of patients treated with the Erwinia c. ASNase had an outcome similar to that of patients treated with the native E. coli ASNase. This was the first study showing the efficacy of Erwinia c. ASNase when used as first-line therapy.40 Albertsen et al. studied the different patterns of ASN depletion between the IV and the IM administration route with the Erwinia c. ASNase given daily at 30,000 IU/m2 for ten days during induction and twice weekly doses for two weeks during the re-induction phase of the NOPHO-92 ALL-protocol.41 Over 92% of the treated patients had trough activity levels > 500 U/L42,43 for both administration routes. Conversely, when the ASNase activity levels were evaluated during the re-induction, lower percentages of patients achieved the target level of > 100 U/L (almost 65% for the IV-treated children vs. 73% for the IM-treated children). The authors concluded that the Erwinia c. ASNase schedule adopted in the induction phase was unnecessarily intense, whereas it was insufficient for both administration routes in the re-induction phase.
In a national randomized study enrolling newly diagnosed patients in the AIEOP ALL-91 study, Rizzari et al. evaluated the therapeutic effects of the Erwinia c. ASNase product given IM to the subjects enrolled in the intermediate risk (IR) group of ALL either at weekly high doses, i.e., 25,000 IU/m2, for a total of 20 doses over 20 weeks (experimental arm) during the reinduction and the early phase of maintenance or at standard doses, i.e., 10,000 IU/m2, every 34 days for a total of four doses over two weeks, during the reinduction phase only (standard arm).44 In this study, the results concerning outcome between the two arms were superimposable, thus proving that the Erwinia c. ASNase product given at standard doses and within a standard treatment schedule was capable of maintaining the outcomes expected from previous Berlin-Frankfurt-Münster (BFM)-based clinical trials using native E. coli ASNase. A similar randomized study embedded in the same AIEOP ALL-91 study but designed for standard risk (SR) ALL patients was conducted in the frame of the IDH international co-operative effort named which included patients from Italy, Hungary and The Netherlands.45 That study aimed to determine the efficacy of a BFM-type modified (i.e., less intensive) chemotherapy regimen including (experimental arm) or not (standard arm) a prolonged use of ASNase given at high doses (i.e., 25,000 IU/m2, for a total of 20 doses over 20 weeks) at the beginning of the continuation
Table 1. Main fi ndings reported in the studies featuring Erwinia c . ASNase given as fi r stor second-line product.
ASNase-associated fi ndings
5-year-DFS Erwinia C . vs . E. coli ASNase: 61% vs . 64% (NS)
Mean Erwinia C . ASNase activity levels: 130 U/L (IM) and 150 U/L (IV)
Mean E. coli ASNase activity levels: 678 U/L (IM) and 553 U/L (IV)
Induction: mean Erwinia C . ASNase levels: 2,360 U/L (IV) and 1,710 U/L (IM)
Re-induction: mean Erwinia C . ASNase levels: 110 U/L (IV) and 83 U/L (IM)
483
62
40
Non-randomized
Non-randomized
Non-randomized
administration
Induction: Erwinia C . or E. coli ASNase given IM at 6,000 IU/m 2 3 x / week x 3 weeks
MRC UK VIII ALL
Induction: Erwinia C . or E. coli ASNase 10,000 IU/m 2 given IV or IM q3 days x 8 doses
Induction: Erwinia C . ASNase 30,000 IU/m 2 given daily IV or IM x 10 days
Re-induction: Erwinia C . ASNase 30,000 IU/m 2 2 x / week for 2 weeks; IV vs . IM
AIEOP ALL-95
NOPHO-92 ALL
Rizzari
5-year-EFS Erwinia C . vs . E. coli ASNase arms: 78% vs . 89% ( P =0.01) SPOTLIGHT REVIEW ARTICLE - The journey of asparaginase Erwinia chrysanthemi W.H. Tong and C. Rizzari
7-year-DFS EXP* vs . SD: 76% vs . 72% (NS) 44
610
MR patients in re-induction and maintenance phase: Erwinia C . ASNase 25,000 IU/m 2 given IM weekly x 20 weeks (EXP) vs . 10,000 IU/m 2 given 2 x / week for 2 weeks (in reinduction only, SD)
6-year-OS Erwinia C vs E. coli ASNase: 75% vs . 84% ( P =0.002) 46
AIEOP ALL-91
494 Pession 45
Erwinia C . ASNase patients were 6.7 times more likely to have worse MRD ( ≥ 10-2) compared to E. coli ASNase patients ( P =0.031)
Panosyan 35
13 cases of 81 showed silent inactivation and had a significantly inferior outcome ( P =0.031) at 30 months (not switched to Erwinia C. ASNase because SI was not detected in real time)
Moghrabi 47 Continued on following page.
700
116 Kwok 48
1,001
Randomized
Randomized
Non-randomized
Randomized
Induction: Erwinia C . vs . E. coli ASNase 10,000 IU/m 2 given IV 2 x / week x 4 weeks
Re-induction: Erwinia C . vs . E. coli ASNase 10,000 IU/m 2 given IV 2 x / week x 2 weeks
EORTC-CLG 58881
SR patients in maintenance phase: Erwinia C. ASNase 25,000 IU/m 2 given IM
weekly x 20 weeks (YES** Erwinia C. ASNase) vs . (NO** Erwinia C. ASNase)
Induction: Erwinia C . vs . E. coli ASNase
10,000 IU/m 2 given IM 2 x / week for 4 weeks
High-risk patients: first-line ASNase product in induction (standard BFM, standard duration): E. coli ASNase given at 6,000 IU/m 2 , IM, 3 x week x 2 weeks vs. E. coli ASNase (3 x week x 4 weeks) standard, BFM increased duration
Re-induction: augmented BFM arm PEGASNase 2,500 IU/m 2 given IM every 2 weeks x 6 vs . 10
Re-induction (routine shift as second-line): Erwinia C . ASNase 6,000 IU/m 2 given IM, 3 x / week for 2 weeks
Re-induction: Erwinia C . vs . E. coli ASNase 25,000 IU/m 2 given IM weekly x 20 weeks
I-BFM-SG (IDH-ALL-91)
NUH
DFCI 95-01
491 Haematologica | 108 - October 2023 2609
Salzer 57
Outcome/main ASNase-associated fi ndings
Tong 21
Vrooman 49
SPOTLIGHT REVIEW ARTICLE - The journey of asparaginase Erwinia chrysanthemi W.H. Tong and C. Rizzari Continued on following page.
No EFS / OS data available
No EFS / OS data available
Patients
Among samples during first two weeks of Erwinia C. ASNase: 96% of samples had at least one trough activity level > 0.1 IU/mL
†
Non-randomized
Re-induction: PEG-ASNase 2,500 IU/m 2 given IV or IM every 2 weeks (frequency depending on risk stratification)
First-line ASP product and schedule in induction: E. coli ASNase 5,000 IU/m 2 given IV q 3 days x 8 doses
Re-induction: PEG-ASNase 2,500 IU/m 2 given IV every 2 weeks x 15
Second-line product: Re-induction: Erwinia C. ASNase 20,000 IU/m 2 given IV 3 x / week for 2 weeks to substitute for one PEG-ASNase dose 59
492
AALL07P2***
DCOG ALL-10****
Ref. Haematologica | 108 - October 2023 2610
Re-induction: SR low PEG-ASNase 2,500 IU/m 2 given IV or IM every 2 weeks x 2 vs . SR low PEG-ASNase (same dose) and schedule: every 2 weeks x 6
10-year-DFS PEG-ASNase (all doses): 86%
10-year-DFS missing PEG-ASNase doses: 77% ( P =0.003)******
Randomized (AALL0331) 5,195 ‡ (AALL0232) 3,001
Re-induction: HD MTX arm no PEG-ASNase vs. Capizzi MTX PEG-ASNase 2,500 IU/m 2
Second-line: re-induction: Erwinia C. ASNase 25,000 IU/m 2 given IM given IV or IM every 2 weeks x 2 3 x / week for 2 weeks to replace one PEGASNase dose
Non-randomized
During 1 st 2 weeks of Erwinia C . ASNase treatment 76% of 48h samples and 24% of 72h samples had trough activity levels > 0.1 IU/mL Phase/dose/schedule/ administration route
Gupta 51 DCOG ALL-11****
37 Kloos 50 Study
IM: intramuscular; IV: intravenous; DFS: disease-free survival; EFS: event-free survival; OS: overall survival; Erwinia c. ASNase: Erwinia chrysanthemi asparaginase; E. coli ASNase: native E. coli asparaginase; PEG-ASNase: pegylated E. coli asparaginase; ID: individualized dose; FD: fixed dose; NS: not significant; MRD: minimal residual disease; CTCAE: Common Terminology Criteria for Adverse Events; NUH: National University Hospital; h: hours. * EXP arm: experimental arm, Erwinia c. ASNase IM (25,000 IU/m2) administered on weekly basis for 20 weeks trespassing to the continuation phase. SD arm: standard arm, Erwinia c. ASNase (10,000 IU/m2) twice a week for 2 weeks during protocol II only. ** YES arm: arm including Erwinia c. ASNase IM (25,000 IU/m2) administered on a weekly basis for 20 weeks during the continuation phase. NO arm: arm without any additional Erwinia c. ASNase during the continuation phase. ***** FD: administration of the E. coli ASNase at the fixed dose of 25,000 IU/m2 (weekly) for a total 30 weeks. ID: administration of the native E. coli ASNase dose starting at 12,500 IU/m2 (weekly) and individualized thereafter based on the results of the pharmacological monitoring (TDM). **** Induction starting with E. coli ASNase 5,000 IU/m2 (eight doses q 3 days). Medium risk (MR) treated with PEG-ASNase 2,500 IU/m2 (every two weeks, 15 doses in total). In case of hypersensitivity reaction (HSR), Erwinia c. ASNase (20,000 IU/m2) was started. DCOG ALL-11 protocol: induction started with PEG-ASNase 1,500 IU/m2 (every two weeks). TDM was applied and the dose of PEG-ASNase could be further lowered. In case of HSR, Erwinia c. ASNase (same dose) was started. *** Erwinia c. ASNase 25,000 IU/m2 (Monday/Wednesday/Friday) schedule for 2 weeks. ****** Number of PEG-ASNase doses varied by trial and strata. Continuation phase did not contain any ASNase doses. †In Vrooman et al.,63 42 patients (out of 215 patients enrolled on DFCI protocol 00-01 between 2000-2002) received Erwinia c. ASNase IM. Twice-weekly Erwinia c ASNase was well tolerated and achieved therapeutically effective trough activity levels in most native E. coli ASNase-allergic patients. It should be noted that the development of an allergy to native E. coli ASNase and subsequent switch to Erwinia c. ASNase did not adversely impact EFS.63 ‡For AALL0232, data were also collected on whether patients receiving Erwinia c. subsequently discontinued it for any reason. Data on subsequent Erwinia c. continuation were not available for patients in AALL0331.
phase. In contrast to the previously described study conducted in IR patients, this randomized clinical trial, conducted in 355 SR ALL children, showed that, compared to the group receiving the standard treatment, the group receiving high-dose ASNase ( Erwinia c.) for a protracted period of time (experimental arm) had a significantly increased 10-year disease-free survival (DFS) (87.5% vs . 78.7%) and a higher overall survival (OS) (93.7% vs. 88.6%). Of note, a 40% relative reduction (RR) in the risk of failure was achieved in the group of patients receiving the protracted high-dose ASNase (Erwinia c.) product, thus proving the beneficial effects of that product and schedule, in that chemotherapy context and in that subgroup of patients.45
Notwithstanding, two randomized clinical trials carried out in the same period (early-mid 1990s) in Europe by the European Organisation for Research and Treatment of Cancer (EORTC)46 and in the US by the Dana-Farber Cancer Institute (DFCI) Consortium (Table 1)47 showed that among ALL children (belonging to any risk group) randomly assigned to receive throughout the whole treatment schedule either the native E. coli ASNase or the Erwinia c. ASNase products (same dosage, administration route and treatment schedule), those treated with the Erwinia c. ASNase had consistently lower toxic effects but poorer EFS. These findings were mainly interpreted as secondary to an inadequate timing and dosage adopted for the Erwinia c. ASNase and therefore to a consequently inadequate asparagine depletion.
In 2006, Kwok et al. reported the results of their investigations on minimal residual disease (MRD) to compare the efficacy of Erwinia c. ASNase and native E. coli ASNase given IM at similar dosage and intervals during a classical four-drug induction therapy. They found that patients treated with the Erwinia c. ASNase were 6.7 times more likely to have higher MRD levels (≥ 10-2) thus reflecting slower lymphoblast clearance, presumably due to insufficient ASNase activity.48
Both the therapeutic and toxic effects associated with the Erwinia c. ASNase used as front-line product may be either very similar or very different from those associated with the native E. coli ASNase products, greatly depending on the dosage and the schedule adopted.
After the first decade of its use, when Erwinia c. ASNase had mainly been used as a first-line ASNase with the findings previously reported, the efficacy of a switch to the Erwinia c. ASNase preparation after HSR to a native E. coli ASNase product became more evident and its use as second-line product became widespread.1 Table 1 summarizes the most relevant details of the various clinical trials in which the Erwinia c. ASNase product has been used as second-line product. (See also below.)
The ASNase activity reduction found among the 280 highrisk pediatric ALL patients treated with Erwinia c. ASNase after HSR to a native E. coli ASNase had an impact on the outcome of the treated patients; in fact, Panosyan et al found an increased rate of events among patients with Ab-positive titers detected during the interim maintenance-1 and the delayed intensification.35 Also, Vrooman et al. showed that patients with SI occurring during treatment with native E. coli ASNase had a worse outcome when not switched to another ASNase product, whilst those with a HSR and switched to Erwinia c. ASNase fully maintained the expected outcome.49
In two further clinical studies conducted by the Dutch Childhood Oncology Group (DCOG), Erwinia c. ASNase was used as a second-line preparation. In the first study, Tong et al. studied ALL children presenting with either HSR or SI to PEG-ASNase during the intensification phase of ALL therapy and who were switched to Erwinia c. ASNase. Such a switch led to effective ASNase activity levels in
most patients.21 In the second study, Kloos et al. studied the role of therapeutic drug monitoring (TDM) within the DCOG ALL-11 protocol.50 After a HSR, 37 patients were started on Erwinia c. ASNase at 20,000 IU/m2 (IV) three times a week for two weeks. During the two weeks, 76% had trough activity levels at 48 hours > 100 U/L but only 24% at 72 hours. Thereafter, the Erwinia c. ASNase product dose varied between 15,000 and 40,000 IU/m2 50 The main finding of that study was that, besides SI, also allergiclike reactions could be identified by using TDM. Finally, the Children’s Oncology Group (COG AALL0232) studied more than 3,000 patients with high-risk ALL comparing the outcome of patients receiving all the planned PEG-ASNase doses (group 1) with that of patients switched to receive the Erwinia c. ASNase to complete the planned ASNase treatment (group 2), and with that of patients who completely missed a variable number of ASNase doses, mainly, but not only, due to HSR (group 3). The authors found that the DFS was similar between groups 1 and 2, whilst in group 3, the DFS was inferior compared to group 1, with a HR of 1.5 (P=0.002).51 The data showed that children with either HSR and/or SI occurring during the administration of E. coli ASNase consistently had a poorer outcome compared to those without such an immunological reactivity; when the Erwinia c. ASNase product was substituted for the E. coli ASNase, outcomes were maintained without unexpected additional toxicities.
Despite the approval of the Erwinia c. ASNase as a second-line ASNase since 2011, important difficulties in the manufacturing process of Erwinia c. ASNase have occurred over the last two decades and this has led to global supply shortages of Erwinia c. ASNase.52,53 For this reason, a recombinant Erwinia c. ASNase (namely JZP-458) utilizing a novel Pseudomonas fluorescent technology expression platform has recently been developed;52 it has the same amino acid sequence as Erwinia c. ASNase and results in no immunologic cross-reactivity to E. coli-derived ASNase products. The recombinant technology-based production allows a stable and enhanced production process thus avoiding the supply problems with the non-recombinant Erwinia c. ASNase.
In 2021, in a phase I study conducted in healthy adult volunteers,54 JZP-458 was used at a starting dose of 25 mg/m2 (IM). Previously, it had been demonstrated that this formulation and dosage was similar in achieving sufficient ASNase activity when compared to the non-recombinant Erwinia c. ASNase (based on the dosage of 25,000 IU/m2).55 Lin et al. concluded that a single IM dose of JZP-458 (25 mg/m2) resulted in similar ASNase activity levels com-
pared to 25,000 IU/m2 of the non-recombinant Erwinia c. ASNase.54 In 2021, based on the clinical and pharmacological data derived from the studies described, the FDA granted the approval of JZP-458.31
In 2022, the results of a phase II/III study conducted within the AALL1931 protocol of the COG were published.31 This study focused on the efficacy and safety of the recombinant Erwinia c. ASNase. Each scheduled dose of PEG-ASNase remaining after an HSR had occurred was replaced by six doses of JZP-458 given IM on a Mon/Wed/Fri basis. Maese et al. thoroughly studied three dosing regimens and concluded that JZP-458 given IM at 25/25/50 mg/m2 on a Mon/Wed/Fri schedule was effective in ALL patients, thus supporting the conclusion that this product with such a dosage, schedule and administration route has an efficacy and a toxicity profile similar to that of the non-recombinant Erwinia c. ASNase, and also of the other ASNase preparations available for clinical use and applied at equivalent dosages. The IM use of the recombinant Erwinia c. ASNase is currently approved by the FDA but additional data deriving from the IV use are under evaluation.
As far as the preferred use of administration route is concerned, ASNase products should be given IV. In fact, the study by Place et al. showed that IV administration of PEG-ASNase was associated with similar outcomes and toxicity pattern but with decreased anxiety compared with IM native E. coli ASNase doses. 56 When focusing on the route of the native form of Erwinia c. ASNase, Tong et al. showed that the administration route might explain the higher median ASNase activity levels found by Salzer et al. 21,57 Of note, previous studies have shown that no differences in mean ASNase activity levels, ASN depletion, and ASNase antibodies were found after IV or IM administration of Erwinia c. ASNase.41,43,58
Currently, the use of Erwinia c. ASNase is limited to the second-line setting and not to the first-line setting because of: i) the results of the previous (old) clinical trials; and ii) its PK characteristics which mandate the drug administration every 48 hour to maintain therapeutic ASNase activity levels ≥ 100 U/L, making it very difficult, if not impossible, to deliver the drug to cover the often several weeks-long ASN depletion needed to exploit its therapeutic efficacy. For example, a huge number of doses would be needed during the induction phase of the AIEOPBFM ALL protocols (and of many others protocols) to achieve the 4 weeks-long ASN depletion needed.
Furthermore, when one considers also the current costs of Erwinia c. ASNase, its use in the first-line setting would be very expensive.59 Therefore, the very few doses needed when PEG ASNase or the calaspargase products are used in the first-line setting make the use of these products the preferred option to be pursued.
Because of the recent frequent global shortages of the Erwinia c. ASNase, the recombinant form of Erwinia c. ASNase represents an opportunity to complement the market availability of the non-recombinant Erwinia c. ASNase preparation. This reassures physicians and parents on being able to complete any E. coli ASNase treatment in case of HSR or SI, and secures adequate opportunities of cure to ALL patients presenting with HSR or SI to the E. coli ASNase products.60
In the wider scenario of childhood ALL treatment, the currently available Erwinia c. ASNase products remain important drugs for the treatment of childhood ALL.8 Even if a number of promising (and also quite expensive) new biological agents (such as blinatumomab and inotuzumab) are currently under investigation to show their efficacy and to deepen their acute and mid-/long-term toxicity profile, it seems quite difficult at the present time to foresee a time when ASNase products will be substituted by such targeted drugs in the short-medium term. Furthermore, some innovative Erwinia c. ASNase formulations aiming to improve its characteristics appear on the horizon, making it even more interesting to look towards the future of this important antileukemia agent.
In this Spotlight Review, we have retraced the journey taken over the last six decades by the Erwinia c. ASNase, a chemotherapy agent particularly important for successfully replacing native or pegylated E. coli ASNase products when they were associated with clinically relevant HSR or SI.
The journey started by the Erwinia c. ASNase product in the 1970s has been characterized by the pioneering experiences conducted as a first-line ASNase preparation and, in more recent decades, as a second-line product after HSR and SI to E. coli ASNase products. Across the subsequent decades, the journey has also been characterized by a lack of continuity in its availability caused by flaws in the production process which, for the moment, seem to have been overcome. In any case, the new recombinant Erwinia c. ASNase product, currently available on the market, secures the continuous availability of this important drug.
The scientific knowledge accumulated over the decades on the pharmacology, biological characteristics and clinical
use of the Erwinia c. ASNase today represents a very important asset to ensure that all children with ALL can be treated with the best dosage and schedule, and that they can attain the expected full benefit from any planned ASNase-based treatment programs.
The title of this Spotlight Review was intuitively derived from the 1985 American film directed by Robert Zemeckis “Back to the Future” starring Michael J. Fox and Christopher Lloyd, considered one of the greatest science-fiction films ever made. When both of us, as co-authors, were working on this scientific review of the Erwinia c. ASNase product, we came to realize even more, and even better, its long journey, marked by different events as far as its use (first- and second-line), schedules (including administration routes and dosages), manufacturing technologies (traditional and recombinant), and availability (shortages) have been concerned. And it somewhat resembled to us the “imaginary” journey made in that famous movie by the two leading actors with their time machine flying from the present to the past and then back to the future. But certainly, the “real” story of Erwinia c. ASNase is not yet over, and there is surely still more to come, just like all the sequels and adaptations made to the original movie after its first appearance in the cinemas.
CR has received reimbursement for travelling expenses and personal fees for scientific talks at scientific meetings sponsored by Servier, Jazz Pharmaceuticals , and Clinigen, companies involved in the production and marketing of different asparaginase products.
WHT and CR performed the literature research and wrote the manuscript. CR supervised this Spotlight Review.
CR thanks the Comitato ML Verga for the unrestricted support to his clinical and research activities. He also thanks physicians, pharmacologists, nurses and technicians as well as parents and patients treated in his and AIEOP (Italian Association of Pediatric Hematology and Oncology) institutions for the contributions given to his research in the field of asparaginase. CR and WHT wish to thank Prof. Stephen Earl Sallan (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA) for his accurate and selfless review of the text which greatly improved its final version.
1. Pieters R, Hunger SP, Boos J, et al. L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer. 2011;117(2):238-249.
2. van der Sluis IM, Vrooman LM, Pieters R, et al. Consensus expert recommendations for identification and management of asparaginase hypersensitivity and silent inactivation. Haematologica. 2016;101(3):279-285.
3. Lanvers-Kaminsky C. Asparaginase pharmacology: challenges still to be faced. Cancer Chemother Pharmacol. 2017;79(3):439-450.
4. Salzer W, Bostrom B, Messinger Y, Perissinotti AJ, Marini B. Asparaginase activity levels and monitoring in patients with acute lymphoblastic leukemia. Leuk Lymphoma. 2018;59(8):1797-1806.
5. Baruchel A, Brown P, Rizzari C, et al. Increasing completion of asparaginase treatment in childhood acute lymphoblastic leukaemia (ALL): summary of an expert panel discussion. ESMO Open. 2020;5(5):e000977.
6. Rizzari C, Conter V, Starý J, Colombini A, Moericke A, Schrappe M. Optimizing asparaginase therapy for acute lymphoblastic leukemia. Curr Opin Oncol. 2013;25 Suppl 1:S1-9.
7. Asselin B, Rizzari C. Asparaginase pharmacokinetics and implications of therapeutic drug monitoring. Leuk Lymphoma. 2015;56(8):2273-2280.
8. Clavell LA, Gelber RD, Cohen HJ, et al. Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N Engl J Med. 1986;315(11):657-663.
9. Figueiredo L, Cole PD, Drachtman RA. Asparaginase Erwinia chrysanthemi as a component of a multi-agent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia who have developed hypersensitivity to E. coli-derived asparaginase. Expert Rev Hematol. 2016;9(3):227-234.
10. Keating GM. Asparaginase Erwinia chrysanthemi (Erwinaze®): a guide to its use in acute lymphoblastic leukemia in the USA. BioDrugs. 2013;27(4):413-418.
11. Maloney KW. Erwinia asparaginase: coming closer to an understanding of its use in pediatric acute lymphoblastic leukemia? Pediatr Blood Cancer. 2010;54(2):189-190.
12. Plourde PV, Jeha S, Hijiya N, et al. Safety profile of asparaginase Erwinia chrysanthemi in a large compassionate-use trial. Pediatr Blood Cancer. 2014;61(7):1232-1238.
13. Salzer W, Seibel N, Smith M. Erwinia asparaginase in pediatric acute lymphoblastic leukemia. Expert Opin Biol Ther. 2012;12(10):1407-1414.
14. Salzer WL, Asselin BL, Plourde PV, Corn T, Hunger SP. Development of asparaginase Erwinia chrysanthemi for the treatment of acute lymphoblastic leukemia. Ann N Y Acad Sci. 2014;1329:81-92.
15. Bussolati O, Belletti S, Uggeri J, et al. Characterization of apoptotic phenomena induced by treatment with Lasparaginase in NIH3T3 cells. Exp Cell Res. 1995;220(2):283-291.
16. Stams WA, den Boer ML, Beverloo HB, et al. Sensitivity to Lasparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood. 2003;101(7):2743-2747.
17. Panosyan EH, Grigoryan RS, Avramis IA, et al. Deamination of glutamine is a prerequisite for optimal asparagine deamination by asparaginases in vivo (CCG-1961). Anticancer Res. 2004;24(2c):1121-1125.
18. Chan WK, Lorenzi PL, Anishkin A, et al. The glutaminase activity
of L-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood. 2014;123(23):3596-3606.
19. Narta UK, Kanwar SS, Azmi W. Pharmacological and clinical evaluation of L-asparaginase in the treatment of leukemia. Crit Rev Oncol Hematol. 2007;61(3):208-221.
20. Tong WH, Pieters R, de Groot-Kruseman HA, et al. The toxicity of very prolonged courses of PEGasparaginase or Erwinia asparaginase in relation to asparaginase activity, with a special focus on dyslipidemia. Haematologica. 2014;99(11):1716-1721.
21. Tong WH, Pieters R, Kaspers GJ, et al. A prospective study on drug monitoring of PEGasparaginase and Erwinia asparaginase and asparaginase antibodies in pediatric acute lymphoblastic leukemia. Blood. 2014;123(13):2026-2033.
22. Asselin BL, Whitin JC, Coppola DJ, Rupp IP, Sallan SE, Cohen HJ. Comparative pharmacokinetic studies of three asparaginase preparations. J Clin Oncol. 1993;11(9):1780-1786.
23. Ortega JA, Nesbit ME Jr, Donaldson MH, et al. L-Asparaginase, vincristine, and prednisone for induction of first remission in acute lymphocytic leukemia. Cancer Res. 1977;37(2):535-540.
24. Ho DH, Brown NS, Yen A, et al. Clinical pharmacology of polyethylene glycol-L-asparaginase. Drug Metab Dispos. 1986;14(3):349-352.
25. Keating MJ, Holmes R, Lerner S, Ho DH. L-asparaginase and PEG asparaginase--past, present, and future. Leuk Lymphoma. 1993;10 Suppl:153-157.
26. Ettinger LJ, Kurtzberg J, Voûte PA, Jürgens H, Halpern SL. An open-label, multicenter study of polyethylene glycol-Lasparaginase for the treatment of acute lymphoblastic leukemia. Cancer. 1995;75(5):1176-1181.
27. Dinndorf PA, Gootenberg J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: pegaspargase (oncaspar) for the firstline treatment of children with acute lymphoblastic leukemia (ALL). Oncologist. 2007;12(8):991-998.
28. Avramis VI, Sencer S, Periclou AP, et al. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2002;99(6):1986-1994.
29. Pieters R, Appel I, Kuehnel HJ, et al. Pharmacokinetics, pharmacodynamics, efficacy, and safety of a new recombinant asparaginase preparation in children with previously untreated acute lymphoblastic leukemia: a randomized phase 2 clinical trial. Blood. 2008;112(13):4832-4838.
30. Li RJ, Jin R, Liu C, et al. FDA Approval Summary: calaspargase Pegol-mknl for treatment of acute lymphoblastic leukemia in children and young adults. Clin Cancer Res. 2020;26(2):328-331.
31. Maese L, Loh ML, Choi MR, et al. Recombinant Erwinia asparaginase (JZP458) in acute lymphoblastic leukemia: results from the phase 2/3 AALL1931 study. Blood. 2023;141(7):704-712.
32. Brigitha LJ, Pieters R, van der Sluis IM. How much asparaginase is needed for optimal outcome in childhood acute lymphoblastic leukaemia? A systematic review. Eur J Cancer. 2021;157:238-249.
33. Woo MH, Hak LJ, Storm MC, et al. Anti-asparaginase antibodies following E. coli asparaginase therapy in pediatric acute lymphoblastic leukemia. Leukemia. 1998;12(10):1527-1533.
34. Woo MH, Hak LJ, Storm MC, et al. Hypersensitivity or development of antibodies to asparaginase does not impact treatment outcome of childhood acute lymphoblastic leukemia. J Clin Oncol. 2000;18(7):1525-1532.
35. Panosyan EH, Seibel NL, Martin-Aragon S, et al. Asparaginase antibody and asparaginase activity in children with higher-risk acute lymphoblastic leukemia: Children's Cancer Group Study CCG-1961. J Pediatr Hematol Oncol. 2004;26(4):217-226.
36. Silverman LB, Gelber RD, Dalton VK, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood. 2001;97(5):1211-1218.
37. Appel IM, Kazemier KM, Boos J, et al. Pharmacokinetic, pharmacodynamic and intracellular effects of PEG-asparaginase in newly diagnosed childhood acute lymphoblastic leukemia: results from a single agent window study. Leukemia. 2008;22(9):1665-1679.
38. Möricke A, Rizzari C, Alten J, et al. Hypersensitivity reactions to native E. coli L-asparaginase in children with acute lymphoblastic leukemia treated in Trial ALL-BFM 2000: impact of treatment schedule and type of glucocorticoid in induction. Hemasphere. 2023;7(6):e888.
39. Rizzari C, Möricke A, Valsecchi MG, et al. Incidence and characteristics of hypersensitivity reactions to PEGasparaginase observed in 6136 children with acute lymphoblastic leukemia enrolled in the AIEOP-BFM ALL 2009 Study Protocol. Hemasphere. 2023;7(6):e893.
40. Eden OB, Shaw MP, Lilleyman JS, Richards S. Non-randomised study comparing toxicity of Escherichia coli and Erwinia asparaginase in children with leukaemia. Med Pediatr Oncol. 1990;18(6):497-502.
41. Albertsen BK, Schrøder H, Jakobsen P, Müller HJ, Carlsen NT, Schmiegelow K. Monitoring of Erwinia asparaginase therapy in childhood ALL in the Nordic countries. Br J Clin Pharmacol. 2001;52(4):433-437.
42. Albertsen BK, Schrøder H, Ingerslev J, et al. Comparison of intramuscular therapy with Erwinia asparaginase and asparaginase Medac: pharmacokinetics, pharmacodynamics, formation of antibodies and influence on the coagulation system. Br J Haematol. 2001;115(4):983-990.
43. Albertsen BK, Schrøder H, Jakobsen P, et al. Antibody formation during intravenous and intramuscular therapy with Erwinia asparaginase. Med Pediatr Oncol. 2002;38(5):310-316.
44. Rizzari C, Valsecchi MG, Aricò M, et al. Effect of protracted highdose L-asparaginase given as a second exposure in a Berlin-Frankfurt-Münster-based treatment: results of the randomized 9102 intermediate-risk childhood acute lymphoblastic leukemia study--a report from the Associazione Italiana Ematologia Oncologia Pediatrica. J Clin Oncol. 2001;19(5):1297-1303.
45. Pession A, Valsecchi MG, Masera G, et al. Long-term results of a randomized trial on extended use of high dose L-asparaginase for standard risk childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23(28):7161-7167.
46. Duval M, Suciu S, Ferster A, et al. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of CancerChildren's Leukemia Group phase 3 trial. Blood. 2002;99(8):2734-2739.
47. Moghrabi A, Levy DE, Asselin B, et al. Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood. 2007;109(3):896-904.
48. Kwok CS, Kham SK, Ariffin H, Lin HP, Quah TC, Yeoh AE. Minimal residual disease (MRD) measurement as a tool to compare the efficacy of chemotherapeutic drug regimens using Escherichia Coli-asparaginase or Erwinia-asparaginase in childhood acute
lymphoblastic leukemia (ALL). Pediatr Blood Cancer. 2006;47(3):299-304.
49. Vrooman LM, Stevenson KE, Supko JG, et al. Postinduction dexamethasone and individualized dosing of Escherichia Coli Lasparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00-01. J Clin Oncol. 2013;31(9):1202-1210.
50. Kloos RQH, Pieters R, Jumelet FMV, de Groot-Kruseman HA, van den Bos C, van der Sluis IM. Individualized asparaginase dosing in childhood acute lymphoblastic leukemia. J Clin Oncol. 2020;38(7):715-724.
51. Gupta S, Wang C, Raetz EA, et al. Impact of asparaginase discontinuation on outcome in childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. J Clin Oncol. 2020;38(17):1897-1905.
52. Maese L, Rizzari C, Coleman R, Power A, van der Sluis I, Rau RE. Can recombinant technology address asparaginase Erwinia chrysanthemi shortages? Pediatr Blood Cancer. 2021;68(10):e29169.
53. Van Trimpont M, Schalk AM, De Visser Y, et al. In vivo stabilization of a less toxic asparaginase variant leads to a durable antitumor response in acute leukemia. Haematologica. 2023;108(2):409-419.
54. Lin T, Hernandez-Illas M, Rey A, et al. A randomized phase I study to evaluate the safety, tolerability, and pharmacokinetics of recombinant Erwinia Asparaginase (JZP-458) in healthy adult volunteers. Clin Transl Sci. 2021;14(3):870-879.
55. Gao C, Ma X, Zhang Z, et al. Asparaginase Erwinia chrysanthemi for acute lymphoblastic leukemia and lymphoblastic lymphoma. Drugs Today (Barc). 2022;58(6):261-271.
56. Place AE, Stevenson KE, Vrooman LM, et al. Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): a randomised, openlabel phase 3 trial. Lancet Oncol. 2015;16(16):1677-1690.
57. Salzer WL, Asselin B, Supko JG, et al. Erwinia asparaginase achieves therapeutic activity after pegaspargase allergy: a report from the Children's Oncology Group. Blood. 2013;122(4):507-514.
58. Rizzari C, Zucchetti M, Conter V, et al. L-asparagine depletion and L-asparaginase activity in children with acute lymphoblastic leukemia receiving i.m. or i.v. Erwinia C. or E. coli L-asparaginase as first exposure. Ann Oncol. 2000;11(2):189-193.
59. Tong WH, van der Sluis IM, Alleman CJ, et al. Cost-analysis of treatment of childhood acute lymphoblastic leukemia with asparaginase preparations: the impact of expensive chemotherapy. Haematologica. 2013;98(5):753-759.
60. Lin T, Dumas T, Kaullen J, et al. Population pharmacokinetic model development and simulation for recombinant Erwinia asparaginase produced in Pseudomonas fluorescens (JZP-458). Clin Pharmacol Drug Dev. 2021;10(12):1503-1513.
61. Inaba H, Pui CH. Advances in the diagnosis and treatment of pediatric acute lymphoblastic leukemia. J Clin Med. 2021;10(9):1926.
62. Maese L, Rau RE. Current use of asparaginase in acute lymphoblastic leukemia/lymphoblastic lymphoma. Front Pediatr. 2022;10:902117.
63. Vrooman LM, Supko JG, Neuberg DS, et al. Erwinia asparaginase after allergy to E. coli asparaginase in children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2010;54(2):199-205.
Correspondence: D.A. Pollyea
Daniel.pollyea@ucdenver.edu
Received: January 3, 2023.
Accepted: April 6, 2023.
Early view: April 13, 2023.
1Division of Hematology, Department of Medicine; 2Center for Cancer and Blood Disorders, Department of Pediatrics; 3Department of Pathology and 4Center for Innovative Design and Analysis, Department of Biostatistics and Informatics, University of Colorado, Aurora, CO, USA
htps://doi.org/10.3324/haematol.2023.282681
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Venetoclax+azacitidine is the standard of care for newly-diagnosed patients with acute myeloid leukemia (AML) for whom intensive chemotherapy is inappropriate. Efforts to optimize this regimen are necessary. We designed a clinical trial to investigate two hypotheses: i) higher doses of venetoclax are tolerable and more effective, and ii) azacitidine can be discontinued after deep remissions. Forty-two newly diagnosed AML patients were enrolled in the investigator-initiated High Dose Discontinuation Azacitidine+Venetoclax (HiDDAV) Study (clinicaltrials gov. Identifier: NCT03466294). Patients received one to three “induction” cycles of venetoclax 600 mg daily with azacitidine. Responders received MRD-positive or MRDnegative “maintenance” arms: azacitidine with 400 mg venetoclax or 400 mg venetoclax alone, respectively. The toxicity profile of HiDDAV was similar to 400 mg venetoclax. The overall response rate was 66.7%; the duration of response (DOR), event-free survival (EFS) and overall survival were 12.9, 7.8 and 9.8 months, respectively. The MRD negativity rate was 64.3% by flow cytometry and 25.0% when also measured by droplet digital polymerase chain recation. MRD-negative patients by flow cytometry had improved DOR and EFS; more stringent measures of MRD negativity were not associated with improved OS, DOR or EFS. Using MRD to guide azacitidine discontinuation did not lead to improved DOR, EFS or OS compared to patients who discontinued azacitidine without MRD guidance. Within the context of this study design, venetoclax doses >400 mg with azacitidine were well tolerated but not associated with discernible clinical improvement, and MRD may not assist in recommendations to discontinue azacitidine. Other strategies to optimize, and for some patients, de-intensify, venetoclax+azacitidine regimens are needed.
Venetoclax with a backbone therapy (hypomethylating agent or low dose cytarabine) is the standard of care for a newly diagnosed patient with acute myeloid leukemia (AML) who is not a candidate for intensive induction chemotherapy. The complete remission (CR)/CR with incomplete hematologic recovery (CRi) rate of 66.4%, duration of response (DOR) of 17.5 months, event-free survival (EFS) of 9.8 months and overall survival (OS) of 14.7 months were all superior to azacitidine alone.1 Measurable residual disease (MRD) negativity as measured by multiparameter flow cytometry was achieved in 41% of responders using this regimen, and these patients had superior outcomes.2
During the phase I study with hypomethylating agents, venetoclax doses greater than 400 mg were administered without dose-limiting toxicity, and the maximum tolerated dose was not reached.3 Furthermore, there were patients who achieved MRD-negative remissions in the >400 mg venetoclax cohorts.4 We, therefore, hypothesized that higher doses of venetoclax could be safely administered, resulting in deeper remissions and better outcomes. Patients who achieve a remission with venetoclax+hypomethylating agent are recommended to continue indefinite treatment with both therapies, although no prospective studies have been performed to evaluate the veracity of this approach. A retrospective study found that for patients who achieved long-term remissions, con-
Jonathan A. Gutman,1 Amanda Winters,2 Andrew Kent,1 Maria Amaya,1 Christine McMahon,1 Clayton Smith,1 Craig T. Jordan,1 Brett Stevens,1 Mohammad Minhajuddin,1 Shanshan Pei,1 Jeffrey Schowinsky,3 Jennifer Tobin,1 Kelly O’Brien,1 Angela Falco,1 Elizabeth Taylor,1 Constance Brecl,1 Katie Zhou,1 Phuong Ho,1 Connor Sohalski,1 Jessica Dell-Martin,1 Olivia Ondracek,1 Diana Abbott4 and Daniel A. Pollyea1tinued treatment had no benefit compared to discontinuation.5 Long-term administration of hypomethylating agents can be challenging due to a multitude of factors, ranging from drug-related toxicity to logistical and qualityof-life obstacles.6,7 Given the established evidence supporting the prognostic significance of MRD negativity in AML,8 as well as the unique ability of venetoclax to target the leukemia stem cell population,9-11 we hypothesized that patients who achieved MRD negativity could discontinue azacitidine and be effectively maintained on venetoclax as a single agent.
In order to test these hypotheses, we designed a singleinstitution, investigator-initiated, phase II study of newly diagnosed older AML patients. Patients were administered 600 mg of venetoclax with the standard dose and schedule of azacitidine. Those who achieved MRD negativity discontinued azacitidine and were maintained on venetoclax alone (High Dose Discontinuation Azacitidine+Venetoclax Study [HiDDAV], clinicaltrials gov. Identifier: NCT03466294). We report here the safety and efficacy outcomes for this clinical trial.
The protocol was approved by the Colorado Multiple Institutional Review Board and is included in the Online Supplementary Appendix; patients were treated in accordance with the Declaration of Helsinki. The primary endpoint was remission duration; at study initiation, the median remission duration for venetoclax with azacitidine in the M14-358 study was 8 months. The alternative hypothesis for this study was a 12-month median remission duration, requiring 42 subjects for 80% power to detect this difference with a significance of 0.10.12 Patients were eligible if they had no prior therapy for non-acute promyelocytic AML, were ≥60 years, ineligible to undergo standard intensive induction chemotherapy due to age or comorbidities or declined to receive this treatment, had an Eastern Cooperative Oncology Group performance status13 of ≤2, adequate organ function, were without central nervous system involvement and had a white blood cell count <25x109/L (hydroxyurea permitted to achieve this).
Patients were admitted to the hospital for “induction”: dose escalation of venetoclax to 600 mg over 4 days, which was continued until day 28, and concomitant azacitidine 75 mg/m2, delivered subcutaneously or intravenously on days 1-7 (Figure 1A). Bone marrow biopsies were performed on cycle 1 on days 8 and 28, with an MRD assessment on day 28. Patients without an overall response (CR/CRi/morphologic leukemia-free state [MLFS]) by day 28 either discontinued the study or, in the setting of a beneficial response short of MLFS (typically a scenario in which a significant decrease in the blast percentage oc-
curred but did not reach <5%), repeated an “induction” cycle, without venetoclax dose escalation or mandatory hospitalization. Those who achieved an overall response and were MRD-positive repeated “induction”, without venetoclax dose escalation or mandatory hospitalization. Up to three “induction” cycles were permitted for non-responders or MRD-positive responders, with bone marrow biopsies at the conclusion of each cycle. Patients without an overall response by the third induction cycle discontinued the study. If patients achieved an overall response but did not achieve MRD negativity after the third induction cycle, they moved to “MRD-positive maintenance”: venetoclax 400 mg days 1-28 with azacitidine on days 1-7. Subsequent bone marrow biopsies were performed at the conclusion of each odd-numbered cycle until cycle nine, at which point bone marrow biopsies occurred after cycle 12 and then every six cycles. From the conclusion of the first “induction” cycle, and continuing with each response assessment, any patient who achieved “full MRD-negativity” (defined below) went to “MRD-negative maintenance”: venetoclax 400 mg days 1-28 and discontinuation of azacitidine. These patients had bone marrow biopsies at the conclusion of every three cycles for the first year and every six cycles thereafter (Figure 1B). MRD-negative patients who had MRD recurrence, or morphologic disease relapse, could resume azacitidine.
Adverse events (AE) were assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. Serious AE (SAE) were defined as AE that were life threatening, or resulted in death, hospitalization or prolongation of hospitalization. Response definitions were performed in accordance with the 2017 European Leukemia Network (ELN);14 for patients who achieved MLFS or CRi, a 14-day treatment-free interval, with or without granulocyte colony-stimulating factor (GCSF), occurred, and “upgraded” responses based on blood count recovery were recorded.15 A hematopathologist (JS) retrospectively reviewed all bone marrow biopsies to assign a French-American-British (FAB) category;16 M0 and M1 were indistinguishable because cytochemical studies are no longer utilized.
In addition to cytogenetic testing, two MRD assessments were employed for patients who achieved CR/CRi/MLFS. First, a multiparameter flow cytometry (flow) approach, analyzing a “different from normal” aberrant immunophenotype17 (Hematologics, Inc, Seattle WA) was utilized; MRDnegative by this modality was defined as no evidence of aberrant myeloid antigen expression or abnormal myeloblasts at a level of detection <0.1% in an adequate sample. In addition, droplet digital polymerase chain reaction (ddPCR) assays were used for MRD. This was accomplished through the application of an institutional baseline nextgeneration sequencing panel utilizing 49 genes (Online Supplementary Table S1); concurrent FLT3 internal tandem
duplications (ITD) was performed by PCR amplification and fragment analysis with a 5% lower limit of detection.18 DNA from fingernails was sequenced and tested alongside the initial bone marrow sample in most patients to exclude germline variants. After gene mutation results were reported, ddPCR assays for as many somatic nonDNMT3A/TET2/ASXL1 (DTA) mutations19 as possible were applied, with a sensitivity of 0.02-0.1%. MRD negativity by this modality was defined as undetectable for all assayed gene mutations. Mutations with large insertions/deletions were unable to be followed by ddPCR. A patient was considered to be “fully MRD-negative” if they were MRDnegative by both flow and ddPCR if eligible for monitoring by ddPCR, or only by flow if not eligible for monitoring by ddPCR.
DOR was defined as time from first response to relapse, per ELN.14 EFS was defined as time from study entry to death from any cause, relapse for responders and the day of treatment initiation for patients who were refractory.20 OS was defined as the time from study entry to death from any cause. Duration of cytopenias in cycle 1 were defined as described elsewhere.21
SAS version 9.4 was used for statistical analyses. Median survival times and associated Hall-Wellner confidence bands were created using Kaplan-Meier product-limit estimates for DOR, EFS, and OS. In order to compare survival times based on MRD status, Mantel-Cox log-rank analysis with a significance threshold of 0.05 was used. Cox regression was used to compute hazard ratios (HR) to assess the effect of variables of interest on the time-to-event out-

comes. Cox regression survival analysis was analyzed both unadjusted for allogeneic stem cell transplantation (ASCT) status and using ASCT status as a time-dependent covariate. Additionally, Cox regression models assessed whether the relationship between survival and the variable of interest differed based on ASCT status; in instances in which the stratified model presented a different effect based on ASCT status, results for Cox regression are reported from the stratified model. Otherwise, the unadjusted Cox regression model results are the same as the modeled results that treat ASCT as a time-dependent covariate. In assessing the effect of variables of interest on response status, logistic regression models were run, and odds ratios (OR) with their 95% confidence intervals (CI) and associated P values are reported.
Baseline data for the 42 patients who were enrolled are summarized in Table 1. Median follow up time is 42 months (95% CI: 29.3-42.5). Patients received a median of four total cycles of therapy (range, 0-40); 19 (45.2%) received
more than one MRD-positive maintenance cycle (median 3; range, 1-18) and nine (21.4%) received more than one MRD-negative maintenance cycle (median 5; range, 1-39). All patients had at least one AE; 700 total AE were reported. Thirty-two patients (76%) had a SAE. Common AE (≥20%) are summarized in Table 2. The most frequently reported hematologic AE grade ≥3 included thrombocytopenia (52%), febrile neutropenia (50%), leukopenia (41%) and neutropenia (38%). Gastrointestinal AE of any grade were common and included constipation (55%), diarrhea (55%), nausea (52%) and emesis (33%). Notable SAE (grade ≥3) included febrile neutropenia (36%), pneumonitis (7%) and bacteremia (5%) (Table 2). In nearly all cycles (230/273, 91.6%), 28 days of venetoclax was administered, according to patient-reported diaries. Decreases in the duration or dose of venetoclax were never prescribed for mitigation of toxicity. Neither laboratory nor clinical tumor lysis syndrome were observed. No patients discontinued treatment due to AE. Mortality at 30 days occurred in five of 42 (11.9%) of patients, all of whom had refractory disease and died of AML.
The overall response rate was 28 of 42 (66.7%); 26 (61.9%) had a CR and two (4.8%) had MLFS as best response (Table 3). GCSF was administered to 17 of 42 (40.5%) of patients; they received a median of two doses (range, 1-17) after cycle 1 (N=13), cycle 2 (N=7), cycle 3 (N=3), cycle 4 (N=3), cycle 5 (N=1), cycle 6 (N=1) and cycle 7 (N=1). During cycle 1, the median time to recover absolute neutrophil count (ANC) to >1.0x109/L was 39 days (range, 7-42), while the median time to recover platelets to >100x109/L was 26 (range, 7-37) days.
Of the responders, 18 of 28 (64.3%) achieved MRD negativity by flow at any point during their treatment course. Twenty-six of 28 (93%) were able to be monitored by ddPCR (2 patients had no baseline mutations); baseline mutations for all patients and those genes that were monitored are shown in the Online Supplementary Table S2. The percentage of all baseline non-DTA genes able to be monitored by ddPCR in responding patients was 70% (45/64). Of patients monitored by ddPCR, nine of 26 (35%) achieved MRD negativity. Full MRD negativity was achieved in seven of 28 (25%) (Table 3).
Forty-one patients came off study; 12 (29.3%) for death, four (9.8%) due to patient decision, four (9.8%) for refractory disease, 11 (26.8%) for relapsed disease and ten (24.3%) for ASCT. Eight (19%) remain alive. Individual patient outcomes are summarized in Figure 2.
AML: acute myeloid leukemia ELN: European Leukemia Network; ITD: internal tandem duplication; TKD: tyrosine kinase domain; MDS: myelodysplastic syndromes.
Thirty-five (83.3%) patients had an interpretable cycle 1 day 8 bone marrow biopsy; 13 (37.1%) had blast clearance to <5% (CRi=1, MLFS=12). Eleven of 13 (84.6%) went on to achieve CR as best response. Fourteen patients without blast clearance on day 8 ultimately responded (N=13 CR, 1 MLFS).
The median DOR was 12.9 months (95% CI: 7.4-17.6).
Table 1. Baseline variables for the 42 patients enrolled in the Higher Dose Venetoclax with Measurable Residual Disease Guided Discontinuation of Azacitidine (HiDDAV) Study.Table 2. Adverse events that occurred with an incidence of ≥20% and serious adverse events that occurred in more than one patient for participants of the High Dose Discontinuation Azacitidine+Venetoclax (HiDDAV) Study.
Median EFS was 7.8 months (95% CI: 2.5-11.2) and median OS was 9.8 months (95% CI: 6.6-14.9) (Figure 3). Patients who achieved MRD negativity by ddPCR, or full MRD negativity, did not have improved OS, EFS or DOR (Online Supplementary Figure S1). However, patients who were MRD-negative by flow had significantly improved EFS (median 18.7 vs. 8.2 months; P=0.047) and DOR (median
16.6 vs. 6.7 months; P=0.037), and an OS of 19.6 vs. 11.2 months, (P=0.16) (Figure 3). Because of the high rate of ASCT in this study (10/42, 24.3%), it was thought this factor may have overcome the impact of more stringent measures of MRD positivity. Therefore, MRD as defined by flow cytometry, ddPCR and both (full MRD negativity) were evaluated as predictors of DOR, EFS and OS after stratify-
Table 3. Responses and incidence of measurable residual disease.
sociated with improved DOR. Predictors for decreased EFS included FAB M5 and TP53 mutation, while IDH1/2 mutations and ASCT were associated with longer EFS. For OS, FAB M5, FLT3 ITD and TP53 mutations were negative predictors while IDH1/2 mutations and ASCT were positive predictors (Online Supplementary Table S7).
measurable residual disease; ddPCR: digital droplet polymerase chain reaction.
ing patients by transplant status. Full MRD negativity, and MRD negativity by ddPCR, did not impact these outcomes in either the transplanted or non-transplanted group, while MRD negativity by flow may be predictive of better DOR, EFS and OS in the non-transplantation group only (Online Supplementary Table S3).
Azacitidine was discontinued in 11 patients; in seven patients this was protocol-mandated due to the achievement of full MRD negativity, and occurred after a median of two cycles (range, 1-6). Four patients chose to stop azacitidine, in violation of the protocol without having achieved MRD negativity, for convenience and/or perceived toxicity, after a median of four cycles (range, 4-6). Five of 11 patients who discontinued azacitidine progressed (N=3 with MRD negativity and N=2 without MRD negativity). At progression, all patients who had repeat cytogenetic and molecular testing showed evidence of clonal evolution (Online Supplementary Table S4). The median DOR, EFS and OS for those who stopped azacitidine with and without MRD guidance was 16.0 months (95% CI: 7.1- not reached [NR]) and 12.9 months (95% CI: 5.5-NR), 17.4 months (95% CI: 8.5-NR) and 14.3 months (95%CI: 6.6-NR), and 17.4 months (95% CI: 8.5NR) and 18.8 months (95% CI: 6.6-NR), respectively (Online Supplementary Table S5).
Six patients who had discontinued azacitidine and continued venetoclax resumed azacitidine after a median of nine cycles (range, 5-20). In two cases this was done for relapse; in three it was done for conversion to MRD positivity from an MRD-negative state, and in one case it was due to patient’s preference. In no cases did the re-institution of azacitidine result in a second remission or decrease in MRD.
Negative predictors for response included FAB M5 and TP53 mutational status; male sex was the sole positive response predictor (Online Supplementary Table S6). Predictors for decreased DOR included FLT3, FLT3 ITD, ELN adverse risk, the need for hydroxyurea to enroll in the study and no blast clearance on cycle 1 day 8, while IDH1/2 mutation and MRD negativity by flow cytometry were as-
The HiDDAV study was conceived and conducted with the dual aims of determining i) whether venetoclax doses >400 mg might result in deeper remissions, and ii) whether MRD could safely guide azacitidine discontinuation. Primarily, we found that “induction” cycles of 600 mg venetoclax, administered for up to the first three cycles of therapy, had a safety profile similar to the definitive VIALE-A study using 400 mg of venetoclax.1 While comparing toxicity between different studies must be done with caution, particularly for studies with different median ages (70 vs. 76-years old for HiDDAV vs. VIALE-A), grade ≥3 thrombocytopenia (52% vs. 45%), neutropenia (38% vs. 42%) and febrile neutropenia (50% vs. 42%) were similar for HiDDAV versus VIALE-A, respectively.1 The 30-day mortality rates were 12% for HiDDAV and 7% for VIALE-A; however, all early mortality events in HiDDAV were from refractory AML. During cycle 1, time to recovery of ANC to >1.0x109/L and platelets to >100x109/L were similar to recently published results using 400 mg venetoclax (39 vs. 35 days and 26 vs. 25 days, for HiDDAV vs. comparator, respectively).21
Just as a comparison of toxicity between studies is fraught, so too are attempts to compare efficacy. However, when this is nonetheless done, there is no suggestion that the HiDDAV study had superior response rates (66.7% overall response rate vs. 66.4% CR+CRi rate for HiDDAV vs. VIALE-A), DOR (12.9 vs. 17.5 months for HiDDAV vs. VIALEA) EFS (7.8 vs. 9.8 months for HiDDAV vs. VIALE-A) or OS (9.8 vs. 14.7 months for HiDDAV vs. VIALE-A1).
In this trial using 600 mg of venetoclax, MRD negativity by flow was numerically higher than previously reported with 400 mg, using a similar methodology (64% vs. 41%);2 conclusions regarding a relationship between venetoclax dose and depth of response would require specific testing in a controlled setting. Consistent with Pratz et al.2 MRD-negativity by flow in HiDDAV was also associated with superior clinical outcomes, here significantly with respect to DOR and EFS. Interestingly, this appeared to be only relevant in patients who did not proceed to ASCT (Online Supplementary Table S3), suggesting ASCT can overcome worse clinical outcomes associated with MRD in the post-venetoclax remission setting.22
MRD negativity by flow is not a panacea; MRD-negative patients using this modality still relapse.2 We were, therefore, interested in investigating an independent and potentially
complementary modality for MRD detection, and applied ddPCR to all responding patients with detectable baseline mutations. With this approach we had 70% coverage of all
baseline non-DTA mutations. As far as we are aware, this represents the first time such comprehensive and prospective ddPCR MRD monitoring has been performed. How-

Figure 3. Time-to-event analyses for subjects on the Higher Dose Venetoclax with Measurable Residual Disease-Guided Discontinuation of Azacitidine (HiDDAV) Study. (A) Duration of response for all patients treated. (B) Event-free survival for all patients treated and (C) overall survival for all patients treated. (D) Responding patients stratified by measurable residual disease by flow cytometry status evaluating duration of response. (E) Event-free survival stratified by measurable residual disease-negative (red) vs. measurable residual disease-positive (blue). (F) Overall survival for measurable residual disease-negative (red) vs. measurable residual disease-positive (blue).

ever, in contrast to flow, MRD negativity by ddPCR was not associated with superior clinical outcomes, when considered alone or in combination with flow. As with the flow MRD assay, we considered whether the presumed deleterious impact of MRD positivity by ddPCR might have been overcome by ASCT, but when this MRD analysis was stratified by ASCT status, we saw no impact on clinical outcomes (Online Supplementary Table S3). For this more comprehensive approach to be predictive, coverage of all non-DTA mutations may need to be >70%. Alternatively, ddPCR may be overly stringent; if persistence of DTA mutations does not impact outcomes,19 this might also be true of other mutations, or for certain mutations that remain detectable below a particular threshold. Finally, it may be that this modality would only show significance with a larger number of patients than were included in our analysis.
Most of the responding patients had full blood count recovery, even with the use of higher doses of venetoclax, and count recovery was not more delayed than recently reported in the setting of venetoclax 400 mg.21 We believe this is due to routine use of breaks from treatment between cycles and the judicious use of GCSF, both of which are standard treatment principles with the use of venetoclax-based regimens.23,24 In addition, responses to this
regimen are typically rapid, but to date little is known about how quickly blast reduction in the bone marrow occurs. This study included cycle 1 day 8 bone marrow biopsies, and a significant percentage of patients had already achieved blast clearance at this early time point; indeed, lack of blast clearance at this time point was associated with a shorter DOR (Online Supplementary Table S6).
Indefinite therapy is a challenge for patients, and efforts to determine when or whether treatment discontinuation is possible are necessary. Our choice to discontinue venetoclax for patients in deep remission was informed by our experience in which long-term responders to venetoclax+azacitidine displayed frequent difficulty receiving indefinite infusions of azacitidine, but were able to manage the orally administered venetoclax. Other designs attempting to maintain patients with azacitidine, perhaps inspired by the data for maintenance oral azacitidine,25 would be reasonable. From our study, we cannot conclude that MRD negativity should lead to a recommendation to discontinue azacitidine; perhaps a larger study would be able to reach this conclusion. A recent retrospective study found no benefit to indefinite treatment in long-term responders;5 other formalized efforts to prospectively study de-escalation, based on MRD or other
factors, should be encouraged. Finally, in our study, after azacitidine was discontinued, we saw no evidence to suggest that its re-introduction could engender a re-response, or a decrease in MRD. Given the well-documented poor outcomes after progression on venetoclax regimens,26-28 novel salvage therapies are needed when patients progress.
Findings from single institution studies can be difficult to extrapolate. Our newly diagnosed AML patients receiving venetoclax+azacitidine do not routinely receive antifungal prophylaxis due to our reported low rates of proven or probable invasive fungal infections in this population.29 We are also likely outliers with respect to our aggressive approach to ASCT in venetoclax+azacitidine patients. 30 Our high ASCT rate explains the relatively low median total number of venetoclax+azacitidine cycles patients received (4 vs. 7 in VIALE-A1). We do not believe that “fitness” for induction chemotherapy has the same relevance it did in the pre-venetoclax era, 31,32 and so we allow patients ≥60 years old who are “fit” for intensive chemotherapy to choose between this treatment and venetoclax+azacitidine, a decision that is informed by biological risk factors and the likelihood of responding to either therapy;33 the HiDDAV protocol specifically allowed for patients who were “unwilling” to receive intensive induction. The HiDDAV study, therefore, likely had younger and fitter patients than other similar studies. In conclusion, venetoclax doses >400 mg with azacitidine were tolerated by this patient population with equivalent toxicity and time to blood count recovery compared with venetoclax 400 mg. Higher doses may result in deeper remissions; deeper remissions appear to have clinical benefi t. However, without controlled data we would not recommend routine use of >400 mg venetoclax with azacitidine, as the clinical outcomes are not clearly superior
1. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617-629.
2. Pratz KW, Jonas BA, Pullarkat V, et al. Measurable residual disease response and prognosis in treatment-naive acute myeloid leukemia with venetoclax and azacitidine. J Clin Oncol. 2022;40(8):855-865.
3. DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;(2):216-228.
4. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7-17.
5. Chua CC, Hammond D, Kent A, et al. Treatment-free remission after ceasing venetoclax-based therapy in patients with acute
to studies using 400 mg. Finally, MRD negativity cannot be used to recommend azacitidine discontinuation at this time; more efforts to optimize venetoclax-based regimens in AML are necessary.
DAP has served as an advisory board member and consultant for Abbvie and Genentech, and receives research funding from Abbvie.
JAG, CSmith, MA, CM, KO, AF, CSohalski, JD-M and OO recruited and/or treated subjects and edited and approved the manuscript. AW, AK, JS, JT, KZ, PH and DA performed study-related analyses and edited and approved the manuscript. CTJ, BS, MM, SP, ET and CB edited and approved the manuscript. DAP recruited and treated subjects, performed study related analyses and wrote, edited and approved the manuscript.
The authors wish to acknowledge the courage and bravery of the patients and their families who participated in this study; without their hope and altruism, progress in this field would not be possible.
This research was supported by the Leukemia and Lymphoma Society’s Scholar in Clinical Research Program, the Robert H. Allen MD Chair in Hematology Research and AbbVie.
Data on individual patients will not be shared. Data from aggregate analyses may be shared upon request.
myeloid leukemia. Blood Adv. 2022;6(13):3879-3883.
6. Zeidan AM, Hu X, Zhu W, et al. Association of provider experience and clinical outcomes in patients with myelodysplastic syndromes receiving hypomethylating agents. Leuk Lymphoma. 2020;61(2):397-408.
7. Zeidan AM, Salimi T, Epstein RS. Real-world use and outcomes of hypomethylating agent therapy in higher-risk myelodysplastic syndromes: why are we not achieving the promise of clinical trials? Future Oncol. 2021;(36):5163-5175.
8. Short NJ, Zhou S, Fu C, et al. Association of measurable residual disease with survival outcomes in patients with acute myeloid leukemia: a systematic review and meta-analysis. JAMA Oncol. 2020;6(12):1890-1899.
9. Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859-1866.
10. Jones CL, Stevens BM, D'Alessandro A, et al. Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood. 2019;134(4):389-394.
11. Jones CL, Stevens BM, D'Alessandro A, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34(5):724-740.
12. Lawless JF. Statistical models and methods for lifetime data. New York: Wiley. 1982.
13. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;(6):649-55.
14. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
15. Abbott D, Cherry E, Amaya M, et al. The propriety of upgrading responses to venetoclax + azacitidine in newly diagnosed patients with acute myeloid leukemia. Leuk Lymphoma. 2021;62(6):1466-1473.
16. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;(4):451-845.
17. Heuser M, Freeman SD, Ossenkoppele GJ, et al. 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138(26):2753-2767.
18. Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111(5):2776-2784.
19. Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189-1199.
20. Norsworthy KJ, Gao X, Ko CW, et al. Response rate, event-free survival, and overall survival in newly diagnosed acute myeloid leukemia: US Food and Drug Administration trial-level and patient-level analyses. J Clin Oncol. 2022;40(8):847-854.
21. Rausch CR, DiNardo CD, Maiti A, et al. Duration of cytopenias with concomitant venetoclax and azole antifungals in acute myeloid leukemia. Cancer. 2021;127(14):2489-2499.
22. Winters AC, Bosma G, Abbott D, et al. Outcomes are similar after allogeneic hematopoietic stem cell transplant for newly diagnosed acute myeloid leukemia patients who received
venetoclax + azacitidine versus intensive chemotherapy. Transplant Cell Ther. 2022;28(10):694.
23. Jonas BA, Pollyea DA. How we use venetoclax with hypomethylating agents for the treatment of newly diagnosed patients with acute myeloid leukemia. Leukemia. 2019;33(12):2795-2804.
24. Pollyea DA, Bixby D, Perl A, et al. NCCN guidelines insights: acute myeloid leukemia, version 2.2021. J Natl Compr Canc Netw. 2021;19(1):16-27.
25. Wei AH, Dohner H, Pocock C, et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N Engl J Med. 2020;383(26):2526-2537.
26. Zainaldin C, Arora S, Bathini S, et al. Dismal survival outcomes of patients with acute myeloid leukemia after failure of venetoclax with hypomethylating agents. Leuk Lymphoma. 2022;63(13):3245-3248.
27. Maiti A, Rausch CR, Cortes JE, et al. Outcomes of relapsed or refractory acute myeloid leukemia after frontline hypomethylating agent and venetoclax regimens. Haematologica. 2021;106(3):894-898.
28. Bewersdorf JP, Shallis RM, Derkach A, et al. Efficacy of FLT3 and IDH1/2 inhibitors in patients with acute myeloid leukemia previously treated with venetoclax. Leuk Res. 2022;122:106942.
29. Zhang A, Johnson T, Abbott D, et al. Incidence of invasive fungal infections in patients with previously untreated acute myeloid leukemia receiving venetoclax and azacitidine. Open Forum Infect Dis. 2022;9(10):ofac486.
30. Pollyea DA, Winters A, McMahon C, et al. Venetoclax and azacitidine followed by allogeneic transplant results in excellent outcomes and may improve outcomes versus maintenance therapy among newly diagnosed AML patients older than 60. Bone Marrow Transplant. 2022;57(2):160-166.
31. Pollyea DA. Acute myeloid leukemia drug development in the post-venetoclax era. Am J Hematol. 2019;94(9):959-962.
32. Pollyea DA. Venetoclax in AML: where we are and where we are headed. Clin Lymphoma Myeloma Leuk. 2020;20 (Suppl 1):S25-S26.
33. Cherry EM, Abbott D, Amaya M, et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv. 2021;5(24):5565-5573.
Correspondence: M. Konopleva mkonople@mdanderson.org
Q. Zhang qzhang9@mdanderson.org
Received: August 22, 2022.
1Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Department of Hematology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China; 3Hematology Research Unit Helsinki, University of Helsinki and Helsinki University Hospital Comprehensive Cancer Center, Helsinki, Finland; 4Translational Immunology Research Program and Department of Clinical Chemistry and Hematology, University of Helsinki, Helsinki, Finland; 5iCAN Digital Precision Cancer Medicine Flagship, Helsinki, Finland; 6Department of Medicinal Chemistry, College of Pharmacy, University of Florida, Gainesville, FL, USA and 7Department of Biochemistry and Structural Biology and Center for Innovative Drug Discovery, Long School of Medicine, University of Texas Health Science Center at San Antonio, San Antonio, TX, USA
Accepted: April 7, 2023.
Early view: April 20, 2023.
htps://doi.org/10.3324/haematol.2022.281915
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
BCL-XL and BCL-2 are key anti-apoptotic proteins and validated cancer targets. 753B is a novel BCL-XL/BCL-2 proteolysis targeting chimera (PROTAC) that targets both BCL-XL and BCL-2 to the von Hippel-Lindau (VHL) E3 ligase, leading to BCLXL/BCL-2 ubiquitination and degradation selectively in cells expressing VHL. Because platelets lack VHL expression, 753B spares on-target platelet toxicity caused by the first-generation dual BCL-XL/BCL-2 inhibitor navitoclax (ABT-263). Here, we report pre-clinical single-agent activity of 753B against different leukemia subsets. 753B effectively reduced cell viability and induced dose-dependent degradation of BCL-XL and BCL-2 in a subset of hematopoietic cell lines, acute myeloid leukemia (AML) primary samples, and in vivo patient-derived xenograft AML models. We further demonstrated the senolytic activity of 753B, which enhanced the efficacy of chemotherapy by targeting chemotherapy-induced cellular senescence. These results provide a pre-clinical rationale for the utility of 753B in AML therapy, and suggest that 753B could produce an added therapeutic benefit by overcoming cellular senescence-induced chemoresistance when combined with chemotherapy.
Acute myeloid leukemia (AML) is a hematopoietic malignancy involving clonal hematopoiesis and defects in differentiation, proliferation, and cell death, resulting in the accumulation of immature blasts, marrow failure, and rapid death of patients if not treated. Therapeutic progress has been slow despite the recent identification of genomic and epigenetic alterations. Fewer than half of adults with AML (and less than 10-20% of elderly [>60 years old] patients with AML) survive long-term. Overexpression of anti-apoptotic BCL-2 family proteins is a core oncogenic property of leukemia, and is associated with disease progression and resistance to chemotherapy by protecting tumor cells from apoptosis.1
Targeting BCL-2 family proteins has been successfully explored as a therapeutic strategy for leukemia, and several small-molecule inhibitors of BCL-2 proteins have been identified.2,3 Venetoclax (ABT-199), a BCL-2–selective inhibitor, has been approved by the US Food and Drug Administration for treatment of chronic lymphocytic leukemia (CLL) and of AML when combined with low-intensity chemotherapy. However, despite high response rates, the majority of the patients treated with hypomethylating agents and venetoclax eventually relapse.4 The upregulation of anti-apoptotic proteins other than BCL-2, such as BCL-XL or MCL-1, have been identified as major determinants of venetoclax resistance in CLL and AML,2,5-7 consistent with the high efficacy of the dual BCL2/BCL-XL inhibitor navitoclax in killing venetoclax-resistant
Yannan Jia,1,2 Lina Han,1 Cassandra L. Ramage,1 Zhe Wang,1 Connie C. Weng,1 Lei Yang,1 Simona Colla,1 Helen Ma,1 Weiguo Zhang,1 Michael Andreeff,1 Naval Daver,1 Nitin Jain,1 Naveen Pemmaraju,1 Kapil Bhalla,1 Satu Mustjoki,3,4,5 Peiyi Zhang,6 Guangrong Zheng,6 Daohong Zhou,7 Qi Zhang1 and Marina Konopleva1CLL cells.7
However, the clinical utility of navitoclax was hampered by the on-target and dose-limiting thrombocytopenia due to dependence of platelet survival on BCL-XL.8 We have previously reported that DT2216, a von Hippel-Landau (VHL)-recruiting proteolysis-targeting chimera (PROTAC) derived from navitoclax, is able to overcome this on-target thrombocytopenia.9 DT2216 selectively ubiquitinated and degraded BCL-XL in a VHL E3 ligase- and proteasomedependent manner in VHL-expressing cells, and was highly effective against tumors that primarily depend on BCL-XL for survival, such as T-cell acute lymphoblastic leukemia (T-ALL).9 Since platelets do not express VHL, DT2216 importantly spares platelets. Although DT2216 binds both BCL-2 and BCL-XL with high affinity, DT2216 degrades only BCL-XL, not BCL-2. Consequently, DT2216 showed minimal efficacy in cancers that co-depend on both BCL-XL and BCL-2 for survival, such as certain subsets of leukemia and mantle cell lymphoma (MCL), unless combined with venetoclax or conventional chemotherapy.10
We recently reported the first-in-class dual BCL-XL/BCL2 PROTAC 753B that induces both BCL-XL and BCL-2 ubiquitination and degradation selectively in cells expressing VHL, and demonstrated its pre-clinical activity in tumor cells that co-depend on BCL-2 and BCL-XL 10 In this study, we characterized its broad activity in AML. In addition, recent findings indicate that chemoresistance in AML is associated with chemotherapy-induced cellular senescence.11,12 Upregulation of both BCL-XL and BCL-2 has been reported essential for senescent cell survival.13 Accordingly, inhibition of BCL-XL and BCL-2 activity may facilitate clearance of senescent cells.14 Here, we sought to evaluate the efficacy of 753B as a senolytic agent in leukemia after chemotherapy. Our study showed that 753B effectively eliminates leukemia cells both in vitro and in vivo, and enhances the efficacy of chemotherapy by targeting senescent cells.
The drugs 753B, DT2216, and QVD (a pan-caspase inhibitor) were kindly provided by Prof. Zhou at the University of Texas Health Science Center and Prof. Zheng at the University of Florida. Venetoclax (ABT-199), Navitoclax (ABT-263), cytarabine (Ara-C) and S63845 were purchased from Selleckchem (Pittsburgh, PA, USA).
Cell lines and primary acute myeloid leukemia samples
Leukemia cell lines were obtained from ATCC (Manassas, VA, USA) or DSMZ (Braunschweig, Germany) and maintained in RPMI 1640 supplemented with 10-20% heat-in-
activated fetal bovine serum (Gibco, Thermo-Scientific) All cell lines were frozen and low passage cells were used for all experiments.
Peripheral blood samples were collected from patients with AML; informed consent was obtained in accordance with the requirements of the Institutional Review Board of the University of Texas MD Anderson Cancer Center. The clinical features of the patients are listed in Online Supplementary Table S1
Western bloting
Cells were lysed and 10-15 mg were prepared,15 resolved by electrophoresis on NuPAGE 4-12% SDS-PAGE gradient gels (Invitrogen), and transferred to polyvinylidene fluoride membranes (Invitrogen). Immunoblotting was performed with primary antibodies: BCL-2 (DAKO), MCL-1 (Santa Cruz Biotechnology), PARP, Caspase-3, Cleaved caspase-3, BCLXL (Cell Signaling Technology), and tubulin (Sigma-Aldrich). Blots were scanned with an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
Fluorogenic senescence-associated β-galactosidase activity assay using 5-dodecanoylaminofluorescein diβ-D-galactopyranoside
Senescence-associated β-galactosidase (SA-β-al) activity was assayed as previously described16 using 20 μM 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG) (Thermo Fisher Scientific). Briefly, C12-FDG was prepared in pre-warmed media and added to cells with gentle mixing of the cell suspension. Cells were then incubated with C12-FDG and protected from light for 2-4 hours (h) at 37°C and 5% CO2. SA-β-gal-positive cells were detected and quantified by flow cytometry.
All mouse experiments were approved by the Institutional Animal Care and Use Committee at the University of Texas MD Anderson Cancer Center. Eight-week-old NOD scid gamma (NSG) mice (The Jackson Laboratory, Bar Harbor, Maine, USA) were injected with 3x106 AML patient-derived xenograft (PDX) #4138550 cells (which have FLT3-ITD, DNMT3A, IDH1, KIT, and NPM1 mutations) via the tail vein. After leukemia engraftment was confirmed by flow cytometry 33 days after injection, mice were randomized to receive vehicle or 753B (5 mg/kg intraperitoneally, every 4 days) for three weeks. Mouse livers, spleens, and femurs were collected after the treatment. Leukemia burden was quantified by hCD45+ flow cytometry.
Statistical analyses were performed using GraphPad Prism version 8. Results are expressed as mean ± Standard Error of Mean (SEM) of three separate replicate experiments, unless otherwise indicated. Two-sided unpaired Student
t-test was used for comparisons between the means of two groups. Kaplan-Meier test was used to analyze the survival rate in the in vivo study and the data were statistically analyzed using log rank (Mantel-Cox) test.
753B is more potent in reducing cell viability in a subset of hematologic cell lines compared with other BCL-XL and/or BCL-2 targeting agents via degradation of BCL-XL and BCL-2
We first evaluated the sensitivity of 24 genetically diverse hematologic cell lines to venetoclax, navitoclax, DT2216 (a BCL-XL selective PROTAC), and 753B (a dual BCL- XL/BCL2 PROTAC). Our cohort of cell lines included 17 AML, five T-ALL, and two AML secondary to myeloproliferative neoplasms (MPN-AML) cell lines. Treatment with 753B for 24 h caused a dose-dependent but variable reduction of cell viability in all leukemia cell lines tested, as determined by CellTiter-Glo© (CTG) assay, with half-maximal inhibitory concentration (IC50) values ranging from 0.01 μM to 27.35 μM (Online Supplementary Table S2). Both BCL-XL-dependent cell lines (CCRF-CEM, TF-1) and BCL-XL/BCL-2-co-dependent cell lines (Kasumi-1, KG-1) were sensitive to 753B (Figure 1A). Median IC50 values of 753B (0.35 μ M) were lower than those of venetoclax (7.69 μM) and navitoclax (0.61 μM) across all cell lines tested (Online Supplementary Figure S1A), indicating that 753B on a molar basis is more potent than other BCL-XL and/or BCL-2 targeting agents. Notably, 12 AML cell lines (KG-1, Kasumi-1, TF-1, HEL 92.1.7, MV4-11, Kasumi-3, UCSD-AML1, HNT37, MOLM-1, CMK, M07e, F-36P) and three T-ALL cell lines (Jurkat, PF832, CCRF-CEM) showed greater sensitivity to 753B than to navitoclax (median value: 0.10 μM for 753B vs. 0.48 μM for navitoclax; P=0.0005) (Figure 1B). The IC50 of 753B in these cell lines was well correlated with that of navitoclax (correlation coefficient: r2=0.6673) (Online Supplementary Figure S1B). Of note, the top seven highly sensitive cell lines to 753B included three AML-EVI-1 (MECOM) rearranged cell lines (Kasumi-3, UCSD-AML1 and HNT37), which represent AML with extremely poor prognosis17 and are resistant to venetoclax (Online Supplementary Table S2, Online Supplementary Figure S1C).
We next analyzed the baseline (pre-treatment) expression of the anti-apoptotic proteins BCL-XL, BCL-2 and MCL-1 in 17 leukemia cell lines by western blotting. BCL-2 was in general highly expressed in AML cell lines with a notable exception for the MPN-AML cell lines SET-2 and HEL 92.1.7 and all T-ALL lines, in which BCL-XL expression was more prevalent. EVI-1 rearranged lines (e.g., Kasumi-3 and UCSD-AML1) co-expressed BCL-XL and BCL-2 (Online Supplementary Figure S1D, E). To quantify BCL-XL/BCL-2 protein degradation by 753B, we performed western blotting
and densitometry analyses in 22 leukemia cell lines treated with 753B. Notably, 753B effectively and potently induced dose-dependent BCL-XL degradation in all tested lines within 24 h, with the concentrations at which 50% of the protein was degraded (DC50) ranging from 0.01 μM to 0.54 μM; in 21 of 22 tested cell lines, the DC50 of BCLXL to 753B was less than 0.15 μM. BCL-2 was also degraded in 21 of the 22 cell lines, but this generally required higher doses of 753B, with DC50 ranging from 0.02 μM to more than 1 μM, with the exception of the T-ALL Loucy cell line (no degradation at 24 h) (Online Supplementary Table S2, Online Supplementary Figure S1E).
To further understand 753B pro-apoptotic effects, we analyzed apoptosis in 753B-treated Kasumi-1 cells harboring AML-ETO rearrangement (BCL-XL and BCL-2 co-dependent). Consistent with rapid degradation of BCL-XL that we observed as early as 4 h (Figure 1C), 753B treatment rapidly induced apoptosis in Kasumi-1 cells as determined by Annexin-V staining after 4 h of treatment (Figure 1D). The results were confirmed by analyzing the cleavage of caspase-3 and PARP after 8 h (Figure 1C). The activation of caspase-3 by 753B was also observed in the HL60 cell line (Online Supplementary Figure S1F). Moreover, the decrease in cell viability induced by 753B was effectively rescued by the pan-caspase inhibitor Q-VD-OPh (QVD) (Figure 1E, Online Supplementary Figure S1G), suggesting that 753B-induced cell death is largely mediated by caspase-dependent apoptosis.
753B is a more potent antitumor agent than DT2216 in both leukemia cell lines and primary patient samples To determine whether 753B is more potent than the firstgeneration BCL-XL-targeted PROTAC DT2216, we exposed 22 cell lines, including AML, T-ALL, and MPN-AML cell lines, to increasing concentrations of DT2216 and 753B for 24 h and then determined their IC50 values. We observed that the sensitivity to DT2216 generally tracked with responsiveness to 753B (Figure 2A). We found that the IC50 of 753B in these cell lines strongly correlated with but were lower than the IC50 of DT2216 (correlation coefficient: r2=0.8732) (Figure 2B). It is notable that in BCL-XL-dependent leukemia cell lines, 753B was more potent than DT2216 (median IC50 : 0.10 vs. 1.16 μM) (Online Supplementary Figure S2A), consistent with higher potency of 753B as BCL-XL degrader due to changing the link-out position on navitoclax and allowing additional lysines on BCL-XL (K20) and BCL-2 (K17) to interact with the E2 enzyme for ubiquitination.10 To better evaluate the antitumor efficacy of 753B, we tested 753B efficacy in the BCL-XL/BCL-2 codependent AML cell line KG-1. Compared with DT2216, 753B more potently induced BCL-XL degradation (DC50, 0.01 vs. 0.06 μM) and additionally induced BCL-2 degradation in KG-1 cells (Figure 2C). Moreover, 753B was the most potent tested inhibitor inducing apoptosis (Figure 2D).
Figure 1. 753B is more potent in reducing cell viability in a subset of hematologic cell lines compared with other BCL-XL and/or BCL-2 targeting agents via degradation of BCL-XL and BCL-2. (A) Representative graphs of cell viability of three acute myeloid leukemia (AML) cell lines (Kasumi-1, TF-1 and KG-1) and one T-cell acute lymphoblastic leukemia (T-ALL) cell line (CCRF-CEM). Cell viability was determined by CellTiter-Glo assay following treatment with DT2216, 753B, venetoclax, or navitoclax at the indicated concentrations for 24 hours (h). (B) Comparison of IC50 values of 753B and navitoclax in 12 AML cell lines (KG-1, Kasumi1, TF-1, HEL 92.1.7, MV4-11, Kasumi-3, UCSD-AML1, HNT37, MOLM-1, CMK, M-07e, F-36P) and three T-ALL cell lines (Jurkat, PF832, CCRF-CEM). Symbols represent individual cell lines. (C and D) Apoptosis in Kasumi-1 cells treated with the indicated concentrations of 753B or with DMSO at the indicated serial time points. Expression of apoptosis-related proteins was detected by western blotting (C) and apoptosis induction was determined by Annexin-V/DAPI staining (D). Tubulin served as loading control. (E) Percentage survival of KG-1 cells treated with 753B at the indicated concentrations for 24 h with or without treatment with the pan-caspase inhibitor Q-VD-OPh (QVD). DMSO: dimethyl sulfoxide. Data are presented as mean ± Standard Error of Mean of three repeats, performed in triplicate. ***P<0.001.
We next tested the sensitivity of 16 primary AML samples to 753B, DT2216, venetoclax, and navitoclax. Our cohort included ten AML samples from patients who were relapsed or refractory (R/R) to venetoclax-based therapies. Consistent with the results in leukemia cell lines, 753B more potently reduced cell viability than DT2216. The median IC 50 value of 753B was 0.23 μ M and ranged from 0.02 μ M to 2.29 μ M with IC 50 values <0.50 μ M in 13 of 16 primary AML patient samples ( Online Supplementary Table S1 ). Of note, the IC50 of 753B in primary samples was lower than that in AML cell lines. 753B was more potent than DT2216 as evident by the extent of BCL-X L degradation, higher apoptosis induction, lower IC 50 with antileukemia potency comparable to that of navitoclax in all tested AML samples, including seven venetoclax-resistant samples (defined as IC 50 >1 μ M) (Figure 2E-G). 753B degraded BCL-XL in all six tested primary samples, and BCL-2 in 4 out of 6 tested samples, with BCL-X L DC 50 of 0.01 to approximately 0.18 μ M, and BCL-2 DC 50 of 0.18 to approximately 3.57 μ M (Figure 2E, H, Online Supplementary Figure S2B-E ). 753B induced apoptosis associated with cleavage of caspase-3 and PARP (Figure 2H).
753B enhances the efficacy of chemotherapy by eliminating senescent leukemia cells
Recent studies suggested that chemotherapy-induced cellular senescence-mediated chemoresistance and disease relapse in AML.18,19 Tumor cells can undergo senescence in response to stress and therapy, characterized by cell-cycle arrest, increased SA-β-gal activity and acquisition of a Senescence-Associated Secretory Phenotype (SASP) that comprises pro-inflammatory cytokines, chemokines and growth factors. Senescent cells can accumulate, especially under chemotherapy-induced immune suppression, and exhibit a deleterious effect on the tissue microenvironment that drives aging phenotype and potentially promotes tumor growth, relapse, metastasis, and resistance to chemotherapy.11,19,20 Navitoclax and other BCL-XL/BCL-2 inhibitors, like ABT-737 and A1331852 are known to function as potent senolytics, small molecules that selectively clear senescent cells in a variety of tissues and solid tumors,13,14,21 help maximize the efficacy of chemotherapy, and prevent tumor relapse and metastasis.13 Based on these findings, we hypothesized that 753B might exhibit senolytic activity in leukemia. To confirm the induction of cellular senescence by Ara-C,

Figure 2. 753B is a more potent antitumor agent than DT2216 in both leukemia cell lines and primary patient samples. (A) IC50 values in leukemia cell lines treated with DT2216 or 753B for 24 hours (h). IC50 values were based on the percentage of viable cells and normalized to controls. (B) IC50 for 753B (y-axis) and DT2216 (x-axis) in hematologic cell lines listed in (A). Linear regression is solid black line. Correlation coefficient is discussed in the text. (C) Representative western blotting showing BCL-2 and BCL-XL protein levels in KG-1 cells treated with the indicated concentrations of DT2216 or 753B for 24 h. (D) Apoptosis in KG-1 cells treated with 0.1 μM or 1 μM DT2216, 753B, venetoclax, or navitoclax for 24 h. Apoptosis was determined using Annexin-V/DAPI staining and flow cytometry. Data are representative of two independent experiments. (E) Western blotting showing protein levels of BCL-2 and BCL-XL in a primary patient sample (#6566444) treated with the indicated concentrations of 753B or DT2216 for 24 h. Tubulin was used as an equal loading control. (F) Apoptosis in a primary patient sample (#6566444) treated with 0.1 μM or 1 μM DT2216, 753B, venetoclax, or navitoclax for 24 h. Apoptosis was determined using Annexin-V/DAPI staining and flow cytometry. Data are presented as mean ± Standard Error of Mean of three repeats, performed in triplicate. (G) IC50 values of DT2216, 753B, venetoclax, and navitoclax in primary patient samples (N=16) treated for 24 h. IC50 values were calculated based on the percentage of viable cells, normalized to control, as determined by CellTiter-Glo assay. (H) Western blotting showing protein levels of BCL-2 and BCL-XL and apoptosis-related proteins in a primary patient sample (#7126060) treated with the indicated concentrations of 753B. Tubulin was used as an equal loading control. DMSO: dimethyl sulfoxide. *P<0.05; ****P<0.0001.

one of the most used chemotherapy agents in AML, we treated MOLM-14 cells with increasing concentrations of Ara-C for three days. Ara-C-treated cells increased in size and cytoplasmic granularity by flow cytometry (Figure 3A), consistent with previous reports. SA-β-gal is a sensitive marker to identify cells in senescence state,22 and its fluorogenic substrate C12-FDG is widely used to quantify senescent cells.16 Consistent with prior reports, the activity of SA- β -gal and C12-FDG median fluorescence intensity (MFI) was drastically increased in viable cells after 0.05 μM Ara-C treatment for 72 h, indicating that Ara-C induced cellular senescence11 (Figure 3B, C). 753B not only inhibited baseline cell senescence (Online Supplementary Figure S3A-C), but also reversed Ara-C-induced cellular senescence, as indicated by reduced SA- β -gal staining and lower MFI of C12-FDG (Figure 3D, E).
To further validate the effect of 753B on cell senescence, we used immunoblotting to analyze the expression of cell cycle regulators such as p16 (CDKN2A), p21 (CDKN1A), and p53 (TP53), which are known to be associated with senescence.23-25 Expression levels of p16 and p21 were increased after Ara-C treatment but abrogated by 753B (Figure 3B). Consistently, Ara-C treatment also increased the expression levels of pro-inflammatory cytokines and chemokines including interleukin (IL)-8, IL-6, IL-1 β , and CCR5, which are SASP markers of senescent cells.26,27 In contrast, 753B co-treatment prevented the Ara-C-induced expression of these cytokines and chemokines (Figure 3G). The inhibitory effect of 753B on chemotherapy-induced senescence was further confirmed in an additional AML cell line, Kasumi-1 (Online Supplementary Figure S3DK). Taken together, these data indicate that 753B can target chemotherapy-induced senescent AML cells.
Chemotherapy-induced senescent cells express higher levels of BCL-XL, representing a therapeutic target for 753B
To explore the molecular mechanism behind the chemotherapy-induced senescence phenotype, we FACS-sorted viable C12-FDG-high (senescent) and C12-FDG-low MOLM14 cells after three days exposure to low doses of Ara-C (0.05 μM or 0.10 μM) (Online Supplementary Figure S4A).
Compared to that in the C12-FDG-low cells, the expression of BCL-XL was significantly higher in the C12-FDG-high senescent cells (Figure 4A). Consistently high expression of BCL-XL was also observed in C12-FDG-high, Ara-C-treated Kasumi-1 cells ( Online Supplementary Figure S4B). This finding suggested that BCL-XL is a key anti-apoptotic protein in senescent cells.
To analyze the clearance of senescent cells upon 753B treatment, MOLM-14 cells were incubated with Ara-C (0.05 μM) for three days to induce senescence, followed by 24 h of treatment with 753B. Compared with control cells without Ara-C treatment (non-senescent cells), se-
nescent cells induced by Ara-C were significantly more sensitive to treatment with 753B, with a 75% death rate at the highest concentration tested (Figure 4B). Besides, the combination of Ara-C and 753B induced higher levels of cleaved caspase-3 and cleaved PARP, indicating increased apoptosis in these cells (Figure 4C). Furthermore, the combination of 753B and Ara-C treatment showed a synergistic growth-inhibitory effect (Figure 4D, E), suggesting that targeting BCL-XL with 753B in combination with chemotherapy may boost chemotherapeutic efficacy in leukemia.
BCL-XL degradation by 753B is associated with MCL-1 upregulation in selected leukemia cells, and the combination of 753B and an MCL-1 inhibitor induced synergistic cell death
MCL-1 is a member of BCL-2 family and a known resistance factor to venetoclax.6,28,29 We have previously reported that in subsets of AML cells, short-term BCL-2 inhibition causes MCL-1 upregulation associated with activation of MAPK signaling.30 The degradation of BCL-XL and BCL-2 by 753B at the intermediate dose (0.11 μ M) was similarly associated with MCL-1 upregulation in 13 of 22 cell lines tested. Out of 13 cell lines, we observed increased expression of MCL-1 at all applied concentrations of 753B in five AML lines (Online Supplementary Table S3, Online Supplementary Figure S1D). MCL-1 dose-dependent upregulation was observed as early as 4 h after 753B exposure (Figure 5A). Co-immunoprecipitation (Co-IP) and BH3 profiling were performed to explore the functional consequences of an increased level of MCL-1. 753B induced an increase in Bim binding to MCL-1 compared with vehicle treated cells as demonstrated by Co-IP (Figure 5B, C). Dynamic BH3 profiling demonstrated higher release of cytochrome C by the MCL-1-specific peptide MS-1 and the MCL-1 inhibitor S63845, indicating increased MCL-1 dependence following 753B exposure (Figure 5D). To further characterize the relationship between expression of BCL2 family proteins and sensitivity to 753B, we quantified the expression levels of BCL-XL, BCL-2 and MCL-1 by western blotting by densitometry of the bands in ten AML cell lines (Online Supplementary Figure S1D). Spearman correlation analysis was performed to evaluate the relationship between IC50 values and the protein change level of BCL-XL, BCL-2 and MCL-1 by 753B. We found that the sensitivity to 753B inversely correlated with MCL-1 level when treated with 753B at the concentration of 1 μM at 24 h (r=0.8182, P=0.0038) but did not correlate with the degradation level of BCL-XL or BCL-2 at the same concentrations (Figure 5E). These data further support MCL-1 dependence following 753B treatment. 753B induced cell death in 50% of OCI-AML-2 cells at a concentration of 0.64 μM and in nearly 100% of the cells, when combined with a low dose of S63845 (0.005 μM) at 24 h by CTG assay; induction of
Figure 3. 753B enhances the efficacy of chemotherapy by eliminating senescent leukemia cells. (A) Flow cytometry forward scatter (FSC) versus side scatter (SSC) plots showing cell size in MOLM-14 cells treated with the indicated concentrations of cytarabine (Ara-C) for three days. (B) Representative images of SA-β-gal activity in MOLM-14 cells exposed to Ara-C for three days and stained with X-gal. Scale bar = 25 µM. (C) Flow cytometry histogram depicting SA-β-gal activity using the fluorogenic β-galactosidase substrate C12-FDG in MOLM-14 cells after three days of Ara-C exposure (0.05 µM, 0.1 µM or 0.2 µM). (D and E) Images of SA-β-gal staining (D) and flow cytometry histogram of C12-FDG (E) showing induction of senescence in MOLM-14 cells gated on viable cells (by DAPI- exclusion) and treated with Ara-C (0.05 µM), 753B (0.037 µM) or the combination (concomitantly). (F and G) Expression of p21, p16, BCL-2, BCL-XL, and MCL-1 (F) and mRNA expression of SASP markers (IL-8, IL-6, IL-1β, CCR5) (G) in MOLM-14 cells exposed to Ara-C, 753B, or the concomitant combination. C12-FDG: 5-dodecanoylaminofluorescein di-β-D-galactopyranoside; SA-βgal: senescence-associated β-galactosidase; SASP: senescence-associated secretion phenotype. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.

cell death was synergistic at multiple concentrations tested (Figure 5F). These data suggest that the dual targeting of MCL-1 and BCL-XL is highly effective in inducing cell death in AML.
Anti-leukemia efficacy of 753B in vivo in acute myeloid leukemia patient-derived xenograf model

To investigate the antileukemia activity of 753B in vivo, we developed a PDX model by injecting NSG mice with AML PDX #4138550 (generated from an AML patient harboring FLT3-ITD, DNMT3A, IDH1, KIT, and NPM1 mutations). After confirming engraftment by peripheral blood flow cytometry analysis, mice were randomized into two groups to receive either vehicle or 753B (5 mg/kg intraperitoneally every 4 days) for three weeks. Mice tolerated 753B therapy well with no significant changes in body weight and no significant normal hematopoietic cells, platelet or white blood cell (WBC) toxicity as measured by blood counts
(Figure 6A, Online Supplementary Figure S5A-C). 753B treatment reduced the circulating leukemia cell burden measured by flow cytometry, reduced liver and spleen weight, and extended overall survival (Figure 6B, C, Online Supplementary Figure S5D). The histological evaluation of Hematoxylin & Eosin (HE) stained sections of the bone marrow (BM), liver, and spleen showed reduction in the tumor burden of the BM, spleen and liver after 753B treatment, with no microscopic evidence of liver or spleen damage (Figure 6D). The reduction of leukemia burden induced by 753B was associated with a notable reduction in BCL-XL expression in leukemia cells harvested after 753B treatment (Figure 6E, F).
Both BCL-XL and BCL-2 are validated therapeutic targets
Figure 4. Chemotherapy-induced senescent cells express higher levels of BCL-XL, representing a therapeutic target for 753B. (A) Western blotting showing BCL-XL, BCL-2, and MCL-1 protein levels in MOLM-14 cells treated with cytarabine (Ara-C) (0.05 μM or 0.1 μM) for 72 hours (h). After treatment, cells were sorted based on C12-FDG staining into two groups: C12-FDG-low (lowest 15%) and C12-FDG-high (highest 15%). Cells were then lysed for western blotting. (B) MOLM-14 cells were treated with dimethyl sulfoxide (DMSO) (non-senescent, control) or Ara-C (0.05 μM) to induce senescent cells (SC) for 72 h followed by treatment with 753B for 24 h. Cell viability was determined by CellTiter-Glo© (CTG) assay. (C) Western blotting showing protein levels of apoptosis markers in MOLM-14 cells treated with 753B (0.037 μM), Ara-C (0.05 μM), or the combination. (D) Heatmap of cell viability of MOLM-14 cells treated with 753B, Ara-C, or the combination at the indicated concentrations for 24 h. Viability was determined by CTG assay. Data are representative of three independent experiments. (E) Combination index (CI) of 753B and Ara-C in MOLM-14 cells treated for 24 h. *P<0.05; ***P<0.001; ****P<0.0001.
with overexpression noted in a wide range of hematologic malignancies.31-33 In addition to MCL-1, BCL-XL is also a key mediator of chemotherapy and venetoclax resistance.4,5,34 Thus, developing a tolerable and effective BCL-XL-targeted or BCL-2/BCL-XL co-targeted therapy is of high priority. Given the caution required in dealing with navitoclax-induced thrombocytopenia, drug discovery efforts have aimed at circumventing this limitation through optimizing navitoclax-based targeting. In this study, we validated the effect of a novel dual BCL-XL/BCL-2 PROTAC 753B, that
was developed using navitoclax as a ligand in genetically diverse leukemia cells. We observed that 753B exhibited a potent anti-leukemia efficacy similar to that of navitoclax, making it a promising therapeutic candidate. We noted that 753B does not degrade BCL-2 as readily as BCL-XL . We speculate that one of the possible mechanisms for the discrepancy in degradation between BCL-XL and BCL-2 is at least partially to be attributed to the differential distribution and orientation of lysines on the protein surfaces, as we reported recently.10 In addition, the

Figure 5. BCL-XL degradation by 753B is associated with MCL-1 upregulation in selected leukemia cells, and the combination of 753B and an MCL-1 inhibitor induces synergistic cell death. (A) Western blotting showing protein levels of MCL-1 in OCI-AML-2 cells treated with 753B (0.111 μM) and collected at the indicated time points. (B) OCI-AML-2 cells were treated with dimethyl sulfoxide (DMSO) or 753B (0.111 μM) for 4 hours (h). Immunoprecipitated Bim lysates demonstrate increased Bim and MCL-1 binding after 753B treatment. Immunoprecipitation was carried out with Bim antibody and detected with anti-MCL-1. S/N: supernatant after immunoprecipitation. (C) Co-immunoprecipitation (Co-IP) of OCI-AML-2 cells treated with DMSO or 753B (0.111 μM) for 4 h. Immunoprecipitation was carried out with MCL-1 antibody and detected with anti-BIM. (D) Column graph showing BH3 profiling of OCI-AML-2 cells treated with DMSO or 0.111 μM 753B for 4 h. (E) Correlation analyses of 753B IC50 values and the protein change levels of MCL-1, BCL-XL and BCL-2 in ten AML cell lines. The changed level of certain protein (MCL-1, BCL-XL or BCL-2) = baseline protein expression level quantified from untreated samples - protein expression level quantified from samples treated with 753B after 24 h at 1 μM. Non-parametric one-tailed Spearman test was used to determine the correlation coefficient r. (F) Heatmap of cell viability of OCI-AML-2 cells treated with 753B, S63845 (MCL-1 inhibitor), or the combination at the indicated concentrations, determined by CTG assay. Data are representative of three independent experiments. *P<0.05; **P<0.01. NS: not significant.
protonated state, flexibility, as well as the other residues around the lysine involved in the E2/POI interface would also be key factors contributing to the difference in lysine ubiquitination effectiveness between BCL-XL and BCL-2. These differences may render BCL-XL more susceptible to 753B-induced protein ubiquitination and degradation than BCL-2. The detailed mechanisms and optimization of the dual degradation will require future studies.
753B induced rapid dose-dependent BCL-XL and BCL-2 degradation in leukemia, which translated into inhibition of cell growth and apoptosis. This effect was observed in 11 BCL-2/BCL-XL co-dependent cell lines (2/2 MPN-AML and 9/17 AML cell lines including the high-risk EVI-1 rearranged cells) and in the majority of the primary AML samples harboring high-risk mutations (such as FLT-ITD, TP53 and NRAS), exhibiting a broader activity than venetoclax. Notably, 10 out of 16 samples were collected from patients, whose disease progressed after venetoclax-containing regimens, and all ten samples responded in vitro to navitoclax and 753B. These data indicate a potential utility of BCL-XL-targeting agents in overcoming resistance to venetoclax. 753B showed preliminary in vivo efficacy in AML-derived PDX harboring FLT3-ITD, DNMT3A, and IDH1 mutations. We noted the rebound in hCD45 cellularity after the last week of 753B treatment and assume that the rebound of leukemia could result from MCL-1 upregulation or due to a sub-optimal dose of 753B used in the in vivo study. Efficacy of 753B or optimized next generation degrader should be tested in additional AML PDX models with diverse genetic backgrounds in future studies. With further optimization of formulation and dosing, 753B has the potential to become the first-in-class platelet-sparing BCL-XL/BCL-2 targeting antitumor agent.
Chemotherapy works in part by inducing cellular senescence; immune cells then clear senescent cancer cells, leading to tumor stasis or regression. However, these senescent cells can escape from immune surveillance and re-enter the cell cycle after escaping from a resting state with accelerated potential for growth.18,35,36 These cells can acquire novel stem-cell and self-renewing features, promote disease relapse, and enhance aggressiveness, as
evidenced in several mouse tumor models, including AML.27 Recent studies identified navitoclax and other BCLXL/BCL-2 inhibitors (ABT-737 and A1331852) as potent senolytics.21,37,38 Because BCL-XL/BCL-2 are key anti-apoptotic proteins in many types of senescent cells, we hypothesized that 753B may eliminate senescent cells before escaping from senescence. In our study, we found that 753B largely reversed Ara-C-induced senescence markers and enhanced the efficacy of Ara-C by targeting senescent AML cells, suggesting an added benefit from targeting BCL-XL/BCL-2 with 753B in combination with chemotherapy for AML treatment. This finding provides a rationale for sequential treatment of chemotherapy followed by 753B, which could reduce the toxicity of concomitant administration, yet avoid relapse by eliminating chemotherapy-induced senescent tumor cells.
Our mechanistic studies suggest that the sensitivity of AML cell lines to 753B inversely correlates with high MCL1 expression. MCL-1 expression commonly accounts for resistance to other BH3 mimetics, including venetoclax.39,40 Our data indicate that 753B has a strong synergistic effect on inducing cell death in MOLM-14 cells when combined with a small molecule inhibitor targeting MCL-1 (Figure 5F). However, the synergy of 753B and MCL1 inhibitors should be tested in additional cells lines or xenograft models in future studies. Notably, co-administration of MCL-1 inhibitor (VU661013) and BCL-2 inhibitor has been reported to have a reasonable safety profile in AML xenografts models.41 Besides, co-targeting BCL-XL and MCL-1 with DT2216 and AZD8055 showed a synergistic effect on inhibiting small-cell lung cancer growth without causing on-target toxicities in mice.42 On the contrary, the simultaneous use of MCL-1 and BCL-XL inhibitors caused acute lethality in mice in lung squamous cell carcinomas and pancreatic cancer in vivo models.43,44 Since 753B is a BCL-2/BCL-XL co-targeted compound, the safety of directly combining an MCL-1 inhibitor with 753B will require careful in vivo safety studies in future experiments. Recognition of the important mechanistic role of BCL-XL in leukemias sparked renewed interest in therapeutically inhibiting BCL-XL. In a recently reported phase I study
of 753B in vivo in acute myeloid leukemia patient-derived xenogra f model . (A) CD34+ bone marrow (BM) cells were sorted from three individual donors (Donor 1-3) by FACS and colony formation was analyzed for CFU-E, CFU-G, CFU-M, CFU-GM or CFU-GEMM cells treated with indicated concentration of 753B. (B) Circulating tumor burden in an acute myeloid leukemia (AML) patient-derived xenograft (PDX) model (#4138550) treated with vehicle (Veh) or 753B (treatment started on day 33 after AML cell injection and was administered intraperitoneally at a dose of 5 mg/kg, every 4 days for 3 weeks). (C) Kaplan-Meier survival curves for mice treated as described in (A) (N=6/arm). Statistical significance was calculated using the log rank (Mantel-Cox) test (P<0.01). (D) Representative Hematoxylin & Eosin (HE) stained sections of BM, spleen and liver from 753B or vehicle-treated mice. Scale bar on the bottom right indicates 200 μm. (E) Immunohistochemical staining of BCL-XL in an AML PDX model from 753B or vehicle-treated mice. Scale bar on the bottom right indicates 100 μm. (F) Western blotting showing BCL-XL protein levels in an AML PDX model (N=2 mice in each group). DMSO: dimethyl sulfoxide; PB: peripheral blood; d: day; w: week.

(clinicaltrials. gov identifier: NCT03181126), co-targeting BCL-XL and BCL-2 by combination of reduced doses of navitoclax and venetoclax showed promising efficacy in patients with relapsed or refractory ALL who progressed after multiple lines of therapy.45 The use of navitoclax as an “add-on” approach to the JAK2 inhibitor ruxolitinib in patients with myelofibrosis produced objective responses in a phase II clinical study (clinicaltrials. gov identifier: NCT03222609), although careful dose titration of navito-
clax was required due to an overall decrease in platelet counts.46 AZD0466 is a nanoparticle-formulated novel dual BCL-XL/BCL-2 inhibitor that is currently in a phase I clinical trial for hematologic malignancies (clinicaltrials. gov identifier: NCT04214093) and it is pharmacologically optimized to minimize thrombocytopenia.47 In contrast, 753B is a first-in-class dual BCL-XL/BCL-2 degrader that avoids platelet toxicity due to selective expression of VHL E3 ligase. Because 753B is derived from navitoclax, 753B
is expected to be a safe strategy to target both BCL-2 and BCL-XL in AML, without resultant thrombocytopenia. Taken together, our study demonstrates that the singleagent 753B has an anti-tumor activity in a subset of hematologic cell lines, primary patient-derived AML blasts, and a xenograft model via BCL-2 family protein degradation. We demonstrated that the senolytic properties of 753B may enhance the efficacy of chemotherapy by targeting BCL-XL-expressing senescent cells. Importantly, the first-in-human dose escalation study of BCLXL PROTAC DT2216 has opened for patient accrual (clinicaltrials. gov identifier: NCT04886622). We anticipate that the clinical use of PROTAC based on our informative pre-clinical findings will lead to safe and effective co-targeting of BCL-2 and BCL-XL in AML.
Disclosures
GZ and DZ are co-founders of and hav e equity in Dialectic Therapeutics, which develops BCL-XL PROTAC to treat cancer. The other authors have no conflict of interests to disclose.
Contributions
YJ conceived, designed and performed most of the biological and biochemical experiments, analyzed and inter-
1. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26(9):1324-1337.
2. Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375-388.
3. Pan R, Hogdal LJ, Benito JM, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4(3):362-375.
4. Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106-1117.
5. Lin KH, Winter PS, Xie A, et al. Targeting MCL-1/BCL-XL forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci Rep. 2016;6:27696.
6. Zhang Q, Riley-Gillis B, Han L, et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct Target Ther. 2022;7(1):51.
7. Oppermann S, Ylanko J, Shi Y, et al. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood. 2016;128(7):934-947.
8. Schoenwaelder SM, Jarman KE, Gardiner EE, et al. Bcl-xLinhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood. 2011;118(6):1663-1674.
9. Khan S, Zhang X, Lv D, et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25(12):1938-1947.
preted data, and wrote the manuscript. LH, CR, ZW, CW, LY, SC and HM performed and analyzed some of the biological experiments. WZ, MA, ND, NJ, NP, KB and SM supervised some of the biological studies and revised the manuscript. PZ synthesized 753B, and prepared the formulation of vehicle and 753B for animal study. GZ conceived, designed and supervised the synthesis of BCL-XL/2 PROTAC, and revised the manuscript. DZ conceived, designed and supervised the synthesis of BCL-XL/2 PROTAC, revised the manuscript, and guided senescence studies . QZ and MK conceived, designed, and supervised the study, analyzed and interpreted data, and wrote the manuscript. All authors discussed the results and commented on the manuscript.
This study was supported by US National Institutes of Health (NIH) grants R01 CA241191 (to GZ, MK and DZ), the Cancer Foundation Finland, and the Academy of Finland (to SM).
Original data and protocols are available to other investigators upon request by contacting the corresponding author or last author.
10. Lv D, Pal P, Liu X, et al. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat Commun. 2021;12(1):6896.
11. Duy C, Li M, Teater M, et al. Chemotherapy induces senescence-like resilient cells capable of initiating AML recurrence. Cancer Discov. 2021;11(6):1542-1561.
12. Milanovic M, Fan DNY, Belenki D, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553(7686):96-100.
13. Yosef R, Pilpel N, Tokarsky-Amiel R, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190.
14. He Y, Zhang X, Chang J, et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat Commun. 2020;11(1):1996.
15. Han L, Zhang Q, Dail M, et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica. 2020;105(3):697-707.
16. Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798-1806.
17. Matsuo H, Kajihara M, Tomizawa D, et al. EVI1 overexpression is a poor prognostic factor in pediatric patients with mixed lineage leukemia-AF9 rearranged acute myeloid leukemia. Haematologica. 2014;99(11):e225-227.
18. De Blander H, Morel AP, Senaratne AP, Ouzounova M, Puisieux A. Cellular plasticity: a route to senescence exit and tumorigenesis. Cancers (Basel). 2021;13(18):4561.
19. Demaria M, O'Leary MN, Chang J, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7(2):165-176.
20. Munoz DP, Yannone SM, Daemen A, et al. Targetable mechanisms driving immunoevasion of persistent senescent cells link chemotherapy-resistant cancer to aging. JCI Insight. 2019;5(14):e124716.
21. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15(3):428-435.
22. Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363-9367.
23. Schmitt CA, Fridman JS, Yang M, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335-346.
24. Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, Watanabe M. Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat Res. 2001;155(1 Pt 2):248-253.
25. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593-602.
26. Elashiry M, Elsayed R, Cutler CW. Exogenous and endogenous dendritic cell-derived exosomes: lessons learned for immunotherapy and disease pathogenesis. Cells. 2021;11(1):115.
27. Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J. 2023;290(5):1362-1383.
28. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16(2):99-109.
29. Delbridge AR, Strasser A. The BCL-2 protein family, BH3mimetics and cancer therapy. Cell Death Differ. 2015;22(7):1071-1080.
30. Konopleva M, Milella M, Ruvolo P, et al. MEK inhibition enhances ABT-737-induced leukemia cell apoptosis via prevention of ERKactivated MCL-1 induction and modulation of MCL-1/BIM complex. Leukemia. 2012;26(4):778-787.
31. Espana L, Fernandez Y, Rubio N, Torregrosa A, Blanco J, Sierra A. Overexpression of Bcl-xL in human breast cancer cells enhances organ-selective lymph node metastasis. Breast Cancer Res Treat. 2004;87(1):33-44.
32. Amundson SA, Myers TG, Scudiero D, Kitada S, Reed JC, Fornace AJ Jr. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res. 2000;60(21):6101-6110.
33. Olopade OI, Adeyanju MO, Safa AR, et al. Overexpression of BCL-
x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am. 1997;3(4):230-237.
34. Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. 2017;58(9):1-17.
35. Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14(6):617-628.
36. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescenceassociated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99-118.
37. Gasek NS, Kuchel GA, Kirkland JL, Xu M. Strategies for targeting senescent cells in human disease. Nat Aging. 2021;1(10):870-879.
38. Zhu Y, Doornebal EJ, Pirtskhalava T, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (Albany NY). 2017;9(3):955-963.
39. Carter BZ, Mak PY, Tao W, et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica. 2022;107(1):58-76.
40. Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell. 2005;8(1):5-6.
41. Ramsey HE, Fischer MA, Lee T, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566-1581.
42. Khan S, Kellish P, Connis N, et al. Co-targeting BCL-X(L) and MCL-1 with DT2216 and AZD8055 synergistically inhibit smallcell lung cancer growth without causing on-target toxicities in mice. Cell Death Discov. 2023;9(1):1.
43. Weeden CE, Ah-Cann C, Holik AZ, et al. Dual inhibition of BCLXL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene. 2018;37(32):4475-4488.
44. Thummuri D, Khan S, Underwood PW, et al. Overcoming gemcitabine resistance in pancreatic cancer using the BCL-XLSpecific Degrader DT2216. Mol Cancer Ther. 2022;21(1):184-192.
45. Pullarkat VA, Lacayo NJ, Jabbour E, et al. Venetoclax and navitoclax in combination with chemotherapy in patients with relapsed or refractory acute lymphoblastic leukemia and lymphoblastic lymphoma. Cancer Discov. 2021;11(6):1440-1453.
46. Harrison CN, Garcia JS, Somervaille TCP, et al. Addition of navitoclax to ongoing ruxolitinib therapy for patients with myelofibrosis with progression or suboptimal response: phase II safety and efficacy. J Clin Oncol. 2022;40(15):1671-1680.
47. Balachander SB, Criscione SW, Byth KF, et al. AZD4320, A dual inhibitor of Bcl-2 and Bcl-xL, induces tumor regression in hematologic cancer models without dose-limiting thrombocytopenia. Clin Cancer Res. 2020;26(24):6535-6549.
1Laboratory of Transfusion Biology, Department of Pathology and Cell Biology, Columbia University Irving Medical Center, New York, NY, USA; 2Department of Translational Medicine and Surgery, Section of General Pathology, Università Cattolica del Sacro Cuore, Rome, Italy; 3Department of Biochemistry and Molecular Genetics, University of Colorado Denver –Anschutz Medical Campus, Aurora, CO, USA; 4University of Virginia School of Medicine, Charlottesville, VA, USA and 5Carter Immunology Center, University of Virginia, Charlottesville, VA, USA
*TAT and AQ contributed equally as co-first authors.
Correspondence: K.E. Hudson
keh2197@cumc.columbia.edu
Received: January 31, 2023.
Accepted: April 12, 2023.
Early view: April 20, 2023.
htps://doi.org/10.3324/haematol.2023.282815
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Although red blood cell (RBC) transfusions save lives, some patients develop clinically-significant alloantibodies against donor blood group antigens, which then have adverse effects in multiple clinical settings. Few effective measures exist to prevent RBC alloimmunization and/or eliminate alloantibodies in sensitized patients. Donor-related factors may influence alloimmunization; thus, there is an unmet clinical need to identify which RBC units are immunogenic. Repeat volunteer blood donors and donors on iron supplements have elevated reticulocyte counts compared to healthy non-donors. Early reticulocytes retain mitochondria and other components, which may act as danger signals in immune responses. Herein, we tested whether reticulocytes in donor RBC units could enhance RBC alloimmunization. Using a murine model, we demonstrate that transfusing donor RBC units with increased reticulocyte frequencies dose-dependently increased RBC alloimmunization rates and alloantibody levels. Transfusing reticulocyte-rich RBC units was associated with increased RBC clearance from the circulation and a robust proinflammatory cytokine response. As compared to previously reported post-transfusion RBC consumption patterns, erythrophagocytosis from reticulocyte-rich units was increasingly performed by splenic B cells. These data suggest that reticulocytes in a donated RBC unit impact the quality of blood transfused, are targeted to a distinct compartment, and may be an underappreciated risk factor for RBC alloimmunization.
Red blood cells (RBC) are transfused to correct anemia from bone marrow failure, hemolytic anemia, and hemorrhage. Although RBC transfusions can be lifesaving, some patients develop alloantibodies against donor RBC blood group antigens. These alloantibodies can be clinically significant, inducing hemolytic transfusion reactions and hyperhemolysis, raising transplantation barriers, and delaying transfusion by making it difficult to find compatible blood for transfusion.1 Once alloantibodies arise, patient care becomes more challenging. Currently, there are few strategies to prevent RBC alloimmunization besides antigen matching or antigen avoidance, and few treatments are available (with limited efficacy) once alloimmunization occurs.1
As opposed to pharmaceuticals, each unit of RBC is
unique as it comes from a distinct individual, undergoes multiple manufacturing steps, and then is followed by a variable duration of refrigerator storage up to 42 days before transfusion.2,3 Increasing evidence suggests that the risk of alloimmunization following an RBC transfusion is influenced by donor factors (e.g., blood group antigen density), component factors (e.g., storage duration), and host factors (e.g., blood group antigen negativity, HLA type, inflammation).4 Identifying new risk factors that can prevent and/or reduce risk of future RBC alloimmunization events is a priority, given the current lack of available treatments for sensitized patients. In both humans and mice, RBC alloimmunization rates increase in transfusion recipients with viral infections or viral-like inflammation.4,5 Viruses induce robust type I interferon (IFN-α/β) production; in mouse models of RBC alloimmunization, blocking IFN receptors prevents alloantibody production, whereas
infusing IFN-α promotes alloantibodies.6,7 Mechanistically, viral-like inflammation stimulates splenic antigen-presenting cells (APC) and alters the consumption of transfused RBC towards immunogenic dendritic cell (DC) subsets and away from red pulp macrophages.8,9
Intriguingly, patients at highest risk for RBC alloimmunization (e.g., those with sickle cell disease [SCD]) have elevated type I IFN levels.10-12 Although the mechanisms underlying type I IFN production are unknown, we and others recently published that mitochondria in human RBC stimulate type I IFN production by immune cells.13,14 Although mitochondrial retention in mature RBC is abnormal, most early reticulocytes have identifiable mitochondria. Some transfusion recipients and repeat volunteer blood donors have elevated reticulocyte frequencies.15,16 Moreover, because compensatory reticulocytosis lasts longer than the interval between donations, RBC units from some repeat volunteer blood donors have reticulocyte counts above the reference range.15 Of note, stressinduced reticulocytes produced in response to anemia (e.g., phlebotomy, hemorrhage) are larger, less deformable, and contain higher numbers of mitochondria.17-23 Thus, we hypothesized that reticulocytes in RBC donor units may enhance RBC alloimmunization.
Using a murine model of RBC alloimmunization, we test the hypothesis that elevated reticulocyte counts in RBC donor units enhance RBC alloantibody production. We demonstrate a direct correlation between reticulocyte counts in an RBC donor unit and RBC alloantibody levels produced in transfusion recipients. Compared to controls, reticulocyte-rich RBC donor units were enriched in mitochondrial proteins and metabolites and, upon transfusion, elicited proinflammatory cytokines. Mechanistically, RBC from reticulocyte-rich units had lower 24-hour posttransfusion recovery (i.e., were cleared more robustly in the first 24 hours after transfusion) and induced significantly higher RBC consumption by splenic APC. Multiple B-cell subsets consumed most of the reticulocytes, representing a departure from normal post-transfusion RBC consumption patterns. The effect of higher reticulocyte frequencies was dramatic because transfusion of short(i.e., 1 day) and medium- (i.e., 6 days) stored RBC donor units similarly induced alloantibodies. Lastly, we show that reticulocyte-mediated enhanced alloimmunization is not Toll-like receptor (TLR)-4 dependent. Together, these data demonstrate that reticulocytes in RBC donor units can enhance alloimmune responses.
Mice
HOD mice,24 expressing an RBC-specific triple fusion protein consisting of hen egg lysozyme, ovalbumin, and
human blood group molecule Duffy, were bred in the Columbia University vivarium. C57BL/6 (B6, strain #027) mice were purchased from Charles River Laboratories. C57BL/6-Tg(UBC-GFP)30Scha/J (GFP; strain #004353) and B6(Cg)-Tlr4tm1.2Karp/J (TLR4-/-, strain #029015) mice were purchased from The Jackson Laboratory. All mice were matintained on a 12:12 light/dark cycle and maintained on a chow diet (ad libitum) unless otherwise specified. All murine experiments were approved by Columbia University’s Institutional Animal Care and Use (IACUC) committee.
Reticulocyte induction and blood bank preparation
Reticulocytosis was induced with phenylhydrazine (PHZ), or by iron deficiency followed by acute iron repletion. PHZ: intravascular hemolysis was induced with two intraperitoneal (i.p.) injections of PHZ (Sigma) 50 mg/kg/day separated by 24 hours or saline control. Iron: weanling (3-week-old) mice were placed on iron-deficient (Harlan Teklad TD.110592) or replete (Harlan Teklad TD.110593) diets for 4 weeks, followed by reticulocytosis induction by i.p. injection of 5 mg of iron dextran (Allergan NDC 00230608-10) and switching to iron-replete chow.25
Whole blood was collected by cardiac puncture into 14% CPDA-1 4 days after PHZ or iron dextran injection, pooled, filter leuko-reduced (Acrodisc WBC syringe filter, Pall), packed to 60% hematocrit, and stored for 1 or 6 days at 4°C in 1.5 mL microcentrifuge tubes.
For reticulocyte quantification 1 μ L of blood was incubated with 2.5 nM of MitoTracker Deep Red FM (ThermoFisher) for 30 minutes (min) at 37°C. After washing with FACS buffer, samples were stained with antibodies against CD45, CD41, and CD71 for 30 min at 4°C ( Online Supplementary Table S1 ).13 After washing with FACS buffer, cells were analyzed using an Attune NxT flow cytometer (ThermoFisher). CD41 + platelets and CD45 + white blood cells were excluded and RBC were identified using a 405 nm filter (i.e., no wash no lyse filter, ThermoFisher) 13 . RBC were distinguished by CD71 expression: CD71 + reticulocytes and CD71 - mature RBC. Data were analyzed with FlowJo software (BD Biosciences).
Red blood cell transfusion, post-transfusion recovery, and red blood cell alloantibody detection
Stored RBC units were spiked 1:5 with fresh biotinylated RBC units before transfusion (200 uL) into recipients, as described.26 Blood samples were collected via tail puncture and post-transfusion recovery (PTR) was measured as the ratio of biotin-positive RBC to GFP-positive RBC circulating 24 hours-post transfusion relative to the same ratio in the spiked blood unit. Sera were collected from recipients and total HOD alloantibodies (IgM + IgG + IgA) were quantified by flow crossmatch.27
Hematocrit and hemoglobin
Whole blood was collected from the submandibular vein into microcentrifuge tubes containing EDTA. Hematocrit was determined following micro-hematocrit capillary tube centrifugation. Hemoglobin was calculated spectrophotometrically (540 nm) using Drabkin’s reagent.
Cytokine production and splenic erythrophagocytosis
Recipients were transfused with 6-day stored PHZ, stored control, or freshly collected GFP RBC units (350 μ L). Plasma collected 2 hours post-transfusion was diluted 1:1 in duplicate. Cytokines were quantified by a fluorescenceencoded bead-based multiplex assay (LEGENDplex Mouse Anti-Virus Response Panel, BioLegend), per manufacturer’s instructions. Spleens were processed into single cells27 and splenocytes were stained with antibodies to delineate leukocyte subsets (Online Supplementary Table S1).8,9,28,29 Results were acquired on an Attune NxT flow cytometer (ThermoFisher) and data analyzed with FlowJo software. Cell images were acquired on an Amnis ImageStreamX MkII cytometer (Luminex); 20,000 CD19+ B cells per file were recorded using INSPIRE software at 60x magnification. Analysis was performed using IDEAS 6.3 software.
Ultra-high-pressure liquid chromatography-mass spectrometry metabolomics
Metabolomics analyses were performed using a Vanquish ultra-high-pressure liquid chromatography (UHPLC) coupled online to a Q Exactive mass spectrometer (ThermoFisher, Bremen, Germany), as described.30 Data were analyzed using Maven (Princeton University) and Compound Discoverer 2.1 (ThermoFisher). Graphs and statistical analyses were prepared with MetaboAnalyst 5.0.31
Protein pellets from RBC units were digested, as described.32
Nano ultra-high-pressure liquid chromatographytandem mass spectrometry proteomics
Sample processing and data collection were performed as described.32
A repeated measures two-way ANOVA with Sidak’s multiple comparisons test was utilized for analysis of alloantibody production over time. For comparison of three or more groups, a one-way ANOVA with Tukey’s multiple comparisons post-test was utilized. An unpaired t test was used to compare two groups; P <0.05 was considered significant. Analyses were performed using GraphPad Prism.
Transfusion of stored reticulocyte-rich blood units enhances red blood cell alloimmunization
To test whether reticulocytes in RBC units modulate alloimmunization, RBC alloantibody production was evaluated in mice transfused with allogeneic RBC units containing elevated reticulocyte counts (experimental design in Figure 1A). In order to generate RBC units enriched in reticulocytes, reticulocytosis was induced by PHZ, which damages RBC, leading to hemolytic anemia followed by stress-induced reticulocytosis.33 A PHZ dose titration revealed that CD71+ reticulocyte frequency peaked in peripheral blood 3-4 days post treatment, with an optimal dose of 50 mg/kg (Online Supplementary Figure S1). Thus, HOD RBC donor mice, which express an RBC-specific HOD alloantigen, were treated with PHZ or saline control to generate RBC units. Compared to control, reticulocyte-rich RBC units had higher reticulocyte levels (2% vs. 50%, P<0.0001; Figure 1B) and the reticulocytes had an increased frequency of mitochondria positivity (51% vs. 64%; P<0.0001; Figure 1C), as determined using Mito-Tracker dye. Allogeneic HOD RBC units were refrigerator-stored 6 days before transfusion into wild-type B6 animals; sera were collected weekly and assessed for anti-HOD alloantibodies by flow crossmatch. Transfusion of stored reticulocyterich RBC units (designated as “reticulocytes”) induced significantly higher HOD alloantibody levels, compared to stored saline control RBC (“control”) (P<0.001; Figure 1D), with an >800-fold difference in RBC alloantibody levels in recipients transfused with reticulocyte-rich RBC units, compared to control (Figure 1E). Additionally, all recipients transfused with reticulocyte-rich RBC units had detectable alloantibodies, compared to some animals in the control transfusion group that did not respond. In order to test whether the enhanced RBC alloimmunization was due to the method of inducing reticulocytosis, RBC units with higher reticulocyte frequencies were also generated using an iron-deficient/iron dextran injection mouse model. As with PHZ-induced reticulocytosis, transfusing reticulocyte-rich RBC units from iron-deficient/iron dextran injection donors induced significantly higher anti-HOD alloantibody levels, compared to controls (Online Supplementary Figure S2). Together, these data demonstrate that transfusing reticulocyte-rich RBC units induces higher RBC alloimmunization rates and alloantibody levels.
In order to test whether transfusing reticulocyte-rich RBC units elicited an inflammatory response, plasma was collected 2 hours post-transfusion and assayed for cytokines. Transfusion of reticulocyte-rich RBC units led to significant increases in MCP-1, CXCL1, CXCL10, and IFN-γ levels,
Figure 1. Transfusing refrigerator-stored reticulocyte-rich blood units enhanced red blood cell alloimmunization. (A) General experimental timeline. HOD mice received 2 intraperitoneal (i.p.) injections of phenylhydrazine (PHZ) (50 mg/kg, referred to as “Reticulocytes”) or saline control (referred to as “Control”) spaced 1 day apart. Four days after treatment, whole blood was co llected by cardiac puncture into 14% CPDA-1, leuko-reduced, packed to a 60% hematocrit, and stored for 6 days at 4°C before transfusion (200 uL). For some experiments, on the day of transfusion, fresh red blood cells (RBC) were collected from biotinylated or GFP mice as an additional control. (B) The percent reticulocytes and (C) the percent of mitochondria-positive reticulocytes generated following PHZ and saline treatment. (D) Post-transfusion sera were analyzed for total anti-HOD RBC alloantibodies by flow crossmatch. (E) Sera collected at 14 days post transfusion were titrated. Naïve B6 sera were run in triplicate to establish the background antibody level. Data are cumulative from 3 independent experiments with 5 mice/group. Data with 2 groups were analyzed with an unpaired t test whereas 3 groups were analyzed by a repeated measures one-way ANOVA with Sidak’s multiple comparisons test and ***P<0.001, ****P<0.0001. Timeline figure created with BioRender.
compared to control (Figure 2). No significant differences were noted in plasma IL-6, CCL5, IL-12, TNF-α, GM-CSF, IFN-α, IFN-β, IL-10, or IL-1β levels (data not shown). Together, these data suggest that transfusing reticulocytes elicited a strong, albeit selective, inflammatory response, characterized by significant chemokine production.
Reticulocyte-rich red blood cell units have elevated levels of mitochondrial metabolites and proteins
Untargeted and targeted metabolomics analyses using UHPLC-MS were performed on 6-day-stored reticulocyte-
rich and control RBC units; detailed results are in the Online Supplementary Table S2. The metabolic phenotypes of reticulocyte-rich blood differed substantially from controls. Hierarchical clustering analysis (Figure 3A) highlighted significant associations of reticulocytes with metabolic pathway differences in lipid (fatty acid, phospholipid, and sphingolipid), amino acid, and purine metabolism. Volcano plots (Online Supplementary Figure S3) from targeted metabolomics analysis highlighted the top metabolites that increased (red) or decreased (blue) in reticulocyte-rich RBC units as compared to controls. Path-

Figure 2. Induction of inflammatory cytokines afer transfusing refrigerator-stored reticulocyte-rich red blood cell units. Reticulocyte-rich and control red blood cell (RBC) units were generated (as in Figure 1) using whole blood collected from GFP animals. RBC units were stored for 6 days and transfused into wild-type B6 animals. Plasma was collected 2 hours post-transfusion and analyzed for cytokines. Data shown are cumulative of 2 independent experiments with 3-4 mice/group. Data were analyzed with an unpaired t-test; ****P<0.0001, ***P<0.001, *P<0.05.
way analysis of untargeted metabolomics data (Figure 3B) revealed altered regulation of metabolic pathways suggesting active mitochondrial function (i.e., β oxidation, tricarboxylic acid cycle [TCA], urea cycle, malate-aspartate shuttle, and glycerol-3-phosphate shuttle) and mitochondrial synthesis (cardiolipin synthesis).
Proteomics analyses were also performed on 6-day-stored reticulocyte-rich and control RBC units. Hierarchical clustering analysis (Figure 3C) highlighted significant associations of reticulocytes with proteins of mitochondrial origin upon gene ontology enrichment analysis for cell localization, with the most differentially expressed proteins involved in mitochondrial fatty acid metabolism, the TCA cycle, electron transport chain, ATP synthesis, mitochondrial structure, and mitochondrial oxidative stress (Figure 3D).
Stored reticulocyte-rich red blood cell units have reduced post-transfusion recovery and are preferentially ingested by splenic B cells
Based on enhanced cytokine production by, and the detectable mitochondrial byproducts in, reticulocyte-rich RBC units, we hypothesized that clearance of transfused RBC from the circulation would be accelerated. In order to test this, recipients were transfused with reticulocyte-rich or control RBC units and PTR (i.e., the percentage of transfused RBC remaining in the circulation for 24 hours) determined. Reticulocyte-rich RBC units had significantly reduced PTR at 24 hours, compared to control (P<0.0001; Figure 4A). To elucidate which cells were responsible for RBC clearance, reticulocyte-rich and control RBC units were generated using GFP mouse donors. Recipient B6 mice were then transfused with stored reticulocyte-rich, stored control, or freshly collected GFP RBC units and spleens harvested from recipients 2 hours later. Total splenocyte numbers were calculated and cells were stained with antibodies to identify antigen presenting cell (APC) subsets. For analysis, T cells and RBC (or cells with RBC attached to their surface) were excluded from total live
splenocytes by excluding those positive for Thy1.2 and/or Ter119; GFP fluorescence provided an indirect measure of RBC consumption.8,9,29 Enumerating leukocytes revealed a significant decrease in total cell numbers and in APC subsets, including F4/80+ macrophages, CD8+ DC, and CD19+ B cells, in recipients transfused with reticulocyte-rich RBC units, compared to stored and fresh control RBC units (Figure 4B, C). No significant numerical differences in CD11b+ DC were noted. Evaluating leukocytes for GFP fluorescence showed a significant increase in the frequency of GFP+ cells, suggesting that transfused reticulocyte-rich RBC units were being consumed by more leukocytes, as compared to controls (P<0.0001; Figure 4D). Individual APC subsets were interrogated for GFP fluorescence to determine which participated in erythrophagocytosis. As previously reported,34 transfusing stored control RBC units induced increased RBC consumption by macrophages, CD8+ DC, and CD11b+ DC, compared to fresh GFP control RBC (Figure 4E). However, as compared to stored control RBC units, RBC consumption from reticulocyte-rich units was significantly increased in CD8+ and CD11b+ DC; nonetheless, consumption by macrophages was not enhanced and was similar to fresh RBC controls. Unexpectedly, B cells markedly consumed RBC from transfused reticulocyte-rich units, compared to stored and fresh controls. These data demonstrate that transfusing reticulocyte-rich RBC units leads to significant changes in splenocyte cell subset composition and RBC consumption patterns. The most striking finding was that >20% of B cells had detectable GFP fluorescence, suggesting that a unique RBC consumption pattern may be associated with stored reticulocyte clearance.
In order to elucidate which splenic B cell subsets were involved in clearing reticulocyte-rich RBC units, B cells were gated into follicular, marginal zone, and B1 B-cell subsets (gating strategy in the Online Supplementary Figure S4).28
Low levels of erythrophagocytosis by B-cell subsets were observed after transfusing stored or fresh control RBC units (Figure 5A). In contrast, there was a marked increase

Figure 3. Omics analysis revealed mitochondrial metabolites and proteins in reticulocyte-rich red blood cell units. Aliquots of 6-day-stored reticulocyte and control red blood cell (RBC) units were subjected to omics analysis. (A) Hierarchical clustering analysis highlighting significant metabolic changes between control and reticulocyte-rich RBC units. (B) Pathway analysis of untargeted metabolomics data. (C) Hierarchical clustering of significantly different proteins between control and reticulocyte-rich RBC units. In (D), zoom in on a subset of mitochondrial proteins (by gene ontology of cell localization) whose levels are higher in reticulocyte-rich RBC units, compared to controls.

in GFP+ B cells following transfusion with reticulocyte-rich units. The most prominent signal was with innate B1 B cells, with approximately 45% positive for GFP. In order to confirm whether B cells actually internalized RBC, samples were analyzed with an imaging cytometer. With reticulocyte-rich samples, three GFP expression patterns emerged: diffuse GFP signal within CD19+ staining, punctate GFP signal along the cell membrane, and clusters of GFP signal within the B cell (Figure 5B, top; Online Sup-
plementary Figure S5). In contrast, in controls, only punctate GFP signal along the B-cell membrane was detectable, along with a few cells that had RBC particulates attached to the B-cell surface (Figure 5B, bottom). In samples from the fresh control RBC transfusion group, very few GFP+ B cells were detected and most positive events reflected RBC particulates attached outside the B cell, as indicated by brightfield images (Online Supplementary Figure S5); this was consistent with the fre-
Figure 4. Transfusion with refrigerator-stored reticulocyte-rich red blood cell units led to increased red blood cell consumption. In order to determine post-transfusion recovery (PTR), reticulocyte-rich and control red blood cell (RBC) units were generated from GFP mice. After 6 days of storage, RBC units were spiked with freshly-biotinylated freshly-obtained RBC (as a tracer) and 350 μL were transfused into wild-type B6 recipients. (A) Whole blood was sampled 24 hours later and the frequency of GFP+ RBC was normalized to biotinylated RBC to determine PTR. In order to evaluate cellular sites of RBC consumption, B6 recipients were transfused with 350 μL of freshly collected GFP+ RBC or 6-day-stored RBC units (i.e., reticulocyte or control). Spleens were collected 2 hours post-transfusion and splenocytes stained with antibodies to delineate APC subsets. GFP signal level was used as an indirect measure of erythrophagocytosis. The total number of cells per spleen were calculated for (B) Thy1.2-Ter119- leukocytes and for (C) individual APC subsets. The frequencies of GFP+ (D) Thy1.2-Ter119- leukocytes and (E) individual APC subsets were determined. The following phenotypes were used to delineate APC subsets: macrophages: CD11c-/loCD11b-/loF4/80+; CD8+ dendritic cells (DC): CD11chiCD8+CD11b-; CD11b+ DC: CD11chiCD11b+CD8-; and B cells: CD19+. Data are aggregated from at least 3 independent experiments with 3-4 mice/group. Data were analyzed with an unpaired t test for 2 groups or one-way ANOVA with Tukey’s multiple comparisons test; ****P<0.0001, ***P<0.001, **P<0.01, *P<0.05.
quency of cells with GFP signal inside the cell membrane (Figure 5C).
Red blood cell alloimmunization is enhanced, even after shorter storage and fewer reticulocytes Because refrigerator storage adversely affects RBC quality, and because reticulocyte counts are much higher in reticulocyte-rich RBC units generated from mice, as compared to volunteer human donors, we generated mouse RBC units with defined reticulocyte frequencies. HOD RBC units were refrigerator-stored for 1 day (equivalent to ~3 days of storage for human RBC units) and then transfused into recipients. Transfusing RBC units with as few as 5% reticulocytes induced significant alloantibody production, as compared to con-
trol (i.e., <2% reticulocytes) (Figure 6A). There was a significant direct correlation between reticulocyte frequency in the RBC donor unit and alloantibody production; this relationship was significant even at low reticulocyte frequencies. Lastly, in order to test whether B-cell subsets consumed RBC from 1-day-stored reticulocyte-rich units, GFP fluorescence of follicular, marginal zone, and B1 B-cell subsets was assessed; significant RBC consumption was observed in each of these subsets following reticulocyte-rich RBC transfusions, compared to controls (Figure 6B). Thus, B-cell consumption patterns were similar between 1- and 6day-refrigerator-stored reticulocyte-rich RBC units. These findings were visually confirmed by imaging cytometry (Figure 6C; Online Supplementary Figure 6S ). Col-

Figure
red blood cells (RBC) or 6-day-stored RBC collected from
or
mice.
were collected 2 hours post-transfusion and splenocytes stained with antibodies to delineate APC subsets. GFP signal level was used as an indirect measure of erythrophagocytosis. (A) Total frequency of GFP+ cells for each B-cell subset was calculated by flow cytometry. (B) Imaging analysis of B cells from mice transfused with refrigerator-stored reticulocyte-rich (top) or stored saline (bottom) RBC units. Cell #4492 demonstrates RBC internalization by a B cell whereas #2527 represents an RBC adhered to the surface of a B cell. (C) Percentage of CD19+GFP+ cells with GFP+ RBC located inside their cell membrane. The phenotypes used to delineate CD19+ B-cell subsets were follicular (FO; B220+IgD+IgMloCD23+CD93-), marginal zone (MZ; B220+IgD+IgM+CD23-CD93-), and B1 (B220lo/CD43+). In order to exclude RBC attached to the external B-cell surface, Ter119+ cells were excluded from parent gates. Data are cumulative from 2 experiments with n=3-4 mice/group. Data were analyzed with one-way ANOVA with Tukey’s multiple comparisons test; ****P<0.0001.

lectively, these data show that even low reticulocyte counts make RBC units more immunogenic, and that prolonged refrigerator storage is not required for this enhanced alloimmunization.
TLR4 is not required for reticulocyte-mediated enhanced alloimmunization
Reticulocytes contain endogenous danger signals that can induce inflammation upon recognition by immune cells. Because stress-reticulocytes are more prone to hemolysis 20,21 (thereby releasing free heme 35 ) and con-
tain mitochondria (enveloped in cardiolipin), 36 we hypothesized that TLR4 was required for the observed enhanced alloimmune responses upon reticulocyte-rich transfusion. To that end, B6 and TLR4 -/- recipients were transfused with reticulocyte-rich or control RBC units; no significant differences in anti-HOD alloantibodies were observed 14 days post-transfusion (Figure 7). Therefore, reticulocyte-mediated enhanced alloimmune responses do not require TLR4 signaling. These data also rule out the potential contribution of LPS contamination in RBC unit production.
Figure 6. Red blood cell alloimmunization is stimulated, even with shorter storage duration and lower reticulocyte frequencies. Red blood cell (RBC) units with defined reticulocyte frequencies were generated by titrating whole blood from phenylhydrazine (PHZ)-treated animals into RBC collected from saline-treated animals. RBC units were leukoreduced and stored at 4°C for 1 day before transfusion into B6 recipient mice. (A) RBC alloantibodies were quantified by flow crossmatch at 14 days post-transfusion and alloantibody mean fluorescence intensity (MFI) was plotted against the RBC unit reticulocyte frequency. Data were analyzed with a one-way ANOVA and tested for linear trend. Data are cumulative of at least 3 independent experiments and each data point is 1 mouse, <2% (N=25), 5% (N=15), 10% (N=13), 33% (N=5), 40% (N=15), 45% (N=5). (B) Flow analysis revealed that multiple B-cell subsets consumed GFP+ RBC from reticulocyte-rich RBC units and this was confirmed visually by (C) imaging analysis. For RBC consumption, data were analyzed with a one-way ANOVA with Tukey’s multiple comparisons test; ****P<0.0001, ***P<0.001.
These results demonstrate that increased reticulocyte count in a refrigerator-stored RBC unit can enhance transfusion-induced RBC alloimmune responses. Reticulocyte-mediated modulation of alloimmunization was dose-dependent, and was not an artifact of PHZ treatment as a similar effects were observed using reticulocytes generated from an iron deficiency/iron repletion model. Moreover, reticulocyte-enhanced alloimmunization was independent of refrigerator storage duration because transfusing reticulocyte-rich RBC units refrigerator stored for either 1 or 6 days enhanced alloimmunization rates and alloantibody levels. In order to explore underlying mechanisms, transfusion of reticulocyte-rich RBC units elicited pro inflammatory cytokines, and transfused RBC were cleared more rapidly, preferentially by splenic B cells. Lastly, we show that the observed reticulocyte-me-

diated enhanced alloimmune responses did not require TLR4 signaling; these data exclude the possibility of LPS contamination and rule out some potentially immunogenic reticulocyte-derived ligands, such as heme and cardiolipin. Together, these findings imply that reticulocytes in RBC units may be immunogenic and could enhance alloimmune responses, even in otherwise stable recipients; this finding may have implications for transfusions into all human patients, particularly those at highest risk for allo-immunization.
Approximately 70% of the blood supply in the USA derives from repeat volunteer blood donors.37,38 Because blood donation typically removes approximately 10% of the total blood volume, compensatory stress erythropoiesis increases production of erythroid precursors and the premature release of reticulocytes; although the minimum inter-donation interval for blood donation in the US is 56 days, elevated reticulocyte counts (over baseline) are ob-
Figure 7. TLR4 is not required for reticulocyte-mediated enhanced red blood cell alloimmunization. Reticulocyte-rich and control red blood cell (RBC) units, generated from phenylhydrazine (PHZ)-treated HOD donor animals, were leuko-reduced and stored 1 day at 4°C before transfusion into wild-type B6 or TLR4-/- mice. Sera was collected 14 days post-transfusion and analyzed for total anti-HOD RBC alloantibodies by flow crossmatch. Data are from 1 experiment with 4-5 mice/group. Data were analyzed with a two-way ANOVA with Sidak’s multiple comparisons test; ns: not significant.
served even after approximately 160 days after donation.15,39 Thus, frequent donation by repeat blood donors can yield RBC units with higher reticulocyte counts. Additionally, current guidance for iron-deficient volunteers is to take iron supplements before donation, which also promote stress erythropoiesis.15 Reticulocytes differ from their mature counterparts by: i) containing immune-enhancing organelles (e.g., mitochondria), lipids, and residual RNA; ii) expressing surface antigens that promote adherence (e.g., CD44, CD36); and iii) being larger and less deformable. Compared to steady-state reticulocytes, stress erythropoiesis-induced reticulocytes also contain significantly more organelles (e.g., mitochondria, ribosomes), express higher levels of surface molecules mediating adherence, and have shorter circulatory lifespans due to their size, decreased deformability, and susceptibility to shear stress.18-23 Thus, reticulocytes, and especially stressinduced reticulocytes, contain and/or express immuneenhancing components.
Recipient inflammation is associated with higher alloimmunization rates and alloantibody levels.4,5 Because RBC alloimmunization correlates with type I IFN levels, and because stress-induced reticulocytes contain ligands that could stimulate type I IFN production (e.g., mitochondrial DNA13,14), we measured plasma IFN- α and IFN- β . Unexpectedly, no significant levels were detectable. This could be due to timing, as plasma was collected 2 hours posttransfusion, or location (e.g., production localized to the spleen); additionally, it could indicate that type I IFN are not required for reticulocyte-enhanced alloimmunization. Nonetheless, transfusion of reticulocyte-rich units elicited high levels of proinflammatory cytokines and chemokines. Other potentially immunogenic reticulocyte components
include mitochondria-derived cardiolipin and heme, both of which are TLR4 ligands. However, transfusing reticulocyte-rich RBC units into TLR4-/- and control recipients led to similar alloantibody levels (Figure 7), demonstrating TLR4 is dispensable for these enhanced alloimmune responses. Future studies will identify which signaling pathways are required for reticulocyte-mediated enhanced alloimmunity.

In a departure from typical RBC clearance patterns,8 B cells preferentially consumed RBC derived from reticulocyte-rich units. Macrophages are typically responsible for steady-state removal of senescent and/or damaged RBC.8,34 In murine models of RBC alloimmunization, with transfusion of stored RBC or in the context of inflammation, macrophages and DC subsets (CD11b+ and CD8+) play critical roles in initiating alloimmune responses.9,34 Although B-cell consumption of transfused RBC is, indeed, a prerequisite for alloantibody production, very few B cells are typically observed in this process. In contrast, B cells play a major role clearing transfused RBC from reticulocyte-rich units. Because of the high frequency of follicular and marginal zone B cells co-localizing with RBC, it is unlikely that RBC phagocytosis is B-cell receptor (BCR)-mediated; however, B1 B cells express polyreactive BCR that bind many autoantigens, including phosphatidylserine (PS), which is expressed at high levels on stress-induced reticulocytes.40,41 As such, future studies will strive to identify which RBC characteristics of RBC (e.g., PS expression, mitochondrial content) are required for their enhanced consumption by B cells and subsequent alloantibody production. Additionally, innate B1 B cells can participate in BCR-independent endocytosis,42 although the underlying mechanisms are not yet defined. As all B cells express complement receptors and Fc receptors,43,44 these pathways may be involved in erythrophagocytosis. Elucidating the key pathways required for B-cell activation following their consumption of RBC in this setting will be an important focus of future experiments.
As anemia of many etiologies induce stress erythropoiesis, one potential limitation of these studies is that various stresses may produce differences in the resulting reticulocytes. Thus, it is possible that reticulocyte-containing RBC units generated from repetitively phlebotomized animals may not enhance RBC alloimmunization. In order to address this concern, the current studies utilized two different models of anemia—chemically-induced hemolytic anemia and iron deficiency anemia followed by iron repletion—to induce reticulocytosis. Transfusing RBC units generated with either model enhanced alloimmunization rates and alloantibody levels, as compared to controls. Another limitation is the inability to distinguish steadystate and stress-induced reticulocytes to assess their relative contributions to RBC alloimmunization; we are optimizing approaches to define a distinct phenotype for
stress-induced reticulocytes, which would be essential for screening RBC donor units and attributing specific functional outcomes in response to post-transfusion clearance. Although most studies presented herein utilized artificially high reticulocyte frequencies in donor RBC units for initial phenomenological studies, clinically relevant enhanced alloimmune responses were observed with as few as 5% reticulocytes (Figure 6A); it is noteworthy that the average reticulocyte frequency in unmanipulated animals in our studies is approximately 2% (Figure 6A), which is approximately twice the average observed in humans; this is likely due to the shorter lifespan of RBC in mice (~45 days) compared to humans (~100-120 days).45 Thus, while these findings may be clinically relevant to human transfusion, studying reticulocyte-directed immune responses is biologically relevant to many other systems (e.g., neocytolysis,46 malaria,47 production of in vitro universal donor RBC48 etc.).
RBC transfusion is an essential therapy in numerous diseases and is the most common inpatient therapeutic procedure requiring consent, with approximately 11 million transfusions per year in the US.49 Although RBC transfusions can be lifesaving, some patients develop alloantibodies against donor RBC blood group antigens. RBC alloimmunization is clinically signi ficant because it can cause adverse events (e.g., hyperhemolysis) and pose barriers to future care (e.g., organ transplantation). In addition, alloantibody prevention and detection, and mitigation of adverse events, currently requires significant medical, financial, and human resources. As such, identifying risk factors that promote alloimmunization is a high priority and would allow for reallocation of resources to patients most at-risk and provide potential therapeutic interventions. Herein, we demonstrate that the reticulocyte count in donor RBC units can enhance RBC alloimmunization, suggesting that increased reticulocyte frequencies may make RBC units more immunogenic and screening for reticulocyte count and/or lengthening the time between donations for repeat blood donors may reduce alloimmunization events.
Although unrelated to this manuscript, KEH has a spon-
1. Tormey CA, Hendrickson JE. Transfusion-related red blood cell alloantibodies: induction and consequences. Blood. 2019;133(17):1821-1830.
2. Roubinian NH, Reese SE, Qiao H, et al. Donor genetic and nongenetic factors affecting red blood cell transfusion effectiveness. JCI Insight. 2022;7(1):e152598.
3. Rapido F, Brittenham GM, Bandyopadhyay S, et al. Prolonged red cell storage before transfusion increases extravascular
sored research agreement with Alpine Immune Sciences. SLS is a scientific advisory board member (Hemanext, Inc. and Alcor, Inc.), consultant (Tioma, Inc. and Team Conveyer Intellectual Properties), executive director for Worldwide Initiative for Rh Disease Eradication (WIRhE), and CEO for Ferrous Wheel Consultants. All other authors have no conflicts of interest to disclose.
TAT, EAH, SLS and KEH designed the studies and experiments. AQ, CYK, DEG, AM, MT, and FDZ collected and analyzed data from murine experiments. MD and AD performed the metabolomics and proteomics analyses. All authors were involved in the interpretation of data. TAT, EAH, SLS, and KEH wrote the manuscript. All authors contributed to the manuscript and approved the submitted version.
The authors would like to thank Michael Kissner, Director of the Columbia Stem Cell Initiative Flow Cytometry Core, for assistance in experimental design and image acquisition with the ImageStreamX MkII imaging cytometer. Research reported in this publication using the ImageStreamX MkII imaging cytometer was performed in the Columbia University Stem Cell Initiative Flow Cytometry core facility at Columbia University Irving Medical Center and was supported by the Ofce of the Director, National Institutes of Health under Award Number S10OD026845. The content is solely the responsibility of the authors and does not necessarily represent the ofcial views of the National Institutes of Health.
This work was supported by the National Institutes of Health R01HL133325 (to KEH) and R01HL14851 (to SLS) by National Heart, Lung, and Blood Institute.
Original data and protocols are available to other investigators upon request by contacting the corresponding author.
hemolysis. J Clin Invest. 2017;127(1):375-382.
4. Ryder AB, Zimring JC, Hendrickson JE. Factors influencing RBC alloimmunization: lessons learned from murine models. Trans Med Hemother. 2014;41(6):406-419.
5. Evers D, van der Bom JG, Tijmensen J, et al. Red cell alloimmunisation in patients with different types of infections. Br J Haematol. 2016;175(5):956-966.
6. Gibb DR, Liu J, Natarajan P, et al. Type I IFN Is Necessary and
sufficient for inflammation-induced red blood cell alloimmunization in mice. J Immunol. 2017;199(3):1041-1050.
7. Lee JY, Madany E, El Kadi N, et al. Type 1 interferon gene signature promotes RBC alloimmunization in a Lupus mouse model. Front Immunol. 2020;11:584254.
8. Richards AL, Hendrickson JE, Zimring JC, Hudson KE. Erythrophagocytosis by plasmacytoid dendritic cells and monocytes is enhanced during inflammation. Transfusion. 2016;56(4):905-916.
9. Richards AL, Sheldon K, Wu X, Gruber DR, Hudson KE. The role of the immunological synapse in differential dffects of APC subsets in alloimmunization to fresh, non-stored RBCs. Front Immunol. 2018;9:2200.
10. Rönnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Curr Opin Rheumatol. 2013;25(2):248-253.
11. Kim M, Hwang S, Park K, Kim SY, Lee YK, Lee DS. Increased expression of interferon signaling genes in the bone marrow microenvironment of myelodysplastic syndromes. PLoS One. 2015;10(3):e0120602.
12. Madany E, Lee J, Halprin C, et al. Altered type 1 interferon responses in alloimmunized and nonalloimmunized patients with sickle cell disease. EJHaem. 2021;2(4):700-710.
13. Moriconi C, Dzieciatkowska M, Roy M, et al. Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br J Haematol. 2022;198(3):574-586.
14. Caielli S, Cardenas J, de Jesus AA, et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell. 2021;184(17):4464-4479.
15. Mast AE, Szabo A, Stone M, Cable RG, Spencer BR, Kiss JE. The benefits of iron supplementation following blood donation vary with baseline iron status. Am J Hematol. 2020;95(7):784-791.
16. Wong E, Rose M, Berliner N. Disorders of red blood cells. Cecil Essentials of Medicine, Tenth Edition. Elsevier. 2022:489-500.
17. Stryckmans PA, Cronkite EP, Giacomelli G, Schiffer LM, Schnappauf H. The maturation and fate of reticulocytes after in vitro labeling with tritiated amino acids. Blood. 1968;31(1):33-43.
18. Come SE, Shohet SB, Robinson SH. Surface remodelling of reticulocytes produced in response to erythroid stress. Nat New Biol. 1972;236(66):157-158.
19. Come SE, Shohet SB, Robinson SH. Surface remodeling vs. whole-cell hemolysis of reticulocytes produced with erythroid stimulation or iron deficiency anemia. Blood. 1974;44(6):817-830.
20. Noble NA, Xu Q-P, Hoge LL. Reticulocytes II: reexamination of the in vivo survival of stress reticulocytes. Blood. 1990;75(9):1877-1882.
21. Robinson SH, Tsong M. Hemolysis of “stress” reticulocytes: a source of erythropoietic bilirubin formation. J Clin Invest. 1970;49(5):1025-1034.
22. Carden MA, Fasano RM, Meier ER. Not all red cells sickle the same: contributions of the reticulocyte to disease pathology in sickle cell anemia. Blood Rev. 2020;40:100637.
23. Sawadogo D, Tolo-Dilkébié A, Sangaré M, Aguéhoundé N, Kassi H, Latte T. Influence of the clinical status on stress reticulocytes, CD36 and CD49d of SSFA 2 homozygous sickle cell patients followed in Abidjan. Adv Hematol. 2014;2014:273860.
24. Desmarets M, Cadwell CM, Peterson KR, Neades R, Zimring JC. Minor histocompatibility antigens on transfused leukoreduced units of red blood cells induce bone marrow transplant rejection in a mouse model. Blood. 2009;114(11):2315-2322.
25. Bandyopadhyay S, Brittenham GM, Francis RO, Zimring JC, Hod EA, Spitalnik SL. Iron-deficient erythropoiesis in blood donors and red blood cell recovery after transfusion: initial studies
with a mouse model. Blood Trans. 2017;15(2):158-164.
26. Kim CY, Johnson H, Peltier S, et al. Deuterated linoleic acid attenuates the RBC storage lesion in a mouse model of poor RBC storage. Front Physiol. 2022;13:868578.
27. Qiu A, Miller A, Zotti FD, et al. FcγRIV is required for IgG2c mediated enhancement of RBC alloimmunization. Front Immunol. 2022;13:972723.
28. Richards AL, Howie HL, Kapp LM, Hendrickson JE, Zimring JC, Hudson KE. Innate B-1 B cells are not enriched in red blood cell autoimmune mice: importance of B cell receptor transgenic selection. Front Immunol. 2017;8:1366.
29. Richards AL, Qiu A, Zotti FD, et al. Autoantigen presentation by splenic dendritic cells is required for RBC-specific autoimmunity. Transfusion. 2021;61(1):225-235.
30. Nemkov T, Reisz JA, Gehrke S, Hansen KC, D'Alessandro A. Highthroughput metabolomics: isocratic and gradient mass spectrometry-based methods. Methods Mol Biol. 2019;1978:13-26.
31. Pang Z, Chong J, Zhou G, et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49(W1):W388-W96.
32. Thomas T, Stefanoni D, Dzieciatkowska M, et al. Evidence of structural protein damage and membrane lipid remodeling in red blood cells from COVID-19 patients. J Proteome Res. 2020;19(11):4455-4469.
33. Shetlar MD, Hill HA. Reactions of hemoglobin with phenylhydrazine: a review of selected aspects. Environ Health Perspect. 1985;64:265-281.
34. Youssef L, Rebbaa A, Pampou S, et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood. 2018;131(23):2581-2593.
35. Janciauskiene S, Vijayan V, Immenschuh S. TLR4 signaling by heme and the role of heme-binding blood proteins. Front Immunol. 2020;11:1964.
36. Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol. 2020;30(11):892-903.
37. Whitaker B, Rajbhandary S, Kleinman S, Harris A, Kamani N. Trends in United States blood collection and transfusion: results from the 2013 AABB Blood Collection, Utilization, and Patient Blood Management Survey. Transfusion. 2016;56(9):2173-2183.
38. Kiss JE, Vassallo RR. How do we manage iron deficiency after blood donation? Br J Haematol. 2018;181(5):590-603.
39. Schotten N, Pasker-de Jong PCM, Moretti D, et al. The donation interval of 56 days requires extension to 180 days for whole blood donors to recover from changes in iron metabolism. Blood. 2016;128(17):2185-2188.
40. Hosseini H, Li Y, Kanellakis P, et al. Phosphatidylserine liposomes mimic apoptotic cells to attenuate atherosclerosis by expanding polyreactive IgM producing B1a lymphocytes. Cardiovasc Res. 2015;106(3):443-452.
41. Baumgarth N. How specific is too specific? B-cell responses to viral infections reveal the importance of breadth over depth. Immunol Rev. 2013;255(1):82-94.
42. Parra D, Rieger AM, Li J, et al. Pivotal advance: peritoneal cavity B-1 B cells have phagocytic and microbicidal capacities and present phagocytosed antigen to CD4+ T cells. J Leukoc Biol. 2012;91(4):525-536.
43. Liu J, Wang Y, Xiong E, et al. Role of the IgM Fc receptor in immunity and tolerance. Front Immunol. 2019;10:529.
44. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20(1):34-50.
45. Krzyzanski W, Brier ME, Creed TM, Gaweda AE. Reticulocyte-
based estimation of red blood cell lifespan. Exp Hematol. 2013;41(9):817-822.
46. Mairbäurl H. Neocytolysis: How to get rid of the extra erythrocytes formed by stress erythropoiesis upon descent from high altitude. Front Physiol. 2018;9:345.
47. Lim C, Pereira L, Saliba KS, et al. Reticulocyte preference and stage development of Plasmodium vivax isolates. J Infect Dis. 2016;214(7):1081-1084.
48. Pellegrin S, Severn CE, Toye AM. Towards manufactured red blood cells for the treatment of inherited anemia. Haematologica. 2021;106(9):2304-2311.
49. Jones JM, Sapiano MRP, Mowla S, Bota D, Berger JJ, Basavaraju SV. Has the trend of declining blood transfusions in the United States ended? Findings of the 2019 National Blood Collection and Utilization Survey. Transfusion. 2021;61 (Supple 2)S1-10.
Miren Lasaga,1* Paula Río,2,3,4* Amaia Vilas-Zornoza,5,6* Nuria Planell,1 Susana Navarro,2,3,4 Diego Alignani,7 Beatriz Fernández-Varas,3,8 Daniel Mouzo,1 Josune Zubicaray,9 Roser M. Pujol,3,1 0,1 1 Eileen Nicoletti,12 Jonathan D. Schwartz,12 Julián Sevilla,3,9 Marina Ainciburi,5,6 Asier UllateAgote,5,6 Jordi Surrallés,3 10 11 Rosario Perona,3,8,1 3 Leandro Sastre,3,8 Felipe Prosper,5,6# David Gomez-Cabrero,1, 14 15 # and Juan A. Bueren2,3,4#
1Translational Bioinformatics Unit, Navarrabiomed, Universidad Pública de Navarra (UPNA), IdiSNA, Pamplona, Spain; 2Hematopoietic Innovative Therapies Division, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), Madrid, Spain; 3Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Madrid, Spain; 4Instituto de Investigaciones Sanitarias, Fundación Jiménez Díaz, Madrid, Spain; 5Area de Hemato-Oncología, Centro de Investigación Médica Aplicada (CIMA), and Servicio de Hematologia y Terapia Celular, Clínica Universidad de Navarra, IDISNA, Pamplona, Spain; 6Centro de Investigación Biomédica en Red de Cáncer, CIBERONC, Madrid, Spain; 7Flow Cytometry Core, CIMA, Universidad de Navarra, Pamplona, Spain; 8Instituto de Investigaciones Biomédicas Alberto Sols, CSIC/UAM, Madrid, Spain; 9Hemoterapia y Hematología Pediátrica, Fundación para la Investigación Biomédica, Hospital Infantil Universitario Niño Jesús, Madrid, Spain; 10Departmento de Genética y Microbiología, Universitat Autónoma de Barcelona, Barcelona, Spain; 11Fundación Instituto de Investigación del Hospital de la Santa Creu y Sant Pau, Barcelona, Spain; 12Rocket Pharmaceuticals Inc., New York, NY, USA; 13Instituto de Salud Carlos III, Madrid, Spain; 14Biological and Environmental Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia and 15Bioscience Program, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
*ML, PR and AV-Z contributed equally as first authors. #JAB, DG-C and FP contributed equally as senior authors.
Correspondence: Juan A. Bueren juan.bueren@ciemat.es
David Gomez-Cabrero david.gomezcabrero@kaust.edu.sa
Felipe Prosper fprosper@unav.es
Received: November 16, 2022.
Accepted: March 29, 2023.
Early view: April 6, 2023.
htps://doi.org/10.3324/haematol.2022.282418
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Clinical trials have shown that lentiviral-mediated gene therapy can ameliorate bone marrow failure (BMF) in nonconditioned Fanconi anemia (FA) patients resulting from the proliferative advantage of corrected FA hematopoietic stem and progenitor cells (HSPC). However, it is not yet known if gene therapy can revert affected molecular pathways in diseased HSPC. Single-cell RNA sequencing was performed in chimeric populations of corrected and uncorrected HSPC co-existing in the BM of gene therapy-treated FA patients. Our study demonstrates that gene therapy reverts the transcriptional signature of FA HSPC, which then resemble the transcriptional program of healthy donor HSPC. This includes a down-regulated expression of TGF-β and p21, typically up-regulated in FA HSPC, and upregulation of DNA damage response and telomere maintenance pathways. Our results show for the first time the potential of gene therapy to rescue defects in the HSPC transcriptional program from patients with inherited diseases; in this case, in FA characterized by BMF and cancer predisposition.
The potential of gene therapy (GT) to correct a variety of human inherited diseases, including primary immunodeficiencies and hemoglobinopathies, has been well demonstrated in previous clinical studies.1 In addition, we have recently shown that lentiviral-mediated GT can also enable correction of more complex diseases, such as Fanconi anemia (FA), in which marked phenotypic defects are already evident in the self-renewing hematopoietic stem cells
(HSC). As previously reported, the infusion of corrected CD34+ cells in non-conditioned FA patients resulted in a marked hematopoietic stem and progenitor cell (HSPC) proliferative advantage, which facilitates the progressive increase in the number of gene-corrected cells in the bone marrow (BM) and peripheral blood (PB).2
Despite significant advances in the field of GT, the full potential of this therapeutic modality will depend on its capacity to re-establish the molecular circuits and the functional potential of diseased cells. This is even more rel-
Figure 1. Experimental design of scRNAseq analyses performed in Fanconi anemia group A patients 2-5 years afer lentiviralmediated gene therapy. Four Fanconi anemia group A (FA-A) patients who had been treated with ex vivo lentiviral-mediated gene therapy in the absence of conditioning were included in this study. At 2-5 years post gene therapy, these patients harbored a chimeric population of corrected and uncorrected hematopoietic stem and progenitor cells (HSPC) in their bone marrow (BM). Aliquots of BM CD34+ cells from these patients were sorted and processed for single-cell RNA-seq. Bioinformatic analyses comparatively investigated changes in the transcriptional program of corrected versus uncorrected HSPC, co-existing in each of the gene therapy-treated patients. HSC: hematopoietic stem cell.
evant in syndromes such as FA, characterized by DNA repair defects resulting in progressive accumulation of DNA damage and additional molecular responses triggered by a defective FA/BRCA pathway. In patients treated in the FANCOLEN-I clinical trial, the presence of a chimeric population of corrected and uncorrected FA HSPC, none of which had been exposed to any conditioning agent, has allowed us to comparatively investigate the differential molecular pathways that characterize each of these populations co-existing in the BM of these patients (Figure 1). This approach allowed us to demonstrate that lentiviralmediated GT not only mediates a progressive engraftment of gene-corrected HSPC in FA patients, but also results in the reprogramming of the transcriptional signature of FA HSPC, which then resembles those of healthy HSPC, thus accounting for the phenotypic correction of FA HSPC.
Fanconi anemia patients included in this study (patients FA02002, FA-02004, FA-02006 and FA-02008) were Fanconi anemia group A (FA-A) patients enrolled in the FANCOLEN1 gene therapy trial (FANCOLEN-1; clinicaltrials.gov NCT03157804; European Clinical Trials Database 2011006100-12) and were 5, 7, 6 and 3 years old, respectively, at the time of transduced cell infusion. Patients complied with all relevant ethical regulations approved by the ethics committees at the Hospital Vall d’Hebron in Barcelona and the Hospital del Niño Jesús in Madrid.
Patients were infused with autologous CD34 + cells after transduction with the therapeutic PGK-FANCA.Wpre* LV.3 Recent clinical data have shown that although no conditioning was given to these patients prior to cell infu-
sion, a progressive engraftment of corrected cells took place over time, implying the presence of a chimeric population of corrected and uncorrected cells in their hematopoietic tissues. 2 For the participation of healthy donors (HD) in this study (median age 20 years), BM aspiration was performed after informed consent. The study was approved by the clinical research ethics committee of the Clínica Universidad de Navarra.
Bone marrow cells from GT-treated patients were obtained in the course of routine follow-up studies of the GT trial and as part of additional exploratory studies. Samples used in these studies were obtained 5 years (FA-02002), 4 years (FA-02004), 3 years (FA-02006), and 2 years (FA-02008) after infusion of transduced CD34+ cells, respectively. Patients FA-02004 and FA-02008 had been treated with eltrombopag to stimulate hematopoiesis 12 and 6 months prior to the evaluation of BM cells, respectively. All samples were processed immediately after BM aspiration.
For the purification of CD34+ cells, erythrocytes were lysed with ammonium chloride lysis solution (0.155 mmol/L NH4Cl + 0.01 mmol/L KHCO3 + 10 4 mmol/L EDTA), washed using PBS (Gibco) + 0.2%BSA (10%) + 2% PenStrep (Gibco) and stained using CD45 APC (clone 2D1; Biolegend) and CD34 PECY7 (clone 4H11; eBiosciences) for 30 minutes at 4°C. DAPI was used at a concentration of 1 μg/mL as a viability marker. CD34+ cells were then sorted in a BD INFLUX™ (BD Biosciences) or BD FACSAria II (BD Biosciencs), as previously shown.2 Purified CD34+ cells were directly used for scRNA seq analysis and small aliquots stored at -80°C for vector copy number analysis.
The diepoxybutane (DEB)-induced chromosomal instabil-

ity test was carried out in PB T cells from the FA patients prior to and in the long-term after infusion of corrected cells. After 24 hours (h) of culture, PB cells were incubated in the absence or the presence of DEB (0.1 μg/mL; Sigma-Aldrich), and 46 h later colcemid was added (Gibco; 0.1 μg/mL). A further two hours later, metaphase spreads were obtained and stained with Giemsa. Fifty metaphases were analyzed from DEB-treated and 25 metaphases from unexposed cultures in a Zeiss Imager M1 microscope coupled to a computer-assisted metaphase finder (Metasystems). Criteria for the determination and quanti fi cation of chromosome breakage have been described previously.4
Additional information on patients and study methods is available in the Online Supplementary Appendix
Gene therapy modifies the hematopoietic stem and progenitor cell transcriptional program in Fanconi anemia patients
Bone marrow CD34+ cells from four FA-A patients (patients FA-02002, FA-02004, FA-02006, and FA-02008) who had respectively been treated by gene therapy (GT) 5, 4, 3, and 2 years previously, were purified and analyzed by single-cell RNA sequencing (scRNAseq) (Figure 1). Quantitative polymerase chain reaction (qPCR) analyses from these samples showed the presence of 0.77 vector copy numbers (VCN) per cell in the BM of patient 02002, and 0.45, 0.26, and 0.29 VCN per cell in purified CD34+ cells in patients FA-02004, FA-02006, and FA-02008, respectively (see Methods). Since the average VCN per corrected cell was of 1.0 copies,2 the proportion of corrected cells in these patients is estimated to be 77%, 45%, 26%, and 29%, respectively, revealing the chimeric nature of the HSPC populations present in their BM.
CD34+ cells from FA patients were classified according to transcriptional profiles described previously,5,6 which identified 12 different HSPC clusters that corresponded to primitive HSC and more committed lympho-hematopoietic progenitor cells. (See analyses from patient FA-02006 in Figure 2A, and patients FA-02002, FA-02004 and FA-02008 in Online Supplementary Figure S1A-D). To investigate the impact that GT had in the transcriptional program of FA HSPC, CD34+ cell subpopulations from GT-treated FA patients were classified as FANCA+ and FANCA-, based on the expression of FANCA by scRNAseq (see Methods). Importantly, we observed that cells with reads mapping to the poly-A sequence of the viral vector, a region only present in gene therapy-treated cells, were enriched for FANCA+ cells (P<0.001 for all patients). As a result, despite the sparsity of single-cell data, we concluded that gene-corrected cells are enriched in the FANCA+ set.
Next, we observed that, in most of the patients, the presence of FANCA+ cells was evident in all HSPC populations (Figure 2B, C; see details for low prevalent cells in Online Supplementary Figure S1E). No significant differences in the proportion of FANCA+ cells present in these subpopulations were observed among the four patients (Online Supplementary Table S1). In addition, a wide range of FANCA expression was observed in the samples analyzed, although higher expression levels were observed in most HSPC subpopulations from patient FA02002 compared with the other patients (Figure 2D).
To investigate the impact that ectopic FANCA expression had in the transcriptional program of HSPC, we performed a differential expression analysis between FANCA+ and FANCA- HSPC present in each GT-treated patient. The ectopic expression of FANCA up-regulated a high number of genes compared to the number of genes that were down-regulated (Figure 2E). As expected, genes with statistically differential expression corresponded to subpopulations with a higher representation (i.e., CD34 + cells committed to the monocyte lineage). Genes with significantly up-regulated and down-regulated expression (P<0.001) are shown in Figure 2E. Remarkably, most of the up-regulated and downregulated genes in FANCA+ versus FANCA- cells showed the same expression pattern in each of the analyzed patients (Figure 2E). To select the genes with a robust differential expression between FANCA + versus FANCAHSPC, we considered those genes that showed a significant differential expression (abs(logFC)>0.25 and adjusted P-value <0.05) in at least one cell type and in at least three patients. In addition, changes in gene expression should be in the same direction in all the four patients. Based on these criteria, a total number of 152 differentially expressed genes were identified ( Online Supplementary Table S2).
Because our analysis is based on the enrichment in FANCA+ cells of gene-corrected cells, we investigated the fingerprint identified when comparing two extreme groups of cells, aiming to characterize the difference between “highly enriched gene-therapy cells (G1) versus non-gene-therapy cells (G2)”. The first group, G1, is enriched with gene-corrected cells by including cells with at least one read mapping to: (i) FANCA; (ii) poly-A sequence of the viral vector; and (iii) PGK1. The second group, G2, is enriched with the opposite: no reads mapping to any of the previous targets. When comparing those two reduced groups of cells per sample, the differences identified by the fingerprint were also observed (Online Supplementary Figure S2).
Taken together, data obtained in these analyses indicate that the ectopic expression of FANCA induced long-term reproducible changes in the gene expression program of HSPC from FA-A patients.

Gene therapy reprograms the transcriptional signature of Fanconi anemia hematopoietic stem and progenitor cells towards the transcriptional profile characteristic of healthy hematopoietic stem and progenitor cells
Next, we investigated whether changes in the transcriptional program of gene-corrected FA-A HSPC resembled profiles
Figure 2. scRNAseq analysis of corrected and uncorrected hematopoietic stem and progenitor cells (CD34+ cells) from Fanconi anemia group A patients 2-5 years afer lentiviral-mediated gene therapy. (A) UMAP plot showing the clustering analysis for BM CD34+ cells from a Fanconi anemia group A (FA-A) patient previously treated by gene therapy (FA-02006 patient as an example; see Online Supplementary Figure S1 for patients FA02002, FA-02004 and FA-02008). A total of 12 clusters were identified, spanning the different hematopoietic stem and progenitor cell (HSPC) CD34+ subpopulations. Identified clusters include an HSC cluster (hematopoietic stem cell; brown). Clusters with megakaryocytic-erythroid identity include MEP (megakaryocyte-erythroid progenitor; purple), erythroid (erythroid progenitor; pink), and basophils (basophil progenitor; light pink). Clusters with lympho-myeloid identity include LMPP (lymphoidprimed multipotent progenitor; light blue), Cycling-LMPP (blue), CLP (common lymphoid progenitor; orange), GMP1 and GMP2 (granulocyte-monocyte progenitor; light green and green), monocytes (monocyte progenitor; red), DC (dendritic cell progenitor; nude), and PreB (B-cell progenitor; light purple). (B) Same UMAP as shown in (A), highlighting the distribution of FANCA+ cells (FANCA mRNA detectable; red) versus FANCA- cells (FANCA mRNA levels are below detection limit; blue). (C) Barplot showing the total number of cells in the different HSPC CD34+ populations corresponding to the four gene therapy-treated patients. In each case, the number of FANCA+ (red) and FANCA(blue) cells is shown. (D) Boxplot of integrated and normalized FANCA gene expression in FANCA+, depicted by cell type and FA individual (N=3). (E) Barplot representation of the differential expression analysis between FANCA+ and FANCA- for each FA patient (N=4) and for each of the HSPC CD34+ populations. In each case, the number of up-regulated and down-regulated genes is shown. Up-regulated genes were defined as logFC>0.25 (orange), up-regulated and significant genes were defined as a logFC>0.25 and adjusted P-value <0.05 (red), down-regulated genes were defined as a logFC<-0.25 (light blue) and down-regulated and significant genes were defined as logFC<-0.25 and adjusted Pvalue <0.05 (dark blue). The number of genes with the same behavior (up-regulated or down-regulated) in the four individuals is shown below each HSPC (labeled as “shared”).
characteristic of healthy donor (HD) HSPC. To this end, scRNAseq data from GT-treated FA CD34+ cells was compared with those obtained from HD CD34+ cells. The ectopic expression of FANCA (FANCA+ cells) was detected in the different CD34+ cell clusters (Figure 3A and Online Supplementary Figure S3). In addition, FANCA expression levels in the

Figure 3. Comparisons of the gene expression signature between corrected and uncorrected hematopoietic stem and progenitor cells co-existing in gene therapy treated Fanconi anemia patients. (A) Left, large panel: UMAP plot showing the clustering analysis of CD34+ bone marrow (BM) cells after integration of data from a gene therapy-treated Fanconi anemia group A (FA-A) patient and a healthy donor (HD). (FA02006 is included as a representative example; see Online Supplementary Figure S3 for the other individuals). Right, small panel: the same UMAP as shown in left panel but highlighting the distribution of HD hematopoietic stem and progenitor cells (HSPC) (yellow), FANCA+ HSPC (red) and FANCAHSPC (blue). Cluster identification as in Figure 1. (B) Boxplot representation of normalized single-cell FANCA expression of FANCA+ cells (red) and HD cells (yellow) by HSPC cluster. For each cell type, differences in the expression levels between the ectopic expression of FANCA from corrected FA CD34+ cells and expression levels corresponding to HD CD34+ cells are shown. *Adjusted P<0.05; **adjusted P<0.01; ***adjusted P<0.001. (C) Results associated to three differential expression contrasts: FANCA+ versus FANCA- HSPC from GT-treated FA patients; HD HSPC versus FANCA- HSPC from GT-treated patients; and HD HSPC versus FANCA+ HSPC from GTtreated patients. Second row shows the twelve different CD34+ cell types; third row shows the sample identification from each of the four GTtreated patients. Up-regulated genes (logFC>0) are shown in orange; those with significant upregulation in red (logFC>0.25 and adjusted P<0.05). Down-regulated genes (logFC<0) are shown in light blue and those with significant downregulation in dark blue (logFC<-0.25 and adjusted P<0.05). Unsupervised hierarchical clustering using Pearson distance and average linkage method was applied for gene classification. Genes included in the heatmap are those that for at least one cell type are identified as differentially expressed (abs(logFC)>0.25 and adjusted P<0.05) in “at least three patients”, and “showing the same direction of the change for the three patients”, when considering the contrast FANCA+ versus FANCA- (N=152; entire list available in Online Supplementary Table S2). FANCA was excluded from the list.

different HSPC subpopulations were highly heterogeneous, not only in the case of GT-treated FA patients, but also in HD (Figure 3B). Despite this heterogeneity, in 8 of the 12 HSPC subpopulation levels of FANCA mRNA were significantly higher in HD HSPC compared with the corresponding values observed in FANCA+ HSPC from GT-treated FA patients. This observation is consistent with the average insertion of one proviral FANCA copy per corrected cell after GT, in comparison with the two copies of WT FANCA per HD cell, and also with the moderate activity of the phosphoglycerate kinase (PGK) promoter that had been selected for the therapeutic vector because of its favorable safety profile.
To investigate if GT reprogrammed the transcriptional signature of FA HSPC towards one corresponding to HD HSPC, additional gene expression analyses were conducted in HD and FA HSPC, focusing on the 152 genes that showed significant expression changes between FANCA+ and FANCA- HSPC in GT-treated patients (Figure 2E and Online Supplementary Table S2). In these studies, three different comparisons were performed in each of the twelve HSPC subpopulations: FANCA+ versus FANCA- HSPC from GT-treated FA patients (GT-FA HSPC); HD HSPC versus FANCA- GT-FA HSPC; and finally, HD HSPC versus FANCA+ GT-FA HSPC (Figure 3C). Significant differences (dark red or blue spots in Figure 3C) were most evident in HSPC subpopulations with a higher representation (i.e., monocyte CD34+ cells; P<0.001) (Figure 2). Regarding the comparison of GT-FANCA+ versus GT-FANCAHSPC, several differentially expressed genes with relevance in FA were identified (Figure 3C). Interestingly, genes involved in functions such as DNA repair or cell cycle were up-regulated in GT-FANCA+ versus GT-FANCA- cells. The gene expression pattern of HSPC from HD was compared with that corresponding to uncorrected HSPC from GT-treated patients (HD vs. GT-FANCA-) (Figure 3C). Strikingly, most of the transcriptional changes observed between these populations resembled changes seen between corrected and uncorrected FA HSPC (Figure 3C; see Online Supplementary Table S3 for detailed analyses per cell type using two complementary statistical analyses). Finally, in contrast to the above-mentioned observations, the comparison of the transcriptional program of HD HSPC versus FANCA+ HSPC from GT-treated FA patients (Figure 3C) showed limited changes, most of which were not significant and not related to differences noted when HD or corrected FA HSPC were compared with uncorrected FA HSPC. Overall, these studies demonstrate that lentiviral-mediated GT reverts the gene expression program of FA HSPC, which then acquire an expression pattern that resembles the signature characteristic of healthy HSPC.
Lentiviral-mediated gene therapy reverts molecular pathways characteristic of Fanconi anemia hematopoietic stem and progenitor cells
Having observed that the ectopic expression of FANCA in
FA HSPC reverts the transcriptional signature of these cells to resemble a healthy HSPC signature, we next performed a gene-set enrichment analysis to determine changes in relevant pathways associated with FA. These include pathways related to DNA damage response and repair, cell cycle checkpoint, cell aging, and telomere maintenance (see Methods). An up-regulated expression of several of these pathways was observed when FANCA+ and FANCA- HSPC present in each of the GT-treated patients were compared (Figure 4 and Online Supplementary Table S4). Moreover, an almost identical upregulation of these pathways was observed when HSPC from HD were compared with FANCA- HSPC from GT-treated patients (Figure 4).
A deeper comparative expression analysis of genes involved in cell cycle control was then performed between FANCA+ and FANCA- HSPC from GT-treated patients (Figure 5A). Similar comparisons were also made between HD HSPC and FANCA- HSPC from GT-treated patients (Figure 5A). As shown, the ectopic expression of FANCA was associated with the downregulation of TGF-β in every patient, and also of p21 (CDKNA1) in 2 of the 4 GT-treated FA patients. On the other hand, a number of cyclins and minichromosome maintenance (MCM) genes were up-regulated in corrected versus uncorrected FA HSPC (FANCA+ vs. FANCA- HSPC). Notably, when similar comparisons were performed between HD HSPC and FANCA- HSPC from GT-treated FA patients, almost identical gene expression changes were observed (Figure 5A), indicating that the behavior of cell cycle and DNA checkpoint pathways in corrected FA-A HSPC resembled the pathways characteristic of healthy HSPC.
Since previous studies have shown that p21 participates in the transcriptional repression of different genes of the FA/BRCA pathway,5-8 changes in the expression of FA/BRCA genes were also comparatively investigated in FANCA+ and FANCA- HSPC from GT-treated patients. In addition to FANCA, several other genes participating in the FA/BRCA pathway, including FANCB, FANCI, FANCD2, FANCG, UB2T (FANCT), BRCA2, PALB2, BRCA1 and BRIP1 (FANCJ) were up-regulated in GT-FANCA+ versus GTFANCA- HSPC (Figure 5B). Again, many of these genes were also up-regulated when HD HSPC were compared with uncorrected GT-FANCA- HSPC.
This set of studies thus demonstrates that GT reverts physiologically-relevant molecular pathways that are severely affected in HSPC from FA patients.
In a final set of experiments, we investigated the functional implications associated with the restored transcriptional program of gene-corrected FA HSPC. First, we investigated
if the restored DNA repair pathways had a direct implication in the correction of the hypersensitivity of FA-HSPC to interstrand crosslinking agents such as mitomycin C (MMC). While colony-forming cells (CFC) from the four patients were highly sensitive to MMC prior to the infusion of corrected HSPC (no colonies were generated when cells were
exposed to 10 nM MMC), survival of these cells to MMC was markedly increased 2-5 years after GT (22.3-67.3% of CFC survived in 10 nM MMC) (Figure 6A). Consistent with these studies, the percentage of T cells with DEB-induced aberrant chromosomes was also markedly decreased after GT of FA-A patients (Figure 6B), which in two patients reached
Figure 4. Enrichment score of restored gene expression pathways associated to the ectopic expression of FANCA in hematopoietic stem and progenitor cells from gene therapy-treated Fanconi anemia patients. Dotplot depicts the outcome of the Gene Set Enrichment Analysis for a selected set of pathways. For each patient, paired comparisons between FANCA+ versus FANCA- hematopoietic stem and progenitor cells (HSPC) from gene therapy (GT)-treated Fanconi anemia (FA) patients and healthy donor (HD) HSPC versus FANCA- HSPC are shown. The dotplot depicts the statistical significance (color) and enrichment score (size). The complete name of the pathways with numbers are: (i) telomere maintenance via semi-conservative replication, (ii) positive regulation of G2/M transition of mitotic cell cycle, (iii) negative regulation of G2/M transition of mitotic cell cycle, (iv) double-strand break repair via nonhomologous end joining, (v) double-strand break repair via homologous recombination, and (vi) DNA damage response, detection of DNA damage.

Figure 5. Restored gene expression of key pathways of CD34+ cells commited to the monocyte lineage in corrected hematopoietic stem and progenitor cells from gene therapy-treated Fanconi anemia patients. (A) Up-regulated and down-regulated genes implicated in cell cycle control. The figure represents the logFC associated to two contrasts (see Contrast box): FANCA+ versus FANCAhematopoietic stem and progenitor cells (HSPC) from gene therapy (GT)-treated Fanconi anemia (FA) patients (internal crowns), and healthy donor (HD) HSPC versus FANCA- HSPC from GT-treated patients (external crowns). Each external and internal crown is divided in four parts (see Sample box), each of them representing one FA patient. Up-regulated genes are shown in red and down-regulated genes in blue. (B) Same representation as in (A) showing changes in the expression of genes participating in the FA/BRCA pathway.

levels comparable to those found in HD. Overall, these studies confirm the functional implications associated with the restoration of the DNA repair pathways observed in the scRNAseq analyses (see Figure 4). Finally, since scRNAseq studies showed upregulation of pathways associated with the telomere elongation and stabilization (Figure 4), including RPA3, FEN1, PARP1 and PRKDC (Online Supplementary Figure S4), we studied changes in the telomere length of PB cells from the four FA patients included in this study (Figure 6C). In particular, we evaluated changes that took place from day 15 post infusion (when the proportion of corrected cells was still undetectable) up to 5 years post infusion. As a negative control, changes in the telomere length of PB cells from three FA-A patients with no significant engraftment of corrected cells (VCN<0.008; control FA group) were investigated at comparable time points. While a clear reduction in the telomere length was observed in PB cells from the negative control group, in 2 of the 4 GT-treated patients (patients 02004 and 02006) an elongation trend was observed in PB cells at 4 years post infusion. Interestingly, the comparison of the te-
lomere length of PB cells with HD of the same age indicated that telomeres from GT-treated patients were still below corresponding HD values. These analyses suggest that lentiviral-mediated gene therapy may be associated with the reactivation of telomere maintenance pathways, and thus with the improved preservation of telomeres from corrected hematopoietic cells.

Overall, the results obtained in this study demonstrate for the first time that GT, and in particular the lentiviral-mediated GT in FA patients, reverts the transcriptional program of diseased HSPC, which then display gene expression signatures, molecular pathways and cellular phenotypes that resemble the physiological status of healthy HSPC.
Gene therapy has emerged as a safe and efficient therapeutic option for diverse monogenic disorders affecting the hematopoietic system.1 In a previous study, we showed for
Figure 6. Functional implications associated to the restoration of DNA repair and telomere biology pathways. (A) Survival to mitomycin C (MMC; 10 nM) of bone marrow (BM) colony forming cells (CFC) from Fanconi anemia group A complementation group (FA-A) patients shown in Figures 1-5 prior and after infusion of gene-corrected cells. (B) Percentage of peripheral blood (PB) T cells with chromosomal aberrations induced by diepoxibutane (DEB), prior to and after gene therapy. (C) Analysis of the telomere length of PB cells from FA-A patients shown in Figures 1-5 at 15th day post infusion, when corrected cells were still undetectable, to the 2nd-5th year post infusion of gene-corrected cells. As a negative control, three FA patients with no significant engraftment of genecorrected cells are included. Dashed lines correspond to telomere lengths from healthy donor (HD) age-matched PB cells. Time points of the MMC, DEB and telomere analyses are the same or the closest ones corresponding to the scRNAseq analyses shown in Figures 1-5. Percentages of survival in (A) are deduced from colony counts in cultures grown in the absence of MMC. Dashed lines (A) and (B) correspond to mean values determined in samples from HD. GT: gene therapy; d: days; y: years.
the first time preliminary clinical evidence of safety and efficacy of GT in a more complex genetic disease, such as FA.2 In that clinical study, we demonstrated that the ectopic expression of FANCA reproducibly conferred engraftment and proliferative advantage of corrected HSC in non-conditioned FA-A patients. Nonetheless, the question of whether corrected FA HSPC acquire the transcriptional profile characteristic of healthy HSPC was not addressed in our previous study, nor in any other GT trial of a monogenic disease.
By leveraging scRNAseq profiling, we have extended the conclusions from our previous study towards the evaluation of changes in the transcriptional program of corrected versus uncorrected HSPC that co-exist in the BM of the same patient. These analyses, and also gene expression comparisons performed with respect to HD HSPC, allowed us to demonstrate for the first time that GT not only corrects the expression of the mutated gene (in this case, FANCA), but also reprograms the molecular circuits of diseased FA-A HSPC, which then acquire a normalized gene expression pattern characteristic of healthy HSPC. Importantly, the restored expression of FANCA decreased the expression of two genes with particular implications in FA: TGF-β and p21. Increased mRNA levels of these two genes have been observed in HSPC from FA patients and murine FA models, and are believed to contribute to the BMF characteristic of the disease.9,10 Notably, previous studies have shown that p21 down-regulates USP1 upon exposure to DNA damage, disrupting FANCD2/L mono-ubiquitination and nuclear foci formation.8 Additional studies have also shown that several genes involved in the telomere biology and in the FA/BRCA pathway are down-regulated via the p21/E2F4 pathway.5,7 Although it might be expected that p53 and Myc, which have been shown to be up-regulated in FA patients,9,11 should be also down-regulated in corrected FA HSPC, our data do not support this hypothesis. These unexpected observations could result either from the enhanced expression of cell cycle genes in corrected FA HSPC, with post-transcriptional regulatory mechanisms involved in p53 expression, or also from the moderate BMF status of the FA patients enrolled in the FANCOLEN I trial.2 Our study also shows that several defective pathways implicated in the FA cellular phenotype were significantly up-regulated as a result of the ectopic expression of FANCA in FA-A HSPC. Collectively, the restoration of DNA repair-related pathways has enormous implications in FA GT since this accounts for the correction of the hypersensitivity of BM progenitors and PB T cells to genotoxic drugs, such as MMC and DEB. Also of note was the observation of the increased expression of mini-chromosome maintenance (MCM) helicase genes in corrected FA-A HSPC. Previous studies have shown that decreased levels of MCM proteins are associated with HSC hypersensitivity to replication stressors,12
and mediate a relationship between FA and accelerated aging.13 Strikingly, our study shows for the first time that corrected FA-A HSPC up-regulates the expression of MCM, emulating the MCM expression observed in HD HSPC, and suggesting that genetic correction should reduce the replication stress and contribute to the rejuvenation of the HSC from FA patients. Importantly, our study also shows that GT up-regulates several genes associated with telomere biology, and reveals for the first time the maintenance or even elongation of the telomere length in PB cells from FA patients after 2-5 years post GT. This observation contrasts with the progressive telomere attrition previously characterized in FA patients,14,15 and also with the negative FA control group included in our study. Although our study suggests that GT may stabilize the telomeres from gene-corrected hematopoietic cells from FA patients, longer follow-up studies should be performed in these patients to confirm the implications of GT to prevent the progressive telomere attrition characteristic of the disease, or even to extend the telomere length of corrected hematopoietic cells to levels observed in HD cells.
Our gene expression analyses in corrected and uncorrected HSPC from patients with a HSC disease, such as FA, has obvious limitations due to the low number of cells that can be analyzed. In addition, due to the limited number of individuals in whom it was possible to perform these studies, it is currently not feasible to conduct correlation studies between the transcriptomic insights and the clinical co-variates.
Taken together, in this study we show for the first time that GT reverts the transcriptional program and defective molecular pathways in corrected FA HSPC. This observation has a particular impact in FA, and possibly in other HSC diseases characterized by DNA repair defects, since here we demonstrate that GT restores the molecular pathways of FA HSPC towards a signature characteristic of healthy HSPC.
The Hematopoietic Innovative Therapies Division at CIEMAT receives funding from and has licensed the PGK-FANCAWPRE* lentiviral vector to Rocket Pharmaceuticals. JAB and JSe are consultants for Rocket Pharmaceuticals. EN and JDS are employees of Rocket Pharmaceuticals. PR, SN, JS and JAB are inventors on patents filed by CIEMAT, CIBERER and Fundación Jiménez Díaz, and may be entitled to receive financial benefits from the licensing of such patents. All other authors have no conflict of interests to disclose.
ML, PR, AV-Z, NP, SN, DA, DM, BF-V, RMJ, MA, AU-A, RP, LS and D-GC performed the experimental studies and analyzed the data. JSe and JZ provided critical materials. ML, PR, D-GC, FP and JAB wrote the manuscript. EN and JDS reviewed the manuscript. PR, SN, D-GC, FP and JAB de-
signed the study. All authors discussed the results and contributed to the preparation of the manuscript.
The authors thank Ramón García-Escudero for careful reading of the manuscript and helpful discussions. The authors also thank A. de la Cal for co-ordinating the delivery of BM samples from patients with FA. The authors are also indebted to the patients with FA, their families, and clinicians from the Fundación Anemia de Fanconi.
This work was supported by grants from the European Commission’s Seventh Framework Program (HEALTH-F5-2012305421) to the EUROFANCOLEN Consortium (to JB and JSe); from Ministerio de Ciencia, Innovación y Universidades and Fondo Europeo de Desarrollo Regional (RTI2018-097125-BI00 and PID2021-125077OB-C21, to PR and SN); from Gobierno de Navarra, Departamento de Desarrollo Económico y Empresarial (AGATA 0011-1411-2020-000010); from Instituto
1. Ferrari G, Thrasher AJ, Aiuti A. Gene therapy using haematopoietic stem and progenitor cells. Nat Rev Genet. 2021;22(4):216-234.
2. Rio P, Navarro S, Wang W, et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat Med. 2019;25(9):1396-1401.
3. Gonzalez-Murillo A, Lozano ML, Alvarez L, et al. Development of lentiviral vectors with optimized transcriptional activity for the gene therapy of patients with Fanconi anemia. Hum Gene Ther. 2010;21(5):623-630.
4. Castella M, Pujol R, Callen E, et al. Chromosome fragility in patients with Fanconi anaemia: diagnostic implications and clinical impact. J Med Genet. 2011;48(4):242-250.
5. Velten L, Haas SF, Raffel S, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19(4):271-281.
6. Zheng GX, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049.
7. Jaber S, Toufektchan E, Lejour V, Bardot B, Toledo F. p53 downregulates the Fanconi anaemia DNA repair pathway. Nat Commun. 2016;7:11091.
8. Rego MA, Harney JA, Mauro M, Shen M, Howlett NG. Regulation of the activation of the Fanconi anemia pathway by the p21 cyclin-
de Salud Carlos III (ISCIII) and co-financed by FEDER CIBERONC (CB16/12/00489 and CB16/12/00225); Redes de Investigación Cooperativa (TERCEL RD16/0011/0005); “Instituto de Salud Carlos III (ISCIII)/Red Espanola de Terapias Avanzadas RICORS/TERAV” (RD21/0017/0027), supported by European Union-NextGenerationEU and from Consejería de Educación, Juventud y Deporte de la Comunidad de Madrid (AvanCell Project; B2017/BMD3692); from Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III, Spain, grant n. PI20/0335 and co-funded by European Regional Development (FEDER) funds. MA was supported by a PhD Fellowship from the Ministerio de Ciencia, Innovación y Universidades (FPU18/05488). CIBERER and CIBERONC are initiatives of the Instituto de Salud Carlos III and Fondo Europeo de Desarrollo Regional.
Data-sharing statement
Data supporting the findings of this study are available at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18 0536
dependent kinase inhibitor. Oncogene. 2012;31(3):366-375.
9. Ceccaldi R, Parmar K, Mouly E, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11(1):36-49.
10. Zhang H, Kozono DE, O'Connor KW, et al. TGF-β inhibition rescues hematopoietic stem cell defects and bone marrow failure in Fanconi anemia. Cell Stem Cell. 2016;18(5):668-681.
11. Rodríguez A, Zhang K, Färkkilä A, et al. MYC promotes bone marrow stem cell dysfunction in Fanconi anemia. Cell Stem Cell. 2021;28(1):33-47.
12. Flach J, Bakker ST, Mohrin M, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512(7513):198-202.
13. Brosh RM Jr, Bellani M, Liu Y, Seidman MM. Fanconi anemia: a DNA repair disorder characterized by accelerated decline of the hematopoietic stem cell compartment and other features of aging. Ageing Res Rev. 2017;33:67-75.
14. Leteurtre F, Li X, Guardiola P, et al. Accelerated telomere shortening and telomerase activation in Fanconi's anaemia. Br J Haematol. 1999;105(4):883-893.
15. Hanson H, Mathew CG, Docherty Z, Mackie Ogilvie C. Telomere shortening in Fanconi anaemia demonstrated by a direct FISH approach. Cytogenet Cell Genet. 2001;93(3-4):203-206.
1Genome Biology Unit, EMBL, Heidelberg, Germany; 2Department of Molecular Life Sciences, University of Zurich, Zurich, Switzerland; 3SIB Swiss Institute of Bioinformatics, University of Zurich, Zurich, Switzerland; 4Medical Faculty Heidelberg, Heidelberg University, Heidelberg, Germany; 5Molecular Therapy in Hematology and Oncology & Department of Translational Oncology, NCT and DKFZ, Heidelberg, Germany; 6Department of Medicine V, Heidelberg University Hospital, Heidelberg, Germany; 7Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China; 8Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden; 9Clinical Genetics, Karolinska University Hospital, Solna, Sweden; 10Department of Internal Medicine, Division of Hematology, The Ohio State University, Columbus, OH, USA and 11Department of Medical Oncology and Hematology, University Hospital Zurich, Zurich, Switzerland.
*AL and JL contributed equally as first authors.
Correspondence: T. Zenz thorsten.zenz@usz.ch
W. Huber wolfgang.huber@embl.org
Received: September 9, 2022.
Accepted: May 18, 2023.
Early view: May 25, 2023.
htps://doi.org/10.3324/haematol.2022.281869
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Understanding the molecular and phenotypic heterogeneity of cancer is a prerequisite for effective treatment. For chronic lymphocytic leukemia (CLL), recurrent genetic driver events have been extensively cataloged, but this does not suffice to explain the disease’s diverse course. Here, we performed RNA sequencing on 184 CLL patient samples. Unsupervised analysis revealed two major, orthogonal axes of gene expression variation: the first one represented the mutational status of the immunoglobulin heavy variable (IGHV) genes, and concomitantly, the three-group stratification of CLL by global DNA methylation. The second axis aligned with trisomy 12 status and affected chemokine, MAPK and mTOR signaling. We discovered non-additive effects (epistasis) of IGHV mutation status and trisomy 12 on multiple phenotypes, including the expression of 893 genes. Multiple types of epistasis were observed, including synergy, buffering, suppression and inversion, suggesting that molecular understanding of disease heterogeneity requires studying such genetic events not only individually but in combination. We detected strong differentially expressed gene signatures associated with major gene mutations and copy number aberrations including SF3B1, BRAF and TP53, as well as del(17)(p13), del(13)(q14) and del(11)(q22.3) beyond dosage effect.
Our study reveals previously underappreciated gene expression signatures for the major molecular subtypes in CLL and the presence of epistasis between them.
Chronic lymphocytic leukemia (CLL) etiology has been linked to abnormal B-cell receptor (BCR) activation and gene mutations targeting multiple pathways, including DNA damage pathways (TP53, ATM), NOTCH signaling (NOTCH1, FBXW7, MED12)1-6 and the spliceosome (SF3B1).7,8 In addition, the immunoglobulin heavy variable (IGHV) gene mutation status, the result of a physiological mutation and maturation process, reflects the tumor’s cell of origin and is one of the strongest predictors of clinical behavior.9 Several genetic subgroups of CLL are known to show profound
differences in clinical course, presentation and outcome,10,11 although considerable variability remains within subgroups. Gene expression profiling can provide a better understanding of the functional role of mutations and may help dissect disease heterogeneity. Indeed, previous studies of CLL transcriptomes found substantial variability,12-17 however, it has been a surprise how little of that variability could be associated with the genetic subgroups or other properties of the disease. For instance, Ferreira et al 12,13 found only a few robust gene expression changes associated with the major cancer driver mutations of CLL. IGHV mutation status only accounted for 1.5% of the overall variance in
Junyan Lu,1,4* Jennifer Hüllein,1 Tatjana Walther,5 Leopold Sellner,5,6 Bian Wu,5,7 Richard Rosenquist,8,9 Christopher C. Oakes,10 Sascha Dietrich,6 Wolfgang Huber1 and Thorsten Zenz5,11their study. Their study reported two gene expressionbased subgroups, termed C1/C2, as a predictor of clinical outcome independent of the known genetic disease groups. However, a later re-analysis of the data suggested a relation of C1/C2 to sample processing.18 Overall, the relations between prominent genetic events that have significant impact on disease course and the gene expression programs of CLL have remained unclear. Among possible explanations for this scarcity of associations are small sample sizes, confounding effects of multiple cytogenetic abnormalities or technical limitations. More recent studies have thus collected larger cohorts with focus on a particular genetic aberration. Abruzzo et al.15 identified a set of dysregulated and potentially targetable pathways in CLL with trisomy 12. Herling et al 16 developed a 17-gene signature that can identify a subset of treatment-naive patients with IGHV-unmutated CLL (U-CLL) who might substantially benefit from treatment with FCR (fludarabine, cyclophosphamide and rituximab) chemoimmunotherapy. These findings underline the importance of transcriptional changes in CLL. Since they were based on focused studies limited to specific, selected subtypes of CLL, one may expect a more systematic picture and additional insights from a comprehensive RNA sequencing-based survey.
In order to understand the impact of genetic and epigenetic subgroups of CLL on gene expression, we profiled 184 CLL samples using RNA sequencing. After careful control of technical variations, and of possible confounding between genetic variants, we searched for transcriptomic signatures and pathway activity changes associated with the major recurrent genetic alterations in CLL. Furthermore, as a step towards gaining a better understanding of functional interdependencies between mutations in a tumor, we used a quantitative model of genetic interactions to identify nonadditive effects of mutations on gene expression profiles.
Data acquisition
RNA sequencing
We selected 184 CLL patient samples for RNA sequencing. One hundred and twenty-three of these patients were used in a prior study.19 The current study is an extension, designed specifically to increase sample sizes of major molecular subgroups and focus on gene expression. The majority of patients (177 of 184) showed the typical CLL phenotype, and five patients were diagnosed with atypical CLL. Ninety-two patients had undergone prior treatment. Patient characteristics are shown in the Online Supplementary Table S1. Total RNA was isolated from blood samples (CD19+ purified n=161) and sequenced using Illumina HiSeq. Sequenced reads were mapped to the Ensembl human reference genome (Homo sapiens GRCh37.75) using STAR20
version 2.5.2a. Mapped reads were summarized in per gene counts using htseq-count.21 version 0.9.0. Further details are provided in the Online Supplementary Appendix.
Somatic variants
Mutation calls for 66 distinct gene mutations and 22 structural variants were generated through targeted sequencing, whole-exome sequencing and whole-genome sequencing as described previously.19 Statistical analyses were restricted to variants found in ≥5 patients, i.e., to 14 gene mutations (BRAF, NOTCH1, SF3B1, TP53, KRAS, ATM, MED12, EGR2, KLHL6, ACTN2, MGA, NFKBIE, PCLO, XPO1), and nine copy number aberrations (CNA): trisomy 12, del(11)(q22.3), del(13)(q14), del(17)(p13), del(8)(p12), gain(8)(q24), gain(2)(p25.3), del(15)(q15.1), gain(14)(q32) (Online Supplementary Figure S2B). In addition, the somatic hypermutation status of the IGHV and a CLL subtype classification defined by global patterns of CpG methylation level22,23 were recorded. Here, we discuss results for variants with >200 differentially expressed genes detected: four CNA, three gene mutations and IGHV mutation status. The somatic mutation information is available in our online repository: https://github.com/almutlue/transcriptome_cll and summarized in the Online Supplementary Table S2
Drug response profiling
For 113 of 184 samples, drug response profiles were reported in a previous study.24
Statistical analyses were performed using R25 version 3.6. We performed quality controls including batch effect estimation26 (Online Supplementary Figure S1), exploratory data analysis and differential gene expression analysis using the Gamma-Poisson generalized linear modeling (GLM) approach of DESeq2, version 1.16.1.27,28 Genetic interactions were identified by testing for an interaction term in the regression of gene expression data on the two variables IGHV mutation status and trisomy 12 using DESeq2. For the validation study, we analyzed microarray data from Abruzzo et al.15 using the R package limma version 3.50.1.29 Gene set enrichment analysis30 was performed using the R package clusterProfiler31 version 3.12.0. Hallmark and KEGG gene set collections version 4.0 were downloaded from MsigDB.32 Transcription factor target genes sets were downloaded from Harmonizome.33 All P values were adjusted for multiple testing using the method of Benjamini and Hochberg.34 Further details are provided in the Online Supplementary Appendix.
The study was approved by the Ethics Committee Heidelberg (University of Heidelberg, Germany; S-206/2011; S356/2013) and Zurich, Switzerland (2019-01744).
Unsupervised analysis reveals major drivers of gene expression variability
We generated RNA-sequencing data from 184 CLL patient samples. In order to obtain a first overview of patterns of gene expression variability in CLL, we performed an unsupervised clustering analysis based on the 500 most variable genes (Figure 1A). This analysis showed a separation of distinct subgroups that coincided very well with IGHV mutation status/methylation epitype and the presence of trisomy 12. The role of IGHV mutation status and trisomy 12 was also reflected in the number of differentially expressed (DE) genes (>3,000 DE genes) (Figure 1B). A similar separation was seen in a principal component analysis (PCA) (Figure 1C, D). The first principal component, which represented 11% of the variance, was associated with IGHV mutation status, while the second component separated samples based on trisomy 12. These results indicate that these two genetic variables shape gene expression in CLL to a previously underappreciated extent. We also considered a classification of CLL based on global DNA methylation levels into three groups according to previous studies,23,24 a refinement of the binary grouping by IGHV mutation status (Figure 1E). The first principal component arranged the DNA methylation subgroups in the consistent order: low, intermediate and high programmed (LP, IP, HP). These results indicate that even though the three-group classification was discovered using DNA methylation data, it is now also apparent at the level of gene expression. Indeed, the global gene expression patterns shown in Figure 1 imply a further refinement into major groups, namely LP, IP and HP each with and without trisomy 12. The results of our unsupervised clustering analysis differ from those of a previous gene expression study, which also used unsupervised clustering of RNA-sequencing data to find novel subgroups of CLL termed C1/C2, marked by 600 differentially expressed genes and associated with BCR activation and outcome.12 In our data, hierarchical clustering of the samples based on the measurements of these 600 genes only, indeed resulted in two main clusters. However, most of these genes showed low variability across samples and only 26 of them were among the 500 most variable genes (Online Supplementary Figure S2).
We performed differential expression analysis to explore the effect of 23 recurrent genetic aberrations and the IGHV mutation status (Online Supplementary Figure S3). In total, we found six additional variants (besides trisomy 12 and the IGHV status) associated with more than 200 differentially expressed genes. These were del(13)(q14), del(17)(p13), del(11)(q22), and SF3B1, TP53 and BRAF mutations (Figure
1B). Complete tables are provided in the computational analysis transcript. We compared previous findings from the literature for single genetic aberrations with differentially expressed genes in this study.
Mutations in the splicing factor SF3B1 gene showed more than 600 associations. Gene sets enriched in CLL with SF3B1 mutations included “Cytokine-cytokine receptor interaction” and “Phosphatidylinositol signaling system” (Online Supplementary Figure S4A). Among differentially expressed genes, we found the chaperone gene UQCC1 (Online Supplementary Figure S4B), which has already been linked to SF3B1 mutations by differential isoform usage.35 Indeed, a differential exon usage analysis using DEXSeq27 showed that UQCC1 had both differential expression and differential exon usage (Online Supplementary Figure S5A, B; Online Supplementary Table S3). There were also instances of genes that had differential exon usage, but for which no gene level differential expression was detected (Online Supplementary Figure S5C, E). These included BRD9, a tumor suppressor whose splicing has also been reported to be regulated by SF3B1. 36,37 A recent proteomic study in CLL confirmed the downregulation of BRD9 in samples with SF3B1 mutations.37 A potential explanation is that the SF3B1 mutation leads to a mis-spliced version of the BRD9 transcript whose level is not detectably altered in gene level RNA-sequencing analysis but whose translation is impaired. Conversely, there were genes detected by gene level differential expression analysis but not by differential exon usage analysis, including PSD2, SRRM5 and TNXB (Online Supplementary Figure S4C-E).
TP53 mutations are recurrent in CLL and are associated with poor prognosis.1 Differentially expressed genes in samples with TP53 mutation were enriched in “Oxidative phosphorylation” and “p53 signaling pathway” (Online Supplementary Figure S6A). The transcriptional regulator CDK12 is upregulated in TP53-mutated samples (Online Supplementary Figure S6C). In order to understand the overlap of genes deregulated by 17p deletion and p53 mutation we analyzed the overlap between those two variants (Online Supplementary Figure S6B). In total, 76 of 272 differentially expressed genes associated with TP53 mutation were also differentially expressed in samples with del(17)(p13). As del(17)(p13) includes the region of the TP53 gene, the overlap is to be expected. Further associations with TP53 include PGBD2 and HYPK (Online Supplementary Figure S6D, E)).
The second highest number of differentially expressed genes was found in the comparison between IGHV-mutated CLL (M-CLL) and IGHV-unmutated CLL (U-CLL): 3,410 genes. This result is in agreement with the PCA of Figure 1C and shows that IGHV mutation status is the main de-
Figure 1. Gene expression variability in chronic lymphocytic leukemia. (A) Heatmap of the gene expression counts. Samples (columns) are ordered in agreement with the hierarchical clustering based on the 500 most variable genes. Gene (rows) counts are row-centered log-transformed (base 2) and split into the 3 main hierarchical clusters. Immunoglobulin heavy variable (IGHV) gene mutation status, methylation subgroups and trisomy 12 align with the clustering result. (B) Number of differentially expressed genes (adjusted [adj] P values <0.01) for genomic markers in chronic lymphocytic leukemia (CLL). Lighter colors indicate genes located on the same chromosome as the respective genetic lesion (potential dosage effects). (C) IGHV status is associated with the first principal component, which explains 11.3% of the variance. (D) Trisomy 12 is associated with thesecond principal component (PC2), which explains 5.1% of the variance. (E) Methylation subgroups split up along principal component 1 (PC1).

terminant of gene expression variability in CLL. It implies a much larger impact of IGHV status on the transcriptome than previously detected (11.3% variance instead of 1.5%12 explained by the associated principal component in PCA) is in line with the key impact of IGHV mutation status on clinical course and biology of disease.9-11 Our observation is also consistent with recent reports that IGHV status is one of the major determinants of the protein expression landscape in CLL.37,38 Genes previously found to be markers related to IGHV mutation status, including CD38, LPL, ZAP70, SEPT10 and ADAM29,39-41 were also detected in our analysis (Online Supplementary Table S4).
In order to understand which pathways were differentially engaged between U-CLL and M-CLL, we performed gene set enrichment analysis. Differentially expressed genes between IGHV groups were enriched in BCR signaling, T-cell receptor signaling and chemokine signaling pathways (Figure 2A). Within the BCR signaling gene set, we identified cell surface molecules (CD19, CD22, CD81) and NFAT and NF-κB to be downregulated in U-CLL. From the “T-cell receptor signaling” gene set, ZAP70, PAK2 and MAPK12 were upregulated in U-CLL, while IL10 and MAP3K8 were downregulated. Within chemokine signaling pathways, we found downregulation of CXCR3 and CXCR5 in U-CLL, while a set of cytokines (CCL24, CCL25) were upregulated.
IGHV genes were also found among the most differentially expressed genes, but showed heterogeneous expression within the U-CLL and M-CLL groups. As expected, commonly used IGHV genes (IGHV1-69 or IGHV4-34) were associated with U-CLL and M-CLL, respectively. Gene expression showed a strong relation to IG gene usage and its variant’s expression (Figure 2B). These data show that RNA sequencing can be used to assess IG gene usage. Further genes associated with IGHV groups were BCAT1, EGR3 and ZAP70 (Figure 2C-E)
In summary, our data are in line with the major biological role of IGHV mutation status in CLL and provide a resource to identify deregulated pathways in the disease.
Based on the global DNA methylation pattern, the stratification of CLL by IGHV mutation status has been refined by introducing a categorization into LP, IP, and HP programmed samples, which are thought to represent the cell of origin.22,23 Based on gene expression data, we found these three groups along the first principle component. The IP group was placed between the LP and HP groups (Figure 1E). Thus, even though the groups were discovered on the basis of DNA methylation, a strong separation was found on the basis of unsupervised PCA of the gene expression data. Previous analysis of methylation groups in CLL suggested a disease-specific role of the transcription factors EGR, NFAT, AP1 and EGF by establishing aberrant methyl-
ation patterns.22,23 In line with this, we found NFATC1 and EGR1 among genes whose expression patterns were associated with methylation groups (Online Supplementary Figure S7A, B). A detailed analysis of the intermediate methylated subgroup revealed multiple genes including SOX11, that were specific for this subgroup (Online Supplementary Figure S7C). SOX11 is a transcription factor whose expression has been associated with adverse prognostic markers in CLL.43
Expression signature in chronic lymphocytic leukemia with trisomy 12
We identified over 5,000 differentially expressed genes (with adjusted P value <0.05) in CLL with trisomy 12 (Figure 1B). Even though chromosome 12 harbors many upregulated genes, the majority of differentially expressed genes were on other chromosomes and, therefore, cannot be ascribed to a simple gene dosage effect (Figure 3A).
Among the differentially expressed genes, we found numerous genes involved in chemokine signaling such as VAV1 (Figure 3B, C). Chemokine signaling pathways are induced by chemokine binding and activate MAPK signaling.44,45 In line with this, we identified differentially expressed genes enriched in MAPK signaling. We also detected an enrichment for the mTOR-signaling pathway, a known modulator of chemokine signaling46 (Figure 3B). Consistent with previous reports, integrins like ITGAM, ITGB2 and ITGA4 were also upregulated in trisomy 12 samples15 (Online Supplementary Table S4). We also found the immune checkpoint gene CTLA4 (Figure 3D) downregulated in trisomy 12 samples. CTLA4 has previously been linked to CLL and is associated with increased proliferation and tumor progression.47,48 By comparison with published protein expression data, we found the protein expressions of VAV1 and ITGB2 were also strongly upregulated in trisomy 12 CLL (Online Supplementary Figure S8). Chemokine signaling and mTORsignaling pathways in trisomy 12 CLL have also been shown to be upregulated on protein level.38
A known mechanism of tumor cells to escape the immune system is to inhibit tumor-specific T cells, and support the conversion of anti-tumor type 1 macrophages to pro-tumor type 2 macrophages by upregulation of ecto-5′-nucleotidase (NT5E), which is necessary to convert extracellular ATP into adenosine. A previous study on gene expression in trisomy 12 patients inferred NT5E to be an important element in a trisomy 12 expression network model.15 This inference was indirect, as the microarray-based study only quantified selected transcripts, excluding NT5E. Here, we directly observed higher expression of NT5E in trisomy 12 and thus can confirm the hypothesis of Abruzzo et al. 15 (Figure 3E). Another study of Tsagiopolou et al.49 studied epigenetic regulatory elements in CLL with trisomy 12 and found several transcription factors in particular RUNX3, which is a master regulator of gene expression during de-
velopment and oncogene in cancer, to be up regulated. We tested differentially expressed genes in trisomy 12 for enrichment of transcription factor target genes sets and found target genes of RUNX3 to be among the top enriched genes sets (Online Supplementary Figure S9). Altogether, these results confirm and expand on the results of existing studies. They suggest that modulation of MAPK

signaling through chemokine signaling and mTOR signaling are important mechanisms in trisomy 12 tumorigenesis.
Epistatic interaction of immunoglobulin heavy variable gene mutation status and trisomy 12
Epistasis describes a phenomenon where the effect of a genetic variant depends on the presence or absence of
another genetic variant.50 There is almost no data on epistasis between cancer mutations. Because of the large effects of each of these variants individually, we asked
whether and to what extent epistatic interactions existed between IGHV mutation status and trisomy 12. We fit a generalized linear model (DESeq2, see Methods) with main
Figure 3. Gene expression in chronic lymphocytic leukemia with trisomy 12. (A) Role of dosage effect: chromosomal distribution of differentially expressed (DE) genes in chronic lymphocytic leukemia (CLL) with trisomy 12. Chromosome 12 has the highest number of DE genes, but the majority of DE genes is distributed across all chromosomes and cannot be ascribed to a dosage effect. (B) DE genes in enriched KEGG pathways of trisomy 12. (C-E) Normalized gene counts of VAV1, CTLA4 and NT5E

and interaction effects for these co-variates. Significant interactions were detected for 893 genes at 10% false discovery rate (FDR) (Figure 4A; Online Supplementary Table S5). For these genes, the effect of trisomy 12 was different in U-CLL and M-CLL. We observed four distinct types of epistatic interactions51,52 and classified the 893 genes into according categories: synergy, where the samples with both variants, i.e., M-CLL samples with trisomy 12, showed a stronger up-regulation than expected from the single variants; bufering, when the presence of both variants led to a strong reduction of gene expression; inversion, when the effects in the single variants were reversed in the double variant; suppression, when a strong expression change (up- or downregulation) of a gene in a single variant was absent in samples with both variants (Figure 4B). Figure 4C-F shows the count data for exemplary genes. EMAPlike 6 (EML6) is upregulated in all trisomy 12 cases, but this effect is on average about 1,000 times stronger in M-CLL patients compared to U-CLL patients (synergy) (Figure 4C). While fibroblast growth factor 2 (FGF2) is consistently upregulated in trisomy 12 cases with U-CLL, this effect is reversed in M-CLL (suppression) (Figure 4D). SYBU shows an inverse expression pattern in M-CLL cases with trisomy 12 compared to U-CLL cases with trisomy 12 (Figure 4E). Lymphoid enhancer binding factor 1 (LEF-1) shows a stable gene expression across samples and has been suggested and tested as a clinical marker for CLL.53 While the presence of either one of the genetic variants (IGHV-M or trisomy 12) does not seem to have an effect, samples with both of them express consistently lower levels of LEF1 (buffering) (Figure 4F). These effects cannot be explained by looking at the genotypes independently or by modeling an additive effect of the variants.
We next asked about the biological functions underlying the epistatic interaction between IGHV status and trisomy 12. We used gene set enrichment analysis on the combined set of all 893 genes. The overall set of genes with an epistasis expression pattern was enriched in TNF α signaling via NF-κB, MYC targets, IL2/STAT5 signaling and G2M checkpoint pathway (Figure 4G). A recent study linked NFκB expression with reduced levels of B-cell signaling.54 Both IGHV status and trisomy 12 are known to affect BCR signaling, but the above data may suggest that signaling in IGHV-mutated CLL is mediated by BCR plus additional survival signals. In order to further investigate these findings, we used an independent cohort of samples from 47 patients with known trisomy 12 and IGHV status assayed on an Illumina microarray with 47,231 probes15 and again screened for epistatic interaction by testing for interaction effects in the linear model. In line with our results, we found multiple probes (100 probes with adjusted P value ≤0.17) that show the same epistatic interaction patterns in their expression for each of the four types of epistasis (Online Supplementary Figure S10A). Furthermore, we investi-
gated the expression of the above-mentioned genes with epistatic interaction (EML6, FGF2, SYBU, LEF-1) in particular (Online Supplementary Figure S10B-E). We found significant epistatic expression patterns for FGF2, SYBU and could assign both genes to the same epistasis groups as in our cohort. While there was no significant epistatic interaction in the expression pattern of EML6 and LEF-1 their expression trends were in line with the expression of the epistatic groups (synergy, buffering) these genes were assigned to in our cohort. The protein expressions of two of those genes, FGF2 and LEF-1, could also be detected in our proteomics dataset38 and were found to have epistatic interactions (Online Supplementary Figure S11).
The epistatic interaction between immunoglobulin heavy variable gene mutation status and trisomy 12 affects ex vivo drug response in chronic lymphocytic leukemia
Ex vivo sensitivity to drugs reflects pathway dependencies of CLL cells. We asked whether the epistatic interaction between IGHV mutation status and trisomy 12 on expression level also affected the drug response phenotype. Previously, we measured the ex vivo sensitivity, as measured by cell viability, of our 184 CLL samples towards 63 compounds.24 Again using two-way ANOVA with an interaction term, we identified six drugs for which there was a significant (10% FDR) interaction between IGHV mutation status and trisomy 12 (Figure 5). For four drugs, namely, vorinostat, NU7441, fludarabine and AZD7762, we observed a suppression effect, where trisomy 12 led to increased drug sensitivity in the U-CLL, but not the M-CLL samples. For the other two drugs, chaetoglobosin A and BIX02188, the samples with trisomy 12 showed decreased sensitivity particularly in M-CLL, but not in U-CLL. The four drugs with the suppression phenotype directly or indirectly target DNA: NU7441 inhibits DNA-dependent protein kinase (DNAPK) and, therefore, potentiate DNA double-strand breaks;55 AZD7762 is a checkpoint kinase (CHEK) inhibitor, which can impair DNA repair and increases cell death;56 fludarabine directly inhibits DNA synthesis and disrupt cell cycle;57 vorinostat, a histone deacetylase (HDAC) inhibitor, was also reported to induce reactive oxygen species and DNA damage in leukemia cells.58 As 42 of 161 patients in the drug screening dataset were treated prior to the acquisition of the samples, we also repeated these analyses only in untreated patient samples. We found consistent results (Online Supplementary Figure S12), suggesting no substantial effect of prior treatment on our findings.
We analyzed 184 CLL transcriptomes and identified gene expression signatures for the most prevalent genetic aberrations. We show that these can be used to capture under-
Figure 4. Mixed epistasis of trisomy 12 and immunoglobulin heavy variable gene mutation status. (A) Heatmap showing the expression of genes affected by the epistatic interactions between trisomy 12 and immunoglobulin heavy variable (IGHV) genemutated chronic lymphocytic leukemia (M-CLL) (adjusted [adj] P value <0.1). (B) Schematic classification of epistasis. (C-F) Types of gene expression epistasis: EML6 (synergy), FGF2 (suppression), SYBU (inversion), LEF1 (buffering). (G) Enriched pathways in genes with epistasis expression pattern.

Figure 5. Ex vivo drug response phenotype related to the epistatic interaction between immunoglobulin heavy variable gene status and trisomy 12. (A) Heatmap plot showing the responses of chronic lymphocytic leukemia (CLL) samples (rows) towards the 6 drugs (columns) for which there was a significant interaction between immunoglobulin heavy variable (IGHV) gene mutation status and trisomy 12. The coloring encodes the column-wise z-scores of sample viabilities after drug treatment. (B) Boxplots of the viabilities (normalized to dimethyl sulfoxide [DMSO] controls) of CLL samples, stratified by their IGHV and trisomy 12 status, towards the 6 drugs shown in the heatmap. The IGHV-unmutated (IGHV-U) and IGHV-mutated (IGHV-M) groups contain unmutated-CLL (U-CLL) and mutated-CLL (M-CLL) samples, respectively, without trisomy 12. The IGHV-U+tri12 and IGHV-M+tri12 groups contain U-CLL and M-CLL samples, respectively, with trisomy 12.
lying pathways. The IGHV mutation status, the three DNA methylation subgroups and trisomy 12 were found as the main drivers of gene expression variability in CLL. This is evident both in unsupervised analyses (clustering, PCA) and in supervised differential-expression analysis. We revealed a much higher impact of IGHV mutation status on the CLL transcriptome than previous reports.12 A further refinement of the disease stratification by IGHV status is provided by the three DNA methylation subgroups. We identified genes whose expression follows an apparent continuum from LP, IP to HP in CLL. This finding supports the biological relevance of these three groups and suggests that although they were discovered based on DNA methylation, they are similarly evident at the level of gene expression. These results highlight the potential of gene expression profiling to increase our understanding of CLL.

In order to avoid potential confounding effects of multiple aberrations, other studies have focused on samples with single abnormalities. While this approach was successfully used to understand trisomy 12 specific gene expression, it is limited to only a subset of the disease and to selected variants.15 Here, we demonstrate an improved approach that employs differential expression analysis with multivariate generalized linear models and blocking factors, and that is able to use the full range of CLL and to investigate a larger number of genetic aberrations.
Genetic interactions, or epistasis, where the effect of one mutation depends on the presence or absence of another mutation, is a well known concept in genetics. However, there is surprisingly little data on such phenomena in cancer. Hence, we used the opportunity to study the combinatorial effects of trisomy 12 and the IGHV mutation
status on gene expression variability. We identified numerous genes (~900) whose expression depended on the presence or absence of these two aberrations in a non-additive manner. We categorized these genes into four categories: buffering, synergy, suppression and inversion, each of which contained dozens to hundreds of genes. This means that there is not a single, simple epistasis phenotype between these two aberrations, but a complex, “mixed” pattern. Mixed epistasis of the gene expression phenotypes of pairs of gene alterations has been described in a yeast model system.52 We employed the genetic interactions between IGHV mutation status and trisomy 12 on gene expression phenotypes to identify pathways, including TNF α signaling via NF-κB and the G2M checkpoint pathway, that mediate the effects of these variants. We reproduced this finding in an independent cohort of 47 patients.15
Our results raise the question whether the pathobiology of trisomy 12 may be different between U-CLL and M-CLL. Based on previous studies, the additional copy of chromosome 12 in CLL cells seems to directly enhance BCR signaling, especially in U-CLL samples: for example, Abruzzo et al. found that NFAT is overexpressed in trisomy 12 CLL, indicating the hyperactivation of calcineurin/NFAT signaling, which is central to BCR signaling.15 On proteomic levels, our previous study suggested that proteins upregulated in trisomy 12 CLL are enriched in BCR signaling pathway.38 In addition, genes located on chromosome 12 and upregulated in trisomy 12 CLL can also give rise to proteins that form protein complexes with BCR components.38 This evidence indicates that trisomy 12 status directly or indirectly modulates BCR signaling. As IGHV status is also a determinant of BCR signaling in CLL, it is reasonable that trisomy 12 regulate the downstream of BCR signaling differentially in IGHV mutated and unmutated CLL cells, i.e., epistasis. We also observed that the epistatic interaction between trisomy 12 and IGHV had an impact on the ex vivo responses to drugs, including those targeting DNA damage response. The effect on drug response levels is currently unclear, but may be in line with the observation on transcriptomic levels that the G2M checkpoint pathway is affected by the epistatic interaction.
Further mechanistic studies are needed to clarify the combinatorial effects of IGHV status and trisomy 12 on the transcriptome, on drug response phenotypes, and the links between these. For studies on CLL biology, the interaction of IGHV and trisomy 12 status need to be considered when gene expression profiles are investigated. This study provides evidence of epistatic interactions in human cancer. Both the phenomenon of epistasis between cancer drivers, and the observation that it can be ‘mixed’ (follow different patterns) for different phenotypes form a new layer of complexity in CLL and in tumor biology more generally. Hence, our study highlights the inherent limitations of studying individual cancer genetic lesions, and
points to the need to also map out and understand how they interact and modify each other’s effects.
Disclosures
RR has received honoraria from Abbvie, AstraZeneca, Janssen, Illumina and Roche. LS is currently an employee of Takeda. TZ has received honoraria from Abbvie, AstraZeneca, Janssen, Beigene, Gilead, Novartis, Janpix and Roche. The remaining authors have no conflicts of interest to disclose.
AL developed the concept, collected and analyzed data, perfomed formal data analysis, visualized the project, developed the methodology, wrote the original draft, and reviewed and edited the manuscript. JL developed the concept, performed formal analysis, developed the methodology, wrote the original draft, reviewed and edited the manuscript. JH and TW collected and analyzed data. LS, BW, CO and SD collected and analyzed data, reviewed and edited the manuscript. RR collected and analyzed data; wrote, reviewed and edited the manuscript. WH and TZ developed the concept and the methology, supervised the project, performed project management, wrote the original draft, and reviewed and edited the manuscript.
We thank members of the Huber and Zenz research teams for valuable discussions. We thank Hanno Glimm, Stefan Fröhling, Daniela Richter, Roland Eils, Peter Lichter, Stephan Wolf, Katja Beck, and Janna Kirchhof for infrastructure and program development within DKFZ- HIPO and NCT POP. For technical support and expertise, we thank the DKFZ Genomics and Proteomics Core Facility.
The work was supported by the European Union (Horizon 2020 project SOUND under grant agreement number 633974) and the German Federal Ministry of Education and Research (TRANSCAN project GCH-CLL 143 under grant agreement number 01KT1610 and CompLS project MOFA under grant agreement number 031L0171A). TZ was supported by the UZH Clinical Research Priority Program “Next Generation Drug Response Profiling for Personalized Cancer Care”, the Swiss Cancer League (KFS-4439-02-2018), The LOOP Zurich (INTERCEPT) and the Monique-Dornonville-dela-Cour Stiftung. RR received funding from the Swedish Cancer Society, the Swedish Research Council, the Knut and Alice Wallenberg Foundation, and Radiumhemmets Forskningsfonder, Stockholm.
RNA-sequencing data are available at European GenomephenomeArchive (EGA) under accession number EGAS00001001746. All code to reproduce this analysis is
available at https://github.com/almutlue/transcriptome_cll (DOI:10.5281/zenodo.4600322). Analysis code and outputs are deployed as a browsable workflowr site: https://almutlue.github.io/transcriptome_cll/index.html (see also: Blis-
1. Campo E, Cymbalista F, Ghia P, et al. TP53 aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. 2018;103(12):1956-1968.
2. Rossi D, Gaidano G. ATM and chronic lymphocytic leukemia: mutations, and not only deletions, matter. Haematologica. 2012;97(1):5-8.
3. Rossi D, Rasi S, Fabbri G, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119(2):521-529.
4. Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. Bcell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118(16):4313-4320.
5. Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108-117.
6. Wu B, Słabicki M, Sellner L, et al. MED12 mutations and NOTCH signalling in chronic lymphocytic leukaemia. Br J Haematol. 2017;179(3):421-429.
7. Zenz T, Mertens D, Küppers R, Döhner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10(1):37-50.
8. Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer. 2016;16(3):145-162.
9. Rosenquist R, Ghia P, Hadzidimitriou A, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: updated ERIC recommendations. Leukemia. 2017;31(7):1477-1481.
10. Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840-1847.
11. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854.
12. Ferreira PG, Jares P, Rico D, et al. Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia. Genome Res. 2014;24(2):212-226.
13. Rosenwald A, Alizadeh AA, Widhopf G, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194(11):16391647.
14. Haslinger C, Schweifer N, Stilgenbauer S, et al. Microarray gene expression profiling of B-cell chronic lymphocytic leukemia subgroups defined by genomic aberrations and VH mutation status. J Clin Oncol. 2004;22(19):3937-3949.
15. Abruzzo LV, Herling CD, Calin GA, et al. Trisomy 12 chronic lymphocytic leukemia expresses a unique set of activated and targetable pathways. Haematologica. 2018;103(12):2069-2078.
16. Herling CD, Coombes KR, Benner A, et al. Time-to-progression after front-line fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy for chronic lymphocytic leukaemia: a
chak JD, Carbonetto P, Stephens M. Creating and sharing reproducible research code the workflowr way. F1000Res. 2019;81749).
retrospective, multicohort study. Lancet Oncol. 2019;20(11):1576-1586.
17. Bloehdorn J, Braun A, Taylor-Weiner A, et al. Multi-platform profiling characterizes molecular subgroups and resistance networks in chronic lymphocytic leukemia. Nat Commun. 2021;12(1):5395.
18. Dvinge H, Ries RE, Ilagan JO, et al. Sample processing obscures cancer-specific alterations in leukemic transcriptomes. Proc Natl Acad Sci USA. 2014;111(47):16802-16807.
19. Dietrich S, Oleś M, Lu J, et al. Drug-perturbation-based stratification of blood cancer. J Clin Invest. 2018;128(1):427-445.
20. Dobin A, Gingeras TR. Mapping RNA-seq reads with STAR. Curr Protoc Bioinformatics. 2015;51:11.14.1-11.14.19.
21. Anders S, Pyl PT, Huber W. HTSeq - a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166-169.
22. Oakes CC, Seifert M, Assenov Y, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016;48(3):253-264.
23. Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236-1242.
24. Dietrich S, Oleś M, Sellner L, et al. Drug perturbation based stratification of lymphoproliferative disorders. Hematol Oncol. 2017;35(S2):56-56.
25. R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/ Accessed 12 June 2023.
26. Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733-739.
27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
28. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
29. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
30. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545-15550.
31. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284-287.
32. Liberzon A, Birger C, Thorvaldsdóttir H, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417-425.
33. Rouillard AD, Gundersen GW, Fernandez NF, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database
(Oxford). 2016;2016:baw100.
34. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289-300.
35. Reyes A, Blume C, Pelechano V, et al. Mutated SF3B1 is associated with transcript isoform changes of the genes UQCC and RPL31 both in CLLs and uveal melanomas. BioRxiv. 2013 July 13. doi: https://doi.org/10.1101/000992 [preprint, not peerreviewed].
36. Inoue D, Chew G-L, Liu B, et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature. 2019;574(7778):432-436.
37. Herbst SA, Vesterlund M, Helmboldt AJ, et al. Proteogenomics refines the molecular classification of chronic lymphocytic leukemia. Nat Commun. 2022;13(1):6226.
38. Meier-Abt F, Lu J, Cannizzaro E, et al. The protein landscape of chronic lymphocytic leukemia. Blood. 2021;138(24):2514-2525.
39. Kienle D, Benner A, Läufle C, et al. Gene expression factors as predictors of genetic risk and survival in chronic lymphocytic leukemia. Haematologica. 2010;95(1):102-109.
40. Rassenti LZ, Jain S, Keating MJ, et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood. 2008;112(5):1923-1930.
41. Benedetti D, Bomben R, Dal-Bo M, et al. Are surrogates of IGHV gene mutational status useful in B-cell chronic lymphocytic leukemia? The example of Septin-10. Leukemia. 2008;22(1):224-226.
42. Beekman R, Chapaprieta V, Russiñol N, et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat Med. 2018;24(6):868-880.
43. Roisman A, Stanganelli C, Nagore VP, et al. SOX11 expression in chronic lymphocytic leukemia correlates with adverse prognostic markers. Tumour Biol. 2015;36(6):4433-4440.
44. Soriano SF, Serrano A, Hernanz-Falcón P, et al. Chemokines integrate JAK/STAT and G-protein pathways during chemotaxis and calcium flux responses. Eur J Immunol. 2003;33(5):1328-1333.
45. Cuesta-Mateos C, López-Giral S, Alfonso-Pérez M, et al. Analysis of migratory and prosurvival pathways induced by the homeostatic chemokines CCL19 and CCL21 in B-cell chronic lymphocytic leukemia. Exp Hematol. 2010;38(9):756–64, 764.
46. Munk R, Ghosh P, Ghosh MC, et al. Involvement of mTOR in CXCL12 mediated T cell signaling and migration. PLoS One.
2011;6(9):e24667.
47. Mittal AK, Chaturvedi NK, Rohlfsen RA, et al. Role of CTLA4 in the proliferation and survival of chronic lymphocytic leukemia. PLoS One. 2013;8(8):e70352.
48. Oh YM, Kwon YE, Kim JM, et al. Chfr is linked to tumour metastasis through the downregulation of HDAC1. Nat Cell Biol. 2009;11(3):295302.
49. Tsagiopoulou M, Chapaprieta V, Duran-Ferrer M, et al. Chronic lymphocytic leukemias with trisomy 12 show a distinct DNA methylation profile linked to altered chromatin activation. Haematologica. 2020;105(12):2864-2867.
50. Fisher RA. The Correlation between relatives on the supposition of mendelian inheritance. Trans R Soc Edinb. 1919;52(02):399-433.
51. van Wageningen S, Kemmeren P, Lijnzaad P, et al. Functional overlap and regulatory links shape genetic interactions between signaling pathways. Cell. 2010;143(6):991-1004.
52. Sameith K, Amini S, Groot Koerkamp MJA, et al. A highresolution gene expression atlas of epistasis between gene-specific transcription factors exposes potential mechanisms for genetic interactions. BMC Biol. 2015;13:112.
53. Menter T, Trivedi P, Ahmad R, et al. Diagnostic utility of lymphoid enhancer binding factor 1 immunohistochemistry in small B-cell lymphomas. Am J Clin Pathol. 2017;147(3):292-300.
54. Meijers RWJ, Muggen AF, Leon LG, et al. Responsiveness of chronic lymphocytic leukemia cells to B-cell receptor stimulation is associated with low expression of regulatory molecules of the nuclear factor-κB pathway. Haematologica. 2020;105(1):182-192.
55. Dong J, Ren Y, Zhang T, et al. Inactivation of DNA-PK by knockdown DNA-PKcs or NU7441 impairs non-homologous endjoining of radiation-induced double strand break repair. Oncol Rep. 2018;39(3):912-920.
56. Zabludoff SD, Deng C, Grondine MR, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7(9):2955-2966.
57. Ricci F, Tedeschi A, Morra E, Montillo M. Fludarabine in the treatment of chronic lymphocytic leukemia: a review. Ther Clin Risk Manag. 2009;5(1):187-207.
58. Petruccelli LA, Dupéré-Richer D, Pettersson F, et al. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One. 2011;6(6):e20987.
Yuqian Wang,1* Zhenzhen Zhang,2* Hanqing He,1 Jinghui Song,3 Yang Cui,1 Yunan Chen,4 Yuan Zhuang,5 Xiaoting Zhang,5 Mo Li,6 Xinxiang Zhang,4 Michael Q. Zhang,2,7,8 Minglei Shi,2 Chengqi Yi,5 and Jianwei Wang1
1School of Pharmaceutical Sciences, Tsinghua University, Beijing, China; 2School of Medicine, Tsinghua University, Beijing, China; 3Department of Bioengineering, University of California San Diego, La Jolla, CA, USA; 4Beijing National Laboratory for Molecular Sciences, Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing, China; 5Department of Basic Medical Sciences, School of Medicine, Institute for Immunology, Beijing Key Laboratory for Immunological Research on Chronic Diseases, THU-PKU Center for Life Sciences, Tsinghua University, Beijing, China; 6Center for Reproductive Medicine, Department of Obstetrics and Gynecology, Peking University Third Hospital, Beijing, China; 7MOE Key Laboratory of Bioinformatics; Division and Center for Synthetic & Systems Biology, BNRist, Department of Automation, Tsinghua University, Beijing, China and 8Department of Biological Sciences, Center for Systems Biology, the University of Texas, Richardson, TX, USA
*YW and ZZ contributed equally as first authors.
Correspondence:
M.Q. Zhang michael.zhang@utdallas.edu
M. Shi shiml79@tsinghua.edu.cn
C. Yi chengqi.yi@pku.edu.cn
J. Wang jianweiwang@mail.tsinghua.edu.cn; wangjianwei@ihcams.ac.cn
Received: October 4, 2022.
Accepted: May 4, 2023.
Early view: May 11, 2023.
htps://doi.org/10.3324/haematol.2022.282211
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Aged hematopoietic stem cells (HSC) exhibit compromised reconstitution capacity and differentiation-bias towards myeloid lineage, however, the molecular mechanism behind it remains not fully understood. In this study, we observed that the expression of pseudouridine (Ψ) synthase 10 is increased in aged hematopoietic stem and progenitor cells (HSPC) and enforced protein of Ψ synthase 10 (PUS10) recapitulates the phenotype of aged HSC, which is not achieved by its Ψ synthase activity. Consistently, we observed no difference of transcribed RNA pseudouridylation profile between young and aged HSPC. No significant alteration of hematopoietic homeostasis and HSC function is observed in young Pus10-/- mice, while aged Pus10-/mice exhibit mild alteration of hematopoietic homeostasis and HSC function. Moreover, we observed that PUS10 is ubiquitinated by E3 ubiquitin ligase CRL4DCAF1 complex and the increase of PUS10 in aged HSPC is due to aging-declined CRL4DCAF1mediated ubiquitination degradation signaling. Taken together, this study for the first time evaluated the role of PUS10 in HSC aging and function, and provided a novel insight into HSC rejuvenation and its clinical application.
Hematopoietic stem cells (HSC) generate all of the blood cells throughout their life-span.1,2 During aging, the function of HSC declines, featured as compromised reconstitution capacity and differentiation skewing towards myeloid lineage.3,4 Although previous studies have identified various molecular signaling pathways promoting HSC aging,5-8 the exact molecular mechanism is still not fully understood. It has been known for several decades that more than 170 different types of chemical modifications to RNA exist.9 Pseudouridine (Ψ), known as “the fifth nucleotide” in RNA, was first identified in 1951 and is the most abundant posttranscriptional RNA modification (with an estimated c/U ratio of 7–9%).10-12 Ψ is generated from isomerization of uridine, which is catalyzed by Ψ synthases.13-15 Ψ plays an important role in various aspects of RNA biology, and,
therefore, participates in many biological process, including translational control,16,17 RNA folding,18-22 protein translation,23-26 and clinical diseases.27-32 A recent study revealed that dysfunction of PUS7 blocks the differentiation of HSC due to the lack of pseudouridylation of mTOG tRF.16 Moreover, the expression of PUS7 is decreased in hematopoietic stem and progenitor cells (HSPC) of patients with myelodysplastic syndrome and delivery of pseudouridylated mTOG to HSPC of myelodysplastic syndromes patients improves their colony formation capacity and differentiation potential.33 In addition, DKC1 is required for accurate HSC differentiation and maintenance of HSC function.34,35 The above-mentioned studies reveal the importance of Ψ in modulating HSC differentiation and malignancies, however, whether Ψ participates in HSC aging has never been investigated.
In this study, we observed that the protein of Ψ synthase
10 (PUS10) is increased in aged HSPC. By conducting in vivo functional assay, we observed that enforced PUS10 impairs the reconstitution capacity of HSPC, which is independent of their Ψ synthase activity. By profiling the Ψ landscape in HSPC, we observed no difference in Ψ between young and aged HSPC at detectable locations. Moreover, we observed that PUS10 interacts with E3 ubiquitin ligase CRL4DCAF1 complex and is ubiquitinated by this complex. Aging-declined CRL4DCAF1 results in the accumulation of PUS10 in HSPC. Taken together, this study for the first time elucidated the role of PUS10 in HSC aging and function, and provided novel insights into HSC rejuvenation and its clinical application.
Mice
C57BL/6 mice (CD45.2), C57BL/6-SJL (CD45.1) mice were obtained from the Jackson Laboratory. Pus10-/- mice were kindly provided by Dr. Mo Li, Peking University Third Hospital, Beijing. Pus10-/- mice on C57BL/6N background were generated by deleting the 2nd exon using CRISPR-Cas9 system. The genomic RNA used to generate Pus10-/-mice are listed in the Online Supplementary Table S2. The genotyping primers are listed in the Online Supplementary Table S3. The recipient mice (CD45.1/2) in the competitive transplantation assays were the first generation of C57BL/6 (CD45.2) and B6.SJL (CD45.1) mice. All mice were housed in specific pathogen-free conditions. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Tsinghua University.
Small RNA demethylase–pseudouridine-sequencing
Small RNA (<200 nt) was extracted and purified using miRNeasy Mini Kit (Qiagen) and RNeasy MinElute Cleanup Kit (Qiagen). Purified small RNA fragments were demethylated by wild-type and mutant AlkB, puri fied by phenol/chloroform extraction and ethanol precipitation, labeled by CMC. The demethylation reaction and CMC labeling were performed as described.14 Briefly, 50 ng small RNA was denatured at 65°C for 5 minutes (min) and demethylated by wild-type and D135S mutant AlkB. The purified small RNA was denatured at 80°C for 5 min, added to BEU buffer with or without CMC, incubated at 37°C for 20 min, then puri fied by ethanol precipitation. The purified RNA was dissolved in sodium carbonate buffer and shaken at 37°C for 6 hours. The library was established as described.14,36 Briefly, small RNA was dephosphorylated with CIP (NEB). The 3' adaptor ligation was added with T4 RNA ligase2, truncated KQ (NEB), followed by 5' Deadenylase (NEB) and RecJf (NEB) digestion. RNA was reverse transcribed by SuperScript III reverse transcriptase (Invitrogen), then digested by RNase H. The 5'
adaptor ligation was added with T4 RNA ligase 1, high concentration (NEB). The ligated cDNA was amplified by Q5 High-Fidelity 2× Master Mix (NEB). The purified libraries were sequenced on Illumina NovaSeq 6000.
Identification of pseudouridine sites and levels on transcribed RNA
For transcribed RNA (tRNA) demethylase-pseudouridine sequencing (DM-Ψ-seq) data, the analysis was performed as described before.14,36 Briefly, the adapter sequences of reads two were trimmed with Trim-galore v0.6.5 (parameters: -q 20--phred33--length 25-e 0.1--stringency 3). Polymerase chain recation (PCR) duplication was removed with Fastx_toolkit v0.0.14 before discarding the random barcode in the 5’ end. Processed reads were further mapped to the genomic tRNA sequences from GtRNA database (http://gtrnadb.ucsc.edu/genomes/eukaryota/Mmusc10/) with Bowtie2 v2.3.5 (parameters: bowtie-a--best--strata-chunkmbs 2000). In order to identify the pseudouridine sites of tRNA, the following criteria were considered: i) the Ψ sites appearing in all independent replicates; ii) stop rate <1% in the BEU sample; iii) CMC coverage >50; iv) stop reads number >5 in the CMC sample; v) stop rate (CMC BEU) difference >4%; vi) Fisher test adjusted P value <0.05. Finally, the Ψ level change for tRNA between young and old HSPC was evaluated and visualized via R package ggplot2.
Data are shown as mean ± standard deviation (SD). Student’s t test (two-tailed unpaired) was used for comparisons between the groups using GraphPad Prism 6.0 software.
Due to the scarcity of HSC, we purified HSPC by using the combination of CD48-LSK(cKit+ Sca1+ Lin-) according to previous reports.37-39 The protein of PUS10 between young and aged HSPC was examined by western blot and it showed that PUS10 is increased upon aging (Figure 1A, B). In order to further confirm this result, we investigated the expression of PUS10 in a database, wherein the researchers compared the proteomic profile between young and aged HSC (CD34-CD150+Flt3-LSK).40 The result shows that PUS10 is indeed increased in aged HSC (Figure 1C).
In order to further investigate whether the increase of PUS10 plays a functional role on HSC, we cloned the cDNA of mouse Pus10 into a lentiviral vector,3 and it exhibited

Figure 1. Aging-activated PUS10 impairs the reconstitution capacity of hematopoietic stem and progenitor cells independently on its enzymatic activity. (A) Representative western blot (WB) showing the expression of PUS10 in hematopoietic stem and progenitor cells (HSPC) between young (3-month-old) and aged (28-month-old) mice. (B) This histogram depicts the protein level of PUS10 in young and aged HSPC from quantitative WB data (N=2). (C) The protein expression of PUS10 in the proteomics dataset of young and old hematopoietic stem cells (HSC). (D) Experimental design of the transplantation assay. (E) These line plots depict the changes in peripheral blood (PB) chimerism in recipients transplanted with vector or PUS10-OE LSK (N=7 mice per group). (F) This histogram displays the lineage distribution of myeloid, T and B cells among donor-derived cells in the PB of vector and PUS10-OE recipients at the 12th week (N=7 mice per group). (G, H) Freshly isolated wild-type (WT) LSK cells were infected by PUS10-carrying lentivirus for 72 hours, and 50 green fluorescent protein-positive (GFP+) HSPC (CD48- LSK) were sorted into 96-well plate and cultured in SFEM medium for 7 days. Then, the clones from vector or PUS10-OE HSPC were photographed and the cell numbers of these clones were analyzed. (G) These images show the expansion of vector or PUS10OE HSPC.(H) The scatter plots show the cell numbers of these clones. (I) The result of liquid chromatography-tandem mass spectrometry shows the Ψ/U ratio in WT and Pus10-/- LK cells with overexpression of WT PUS10 or PUS10D342A. (J) These line plots display the changes in PB chimerism in recipients transplanted with vector, PUS10-OE or PUS10D342A-OE LSK (N=7-8 mice per group). All data are shown as mean ± standard deviation; *P<0.05, **P<0.01, ***P<0.001.
efficient overexpression of PUS10 (Online Supplementary Figure S1A, B). Freshly isolated WT LSK cells were infected by PUS10-carrying lentivirus; 72 hours later, 2×104 green fluorescent protein-positive (GFP+) cells were purified and transplanted into lethally irradiated recipients together with 2×105 competitor cells (Figure 1D). Chimera in peripheral blood was evaluated every 4 weeks until the 12th week by using this gating strategy (Online Supplementary Figure S1C, D). The results showed that enforced PUS10 severely impairs the reconstitution capacity of HSPC (Figure 1E). Moreover, enforced PUS10 promotes HSPC differentiation bias towards the myeloid lineage (Figure 1F), which is a classical phenomenon of aged HSC. Consistently, enforced PUS10 significantly inhibits HSPC expansion in vitro (Figure 1G, H).
In order to investigate whether the inhibitory function of enforced PUS10 on HSPC depends on its Ψ catalytic activity, we mutated the key enzyme site of PUS10 to generate catalytic dead PUS10D342A according to a previous study.14 We first measured the Ψ/U ratio in WT and Pus10-/- lineage-cKit+ (LK) cells using liquid chromatography-tandem mass spectrometry (LC-MS/MS). The result showed that targeted dysfunction of Pus10 leads to a significant decrease of the Ψ/U ratio. In order to test whether PUS10D342A is an inactive Ψ synthase, we reintroduced WT PUS10 and PUS10D342A into Pus10-/- LK cells, and measured the Ψ/U ratio for them. The result showed that the decrease of the Ψ/U ratio upon Pus10 deletion is rescued by the reintroduction of WT PUS10, but not PUS10D342A (Figure 1I). This result indicates that the D342 residue is the key enzyme site for its Ψ synthase activity.
In order to investigate whether the Ψ synthase activity of PUS10 modulates HSC aging, freshly isolated WT LSK cells were infected with either PUS10 or PUS10D342A-overexpressing lentivirus for 72 hours, and 2×104 GFP+ cells were transplanted into lethally irradiated recipients together with 2×105 competitor cells. The chimera in peripheral blood was evaluated every 4 weeks until the 12th week. The results revealed that both enforced PUS10 and PUS10D342A significantly impair the reconstitution capacity
of HSPC (Figure 1J), indicating that the destructive role of PUS10 on HSPC is independent on its enzymatic activity.
No difference of pseudouridylation profile between young and aged hematopoietic stem and progenitor cells We then set out to investigate whether aged HSC exhibit aberrant Ψ profile compared to young ones. Due to the limited cell number of HSC and the large amount of cells required for Ψ sequencing, we performed DM-Ψ-seq by using freshly isolated lineage- cells, which are hematopoietic stem and progenitor-enriched cells, from 3-monthold and 29-month-old mice according to an elegant approach14,36 (Figure 2A). The result revealed no difference of the bulk Ψ profile between young and aged HSPC (Figure 2B). We then wondered whether the percentage of Ψ on certain sites of the tRNA exhibits a difference between them. In order to address this question, we evaluated all of the Ψ on detectable tRNA. The results revealed that the percentage of Ψ28 , Ψ32 , Ψ53 and Ψ54 of tRNAHis-GTG is static in young and aged HSPC (Figure 2C, D), and identical to other detected tRNA (Online Supplementary Figure S2). Briefly, these results indicate that not only the bulk Ψ profile but also the percentage of each Ψ on tRNA exhibits no difference between young and aged HSPC, which is consistent with the data that the toxicity of enforced PUS10 on HSPC is independent on its enzymatic activity.
Given that PUS10 is increased in aged HSPC and enforced PUS10 impairs the reconstitution capacity of HSPC, we then wondered how PUS10 is increased with aging. Firstly, we examined the mRNA level of Pus10 in young and aged HSPC (CD48-LSK) by reverse transcription PCR (RT-PCR), and the result revealed no difference of Pus10 mRNA between them (Figure 3A). In order to confirm this observation, we examined the mRNA level of Pus10 between young and aged HSC by exploring published RNA-sequencing data, and the result revealed that the mRNA level of Pus10 remains static in young and aged HSC (Online Supplemen-
Figure 2. No difference of pseudouridine modification profile between young and aged hematopoietic stem and progenitor cells. (A-D) Lineage- cells were isolated from young (3-month-old) and aged (29-month-old) mice. Small RNA (<200 nt) was extracted and purified to perform demethylase-pseudouridine sequencing (DM–Ψ-seq). (A)
Experimental design. (B) The scatter plot depicts Ψ levels in transcribed RNA (tRNA) between young and aged hematopoietic stem and progenitor cells (HSPC) (N=2). (C) Schematic of tRNA-His-GTG-2-1 shows Ψ sites Ψ28 , Ψ32 , Ψ53 and Ψ54. (D) Ψ sites (red arrows) and levels of tRNA-His-GTG-2-1 are identified in young and aged HSPC. The x axis represents nucleotide position. The y axis represents Ψ levels.

Figure 3. Aging-declined CRL4DCAF1mediated ubiquitination degradation signaling leads to the increase of PUS10. (A) This histogram depicts the mRNA expression of Pus10 in young (2 months) and aged (31 months) hematopoietic stem and progenitor cells (HSPC). (B) Affinity puri fication of PUS10 protein from HEK293T cells stably expressing Flag-tagged PUS10. Proteins identified by mass spectrometry are listed. The bait protein is marked in bold letters. (C) HEK293T cells were co-transfected with plasmids encoding SFBtagged PUS10 and Myc-tagged DDB1, DCAF1, CUL4B followed by co-immunoprecipitation using anti-Flag, anti-Myc antibody. Representative western blot (WB) shows that PUS10 interacts with DDB1, DCAF1 and CUL4B. (D) HEK293T cells were co-transfected with plasmids encoding SFBtagged PUS10, Myc-tagged DDB1, DCAF1, CUL4B and HA-tagged UbWT or Ub-K48R followed by coimmunoprecipitation using anti-HA, anti-Flag, anti-Myc antibody. Representative WB shows that PUS10 is ubiquitinated by the CRL4DCAF1 complex. (E) Representative WB shows the expressions of DDB1 and CUL4B in young (3 months) and aged (28 months) HSPC. (F) 32D cells were infected by lentivirus carrying Ddb1-shRNA. 72 hours later, green fluorescent protein-positive (GFP+) cells were sorted for WB to validate the expression of PUS10 and DDB1. Representative WB shows the expression of PUS10 and DDB1 in vector and DDB1-KD 32D cells. (G) This histogram depicts the protein level of PUS10 in vector and DDB1KD 32D cells from quantitative WB data (N=2). (H) This histogram depicts the protein expression of DDB1 in vector and DDB1-KD 32D cells from quantitative WB data (N=3).

tary Figure S3). Then, it is conceivable that the increase of PUS10 might be modulated via post-transcriptional modification manner. In order to test this hypothesis, we purified proteins interacting with PUS10 via affinity purification and we observed that the CRL4DCAF1 complex, including DDB1, DCAF1 and CUL4B, interacts with PUS10 (Figure 3B). CRL4DCAF1 complex is E3 ubiquitin ligase targeting substrate for protein degradation.41 In order to confirm this observation, we performed co-immunoprecipitation (Co-IP) assay by infecting HEK293T cells with S-protein, FLAG, and streptavidin-binding peptide (SFB)-tagged PUS10 together with Myc-tagged DDB1, DCAF1 or CUL4B respectively. Cell lysates were incubated with S-protein beads and probed with anti-Flag, anti-Myc antibodies. The result showed that PUS10 exhibits strong interaction with DDB1, DCAF1 and CUL4B (Figure 3C).
Previous study has shown that CRL4DCAF1 complex participates in ubiquitin dependent degradation,41 we then set out to determine whether CRL4DCAF1 regulates PUS10 ubiquitination. Plasmids encoding SFB-tagged PUS10, Myctagged DDB1, DCAF1, CUL4B and HA-tagged wild-type ubiquitin (Ub-WT), mutant ubiquitin (Ub-K48R) were cotransfected into HEK293T cells; 24 hours later, cell lysates were collected to detect the ubiquitination of PUS10. The result revealed that CRL4DCAF1 vigorously promotes the ubiquitination of PUS10 in cells expressing WT ubiquitin (Figure 3D). Compared with WT ubiquitin, the ubiquitination of PUS10 was completely abolished in cells expressing K48R ubiquitin, indicating that CRL4DCAF1 promotes polyubiquitination of PUS10 via the formation of the K48 linkage.
Next, we set out to investigate whether the increase of PUS10 is due to the alteration of CRL4DCAF1 in aged HSPC. We first evaluated the expression of CRL4DCAF1 complex in young and aged HSPC. The result showed that the expression of DDB1 and CUL4B is decreased in aged HSPC (the expression level of DCAF1 is too low to be detected) (Figure 3E), which is negatively correlated with the alteration of PUS10 between young and aged HSPC (Figure 1A). Given that the protein level of PUS10, but not mRNA level, is elevated in aged HSPC (Figures 1A and 3A), and that PUS10 is poly-ubiquitinated by CRL4DCAF1 complex (Figure 3D), and that DDB1 and CUL4B are decreased in aged HSPC (Figure 3E), we then wondered whether aging-declined CRL4DCAF1 complex leads to the increase of PUS10. In order to test this hypothesis, we generated two efficient small hairpin RNA (shRNA) against Ddb1 (Figure 3F, H; Online Supplementary Table S2), which is the key linker protein of CRL4DCAF1 complex.41 32D cells were infected by shDdb1 carrying lentivirus for 72 hours, and GFP+ cells were subjected to detect the protein level of PUS10 by western blot. The result showed that PUS10 is elevated upon the knockdown of Ddb1 (Figure 3F, G). Taken together, these data suggest that aging-declined
CRL4DCAF1-mediated ubiquitination degradation signaling leads to the accumulation of PUS10.
Young Pus10-/- mice exhibit no influence on hematopoietic homeostasis and hematopoietic stem cell function
The above-mentioned results revealed the functional role of Pus10 in modulating HSC aging, we then wondered whether targeted dysfunction of Pus10 plays a role in regulating hematopoietic homeostasis and HSC function. In order to address this question, we generated Pus10 knockout (Pus10-/-) mice on C57BL/6N background by deleting the 2nd exon using CRISPR-Cas9 system (Figure 4A, see details in the Material and Methods) and we achieved efficient deletion of PUS10 in LSK cells (Figure 4B, C). We then performed complete blood count assay for Pus10 -/- and age-matched control mice. The result revealed no difference of white blood cell (WBC), lymphocyte (LYM), neutrophil (NEUT), red blood cell (RBC) and platelet (PLT) count between Pus10-/- and WT mice (Figure 4D). We then sought to investigate the lineage composition in peripheral blood (PB) and bone marrow (BM) of Pus10-/- mice, including T cells, B cells and myeloid cells (Online Supplementary Figure S4A). The results revealed no difference of Pus10-/- mice compared to WT in PB (Figure 4E) and BM (Figure 4F).
We next analyzed HSPC of Pus10-/- mice, including common myeloid progenitors (CMP), granulocyte-macrophage progenitors (GMP), megakaryocyte-erythroid progenitors (MEP), common lymphoid progenitors (CLP), multipotent progenitor cells (MPP) and HSC ( Online Supplementary Figure S4B). The results revealed no significant difference of the above-mentioned populations between Pus10-/- and WT mice (Figure 4G-K).
In order to further investigate the reconstitution capacity of Pus10-/- HSC, 20 freshly isolated Pus10-/- and WT HSC were transplanted into lethally irradiated recipients together with 3×105 competitor cells (Figure 5A). The chimera in the PB of recipients was evaluated every 4 weeks until the 16th week (Online Supplementary Figure S4C, D). The results showed that the reconstitution capacity of Pus10-/- HSC is comparable with WT ones (Figure 5B), while dysfunction of Pus10 promotes the differentiation bias towards lymphoid lineage (34.31% vs. 48.43%; Figure 5C). Donor-derived HSC of recipients revealed no significant difference between Pus10-/- and WT mice (Figure 5D-F).
We then investigated the phenomenon of aged Pus10-/mice. We performed complete blood count assay for aged WT and Pus10-/- mice (26-month-old). The result showed no significant difference of WBC, LYM, NEUT, RBC and PLT
Figure 4. Young Pus10-/- mice exhibit no influence on hematopoietic homeostasis.(A) Schematic illustration of the Pus10 knockout (Pus10-/-) mice. (B) Representative western blot (WB) shows the expression of PUS10 in wild-type (WT) and Pus10-/- LSK cells. (C) This histogram depicts the protein expression of PUS10 in WT and Pus10-/- LSK cells from quantitative WB data (N=2). (D) The scatter plots show the numbers of white blood cells (WBC), lymphocytes (LYM), neutrophils (NEUT), red blood cells (RBC) and platelets (PLT) between WT and Pus10-/- mice. (E, F) The scatter plots depict the frequency of B cells, T cells and myeloid cells in peripheral blood (PB) (E) and bone marrow (BM) (F) of WT and Pus10-/- mice. (G) The scatter plots show the cell numbers of BM in WT and Pus10-/- femurs. (H-K) The scatter plots depict the cell numbers and frequency of common myeloid progenitors (CMP), granulocyte/macrophage progenitors (GMP), megakaryocytic/erythroid progenitors (MEP), common lymphoid progenitors (CLP), multipotent progenitor cells (MPP) and HSC in WT and Pus10-/- femurs. All data are shown as mean ± standard deviation; *P<0.05, **P<0.01, ***P<0.001.

Figure 5. Young Pus10-/- mice exhibit no influence on hematopoietic stem cell function. (A) Experimental design of the competitive transplantation strategy. (B) These line plots depict the changes in peripheral blood (PB) chimerism in recipients transplanted with wild-type (WT) or Pus10 knockout (Pus10-/-) hematopoietic stem cells (HSC) (N=5 mice per group). (C) This histogram displays the lineage distribution of myeloid, T and B cells among donor-derived cells in PB of the recipients at the 16th week (N=5 mice per group). (D) The gating strategies for the frequency of the test donor-derived HSC. (E, F) The scatter plots depict the cell numbers (E) and frequency (F) of donor-derived HSC in recipients transplanted with WT or Pus10-/- HSC (N=5 mice per group). All data are shown as mean ± standard deviation; *P<0.05, **P<0.01, ***P<0.001.


Figure 6. Aged Pus10-/- mice exhibit mild alteration of hematopoietic homeostasis and hematopoietic stem cell function. (A) The scatter plots show the numbers of white blood cells (WBC), lymphocytes (LYM), neutrophils (NEUT), red blood cells (RBC) and platelets (PLT) between aged wild-type (WT) and Pus10 knockout (Pus10-/-) mice. (B-E) The scatter plots depict the frequency of B cells, T cells and myeloid cells in the peripheral blood (PB) (B), bone marrow (C), spleen (D) and thymus (E) of aged WT and Pus10-/- mice. (F) The scatter plots show the cell numbers of bone marrow in WT and Pus10-/- femurs. (G-J) The scatter plots indicate the cell numbers and frequency of common myeloid progenitors (CMP), granulocyte/macrophage progenitors (GMP), megakaryocytic/erythroid progenitors (MEP), common lymphoid progenitors (CLP), multipotent progenitor cells (MPP) and hematopoietic stem cells (HSC) in WT and Pus10-/- femurs. (K) Experimental design of the transplantation assay. (L) These line plots depict the changes in PB chimerism in recipients transplanted with aged WT or Pus10-/- HSC (N=8-10 mice per group). (M) This histogram displays the lineage distribution of myeloid, T and B cells among donor-derived cells in the PB of the recipients at the 12th week (N=8-10 mice per group). All data are shown as mean ± standard deviation; *P<0.05, **P<0.01, ***P<0.001.
between aged WT and Pus10-/- mice (Figure 6A). We then analyzed the frequency of T cells, B cells and myeloid cells in PB, BM, spleen and thymus of aged Pus10-/- and WT mice. The results revealed no significant difference between them in the PB (Figure 6B) and thymus (Figure 6E). However, the percentage of T cells in the BM (Figure 6C) and the percentage of myeloid cells in the spleen (Figure 6D) of aged Pus10-/- mice are increased. We next investigated HSPC of aged Pus10-/- and WT mice. The results indicated no significant difference of the CMP, GMP, MEP, CLP and MPP, while the frequency of HSC is increased in aged Pus10-/- mice compared to WT (Figure 6F-J). In order to further explore the reconstitution capacity of aged Pus10-/- HSC, 150 freshly isolated aged Pus10-/- and WT HSC were transplanted into lethally irradiated recipients together with 3×105 competitor cells (Figure 6K). The chimera in the PB of recipients was evaluated every 4 weeks until the 12th week. The results revealed that the reconstitution capacity of aged Pus10-/- HSC is comparable with WT ones, while dysfunction of Pus10 promotes the differentiation bias towards B-cell lineage (Figure 6L, M). In brief, our study for the first time revealed that enforced PUS10 impairs the reconstitution capacity of HSPC. Howerer, the hematopoietic homeostasis and reconstitution capacity of young Pus10-/- mice is comparable with control mice, while aged Pus10-/- mice exhibit mild alteration of hematopoietic homeostasis and HSC function. In summary, these data suggest that aging-diminished CRL4DCAF1mediated ubiquitination degradation signaling leads to the accumulation of PUS10, which impairs HSPC (Figure 7).
Our study for the first time evaluated the role of PUS10 in HSC aging and function. This study will expand our understanding of RNA modification on HSC function regulation.

An elegant study reported a proteomics resource from mass spectrometry of mouse young and aged HSC, and identified a subset of genes with apparent post-transcriptional alteration during aging.40 This indicates that tran-
scriptomic levels may not reflect the functional change of aged HSC. The alteration of the protein level achieved either by RNA or protein modification in aged HSC might play an essential role in promoting HSC aging. Our unpublished data confirmed this observation by identifying a group of RNA modification genes which modulate HSC aging. In addition, our study also identified that CRL4DCAF1mediated ubiquitination participated in regulating HSC aging by degrading PUS10 and other important proteins (unpublished). Whether there are other proteins modified by ubiquitination which lead to their changes during aging, thereby regulating HSC aging and other cell aging is a question worthy of study. It is also intriguing to investigate the molecular mechanism by which ubiquitin ligases are altered during aging.
Therefore, exploring the functional role of post-transcriptional modification (PTM) in aging might strengthen the understanding of aging on HSC and its clinical relevance.
Figure 7.
Aging-enforced PUS10 mediated by the decrease of ubiquitination degradation impairs hematopoietic stem and progenitor cells. The proposed model diagram illustrates the accumulation of PUS10 mediated by the decrease of ubiquitination degradation during aging impairs hematopoietic stem and progenitor cells (HSPC).
RNA epigenetics versus hematopoietic stem cell aging
In our study, we observed the Ψ profile is not changed in aged HSPC and the aging-increased PUS10 promotes HSC aging, which is not achieved by its Ψ synthase activity. Up to date, more than 170 RNA modifications have been identified and some of them play an essential role in various biological process and clinical diseases.42,43 Whether other RNA modification profiles are altered in aged HSC and whether the corresponding enzymes are involved in regulating HSC aging is a question worthy of investigation. In this study, we performed Ψ profiling by using HSPC, but not pure HSC, due to the limitation of HSC number. Whether the Ψ profile of aged HSC is identical as we observed in HSPC is also a question worth investigating, which depends on the development of sequencing technology.
Disclosures
No conflicts of interest to disclose.
Contributions
JW and CY developed the concept. JW and CY developed the methodology. YW, ZZ, HH, JS , YC, YC, YZ and XZ carried
1. Dzierzak E, Bigas A. Blood development: hematopoietic stem cell dependence and independence. Cell Stem Cell. 2018;22(5):639-651.
2. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327-334.
3. He H, Xu P, Zhang X, et al. Aging-induced IL27Ra signaling impairs hematopoietic stem cells. Blood. 2020;136(2):183-198.
4. Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208(13):2691-2703.
5. Wang J, Sun Q, Morita Y, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012;148(5):1001-1014.
6. Mohrin M, Shin J, Liu Y, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347(6228):1374-1377.
7. Norddahl GL, Pronk CJ, Wahlestedt M, et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8(5):499-510.
8. Sun D, Luo M, Jeong M, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14(5):673-688.
9. Boccaletto P, Stefaniak F, Ray A, et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022;50(D1):D231-D235.
10. Cohn WE, Volkin E. Nucleoside-5′-phosphates from ribonucleic acid. Nature. 1951;167(4247):483-484.
11. Charette M, Gray MW. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000;49(5):341-351.
12. Ge J, Yu YT. RNA pseudouridylation: new insights into an old modification. Trends Biochem Sci. 2013;38(4):210-218.
13. Spenkuch F, Motorin Y, Helm M. Pseudouridine: still mysterious,
out the investigation. JW, CY, MS , MQZ, XZ and ML performed the formal analysis. JW, CY, MS and MQZ provided resources. JW and CY wrote the manuscript. JW and CY acquired funding. JW, CY, MS and MQZ supervised the study.
We thank the Tsinghua-Peking Center for Life Sciences for facility and financial support.
This work was supported by grant numbers Z200022, 82250002, 81870118 and 2018YFA0800200 to JWW, and 2019YFA0110900, 2019YFA0802200 to CQY, and 81890991 to MS from the National Key R&D Program of China or the Beijing Municipal Science & Technology Commission and the National Natural Science Foundation of China.
The data are available on request. All sequencing raw data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus . The accession code is GSE213422 with the enter token atstusiqpxmhvmj.
but never a fake (uridine)! RNA Biol. 2014;11(12):1540-1554.
14. Song J, Zhuang Y, Zhu C, et al. Differential roles of human PUS10 in miRNA processing and tRNA pseudouridylation. Nat Chem Biol. 2019;16(2):160-169.
15. Li X, Zhu P, Ma S, et al. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol. 2015;11(8):592-597.
16. Guzzi N, Cieśla M, Ngoc PCT, et al. Pseudouridylation of tRNAderived fragments steers translational control in stem cells. Cell. 2018;173(5):1204-1216.
17. Levi O, Arava YS. Pseudouridine-mediated translation control of mRNA by methionine aminoacyl tRNA synthetase. Nucleic Acids Res. 2021;49(1):432-443.
18. Bernick DL, Dennis PP, Hochsmann M, Lowe TM. Discovery of Pyrobaculum small RNA families with atypical pseudouridine guide RNA features. RNA. 2012;18(3):402-411.
19. Ni J, Tien AL, Fournier MJ. Small nucleolar RNAs direct sitespecific synthesis of pseudouridine in ribosomal RNA. Cell. 1997;89(4):565-573.
20. Keffer-Wilkes LC, Veerareddygari GR, Kothe U. RNA modification enzyme TruB is a tRNA chaperone. Proc Natl Acad Sci U S A. 2016;113(50):14306-14311.
21. Leppik M, Liiv A, Remme J. Random pseuoduridylation in vivo reveals critical region of Escherichia coli 23S rRNA for ribosome assembly. Nucleic Acids Res. 2017;45(10):6098-6108.
22. Zhao X, Patton JR, Davis SL, Florence B, Ames SJ, Spanjaard RA. Regulation of nuclear receptor activity by a pseudouridine synthase through posttranscriptional modification of steroid receptor RNA activator. Mol Cell. 2004;15(4):549-558.
23. Anderson BR, Muramatsu H, Nallagatla SR. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010; 38(17):5884-5892.
24. Kariko K, Muramatsu H, Welsh FA, et al. Incorporation of
pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16(11):1833-1840.
25. Svitkin YV, Cheng YM, Chakraborty T, Presnyak V, John M, Sonenberg N. N1-methyl-pseudouridine in mRNA enhances translation through eIF2alpha-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017;45(10):6023-6036.
26. Borchardt EK, Martinez NM, Gilbert WV. Regulation and function of RNA pseudouridylation in human cells. Annu Rev Genet. 2020;54:309-336.
27. Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet. 2004;74(6):1303-1308.
28. Shaheen R, Han L, Faqeih E, et al. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum Genet. 2016;135(7):707-713.
29. Festen EA, Goyette P, Green T, et al. A meta-analysis of genomewide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn's disease and celiac disease. PLoS Genet. 2011;7(1):e1001283.
30. de Brouwer APM, Abou Jamra R, Körtel N, et al. Variants in PUS7 cause intellectual disability with speech delay, microcephaly, short stature, and aggressive behavior. Am J Hum Genet. 2018;103(6):1045-1052.
31. Darvish H, Azcona LJ, Alehabib E, et al. A novel PUS7 mutation causes intellectual disability with autistic and aggressive behaviors. Neurol Genet. 2019;5(5):e356.
32. Arroyo JD, Jourdain AA, Calvo SE, et al. A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 2016;24(6):875-885.
33. Guzzi N, Muthukumar S, Ciesla M, et al. Pseudouridine-modified tRNA fragments repress aberrant protein synthesis and predict
leukaemic progression in myelodysplastic syndrome. Nat Cell Biol. 2022;24(3):299-306.
34. Bellodi C, McMahon M, Contreras A, et al. H/ACA small RNA dysfunctions in disease reveal key roles for noncoding RNA modifications in hematopoietic stem cell differentiation. Cell Rep. 2013;3(5):1493-1502.
35. Gu BW, Fan JM, Bessler M, Mason PJ. Accelerated hematopoietic stem cell aging in a mouse model of dyskeratosis congenita responds to antioxidant treatment. Aging Cell. 2011;10(2):338-348.
36. Cui Q, Yin K, Zhang X, et al. Targeting PUS7 suppresses tRNA pseudouridylation and glioblastoma tumorigenesis. Nat Cancer. 2021;2(9):932-949.
37. Termini CM, Pang A. Syndecan-2 enriches for hematopoietic stem cells and regulates stem cell repopulating capacity. Blood. 2022;139(2):188-204.
38. Akinduro O, Weber TS, Ang H, et al. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat Commun. 2018;9(1):519.
39. Tadokoro Y, Hoshii T, Yamazaki S, et al. Spred1 safeguards hematopoietic homeostasis against diet-induced systemic stress. Cell Stem Cell. 2018;22(5):713-725.
40. Zaro BW, Noh JJ, Mascetti VL, et al. Proteomic analysis of young and old mouse hematopoietic stem cells and their progenitors reveals post-transcriptional regulation in stem cells. eLife. 2020;9:e62210.
41. Jackson S, Xiong Y. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci. 2009;34(11):562-570.
42. Shi H, Chai P, Jia R, Fan X. Novel insight into the regulatory roles of diverse RNA modifications: re-defining the bridge between transcription and translation. Mol Cancer. 2020;19(1):78.
43. Delaunay S, Frye M. RNA modifications regulating cell fate in cancer. Nat Cell Biol. 2019;21(5):552-559.
1Institute for Clinical and Experimental Transfusion Medicine, Medical Faculty of Tübingen, University of Tübingen and 2Center for Clinical Transfusion Medicine, Tübingen, Germany
Correspondence: T. Bakchoul tamam.bakchoul@med.uni-tuebingen.de
Received: October 17, 2022.
Accepted: April 19, 2023.
Early view: April 27, 2023.
htps://doi.org/10.3324/haematol.2022.282275
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Heparin-induced thrombocytopenia (HIT) is a severe immune-mediated prothrombotic disorder caused by antibodies (Ab) reactive to complexes of platelet factor 4 and heparin. Platelets (PLT) and their interaction with different immune cells contribute to prothrombotic conditions in HIT. However, the exact mechanisms and the role of different PLT subpopulations in this prothrombotic environment remain poorly understood. In this study, we observed that HIT patient Ab induce a new PLT population that is characterized by increased P-selectin expression and phosphatidylserine (PS) externalization. Formation of this procoagulant PLT subpopulation was dependent on engagement of PLT Fc-γ-RIIA by HIT Ab and resulted in a significant increase of thrombin generation on the PLT surface. Using an ex vivo thrombosis model and multi-parameter assessment of thrombus formation, we observed that HIT Ab-induced procoagulant PLT propagated formation of large PLT aggregates, leukocyte recruitment and most importantly, fibrin network generation. These prothrombotic conditions were prevented via the upregulation of PLT intracellular cAMP with Iloprost, a clinically approved prostacyclin analogue. Additionally, the functional relevance of P-selectin and PS was dissected. While inhibition of P-selectin did not affect thrombus formation, the specific blockade of PS prevented HIT Ab-mediated thrombin generation and most importantly procoagulant PLT-mediated thrombus formation ex vivo. Taken together, our findings indicate that procoagulant PLT are critical mediators of prothrombotic conditions in HIT. Specific PS targeting could be a promising therapeutic approach to prevent thromboembolic events in HIT patients.
Heparin-induced thrombocytopenia (HIT) is an immunemediated prothrombotic disorder that is characterized by low platelet (PLT) counts and high risk of thromboembolic events.1-3 HIT is caused by antibodies (Ab) of the immunoglobulin G (IgG) subclass that recognize complexes of the endogenous protein platelet factor 4 (PF4) and heparin.4,5 These immune complexes evolve 4-14 days after heparin exposure and harbor the potential to elicit prothrombotic conditions due to their interaction with PLT, neutrophils and monocytes via immune receptor Fc- γ -RIIA.6-8
Despite low PLT counts, HIT is associated with an increased risk of thrombosis.9,10 In fact, Fc-γ-RIIA-mediated PLT activation by HIT immune complexes is associated with intense thrombin generation and prothrombotic po-
tential.7,11-13 Additionally, binding of HIT Ab to PF4 and glycosaminoglycans on the surface of monocytes has been reported to result in increased tissue factor and microparticle release which promotes thrombin generation and PLT activation.6,14,15 Furthermore, HIT immune complexes can activate neutrophils via Fc- γ -RIIA and lead to the formation of PLT-neutrophil aggregates as well as the release of prothrombotic neutrophil extracellular traps (NET).16 Although this evidence directs towards a prominent role of PLT in the pathogenesis of HIT, a better understanding of HIT Ab-mediated PLT changes and the potential relevance of different PLT subpopulations in prothrombotic conditions in HIT patients is needed. Procoagulant PLT, a distinct subpopulation of activated PLT, are characterized by an increased externalization of the negatively charged phospholipid phosphatidylserine (PS).17,18 High levels of PLT PS are efficient to promote as-
sembly of coagulation complexes and subsequent thrombin burst.19,20 Recently, we showed that procoagulant PLT might contribute to the prothrombotic state in patients with COVID-19 and vaccine-induced immune thrombotic thrombocytopenia (VITT).21,22
In the current study, we hypothesized that anti-PF4/heparin HIT IgG (HIT Ab) induce a procoagulant PLT response that contributes to the hypercoagulable state in HIT. Using an ex vivo model for thrombosis we analyzed the molecular mechanisms of HIT Ab-induced thrombosis. Our data indicate that anti-PF4/heparin IgG Ab can generate procoagulant PLT via the crosslinking of Fc-γ-RIIA leading to increased intracellular calcium and subsequent P-selectin (CD62p) expression as well as PS externalization. Most importantly, thrombus formation by HIT Ab was mediated by PLT PS rather than CD62p expression.
Patients and sera
Experiments were performed using serum material from HIT patients who were referred to our laboratory between March 2019 and April 2022. Where indicated, IgG fractions were isolated from HIT as well as from control sera using a commercially available IgG purification kit. Studies involving human material were approved by the Ethics Committee of the Medical Faculty, Eberhard-Karls University, Tübingen, Germany (application number: 140/2022BO2) and were conducted in accordance with the Declaration of Helsinki. Additional details are available in the Online Supplementary Appendix.
Testing for anti-PF4/heparin antibodies
A commercially available IgG enzyme-immune assay was used in accordance to manufacturer’s instructions (Hyphen Biomed, Neuville-sur-Oise, France). The ability of sera to activate PLT was tested using HIPA as previously described.23 Additional details are available in the Online Supplementary Appendix.
Preparation of washed platelets
Washed PLT (wPLT) were prepared from venous blood samples as previously described.23,24 Additional details are available in the Online Supplementary Appendix
Treatment of platelets with sera/immunoglobulins
A total of 37.5 μL of wPLT or platelet-rich plasma (PRP) were supplemented with 5 μ L serum/IgG from HIT patients or controls and incubated for 1 hour under rotating conditions at room temperature. Changes in the expression of CD62p and PS were assessed via flow-cytometry. Additional details are available in the Online Supplementary Appendix
Analysis of heparin-induced thrombocytopenia antibodyinduced mechanomolecular signaling mechanisms
In order to investigate the underlying mechanisms that result in HIT Ab-induced procoagulant PLT effects, PLT were pretreated with the Fc-γ-RIIA blocking monoclonal Ab IV.3 or a monoclonal isotype control, prior to HIT serum/IgG treatment. In order to investigate the role of upregulated intracellular levels of cAMP, PLT were pretreated with the prostacyclin analogue Iloprost or vehicle prior to incubation with IgG from HIT patients or healthy controls (HC). Additional details are available in the Online Supplementary Appendix.
Specific blockade of platelet surface P-selectin and phosphatidylserine
P-selectin (CD62p) was blocked via the pretreatment of PLT with 5 μg/mL anti-SELP humanized Ab (anti-CD62p, 15 minutes [min] at room temperature; ProteoGenix, Schiltigheim, France). Specific inhibition of externalized PS on the surface of procoagulant PLT was performed via the incubation of PLT with Lactadherin, a calcium-independent ligand of PS.25,26
HIT Ab-induced thrombin generation on PLT was detected using CAT (Stago, Maastricht, the Netherlands) according to the manufacturer’s instructions. Additional details are available in the Online Supplementary Appendix
In order to investigate the effect of Ab-induced PLT alterations on the coagulation cascade and subsequent thrombus formation, an ex vivo model for thrombus formation (BioFlux 200, Fluxion Biosciences, Alameda, USA) was used according to the recommendations of the ISTH Standardization Committee for Biorheology.27 Images were processed identically using adjusted threshold settings and exclusion of image artefacts by using Fiji image processing software.28 Additional details are available in the Online Supplementary Appendix.
Statistical analysis was performed using GraphPad Prism, Version 8.0 (GraphPad, La Jolla, USA). Comparison between groups was performed by Mann-Whitney U test for unpaired data sets and Wilcoxon matched-pairs signed-rank or paired sample t test for paired data sets. P values <0.05 were considered statistically significant.
In this study, we used serum samples from 37 patients with suspected HIT (Table 1). Twenty-three of 37 (62%) pa-
tients were male and the mean age was 65 years (range, 3787 years). Twenty-eight of 37 (76%) patients tested positive for anti-PF4/heparin IgG Ab in enzyme-immune assay (mean optical density [OD] 2.12; range, 0.51-3.61). The diagnosis of HIT was confirmed in 20 of 37 (54%) using heparin-induced PLT activation assay (HIPA, median time to platelet aggregation in the presence of 0.2 IU/mL heparin: 5 min; range, 5-
25 min]). In the HIT-positive cohort, thrombocytopenia (PLT count <150x106/mL) occured in 19 of 20 (95%) and clinical overt thrombosis was observed in nine of 20 (45%) patients, including six pulmonary embolisms, two occlusions of arterial vessels and one disseminated intravascular coagulation (for more details see the Online Supplementary Table S1).
ID: identification; PLT: platelet; M: male; F: female; OD: optical density; EIA: enzyme-immune assay; HIPA: heparin-induced platelet activation assay; Pos.: positive; Neg: negative. †Reference range PLT count (150-450x109/L); #1-17: patients with suspected heparin-induced thrombocytopenia (HIT); #18-37: patients with confirmed HIT.
Membrane phospholipid symmetry of platelets is rearranged by sera from heparin-induced thrombocytopenia patients
In order to investigate the ability of HIT Ab to induce new PLT populations, wPLT from healthy individuals were incubated with sera from 37 patients with suspected HIT in the presence of buffer, low- (0.2 IU/mL) or high-dose (100 IU/mL) heparin. As shown in Figure 1A, marked changes in CD62p expression and PS externalization were induced by sera from HIT patients (HIPA-positive) in the presence of

low-dose (0.2 IU/mL) heparin but not in the presence of high-dose (100 IU/mL) heparin (mean percent of CD62p and PS double-positive PLT [mean %] ± standard error of the mean [SEM]: 38.65±5.79% vs. 1.14±0.26%; P<0.0001; Figure 1A). In contrast, no significant alteration in the expression levels of PLT CD62p and PS was observed by testing sera from patients with suspected but not confirmed HIT (HIPA-negative) compared to sera from HC in the presence of low-dose (0.2 IU/mL) heparin (mean % ± SEM: 0.63±0.12% vs. 1±0%; P=0.0565; Figure 1A). In order
Figure 1. Heparin-induced thrombocytopenia antibodies induce procoagulant platelets and thrombin generation in a heparindependent manner. (A) Platelets (PLT) were incubated with sera from patients with suspected heparin (Hep)-induced thrombocytopenia (HIT, N=37), control sera (healthy controls [HC], N=6) or (B) with corresponding immunoglobulin G (IgG) isolates and tested for changes in the expression levels of P-selectin (CD62p) and phosphatidylserine (PS) via double staining in flow cytometry. Where indicated, PLT were co-incubated with low- (0.2 IU/mL) or high-dose (100 IU/mL) heparin. (C) Representative thrombin generation curve induced on PLT after incubation with IgG from HC (black lines) or HIT patients (red lines) in the presence of buffer or heparin (0.2 IU/mL). Each curve represents the amounts of generated thrombin over time in the presence of buffer (dashed lines) or heparin (0.2 IU/mL, [solid lines]). (D) Data were quantified as peak thrombin generated (nM) using Thrombinoscope software and Graphpad prism. The number of patients/IgG tested is reported in each graph. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. *P<0.05, **P<0.01, ***P<0.001, and ****P< 0.0001. ns: non-significant; CAT: calibrated automated thrombogram; pos.: positive; neg.: negative.
to investigate whether our findings were solely due to IgG-PLT interactions and not induced by unspecific serum-mediated effects, we next investigated IgG fractions from corresponding HIT patients ( Online Supplementary Figure S1 ). Compared to sera, HIT IgG isolates not only showed similar binding properties to PF4/heparin complexes (Online Supplementary Figure S2) but also induced a significant increase in procoagulant PLT formation in a heparin-dependent manner (mean % ± SEM: 33.71±3.88% vs . 4.30±1.13%; P =0.0039; and vs 1.40±0.84%; P=0.0091, respectively; Figure 1B).
Heparin-induced thrombocytopenia antibodies mediate increased thrombin generation and thrombus formation When IgG from HC were incubated with test PLT in the presence of low-dose (0.2 IU/mL) heparin, no significant increase in thrombin generation was observed compared to buffer (mean peak thrombin [nM] ± SEM: 302.70±17.47 vs. 297.90±26.20; P=0.3258; Figure 1C, D). In contrast, IgG from confirmed HIT cases were able to induce higher
levels of thrombin on the PLT surface (mean peak thrombin [nM] ± SEM: 452.70±15.67 vs. 312.00±10.76; P=<0.0001; Figure 1C, D).
Next, we established an ex vivo HIT thrombosis model that utilizes tetra staining to visualize the contribution of procoagulant PLT (Annexin V-positive, [red]), non-procoagulant PLT (DiOC6-positive, [green]), leukocytes (Hoechst 33342-positive, [blue]) and Fibrin -(ogen) (magenta) to HIT Ab-induced thrombus.29-31 In order to resemble venous shear conditions, experiments in the ex vivo thrombosis model were performed at venous shear rates as venous thrombosis is common in HIT patients.3,32,33 In a first proof of concept experiment, we tested whether our thrombosis model resembles the pathophysiology of HIT, namely heparin-dependent multi-cellular activation and subsequent vessel occlusion. As shown in Figure 2, the presence of heparin (0.2 IU/mL) resulted in a significant increase of thrombus formation while such changes were not detectable under buffer conditions (mean cumulative area of thrombus total surface area coverage [%SAC]:
Figure 2. Heparin dependency of the ex vivo heparin-induced thrombocytopenia thrombosis model. Platelet-rich plasma from healthy individuals was incubated with Immunoglobulin G (IgG) from heparin (Hep)-induced thrombocytopenia (HIT) patients in the presence of buffer (upper panel) or heparin (lower panel) prior to labeling of platelets (PLT) with DiOC6 (green), procoagulant PLT with AF647 Annexin V (red), AF546 Fibrinogen (magenta) and leukocytes with Hoechst 33342 (blue). After labeling, samples were reconstituted into autologous whole blood. Samples were then recalcified and perfused through microfluidic channels at a venous shear rate of 250s-1 (10 dyne) for 10 minutes. Images were acquired at x40 magnification in different fluorescence channels using a Zeiss Axio Observer 7 microscope. Scale bar 20μm. Images were processed identically using adjusted threshold settings and exclusion of image artefacts using Fiji image processing software. Violin plots showing the percentage of total surface area coverage (% SAC) by DiOC6, phosphatidylserine (PS), Fibrin (-ogen), number of Hoechst-positive labeled cells and cumulative area with DiOC6, PS and Fibrin (-ogen) labeled thrombus captured in the microfluidic channel. *P<0.05, **P<0.01 and ***P<0.001. ns: non-significant.

27.20±5.02% vs. 4.31±1.87%; P=0.0110; Figure 2). Interestingly, procoagulant PLT were observed to circulate and attach to the collagen surface of the microfluidic system from the beginning in our experiments (mean %SAC Annexin V ± SEM: 3.57±0.29% vs. 0.72±0.19%; P=0.0012; Figure 2; Online Supplementary Movie S1).
Furthermore, circulating procoagulant PLT incorporated later into the growing thrombus (Online Supplementary Movie S2). Additionally, heparin-dependent deposition of non-procoagulant PLT was observed in the presence of HIT Ab (mean % SAC DiOC6 ± SEM: 14.69±3.86% vs. 2.21±1.48%; P=0.020; Figure 2; Online Supplementary Movie S3). Moreover, the multicellular composition of HIT Ab-mediated thrombosis in our microfluidic system was confirmed by an increased heparin-dependent recruitment of leukocytes to the thrombi (mean number of Hoechst-positive cells ± SEM: 20±13 vs. 2±0; P=0.141; Figure 2). Finally, HIT Ab were able to activate the plasmatic coagulation system in our assays as indicated by increased Fibrin (-ogen) network generation (mean % SAC Fibrin (-ogen) ± SEM: 8.93±1.09% vs. 1.38±0.54%; P=0.0047; Figure 2; Online Supplementary Movie S4). No increased thrombus formation was induced
by IgG from HC in the presence of low-dose (0.2 IU/mL) heparin (Online Supplementary Figure S3).
Fc-γ-RIIA signaling induces procoagulant platelets in heparin-induced thrombocytopenia

We next aimed to dissect the underlying molecular mechanisms leading to HIT Ab-induced procoagulant PLT formation and increased prothrombotic potential. Pretreatment of PLT with Fc-γ-RIIA blocking monoclonal Ab IV.3 signi fi cantly reduced HIT Ab-induced procoagulant PLT formation (mean % ± SEM: 46.74±4.97% vs . 5.48±1.12%, P=0.0313; Online Supplementary Figure S3A) as well as thrombin generation on test PLT (mean peak thrombin [nM] ± SEM: 423.70±18.92 vs . 283.3±24.95; P=0.0085; Online Supplementary Figure S3B, C). Most importantly, blockade of PLT Fc- γ -RIIA inhibited HIT Ab-induced procoagulant PLT deposition (mean % SAC Annexin V ± SEM: 4.83±0.23% vs . 1.12±0.43%; P =0.0042; Figure 3). Similarly, a significant reduction in the deposition of non-procoagulant PLT was observed in the presence of monoclonal Ab IV.3 (mean % SAC DiOC6 ± SEM: 20.99±0.97% vs . 2.69±1.15%; P =0.0062; Figure 3). Fur-
Figure 3. Fc-γ-RIIA inhibition prevents thrombus formation by heparin-induced thrombocytopenia immunoglobulin G. Platelets (PLT) from healthy individuals were incubated with immunoglobulin G (IgG) from heparin (Hep)-induced thrombocytopenia (HIT) patients in the presence of heparin (0.2 IU/mL), and monoclonal antibody IV.3 (lower panel) or isotype control (upper panel) before reconstitution into whole blood and perfusion through microfluidic channels at a venous shear rate of 250s-1 (10 dyne) for 10 minutes. After perfusion, images were acquired at x40 magnification. Scale bar 20μm. Violin plots showing the percentage of total surface area coverage (% SAC) by DiOC6, phosphatidylserine (PS), Fibrin (-ogen), count of Hoechst-positive labeled cells and cumulative total % SAC with DiOC6, PS and Fibrin(-ogen) labeled thrombus captured in the microfluidic channel. *P<0.05, **P<0.01 and ***P<0.001.
thermore, blockade of PLT Fc-γ-RIIA resulted in a marked inhibition of fibrin deposition and leukocyte attachment to the thrombus surface compared to isotype control (mean % SAC Fibrin (-ogen) ± SEM: 13.90±1.59% vs. 2.95±1.49%; P=0.0317; and mean number of Hoechst-positive cells ± SEM: 19±4 vs. 5±1; P=0.0257, respectively; Figure 3).

cAMP elevation using Iloprost prevents heparin-induced thrombocytopenia antibody-induced thrombosis ex vivo Sustained high levels of intracellular calcium are a typical feature of procoagulant PLT.34 Prostacyclins are known to be able to inhibit intracellular calcium release via binding of PLT prostacyclin receptor (IP-R), activation of membrane bound adenylyl cyclase and subsequent elevation of intracellular cAMP.35 The pretreatment of PLT with the prostacyclin analogue Iloprost clearly inhibited HIT Ab-induced procoagulant PLT formation (mean % ± SEM: 35.99±5.98% vs. 4.20±1.08%; P=0.0313; Online Supplementary Figure S4A). Iloprost pretreatment also reduced procoagulant PLT-induced thrombin generation (mean peak thrombin [nM] ± SEM: 295.80±17.70 vs. 125.80±7.08; P=0.0026; Online
Supplementary Figure S5B, C). Most importantly, the deposition of non-procoagulant PLT and procoagulant PLT was significantly reduced when PLT were pretreated with Iloprost prior to HIT IgG incubation (mean % SAC DiOC6 ± SEM: 17.97±2.25% vs. 2.19±0.76%; P=0.0054; and mean % SAC Annexin V ± SEM: 4.09±0.24% vs. 1.42±0.19%; P=0.0003, respectively; Figure 4). Additionally, Iloprost pretreatment reduced Fibrin (-ogen) deposition and prevented the recruitment of leukocytes to thrombi (mean % SAC Fibrin (ogen) ± SEM: 10.40±1.41% vs. 3.49±0.90%; P=0.0037; and mean number of Hoechst-positive cells ± SEM: 14±5 vs. 2±0; P=0.0257, respectively; Figure 4).
Phosphatidylserine but not P-selectin is essential to propagate HIT antbody-mediated thrombosis
HIT Ab-induced procoagulant PLT formation and subsequent prothrombotic changes were significantly inhibited via PLT Fc-γ-RIIA blockade as well as intracellular cAMP elevation. However, these data do not allow a discrimination, whether increased CD62p or high PS on the surface of Ab-induced procoagulant PLT is the causing factor for an increased prothrombotic potential.
Figure 4. Upregulation of platelet cAMP protects from heparin-induced thrombocytopenia antibody-induced thrombus formation. Platelets (PLT) from healthy individuals were incubated with immunoglobulin G (IgG) from heparin-induced thrombocytopenia (HIT) patients in the presence of vehicle (upper panel) or Iloprost (20 nM, [lower panel]) and heparin (Hep) (0.2 IU/mL). After reconstitution into autologous whole blood and recalcification, samples were perfused through microfluidic channels at a venous shear rate of 250s-1 (10 dyne) for 10 minutes. After perfusion, images were acquired at x40 magnification. Scale bar 20µm. Violin plots showing the percentage of total surface area coverage (% SAC) by DiOC6, phosphatidylserine (PS), Fibrin (ogen), count of Hoechst-positive labeled cells and cumulative % SAC with DiOC6, PS and Fibrin (-ogen) labeled thrombus captured in the microfluidic channel. *P<0.05, **P<0.01 and ***P<0.001.
We observed a slight inhibition of thrombin generation in the presence of the CD62p blocking Ab when PLT were incubated with IgG from HC as well as from HIT patients. However, HIT IgG were still able to induce a significantly higher increase in thrombin generation compared to HC despite the inhibition of CD62p (Figure 5A, B). In our ex vivo thrombosis model, anti-CD62p Ab was able to reduce leukocyte deposition on the thrombus surface (mean number of Hoechst-positive cells ± SEM: 16±3 vs. 4±1; P=0.0434; Figure 6). The reduction of leukocyte recruitment did not affect thrombus formation as no significant alterations in the deposition of procoagulant and non-procoagulant PLT was ob-

served compared to control (mean % SAC Annexin V ± SEM: 4.80±0.67% vs. 5.27±0.21%; P=0.262; and mean % SAC DiOC6 ± SEM: 17.85±2.91% vs. 19.78±1.78%; P=0.124, respectively; Figure 6). Moreover, no changes in plasmatic coagulation, namely Fibrin (-ogen) desposition, were observed in the presence of anti-CD62p (mean % SAC Fibrin (-ogen) ± SEM: 9.71±1.42% vs. 11.68±2.31%; P=0.256; Figure 6).
Finally, we investigated the prothrombotic role of PS on the surface of HIT Ab-induced procoagulant PLT. Notably, blocking PS with Lactadherin resulted in a significant inhibition of HIT Ab-induced procoagulant PLT-mediated thrombin generation compared to vehicle (mean peak
Figure 5. Increased platelet phosphatidylserine and not CD62p causes higher thrombin generation in heparin-induced thrombocytopenia. Panel (A) shows thrombin generation potential on platelets (PLT) after incubation with wit immunoglobulin g (IgG) from heparin-induced thrombocytopenia (HIT) patients (red line) or healthy controls (HC) (black line) in the presence of vehicl e or anti-CD62p blocking antibody (Ab). (C) Thrombin generation on PLT that were incubated with different HIT patient IgG (red line) or HC IgG (black line) and treated with Lactadherin (Lact.) or vehicle, before calibrated automated thrombogram (CAT) analysis was performed. Each curve represents the amounts of generated thrombin over time induced by HIT IgG in the presence of vehicle (solid lines) or anti-CD62p blocking Ab or Lactadherin (dashed lines), respectively. (B and D) Data were quantified as peak thrombin generated (nM) using Thrombinoscope software and Graphpad prism. *P<0.05, **P<0.01 and ***P<0.001. ns: nonsignificant; CD62p: P-selectin.
thrombin [nM] ± SEM: 305.60±22.24 vs. 151.80±30.28; P=0.0040; Figure 5C, D; Online Supplementary Table S2). Our hypothesis that increased PS could be the critical mediator of increased thrombus formation was further reinforced as PS blockade with Lactadherin resulted in significant reduction of HIT Ab-induced thrombus formation (mean cumulative area of thrombus % SAC: 33.37±.1.97% vs. 6.00±1.97%; P=0.0005; Figure 7). Treatment of PLT with HIT IgG upon Lactadherin incubation resulted in marked reduction of procoagulant PLT deposition (mean % SAC Annexin V ± SEM: 4.29±0.82% vs. 0.912±0.20%; P=0.0357; Figure 7). Additionally, the presence of Lactadherin prevented non-procoagulant PLT deposition on the collagen surface (mean % SAC DiOC6 ± SEM: 18.25±2.58% vs. 2.20±0.58%; P=0.0080; Figure 7). Most importantly, PS inhibition interfered with plasmatic coagulation as a nearly complete inhibition of fibrin (-ogen) network deposition was observed (mean % SAC Fibrin (-ogen) ± SEM: 10.83±0.53% vs 2.88±1.30%; P=0.0067; Figure 7). Interestingly, also multicellular thrombus composition was affected, as a reduction of leukocytes, although not significant, was observed in the presence of Lactadherin (mean number of Hoechst positive cells ± SEM: 14±6 vs. 1±1; P=0.0602; Figure 7).
Thromboembolic events leading to high morbidity and mortality are frequent and still unpredictable complications in HIT.3 Of all laboratory findings, thrombocytopenia seems to be the most pronounced change associated with thrombosis in HIT.9,10 Although recent studies indicate the involvement of multicellular effector mechanisms, the question whether PLT alone are capable of initating a prothrombotic response in HIT, remains elusive. In this study, we found that HIT IgG Ab isolated from serologically and clinically confirmed HIT cases induce a new PLT subpopulation, namely procoagulant PLT. Our data indicate that Fc-γ-RIIA and subsequent intracellular calcium-dependent signaling pathways mediate the generation of procoagulant PLT in HIT. Most importantly, we found that procoagulant PLT-induced prothrombotic conditions are mediated by externalized PS rather than P-selectin. These findings direct towards an essential role of procoagulant PLT in the pathophysiology of thromboembolic complications in HIT. Our findings might have potential therapeutic as well as diagnostic relevance for the management of patients with suspected HIT.
Figure 6. Heparin-induced thrombocytopenia antibodies can induce thrombus independent of CD62p. Platelets (PLT) from healthy individuals were incubated with heparin-induced thrombocytopenia (HIT) patient immunoglobulin G (IgG) and treated with vehicle (upper panel) or anti-CD62p blocking antibody (lower panel). After reconstitution into autologous whole blood and recalcification, samples were perfused through microfluidic channels at a venous shear rate of 250s-1 (10 dyne) for 10 minutes. After perfusion, images were acquired at x40 magnification. Scale bar 20 µm. Violin plots showing the percentage of total surface area coverage (% SAC) by DiOC6, phosphatidylserine, Fibrin (-ogen), count of Hoechst-positive labeled cells and cumulative area of DiOC6, PS and Fibrin (-ogen) labeled thrombus in the microfluidic channel. *P<0.05, **P<0.01 and ***P<0.001. ns: non-significant. Hep: heparin.

We observed that the ability to generate procoagulant PLT is restricted to sera from patients with laboratory- and clinically-confirmed HIT. These data confirm recent findings by Lee et al. who showed that HIT patient plasma samples have the ability to induce a PLT phenotype that shows increased signal of CD62p (P-Selectin) and the arsenic-based PLT necrosis marker 4-(N-(S-glutathionylacetyl)amino) phenylarsonous acid (GSAO) using whole blood and patient plasma. The most significant difference in our setting was the absence of any additional PLT agonist (SFLLRN, thrombin analogue) that was necessary to induce the HIT patient-plasma induced PLT phenotype observed by Lee et al. 36 In fact, in our study IgG fractions from these sera modulate the expression of CD62p and PS on the surface of PLT. Hence, the ability of HIT immune complexes to induce a procoagulant PLT phenotype seems to be independent of costimulatory signals i.e., additional PLT activation by extracellular agonists like thrombin in patient sera.6,36 The discrepancy could be also due to different experimental settings. We incubated isolated wPLT with HIT Ab in the absence of other cells in our experiments.37 Preactivation of PLT during preparation of wPLT has also to be considered as a preanalytical factor
that could further explain why dual receptor activation for procoagulant PLT formation was not necessary in our studies.38,39 Additionally, a higher concentration of PF4/heparin-HIT Ab immune complexes in the fluid phase could interact directly with PLT Fc- γ -RIIA in our experimental conditions, which may have amplified Fc-γ-RIIA-mediated signaling and resulted in increased procoagulant PLT formation despite the lack of other costimulatory signals. However, and despite the use of wPLT and IgG isolates, we cannot exclude potential contribution of other cofactors.

In order to evaluate the biological relevance of HIT Ab-induced procoagulant PLT, we analyzed the interaction with the plasmatic coagulation system and other blood cells using a PLT-based thrombin generation assay and an ex vivo thrombosis model, respectively. HIT Ab-mediated significant increase in thrombin generation on PLT in a heparin-dependent fashion. Previously, HIT IgG-activated monocytes have been shown to be a main source of tissue factor release which results in the activation of factor VII and subsequent increased thrombin generation.6 Our finding directs towards the ability of HIT Ab to induce a procoagulant PS containing PLT surface that provides an
Figure 7. Platelet phosphatidylserine externalization is essential for heparin-induced thrombocytopenia antibody-induced thrombus formation. Platelets (PLT) from healthy individuals were incubated with heparin-induced thrombocytopenia (HIT) patient immunoglobulin G (IgG) and treated with vehicle (upper panel) or Lactadherin (lower panel). After reconstitution into autologous whole blood and recalcification, samples were perfused through microfluidic channels at a venous shear rate of 250s-1 (10 dyne) for 10 minutes. Images were acquired at x40 magnification. Scale bar 20μm. Violin plots showing the percentage of total surface area coverage (% SAC) by DiOC6, phosphatidylserine (PS), Fibrin (-ogen), count of Hoechst-positive labeled cells and cumulative area with DiOC6, PS and Fibrin (-ogen) positive labeled thrombus in the microfluidic channel. *P<0.05, **P<0.01 and ***P<0.001. ns: non-significant. Hep: heparin.
assembly site for increased thrombin generation. Furthermore, these results indicate a potential capability of HIT Ab-induced procoagulant PLT to cause increased ex vivo thrombosis. We optimized our thrombosis model to allow specific analysis of PLT-mediated thrombosis. In this model, PLT that were incubated with IgG from HIT patients but not HC, induced increased thrombus formation in the presence of heparin. Interestingly, PLT aggregates showed two different PLT phenotypes, namely procoagulant PLT (Annexin V-positive/DiOC6-negative) and non-procoagulant PLT (Annexin V-negative/DiOC6-positive).30,31 Recent evidence highlights that procoagulant PLT primarily evolve on the surface of the growing thrombus where they promote thrombin generation.40 In our model, circulating HIT Ab-induced procoagulant PLT attached directly to the collagen surface and later incorporated directly into the growing thrombus. Apart from this PS-positive PLT population, non-procoagulant PLT also adhered to collagen in our model. The fact that the recruitment of non-procoagulant PLT and leukocytes was inhibited by PS blockade suggests that procoagulant PLT are not only an important component in the thrombus structure but also propagate the deposition of other PLT subpopulations as well as immune cells into the growing thrombus body.41
Of potential therapeutic interest is the observation that the prostacyclin analogue Iloprost signi ficantly reduces HIT Ab-induced procoagulant PLT and thrombus formation. Interestingly, in a recent clinical study patients with acute HIT undergoing cardiac surgery were treated with Iloprost in addition to heparin.42 HIT Ab levels remained stable and no HIT Ab-induced reactivity on PLT was observed during the procedure, despite the presence of heparin. Most importantly, compared to the non-HIT control group, the incidence of thrombotic and thromboembolic events was similar (5.4% vs. 5.1%, respectively). Nevertheless, according to current HIT therapy guidelines, the use of prostacyclins is limited to cardiac surgery patients with acute HIT where surgery delay is not feasible.43 Our results suggest that Iloprost may have a potential role in the treatment of a broader HIT patient collective. However, further clinical studies are needed to test the effectiveness and safety of Iloprost in patients with HIT.
HIT Ab-induced PLT-neutrophil interplay via P-selectin/Pselectin glycoprotein ligand-1 was reported to result in NET formation and subsequent increased thrombosis in mice.16,44 In our experiments, blocking P-selectin reduced the recruitment of leukocytes to the thrombus, but did not affect procoagulant PLT-mediated thrombin generation or thrombus formation. This finding indicates that HIT Ab-induced procoagulant PLT can cause thrombus formation in the absence of direct PLT-leukocyte interactions. In contrast, the blockade of PS with Lactadherin significantly impaired the ability of HIT Ab-induced procoagulant PLT to promote thrombin generation and
thrombus formation. These data indicate an indispensable role of PS on HIT Ab-induced procoagulant PLT to link cellular with plasmatic components of the coagulation cascade, leading to thrombin burst and subsequent Fibrin (-ogen) network formation. Lactadherin also prevented the deposition of DiOC6-positive labeled PLT in the thrombus structure. This observation enforces our theory that procoagulant PLT might initiate the deposition of other PLT phenotypes such as activated PLT. The findings of our study might have several important clinical implications. Currently no predictive biomarker exists that distinguish between HIT patients with high risk for thrombosis and those with ‘‘only’’ thrombocytopenia. The detection of procoagulant PLT in HIT patients via FC could help in risk stratification for prophylactic or therapeutic anticoagulation. Without any doubt, targeting PS to inhibit propagation of the prothrombotic condition in HIT is another promising clinical aspect of our results. Although our data regarding the potential use of PS blockade to prevent thrombosis might be promising, it has to be considered that hemostatic defects with increased tail bleeding has been reported when Lactadherin was investigated in an animal model. 26 However, in this study healthy mice were used. The prothrombotic nature of HIT might lead to different findings. Therefore, further investigations that assess the effect of Lactadherin in different animal models (e.g., HIT mouse model) might shed light whether the inhibition of Ab-induced thrombus formation is associated with an increased bleeding risk. As our data provide further insights regarding the molecular mechanisms of thromboembolic complications in HIT, some technical aspects might limit immediate translation. First, our data are based on testing sera from a relatively small retrospective patient cohort. Despite significant formation of procoagulant PLT was induced by all sera from patients with confirmed HIT, thrombosis was only detected in approximately 50% of our patient cohort. In our study, we did not perform deeper examinations to detect asymptomatic thrombosis during the follow-up period (e.g., routine duplexsonography etc.). Another explanation might be a high clinical awareness of treating physicians in our specialized center which may have resulted in prompt discontinuation of heparin and advanced treatment with alternative antiocoagulants. Second, our study does not provide data on the dynamics of thrombus formation. Future research attempts should focus on the sequence of events and the relevance of intercellular interactions to enable better identification of procoagulant PLT role in the initiation and propagation of thrombus formation. Additionally, procoagulant PLT-leukocyte interactions have to be investigated in future research attempts to conclude whether neutrophils and other immune cells are required for procoagulant PLT-mediated thrombus formation. Finally, while our ex vivo thrombosis
model might provide information on the interaction between blood cells, further investigations are needed to assess the influence of other cell types (e.g., endothelial cells). Additional in vivo approaches that utilize Fc-γ-RIIA transgenic mice or a humanized PLT transfusion mouse model would allow robust confirmation of our results. In conclusion, our study suggests an indispensable role of procoagulant PLT in the pathophysiology of HIT-associated thrombosis. The inhibition of HIT Ab-induced procoagulant PLT formation with Iloprost or the inhibition of prothrombotic effects with PS targeting specific therapeutics could be a promising approach to prevent the onset of thromboembolic events in HIT patients.
Disclosures
TB has received research funding from CoaChrom Diagnostica GmbH, DFG, Robert Bosch GmbH, Stiftung Transfusionsmedizin und Immunhämatologie e.V.; Ergomed, DRK Blutspendedienst, Deutsche Herzstiftung, Ministerium für Wissenschaft, Forschung und Kunst Baden-Wuerttemberg; has received lecture honoraria from Aspen Germany GmbH, Doctrina Med AG, Meet The Experts Academy UG, Mattsee, Stago GmbH, Mitsubishi Tanabe Pharma GmbH, Novo Nordisk Pharma GmbH; has provided expert witness testimony relating to heparin-induced thrombocytopenia (HIT) and non-HIT thrombocytopenic and coagulopathic disorders. All of these are outside the current work. TB, JZ and KA submitted a patent for the detection of procoagulant pla-
1. Chong BH, Fawaz I, Chesterman CN, Berndt MC. Heparininduced thrombocytopenia: mechanism of interaction of the heparin-dependent antibody with platelets. Br J Haematol. 1989;73(2):235-240.
2. Greinacher A. CLINICAL PRACTICE. Heparin-induced thrombocytopenia. N Engl J Med. 2015;373(3):252-261.
3. Arepally GM, Padmanabhan A. Heparin-induced thrombocytopenia: a focus on thrombosis. Arterioscler Thromb Vasc Biol. 2021;41(1):141-152.
4. Amiral J, Bridey F, Dreyfus M, et al. Platelet factor 4 complexed to heparin is the target for antibodies generated in heparininduced thrombocytopenia. Thromb Haemost. 2018;68(01):95-96.
5. Cai Z, Zhu Z, Greene MI, Cines DB. Atomic features of an autoantigen in heparin-induced thrombocytopenia (HIT). Autoimmun Rev. 2016;15(7):752-755.
6. Tutwiler V, Madeeva D, Ahn HS, et al. Platelet transactivation by monocytes promotes thrombosis in heparin-induced thrombocytopenia. Blood. 2016;127(4):464-472.
7. Reilly MP, Taylor SM, Hartman NK, et al. Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcgammaRIIA. Blood. 2001;98(8):2442-2447.
8. Lhermusier T, van Rottem J, Garcia C, et al. The Syk-kinase inhibitor R406 impairs platelet activation and monocyte tissue factor expression triggered by heparin-PF4 complex directed antibodies. J Thromb Haemost. 2011;9(10):2067-2076.
telets as a diagnostic tool for HIT and vaccine-induced thrombotic thrombocytopenia. All other authors have no conflicts of interest to disclose.
Contributions
JZ and TB designed the study. KA and TB were responsible for the treatment of the patients . JZ and KA collected and analyzed the clinical data. JZ, AS, KW, HJ, LP, and KA performed the experiments. JZ, AS, KA, GU and TB analyzed the data, interpreted the results and wrote the manuscript. All authors read and approved the manuscript.
We thank Flavianna Rigoni and Andreas Witzemann for their excellent technical support. The authors thank Susanne Staub for language editing of the article.
This work was supported by grants from the German Red Cross and the Herzstiftung (to TB) (BA5158/4 and TSGStudy). This project was supported by the German Research Foundation (DFG) - project number 374031971 - TRR 240 and the Rudolf Marx Scholarship from the Society for Thrombosis and Haemostasis Research (GTH).
Data generated from this study are available from the corresponding author upon reasonable request.
9. Greinacher A, Farner B, Kroll H, Kohlmann T, Warkentin TE, Eichler P. Clinical features of heparin-induced thrombocytopenia including risk factors for thrombosis. A retrospective analysis of 408 patients. Thromb Haemost. 2005;94(1):132-135.
10. Warkentin TE, Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med. 1996;101(5):502-507.
11. Tardy-Poncet B, Piot M, Chapelle C, et al. Thrombin generation and heparin-induced thrombocytopenia. J Thromb Haemost. 2009;7(9):1474-1481.
12. Warkentin TE, Hayward CP, Boshkov LK, et al. Sera from patients with heparin-induced thrombocytopenia generate platelet-derived microparticles with procoagulant activity: an explanation for the thrombotic complications of heparininduced thrombocytopenia. Blood. 1994;84(11):3691-3699.
13. Greinacher A, Eichler P, Lubenow N, Kwasny H, Luz M. Heparininduced thrombocytopenia with thromboembolic complications: meta-analysis of 2 prospective trials to assess the value of parenteral treatment with lepirudin and its therapeutic aPTT range. Blood. 2000;96(3):846-851.
14. Kasthuri RS, Glover SL, Jonas W, et al. PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcgammaRI. Blood. 2012;119(22):5285-5293.
15. Khandelwal S, Barnes A, Rauova L, et al. Complement mediates binding and procoagulant effects of ultralarge HIT immune
complexes. Blood. 2021;138(21):2106-2116.
16. Perdomo J, Leung HHL, Ahmadi Z, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparininduced thrombocytopenia. Nat Commun. 2019;10(1):1322.
17. Reddy EC, Rand ML. Procoagulant phosphatidylserine-exposing platelets in vitro and in vivo. Front Cardiovasc Med. 2020;7:15.
18. Dale GL, Friese P, Batar P, et al. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002;415(6868):175-179.
19. Heemskerk JWM, Vuist WMJ, Feijge MAH, Reutelingsperger CPM, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood. 1997;90(7):2615-2625.
20. Agbani EO, van den Bosch MTJ, Brown E, et al. Coordinated membrane ballooning and procoagulant spreading in human platelets. Circulation. 2015;132(15):1414-1424.
21. Althaus K, Moller P, Uzun G, et al. Antibody-mediated procoagulant platelets in SARS-CoV-2-vaccination associated immune thrombotic thrombocytopenia. Haematologica. 2021;106(8):2170-2179.
22. Althaus K, Marini I, Zlamal J, et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood. 2021;137(8):1061-1071.
23. Greinacher A, Michels I, Kiefel V, Mueller-Eckhardt C. A rapid and sensitive test for diagnosing heparin-associated thrombocytopenia. Thromb Haemost. 1991;66(6):734-736.
24. Zlamal J, Althaus K, Jaffal H, et al. Upregulation of cAMP prevents antibody-mediated thrombus formation in COVID-19. Blood Adv. 2022;6(1):248-258.
25. Shi J, Gilbert GE. Lactadherin inhibits enzyme complexes of blood coagulation by competing for phospholipid-binding sites. Blood. 2003;101(7):2628-2636.
26. Shi J, Pipe SW, Rasmussen JT, Heegaard CW, Gilbert GE. Lactadherin blocks thrombosis and hemostasis in vivo: correlation with platelet phosphatidylserine exposure. J Thromb Haemost. 2008;6(7):1167-1174.
27. Mangin PH, Gardiner EE, Nesbitt WS, et al. In vitro flow based systems to study platelet function and thrombus formation: recommendations for standardization: communication from the SSC on Biorheology of the ISTH. J Thromb Haemost. 2020;18(3):748-752.
28. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an opensource platform for biological-image analysis. Nat Methods. 2012;9(7):676-682.
29. Agbani EO, Williams CM, Hers I, Poole AW. Membrane ballooning in aggregated platelets is synchronised and mediates a surge in microvesiculation. Sci Rep. 2017;7(1):2770.
30. Jobe SM, Wilson KM, Leo L, et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood. 2008;111(3):1257-1265.
31. Dale GL. Procoagulant platelets: further details but many more questions. Arterioscler Thromb Vasc Biol. 2017;37(9):1596-1597.
32. Coenen DM, Mastenbroek TG, Cosemans JMEM. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood. 2017;130(26):2819-2828.
33. Lipowsky HH, Kovalcheck S, Zweifach BW. The distribution of blood rheological parameters in the microvasculature of cat mesentery. Circ Res. 1978;43(5):738-749.
34. Abbasian N, Millington-Burgess SL, Chabra S, Malcor JD, Harper MT. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv. 2020;4(1):154-164.
35. Smolenski A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J Thromb Haemost. 2012;10(2):167-176.
36. Lee CSM, Selvadurai MV, Pasalic L, et al. Measurement of procoagulant platelets provides mechanistic insight and diagnostic potential in heparin-induced thrombocytopenia. J Thromb Haemost. 2022;20(4):975-988.
37. Rauova L, Hirsch JD, Greene TK, et al. Monocyte-bound PF4 in the pathogenesis of heparin-induced thrombocytopenia. Blood. 2010;116(23):5021-5031.
38. Reddel CJ, Pennings GJ, Curnow JL, Chen VM, Kritharides L. Procoagulant effects of low-level platelet activation and its inhibition by colchicine. Thromb Haemost. 2018;118(4):723-733.
39. Lindahl T, Rånby M, Ramström S. Platelet phosphatidylserine exposure and procoagulant activity in clotting whole blood –different effects of collagen, TRAP and calcium ionophore A23187. Thromb Haemost. 2017;89(01):132-141.
40. Munnix IC, Kuijpers MJ, Auger J, et al. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. 2007;27(11):2484-2490.
41. Agbani EO, Hers I, Poole AW. Temporal contribution of the platelet body and balloon to thrombin generation. Haematologica. 2017;102(10):e379-e381.
42. Palatianos G, Michalis A, Alivizatos P, et al. Perioperative use of iloprost in cardiac surgery patients diagnosed with heparininduced thrombocytopenia-reactive antibodies or with true HIT (HIT-reactive antibodies plus thrombocytopenia): an 11-year experience. Am J Hematol. 2015;90(7):608-617.
43. Cuker A, Arepally GM, Chong BH, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: heparin-induced thrombocytopenia. Blood Adv. 2018;2(22):3360-3392.
44. Gollomp K, Kim M, Johnston I, et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight. 2018;3(18):e99445.
Correspondence: N. Kalleda
natarajaswamy.kalleda@viforpharma.com
Received: October 28, 2022.
Research and Non-clinical Development, CSL Vifor, St. Gallen, Switzerland
°GI current address: Avelo AG, Schlieren, Switzerland. °CD current address: Anjarium Biosciences, Zurich, Switzerland.

Accepted: May 4, 2023.
Early view: May 11, 2023.
htps://doi.org/10.3324/haematol.2022.282328
Published under a CC BY license
β-thalassemia is an inherited anemia characterized by ineffective erythropoiesis. Blood transfusions are required for survival in transfusion-dependent β-thalassemia and are also occasionally needed in patients with non-transfusion-dependent βthalassemia. Patients with transfusion-dependent β-thalassemia often have elevated transferrin saturation (TSAT) and nontransferrin-bound iron (NTBI) levels, which can lead to organ iron overload, oxidative stress, and vascular damage. Vamifeport is an oral ferroportin inhibitor that was previously shown to ameliorate anemia, ineffective erythropoiesis, and dysregulated iron homeostasis in the Hbbth3/+ mouse model of β-thalassemia, under non-transfused conditions. Our study aimed to assess the effects of oral vamifeport on iron-related parameters (including plasma NTBI levels) and ineffective erythropoiesis following blood transfusions in Hbbth3/+ mice. A single dose of vamifeport prevented the transient transfusion-mediated NTBI increase in Hbbth3/+ mice. Compared with vehicle treatment, vamifeport significantly increased hemoglobin levels and red blood cell counts in transfused mice. Vamifeport treatment also significantly improved ineffective erythropoiesis in the spleens of Hbbth3/+ mice, with additive effects observed when treatment was combined with repeated transfusions. Vamifeport corrected leukocyte counts and significantly improved iron-related parameters (serum transferrin, TSAT and erythropoietin levels) versus vehicle treatment in Hbbth3/+ mice, irrespective of transfusion status. In summary, vamifeport prevented transfusion-mediated NTBI formation in Hbbth3/+ mice. When given alone or combined with blood transfusions, vamifeport also ameliorated anemia, ineffective erythropoiesis, and dysregulated iron homeostasis. Administering vamifeport together with repeated blood transfusions additively ameliorated anemia and ineffective erythropoiesis in this mouse model, providing preclinical proof-of-concept for the efficacy of combining vamifeport with blood transfusions in β-thalassemia.
β-thalassemia is an inherited anemia caused by mutations in the β-globin gene of hemoglobin, which decrease the synthesis of β-globin chains, resulting in an imbalance in α- and β-globin chains and an accumulation of insoluble α-globin aggregates on the membranes of red blood cells (RBC).1-3 These α-globin aggregates contain free heme and iron, which can cause the formation of reactive oxygen species that reduce the lifespan of RBC and result in anemia and tissue hypoxia. In order to compensate for this, erythropoiesis is stimulated in patients with β-thalassemia, which leads to the increased proliferation but decreased differentiation of erythroid precursors (so-called ineffective erythropoiesis).1,2,4-6 As levels of the ferropor-
tin-blocking hormone hepcidin are suppressed by erythropoiesis, iron deficiency, and hypoxia, β-thalassemia is also associated with organ iron overload due to increased dietary iron absorption via ferroportin-mediated iron export from enterocytes, as well as iron efflux from spleen macrophages recycling damaged erythrocytes.1,3,7,8
Thalassemia is often classified based on the transfusion requirement of the patient. Those with non-transfusiondependent thalassemia usually receive only occasional blood transfusions,9 while patients with the most severe form, transfusion-dependent β-thalassemia, require regular blood transfusions for survival, which can lead to secondary iron overload that requires iron chelation therapy.10 Moreover, these patients often show elevated transferrin saturation (TSAT) and non-transferrin-bound iron (NTBI),
Natarajaswamy Kalleda, Anna Flace, Patrick Altermatt, Giada Ingoglia, Cédric Doucerain, Naja Nyffenegger, Franz Dürrenberger and Vania Manolovatriggered by ferroportin-mediated iron release from macrophage recycling of damaged RBC, which can lead to oxidative stress, vascular injury and organ iron overload.8,11 In addition, the accelerated recycling of RBC due to blood transfusions results in a high serum iron concentration and an increase in NTBI that correlates with the occurrence of heart disease.12
One promising therapeutic approach to correcting the dysregulated iron metabolism seen in patients with β-thalassemia is to target the hepcidin-ferroportin axis,13,14 for example by substituting for the reduced endogenous hepcidin levels by administering a synthetic small-molecule ferroportin inhibitor. Vamifeport (CSL Vifor, St. Gallen, Switzerland), also known as VIT-2763, is an oral ferroportin inhibitor that is currently being investigated as a potential treatment option for patients with β-thalassemia and sickle cell disease.8,15 Limiting iron availability with vamifeport ameliorated anemia, ineffective erythropoiesis, and dysregulated iron homeostasis in the Hbbth3/+ mouse model of β -thalassemia, under non-transfused conditions.1 This mouse model closely resembles the pathophysiology of patients with β-thalassemia intermedia.16
It is characterized by ineffective erythropoiesis, splenomegaly, and secondary iron overload and has been used to test the efficacy of other candidate drugs for β-thalassemia.17 The aim of the present study was to determine the effect of vamifeport on plasma NTBI levels and ineffective erythropoiesis following a single transfusion or repeated blood transfusions in the Hbbth3/+ mouse model of β-thalassemia.
Ethics statement
Experiments complied with Swiss law and associated animal experimentation guidelines were approved by the responsible authority (Veterinary Department, Canton Zurich; License Number ZH108/2017). Studies complied with the CSL Vifor Code of Conduct and the principles of 3R (Replace, Reduce, Refine). Experimental mice were scored daily for body weight and physical condition.
Animal model
Hbbth3/+ mice16 were bred in the CSL Vifor animal facility, which employs 12-hour dark/12-hour light cycles. These non-blinded studies utilized 12–14-week-old Hbbth3/+ male and female mice (7-8 per group). Mice were housed 3–5 per cage and provided with standard chow (containing 250 ppm iron) and water ad libitum. Experiments were conducted in the experimental room; animals were taken to the holding room after each procedure.
In the single-transfusion studies, mice received singledose vamifeport (120 mg/kg; 10 mL/kg), or vehicle (0.5%
methylcellulose in Milli-Q purified water), by oral gavage. Thirty minutes post dose, mice received a 200 μL tail vein transfusion of blood from preterminally anesthetized sexmatched wild-type mice, ubiquitously expressing green fluorescent protein (GFP).18 Animals were terminally anesthetized using isoflurane 3 or 6 hours post transfusion and peripheral blood was collected. Two studies was conducted, one in males, one in females, and data was pooled.
In the repeated-transfusion studies, transfusions were administered on days 1, 9, 16, and 22 or 23. Hbbth3/+ mice received once-daily oral vamifeport 120 mg/kg or vehicle (on transfusion days, dosing was 30 minutes before transfusion). Animals were terminally anesthetized approximately 24 hours after the fourth transfusion. Non-transfused, vehicle-treated Hbbth3/+ mice or wild-type littermates acted as controls.

Serum NTBI concentrations were measured19 in all studies; the remaining parameters were assessed after repeated transfusion. Serum iron (Abbott) and serum transferrin (Abcam) were analyzed using commercial kits according to manufacturer’s instructions. TSAT levels were calculated according to the formula:
Liver Hamp1 and spleen erythroferrone (Erfe) messenger RNA (mRNA) levels were determined by quantitative polymerase chain reaction using PrimePCR™ SYBR® Green Assays (BIO-RAD). Plasma hepcidin levels were determined using enzyme-linked immunosorbent assay (Intrinsic Lifesciences). Organ iron levels were determined as previously described.1
All hematological parameters were analyzed using a ProCyte analyzer (Idexx Bioresearch). Spleen erythroid cells were analyzed by flow cytometry.1 Distinct stages of RBC precursors were identified based on TER119, CD44, and CD71 expression levels, and using the forward scatter to measure cell size. Donor-derived GFP-positive RBC were excluded from the analysis.
At least 450 μL of blood were needed for the preparation of membrane cytoskeletons and so blood samples were pooled from two mice from the same treatment group in these analyses, meaning that each data point was obtained from two biological replicates. Harvested membrane cytoskeletons were snap frozen and analyzed using TAU polyacrylamide gel electrophoresis.1
Assessment of parameters over time used two-way analysis of variance (ANOVA), with repeated measures for time course effects. Where significant effects were observed, post hoc tests were performed using Dunnett’s adjustment for multiple comparisons, where each group was compared with vehicle-treated non-transfused or vehicletreated transfused controls. Endpoint parameters were analyzed using one-way ANOVA with Dunnett’s multiple comparison test. All analyses were performed using GraphPad Prism, Version 8.4.2 (San Diego, CA).
Vamifeport reduced plasma non-transferrin-bound iron levels afer a single blood transfusion in Hbbth3/+ mice
A single blood transfusion resulted in significantly higher plasma NTBI levels (mean [standard deviation] 1.35 [±0.55] μ M) compared with those observed in non-transfused Hbbth3/+ mice (mean [standard deviation] 0.55 [±0.18] μM) (Figure 1). NTBI levels were transiently elevated 3 hours after blood transfusion in vehicle-treated transfused mice. A single oral dose of vamifeport prevented the plasma NTBI increase seen in transfused vehicle-treated Hbbth3/+ mice at 3 hours post transfusion, with levels remaining similar to those observed in non-transfused controls, demonstrating the potential of vamifeport to sequester iron recycled from transfused RBC. Non-trans-
fused, vehicle-treated Hbbth3/+ mice had low NTBI levels (mean [standard deviation] 0.55 [±0.185] μ M). NTBI returned to baseline levels by 6 hours in vehicle-treated transfused mice, suggesting that damaged RBC are cleared from the circulation and their heme iron recycled by macrophages in the first few hours after transfusion in this mouse model. Similar results were observed for donor blood collected 1 day or 15 days before transfusion, suggesting that prolonging the storage of blood before transfusion does not appear to increase the levels of NTBI. In vehicle-treated mice after repeated blood transfusions, no significant effect was observed on plasma NTBI levels, supporting an acute increase in NTBI post transfusion and reflecting the late time point of blood sampling (24 hours post blood transfusion). As anticipated, vamifeport alone or combined with repeated transfusions had no significant effects on chronic NTBI levels (Online Supplementary Figure S1).

Vamifeport improved hematological parameters in Hbbth3/+ mice receiving repeated blood transfusions Hbbth3/+ mice have lower levels of hemoglobin than their wild-type counterparts. Two days after the first blood transfusion, significantly higher hemoglobin levels were observed in Hbbth3/+ mice compared with non-transfused controls, both in the presence or absence of vamifeport (Figure 2). Vamifeport alone increased hemoglobin levels starting from study day 11 in Hbbth3/+ mice. Hemoglobin levels were significantly higher on study day 25 in the vamifeport-
Figure 1. Non-transferrin-bound iron levels in Hbbth3/+ mice afer vamifeport treatment alone or in combination with a single blood transfusion. Representative scatter plots of individual values (with mean and standard deviation) showing the effects of transfusion, vehicle, and vamifeport treatment. Donor blood was collected (A) 1 day or (B) 15 days before the blood transfusion. Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison test. Significant differences compared with the Hbbth3/+ vehicle transfused group are indicated as: *P<0.05, **P<0.01, and ***P<0.001. NTBI: non-transferrinbound iron.
reticulocyte counts in this mouse model. Vamifeport treatment significantly increased RBC counts in Hbbth3/+ mice, independent of blood transfusion status. Vamifeport treatment also signi fi cantly increased hematocrit levels and decreased reticulocyte counts, independent of blood transfusion status. Furthermore, the transfusionmediated rise in hematocrit was additively increased when transfusion was combined with vamifeport treatment.
Haematologica | 108 - October 2023 2706 ARTICLE - Vamifeport in β-thalassemia model with transfusion N. Kalleda et al. D

Vamifeport reduced the formation of α-globin aggregates in the membranes of red blood cells in Hbbth3/+ mice receiving repeated blood transfusions
The imbalanced synthesis of α- and β-globin chains of hemoglobin in Hbbth3/+ mice compared with wild-type mice leads to the formation of α -globin aggregates in RBC membranes.16 Vamifeport, alone or in combination with repeated blood transfusions, decreased α-globin aggregates in the RBC of Hbbth3/+ mice (Figure 3). Repeated blood transfusions alone caused a less pronounced reduction of α -globin aggregates in RBC compared with vamifeport plus repeated transfusion.

Vamifeport combined with repeated blood transfusions additively improved ineffective erythropoiesis in Hbbth3/+ mice
Non-transfused, vehicle-treated Hbbth3/+ mice exhibited ineffective erythropoiesis with increased proliferation of
immature erythroid cells (proerythroblasts, basophilic erythroblasts, polychromatic erythroblasts, and orthochromatic erythroblasts) and decreased differentiation of erythroid progenitors to mature erythrocytes in the spleen, compared with their wild-type counterparts (Figure 4). Treatment with either vamifeport, or repeated blood transfusions alone, significantly improved ineffective erythropoiesis in the spleens of recipient Hbbth3/+ mice. Combining vamifeport with blood transfusions resulted in a lower proportion of immature erythroid cells and a higher percentage of mature erythrocytes, compared with either treatment alone, suggesting additive effects of vamifeport and repeated blood transfusions on erythropoiesis (donor-derived RBC were excluded from the erythropoiesis analysis).
Hbbth3/+ mice have increased spleen weight due to ineffective erythropoiesis in their spleens. In agreement with the improvement in ineffective erythropoiesis, repeated
Figure 3. α-globin aggregates in red blood cell membranes of Hbbth3/+ mice afer vamifeport treatment alone or in combination with repeated blood transfusions. (A) TAU gel electrophoresis of membrane-bound globins in red blood cells from Hbbth3/+ and wild-type (WT) mice. Soluble α and β hemoglobin from wild-type red blood cells are shown for reference. (B) Quantification of the signal intensity of the TAU gel α bands by densitometry represented as a scatter plot of individual values (with mean and standard deviation).
Figure 4. Erythropoiesis in the spleens of Hbbth3/+ mice afer vamifeport treatment alone or in combination with repeated blood transfusions. (A) Gating strategy used to identify erythroid progenitors in the spleen by flow cytometry: gate I, proerythroblast; gate II, basophilic erythroblast; gate III, polychromatic erythroblast; gate IV, orthochromatic erythroblast; gate V, mature erythrocytes. Green fluorescent protein-positive (GFP+) transfused donor cells are excluded from the analysis. The remaining figures are representative scatter plots of individual values (with mean and standard deviation) showing the effects of transfusion, vehicle, and vamifeport treatment at the end of the study on levels of: (B) proerythroblasts, (C) basophilic erythroblasts, (D) polychromatic erythroblasts, (E) orthochromatic erythroblasts, and (F) mature erythrocytes. WT: wild-type. Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison test. Significant differences compared with the Hbbth3/+ vehicle non-transfused group (black) and vehicle-transfused group (purple) are indicated as: *P<0.05, **P<0.01, and ***P<0.001. Significant differences between the vamifeport-treated transfused group compared with the vamifeport-treated non-transfused group are indicated in red (unpaired t test).

transfusions significantly reduced splenomegaly; splenomegaly was further reduced with vamifeport alone or combined with transfusion (Online Supplementary Figure S2).
Vamifeport corrected leukocyte counts in Hbbth3/+ mice receiving repeated blood transfusions
Non-transfused, vehicle-treated Hbbth3/+ mice had significantly elevated overall leukocyte counts, including neutrophil, lymphocyte, and monocyte counts compared with their wild-type counterparts (Figure 5). Repeated blood transfusions significantly reduced overall leukocyte, neutrophil, and lymphocyte counts, but did not significantly impact monocyte numbers in Hbbth3/+ mice. Vamifeport, alone or in combination with repeated blood transfusions,
significantly reduced overall leukocyte, neutrophil, and lymphocyte counts, and also significantly reduced monocyte counts in Hbbth3/+ mice. Levels of all these cells were similar to those observed in wild-type mice, demonstrating that iron restriction by vamifeport not only corrects ineffective erythropoiesis, but also leads to amelioration of abnormal myelopoiesis (both recipient and donor blood cells were included in these analyses).
Vamifeport improved iron-related parameters in Hbbth3/+ mice receiving repeated blood transfusions
Hbbth3/+ mice had significantly lower serum iron and TSAT levels and significantly higher serum transferrin and erythropoietin levels than their wild-type counterparts (Figure 6). Repeated blood transfusions had no signifi cant
Figure 5. Leukocyte, neutrophil, lymphocyte, and monocyte counts in Hbbth3/+ mice afer vamifeport treatment alone or in combination with repeated blood transfusions. Representative scatter plots of individual values (with mean and standard deviation) showing the effects of transfusion, vehicle, and vamifeport treatment at the end of the study on counts of: (A) leukocytes, (B) neutrophils, (C) lymphocytes, (D) monocytes. WT: wild-type. Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison test. Significant differences compared with the Hbbth3/+ vehicle non-transfused group (black) are indicated as: *P<0.05, **P<0.01, and ***P<0.001. There were no significant differences between any other treatment group compared with the vehicle-transfused group, for any of these parameters.

Figure 6. Iron-related parameters in Hbbth3/+ mice afer vamifeport treatment alone or in combination with repeated blood transfusions. Representative scatter plots of individual values (with mean and standard deviation) showing the effects of transfusion, vehicle, and vamifeport treatment at the end of the study on: (A) serum iron, (B) transferrin, (C) transferrin saturation, (D) erythropoietin, (E) Hamp1 mRNA, (F) serum hepcidin. WT: wild-type. Analysis was performed by using one-way analysis of variance with Dunnett’s multiple comparison test. Significant differences compared with the Hbbth3/+ vehicle nontransfused group (black) and vehicle-transfused group (purple) are indicated as: *P<0.05, **P<0.01, and ***P<0.001.

effect on serum iron or plasma erythropoietin levels but significantly decreased serum transferrin levels and significantly increased TSAT, liver Hamp expression, and plasma hepcidin levels. Vamifeport, alone or in combination with transfusions, significantly decreased serum iron, serum transferrin and TSAT levels in Hbbth3/+ mice when compared with vehicle-treated animals, reflecting the ability of vamifeport to inhibit ferroportin-mediated iron efflux into the plasma. This iron-lowering effect was transient, with the effect reversed 4–6 hours post dose, reflecting the pharmacodynamic activity of vamifeport inhibiting ferroportin. Vamifeport also significantly decreased plasma erythropoietin levels, both alone and when combined with repeated transfusions. Non-transfused, vehicle-treated Hbbth3/+ mice had similar liver Hamp1 mRNA levels and serum hepcidin levels as compared with wild-type mice. As expected, repeated blood transfusions significantly increased liver Hamp1 mRNA levels and serum hepcidin levels, compared with nontransfused, vehicle-treated controls. Vamifeport alone or combined with transfusion significantly decreased liver Hamp1 expression compared with vehicle-treated transfused Hbbth3/+ mice. Serum hepcidin levels in transfused Hbbth3/+ mice also reduced following vamifeport treatment, but this did not reach statistical significance.
Non-transfused, vehicle-treated Hbbth3/+ mice had elevated liver and kidney iron concentrations compared with their wild-type counterparts; heart iron concentration was significantly lower in Hbbth3/+ mice compared with wildtype mice (Online Supplementary Figure S3). Repeated blood transfusions had no effects on iron concentrations in liver or heart, but increased kidney iron levels in Hbbth3/+ mice, likely due to iron derived from transfused RBC.11 In transfused or non-transfused Hbbth3/+ mice, vamifeport had no significant effects compared with vehicle treatment on liver and heart iron content, suggesting that vamifeport does not remove existing iron, but rather prevents further iron accumulation in cells from these tissues.1 In contrast, vamifeport significantly reduced spleen iron content in non-transfused Hbbth3/+ mice (Online Supplementary Figure S2). Although transfusion alone did not affect spleen iron content, it did signi ficantly increase spleen iron concentration, which might be due to iron originating from transfused RBC. Vamifeport alone significantly increased spleen iron concentration and combining vamifeport with transfusion resulted in an additive effect. This additive effect most likely reflects iron derived from recycled RBC. Hbbth3/+ mice have significantly increased spleen Erfe expression compared with wild-type control mice. Repeated transfusions had no effect on spleen Erfe expression, but when transfusion was combined with vamifeport treatment, Erfe expression was significantly reduced. Vamifeport alone had no effect on spleen Erfe expression in this experimental setting in Hbbth3/+ mice.
After a single blood transfusion, vamifeport prevented the formation of blood transfusion-mediated NTBI in the plasma of Hbbth3/+ mice, suggesting that the iron restriction resulting from ferroportin inhibition prevents the formation of potentially harmful reactive plasma iron species. In the present study, plasma NTBI levels returned to baseline levels at 6 hours post blood transfusion; one possible explanation for this is that blood transfusion induces hepcidin (Hamp1) expression, which prevents the formation of NTBI. Another possible explanation for this is that NTBI is rapidly cleared from plasma by parenchymal tissues, as has previously been demonstrated in rodent models.20 We also assessed the longer-term effects of vamifeport after repeated blood transfusions; these studies did not use the same time points as the single blood transfusion study (3 hours and 6 hours post transfusion) due to ethical concerns with repeated blood sampling in mice so soon after tail vein blood transfusion. NTBI did not accumulate in Hbbth3/+ mice after four repeated blood transfusions, hence we did not observe any effect on NTBI levels with vamifeport treatment, most likely due to the timing of the blood sampling post transfusion (24 hours). These observations are of interest as patients with transfusion-dependent β-thalassemia receiving regular transfusions and iron chelation therapy have been shown to have elevated NTBI levels.12 Those with heart disease reportedly also had significantly higher NTBI levels than those without,12 suggesting a potential link between transfusional iron overload and heart disease. Our findings in this mouse model suggest that vamifeport might prevent such adverse effects by sequestrating NTBI in macrophages, thereby interrupting a vicious cycle in β-thalassemia.1
Vamifeport ameliorated anemia and improved ineffective erythropoiesis and dysregulated iron homeostasis in Hbbth3/+ mice, either when administered alone or in combination with repeated blood transfusions. Importantly, when given along with blood transfusions, vamifeport additively ameliorated ineffective erythropoiesis in Hbbth3/+ mice. The effects of vamifeport on erythropoiesis were also associated with a significant reduction in splenomegaly. These results are consistent with our previous observations in Hbbth3/+ mice under non-transfused conditions.1 β-thalassemia is associated with several abnormalities of the innate immune system. The Hbbth3/+ mice show substantially elevated leukocyte counts compared with wild-type mice, as we have previously reported.21 Furthermore, vamifeport normalizes the myeloid spleen composition by reducing the proportion of immature myeloid cells and inflammatory monocytes.1 It has also been shown that the proportion of immature myeloid cells is significantly increased in Hbbth3/+ mice compared
with wild-type mice,22 suggesting that abnormal myeloid cell maturation could contribute to the increased levels of circulating granulocytes seen in Hbbth3/+ mice. We have recently demonstrated that vamifeport reduces systemic and vascular inflammation in Townes mice (a sickle cell disease model) as shown by lower levels of Cxcl1, RANTES and soluble P-selectin levels.15 Vamifeport treatment also reduced peripheral neutrophil counts in this model. Furthermore, plasma iron has been shown to control neutrophil production and function, and neutrophil production is much more iron-demanding than the generation of other white blood cell types.23 Hence, we hypothesize that iron restriction by vamifeport-induced hypoferremia in Hbbth3/+ mice, could be a possible mechanism for the observed normalization of white blood cell counts during vamifeport treatment.
In our study, vamifeport also reduced the transfusion-mediated increase in hepcidin (Hamp1) expression, which led to an associated reduction in serum hepcidin levels. Although hepcidin levels increase following transfusions in patients with transfusion-dependent β-thalassemia,7 hepcidin levels may decrease during the intervals between blood transfusions, which could result in the formation of NTBI via ferroportin. It is possible that the ferroportin inhibitor vamifeport might prevent these noxious effects by two mechanisms: i) sequestration of iron from recycled RBC in macrophages; ii) prevention of increased intestinal iron absorption during the intervals between blood transfusions. Based on the data from the present study, it seems most likely that vamifeport inhibits ferroportin on macrophages recycling iron from engulfed RBC, and thereby prevents the generation of plasma NTBI. The effects of other candidate drugs for β -thalassemia have been investigated in Hbbth3/+ mice, supporting the validity of this model. In β-thalassemia, increased erythropoietin production leads to persistent phosphorylation of JAK2, activated STAT5, and increased erythroid progenitor levels in the spleen, which result in splenomegaly and contribute to ineffective erythropoiesis.6 In Hbbth3/+ mice, administration of JAK2 inhibitors has been shown to decrease the number of erythroid nucleated cells in the spleen and to reduce splenomegaly, but was also associated with a slight reduction in hemoglobin levels.17 Coadministration of JAK2 inhibitors with blood transfusion further reduced splenomegaly in both Hbbth3/+ mice and mice requiring chronic transfusions (Hbbth3/th3).17 However, co-administration did not significantly affect hemoglobin or circulating RBC levels. Combining vamifeport with blood transfusions in the current study produced additive effects on erythropoiesis, with reduced proportions of immature erythroid cells and increased proportions of mature erythrocytes observed, and a reduction in splenomegaly, compared with either treatment alone. In contrast to findings with JAK2 inhibitors, however, vamifeport sig-
nificantly increased hemoglobin levels when given alone or combined with transfusion. The effects of apo-transferrin on NTBI levels have also been assessed in the Hbbth3/+ mouse model.24 The rapid reduction in NTBI levels observed after vamifeport treatment in the present study are in line with data reported after the administration of apo-transferrin. The effects of the SMAD2/3 signaling inhibitor RAP-536 (the mouse version of luspatercept) have been investigated in a different mouse model of β-thalassemia intermedia (Hbbth1/th1).25,26 In this model, RAP-536 had similar effects to those reported here for vamifeport: it reduced α-globin aggregates in RBC, promoted differentiation of late-stage erythroid precursors, reduced anemia, and improved iron overload, splenomegaly, and erythrocyte life span.25,26
Due to the very low levels of hemoglobin and RBC production and their short lifespan, it is not possible to fully assess the effects of some candidate thalassemia treatments in the Hbbth3/th3 mouse chronic transfusion model. Casu et al., therefore, developed the C57-FLCth1/th2 mouse model, which more closely resembles the phenotype of human transfusion-dependent thalassemia.27 In the C57FLCth1/th2 model, minihepcidins alone improved RBC lifespan, ineffective erythropoiesis, and splenomegaly. When combined with chronic RBC transfusion, ineffective erythropoiesis and splenomegaly were further improved and iron overload was reversed in this mouse model. As similar results were observed in the present study, this may provide some validation for the repeated-transfusion model used here. In contrast to our results in the Hbbth3/+ model, however, minihepcidins did not improve anemia but did decrease liver iron content when combined with transfusions in the C57-FLCth1/th2 mouse model.27 As mentioned previously, Hbbth3/+ mice are a well-established disease model of β-thalassemia intermedia.16 Currently, the published disease models for transfusion-dependent thalassemia are complex and difficult to access. Although Hbbth3/+ mice do not require blood transfusions for survival, they are anemic and have reduced RBC counts, which makes this model suitable for studying the effects of vamifeport combined with transfusions. Furthermore, some patients with thalassemia intermedia receive sporadic blood transfusions if their blood parameters and physical condition deteriorate. Although this and previously published studies17 demonstrated that it is possible to establish a model for repeated blood transfusions in Hbbth3/+ mice, the system presented here is not a model for transfusion-dependent β-thalassemia. Due to its positive effects on ineffective erythropoiesis, splenomegaly, and anemia in Hbbth3/+ mice, it would be of interest to also assess the effects of vamifeport in the C57-FLCth1/th2 mouse model of transfusion-dependent thalassemia. The pharmacodynamic effects of vamifeport have also recently been evaluated in phase I and II clinical trials.
Vamifeport reduced serum iron and TSAT levels in healthy volunteers28 and more recently has been shown to reduce serum iron and TSAT levels in patients with non-transfusion-dependent β-thalassemia.29 In addition to β-thalassemia, vamifeport has been shown to correct ineffective erythropoiesis and to ameliorate abnormal myelopoiesis in a mouse model of myelodysplastic syndrome.30 Moreover, vamifeport prevented further liver iron overload in a mouse model of hereditary hemochromatosis,31 improved hemodynamics in a mouse model of sickle cell disease,15 and corrected hematocrit and RBC levels in a mouse model of polycythemia vera.32,33 The clinical relevance of these observations in rodent disease models now needs to be assessed in controlled clinical trials. In summary, in the present study in Hbbth3/+ mice, vamifeport prevented the formation of transfusion-mediated NTBI and ameliorated anemia, ineffective erythropoiesis, and dysregulated iron homeostasis, either when given alone or when combined with repeated blood transfusions. Importantly, administering vamifeport together with blood transfusions additively ameliorated anemia and ineffective erythropoiesis in this mouse model. The clinical utility of combination treatment with vamifeport and transfusions in patients with β -thalassemia receiving blood transfusions remains to be investigated in controlled clinical trials.
Disclosures
NK, AF, PA, GI*, CD*, NN, FD, and VM are all current or past* employees of CSL Vifor and may own equities. PA, NN, VM, and FD are inventors in patents related to this publication.
1. Manolova V, Nyffenegger N, Flace A, et al. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of βthalassemia. J Clin Invest. 2019;130(1):491-506.
2. Jaing TH, Chang TY, Chen SH, Lin CW, Wen YC, Chiu CC. Molecular genetics of β-thalassemia: a narrative review. Medicine (Baltimore). 2021;100(45):e27522.
3. Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155-167.
4. Fibach E, Rachmilewitz EA. Pathophysiology and treatment of patients with beta-thalassemia - an update. F1000Res. 2017;6:2156.
5. Khandros E, Kwiatkowski JL. Beta thalassemia: monitoring and new treatment approaches. Hematol Oncol Clin North Am. 2019;33(3):339-353.
6. Ginzburg Y, Rivella S. β-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118(16):4321-4330.
7. Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: a longitudinal study. Blood.
NK contributed to the study design, data collection, and data analysis/interpretation. AF, PA, GI, and CD contributed to data collection and data analysis/interpretation. NN, FD, and VM contributed to the study design and data analysis/interpretation. All authors reviewed and critically revised the article content, approved the final draft for submission, and agree to be accountable for the article’s accuracy and integrity.
The authors thank the following colleagues from CSL Vifor for their valuable contributions: Stefan Reim and the chemical development team for providing vamifeport; and Maria Wilhelm, Anna-Lena Steck, Martin Khoeiklang, and the analytical team for tissue iron analyses. Special thanks to the animal facility head Marco Franchini and animal caretaker Martin Haenggi. Medical writing support, including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking, and referencing, was provided by Dawn Batty, PhD, at Aspire Scientific Limited (Bollington, UK).
This study was funded by CSL Vifor (St. Gallen, Switzerland). Funding for medical writing support for this article was provided by CSL Vifor.
Data can be shared upon request to the corresponding author.
2013;122(1):124-133.
8. Porter J, Taher A, Viprakasit V, et al. Oral ferroportin inhibitor vamifeport for improving iron homeostasis and erythropoiesis in β-thalassemia: current evidence and future clinical development. Expert Rev Hematol. 2021;14(7):633-644.
9. Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Nontransfusion-dependent thalassemias. Haematologica. 2013;98(6):833-844.
10. Olivieri NF. The beta-thalassemias. N Engl J Med. 1999;341(2):99-109.
11. Baek JH, Shin HKH, Gao Y, Buehler PW. Ferroportin inhibition attenuates plasma iron, oxidant stress, and renal injury following red blood cell transfusion in guinea pigs. Transfusion. 2020;60(3):513-523.
12. Piga A, Longo F, Duca L, et al. High nontransferrin bound iron levels and heart disease in thalassemia major. Am J Hematol. 2009;84(1):29-33.
13. Schmidt PJ, Fleming MD. Modulation of hepcidin as therapy for primary and secondary iron overload disorders: preclinical models and approaches. Hematol Oncol Clin North Am.
2014;28(2):387-401.
14. Ginzburg YZ. Hepcidin-ferroportin axis in health and disease. Vitam Horm. 2019;110:17-45.
15. Nyffenegger N, Zennadi R, Kalleda N, et al. The oral ferroportin inhibitor vamifeport improved hemodynamics in a mouse model of sickle cell disease. Blood. 2022;140(7):769-781.
16. Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O. A mouse model for beta 0-thalassemia. Proc Natl Acad Sci U S A. 1995;92(25):11608-11612.
17. Casu C, Presti VL, Oikonomidou PR, et al. Short-term administration of JAK2 inhibitors reduces splenomegaly in mouse models of β-thalassemia intermedia and major. Haematologica. 2018;103(2):e46-e49.
18. Huo Y, McConnell SC, Ryan TM. Preclinical transfusiondependent humanized mouse model of beta thalassemia major. Blood. 2009;113(19):4763-4770.
19. Jenkitkasemwong S, Wang CY, Knutson MD. Measurement of transferrin- and non-transferrin-bound iron uptake by mouse tissues. Bio Protoc. 2016;6(17):e1922.
20. Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J. Tissue distribution and clearance kinetics of nontransferrin-bound iron in the hypotransferrinemic mouse: a rodent model for hemochromatosis. Proc Natl Acad Sci U S A. 1987;84(10):3457-3461.
21. Nyffenegger N, Flace A, Doucerain C, Dürrenberger F, Manolova V. The oral ferroportin inhibitor VIT-2763 improves erythropoiesis without interfering with iron chelation therapy in a mouse model of β-thalassemia. Int J Mol Sci. 2021;22(2):873.
22. Siwaponanan P, Siegers JY, Ghazali R, et al. Reduced PU.1 expression underlies aberrant neutrophil maturation and function in β-thalassemia mice and patients. Blood. 2017;129(23):3087-3099.
23. Frost JN, Wideman SK, Preston AE, et al. Plasma iron controls neutrophil production and function. Sci Adv. 2022;8(40):eabq5384.
24. Gelderman MP, Baek JH, Yalamanoglu A, et al. Reversal of hemochromatosis by apotransferrin in non-transfused and transfused Hbbth3/+ (heterozygous B1/B2 globin gene deletion)
mice. Haematologica. 2015;100(5):611-622.
25. Suragani RN, Cawley SM, Li R, et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine β-thalassemia. Blood. 2014;123(25):3864-3872.
26. Suragani RN, Cadena SM, Cawley SM, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408-414.
27. Casu C, Chessa R, Liu A, et al. Minihepcidins improve ineffective erythropoiesis and splenomegaly in a new mouse model of adult β-thalassemia major. Haematologica. 2020;105(7):1835-1844.
28. Richard F, van Lier JJ, Roubert B, Haboubi T, Göhring UM, Dürrenberger F. Oral ferroportin inhibitor VIT-2763: first-inhuman, phase 1 study in healthy volunteers. Am J Hematol. 2020;95(1):68-77.
29. Taher A, Kourakli-Symeonidis A, Tantiworawit A, Wong P, Szecsödy P. Safety and preliminary pharmacodynamic effects of the ferroportin inhibitor vamifeport (VIT-2763) in patients with non-transfusion-dependent beta-thalassemia (NTDT): results from a phase 2A study. HemaSphere. 2022;6(Suppl 3):350.
30. Vance SZ, Antypiuk A, Sharma S, Dürrenberger F, Manolova V, Vinchi F. Alteration of iron homeostasis through genetic and pharmacologic modulation of ferroportin modifies MDS pathophysiology in a preclinical mouse model. HemaSphere. 2022;6(Suppl 3):356-357.
31. Nyffenegger N, Flace A, Varol A, et al. Oral ferroportin inhibitor prevents iron overload in the HFE C282Y mouse model of hereditary hemochromatosis. European Iron Club Annual Meeting: Oral Sessions and Posters. 2018; February:54.
32. Kubovcakova L, Manolova V, Nyffenegger N, et al. Efficacy of oral ferroportin inhibitor in a mouse model of polycythemia vera. European Iron Club Annual Meeting: Oral Sessions and Posters. 2018; February:144.
33. Stetka J, Usart M, Kubovcakova L, et al. Iron is a modifier of the phenotypes of JAK2-mutant myeloproliferative neoplasms. Blood. 2023;141(17):2127-2140.
Correspondence: D. Starczynowski
Daniel.Starczynowski@cchmc.org
T. Muto
Tomoya.Muto@chiba-u.jp
1Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA; 2Department of Hematology, Chiba University Hospital, Chiba, Japan; 3Division of Hematology and Oncology, University of Cincinnati, Cincinnati, OH, USA; 4Department of Cancer Biology, University of Cincinnati, Cincinnati, OH, USA; 5Department of Pharmaceutical Science and Research, Marshall University, Huntington, WV, USA; 6Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA and 7UC Cancer Center, Cincinnati, OH, USA
Received: November 2, 2022.
Accepted: April 17, 2023.
Early view: April 27, 2023.
htps://doi.org/10.3324/haematol.2022.282349
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Inflammation is associated with the pathogenesis of myelodysplastic syndromes (MDS) and emerging evidence suggests that MDS hematopoietic stem and progenitor cells (HSPC) exhibit an altered response to inflammation. Deletion of chromosome 5 (del(5q)) is the most common chromosomal abnormality in MDS. Although this MDS subtype contains several haploinsufficient genes that impact innate immune signaling, the effects of inflammation on del(5q) MDS HSPC remains undefined. Utilizing a model of del(5q)-like MDS, inhibiting the IRAK1/4-TRAF6 axis improved cytopenias, suggesting that activation of innate immune pathways contributes to certain clinical features underlying the pathogenesis of low-risk MDS. However, low-grade inflammation in the del(5q)-like MDS model did not contribute to more severe disease but instead impaired the del(5q)-like HSPC as indicated by their diminished numbers, premature attrition and increased p53 expression. Del(5q)-like HSPC exposed to inflammation became less quiescent, but without affecting cell viability. Unexpectedly, the reduced cellular quiescence of del(5q) HSPC exposed to inflammation was restored by p53 deletion. These findings uncovered that inflammation confers a competitive advantage of functionally defective del(5q) HSPC upon loss of p53. Since TP53 mutations are enriched in del(5q) AML following an MDS diagnosis, increased p53 activation in del(5q) MDS HSPC due to inflammation may create a selective pressure for genetic inactivation of p53 or expansion of a pre-existing TP53-mutant clone.
Myelodysplastic syndromes (MDS) are clonal hematopoietic stem and progenitor cell (HSPC) malignancies defined by blood cytopenia, myeloid cell dysplasia, and ineffective hematopoiesis. Another underlying feature of MDS is the contribution of inflammation.1-3 The MDS bone marrow (BM) niche exhibits increased inflammatory signaling, including elevated cytokines, chemokines, and alarmins. Microenvironmental alterations are partly due to aging, but there is also evidence that MDS hematopoietic cells themselves alter the stem-cell niche through activation of the innate immune system and related inflammatory signaling. Indeed, inflammation can suppress HSPC fitness, such as through reducing self-renewal or promoting differentiation, leading to selection of various oncogenic events during myeloid leukemogenesis.4-7 Re-
cently, several studies have implicated that low-grade inflammation favors the expansion of pre-leukemic or MDS hematopoietic stem cells (HSC) over normal HSC.8-12 Thus, inflammation in MDS has pleiotropic effects on the etiology of the disease.
Hemizygous deletion of the long arm of chromosome 5q (del(5q)) is the most common cytogenetic alteration in MDS. Del(5q) is also found in 25% of therapy-related MDS cases and is strongly correlated with progression to AML.13-15 Patients with a sole deletion of 5q exhibit macrocytic anemia, leukopenia, and increased platelets. BM from these patients show dysplastic mononuclear megakaryocytes, erythroid hypoplasia, and low blasts (<5%). Haploinsufficiency of several chr 5q genes have been implicated in del(5q) MDS, including genes that play important roles in ribosome function (RPS14), cell cycle regulation (CDC25c/PP2A), innate immune signaling (miR-
Tomoya Muto,1,2 Callum S. Walker,1 Puneet Agarwal,1 Eric Vick,3 Avery Sampson,1 Kwangmin Choi,1 Madeline Niederkorn,1 Chiharu Ishikawa,1,4 Kathleen Hueneman,1 Melinda Varney5 and Daniel T. Starczynowski1,4,6,7145, miR-146a, TIFAB, and DIAPH), stress response signaling (EGR1), and β-catenin signaling (APC, CSNK1A1). Deletion of RPS14 results in a p53-dependent defect in erythropoiesis, but not in a competitive advantage of HSPC.16 Moreover, loss of RPS14 coincides with increased expression of alarmins and hematopoietic defects associated with non-cell autonomous innate immune signaling.17,18 Genetic barcoding revealed that CSNK1A1 haploinsufficiency is a driver of clonal expansion of del(5q) MDS HSPC.19 Deletion of miR-145, miR-146a, mDia1, and TIFAB collectively contribute to increased innate immune signaling in MDS HSPC, albeit through distinct mechanisms. Deletion of mDia1 increases innate immune signaling in neutrophils due to upregulation of CD14 and results in an MDS-like phenotype in mice.20 Deletion of miR-145, results in derepression of Mal/TIRAP, which is important for an initial step of signaling by the Toll-like receptor (TLR) superfamily.21,22 Deletion of miR-146a increases TRAF6 and IRAK1 mRNA and protein translation, while loss of TIFAB increases TRAF6 protein stability, thus resulting in activation of TRAF6 and IRAK1 in MDS HSC even in the absence of ligand-mediated activation of TLR.22-30 Reduced TIFAB expression also results in diminished USP15 de-ubiquitinase function and consequently p53 activation in hematopoietic cells.31 Concurrent hematopoietic-specific deletion of TIFAB and miR-146a results in higher levels of TRAF6 expression and innate immune pathway activation, which coincides with a highly penetrant BM failure, a diseased phenotype that more faithfully recapitulates human del(5q) MDS.25
Despite overwhelming evidence of chronic inflammation in del(5q) MDS patients, the effects of low-grade inflammatory signals on del(5q) MDS HSPC and disease progression remains uncharacterized. In this study, we focused on the effects of low-grade inflammation in del(5q) MDS by utilizing a mouse model with co-deletion of miR-146a and TIFAB, two mediators of aberrant innate immune signaling in del(5q) MDS HSPC. We found that low-grade inflammation suppresses del(5q) MDS HSPC via p53 and provide a potential explanation for the high rate of TP53 mutations in patients with del(5q).
Lipopolysaccharide (LPS) (L6529) and LPS-EB Ultrapure (TLRL-PEKLPS) were purchased from Sigma and InvivoGen, respectively. The IRAK1/4 inhibitor (NCGC-1481) has been previously described.32,33 UBE2N inhibitor (UC764865) has been previously described.34 MDS-L cells have been previously described.35,36
Mice
miR-146a-/- C57BL/6 mice were obtained from Dr. David Baltimore as previously described.29 Generation of Tifab-/-;miR146a-/- mice was previously described.25,26 Tifab-/-;miR-146a-/mice were crossed with Trp53-/- mice (Jackson Laboratories, 002101). All mouse experiments were performed in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility of Cincinnati Children’s Hospital. Additional information on the mice can be found in the Online Supplementary Appendix.
For non-competitive BM transplantations, CD45.2+ BM cells were transplanted into lethally-irradiated recipient mice (CD45.1+ B6.SJLPtprca Pep3b/Boy; 6-10 weeks of age). For competitive transplantations, BM cells from 8-week-old CD45.2+ BM cells were transplanted with WT CD45.1+ BM cells (10:1 ratio) into lethally irradiated CD45.1+ recipient mice. For serial transplantation, BM cells were collected from all recipient mice 3 months after transplantations, pooled together, and then BM cells were transplanted into lethally-irradiated recipient mice (CD45.1+ B6.SJLPtprca Pep3b/Boy; 6-10 weeks of age). Detailed information on BM transplantations has been previously reported.37-39
Bromouridine (BrdU) (Sigma-Aldrich) was administered continuously to mice via drinking water (0.5 mg/mL). After 1 week, BrdU incorporation was analyzed using a BrdU Flow Kit (559619, BD Biosciences) according to the manufacture’s recommendation. Annexin V viability staining was carried out according to manufacturer’s instructions (C0556419, BD Biosciences).
Additional methods and materials can be found in the Online Supplementary Appendix
Inhibition of IRAK1/4-TRAF6 alleviates cytopenias in Tifab-/-;miR-146a-/- mice
TIFAB and miR-146a are co-deleted in ~80% of del(5q) MDS patients and are both implicated in innate immune and inflammatory signaling. Deletion of miR-146a results in de-repression of TRAF6 and IRAK1 mRNA, while loss of TIFAB increases expression of TRAF6 protein in MDS HSPC (Figure 1A), thus resulting in activation of innate immune pathways.22-30 We utilized a mouse model in which Tifab and miR-146a were simultaneously deleted (Tifab-/-;miR146a-/-), as previously described.25 This model when evaluated in bone marrow (BM) chimeras recapitulates several aspects of del(5q) MDS, including HSPC defects primarily affecting the myeloid lineage, progressive peripheral blood
cytopenias, myeloid dysplasia, and a fatal BM failure in a subset of mice. Moreover, the BM plasma of Tifab-/-;miR146a-/- mice exhibit an altered cytokine milieu as indicated by elevated expression of IL1a, CXCL1, CXCL2, CXCL5, and CXCL9 and reduced expression of IL15 as compared to BM plasma from wild-type (WT) mice (Online Supplementary Figure S1A). As in human del(5q) MDS HSPC, deletion of Tifab and miR-146a results in activation of IRAK1 and TRAF6 signaling, which directly lead to aberrant HSPC
function.25 Therefore, to determine whether suppressing IRAK1/4-TRAF6 signaling can restore normal blood counts in Tifab-/-;miR-146a-/- mice with cytopenias, we utilized an IRAK1/4 inhibitor (NCGC-1481) and UBE2N inhibitor (UC764865, or “UC-65”) in vivo. 32,34 The IRAK1/4 inhibitor targets the kinase functions of IRAK1 and IRAK4, while the UBE2N inhibitor targets the ubiquitin-conjugating enzyme (UBE2N) utilized by TRAF6 for signaling. As expected, activation of IRAK1 and NF-κB signaling in Tifab-/-;miR-146a-/- BM cells
Figure 1. Inhibition of the TRAF6-IRAK1/4 axis restores blood counts in Tifab-/-;miR-146a-/- mice. (A) Overview of del(5q) myelodysplastic syndromes (MDS) resulting in haploinsufficient expression of the 5q- genes miR-146a and TIFAB and a corresponding increase in expression of their targets, TRAF6 and IRAK1. (B) Immunoblotting of Tifab-/-;miR-146a-/- bone marrow (BM) cells treated with lipopolysaccharide (LPS) (1 mg/mL) and the IRAK1/4 (1 mM) or UBE2N (5 mM) inhibitor for 30 minutes. (C) Outline of BM transplantations using wild-type (WT) or Tifab-/-;miR-146a-/- BM cells. Peripheral blood (PB) analysis was performed monthly on recipient mice to monitor for cytopenias. At onset of cytopenias in the Tifab-/-;miR-146a-/- recipient mice, an IRAK1/4 inhibitor (NCGC-1481 at 30 mg/kg) or UBE2N inhibitor (UC-65 at 20 mg/kg) was administered daily (or phosphate-buffered saline [PBS], vehicle control). PB counts were performed weekly after the treatment was initiated. (D) PB counts of the recipient mice before (20 weeks post BM transplantation) and after treatment (post-Tx) with the IRAK1/4-inhibitor (N=4-5 per group). (E) PB counts of the recipient mice before (20 weeks post BM transplantation) and post-Tx with the UBE2N-inhibitor (N=4-5 per group). Significance for panels (D and E) was determined with a Student’s t test (*P<0.05) between treated and vehicle-treated groups.

was suppressed with the IRAK1/4 or UBE2N inhibitors (Figure 1B). BM cells from Tifab-/-;miR-146a-/- or WT mice were transplanted into lethally-irradiated CD45.1 WT mice. Once the Tifab-/-;miR-146a-/- recipient mice developed peripheral blood (PB) cytopenias (~20 weeks post engraftment), the WT and Tifab-/-;miR-146a-/- recipient mice were randomized and treated daily for 2 weeks with the IRAK1/4 inhibitor, UBE2N inhibitor, or vehicle (phosphate-buffered saline [PBS])32,33 (Figure 1C). The Tifab-/-;miR-146a-/- recipient mice treated with PBS progressively developed anemia and neutropenia (Figure 1D, E). However, treatment of the Tifab-/-;miR-146a-/- recipient mice with the IRAK1/4 inhibitor or UBE2N inhibitor restored red blood cell counts and hematocrit, and resulted in a modest improvement in neutrophils (Figure 1D, E), suggesting that the cytopenias associated with deletion of TIFAB and miR-146a in del(5q) MDS are partly attributed to increased IRAK1/4-TRAF6 signaling and can be rescued by targeting IRAK1/4 or TRAF6 activation with small molecule inhibitors.
Low-grade inflammation has pleotropic effects on the disease phenotype of Tifab-/-;miR-146a-/- mice
Since IRAK1/4-TRAF6 signaling mediates inflammatory signals from various immune-related receptors, we next investigated the impact of chronic low-grade inflammation on the pathogenesis of del(5q) MDS. Low-grade inflammation via the TLR/IL1R superfamily is observed in MDS and implicated in the pathogenesis of disease progression. As such, an inflammatory milieu was established using the gram-negative bacterial component LPS, a ligand for TLR4 and activator of IRAK1/4-TRAF6 signaling, which is freely in circulation of leukemia patients, induces systemic inflammation and affects HSPC function.40,41 Chronic lowdose (LD) treatment with LPS (1 mg/g, hereafter LD-LPS) was administered via intraperitoneal (i.p.) injection twice a week for 30 days into Tifab-/-;miR-146a-/- or WT mice (Figure 2A). After the last dose of LD-LPS or vehicle, hematopoietic cell chimeric mice were then established by transplanting BM cells from the treated Tifab-/-;miR-146a-/or WT mice into lethally-irradiated CD45.1 WT mice. As we observed above, mice engrafted with Tifab-/-;miR-146a-/- BM cells developed multilineage cytopenias, including neutropenia, anemia and thrombocytopenia 4 months posttransplant as compared to mice engrafted with WT BM cells (Figure 2B). As expected, the severity of cytopenias following LD-LPS administration in mice engrafted with Tifab-/-;miR-146a-/- BM cells was worse as compared to LDLPS treated mice engrafted with WT BM cells (Figure 2B). The proportion of myeloid cells (CD11b+Gr1-) and lymphoid cells (CD3+ and B220+) in the PB was also significantly reduced following LD-LPS treatment in mice engrafted with Tifab-/-;miR-146a-/- BM cells as compared to WT mice (Figure 2C). However, LD-LPS administration did not contribute to more severe cytopenias in mice engrafted with
Tifab-/-;miR-146a-/- BM cells as compared to PBS-treated mice (Figure 2B).
In order to evaluate the effects of chronic low-grade inflammation on disease development, we monitored the mice for over 1 year and found that all mice engrafted with Tifab-/-;miR-146a-/- BM cells that were either treated with LD-LPS or vehicle (PBS) succumbed to disease by ~500 days (median survival ~400 days), while the majority of WT mice treated with LD-LPS or PBS were alive at the conclusion of the experiment (median survival not reached)(Figure 2D). Surprisingly, the disease latency or severity were not impacted by LD-LPS in mice with Tifab-/;miR-146a-/- BM cells. The mice transplanted with Tifab-/-;miR-146a-/- BM cells that were treated with PBS or LD-LPS developed multi-lineage cytopenias, reduced BM cellularity, and splenomegaly, which are consistent with findings of BM failure (Figure 2E; Online Supplementary Figure S1B-E). Administration of LD-LPS resulted in slightly lower blood counts in mice transplanted with Tifab-/-;miR146a-/- BM cells as compared to vehicle-treated mice, but these differences were not significant (Figure 2E). Examination of the hematopoietic stem and progenitor cells showed that Tifab-/-;miR-146a-/- BM cells produce fewer HSC (Lin-cKit+Sca1+CD150+CD48-) and hematopoietic progenitor cells (HPC) (Lin-cKit+Sca1+ and Lin-cKit+) than WT BM cells. Treatment with LD-LPS resulted in a slight, albeit not significant, reduction in Tifab-/-;miR-146a-/- HSC and HPC (Figure 2F). In contrast, LD-LPS induced an expansion of WT HPC as expected based on previous studies. Altogether, these observations suggest that chronic low-grade inflammation does not meaningfully contribute to a more severe disease in the del(5q)-like MDS model.
Low-grade inflammation results in impaired Tifab-/-; miR-146a-/- hematopoietic stem and progenitor cells In order to explore the effects of LD-LPS on Tifab-/-;miR146a-/- HSPC, we isolated LSK from WT and of Tifab-/-;miR146a-/- mice treated in vivo with chronic LD-LPS, and performed hematopoietic progenitor cell assays in methylcellulose. LD-LPS had minimal effects on WT LSK as compared to vehicle-treated cells but resulted in a significant reduction of colony formation by Tifab-/-;miR-146a-/- LSK (Figure 3A). One of the consequences of low-grade inflammation on HSC is reduced quiescence due to increased cell proliferation.42,43 Therefore, we next evaluated the effect of LD-LPS on the total number and cell cycle status of mice injected with LD-LPS (Figure 3B). LD-LPS resulted in significantly reduced number of Tifab-/-;miR-146a-/- HSC (Lin-Sca1+cKit+CD150+CD48-) in the BM as compared to PBS-treated Tifab-/-;miR-146a-/- mice or WT mice treated with LD-LPS (Figure 3C). The reduction in Tifab-/-;miR146a-/- HSC in the BM treated with LD-LPS correlated with an increased frequency of BrdU+ Tifab-/-;miR-146a-/- HSC
Figure 2. Low-dose lipoploysaccharide does not significantly impact the phenotype of Tifab-/-;miR-146a-/- mice. (A) Outline of bone marrow (BM) transplantations using wild-type (WT) or Tifab-/-;miR-146a-/- mice in the presence of low-dose lipopolysaccharide (LD-LPS). (B) Peripheral blood (PB) counts of the recipient mice at 4 months after transplantation (N=9-10 per group). (C) Summary of lymphoid (CD3+ and B220+), myeloid (CD11b+Gr1-) proportions within the PB of the recipient mice (N=9-10 per group). (D) Overall survival of mice transplanted with WT or Tifab-/-;miR-146a-/- mice treated with either PBS or LPS (N=9-10).
(E) PB counts of the moribund Tifab-/-;miR-146a-/- mice (N=4-5 per group) at 10-13 months after transplantation. (F) Proportion of the indicated hematopoietic stem and progenitor cells in the BM from moribund Tifab-/-;miR-146a-/- mice (N=5-7 per group). Significance for panels (B, C, E, and F) was determined with a Student’s t test (*P<0.05; **P<0.01; ***P<0.001).

cells treated with LD-LPS compared to WT BM cells treated with or without LPS and PBS-treated Tifab-/-;miR146a-/- BM cells (Figure 3C, D). The reduction in Tifab-/;miR-146a-/- HSC in the BM after LD-LPS was not due to decreased cell viability. The percent of apoptotic HSC (7AAD/AnnexinV) from Tifab-/-;miR-146a-/- mice treated with LD-LPS was similar to PBS-treated mice or to WT treated with LD-LPS (Figure 3E).
Low-grade inflammation suppresses the long-term competitive advantage of Tifab-/-;miR-146a-/-
hematopoietic stem cells
We next investigated the consequences of chronic lowgrade inflammation on hematopoiesis and the long-term function of Tifab-/-;miR-146a-/- HSPC in vivo. Total BM cells isolated from CD45.2 Tifab-/-;miR-146a-/- or CD45.2 WT mice injected i.p. with LD-LPS twice a week for 30 days and serially transplanted every 4 months with CD45.1 WT competitor BM cells into lethally-irradiated CD45.1 WT
mice (Figure 4A). Exposure to LD-LPS resulted in reduced proportion of CD45.2+ Tifab-/-;miR-146a-/- in the PB compared to PBS-derived CD45.2+ Tifab-/-;miR-146a-/- PB cells (Figure 4B). The reduction in PB chimerism is mainly due to impaired lymphopoiesis of Tifab-/-;miR-146a-/- cells exposed to LD-LPS (Figure 4C). At this time point, the production of myeloid cells in the PB was increased for Tifab-/-;miR-146a-/- cells exposed to LD-LPS as compared to the control PBS groups following the primary BM transplantation (Figure 4C). Exposure of CD45.2+ WT HSPC to LD-LPS resulted in negligible effects at month 3 upon primary BM transplantation (Figure 4C). However, exposure to LD-LPS resulted in reduced proportion of CD45.2+ WT cells in PB and CD45.2+ WT CD150+CD48-LSK (LT-HSC) in the BM compared to PBS-derived CD45.2+ WT cells at month 3 of the secondary BM transplantation (Figure 4EG). In stark contrast, CD45.2+ Tifab-/-;miR-146a-/- BM cells exposed to LD-LPS resulted in significantly reduced proportions of BM cells compared to PBS-derived CD45.2+ BM

Figure 3. Tifab-/-;miR-146a-/- hematopoietic stem and progenitor cells are less quiescent following low-dose lipoploysaccharide.
(A) Colony assay of wild-type (WT) or Tifab-/-;miR-146a-/- LSK treated with low-dose lipopolysaccharide (LD-LPS) (N=3 per group).
(B) Outline of in vivo bromouridine (BrdU) incorporation assay using WT and Tifab-/-;miR-146a-/- mice in the presence of low-dose chronic inflammation. (C) Absolute number of hematopoietic stem cells (HSC) from WT and Tifab-/-;miR-146a-/- mice treated with LPS (N=6-10). Error bars represent the standard error of the mean (SEM). (D) Proportion of BrdU-positive cells within HSC from WT and Tifab-/-;miR-146a-/- mice treated with LPS (N=6-8). Error bars represent the SEM. (E) Proportion of 7AAD+ AnnexinV+ cells within HSC from WT and Tifab-/-;miR-146a-/- mice treated with LPS (N=3-5). Error bars represent the SEM. Significance for panels (C, D, and E) was determined with a Student’s t test (*P<0.05; **P<0.01; ***P<0.001).

Figure 4. Tifab-/-;miR-146a-/- hematopoietic stem and progenitor cells are functionally defective following low-dose lipoploysaccharide. (A) Outline of competitive bone marrow (BM) transplantations using wild-type (WT) or Tifab-/-;miR-146a-/mice in the presence of low-dose lipopolysaccharide (LD-LPS). (B) Summary of donor-derived peripheral blood (PB) proportions at the indicated time points (N=5-6 per group). Error bars represent the standard error of the mean (SEM). Statistical analysis was performed between Tifab-/-;miR-146a-/--phosphate-buffered saline (PBS) and -LPS. (C) Proportion of donor-derived PB cells from primary recipient mice (N=5-6 per group). Error bars represent the SEM. (D) Proportions of donor-derived BM cells from primary recipient mice is reported 3 months after transplantation. Error bars represent the SEM (N=5-6 per group). (E) Proportion of donor-derived PB cells from secondary recipient mice (N=6 per group). Error bars represent the SEM. (F) Proportions of donorderived BM cells from secondary recipient mice is reported 3 months after transplantation. (G) Representative flow cytometric profiles of Tifab-/-;miR-146a-/- and WT BM hematopoietic stem cells (HSC) after the tertiary transplantation. Error bars represent the SEM (N=6 per group). LK: Lin-cKit+; LSK: Lin-cKit+Sca1+; MPP: multipotent progenitor (Lin-cKit+Sca1+CD48+CD150-); ST-HSC, Lin-cKit+Sca1+CD48-CD150-; LT-HSC, Lin-cKit+Sca1+CD48-CD150+. Significance for panels (B, D, and E) was determined with a Student’s t test (*P<0.05; **P<0.01; ***P<0.001).
cells following the primary BM transplantation (Figure 4E). Furthermore, the Tifab-/-;miR-146a-/- BM cells exposed to LD-LPS were unable to recover in the secondary BM transplantation as the proportion of all HSPC subsets were further out-competed by WT cells (Figure 4D-G). Collectively, these results suggest that deletion of Tifab and miR-146a reduces the long-term repopulating potential of HSPC immediately following inflammation due to loss of cellular quiescence.
Low-grade inflammation results in p53 expression and activation in Tifab-/-;miR-146a-/- hematopoietic stem and progenitor cells
In order to identify the molecular basis for the impaired function of Tifab-/-;miR-146a-/- HSPC during low-grade inflammation, we performed RNA-sequencing in Tifab-/-; miR-146a-/- and WT LSK treated with LD-LPS in vitro for 90 min (Figure 5A). LPS treatment resulted in expression of 90 differentially upregulated genes and 212 downregulated genes in Tifab-/-;miR-146a-/- LSK cells compared to vehicletreated cells (Figure 5B,C; Online Supplementary Table S1). In contrast, LPS treatment resulted in 79 upregulated and 125 downregulated genes in WT LSK cells compared to vehicle-treated cells (Figure 5B, C; Online Supplementary Table S2). Moreover, there was minimal overlap in the identity of differentially expressed genes in LPS-treated Tifab-/-;miR-146a-/- LSK and WT LSK cells (Figure 5C). Pathway analysis of differentially expressed genes revealed changes in megakaryocyte/platelet- and nicotinic acetylcholine receptor-related pathways in Tifab-/-;miR-146a-/LSK cells (Online Supplementary Figure S2). WT cells treated with LPS exhibited enrichment in pathways related to epigenetic regulation, including histone methylation and acetylation (Online Supplementary Figure S2). In order to identify regulatory factors that drive the Tifab-/-;miR-146a-/HSPC phenotype during low-dose inflammation, we assessed genes that are overexpressed or downregulated in Tifab-/-;miR-146a-/- versus WT LSK following LPS stimulation (Figure 5D). We identified significant enrichment of binding motifs for TP53 (P=0.01), GATA2 (P=0.006), and RUNX1 (P=0.01) in the overexpressed genes from LPSstimulated Tifab-/-;miR-146a-/- as compared to WT LSK
(Figure 5E). Downregulated genes were not significantly enriched for any specific motif (Figure 5E). These results indicate TLR stimulation of Tifab-/-;miR-146a-/- and WT HSPC results in a distinct effect on gene expression programs, pathways, and cellular states.
We next focused on the potential effects of TP53 signaling in LPS-stimulated Tifab-/-;miR-146a-/- HSPC. In addition to regulating TRAF6, TIFAB also directly binds USP15. One of the key substrates regulated by the TIFAB-USP15 axis includes p53.31 Deletion of TIFAB sensitizes hematopoietic cells to a variety of cellular stressors, which are dependent on p53 activation.31 Gene set enrichment analysis (GSEA) revealed that a p53-related gene signature was positively enriched in Tifab-/-;miR-146a-/- LSK cells treated with LPS compared to vehicle-treated Tifab-/-;miR-146a-/cells, while there was no significant enrichment of a p53related signature in WT cells treated with LPS (Figure 5F). Baseline expression of p53-related genes were comparable between vehicle-treated WT and Tifab-/-;miR-146a-/- HSPC, suggesting that p53 activation occurs during inflammation in del(5q)-like MDS HSPC.
In order to evaluate p53 protein levels in Tifab-/-;miR-146a-/HSPC exposed to chronic inflammation, we purified cKit+ BM cells from WT or Tifab-/-;miR-146a-/- mice that were treated with LD-LPS for 30 days or PBS (Figure 6A). Baseline p53 protein levels were slightly increased in Tifab-/-;miR146a-/- HSPC as compared to vehicle-treated Tifab-/-;miR146a-/- HSPC or WT HSPC (Figure 6B). Importantly, p53 protein was further increased upon treatment with LD-LPS in Tifab-/-;miR-146a-/- HSPC as compared to vehicle-treated Tifab-/-;miR-146a-/- HSPC or LPS-treated WT HSPC (Figure 6B). The effects of LPS on p53 expression in Tifab-/-;miR146a-/- HSPC is dependent on IRAK1/4-TRAF6 activation. In vitro-treated Tifab-/-;miR-146a-/- HSPC with LPS resulted in increased p53 protein expression, which was suppressed with the IRAK1/4 or UBE2N inhibitors (Figure 6C). Lastly, to determine whether loss of TIFAB and miR-146a in human MDS HSPC exhibit activation of p53, we treated a human del(5q) MDS cell line (MDSL) with LPS. LPS stimulation of MDSL cells resulted in an increase in p53 protein levels (Figure 6D). Moreover, the IRAK1/4 inhibitor suppressed p53 expression upon LPS stimulation (Figure 6E), indicat-
Figure 5. Differential TLR4 stimulation and p53 activation in Tifab-/-;miR-146a-/- hematopoietic stem and progenitor cells with low-dose lipoploysaccharide. (A) Outline of RNA-sequencing using hematopoietic stem and progenitor cells (HSPC) from wildtype (WT) and Tifab-/-;miR-146a-/- mice in the presence of low-dose lipopolysaccharide (LD-LPS). (B) Heatmap of differentially expressed genes in WT or Tifab-/-;miR-146a-/- LSK cells treated with LPS or phosphate-buffered saline (PBS) (1.5-fold; P<0.05; N=3 per group). (C) Venn diagram of upregulated/downregulated genes (1.5-fold; P<0.0.5) in WT (relative to WT LSK cells treated with PBS) or Tifab-/-;miR-146a-/- LSK cells (relative to Tifab-/-;miR-146a-/- LSK cells treated with PBS). (D) Heatmap of differentially expressed genes in LPS-stimulated WT or Tifab-/-;miR-146a-/- LSK cells (1.5-fold; P<0.05; N=3 per group). (E) Enrichment of transcription factors was determined with the ENCODE and CHIP Enrichment Analysis (ChEA) libraries using genes that are overexpressed (left panel) or downregulated (right panel) in LPS-stimulated Tifab-/-;miR-146a-/- vs. WT LSK. (F) Gene set enrichment analysis plots for TP53 targets in LPS-stimulated WT LSK cells (relative to WT LSK cells treated with PBS) and LPSstimulated Tifab-/-;miR-146a-/- LSK cells (relative to Tifab-/-;miR-146a-/- LSK cells treated with PBS). NES: normalized enrichment score.
ing that chronic inflammation can directly induce p53 expression via the IRAK1/4-TRAF6 axis. Collectively, these findings suggest that del(5q) MDS HSPC are primed to activate p53 signaling via IRAK1/4-TRAF6 signaling.

Deletion of p53 restores the functional defect of Tifab-/-; miR-146a-/- hematopoietic stem and progenitor cells during low-grade inflammation
TP53 mutations are highly enriched in del(5q) AML patients following an initial MDS diagnosis,44 therefore, increased p53 signaling in del(5q) MDS HSPC due to low-grade inflammation may create a selective pressure that eventually leads to genetic inactivation of p53 in the MDS HSPC. In order to examine the role of p53 in Tifab-/-;miR-146a -/-
HSPC during low-grade inflammation, we generated Tifab-/-;miR-146a-/- mice in which one copy of p53 is deleted (Tifab-/-;miR-146a-/-;p53+/-), which mimics MDS patients with a monoallelic mutation in TP53 45 ( Online Supplementary Figure S3 ). WT, Tifab -/- ;miR-146a -/- or Tifab-/-;miR-146a-/-;p53+/- mice were treated with LD-LPS or PBS twice a week for 30 days (Figure 7A). After 30 day exposure to LD-LPS, we confirmed that p53 expression was suppressed in Tifab -/- ;miR-146a -/- ; p53 +/- HSPC as compared to Tifab-/-;miR-146a-/- HSPC (Figure 7B). Following the last treatment with LD-LPS, BM cells (CD45.2 ) were isolated and co-transplanted with WT competitor BM cells (CD45.1) (one to one ratio) into lethally-irradiated CD45.1 WT mice (Figure 7A). At 5 months post-transplant,
LD-LPS treatment resulted in reduced proportion of Tifab-/-;miR-146a-/- cells in the PB due to impaired production of myeloid cells (Figure 7C, D), as observed in Figure 2C and Figure 4D. In contrast, deletion of p53 restored the PB chimerism of Tifab -/- ;miR-146a -/- ; p53 +/- myeloid cells exposed to LD-LPS (Figure 7C, D). Furthermore, Tifab-/-;miR-146a-/-;p53+/- HSC did not decrease in the BM upon LD-LPS treatment as compared to PBS-treated Tifab-/-;miR-146a-/-;p53+/- HSC, or LD-LPS treated Tifab-/-; miR-146a-/- HSC (Figure 7E). We observed that there were fewer BrdU+ Tifab-/-;miR-146a-/-;p53+/- HSC upon LD-LPS treatment as compared to Tifab-/-;miR-146a-/- HSC (Figure 7F), suggesting that p53 is responsible for the excessive proliferation of Tifab-/-;miR-146a-/- HSC exposed to lowgrade inflammation. Similar findings were observed upon chronic elevation of p53 protein in HSPC.46 Although deletion of p53 provides a competitive advantage of Tifab-/-;miR146a-/- HSPC during low-grade inflammation, we did not observe evidence of aberrant BM morphology nor overt AML in these mice after 1 year (Online Supplementary Figure S4
and data not shown). Therefore, we posit that p53 mutations preserve the competitive advantage of del(5q) HSPC during chronic low-grade inflammation and that additional cooperating mutations contribute to the development of AML in del(5q) MDS.
Inflammation has been shown to favor the expansion of pre-leukemic or MDS HSC over normal HSC and contribute to the pathogenesis of MDS. Based on cytokine profiling of patients with pre-leukemic conditions and MDS, inflammatory cytokine signatures are involved in the early stages of pathogenesis, well before the onset of MDS.47 More recently, microbial components, including circulating LPS, have been reported in patients with MDS.48 Our study investigated the effects of low-grade chronic infl ammation on the function of del(5q)-like mouse MDS HSPC and contribution to disease. Although
Figure 6. Low-dose lipoploysaccharide induces p53 expression in Tifab-/-;miR-146a-/- hematopoietic stem and progenitor cells via IRAK1/4-TRAF6. (A) Outline of in vivo administration of lipopolysaccharide (LPS) following by immunoblot analysis. (B) Immunoblot analysis of wild-type (WT) and Tifab-/-;miR-146a-/- c-Kit+ bone marrow (BM) cells from WT and Tifab-/-;miR-146a-/- mice treated with low-dose LPS (LD-LPS) (1 μg /g) or vehicle twice a week for 30 days. (C) Immunoblotting of Tifab-/-;miR-146a-/- BM cells treated with LPS (1 mg/mL) and the IRAK1/4 (1 mM) or UBE2N (5 mM) inhibitor for 60 minutes. (D) Immunoblot analysis of a patient-derived del(5q) myelodysplastic syndromes (MDS) cell lines (MDSL) was treated with LPS (100 ng/mL) for the indicated time points. (E) Immunoblot analysis of MDSL cells treated with LPS (100 ng/mL) and the IRAK1/4 inhibitor (1 mM) for 2 hours.

Figure 7. Deletion of p53 restores exhaustion of Tifab-/-;miR-146a-/- hematopoietic stem cells following low-dose lipoploysaccharide. (A) Outline of competitive bone marrow (BM) transplantations using Tifab-/-;miR-146a-/- or Tifab-/-;miR146a-/-;p53+/- mice in the presence of low-dose chronic inflammation. (B) Immunoblot analysis of Tifab-/-;miR-146a-/- or Tifab-/-;miR146a-/-;p53+/- c-Kit+ BM cells from wild-type (WT) and Tifab-/-;miR-146a-/- mice treated with low-dose lipopolysaccharide (LD-LPS) (1 μg/g) or vehicle twice a week for 30 days. (C, D) Proportion of donor-derived peripheral blood (PB) cells from the recipient mice transplanted with Tifab-/-;miR-146a-/- or Tifab-/-;miR-146a-/-;p53+/- mice treated with either phosphate-buffered saline (PBS) or LPS. Error bars represent the standard error of the mean (SEM). (E) Proportion of donor-derived SLAM hematopoietic stem cells (HSC) in total cells from the recipient mice transplanted with Tifab-/-;miR-146a-/- or Tifab-/-;miR-146a-/-;p53+/- mice treated with either PBS or LPS. Error bars represent the SEM. (F) Proportion of bromouridine (BrdU)-positive cells within HSC from Tifab-/-;miR-146a-/- or Tifab-/-;miR-146a-/-;p53+/- mice treated with LPS (N=3 per group). Error bars represent the SEM. (G) Summary of findings. Del(5q) myelodysplastic syndromes (MDS) hematopoietic stem and progenitor cells (HSPC) (blue) exhibit impaired hematopoiesis and increased innate immune signaling because of derepression of TRAF6 and IRAK1 following deletion of miR146a and TIFAB. Loss of TIFAB also results in increased p53 activation due to diminished USP15 activation. During inflammation, del(5q) MDS HSPC activated IRAK-TRAF6 signaling and induce p53, which results in clonal depletion. Loss-offunction mutations (LOF) or deletion of p53 permits clonal expansion of del(5q) MDS HSPC during inflammation. Significance for panels (C, D, E, and F) was determined with a Student’s t test (*P<0.05).

suppression of innate immune signaling by targeting IRAK1/4 or TRAF6-UBE2N improved cytopenias, we observed that low-grade inflammation via TLR/IL1R superfamily in the del(5q)-like MDS model did not contribute to more severe disease but instead impaired the function of del(5q)-like HSPC. Unexpectedly, the functional defect of del(5q) HSPC exposed to the TLR4 ligand LPS, including their attrition following excessive proliferation, was restored by p53 deletion. Since TP53 mutations are enriched in del(5q) AML following an MDS diagnosis, increased p53 activation in del(5q) MDS HSPC due to inflammation may create a selective pressure for genetic inactivation of p53 or expansion of a pre-existing TP53mutant clone (Figure 7G).
The prevalence of TP53 mutations in all MDS subtypes is low, averaging 5–10%, but is highly associated with isolated del(5q) or complex karyotypes with -5/5q-.44,49 TP53 mutations are a predictor of poor prognosis in both de novo and secondary MDS and are associated with higherrisk disease, p53 protein overexpression, increased blast count and AML progression.44,50 An isolated del(5q) in MDS has a better prognosis as compared to the other MDS cytogenetic subtypes. However, TP53 mutations occur in lower-risk del(5q) MDS at a frequency of approximately 20% and are associated with a dismal prognosis.44 Disease progression of lower-risk del(5q) MDS is related to the evolution of pre-existing or emerging subclones carrying a TP53 mutation.51 TP53 mutations at diagnosis are predictive of disease progression in lower risk del(5q) MDS and disease progression mostly occurred in patients with TP53 mutations emerging before or at the time of progression. However, it was observed in certain cases that the absence of a TP53 mutation at diagnosis suggesting that the TP53 mutations are secondary events in del(5q) MDS patients. In line with our reported findings, increased p53 protein expression in lower risk del(5q) MDS predicts shorter overall survival and disease progression.52 The del(5q) MDS patients with the highest level of p53 protein expression reflect mutant TP53, suggesting there are selective pressures that lead to TP53 inactivating mutations in the del(5q) MDS clones.
It is also well established that p53 mutations are acquired in hematopoietic cells in patients receiving genotoxic therapy, leading to therapy-related MDS or AML. In this scenario, the genotoxic stress is a selective pressure for loss of p53-mediated apoptosis in the hematopoietic cells. However, the reasons for the high frequency of p53 mutations in del(5q) and which of the p53 functions are essential in del(5q) MDS is insufficiently understood. Moreover, it also remains unresolved why these p53 functions are selected against during clonal evolution and disease progression. Paradoxically, elevated p53 levels in del(5q)-like MDS HSPC following inflammation correlate with increased HSC proliferation and a reduced competitive ad-
vantage, while monoallelic loss of p53 in these cells results in reduced HSC proliferation and an increased competitive advantage. A similar effect was observed in a mouse model of hyperactive p53.46 Hyperactive p53 results in reduced numbers of proliferating HSC in aged mice. These paradoxical observations related to p53 activity and HSPC function may be explained by the unique dependency of p53 in the context of del(5q)-like MDS HSPC within an aged and/or inflammatory milieu. Low-grade inflammation following LPS administration results in fewer number and more proliferative Tifab-/-;miR146a-/- HSC. In order to overcome the negative effects of inflammation, we posit that Tifab-/-;miR-146a-/- HSC have a proclivity to gain a competitive advantage upon loss of p53. TIFAB has previously been implicated in p53 regulation by directly regulating USP15 activity.31 Expression of TIFAB in HSPC permits USP15 signaling to substrates, including MDM2, and mitigates p53 expression in leukemic myeloid cells. As such, TIFAB-deficient HSPC exhibit compromised USP15 signaling and are sensitized to a variety of hematopoietic stressors by derepression of p53. Thus, our finding that loss of miR-146a and TIFAB in HSC results in increased p53 expression during inflammation is likely due to a cooperative effect through both del(5q) genes. Interestingly, deletion of p53 in Tifab-/-;miR-146a-/- HSC did not result in overt leukemia, suggesting that the primary consequence of monoallelic p53 loss is to provide del(5q) MDS HSC a competitive advantage during low-grade inflammation. Additional del(5q) genes could cooperate with loss of p53 to manifest overt leukemia. As the current study focused on two del(5q)-related genes, miR-146a and TIFAB, it likely that additional genes within the deleted segment on chr 5q also contribute to the functional consequences of inflammation in del(5q) MDS.
Increased innate immune signaling in del(5q) MDS HSPC is attributed to several haploinsufficient genes. The individual contribution of each of these haploinsufficent genes to MDS phenotypes has been extensively evaluated in mouse models.1-3 More recently, various models were generated to examine the cooperation of multiple del(5q) genes. Mice lacking miR-143 and miR-145 have impaired HSPC activity with depletion of functional HSC, but activation of progenitor cells.53 Lam et al. identi fied components of the transforming growth factor β (TGF β ) pathway as key targets of miR-143 and miR-145. As expected, the combined deletion of miR-146a with RPS14 and CSNK1A1 recapitulated many cardinal features of del(5q) MDS, including more severe anemia with faster kinetics than Rps14 haploinsufficiency alone and pathognomonic megakaryocyte morphology.18 Combined hematopoietic-specific deletion of Tifab and miR-146a resulted in more rapid and severe cytopenia, and progression to a fatal BM failure-like disease as compared with Tifab- or miR-146a-deficiency alone.25 Dual deficiency of mDia1 and
miR-146a caused an age-related anemia and ineffective erythropoiesis mimicking human MDS.54 It was also demonstrated that the aging BM microenvironment was important for the development of ineffective erythropoiesis in these mice. Of note, loss of miR-146a alone is sufficient to induce HSC defects and hematopoietic disease.55,56 Deletion of miR-146a in HSC promotes premature HSC aging and inflammation, preceding development of agingassociated myeloid malignancy. These effects are mediated in part by excessive signaling through its targets TRAF6 and IRAK1. Collectively, these findings confirm that aberrant innate immune signaling in del(5q) MDS HSC and altered responses to the inflammatory microenvironment play a critical role in the pathogenesis and complex phenotype of myeloid malignancies.
Several attempts are being pursued to restore normal innate immune signaling in MDS HSC. One approach has been to inhibit IRAK4.57 There are ongoing clinical studies evaluating IRAK4 inhibitors for low-risk MDS patients, which will provide insight into the clinical benefit of suppressing cell-intrinsic innate immune signaling pathways downstream of IRAK-TRAF6. In our study, we utilized IRAK1/4 and UBE2N inhibitors, which individually improved the anemia in the del(5q)-like MDS model. Moreover, it is also possible that suppressing chronic inflammation in low-risk del(5q) MDS patients may diminish the selective pressures leading to acquired p53 mutations. Such an approach has been demonstrated experimentally wherein the selective pressure against p53 in models of lymphoma can be mitigated by targeting the apoptotic pathway.58 Therefore, future studies should determine whether mitigating inflammation in del(5q) MDS may reduce the likelihood of secondary p53 mutations and progression towards more aggressive disease.
Disclosures
DTS serves on the scientific advisory board at Kurome Therapeutics and is a consultant for and/or received funding
1. Trowbridge JJ, Starczynowski DT. Innate immune pathways and inflammation in hematopoietic aging, clonal hematopoiesis, and MDS. J Exp Med. 2021;218(7):e20201544.
2. Barreyro L, Chlon TM, Starczynowski DT. Chronic immune response dysregulation in MDS pathogenesis. Blood. 2018;132(15):1553-1560.
3. Stubbins RJ, Platzbecker U, Karsan A. Inflammation and myeloid malignancy: quenching the flame. Blood. 2022;140(10):1067-1074.
4. Cai Z, Kotzin JJ, Ramdas B, et al. Inhibition of inflammatory signaling in Tet2 mutant preleukemic cells mitigates stressinduced abnormalities and clonal hematopoiesis. Cell Stem Cell. 2018;23(6):833-849.
5. Higa KC, Goodspeed A, Chavez JS, et al. Chronic interleukin-1 exposure triggers selection for Cebpa-knockout multipotent
from Kurome Therapeutics, Captor Therapeutics, Treeline Biosciences, and Tolero Therapeutics. DTS has equity in Kurome Therapeutics. The other authors have no conflicts of interest to disclose.
Contributions
TM performed experiments, analyzed and interpreted data, and wrote the manuscript. CSW performed experiments, analyzed and interpreted data, and contributed to writing the manuscript. PA, EV, KC, MN, CI, KH, AS and MV performed experiments and analyzed and interpreted data. TM and DTS conceived and directed the study, analyzed and interpreted data, and wrote and/or edited the manuscript. All authors approved the final version of the manuscript.
This work was supported in parts by the National Institute of Health (U54DK126108, R35HL166430, R01CA271455, R01CA275007), Cincinnati Children’s Hospital Research Foundation, Cancer Free Kids, and Blood Cancer Discoveries Grant program through The Leukemia & Lymphoma Society, The Mark Foundation for Cancer Research and The Paul G. Allen Frontiers Group to DTS. TM was supported by The Uehara Memorial Foundation, The Waksman Foundation of Japan, The Mochida Memorial Foundation for Medical and Pharmaceutical Research, Japan Society for the Promotion of Science, and Ohio State Univ ersity Comprehensive Cancer Center. TM was a Leukemia and Lymphoma Society Special Fellow. EV was supported by an NIH Institutional Research Training Grant (T32CA236764).
Cell lines and mouse models used in these studies are publicly available through commercial sources or may be made available from the authors upon written request and material transfer agreement approval. The authors are also glad to share guidance regarding protocols and assays used in these studies upon written request.
hematopoietic progenitors. J Exp Med. 2021;218(6):e20200560.
6. Reynaud D, Pietras E, Barry-Holson K, et al. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20(5):661-673.
7. Hemmati S, Haque T, Gritsman K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front Oncol. 2017;7:265.
8. Avagyan S, Henninger JE, Mannherz WP, et al. Resistance to inflammation underlies enhanced fitness in clonal hematopoiesis. Science. 2021;374(6568):768-772.
9. Muto T, Walker CS, Choi K, et al. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome
HSCs. Nat Immunol. 2020;21(5):535-545.
10. Liao M, Chen R, Yang Y, et al. Aging-elevated inflammation promotes DNMT3A R878H-driven clonal hematopoiesis. Acta Pharm Sin B. 2022;12(2):678-691.
11. Zhang CR, Ostrander EL, Kukhar O, et al. Txnip enhances fitness of Dnmt3a-mutant hematopoietic stem cells via p21. Blood Cancer Discov. 2022;3(3):220-239.
12. Hormaechea-Agulla D, Matatall KA, Le DT, et al. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNgamma signaling. Cell Stem Cell. 2021;28(8):1428-1442.
13. Qian Z, Joslin JM, Tennant TR, et al. Cytogenetic and genetic pathways in therapy-related acute myeloid leukemia. Chem Biol Interact. 2010;184(1-2):50-57.
14. Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol. 2008;87(7):515-526.
15. Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110(13):4385-4395.
16. Ebert BL, Pretz J, Bosco J, et al. Identification of RPS14 as a 5qsyndrome gene by RNA interference screen. Nature. 2008;451(7176):335-339.
17. Schneider RK, Schenone M, Ferreira MV, et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med. 2016;22(3):288-297.
18. Ribezzo F, Snoeren IAM, Ziegler S, et al. Rps14, Csnk1a1 and miRNA145/miRNA146a deficiency cooperate in the clinical phenotype and activation of the innate immune system in the 5q- syndrome. Leukemia. 2019;33(3):1759-1772.
19. Stalmann USA, Ticconi F, Snoeren IAM, et al. Genetic barcoding systematically compares genes in del(5q) MDS and reveals a central role for CSNK1A1 in clonal expansion. Blood Adv. 2022;6(6):1780-1796.
20. Keerthivasan G, Mei Y, Zhao B, Zhang L, Harris CE, Gao J et al. Aberrant overexpression of CD14 on granulocytes sensitizes the innate immune response in mDia1 heterozygous del(5q) MDS. Blood. 2014;124(5):780-790.
21. Kumar MS, Narla A, Nonami A, et al. Coordinate loss of a microRNA and protein-coding gene cooperate in the pathogenesis of 5q- syndrome. Blood. 2011;118(17):4666-4673.
22. Starczynowski DT, Kuchenbauer F, Argiropoulos B, et al. Identification of miR-145 and miR-146a as mediators of the 5qsyndrome phenotype. Nat Med. 2010;16(1):49-58.
23. Fang J, Barker B, Bolanos L, et al. Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-κB gene network. Cell Rep. 2014;8(5):1328-1338.
24. Rhyasen GW, Bolanos L, Fang J, et al. Targeting IRAK1 as a therapeutic approach for myelodysplastic syndrome. Cancer Cell. 2013;24(1):90-104.
25. Varney ME, Choi K, Bolanos L, et al. Epistasis between TIFAB and miR-146a: neighboring genes in del(5q) myelodysplastic syndrome. Leukemia. 2017;31(2):491-495.
26. Varney ME, Niederkorn M, Konno H, et al. Loss of Tifab, a del(5q) MDS gene, alters hematopoiesis through derepression of Tolllike receptor-TRAF6 signaling. J Exp Med. 2015;212(11):1967-1985.
27. Fang J, Bolanos LC, Choi K, et al. Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nat Immunol. 2017;18(2):236-245.
28. Starczynowski DT, Morin R, McPherson A, et al. Genome-wide identification of human microRNAs located in leukemiaassociated genomic alterations. Blood. 2011;117(2):595-607.
29. Boldin MP, Taganov KD, Rao DS, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice.
J Exp Med. 2011;208(6):1189-1201.
30. Zhao JL, Rao DS, Boldin MP, Taganov KD, O'Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146adeficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A. 2011;108(22):9184-9189.
31. Niederkorn M, Hueneman K, Choi K, et al. TIFAB regulates USP15-mediated p53 signaling during stressed and malignant hematopoiesis. Cell Rep. 2020;30(8):2776-2790.
32. Melgar K, Walker MM, Jones LM, Bolanos LC, Hueneman K, Wunderlich M et al. Overcoming adaptive therapy resistance in AML by targeting immune response pathways. Sci Transl Med. 2019;11(508):eaaw8828.
33. Jones LM, Melgar K, Bolanos L, et al. Targeting AML-associated FLT3 mutations with a type I kinase inhibitor. J Clin Invest. 2020;130(508):2017-2023.
34. Barreyro L, Sampson AM, Ishikawa C, et al. Blocking UBE2N abrogates oncogenic immune signaling in acute myeloid leukemia. Sci Transl Med. 2022;14(635):eabb7695.
35. Fang J, Liu X, Bolanos L, et al. A calcium- and calpaindependent pathway determines the response to lenalidomide in myelodysplastic syndromes. Nat Med. 2016;22(7):727-734.
36. Rhyasen GW, Wunderlich M, Tohyama K, Garcia-Manero G, Mulloy JC, Starczynowski DT. An MDS xenograft model utilizing a patient-derived cell line. Leukemia. 2014;28(5):1142-1145.
37. Fang J, Muto T, Kleppe M, et al. TRAF6 mediates basal activation of NF-kappaB necessary for hematopoietic stem cell homeostasis. Cell Rep. 2018;22(5):1250-1262.
38. Muto T, Guillamot M, Yeung J, et al. TRAF6 functions as a tumor suppressor in myeloid malignancies by directly targeting MYC oncogenic activity. Cell Stem Cell. 2022;29(2):298-314.
39. Chlon TM, Stepanchick E, Hershberger CE, et al. Germline DDX41 mutations cause ineffective hematopoiesis and myelodysplasia. Cell Stem Cell. 2021;28(11):1966-1981.
40. Schuettpelz LG, Link DC. Regulation of hematopoietic stem cell activity by inflammation. Front Immunol. 2013;4:204.
41. Wang R, Yang X, Liu J, et al. Gut microbiota regulates acute myeloid leukaemia via alteration of intestinal barrier function mediated by butyrate. Nat Commun. 2022;13(1):2522.
42. Takizawa H, Fritsch K, Kovtonyuk LV, et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. 2017;21(2):225-240.
43. Esplin BL, Shimazu T, Welner RS, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol. 2011;186(9):5367-5375.
44. Zhang L, McGraw KL, Sallman DA, List AF. The role of p53 in myelodysplastic syndromes and acute myeloid leukemia: molecular aspects and clinical implications. Leuk Lymphoma. 2017;58(8):1777-1790.
45. Bernard E, Nannya Y, Hasserjian RP, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26(10):1549-1556.
46. Dumble M, Moore L, Chambers SM, et al. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736-1742.
47. Nielsen AB, Hansen JW, Orskov AD, et al. Inflammatory cytokine profiles do not differ between patients with idiopathic cytopenias of undetermined significance and myelodysplastic syndromes. Hemasphere. 2022;6(5):e0713.
48. Riello GBC, Mendonça da Silva P, da Silva Oliveira FA, et al. Gut microbiota composition correlates with disease severity in myelodysplastic syndrome. medRxiv. 2022 Apr 18.
doi: 10.1101/2022.04.18.22273768 [preprint, not peer-reviewed]
49. Hosono N, Makishima H, Mahfouz R, et al. Recurrent genetic defects on chromosome 5q in myeloid neoplasms. Oncotarget. 2017;8(4):6483-6495.
50. Jadersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol. 2011;29(15):1971-1979.
51. Lode L, Menard A, Flet L, et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica. 2018;103(4):e143-e146.
52. Saft L, Karimi M, Ghaderi M, et al. p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica. 2014;99(6):1041-1049.
53. Lam J, van den Bosch M, Wegrzyn J, et al. miR-143/145 differentially regulate hematopoietic stem and progenitor activity through suppression of canonical TGFβ signaling. Nat
Commun. 2018;9(1):2418.
54. Mei Y, Zhao B, Basiorka AA, et al. Age-related inflammatory bone marrow microenvironment induces ineffective erythropoiesis mimicking del(5q) MDS. Leukemia. 2018;32(4):1023-1033.
55. Grants JM, Wegrzyn J, Hui T, et al. Altered microRNA expression links IL6 and TNF-induced inflammaging with myeloid malignancy in humans and mice. Blood. 2020;135(25):2235-2251.
56. Zhao JL, Rao DS, Boldin MP, Taganov KD, O'Connell RM, Baltimore D. NF-kappaB dysregulation in microRNA-146adeficient mice drives the development of myeloid malignancies. Proc Natl Acad Sci U S A. 2011;108:9184-9189.
57. Bennett J, Starczynowski DT. IRAK1 and IRAK4 as emerging therapeutic targets in hematologic malignancies. Curr Opin Hematol. 2022;29(1):8-19.
58. Lu X, Yang C, Yin C, Van Dyke T, Simin K. Apoptosis is the essential target of selective pressure against p53, whereas loss of additional p53 functions facilitates carcinoma progression. Mol Cancer Res. 2011;9(4):430-439.
Srdan Verstovsek,1° Lynda Foltz,2 Vikas Gupta,3 Robert Hasserjian,4 Taghi Manshouri,1 John Mascarenhas,5 Ruben Mesa,6 Olga Pozdnyakova,7 Ellen Ritchie,8 Ivo Veletic,1 Katia Gamel,9 Habib Hamidi,10 Lyrialle Han,10 Brian Higgins,10 Kerstin Trunzer,9 Marianne Uguen,9 Dao Wang,9 Tarec Christoffer El-Galaly,9° Boyan Todorov9 and Jason Gotlib11
1Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2St Paul’s Hospital, University of British Columbia, Vancouver, British Columbia, Canada; 3Princess Margaret Cancer Center, University of Toronto, Toronto, Ontario, Canada;
4Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; 5Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 6Mays Cancer Center at UT Health San Antonio MD Anderson, San Antonio, TX, USA; 7Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA; 8Weill Cornell Medicine, New York, NY, USA; 9F. Hoffmann-La Roche, Ltd., Basel, Switzerland; 10Genentech Inc, South San Francisco, CA, USA and 11Stanford Cancer Institute/Stanford University School of Medicine, Stanford, CA, USA
°SV current address: Kartos Therapeutics Inc, Redwood City, CA, USA.
°TCE-G current address: Department of Hematology, Aalborg University Hospital, Aalborg, Denmark.
Correspondence: S. Verstovsek srdanverstovsek@gmail.com
Received: November 11, 2022.
Accepted: May 3, 2023.
Early view: May 11, 2023.
htps://doi.org/10.3324/haematol.2022.282411
Published under a CC BY license
Pentraxin 2 (PTX-2; serum amyloid P component), a circulating endogenous regulator of the inflammatory response to tissue injury and fibrosis, is reduced in patients with myelofibrosis (MF). Zinpentraxin alfa (RO7490677, PRM-151) is a recombinant form of PTX-2 that has shown preclinical antifibrotic activity and no dose-limiting toxicities in phase I trials. We report results from stage 1 of a phase II trial of zinpentraxin alfa in patients with intermediate-1/2 or high-risk MF. Patients (n=27) received intravenous zinpentraxin α weekly (QW) or every 4 weeks (Q4W), as monotherapy or an additional therapy for patients on stable-dose ruxolitinib. The primary endpoint was overall response rate (ORR; investigatorassessed) adapted from International Working Group-Myeloproliferative Neoplasms Research and Treatment criteria. Secondary endpoints included modified Myeloproliferative Neoplasm-Symptom Assessment Form Total Symptom Score (MPN-SAF TSS) change, bone marrow (BM) MF grade reduction, pharmacokinetics, and safety. ORR at week 24 was 33% (n=9/27) and varied across individual cohorts (QW: 38% [3/8]; Q4W: 14% [1/7]; QW+ruxolitinib: 33% [2/6]; Q4W+ruxolitinib: 50% [3/6]). Five of 18 evaluable patients (28%) experienced a ≥50% reduction in MPN-SAF TSS, and six of 17 evaluable patients (35%) had a ≥1 grade improvement from baseline in BM fibrosis at week 24. Most treatment-emergent adverse events (AE) were grade 1–2, most commonly fatigue. Among others, anemia and thrombocytopenia were infrequent (n=3 and n=1, respectively). Treatment-related serious AE occurred in four patients (15%). Overall, zinpentraxin alfa showed evidence of clinical activity and tolerable safety as monotherapy and in combination with ruxolitinib in this open-label, non-randomized trial (clinicaltrials gov. Identifier: NCT01981850).
Myelofibrosis (MF) is a rare and often fatal hematologic neoplasm characterized by a chronic pro-inflammatory state and progressive bone marrow (BM) failure.1-3 The pressures of chronic inflammation, such as pro-inflammatory cytokine and chemokine release, allow for the selection, clonal expansion, and evolution of mutant
hematopoietic stem cells (HSC).2,4-6 Mutant HSC proliferate in the BM, driving cytokine release, myeloid hyperproliferation, and BM fibrosis.2,5 This chronic inflammation also results in remodeling of the BM microenvironment, perturbing the BM niche, and in turn disturbing normal hematopoiesis.7 The resulting ineffective hematopoiesis and activation of inflammatory pathways have been linked to low hemoglobin levels, high leukocyte count, progress-
ive thrombocytopenia, and circulating blast cells, which have predicted shortened survival of patients with MF.3,8 Pentraxin 2 (PTX-2, also known as serum amyloid P component) is a circulating, endogenous regulator of the inflammatory response to tissue injury that is constitutively produced by the liver (Figure 1).9-12 At the sites of tissue injury, PTX-2 binds to damaged cells and nuclear components (damage- and pathogen-associated molecular patterns) in the early stages of apoptosis, and facilitates their safe removal in a nonimmunogenic manner.9,10,13-16 By binding to Fcγ receptors and DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), PTX-2 promotes immunoregulatory and phagocytic polarization of monocytes and inhibits their differentiation into fibroblast-like, collagen-producing cells called fibrocytes.9,10,12-15 Consequently, low PTX-2 levels may contribute to fibrotic diseases.14 Supporting this hypothesis, plasma levels of PTX-2 were shown to be lower in patients with primary MF17 and idiopathic pulmonary fibrosis (IPF) than in healthy controls.15

Zinpentraxin alfa is a recombinant human form of PTX-2 with a mechanism of action that has shown broad anti-fibrotic and anti-inflammatory activity in preclinical models of fibrotic disease.17-19 Zinpentraxin alfa prevented BM fibrosis through its inhibition of primary MF BM-derived fibrocyte differentiation in vitro, and prevented and reversed fibrosis in animal models of MF.17 In addition, zinpentraxin alfa significantly improved survival in a mouse model with a lethal MF-like phenotype.17 Clinically, zinpentraxin alfa has shown no dose-limiting toxicities in phase I trials in healthy volunteers and patients with IPF.20 Evaluation of zinpentraxin alfa in a phase II trial (clinicaltrials gov. Identifier: NCT02550873) in patients with IPF has demonstrated clinical activity and a generally well-tolerated safety profile.21-23
Currently, there are three approved drug therapies for controlling the symptoms of MF, all of which are Janus kinase (JAK) inhibitors: ruxolitinib, fedratinib, and pacritinib.24-26 Clinically, JAK inhibitors have consistently demonstrated the ability to improve symptoms and reduce spleen size in patients with MF25,27-31; however, ruxolitinib is associated with suppression of hematopoiesis, and the resulting anemias often limit the duration of therapy in real-world settings.32-34 The effect of JAK inhibition on BM fibrosis remains controversial: an analysis of BM biopsies from patients with MF in the phase III COMFORT-II study showed no improvements in histopathological abnormalities at 6 months of therapy with ruxolitinib,35 and no consistent pattern of fibrosis grade improvement was observed with prolonged ruxolitinib therapy for up to 5 years.33 Therefore, there remains an unmet medical need for disease-modifying therapies for patients with MF with improved tolerability, and potential targeting of not only the MF HSC but also the malignant BM niche.7,36,37 Supplementation of ruxolitinib with a therapy that can deplete affected monocytes, and,
thus reduce fibrosis and inflammation, is anticipated to be a promising approach in MF.38
We conducted a phase II trial (clinicaltrials gov. Identifier: NCT01981850) to explore the efficacy and safety of zinpentraxin alfa in patients with MF. Here, we report the efficacy and safety findings from stage 1 of this trial, which investigated zinpentraxin alfa as monotherapy and as an additional therapy for patients on stable-dose ruxolitinib, each with two different dosing schedules, in patients with primary MF, post-essential thrombocythemia (post-EV) MF or post-polycythemia vera (post-PV) MF, with primary analysis at week 24, followed by an open-label extension (OLE) for patients with a clinical benefit from treatment.
The trial was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice of the International Council for Harmonization. The protocol was approved by the relevant Institutional Review Board/Independent Ethics Committee at each site. All patients provided informed written consent.
This is an open-label, phase II trial comprising a 4-week screening period, 24-week treatment period (6×4-week
cycles), and 4-week follow-up period (Online Supplementary Figure S1). An OLE was available for patients without disease progression or toxicity warranting treatment discontinuation, and with clinical benefit as judged by the investigator, for as long as there was a benefit. Patients in the monotherapy cohorts received zinpentraxin alfa weekly (QW; cohort 1) or every 4 weeks (Q4W; cohort 2). Patients in the combination cohorts (on a stable dose of ruxolitinib for ≥12 weeks) continued with oral ruxolitinib twice daily at the same dose plus zinpentraxin alfa QW (cohort 3) or Q4W (cohort 4). Dosing frequency allocation was based on patients’ availability for weekly visits. Zinpentraxin alfa 10 mg/kg was administered as a 60-minute intravenous infusion. Dosing and administration are further described in the Online Supplementary Appendix
Patients
Key eligibility criteria included: aged ≥18 years with primary, post-ET, or post-PV MF; intermediate-1, intermediate-2, or high-risk disease per the International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) Dynamic International Prognostic Scoring System (DIPSS39); and locally assessed MF grade 2 or 3 BM fibrosis according to the European Consensus on Grading of Bone Marrow Fibrosis. A BM biopsy was required within 4 weeks prior to treatment initiation to establish the baseline fibrosis grade by central review.40,41 Patients in the monotherapy arms must have received no treatment for MF for ≥2 weeks.
The overall response rate (ORR) at week 24 was the primary endpoint. ORR was defined as the percentage of patients with a response (assessed by the investigator) as follows (Online Supplementary Table S1): clinical improvement, partial remission, or complete remission per IWGMRT criteria42 at a post-baseline assessment of treatment response OR stable disease for three consecutive end-ofcycle response assessments (i.e., day 1 of the subsequent cycle) along with an improvement in the BM fibrosis score (determined by central review, blinded to subject, treatment, and time point) relative to baseline by ≥1 grade at any time point during the period of stable disease. Key secondary endpoints included: modified Myeloproliferative Neoplasm-Symptom Assessment Form Total Symptom Score (MPN-SAF TSS) change from baseline over time and proportion of patients with ≥50% relative reduction versus baseline at the beginning of each cycle (cycle 2 onward); BM MF grade reduction based on reticulin and trichrome staining40,41 relative to baseline at week 24; pharmacokinetics (PK); and safety.
Response assessments in the OLE were performed on day 1 of every third cycle (every 12 weeks), beginning on cycle 10 day 1 (except ORR: best response at any time).
Additional endpoints and analyses (palpable spleen size, fibrocyte quantification, cytokine levels, and gene expression analyses), procedures, and statistical methods are described in the Online Supplementary Appendix.
In total, 27 patients were enrolled at seven sites in the USA and Canada and assigned to one of the four treatment cohorts: cohort 1, n=8; cohort 2, n=7; cohort 3, n=6; cohort 4, n=6 (Online Supplementary Figure S1). Baseline demographics and clinical characteristics are shown in the Online Supplementary Table S2. The median age of patients was 67.0 (range, 51–85) years, with 66.7% of patients aged ≥65 years. The majority of patients were anemic (hemoglobin <100 g/L), around half of the patients were thrombocytopenic (platelets ≤100×109/L), and a considerable proportion had severe thrombocytopenia at baseline (37.0%; platelets ≤50×109/L). By central BM review, around a third of patients had BM fibrosis grade 2 and approximately half had grade 3 at baseline; over twothirds of patients had intermediate-2 or high-risk MF per IWG-MRT DIPSS. Approximately half of patients had primary MF, around one-third of patients had post-PV MF, and the remaining patients had post-ET MF. Five patients were categorized as platelet transfusion–dependent at baseline (≥2 platelet transfusions in any 12 weeks prior to cycle 1 day 1, regardless of baseline platelet level). Patient disposition, including reasons for discontinuations, is shown in the Online Supplementary Figure S2. Overall, 20 patients completed the main trial and were evaluable at week 24. A total of 18 patients continued into the OLE; two patients did not continue based on patient decision. Median treatment duration in the OLE following completion of the main study was 24.4 months, up to a maximum of 70 months (cycle 83).
Efcacy
Overall, nine of 27 (33.3%; 90% confidence interval [CI]: 18.6–51.0) patients were responders during the main phase of the trial (up to week 24 of treatment); these patients included three of eight (37.5%) patients receiving zinpentraxin alfa QW in cohort 1, one of seven (14.3%) receiving zinpentraxin alfa Q4W in cohort 2, two of six (33.3%) receiving zinpentraxin alfa QW plus ruxolitinib in cohort 3 and three of six (50.0%) receiving zinpentraxin alfa Q4W plus ruxolitinib in cohort 4 (Table 1). During the OLE, the ORR increased to 59.3% of patients (n=16/27), which included clinical improvement in 33.3% of patients (n=9/27), and stable disease with BM improvement in 51.9% (n=14/27).
Five of 18 evaluable patients (27.8%) experienced a ≥50%
Figure 2. Percentage change from baseline in Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score and palpable spleen size at week 24. (A) Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPNSAF TSS)a; (B) palpable spleen sizeb; all treated population. aData shown for 18 patients in total; 7 patients withdrew from the trial before week 24 and 2 patients had missing values at week 24. bData shown for 15 patients in total; 7 patients withdrew from the trial before week 24 and 5 patients had missing values at week 24. Q4W: every 4 weeks; QW: weekly.

Table 1. Overall response rate in the all-treated population at week 24 (primary endpoint).
BM: bone marrow; CI: confidence interval; ORR: overall response rate; Q4W: every 4 weeks; QW: weekly; SD: stable disease.
reduction in MPN-SAF TSS at week 24, most of whom were in cohort 1 (n=3/8; 37.5%; Figure 2A). In total, 15 of 27 patients (55.6%) had a ≥50% reduction in MPN-SAF TSS at any time during the trial (Online Supplementary Figure S3A). At week 24, 35.3% (6/17) of patients with evaluable BM fibrosis grade demonstrated reduction (improvement) of ≥1 grade compared with baseline, and one of two patients with baseline fibrosis grade 1 had a one-grade increase (Table 2). Notably, one patient had complete resolution of grade 3 fibrosis; however, there was no follow-up to determine if this was sustained. Percentage change from baseline to week 24 in palpable spleen size is shown in Figure 2B. At week 24, two of 15 patients (13.3%) had a ≥35% reduction in palpable spleen size. In total, six of 21 patients (28.6%) had a ≥35% reduction in palpable spleen size at any time during the trial (Online Supplementary Figure S3B).
In total, 96.3% (n=26/27) of patients experienced ≥1 treatment-emergent adverse event (TEAE) in the main phase of the trial (100%, n=8 in cohort 1, n=6 in each of cohorts 3 and 4; 85.7%, n=6 for cohort 2) (Table 3). The majority of TEAE in the main phase of the trial were grade 1 or 2 in intensity, the most common being fatigue, cough, diarrhea, nausea, and oral herpes. New onset or exacerbation of anemia and thrombocytopenia were infrequently reported in all treatment cohorts (anemia, n=3 [1 patient in each of cohorts 1, 3, and 4]; thrombocytopenia, n=1 in cohort 3). Grade 3–4 TEAE were observed in three patients (11.1%; n=1 in each of cohorts 1, 2 and 3; all grade 3): anemia in two patients (7.4%; n=1 in each of cohorts 1 and 3), and abdominal pain, sialadenitis, urosepsis, hypoxia, nerve compression, sciatic nerve neuropathy, post procedural hematoma, and iron overload (3.7%, 1 patient each); grade 3 anemia (n=1), abdominal pain, and sialadenitis were considered related to zinpentraxin alfa or ruxolitinib treatment.
Serious TEAE and treatment-related serious TEAE are summarized in Table 3. Fourteen serious TEAE were observed in five patients (18.5%; n=2 in each of cohorts 1 and
3, n=1 in cohort 2) in the main trial phase. None of the individual serious TEAE were reported in more than one patient. Five serious treatment-related TEAE occurred in four patients (14.8%; n=1 in each of cohorts 1 and 2, n=2 in cohort 3): abdominal pain, sialadenitis, viral pneumonia, gastroenteritis, and respiratory syncytial virus infection (1 patient each; 1 patient experienced 2 different events). Three patients (11.1%; n=2 in cohort 1, n=1 in cohort 3) experienced a fatal TEAE during the main phase of the trial. One of these patients (cohort 1) experienced grade 5 gastroenteritis and viral pneumonia, which the investigator considered related to treatment. None of the other grade 5 events in the other two patients (cardiac arrest, organ failure, metabolic encephalopathy and renal failure in 1 patient, and pneumonia in another patient) were considered related to study treatment. An overview of adverse events during the OLE can be found in the Online Supplementary Table S3. Notably, one patient in the OLE continued until cycle 83.
During the main phase of the trial, infusion-related reactions (IRR) were reported in one patient (cohort 4; grade 2) after fi ve doses of zinpentraxin alfa ; no action was taken on study treatment and the event was resolved. During the OLE, two patients experienced IRR: one patient zinpentraxin alfa experienced grade 2 rash pruritic after 11 doses of zinpentraxin alfa ( infusion was interrupted and restarted on the same day without reoccurrence of IRR after administration of diphenhydramine and dexamethasone) and one patient experienced a grade 1 IRR after ten doses of zinpentraxin alfa (no action was taken on study treatment and the event resolved ). All three patients with an IRR tested positive for treatmentemergent anti-drug antibodies (ADA) in the main phase or the OLE of stage 1. However, there was no overall evidence of an impact of ADA status on the safety and tolerability of
Results of the pharmacokinetics analysis are provided in the Online Supplementary Appendix.
aA total of 25/27 patients in the study underwent a bone marrow (BM) biopsy. bOne patient had missing BM fibrosis grade at baseline. cEight patients had missing BM fibrosis grade at week 24. BM fibrosis grades according to World Health Organization criteria (as determined by central review). Patients were initially enrolled based on having grade ≥2 BM fibrosis according to local assessment (2 patients were scored as baseline grade 1 fibrosis after central review). Key: italics: reduction in grade; bold underline: increase in grade.
zinpentraxin alfaHemoglobin, platelet, and transfusions
Hemoglobin (g/L) and platelet count (109/L) mean changes from baseline and the proportion of patients who received red blood cell and platelet transfusions during the main phase of the trial by dosing regimen/treatment group are shown in Figure 3 and the Online Supplementary Figure S4. The full details of this analysis are included in the Online Supplemental Appendix. Hemoglobin and platelet count levels fluctuated over the course of the trial and there was no obvious trend observed. Likewise, no obvious trend was observed in the proportion of patients requiring transfusions.
BM fibrocytes were evaluated by immunostaining in eight patients from a single center, who each had completed ≥12 cycles of treatment with zinpentraxin alfa, as part of a post hoc analysis. Mean fibrocyte count declined from 378.0 cells/mm3 at baseline to 115.1 cells/mm3 at week 24,
with a corresponding reduction in the proportion of fibrocytes as a percentage of total BM cells, from 11.2% to 3.0% (Figure 4). Plasma samples from five of these patients were also available for post hoc cytokine analysis. Changes in cytokine levels in these patients over time are shown in Figure 5 and the Online Supplementary Figure S5. Of 77 analyzed cytokines, downregulation in certain cytokines was observed, notably interleukin (IL)-8 (Figure 5). A correlational analysis of mean fibrocyte count and percentage of fibrocytes over time stratified by bone marrow fibrosis grade did not find any significant correlation, perhaps due to the relatively small sample size.
In an exploratory analysis, no significant changes over time with zinpentraxin alfa treatment were observed in gene expression from whole blood mRNA sequencing ( data not shown ; based on 119/124 samples from 25 patients; in total, 20,776 of 58,303 genes had sufficient expression for analysis).
aIn total safety population (N=27). bRelated to zinpentraxin alfa ALT: alanine aminotransferase; Q4W: every 4 weeks; QW: weekly; TEAE: treatment-emergent adverse event; URTI: upper respiratory tract infection.
Figure 3. Effect of treatment with zinpentraxin alfa on hemoglobin and red blood cell transfusions. Mean hemoglobin (g/L) levels and proportion of patients with red blood cell (RBC) transfusions over timea; all-treated population. *Zero transfusions recorded at scheduled time point. a(A and C) Time points are day 1, 8, 15, and 22 in each cycle, plus day 29 for cycle 6; (B and D) time points are day 1 and 15 in cycle 1, day 1 in cycles 2 to 5, and days 1 and 29 in cycle 6. Q4W: every 4 weeks; QW: weekly; Rux: ruxolitinib.

In this open-label, non-randomized trial, clinical activity with zinpentraxin alfa alone and in combination with ruxolitinib was observed in patients with MF at week 24 and through the OLE for up to 332 weeks (>6 years). During the main phase of the trial, one-third of patients with MF receiving zinpentraxin alfa as monotherapy or in combination with ruxolitinib had a response per protocol-specified criteria, the majority of whom had stable disease with BM improvement, and the ORR further increased during the OLE phase to 59.3% (n=16/27). This trial used an ORR adapted from the IWG-MRT and European LeukemiaNet criteria, which aims to assess the value of new drugs in inducing morphologic remission or improvements in MF-associated signs and symptoms.42 The IWG-MRT criteria (introduced at the time of protocol development) have not, to our knowledge, been used in other MF clinical trials to date, making it difficult to draw direct comparisons.
Improvements in symptoms were noted following treatment with zinpentraxin alfa, with nearly 20% of evaluable
patients experiencing a ≥50% reduction in MPN-SAF TSS at week 24. Six patients (35.3%) had an improvement from baseline in BM fibrosis grade at week 24 and reductions in palpable spleen size were observed in two of 15 patients (13.3%). However, resolution of fibrosis requires complete remodeling of the BM niche, with removal of excessive fi brosis, extracellular matrix remodeling, and cellular migration back to the primary hematopoietic sites, which would be unlikely to occur within 24 weeks; as such, substantial clinical impact on the extra-medullary hematopoiesis and spleen size may not be apparent during this time frame.43,44 Furthermore, the lack of a placebo arm in this trial prevents comparisons to the natural course of BM fibrosis. Therefore, we were unable to assess the natural variability in BM fibrosis that may be reflected in inter-sample variability (i.e., heterogeneity due to sampling location, or different amounts of material in each sample), though inter-assessor variability was accounted for by the use of central review. In addition, spleen size measured by palpation is less accurate compared with magnetic resonance imaging, even though palpation is util-
(A) Mean fibrocyte count and percentage of fibrocytes as a proportion of total cells in the bone marrow (BM) at baseline and week 24a (N=8); (B) representative fluorescence micrographs of BM biopsies of a patient at baseline and at week 24. In (A) data shown for 8 patients from a single center, who had each completed ≥12 cycles of treatment. Error bars represent standard error. In (B), depicted for 1 patient are DNA (Hoechst 33258; blue), CD45 (Opal 690; red), CD68 (Opal 520; green), and procollagen-I (Opal 570; white). Images are shown in pseudo-colors corresponding to individual channel intensities and as a composite of all 4 channels (Merge). Scale bar, 100 μm. Micrographs are provided at a resolution of 600 px/in. aIncludes 1 patient with data from week 20.

ized frequently in the routine clinical setting. However, these results can be considered promising given this population’s likely poor prognosis: the majority of patients had intermediate-2 or high-risk MF, had received prior JAK inhibitor therapy, and had a low platelet count, and some were platelet-transfusion-dependent. Low hemoglobin and platelet counts are both associated with a poor prognosis in patients with MF, and these patients have very limited treatment options.3,45-47 While the hemoglobin, platelet count, and transfusions fluctuated during therapy, there were no notable downward trends in these hematologic parameters, contrary to the reported anemia and thrombocytopenia progression in patients with such advanced disease.48 These observations are supported by the number of patients requiring platelet or red blood cell transfusions not increasing during the trial. This is a significant finding as patients with MF often have progressive anemia and thrombocytopenia that are further exacerbated by the use of JAK inhibitors such as ruxolitinib,49 although the underlying reason for the observed results in the current study cannot be isolated based on the data available. The safety data from this trial suggest an overall good tolerability of zinpentraxin alfa as monotherapy, as well as in combination with ruxolitinib. The incidence of serious TEAE was numerically higher in the QW cohorts (with or without ruxolitinib) than in the Q4W cohorts (with or with-
out ruxolitinib). However, the small number of patients in each cohort and lack of randomization in this study preclude conclusions regarding any potential differences in the safety of these dosing regimens. Overall, a total of three IRR were reported. All IRR were grade 1–2, non-serious, and resolved, and patients continued study treatment.
Of note, while anemia and thrombocytopenia frequently result from the use of JAK inhibitors as well as MF disease progression itself,8,27-29,48 zinpentraxin alfa is not anticipated to have myelosuppressive effects based on its mechanism of action. Consistent with this expectation, new onset or exacerbation of anemia was infrequent in all treatment cohorts, occurring in only three patients (one patient each in cohorts 1, 3, and 4) in the main phase of the trial. Treatment-emergent thrombocytopenia was not observed with zinpentraxin alfa monotherapy and occurred in only one patient receiving zinpentraxin alfa in combination with ruxolitinib. Moreover, prolonged administration of zinpentraxin alfa may alleviate the progressive anemia and thrombocytopenia resulting from the BM-associated fibrosis,8 as well as from the myelosuppressive activity of ruxolitinib and other JAK inhibitors.27-29 The nonoverlapping safety profiles of zinpentraxin alfa and JAK inhibitors with respect to hematologic AE25,27-29 also provide support for investigating rational combinations of zin-
Figure 5. Effect of treatment with zinpentraxin alfa on cytokines. Mean levels of selected cytokines across the treatment period from baseline (BSL) to week 130, showing a significant decrease over the course of treatment (N=5). Plasma was analyzed by combined 40- and 37-plex magnetic bead-based immunoassays. Graphs show mean levels of 8 selected cytokines of interest over time, over the period on treatment (time points T1–7 are defined in the Online Supplementary Figure S5). The data points in green denote significant (P<0.05) on treatment changes relative to BSL in the 5 patients. P values were calculated for each time point using an empirical Bayes moderated t test with a Benjamini & Hochberg adjustment for multiple testing.

pentraxin alfa with currently available treatments such as JAK inhibitors. Additionally, the similarity in PK parameters across the four treatment cohorts and the lack of accumulation with more frequent dosing suggests that pentraxin exposure differences across the different dosing schedules were unlikely to impact on zinpentraxin alfa’s safety and tolerability profile. Furthermore, PK parameters were comparable between patients with MF in this analysis and patients with IPF in previous research.50
In preclinical studies of primary MF, zinpentraxin alfa suppressed the formation of fibrocytes cultured from patients’ BM CD14+ monocytes and, in a xenograft murine model, slowed the development of BM fibrosis and prolonged survival.17 In the current trial, quantification of fibrocytes in BM demonstrated a reduction at week 24 compared with baseline, suggesting that zinpentraxin alfa might induce similar effects in patients with MF. Furthermore, plasma cytokine profiling in a subset of patients showed time-dependent reduction of cytokines associated with differentiation and function of fibrocytes and profibrotic macrophages, such as interleukins (i.e., IL-8), which is in line with previously described anti-inflammatory effects of zinpentraxin alfa.9,13-15 Both the observed reduction in BM fibrocyte counts and changes in plasma cytokine levels indicate a zinpentraxin alfa drug effect in line with preclinical findings. However, these results are from a post hoc analysis in only a small subset of patients from this trial and so further investigation in a larger group of patients is required to draw any firm conclusions, particularly as no obvious differences were observed in the gene expression analysis.
A major limitation of this trial is that there was no randomization of cohorts: patients were assigned zinpentraxin alfa as monotherapy versus combination therapy depending on whether they were already receiving ruxolitinib, and the frequency of dosing (QW vs . Q4W) was based on the patient’s availability for trial visits. As such, there is the possibility of significant investigator bias in assigning patients to cohorts, precluding any meaningful inter-cohort comparisons. Also, for the cohorts receiving concomitant ruxolitinib, ruxolitinib administration was heterogenous, ranging from 5 to 25 mg twice daily, with variability between patients as well as for individual patients throughout the study. Additionally, the absence of a control arm meant that the isolated effect of zinpentraxin alfa could not be delineated. Due to the inclusion criteria permitting the participation of patients with a wide range of prognosis risk scores, there was marked variation in disease severity in the population at baseline leading to heterogeneity within each cohort. Furthermore, few patients in each cohort were treatment-naïve; many patients had taken prior therapies or were transfusiondependent, indicating they had more advanced disease. Since there were no obvious differences in effi cacy or
safety between treatment cohorts, and given the small patient numbers and lack of randomization, the results of this phase II trial are difficult to interpret. Hence, any potential differences between zinpentraxin alfa monotherapy and zinpentraxin alfa in combination with ruxolitinib, or differences related to the dosing frequency cannot be determined. Although it is possible that an improvement in hematologic parameters in patients treated with zinpentraxin alfa could have also led to improved tolerably and/or changes in ruxolitinib dosing, no obvious differences were noted between the two combination cohorts. However, further investigations were precluded due to the small number of patients, the significant baseline heterogeneity in the population, lack of randomization, and absence of a ruxolitinib monotherapy control cohort. However, given the consistent ORR across all cohorts, the Q4W schedule was carried into stage 2 of the trial for further monotherapy testing as it offered optimal flexibility and patient convenience. Last but not least, accurate quanti fi cation of BM fi brosis is critical for the development of novel compounds.51 However, accuracy may be compromised due to heterogeneity in fi brosis grade and megakaryocyte clusters within the BM that can lead to inconsistency between different samples and assessments.41 More global quantitative assessments of changes in BM fibrosis using less invasive tools such as magnetic resonance imaging or positron emission tomography/computed tomography scanning of the skeleton may be useful to study anti-fi brotic effects of novel agents in MF.
In summary, zinpentraxin alfa has the potential to be a valuable treatment as monotherapy or in combination with anti-proliferative drugs such as ruxolitinib, through specifically targeting the BM microenvironment to reduce inflammation and fibrosis with no added toxicity. These factors could enable a long-term treatment plan that remains tolerable, evidenced by one patient continuing up to 83 cycles in the OLE. Notwithstanding the small patient numbers, results from stage 1 of this phase II trial showed evidence of clinical activity and a tolerable safety profile of zinpentraxin alfa as single agent and in combination with ruxolitinib, with QW or Q4W dosing schedules. Downward shifts in the BM fibrosis score in some patients and preliminary post hoc subpopulation analyses showing fibrocyte reductions and decreased levels of proinflammatory cytokines support the postulated mechanism of action of zinpentraxin alfa. The results from this trial were used to inform the zinpentraxin alfa monotherapy dosing schedule in stage 2 of this trial, which will be reported separately. These results will advise future investigations of zinpentraxin alfa in select populations and using treatment combinations that have the best chance to ameliorate the disease course for patients.
Disclosures
SV has received research support from BMS, Constellation, CTI BioPharma, Galecto, Geron, Incyte, Kartos, Novartis, NS Pharma, Protagonist, PharmaEssentia, Roche, and Sierra, and consulting fees from BMS, Celgene, Constellation, Incyte, and Novartis. LF has received research funding paid to her institution from BMS, Constellation, CTI BioPharma, Incyte, and Sierra Oncology, and honoraria from BMS and Novartis, and has participated on an advisory committee for Incyte and Paladin Labs. VG has received consulting fees from AbbVie, BMS Celgene, Constellation Biopharma, Novartis, Pfizer, Sierra Oncology; payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing, or educational events from BMS Celgene, Constellation Biopharma, and Novartis; and has participated on a Data Safety Monitoring Board or Advisory Board for AbbVie, BMS Celgene, Pfizer, and Roche. RH has received consulting fees from Daiichi Sankyo, Jazz Pharmaceuticals, and Promedior, Inc., acquired by Roche in February 2020. TM has no conflicts of interest to declare. JM has received research funding paid to his institution from AbbVie, BMS, Celgene, CTI BioPharma, Geron, Incyte, Kartos, Merck, Novartis, PharmaEssentia, and Roche; and consulting fees from AbbVie, BMS, Celgene, Constellation, CTI Bio, Galecto, Geron, Incyte, Kartos, Karyopharm, Novartis, PharmaEssentia, Roche, and Sierra Oncology. RM has received research support from AbbVie, Blueprint Medicines, BMS, CTI BioPharma, Genentech, Incyte, MorphoSys, and Sierra; and honoraria from AbbVie, Blueprint, BMS, CTI BioPharma, Genentech, Geron, GSK, Incyte, Novartis, Sierra, Sierra Oncology and Telios. OP has received consulting fees from Roche and Scopio. ER has received research funding from Jazz and Pfizer, and consulting fees from AbbVie, BMS, Celgene, Incyte, and Novartis. IV has received research funding through his institution from Avilect Biosciences. KG is an employee of Roche and owns equity. HH is an employee of Roche/Genentech. LH is an employee of Genentech and owns stocks. BH is an employee of Roche/Genentech and owns stocks. KT and MU are employees of Roche and own stocks. DW is an employee of Roche. TCE-G was previously an employee of Roche. BT is contracted by Roche. JG has received research funding from AbbVie, Blueprint Medicines, Celgene/BMS, Cogent Biosciences, Deciphera Pharmaceuticals, Incyte, Kartos, Novartis, and Protagonist Therapeutics. He has also served on Advisory Boards and received consulting fees from AbbVie, Blueprint Medicines, Cogent Biosciences, Deciphera Phar-
1. Moulard O, Mehta J, Fryzek J, Olivares R, Iqbal U, Mesa RA. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur J Haematol. 2014;92(4):289-297.
2. Gleitz HFE, Benabid A, Schneider RK. Still a burning question:
maceuticals, Incyte, Kartos, Novartis, PharmaEssentia, and Protagonist Therapeutics.
SV, TM, IV, RM and MU developed the concept and designed the study. JM, SV, TM, IV, LF, ER and RM acquired data. BH, LH, TCE-G, KG, OP and RH (BM morphologic analysis/review), SV, TM, IV, VG, KT, MU, HH, DW, JG and RM analyzed data. BH, LH, JM, TCE-G, KG, SV, TM, IV, VG, BT, LF, KT, MU, HH, RM, ER, DW and JG interpreted results. All authors were involved in the review/revision of the manuscript, approved the final version, and vouch for the accuracy of the content included in the manuscript.
We would like to thank the patients, their family members, participating staff at all of the study centers, and the Promedior study team. We would also like to thank Susanne Muehlbauer for her input into drafting the content of this manuscript and Sudhakar Katakam for providing the statistical analysis for figures . Medical writing support, under the guidance of the authors, was provided by Harriet Oxford and Fiona Scott, PhD, on behalf of CMC AFFINITY, a division of IPG Health Medical Communications , funded by F. Hoffmann-La Roche, Ltd, in accordance with Good Publication Practice (GPP 2022) guidelines. Fibrocyte imaging studies were partially performed in the MD Anderson Cancer Center Flow Cytometry & Cellular Imaging Core Facility, which is supported in part by the National Institutes of Health through Cancer Center Support Grant CA016672.
This study was sponsored by Promedior (original sponsor) and F. Hofmann-La Roche, Ltd. (current sponsor).
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org). Further details on Roche’s criteria for eligible studies are available here: https://vivli.org/ourmember/roche/. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https:www.roche.com/innovation/process/clinicaltrials/data-sharing/.
the interplay between inflammation and fibrosis in myeloproliferative neoplasms. Curr Opin Hematol. 2021;28(5):364-371.
3. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the
International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895-2901.
4. Cervantes F. How I treat myelofibrosis. Blood. 2014;124(17):2635-2642.
5. O'Sullivan JM, Harrison CN. Myelofibrosis: clinicopathologic features, prognosis, and management. Clin Adv Hematol Oncol. 2018;16(2):121-131.
6. Mead AJ, Mullally A. Myeloproliferative neoplasm stem cells. Blood. 2017;129(12):1607-1616.
7. Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285-299.
8. Mughal TI, Vaddi K, Sarlis NJ, Verstovsek S. Myelofibrosisassociated complications: pathogenesis, clinical manifestations, and effects on outcomes. Int J Gen Med. 2014;7:89-101.
9. Castano AP, Lin SL, Surowy T, et al. Serum amyloid P inhibits fibrosis through FcγR-dependent monocyte-macrophage regulation in vivo. Sci Transl Med. 2009;1(5):5ra13.
10. Zahr AA, Salama ME, Carreau N, et al. Bone marrow fibrosis in myelofibrosis: pathogenesis, prognosis and targeted strategies. Haematologica. 2016;101(6):660-671.
11. Mantovani A, Garlanda C, Doni A, Bottazzi B. Pentraxins in innate immunity: from C-reactive protein to the long pentraxin PTX3. J Clin Immunol. 2008;28(1):1-13.
12. Pilling D, Gomer RH. The development of serum amyloid P as a possible therapeutic. Front Immunol. 2018;9:2328.
13. Pilling D, Roife D, Wang M, et al. Reduction of bleomycininduced pulmonary fibrosis by serum amyloid P. J Immunol. 2007;179(6):4035-4044.
14. Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;171(10):5537-5546.
15. Murray LA, Chen Q, Kramer MS, et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by serum amyloid P. Int J Biochem Cell Biol. 2011;43(1):154-162.
16. Familian A, Zwart B, Huisman HG, et al. Chromatin-independent binding of serum amyloid P component to apoptotic cells. J Immunol. 2001;167(2):647-654.
17. Verstovsek S, Manshouri T, Pilling D, et al. Role of neoplastic monocyte-derived fibrocytes in primary myelofibrosis. J Exp Med. 2016;213(9):1723-1740.
18. Murray LA, Rosada R, Moreira AP, et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS One. 2010;5(3):e9683.
19. Nakagawa N, Barron L, Gomez IG, et al. Pentraxin-2 suppresses c-Jun/AP-1 signaling to inhibit progressive fibrotic disease. JCI Insight. 2016;1(20):e87446.
20. Dillingh MR, van den Blink B, Moerland M, et al. Recombinant human serum amyloid P in healthy volunteers and patients with pulmonary fibrosis. Pulm Pharmacol Ther. 2013;26(6):672-676.
21. Raghu G, van den Blink B, Hamblin MJ, et al. Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA. 2018;319(22):2299-2307.
22. Raghu G, Hamblin MJ, Brown AW, et al. Long-term evaluation of the safety and efficacy of recombinant human pentraxin-2 (rhPTX-2) in patients with idiopathic pulmonary fibrosis (IPF): an open-label extension study. Respir Res. 2022;23(1):129.
23. Raghu G, van den Blink B, Hamblin MJ, et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: an open-label extension study. Lancet Respir Med. 2019;7(8):657-664.
24. Singer JW, Al-Fayoumi S, Ma H, Komrokji RS, Mesa R, Verstovsek S. Comprehensive kinase profile of pacritinib, a nonmyelosuppressive Janus kinase 2 inhibitor. J Exp Pharmacol. 2016;8:11-19.
25. Bewersdorf JP, Jaszczur SM, Afifi S, Zhao JC, Zeidan AM. Beyond ruxolitinib: fedratinib and other emergent treatment options for myelofibrosis. Cancer Manag Res. 2019;11:10777-10790.
26. Food and Drug Administration. VONJO™ (pacritinib) capsules, for oral use. US Prescribing Information. 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/20 8712s000lbl.pdf. Accessed March 2022.
27. Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643-651.
28. Pardanani A, Tefferi A, Masszi T, et al. Updated results of the placebo-controlled, phase III JAKARTA trial of fedratinib in patients with intermediate-2 or high-risk myelofibrosis. Br J Haematol. 2021;195(2):244-248.
29. Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebocontrolled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799-807.
30. Harrison CN, Schaap N, Vannucchi AM, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017;4(7):e317-e324.
31. Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4(5):652-659.
32. Palandri F, Breccia M, Bonifacio M, et al. Life after ruxolitinib: reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer. 2020;126(6):1243-1252.
33. Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):17011707.
34. Verstovsek S, Mesa RA, Gotlib J, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10(1):55.
35. Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787-798.
36. Vannucchi AM, Barbui T, Cervantes F, et al. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(Suppl 5):v85-99.
37. Pardanani A, Tefferi A. How I treat myelofibrosis after failure of JAK inhibitors. Blood. 2018;132(5):492-500.
38. Ozono Y, Shide K, Kameda T, et al. Neoplastic fibrocytes play an essential role in bone marrow fibrosis in Jak2V617F-induced primary myelofibrosis mice. Leukemia. 2021;35(2):454-467.
39. Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703-1708.
40. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405.
41. Kvasnicka HM, Beham-Schmid C, Bob R, et al. Problems and
pitfalls in grading of bone marrow fibrosis, collagen deposition and osteosclerosis - a consensus-based study. Histopathology. 2016;68(6):905-915.
42. Tefferi A, Cervantes F, Mesa R, et al. Revised response criteria for myelofibrosis: International Working GroupMyeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013;122(8):1395-1398.
43. Glasser SW, Hagood JS, Wong S, Taype CA, Madala SK, Hardie WD. Mechanisms of lung fibrosis resolution. Am J Pathol. 2016;186(5):1066-1077.
44. Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest. 2018;128(1):97-107.
45. Masarova L, Alhuraiji A, Bose P, et al. Significance of thrombocytopenia in patients with primary and postessential thrombocythemia/polycythemia vera myelofibrosis. Eur J Haematol. 2018;100(3):257-263.
46. Al-Ali HK, Vannucchi AM. Managing patients with myelofibrosis and low platelet counts. Ann Hematol. 2017;96(4):537-548.
47. Patnaik MM, Caramazza D, Gangat N, Hanson CA, Pardanani A, Tefferi A. Age and platelet count are IPSS-independent prognostic factors in young patients with primary myelofibrosis and complement IPSS in predicting very long or very short survival. Eur J Haematol. 2010;84(2):105-108.
48. Tefferi A, Lasho TL, Jimma T, et al. One thousand patients with primary myelofibrosis: the Mayo Clinic experience. Mayo Clin Proc. 2012;87(1):25-33.
49. Mascarenhas J, Mehra M, He J, Potluri R, Loefgren C. Patient characteristics and outcomes after ruxolitinib discontinuation in patients with myelofibrosis. J Med Econ. 2020;23(7):721-727.
50. van den Blink B, Dillingh MR, Ginns LC, et al. Recombinant human pentraxin-2 therapy in patients with idiopathic pulmonary fibrosis: safety, pharmacokinetics and exploratory efficacy. Eur Respir J. 2016;47(3):889-897.
51. Thiele J, Kvasnicka HM. Grade of bone marrow fibrosis is associated with relevant hematological findings-a clinicopathological study on 865 patients with chronic idiopathic myelofibrosis. Ann Hematol. 2006;85(4):226-232.
Chatphatai Moonla,1,2 Chantana Polprasert,1,2 Patcharee Komvilaisak,3 Thanawat Rattanathammethee,4 Sunisa Kongkiatkamon,1,2 Kitsada Wudhikarn,1,2 Sirorat Kobbuaklee,2 Pitchayut Boonyabaramee,1 Nuanrat Tangcheewinsirikul,1,2 Samart Pakakasama,5 Piya Rujkijyanont,6 Chane Choed-Amphai,7 Kamon Phuakpet,8 Saranya Pongudom,9 Udomsak Bunworasate,1,2 Narittee Sukswai,10 Darintr Sosothikul11 12 and Ponlapat Rojnuckarin1,2
1Department of Medicine, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Bangkok; 2Center of Excellence in Translational Hematology, Faculty of Medicine, Chulalongkorn University, Bangkok; 3Division of Hematology-Oncology, Department of Pediatrics, Faculty of Medicine, Khon Kaen University, Khon Kaen; 4Department of Medicine, Faculty of Medicine, Chiang Mai University, Chiang Mai; 5Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok; 6Department of Pediatrics, Phramongkutklao Hospital and Phramongkutklao College of Medicine, Bangkok; 7Department of Pediatrics, Faculty of Medicine, Chiang Mai University, Chiang Mai; 8Department of Pediatrics, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok; 9Department of Medicine, Udon Thani Medical Education Center, Udon Thani Hospital, Udon Thani; 10Department of Pathology, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Bangkok; 11Department of Pediatrics, Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Bangkok and 12Integrative and Innovative Hematology/Oncology Research Unit, Department of Pediatrics, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
Correspondence: C. Polprasert chantana.po@chula.ac.th jeedchantana@gmail.com
Received: November 14, 2022.
Accepted: April 5, 2023.
Early view: April 13, 2023.
htps://doi.org/10.3324/haematol.2022.282419
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Germline HAVCR2 mutations are frequently detected in subcutaneous panniculitis-like T-cell lymphoma (SPTCL) patients with/without hemophagocytic lymphohistiocytosis (HLH) but factors associated with variable manifestations remain undetermined. To evaluate clinical variations and associated factors in SPTCL and/or HLH with/without HAVCR2 mutations, we performed direct sequencing of HAVCR2 exon 2 using DNA from patients with SPTCL or idiopathic HLH/HLH-like systemic illnesses, defined by HLH alone without secondary causes. The systematic review and individual patient data (IPD) level meta-analysis which included the present and previously published studies reporting HAVCR2 mutations in SPTCL with/without HLH populations was subsequently conducted using random-effects meta-analysis and multivariate logistic regression. Among 34 patients enrolled, ten of 28 SPTCL patients developed HLH/HLH-like systemic illnesses. Six cases with HAVCR2Y82C mutation manifested with HLH without panniculitis. Male sex (P=0.03) and age <18 years (P=0.04) were associated with HLH, corresponding to the inverse correlation between age and HLH-2004 score (r=-0.40; P=0.02). Homozygous HAVCR2Y82C mutation was more common in the presence of HLH compared with the absence (75.0% vs 44.4%; P=0.02). Using IPD from the present and the other three eligible cohorts (N=127), male sex, heterozygous and homozygous/compound heterozygous HAVCR2 mutations were associated with HLH by the adjusted odds ratio of 2.93 (95% confidence interval [CI]: 1.22-7.06), 4.77 (95% CI: 1.05-21.63) and 8.48 (95% CI: 2.98-24.10), respectively. Patients with male sex and/or germline HAVCR2 mutations showed an increased risk of developing HLH. Younger patients tended to manifest with HLH, while older patients typically presented with SPTCL with less frequent HLH/HLH-like systemic illnesses.
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare subtype of T-cell non-Hodgkin lymphoma classically presenting with subcutaneous nodules. The pathological hallmark of SPTCL is an adipocyte rimming by atypical lymphoid cells expressing CD3, CD8, T-cell intracytoplasmic antigen 1 (TIA-1) and T-cell receptor β F1 (BF1). Recently, germline mutations of hepatitis A virus cellular receptor 2 (HAVCR2) gene have been discovered to be an important predisposing factor for primary SPTCL, while secondary SPTCL could be associated with several conditions, including viral infections and autoimmune diseases.1
Hemophagocytic lymphohistiocytosis (HLH) is a lifethreatening syndrome commonly observed in patients with SPTCL as well as germline HAVCR2 mutations.2,3 These mutations result in misfolding of T-cell immunoglobulin and mucin-domain containing-3 (TIM-3) protein, leading to persistent immune activation and cytokine release, which are responsible for the pathogenesis of HLH. In general, HLH is often secondary to several conditions, including autoimmune diseases, cancers, and infections. Nonetheless, HLH without identifiable primary cause or idiopathic HLH has been observed.4 Germline HAVCR2 variants in this entity have never been explored. For clinical studies, HLH is customarily diagnosed by fulfilling the HLH-2004 classification criteria,5 but in clinical practice, patients with high-grade fever, cytopenias and presence of hemophagocytic activity in bone marrow (BM) can be presumptively diagnosed as having hemophagocytic syndrome (HPS). These clinical phenotypes should be categorized as HLH-like systemic illnesses where treatments are urgently required.
The previously reported germline HAVCR2 mutations included p.Y82C, p.I97M and p.T101I substitutions.1-3 Although the pathogenic consequences of homozygous or compound heterozygous HAVCR2 mutations have been described, the effects of heterozygous HAVCR2 mutations on clinical manifestations of SPTCL and associated HLH remain undetermined. SPTCL shows a wide array of presentations and can be observed in both male and female sex, and in all age groups, ranging from children, young adults, to the elderly. Factors that influence the phenotypic variations are not yet well defined. Therefore, this multicenter study aims to explore the associations between clinical manifestations of SPTCL and/or HLH/HLHlike systemic illnesses and several factors, including HAVCR2 mutational statuses, sex and age groups. In order to enhance better understanding of the clinical spectrum of the rare disorder, a systematic review and meta-analysis which incorporates data from previously published studies reporting HAVCR2 mutations in SPTCL/HLH populations is also conducted.
Patients with SPTCL with/without HLH or patients with idiopathic HLH were enrolled from nine hematology centers in Thailand from January 2009 to June 2022 (Online Supplementary Appendix; Online Supplementary Figure S1). Idiopathic HLH included patients manifested with HLH alone without any conditions contributing to HLH, i.e., infections, autoimmune disorders, and malignancies. Pathological tissues designating SPTCL/HLH were revalidated by the hematopathologists. SPTCL clonality was identified by immunohistochemistry staining and T-cell receptor gene rearrangement analyses, as previously described.1 HLH was defined by the HLH-2004 criteria.5 Those incompletely fulfilling the criteria but being clinically consistent with HLH (i.e., fever, cytopenias, serum ferritin ≥500 μg/L, and the presence of hemophagocytosis in BM) were accounted for ‘HLH-like systemic illnesses’.3 The study protocol was ethically approved by the Institutional Review Boards of the respective institutions.
Sanger sequencing to detect germline HAVCR2 exon 2 mutations was performed in all subjects using DNA extracted from bone marrow (BM) or peripheral blood. The designed primer could cover pathogenic HAVCR2 variants of p.Y82C, p.I97M and p.T101I. Whole exome sequencing (WES) following the standard pipeline (Novogene Co. Ltd., Beijing, China) was explored in the selected cases (NovaSeq 6000 platform, Illumina Inc., San Diego, CA). The methodology on HAVCR2 sequencing and WES is fully outlined in the Online Supplementary Appendix and the Online Supplementary Figures S2, S3.
Due to the disease rarity, a systematic review and metaanalysis of previously published series of SPTCL with/without HLH/HLH-like systemic illnesses, including one from our group,1 combined with our present cohort, was performed. The prespecified individual patient data (IPD) level meta-analysis would be analyzed if the included studies provided sufficient outcomes per each individual. The primary outcome was the risk factors associated with HLH/HLH-like systemic illnesses, including HAVCR2 mutations, sex and age at diagnosis. The prevalence of HAVCR2 mutations was the secondary outcome. This systematic review and meta-analysis were proceeded following the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) guidelines and the International Prospective Register of Systematic Reviews (PROSPERO) registration (CRD42022341310).6 Study search and selection, data extraction, and quality assessment of the included studies are fully described in the Online Supplementary Appendix
For the present cohort, STATA version 15.1 (StataCorp, Col-
lege Station, TX) and descriptive statistics were employed, considering P<0.05 for statistical significance. For the conventional meta-analysis, Comprehensive Meta-Analysis version 3.0 (Biostat Inc., Englewood, NJ) was applied. The pooled odds ratios (pOR) or pooled differences in means (pMD) with 95% confidence intervals (CI) for risk factors associated with HLH/HLH-like systemic illnesses were estimated by the DerSimonian and Laird random-effects meta-analysis. The weighted mean prevalence of HAVCR2 mutations and HLH/HLH-like systemic illnesses was also reported. The influence of factors on the synthesized estimates were determined by meta-regression analysis, if appropriate.7 The publication bias and inter-study heterogeneity were assessed systematically (Online Supplementary Appendix).
For the IPD level meta-analysis, the risk factors associated with HLH/HLH-like systemic illnesses were determined by logistic regression. Multivariate models were developed by adjusting for covariates with P<0.1 in univariate models and stepwise backward logistic regression to select the final models. The adjusted odds ratios (aOR) were estimated alongside 95% CI.
Among the 34 cases enrolled, 28 cases presented with SPTCL with/without HLH/HLH-like systemic illnesses, while six cases presented with idiopathic HLH/HLH-like systemic illnesses. The median age of the patients at the time of diagnosis was 20.5 years (interquartile range [IQR], 12-32), with female sex predominance (64.7%). Homozygous HAVCR2Y82C mutation was detected in 20 cases (58.8%), heterozygous HAVCR2Y82C mutation in seven cases (20.6%), while no HAVCR2Y82C mutation was detected in 7 cases (20.6%; Table 1; Online Supplementary Table S1). No HAVCR2 p.I97M or p.T101I mutations were detected in our cohort. Patients younger than 18 years showed more HLH/HLHlike systemic illnesses than those 18 years or older. There was a higher proportion of male patients in patients who had HLH/HLH-like symptoms compared to those who did not (56.3% vs. 16.7%; P=0.03). In patients with HLH/HLHlike events, the frequency of homozygous HAVCR2Y82C mutation was significantly higher than those without HLH/HLH-like systemic illnesses (75.0% vs. 44.4%; P=0.02; Table 2).
When analyzing HLH-2004 score according to the HLH2004 criteria, there was an inverse correlation between patient age and HLH-2004 score (r=-0.40; P=0.02; Figure 1A). Patients harboring HAVCR2Y82C mutations were younger than those with no HAVCR2Y82C mutation (median age of 15 vs. 19 vs. 36 years for homozygous mutation, heterozygous mutation, and no HAVCR2Y82C mutation, respectively;
HLH:
P=0.02). Patients with HAVCR2Y82C mutations had higher median HLH-2004 scores than those with no HAVCR2Y82C mutation (median HLH-2004 score of 4 for homozygous mutation, 5 for heterozygous mutation, and 0 for no HAVCR2Y82C mutation; P=0.003; Online Supplementary Table S2). There were no clinical factors associated with disease relapse and mortality rates.
WES was done in one heterozygous HAVCR2Y82C mutation case (case no. 23) and in five homozygous HAVCR2Y82C mutation cases with HLH/HLH-like systemic illnesses (case no. 2, 3, 12, 18, and 20). There were no other pathogenic HAVCR2 mutations in a case with heterozygous HAVCR2Y82C mutation. In the five cases with a homozygous HAVCR2Y82C mutation, although no mutations in other familial HLH genes were identified, mutated genes associated with primary immune deficiency disease (PIDD) and dysregulated immune activation or proliferation (DIAP)8 were observed in all cases (Online Supplementary Table S3; Online Supplementary Figure S4).
Online databases including MEDLINE, Embase and Cochrane Library were systematically searched between the inception date of each database and July 15, 2022. Our present cohort was integrated into 312 results from the initial search. After removing 60 duplicates, 237 studies were excluded by screening through the titles and abstracts. Three of 16 studies were excluded due to studying in the same populations with other more mature studies. Thirteen studies (6 cohorts and 7 case reports) were eli-
gible for the systematic review (Table 3).1-3,9-17 The PRISMA flow diagram for study screening and the selection process is illustrated in the Online Supplementary Figure S5. The risks of bias for cohort studies were evaluated by the Newcastle-Ottawa scale as outlined in the Online Supplementary Table S4. 18
Of 13 studies, six cohorts with at least ten participants were finally included in the meta-analysis, representing 224 pa-
tients with SPTCL with/without HLH/HLH-like systemic illnesses and six idiopathic HLH/HLH-like systemic illnesses.13,9-10 From which, 207 cases had been documented for both statuses in HLH/HLH-like systemic illnesses and HAVCR2 mutation. Overall, the pooled prevalence of HLH/HLH-like systemic illnesses was 31.9% (95% CI: 18.2-49.7; I2=81%), while the pooled prevalence of HAVCR2 mutation was 51.6% (95% CI: 30.3-72.4; I2=87%). HAVCR2Y82C variant was the most
HLH: hemophagocytic lymphohistiocytosis; IQR: interquartile range. *There were 4 cases with unknown HLH status and 4 cases with unknown mutational status. In which, there were 2 cases with unknown both HLH and mutational statuses.

commonly detected (54.5%; 95% CI: 29.1-77.8; I2=89%). The weighted mean age at diagnosis was 30.4 years (95% CI: 25.3-35.5; I2=8%) and approximately one third of the patients were men (34.8%; 95% CI: 26.8-43.7; I2=0%). Regarding histopathology, necrosis was a typical feature found in BM among the overall population (44.0%; 95% CI: 32.5-56.1; I2=0%). However, granulomatous inflammation was less frequently observed in patients with HAVCR2 mutation, compared with those in the HAVCR2 wild-type group (9.6% vs 49.0%; pOR=0.06; 95% CI: 0.01-0.53; I2=0%; P=0.01). The pooled data on participant characteristics are displayed in the Online Supplementary Tables S5, S6. Notably, patients with HLH/HLH-like systemic illnesses harbored HAVCR2 mutations substantially higher than those without HLH/HLH-like systemic illnesses (76.6% vs 33.5%; pOR=6.75; 95% CI: 1.65-27.64; I2=56%; P=0.008), particularly homozygous/compound heterozygous mutation (71.2% vs. 31.3%; pOR=4.67; 95% CI: 1.07-20.35; I2=65%; P=0.04; Figure 2A, B; Online Supplementary Table S7) and HAVCR2Y82C variant (75.5% vs. 35.3%; pOR=7.06; 95% CI: 1.05-47.51; I 2=65%; P=0.04). Compared with no HAVCR2 mutation, heterozygous HAVCR2 mutation showed a ten-
dency to be associated with HLH/HLH-like systemic illnesses (pOR=6.41; 95% CI: 0.94-43.58; I2=0%; P=0.06; Figure 2C; Online Supplementary Tables S7, S8), despite its low prevalence (7.5%; 95% CI: 2.8-18.6; I2=54%) among studies. Furthermore, the meta-regression confirmed a positive correlation between allelic numbers of HAVCR2 mutation and occurrence of HLH/HLH-like systemic illnesses (P=0.01; Figure 3). Male sex was also correlated with HLH/HLH-like systemic illnesses (pOR=2.97; 95% CI: 1.28-6.85; I2=3%; P=0.01; Figure 2D). The pOR of HAVCR2 mutational statuses and male sex as risk factors for the presence of HLH/HLHlike systemic illnesses are illustrated as the forest plots in Figure 2.
Patients with HLH/HLH-like systemic illnesses tended to develop the disease at a younger age, compared with those without HLH/HLH-like systemic illnesses (26.5 vs 32.7 years), but did not reach statistical signi ficance (pMD=-6.02 years; 95% CI: -15.40 to 3.35; I2=54%; P=0.21; Online Supplementary Table S7). Nonetheless, by excluding Polprasert et al. 1 which comprised only one patient younger than 18 years old as a sensitivity analysis, patients with HLH/HLH-like systemic illnesses manifested their
CR: case report; HLH: hemophagocytic lymphohistiocytosis; IQR: interquartile range; NA: not available; RCS: retrospective cohort study; SPTCL, subcutaneous panniculitis-like T-cell lymphoma. *Only 53 cases had available data on HAVCR2 mutational status. **There were 4 cases with unknown HLH status and 4 cases with unknown mutational status. In which, there were 2 cases with unknown both HLH and mutational statuses. †Omental panniculitis was found in the reported case without detectable monoclonal T-cell receptor gene rearrangement.

Figure 2. Association of HAVCR2 mutational statuses, male sex and hemophagocytic lymphohistiocytosis (HLH)/HLH-like systemic illnesses. The forest plots demonstrate the pooled odds ratios with 95% confidence intervals of any HAVCR2 mutations (A), homozygous/compound heterozygous HAVCR2 mutation (B), heterozygous HAVCR2 mutation in comparison with HAVCR2 wild-type (C), and male sex (D) in significant associations with presence of HLH/HLH-like systemic illnesses among patients with subcutaneous panniculitis-like T-cell lymphoma or idiopathic HLH.

disease phenotypes significantly earlier than those without HLH/HLH-like systemic illnesses (pMD=-10.31 years; 95% CI: -17.04 to -3.57; I2=0; P=0.003). Since fewer than ten studies were aggregated in the meta-analysis, evaluation of the publication bias was not indicated.
Three previously published cohorts provided individual data of their participants.1-3 We therefore conducted the IPD level meta-analysis using data from those three studies and our present cohort, resulting in a total of 127 cases. Based on 123 cases with HAVCR2 mutational statuses available, it was found that 65 cases harbored homozygous HAVCR2 mutations (63 cases with p.Y82C and 2 cases with p.I97M), two cases harbored compound heterozygous HAVCR2 mutations (p.Y82C with p.T101I and p.Y82C with p.I97M), 12 cases harbored heterozygous HAVCR2 mutations (11 cases with p.Y82C and 1 case with p.I97M), while 44 cases showed no HAVCR2Y82C mutation. Diagram of case distribution based on SPTCL/HLH phenotypes and mutational statuses is shown in the Online Supplementary Figure S6
Patients who manifested HLH/HLH-like systemic illnesses were younger than patients without HLH/HLH-like systemic
illnesses (median age of 23 vs. 32 years; P=0.005). There were more men in cases with HLH/HLH-like systemic illnesses than without HLH/HLH-like systemic illnesses (50.0% vs. 25.4%; P=0.005). The homozygous/compound heterozygous HAVCR2 mutation group showed more HLH/HLH-like systemic illnesses than the others (78.9% vs. 9.6% [heterozygous] vs. 11.5% [no mutation]; P<0.001; Table 2).
Since the correlation between age at diagnosis and HLH2004 score could not be estimated by the conventional meta-analysis due to lack of reported data based on age groups, we re-evaluated this outcome in the IPD level meta-analysis. Remarkably, an inverse correlation between age at diagnosis and HLH-2004 score was demonstrated (r=-0.29; P=0.02; Figure 1B). Patients with no HAVCR2Y82C mutation were significantly older (P<0.001) and associated with lower HLH-2004 scores (P=0.002; Online Supplementary Table S2). By the multivariate analysis, male sex (aOR=2.93; 95% CI: 1.22-7.06; P=0.02), heterozygous HAVCR2 mutation (aOR=4.77; 95% CI: 1.05-21.63; P=0.04) and homozygous/compound heterozygous HAVCR2 mutation (aOR=8.48; 95% CI: 2.98-24.10; P<0.001) were significantly associated with HLH/HLH-like systemic illnesses (Table 4).
Table
Risk factors associated with hemophagocytic lymphohistiocytosis (HLH)/HLH-like systemic illnesses by univariate and multivariate analyses using the individual patient data from 4 cohorts (N=127)
aOR: adjusted odds ratio; CI: confidence interval; HLH: hemophagocytic lymphohistiocytosis; OR: odds ratio; Ref: reference.
SPTCL is a unique hematologic malignancy that is associated with germline HAVCR2 mutations (25-60%) 2,9 and concomitant systemic inflammation which may be as severe as HLH. In this multicenter study, we demonstrated that patients with SPTCL-associated and idiopathic HLH/HLH-like systemic illnesses were associated with a younger age group, male sex, and the presence of germline HAVCR2 mutation. The results were substantiated by the systematic review and IPD level metaanalysis which included all existing cohorts with available data.
Although HAVCR2 mutation is a congenital mutation, SPTCL with or without HLH can occur later in life. In addition, a positive family history in SPTCL and idiopathic HLH is unusual. Therefore, additional factors, such as infections, additional somatic mutations, or vaccines, are generally required to trigger disease manifestations.1 Furthermore, our preliminary WES data suggest possible roles of other concomitant immunoregulatory gene mutations in the development of HLH/HLH-like systemic illnesses, but the known variants in the genes causing familial HLH (i.e., perforin 1 [PRF1], UNC-13 homolog D [UNC13D], RAS-related protein Rab-27A [RAB27A], syntaxin 11 [STX11] and syntaxin binding protein 2 [STXBP2] genes) were not observed. In this study, older patients were more likely to present with SPTCL alone, while younger patients usually had SPTCL with concurrent HLH/HLH-like systemic illnesses or even HLH/HLH-like systemic illnesses alone. The earlier onset of illnesses was associated with homozygous or compound heterozygous HAVCR2 mutations. This strong genetic predisposition may make them more susceptible to developing HLH.
The germline HAVCR2 mutation in patients with HLH/HLH-
like systemic illnesses without clinical panniculitis has never been reported. In our cohort, there were six patients who manifested with idiopathic HLH/HLH-like systemic illnesses without SPTCL and no evidence of infections or other malignancies. Five of the six cases harbored a homozygous HAVCR2Y82C mutation, while one case showed a heterozygous HAVCR2Y82C mutation. Patients with a HAVCR2 mutation may have clinical manifestations similar to other familial HLH. Most familial HLH occurs at a very young age of approximately 1 year, and germline mutations in PRF1, UNC13D, RAB27A, STX11 and STXBP2 genes were responsible for this entity.4,8 The germline HAVCR2 mutation is probably the first genetic factor that predisposes the patients to develop late childhood- and young adult-onset familial HLH. In our series, two fatal cases with HLH were treated with chemotherapy, while five cases with HLH who received only immunosuppression survived (Online Supplementary Table S1). Hence, the detection of HAVCR2 mutation in the patients with unknown causes of HLH/HLH-like systemic illnesses is crucial since chemotherapy might be avoided in this setting to prevent serious complications of cytotoxic agents, particularly BM suppression. Due to the fact that germline HAVCR2 mutations led to persistent immune activation,2 immunosuppressive agents should provide potential therapeutic benefits for these patients. As supported by our cohort, most patients (58.8%), with or without HLH, responded well to corticosteroids and/or cyclosporine.
Based on this multicenter study and IPD level meta-analysis, we proposed the age-dependent clinical spectrum from HLH/HLH-like systemic illnesses with or without SPTCL in children and young adults to SPTCL alone in elderly patients. Of note, the age-dependent effect on the phenotype of HLH/HLH-like systemic illnesses was masked by a study from Polprasert et al. 1 which comprised mainly adult pa-
tients in the conventional meta-analysis but could demonstrate its significance when analyzed at the IPD level. The presence of HLH is also more prevalent in male patients and patients with a homozygous HAVCR2 mutation (Figure 4). Although a heterozygous HAVCR2 mutation was significantly associated with an increased risk of HLH at the IPD level (Table 4), supported by the meta-regression in which the occurrence of HLH increased when the allelic number of HAVCR2 mutation increased (Figure 3), the small number of cases per study limited its interpretation in the conventional meta-analysis.
As a limitation of the retrospective study, we could not exclude the presence of internal organ panniculitis in our six patients with idiopathic HLH/HLH-like systemic illnesses since the computed tomography scan of the whole abdomen was not performed among those without abdominal symptoms, although omental panniculitis has been reported in a patient with a germline HAVCR2 mutation presenting with HLH without skin lesion.17 Additionally, we could not demonstrate any factors associated with disease relapse or mortality, possibly due to the heterogeneity of the treatments among cohorts. WES was not performed in the patients with no HAVCR2 mutation to identify other potential genetic alterations contributing to HLH/HLH-like systemic illnesses. Nevertheless, data on other additional immune regulatory diversities from our study were small and therefore deserved further exploration in a larger study.
In conclusion, the clinical manifestations of patients with SPTCL and associated HLH/HLH-like systemic illnesses are sex- and HAVCR2 mutation-dependent. The presence

of HLH/HLH-like systemic illnesses is more common in younger patients, while SPTCL alone is more prevalent in the older age group.
Disclosure
No conflicts of interest to dislcose.
Contributions
CM designed and performed the systematic review and meta-analysis, analyzed data and wrote the manuscript. CP collected samples and clinical data, designed and performed the systematic review and meta-analysis, analyzed data, wrote the manuscript, and conceptualized the overall research. PK, TR, PB, PR, KP, SarP, UB, DS provided samples and clinical data. SuK and KW wrote the manuscript. SiK extracted DNA and performed PCR. NT performed the meta-analysis and wrote the manuscript. SamP and CC provided samples, clinical data and wrote the manuscript. NS conducted histopathological studies . PR supervised, conceptualized the research, and wrote the manuscript. All authors read and approved the final manuscript.
The authors thank the research team of the Department of Medicine, Faculty of Medicine, Chulalongkorn University for editing the final manuscript. We extend our gratitude to Jiratchaya Sophonphan, M.Sc., of HIV-NAT, the Thai Red Cross AIDS Research Center, Bangkok, for statistical consultation; and Asst. Prof. Teerada Daroontum, M.D., of the Department of Pathology, Faculty of Medicine, Chiang Mai University for pathological consultation.
Funding
This research was supported by grants from the Anandamahidol Foundation (to CP), the Center of Excellence in Translational Hematology, Faculty of Medicine, Chulalongkorn University (to CP), the Ratchadapiseksompotch Fund, Faculty of Medicine, Chulalongkorn University (RA64/004; to CP), and
1. Polprasert C, Takeuchi Y, Kakiuchi N, et al. Frequent germline mutations of HAVCR2 in sporadic subcutaneous panniculitislike T-cell lymphoma. Blood Adv. 2019;3(4):588-595.
2. Gayden T, Sepulveda FE, Khuong-Quang DA, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet. 2018;50(12):1650-1657.
3. Koh J, Jang I, Mun S, et al. Genetic profiles of subcutaneous panniculitis-like T-cell lymphoma and clinicopathological impact of HAVCR2 mutations. Blood Adv. 2021;5(20):3919-3930.
4. Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: An update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34(4):101515.
5. Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131.
6. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
7. Thompson SG, Higgins JP. How should meta-regression analyses be undertaken and interpreted? Stat Med. 2002;21(11):1559-1573.
8. Chinn IK, Eckstein OS, Peckham-Gregory EC, et al. Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis. Blood. 2018;132(1):89-100.
9. Sonigo G, Battistella M, Beylot-Barry M, et al. HAVCR2 mutations are associated with severe hemophagocytic syndrome in subcutaneous panniculitis-like T-cell lymphoma. Blood. 2020;135(13):1058-1061.
10. Cheng J, Xi L, Jang Y, et al. Germline variants of HAVCR2 in a North American Consult Practice Cohort of subcutaneous panniculitis-like T-cell lymphoma [abstract]. In: Abstracts from USCAP 2020: Hematopathology (1316-1502). Mod Pathol. 2020;33(Suppl 2):S1266-S1267.
11. Wegehaupt O, Gross M, Wehr C, et al. TIM-3 deficiency presenting with two clonally unrelated episodes of mesenteric and subcutaneous panniculitis-like T-cell lymphoma and
the Thailand Science Research and Innovation Fund, Chulalongkorn University (CU_FRB65_hea(24)_031_30_12; to CP).
Data-sharing statement
WES results have been deposited in the European Genomephenome Archive under accession number: EGAS00001006740.
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2020;67(6):e28302.
12. Bauman B, Pittaluga S, Zhang Y, et al. Unusual presentation of subcutaneous panniculitis-like T cell lymphoma in patients with BENTA disease. In: Selected Abstracts from the 12th Annual Meeting of the Clinical Immunology Society: 2021 Virtual Annual Meeting: Immune Deficiency and Dysregulation North American Conference. J Clin Immunol. 2021;41(Suppl 1):S12-S13.
13. Chaweephisal P, Sosothikul D, Polprasert C, et al. Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) with hemophagocytic lymphohistiocytosis (HLH) syndrome in children and its essential role of HAVCR2 gene mutation analysis. J Pediatr Hematol Oncol. 2021;43(1):e80-e84.
14. Frederiks AJ, Spagnolo DV, Ramachandran S, et al. Subcutaneous panniculitis-like T-cell lymphoma in a 14-yearold female homozygous for HAVCR2 mutation. Australas J Dermatol. 2021;62(4):e576-e579.
15. LeBlanc RE, Lansigan F. Unraveling subcutaneous panniculitislike T-cell lymphoma: An association between subcutaneous panniculitis-like T-cell lymphoma, autoimmune lymphoproliferative syndrome, and familial hemophagocytic lymphohistiocytosis. J Cutan Pathol. 2021;48(4):572-577.
16. Sheng G, Garcia-Lloret M, Butte M. A case of subcutaneous panniculitis-like T-cell lymphoma with hemophagocytic lymphohistiocytosis independent of germline variants in HAVCR2. In: Selected Abstracts from the 12th Annual Meeting of the Clinical Immunology Society: 2021 Virtual Annual Meeting: Immune Deficiency and Dysregulation North American Conference. J Clin Immunol. 2021;41(Suppl 1):S121.
17. Tromp SAM, Gillissen MA, Bernelot Moens SJ, et al. Treatment of an HLH-mimic disease based on HAVCR2 variants with absent TIM-3 expression. Blood Adv. 2022;6(15):4501-4505.
18. Wells G, Shea B, O’Connell D, et al. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. The Ottawa Hospital Research Institute. http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp Accessed 18 July 2022.
Borja Puertas,1* Verónica González-Calle,1* Anna Sureda,2 María José Moreno,3 Albert Oriol,4 Esther González,5 Laura Rosiñol,6 Jordi López,7 Fernando Escalante,8 Joaquín Martínez-Lopez,9 Estrella Carrillo,10 Esther Clavero,11 Rafael Ríos-Tamayo,12 Beatriz Rey-Bua,1 Ana Pilar González-Rodríguez,13 Victoria Dourdil,14 Felipe de Arriba,15 Sonia González,16 Jaime Pérez-de-Oteyza,17 Miguel T. Hernández,18 Aránzazu García-Mateo,19 Joan Bargay,20 Joan Bladé,6 Juan José Lahuerta,9 Jesús F. San Miguel,21 Enrique M. Ocio22 and María-Victoria Mateos1
1Hematology Department, University Hospital of Salamanca/IBSAL/Cancer Research CenterIBMCC (USAL-CSIC), CIBERONC, Salamanca; 2Hematology Department, Institut Català D'Oncologia L'Hospitalet, Barcelona; 3Hematology Department, Hospital Clínico Universitario Virgen De La Arrixaca, Murcia; 4Hematology Department, Institut Josep Carreras and Institut Catala d'Oncologia, Hospital Germans Trias i Pujol, Badalona; 5Hematology Department, Hospital De Cabueñes, Gijón; 6Department of Hematology, IDIBAPS, Hospital Clinic, Barcelona; 7Hematology Department, Hospital De La Santa Creu i Sant Pau, Barcelona; 8Department of Hematology, University Hospital of Leon, Leon; 9Hematology Department, Hospital Universitario
12 de Octubre, Universidad Complutense, CNIO, Madrid; 10Department of Hematology, University Hospital Virgen del Rocio, Instituto de Biomedicina de Sevilla (IBIS/CISC), Sevilla; 11Hematology
Department, Hospital Universitario Virgen De Las Nieves, Granada; 12Hospital Universitario Puerta de Hierro Majadahonda, Madrid; 13Hematology Department, Hospital Universitario Central De Asturias, Oviedo; 14Hematology Department, Hospital Clínico Universitario Lozano Blesa, IIS Aragón, Zaragoza; 15Servicio de Hematología y Oncología Médica, Hospital Universitario Morales Meseguer, IMIB-Pascual Parrilla, Universidad de Murcia, Murcia; 16Hematology Department, Complexo Hospitalario Universitario De Santiago, Santiago de Compostela; 17Hematology Department, Hospital Universitario Madrid Sanchinarro, Madrid; 18Hematology Department, Hospital Universitario De Canarias, San Cristóbal de La Laguna, Tenerife; 19Hematology Department, Hospital General De Segovia, Segovia; 20Hematology Department, H. Universitario Son Llàtzer, IdIsBa, Mallorca; 21Hematology Department, Clínica Universidad de Navarra, CIMA, IDISNA, CIBERONC, Pamplona; 22Hematology Department, Hospital Universitario Marqués De Valdecilla (IDIVAL), Universidad De Cantabria, Santander, Spain
*BP and VGC contributed equally as first authors.
Correspondence: M.V. Mateos mvmateos@usal.es
Received: November 29, 2022.
Accepted: April 17, 2023.
Early view: April 27, 2023.
htps://doi.org/10.3324/haematol.2022.282490
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
In this randomized phase II study (GEM-KyCyDex, clinicaltrials gov. Identifier: NCT03336073), the combination of weekly carfilzomib 70 mg/m2, cyclophosphamide and dexamethasone (KCd) was compared to carfilzomib and dexamethasone (Kd) in relapsed/refractory multiple myeloma (RRMM) after 1-3 prior lines (PL). One hundred and ninety-seven patients were included and randomized 1:1 to receive KCd (97 patients) or Kd (100 patients) in 28-day cycles until progressive disease or unacceptable toxicity occurred. Patient median age was 70 years, and the median number of PL was one (range, 1-3). More than 90% of patients had previously been exposed to proteasome inhibitors, approximetely 70% to immunomodulators, and approximetely 50% were refractory to their last line (mainly lenalidomide) in both groups. After a median follow-up of 37 months, median progression-free survival (PFS) was 19.1 and 16.6 months in KCd and Kd, respectively (P=0.577). Of note, in the post hoc analysis of the lenalidomide-refractory population, the addition of cyclophosphamide to Kd resulted in a significant benefit in terms of PFS: 18.4 versus 11.3 months (hazard ratio =1.7, 95% confidence interval: 1.1-2.7; P=0.043). The overall response rate and the percentage of patients who achieved complete response was around 70% and 20% in both groups. The addition of cyclophosphamide to Kd did not result in any safety signal, except for severe infections (7% vs. 2%). In conclusion, the combination of cyclophosphamide with Kd 70 mg/m2 weekly does not improve outcomes as compared with Kd alone in RRMM after 1-3 PL, but a significant benefit in PFS was observed with the triplet combination in the lenalidomide-refractory population. The administration of weekly carfilzomib 70 mg/m2 was safe and convenient, and, overall, the toxicity was manageable in both arms.
The introduction of agents such as bortezomib and lenalidomide in the upfront treatment of multiple myeloma (MM) drastically improved the survival of MM patients.1 However, most patients end up relapsing. Novel rescue therapies are emerging for the treatment at the moment of the relapse, some of which have a different mechanism of action from bortezomib or lenalidomide, while others belong to the same class.
Carfilzomib is a tetrapeptide epoxyketone second-generation proteasome inhibitor that irreversibly inhibits the chymotrypsin-like (CT-L) activities of the constitutive proteasome and immunoproteaseome.2 Several studies and clinical trials have been designed to improve the efficacy of carfilzomib by increasing its dose,3 combining it with another agent,4,5 reducing infusion-related events and toxicity, as well as developing a most convenient schedule.6,7
The results of the randomized, open-label, multicenter phase III study (ENDEAVOR trial)5 have led to the combination of carfilzomib plus dexamethasone becoming one of the backbones of relapsed/refractory MM (RRMM) treatment. In this trial, patients with RRMM after 1-3 prior lines (PL) of therapy were randomized to receive carfilzomib at a dose of 56 mg/m2 twice-weekly in a 30-minute infusion or bortezomib at a dose of 1.3 mg/m2 intravenously or subcutaneously, plus low-dose dexamethasone in both arms.
The carfilzomib group showed an improved overall response rate (ORR) compared to the bortezomib group (77% vs. 63%). Substantial numbers achieved at least very good partial response (VGPR) (54% vs. 29%) and even complete response (CR) (13% vs. 6%), with a good safety profile. Overall, these responses in the carfilzomib group led to significantly better progression-free survival (PFS) (18.7 vs 9.4 months) and overall survival (OS) (47.6 vs. 40.0 months).8 However, the twice-weekly schedule of carfilzomib treatment is likely to be burdensome. For this reason, phase I/II and phase III studies, the CHAMPION-1 and A.R.R.O.W. trials, respectively, were developed to adapt the treatment to allow once-weekly administration.6,7 The A.R.R.O.W. trial, which compared car filzomib 27 mg/m2 given twice weekly with carfi lzomib 70 mg/m2 once weekly in RRMM after 2-3 PL, showed significant statistically differences in ORR (62.9% vs . 40.8%) and in PFS (11.2 vs. 7.6 months) that favored the once-weekly arm.
Cyclophosphamide (Cy) is a convenient alkylating agent with a well-known anti-tumoral activity which has been widely used in combination with bortezomib9–12 in MM patients, improving the efficacy with a good safety profile. Based on this background, the Spanish Myeloma Group (GEM/PETHEMA) designed a randomized, open-label, multicenter phase II study (GEM-KyCyDex) to evaluate the safety and efficacy of the combination of once-weekly
carfilzomib 70 mg/m2 and dexamethasone plus/minus cyclophosphamide in RRMM patients. Here we report, the results of this trial.
Patients were recruited from 24 Spanish hospitals, and had to meet the following inclusion criteria: i) age ≥18 years; ii) Eastern Cooperative Oncology Group (ECOG) performance status ≤2; iii) RRMM after 1-3 PL of therapy; and iv) measurable disease according to the International Myeloma Working Group (IMWG) criteria.13 Refractoriness to proteosome inhibitors (PI) was an exclusion criterion. All participants provided written informed consent and the study protocol was approved by the Institutional Review Boards or Ethics Committees of all participating institutions. The trial is registered at clinicaltrials gov. Identifier: NCT03336073.
Patients were randomized (1:1) to receive either carfilzomib, Cy and dexamethasone (KCd group) or carfilzomib and dexamethasone (Kd group).
Patients received a 30-minute infusion carfilzomib at a dose of 20 mg/m2 on day 1 of cycle 1, and of 70 mg/m2 given thereafter on days 1, 8, and 15. Patients older than 75 years received 56 mg/m2 during cycles 1 and 2, and 70 mg/m2 thereafter if it was well-tolerated. Patients also received dexamethasone (20 mg orally or intravenously; 10 mg for those >75 years old) on the day carfilzomib was administered and the day after, plus/minus Cy at a dose of 300 mg/m2 intravenously on days 1, 8 and 15 during the first 12 cycles. After the 12th cycle, treatment was administered every other week. The treatment was administered in 28-day cycles until disease progression, the occurrence of unacceptable toxicity, a physician’s decision, or the revocation of a participant’s informed consent. Dose reductions were allowed to manage the toxicity.
Adverse events (AE) were reported until 30 days after the final dose of the study treatment and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events 4.0.
Cytogenetic risk status was assessed using fluorescence in situ hybridization in a central laboratory.14 Cytogenetic high risk was defined based on International Myeloma Working Group (IMWG) criteria.15
Responses were assessed monthly according to 2014 IMWG criteria.16 A bone marrow aspiration was performed in central laboratories to confirm suspected CR and to measure the minimal residual disease (MRD).17
The primary endpoint was PFS, defined as the time from randomization to disease progression or death from any cause. Secondary endpoints were ORR, defined as the attainment of a partial response (PR) or better; percentage of immunophenotypic CR; time to progression (TTP), defined as the time from randomization to disease progression; OS, defined as the time from randomization until death from any cause; incidence and grade of AE; and discontinuation of treatment in both groups.
The necessary sample size was estimated using the onesample survival method, based on the primary endpoint of the study. A median PFS of 23 months in the KCd arm would be considered effective based on the 18-month median PFS of Kd previously reported.8 Two hundred and fifty-six patients were required to provide a power of 80% to demonstrate the efficacy of the experimental arm at a significance level (α) of 0.05.
The distributions of PFS, TTP and OS were estimated using the Kaplan–Meier method. The differences between survival in the two arms were defined using the log-rank test, and the corresponding hazard ratio (HR) and 95% confidence interval (CI) were estimated using by Cox regression. χ2 tests identified statistically significant differences between qualitative variables and the associated odds ratio (OR) and 95% CI were estimated by logistic regression.
The analysis included all randomized patients who had received at least one dose of a study treatment.
Values of P<0.05 were considered significant. Statistical analysis was performed with IBM SPSS Statistics, version 26.
Between February 2018 and April 2020, 229 patients were considered for inclusion in the trial. The 197 of them who met the inclusion criteria were randomly allocated to the Kcd (N=97) or the Kd (N=100) groups (Figure 1). There were no significant statistically differences between the characteristics of the two groups (Table 1). The median number of PL of therapy was 1 (range, 1-3), 90% and 12% of patients had previously been exposed to PI and to Cy, respectively, 60% had received immune-modulatory drugs (IMID), and approximately 50% were refractory to lenalidomide (43 [44.3%] and 46 [46.0%] patients in the KCd and Kd groups, respectively).
At the cut-off date (April 2022), the median follow-up was 37 months (range, 4.7-50.2 months), by which time 173 events had been reported, 72 (74.2%) in the KCd arm and 78 (78.0%) in the Kd arm. Twenty-four patients were still under treatment. At least 12 cycles were administered in 51% of patients of the KCd group, and in 49.5% of the Kd
group (P=0.821), with a median of 12 (range, 1-45) and 11 (range, 1-40) cycles (P=0.833), respectively. Ef
Median PFS was 19.1 months in the KCd group, and 16.6
MM: multiple myeloma; Ig: immunoglobulin; ISS: International Staging System; LDH: lactate dehydrogenase; CTG: cytogenetic; SR: standardrisk; HRCA; high-risk cytogenetic abnormalities; ECOG: Eastern Cooperative Oncology Group; LVEF: left ventricular ejection fraction; PL: prior line; PI: proteasome inhibitor; V: Bortezomib; Ixa: ixazomib; Tal: thalidomide; Len: lenalidomide; Pom: pomalidomide; IMID: immunomodulator; mAb: monoclonal antibody; KCd: carfilzomib, cyclophosphamide and dexamethasone; Kd: carfilzomib and dexamethasone; ASCT: autologous stem cell transplantation.
months in the Kd group (HR=1.1; 95% CI: 0.8-1.5; P=0.577) (Figure 2A). The entire post hoc subgroup analysis is shown in Table 2. It is worth mentioning that patients who received KCd and who were refractory to the last line of therapy (regardless of the treatment received) achieved better PFS than patients treated with Kd (19.1 vs. 11.7 months; HR=1.8; 95% CI: 1.1-2.8; P=0.014). It is also of note that the addition of Cy resulted in better PFS in the lenalidomide-refractory population compared with the Kd group, with a median PFS of 18.4 versus 11.3 months (HR=1.7; 95% CI: 1.1-2.7; P=0.043) (Figure 2B). This improvement was focused on lenalidomide-refractory patients after one PL.
No differences were observed in the ORR, CR or MRD rates between the KCd and Kd groups (Table 3). However, 11.3% and 12.2% of patients achieved MRD negativity in the KCd and Kd groups, respectively, and PFS was significantly better in patients in the triplet arm who reached this response (not estimated, HR=8.9; 95% CI: 1.1-73.6; P=0.044) (Online Supplementary Figure S1). In addition, lenalidomide-refractory patients allocated to KCd were significantly more likely to reach stringent CR (22.0% vs. 6.5%; OR 4.0; 95% CI: 1.0-16.1; P=0.037) and MRD negativity (22.0% vs. 2.1%; OR 54.0; 95% CI: 2.8-1,040.0; P=0.005) (Table 3).
With regard to TTP, 58 (59.7%) and 73 (73%) patients experienced progressive disease in the KCd and Kd groups, respectively. However, no differences were observed between patients treated with the alkylator-containing versus alkylator-free regimen, with TTP of 25.5 versus 17.1

months (HR=1.3; 95% CI: 0.9-1.8; P=0.168) (Figure 3A). Turning, finally, to OS, 46 (47.4%) and 36 (36.0%) events occurred in the KCd and Kd arms, respectively. No statistically significant differences were noted. The median OS of the triplet regimen was 39.7 months, and it was not reached in the doublet scheme (HR=1.4; 95% CI: 0.9-2.2; P=0.111) (Figure 3B). In addition, no difference in OS was observed in lenalidomide-refractory patients between KCd and Kd groups (37.0 vs. 38.0 months, HR=1.2; 95% CI: 0.7-2.2; P=0.574). A post hoc analysis of PFS2, defined as the time from randomization to disease progression after the next line of treatment or death from any cause, was performed. Overall, 49 (50.5%) and 65 (65.0%) patients in the KCd and Kd groups received next line of therapy (P=0.255). No differences were observed in the subsequent line of treatment received (Online Supplementary Table S1), and the majority of patients in both arms were rescued by daratumumab-based combinations (37 (69.4%) patients in the KCd arm and 44 (67.7%) patients in the Kd arm (P=0.847)). The probability of achieving PR or better was 73.3% and 77.4% in the KCd and Kd arms, respectively (P=0.627). However, the median PFS2 was shorter in the KCd arm compared to the Kd arm: 23.7 months versus 36.3 months (HR=1.5; 95% CI: 0.9-2.3; P =0.073) (Online Supplementary Figure S2).
The most frequent AE and the AE of interest in the safety population are summarized in Tables 4 and 5, respectively.
Figure 2. Primary endpoint: progression-free survival. (A) Progression-free survival in the entire cohort. (B) Progression-free survival in the lenalidomide-refractory patients. CI: confidence interval; HR: hazard ratio; KCd: carfilzomib, cyclophosphamide and dexamethasone; Kd: carfilzomib and dexamethasone.

Table 2. Progression-free survival: post hoc subgroup analysis.
KCd: carfilzomib, cyclophosphamide and dexamethasone; Kd: carfilzomib and dexamethasone; Cy: cyclophosphamide; HR: hazard ratio; CI: confidence interval; PFS: progression-free survival; ISS: International Staging System; SR: standard-risk; HRCA: high-risk chromosomal abnormalities; PL: prior lines; PI: proteasome inhibitor; IMID: immunomodulators; NR: not reached.
AE grade ≥3 were reported in 53.6% (44/97) and 43% (43/100) of patients in the KCd and Kd groups, respectively. After 12 cycles, the median dose intensity of carfilzomib in the KCd arm was 90% (interquartile range, 76.1-100.0) and 89.2% (interquartile range, 76.4-97.2) in the Kd arm. In general terms, the addition of cyclophosphamide to Kd did not result in any new safety signal, but there was a trend towards higher toxicity in the triplet group. With regard to hematological toxicity, no differences were observed be-

tween both groups, except for neutropenia (Table 4). The incidence of any grade of neutropenia was higher in the KCd group (24.7% vs. 16.0%), especially the grade ≥3 AE (13.4% vs. 5.0%; OR 3.0; 95% CI: 1.1-8.6; P=0.049). Considering solely the non-hematological AE, asthenia was the only AE reported in KCd with a statistically significant difference compared with Kd (32.9% vs. 20.0%; OR 2.0; 95% CI: 1.1-3.8; P=0.040). Infections were the main type of non-hematological toxicity reported in the trial, occurring in roughly equal
*Response not evaluated in 3 patients in the KCd group and in 2 patients in the Kd group. KCd: carfilzomib, cyclophosphamide and dexamethasone; Kd: carfilzomib and dexamethasone; CR: complete response; MRD: minimal residual disease; neg: negative; pos: positive; VGPR: very good partial response; PR: partial response; MR: minimal response; SD: stable disease; PD: progression disease; ORR: overall rate response; NS: non-significant.
Table 3. Responses achieved in the trial: all randomized and lenalidomide-refractory patients.proportions in the two groups (Table 4). However, it is worth noting that grade 4 infections (sepsis) were more frequent in patients treated with KCd (7.2% vs. 2.0%; P=0.080). Although no differences in any grade or grade ≥3 of hypertension between groups, the addition of cyclophosphamide showed a numerical increase in cardiovascular events (including atrial fibrillation, cardiac failure, acute pulmonary edema, ischemic cardiac disease and pulmonary hypertension) (12.4% vs. 5.0%; P=0.065). In addition, it is important to mention that nine grade 5 AE were notified, five in the KCd (one due to sudden death, and the others due to various types of pneumonia (aspiration, nosocomial, Influenza A, and bilateral SARS-CoV2 pneumonia) and four in the Kd group (two due to acute respiratory distress syndrome, one to Legionella pneumoniae sepsis, and one to uremic hemolytic syndrome).
There were no differences in the percentages of patients who delayed their treatment between the KCd (44 patients, 44.5%) and Kd (41 patients, 41.0%) groups (Online
Supplementary Table S2). In the triplet regimen, the most common reason for delay were upper respiratory infections (12.4%), neutropenia (8.2%), diarrhea (6.2%) and asthenia (6.2%). However, upper respiratory tract infections (12.0%), other infections (7.0%) and pneumonia (5.0%) occurred in the doublet regimen. There were also no significant differences in patients who required dose reductions due to AE, 23 (23.7%) patients with KCd, and 22 (22.0%) patients with Kd. The foremost causes of treatment dose reduction were asthenia (5.2%) in the KCd group and arterial hypertension (8.0%) in the Kd group. Carfilzomib was the drug most often adjusted after the development of AE in both arms of the study.
During treatment, 173 patients discontinued, mainly because of progressive disease. Discontinuation due to progression was less frequent in the KCd (47.0%) than the Kd group (67.8%) (OR 2.4; 95% CI: 1.3-4.4; P=0.006). Toxicity was the second most common cause of discontinuation and was higher in the KCd than the Kd group, with 16 ver-
sus seven patients (19.3% vs. 7.8%; OR 2.8; 95% CI: 1.1-7.3; P=0.031). Within KCd group, three patients discontinued due to renal impairment, three due to cardiac failure, two due to bad tolerance, and eight (1 patient each severe AE) due to neutropenia, atrial fibrillation, myocardial infarction, acute pulmonary edema, pulmonary hypertension, thrombotic microangiopathy, hemolytic anemia and hepatic toxicity. In contrast, three patients discontinued Kd due to bad tolerance, two due to cardiac failure, and one due to uremic hemolytic syndrome and one due to posterior reversible encephalopathy syndrome. No differences were observed in the occurrence of cardiotoxicity as a cause of discontinuation (8 patients in KCd and 3 in Kd). Sixty percent of patients who presented this unacceptable toxicity were over 70 years of age. However, the timing of the occurrence of toxicity differed between the arms. Patients treated with KCd developed AE, resulting in earlier discontinuation than in those treated with Kd, after a median of five (1-26) versus 17 cycles (10-20) cycles. After 37 months of follow-up, 56% and 66% of patients in KCd and Kd, respectively, were still alive. During the clinical trial, and within 30 days of receiving the last dose of the study treatment, ten patients died in the KCd group (3 from disease progression, 4 from an unrelated cause, and 3 due to infections), and seven in the Kd group (3 from disease progression, 3 due to infections and 1 due to uremic hemolytic syndrome).
In this randomized phase II trial, a 70 mg/m2 dose of carfilzomib once weekly with dexamethasone plus/minus Cy was effective in RRMM after 1-3 PL. The addition of Cy to Kd showed no significant statistical differences compared with
patients treated with Kd in PFS, TTP, OS, ORR or the other response categories when all randomized patients were analyzed. However, Kd alone was suboptimal in patients who were refractory to lenalidomide with a median PFS of 11.3 months, whereas these patients treated with KCd achieved a median PFS of 18.4 months, similar to all randomized subjects. Moreover, the addition of Cy to Kd in this subgroup of patients resulted in a significantly higher percentage of stringent CR and MRD-negative rates. The results of GEM-KyCyDex study are consistent with those of other clinical trials that tested the KCd combination. The phase II MUKfive trial18 showed greater efficacy of KCd over VCd (bortezomib, cyclophosphamide, dexamethasone) in RRMM after one PL. In this study, carfilzomib was administered twice weekly at a dose of 36 mg/m2 and oral Cy was given weekly (on days 1, 8 and 15) at a fixed dose of 500 mg. In addition, after six cycles, patients were randomized 1:1 to receive carfilzomib maintenance on days 1 and 15, or nothing. This twiceweekly KCd yielded an ORR of 84.0%, which is slightly higher than that reported in our study. However, the population of the MUKfive study was RRMM after one PL, less exposed to PI and IMID (1/5 patients to each drug), if we focus on those patients who received maintenance with Kd, PFS and OS were not different to those reported in our study (19 and 32 months, respectively). In the single-arm, multicenter, Canadian phase II trial ( clinicaltrial gov. Identifier: MCRN-003/CCTGMYX. 1),19 the target population was similar to that of the patients enrolled in our trial: RRMM after 1-3 PL, almost 90% and 80% of patients exposed to PI and IMID, respectively; and similar drug schedule: weekly Kd 70 mg/m2 and weekly oral dose of 300 mg/m2 Cy (capped at 500 mg and discontinued after the 12th cycle). This combination resulted in a similar ORR (85%) but the 29% of patients achieving at least CR was
higher than in our study. However, this did not translate into a more prolonged PFS or OS than that reported in our study, 17.2 and 27.4 months, respectively.
One finding to be highlighted is the potential benefit of the addition of Cy to Kd to overcome the dismal prognosis of the lenalidomide-refractory population. In our trial, Kd alone was suboptimal in this population with a PFS of 11.3 months. This discouraging result is consistent with most experimental treatments explored in phase III trials of RRMM after 1-3 PL, 20–23 except for the combination of anti-CD38 monoclonal antibodies and Kd.24,25 Our results, with a median PFS of 18.4 months in the lenalidomide-refractory population, argue in favor of this combination. Therefore, KCd once weekly could be a cost-effective treatment in these populations, especially if the anti-CD38 monoclonal antibody is not a valid option because of safety or other issues. Although the majority of the patients in this study were naïve for anti-CD38 monoclonal antibodies, this combination could represent an option in the future for patients already exposed to both lenalidomide and anti-CD38 monoclonal antibodies.
The twice-weekly Kd combination had been investigated in several phase III trials. However, in the present study, weekly Kd was explored with the aim of conciliating efficacy and patient quality of life. Our Kd arm resulted in a median PFS of almost 17 months, slightly lower than that of the twice-weekly Kd of the ENDEAVOR trial (18.7 months). 5 Notably, patients enrolled in the ENDEAVOR study were less exposed to bortezomib (54%), and less exposed and refractory to lenalidomide (38% and 24% respectively). Also, Kd 56 mg/m 2 twice weekly was the control arm of the phase III CANDOR and IKEMA trials. 24,26 Both studies enrolled patients with similar characteristics: RRMM after 1-3 PL (50% after ≥2 PL), 8090% and 70-80% have been previously exposed to PI and IMiD, and one of three patients were refractory to PI or IMiD. In the CANDOR trial, twice-weekly Kd showed consistent results to our weekly Kd in ORR (73.0%), at least CR (13.0%) and PFS (15.2 months). The results showed in the IKEMA trial were slightly better in response (ORR: 83.7% and CR rate: 28.5%) and PFS (19.2 months). Hence, weekly 70 mg/m 2 Kd presented comparable efficacy to twice-weekly 56 mg/m 2 Kd and this schedule could be a suitable and convenient option for Kd-eligible patients, as previously reported in the A.R.R.O.W study although it was only focused in patients after 2-3 prior lines.7 In addition, since no differences were noted between both arms of our trial, some subset of patients might benefit from Kd alone, such as elderly patients who are not refractory to lenalidomide or patients with a poor bone marrow reserve.
The trend towards worse OS with KCd in our study was an unexpected result. No differences were observed in
the death rates during the clinical trial, or within 30 days following the last dose of the study treatment. In the post hoc analysis of PFS2, fewer patients received rescue therapies in the KCd group compared with the Kd group. Although both groups were treated with similar salvage therapies, the PFS2 in patients assigned to the KCd group was shorter. There is no clear explanation for this exploratory finding, but cumulative toxicity with KCd might have had a role in this poorer outcome. In addition, a speculative hypothesis could be that the addition of Cy may have caused further DNA damage in the tumor cell, with consequent genomic instability, resulting in a more aggressive and difficult-to-treat disease. No new safety signals were reported in the GEM-KyCyDex trial. As expected, the triplet was more toxic than the doublet scheme. This fact resulted in a significant higher discontinuation rate in the KCd arm, especially in older patients (≥70 years) and during the early cycles. In this regard, we propose that the dose of carfilzomib in the KCd group could be raised to 56 mg/m 2 instead of 70 mg/m2 considering the efficacy as well as the safety profile. In addition, it is unclear whether a medium-high dose of cyclophosphamide produces the same tumor activity as a daily low dose. Switching to daily oral lowdose cyclophosphamide could improve safety without substantially changing effectiveness. Both strategies are worth investigating to mitigate toxicity and individualize the dose, especially in the elderly population.
Our trial has some limitations such as being a phase II randomized trial, the completion of recruitment earlier than expected because of the SARS-Cov-2 pandemic, and the study population. Enrolled patients do not represent the current RRMM after 1-3 PL because most of them have been already exposed to PI, IMiD and antiCD38 monoclonal antibodies, whilst only 5% of patients in our study were exposed to an anti-CD38 drug, and those refractory to PI were not included. Therefore, the real-world candidates to receive weekly KCd or Kd are unrepresented in this trial.
In summary, this study has not met its primary endpoint, and in general terms, there were no differences between KCd and Kd. However, the GEM-KyCyDex trial has shown how weekly Kd plus/minus Cy is a feasible, effective, and well-tolerated regimen for RRMM patients after 1-3 PL. Our data points to the addition of Cy to Kd could result in a significant clinical impact in the lenalidomide-refractory population. KCd could be used as cost-effective salvage therapy in places where access to anti-CD38 monoclonal antibodies is limited, or as a rescue tool until new immunotherapies are approved for treating patients with earlier relapses.
Disclosures
BP has received honoraria from Janssen and Aptitude
Health. VGC has received honoraria from Janssen and Celgene, research funding from Janssen; has a consulting or advisory role at Prothena and Janssen. AS has received honoraria derived from lectures and consulting from Takeda, BMS, MSD, Janssen, Amgen, Novartis, Gilead and Sanofi. LR has received honoraria from Janssen, BMS-Celgene, Amgen, Takeda, Sanofi, GSK. FE has received speaker’s fees from Janssen, Sanofi, Amgen and Sanofi, and honoraria for a consulting or advisory role from Janssen, Biogene, Sanofi, GSK, Takeda, Amgen y BMS. BRB has received speaker’s fee from Janssen. APGR has acted as a consultant from Celgene, Sanofi, Janssen and BMS, and has received speaker’s fees from BMS, Janssen, Celgene, Alexion, Takeda and Astra-Zeneca. FdA has received honoraria from Janssen, BMS, Amgen, GlaxoSmithKline, Sanofi and Takeda. JBlade has received honoraria for lectures from Janssen, Amgen, Celgene/BMS, Takeda and Oncopeptides. JJL has received honoraria for lectures or advisory role from Celgene/MBS, Sanofi, Takeda, Amgen and Janssen-Cilag. JFSM reports consultancy for BristolMyers Squibb, Celgene, Novartis, Takeda, Amgen, MSD, Janssen, and Sanofi and membership on board of directors or advisory committees of Takeda. EMO has received honoraria derived from lectures and advisory boards from Amgen, BMS/Celgene, GSK, Janssen, Pfizer, Oncopeptides, Sanofi, Takeda and Karyopharm. MVM has received honoraria derived from lectures and advisory boards from Janssen, BMS-Celgene, Amgen, Takeda, Abbvie, Sanofi, On-
1. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516-2520.
2. Muchtar E, Gertz MA, Magen H. A practical review on carfilzomib in multiple myeloma. Eur J Haematol. 2016;96(6):564-577.
3. Papadopoulos KP, Siegel DS, Vesole DH, et al. Phase I study of 30-minute infusion of carfilzomib as single agent or in combination with low-dose dexamethasone in patients with relapsed and/or refractory multiple Myeloma. J Clin Oncol. 2015;33(7):732-739.
4. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372(2):142-152.
5. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38.
6. Berenson JR, Cartmell A, Bessudo A, et al. CHAMPION-1: a phase 1/2 study of once-weekly carfilzomib and dexamethasone for relapsed or refractory multiple myeloma. Blood. 2016;127(26):3360-3368.
7. Moreau P, Mateos MV, Berenson JR, et al. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): interim analysis results of a randomised, phase 3 study. Lancet Oncol.
copeptides, Adaptive, Roche, Pfizer, Regeneron, GSK, Bluebird Bio, Sea-Gen. All other authors have no conflicts of interest to disclose.
MVM and EMO developed the concept of the study. MVM, BP and VGC developed the methodology and performed the research, wrote the original draft, reviewed and edited the manuscript, and performed formal analysis and visualization. All other authors performed the research, reviewed and edited the manuscript.
The authors would like to thank to Roberto Maldonado for his help with data management and Philip Mason for language revision of the manuscript.
The PETHEMA Foundation sponsored the trial. The manufacturer of carfilzomib (Amgen) supplied the drugs free of charge and financially supported the trial, but was not involved in the analysis, interpretation, or decision to publish the trial. Additionally, this study was supported by the International Myeloma Society.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
2018;19(7):953-964.
8. Dimopoulos MA, Goldschmidt H, Niesvizky R, et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an openlabel, randomised, phase 3 trial. Lancet Oncol. 2017;18(10):1327-1337.
9. Kropff M, Bisping G, Schuck E, et al. Bortezomib in combination with intermediate-dose dexamethasone and continuous lowdose oral cyclophosphamide for relapsed multiple myeloma. Br J Haematol. 2007;138(3):330-337.
10. Fu W, Delasalle K, Wang J, et al. Bortezomibcyclophosphamide-dexamethasone for relapsing multiple myeloma. Am J Clin Oncol. 2012;35(6):562-565.
11. de Waal EGM, de Munck L, Hoogendoorn M, et al. Combination therapy with bortezomib, continuous low-dose cyclophosphamide and dexamethasone followed by one year of maintenance treatment for relapsed multiple myeloma patients. Br J Haematol. 2015;171(5):720-725.
12. Kropff M, Vogel M, Bisping G, et al. Bortezomib and low-dose dexamethasone with or without continuous low-dose oral cyclophosphamide for primary refractory or relapsed multiple myeloma: a randomized phase III study. Ann Hematol. 2017;96(11):1857-1866.
13. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet
Oncol. 2016;17(8):e328-e346.
14. López-Corral L, Gutiérrez NC, Vidriales MB, et al. The Progression from MGUS to smoldering myeloma and eventually to multiple myeloma Involves a clonal expansion of genetically abnormal plasma cells. Clin Cancer Res. 2011;17(7):1692-1700.
15. Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210-2221.
16. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548.
17. Flores-Montero J, Sanoja-Flores L, Paiva B, et al. Next generation flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia. 2017;31(10):2094-2103.
18. Yong KL, Hinsley S, Auner HW, et al. Carfilzomib or bortezomib in combination with cyclophosphamide and dexamethasone followed by carfilzomib maintenance for patients with multiple myeloma after one prior therapy: results from a multicenter, phase II, randomized, controlled trial (MUKfive). Haematologica. 2021;106(10):2694-2706.
19. Venner CP, LeBlanc R, Sandhu I, et al. Weekly carfilzomib plus cyclophosphamide and dexamethasone in the treatment of relapsed/refractory multiple myeloma: final results from the MCRN 003/ MYX .1 single arm phase II trial. Am J Hematol. 2021;96(5):552-560.
20. Moreau P, Joshua D, Chng WJ, et al. Impact of prior treatment on patients with relapsed multiple myeloma treated with carfilzomib and dexamethasone vs bortezomib and dexamethasone in the phase 3 ENDEAVOR study. Leukemia. 2017;31(1):115-122.
21. Mateos MV, Sonneveld P, Hungria V, et al. Daratumumab, bortezomib, and dexamethasone versus bortezomib and dexamethasone in patients with previously treated multiple myeloma: three-year follow-up of CASTOR. Clin Lymphoma Myeloma Leuk. 2020;20(8):509-518.
22. Dimopoulos MA, Terpos E, Boccadoro M, et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22(6):801-812.
23. Richardson PG, Oriol A, Beksac M, et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): a randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(6):781-794.
24. Usmani SZ, Quach H, Mateos MV, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2022;23(1):65-76.
25. Moreau P, Dimopoulos MAC, Mikhael J, et al. VP5-2022: updated progression-free survival (PFS) and depth of response in IKEMA, a randomized phase III trial of isatuximab, carfilzomib and dexamethasone (Isa-Kd) vs Kd in relapsed multiple myeloma (MM). Ann Oncol. 2022;33(6):664-665.
26. Moreau P, Dimopoulos MA, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet. 2021;397(10292):2361-2371.
Correspondence: J.R. Cook cookj2@ccf.org
Received: November 15, 2022.
Accepted: March 30, 2023.
Early view: April 6, 2023.
htps://doi.org/10.3324/haematol.2022.282389
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
A subset of patients with immunoglobulin M (IgM) monoclonal gammopathy of undetermined significance (MGUS) develop IgM-related disorders (IgM-RD) including peripheral neuropathy, cryoglobulinemia and/or cold agglutinin disease (CAD). We examined the clinical and bone marrow pathologic findings in 191 IgM MGUS patients (2016 World Health Oragnization criteria). Clonal plasma cells were identified in 41 of 171 (24%) cases by immunohistochemistry (IHC) and clonal B cells in 43 of 157 (27%). IgM-RD was identified in 82 (43%) cases, including peripheral neuropathy (n=67, 35%), cryoglobulinemia (n=21, 11%), and CAD (n=10, 5%). Cases of CAD showed distinctive features including lack of MYD88 mutations (P=0.048), supporting the concept of primary CAD as a distinct clinicopathologic disorder. Following exclusion of CAD, comparison of the remaining cases with (n=72) or without (n=109) IgM-RD showed IgM-RD to be more frequent in men than women (P=0.02) and to be more highly associated with MYD88 L265P (P=0.011). Cases with and without IgM-RD otherwise showed similar features including serum IgM concentrations, presence of lymphoid aggregates, clonal B cells by flow cytometry or clonal plasma cells by IHC. No differences were observed in overall survival between cases with and without IgM-RD. No cases in this series met criteria for plasma cell type IgM MGUS as defined in the 2022 International Consensus Classification of lymphoid neoplasms. These results show IgM-RD to be common in patients with IgM MGUS. While CAD shows distinctive features, the remaining cases of IgM-RD largely show pathologic findings similar to IgM MGUS without IgM-RD.
In contrast to immunglobulin G (IgG) or IgA monoclonal gammopathy of undetermined significance (MGUS), which typically progress to multiple myeloma or other plasma cell neoplasms, IgM MGUS characteristically shows progression to lymphoplasmacytic lymphoma (LPL) or other B-cell lymphoproliferative disorders.1–4 IgM multiple myeloma is rare, but also presumably develops through a precursor phase of IgM MGUS.5–7 In the revised 4th edition of the World Health Organization (WHO) classification of hematolymphoid neoplasms, IgM MGUS is defined by the presence of an IgM paraprotein with a serum concentration of less than 3 g/dL, less than 10% clonal lymphoplasmacytic cell infiltration in the bone marrow, and lack of myeloma-defining clinical features.8
The criteria for diagnosis of IgM MGUS and distinction from LPL, however, has been somewhat controversial. Some cases meeting revised 4th edition WHO criteria for IgM MGUS would be classified as LPL by the criteria of the International Workshop on Waldenström’s macroglobulinemia9 or the recently published International Consensus Classification (ICC) of lymphoid neoplasms.10 The ICC further distinguishes between IgM MGUS of plasma cell type (defined as having clonal plasma cells without clonal B cells and lacking MYD88 mutations) versus IgM MGUS, not otherwise specified (all other cases) in an effort to distinguish cases that may progress to IgM myeloma versus those more likely to progress to lymphoproliferative disorders.10
It has also increasingly been recognized in recent years that patients fulfilling criteria for IgM MGUS, despite hav-
ing relatively low disease burden in the marrow, may in some cases have significant symptoms which require therapy.11–15 The most commonly reported symptoms include neuropathies, cryoglobulinemia, or cold agglutinin disease (CAD). If these symptoms secondary to an IgM paraprotein are present, the term “IgM-related disorder” (IgM-RD) has been proposed to recognize the potential need for treatment in these patients. 9 Recent studies have shown that primary CAD represents a distinct lymphoproliferative disease with clinicopathologic findings differing from LPL or other IgM MGUS,16–18 and the 2022 ICC recognizes primary CAD as a distinct clinicopathologic entity.10 It is currently unclear whether other forms of IgM-RD may show distinctive clinicopathologic findings compared to asymptomatic IgM MGUS patients. In this study, we examine the detailed clinicopathologic findings in a cohort of patients meeting 2016 WHO criteria for IgM MGUS and compare the findings in patients with and without IgM-RD. We also examine the impact of the new 2022 ICC diagnostic criteria on diagnosis of IgM versus the 2016 WHO system. These results provide detailed guidance for diagnosis of IgM MGUS and have implications for the most appropriate classification of these clinically heterogeneous cases.
After Cleveland Clinic Institutional Review Board approval, we retrospectively reviewed patients with an IgM serum paraprotein seen at our institution from 20022021 and identified those who underwent bone marrow biopsies. Formalin-fixed, paraffin-embedded, and hematoxylin and eosin stained tissue sections from bone marrow trephine biopsies and aspirate clot sections as well as Wright-Giemsa stained bone marrow aspirate smears were retrieved from the archives. Previously performed immunohistochemistry was reviewed, as were available results of prior flow cytometry, metaphase cytogenetics, fluorescence in situ hybridization (FISH) for plasma cell associated abnormalities, and MYD88 L265P mutation testing.
Clinical data was obtained by retrospective review of the electronic medical record. The presence or absence of neuropathy not attributable to other causes per the treating physician was noted, as was previously documented cryoglobulinemia or CAD. Where previously performed, results of anti-MAG antibody testing were noted (cutoff for positive results defined as >1,000 titer units). For clinical follow up analysis, progression events were defined as the development of an overt lymphoid or plasma cell neoplasm, amyloidosis, or death.
In cases where immunohistochemistry (IHC) had not been previously performed and paraffin blocks were available, immunohistochemical stains for CD138, κ and λ immunoglobulin light chains, and CD20 were performed. IHC staining was performed on the Ventana Benchmark automated immunostainer (Roche Diagnostics, Indianapolis, IN), using the following antibodies (concentration): CD20 mouse monoclonal L26 antibody (Dako, 1:200); CD138 mouse monoclonal B-A38 (Biocare, 1:200); κ light chains rabbit polyclonal (Dako, 1:16,000); λ light chains rabbit polyclonal (Dako, 1:32,000). Blinded scoring was performed by two pathologists (FKB, JRC); discrepancies were resolved by a third reviewer (MON).
Statistical tests including two-tailed Student’s t-test and Kruskal-Wallis test for continuous variables and Χ 2-test and Fisher exact test, when applicable, for categorical variables were performed with R (R Core Team, Vienna, Austria) and a significance threshold of P≤0.05 was used.
Clinicopathologic features of immunoglobulin M monoclonal gammopathy of undetermined significance as defined by 2016 World Health Organization criteria Out of 1,222 patients with an IgM paraprotein detected at our institution from 2002-2021, 238 patients had available bone marrow biopsies and fulfilled 2016 WHO IgM MGUS criteria. Twelve patients with concurrent or prior myeloid neoplasms, 15 patients with amyloidosis, and 20 patients with previously diagnosed and treated lymphoproliferative disorders were excluded for a final cohort of 191 patients. The clinicopathologic features of this cohort are detailed in Table 1. Overall, the average age at bone marrow biopsy was 69.6 years and there was a male predominance (male: female, 1.5:1). The median IgM serum paraprotein was 452 mg/dL and the median hemoglobin concentration was 13 g/dL.
Clonal plasma cells were identified by light chain IHC in 41 of 171 (24%) cases that could be assessed. Flow cytometric studies, performed in 157 cases, are summarized in Figure 1. Flow cytometry identified a clonal B-cell population in 43 (27%) cases including 30 cases with B cells negative for CD5 and 13 cases positive for CD5. In each of the 30 cases (100%) with CD5-negative clonal B cells, the monotypic light chain expressed on B cells matched the IgM paraprotein light chain, while the B-cell light chains and IgM paraprotein light chains were concordant in only seven of 13 (54%) CD5-positive B-cell cases (P=0.0003, Fisher exact test).
Lymphoid aggregates were present in 61 of 191 (32%)
MGUS: monoclonal gammopathy of unknown significance; IgM: immunoglobulin M; w/o: without; RD: related disorders; CAD: cold agglutinin disease; Bmbx: bone marrow biopsy; IFE: immunofixation electrophoresis; SD: standard deviation; Hgb: hemoglobin; PD: poorly defined; Lymphs: lymphocytes; PC: plasma cells; IHC: immunohistochemistry; FC: flow cytometry; MAG: myelin-associated glycoprotein.
cases, one case displayed T-cell-rich aggregates interpreted as being reactive, two case showed clonal plasma cells plus a CD5-positive B-cell population with light chains that did not match the IgM paraprotein, consistent with synchronous IgM MGUS and monoclonal B lymphocytosis, and three cases contained B-cell-rich aggregates representing <10% of the core biopsy as well as CD5negative B-cell populations with light chains that match clonal plasma cells by IHC and the IgM paraprotein light chain. These latter three cases (3/191, 1.6%) are compatible with a diagnosis of LPL using 2022 ICC criteria or Waldenstrom Workshop criteria (Figure 2).9,10 Additional cases with abnormal, interstitial lymphoplasmacytic infiltrates but without aggregates, which would meet the Waldenstrom Workshop definition of LPL but not the 2022 ICC definition, were not identified in this cohort.

samples, but only six cases contained lymphoid aggregates in the core biopsy as well as clonal plasma cells by IHC and clonal B cells by flow cytometry. Of these six
Metaphase karyotyping was performed in 173 cases (90.6%); 22 (13%) cases had an abnormal karyotype (Online Supplementary Table S1). FISH testing for myeloma related abnormalities was performed in 86 of 191 (45%)
Figure 2. Histologic features in a case meeting 2016 World Health Organization criteria for immunglobulin M monoclonal gammopathy of unknown significance but diagnostic of lymphoplasmacytic lymphoma using 2022 International Consensus Classification criteria. The aggregate in this case (A) (hematoxylin and eosin staining, 40X magnification) contains numerous CD20-positive small cells (B) (40x magnification) representing <5% of bone marrow cellularity admixed with numerous κ-positive (C) (40x magnification) and only rare λ-positive (D) (40x magnification) plasma cells. This case also displayed κ monotypic B cells by flow cytometry.

cases, of which 27 (31.4%) yielded insufficient plasma cells. Among 59 cases that were assessed, one case of IgM MGUS with a normal karyotype showed loss of chromosome 15 in 70 of 100 plasma cells. No myeloma associated IGH translocations were identified. The MYD88 L265P mutation was detected by allele specific polymerase chain recation (PCR) in 35 of 56 (63%) cases analyzed. Clinically, IgM-RD was identi fied in 82 of 191 (43%) patients, including neuropathy in 67 of 191 (35%) cases, followed by cryoglobulinemia in 21 of 191 (11%) and CAD in ten of 191 (5%) (Figure 3). Patients with cryoglobulinemia presented with concurrent neuropathy in 14 of 21 cases (67%), and rarely with concurrent cryoglobulinemia and CAD (2 cases, 1%). Neuropathy and CAD were not observed in the same patients in our cohort.
The ten cases with clinical and laboratory evidence of CAD were considered separately (Table 1): compared to cases without CAD, the IgM serum concentration and hemoglobin level were significantly lower (P=0.034 and P=0.007, respectively). Clonal plasma cells were found by IHC in three of eight (38%) of cases. Flow cytometry showed a
clonal B-cell population in four of nine cases (44%) including two CD5-positive and two CD5-negative B-cell clones. Cases with CAD showed lymphoid aggregates more frequently than cases without CAD (60% vs. 30.4%), although the difference did not reach statistical significance (P=0.077). None of the three CAD patients tested showed the MYD88 mutation (P=0.048, Fisher exact test).
gammopathy of undetermined significance with and without immunoglobulin M-related disease
After exclusion of primary CAD cases, patients with IgMRD represented 40% (72/181) of the cohort versus 60% (109/181) without IgM-RD (Table 2). Patients with IgM-RD showed a greater male predominance (P=0.020) and had higher hemoglobin levels compared to those without IgMRD (13.4 g/dL vs. 12.3 g/dL; P<0.001). Anti-MAG antibodies were identified in 52% (16/31) of tested patients, all of whom presented with IgM-RD due to neuropathy (16/27, 59%) and three of whom also had cryoglobulinemia. Four patients without neuropathy or cryoglobulinemia tested negative for MAG antibodies (P=0.043). MYD88 L265P mutations were detected in 84% (21/25) of IgM-RD cases
Figure 3. Venn diagram of the symptoms of patients with immunglobulin M monoclonal gammopathy of unknown significance wit h and without immunglobulin M-related disorders and patients with cold agglutinin disease.

compared to 50% (14/28) in those without IgM-RD (P=0.011).
Comparison of immunoglobulin M monoclonal gammopathy of undetermined significance with and without clonal B cells and/or plasma cells
We next examined the clinicopathologic features of IgM MGUS (excluding CAD) based on the presence or absence of clonal B cells and/or clonal plasma cell populations. This analysis was restricted to the 148 patients where results of flow cytometry and immunohistochemistry was available for review (Table 3). Cases with clonal plasma cells, with or without clonal B cells, were associated with higher levels of IgM serum concentration (P<0.001) and were more likely to contain lymphoid aggregates in the bone marrow (P=0.086). Cases with monotypic plasma cells, regardless of the presence of clonal B cells, showed the MYD88 L265P mutation in 100% of cases analyzed. The subset with both clonal B cells and monotypic plasma cells showed the highest IgM serum concentration, highest incidence of IgM-RD (62%), and all patients were male. Cases lacking both detectable clonal plasma cells and clonal B-cells showed the lowest IgM serum paraprotein concentrations, infrequently harbored lymphoid aggregates, showed the lowest incidence of an abnormal karyotype, yet showed the MYD88 L265P mutation in 57% (12/21) of cases. Abnormal karyotypes were present at similar levels in all subsets of cases.
The mean follow-up time post bone marrow biopsy for the cohort including cases with IgM MGUS with and without IgM-RD (excluding CAD) was 4.17 years (median 2.78 years; range, 0.03-19.48 years). Only two patients, both IgM MGUS without RD, showed progression to an overt disease on available follow-up. One patient (with polyclonal B cells and polyclonal plasma cells in the bone marrow) was diagnosed with diffuse large B-cell lymphoma and a second patient (with polytypic plasma cells and no flow cytometry data) developed renal amyloidosis with a different immunoglobulin light chain. Overall survival (OS) was similar between IgM MGUS and IgM-RD (P=0.43, Figure 4). MYD88 mutation status, available in 56 patients across the entire cohort, was not significantly associated with survival (P=0.27). Within CAD patients, no progression events occurred with a mean follow-up of 3.16 years (median 3.16 years).
MGUS has long been recognized as a precursor condition that may progress to multiple myeloma, other plasma cell neoplasms, or lymphoproliferative disorders, but the approach to diagnosis and classi fication of MGUS has changed over recent years.1–4 Recognizing the biologic differences between IgM MGUS, which typically progresses
Table 2.
M
significance and with and without IgM-related disorders (after exclusion of cold agglutinin disease).
of
MGUS: monoclonal gammopathy of unknown significance; w/o: without; RD: related disorders; CAD: cold agglutinin disease; Bmbx: bone marrow biopsy; IFE: immunofixation electrophoresis; SD: standard deviation; Hgb: hemoglobin; PD: poorly defined; Lymphs: lymphocytes; PC: plasma cells; IHC: immunohistochemistry; FC: flow cytometry; MAG: myelin associated glycoprotein.
to LPL or other lymphoproliferative disorder, versus nonIgM (i.e., IgA or IgG) MGUS which generally progress to multiple myeloma, these two entities were separately defined in the 2014 update to the International Myeloma Working Group diagnostic criteria,19 and these changes were incorporated into the 2016 WHO classi fication of plasma cell neoplasms.20 In addition, there has been increasing interest in recognizing the subset of patients who meet criteria for diagnosis as MGUS but who may nevertheless have clinically significant symptoms that may require treatment.11–15 In patients with IgM MGUS, the most common symptoms include neuropathy, cryoglobulinemia and CAD, which have collectively been described under the term IgM-RD.21,22 Prior studies of patients with IgM-RD have primarily focused on clinical manifestations, and it has been unclear to what extent the bone marrow pathologic features may differ in patients with and without IgMRD.
In the current study, we have examined the detailed clinicopathologic findings in a large series of patients with IgM
MGUS. Overall, IgM-RD (including CAD) was identified in 82 of 191 (43%) of cases. This finding is similar to that reported in the literature, where peripheral neuropathy alone has been reported in approximately 30-50% of IgM MGUS patients, emphasizing that symptomatic disease is not rare in IgM MGUS.23,24 Indeed, this study likely represents an underestimate of symptomatic disease as we limited our definition of IgM-RD to neuropathy, cryoglobulinemia and CAD. We elected to focus on these manifestations of IgM-RD as these conditions are reported to be relatively common, and because cryoglobulinemia and CAD may be readily documented by routine laboratory testing. This analysis did not include other conditions considered part of the spectrum of IgM-RD by some investigators such as autoimmune thrombocytopenia or IgM-related nephropathy.1,15,25 Similarly, amyloidosis was excluded from this analysis as it is recognized as a distinct clinicopathologic entity in the 2016 WHO classification8 and the 2022 ICC system.10 Prospective studies of IgM MGUS patients with systematic assessment for all po-
B: B cells; PC: plasma cells; IFE: immunofixation electrophoresis; SD: standard deviation; Hgb: hemoglobin; PD: poorly defined; Lymphs: lymphocytes; PC: plasma cells; IHC: immunohistochemistry; FC: flow cytometry; MAG: myelin-associated glycoprotein.
tential IgM-related conditions may be helpful to further refine the incidence of symptomatic disease
The prior literature contains limited information regarding the flow cytometric findings in IgM MGUS, primarily focusing on the number of total B cells and plasma cells identified rather than the characteristics of clonal B-cell or plasma cell populations.26,27 Using a complex gating strategy involving 17 different antigens, Pavia et al. reported clonal B-cell populations detectable in 15 of 20 (75%) MGUS patients.28 In this series, clonal B-cells were identified in 43 of 157 (27%) patients using a standard clinical flow cytometry approach, including 13 cases positive for CD5 and 30 cases negative for CD5. Importantly, in all cases of CD5-negative clonal B-cell populations, the light chain identified by flow cytometry matched the monoclonal protein’s light chain. In contrast, for cases with CD5-positive B-cell clones by flow cytometry, the light chain on B cells matched the monoclonal protein in only seven of 13 (54%) cases. This observation suggests that at least many CD5 positive clonal B-cell proliferations detected in IgM MGUS are unrelated to the monoclonal
protein. In such cases, a diagnosis of concurrent IgM MGUS and monoclonal B lymphocytosis may be appropriate. Definitively evaluating the relationship between a CD5-positive B-cell population and clonal plasma cells expressing the same light chain would require molecular studies on sorted B-cell and plasma cell populations and such studies are rarely if ever available in routine clinical practice. In this situation, the possibility that the CD5positive B-cell population is unrelated to the paraprotein should be noted and staging studies are appropriate to rule out an overt CD5-positive B-cell lymphoproliferative disorder elsewhere.
CAD has been recently reported to show characteristic findings that distinguish these cases from LPL/IgM MGUS including lack of MYD88 L265P mutations and the presence of recurrent chromosomal trisomies of chromosomes 3, 12 and 18.16–18 CAD was identified in ten of 191 (5%) cases in this cohort. These patients showed a lower hemoglobin level (P=0.007) and a trend towards more frequent lymphoid aggregates (P=0.077) compared to other cases of IgM MGUS and MYD88 L265P was absent in all
tested CAD cases. Our results confirm the distinctive nature of CAD compared to IgM MGUS and support the approach of the 2022 ICC system which separately recognizes primary CAD as a distinct lymphoproliferative disorder.10 FISH studies for chromosomal trisomies were not performed as part of this study, and additional studies are warranted to determine whether cases of CAD may be more similar to cases of marginal zone lymphoma than they are to cases of LPL/IgM MGUS. After exclusion of CAD, we compared the findings in the 181 patients with IgM MGUS with (n=72) or without (n=109) IgM-RD. These cases were similar in terms of the serum IgM concentrations, presence of lymphoid aggregates, clonal B-cell populations by flow cytometry, and clonal plasma cells by IHC. We also saw no differences in overall survival in patients with or without IgM-RD, although the follow-up data was limited for this cohort. These findings suggest the symptomatology in patients with IgM-RD is a function of the specificity of the IgM antibody produced rather than a result of distinctive underlying lymphoproliferative disorders in the bone marrow. This data supports the approach of the 2022 ICC system which does not recognize IgM-RD as a distinct disease, but instead encourages the use of terms such as “monoclonal gammopathy of clinical significance” as descriptors which may be appended to a pathologic diagnosis.10 Intriguingly, however, the MYD88 L265P mutation was detected in a higher proportion of patients with IgM-RD compared to those without IgM-RD (P=0.011). It is unclear whether this result may reflect a quantitative distinction, i.e., a lower burden of clonal cells in IgM MGUS without IgM-RD such that the MYD88 mutations may not be detectable within the limits of the allele specific assay employed, or whether this may
Figure 4. Overall survival of immunglobulin
M monoclonal gammopathy of unknown significance patients with or without immunglobulin M-related disorders. MGUS: monoclonal gammopathy of unknown significance; IgM: immunoglobulin M; RD: related disorders.
reflect a qualitative distinction with MYD88 wild-type MGUS cases being less likely to develop these specific forms of IgM-RD. Additional studies on sorted populations of B cells and plasma cells may be necessary to further examine this phenomenon.

In the 2022 ICC system, two subtypes of IgM MGUS were recognized.10 IgM MGUS of plasma cell type, the presumed precursor of IgM myeloma, is defined as cases with myeloma-associated cytogenetic abnormalities or as cases with clonal plasma cells but lacking clonal B cells by flow cytometry and lacking MYD88 mutations. IgM MGUS, NOS, on the other hand, encompasses all other cases of IgM MGUS which are presumed to progress to LPL or other lymphoproliferative disorder rather than myeloma. In an effort to understand the frequency and signi ficance of these subsets, we compared cases with and without clonal B cells and/or plasma cells. Among cases with clonal plasma cells but no clonal B cells, MYD88 L265P was detected in all cases tested (8/8, 100%). Similarly, none of the 59 cases of IgM MGUS tested by FISH showed an IGH::CCND1 rearrangement or other myeloma-associated IGH translocation. While our data is limited due to MYD88 and FISH testing being performed in only a subset of patients, the available data suggests that IgM MGUS of plasma cell type is very uncommon and no cases of this subtype were identified in this cohort.
The diagnostic criteria for distinguishing IgM MGUS from LPL have been controversial. In the 2016 WHO system, a diagnosis of LPL requires a clonal lymphoplasmacytic infiltrate representing ≥10% of bone marrow cellularity, while cases showing less than 10% of an infiltrate may be classified as an IgM MGUS if other remaining criteria are met.8 In contrast, the International Workshop on Walden-
ström’s macroglobulinemia criteria for diagnosis of LPL includes cases with clonal lymphoplasmacytic infiltrates of any extent,9 and the 2022 ICC system defines LPL based on the presence of neoplastic lymphoplasmacytic aggregates at any percentage of bone marrow infiltration.10 The cohort of cases in this study was defined using 2016 WHO criteria because the 10% threshold is relatively easy to objectively apply, and because cases with ≥10% infiltration would fulfill criteria for LPL in 2016 WHO, 2022 ICC, and Waldenström Workshop criteria. Of the 191 cases, only three of these showed abnormal aggregates of clonal B cells and clonal plasma cells meeting criteria for LPL by 2022 ICC criteria or Waldenström Workshop criteria. This observation provides reassurance that most cases historically classified as IgM MGUS in recent years would still remain best classified as IgM MGUS in the recently published criteria of the 2022 ICC system. Nevertheless, lymphoid aggregates were identi fied on hematoxylin and eosin sections in 61 of 191 (32%) of the entire cohort, and a multiparameter approach including flow cytometry and immunohistochemistry is required to distinguish these relatively common reactive aggregates from cases diagnostic of LPL by 2022 ICC criteria.
In conclusion, this study has provided detailed clinicopathologic findings in a large series of IgM MGUS as defined by 2016 WHO criteria. The precise incidence of IgM-RD is difficult to determine as we did not assess for all conditions that may be secondary to an IgM paraprotein, leading to a likely underestimate of IgM-RD, while at the same time, as our institution represents a tertiary referral center, there may be referral bias leading to overrepresentation of symptomatic patients. Nevertheless,
1. Mouhieddine TH, Weeks LD, Ghobrial IM. Monoclonal gammopathy of undetermined significance. Blood. 2019;133(23):2484-2494.
2. Go RS, Rajkumar SV. How I manage monoclonal gammopathy of undetermined significance. Blood. 2018;131(2):163-173.
3. Ho M, Patel A, Goh CY, et al. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Leukemia. 2020;34(12):3111-3125.
4. Seth S, Zanwar S, Vu L, et al. Monoclonal gammopathy of undetermined significance: current concepts and future prospects. Curr Hematol Malig Rep. 2020;15(2):45-55.
5. Castillo JJ, Jurczyszyn A, Brozova L, et al. IgM myeloma: a multicenter retrospective study of 134 patients. Am J Hematol. 2017;92(8):746-751.
6. Feyler S, O’Connor SJM, Rawstron AC, et al. IgM myeloma: a rare entity characterized by a CD20-CD56-CD117immunophenotype and the t(11;14). Br J Haematol. 2008;140(5):547-551.
7. Lu H, Durkin L, Zhao X, et al. IgM plasma cell myeloma. Am J Clin Pathol. 2022;157(1):47-53.
this study shows that IgM-RD, defined here as neuropathy, cryoglobulinemia or CAD, is common. While cases of CAD show distinct features warranting their recognition as a separate clinicopathologic entity, other cases of IgM MGUS with and without IgM-RD are not clearly pathologically distinguishable. The presence of IgM-RD is therefore best designated as a clinical descriptor attached to the underlying pathologic diagnosis of IgM MGUS, rather than being recognized as a distinctive entity. Finally, IgM MGUS of plasma cell type, defined by the recent 2022 ICC system as the putative precursor of IgM myeloma, is rare with no cases of that subtype identified in this cohort.
No conflicts of interest to disclose.
JC and MN designed the study. FB, PM, EB and MN collected data. FB, PM, MN, and JC analyzed and interpreted the data. FB wrote the draft. All authors provided important scientific insights, critically revised and edited the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by an internal research grant from the Robert J. Tomsich Pathology & Laboratory Medicine Institute, Cleveland Clinic, Cleveland, OH, USA.
Data-sharing statement
Data are available from the corresponding author upon reasonable request.
8. Cook JR, Harris NL, Swerdlow SH, et al. IgM monoclonal gammopathy of undetermined significance. In: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Fourth Edition. World Health Organization Classification of Tumours. International Agency for Research on Cancer. 2017:236.
9. Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30(2):110-115.
10. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood. 2023;141(4):437.
11. Fermand JP, Bridoux F, Dispenzieri A, et al. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. 2018;132(14):1478-1485.
12. Dispenzieri A. Monoclonal gammopathies of clinical significance. Hematology. 2020;2020(1):380-388.
13. Moreno DF, Paz S, Mena MP, et al. Prognostic impact of MYD88 L265P mutation by droplet digital PCR in IgM MGUS and
smoldering Waldenström macroglobulinemia. Blood. 2021;138(Suppl 1):S462.
14. Leung N, Bridoux F, Batuman V, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15(1):45-59.
15. Leung N, Bridoux F, Nasr SH. Monoclonal gammopathy of renal significance. N Engl J Med. 2021;384(20):1931-1941.
16. Randen U, Trøen G, Tierens A, et al. Primary cold agglutininassociated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica. 2014;99(3):497-504.
17. Małecka A, Trøen G, Tierens A, et al. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br J Haematol. 2018;183(5):838-842.
18. Małecka A, Delabie J, Østlie I, et al. Cold agglutinin-associated B-cell lymphoproliferative disease shows highly recurrent gains of chromosome 3 and 12 or 18. Blood Adv. 2020;4(6):993-996.
19. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-548.
20. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
21. Khwaja J, D’Sa S, Minnema MC, et al. IgM monoclonal gammopathies of clinical significance: diagnosis and
management. Haematologica. 2022;107(9):2037-2050.
22. Girard LP, Soekojo CY, Ooi M, et al. Immunoglobulin M paraproteinaemias. Cancers (Basel). 2020;12(6):1688.
23. Nobile-Orazio E, Barbieri S, Baldini L, et al. Peripheral neuropathy in monoclonal gammopathy of undetermined significance: prevalence and immunopathogenetic studies. Acta Neurol Scand. 1992;85(6):383-390.
24. Talamo G, Mir MA, Pandey MK, et al. IgM MGUS associated with anti-MAG neuropathy: a single institution experience. Ann Hematol. 2015;94(6):1011-1016.
25. Girard LP, Soekojo CY, Ooi M, et al. Immunoglobulin M monoclonal gammopathies of clinical significance. Front Oncol. 2022;12:905484.
26. Paiva B, Montes MC, García-Sanz R, et al. Multiparameter flow cytometry for the identification of the Waldenström’s clone in IgM-MGUS and Waldenström’s Macroglobulinemia: new criteria for differential diagnosis and risk stratification. Leukemia. 2014;28(1):166-173.
27. Ocio EM, del Carpio D, Caballero Á, et al. Differential diagnosis of IgM MGUS and WM according to B-lymphoid infiltration by morphology and flow cytometry. Clin Lymphoma Myeloma Leuk. 2011;11(1):93-95.
28. Paiva B, Corchete LA, Vidriales MB, et al. The cellular origin and malignant transformation of Waldenström macroglobulinemia. Blood. 2015;125(15):2370-2380.
Alexis Talbot,1* Arthur Bobin,2* Léa Tabone,3 Jérôme Lambert,4 Catherine Boccaccio,3 Cécile Deal,3 Marie-Odile Petillon,5 Olivier Allangba,6 Philippe Agape,7 Pierre Arnautou,8 Rakiba
Belkhir,9 Sylvie Cailleres,10 Driss Chaoui,11 Marie-Lorraine Chrétien,12 Olivier Decaux,13
Samantha Schulmann,14 Laurent Frenzel,15 Lauris Gastaud,16 Antoine Huart,17 Cyrille Hulin,18
Lionel Karlin,19 Kamel Laribi,20 Ronan Le Calloch,21 Pascal Lenain,22 Margaret Macro,23
Salomon Manier,24 Lydia Montes,25 Stéphane Moreau,26 Philippe Moreau,27 Véronique
Morel,28 James Norwood,29 Frédérique Orsini Piocelle,30 Aurore Perrot,31 Gian Matteo Pica,32
Philippe Rey,33 Anna Schmitt,34 Anne-Marie Stoppa,35 Mourad Tiab,36 Cyrille Touzeau,27
Valérie Vidal,37 Marguerite Vignon,38 Laure Vincent,39 Zoé Van De Wyngaert,40 Charles
Zarnitsky,41 Naima Kerbouche,42 Prani Paka,43° Xavier Leleu,2 Bertrand Arnulf1 and Hervé Avet-Loiseau44
1Hôpital Saint Louis, APHP, Immuno-Hématologie, INSERM U976, équipe 5, Paris, France; 2CHU de Poitiers, Service d'Hématologie et Thérapie Cellulaire, CIC U1402, Poitiers, France; 3IFM Intergroupe Francophone du Myélome, Paris, France; 4ECSTRA, Centre de Recherche en Epidémiologie et Statistiques, INSERM UMR 1153, Paris, France; 5Hôpital Claude Huriez, Hématologie, Lille, France; 6Centre Hospitalier Yves Le Foll, HématologieOncologie, Saint Brieuc, France; 7Centre Hospitalier de Laval, Hématologie, Laval, France; 8Hôpital d'Instruction des Armées Percy, Hématologie, Clamart, France; 9APHP Bicêtre, Rhumatologie, Kremlin-Bicetre, France; 10Centre Hospitalier du Pays d'Aix, Service Hématologie Oncologie, Aix-en-Provence, France; 11Centre Hospitalier Victor Dupouy, Hématologie, Argenteuil, France; 12CHU Dijon Bourgogne, Hématologie, Dijon, France; 13CHRU Hôpital de Pontchaillou, Hématologie, Rennes, France; 14HRU Hôpitaux de Brabois, Hématologie, Nancy, France; 15PHP, Hôpital Universitaire Necker Enfants Malades, Hématologie Adultes, Paris, France; 16Centre Antoine Lacassagne, Service OncoHématologie, Nice, France; 17CHU Toulouse - Hôpital de Rangueil, Néphrologie, Toulouse, France; 18CHU Bordeaux, Hématologie et Thérapie Cellulaire, Bordeaux, France; 19Centre 2774 Hospitalier Lyon Sud, Hématologie, Lyon Sud, France; 20Centre Hospitalier du Mans, Hématologie clinique, Le Mans, France; 21Centre Hospitalier de Quimper Cornouaille, Service d’Hématologie, Quimper, France; 22Centre Henri Becquerel, Hématologie, Rouen, France; 23CHU Caen, Hématologie, Caen, France; 24CHRU Hôpital Claude Huriez, Maladie du Sang, Lille, France; 25CHU Amiens, Hématologie Clinique, Amiens, France; 26CHU de Limoges, Hématologie Clinique et Thérapie Cellulaire, Limoges, France; 27CHU de NantesHôtel Dieu, Hématologie Clinique, Nantes, France; 28APHP Pitié-Salpêtrière, Hématologie Clinique, Paris, France; 29Hôpital du Scorff, Hématologie, Lorient, France; 30CH Annecy Genevois, Hématologie, Annecy, France; 31CHU de Toulouse, IUCT-O, Service Hématologie, Université de Toulouse UPS, Toulouse, France; 32Centre Hospitalier Métropole Savoie, Hématologie, Chambéry, France; 33Centre Léon Bérard, Onco-Hématologie, Lyon CAC, France; 34Bordeaux Unicancer, Onco-Hématologie, Bordeaux, France; 35IPC Unicancer Marseille, Hématologie, Marseille, France; 36CHD Vendée, Médecine Interne, La Roche-surYon, France; 37APHP Avicenne, Hématologie Clinique, Paris, France; 38APHP Cochin, Hématologie Clinique, Paris, France; 39CHU Montpellier - Hôpital Saint Eloi, Hématologie, Montpellier, France; 40APHP Saint Antoine, Hématologie Clinique et Thérapie Cellulaire, Paris, France; 41Hôpital Jacques Monod, Rhumatologie, Le Havre, France; 42GlaxoSmithKline, GlaxoSmithKline, Rueil Malmaison, France; 43GlaxoSmithKline, Philadelphia, PA, USA and 44UC-T Oncopole, Unité de Génomique du Myélome, Toulouse, France
*AT and AB contributed equally as co-first authors.
°Current address: Janssen Pharmaceuticals , Somerville, NJ, USA.
Correspondence: A. Talbot alexis.talbot.fr@gmail.com
Received: October 14, 2022.
Accepted: April 11, 2023.
Early view: April 20, 2023.
htps://doi.org/10.3324/haematol.2022.281772
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Belantamab mafodotin (BM) is an anti-BCMA antibody-drug conjugate (GSK2857916) that represents an alternative option in multiple myeloma. We sought to assess the efficacy and safety of BM in a real-world setting in patients who benefited from an early access program. We conducted an observational, retrospective, multicenter study. Eligibility criteria were treatment of relapsed or refractory multiple myeloma (RRMM) in monotherapy in adult patients who have received at least three lines of therapy previously, including at least one immunomodulatory agent (IMiD), a proteasome inhibitor (PI) and an anti-CD38 monoclonal antibody, and whose disease progressed during the last treatment period. The primary endpoint of the study is to assess the overall survival (OS). Between November 2019 and December 2020, 106 patients were treated with BM; 97 were eligible for the efficacy evaluation and 104 for safety. The median age was 66 (range, 37–82) years. High-risk cytogenetics were identified in 40.9% of patients. Fifty-five (56.7%) patients were triple-class refractory and 11 (11.3%) were penta-class refractory. The median number of prior lines of treatment was five (range, 3–12). The median number of BM cycles administered was three (range, 1–22). The overall response rate at best response was 38.1% (37/97). The median OS was 9.3 months (95% confidence interval [CI]: 5.9-15.3), and median progression-free survival was 3.5 months (95% CI: 1.9-4.7). The median duration of response was 9 months (range, 4.65-10.4). Treatment was delayed for 55 (52.9%) patients including 36.5% for treatment-related toxicity. Ophthalmic adverse events, mainly grade ≤2, were the most common toxicity (48%). The occurrence of keratopathy was 37.5%. Overall, our data are concordant with the results from DREAMM-2 in terms of efficacy and safety on a non-biased population.
Effective and safe novel therapies are needed for the treatment of patients with relapsed or refractory multiple myeloma (RRMM). The course of the disease consists in multiple relapses, as MM is still not curable. The treatment of RRMM is particularly challenging after the use of the three principal MM drug classes: immunomodulatory drugs (IMiD), proteasome inhibitors (PI) and anti-CD38 monoclonal antibodies (mAb). Even though the range of options has widened in recent years, especially with the emergence of immunotherapy, RRMM still has a poor prognosis, and, therefore, new drugs with innovative mechanisms are necessary.
Belantamab mafodotin (GSK2857916) (BM) is a first-inclass IgG1 humanized anti-BCMA (B-cell maturation antigen) mAb conjugated to a cytotoxic agent, monomethyl auristatin F (MMAF), via a protease-resistant maleimidocaproyl linker. BM binds to BCMA on the surface of plasma cells (PC) and delivers MMAF directly into PC, thereby promoting apoptosis. BM has recently proven effective for heavily pretreated RRMM in the first-in-human phase I DREAMM-1 study, which led to a phase II study called DREAMM-2.1–4 This study was an open-label two-arm study for RRMM patients whose disease had progressed after ≥3 prior lines of treatment and who were refractory to IMiD, PI and anti-CD38 mAb. In DREAMM-2, 196 patients received single-agent intravenous BM at either 2.5 mg/kg (97 patients) or 3.4 mg/kg (99 patients) for a 3-week cycle until disease progression or unacceptable toxicity. BM demonstrated a promising anti-tumor activity with an overall response rate (ORR) of 31% (97.5%; 95% confidence interval [CI]: 20.8–42.6) in the 2.5 mg/kg cohort and 34% (97.5%;
95% CI: 23.9–46.0) in the 3.4 mg/kg cohort. After a median follow-up of 6 months, the median progression-free survival (PFS) was 2.9 months (95% CI: 2.1–3.7) and 4.9 months (range, 2.3–6.2) in the two groups, respectively. Regarding tolerance, the main toxicity was corneal toxicity, with one quarter of patients exhibiting grade 3 or 4 disorders. After considering the benefit-risk balance, the 2.5 mg/kg dose was recommended for clinical use.
Based on these results, temporary utilization of BM was approved in France by the French National Agency for the Safety of Medicine and Health Products (ANSM) under an ATU (“autorisation temporaire d’utilisation”) format, thus providing early access to BM for RRMM patients. We designed a retrospective study using data from this temporary utilization, so-called “nominative ATU”, to further explore the efficacy and safety of BM in RRMM in a real-life context.
This study (IFM 2020-04) was an observational, retrospective, multicenter study that enrolled patients treated with BM as part of the ATU program in France from when the authorization began on September 17, 2019. Eligible patients had received at least one cycle of BM (GSK2857916) monotherapy whose indication as part of the ATU was the treatment of RRMM in adult patients who had received at least three lines of therapy previously, including at least one IMiD, a PI and an anti-CD38 mAb, and whose disease progressed during the last treatment period. The trial was sponsored by the French group IFM (“Intergroupe Francophone du Myélome”) and supported by GSK. This study was
conducted in full compliance with the principles of the Declaration of Helsinki with non-opposition of the patients and complies with the regulations in force in France for this type of clinical investigation on patients.
The primary outcome was to evaluate the efficacy and safety of BM (GSK2857916) in a real-world context as part of the nominative ATU for patients with RRMM who had received at least three previous lines of therapy. The safety population includes all patients who received at least one cycle of BM. The efficacy population includes all patients of the safety population with the evaluation of the main criterion (non-missing status: alive or dead at the last news) and satisfying inclusion criteria. The primary goal of the study was to assess the overall survival (OS) of patients following BM treatment. The secondary goals were to analyze real-world efficacy of BM based on the response rate (overall response rate [ORR], according to IMWG definition5), the time to response and duration of response (DOR), to proceed to a survival analysis with the progression-free survival (PFS) and event-free survival (EFS, calculated as the time from the first administration treatment to the date of disease progression event or permanent treatment discontinuation or death, whichever occurred earlier), to determine the time-to-next treatment (TTNT) defined as the time from first administration of BM to the date of the new line of treatment or death, time to progression (TTP), time to discontinuation and to focus on specific subset of patients such as high-risk (HR) or frail

patients, and toxicity (according to NCI-CTC V5.0 criteria) taking into account the results of the DREAMM-2 phase II clinical trial. HR patients were displaying adverse cytogenetics features such as del(17p), t(4;14), del(1p32) or 1q gain.
Quantitative variables will be described in terms of extreme values, quartiles, median, or mean and standard deviation for data with a normal distribution (95% CI). Qualitative variables will be described in terms of headcount and proportions (with a 95% CI, binomial law). Survival data (PFS and OS) are estimated using the Kaplan-Meier method. Median survival times with a 95% CI and median follow-up are estimated using the inverted Kaplan-Meier method. The duration of response is described in responding subjects using descriptive statistics (median, range). The database is reported in compliance with the General Data Protection Regulation (GDPR).
The IFM 2020-04 study enrolled 106 patients overall. Patients were treated between November 2019 and December 2020. The safety and efficacy populations were composed of 104 and 97 patients, respectively (Figure 1). Baseline characteristics of the patients are presented in Table 1. The median age at diagnosis was 66 years (range, 7–82), and 49 patients (50.5%) were male. The median time
ISS: international staging system; ECOG: eastern cooperative oncology group; BM: belantamab mafodotin; GFR: glomerular filtration rate.
from MM diagnosis was 6.37 years (range, 1.19–19.61), 57 of 82 (69.5%) patients were fit as defined by Eastern Cooperative Oncology Group (ECOG) performance score ≤1, and three of 82 (3.9%) were frail as defined by ECOG ≥2 and ≥75 years old at treatment initiation. Concerning MM disease, 27 of 66 (40.9%) patients had HR cytogenetics and 13 of 93 (14%) had an extra-medullary disease (EMD). Patients had received a median of five (range, 3–12) prior lines of MM treatment. Regarding the treatments before BM, 55 (56.7%) patients were triple-class refractory (IMiD, PI, antiCD38 mAb) and 11 (11.3%) were refractory to bortezomib, lenalidomide, carfilzomib, pomalidomide, and daratumumab (pentarefractory). Besides, 70 (72.2%) patients under-
went prior autologous stem cell transplant (ASCT). Based on the safety population (data from 100/104 patients), the treatment criteria were a biological relapse for 44 patients (44.0%), a clinical relapse for 29 (29.0%), and both a biological relapse and a clinical relapse for 27 (27.0%).
The median number of BM cycles administered was three (range, 1–22). The ORR at best response was 38.1% (37/97). The best response rates are summarized in Table 2. At 12±3 weeks, ORR was 35.6 % (26/73), including five patients (6.8%) with a complete response (CR), seven (9.6%) with a very good partial response (VGPR), and 14 (19.2%) with a partial response (PR), and eight (11.0%) patients had a stable disease (SD). The median time to achieve the best response was 2.05 months (range, 0–14.03). The median duration of response (DOR) was 9 months (range, 4.6510.4). The DOR according to the best response is shown in Figure 2. The median TTNT was 4.25 months (95% CI: 3.176.6). The estimated incidence of treatment response was 21.8% at 3 months, 56.2% at 6 months, and 57.8% at 1 year.
The median OS was 9.3 months (95% CI: 5.9-15.3) (Figure 3A), and median PFS was 3.5 months (95% CI: 1.9-4.7) (Figure 3B). The median EFS was 2.4 months (range, 1.4–3.5). Similarly, median OS for both high-risk (n=27/66) and standard-risk (n=39/66) patients was 9.2 months. The median OS was 14 months in patients with EMD (n=13/93), and 9.1 months in patients without EMD (n=80/93) (hazard ratio [HR]=1.04, 95% CI: 0.49-2.20). The median OS was 8.5 months in patients previously treated by less than 6 lines (n=51) and 10.2 months in patients previously treated by ≥6 lines (n=46) (HR=0.80, 95% CI: 0.47-1.35). The median OS was 16.8 months in fit patients and 6 months in unfit patients (HR=2.16, 95% CI: 1.18-3.95); two-tailed P=0.0103) (Online Supplementary Table S1). Kaplan-Meier curves of OS and PFS according to the best responses are presented in Figure 4. The median PFS was 2.7 months (95% CI: 1.6-4.3) in patients with triple-class refractory MM and 3.3 months (95% CI: 1.4-26.0) in patients without triple-class refractory MM (two-tailed P=0.7686). The estimated incidence of disease progression was 30.4% at 3 months, 47.8% at 6
months and 67.6% at 1 year. At data cut-off (March 10th, 2022), 69 (67%) patients in the cohort had passed away; for 47 (68%) this was due to disease progression, for 12 (17%) it was due to infection and for two (3.0%) it was due to both progression and infection.
BM was administered every 21 days intravenously in monotherapy. The treatment was stopped for 55 (52.9%) patients, comprising 38 (36.5%) for treatment-related toxicity, and 14 of them experienced a treatment inter-

ruption more than once. The median duration of BM treatment was 1.4 months and patients received a median of three treatment cycles (range, 1-22). The median time for the first interruption since the start of BM was 1.6 months (range, 0–13.3). For 42 patients, the treatment was restarted after a median time of interruption of 29 days (range, 1–300). Dose reductions were reported for 23 (22.1%) patients, mostly from 2.5 mg/kg to 1.9 mg/kg, as the first dose reduction. The minimal dose used was 1.4 mg/kg for one patient. Ophthalmic toxicity was observed for 49 of 102 (48%) patients (Table 3).
 Of the patients who
Of the patients who
experienced ophthalmic toxicity, the keratopathy percentage was 81,3% (n=39/48) and the percentage of reduced visual acuity was 4.8%. Ophthalmic toxicity was mainly grade ≤2 (55%) and no grade 4 was observed. Ocular events resulted in delayed treatment administration or in a dose reduction in 30% of concerned patients. Premedication before BM was used for 91.3% of patients and 64 (61.5%) patients received premedication with eye drops to limit ocular toxicity before the infusion of BM. Besides, 16 (15.4%) patients developed grade 3 or 4 hematologic toxicity. Hematologic toxicity was principally marked by thrombocytopenia (43.8%). Grade 3 or 4 non-hematologic toxicity was reported in nine (8.7%) patients. Ten (9.6%) patients had infusion-related reactions (IRR) of any grade (80% of grade ≤2), mostly at the first two cycles. One pa-

tient had a grade 3 bronchospasm at cycle 6. Of note, nearly 40% of patients received paracetamol or antihistaminic agent before BM was administered.
The clinical course of MM is characterized by multiple relapses and finally patients lack treatment options and therefore their survival is impacted.6 The BCMA was recently found as a promising target for the development of naïve and armed immunotherapies, which extended the options in the relapse setting. Currently, several antiBCMA therapies are emerging and being investigated, including bispecific antibodies (bsAb), chimeric antigen receptor T (CAR T) cells, and antibody-drug conjugates (ADC). Belantamab mafodotin proved effective in the phase II DREAMM-2 trial. Based on the data from this trial, a temporary utilization program (ATU program) in France was authorized for patients with RRMM to benefit to an early access to the molecule.
Our real-life cohort included 106 patients, with 97 in the efficacy population which is the same number as in the DREAMM-2 2.5 mg/kg cohort. We present the highest percentage of responding patients compared to the literature (ORR 38.1%). Survival rates are also notable with a median OS of 9 months and median PFS of 3.5, which is in line with previous studies on BM. As a comparison, the last line of treatment before BM was administered for five cycles in median and the best response rate were: seven CR (7.8%); 17 VGPR (18.9%); 27 PR (30.0%), and 39 SD (43.3%), ORR at 56.7%. In the best response related to the next treatment following BM (n=36) 23 patients (63.9%) had stable disease, seven PR (19.4%), three VGPR (8.3%)
and three CR (8.4%), ORR was 36.1%. Moreover, the toxicity profile of BM was similar to the previous studies that investigated it, as it essentially consisted of ophthalmological and hematological adverse events (AE). The keratopathy rate was 37% and mainly low grade. Ocular toxicity was reversible, as the median duration of delay was 29 days, which means in “real-life” the management of ophthalmological AE seems manageable. We did not report any unexpected AE compared to other BM studies.3
The better response rates might be explained by the lower number of previous line (median of 5 prior lines) in comparison to the DREAMM-2 (median of 7 prior lines) cohort and another real world study from the mayo clinic (median of 8 prior lines).8 Table 4 compares the different variables from our study, DREAMM-2 and the Mayo Clinic study. One major difference is that in our study, not all patients (55%) were triple-class refractory (refractory to an IMiD, a PI, and an anti-CD38). Patients had to be exposed to all three classes to benefit from the ATU program. Even though patients from our study had better responses, the median OS is longer in the extended 13month follow-up of DREAMM-2. Interestingly, in subgroup analysis, no significant difference was observed in
terms of OS, PFS and TTNT for patients with extramedullary disease (Online Supplementary Table S1; Online Supplementary Figure S1) as well as with ORR (Online Supplementary Figure S2). In addition, the keratopathy rate was nearly similar to the real-life one reported by the Mayo clinic and were both lower than in DREAMM-2. We can hypothesize either that AE were less reported than in a clinical trial or that the management of ocular toxicity has improved since DREAMM-2.
Anti-BCMA treatments are increasingly being used for patients with RRMM, as most of patients will eventually be exposed and refractory to anti-CD38 mAb.6 A novel target was, therefore, needed for advanced patients so that they can still benefit from immunotherapy.9 Currently, innovative immunotherapies targeting BCMA include CAR T cells, bsAb and ADC.10–14 Even though CAR T and bsAb might seem more appealing because of their efficacy,15 BM still represent an alternative anti-BCMA treatment. Nonimmunologic agents, although still necessary, are usually associated with toxicities such as selinexor16,17 or melflufen,18 whose development has been put on hold, and therefore immune-based treatment such as BM offer innovative strategies. To contextualize median OS of 9.3 months, we found a median OS of about 3-4 months for
autologous stem cell transplant; OS: overall survival; PFS: progression-free survival; ORR: overall response rate; CR: complete response; VGPR: very good partial response; PR: partial response.
MAMMOTH-like MM patients in a French national healthcare database OS study.6,19 BM represents a promising alternative option for these patients with its unique mechanism of action for the delivery of the cytotoxic agent directly into MM cells after binding BCMA. Real-world patients are not those selected according to the strict inclusion and exclusion criteria for a clinical trial. Most fragile patients with multiple comorbidities and particularly aggressive diseases are generally not included in the trials. The results of the trials, therefore, have strong internal validity, but cannot be extrapolated for the wider and clinically more heterogeneous population. Reallife data, though not collected in an international setting, are now considered a major issue to confirm or refute the outcome of patients actually exposed to the treatment. We note several limitations of this study. It is limited by its retrospective nature, and, therefore, some data are lacking for several patients. Nevertheless, we present the largest real-life cohort of patients, which is comparable to the DREAMM-2 study. No exclusion criteria were defined, and, therefore, this study is composed of patients who might have presented with renal failure and with an impaired performance status.
Overall, the results of our retrospective real-life study are concordant with the previously reported results of the phase II clinical trial DREAMM-2 in terms of response, survival, and toxicities. Although the ophthalmological issues are a concern,20–22 BM remains an alternative option for previously highly treated patients that can thus benefit from an anti-BCMA therapy in the late course of disease. We expect that the management of AE will improve with increased use of the drug.23
Disclosures
AP is part of the advisory board of AbbVie, Amgen, BMS/Celgene, GSK, Janssen, Pfizer, Sanofi and Takeda. LG is part of the advisory board of Abbvie, Pfizer, BMS, GSK
1. Trudel S, Lendvai N, Popat R, et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019;9(4):37.
2. Trudel S, Lendvai N, Popat R, et al. Targeting B-cell maturation antigen with GSK2857916 antibody–drug conjugate in relapsed or refractory multiple myeloma (BMA117159): a dose escalation and expansion phase 1 trial. Lancet Oncol. 2018;19(12):1641-1653.
3. Lonial S, Lee HC, Badros A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a twoarm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21(2):207-221.
4. Lonial S, Lee HC, Badros A, et al. Longer term outcomes with single-agent belantamab mafodotin in patients with relapsed or refractory multiple myeloma: 13-month follow-up from the
and Immunocore. LK is part of the advisory board of AbbVie, Janssen, Novartis and Takeda; received personal fees from AbbVie, AstraZeneca, BeiGene, iQone, Novartis, Seagen and Takeda. PM is part of the advisory board and received honoraria from Janssen, Celgene, Amgen, Takeda, Sanofi and Abbvie. RLC is part of the advisory board of Takeda and Abbvie; acts as a consultant for Takeda, Abbvie and Gilead. RP is part of the advisory board of GSK. XL received honoraria from Janssen, Bms, Novartis, Amgen, Sanofi, Takeda, Pfizer, GSK and Roche.
AT, AB, XL, BA and HAL developed the concept and designed the study. AT, AB, LT, CB, CD, MOP, OA, PA, PA, RB, SC, DC, MLC, OD, PF, LF, LG, AH, CH, LK, KL, RLC, PL, MM, SM, JPM, SM, PM, VM, JM, FOP, AP, GMP, PR, AS, AMS, PT, CT, VV, MV, LV, ZVDW, CZ, XL, BA and HAL collected and assembled the data. AT, AB, LT and HAL analyzed and interpreted the data. AT, AB, LT and HAL wrote the article. All authors read and approved the final version of this manuscript.
The authors wish to thank the patients and families, the centers that participated to the study, the IFM teams and the statistical team (Statitec - MultiHealth Group).
The trial was sponsored by the French group IFM and supported by GSK.
The original contributions presented in the study are included in the article and its Online Supplementary Appendix. Further inquiries can be directed to the corresponding author.
pivotal DREAMM-2 study. Cancer. 2021;127(22):4198-4212.
5. Kumar S, Paiva B, Anderson KC, et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17(8):e328-e346.
6. Gandhi UH, Cornell RF, Lakshman A, et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33(9):2266-2275.
7. Lassiter G, Bergeron C, Guedry R, et al. Belantamab mafodotin to treat multiple myeloma: A comprehensive review of disease, drug efficacy and side effects. Curr Oncol. 2021;28(1):640-660.
8. Vaxman I, Abeykoon JP, Dispenzieri A, et al. “Real-life” data of the efficacy and safety of belantamab mafodotin in relapsed multiple myeloma- the Mayo Clinic experience. Blood. 2021;11(12):196.
9. Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34(4):985-1005.
10. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 openlabel study. Lancet. 2021;398(10297):314-324.
11. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705-716.
12. Sebag M, Raje NS, Bahlis NJ, et al. Elranatamab (PF-06863135), a B-cell maturation antigen (BCMA) targeted CD3-engaging bispecific molecule, for patients with relapsed or refractory multiple myeloma: results from Magnetismm-1. Blood. 2022;387(6):495-505.
13. Zhao WH, Liu J, Wang BY, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141.
14. Moreau P, Garfall AL, van de Donk NWCJ, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495-505.
15. Rodriguez-Otero P, Ayers D, Cope S, et al. Matching adjusted indirect comparisons of efficacy outcomes for idecabtagene vicleucel (ide-cel, bb2121) versus selinexor + dexamethasone and belantamab mafodotin in relapsed and refractory multiple myeloma. Leuk Lymphoma. 2021;62(10):2482-2491.
16. Gavriatopoulou M, Chari A, Chen C, et al. Integrated safety profile of selinexor in multiple myeloma: experience from 437 patients enrolled in clinical trials. Leukemia.
2020;34(9):2430-2440.
17. Grosicki S, Simonova M, Spicka I, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-perweek bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open-label, phase 3 trial. Lancet. 2020;396(10262):1563-1573.
18. Richardson PG, Oriol A, Larocca A, et al. Melflufen and dexamethasone in heavily pretreated relapsed and refractory multiple myeloma. J Clin Oncol. 2021;39(7):757-767.
19. Touzeau C, Quignot N, Meng J, et al. Survival and treatment patterns of patients with relapsed or refractory multiple myeloma in France - a cohort study using the French National Healthcare database (SNDS). Ann Hematol 2021;100(7):1825-1836.
20. Farooq A V., Degli Esposti S, Popat R, et al. Corneal epithelial findings in patients with multiple myeloma treated with antibody–drug conjugate belantamab mafodotin in the pivotal, randomized, DREAMM-2 study. Ophthalmol Ther. 2020;9(4):889-911.
21. Popat R, Warcel D, O’Nions J, et al. Characterization of response and corneal events with extended follow-up after belantamab mafodotin (GSK2857916) monotherapy for patients with relapsed multiple myeloma: A case series from the first-timein-human clinical trial. Haematologica. 2020;105(5):e261-e263.
22. Patel S V., Joshi N, Thareja T, Jhanji V. Corneal epithelial toxicity induced by belantamab mafodotin. Clin Exp Ophthalmol. 2021;49(9):1113-1115.
23. Lonial S, Nooka AK, Thulasi P, et al. Management of belantamab mafodotin-associated corneal events in patients with relapsed or refractory multiple myeloma (RRMM). Blood Cancer J. 2021;11(5):103.
Alexandra Schifferli,1 Guillaume Moulis,2 Bertrand Godeau,3 Thierry Leblanc,4 Nathalie Aladjidi,5 Marc Michel,3 Guy Leverger,6 Mohsen Elalfy,7 John Grainger,8 Meera Chitlur,9 Andrea Heiri,1 Susanne Holzhauer,10 Gautier le Gavrian,1 Paul Imbach1 and Thomas Kühne1
1Department of Hematology/Oncology, University Children’s Hospital Basel, Basel, Switzerland; 2Service de Médecine Interne and Centre d’Investigation Clinique 1436, équipe PEPSS, Centre Hospitalier Universitaire de Toulouse, Toulouse, France; 3Department of Internal Medicine, National Reference Center for Adult Immune Cytopenias, Henri Mondor University Hospital, Assistance Publique Hopitaux de Paris, Université Paris-Est Créteil, France; 4Department of Hematology, APHP-Robert Debré Hospital, Paris, France; 5Centre de Référence National des Cytopénies Autoimmunes de l’Enfant (CEREVANCE), Pediatric Hematologic Unit, Centre d’Investigation Clinique Plurithématique (CICP) INSERM 1401, University Hospital of Bordeaux, Bordeaux, France; 6Hôpital Armand-Trousseau AP-HP, Sorbonne Université, Service d’Hémato-oncologie Pédiatrique, Paris, France; 7Department of Pediatric Hematology/Oncology, Ain Shams University, Cairo, Egypt; 8Royal Manchester Children’s Hospital, Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Science Center, Manchester, UK; 9Carmen and Ann Adams
Department of Pediatrics, Division of Hematology/Oncology, Children’s Hospital of Michigan, Central Michigan University, Detroit, MI, USA and 10Department of Pediatric Hematology and Oncology, Charité, Berlin, Germany
Correspondence: A. Schifferli alexandra.schifferli@ukbb.ch
Received: December 13, 2022.
Accepted: April 4, 2023.
Early view: April 13, 2023.
htps://doi.org/10.3324/haematol.2022.282524
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Current immune thrombocytopenia (ITP) guidelines target children and adults, leading to oversimplification. Adolescents and young adults (AYAS) comprise a separate group with distinct health and psychosocial issues. This study aimed to describe the clinical presentation and therapeutic strategies of ITP among AYAS. We analyzed data from two large ITP registries (PARC-ITP; CARMEN-France) and included newly diagnosed ITP patients (aged 12–25 years) with an initial platelet counts of <100×109/L. Patients with secondary ITP or non-immune thrombocytopenia (n=57) and pregnant women (n=10) were excluded. Of the 656 cases of AYAS with primary ITP registered from 2004 up to 2021, 12-month follow-up data were available for 72%. The initial median platelet count was 12×109/L. In 109 patients (17%), the diagnosis was incidental, without documented bleeding. Apart from gynecological bleeding, the clinical and therapeutical characteristics of females and males were similar. Platelet-enhancing drugs were reported in 66%, 45%, and 30% of patients at diagnosis, 1–6 months, and 6–12 months after diagnosis, respectively. Corticosteroids were the preferred treatment at all time points. At 12 months, 50% of all patients developed chronic ITP. In the subgroup of patients with initial severe thrombocytopenia (<20×109/L), those receiving frontline treatment had a higher remission rate at 1 year than those who followed an initial watch-and-wait strategy (53% and 32%; P<0.05). Our analysis indicates that the remission rate at 1 year may be associated with the initial treatment strategy. This hypothesis must be confirmed in prospective studies.
Primary immune thrombocytopenia (ITP) is an autoimmune disease characterized by increased platelet destruction and impaired production. Most textbooks and practice guidelines describe pediatric ITP as acute and profound but self-limiting, with a low risk of life-threatening bleeding in the majority of patients.1-4 In contrast, adult ITP is a chronic disease with an insidious onset of bleeding that is sometimes diagnosed incidentally. Adult ITP carries a high risk of severe bleeding in patients with a platelet
count of <20–30×109/L, with increased morbidity and mortality, particularly in patients aged >60 years.5 Platelet-enhancing therapy is usually advised for adults with severe thrombocytopenia, especially the elderly or those with comorbidities.6,7 In contrast, a watch-and-wait strategy is recommended for children with non-severe bleeding.2,3 Recently, comparative studies in children and adults demonstrated unexpected similarities in the clinical and laboratory findings of both groups, particularly at disease onset, although differences in long-term prognosis and some aspects of patient management have been confirmed.8-10
Adolescents and young adults (AYAS) are recognized as a specific age group with symptoms, disease course, treatment goals, side effects, needs, and expectations that differ from children and adults. AYAS are of particular interest in oncology and transition medicine.11,12 Characteristic features of AYAS include a high disease burden and poor compliance. The disease and treatment may negatively affect their personal development.11,12 Some treatment difficulties in this age group can be partly attributed to their physiological development. Adolescents have rapid hormonal, behavioral, and social changes, all of which can potentially modify the disease or symptoms. There is no international consensus on the definition of AYAS. In the literature, age differs from 12–16 years at the lower and 25–40 years at the upper limit.11
AYAS are poorly studied in ITP-related literature13,14 and clinical studies in this specific age group are lacking. Recommendations for AYAS with ITP are usually not covered by practice guidelines; therefore, the institutions providing care guide treatment options, are often designed exclusively for children or adults. However, in most ITP studies, the median age of patient cohorts is >40–50 years for adults13,15 and <10 years for children. Thus, treatment goals and guidelines for AYAS are biased.
Many pediatric studies have revealed that children over the age of 10 years are at risk for chronic ITP.16 Therefore, it is reasonable to suppose that AYAS have a major risk of developing chronic ITP. However, assuming that the immune system of AYAS has a high potential to reverse immune dysregulation and restore self-tolerance, we hypothesize that therapeutic strategies involving immunomodulation should result in better long-term outcomes in these individuals than in adults. Most ITP treatment strategies currently focus on preventing premature platelet destruction (immunosuppression and splenectomy) and promoting platelet production (thrombopoietin-receptor agonist [TPO-RA] treatment).
It is unclear whether the initial presentation, risk of chronicity, and response to ITP treatment in AYAS are more similar to adults or children. It is important to understand the disease characteristics in AYAS to use appropriate diagnostic tools, treatment strategies, and goals. This study aimed to describe the clinical and laboratory presentation of ITP and its corresponding treatment approach for AYAS.
Data source
We extracted data of AYAS from the Pediatric and Adult Registry on Chronic ITP (PARC-ITP) registered between May 2004 and May 2021 and CARMEN-France registry (Cytopénies Auto-immunes Registre Midi-Pyrénéen) be-
tween January 2013 and May 2021. Data extraction from PARC-ITP and CARMEN registries was approved by the local Review Boards and national or regional Ethics Committees. Informed consent was obtained from all participants registered in PARC-ITP. All of the patients in the CARMEN-France registry were informed; no one opposed data collection. See the Online Supplementary Appendix for more information.
Inclusion criteria were: patients aged between 12 and 25 years at the time of diagnosis of primary ITP as defined by international guidelines, i.e., an initial platelet count of <100×109/L and no other conditions that may cause thrombocytopenia.4 Patients with a subsequent diagnosis of secondary ITP or of non-immune thrombocytopenia and pregnant women in the study period were excluded.
Treatment-free remission at 1-year FU was defined as a platelet count ≥100×109/L at the 12-month FU without treatment for 6 months before, around, or after this FU. This definition was chosen in order to ensure the trueness of remission and not to include patients with prolonged response to treatment, rather than real remission. Chronic disease was defined as a platelet count of <100×109/L at the 12-month FU or platelet count of 100×109/L under therapy at this time point. For patients with borderline platelet counts (100–149×109/L), all available FU were studied to identify patients with relapse and reclassify them as chronic disease. Accurate diagnoses of remission versus chronic ITP were achieved through a precise decision tree (Online Supplementary Figure S1). See the Online Supplementary Appendix for further details.
Initial clinical data and FU information up to 12 months were analyzed. Continuous variables are reported as medians with the corresponding interquartile range (IQR). We analyzed the different subgroups based on sex, age (adolescents, 12–18 years vs. young adults, 18–25 years), and remission status at 12 months. We performed a multivariable logistic regression analysis to predict remission using sex, age, initial platelet count, and initial treatment at diagnosis (first-line drugs vs. watch-and-wait approach) as predictors. We hypothesized that initial treatment depends on initial platelet counts, and this possible interaction was considered in the model. Additionally, continuous data (age and initial platelet count) were modeled non-linearly using a three-knot restricted cubic spline. Since the non-linear contribution was not significant, these predictors were modeled linearly. We also tested whether initial bleeding moderated the effect of the above predictors on remission after 1 year. Comparing the two models (the initial model and the model with initial bleeding as a covariate) with the likelihood ratio test
shows that adding initial bleeding does not improve the model fit (χ2 test value =0.99; P=0.32). Thus, initial bleeding was not included as a predictor. Odds ratios for selected initial platelet counts were reported with their corresponding 95% confidence interval (CI). For better understanding, the results of the logistic regression are presented as the probability of remission after 1 year. P values were calculated with the χ2 test , Fisher’s exact test, or the Mann–Whitney U test. Fisher´s exact test was used when the expected frequency of one or more cells was < 5. A P value <0.05 was significant. All analyses were performed using R version 4.1.2.

A total of 652 AYAS with initial diagnoses of primary ITP were recorded in the PARC-ITP and 71 in the CARMENFrench registries. Overall, 67 patients were excluded due to secondary ITP or non-immune thrombocytopenia (initially misdiagnosed) (n=57) or pregnancy (n=10). Finally, 656 AYAS with primary ITP were selected for analysis. The AYAS were predominantly females (61%), particularly in the 18–25-year subgroup (70%). However, clinical characteristics were similar among the sexes. Males and females shared similar initial diagnostic procedures, comorbidities, bleeding symptoms (other than gynecological bleeding), median platelet counts, percentages of severe thrombocytopenia (<20×109/L), treatment requirements (except for red blood cell transfusion), and drug
choices. Bone marrow puncture was performed at the time of diagnosis in 291 patients (44%), which supported the diagnosis of primary ITP. Comorbidities were reported in only 100 patients (15%), and two women but no men reported a thromboembolic event. At diagnosis, red blood cell transfusion rates differed between sexes (7% in females vs. < 1% in males). Females receiving a blood transfusion (n=27) had a mean hemoglobin of 7.5 g/dL, and 67% suffered active gynecological bleeding. Antiphospholipid antibodies were positive in 11 of 179 patients (7 females and 4 males, rate of positivity 6% for both). The initial positive results for ANA were higher in females (66/236; 28%) than in males (13/132; 10%). Similarly, the rate of reported thyroid disease was higher in females (2·2%) than in males (0.4%). The initial median platelet count for all AYAS was 12×109/L (interquartile range [IQR], 5-28). ITP was diagnosed as incidental (no reported bleedings) in 109 (17%) patients, whereas 538 (82%) suffered from bleeding symptoms at diagnosis; no information was provided for nine patients. AYAS without bleeding symptoms at diagnosis had a median platelet count of 38×109/L (IQR, 16-61) (Figure 1), whereas those with cutaneous and wet or internal bleeding had counts of 15×109/L (IQR, 7-26) and 8×109/L (IQR, 4-17), respectively. The initial clinical characteristics of patients are shown in Table 1. Intracranial hemorrhage occurred in four patients; two were diagnosed with ITP at the initial visit and two within the first 6 months of the disease. None of these patients reported hematuria, but it is unclear if microhematuria was tested. Their clinical descriptions and treatment approaches are
Figure 1. Occurrence of bleeding symptoms at diagnosis in patients with initial profound versus moderate thrombocytopenia. Median platelet count for patients without bleeding at initial diagnosis was 37.5×109/L and 11×109/L for patients with some bleeding symptoms (P<0.001).
shown in the Online Supplementary Table S1
FU data was available for 547 (83%) patients at 6 months and 470 (72%) at 12 months. The median platelet counts and the distribution of severity (severe, moderate, and mild thrombocytopenia) at 6 and 12 months of FU were similar. Platelet counts <100×109/L were present in 49% of patients at 6 months and 42% at 12 months. However, bleeding propensity and treatment requirements decreased equally (Table 1).
We found only minor clinical differences between adolescents (12–18 years; 59% girls) and young adults (18–25 years; 70% women), with young adults presenting more
mild-to-moderate thrombocytopenia (platelet count between 20–99×109/L) at diagnosis than adolescents (47% and 32%, respectively) and less initial bleeding (70% and 85%, respectively). However, the remission rate was identical in both age groups (57%). The higher median platelet count at 6 months FU in young adults compared to adolescents (114×109/L; IQR, 55-222 and 93×109/L, IQR 40-197, respectively) could be the result of a higher treatment rate at this time point (58% and 42%, respectively) (Table 2). Drug treatments were reported for 66% at diagnosis, 45% after initial treatment until 6 months, and 30% at 6–12 months after diagnosis. Corticosteroids were most fre -
FU: follow-up; IQR: interquartile range ; NK: not known; SD: standard deviation; IVIG: intravenous immunoglobulin. *Platelet counts are values recorded at diagnosis and follow-up visits. The platelet count at 6 months was defined as the value at 3–9 months; platelet count at 12 months was defined as the value at 11–18 months. ƒInformation about bleeding is clinical data between follow-up visits (6 or 12 months). †“Initial” treatment is the treatment lasting over a maximum of 4 weeks; “6-mo FU” treatment is the treatment lasting until 6 months, excluding initial therapy; “12mo FU” treatment is the treatment between 6 and 12 months. Patients with a combination of different corticosteroids are counted once. Patients treated with a combination of different second- or third-line treatments are counted once. Patients treated with a combination of different immunoglobulins are counted once. **Seven patients received recombinant thrombopoietin-receptor agonists, probably as part of a study (China, Egypt). ƒƒSplenectomy are only counted once during follow-up. At 12 months FU, in total 13 patients were splenectomized.
quently used at all time points. Among the treated patients, 74% received corticosteroids during the first 6 months (excluding initial treatment) and 68% between 6 and 12 months. Second-line treatments were heterogeneous and administered to 28% of patients receiving treatment beyond 6 months (Table 3).
At the 12-month FU 236 of 470 patients (50%) suffered from chronic disease, 38% were in remission, and 12% had an unclear status. Chronic disease at 12 months was identical for both sexes, and the median platelet count in chronic ITP was 57×109/L (IQR, 31-85). Asymptomatic chronic ITP (no bleeding between 6 and 12 months) characterized 109 of 236 (46%) patients with chronic disease, irrespective of the treatment strategy. At 12 months, 56
patients not undergoing treatment in the last 6 months had borderline platelet counts ranging from 100×109/L to 149×109/L. Of these, at least 16 (29%) had a relapse (platelet count <100×109/L) at a later date, with seven of 16 harboring platelets <50×109/L (available FU differed greatly in this group of patients).
The characteristics of patients in remission compared to those with chronic ITP are shown in Table 4. Overall, 23% of chronic ITP and 15% of patients in remission tested positive for ANA at their initial visit. However, this blood test was performed in only half of the cohort, and definition of positivity may show strong variation. An initial diagnosis of severe thrombocytopenia was more prevalent among patients in remission at 12 months than those
platelet count ×109/L, (IQR)*
% patients receiving coticosteroids
% patients receiving IVIG
% patients receiving second- or third- line** 6–12-month
%
FU: follow-up; IQR: interquartile range; IVIG: intravenous immunoglobulin; ANA: antinuclear antibody. *Platelet count at 6 months was defined as the value at 6±3 months; platelet count at 12 months was defined as the value at 11–18 months. ƒNo information for 40 and 49 patients at 6-month FU and 12-month FU, respectively. †Initial treatment is treatment lasting a maximum of 4 weeks; 6-month follow-up treatment is defined as the treatment lasting until the 6-month follow-up, excluding initial therapy. No information for 25 and 36 patients at 6-month and 12-month FU, respectively. **Patients with a combination of different second- or third-line treatments are counted once.
with chronic ITP (69% vs. 56%). However, patients with initial platelet counts <20×109/L (n=256) showed a similar tendency to develop chronic disease (51%) as to go into remission (49%) at 12 months. In contrast, among the 159 patients with an initial platelet count ≥20×109/L, only 55 (35%) were in remission at 12 months (Figure 2). The initial platelet count was a significant factor of remission in the analysis of predictors presented further down. Adolescents (12–18 years) and young adults (18–25 years) showed identical remission rates (43%). Further analyses
of the different age groups (12–<15, 15–<18, and 18–<21, 21–<25 years) are shown in Figure 2A. The worst outcome, i.e., the lowest remission rate of 28%, was observed in the 15–<18-year subgroup with an initial platelet count of ≥20×109/L.
Among patients with initial severe thrombocytopenia (< 20×109/L, n=256), the remission rate at 1 year depended on the initial treatment strategy, i.e., remission rates were 53% and 32% for patients receiving any frontline treatment and watch-and-wait treatment, respectively
FU: Follow-up; IVIG: intravenous immunoglobulin; TPO-RA: thrombopoietin receptor agonist. *Treatment “6-mo FU”: until 6 months, excluding initial therapy; treatment “12-mo FU”: between 6 and 12 months. ƒPatients may receive more than one second-line treatment. †e.g., Dapsone, hydroxychloroquine, belimumab, and probably more study drugs.
ITP: immune thrombocytopenia; FU: follow-up; IQR: interquartile range; ANA: antinuclear antibody. *Definition of remission at 12 months: platelet count > 100 × 109/L and no treatment at least for 6 months surrounding the 12-month FU (the time without treatment could be before or after the 12-month FU (see also Online Supplementary Figure S1). ƒThe platelet count at 12 months was defined as the value at 11–18 months.
(P<0.05) (Figures 2B, 3). These two treatment subgroups did not differ in age, sex, and bleeding occurrence. In contrast, among patients with initial moderate thrombocytopenia, those followed with the watch-and-wait approach had a higher remission rate (37/90, 41%) than those receiving any first-line treatment (18/69, 26%), particularly intravenous immunoglobulin (IVIG) (4/24, 17%) (Figure 2B). However, the size of the last subgroup was small. Figure 3 shows the probability of remission according to the initial platelet count and initial treatment strategy, with the curves crossing at a platelet count of 30×109/L (interaction). This means that the effect of initial treatment on remission depends on the magnitude of the initial platelet count.

In our analysis, AYAS exhibited a clinical pattern that reflected overlapping characteristics of children and adults. As for children AYAS presented with infrequent comorbidities, high initial bleeding rate, and a very low initial pla-
telet count. Conversely, and similar to adults we found more females, a high risk of chronicity >50%, and a prolonged need for treatment between 6-12 months in 30% of cases (Online Supplementary Table S2).9 The AYAS group was clinically very homogenous, and we found no reason to clinically separate young adults from adolescents given that the course of disease and risk of chronicity are similar (Table 2). Our analysis highlights that AYAS are mainly treated with corticosteroids throughout the 1-year observation period, probably leading to considerable side effects (Table 1). However, we do not have information on dose and duration. The reason for this treatment may be drug convenience, offering the possibility of on-demand treatment (especially for menstruating women); moreover, the availability of alternative treatments such as TPO-RA was limited (especially for children aged <16 years) in the study period, and such treatments are costly. The high rate of chronic disease in AYAS confirms the poor recovery rates among older children. Many pediatric studies reveal that age >10 years is a major risk factor for chronicity.16
According to the ICIS I registry, the remission rates of patients aged 10–16 and 1–10 years were 53% and 72%, re-
Figure 2. Rate of remission. (A) Rate of complete remission off-treatment, depending on different adolescents and young adults (AYAS) age groups. (B) Rate of remission, depending on initial platelet count (×109/L) and initial choice of treatment strategy. Remission rate shows no significant differences among age groups within AYAS (A). Note: percentages are calculated for patients with known remission status. No or insufficient information about remission state for 124/338 (37%) of patients 12–<15years, 70/179 (39%) of patients 15–<18 years, and 47/139 (34%) of patients 18 years. Rate of complete remission (Tc >100×109/L, off-treatment) was calculated according to initial treatment and initial platelet count (B). Treated: initial treatment (intravenous immunoglobulin [IVIG] and/or corticosteroids), W&W: initial watch-and-wait strategy. * Remission rate for patients that initially received IVIG monotherapy (N=35/60) (58%), IVIG + corticosteroids (N=25/49) (51%), corticosteroid monotherapy (N=45/89) (51%), other (N=3/5). ** Remission rate for patients that initially received IVIG monotherapy (N=2/16) (13%), IVIG + corticosteroids (N=2/8) (25%), corticosteroid monotherapy (N=14/43) (33%), other (N=0/2).
spectively.18 Similarly, Donato et al. reported recovery rates of 49% among children aged 9–18 years and 71% in those aged 1–8 years at diagnosis.19 Among patients diagnosed with ITP at 10–18 years from 1976 to 2000, Lowe and Buchanan observed 57% with chronic disease, 27% in recovery, and 15% with unclear results.14 These values are similar to our AYAS cohort aged between 12 and 25 years. However, it is to note that definition of remission and chronicity varies in the literature and in published guidelines. The term “chronic disease” was corrected in 2009 to designate a disease lasting ≥12 months and with a platelet count <100×109/L. In our analysis, adolescents aged 15–18 years had a slightly higher chronic disease rate (61%) than those aged 12–15 years (55%). We found no differences in the remission rates of males and females. This finding agrees with Lowe and Buchanan.14 Among the factors that can modify chronicity, we found that initial ANA was more frequently positive in patients with chronic disease (23%) than those in remission at 1
year (15%) (Table 4). However, interpretation is limited given the heterogeneous definition of ANA positivity among institutions and the low testing frequency. In the literature, initial ANA results have been reported as an indicator of ITP chronicity, although the data on children and adults remains controversial.15,20-22 We also found that AYAS in remission had lower initial platelet counts than those with later chronic disease. These results are similar to previous publications of outcome predictors in children and adults.16,21 Surprisingly, AYAS with severe thrombocytopenia who initially followed a watch-and-wait strategy had a higher rate of chronic disease (68%) than those initially treated with corticosteroids and/or IVIG (47%) (Figure 2B). Our results suggest that early treatment benefits AYAS suffering from "pediatric-like" severe ITP. However, data from a meta-analysis and a prospective trial yielded contradictory results regarding initial IVIG and long-term remission in children.16,23 Our findings show that AYAS with moderate and mild thrombocytopenia who in-
Figure 3. Logistic regression predicting remission versus no remission. Logistic regression was performed to predict remission. Predictors were age, sex, initial platelet count, and initial therapy (presence or absence). The influence of age and sex was not significant (P=0.15; P=0.78, respectively). The interaction between treatment and platelet count was significant (P=0.002). The graph (on the probability scale) shows the curves crossing at about 30×109/L. As an example, a patient with an initial platelet count of 10× 109/L AND receiving upfront treatment has a 55% chance of remission (brown line), and the same patient receiving NO treatment has a 29% chance of remission (black line), (odds ratio=2.07, 95% 95% confidence interval [CI]: 1.15- 3.74; P=0.020). Conversely, a patient with an initial platelet count of 60×109/L AND receiving upfront treatment has a 12% chance of remission (brown line), and the same patient receiving NO treatment has a 44% chance of remission (black line) (odds ratio=0.28, 95% CI: 0.080.91; P=0.03). The odds ratios in the table below indicate the change of the risk dependent on therapy and platelet count. Odds ratios were calculated for several platelet count level.

itially received frontline treatment have a lower remission rate (26%) than the watch-and-wait subgroup (41%) (Figure 2B). Several hypotheses can be considered, such as the possibility that those patients requiring medical intervention for bleeding despite only moderate thrombocytopenia might have been misdiagnosed with ITP. They may have hereditary thrombocytopenia, secondary ITP, or a combination of pathologies with a higher propensity to bleed despite moderate thrombocytopenia. Therefore, this special subgroup would have high rates of “chronic disease,” if the diagnosis was wrong and the treatment was inadequate. Further investigations are warranted to understand the underlying disease mechanism of these patients.
We found a high relapse rate among patients in remission at 12 months harboring borderline platelet values (100149×109/L). This finding raises the question of whether the threshold of 100×109/L to define ITP remission is correct for AYAS.
Current practice guidelines do not address AYAS; diagnostic and therapeutic recommendations focus only on children and adults. According to the literature, ITP in children and adults presents clinical differences that give the impression of two different diseases, with adults presenting with more moderate thrombocytopenia and less initial bleeding.10 However, using a temporal perspective, ITP may differ only in disease initiation and cessation, i.e., the trigger of the disease and the intrinsic capacity to induce tolerogenic mechanisms. Young children have “favorable pathophysiological and physiological features” at both time points, whereas adults have “unfavorable features.” In most children, the trigger seems to be a viral infection that causes transient cross-reactivity with platelet antigens, ultimately corrected by the immune system. In adults, the trigger is less clear, and immune dysregulation progresses to a complex disease that is not easily or rapidly corrected, probably due to immunosenescence, comorbidities, and drugs.24,25 Herein, we showed that the course of ITP in AYAS is similar to “adult-type” ITP. Nevertheless, AYAS may benefit from new treatment goals focusing on restoring a durable immune balance and curing the disease rather than a symptomatic strategy, e.g., elevating the platelet count, limiting patient activities, and risk management.26 Much interest remains in whether more aggressive therapy during the early phase of ITP mitigates persistent or chronic disease.27 Novel approaches involving early combined therapies against T and B cells with and without TPO-RA have provided promising results.28-30 Some smallscale trials focusing on young adults (iROM-study, clinicaltrials gov. Identifier: NCT02760251, and iROM2-study, clinicaltrials gov. Identifier: NCT04812483) are still ongoing. The use of rituximab early in the disease course, especially in young women, could be a promising strategy.31 Restoring immune tolerance appears to be a promising
early treatment goal for AYAS. However, intense therapies require early identification of those that elicit a refractory response.
The loss of FU and missing data may have biased our results, particularly the remission rate. In a previous analysis of the PARC, patients of all ages lost to FU were predominantly those with remission in the first months of the disease.9 In addition, our efforts to accurately classify patients in remission and chronic disease have generated unequal observation periods. This could be another source of bias, especially for patients with relapses later at FU but missing data. Specific limitations of both registries have been published elsewhere15,32 and will not be discussed in detail here; however, the difficulties of making an accurate diagnosis of primary ITP could be the explanation for some unexpected results.
Defining ITP in only two age categories - children and adults - seems inappropriate and oversimplifies the clinical characteristics and needs of AYAS. AYAS have a long life expectancy and high quality of life expectations; thus, treatment strategies and goals must be reconsidered for this age group. Upfront immune modulation could be a promising strategy for AYAS and an approach that requires further investigation. Future ITP trials should have the following aims: i) discern AYAS ITP as an entity distinct from pediatric and adult ITP, ii) adopt treatment goals that reduce the rate of chronic disease, and iii) dissuade the use of corticosteroids beyond initial management.
AS reports Novartis honoraria and research funding; Sobi and Takeka honoraria; Platelet Disorder Support Association (PDSA) grant recipient. GM received meeting attendance grants from Amgen, Grifols, and Novartis and is the coordinator of research studies granted by Amgen, CSL Behring, Grifols, Novartis, and Sanofi. He participated in educational sessions funded by Amgen, Grifols, and Novartis, and on boards of Amgen, Argenx, Novartis, and Sobi. MM reports Novartis honoraria. JG reports honoraria from Amgen, Novartis, Dova, ONO Pharmaceuticals and Bitest; ITP Support Association medical advisor and grant recipient. MC reports Novo Nordisk honoraria; participated on boards of Novo Nordisk, Takeda Inc, BPL Pharmaceuticals , Octapharma, Genzyme Corp, Emerging Therapeutics Inc, Guidepoint Global, Neri Science. TK reports Amgen and Novartis research funding and honoraria; Sobi and UCB honoraria. All other authors have no conflicst of intererest to dislose.
Conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision and writing by AS. Conceptualization, methodology, review, and editing, investigation, data curation and supervision by GM. Conceptualization, review and
editing, and supervision by BG. Conceptualization, review, and editing by TL, NA and MM. Review and editing, investigation, and data curation by GL and JG. Review and editing, and investigation by ME, MC, AH and SH. Review and editing, and funding acquisition by GlG. Review and editing by PI. Conceptualization, methodology, supervision, review and editing, and funding acquisition by TK.
We thank all the investigators of the PARC and CARMEN study groups, as well as Andy Schötzau for the statistical analysis.
1. Kühne T. Immune Thrombocytopenia. 1st ed. Bremen. UNI-MED, 2010.
2. Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):37803817.
3. Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829-3866.
4. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386-2393.
5. Moulis G, Palmaro A, Montastruc JL, Godeau B, Lapeyre-Mestre M, Sailler L. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood. 2014;124(22):3308-3315.
6. Cohen YC, Djuibegovic B, Shamai-Lubovitz O, Mozes B. The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. Arch Intern Med. 2000;160(11):1630-1638.
7. Cortelazzo S, Finazzi G, Buelli M, Molteni A, Viero P, Barbui T. High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenic purpura. Blood. 1991;77(1):31-33.
8. Kühne T, Berchtold W, Michaels LA, et al. Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica. 2011;96(12):1831-1837.
9. Schifferli A, Holbro A, Chitlur M, et al. A comparative prospective observational study of children and adults with immune thrombocytopenia: 2-year follow-up. Am J Hematol. 2018;93(6):751-759.
10. Despotovic JM, Grimes AB. Pediatric ITP: is it different from adult ITP? Hematolology Am Soc Hematol Educ Program. 2018;2018(1):405-411.
11. Bibby H, White V, Thompson K, Anazodo A. What are the unmet needs and care experiences of adolescents and young adults with cancer? A systematic review. J Adolesc Young Adult Oncol. 2017;6(1):6-30.
12. Nass SJ, Beaupin LK, Demark-Wahnefried W, et al. Identifying and addressing the needs of adolescents and young adults with cancer: summary of an Institute of Medicine workshop. Oncologist. 2015;20(2):186-195.
13. Neylon AJ, Saunders PW, Howard MR, Proctor SJ, Taylor PR,
Funding was receieved from the Platelet Disease Support Association (PDSA), Ohio, USA; Stiftung hämatologische Forschung, Basel, CH; Stiftung zur Förderung medizinischer und biologischer Forschung, Arlesheim, CH. The PARC registry received financial support from the Intercontinental Cooperative ITP Study Group (ICIS). The CARMEN registry received support from the French National Society of Internal Medicine, Toulouse Referral Center for Autoimmune Cytopenias, Toulouse University Hospital, Amgen, CSL Behring, Grifols, and Novartis.
Northern Region Haematology Group. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br J Haematol. 2003;122(6):966-974.
14. Lowe EJ, Buchanan GR. Idiopathic thrombocytopenic purpura diagnosed during the second decade of life. J Pediatr. 2002;141(2):253-258.
15. Moulis G, Germain J, Comont T, et al. Newly diagnosed immune thrombocytopenia adults: clinical epidemiology, exposure to treatments, and evolution. Results of the CARMEN multicenter prospective cohort. Am J Hematol. 2017;92(6):493-500.
16. Heitink-Pollé KM, Nijsten J, Boonacker CW, de Haas M, Bruin MC. Clinical and laboratory predictors of chronic immune thrombocytopenia in children: a systematic review and metaanalysis. Blood. 2014;124(22):3295-3307.
18. Kühne T, Buchanan GR, Zimmerman S, et al. A prospective comparative study of 2540 infants and children with newly diagnosed idiopathic thrombocytopenic purpura (ITP) from the Intercontinental Childhood ITP Study Group. J Pediatr. 2003;143(5):605-608.
19. Donato H, Picón A, Martinez M, et al. Demographic data, natural history, and prognostic factors of idiopathic thrombocytopenic purpura in children: a multicentered study from Argentina. Pediatr Blood Cancer. 2009;52(4):491-496.
20. Altintas A, Ozel A, Okur N, et al. Prevalence and clinical significance of elevated antinuclear antibody test in children and adult patients with idiopathic thrombocytopenic purpura. J Thromb Thrombolysis. 2007;24(2):163-168.
21. Grimaldi-Bensouda L, Nordon C, Michel M, et al. Immune thrombocytopenia in adults: a prospective cohort study of clinical features and predictors of outcome. Haematologica. 2016;101(9):1039-1045.
22. Moulis G, Comont T, Germain J, et al. Significance of antinuclear antibodies in primary immune thrombocytopenia: results of the CARMEN registry. Blood Adv. 2020;4(9):1974-1977.
23. Heitink-Pollé KMJ, Uiterwaal CSPM, Porcelijn L, et al. Intravenous immunoglobulin vs observation in childhood immune thrombocytopenia: a randomized controlled trial. Blood. 2018;132(9):883-891.
24. Duggal NA, Upton J, Phillips AC, Sapey E, Lord JM. An agerelated numerical and functional deficit in CD19(+) CD24(hi) CD38(hi) B cells is associated with an increase in systemic autoimmunity. Aging Cell. 2013;12(5):873-881.
25. Enger C, Bennett D, Forssen U, Fogarty PF, McAfee AT. Comorbidities in patients with persistent or chronic immune
thrombocytopenia. Int J Hematol. 2010;92(2):289-295.
26. Cooper, N, Kruse, A, Kruse, C, et al. Immune thrombocytopenia (ITP) World Impact Survey (I-WISh): Impact of ITP on healthrelated quality of life. Am J Hematol. 2021;96(2):199-207.
27. Schifferli A, Kühne T. Thrombopoietin receptor agonists: a new immune modulatory strategy in immune thrombocytopenia? Semin Hematol. 2016;53 Suppl 1:S31-34.
28. Gómez-Almaguer D, Colunga-Pedraza PR, Gómez-De León A, Gutiérrez-Aguirre CH, Cantú-Rodríguez OG, Jaime-Pérez JC. Eltrombopag, low-dose rituximab, and dexamethasone combination as frontline treatment of newly diagnosed immune thrombocytopaenia. Br J Haematol. 2019;184(2):288-290.
29. Zhang L, Zhang M, Du X, Cheng Y, Cheng G. Safety and efficacy of
eltrombopag plus pulsed dexamethasone as first-line therapy for immune thrombocytopenia. Br J Haematol. 2020;189(2):369-378.
30. Bradbury CA, Pell J, Hill Q, et al. Mycophenolate mofetil for first-line treatment of immune thrombocytopenia. N Engl J Med. 2021;385(10):885-895.
31. Bussel JB, Lee CS, Seery C, et al. Rituximab and three dexamethasone cycles provide responses similar to splenectomy in women and those with immune thrombocytopenia of less than two years duration. Haematologica. 2014;99(7):1264-1271.
32. Kühne T. Update on the Intercontinental Cooperative ITP Study Group (ICIS) and on the Pediatric and Adult Registry on Chronic ITP (PARC ITP). Pediatr Blood Cancer. 2013;(60 Suppl 1):S15-18.
Treatment of patients with acute myeloid leukemia (AML) fit to receive intensive treatment and under 65 years of age consists of one or two cycles of high-dose induction (“3+7”) chemotherapy (C1 or C2) followed by different options of post-remission treatment.1 These options include allogeneic stem cell transplantation (allo-SCT), continuation with chemotherapy or high-dose chemotherapy with autologousSCT (auto-SCT).2 Choosing the right consolidation treatment is a trade-off between anti-leukemic effect and treatment safety. Relapse chances are lowest after allo-SCT, but this treatment modality is also associated with considerable therapy-related morbidity (e.g., graft-versus-host disease), reduced quality of life, and higher procedure-related mortality.3,4 Therefore, for patients with a relatively favorable outcome (i.e., core-binding factor AML), allo-SCT is often avoided as first-line consolidation treatment. In contrast, for patients with an adverse-risk disease (if deemed feasible) this additional anti-leukemic effect is needed, making alloSCT the preferred post-remission treatment option. For patients with intermediate-risk AML, the optimal post-remission therapy is still a subject of debate.5 Measurable residual disease (MRD) assessment, by multiparameter flow cytometry (MFC) and/or by NPM1 gene mutation reverse transcriptase polymerase chain reaction (RT-PCR), has been proposed to guide this decision due to the strong prognostic value and the ability to predict relapse when applied in complete remission (CR) (or CR with incomplete hematologic recovery [CRi], according to the 2017 European LeukemiaNet [ELN] classification) after C2.1,6 Therefore, the presence of MRD at this time point may warrant an allo-SCT as additional intensive therapy.2 In contrast, patients in CRi without MRD before transplant have a relatively low risk of relapse and therefore allo-SCT may be omitted.7 According to the protocol of the HOVON-SAKK132 trial (HO132: registered as NTR4376; available from the Netherlands Trial Register at https://trialsearch.who.int/), the choice of post-remission therapy in intermediate-risk patients was guided by MRD status defined by MFC (>0.1%) and mutant NPM1 (>10-4). Patients with MRD were recommended to receive allo-SCT while patients without MRD were recommended to proceed with less intensive non-allo treatment (auto-SCT or a third cycle of chemotherapy).1 Notably, the previously reported analysis on the HO132 trial showed no difference in relapsefree survival for the ELN-2017 intermediate-risk category,
which may suggest the positive effect of MRD guidance.8 In order to better understand the influence of MRD guidance, we present a more detailed analysis of the results of MRDguided post-remission therapy for intermediate-risk AML or high-risk myelodysplastic syndrome (MDS) patients in relation to treatment outcome in the HO132 trial, including a per protocol analysis. In addition, since the HO132 was guided by MRD status, we compared this MRD-guided cohort to an unguided cohort using a propensity score match (PSM) analysis. This unguided matched control group was derived from previous HOVON-SAKK trials that had no MRD-guided post-remission therapy.
A total of 153 ELN intermediate-risk patients in the HO132 trial (also including patients enrolled in the run-in phase) were in CRi and had a MRD result after C2 as assessed by either leukemia-associated immunophenotype detection with MFC and/or by RT-PCR for mutated NPM1, according to earlier published guidelines.2,9 Of these 153 patients, 110 (72%) were MRD-negative (by both techniques), of which 44/110 (40%) received, as advised per protocol, non-allo-SCT consolidation therapy and 48/110 (44%) patients received an allo-SCT. The other 18 patients (16%) received no consolidation therapy, mainly due to an early relapse. Reasons for deviating from the advised protocol treatment were not systematically collected, but MRD-negative patients who received allo-SCT had significantly more complex karyotype (45.8% vs. 4.5%) and were more often in first CR after C2 instead of C1 (33.3% vs. 4.5%), compared to MRD-negative non-allo consolidated patients. Protocol adherence was better for MRD-positive patients, with 36/43 (84%) receiving the per protocol recommended allo-SCT. Survival differences were analyzed using Kaplan-Meier curves for eventfree survival (EFS) and overall survival (OS), with Cox regression accounting for clustering. There was no significant difference in EFS, defined as the time between MRDassessment after C2 in CRi and relapse or death, between the patients with and without MRD (hazards ratio [HR]: 1.24; 95% CI: 0.75-2.00; P=0.42) (Figure 1A), with an EFS after 36 months of 47% compared to 54%, respectively. OS (defined as the time between MRD-assessment in CRi and death or censoring) (HR: 1.50; 95% CI: 0.85-2.64; P=0.16) (Figure 1B) seems to be slightly worse for MRD-positive patients (5-year OS 54% compared to 65% for MRD-negative), although not statistically significant. Both EFS and OS were in line with
disease-guided therapy in intermediate-risk acute myeloid leukemia patients is a valuable strategy in reducing allogeneic transplantation without negatively affecting survival
recently published HOVON-SAKK trials.8,11 For MRD-negative patients, we also compared the patients who, contrary to trial protocol, received allo-SCT with patients who received non-allo-SCT treatment according to protocol. Between these two groups, there were no apparent significant differences in EFS (HR: 0.69; 95% CI: 0.37-1.29; P=0.24) (Figure 1C)
nor in OS (HR: 1.24; 95% CI: 0.59-2.63; P=0.57) (Figure 1D). However, the sequence of events did differ between MRDnegative patients treated with allo-SCT versus non-allo-SCT.
A total of 15/48 (31%) of allo-SCT treated patients relapsed within three years after CR, of whom most (93%, 14/15) died within ten months after relapse. Although in the non-all-SCT
Figure 1. Survival of European LeukemiaNet (ELN) intermediate-risk patients in the measurable residual disease (MRD)-guided HO132 trial and subgroup analyses for MRD-negative patients afer two cycles of induction chemotherapy. (A) In the HO132 trial, a total of 153 patients with an MRD result were ELN intermediate-risk, of which 110 (72%) were (MRDneg) after two cycles of chemotherapy. There was no significant difference in event-free survival (EFS) assessed using Cox regression between MRDneg and MRDpos patients (HR: 1.24; 95% CI: 0.75-2.00; P=0.42). (B) Neither was there any significant difference in overall survival (OS) between MRDneg and MRDpos patients (HR: 1.50; 95% CI: 0.85-2.64; P=0.16). (C) A subgroup analysis of the intermediate-risk MRDneg patients in the HO132 trial showed no difference in EFS between 44 patients who received the recommended non-allo (cycle 3 or auto-SCT) consolidation treatment compared to 48 patients who received an allo-SCT (HR: 0.69; 95% CI: 0.37-1.29; P=0.24). (D) The same subgroup also showed no difference in OS between patients treated with non-allo and allo-SCT (HR: 1.24; 95% CI: 0.59-2.63; P=0.57). (E) Swimmer plot of MRDneg patients in the HO132 (MRD-guided) study ordered by first post-remission therapy (y-axis) and OS (x-axis). The non-allo-stem cell transplant (SCT) group consists of 30 patients who received an autoSCT and 14 patients who received a third cycle of chemotherapy. The majority (67%, 12/18 patients) of the relapsed patients (symbolized by a triangle) who initially received a non-allo consolidation therapy were able to undergo a delayed allo-SCT after successful salvage therapy (red beam). (The HO132 trial is registered as NTR4376; available from the Netherlands Trial Register at https://trialsearch.who.int/)

group 18/44 (41%) relapsed, 12 patients could be successfully salvaged with a (delayed) allo-SCT, which was followed by a long leukemia-free follow-up for 10/18 (55%) patients (Figure 1E). Importantly, for MRD-negative patients, 32 allo-SCT could be averted and 12 postponed without negatively effecting EFS and OS compared to the patients treated with allo-SCT. Therefore, based on these results from the HO132 trial, non-allo-SCT treatment options seem to be justified for intermediate-risk MRD-negative patients. Although benefits of MRD-guided therapy would preferably be evaluated in a randomized controlled trial (RCT), we do not consider this realistic due to the extensive use of MRD in daily practice and the current evidence for the prognostic value of MRD. In addition, no such AML trials are currently reported to be ongoing or planned. Hence, we simulated the analysis by comparing survival of the MRD-guided intermediate-risk patients from the HO132 trial (conducted from 2014
to 2017) to an MRD-unguided cohort from HOVON-SAKK phase II/III trials (HO42A: NTR230; HO81: NTR904; HO92: NTR1446; HO102: NTR2187; all available from the Netherlands Trial Register at https://trialsearch.who.int/), conducted from 2006 to 2013.7,10-12 The principle of measuring MRD and gating strategy remained the same across the studies and followed a strict protocol.9 Via PSM,13 the intermediate-risk patients derived from the unguided studies in CR with MRD measurement after C2 (n=150) were matched to the HO132 MRDguided patients using six baseline variables that are associated with survival: age, white blood cell count at diagnosis, WHO classification, karyotype, NPM1 status, and FLT3-ITD status (Online Supplementary Figure S1). The MRDguided and unguided studies included in our analyses were randomized for an investigational treatment, but no significant differences in EFS or OS were observed between the standard and the investigational arms for included patients

Figure 2. Survival by propensity score match analysis between measurable residual disease (MRD)-unguided and MRD-guided groups, and subgroup analysis of only MRD-negative patients. (A) Event-free survival (EFS) after 36 months was 47% for the MRD-unguided group and 54% for the MRD-guided group (Hazard Ratio [HR]: 0.87; 95% confidence interval [CI]: 0.60-1.26; P=0.47).
(B) Overall survival (OS) after 60 months was 56% in the MRD-unguided group and 61% in the MRD-guided group (HR: 0.80; 95% CI: 0.52-1.24; P=0.32). (C) EFS for MRD-negative patients after 36 months was 48% in the unguided group compared to 56% in the MRD-guided group (HR: 0.86; 95% CI: 0.56-1.33; P=0.50). (D) OS after 60 months was 60% in the unguided group, compared to 64% in the MRD-guided group (HR: 0.84; 95% CI: 0.50-1.40; P=0.50).
(Online Supplementary Figure S2); hence, investigational treatment was omitted in the matching. We used the ‘nearest neighbor’ matching technique with a caliper (maximum distance between cases) of 0.25, because this rendered the lowest standardized mean difference of 0.09.14 This resulted in 110 matches with similar patient characteristics. All clinical features between the MRD-guided and matched control group were comparable, except for more karyotype abnormalities in the MRD-guided cohort (Online Supplementary Table S1). EFS after 36 months was comparable between the MRD-guided group (54%) and the historical control cohort (47%) (HR: 0.87; 95% CI: 0.60-1.26; P=0.47) (Figure 2A). In addition, the same comparable results were found for OS with a 61% survival rate for MRD-guided and 56% for the unguided cohort after 60 months (HR: 0.80; 95% CI: 0.52-1.24; P=0.32) (Figure 2B). Between the two cohorts, preferred consolidation treatment had only changed for MRD-negative patients. In former HOVON-SAKK trials, all intermediate-risk patients had been advised to receive an allo-SCT, which changed to only for the MRD-positive patients in the HO132 trial. Therefore, a separate subgroup analysis was carried out for only MRD-negative patients, which showed that there was no significant difference in EFS after three years (HR: 0.86; 95% CI: 0.56-1.33; P=0.50) or OS after five years (HR: 0.84; 95% CI: 0.50-1.40; P=0.50) between unguided and guided MRDnegative patients (Figure 2C, D). These results again suggest that MRD-guidance for consolidation selection in intermediate-risk patients allows allo-SCT treatment to be safely curcumvented without having a negative impact on EFS or OS, which is in accordance with previous data provided by the GIMEMA AML1310 trial.15
We wish to add that the PSM method is a valuable alternative for RCT to compare two groups, although it is less preferred because of possible unequal distributions of unknown confounding factors. In addition, due to matching with historical data, the time frame in which patients were treated differed, which may have influenced survival due to changes in patient care, such as supportive care. Here, these effects seem relatively limited since OS did not deviate between patients included from different studies (Online Supplementary Figure S2). Unfortunately, for both cohorts, the exact reasons for choosing a specific consolidation treatment are not known. The conclusions of the retrospective PSM-based comparison with historical non-guided data are based on non-statistical significant data with a broad confidence interval. Nevertheless, these conclusions are substantiated by the results from the in-depth subgroup
analysis of the MRD-guided patients in the HO132 trial, which also support the value of MRD-negativity for selecting a less intensive consolidation treatment than allo-SCT for intermediate-risk patients. Future improvements to MRD assays can potentially further increase appropriate MRD-guided post remission therapy.
Jesse M. Tettero,1,2 Lok Lam Ngai,1,2 Costa Bachas,1,2 Dimitri A. Breems,3 Thomas Fischer,4 Bjorn T. Gjertsen,5 Patrycja Gradowska,6 Laimonas Griskevicius,7 Jeroen J.W.M. Janssen,1,2 Gunnar Juliusson,8 Johan Maertens,9 Markus G. Manz,10,11 Thomas Pabst,11,12 Jakob Passweg,11,13 Kimmo Porkka,14 Peter J.M. Valk,15 Bob Löwenberg,15 Gert J. Ossenkoppele1,2 and Jacqueline Cloos1,2
1Amsterdam University Medical Centers, Location VUmc, Amsterdam, The Netherlands; 2Imaging and Biomarkers, Cancer Center Amsterdam, Amsterdam, The Netherlands; 3Ziekenhuis Netwerk Antwerpen, Antwerp, Belgium; 4Otto von Guericke University Hospital Magdeburg, Magdeburg, Germany; 5Haukeland University Hospital, Bergen, Norway; 6Dutch-Belgian Hemato-Oncology Cooperative Group Data Center-Erasmus MC Cancer Institute, Rotterdam, The Netherlands; 7Vilnius University Hospital Santaros Klinikos and Vilnius University, Vilnius, Lithuania; 8Skanes University Hospital, Lund, Sweden; 9University Hospital Gasthuisberg, Leuven, Belgium; 10University Hospital, Zurich, Switzerland; 11Swiss Group for Clinical Cancer Research (SAKK), Bern, Switzerland; 12Department of Medical Oncology, Inselspital; University Hospital, Bern, Switzerland; 13University Hospital, Basel, Switzerland; 14Helsinki University Hospital Cancer Center, Helsinki, Finland and 15Erasmus University Medical Center (MC) and Erasmus MC Cancer Institute, Rotterdam, The Netherlands
Correspondence:
J. CLOOS - j.cloos@amsterdamumc.nl
https://doi.org/10.3324/haematol.2022.282639
Received: December 27, 2022.
Accepted: March 24, 2023.
Early view: April 6, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
BG serves as a consultant for BerGenBio, Phizer Inc., and Novartis, and holds stock options in Alden Cancer Therapy and KinN Therapeutics. JJ serves as a consultant for Bristol-Myers Squibb, Novartis, Pfizer Inc., has received research funding from BristolMyers Squibb, Novartis, Incyte Biosciences Benelux BV, Uppsala County Council, Glycomimetics, Avillion and Ellipses Pharma, is a Speaker Bureau member for Incyte Biosciences Benelux BV, Roche and Celgene, and holds a membership on an entity's Board of Directors for Celgene. MM serves as a consultant for CDR-Life Inc., holds stock options in CDR-Life Inc., and has a patent licensed to the University of Zurich. BL holds a membership on an entity's Board of Directors for AbbVie, Astellas, Catamaran Bio Inc., Bristol-Myers Squibb, Celgene, and F. Hoffmann La Roche, serves as a consultant for Clear Creek Bio, and has received honoraria from Clear Creek Bio. GO serves as a consultant for Novartis, Pfizer Inc., Celgene, Janssen, AGIOS, Amgen, Gilead, Astellas, Roche, Jazz Pharmaceuticals and Merus, has received honoraria from Novartis, Celgene, AGIOS, Gilead and Astellas, has received research funding from Novartis, and holds a membership on an entity's Board of Directors for Roche. JC serves as a consultant for Novartis, and has received research funding from Takeda, DC-one, Genentech, Janssen, Novartis and Merus.
1. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
2. Heuser M, Freeman SD, Ossenkoppele GJ, et al. 2021 update measurable residual disease in acute myeloid leukemia: European LeukemiaNet Working Party Consensus Document. Blood. 2021;138(26):2753-2767.
3. Versluis J, Kalin B, Zeijlemaker W, et al. Graft-versus-leukemia effect of allogeneic stem-cell transplantation and minimal residual disease in patients with acute myeloid leukemia in first complete remission. JCO Precis Oncol. 2017;1:1-13.
4. Andersson I, Ahlberg K, Stockelberg D, Persson LO. Patients' perception of health-related quality of life during the first year after autologous and allogeneic stem cell transplantation. Eur J Cancer Care (Engl). 2011;20(3):368-379.
5. Ganzel C, Rowe JM. Revisiting autologous transplantation in acute myeloid leukemia. Curr Opin Hematol. 2018;25(2):95-102.
6. Short NJ, Zhou S, Fu C, et al. Association of measurable residual disease with survival outcomes in patients with acute myeloid leukemia: a systematic review and meta-analysis. JAMA Oncol. 2020;6(12):1890-1899.
7. Terwijn M, van Putten WL, Kelder A, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol. 2013;31(31):3889-3897.
8. Löwenberg B, Pabst T, Maertens J, et al. Addition of lenalidomide to intensive treatment in younger and middleaged adults with newly diagnosed AML: the HOVON-SAKK-132 trial. Blood Adv. 2021;5(4):1110-1121.
JT, LN, CB, GO and JC contributed to the conception and design of the study. DB, TF, BG, LG, JJ, GJ, JM, MM, TP, JP, KP, BL and GO collected the data. JT and LN organized the database. JT performed the statistical analysis. PG provided statistical consultation. JT wrote the first draft of the manuscript. All authors contributed to the revision of the manuscript, and read and approved the final version for publication.
We would like to thank the MRD-team of the Amsterdam UMC and Erasmus MC for their help in measuring all samples.
This study was supported by the Dutch Cancer Society (n. ALPE2013-6371).
The datasets used and/or analyzed during the study are available from the HOVON/SAKK AML Group upon reasonable request to the corresponding author.
9. Cloos J, Harris JR, Janssen JJWM, et al. Comprehensive protocol to sample and process bone marrow for measuring measurable residual disease and leukemic stem cells in acute myeloid leukemia. J Vis Exp. 2018;(133):56386.
10. Ossenkoppele GJ, Stussi G, Maertens J, et al. Addition of bevacizumab to chemotherapy in acute myeloid leukemia at older age: a randomized phase 2 trial of the Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) and the Swiss Group for Clinical Cancer Research (SAKK). Blood. 2012;120(24):4706-4711.
11. Löwenberg B, Pabst T, Maertens J, et al. Therapeutic value of clofarabine in younger and middle-aged (18-65 years) adults with newly diagnosed AML. Blood. 2017;129(12):1636-1645.
12. Randomized study to assess the added value of laromustine in combination with standard remission-induction chemotherapy in patients aged 18-65 years with previously untreated acute myeloid leukemia (AML) or myelodysplasia (MDS) (RAEB with IPSS >= 1.5). 2013. https://trialsearch.who.int/Trial2.aspx? TrialID=EUCTR2008-000404-92-NL. Accessed January 14 2022.
13. Cenzer I, Boscardin WJ, Berger K. Performance of matching methods in studies of rare diseases: a simulation study. Intractable Rare Dis Res. 2020;9(2):79-88.
14. Lunt M. Selecting an appropriate caliper can be essential for achieving good balance with propensity score matching. Am J Epidemiol. 2014;179(2):226-235.
15. Venditti A, Piciocchi A, Candoni A, et al. GIMEMA AML1310 trial of risk-adapted, MRD-directed therapy for young adults with newly diagnosed acute myeloid leukemia. Blood. 2019;134(12):935-945.
Chimeric antigen receptor (CAR) T-cell therapy showed enormously promising results in patients with relapsed or refractory multiple myeloma,1,2 leading to approval of the first two products by the American Food and Drug Administration and the European Medicines Agency.3,4 Current responses seem to be comparable across CAR T-cell products and trials (both commercial and academic), with an overall response of ~80%.5 However, translation into sustained responses and thus, survival remain unclear, with relapse rates of ~45% among initial responders. Despite the promises, CAR T-cell therapy is a very complex and expensive treatment, which poses challenges to healthcare systems and society in general.6,7 Therefore, and in the wake of current efforts to study the effects of CAR T-cell therapy even in earlier lines of the treatment algorithm, patient selection becomes crucial. In this regard, the identification of high-risk patients is of utmost importance in order to be able to provide the most accurate counseling and choose the most effective treatment strategy that optimizes the outcome.8 Recent analyses suggested that the incorporation of novel monoclonal antibodies such as daratumumab as a backbone for regimens in relapsed or refractory high-risk multiple myeloma, defined as the presence of t(4;14), t(14;16), or del(17p), may be associated with improved progressionfree survival compared to that achieved with other regimens, while absolute outcome is still worse compared to that of patients with standard-risk features.9 In contrast, for patients with other high-risk disease features such as extramedullary disease, results are still generally disappointing.10 Sufficient reports on CAR T-cell therapy in these high-risk groups are lacking. Here, we summarize the current body of evidence on the role of novel CAR-T cell therapies for relapsed or refractory multiple myeloma and high-risk disease, focusing on the identification of patients who may benefit and those who may not.
We performed a systematic literature review to identify all fully published prospective trials in accordance with current guidelines.11 We searched Medline, EMBASE, Cochrane trials registry, and www.clinicaltrials.gov. A conventional meta-analysis was conducted using R statistical software.5 Risk ratios and 95% confidence intervals (95% CI) were calculated within a random-effects framework using the Mantel-Haenszel method. Heterogeneity across trials was measured using I². The I² statistic describes the percentage of variation across studies that is due to het-
erogeneity rather than chance. More than 50% is judged to show moderate heterogeneity, whereas >75% indicates severe heterogeneity.11 The quality of evidence was documented according to the GRADE system, using the following levels of evidence: high, moderate, low and very low.12 Because all trials conducted to date are non-randomized single-arm trials, the risk of bias was judged a priori to be serious.
The main efficacy outcomes were overall response rate, measurable residual disease, mortality, and relapse or progression. High-risk disease features were defined as the presence, at the time of CAR T-cell infusion, of cytogenetic high-risk, defined as at least either del(17p), t(14;16) or t(4;14), or disease-specific risk, defined as the presence of extramedullary disease or revised International Staging System (R-ISS) stage III disease.13
Out of a total of 769 screened articles, 17 trials comprising a total of 723 patients with heavily pretreated relapsed or refractory multiple myeloma were included in quantitative analyses (Online Supplementary Figure S1; all references are listed in the Online Supplementary Appendix). Overall, patients had received a median of five prior lines of treatment (such as proteasome inhibitors, immunomodulatory drugs, monoclonal antibodies), including autologous stem cell transplantation in 51% of patients. The median age of the patients was 59 years (Online Supplementary Table S1). The patients’ characteristics, such as age and number of prior lines of therapy including autologous transplantation, were affected by trial origin. The median age and number of prior lines of therapy were 61 years and seven lines in USA-based trials versus 57 years and four lines in China-based trials. Ninety-two percent of patients had undergone autologous transplantation before receiving CAR T-cell therapy in USA/European trials, whereas only 28% of patients in China-led trials had undergone prior autografts.
The most frequent single target was B-cell maturation antigen (BCMA). Four trials used tandem CAR, including two trials targeting both BCMA and CD38 and two trials targeting both BCMA and CD19. One trial only targeted CD19. The co-stimulatory domain was 4-1BB in most trials, and fludarabine and cyclophosphamide constituted the most common lymphodepletion regimen.
Regarding disease risk and outcomes, extramedullary disease was not significantly associated with a worse overall response rate, showing a risk ratio of 0.97 (95% CI: 0.921.02; P=0.26). The quality of evidence was moderate (Figure
1A). In terms of depth of response, no significant association with measurable residual disease was observed (P=0.84), with a risk ratio of 0.93 (95% CI: 0.73-1.19); and the quality of evidence was moderate (Online Supplementary Figure S1). In contrast, in terms of progression-free survival, the presence of extramedullary disease was significantly associated with worse outcome, showing a risk ratio of 1.44 (95% CI: 1.24-1.67; P<0.001) in favor of patients without extramedullary disease. Thus, presence of extramedullary disease at the time of CAR T-cell therapy was associated with a 44% increased risk of relapse/progression or death after treatment, and the quality of evidence was moderate (Figure 1B). This also translated into significantly worse overall survival. The presence of extramedullary disease was associated with a 96% increased risk of death from any cause, showing a risk ratio of 1.96 (95% CI: 1.48-2.58; P<0.001) (Online Supplementary Figure S1). Furthermore, R-ISS stage III was significantly associated with a worse overall response rate (P<0.001).

In terms of cytogenetic risk and outcomes, a high-risk
cytogenetic profile was significantly associated with worse overall response rate, showing a risk ratio of 0.86 (95% CI: 0.76-0.97) in favor of standard-risk cytogenetics (P=0.01). Correspondingly, the presence of high-risk cytogenetics at the time of CAR T-cell infusion was associated with a 14% increased risk of lack of response, and the quality of evidence was low (Figure 2A). For depth of response, the presence of high-risk cytogenetics appeared to be significantly associated with a 23% increased risk of measurable residual disease-positivity, showing a risk ratio of 0.78 (95% CI: 0.60-1.01; P=0.06) (Online Supplementary Figure S1). In terms of progression-free survival, high-risk cytogenetics were significantly associated with worse outcome, showing a risk ratio of 1.70 (95% CI: 1.292.25; P<0.001) in favor of standard-risk cytogenetics. Thus, the presence of high-risk cytogenetics was associated with a 70% increased risk of progression/relapse or death (Figure 2B). Correspondingly, high-risk cytogenetics were associated with significantly worse overall survival, showing a risk ratio of 2.11 (95% CI: 1.27-3.52; P=0.004) (Online
Supplementary Figure S1). Heterogeneity is summarized in Online Supplementary Table S2; no heterogeneity was found for most outcomes.
There are currently several approved and ongoing T-cell redirection strategies in multiple myeloma (bispecific antibodies and CAR T-cell therapy) and understanding the immune microenvironment prior to treatment and at relapse or progression could provide insights into rational sequencing of certain treatments. For example, radiotherapy for accessible extramedullary manifestations represents a frequently used option for bridging in clinical practice. A recent study using bridging radiotherapy before CAR T-cell therapy showed that bridging appeared to be safe and feasible in relapsed/refractory patients.14 This could be an option to reduce tumor burden during the currently long turnaround times required to provide the cell product, especially for patients who are in need of immediate symptomatic relief, who present with functional deficits, pathological fractures, or involvement of eyes and the central nervous system. Regarding competitive and new treatments with bispecific antibodies, response rates for the newly approved bispecific antibody teclistamab were lower in patients with extramedullary
disease and R-ISS stage III disease, whereas high response rates were consistent across patients with highrisk cytogenetic abnormalities and those with penta-drug refractory disease.15 Thus, together with results presented in our evidence synthesis, studies should evaluate the preferred treatment sequence or even a combination approach of bispecific antibody and CAR T cells for patients with high-risk cytogenetics and for those with extramedullary disease.16 For high-risk patients, earlier treatment with CAR T cells and consolidation/maintenance after the CAR T-cell therapy showed promising results in a recent small study.17 Whether this could overcome an initial poor prognosis and induce better immune surveillance should be investigated. In this regard, longitudinal assessment of changes of the immune microenvironment and timing of disease progression is urgently needed to understand the mechanisms of relapse and immune escape, especially in extramedullary disease.
In conclusion, high-risk cytogenetics were significantly associated with worse outcomes after CAR T-cell therapy for relapsed or refractory multiple myeloma. Although extramedullary disease showed promising initial responses, including decreases in measurable residual dis-

ease, the risk of relapse and mortality was increased significantly, so the results should be interpreted with caution and underscore the need for future research into mechanisms of relapse, the design of innovative treatment sequencing studies, and careful follow-up of patients even after an initial response.
Nico Gagelmann, Francis A. Ayuk, Evgeny Klyuchnikov, Christine Wolschke, Susanna Carolina Berger and Nicolaus Kröger
Department of Stem Cell Transplantation, University Medical Center
Hamburg-Eppendorf, Hamburg, Germany
Correspondence:
N. KRÖGER - nkroeger@uke.de
https://doi.org/10.3324/haematol.2022.282510
1. Bruno B, Wäsch R, Engelhardt M, et al. European Myeloma Network perspective on CAR T-cell therapies for multiple myeloma. Haematologica. 2021;106(8):2054-2065.
2. Gagelmann N, Riecken K, Wolschke C, et al. Development of CAR-T cell therapies for multiple myeloma. Leukemia. 2020;34(9):2317-2332.
3. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705-716.
4. Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 openlabel study. Lancet. 2021;398(10297):314-324.
5. Gagelmann N, Ayuk F, Atanackovic D, Kröger N. B cell maturation antigen-specific chimeric antigen receptor T cells for relapsed or refractory multiple myeloma: a meta-analysis. Eur J Haematol. 2020;104(4):318-327.
6. Gagelmann N, Sureda A, Montoto S, et al. Access to and affordability of CAR T-cell therapy in multiple myeloma: an EBMT position paper. Lancet Haematol. 2022;9(10):e786-e795.
7. Hillengass J, Cohen AD, Delforge M, et al. MM-183 CARTITUDE-2 cohort A: updated clinical data and biological correlative analyses of ciltacabtagene autoleucel (cilta-cel) in lenalidomide-refractory patients with progressive multiple myeloma (MM) after 1–3 prior lines of therapy (LOT). Clin Lymphoma Myeloma Leuk. 2022;22:S411.
8. Goldman-Mazur S, Kumar SK. Current approaches to management of high-risk multiple myeloma. Am J Hematol. 2021;96(7):854-871.
9. Giri S, Grimshaw A, Bal S, et al. Evaluation of daratumumab for
Received: December 4, 2022.
Accepted: February 17, 2023.
Early view: February 23, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
NG conceived the study design, searched literature, performed analyses, and wrote the first draft of the manuscript. NK searched the literature, performed analyses, interpreted results and wrote the first draft of the manuscript. FA, EG, CK and SCB interpreted results and wrote the manuscript. All authors approved the final version of the manuscript.
Data-sharing statement
Data can be made available if requested by e-mail to the corresponding author.
the treatment of multiple myeloma in patients with high-risk cytogenetic factors: a systematic review and meta-analysis. JAMA Oncol. 2020;6(11):1759-1765.
10. Moreno DF, Clapés V, Soler JA, et al. Real-world evidence of daratumumab monotherapy in relapsed/refractory multiple myeloma patients and efficacy on soft-tissue plasmacytomas. Clin Lymphoma Myeloma Leuk. 2022;22(8):635-642.
11. Higgins JPT, Altman DG, Gøtzsche PC, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ. 2011;343(7829):d5928.
12. Guyatt GH, Oxman AD, Vist G, et al. GRADE guidelines: 4. Rating the quality of evidence--study limitations (risk of bias). J Clin Epidemiol. 2011;64(4):407-415.
13. Sonneveld P, Avet-Loiseau H, Lonial S, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127(24):2955-2962.
14. Manjunath SH, Cohen AD, Lacey SF, et al. The safety of bridging radiation with anti-BCMA CAR T-cell therapy for multiple myeloma. Clin Cancer Res. 2021;27(23):6580-6590.
15. Moreau P, Garfall AL, van de Donk NWCJ, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495-505.
16. Mouhieddine TH, van Oekelen O, Melnekoff DT, et al. Sequencing T-cell redirection therapies leads to deep and durable responses in relapsed/refractory myeloma patients. Blood Adv. 2023;7(6):1056-1064.
17. Shi X, Yan L, Shang J, et al. Anti-CD19 and anti-BCMA CAR T cell therapy followed by lenalidomide maintenance after autologous stem-cell transplantation for high-risk newly diagnosed multiple myeloma. Am J Hematol. 2022;97(5):537-547.
Antithrombin (AT) is a key anticoagulant serpin that efficiently inhibits multiple procoagulant proteases, mainly thrombin and FXa. Congenital AT deficiency is the strongest thrombophilia and a complete absence of AT causes embryonic lethality.1
The prevalence of AT deficiency, which is evaluated mainly by chromogenic methods using anti-FXa functional assays,2 is estimated at 0.02% to 0.2% of the general population,3 and the annual risk of venous thromboembolism (VTE) in this patient group is 2.3%.4
Most cases with AT deficiency are caused by SERPINC1 defects (chromosome 1q25.1). Nearly all changes affecting this sensitive serpin have functional or structural consequences, making them deleterious, and only few missense polymorphisms with low frequency have been described until now.1

Of note, the majority of variants causing AT deficiency are private or found only in a single family and recurrent variants are very rare among patients. The identification and characterization of SERPINC1 may help to understand the pathogenic mechanisms, and their prognostic relevance.1 Here we report the genetic identification and clinical characterization of a recurrent SERPINC1 variant with a founder effect in Polish patients with AT deficiency. Figure 1 shows the results of the molecular characterization of the Polish cohort of 71 unrelated cases with AT deficiency, recruited within 9 years between 2013 and
2022. Relatives of selected probands were also assessed. Most cases are explained by single nucleotide variants (SNV); 34 causing missense, four nonsense, seven splicing, and six regulatory defects. We observed a considerable molecular heterogeneity as most variants were private or present in a single family and only four variants were identified in two or more unrelated probands. However, SERPINC1 (NM_000488.4): c.1157T>C (p.Ile386Thr) variant was found recurrently in ten Polish patients with AT deficiency (14.1% of the total cohort), always in a heterozygous state (Figure 2).
This variant was reported in the Human Gene Mutation database (HGMD): CM113684 (https://www.hgmd.cf.ac.uk/ ac/all.php). The variant was first identified in two patients from a German cohort of patients with AT deficiency,5 but it has also been sporadically reported in single Hungarian,6 Czech7 and French8 individuals, but not found it in 275 unrelated Spanish patients with AT deficiency. This distribution may be explained by the migration flow of the Polish population. All reported carriers belong to the CentralEastern European population mainly consisting of Slavs. Archaeological, historical, and genetic evidence shows that Slavs arrived in the land of contemporary Poland in the sixth century.9 The Malopolska region where all our patients live did not undergo significant population movements during the last 300 years. The latest infiltration of
about 18,000 inhabitants (3,000 families) from Germany occurred at the end of the eighth century,10 but it accounts for only 1% of inhabitants of this region and certainly had no influence on this population gene pool. Interestingly, this variant was very rare in the general population, (rs1449772752: 1/251466 alleles; MAF: 3.98e-6) is reported as pathogenic/likely pathogenic.11 According to the American College of Medical Genetics (ACMG), the in silico predictions for the SERPINC1 c.1157T>C (p.Ile386Thr) variant supported a likely pathogenic effect (PP5 strong; PP3 moderate and PM2 supporting) (Online Supplementary Table S1). These data and the high prevalence of this variant in the Polish cohort of patients with AT deficiency strongly suggest a founder effect i.e., a genetic variant observed with a relatively high frequency in a specific population, in which one or more of the ancestors carried the altered version of the gene. A founder mutation is always embedded in a longer stretch of DNA identical to that of the founder, i.e., a haplotype and is often responsible for more or less infrequent monogenic disorders.12 In order to verify this hypothesis, we evaluated the SERPINC1 gene in carriers of this variant by nanopore sequencing using two approaches: whole genome sequencing and sequencing of the whole gene using long-range polymerase chain reaction (PCR). Sequencing of long fragments using this method allows reading all variants of the same allele, so
phasing can be done to determine the haplotype linked to the variant. This study demonstrated that all carriers of the c.1157T>C (p.Ile386Thr) variant shared a common haplotype linked to the variant defined by six additional SNV, rs2227612, rs677, rs61827936, rs1799876, rs5878 and rs5877 (Figure 2C), which confirms the founder effect.

The c.1157T>C (p.Ile386Thr) variant causes a moderate AT deficiency according to the anticoagulant activity of carriers, with anti-FXa of 62.5±10.2% and anti-FIIa of 69.0±6.5%. Antigen levels were concordant with activity values 67.7±8.3% supporting a mild type I deficiency. However, an aberrant AT with slower electrophoretic mobility was observed in native gels (Online Supplementary Figure S1). Crossed immunoelectrophoresis confirmed the mild type I deficiency without increased levels of forms with low heparin affinity (Online Supplementary Figure S1). The variant affected one relatively conserved residue among the serpin superfamily located in the loop connecting helix I with strand s5A (Online Supplementary Figure S2).
Clinical data of ten unrelated probands (representing 14.1% of the whole cohort of Polish patients with this disorder) is shown in Table 1.
The mean age of the first thromboembolic event was 34.3±12.3 years. Nine patients were without any other thrombophilia and one patient (patient 3) was heterozygous for the factor V Leiden polymorphism. Positive family
Figure 2. Identification of the p.Ile386Thr variant. (A) Electropherogram of exon 6 of SERPINC1 in a patient carrying the variant and in a healthy control. In the patient, an arrow points the c.1157T>C variant. (B) Genotyping of the c.1157T>C variant in a representative family by polymerase chain reaction and and allele-specific restriction analysis (PCR-ASRA) using Rsa I. The restriction pattern of the wild-type allele is indicated by black arrows, while the presence of the variant generates a smaller fragment (red arrow). The proband is indicated by a green arrow. (C) Six polymorphisms defining the common haplotype shared by carriers of the variant of interest and the variant of interest itself plotted on the schematic representation of SERPINC1 gene; grey boxes – untranslated regions, white squares boxes – coding regions.
history was found in eight (80.0%) patients. At the time of diagnosis, five (50.0%) probands had a history of one thromboembolic event, while the remainder experienced two (n=4, 40.0%) or three (n=1, 10%) recurrent VTE episodes. Five (50%) patients had provoked thromboembolic events. Nine (90.0%) probands received non-vitamin K antagonist oral anticoagulants (NOAC), most commonly apixaban (n=5, 50.0%), followed by rivaroxaban (n=3, 30.0%) and dabigatran (n=1, 10.0%). One (10%) patient received warfarin. During
the median follow-up of 43 (interquartile range [IQR], 15.5-74.5) months, no thrombotic events were recorded. Three patients (patients 1, 3, 8) had COVID-19 with uninterrupted anticoagulation in March 2021, October 2020, and January 2022, respectively, without recurrent thromboembolism.
Analysis of 21 relatives carriers of the c.1157T>C (p.Ile386Thr) variant, showed that three (14%) experienced thromboembolic events (Table 1)
This study is the first to show that the c.1157T>C
CVST: cerebral venous sinus thrombosis; DVT: deep vein thrombosis; MI: myocardial infarction; OAT: oral anticoagulation therapy; PE: pulmonary embolism; TIA: transient ischemic attack; VTE: venous thromboembolism; F: female; M: male, yrs: years; mths: months; bid: twice daily; d: day. aNormal range: 79-112%/ 83-118%; bnormal range: 0.19-0.31 g/L. The AT activity was measured using an assay based on factor Xa inhibition (INNOVANCE ATIII, Siemens Healthcare Diagnostics, Marburg, Germany) or thrombin inhibition assays (Siemens Healthcare Diagnostics), the reference range for both was from 83% to 118%. The AT antigen was measured nephelometrically (Siemens Healthcare Diagnostics; reference range, 0.19–0.31 g/L). Results were verified in the Spanish laboratory by different assays: anti-FXa (Werfen, Bedford, USA) anti-FIIa evaluating both progressive and heparin cofactor activity using a home-made chromogenic assay.
(p.Ile386Thr) variant in SERPINC1, causing AT deficiency, seems to be restricted, or at least mainly found, in a Polish population. Indeed, gnomAD data revealed that this variant has been identified with a very low frequency (0.000008792) only in non-Finish Europeans and is completely absent in all other tested populations.
Our study supplies further data supporting the origin of this variant in the Polish population, specifically the presence of the common haplotype, which was obtained by a new method of long-range based sequencing with nanopore.13 Only three founder variants have been described as causative of AT deficiency, including two in Caucasian populations such as AT Budapest 3 (p.Leu131Phe) found in the Roma population14 and AT Basel (p.Pro73Leu) very frequent in the Finish population,15 and one African, p.Thr147Ala.16 These three previously described founder variants all causes type II deficiencies, which are characterized by a lower thrombotic risk than type I deficiencies.1,2 The Polish c.1157T>C (p.Ile386Thr) founder variant is the first one causing type I deficiency. However, we must point out that the deficiency caused by this variant is unusual, since the reduction of antigen (and anticoagulant activity) is not very severe (> 60%). Probably the variant caused an impaired secretion of the variant molecule having nearly normal anticoagulant activity. Indeed, the identification of traces of aberrant AT of lower electrophoretic mobility in plasma of the carriers and the relative conservation of the Ile386 residue in the serpin superfamily along with its structural localization (in the long loop connecting helix I with strand s5A) strongly suggest that this variant might have mild conformational consequences potentially favoring formation of oligomers that might be retained intracellularly. This hypothesis, which should be confirmed by further studies, could also explain the severe risk of thrombosis observed in carriers. Carriers of other conformational variants identified in SERPINC1 usually have a severe clinical phenotype with early and recurrent thrombotic events that usually require exogenous triggering factors to exacerbate the pathogenic consequences of the variant.16 We speculate that under normal conditions, the deficiency caused by the c.1157T>C (p.Ile386Thr) variant is mild and only increases the prothrombotic potential, which might be beneficial under certain conditions, thus representing a selective advantage for carriers that could also explain the expansion of the variant. This hypothesis has also been reported for the most common prothrombotic variant Factor V Leiden.16 However, under particular circumstances exacerbating the pathogenic consequences of the variant, the c.1157T>C (p.Ile386Thr)-related AT deficiency could probably lead to much higher risk of thrombosis, similar to that seen in the case of other SERPINC1 variants.18
The clinical characteristics of Polish patients with the recurrent c.1157T>C (p.Ile386Thr) variant differ from those
of subjects carrying the previously described variants with a founder effect in the Hungarian, Finnish and African origin populations.13,15,19 First, in our cohort, in addition to dominant VTE episodes in typical locations, we observed one uncommon thrombosis, i.e., cerebral venous sinus thrombosis (CVST) in a very young female patient who is a smoker and is on oral contraceptives. Secondly, in our carriers, arterial thrombosis was less common. We recorded one (10%) transient ischemic attack in a 49year-old patient, observed following major surgery despite thromboprophylaxis. In the study by Gindele et al., 19 in which the median age at the first thrombotic event in Hungarian heterozygous carriers of the p.Leu131Phe variant was similar to our cohort (33.0 vs. 34.4 years, respectively), 19.5% of all events were related to arterial thrombosis. Puurunen et al 15 in the study of large population of Finnish patients recorded a similar frequency of arterial thromboses, i.e., ten events (22% of all thrombotic episodes), including five strokes at a young age. Moreover, in our patients, obstetric complications were observed less frequently as well.
In conclusion, we report here that the c.1157T>C (p.Ile386Thr) variant is a pathogenic founder variant for AT-deficient Polish patients. The mutant allele shared a common haplotype involving six SERPINC1 SNV. It seems that under normal conditions, the deficiency caused by the p.Ile386Thr variant is mild and only increases the prothrombotic potential; however, when additional triggering factors are present, carriers of the p.Ile386Thr variant usually have a severe clinical phenotype with early and recurrent thrombotic events.
Anna Weronska,1* Belen de la Morena-Barrio,2* Sarah GoldmanMazur,3 Maria Eugenia de la Morena-Barrio,2 José Padilla,2 Antonia Miñano,2 Pedro Garrido-Rodriguez,2 Jacek Treliński,4 Daniel Piotr Potaczek,5,6,7 Anita Szczepanek,8,9 Anetta Undas,10,1 1 Javier Corral2# and Ewa Wypasek1,11#
1Faculty of Medicine and Health Sciences, Andrzej Frycz Modrzewski Krakow University, Kraków, Poland; 2Servicio de Hematología, Hospital Universitario Morales Meseguer, Centro Regional de Hemodonación, IMIB, CIBERER-ISCIII, Universidad de Murcia, Murcia, Spain; 3Department of Hematology, Cellular Therapy and Hemostaseology, Leipzig University Hospital, Leipzig, Germany; 4Department of Hemostasis Disorders, Medical University of Lodz, Łódź, Poland; 5Translational Inflammation Research Division and Core Facility for Single Cell Multiomics, Medical Faculty, Philipps University of Marburg, Marburg, Germany; 6Center for Infection and Genomics of the Lung (CIGL), Universities of Gießen and Marburg Lung Center (UGMLC), Gießen, Germany; 7Bioscientia MVZ Labor Mittelhessen GmbH, Gießen, Germany; 8Institute of Archaeology and Ethnology,
Polish Academy of Sciencies, Kraków, Poland; 9Department of Anatomy, Jagiellonian University Medical College, Kraków, Poland; 10Institute of Cardiology, Jagiellonian University Medical College, Kraków, Poland and 11John Paul II Hospital, Kraków, Poland
*AW and BdlM-B contributed equally as first authors. #JC and EW contributed equally as senior authors.
Correspondence:
E. WYPASEK - ewa.wypasek@wp.pl
https://doi.org/10.3324/haematol.2022.282459
Received: November 22, 2022.
Accepted: March 30, 2023.
Early view: April 6, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
1. Corral J, de la Morena-Barrio ME, Vicente V. The genetics of antithrombin. Thromb Res. 2018;169:23-29.
2. Van Cott EM, Orlando C, Moore GW, Cooper PC, Meijer P, Marlar R. Subcommittee on Plasma Coagulation Inhibitors. Recommendations for clinical laboratory testing for antithrombin deficiency; communication from the SSC of the ISTH. J Thromb Haemost. 2020;18(1):17-22.
3. Refaei M, Xing L, Lim W, Crowther M, Boonyawat K. Management of venous thromboembolism in patients with hereditary antithrombin deficiency and pregnancy: case report and review of the literature. Case Rep Hematol. 2017;2017:9261351.
4. Croles FN, Borjas-Howard J, Nasserinejad K, Leebeek FWG, Meijer K. Risk of venous thrombosis in antithrombin deficiency: a systematic review and Bayesian meta-analysis. Semin Thromb Hemost. 2018;44(4):315-326.
5. Luxembourg B, Delev D, Geisen C, et al. Molecular basis of antithrombin deficiency. Thromb Haemost. 2011;105(4):635-646.
6. Gindele R, Selmeczi A, Oláh Z, et al. Clinical and laboratory characteristics of antithrombin deficiencies: a large cohort study from a single diagnostic center. Thromb Res. 2017;160:119-128.
7. Provazníková D, Matýšková M, Čápová I, et al. Seventeen novel SERPINC1 variants causing hereditary antithrombin deficiency in a Czech population. Thromb Res. 2020;189:39-41.
8. Alhenc-Gelas M, Plu-Bureau G, Hugon-Rodin J, Picard V, Horellou MH. GFHT study group on genetic thrombophilia. Thrombotic risk according to SERPINC1 genotype in a large cohort of subjects with antithrombin inherited deficiency. Thromb Haemost. 2017;2;117(6):1040-1051.
9. Rębała K, Mikulich AI, Tsybovsky IS, et al. Y-STR variation among Slavs: evidence for the Slavic homeland in the middle Dnieper basin. J Hum Genet. 2007;52(5):406-414.
10. Wieloucha A. Kolonizacja józefińska w galicyjskich Karpatach,
Disclosures
No conflicts of interest to disclose.
Contributions
MM, DP and JC performed the research. JP, AM, PG and AS performed genetic analysis. JT, EW and AW performed data analysis. AW, SG and BM wrote the manuscript. JC, EW and AU supervised the study.
Funding
This study was supported by grants (N41/DBS/000682 to AU) from Jagiellonian University School of Medicine and from Instituto de Salud Carlos III, FEDER and Next Generation UE (PI21/00174 & PMP21/00052).
Data-sharing statement
Original data can be made available upon request to the corresponding author.
PŁAJ nr 19, wyd. Warszawa: Towarzystwo Karpackie; 1999.
11. National Center for Biotechnology Information. ClinVar; [VCV000863657.4], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000863657.4. Accessed Aug. 25, 2022.
12. Evans JA. Old meets new: identifying founder mutations in genetic disease. CMAJ. 2015;187(2):93-94.
13. Orlando C, de la Morena-Barrio B, Pareyn I, et al. Antithrombin p.Thr147Ala: the first founder mutation in people of African origin responsible for inherited antithrombin deficiency. Thromb Haemost. 2021;121(2):182-191.
14. Bereczky Z, Gindele R, Fiatal S, et al. Age and origin of the founder antithrombin Budapest 3 (p.Leu131Phe) mutation; its high prevalence in the Roma population and its association with cardiovascular diseases. Front Cardiovasc Med. 2021;5;7:617711.
15. Puurunen M, Salo P, Engelbarth S, Javela K, Perola M. Type II antithrombin deficiency caused by a founder mutation Pro73Leu in the Finnish population: clinical picture. J Thromb Haemost. 2013;11(10):1844-1849.
16. Corral J, Vicente V, Carrell RW. Thrombosis as a conformational disease. Haematologica. 2005;90:238-246.
17. Van Mens TE, Levi M, Middeldorp S. Evolution of Factor V Leiden. Thromb Haemost. 2013;110(1):23-30.
18. De la Morena-Barrio ME, Suchon P, Jacobsen EM, et al. Two SERPINC1 variants affecting N-glycosylation of Asn224 cause severe thrombophilia not detected by functional assays. Blood. 2022;14;140(2):140-151.
19. Gindele R, Oláh Z, Ilonczai P, et al. Founder effect is responsible for the p.Leu131Phe heparin-binding-site antithrombin mutation common in Hungary: phenotype analysis in a large cohort. J Thromb Haemost. 2016;14(4):704-715.
Nijmegen breakage syndrome (NBS) is an inherited DNA repair disorder characterized by a high predisposition to develop lymphoid malignancies during childhood, with diffuse large B-cell lymphoma (DLBCL) being one of the leading types.1 Due to concomitant immunodeficiency and an increased risk of chemotherapy-induced toxicity, NBS patients often require modified and individualized cancer treatment.2 Nevertheless, they rarely achieve progressionand relapse-free long-term survival rates without hematopoietic stem cell transplantation.3 Unfavorable outcome of lymphomas in NBS may result not only from the reduction of drug dosages but could be associated with molecular aberrations occurring on the background of chromosomal instability. Therefore, we aimed to investigate clinical outcome, histopathological features, and genomic alterations of DLBCL in pediatric patients with NBS. The algorithm of the study is shown in the Online Supplementary Figure S1A
First, we estimated the probability of overall survival (OS) depending on NBS status in 50 children with newly diagnosed DLBCL, including nine (18%) NBS patients. They were treated in the 13 centers of the Polish Pediatric Leukemia/Lymphoma Study Group between 2014 and 2021 (NonNBS) and between 1993 and 2021 (NBS). All of the individuals with NBS carried the germline biallelic founder mutation c.657_661del5, p.Lys219Asnfs in the NBN gene (NM_001024688.3). The median age at diagnosis of lymphoma was 12.97 years (interquartile range [IQR], 7.9815.64) and was similar in NBS and nonNBS individuals: 13.01 (IQR, 6.73-14.85) versus 12.94 (IQR, 8.39-15.64; P=0.11). The median observation time was 2.42 (IQR, 0.89-4.70) years. During observation time, nine children died: seven (78%) NBS and two (22%) NonNBS (P<0.001). The probability of 5-years OS was significantly lower in patients with NBS as compared to NonNBS individuals: 33.33% versus 95.12%, respectively (P<0.001) (Figure 1A). Regarding the causes of death in NBS patients, lymphoma progression was the most common (n=4) and was followed by: excessive toxicity (n=1), and infectious complications (n=2). While both NonNBS children experienced fatal treatmentrelated toxicity.
Next, we performed a two-step pathology review of tumor tissues, by the local and the central hematopathologists, following criteria for aggressive B-cell lymphomas included in the 4th World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissues.4 Six-
teen pediatric patients with diagnosis of DLBCL had available formalin-fixed paraffin-embedded (FFPE) tissue blocks for extended pathological analysis including nine NonNBS (56.2%) and seven with NBS (43.8%). Our histopathological analysis demonstrated that NBS-positive lymphomas were more similar to adult-type than to pediatric-type DLBCL NOS. The tumors were predominantly classified as ABC and non-double expressors (NonDE) had a high Ki67 proliferation index and presented heterogenous morphology. Of note, none of the NBS lymphomas displayed positivity for CD10, and, in contrast, six (n=6/9, 66.7%) NonNBS tumors were positive for CD10; P=0.010. (Table 1A). CD10-negativity and other pathological features might be regarded as indicators of poor prognosis of NBS DLBCL when compared to non-NBS DLBCL.5
In order to delineate the entire spectrum of genetic alterations in DLBCL in NBS, whole genome sequencing (WGS) using Illumina TruSeq Nano 550 polymerase chain reaction (PCR)-free library preparation protocol was executed. The 150 bp paired-end sequences from the tumor and matched normal genomes were generated corresponding to an average per-base sequencing coverage of 107-fold and 32-fold, respectively. After applying a filtering algorithm, we detected an average of 8.763 somatic sequence alterations (median: 5.735; resulting in a tumor mutation burden [TMB] of 2.89 mutations/Mb/sample on average), including in total 39.021 single nucleotide variants (SNV) and 13.558 insertions and deletions, alongside 158 structural variants (SV). DLBCL in NBS patients shared a common predominant mutational signature (COSMIC SBS 9; see Figure 2), which has been frequently reported in malignant B-cell lymphomas.6 It is associated with hyperactivation of AID enzyme and aberrant activity of DNA polymerase η during illegitimate somatic hypermutation events outside of immunoglobulin loci.7 In two DLBCL cases an aggressive course of the disease could be explained by the presence of TP53 lesions or high TMB concomitant with PDL1 amplification, respectively. In PDL1-amplified sample we additionally observed kataegis at 7qter. The number of SNV and TMB in this DLBCL was n=22,334 and 7.9%, respectively compared to the median number of SNV and TMB n=1,999 (IQR, 1,328-6,016) and 1.97% (IQR, 1.145-2.6%) in the remaining genomes. TP53 is affected in 8.9-31.7% of DLBCL and in the majority of studies predicted worse outcome.8 PD-L1 gene translocations and amplification, were observed in ~10% of DLBCL
Figure 1. Patients with Nijmegen breakage syndrome, who developed diffuse large B-cell lymphoma (DLBCL), showed not only significantly lower 5-year overall survival, but also different tumor-gene expression profile when compared to non-syndromic DLBCL patients. (A) Kaplan–Meier curves displaying the probability of overall survival among pediatric Nijmegen breakage syndrome (NBS) patients with DLBCL (the red line) and non-syndromic individuals (the blue line): 33.33% vs. 95.12%, respectively (P<0.001). The comparison of tumor gene expression profile between (B) NBS vs. NonNBS, (C) germinal center B cell (GCB) vs. NonGCB, and (D) NBS vs NonNBS within the subgroup of NonGCB DLBCL. Differences are shown with respect to the top of 25 differentially expressed genes sorted by weight*PLSDA coefficient for LV1 (group: NBS vs. NonNBS) and LV2 (molecular subtype: GCB vs. NonGCB) for the comparison between: (E) NBS vs. NonNBS, (F) GCB vs. NonGCB, and (G) NBS vs. NonNBS among NonGCB patients. These genes were further grouped based on their functions (see the Online Supplementary Figure S1B).

patients,9,10 especially in patients less than 60 years old.11 Although in all but one sample driver mutations were dissected, only a slight overlap of affected genes was present between the lymphoma samples, e.g., STAT3, KMT2D,
HIST1H1B was found in two samples for each of the genes (Table 1B). Based on the knowledge that genomic instability is an intrinsic feature of NBS, we also searched for somatic SV that could have evolved due to inheritance.
(A) The pathological characteristic of DLBCL samples
DLBCL _NBS1
(B) Pathogenic somatic alterations detected in NBS DLBCL
Genes with identi
seq[hg19]11 q24.2-q25(127130254-131045787)x5;
seq[hg19]5p15.33(11881-1998109)x6;
seq[hg19]7q21.3-q36.3(97119555-15512689)x7-8; seq[hg19]9p24.3-p23(1280059-9700448)x5-16 (amplification of 9p24.3-p23 involving locus PDL1)
DLBCL _NBS2 duplication:
DLBCL _NBS3
DLBCL _NBS4
DLBCL _NBS5
DLBCL _NBS7
seq[hg19]2p16.1(60753592-61234194)x3
PTEN, KMT2D, BTG1, BTG2, PIK3CG, BTX, STAT3, FAT1, SMARCE1
STAT3 deletion; PTPRD truncating translocation
not present inverted duplication of BCL11A
seq[hg19]3q12.1-q13.2(98567732-117599362)x3, seq[hg19]11p15.5-p11.2(48684843-48597576)x3 LYN, KMT2D, FAS possible fusion DOPEY2-TTC3
seq[hg19]11p22.1-q22.2 (100890992-102613285)x14-16
(possible hyperactivation of YAP1 and BIRC3); seq[hg19]18p21.2(48430066-49202094)x6; seq[hg19]Xq22.3-26.2(107296028-133033076)x5; seq[hg19]Xq28(148131415-154929412)x5
TP53 (+ TP53 LOH), HIST1H1B, FAT4
SPEN deletion Chromoanagenesis on chromosome 6q
IRF4 translocation, DLEU-KLF12 fusion Chromoanagenesis on chromosome 6p
(A) Differences regarding histopathological features of diffuse large B-cell lymphoma (DLBCL) between children with Nijmegen breakage syndrome (NBS) and non-syndromic pediatric individuals. The qualitative variables were presented as numbers followed by percentages of respective subgroups and quantitative variables were presented as medians followed by quartiles in brackets. #Pearson’s X2 test, ^two-tailed Fisher’s X2 test, *Mann Whitney U-test. (B) Pathogenic somatic alterations detected in DLBCL samples in patients with NBS. DLBCL NOS: diffuse large B-cell lymphoma not-otherwise specified. GCB: germinal center B cell; ABC: activated B cell-like; IQR: interquartile range; SNV: single nucleotide variant; Indels: insertions and deletions.
Beside of PDL1 amplification, the following SV were detected: amplification within the chromosome 11p22.1p22.2 leading to hyperactivation of YAP1 and BIRC3, inverted duplication of BCL11A, and in two DLBCL samples the possible incidence of chromoanagenesis on chromosome 6. Complex structural variants events including chromothripsis, chromoplexy, and templated insertion has been observed in non-syndromic, newly diagnosed and relapsed DLBCL patients. However, only chromothripsis co-occurred with APOBEC signature and poor outcome.12 We further investigated the difference in gene expression profiles between DLBCL in patients with NBS and nonsyndromic individuals according to cell-of-origin (COO) subtype (germinal center B cell [GCB] or activated B cell-
like [ABC]). In order to enrich the study group in ABC cases represented the minority among children without NBS, we additionally included 28 adult DLBCL samples representing both DLBCL subtypes (ABC n=13, GCB n=13) in the transcriptomic assessment. RNA sequencing was performed using SureSelect XT RNA Direct Human All Exon V6+UTR (Agilent Technologies, USA) and NextSeq™ 500/550 High Output Kit v2.5 (300 cycles; Illumina, USA). Differential expression analysis using DESeq2 revealed significant downor upregulation of 195 (23.01%) genes between NBS versus NonNBS samples. However, the transcriptomic differences were partly affected by the representation of COO subtypes within both groups, GCB versus ABC DLBCL (n=524 genes, 22.45%). Therefore, we narrowed our analysis to

ABC patients, a frequent subtype in NBS, and found differential expression of 197 genes between NBS and nonsyndromic tumors (11.24%). We employed the PLS-DA approach to visualize the differences between NBS versus NonNBS, GCB versus ABC, as well as NBS versus NonNBS within the group of ABC patients. The top 25 differentiating genes for these comparisons are shown in Figure 1B-G. We subsequently grouped these genes according to their cellular function (Online Supplementary Figure S1B). Among several groups of genes, NBS tumors showed downregulation of cAMP (PRKAR1A) which has been described as an aberrations promoting tumorgenesis in different types of malignancies, such as melanoma, lung cancer, and pancreatic tumors.13 Additionally, targeting cAMP and its effectors may be a possible cancer treatment strategy.14
Considering relatively frequent incidence of Epstein Barr virus (EBV) infection or reactivation that precedes DLBCL development in NBS patients and the fact that EBV is one of the prominent infection-related causes of cancer, we aimed to investigate viral DNA incorporation into the tumor genomes.15,16 Three of seven NBS DLBCL samples showed an enrichment of the number of EBV integration sites in somatic genomes with respect to non-cancer DNA, using the threshold of ten times difference between tumor and normal tissues. This reflected the median number of EBV-human split reads reaching n=2,047 (range, 62-6,743) in EBV-positive lymphomas as compared to n=12 (range, 9-19) in the remaining four EBV-negative DLBCL cases ( P=0.05). Next, we searched for recurrent sites of EBV incorporation in the positive DLBCL samples and found five common EBV locations including three sites that were present in each of the tumor: on chromosome 2 (33141296-33141626, within the LINC00486), on chromosome 7 (105741882-105742671; within the intron 1 of SYPL1) and on chromosome 14 (99887280-99887288; within the intron 6 of SETD3) (Online Supplementary Table S1A). However, all of these recurrent EBV integration sites exclusively affected non-coding regions including simple repeats (polyG, polyT, polyA), introns, and Alu elements, the latter ones also being binding sites for several B-cell specific transcription factors (data not shown). This observation is in the line with WGS results obtained by Zapatka et al. who studied viral integration in more than 2,500 cancers across 38 tumor types in which it was found only 3.4% of integrations located in gene coding sequences.16 Interestingly, one of the recurrent EBV incorporation in DLBCL genomes occurred at the short arm of chromosome 2 within the poly-G tract of LINC00486. We analyzed the incidence of similar long poly-G tracts (>100 nt and G content >85%) in the hg19 reference genome that could be incorrectly assigned to the site on chromosome 2. However, this genomic region showing preferential EBV incorporation turned out to be a unique sequence in the human genome regarding the length and high
amount of G content. Moreover, it has been already described as an integration site of hepatitis B virus into the somatic genome of intrahepatic cholangiocarcinoma.17 In order to investigate whether identified recurrent sites of EBV integration are unique for tumors in NBS patients, we performed deep WGS of genomes extracted from four non-syndromic and two NBS-positive EBV-immortalized cell lines. All of them showed the enrichment of viral incorporation in the previously observed chromosomal regions in NBS tumors (Online Supplementary Table S1B). These observations raise the question whether and why this region could be a preferential site of viral integration and what the functional consequences of the specific EBV incorporation could be.
To the best of our knowledge this is the first study providing a comprehensive analysis of tumor morphology and molecular aberrations that could contribute to tumor development in NBS patients. However, one of the essential limitations of this work is the limited number of DLBCL in NBS included in WGS due to unavailability of fresh frozen tumor tissue. This fact, combined with a significant molecular heterogeneity of the DLBCL, positions our results as a preliminary report requiring further investigation and improvements.
Agata Pastorczak,1 Bartosz Szmyd,1 Marcin Braun,2 Joanna Madzio,1 Kamila Wypyszczak,1 Pawel Sztromwasser,3 Wojciech Fendler,3 Marzena Wojtaszewska,4 Jedrzej Chrzanowski,3 Wieslawa Grajkowska,5 Hanna Gregorek,6 Anna Wakulinska,7 Bernarda Kazanowska,8 Zdenka Krenova,9,10 Dilys D. Weijers,11 Roland P. Kuiper11,12 and Wojciech Mlynarski1
1Department of Pediatrics, Oncology and Hematology, Medical University of Lodz, Lodz, Poland; 2Department of Pathology, Medical University of Lodz, Lodz, Poland; 3Department of Biostatistics and Translational Medicine, Medical University of Lodz, Lodz, Poland; 4Department of Hematology, Frederic Chopin Provincial Specialist Hospital, Rzeszow, Poland; 6Department of Microbiology and Clinical Immunology, Children's Memorial Health Institute, Warsaw, Poland; 5Department of Pathology, The Children's Memorial Health Institute, Warsaw, Poland; 7Department of Oncology, The Children's Memorial Health Institute, Warsaw, Poland; 8Department of Pediatric Hematology, Oncology and Transplantology, Medical University of Wroclaw, Wroclaw, Poland; 9Department of Pediatric Oncology, University Hospital and Faculty of Medicine, Masaryk University, Brno, Czech Republic; 10Department of Pediatric Oncology, University Hospital Brno and Faculty of Medicine, Masaryk University, Brno, Czech Republic; 11Princess Máxima Center for Pediatric Oncology, Utrecht, the Netherlands and 12Department of Genetics, Utrecht University Medical Center, Utrecht University, Utrecht, the Netherlands
Correspondence:
A. PASTORCZAK - agata.pastorczak@umed.lodz.pl
https://doi.org/10.3324/haematol.2022.282325
Received: November 20, 2022.
Accepted: March 29, 2023.
Early view: April 6, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
AP and WM designed the research. AP and BSz analyzed the data and wrote the paper. JM, KW and BSz performed RNA sequencing experiments. PS and MW provided bioinformatic support in WGS
1. Szmyd B, Mlynarski W, Pastorczak A. Genetic predisposition to lymphomas: Overview of rare syndromes and inherited familial variants. Mutat Res Rev Mutat Res. 2021;788:108386.
2. Pastorczak A, Attarbaschi A, Bomken S, et al. Consensus recommendations for the clinical management of hematological malignancies in patients with DNA double stranded break disorders. Cancers. 2022;14(8):2000.
3. Wolska-Kusnierz B, Pastorczak A, Fendler W, et al. Hematopoietic stem cell transplantation positively affects the natural history of cancer in Nijmegen breakage syndrome. Clin Cancer Res. 2021;27(2):575-584.
4. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
5. Gutiérrez-García G, Cardesa-Salzmann T, Climent F, et al. Geneexpression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Blood. 2011;117(18):4836-4843.
6. Ye X, Ren W, Liu D, et al. Genome-wide mutational signatures revealed distinct developmental paths for human B cell lymphomas. J Exp Med. 2021;218(2):e20200573.
7. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415-421.
8. Lopez-Santillan M, Lopez-Lopez E, Alvarez-Gonzalez P, et al. Prognostic and therapeutic value of somatic mutations in diffuse large B-cell lymphoma: a systematic review. Crit Rev
analysis. DDW and RPK performed WGS and interpreted data. WG and MB conducted histopathological analysis. JCh and WF interpreted data analysis and visualization. WG, MB, HG, AW, BK and ZK provided patients samples and outcome data. WM supervised the project. All authors have read and agreed to the published version of the manuscript.
Funding
AP, BSz and WM were supported by the National Science Center grants 2017/26/D/NZ5/00811 (to AP) and 2020/39/O/NZ2/02954 (to BSz, WM). AP, ZK, RPK and WM were supported by the LEukemia GENe Discovery (LEGEND) by data sharing, mining and collaboration (COST Action - CA16223) 2017-2021. BSz was supported by the Ministry of Education and Science “Diamond Grant” (0136/DIA/2020/49). DDW is supported by grant KWF12090 from the Dutch Cancer Society.
Data-sharing statement
Data are available upon request to the corresponding author.
Oncol Hematol. 2021;165:103430.
9. Huang S, Nong L, Liang L, et al. Comparison of PD-L1 detection assays and corresponding significance in evaluation of diffuse large B-cell lymphoma. Cancer Med. 2019;8(8):3831-3845.
10. Kwon HJ, Yang JM, Lee J-O, Lee JS, Paik JH. Clinicopathologic implication of PD-L1 and phosphorylated STAT3 expression in diffuse large B cell lymphoma. J Transl Med. 2018;16(1):320.
11. Wang Y, Wenzl K, Manske MK, et al. Amplification of 9p24.1 in diffuse large B-cell lymphoma identifies a unique subset of cases that resemble primary mediastinal large B-cell lymphoma. Blood Cancer J. 2019;9(9):73.
12. Jain MD, Ziccheddu B, Coughlin CA, et al. Genomic drivers of large B-cell lymphoma resistance to CD19 CAR-T therapy. Blood. 2021;138(Suppl 1):S42.
13. Sapio L, Di Maiolo F, Illiano M, et al. Targeting protein kinase a in cancer therapy: an update. EXCLI J. 2014;13:843-855.
14. Ahmed MB, Alghamdi AAA, Islam SU, Lee J-S, Lee Y-S. cAMP Signaling in cancer: a PKA-CREB and EPAC-centric approach. Cells. 2022;11(13):2020.
15. Georgiou K, Chen L, Berglund M, et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood. 2016;127(24):3026-3034.
16. Zapatka M, Borozan I, Brewer DS, et al. The landscape of viral associations in human cancers. Nat Genet. 2020;52(3):320-330.
17. Li M, Du M, Cong H, et al. Characterization of hepatitis B virus DNA integration patterns in intrahepatic cholangiocarcinoma. Hepatol Res. 2021;51(1):102-115.
Post-transplant lymphoproliferative disorders (PTLD) are a heterogeneous group of conditions that involve uncontrolled proliferation of lymphoid cells developing as a consequence of extrinsic immunosuppression after solid organ or hematopoietic stem cell transplantation.1 The 2022 World Health Organization (WHO) classification distinguishes lymphomas from other proliferations arising in the setting of immune deficiency and further categorizes them as in immunocompetent patients, the most frequent being diffuse large B-cell lymphoma (DLBCL).
Based on several phase II trials,2,3 rituximab-based therapy following reduction of immunosuppressive therapy (RIS) is now standard of care for most CD20+ PTLD. Sequential therapy (4 weekly courses of rituximab either completed by 4 more courses of rituximab in responders [complete remission [CR] and low-risk partial remission [PR] group: 25-26%] or by 4 cycles of rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone every 21 days [R-CHOP-21] in patients not responding to rituximab monotherapy sufficiently [74-75%]) showed encouraging results for CD20+ B-cell PTLD with 7-8% treatment-related-mortality (TRM), 2-year progression-free survival (PFS) of 56-67% and 2-year overall survival (OS) of 6872%.4,5 Current PTLD guidelines encourage sequential therapy in CD20+ polymorphic and monomorphic DLBCL-type PTLD that fail to respond to upfront RIS or if RIS is not feasible.6
Burkitt lymphoma (BL) PTLD (BL-PTLD) is a rare subtype of post-transplant lymphoma representing less than 10% of adult PTLD.7 Its immune-competent counterpart is characterized by a highly aggressive clinical course, frequent involvement of bone marrow (BM) and central nervous system (CNS), and is biologically hallmarked by MYC gene translocation to an immunoglobulin locus. Due to the reduced number of BL-PTLD cases, their clinical characteristics, outcome, and most adequate therapeutic options are not well established.8–10 We conducted a multicenter retrospective study in order to better characterize these elements.
Our study included adult patients diagnosed with BL-PTLD between 1989 and 2022 who were identified using the French registry of PTLD after kidney transplantation, the K-Virogref network, the Lymphopath database and the prospective German PTLD registry. After the review of medical and pathological records, 55 patients treated in 20 French and eight German centers were included. The study was approved by national (CNIL 913611) and local (CPP Ile-De-France PP 13-022) ethics committees. Diagno-
sis of BL-PTLD followed criteria from the 2022 revised edition of the WHO classification,11 taking into account morphology, immunohistochemistry (CD20, CD10, BCL2, BCL6, MUM-1, Ki67 data) and cytogenetics (C-MYC, BCL2, BCL6 in fluorescence in situ hybridization [FISH] and/or karyotype). Association with Eppstein Barr virus (EBV) was assessed by in situ hybridization for Epstein Barr-encoded small RNA (EBER). Among different therapeutic options, the most frequently used chemotherapies were: CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), EPOCH (etoposide, doxorubicin, vincristine, cyclophosphamide, prednisone), COPADEM (vincristine, methotrexate, cyclophosphamide, doxorubicin, prednisone 60 mg/m2) and hyperCVAD (hyperfractioned cyclophosphamide, vincristine, doxorubicin, dexamethasone). Response to treatment followed standard international criteria.12,13 Subgroup comparisons were performed using Χ2 of Fisher’s exact test for categorical variables, and Student’s t-test for continuous variables. Time-to-event outcomes were estimated using Kaplan-Meier curves, and were compared using the logrank test. The level of significance in all analyses was set at P<0.05. Statistical analysis was performed using R Studio 2021.09.1.
The study population comprised 55 patients, with a median age at diagnosis of 40.3 years and the most frequently transplanted organs were kidney (n=37) and liver (n=10) (Table 1). Median time from transplant to BL-PTLD diagnosis was 5.4 years (range, 0.2-24). Most patients presented with an aggressive and advanced stage of disease at diagnosis: Eastern Cooperative Oncology Group (ECOG) score ≥2 in 44.6% (n=21/47 cases), elevated lactate dehydrogenase (LDH) in 84% (n=41/49), Ann Arbor stage IV in 84% (n=46/55) patients. Extranodal disease was frequent (n=49, 89%), with the digestive tract being the most frequently involved organ (n=29, 53%). CNS infiltration was observed in seven patients (13%). BL-PTLD tumor samples were all CD20+, CD10+, BCL2-, expressed Ki67 in more than 90% of cells and harbored MYC rearrangement detected by FISH. EBER was positive in 12 of 29 samples (41%) and LMP was positive in one of nine (11%).
Fifty-three patients received curative-intended therapy while two patients received supportive care only. RIS never was proposed as a unique initial strategy but was performed in 42 of 53 (79%) patients. Fourteen of 53 (25%) patients were treated according to sequential strategies. These patients received up to four weekly courses of rituximab followed either by: (i) rituximab consolidation (4 3-weekly courses) in a single case of CR (1/14, 7%) or (ii)
BL-PTLD: Burkitt lymphoma post-transplantation lymphoproliferative disorder; ECOG: Eastern Cooperative Oncology Group; LDH: lactate dehydrogenase; CNS: central nervous system; IQR: interquartile range.
subsequent chemo(immuno)therapy (C(I)T) (n=13/14, 93%): CHOP (n=2), R-CHOP (n=9, including one with concomitant high dose methotrexate [MTX]), R-COPADEM followed by R-CYM (n=1), unknown (n=1). Seven (13%) patients received frontline polychemotherapy (CT) (COPADEM [n=6] and CHOP [n=1]) and 32 (60%) were treated with CIT (R-COPA-
DEM [n=12], R-CHOP [n=13, 7 without MTX, 6 with sequential or concomitant MTX], R-EPOCH [n=5], R-hyperCVAD [n=1], rituximab with whole brain radiotherapy [WBRT] and 90Y-ibritumomab tiuxetan [n=1]) (Online Supplementary Figure S1). There was no significant difference between patients who had received sequential therapy or frontline CT/CIT, except that patients with CNS involvement were all treated with CT/CIT and those treated with sequential therapy had a lower ECOG score (Online Supplementary Table S1).
Median follow-up was 7.5 years (95% confidence interval [CI]: 5.8-10.5). Median OS and PFS were 7.1 (95% CI: 1.7-not applicable [NA])and 4.9 years (95% CI: 0.7-NA) respectively (Figure 1A, B). Thirty-eight patients (70%) attained CR after first-line treatment, including four of seven (57%) who received CT, 25 of 32 (78%) after CIT, and nine of 14 (64%) after completing sequential treatment. With sequential treatment, the overall response rate (CR + PR) to four doses of rituximab was 21% (1 CR, 2 PR). Among patients receiving frontline CIT, CR rates were 61%, 100% and 83% for patients treated with R-CHOP, R-EPOCH and R-COPADEM respectively, without significant statistical difference (P=0.26). Relapses occurred in four patients who had attained CR after a median of 6 months. Two had received upfront CIT and two sequential therapy. Out of these, only a patient relapsing after sequential therapy containing RCHOP was successfully rescued by another CIT regimen. Three-year OS and PFS for the whole cohort were 58% (95% CI: 45.8-73.1) and 54% (95% CI: 41.9-68.9), respectively. For patients receiving upfront CIT, 3-year OS and PFS were 59% (95% CI: 42.9-80.1) and 58% (95% CI: 43-78.5). For patients receiving sequential therapy, 3-year OS and PFS were 64% (95% CI: 43.5-95.0) and 50% (95% CI: 29.684.4) (Figure 1C, D). OS and PFS after CIT and sequential therapy were not statistically different. There was no significant difference in PFS or OS between patients receiving upfront R-CHOP or R-COPADEM regimens (Figure 1E, F). Patients with CNS involvement (n=7) were treated with intrathecal (n=3) or high-dose intravenous MTX (n=2) or both (n=1) or WBRT (n=1) and five of them achieved CR. Twentytwo of 48 (46%) patients without CNS involvement received CNS prophylaxis during first-line therapy (either by intrathecal or systemic chemotherapy containing cytarabine or MTX). CNS prophylaxis had no impact on OS or PFS (data not shown).
Twenty-nine deaths occurred during follow-up mainly due to progressive disease (n=11, 38%). Treatment-related deaths (n=7, 24%; including sepsis, n=6; cerebral hemorrhage, n=1) occurred during frontline CIT treatment in five patients and in the relapse setting in two patients (Online Supplementary Table S2).
Univariate analysis showed that age over 50 years, ECOG score and BM involvement were significant predictors for poorer OS whereas complete response to first-line therapy
Figure 1. Survival for the whole cohort, according to the type of therapy and according to the type of chemoimmunotherapy. (A) Overall survival (OS) and (B) progression free-survival (PFS) for the whole Burkitt lymphoma post-transplantation lymphoproliferative disorder (BL-PTLD) cohort. The dashed lines indicate the respective times of median survival. (C) OS and (D) PFS by type of therapies used: chemotherapy (CT, N=7) (blue line), chemoimmunotherapy (CIT, N=32) (red line) and sequential therapy (sequential, N=14) (green line). (E) OS, (F) PFS according to the type of CIT regimen: R-CHOP (N=13) (blue line), R-COPADEM (N=12) (red line). R-CHOP: cyclophosphamide, doxorubicin, prednisone, rituximab and vincristine; R-COPADEM: rituximab, cyclophosphamide, vincristine, prednisone, doxorubicin, and methotrexate.

was associated with longer survival and was the only variable retaining significance in multivariate analysis (Table 2). Complete response to first-line therapy was not significantly associated with other variables (i.e., age, transplanted organ, EBER, ECOG score, LDH levels, stage, BM or CNS involvement, or type of treatment).
To our knowledge, this is the largest retrospective series of BL-PTLD to date. BL-PTLD is a late-onset PTLD (likePTLD5) with an aggressive presentation, frequent advanced disease, altered performance status and CNS involvement. In our series, EBV association (41%) appears to be similar to what is observed in DLBCL-PTLD (around 50%) and differs from previous series of BL-PTLD,7,9 in keeping with publications suggesting that late-onset PTLD are more frequently EBV-negative.1 Reflecting the lack of consensus, our retrospective BL-PTLD cohort used a variety of therapeutic approaches, ranging from high-dose chemotherapy and R-CHOP-based regimens to rituximab monotherapy.
In a sequential strategy setting, the CR rate after the first four courses of rituximab monotherapy was very low (7%), but subsequent CIT provided disease control similar to the frontline CIT strategy (similar CR rate, OS and PFS). Although rituximab monotherapy does not appear to be a valuable option in BL-PTLD, sequential therapy could be discussed to avoid early complications associated with upfront C(I)T, considering the relatively high TRM observed in this series (13%). R-CHOP-based regimens yielded similar CR rates, PFS and OS compared to more dose-intensive regimens such as R-COPADEM and represent a valuable option in BL-PTLD, even with only four cycles as part of sequential therapy. In univariate analyses, age, ECOG score and BM involvement were significantly associated with poorer OS as in the immunocompetent BL cohort from which the BL-IPI was derived,14 while complete response to first-line therapy was the strongest prognostic factor for longer OS in univariate and multivariate analyses. One
Table 2. Predictors of overall survival (Cox model, univariate and multivariate).
*Late and early were defined as BL-PTLD occurring before and after 12 months post-transplantation respectively. **For sequential strategy, response was evaluated at the end of the procedure (which means after CIT in case of insufficient response to initial rituximab monotherapy). CI: confidence interval; PTLD: post-transplantation lymphoproliferative disorder; EBER: Epstein Barr-encoded small RNA; ECOG: Eastern Cooperative Oncology Group; LDH: lactate dehydrogenase; CIT: chemoimmunotherapy; CNS: central nervous system.
of the main limitations of our work is its retrospective nature and the use of different sources for patient selection. The fact that the majority of cases occurred after kidney transplantation is probably partly related to selection bias, but also to the fact that kidney transplantation is by far the most frequent transplantation in adults. This may partly explain the contrast between our data and a recently published pediatric cohort of BL-PTLD,15 in which most patients had received heart or liver transplantation, although other elements (such as higher EBV association) may suggest a different disease biology.
In conclusion, our study confirms that BL-PTLD is an aggressive, late-onset PTLD with frequent extranodal and CNS localization. Although initial rituximab monotherapy is not as effective as for non-Burkitt PTLD, sequential strategy was as effective as frontline CIT regimens among which R-CHOP and more dose intensive regimens showed similar outcomes.
Pierre Walczak,1 Sylvain Choquet,1 Jacques Dantal,2 David Boutboul,3
Felipe Suarez,4 Marine Baron,1 Véronique Morel,1 Thomas Cluzeau,5 Mohamed Touati,6 Michelle Elias,7 Emmanuel Bachy,8 Emmanuelle
Nicolas-Virelizier,9 Roch Houot,10 Geoffroy Venton,11 Caroline Jacquet,12
Marie-Pierre Moles-Moreau,13 Fabrice Jardin,14 Eric Durot,15 Noureddine
Balegroune,1 Laure Ecotière,16 Romain Guièze,17 Nassim Kamar,18 Loïc
Ysebaert,19 Lionel Couzi,20 Hugo Gonzalez,21 Louise Roulin,22 Kevin Ou,23 Sophie Caillard,24 Heiner Zimmermann,25,26 Ralf Ulrich Trappe,26,27 and Damien Roos-Weil1 for the K-VIROGREF study group
1Sorbonne Université, Service d'Hématologie Clinique, Hôpital PitiéSalpêtrière, APHP, Paris, France; 2Institut de Transplantation Urologie Néphrologie (ITUN), Service de Néphrologie et Immunologie clinique, CHU Nantes, Nantes, France; 3Service d’Immunologie Clinique, Hôpital Saint-Louis, APHP, Paris, France; 4Service d’Hématologie Clinique, Hôpital Necker, APHP, Paris, France; 5Service d'Hématologie Clinique, CHU de Nice, Nice, France; 6Service d’Hématologie Clinique, CHU Limoges, Limoges, France; 7Service de Néphrologie, Hôpital Saint-Louis, APHP, Paris, France; 8Service d'Hématologie Clinique, Hospices Civils de Lyon, Pierre-Bénite, France; 9Service
d’Hématologie, Centre Léon Bérard, Lyon, France; 10Service
d'Hématologie, CHU Rennes, Université de Rennes, INSERM U1236, Rennes, France; 11Service d’Hématologie et Thérapie Cellulaire, Hôpital universitaire de la Conception, Marseille, France; 12Service
d’Hématologie, CHU de Nancy, Vandoeuvre les Nancy, France; 13Service d’Hématologie, CHU d’Angers, Angers, France; 14Service
d’Hématologie, Centre Henri Becquerel, Rouen, France; 15CHU Reims, Hématologie Clinique, Reims, France; 16Service de Néphrologie, Hémodialyse et Transplantation Renale, Centre Hospitalier Universitaire de Poitiers, Poitiers, France; 17Service d’Hématologie Clinique et Thérapie Cellulaire, CHU Clermont-Ferrand, Clermont-
Ferrand, France; 18Service de Néphrologie et Transplantation d'Organes, CHU Rangueil, Toulouse, France; 19Service d’Hématologie, Institut Universitaire du Cancer Toulouse-Oncopole, Toulouse, France; 20Service de Néphrologie, Transplantation, Dialyse et Aphérèses, CHU Bordeaux, France; 21Service d’Hématologie, Centre Hospitalier René-Dubos, Pontoise, France; 22Service Unité Hémopathies Lymphoides, Groupe Hospitalo-universitaire Chenevier Mondor, APHP Créteil, France; 23Service de Néphrologie et Transplantation Rénale, Hôpital Foch, Suresnes, France; 24Service de Néphrologie et Transplantation Rénale, CHU de Strasbourg, Strasbourg, France; 25Department of Internal Medicine-Oncology, Carl v. Ossietzky University of Oldenburg, Pius-Hospital, Oldenburg, Germany; 26Department of Hematology and Oncology, DIAKO Ev. Diakonie-Krankenhaus Bremen, Bremen, Germany and 27Department of Internal Medicine II: Hematology and Oncology, University Medical Center Schleswig-Holstein, Campus Kiel, Kiel, Germany
Correspondence:
D. ROOS-WEIL - damien.roosweil@aphp.fr.
https://doi.org/10.3324/haematol.2022.282297
Received: November 8, 2022.
Accepted: March 1, 2023.
Early view: March 9, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
PW, SC and DRW designed the research. PW and DRW analysed data and wrote the manuscript. PW, SC, JD, DB, FS, MB, VM, TC, MT, ME, EB, ENZ, RH, GV, CJ, MPM, FJ, ED, NB, LE, RG, NK, LY, LC, HG, LR, KO, SC, HZ, RT and DRW recruited patients. All authors reviewed and approved the manuscript.
Acknowledgments
The authors thank Camille Laurent, Nadia Amara, Elise Chapiro, Laura Rozalska, Patricia Brugel and Hervé Sartelet for providing histological and cytogenetic information.
Funding
This study was supported by grants from INCA-DGOS-INSERM-12560 (SiRIC CURAMUS is financially supported by the French National Cancer Institute, the French Ministry of Solidarity and Health and INSERM with financial support from ITMO Cancer AVIESAN).
The data that support the findings of this study are available from the corresponding author upon reasonable request.
1. Dharnidharka VR, Webster AC, Martinez OM, et al. Posttransplant lymphoproliferative disorders. Nat Rev Dis Primers. 2016;2:15088.
2. Choquet S, Leblond V, Herbrecht R, et al. Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood. 2006;107(8):3053-3057.
3. Trappe R, Oertel S, Leblond V, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell posttransplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13(2):196-206.
4. Trappe RU, Dierickx D, Zimmermann H, et al. Response to rituximab induction is a predictive marker in B-cell posttransplant lymphoproliferative disorder and allows successful stratification into rituximab or R-CHOP consolidation in an international, prospective, multicenter phase II trial. J Clin Oncol. 2017;35(5):536-543.
5. Zimmermann H, Koenecke C, Dreyling MH, et al. Modified riskstratified sequential treatment (subcutaneous rituximab with or without chemotherapy) in B-cell post-transplant lymphoproliferative disorder (PTLD) after solid organ transplantation (SOT): the prospective multicentre phase II PTLD-2 trial. Leukemia. 2022;36(10):2468-2478.
6. Shah N, Eyre TA, Tucker D, et al. Front-line management of post-transplantation lymphoproliferative disorder in adult solid organ recipient patients - a British Society for Haematology guideline. Br J Haematol. 2021;193(4):727-740.
7. Zimmermann H, Reinke P, Neuhaus R, et al. Burkitt posttransplantation lymphoma in adult solid organ transplant recipients: sequential immunochemotherapy with rituximab (R)
followed by cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or R-CHOP is safe and effective in an analysis of 8 patients. Cancer. 2012;118(19):4715-4724.
8. Xicoy B, Ribera J-M, Esteve J, et al. Post-transplant Burkitt’s leukemia or lymphoma. Study of five cases treated with specific intensive therapy (PETHEMA ALL-3/97 trial). Leuk Lymphoma. 2003;44(9):1541-1543.
9. Bobillo S, Abrisqueta P, Sánchez-González B, et al. Posttransplant monomorphic Burkitt’s lymphoma: clinical characteristics and outcome of a multicenter series. Ann Hematol. 2018;97(12):2417-2424.
10. Dierickx D, Tousseyn T, Gheysens O. How I treat posttransplant lymphoproliferative disorders. Blood. 2015;126(20):2274-2283.
11. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720-1748.
12. Younes A, Hilden P, Coiffier B, et al. International Working Group consensus response evaluation criteria in lymphoma (RECIL 2017). Ann Oncol. 2017;28(7):1436-1447.
13. Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579-586.
14. Olszewski AJ, Jakobsen LH, Collins GP, et al. Burkitt lymphoma International Prognostic Index. J Clin Oncol. 2021;39(10):1129-1138.
15. Afify Z, Orjuela-Grimm M, Smith CM, et al. Burkitt lymphoma after solid-organ transplant: treatment and outcomes in the paediatric PTLD collaborative. Br J Haematol. 2023;200(3):297-305.
Oral azacitidine (Oral-AZA) is a hypomethylating agent approved for the treatment of adult patients with acute myeloid leukemia (AML) in first remission after intensive chemotherapy (IC).1,2 In the phase III, randomized, doubleblind, placebo-controlled QUAZAR AML-001 trial (clinicaltrials gov. identifier: NCT01757535), Oral-AZA significantly prolonged relapse-free survival (RFS) and overall survival (OS) compared with placebo in patients with AML in first complete remission (CR) or CR with incomplete blood count recovery (CRi) after IC (induction ± consolidation) who were not candidates for hematopoietic stem cell transplantation (HSCT).3 The primary goal of QUAZAR AML001 was to evaluate the effect of maintenance therapy with Oral-AZA for patients in remission after induction. While there were no protocol-specified criteria regarding prior chemotherapy used before study entry, including the use or number of consolidation cycles received, it is of clinical interest to assess whether the amount of pre-study chemotherapy may have influenced survival outcomes in this trial. Here, we present RFS and OS outcomes in patient subgroups defined by the use of consolidation and number of chemotherapy courses received prior to study entry. IC is the cornerstone of initial AML therapy for patients fit enough to receive it, and most patients achieve CR with induction. Once in remission, patients may receive subsequent consolidation chemotherapy, but the optimal number of consolidation cycles is not well-defined, especially for older patients. After IC, the primary therapeutic goals for patients with AML in remission who are not eligible for HSCT are to delay relapse and prolong survival. Until Oral-AZA, no agent studied in the remission maintenance setting had significantly prolonged both RFS and OS.1-8 Study design and key eligibility criteria of QUAZAR AML-001 have been reported in detail elsewhere.3 Briefly, eligible patients were aged ≥55 years with newly diagnosed AML in first remission after IC, had intermediate- or poor-risk cytogenetics (NCCN 2011 criteria9) and an Eastern Cooperative Oncology Group performance status (ECOG PS) score ≤3, and were HSCT-ineligible. Induction and consolidation regimens were administered at the discretion of the treating physician before study screening; trial eligibility was not contingent on the use of consolidation chemotherapy or amount of consolidation cycles received, but patients must have been screened for eligibility within 4 months of
achieving initial CR/CRi during induction. Eligible patients were randomized 1:1 to Oral-AZA 300 mg or placebo once daily for 14 days per 28-day treatment cycle. Measurable residual disease (MRD) was assessed centrally via multiparameter flow cytometry, with a positivity threshold of 0.1% in the bone marrow for aberrant cells (different from normal or leukemia aberrant phenotype).
The primary trial endpoint was OS, defined as the time from randomization until death, and the key secondary endpoint was RFS, the time from randomization until relapse or death. Comparisons of OS and RFS between OralAZA and placebo within patient subgroups defined by use of consolidation therapy after induction (yes or no) were prospective exploratory endpoints in the trial protocol. Additional post hoc analyses were performed to assess survival outcomes in subgroups defined by the number of consolidation courses received (0, 1, or ≥2) and total number of induction and consolidation cycles. Induction courses were defined as AML-directed chemotherapy regimens administered prior to the date of first CR/CRi recorded on the electronic case report form and consolidation regimens were those given after that date. Survival endpoints were estimated using Kaplan-Meier methods and compared between treatment arms using hazard ratios (HR) and 95% confidence intervals (CI) from stratified Cox proportional hazards models and P values from stratified log-rank tests. The post hoc survival analyses by number of consolidation cycles and total cycles of induction and consolidation were not sufficiently powered to determine statistically significant differences within or between treatment arms, precluding meaningful interpretation of P values; HR point estimates and 95% CI in these subgroups are provided for informational purposes only. The data cutoff was performed in July 2019.
The trial enrolled 472 patients (Oral-AZA 238, placebo 234) (Figure 1). Prior to enrollment, the most common agents used for induction and consolidation were cytarabine (99% and 80%, respectively), idarubicin (55% and 20%), and daunorubicin (33% and 8%); use of these agents was similar between the Oral-AZA and placebo arms. Most patients (80% [378/472]) received consolidation after induction, and use of consolidation was similar between treatment arms (Oral-AZA 78% [186/238], placebo 82% [192/234]) (Figure 1). Nearly half of the patients in the Oral-AZA (n=110 [46%]) and
placebo (n=102 [44%]) arms received one prior consolidation, and 32% (n=76) and 38% (n=90) of patients, respectively, received ≥2 prior consolidation cycles. The remaining 20% of patients (n=94) did not receive consolidation, including 52 patients (22%) in the Oral-AZA arm and 42 (18%) in the placebo arm. Baseline characteristics were generally similar among consolidation-defined subgroups within and between treatment arms (Online Supplementary Table S1). In both arms, patients who did not receive consolidation tended to be older than those who did. Rate of measurable residual disease (MRD) negativity at screening was similar between consolidation-defined cohorts within the Oral-AZA arm, whereas in the placebo arm, a larger proportion of patients who received consolidation were MRD-negative compared with those who did not (50% vs. 36%, respectively). Oral-AZA significantly prolonged both RFS and OS from the time of randomization compared with placebo, regardless of whether patients received consolidation prior to study entry. For patients who did not receive consolidation, median RFS was prolonged with Oral-AZA by 4.5 months versus placebo (median 8.4 vs. 3.9 months, respectively; HR=0.58; 95% CI: 0.36-0.94; P=0.0258) and the estimated 1-year RFS rate was 18.7% higher with Oral-AZA (40.8% vs. 22.0%) (Figure 2A; Table 1). Oral-AZA also prolonged median OS in this subgroup by approximately 12 months compared with placebo (median 23.3 vs. 10.9 months, respectively; HR=0.54; 95% CI: 0.33-0.87; P=0.0103) and improved 1-year
survival rate by 30.7% (71.2% vs. 40.5%) (Figure 2B; Table 1). For patients who did receive consolidation following initial induction, median RFS was prolonged more than 2-fold with Oral-AZA versus placebo - 10.2 versus 5.0 months, respectively (HR=0.67; 95% CI: 0.53-0.85; P=0.001) - and 1year RFS rates were 45.9% and 28.6%, respectively (Figure 2A; Table 1). Median OS was 24.7 months with Oral-AZA and 15.4 months with placebo (HR=0.74; 95% CI: 0.58-0.94; P=0.0147) and estimated 1-year survival rates were 73.2% and 59.2%, respectively (Figure 2B; Table 1). Estimated median RFS was approximately twice as long with Oral-AZA compared with placebo in both the one consolidation and ≥2 consolidation cohorts, and Oral-AZA increased 1-year survival rates in these cohorts by 17.3% and 19.6%, respectively (Table 1). Oral-AZA nominally improved OS regardless of the number of prior consolidation cycles received (0, 1, or ≥2), with median OS estimates ranging from 21.0 to 28.6 months in the Oral-AZA arm and 10.9 to 17.6 months in the placebo arm (Table 1). Intriguingly, median RFS appeared favorable for patients receiving Oral-AZA without any prior consolidation therapy (8.4 months), compared with patients receiving consolidation therapy but no maintenance in the placebo arm (5.0 months). Analogously, median OS was also longer for patients receiving Oral-AZA and no prior consolidation therapy (23.3 months), compared with patients receiving consolidation therapy but no maintenance treatment in the placebo arm (15.4 months).
 Figure 1. Patient enrollment and prior chemotherapy details. *The ≥2 consolidations cohort included 19 patients (oral azacitidine [Oral-AZA] 6, placebo 13) who received 3 consolidation cycles. CR: complete remission; CRi: CR with incomplete blood count recovery.
Figure 1. Patient enrollment and prior chemotherapy details. *The ≥2 consolidations cohort included 19 patients (oral azacitidine [Oral-AZA] 6, placebo 13) who received 3 consolidation cycles. CR: complete remission; CRi: CR with incomplete blood count recovery.
Overall, 79% of patients (n=375) received a single induction course before achieving remission and 21% (n=97) received ≥2 inductions (Figure 1). When accounting for total chemotherapy received before study entry (i.e., number of induction and consolidation courses), median RFS was numerically prolonged by 1.5 to 8.5 months with Oral-AZA versus placebo across all induction/consolidation cohorts (Online Supplementary Table S2). Patients who received a single induction followed by ≥2 cycles of consolidation appeared to have the most favorable survival outcomes within each treatment arm, whereas the small subgroup of patients who received ≥2 courses of induction and no consolidation generally had poor outcomes, but sample sizes prevent meaningful interpretation.

The overall safety profile of Oral-AZA was similar among consolidation groups and was aligned with the overall QUAZAR population. No associations were found between the number of consolidation cycles received and Oral-AZA dose modifications (data not shown).
As mentioned, the primary objective of the QUAZAR AML001 trial was to determine the efficacy of Oral-AZA as maintenance therapy subsequent to chemotherapy for patients already in remission. A broad assessment of the impact of consolidation therapy in the front-line management of AML is beyond the scope of this trial, and Oral-AZA is not meant to replace consolidation chemotherapy for patients who can receive it. Overall, Oral-AZA maintenance significantly prolonged both RFS and OS compared with placebo re-
gardless of whether patients received consolidation after initial induction. With the caveat regarding small sample sizes and lack of statistical power, post hoc analyses in subgroups defined by number of consolidation cycles received suggest that Oral-AZA may prolong RFS and OS compared with a “watch-and-wait” approach (emulated with placebo) for patients with AML in first remission after IC, independent of the number of induction and consolidation courses received before beginning maintenance treatment. A previous analysis examining the relationship between survival outcomes and MRD in QUAZAR AML-001 found that although patients with MRD responses (i.e., conversion from MRD-positive at baseline to MRDnegative) were more likely to have received consolidation chemotherapy before study entry than those who remained MRD-positive on-study, the number of chemotherapy cycles received before study entry was not significantly predictive of MRD response or duration on-
study with MRD-negative status.10 Overall, these findings indicate that intensive induction chemotherapy followed by Oral-AZA maintenance therapy is effective regardless of the amount of prior consolidation delivered, and represents an important component of therapy in patients with intermediate- or poor-risk AML in remission not candidates for HSCT.
Victoria, Australia; 2Australian Center for Blood Diseases, Monash University, Melbourne, Victoria, Australia; 3Weill Cornell Medicine, New York, NY, USA; 4New York Presbyterian Hospital, New York, NY, USA; 5Hematology, Hôpital Saint-Louis, Assistance Publique – Hôpitaux de Paris (AP-HP), Paris, France; 6Institut de Recherche Saint-Louis, Université Paris Cité, Paris, France; 7Department of Internal Medicine
III, Ulm University Hospital, Ulm, Germany; 8Princess Margaret Cancer Center, Toronto, Ontario, Canada; 9Hospital Universitario y Politécnico La Fe, Valencia, Spain; 10AZ Sint-Jan Brugge-Oostende AV, Bruges, Belgium; 11RM Gorbacheva Research Institute, Pavlov University, St. Petersburg, Russia; 12Bristol Myers Squibb, Summit, NJ, USA; 13University of Kansas Cancer Center, Kansas City, KS, USA and 14Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
°Current address
Department of Clinical Hematology, Peter MacCallum Cancer Center and Royal Melbourne Hospital, Melbourne, Vicoria, Australia.
Correspondence:
A.H. WEI - andrew.wei@petermac.org
https://doi.org/10.3324/haematol.2022.282296
Received: October 25, 2022.
Accepted: March 16, 2023.
Early view: March 23, 2023.
Published under a CC BY-NC license
Disclosures
BS and CLB were employed by Bristol Myers Squibb at the time the study was conducted. AHW has served on advisory boards for AbbVie, Agios, Amgen, Celgene/Bristol Myers Squibb, Gilead, Janssen, MacroGenics, Novartis, Pfizer, Roche, and Servier; has received research funding to his institution from AbbVie, Amgen, AstraZeneca, Celgene/Bristol Myers Squibb, Novartis, and Servier; has served on a speakers bureau for AbbVie, Celgene, and Novartis; and is eligible for royalty payments from the Walter and Eliza Hall Institute of Medical Research related to venetoclax. GJR reports receiving research support from Janssen and has served in an advisory position for AbbVie, Agios, Amgen, Astellas, AstraZeneca, Bluebird Bio, Blueprint Medicines, Bristol Myers Squibb, Catamaran, Celgene, Glaxo SmithKline, Helsinn, Janssen, Jasper Therapeutics, Jazz Pharmaceuticals, Mesoblast, Novartis, Pfizer, Roche, Syndax, and Takeda (IRC Chair). HDo reports receiving honoraria from Incyte and Servier. HDö has served in a consultancy position for AbbVie, Agios, Amgen, Astellas, Berlin-Chemie, Bristol Myers Squibb, Daiichi Sankyo, Gilead, Janssen, Jazz Pharmaceuticals, Novartis, Servier, and Syndax; reports receiving research funding from AbbVie, Agios, Amgen, Astellas, Bristol Myers Squibb, Jazz Pharmaceuticals,
Kronos Bio, Novartis, and Pfizer; and reports receiving honoraria from AbbVie, Agios, Amgen, Astellas, AstraZeneca, Berlin-Chemie, Bristol Myers Squibb, Daiichi Sankyo, Gilead, Janssen, Jazz Pharmaceuticals, Novartis, Servier, and Syndax. ACS has served on an advisory committee for AbbVie, Agios, Amgen, Astellas, Bristol Myers Squibb, Jazz, Pfizer, Teva, and Servier; reports receiving research funding from AbbVie, Agios, Amgen, Astellas, Bristol Myers Squibb, GlycoMimetics, Kite/Gilead, Pfizer, and Servier; and reports receiving honoraria from AbbVie, Agios, Amgen, Astellas, Bristol Myers Squibb, Pfizer, Teva, and Servier. PM has served in a consultancy position for Menarini/Stemline, Gilead, Otsuka, Kura Oncology, AbbVie, Bristol Myers Squibb, Novartis, Jazz Pharmaceuticals, BeiGene, Astellas, Pfizer, Incyte, Takeda, Ryvu, and Nerviano; reports receiving research funding from AbbVie, Bristol Myers Squibb, Jazz Pharmaceuticals, Menarini/Stemline, Novartis, Pfizer, and Takeda; and has served on a speakers bureau for AbbVie, Astellas, Bristol Myers Squibb, Gilead, Jazz Pharmaceuticals, and Pfizer. DS reports receiving grants or contracts, honoraria, consulting fees, and travel support from AbbVie, Bristol Myers Squibb, Novartis, and Pfizer; and has served in a leadership role for the Belgian College for Reimbursement of Orphan Drugs. TP reports employment and stock ownership with Bristol Myers Squibb. YL reports employment with Bristol Myers Squibb and Eli Lilly stock ownership. BS reports prior employment with Celgene/Bristol Myers Squibb. CLB reports prior employment and stock ownership with Bristol Myers Squibb. FR reports receiving research funding from Amgen, Astellas, Astex/Taiho, Biomea Fusion, Celgene/Bristol Myers Squibb, Prelude, Syros, and Xencor; and honoraria from AbbVie, Astellas, AstraZeneca, Celgene/Bristol Myers Squibb, Novartis, and Syros. AstraZeneca, Celgene/Bristol Myers Squibb, Novartis, and Syros.
The sponsors collected and analyzed data in conjunction with all authors. AHW wrote the first draft of the manuscript. All authors revised the manuscript and reviewed and approved the final version for submission.
The authors would like to thank the patients, families, investigators, staff, and clinical study teams who participated in the QUAZAR AML-001 trial.
Writing and editorial support was provided by Korin Albert, PhD, of Excerpta Medica, funded by Bristol Myers Squibb. The study was sponsored by Bristol Myers Squibb.
Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independentresearch/data-sharing-request-process.html.
1. Celgene Corporation. ONUREG® (azacitidine tablets) prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/ label/2020/214120s000lbl.pdf Revised September 2020. Accessed September 22, 2022.
2. European Medicines Agency, Committee for Medicinal Products for Human Use. ONUREG® (azacitidine). https://www.ema. europa.eu/en/documents/smop-initial/chmp-summary-positiveopinion-onureg_en.pdf. Accessed September 22, 2021.
3. Wei AH, Döhner H, Pocock C, et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N Engl J Med. 2020;383(26):2526-2537.
4. Palva IP, Almqvist A, Elonen E, et al. Value of maintenance therapy with chemotherapy or interferon during remission of acute myeloid leukaemia. Eur J Haematol. 1991;47(3):229-233.
5. Huls G, Chitu DA, Havelange V, et al. Azacitidine maintenance after intensive chemotherapy improves DFS in older AML patients. Blood. 2019;133(13):1457-1464.
6. Blum W, Sanford BL, Klisovic R, et al. Maintenance therapy with decitabine in younger adults with acute myeloid leukemia in first remission: a phase 2 Cancer and Leukemia Group B Study (CALGB 10503). Leukemia. 2017;31(1):34-39.
7. Lowenberg B, Suciu S, Archimbaud E, et al. Mitoxantrone versus
daunorubicin in induction-consolidation chemotherapy-the value of low-dose cytarabine for maintenance of remission, and an assessment of prognostic factors in acute myeloid leukemia in the elderly: final report. European Organization for the Research and Treatment of Cancer and the Dutch-Belgian Hemato-Oncology Cooperative Hovon Group. J Clin Oncol. 1998;16(3):872-881.
8. Lowenberg B, Beck J, Graux C, et al. Gemtuzumab ozogamicin as postremission treatment in AML at 60 years of age or more: results of a multicenter phase 3 study. Blood. 2010;115(13):2586-2591.
9. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology for Acute Myeloid Leukemia. National Comprehensive Cancer Network website. http://www.nccn.org/professionals/physician_gls/PDF/aml.pdf. Accessed 01 Mar 2011.
10. Roboz GJ, Ravandi F, Wei AH, et al. Oral azacitidine prolongs survival of patients with AML in remission independently of measurable residual disease status. Blood. 2022;139(14):2145-2155.
For more than four decades, conventional therapy for acute myeloid leukemia (AML) has been cytarabine/anthracycline-containing regimens, followed by consolidation therapy, including allogeneic hematopoietic stem cell transplantation (HSCT). In recent years, the approach to the treatment of AML has shifted significantly toward the use of novel and effective, target-directed therapies, including the anti-CD33 immunoconjugate, gemtuzumab ozogamicin (GO), and an inhibitor of mutant FMS-like tyrosine kinase 3 (FLT3), midostaurin.
A large meta-analysis evaluating data from five randomized controlled trials of GO in addition to induction chemotherapy among cytogenetically favorable-risk and intermediate-risk AML patients revealed a significant reduction in the risk of relapse and improved long-term overall survival.1 The definitive favorable benefit/risk ratio of GO added to standard intensive chemotherapy (IC) in the treatment of AML was confirmed in the final analysis of the ALFA-0701 randomized trial.2 Similarly, the multitargeted kinase inhibitor midostaurin in addition to standard chemotherapy provided a significant beneficial effect on prolonged overall and event-free survival among patients with FLT3-mutated AML across all FLT3 subtypes.3 Moreover, the toxicity profile of a midostaurin-based combination therapy was favorable, non-additive, and comparable to that of standard intensive treatment. However, with regard to the interaction of gender, European LeukemiaNet (ELN) risk group, and FLT3 subtype, certain subgroups of patients did not derive significant benefit from combined treatment with midostaurin.3 Based on study results, both midostaurin and GO were approved by the European Medicines Agency in 2017 and 2018, respectively, for the treatment of AML.4,5 In literature, robust data on a midostaurin plus GO combination with standard IC in newly diagnosed FLT3-mutated/CD33+ AML are lacking. Only limited conference abstracts of ongoing studies have been published.6-8 Since both drugs were available at once for newly diagnosed AML patients in real life, and there was no apparent pharmacological contraindication to combining GO plus midostaurin with standard IC, we decided to employ and retrospectively evaluate the effectiveness and safety of GO + midostaurin + standard IC in patients treated at two Czech hematology centers.
Our retrospective study included random successive, newly
diagnosed patients with AML, CD33 positivity plus FLT3 mutation and good- or intermediate-risk cytogenetics, fit for intensive therapy, treated with GO + midostaurin + standard induction IC (7+3 regimen using cytarabine 200 mg/m2 continuous infusion days 1-7 plus daunorubicin 60 mg/m2 days 1-3). No other prespecified criteria were applied. Midostaurin was given orally at a dose of 50 mg BID during days 8-21, and GO was administered at a dose of 3 mg/m2 QD intravenously on days 1, 4, and 7, according to the Summary of Product Characteristics. Subsequently, patients in complete response without serious adverse events in reaction to GO after induction chemotherapy received one or two courses of consolidation with GO + midostaurin (GO 3 mg/m2 on day 1 intravenously plus midostaurin 50 mg BID from day 8 to day 21 orally plus daunorubicin 60 mg/m2 days 1 and 2 intravenously plus cytarabine 1 g/m2 BID on days 1 to 4 intravenously).
Patients were diagnosed in the period from July 13, 2020 through June 9, 2022 at two Czech hematology centers. Data were obtained from a detailed database of real-world AML patients - DATOOL-AML (Database of Acute LeukemiaTool) on behalf of the CELL group (Czech Leukemia Study Group for Life) covering epidemiology, AML characteristics, therapy, safety, and outcome.
This research was undertaken in respect of relevant guidelines and regulations with project approval by the Multicenter Ethics Committee of Brno University Hospital (Number 01-191022/EK). All patients involved signed informed consent.
Basic statistical methods were applied to describe absolute and relative frequencies for categorical variables, and median, mean, minimum, and maximum values for continuous variables.
A total of 11 patients with FLT3+/CD33+ AML (64% men; median age 44 years) were evaluated with a median 53week follow-up (mean, 52 weeks; range, 6-104) since the diagnosis of AML. Most patients had AML with recurrent genetic abnormalities (46% NPM1; 9% RUNX1::RUNX1T1; 9% RUNX1), two had AML not otherwise specified (18%), and two had therapy-related AML (18%). A FLT3-internal tandem duplication (ITD) was present in six patients (55%), FLT3tyrosine kinase domain (TKD) mutations in three cases (27%), and both FLT3-ITD and FLT3-TKD mutations in two patients (18%). The majority had normal cytogenetic find-
ings (82%). The median baseline white blood count was 50x109/L (range, 1.4-426.0). Regarding ELN 2017 risk stratification, seven cases were classified as favorable risk (64%), one as intermediate risk (9%), and three as adverse risk (27%). The patients’ characteristics are shown in Online Supplementary Table S1.
The median number of days from diagnosis to initiation of combined intensive induction treatment was 8 days (range, 4-12). Initially, nine (82%) patients received cytoreduction with hydroxyurea and leukapheresis was additionally undertaken in three (27%) of them. After GO + midostaurin + IC induction, most of the cohort attained complete remission (n=10; 91%), among whom one patient with NPMmutated AML and two patients with FLT3-ITD/FLT3-TKD mutations reached molecular minimal residual disease negativity (27%). In one patient with refractory disease (9%), a FLAG (fludarabine, cytarabine and filgrastim) salvage regimen resulted in hematologic complete remission which was followed by allogeneic HSCT. A total of six (55%) patients continued with one or two courses of consolidation including GO + midostaurin, and three (27%) of them subsequently underwent allogeneic HSCT. Regarding the use of GO + midostaurin in consolidation, two (18%) patients were not evaluable because the data cutoff was exactly at the time of response assessment after induction, and two (18%) patients did not continue GO at the discretion of their treating physicians, because of toxicity. Finally, all patients achieved complete remission, with all NPM1mutated patients reaching a molecular complete remission (46%) (Online Supplementary Table S2). The median time to recovery of the neutrophil count to 1.0x109/L and the platelet count to 50x109/L following the start of induction treatment was 38 days and 29 days, re-

spectively, whereas after the first consolidation with GO + midostaurin, it was 19 days and 22 days, respectively. Adverse events after induction treatment comprised one case of infection grade 3 (9%), one case of sinusoidal obstruction syndrome grade 2 (9%), two cases of bleeding (gastrointestinal grade 3, gynecological grade 2) (18%), one case of Clostridium difcile infection grade 2 (9%), eight cases of elevated bilirubin/liver transaminases grade 1-2 (73%), and one case of elevated liver transaminases grade 3 (9%). Following consolidation therapy, we noted two cases of infection grade 3 (18%), and three cases of elevated liver transaminases grade 1-2 (27%). In two patients (18%), GO was not added to consolidation because of serious infectious complications and sinusoidal obstruction syndrome after induction, with a high probability of these being related to GO. Subsequently, within the second consolidation, GO was not administered in two (19%) cases because of allogeneic HSCT (Online Supplementary Table S2). Midostaurin was not interrupted or discontinued due to toxicity in any case.
At the last follow-up, a total of ten patients (91%) were still alive; one patient with therapy-related AML in molecular complete remission (NPM1) died from infectious complications (9%) during aplasia after haploidentical HSCT (Figure 1). Complete remission was maintained in seven patients (7/11; 64%); three patients relapsed (3/11; 27%): two of them had molecular relapses (2/11; 18%) and one had a hematologic relapse (1/11; 9%).
Our study highlights the excellent effectiveness of GO plus midostaurin in addition to standard IC for newly diagnosed FLT3-mutated/CD33+ AML patients: the tolerability of the combination was good and there was not an unusual increase in toxicity.
Figure 1. Overall survival of patients with newly diagnosed acute myeloid leukemia treated with the combination regimen. Overall survival of patients in weeks since the start of treatment with the combination of gemtuzumab ozogamicin + midostaurin + intensive chemotherapy induction.
A number of studies published in the literature have separately evaluated the efficacy and safety of either GO or midostaurin added to standard induction chemotherapy in newly diagnosed AML.1-3 However, to the best of our knowledge, an original article analyzing GO-based intensive treatment in combination with midostaurin and standard chemotherapy has not yet been published. In the last 2 years, results of ongoing randomized studies have been presented in the form of conference abstracts. One such study was that by Röllig et al., 6 who reported a phase I trial of 11 patients with newly diagnosed FLT3-mutated AML combining standard induction chemotherapy with midostaurin and GO. Similar to our results, the 30-day mortality among all enrolled patients was 0%, notwithstanding one case of sinusoidal obstruction syndrome. Similarly, a composite complete remission was reached by 91% of the patients. The authors defined the standard GO dose on days 1+4 and the standard midostaurin dose on days 8-21 of induction treatment as the maximum tolerable dose which could be safely combined with standard IC in newly diagnosed AML. Another phase I dose-finding study by Borate et al.7 assessing the safety and preliminary efficacy of GO + midostaurin + IC in eight patients with newly diagnosed FLT3-mutated AML yielded promising responses and the tolerability of the combination was good, with no doselimiting toxicity. The composite complete remission rate was 75%. Moreover, no treatment-related deaths were documented in the first 30 days. One patient had a serious adverse event designated as GO-related grade 4 sinusoidal obstruction syndrome. Finally, the third published abstract concentrated on the results of the MIDOTARG pilot trial evaluating the safety of GO + midostaurin + IC in 59 patients with newly diagnosed FLT3+ AML and the impact on minimal residual disease kinetics.8 Compliance with the induction treatment was comparable to that in our study (100% and 100%, respectively). The difference between the MIDOTARG pilot trial and our study was the number of GO doses during induction treatment (2 vs. 3, respectively). Day 60 mortality 0% was the same as in our study. No cases of sinusoidal obstruction syndrome were reported in the MIDOTARG trial, compared to one case in our study (0% vs. 9%, respectively). Time to neutrophil recovery to 1.0x109/L was 32 days compared to 38 days in our cohort. The longer time to neutrophil recovery may be associated with the higher total dose of GO administered within induction in our cohort compared to that given to patients in the MIDOTARG trial. Similarly, time to platelet recovery to 100x109/L was 29 days, compared to 32 days in our study and 35 days in ALFA-0701.2,8 Complete remission with or without recovery of blood counts was achieved in 88% compared to 91% in our study.8 In our study, only one patient died due to infection in molecular complete remission (9%).
Our study's major strength is its unique focus on a homogeneous cohort of patients with FLT3-mutated/CD33+ AML
treated with GO + midostaurin + IC at two academic hematology centers. The study limitations concern only the small number of evaluated cases.
In summary, our data highlighted a high response rate and good tolerability with no evidence of increased toxicity (with the exception of slightly prolonged recovery of neutrophil count) of GO plus midostaurin added to standard IC in patients with newly diagnosed FLT3-mutated/CD33+ AML. Our results should be validated in a larger group of patients with a longer follow-up.
Barbora Weinbergerová,1,2+ Martin Čerňan,3 Tomáš Kabut,1,2 Lukáš Semerád,1,2 Natália Podstavková,1,2 Tomáš Szotkowski,3 Ivana Ježíšková1,2 and Jiří Mayer1,2+
1Department of Internal Medicine, Hematology and Oncology, Masaryk University, Brno; 2Department of Internal Medicine, Hematology and Oncology, University Hospital Brno, Brno and 3Department of Hemato-Oncology, Faculty of Medicine and Dentistry, Palacký University Olomouc and University Hospital Olomouc, Olomouc, Czech Republic
+BW and JM contributed equally to this work.
Correspondence:
B. WEINBERGEROVÁ - Weinbergerova.Barbora@fnbrno.cz
https://doi.org/10.3324/haematol.2022.282263
Received: October 21, 2022.
Accepted: February 15, 2023.
Early view: February 23, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
BW and JM contributed to study conception and design, implemented preparation of the material, data collection and analysis and wrote and revised the manuscript. MČ, TK, LS, NP, TS, and IJ contributed to the data collection, commented on previous versions of the manuscript and reviewed and approved the final version.
Funding
This study was supported by the Czech Republic Ministry of Healthconceptual development of research organization (FNBr, 65269705)
and the National Institute for Cancer Research Project (Programme EXCELES, ID Project N. LX22NPO5102) funded by the European Union-Next Generation EU.
1. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
2. Lambert J, Pautas C, Terré C, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica. 2019;104(1):113-119.
3. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454-464.
4. European Medicines Agency/Rydapt (midostaurin). https://www.ema.europa.eu/en/medicines/human/EPAR/rydapt Accessed September 20, 2022.
5. European Medicines Agency/Mylotarg (gemtuzumab
Data-sharing statement
Complete data can be shared upon request.
ozogamicin). https://www.ema.europa.eu/en/medicines/human/EPAR/mylotar g-0 Accessed September 20, 2022.
6. Röllig C, Schliemann C, Mikesch JH, et al. Gemtuzumab ozogamicin plus midostaurin in combination with standard intensive induction therapy in newly diagnosed AML: results from a phase-I study. Blood. 2021;138(Suppl 1):2324.
7. Borate U, Kaempf A, Minnier J, et al. FLT3-mutated acute myeloid leukemia using a novel regimen of gemtuzumab ozogamicin and midostaurin in combination with standard cytarabine and daunorubicin induction therapy. Blood. 2021;138(Suppl 1):1269.
8. Russell N, Wilhelm-Benartzi C, Othman J, et al. Gemtuzumabbased induction chemotherapy combined with midostaurin for FLT3 mutated AML. Results from the NCRI AML19 “MIDOTARG” pilot trial. Hemasphere. 2022;6(s126):27-28.
Only a few humanized monoclonal antibodies (mAb) are available to treat patients with peripheral T-cell lymphomas (PTCL), with the exception of anti-CCR4 (mogamulizumab) and anti-CD30 antibody-drug conjugate (Brentuximab-vedotin). Lacutamab, a first-in-class humanized anti-KIR3DL2 cytotoxicity-inducing mAb which has demonstrated in vitro antitumor activity by antibody-dependent cell-mediated cytotoxicity (ADCC) and encouraging clinical activity in a phase I clinical trial (dose-escalation plus expansion cohort) that enrolled relapsed/refractory advanced cutaneous Tcell lymphomas (CTCL) patients (clinicaltrials gov. Identifier: NCT02593045),1 is under phase I evaluation (clinicaltrials gov. Identifier: NCT03902184). Aside from adult T-cell leukemia lymphomas (ATL) and CTCL, KIR3DL2 expression has not yet been investigated in large cohorts of PTCL patients and the possible modulation of KIR3DL2 by therapeutic agents has not been addressed in this context. We found that KIR3DL2 was expressed at the protein level in several aggressive PTCL entities and could be efficiently targeted using lacutamab in vitro. The regulation of KIR3DL2 expression is controlled by DNA methylation in PTCL, and chemotherapy (gemcitabine and oxaliplatin) could increase lacutamab efficacy through the modulation of KIR3DL2 expression. KIR3DL2 is an inhibitory receptor of the killer immunoglobulin-like receptor (KIR) family, clonally expressed in normal NK cells and sub populations of αβ and γδ T cells, as are the other KIR.2 KIR3DL2 is expressed by the neoplastic T cells of Sézary syndrome (SS)3 and other CTCL.4 We have also recently shown that KIR3DL2 is the only KIR recurrently expressed in ATL, mostly in acute subtypes.5
Following the finding of KIR3DL2 mRNA overexpression in some PTCL, especially in PTCL-not otherwise specified (PTCL-NOS) (n=16/50) and ALK-anaplastic large cell lymphomas (ALCL) (n=6/25), as compared to controls (data not shown and unpublished) using RNA sequencing, we performed immunohistochemistry and flow cytometry using specific anti-KIR3DL2 mAb. The anti-KIR3DL2 13E4 mAb (Innate Pharma, Marseille, France) was used for flow cytometry. Several anti-KIR3DL2 antibodies were used for immunohistochemistry: two specific anti-KIR3DL2 mAb (clones MOG1-MK323-12B11 [12B11] and MOG1l-P3-R4D-H5 [H5]; Innate Pharma), allowing the staining of frozen and formalin-fixed paraffin-embedded (FFPE) tissue sections, respectively, or the commercially available 5.133 clone, which recognizes KIR3DL2, as well as KIR3DL1 and other KIR (Miltenyi Biotec, Paris, France), for frozen sections only. We showed KIR3DL2 protein expression in 95 of 252 (38%) PTCL (Figures 1A-D; Table 1; Online Supplementary Figure
S1), including 20 of 88 (23%) angio-immunoblastic TCL (AITL), 21 of 51 (42%) PTCL-NOS, nine of 29 (31%) ALCL (including 5/13 ALK+ and 5/16 ALK- ALCL), nine of 20 (45%) enteropathy-associated TCL (EATL), nine of nine (100%) monomorphic epitheliotropic intestinal TCL (MEITL), eight of 21 (38%) nasal-type NK/T-cell lymphomas (NK/TCL), 13 of 17 (76%) hepatosplenic TCL (HSTL), and none of eight T-cell prolymphocytic leukemia (TPLL) samples. Nearly half of the 92 KIR3DL2+ PTCL (44 cases) showed high expression (>50% KIR3DL2+ cells) by immunohistochemistry, a feature common to HSTL (9/10), MEITL (6/9) and PTCL-NOS (11/38). The neoplastic nature of two AITL and two HSTL cases could be demonstrated by double immunofluorescence tests (Figure 1C). We conducted a concordance study between three observers and three centers on 12 selected cases to test the validity of the H5 clone (see legend of Table 1 for details) to address KIR3DL2 expression on FFPE samples. The interobserver agreement was considered to be good with an estimated weighted κ ranging from 0.65 to 0.78 depending on the readers, and an excellent International Consensus Classification (0.9, 95% confidence interval [CI]: 0.82-0.95), as detailed in the Online Supplementary Figure S2.
The role of KIR3DL2 in lymphomagenesis is a subjet of debate, as it has been suggested that it may increase cell survival in SS by inhibiting the activation-induced cell death (AICD)6 or conversely may lead to apoptosis through the engagement of CpG oligodeoxynucleotides (ODN CpG).7 In PTCL, KIR3DL2 expression may reflect the proposed derivation of NKTCL, MEITL, and HSTL from innate immune cells with constitutive expression of KIR3DL2. However, the reason why KIR3DL2 may be found in nodal PTCL, such as AITL or PTCL-NOS, likely derived from cells of the adaptive immune system, as are SS and ATL, is still unclear. Several lines of evidence suggest that KIR are induced upon T-cell activation. KIR+ CD4+ T cells can be identified in various chronic inflammatory diseases such as rheumatoid arthritis, coronaritis or spondylarthritis.2,8 In these T-cell subsets, KIR were shown to act as costimulatory or co-inhibitory receptors of the T-cell receptor (TCR), as they may do in SS cells.9 In accordance with these findings, inhibitory KIR have been shown to downregulate the TCR-signaling pathway and promote resistance of human and mouse T cells to AICD.10 Consistent with this hypothesis, murine PTCL showed NK cell–like reprogramming of neoplastic murine T cells toward NK cells, with expression of various NK receptors due to chromatin remodeling in a mouse model of PTCL, driven by chronic TCR stimulation based on homeostatic prolif-
PTCL: peripheral T-cell lymphoma; FCM: flow cytometry; IHC: immunohistochemistry; PTCL,NOS: PTCL not otherwise specified; AITL: angioimmunoblastic T-cell lymphoma (TCL); ALCL: anaplastic large-cell lymphoma; EATL: enteropathy-associated TCL; MEITL: monomorphic epitheliotropic intestinal TCL; NKTCL: nasal-type NK/TCL; HSTL: hepatosplenic TCL; LGL: large granular lymphocytic; TPLL: T-cell prolymphocytic leukemia. FCM was applied to viable cell suspensions of frozen PTCL samples (spleen, lymph nodes, liver, blood) from the CeVI collection of the Carnot Institute CALYM (N=55), maintained in dimethyl sulfoxide and fresh blood samples of patients in a leukemic phase from our institution (N=8). Cases were considered to be positive if the Δ mean fluorescence intensity (MFI) between the specific antiKIR3DL2 13E4 monoclonal antibody (Innate Pharma) and isotype control was superior to 100. For IHC, frozen (N=73) or formalin-fixed paraffin-embedded (FFPE) (N=122) PTCL tissue samples (N=195 samples from 189 patients), including 6 for which there were both frozen and FFPE material, were retrieved from the biobank of the multicentric T-cell lymphoma consortium (TENOMIC) of the Lymphoma Study Association (LYSA). The positivity threshold for KIR3DL2 expression was set to 5% of positive tumor cells.
eration of p53–/– T cells.11 Moreover, we recently demonstrated that HTLV-1 infection triggered KIR3DL2 expression by normal CD4+ cells after 7 days of cell culture, suggesting that HTLV-1 influences KIR3DL2 expression either through infection-dependent chronic antigenic stimulation, viral integration, or the transcriptional activity of viral proteins.5 Thus, in PTCL, it is conceivable that KIR3DL2, expressed either because of lineage properties or downstream of chronic antigenic stimulation, promotes PTCL cell survival through its inhibitory functions on the TCR pathway leading to reduced AICD.
We performed heterologous ADCC assays using lacutumab on various PTCL cell lines (KIR3DL2+ HSTL lines DERL2, DERL7 and KIR3DL2- NKTCL-derived SNK6 line), as well as primary cells from KIR3DL2+ AITL (n=2), HSTL (n=2), PTCL-NOS (n=1), one KIR3DL2- HSTL together with SS cells used as positive controls. We observed effector/target ratio-dependent cytotoxic activity against KIR3DL2+ DERL2 and DERL7 HSTL-derived cells, but not against KIR3DL2- SNK6 cells and the primary KIR3DL2cells from a HSTL. Similar results were obtained with the primary KIR3DL2+ lymphoma cells from AITL, PTCL-NOS and HSTL patients (Figures 2A, B). Unfortunately, autologous assays from the primary cell samples could not be performed due to insufficient quantity of cells.
Deciphering the molecular mechanisms that drive KIR3DL2 expression in neoplastic T cells of PTCL and CTCL is important, because it may pave the way to innovative combination therapies, thus improving the efficiency of
lacutamab. In normal NK and T lymphocytes, KIR expression was previously shown to be regulated at the epigenetic level, mainly through DNA methylation of CpG islands.12 However, the mechanisms that drive KIR3DL2 expression in other PTCL have never been explored. We, thus, investigated the potential role of DNA methylation in modulating KIR3DL2 expression in PTCL cells and tumors. We found lower promoter methylation of KIR3DL2 CpG islands in KIR3DL2+ (HUT78, DERL2, DERL7) than KIR3DL2- (MyLa and SNK6) cells by bisulfite sequencing. We obtained similar results for primary lymphoma cells and patients tumors (HSTL and EATL tumor samples and PBMC from one PTCL-NOS) (Figures 2C, D). Overall, the median level of methylated CpG islands was significantly lower in KIR3DL2+ than in KIR3DL2- samples (49.2 vs. 83.9%; P<0.0001). Though a more extensive analysis of all different PTCL subtypes is needed, our data suggest that CpG methylation at the promoter locus may participate in the regulation of KIR3DL2 expression. The ability of 5-azacytidine to induce or increase KIR expression in normal NK cells13 and both CD4+ and CD8+ T cells12 in vitro has been previously shown. Using a similar approach, 5-azacytidine treatment allowed us to increase KIR3DL2 expression at the cell surface of HUT78 cells, expanding our findings in ATL,5 and to induce it in the MyLa (Figures 2E, F) but not SNK6 cell lines, the former being constitutively KIR3DL2+ and the two later KIR3DL2-. This suggests that KIR3DL2 silencing by promoter methylation in PTCL may be reversible. Importantly, the level of CpG methylation post-treatment
Table 1. KIR3DL2 expression using immunohistochemistry and flow cytometry in the peripheral T-cell lymphoma cohort.Figure 1. KIR3DL2 expression in peripheral T-cell lymphoma subtypes. (A) KIR3DL2 protein expression (red line) by flow cytometry using the specific anti-KIR3DL2 13E4 monoclonal antibody (mAb) (Innate Pharma) in the DERL2 and DERL7 hepatosplenic T-cell lymphoma (HSTL)-derived and SNK6 nasal-type NK/TCL (NKTCL)-derived cell lines and peripheral blood mononuclear cells (PBMC) from 3 peripheral TCL (PTCL) patients (angio-immunoblastic TCL [AITL], HSTL, PTCL not otherwise specified [PTCL-NOS]) in a leukemic phase, on which antibody-dependent cell-mediated cytotoxicity (ADCC) assays could be performed, compared to staining with an isotype-matched control mAb (green line). (B) Violin plots of KIR3DL2 protein expression using flow cytometry in the 63 PTCL. (C) KIR3DL2 protein expression by immunohistochemistry (IHC) in formalin-fixed paraffin-embedded (FFPE) samples from representative patients with AITL, HSTL, monomorphic epitheliotropic intestinal TCL (MEITL), and PTCL-NOS, showing scattered positive atypical cells in the AITL sample, with co-expression of PD1 (green) in a few cells (double immunofluorescence). Diffuse expression of KIR3DL2 by neoplastic cells was observed by IHC in the HSTL, MEITL and PTCL-NOS samples, as confirmed by double immunofluorescence with co-expression of KIR3DL2 (red) and TCR- δ ( green ) in most cells of the HSTL sample. Cell nuclei counterstained with DAPI appear in blue. Double immunofluorescence and chromogenic staining experiments (PD-1/KIR3DL2 and TCR- δ / KIR3DL2) were performed on FFPE tissue using the Opal™ Reagent Pack system (Akoya Biosciences, Marlborough, USA) or BOND -III Autostainer (Leica Microsystems, Newcastle upon Tyne, UK), respectively. (D) Violin plots of KIR3DL2 protein expression obtained from IHC analyses of 186 PTCL tissue samples. The semi-quantitative evaluation of the proportion of positive cells was independently performed by 2 pathologists (JB and NO).


Figure 2. In vitro assays for lacutamab and KIR3DL2 promoter methylation levels in peripheral T-cell lymphoma samples and cell lines. (A) Targeted ex vivo antitumor efficiency of lacutamab against several peripheral T-cell lymphoma (PTCL) cell lines (upper panel) and primary PTCL cells (lower panel) in antibody-dependent cell-mediated cytotoxicity (ADCC) assays at various effector/target (E/T) ratios with either lacutamab or isotype control monoclonal antibody (mAb) (1 μg/mL each). The results are expressed as the percentage of dead cells in the presence of lacutamab relative to dead cells in the presence of the isotype control for each population at a given E/T ratio. Dead cells were measured using the Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific, Inc., Waltham, MA, USA) on a LSR Fortessa X20 instrument (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo software (TreeStar). All experiments were performed in triplicate for cell lines. For each set of data at different E/T ratios we used the same donor, but different donors were used across the different cell lines and primary samples studied. (B) Targeted ex vivo antitumor efficacy of lacutamab against one KIR3DL2+ (upper panel) and one KIR3DL2- (lower panel) hepatosplenic TCL (HSTL) primary samples, assessed with the same donor. (C) Percentage of methylated CpG islands of the KIR3DL2 gene promoter in KIR3DL2+ PTCL samples (3 cell lines, peripheral blood mononuclear cells [PBMC] from 1 Sézary syndrome (SS), 7 HSTL and 2 enteropathy-associated TCL [EATL] frozen tumor samples) relative to that of KIR3DL2- cases (2 cell lines, PBMC from 1 SS, and 2 EATL frozen tumor samples). Analysis of the KIR3DL2 promoter methylation patterns was performed after bisulfite treatment of the KIR3DL2 promoter followed by plasmid ligation for clonal amplification and direct Sanger sequencing, as previously decribed,12 using primers generating a 353-base pair amplicon covering the KIR3DL2 promoter region (forward primer ATTTTATGTGTGAGAGGTTGGATTTGAG; reverse primer CTCCATCCCCACTCTCCCTCCCTCTAT).
The mean percentage of methylation across all CpG island from each clone (sum of %mCpG1-15/15) is shown for each sample. The median level of CpG island methylation was compared between KIR3DL2+ and KIR3DL2- samples and the statistical significance assessed using the non-parametric Mann-Whitney U test with Prism 6.0 software (GraphPad). (D) CpG island methylation in tumor samples from examples of KIR3DL2+ PTCL patients (left panels), and the KIR3DL2- SNK6 and KIR3DL2+ DERL PTCL cell lines (right panels). CpG island methylation is higher in KIR3DL2- samples. (E) Increase in KIR3DL2 expression in the MyLa cell line after pretreatment with 5-azacytidine for 7 days (at 2, 5, and 10 μM), as determined by flow-cytometry. (F) CpG island methylation of the KIR3DL2 gene promoter at baseline and after treatment with 5-azacytidine (5 μM), determined after bisulfite treatment.
did not change in the SNK6 line, suggesting resistance to the 5-azacitidine hypomethylation effect ( data not shown). To note, although weak KIR3D2 expression could be induced in MyLa cell line, this did not increased its sensitivity to lacutamab. KIR3DL2 expression is, at least in part, epigenetically regulated at the level of the gene promoter in PTCL, as in normal NK and T cells.12 Of note, we recently demonstrated that KIR3DL2 is also upregulated by gene promoter hypomethylation, but required TAX viral protein expression in ATL neoplastic T cells.5 The role of other gene expression regulators, including transcription factors and other key epigenetic players, such as chromatin remodeling systems and histone modifications, or signal transducers is yet to be studied. Importantly, the role of histone acetylation and methylation was shown to be involved in KIR expression in normal NK cells.14 Targeting KIR3DL2 may, thus, represent a promising new therapeutic approach for patients with KIR3DL2-expressing PTCL that is refractory/resistant to conventional chemotherapy. We also provide a rationale for combination approaches with epigenetic modifiers, such as the Food and Drug Administration-approved 5-azacytidine, currently being assessed in a phase III study for relapsed AITL patients (clinicaltrials gov. Identifier: NCT03593018). Chemotherapy regimens, especially with gemcitabine and oxaliplatin, which are widely used in relapsed/refractory PTCL patients, are already known to modulate cell-surface receptors expression.15 We found that pretreatment of the KIR3DL2+ HUT78 cell line with gemcitabine + oxaliplatine (GemOx) increased the level of KIR3DL2 expression and sensitivity to lacutamab (Online Supplementary Figure S3). In this respect, the KILT phase II clinical trial will aim to evaluate the combination of lacutamab with gemcitabine + oxaliplatine (Gemox) in patients with KIR3DL2-ex-
pressing relapsed/refractory PTCL (clinicaltrials gov. Identifier: NCT04984837).
Amandine Decroos,1* Morgane Cheminant,2,3* Julie Bruneau,2,4+ Sylvain Carras,5+ Vincent Parinet,1+ Laura Pelletier,1 Laetitia Lacroix,1,2 Nadine Martin, 1 Jérôme Giustiniani,1 Ludovic Lhermitte,6 Vahid Asnafi, 6 Maxime Battistella,7 François Lemonnier,1,8 Laurence de Leval,9 Hélène Sicard, 10 Cécile Bonnafous,10 Laurent Gauthier,10 Laurent Genestier,11,12 Stefano Caruso,13 Philippe Gaulard,1 13# Olivier Hermine2,3# and Nicolas Ortonne 1 13#
1Paris-Est Créteil University (UPEC), Institut Mondor de Recherche Biomédicale (IMRB), INSERM U955, Créteil, France; 2Université de Paris, Institut Imagine, Laboratory of Hematological Disorders, INSERM UMR1163, Paris, France; 3Necker-Enfants Malades University Medical Center, AP-HP Clinical Hematology, Paris, France; 4NeckerEnfants Malades University Medical Center, AP-HP, Department of Pathology, Paris, France; 5Institute for Advanced Biosciences, "Translational Epigenetics" Team, UMR 1209/CNRS 5309, La Tronche, France; 6Université de Paris, Institut Necker-Enfants Malades, Laboratory of Onco-Hematology, AP-HP, INSERM UMR 1151, Paris, France; 7Université Paris Cité, INSERM U976, AP-HP, Department of Pathology, Saint-Louis Hospital, Paris, France; 8Henri Mondor Hospital, Clinical Hematology, Unité Hémopathies Lymphoïdes, APHP, Créteil, France; 9Institute of Pathology, Department of Laboratory Medicine and Pathology, Lausanne University Hospital and Lausanne University, Switzerland; 10Innate Pharma, Marseille, France; 11Claude Bernard Lyon 1 University, ENS de Lyon, CNRS, UMR5308, INSERM U1111, CIRI, Centre International de Recherche en Infectiologie, Lymphoma Immunobiology Team (LIB), Equipe
Labellisée Ligue Contre le Cancer, Lyon, France; 12Hospices Civils de Lyon, Lyon, France and 13Henri Mondor Hospital, AP-HP, Department of Pathology, Créteil, France
*AD and MC contributed equally as first authors. +JB, SC and VP contributed equally.
#PG, OH and NO contributed equally as senior authors.
Correspondence:
N. ORTONNE - nicolas.ortonne@aphp.fr
https://doi.org/10.3324/haematol.2022.282220
Received: October 4, 2022.
Accepted: May 3, 2023.
Early view: May 11, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
HS, CB, VP, FG and LG are employees of Innate Pharma and Amandine Decroos received financial support from Innate Pharma for her PhD. HS, LG and CB are senior directors and have an ownership interest (including patents) in Innate Pharma. AD, MC, JB, NO, PG and OH received research grants from Innate Pharma. The other authors have no conflicts of interest to disclose.
Contributions
AD performed the ADCC experiments and part of the KIR3DL2 immunostaining (TENOMIC Cohort), participated in the analyses of KIR3DL2 promoter methylation, analyzed the data, and revised the paper. MC participated in the design of the study, KIR3DL2 immunostaining, analyzing of the data, and writing and revision of the paper. JB performed part of the KIR3DL2 immunostaining, analyzed the results, and revised the paper. LLh and VA performed and analyzed the results of the flow cytometry studies. MB participated in the interobserver correlation study on KIR3DL2 expression in FFPE samples. SC performed and analyzed the results of the flow cytometry studies (KIR3DL2 and KIR3DL1 on the CeVi
References
1. Bagot M, Porcu P, Marie-Cardine A, et al. IPH4102, a first-inclass anti-KIR3DL2 monoclonal antibody, in patients with relapsed or refractory cutaneous T-cell lymphoma: an international, first-in-human, open-label, phase 1 trial. Lancet Oncol. 2019;20(8):1160-1170.
2. Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5(3):201-214.
3. Ortonne N, Huet D, Gaudez C, et al. Significance of circulating Tcell clones in Sezary syndrome. Blood. 2006;107(10):4030-4038.
4. Battistella M, Leboeuf C, Ram-Wolff C, et al. KIR3DL2 expression in cutaneous T-cell lymphomas: expanding the spectrum for KIR3DL2 targeting. Blood. 2017;130(26):2900-2902.
cohort) and revised the paper. VP and LP designed the experiments and performed the analyses of KIR3DL2 promoter methylation. LLa and NM provided technical support for the KIR3DL2 staining by immunohistochemistry with the 5.133, MOG1-MK323-12B11, and MOG1l-P3-R4D-H5 mAb and LLa performed the double staining experiments by immunofluorescence for confocal imaging. JG provided technical support for the ADCC experiments. HS, CB, and LGa provided the specific antibodies to KIR3DL2 for the immunohistochemistry, flow cytometry, and ADCC experiments and participated in writing and revising the paper. LGe participated in the design of the study and in writing and revising the paper. SCa performed the statistical analysis regarding interobserver correlations on KIR3DL2 expression on FFPE samples. LdL reviewed PTCL included in the study. PG participated in the design of the study and writing and revising the paper. OH participated in the design of the study and writing and revising the paper. NO designed the study, analyzed the data, and wrote the paper.
The authors wish to thank the TENOMIC consortium (see Online Supplementary Appendix) and the Etablissement Français du sang (EFS) for providing blood samples from healthy donors for the ADCC experiments. We thank Innate Pharma for providing the antiKIR3DL2 monoclonal antibodies used for immunohistochemistry, flow cytometry, and ADCC experiments. We thank Valentine Peri and Franceline Guillot, who performed the in vitro experiments using gemcitabine and oxaliplatin. We also thank the staff of the IMRB’s platforms who provided important support for flow cytometry analyzes and molecular biology (plateformes de cytométrie et de séquençage) and Bruno Tesson for providing RNA-sequencing data (Institut Carnot CALYM, Paris, France).
Funding
This study was financially supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), Innate Pharma (Marseille, France), and the Institut National du Cancer (INCa). AD is the recipient of a grant from Innate Pharma and MC is the recipient of INCa.
Data-sharing statement
Data presented in this manuscript are available upon request.
5. Cheminant M, Lhermitte L, Bruneau J, et al. KIR3DL2 contributes to the typing of acute-type adult T-cell leukemia and is a potential therapeutic target. Blood. 2022;140(13):1522-1532.
6. Thonnart N, Caudron A, Legaz I, et al. KIR3DL2 is a coinhibitory receptor on Sézary syndrome malignant T cells that promotes resistance to activation-induced cell death. Blood. 2014;124(22):3330-3332.
7. Ghazi B, Thonnart N, Bagot M, Bensussan A, Marie-Cardine A. KIR3DL2/CpG ODN interaction mediates Sézary syndrome malignant T cell apoptosis. J Invest Dermatol. 2015;135(1):229-237.
8. Snyder MR, Lucas M, Vivier E, Weyand CM, Goronzy JJ. Selective activation of the c-Jun NH2-terminal protein kinase signaling
pathway by stimulatory KIR in the absence of KARAP/DAP12 in CD4+ T cells. J Exp Med. 2003;197(4):437-449.
9. Marie-Cardine A, Huet D, Ortonne N, et al. Killer cell Ig-like receptors CD158a and CD158b display a coactivatory function, involving the c-Jun NH2-terminal protein kinase signaling pathway, when expressed on malignant CD4+ T cells from a patient with Sezary syndrome. Blood. 2007;109(11):5064-5065.
10. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486-499.
11. Carras S, Chartoire D, Mareschal S, et al. Chronic T cell receptor stimulation unmasks NK receptor signaling in peripheral T cell lymphomas via epigentic reprogramming. J Clin Invest. 2021;131(13):e139675.
12. Li G, Yu M, Weyand CM, Goronzy JJ. Epigenetic regulation of killer immunoglobulin-like receptor expression in T cells. Blood. 2009;114(16):3422-3430.
13. Chan H-W, Kurago ZB, Stewart CA, et al. DNA methylation maintains allele-specific KIR gene expression in human natural killer cells. J Exp Med. 2003;197(2):245-255.
14. Santourlidis S, Graffmann N, Christ J, Uhrberg M. Lineagespecific transition of histone signatures in the killer cell Ig-like receptor locus from hematopoietic progenitor to NK cells. J Immunol. 2008;180(1):418-425.
15. Gravett AM, Dalgleish AG, Copier J. In vitro culture with gemcitabine augments death receptor and NKG2D ligand expression on tumour cells. Sci Rep. 2019;9(1):1544.
Acute myeloid leukemia (AML) represents a genetically heterogeneous group of myeloid neoplasms derived from early hematopoietic progenitors. Mutations in the NPM1 gene and chromosomal rearrangements of the gene encoding the histone 3 lysine 4 (H3K4) methyltransferase MLL (KMT2A) represent recurrent genetic abnormalities that define distinct AML subtypes and predict treatment outcomes.1,2 Both genetic alterations in AML are associated with activating a particular leukemogenic transcriptional program, including the aberrant expression of the self-renewal associated homeobox (HOX), MEIS1, and PBX3 transcription factor genes and their target genes FLT3 and BCL2. 3 The interaction of MLL with the adaptor protein menin is required for MLL-rearranged (MLL-r) leukemogenesis,3 and we discovered that NPM1mut AML also depends on the menin binding motif in MLL.4 Small-molecule inhibitors that block the menin-MLL interaction cause downregulation of leukemic gene expression, induce differentiation, and have anti-leukemic ac-
tivity against NPM1mut and MLL-r leukemia models (reviewed in5). Ziftomenib is one of five menin inhibitors currently assessed in clinical phase I/II trials (clinicaltrials gov. Identifiers: NCT04067336, NCT04065399, NCT05153330, NCT04811560, NCT04988555) with explorative single-agent efficacy of ziftomenib reported in relapsed or refractory AML. Here, we selectively screened for synergistic treatment partners using a combinatorial drug screen and deciphered synergistic effects of ziftomenib on MLL-r and NPM1mut AML. In order to characterize the single-drug treatment effects of ziftomenib, we first assessed its anti-proliferative activity in various MLL-r (MOLM13, MV411, OCI-AML2) and NPM1mut (OCI-AML3) human AML cell lines. Similar to other menin inhibitors such as VTP-50469, we observed strong and dose-dependent inhibitory effects in all MLL-r and NPM1mut AML at low nanomolar concentrations upon 7 days of treatment (half-maximal inhibitory concentration [IC50] <25 nM; Figure 1A, B). HL60 and NB4 AML cells with-

Figure 1. Zifomenib acts highly selective in MLL-r and NPM1mut acute myeloid leukemia and synergizes with a variety of targeted drugs in vitro. (A) Dose-response curve from cell viability assays in human acute myeloid leukemia (AML) cell lines after 7 days of treatment. (B) Ziftomenib half-maximal inhibitory concentration (IC50) values determined after 7 days of treatment by GraphPad Prism software. (C) Volcano plots of RNA-sequencing data after 150 nM ziftomenib treatment in OCI-AML3, MOLM13 (4 days) and MV411 (3 days). Negative log2 values represent downregulated genes with ziftomenib compared with dimethyl sulfoxide (DMSO). Selected MLL targets are labeled. (D) Drug synergy screen of ziftomenib with 37 targeted drugs. Depicted are weighted combination indices (CI) calculated after 2 days pretreatment with ziftomenib followed by 2.5 days combined treatment with small molecule inhibitors. Viable (DAPI-negative) cells were assessed by flow cytometry. CI values were calculated using CompuSyn software and weighted for IC75, IC90 and IC95 values ((1xIC75+2xIC90+3xIC95)/6) based on recommendations from Chou Talalay for anti-cancer drugs because strong inhibitory effects are a necessity in the treatment of cancer. For MLL-r cell lines, the mean CI of MV411 and MOLM13 was calculated. (E) Venn-Diagram of common top 15 synergistic drugs based on highest CI values. Data in panels (A, B) represent the mean of 3 independent experiments, each in technical triplicates, error bars represent standard deviation. RNA sequencing of each cell line was performed in triplicates. The drug screens were performed twice in each cell line, CI values represent the mean of these independent experiments.
out MLL-r or NPM1mut served as negative controls and exhibited only minor responses to very high concentrations (IC50 >2,500 nM) consistent with a high selectivity of ziftomenib (Figure 1A, B). Next, we assessed gene expression changes by RNA sequencing. Ziftomenib uniformly induced transcriptional downregulation of MEIS1, PBX3, FLT3, and BCL2 in all NPM1mut and MLL-r cells (Figure 1C; Online Supplementary Figure S1A) and demonstrated high consistency with the effects of other menin inhibitors3,6–10 (Online Supplementary Figure S1E). Ziftomenib also induced differentiation as evidenced by upregulation of the monocytic cell
surface markers CD11b and CD14 and cytomorphologic analysis (Online Supplementary Figure S1B, C). Also, the genes upregulated with ziftomenib treatment were highly enriched for genes associated with hematopoietic cell differentiation (Online Supplementary Figure S1D).
As single-drug treatment is generally unlikely to induce long-term remissions in AML, we sought to screen ziftomenib for effective drug combination partners. Therefore, we designed a screen assessing single and combined drug effects of ziftomenib and 37 targeted compounds, each at four concentrations in a constant 1:4 ratio. Com-

pounds were selected based on previously reported (pre)clinical activity against AML (Online Supplementary Figure S2A). Because treatment with menin inhibitors affect proliferation with a latency of several days,4,8 we pretreated leukemic cells with ziftomenib for 2 days and then added the other compounds for another 2.5 days. Drug synergy was calculated based on viable cell count as previously described9 (Online Supplementary Figure S2B). The screen revealed synergistic effects of ziftomenib with many of the preselected drugs. There was a strong overlap of most synergistic combination partners between MLL-r and NPM1mut AML subtypes. Of the top 20 synergistic compounds from each AML subtype, 15 overlapped and were
enriched for agents targeting epigenetic regulation and DNA damage (e.g., LSD1 [ORY-1001], PRMT5 [GSK3326595, JNJ-64619178], and PARP [olaparib, talazoparib]) as well as apoptosis and cell cycle (e.g., BCL2 [venetoclax], AKT [MIK-2206], and CDK4/6 [ribociclib, palbociclib]) (Figure 1D, E; Online Supplementary Figure S2C, D). All-trans-retinoic acid (ATRA) was highly synergistic in both AML subtypes, which was confirmed in cell viability assays. The combination induced strong differentiation as evidenced by upregulation of CD11b (Figure 2A, B; Online Supplementary Figure S2E). As previously reported, menin inhibitors synergize with the FLT3 kinase inhibitor gilteritinib.9 This was confirmed for ziftomenib against the MLL-r AML cells

Figure 2. Characterization of the highly synergistic effects of zifomenib shows differentiation with ATRA and a pro-apoptotic signature with venetoclax. (A) Dose-response curves from combinational cell viability assays of MV411, MOLM13, and OCI-AML3 comparing 150 nM ziftomenib (5 days), 100 nM ATRA (5 days), and combinational treatment (5 days / 5 days). (B) Surface CD11b expression of all viable cells was assessed by flow cytometry after treatment described in (A). (C) Dose-response curves from combinational cell viability assays of MV411, MOLM13, and OCI-AML2 comparing ziftomenib (5 days), venetoclax (24 hours), and combinational treatment (4 days ziftomenib pretreatment and additional 24 hours combined treatment with venetoclax). For OCI-AML3, treatment duration was 5 days with both drugs. (D) Percentage of apoptotic (Annexin V) and dead (DAPI-stained) cells after single and combinational treatment with ziftomenib (150 nM) and venetoclax (100 nM; OCI-AML3 500 nM, OCI-AML2 25 nM) in human cell lines. (E) Affinity of BCL2 proteins to different BH3 activating or sensitizing peptides. (F) Schematic of BH3 profiling: After permeabilization with 0.002% digitonin, cells were exposed to BH3-peptides and then fixed. As readout, loss of cytochrome c was evaluated by flow cytometry as surrogate for apoptotic priming. (G) BH3 profiling was conducted in cells treated with 75 nM ziftomenib for 48 hours. Cells were exposed to the BH3-peptides (BIM 0.1 μM, BAD 1 μM, PUMA 1 μM). Panels (A-D, G) represent the mean of 3 independent experiments, each performed in technical triplicates. Error bars represent standard deviation.
harboring an FLT3-internal tandem duplication (ITD) mutation (MOLM13 and MV411) in our screen and in more detailed cellular assays (Online Supplementary Figure S2F). As mentioned above, BCL2 was uniformly downregulated with ziftomenib, and the specific BCL2 inhibitor venetoclax appeared among the top synergistic combination partners in our screen (Figure 1C, D).
In order to explore this combination in more detail, we first assessed venetoclax in single-agent treatment assays to determine IC50 values across the respective AML cell lines. We found remarkable growth inhibition in the MLL-r cells and resistance of the NPM1mut OCI-AML3 cells (Online Supplementary Figure S3A), as previously described.11 Assessment of combined venetoclax and ziftomenib treatment in cellular response assays confirmed synergistic growth inhibition in the MLL-r cells and also less pronounced in the NPM1mut OCI-AML3 cells (Figure 2C) as reported.12–14 No growth inhibition was detected in the NPM1 and MLL wildtype NB4 cells (Online Supplementary Figure S3B, C). It is of interest to note that drug synergy in OCI-AML3 cells was only observed with a prolonged 5-day combined drug exposure of ziftomenib and venetoclax (Figure 2C). No synergy was found in OCI-AML3 when venetoclax was added for 24 hours (h) (including much higher concentrations) after a four-day ziftomenib pretreatment (Online Supplementary Figure S3D, E). In order to confirm the enhanced anti-leukemic activity of menin targeting with venetoclax treat-

ment, we knocked down MEN1 (menin) using two validated small hairpin RNA (shRNA) in MLL-r MOLM13 cells (Online Supplementary Figure S3F). MEN1 knock-down increased venetoclax-mediated cell killing, phenocopying the effects of pharmacological menin inhibition. Next, we performed Annexin V staining demonstrating substantial apoptosis induction with venetoclax that was even more pronounced in the combination (significant in 2 of 4 cell lines) (Figure 2D).
In order to assess potential apoptotic priming of ziftomenib we performed selected BCL2 homology (BH) 3 screening exemplarily in the MLL-r MV411 and NPM1mut OCI-AML3 cells. Therefore, cells were treated with ziftomenib for 48 h versus vehicle control and then exposed to different concentrations of various BH3-peptides (Figure 2E, F). We found a significantly enhanced cytochrome c release in ziftomenibtreated cells in response to the pro-apoptotic peptides BIM, PUMA and BAD (Figure 2G). This may partially be explained by the downregulation of BCL2 leading to an increased sensitivity to venetoclax. It is worth mentioning that apoptotic priming was only detected at higher BIM concentrations (0.1 uM), which may explain discrepancies with published data assessing other menin inhibitors, where lower peptide concentrations were used.10 While more detailed studies are required to determine the net response of BCL2 family proteins upon ziftomenib treatment, these findings suggest that apoptotic priming may contribute to drug synergy with venetoclax in these leukemias.

Figure 3. The combination of zifomenib and venetoclax demonstrates efficacy in NPM1mut primary patient samples and in vivo. (A) Patient characteristics of NPM1mut acute myeloid leukemia (AML) patient samples. (B) AML patient samples were seeded with irradiated HS27 cells and treated with dimethyl sulfoxide (DMSO), ziftomenib (75 nM, 5 days), venetoclax (10 nM, 24 hours), or both compounds. Readout was flow cytometry based human CD45-positive, viable cell count compared to DMSO. (C) Viable human CD45-positive cells upon drug treatment of NPM1mut AML samples as described above. (D) Experimental design of the assessment of leukemia burden in MV411-derived leukemic xenograft mice (N=4). Engraftment was confirmed on day 11 by flow cytometry before start of treatment. (E) Assessment of leukemia burden defined as human CD45-positive bone marrow cells (N=3 and 4 mice/group) upon treatment with drug vehicles, ziftomenib (50 mg/kg; by mouth [PO]; once daily), venetoclax (100 mg/kg; PO; once daily), or combination. (F) Experimental design of the assessment of overall survival (N=8). Engraftment was confirmed on day 11 by flow cytometry before start of treatment. (G) Kaplan-Meier estimates of MV411-derived leukemic xenograft mice (N=8 mice/group) upon treatment with drug vehicles, ziftomenib (days 12-35, 50 mg/kg; PO; once daily), venetoclax (days 16-35, 100 mg/kg; PO; once daily) or both. The log-rank (Mantel-Cox) test was used to calculate P values from the Kaplan-Meier estimates. In panels (C, E) significance was calculated by one-sided student t test. Error bars represent standard deviation.
We next investigated combinatorial ziftomenib and venetoclax treatment in four primary NPM1mut AML patient samples (Figure 3A-C). We selected AML samples harboring this particular gene mutation as - unlike the venetoclax-resistant NPM1mut OCI-AML3 cells - NPM1mut patients responded particularly well to venetoclax-based treatment in clinical trials.15 NPM1mut AML cells were treated for 5 days in a human stromal cell co-culture assay as previously described9 (Figure 3B). All four samples showed an enhanced reduction of viable AML cells with combinatorial versus single-drug treatment with statistical significance in two samples (Figure 3C).
In order to assess the therapeutic potential of combined ziftomenib and venetoclax treatment in vivo, we used a disseminated MLL -r MV411 xenotransplantation model. We transplanted MV411 cells into NSG mice via tail vein injection and randomly assigned the animals into four groups to receive either drug vehicle, ziftomenib (50 mg/kg), venetoclax (100 mg/kg), or their combination. Treatment was initiated on day 12 after transplantation. In order to assess leukemic burden - as defined by the percentage of human CD45-positive cells in the bone marrow - the animals were euthanized on day 26. We observed a signi fi cant reduction within the combinationtreated animals compared to venetoclax and ziftomenib single-drug treatment (Figure 3D, E). In a separate experiment, we explored the survival of MV411 xenograft mice. Treatment was started on day 12 and continued until day 35. Animals receiving venetoclax and ziftomenib exhibited the most extended median survival across all treatment groups. The survival benefit of the combination was signi fi cant compared to treatment with drug vehicle or venetoclax alone but not to single-agent ziftomenib, potentially due to the small sample size, the long interval after the end of treatment, and the high potency of ziftomenib alone (Figure 3F, G). When assessing the combination treatment in a murine xenotransplantation model of the venetoclax-resistant OCI-AML3 cells, we found a trend toward superior reduction of leukemic burden compared to single-drug treatment. While these results are consistent with the above-reported in vitro data, this trend did not reach statistical significance. Of interest,
the ziftomenib-treated animals exhibited strong differentiation effects in nearly all engrafted AML cells (Online Supplementary Figure S3G, H).
In summary, the menin inhibitor ziftomenib has significant activity against NPM1mut and MLL-r AML, suppresses specific leukemogenic gene expression, and induces differentiation. Ziftomenib exhibited synergistic leukemia cell killing in combination with drugs from various classes, many of them targeting chromatin regulation and DNA damage as well apoptosis and cell cycle. While additional studies will be required to characterize these combinations in detail, we have focused here on the promising combinations with ATRA and venetoclax, the latter being currently considered the standard of care for non-fit AML patients. Consistent with published data,12–14 we demonstrated that ziftomenib has profound anti-proliferative activity in combination with venetoclax against MLL-r and NPM1mut AML and is already available for assessment in clinical trials. Our data suggest that ziftomenib may induce apoptotic priming and (re-)sensitize AML cells to venetoclax. If confirmed, this finding could support further evaluation as a potential treatment option for AML patients harboring these genotypes that failed venetoclax-based regimens and have few further treatment options.
Johanna Rausch,1,2,3 Margarita M. Dzama,1 Nadezda Dolgikh,1,2,3 Hanna L. Stiller,1,2,3 Stephan R. Bohl,4 Catharina Lahrmann,1,2,3 Kerstin Kunz,1,2,3 Linda Kessler,5 Hakim Echchannaoui,1,2,3 Chun-Wei Chen,6 Thomas Kindler,1,2,3 Konstanze Döhner,7 Francis Burrows,5 Matthias Theobald,1,2,3 Daniel Sasca1,2,3 and Michael W. M. Kühn1,2,3
1Department of Hematology and Medical Oncology, University Medical Center, Johannes Gutenberg-University, Mainz, Germany; 2German Cancer Consortium (DKTK) partner site Frankfurt/Mainz and German Cancer Research Center (DKFZ) Heidelberg, Germany; 3University Cancer Center Mainz, Mainz, Germany; 4Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA;
5Kura Oncology Inc., San Diego, CA, USA; 6Department of Systems Biology, Beckman Research Institute City of Hope, Duarte, CA, USA and 7Department of Internal Medicine III, Ulm University Hospital, Ulm, Germany
Correspondence:
M. W. M. KÜHN - mickuehn@uni-mainz.de
https://doi.org/10.3324/haematol.2022.282160
Received: September 25, 2022.
Accepted: April 14, 2023.
Early view: April 27, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
MWMK receives honoraria and is a consultant for Pfizer, Kura Oncology, Jazz Pharmaceuticals, Bristol-Myers Squibb / Celgene and Abbvie; is on the speakers bureau of Gilead and receives travel support from Daiichi Sankyo. JR has received travel support from Abbvie. LK and FB are employed by Kura Oncology and are current equity holders. DS receives honoraria from Bristol-Myers-Squibb, Astra Zeneca and Abbvie. KD receives honoraria and is on the advisory board of Novartis, Janssen, BMS/Celgene, Daiichi Sankyo, Jazz and Roche; is member of advisory board of Abbvie and receives research funding from Novartis, BMS/Celgene, Agios and Astellas. The remaining authors have no conflicts of intererest to disclose.
1. Döhner H, Wei AH, Appelbaum FR, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345-1377.
2. Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. New Engl J Med. 2005;352(3):254-266.
3. Krivtsov AV, Evans K, Gadrey JY, et al. A Menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer Cell. 2019;36(6):660-673.
4. Kühn MWM, Song E, Feng Z, et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 2016;6(10):1166-1181.
5. Issa GC, Ravandi F, DiNardo CD, Jabbour E, Kantarjian HM, Andreeff M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia. 2021;35(9):2482-2495.
6. Uckelmann HJ, Kim SM, Wong EM, et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science. 2020;367(6477):586-590.
7. Klossowski S, Miao H, Kempinska K, et al. Menin inhibitor MI3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J Clin Invest. 2020;130(2):981-997.
8. Borkin D, He S, Miao H, et al. Pharmacologic inhibition of the menin-MLL interaction blocks progression of MLL leukemia in
MWMK designed the research, analyzed and interpreted the data, wrote and revised the manuscript, and supervised the study. JR performed the experiments, analyzed and interpreted the data, wrote the original draft of the manuscript and edited the manuscript. MMD and ND performed experiments, analyzed and interpreted the data. SRB provided experimental support for BH3 profiling and revised the manuscript. KK, CL and HLS performed experiments and analyzed the data. DS analyzed and interpreted the RNA-sequencing data. FB und LK provided the menin-MLL inhibitor ziftomenib and revised the manuscript. HE provided NSG mice and revised the manuscript. MT and TK provided administrative support and revised the manuscript. KD provided genetic characterization of AML samples.
The authors would like to thank Marlene Steiner for technical help; the Translational Animal Research Center (TARC) of the University Medical Center, Mainz, for providing the animals; and Basepair for bioinformatics support.
This work was supported by a grant from the German Research Foundation (DFG) (KU-2688/2-1 and DFG 318346496, SFB1292/2 TP12).
Data-sharing statement
The RNA-sequencing data reported in this article have been deposited on Gene Expression Omnibus database (accession number GSE228307). Original data and methods are available by email request to the corresponding author.
vivo. Cancer Cell. 2015;27(4):589.
9. Dzama MM, Steiner M, Rausch J, et al. Synergistic targeting of FLT3 mutations in AML via combined menin-MLL and FLT3 inhibition. Blood. 2020;136(21):2442-2456.
10. Aubrey BJ, Cutler JA, Bourgeois W, et al. IKAROS and MENIN coordinate therapeutically actionable leukemogenic gene expression in MLL-r acute myeloid leukemia. Nat Cancer. 2022;3(5):595-613.
11. Pan R, Hogdal LJ, Benito JM, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4(3):362-375.
12. Carter BZ, Tao W, Mak PY, et al. Menin inhibition decreases Bcl-2 and synergizes with venetoclax in NPM1/FLT3-mutated AML. Blood. 2021;138(17):1637-1641.
13. Fiskus W, Boettcher S, Daver N, et al. Effective Menin inhibitorbased combinations against AML with MLL rearrangement or NPM1 mutation (NPM1c). Blood Cancer J. 2022;12(1):5-11.
14. Fiskus W, Daver N, Boettcher S, et al. Activity of menin inhibitor ziftomenib (KO-539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia. 2022;36(11):2729-2733.
15. Chua CC, Hammond D, Kent A, et al. Treatment-free remission after ceasing venetoclax-based therapy in patients with acute myeloid leukemia. Blood Adv. 2022;6(13):3879-3883.
Diffuse large B-cell Lymphoma (DLBCL) is the most common type of lymphatic neoplasia with an incidence in Europe of 4.92 cases per 100.000 population per year.1,2 Treatment for patients with DLBCL includes conventional chemotherapy like the CHOP protocol (cyclophosphamide, doxorubicin, vincristine [VCR], prednisone).3 The addition of the anti-CD20 antibody rituximab (R) to chemotherapy improved overall survival (OS) and disease-free survival of patients with DLBCL, leading to long-term survival rates of up to 70% for all patients.4,5 Since the publication of the POLARIX trial, which reported a progression-free survival (PFS) benefit for polatuzumab vedotin as replacement for vincristine in R-CHOP (pola-R-CHP) while showing similar rates of adverse events, this vincristine-free regimen represents an additional treatment option.6 R-CHOP is given in four to eight cycles depending on patient’s age and the individual risk score according to the International Prognostic Index (IPI) with intervals of 14 or 21 days.7
Even though R-CHOP achieves excellent response rates, treatment-related side effects can have severe impact on quality of life (QoL).8 One of the most common side effects with relevant dose-limiting toxicity is peripheral polyneuropathy (PNP), mainly caused by VCR and symptoms can be categorized into sensory, motor, autonomic and cranial neuropathy.9 VCR is a vinca alkaloid, whose pharmacodynamics relies on anti-microtubule activity.10 Unfortunately, only very limited evidence of effective treatment options for chemotherapy-induced PNP exists and physicians often face the dilemma of dose reduction or omission of VCR while fearing attenuated efficacy.11 Hence, dose reduction of VCR is performed regularly in clinical practice but its impact on patient outcomes is unclear.12–14 In order to provide more evidence regarding this field of investigation, we performed a post hoc analysis of the RICOVER-60 trial, investigating the impact of dose reduction/omission of VCR on the outcome of older patients with aggressive B-cell lymphoma. The RICOVER-60 trial (clinialcaltrials gov. Identifier: NCT00052936, EU-20243) randomly assigned (1:1:1:1) 1,222 older patients (aged 61–80 years) with newly diagnosed CD20-positive aggressive B-cell lymphoma to receive six or eight cycles of CHOP chemotherapy every 2 weeks with or without eight biweekly doses of rituximab. Prephase therapy consisted of 1 mg VCR as absolute dose and 100 mg per day of prednisone over 7 days. Decisions concerning VCR dose reduction or omission in any particular treatment cycle were left to the local investigator. The primary end-
point of the RICOVER-60 trial was event-free survival (EFS), secondary endpoints were PFS and OS.
Patients who received less or more than the planned number of treatment cycles (6 or 8), who received VCR not according to protocol (more than 2 mg or vinblastine instead of VCR), for whom the VCR dose was unknown or patients with missing documentation were excluded from further analyses. Subgroup analyses were performed for patients of the best treatment arm of the RICOVER-60 trial, which was six cycles of CHOP and eight cycles of rituximab (6xCHOP-14 + 8xR).
VCR dose reductions were common and were observed in 51% of all patients. VCR was reduced significantly more often in female (P<0.001) and older patients (P=0.010). Other disease characteristics were equally distributed between patients with and without VCR dose reductions (Table 1). These differences in age and sex distribution for patients with VCR reduction were observed across all levels of dose reductions (≥80-<100%, >50-80%, 50% or <50% of planned VCR dose; data not shown). Patient characteristics of the group that was treated with the best treatment arm, i.e. 6xCHOP-14 + 8xR, and their relation to VCR reduction showed matching results to the whole study population (data not shown). As expected from the typical VCR toxicity profile, VCR dose reductions were more common in patients with higher grades of reported PNP (Online Supplementary Table S1).
With regard to outcome, patients who received 2 mg of VCR in each of the six of eight cycles (n=391) were compared for survival differences to patients with any reduction in VCR dosage (n=405). EFS, PFS and OS were not different between both groups. These results were confirmed in multivariable analyses adjusted for IPI factors, sex and age (Figure 1A-C) and were also observed for the subgroup treated within the 6xCHOP-14 + 8xR study arm (Figure 1DF). In further analyses, EFS, PFS and OS were compared between patients receiving 100% of the intended VCR dose and the subgroups that received ≥80-<100%, >50-<80%, 50% or <50% of planned VCR dose. There were no differences in EFS, PFS and OS between these VCR dosing subgroups for all analyzed patients (Online Supplementary Figure S1A-C) and for patients of the 6xCHOP-14 + 8xR arm (Online Supplementary Figure S1D-F), even if VCR was reduced to less than 50% of the intended dose.
Since some study centers used 1 mg of VCR in every cycle as standard dose, we compared outcomes of patients receiving 2 mg of VCR in all cycles to outcomes of patients
Table 1. Patient characteristics according to administered vincristine dose.
*Some missing values; #sex: P<0.001, age groups: P=0.010 for patients without VCR reduction (n=391) vs. patients with VCR reduction (n=405). VCR: vincristine; LDH: lactate dehydrogenase; UNV: upper normal value; ECOG: Eastern Cooperative Oncology Group performance status; IPI: International Prognostic Index; BM: bone marrow; extralymph.: extralymphatic; inv.: involvement; DLBCL: diffuse large B-cell lymphoma.
who received 1 mg of VCR in each cycle. EFS, PFS and OS did not differ between both groups when analyzing all patients (Figure 2A-C) and patients of the 6xCHOP-14 + 8xR treatment arm (Figure 2D-F), supporting the notion that 1.4 mg/m2 of VCR may be an excessive dose for most patients and not only for patients developing VCR-induced side effects.
Because the IPI score correlates directly with prognosis,15 we speculated that patients with higher IPI scores might have an additional benefit of higher cumulative VCR doses. However, when analyzing EFS, PFS and OS according to IPI groups (1, 2, 3, 4–5) of all 796 patients, no differences were found between patients with and without VCR reduction/omission (Online Supplementary Figure S2A-D; data for EFS and OS not shown). This indicates that even in patients with high-risk aggressive B-cell lymphoma, dose reduction of VCR has no bearing on survival outcomes.
When analyzing the age groups according to VCR dose reduction, no impact of VCR dosage on survival outcomes was observed within age groups of 61-65 years, 66-70 years and 71-75 years. Interestingly, a significant benefit in survival rates occurs for patients from 76–80 years of age in whom VCR was reduced or omitted at any time (Online Supplementary Figure S2E-H; data for EFS and OS not shown). These findings suggest that the reduction of potential VCR toxicity may have had a higher impact on survival than its additional cytostatic effect in 76–80-year-old patients with aggressive B-cell lymphoma. This is of special interest as this patient group (>75 years) is usually underrepresented in clinical trials and our data therefore support attempts of VCR dose reduction in >75-year-old patients as full dosing of VCR might be harmful in this vulnerable patient group.
In a sensitivity analysis, we compared survival outcomes of
Figure 1. Survival outcomes for patients with and without vincristine reduction. (A, D) Event-free, progression-free (B, E) and overall survival (C, F) of all 796 patients (A–C) and of 234 patients treated with the best treatment arm 6xCHOP-14 + 8xR (D–F) participating in the RICOVER-60 trial according to their vincristine (VCR) dose (not reduced vs. reduced) are shown. Log-rank P values, hazard ratios adjusted for International Prognostic Index factors, sex and age, 95% confidence intervals and the P value from the multivariable Cox Model for VCR dose are presented. CHOP: cyclophosphamide, doxorubicin, vincristine, prednisone; 6xCHOP-14 + 8xR: 6 cycles of CHOP and 8 cycles of rituximab.

Figure 2. Comparison of survival outcomes in patients who received 1 mg versus 2 mg of vincristine in all cycles. (A, D) Eventfree, (B, E) progression-free and (C, F) overall survival of 438 patients treated with 6/8xCHOP-14 ± 8xR (A–C) and of 151 patients treated with 6xCHOP-14 + 8xR (D–F) participating in the RICOVER-60 trial according to their vincristine (VCR) dose (2 mg vs. 1 mg in each of the 6/8 cycles) are shown. Log-rank P values are presented. CHOP: cyclophosphamide, doxorubicin, vincristine, prednisone; 6CHOP-14 ± 8xR: 6 cycles of CHOP and 8 cycles of rituximab.

patients from the two dosing subgroups (with or without VCR dose reduction) separately, depending on the application of VCR during prephase (>0 and ≤1 mg or >1 mg), since this may act as confounding factor. We found no differences in PFS, EFS or OS (data not shown) between both groups regardless of VCR dosing during the prephase.
We further investigated the influence of toxicities caused by co-administered chemotherapeutic agents. Considering only patients of the 6xCHOP-14 + 8xR arm who received fully dosed chemotherapy (cyclophosphamide ≥95%), there was no difference in PFS related to VCR reduction. There was, however, a non-significant (P=0.114) PFS difference with regard to VCR reduction in the small subgroup of patients in whom additional reduction of chemotherapy (cyclophosphamide <95%) was necessary, indicating that only after reduction of other chemotherapeutic agents, reduction of VCR might have an influence on outcomes.
When analyzing the influence of body surface area (BSA) on PNP, VCR reduction and survival outcomes, no correlation between BSA and maximal developed PNP grade was found. Interestingly, VCR was more often reduced in patients with lower BSA. However, VCR reduction (yes vs. no) had no impact on survival endpoints (EFS, PFS and OS) regardless of BSA (data not shown).
Due to the unclear benefit VCR adds to the CHOP regimen, VCR seems to be a reasonable choice as an exchange partner in future studies investigating the efficacy and safety of new drugs in the treatment of aggressive lymphomas. This is supported by the recently published, international phase III POLARIX trial, which evaluated the replacement of vincristine (as part of standard R-CHOP) with polatuzumab vedotin in patients with previously untreated intermediate- or high-risk DLBCL.6
Taken together, we here present the largest analysis of a phase III trial regarding VCR reduction in patients with aggressive B-cell lymphoma so far. Dose reduction of VCR due to toxicity in older patients with aggressive B-cell lymphoma treated with CHOP-14 seems to have no impact on survival outcomes (EFS, PFS and OS).
Major conclusions are: i) VCR reduction due to toxicity had no impact on outcomes of patients with aggressive B-cell lymphoma treated within the RICOVER-60 trial, ii) even in patients of the highest IPI risk group, VCR reduction had no influence on outcomes, iii) no survival differences were observed for patients who received only 1 mg VCR by institutional standards as compared to patients who received 2 mg of VCR in every cycle.
Thomas Bittenbring,1 Norbert Schmitz,3 Lorenz Truemper,4 Michael Pfreundschuh,1† Konstantinos Christofyllakis,1 Markus Loeffler,5 Bettina Altmann5# and Marita Ziepert5#
1Department of Internal Medicine 1 (Oncology, Hematology, Clinical Immunology, and Rheumatology), Saarland University Medical Center, Homburg/Saar; 2Department of Internal Medicine 1, Westpfalz-Klinikum, Kaiserslautern; 3Department of Medicine A, University Hospital Muenster, Muenster; 4Department of Hematology and Oncology, Georg August University of Goettingen, Goettingen and 5Institute for Medical Informatics, Statistics and Epidemiology, University Leipzig, Leipzig, Germany
†Posthumous.
*MB, DKM and IAK contributed equally as first authors. #BA and MZ contributed equally as senior authors.
Correspondence:
M. BEWARDER - moritz.bewarder@uks.eu
https://doi.org/10.3324/haematol.2022.282126
Received: September 18, 2022.
Accepted: February 15, 2023.
Early view: February 23, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
MB received honoraria and travel support from AstraZeneca and Janssen. VL received honoraria from Abbvie. KC received travel grants from Hexal, Celgene, Jazz Pharmaceuticals, Abbvie and Novartis; honoraria from Roche and research funding from AstraZeneca. VP received institutional grants from Acrotech, Amgen, BMS, Chugai, Spectrum, Roche, Talon Therapeutics; travel support from Abbvie, Amgen, BMS and Roche. DKM received honoraria for speaker bureaus from Novartis, ViiV Healthcare, Gilead and Hexal Ratiopharm; travel support from Janssen and Gilead; payment for advisory boards from Janssen and Sanofi. All other authors have no conflicts of interest to disclose.
Contributions
MP, MZ, BA, DKM, MB and IK developed the concept and design of the study. BA and MZ performed statistical analysis. All authors interpreted the data. MB, DKM, BA, IK, KC and MZ wrote the initial draft of the manuscript. MB, NS, VL, MZ, ML and BA reviewed the first draft. All authors gave their final approval of the manuscript. Data analysis and interpretation, writing of the paper, and decision to submit were left to the authors’ discretion and were not influenced by third parties.
The authors are grateful to all patients and their relatives as well as to the participating investigators and sub-investigators of the
Moritz Bewarder,1* Dominic Kaddu-Mulindwa,1* Igor Age Kos,1* Vadim Lesan,1 Gerhard Held,2 Viola Poeschel,1 Lorenz Thurner,1 JoergRICOVER-60 for their continued support. We would like to thank Katja Rillich (IMISE Leipzig) for technical assistance. F. Hoffmann La Roche Ltd. provided rituximab during the first year of recruitment.
Funding
The RICOVER-60 study was financially supported by Deutsche
1. Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107(1):265-276.
2. Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood. 2010;116(19):3724-3734.
3. Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with 3hree intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med. 1993;328(14):1002-1006.
4. Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235-242.
5. Pfreundschuh M, Trümper L, Österborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-Bcell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 2006;7(5):379-391.
6. Tilly H, Morschhauser F, Sehn LH, et al. Polatuzumab vedotin in previously untreated diffuse large B-cell lymphoma. N Engl J Med. 2022;386(4):351-363.
7. Sehn LH, Salles G. Diffuse large B-cell lymphoma. N Engl J Med. 2021;384(9):842-858.
8. Doorduijn J, Buijt I, Van Der Holt B, Steijaert M, Uyl-De Groot C,
Krebshilfe.
Data-sharing statement
Additional data can be made available upon reasonable request to the corresponding author.
Sonneveld P. Self-reported quality of life in elderly patients with aggressive non-Hodgkin’s lymphoma treated with CHOP chemotherapy. Eur J Haematol. 2005;75(2):116-123.
9. Legha SS. Vincristine neurotoxicity: pathophysiology and management. Med Toxicol. 1986;1(6):421-427.
10. Swain SM, Arezzo JC. Neuropathy associated with microtubule inhibitors: diagnosis, incidence, and management. Clin Adv Hematol Oncol. 2008;6(6):455-467.
11. Loprinzi CL, Lacchetti C, Bleeker J, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: ASCO guideline update. J Clin Oncol. 2020;38(28):3325-3348.
12. Marshall S, Nishimura N, Inoue N, et al. Impact of omission/reduction of vincristine from R-CHOP in treatment of DLBCL. Clin Lymphoma, Myeloma Leuk. 2021;21(3):162-169.
13. Mörth C, Valachis A, Sabaa AA, Molin D, Flogegård M, Enblad G. Does the omission of vincristine in patients with diffuse large B cell lymphoma affect treatment outcome? Ann Hematol. 2018;97(11):2129-2135.
14. Utsu Y, Takaishi K, Inagaki S, et al. Influence of dose reduction of vincristine in R-CHOP on outcomes of diffuse large B cell lymphoma. Ann Hematol. 2016;95(1):41-47.
15. International Non-Hodgkin's Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin’s lymphoma. N Engl J Med. 1993;329(14):987-994.
Acute myeloid leukemia (AML) is an aggressive hematological malignancy more common in the elderly. While selected older patients can receive intensive chemotherapy resulting in a 50-60% complete remission (CR) rate, they are generally not eligible for hematopoietic stem cell transplantation (HSCT) and median overall survival (OS) is less than 1 year due to frequent relapse. Therefore, renewed interest in maintenance strategies recently emerged to improve survival among this population. Several maintenance approaches including treatment with histamine dihydrochloride-IL2, gemtuzumab ozogamycin, cytarabine or norethandronolone showed limited impact.1–3 Hypomethylating agents (azacitidine [AZA] and decitabine) are DNA methyltransferase inhibitors with a favorable efficacy/safety profile in the treatment of myelodysplastic syndromes and AML.4 Recently, AZA maintenance demonstrated survival gain in AML, either using a conventional subcutaneous administration route or a newly available oral formulation (CC-486) in clinical trials.5–7 In this study, we report our single-center experience on the use of subcutaneous AZA maintenance in a real-world cohort of 39 patients with AML in first CR after intensive induction chemotherapy, with a focus on the impact of leukemic cell genotype on survival. We retrospectively investigated a cohort of AML patients treated with intensive chemotherapy frontline, who received subcutaneous AZA maintenance after CR achievement from 2012 to 2022.
Characteristics of the 39 patients treated from 2012 to 2022 are reported in Table 1. Median age was 72 years (range, 2378) and 44% had secondary AML. Conventional cytogenetic analysis was available at diagnosis for 36 patients, who were classified according to the national comprehensive cancer network (NCCN) cytogenetic risk categories8 as favorable, intermediate or adverse in 5%, 72% and 15%, respectively. Moreover, most patients were classified as intermediate (41%) or adverse (44%) risk according to the 2017 European Leukemia Network (ELN) guidelines.9 The genomic landscape of leukemic samples at diagnosis is provided (Online Supplementary Figure S1). All patients were in CR after one (87%) or two (13%) cycles of intensive induction chemotherapy with idarubicin (64%) or daunorubicin (26%) combined with cytarabine, and 33% received at least one consolidation cycle before AZA maintenance initiation (Table 1). During AZA maintenance, 14 patients received other treatments including venetoclax (n=5), tyrosine kinase inhibitor (n=4)
and other (n=5), and two patients had HSCT after one and two AZA cycles. Patients received a median number of six (range, 1-38) AZA maintenance cycles. Initial AZA dosing was 75 mg/m2/day during 7 days in all patients, and dose-reduction occurred in 36% of patients during follow-up (Online Supplementary Table S1). The most common adverse events during AZA maintenance were thrombocytopenia and/or neutropenia, leading to AZA dose reduction and discontinuation in 23% and 8% of patients, respectively (Online Supplementary Table S1). After a median follow-up of 20.2 (range, 4-102) months, only 10% patients remained on-therapy and the main causes of AZA discontinuation were relapse (36%) and persistence of CR (18%) (Figure 1; Online Supplementary Table S1).
From AZA maintenance onset, median RFS and OS were 18 and 30 months, respectively, for the whole cohort (Figure 2A, B). We performed subgroup analyses and observed using a univariate Cox model that presence of a TP53 alteration (mutation and/or deletion), adverse ELN 2017 or cytogenetic NCCN risk categories, and administration of less than six cycles of AZA maintenance were significantly associated with decreased RFS and OS probabilities (Figure 2C, D). Particularly, ELN 2017 risk classifier, which aggregates several of these variables was highly predictive of survival (Figure 2E, F). Finally, comparison of variant allele frequency at diagnosis and post-AZA relapse suggested patterns of clonal evolution with reappearance of the initial clone in most cases and occasional subclone loss or gain (Online Supplementary Figure S2).
In our single-center retrospective real-world study, we found that subcutaneous AZA maintenance therapy was feasible in selected elderly patients with AML in CR after intensive induction chemotherapy. Moreover, the 2-year survival probabilities were similar to those observed in prospective clinical trials including the HOVON97 (subcutaneous AZA, 2-year RFS 44%) and QUAZAR-AML-001 (CC486, median OS 24 months) trials.5,7 In contrast to a recent large real-world study on AZA treatment in MDS/AML showing inferior survival in comparison with results from clinical trials,10 our retrospective study confirmed the results of two large prospective phase III trials of AZA maintenance in AML, suggesting that the benefits of this strategy is also found in the routine practice. This could also have reflected an important selection of patients eligible for this strategy both in clinical trials and in routine practice due to the use of intensive chemotherapy induc-
#miniCIA (Idarubicin 12/mg/m2 day 1+ subcutaneous cytarabine 40 mg/m2/d day 1-5) or CPX-351. CRi: complete response with incomplete hematologic recovery defined according to the European LeukemiaNet (ELN) as CR criteria except for residual neutropenia (<1.0×109/L ) or thrombocytopenia (<100×109/L); AML: acute myeloid leukemia; WHO: World Health Organization; NOS: not otherwise specified; NCCN: National Comprehensive Cancer Network; MRD: minimal residual disease; AZA: azacitidine.
tion. Representing a potential bias to this study, 36% patients received concomitant treatment during AZA maintenance including 13% with venetoclax. However, these time-limited associations had no impact on survival in this small cohort (Figure 2C, D).

HOVON97 and QUAZAR-AML-001 trials focused on the comparison of AZA maintenance with observation or placebo, respectively, and did not specifically investigate variables predicting AZA efficacy. Here, we showed that adverse ELN 2017 risk group predicted shorter survival upon AZA maintenance therapy even in a small-sized cohort of patients with AML. While ELN 2017 risk categories are strongly predictive of treatment response in patients with AML undergoing intensive chemotherapy,11 the validation of our finding in larger cohorts could represent an important predictive tool in the context of AZA mainten-
Figure 1.
representing time from CR (complete remission) to initiation of azacitidine (AZA), duration of AZA maintenance (beginning on day 1 of cycle one and ending at day 1 of the last cycle), survival afer AZA discontinuation and time of confirmed relapse. Some patients who were lost to followup may have relapsed, and some may have relapsed after the end of follow-up (relapse date unknown).
Figure 2. Survival analysis of patients with acute myeloid leukemia receiving azacitidine maintenance. (A, B) Survival analyses: from the onset of azacitidine (AZA) maintenance until the next event, defined as death for overall survival (OS) (A), and as death or disease progression for relapse-free survival (RFS) (B), and estimated using the Kaplan-Meier method. Patients were censored at the time of allogenic stem cell transplantation. (C, D) Univariate subgroup analysis of OS (C), and RFS (D). HR: hazard ratio; CI: confidence interval, 1: mutation in IDH1, IDH2, TET2 or DNMT3A; 2: mutation in ASXL1, EZH2, BCORL1, BCOR or STAG2; 3: mutation in SRSF2, SETBP1, SF3B1, ZRZR2 or U2AF1; 4: mutation in FLT3, KRAS, NRAS, PTPN11, CBL, JAK2, CSF3R, MPL or RIT1; 5: mutation in CEBPA, RUNX1, GATA2, ETV6 or WT1; 6: mutation/deletion in TP53; 7: NPM1 mutation; >6 AZA: patients who received more than 6 AZA maintenance cycles; MRD+: positivity of minimal residual disease. (E, F) Survival stratified on ELN 2017 risk category. Kaplan Meyer representations for OS (E) and RFS (F).

ance as well. We also identified that patients receiving more than six cycles of AZA maintenance had improved survival, suggesting either that early treatment failure led to AZA discontinuation, or that the anti-leukemic activity of AZA required sufficient exposure to reach significant clinical benefit in patients. In addition, we observed that mutational status correlated with survival in our cohort, especially the presence of TP53 alterations. We also observed that NPM1 mutations were associated with a trend to improved RFS, in agreement with the results of the QUAZAR-AML-001 trial.12 Together these results suggest that cytogenetic and molecular biomarkers, such as aggregated in the ELN 2017 risk stratification represent important predictive tools in the context of AZA maintenance therapy in AML. Recently, the combination of AZA with venetoclax (VEN), a BCL-2 inhibitor demonstrated a high activity in elderly patients with AML.13 This strategy opens new perspectives currently evaluated in clinical trials, including the comparison of single-agent oral AZA maintenance versus oral AZA+VEN maintenance in older patients and the comparison of subcutaneous AZA+VEN in patients under 65 years old versus observation after CR (clinicaltrials gov. Identifiers, respectively: NCT04102020, NCT05404906), and will represent new opportunities to investigate predictive biomarkers as initiated in our current study.
To summarize, we showed that subcutaneous AZA maintenance is an effective option in patients with AML achieving CR in a real-world setting. We also observed that the ELN 2017 classifier efficiently predicted survival during AZA maintenance. While the duration of maintenance remains controversial, our data suggest that at least six cycles of AZA should be given to patients with AML achieving postinduction CR. The recent availability of oral AZA will probably result in a switch to a large use of this more convenient formulation, and future studies should investigate the impact of cytogenetic and molecular markers in large cohorts of patients with AML treated with oral AZA.
Natacha
1. Brune M, Castaigne S, Catalano J, et al. Improved leukemia-free survival after postconsolidation immunotherapy with histamine dihydrochloride and interleukin-2 in acute myeloid leukemia: results of a randomized phase 3 trial. Blood. 2006;108(1):88-96.
2. Löwenberg B, Beck J, Graux C, et al. Gemtuzumab ozogamicin as postremission treatment in AML at 60 years of age or more: results of a multicenter phase 3 study. Blood. 2010;115(13):2586-2591.
3. Pigneux A, Béné MC, Guardiola P, et al. Addition of androgens
Rudy Birsen,1 Marguerite Vignon,1 Paul Deschamps,1 Patricia Franchi,1 Johanna Mondesir,1 Benedicte Deau-Fischer,1 Elsa Miekoutima,1 Ismaël Boussaid,2 Nicolas Chapuis,2,3 Olivier Kosmider,2,3 Didier Bouscary,1,3 Jerome Tamburini1,3,4,5 and Justine Decroocq1
1Assistance Publique-Hôpitaux de Paris, Centre-Université de Paris, Service d'Hématologie Clinique, Hôpital Cochin, Paris, France; 2Hematology Laboratory, Assistance Publique-Hôpitaux de Paris, Centre-Université de Paris, Cochin Hospital, Paris, France; 3Université de Paris, Institut Cochin, CNRS UMR8104, INSERM U1016, Paris, France; 4Translational Research Center in Onco-hematology, Faculty of Medicine, University of Geneva, Geneva, Switzerland and 5Swiss Cancer Center Leman, Geneva, Switzerland
Correspondence:
J. DECROOCQ - justine.decroocq@aphp.fr
https://doi.org/10.3324/haematol.2022.282009
Received: November 30, 2022.
Accepted: March 23, 2023.
Early view: March 30, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
NJ collected clinical data. NJ and MT collected biological data. NJ, JT and JD performed statistical analyses and contributed to the analysis of the results. NJ, CF, IB, JT and JD designed the figures. NJ, JT and JD drafted the manuscript. All authors have reviewed and approved the final manuscript.
To the memory of our dear colleague Eugenie Duroyon, who extensively contributed to this work and many others.
Data-sharing statement
The data that support the findings of this study are available on request from the corresponding author.
improves survival in elderly patients with acute myeloid leukemia: A GOELAMS Study. J Clin Oncol. 2017;35(4):387-393.
4. Dombret H, Seymour JF, Butrym A, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126(3):291-299.
5. Huls G, Chitu DA, Havelange V, et al. Azacitidine maintenance after intensive chemotherapy improves DFS in older AML
Johnson,1 Marie Templé,2 Chloe Friedrich,2,3 Lise Willems,1patients. Blood. 2019;133(13):1457-1464.
6. Burnett A, Russell N, Freeman S, et al. A comparison of limited consolidation chemotherapy therapy or not, and demethylation maintenance or not in older patients with AML and high risk MDS: long term results of the UK Ncri Aml16 Trial. EHA Library. Burnett A. 06/13/15;103225;S513.
7. Wei AH, Döhner H, Pocock C, et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N Engl J Med. 2020;383(26):2526-2537.
8. Pollyea DA, Bixby D, Perl A, et al. NCCN guidelines insights: acute myeloid leukemia, version 2.2021. J Natl Compr Cancer Netw. 2021;19(1):16-27.
9. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424-447.
10. Mozessohn L, Cheung MC, Fallahpour S, et al. Azacitidine in the
‘real-world’: an evaluation of 1101 higher-risk myelodysplastic syndrome/low blast count acute myeloid leukaemia patients in Ontario, Canada. Br J Haematol. 2018;181(6):803-815.
11. Herold T, Rothenberg-Thurley M, Grunwald VV, et al. Validation and refinement of the revised 2017 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia. 2020;34(12):3161-3172.
12. Döhner H, Wei AH, Roboz GJ, et al. Prognostic impact of NPM1 and FLT3 mutations at diagnosis and presence of measurable residual disease (MRD) after intensive chemotherapy (IC) for patients with acute myeloid leukemia (AML) in remission: outcomes from the QUAZAR AML-001 trial of oral azacitidine (Oral-AZA) maintenance. Blood. 2021;138(Suppl 1):S804.
13. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629.
Glanzmann thrombasthenia (GT) is a rare, inherited, autosomal-recessive platelet disorder resulting in absent, reduced or defective glycoprotein (GP) IIb/IIIa (also known as integrin αIIbβ3).1 As GPIIb/IIIa is a receptor required for platelet aggregation, patients with GT are unable or have reduced ability to form clots and are at risk of spontaneous and increased bleeding following trauma and surgery, with potentially serious consequences.1 Standard-of-care GT treatment involves platelet transfusion,1 ideally human leukocyte antigen (HLA)-matched single-donor and leukocytereduced apheresis platelets; however, this therapy may be associated with alloimmunization leading to refractoriness to future platelet transfusion and/or allergic reactions.1 This report describes the safety of platelet-based therapy in patients with GT included in the Glanzmann Thrombasthenia Registry (GTR) (clinicaltrials gov. Identifier: NCT01476423). Platelet-refractoriness is a variable term generally referring to the failure of platelets to increase after transfusion or platelet functional ineffectiveness to control bleeding.1 Antiplatelet antibodies against HLA and/or GPIIb/IIIa may cause platelet ineffectiveness.1 The presence of anti-platelet antibodies does not always lead to platelet-refractoriness, whilst the absence of antibodies may occur in patients experiencing platelet-refractoriness.1
Recombinant activated factor VII (rFVIIa, NovoSeven®, Novo Nordisk A/S, Bagsværd, Denmark)2 is an alternative therapy in patients with GT with refractoriness to platelet transfusions and/or platelet antibodies, or where platelets are not readily available.1 The GTR was a prospective, observational, international registry, which assessed effectiveness and safety of available treatment options in patients with GT.3,4 Treatment was administered as per local practices and included rFVIIa (F7), platelets (P) and antifibrinolytics/other hemostatic treatments (OH) alone or in combination. Treatment data were recorded for 218 patients of all ages with a diagnosis of congenital GT enrolled from 45 sites in 15 countries between May 10, 2007 and December 16, 2011.3,4 Following a protocol amendment (January 10, 2008), cases generated from 2004 could be registered retrospectively.5 Patients with acquired thrombasthenia due to medication or autoimmune disease were excluded.
A complementary study from the GTR was performed to identify patients with a recorded change in platelet-refractoriness and/or anti-platelet antibody status during the follow-up period, confirmed either as an adverse event (AE) during hospital admission, or through data collected at the
next admission. Clinical criteria for platelet-refractoriness have been defined3,4 and include bleeding during surgery and/or persistence of bleeding and/or rebleeding within 24 hours (despite adequate platelet infusion).4 The GTR captured data which was entered at patient inclusion and, where available, relevant historical treatment and plateletrefractoriness/antibody status data was added. Effectiveness was assessed using a previously defined three-point scale.3,4 Statistical analyses were descriptive.
Overall, 218 patients were included in this sub-analysis, of whom 34 (16%) had ≥1 instance of platelet-refractoriness recorded and 65 (30%) had anti-platelet antibodies. GPIIb/IIIa antibodies were recorded in 47 (22%) patients, HLA antibodies in 21 (10%) patients and 13 patients (6%) had both GPIIb/IIIa and HLA antibodies.
During the GTR study period, a change in platelet refractoriness and/or antibody status was recorded in five (2%) registered patients (referred to as patients 1–5) (Online Supplementary Table S1). In these patients, platelet-based regimens had been used before the beginning of the study and were used in 64 of 77 (83%) of study admissions.
Patient 1 was an adult male with a negative-to-positive change in antibody status for HLA antibodies over three admissions in 10 months. At the first admission, treatment with F7POH was evaluated as effective, with a negative follow-up test for HLA antibodies approximately 5 months later. At the following two admissions, treated with POH and P, the patient tested positive for HLA antibodies. Both treatments were recorded as effective.
Patient 2 was a male pediatric patient with five admissions over 1 year. At admissions 1 and 2, the patient received F7OH and F7POH, respectively. Both treatments were effective, and the patient was negative for platelet-refractoriness. All subsequent admissions were treated with F7OH effectively but were declared positive for platelet-refractory status by the investigator. Antibody status was negative at admission 2; however, 5 months after the fourth admission the patient was declared GPIIb/IIIa antibody-positive.
Patient 3 was a male pediatric patient with a previous history of platelet transfusion and 11 admissions over 7 years. Various treatments were administered throughout. Although POH was received at the first admission, the patient was not positive for refractoriness or antibodies until admission 7. The patient received platelets for admission 5 that were effective but was declared anti-platelet antibody-positive 5 months later. The nature of the platelet antibodies was
unavailable. F7POH was administered during admissions 8 (effective) and 9 (partially effective).
Patient 4 was a male pediatric patient with three admissions over <1 year, treated effectively with OH and POH at first and second admissions, respectively, without refractoriness. At the third admission, POH was administered but platelet transfusion was ineffective (platelet refractoriness positive) and the patient underwent surgical repair for hemostasis. The nature and status of the platelet antibodies were unavailable.
Patient 5 was a female pediatric patient with 55 admissions over 3 years and a history of platelet transfusion. Refractoriness status was recorded for 34 of 55 admissions and was positive at the first admission (POH treatment). Despite this, P and POH-based regimens were administered in 52 of 55 admissions; F7POH was administered during admission 12. At admission 50, the patient was negative for plateletrefractoriness. At admission 51, she was positive, switching to negative at admissions 52–54. Treatment effectiveness was recorded as ineffective on one occasion of platelet transfusion (admission 32) and only partially effective or effective in multiple admissions treated with P or POH. Antibody status was documented at admissions 5–21; the nature of platelet antibodies was unavailable, and a positive result was observed in all instances except admission 16, which was entered as negative.
Of 581 hospital admissions in 152 patients who received P or POH, 14 AE (13 minor) related to treatment for bleeding events were reported in seven patients. A single serious AE
(generalized hives) occurred in a patient receiving POH (Table 1). The limited number of patients in this sub-study and the limited records on platelet refractoriness status after platelet transfusion means safety data must be interpreted with caution.
The development of anti-platelet antibodies after P or POHbased treatments may change the platelet refractoriness status and lead to the inefficacy of platelets to prevent or stop a bleed. This was observed in patient 4 at admission 3 where POH treatment failed to achieve hemostasis and surgical intervention was required. Additionally, anti-HLA antibodies may either decrease with time in the absence of a new P or POH treatment,6 or may persist even without transfusion. Anti-HLA antibodies may result in serious posttransfusion complications including lung injury.7 Therefore, to facilitate optimal treatment decisions from the available therapeutic tools, identifying platelet refractoriness/antibody status is crucial. Analogous to inhibitor assessment in hemophilia patients, baseline testing for antibodies to HLA and GPIIb/IIIa should be performed upon GT diagnosis, and periodically monitored following exposure to blood products containing platelets, which may include unwashed red-cell concentrates.1,8 Thus, treatment choices can be adapted to minimize the risk of uncontrolled bleeding. The follow-up of anti-GPIIb/IIIa is also particularly important for women, as anti-platelet antibodies may lead to fetal/neonatal thrombocytopenia.
Despite limited data, several studies have reported antiGPIIb/IIIa antibodies in patients with GT.3,4,9-12 To date, the
Table 1. Adverse events occurring during hospital admissions of patients in the Glanzmann Thrombasthenia Registry receiving platelet-based hemostatic treatment not including recombinant activated factor VII either for a bleeding episode or for treatment related to surgery.
Multiple adverse events could be recorded for a patient during a single admission. Antibody status was not recorded for all patients experiencing an adverse event. *One patient experienced both a non-serious adverse event and a serious adverse event. †Related to treatment for a bleeding event. ‡Platelet refractoriness was defined as: bleeding during surgery despite an adequate amount (as determined by the treating clinician) of platelet infusion, and/or persistence of bleeding despite an adequate amount (as determined by the treating clinician) of platelet infusion, and/or rebleeding within 24 hours despite an adequate amount (as determined by the treating clinician) of platelet infusion.3,4 OH: other hemostatic treatment (mostly antifibrinolytics); P: platelets; Pos: positive; Neg: negative; NR: not recorded; POH: platelets and other hemostatic treatments.
GTR has the largest GT cohort in which alloantibody proportion was assessed.3,4 Nevertheless, as a change in antibody/platelet-refractoriness status was not an objective of the GTR, it was not recorded for every admission, making interpretation difficult.3,4 Additionally, the GTR was not specifically designed to record platelet-related AE.
Not all centers participating in the GTR conducted platelet antibody studies due to the specialized nature, and potentially restricted availability, of testing. Indeed, in patients 3–5, the nature of the platelet antibodies (anti-HLA or GPIIb/IIIa) was unavailable. Thus, antibody status is likely underreported.10 Additionally, as antibodies can be cleared if no further transfusions are received, delayed assessment may give misleading negative results.12 Conversely, platelet transfusion may remain effective in previously alloimmunized GT patients as transfused platelets may not be removed immediately (potentially dependent on antibody isotype).10,11 Non-neutralizing antibodies (detected by immunological assays) that do not inhibit platelet aggregation may explain some persistent clinical efficacy of transfused platelets. Fluctuations in platelet refractoriness status, as observed in patient 5, suggest that some database entries may be inaccurate. Therefore, misinterpretation of the platelet refractoriness definition or incorrect data entry cannot be excluded.
Despite positive platelet refractoriness/antibody status, administration of platelet-based regimens continued in most admissions instead of transitioning to rFVIIa-based regimens. rFVIIa may not have been readily available during the timeframe of the GTR, either because of not yet being locally approved, or not being approved by the hospital or the patient’s insurance. Notably, rFVIIa was administered in patients 3 and 5 when platelet antibodies were positive. On two of these occasions in patient 3 and once in patient 5, treatment with F7POH was effective/partially effective. Hence, rFVIIa could have contributed to the outcome. Due to the real world nature of the database, admission records were often incomplete, making interpretation difficult. Importantly, bleeding event type and/or platelet refractoriness/antibody status can affect treatment efficacy and impact the patient treatment journey. This highlights the importance of comprehensively recording patient data, outcomes, platelet efficacy and antibody testing in patients with GT. Hence, there is an unmet need to initiate clinical practice changes to ensure routine antibody testing is performed within weeks of transfusion to improve patient management decisions.
1Departments of Medicine, Pediatrics and Oncology, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada;
2Southern Alberta Rare Blood and Bleeding Disorders
Comprehensive Care Program, Foothills Medical Center, Calgary, Alberta, Canada; 3Centre de Référence de l’Hémophilie et des Maladies Hémorragiques Constitutionnelles, Hôpital Bicêtre, AP-HP, and HITh, UMR_S1176, INSERM, Université Paris Saclay, Le KremlinBicêtre, France; 4Biostatistics and Programming GD GBS, Novo Nordisk Service Center India Private Ltd, Whitefield, Bangalore, India; 5Institute for Laboratory Medicine, Blood Coagulation and Transfusion Medicine (LBT), Düsseldorf, Germany; 6Department of Hemostasis, Hemotherapy and Transfusion Medicine, Heinrich Heine University Medical Center, Düsseldorf, Germany and 7Department of Clinical Medicine and Surgery, Regional Reference Center for Coagulation Disorders, Federico II University, Naples, Italy
†Posthumous.
Correspondence:
M.-C. POON - mcpoon@ucalgary.ca
https://doi.org/10.3324/haematol.2022.281973
Received: September 2, 2022.
Accepted: March 6, 2023.
Early view: March 16, 2023.
Published under a CC BY license
Disclosures
MCP was the chair of Novo Nordisk’s expert panel on the Glanzmann Thrombasthenia Registry, has been an ad hoc speaker for Bayer, Novo Nordisk, and Pfizer, attended advisory board meetings of CSL Behring, KVR Pharmaceuticals, Novo Nordisk, Octapharma, Pfizer, Bayer, Roche, Sobi and Takeda, and received grant funding from Bayer and CSL Behring. RdO was a member of Novo Nordisk’s expert panel on the Glanzmann Thrombasthenia Registry, has received research support for clinical trials or fees for advisory board honoraria or invitation as speaker in symposia from: Baxalta/Shire/Takeda, Biomarin, CSL Behring, LFB, Novo Nordisk, Octapharma, Pfizer, Roche, Sobi and Spark Therapeutics. SB is an employee of Novo Nordisk. RBZ was a member of Novo Nordisk’s expert panel on the Glanzmann Thrombasthenia Registry, has been a speaker in symposia for Aspen, AstraZeneca, Bayer, Biotest, Boehringer Ingelheim, CSL Behring, GlaxoSmithKline, LEO Pharma, MEDA, Novartis, Novo Nordisk, Octapharma, Pfizer, Sanofi-Aventis, has been a member of advisory boards at Bayer, BMS, Novo Nordisk, Pfizer and he has received research funding from CSL Behring. GDM was a member of Novo Nordisk’s expert panel on the Glanzmann Thrombasthenia Registry.
Contributions
MCP, RdO, RBZ and GDM, as members of Novo Nordisk’s expert panel on the Glanzmann Thrombasthenia Registry (GTR), participated in the planning and data monitoring of the GTR. All
Man-Chiu Poon,1,2 Roseline d’Oiron,3 Sibin Baby,4 Rainer B Zotz5,6† and Giovanni Di Minno7authors participated in data analysis, with SB providing the statistical analytical expertise. All authors contributed equally to the revision, finalization and approval of the manuscript.
Acknowledgments
This publication is dedicated to the memory of Dr Rainer B. Zotz, who sadly passed away during the preparation of the manuscript. We would like to acknowledge his important contributions to the field of hematology. The authors thank all investigators and patients participating in the Glanzmann Thrombasthenia Registry (GTR). Novo Nordisk Health Care AG (Zurich, Switzerland)
1. Poon MC, d'Oiron R. Alloimmunization in congenital deficiencies of platelet surface glycoproteins: focus on Glanzmann's thrombasthenia and Bernard-Soulier's syndrome. Semin Thromb Hemost. 2018;44(6):604-614.
2. Novo Nordisk. NovoSeven®, Summary of product characteristics. 2023. https://www.ema.europa.eu/ en/documents/product-information/novoseven-epar-productinformation_en.pdf. Accessed 20 February 2023.
3. Di Minno G, Zotz RB, d'Oiron R, et al. The international, prospective Glanzmann Thrombasthenia Registry: treatment modalities and outcomes of non-surgical bleeding episodes in patients with Glanzmann thrombasthenia. Haematologica. 2015;100(8):1031-1037.
4. Poon MC, d'Oiron R, Zotz RB, et al. The international, prospective Glanzmann Thrombasthenia Registry: treatment and outcomes in surgical intervention. Haematologica. 2015;100(8):1038-1044.
5. EMA. Assessment report NovoSeven. https://www.ema.europa. eu/en/documents/variation-report/novoseven-h-c-000074-ii0104-epar-assessment-report-variation_en.pdf. Accessed 16 March 2023.
6. Murphy MF, Metcalfe P, Ord J, Lister TA, Waters AH. Disappearance of HLA and platelet-specific antibodies in acute leukaemia patients alloimmunized by multiple transfusions. Br
sponsored the GTR and was responsible for its conduct, including engaging an external panel to provide advice. Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Ashfield MedComms GmbH, an Inizio company, and financially supported by Novo Nordisk Health Care AG.
Data-sharing statement
The subject level analysis data sets for the research presented in the publication are available from the corresponding author on reasonable request.
J Haematol. 1987;67(3):255-260.
7. Weinstock C, Schnaidt M. Human leucocyte antigen sensitisation and its impact on transfusion practice. Transfus Med Hemother. 2019;46(5):356-369.
8. Huguenin Y, Levoir L, Bouton M, Fiore M. High rates of antialphaIIbbeta3 antibodies produced by a Glanzmann thrombasthenia patient after first and unique red blood cells administration. Acta Haematol. 2023;146(1):44-46.
9. Fiore M, d'Oiron R, Pillois X, Alessi MC. Anti-alpha(IIb) beta(3) immunization in Glanzmann thrombasthenia: review of literature and treatment recommendations. Br J Haematol. 2018;181(2):173-182.
10. Fiore M, Firah N, Pillois X, Nurden P, Heilig R, Nurden AT. Natural history of platelet antibody formation against alphaIIbbeta3 in a French cohort of Glanzmann thrombasthenia patients. Haemophilia. 2012;18(3):e201-209.
11. Nurden A, Combrie R, Nurden P. Detection of transfused platelets in a patient with Glanzmann thrombasthenia. Thromb Haemost. 2002;87(3):543-544.
12. Santoro C, Rago A, Biondo F, et al. Prevalence of alloimmunization anti-HLA and anti-integrin alphaIIbbeta3 in Glanzmann Thromboasthenia patients. Haemophilia. 2010;16(5):805-812.
B-cell acute lymphoblastic leukemia (B-ALL) is often characterized by gene fusions of transcription factors such as ETV6-RUNX1 or tyrosine kinases such as BCRABL1. Novel gene fusions harboring the transcription factor MEF2D with various partner genes have been identi fi ed in 2016.1 The estimated frequency of MEF2D fusions in children and adolescents with newly diagnosed ALL is 2-6%, of which the MEF2D-BCL9 fusion is most commonly found.1–4 Patients harboring a MEF2D fusion have a poor prognosis (5-year event-free survival of ~55%). They present with unfavorable clinical characteristics including an older age of onset (median ranging between 9 and 14 years) and a high white blood cell count (median 27.3x106 cells/mL).1,3,4 Molecular characteristics are differential CD5 and CD10 expression compared to other B-ALL subtypes, along with increased HDAC9 expression and a higher frequency of deletions and mutations in CDKN2A/B and PHF6 , respectively.1,3,4 Primary leukemic cells of B-ALL patients generally do not replicate ex vivo , which makes functional studies diffi cult. However, occasionally prolonged culturing of leukemic cells is successful; well-known examples are Reh (ETV6RUNX1), SupB15 (BCR-ABL1), and RCH-ACV (TCF3-PBX1)5–7 cell lines. A retrospective study recently identified MEF2D fusions in 19 non-publicly available B-ALL cell lines with limited molecular and functional characterization.2 In this study, we characterize an in-house made patient-derived MEF2D fusion positive B-ALL cell line, named M4A1-M2B9, and compare the characteristics to its corresponding primary material. The patient-derived cell line is comparable to the primary material when examining mutations in the exome, gene expression, the immunophenotype, and sensitivity to drugs that do not affect division and/or cell cycle. These results imply that M4A1-M2B9 is a valuable resource to examine novel therapeutics targeting MEF2D fusion proteins or downstream targets activated by MEF2D, as well as to study immunotherapy options for B-ALL.
In accordance with the Declaration of Helsinki, written informed consent to use excess diagnostic material for research purposes was obtained from parents or guardians, as approved by the medical Ethics Committee of the Erasmus Medical Center. All used reagents and primers are described in the Online Supplementary Table S1. We used paired-end stranded total RNA illumina sequencing (RNAseq) after riboRNA depletion with fragments of 150 bp to detect gene expression differences. Paired-end whole exome sequencing (WES) was used to detect vari-
ants and copy number alterations in the coding regions. Sequencing data are deposited at the European Genome-Phenome Archive (EGA) (RNA sequencing: EGAD00001009759, WES: EGAD00001009758). Primary cells were expanded after intrafemoral injection in NOD scid γ mice, leukemic burden was tested every 2-4 weeks in peripheral blood and the cells were harvested from the spleen upon overt leukemia. Cells were seeded in a 96-wells plate and medium was replaced every 3-4 days until visible cell growth. Afterwards, cells were routinely cultured. Cell survival of primary human mesenchymal stromal cells (MSC derived from a B-ALL patient at day 79) was measured after 4 days of co-culturing by harvesting and staining the cells followed by flow cytometry (Beckman Coulter). The viability after 4 days of drug or ligand exposure was measured using MTT. In all experiments, the blast percentage of the primary samples was ≥90% at the start of the experiment. M4A1-M2B9 has been deposited at DSMZ for future distribution.
A 15-year-old male patient was diagnosed with B-ALL harboring a MEF2D-BCL9 fusion, a biallelic CDKN2A/B deletion, and presenting a high white blood cell count of ~150x106 cells/mL (Online Supplementary Figure S1A-C).
The leukemic cells had a pretreatment karyotype of 46~48,XY,del(1)(q21),-9,del(9)(p1?2),+1~3mar,inc[6]/46,XY [15] and a cytoplasmic μ chain-positive pre-B immunophenotype (CD19 + /CD20 - /CD10 + /CD45 dim /CD34 - /CD38 + ).
The patient was treated according to ALL10 high-risk protocol. The patient initially responded poorly to therapy having a high minimal residual disease (MRD) level (>1%) at day 33, however, at day 79 the MRD level was positive but not quanti fi able. The patient received stem cell transplantation and has been in continuous complete remission for >6 years till last follow-up.
We established a patient-derived cell line in which we studied the dependence on external factors, specifically serum dependence, stromal support, and addition of cytokines. M4A1-M2B9 was derived from bone marrow leukemic cells which were used for intrafemoral injection, the mouse was sacrificed after 53 days and 86% of the harvested spleen-derived cells were blasts. Within 1 month of culturing in primary medium, the spleen-derived cells were dividing and were split every 3-4 days. In 2.5 months, the cells grew from 0.4 million to 80 million (Figure 1A). The presence of the MEF2D fusion was confirmed by reverse transcriptase polymerase chain reaction (PCR). We compared the growth speed of M4A1-M2B9 in our primary medium, containing 20% fetal calf serum
(FCS) and supplemented with insulin, transferrin, and selenium (ITS), with our standard cell line medium con-
taining either 10% or 20% FCS lacking ITS. M4A1-M2B9 is serum-dependent, the cells died in the culture with 10%

A B
Figure 1. Growth and survival of patient-derived xenograf MEF2D-BCL9 cell line. (A) The growth curve of cells freshly obtained from a patient-derived xenograft (PDX) when cultured on primary medium (RPMI 1640 medium supplemented with 20% fetal calf serum [FCS], insulin, transferrin and sodium selenite [ITS], glutamine and gentamycin) in the first 2.5 months. For the first month half of the medium was replaced every 3-4 days, afterwards the cell concentration was determined every 3-4 days by manual cell count using trypan blue staining and the cells were split accordingly. (B) Examining the dependence of the cells on factors present in primary medium by comparing growth in primary and cell line medium (RPMI 1640 medium supplemented with 20% or 10% FCS, penicillin-streptomycin and Amphotericin). Cells cultured on primary medium were spun down and placed on cell line medium containing either 20% or 10% FCS. Cells were counted manually every 3-4 days and cultured at a density of 0.5x106 cells/mL. The average doubling time of the cells cultured in primary medium is 42.2 hours, the average doubling time of cells cultured in the cell line medium with high FCS is 56.8 hours. (C, D) Examination of the dependence of mesenchymal stromal cells (MSC) support using primary material and the cell line. Primary material and the cell line were cultured in a 48-wells plate for 4 days at 37°C and 5% CO2 either in monoculture or on a MSC layer (16.500 cells/well seeded at day -1) by replacing MSC medium with 410 µL cell suspension at a concentration of 1x106 cells/mL or 0.5x106 cells/mL, respectively. The number of viable leukemic cells after 4 days was measured using flow cytometry (C) and the percentage survival relative to the monoculture (D) are plotted. Bars show mean ± standard error of the mean (SEM) of 2 independent experiments. (E) Examination of the benefit of cytokines using primary material and M4A1-M2B9. Primary material and the cell line were cultured for 4 days at 37°C and 5% CO2 in the presence or absence of cytokines (IL7 at 8 ng/mL, TSLP at 20 ng/mL or FLT3L at 32 ng/mL or a combination thereof) at a concentration of 1.6x106 cells/mL and 0.5x106 cells/mL, respectively. The metabolic activity relative to unstimulated cells was measured after addition of MTT, incubation for 2-6 hours, dissolving the crystals using acidified isopropanol and subsequently the absorbance at 562 nm and 720 nm was obtained using the VersaMax plate reader. Bars show mean ± SEM of 2 independent experiments.

Figure 2. Comparison of gene expression, genomic variants and cell surface protein between primary material and cell line. (A) Global gene expression comparison between primary material and cell line by plotting the log10 FPKM (fragments per kilobase million) + 0.01 per gene. (B) Heatmap of Z-scores of up- and down-regulated genes compared to the control B-cell acute lymphoblastic leukemia (B-ALL) cohort (N=424, derived from various treatment protocols). The up- (red) or down- (blue) regulated genes are selected based on the gene expression signature of MEF2D fused patients described by Ohki et al.4, gene names are colored accordingly. IGHG1 and IGHV5-51 were not plotted due to the lack of expression in our cohort. Heatmap was made using the ComplexHeatmap package in R. Z-score was calculated, Z-scores >3 are set to 3, Z-score <-3 are set to -3. (C) Variant analysis of the cell line was compared with the germline and the primary tumor. First, the reads were filtered using Mutect2. Subsequently, the variants were filtered with a cut-off of at least 10 variant reads and the variant should be present in a transcript with one of the following biotypes: protein coding region, long non-coding RNA, nonsense-mediated decay, or intron retention. Next, all remaining variants were filtered based on their variant allele fraction (VAF) ≥10. After that, the remaining variants were checked manually in Integrative Genomics Viewer and if absent in the germline and present in tumor and/or cell line the variants and estimated VAF were noted. In the primary sample, there are 12 variants with a VAF <25% and 15 variants with a VAF >25%. All variants with a VAF >25% were also found in the cell line with a VAF of 34-58% (see Table, blue). In addition, 1 variant which was present in the primary sample with a VAF <25% was also found in the cell line with a VAF >10% (see Table, orange). Moreover, 2 variants were found in the cell line, which were not present or below the detection level in the primary material (see Table, red). Of these variants one originates from the patient-derived xenograft (PDX), while the other is only in present in the cell line. (D-E) Copy number alteration analysis for the primary material and the cell line, respectively. The upper figure shows the copy number of the primary material per chromosome and the lower figure the copy number of the cell line. The alterations, such as a deletion on chromosome 9 and 14, were unchanged. (F) Histograms of primary material (blast percentage of 93%) and cell line material showing the cell surface markers as determined by flow cytometry, primary cells in dark gray, cell line in light gray. Debris was discarded and alive cells were selected. X-axis, fluorescence intensity; Y-axis, modal.
FCS but grew in primary medium and cell line medium containing 20% FCS (Figure 1B). In addition, we studied the survival and cell proliferation of the primary material and M4A1-M2B9 in an ex vivo leukemic niche using a coculture with MSC.8 As expected, the primary material showed improved survival after co-culture on MSC, whereas M4A1-M2B9 showed equal survival with or without MSC support (Figure 1C, D). Moreover, we evaluated the effect of adding cytokines to M4A1-M2B9 and the primary material. IL7, TSLP and FLT3L were added as single supplement or in combination, as these cytokines stimulate common B-ALL activated pathways. 9 Both M4A1M2B9 and primary material benefited up to 30% from the addition of FLT3L, however, the primary material had a benefit of 70% from the cytokine cocktail and benefited from multiple cytokine combinations (Figure 1E). In addition to the dependence on external factors, we studied genetic and immunophenotypic differences between the primary material and M4A1-M2B9. Global gene expression was similar for primary material and the cell line (Figure 2A) and resembled the MEF2D-specific gene expression signature identified by Ohki et al.4 (Figure 2B). Moreover, high expression of HDAC9 (Z-score=3.96/4.27), BCL9 (Z-score=3.01/1.98), and CD5 (Z-score=1.0 9/0.91), and low expression of CD10 (Z-score=-1.04/-1.20) was observed in the primary material and M4A1-M2B9 (Figure 2B; Online Supplementary Figure S1D). WES was used to detect leukemia-speci fi c sequence variants and copy number alterations. Based on the variants and their allele frequencies, we conclude that the major clone of the initial material expanded in the patient-derived xenograft (PDX) (Figure 2C). The copy number alterations of the primary material and M4A1-M2B9 were comparable, and both contained a bi-allelic CDKN2A/B deletion (Figure 2D, E). In order to determine whether the cell surface marker
expression remained stable upon cell line establishment, we performed immunophenotyping on viable cells using the cell surface markers CD5, CD10, CD19, CD22 and CD45. M4A1-M2B9 was 100% CD5-positive, while the primary material harbored 75% CD5-positive and 25% CD5negative cells (Figure 2F). CD5 is often highly expressed on the cell surface of MEF2D fused B-ALL cells.4 In addition, CD10 and CD45 were expressed in the primary material, but were weaker and partially lost in the cell line. Expression of CD19 and CD22 varied slightly between the primary material and M4A1-M2B9 but both were stably expressed (Figure 2F). Together these results suggest that our MEFD2-BCL9-positive cell line represents the major clone of the primary material. In order to gain a better understanding of the state of cells in which the establishment of clonal selection occurred, the experiments should have been performed with the cells directly harvested from the PDX as well. Unfortunately, the number of blasts yielded from the spleen was insufficient to perform these experiments.
Finally, we determined whether the drug sensitivity of M4A1-M2B9 resembles the drug sensitivity of the primary leukemic cells. Sensitivity to drugs that do not directly affect the cell cycle nor DNA or RNA synthesis, such as prednisolone, dexamethasone, asparaginase and different pan-HDAC inhibitors, differed minimally between the primary material and M4A1-M2B9 (Figure 3A-D; Online Supplementary Figure S2 ). As expected, drugs which influence cell cycle or DNA/RNA synthesis such as vincristine, cytarabine, and daunorubicin affected the proliferating M4A1-M2B9 more than the primary material (Figure 3E-G).
In conclusion, the patient-derived MEF2D-BCL9-positive cell line is highly representative for the major clone of the primary material, showing similar gene expression, mu-
Figure 3. Drug sensitivity comparison between primary material and the cell line. Primary material and cell line were cultured for 4 days at 37°C and 5% CO2 either in the presence or absence of the drug starting at a concentration of 1.6x106 cells/mL and 0.5x106 cells/mL, respectively. Survival was measured using MTT and the metabolic activity was compared to untreated cells. Primary material is shown in dark gray and the cell line in light gray. (A-D) Comparison of drug sensitivity using compounds that do not affect the cell cycle or DNA/RNA synthesis between the primary material and cell line. These drugs include 2 glucocorticosteroids, dexamethasone (1:8 dilution) and prednisolone (1:8 dilution), a metabolic drug, asparaginase (1:5 dilution), and a pan-HDAC inhibitor panobinostat (1:2 dilution). (E-G) Comparison of drug sensitivity using compounds that affect the cell cycle or DNA/RNA synthesis between the primary material and the cell line. These drugs include AraC (1:4 dilution), daunorubicin (1:4 dilution), an anthracycline, and vincristine (1:4 dilution), which inhibits mitotic spindle formation. (A, B) X-axis, concentration of the drug, Y-axis, metabolic activity relative to untreated cells. For patient material and cell line the graphs show mean ± standard error of the mean (SEM) of 2 or 2-3 independent experiments, respectively. The bar plots show the mean lethal concentration 50 (LC50) ± SEM of 2 or 2-3 independent experiments, respectively, the blue line in the half maximal inhibitory concentration (IC50) plots indicate the lowest measured concentration. An unpaired t-test was used to compare the LC50 values between the primary material and the cell line (ns: not significant P> 0.05; * P≤0.05, **P≤0.01, ***P≤0.001).

tations, copy number alterations, and immunophenotype. Because the cell line proliferates, M4A1-M2B9 may better represent the drug sensitivity of leukemic cells in the patient, especially of drugs that affect cell cycle or DNA/RNA synthesis. M4A1-M2B9 facilitates functional studies, for example the development of targeted therapies directed against the MEF2D fusion protein or downstream targets, or CAR T products directed against CD5, which are currently being studied for patients with T-cell ALL.10 As MEF2D fusion protein-positive pediatric B-ALL is associated with high-risk features, such as high age, high white blood cell count, high MRD, and poor outcome,1,3 targeted therapy might improve the treatment of patients with a MEF2D fusion.
Inge van Outersterp,1,2* Femke M. Hormann,1,2,3* Alex Q. Hoogkamer,1,2 Aurélie Boeree,1,2 Stijn A. van den Broek,1,2 Monique L. den Boer1,2,3 and Judith M. Boer1,2
1Princess Máxima Center for Pediatric Oncology, Utrecht; 2Oncode Institute, Utrecht and 3Erasmus MC - Sophia Children's Hospital, Department of Pediatric Oncology and Hematology, Rotterdam, the Netherlands
*IvO and FMH contributed equally as first authors.
Correspondence:
J.M. BOER - j.m.boer-20@prinsesmaximacentrum.nl
1. Suzuki K, Okuno Y, Kawashima N, et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J Clin Oncol. 2016;34(28):3451-3459.
2. Akahane K, Yasuda T, Tsuzuki S, et al. High prevalence of MEF2D fusion in human B-cell precursor acute lymphoblastic leukemia cell lines. Hematol Oncol. 2020;38(4):614-617.
3. Gu Z, Churchman M, Roberts K, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:1-10.
4. Ohki K, Kiyokawa N, Saito Y, et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica. 2019;104(1):128-137.
5. Rosenfeld C, Goutner A, Venuat AM, et al. An effective human leukaemic cell line: Reh. Eur J Cancer. 1977;13(4):377-379.
https://doi.org/10.3324/haematol.2022.281712
Received: November 18, 2022.
Accepted: March 31, 2023.
Early view: April 13, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
This project was conceived by JMB and MLdB and conceptualized together with IvO and FMH. Experimental and computational analyses were performed by IvO, FMH, AQH, AB and SvdB. Data interpretation was performed by IvO, FMH, MLdB and JMB. The manuscript was drafted by IvO, FMH, MLdB and JMB. All authors approved the final version of the manuscript.
Funding
The authors would like to thank Foundation Pediatric Oncology Center Rotterdam (SKOCR), the Dutch Cancer Society grant KWF10482, and the KiKa Foundation Kika-264 grant for their support.
The data that support the findings of this study are available on request from the Data Access Committee Princess Maxima Center (e.g. https://ega-archive.org/datasets/EGAD00001009759 [RNAseq], https://ega-archive.org/datasets/EGAD00001009758 [WXS]). The data are not publicly available due to them containing information under controlled access.
6. Naumovski L, Morgan R, Hecht F, Link MP, Glader BE, Smith SD. Philadelphia chromosome-positive acute lymphoblastic leukemia cell lines without classical breakpoint cluster region rearrangement. Cancer Res. 1988;48(10):2876-2879.
7. Jack I, Seshadri R, Garson M, et al. RCH-ACV: a lymphoblastic leukemia cell line with chromosome translocation 1;19 and trisomy 8. Cancer Genet Cytogenet. 1986;19(3-4):261-269.
8. Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, Campana D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood. 1992;79(9):2370-2377.
9. Harrison CJ. Targeting signaling pathways in acute lymphoblastic leukemia: new insights. Hematology. 2013;2013(1):118-125.
10. Hill LC, Rouce RH, Smith TS, et al. Safety and anti-tumor activity of CD5 CAR T-cells in patients with relapsed/refractory T-cell malignancies. Blood. 2019;134(Suppl 1):S199.
KMT2A translocations, the commonest abnormalities in acute lymphoblastic leukemia (ALL) in <1-year-old infants, are associated with poor outcomes,1-5 yet the prognostic significance of KMT2A-rearranged (KMT2A-R) versus KMT2A-germline (KMT2A-G) status and KMT2A partner genes is poorly understood in the context of demographic and clinical covariates. We investigated these variables in the Children’s Oncology Group (COG) P9407 trial for newly diagnosed infant ALL (clinicaltrials gov. Identifier: NCT00002756). This trial, designed to reduce early relapses by induction intensification, enrolled 221 infants (209 treatment-eligible) from June 1996-June 2006, and was amended in cohorts 2 and 3 for toxicities and hematopoietic stem cell transplantation lacking benefit.3 Specimens were collected under Pediatric Oncology Group (POG) 9900 or COG AALL03B1 with separate consent and individual Institutional Review Board approvals.3
Of 209 (199 treatment-eligible) cases analyzed, 157 (75.1%) (148/199 treatment-eligible; 74.4%) were KMT2A-R, and the rest were KMT2A-G. Partner genes in 5’-KMT2Apartner-3’ fusions were assigned to five categories similar to Interfant-99/Interfant-062,4 and COG AALL06315: AFF1 (n=78, 49.7%; 73/78 treatment-eligible); MLLT1 (n=33, 21.0%; 31/33 treatment-eligible); MLLT3 (n=14, 8.9%; 13/14 treatment-eligible); ‘other’ (n=20, 12.7%; all treatment-eligible); ‘unknown’ (n=12, 7.6%; 11/12 treatment eligible).
‘Other’ included all non-AFF1/non-MLLT1/non-MLLT3 partner genes: EPS15 (n=4), MLLT10 (n=1), ACER1 (n=1), ACTN4 (n=1), non-AFF1/non-MLLT1/non-MLLT3 not further classified (n=13). ‘Unknown’ included non-AFF1 not further classified (n=5; 4/5 treatment-eligible) and KMT2A-R not further classified (n=7; all treatment-eligible). COG P9407 used leukemia classification methods of the trial era including karyotype, fluorescence in situ hybridization, Southern blot and/or conventional polymerase chain reaction (PCR), and panhandle PCR analyses for unknown partner genes.6 While advanced genomics may detect covert rearrangements and improve partner gene assignments,7,8 the 75.1% KMT2A-R in this study is comparable to 79%, 74%, and 70% in other large infant ALL clinical trials.2,4,5
Limitations notwithstanding, breakpoint heterogeneity
and partner gene diversity were uncovered: one case had a 5’-KMT2A exon 9-AFF1 exon 11-3’ transcript outside the usual AFF1 exon 3-6 breakpoint region.9 Another had the partner genes ACTN4 and RYR1, both from 19q13.2, in 5’KMT2A-partner-3’ and 5’-partner-KMT2A-3’ fusions, suggesting a new unbalanced t(11;19). ACTN4 and RYR1 have not occurred as partner genes in KMT2A-R infant ALL, but ACTN4 was reported in separate cases of treatment-related ALL and treatment-related MDS.10 ACER1 from 19p13.3 was reported in one case of infant ALL.11
Five-year event-free survival (EFS) was: MLLT1, 25±9%; AFF1, 34±7%; ‘other’, 40±14%; MLLT3, 68±17%; KMT2A-G, 69±9% (log-rank test; P<0.0001) (Figure 1A). After treatment modifications ameliorated excessive toxicities, in cohort 3 5-year EFS was: MLLT1, 15±10%; AFF1, 33±10%; ‘other’, 39±15%; MLLT3, 73±27%; KMT2A-G, 70±13% (P=0.0004) (Online Supplementary Figure S1A), suggesting consistent impact of genetic subtypes.
The COG uses ≤90 days to define high-risk KMT2A-R infant ALL.5 Our microarray studies had revealed AFF1 case separation at ~90 days, with elevated expression of B-cell maturation genes in older versus interleukin, HSP, and HLA genes in younger infants,12 providing biological grounds for this cut-off. Consistently, in ≤90-day-old versus >90-dayold infants, 5-year EFS was worse among KMT2A-R overall (6±4% vs. 47±6%; P<0.0001); AFF1 (5±5% vs. 46±9%; P<0.0001); and MLLT1 (0% vs. 37±12%; P=0.0008) (Figure 1B). Using National Cancer Institute risk groups of white blood cell (WBC) count <50x109/L versus ≥50x109/L,13 5year EFS was 76±7% versus 33±5% (P<0.0001) in treatment-eligible overall (Table 1); 67±10% versus 29±5% (P=0.0002) in all KMT2A-R; 67±22% versus 15±8% in MLLT1 (P=0.029); and 88±9% versus 52±14% in KMT2A-G (P=0.013) (Figure 1C).
Using Interfant-99 risk groups,2 5-year EFS was: high-risk (KMT2A-R with age <6 months and WBC count >300x109/L), 24±9%; intermediate-risk (IR) ( KMT2A-R without these features), 41±6%; low-risk (LR) (KMT2A-G), 69±9% (overall P<0.0001; high-risk vs. IR; P=0.047) (Online Supplementary Figure S1B), suggesting better KMT2A-R separation when analyzed by partner gene, age ≤/>90 days, or WBC count </≥50x109/L (Figure 1A-C).
partner genes in infant acute lymphoblastic leukemia have prognostic significance and correlate with age, white blood cell count, sex, and central nervous system
Figure 1. Kaplan-Meier plots of event-free survival in COG P9407 trial. Event-free survival (EFS) in treatment-eligible infants estimated by Kaplan-Meier method as a function of: (A) KMT2A-G and KMT2A partner genes (11 treatment-eligible in category ‘unknown’ partner gene excluded); (B) age ≤/>90 days in KMT2A-R overall (left), AAF1 (middle), and MLLT1 (right); (C) white blood cell (WBC) count </≥50×109/L in KMT2A-R overall (left), MLLT1 (middle), and KMT2A-G (right); (D) male vs. female sex in KMT2A-R overall (left), and AAF1 (right); (E) central nervous system (CNS) status in KMT2A-R overall, and AAF1 (right). CNS1 (CNSnegative), CNS2 (positive cytomorphology but <5 WBC/µL), CNS3 (≥5 WBC/µL and positive cytomorphology). (A-E) EFS calculated as time from diagnosis to first event (induction failure, relapse, secondary malignancy, remission death). Patients having no event were censored at last contact. Numbers of patients are indicated in the plots. P values calculated by log-rank test are at top right in the plots.

Male sex adversely affected 5-year EFS in KMT2A-R overall (28±6% vs. 45±7%, girls; P=0.0147) and AFF1 (24±9% vs. 42±10%, girls; P=0.0198) (Figure 1D). Five-year EFS for CNS1, CNS2, and CNS3 was 54±6%, 41±8%, and 33±10% (P=0.025) in treatment-eligible overall (Table 1); 48±7%, 27±8%, and 30±9% (P=0.0155) in all KMT2A-R; and 53±12%, 18±10%, and 23±12% (P=0.022) in AFF1 (Figure 1E).
The impact of KMT2A-R versus KMT2A-G, partner genes, and demographic and clinical covariates was further studied using multivariable Cox regression models. After adjusting for sex, age, WBC, and central nervous system (CNS) disease at diagnosis, AFF1 (hazard ratio [HR]=2.51; P=0.0032), MLLT1 (HR=3.30; P=0.0007), ‘other’ (HR=2.50; P=0.021), and ‘unknown’ (HR=2.81; P=0.048) partner genes were significant for high risk, and MLLT3 had similar risk (HR=1.04; P=0.95) compared to KMT2A-G as reference. Age ≤90 days (HR=2.58; P<0.0001), WBC count ≥50x109/L (HR=2.70; P=0.0027), and male sex (HR=1.59; P=0.023) were associated with significantly higher risk (Table 1). Separate multivariable analysis of KMT2A-R subtypes without KMT2A-G (Online Supplementary Table S1) re-
vealed similar effects of age, WBC count, sex, and CNS disease, with all except CNS disease independently impacting outcome. Using AFF1 as reference, MLLT3 had lower observed risk (HR=0.41; P=0.15), and MLLT1 had higher observed risk (HR=1.34; P=0.27), but KMT2A-R subtypes did not reach signifi cance. We also explored a model containing age as a continuous variable; age proved to be significant, and effects of all other covariates were similar to both models described above (Online Supplementary Table S1). Another model controlling for cohort 3 after therapy adjustments did not show an effect of treatment; KMT2A partner genes, age, WBC count, and sex retained independent impact on prognosis (Online Supplementary Table S1). We then asked whether demographic or clinical covariates differed by genetic subtype. Age distribution differed among AFF1, MLLT1, MLLT3, ‘other’, and KMT2A-G (P<0.0001) (Figure 2A). Age ≤90 days was more common in KMT2A-R overall (40/157; 25.5%) than KMT2A-G (6/52; 11.5%) (P=0.036). Nineteen of 20 ‘other’ (i.e., all nonAFF1/non-MLLT1/non-MLLT3) (95%) occurred in >90-day-
**P value for the log-rank test on the difference between subgroups. #Data are hazard ratio (95% confidence interval). ##Calculated with Wald tests in joint analysis of sex, age, white blood cell (WBC) count, KMT2A partner gene category vs. KMT2A-G, and central nervous system (CNS) status. ‘Other’ partner gene includes: EPS15, MLLT10, ACTN4, ACER1, and non-AFF1/non-MLLT1/non-MLLT3 not further classified. ‘Unknown’ partner gene includes: non-AFF1 not further classified, and KMT2A-R not further classified. CNS status: CNS1, negative; CNS2, positive cytomorphology/and <5 WBC/µL; CNS3, ≥5 WBC/µL/and positive cytomorphology. Analysis does not include CNS status unknown stratum (N=2). CNS: central nervous system; EFS: event-free survival; SE: standard error.
Figure 2. Correlations of demographic and clinical covariates with KMT2A-G and KMT2A partner genes. Distributions among KMT2A-G and AFF1, MLLT1, MLLT3, and ‘other’ KMT2A-R genetic subtypes for all treatment-eligible and treatment-ineligible infants plotted by: (A) age at diagnosis ≤90 days (d) vs. >90 d (top left), and age as a continuous variable (top right). Box and whisker plots (bottom) indicate that distribution by age in days as a continuous variable differs among genetic subtypes (χ2 statistic 30.2; degrees of freedom (DF) 4; P<0.0001). Error bars represent range; horizontal lines, quartiles; and small diamond, the mean. (B) Presenting white blood cell (WBC) count <50x109/L vs. ≥50x109/L (top left), and as a continuous variable (top right). Box and whisker plots (bottom) indicate that WBC distribution as a continuous variable differs among genetic subtypes (χ2 statistic 39.4; DF 4; P<0.0001). (A, B) Distributions among genetic subtypes were compared by Kruskal-Wallis χ2 square-test; (C) sex; (D) central nervous system (CNS) status at diagnosis. (A-D) Cases in ‘unknown’ partner gene category were excluded.

old infants versus 88 of 125 (70.4%) with any of these partner genes (P=0.026); 55 of 78 (70.5%) AFF1 (P=0.022); and 22 of 33 (66.6%) MLLT1 (P=0.020) (Online Supplementary Table S2).
WBC count differed among AFF1, MLLT1, MLLT3, ‘other’, and KMT2A-G (P<0.0001) (Figure 2B). WBC count was ≥50x109/L in 123 of 155 (79.4%) KMT2A-R versus 27 of 52 (51.9%)
KMT2A-G (P=0.0003). WBC count was ≥50x109/L in higher proportions of AFF1 (71/77; 92.2%) versus any non- AFF1 partner gene (47/71; 66.2%) (P<0.0001); AFF1 versus MLLT3 (6/14; 42.9%) (P<0.0001); MLLT1 (26/32; 81.3%) versus MLLT3 ( P=0.015); and any non-MLLT3 partner gene (110/129; 85.3%) versus MLLT3 (P=0.0008) (Online Supplementary Table S2).
The 45.9% male (n=72)/54.1% (n=85) female KMT2A-R differed from 63.5% male (n=33)/36.5% female (n=19) KMT2A-G (P=0.037). The 46.2% (n=36) male/53.8% (n=42) female AFF1 did not differ statistically from KMT2A-G (P=0.073) or any other partner subtypes (Figure 2C; Online Supplementary Table S2).
CNS1 occurred in 32 of 52 (61.5%) KMT2A-G versus 71 of 155 (45.8%) KMT2A-R overall (P=0.056); 32 of 77 (41.6%) AFF1 (P=0.032); and 13 of 33 (39.4%) MLLT1 (P=0.074). A higher proportion of KMT2A-G were CNS1 or CNS2 (47/52; 90.4%) versus KMT2A-R overall (117/155; 75.5%) (P=0.029); AFF1 (57/77; 74.0%) (P=0.024); and MLLT1 (23/33; 69.7%) (P=0.020) (Figure 2D; Online Supplementary Table S2). In summary, this study demonstrates the impact of KMT2A partner genes on prognosis, and partner gene associations and interactions with demographic and clinical covariates. Univariate and multivariable analyses identify KMT2A-R subtypes (AFF1, MLLT1, ‘other’) with significantly decreased survival, and survival for MLLT3 equivalent to KMT2A-G, establishing independent prognostic significance of KMT2A partner genes compared to KMT2A-G. A separate multivariable model excluding KMT2A-G revealed non-significantly increased and decreased observed risks versus AFF1 for MLLT1 and MLLT3, respectively. The study further shows that age, WBC count, sex, and CNS disease are unevenly distributed and, in univariate analyses, partition EFS within genetic subtypes. Moreover, age, WBC count, and sex have independent prognostic significance in all multivariable models tested. Therefore, KMT2A partner genes, demographic and clinical covariates, and outcome in infant ALL are interrelated.
MLLT10 and the YEATS-domain-containing proteins MLLT1 and MLLT3 are DOT1L complex members. MLLT3 and AFF1 are Super Elongation Complex (SEC) members.14 Yet AFF1, MLLT1, and ‘other’ partner genes proved high-risk compared to KMT2A-G whereas, similar to pediatric AML,15 MLLT3 was favorable.
In contrast, CCG 1953 and Interfant-99 suggested poorer outcomes for all KMT2A-R including MLLT3, 1,2 and Interfant-06 found higher risk for t(4;11)+t(11;19) together and
t(9;11)+‘other’ together.4 Survival was worse among KMT2A-G with WBC count ≥50x109/L in our study, but not WBC count >300x109/L in Interfant-99/Interfant-06.16 In our study male sex adversely impacted EFS in KMT2A-R and AFF1 in univariate analysis, and in multivariable models, whereas Interfant-06 found higher EFS in boys than girls for KMT2A-R and KMT2A-G together by univariate analysis but no difference by sex in multivariable analysis.4
The disproportionate ≤90-day-old infants among AFF1 and MLLT1, and >90-day-old infants among ‘other’ here, and the 67% of t(11;19), but 31% of t(9;11) in <6-month-old infants in Interfant-99,2 agree with variable leukemia latencies by partner gene in murine models.17
Improved understanding of the spectrum of KMT2A partner genes relative to their complex interplay with demographic and clinical covariates is critical to discern high-risk infants. KMT2A fusion proteins involving members of the AF4 (AFF1) and ENL (MLLT1, MLLT3) protein families constitutively activate transcription by forming AEP (AF4/ENL/P-TEFβ) complexes, whereas different KMT2A fusion proteins alter transcription by varied mechanisms.18 Considering the partner gene heterogeneity and paucity of infant ALL with some ‘other’ partner genes, advanced genomics classification7,8 and pooling across trials are essential to discern and validate the prognostic importance of partner genes and demographic and clinical covariates, especially as transcription-targeting agents18 continue advancing.
Blaine W. Robinson,1° John A. Kairalla,2 Meenakshi Devidas,3 Andrew J. Carroll,4 Richard C. Harvey,5 Nyla A. Heerema,6 Cheryl L. Willman,7 Amanda R. Ball,1° Elliot C. Woods,1° Nancy C. Ballantyne,1° Karen A. Urtishak,1° Frederick G. Behm,8 Gregory H. Reaman,9° Joanne M. Hilden,10 Bruce M. Camitta,11 Naomi J. Winick,12 Jeanette Pullen,13 William L. Carroll,14 Stephen P. Hunger,1,15 ZoAnn E. Dreyer16 and Carolyn A. Felix1,15
1Division of Oncology and the Center for Childhood Cancer Research, Children’s Hospital of Philadelphia, Philadelphia, PA; 2Department of Biostatistics, University of Florida College of Public Health and Health Professions and College of Medicine, Gainesville, FL; 3Department of Global Pediatric Medicine, St Jude Children’s Research Hospital, Memphis, TN; 4Department of Genetics, University of Alabama at Birmingham, Birmingham, AL; 5University of New Mexico Cancer Center and Department of Pathology, Albuquerque, NM; 6Department of Pathology, The Ohio State University Comprehensive Cancer Center, Columbus, OH; 7Mayo Clinic Comprehensive Cancer Center, Rochester, MN; 8Department of Pathology, University of Illinois at Chicago, Chicago, IL;
9Children's National Medical Center, Washington DC; 10Center for Cancer and Blood Disorders, Children’s Hospital Colorado, Aurora, CO; 11Medical College of Wisconsin, Milwaukee, WI; 12Division of Pediatric Hematology/Oncology, University of Texas Southwestern School of Medicine, Dallas, TX; 13Pediatric Hematology/Oncology, University of Mississippi Medical Center, Jackson, MS; 14Department of Pediatrics and Perlmutter Cancer Center, NYU Langone Health, New York, NY; 15Department of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA and 16Texas Children's Cancer Center, Houston, TX, USA
°Current address
BWR: Therapy Acceleration Program, The Leukemia & Lymphoma Society, Rye Brook, NY; ARB: Division of Neonatology, Children's Hospital of Philadelphia, Philadelphia, PA; ECW: Department of Medicine, Massachusetts General Hospital, Boston, MA; NCB: General Radiology, Wake Forest Baptist Medical Center, Greensboro, NC; KAU: Oncology Translational Research, Janssen Research and Development, Spring House, PA and GHR: Childhood Cancer Data Initiative, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
Correspondence:
C. A. FELIX - felix@chop.edu
https://doi.org/10.3324/haematol.2022.281552
Received: June 15, 2022.
Accepted: February 23, 2023
Early view: March 2, 2023.
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
Disclosures
CAF owns patented methodology (Methods and Kits for Analysis of Chromosomal Rearrangements Associated with Leukemia - US Patent # 6,368,791) that has not been licensed, which was used in the performance of this research. SPH received consulting fees
1. Hilden JM, Dinndorf PA, Meerbaum SO, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children's Oncology Group. Blood. 2006;108(2):441-451.
2. Pieters R, Schrappe M, De Lorenzo P, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370(9583):240-250.
3. Dreyer ZE, Hilden JM, Jones TL, et al. Intensified chemotherapy without SCT in infant ALL: results from COG P9407 (Cohort 3). Pediatr Blood Cancer. 2015;62(3):419-426.
4. Pieters R, De Lorenzo P, Ancliffe P, et al. Outcome of infants younger than 1 Year with acute lymphoblastic leukemia treated
from Novartis and honoraria from Amgen, Jazz, and Servier, and owns common stock in Amgen. All other authors have no conflicts of interest to disclose.
Contributions
CLW, JMH, BMC, NJW, WLC, SPH, ZED and CAF developed the concept. BWR, JAK, MD and CAF developed the methology. BWR, JAK, MD and CAF performed fromal data analysis. BWR, AJC, RCH, NAH, ARB, ECW, NCB, FGB and JP carried out the research. BWR and CAF wrote the original draft. CAF and JAK revised the draft. BWR, JAK, MD, GHR, NJW and SPH reviewed and edited the manuscript. BWR, JAK, MD, KAU and CAF created figures for data visualization. CAF, CLW, MD, GHR and SPH provided funding. CAF supervised the research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
SPH is the Jeffrey E. Perelman Distinguished Chair in the Department of Pediatrics. CAF was the Joshua Kahan Endowed Chair in Pediatric Leukemia Research. The authors would like to thank Dr. Mignon Loh for critical review and comments, and Juliet Kilcoyne for assistance with figure preparation.
This investigation was supported by SCOR grant 7372-07 to CAF, CLW, MD, GHR and SPH from the Leukemia & Lymphoma Society, USA; R01CA80175 to CAF from the National Cancer Institute (NCI), National Institutes of Health (NIH), USA; and grants to Children’s Oncology Group (COG) including U10CA98543 (COG Chair’s Grant), U10CA98413 (COG Statistics and Data Center), U24CA114766 (COG Specimen Banking), U10CA180886 (NCTN Operations Center Grant), U10CA180899 (NCTN Statistics and Data Center), and U24CA196173 (COG Biospecimen Bank Grant) from the NCI, NIH, USA, and a grant from the St. Baldrick’s Foundation, USA.
Requests for access to COG protocol research data should be sent to: datarequest@childrensoncologygroup.org.
with the Interfant-06 Protocol: results from an international phase III randomized study. J Clin Oncol. 2019;37(25):2246-2256.
5. Brown PA, Kairalla JA, Hilden JM, et al. FLT3 inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2Arearranged infant acute lymphoblastic leukemia: Children's Oncology Group trial AALL0631. Leukemia. 2021;35(5):1279-1290.
6. Robinson BW, Felix CA. Panhandle PCR approaches to cloning MLL genomic breakpoint junctions and fusion transcript sequences. Methods Mol Biol. 2009;538:85-114.
7. Andersson AK, Ma J, Wang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47(4):330-337.
8. Meyer C, Lopes BA, Caye-Eude A, et al. Human MLL/KMT2A gene exhibits a second breakpoint cluster region for recurrent MLL-USP2 fusions. Leukemia. 2019;33(9):2306-2340.
9. Nilson I, Reichel M, Ennas MG, et al. Exon/intron structure of the human AF-4 gene, a member of the AF-4/LAF-4/FMR-2 gene family coding for a nuclear protein with structural alterations in acute leukaemia. Br J Haematol. 1997;98(1):157-169.
10. Yang JJ, Park TS, Lee ST, et al. Molecular characterization and clinical course of MLL-ACTN4 rearrangement in therapy-related hematologic malignancies. Haematologica. 2014;99(4):e49-51.
11. Meyer C, Burmeister T, Groger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32(2):273-284.
12. Kang H, Wilson CS, Harvey RC, et al. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children's Oncology Group study. Blood. 2012;119(8):1872-1881.
13. Smith M, Arthur D, Camitta B, et al. Uniform approach to risk classification and treatment assignment for children with acute
lymphoblastic leukemia. J Clin Oncol. 1996;14(1):18-24.
14. Luo Z, Lin C, Guest E, et al. The super elongation complex family of RNA polymerase II elongation factors: gene target specificity and transcriptional output. Mol Cell Biol. 2012;32(13):2608-2617.
15. Balgobind BV, Raimondi SC, Harbott J, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114(12):2489-2496.
16. van der Linden MH, Boer JM, Schneider P, et al. Clinical and molecular genetic characterization of wild-type MLL infant acute lymphoblastic leukemia identifies few recurrent abnormalities. Haematologica. 2016;101(3):e95-98.
17. Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21(21):2762-2774.
18. Yokoyama A. Leukemogenesis via aberrant self-renewal by the MLL/AEP-mediated transcriptional activation system. Cancer Sci. 2021;112(10):3935-3944.
Erythropoiesis is a tightly regulated and complex process originating in the bone marrow from a multipotent stem cell and terminating in a mature, enucleated erythrocyte.1 Erythropoiesis is controlled by erythropoietin (EPO) and signaling through its receptor, EPOR. Defects in erythropoiesis can lead to erythrocytosis.2 Erythrocytosis embraces disorders characterized by an increase in the red cell mass, hemoglobin, hematocrit, or red blood cell count.3 Erythrocytosis can be distinguished as familial or acquired and can be divided into primary (due to an intrinsic defect in the erythroid cell) and secondary (extrinsic to the erythroid cell).4,5 Primary familial erythrocytosis (ECYT1) is usually an autosomal dominant condition, associated with low EPO levels due to germline EPOR variants. Secondary familial erythrocytosis (ECYT2-5) is caused by germline pathogenic variants in different genes involved in the hypoxia-sensing pathway, such as VHL (ECYT2), EGLN1 or PHD2 (ECYT3), EPAS1 or HIF-2α (ECYT4), and EPO (ECYT5). Secondary erythrocytosis can result from defects causing tissue hypoxia, such as hemoglobin variants with high oxygen affinity, due to mutations in the globin genes (HBB or HBA) or defective bisphosphoglycerate mutase leading to 2,3-bisphoshoglycerate deficiency.6 These genes are involved in key mechanisms of erythropoiesis including the HIF-EPO pathway in kidneys, the EPO-EPOR signaling pathway in the bone marrow, and the regulation of hemoglobin-oxygen affinity in red blood cells.6 Indeed, mutations in these genes are associated with hemoglobin with high oxygen affinity and with augmented EPO production.7 In normoxia, the VHL protein binds to HIF-1 α or HIF-2 α (product of the EPAS1 gene) when hydroxylated by PHD2 (product of the EGLN1 gene). This complex is then degraded by ubiquitination.8 Any variation in any one of these genes can induce either loss or gain of function and results in the specific phenotype of erythrocytosis.
EPAS1 is the causative gene of ECYT4, a dominantly inherited disorder. It encodes a transcription factor involved in the induction of oxygen-regulated genes in hypoxic conditions. Most of the EPAS1 causative variants are located in exon 12, encoding the oxygen-dependent degradation domain.9 These variants prevent the binding of PHD2 hydroxylase to the EPAS1 protein and subsequent hydroxylation and binding of the VHL protein. This leads to an increase in EPO production, resulting in a gain-offunction phenotype. Interestingly, most of the ECYT4 cau-
sative variants are missense, possibly because stop gain variants would lead to deletion of the C-terminus.9 On the other hand, common loss-of-function variants in EPAS1 have been associated with physiological adaptation to low oxygen in healthy individuals living at high altitudes.10 These polymorphic variants are mainly located in the noncoding region, probably affecting the regulation of EPAS1 expression.
We herein describe an 8-year-old male proband referred to our Medical Genetics Laboratory Unit from the Hematology Unit of Ancona (Hospital Riuniti, Ancona, Italy) in 2021. At 4 years, the patient was firstly evaluated for microcytic anemia (hemoglobin 9.6 g/dL, mean corpuscolar volume 76 fL) unresponsive to iron treatment. Firstline investigations revealed that he was negative for fecal occult blood, IgA anti-transglutaminase, and Helicobacter pylori antigens. The levels of vitamin B12 and folic acid were in the normal range. Hemoglobin electrophoresis and HBB and HBA genetic testing did not identify alterations. As all performed tests were almost normal, a primary bone marrow defect was suspected. Diepoxybutane and erythrocyte adenosine deaminase tests were performed. Negative results excluded Fanconi anemia and Diamond-Blackfan anemia. Subsequently, bone marrow biopsy showed a slight hypocellularity and abnormalities of the erythroid lineage, which was mostly immature, suggesting a congenital bone marrow deficiency. Thus, the patient was originally suspected to have a myelodysplastic syndrome.
When he was referred to our Medical Genetics Unit, he had chronic normocytic anemia with a reduced red blood cell count (Table 1). The family history revealed that the proband’s father (I.1, 45 years old) and sister (II.1, 11 years old) also suffered from anemia, suggesting a dominant condition. The mother (I.2, 48 years old) and older brother (II.2, 15 years old) were healthy (Figure 1A, Table 1). All the affected subjects had variable degrees of anemia (hemoglobin levels ranging from 10.2 g/dL to 12.5 g/dL) and low hemocrit (ranging from 31.2% to 38.4%). The reticulocyte count was almost normal in all family members, as were the leukocyte and granulocyte counts, while the red blood cell count was slightly reduced in all three affected subjects (proband 3.8x1012/L, father 4.2x1012/L, and sister 4.1x1012/L). Similarly, slightly reduced EPO levels were observed in both the proband (4.7 U/L) and father (3.8 U/L), while EPO levels close to the lower range value (7.6 U/L)
Table 1. Clinical and genetic features of the proband and his affected/unaffected family members.
*Reference ranges from AOU Ospedali Riuniti, Ancona, Italy. EPAS1 reference sequence, NM_001430.4. RBC: red blood cell count; MCV: mean corpuscular volume; F: female; M: male; yrs: years; retic.: reticulocyte.
were observed in the sister (Table 1).
The proband (II.3) was investigated through a custom gene panel for hereditary red blood cell defects.11,12 Genomic DNA preparation, genetic testing by targeted next-generation sequencing for hereditary red blood cell defects, and validation of the variant by Sanger sequencing were performed as previously described.13 Next-generation sequencing revealed the presence of a novel heterozygous deletion NM_001430.4:c.(61del) in the EPAS1 gene that is predicted to affect the protein sequence by the introduction of a premature stop codon p.(Asp21Metfs*27) (Figure 1B, C). According to the American College of Medical Genetics and Genomics guidelines, this variant was predicted as pathogenic. To confirm the pathogenicity of the variant, sequencing was also carried out on the other family members. In agreement with the dominant inheritance, both the father and the sister presented the EPAS1 variant in the heterozygous state, while the mother and the brother were negative for it (Figure 1B, C).
To characterize the effect of the EPAS1 variant, functional studies on blood samples obtained from family members were carried out. Samples were obtained after signed informed consent according to the Helsinki Declaration, allowing, by the way, anonymous use of clinical, genetic, and functional data for research purposes. The local University Ethical Committee approved the collection of the
data (DAIMedLab, ‘Federico II’ University of Naples; N. 252/18, October 2018). First, total RNA and proteins were extracted from peripheral blood leukocytes as previously described.12 We searched for the c.(61del) variant at the cDNA level by amplification of the specific exon region encompassing the mutation. We observed the presence of the deletion as heterozygous, suggesting that it does not account for nonsense-mediated mRNA decay. Accordingly, mRNA expression of EPAS1 showed no variations in patients compared to healthy controls (n=15) (Figure 1D). Additionally, we tested the effect of the variant on protein structure and synthesis. Computational predictions of the variant (phyloP100way, http://hgdownload.soe.ucsc.edu/goldenPath/hg38/phyloP100way/) revealed that the mutated residue is highly conserved among vertebrates and is comprised among a helix-loop-helix domain, necessary for heterodimerization. As shown by the protein sequence prediction and the general protein model in Figure 2A, B, the deletion introduces a premature stop producing a mutated protein of 46 amino acids. To evaluate the effect of the mutation on the expression of EPAS1 protein, we measured the amount of wildtype protein in patients and compared it to the amount in healthy controls using a multiple reaction monitoring approach.14 Briefly, 50 μg of protein extracts were obtained from three patients and three controls. For each protein, at least three double-
Figure 1. Genetic analysis and functional assessment of the identified EPAS1 loss-of-function variant. (A) Family pedigree of the proband. Squares, males; circles, females; solid symbols, affected subjects; a black arrow indicates the proband. I.1, II.2, II.2 show the subjects carrying the heterozygous variant c.(61del), p.(Asp21Metfs*27) in the EPAS1 gene. (B) Genome browser visualization of the EPAS1 reference sequence and heterozygous mutated sequence of the affected proband and relatives (Alissa Interpret software v5.2.10; Agilent Technologies). (C) Representative Sanger sequence of the wildtype EPAS1 gDNA (upper), mutated gDNA (middle), and mutated cDNA sequence (lower). The red arrow indicates the deleted nucleotide. (D) EPAS1 gene expression in peripheral blood leukocytes isolated from the proband and healthy controls (HC, N=15). No significant changes in EPAS1 gene expression were observed in patients compared to HC (P=0.127 Kruskal-Wallis test; HC vs. I.1 P>0.99; HC vs. II.2 P=0.21; HC vs. II.3 P>0.99, post-hoc correction by the Dunn multiple comparisons test). Data are presented as the median with 95% confidence interval. WT: wildtype; HC: healthy controls.

charged prototypic peptides were selected with at least three transitions (Figure 2A), in silico predicted by using Skyline. Fold changes were determined by normalization to GAPDH-derived peptides. Interestingly, no EPAS1-mutated protein was detected in either the healthy controls or affected subjects. Accordingly, we observed a strong reduction of EPAS1 wildtype protein in patients compared to healthy controls (Figure 2C).
Although gain-of-function variants in EPAS1 are classically associated with ECYT4, hematochemical and biochemical data from this family did not recapitulate the clinical picture of erythrocytosis but an opposite phenotype due to
a loss-of-function variant. Indeed, our data suggest that the deletion herein described accounts for EPAS1 haploinsufficiency. Interestingly, EPAS1/HIF-2α null mice presented a similar phenotype, with normocytic anemia in the presence of reduced hematocrit, normal reticulocyte count, and reduced erythrocyte count, accompanied by pancytopenia.15
To the best of our knowledge, this is the first report of a null variant in the EPAS1 gene that accounts for an anemic phenotype. This case underlines once again the relevance of next-generation sequencing in the diagnostic workflow of the complex landscape of hereditary erythrocyte de-
Figure 2. The EPAS1 deletion variant accounts for EPAS1 haploinsufficiency. (A) Predicted protein reference (upper) and mutated (lower) sequence by Mutalyzer (v 3.0.4, https://mutalyzer.nl/). The three EPAS1 wildtype-derived peptides for multiple reaction monitoring analysis are highlighted in light blue. (B) General protein model of EPAS1 representing EPAS1 wildtype (upper) and hypothesized mutated (lower) protein structure. (C) Boxplot showing normalized mean areas for the amount of EPAS1 wildtype protein in patients compared to that in healthy controls. The mean of the total transition area relative to each peptide of the EPAS1 protein was normalized to the mean of the total transition area of the GAPDH protein. All samples were run in technical duplicates. **P<0.01. P value by the Mann-Whitney test.

fects. Moreover, this paradigmatic clinical case highlights how the effect of genetic variants strictly depends on the variant itself, suggesting that the gene-to-disease paradigm should be replaced by the variant-to-disease one in the current era of precision medicine.
Barbara Eleni Rosato,1,2 Roberta Marra,1,2 Federica Del Giudice,2 Antonella Nostroso,1,2 Simona Gobbi,3 Barbara Bruschi,3 Paola Coccia,3 Vittoria Monaco,2,4 Maria Monti,2,4 Achille Iolascon,1,2 Immacolata Andolfo1,2 and Roberta Russo1,2
1Dipartimento di Medicina Molecolare e Biotecnologie Mediche,
Università degli Studi di Napoli Federico II, Naples; 2CEINGE
Biotecnologie Avanzate Franco Salvatore, Naples; 3Oncoematologia Pediatrica, AOU Ospedali Riuniti, Ancona and 4Dipartimento di Scienze Chimiche, Università degli Studi di Napoli Federico II, Naples, Italy.
Correspondence:
I. ANDOLFO - immacolata.andolfo@unina.it
R. RUSSO - roberta.russo@unina.it
https://doi.org/10.3324/haematol.2022.282457
Received: November 22, 2022.
Accepted: April 14, 2023.
Early view: April 27, 2023.
©2023
Published under a CC BY-NC license
Disclosures
No conflicts of interest to disclose.
Contributions
AI, IA, and RR designed and conducted the study. BER prepared the initial draft of the manuscript. RM, FDG, and AN prepared the library enrichment, and performed the Sanger sequencing analysis and gene expression analysis. SG, BB, and PC cared for the patient. VM and MM performed the multiple reaction monitoring analysis. AI critically revised the manuscript.
1. Zivot A, Lipton JM, Narla A, Blanc L. Erythropoiesis: insights into pathophysiology and treatments in 2017. Mol Med. 2018;24(1):11.
2. McMullin MF. Investigation and management of erythrocytosis. Curr Hematol Malig Rep. 2016;11(5):342-347.
3. Patnaik MM, Tefferi A. The complete evaluation of erythrocytosis: congenital and acquired. Leukemia. 2009;23(5):834-844.
4. McMullin MF. Diagnosis and management of congenital and idiopathic erythrocytosis. Ther Adv Hematol. 2012;3(6):391-398.
5. Prchal JF, Prchal JT. Molecular basis for polycythemia. Curr Opin Hematol. 1999;6(2):100-109.
6. Gaspersic J, Kristan A, Kunej T, Zupan IP, Debeljak N. Erythrocytosis: genes and pathways involved in disease development. Blood Transfus. 2021;19(6):518-532.
7. Chandrasekhar C, Pasupuleti SK, Sarma P. Novel mutations in the EPO-R, VHL and EPAS1 genes in the congenital erythrocytosis patients. Blood Cells Mol Dis. 2020;85:102479.
8. Franke K, Gassmann M, Wielockx B. Erythrocytosis: the HIF pathway in control. Blood. 2013;122(7):1122-1128.
9. Kristan A, Debeljak N, Kunej T. Genetic variability of hypoxiainducible factor alpha (HIFA) genes in familial erythrocytosis: analysis of the literature and genome databases. Eur J
The authors thank the NGS sequencing facility and Sanger sequencing facilities of CEINGE Biotecnologie Avanzate Franco Salvatore. The authors also thank Anthony Iscaro for his contribution to the EPAS1 gene expression analysis.
Funding
This research was funded by PRIN 2020 to AI (E67G21000010001Italian Minister of University and Research).
The original data and protocols are available to other investigators upon request.
Haematol. 2019;103(4):287-299.
10. Beall CM, Cavalleri GL, Deng L, et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U S A. 2010;107(25):11459-11464.
11. Russo R, Andolfo I, Manna F, et al. Multi-gene panel testing improves diagnosis and management of patients with hereditary anemias. Am J Hematol. 2018;93(5):672-682.
12. Russo R, Marra R, Rosato BE, Iolascon A, Andolfo I. Genetics and genomics approaches for diagnosis and research into hereditary anemias. Front Physiol. 2020;11:613559.
13. Rosato BE, Alper SL, Tomaiuolo G, Russo R, Iolascon A, Andolfo I. Hereditary anemia caused by multilocus inheritance of PIEZO1, SLC4A1 and ABCB6 mutations: a diagnostic and therapeutic challenge. Haematologica. 2022;107(9):2280-2284.
14. Andolfo I, Monaco V, Cozzolino F, et al. Proteome alterations in erythrocytes with PIEZO1 gain-of-function mutations. Blood Adv. 2023;7(12):2681-2693.
15. Scortegagna M, Morris MA, Oktay Y, Bennett M, Garcia JA. The HIF family member EPAS1/HIF-2alpha is required for normal hematopoiesis in mice. Blood. 2003;102(5):1634-1640.
Charnise Goodings,1* Xujie Zhao,1* Shannon McKinney-Freeman,2 Hui Zhang3 and Jun J. Yang1,4
1Department of Pharmaceutical Sciences, St. Jude Children's Research Hospital, Memphis, TN, USA; 2Department of Hematology, St. Jude Children's Research Hospital, Memphis, TN, USA; 3Department of Hematology/Oncology, Shanghai Children’s Medical Center, Shanghai, China and 4Department of Oncology, St. Jude Children’s Research Hospital, Memphis, TN, USA.
*CG and XZ contributed equally as first authors.
After the publication of our article “ARID5B influences B-cell development and function in mouse” in the February 2023 issue of Haematologica, 1 all authors are signatories to the request to add the name of Laura Janke to the list of authors of this work for her substantial contribution in performing experiments, and interpreting the data and research findings. The updated list of authors together with their affiliations for the work in question1 is, therefore, as follows:
Charnise Goodings,1* Xujie Zhao,1* Laura Janke,2 Shannon McKinney-Freeman,3 Hui Zhang4 and Jun J. Yang1,5
1Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, Memphis, TN, USA; 2Department of Pathology, St. Jude Children’s Research Hospital, Memphis, TN, USA; 3Department of Hematology, St. Jude Children’s Research Hospital, Memphis, TN, USA; 4Department of Hematology/Oncology, Shanghai Children’s Medical Center, Shanghai, China and 5Department of Oncology, St. Jude Children's Research Hospital, Memphis, TN, USA
*CG and XZ contributed equally.
1. Goodings C, Zhao X, McKinney-Freeman S, Zhang H, Yang JJ. ARID5B influences B-cell development and function in mouse. Haematologica. 2023;108(2):502-512.
Correspondence: J.J. Yang
jun.yang@stjude.org
Received: June 5, 2023.
Accepted: June 5, 2023.
htps://doi.org/10.3324/haematol.2023.283687
©2023 Ferrata Storti Foundation
Published under a CC BY-NC license
