Rapid Evolution of the Biotech Industry

2023 and Beyond
Developing Robust Analytical Methods for Early-Stage Biologics


Accelerating Biologics Manufacturing Strategies for Efficient Production Timelines
Using a LIMS to Improve Bottom Line Profitability
Sponsor Company: Supporting Exhibitions



Peer Reviewed
www.international-biopharma.com

Volume 6 Issue 3
II INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Learn more at www.sygnaturediscovery.com #anewdiscovery With the scale, technology and talent to deliver on complex drug discovery programs efficiently and on time Sygnature Discovery is now one of North America’s largest drug discovery CROs
DIRECTOR: Mark A. Barker
INTERNATIONAL MEDIA DIRECTOR: Anthony Stewart anthony@senglobalcoms.com
EDITORIAL MANAGER: Beatriz Romao beatriz@senglobalcoms.com
DESIGN DIRECTOR: Jana Sukenikova www.fanahshapeless.com
FINANCE DEPARTMENT: Akash Sharma accounts@senglobal.co.uk
RESEARCH & CIRCULATION: Jessica Chapman info@senglobalcoms.com
COVER IMAGE: iStockphoto ©
PUBLISHED BY: Senglobal ltd.
Unit 5.02, E1 Studios, 7 Whitechapel Road, E1 1DU, United Kingdom
Tel: +44 (0)20 4541 7569 Email: info@senglobalcoms.com www.international-biopharma.com
All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IBI will be published in Winter 2023. ISSN No.International Biopharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
2023 Senglobal ltd.
Volume 6 Issue 3 – Autumn 2023
04 Foreword
TALKING POINT
06 Cloud-based Drug Discovery Technology
IBI speaks with Ed Champnessin of Optibrium, on cloud-based drug discovery technology, and how continuous developments to its StarDrop platform are enabling scientists to tackle today's challenges, with powerful software for predictive modelling, 3D drug design and beyond.
REGULATORY & COMPLIANCE
10 Rapid Evolution of the Biotech Industry: 2023 and Beyond
The biopharmaceutical industry has embraced the adage of “Don’t be afraid of change, welcome it.” In doing so, the industry has fuelled a staggering transformation over the past 20 years. Extremely complex nucleic acid molecules and recombinant proteins with highly defined and functionally sensitive characteristics have been scaled to thousands of litters in commercial-scale bioreactors. The emergence and commercialisation of “living medicines” have allowed reprogramming patient’s T cells ex vivo to exhibit drastic tumour regression and long-term tumour surveillance, developed gene therapies to reverse monogenic diseases that otherwise have little or no treatment options. Jeff Briganti of Aldevron, LLC, discusses how novel therapeutic development is moving fast and the biotech industry is rapidly evolving.
RESEARCH / INNOVATION / DEVELOPMENT
14 A Summary of the Pharmaceutical Development Process, the Changes and Challenges and Future Opportunities
The drug development process has developed considerably over the years to meet the demands of the pharmaceutical company, regulators, and the patient. The strive to outsource has gathered huge pace as an attempt to cut costs and free up resource. Dean Hatt of Broughton summarises the ‘historical’ development plan for pharmaceuticals, the changes that have occurred with regards to differential development including the requirements for biologicals, the typical challenges the industry faces now and, in the future, the opportunities this brings.
20 Developing Robust Analytical Methods for Early-Stage Biologics
Biologics are a broad class of therapeutic agents, encompassing vaccines, monoclonal antibodies, therapeutic proteins, nucleic acid-based therapies, blood components, tissue therapies, and cellular therapies. Biologics, however, present unique challenges in the journey from discovery to commercialisation due to their inherent complexity, size and charge heterogeneity and susceptibility to changes that could potentially impact efficacy and the patient safety. Pavan Kumar Kunala of Almac Group shows how developing robust analytical methods for biologics during early-stage development is a crucial but a challenging task.
26 How to Develop a Successful in vitro Screening Strategy
Developing a drug can take years and comes with significant costs. During the early phases of this journey, it is vital to have a robust process in place to allow rapid and informed decision making. A fit-for-purpose drug screening cascade is core to this, and this system must have the capability to test your hypotheses in biological systems to ensure you stay on the right course. Stuart Thomson & Graham Trevitt of Sygnature Discovery shows how developing a dynamic,
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 1 www.international-biopharma.com
Contents
purpose-built, and robust in vitro screening cascade, encompassing both biology and DMPK, enhances the likelihood of discovering new drugs efficiently or making timely project terminations.
MANUFACTURING & PROCESSING
32 Accelerating Biologics Manufacturing: Strategies for Efficient Production Timelines
Optimised development and manufacturing processes that accelerate biologic production timelines while keeping costs down are critical to meeting the market demand. Streamlining drug product (DP) manufacture helps shorten timelines for biologic production, but it requires expertise and experience. As a result, an estimated 26% of biotech companies outsource activities to contract development and manufacturing organisations (CDMOs), forming a trusted partnership model to produce biologics. Jinhyeok Jeong and John Thomas at Samsung Biologics, explore the major areas of DP manufacturing that can accelerate timelines along with the necessary qualities that should be sought after in a specialist service-providing partner.
MARKET REPORT
36 How Can the Pharmaceutical Industry Overcome Barriers to Reducing its Carbon Footprint?
As a result of centuries of carbon and other greenhouse gas (GHG) emissions from human industrial activity, the Earth is already about 1.2°C warmer than it was in the late 1800s. Worryingly, atmospheric CO2 levels continue to rise despite efforts over recent decades to address the issue. The pharmaceutical industry has a key part to play in supporting the effort to tackle climate change by reducing its own carbon emissions. Åsa Bergström at Recipharm explores how pharmaceutical companies can address challenges to making a meaningful contribution to global carbon emissions reduction.
TECHNOLOGY
40 Innovative Laboratory Testing Methods for Clinical Monitoring of Cell Therapies Using Flow Cytometry ddPCR and qPCR Assays
Gene-modified cell therapy for the treatment of cancer and other diseases is a rapidly growing area of clinical research. The therapeutic use of engineered cells has necessitated the development of novel lab tests for assessing patient safety and efficacy. The rapid and continued growth of gene-modified cell therapies shows great promise for those suffering from both haematological and solid tumours. Dr. Chad Galderisi of ICON discuss that, to support that promise, biomarker assays will be needed to monitor cellular kinetics and safety, and patient selection assays may be needed for therapies designed to attack cancer cells with more unique targets.
52 Using a LIMS to Improve Bottom Line Profitability
Whether manufacturing cars, supplying clean water, or manufacturing pharmaceuticals, all repeated processes must include quality control to ensure products meet their defined requirements. Quality management is very often about measuring and minimising variance within and across batches to ensure adherence to acceptable limits. This in turn is all about keeping meticulous records, as well as managing and accessing that data. LIMS help QC laboratories adopt good manufacturing practice and ensure that procedures and standards are followed. Tim Daniels of Autoscribe Informatics provides examples of how the LIMS will help drive those standards, but there are many more aspects to both Matrix Gemini LIMS, and QC management generally.
APPLICATION NOTES
44 Advancing Diagnostics with the o~pal Reader
The nature of the o~pal rapid test system and the digitalised service, captures test results at the point of the sample collection so anonymised health information can be recorded at scale. This capability extends to a wide range of applications, including inflammation detection, female health, and hormone testing, and acting as a measure around antibiotic resistance. Paul Ko Ferrigno, et al at e’clateral discuss how, applying these capabilities across many other areas of health and wellness whilst bringing clinical grade diagnostic tests into the home.
47 Monodisperse PEGs for Therapeutics
In the world of therapeutics, several challenges have hindered development and overall success of drugs and vaccines. One pressing issue is the short half-life of therapeutics, with drugs rapidly breaking down and being eliminated from the body. Maintaining drug stability during storage and administration is also crucial as many compounds. Addressing these problems is essential for optimal therapeutic outcomes and clinical approval. Erik Agner of Polypure discuss a strategy for increasing the stability of drugs before and after administration. This process has been critical in new drug discovery as well as in generating new therapeutics from previously known drugs.
57 Via Nova Therapeutics Uses CDD Vault to Support Global Collaboration
Collaborative Drug Discovery provides a modern approach to drug discovery informatics that is trusted globally by thousands of leading researchers. Our CDD Vault is a hosted informatics platform that securely manages both private and external biological and chemical data. It provides core functionality including chemical registration, structure activity relationship, inventory, visualization, and electronic lab notebook capabilities. Benjamin R. Taft at Via Nova Therapeutics discusses how they were able to achieve centralized data repository needs with CDD Vault – without having to invest in infrastructure.
60 Antibody Isotypes – The Devil in the Details
The immune system serves as the body's primary defence mechanism against a wide array of infectious agents, ranging from bacteria and viruses to parasites and fungi. It is an intricate network of specialised cells, tissues, and molecules that work in concert to recognise and eliminate foreign invaders while preserving the integrity of the host organism. Kevin Bittman of Sengenics explains that circulating antibodies are among the first manifestations of disease, furnishing clinicians with unheralded details about patient status, often before symptoms. Identifying isotype and antigen specificity provides highly specific metrics for detecting and understanding disease pathology that allows clinicians to develop tailored strategies.
66 Reagents for Biomedical Research
PELOBIOTECH GmbH is a supplier of reagents for biomedical research. With a focus on primary cell culture the Munich Cell Specialist distributes one of the broadest portfolios of cell culture products & media from their exclusive partners all over the world. The portfolio currently offers 5,000+ products in the field of human &animal primary cells, adult stem & iPS cells, media & tools. Dr. Lothar Steeb & Dr. Peter Frost of PELObiotech explain the offerings of customised solutions with a choice of prime media in R&D, GMP-like and GMP quality, and keep an eye on our xeno-free, serum-free and defined solutions.
2 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Contents
nature innovation + our reputation

We are Scientific Protein Laboratories. With over 40 years of expertise in development and cGMPcompliant manufacturing, we have become a trusted global source for innovation, customization, and the manufacturing of high quality API’s and naturally derived pharmaceutical products.
• Custom process development and formulations

• Traceable supply chain of natural ingredients

• Scale up and cGMP production

• Worldwide regulatory support
• Decades of experience manufacturing naturally derived materials including heparin and pancreatic enzymes
Put our quality team to work on your product solution. Visit
splpharma.com
700 E. Main Street Waunakee, WI 53597 USA
+1 (608) 849-5944
us at CPhl Worldwide 2023 in October in Barcelona. Booth #7J60
Scientific Protein Laboratories LLC part of Shenzhen Hepalink Pharmaceutical Group Co.,Ltd.
I was writing this foreword whilst travelling home from a conference about Oligonucleotide and mRNA-based drugs and vaccines. During the presentations and the conversations that subsequently followed at coffee breaks, there was a wave of optimism of a ‘we-can-do-this’ attitude among all participants, it was truly inspiring!
I think this new optimism has infected the complete industry and this is reflected in one of the first articles of this issue. Jeff Briganti of Aldevron, LLC, gives a very comprehensive overview over rapid development in the pharma landscape from past achievement like monoclonal antibodies which, I believe still have a great future ahead, and the awe-inspiring development in the fields of gene and cell therapies. Along with new therapies come development in analytical techniques and compliance strategies to safeguard quality, efficacy and safety.
In this we-can-do spirit there are six articles talking about how we can do things better to save resources and time. Drug development often starts with a set of screening assays, enabling a fast and reliable elimination of unsuitable candidates and a selection of druggable compounds. Stuart Thomson & Graham Trevitt of Sygnature Discovery, shows strategies to set up robust in vitro screening cascade to make timely and informed decisions about possible drug candidates. Dean Hatt of Broughton looks at the biopharmaceutical development process and how we have gone about this in the past and what we can learn from this for the future. On a similar note, Jinhyeok Jeong and John Thomas at Samsung Biologics explore how to accelerate the biologics manufacturing process. They propose to leverage on the experience and expertise of experts like CMOs, CDMOs and CROs. The use of such organizations frees up capacity which would otherwise be used to fulfill tasks which are absolute routine to expert organization. An important tool to integrate different organizations along the value chain are LIMS systems. They can help to speed up the data transfer in which, e.g., CROs directly report in their clients LIMS instead of issuing reports from which information needs to be transferred and
IBI – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Senior Medical & Regulatory Writer, Clarivate Analytics
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organization (WHO) Expert in ethics
• Hermann Schulz, MD, Founder, PresseKontext
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
reformatted. Apart for that fact that this is not a very rewarding task, it also bares the risk of errors and misinterpretation of results. Tim Daniels of Autoscribe Informatics gives an insight into different LIMS applications and their value. Taking this idea one step further are global collaborative drug development programs. This requires the possibility to share and securely store a wide range of information from different sources.
Benjamin R. Taft at Via Nova Therapeutics explains how the use of CDD Vault supports their global collaborative drug discovery projects. Further on in the topic of how things can be improved, is an article by Erik Agner of Polypure who talks about monodisperse PEGs for pharmaceuticals. Anyone who has to analyze PEGylated proteins or other PEG-based drug substances and excipients knows the challenges these polymeric substances create in QC analysis. Of course, PEGs with high heterogeneity and batch to batch inconsistency may also have implications on drug stability and efficacy.
With all the hype about new drug modalities and their capability to treat and possibly heal diseases, we should not forget the importance of diagnostics to identify diseases and monitor disease progression. Paul Ko Ferrigno, et al at e’clateral discuss how the o~pal rapid test system and digitalized service can be used to collect and analyze a multitude of health-related information for individuals and how this can be used to monitor the overall health status. Kevin Bittman of Sengenics explores how naturally occurring antibodies in patients can give insight to disease onset, progression and even may be used to personalize therapies. Dr. Chad Galderisi of ICON discusses how Cytometry, ddPCR and qPCR Assays can be used to measure biomarkers to test gene and cell therapies and how these techniques can be applied to individualize these therapies.
I hope you all enjoy this issue of IBI, and I look forward to perhaps meeting most of you at CPHI in Barcelona, Drug Discovery at Liverpool and DDL in Edinburgh.
Dr. Steven A. Watt, Head of Business Development at A&M STABTEST GmbH
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Lorna. M. Graham, BSc Hons, MSc, Director, Project Management, Worldwide Clinical Trials
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, Asia-Pacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc

4 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Foreword
We have helped life science companies raise over $200 M by delivering high-end visuals for science and medical communication with investors, clients and patients. We transform your technology into visual stories that make impact!






















INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 5 www.international-biopharma.com www.reciprocal.space VISUALS FOR BIO
MED · TECH
·
PITCH DECK AND DATA PRESENTATION
DESIGN
AND INFOGRAPHICS
AND VIDEO
3D AND 2D ANIMATION
WEBSITE
ILLUSTRATIONS
BRANDING PHOTOGRAPHY
Cloud-based Drug Discovery Technology
Q: What are some of the key challenges scientists face in early-stage discovery projects?
A: In drug discovery, the overarching challenge is to discover and bring to market a safe, effective drug in the most efficient way, minimising the time, resources and money required. For early-stage discovery, this means identifying compounds with the right balance of properties for a particular project. It also means prioritisation, knowing when to drop a compound and where to focus efforts. These challenges are compounded by the limited data available: typically sparse, highly complex and noisy. The expanse of chemical space and numerous properties of interest make it nearly impossible to explore every parameter for every compound.
On top of these scientific challenges come the more general challenges associated with a drug discovery career. For example, how to communicate your complex results or how to work in an interdisciplinary team with lots of different technology, software and data. Researchers face a wide range of challenges.

Q: Optibrium offers a range of drug discovery software, such as StarDrop and Cerella. How are these enabling scientists to overcome today’s challenges?
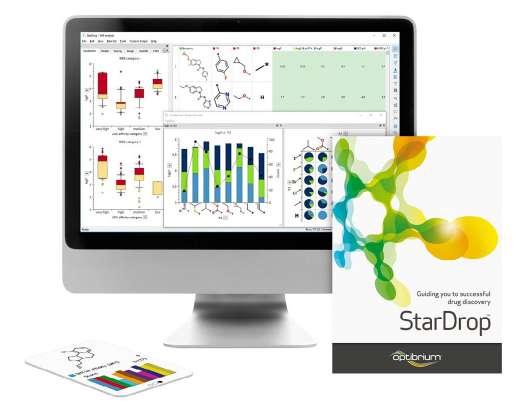
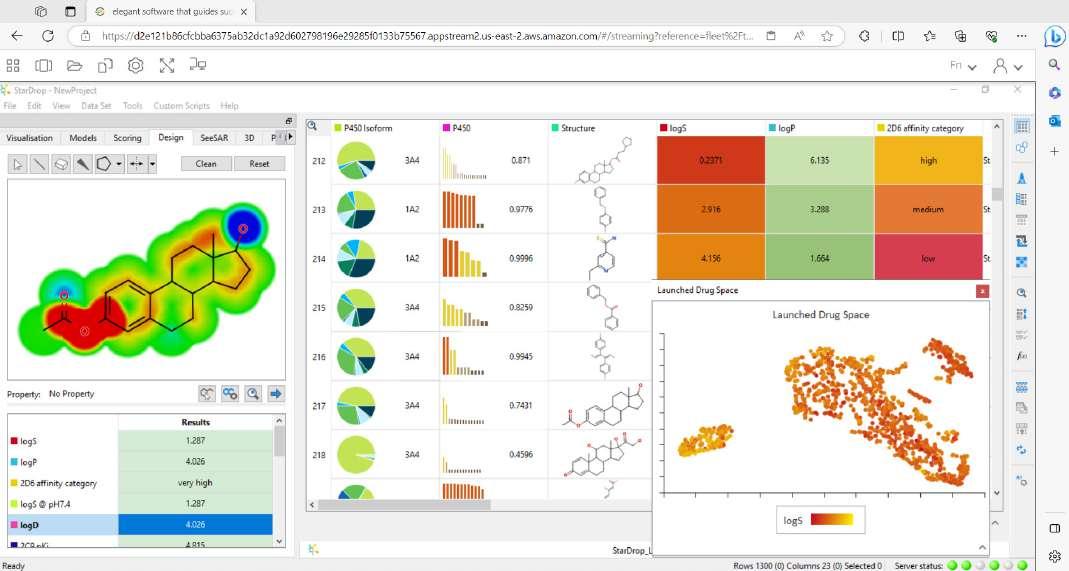
A: Everything we develop, we create with discovery scientists in mind. StarDrop™ was built as a comprehensive discovery platform for small molecule design, optimisation and data analysis. StarDrop’s core features include Probabilistic Scoring, an approach designed to support multi-parameter optimisation across the many properties a drug designer needs to consider. Unique visualisation abilities, including Card View® and the Glowing Molecule™, allow researchers to better see structureactivity relationship (SAR) information, with additional built-in SAR analysis tools to support this exploration.
StarDrop provides a seamless workflow from predictive modelling to generative chemistry to 3D drug design and more, with optional modules extending its capabilities, so scientists can tailor the platform to their specific needs. This is all within a highly visual, user-friendly interface to make use as easy and efficient as possible.
We have integrated StarDrop with many other platforms, so researchers can have access to everything they need in one smooth workflow. These include external platforms, such as
CDD Vault or PostEra’s Manifold, and in-house software, such as Cerella™, our AI drug discovery platform.
Cerella helps overcome the problems associated with limited experimental data by using deep learning imputation. It augments the user’s data, filling in gaps in sparse data sets and providing estimates of uncertainty, so users can identify the compounds which are most certain to be successful in their project. These uncertainties are extended to the experimentally measured data too, to highlight potential outliers or erroneous values, which should be rechecked; these values may represent hidden opportunities. Cerella works across multiple complex endpoints, capturing relationships where other methods fail, and can be applied at any scale, from individual projects to global data repositories.
Q: Optibrium is taking StarDrop to the cloud in a new and innovative way. How will this development benefit your customers?
A: The approach we are taking in making StarDrop accessible via the cloud ensures that our customers will be able to access the same level of rich interactivity as in the desktop application, but with flexibility, from any internet-connected computer, anywhere. StarDrop in the cloud customers will be using exactly the same software as our desktop users; only we’ll be hosting it for them.
Data security is one of our top priorities, and we chose Amazon Web Services (AWS) to host StarDrop in the cloud due to their world-leading security model. All data are protected behind a firewall and encrypted, meaning files can be uploaded and downloaded securely. Our ISO 27001-accredited information security management system also gives our customers confidence that their confidential information is safe and secure.
6 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Talking Point
Figure 1: StarDrop, Optibrium’s comprehensive platform for small molecule design, optimisation and data analysis
IBI in conversation with Optibrium on cloud-based discovery technology, and how continuous developments to its StarDrop platform are enabling scientists to tackle today's challenges, with powerful software for predictive modelling, 3D drug design and beyond.
A major benefit of this innovation to our customers is that StarDrop deployment and support will be much simpler for users, with customers automatically upgraded upon each release and Optibrium responsible for all maintenance too. This will significantly reduce the cost of ownership for our customers, enabling them to focus on the science.
Q: What other scientific developments are Optibrium releasing this year?
A: One particularly exciting impending release is StarDrop’s Metabolism module, the culmination of more than six years
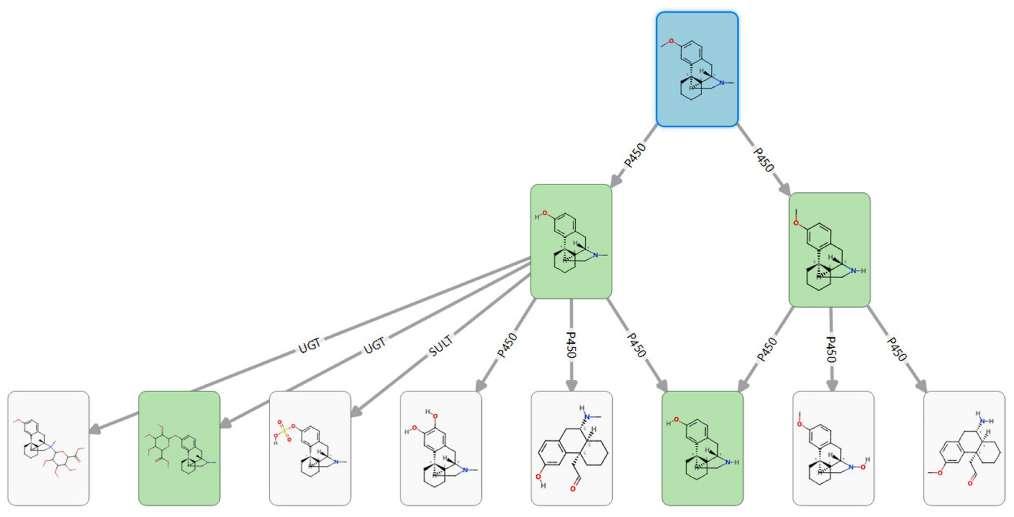
of research and development into drug metabolism and how to predict it. With around 40 % of late-stage drug failures being attributed to ADME issues, predicting drug metabolism can bring huge benefits in terms of cost, time and resource savings. The Metabolism module contains predictive models covering 80% of human Phase I and 60% of human Phase II metabolism, supporting the identification of which enzyme is most likely to metabolise a compound and at which site. Heuristics combining our quantum mechanics and machine learning models enable the identification of multiple generations of metabolites with precision, minimising overprediction to suggest the most likely experimentally
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 7 www.international-biopharma.com Talking Point
Figure 3: Metabolite pathway for Dextromethorphan (in blue), with arrows identifying the enzymes responsible for metabolism. The green cards are the experimentallyobserved metabolites
Figure 2 : Screenshot of StarDrop in the cloud, accessed via a computer web browser
observed metabolites. In addition, models for rat, mouse and dog cytochrome P450 enzymes help users to choose the most appropriate species for preclinical studies.
Q: What do you predict for the future of cloud-based drug discovery software?
A: Cloud-based drug discovery is the future, and as computing power and technology advances, I only expect cloud platforms to become more prevalent.
It is in the nature of the game; cross-disciplinary collaboration accelerates discovery and partnerships from academia, industry and beyond need to come together to tackle common goals. Therefore, having platforms with flexible global access to enable collaboration will only become more important. As well as accessibility, the flexibility of the cloud to scale resources on demand, whether handling large volumes of data or performing complex calculations, also opens up new possibilities to put
the analyses and results into the hands of decision-makers more efficiently.
Q: Beyond StarDrop in the cloud, what’s next for Optibrium?
A: We have a lot of exciting developments in the pipeline. As mentioned earlier, exploring chemical space to find a compound with the right balance of properties for a project can be a real challenge. However, generative chemistry is evolving at speed to tackle this head-on and is a field Optibrium has been involved in for many years, as exemplified by our Nova™ and Inspyra™ software. You can expect to see some innovative artificial intelligence-driven generative chemistry updates coming from us soon. We’ve also been thinking about how best to facilitate drug discovery workflows for project teams and have some exciting ideas around tracking project progress and easing and improving collaboration to make drug discovery more efficient. Stay tuned!
Ed Champness
After graduating with a degree in Mathematics in 1995, Ed joined GlaxoWellcome working as part of a pioneering team building predictive pharmaceutical tools. He developed the first graphical user-interfaces for working with predictive models which were adopted globally within GlaxoWellcome. He was a core member of the team which established the UK operation of Camitro in 2001 and remained with that company (now operating within BioFocus DPI following merger and acquisition) until 2008. During this time, he designed and built the StarDrop software and, in 2009, co-founded Optibrium.

8 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Talking Point
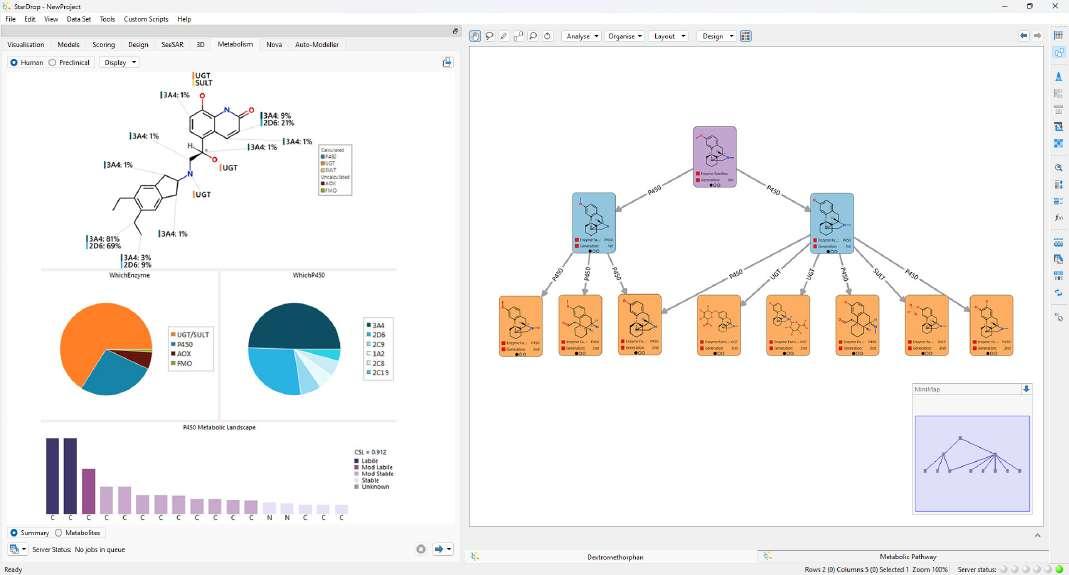
Figure 4: StarDrop's Metabolism module, with labelled molecules depicting the sites of metabolism for different enzymes, pie charts showing which enzyme families and isoforms are most likely to metabolise the compound and a metabolic pathway diagram in Card View.
Figure 5: StarDrop enables a seamless flow from the latest data through to predictive modelling and decision-making regarding the next round of synthesis and research
Global CRO: Pioneering Your Journey From Discovery to Delivery
• Central Lab: Kitting, Logistics, and Biostorage With Virtual Sample Inventory Management
• Biospecimens: Curated Inventory and Analysis With Advanced Biobanking
• Preclinical: Advanced Cell-Based Assays and Target Validation
• Genomics: Single-Cell to Multiplex Studies
• Bioanalytics: Immunogenicity and PK Testing
• Immune Monitoring: Comprehensive Cell Phenotyping and Profiling
• Tissue and Liquid Biopsy: Rare Cell and CTC Isolation and Analysis
• Clinical Trials: Design, Strategy, and Full-Spectrum Support

• Diagnostics and CDx: Regulatory Consulting, Companion Dx, and NGS
• Data Sciences: Biometrics and Biostatistics via QuartzBio® precisionformedicine.com
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 9 www.international-biopharma.com
Data Sciences Biospecimens, Biostorage, Sample Protection IV, Transfusion Administration Clinical Trial Support Commercialization Circultaing Tumor Cells (CTCs) Protein Molecule DNA RNA Assays Flow Cytometry Sample Collection Sample Collection II Sample Processing Data Solutions Online Management, Tracking, and Reporting Biobanking Regulatory Global CRO Manufactiring Specialty Labs Central Lab Services Data Sciences Biospecimens, Biostorage, Sample Protection IV, Transfusion Administration Liquid Biopsy Tissue Custom Biospecimen Collections Circultaing Tumor Cells (CTCs) Protein Molecule DNA RNA Sample Collection Sample Processing Data Solutions Cells Clinical Site Training Global Shipping and Logistics Local Logistics Supply Chain Management Online Management, Tracking, and Reporting Global CRO Manufactiring Specialty Labs Central Lab Services Companion Diagnostic Blood, biofluids and derivatives Kitting, Custom Kit Production Liquid Biopsy Tissue Custom Biospecimen Collections Cells Clinical Site Training Global Shipping and Logistics Local Logistics Supply Chain Management Global CRO Manufactiring Specialty Labs Central Lab Services Companion Diagnostic Blood, biofluids and derivatives Kitting, Custom Kit Production Liquid Biopsy Tissue Custom Biospecimen Collections Cells Clinical Site Training Global Shipping and Logistics Local Logistics Supply Chain Management Global CRO Manufactiring Specialty Labs Central Lab Services Data Sciences Biospecimens, Biostorage, Sample Protection Companion Diagnostic Blood, biofluids and derivatives Kitting, Custom Kit Production Liquid Biopsy Tissue Custom Biospecimen Collections Circultaing Tumor Cells (CTCs) Protein Molecule Sample Collection Cells Clinical Site Training Global Shipping and Logistics Local Logistics Supply Chain Management Online Management, Tracking, and Reporting Global CRO Manufactiring Specialty Labs Central Lab Services Data Sciences Biospecimens, Biostorage, Sample Protection Companion Diagnostic Blood, biofluids and derivatives Kitting, Custom Kit Production Liquid Biopsy Tissue Custom Biospecimen Collections Circultaing Tumor Cells (CTCs) Protein Molecule Sample Collection Cells Clinical Site Training Global Shipping and Logistics Local Logistics Supply Chain Management Online Management, Tracking, and Reporting Biospecimens Specialty Labs Companion Diagnostics Clinical Trials Central Lab Services Data Sciences
Rapid Evolution of the Biotech Industry: 2023 and Beyond
The Fast Pace of Biotech
The biopharmaceutical industry has embraced the adage of “Don’t be afraid of change, welcome it.” In doing so, the industry has fuelled a staggering transformation over the past 20 years. Extremely complex nucleic acid molecules and recombinant proteins with highly defined and functionally sensitive characteristics have been scaled to thousands of litters in commercial-scale bioreactors. The emergence and commercialisation of “living medicines” have allowed reprogramming patient’s T cells ex vivo to exhibit drastic tumour regression and long-term tumour surveillance, developed gene therapies to reverse monogenic diseases that otherwise have little or no treatment options, and fast-tracked RNA-based vaccines at a scale never attempted to overcome a global health crisis.
Accompanying this exponential growth in developing novel therapeutic modalities, there appears to be a dramatic shift in the underlying occupational market structure. In the past, graduate students who successfully defended their Ph.D. thesis were expected to enroll for additional training as a post-doctoral researcher that would eventually catapult their career to a tenured position within a prestigious university or see them placed in a commercial organisation. However, it now appears that both the National Institutes of Health (NIH) as well as other exclusive academic institutes are struggling to recruit and retain post-doctoral researchers. From 1990–1999, the number of postdoctoral appointees holding non-faculty research positions in science grew 29.8% and likewise, from 2000–2009 grew 27.7%.1 The following decade, 2010–2019, this metric shrank to a growth of only 3%.1 Even more alarming, a 4.1% reduction in postdocs was seen from 2020–2021.1
Similarly, trends in funding may also indicate a shift in the overall behaviour of the biopharmaceutical ecosystem. Large, more traditional multi-year grants such as the R01 and R21 are ideally suited for academic research, and over the years have served their purpose well. However, more novel mechanisms of funding research, specifically for the development of commercial technologies, have emerged and the frequency of these translational funding mechanisms has surged dramatically.
In fact, a recent study found that programs that include the Small Business Innovation Research (SBIR) and the Small Business Technology Transfer (STTR) programs have provided small businesses with what amounts to 60–65% of the overall U.S. seed funding.2 Other translational private funding mechanisms, such as angel investors and venture capital (VC), have also surged in popularity over the past decade. In 2016, VCs invested in 2,200 biotech startups globally.3 In 2021, the number of VC−funded biotech companies jumped 40%, with an estimated raised capital of more than $34 billion globally.3
The role of large biopharmaceutical companies has shifted along with these changing times as well. Even with considerable fiscal budgets to spend, large pharma is not leading the pack in this massive push of innovative drug development. In the case of Pfizer, 34 products developed by third parties accounted for 86% of their revenue generated in 2019.4 Likewise, at Johnson & Johnson, 16 of their 18 portfolio products were not internally developed, which accounted for 89% of their $31.4 billion revenue.4 So, who is leading the pack in developing all these innovative, revenue generating drugs?
The answer to these three seismic shifts in the biotech industry is emerging tech companies, which are companies with less than $200 million in annual R&D expenditures, and annual sales less than $500 million. These companies were responsible for 67% of approved drugs in 2022.5 This figure is up by 4% compared to the prior year, and up 33% from a decade earlier.5 All in all, this staggering innovation push from biotech companies has been steadily increasing year over year at a rate of 4% and is expected to rise as high as 80% within the next five years.5
The Innovators
This rapidly evolving landscape is ultimately shifting the identity of the movers and shakers of the industry. Fuelled by these changes in funding mechanisms and uninhibited access to freshly graduated doctorate-level talent motivated by perks not heard of in academia, small biotechs cumulatively form the innovation engine that is driving this drastic global market transformation in the biopharmaceuticals industry. These emerging biotech companies share a somewhat common lifecycle and the desire to pursue exit strategies that are advantageous to them, big pharma, as well as the public.
Small biotech companies commonly spin out of universities or are created through licensing of a significantly enabling technology from a university or research centre.6 At this stage, many are funded by more traditional grant mechanisms or pre-seed VC investment. Their focus is on conducting significant preclinical research and development work to define their agent’s mechanism of action in vitro with the goal of conducting subsequent in vivo studies in animal models to test specific target disease applications.
A few years into this stage, where their technology is still under development and traditional grant resources have typically been exhausted, companies require additional investment to fill the financial gap and provide runway to move their technology forward. Emerging biotech may receive additional SBIR/STTR funds but are increasingly likely to seek angel, pre-seed, or seed round VC investments because these funding mechanisms can be more substantial, timelier, and often come with business advisement as well as liquid funds. As a measure of how prevalent this is, $21.0 billion in angel and seed funds were invested across an estimated 7,261 deals, and
10 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Regulatory & Compliance
PEER REVIEWED
a total of $68.4 billion was invested by early stage VCs in 2022.7 These investments, and often later round investments (i.e., B and C rounds), are typically used by companies to generate sufficient pre-clinical and Phase I clinical data to attract large scale investment.
Regardless of where they find funding, the focus of an emerging biotech is now on generating a product worthy of investing in a clinical trial while being conscious of both timeline and scalability. At this point, they must consider if they take this all on themselves or enlist the partnership services of a contract development and manufacturing organisation (CDMO). However, they get there, once enough promising and enticing clinical data has been generated to start convincing the market that the technology can deliver on the promises of their drug candidate, the time has arrived to consider and execute an exit strategy.
The emerging biotech may choose to fund later phase clinical trials of a single candidate or fund a therapeutic pipeline while continuing to grow organically. This strategy typically involves making an initial public offering (IPO) to finance the overarching goal of taking drug products to market. Alternatively, and not atypical, the company may choose to exit through a relationship with large pharma. Typical outcomes include partnering, licensing, or full acquisition by a large biopharmaceutical company that will have available resources to enable completion of later phase clinical trials and subsequent commercialisation activities.
Even though this emerging biotech lifecycle has a high rate of attrition, several recent acquisitions, such as the Asklepios Bio acquisition by Bayer, the AveXis acquisition by Novartis, and the Translate Bio acquisition by Sanofi, suggests that this model is successful enough to win continued attention from both VCs and big pharma. A proof-point of the popularity and viability of this approach is the fact that in 2019 the 10 largest biopharma merger and acquisition (M&A) deals were $200 billion in value.8 In addition, these M&A and partnering activities with small biotech companies have been increasingly used as a growth and sustainment strategy by large pharma. For 2023, this sustainment strategy, combined with a marketinduced resetting of biotech valuations, is expected to drive a big year for M&A activities.8 Further evidence of this strategy by large pharma is the growing number of corporate and academic partnerships leveraged for drug discovery. Here, academic labs are essentially serving the strategic role of a biotech for big pharma. From 2012 to 2016, the number of large pharma academic collaborations has more than doubled to over 25,000.9
The Journey of an Innovator
With the goal being IPOs or attracting the attention of a suitor from large pharma, the road to biotech success is riddled with potholes, hurdles, and dead ends. Many biotech companies fail on their journey from conception to partnering with large pharma for multiple reasons and, unfortunately, good science doesn’t always translate to the successful management of a program.10 Clinical failure because of not meeting primary end points in a study or excessive adverse events can mean certain death for an emerging biotech. Chemistry, manufacturing, and controls (CMC) issues that can manifest in drug product
heterogeneity or limited drug product availability can also cause companies to fold. In other instances, failure to meet the expectations of the regulators can delay, or all together prevent, clinical studies and push a small company past the reach of its cash runway. Finally, in the case of angel investors or VCs, primary investors may withdraw funding for a variety of reasons, causing cash shortages and failure.
With all the looming obstacles they face, emerging biotechs are motivated to be extremely reactive and agile as speed is the key to success. Time is of the essence since the market and the players know that the attrition rate is high. The C−suite executives of these companies are almost always themselves innovators and early adopters of new technologies, with strong entrepreneurial tendencies and habits. The technical teams they assemble are also highly translational and entrepreneurial. The facilities where these small biotechs operate are usually extremely modest with limited wet lab capabilities. Select companies are fortunate enough to operate out of startup incubators that can offer critical core services including flow cytometry, mass spectrometry, and other analytical methods that typically require dedicated staff and facilities to operate. Others are completely virtual and totally lack wet lab space. All in all, these groups are not stereotypically investing in long-term commercialisation activities, but instead are focused on short-term milestones and goals that will hopefully deliver the next round of funding to lengthen their cash runway and thus ensure survival for a while longer.6
CDMOs – an Enabling Resource for Emerging Biotech
Within the described startup lifecycle, these agile emerging biotech companies will at some point need to consider investing in the infrastructure required to scale up, manufacture, and commercialise their innovative drug. Typical facilities and expertise in the areas of process development, manufacturing and quality, and regulatory support will all be required to progress the drug through clinical development. Thus, a critical choice must be made to either build each of these capabilities (staff, equipment, and facilities) inhouse or to buy it through establishing relationships with contract development and manufacturing organisations (CDMOs), such as Aldevron.
Smaller biotech companies, who are already facing a fair amount of risk, are increasingly gravitating toward establishing relationships with CDMOs to improve the probability of success, regardless of intended exit strategy.11 This trend toward partnering with CDMOs is evidenced in the forecasted growth of CDMO revenues, which are expected to surpass $46 billion by 2025, an increase of more than 40% from 2020.12 From a risk-mitigation perspective, CDMOs can prevent common issues in both the preclinical and clinical phases of development. In the preclinical phase, processes that are manufacturable and compliant can be efficiently and rapidly scaled up. From a manufacturing perspective, CDMOs offer state of the art facilities and equipment to ensure manufacturing efficiency, consistency, and ultimately successful release of their client’s drug product. Operating and maintaining these facilities takes significant resources that an emerging biotech does not have to fully support. As a result, the cash runway can be significantly extended to maximise the chances of survival and eventual success. In addition, small companies may lack adequate experience in all areas of drug product
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 11 www.international-biopharma.com Regulatory & Compliance
manufacturing. Thus, leveraging the history of success of a CDMO can facilitate product development through accessing deep expertise and end-to-end capabilities in all facets of drug development. Whether it be through regulatory support during agency engagement, or through accessing advisory support during different phases of product development, CDMOs can offer extensive advice to their clients to avoid potential pitfalls and delays. Finally, establishing a relationship with a CDMO can increase visibility to potential buyers in addition to making the acquisition process smoother overall.
An experienced CDMO partner with varying capabilities can provide tangible value for any company developing a novel nucleic acid-based therapy by sharing valuable lessons learned over the course of hundreds or thousands of clinical trials. Aldevron, for example, has produced more than 165,000 lots of plasmid DNA with nearly 5,000 of them produced at Good Manufacturing Practice (GMP) or GMP-Source® quality over the course of 25 years. Since 2017 Aldevron has also produced more than 70 GMP lots of mRNA drug substance. Altogether, this translates to supporting clients though more than 1,100 clinical trials, forging an understanding of regulatory agencies, fostering the development of new innovations, and incorporating the newest state-of-the-art technologies, which translates to enhanced capabilities for the development of new therapies through the clinical trial path. Where else can an emerging biotech find this level of expertise, experience, and support?
Summary and Conclusions
Novel therapeutic development is moving fast and the biotech industry as a whole is rapidly evolving. Small agile biotech companies are attracting a plethora of new doctorate-level talent and are being sustained through multiple avenues of novel funding mechanisms geared at early phase investment. As a result, emerging biotechs are now the leading innovators of the industry, creating the next generation of life-saving medicines. The life cycle of these companies is equally fast paced with their success dependent on them demonstrating value before their funding is exhausted. Given their focus on demonstrating value quickly to attract additional funding or an exit, these biotech companies are either seeking partnerships with pharma companies in their development, or leveraging the experience, facilities, and expertise of CDMOs to improve their chances of success of drug product commercialisation. Partnering with the right CDMO, such as Aldevron, can provide access to the industry-leading facilities and expertise, which can reduce cost and timelines in multiple facets of product development.
REFERENCES
1. Survey of Graduate Students and Postdoctorates in Science and Engineering: Fall 2021. 2023.
2. David Lee ES and NG. SBIR/STTR Grants: Introduction and Overview [Internet]. 2019 [cited 2023 May 17]. Available from: https:// academicentrepreneurship.pubpub.org/pub/1elox915/release/3
3. Olivier Leclerc MS and LT. What are the biotech investment themes that will shape the industry? [Internet]. 2022 [cited 2023 May 17]. Available from: https://www.mckinsey.com/industries/life-sciences/ our-insights/what-are-the-biotech-investment-themes-that-willshape-the-industry#/
4. Emily H. Jung AE and ASK. Do large pharma companies provide
drug development innovation? Our analysis says no [Internet]. Stat. 2019 [cited 2023 May 18]. Available from: https://www.statnews. com/2019/12/10/large-pharma-companies-provide-little-newdrug-development-innovation/
5. Gabrielle Masson. “Stunning” 4% yearly rise in R&D share has emerging biopharma dominating pipeline [Internet]. 2023 [cited 2023 May 17]. Available from: https://www.fiercebiotech.com/ biotech/emerging-biopharma-companies-dominate-rd-pipeline22-iqvia-finds
6. Tajonar A. How to start a biotech company. Vol. 25, Molecular Biology of the Cell. American Society for Cell Biology; 2014. p. 3280–3.
7. Q4 Venture Monitor [Internet]. [cited 2023 May 18]. Available from: https://nvca.org/wp-content/uploads/2023/01/Q4_2022_ PitchBook-NVCA_Venture_Monitor.pdf
8. Eric Sagonowsky KDFKALZB. The top 10 biopharma M&A deals of 2022 [Internet]. 2023 [cited 2023 May 18]. Available from: https:// www.fiercepharma.com/pharma/top-10-ma-deals-2022

9. Crew B. Top 5 corporate–academic collaborations in biomedical sciences [Internet]. 2019 [cited 2023 May 23]. Available from: https://www.nature.com/nature-index/news-blog/top-corporateacademic-collaborations-biomedical-sciences
10. Dutton G. Good Science Doesn’t Guarantee Success: Why Some Biotech Startups Fail [Internet]. 2022 [cited 2023 May 18]. Available from: https://www.biospace.com/article/good-science-doesn-t-guaranteesuccess-here-s-why-some-biotech-startups-fail/#:~:text=Biotech%20 startups%20fail%20for%20a,several%20unanticipated%20 commonalities%20still%20remain.
11. Kurata H, Ishino T, Ohshima Y, Yohda M. CDMOs Play a Critical Role in the Biopharmaceutical Ecosystem. Vol. 10, Frontiers in Bioengineering and Biotechnology. Frontiers Media S.A.; 2022.

12. Joachim Bleys EFHM and LT. CROs and biotech companies: Fine-tuning the partnership [Internet]. 2022 [cited 2023 May 18]. Available from: https://www.mckinsey.com/industries/life-sciences/ our-insights/cros-and-biotech-companies-fine-tuning-thepartnership
Jeff Briganti
Jeff Briganti is Sr. Director, Global Strategic Marketing for Aldevron, LLC. He is responsible for developing and executing strategies to promote Aldevron’s market-leading custom development and manufacturing of plasmid DNA, RNA, and protein, supporting cell and gene therapies. Briganti joined Aldevron in 2021, bringing nearly 30 years of experience. He spent the previous 10 years at Thermo Fisher Scientific, in various roles in Molecular Biology and Molecular Spectroscopy, most recently as Director of Strategy and Business Development, Molecular Biology. He has an MBA and a BS in Genetics, both from the University of WisconsinMadison.
12 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Regulatory & Compliance

• • • S • • •
A Summary of the Pharmaceutical Development Process, the Changes and Challenges and Future Opportunities
In this white paper, Broughton summarises the ‘historical’ development plan for pharmaceuticals, the changes that have occurred with regards to differential development including the requirements for biologicals, the typical challenges the industry faces now and, in the future, the opportunities this brings.
This white paper forms a simplified reference point for developing pharmaceuticals both now and the future and some of the choices/challenges companies have.
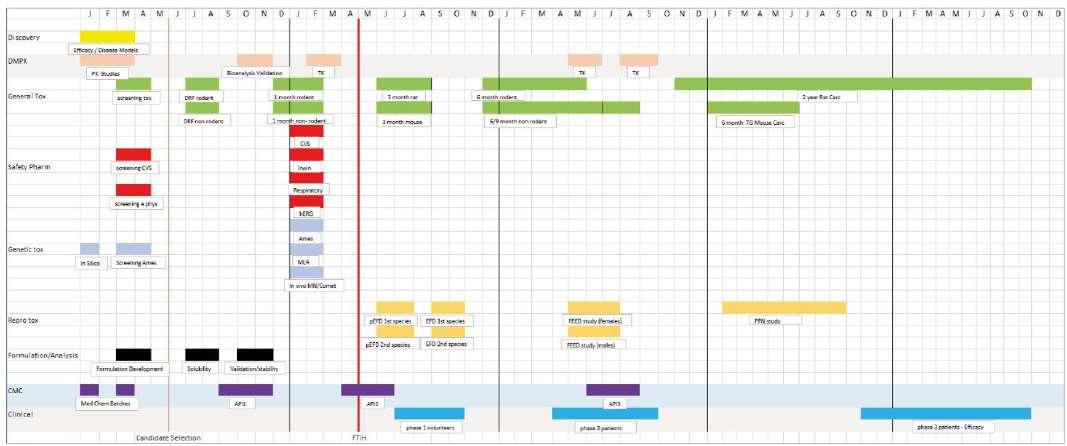
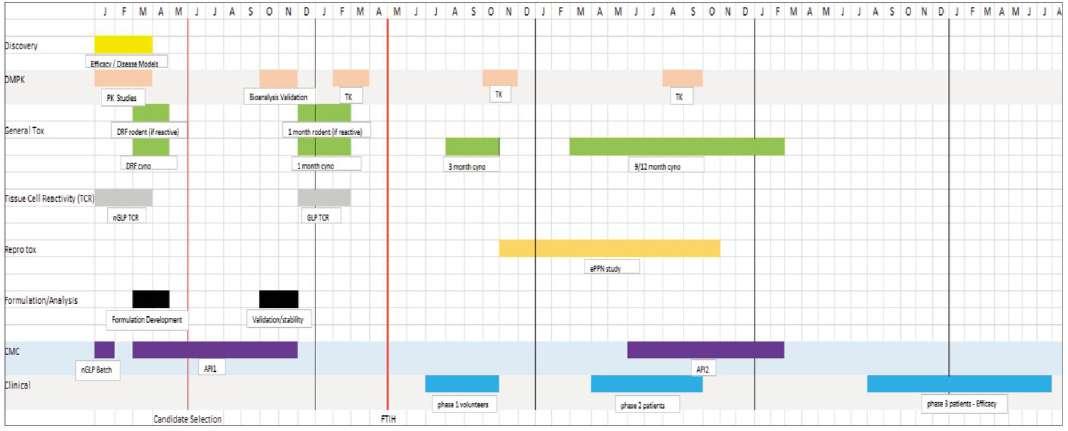
Many scientists working within the pharmaceutical sector over the past 30 years will recognise the typical pre-clinical safety development plan (see figure 1).
The duration of discovery activities is defined by the speed of success and the amount of screening conducted, therefore they are included here for completeness only. Once promising targets are identified via efficacy studies, disease models and Pharmacokinetics (PK) studies, the initial safety screening occurs with in-silico structure activity modelling and in vitro mutagenicity assays to candidate selection with in vivo screens to identify target organ toxicity and cardiovascular endpoints.
Once candidate selected, and in many cases, this may involve multiple compounds of the same class, development begins in earnest with dose-range general toxicology studies re-assessing the previously identified target organ toxicity with an aim to defining early exposure safety margins in both rodent and non-rodent species. Concurrently, costly drug synthesis is green lit alongside key formulation development activities, analytical method development and validation exercises. To coincide with the arrival of the GMP drug substance with appropriate
certificate of analysis, the safety assessment studies comprising a battery of general toxicology, safety pharmacology, genetic toxicology and formulation stability will commence to support entry into the clinic. The readout of these studies, subsequent regulatory submission and internal safety board swiftly leads into the first clinical trial, typically in healthy volunteers.
The development of a drug does not cease during these short First Time In Human (FTIH) trials, moving into early reproductive toxicology studies looking at Embryofoetal Development (EFD) and also starting longer term general toxicology which not only serves as a means to further investigate target organ toxicity and margins of safety but also to act as dose range finders for the lifetime carcinogenicity studies, the protocols for which require advanced Food and Drugs Administration (FDA) approval. Chronic toxicity studies in rodent and non-rodent species continue the comprehensive safety assessment battery of studies alongside and coinciding with fertility studies in both male and female rats and also phase 2 clinical trials, which are multi layered trials in patients. Lifetime carcinogenicity studies in rats and mice (the latter now benefiting from a shorter 6 month study in transgenics) commence alongside a multi generation peri and postnatal development study which now concludes the reproductive and developmental process from pre-mating to sexual maturity of offspring.
Once the phase 3 efficacy clinical trial commences, the road to Marketing Approval Application (MAA)/New Drug Application (NDA) submission and marketing is almost over, assuming proof of concept and no carcinogenicity is proven, not withstanding the timely and arduous regulatory submission process.
The Challenges
The timescale for this development process is rarely as little as the 5.5 years illustrated in Figure 1 since the key phases and
14 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Research / Innovation / Development
Figure 1. Classic Development Plan for new medicine
decisions made during discovery, candidate selection, FTIH and prior to each large spending milestone (carcinogenicity studies, phase 2, 3 clinical trials) often require high boardroom-level cost-benefit discussions which prioritise a portfolio leading to acceleration or deceleration of the process for a particular drug. This timescale also assumes timely method development and validation of analytical work, drug synthesis, stability, analysis and release and no additional studies, investigations or delays due to toxicity. A popular school of thought suggest if the development has no challenges, the drug’s efficaciousness will!
This is where the first dichotomy begins, do you try to maximise return on investment by speeding up the development plan thereby beating competitors to market and maximising the patency for exclusivity or do you take the conservative approach, preventing costly delays to studies or cancellation charges thus minimising unnecessary spend on new drugs that may never make it to market?
Pharmaceutical companies have attempted to answer this question by the landscape over the last 20 years. The main change has been selection of the in vivo Contract Research house (CRO) more often, which at its best involves using experienced ‘contractor’ staff only when you need them thereby freeing up your own staff for the more complex or confidential science. This also leads to minimising the spend and maintenance on building new labs and animal houses, that you would rarely fill. At its worst, it involves an equally expensive monitoring oversight ensuring the CRO’s run the studies exactly as the sponsor wishes, and ultimately as there is greater reliance on CRO’s who largely now own the expertise, demand has driven the price upwards and consequently, the CRO availability, not to mention animal availability, has reduced. Ultimately, there are often now delays waiting for studies to start at a CRO which may have been prevented if they were conducted in house. This is the second dichotomy. Do you save ‘apparent’ costs and staff by outsourcing your most resource intensive work but in doing so lose control of the timing, or do you keep control of the timings yet potentially have empty buildings and under-utilised staff? A flexible model, that allows studies to be conducted in house when CRO slots do not exist seems a logical move but one that pharmaceutical companies have largely opted against.
To overcome this, is to plan well ahead of time and selecting carefully your CRO partners (preferred suppliers) that can give you those study slots at a cost you have planned for and at a time you need them. Herein lies the bigger problem. There is always a delay somewhere in the development process. Whether it’s a funding decision, which is heightened when you are paying an external customer, an ongoing toxicology investigation, a problem with the drug manufacture, release or analysis or more recently, the availability of animals, delays are inevitable. In vivo CRO’s and manufacturing/analytical CRO’s also need to avoid empty space, so will typically charge delay/ cancellation fees or at best, re-use your study slot for another client. So having back-up CRO’s with a good understanding of the development pitfalls is a must.
Despite these challenges, the outsourcing strategy has gathered pace with a greater desire to externalise many more activities to reduce internal burden. However, these
decisions are often taken within departments, at a level where inter-departmental needs are not balanced against intra-departmental gain. This has resulted in expertise being lost from the pharmaceutical companies and further delays as the lead time for some activities can no longer be conducted instantaneously.
Gaps in the Pharmaceutical Armoury
The space between Safety Assessment and Pharmaceutical Development is a clear example of gaps in the pharmaceutical armoury. Having expertise and industry-leading technologies to develop clinical formulations is pivotal to pharmaceutical companies, however, this is prioritised significantly higher than expertise required for developing pre-clinical formulations, which are typically very different.
Developing a poor pre-clinical formulation can lead to de-selection of a perfect drug candidate and the subsequent loss of profit to the pharmaceutical company and potentially the loss of clinical benefit to the patient. It also leads to ethical concerns with adverse clinical effects on the animals and ultimately wasted and repeated studies. Developing a pre-clinical formulation uses a different set of skills to develop a clinical formulation, as you have to consider the needs of the studies to be supported e.g. dose versus dose volume versus concentration versus duration, the nuances associated with each pre-clinical species (pH, osmolality etc) and the capabilities of each CRO. You also need to understand specifics like scalability, compatibility with infusion equipment or perhaps just importance of short-term stability knowing when each formulation will be dosed. Most importantly, you must understand the toxicity of excipients in each species by each route and in combination with each other. Every excipient is both safe and lethal in equal measure, it’s just the quantities that determine where on that spectrum you sit. By developing a good formulation, you get to maximise the power of each study, optimised enough to provide the required exposure margins, without having any excipient toxicity, but crucially, being simple enough that it is easily transferable and not unnecessarily arduous to prepare.
Some clinical formulations are the same as those used pre-clinically, e.g. biologicals, long acting injectables. However, it is imperative that conversations between the biologists and chemists occur to ensure clinical formulations are fit for purpose for the needs of the pre-clinical studies.
Similarly, when it comes to issues with the test substance and documentation, the acceptability of impurities and degradation products (including nitrosamines and extractables and leachables), the knowledge surrounding salt (counter ion) selection, not to mention endotoxins (for biologicals), it is unclear where the responsibility lies. An example of this is with the development of a nitrate salt. This may provide for a stable and cost-efficient drug but when it causes renal toxicity not associated with the active substance, those decisions seem poor. It should also be noted that many challenges exist that are associated with the regulations. The in vivo CRO’s work to GLP and whilst the assumption from chemists that GMP is of a higher standard than GLP is often made, the differences are only realised when comments are made by the regulatory agencies (FDA/Medicines and Healthcare Regulations Agency (MHRA))
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 15 www.international-biopharma.com Research / Innovation / Development
detailing deficiencies and why a study will need to be repeated. GLP and GMP are different standards both utilised for specific purposes.
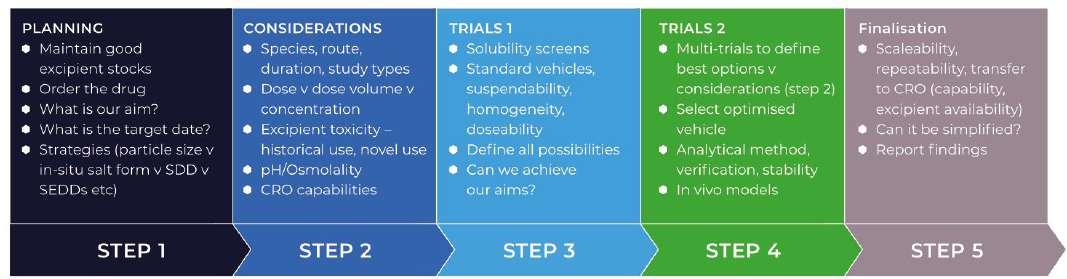
In figure 2 below, you will see a step-wise approach to pre-clinical formulation design, demonstrating the need for a comprehensive safety assessment and GLP knowledge.
Another couple of examples also demonstrate the need for a biological mindset over a chemical one.
Inhalation characterisation, which is required prior to costly pre-clinical inhalation studies to ensure accurate dose delivery, whether it is a dry powder system where the selection of packing pressures, blend strength or canister size determining the aerosol concentration or a liquid formulation where the concentration and constituents determine the durability in solution in a pressurised nebuliser system; both require deep knowledge of the delivery systems and dosing practicalities to ensure success.
Another example is the development of a hERG in vitro formulation. Considering the aim to maximise exposure, the use of various acceptable solvent/buffer systems and the fine line between a solution that has microscopically insoluble particles, hardly visible opacity or a concentration so low that insolubility is invisible, all at a concentration where stability is difficult to prove, understanding the aims of the study is of utmost importance.
In all these examples, it is also important to have a thorough understanding of both chemistry and biology needs to calculate drug requirements. Sufficient to successfully run each study with a sensible overage but not too much to make manufacture unnecessarily expensive and wasteful.
Strangely, whether its planned or unplanned delays to development, pharmaceutical companies when looking at the cycle time of a drug to market, overlook the reasons for each delay in favour of the means. These metrics point towards a lack of compliance in meeting study targets, however, a slightly deeper dive will reveal poor choices the companies have made.
Differential Development
One improvement over the past 10–15 years is the concept of differential development and the remarkably large reductions in animal use as a result of questioning study design. In some cases, whole studies have been removed, certainly shortened in duration, and in some case a study bolted on to another one.
The table below lists a few of these changes:
Study Type Description
Study Type Description
28-day general tox study
In vivo genotoxicity micronucleus/ COMET assays
Routinely now often 1–14 days depending on FTIH strategy and therapeutic use
Irwin study
Maybe combined with a CVS study with multiple endpoints
2-year mouse carcinogenicity study
Often now bolted on to definitive tox study
EFD/FEED (embryofoetal development/ fertility studies)
These maybe combined with the Peri and Postnatal study to create an ePPN reproductive tox and developmental study
Rat screening assay
Now universally replaced by a 6-month transgenic mice assay
Now doubled up to act as a rat range finding study
Dual routes of administration for general tox
Typically, the clinical route of administration plus a top up route to increase exposure
FEED (fertility study)
Often run as part of a general tox study
In essence, this concept of differential development has already been in place for anticancer drugs where late stage or advanced disease treatment morally outweighs the comprehensive safety assessments needed. This has resulted in the removal of studies to test for genotoxicity, safety pharmacology, reproductive toxicology and carcinogenicity.
Another change has occurred with the introduction and proliferation of biologicals including monoclonal antibodies, recombinant proteins, oligonucleotides, etc, where previously accepted convention is replaced with a bespoke fit for purpose safety plan. Here, drug product is often dosed intermittently and the need for in vitro assays/genotoxicity/carcinogenicity are unwarranted and safety pharmacology is assessed within the general toxicology study. An example of a development plan for these large molecules is shown below:
16 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Research / Innovation / Development
Figure 2. A Formulation Optimisation process
Table 1. Examples of Pre-Clinical Study enhancements
CONTRACT DEVELOPMENT AND MANUFACTURING OF BIOPHARMACEUTICALS
Richter-Helm is a Germany-based GMP manufacturer specialized in products derived from bacteria and yeasts, with a proven 30-year track record.

Count on us to flexibly provide a comprehensive range of services and customized solutions. Clients worldwide have already benefited from our commitment to good manufacturing practice and total transparency. Our work focuses on recombinant proteins, plasmid DNA, antibody fragments, and vaccines.
Richter-Helm consistently works to the highest standards of pharmaceutical quality.
Contact us
+49 40 55290-801
www.richter-helm.eu
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 17 www.international-biopharma.com
SERVICES AND
LEARN MORE ABOUT OUR
CAPABILITIES
MICROBIAL PRODUCTION? ARE YOU LOOKING FOR EXPERTS IN
Figure
Here, the discovery phase is most definitely longer than small molecules and the animal studies are largely restricted to species where the desired epitope is expressed. Where no suitable species exists, a relevant transgenic subset is selected as a surrogate. Suitability of species involves an in vitro tissue cell reactivity assay to define the relevancy and then an abridged plan developed, typically involving one species (commonly non-human primates) being dosed weekly or monthly (with immunogenicity as a prime focus) and a single extended pre- and post-natal development (ePPND) study in the same species handling the reproductive liability. Whilst the development plan has far fewer components, each study is more complicated and costly and the timelines are lengthened by the much longer biotechnology manufacture and testing times which can easily take 8–9 months.
Cell and gene therapy is another clear example where bespoke development plans are designed. Here, adenoviruses act as vectors to deliver the product or analogous substances into ‘appropriate’ animal species often genetically diseased to better understand the relationship of dose to activity and toxicity. Early communication with the regulators is of paramount importance here to ensure aligned thinking for studies to assess pre-clinical safety (USFDA, 2013).
In essence, there now seems to be a greater acceptance from regulatory authorities to accept differential development including hybrid or novel bespoke studies both in the name of reducing animal usage but also expedition of the process and the responsibility is now on the pharmaceutical company to navigate an appropriate path. A recent example of this is with long acting injectables where a monthly injection requires a case by case approach but one which bears no resemblance to any ICH guidance.
With this onset of fit for purpose drug development, it is important that the most appropriate regulatory submission pathway is also chosen. Whilst the hybrid (reg 52) or generic (reg 51) submissions are the clear alternatives to the full application (reg 50) in the UK, and the Abbreviated New Drug Application (ANDA) approach an alternative to the NDA in the US, less is known about facilitated regulatory pathways
(Fast Track (FT)/ Breakthrough Therapy (BTD)/Priority Review (PT) /Accelerated Approval (AA)) to expediate drugs with high benefit or ‘orphan drug’ status for those with high benefit yet low potential profitability, Innovative Licensing and Access Pathways (ILAP’s) to accelerate drugs to market for innovative medicines, those claiming ‘Well Established Use’ or those more commonly used for combination products or for ‘informed consent’ amongst others. An engineered development and submission pathway is a must to maximise a drugs clinical and earnings potential.
The Future
Very recently, in the US, the FDA modernization Act 2.0 has been passed which refutes the mandate for animal testing (PubMed, 2023).
Whilst there is no expectation this will lead to immediate wholesale changes to development plans, it does give companies the legal framework to challenge the regulators and replace animal testing with novel in vitro methodologies and artificial intelligence. Many of these in vitro models have been around a while, currently utilised alongside in vivo testing for screening and mechanistic investigations.
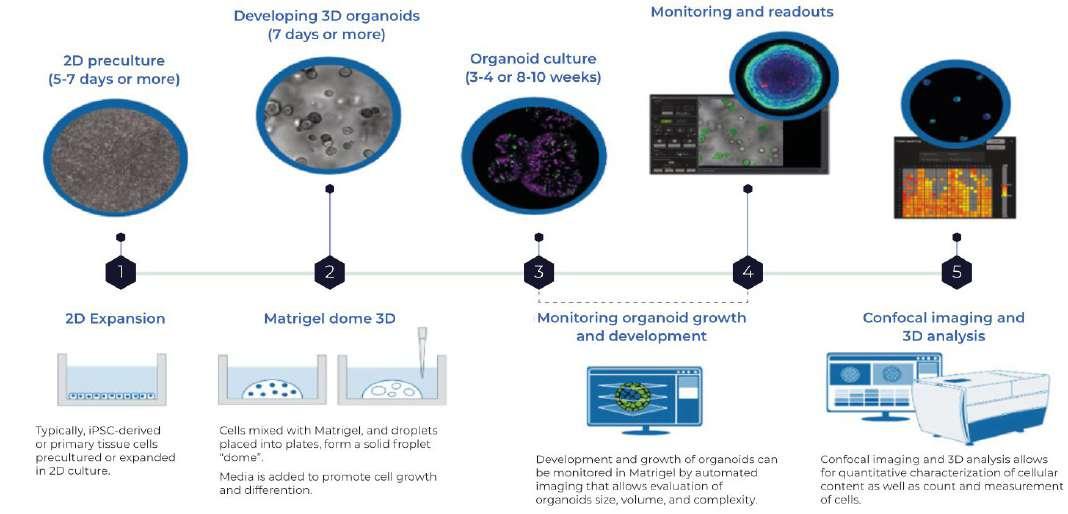
However, the change in the law may allow models to be accepted in isolation, where justified. An example of this is within the field of inhalation toxicology, where in vitro human models have been demonstrated to be better indicators of human toxicity than in vivo animal studies. The Epi airways and Immulung 3D models are clear examples of this. Alongside this are the advances in organoids, where embryonic or pluripotent stem cells create miniature but functional and complex organ systems. The diagram below shows a schematic of the process for an organoid system.
The advent of 3D organoids, spheroids, mini organ cultures and organ-on-chip MPS Micro Physiological Systems (MPS) only demonstrate the huge amount of research that is already happening in this field, and companies ready to understand the possibilities will also be those ready to reap the benefits. A human organoid re-creates the architecture and physiology of human organs in remarkable detail and therefore should
18 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Research / Innovation / Development
3. Development Plan for a biopharmaceutical
address the concerns that reliance on animal models on human diseases and treatment have (Kim et al.,2020). The schematic below shows the potential and direction of an array of organ-on-chip systems.
Summary
The drug development process has developed considerably over the years to meet the demands of the pharmaceutical company, regulators and the patient. The strive to outsource has gathered huge pace as an attempt to cut costs and free up resource, however, this has left some big gaps in expertise at pharmaceutical companies, most specifically where chemistry meets biology. The future of life after animal testing also begins here, it won’t be instantaneous, but it will ultimately be beneficial for the ‘forward-thinking’ companies and of course ethically. In addition, navigating the regulatory pathways and understanding how or when the regulators will accept the novel approaches will be key to obtaining regulatory approval quicker.
Here at Broughton, we have built a reputation for delivery of projects with high customer focus and satisfaction. We have a great team of consultants from toxicologists to chemists and regulatory experts, all willing to go the extra mile for our clients and modern GMP/GLP facilities for comprehensive testing. Knowing all the development options, pitfalls and
future opportunities and having expertise which fills the gaps between the pharmaceutical companies and the in vivo CRO’s, places Broughton at the forefront of these exciting times.
REFERENCES
1. PubMed. 2023. Han J. FDA Modernization Act 2.0 allows for alternatives to animal testing. 2023 Mar;47(3):449-450.doi: 10.1111/aor.14503. Epub 2023 Feb 10. https://pubmed.ncbi.nlm. nih.gov/36762462/.
2. USFDA. 2013. Preclinical Assessment of Investigational Cellular and Gene Therapy Products. https://www.fda.gov/media/87564/ download
3. Kim, J., Koo, BK. & Knoblich, J.A. Human organoids: model systems for human biology and medicine. Nat Rev Mol Cell Biol 21, 571–584 (2020). https://doi.org/10.1038/s41580-020-0259-3
Dean Hatt
Dean joined Broughton having enjoined a successful career in the pharmaceutical industry with over 30 years of experience with GlaxoSmithKline. Senior Toxicology Consultant; Formulation, Analysis and Test Article Expert Significant GLP, Safety and Controlled drug experience; Drug development expertise across all study types (in vitro/ in vivo); He started his career as an in vivo Toxicologist, becoming a Study Director, Study Monitor, and Project Toxicologist, before finally managing an industry-leading Formulation and Analysis department. He has extensive expertise in vitro and in vivo drug delivery including inhalation and drug development of both new chemical entities and biologicals. Has worked as a Project Specialist providing key opinions relating to test articles and off-target toxicity (quality, documentation, excipients, impurities, counter ions, etc.), to meet GLP regulatory guidelines and expectations. He is a problem solver with great attention to detail. A great communicator with a can-do attitude who is focused on meeting customer needs.

INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 19 www.international-biopharma.com
Figure 4. 3D Organoid Development
Research / Innovation / Development
Figure 5. Organ-on-chip Schematic
Developing Robust Analytical Methods
for Early-Stage Biologics
Biologics are a broad class of therapeutic agents, encompassing vaccines, monoclonal antibodies, therapeutic proteins, nucleic acid-based therapies, blood components, tissue therapies, and cellular therapies. Recently, the US Food and Drug Administration (FDA) revised the definition of term “protein” to include peptides containing more than 40 amino acids, therefore categorising them as biologics.1 The search for new and effective treatments for serious and life-threatening diseases has led to the growing interest in biologics in the last two decades. Biologics have shown promising clinical outcomes compared to traditional small molecules due to their high specificity, profoundly transforming the treatment strategies for cancer and autoimmune disorders. The COVID-19 pandemic has also accelerated the adoption of biologics within the healthcare sector. It is estimated that by 2027, the biologics market will have significant growth to $666 billion from $474 billion in 2023.2 This remarkable shift can be attributed to several factors, including the rising prevalence of chronic diseases, approval of several Advanced Therapy Medicinal Products (ATMPs), the increasing availability of biosimilars to patients, and the growing recognition of the benefits of biologics over small molecules. Emerging biopharmaceutical companies1 have also contributed to the growth of the biologics market. Approximately 65% of molecules in the research and development pipeline, including biologics and small molecules, are from emerging biopharmaceutical companies without the involvement of larger biopharmaceutical organisations. This share has seen a steady growth from 34% in 2001 to 50% in 2016, due to increased funding and investments.3
Biologics, however, present unique challenges in the journey from discovery to commercialisation due to their inherent complexity, size and charge heterogeneity and susceptibility to changes that could potentially impact efficacy and the patient safety. Therefore, development and implementation of robust analytical methods to monitor critical quality attributes (CQAs) during early-stage is vital for successful biologic development. At early-stage, analytical methods play an important role in candidate screening, process development, formulation screening, stability determination and release testing for first-in-human (FIH) studies. However, developing sensitive and robust analytical methods is challenging in the early-stage of biologics development as there is often insufficient product knowledge. The draft ICH Q14 guideline4 provides a comprehensive framework based on science and risk-based approaches to overcome some of these challenges and streamlines the post-approval change management of analytical methods. This article primarily focuses on developing analytical methods for release and stability testing of biologics, while developing methods required for characterisation is out-of-scope.
Analytical Target Profile (ATP) –Setting the Analytical Benchmarks
ICH Q14 defines the ATP as a “prospective summary of the performance characteristics of the analytical procedure with anticipated performance criteria to ensure the results are appropriate for the intended use.” In other words, the ATP is a pre-defined set of analytical benchmarks that a method should meet to be considered suitable for its intended purpose. The ATP is analogous to the quality target product profile (QTPP) described in ICH Q85 and help guide the selection of analytical technology and the method development process.
Product Development
Quality Target Product Profile (QTPP)
Critical Quality Attributes (CQAs)
Risk Assessment
Design Space
Control Strategy
Continued Process Verification
Analytical Method Development
Analytical Target Profile (ATP)
Critical Analytical Method Attributes
Risk Assessment
Method Operable Design Region (MODR)
Analytical Method Control Strategy
Continued Analytical Method Verification
Table adapted from reference6
The ATP is written based on the product's needs as summarised in the QTPP and the analytical knowledge based on the physical and chemical properties of the drug substance and drug product. However, during the early stages of biologic development, there is often insufficient knowledge about the product and analytical method benchmarks that need to be achieved. As a result, establishing an appropriate ATP can be challenging and will remain as a living document until more knowledge is gathered. Similarly, when changes happen to QTPP warranting improved benchmarks, the ATP should be updated to re-examine method selection. For example, establishing an ATP for CQAs such as dimer and oligomer species is less complicated than determining plasmid DNA isoforms, as there is more knowledge available in the public domain. Therefore, knowledge management becomes a crucial aspect of the method development process – knowledge regarding the physical and chemical properties of the drug substance and drug product is necessary.
20 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Research /
/ Development
Innovation
PEER REVIEWED




PHARMACEUTICAL INDUSTRY 21
Stage of Product – Phase I
Intended Purpose
Link to CQA
Plasmid DNA exists in several topological forms such as supercoiled, linear, and open circular. As supercoiled plasmid shows the highest efficiency in transfecting eukaryotic cells, the content of supercoiled plasmids becomes an important indicator of plasmid quality.
Characteristics of the reportable result
Characteristic Acceptance Criteria
Specificity
Linearity
Precision
Accuracy
The analytical method is specific for determination of plasmid DNA isoforms and shows no matrix interference from the formulation components. Any carryover observed should be less than 0.1%
The analytical method should exhibit linearity from 50% to 150% of nominal protein concentration
Supercoiled peak: ≤ 3.0% RSD repeatability
Linear or open circular peak: ≤ 0.4 SD repeatability
Total analytical error (TAE) ≤ 50% of allowable process capability to achieve Ppk 1.0 in QTPP range (SD 0.4%)
Supercoiled peak: 95.0 – 105% recovery
LOQ QL ≤ 1.0%
Risk Assessment and Technology Selection
Analytical method risk assessments are performed to identify and assess critical method variables and parameters that can impact the ATP. This assessment is highly analogous to identification of critical process parameters (CPPs) during process method development. The structured risk assessments provided in the ICH Q14 include Ishikawa diagrams, Failure Mode Effects Analysis (FMEA) and preliminary hazard analysis.
Technology selection is entirely based on the analytical method performance characteristics drafted in the ATP. If there are several technologies that meet potential requirements, factors including throughput, costs and knowledge of analytical technology should be considered. For the determination of plasmid isoforms, several techniques are available, such as slab gel electrophoresis, HPLC, and capillary electrophoresis.
However, capillary electrophoresis is the preferred technique due to the performance characteristics that meet criteria in the ATP.
Method Development Strategies
One of the primary goals of developing analytical methods for early-stage biologics is to determine specific quality attributes of the drug substance and drug product to ensure that they meet applicable standards of identity, strength, quality, and purity as defined in the QTPP. The analytical results generated are then used to support process development, formulation screening, shelf-life determination and demonstrate manufacturing consistency. A well-developed analytical method should achieve the required accuracy and precision in alignment with the Analytical Target Profile (ATP) to support product development.
22 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
An example ATP for determination of plasmid DNA isoforms
An example Ishikawa diagram for assessing risk determination of plasmid DNA isoforms
/
/ Development
Research
Innovation
The traditional (minimal as per ICH Q14) approach largely focuses on the determination of quality attributes the analytical method based on the iterative approach. However, the enhanced approach adopts the principles of Analytical Quality-by-design (AQbD) to identify and minimise factors that could potentially introduce variability into the final reportable result. AQbD begins with building specific objectives through sound–science and quality risk management to achieve the ATP method characteristics.
Many biopharmaceutical companies that develop and manufacture biologics still use the traditional approach to method development because they often lack the resources and knowledge using the AQbD approach.

Method Qualification and Validation
Method qualification is the process of determining whether an assay is fit for its intended purpose. It is typically not a pass/ fail process, but rather an iterative process of optimisation until the method meets the performance acceptance criteria defined in the ATP. In general, method qualification is carried out after determining potential analytical method parameters and before method validation. Typically, method qualification differs from validation in that qualification does not have pre-set specifications (acceptance criteria) and may not be carried out in a GMP environment. As a minimum, specificity, linearity, range, precision in terms of reproducibility, needs to be evaluated to ensure the method is scientifically sound. Method qualification is required for early clinical phases while assays intended to ensure patient's safety should be validated.
Method validation is the process of demonstrating that analytical procedures are suitable for their intended use. The term “validation” is specifically linked to the product release either lot-release or stability in a GMP environment. It accounts for strict pre-determined acceptance criteria and requires QA approval of parameters in alignment with ICH Q2R1.7 Validation involves documenting, using specific laboratory investigations, that performance characteristics of the method are suitable for intended analytical applications and are reliable. The acceptability of analytical data corresponds directly to the ATP criteria used to validate the method.
Stability-Indicating Methods for Biologics
Stability studies are an essential and vital part of drug development. A stability-indicating method is an analytical test that can detect meaningful changes in quality attribute(s) during storage. For cell and gene therapies, the assessment of product functionality/potency represents a stability indicating test method. They typically start at the preclinical stage and continue through Phase I–Phase III clinical trials to support formulation development, and to satisfy the regulatory requirements for clinical trials.
During early-stage development of biologics, degradation pathways are elucidated, and the drug substance and drug product are tested under various storage conditions to determine their shelf-life. To ensure reliable data interpretation, robust stability indicating methods are needed, which allow for the detection of physical, chemical, or microbiological changes that affect safety, purity and efficacy of biologics. To demonstrate a method is stability indicating, forced degradation
samples should be assessed during method qualification. If the biologic is found to be stable, spiking of known impurities or variants could be performed to demonstrate method suitability.
Reference Standard Qualification
In accordance with ICH Q6A guidelines, “a reference standard or reference material is a substance prepared for use as the standard in an assay, identification, or purity test. It should have appropriate quality for its intended use”. Reference standards are a critical component of a biologic development and are part of analytical control strategy. Due to complexity of biologics, a well characterised reference standard is essential and should be introduced early in the development program. The criteria for selection of reference standards are that they should be stable, uniform and represent the properties of test material with respect to the quality attributes.
Often primary reference standards are not available from an official recognised source and therefore “in-house reference standards” should be established. Recently, National Institute of Standards and Technology (NIST) has released an extensively characterised IgG1k monoclonal antibody called the NISTmAb reference material to represent an industry standard for evaluating the performance of methods that determine physicochemical and biophysical attributes of monoclonal antibodies.8
If the reference standards are not available from an official source, they should derive from the toxicology material and evaluated for its intended purpose using extended characterisation techniques along with release methods. However, the primary reference standard is usually manufactured from process performance qualification (PPQ) batches most importantly, from a batch representative of the pivotal batch. Pooling of batches may be used to represent an average of properties (mostly commonly for vaccines).
The qualified reference standard should serve as an analytical control to monitor method performance characteristics as defined in the ATP.
Conclusion
Developing robust analytical methods for biologics during early-stage development is a crucial but a challenging task. ICH Q14 guideline provides a detailed framework for this process. As the landscape continues to evolve for biologics, the principles of ICH Q14 will remain instrumental in advancing analytical methods and facilitating the development of safe and effective biologic therapies.
REFERENCES
1. Definition of the Term “Biological Product” – https://www. federalregister.gov/documents/2020/02/21/2020-03505/ definition-of-the-term-biological-product
2. The Global Use of Medicines 2023: OUTLOOK TO 2027. IQVIA
23 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
/ Innovation / Development
Research
Institute for Human Data Science, January 2023
3. Emerging Biopharma’s Contribution to Innovation. IQVIA Institute for Human Data Science, June 2022

4. ICH Q14 Analytical procedure development (pending publication)
5. ICH Q8 (R2) pharmaceutical development - scientific guideline
6. Verch, T., Campa, C., Chéry, C.C. et al. Analytical Quality by Design, Life Cycle Management, and Method Control. AAPS J 24, 34 (2022)
7. ICH Q2 (R1) Validation of Analytical Procedures
8. Schiel JE, Turner A, Mouchahoir T, Yandrofski K, Telikepalli S, King J, DeRose P, Ripple D, Phinney K. The NISTmAb Reference Material 8671 value assignment, homogeneity, and stability. Anal Bioanal Chem. 2018
9. A. Mire-Sluis, N. Ritter, B. Cherney, D. Schmalzing, and M. Blümel, “Reference Standards for Therapeutic Proteins,” Bioprocess Int., 2014.
Pavan Kumar Kunala is BioPharma Lab Manager at Almac Group and has a strong background in the field of analytical development for biologics, with more than 10 years of experience. In his previous roles at Intas Biopharmaceuticals and Pfizer, he was responsible for leading a team of scientists to support analytical method development, validation and overall CMC strategy for advancement of early, late and commercial biologics. Pavan holds a M.S. degree in Biological Sciences from Arkansas State University.

24 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Pavan Kumar Kunala
Different types of reference standards used throughout the lifecycle of biologic development, Image adapted from reference 9
Research / Innovation / Development
The biotechnology industry is poised to deliver amazing products and solutions that will dramatically change millions of lives for the better. Aldevron is ready.

As a globally trusted CDMO, our years pioneering ground-breaking solutions to advance biologic manufacturing for research use, as well as cGMP for clinical and commercial applications, has enriched us with a body of knowledge that is critical to support these next series of breakthroughs. Contact
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 25 www.international-biopharma.com
now and
Supplying what’s needed
for what’s next
day 23-ALD-AED-G2A
us to advance your program. +1 (701) 297-9256 | Toll-free (U.S. and Canada): +1 (877) 787-3362 aldevron.com/advancing-every-day 25 years advancing every
How to Develop a Successful in vitro Screening Strategy
Developing a drug can take years and comes with significant costs. During the early phases of this journey, it is vital to have a robust process in place to allow rapid and informed decision making. A fit-for-purpose drug screening cascade is core to this and this system must have the capability to test your hypotheses in biological systems to ensure you stay on the right course.
As you embark on designing an appropriate screening cascade, it is imperative to define the fundamental properties you seek in your prospective drug candidates. These attributes will significantly influence the selection of assays to be incorporated into your screening cascade. Questions such as the importance of selectivity for your target, whether you are targeting orthosteric or allosteric binding sites, the need for blood-brain barrier permeability, the specificity of target expression in certain cell types, and concerns about on-target toxicity must all be addressed early in the discovery process. Early discussions among your team are vital to ensure the right assays can be strategically implemented as your project progresses.
The primary objective of a screening cascade, or the design-make-test cycle, is to enable swift decision-making. Screening cascades evolve and adapt as molecules advance, and a cascade developed during the hit-to-lead phase may differ significantly from one in the lead optimisation phase. Nonetheless, at each stage, the cascade should swiftly provide answers to the most pressing questions facing the project team at that particular juncture. This rapid decision-making can accelerate compound progression, facilitate timely intellectual property filings for novel chemical matter, and, importantly,
lead to cost savings in the long run. Recognising when to halt a program and reconsider the strategy is as crucial as striving to achieve key milestones.
Screening cascades can vary widely depending on the stage of drug discovery. In the following sections, we will outline key considerations for constructing an effective and robust screening cascade, with a focus on the hit-to-lead phase, followed by insights into how screening cascades evolve during the lead optimisation phase.
Hit-to-Lead Phase
During the early stages of drug discovery, the primary objective is to identify one or ideally several chemical series that effectively engage the target in the desired manner. While these series may still have a long journey ahead before becoming drug candidates, they serve as the foundational starting points. Consequently, the screening cascade at this stage should be simple and facilitate rapid data generation. This enables the chemistry team to develop a strong structure-activity relationship (SAR) and a grasp of the drug-like characteristics of the initial chemical compounds. This understanding is vital for instilling confidence in the selected chemical series.
A typical cascade for this phase may include a primary assay, an orthogonal assay, a cell-based assay, and a selection of in vitro DMPK (drug metabolism and pharmacokinetics) assays. It's important to emphasize that these are guiding principles, and customising the cascade often becomes necessary to meet project-specific requirements.
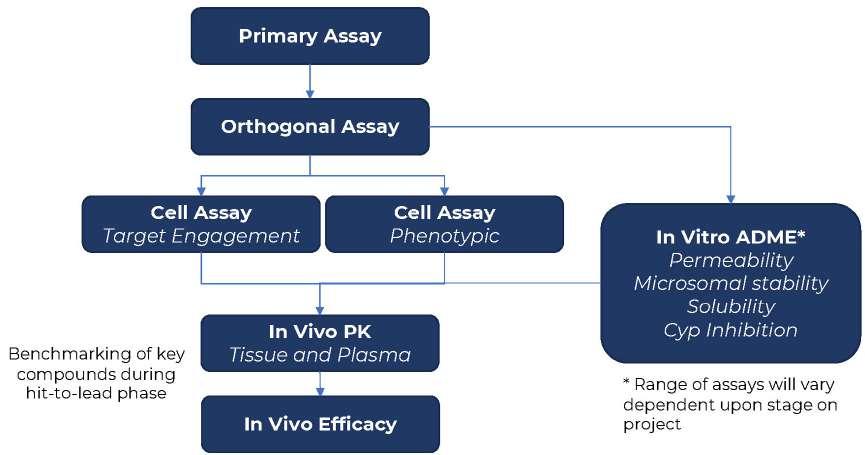
Primary Assay
The selection of an appropriate primary screen is pivotal to the success of a drug discovery project, as it serves as the initial
26 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Research / Innovation / Development
Standard hit-to-lead screening cascade
PEER REVIEWED
Recombinant Insulin for upstream processes
Enabling better medicines Enabling better lives
Grow your cells with Recombinant Insulin
As the leading supplier of Recombinant Insulin for serum-free and chemically defined cell culture media, we can help you keep development on track and production flowing.
Novo Nordisk Pharmatech A/S remains the best supplier of Recombinant Insulin – beginning with the quality of our Insulin sourced directly from Novo Nordisk, the world’s largest producer.

Manufacturing and quality control, precision delivery and a risk mitigation strategy that assures continuous availability are a few reasons why you should grow your cells with our Recombinant Insulin.
Learn more about our Recombinant Insulin at novonordiskpharmatech.com

Are you attending Cell 2023 in London? Visit us at booth 16

INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 27 www.international-biopharma.com
Primary assay selection considerations
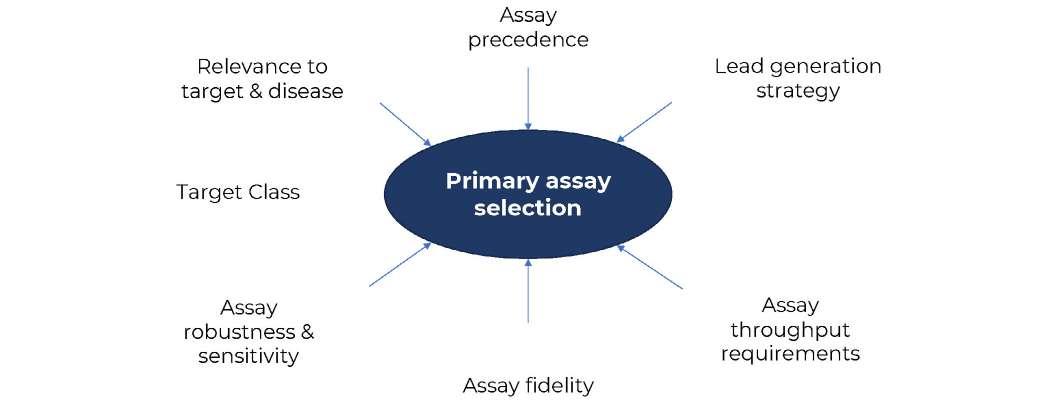
filter to identify compounds with potential therapeutic activity. Several considerations come into play when determining the optimal primary screen for a project. These considerations include the nature of the biological target, the degree of existing knowledge about target modulation, and the target's suitability for recombinant protein-based biochemical assays. Enzyme targets, for example, are often amenable to such assays. However, the choice of primary screen should align with the chosen hit-finding strategy. For instance, fragment-based lead generation typically requires biochemical or biophysical primary screens due to the need to test compounds at high concentrations.
Primary screen quality assessment is essential to ensure it serves as an effective initial filter. This involves evaluating assay sensitivity and specificity. A sensitive assay can detect subtle changes in compound activity, enhancing the likelihood of identifying hits. On the other hand, high assay specificity minimises the proportion of false-positive hits. Biochemical and biophysical screens are generally less prone to off-target effects compared to cell assays, but both types can be susceptible to compound interference mechanisms, necessitating a tailored triage strategy downstream of the primary screen.
To support high-volume screening, primary assays should be designed to be as simple as possible. The use of homogeneous assay detection technologies is advantageous in these cases, minimising the number of steps required and allowing for efficient miniaturisation.
In many instances, assays for the target of interest may already be available from previous screening campaigns or literature sources. However, these assays often require development and optimisation to suit the needs of a large-scale drug screening campaign. Parameters such as assay signal-tobackground, inter- and intra-plate signal uniformity, and overall sensitivity and specificity of the screen should be fine-tuned. While IC50 values are commonly used to assess compound potency, it's crucial to evaluate concentration-response curves comprehensively. Parameters such as top and bottom values of the curve, hill slopes, and compound solubility must also be considered. Ignoring these aspects can lead to misleading results and impact project timelines and costs.
Orthogonal Assays
Orthogonal assays serve two vital roles within a screening cascade: confirmation of primary data and generation of additional data that cannot be obtained in the primary assay format. These assays complement the data from the primary assay and build confidence in the results while providing further insights into the binding interaction. Orthogonal assays are instrumental throughout the drug development process, from initial hit identification to optimisation and characterisation of therapeutic candidates.
Orthogonal assays can take various forms, often being biophysical in nature to directly observe the binding of interacting molecules. While they generally have lower throughput than primary assays, they offer a deeper understanding of the structure-activity relationship (SAR) and behaviour of potential therapeutics. These assays also possess the ability to detect binding over a broader affinity range, from millimolar binders to picomolar interactions.
Confirming target engagement and generating affinity data using orthogonal techniques are essential to support IC50 data derived from primary assays. Some technologies, such as surface plasmon resonance (SPR), enable the determination of kinetic parameters and stoichiometry, providing crucial insights into binding modes and behaviour.
It's important to note that no assay is flawless, including orthogonal assays. While they offer valuable data, they still represent a departure from natural biological environments. This is where cellular assays come into play, armed with primary and orthogonal data, to further our understanding of how candidate molecules influence specific biology.
Cell-Based Assay
Gaining insight into cellular activity during the hit-to-lead phase is desirable. Developing a cell model that allows the evaluation of compounds in a relevant context is a pivotal component of the early drug discovery cascade. The choice of cell model and the endpoint to be measured should align with the target biology. For example, anti-cancer programs may seek a cell-killing phenotype, while those targeting inflammatory diseases may focus on cytokine profile modulation.
28 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Research / Innovation / Development
The complexity of the cell assay deployed at this stage should be considered in light of the need for rapid decisionmaking. Simplifying the system to provide information on cellular activity at weekly or bi-weekly intervals allows for timely decisions.
Cell activity can be measured as a phenotypic endpoint (e.g., cell proliferation) or through mechanistic assays confirming target engagement within the cell. Ideally, both types of assays should be included in the screening cascade, as they provide complementary insights.
Drug Metabolism and Pharmacokinetics (DMPK)
A successful drug candidate must balance biological activity and pharmacological effects with human pharmacokinetics to ensure adequate drug delivery to the target site for clinical efficacy. During the hit-to-lead phase, compounds from different series are assessed across a range of in vitro assays to understand the properties that need to be optimized in parallel with pharmacological profile evaluation.
Interpreting data from various assays during the hit-tolead phase is crucial for making informed decisions. Assessing properties like LogD7.4, metabolic stability, permeability, P450 inhibition, and solubility helps identify potential issues and prioritize series for progression.

The information gathered during this initial profiling phase informs the screening cascade in the lead optimization phase, focusing on properties with significant associated risks. Screening for permeability, for example, may be unnecessary if initial hits already exhibit high permeability, and the chemistry plan aligns with similar physicochemical characteristics. Flexibility is key, and adjustments should be made based on the project's specific needs.
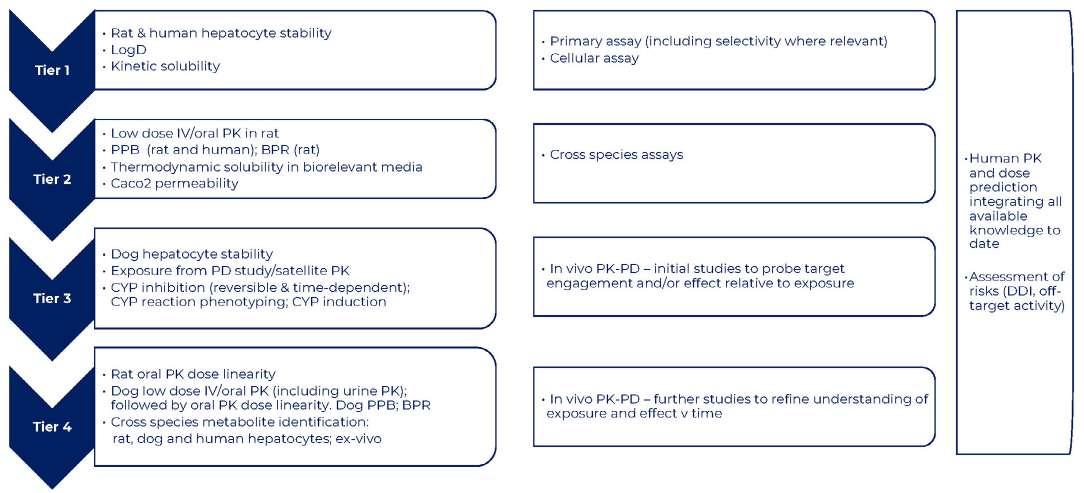
Lead Optimisation (LO) Phase
The transition to the LO phase occurs when one or more chemical series are considered suitable for progression towards drug candidate status. At this stage, there's a deeper understanding of the liabilities associated with each series,
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 29 www.international-biopharma.com
Research / Innovation / Development
Typical LO screening cascade
allowing for modifications to the screening cascade to prioritise these challenges.
While the primary assays developed during the hit-to-lead phase remain essential, additional assays may be introduced. These could include assessments of selectivity against closely related targets, cross-species evaluation, and more complex cell-based assays. The focus on the lead chemical series may evolve, and each series presents unique challenges, necessitating continuous evaluation and adaptation of the screening cascade.
In the LO phase, there's an increased emphasis on assessing the overall profile of molecules, beyond potency alone.
Data Analysis
With the substantial volume of data generated during the hit-to-lead phase, robust mechanisms for data analysis are essential. Correlation analysis plays a pivotal role in ensuring data consistency across different assay platforms. Correlations should be tracked between primary assays, orthogonal assays, and cell-based readouts to confirm that target engagement is driving the desired biological effect. Deviations from linear correlations warrant further investigation, as they may indicate issues with compound physicochemical properties or target inhibition strategies.
Bringing it All Together
Developing a dynamic, purpose-built, and robust in vitro screening cascade, encompassing both biology and DMPK,

enhances the likelihood of discovering new drugs efficiently or making timely project terminations. Efficient assays, co-located within the design-make-test-analyse process, provide scientists with the necessary data for informed decision-making. Drug discovery thrives on collaboration, and isolated efforts are more likely to falter. his is where expert input in DMPK becomes indispensable.
Stuart Thomson

Stuart is Director of Bioscience at Sygnature Discovery, a translatlantic integrated drug discovery CRO. He has 20+ years experience working in the UK and the USA for both pharmaceutical companies and biotechs focused on oncology drug discovery and translational research. Stuart holds a PhD in cell and molecular biology and has carried out post-doctoral research at the University of Oxford and King's College London.
Graham Trevitt
Graham is the Director of DMPK Projects at Sygnature Discovery, with over 20 years of experience in DMPK. His professional background includes eight years at UCB, five years at Almac Discovery, with academic studies at Imperial College London and a PhD in Organic Chemistry from the University of Nottingham.

30 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Research / Innovation / Development
Lyophilization and Sterile Manufacturing
Driving development and connecting commercialization of sterile and lyophilized drug products, we are dedicated to your success in bringing life-changing therapies to patients.


OUR END-TO-END BIOLOGIC SOLUTIONS INCLUDE:

• Sterile Formulation & Lyophilization Cycle Development
• Lyophilization and Sterile Fill-Finish Manufacturing
• Aseptic Robotic Technologies
• Analytical Support
• Clinical & Commercial Labeling & Packaging
• Refrigerated/Frozen Storage & Distribution
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 31
Your sterile drug product destination. Our world.
www.pci.com talkfuture@pci.com
Accelerating Biologics Manufacturing: Strategies for Efficient Production Timelines
The main aspects of streamlining manufacturing speed include:
Optimised development and manufacturing processes that accelerate biologic production timelines while keeping costs down are critical to meeting the market demand. Streamlining drug product (DP) manufacture helps shorten timelines for biologic production, but it requires expertise and experience.
As a result, an estimated 26% of biotech companies outsource activities to contract development and manufacturing organisations (CDMOs),1 forming a trusted partnership model for the production of biologics.2 Working with a biologics-focused CDMO partner gives companies access to specialist facilities and expertise, with the capacity to streamline and accelerate production timelines.
In this article, Jinhyeok Jeong and John Thomas, Senior Directors of DP Inspection & Packaging and DP MSAT, respectively, at Samsung Biologics, explore the major areas of DP manufacturing that can accelerate timelines along with the necessary qualities that should be sought after in a specialist service-providing partner.
Overcoming Challenges to Market
Developing a drug product is complex and comes with scientific, technical, and regulatory challenges potentially hindering progress to market. Companies are increasingly outsourcing activities to CDMOs to overcome development and manufacturing barriers. These barriers include scaling up biologic production while maintaining quality and the need for specialised equipment and expertise in drug product fill/ finish to ensure clinical studies are supplied with product on time, the PPQ batches effectively challenge critical process parameters, leading to commercialisation and continuous monitoring of commercial batches. Specialised CDMOs offer the proficiency and knowledge to overcome these obstacles, helping to streamline production processes. The capability of CDMOs to streamline workflows reduces production costs while increasing efficiency and capacity to help organisations gain a competitive market advantage.
The CDMO sector’s capabilities to handle increased demand in the future are reflected in the projected CDMO outsourcing market development, which predicts a compound annual growth rate of 9.2% and an increase of $287 billion between 2023 and 2030.3
A Strategy for Accelerated Timelines
In the highly competitive biopharmaceutical industry, bringing a product to market at speed positions an organisation for commercial success while potentially improving patient outcomes. To reach milestones quickly and demonstrate to investors the potential for return on investment, drug developers in DP manufacturing require a key set of attributes.
1. Effective Communication and Alignment of Goals
Engaging with a CDMO early in the development process ensures that goals are clearly defined, providing full awareness of the needs of the project and the desired product outcomes. Establishing a timeline that outlines the required capabilities and embeds strict milestones facilitates streamlined production and upholds project momentum.
Communication between the CDMO and the client should be built and baked into the collaboration from the beginning. Working together as a single unit allows production goal alignment and incorporates originator client and CDMO expertise into technical and strategic planning workflows, positioning the project for success.
2. Minimising Production Risk
When outsourcing biologic manufacturing, the early conversations help identify time constraints and process limitations that could lead to delays further down the development pipeline. Earlier gap assessment of components and processes enhances available processes and systems that are then gauged against production goals to identify any areas that require additional attention to detail.
Having a proactive approach and addressing these potential issues at an early stage allows for a mitigation plan to be put in place. Implementing preemptive strategies can prevent manufacturing challenges and outline actions that can be added to the manufacturing process to prevent roadblocks. With this plan in place, the risks of delays to drug production are significantly reduced.
3. Building a Solid Manufacturing Process
DP manufacture is complex, made up of numerous procedures and processes that need to run smoothly for accelerated production timelines. Ensuring that all essential factors have been optimised before manufacture can prevent potential delays. These considerations include:
• A robust supply chain: Reliable access to materials (such as raw materials, single-use items, primary packages, etc.) and the equipment required for production can minimise delays caused by long lead times.
• Prepared documents: All documentation should be ready early in collaboration with the client. Standardisation of documentation ensures that all GMP tech transfer activities are completed accurately and quickly to meet demanding timelines. At the same time recognising the drug being transferred to the CDMO may need specialised processes should be identified early in the process.
• Skilled workforce: A comprehensive training plan ensures that all staff from manufacturing, quality control/assurance to validation have been fully trained for manufacturing,
32 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Manufacturing & Processing
PEER REVIEWED
testing, and documentation filing required. Prior to manufacturing, additional training is often provided by DP MSAT to provide operator’s with a review of the drug (the why?) and specific action required per pre-approved protocols.
• Readiness of equipment: Pre-testing of all required equipment guarantees it will run as intended with the necessary materials for successful batch production.
The combination of measures to increase manufacturing speed helps to minimise potential bottlenecks, reduce delays and ensure efficient production processes.
4. Efficient and Timely Batch Release
To continue to meet stringent timeframes, once a commercial batch has been manufactured, it should be released within the defined time frame of 30 days. Quality control (QC) testing is completed to prove that a product meets the defined good manufacturing practice (GMP) guidelines before release, fulfills the client’s needs and is safe for patients. As part of this process, the quality and manufacturing teams review the master batch release (MBR) documentation, thus ensuring that a batch can be released quickly. Information spanning all processes and procedures, drug formulation and equipment details, including the electronic audit trail of all software systems used, should be included in the MBR documentation. Having all the required elements in place upon final DP completion means no time is wasted and documentation can be filed and a batch released immediately. Each commercial batch is reviewed by DP MSAT and the Continuous Process Verification (CPV) begins for each commercial batch, the Critical Quality Attributes are monitored to ensure the commercial production process remains in control.
5. Meeting Client Needs for Capacity and Flexibility
Having the capacity and capability to manufacture both the drug substance (DS) and DP at a single site can significantly accelerate drug production timelines. When this occurs, the DS can be conditionally released for continued manufacture and funnelled into subsequent processes for DP fill/finish, reducing the overall batch release timeline. As QC testing on the DS must be performed within 45 days of release, a small proportion of the DS batch could be held back for this testing while the remainder is used to complete the fill/finish process. Standard processing times in releasing the DS will work in concert with the progression to DP manufacture and release. The flexibility instilled through conditional release allows DS batch release and DP manufacture to run in parallel, helping to balance the demand for critical treatments with patient safety. The final batch release of both the DS and DP are often run in parallel to demonstrate the competitive advantage for end-to-end batch manufacturing.
6. Regulatory Excellence and Meeting Changing Regulations
Throughout manufacture, all processes and procedures must abide by regulations. To ensure regulations are met, CDMOs are required to integrate a strict quality management system (QMS) so that every raw material, component of drug production, and manufacturing process is well characterised and defined to meet all GMP regulations. Integration of QMS acts as a framework for quality, safety and efficacy, aiming to instil consistent and regulatory-compliant standards.
Part of the QMS is producing a quality policy, which is a statement that defines the organisation’s commitment to quality. Guiding decision-making and actions across the business, the quality policy outlines the overall quality objectives, guiding principles and values. A built-in and well established culture of compliance within a company demonstrates employees actions that embrace and strive for high-quality and regulatory-compliant products at all times.
As part of the QMS, regular audits are performed to highlight any areas for improvement, which are constantly under revision to ensure quality standards remain high. Having a QMS in place eases the process of proving regulatory compliance when bringing a product to market as quality is built into the manufacturing process, streamlining approval and timelines to market.
The success of bringing a DP to market and to patients rests on the ability of an organisation to navigate the ever-changing regulatory landscape. As demonstrated by the recent updates to Annex 1 of the GMP, which saw changes to the way sterile products are handled, regulatory guidelines are constantly revised to enforce the highest safety standards. An organisation must maintain regulatory awareness so that any changes to regulations are flagged early, allowing the time to incorporate any modifications into practice. Failure to respond could result in delays through non-compliance or lead to batch rejection. Outsourcing to a CDMO can provide regulatory expertise thanks to its experience in progressing DPs throughout the regulatory lifecycle.
Having regulatory expertise is a prerequisite for a CDMO to deliver client products on time. The regulatory landscape for drug development and manufacturing becomes increasingly complex as regulatory agencies continuously improve stringent requirements that evolve with the advancement of the sector. In-depth knowledge and compliance with guidelines are essential for a CDMO to ensure that necessary approvals and documentation are in place, minimising disruptions during the approval process. On-time delivery is of significant importance to many stakeholders:
• Clients: Ensuring market competitiveness and revenue potential by reaching the market on schedule.
• Patients and their families: Quick access to life-changing therapies that can improve outcomes and give family and friends reassurance of a patient’s prompt treatment.
• Healthcare professionals: Facilitating seamless healthcare planning and continuity of care.
The implementation of regulatory expertise and timely product delivery enhances patient well-being and meets healthcare demands effectively.
7. Experience and Expertise in Reaching Commercialisation Technology transfer is an essential process in preparing a production line for commercialisation, facilitating the transformation of small-scale production into scaled-up operations. As part of this strategy, an assessment of process needs evaluates the requirements for the successful transfer of technology. Early planning helps to identify and mitigate any potential roadblocks that could occur while introducing
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 33 www.international-biopharma.com Manufacturing & Processing
Manufacturing & Processing
progress which leads to on-time, right-the-first-time production capability, and mindset.
The Future of Biologics Manufacturing Through Collaboration


Forming a supportive and free-flowing communicative partnership with a CDMO can propel the manufacture of biologics. Access to expert knowledge and specialised equipment can be gained through collaboration with a CDMO, which helps streamline production processes. In addition, DP manufacture disruption risks are mitigated by forming a partnership early and proactively incorporating preventative measures.
changes. The insight provided by a CDMO with expertise in technology transfer can be essential in ensuring the transfer process runs smoothly.
Key assets a CDMO must have to bring DP success to clients include:
• Dedicated Single Use Systems designed to reduce timelines, define and harmonise cleaning validation evaluations, and smoother CTD Regulatory processes
• Fill-volume accuracy achieved through cycle development, engineering batches leading up to the GMP production process
• Process robustness based on MSAT experience leads to standardisation across product platforms.
Having a clear understanding of both the product and the process beforehand is essential as it can preempt any challenges that are identified when a project is in the early stages through the product lifecycle. The impact of successful technology transfer on biologics production cannot be overvalued, as a small change to manufacturing could result in unexpected impact on the product.
Comprehensive standard operating procedures are detailed guidelines that outline specific processes and protocols and can be invaluable for the efficient transfer of technology by providing a structured framework. This helps to ensure consistency, facilitate knowledge transfer and assure compliance with regulations through effective quality control. Comprehensive documentation also aids regulatory compliance and assures process traceability.
A CDMO should have the flexibility to scale up and scale down production based on the diverse needs of the client and the market. Building this flexibility into production lines is key for adapting manufacturing systems without hindering
Partnering with a CDMO can help drive forward biologics manufacture thanks to continuous innovation to accelerate timelines to market and gain a competitive advantage. Technological advancements aid accelerated production outcomes. Automated manufacturing and AI systems are streamlining processes and data analysis, providing real-time insights into procedures to continuously improve current systems and manufacture. Where manufacturing traditionally relied heavily on manual input, biologic production is being streamlined through the incorporation of technology into production systems. As technological advances continue, their impact on the pharmaceutical industry, with a particular focus on biologic manufacture, will keep accelerating.
REFERENCE
1. Contract Development and Manufacturing Organization Market Analysis (delveinsight.com) https://www.delveinsight.com/blog/ cdmo-services-and-major-players
2. Kurata H, Ishino T, Ohshima Y, Yohda M. CDMOs Play a Critical Role in the Biopharmaceutical Ecosystem. Front Bioeng Biotechnol. 2022 Mar 21;10:841420. DOI: 10.3389/fbioe.2022.841420. PMID: 34631751; PMCID: PMC9345064.
3. Global Contract Development And Manufacturing Organization (CDMO) Outsourcing Market (skyquestt.com) https://www.skyquestt.com/ report/contract-development-and-manufacturing-organizationoutsourcing-market
John Thomas
John Thomas, Senior Director of Drug Product MSAT, Samsung Biologics, has 24 plus years of industry experience in manufacturing across multiple manufacturing platforms including lyophilization, vial, PFS, PFS-S and PFP systems, automatic inspection processes, and blister-packing. Prior to his current role, he served as the Associate Director for Drug Product Project Management, External Manufacturing at Regeneron Pharmaceuticals and Senior Technology Transfer Process Engineer role at Fresenius Kabi USA.
Jinhyeok Jeong
Jinhyeok Jeong, Senior Director of Drug Product Inspection & Packaging, Samsung Biologics, has more than 19 years of experience in drug product operation, Before joining Samsung Biologics in 2012, he worked at Dongwha pharmaceutical company focusing on drug product and ointment.

34 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
MISSION
#OneStopShop for stability and release testing
Since 1995 A&M STABTEST has been a household name for highest quality analytical services “Made in Germany”. Our 340 strong team of dedicated specialists and stateof- the - art equipment get your projects done on time and in budget. We are your #OneStopShop solution for:

• Stability and release testing of small molecule and biological drug substances and drug products
• Analytical development and method validation

• mRNA -LNP ready methods acc. to draft USP guideline
• Dedicated cell-based potency assay group
• ICH- stability storage and logistics service



• Laboratory dedicated to inhaled drug testing (DPI, MDI, Nebulizer)

• GMP compliant LC -MS service for extractable and leachable testing, protein characterization, identification of unknowns and quantification of impurities and excipients
• Functional testing of container closure systems, prefilled syringes and injection devices (CCIT, force measurements)
• Analysis of ATMPs
A&M STABTEST; Kopernikusstrasse 6; 501026 Bergheim; Germany
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 35 www.international-biopharma.com
#CONTACT www.am-labor.de
anfragen.BM@am-labor.de
How Can the Pharmaceutical Industry Overcome Barriers to Reducing its Carbon Footprint?
Åsa Bergström, Director of Sustainability, Recipharm explores how pharmaceutical companies can address challenges to making a meaningful contribution to global carbon emissions reduction.
As a result of centuries of carbon and other greenhouse gas (GHG) emissions from human industrial activity, the Earth is already about 1.2°C warmer than it was in the late 1800s. Worryingly, atmospheric CO2 levels continue to rise despite efforts over recent decades to address the issue.1
To keep global warming under 1.5°C (to minimise the worst effects of climate change), in 2015, some 195 countries, including the European Union (EU), signed the Paris Agreement. In doing so, they committed to reducing their economies’ GHG emissions by 45% by 2030 and to reaching net zero by 2050.2
The pharmaceutical industry has a key part to play in supporting the effort to tackle climate change by reducing its own carbon emissions. However, this is easier said than done. There is more to addressing a company’s carbon footprint than drawing up a list of energy reduction targets for its own sites. Every aspect of a company’s operations and its supply chain needs to be explored to identify how CO2 and other GHGs can be removed from key processes, how to mitigate remaining emissions and where energy savings can be made. Moreover, the greater the coordination with the wider industry, the more effective efforts will be.
How can pharmaceutical companies minimise emissions? How can they set effective targets to make a meaningful reduction in their greenhouse gas emissions and how can they coordinate with other companies to amplify their impact?
The Importance of Target Setting
Multiple factors have made it pressing for pharmaceutical companies not just to set ambitious GHG emissions reduction targets, but to report on progress regularly.
National governments are drawing up domestic legislation demanding reductions in GHG emissions. This is needed to meet their commitments to the Paris Agreement and related international treaties, such as the Kigali Amendment to the Montreal Protocol to phase down the use of F-gases with high global warming potential (GWP).3 To keep operating within their home and overseas markets, pharmaceutical companies must revise their operations and governance practices to comply with these new regulations. Under upcoming EU legislation that will be in scope from 2024, companies will be required to report annually on their emissions, which increases pressure to take meaningful action to minimise them.
In light of this, increasing numbers of pharmaceutical companies are requesting that their suppliers in the value chain
set and report on strict targets to reduce their GHG emissions. Setting ambitious and measurable goals demonstrates to partners and customers, as well as investors, that they can rely on the company in question to help them achieve their own carbon-reduction targets and align with upcoming regulations.
In addition, reductions in an organisation’s GHG emissions can actually help minimise operating costs. By enhancing energy efficiency and reducing gas consumption, for instance, a pharmaceutical manufacturer not only contributes to a reduction in CO2 emissions but also saves money. In the EU, companies can save up to €120 in carbon tax for every tonne of CO2 they cut from their overall emissions.4
An often overlooked reason for setting more ambitious and measurable GHG emissions reduction targets is to continue to attract and hold on to talent. As in other areas of the global economy, the pharmaceutical labour market is increasingly dominated by Millennial and Generation-Z professionals. Large numbers of individuals in these cohorts are passionate about environmental and climate issues and want their employers to share these values. According to a recent report, some two-fifths of Millennials select new roles based on the company’s sustainability credentials.5 Failure to take this into account could mean a company loses out on the best talent in the future.
The Pitfalls Facing Companies in Achieving Paris Agreement Targets
There are several key challenges that may prevent companies from fully playing their role in achieving the Paris Agreement goals. These include:
Uncertainty over what precisely qualifies as a company’s own GHG emissions: It remains the case that pharmaceutical companies often do not understand the scope of their own GHG emissions, meaning many organisations underestimate their impact. Many still consider the emissions from their own operations – their facilities and their own transport networks –to be the full scope of their carbon footprint.
Every part of the value chain contributes its own emissions, from external suppliers providing raw materials, to contract development and manufacturing organisations (CDMOs), to end patients and clinics disposing of waste products.
As such, the emissions across the entire supply chain should be considered when calculating GHG emissions for a company. Targets should then be set both for the emissions directly linked to the company's own operations – known as scope 1 and 2 –and for emissions in the value-chain – scope 3 – to really make an impact on efforts to meet the Paris agreement.
Confusion
over terminology and definitions:
A variety of terms are used when it comes to communications around GHG emissions, including:
36 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Market Report
• Carbon neutral: Referring to a specific product or business and indicating that all CO2 emissions for the reporting period have been compensated for through carbon offsets. This can be done in combination with CO2 reduction targets.
• Carbon negative: Going beyond carbon neutral through the purchase of additional carbon offsets once the full carbon footprint has been compensated for.
• Net-zero: Committing to purchasing carbon offsets only for the remaining emissions after reducing to the best of your ability.
For many in the pharmaceutical sector, these terms are new. As a result, there is confusion in distinguishing this terminology, which can hinder companies’ efforts to set effective goals.
For example, all these terms rely on compensating for emissions by purchasing offsets – even though net-zero allows for it only after actual reductions to the best of the company’s ability. When the offsets are allocated to a specific product or operation, without first reducing your own emissions, a carbonneutral claim can be made when offsets equal emissions. To address any ambiguity regarding efforts to reduce carbon emissions, it is essential to be clear on the activities undertaken, and their impact in reducing your carbon footprint.
All this vagueness has ramifications for transparency – if the words used by a company don’t have clear, commonly understood definitions, how can its partners understand its efforts and goals? This can undermine companies’ efforts to
15th Annual
SAVE €450! Register by 8 September
identify partners capable of truly supporting their sustainability goals. So, part of your sustainability work is also transparency of the definitions you use – not only of the KPIs you report.
Poor industry Coordination Over Emissions Reduction Targets:
Insufficient visibility across the supply network makes it hard for companies to work together to determine which links in the value chain are the largest carbon contributors, highlighting which points in the chain require the most attention. This uncertainty extends to product disposal, with a lack of understanding of how drug products including packaging are disposed of at the end of their lifecycle and the role this plays in contributing to GHG emissions. This means that many companies are setting goals in isolation, limiting their impact and the industry’s overall GHG reduction efforts.
The sector also needs to work on improving cross-industry communication so that best practices can be shared more effectively. Companies need to learn from each other to improve their emissions reduction initiatives. Increasing visibility would help address these issues, enabling companies to work together to identify which areas to target to better address carbon emissions.
Coordination and Science-based Targets Can Overcome These Challenges
These issues are serious, but they shouldn’t be seen as a daunting barrier to committing to reducing GHG emissions for pharmaceutical companies. Targets need to be ambitious, yet achievable. They need to be measurable and transparent, and the contribution of each site to the company’s group-wide
- 16 NOVEMBER 2023
Protein & Antibody Engineering Summit
Described as “the best biologics technology meeting in Europe”, PEGS Europe is the leading European event covering all aspects of protein and antibody engineering, and will feature:

■ 1,500+ Attendees
■ 250 Speakers
■ 150+ Industry-Leading Sponsors & Exhibitors
■ 60 Talks from Top Biopharma
■ 150+ Scientific posters
■ 20 Keynotes
■ Dedicated Networking Opportunities
■ Exclusive Exhibit & Poster Viewing Hours
■ Interactive Roundtable, Breakout & Panel Discussions
ADDITIONAL HIGHLIGHTS
INCLUDE a Plenary Keynote address from Dr. Rebecca Croasdale-Wood, Director, Augmented Biologics Discovery & Design, Biologics Engineering, Oncology at AstraZeneca, who will discuss benchmarking the impact of AI biologics discovery and optimisation for pharma.

Market Report
14
LISBON CONGRESS CENTER | LISBON, PORTUGAL PEGSummitEurope.com
targets needs to be defined. Ideally, they should also be strategically coordinated with other companies up and down the pharmaceutical value chain.
With this in mind, global organisations and initiatives, such as the United Nations Global Compact, World Wildlife Fund, CDP (formerly known as the Carbon Disclosure Project) and the World Resource Institute have united in a partnership to support industry in setting emission-reduction targets in line with current science from the Intergovernmental Panel on Climate Change. This initiative has been named the Science Based Targets initiative (SBTi).
Setting science-based targets via the SBTi can help ensure climate goals are truly meaningful in reducing a company’s carbon footprint. Coordinating with partners and other businesses across the pharmaceutical industry can help amplify the impact of the sector’s climate-action efforts.
Today, over 3,000 companies around the world, including Recipharm, are committed to science-based target with the SBTi, with clearly defined paths to meet the Paris Agreement.
Expert Support to Make Targets Meaningful and Impactful

The SBTi provides independent third-party verification of a company’s climate-related targets. It requires a company to calculate its emissions across three different areas:
• Scope 1: Direct GHG emissions from sources on site (such as fuel combustion in boilers, leaks from refrigeration, process emissions).
• Scope 2: Emissions from consumption on site but emitted elsewhere (such as electricity or district cooling).

• Scope 3: Emissions from activities in the value chain, but outside of the direct control of the company (such as carbon footprint from material to products, emissions or waste from material manufacturers’ operations and waste from used products).
Calculations must be made according to the requirements of the Greenhouse Gas Protocol, an initiative that provides stringent standards for measuring emissions.
Once a company has made its calculations, it must submit them to the SBTi, which will then verify the calculations as well as the targets. The SBTi looks globally at the amount of CO2 currently in the atmosphere and uses this data to determine how much each signatory company needs to reduce their emissions to do their part to meet the Paris Agreement’s 1.5°C global temperature rise target. The later companies sign up to the SBTi, the less time they will have to reduce their emissions.
After emissions are calculated and targets are set, signatory companies must transparently report progress annually, through the CDP, for example, in accordance with the SBTi requirements. The reporting should be externally audited and verified to ensure accuracy and reliability.
All of this ensures that each company signed up to the SBTi is able to set and commit to targets designed to make a meaningful contribution to global efforts to meet the Paris Agreement. The initiative’s reporting processes optimise transparency, enabling companies to hold their partners in the value chain to account,
amplifying the effects of efforts across the industry and the wider economy. It also provides a space for companies in all sectors to share best practices to learn from each other and enhance the quality of industry initiatives.
With these kinds of partnerships, it is possible to address challenges in setting effective GHG targets, increase transparency and hold each other to account not just for companies in the pharmaceutical sector, but across the global economy.
Looking Ahead
The goals set by initiatives like SBTi are highly ambitious, which is necessary if the pharmaceutical industry and other sectors are to make a real contribution to efforts to limit global temperature increases to below 1.5°C.
The more pharmaceutical companies join the SBTi and other similar partnerships, the more impactful these efforts will be. Companies that join such initiatives will also find it easier to set, stick to and achieve goals, benefiting from expert advice and support. As we saw with the global fight against the COVID-19 pandemic, collaboration and partnership have helped the pharmaceutical sector address major challenges. They can help the industry do the same for climate change.
REFERENCES
1. https://www.un.org/en/climatechange/net-zero-coalition#:~: text=Currently%2C%20the%20Earth%20is%20already,reach%20 net%20zero%20by%202050.
2. https://www.un.org/en/climatechange/paris-agreement
3. https://treaties.un.org/Pages/ViewDetails.aspx?src=IND&mtdsg_ no=XXVII-2-f&chapter=27&clang=_en
4. https://www.ecb.europa.eu/pub/economic-bulletin/articles/2022/ html/ecb.ebart202206_01~8324008da7.en.html%C2%A0
5. https://www.peoplemanagement.co.uk/article/1746235/ why-millennials-driving-force-behind-corporate-policies#:~:text= Millennial%20generation&text=Of%20this%20demographic% 2C%2040%20per,with%20a%20robust%20sustainability%20plan.
Åsa Bergström
Åsa Bergström owns Recipharm's sustainability strategy, and is responsible for understanding and interpreting all stakeholder requirements on sustainability and build the framework for our internal work. Sustainability is a broad topic – spanning over social and HR related matters, environment, business ethics and finance – so interacting with all functions to align the Recipharm strategy is key for success. Åsa is also responsible for all formal sustainability external reporting for Recipharm, and owns our sustainability data. With 30 years in the company, in various central roles, Åsa has a wide network and understanding of the Group.
38 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Market Report
SGS Biosafety Solutions
Health Inspired, Quality Driven.
Our solutions
Our Center of Excellence for biosafety based in Glasgow provides:

• Electron microscopy
• GMP Sanger sequencing
• Viral vaccines Adventitious agents and species specific viruses
• Replication of competent vectors
• Retrovirus
• Process impurities
• Genetic stability
• Other microbial contaminants
"Most notably, the Biosafety Center of Excellence in Glasgow has participated in the batch-testing and release of over 3 billion doses of COVID-19 vaccine, helping to increase vaccine access and bring the global population out of the pandemic."
Archie Lovatt, Site Manager & Scientific Director at SGS
Contact us
To discuss your biosafety requirements, contact us today +44 141 952 0022
healthscience@sgs com sgs com/services/biosafety sgs com/linkedin
Supporting a variety of molecules
Our team of experts can support manufacturers of:
• Monoclonal antibodies
• Recombinant proteins
• Viral vaccines
• Cell therapies
• Gene therapies

Why SGS?
• More than 25 years of experience
• Direct access to 155 employees, from study directors to scientists
• Regulatory compliant and recognized quality systems based on cGMP/GLP
• Accurate and reliable report
• Fast turn around times
• Online digital communication platform (booking GMP slot, protocols, results, invoices )
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 39 www.international-biopharma.com
1
Innovative Laboratory Testing Methods for Clinical Monitoring of Cell Therapies Using Flow Cytometry
ddPCR and qPCR Assays
Gene-modified cell therapy for the treatment of cancer and other diseases is a rapidly growing area of clinical research. The therapeutic use of engineered cells has necessitated the development of novel lab tests for assessing patient safety and efficacy. We’ll explore how unique applications of PCR and flow cytometry can be used to provide critical and complementary data that is needed to bring new life saving treatment options to patients.
Cell therapy involves employing human cells to treat genetic diseases, cancer and infectious diseases; the manipulated material can either be from the patient’s own cells (autologous) or from a donor (allogeneic). Gene-modified cell therapies involve engineering the donor cells using customised lentiviral or retroviral vectors that have been altered to remove most of the viral genes. The altered viral vectors allow a functioning human gene to be integrated into the genome of the donor cells.
What is CAR-T?
A common gene-modified cell therapy is chimeric antigen receptor T cell therapy (CAR-T), a progressive new immune cell therapy used to treat cancer. CAR-T cells are engineered ex vivo to express a functioning receptor that recognises tumour antigens, resulting in attack and destruction of the tumour cells. Following the gene modification and manufacturing process, the CAR-T cells are infused into the patient. Several CAR-T therapies have been approved for the treatment of haematological malignancies.
Biomarkers of CAR-T Response
Since its development, CAR-T therapy has transformed the treatment landscape for haematological malignancies. As with any novel treatment, CAR-T therapy isn’t without its challenges. Not all patients respond to treatment. Moreover, the duration of response to CAR-T therapy can be variable. Various factors could be potential predictors of response and the duration of remission such as the patient age, tumour burden and CAR-T dose. More recently the cellular kinetics of the CAR-T cells during the first 3-6 months have been shown to be promising potential biomarkers of response. As a “living drug”, CAR-T cells exhibit the phases of distribution, expansion, contraction, and persistence following administration. Measuring the cellular kinetics of a “living drug” requires novel assay designs.
Tools to Monitor CAR-T Expansion and Persistence –PCR and Flow Cytometry
Blood and/or bone marrow samples are taken at frequent timepoints following the infusion of the CAR-T therapy. These samples are used to assess the amount of CAR-T cells that are in circulation, i.e. whether the CAR-T cells are proliferating and persisting. One tool or methodology used to monitor the level of CAR-T cells in circulation is polymerase chain reaction (PCR). Here at ICON Laboratories, we have developed multiple custom
PCR-based assays for various CAR-T and other gene-modified cell therapy programs, for use in baseline assessments and pharmacokinetic (PK) monitoring of the cell therapy product.
Using DNA extracted from the blood or bone marrow samples, both real time quantitative PCR (qPCR) and droplet digital PCR (ddPCR) can be used to amplify and quantify a specific segment of DNA. For the purposes of monitoring the kinetics of CAR-T cells, PCR-based vector copy number (VCN) assays are designed to detect, amplify and quantitate specific vector sequences that are unique to the cell therapy product.
qPCR requires the use of calibrators and a standard curve to calculate copy numbers of the targeted sequence. Unlike qPCR, ddPCR offers absolute quantitation without the need for calibrators and is ideal for VCN analysis. ddPCR is also more sensitive than qPCR, which improves the precision and reproducibility of quantification of lower levels of therapeutic cells. The custom ddPCR assays are a multiplex design, allowing for the detection of the target vector sequence and a reference gene sequence in the same reaction.
One challenge with the detection and quantification of therapeutic cells using VCN assays is especially notable when testing samples that are collected within the first 3 months post-infusion. Patients must undergo a short course of chemotherapeutic treatment prior to receiving the CAR-T cell infusion. The purpose of the conditioning, aka lymphodepletion, is to reduce the number of immune cells in the patient’s body in order to increase the chances of survival of the therapeutic T cells.
The quantity and quality of the DNA extracted from blood and bone marrow samples that are collected in the first few months following conditioning is typically low. The overall DNA yield is low due to lower than normal cell counts and the DNA can be fragmented due to apoptosis caused by the chemotherapeutics. Therefore, our researchers have developed and implemented different strategies to overcome the sample quality issues that are seen at this early, critical timepoints. First, efficient and reliable extraction methods have been validated to maximise DNA yields from blood or bone marrow samples. Second, the DNA is quantified using Tapestation or Qubit methods rather than Nanodrop. These methods are better suited for samples with a lower DNA concentration, and they enable a more accurate calculation of expected reference gene copies in the sample. Third, guidelines for reporting results have been developed based on the limit of detection (LoD), lower limit of quantification (LLoQ) and the expected reference gene copies in the sample. This approach maximises the number of data points that can be reported, allowing the drug sponsor to gain a more complete picture of the therapy’s cellular kinetics in each patient. Finally, experiments are performed during assay validation to determine the selectivity (recovery) of the target using samples that are created to
40 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Technology









INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 41 www.international-biopharma.com *In addition to an air bubble and overfill Aidaptus® is a registered trademark of Owen Mumford Ltd, ©️2023 OMPS/ibi/ad/ob/0923/7
fill volume may change, with Aidaptus® auto-adjust plunger technology your auto-injector doesn’t need to 0.3mL - 1mL* 0.5mL - 2.0mL*
intuitive delivery Auto-adjust plunger technology 2.25mL 1mL Auto-adjust plunger technology In collaboration with See our auto-adjust plunger technology in action
the QR code or visiting ompharmaservices.com/ibi-sept2023
Your
Versatile design
Find out more by scanning
production
in the
Now in
Accommodates both 1mL and 2.25mL glass syringes
same device
simulate the lymphodepleted timepoints. These experiments also allow the determination of the lowest possible sample DNA input that can be used without compromising the safety or the reliability of the data.
While PCR methods can be used to detect and quantify vector DNA sequences that are found in the therapeutic cells, flow cytometry can be used to detect, quantify and subtype the therapeutic T cells at a cellular level based on the presence of cell-specific proteins.

Flow cytometry is a laboratory tool that is used to identify and measure the physical and chemical characteristics of cells or particles. The flow cytometry platforms that we use allow for up to 25-colour multiplexing. This capability enables comprehensive profiling of the CAR cells; measurement of CAR expression as well as the expression of other T cell subsetspecific protein markers such as CD4 and CD8. Detailed information about the CAR-T cell subsets that are proliferating and persisting in the first 3–6 months provides additional information that could be critical for predicting overall patient response that cannot be obtained using PCR methods. High parameter panels can also be designed to characterise the cancer cells as well as the patient’s own immune cells, providing additional data related to efficacy and response.
The reagent of use in flow cytometry to detect cellular proteins is critical to the success of an assay. There are two reagent approaches: generic or specific. Generic reagents such as a protein L, enable the researcher to bind to the LG light chain and detect multiple CARs. CAR specific reagents are preferable because they give precise results and low background; however, the availability of these reagents is limited and more costly.
As with any flow cytometry assay, the panel design should be thoroughly evaluated to avoid spectral overlap with the CAR detection reagent, which should have a bright fluorophore.
A panel designed to monitor the therapeutic CARs and the patient’s own cell subsets cannot use CD19 protein for CAR detection and anti-CD19 antibodies on the same panel because they will bind and interfere with the study’s results.
The selection and creation of an appropriate positive control must be discussed with the trial’s sponsor. It is preferable to use stably expressing CAR cell lines and Jurkat cells in the case of a CAR-T cell, both of which take time; however, they are critical to the development of the assay. As a negative control, researchers may consider using a control cell line or a mock-transfected cell line.
Conclusion
The rapid and continued growth of gene-modified cell therapies shows great promise for those suffering from both haematological and solid tumours. To support that promise, biomarker assays will be needed to monitor cellular kinetics and safety, and patient selection assays may be needed for therapies designed to attack cancer cells with more unique targets or to identify other predictive biomarkers of response.
Dr. Galderisi currently serves as Vice President and Global Medical Director for ICON Specialty Laboratories. He has over 15 years of experience in diagnostic surgical and molecular pathology including the oversight of laboratory medical device testing for the FDA submission and approval of multiple companion diagnostics. Dr. Galderisi completed his residency in Anatomic Pathology at Penn State/MS Hershey Medical Center and a fellowship in Molecular Genetic Pathology at Oregon Health and Science University. He is licensed to practice medicine in California, Washington, Oregon, Massachusetts, and New York states.

42 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Dr. Chad Galderisi
Technology
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 43 www.international-biopharma.com y archive Independent Dedicated Secure Contact us today to find out more 01933 357953 | archives@qualogy.co.uk | www.qualogy.co.uk The Archivist, Qualogy Ltd, Po Box 6255, Thrapston, Northamptonshire, NN14 4ZL Compliant
Advancing diagnostics with the o~pal reader
Who we are
éclateral is an electrochemical lateral flow test (LFT) medical device company that was founded in 2021 by Paul Ko Ferrigno (CEO), Uroš Zupančič (CSO) and Jean Dumont. They realise that while LFTs are a powerful diagnostic tool at the point-ofcare, their qualitative results and lack of data security limit their use. éclateral has further grown and now comprises a team of dedicated engineers, chemists, and biologists with the aim of making the lateral flow test electric.


LFT Introduction
First generation lateral flow tests (LFTs; see the schematic in (figure 1) were developed 50 years ago and quickly found use in pregnancy testing and glucose monitoring. A lateral flow test separates the components in a biological sample through simple chromatography across a nitrocellulose test strip. Antibodies (or other detection reagents) are printed in two lines at right-angles to the sample flow, so that analytes can be captured as they flow past. In first generation LFTs, captured analytes are visualised with a second antibody that flows with the sample and carries a gold nanoparticle label. It is the reflection of light from the gold nanoparticles accumulating on the captured analyte that gives rise to the familiar pink or blue test or (T) line. The second line, known as the control or C line, develops colour in a similar way only when a sample has been applied to the LFT, so tests are only recorded as positive if both lines develop a colour.
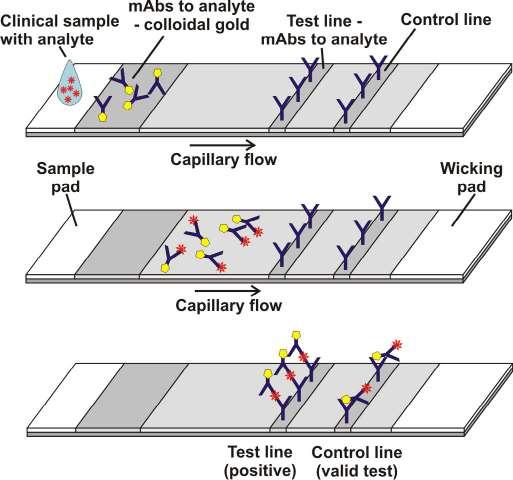
Lateral flow devices are now ubiquitous in healthcare and have also found applications in environmental monitoring. LFTs became seemingly ubiquitous through the Covid-19 pandemic as governments sought to understand the scale of infections in their populations. As LFTs gained widespread acceptance during the Covid-19 pandemic, the tests emerged as an inexpensive and accessible method for monitoring the spread of the virus. In the UK, with a few simple steps on the Government's website, individuals could order free, home delivered LFTs. Users were encouraged to upload their results through a website so that infections rates could be measured, but public health efforts using Covid-19 LFTs in the UK still relied heavily on visual interpretation and honesty in reporting results through health service websites.
Despite being used as a rapid diagnostic tool for a wide range of medical biomarkers, LFTs continue to face criticism from healthcare professionals concerned about their lack of a sample control and the subjective nature of the visual readout. To put it bluntly- many clinicians have been concerned that patients cannot reliably take a sample at home, and that patients with poor eyesight might struggle to detect a faint signal. The control line in first generation LFTs provides some confidence that a sample has been taken and has flowed along the test strip, but this is not foolproof, as evidenced by the anecdotal enterprising children using orange juice to guarantee a positive Covid test
that would allow them to skip school- which does validate the clinician’s concern that patients may not use the right sample. On the other hand, the subjectivity issue has been to some extent addressed using light emitting diodes (LEDs), such as those found in digital pregnancy tests from ClearBlue, although these bring increased costs.
At éclateral, our mission is to address these challenges with sample controls, objective readouts with third generation LFTs that foster greater accessibility, improved healthcare, and enhanced privacy through our innovative solutions – all without significantly increasing the costs to users or buyers.
éclateral Technology
The Need for Transformation:
The experience during the pandemic was that people took the test and then threw it away without logging it online, so the
44 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Application Note
Figure 1. Lateral flow assay with colloidal gold as label (Reproduced from Lee et al.).
population-level data was flawed due to inaccurate reporting. Recognising the need to capture data at the point of test our development includes the automated upload of sample data to a secure server: if the medical need justifies it, every test taken can be recorded. If privacy concerns are paramount e.g., in pregnancy testing then test results will not be recorded.

This transformation is enabled because our innovation allows the lateral flow test to be entirely digital- which also increases test precision beyond that possible with visual reporting. The éclateral digitalised health test integrates electrical components into the chemical matrix of a conventional lateral flow test. This feature translates the traditional visual lateral flow device results to an electrical measurement, using our o~pal mobile handheld reader. This has resulted in a digital health test device that is user-friendly for at-home use, delivering results in thirty minutes or less, depending on the analyte.
The Digitalisation Process:
The o~pal reader is at the core of the service, a low cost and clinical grade device which provides the electrical current and captures the electrical measurement of the analyte at the T line, and the control nanoparticles at the C line. Our o~pal tests will also incorporate an S line, which will measure a non-varying analyte in the sample to control for the amount and type of sample that was loaded into the test cassette. The Reader transmits test information by Bluetooth to the user’s smartphone, where the dedicated o~pal App can transmit the result to a healthcare professional at a remote location or display the result to the user. By design, the use of our smartphone App enables accessibility to people who have not been able to use LFTs before, such as blind and partially sighted users. In addition, for all users, digitalisation provides an extra layer of accuracy and convenience, including for clinicians and healthcare teams who can prescribe tests to be taken at home, but can interpret the results from their offices.When showcasing this technology’s effectiveness with SARS-CoV-2 samples, the potential benefits of digitalisation become evident. Every test taken gets reported, and the number of false negatives drops by more than 40%, resulting in improved clinical sensitivity. The innovative solution holds promise for the detection of analytes and antigens at levels that may not be visible to the naked eye for a diverse array of diagnostic applications (see reference 1). Our areas of focus extend to the CRP test, aiming to reduce unnecessary antibiotic usage, as well as female-centric health concerns like menopausal hormone testing or even a simple pregnancy test that can be used by blind women who cannot read first or second generation LFTs, but who will be able to hear the result using the accessibility
settings on their smartphone together with the o~pal app. We are taking steps to address the associated privacy concerns that have led to an investigation by the UK’s Information Commissioner’s Office into data collection by for example, fertility Apps. We clearly distinguish between medical tests, where data needs to be shared with healthcare professionals, and wellness tests where the data solely belong to the user and can only be seen by them.
Digitalisation Benefits
Data Integrity
Our lateral flow tests are encased in a cassette containing electrodes to engage communication between each lateral flow test strip and the o~pal reader. Each cassette carries a unique barcode, is strictly designed for single-use and is linked exclusively to the user's account when they scan the barcode. Any attempt to reuse a cassette or for it to be read by another individual is prevented both physically – the reader includes a mechanism that renders the cassette unusable – and digitally: each cassette's barcode is unique and cannot be reused. As the system records the initiation of each test, and a cassette cannot be used twice, this ensures complete traceability. All test results are captured and associated with the specific user and dual barcode scanning means that even the Reader used for each test is known, which can be important in high-volume settings if devices are being used in healthcare or residential care settings.
Applications
Healthcare Professional
The unambiguous nature of electrochemical measurements allows for the addition of sample controls to each lateral flow test, enabling both clinical confidence that the test was taken correctly but also enabling absolute quantification of the analyte against an invariant biomarker- although the nature of each sample loading biomarker may need to vary from test to test. This level of precision can be invaluable in healthcare settings, particularly when monitoring disease progression or treatment efficacy and here it is important to note that the user or patient can be prevented from seeing the result until they have been spoken to by a healthcare professional if the result means that there has been a change in their condition, or will be a change in their medical treatment.
For patients using a test at their home, each test result can be assessed by a clinician remotely, providing peace of mind to individuals. This telemedicine approach reduces the need for in-person healthcare visits, streamlining the diagnostic process and potentially reducing the burden on healthcare systems.
Clinical Trials
This secure data handling process, in conjunction with the elimination of optical readouts and data anonymisation is suitable for double-blinded clinical trials.
The digitalisation of test results, incorporating pseudonymisation, combined with our robust traceability strategy, ensures the integrity and accessibility of data to support a wide spectrum of applications (see reference 1), including clinical trials or for personal use at home. There is an enhanced traceability strategy with a single-use cassette being assigned to each user before commencing the test. The
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 45 www.international-biopharma.com
45
Application Note
test results remain confidential until they are uploaded to a secure server, which is password protected, encrypted, and IP locked.
This secure data handling process, in conjunction with the elimination of optical readouts and data anonymisation is suitable for double-blinded clinical trials.
Home Users
There is a growing need for clinical grade diagnostic tests from the comfort of one's own home. Clinical-grade, user-friendly solutions are an advantage of the éclateral technology whilst ensuring its affordability.
Since LFTs are visually interpreted they are ultimately subjective and are therefore difficult to analyse for non-specialists. Analysis becomes even more difficult for users who are visually impaired which in turn reduces the accessibility of LFTs. Replacing the visual aspect of testing with digital access to results will encourage individuals to have the confidence to track their wellness, monitor their progress and gather health data over time, all within the privacy of their own home.
LFT data is also not private in its current form as it can easily be visually interpreted by others, preventing users from having sole access to their medical device records. Because prioritising data privacy is paramount, privacy and data control are a critical aspect of éclateral’s offering. A home users’
personal wellness information is protected and can only be accessed in ways they permit.
Future uses
Providing the ability for early disease detection we look ahead to support various fields in healthcare, wellness, and disease monitoring. The possibility to function as a global health tracking tool for infectious diseases and future pandemics, and for localised public health management, will enable the identification and management of pockets of reinfection before they escalate into widespread outbreaks.
The nature of the o~pal rapid test system and the digitalised service, captures test results at the point of the sample collection so anonymised health information can be recorded at scale. This capability extends to a wide range of applications, including inflammation detection, female health, and hormone testing, and acting as a measure around antibiotic resistance.


Applying these capabilities across many other areas of health and wellness whilst bringing clinical grade diagnostic tests into the home, we hope to help relieve pressures in primary care and on healthcare systems globally.
REFERENCES
Paul Ko Ferrigno
Paul is the CEO and Co-founder of éclateral. He has been involved in commercialising technological innovations since 2008 when he founded Aptuscan (acquired by Avacta Group in 2012). He also invests in the future through work on multiple Boards for charitable, educational and grassroots sports organisations.

Bethan Larkin
Beth has recently graduated with a First Class Degree in Biological Sciences BSc (Hons). She is one of the team of scientists, passionate about making a difference is people's lives.

Benjamin Edwards
Ben (M.Chem, PhD), a scientific researcher at éclateral has helped contribute to two patents while developing the electrochemical methods and assay chemistry needed for technology development.

Sara Monem
Sara, Business manager at éclateral, brings with her experience from the Met Office in sales and digital marketing.

46 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Application
Note
1. Lee, L.G., Nordman, E.S., Johnson, M.D. and Oldham, M. F. A Low-Cost, High-Performance System for Fluorescence Lateral Flow Assays. Biosensors 3(4):360-73 (2013).
Monodisperse PEGs for Therapeutics
In the world of therapeutics, several challenges have hindered development and overall success of drugs and vaccines. One pressing issue is the short half-life of therapeutics, with drugs rapidly breaking down and being eliminated from the body. This leads to poor drug efficacy and consequently having to introduce frequent dosing, and potential issues with patient compliance. Additionally, some drugs face problems with solubility, making it difficult for poorly soluble compounds to be effectively absorbed. Maintaining drug stability during storage and administration is also crucial as many compounds, in particular biologics tend to degrade or undergo chemical changes. Addressing these problems is essential for optimal therapeutic outcomes and clinical approval.1 One strategy for increasing the stability of drugs before and after administration, as well as increasing water solubility, is to chemically incorporate inert polyethylene glycol (PEG) into the drugs. This process has been critical in new drug discovery as well as in generating new therapeutics from previously known drugs.
PEGylation is a bioconjugation technique, which involves attaching PEG chains to therapeutic agents, such as drugs or vaccines. This process enhances the pharmacokinetic and pharmacodynamic properties of the molecules, leading to prolonged circulation time in the body, better solubility, and improved stability. PEGylation has been a successful strategy for new drug development in many instances but until now it has been performed with commercially available off-the-shelf chemicals that are composed of polymers with non-uniform chain length, known as polydisperse PEGs.2
Over the last 25 years Polypure has developed techniques to synthesise and purify PEGs to a distinct chain length, known as monodisperse, uniform or single-length PEGs. These high-quality PEGs lead to higher reproducibility in production, thorough understanding of reaction dynamics, and grant greater efficiency in purification and analysis of products. The nature of Polypure’s PEG derivatives facilitates quality control, simplifies monitoring, and optimisation of the chemical processes.
Many companies are starting to choose uniform PEG alternatives for their product development and manufacturing. The consistency of raw materials contributes to reliable production, it increases reproducibility, and dramatically reduces batch-to-batch variations. This not only favours high-quality final products, but also streamlines research and development activities by reducing the consecutive complex impurity profile associated with polydisperse materials. Ultimately, the use of the monodisperse PEGs contributes to more precise and reliable outcomes for pharmaceutical manufacturing.
R&D projects
Polypure is actively engaged in two projects that center around
PEGylation, both of which are supported by the European Union's Horizon 2020 Research and Innovation Programme through the Marie Skłodowska-Curie Actions. Polypure’s role is to provide tailor-made PEG derivatives and to purify compounds from complex mixtures.

The NOVA-MRI (MSCA Grant #859908) project aims to improve the stability of PLGA nanoparticles for the development of novel 19F MRI contrast agents. This is achieved by attaching monodisperse PEG derivatives onto fluorine labelled nanoparticles. Polypure is actively investigating the impact of PEG length and the choice of end-group on the binding of nanoparticles to various types of immune cells for improved biocompatibility.
The success of the SARS-CoV-2 mRNA vaccine has showcased the substantial contribution that PEGs can make in advancing vaccine development. The PAVE project (MSCA Grant #861190) seeks to establish immunotherapy-driven strategies for the treatment of pancreatic cancer. Polypure’s responsibility in the PAVE network is to develop PEG products that can be used for vaccine technologies such as lipid nanoparticles (LNPs) and peptide-based vaccines.
PEG-peptides
Peptide-based cancer vaccines are made up of sequences of amino acids derived from tumor antigens (Figure 1). These peptide sequences are typically lengthy and comprise blocks of hydrophobic amino acids, making the synthesis difficult and requiring rigorous purification to obtain a high-quality product.3 The contribution of Polypure is to assist in PEGylating peptides to improve solubility and simplify downstream processing of vaccine. We have shown that incorporation of PEG chains in peptide sequences reduces synthesis, purification, and solubility problems frequently encountered with hydrophobic peptides.
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 47 www.international-biopharma.com 47 Application Note
Figure 1: PEGylation of peptide-based cancer vaccines
PEG Lipids
Another area of research that Polypure has been exploring is the development of synthesis methods for acquiring monodisperse PEG lipids utilized in LNPs. Incorporating PEG lipids in vaccine LNPs, extends circulation time and thereby improves vaccine efficacy (Figure 2). Commonly polydisperse DMG-PEG 2000 has been used in the rapid development of new vaccines.4,5 We intend to provide similar structures but with defined chain lengths for reliable reproducibility and better outcomes.
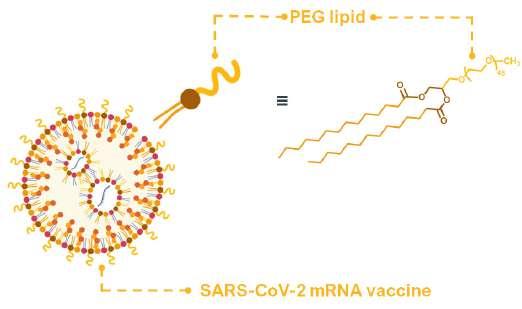
The technology of producing uniform PEGs utilised at Polypure has been previously employed on a commercial scale to provide multi-kilogram quantities. This will be particularly important when incorporating monodisperse PEG in LNPs manufacturing.
of PEG and belongs to a group of polyethers finding a variety of applications in biomedical formulations. The presence of an additional methylene group in each monomer unit in the chain significantly alters its physicochemical properties (Figure 3).6 This results in increased hydrophobicity and unique properties in terms of thermosensitive character, micellar behavior, and stereochemistry.6–8
Single-length 2-5kDa PEGs in Commercial Applications
Polydisperse PEG materials of molecular weights in the range of 2-5 kDa have been previously used in drug delivery. In order to develop precise treatments with reproducible properties, the need for elimination of impurities and high control of the quality of PEG is essential. Therefore, single-length materials have been increasingly attractive for the medical industry. The incorporation of PEG chains of 2–5 kDa can significantly influence release kinetics, providing control over the gradual and sustained release of the drug over a specified period. This capability is particularly valuable when designing medications that require a controlled release profile for optimal therapeutic outcome.2
Polypure has been actively working with a wide range of oligomer lengths and extending the ethylene glycol chain. Our uniform PEG derivatives of 1.6 kDa have been available in the drugs since 2002. While the feasibility of achieving longer PEG chains has been repeatedly demonstrated in our laboratory, the primary challenge revolves around effective production methods for larger-scale production. As a part of our ongoing development, Polypure has recently expanded its portfolio by introducing PEG45 and its derivatives, with the aim of making them readily available in multikilogram quantities without compromising quality. Emphasis is placed on developing clinically attractive PEG derivatives similar to those that have been recently used in vaccine formulations. This includes the monodisperse PEG lipid DMG-PEG 45.
PPG – A Close Cousin of PEG
A great novelty in Polypure’s R&D department is a new class of products, poly(propylene glycol) (PPG). PPG is a close cousin
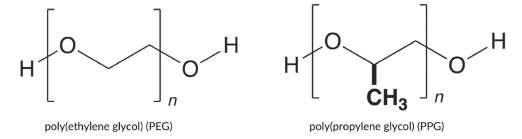
PPG development has been funded by European Union’s Horizon 2020 Research and Innovation Programme through the Marie Skłodowska-Curie Actions project PIANO (MSCA Grant #956477). Polypure’s primary focus in this project is to develop multicomponent hydrogel formulations as a potential drug delivery system. The formulations make use of monodisperse PPG and PEG derivatives, as well as defined thermoresponsive PEG-based block copolymers.
Applications of PPG in Biomedical Formulations
Just like PEG, PPG is an attractive candidate for a variety of nanoformulations. It can be utilised for the coating of nanoparticles to ensure their low immunogenic activity, to increase solubility and biocompatibility and consequently prevent opsonisation by blood proteins. 7,9–11 PPG is also an attractive additive for formulations of poorly soluble compounds, such as chemotherapy drugs. PPG can assist in solubilising a hydrophobic drug whilst increasing chemical affinity towards the therapeutic agent.11
There are several established applications of PPG in drug delivery systems, such as surface self-assembling polymers. This particularly refers to copolymers where PPG acts as hydrophobic unit, which leads to micellar self-organisation in aqueous solution and formation of nanogels. Due to the temperature sensitivity of the PPG block, a change in polymer chain conformation can occur. This feature allows for better control of drug release kinetics and eliminating burst release as it is observed in simple physical encapsulation of drugs. This approach does not require any covalent bonding between the drug and the polymer. The release of the therapeutic cargo is determined by diffusion and the degradation rate of the copolymer structure.9–11
Why is PPG alone not (yet) widely used in biomedical formulations?
The use of PPG in the biomedical industry has not yet been widely popularised due to inconsistent composition and presence of unsaturated impurities. These stem from the polymerization process of the commercially available products (Figure 4). Approaches to minimise the degree of unsaturation are not fully effective and render PPG of high polydispersity.6 Thus, working with high molecular weight PPG is challenging. Despite this complexity, several examples of PPG use in bioformulations can be found in literature. Therefore, we see an increasing need for high purity PPG derivatives delivered in multi-kilogram scale. This is particularly important to
48 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Application Note
Figure 2: PEG-lipid (DMG-PEG 2000) in the COVID-19 mRNA vaccine
Figure 3: PEG vs PPG structure
enable reproducible drug manufacturing and support effective bench-to-bedside translation of preclinical solutions developed by the pharmaceutical industry.
Our Expertise – High Purity PPG Oligomers
At Polypure, we have expanded our established SDC purification technology from PEG to PPG.12 Our technology allows us to utilise commercial PPG as a raw material and we can efficiently purify it to over 98 % oligomer purity. Our purification process relies exclusively on green solvents with continuous recycling to minimize overall consumption.
Twenty years of experience with PEG chemistry paves the way to transfer this knowledge to derivatisation of PPG. This has led us to develop a new library of PPG compounds

Application Note
with high purity, increased reactivity, and defined chemical composition. These include maleimide, thiol or amino functionalized PPG of varying lengths. Our dedication is in ensuring the highest reproducibility and control over chemistry of the PPG derivatives for pharma and biotech industries.
REFERENCES
1. Harris, J. M., & Chess, R. B. (2003). Effect of pegylation on pharmaceuticals. Nature reviews Drug discovery, 2(3), 214-221.
2. Székely, G., Schaepertoens, M., Gaffney, P. R., & Livingston, A. G. (2014). Beyond PEG2000: synthesis and functionalisation of monodisperse PEGylated homostars and clickable bivalent polyethyleneglycols. Chemistry–A European Journal, 20(32), 10038-10051.
3. Harris, J. M., & Chess, R. B. (2003). Effect of pegylation on pharmaceuticals. Nature reviews Drug discovery, 2(3), 214-221.
4. Hou, X., Zaks, T., Langer, R., & Dong, Y. (2021). Lipid nanoparticles for mRNA delivery. Nature Reviews Materials, 6(12), 1078-1094.
5. Tenchov, R., Bird, R., Curtze, A. E., & Zhou, Q. (2021). Lipid nanoparticles – from liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS nano, 15(11), 16982-17015.
6. Herzberger, J., Niederer, K., Pohlit, H., Seiwert, J., Worm, M., Wurm, F. R., & Frey, H. (2016). Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: synthesis, novel polymer architectures, and bioconjugation. Chemical reviews, 116(4), 2170-2243.
7. Ajiro, H., & Akashi, M. (2010). Cell proliferation on stereoregular isotactic-poly (propylene oxide) as a bulk substrate. Biomacromolecules, 11(11), 2840-2844.
8. Bordat, A., Boissenot, T., Nicolas, J., & Tsapis, N. (2019). Thermoresponsive polymer nanocarriers for biomedical applications. Advanced drug delivery reviews, 138, 167-192.
9. Al-Nadaf, A. H., Dahabiyeh, L. A., Bardaweel, S., Mahmoud, N. N., & Jawarneh, S. (2020). Functionalized mesoporous silica nanoparticles by lactose and hydrophilic polymer as a hepatocellular carcinoma drug delivery system. Journal of Drug Delivery Science and Technology, 56, 101504.
10. Cheng, C. C., Liang, M. C., Liao, Z. S., Huang, J. J., & Lee, D. J. (2017). Self-Assembled Supramolecular Nanogels as a Safe and Effective Drug Delivery Vector for Cancer Therapy. Macromolecular Bioscience, 17(5), 1600370.
11. Al-Nadaf, A. H., Dahabiyeh, L. A., Bardaweel, S., Mahmoud, N. N., & Jawarneh, S. (2020). Functionalized mesoporous silica nanoparticles by lactose and hydrophilic polymer as a hepatocellular carcinoma drug delivery system. Journal of Drug Delivery Science and Technology, 56, 101504.
12. Agner, E. (2003). U.S. Patent No. 6,576,134. Washington, DC: U.S. Patent and Trademark Office.
Erik Agner
Erik Agner, CEO and founder of Polypure, has an academic background in chemistry from the University of Linköping in Sweden. Subsequently, he pursued his career by joining pharmaceutical companies in both Sweden and Norway where he engaged in peptide related projects. Through early work experiences, Erik Agner realized the challenges in generating high-quality materials for advancing pharmaceuticals and biotechnology. With interest in purification technology, he initiated a startup and filed patents based in displacement chromatography. From that novel technology he founded Polypure in 1999.
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 49 www.international-biopharma.com 49
Figure 4: Performed HPLC analysis of commercial PPG
The Most Important Drug Discovery Event of The Year is Just Around the Corner
ELRIG (The European Laboratory Research & Innovation Group) are looking forward to delivering a truly unmissable knowledge-sharing and career-boosting conference in Liverpool at the ACC Exhibition Centre, on Oct 18th & 19th 2023.
This year, Europe’s leading event for the drug discovery community, Drug Discovery 2023, focuses on the sheer power of diversity – the lifeblood of the life sciences innovation ecosystem –highlighting its role in propelling the sector forward.
So, what can you expect from the event?
The Latest World-class Science, from a Diversity of Leading Thinkers
ELRIG have put together an unparalleled series of 8 actionpacked tracks to keep you at the forefront of life sciences knowledge – all from some of the world’s leading thinkers and institutions).
Tracks will cover a variety of fascinating themes:
Whether you’re involved in early drug discovery research or translating your promising therapy to the clinic, you’ll be sure to discover something that can help you carve a smoother path to success at this year’s event!
Hands-on Access to the Latest Path-shaping Technologies And the innovation and diversity doesn’t stop there.
More than 150 suppliers and service providers will flood the exhibition floor, showcasing the latest tools and instruments that could be just what you need to overcome your stickiest drug discovery challenges.

Don’t miss your chance to be among the first in the world to get acquainted with the next generation of tools and techniques that could propel your research forward, several companies will be launching new products.
And, as if that wasn’t enough, you’ll also get the chance to see some of this tech in action, with our Tech Theatre – with an exciting programme for Drug Discovery 2023! Learn all about implementing robotics and automation in drug discovery, as well as practical ways in which your lab can become more sustainable.
Accessible
and Inclusive: Removing Barriers to the Best Science
The only thing better than path-shaping science and technologies? FREE access to path-shaping science and technologies.
That’s right, at ELRIG, we’re committed to removing as many barriers to conference attendance and world-class science as possible. So, as part of our commitment to accessibility, all our
events, including Drug Discovery 2023 are completely free of charge! You won’t have to pay a penny for registration, and lunch and refreshments will be provided. (Who said there’s no such thing as free lunch?)
And as with all our events, we’ll provide our diverse and vibrant community with a dedicated prayer room, family room and quiet space.
Advance Your Career and Unlock New Opportunities
New to the drug discovery sector and want to supercharge your career development? Then Drug Discovery 2023 has got you covered. This is Europe’s most well-attended face-toface networking event – offering you unrivalled networking opportunities. What’s more, we’ve created a host of activities tailored specifically to early career professionals (ECPs) –a critical part of our strong and vibrant community.
Explore our ECP Learning Zone and partake in friendly, no-pressure meet-and-greets, our ever-popular ‘Network Like a Boss” session to get world-class face-to-face mentoring with industry veterans who want to help you take your next step.
Oh, and you’ll be able to source expert advice on how to craft a show-stopping CV, as well as a mountain of top tips to accelerate your career growth.
Get Recognised, Get Rewarded
We love celebrating the power of diverse science. And that means we love celebrating YOUR successes.
At the conference, there’ll also be ample opportunity for you to share your work and get the recognition you deserve: poster presentations with prizes and our renowned Early Career Professional Impact Award are just a couple of ways you can raise your profile and get rewarded at the event.
Secure Your Spot Today!
Drug Discovery 2023 is set to be the event of the year for the life sciences community. Find out more or register now at elrig.org.
50 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Elrig Inspired
2023: DIVERSE THINKINGNEW PERSPECTIVES
18TH & 19TH OCTOBER 2023
ACC, LIVERPOOL
REGISTER NOW!
To find out more and to register please visit elrig.org
THE MOST IMPORTANT DRUG DISCOVERY CONFERENCE OF THE YEAR!

Drug Discovery 2023, is a two day event providing stimulating scientific talks and posters, the wide range of technologies and applied research to spark ideas.
CONFERENCE TRACKS
Day 1 Celullar Technologies for Drug Discovery
Day 2 Biopharmaceutical discovery
SCIENTIFIC PARTNERS

CRUK - Horizonsoncology, a diverse thinking theme


Translating ideas into therapies
Delving into the diversity of chemistry, driving molecules from concept to clinic
SLAS - HIT identification and screening



Augmenting R&D with AI
Chemistry enabling drug discovery from start to clinic

INDUSTRY PARTNERS
MEDIA PARTNERS
www.international-biopharma.com DRUG
DISCOVERY
elrig @ELRIG_UK elrig.org
Using a LIMS to Improve Bottom Line Profitability
The Model T Ford is still discussed in business schools today because of how Henry Ford relentlessly drove quality and innovation. When first introduced in 1908 the Model T cost $825; by 1916 it cost $360. While reducing the cost by more than half, Ford increased the safety, reliability, and speed of the product. His laser-like focus on quality was legendary and his quote “Quality means doing it right when no one is looking”, is still a mainstay of quality theory and practice.
Whether manufacturing cars, supplying clean water, or manufacturing pharmaceuticals, all repeated processes must include quality control to ensure products meet their defined requirements. Quality management is very often about measuring and minimising variance within and across batches to ensure adherence to acceptable limits. This in turn is all about keeping meticulous records, as well as managing and accessing that data.
Pharmaceutical Manufacturing Issues
The FDA (U.S. Food and Drug Administration) keep a public record of the warning letters that they send out to Companies and the citations they contain.1 Looking at the last five years of data provides a useful insight as to the main issues they see when inspecting quality control regimes in pharmaceutical manufacturing companies. The data shown here is sifted to report only on the primary GMP regulations for drugs (FDA 21 CFR 211). The FDA data dashboard2 also has some caveats regarding completeness of the data. Nevertheless, the data provides good insight as to their overall findings.
The top 10 GMP Drug Inspection citations for FDA financial years 2018–2022 are shown in Figure 1. This provides a summary of the issues they found. A description of each CFR number is provided in Table 1.
CFR 211.194(a) – Laboratory records shall include complete data derived from all tests necessary to assure compliance with specifications...
Table 1: Description of Top 10 CFR Numbers
Laboratory Information Management Systems
Laboratory Information Management Systems (LIMS) help manage quality control in tightly regulated industries, such
52 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
# Top 10 CFR Number - Description Qty LIMS Module 1 21 CFR 211.22(d) – The responsibilities and procedures applicable to the quality control unit shall be in writing; ...and followed... 535 Document Manager 2 21 CFR 211.192 – All production and control records, shall be reviewed and approved... 518 Electronic Approval
21 CFR 211.160(b) – Laboratory controls shall include the establishment of scientifically sound and appropriate specifications, standards, sampling plans, and test procedures... 458 Test Limits 4 21 CFR 211.100(a) – Procedures ...to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess... 285 LES 5 21 CFR 211.166(a) – Testing program designed to assess the stability characteristics (shelf-life etc) of drug products... 278 Stability Manager 6 21 CFR 211.67(b)
be
and
for cleaning and maintenance of equipment... 259 Instrument Calibration and Maintenance 7 21 CFR
and
records shall be prepared for
of
product produced... 244 Batch Manager 8 21
over computer or
to assure
are
only by authorized personnel... 236 Access Control
21
215 Training Manager
181 Records and Reports
Figure 1: Top 10 GMP Drug Inspection Citations: FY2018–FY2022
3
– Procedures shall
established
followed
211.188 – Batch production
control
each batch
drug
CFR 211.68(b) – Appropriate controls shall be exercised
related systems
...records
instituted
9
CFR 211.25(a) – Each person ... shall have education, training, and experience, ...to enable that person to perform the assigned functions...
10 21
Elrig Inspired




www.discovery-park.co.uk
as pharmaceutical manufacturing. Much like Henry Ford, the built in controls and checks help drive the quality control of excipients and active ingredients within the manufactured product, and consistently manage quality over time. The quality control procedure in drug manufacturing consists of taking regular samples to ensure consistent quality of raw, intermediate, and final products. Here we outline one aspect of how using a LIMS can help prevent each of the top 10 quality issues highlighted by FDA inspections.
Document Management
The top issue, with 535 citations found through FDA inspections, is one of documenting and following procedures. A QC laboratory LIMS will usually manage the documentation concerning sample collection, testing, and result reporting. New document versions can be released to appropriate staff to electronically sign as proof they have read and understood them. A record of this information can be kept on their training record and is available for audit purposes. Document manager enables QC procedures to be put in place and followed by all staff, and is a key part of implementing 21CFR211 22(d). It can also be part of a competency management process that prevent staff undertaking specific tasks if a specific competency assigned to them is not up to date.

Electronic Approval
Production environments demand that QC checks are performed and formally approved on each batch of product. And yet this is the second most common inspection issue with 518 citations identified. A central theme of a LIMS is that everything you do is logged. Managing the review and approval process of all sample records becomes much simpler using a LIMS. Only authorised personnel may review and approve QC results, and approval can be performed by adding an electronic signature (username and password) to the results approval screen. Once this approval step is done the batch of material (either raw ingredients, or intermediate or final product) may be released from quarantine and used. Any future audit will be able to show who approved the product batch and when, so an accurate record is always kept for the purposes of 21 CFR 211.192.
Test Limits
The third most common FDA issue during inspection, with 458 citations, is that of applying appropriate specifications, standards, sampling plans and test procedures. Test results can be automatically checked within the LIMS against pre-built product rules defining, for instance, maximum and minimum limits outside which the product fails. Statistical process control
54 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Elrig Inspired




INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 55 www.international-biopharma.com
(SPC) may also be used across batches of the same product to ascertain process drift, or other key factors, in the production environment. Understanding specification values and applying these rigorously to production and control samples enables a good understanding of what ‘good’ looks like in terms of production quality.
Lab Execution System
21 CFR 211.100(a) covers the written procedures that ensure that drug products ‘do what they say on the tin’ (to quote Ronseal). Within the QC laboratory test procedures should always be documented and followed to ensure the same outcome every time. Steps however can easily be missed when under pressure and this is where a laboratory execution system (LES), which many LIMS include, can help. An LES breaks down a written test procedure into a step-by-step approach, ensuring that the laboratory technician does not miss a step. Following procedure, automatically comparing test results to specifications, and ensuring QC laboratory results are never ignored in preference to ‘shipping product’, will all help drive the number of citations down in this area of GMP.
Stability Studies
Pharmaceuticals have active ingredients that may decay over time. It is therefore important that drugs are subject to a stability study programme to assess the shelf-life of each. The fifth most common citation (with 278) is the absence of a stability study programme. Matrix Stability Study Manager, a LIMS module, supports this complex area of analysis. Study programmes can be set up to define storage conditions and time points and identify when samples should be taken for testing. Study programmes can run for many years improving the knowledge of the drug’s stability over time to determine both the storage conditions and the shelf life appropriate for the product.
Instrument Calibration and Maintenance (ICMS)
The QC laboratory bristles with test instruments, each of which needs to be regularly maintained and calibrated. The LIMS instrument calibration and maintenance module allows the laboratory to document and record maintenance and calibration events on each instrument, as well as preventing instruments being used if the events have not been completed as required, or calibration identifies a problem. Demonstrating that analytical instruments are properly maintained and calibrated is a key QC laboratory function, and an area that attracted 259 citations over the 5-year period.
Batch Manager
The batch manufacturing functionality within a LIMS must include batch genealogy. This links together the raw materials, intermediates, and final product batches, allowing a complete view of exactly what is in every final product batch. In addition, if an issue is found with a specific raw material, or intermediate, any final product they have been used in can be identified. This is one way QC labs can comply with 21 CFR 211.188 which is number 7 on our list with 244 citations.
Access Control
21 CFR 211.68(b) demands that records are only created by authorized personnel. LIMS do this by managing access via username and password. In this way only authorized personnel can create or manage records. Different access rights can be
allocated to different staff, so that some may only have access to sample registration functionality, for instance, while others have the ability to approve batches for release. In all cases a record is kept of who logged in, along with what records they created or changed, and when.
Training Manager
The training manager module keeps a record of each staff member’s education, training, and experience. The LIMS can use this so that only qualified staff can perform certain functions, enter certain results, review and approve results and so forth. This enables tight control in a QC laboratory and ensures procedures are always seen to be followed, which was number 9 in our top 10 GMP issues.
Records and Reports
With 181 citations, the ability for a laboratory to properly record and recall all test data is the last in our top 10 GMP issues. The function of a LIMS is to ensure all test records are complete. For example, during registration the source of the sample, along with its lot number, date sampled, and other relevant details may be taken. Test results can be updated, and new revision saved, but the original result will always remain within the system, and available for inspection (unlike a spreadsheet or paper and pencil). A LIMS ensures the audit and chain of custody functions are available, allowing laboratories to work to, and fulfil, GMP standards.
Summary
LIMS help QC laboratories adopt good manufacturing practice, and ensure that procedures and standards are followed. The above provides examples of how the LIMS will help drive those standards, but there are many more aspects to both Matrix Gemini LIMS, and QC management generally. If you are a QC laboratory in pharma/biopharmaceutical manufacturing, then you should be using a properly implemented and validated LIMS to help drive quality improvement cycles. Remember that “Quality means doing it right when no one is looking”.
REFERENCES
1. FDA warning letters https://www.fda.gov/inspections-complianceenforcement-and-criminal-investigations/compliance-actions-andactivities/warning-letters
2. FDA data dashboard (data source) https://datadashboard.fda.gov/ ora/cd/inspections.htm
Tim Daniels
Tim has over 30 years of experience working in national and international markets across a range of software/high-technology products. He is currently the Marketing Manager at Autoscribe Informatics, based in the UK. Autoscribe Informatics provides database management solutions such as Laboratory Information Management Systems (LIMS) that, uniquely, are graphically configurable requiring no scripting or custom coding to configure solutions. Tim‘s broad background provides a unique blend of practical knowledge and insight to drive all aspects of product and corporate marketing within Autoscribe Informatics.

56 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Elrig Inspired
Via Nova Therapeutics Uses CDD Vault to Support Global Collaboration


“We’re able to achieve our centralized data repository needs with CDD Vault – without having to invest in infrastructure. We see CDD Vault as a huge value.”
Benjamin R. Taft, PhD, Executive Director, Chemistry, Via Nova Therapeutics
Situation
Via Nova Therapeutics deploys state-of-the-art drug discovery modalities to identify novel, first-in-class antivirals for the individual infections it studies. These modalities include phenotypic screening in infected cultured cells, biochemical screening in vitro on virus-encoded enzymatic targets and structure-based drug design, as well as combinations of the above approaches. Where appropriate, the company also targets host factors essential for viral infection.
The application of these approaches has generated specific antiviral programs targeting influenza, rhinovirus, BK virus, and adenovirus.
As a recently launched startup with funding from Aditum Bio, Via Nova prides itself in its tightly focused lean operations and making efficient use of a dispersed workforce including contract research organisations.
Initially the company uploaded spreadsheets into a shared repository, but soon outgrew this solution and required a secure
and robust central repository to support collaboration across the globe.
Solution
Via Nova Therapeutics deployed Collaborative Drug Discovery’s CDD Vault, the hosted drug discovery informatics platform that securely manages both internal and external biological and chemical data.
“Prior to CDD Vault, if someone would want to dive into the data, we would waste time trying to find the exact data sets we needed, recalls Benjamin R. Taft, PhD, Executive Director, Chemistry, at Via Nova Therapeutics. “CDD Vault really helps us get organized and have all of our data in one place where we consider it published and final, searchable, and sortable.”
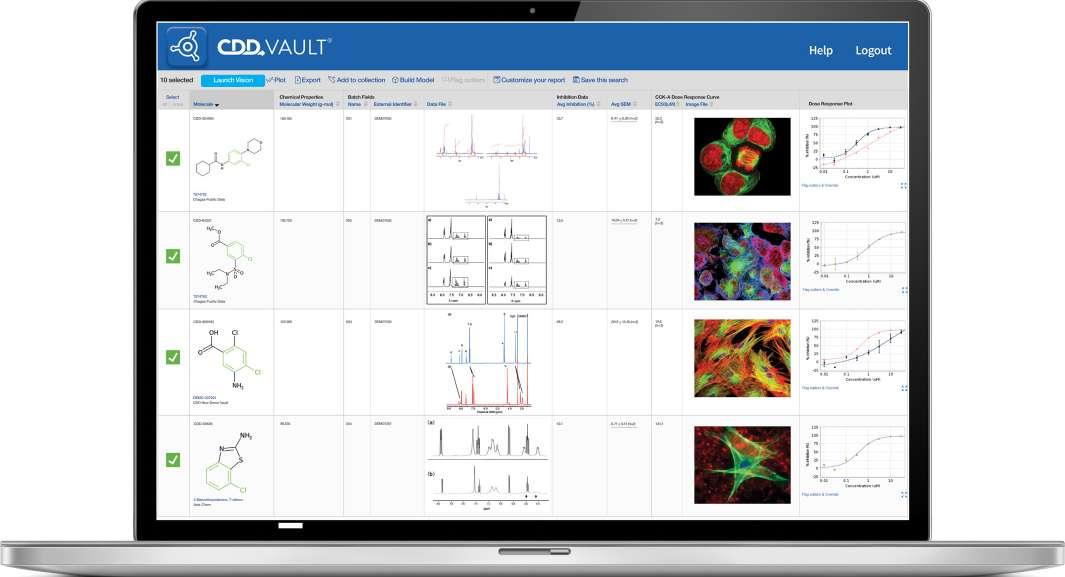
CDD Vault: Highly Recommended
Via Nova Therapeutics immediately gravitated to CDD Vault –especially after checking in with colleagues at other companies that were using it.
“When we talked to others about CDD Vault they all had the same thing to say: It’s safe, secure, supports collaboration, and has great visualization tools,” Taft says. “People we spoke with said they liked the flexibility, and the ability to provide securely controlled granular access to CROs and other parties. Everyone also said CDD Vault represented a tremendous value. So it seemed like a no brainer for us to give it a shot and evaluate it – and once we tried it we knew it was the solution we needed.”
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 57 www.international-biopharma.com 57 Elrig Inspired
Used by Biologists and Chemists
Via Nova biologists and chemists use CDD Vault to store data on small molecules of interest, along with related data.
“Both groups are equally using the Vault,” Taft says. “The chemists are registering all small molecule structures, and making sure they're all updated and annotated, and that the new batches coming in are updated and annotated. Our biologists are uploading their data, and both the chemists and biologists are using the CDD Vault visualization tools.”
Benefits
Via Nova Therapeutics has found a number of benefits since adopting CDD Vault, including:
• Gaining “one true secure data repository”
• Supporting a globally dispersed workforce
• Facilitating collaboration
• Flexibility to upload rich data
• Integrated visualization tool
• Pricing that represents a “huge value”
• CDD is a great company to work with and “feels like a partnership”
Gaining “One True Secure Data Repository”
After trying to store and track data within its previous shared repository, Via Nova Therapeutics has deep appreciation for the ease of use and precision of CDD Vault.
“We’ve gained something mission critical from a business standpoint – having one true secure data repository,” Taft says. “All the new data we generate as a company gets uploaded into our CDD Vault. From there we can sort it, query the most important data really effectively and efficiently to find and then visualize it – if needed, with plots and graphics. And we can do this from wherever we have connectivity. I even look at data from my phone at times.”
Taft says the value of one true, secure, data repository extends to enhancing productivity. “Some organizations might underestimate the value of one true source of data, but scientists are much more productive when they don’t have to constantly look for the final version of a file. The same goes for preparing for leadership meetings. With CDD Vault we always have our most current data immediately accessible, including when someone needs to take a deep dive into a particular data set.”
Supporting a Globally Dispersed Workforce
With its globally dispersed workforce, Via Nova Therapeutics values the unifying force of its CDD Vault.
“We're rarely in the same building, let alone the same room, but we're all working together using CDD Vault as our one true source of data,” Taft says. “Our people can access data from anywhere in the world – which is critical for our work.”
The company also appreciates that it can provide granular access to data to CROs and contractors.
“We have contractors and part-time employees that are supporting some of our projects,” Taft says. “We can give them access to the Vault, with layers of security so that they can
only access the data required for their work, allowing them to register structures, upload data, as well as download their allowed data into other tools as needed. This helps them work more efficiently, and it frees us from having to always be doing things for them.”
“Our people can access data from anywhere in the world –which is critical for our work.”
Benjamin R. Taft, PhD, Executive Director, Chemistry, Via Nova Therapeutics
Facilitating Collaboration
Via Nova Therapeutics values the ways in which CDD Vault facilitates collaboration across its operations.
“We have files come in almost every morning from our contract labs in Asia,” Taft says. “I can review the data from my home, send over a note to a colleague in a different county, who will QC the data and upload it into the Vault. I can then pull the data into a query and visualize it and create some slides and take it to our executive leadership team meeting and show visualizations of the data to our executive team. And it's all pretty seamless, because we can do that workflow, whether we're at home or in an office space somewhere or traveling it really just allows us to get our work done super efficiently wherever we are. The collaboration that CDD Vault enables is awesome.”
Flexibility to Upload Rich Data
Via Nova Therapeutics is impressed with the wealth of supplementary information they can store within CDD Vault.
“We’re doing exclusively small molecules, but for some of our more advanced programs, we have lots of additional data associated with each molecule,” Taft explains. “One of our favorite features is how flexible CDD Vault is. You can pretty much upload any data you want.”
Taft was impressed by the ease with which the extra data was uploaded into CD Vault.
“We recently got some really complex PK data, analyzing three different tissue types across different time points,” Taft says. “It was a huge web of complicated data. My colleague, who does most of the uploading was able to do it within half an hour. This included plots of the PK timecourse curves, dose response curves, in vitro, in vivo, and DMPK. It's all in there. CDD Vault allows us to put everything we want in there very easily.”
Integrated Visualization Tool
The Visualization tool built into CDD Vault has proven popular with scientists at Via Nova Therapeutics.
“We really like that visualization is built into CDD Vault,” Taft says. “It’s already there, so if I'm querying some data, visualization is just one click away. Something as simple as a scatter plot can sometimes really help you look at the data in a way you need to. The Visualization tool is convenient, fast, and it allows you to do a lot of the visualization that you need to do on a day-to-day basis, in a very convenient, streamlined way.”
58 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Elrig Inspired
The company also likes that CDD Vault integrates seamlessly with third-party visualisation tools.
And we are getting this at a great price point. We see CDD Vault as a huge value.”
“We really like that visualization is built into CDD Vault. It’s already there, so if I'm querying some data, visualization is just one click away.”
 Benjamin R. Taft, PhD, Senior Director, Chemistry, Via Nova Therapeutics
Benjamin R. Taft, PhD, Senior Director, Chemistry, Via Nova Therapeutics

“If I need to do something that's a little more complex, I can easily export the data into our StarDrop™ software. And it's pretty seamless. It's really nice it has that function to interface with other software because you can't expect one piece of software to do everything. There's always going to be other things you want to do.”
CDD Pricing Represents a “Huge Value”
Via Nova Therapeutics wants to dedicate its resources toward research – not infrastructure.
“We want to stay lean as we move toward our next value inflection point in our company,” Taft says. “Coming from a large pharmaceutical background, you have a good idea of the kind of immense infrastructure they have for data storage. We’re able to achieve our centralized data repository needs with CDD Vault – without having to invest in infrastructure.
CDD is a Great Company to Work with “Feels Like a Partnership”
Via Nova Therapeutics enjoys working with CDD, including the company’s excellent Customer Support.
“The CDD support team was extremely helpful in getting us up and running,” Taft says. “They helped us standardize formats for our first few hundred Vault entries, getting them stored the way we needed them to be. They have great knowledge and are dedicated to helping us in our drug discovery efforts. Working with CDD feels like a partnership.”
About Collaborative Drug Discovery
Collaborative Drug Discovery provides a modern approach to drug discovery informatics that is trusted globally by thousands of leading researchers. Our CDD Vault is a hosted informatics platform that securely manages both private and external biological and chemical data. It provides core functionality including chemical registration, structure activity relationship, inventory, visualization, and electronic lab notebook capabilities. For more information, visit us at www. collaborativedrug.com.
Benjamin R. Taft
Benjamin R. Taft, PhD is Executive Director and Head of Chemistry at Via Nova Therapeutics, a biotech company with a pipeline of antiviral drugs in development. Prior to that, he had 10 years of experience as Principal Scientist in the area of infectious disease at Novartis Institutes for BioMedical Research.
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 59 www.international-biopharma.com 59 Elrig Inspired
Antibody Isotypes –The Devil in the Details

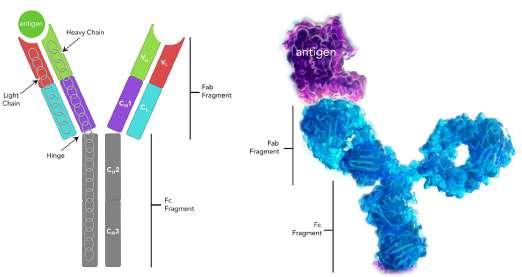
The immune system serves as the body's primary defence mechanism against a wide array of infectious agents, ranging from bacteria and viruses to parasites and fungi. It is an intricate network of specialised cells, tissues, and molecules that work in concert to recognise and eliminate foreign invaders while preserving the integrity of the host organism. Coordination of the activities among the different immune cells is critical. Antibodies, products of B lymphocytes, are a unique soluble component of the immune system that engage with both foreign bodies and host innate immune cells.
Antibodies consist of two distinct regions, the Fab region, involved in antigen specificity and binding, and the Fc region involved in effector functions via Fc receptors expressed largely by innate immune cells (Figure 1). Both regions contain valuable information for research and discovery: the Fab region identifies disease-relevant proteins, including autoantigenic, and the Fc region provides information related to specific antibody functionality. While the Fab region possesses enormous diversity for identification of innumerous antigens,4 the Fc region can assume five structurally and functionally different structures, each characteristic of a different isotype: IgM, IgD, IgA, IgG, and IgE (Table 1, Figure 1). Through effector functions from both domains, antibodies help coordinate the activities of the innate and adaptive immune system, directing pathogen removal with different strategies for different pathogens. These strategies include neutralisation, agglutination, opsonisation and phagocytosis, degranulation, complement activation, and antibody-dependent cellular cytotoxicity. Pathogenesis-specific antibodies are often elicited years before a disease becomes clinically apparent. With high disease specificity, early detection, ease of obtaining, and durability during storage, antibodies make excellent biomarkers. Historically, antibodies were among the first pragmatic biomarkers used by clinicians. They make up a unique subset of the proteome, with over 1 trillion potential combinations.4 Identifying antibody specificity and isotype class within patients can improve diagnostic accuracy at early stages of disease prior to overt symptoms, has the potential to indicate anatomical/tissue compartment, most affected can provide
greater precision for therapeutic intervention and improves our understanding of pathogenic mechanisms. Functional protein antigen microarrays are emerging with the capacity to simultaneously measure antibody specificity and isotypes using miniscule volumes of patient-derived biofluid. This provides a solid platform for disease understanding, antigen discovery, vaccine development, biomarker discovery, development of in vitro diagnostics, and true precision medicine.
Antibody Structure
Antibodies are produced and secreted by B lymphocytes, or B cells. Each B cell (referred to as a B cell clone) produces a unique antibody specificity that recognises a particular cognate antigen. Most antibodies can be membrane bound or secreted into the lymph and blood. Each symmetric Y-shaped immunoglobulin is composed of two heavy and two light chains, held together by disulfide bonds. Heavy chain C-termini form the Fc region (the stem of the Y), while light chains together with heavy chain N-termini form the Fab region. Antibodies were originally classified into groups, or isotypes, by electrophoresis using Greek letters to designate each fraction.5 In addition to five distinct isotypes, four subclasses of IgG and two of IgA are also recognised. Some isotypes act as multimers, IgM can form pentamers, and IgA dimers (Figure 2). The most enigmatic isotype, IgD, exists predominantly in membrane bound form.
60 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Elrig Inspired
Antibody Subclasses Primary Function Serum Level (g/L) IgA IgA1–2 Pathogen neutralisation, anti-inflammatory, mucosal 0.6–4 IgG IgG1–4 Circulating and tissue immunity 7–15 IgD None Upper aerodigestive immunity, B-cell development, immune regulation 0–0.14 IgE None Tumour surveillance, anti-venom defence, anti-parasitic defence, type I hypersensitivity Trace IgM None Immune surveillance, acute response (esp. agglutination, complement activation) 0.6–3
Table 1. Antibody Primary Functions and Adult Reference Ranges2,6,7
Figure 1
At the N-terminus of the Fab region are three hypervariable loops produced through random gene segments recombination and somatic hypermutation. These loops help determine specificity of the paratope, the region of the antibody that makes contact with a cognate epitope on an antigen. In the case of protein antigens, antibodies typically recognise non-continuous epitopes resulting from protein 3D conformational characteristics. 8–10 Random recombination events result in an enormous potential antibody repertoire, up to 1018 combinations.4,11,12 While the Fab region confers antigen specificity, the Fc region interacts will soluble and cellular host components, determining antibody function. The Fc region constant domains are encoded by different genes that undergo alternative splicing and class switching during B cell development to generate the various isotypes. Class switching is irreversible, involving constant region deletional recombination.1 This region is involved in anchoring the antibody in the B cell membrane, transducing antigen stimulated signaling, and when secreted, acts as ligand to Fc receptors to bind with and regulate other immune cells.
Antibody Effector Functions
The immune response is somewhat tailored by the inciting stimulus. The environment, type of stimulus (pathogen, trauma, toxin, etc.), and the encounter location within the body influence the humoral system response resulting in class switching to
antibody isotypes matched to handle the insult.2 The isotypes bind with cognate Fc receptors expressed on other cell types to modulate the appropriate immune response (Table 2). Fc receptor nomenclature incorporates the isotype-corresponding lowercase Greek letter. For example, FceRI refers to the first IgE receptor discovered, expressed by mast cells, basophils, and eosinophils. Often there are multiple Fc receptor subtypes for an individual isotype with differing affinities and differential expression across different immune cells. Altogether, the antibody, antigen and innate immune effector cell form a complex that results in a specific defence strategy determined by the Fc receptor binding, the binding affinity, and the cell types involved.3 As an example, IgG1 opsonises virus and via FcgRI receptor signalling, stimulates macrophage phagocytosis of virus. Similarly, IgM influences macrophages, but IgM activates complement that then activates macrophages. IgD can bind directly to basophils, promoting an inflammatory response via IL-4 release.13 The antibody isotype and subclass, including glycosylation status, play a crucial role in shaping the immune response, impacting various aspects of immune defence that when known can provide clues to researchers regarding the antigens encountered, tissues involved, immune systems engaged, potential for inflammation, and disease status.
B Cell Development, Activation and Antibody Production
Initially, IgM is the first isotype expressed by B cells. During
Cytotoxicity
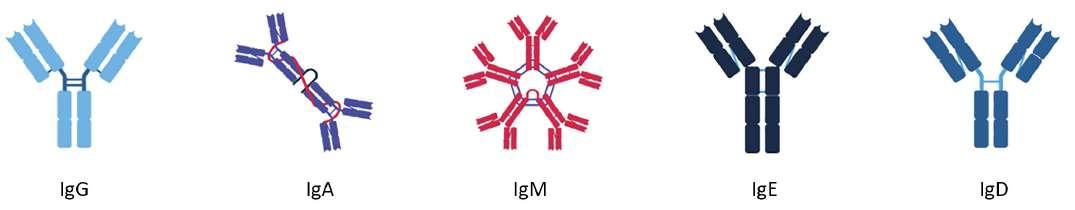
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 61 www.international-biopharma.com Elrig Inspired Protects From Effector Function Isotypes FC Receptors Effector Cells Examples Intracellular Organisms Opsonisation IgA1 αRI Macrophages IgG3 γRI, γRIIa, γRIIIa Helminths Degranulation IgG4 γRIIa Neutrophils, Mast Cells, Basophils, Eosinophils IgE εRI Mast Cells, Basophils, Eosinophils Extracellular Organisms Opsonisation, Degranulation IgG2 γRIIa Macrophages, Neutrophils Opsonisation, Degranulation IgA2 αRI Macrophages, Neutrophils Neutralisation, Degranulation IgD Galectin-9/CD44 Mast Cells, Basophils Neutralisation, Agglutination IgA, IgM pIgR Epithelial Cells of Mucosal Membranes IgE εRII Intestinal Epithelial Cells, Antigen Presenting Cells Viruses Opsonisation, Phagocytosis IgG1 γgRI, γgRIIa, γgRIIIa Macrophages, Monocytes, Natural Killer Cells IgA1 αRI Macrophages, Monocytes, Natural Killer Cells Complement Activation IgM C1q Macrophages Tumor Growth Antibody-Dependent Cell-mediated
IgG1, IgG3 γRIIIa Natural Killer Cells
2
Table
Figure
2
61
B cell development, immature bone marrow B cells express membrane-bound IgM, forming part of the B-cell receptor (BCR) complex, with the Fc region inserted into the cell membrane and the Fab region exposed extracellularly to interact with antigen (Figure 3). A signal transduction region (CD79A, CD79B) couples with the IgM to complete the complex. Early during development, BCRs with strong affinity for self-antigens will usually undergo clonal deletion via apoptosis, decreasing the likelihood of autoreactive antibodies. Surviving B cells undergo further maturation in the spleen or lymph nodes where they express transmembrane bound IgD or IgM antibodies. When B cells encounter an unrecognised antigen for the first time, the initial antibody response comprises IgM released into the serum predominantly as a pentamer. Although short lived, IgM is the first circulating antibody, marking disease early in infection. Among other functions, IgM can neutralise and agglutinate extracellular organisms, for example preventing bacteria from accessing the gut lumen, or activate complement in response to viruses. As the initial wave of IgM mounts and peaks, class switching and somatic hypermutation occur to tailor the immune response to the inciting event at a slight delay. Specificity remains intact, but the isotype switches to strategically match the inciting context, influenced by the specific biological compartment. B cells then form germinal centres expressing high levels of secreted antibody cognate to the stimulating antigen.12,14,16
The Value of Isotype Screening in Understanding Disease
Antibodies are excellent biomarkers for diseases not only infectious diseases, but all diseases. They are highly specific to antigens, are easy to obtain from serum, and are highly stable frozen for years. However, most analyses have focused on IgG and IgM antibodies.6 Isotype identification has been underutilised,
but the isotype carries quite valuable information. The different isotypes are expressed at different times during disease, are privy to different tissue compartments, and interact with different immune mechanisms, resulting in unique spatial and temporal expression patterns that can delineate disease. For example, while IgM and IgG are the most abundant isotypes in nearly all autoimmune diseases, IgA autoantibodies are prominent in antiphospholipid syndrome of the vascular system and IgE is prevalent in SLE.17 Taken together, the advantages of antibody and isotype screening for medicine include improved early disease detection, disease monitoring, outcome prediction, patient stratification, and greater understanding of immune mechanisms.
There are two basic approaches to antibody screening, targeted and untargeted. In a targeted approach, often used to refine diagnostics, specific antigens are assessed for antigenicity related to disease. In a meta-analysis of Rheumatoid Arthritis (RA) patients, Motta et. al. evaluated the diagnostic potential of different antibody isotypes to rheumatoid factor (RF). While IgM and IgG to RF are known to dominate serum, IgA emerged with the highest specificity to RA of 91%, but the sensitivity was only 49%.18 Many of the studies in the analysis did not measure multiple isotypes simultaneously, which could have raised both specificity and sensitivity. A multiple monitoring approach was used by Sieghart et. al. By combining quantification of antibody isotypes IgG, IgM, and IgA with antigen specificities to rheumatoid arthritis related antigens (RF, ACPA and RA33) they found a 30% improvement in diagnoses.19
Targeted antibody screening is often used in infectious diseases where the antigen is known. Assessing the ratios of
62 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Elrig Inspired
62 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Figure 3
isotypes expressed in these cases has merit beyond diagnosis. It may help predict predisposition to future disease. In a study of human papillomavirus (HPV), anti-HPV IgA and IgG1 were the most abundant isotypes found in patient serum, but with different pathologies. IgA was present early in disease while IgG1 persisted long-term, suggesting lifetime cumulative exposure to virus in some patients. Further, IgG1 expression and titer correlated with future risk of cervical cancer while the early presence of IgA correlated with reduced risk.20 These data indicate that the isotype expressed may help assess patient risk. By looking for antibody specificities and isotypes, researchers can also identify patient liabilities and stratify patients into appropriate treatment routines. In another example, HIV patients infected with SARS-CoV-2 lacked anti-Spike Protein IgA, but not IgG1.21 IgA is one of the first lines of defence against respiratory infections. In this case, infection with HIV may affect susceptibility and severity of future novel respiratory infections. With this information, clinicians can better direct care for HIV patients.
A better understanding of disease pathology can be achieved when examining multiple isotypes. In dengue virus (DENV), secondary infections are much more severe than initial infections. In the past, DENV research focused mainly on IgM and IgG antibody responses to DENV. More recently, Waickman et. al. found that following primary DENV infection, IgM, IgG, and IgA are expressed in near equal proportions. Further IgG1 and IgA dominated secondary infections. The researchers suggested that IgA may be competing with IgG1 for binding with DENV, preventing the IgG mediated immune responses and enabling the virus to persist in secondary infections.22 In this case, the virus appears to take advantage of the IgA response.
IgA is the most abundant isotype in the body (Table 1) found mostly in mucosal membranes. This isotype is one of the first to combat respiratory infections. Interestingly, IgA has access to the gut microbiome and it is believed that through regulating the gut flora, IgA may have strong influence on diseases such as cancer and neurodegenerative.23–26 IgA is emerging as a particularly specific isotype for disease detection across a variety of diseases, but it is not often studied.
These targeted approaches are straightforward, simple, and target known antigens. ELISA and protein microarrays are most suitable for this methodology. However, no new antigens or antigenic pathways are discovered. In an untargeted approach, the researcher compares antigen presence or absence between patients and control subjects against a massive library of potential antigens, numbering in the thousands. The approach is exploratory and often utilises a second validation study with a new group of patients to verify the results. This approach can uncover novel antigens related to disease. New disease related protein pathways can also be discovered with the potential for uncovering novel druggable targets. Functional protein microarrays are among the best technologies for this approach.
Large scale, untargeted antibody screening has been used to detect complex diseases such as cancer and neurodegenerative diseases, both of which produce aberrant protein expression patterns that humoral immune system recognises as foreign.27,28 These complex diseases can affect multiple organs resulting in
production of several disease related antibodies with unique specificities and different tissue specific isotypes. In a recent, comprehensive study by Patel et. al., 60 different IgG antibodies of interest were uncovered from a screen of more than 1600 antigens across a cohort of 157 patients with non-small cell lung cancer (NSCLC). Eighteen of the 60 antibodies correlated with post-resection survival rates. Evaluating various permutations of these 18 antibodies revealed that 13 strongly correlated with 5-year patient survival in both the study and validation cohorts.29 The repertoire of antibodies induced by these complex diseases can not only accurately detect disease early, ahead of many other diagnostics, but also indicate potential druggable protein pathways.30 In this case, Cancer Testis Antigens (CTA) featured prominently. CTAs are typically expressed in male germ cells and during embryogenesis. Thus, their ectopic expression in NSCLC patients (male and female) may provide an excellent well aimed therapeutic target.
For diseases such as cancer and Parkinson’s, researchers have yet to examine multiple isotypes. These diseases remain difficult to diagnose. Future research evaluating different antibody isotypes and their antigen specificities is likely to provide greater understanding of these diseases and better diagnostics.
The Role of Antibody Isotypes in Drug Development
Detecting the isotypes present in patient sera can provide valuable insights regarding inflammatory status that can lead to undesired drug reactions. Some isotypes can react with complement, promoting inflammation. IgG1, IgG3 and IgM are examples, and can induce macrophage activation leading to inflammatory cytokine release.1 Others, such IgG2 and IgG4 are anti-inflammatory. Examining patient serum for various isotypes and antigen specificities can help researchers identify autoantigens that induce inflammation. Further, this can help clinicians monitor patients and may provide valuable insight into adverse events from treatments.31 For example, distinct autoantibody signatures predict adverse events in patients receiving immune checkpoint inhibitor monoclonal antibody therapy.32,33 In neurodegenerative research, a promising finding regarding the effector function of IgG antibodies on microglial cells has promoted research into monoclonal antibody therapy for Alzheimer’s and Parkinson’s. Microglial cells increase uptake and degradation of tau and a-synuclein particles in vitro in response to FcgR activation, an important finding that could lead to new treatments for neurodegenerative diseases involving protein aggregates (Parkinson’s, Alzheimer’s, Huntington’s). However, FcgR activation results in inflammation.34 Further, most monoclonal antibody therapies utilise IgG1 antibodies that can react with FcgR. For future monoclonal antibody therapies, researchers have started examining IgG4, an isotype with low affinity for FcgR and that does not react with complement.1,35 Patient immunoprofiling may help determine the likelihood of inflammatory adverse events based off the repertoire of antibodies and isotypes present in serum. Further, selecting antibody isotypes will become an important decision to ensure the safety and efficacy of future monoclonal antibody therapies.
Simultaneous Measurement of Antibody Isotypes
Isotype screening has traditionally been done with immunochemistry using ELISA technology and isotype specific secondary antibodies. However, ELISA lacks the throughput and
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 63 www.international-biopharma.com Elrig Inspired
63
sensitivity of more modern protein microarrays. For biomarker discovery, functional protein microarrays are an excellent, cost effective, reliable, high throughput means of quickly profiling the myriad of antibodies present in an individual’s serum, and uncovering those related to disease. In these arrays, thousands of proteins are printed onto a slide surface. Antibodies from patient serum bind cognate proteins that are illuminated with indirect immunofluorescence. Secondary antibodies directed to IgG, IgA, IgM, etc. can be applied singly or in pairs to recognise both specificity and isotype from a single sample. Done properly, protein microarrays can accurately determine disease related antibodies and isotypes from small serum samples while also retaining function, allowing examination of post translational modifications, such as glycosylation and citrullination. Specificity, isotype and post translational activity
can be studied from a single sample. However, specificity is tightly linked to antigen shape, not protein sequence. 8,10 Sengenics is one of the few companies that offers a validated, high-quality dual colour detection analysis that considers antigen shape. Using the patented KREX technology, only correctly folded proteins with discontinuous epitopes intact are present on the array. This results in highly specific, highly reproducible antibody-antigen binding, a necessity for dual isotype profiling where non-specific binding could seriously confound the results (Figure 4). With this technology, distinct IgA and IgG profiles were observed among individuals with autoimmune disease, thus providing greater detail regarding the disease and possibly more accurate diagnosis (Figure 5).
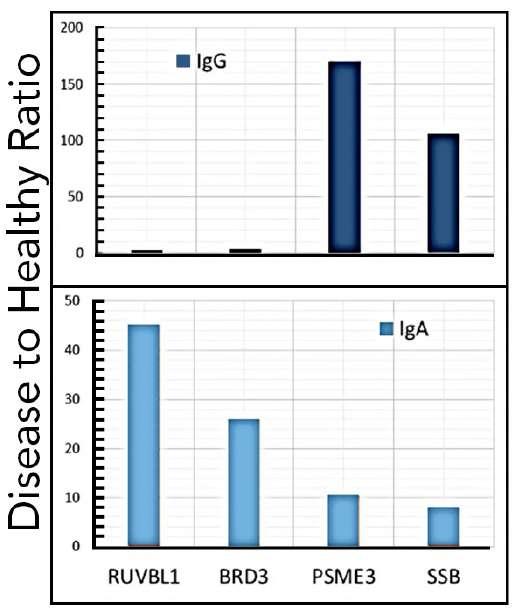
Conclusions
Circulating antibodies are among the first manifestations of disease, furnishing clinicians with unheralded details about patient status, often before symptoms. Identifying isotype and antigen specificity provides highly specific metrics for detecting and understanding disease pathology that allows clinicians to develop tailored strategies for different patients, monitor progress, and watch for adverse events. Study of different antibody isotypes in disease, especially complex diseases like cancer and neurodegenerative diseases, is in the early stages. Newer technologies such as next generation sequencing, modern protein microarrays, and machine learning have begun to enable researchers to capture this information.
REFERENCES
1. James, L.K., B cells defined by immunoglobulin isotypes. Clin Exp Immunol, 2022. 210(3): p. 230-239.
2. Hu, W.C., A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front Immunol, 2020. 11: p. 1992.
3. Lu, L.L., et al., Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol, 2018. 18(1): p. 46-61.
4. Briney, B., et al., Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature, 2019. 566(7744): p. 393-397.
5. Black, C.A., A brief history of the discovery of the immunoglobulins and the origin of the modern immunoglobulin nomenclature. Immunol Cell Biol, 1997. 75(1): p. 65-8.
6. Volkov, M., et al., Comprehensive overview of autoantibody isotype
64 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Elrig Inspired
Figure 4
Figure 5
and subclass distribution. J Allergy Clin Immunol, 2022. 150(5): p. 999-1010.
7. Gómez Román, V.R., J.C. Murray, and L.M. Weiner, Chapter 1Antibody-Dependent Cellular Cytotoxicity (ADCC), in Antibody Fc, M.E. Ackerman and F. Nimmerjahn, Editors. 2014, Academic Press: Boston. p. 1-27.
8. Van Regenmortel, M.H.V., Mapping Epitope Structure and Activity: From One-Dimensional Prediction to Four-Dimensional Description of Antigenic Specificity. Methods, 1996. 9(3): p. 465-72.
9. Muro, Y., et al., Synthetic compound peptide simulating antigenicity of conformation-dependent autoepitope. J Biol Chem, 1994. 269(28): p. 18529-34.
10. Barlow, D.J., M.S. Edwards, and J.M. Thornton, Continuous and discontinuous protein antigenic determinants. Nature, 1986. 322(6081): p. 747-8.
11. Hozumi, N. and S. Tonegawa, Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc Natl Acad Sci U S A, 1976. 73(10): p. 3628-32.
12. Tonegawa, S., Somatic generation of antibody diversity. Nature, 1983. 302(5909): p. 575-81.
13. Shan, M., et al., Secreted IgD Amplifies Humoral T Helper 2 Cell Responses by Binding Basophils via Galectin-9 and CD44. Immunity, 2018. 49(4): p. 709-724 e8.
14. Melamed, D., et al., Developmental Regulation of B Lymphocyte Immune Tolerance Compartmentalizes Clonal Selection from Receptor Selection. Cell, 1998. 92(2): p. 173-182.
15. LeBien, T.W. and T.F. Tedder, B lymphocytes: how they develop and function. Blood, 2008. 112(5): p. 1570-80.
16. Mårtensson, I.-L., et al., The pre-B cell receptor checkpoint. FEBS Letters, 2010. 584(12): p. 2572-2579.
17. Suurmond, J. and B. Diamond, Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest, 2015. 125(6): p. 2194-202.
18. Motta, F., et al., Rheumatoid factor isotypes in rheumatoid arthritis
diagnosis and prognosis: a systematic review and meta-analysis. RMD Open, 2023. 9(3).
19. Sieghart, D., et al., Determination of Autoantibody Isotypes Increases the Sensitivity of Serodiagnostics in Rheumatoid Arthritis. Front Immunol, 2018. 9: p. 876.
20. Wang, Z.H., et al., Type specificity and significance of different isotypes of serum antibodies to human papillomavirus capsids. J Infect Dis, 2000. 181(2): p. 456-62.
21. Smith, M., et al., Longitudinal IgA and IgG Response, and ACE2 Binding Blockade, to Full-Length SARS-CoV-2 Spike Protein Variants in a Population of Black PLWH Vaccinated with ChAdOx1 nCoV-19. Viruses, 2023. 15(448): p. 11.
22. Waickman, A.T., et al., Transcriptional and clonal characterization of B cell plasmablast diversity following primary and secondary natural DENV infection. EBioMedicine, 2020. 54: p. 102733.
23. Pruss, H., Autoantibodies in neurological disease. Nat Rev Immunol, 2021. 21(12): p. 798-813.
24. Li, T. and W. Le, Biomarkers for Parkinson's Disease: How Good Are They? Neurosci Bull, 2020. 36(2): p. 183-194.
25. Pu, A., et al., The Impact of IgA and the Microbiota on CNS Disease. Front Immunol, 2021. 12: p. 742173.
26. Zhong, Z., et al., Pro- and Anti- Effects of Immunoglobulin AProducing B Cell in Tumors and Its Triggers. Front Immunol, 2021. 12: p. 765044.
27. Kathrikolly, T., et al., Can serum autoantibodies be a potential early detection biomarker for breast cancer in women? A diagnostic test accuracy review and meta-analysis. Syst Rev, 2022. 11(1): p. 215.
28. DeMarshall, C.A., et al., Detection of Alzheimer's disease at mild cognitive impairment and disease progression using autoantibodies as blood-based biomarkers. Alzheimers Dement (Amst), 2016. 3: p. 51-62.
29. Patel, A.J., et al., A highly predictive autoantibody-based biomarker panel for prognosis in early-stage NSCLC with potential therapeutic implications. Br J Cancer, 2022. 126(2): p. 238-246.
30. Duarte, J.S., J; Mulder, N; Blackburn, J., Protein Functional Microarrays: Design, Use and Bioinformatic Analysis in Cancer Biomarker Discovery and Quantitation, in Bioinformatics of Human Proteomics, X. Wang, Editor. 2013, Springer Science+Business Media Dordrecht. p. 39-74.
31. Da Gama Duarte, J., et al., Autoantibodies May Predict ImmuneRelated Toxicity: Results from a Phase I Study of Intralesional Bacillus Calmette-Guerin followed by Ipilimumab in Patients with Advanced Metastatic Melanoma. Front Immunol, 2018. 9: p. 411.
32. Johannet, P., et al., Baseline Serum Autoantibody Signatures Predict Recurrence and Toxicity in Melanoma Patients Receiving Adjuvant Immune Checkpoint Blockade. Clin Cancer Res, 2022. 28(18): p. 4121-4130.
33. Ibrahim, M., et al., Determinants of racial disparities in immunerelated adverse events (irAE) with checkpoint inhibition (ICI) in melanoma. Journal of Clinical Oncology, 2023. 41(16_suppl): p. 9549-9549.
34. Cao, S., D.G. Standaert, and A.S. Harms, The gamma chain subunit of Fc receptors is required for alpha-synuclein-induced pro-inflammatory signaling in microglia. Journal of Neuroinflammation, 2012. 9(1): p. 259.
35. Katsinelos, T., et al., The Role of Antibodies and Their Receptors in Protection Against Ordered Protein Assembly in Neurodegeneration. Front Immunol, 2019. 10: p. 1139.
Kevin Bittman
Kevin Bittman is a Senior Content Manager for Sengenics. Kevin has an MS and Ph.D. in Physiology awarded from the University of Connecticut. With 13 years as a bench scientist and 15 years as a Field Applications Scientist, Kevin uses his experience to help others understand the tools available to accelerate their research.

INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 65 www.international-biopharma.com Elrig Inspired
65
Reagents for Biomedical Research
What are Exosomes?
Exosomes are small, membrane – bound extracellular vesicles (EVs) that are released by cells, such as adipose –derived stem cells/stoma cells, mesenchymal stem cells/ stroma cells (MSC), dendritic cells, T and B cells. These vesicles can contain a wide variety of molecules, such as proteins, lipids and nucleic acids, that are derived from the cell of origin.
They play a key role in intercellular communication, as they can transfer contents to other cells and influence their behaviour. They have been implicated in a wide variety of physiological and pathological processes, including immune regulation, tissue repair, and cancer progression. They are also used as a tool for drug delivery and as a biomarker for disease diagnosis and prognosis.
How are EVs produced?
One approach to produce exosomes for therapeutic purposes involves mobilizing hematopoietic stem cells (hSC) from the patient's peripheral blood, followed by differentiation into dendritic cells (DCs). These DCs are then genetically engineered with tissue-specific exosomal membrane protein plasmids, which allows the resulting exosomes to express the same tissue-specific proteins on their surface.
Once the exosomes are produced, they are loaded with therapeutic cargo such as mRNA or siRNA, which are designed to target specific disease-causing genes or pathways. These exosomes are then subjected to quality control measures to ensure purity, yield, and functional activity, before they are administered to patients.
PELOBIOTECH GmbH is a supplier of reagents for biomedical research. With a focus on primary cell culture the Munich Cell Specialist distributes one of the broadest portfolios of cell culture products & media from their exclusive partners all over the world. The portfolio currently offers 5,000+ products in the field of human &animal primary cells, adult stem & iPS cells, media & tools. Scientists may choose from a range of 18,000 cell culture products. PELOBiotech offers modified and tagged primary & stem Cells, diseased cells, and 2D&3D cell culture systems. Researchers also use our comprehensive Cell Sourcing Service from healthy and diseased donors (tissue, cells, plasma…).
As one of our key features we offer customized solutions with a choice of prime media in R&D, GMP-like and GMP quality, and keep a special focus on xeno-free, serum-free and defined solutions. We also present our own prime product line Cellovations®, based on heart, experience and knowledge, which researchers need for a successful and accelerated set-up.
Furthermore, PELOBIOTECH develops and produces special cell culture media for human primary cells, stem cells and embryonic stem cells as well as special supplements for tumor stem cells.
We focus on our customers and their research first. At PELOBiotech you get everything from one shop, talking from scientist to scientist. This makes PELOBIOTECH a leading provider of excellent research tools worldwide. Our mission is to provide all scientists with high-quality cell culture products to bridge the gap from bench to bedside with top notch solutions for the research project.
PELOBiotech GmbH – More than Competence4Cells

Tools for Exosome Production
Cultivation and expansion of mesenchymal stromal cells/stem cells (MSCs) and production of extracellular vesicles (EVs) have gained much importance in both basic research and regenerative medicine. One factor for success is the choice of appropriate cell culture media that supports both the expansion of MSCs and the production of functional EVs.
Ready-to-use media saves time and offers the advantage of standardized cultivation of MSCs. They contain all necessary nutrients, growth factors, hormones, and buffer systems for optimal expansion and/or production of exosomes. The quality of EVs produced is highly dependent on culture conditions and media. Ready-to-use systems optimized specifically for EV production can increase or enable EV numbers and functionality.
One of the highest quality supplements on the market is the human platelet lysate UltraGRO Pure GI GMP Grade or even the exosome-depleted UltraGro-PURE for EV production with MSCs.
Those looking for an alternative rely on chemically defined media: CellCor EXO CD or CellCor CD MSC medium is also used very successfully for isolation, expansion and MSC-dependent EV production. No media change from culture to isolation is required, and exosomes obtained with CellCor EXO CD provide high functionality and regenerative effects.
66 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Elrig Inspired
Why us?
• Get your answers in a blink
• Fast Delivery & Skilled Logistics
• More than 1,000 Human & Animal Primary Cells straight off the shelf

• Unique Cell & Tissue Sourcing
• Established workflows guarantee reliable timeline
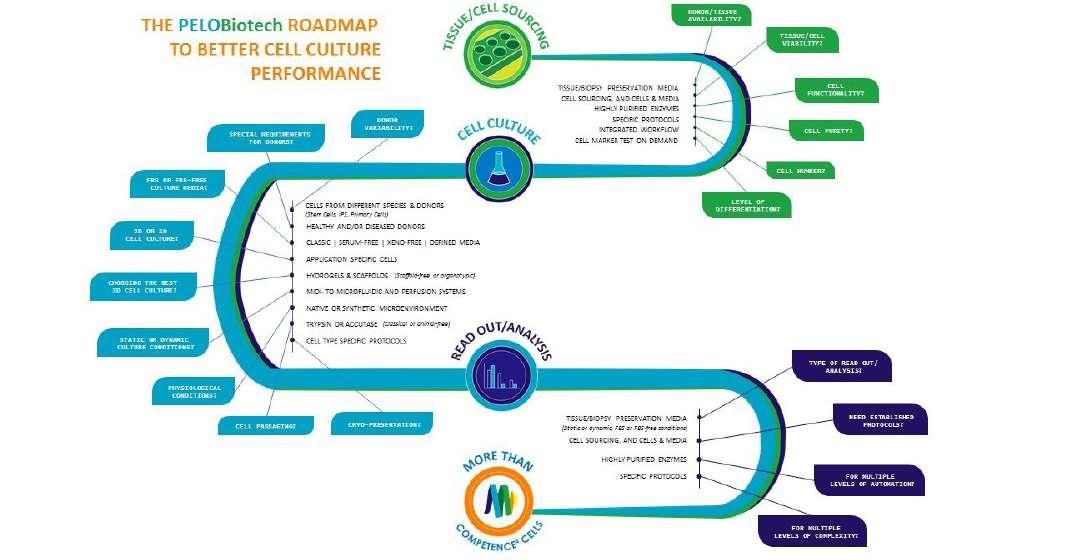
• Progressive Portfolio&Global Network to get predictive results
• Easy switch from Research to cGMP products in a second
With 18,000+ products offers your competent partner PELOBiotech one of the broadest portfolio of Primary Cells, Stem Cells and corresponding Media for 2D & 3D Cell Culture worldwide.

Tissue Dissociation
Highly Purified Enzymes for High Quality Cell Isolation.
• Collagenase HA & MA,
• Gold & Gold Plus (GMP)
• rec. Collagenase (ACF)
• DE Collagenase
• Enzyme Blends
• BP Protease (GMP & ACF)
• Thermolysin (GMP & ACF)

Extracellular Matrices & Hydrogels
Gelatin, Speed Coating Solution, PhenoDrive, Collagen (I, II, III, IV), Fibronectin, Lamin, Vitronectin, ECM-Mixtures, EHS Matrix, alternatives to Matrigel and organ & cell culture derived ECMs.
3D Technologies & 3D Models
• Natural & Synthetic Hydrogels
• Natural & Synthetic Scaffolds
• SpheroSeev – Spheroid formation tool



• Scaffold-based static 3D Cell Culture
• 3DCoSeedisTM for scaffoldfree organoid formation
Cellular Systems / Human & Animal



• Neural Cells
• Skin Cells
• Hair Cells
• Oral and Nasal Cells
• Ocular and Retinal Cells
• Lung/Airway Cells
• Gastrointestinal and

• Endocrinal Cells
• Cardiac Cells
• Cardiomyocytes
• Renal Cells
• Prostate Cells
• Liver Cells
• Bladder and Urethral Cells
• Breast Cells
INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY 67 www.international-biopharma.com 67 Elrig
Inspired
Dr. Lothar Steeb & Dr. Peter Frost, CEOs: Competence of 45+ yrs Cell Culture experience
• Adipose Tissue Cells
• Musculoskeletal Cells
• Reproductive Organ Cells
• Lymphatic Cells
• Blood Cells & Bone Marrow Cells
• Diseased Cell Systems
• (Diabetic, Cancer and more)

• ES/iPS & iPS cell derived Cells
• Adult Stem Cells
Adipocytes, Astrocytes, Chondrocytes, Endothelial Cells, Epithelial Cells, Fibroblasts, Hepatocytes, Kupffer Cells, Mesenchymal Stroma/Stem Cells, Microglia Cells, MNCs, Osteoblasts, Skeletal Muscle Cells, Smooth Muscle Cells, Stellate Cells, Tenocytes and more.
Cell Culture Media Systems
• Media for Special Applications e.g.
• Extracelluar Vesicle (EV) Production
• Media for ES/iPS Cells & Adult Stem Cells

• Media for Primary Cells (from FBS to defined)
• Supplements for improved Cell Culture (AB serum, A serum, BIT, hPL, ECGS)
3D Dynamic Fluidic in-vitro Models
• Midifluidic & Microfluidic Systems
• Multi Organ Perfusion Systems
Antibodies, Proteins, Dyes & TR-FRET
• Antibodies
• Cytokines, Growth Factors & Chemokines
• Dyes & Europium Chelates
Arrays & Assays
• CFC-Assay Media
• MicroMatrix Array
• Proliferation Assay

• Mycoplasma Detection Assays

• Chimerism Assay & Cell Purity Assay
Sample Purification & Preparation

• Using Magnetic Forces and High Capacity Microspheres (TI-IMAC, Zr-IMAC, HILIC, Trypsin, Chemotrypsin, Prot A, Prot G, Streptavidin …..)
Dr. Lothar Steeb
Dr. Lothar Steeb is our expert in the field of cell culture research and media development, and is leading the R&D and production departments at PELOBiotech as well as numerous research projects with our academic partners.


Dr. Peter Frost
Dr. Peter Frost has more than 25 years of experience in marketing and sales of life science products and service solutions. He consults our customers about the variety and possibilities of our products, and is our expert for GMP products and 3D technology.
68 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 Elrig Inspired
7 December 2023,1 Wimpole Street, London
Attend London’s premier Life Science networking conference.
Under a headline theme of Maximising Returns from Life Science Innovation, the event will feature:

• Plenary Presentations and Panel Debates
• A digital programme, including live online innovation workshops
• Pre-scheduled 1-2-1 Partnering via a dedicated conference app
• 300 delegates from across the Life Science & Healthcare industry


• Exhibition
• Genesis Fringe: focused events around Genesis

• 2023 Genesis BioNewsRound Award
genesisconference.com
INTERNATIONAL
#genesislondon23 @OneNucleus genesis 2023
70 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3 The Premier International Conference covering all aspects of respiratory drug delivery Edinburgh International Conference Centre and Virtual 6/7/8th December 2023 The Premier International Pulmonary and Drug Delivery Conference • 3 days of expert lectures • 1000+ attendees expected • 100+ scientific posters • Large Industry exhibition • Free registration for students • Networking opportunities • Awards and grants for young researchers www.ddl-conference.com info@ddl-conference.com Visit the website for full details about the conference
MUNICH IS THE PLACE TO BE THIS FALL!

NOVEMBER 68, 2023 | MUNICH, GERMANY
BIO-Europe®, recognized as Europe’s flagship partnering event, is on track to be the largest yet this fall. Registration patterns not only support impressive growth, but they also reinforce the event’s international appeal: it is indeed ‘your gateway to the global biopharma community.’ Over 5,000 delegates will convene in Munich. They will be joined by an additional 800 delegates for digital partnering from November 14–15, 2023, hosted on partneringONE®.
You must be there to experience it!
REGISTER NOW: bioeurope.com
In collaboration
Produced by:
with:
Page 25
Page 35
Page 55
Aldevron LLC
A&M STABTEST
Autoscribe Informatics UK
Page 71 BIO Europe
Page 57–59
Page 21
Page 70
Page 53
Page 44–46
Page 50–51
OBC
Page 69
Page 13
Page 27
Page 6–8
Page 41
Page 31
Page 66–68
Page 47–49
Page 9
Page 35
Page 43
Page 5
Collaborative Drug Discovery Inc.
CRYOPDP
DDL 2023 – Drug Delivery to the Lungs
Discovery Park
eclateral
ELRIG UK
FUJIFILM Wako Chemicals USA
Genesis 2023
GenXPro GmbH
Novo Nordisk Pharmatech A/S
Optibrium Ltd.
Owen Mumford Ltd.
PCI Pharma Services
PELOBIOTECH GmbH
POLYPURE AS
Precision for Medicine
Protein & Antibody Engineering Summit
Qualogy Ltd.
Reciprocal Space
IBC ReciBioPharm
Page 17
Page 3
Page 60–65
Page 39
Richter-Helm Biologics GmbH & Co. Kg
Scientific Protein Laboratories
Sengenics Corporation Pte Ltd
SGS
IFC Sygnature Discover
I hope this journal guides you progressively, through the maze of activities and changes taking place in the biopharmaceutical industry
IBI is also now active on social media. Follow us on: www.facebook.com/Biopharmaceuticalmedia www.plus.google.com/biopharmaceuticalmedia

72 INTERNATIONAL BIOPHARMACEUTICAL INDUSTRY Autumn 2023 Volume 6 Issue 3
Subscribe today at www.international-biopharma.com or email info@senglobalcoms.com
www.twitter.com/biopharmace www.biopharmaceuticalmedia.tumblr.com/ Ad Index
Delivered with certainty
Our worldwide reach, localised market knowledge and ever-expanding alliance of expertise allow us to anticipate your needs and provide tailored, reliable services to consistently deliver on time and in full. Discover a global CDMO dedicated to meeting your evolving needs. Meet the Recipharm team at CPHI, booth 3N60 in hall 3.0.


Endotoxin Detection Reagent
Convenient and
www.wakopyrostar.com ~ wkuspyrostarinfo@fujifilm.com FUJIFILM Wako Chemicals U.S.A. Corp. © FUJIFILM Wako Chemicals U.S.A. Corp. - 2023 PYROSTAR ™ Neo is a new endotoxin detection reagent developed by recombinant technology.
Neo PYROSTAR ™ Recombinant
A new synthetic colorimetric method that can be run using a standard absorbance microplate reader.
Easy to Use.
