





Sponsor Company: www.international-pharma.com Volume 15 Issue 2 Peer Reviewed The Four Trends Set to Disrupt Highly Regulated Industries in 2023 Individual Device Identification Coding and Marking for the New Dimension in Patient Health and Safety Why an Outcome-driven Approach to Supply Chain Is a Strategic Advantage for Pharmaceutical Businesses Advancements of Gene Editing Technologies (CRISPR/Cas9)
PROTECT YOUR PHARMA PRODUCT WITH OUR QUALIFIED REFRIGERATED AND DEEP FREEZER CONTAINERS.
REDUNDANT REFRIGERATED & -70°C DEEP FREEZER CONTAINERS FOR PHARMA PRODUCTS
Klinge Corporation has been safely transporting and storing pharmaceuticals for nearly 40 years.
Avoid Temperature Excursions with Redundant Reefer/ Freezer Containers


Keep pharma products at the required temperature from +25°C down to -70°C
Meet international transport regulations
Safeguard pharma products with temperature recorder & online temperature tracking

inquiry@klingecorp.com


www.klingecorp.com
USA: + 1-717-840-4500
DIRECTOR: Mark A. Barker
BUSINESS DEVELOPMENT: Eliza Sarfaraz eliza@senglobalcoms.com
EDITORIAL: Virginia Toteva virginia@senglobalcoms.com
DESIGN DIRECTOR: Jana Sukenikova www.fanahshapeless.com
FINANCE DEPARTMENT: Akash Sharma accounts@senglobal.com
RESEARCH & CIRCULATION: Jessica Chapman jessica@senglobalcoms.com
COVER IMAGE: iStockphoto ©
PUBLISHED BY: Senglobal Ltd.
Unit 5.02, E1 Studios, 7 Whitechapel Road, E1 1DU, United Kingdom
Tel: +44 (0) 2045417569
Email: info@senglobalcoms.com www.international-pharma.com
All rights reserved. No part of this publication may be reproduced, duplicated, stored in any retrieval system or transmitted in any form by any means without prior written permission of the Publishers.
The next issue of IPI will be published in Autumn 2023. ISSN No.International Pharmaceutical Industry ISSN 1755-4578.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
2023 Senglobal Ltd./Volume 15 Issue 2 – Summer– 2023
06 Editor’s Letter
TALKING POINTS
08 Introducing a New Era in HPLC
The Alliance iS HPLC System and Empower 3.8.0 release also support Waters new eConnect HPLC Column technology, designed to ensure complete column traceability, and facilitate post-run troubleshooting. Readyto-use from the box, eConnect columns are securely fitted with a NFC-enabled eConnect device tag, ensuring they are always automatically identified, verified, and tracked by the system. Analysts and managers can easily track column usage and make sure they’re always using the right column. IPI speaks with Fraser McLeod, Vice President of QA/QC for Waters Corporation over, Waters next generation Alliance iS HPLC SystemTM which will revolutionise productivity and efficiency for modern QC labs.
10 IPI Speaks with Pharmalex’s Local Affiliate Services Lead on The Rapidly Changing Global Market
There is rising demand from clients for services at the local level to manage mandatory activities that are normally handled by their affiliates. The need was initially greatest at large organisations, which were looking to streamline the business and focus on their strategic development projects. To achieve that, they needed support to ensure they met their local compliance obligations, especially for their mature products. IPI speaks with Dr. Stefanie Lietsch-Dallwig, Senior Director Global Program Management – Service Solution Lead Local Affiliate Services, at PharmaLex, over why, a more all-encompassing approach to managing the local affiliate important.
REGULATORY & MARKETPLACE
14 The Four Trends Set to Disrupt Highly Regulated Industries in 2023
Businesses and manufacturers in highly regulated markets, in particular pharmaceutical, food and beverage, cosmetics and consumer packaged goods (CPG), have experienced huge changes in recent years, most significantly in the technologies they use, the regulations they face, and the changing demands from their customers. Bob Tilling at Kallik identifies four trends set to disrupt manufacturers in highly regulated industries in 2023: the industrial metaverse, the need to get a tighter arm on cybersecurity, the rise of eco-anxiety as the 2025 sustainable packing deadline looms, and a focus on data centricity.
16 Vaccine Regulation in India
Vaccines are the most important health intervention. Vaccination stimulates the immune system to identify and battle invaders such as viruses or bacteria. A vaccination encourages our immune system to manufacture antibodies the same way as exposure to the illness would. A stringent administrative method must be followed during the creation of a vaccine to determine its safety, effectiveness, and quality. In the next lines, Dr. M P Venkatesh, Mr Arjun HR and Mr Praveen Halagali at JSS College of Pharmacy explain how vaccine regulations work in India.
DRUG DISCOVERY, DEVELOPMENT & DELIVERY
22
Serving the 0.05% of Patients Living with a Rare Disease
Though estimates show that there are now up to 30 million people in the European Union living with a rare disease, progress with diagnosis remains worryingly slow, with patients suffering as a result. Healthcare professionals (HCPs) must consider between 6,000 to 8,000 rare diseases when diagnosing a patient, with the result being that it is often common
INTERNATIONAL PHARMACEUTICAL INDUSTRY 1 www.international-pharma.com Contents
for them to fail to spot the symptoms within relatively small patient populations (five or fewer in 10,000). This means patients commonly wait years for a diagnosis or may never be diagnosed at all. In this article, Chris Moore at Veeva Europe reveals how some leaders are transforming their approach to data and technology to speed the delivery of new treatments to patients.
24 Unlocking the Potential of Drug Discovery Labs with Automation
Trying to achieve more without increasing costs, sacrificing quality, or slowing down growth is a constant mission for any expanding business or industry – but it is particularly acute when it comes to drug discovery. The COVID-19 pandemic highlighted the pressing need to rapidly develop effective drugs and treatments, and the growing industry has made remarkable progress in recent years – with the government recently investing £227 million into the sector. Areas like precision medicine, novel vaccines, cell and gene therapies and synthetic biology are all driving healthcare forwards – but there are challenges for drug discovery companies to effectively scale and bring new cutting-edge products to market. Russell Green at Automata explains that the three main challenges are the finite amount of lab space for expansion in the UK, manual processes impeding scaling and reproducibility, and scientists spending too much time on manual tasks rather than research and discovery.
26 How to Harness the Benefits of Inhalation When Re-formulating Biologics
Reformulating an existing drug product’s route of administration can extend exclusivity. This, coupled with breakthrough innovation in targeted therapies for chronic and orphan lung diseases, means that companies are shifting towards delivery via the respiratory tract. In this article, Bernhard Müllinger, of Resyca® and Kris Brosig at Recipharm, discuss the current challenges of inhaled biologics, exploring how formulation and device innovation may be the answer to overcome them.
30 Advancements of Gene Editing Technologies (CRISPR/Cas9)
The dynamic promise of gene editing for humans is the ability to precisely manipulate the sequence of the cell genome to overcome genetic diseases. CRISPR nucleases have revolutionary potential to enable major medical breakthroughs. Dr. Inbar Friedrich Ben-Nun and Fatma Aybegum Senkesen at Lonza explain that gene editing allows for the modification of existing DNA in a cell, where genetic material is added, removed, or replaced at precise points within the genome.
34 Gene Edited iPSCs May Find a Place in Allogeneic Therapies
The pluripotent state has been proven time and again using various protocols for generating cell types and organoids covering nearly
every system in the human body. While the iPSC may be considered a “finicky” cell type to work within the lab and costly to grow due to reliance on complex growth media and matrices, it is unique in that it is a patient (or donor) sourced cell type, but with the added potential for long-term growth in culture. Easily derived from primary skin biopsies or blood, iPSCs maintain the existing genetic and epigenetic makeup of the human from which they came without the ethical dilemmas surrounding the use of embryonic stem cells. The author Amanda Haupt at Revvity discusses that nearly as important as their differentiation potential and proximity to the primary source is the ability of iPSC cultures to be scaled and banked for manufacturing gene and cell therapies.
CLINICAL & MEDICAL RESEARCH
38 Accessibility in Rare Disease Paediatric Clinical Trials
Approximately 360 million people globally are living with a rare disease. Over half of these are children. Many of the diseases are life-threatening, and only 5% have an approved treatment. Trials in rare diseases are typically conducted at fewer sites, are longer in duration and are more frequently terminated than trials with more common indications. Kirsten Sherman Cervati and William C. Maier at ICON reveal that while there is continued growth in the development of drugs to treat rare diseases, clinical trial accessibility for patients remains challenging due to the requirements inherent in these studies.
TECHNOLOGY
40 In-flight Data Control: How to Approach the Next Frontier in Transforming Life Sciences Business Process Efficiency
The trouble with non-standardised data, from an internal company perspective, is that it hampers agility and the ability to innovate. If each department uses slightly different terminology for a product and has its system and way of logging information, the scope for coordinating associated insights, identifying opportunities, and accelerating processes will be compromised. Here, Max Kelleher, Chief Operating Officer at Generis and Remco Munnik, a Director at Iperion, a Deloitte business, offer practical tips on how companies can systematically control and harness the flow of data between functional silos and the potential benefits this could have.
44 The Future of AI in Biotechnology On the Precipice of the New Technological Revolution
The artificial intelligence revolution kicked into high gear seemingly overnight. Generative AI platforms like ChatGPT and Google Bard have dominated headlines for months as we explore the possibilities for this powerful new technology, which has potentially significant implications for nearly every industry on the planet. Eduardo Abeliuk at TeselaGen says, although it’s still early days, there are many indications that we’re on the precipice of a technology revolution, unlike anything we’ve ever seen before. AI is allowing biotechnologists to augment the very building blocks of life in service of a healthier future for humanity. We are in uncharted territory, and how this shakes out will have world-changing ramifications.
MANUFACTURING
46 Cutting-edge Formulation is Needed to Enable the Next Generation of Oral Biologics
Despite OSD popularity, certain active pharmaceutical ingredients (APIs) are incompatible with traditional OSD formulation techniques, as is the case for biologics. This is due to a myriad of reasons that
2 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Contents







INTERNATIONAL PHARMACEUTICAL INDUSTRY www.international-pharma.com
stem from biological physicochemical properties, stability, absorption, and immunogenicity. In this article, Dr. Uwe Hanenberg of Recipharm, explores recent advances in formulation technologies for both OSD and oral liquid dosage forms designed to enable the creation of oral biologics.
48 Individual Device Identification: Coding and Marking for the New Dimension in Patient Health and Safety
Counterfeit products and product piracy are serious issues for the pharmaceutical industry – but recent product marking requirements in developed economies, including the US and the European Union, has gone some way towards securing legitimate pharmaceutical supply chains. The WHO estimates that the share of falsified medicine in global marketplaces ranges from 10% in certain low- and middle-income economies to as little as 1% in developed countries. Ian Chapman at Domino states that while there is still much to be done to increase the scope and reach of current legislation and improve safe access to pharmaceuticals in less developed economies, regions that have already implemented compulsory serialisation of pharmaceuticals should also prepare for change.
50 Potent Product Manufacturing in a Multi-product Facility
Highly potent drugs are becoming increasingly common in the drug development pipeline. The focus on oncology, rare diseases, and targeted therapies is growing even more acute. While highly potent compounds have benefits in treating many medical conditions, companies with promising highly potent active pharmaceutical ingredients (HPAPIs) can face significant challenges when it comes to using them in development and manufacture. Key challenges in bringing them to market, reckons Anshul Gupte at Catalent, include ensuring that workers and the environment are protected from exposure and, in a multi-product facility, delivering adequate, compliant controls to mitigate the risk of cross-contamination.
PACKAGING
52 Effective Operations for Risk Reduction and Supply Chain Resilience
There is no denying that the market for injectable drug delivery devices is growing rapidly, with forecasts predicting an increase from $39.93 billion in 2022 to $43.54 billion in 2023 at a compound annual growth rate (CAGR) of 9.0%. With 60% of drugs in the R&D pipeline designed for injectable delivery, there are no signs of this trend slowing shortly. However, bringing a new drug delivery device to market is a long and complex process – from defining user requirements and early design concepts, going through building and testing prototypes, human factors testing and finally device verification and validation. John Swift at Owen Mumford Pharmaceutical Services states that producing a device which can be successfully used for drug delivery by different patient groups at each stage of the industrialisation process must be carefully considered.
HEALTH OUTCOMES
54 Choosing and Developing User-friendly Osmotic Laxatives for a More Patient-centric Portfolio
At least 1 in 10 people worldwide suffer from constipation at some point in their lives. It affects people of all ages and has many causes. The symptoms of constipation include pain in the lower abdomen and irregular and painful bowel movements. Laxatives are often needed in addition to dietary changes to treat constipation. There are many laxatives to choose from, each with different mechanisms of
action and, consequently, different advantages and disadvantages. Dr. Martin Koeberle and Dr. Verena Garsuch at HERMES PHARMA look at the important role of osmotic laxatives and how they overcome many of the side effects and drawbacks of other constipation treatments.
58 Artificial Intelligence is a Core Pillar in the Evolution of Digital Health and Patient-centric Solutions
The US Food & Drug Administration has stated that digital technology is driving a revolution in healthcare. The lines between healthcare delivery and clinical research are blurring as the patient becomes a key partner and focus. We see a rapid expansion in the use of mobile and patient-centric devices, exponential growth in the volume and diversity of life sciences data and acceleration in the use of data-dependent computation to gain insight and automate, loosely called artificial intelligence (AI). Michael Phillips and Gerard Quinn at ICON discuss Artificial intelligence as a core pillar in the evolution of digital health and patient-centric solutions.
LOGISTICS & SUPPLY CHAIN MANAGEMENT
60 Net Zero Healthcare –Priorities for Decarbonising the Pharma Supply Chain
The climate and our health are inextricably linked. The effects of climate change on global health systems and outcomes are already clear, with WHO proclaiming it to be the biggest health threat facing humanity today. And it’s projected to get worse over time. Every year between 2030 and 2050, climate change is expected to cause an additional 250,000 deaths. Here, Steve Brownett-Gale at Origin, discusses that pharma, as one of the largest global industries, is both part of the problem and the solution for minimising the adverse effects.
63 Harnessing the Power of cMaaS in Pharma Logistics: Ensuring Regulatory Compliance and Sustainable Supply Chain Management
End-to-end digital visibility across the supply chain can be used to identify hotspots, support real-time decision making and improve demand forecasting. Pharma companies, acting in collaboration, can incentivise third party organisations to set their own targets for improving their environmental footprint, both upstream and downstream from their own operations. Modern technology such as cMaaS offers many advantages. With realtime monitoring and control capabilities, cMaaS can help address the challenges of temperature excursions and sudden changes in environmental conditions. Charles Bourbonnais of Hive-Zox International S.A, explains that, by providing real-time information and automated responses, cMaaS can help logistics providers to optimize their supply chain, reduce costs, and ensure the safety and efficacy of pharmaceutical products.
66 Why an Outcome-driven Approach to Supply Chain is a Strategic Advantage for Pharmaceutical Businesses
Since 2020, the pharmaceutical industry has been grappling with frequent and rapid changes, shifting customer demands, and increasing costs. In response, organisations have stepped up and adapted quickly, recognising opportunities to discover and trust new technology and approaches to create resilience. However, for pharma businesses, the fact remains that most supply chains are still driven by activity and necessity instead of outcomes and possibilities. So why is this happening? Philip Ashton at 7bridges explains that due to the complex and specialist nature of the pharmaceutical industry, often results in the siloing of data across different platforms and partners, which limits visibility and reduces resilience.
4 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Contents



INTERNATIONAL PHARMACEUTICAL INDUSTRY 5 www.international-pharma.com • Simple, one-person operation • Low profile • Eliminates floor damage • Low friction/vibrationless • Unlimited maneuverability • Precise positioning • Auto-loading • Pharmaceutical grade construction Call: 1-800-426-4757 www.aerogo.com/applications/pharmaceutical-biotech/ info@aerogo.com Pharmaceutical processing equipment moves are made easier by capturing the power of air
2023 is shaping up to be a transformative year for the pharma industry, even more so than in years past. While the industry may endure a reduction in some corners, following years of growth, it will also see rapid expansion in burgeoning areas. The market has been tough for most industries, especially tech where stocks have dipped significantly, and capital has dried up. Jobs have been cut across the board and there is a new realization that large companies can survive and be effective with a smaller workforce. Other industries have taken notice with banks, retail and media following suit. We saw some examples of this in the pharma industry as well and expect this to continue to be a theme in 2023.
In the last 20+ years, pharma has been on a massive acquisition spree. Mergers and acquisitions (M&As) had been a key strategy for expansion and growth. Not anymore. Big mergers are going to dry up for three reasons. The acquisition spree has not left that many targets that are up for grabs. The ticket size has gone up significantly for the ones that are acquisition targets and the risk involved would keep companies on the fence and the capital to fund these deals has dried up. But the industry will still see some large mergers. This M&A activity will focus on acquiring speciality, rare, AI and platform-driven therapeutics.
In 2022, there were 18 AI-created drugs in the clinical phase, up from zero in 2020. According to Gartner, by 2026, AI-based discovery will overtake traditional bench-based research. On the other hand, digital therapeutics, apps, and smart devices are being launched in rapid succession.
Pharma companies are finally learning to speak the language of patients, who at the end of the day are consumers of healthcare products and services. Expect the purpose statements to shift from ‘improving patient lives’ to ‘health and wellbeing’ of consumers with more focus on prevention.
Editorial Advisory Board
Bakhyt Sarymsakova, Head of Department of International Cooperation, National Research, Center of MCH, Astana, Kazakhstan
Catherine Lund, Vice Chairman, OnQ Consulting
Deborah A. Komlos, Principal Content Writer, Clarivate

Diana L. Anderson, Ph.D president and CEO of D. Anderson & Company
Franz Buchholzer, Director Regulatory Operations worldwide, PharmaNet development Group
Francis Crawley. Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organization (WHO) Expert in ethics
Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
The ongoing trend of new drugs becoming ever more complex is sure to continue. The number of synthetic steps for making small molecule APIs has grown by two-thirds in the past 20 years, as has the number of chiral centres. Average molecular weights continue to rise, and maybe three-quarters have poor solubility. With so many NMEs being approved via some form of expedited approval pathway, there is bound to be continued high demand for CDMO services, and a demand to meet ever-shorter timelines. Innovation will be vital, and digital tools that support the development of robust processes will be increasingly important.
As you can see, there are several new factors coming into play in our market this year. Pharma companies must not only stay abreast of these trends, but they must make earnest efforts to enter these spaces as well. They must be willing to get on board, or they will be left behind at the station.
Welcome to the Summer Issue of IPI. Keeping in mind all the new developments in the industry we have brought you an exciting array of articles and features to guide you within this dynamic market.
We start off with an interview with Fraser McLeod, Vice President of QA/QC for Waters Corporation, explains how Waters's nextgeneration Alliance™ iS HPLC System will revolutionise productivity and efficiency for modern QC labs.
Vaccines are the most important health intervention. A stringent administrative method must be followed during the creation of a vaccine to determine its safety, effectiveness, and quality. Regulatory authorities in various nations oversee vaccine development, submission assessment, approval, and post-approval operations. Indian National Regulatory Authority (CDSCO) is the supervisory agency in charge of vaccine assembly and import. The Regulatory Section features an article by, Dr. M P Venkates and his colleagues, at JSS College of Pharmacy explaining how vaccine regulations work in India.
In the Clinical Research section, Kirsten Sherman Cervati and William C. Maier at ICON discuss that while there is continued growth in the development of drugs to treat rare diseases, clinical trial accessibility for patients remains challenging due to the requirements inherent in these studies.

I hope you all enjoy this edition of IPI. Our next edition due in September will be the official edition for CPHI Worldwide in Barcelona. I hope to see you all there. Have a wonderful summer.
Virginia Toteva, Editorial Manager – IPI
Georg Mathis Founder and Managing Director, Appletree AG
Jagdish Unni, Vice President – Beroe Risk and Industry Delivery Lead – Healthcare, Beroe Inc.
Jeffrey Litwin, M.D., F.A.C.C. Executive Vice President and Chief Medical Officer of ERT
Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma
Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
Stanley Tam, General Manager, Eurofins MEDINET
(Singapore, Shanghai)
Steve Heath, Head of EMEA – Medidata Solutions, Inc
Patrice Hugo, Chief Scientific Officer, Clearstone Central Laboratories
Heinrich Klech, Professor of Medicine, CEO and Executive Vice President, Vienna School of Clinical Research
Robert Reekie, Snr. Executive Vice President
Operations, Europe, Asia-Pacific at PharmaNet Development Group
Sanjiv Kanwar, Managing Director, Polaris BioPharma
Consulting
Stefan Astrom, Founder and CEO of Astrom Research International HB
T S Jaishankar, Managing Director, QUEST Life Sciences
6 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Editor's Letter
Say ‘hi’ to your new ,
•Prevent up to 40% of common errors
•Fast, simple guidance at the point of need
•Boost productivity and capacity
•Drive workflow e ciencies and quality improvements
Welcome to a new era of intuitive simplicity. Find out more at waters.com/AllianceiS

INTERNATIONAL PHARMACEUTICAL INDUSTRY 7 www.international-pharma.com
Introducing a New Era in HPLC
Fraser McLeod, Vice President of QA/QC for Waters Corporation, explains how Waters next-generation Alliance™ iS HPLC System will revolutionise productivity and efficiency for modern QC labs.
Q: Waters Corporation has a rich history of supporting QC laboratories. Has the business observed any changes in the challenges faced by QC labs over the years?
A: Waters introduced its first commercial HPLC system 50 years ago, helping pharmaceutical QC laboratories to ensure tens of thousands of prescription drugs on the market are pure, safe, and work as expected.
At this time, many of the biggest challenges experienced by QC labs were based on the scientific capabilities of the separation technology, such as the kinds of columns that can help meet their needs. For modern QC labs, the story is slightly different.
In recent years, we closely collaborated with hundreds of QC managers and bench analysts to fully understand their challenges and find out specifically what they need today from a state-of-the-art HPLC system to help their labs perform at their best.
We learned that the challenges experienced most often are based on the relentless demands faced from both a scientific and business perspective to do more with less, manage high staff turnover, and deal with growing levels of sample complexity.
All these factors, and more, can increase the chance of errors, such as forgotten samples, incorrect solvent levels, incorrect tightening of system fittings, or not verifying correct system performance. These errors can have a significant impact on efficiency and productivity in the lab, and the associated costs incurred when investigating them.
We also heard a lot about the desire for improved usability and simplicity, so that managers can help analysts get up to speed quickly with new instruments, especially as they navigate from legacy methods and onto new technology.
At a time when challenges like these are mounting, we recognized that the need for simplicity in the QC lab has never been greater, to support their evolving scientific and business needs.
Q: How does the newest HPLC launch from Waters address these challenges in QC labs?
A: The Alliance iS HPLC System has been developed by Waters specifically for QC labs, with a suite of features and solutions designed to minimise errors and improve system usability.
The system prevents up to 40% of common errors* by performing pre-run verification of the correct column, vials, and plates, as well as solvent levels and expiration dates. Tool-free fittings prevent leaks, and clearly labelled and organised solvent lines and auto-recognition of Waters eConnect HPLC Columns prevent user errors when setting up the system for analysis.
We have also added several new hardware components designed to maximise reliability and ease of use. The intuitive touchscreen interface provides guided workflows, troubleshooting and maintenance, ensuring consistence and ease of operation. Quick and easy access to the Waters Help Center, directly from the instrument touchscreen, also makes it much easier to address errors in real-time and minimise unscheduled downtime.
The usability elements also address a big piece of the efficiency puzzle: training new hires. Modern QC labs can experience high staff turnover, as increasing numbers of analysts move onto other roles, creating a constant need to train new hires. The intuitive simplicity built into the Alliance iS HPLC System makes it easy for new hires to quickly become proficient in executing routine processes with minimal avoidable errors.
Overall, this suite of features will save QC labs valuable time and money, plus protect them from the risk of investigations.
Q: Does the Alliance iS HPLC System work with other products from the Waters ecosystem?
A: It was important to us to develop a system that could connect with other key solutions in the Waters portfolio to offer a cohesive and seamless operational experience to QC labs.
The Alliance iS HPLC System is uniquely built for Empower Software, which is the most used software in QC labs today. Waters Empower Software Suite helps the entire laboratory operate more efficiently, with the industry’s most advanced data acquisition, management, processing, analytics, and reporting capabilities. The Empower 3.8.0 release provides exclusive capabilities for the Alliance iS HPLC System, including:
• Method Matching
• Column Tracking & History (with eConnect HPLC Columns)
• Pre-run Checks
• Empower System Audit Trail
• Touchscreen Interactivity
Additionally, waters_connect System Monitoring provides intuitive and centralised access to detailed Empower Software instrument information via the waters_connect Cloud Platform, without compromising security or compliance. This helps QC labs to maximise productivity and utilise resources more effectively from anywhere, anytime.
The Alliance iS HPLC System and Empower 3.8.0 release also support Waters new eConnect HPLC Column technology, designed to ensure complete column traceability and facilitate post-run troubleshooting. Readyto-use from the box, eConnect columns are securely fitted with a NFC-enabled eConnect device tag, ensuring they are always automatically identified, verified, and tracked by the system. Analysts and managers can easily track column usage and make sure they’re always using the right column.
Using the combination of status lights, touchscreen interface, Empower Software, integration with waters_connect System
8 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Talking
Point
Monitoring, and smart column technology; the entire QC lab can access an instant view of system status and access real-time information for informed decision-making.
Q: How can the Alliance iS HPLC System reduce the compliance burden felt by QC analysts and managers?
A: QC labs need their systems to support both legacy and new methods that can be operated repeatably anywhere in the world, all while meeting essential regulatory requirements. This inevitably places a large burden on the analysts to ensure errors are minimal and processes are followed meticulously.

To address this and help QC labs meet global compliance needs more effectively, the Alliance iS HPLC System includes features that are designed to reduce the number of quality incidents that occur in their labs, as well as the amount of time spent investigating quality incidents – all the while empowering analysts to access results much more quickly.
QC labs can rely on repeatable global method performance with enhanced temperature control in the sample manager, column manager, and detector, plus reduced adsorptive carryover and eliminated volumetric carryover.
When coupled with Empower Software Solutions and eConnect HPLC Columns, analysts can access new features such as the Intelligent Method Translator App (iMTA). This enables analysts to create an Alliance iS HPLC System Instrument Method directly from a Waters or other third-party LC system. Basic chromatographic conditions are automatically
transferred, and the new Empower Software instrument method can be reported, providing trace-ability to the originator method for compliance. This is complemented by Method Matching technology, which can warn users if the column installed is not matched to the selected method.
For enhanced compliance tracking, users can access a System Audit Trail, where actions performed on the touchscreen interface are automatically logged into Empower Software.
Q: How would you say the Alliance iS HPLC System differs from other HPLC innovations?


A: The two biggest differentiators of this system are its intuitively simple operation, and ability to reduce errors by up to 40%*. All the features we included in the design were geared specifically towards addressing usability challenges and error reduction.
To make sure interaction is as smooth and seamless as possible for users, a considerable amount of industrial design research and planning went into this product, reflected in features such as the color-coded solvent tubing clips, status lighting, and intuitive touchscreen. This instrument represents the highest level of usability we have achieved for a single product at Waters.
We were very mindful about in-corporating features targeted at reducing errors but without putting any extra burden on analysts with extra steps, and instead reducing errors in a way that naturally supports the user's workflow. The system is designed with intuitive mechanisms that provide fast feedback, plus health monitoring tools that enable teams to dive deeper into crucial performance parameters in real time.
Ultimately, our priority was making sure that operating the Alliance iS HPLC System would be as seamless as possible. We see this as perhaps the most innovative overall element we’re introducing to the HPLC market with this instrument.

Q: How do you think the future looks for Waters in terms of HPLC innovation?
A: Waters has been established in the chromatography field for decades, and
we know how to create instruments that perform. However, we believe the new vector of usability, simplicity, and error reduction we have achieved with the Alliance iS HPLC System will help to take HPLC innovation to the next level.
We have made great strides towards demonstrating our longstanding commitment to solving the problem that matters to our customers by creating an instrument that can reduce common errors in QC labs by an impressive 40%*. However, we know that this still leaves the door open to reduce errors by a further 60%. This is where we will continue to focus our efforts and is where we expect to make many more great strides in the years to come.
* Estimate based on Waters' market research in 2022 after surveying 56 global pharmaceutical company QC labs.
Fraser McLeod is the Vice President and General Manager for Waters QA/QC Business Segment. In his role, he runs a team that develops and commercializes solutions that are predominantly used in QC labs and is responsible for all Waters chromatography instruments as well as the NuGenesis and Empower Software Solutions. Fraser has worked in the life sciences industry for more than 25 years focusing on new product development and business transformation strategies.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 9 www.international-pharma.com Talking Point
PHARMACEUTICAL INDUSTRY www.international-pharma.com
Fraser McLeod
IPI Speaks with PharmaLex’s Local Affiliate Services Lead on the Rapidly Changing Global Market
Q: Can we start with some insights as to why the Local Affiliate Services solution was established and what its overarching objectives are?
A: There is rising demand from our clients for services at the local level to manage mandatory activities that are normally handled by their affiliates. The need was initially greatest at large organisations, which were looking to streamline the business and focus on their strategic development projects. To achieve that, they needed support to ensure they met their local compliance obligations, especially for their mature products.
There is now a growing trend amongst mid-sized companies wishing to expand into new geographic regions, but without having to invest in the setup of the required local roles. As an example, a Japanese client that was launching products in Europe needed local affiliate support to meet their obligations in various EU markets but decided not to invest in hiring their own people in each country because they would not have enough work for a full-time role.
We have also found that many clients who have been working with a diverse set of local services providers welcome that PharmaLex is in a position to offer them consolidation and all the support they need.
Q: A lot is said about the challenges companies have with managing compliance requirements at the affiliate level. Why is a more all-encompassing approach to managing the local affiliate important?
A: Compliance requirements often relate to more than one country, for example, the update and controlled implementation of a new side effect in a leaflet, an update to packaging material or the reporting of cases. If such activities are handled by several service providers, the sponsor company would have to oversee the status of each activity for every concerned country.
Having an all-encompassing approach with a program leader and function-related workstream leads at one service provider minimises oversight and control for the client company, while giving them confidence and ensuring compliance.
Another important consideration is the growth in mergers and acquisitions or portfolio acquisitions, which can result in short-term joint ventures. These require quick setup of local mandatory and operational roles.
Sometimes, contracts preclude having these joint venture activities handled by either company involved in the joint venture, so a third party needs to take over local tasks. These might include changes in packaging materials and ensuring the right information is included to facilitate the transfer of the product/s or venture.
Q: Conversely, what are the biggest challenges and limitations that local affiliate resources face in a rapidly changing regulatory environment?
A: At the local level, you need to comply with national regulations and maintain a
trusted relationship with your local health authorities, ensuring that established products remain compliant.
On the other hand, you are striving to ensure you follow headquarter guidance and meet the work schedule, particularly on priority and strategic development activities.
Also, the follow-up of new local requirements needs to be fed back to headquarters to be included in the overall global strategy and plan.
This situation of different tasks and priorities may be a stretch in terms of workload, availability, adequate response times and cost for the local staff. However, this local work is of major importance to keep the business steady and growing, while meeting compliance requirements.
Q: What do companies need to do to both meet their local affiliate compliance requirements while retaining a single, unified global perspective? (Is it even possible?)

As previously mentioned, the local affiliate needs to comply with local regulations and maintain a trusted relationship with the local health authorities to keep established products on the market. These demands may prevent them from having the time to focus on the company’s strategic development projects.
In order to both meet compliance requirements and support the business objectives, it may make sense to separate these responsibilities through an external partner to support and manage daily routine tasks.
The added benefit of this is it allows companies to take advantage of faster, more streamlined processes. While local affiliates are highly professional, there can often be resistance to changes to established ways of working. This is at odds with a global push to get products to market faster and at less cost, both to the benefit of the company and to patients.
10 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Talking Point
Q: What are the biggest limitations facing the local affiliate model and how can they be mitigated?
A: Especially for smaller or mid-size companies expanding their territories, it may be very cost intensive to install a full affiliate model with all the locally required functions and tasks, for example a designated person for health authority communication, the screening of local literature, the handling of complaints or a 24/7 contact, sometimes even before the launch of their product and having actual revenue.
For large pharma companies, on the other hand, a focus on routine tasks can make it hard for affiliates to concentrate on strategic work or require them to bring in additional resources at added cost.
Sometimes there is also no need for a full-time employee, but there is still a requirement to have a local person with specific qualifications to manage certain activities, such as pharmacovigilance obligations. Hiring someone with the required education and background for just 20 hours a month can be extremely difficult, if not impossible, especially in markets where there is competition for a limited number of professionals. However, a service provider can handle those tasks efficiently and more cost effectively because those
local staff members manage compliance activities for several clients.
Q: Are there cultural barriers holding companies back from improving the collaboration between the local affiliate and headquarters? And, if so, what are these?
A: Open reflection on long-established processes and the acknowledgement that some practices do not work efficiently any longer may be difficult in certain cultures, preventing the improvement and streamlining of activities.
In addition, local staff are often so busy with their daily tasks and routine that they cannot see possibilities for reducing burden and concentrating on strategic tasks. This is accompanied by the fear of losing control if they don’t manage everything themselves.
Bringing in an external perspective can break such obstacles and lead to more efficient ways of working.
When implementing local affiliate services in one region for a large pharmaceutical client, we have encountered resistance to adopting new, more streamlined procedures. At the same time, due to cultural nuances, the affiliate has been uncomfortable pushing back. With

careful management, it was possible to gain confidence, openly address concerns and finally have a willingness to adapt.
Q: What role can/does technology play both in creating more barriers between HQ and affiliate and, conversely, driving a more harmonized approach?
A: It is important to consider both the advantages and potential barriers that technology offers. There is certainly huge potential for harmonisation. For example, technology enables real-time collaboration, allowing headquarters and affiliates to work together on shared platforms, such as project management systems or document collaboration tools. This facilitates faster decision-making, enhances efficiency, and promotes a sense of unity among teams.
Technology can also offer substantial support and access to resources, for example, by screening intelligence developments using an automated approach and offering easy access to training materials and best practices. This allows affiliates to tap into headquarter expertise and leverage shared resources, fostering a harmonized approach in terms of processes, quality standards, and regulatory compliance. It also allows headquarters to anticipate affiliate needs and consider them early in the planning process.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 11 www.international-pharma.com Talking Point www.international-pharma.com
At the same time, there is also a risk of information overload, so headquarters needs to be careful not to flood affiliates with excessive data and instructions, as this can hinder effective decision-making and cause confusion, and potentially even lead to inconsistencies and mistakes in interpreting data.
An overreliance on technology for communication can also lead to misunderstandings or misinterpretations. Where there are cultural and language barriers, technology-mediated communication can make it challenging for HQ and affiliates from different regions or countries to understand each other's perspectives and work together seamlessly.
Q: What does the term Glocal mean to you, and how can its vision be realized?
A: Glocal for me is a PharmaLex attitude: we think globally and holistically. Most of us come from the pharma industry or health authorities and understand the responsibilities of central functions. On the other hand, we respect and recognise the importance of the local national requirements and of having adequate representation and responsibility on the ground. It means having a global vision supported by local people, including having native language speakers who have specific local knowledge and connections.
With our own affiliates or team members, as well as with qualified local consultants and partners, our professionals around the world have relationships with the regulators and, where relevant, can attend training sessions provided by the regulators and give input to local or regional industry trade associations.
They are able, by way of example, to compile a regulatory dossier according to local requirements, to follow up the process with the local health authority, or to take the responsibility of a national contact person for pharmacovigilance.
At PharmaLex, because we work in many regions, with many different companies and products, we tend to attract highly motivated employees who are eager to take on new opportunities and apply different skill sets.
Q: PharmaLex’s Local Affiliate Services solution seeks to help companies navigate a more complex global regulatory environment through a tailored model. What does that look like in practice and why is it important?

A: At PharmaLex, we listen to the needs of our clients. We never apply a standardised one-size-fits-all model of support. Instead, we develop a setup that is best for the client and agree on a model for collaboration, reporting and issue escalation together. Depending on the scope of activities, we may have a program manager as the primary point of contact for the client, appoint specific workstream leads and regional hub teams, or, in some cases, have a very lean collaboration structure where the client can work directly with our local experts, without additional overlay.
Dr. Stefanie Lietsch-Dallwig is Senior Director Global Program Management – Service Solution Lead Local Affiliate Services, at PharmaLex, where she draws on her vast experience with registration and launch planning for new applications and variations in multiple markets. Stefanie is an international Regulatory Affairs professional, who has held senior positions at large pharmaceutical companies, including as Director Regulatory International & Emerging Markets and Head of Regulatory Intelligence.

12 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Talking Point
Dr. Stefanie Lietsch-Dallwig



Harnessing decades of global drug product development and commercialization, you can rely on our integrated speed solutions to simplify your supply chain, spanning the cycle from study to launch. Introducing speedsolutions™ Your bridge between life-changing therapies and patients speedtostudy™ speedtopatient™ speedtoapproval™ speedtolaunch™ PH II PH III COMMERCIALIZATION PH I Let’s talk future™ talkfuture@pci.com | pci.com Accelerating your product through development to commercialization and beyond Development & Manufacturing | Clinical Trial Services | Commercial Packaging
The Four Trends Set to Disrupt Highly Regulated Industries in 2023
Bob Tilling, VP Global Sales at Kallik identifies four trends set to disrupt manufacturers in highly regulated industries in 2023: the industrial metaverse, the need to get a tighter arm on cybersecurity, the rise of ecoanxiety as the 2025 sustainable packing deadline looms, and a focus on data centricity.
Businesses and manufacturers in highly regulated markets, in particular pharmaceutical, food and beverage, cosmetics and consumer packaged goods (CPG), have experienced huge change in recent years, most significantly in the technologies they use, the regulations they face, and the changing demands from their customers.
In 2023 these macro level changes are going to have a serious impact on regulated industries – with technology-driven change exploiting the industrial metaverse right down to the packaging, labelling and artwork used when manufacturing each individual product.
1. The Industrial Metaverse Will Blur Digital and Physical Like Never Before
The global metaverse market is set to grow at an annual compound rate of 39.8% between 2022–2030. As part of the wider metaverse umbrella, the industrial metaverse combines a mix of immersive technologies including physical-digital fusion and human augmentation to create digital representations of a physical environment. Early adopters are already seeing benefits in terms of streamlining logistics and processes, achieving tangible return on investment (ROI), and delivering high-quality products across multiple industrial applications.
Many companies are still trying to envision the future of the industrial metaverse, but its potential to transform design, manufacturing, and interactions across global ecosystems is gaining significant interest. We have already seen the introduction of digital twins during Industry 4.0, a virtual model designed to
accurately represent a physical object. When supported by other innovative technologies such as artificial intelligence (AI), Internet of Things (IoT) and 3D rendering, the true powers of digital twins can be felt and the road to the full impact of the industrial metaverse becomes nearer.
In conjunction with digital twins, we can expect to see the use of 3D rendering rapidly increase in 2023. 3D rendering is the process of using a computer to generate a 2D image from a digital three-dimensional scene. In fact, 3D rendering for artwork and labelling is already a core industry focus – helping to generate labels or artwork to put on the product and produce a 360-degree view of what it will look like before it goes to market.
2. Get to Grips with Cybersecurity – Put Data at the Heart of Operations
Sophisticated hackers are increasingly finding ways through business security defences, so cybersecurity will become a clear focus in 2023. In the UK, nearly two-fifths of businesses experienced a cyberattack in the year leading up to July 2022. For the healthcare sector, cyberattacks have been a long standing issue – it received 20% of the UK’s cyberattacks in 2021. Its vast amount of personal data combined with a reliance on outdated, legacy technology has made the healthcare market a soughtafter target and unfortunately, medical devices have become an easy entry point for attackers. Medical device manufacturers are on the front line and must integrate an effective cybersecurity plan throughout the entire product development lifecycle, from pre-market and post-market phases, to device disposal.
Prevention all comes down to data management – companies stand a much higher chance of warning off unwelcomed attackers by putting data at the core of operations. This applies right down to access control for critical recipes and formulas in the manufacturing supply chain – and that includes the label and artwork management (LAM) solution, where security is of upmost importance. LAM solutions are accessed by multiple parties across the manufacturing and distribution process, so product owners need to hold one single source of truth and
be able to be sure there is no unwanted data sharing. They must ensure that not only users of the platform are only able to see information relevant to them, but also that out-of-date files are prohibited to reduce risk. Users must only be able to view and edit one version of a label that will need rigorous approvals thereafter. This will provide the necessary reassurance that a consistent audit trail has been followed and minimises the likelihood of data leakage, businesses can be confident that attackers are unable to edit or share data without access or permission.
3. The Rise of “Eco-anxiety” – Sustainable Packaging Remains at the Front of Consumer Minds
Environmental, social and governance (ESG) may be no new phenomenon, but it is one that requires urgent action as pressure mounts daily from investors and an ecoconscious society. In 2023, this focus isn’t going to die down – organisations can even expect to lose their competitive edge or market share if ESG is not taken seriously. In a recent report by Kantar, 97% of consumers globally reported that they are prepared to take action and live a more sustainable life, with another 79% keen to purchase more sustainable products.
Most recently, microplastics have found themselves under the microscope – every minute, over seven kilos of microplastics from cosmetics and personal care products end up in the European environment. The cosmetics industry is at the forefront of this issue, as 87% of products from the ten bestselling cosmetics brands contain microplastics. The growing issue has sparked major interest – and consumers have called upon EU regulators to action new laws against the use of microplastics. If approved, the restrictions will have huge implications for manufacturers, not only to product composition but to a major uphaul of existing labels to reflect the ingredients change. Editing such a sheer number of labels will need an advanced and sophisticated LAM (Label & Artwork Management) solution to match – a LAM solution that can manage vast amounts of data, symbols, words, and phrases on a large scale.
14 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Regulatory & Marketplace
Exacerbating these pressures is the real and unavoidable 2025 deadline for sustainable packaging. Although market challenges such as cost of living increases and supply chain shortages will wreak havoc for organisations, up to 70% of consumers are willing to pay more for products with sustainable packaging. It will require business leaders to rethink both operations and timeframes, including the use of sustainable materials, and how this will affect printing operations. Here, technology will once again prove its value, reducing long manufacturing development lifecycles and waste in label and artwork management processes by easily allowing adaptability to fit new packaging sizes and types. LAM solutions help companies reduce labelling and artwork completion times by 50%, and many major consumer goods companies are already undergoing the migration from plastic packaging to cardboard packaging in 2023.
4. Become a Data-centric Business –Growth and Success on Scale will Follow Finally,
in 2023, more organisations will realise the true power of data when analysed and used at scale. Despite investment of trillions of dollars by U.S. companies into data analytics integration at scale, up to
now only 8% of organisations are capturing real value. From a labelling and artwork management perspective, effectively harnessing data can unlock a whole host of opportunities, and help prepare for changes in regulations, product updates, and react in the event of product recalls.
In the artwork and labelling space, being able to react to changing market conditions will be pivotal to stay compliant and operational – and companies must invest in a data heavy system, capable of examining, storing, and printing mass amounts of data, quickly. Take the postponed UKCA certification deadline. By end of year 2024, companies must have changed all labels to reflect the new UKCA marking to remain compliant. Or consider the move to sustainable packaging highlighted earlier, where companies will need to alter recycling symbols for each product based on the material type, regional recycling regulations, as well as the storage and recycling instructions for every region and every language – each of these will need an LAM solution to change speedily, safely and at scale.
Analysing data at scale and actioning cross-organisational change across thousands of product lines will be a
long, arduous, and error-prone task with a manual or paper system. It is simply impossible now to adapt to change and to grow without the help technology.
With bespoke LAM software, companies can reduce artwork changes down to five minutes – a 55-minute saving per change compared to other solutions. LAM databases and rules engine functions keep products and predetermined phrases stored in 3040 different languages, to allow users to quickly search and find the relevant data that can be used for artwork – saving time and risk of human error. While many large companies have been found to pass artwork and labelling tasks on to outbound agencies, LAM software can provide huge savings on third-party artwork agency fees for manufacturers.
The proof is in the Numbers – Make Sure you Follow them in 2023
Despite previous years being filled with uncertainty, there is still much externally enforced change for manufacturers in highly regulated markets in 2023 – whether it be the rise of the industrial metaverse, preparing for upcoming regulations, tightening cybersecurity defences or an increased focused on ESG. This will give rise to an increased reliance on digital LAM solutions to ensure product packaging and labelling keeps pace with these developments.
For all manufacturers but in particular for those businesses in regulated industries, the label may be the last part of the production cycle, but it will be one of first parts to cause regulatory issues and even impact consumer safety.
 Bob
Bob
Tilling
Bob Tilling is the VP of Global Sales at Kallik, an enterprise labelling and artwork management company. He has a wealth of knowledge when it comes to the life sciences industry, particularly regarding medical devices. Bob helps businesses in highly regulated industries begin their journey of transforming their labelling and artwork management.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 15 www.international-pharma.com Regulatory & Marketplace
Email: bob.tilling@kallik.com Linkedin: ww.linkedin.com/in/bob-tilling
Vaccine Regulation in India
Vaccines are the most important health intervention. Vaccination stimulates the immune system to identify and battle invaders such as viruses or bacteria. A vaccination encourages our immune system to manufacture antibodies in the same way as exposure to the illness would. A stringent administrative method must be followed during the creation of a vaccine in order to determine its safety, effectiveness, and quality. Regulatory authorities in various nations oversee vaccine development, submission assessment, approval, and post-approval operations. Indian National Regulatory Authority (CDSCO) is the supervisory agency in charge of vaccine assembly and import. CDSCO carries out tasks delegated by the federal government under the D & C act 1940 and the Guidelines of 1945. The CDSCO vision.
Introduction
Vaccines are one of science's and public health's most significant achievements. Edward Jenner defined vaccine in 1796 as the use of cowpox (Latin variola vaccinia derived since the Latin vaccine-us, from vacca, cow) to immunise individuals and give protection against chickenpox. Vaccines are used to protect against infectious illnesses.1
The medical regulatory framework in India is split between national and state bodies. The Central Drugs Standard Control Organization (CDSCO), headquartered in New Delhi, is the primary national drugcontrol authority. DCGI is in charge of it. CDSCO collaborates with Health Canada, the US Food and Drug Administration, Brazil, and South Africa to improve its efforts. It also collaborates with WHO to promote worldwide regulatory harmonisation and good manufacturing practises. The Drugs and Cosmetics Act of 1940 and the Rules of 1945 established specific requirements for CDSCO. Only pharmaceuticals that fulfil these standards may be imported, manufactured, stocked, sold, or distributed in India; specific statutory requirements for pharmacovigilance in India are listed in Schedule Y of this Act. To summarise, the most recent modification.2
• Research the different regulatory organisations involved in vaccination registration.
• Research the ADR reporting procedure in India.

Vaccine Regulatory Issues in India
CDSCO in India is the National Administrative expert who evaluates the protection, effectiveness, and quality of drugs in the country. DBT is responsible for creation and pre-clinical assessment of recombinant biologics through the RCGM. Various firms are now active in the marketing and production of vaccines in India. So far, RCGM and CDSCO have authorised these biologics products based on a simplified version of the route material to new drugs. Because there are multiple similar goods in development in India, both regulatory bodies assess the requirements to determine the regulatory path that will assure equivalent product safety, efficacy, and quality.2
Multiple authorities have developed regulatory regulations for vaccination registration. They are as follows: Ministry of Health and Family Welfare, NTAGI, ICMR, CDSCO, CLAA
Ministry of Health and Family Welfare
The Ministry of Health and Family Welfare is organised into two departments, each managed by a Government of India Secretary: the Department of Health and Family Welfare and the Department of Health Research.2
• Department of Health: MCI, Food Safety and Standards Authority of India and CDSCO
• Department of Family Welfare: CDRI, Lucknow and ICMR, New Delhi
Health Research Department
• HRD
• Establishment of a laboratory network for controlling epidemics and natural disasters.
• Grant-in-aid (GIA) scheme for intersectoral convergence and cooperation for health research promotion and advice.
The formation of multifunctional research units (MRU) at government medical college research facilities.2
NTAGI
The national technical advisory group of immunisation takes the decision on the introduction of new vaccinations and the expansion of the Universal Immunisation Programme (UIP). NTAGI's mission and membership have evolved throughout time to meet the evolving demands and goals of the Government of India. Current issues include institutionalising processes for following up on and monitoring suggestions, funding research to remedy knowledge gaps, and providing technical help for monitoring and regularly evaluating the UIP.3
ICMR
The Indian Council of Medical Research, based in New Delhi, is the country's leading organisation for the development, coordination, and promotion of biomedical research. It is one of the oldest medical research organisations in the world. The Indian Council for Medical Research has always attempted to meet the rising demands of scientific advances in biomedical research on the one hand, and the need to find practical solutions to the country's health challenges on the other. The Indian Council for Medical Research, formerly known as the IRFA, recognises that it still has a long way to go in terms of scientific breakthroughs and health goals.4
CDSCO
CDSCO is an Indian regulatory agency. It carries out the tasks delegated to it by the Central Government under the Drugs and Cosmetics Act. CDSCO's objective is to safeguard and improve public health in India, and its mission is to ensure the safety, effectiveness, and quality of pharmaceuticals, cosmetics, and medical devices.
The Drugs and Cosmetics Act of 1940 and the Rules of 1945 established CDSCO standards. Only pharmaceuticals that fulfil these requirements are allowed for import, production, stocking, sales, and distribution. It is led by the Drugs Controller General of India in India (DCGI). CDSCO has created zonal offices in partnership with the Central Government of India, which work in collaboration with the State Drugs Control Administration and assist DCGI in carrying
16 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Regulatory & Marketplace
out its operations. The DCGI is in charge of the headquarters, zonal offices, sub-zonal offices, port offices, and so on.5

Central Licensing Approval Authority (CLAA)
This authority issues or renews licences for the importation, sale, manufacturing, and distribution of pharmaceuticals and cosmetics. Whether or not the application meets the stipulated standards, the licencing body may or may not issue a licence. The licencing authority is also entitled to suspend or terminate licences issued by them if the licensee fails to comply with any of the license's terms after giving him an opportunity to explain why.5
Quality Assessment1
The quality ratings are based on four characteristics.
1. Methods of Analysis: Techniques are chosen based on essential quality traits of the product many methods can employed to examine characteristic.
2. Product Characterisation: Tests for physiochemistry, biological activity, immunological characteristics, functional analysis, purity, uncleanness, and strength.
3. Specifications: Created about important product eminence aspects; and Product quality consistency and comparability to reference biologics have been confirmed.
4. Stability: Vaccine shelf-life and stowing condition testing will be performed.
Figure 1. Adverse
INTERNATIONAL PHARMACEUTICAL INDUSTRY 17 www.international-pharma.com Regulatory
& Marketplace
Stage Agency Application Approval License for testing, manufacturing, and analysis CDSCO Form Number 30 Form Number 29 Preclinical approval RCGM Form Number C3 Form Number C4 Preclinical report submission RCGM Form Number C5 Form Number C6 Clinical studies CDSCO Form Number 44 Permission letter Manufacturing and distribution CDSCO Form Number 44 Form Number 46 Manufacturing authorisation State authority Form Number 27D Form Number 28D Import and registration CDSCO Form Number 8 Form Number 10 Table 1: Agency, Approval, Application and Stage1 SUSPECTED ADVERSE DRUG REACTION REPORTING FORM For VOLUNTARY reporting of ADRs by Healthcare Professionals INDIAN PHARMACOPOEIA COMMISSION (National Coordination Centre - Pharmacovigilance Programme of India) Ministry of Health & Family Welfare, Government of India, Sector- 23, Raj Nagar, Ghaziabad - 201002 PvPI Helpline (Toll Free) :1800 - 180- 3024 (9:00 AM to 5:30 PM, Monday - Friday) Version 1.4 Initial Case Follow - up Case FOR AMC / NCC USE ONLY A. PATIENT INFORMATION * Reg. No. / IPD No. / OPD No. / CR No. : 1. Patient Initials : 2. Age or date of birth : AMC Report No. : 3. Gende r: M F Other 4. Weight (in Kg.) Worldwide Unique No. : 12 Relevant investigations with dates : B. SUSPECTED ADVERSE REACTION * 5. Event / Reaction start date (dd/mm/yy yy) 6. Event / Reaction stop date (dd/mm/yy yy) 7. Describe Event/Reaction management with details , if any 13. Relevant medical / medication history (e.g. allergies, pregnancy, addiction , hepatic , renal dysfunction etc.) 14. Seriousness of the reaction : No if Yes ( please tick anyone ) Death (dd/mm/yyyy) Congenital - anomaly Life threatening Disability Hospitalization- Initial/Prolonged Other Medically important 15. Outcome: Recovered Recovering Not Recovered Fatal Recovered with sequelae Unknown C. SUSPECTED MEDICATION(S) * S. No. 8. Name (Brand/ Generic) Manufactu rer (if known) Batch No. / Lot No. Expiry Date (if known) Dose Route Frequency Therapy Dates Indication Causality Assessment Date Started Date Stopped i ii iii i v # 9. Action t aken after reaction ( please tick ) 10. Reaction reappeared after reintroduction of suspected medication ( please tick ) S. No. as per C Drug withdrawn Dose increased Dose reduced Dose not changed Not applicable Unknown Yes No Effect unknown Dose (if reintroduced) i ii iii i v 11. Concomitant medical produ ct including self- medication and herbal remedies with therapy dates (Exclude those used to treat reaction) S. No. Name (Brand / Generic) Dose Route Frequency (OD, BD, etc.) Therapy Dates Indication Date Started Date Stopped i ii iii # Additional Information : D. REPORTER DETAILS * 16. Name & Address : __________________ Pin : __________ Email : _______ Contact No- : __ Occupation : _______________________Signature : ___ 17. Date of this report (dd/mm/ y yyy) : Signature and Name of Receiving Personnel : Confidentiality : The patient’s identity is held in strict confidence and protected to the fullest extent. Submission of a report does not constitute an admission that medical personnel or manufacturer or the product caused or contributed to the reaction. Submiss ion of an ADR report does not have any legal implication on the reporter. # Use separate page for more information * Mandatory Fields for suspected ADR Reporting Form
Drug Reaction
Form6
Reporting
Conceptualising the vaccine
Institutional biosafety committee (IBSC)
• Approval of project on biosafety grounds for recommendation to RCGM
Reporting of ADRs
ADRs must be reported as soon as possible after they occur, including suspected ADRs, unexpected ADRs, serious reactions, an increase in the frequency of reactions, ADRs due to interactions with food, food supplements, or other drugs, ADRs due to drug abuse, drug use in pregnancy, during pregnancy, during lactation, ADRs due to over dosage or medication errors, and so on. It is the obligation of health care professionals (doctors, pharmacists, nurses, and so on), medication makers or product registrants, and health facility administrators to report the incidence of such ADRS.7
Requirements for Reporting ADRs7 Patient Information
The patient's name or record number, date of birth, gender, and weight must be recorded.
Details of the Adverse Reaction
The date of beginning of the response, the time interval between the date of drug administration and the occurrence of the ADR, and a brief description of the ADR, including the afflicted body part, intensity of the reaction, and so on, are all necessary. Other pertinent information, such as the patient's medical history, laboratory tests performed, and the findings received, should also be carefully mentioned.
Information Related to Suspected Drugs 7
Details on the substance suspected of causing an adverse reaction are required.
a) The brand or generic name, whichever is available, the strength of the drug, the
Review committee on genetic manipulations (RCGM) (Under DBT, GOI)
• Deals with any research involving genetic manipulation.
Genetic engineering approval committee (GEAC) (Under Ministry of Environment and Forests, GOI)
• Deals with any research involving genetic manipulation.
Drugs controller general of India (DCGI) (final approval and licensing authority in India)
• Examines the animal toxicity and quality control data.
• Approves the protocol and recommends conduct of human clinical trial.
• The examination and approval of clinical trial report
• Approval for production of trial batches.
• Acceptance of testing reports from central drug laboratory and final approval to manufacture and market also granted by the DCGI.
Registration
Registration of Vaccines in India
Phase I (10-30 subjects
• Initial Human Studies
• Safety & immunological studies
Vaccine manufacturer
Conduct clinical trials.
Phase II (100 Subjects)
• Dose ranging studies.
Submit result to NRA for approval.
GCP & ethical guidelines (by ICMR) approvals are required.
Phase III (1000 subjects)
• Documentation of effectiveness & safety
Central licensing approval authority is responsible for central license of vaccines.
DTAB approves introduction of vaccines into the immunisation services.
All vaccines approval & clinical trials are done by CLAA
Conduct phase IV clinical trials (Post marketing surveillance)
Any compliant regarding safety & efficiency should be directed to NRA
batch number, the expiry date, the reason for using the drug, the dosage form used, the route of administration, the dosing frequency, the duration of drug therapy, and any other drugs used concurrently with the suspected drug.7
Information about Management of ADR
Reasons for the ADR's association with the suspected drug should be given. Reasons for classifying the ADR as serious should be included, as well as the treatment used to decrease the response and its outcomes.7
18 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Regulatory & Marketplace
Figure 2. The Schematic representation of conceptualising the vaccines7
Figure 2. The Schematic representation of conceptualising the vaccines (7)
Figure 3. Regulatory approval process of vaccine in India (7)
of vaccines in India
Figure 3. Regulatory approval process of vaccines in India7
Regulatory & Marketplace
FORM C5
FORMAT FOR SUBMISSION OF PRECLINICAL OR OTHER SAFETY STUDIES REPORT OF rDNA PRODUCTS DEVELOPED USING GENETICALLY MODIFIED ORGANISMS (GMOs)/ LIVING MODIFIED ORGANISMS (LMOs) FOR HEALTHCARE, INDUSTRIAL OR ANY OTHER USE

1. Name of the Applicant:
Designation:
Address:
Telephone No.:
Fax No.:
e‐mail:
2. DBT Office Memorandum No.:
3. Objectives of the proposal:
4. Summary of the products characteristics and process of development:
5. List of preclinical study protocols approved by RCGM: (please attach a copy of the approval letter)
6. Preclinical study reports:
6.1 List of studies completed and deviations, if any from the approved protocols
6.2 Dose calculation for conduct of safety studies
6.3 Study reports (Each study report would reflect all the issues approved in the protocols). In addition, the following to be included:
• RCGM approval of protocol
• IBSC approval of report
• IAEC approval for animal use and for the p rocedure
• Individual animal data, summary data and any other data like computer analysis outputs etc.
• Conclusion
6.4 Address and accreditation status of the labs where these studies were conducted.
7. Measures taken for containment:
8. Decontamination and disposal mechanisms:
9. Risk management (Emergency plan):
10. Any other relevant information:
11. Declaration:
I declare that the information provided in the above form is correct and accurate to the best of my knowledge.
Reporter Information
Name, contact number, and signature of the reporter are necessary. The date of reaction reporting and the address of the hospital, institution, or other location where the therapy was administered are also required.7
Current System of Reporting ADRs in India
All reports, including safety reports, postmarketing surveillance reports, serious adverse events (SAEs) from clinical trials, reports from zonal pharmacovigilance centres, and individual reports from hospitals, physicians, patients, and others, are submitted to the office of the Drug Controller General of India (DCGI), where they are saved in the form of databases. These are provided to research organisations and pharmaceutical businesses. The DCGI office submits findings to the National Pharmacovigilance Advisory Committee, which makes decisions on safe medication usage.7
Conclusion
Vaccines are developed, tested, and monitored in the same manner as other drugs are. Vaccines are frequently investigated more thoroughly than nonvaccine drugs because vaccine clinical trials generally involve a greater number of human volunteers. Furthermore, regulatory organisations regularly monitor vaccination post-licensure surveillance. The major
Date:
Forwarded:
Signature of the applicant
The proposal set out above has been considered and approved by the “Institutional Biosafety Committee” in its meeting held on and is forwarded to RCGM for further necessary action. Figure
INTERNATIONAL PHARMACEUTICAL INDUSTRY 19 www.international-pharma.com
4. Form C58
Form 27 -D [See Rule 75]
Application for grant [ * * ] of a licence to manufacture for sale or for
distribution of Large Volume Parenterals/Sera and Vaccines excluding those specified in Schedule X
1. I/We.......... ....... of ....
............ hereby apply for the grant [ * * ] of a licence to
manufacture for sale or distribution on the premises situated at ............................................................. the under mentioned Large Volume Parenterals / Sera and Vaccines, specified in Schedule C and C(1), to the Drugs and Cosmetics Rules,1945.
2. Name(s) of Drug(s) (each item to be separately specified).
3. The name(s), qualification and experience of the competent technical staff responsible for the manufacture of the above-mentioned drugs.
(a) Name(s) of staff responsible for testing:
(b) Name(s) of staff responsible for manufacturing:
by B.Pharmacy VIII-Sem PCI Sia publication author Annavarapu Tirupathi Rao.
8. Form 27 [Internet] (cited on 2021 Oct 07) Available from: https://www.google.com/ url?sa=t&source=web&rct=j&url=https:// drugscontrol.org/pdf/Form27D.pdf&ved= 2ahUKEwihk-zh_7bwAhX0yzgGHY_iCmIQFjA BegQIAxAC&usg=AOvVaw2ABpyhDlSZb 8ip5je7qB
9. Form C5 [Internet](cited on 2021 Aug 27) Available from; https://www.google.com/ url?sa=t&source=web&rct=j&url=http:// dbtindia.gov.in/sites/default/files/ uploadfiles/C5.pdf&ved=2ahUKEwjBLbQ-7bwAhUClEsFHYfpCXwQFjAAegQIBBAC&us g=AOvVaw29KsnPFnr6CDKFK1cbRsJL
Arjun HR
4. The premises and plant are ready for inspection/will be ready for inspection on
5. A fee of rupees and an inspection of rupees .........has been credited to Government under the head of account
Mr. Arjun HR holds Master’s degree in Pharmacology from JSS College of Pharmacy, Mysuru, India. His interest lies in regulatory aspects of biologicals/ vaccines.

Email: arjunglass@gmail.com
Date:.................
* * Omitted by GSR 1337 (E) dt. 27.10.2017
Signature.................. Designation ..............
Figure 5. Form 27-D9 role of regulatory bodies is to ensure that vaccination quality, safety, and effectiveness are not jeopardised. CDSCO establishes pharmacological, cosmetic, diagnostic, and device standards. It also establishes regulatory measures, amends Acts and Rules, and regulates new medication market authorisation and clinical research in India. The CDSCO uses recognised bodies to do conformity assessments for all drug and device applications.
REFERENCES
1. International Journal of drug Regulatory Affairs
Published by Diva Enterprises Pvt. Ltd., New Delhi. Associated with Delhi Pharmaceutical Sciences and Research University [Internet]
Available on :2020 Mar 15 (cited on: 2021 June 16) Available from: https://ijdra.com/index. php/journal (Abstract, introduction, regulatory aspects of vaccine in India, quality assessment, conceptualization of vaccines, application and approval of vaccines in India, registration process of vaccines in India)
2. Ministry of Health and Family Welfare [Internet] (cited on: 2021 Aug 06) Available from: https:// en.wikipedia.org/wiki/
3. NTAGI [Internet] (cited on: 2021 Sep 06) . Available from: https://www.nitag-resource. org/media-center/document/158-indias-national-technical-advisory-group-onimmunisation
4. ICMR [Internet] (cited on :2021 May 06) Available from: https://main.icmr.nic.in/ content/about-us
5. CDSCO CLAA: A Textbook of Industrial Pharmacy-II, B.Pharmacy VII-Sem PCI (As Per The Revised 2016-17 Regulations of The Pharmacy Council of India) 2020-21 Edition, Dr. Shaik Harun Rasheed, SIA Publishers & Distributors Pvt Ltd [Text Book]
6. Adverse drug reaction reporting [Internet] (cited on: 2021 May 07) Available from: https://www.google. com/url?sa=t&source=web&rct=j&url=https:// cdsco.gov.in/opencms/export/sites/CDSCO_ WEB/Pdf-documents/Consumer_Section_PDFs/ ADRRF_2.pdf&ved=2ahUKEwjJ1d-Tk7fwAhVlyjgG HSBtAQIQFjALegQIEhAC&usg=AOvVaw39IhYZeB BnKToInXFZAKsv
7. ADR reporting: A Textbook of Pharmacovigilance, As per the Syllabus Prescribed
Dr. M P Venkatesh
Dr. M P Venkatesh is currently working as Associate Professor, Dept. of Pharmaceutics at JSS College of Pharmacy, Mysuru, India. He is the Coordinator of Regulatory Affairs Program in the Institution. Dr. Venkatesh is serving as Guest Assistant Professor in the Faculty of Pharmaceutical Sciences at UCSI University, Malaysia.

Email: venkateshmpv@jssuni.edu.in
Praveen Halagali

Mr. Praveen Halagali holds master’s degree in Industrial Pharmacy from JSS College of Pharmacy, Mysuru, India. His areas of interest are pharmaceutics and regulatory sciences.
Email: praveenhalagali8@gmail.com
20 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Regulatory
& Marketplace



INTERNATIONAL PHARMACEUTICAL INDUSTRY 21 www.international-pharma.com RECYCLABLE THERMAL BLANKETS For Healthcare Airfreight Temperature Qualifications & Validations - Multilayer thermal blanket for PMC-ULD – Euro and Block pallets - Stress tested in summer (+46°C) and winter ( -15°C) profiles - Airfield Tarmac tested on solar power and greenhouse effects Recycling Qualification - NOT laminated = Dismantlable = Recyclable = Remanufacturing Available worldwide at your freight forwarder KRAUTZ - TEMAX Group Tel: (Belgium) +32-11.26.24.20 / Website www.krautz.org / Email info@krautz.org Temperature protection of pharmaceuticals and healthcare products in airfreight (+15°C +25°C) and (+2°C +30°C)
Serving the 0.05% of Patients Living with a Rare Disease
Increasing adoption of digital trials and data-driven commercial strategies has laid the foundations for improved pinpointing of treatments and speedier diagnoses.
Though estimates show that there are now up to 30 million people in the European Union living with a rare disease, progress with diagnosis remains worryingly slow, with patients suffering as a result.1 Healthcare professionals (HCPs) must consider between 6,000 to 8,000 rare diseases when diagnosing a patient, with the result being that it is often common for them to fail to spot the symptoms within relatively small patient populations (five or fewer in 10,000). This means patients commonly wait years for a diagnosis or may never be diagnosed at all.
Dignosis Pharma companies trying to deliver effective treatments for rare diseases are searching for the proverbial needle in the haystack. During development, it's a challenge to find, recruit, and retain patients for trials. Getting to a viable product requires seamless coordination of clinical data, document review, and regulatory submissions across multiple functions. In the commercial phase, medical science liaisons (MSLs) have to identify and engage the right experts to shape their medical strategy. After launch, many field teams struggle with access: 65% of accessible HCPs meet with three or fewer pharma companies in Europe.2 It's unsurprising that 95% of rare diseases have no approved treatment.3
With the odds stacked against them, pharma companies of all sizes need to unlock the clinical and commercial value of their vital work swiftly. Here's how some leaders are transforming their approach to data and technology to speed the delivery of new treatments to patients.
Boosting the Development Pipeline
To run successful clinical trials, CROs must be able to quickly and cost-effectively recruit participants with rare diseases across diverse locations. Fortunately, the industry has increasingly leveraged digital trials to find patients situated across the globe
whilst minimising expenses. That being said, difficulty arises when it comes to retaining their involvement in a trial, especially if the trial is site-based and their health worsens.
To improve retention, patients deserve more choice over how to participate in studies from one day to the next, whether in person or through digital means, based on their daily health status and personal preferences. Wearable technology, electronic Patient-Reported Outcomes (ePRO), and remote monitoring devices can help track patients' health status. But technology needs to keep pace with these scientific developments. Traditional electronic data capture (EDC) systems no longer give study teams the whole picture, and data managers lack the tools to manage non-traditional data at scale.
Once the trial is over, companies need to find ways to cut the time and cost of developing a safe and effective treatment for a rare disease. An integrated approach to technology and data could help to accelerate their product pipelines by improving crossfunctional collaboration and establishing a single source of truth across clinical, regulatory, safety, and quality.
For example, Alexion's rapid growth trajectory required a fourfold increase in clinical trials over three years, without overwhelming its people, processes, or systems. The company was able to build a flexible foundation to support its fastgrowing development pipeline by improving clinical data management and trial oversight, consolidating regulatory information, and boosting efficiency in quality document management. Michael Sauter, senior director of global regulatory operations, notes: "Stakeholders are starting to realise the advantage of having a single system where everything is located."
Utilising Data to Enable Precision Engagement
After a product has been authorised, the next priority for pharma companies is to identify and cultivate relationships with key people whose insights will shape medical strategy. Finding the appropriate people focused on rare diseases is difficult, and
teams with limited resources are often forced to prioritise only a handful of experts. As Malcolm Crooks, chief operations officer at COUR Pharmaceuticals, states: "I worked on a program in the U.K. where there were only 300 patients in the whole country suffering from a specific disease, with data held in one registry in Salford, near Manchester... It was a unique challenge to work with one key opinion leader, but also an opportunity to understand their ecosystem and discover how to support patients' needs with our solutions."
This high-value engagement necessitates meticulous planning. Despite this, MSLs are under constant pressure to identify and approach digital opinion leaders, community leaders and experts as fast as possible. For MSLs to most effectively conduct outreach, they should be provided with a single point of access to relevant, upto-date information on key people's clinical and academic interests. With large amounts of clinical research data being rapidly exchanged and debated on social media, medical affairs teams that still use a manual approach could easily lose track of the latest developments. To mediate this, they need compliant access to both social media feeds and alternatively published resources to make sure they are fully prepared before engaging with an expert.
After launch, some companies get a head start on market access and prioritise field activity by HCP/KOL types, primary centres, and specialty. One biotech focused on rare and serious diseases was able to hit the ground running in countries where launches were live because its management team had access to dashboards based on clean, realtime customer reference data on the full ecosystem surrounding its drug. Through early visibility of market potential, it could even hire key roles in markets yet to launch.
With few customers for any given product, companies need to effectively marshal their limited resource capacity. This makes it critical for the field force to appreciate the value of new technology and start using it. Elodie Privat, sales analytics and operations lead at Novartis Gene Therapies, describes the situation when launching a product for
22 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Drug Discovery, Development & Delivery
spinal and muscular atrophy: "Whenever we launch in a country, we need accurate data for HCPs and specialties. If you don't have accurate data, you won't build trust, and users won't adopt the technology."
Data can pinpoint the path forward, even when patient journeys are complex and varied (like in rare autoimmune diseases). In North America, end-to-end patient data is increasingly used to map the most likely patient pathways from diagnosis to treatment. For example, ANI Pharmaceuticals used advanced customer segmentation and technology to build specialised field teams in months, empowering them with information on different patient segments, therapeutic areas, and HCP customers. Field teams succeed because they prioritise engagement based on which doctors have the most appropriate patient visits and what they tend to prescribe.
Finally, improving HCP awareness of a rare disease is key to reducing diagnosis time. For this, HCPs need seamless access to information to make an early diagnosis – for example, education about symptoms they may be unaware of and at risk of ignoring. At one company, field teams have shared insights into the patient pathway (including surveys on the patient experience) and other real-world evidence with HCPs. By combining CRM data (e.g., when a field team member last met a customer) with external data sources (e.g., HCP demographics, online journeys, and activity), the field team has developed impactful relationships with the most relevant doctors in the health ecosystem. It can now deliver innovative treatments to patients faster because of its established connections with HCPs.
Digital Transformation Paves the Way to Treatment

The challenges presented by rare diseases
mean that a unique approach is necessary across the end-to-end process of bringing a treatment to market. Technology and accurate data must be fully leveraged together at every stage. As clinical, commercial, and market access teams are able to cohesively share insights and collaborate, the launch process will be accelerated.
With no margin for error, companies of all sizes focused on this area need an early view of market viability to commercialise new treatments at scale. They also need to equip MSLs with the right tools to work in a hybrid way, based on HCPs' changing preferences and needs.
Despite the barriers, leading companies are getting so much closer to discovering and delivering for the five in 10,000 affected by rare diseases. For patients and HCPs alike, an accelerated pathway to rare disease treatment cannot come soon enough.
REFERENCES
1. European Commission, Research and Innovation, 'EU Research on Rare Diseases'
2. Veeva Pulse Field Trends Report
3. The Lancet Diabetes & Endocrinology, 'Spotlight on Rare Diseases', Vol. 7, Issue 2, February 2019
Chris Moore
As president of Veeva Europe, Chris is responsible for growing the business in the region. A 29-year veteran of the life sciences industry, Chris started his career at ICI Pharmaceuticals (now AstraZeneca). Chris then joined a start-up called Kinesis, building a team delivering document management solutions for pharmaceutical companies. Through a series of mergers and acquisitions, Kinesis ultimately became PwC; Chris made partner with PwC in 2001. Chris went on to run both European and US (West Coast) life sciences business for IBM before leading the IBM global life sciences consulting Business Analytics and Optimization unit. Most recently, Chris was the lead partner for life sciences for Europe, the Middle East, and Africa at EY.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 23 www.international-pharma.com Drug Discovery, Development & Delivery
Unlocking the Potential of Drug Discovery Labs with Automation
Trying to achieve more without increasing costs, sacrificing quality, or slowing down growth is a constant mission for any expanding business or industry – but it is particularly acute when it comes to drug discovery. The COVID-19 pandemic highlighted the pressing need to rapidly develop effective drugs and treatments, and the growing industry has made remarkable progress in recent years –with the government recently investing £227 million into the sector. Areas like precision medicine, novel vaccines, cell and gene therapies and synthetic biology are all driving healthcare forwards – but there are challenges for drug discovery companies to effectively scale and bring new cutting-edge products to market.
The three main challenges are the finite amount of lab space for expansion in the UK, manual processes impeding scaling and reproducibility, and scientists spending too much time on manual tasks rather than research and discovery.
Consultancy firm, Bidwells, recently found that the demand for lab space in Oxford and Cambridge is significantly surpassing what is available – with 10 thousand sq. ft available in Cambridge compared to two million sq. ft of demand. This means that drug discovery is at risk of facing a slowdown simply because there is not enough space to conduct cuttingedge research – and investors may turn their heads to other cities, such as Boston in the US, that have millions of square feet in lab space readily available. To meet this demand, there has even been a rise in the number of non-traditional spaces being converted into labs – such as refitting closed high-street shops.
The ability to scale throughput can also be difficult. As drug discovery labs seek to expand, manual processes struggle to keep pace and maintain accuracy. Being able to reproduce highly accurate results is vital when researching new drugs, as is the ability to test out as many leads as possible. With scientists carrying out many processes by hand, there is a limit to how fast it is possible to scale.
And finally, highly skilled and intelligent scientists in drug discovery aren’t fully utilising their expertise due to the need to perform routine tasks in the lab, rather than spending time designing new experiments.
However, there are ways to combat these challenges and drive drug discovery forwards. Automation is one tool which has been used in laboratories for several years and is continually evolving. Increasingly, labs are embracing automation to help scale, free up scientists time and work more efficiently.
Achieving More in the Same Space
Expanding into additional lab space is not always an option for biotech and life sciences businesses for two reasons: firstly, the scarcity of available lab space, and secondly, the cost of doing so. They need to make their existing resources work harder now more than ever. Thus, the ability to optimise existing space becomes a crucial alternative for scaling up, especially for smaller or oddly-shaped labs – the latter of which is more common in re-purposed buildings such as shops and older buildings.
To scale-up, workflows need to be flexible and able to adapt depending on the needs of the lab. Therefore, flexibility in implementing automation to lab benches where and when it’s required for each specific segment of a lab experiment can help ensure that automation can operate across multiple functions and processes, and achieve efficiency, accuracy, and throughput. With recent developments in automation technology for flexible lab benches, labs can now reconfigure and modify facilities to suit any space as their needs and functions change – no matter the stage of their automation journey – enabling them to efficiently perform multiple processes –from liquid handling to thermocycling – and maximise equipment capabilities, helping to scale without manual intervention.
One organisation that has been able to boost throughput and maximise space through automation is the Advanced Sequencing Facility (ASF) at the Francis Crick Institute. Genomics sample preparation is ideally suited for automation, given the need for the process to be robust and scalable.
The ASF has successfully automated three essential workflows for genomics sample preparation. This has enabled the facility to eliminate manual touchpoints, increase walkaway time, and improve sample quality – which has allowed it to work at a faster pace, scale up, and expand its genomics research for early-stage drug discovery within its existing space more efficiently.
Relieving Scientists’ Workload
Automation is also valuable in drug discovery as it can reduce the manual workload that scientists must carry out each day, like plate handling and data uploading to LIMS systems. Instead of manually retrieving and reading data from the equipment, automating the process allows scientists to directly access data from a plate via cloud software, freeing up more time to focus on tasks like experimental design and data analysis, which require the skill of a trained scientist. This increased walkaway time can be a game-changer in the lab, allowing scientists to dedicate time towards making significant strides in drug discovery.
Although some automated processes require human interaction, many can also run entirely independently. This can relieve scientists from having to come into labs out of hours and means that experiments can run continuously over extended periods. For example, drug discovery organisations may need stem cells to test new compounds or potential new drugs on – but these are very needy and must be tended to every day. With an automated process, scientists are freed up from tending to them every day while still being able to deliver results at pace.
Boosting Accuracy and Repeatability
Finally, automation has an important role to play in maintaining accuracy and repeatability – a critical aspect of scientific experimentation – such as during cell manufacturing which is an important part of early-stage drug discovery when testing how small-molecule drug compounds would respond in human cells.
Replicating experiments with precision remains a challenge that can hamper drug discovery. This is where automation
24 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Drug Discovery, Development & Delivery
can really bring value – by facilitating the replication of complex experiments multiple times, generating more extensive and welldocumented data than a person could in the same timeframe. This not only increases the quantity of data available, but also its quality and reliability, reducing the risk of human error, standardising workflows, and leading to more robust scientific findings. This is essential to drug discovery, as scientists need strong and well-supported conclusions to develop safe and effective drugs.
Automation also allows labs to ‘widen the funnel’ when developing new treatments and cell therapies, which could be transformational to R&D efforts. Typically, a lab might start with thousands – or even millions – of potential leads in the discovery process. Current approaches are aimed at quickly reducing this to a small number in order to move forward to the next phase as efficiently as possible. This means that

potential leads that did not meet some limited criteria may be missed, or those that are taken forwards might have characteristics that might make them unsuitable. Either scenario may mean stepping back in the process in order to move forwards.
Instead, automation makes it possible to keep the funnel wider for longer, testing out a higher number of leads in the same amount of time in order to qualify the best drug options, while also gathering deeper and broader datasets for potential hits. This allows scientists to generate a larger pool of leads with greater data attached and ultimately transform the drug discovery process for the better.
Closing Thoughts
Empowering labs with modular, integrated automation can help leading life science companies in the UK within the drug discovery industry to scale without compromising on
accuracy or speed, make the most of existing lab space and equipment, free up scientists to work on more skilled tasks, and unlock untapped value within their business. With a modular approach, automation can be applied flexibly and adapted when needed to be the right fit for every lab space at any point in their journey.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 25 www.international-pharma.com Drug Discovery, Development & Delivery
Russell Green
Russell Green is Director of Product Growth for Cell Culture at Automata, the leading automation provider for the life science industry, which is transforming the way labs work with open, integrated automation.
Drug Discovery, Development & Delivery
How to Harness the Benefits of Inhalation When Re-formulating Biologics
The biologics market is growing rapidly, and is on track to be worth $719.14 bn by 2030, up from $366.6 bn in 2021.¹ This growth is being driven primarily by the rising prevalence of chronic diseases, such as cancer, diabetes, and heart disease, which demand sophisticated diagnostics and therapeutic treatments.²
Many of these life-changing biologics –used to treat conditions such as rheumatoid arthritis, Crohn’s disease, and cancer – are reaching blockbuster status, providing treatment for millions of chronic disease patients around the world. However, many of these therapies were brought to market during the first wave of innovation, and their patents will expire in the coming years. With exclusivity only lasting approximately 20 years, 230 biologics are expected to fall out of patent in the next few years up to 2025.³,⁴
After the loss of exclusivity biopharma companies will be able to develop generic versions of these treatments to compete with the original branded product. This is already reflected in the biosimilar market, which is expected to reach $143.6 bn in 2031, growing at a compound annual growth rate of 24.7% from 2021.⁵ The biosimilar product will be cheaper than the original, leading to the original innovator losing market share. If pharmaceutical companies want to compete with standard generic products, they will have to reformulate their product to extend its life cycle.
Reformulating an existing drug product’s route of administration can extend exclusivity. This, coupled with breakthrough innovation in targeted therapies for chronic and orphan lung diseases, means that companies are shifting towards delivery via the respiratory tract. In this article, Bernhard Müllinger, General Manager and COO, Resyca® and Kris Brosig, Director of Business Development discuss the current challenges of inhaled biologics, exploring how formulation and device innovation may be the answer to overcome them.
The Benefits of Biologic Reformulation
Traditionally, biologics have been administered parenterally, relying on
healthcare professionals (HCPs) to deliver these essential medications to patients. Diversifying the delivery route of biologics to self-administered alternatives, such as oral, inhaled, or self-administered injectable drugs, can help reduce the load on HCP resources while improving patient convenience. This is because medication can be taken at home rather than in a clinical setting. In addition, up to 10% of the population suffers from injection anxiety.⁶ Moving away from injectables, coupled with easing treatment access, can help to improve patient adherence, moving towards more patient-centric healthcare.
Choosing a Delivery Method for Biologic Reformulation
There are many options for administering a drug product, and biologic oral solid dose (OSD) formulation is a method that has also gained interest in recent years. As with formulating any drug product, there are challenges that need to be overcome. For orally administered biologics, this stems from the fact that they must travel through the gastrointestinal tract to be absorbed by the body and reach the desired target. Biologics are sensitive materials that undergo degradation under acidic conditions and when exposed to stress. Drug formulations must make the biologic robust enough to survive the stress associated with each route of administration or encapsulate it for protection. Recent advancements in formulation technology have led to breakthroughs in this area, enhancing product stability and bioavailability even for molecules with a larger molecular weight. Nevertheless, oral administration may not be suitable for all biologic drug products, and so other methods of delivery are also being investigated.
Inhaled biologics are of particular interest as they promote patient centricity, delivering the drug directly to the target site within the lungs, or other parts of the respiratory system, such as the nasal passage or upper airways. With pulmonary diseases still being a leading cause of mortality globally, novel drug products and biologic reformulation are being investigated for inhaled delivery, allowing administration direct to the desired part of the respiratory system.
Inhaled biologics offer numerous advantages that make them a viable option for biologic administration:
• Reduced side effects: Inhaled biologics reduce off-target toxicity that leads to adverse effects at locations within the body other than the desired target.⁷
• Large absorption area: The substantial absorption area (130 m²) of the respiratory system aids drug product bioavailability, reduces dosage requirements, and offers similar permeability to the small intestinal mucosa.⁸
• Improved efficacy: For local treatment, inhaled biologics have shown improved efficacy compared with traditional parenteral delivery.⁹
• Enhanced nervous system targeting: Potential delivery of biologics to the central nervous system would result in rapid response upon administration.¹⁰
Although inhalation offers a number of benefits, inhaled biologics are still in the early stages of development, and a number of challenges must be overcome to successfully use this method to produce, or reformulate, a drug product.
The Challenges Associated with Inhaled Biologics
Biologics remain highly sensitive and complex materials, and therefore a number of challenges need to be addressed to develop inhaled biotherapeutics. Inhaled biologics as a segment remains in its infancy, and so it has had, as yet, limited regulatory exposure.
This limited regulatory exposure may lead to additional scrutiny when seeking approval, as there is still ambiguity regarding dosing, efficacy, and safety that needs to be addressed, and will require extensive analysis and evidential documentation.
Many challenges arise throughout the development, formulation, and delivery of a desired biologic, requiring innovative solutions to ensure success. This is the case for both novel biologic treatments and reformulated products.
When developing and formulating an inhaled biologic, challenges to overcome include:
26 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
UniSafe® Platform
Delivering the future with safety and simplicity
Developed around UniSafe®, our platform meets the needs of you, our partner, and your varying patients’ needs both now and in the future.
A spring-free, passive safety device for 1mL pre-filled syringes, designed for simple assembly and use.
A spring-free, passive safety device for 2.25mL pre-filled syringes, designed for simple assembly and use.

Proven, on market, in patient use
Fully industrialised and ready to supply
A reusable companion auto-injector for UniSafe® 1mL.

A reusable connected companion auto-injector for UniSafe® 1mL.


INTERNATIONAL PHARMACEUTICAL INDUSTRY 27 www.international-pharma.com
UniSafe® is a registered trademark of Owen Mumford Ltd. © 2023 OMPS/ipi/ad/ob/0623/7
soon Want to know more? Visit ompharmaservices.com/ipi-june2023 or email pharmaservices@owenmumford.com
Coming soon Coming
• Bioavailability issues: Molecules with a large molecular weight have limited permeability across the membrane and despite the large absorption area, this is also true for inhaled biologics. This poses questions about possible drug build-up in the lungs, leading to toxicity and potential immunogenicity.
• Formulation and delivery challenges: A large number of excipients are used when formulating a drug product for parenteral delivery. However, for an inhaled product it is imperative that the formulation allows for aerosolised delivery of the active ingredient, ensuring delivery to the target area of the respiratory system. Consideration must be given to stability of the molecule over the shelf life of the product in the combination device and also stabilising the molecule to ensure any delivery shear forces do not adversely impact the activity of the molecule. This means novel excipients may need to be added to the formulation, which can add an additional hurdle to regulatory approval, and is considered on a case-by-case basis.¹² In addition, both the active component and the excipients used must be compatible with the device materials.
With over 40 inhaled biopharmaceuticals currently in the early phases of the drug development pipeline, overcoming the inhaled biologic challenges is a priority.¹³ The limited range of possible inhaled dosage forms – from MDIs to nebulisers and dry powder inhalers (DPIs) – add to the complexity, as the biologic formulation must be adapted to meet the needs of these formats.
Advances in Inhaled Devices and Formulation
Drug product formulation plays a large part in overcoming the challenges associated with biologic drug products. Adding excipients into the product formulation can be used to help overcome the bioavailability and sensitivity issues while also helping to better formulate a drug for aerosolised delivery.
The design of the delivery device – either nasal or oral form – can be a powerful tool to overcome drug development and delivery challenges. Although in the past devices required extensive product customisation to ensure effective, safe, and efficacious delivery, recent innovations in soft mist inhaler (SMI) technology have significantly reduced customisation needed for a specific
biologic formulation, reducing project costs and accelerating timelines. These nextgeneration SMIs and nasal sprays produce a slow-moving aerosol cloud that can provide more effective targeted delivery while minimising stress and damage to the biologic.
SMIs with novel spray technologies are well suited to deliver sensitive compounds as they limit the shear stress exposed to the drug during delivery. This is particularly important for sensitive biologics (e.g. mRNA) or sensitive formulations such as lipid nanoparticles (LNPs). Using novel spray technologies can reduce degradation and aggregation of biologicals upon administration.
In addition, easy alteration of the spray cone angle of these innovative devices enhances delivery precision, expertly targeting the desired site at high doses while enhancing design uniformity, both of which can result in improved drug performance. The enhanced therapeutic performance per inhalation leads to lower dosage requirements – 20 times smaller than for parenteral delivery – making this mode of delivery much more cost effective. The novel design of these new SMI technologies also requires less pressure to actuate than traditional devices, making them even easier to administer.
Working with Experts
We are at the very beginning of the inhaled biologic story. This means that we have few resources to guide new drug development or re-formulation projects to regulatory approval. Nevertheless, the rewards of pioneering this approach to successfully deliver essential medication to patients via the respiratory tract are many.
Forming a supportive partnership with an expert in inhaled biologic formulation and in innovative devices – such as SMIs – can ease this process. Access to a bank of knowledge and expert advice will help drive a project forward at speed so companies can stay ahead of the patent expiry curve and ensure future exclusivity – or bring their new biologic to market with the best chance of success.
REFERENCES
1. Biologics Market Size, Share | Industry Forecast by 2030 (emergenresearch.com)
2. Chronic illnesses: UN stands up to stop 41 million avoidable deaths per year | UN News
3. Patent expiry dates for biologicals: 2018 update – GaBI Journal (gabi-journal.net)
4. List – 100 Biologics Patents Expiring Between 2022 to 2027 - GreyB
5. Biosimilars Market Size & Share | Statistics Report [2021–2031] (alliedmarketresearch.com)
6. Injection Phobia – Anxiety UK
7. Dry powder pharmaceutical biologics for inhalation therapy – PubMed (nih.gov)
8. Alveoli Function, Structure, and Lung Disorders (healthline.com)
9. Fröhlich, E. and Salar-Behzadi, S. (2021) “Oral inhalation for delivery of proteins and peptides to the lungs,” European Journal of Pharmaceutics and Biopharmaceutics, 163, pp. 198–211. Available at: https://doi.org/10.1016/j. ejpb.2021.04.003.
10. Tai, W. and Kwok, P.C. (2022) “Recent advances in drug delivery to the central nervous system by inhalation,” Expert Opinion on Drug Delivery, 19(5), pp. 539–558. Available at: https://doi.org /10.1080/17425247.2022.2074975.
11. Hall, P., Vahle, J. L., & Colman, K. (2021). “Inspiration and Exasperation: The Challenges of Inhaled Biologics”, Toxicologic Pathology, 49(2), pp. 232–234. doi:10.1177/0192623320984715
12. Taking a Breath: Advances in Inhaled Biologics (biopharminternational.com)
13. Inhaled Biologics Delivery: A Resurgent Market? – Oxford Global
Bernhard Müllinger
Bernhard Müllinger is the General Manager and Chief Operations Officer of Resyca® and is based in Munich. Bernhard has experience with smart nebuliser devices and has worked in this industry for most of his career. He has extensive knowledge in medical device development and clinical development of combination products. Prior to joining Resyca®, Bernhard worked at Vectura, Activaero, Inamed-CRO, Asklepios Clinic and Helmholtz-Zentrum.

Kris Brosig is Director of Business Development for the Advanced Delivery Systems business unit at Recipharm. Based in Germany, he has over five years of experience in the pharmaceutical industry. Before joining Recipharm, he worked as a Sales Manager for Harro Höfliger, selling capsule filling and inhalation equipment. He holds a master's degree in Leadership in Industrial Sales and Technology from the University of Aalen and Tec de Monterrey, Mexico.

28 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Drug Discovery, Development & Delivery
Kris Brosig
Bring your vision to life
Design, development and manufacturing on a global scale with lower risk.
Phillips-Medisize, a Molex company, helps you get from concept to creation quickly while reducing the inherent risks of any new product journey. We act as a singular resource for your company, offering robust end-to-end capabilities, including drug delivery device platforms. For over 60 years, pharma, diagnostic, and med tech companies have trusted us to deliver quality products that help people live healthier lives.
Get started today at phillipsmedisize.com

INTERNATIONAL PHARMACEUTICAL INDUSTRY 29 www.international-pharma.com
Advancements of Gene Editing Technologies (CRISPR/Cas9)
An Introduction to Gene Editing –CRISPR-Cas9
The dynamic promise of gene editing for humans is the ability to precisely manipulate the sequence of the cell genome to overcome genetic diseases.
Previously, gene therapy techniques focused upon introducing new genetic material within cells to provide a copy of faulty or lost proteins to reinstate their original function or provide a new function.1 These are primarily mediated by viral vectors, allowing the integration of desired gene copy into the cell genome. Gene editing allows for the modification of existing DNA in a cell, whereupon genetic material is added, removed, or replaced at precise points within the genome.1
CRISPR nucleases have revolutionary potential to enable major medical breakthroughs. Originating in the 1990s,1 and culminating in the discovery of the tracrRNA mechanism for successful targeting by Dr. Emmanuelle Charpentier and her team, CRISPR is now a promising option for many diseases.
CRISPR-Cas9 was discovered accidentally. Back in 1987, a team led by Yoshizumi Ishino inadvertently cloned a series of repeated sequences interlinked with spacer sequences whilst analysing alkaline phosphatase.2 A mystery at the time, it was not until 2000 when a team led by Francisco Mojica recognised reported disparate repeat sequences shared common features.2 This led to their definition as CRISPRs, or Clustered Regularly Interspaced Short Palindromic Repeats, and marked a distinct step in the journey from research to clinical use.
Targeting the nuclease to cut in a specific place in the genome is mediated by the nuclease interaction with a guide ribonucleic acid (gRNA). The gRNA has to bind the DNA area of interest for editing. Upon interaction of the nuclease with the genome, a cut is performed. The intrinsic
cellular gene repair mechanism are then induced, and the cut is closed, not without leaving out part of the sequence. This technique is used in the lab to disrupt gene expression (gene knockout).1
When providing a donor DNA sequence with homology arms to the cut area, an event of DNA insertion could happen (gene knock in). Now, imagine a gene with a mutation that leads to a certain disease. The ability to replace the mutated gene, or the mutated area with a correct sequence, will ultimately lead to gene correction and disease cure.1
Furthermore, there is base editing, a technology allowing the introduction of single base replacement in DNA without generating double strand breaks (DSBs). Two major classes have been developed – cytidine base editors (CBEs) to allow C>T conversions, and adenine base editors (ABEs) allowing A>G conversions. Now expanded, base editing is a promising therapeutic strategy.4
Gene editing can be performed in an ex vivo or in vivo manner. Ex vivo is performed on cells in the lab with cells sourced from the patient or a healthy donor. In vivo is performed in the patient. When performing ex vivo gene editing, the modified cells are isolated. As such, targeting the delivery to a specific cell type is not necessary. Ex vivo gene editing provides the option to select the modified cells before administration. It enables performing several, sequential gene editing events, leading to the generation of an off-the-shelf therapeutic cell line. In vivo gene editing does not involve removal of cells from patients and does not rely on obtaining cells from healthy donors. In vivo, however, has a need to direct the vehicle that carries the nuclease cargo to the target cells, or the nuclease will be introduced to various cell types, reducing effectivity.
The differences in the requirements and attributes of ex vivo and in vivo gene editing highly determine the delivery method of the gene editing components to the cells, the need for targeting to a specific cell type, and additional considerations. While CRISPR holds great promise, there is still much work needed to make it translatable to therapeutic and clinical settings.
Within cell and gene therapy (CGT), there is a significant role for contract development & manufacturing organisations (CDMOs) to enable the manufacturing of gene editing-based therapies. CDMOs can bring the experience, flexibility, expertise and infrastructure to manufacture innovative new therapies, and provide the tools and consumables necessary to enable high quality, good manufacturing practice (GMP) of gene therapy solutions such as CRISPR gene editing.
The Challenges of the CRISPR Gene Editing Landscape
CRISPR technology has several challenges, that should be addressed in order to utilize its benefits.
First, there is delivery. The CRISPR vehicles of delivery could be either viral and non-viral. As mentioned previously, ex vivo or in vivo plays a key role in determining vehicle and cargo.5 The selection of vehicle will determine if the cargo is delivered in the form of a protein, DNA or RNA.5
Non-viral delivery could be divided to chemical and physical techniques. Chemicalbased delivery includes encapsulation of the material to be delivered in lipid vesicles that will be taken up by the cells. Nano lipid particles are an example of a chemical-based delivery method that could be used in order to deliver DNA and RNA into cells.5 An example of a physical delivery is generating pores in the cell membrane using electric current (electroporation). This enables introduction of DNA, RNA or proteins into the cell. While nonviral physical delivery necessitates ex vivo cell modification, chemical and no-replicative viral delivery could, in principle, be applied in vivo too.
Several types of non-replicative viruses are known to mediate gene delivery into cells. Some carry DNA, some RNA. Viruses of the retroviruses family lead to random integration of the insert sequence into the genome, while other viruses, such as adeno-associated viral vectors (AAVs) could direct the integration to a specific place in the genome by utilising the virus’s own machinery,6 or by having the virus carrying a precise gene editing tool, such as CRISPR.
30 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Drug Discovery, Development & Delivery

www.international-pharma.com www.wakopyrostar.com ~ wkuspyrostarinfo@fujifilm.com FUJIFILM Wako Chemicals U.S.A. Corp. © FUJIFILM Wako Chemicals U.S.A. Corp. - 2023 PYROSTAR™ Neo is a new endotoxin detection reagent developed by recombinant technology. Neo PYROSTAR ™ Recombinant Endotoxin Detection Reagent Stable storage after dissolution - Offers longer shelf life for less reagent waste (4 hrs. at 2-8°C and 2 weeks at -30°C)
Viral vectors are known to efficiently deliver genetic material to host cells.
Viral mediate delivery, however, could have disadvantages, such as immunogenicity caused by viruses and the difficulty of manufacturing them at large scales and of high quality. Additionally, adeno-associated viral vectors (AAVs) can be limited in terms of the size of cargo which can be placed within them.5 Elsewhere, full-size adenoviral vectors (AdVs) may cause off-target effects, namely when a treatment binds to targets in the body other than those for which the treatment was intended.5
Precision is another challenge. Gene editing mediated by nucleases involves making cuts in the genome, with a risk of non-direct cut events or “off-target events”. While the nuclease is directed to a specific location in the genome by the gRNA, off-target cleavage activity does exist.7 Eradicating off-target events is crucial for enhancing the safety of gene editingbased therapeutics. Several advancements were made in the field to engineer CRISPR nucleases to provide highly efficient genome editing with reduced off-target events.9–10
Additionally, the issue of off-target events remains. As noted previously, there is a risk of therapy binding to targets in the body beyond those for which it was intended across the scope of CRISPR nucleases, thus requiring constant focus and evolution of delivery to minimise such occurrences.5 Furthermore, this must be aligned to address concerns about lacking or ineffectual specificity of CRISPR-gene editing tools, compared to either initial expectations of results or other technologies.
Another key challenge is quality and consistency of the gene therapy product, regardless of delivery. Manufacturing reproducibility and reducing batch to batch differences is highly dependent on process understanding, process monitoring and final product characterisation tools. Ensuring uniformity is just as key for precision as addressing off-targeting and specificity, requiring robust analytical methods for quality control. We may not be able to always influence precision within a selected CRISPR method, as this can be dependent on delivery. However, where we can influence, so too remain challenges ensuring precision.
Next, we come to intellectual property (IP). Historically, IP for CRISPR nucleases
resided with only a handful of companies. Furthermore, as CRISPR became recognised as a gene therapy solution with immense potential, legal battles soon emerged as companies and institutions competed for rights.11
This came in tandem with the establishment of a series of biotech-academic partnerships to convert CRISPR technology into new treatments for a broad range of diseases. Despite the broad focus, the IP nevertheless remained contained to this small group for much of the 2010s. Nowadays, proliferation of IP for CRISPR technology has improved. The field has evolved with nucleases such as MAD-7, STARNuclease and Cas12, meaning that there are alternative sequences to Cas9, allowing for separated IP.
However, patent disputes have not gone away. As recently as last year, the longrunning dispute between the Broad Institute and the team of the University of California, Berkeley and the University of Vienna over the original grant of CRISPR-Cas9 patent to Broad entered another round of appeals and legal challenges.10 This is just one example amongst many ongoing IP legal cases within the CRISPR field, and so we must contend with how to protect IP and patents amidst such an uncertain landscape.
Safety is paramount, given the need to ensure no off-target events generally, as well as for ex vivo and, specifically, for in vivo. For ex vivo, it is key to show there are no nuclease sequences or proteins left in the cells before patient administration, while for in vivo it is a matter of ensuring direct delivery.7 While CRISPR-based therapy is relatively new, regulatory agencies are publishing guidelines to facilitate the viability and commercialisation of gene editing by industry.10
Generally, whilst progress is being made in safety and detection, there remain challenges in fully assessing potential offtarget events. This needs to be solved, as regulators seek longer-term evidence bases focused on efficacy. It can be difficult to achieve for CRISPR based therapies in some instances, given it is still a new field, thus making the establishment of long-term efficacy a challenge. A balance is required between regulators and providers but this is not always simple to achieve.
Patient safety is the hallmark of CGTs. Several CRIPSR-based gene editing drugs
are being tested clinically. Among those are CRISPR-Cas9 in vivo Gene Editing for Transthyretin Amyloidosis,13 and ex vivo CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia.14
Scalability is the final challenge. As with any innovation, there is the age-old question of how best to translate its potential to many different healthcare systems and a wide range of patient populations. From manufacturing and development through to application and usage, it is a challenge across the value chain.
Significantly, there are concerns surrounding the availability of gene editing technology in less economically developed countries. For example, across a considerable number of laboratories, scientists could face issues in creating CRISPR pipelines at scale, as the design and build process requires the building of gRNAs and nucleases.
This is a complex process and could prove to be insurmountable without help, as laboratories can lack the infrastructure required.
The Need for Experienced Partners
With such challenges, where can solutions be found? We believe CDMOs are in a unique position to advance the development and adoption of gene editing tools.
CDMOs can offer solutions, benefits and guidance across the entire gene editing value chain. This includes:
• Ensuring precision and consistency in tool development, enhanced through experience and effectively measured steps and outcomes.
• A great degree of flexibility through providing a whole suite of delivery methods for CRISPR-based gene editing.
• Significant volume and breadth of different approaches for development, delivery and tracking.
• An agnostic approach to new methods, recognising that gene editing is a constantly developing field.
• A proactive approach to partnering, where CDMOs can advise on the methods of gene editing to support the ambitions of a company in a therapeutic area.
Considering the challenges, CDMOs can leverage their specialised expertise with development, management, tools and manufacturing methods.
32 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Drug
& Delivery
Discovery, Development
Drug Discovery, Development & Delivery
On delivery, for example, CDMOs have the flexibility and capabilities to create and provide the mechanisms of delivery required for gene editing, whilst similarly using their expertise to advise on which methods would be most effective for the therapeutic area in question. A similar point applies to precision challenges, as through the experience of dealing with off-target events, lacking specificity and time intensive gRNA processes, CDMOs can provide advice and solutions to biotech companies looking for guidance.
CDMOs can leverage knowledge of more mature fields, such as bio production, and apply it to CGT. This includes viral manufacturing at scale, advance characterisation tools for the quality of the virus, and high edge techniques for identification of off-target events and genome integrity assessment. All these provide better understanding and identification of the therapy, contributing to efficiency and safety.
CDMOs can consult on different methods and available solutions. Such functionality is built into their operations when consulting biotech companies about the challenges of CRISPR-based gene editing therapies beyond production and manufacturing. With the regulatory landscape too, CDMOs can develop solid evidence bases to satisfy concerns about long-term efficacy and comparability. These come from effective processes to ensure well measured steps and outcomes throughout testing.
With respect to scalability, CDMOs have a global reach and considerable capabilities and resources to advance gene editing solutions to market and clinic.
Conclusion
Gene editing is a constantly evolving field with a significant range of challenges. CDMOs are uniquely placed to use their expertise and experience with tools and manufacturing
methods, alongside their ability to produce and provide these, to enable high quality good manufacturing practice for gene therapy products. This can help to quickly and efficiently advance solutions from inception through to treating the patient populations that need them most.
Moving forward, as the entire gene therapy field considers how to respond to the ongoing challenges faced, the role of CDMOs will be vital in helping utilise the immense potential benefits of gene therapy for the world.
REFERENCES
1. Luo, S, et al. CRISPR/Casi-Mediated in vivo Genetic Correction in a Mouse Model of Hemophilia A. Frontiers in Cell and Developmental Biology. 2021. 9:672564. Available at: https://www.frontiersin.org/ articles/10.3389/fcell.2021.672564/full
2. Broad Institute. CRISPR Timeline. Broad Institute Website. 2023. Available at: https:// www.broadinstitute.org/what-broad/areasfocus/project-spotlight/crispr-timeline
3. Ishino, Y, et al. History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology. Journal of Bacteriology. 2018. 12;200(7):e0058017. Available at: https://pubmed.ncbi.nlm.nih. gov/29358495/
4. Antoniou, P, et al. Base and Prime Editing Technologies for Blood Disorders. Frontiers in Genome Editing. 2021. 3:618406. Available at: https://www.frontiersin.org/articles/10.3389/ fgeed.2021.618406/full
5. Roberts, R. Delivery of CRISPR-Cas9: Cargo, Vehicles, Challenges and More. Synthego. 2023. Available at: https://www.synthego.com/blog/ delivery-crispr-cas9
6. Xu, L, et al. Lipid Nanoparticles for Drug Delivery. Advanced NanoBiomed Research. 2021. 2:2;2100109. Available at: https:// onlinelibrary.wiley.com/doi/full/10.1002/ anbr.202100109
7. Bijlani, S, et al. The Role of Recombinant AAV in Precise Genome Editing. Frontiers in Genome Editing. 2022. 3:799722. Available at: https://www.frontiersin.org/articles/10.3389/ fgeed.2021.799722/full
8. Zhang, X-H, et al. Off-target events in CRISPR/ Cas9-mediated Genome Engineering. Molecular Therapy Nucleic Acids. 2015. 4:e264. Available at: https://www.sciencedirect.com/science/ article/pii/S216225311630049X
9. Kleinstiver, B, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide offtarget effects. Nature. 2016. 28:529(7587):4905. Available at: https://pubmed.ncbi.nlm.nih. gov/26735016/
10. Kleinstiver, B, et al. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nature Biotechnology. 2019. 37(3):276282. Available at: https://pubmed.ncbi.nlm.nih. gov/30742127/
11. Balakrishnan, V & Jewell, C. The battle to own
the CRISPR-Cas9 gene-editing tool. World Intellectual Property Organisation (WIPO) Magazine. April 2017. Available at: https:// www.wipo.int/wipo_magazine/en/2017/02/ article_0005.html
12. FDA. Human Gene Therapy Products Incorporating Human Genome Editing – Draft Guidance for Industry. 2022. Available at: https://www.fda.gov/media/156894/download
13. Gillmore, J, et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. New England Journal of Medicine. 2021. 5:385(6):493502. Available at: https://pubmed.ncbi.nlm.nih. gov/34215024/
14. Frangoul, H, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. New England Journal of Medicine. 2021. 21:384(3):252260. Available at: https://pubmed.ncbi.nlm.nih. gov/33283989/
Inbar Friedrich Ben-Nun

Dr. Inbar Friedrich Ben-Nun is Director of Cell Therapy Research and Development, at Lonza. Dr. Friedrich Ben-Nun holds a Ph.D. degree in Molecular and Structural Biochemistry (The Hebrew University of Jerusalem, Israel). She joined Lonza 11 years ago as a Scientist, after completing a Postdoctoral research on human embryonic stem cells (The Hebrew University of Jerusalem, Israel) followed by research at the Scripps Research Institute, La Jolla, where she obtained an extensive experience with iPSCs. In her current role, Inbar is driving and executing innovative solutions for allogeneic cell therapy processes and platforms.

Fatma Aybegum Senkesen
Fatma Aybegum Senkesen is Head of Divisional Projects in Lonza's Cell & Gene division, where she spearheads the development of strategic initiatives and oversees the evaluation of key projects for the division. She has held various roles at Lonza, including Market Intelligence, Marketing, and Commercial Development. Prior to joining Lonza, Fatma worked for companies specialising in market research, product forecasting and data analytics consulting for the Pharma & Biotech sector. She holds a BSc in Chemistry from Bogazici University and an MBA from the University of Miami.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 33 www.international-pharma.com
Dr.
Gene Edited iPSCs May Find a Place in Allogeneic Therapies
An Introduction to Pluripotency
Pluripotent: adjective; capable of giving rise to many different cell types. This is the hallmark characteristic of the induced pluripotent stem cell, or iPSC. Since the discovery of reprogramming adult human fibroblasts into an induced state of pluripotency in the early 2000s, the scientific community has been enamored with the possibility of this dynamic cell type.1 A seemingly vast universe of possibilities exists for the iPSC from the corners of basic research to the depths of clinical medicine.
The pluripotent state has been proven time and again using various protocols for generating cell types and organoids covering nearly every system in the human body. Industry and pharma have also come to favor the iPSC for many of its other qualities. While the iPSC may be considered a “finicky” cell type to work with in the lab, and costly to grow due to reliance on complex growth medias and matrices, it is unique in that it is considered to be a patient (or donor) sourced cell type, but with the added potential for long-term growth in culture. Easily derived from primary skin biopsies or blood, iPSCs maintain the existing genetic and epigenetic makeup of the human from which they came without the ethical dilemmas surrounding the use of embryonic stem cells. Nearly as important as their differentiation potential and proximity to the primary source is the ability of iPSC cultures to be scaled and banked for the purposes of manufacturing gene and cell therapies.
In the undifferentiated state iPSCs have been broadly studied across research fields. They have been pivotal instruments for understanding basic cell biology, advancing drug discovery efforts, and have served as the foundation of clinical therapies. Differentiated but otherwise unmodified iPSCs already began to have a clinical impact within the first decade of their discovery. In 2017, somatic cells harvested from a patient with macular degeneration were converted to iPSCs and differentiated into
retinal pigment epithelial cells, which were then used in the same patient in an effort to restore vision as an autologous (self) cell therapy.2 Since then, other clinical uses of autologous stem cell therapies have been investigated for regenerative medicines in the fields of diabetes and cardiology, among others.3,4
Broadening Therapeutic Potential
Today, the use of iPSCs has progressed from regenerative therapies to immunotherapies and other precision medicines. We have already seen the impact of autologous chimeric antigen receptor (CAR) T cells in the clinic where they are used to address hematological malignancies. Autologous therapies avoid concerns over undesired immune response in patients, because typically, even if the cells are engineered ex vivo they are still recognised as “self” once returned to the patient’s body. However, engineering any cells ex vivo to enable autologous personalised medicine is a time consuming and expensive process because each dose of the therapy must be engineered on-demand for every patient.
Limitations exist even for immunotherapies where both engineered T and natural killer (NK) cells have proven to be robust therapeutic agents. Therapies that rely on autologous primary cells have lengthy production times, sometimes longer than the patient can survive. There is also a substantial burden on the cells during the manufacturing process resulting in variable functionality of the end product. The alternative is an allogeneic (non-self) therapy which could be manufactured in large batches and readily given as an “off-the-shelf” universal product. Given the previously mentioned benefits of iPSCs having the capacity for expansion and the ability to differentiate into other cell types, including those of the immune system, the allogeneic iPSC holds special promise for a wide range of advanced therapies.
The Immune Response Problem
To realise the promise of iPSC-derived T cells for universal immune oncology therapies the allogeneic product must not only harbor the cancer-specific payload (usually an engineered T cell receptor), but also be immune-neutral to the host
to avoid debilitating graft vs. host disease (GvHD), where the iPSC-derived transplanted immune cells regard the recipient’s body as foreign and begin to attack. Being immuneneutral is also important for iPSC or iPSCderived transplanted non-immune cells in regenerative therapies to ensure they are not simply cleared from the body by the host immune system before they can have an effect.
The immune system uses a sophisticated complex of genes, expressed on the cell surface as peptides, to recognize self from non-self. Most notably the major histocompatibility complex (MHC) class I and class II glycoproteins present peptides to the T cell receptors. In humans, MHC class I and MHC class II are encoded by the human leukocyte antigen system (HLAI and HLAII, respectively). Research in the use of iPSCs for regenerative medicine has shown that knocking out β2 microglobulin (B2M) and class II major histocompatibility class transactivator (CIITA), which are critical for the HLAI/II display on the cellular surface, can render iPSC-derived cells less susceptible to immune rejection in an allogeneic therapy.5 To further improve compatibility iPSCs can be engineered to express HLA-E as their only surface HLA class I molecule, which prevents NK cell lysis of B2M -/- cells.6
Allogeneic primary T cells can be engineered to avoid GvHD by removal of the T cell receptor α (TCR) and CD52 genes.7 Further engineering to knock out B2M and programmed cell death protein 1 (PDCD1) can additionally enhance T cell therapy function.8 Still, the expansion protocols required for editing primary T cells can also result in exhausted cells and less effective products. Although NK cells naturally perform their function regardless of the patient’s HLA haplotype, they have a limited proliferative capacity that makes genetic engineering of this cell type ex vivo extremely challenging. Because both T and NK cells can be derived from iPSCs, the logic holds that iPSCs edited to be allogeneic medicines could serve as a better universal starting point for these oncology treatments.
CRISPR Revolution
With the discovery and adoption of the
34 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Drug Discovery, Development & Delivery


































INTERNATIONAL PHARMACEUTICAL INDUSTRY 35 www.international-pharma.com KahleAutomation.com We’ve merged with BBS Automation Our ideas just got a whole lot brighter Announcing a partnership in automation equipment that brings a whole new light to medical and pharmaceutical device manufacturing. Visit www.KahleAutomation.com or contact Kahle@KahleAutomation.com U.S.A. | ITALY | CHINA Kahle® is dedicated to providing custom automation machinery solutions for the Medical Device, Pharmaceutical, and Healthcare Industries around the world.
robust CRISPR-Cas9 gene editing system, knockout and knock-in capabilities are well understood and have brought the promise of allogeneic products within reach, though this method of gene editing is not without limitations. Type II nucleases like Cas9 introduce knockout through a targeted double strand DNA (dsDNA) break in the genome. When the cell uses inherent DNA repair pathways to repair the damage the dsDNA is often repaired incorrectly. The resulting insertions and deletions (INDELS) cause the target protein to be non-functional or not transcribed at all, which knocks out the protein function. Following the same process, the targeted dsDNA break can be supplemented with an exogenously delivered DNA repair template, which can incorporate new sequence into the broken locus through endogenous homology directed repair mechanisms and thereby knock in new protein function. Sequencespecific targeting limitations and off-target INDELS aside, the dsDNA break induced by Cas9 is a catastrophic and often fatal event for the cell, particularly for the sensitive iPSCs. Furthermore, when multiple dsDNA breaks are introduced in the same cell at the same time (whether it’s two on-target genes in a multiplex knockout experiment or one on-target and one off-target gene in a singleplex knockout experiment) the cut ends can translocate, resulting in large genomic rearrangements that can lead to oncogenic phenotypes.9
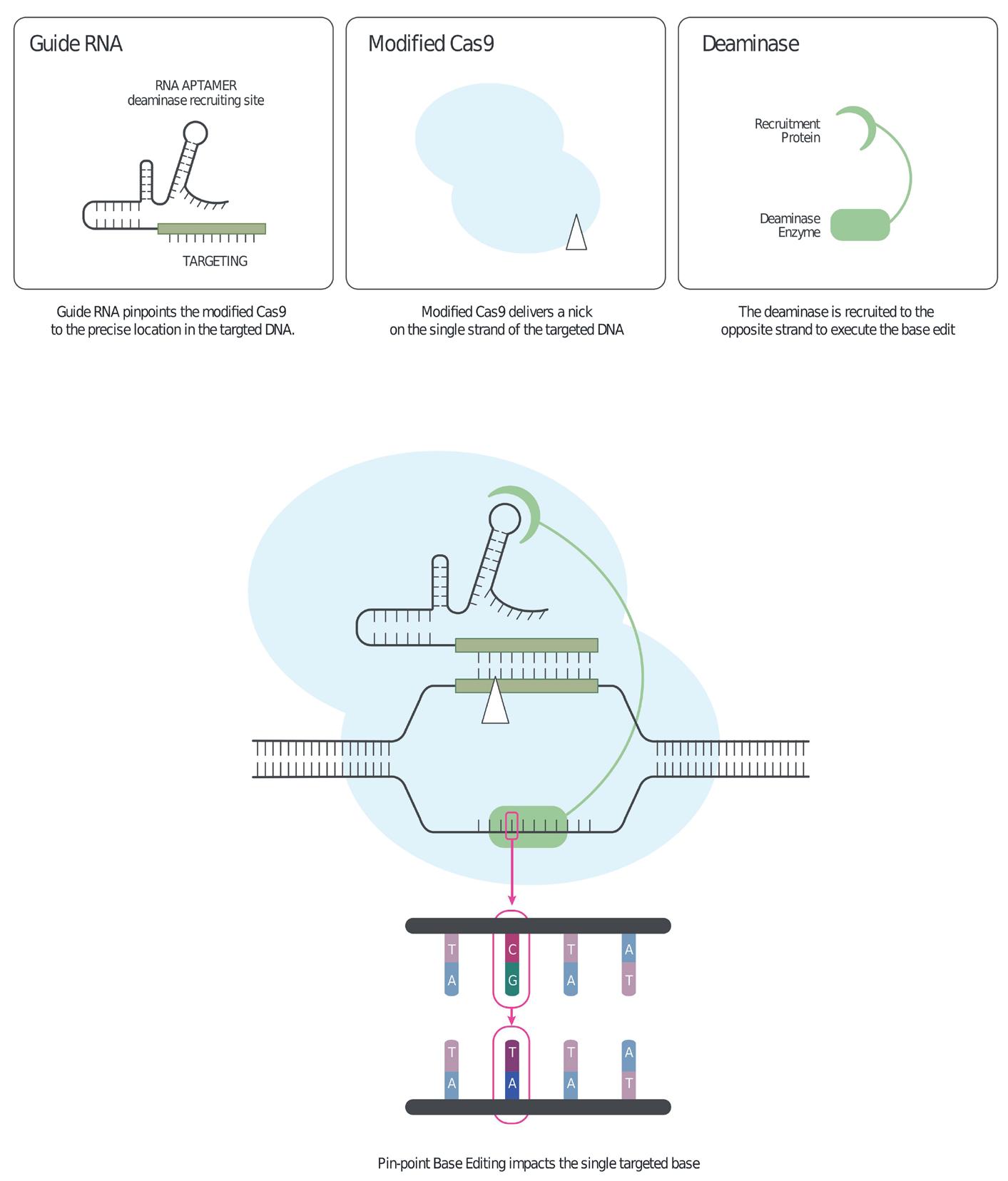
One way to get around this potentially fatal flaw draws on the iPSC’s ability to persist through many divisions in culture. Gene editing with CRISPR-Cas9 is done sequentially and at each round, clonal cell lines are isolated, grown, and analysed for precise editing, genomic integrity, and stem cell health. This intensive effort plays out over weeks to months and is in theory undertaken four times in the case of the sequential B2M/CD52/TRAC/ PDCD1 knockouts required to create the allogeneic iPSC-derived T cell product. While the iPSCs are capable of significant expansion in culture, each round of division (roughly every 24 hours) may have additive negative impact on the health of the cell culture where spontaneous karyotypical abnormalities and loss of pluripotency tend to arise over time, even in clonal cell lines.
Base Editing: Enhancing iPSC Engineering
To speed up and de-risk the generation of allogeneic iPSC products one could consider
next-generation gene editing approaches which do not introduce dsDNA breaks, such as base editing. Avoidance of dsDNA breaks is an improved safety feature that enables multiplex editing in a single step. Compared to CRISPR-Cas9 knockout which relies on INDEL formation, base editing introduces nucleotide-specific changes to the DNA and can cause a knockout phenotype through the introduction of a premature stop codon or a splice site disruption.10,11
Base editing (Figure 1) also utilises an RNA-guided nuclease like Cas9, but the nuclease is modified in one of two ways: (i) The DNA cleavage domains are either fully deactivated (dCas9) or (ii) one domain is deactivated to create a nuclease that can nick but not cut DNA (nickase, nCas9). While the deactivated enzyme is considered to have less unintended effects, the nickase gives increased efficiency.12 The deamination of the target base happens only on one strand, so by nicking the single DNA strand opposite to the edited base the cell is
forced to repair the DNA using the edited strand as the template, thus improving the likelihood of the edit being incorporated in the next round of DNA replication. Utilising base editing for multiplexed knockout is particularly advantageous as there is less damage to the genome and less chance for genomic translocations between edited loci when no dsDNA breaks occur.13 This results in healthier cells in a shorter amount of time at reduced cost.
Beyond its application in the creation of next-generation adoptive T cell therapies, it is expected that base editing will offer similar opportunities for the engineering of a wide range of allogeneic cell therapies with increasingly advanced safety and functionality profiles. The continued development of advanced base editors will set the stage for affordable off-the-shelf allogeneic treatments for a broad range of indications. With advances in gene editing technologies, the iPSC may have found its place in allogeneic therapies.
36 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Drug Discovery, Development & Delivery
Figure 1: Base editing technology utilising a nickase form of Cas9 to elicit precise single strand cuts at target sites in the genome. Illustration by Revvity, inc.
Drug Discovery, Development & Delivery

REFERENCES
1. Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126(4):663-676. doi:10.1016/j. cell.2006.07.024
2. Mandai M, Watanabe A, Kurimoto Y, et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. New England Journal of Medicine. 2017;376(11):1038-1046. doi:10.1056/nejmoa1608368
3. Kondo Y, Toyoda T, Inagaki N, Osafune K. iPSC technology-based regenerative therapy for diabetes. J Diabetes Investig. 2018;9(2):234-243.
doi:10.1111/jdi.12702
4. Kadota S, Tanaka Y, Shiba Y. Heart regeneration using pluripotent stem cells. J Cardiol. 2020;76(5):459-463. doi:10.1016/j. jjcc.2020.03.013
5. Mattapally S, Pawlik K, Fast V. Human Leukocyte Antigen Class I and II Knockout Human Induced Pluripotent Stem Cell–Derived Cells: Universal Donor for Cell Therapy. J American Heart Assoc. 2018;7:e010239. doi/10.1161/JAHA.118.010239
6. Gornalusse G, Hirata R, Funk S. HLA-Eexpressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. 2017;35(8):765-772. doi: 10.1038/ nbt.386
7. Qasim W, Zhan H, Samarasinghe S. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9(374):eaaj2013. doi: 10.1126/ scitranslmed.aaj2013
8. Rupp L, Schumann K, Roybal K. CRISPR/Cas9mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737. DOI: 10.1038/s41598017-00462-8
9. Nahmad A, Reuveni E, Goldschmidt E. Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat Biotechnol. 2022;40(12):1807-1813. doi: 10.1038/s41587-02201377-0
10. Billon P, Bryant E, Joseph S. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol Cell. 2017;67(6):1068-1079.e4. doi: 10.1016/j.molcel.2017.08.008
11. Webber B, Lonetree C, Kluesner M. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun. 2019;10(1):5222. doi: 10.1038/s41467-019-13007-6
12. Gaudelli N, Komor A, Rees H. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464-471. doi: 10.1038/ nature24644
13. Diorio C, Murray R, Naniong M. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140(6):619-629. doi: 10.1182/blood.2022015825
Dr. Amanda Haupt
Dr. Amanda Haupt is the Base Editing Product Manager at Revvity where she has contributed as an R&D Scientist to the company’s portfolio of gene editing reagents. Today, Amanda supports the company’s Pin-point™ base editing technology. Before joining Revvity, Amanda worked at the Allen Institute for Cell Science in Seattle, WA where she contributed to the generation of the Allen Cell Collection of engineered iPSC lines.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 37 www.international-pharma.com
Clinical and Medical Research
Accessibility in Rare Disease Paediatric Clinical Trials
Approximately 360 million people globally are living with a rare disease. Over half of these are children. Many of the diseases are life threatening and only 5% have an approved treatment. Trials in rare diseases are typically conducted at fewer sites, are longer in duration and are more frequently terminated than trials with more common indications.1 While there is continued growth in the development of drugs to treat rare diseases, clinical trial accessibility for patients remains challenging due to the requirements inherent in these studies.
Participation in a paediatric rare disease trial involves not just the child, but the whole family and may be the only potential treatment available. It is critical to consider the experience of the child and their caregivers not only at the beginning but also throughout the study, which can be many years in length.
There are physical and logistical, financial, and psychological and emotional pressures that come into play when participating in a clinical trial.
Physical and Logistical
Most patients with a rare disease already visit specialists on a regular basis. Participation in a clinical trial requires additional travel, time away from school and work and the need to arrange other family care while away from home. Depending on the trial site location, families may travel significant distances by car, train or air. Air travel can be particularly challenging due to the length and discomfort of the journey and difficulty accommodating wheelchair bound travellers.
Rare disease trial sponsors frequently employ organisations that specialise in the provision of patient travel support during trials. These organisations can coordinate travel to meet the specific requirements of each family taking into consideration work and school commitments for parents and children, respectively.
These organisations can also work with trial sites to ensure that specific
accommodations meet the needs of patients during site attendance. This may include the selection of residential style lodging with suitable patient and family accessibility located near the study site.
Study site accommodations should include a child-friendly environment for entertainment for the patient and additional space for siblings if they are attending. For those sites without sufficient space, these travel support services could arrange alternative facilities for waiting and caring for patient siblings.
Financial
Families incur additional costs and not all expenses are reimbursed, such as loss of a parent’s wages to attend study visits. Often parents are expected to pay up front for trial related expenses and submit receipts which can be inconvenient and burdensome.
Additional expenses can include care for siblings, pets and in some cases elders while primary carers are away from home.
To address some of these financial barriers, trial sponsors should incorporate anticipated patient costs into the clinical study budget allowing for stipends before costs are incurred. Support services can include provision of renewable gift cards and serve as a central point of contact to help manage patient expenses.
Psychological and Emotional
The emotional impact on children with rare diseases and their families is noteworthy. The requirements for participation in a clinical trial add to time away from home, school and work. Children often fall behind in school and the pressures to balance work, caregiving and the logistics of attending study visits, often far from home, are challenging. Siblings are commonly affected as all of this disrupts the family’s routine.
Sponsors can reduce some of these burdens by incorporating decentralised elements into their trial design resulting in a reduced number of on-site visits. Home health nurses can often conduct study procedures at the patient’s residence or school. Identifying which study activities
can take place remotely, such as follow up communication through telemedicine, can further reduce the inconvenience and disruption to participants and their families.
Patient advocacy groups can be a good source for education and connecting families with similar experiences. Provision of counselling services and support groups for siblings can also help support patients and their families.
Patient Insights
Previous research on paediatric rare disease patients and their caregivers has described some of the key motivating factors for participation in a clinical trial. In 2021, we conducted an online survey through email and social media outreach to North American Rare Disease patient communities. Respondents included (n=126) patients with 69 different rare diseases. Thirty-seven percent (37%) of survey respondents (n=46) reported they had taken part in at least one clinical trial; however, only 67% reported that they had received any participation support services, such as travel support or financial assistance. Of those who did receive participation support services, 71% reported that these services somewhat or definitely impacted their ability to participate in and complete the trial(s).
Satisfaction with the services provided was significant. Approximately half of the survey respondents indicated they were very satisfied with the services they received. Overall, 84% of survey respondents, including those who had and had not participated in a trial, stated that receiving participation support services would somewhat or definitely impact their ability or willingness to take part in a future trial. A key implication is that participation support services are highly relevant to reducing risks to the efficiency of rare disease clinical trials in areas such as enrolment and retention, and the resulting timeline delays.
Patient Journey
The following illustrates a representative patient journey and some of the challenges that had to be overcome to participate in a study. The family in this example had a daughter with a rare disease involving severe neurological complications.
38 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
The daughter experienced frequent status seizures that were difficult to control and required emergency treatment in a hospital. The solution employed by a travel coordinator was to customise the route from their home to the study site to ensure they remained close to a hospital along the way as well as to notify them of any route delays or problems during their trip to the site. This allowed for rapid access to appropriate care, if needed.
Despite this support, there were problems being reimbursed for travel expenses. Because only one parent could travel with the child, it was often not possible to obtain receipts. The reimbursement process required the parent to save receipts from the gas station. The parent could not leave the child alone and outdoor payment systems were unable to dispense receipts resulting in substantial out of pocket travel costs.
In addition, this parent, who was the primary day time caregiver for all children in the family, had experienced trouble getting care for her daughter’s siblings. As a result, the family had to make local arrangements for childcare and these expenses were not part of the reimbursement plan for study participants. These financial constraints may have deterred the participation of other patients and families. Earlier consideration of
Clinical and Medical Research
these significant obstacles and incorporation of these into the reimbursement plan would have eased this burden on this family.

Conclusion
Participation in paediatric rare disease trials can be burdensome for patients and their families as it requires significant commitment but is sometimes the only potential for a treatment. There are numerous physical, logistical and financial challenges, in addition to substantial psychological and emotion impact on child participants and their families.
Many of these barriers to trial access can be lessened by taking the perspective of the patients and their families to understand their journey at the outset. By incorporating their experience, support services to make it easier for them to participate in a clinical trial can be tailored to their needs. For this to be successful, it is critical that all stakeholders including product developers, researchers and advocacy groups are engaged in this process.
REFERENCES
1. Bell SA, Tudur Smith C. A comparison of clinical trials in rare versus non-rare diseases: an analysis of ClinicalTrials. gov. Orphanet J Rare Dis. 2014; 9: 170. doi: 10.1186/s13023-014-0170-0
Kirsten Sherman Cervati

Kirsten Sherman Cervati, BA, CCRPS is a Senior Director, Center for Pediatric Clinical Development and Head of ICON’s Pediatric Research Partners at ICON. She has 20+ years of experience in the industry, having held operations and leadership positions in academic, commercial research and contract research organisational settings. Prior to joining PRA in November 2017 to develop and lead ICON’s unique Pediatric Research Partners, Ms Sherman Cervati led global teams in the conduct of protocol and site feasibility and strategic site partnerships. She is a fierce advocate for children’s healthcare and rights in both her professional and personal life, as she is involved in the International Children’s Advisory Network (iCAN) as a youth chapter leader and serves as the parent representative for the American Academy of Pediatrics’ Section on the Advances of Therapeutics and Technology Executive Committee.
William Maier
William Maier, MD, PhD is Vice President, Rare Disease, Drug Development Sciences at ICON. Dr. Maier has over 25 years of experience in drug development and commercialisation at pharmaceutical companies in Europe, Canada, the United States and Asia. At ICON he works with pharmaceutical companies throughout the world to provide regulatory, strategic and scientific guidance on medical treatment development and commercialisation. He is a member of the EMEA’s European Network of Centres for Pharmacoepidemiology and Pharmacovigilance (www.encepp.eu). In addition, he is a frequent speaker at medical conferences, has had academic appointments in the UK (Dundee) and the USA (North Carolina) and is a member of the Royal Society of Medicine in the UK.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 39 www.international-pharma.com
In-flight Data Control: How to Approach the Next Frontier in Transforming Life Sciences Business Process Efficiency.
The trouble with non-standardised data, from an internal company perspective, is that it hampers agility and the ability to innovate. If each department uses slightly different terminology for a product and has its own system and way of logging information, the scope for coordinating associated insights, identifying opportunities, and accelerating processes will be compromised.
As agility becomes a focus for the European Medicines Agency, and pharma companies comprehend more fully the benefits of a continuous, reliable, harmonised data flow across their operations, the focus is turning to ‘in-flight’ data control. This is about ensuring that information captured and used in one function or part of an extended process can be matched and reconciled with related information elsewhere. The idea is to create a rich, combined and current narrative which all functions can access and add to, to support a range of use cases.
Here, Max Kelleher, Chief Operating Officer at Generis and Remco Munnik, a Director at Iperion, a Deloitte business, offer practical tips on how companies can systematically control and harness the flow of data between functional silos, and the potential benefits this could have.
At the recent DIA Europe event in Switzerland, conversations abounded about how to leverage live company master data more effectively and strategically across the Life Sciences R&D lifecycle. Rather than focusing solely on the information logged formally (and often in duplicate or worse) in separate departmental systems, this is about the flow of broader data and insights between functions, potentially serving a range of different use cases. Much of this ‘in-flight’ data is incidental information captured as part of a task, yet its value in providing oversight, traceability and impact assessment to senior management could be considerable – if only companies could
find a way to harness and control it more systematically.
Today, ‘point’ software solutions –regulatory systems (RIMS), clinical trial management (CTMS), pharmacovigilance (PV), or whatever – have become commodities, whose value is largely restricted to the immediate use case. And it is in the handover of the data between these departments that gaps and discrepancies in information between systems occur, leading to operational blind-spots and strategic oversights at best, or regulatory incompliance at worst. This makes hard work of change management, and could mean that product development information, and indeed patient safety events, aren’t fully traceable. Overcoming the silos, interconnecting the data, and keeping those connections dynamic and smart, is the next big opportunity – and provide the key to using everyday operational data to drive business improvements.
But how?
The answer lies in understanding where key data is generated, and how the supply and demand of that data looks across the ‘chain of custody’, as that data is re-used in different ways. Then a plan can be devised for improving the connection and flow of more unified data (one enhanced source, rather than inconsistent duplicates) across departmental divides.
For young biotech companies starting from scratch, there is a clear opportunity to establish clean, consistent and definitive data from the outset, whereas for larger and more established companies the best options may be around intelligently mapping existing data sources and data flow. Then interconnections and interdependencies can be identified and managed more effectively, until such time as data remediation and endto-end standardisation can be achieved (e.g. to bring data fields and formats into line).
It’s in this context that leveraging Ontologies is attracting interest as an option, for instance – allowing inconsistentlyformatted data to coexist, while recognising that the items referenced are the same, and linked. This is a useful first step in the move
to treat all data as one joined-up resource, so that it can drive new actionable insights, decisions and processes. A more thorough overhaul of data can then happen more gradually over time.
With all of this in mind, here are some considerations and tips for tackling internal data transformation.
Establishing Priority ‘Wins’ to Aim for
Unless the company is a young biotech with a largely greenfield tech set-up, Life Sciences companies will be approaching the road to data-based operational agility with a considerable amount of baggage.
Large legacy systems, vast volumes of data, and the variable quality and availability of that data, will make it hard to know where to start in transforming its contribution and value. Rather than try to tackle everything everywhere all at once, the prudent choice involves identifying some tangible gains from higher-quality, interconnected data which, once cleaned and combined, will tell a fuller story. That might be linking supply chain data to Regulatory data, to enable serialisation, (semi)automated batch release, and mitigation of shortage reporting, for instance. Or perhaps the aim is to shave a week off clinical development timescales, or complete eCTD applications or submit variations more speedily. All depending on the priority and size of the company.
Draw a Map
Mapping what data exists, and where, is the best place to start with all of this. It is only through visualising the current spread of information assets and associated use cases that companies will appreciate the potential for greater uniformity and fluidity of data use between the different departments.
This will help the company establish key processes to transform, for quick yet potentially far-reaching wins for the business. An effective map will chart where given data is used along a process including creation, modification, and re-use by different teams and systems.
Where there is an existing process optimisation or digital transformation team
40 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Technology
in place, or consultants that are advising on associated initiatives, these professionals would be the ideal drivers of a crossfunctional data map – in partnership with key functions such as Regulatory Affairs, Quality, and so on. Companies that have already appointed Chief Data Officers, or equivalents, will have a head start as these roles typically take more of a view of the commercial value of data, where Regulatory Affairs might not be the direct creator of the data, but more the guardian – as the spider in the web – providing a more detailed perspective of data’s links and touch points.
Make Ongoing Data Governance Everyone’s Responsibility
A lot has been said and written already about the importance of improving and maintaining high data quality, as its day-today value in supporting real-time business processes increases.
While some arguments favour a strong sense of data ownership within specific functions with the most involvement with the given data, it can be more powerful to encourage everyone across the company to buy into the value of consistent data so that all functions and teams play their own part in keeping data clean, compliant, comprehensive and current.
Effective strategies here involve strong, broad communication of the associated benefits of robust data, and incentives (recognition and reward) for those who actively play their part. Instead of data ‘ownership’, think in terms of a ‘chain of data custody’ spanning multiple groups of data processors and guardians over time.
Establish the Company’s Most Effective Path to Data Nirvana
Once companies can more readily visualise
their current data position and the full scale of the task ahead of them – to make their data work harder for the organisation -it’s time to decide the most prudent way forward.
In the case of large pharma companies with extensive product portfolios and vast system and data legacies, comprehensive data remapping and/or investing in master data management is likely to be an overwhelming undertaking that could take many years.
Ontologies, now being championed through the Pistoia Alliance Ontologies Mapping project (a non-profit initiative), offer a more practical middle ground. They allow companies to assess the diversity in their current data estate and arrive at a workaround wherever the legacy complexity is too great to tackle straight off.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 41 www.international-pharma.com Technology
Although the end goal is that all active data will be harmonised, and this should be the ambition where there are sufficient similarities in the formats used between functions, ontologies allow overly inconsistently recorded data to be linked reliably. The idea is that the same fundamental information, even if formatted differently, can be recognised and treated as the same across respective departmental systems.
Where there is a good level of similarity between respective data entries, the involved groups in the chain of data custody (e.g. Regulatory and Quality functional representatives) can negotiate a more standardised way of recording information, as they reach a mutual understanding for why this is important and how both parties might compromise for the greater good.
Harness External Standards for Internal Benefit
Through data standards (e.g., ISO IDMP), regulators including EMA and more recently the FDA, plus the World Health Organization (WHO) on a wider scale, are championing global identifiers for medicinal products and their active substances. Life Sciences companies that are inventing and developing these products and active substances would benefit greatly from adopting data standards consistently
from early development, and throughout their marketing authorisation/registration information and variations submissions.
But there is a broader opportunity for Life Sciences companies to build on those standards internally, to smooth the flow of consistently formatted data across their own processes, to steadily eliminate any overlap or discrepancies in the way information is recorded. The more that data can be recorded uniformly and shared reliably between functions along an extended process, the richer the overall picture and the more effectively and efficiently it can be harnessed with a view to greater productivity, process agility and innovation potential.
Finally, going back to the need to foster a culture of data quality across an organisation, there also needs to be a sense of shared purpose here – for example the role of improved process efficiency in staying agile, enabled by the ability to speak the same language internally about products right across the organisation. As long as individual departments all mean something different with their definitions, there is the potential for risk and delayed innovation.
Adopting an agile mindset isn’t just about prioritising data quality, of course, though. It is also about being prepared
to try something, fail at it, learn from that and move forward – in increments. So that is a further consideration for Life Sciences, which has traditionally been very conservative as an industry: how companies here can simultaneously trust data yet allow for mistakes to foster faster progress.


Networking with peers from other companies, or looking to other industries for inspiration, can help determine the most effective way to approach this.
Max Kelleher is Chief Operating Officer at Generis and formerly the company’s Head of European Operations. He is passionate about providing a viable, pragmatic path for modernising enterprise information management in regulated industries. His close work with both pharma companies and specialist solution partners has afforded him deep insight into the critical modern-day challenges that traditional approaches to business processes and information use in complex industries like Life Sciences do not fulfil.
Email: max.kelleher@generiscorp.com Web: www.generiscorp.com
Remco Munnik is a Director at Iperion, a Deloitte business, and a respected subject matter expert in RIM, eCTD, xEVMPD and ISO IDMP. He is Chair of Medicines for Europe Telematics group; and President of the IRISS Forum, a global, open, multidisciplinary, non-profit networking organisation for life science professionals by life science professionals. Iperion, a Deloitte business is a globally-operating life sciences consultancy firm which is paving the way to digital healthcare, by supporting standardisation and ensuring the right technology, systems and processes are in place to enable insightful business decision-making and innovation.
Email: remunnik@deloitte.es

42 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Technology
Max Kelleher
Remco Munnik
Media and Communications
IPI

Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.

www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.

www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.

www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.

www.international-biopharma.com

PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.

www.pharmanaturepositive.com
PHARMA’S DNA
Listen to industry experts on the latest in drug discovery, development, research, industry regulations and much more at Pharma,s DNA, the podcast channel by Senglobal Ltd., available on Sound Cloud, Spotify, iTunes and YouTube.
senglobalcoms.com
The Future of AI in Biotechnology On the Precipice of the New Technological Revolution
The artificial intelligence revolution kicked into high gear seemingly overnight. Generative AI platforms like ChatGPT and Google Bard have dominated headlines for months as we explore the possibilities for this powerful new technology, which has potentially significant implications for nearly every industry on the planet.

One of the most promising uses for artificial intelligence is its applications in biotechnology. Doctors and scientists are leveraging AI and machine learning to devise entirely new treatment solutions for diseases and chronic conditions affecting millions around the world. AI is even changing how we interact with our very DNA, not only helping scientists and researchers build safer, more effective drugs and vaccines, but better understanding how biology works and how species evolve over time.
Although it’s still early days, there are many indications that we’re on the precipice of a technology revolution unlike anything we’ve ever seen before. AI is allowing biotechnologists to augment the very building blocks of life in service of a healthier future for humanity. We are in uncharted territory, and how this shakes out will have world-changing ramifications.
Developing New Drugs and Vaccines
Biological systems are immensely complex and contain far more data than human beings could ever possibly keep track of. And so historically, drug development has been a protracted, expensive process primarily relying on trial and error. Artificial intelligence can rapidly process this data, make informed predictions about pathways and pathogen targets, and design new drugs to combat potential health threats.
This allows pharmaceutical companies to significantly reduce the time to market for biologically derived products. Given that biological systems contain so much data, the drug development process involves a great deal of experimentation and repetition. Artificial intelligence gives researchers the power to automate many of those tasks. We saw this firsthand with the development
of the COVID-19 mRNA vaccine, which was devised and released to the public faster than any other vaccine in history thanks in large part to AI and machine learning technology.
“We are able to control biology in ways we never have in 4 billion years,” said Michael Specter, author of Higher Animals: Vaccines, Synthetic Biology, and the Future of Life on a recent episode of Amanpour and Company.
“We’re able to make things, alter things. The COVID vaccine was basically assembled in a couple of days once it was downloaded from the internet. And those words ought to be profound – we downloaded the blueprints from the internet. When you can do that, you can do a lot of things. It means biology moves at the speed of light now.”
The breakneck speed of biotechnology’s evolution was also on full display with the recent release by DeepMind’s AlphaFold about the 3D structure of 200 million proteins. AlphaFold, a subsidiary of Google’s parent company Alphabet, had a longstanding grand challenge of accurately predicting the 3D structure of every known protein sequence. AlphaFold could provide a sharp view about
the 3D structure of malaria surface proteins, allowing researchers to test new vaccines targeting malaria. DeepMind’s release was a striking example of artificial intelligence’s ability to assist researchers in the process of discovering new drugs.
Developing new bioproducts can take years, and typically costs millions of dollars. Biotech leader TeselaGen serves as an operating system for biotechnology, leveraging AI and machine learning to reduce both cost and time to market. “We provide an end-to-end integration combining powerful software modules for designing, building and optimizing different biological products,” said Teselagen Founder & CEO Eduardo Abeliuk. “Our AI-enabled enterprise platform radically accelerates the development of everything from therapeutics, high-value chemicals, and agricultural products. “Teselagen’ highthroughput capabilities greatly shortens development times. The platform is not just a set of tools, but an Operating System for Biotechnology”, he added.
Transforming Genetics for a Healthier World Surprisingly, a big portion of the information we need to treat the world’s worst diseases
44 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Technology
is contained within DNA. It’s at the heart of scientists’ understanding of not only what causes disease, but how they can be prevented and cured. We now have the ability to manipulate DNA in ways that allow us to alter the basic genetic structure of both animals and humans, clearing the way to developing new and more effective therapies.
Malaria, for example, kills millions of people every year, particularly in the poorest regions of the globe. In recent years, researchers have figured out how to alter the DNA of mosquitoes so the females are unable to produce viable offspring. Such genetic modifications are an alternative to vaccine development and have the potential to wipe out the gene pool of malaria-carrying mosquitoes, which could result in millions of lives saved. Eradicating diseases like malaria that chiefly affect the world’s most vulnerable populations is a core focus of the Gates Foundation. In a recent blog post, Bill Gates cited AI as a technology that will “dramatically accelerate the rate of medical breakthroughs.” He says the upcoming generation of solutions “will be much more efficient, and they’ll be able to predict side effects and figure out dosing levels.”
Gene editing can also be used to help save the lives of endangered wildlife. The black-footed ferret is one of North America’s most endangered animals, due largely to their high susceptibility to the plague. Researchers are working on a way to embed the plague vaccine into an animal’s germ cells so its offspring will be immune to the disease. “Those progenies will basically be born with a heritable vaccine,” says Specter.
TeselaGen’s platform takes this ability to edit and assemble genes to the next level, while allowing researchers and enterprises to automate, manage, and track everything in the lab. Our design tools are built for industrial biotech and biopharma companies to digitally design modified biomaterials including DNA, oligos, strains, microbial materials, proteins, enzymes, or any other configurable, domain-specific biomaterial type. Our Build toolkit then lets users execute on the Design protocols and optimize their product with lightning speed.
We’ve entered an exciting and unprecedented new era for AI-enabled drug discovery and disease prevention. As the


















science advances further, expect artificial intelligence and biotechnology to be at the forefront of efforts to eradicate the world’s diseases and ensure a healthier future for us all.
Dr. Eduardo Abeliuk is a U.S. based entrepreneur and technologist with over twenty years of experience driving technology development and innovation at various high-tech companies in the US. He is the Chairman and CEO at TeselaGen Biotech., a Silicon Valley based company working at the interface between Biotechnology, Artificial Intelligence and Enterprise Software. Dr. Abeliuk holds an M.S. in Bioengineering and a Ph.D. in Electrical Engineering from Stanford University. He holds multiple U.S. patents on computational biology and AI.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 45 Pressure regulators Control valves Pipeline ancillaries Steam traps Special equipment STEAMING SOLUTIONS FOR ALL INDUSTRIES adca@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467, Guia PBL PORTUGAL PRODUCTS MANUFACTURED IN PORTUGAL AV 001 IN EN 01.20 Technology
Dr. Eduardo Abeliuk
Cutting-edge Formulation is Needed to Enable the Next Generation of Oral Biologics
An Introduction to Oral Solid Dose
Traditionally, the pharmaceutical market has been dominated by oral drug delivery – whether oral solid dose (OSD) or liquid suspensions and solutions.
Oral drugs ease access by enabling patients to take medication at home with little discomfort or complexity. This differs from other dosage forms, such as parenteral, which often rely on healthcare professionals for their delivery. Additional OSD advantages include extended shelf-life, which eases transport and long-term storage.
Despite OSD popularity, certain active pharmaceutical ingredients (APIs) are incompatible with traditional OSD formulation techniques, as is the case for biologics. This is due to a myriad of reasons that stem from biologic physicochemical properties, stability, absorption, and immunogenicity.
Many pharmaceutical industry research programmes are exploring new strategies and technologies to propel OSD biologic formulations. These novel approaches aim to overcome associated obstacles and include the use of prodrugs, nanoparticle-based delivery systems, and innovative methods to optimise absorption and stability.
In this article, Dr. Uwe Hanenberg, Head of Product Development in Oral Solid Dose at Recipharm, explores recent advances in formulation technologies for both OSD and oral liquid dosage forms designed to enable the creation of oral biologics.
The Driving Force for Solid Dose Biologics
By the end of 2022, biologic approvals had narrowly outpaced those of new smallmolecule entities for the first time, setting a new precedent.¹ The biologic market is predicted to continue experiencing rapid growth with a compound annual growth rate of 4.1% between 2023 and 2028, reaching an estimated value of $415 billion by 2026.²
This rise in biologic approvals could contribute to the burden on the healthcare
system, as biologics are often limited to parenteral administration, requiring healthcare professionals’ (HCPs) involvement. The way the biopharma industry can help reduce the HCPs’ load is by working towards self-administered therapeutics. Oral dosage forms seem an obvious alternative, but to make this delivery system viable, innovation is needed to improve biologic solubility, absorption, and stability. Patient centricity is also a priority for many pharmaceutical companies, and expanding the range of selfadministered drugs increases patient access.
Many pharmaceutical companies are investing in OSD formulation research to find innovative solutions to current problems hindering oral biologic development. Overcoming these challenges requires a strong understanding of the issues that may arise during OSD biologic formulation.
What are the main challenges associated with OSD biologics?
The many difficulties developers and manufacturers face when taking on the challenges of formulating a biologic OSD include:
• Instability: As biopharmaceuticals are highly sensitive materials, they often degrade in acidic environments, such as in the stomach, as well as under the stress and harsh conditions of the intestinal tract. Upon degradation, the biologic is no longer medicinally beneficial due to loss of activity.
• Physicochemical properties: The inherently complex structures and properties of biologics, such as large molecular weight, high hydrophilicity, and sensitivity, make them difficult to formulate into oral drugs.
• Absorption:
Larger molecules, such as biologic APIs, often struggle to be absorbed through the intestinal wall and into the bloodstream. This can further limit their bioavailability and effectiveness when taken orally.
• Immune response: Biopharmaceuticals may elicit an immune response in the body. The resulting formation of antibodies that specifically recognise and target the
biologic API can reduce the drug’s efficacy and increase the risk of adverse reactions.
• Regulatory requirements:
The regulatory requirements for the development and approval of oral biologics are stringent, and the formulation must meet strict standards for quality, safety, and efficacy.
How Excipients Can Help to Overcome Biologic OSD Challenges
There are several approaches to overcoming the challenges surrounding biologic OSDs, and extensive research concentrates on developing strategies and techniques to enhance product stability and bioavailability. Adding carefully selected excipients during product formulation – a common practice for small-molecule oral drugs – can beneficially alter the characteristics of the API to achieve the desired result, including bestowing attributes such as extended shelflife or improved solubility. Biologic OSDs can utilise the same approach.
Strategies to achieve this include using coatings or capsules to protect biologics from harsh stomach and gastrointestinal tract conditions. These coverings act as a shield, protecting the product from degradation. They also allow the controlled release of the drug, increasing efficacy and safety for the patient by programming a predetermined release rate over a specified time period.³
Alternatively, using nanoparticle-based oral delivery, the biologic drug can be encapsulated within a nanoparticle, which also controls drug product release. The outer surface of the nanoparticle can be designed to target receptors within the intestinal barrier. This increases the bioavailability of the drug, helping it to be transported across the membrane.³
Although research is still ongoing, the following excipients have been explored for their impact on biologic properties:
• Permeation enhancers and absorption promoters to increase bioavailability. Examples of such excipients include
46 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Manufacturing
sodium caprate, chitosan, and bile salts.
• Surfactants, co-solvents, and complexing agents that can be used to improve solubility and enhance absorption. Examples include polysorbate 80, cyclodextrins, and polyethylene glycol (PEG).
• Stabilisers and antioxidants that can be added to the formulation to protect the drug from degradation. Examples of such excipients include trehalose, mannitol, and vitamin E.
• Immunomodulators and adjuvants that can be added to the formulation to reduce the immune response. Examples include aluminium hydroxide, MF59, and monophosphoryl lipid A.
• Mucoadhesive patches that can be placed in capsules to bind the intestinal mucus, prolonging residence time and localising biologics near the mucosal interface to enable unidirectional diffusion.
• pH-sensitive hydrogel patches for controlled release.
Formulated to form the final drug product, the excipient and API mixture must adhere to strict regulatory requirements, and the excipients used are carefully chosen to comply with regulations. For example, bulking agents, fillers, and binders, such as microcrystalline, cellulose, lactose, and magnesium stearate, can be added to the formulation to improve manufacturing and meet regulatory requirements.
A Strategy for Determining Excipients
Researching excipients to improve biologic OSD formulations is still in its early stages. Upon excipient addition, an in-depth analysis of product activity, stability, and bioavailability is essential to monitor the impact on the biologic formulation. A number of steps should be followed to ensure the appropriate excipient is selected for the required characteristic:
1. Define the objective of the study: Outline the desired characteristics that must be achieved with the chosen excipient, highlighting potential options.
2. Select the appropriate excipient: Quantify the effect of potential excipients on the stability, solubility, bioavailability, or pharmacokinetics of the biologic. Select an excipient that meets the intended application without altering biologic structure or function.
3. Conduct a pre-formulation study: Determine the physicochemical properties of
the biologic and excipient. This includes particle size, morphology, surface area, and surface charge.
4. Formulate the biologic with the excipient: Use the appropriate techniques for product formulation, such as spray drying, freeze-drying, or hot melt extrusion.
5. Conduct in vitro testing: Analyse the impact of the excipient on the solubility, stability, and release of the biologic. This includes dissolution testing, stability testing, and particle size analysis.
6. Conduct in vivo testing: Determine the impact of the excipient on the pharmacokinetics and bioavailability of the biologic. This includes animal studies and clinical trials.
7. Draw a conclusion: Analyse and interpret data from in vitro and in vivo studies to determine the excipient’s impact on the biologic. Compare it to a control group for statistical significance and, based on the results, draw a conclusion on what impact the excipient use has on the biologic.
Following the steps above is critical to ensure product efficacy and safety. However, extensive testing can be time consuming and lead to delays in bringing a desired biologic to market. To help ease this process, forming a communicative partnership with a contract development and manufacturing organisation (CDMO) with experience and expertise in biologic OSD formulation can be invaluable.
How can a supportive partner help overcome these challenges?
Access to an experienced partner can ease biologic OSD formulation, helping to overcome the challenges of bringing a product to market successfully.
CDMOs can irrevocably aid and accelerate drug production by providing expert sector knowledge, regulatory requirement insights, and scientific expertise. Forming a free-flowing partnership with a CDMO can ease OSD formulation, with access to highend equipment, the latest technologies, continuously harnessing innovation to formulate a desired biologic OSD target.
Looking to the Future
Orally-taken biologic drugs offer a patientcentric approach to treating diseases. As our understanding of overcoming challenges associated with oral biologics grows, we can work to harness coatings, capsules, and
excipients to drive oral biologic formulation from lab to market. Aided by a supportive CDMO partnership, innovative biologic OSD strategies can be used to accelerate development.

Biologic OSD development is expected to significantly impact the biopharma space in the coming years. As these new therapies become standard practice, we can anticipate increased patient compliance, improved biologic stability and efficacy, and enhanced drug solubility, leading to increased bioavailability and improved therapeutic outcomes. The market will likely also experience a shift as drugs only previously available via parenteral administration will now be available in OSD.
Biologic OSDs are now at the forefront of biologic development, helping manufacturers increase patient access by bringing vital medicines directly to patients.
REFERENCES
1. Senior, M. Fresh from the biotech pipeline: fewer approvals, but biologics gain share. Nat Biotechnol 41, 174–182 (2023). https:// doi.org/10.1038/s41587-022-01630-6
2. Biologics Market Size, Share, Price, Demand, Growth, Forecast 2023-2028 (expertmarketresearch.com)
3. Anselmo, A., Gokarn, Y. & Mitragotri, S. Noninvasive delivery strategies for biologics. Nat Rev Drug Discov 18, 19–40 (2019). https://doi.org/10.1038/nrd.2018.183
Dr. Uwe Hanenberg, Head of Product Development, Oral Solid Dose (OSD) at Recipharm. Uwe is responsible for implementing and executing the OSD Product Development strategy that assures science driven, timely development of new products or services. Uwe has 25 years of experience in the pharmaceutical industry with Bayer, Altana, Grünenthal and Catalent. He has held several leading positions in quality, manufacturing and packaging, product development, project management within the Science & Technology sphere. His areas of expertise are oral formulation development, oral manufacturing technologies, stick pack technologies and pharmaceutical contract services and project management.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 47 www.international-pharma.com Manufacturing
Uwe
Hanenberg
Individual Device Identification: Coding and Marking for the New Dimension in Patient Health and Safety
Counterfeit products and product piracy are serious issues for the pharmaceutical industry – but recent product marking requirements in developed economies, including the US and the European Union, have gone some way towards securing legitimate pharmaceutical supply chains. The WHO estimates that the share of falsified medicine in global marketplaces ranges from 10% in certain low- and middle-income economies to as little as 1% in developed countries.1
While there is still much to be done to increase the scope and reach of current legislation and improve safe access to pharmaceuticals in less developed economies, regions that have already implemented compulsory serialisation of pharmaceuticals should also prepare for change.
In economies governed by regulatory serialisation requirements, hospitals, pharmacies, and healthcare providers are realising the benefit of data in helping to protect patient safety. These key stakeholders are now placing pressure on pharmaceutical manufacturers to provide more granular information on pharmaceutical products to protect patients from the risk of unsafe medication practices and medication errors.
Medication errors are one of the leading causes of avoidable harm in healthcare worldwide. On a global level, it is estimated that the total annual cost for medication errors is USD 42 billion; some 5% of all patients admitted to hospital experience a medication error; and an average hospital will have one medication error every 23 hours or every 20 admissions.2
Stricter serialisation practices – requiring identification down to the individual dose of a pharmaceutical product – could help to mitigate unsafe medication practices and medication errors.
Tackling Medication Errors
Errors can occur at different stages of the medication use process – from preparation to patient monitoring – and may be due to
inefficient in-house systems or human factors such as fatigue, poor environmental conditions, or staff shortages. A simple mistake can have potentially devastating effects, resulting in severe patient harm, disability, and even death.
Stricter serialisation measures – with individual identification down to the specific dose – can play a crucial role in helping to overcome the risk of medication errors in pharmacies and healthcare providers and ensure that the right patient gets the right dose of the right medication at the right time.
Adding more granular data to strips and blister packs of medicines, for example, can allow for better control within hospitals, care homes, and other healthcare facilities to improve medicine distribution. Scanning serialised 2D codes can allow for automatic electronic validation of medicines to ensure that patients receive the correct medicine and dose. This level of detail decreases dispensing errors and can improve inventory management and stock control.
Individual Dose Identification
Today, products are typically produced with a 2D code that contains product, dose, batch code, and expiry date information on secondary boxes and packs. The next level will require solutions for coding each product dose – which could include vials, ampules, and individual blister pockets containing a tablet.
While the benefits of individual dose serialisation are obvious, the industry needs the buy-in of pharmacies, doctors, hospitals, and other healthcare providers. Existing initiatives involve scanning individual items to demonstrate the value of individual dose identification and drive change. Healthcare providers and patients see the benefits of this next step and coding each medicine dose.
To get involved in such initiatives and prepare for a future where individual dose identification is the norm, pharmaceutical manufacturers may need to update their current coding and marking capabilities.
Product Handling Requirements
Manufacturers must print serialised 2D codes correctly to ensure that they can be effectively scanned – this is particularly crucial when
using 2D codes for regulatory purposes, such as those used in pharmaceutical applications.
Product handling or the ‘presentation of the product’ to the coding device is fundamental to achieving high-quality codes – this will become more crucial for manufacturers looking to explore individual dose identification. Manufacturers that attempt to code products in-line without effective product handling will be subject to production line variations that can affect final code quality, including:
• Product position: Small variations in the position of products on a production line may result in codes applied in the wrong area or missing or incomplete codes.
• Product distance from the printer: Positioning too close or too far from the coding device can result in blurry or unreadable codes.
• Product angle: A slight rotation in product positioning, even by just a few degrees, can result in deformed codes.
• Line speed: Minimal speed fluctuations will affect the quality of the code, leading to squeezed or stretched codes.
• Conveyor vibrations: At high speeds, vibrations can affect code quality leading to low-quality, blurred, or wavy codes.
• Challenging product geometry: Certain packaging types can be challenging for a standard coding set-up.
At best, a poor quality 2D code resulting from inadequate product handling will cause rejections, rework, and defective stock. The repercussions will be even more severe if an unreadable 2D code leaves the factory unnoticed. Pharmaceutical brands may face financial penalties such as fines, loss of business, product recalls, and potential legal implications.
A bespoke product handling solution can solve all issues concerning code quality by ensuring optimal and consistent product delivery to the coding equipment. The optimal solution will be designed based on several different considerations, including:
• Product and packaging type: Factors such as the shape of a piece of packaging, substrate type, and weight of a filled pack can be crucial. In pharmaceuticals,
48 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Manufacturing
for example, boxes of blister packs are light and have regular forms, but glass or plastic medicine bottles will be more challenging to code.
• Code requirements: Code type, placement, and resolution are determining factors. Many machine-readable codes have minimum size requirements and necessitate the use of high-resolution printing technologies.
• Existing production specifications: If a coding solution is integrated into an existing production line, it must be designed with existing specifications in mind so as not to slow down production or reduce overall equipment effectiveness (OEE).
Depending on the product type, a bespoke solution may use multiple types of technology to handle finished products and present them to the chosen coding device for final printing. These devices can use different methods to handle products with varying levels of force for delicate and robust products alike. A bespoke handler could be developed with pneumatic or servo-electric driven side, top, or bottom belts, or even magnetic or vacuum solutions.
In-line Digital Printing for Blister Packs
Pharmaceutical manufacturers managing products packed in blister packs could find themselves well-placed to update their current coding and marking equipment with an in-line digital printing solution specifically for blister pack foils.

In-line digital coding allows manufacturers to migrate from the traditional embossed batch and date coding to full individual serialised data at the individual blister pocket level. This not only gives improved visibility of the codes but can also allow for full product printing inhouse, using plain foils. Indeed, when investing in an in-line digital printing solution, the price difference between a narrow printhead that is only suitable for printing variable information and a wider printhead capable of managing larger graphics, including artwork, is often negligible compared with the possible savings.
This new approach can allow manufacturers to create short-run variations of products and
afford significant time savings, by removing the need to purchase pre-printed foils. To the same end, wide-web digital printing can also enable considerable waste reduction – as manufacturers typically order 10–20% extra when purchasing pre-printed foil to allow for unforeseen issues or wastage from set-up and product changeovers. In practice, much of this excess will often go to waste; this is not an issue with plain foils, as any excess can be reused for subsequent print runs.
Data Requirements
Though the introduction of individual dose identification significantly enhances traceability and can aid in error reduction, there are challenges in the amount of data that now needs to be generated, tracked, and managed for both the manufacturer and at all stages along the supply chain,
All modern printing technology will ‘print’ the images delivered to it and can cope with thousands, millions, and billions of data changes; however, this needs to be handled, stored, and managed to ensure traceability remains intact.
For example, imagine a blister pack containing seven tablets and a production run of one million individual packs. Typically, the manufacturer must create, store, and print one million codes to comply with the serialisation requirements of one code per pack. When tracking down to the individual tablet, this increases the data requirement to seven million codes. It is likely that this data already exists, but it doesn’t need to be stored until the point at which it is printed. In this instance, serialised coding at the tablet level increases the data storage requirements by six million pieces of data.
Therefore, the step to individual dose identification comes with a burden of data volume and storage requirements. However, the benefits of full traceability provide significant potential for stock control accuracy. Manufacturers can trace where every item or tablet has moved, know the expiry date, and, most importantly, ensure dispensing accuracy.

Globally, the legislative requirements of serialisation are well known, and manufacturers are the key to implementing a solution. Unlike in the past, the cost is no longer a barrier, with more advanced labelling and serialisation systems available at lower price points. Ultimately with patient safety a key driver, demand for this data and pressure to provide it can only increase.
Upgrade Today
When it comes to individual dose identification, it is not a matter of if manufacturers will have to comply; the key question is when.
Demand for greater levels of traceability and accountability in the pharmaceutical industry is only set to increase. Manufacturers will be well placed to ensure that their coding and marking capabilities are equipped to enable accurate and reliable identification of each dose.
By doing so, manufacturers can enhance patient safety, reduce the risk of counterfeiting, and ensure compliance with the latest regulatory standards. By ensuring that their lines are equipped with the latest coding and marking technologies, including accurate product handling, or in-line digital printing solutions, manufacturers can ensure they are prepared for any future serialisation requirements.

REFERENCES
1. Substandard and falsified medical products https://www.who.int/news-room/fact-sheets/ detail/substandard-and-falsified-medicalproducts, visited 17th March 2023.
2. Medication Without Harm https://www.who.int/ initiatives/medication-without-harm, visited 17th March 2023.
Domino Printing Sciences’ Strategic Manager for Digital Coding, Ian Chapman, provides technical and development support in digital coding for life sciences. Ian has an extensive background in product development of digital printing control systems, and joined Domino in 2007. He has a degree in information and business systems technology from the University of Essex.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 49 www.international-pharma.com Manufacturing
Ian Chapman
Potent Product Manufacturing in a Multi-product Facility
Highly potent drugs are becoming increasingly common in the drug development pipeline. The focus on oncology, rare diseases, and targeted therapies is growing even more acute. While highly potent compounds have benefits in treating many medical conditions, companies with promising highly potent active pharmaceutical ingredients (HPAPIs) can face significant challenges when it comes to using them in development and manufacture. Key challenges in bringing them to market include ensuring that workers and the environment are protected from exposure, and, in a multi-product facility, delivering adequate, compliant controls to mitigate the risk of crosscontamination.
Historically, most of these potent compounds have demanded dedicated facilities, and separate dedicated equipment to manufacture them safely. But, with more of these compounds entering the market, the economics that supported this strategy are not always financially viable, especially for developers of novel compounds for smaller patient groups. Fortunately, pharma’s innovators have access to the flexible costefficient multi-use capacity they need from the contract development and manufacturing organisation (CDMO) industry. However, introducing and transferring a HPAPI compound to a CDMO partner’s multi-use facility presents unique challenges that need to be overcome in order to sustain worker safety and prevent cross-contamination.
Highly Potent Products require a Rigorous Containment Strategy
An important initial step when developing and manufacturing any HPAPI drug substance and drug product is to properly assess and categorise these complex compounds, and then to develop an appropriate containment strategy to manufacture them safely. Unlike in a dedicated facility where the containment strategy is set for a single compound, in a multi-use facility each new program demands this initial and critical risk-based assessment before work can commence.
Although the characteristics that define what constitutes a HPAPI compound are commonly shared by manufacturers, the systems for rating their potency to assess risk are varied. The rating system used - and how it is applied – will also differ from site to site, and between host manufacturers and their CDMO partners. And this is where conflict can occur. Some negotiation is required to arrive at the correct classification of the HPAPI material in question. Most pharma manufacturers use either a five-, or six-band system with the containment numbers between company A and company B differing slightly.
The HPAPI landscape is diverse and the active ingredients a CDMO might encounter can range from a novel or research compound to a new chemical entity (NCE) out of R&D. These potent compounds possess an API with a therapeutic daily dose of less than 10 milligrams and an occupational exposure limit (OEL) that is less than or equal to 10 micrograms per cubic meter.
Uncharted Development Territory Ahead
It is crucial for a manufacturer to establish the potency and other properties of active compounds, to develop plans and take necessary precautions to address any potential exposure risks. It is critical to environmental, health and safety (EHS) because these compounds often have the potential to cause serious illnesses such as cancers, developmental defects, and reproductive issues. One thing to keep in mind is that any biological effect from exposure to a highly potent active in any “healthy worker” population is considered an adverse effect.
For certain compounds, toxicologists and industrial hygienists have open access to data from the Safety Data Sheet (SDS) or the sponsor’s brochure. These are data-rich compounds because there is plenty of data available to assign EHS risk to the compound. However, when no data exists, its corollary, a data-poor compound, is often sent to an external toxicology firm with the expertise and analytical means to band data-poor compounds accurately.
Under these circumstances, data-poor actives should be classified in a much higher band in the rating scheme until the necessary
additional information becomes available to move it up or down. It is essential to the efficiency of the program for drug sponsors and intellectual property owners to provide all relevant data to the product’s external manufacturing partner. It is a key aspect of collaboration, and necessary to uncover as much relevant data as possible and drive proper classification at each phase. With datapoor compounds, classification requires a riskbased approach and extra due diligence to assess properly. There are no cutting corners at this stage.
One way to judge the quality and capabilities of a potential HPAPI manufacturer is how it treats the intake of these compounds as part of an overall cGMP frame of operations. In a contemporary CDMO setting, a proper New Product Introduction (NPI) strategy considers several factors:
HPAPI Categorisation and Risk Classification
To assure worker safety and product quality, the process for classification of HPAPIs must be robust, systematic and capable of being validated. Depending on the supplier, the EHS risk assessment banding process and who conducts it may vary. Regardless, the process needs to be implemented within the context of the organization, its teams, and the integrity of the process.
When comparing different suppliers, it is crucial to consider the discrepancies and variations in their rating systems, as well as any conflicting information. For the most part, any variations must be resolved to address issues that may arise. This is typically accomplished during the pre-bid stage of the proposed program. Ranking the compound accurately is probably the most critical aspect of introducing a HPAPI into a multi-product facility.
Much of a project’s success hinges on the classification because it has such a tremendous impact on the programme's schedules, timelines, costs, and more. Generally, the more data-poor a compound is, the higher risk or band it receives, unless and until data is obtained that suggests downgrading its risk to a lower band. Banding dictates the levels and types of containment needed. The higher the band, or level or classification, the stricter
50 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Manufacturing
handling guidance and containment protocols become.
Containment and Process Flow
It is important that process flow and containment strategy are harmonised to satisfy EHS requirements by eliminating cross-contamination and exposure for oral solid dose drug substance and drug product. More specifically, it is important to understand how a potential supplier approaches the process flow and containment strategy for a given HPAPI compound. Best practice calls for a riskbased approach focusing on data and the physical aspects of the plant to define the process and containment the compound calls for.
The containment strategy must consider manufacturing scale and process flow. Equipment and scale can vary wildly with HPAPIs but, in general, as the scale of the process increases so does the difficulty in containing it. Containment can be achieved through a broad range of technologies and protocols, but a proper risk assessment will determine its scope best and suggest the most cost-efficient and effective means to achieve the measures the assessment identifies.
When thinking about scaling up production, combining larger batch sizes with multiple products can be difficult. It's important to remember to remain adaptable and not to lose sight of the importance of flexibility. In a multi-product facility, it is essential to set up processes in such a way that safety is maximised, and cross-contamination is minimised.
Focused Cross-contamination Risk Mitigation

Within a multi-product facility, regulatory guidance calls for appropriate controls to protect staff, and a risk-based management approach to control cross-contamination risks and protect other products. Experience has shown that with the right combination of controls, a CDMO can support the simultaneous manufacture of cytotoxic prostaglandins, non-reproductive hormones, and other highly sensitising or genotoxic or teratogenic materials in one multi-product facility.
How potential CDMOs approach controlling cross-contamination will vary, but cGMP guidance calls for using riskbased approaches to assess and regulate it effectively. One good source of guidance comes from the International Society of Pharmaceutical Engineering (ISPE). The ISPE’s
“Risk Based Manufacture of Pharmaceutical

Products” guidance provides a method to map risk and describes a process that allows manufacturers to assess risk accurately and determine which strategies will control exposure limits and cross-contamination.
With attention to the abovementioned factors, EHS safety is assured. However, containment performance must be proven. One compliant way is to conduct industrial hygiene air monitoring with a validatable system that provides data to prove that the containment strategy and systems approach you apply is, in fact, hitting the performance targets that have been set. For example, if your containment performance target was one microgram per cubic meter, regulators would want all of the air samples to be below that level as determined by the containment performance target.
Creating an Efficient Space for Potent Compound Development
From assessing potency, assigning risk, and configuring containment and processes, there are a lot of moving parts necessary to create a safe and efficient space for HPAPI manufacturing in a multi-product facility. A programs commercial viability can hinge on a given compound’s potency. Too potent, and it may be too expensive to contain and manufacture safely, something that would likely put an abrupt halt to commercial development. Fortunately, from both an operational and technical perspective, pharma and its manufacturers have the tools and techniques to remove that risk for most HPAPI drug development.
Anshul Gupte, Ph.D., RAC Drugs joined Catalent in 2022 and currently serves as Senior Director, Scientific and Technical Affairs. He has over 15 years of experience in product and analytical drug product development. He has contributed to several branded and generic regulatory submissions for US and worldwide markets. He has experience in working on drug product concept to commercialization from both a CDMO and sponsor perspective for a variety of solid oral and topical dosage form delivery systems.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 51 www.international-pharma.com Manufacturing
Anshul Gupte
Effective Operations for Risk Reduction and Supply Chain Resilience
There is no denying that the market for injectable drug delivery devices is growing rapidly, with forecasts predicting an increase from $39.93 billion in 2022 to $43.54 billion in 2023 at a compound annual growth rate (CAGR) of 9.0%.1 With 60% of drugs in the R&D pipeline designed for injectable delivery,2 there are no signs of this trend slowing in the near future.
However, bringing a new drug delivery device to market is a long and complex process – from defining user requirements and early design concepts, going through building and testing prototypes, human factors testing and finally device verification and validation. To produce a device which can be successfully used for drug delivery by different patient groups, each stage of the industrialisation process must be carefully considered.
In addition, manufacturers must also be able to scale up production of the device in large quantities, while ensuring that quality and performance are consistently and rigorously maintained.
Against this background, this article will explain how to develop an effective operations plan to secure supply resilience alongside product development projects and programmes, to predict and eliminate potential supply chain vulnerabilities.
Planning Ahead: Starting From Lifecycle Management & Design for Manufacture

The importance of lifecycle management has long been acknowledged in the delivery device industry. Strategic planning is key in this fast-paced market, as demand for medical devices typically changes throughout the product’s lifecycle. Manufacturers must be able to adapt rapidly with carefully planned and effectively executed operations strategies that prevent any disruptions to product supply.
Lifecycle planning begins at the design phase, and risk management should be factored into decisions on manufacturing and assembly processes. New products should
be designed for both low and high-volume manufacturing, ranging from single cavity moulding and low volume manual to semiautomated assembly for smaller scale, low volume opportunities and high cavitation moulding with fully-automated production for large scale manufacturing.
Effective lifecycle management also means utilising the manufacturing network effectively, assessing the equipment and resources available at each production facility, as well as related cost implications, to ensure that the most suitable site is used at each stage of the product lifecycle. The ability to dual source – and consequent reduction in time required to execute alternate sourcing strategies – is another option for reducing supply chain risk. This contributes to establishing supply at the right time and in the right place, in the appropriate volumes for the locations in question.
Safeguarding Assets
Optimising asset management is a key component in ensuring business continuity. It is vital for supporting capacity growth while ensuring the maintenance, refurbishment, and replacement of key assets. Asset management strategies must take into account the planned usage of critical equipment and changes to the product lifecycle, particularly in product launch and growth phases, and at end of life, in order to cope with changes in demand.
In addition, asset management programmes must provide mitigation strategies for potential external supply incidents, including geopolitical events that may impact supply chains. The COVID19 pandemic and the war in Ukraine are two recent examples of world events that highlighted risk areas across many industries, stressing the importance of optimised strategies for ensuring continuity of supply.
Strengthening Collaboration with Your Suppliers
Managing suppliers effectively requires clear roles and responsibilities, governance, and escalation procedures to be laid out in advance. This is essential for ensuring product quality and consistent supply.
Supplier audits allow manufacturers to ensure that appropriate systems, equipment, methods, and skill sets are in place so that the agreed level of quality is maintained. They can also reveal any potential issues with key suppliers’ performance, capability, or strategies, allowing for corrective measures to be taken. A collaborative approach is key to successful supplier relationships, particularly where guidance from manufacturers can help to align methodologies between companies and reduce the risk of differences between components, measurements, processes or settings where direct comparison is necessary.
52 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Packaging
New requirements regarding sustainability reporting may have positive knockon effects for supply chain resilience, as regular sustainability audits and a more holistic approach may reveal areas of risk and result in improvements to logistics and processes.
At Owen Mumford, we have developed a ‘virtual factory management’ concept to facilitate ongoing supplier management. This was prompted by recent world events, namely Brexit and the COVID19 pandemic, which led us to review how we monitor supply chain vulnerability. The virtual factory management approach entails setting, sharing and reviewing KPIs to detect and correct potential vulnerabilities.
Grasping Opportunities from Continuous Tracking and Improvement

Once lifecycle, asset and supplier management strategies have been established, and in order to reduce risks as far as possible, manufacturers can make use of various tools including Process Failure Mode Effects Analysis (PFMEAs). These help to track and mitigate residual manufacturing risks, changes and potential impacts. Teams can proactively maintain and update risk registers to assess the likelihood of risks impacting project delivery timescales and take any necessary corrective actions rapidly, to manage risk appropriately, and to ensure supply continuity.

Another way to improve efficiency and performance, thus protecting organisations from risk, is the creation of Centres of Excellence. These hubs for key engineering skills and expertise allow teams to focus on specific areas of operations such as moulding, assembly, automation, or another key function. By clearly defining the core skills and tools needed for each individual Centre of Excellence, it is possible to create tailored training programs that support staff development. In addition to benefits for quality and efficiency, strong training programs also contribute to increased staff retention, which in turn reduces the risks that come with high staff turnover. Operations teams are also able to develop and implement best practices in these hubs, building on the range of skills and expertise offered by people from across the organisation.
At Owen Mumford, we have been building our own Centre of Excellence in Witney, Oxfordshire, England. When completed, this bespoke production site will become a Centre of Excellence for automation and
assembly, setting a benchmark for worldclass design and development in medical devices. The new building is set to be included in the top 25 percent of buildings assessed through the Building Research Establishment Environmental Assessment Method (BREEAM), a leading sustainability assessment for the building environment. This certification recognises the highest levels of environmental, social and economic sustainability performance, in line with the company’s strong sustainability agenda.
A Proactive Approach to Operations
The common thread linking all of these strategies is the need for a proactive approach to optimising operations during device design, development and manufacturing, that in turn ensures risk reduction and supply chain resilience. To take full advantage of technology investments, product developments and partnerships, Operations need to support product launches and growth in parallel to the phased design approvals from Research & Development. This approach helps to avoid risks in supply during the early launch phases and subsequent product growth, allowing manufacturers to ensure supply of existing products without limiting their capacity to innovate.
REFERENCES
1. The Business Research Company. (2023). Injectable Drug Delivery Devices Global Market Report 2023 – By Type (Conventional Injectable, Pre-Filled Syringes, Auto-Injectors, Pen-
Injectors), By Application (Autoimmune Diseases, Hormonal Disorders, Oncology, Orphan Diseases, Pain Management, Respiratory Therapy, Other Applications), By End User (Hospitals And Clinics, Home Healthcare Settings, Pharmaceutical And Biotechnological Companies, Research Laboratories, Other End Users) – Market Size, Trends, And Market Forecast 2023-2032. https:// www.thebusinessresearchcompany.com/report/ injectable-drug-delivery-devices-globalmarket-report
2. Statista. (2022). Percentage of drugs in R&D pipeline worldwide by delivery route as of 2022. https://www.statista.com/statistics/791731/shareof-pipeline-drugs-worldwide-by-delivery-route/
John Swift is Head of Supply Chain at Owen Mumford Ltd. He is an experienced operations program manager with a successful track record working throughout the supply chain, covering procurement, supplier management, invention, development and manufacture, as well as promotion, sales and distribution. He is experienced in applying and adapting skills across both large corporations corporations – such as Abbott, Abbvie and Tyco – and SMEs, and has worked in multiple industries, including medical device, aerospace and defence, rail, chemical, automotive, and printing.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 53 www.international-pharma.com
John Swift
Packaging
Choosing and Developing User-friendly Osmotic Laxatives for a More Patient-centric Portfolio

At least 1 in 10 people worldwide suffer from constipation at some point in their lives.1,2 It affects people of all ages and has many causes. The symptoms of constipation include pain in the lower abdomen and irregular and painful bowel movements. Laxatives are often needed in addition to dietary changes to treat constipation. There are many laxatives to choose from, each with different mechanisms of action and, consequently, different advantages and disadvantages. Here we look at the important role of osmotic laxatives, how they overcome many of the side effects and drawbacks of other constipation treatments, and the manufacturing expertise needed to make user-friendly laxatives part of a patient-centric product portfolio.
Constipation:
Prevalence, Causes and Treatment
Constipation affects many of us, but it is more common in older people, women and children.2 The prevalence of constipation
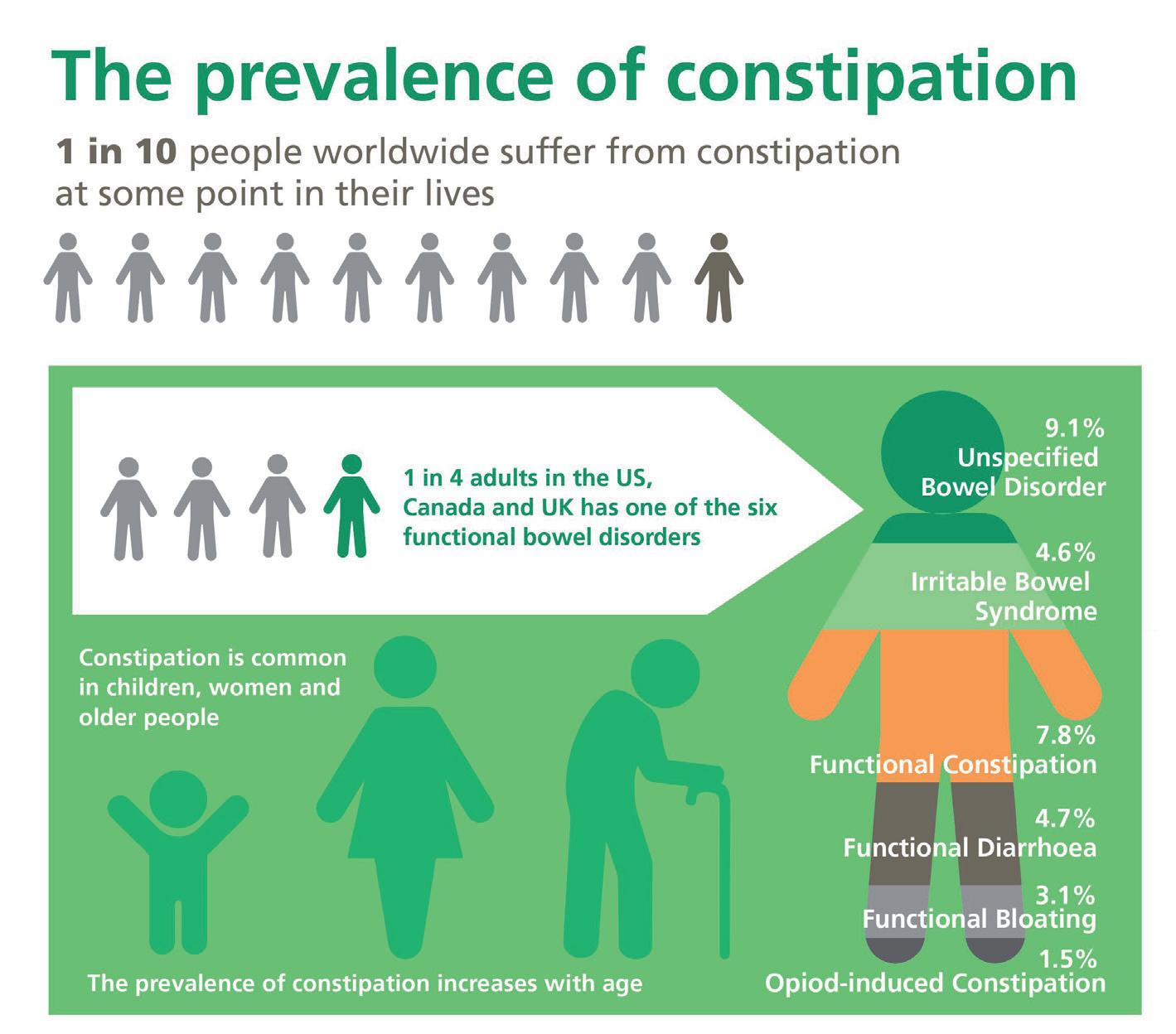
increases with age because of factors such as medicine use, underlying disease, change in drinking and diet habits, weakened pelvic floor muscles and long-term hospitalisation or institutionalisation. Women are twice as likely to be affected by constipation as men, especially during pregnancy where hormones influence bowel muscle movement.2,3
For most people, constipation is caused by not drinking enough water or not eating enough fiber, but it is also a common side effect of taking certain medicines (e.g., opioids and diuretics), and can be associated with conditions such as anxiety or depression and gastrointestinal disease.
Some people experience acute constipation, which lasts for only a few hours and often results from lifestyle changes, such as travel or stress. For others, constipation becomes a chronic condition (obstipation) where symptoms persist for more than three months and have a significant impact on quality of life. They may not be able to pass stools at all, or they may pass stools that are hard, cause pain and lead to secondary
issues such as anal tears (fissures) or even fainting or heart attacks in the elderly.2
Treatment for adults usually involves increasing water and fiber intake as well as upping exercise levels, and laxatives may be offered if symptoms don’t improve. In children, laxatives are usually offered as first-line standard care because constipation can worsen quickly.
Types of Laxatives
There are many different laxatives available, each varying in their mechanisms of action, ease of administration and the side effects they can cause:
54 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Health
Outcomes
Illustration 1: The prevalence of constipation
Type of laxative Mechanism of action
Contact stimulant laxatives Increase fluid secretion into the bowel and prevent resorption of water
Stimulate nerves in the bowel muscles, which increases bowel movement (peristalsis)
Softeners and emollients Lubricate the intestine wall and soften stools to make them easier to pass
Examples
Bisacodyl (available as oral tablets and as suppositories)
Sodium picosulfate (given as oral tablets often before a colonoscopy or bowel surgery)
Anthraquinones found in plants such as Senna
Paraffin
Glycerol
Sorbitol
Considerations/restrictions on use
Side effects include irritation of the bowel lining and gastrointestinal spasms
Can cause heart dysfunction
May not be suitable for diabetics as they reduce the effectiveness of insulin
Can lead to steatorrhea (fatty stools) and diarrhea
Can reduce absorption of fat-soluble vitamins, such as vitamin K
Some (e.g., glycerol) need to be administered as suppository or clysta (enema) so are fast-acting but only work within the rectum and not the large bowel
In some countries, paraffin is not recommended for use in children under 18 or during pregnancy
Bulk-forming laxatives Contain non-digestible substances that add bulk to stool
Absorb water to increase the volume of stools
Larger, softer stools stretch the internal bowel wall and trigger bowel movements
Osmotic laxatives Facilitate the drawing and retention of water into the gut, increasing stool volume and softness and triggering bowel movement
Different types of osmotic laxatives stimulate water release into the gut in different ways
In the rest of this article, we will look more closely at osmotic laxatives and discuss why these are considered one of the most patient-friendly and effective treatments for constipation in all age groups.
Osmotic Laxatives –A Patient-friendly Solution
Osmotic laxatives can be further divided into four groups: lactulose, sugar alcohols, saline laxatives and macrogols (polyethylene glycol).
• Lactulose is a non-absorbable disaccharide that is metabolised in the
Psyllium husk (taken as a drink)
Flaxseed
Additional stool bulk can cause bloating and abdominal pain
Essential to drink plenty of water to avoid risk of bowel obstruction
Lactulose
Sugar alcohols
Saline laxatives
Macrogols
Some osmotic laxatives are metabolised, and others are not, leading to differences in side effects
Most are given orally, but some are also available as enemas
gut. The metabolites bind water and draw it into the colon, softening stools.
• Sugar alcohols work in a similar way to lactulose: they are metabolised by intestinal bacteria to form acids which stimulate peristalsis, and the metabolites draw water from the gut mucosa, softening stools.
• Saline laxatives contain magnesium or potassium and work primarily by drawing water into the bowel and making stools softer and easier to pass.
• Macrogols (polyethylene glycol) work by binding the water they are consumed with to make stools slightly larger (which stimulates bowel movements) and softer (which makes stools easier to pass). There are different types of macrogols available, characterised by their mean chain-length. Macrogol 3350 and Macrogol 4000, which are mainly used, have approximately 3350 and 4000 ethylene units respectively, and they have virtually the same properties.
INTERNATIONAL PHARMACEUTICAL INDUSTRY 55 www.international-pharma.com
Table 1. Types of laxatives and considerations for use
Health Outcomes
need to take more laxatives. In addition, when stools are hard, the body sends fewer reflexes to go to the toilet, which further worsens constipation. Thus, the patient needs greater laxative levels to achieve a bowel movement and the vicious cycle continues.
Some laxatives include salts to mitigate for this issue but taking these can be problematic for patients who have cardiovascular or renal disease and need to be on a low potassium or sodium diet. The impact of losing salts is also much worse for these patients. For example, in someone with cardiovascular disease, the heart already has a high workload, but an electrolyte imbalance requires it to work even harder.
Because macrogols have only a physical mode of action they avoid this issue with salt loss. Some macrogol products do contain electrolytes, but these are added in such a way as to balance the electrolyte
These four different classes of osmotic laxatives have different advantages and disadvantages, as summarised in Table 2.
From Table 2, macrogols stand out as the osmotic laxative that can be most widely used and that causes the fewest side-effects. They are easy to take as an oral solution and are also available as a dosage strength that is suitable for children from 6 months to 14 years. Further, they do not cause fermentation in the gut, so they avoid bloating and abdominal pain. Unlike softeners and emollients, macrogols do not affect the uptake of nutrients and, because they don’t cause any changes to the gut microbiome, they can be taken long-term.
Macrogols can be classed as either a medicine or a medical device in the EU because it only has a physical mode of action and is non-resorbable. This makes it faster and easier for such products to be registered in Europe as the process
is centralised rather than being country specific. For the patient, the benefit comes through a simple physical mechanism of action that also ensures there is no loss of effectiveness over time. It is easy to add other substances to macrogol-containing products such as electrolytes to maintain salt balance, or prebiotics such as inulin, citrus fibers, or partially hydrolysed guar gum (PHGG) to improve intestinal flora, all of which contributes to improving bowel function long-term.
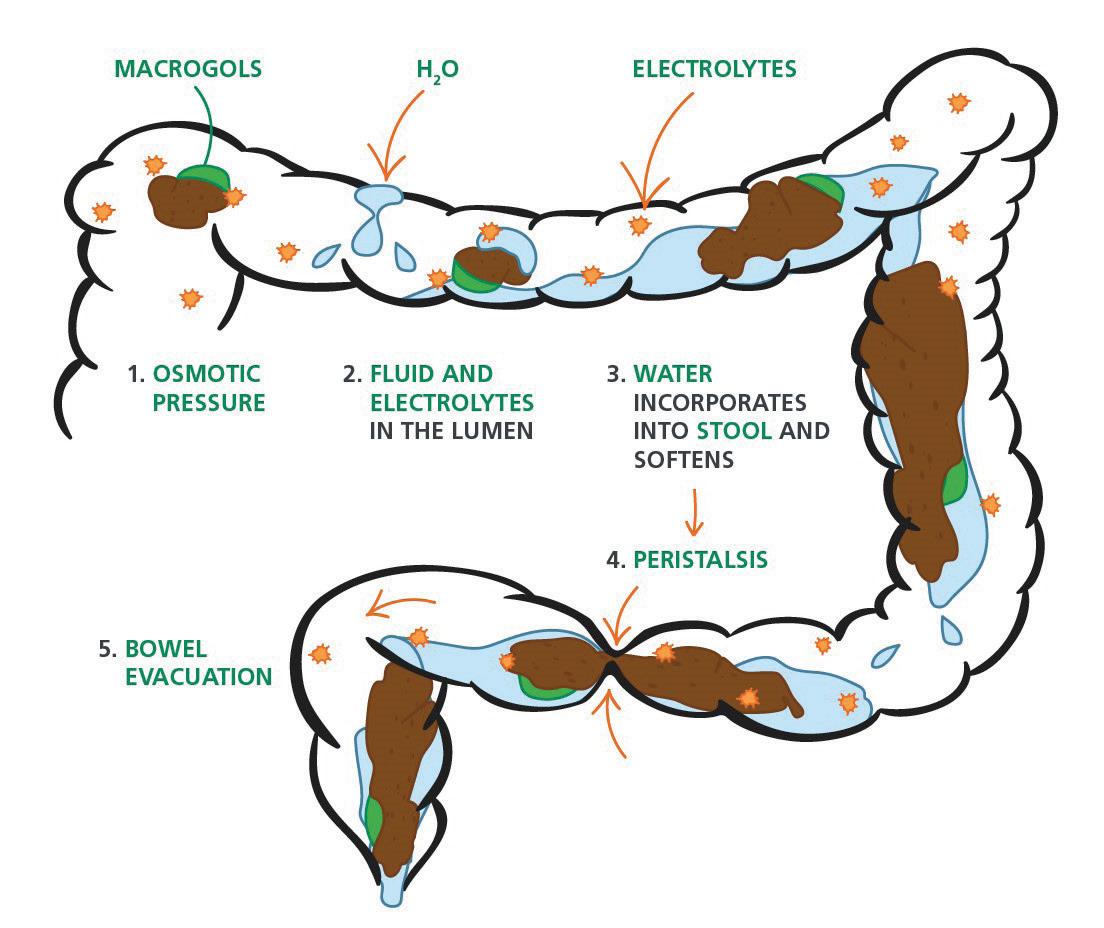
Avoiding Electrolyte Loss
A key general drawback with laxatives is the vicious cycle that occurs when they are used over a long time. Many laxatives cause electrolyte loss from the body into the stool, and this can cause stools to thicken. In turn, this leads to increased constipation and a
levels in the body and in the stool. This reduces the risk that electrolytes move from the body into the stool and keeps the patient in control of their condition.
The fact that these products are also classed as medical devices rather than medicines indicates that the notified bodies accept that the electrolytes are not absorbed by the body but simply balance electrolyte levels and have no pharmacological effect.
And that’s Not All
Beyond constipation, a physical laxative has several other applications. They can be used to empty the intestine before surgery, to induce labor when babies are overdue, and to flush the system ready for colonoscopy. The doses used will vary depending on the objective of treatment. For constipation and
56 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Health Outcomes
Illustration 2: Mechanism of action of macrogols in the large intestine
Table 2. Advantages and disadvantages of osmotic laxatives.
Ease of administration Causes bloating Causes abdominal pain Affects microbiome or nutrient uptake Safe for children? Safe for those with renal impairment or diabetes? Safe and effective for longterm use? Lactulose Oral Y Y Y Y* N N** Sugar alcohols Oral or rectal Y Y Y Y* N N Saline laxatives Oral or rectal Y Y Y N N N Macrogols Oral (as a drink) N N N Y Y Y
* For short term use only ** Although lactulose is often used long-term with patients in retirement/care homes.
Health Outcomes
stool softening, a daily dosage of around 10g for adults is sufficient. To clear the bowel before a colonoscopy, a dosage of around 40g is used.
Overcoming Formulation and Manufacturing Hurdles
Despite their many advantages for the patient, user-friendly macrogol products can be challenging to create, requiring a thorough understanding of the product and its components, as well as deep technical expertise in manufacturing and formulation. Product development should focus on end-product homogeneity and content uniformity. Specifically, the particle size distributions of macrogol and other ingredients must match to ensure good blend homogeneity. Even minor variability in particle size distribution can lead to dust formation, which compromises proper sachet sealing and risks ingress of moisture. Resultant product agglomeration can then create poor dissolution characteristics when mixed with water, or a change in the product’s appearance when still in the sachet. Products suffering from such issues risk not meeting patient expectations, compromising brand reputation.
Flavoring macrogol products is not straightforward either, primarily because the large volume of macrogol dominates product composition. Creating a palatable and enjoyable product that patients are happy to buy repeatedly thus requires deep expertise in flavoring. For example, in macrogol products containing inulin or PHGG, which are generally included in the composition at doses above 500 mg, knowledge of which flavors will work optimally can avoid unnecessary trial and error, and accelerate product development and time to market.
Macrogol is also incompatible with several excipients, as they can cause the formation of formaldehyde upon storage, impacting product quality, reducing shelf life, and necessitating inconvenient storage restrictions (storing below 25°C, for example). Knowledge of these incompatibilities not only reduces product development time but also eliminates the need for additional storage restrictions, offering better convenience for the patient and additional selling points to differentiate from competing products.
To obtain high product quality, managing excellent supplier relations can’t be overstated. Access to the right, highquality raw materials and excipients, as well as reliable and consistent delivery performance, must be secured in order to deliver On Time and In Full (OTIF) – reducing the chances of being impacted by stock shortage.
Conclusion
Of the many types of laxatives available to treat constipation, osmotic laxatives are some of the easiest to use by virtue of their easy route of administration. Within this class, there are options that avoid the side effects that are common with other types of constipation treatment – namely, bloating, abdominal pain, and flatulence.
We know that patients are more likely to use laxatives if they are user-friendly: easy to take and avoid the discomfort and embarrassment of these common side effects, especially when they are needed for long term use.
As an example, macrogol is an easyto-use alternative available in a range of formulations and doses that are safe for children as young as six months, and for older adults, including those with long-term health complications such as heart and renal conditions, and diabetes. It’s wide applicability to all patients with constipation and the ability to register as a medical device rather than a medicinal product in the EU makes it a straightforward and patient-friendly addition to any company’s portfolio of constipation treatments.
Although easy to use, macrogols are challenging to formulate, and so technical expertise in their formulation and manufacturing is key to ensuring they are effective, user-friendly and purchased again and again.
REFERENCES
1. Suares NC, Ford AC. Prevalence of, and risk factors for, chronic idiopathic constipation in the community: systematic review and metaanalysis. Am J Gastroenterol. 2011;106(9):15821592. doi:10.1038/ajg.2011.164
2. Camilleri M, Ford AC, Mawe GM, et al. Chronic constipation. Nat Rev Dis Primers. 2017;3:17095. Published 2017 Dec 14. doi:10.1038/nrdp.2017.95
3. https://www.nhsinform.scot/illnesses-andconditions/stomach-liver-and-gastrointestinaltract/constipation
4. Gandell D, Straus SE, Bundookwala M, Tsui V, Alibhai SM. Treatment of constipation in older people. CMAJ. 2013;185(8):663-670. doi:10.1503/ cmaj.120819
Dr. Martin Koeberle is Head of Analytical Development & Stability Testing at HERMES PHARMA, where he is responsible for the analytical and stability aspects of all products developed and manufactured by the company. Koeberle has deep know-how in working with a broad range of APIs, vitamins, and minerals, and is an expert in developing user-friendly dosage forms. He is experienced in defining and justifying product specifications against international regulatory scrutiny, and has authored numerous technical articles in leading pharmaceutical publications.

Dr. Verena Garsuch is Manager Analytical and Clinical Development & Stability Testing at HERMES PHARMA, where she organizes and evaluates stability studies of food supplements and manages GCP activities. Garsuch has profound knowledge of the formulation and production of user-friendly dosage forms. She is also proficient in assessing stability data, setting up and managing bioequivalence studies, and has authored numerous technical articles in leading pharmaceutical publications. Prior to HERMES PHARMA, Garsuch held formulation development roles at Hexal/ Novartis and Acino Pharma.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 57 www.international-pharma.com
Dr. Martin Koeberle
Dr. Verena Garsuch
Artificial Intelligence is a Core Pillar in the Evolution of Digital Health and Patient-centric Solutions
The US Food & Drug Administration has stated that digital technology is driving a revolution in healthcare. The lines between healthcare delivery and clinical research are blurring, as the patient becomes a key partner and focus. We are seeing a rapid expansion in the use of mobile and patient-centric devices, exponential growth in the volume and diversity of life sciences data and acceleration in the use of data-dependent computation to gain insight and automate – loosely called artificial intelligence (AI). These digital health trends are naturally combining to transform the patient experience and the application of new scientific ideas and breakthroughs.
The Covid-19 pandemic taught us that rapid and innovative responses are not only possible but extremely effective. AI has the potential to permeate all aspects of clinical
research and support the implementation of more patient-centric, decentralised clinical trials. Patient centricity has been driving innovation in clinical trials for some time, but ever-increasing levels of digitisation and the availability of advanced tools and expertise are accelerating the process. Against the backdrop of this increased use of AI in clinical trial design, patient identification, mobile technology, remote monitoring and clinical data management, we also see a corresponding rise in the need to regulate AI.
Enabling Greater Patient Centricity
Clinical trial protocols increasingly need to be more patient centric, and this means incorporating more remote monitoring and virtual trial elements, as well as making healthcare a core component of a trial. Machine learning models can be trained to predict the impact of a particular trial design, and unsupervised methods can be used to provide deep insight into the possible outcomes of a particular approach. The
application of AI to the design of a clinical trial can optimise the number and diversity of patients needed to reach the desired endpoints and give the patients who do participate a higher value experience.
Using real world evidence such as health insurance claims data, AI can foster a ‘right patients right time’ philosophy for each trial. Where the human mind finds it impossible to make connections, AI algorithms can combine the meaning of a health insurance code with that of a medication code, a social media event and many other data points to establish a robust country/site mix that drives the overall strategy for the trial. Identifying the right patients ensures better diversity and inclusion and a better patient experience, as well as scientific success. Diversity objectives range from reaching all patients who can benefit from participation in a clinical trial to ensuring that the trial collects the maximum range of data within the scientific inclusion/exclusion criteria. Identifying the right patients also means you identify the right investigators and expedite

58 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Health
Outcomes
study start up activities. The new era of patient centricity, combined with the advance of AI, expands the horizon for patient identification beyond traditional site-based models.
Increased Data Collection
More and more ways of collecting data directly from patients are being implemented, and this march of digitisation, aided by the increasing sophistication and connectedness of mobile devices, creates a positive virtual cycle with AI. More data means more opportunity to develop smarter risk monitoring processes that use AI to detect emerging patient risk early and recommend mitigation. This is important because it enables more patientcentric approaches that allow patients to participate in clinical trials from their own homes and derive a direct healthcare benefit at the same time. Even more exciting is the prospect of combining data collected by remote devices with electronic clinical outcomes assessment data collected through questionnaires to develop new digital endpoints or predict placebo effects. What was once seen as aspirational innovation is becoming reality as more and more data are collected and increasing numbers of data science professionals apply AI to those data sets. It will require effort and courage to maintain momentum in the development and validation of these challenging initiatives but the advantages to patients provide a clear incentive.
The traditional tasks of clinical data management, where clinical trial databases

must be carefully curated, cleaned and standardised for statistical analysis, are coming under the eye of data scientists. Machine learning and natural language processing techniques are transforming the way we look at clinical data, allowing us to take a more risk-based and patient-centric approach. Furthermore, the diversification of data sources, such as the data from remote monitoring devices, is driving the need for smarter, more efficient ways of conducting data management. This need is matched by the increased focus on AI as a means of optimising all aspects of the clinical research value chain.
Synergies Between Artificial Intelligence and Human Insight
AI is a key element in the evolution of digital health and patient-centric solutions. There are plenty of definitions of AI, and many are quite specific. However, it is the more vague, general sense of a machine being able to do something previously thought to be an exclusively human activity that is gaining practical acceptance by business leaders. It doesn’t matter if the solution is an advanced deep learning model or a traditional piece of procedural programming, if the result is better insights or more optimised processes.
The digitisation of all aspects of life is driving business leaders to restructure processes around new digital technologies, and data scientists are listening to those business leaders to develop AI solutions. This is fundamentally an activity driven by data availability and data literacy. A key sustainer of the relationship between data scientist and business leader is model transparency and direct end user involvement in any AI solution. Transparency and the dynamic injection of human decision making into AI models not only supports real-world validation and accountability, but it also generates a key synergy between AI and HI (human insight).
The Importance of Regulation
One area of the AI landscape that is going to impact and change how AI is applied across the healthcare arena is the introduction of AI regulations. This is a welcome development and will provide guidance and frameworks that should be used as the industry continues to explore the potential of these technologies. With the impending introduction of the EU Artificial Intelligence Act, business leaders, data scientists and legal experts need to establish the appropriate AI policy, identify the risks, and associated remedial actions for each AI implementation and understand what
implications the AI has on data protection requirements.
Clinical research, digitalisation and AI are at an important juncture as we emerge from the Covid-19 pandemic. There is much to glean from the experience of the past two years and even more to gain in the future as the industry continues to transform the clinical trial into a more patient-centric, decentralised, dynamic, data-driven, AI-powered activity. Clinical research is increasingly becoming a healthcare option, and with the introduction of AI regulations, trust in AI and its impact on clinical trial and care options will be paramount within the patient population. AI and the accompanying technology solutions will find their way into mainstream healthcare practices, decreasing the need for expensive and inconvenient in-patient care. The benefits will be felt by scientists, clinicians, commercial interests and patients alike.
Gerard Quinn

Gerard Quinn, VP, IT Innovation & Informatics, has worked at ICON for over seven years and has over 25 years in the life science industry, with experience in innovation, strategy, process improvement and IT. Gerard leads his team’s work on the innovative activities in technology and data science. They identify and build key partnerships to co-develop services by evaluating joint capabilities with the aim of driving efficient clinical trials for sponsors and patients. The team also improve cycle times for ML/AI solutions across the clinical research value chain.
Dr. Michael Phillips
Dr. Michael Phillips is a director of innovation & informatics at ICON. He has over 20 years’ experience in IT, business intelligence, data analytics and e-clinical innovation, as well as broader experience in biomedical research and management roles in general science publishing. His team of data scientists at ICON designs and builds AI solutions for a diverse range of business and clinical applications.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 59 www.international-pharma.com
Health Outcomes
Net Zero Healthcare – Priorities for Decarbonising the Pharma Supply Chain
The climate and our health are inextricably linked. The effects of climate change on global health systems and outcomes are already clear, with WHO proclaiming it to be the biggest health threat facing humanity today.
And it’s projected to get worse over time. Every year between 2030 and 2050, climate change is expected to cause an additional 250,000 deaths.
Pharma, as one of the largest global industries, is both part of the problem and the solution for minimising the adverse effects.
A first-of-its-kind study by environmental engineers found the pharmaceutical industry is significantly more emission-intensive (13 percent more) than the automotive industry despite the sector being 28 percent smaller. Over half of these emissions are produced by supply chains.
Given this link and the seriousness of the situation we find ourselves faced with in the near future, the pharmaceutical industry has a unique responsibility to act. And act fast.
So, what progress is being made to decarbonise the pharma supply chain and help combat the negative health impacts of the climate emergency?
Latest Progress and the Health Systems Taskforce
As part of the Sustainable Markets Initiative, the Health Systems Taskforce (HST) was formed in 2021 at the 26th United Nations Climate Change Conference (COP26) in Glasgow, UK.
The HST is a public-private partnership set up with the collective and ambitious goal of decarbonising supply chains to help pharma organisations reach net zero. Taskforce members include senior pharma leaders and experts from NHS England, GSK, Roche, AstraZeneca, WHO and Unicef.
Actions focus on three priority areas: Supply Chain and Patient Care Pathways,
Decarbonisation, and the use of Digital Innovation in Clinical Research.
Within these areas, the HST recommends eight levers to create low-carbon, climateresilient health systems. These include product and packaging redesign to reduce material and energy use, increasing process efficiency to cut emissions and save costs with smarter data use, and cleaner transport, shifting to sea, road and rail freight instead of air and transitioning to electric or bio-based fuels within the fleet.
In November 2022, at COP27 in Sharm El Sheikh there was a call for greater crosssector partnership to accelerate action on climate as the world faces a critical juncture. Active Pharmaceutical Ingredient (API) supply chains were a key focus, addressing this shared challenge through the newly launched Activate programme.
COP27 also marked a major milestone for the Energize programme and the first buyers’ cohort for renewable electricity was announced. By enabling suppliers to reduce their Scope 2 emissions, the programme assists pharma manufacturers to reduce their Scope 3 emissions too. These are indirect emissions that occur in the upstream and downstream activities of an organisation.
It was also an opportunity for Big Pharma to engage with key stakeholders about their personal flagship decarbonisation programmes, and present how they are delivering this in key countries and through partnership.
Towards a whole Lifecycle View
All products and services have lifecycles. The lifecycle refers to the period from the product’s first launch into the market until its final withdrawal.
Traditionally, Product Lifecycle Management (PLM) has first and foremost been used to help companies understand and realise its position in the market compared to competitors and a product’s success or failure.
But having a unified view of the entire product development lifecycle with the ability to view and trace every detail throughout the
entire process can be hugely valuable from a sustainability standpoint.
It’s important to consider the whole lifecycle of a medicine, from design and development and production to sale, consumption and disposal, to reduce pharma’s negative impact on the environment.
Different stages of the lifecycle produce different levels of carbon and other waste.
Product Development and Manufacturing
Pharmaceutical development and manufacturing require huge amounts of energy to output a comparatively small amount of Active Pharmaceutical Ingredient (API). Like any industrial process, where there is significant energy, there also tends to be significant waste.
Solutions to reduce energy-related emissions in the manufacturing process include on-site anaerobic digestion plants to treat hybrid waste, energy-efficient lighting and solar panels to power facilities using renewable energy.
Firms can also choose to install rainwater harvesting systems, solar panels, inverter driven machinery and reactive lighting designed to maintain a consistent lux output whenever an area is occupied, to robotics which increase production yields and accuracy with reduced input.
Packaging Design
Prioritising waste prevention from the outset of packaging design can also improve pharma’s environmental footprint.
3D visualisation and printing technologies are helping manufacturers meet their sustainability goals by being more mindful of waste prevention strategies and improving the quality of design. From the outset, manufacturers can plan and test their products’ efficiency to the highest standard, reducing the volume of substandard defunct packaging.
At this stage, packaging designers can also integrate key safety features into the core of their designs to limit the use of multiple packaging materials. In doing so,
60 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Logistics & Supply Chain Management
packaging is both more efficient and easier to recycle. For example, printing product information directly onto the secondary packaging can reduce labelling materials while QR codes can allow patients to access their private information and specific dosage requirements without the need for excessive labelling.
Manufacturers are also experimenting with more sustainable materials for their packaging, with plant-based plastics becoming more readily adopted by pharmaceutical companies.
Astellas Pharma, for example, made waves switching to sugarcane-derived blister packaging in 2021 – a world first for biomassbased plastic for blister packages. Plantbased materials made up 50 percent of the raw materials used in its development while still providing the same protection function and usability.
The move to biomass-based materials could prove more common in the coming years as technologies evolve and consumer demand for sustainable alternatives grows.

Chain Management
Distribution
While manufacturing carries a large carbon footprint, the distribution of medicines from the factory and into patients’ hands also has a significant impact on the environment.
One of the most energy-intensive processes is delivering temperature-sensitive products, like insulin and some vaccines, from the point of manufacture to the patient within the cold chain. Refrigerated vehicles require additional energy to power the cooling systems, known as transport refrigeration units.








The simplest way to reduce the carbon footprint of shipping is to use cleaner fuels and transport types, such as rail, road and sea freight instead of air.
Hydrotreated vegetable oil, a renewable, bio-based fuel that can be used in diesel engines, can reduce greenhouse gas emissions by up to 90 percent compared with diesel. Other alternative fuels include: compressed natural gas, liquefied natural gas, liquefied petroleum gas (LPG), and their renewable counterparts - biomethane and bio-LPG.
Localised manufacturing, or re-shoring, can also reduce the carbon footprint by reducing the miles the final product has to travel to reach the consumer. This approach optimises the amount of time a product needs to be maintained within a storage environment or shipped amongst the different transportation solutions.

Use and Disposal





Once waste and carbon emissions have been minimised in the product’s production and distribution, attention should focus on to how it will be used and ultimately discarded by the end consumer.
Currently, most medicine packaging –particularly primary - ends up in a landfill or is incinerated, losing its value as a material resource. So, brands must continue to think about not only innovative recycling methods but upcycling and repurposing materials too.
End users play a key role in the lifecycle of these products and must be committed to act alongside manufacturers. But this requires education initiatives, clear instructions and simple steps to follow to maximise compliance.

INTERNATIONAL PHARMACEUTICAL INDUSTRY
&
Logistics
Supply
HIGH PURITY. HIGH DEMANDS. Pressure regulators Control valves Pipeline ancillaries Special equipment Steam traps Safety valves High purity equipment for clean steam adca@valsteam.pt www.valsteam.com +351 236 959 060 Zona Ind. da Guia, Pav. 14 - Brejo 3105-467, Guia PBL PORTUGAL PRODUCTS MANUFACTURED IN PORTUGAL AV 001 AP EN 01.20
Logistics & Supply Chain Management
If materials cannot be recycled and reintroduced in the lifecycle, manufacturers must invent innovative ways to repurpose the discarded materials.
For example, Novo Nordisk has innovated a way to repurpose its insulin pens. Despite being mostly made of plastic, they cannot be put in plastic recycling bins. In response, Novo created a system that sorts the pens into many component parts and partnered with a Danish design company to make office chairs using the waste plastic and lamps using the discarded glass.
Data-driven Digital Transformation to Unlock Further Gains
Pharma is one of many industries trying to become smarter in collecting, analysing and leveraging the power of data to make decision-making faster, solving inefficiencies and meeting sustainability targets.
Historically, the transition from industrialage to digital-era operating models has been slow.
Inflexible IT infrastructure is a barrier to digitisation, particularly making old and new systems interoperable. Pharma 4.0 demands the gap between the digital and physical is closed, allowing for a 365-degree view of business operations. In a global supply chain, this can be difficult to achieve.

Supply chains are healthcare’s climate Achilles heel - technology that connects the lab to the marketplace is lagging. But recent
advancements in big data technologies, machine learning and artificial intelligence are propelling digital strategies forward, unlocking sustainability gains.
Big data and AI have a synergistic relationship. AI requires large volumes of high-quality data to learn and improve decision-making processes. Over time, the more data the algorithm receives, the more accurate and efficient it can become – and so can pharma.
With greater automation and real-time data accessibility, combined with AI, pharma firms can collect, analyse and act on data insights before an issue occurs. It can also minimise human error, provide end-to-end visibility, and protect the integrity of supply chains.
Over time, AI will transform the industry’s operating models and help it achieve its sustainability targets. However, full digitalisation takes time and strategic thinking, and involves a fundamental shift from linear supply chains to dynamic, interconnected and open AI-enabled digital supply networks (DSNs).
The workforce will also need further diversification to ensure the industry has the skillsets and knowledge that matches its ambitious digital-driven aims.
Today, manufacturers that want to move into an end-to-end digital supply chain are increasingly leaning on external experts given
Tomorrow, the talent pipeline needs to be agile, digitally literate and open to continuous learning to maximise the opportunities these new technologies present pharma.
Steve Brownett-Gale is a marketing professional with a career spanning both communications and products in B2B and B2C markets across Manufacturing and Services sectors. At Origin, in his role as Marketing Lead, Steve is responsible for positioning the company as a worldleading supplier of innovative and groundbreaking pharmaceutical packaging devices, as well as offering a unique and disruptive supply chain model. Established 60 years ago, Origin offers customers a remarkable range of versatile packaging solutions that respond to the unique needs of the global pharmaceutical marketplace. Origin engages in the design, manufacture, and consolidated supply of pharmaceutical packaging, partnering with licence holders and CMOs.
Web: www.originltd.com

62 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Steve Brownett-Gale
the lack of dedicated teams with established knowledge on AI design thinking.
Logistics & Supply Chain Management
Harnessing the Power of cMaaS in Pharma Logistics: Ensuring Regulatory Compliance and Sustainable Supply Chain Management
End-to-end digital visibility across the supply chain can be used to identify hotspots, support real-time decision making and improve demand forecasting. Pharma companies, acting in collaboration, can incentivise third party organisations to set their own targets for improving their environmental footprint, both upstream and downstream from their own operations’.
source: Deloitte report
Pharmaceuticals are a critical component of modern healthcare, and ensuring their safety and efficacy is paramount. This is particularly true when it comes to logistics, where pharmaceuticals must be transported across great distances and under a variety of conditions. Regulatory requirements for logistics, such as Good Distribution Practice (GDP), have become increasingly stringent in recent years, requiring that pharmaceuticals be transported under specific temperature and humidity conditions.
The Pharmaceutical logistics is estimated to be responsible for 2–3% of global greenhouse gas emissions, which is significant considering that the pharmaceutical industry as a whole accounts for 4.5% of global emissions. The main source of these emissions come from Transportation, packaging and waste.
In this article, we will examine the importance of temperature control in pharmaceutical logistics, and how modern technology such as connected Monitoring as a Sservice (cMaaS) can help address some of the key challenges in this field.
Pharmaceutical Logistics Issues
It is challenging to determine the exact annual waste of medicine due to spoilage during logistics, as various factors contribute to the loss, and the data may vary across countries and supply chains. However, it is estimated that globally, around 15–20% of temperaturesensitive pharmaceuticals are wasted annually and 30% of the scrapped products are due to inadequate temperature control during transportation and storage, with a value of over $35 billion per year (Statista).
Estimating the exact number of lives lost annually due to poorly maintained medicines is challenging, as the impact can vary significantly across countries and medical conditions. Spoilage and degradation of pharmaceutical products can lead to reduced efficacy or increased risk of adverse effects, both of which can have severe consequences for patient health.
In developing countries, where access to quality healthcare and medicines is limited, the impact of improperly maintained medicines may be more pronounced. For example, the World Health Organization (WHO) has reported that up to 35% of vaccines are wasted globally, primarily due to inadequate temperature control during transportation and storage. This waste contributes to millions of unvaccinated children and adults, increasing the risk of preventable diseases and associated morbidity and mortality.
Challenges in Pharmaceutical Logistics End-to-end Digital Visibility
The lack of end-to-end digital visibility in the supply chain can lead to avoidable losses, poor demand forecasting, and inefficient decision-making. Moreover, billions of medicines are discarded and unused annually, which could be provided to the two billion people that lack access to basic medicines (Sustainable Medicines Partnership, SMP).
Reliance on Paper-based Information
Traditional logistics practices in the pharmaceutical industry have long depended on paper-based information for tracking and managing medicines. This approach not only consumes valuable resources, but also often results in vital information going unread. As a consequence, the industry faces significant inefficiencies and an increased environmental footprint.
Paper-based systems are prone to human error, leading to inaccuracies, mismanagement, and delays in the supply chain. Additionally, the reliance on physical documentation can make it challenging to share and access vital information in realtime, hampering decision-making processes
and hindering timely responses to issues that may arise during transportation.
Datalogging Limitations and the Emergence of cMaaS
One of the main regulatory requirements for pharmaceutical logistics is maintaining temperature control. Almost all pharmaceutical products are sensitive to temperature and must be stored and transported under specific conditions. Temperature excursions, even brief ones, can lead to product degradation and reduced efficacy. Datalogging is commonly used to monitor temperature conditions during transit, however, this technology has limitations as the information is only provided after the shipment has been completed. This postmortem analysis means that shipments with excursions are sent into a disposition process to be analysed and determine if the product integrity is intact or if the product and shipment needs to be discarded. This is a very time and cost consuming process. These events then form the basis of a root cause analysis and eventual corrective or preventive actions to put in place to see if it can be avoided in the future.
cMaaS is a relatively new technology that can help address all of these challenges. cMaaS allows for real-time monitoring and control of shipments, providing pharmaceutical logistics operators with real-time actionable information to take immediate corrective actions if a temperature excursion occurs or preventive actions if it is about to occur. The system uses advanced sensors to monitor environmental conditions (temperature and humidity) and physical conditions (shock, tilt, pressure) and other events such as light exposure and geo-location all in real-time, and provides alerts if any of these variables deviate from specified ranges.
Quality Requirements: Good X Practice (GxP)
Pharmaceutical logistics are subject to rigorous regulatory requirements, such as Good Manufacturing Practice (GMP) and Good Distribution Practice (GDP), which provide the minimum requirements and standards that manufacturers and distributors must meet to ensure the quality and integrity of
INTERNATIONAL PHARMACEUTICAL INDUSTRY 63 www.international-pharma.com
Logistics
Supply Chain Management
the medicines throughout the supply chain, from production to the patient. Maintaining strict temperature control is of paramount importance throughout this process, as most pharmaceutical products are highly sensitive to temperature variations. Deviations from the specified temperature ranges can result in the degradation of the active ingredients in these medications, ultimately leading to a decrease in their efficacy and potential harm to patients.
Transporting time and temperaturesensitive pharmaceutical products poses a significant challenge for logistics service providers, as they must ensure the continuous monitoring and maintenance of the specified temperature and humidity conditions throughout the entire supply chain, including not storage facilities and transportation between various stages; the manufacturer to the wholesaler or from the wholesaler to the pharmacy, hospital or clinic and event direct to patient.
Failure to adhere to these stringent temperature and humidity requirements can have severe consequences, including compromised product quality, regulatory penalties, increased product recalls, and ultimately, jeopardised patient safety. In order to ensure compliance with regulatory standards and safeguarding the integrity of medicines, logistics providers must invest in advanced technologies, such as temperaturecontrolled storage and transportation equipment, real-time monitoring systems, and data-driven decision-making tools.
Benefits of cMaaS in Pharmaceutical Logistics
Real-time Response to Temperature and Humidity Changes
cMaaS provides instant notifications to freight forwarders, couriers, carriers, and manufacturers about sudden changes in the conditions of the shipments and products. The real-time responses allow for prompt action to be taken to prevent further degradation of shipped goods and save valuable pharmaceutical products, bringing pharma and cold chain logistics towards zero waste supply chain management. With cMaaS, parties responsible for the shipment can prevent losses due to unexpected conditions during storage and transportation.
by cMaaS provides for real-time actionable information allowing logistics and quality assurance operators and managers significant time and cost saving possibilities.
For logistics operators, the setup, retrieval and completion time is 1/10th of that needed with traditional dataloggers. The process is significantly more efficient as the cMaaS devices are always monitoring and receive their instructions directly from platforms which are connected to the customers. A simple scan, click and go and you are on your way to receiving vital real-time shipment information.
For quality assurance personnel, cMaaS platforms provide overall risk management tools with timely information to prevent the loss of medicines, allow for automated release of shipments, and no need for lengthy and costly disposition processes, improving overall efficiency. No more security risks associated with the plugging in of USB device into corporate computers.
The real-time data intelligence provided allows for supply chain optimisations beyond those already mentioned. It can also help in packaging analysis and improvements, provide predictive times of arrival, route selection and changes required due to events, lane validation and continuous lane risk score updates.
Sustainability and ESG Targets
cMaaS provides the real-time data intelligence to reduce, and eventually eliminate, the waste caused in pharmaceutical logistics from using antiquated technologies. Improvements in real-time preventive actions, using reusable non-lithium devices, and analysis, and recommendations provided all help to reduce the 2–3% of global emissions caused by pharmaceutical logistics. Helping pharmaceutical companies and logistics providers to improve sustainability and achieve ESG targets.
Time and
Cost
Reduction
in TemperatureControlled Logistics
Real-time data on shipment status captured
Enhanced Supply Chain Efficiency cMaaS reduces the need for manual temperature monitoring or additional quality control measures, contributing to more efficient supply chain management.
While datalogging has long been the standard for tracking temperature conditions during transit, its inability to provide realtime information hampers effective corrective actions, and more importantly preventive actions, when temperature excursions occur or will occur. Thereby, simply being a tool to confirm if the shipment and products are intact. If they have excursions a long and

64 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
&
Logistics & Supply Chain Management

costly disposition process starts to determine if they can be kept and used safely or need to be disposed. With cMaaS, logistics providers can now monitor and control shipments in real-time, ensuring the safety and efficacy of transported pharmaceutical products.
Conclusion
In conclusion, temperature control is a critical component of pharmaceutical logistics, and regulatory requirements have become increasingly stringent in recent years. Datalogging has traditionally been used to monitor temperature conditions during transit, but modern technological solutions such as cMaaS offers many advantages. With real-time monitoring and control capabilities, cMaaS helps address the challenges of temperature excursions and sudden changes in environmental conditions. By providing realtime data intelligence cMaaS help logistics and quality assurance operators and managers to optimise their supply chain, reduce waste, reduce costs, improve their environmental footprint all whileensuring the safety and efficacy of pharmaceutical products.
REFERENCE
1. FDA: Guidance for Industry: Non-Penalty Regulatory Compliance Policy for Certain Food, Drug, Device, and Cosmetic Act Requirements During the COVID-19 Public Health Emergency, https://www.regulations. gov/document/FDA-2020-D-1138-0105
2. EMA: Good Distribution Practice of medicinal products for human use, https://www. ema.europa.eu/en/human-regulatory/ post-authorisation/compliance/gooddistribution-practice
3. WHO: Guidance on Good Distribution Practices (GDP) for pharmaceutical products, https://www.gmp-compliance. org/guidelines/gmp-guideline/who-gooddistribution-practice-for-pharmaceuticalproducts
4. ISPE: Good Practice Guide: Cold Chain Management, https://ispe.org/publications/ guidance-documents/good-practice-guidecold-chain-management
5. EU-GDP: Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use, chrome-extension:// efaidnbmnnnibpcajpcglclefindmkaj/https:// eur-lex.europa.eu/LexUriServ/LexUriServ. do?uri=OJ:C:2013:343:0001:0014:EN:PDF
Charles Bourbonnais is the CEO and CoFounder of Hive-Zox International S.A., an innovative Swiss technology company focusing on autonomous logistics and supply chain management solutions. Using the latest in IoT technology, Charles aims to track and monitor every product to optimize logistics and ensure customer safety & guarantees. Prior to founding Hive-Zox, Charles had a successful career in finance. He is a Canadian who has become naturalized Swiss citizen and believes in incorporating international experiences and cultural diversity to create innovative solutions. As a father and husband, Charles's commitment to sustainability and creating a better future is reflected in Hive-Zox's mission.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 65 www.international-pharma.com
Charles Bourbonnais
Why an Outcome-driven Approach to Supply Chain is a Strategic Advantage for Pharmaceutical Businesses
Since 2020, the pharmaceutical industry has been grappling with frequent and rapid changes, shifting customer demands and increasing costs. In response, organisations have stepped up and adapted quickly, recognising opportunities to discover and trust new technology and approaches in an attempt to create resilience. However, for pharma businesses, the fact remains that most supply chains are still driven by activity and necessity instead of outcomes and possibilities.
So why is this happening? Due to the complex and specialist nature of the pharmaceutical industry, it often results in the siloing of data across different platforms and partners, which limits visibility and reduces resilience. When medicines are being transported to patients across the globe it is imperative that operators have a holistic view across a fully responsive supply chain. This is necessary to ensure that they can minimise or avoid disruption altogether. In order to achieve this, they need to utilise a data-led approach, that can facilitate a smoother transition from siloed and reactive to more proactive operational processes; which in turn, can increase resilience and ensure that businesses are well placed to satisfy customers and ultimately recognise revenue.
What we’ve seen therefore is that pharma businesses are not set up to be flexible and adaptable to changing market conditions, customer demand, and supply chain disruptions. From our analysis of the industry, we can conclude that there are 3 key priorities that need to be addressed in order to create truly resilient, outcome-driven supply chains:

• The need for end-to-end visibility
• Operational resilience
• Making your SLAs work for you
The Need for End-to-end Visibility Visibility can be a tricky concept to define within the context of a supply chain, as its meaning can vary depending on the specific situation. While there are certain types of data that are generally useful, the key is

to understand the ultimate objectives that organisations are aiming to achieve.
What we’ve found is that what organisations think of as a visibility problem is ultimately more specifically around missing revenue or costs, and a lot of businesses believe this is tied to inventory. Therefore, it’s crucial to focus on the outcomes that your business wants to achieve, and then determine what data is required to make that happen. In some cases, the necessary data may already exist outside the typical supply chain data path, and it may be necessary to obtain additional information to gain a more nuanced understanding of the situation. For example, as you’ll know in the pharmaceutical industry, it’s essential to have full visibility of temperature control data in order to ship any drug with a temperature-sensitive component, as this ensures the quality and safety of the product.
In general, when it comes to visibility, it’s essential to consider how it will ultimately impact and benefit the organisation. Simply striving for greater visibility without a clear understanding of how it will deliver tangible value can result in disappointment.
Operational Resilience
Maintaining operational resilience is imperative for businesses that want to take a proactive approach in order to mitigate risk. With disruptive events from the RussiaUkraine war and earthquake in Turkey, to medical outbreaks like the recent eruptions of the Marburg virus happening more and more frequently, organisations must consider their options and put measures in place to protect themselves.
This goes beyond merely identifying disruption thanks to having visibility across the supply chain, but positively impacting the outcome of how you deal with those events. Being able to rapidly identify a challenge and secure the right carrier that can help get the product from a to b can be the difference between life and death, and without resilience baked into every layer of your operations your ability to do that is restricted.
As an example, for many pharmaceutical companies, things like carrier selection have often been relationship driven. Remaining wedded to a handful of carriers limits business agility and proactivity, minimising operators’ ability to maintain resilience and respond to changes quickly if needed. Implementing tools and systems that assess the best carrier for the job by comparing the performance of a current provider against competitors places operators in a better position to negotiate a more efficient, costeffective service, improving performance without increasing risk.
Making Your SLAs Work for You
This brings us to the third major issue facing pharmaceutical companies – ensuring that their carrier SLAs are maintained and reflective of the kind of service they’re paying
66 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2 Logistics & Supply Chain Management
for. Pharma companies are all too aware of the risks that come with mistakes being made, ranging from reputational damage on one hand to loss of life at the extreme end. Under these circumstances, operators are less inclined to try new methods and instead choose to stick with suppliers that they’ve worked with previously, who they can rely on to deliver medications on time.
However, this caution often results in companies missing out on the opportunity to receive a higher quality of service at a lower cost. Specialty carriers in particular, given how time and temperature sensitive, and how heavily regulated the goods they move are, can be a particular culprit in this regard. Without being able to effectively assess and monitor performance you don’t know if the service you’re receiving is actually at the level you’re paying for and given the costs of moving some goods across the world, those savings can be substantial.
This is where visibility once again becomes a significant item, because it’s through ensuring complete data visibility across your carriers that you’re able to track and measure if SLAs are being adhered to, and actively identify carriers that are failing to maintain them. With a more resilient supply chain, you’re also able to secure rates and contracts from other carriers that can deliver for you while more actively balancing performance and cost, meaning you don’t become reliant on a relationship that you aren’t deriving the most value from.
Simulations a Stepping Stone Towards an Outcome-driven Supply Chain
Monitoring products in real-time can remove uncertainty and increase the accuracy and agility of the supply chain. The efficacy of such technology has been evidenced in recent years via the blockage of the Suez Canalan outcome-driven supply chain would enable businesses in a similar situation to reroute their shipment to alternative carriers,

reducing the impact of the disruption on their individual operations. Direct and indirect costs could have been mitigated, from the loss of materials to missed opportunities to exceed customer expectations.
Simulation tools can also prove crucial in aiding operators’ response to disruption. The global pharmaceutical logistics market is one of the most regulated, expensive and fragile cargo markets in the world today. A recent Pharmaceutical Logistics Market report by Mordor Intelligence valued the market at $83.5 billion and found an increasing demand for sea and air freight transport within the industry. Whilst road is the most widespread method, intercontinental distribution often occurs via air freight, despite high levels of cost and emission production. Simulations can help operators make the best choice regarding mode of transport, allowing them to balance a reduction in carbon emissions with minimal cost increases. For instance, they can identify cases where sea freight – a safer, inexpensive and more reliable alternative to air – is a practical option for a leg of the product’s journey.
Alternatively in the case of a canal blockage, by analysing the impact of an event before it happens, along with various potential responses, the outcome of various business decisions can be simulated to illustrate how this would impact the overall supply chain. Should said event then occur, the real-time data can be inputted, such as the nearest deployment centre, most optimum carriers and alternate routes, alongside any other specific chosen parameters such as emission levels and cost to inform operators of the best way to resolve the situation at hand. When lives are at stake, as is the case in pharma supply chains, such preventative modelling and quick reactivity could be the decider between life and death.
Take the recent antibiotic shortages in much of Europe at the end of last year. We conducted data investigating the impact of shortages and found that 25% of UK
consumers believe that pharmaceuticals has been the most impacted industry when it comes to supply chain disruptions. Additionally, 11% said they were unable to get hold of the medicine they needed in their local pharmacy. An excellent example of how end patients have been affected by inefficient, activity-driven supply chains.

What next?
So, creating an outcome-driven supply chain is imperative for the pharmaceutical industry. But where should you start?
Businesses will need to use AI for scenario planning and stress testing to optimise their supply chain – this is particularly vital as costs, pressures and disruptions continue to increase globally. As AI and machine learning become more advanced, the ability of technology to rapidly recalculate various factors to overcome disruption will increase. Simulations are a strategic tool, and when paired with data insight and management platforms, can make the application of informed business decisions more seamless.
The outcome-driven supply chain model is designed to be flexible and adaptable to changing market conditions, demand, and supply chain disruptions. It focused on achieving business outcomes through a collaborative and integrated approach that prioritises continuous improvement and responsiveness. It utilises data analytics, advanced technology, and process improvements to optimise the flow across the supply chain, reduce waste, and increase efficiency.
Philip Ashton is the CEO and Co-Founder of 7bridges, the AI-powered logistics platform. From a background in analysis and development in management consultancy (Oliver Wyman) and specialist logistics provision (World Courier), Philip co-founded 7bridges in 2016 to apply customised AI to logistics operations. 7bridges works with leading brands across retail, pharmaceutical, ecommerce and manufacturing and is committed to reinventing supply chain management because smarter supply chains benefit everyone.

INTERNATIONAL PHARMACEUTICAL INDUSTRY 67 www.international-pharma.com
Philip Ashton
Logistics & Supply Chain Management


Summer 2023 Volume 15 Issue 2 7TH EDITION The European partnering event for innovation partnerships and investment rounds in the MedTech, Diagnostic and Digital Health sectors Contact: Yingjie Weng yweng@eurasante.com +33 (0)9 78 31 55 17 www.medfit-event.com MedFIT Event ONE-ON-ONE MEETINGS PITCH SESSIONS CONFERENCES EXHIBITION PROJECT - CEO MATCHMAKING October 17th, 2023 ONLINE MEETINGS October 10th & 11th, 2023 Strasbourg, FRANCE IN-PERSON Register now and benefit from 10% off the full pass with the code: MF23_IPI_2706 Applied on the regular fee


www.international-pharma.com At the heart of Pharma 24-26 October 2023 Barcelona, Spain Book Your Stand Now
Pharma’s largest event is making an exciting return to Barcelona on 24-26 October 2023. SCAN ME
Exclusive Entry Into The Global Pharma Market
REGISTERSTUDENTSFORFREE!
Edinburgh International Conference Centre
Emerging Scientist 2023 – 30th June 2023 – a chance to present on the podium at DDL2023.
Pat Burnell Young Investigator Award
– 14th July 2023 – young researcher finalists compete on the podium at DDL2023.
REGISTRATION NOW OPEN
70 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
HEALTHCARE EVENTS
The ultimate events for the pharmaceutical and biotechnology community to discover how to excel in clinical supply strategy as well as form key connections for long-term success


A great opportunity to get together and network face to face, hear from our expert speaker line-up and discuss the latest innovations and regulatory updates in the industry.

This exclusive event brings together attendees from established pharma, large and small, alongside biopharmaceutical companies and gives opportunity to dive into the operational challenges and innovations in clinical development found within the UK & Ireland region.
Use Reference Code: IBI
This year the conference will be focusing on interactive discussions to provide insight into the current major operational issues with running clinical trials in the coming years.
The conference will be focused on interactive discussions to provide insight into the current major operational issues with running clinical trials in the coming years.


Use Reference Code: IBI
Clinical Trial Supply East Coast offers opportunities for both learning and networking with peers from across the CTS sphere, via informal networking breaks and lunches, as well as a relaxed drinks reception at the end of Day 1.
Our conference includes speakers from Pfizer, Merck, Janssen and many more who will be covering the latest innovations in the clinical supply industry Use Reference Code: IBI
INTERNATIONAL PHARMACEUTICAL INDUSTRY 71 www.international-pharma.com Register
Now
Register
Now
Register
Now
Page 5
Page 71
Page 3
Page 69
Page 70
Page 31
Page 35
AeroGo Inc.
Arena – Healthcare Event
ASEPTCONN AG
CPHI Barcelona
DDL 2023
FUJIFILM Wako Chemicals U.S.A. Corporation
Kahle Automation
IFC Klinge Corporation
Page 21
Krautz Temax
Page 68 MedFit
BC Natoli Engineering Company
IBC Nipro
Page 27
Owen Mumford
Page 13 PCI Pharma Services

Page 29
Page 43
Page 45 & 61
Phillips Medisize
Senglobal Ltd.
Valsteam ADCA
Page 7 Waters Subscribe today at www.international-pharma.com or email info@senglobalcoms.com
I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry
IPI is also now active on social media. Follow us on:
www.twitter.com/ipimediaworld www.facebook.com/ipimediaworld www.plus.google.com/+ipimediaworldmagazine www.ipimediaworld.tumblr.com
72 INTERNATIONAL PHARMACEUTICAL INDUSTRY Summer 2023 Volume 15 Issue 2
Advertisers Index
Hypodermic Needles in Hard Plastic Unit Packaging

Ideal to by-pack with drug products!
Each needle is sterile packed individually
•
Packaging design supports compatibility with automated pick-and-place systems/feeders
•
Color-coded labels and barcode on label enable in-line product identification
73 INTERNATIONAL PHARMACEUTICAL INDUSTRY NIPRO PHARMAPACKAGING INTERNATIONAL Blokhuisstraat 42, 2800 Mechelen, Belgium | pharmapackaging@nipro-group.com | www.nipro-group.com





scan the QR to receive your free digital copy. Request your Multi-tip Tooling Guide Today, Dramatically increase your production without adding a new tablet press or extending press run time. With innovative tooling designs, superior customer service, and incomparable construction, Natoli is your number one choice. Natoli Multi-Tip Tooling natoli.com
