NEUROLOGÍA
Christiam Ochoa
Í NDICE 01 Aspectos generales Coma pág. 197 Migraña Cefalea Tensional Cefaleas 02 pág. 199 03 pág. 203 Aspectos generales Diagnóstico Tratamiento Enfermedad cerebro vascular (ECV) 04 pág. 209 Meningitis 07 Trastornos Hipercinéticos Trastornos Hipocinéticos Disquinesias (trastornos del movimiento) pág. 217 Aspectos generales Diagnóstico Tratamiento 08 pág. 219 Enfermedad de Parkinson Diagnóstico Tratamiento Cefalea de Horton Neuralgia del trigémino Aspectos generales Diagnóstico Tratamiento 09 pág. 221 Enfermedad Alzheimer Aspectos generales Diagnóstico Tratamiento 10 pág. 223 Epilepsia Aspectos generales 11 pág. 227 Crisis epiléptica y estatus epiléptico Intoxicaciones Miastenia gravis Síndrome de Eaton-Lambert Botulismo 05 pág. 213 Enfermedad de placa motora Aspectos generales Diagnóstico Tratamiento 12 pág. 231 Crisis febriles Circulación cerebral Ictus ECV Isquémico ECV Hemorrágico Hemorragia Subaracnoidea (HSA) Aspectos generales Diagnóstico 06 pág. 215 Síndrome de guillian barré (SGB) Diagnóstico Tratamiento


NEUROLOGÍA Neurología es considerado un capítulo mayor, representa en promedio el 3% (3 de 100 preguntas) del Examen de ESSALUD. 11 Enfermedad cerebrovascular (ECV) 02 Cefalea 06 Neuropatías 04 Disquinesias 10 Meningitis 05 Coma 01 Pares craneales 01 Convulsión febríl 01 Esclerosis múltiple 06 Epilepsia 01 Placa motora 01 Tumores ESSALUD 2001-2022 Número de preguntas por temas en el capitulo de Neurologia 3%
Aspectos generales
Se define como el estado en el que la persona no responde ni despierta ante los estímulos. Se recomienda no usar términos ambiguos como somnolencia o estupor. Es una emergencia neurológica. Se debe a la lesión de sistema reticular activador ascendente o a los dos hemisferios cerebrales juntos. La causa más frecuente de coma es el traumatismo encefalocraneano y en segundo lugar el coma metabólico (ENARM 2020-B)
Diagnóstico
Recuerda
La causa más frecuente de coma es el traumatismo encefalocraneano y en segundo lugar el coma metabólico.
El reflejo fotomotor que sirve para diferenciar lesión del par craneal II del III.
El objetivo es encontrar el “nivel” de lesión que tiene un paciente en coma para abordar la posible etiología. Para ello se requiere revisar los reflejos del tronco encefálico.
Pupilas: reflejo fotomotor que sirve para diferenciar lesión del par craneal II del III. Motor Ocular: roving., reflejo óculocefálico, reflejos óculovestibulares, bobbing y desviación conjugada de la mirada.
Reflejo Corneal: evalúa la vía aferente del par V1 y la eferente del par VII.
Respuesta al Dolor: decorticado o flexión patológica si la lesión esta por encima del núcleo rojo y descerebrado o extensión patológica si la lesión es por debajo. Si hay hipotonía se sospecha lesión bulbar. Si la respuesta es asimétrica se sospecha focalización.
Ritmo Respiratorio: no son signos específicos y pueden llevarnos a confusión. Sin embargo, el patrón más característico que representa una lesión diencefálica o cortical bilateral es la respiración de Cheyne Stokes. Otros patrones son la hiperventilación neurógena central, apnéustica y atáxica. La bradipnea se asocia a sedación. Una vez identificado el “nivel” se debe evaluar la severidad con la escala de Glasgow. Menos de 8 puntos, indica severidad. El descenso de 3 o más puntos, en exámenes repetidos, independientemente de la cifra inicial, orienta hacia una lesión grave.
Recuerda
El reflejo corneal evalúa la vía aferente del par V1 y la eferente del par VII.
En la respuesta al dolor, la posición en decorticado o flexión patológica indica lesión por encima del núcleo rojo y descerebrado o extensión patológica, la lesión es por debajo.
1. APERTURA DE LOS OJOS Espontánea – 4p
A la voz – 3p Al dolor – 2p Nula – 1p
www.qxmedic.com PAG. 197
01. COMA
Recuerda
Una puntuación de menos de 8 puntos, indica severidad en la escala de Glasgow.
2. RESPUESTA A ESTÍMULOS
Obedece – 6p
Localiza – 5p Retira – 4p Respuesta flexora – 3p Respuesta extensora – 2p Nula – 1p
3. RESPUESTA VERBAL Orientado – 5p Confuso – 4p Inadecuado – 3p
Incomprensible – 2p Nula – 1p
Recuerda
En toda emergencia, primero se aplica el ABCD. Si la escala de Glasgow es igual o menor a 8 se intuba.
Tratamiento Intoxicaciones
Como toda emergencia, primero se aplica el ABCD. Si la escala de Glasgow es igual o menor a 8 se intuba. Si hubo TEC se coloca collarín. Si desatura se aplica O2 suplementario.
Todo paciente en coma recibe un bolo de glucosa 50% EV 50mL y se añade tiamina 100 mg EV si se sospecha que el paciente es alcohólico. Las convulsiones en el coma se deben tratar con fenitoína o equivalente. Se inicia tratamiento empírico con antibióticos si se sospecha infección del SNC o manitol si se sospecha HTEC.
En los pacientes con coma se debe sospechar intoxicación por sustancias e iniciar su tratamiento lo antes posible. Las sustancias más frecuentes causantes de coma son:
Recuerda
En la intoxicación por organofosforados – carbamatos, el paciente presenta bradicardia, hipotensión arterial, bradipnea, miosis, fasciculación, aumento de secreciones.
Organofosforados – Carbamatos: El paciente presenta bradicardia, hipotensión arterial, bradipnea, miosis, fasciculación, aumento de secreciones. (ENARM 2021 – A) Se trata con atropina (ENARM 2019-B, ENARM 2021 – B) y en casos severos se aplica el antídoto del organofosforado que es la pralidoxima. Los carbamatos no tienen antídoto.
Opiodes: Se presenta por consumo de opioides como morfina, oxicodona, fentanilo, heroína, etc. Ocurre en pacientes con factores de riesgo de drogadicción o personal de salud con acceso a opioides. Es frecuente encontrar lesiones de venopunción en los brazos. La clínica característica es agitación, disforia, miosis puntiforme, bradipnea (apnea en casos extremos), bradicardia y disminución de los ruidos intestinales.

Antimuscarínicos (belladona, atropina): Presenta taquicardia, hipertensión arterial, midriasis, sequedad de piel y mucosas, rubor y calor. Se trata con inhibidores de la acetilcolinesterasa (ENARM 2022) (ESSALUD 2018)
Cocaína: Presenta midriasis, diaforesis, pero con boca seca. Taquicardia, hipertensión arterial y agitación psicomotriz. El manejo es de soporte y se deben evitar los calcio antagonistas o beta bloqueadores (ENARM 2018-A) (ESSALUD 2018)
Sedantes: Se presenta tras intoxicarse con benzodiacepinas, alcohol o barbitúricos. La clínica consiste en sedación, bradipnea (apnea en casos severos), hiporreflexia y en algunos casos miosis. No hay otros hallazgos y suele ser por descarte. El manejo es con Flumazenilo como antídoto en caso de sospecha de benzodiacepinas. Para el resto se recomienda observación y vigilancia de la ventilación. En casos severos de intoxicación alcohólica se puede requerir diálisis.
www.qxmedic.com NEUROLOGÍA PAG. 198
02. CEFALEAS
Migraña
Aspectos generales
Es la cefalea primaria más frecuente motivo de consulta al neurólogo. Afecta a mujeres jóvenes con antecedentes familiares de migraña. Tiene dos tipos:
Común: La migraña más frecuente que no presenta aura (ENARM 2021 – A)
Clásica: Afecta a un 15% de migrañosas y presenta el fenómeno de aura.
Recuerda
El tipo de migraña más frecuente que no presenta aura es la llamada ¨Común¨.
Recuerda
- La cefalea cumple al menos dos entre:
• Unilateral
• Pulsátil
• Intensidad moderada a grave
• Aumenta con la actividad física
Diagnóstico
Para establecer el diagnóstico es crucial la cronopatología de la historia clínica y la ausencia de signos que orienten a otras cefaleas secundarias. Los exámenes auxiliares son normales y no suelen ser de ayuda para el diagnóstico.
CRITERIOS DIAGNÓSTICO DE MIGRAÑA SEGÚN LA IHS (Internacional Headache Society) (ENARM 2018-A)
Historia, exploración clínica y exámenes complementarios negativos (de otro proceso cerebral).
Ataques múltiples (al menos 5) de cefalea.
Duración de horas o días (4-72h).
La cefalea cumple al menos dos de los siguientes:
♦ Unilateral
♦ Pulsátil
♦ Intensidad moderada a grave
♦ Aumenta con la actividad física
Al menos una de las siguientes características:
♦ Náuseas, vómitos o ambos
♦ Fotofobia o sonofobia
No existe otra enfermedad que explique mejor la cefalea.
www.qxmedic.com PAG. 199
Figura 1. Fisiopatología de la migraña
Otras formas de migraña
Existen otras formas de migraña como:
Migraña basilar: dolor de predominio occipital acompañado de síntomas vertebro-basilares como: borrosidad visual, disartria, vértigo, diplopía, parestesias o debilidad bilateral o alteraciones del nivel de conciencia.

Aura migrañosa sin cefalea o equivalente migrañoso.
Migraña hemipléjico familiar: El aura es una hemiparesia de predominio en la cara y extremidad superior.
Migraña complicada, infarto migrañoso o migraña-stroke: Los síntomas del aura persisten más allá del fin de la cefalea y hay lesión isquémica en la resonancia.
Estado de mal migrañoso (status migrañoso): Episodios de migraña que duran más de 72 horas a pesar del tratamiento.
Migraña transformada o crónica criterios diagnósticos: Cefalea tensional y/o migraña al menos 15 días al mes durante al menos 3 meses. Dolor de cabeza que cumpla los criterios de migraña sin aura, más de 8 días/mes durante al menos 3 meses.
Tratamiento
Recuerda
El manejo inicia en la migraña es con AINES, si no mejoran se añade TRIPTANOS como el Sumatriptán.
El manejo de la migraña es escalonado, se recomienda siempre encontrar el desencadenante del ataque migrañoso para evitarlo en el futuro. El manejo inicia administrando AINES, si no mejoran se añade triptanos como el Sumatriptán. Casos extremos como el “status migrañoso” requiere la aplicación de drogas endovenosas y añadir corticoides.
Pacientes que tienen 4 o más episodios de migraña al mes requieren tratamiento profiláctico con betabloqueantes (ENARM 2022) (ESSALUD 2018), calcio antagonistas, antidepresivos tricíclicos o anticonvulsivantes.
Cefalea tensional
Aspectos generales
Es la cefalea más frecuente pero no suelen acudir al neurólogo. El 50% se asocia a estrés, ansiedad y depresión. También, se ha relacionado a traumatismos cervicales.
Diagnóstico
Sensación de "pesadez" en la cabeza, opresión en banda o en casco. Bilateral que no impide las tareas del día. Empeora con el paso del día y con el estrés.
www.qxmedic.com NEUROLOGÍA PAG. 200
Figura 2. Patrón de la cefalea tensional en Vincha
Recuerda
El dolor presenta al menos 2 de las siguientes características en la cefalea tensional:
- Bilateral
- Calidad opresiva
- Intensidad levemoderada
- No se agrava con la actividad física
Manejo inicial con AINES como el paracetamol.
CRITERIOS DIAGNÓSTICOS DE CEFALEA TENSIONAL (ENARM 2020-A)
Mínimo de 10 crisis que cumplan los criterios 2-4. Cefalea que dura entre 30 min y 7 días.
El dolor presenta al menos 2 de las siguientes características:
♦ Bilateral
♦ Calidad opresiva
♦ Intensidad leve-moderada
♦ No se agrava con la actividad física Cumple las siguientes características
♦ Sin nauseas /vómitos
♦ Puede asociar sono/fotofobia, pero no ambas. Cefalea no atribuible a otras causas
Tratamiento
Manejo inicial con AINES como el paracetamol. (ENARM 2017 – B). Si no mejora se considera uso de antidepresivos en caso de asociarse a clínica psiquiátrica. Si hay componente traumático, los miorrelajantes pueden ayudar junto a la fisioterapia.
Cefalea de Horton – en racimos o “Cluster”
Aspectos generales
Recuerda
- La cefalea se acompaña de al menos uno de los siguientes signos:
• Inyección conjuntival ipsilateral, lagrimeo o ambos.
• Congestión nasal ipsilateral, rinorrea o ambos.
• Edema palpebral ipsilateral.
• Sudación en la frente y cara ipsilateral.
• Miosis o ptosis ipsilateral o ambos.
• Inquietud motora y desasosiego.
• Frecuencia de las crisis entre 1 cada 2 días y 6 al día.
Es una cefalea trigémino-autonómica. Frecuente en varones de edad media. Afecta por temporadas y se asocia a ansiedad, depresión y suicidio en casos severos mal diagnosticados.
Diagnóstico
CRITERIOS DIAGNÓSTICOS (ENARM 2016 -A)
Se necesitan al menos 5 crisis que cumplan los criterios. Dolor intenso o muy intenso estrictamente unilateral, orbitario, supraorbitario, temporal o ambos, que dura entre 15 y 180 minutos sin tratamiento.
La cefalea se acompaña de al menos uno de los siguientes signos:
♦ Inyección conjuntival ipsilateral, lagrimeo o ambos.
♦ Congestión nasal ipsilateral, rinorrea o ambos.
♦ Edema palpebral ipsilateral.
♦ Sudación en la frente y cara ipsilateral.
♦ Miosis o ptosis ipsilateral o ambos.
♦ Inquietud motora y desasosiego.
♦ Frecuencia de las crisis entre 1 cada 2 días y 6 al día.
La dolencia no se puede atribuir a otra enfermedad.
Tratamiento
Las crisis se manejan con oxígeno alto flujo de hasta 7-10 litros/min. También se usa Sumatriptán SC o intranasal. Requiere siempre profilaxis, se inicia con prednisona y verapamilo. Luego se usa litio.
www.qxmedic.com NEUROLOGÍA PAG. 201
Neuralgia del trigémino

Aspectos generales
Frecuente en mujeres mayores de 50 años, si se presenta en menores sospecha esclerosis múltiple. Es una cefalea trigémino-autonómica idiopática.
Diagnóstico
Dolores lancinantes, intensos, de segundos de duración, en territorios de alguna de las ramas del trigémino (V2 y V3, sobre todo). Tras un episodio doloroso hay un periodo refractario. Se repite decenas de veces al día.
El 95% es unilateral. Es típica una zona "gatillo" que desencadena el dolor. El examen neurológico es normal, no hay alteraciones sensitivas. El dolor respeta el sueño.
Tratamiento
Las carbamazepinas son de elección. Como segunda línea se usa baclofeno, lamotrigina, fenitoína, o gabapentina. En casos refractarios, técnicas quirúrgicas ablativas del ganglio de Gasser.
www.qxmedic.com NEUROLOGÍA PAG. 202
Figura 3. Neuralgia del trigémino
03. ENFERMEDAD CEREBRO VASCULAR
Topografía y territorios vasculares
Circulación anterior
Recuerda
- La obstrucción de la arteria cerebral anterior puede ocasionar déficit motor o sensitivo a predominio de miembros inferiores.
- La obstrucción de la arteria cerebral media en el hemisferio dominante puede ocasionar afasia motora (frontal), afasia sensitiva (temporal) o afasia global (ambos), déficit motor o sensitivo a predominio de la cara y miembros superiores.
Recuerda
La obstrucción de la arteria cerebral posterior ocasiona hemianopsia homónima contralateral con preservación de la visión central.
Además del síndrome de Déjerine-Roussy (pérdida sensorial, coreoatetosis y dolor talámico).
Arteria cerebral anterior:
Irriga la parte medial de los hemisferios cerebrales (frontal y parietal) y la parte anterior de los ganglios basales.
Su obstrucción puede ocasionar déficit motor o sensitivo a predominio de miembros inferiores. Aparición de reflejos primitivos, abulia, rigidez paratónica y apraxia de la marcha.
Arteria cerebral media (ENARM 2021 – B)
Irriga los ganglios basales y la mayor parte de la cápsula interna, el lóbulo de la ínsula y la parte lateral de los hemisferios cerebrales (frontal, temporal y parietal).
Su obstrucción en el hemisferio dominante puede ocasionar afasia motora (frontal), afasia sensitiva (temporal) o afasia global (ambos), déficit motor o sensitivo a predominio de la cara y miembros superiores, hemiplejía completa contralateral, hemianopsia homónima contralateral sin preservación de la visión central.
Su obstrucción en el hemisferio no dominante puede ocasionar negligencia, anosognosia y no se produce afasia.
Circulación posterior
Arteria cerebral posterior (ENARM 2020-B) (ESSALUD 2014)
Irriga el mesencéfalo, el diencéfalo, la parte profunda de los ganglios basales, la parte basal del lóbulo temporal y casi la totalidad del lóbulo occipital.
Su obstrucción ocasiona hemianopsia homónima contralateral con preservación de la visión central, alexia sin agrafia (si afecta al hemisferio dominante), alucinaciones visuales, perseveraciones. Ocasiona el síndrome de Déjerine-Roussy (pérdida sensorial, coreoatetosis y dolor talámico), síndrome alterno de Weber (parálisis III, hemiplejía contralateral) y en ocasiones paresia mirada vertical.
Arteria cerebelosa posterior inferior – PICA:
Irriga el cerebelo posterior y la parte laterodorsal del bulbo raquídeo (componente sensitivo principalmente).
Su obstrucción ocasiona el síndrome alterno de Wallemberg (parálisis del V3, IX, X, síndrome de Horner y ataxia ipsilaterales con hemianestesia termoalgésica contralateral). Si se lesiona solo la parte cerebelosa ocurre dismetría, temblor de intención, disartria y alteraciones de la coordinación fina.
Arteria basilar:
Irriga el puente y la parte anterior del cerebelo. Su lesión se presenta con trastorno del sensorio, déficit de pares craneales y motores. (ENARM 2021 – B)
Es mortal la mayoría de las veces.
www.qxmedic.com PAG. 203
Recuerda
Ante una focalización o ictus se debe sospecha siempre compromiso vascular e iniciar el estudio de este con una tomografía sin contraste.
ICTUS ECV Isquémico

Se define como la focalización no convulsiva debido a déficit de aporte vascular de un grupo de neuronas que compromete la función asignada a ellas. Las convulsiones no se consideran focalización.
Ante una focalización o ictus se debe sospecha siempre compromiso vascular e iniciar el estudio de este con una tomografía sin contraste. Las causas vasculares del ictus se clasifican en isquémicas (85%) o hemorrágicas (15%) según los hallazgos tomográficos.
Aspectos generales
El 70% se produce en mayores de 70 años. La mejor medida preventiva es tratar la Hipertensión Arterial (HTA). Globalmente fallecen 15%. Los infartos de territorio posterior son de peor pronóstico.
Factores de riesgo
Los factores de riesgo principales son: Edad mayor de 55 años y sexo masculino Hipertensión arterial (principal factor de riesgo)
Diabetes
Estenosis carotidea asintomática, cardiopatías, arritmias (fibrilación auricular), estenosis mitral, válvulas protésicas, dilatación ventricular Tabaco, dislipidemias, alcoholismo, drogas (Ej. cocaína), TIA previos, anticonceptivos orales.

www.qxmedic.com NEUROLOGÍA PAG. 204
Figura 4. Patrón de irrigación de la arteria cerebral anterior, media y posterior
Arteria cerebral anterior Arteria cerebral media
Arteria cerebral posterior
Tipos de ictus isquémicos
Recuerda
El ataque isquémico transitorio (AIT o TIA) consiste en déficit neurológicos de duración corta, cuya recuperación ocurre en la mayoría de los casos antes de la hora de instaurados y en el 100% de los casos antes de las 24 horas.
Recuerda
- La causa principal del infarto cardioembólico son las arritmias.
- La causa principal del infarto lacunar es la HTA mal controlada y la edad avanzada. Se diagnostican mejor con RMN.
Recuerda
- El tiempo de ventana desde el ictus hasta el inicio de la trombólisis es de 3 HORAS (Recomendación A) o de 3-4.5 HORAS (Recomendación B).
- Para iniciar la trombólisis la presión arterial debe estar por debajo de PAS 185mmHg y PAD 110mmHg.
Global: Cuando hay interrupción generalizada del riego sanguíneo. Como casos de shock o hipoxia. Se afecta las zonas “frontera” donde terminan los territorios vasculares.
Focal: Cuando se obstruye una arteria con irrigación terminal. Se compromete una zona específica del parénquima cerebral que explica topográficamente la sintomatología. Los ictus focales pueden ser:
♦ Ataque isquémico transitorio (AIT o TIA): Consiste en déficit neurológicos de duración corta, cuya recuperación ocurre en la mayoría de los casos antes de la hora de instaurados y en el 100% de los casos antes de las 24 horas. (ENARM 2017 – B). No deben tener lesión de isquemia en la imagen (RMN o TAC). El 40% de AIT tendrán un ictus establecido o infarto a los 3 meses; por lo tanto, es importante realizar prevención primaria de infarto ante cualquier AIT. La etiología principal son émbolos arterio-arteriales provenientes de la carótida y en segundo lugar cardio émbolos en pacientes con cardiopatías o arritmias. La sintomatología más frecuente es la amaurosis fugax por obstrucción de la arteria oftálmica (rama de la carótida interna).
♦ Infarto: Se define infarto cerebral como la muerte neuronal por falta de aporte sanguíneo. El diagnóstico final se hace mediante imágenes RMN o TAC. Se prefiere la RMN para fases hiperagudas de infarto, la TAC es específica pasada las 48 horas del ictus. Según su frecuencia, las causas se clasifican en las siguientes:
Aterotrombótico: obstrucción debido a una placa de ateroma inestable que forma un trombo en su superficie. Suele afectar a vasos pequeños y presentan focalizaciones menores. La clínica suele ser fluctuante y puede durar días en establecer el nivel de obstrucción final.
Cardioembólico: obstrucción debido a un émbolo que viaja del corazón o de una arteria grande. El tamaño del vaso obstruido es mayor y por ende la clínica más significativa. La evolución de la focalización es tórpida y alcanza el 100% de clínica en pocas horas. Igualmente, de eliminarse el trombo, la mejora es muy rápida también. La causa principal son las arritmias cardiacas (ENARM 2022) – Lacunar: infartos pequeños de máximo 1,5 cm, ocurren en la profundidad de los hemisferios cerebrales o del tronco cerebral afectando territorios estratégicos que dan clínica desproporcional al daño neurológico. Representan el 20% de los ictus y su causa principal es la HTA mal controlada y la edad avanzada. Se diagnostican mejor con RMN.
Inhabitual: causas raras como coagulopatías, malformaciones, infecciones, enfermedades autoinmunes, paraneoplásicos, etc.
– Indeterminado: no se llega a determinar la causa del infarto.
Tratamiento
Pre Hospitalario:
♦ Promoción de la salud en la comunidad y en pacientes con factores de riesgo para infarto. El acrónimo “FAST” – Face, Arm, Speech, Time – ayuda a reconocer los síntomas iniciales más frecuentes.
♦ Activar la alarma rápido a los teléfonos de emergencia para poder reducir el tiempo de llegada al hospital. Los paramédicos deben estar capacitados, deben avisar el hospital más cercano y se debe tener redes de centros de salud con capacidad resolutiva que reduzcan el tiempo de llegada al hospital.
Hospital:
♦ Debe tener algún certificado de calidad como la JCI. Los pacientes tienen mejor pronóstico si son tratados por un equipo especializado que se conocen y trabajan siempre en el mismo equipo de especialistas. El hospital debe contar con protocolos de STROKE.
♦ La telemedicina ha sido aprobada por la FDA para asistir casos de infarto en centros que carecen de equipo especializado.
♦ Siempre se debe usar la escala NIHSS para evaluar la severidad de cuadro.
♦ El tiempo desde que ingresa al hospital y recibe la primera dosis de t-PA (DTN – door to needle) no debe exceder los 60 minutos. Se debe capacitar a todo el personal de la emergencia para llegar a esta meta. La tomografía o RMN debería estar lista en menos de 20 minutos. El único examen que se toma antes de trombolizar es glucosa.
www.qxmedic.com NEUROLOGÍA PAG. 205
–
–
–
Recuerda
El tiempo desde que ingresa al hospital y recibe la primera dosis de t-PA (DTN – door to needle) no debe exceder los 60 minutos.
El acrónimo “FAST” – Face, Arm, Speech, Time – ayuda a reconocer los síntomas iniciales más frecuentes.
♦ Tratamiento específico del infarto cerebral:
Ventilación mecánica si la saturación de oxígeno es menor de 94%. Se debe tratar la hipotensión arterial si la hubiera. La temperatura debe mantenerse por debajo de T°38 y la glucosa entre 60mg/dl y 140-180mg/dl como máximo.
– Para iniciar la trombólisis la presión arterial debe estar por debajo de PAS 185mmHg y PAD 110mmHg (ENARM 2018-B). Durante la trombólisis se debe mantener en 180/105 mmHg y se controla de preferencia con labetalol.
– Trombólisis con T-PAr (activador del plasminógeno tisular recombinante – Alteplase): Es el manejo de elección si se cumplen los criterios de inclusión y exclusión (ver tabla). La dosis es de 0.9mg/kg y se aplica en 60 minutos (máxima dosis de 90mg) el 10% se da en bolo en 1 min. El tiempo de ventana desde el ictus hasta el inicio de la trombólisis es de 3 horas (Recomendación A) o de 3 - 4.5 horas (Recomendación B). Efectos adversos principales: hemorragia, angioedema, no administrar junto a Abciximab (ENARM 2022)
Si el paciente requiere anticoagulación ésta debe interrumpirse durante la trombólisis y reiniciarse en casos leves a los 3 días, en casos moderados a los 6 días y en casos severos a los 12 días de aplicado la trombólisis (ENARM 2018-B). La severidad del infarto se mide con la escala NIHSS.
Como prevención secundaria del infarto se recomienda aspirina (ENARM 2022), hipolipemiantes, manejo de los factores de riesgo, control glicémico, cambios del estilo de vida.
Criterios de Inclusión
– Síntomas neurológicos por periodo inferior a 270 min de evolución, con hora de inicio definida
Déficit neurológico entre 5 y 23 puntos en la escala de NIHSS y por más de 30 min
– Edad mayor de 18 años
Tomografía axial de cerebro sin evidencias de hemorragia intracraneal
– Ausencia de los criterios de exclusión
Criterios de Exclusión
Hora inicio de síntomas desconocida, o mayor a 270 min al inicio de la infusión. Si inician durante el sueño se considera la hora en que paciente fue visto asintomático por última vez
– NIHSS > 23 puntos: déficit neurológico severo o <5 puntos: déficit leve
ACV extenso en los últimos 3 meses
– Traumatismo craneal encefálico o cirugía del sistema nervioso central en los últimos 3 meses
IAMc dentro de últimos 21 días, a excepción del que ocurre de la manera concomitante con el ACV
– Cirugía mayor o biopsia de órgano no compresible en los últimos 14 días
Antecedentes de hemorragia intracraneana
– Síntomas sugerente de hemorragia subaracnoidea
Malformación arteriovenosa o aneurisma cerebral
– Antecedente de hemorragia gastrointestinal o urinaria en los últimos 21 dias
Antecedentes de coagulopatía (ej: hemofilia)
– Punción arterial en sitio no compresible o punción lumbar en los últimos 7 dias
PAS > 185 mmHg y PAD > 110 mmHg refractaria a la administración de labetalol y/o nitroprusiato
– Signos neurológicos que revierten rápidamente
INR > 1,5; TTP > 15 segundos por sobre el límite superior. Uso de heparina de bajo peso molecular en las últimas 24 h. Recuento plaquetario < 100.000/mm
– Glicemia < 50 o > 400 mg/dl
TAC de encéfalo sin contraste con ASPECT<7
– Otros: antecedentes de varices esofágicos, colitis ulcerosa, diverticulitis o pancreatitis aguda, retinopatia diabética con riesgo de hemorragia, neoplasia con riesgo de hemorragia, trauma agudo (fracturas), enfermedad sistémica o infecciosa grave, embarazo o parto dentro del último mes, evidencia de sangrado, sospecha de embolia séptica o de endocarditis infecciosa.
www.qxmedic.com NEUROLOGÍA PAG. 206
–
–
–
–
–
–
–
–
–
–
–
–
–
–
Tabla 1. Criterios de inclusión y exclusión
ECV hemorrágico
Aspectos generales
Recuerda
La principal causa de ECV hemorrágico es el traumatismo encefalocraneano, considerado como causa secundaria. De las causas primarias, la más frecuente es la hipertensión arterial mal controlada que causa hemorragia intracerebral en ancianos.
Representa el 15% de los ECV y se deben a ruptura de un vaso intracraneal. La principal causa de ECV hemorrágico es el traumatismo encefalocraneano, considerado como causa secundaria. En neurología estudiamos las causas primarias, siendo la más frecuente la hipertensión arterial mal controlada que causa hemorragia intracerebral en ancianos (ESSALUD 2013). Otras causas más raras son la rotura de aneurismas en gente joven que genera hemorragia subaracnoidea, hemorragia intraventricular en recien nacidos extremadamente prematuros y angiopatía amiloide en ancianos de edad muy avanzada. La hemorragia intracerebral en adultos se debe principalmente a hipertensión arterial mal controlada que rompe las arterias profundas del segmento M1 de la cerebral media. Se conoce como aneurisma de Charcot Bouchard al que se forma producto de la hipertensión en estos pacientes y es mucho más frecuente en territorios profundos cerca del putámen. El segundo lugar de sangrado es el lóbulo frontal o parietal y se debe a malformaciones arteriales, principalmente. Finalmente, las hemorragias de territorios posteriores son mucho más catastróficas y letales, pero solo representan menos de la quinta parte de sangrados primarios.
Diagnóstico
Al igual que el ECV isquémico, la prueba inicial siempre debe ser la neuroimagen sin contraste. La tomografía sin contraste es el gold estándar diagnóstico y es mucho más sensible y específica que la resonancia para detectar sangrado.
Hemorragia subracnoidea (HSA)
Aspectos generales
Recuerda
La rotura de los aneurismas intracraneales es más frecuente en el territorio de la arteria cerebral anterior (comunicante).
La causa no traumática más frecuente es la rotura de aneurismas intracraneales cerebrales. Los aneurísmas más frecuentes son del territorio de la arteria cerebral comunicante anterior (ENARM 2022) seguido de la carótida interna, la comunicante posterior y la arteria cerebral media. Otra causa es rotura de una malformación arteriovenosa o angiomas cavernosos. Los factores de riesgo para tener aneurismas cerebrales son la enfermedad renal poliquística, la displasia fibromuscular y la coartación de aorta.
Diagnóstico
La clínica se debe a hipertensión endocraneada súbita que se manifiesta por cefalea súbita intensa (la peor de la vida del paciente), seguido de trastorno de conciencia. Hay vómitos, oftalmoparesia del VI par, signos de irritación meníngea a las horas del evento. Las convulsiones y focalización son raras.
Para establecer el diagnóstico se hace una tomografía sin contraste y si sale negativa para sangrado, pero la clínica es muy sugestiva, se realiza punción lumbar diagnóstica.
Las escalas para clasificar la severidad de la HSA tanto clínica como radiológicamente son las escalas de Hunt - Hess y la escala de Fisher respectivamente.
www.qxmedic.com NEUROLOGÍA PAG. 207
Clasificación de Hunt y Hess
Grado Descripción
I Asintomático o mínima cefalea, ligera rigidez de nuca
II Cefalea moderada o aguda, sin defecto
III Somnolencia, confusión o defecto neurológico foca leve
IV Estupor, hemiparesia moderada o grave, posible rigidez de descerebración o trastornos vegetativos
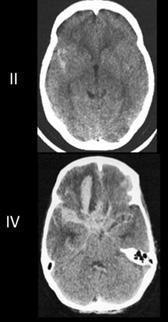
V Coma, rigidez de descerebración, aspecto moribundo
Escala de Fisher
Grado Descripción
I Sin evidencia de sangrado en cisternas ni ventrículos
II Sangre difusa fina, con una capa <1 mm en cisternas medida verticalmente
III Coágulo grueso cisternal, >1 mm en cisternas medido verticalmente
IV Hematoma intraparenquimatoso, hemorragia intraventricular, +/- sangrado difuso.
V Coma, rigidez de descerebración, aspecto moribundo
Recuerda
La clínica se debe a hipertensión endocraneada súbita que se manifiesta por cefalea súbita intensa (la peor de la vida del paciente), seguido de trastorno de conciencia.
Tratamiento
Los pacientes deben ingresar a UCI. Guardar reposo absoluto, brindar analgesia potente. El resangrado es la principal causa de muerte, representa el 40% de los casos y suele ser en la primera semana del evento. La única medida que lo previene es la cirugía precoz en las primeras 24 a 48 horas.
Otras causas de muerte son las complicaciones como la acidosis metabólica, hiperglicemia, arritmias e hiponatremia (SIADH). Se recomienda mantener la presión arterial media menor de 160 para evitar resangrado y se aplica nimodipino 60mg/cada 4 horas como neuroprotector para evitar el vasoespasmo.
www.qxmedic.com NEUROLOGÍA PAG. 208
Tabla 2. Clasificación de Hunt y Hess
Tabla 3. Escala de Fisher
Figura 6. TAC: Hemorragia subaracnoidea (HSA) (Escala de Fisher)
Aspectos generales
La meningitis es la inflamación infecciosa de las meninges que puede complicarse y causar encefalitis por contigüidad. Las causas más frecuentes en pediatría son las meningitis virales por Echovirus, Coxsackie y otros enterovirus. En el adulto predominan las meningitis bacterianas siendo la bacteria más frecuente el neumococo. Algunos parásitos pueden causar meningitis como las amebas de vida libre (ENARM 2019 B) y en pacientes con VIH, estadio SIDA, el hongo Cryptococcus neoformans es una causa frecuente de meningitis.
Aponeurosis
Recuerda
Las causas más frecuentes en pediatría son las meningitis virales por Echovirus, Coxsackie y otros enterovirus.
Meninges


Duramadre
Aracnoides
Piamadre
De entre todos los patógenos, por su frecuencia y letalidad, las bacterias son las más importantes y su frecuencia tiene un patrón según las edades de presentación.
En recién nacidos el Streptococo agalactiae y la E. coli son los más frecuentes. También podría haber casos de Listeria monocytogenes.


En lactantes aparece el Haemophilus tipo B si no se vacunaron.


Los niños (sobretodo mayores de 10 años) tienen Meningococo como causa principal.
Los adultos a partir de 18 años tienen Neumococo (ESSALUD 2015) como causa más prevalente, y en casos de inmunosupresión celular o edad superior a 50 años, aparece Listeria o E. coli nuevamente.
Recuerda
- En lactantes aparece el Haemophilus tipo B si no se vacunaron.
- Los adultos a partir de 18 años tienen Neumococo como causa más prevalente.
Los factores de riesgo para meningitis son la falta de vacunación, asplenia, déficit de complemento o anticuerpos, uso de glucocorticoides, bacteriemia o TEC de base de cráneo.
www.qxmedic.com PAG. 209
MENINGITIS
04.
Figura 7. Anatomía de Meninges
Piel
Periostio
aracnoidal Seno venoso dural Hueso 0-4 semanas 4-12 semanas H. influenza 3m-18 años 18 a 50 años Neumococo Más de 50 años Listeria
Vellosidad
Diagnóstico
La sospecha de meningitis siempre es clínica y debe considerarse como tal hasta que no se demuestre lo contrario. Se sospecha de meningitis en pacientes con fiebre alta, cefalea intensa del tipo hipertensión endocraneana y con signos meníngeos como rigidez de nuca, Kernig y Brudzinsky. El sensorio se puede afectar en casos severos y aparecer focalización de pares craneales. (ESSALUD 2018)
Recuerda
Se sospecha de meningitis en pacientes con fiebre alta, cefalea intensa del tipo hipertensión endocraneana y con signos meníngeos como rigidez de nuca, Kernig y Brudzinsky.
meníngeos:
A. Brudzinsky
B. Kernig
Ante toda sospecha clínica de MEC se debe hacer una punción lumbar (PL) y hemocultivos de forma inmediata. Si el paciente presenta trastorno severo del sensorio, inmunosupresión, convulsiones, antecedente de enfermedad del SNC, papiledema o focalización se solicita una TAC previo a la PL. Luego se debe iniciar con dexametasona y tratamiento antibiótico empírico hasta conformar la etiología con los estudios de líquido cefalorraquídeo LCR.

Recuerda
Podemos encontrar en el análisis del LCR por infección bacteriana los siguientes hallazgos:
- Presión: alta
- Aspecto: turbio
- Células: 10002000 PMN
- Proteínas: 1001000
- Glucosa: muy baja
2015)
BACTERIA ATÍPICA (ENARM 2020-B, ENARM 2018-A, ENARM 2022)
www.qxmedic.com NEUROLOGÍA PAG. 210
LIQUÍDO PRESIÓN ASPECTO CÉLULAS PROT GLU NORMAL 8-12 cmH2O Claro 5 mm3 15-45 mg/dl 65-80% INFECCIÓN BACTERIANA (ENARM
Alta Turbio 1000-2000 PMN 100-1000 Muy baja
VIRAL (ENARM 2019-B) Normal/alta Claro 300 MN 40-100 Normal
Alta Xantocrómico 50-300
60-700
2022) (ESSALUD
INFECCIÓN
INFECCIÓN
MN
Baja
Tabla 4. Comparativa de características del líquido cefalorraquídeo
Figura 8. Signos
Tratamiento
Tratamiento empírico - adultos:
Neumococo: vancomicina 30-60mg/kg (8-12h) y, además una cefalosporina de tercera generación (CFTX 4g - 12h) o fluoroquinolona
Meningococo: cefalosporina de tercera generación o como alternativa penicilina G, ampicilina, cloranfenicol, fluoroquinolona o aztreonam.
Listeria monocitógenes: ampicilina 12g (4h) o como alternativa penicilina G, cotrimoxazol o meropenem
H. influenzae: cefalosporina de tercera generación o como alternativa cloranfenicol, cefepime, meropenem o fluorquinolona
Dexametasona para reducir secuelas en caso de infecciones por H. Influenzae (ENARM 2020-B)
♦ Dar antes de la primera dosis de antibiótico.
♦ Dosis: 0.6mg/kg/día en 4 dosis por 2-4 días
Tratamiento empírico - pediatría: (ENARM 2021 – A)
Cefotaxima o ceftriaxona + vancomicina + aminoglucósido
Profilaxis meningitis:
Neumococo, H. Influenzae tipo B: Vacuna
Menigococo: Vacuna, rifampicina por 2 días, ciprofloxacino (ESSALUD 2018)
Tratamiento meningitis – TBC
Aplicar el esquema 1 pero por 12 meses.
Tratamiento meningitis - Inmunosuprimido
Vancomicina + ceftazidima + ampicilina
Tratamiento meningitis – Hongo
- Anfotericina B + flucitosina y fluconazol profiláctico hasta mejorar recuento de CD4+ (ENARM 2016 – B)
Tratamiento de encefalitis viral por herpes
Aciclovir EV 10mg/K
Recuerda
El tratamiento para neumococo es vancomicina 30-60mg/kg (8-12h) y, además, una cefalosporina de tercera generación (ceftriaxona 4g - 12h).
Recuerda
Se debe administrar dexametasona antes de la primera dosis de ATB, esto para reducir las secuelas en caso de infecciones por H. influenzae.
www.qxmedic.com NEUROLOGÍA PAG. 211
05. ENFERMEDA DE PLACA MOTORA
ASPECTOS LAMBERT EATON BOTULISMO MIASTENIA
ETIOLOGÍA
EPIDEMIOLOGÍA
Anticuerpos anti-canales de calcio presináptico. Síndrome paraneoplásico de cáncer de pulmón.
Edad promedio 40 años. Afecta por igual ambos sexos.
MÚSCULOS AFECTADOS
MEJORAMIENTO DE CLÍNICA
PATOLOGÍA ASOCIADA
TRATAMIENTO
Extraoculares en el 70% de casos. Proximales de MMII.
Ejercicio, avance del día. Uso de guanidina.
Cáncer pulmonar (ESSALUD 2015) microcítico “células de avena”
Plasmaféresis en caso de crisis. Piridostigmina + Guanidina Inmunosupresores. Extirpación del tumor de fondo.
Toxina botulínica del Clostridium botulinum. Causa déficit de acetilcolina presináptico por inhibición de la liberación vesicular.
Afecta principalmente a lactantes de 6 meses de edad. Asociado a la ablactancia y uso de conservas.
Afectación precoz de pares craneales. Clínica descendente simétrica. Puede llegar al paro respiratorio.
Anticuerpos anti-receptor nicotínico muscular postsináptico. (ENARM 2017 – B, ENARM 2019-A). Se han reportado anticuerpos antiMUSK (músculo liso) en casos severos.
El 66% de los casos afectan a mujeres de 28 años. Un 33% a varones de 50 años.
Músculos extraoculares, faciales, bulbares y finalmente axiales de MMSS.

EMPEORA: avance del día, ejercicio, infecciones, gestación, calor, aminoglucósidos, betabloqueantes y derivados del curare.
Frío, reposo, test de edrofonio (inhibidor de la acetilcolinesterasa de acción ultracorta)
Mala conservación de alimentos. Heridas contaminadas.
Soporte ventilatorio. Antitoxina.
En casos de mujeres jóvenes se asocia a hiperplasia de timo 65% y timomas en un 10%. Los casos de varones de edad media se asocian a cáncer de pulmón (paraneoplásico)
Piridostigmina como primera medida. Inmunosupresores en casos de resistencia. La timectomía entre los 15 y 55 años siempre que haya afectación del timo. Plasmaféresis o terapia hiperinmune IgG en caso de crisis miasténica (disnea o disfagia). Ventilación mecánica en casos severos.
www.qxmedic.com PAG. 213
Tabla 5. Resumen de los síndromes de placa motora más importantes
Figura 9. Comparación entre la miastenia gravis y el síndromes de Lambert-Eaton
Síndrome miasténico de Lambert-Eaton
Miastemia gravis (MG)
FACIE MIASTÉNICA
Lambert-Eaton
06. SÍNDROME DE GUILLAIN BARRÉ
Aspectos generales
Este síndrome es una poliradiculoneuropatía (afecta varias raíces nerviosas a la vez), aguda (se instala en menos de 4 semanas), inflamatoria (se pueden ver signos de activación de macrófagos sobre los axones neuronales), ascendente (inicia en MMII y sube progresivamente), predominio motor (mayor afectación de motoneuronas alfa por su grosor) y autoinmunitaria (presencia de autoanticuerpos contra los gangliósidos GM1, GD1, Gq1b, entre otros).
El 66% se debe a una infección previa por bacterias como el Campylobacter jejuni o virales como enterovirus, herpes, hepatitis o VIH. A la anatomía patológica se evidencian signos de desmielinización inflamatoria.
Diagnóstico
La triada clásica del Guillain Barre es: (ENARM 2021 – B) (ESSALUD 2011)
1º Parestesias de miembros inferiores (al inicio se afecta la zona lumbar).
2º Paresia simétrica ascendente con clínica de segunda neurona.

3º Arreflexia profunda, este signo suele ser tardío.
Son signos de apoyo: ausencia de fiebre, líquido cefalorraquídeo (LCR) alterado a partir de la segunda semana de iniciado el cuadro y electromiografía (EMG) con patrón de desmielinización
Existen variantes clínicas del tipo motora pura, sensitiva pura, entre otros.
www.qxmedic.com PAG. 215
Figura 10. Fisiopatología del Síndrome de Guillain Barré
Recuerda
La triada clásica del Guillain Barre es:
– 1º Parestesias de miembros inferiores (al inicio se afecta la zona lumbar).

– 2º Paresia simétrica ascendente con clínica de segunda neurona.
– 3º Arreflexia profunda, este signo suele ser tardío.
Un tipo especial de variante es el síndrome de Miller Fisher que presenta oftalmoparesia, ataxia y arreflexia. Se debe a una afectación del cordón espinal posterior y los nervios de los pares craneales extraoculares. Los anticuerpos reconocen el gangliósido Gq1b.
Tratamiento
Recuerda
El manejo actual de primera línea es el uso de inmunoglobulinas IgG.
El manejo actual de primera línea es el uso de inmunoglobulinas IgG para neutralizar los autoanticuerpos. Como segunda opción queda la plasmaféresis.
Todo paciente debe tener acceso a una UCI y se debe buscar el sostén de funciones vitales hasta que la enfermedad remita.
Un 85% presentan recuperación completa, el 10% queda con algún grado de déficit motor y un 5% mueren. Hay un 10% de casos que recurren y en ellos debemos sospechar síndrome paraneoplásico.
www.qxmedic.com NEUROLOGÍA PAG. 216
Figura 11. Variantes de la sintomatología del Síndrome de Guillian Barré
07. DISQUINESIAS (TRANSTORNOS DEL MOVIMIENTO)
Los trastornos de movimientos involuntarios o disquinesias se deben a la afectación del equilibro en el sistema extrapiramidal. Los aspectos clínicos se clasifican en trastorno hiper e hipocinéticos.
Aspectos generales
Temblor esencial: el más frecuente de todas las hipercinesias. Se trata de una afectación idiopática con marcado componente familiar que inicia en la edad adulta, en manos bilateralmente que empeora con la edad. Suele ser postural y muy fino. Si causa dificultades para la vida diaria se indican betabloqueantes.
Distonía: se deben principalmente al uso de drogas antidopaminérgicos de forma aguda y de elevada potencia como el haloperidol. La distonía más frecuente es el blefaroespasmo y luego el tortícolis. Su manejo es retirar la droga causante del problema y esperar. Los casos severos requieren uso de diazepam o incluso toxina botulínica para relajar el musculo.
Hemibalismo: debido a lesiones del núcleo subtalámico de Luys contralateral al lado afectado.
Recuerda
Hemibalismo se debe a lesiones del núcleo subtalámico de Luys contralateral al lado afectado.
Corea: la más representativa es la corea de Huntington que se debe a una alteración autosómica dominante que aumenta la expresión de la proteína Huntingtina por acumulación de tripletes CAG. Esta enfermedad se presenta con una triada clásica de corea de codos y rodillas, demencia y trastornos psicóticos hereditarios. No tiene cura y su pronóstico es peor en los hijos que en los padres.
TICS: son movimientos estereotipados, involuntarios y repetitivos que no pueden controlarse. Uno característico es el síndrome de Guilles de la Tourette, donde los tics se asocian a coprolalia y trastorno obsesivo compulsivo de adulto.
Transtornos hipocinéticos o parkinsonismos
Recuerda
- Las causas del parkinsonismo idiopático más común es la enfermedad del Parkinson.
- Las causas de parkinsonismo se dividen en primarias o idiopáticas y las secundarias.
- Las causas del parkinsonismo secundario más común es por fármacos (anti dopaminérgicos D2), y el fármaco más frecuente es metoclopramida.
Se caracterizan por presenta temblor de reposo, rigidez en “rueda dentada”, bradicinesia o lentitud al movimiento y al final alteración de la marcha con arrastre de los pies conocido como marcha festinante.
Las causas de parkinsonismo se dividen en primarias o idiopáticas y las secundarias.
Idiopáticos:
♦ Enfermedad de Parkinson (causa más frecuente) (ESSALUD 2011)
♦ Atrofia multisistémica
♦ Parálisis supranuclear progresiva
♦ Degeneración cortico basal
♦ Enfermedad de Huntington
♦ Enfermedad de Alzheimer
♦ Demencia a cuerpos de Lewy
Secundarios:
♦ Post encefálico
♦ Fármacos: antidopaminérgicos D2 (metoclopramida) (causa secundaria más frecuente)
♦ Tóxicos
♦ Hidrocefalia normotensiva
www.qxmedic.com PAG. 217
08. ENFERMEDAD DE PARKINSON
Aspectos generales
La etiología es desconocida. Cuya prevalencia aumenta con la edad. No tiene factor hereditario importante. La cafeína protege. Hay lesión en la pars compacta de la sustancia negra del mesencéfalo, aparecen inclusiones neuronales llamadas “cuerpos de Lewy”. Esto genera un déficit de células dopaminérgicas en el sistema nigroestriado (zona A9 mesencefálica). Por lo tanto, baja la dopamina (ENARM 2022), se eleva compensatoriamente la acetilcolina y se ha visto que baja la noradrenalina y serotonina también.


www.qxmedic.com PAG. 219
Figura 12. Fisiopatología del Parkinson, degenerción de la sustancia negra
Figura 13. Cuerpos de Lewy Microscopía de la sustancia negra de un paciente con enfermedad de Parkinson
Sustancia negra
Enfermedad de Parkinson
El diagnóstico es clínico, existen varios criterios para establecerlo, pero en resumen se debe tener al menos la presencia de dos de los signos cardinales de la enfermedad durante un año y, además, la respuesta al tratamiento con L-dopa de al menos 1 año (ENARM 2020-A)
Dentro de los signos cardinales de la enfermedad están el temblor, la bradicinesia, la rigidez y la marcha festinante (ENARM 2022).
Recuerda
La bradicinesia consiste en la lentitud de movimientos, dificultad para movimientos finos, amimia, voz monótona, micrografía, es el síntoma más incapacitante.
Diagnóstico Tratamiento
El temblor es una oscilación involuntaria a + 6Hz rítmica, algunos pacientes no tienen temblor. El comienzo es unilateral, suele iniciar en una mano, pasa a la pierna ipsilateral y luego a la otra mano. Es temblor de reposo, predomina en los dedos (contar monedas). En raros casos se asocian a temblor postural o de intención.

La rigidez es la resistencia a la movilización pasiva. Se llama rigidez en "rueda dentada" o rigidez "cérea”. Incrementa con el movimiento voluntario de la mano contralateral (signo de Froment).
La bradicinesia consiste en la lentitud de movimientos, dificultad para movimientos finos, amimia, voz monótona, micrografía. Es el síntoma más incapacitante. Marcha festinante o alteraciones en la marcha consiste en una marcha lenta, a pequeño paso, arrastrando los pies (festinación). Falta de braceo al caminar. Caídas frecuentes. No mantiene la postura si se le empuja (retropulsión) y no puede caminar hacia atrás. Existen síntomas no motores de la enfermedad de Parkinson como dolores osteomusculares inespecíficos, hiposmia, hipotensión ortostática, estreñimiento, disfunción sexual, síntomas urinarios, síntomas depresivos, síndrome de piernas inquietas al dormir. Alucinaciones visuales o delirios. Deterioro cognitivo en mayores de 80 años.
El Gold estándar en mayores de 70 años es administrar L-DOPA asociada a Carbidopa o Benseracida, que son inhibidores de la dopa decarboxilasa periférica (para evitar efectos secundarios como náuseas, vómitos, hipotensión, arritmias.
Recuerda
El Gold estándar en mayores de 70 años es administrar L-DOPA asociada a Carbidopa o Benseracida.
Para pacientes menores de 70 años, a fin de evitar los efectos adversos de este fármaco de forma precoz, se recomienda iniciar con agonistas dopaminérgicos postsinápticos como ropirinol, pramipexol, bromocriptina y cabergolina. El progreso de la enfermedad es lento pero inexorable. Los fármacos aumentan sobrevida entre 10-20 años. Un 25% de pacientes están severamente incapacitados a los 5 años del diagnóstico, un 65% a los 10 años y un 80% a los 15 años. No se sabe la evolución futura al inicio.
www.qxmedic.com NEUROLOGÍA PAG. 220
09. DEMENCIAS – ENFERMEDAD ALZHEIMER
Aspectos generales
Desde un punto de vista académico las demencias pueden clasificarse en 2 grupos.
Cortical: en este tipo de demencias predomina al inicio la clínica de afectación de funciones cerebrales superiores como la afasia, apraxia, agnosia, acalculia. Entre sus ejemplos tenemos a la enfermedad de Alzheimer (90%), demencia alcohólica (10%), demencia frontotemporal de Pick y la enfermedad de Creutzfeldt-Jakob.
Subcortical: donde al inicio predomina clínica subcortical como retardo psicomotor, movimientos anormales, disartria, depresión endógena, parkinsonismo, entre otros. Algunos ejemplos son la enfermedad de Huntington, enfermedad de Parkinson, enfermedad de Wilson, demencia vascular (10%), VIH, demencia pugilística o post traumática.
En cuando a la enfermedad de Alzheimer, el 95% de casos son espontáneos sin antecedentes familiares directos, se ha visto implicado la mutación del alelo E4 de APO E (cromosoma 19). Son factores de riesgo la edad mayor a 60 años, ser mujer, TEC previo. Se conocen factores protectores como la presencia del APOE2, uso crónico de AINES, exposición a estrógenos y educación permanente.
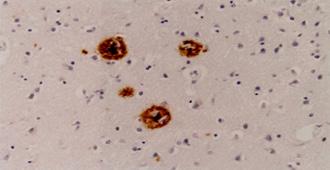
El aspecto patogénico característico de la esta enfermedad es la degeneración de las neuronas colinérgicas, sobre todo del hipocampo. Se encuentra en la anatomía patológica los ovillos neurofibrilares por depósito de la proteína tau dentro de los somas neuronales y también se detectan las placas beta amiloide por el depósito anormal de esta proteína en las sinapsis, esta placa activa el sistema inflamatorio que terminan destruyendo la sinapsis. Con el progreso de la enfermedad la degeneración se extiende a toda la corteza.
Diagnóstico
Para establecer el diagnóstico seguimos los criterios de la DSMV.
DSMV -DESORDEN NEUROCOGNITIVO MAYOR (DNM)
Deterioro cognitivo en 1 o más de:
♦ Aprendizaje y memoria.
♦ Lenguaje.
♦ Funciones ejecutivas.
♦ Atención compleja.
♦ Motor-percepción.
♦ Cognición social.
www.qxmedic.com PAG. 221
Figura 15. Placas amiloides en la enfermedad de Alzheimer
Dependencia en actividades diarias. No hay delirium o trastorno del sensorio. No hay otros trastornos mentales que expliquen el deterioro mental. Adicionalmente, para especificar que el DNM se debe a la enfermedad de Alzheimer se debe cumplir:

♦ Inicio gradual del cuadro y deterioro cognitivo continuo.
♦ No hay otra causa que lo explique mejor.
Tratamiento
(A y D: persona sana; C y D: persona con enfermedad de Alzheimer)
El manejo no altera el curso inexorable de la enfermedad, pero sí puede mejorar la calidad de vida. Se aplica inhibidores de la acetilcolinesterasa de acción central como la rivastigmina, donapezilo y galantamina. Para casos severos se recomienda memantina, un antagonista no competitivo del receptor NMDA.
Recientemente se aprobó el uso de aducanumab como tratamiento de Alzheimer en fases iniciales para pacientes selectos. Se vio que puede retrasar la progresión de la enfermedad.

www.qxmedic.com NEUROLOGÍA PAG. 222
Figura 16. Masa encefálica normal vs pacientes con Alzheimer
Figura 15. Atrofia cerebral en la enfermedad de Alzheimer.
Aspectos generales
La epilepsia es una enfermedad que afecta al 0.5 a 1% de la población. Se define como la presencia de 2 o más crisis espontáneas sin una causa desencadenante inmediata que las explique (no provocadas). Las epilepsias tienen una edad de presentación, una causa, factores de riesgo, desencadenantes, patrones de electroencefalograma entre otras características que permiten agruparlas como síndromes epilépticos. Así clasificamos estos síndromes epilépticos según su origen en:
Idiopáticos: cuando la causa de origen orgánica no es conocida ni se puede demostrar en imágenes. Sin embargo, se asocian familiarmente a un patrón hereditario de canalopatías y otros trastornos de conducción sináptica.
Sintomáticos: son los síndromes que ocurren con lesión orgánica evidente, la cual explica la epilepsia. Para ello se deben evidenciar las lesiones con neuroimágenes.
Criptogénicos: cuando el origen es muy probablemente orgánico por historia clínica pero aún no se puede demostrar la lesión con las neuroimágenes.
Diagnóstico
Para diagnosticar un síndrome epiléptico se requiere una adecuada historia clínica, un electroencefalograma y una resonancia magnética con contraste (ENARM 2022)


Una vez confirmado la epilepsia, el cuadro clínico y el patrón electroencefalográfico podemos clasificar los síndromes de forma más detallada según el siguiente cuadro de la Liga Internacional de lucha contra la Epilepsia (ILAE).
Las crisis de ausencia se caracterizan por presentar pérdida del sensorio sin alteraciones motoras. Ocurren en niños menores de 10 años y los episodios se repiten varias veces durante el día (ENARM 2018 - B, ENARM 2019 - B, ENARM 2018 – A)
www.qxmedic.com PAG. 223
EPILEPSIA
10.
Figura 18. Actividad cerebral registrada por electroencefalograma
Crisis Generalizada
Crisis Parcial
Periodo neonatal
Clasificación de los síndromes epilépticos (ILAE 2010) - RESUMEN
– Crisis neonatales benignas
– Epilepsia familiar neonatal benigna
– Síndrome de Ohtara
Lactancia
Crisis febriles/crisis febriles plus
– Epilepsia benigna de la infancia (de la lactancia)
– Epilepsia benigna familiar de la infancia
– Síndrome de West
Síndrome de Dravet
Epilepsia mioclónica de la infancia (de la lactancia
Infancia
– Crisis febriles/crisis febriles plus
– Epilepsia occipital de la infancia de inicio temprano (Síndrome de Panylotopoulos)
– Epilepsia con crisis mioclónicas atónicas (previamente astáticas)
Epilepsia en ausencia infantil
Epilepsia benigna con puntas centrotemporales
– Síndrome de Lennox-Gastaut
Adolescencia/edad adulta
– Epilepsia en ausencia juvenil
Epilepsia mioclónica juvenil
Epilepsia con crisis generalizadas tónico-clónicas solamente
– Epilepsia autosómico dominante con características auditivas
Edad de inicio variable
– Epilepsia focal familiar con focos variables
Epilepsia mioclónica progresiva
Constelaciones específicas/síndromes quirúrgicos
– Epilepsia temporal mesial con esclerosis del hipocampo
– Síndrome de Asmussen
– Crisis golásticas con hemartoma hipotalámica
– Epilepsia con hemiconvulsión-hemiplejia
Constelaciones específicas/síndromes quirúrgicos
Epilepsia temporal mesial con esclerosis del hipocampo
– Síndrome de Rasmussen
Epilepsias no sindrómicas
– Epilepsias atribuidas a causas estructurales-metabólicas y organizadas de acuerdo con ellas
• Malformaciones de desarrollo cortical (hemimegalencefalia, heterotópicas, etc.)
• Síndromes neuro cutáneos (complejo esclerosis tuberosa, Stuger Weber, etc.)
• Tumor, infección, trauma, angioma, lesiones prenatales y perinatales, accidente cerebrovascular, etc.
– Epilepsia de causa desconocida
www.qxmedic.com NEUROLOGÍA PAG. 224
–
–
–
–
–
–
–
–
–
Tabla 6. Clasificación de los síndromes epilépticos (ILAE 2010)
Valproato Nauseas, vomitos, temblor, peso, alopecia.
Fenitoina Nistagmo, hipertrofia gingival, hipertricosis, rasgos faciales.
Hepatotoxicidad, pancreatitis, “penias”.
Discrasias, S. Johnson, hepatoxicidad, polineuropatías, ataxia, LES.
Carbamazepina Diplopía, hiponatrémia, neutropenia, rash Agranulocitosis, anemia aplásica , S. Johnson, ataxia
Fenobarbital Sedación, impotencia.
Etosuccimida Nauseas, vomitos, cefalea, sedación, agitación.
Tratamiento
Recuerda
El efecto adverso mas frecuente del uso de carbamazepina es diplopía, hiponatrémia, neutropenia, rash; y los efectos adversos más raros son: agranulocitosis, anemia aplásica, S. Johnson, ataxia.
Necrólisis epidermolítica tóxica, hepatotoxicidad, distrofia escapulohumeral.
Eritema multiforme, S.
El manejo crónico de la epilepsia debe procurar administrar la dosis más baja de solo un fármaco antiepiléptico (FAE), es decir se prefiere la monoterapia siempre. Todo cambio o supresión de FAE debe hacerse de forma gradual. Recomendar siempre evitar el alcohol y mantener el sueño nocturno adecuado. (ESSALUD 2019)
En general los síndromes de epilepsia generalizada idiopática se tratan con: acido valproico (ENARM 2017 – A) (ESSALUD 2019), lamotrigina, topiramato o levetiracetam. De todos ellos el valproato es el más usado. Un caso especial es la crisis generalizada de ausencia, la cual puede tratarse con etosuximida (ENARM 2019-B) que es un fármaco de espectro reducido (bloquea solo el canal de calcio) pero es eficaz en niños con crisis típicas, para crisis atípicas se debe usar valproato (ENARM 2018 A).
Para los síndromes epilépticos que cursan con crisis focales, los fármacos principales son: carbamazepina (ENARM 2017 – B, ENARM 2022) y oxcarbacepina, fenitoína, o gabapentina.
Nomenclatura abreviada de fármacos antiepilépticos (FAE):
CBZ: Carbamazepina
VPA: Valproato
ETX: Etosuximida
LTG: Lamotrigina
LEV: Levetiracetam
TPR: Topiramato
PHT: Fenitoína
PB: Fenobarbital
GBP: Gabapentina
www.qxmedic.com NEUROLOGÍA PAG. 225
EFECTO
FÁRMACO ANTIEPILÉTICO
ADVERSO FRECUENTE EFECTO ADVERSO RARO
Johnson.
Tabla 7. Efectos Adversos de los antiepiléptico
CLASIFICACIÓN DE FAE SEGÚN ESPECTRO
Amplio espectro: Clobazam - Felbamato –Lamotrigina –LevetiracetamPerampanel -Rufinamida – Topiramato – Valproato – Zonisamida
Espectro reducido: Carbamazepina – Gabapentina - Lacosamida –Oxcarbazepina – Fenobarbital – Fenitoína (ENARM 2018-A) – Pregabalina – Tiagabina – Vigabatrina – Etosuximida (ausencia)
Aproximadamente un 20-30 % de los pacientes con epilepsia son resistentes al tratamiento médico y se procede a la cirugía. Seccionar el cuerpo calloso se ha mostrado eficaz en las convulsiones tónicas clónicas intratables.
Recuerda
Las crisis generalizada de ausencia, puede tratarse con etosuximida que es un fármaco de espectro reducido (bloquea solo el canal de calcio) pero es eficaz en niños con crisis típicas.
www.qxmedic.com NEUROLOGÍA PAG. 226
Tabla 8. Clasificación de FAE según espectro
4 2 3 1
Figura 19. Posición de seguridad antes una epilepsia
11. CRISIS EPILÉPTICA Y ESTATUS EPILÉPTICO
Aspectos generales
Las crisis epilépticas son episodios provocados por una descarga brusca anormal de determinada población neuronal. Puede ocurrir por muchas causas distintas y llega a afectar al menos una vez en la vida de casi el 10% de la población. Las causas de las crisis pueden clasificarse según la edad de la siguiente manera:
0-2 años: hipoxia, trastornos metabólicos, infecciones del SNC, crisis febriles.
2-12 años: idiopáticas, crisis febriles.
12-18 años: idiopáticas, traumatismo encefalocraneano.
18-35 años: TEC, neurocisticercosis (zonas endémicas)
35-55 años: tumoral de fosa anterior.

55+ años: enfermedades vasculares, patología degenerativa, TEC, y tumores. El sufrir una o varias crisis en la vida de forma provocada (por alguna noxa directa) no supone ser epiléptico necesariamente, para ello se requieren que las crisis sean no provocadas. Independientemente si una persona es epiléptica o no, una crisis puede evolucionar a un “status epiléptico” el cual se considera una emergencia neurológica, por lo tanto, debe ser tratada inmediatamente. Se define “status epiléptico” a la actividad epiléptica continua durante un tiempo igual o mayor de 30 min o al conjunto de 3 o más crisis epilépticas repetidas sin recuperación de la conciencia (ENARM 2017 – B). El 60% de pacientes con “estatus” queda con secuelas neurológicas, por ello se inicia el tratamiento empírico preventivo mucho antes de que se establezca el daño.
Para el caso de las crisis tónico-clónicas generalizadas se considera que, tras 5 minutos de duración de la crisis, la probabilidad de estatus es muy alta y corresponde iniciar tratamiento empírico. Para otros tipos de crisis a partir del minuto 10 se debe iniciar manejo empírico del estatus.
Recuerda
Se define “status epiléptico” a la actividad epiléptica continua durante un tiempo igual o mayor de 30 min o al conjunto de 3 o más crisis epilépticas repetidas sin recuperación de la conciencia.
www.qxmedic.com PAG. 227
c
Figura 20. Convulsión tónico-clónica generalizada.
Diagnóstico
El diagnóstico de las crisis es solamente clínico cuando hay componente motor evidente, a ellas se les conoce como crisis convulsivas.
Para las crisis no convulsivas se necesita un electroencefalograma junto con la anamnesis para establecer el diagnóstico exacto del tipo de crisis. Los tipos de crisis se clasifican de la siguiente manera.
Inicio focal
Inicio generalizado
Inicio desconocido
Conciencia alterada Conciencia preservada
Inicio motor Automatismos
Atónica
Clónica
Espasmo epiléptico
Hiperquinética
Mioclónica
Tónica
Inicio no motor Autosómica
Detención del comportamiento
Cognitiva
Emocional
Sensorial
Motora
Tónica clónica
Tónica
Clónica
Mioclónica
Mioclónica - TónicaClónica
Mioclónica - atónica
Atónica
Espasmos epiléptico
No motora
(ausencia)
Típica
Atípica
Mioclónica
Mioclónica palpebral
Motora
Tónica clónica
Otro Motor
No motor
Detención del comportamiento
No clasificada
Focal a bilateral Tónica-Clónica
Tratamiento
El tratamiento de las crisis solo es con fines preventivos de un posible “estatus”, la mayoría de las crisis no se tratan y remiten espontáneamente.
Hay que recordar que el manejo de epilepsia es totalmente distinto y para ello se requiere haber diagnosticado al paciente como tal. En este apartado veremos el manejo de un “estatus epiléptico”.
www.qxmedic.com NEUROLOGÍA PAG. 228
Figura 21. Clasificación operacional de los tipos de crisis, versión extendida ILAE 2017.
Recuerda
El manejo inicial de las crisis es colocar en un primer acceso venoso: lorazepam 4mg (0.1mg/kg) o diazepam 10mg (0.15mg/k) por 1 min
Evaluación inicial
Aplicar el ABCDE y un examen neurológico completo (ENARM 2019-A) Aportar O2 si desatura, ventilación mecánica si requiere y monitorizar los signos vitales.
Dosar glucosa, electrolitos, tóxicos en sangre, concentración de fármaco antiepiléptico (en caso sea paciente epiléptico en tratamiento).
Administrar en bolo glucosa más tiamina (solo si sospecha de alcoholismo crónico).
Manejo inicial de la crisis
En un primer acceso venoso colocar:
♦ Lorazepam 4mg (0.1mg/kg) o diazepam 10mg (0.15mg/kg)
♦ Si no es posible el acceso venoso: midazolam 10mg (40kg) de forma intramuscular: Si el paciente no mejora podemos repetir la dosis y, además, en un segundo acceso venoso colocar:
♦ Fenitoína 20mg/kg (25-50mg/min) o valproato 30mg/kg (10mg/min) o levetiracetam 50mg/ kg en 15min
♦ Corregir cualquier trastorno metabólico que se sospecha Si, a pesar de estas medidas, el paciente no mejora se cataloga como estatus refractario.
♦ Para estos casos se procede a intubar, pasar a UCI y colocar monitoreo continuo de electroencefalograma
♦ Iniciar fármacos de segunda línea: midazolam o pentobarbital (endovenoso)
♦ Ver evolución
www.qxmedic.com NEUROLOGÍA PAG. 229
12. CRISIS FEBRILES
Aspectos generales
Las crisis febriles no se consideran un tipo de epilepsia y usualmente no incrementa el riesgo de esta. Representan el 50% de las crisis en los primeros 5 años. Afectan al 2-3% de la población general de niños y la edad de presentación oscila entre los 6 meses y los 5 años, siendo el pico máximo alrededor de los 2 años de vida. Afecta más a varones, hay predisposición genética asociada a los genes FEB1,2,3,4.
Se clasifican en crisis febriles simples (sin riesgo de epilepsia a futuro) y complejas (con ligero riesgo de epilepsia que requiere monitoreo).
Diagnóstico
Para establecer el diagnóstico se requiere lo siguiente:
Niño neurológicamente sano
Edad de presentación entre 6 meses y 5 años
Fiebre mayor de 38°
La convulsión aparece al inicio del cuadro febril o incluso lo antecede
Las crisis febriles simples representan el 80% y son crisis generalizadas , de duración menor a 15 minutos, ocurren 1 vez al día y no presentan riesgo incrementado de epilepsia a futuro del 3% (ENARM 2018 – A, ENARM 2017 – B) (ESSALUD 2019)
Las crisis febriles complejas representan el 20% y son crisis focalizadas , de duración mayor a 15 minutos, ocurren varias veces al día y sí presentan riesgo incrementado de epilepsia a futuro (ENARM 2017 – B, ENARM 2018 - B, ENARM 2019 - A, ENARM 2018-B, ENARM 2021-B, ENARM 2022)
Tratamiento
Recuerda
Las crisis febriles complejas representan el 20% y son crisis focalizadas, de duración mayor a 15 minutos, ocurren varias veces al día y sí presentan riesgo incrementado de epilepsia a futuro.
Bajar la fiebre
El uso de diazepam es opcional
No requieren manejo continuo a largo plazo
Consejería familiar si hay ansiedad
Solo hacer seguimiento si son crisis febriles complejas
www.qxmedic.com PAG. 231

