
Journal for Clinical Studies PEER REVIEWED Volume 15 Issue 2 Exploring the Limb Girdle Muscular Dystrophy Clinical Trial Landscape Dispelling Doubt Effective Scientific Journalism in a Time of Polarisation Ensuring Unbiased Oversight The Case for Independent Expert Committee Management Enhancing Health Care Through Improved Patient Labelling www.journalforclinicalstudies.com
LISTEN, DEVISE BREAKTHROUGH
Our approach to advancing your therapies beyond milestones to transforming lives


Discover Catalyst
A biotech-focused Oncology CRO
helping to bring next-generation therapies to cancer patients in need
Resourcing and functional solutions
helping to drive healthcare innovations for patients in need

Journal for Clinical Studies
MANAGING DIRECTOR
Mark A. Barker
BUSINESS DEVELOPMENT info@senglobalcoms.com
EDITORIAL MANAGER
Beatriz Romao beatriz@senglobalcoms.com
DESIGNER
Jana Sukenikova www.fanahshapeless.com
RESEARCH & CIRCULATION MANAGER
Jessica Chapman jessica@senglobalcoms.com
ADMINISTRATOR
Barbara Lasco barbara@senglobalcoms.com
FRONT COVER istockphoto
PUBLISHED BY Senglobal Ltd.
Unite 5.02, E1 Studios, 7 Whitechapel Road, E1 1DU, United Kingdom
Tel: +44 (0) 2045417569
Email: info@senglobalcoms.com www.journalforclinicalstudies.com
Journal for Clinical Studies – ISSN 1758-5678 is published quarterly by Senglobal Ltd.
4 FOREWORD
WATCH PAGES
6 Clinical Trial Technology Trends: The Top 5 Innovations Shaping Clinical Trials in 2023

Clinical trials play a crucial role in the development and approval of new medical treatments and drugs. As technology continues to advance, Clinical Trial Technology is becoming a more important factor in shaping the way trials are conducted. Rajesh Pothula at Clinion shows the top 5 innovations shaping clinical trials in 2023.
8 Enhancing Health Care Through Improved Patient Labelling
When patients receive a prescription drug or biological product in an outpatient setting, it may be accompanied by one or more types of written information approved by the US Food and Drug Administration (FDA). The quality of that information, known as the patient labelling, plays a decisive role in whether patients take their treatment properly, or at all. Deborah Komlos at Clarivate outlines the various approaches taken will assist with improving access to medication information for all patients who may be dispensed a prescription drug product in an outpatient setting.
REGULATORY
10 Comparing Treatment Performance through Evidence Synthesis
If there is a variety of treatment options in a specific disease area, decision makers require evidence of efficacy and safety of novel interventions in comparison to established treatments. One randomised controlled trial (RCT) is not sufficient to come to a conclusion, especially since RCTs often present contradictory results. Katrin Haeussler, Matthias Hunger & Nathan Green at ICON discuss a novel method, that uses individual patient data to adjust for differences in effect modifiers between studies.
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
Volume 15 Issue 2 Summer 2023 Senglobal Ltd.
Journal for Clinical Studies 1 www.journalforclinicalstudies.com
Contents
13 The Role of LIMS in Supporting ISO 17025 Accreditation
ISO/IEC17025:2017 is the international standard that specifies the general requirements for the competence, impartiality and consistent operation of laboratories. It can be used by laboratory customers, accreditation bodies, and other organizations to recognise the competence of laboratories. The key element of the standard is that it requires laboratories to demonstrate that they operate competently and can generate valid results. Tim Daniels at Autoscribe Informatics explores the ways in which an integrated Laboratory Information Management System (LIMS) can play a key role in achieving, maintaining, and benefiting from ISO17025 accreditation.
18 WhyzeHealth: Improving Access and Participation in Clinical Trials in Ireland, a Patient-Centred Digital Health Platform
Across Europe, we continue to see a declining trend in the number of clinical trials taking place, most notably since the European Union Clinical Trials Directive (CTD) was established in 2004. The decline in clinical trials in Ireland is a major concern, as it means that fewer people are given access to potentially life-saving treatments. This can have long-term effects on the quality of care being provided, as well as making it harder for new treatments to be developed and tested. However, the CTD has long-felt ramifications for European patients. Prof. Frank Sullivan at Whyze Health explains how to improve access and participation in clinical trials in Ireland.
20 How Lilly is Streamlining Regulatory Operations
In 2016, a cross-functional team at Lilly began to re-evaluate its approaches to global submissions and determine if newer technologies or processes could help improve efficiency. Reliance on traditional RIM software and manual processes hindered the regulatory team’s ability to keep up with submission volumes. Marc Gabriel at Veeva Systems talks about how Lilly is streamlining regulatory operations.
RESEARCH & DEVELOPMENT
22 Ensuring Unbiased Oversight: The Case for Independent Expert Committee Management
Expert committees are a critical component in the success of a clinical trial. By turning to third-party providers to manage these services, sponsors can enhance trial credibility, mitigate risks, reduce bias and the perception of bias, and drive operational efficiencies. Data Monitoring and Endpoint Adjudication Committees are groups of independent medical and/or statistical experts charged with a specific set of oversight activities related to a development program. David Cutler at WCG explains how the right partner helps sponsors ensure
clinical integrity and regulatory compliance, attain operational and financial efficiencies, and achieve clinical trial success.

MARKET REPORT
26 Dispelling Doubt: Effective Scientific Journalism in a Time of Polarisation
In an era of rapidly evolving information and ever-burgeoning scientific innovations, it can be disheartening to witness the persistent lack of trust in the scientific process and establishment. Science journalists must become beneficent mediators to help create an understandable and comprehensible stream of information for consumers to digest with the goal of humanising findings and both engaging and enticing the public toward. Lucas Riordan, a rising Junior at University of Delaware and esteemed author Henry Riordan discuss how rebuilding trust in science is a continuous endeavour that will require efforts from the scientific community, science writers and the public.
THERAPEUTICS
30 Exploring the Limb Girdle Muscular Dystrophy Clinical Trial Landscape
With the recent advent of adeno-associated, virus-based gene therapy treatments, limb girdle muscular dystrophy (LGMD) is currently attracting the attention of the biopharmaceutical industry, especially with the goal of restoring full or partial proteins that are otherwise dysregulated. The diagnosis of limb girdle muscular dystrophy is often challenging because of significant disease heterogeneity. Raymond A. Huml of IQVIA, et al, discusses that with further LGMD feasibility analyses, we will gain a better grasp on investigator interest and experience with the potential to allow all patients with LGMD, regardless of subtype or geography – to enrol in a clinical trial to explore products that could provide desperately needed disease-modifying treatments or a cure.
LOGISTICS AND SUPPLY CHAIN
38 Shipping Clinical Trials, Robustly and Reliably
According to recent forecasts by Fortune Business Insights, the global clinical trials market is projected to reach nearly $100 billion in 2030. That represents huge growth yet achieving it simply won’t be possible without sensible investment across the supply chain. When it comes to the transportation of clinical trial samples, the industry faces the same problems as the wider pharmaceutical market, but further intensified and complicated by the very nature of clinical trials. Nick Gilmore at Tower Cold Chain analyses why it is important to understand both the similarities with the pharmaceutical cold chain, and the very different dynamics involved in transporting clinical trial samples, to evolve a robust and reliable system for shipping samples around the world.
2 Journal for Clinical Studies Volume 15 Issue 1 Contents


The clinical trial industry is entering a new era of possibilities in 2023. With the rise of transformative technologies and process automation in the market, there are more opportunities than ever to drive efficiencies across the clinical trial lifecycle. From hybrid trials to more sustainable practices at sites, how will these developments shape the landscape for clinical research in the future?
Clinical trials play a crucial role in the development and approval of new medical treatments and drugs. As technology continues to advance, Clinical Trial Technology is becoming a more important factor in shaping the way trials are conducted. We can expect to see several technology trends that are set to further revolutionize the design, conduct, and analysis of trials. Some of these trends include decentralized trials, wearable devices, machine learning, and RiskBased Quality Management (RBQM).
Over time, the clinical trial industry has witnessed an increasing number of eClinical technology tools. Across different phases of the life cycle, solutions have been introduced to solve meaningful business problems; yet it is solution interoperability and workflow integration that can streamline trial operations, promote trust across stakeholders, and ultimately reduce white space.
AI and Machine learning are becoming increasingly important in clinical trials, as they provide a way to analyse large amounts of data and make predictions about outcomes. From identifying potential trial participants to predicting patient responses to treatment, Machine Learning can help make clinical trials more efficient and effective. Additionally, these technologies can help automate routine tasks, freeing up time and resources that can be used to focus on other aspects of the trial. Some of the AI & ML applications in clinical trials include eProtocol Design, eCRF Design, Medical Coding, DB Creation, Data Analytics, CSR Automation, SDV, Site Selection, SDTM Mapping, RBM, Query Management and Chatbots.
The clinical trial industry has always been reliant on data and insights to improve the drug development process. However, this year the industry is expected to become even more focused on harnessing big data and applied data science to optimise financial planning, increase predictability, and proactively pinpoint areas of risk.
JCS – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Principal Content Writer, Clarivate
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organization (WHO) Expert in ethics
• Georg Mathis, Founder and Managing Director, Appletree AG
Risk-based monitoring (RBM) is becoming increasingly popular in clinical trials, as it allows for a targeted and efficient approach to monitor trial data. However, RBQM (Risk-Based Quality Management) takes this a step further, incorporating a risk-based approach into all aspects of trial management, from protocol development to data management and analysis. RBQM ensures that resources are targeted where they are needed most, resulting in a more efficient and effective clinical trial process.
In 2023, the mission to introduce further sustainability in clinical trials will increase. Advancements in technologies will help connect and simplify workflows, enabling industry stakeholders to reduce resource leakage, while improving the accuracy and efficacy of their research outcomes. The use of remote patient monitoring systems and virtual technology will allow for more accessible data collection.
Welcome to the summer issue of JCS. We have a huge selection of exciting articles for you.
Rajesh Pothula at Clinion shows the top 5 innovations shaping clinical trials in 2023. Katrin Haeussler, Matthias Hunger & Nathan Green at ICON discuss a novel method, that uses individual patient data to adjust for differences in effect modifiers between studies.
An exciting article by Prof. Frank Sullivan at Whyze Health explains how to improve access and participation in clinical trials in Ireland, and Marc Gabriel at Veeva Systems talks about how Lilly is streamlining regulatory operations.
In an era of sensationalist journalism, but rapidly evolving information and ever-burgeoning scientific innovations, it can be disheartening to witness the persistent lack of trust in the scientific process and establishment. Lucas Riordan, a rising Junior at University of Delaware and esteemed author Henry Riordan within the article “Dispelling Doubt: Effective Scientific Journalism in a Time of Polarisation” discusses how rebuilding trust in science is a continuous endeavour that will require efforts from the scientific community, science writers and the public.
I hope you all enjoy this edition, and I look forward to coming to you in September, with another array of topics to keep you updated in this extraordinary field of work.
Beatriz Romao, Editorial Manager, Journal for Clinical Studies

• Hermann Schulz, MD, Founder, PresseKontext
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, AsiaPacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
4 Journal for Clinical Studies Volume 15 Issue 1 Foreword
Ramus Corporate Group
is a union between Ramus Medical, Medical Diagnostic Laboratory Ramus and Medical Centre Ramus. All the companies are situated in the Ramus building in Sofia, Bulgaria. They are certified in compliance with the requirements of ISO 9001:2015.
Ramus Medical is full service CRO, working CTs in a variety of therapeutic areas and medical device.
• Medical writing for drugs and devices
• Scientific review of documentation
• Clinical trial management
• Monitoring
• Data management
• Regulatory advising and services during clinical trial

Medical Diagnostic Laboratory Ramus (SMDL-Ramus)
• 30 clinical laboratories in Bulgaria and North Macedonia
• 325 affiliates for sampling in Bulgaria and North Macedonia
• More than 20 years’ experience in the CT field as central and safety laboratory;
• Largest PCR laboratory in Bulgaria

• Laboratory System integrates cluster generation, sequencing, and data analysis
• Total laboratory automation with Abbott GLP-System
• Bioanalytical laboratory – ISO/IEC 17025:2017 accredited
Medical Centre Ramus with Phase I Unit
• PK/PD studies
• Medical devices investigations
• Phase I–IV
• Non-interventional studies
Others:
• Readability user testing
• Bridging report
• Carriage and storage of dangerous goods in compliance with ADR principles
Medical Diagnostic Laboratory Ramus Ltd
26 Kapitan Dimitar Spisarevski Street, 1592 Sofia, Bulgaria
Tel/Fax: +359 2 944 82 06 www.ramuslab.com email: info@ramuslab.com
Ramus Medical Ltd


26 Kapitan Dimitar Spisarevski Street, 1592 Sofia, Bulgaria
Tel./Fax: +359 2 841 23 69 www.ramusmedical.com email: office@ramusmedical.com
Mihaylov Marketing Director

Corporate Profile
Journal for Clinical Studies 5
! S ct a e f r e r , o f c ast, to V i e C r o e Tut
Dimitar
www.journalforclinicalstudies.com
Clinical Trial Technology Trends: The Top 5 Innovations Shaping Clinical Trials in 2023
Expansion of Decentralised Trial Models
Clinical trials play a crucial role in the development and approval of new medical treatments and drugs. As technology continues to advance, Clinical Trial Technology is becoming a more important factor in shaping the way trials are conducted. In 2023, we can expect to see several technology trends that are set to further revolutionise the design, conduct, and analysis of trials. Some of these trends include:
• Growing Use of Machine Learning and AI

• Expansion of Decentralized Trial Models

• Adoption of Digital Patient Engagement Tools
• Rise of Wearable Devices and Sensors
• RBM to RBQM: A Step Forward in Clinical Trials
Growing Use of Machine Learning and AI
AI and Machine learning are becoming increasingly important in clinical trials, as they provide a way to analyse large amounts of data and make predictions about outcomes. From identifying potential trial participants to predicting patient responses to treatment, Machine Learning can help make clinical trials more efficient and effective. Additionally, these technologies can help automate routine tasks, freeing up time and resources that can be used to focus on other aspects of the trial. Some of the AI & ML applications in clinical trials include eProtocol Design, eCRF Design, Medical Coding, DB Creation, Data Analytics, CSR Automation, SDV, Site Selection, SDTM Mapping, RBM, Query Management and Chatbots.To take advantage of these benefits, CROs and Pharma companies should consider partnering with technology companies that specialise in Machine Learning and developing or integrating Machine Learning algorithms into their clinical trials.
Decentralised trials, also known as remote trials, are changing the way clinical trials are being conducted by allowing for more flexible and patient-centred study designs that facilitate the collection of data from patients remotely in real-time. By using technologies like telemedicine, remote monitoring, and electronic consent, patients can participate in clinical trials from the comfort of their own homes, without the need for frequent visits to a physical trial site. This not only makes it easier for patients to participate in trials, but it also enables trials to be conducted more efficiently, with fewer missed visits and less time and money spent on travel and site visits. CROs and Pharma companies can leverage decentralised trials to increase patient engagement and participation, streamline trial processes, and reduce costs. By investing in telemedicine and remote monitoring technologies, they can help ensure that trials are conducted as efficiently and effectively as possible, while still providing effective patient care and safety.
Adoption of Digital Patient Engagement Tools
Clinical trials have greatly evolved with the integration of digital patient engagement tools. These tools have proven to be invaluable in enhancing the patient experience while simultaneously providing valuable insights into patient behaviour and their experiences. Not only do they increase patient engagement and satisfaction, but they also simplify trial processes and promote better compliance and patient retention. To fully harness the potential of these advanced engagement tools, pharmaceutical companies and CROs should consider collaborating with technology firms that are experts in digital patient engagement solutions. It’s also important for these companies to prioritise user-friendliness and engagement when developing or selecting these tools, so that patients are eager to participate and provide valuable data.

6 Journal for Clinical Studies Volume 15 Issue 1
Watch Pages
Rise of Wearable Devices and Sensors

Wearable devices, such as fitness trackers and smartwatches, are becoming increasingly important in clinical trials, as they provide a way to collect real-time data on patient behaviour and health. From tracking physical activity to monitoring vital signs, wearable devices can help researchers gather valuable data that can be used to improve the efficacy and safety of new medical treatments. To leverage the potential of wearable devices, CROs and pharmaceutical firms may consider partnering with wearable device manufacturers or integrating wearable device technology into their clinical trial.
RBM to RBQM: A Step Forward in Clinical Trials


Risk-based monitoring (RBM) is becoming increasingly popular in clinical trials, as it allows for a targeted and efficient approach to monitor trial data. However, RBQM (Risk-Based Quality Management) takes this a step further, incorporating a risk-based approach into all aspects of trial management, from protocol development to data management and analysis. RBQM ensures that resources are targeted where they are needed most, resulting in a more efficient and effective clinical trial process.
Rajesh Pothula is a seasoned product marketing manager with a wealth of experience in the eClinical platform industry. He currently works at Clinion, a leading provider of innovative clinical trial management solutions. Rajesh has a deep understanding of the challenges faced by sponsors, CROs, and clinical research professionals, and is committed to helping them overcome these challenges through effective marketing strategies and solutions.

Journal for Clinical Studies 7 www.journalforclinicalstudies.com Watch Pages
Rajesh Pothula
Enhancing Healthcare Through Improved Patient Labeling
When patients receive a prescription drug or biological product in an outpatient setting, it may be accompanied by one or more types of written information approved by the US Food and Drug Administration (FDA). The quality of that information, known as the patient labelling, plays a decisive role in whether patients take their treatment properly – or at all.
“Studies have found that the current system for written information for prescription drugs and certain biological products can be confusing, conflicting, incomplete, or repetitive,” Robert M. Califf, MD, MACC, the Commissioner of Food and Drugs, said in a statement issued in May 2023.1 He noted that difficulty in comprehending information can cause patients to become frustrated, stop taking their medications, or not take them as directed. Medication nonadherence, Califf said, “can contribute to nearly 25% of hospital admissions, 50% of treatment failures, and approximately 125,000 deaths” yearly in the US.
Labelling for prescription medicines is the FDA’s primary tool for communicating drug information to healthcare professionals and to patients and their caregivers. The three most common types of FDA-approved patient labelling are patient package inserts, Medication Guides, and Instructions for Use. These documents are proposed by the sponsor and reviewed and approved by the FDA, and their content is based on the Prescribing Information.
With the intent to “improve health outcomes,” the FDA has proposed to require Patient Medication Information (PMI),2 a new type of Medication Guide for prescription drugs and certain biological products (both brand name and generic) that are used, dispensed, or administered on an outpatient basis, as well as for blood and blood components transfused in an outpatient setting.
As paper handouts that accompany many prescription medicines, Medication Guides address issues that are specific to particular drugs and drug classes, and they contain FDA-approved information that can help patients avoid serious adverse events (SAEs). The agency might require that Medication Guides be issued with certain prescribed drugs and biological products when it determines that 1) specific information is necessary to prevent SAEs, 2) patient decision-making should be informed by information about a known serious risk or risks, or 3) patient adherence to directions for the use of a product are essential to its effectiveness. The FDA has a Medication Guides database3 that houses these documents (but excludes FDA-approved allergenic or cellular/tissue products).
In the May 2023 statement, Califf noted that PMI "would provide patients with clear, concise, accessible, and useful written information for prescription drugs and certain biological products and would be delivered in a consistent and easy-to-understand format" to help patients use these products safely and effectively. PMI would consist of one-page documents highlighting critical information that patients should know in a standardised format, including the drug/biological product name, a concise summary of the indications and uses, important safety information, common
side effects, and directions for use. This information would be provided along with the medication in person and available online for public access. The FDA has provided an example of PMI for a fictitious drug, Rheutopia (see below).

The Proposed Rule
To implement use of PMI, the FDA has proposed to amend its human prescription drug product labelling regulations for Medication Guides. As outlined in the Federal Register4 announcement of the proposed rule, the FDA is intending to revise the part heading and all subparts of the current regulation under 21 Code of Federal Regulations (CFR) part 208.5 The heading part would be revised to “Medication Guides” (from “Medication Guides for Prescription Drug Products”). For the purposes of the proposed rule, a drug product also includes a biological product licensed under section 351(a) or (k) of the Public Health Service (PHS) Act.
Medication Guides are a potential element of a risk evaluation and mitigation strategy (REMS), as explained in the FDA’s final Guidance: Medication Guides – Distribution Requirements and Inclusion in Risk Evaluation and Mitigation Strategies (REMS), 6 from November 2011. Per section 505-1 of the Federal Food, Drug, and Cosmetic Act (FD&C Act), the FDA is authorized to require a REMS when necessary to ensure that the benefits of a drug outweigh the risks. Under the proposed rule, Medication Guides would continue to be available as a possible REMS component until the FDA has approved PMI for the prescription drug product.
8 Journal for Clinical Studies Volume 15 Issue 1 Watch Pages
According to the FDA, “it is important that patients continue receiving FDA-approved patient information for prescription drug products that we previously determined posed a serious and significant public health concern during the implementation of the final rule.” Once a prescription drug product has FDA-approved PMI, its current Medication Guide requirement (if any) would no longer be applicable. The agency would withdraw the current regulations governing Medication Guides in 21 CFR part 208 after all prescription drug products that had Medication Guides have FDA-approved PMI.
Provisions of PMI
The FDA has proposed a 5-year implementation schedule for PMI. Under the proposed rule, once it is finalized, applicants of all new and approved new drug applications (NDAs) and biologics license applications (BLAs) would be required to create PMI for prescription drug products that are to be used, dispensed, or administered on an outpatient basis. These PMI would need to be submitted to the FDA for approval. It would also require applicants of new and approved abbreviated NDAs (ANDAs) that refer to a listed drug for which the FDA has approved PMI to have PMI that is the same as that of the reference listed drug (RLD) apart from certain differences in labelling permitted under the law. Among the many requirements and details regarding the proposed rule are the following:
• PMI would be stored in an online central repository managed by the FDA.
• Authorised dispensers would be required to provide PMI to patients each time a prescription drug product for which an FDA-approved PMI exists is used, dispensed, or administered

on an outpatient basis. The default method of distribution for PMI would be paper form.
• Electronic dissemination of PMI instead of the paper form would be permitted upon a patient’s request and would accommodate future technological advances in the methods used to provide PMI upon a patient’s request.
• PMI would be available for distribution to transfusion services of blood and blood components unless a waiver applies.
• PMI for NDAs and BLAs would need to be updated when new information becomes available that would cause PMI to become inaccurate, false, or misleading.
In the FDA’s view, the various approaches taken will assist with improving access to medication information for all patients who may be dispensed a prescription drug product in an outpatient setting. The agency is seeking input “on whether the proposed format and content requirements support the accessibility of patient medication information for all intended users, including patients with low health literacy.” Comments on the proposed rule may be submitted to Docket No. FDA-2019-N-5959 by November 27, 2023.

REFERENCES
1. FDA Statement. FDA Proposes New, Easy-to-Read Medication Guide for Patients, Patient Medication Information. Food and Drug Administration webpage. May 30, 2023. https://www.fda.gov/news-events/pressannouncements/fda-proposes-new-easy-read-medication-guide-patientspatient-medication-information
2. Patient Medication Information. Food and Drug Administration webpage. https://www.fda.gov/drugs/fdas-labeling-resources-human-prescriptiondrugs/patient-medication-information
3. Medication Guides. Food and Drug Administration webpage. https://www. accessdata.fda.gov/scripts/cder/daf/index.cfm?event=medguide.page
4. Medication Guides: Patient Medication Information. Federal Register, May 31, 2023. https://www.federalregister.gov/documents/2023/05/31/2023-11354/ medication-guides-patient-medication-information
5. 21 CFR part 208. https://www.ecfr.gov/current/title-21/chapter-I/ subchapter-C/part-208
6. Guidance: Medication Guides—Distribution Requirements and Inclusion in Risk Evaluation and Mitigation Strategies (REMS), November 2011. Food and Drug Administration. https://www.fda.gov/media/79776/download
Deborah Komlos, MS, is a Principal Content Editor for the Cortellis suite of life science intelligence solutions at Clarivate. In this role, her coverage centres on FDA advisory committee meetings, workshops, and product approvals. Her previous positions have included writing and editing for magazines, newspapers, online venues, and scientific journals, as well as publication layout and graphic design work.
Email: deborah.komlos@clarivate.com

Journal for Clinical Studies 9 www.journalforclinicalstudies.com Watch
Pages
Deborah Komlos
Comparing Treatment Performance through Evidence Synthesis A Solution for Sparse Evidence, Heterogeneous Studies, and Disconnected Networks
If there is a variety of treatment options in a specific disease area, decision makers require evidence of efficacy and safety of novel interventions in comparison to established treatments. One randomised controlled trial (RCT) is not sufficient to come to a final conclusion, especially since RCTs often present contradictory results.

In certain situations, it is possible to synthesise existing evidence from multiple studies to calculate a pooled treatment effect and thus demonstrate the comparative performance of a novel treatment against established interventions. Standard models for evidence synthesis work well when there is a large evidence base, the absence of any effect modifiers, and connected networks of studies have a common comparator linking two interventions of interest. However, even when there are no direct, head-to-head comparisons available, it is nonetheless possible to estimate effects through indirect comparisons using specific methods of evidence synthesis. We’ll explore these options in this article.
Evidence Synthesis – An Overview
Evidence synthesis implies using classical frequentist or Bayesian statistical methodology to estimate a pooled treatment effect (for example an odds ratio or a risk difference) to demonstrate the comparative performance of a novel treatment against established interventions. To ensure that all relevant evidence is identified, prior to conducting evidence synthesis, a systematic literature review (SLR) has to be conducted. As a next step, all RCTs of high quality are pooled. If head-to-head studies comparing an experimental intervention to the same comparator are available, the simplest approach to evidence synthesis is a so-called pairwise meta-analysis. In the absence of headto-head studies, more complex approaches are used, such as network meta-analysis (NMA) based on generalised linear models.
When assessing the feasibility of indirect treatment comparisons, as a first step, a so-called network of evidence is drawn (see Figure 1). This is to assess data availability per outcome of interest and to investigate the presence of a common comparator. Each intervention is shown as a node, whereas links between nodes are shown through lines (dotted for indirect comparisons).
Standard methodology for NMA works well if there is a large evidence base, if the network of studies is connected through common comparators linking the interventions of interest, and in the absence of any effect modifiers, i.e. variables that alter the effect of treatment on outcomes.
There are, however, a variety of situations in which these analytical methods may not be sufficient, such as when:
• There is a sparse network of evidence such that fewer than five studies inform one outcome, or that only one study informs each direct comparison of treatments in the network of studies. This is especially an issue if Bayesian methods with non-informative priors on between-study standard deviation parameters are used. A non-informative prior implies that nothing is known about the parameter in advance, and a dynamic process of learning from the data is conducted. If data are sparse, there is not enough information to update the prior into the posterior
• A large amount of heterogeneity exists between the studies
• The network is disconnected – in other words when there is no common comparator linking two interventions of interest
Prior to conducting evidence synthesis, the feasibility of pooling the data has to be assessed. The main aim is to evaluate whether there are issues with an excessive amount of heterogeneity, which would be a violation of the similarity assumption.

Performing an Evidence Synthesis Feasibility Assessment
The key assumption for indirect treatment comparisons is the socalled “similarity assumption”. This implies that one would expect the relative effect of treatment A vs. placebo to remain unchanged if the study was conducted under the conditions of the treatment B vs. placebo study (and vice versa). Similarity means that the pairs of trials analysed are comparable regarding potential treatment effect modifiers.

When the patients in all the trials match the target population for the treatment decision, the data can be pooled. If, on the other hand, there is a match between only a subset of patients in the studies, one must consider whether there are also differences in the treatment effect, applying a decision tree as illustrated in Figure 2.
There are several ways to address treatment effect modifiers, such as excluding studies with outlier data, conducting subgroup
10 Journal for Clinical Studies Volume 15 Issue 1 Regulatory
Figure 1: Indirect comparisons of treatments A and B through a common comparator placebo
analyses on specific populations, incorporating relevant covariates into meta-regression models, or using matching-adjusted indirect comparison (MAIC) or multi-level network meta-regression (MLNMR).
Standard, Sparse and Disconnected Networks of Evidence

To perform a standard Bayesian NMA using non-informative priors, it is ideal to have a large network of evidence incorporating several studies for each link between the interventions, as seen in the upper left corner of Figure 3. Otherwise, the results could become unrealistic, such as incorporating a large amount of uncertainty in the credible intervals. Using frequentist methods, sparse networks as shown in the upper right corner of Figure 3 do not have such a large effect on uncertainty in results.
At times, the network is disconnected (meaning that one or several studies or a subnetwork of studies cannot be linked to the main network of evidence) as shown in the bottom right corner. In such situations, matching-adjusted indirect comparison (MAIC)
could be a valid alternative to conducting an indirect comparison of the interventions of interest. In addition, if effect modifiers are present in the data, MAIC or multi-level network meta-regression (ML-NMR) could be used instead of standard NMA methods to account for these.
Choosing Suitable Priors
In standard approaches to Bayesian NMA, non-informative prior distributions are assigned to the parameter on the between-study standard deviation. When a large volume of data can be used to update these non-informative priors into the posteriors, the result is realistic posteriors and as a consequence robust findings of the NMA (with realistically wide credible intervals). If, however, the amount of available study data in the network of evidence is sparse, updating the priors into the posteriors generates a high amount of uncertainty in the analysis.
There are various approaches to prior elicitation, such as from the literature or expert clinician opinion. A suitable prior can be

Journal for Clinical Studies 11 www.journalforclinicalstudies.com Regulatory
Figure 2: Determining the feasibility of conducting an NMA
Figure 3: The standard network versus sparse and disconnected networks of evidence
identified through posterior predictive checks, which evaluate the model fit by comparing simulated data to the actual study data.
Matching-Adjusted Indirect Comparisons
MAICs can be used in both connected (with common comparator) and disconnected networks (see Figure 4). In an anchored comparison (a comparison where there is a common comparator), they can be especially helpful to adjust for bias due to the presence of effect modifiers. MAICs can also remove some of the biases that unadjusted (naïve) direct comparisons of outcomes can have when a common comparator arm is missing (an unanchored comparison). (See Figure 4.)
heterogeneity parameters in a Bayesian NMA represents a valid alternative to the use of non-informative priors in the standard case. For disconnected networks, MAICs are often used to still enable indirect comparisons, adjusting for effect modifying and population imbalance. These can also be used in connected networks accounting for a large amount of heterogeneity. Finally, ML-NMR is a novel method and a direct extension of the standard network meta-analysis framework that uses individual patient data to adjust for differences in effect modifiers between studies.
Typically, MAICs are employed using individual patient data from one clinical trial for the company’s own treatment and using published, aggregate data on baseline characteristics and the outcomes of interest for the comparator treatment. MAICs match the baseline characteristics (including prognostic factors and/or effect modifiers) from Study A for which there is individual patient data to the same characteristics from Study B for which there is only aggregate data. This is done by weighting the individual patient data.
Multi-Level Network Meta-Regression (ML-NMR)

The MAIC method is not practical for large networks of trials, nor is it able to achieve estimates in a population other than that of the aggregate study. Thus, there remains a need to synthesise data from large treatment networks with an arbitrary population (not constrained by the aggregate study) while also avoiding aggregation bias.
Multi-level network meta-regression (ML-NMR) is a possible solution. The ML-NMR method embeds a probabilistic approach to population adjustments within the aggregate NMA model. It synthesises mixtures of individual patient data and aggregated data to perform a population adjustment in networks of any size. Fundamentally, ML-NMR consists of two steps:
1. Define a regression model on the individual patient data
2. Form an aggregate-level model by averaging (or integrating) the aggregate study population
ML-NMR devises a relationship between the input covariates and the outcome, which simultaneously incorporates the information from both the individual patient data and the aggregate data.
Demonstrating the comparative efficacy and safety of a novel treatment and established interventions is not always straightforward, depending on data availability. If the network of evidence is sparse, using informative priors on between-study


Katrin Haeussler
Katrin Haeussler, MSc, PhD, Senior Health Economist, ICON, works in the area of evidence synthesis, conducting analyses in both Bayesian and frequentist settings. She is experienced in implementing network metaanalysis models in R and SAS, and scientific writing. Therapeutic area experience includes chronic obstructive pulmonary disease, breast cancer, hereditary angioedema, diabetes and herpes zoster.

Matthias Hunger
Matthias Hunger, MSc, Principal, ICON, has more than 10 years of experience in health economics and outcomes research. At ICON, his work includes database studies and posthoc clinical trial analyses, and he was the Lead Statistician in numerous matching-adjusted indirect comparisons and external control arm projects. He has extensive knowledge in the analysis of patient level data in SAS or R, including statistical analyses of health-related quality of life, utility, healthcare cost or survival data. Matthias has experience in various disease areas including cancer, diabetes and psoriatic arthritis.

Nathan Green
Nathan Green, PhD, Senior Research Fellow, University College London, has a number of years of experience working on a wide range of projects across government and academia in defence and health. He currently works in the Department of Statistical Science at UCL. His research interests focus on Bayesian statistical modelling, including cost-effectiveness analysis, survival analysis and decision-theoretic approaches.
12 Journal for Clinical Studies Volume 15 Issue 1 Regulatory
Figure 4: Treatments of interest without a common comparator
The Role of LIMS in Supporting ISO 17025 Accreditation
ISO/IEC17025:20171 is the international standard that specifies the general requirements for the competence, impartiality and consistent operation of laboratories. It can be used by laboratory customers, accreditation bodies, and other organizations to recognize the competence of laboratories. The key element of the standard is that it requires laboratories to demonstrate that they operate competently and can generate valid results. However, the latest version of the standard (2017) does not prescriptively state how this should be done; rather it encourages a risk-based approach. This requires organizations to assess the risks associated with the provisions of the standard and to show how the identified risks have been minimized. Key to this is the management of the laboratory process, the management of laboratory resources and the management of data that exists in, or is created by, the laboratory.

This white paper will explore the ways in which an integrated Laboratory Information Management System (LIMS) can play a key role in achieving, maintaining, and benefiting from ISO17025 accreditation.
Managing Sample Handling and Testing Processes
In some circles there is still an impression LIMS focus on the management of samples, tests, and results with, potentially, some workflow management functionality. However, ISO17025 contains surprisingly little on this aspect of the laboratory operation. Section 7.5 (Technical records) is short; it states that technical records shall include the date they were taken, the person responsible for the recording, any calculations involved in arriving at the results and the identity of the person checking the data and results. It also goes on to say that any amendment to those technical records needs to be checked as well. All of this is basic LIMS functionality. As far as supporting ISO 17025 it is more interesting to look at other sections of the standard and how an integrated fully functional LIMS supports these.
Resources Requirements
Section 6 of ISO17025 provides considerable details on the management of laboratory resources. It states specifically that the laboratory shall have available the personnel, facilities, equipment, systems, and support services necessary to manage and perform its laboratory activities. A fully integrated LIMS has a key role in helping the laboratory meet the requirements for this as detailed in a number of the subsections to Section 6.
A.2 Personnel (Section 6.2)
Section 6.2 covers personnel and, in particular, staff competency and training. As an example of the risk-based approach, the standard stipulates that the laboratory must document competency requirements for functions that influence the results of the laboratory activities. However, it does not state in exact detail what these functions are, although it does go on to state that personnel must have the competence to perform the activities they are responsible for. While staff competency and training may initially be perceived as outside the scope of LIMS, this is far from the case. An integrated
LIMS will have the ability to manage staff training and competency records, including the scheduling of training and retraining activities. To take this further, however, the LIMS must be able to link these records to specific activities; preventing personnel not trained or certified in an activity or task from carrying it out. A common example is competency to carry out specific tests or methods within the lab. The system must be able to check the competency of the user at the time that they are performing the analysis. If they do not have the required training or competency, or if it has lapsed, they will be prevented from entering results into the LIMS. The same principle can be applied to other activities such as instrument maintenance and calibration.
A.1 Facilities and Environmental Conditions (Section 6.3)
At first sight it might seem unlikely that LIMS could have a major part to play in helping organizations with the management of their facilities and environmental conditions within the context of ISO17025. When it is understood that this covers the monitoring, control and recording of environmental conditions as required, including where environmental conditions may influence the validity of results, the role of LIMS becomes clearer. However, this role can extend beyond the basic recording of results of environmental monitoring, to the management of the monitoring itself.
Journal for Clinical Studies 13 www.journalforclinicalstudies.com
Regulatory
An integrated environmental monitoring function will allow sampling locations within the facilities to be defined and mapped. A sampling schedule may also be defined based on the frequency of sampling and the type of testing needed for those locations. If testing gives a result that exceeds the specified contamination limit, that result can be flagged as such. Results can also be tracked and charted over time to show if there are any significant data trends, even if limits have not been exceeded. This type of functionality is particularly useful for tracking microbial or particulate contamination within clean environments.


A.3 Equipment (Section 6.4)
The management of equipment in the laboratory is a key aspect of ISO17025. Not surprisingly, maintenance and calibration plans, and the ability to prevent equipment being used if it has not been serviced, is not in calibration or is out of service for some other reason, is emphasized. An integrated LIMS will have an instrument management system that allows maintenance and calibration plans to be defined, managed, and enforced. These plans cover maintenance and calibration requirements, as well as frequency, and allow the results of all calibration and maintenance events
14 Journal for Clinical Studies Volume 15 Issue 1
Example of Tracking Staff Competency in Matrix Gemini LIMS
Example of Environmental Monitoring in Matrix Gemini LIMS
Regulatory
to be recorded. The maintenance and calibration history of any equipment can therefore be fully tracked. Equipment status can be checked at the time of use and, in the same way that an analyst can be prevented from recording results for a test if their certification is not current, the use of the instrument can be prevented if the status is not correct.

ISO 17025 also defines standards, reference materials, reference data, reagents, and consumables as equipment. Here the inventory management functionality of LIMS comes into play. Inventory
Example of Instrument Calibration and Maintenance in Matrix Gemini LIMS management will allow the receipt of equipment of this type to be recorded together with other key data such as the supplier, amount, use by dates and Certificates of Analysis. Data such as the certified values of standards can also be recorded. Amounts (or stock levels) can be managed and inventory used for specific purposes can be recorded, for example the consumables and reagents used as part of an analytical run. Linking inventory to specific tasks in this way also allows for the amount used to be automatically decremented from the available stock, allowing stock levels to be monitored and stock to be reordered when it falls below defined levels.

Journal for Clinical Studies 15 www.journalforclinicalstudies.com
Regulatory
Example of Tests Dictated by Chosen Substance in Matrix Gemini LIMS
Process Requirements
It is clear that ISO17025 requires proper management of the resources within the organization, but the processes within the organization are as important. Processes are covered in section 7 of the standard and again provides an opportunity for LIMS to prove its value by supporting process definition and enforcement.
B.1 Review of Requests, Tenders, and Contracts (Section 7.1)
Section 7.1 covers the review of requests logged by customers, which may be required before work can be started. A LIMS can prevent the processing of a request until it has been reviewed and approved by a qualified person. As systems become more open through the use, for example, of customer portals that allow them to log requests on-line, this type of review is becoming more important to ensure that the customer has asked for something which is possible and covered by any contract that is in place.
B.2 Selection of Methods (Section 7.2)
In Section 7.2 emphasis is placed on the selection and verification of the appropriate tests to be used. LIMS can play a big role in ensuring that this requirement can be met by, for example, automatically assigning methods and tests based on the material (or substance) under test, or other factors such as the submitter of the testing request. The system ensures that the latest version of the method is applied and, if appropriate, relevant limits are assigned to check the validity of results. Where validation of methods is required projects can be set up within LIMS to help manage the process and record the results of the validation process.

B.3 Sampling (Section 7.3)
Sampling, and the way that sampling is carried out, is a key element in ensuring consistency of operation within a calibration or testing laboratory. As well as helping to define sampling plans or interfacing with complex sample planning software such as used in the UK water industry, LIMS records and retains the sampling data that can form part of the testing or calibration that the laboratory does, as defined in section 7.3 of the standard. This can include information such as date and time of sampling, id of the person who carried out the sampling as well as recordings of any deviations from the defined sampling plan. The LIMS will, of course, also provide unique identification for the samples. It is important however that the LIMS is flexible enough to support different information for different types of samples and, in addition, easily supports the needs of new sampling requirements that may be required.
B.4 Measurement Uncertainty and Validity of Results
(Section 7.6 & 7.7)
Measurement uncertainty is a complex area, especially for calibration laboratories, and again one where LIMS can have a role to play. Where a test method used by a testing laboratory specifies limits these can be applied automatically by the LIMS at the time of result entry. More complex calculations can be implemented where these types of limits are not applicable. For example, measurement uncertainty for a run, or batch, of samples that includes QA/QC samples (controls, spikes, duplicates, replicates etc.) can be determined from the results recorded for the run. Linked to this is ensuring the validity of the results where many other aspects of LIMS functionality have a role to play. Reference materials can be managed and tracked using inventory management functionality, calibration of instruments can be tracked using instrument calibration and maintenance options, retesting and replicate testing can be controlled, and control charts produced as required. Reporting functions allow the review of reported results. Correlation of results for characteristics of an item across different batches is possible because the required data is all in the same place.
B.5 Reporting of Results (Section 7.8)
Reporting is a vital part of any testing or calibration laboratory as it is the point at which the product of the laboratory, that is the data and information produced, is delivered to the consumer or customer of that product. Clearly it is essential that the correct information is delivered and reports, in whatever format, must include all the information agreed with the customer. The ISO 17025 section on reporting (7.8) also requires results to be reviewed and authorized prior to their release. LIMS supports the review and authorization steps required, so that reports cannot be issued until reviewed or authorized by an approved user. Flexible reporting options allow the creation and management of customer-specific reports which contain all the required information in the format or formats required for specific substances under test. With all the information required coming from the LIMS, reports can be automatically generated and made available for review once the results of the testing have been approved. This speeds report creation and review, and therefore delivery of the results to the customers.
B.6 Managing Complaints and Non-conforming Work (Section 7.9 & 7.10)
Sections 7.9 and 7.10 refer to complaints and nonconforming work. Complaints must be managed and tracked, and procedures must be in place for handling any nonconforming work that is identified. Complaints and non-conformances can be managed within a LIMS provided it has an integrated Corrective Action, Preventive Action (CAPA) management facility. Such a facility allows the tracking and management of the CAPA from the time it is created, through to the time that it is resolved. The actions associated with the CAPA are recorded together with the corrective actions identified and implemented allowing full traceability of the CAPA process.
16 Journal for Clinical Studies Volume 15 Issue 1
Regulatory
B.7 Control of Data and Information Management (Section 7.11)

Section 7.11 Control of data and information management is not especially long but it may have far reaching consequences. It firstly states that the laboratory shall have access to the data and information needed to perform laboratory activities. While this may seem like a statement of the obvious it is perhaps worthwhile asking how easy it is for your lab staff to access the data and information they require to do their jobs. Can an analyst easily access what can be proven to be the latest version of a Standard Operating Procedure to ensure they are carrying out a technique correctly? How easy is it to check the calibration or maintenance status of a specific piece of equipment that is needed particularly if, for example, is it located in a different facility or building? How simple is it to check there is enough reagent in stock to carry out a specific technique, is it still within its use by date and where is it? All of these are part of the data set that a laboratory must be able to access in order to function, in addition to all the information about customers, requests, contracts, training, environment and technical records. By implementing an integrated LIMS this data can be brought into a single place.
Another key aspect of 7.11 is the concept of validation2 of the laboratory information management system (or systems) that are in use. It must be remembered that laboratory information management systems may be computerized or non-computerized systems, meaning that paper records count. The pharmaceutical industry has had to face the challenge of validation for many years and experience has shown that paper-based systems, and even spreadsheet-based systems, can be difficult to validate. LIMS are well suited to validation; integrating information and functionality into a LIMS minimizes the number of different systems that need to be validated and, because of their long history of use in the pharmaceutical industry, many LIMS come with supporting material, such as validation scripts and packs. These are designed to help the process of validating the information management system.
Summary
ISO 17025 at its heart is a quality management system, and while an integrated LIMS will support the quality management system (QMS),
it cannot replace management commitment to making a QMS work. However, investing in and implementing a LIMS provides evidence of management commitment as it supports so many aspects of the QMS. It can help ease the burdens of compliance by showing that the processes and operational requirements defined as part of the QMS are in place and are being adhered to. The LIMS can also reduce audit and inspection overheads by integrating information in a single place. Equally importantly it gives confidence and assurance to customers or collaborators that work is carried out competently and that the data and the information produced is valid. The capabilities of modern integrated LIMS make them a vital support system for successfully achieving, maintaining, and gaining benefit from ISO 17025 accreditation.

REFERENCE
1. ISO/IEC 17025:2017 General requirements for the competence of testing and calibration laboratories https://www.iso.org/standard/66912.html
2. LIMS System Validation White Paper https://www.autoscribeinformatics. com/resources/white-papers/lims-system-validation
Tim Daniels
Tim Daniels has over 30+ years of experience working in national and international markets across a range of software/high-technology products. As worldwide Marketing Manager at Autoscribe Informatics Tim works with product development, technical services, sales, and management teams across the company to drive marketing goals and achieve growth plans. Autoscribe Informatics provides database management solutions such as Laboratory Information Management Systems (LIMS) that, uniquely, are graphically configurable, requiring no scripting or custom coding to configure solutions. Tim‘s broad background in marketing and technical roles provides a unique blend of practical knowledge and insight to drive all aspects of product and corporate marketing at Autoscribe Informatics.
Journal for Clinical Studies 17 www.journalforclinicalstudies.com
Example of CAPA Management in Matrix Gemini LIMS
Regulatory
Improving Access and Participation in Clinical Trials in Ireland, a Patient-centred Digital Health Platform
Across Europe, we continue to see a declining trend in the number of clinical trials taking place, most notably since the European Union Clinical Trials Directive (CTD) was established in 2004. The decline in clinical trials in Ireland is a major concern, as it means that fewer people are given access to potentially life-saving treatments. This can have long-term effects on the quality of care being provided, as well as making it harder for new treatments to be developed and tested. However, the CTD has long-felt ramifications for European patients. The regulatory burden and insurance requirements grew even more burdensome. At the same time, the global COVID-19 pandemic also affected the number of clinical trials, which decreased by 19.6% in Europe between August and October of 2020 compared to 2019. However, the effects of this decrease in access to clinical trials are being felt more severely in countries like Ireland which have far fewer trials than their peers. In this article, we explore the causes of decreased access to ground-breaking trials in Ireland and how to improve the quality of health care to create a healthier, more efficient system that better serves the Irish population.
The Current Landscape of Research in Europe and Home in Ireland
Despite Ireland's significant spend in its healthcare system, diverse population, and heavy inbound investment from the pharma and med-tech sectors, Ireland is not able to keep up with its European counterparts in terms of clinical trial participation. According to the Central Statistics Office (CSO) of Ireland, the foreign direct investment (FDI) in Ireland increased by €109 billion to €1,208 billion in 2021, a large proportion of that coming from the pharmaceutical and med tech sectors. Compared to Finland and Denmark, which both have similar populations and economic wealth, Ireland has only seen 18% of the 2,290 clinical trials conducted between 2013 to 2021 in the three countries. In contrast, Finland and Denmark have respectively seen 29% and 53% of these clinical trials.
Ireland is not alone. The number of clinical trials in the UK has also declined significantly in recent times, with the Association of British Pharmaceutical Industry reporting that the number of industry-backed clinical trials started in the UK each year fell by 41% between 2017 and 2021. This steep drop is due largely to a combination of factors, including slow set-up times, increased staff fatigue and turnover, and a reduction in the NHS’s research capacity. Unfortunately, this means that UK and Ireland are lagging behind many of their European counterparts in attracting clinical trials.
The main barrier for Ireland when it comes to clinical trials is the need for more resources, both in terms of funds and personnel. Clinical trials require a significant amount of time from both researchers and practitioners to be successful. However, due to budgetary constraints, they are often forced to make do with limited personnel or wait for additional funding. Additionally, the infrastructure needed to support clinical trials is often expensive and difficult to obtain.
Another barrier in Ireland is the public's lack of awareness and understanding regarding clinical trials. Many people are unaware of both their purpose and potential benefits, so they do not see them as a worthwhile investment of time and effort. As such, recruitment for clinical trials becomes more difficult as fewer people are willing to participate. Finally, there is the problem of bureaucracy in Ireland when it comes to starting up clinical trials. It can be a lengthy process that requires navigating complex legal structures and regulatory requirements, which can be challenging and time-consuming, and “off-putting” for established healthcare providers.
A further barrier to trials expansion in Ireland is the lack of digitization and coordination in the health records. Patient records are often still paper based, and fragmented and silo’d, which makes the process of identifying suitable trial candidates difficult, time consuming and expensive. There is urgent need to reform Ireland’s digital health landscape.
Overall, the current landscape of research and clinical trials in Ireland is hindered by these various factors. If these barriers are not addressed, it will become increasingly difficult for Ireland to keep up with its European and global counterparts regarding clinical trial participation and access to ground-breaking treatments. Dr Rebecca Cramp has been appointed scientific and regulatory affairs manager with the Irish Pharmaceutical Healthcare Association (IPHA), which represents the international research-based pharmaceutical industry in Ireland. She believes that Ireland should be able to attract more clinical trials given the size of its biopharmaceutical manufacturing footprint. To make this a reality, she believes that further standardisation in the clinical trials space is needed to help Ireland catch up with Finland and Denmark, and improve its ability to host important studies that give people access to life-saving treatments.
Addressing Barriers in the Current Clinical Trial System Bureauocratic Impediments:
To reverse the trend of declining clinical trials in Ireland, the government has taken steps to address the administrative barriers to clinical trial participation in Ireland. One significant improvement is the centralisation of ethics and standardising the clinical trial process. The National Clinical Trials Office (NCTO) was established to facilitate clinical trial research in Ireland. The NCTO provides a centralised infrastructure to support the set-up and management of clinical trials, including a Clinical Trial Management System (CTMS), which allows researchers to track patient recruitment, trial progress, and data management.
Private-public Expansion:
Through the Health Research Board (HRB) infrastructure, Ireland has invested heavily in Clinical Research facilities, generally based in and around the public hospital university system, but penetration in the private healthcare delivery system is patchy at best. Up to one half of the irish population carries health care insurance, and receives some or all of its healthcare in the private system. There is clear scope to expand the pool of patients accessing trials, through measures designed to include all eligible patients, treated in both the private as well as public sectors in Ireland.
18 Journal for Clinical Studies Volume 15 Issue 1 Regulatory WhyzeHealth:
Data Issues:
Finally, there is a clear need to modernise the data infrastructure, to allow for advances in artificial intelligence (AI) and machine learning to bring further increases in access and retention to clinical trials. We will address this issue here.

Changing Clinical Trials in Ireland with Whyze Health

Of crucial importance to the issues discussed in the previous section, a significant barrier to clinical trial participation in Ireland is the lack of patient access to digitised personal health records and control over their own medical data. Patients should have access to their medical records and be able to move from one treatment centre to another and to obtain access to trials. This is particularly important for patients with rare diseases, who may need to travel outside their local area to access the best treatments and trials.
The Whyze Health (WH) digital platform provides a solution to this problem by allowing patients to access their personal medical records and connect with their healthcare providers to learn about new treatments and clinical trials. The platform also enables realtime (real-world) monitoring of patients' conditions, which can help patients and healthcare providers make more informed decisions about treatment options. Furthermore, the platform's digital asset management system encourages long-term patient participation in trials, providing a unique opportunity for healthcare providers to extend their reach and increase their revenue through research and clinical trial involvement.
There is good clinical reasons to promote this activity. Evidence suggests that being treated in a centre where trials are offered is associated with better patient outcomes. Patients who are treated at centres with clinical trials have access to the latest treatments and technologies, which can improve their health outcomes. Better outcomes lead to better value to the overall health sector. Therefore, improving access to clinical trials in Ireland is critical for improving the quality and value of care provided to Irish patients.
The WH platform is designed to unify health and research to advance both knowledge areas. The patient-centered solution encourages patients to connect with their healthcare providers, allowing them access to information about new treatments and clinical trials and real-time monitoring of their conditions. This access is particularly beneficial for those who may not have been previously aware that participation in clinical trials was a viable option, or who had yet to be invited by their healthcare providers. Increasing the quality of the health interventions through clinical trials improves patients’ outcomes. Improved outcomes lead to improved value to the overall system. Improved value through clinical trials leverages the substantial FDI incoming to Ireland through the pharmaceutical and med tech investments.
In addition to providing resources and information to patients, the Whyze platform also provides a unique opportunity for healthcare providers to extend their reach and increase their revenue through research and clinical trial involvement. Healthcare providers can now link their patients more directly to relevant studies and trials, enabling more people to benefit from state-of-the-art treatments.
Real World Evidence Driving Quality and Value in Healthcare
In closing, the Whyze Health Platform collects data in a digital health & research data lake that is used for post-market surveillance of medical products, further improving patient outcomes, and increasing the overall quality of the healthcare delivered in Ireland. As new treatments are used in the country, the payers can track the longer term effects of these interventions, to ensure only the best
treatments continue to receive financial support. As the platform grows and continues to incentivise patient participation, it can help bridge the gap between high-performing healthcare systems in Europe and their counterparts in America and Asia, allowing patients access to ground-breaking treatments and creating improved health outcomes for all. This amplify’s the return on investment Ireland derives from the substantial FDI from pharmaceutical and med tech sectors. Through its unique patient-centred approach, Whyze has provided a much-needed solution to the problems posed by a lack of European access to clinical trials. This is truly the time for a change, and Ireland can rise in the forefront meeting these challenges.
Conclusion
Increasing clinical trials activity in Ireland is a healthcare priority. Direct benefits include improved outcomes and value for the patients and the healthcare system. By products are increase in return on FDI investment for Ireland. Improvements in digital health platforms including AI and machine learning can help drive this important change.
Frank Sullivan
Professor Frank Sullivan is a Co-Founder and Chief Medical Officer of Whyze Health, a Consultant Radiation Oncologist, mainly based at the Galway Clinic, with over forty years’ experience in healthcare, and a career rich in organizational development. He has held key leadership roles in both public as well as private healthcare organizations, in Ireland and the US. He is currently based mainly at the Galway Clinic, where in addition to his active Cancer Practice, he serves on several internal committees, including the Medical Advisory Committee (MAC) as well as a Senior management Working Group helping to make management recommendations to the Board, regarding the safe and effective running of the Clinic, and improving working relations with the practicing Consultants. He also presently sits on the Clinical Governance Committee, and for almost two years served as a Board Member.
Journal for Clinical Studies 19 www.journalforclinicalstudies.com Regulatory
How Lilly is Streamlining Regulatory Operations

Using agile methods, and starting with CMC post-approval submissions, an innovative team has brought the benefits of cloud-based RIM to users throughout the world.
In 2016, a cross-functional team at Lilly began to re-evaluate its approaches to global submissions and determine if newer technologies or processes could help improve efficiency. Reliance on traditional RIM software and manual processes hindered the regulatory team’s ability to keep up with submission volumes. This was especially true in the CMC space, explains Paula Hudson, senior director of global regulatory affairs, who led the team with Amber Karns, senior director of information and digital solutions.
The CMC portion of any submission is crucial to show regulators that the process for making a new therapy will consistently deliver safe and effective products. Getting it right is essential. Errors or
data omissions, whether in pre-approval applications, post-approval supplementary applications, or annual reports, will delay patient access to therapies and compromise confidence in drug supply chains. However, collecting the data required can be time-consuming and costly, prompting the industry to study new ways to streamline the process by shifting from narrative to data-based applications.1
Starting with Post-approval CMC Submissions
Hudson and Karns started the project by evaluating the way postapproval CMC submissions were handled. The department that focused on these was relatively small but had struggled to keep up with increasing workloads. “We needed better tools for audit readiness and strategic forecasting,” Hudson recalls. In addition, the team had never used collaborative authoring tools before, and leaders wanted them to be trained.
Close analysis of processes and performance gaps revealed the need to standardise processes and data, integrate systems, and
20 Journal for Clinical Studies Volume 15 Issue 1 Regulatory
automate. The company also needed to consolidate fragmented data and documents, which were dispersed across two different authoring systems – one for commercial products and another for products that were still in development.
Bringing the two systems together, connecting them with the publishing system, and retrieving correspondence from the submissions archive required manual processes. “Anyone trying to trace the history of activities related to a product submission had to go to multiple places to find information, which made it difficult to respond to questions, manage institutional knowledge, and handle other daily work responsibilities efficiently,” Hudson recalls.
Lilly’s team decided to focus efforts on integrating processes and data and standardising across its different submission groups. It implemented a suite of advanced regulatory applications on a single cloud platform for post-approval CMC so that submissions could be written, reviewed, validated, and archived within one central location. Viewed as setting the foundation for their transformation, this stage of the project had a large scope and took more than nine months to complete but yielded positive results.
Speeding Up and Extending Implementation
Hudson and Karns then moved to speed up the new RIM system’s implementation throughout Lilly’s commercial and development portfolio. They moved on from post-approval into pre-approval CMC and safety periodic report submissions, which required working with different groups of people. At the same time, they extended the work into new functionalities such as publishing.
To help speed deployment, their team shifted from a waterfall to an agile project management approach. They planned for three incremental software releases each year to address internal enduser needs. Each had very clearly defined milestones. To avoid any delays, they synchronised these updates with software updates that the vendor had already scheduled for the year. Any scope that didn’t make it into a release was included in the next one.
Employee training was key to success, Karns said, and team members gained a detailed understanding, not only about the new RIM environment and its capabilities but the specifics of Lilly’s configuration. Agile teams, called Discovery Action Teams, were set up to maximise the value obtained from each new release and to help refine capabilities. “We named the teams for their mission. Discovery, because they were really out ahead of the rest of us, understanding user requirements, and action, because they were taking the steps needed to ensure that we had those user requirements ready, and were prepared for configuration and testing,” says Karns.
The teams helped optimise key project parameters by defining requirements and managing releases, configuration, testing, and enduser communications, all of which improved efficiency and shortened timelines. They also established dashboards, and, to minimise rework, they set up weekly reviews and end-to-end walkthroughs before configuration locks. Closer attention to detail with day-to-day issues helped the team resolve technical, resource, and scope problems quickly.
Together, these strategies helped Lilly get more value from its new RIM system. The company’s RIM user base has grown to over 7,000 users since implementation began in late 2018.
The Benefits of Connected Data and Documents
The implementation has improved efficiencies by enabling
information to be accessed and shared quickly. “We can now go into an application record within our RIM system and see the entire history of our submissions, correspondence, commitments, and health authority questions. Being able to share and pull together our metrics from one system is so much easier than having to hunt through different applications,” says Hudson.
The ability to automate processes has also been a coup, adds Karns. End-to-end data flows and connectors will enable further efficiency gains by linking cross-functional data. “We won’t have to go look for something in another database and re-enter data. Over time, our RIM system will allow us to improve performance in other areas such as product quality,” she comments.
With collaborative authoring capabilities, writing and reviewing submission documents is much easier. Using traditional software, each author or reviewer would have to add content or comment on the draft individually, often without context from other reviewers. Given the size of the teams involved, this could be a major time challenge.
Hudson recalls on-site meetings where one person would be designated to collect and input all group members’ revisions or additions and address all their comments, a tedious process and effort that could require several rounds to complete. Now, she says, 20 people can review and mark their revisions in the document in real-time, responding to each other’s questions, with a full understanding of context.
For CMC submissions, where the project began, the new solution allows the company to save substantial amounts of time gathering the information they need for writing and publishing annual reports. According to Hudson, metrics collection, which could take someone a week to complete using the company’s previous software, can now be done in minutes.
Using an advanced regulatory system is also helping teams understand data on a much deeper level than they had before. Since the implementation, Lilly’s regulatory affairs department has established a data council with representatives from medical, safety, and commercial to ensure consistency in the way that data are defined. Regulatory leaders now see more opportunities to share information, not only across global regulatory affairs, but with colleagues in manufacturing, clinical, and marketing. “We are just beginning this part of the journey, but we can already see the potential,” says Hudson.
REFERENCES
1. Eglovitch, J.S., “Global Initiatives to Standardize CMC Quality Data Gaining Steam,” RAPS Journal, June 22, 2022
Marc Gabriel is vice president of regulatory strategy at Veeva Systems. Before joining Veeva in June 2017, he served as a client partner and RIM consulting lead at Kinapse. Marc also spent fourteen years in Accenture's pharmaceutical R&D practice, leading the organization's regulatory advisory, business development, and alliance efforts.
Email: marc.gabriel@veeva.com

Journal for Clinical Studies 21 www.journalforclinicalstudies.com Regulatory
Marc Gabriel
Ensuring Unbiased Oversight: The Case for Independent Expert Committee Management
Expert committees are a critical component in the success of a clinical trial. By turning to third-party providers to manage these services, sponsors can enhance trial credibility, mitigate risks, reduce bias and the perception of bias, and drive operational efficiencies.
Data Monitoring and Endpoint Adjudication Committees are groups of independent medical and/or statistical experts charged with a specific set of oversight activities related to a development program.
Data Monitoring Committees (DMCs) go by a variety of names, including Data Safety Monitoring Boards and Data Safety Boards. All clinical trials require safety monitoring, but not all require a DMC.
The DMC experts sit at arm’s length from a clinical trial and look at the overall trial or clinical program data as it’s unfolding. Unlike the sponsors and study CROs (contract research organisations), the DMC has access to unblinded data that it reviews for early indicators of safety problems.

An Endpoint Adjudication Committee (EAC) – also called a Clinical Endpoint Committee – adjudicates suspected events to determine if they meet event definitions, per the Charter. This panel of independent adjudicators often reviews blinded data to give their expert opinion on patient-level safety or efficacy events of interest according to predefined rules outlined within the Charter.
Sponsors may opt to manage these committees in-house, outsource to a CRO, or outsource to a specialty provider. While the first two options are viable operationally, they can create unnecessary risk, increased cost, and operational inefficiencies. Outsourcing expert committee management to a specialty provider with committee management expertise can be the best option from a regulatory, ethical, and business perspective.
The Value of Independent Review: Eliminating Bias
While it can prove challenging to keep the EAC and DMC independent from the clinical trial sites, participants, and the sponsor, it is critically important to do so. Maintaining third party independence between the committee and sponsor can greatly reduce the risk of bias and perceived bias compared to a sponsor managing a committee directly. Committee management and the related processes also add a level of complexity to trial conduct and benefit from expert management.
This is especially true for DMCs, which have access to unblinded data. Each person involved with unblinded data must be separate from clinical trial operations because the information could – even
unintentionally – influence their actions and put the trial’s integrity at risk. Relying on a CRO to manage the DMC could increase risk in that domain because the CRO might also be responsible for monitoring and running the trial and developing the final analysis.
Protecting Sponsor/Expert Relationships
This firewalled approach can help sponsors maintain relationships with industry experts and key opinion leaders, that often serve on independent committees. Since it is important to preserve these relationships, there are clear benefits to engaging an independent specialty provider to set expectations, manage disputes, and handle compensation. This approach enables sponsors to protect their relationships and the integrity of the trial.

Quality Control Through Committee Management
Independent specialty providers can offer an additional level of quality control for trials. For example, DMCs review study data and can provide quality control checks on the trial itself in areas such as data timeliness and cleanliness. Specialty providers focus on trends in data reporting and study progress, allowing for timely recommendations or midcourse corrections and early identification of risks.
Selecting the Right Provider to Achieve Efficiencies
Working with an experienced provider can save time and money. Building the committee infrastructure – such as a web-based adjudication platform, process guidelines, and charters – requires a significant investment of time and financial resources. Sponsors can save time and money by partnering with a provider that has existing infrastructure, systems, and templates.
Conducting due diligence during the partner selection process will ensure the provider has the qualifications, experience, and resources to effectively manage the EAC or DMC.
Successful vetting begins with asking the prospective provider some key questions:
• Timeline Management: What mechanisms do you have to monitor timelines and ensure timeline compliance? Do you have additional resources to expedite processes that will enable the sponsor to hit key milestones?
• Member Management: Can you describe your experience and strategies to manage the actions and tasks required of the members to ensure critical timelines are met? What mechanisms do you have to measure committee member satisfaction and engagement? Managing the committee members and the required actions of the members is critical.
• Audit Support: What mechanisms do you have to answer regulatory agency questions? What is your experience hosting
22 Journal for Clinical Studies Volume 15 Issue 1 Research & Development
audits of EACs and DMCs? What level of support will you provide for audits? Look for a provider that has a depth of expertise supporting and hosting audits, specifically those related to expert committee management.

• Operational Efficiencies: What operational efficiencies are available as we grow this program? Look for a provider who can find operational efficiencies and cost savings for a highvolume program. As programs grow or protocols are added, the right provider should be able to scale the program leveraging existing systems, processes, or documents.
• Reporting & Data Access: How will I view status reports and access the results of my committee? What is the turnaround time and how will it be measured? How do you manage member spend reporting and payments? Having access to your data when you need it is critical and working with a provider that understands your reporting needs is essential.
• Staff Expertise: Can you tell me about the project team that will be managing the expert committee? Can you tell me about coverage plans and staffing size to support this project as it scales? Will the staff working on this DMC or EAC work exclusively on expert committee projects? When outsourcing
expert committees, look for a provider who is as much an expert consultant as they are a provider managing the operational tasks of the project. Qualified providers should have a depth of experience managing expert committees and ensuring the project team has the required expertise and training is critical.
Ensure Alignment Before Startup
Early alignment between the sponsor and vendor is crucial. Developing high quality systems, processes and documentation up front will increase the project quality and efficiency and help to mitigate downstream issues. Successful partnerships are built on strong relationships, open communication, clear goals, and accountability.
• What are the critical time points (e.g., interim analyses, database locks, DMC meetings)? Be clear on key milestones at each step of the journey, making sure they are documented with expectations. This ensures all stakeholders are aware of and working towards meeting critical deliverables throughout the lifecycle of the project.
• Who are the key decision makers? When developing an expert committee there are numerous documents, forms, and
Journal for Clinical Studies 23 www.journalforclinicalstudies.com Research & Development
Research & Development
systems that need to be reviewed and aligned between the sponsor and vendor. Ensure the right people are in the right discussions and approval processes up front, to eliminate downstream issues.
• Who is responsible for what tasks? Roles and responsibilities should be well documented in the Charter or project guidelines. Strategically delineating task ownership between the sponsor and vendor is a critical discussion that should be held early. When making these decisions, aim to have the parties with the most experience and expertise owning each task or process to ensure operational success. For example, leveraging an EAC vendor with a depth of experience in EAC event identification can help to ensure high accuracy, quality, and a clear strategy that can be documented for regulatory authorities.
• What are the communication and escalation pathways between the sponsor and vendor? Define escalation pathways upfront and revisit them often. Is the process working as expected? Do the right people have access to vital information? Are delays or obstacles escalated to the appropriate parties to drive results? How is success measured and what does success look like? Use these data to drive results.
• Who requires access to the adjudication results? It’s important to make this clear from the beginning. Who needs the data? Are data transfer agreements in place to ensure the appropriate party receives the data?
Managing Committee Members
The right partner not only knows how to manage a committee; they know who to select to join it. An expert provider should have a vast network of committee members with a wide range of therapeutic expertise to draw upon.

The member selection process should ensure that the right members are chosen from the outset. The vendor should ensure the members have appropriate licensing, training, and expertise, with no conflicts of interest. Additionally, the members must have sufficient availability to be able to commit to the requirements of the committee and necessary turnaround times. Often members participate in a committee in addition to their job as a physician, so ensuring they have enough time to participate is critical for the project's long-term success and retention of members.
Expert committees should always be designed, structured, and managed with the members' experience in mind. Members want to be part of a high functioning, organised committee, engaged in interesting work and scientific problems. To drive results with the committee in mind, consider the following:
• Set expectations: Give committee members detailed guidance on roles, responsibilities, and operating procedures. This information should be well documented in the Charter. Make sure each member understands and agrees to the expectations and requirements – including availability – well before the first meeting or start of adjudication.
• Prepare: Provide the right data, in the right format, at the right time. Deliver data and meeting materials far enough in advance to allow members to thoroughly prepare for the meeting.

• Respond: Promptly answer committee questions and concerns, ensure clear escalation procedures are defined, and that committee members have access to staff when they need
them. This not only reduces member frustration, but it can also reduce delays.
• Provide seamless administration: In short, make things easy for the committee members. Committee members should be able to focus on the key responsibilities outlined in the Charter leveraging their medical and statistical expertise with minimal time spent navigating administration or technology issues. The members should receive seamless onboarding, contracting, and compensation.
• Seek feedback: Leverage member experience and feedback to drive efficiency and improve the process. Seek feedback and engage with the committee members throughout the project's lifecycle.
Efficient, Safe and Unbiased
Sponsors rely on expert committees and when managed well, these committees enhance trial integrity, bolster efficiency, mitigate risks, and safeguard patient safety. Managing expert committees well takes expertise, effort, and experience. EACs and DMCs are complex, and sponsors need a partner they can consult with, not merely a vendor who performs outsourced tasks. Sponsors need a specialty provider with committee expertise, and years of regulatory and operational experience, who can navigate those challenges, make recommendations, and provide guidance.
The right partner helps sponsors ensure clinical integrity and regulatory compliance, attain operational and financial efficiencies, and achieve clinical trial success.
David Cutler is Director of Project Management for the EAC service line at WCG and has over a decade of industry experience. Prior to joining WCG, David worked in a variety of leadership roles in healthcare, academic, and non-profit organisations leading clinical research programs and health care delivery systems. David has a BA in Psychology from the University of California Santa Cruz and an MA in Counseling, Health Psychology from Santa Clara University.
24 Journal for Clinical Studies Volume 15 Issue 1
David Cutler
VISIT DRUG DISCOVERY 2023: DIVERSE THINKING – NEW PERSPECTIVES

JOIN US IN THE TECH THEATRE FOR SUSTAINABILITY PRACTICES IN DRUG DISCOVERY RESEARCH
18TH & 19TH OCTOBER 2023
ACC LIVERPOOL EXHIBITION CENTRE
To find out more and to register please scan the QR code or visit elrig.org.
ADVANCEMENTS IN CELL & GENE THERAPY: NEW THERAPEUTIC

HORIZONS FORUM
15TH SEPTEMBER 2023
BIOZENTRUM UNIVERSITY OF BASEL
To find out more and to register please scan the QR code or visit elrig.org.
Journal for Clinical Studies 25 www.journalforclinicalstudies.com
elrig elrig_uk @ELRIG_UK elrig.org
Dispelling Doubt: Effective Scientific Journalism in a Time of Polarisation


In an era of rapidly evolving information and ever-burgeoning scientific innovations, it can be disheartening to witness the persistent lack of trust in the scientific process and establishment particularly in regard to the COVID-19 pandemic. The need to communicate information regarding the virus, especially ways to prevent and treat symptoms in a simple and robust manner, was paramount, at the beginning of the pandemic. To address this need the authors wrote a short piece stressing the importance of scientific journalism in this endeavour suggesting that...
Science journalists have a fundamental responsibility in being accurate and unbiased but also have a social responsibility to inform, enlighten, and to serve as a bridge between the scientific establishment and the public. Science journalists must become beneficent mediators in order to help create an understandable and comprehensible stream of information for consumers to digest with the goal of humanising findings and both engaging and enticing the public toward action – even if that action is a simple acknowledgment of the truth.1
With the new demands of the pandemic scientific reporting adjusted, with contributions becoming more frequent, timely, readily digestible and accessible. Despite this, public skepticism about science has remained a constant and significant hurdle to overcome, and in that sense scientific journalism has not met the simple goal of moving the public toward action and the acknowledgment of facts. The main reason for this failure was that those who were disseminating scientific information largely were ignorant of the powerful forces that undermined their efforts, and when they were aware they were ill-equipped to address these deleterious efforts. Furthermore, science writers are often unfamiliar with effective communication styles that take into account the psychology of persuasion, social identity and epistemic differences amongst their potential audience members.
Prior to the epidemic there was a large push to educate lay people in regard to general scientific literacy in hopes of building a public trust in scientific institutions and ultimately improving public health. This was mainly accomplished by science writers who became very proficient in interpreting and translating complex technological concepts into simple, understandable and consumable language. More recently those undertaking this lofty task have vastly underestimated the role of social identity and partisan politics in both information consumption, evaluation and adoption. The focus of science writers must now shift to making scientific information not only more easily consumable but also more relatable and relevant to a politically hyperpolarised society. By employing clear and concise language, to providing relatable examples, engaging commentary, acknowledging differences while focusing on a shared set of values, desires and concerns that emphasize self-benefit, and matching the delivery of their message to the epistemic style of the recipient,
science writers will be better able to convey complex scientific concepts in a manner that resonates with diverse audiences across a wide spectrum of political ideologies and hopefully rebuild trust in the process.
One major challenge in building this public trust continues to lie in the public's limited understanding of the prolonged and complicated nature of the scientific process. Science is a repetitive and self-correcting endeavour, subject to rigorous scrutiny and advancing from constant review and revision. Inappropriately, many lay individuals expect immediate answers and highly conclusive results, which of course can lead to hasty judgments and premature conclusions. Incomplete findings or contradictory information are often misinterpreted as confusion, a lack of credibility or deliberate deception, creating a loop of confirmation bias in which recipients only access information that supports their preexisting beliefs, thus perpetuating a distrust in science as a whole. It is the responsibility of scientific journalism to educate the public about research methodology, fostering patience and appreciation for the meticulous process that underpins scientific progress.
One beneficial aspect of the pandemic is that the general public has become more interested and knowledgeable about the scientific process as whole. When the pandemic was still in its infancy there were concerns about studies being untrustworthy and not having adequate data, or that studies were being conducted hastily with small numbers of participants. Beyond that, there was controversy from conflicting results reported early on in the pandemic. To the scientifically literate eye, this was nothing more than what can be expected in the scientific process, but to lay people this was more concerning. Many lay people were unsure as to what information was trustworthy and whether they should adopt this information into their routine lives. This concern was warranted, as it was not unusual to see conflicting news reports making opposing claims: certain masks work while others don't, debating whether disinfecting groceries was necessary, touting unproven remedies, and even reporting/ projecting mortality rates for different strains of COVID-19. The influx of the large volume of often disparate information that the public was expected to stay current on became simply overwhelming for most.
In an attempt to provide the public with quick and easy access to the most up to date findings, and to promote transparency, more formal scientific reporting adapted rapidly. Part of this involved ushering in a new era of unprecedented access to scientific preprints, allowing virtually anyone to observe the ongoing methodology within the scientific community. Up until this time this type and extent of information was reserved solely for those few researchers working on these studies, those reviewing these studies as journal referees, and journal editors. Although welcomed, this type of access also raised concerns about the credibility of many of these preprints, as they had not yet undergone the rigorous scrutiny of peer review.
Unfortunately, this concern seemed to generalise to all scientific studies and not just unreviewed preprints. The release of preprints
26 Journal for Clinical Studies Volume 15 Issue 1 Market Report
aimed to provide a glimpse into the ongoing efforts and newest findings on a wide variety of topics regarding the virus, whether these studies focused on mortality rates, the gestation period of the virus, or the effectiveness of masks. Importantly it is noteworthy that these studies, although not yet peer-reviewed, were still fundamentally valid and the overall findings did not vary substantively after review. Nelson et al., compared COVID-19 evidence presented in one hundred preprints to changes after peer review in subsequent journal publication. They reported a point estimate value change of an average of 6% during review; that the correlation between estimate values before and after review was high, and significantly that there was no systematic trends - which all support the value and robustness of evidence in preprints during the COVID-19 pandemic.2
While scientific journalism continued to evolve in its quest to disseminate information as rapidly as the spread of the virus, some peculiar geographic patterns began to emerge within different demographics, such as those observed across various counties within the United States (US). In the initial wave of COVID-19, it was noted that a majority of infections occurred in geographic regions characterized by a more politically liberal population. This pattern might be attributed to the higher population density of these areas, as many large cities in the US tend to lean leftward politically.3 However, as more scientific knowledge became available and precautions like masks, social distancing, and eventually vaccines became more widespread and promoted by good science reporting, this trend began to diminish.
Another distinct pattern emerged indicating higher infection rates in US counties that were relatively more politically conservative. There are multiple factors contributing to this trend, but one leading explanation suggests that this stems from the intense partisan nature of COVID-19 with disparate messages coming from media influencers and politicians who were unable or unwilling to provide accurate and reliable information about the ongoing pandemic. A particularly noteworthy correlation was observed between vaccination rates and subsequent mortality rates in these predominantly conservative counties, compared to counties characterised by opposing political views, which had higher vaccination rates and consequent lower mortality rates.3 This pattern suggests leading to disproportionate infections and deaths based on political ideology. Long gone are the days when beliefs about science and those about politics were seen as being independent. Unfortunately, science and politics now appear to be intrinsically intertwined.
This reckless disregard for science has been dismaying to the scientific community as it subverts the established efforts it has gone through to create an effective way to inform the public. The intersection of political/media influences with a once in a lifetime pandemic has created the opportunity for the proliferation of misguided misinformation but also the active spread of deliberate disinformation. This has created an environment that necessitates more efficient and effective science reporting to not educate the masses but to ensure that this information is presented in a manner that matches a person’s epistemic style of thinking, and sociopolitical camp. This approach acknowledges the fact that it is often easier to simply reject novel scientific information off-hand than to reject one’s own pre-existing political ideology, beliefs, attitudes and social identity. This rejection of science is an effective way to decrease the cognitive dissonance brought on by the novel scientific information often delivered by those seen as having unknown or poor credibility from another camp.
Therefore it is not enough simply to put out scientifically valid information and expect this to be readily adopted in this time of
heightened political polarisation. Of note researchers reported strong evidence of polarisation in beliefs about COVID-19 treatments even among highly trained professionals who were making treatment decisions regarding hydroxychloroquine and ivermectin.4 It appears that not only do people consume different types of information (conformation bias), but they also evaluate and interpret this information in a politically biased manner, and the scientists themselves are not immune to this tendency.
Findings such as this may provide some insight into the causes of regional or even country wide variation in the use of putative politicised treatments, and suggest that this variation is driven not solely by patient preference but also by physician beliefs. This research implies that there are apparent limits of expertise and exposure to scientific evidence in the presence of such implying that those with more scientific literacy may simply be more sophisticated at bolstering their pre-existing beliefs by hand picking information that adopts to their established views/values. Therefore, journalists will need to take this into account and be sure to appeal to the common values of both liberals and conservatives by emphasizing desirable actions and consequences that are important across the spectrum.
Although a bit surprising the above findings are really driven home by researchers who reported that higher exposure to conservatism in a given congressional district was associated with higher COVID-19 age-standardized mortality rates, a finding that held even after taking into account things like social characteristics, and hospital ICU capacity.5 Using models adjusting for political/social metrics and vaccination rates, a more conservative voter political lean independently remained significantly associated with a higher COVID-19 mortality rate. This was seen for the overall time period for COVID as well as separately for both Delta and Omicron variant waves, and even for models that adjusted for baseline heath and obesity/diabetes. This distressing data suggests that it is not enough for health organisations or journalists to simply put out reliable and valid information to their intended audience but that their messages need to be tailored to the recipient’s ideology making self-benefit appeals in an effort to diminish political differences in ideology.6
The issue of weeding out accurate and reliable studies and parsing scientific information remains a challenge for most science consumers. Science is still actively being conducted, and there are still new findings almost daily regarding COVID-19; however, we are no longer "in the dark," as some would say in terms of some basic facts. The data is clear that masks do help reduce the spread of the virus; that infections from contaminated surfaces is substantially less within 24 hours; and the mortality rate of COVID is higher than that of the seasonal influenza. However, misinformation and disinformation remain so prevalent that the FDA has found it necessary to dedicate an official webpage to stopping the spread of misinformation related to COVID by debunking falsehoods related to vaccine impurities, effectiveness, linkages to monkey pox and repeated vaccination, amongst other topics.7
Despite efforts such as this and the abundance of accurate, reliable, and accessible information from credible sources, large portions of the public still do not have trust in basic facts. This leads to what could be an even bigger issue that is yet to be truly faced: the lack of trust in scientists themselves. This distrust is not new, tracing back long before the pandemic emerged. Science was once practiced by a select elevated to near-mythical status, admired for their incomprehensible measurements and awe-inspiring adventures like Darwin's voyage or Humboldt’s climate experiments. Scientists were often romanticized along with their studies. However, science
Journal for Clinical Studies 27 www.journalforclinicalstudies.com Market Report
shifted in the twentieth century with the rise of “Big Science”, funded largely by wealthy nations with science being taught in massive lecture halls and becoming more mainstream. While some scientists pursued societally beneficial endeavours, others focused on more deleterious or purely fiscal applications, and this shift may have altered the public perception of science primarily from exploration to gain, transforming scientists from “Gentleman Naturalists” to government-funded lab coats, inadvertently leading to issues of source credibility with many refusing to believe the objectivity and credibility of those delivering the message.
Rebuilding trust in science is a continuous endeavour that will require efforts from the scientific community, science writers and the public. In a world flooded with information, it is imperative to make scientific media more accessible and comprehensible to the everyday

person. It is vital to educate the public about the intricacies and iterative nature of the scientific process, dispelling misconceptions and fostering patience. But it is not enough to simply convey knowledge in an accurate and easily digestible manner. By employing relatable examples, engaging rhetoric, and appealing to a shared selfbeneficence and values while acknowledging an understanding of competing viewpoints, science writers must bridge the gap between complex scientific concepts and public understanding/acceptance in a highly partisan enviroment.8 Effective science reporting must acknowledge and address the confirmation bias and differences in data evaluation and interpretation as being an inexplicable result of a hyperpolarised society. To address this, it is important to utilise what is known about the psychology of persuasion and communication, which often means communicating in a calm, warmer, respectful and inclusive manner emphasizing common ground and shared identity, and importantly to craft both the content of messages and the delivery approach to the intended recipients.
REFERENCES
1. Riordan, H. and Riordan, L. (2020). Truth Matters: Why Scientific Journalism Has Never Been So Important. Journal of International Pharmaceutical Industry, Winter Issue Vol 12 (4) pp 14-15. https://issuu. com/senglobal1/docs/2020-ipi-winter-web_compressed/s/15200479
2. Nelson L, Ye H, Schwenn A, Lee S, Arabi S, Hutchins BI. Robustness of evidence reported in preprints during peer review. Lancet Glob Health. 2022 Nov;10(11):e1684-e1687.
3. Sehgal N., Yue D., Pope E., et al. The Association Between COVID-19 Mortality And The County-Level Partisan Divide In The United States. Health Affairs. 2022. Vol. 41(6).
4. Levin JM, Bukowski LA, Minson JA, Kahn JM. The political polarization of COVID-19 treatments among physicians and laypeople in the United States. Proc Natl Acad Sci U S A. 2023 Feb 14;120(7).
5. Krieger N, Testa C, Chen JT, Hanage WP, McGregor AJ. Relationship of political ideology of US federal and state elected officials and key COVID pandemic outcomes following vaccine rollout to adults: April 2021-March 2022. Lancet Reg Health Am. 2022 Dec;16.
6. Cakanlar, A., Trudel, R., & White, K. (2022). Political ideology and the perceived impact of coronavirus prevention behaviors for the self and others. Journal of the Association for Consumer Research, 7(1), 36–44.
7. FDA Webpage. (2023). Rumor control: Learn and share FDA facts to help stop the spread of misinformation . U.S. Food and Drug Administration. https://www.fda.gov/news-events/rumor-control
8. Philipp-Muller A, Lee SWS, Petty RE. Why are people antiscience, and what can we do about it? Proc Natl Acad Sci U S A. 2022 Jul 26;119(30).
Lucas Riordan
Lucas Riordan is a rising Junior at University of Delaware majoring in psychology with plans to continue his instruction in graduate school and expand his education in both psychology and writing.

Email: lucas.riordan03@gmail.com
Henry Riordan
Dr. Riordan has been involved in the assessment, treatment and investigation of various neuroscience drugs and disorders in both industry and academia for the past 30 years.
Email: hank.riordan@yahoo.com

28 Journal for Clinical Studies Volume 15 Issue 1
Report
Market
HEALTHCARE EVENTS
The ultimate events for the pharmaceutical and biotechnology community to discover how to excel in clinical supply strategy as well as form key connections for long-term success


A great opportunity to get together and network face to face, hear from our expert speaker line-up and discuss the latest innovations and regulatory updates in the industry.

This exclusive event brings together attendees from established pharma, large and small, alongside biopharmaceutical companies and gives opportunity to dive into the operational challenges and innovations in clinical development found within the UK & Ireland region
Register Now
Use Reference Code: IBI
This year the conference will be focusing on interactive discussions to provide insight into the current major operational issues with running clinical trials in the coming years.
The conference will be focused on interactive discussions to provide insight into the current major operational issues with running clinical trials in the coming years.


Clinical Trial Supply East Coast offers opportunities for both learning and networking with peers from across the CTS sphere, via informal networking breaks and lunches, as well as a relaxed drinks reception at the end of Day 1.
Our conference includes speakers from Pfizer, Merck, Janssen and many more who will be covering the latest innovations in the clinical supply industry.
Register Now
Use Reference Code: IBI
Register Now
Use Reference Code: IBI
Journal for Clinical Studies 29 www.journalforclinicalstudies.com
Exploring the Limb Girdle Muscular Dystrophy Clinical Trial Landscape
With the recent advent of adeno-associated, virus-based gene therapy treatments, limb girdle muscular dystrophy (LGMD) is currently attracting the attention of the biopharmaceutical industry, especially with the goal of restoring full or partial proteins that are otherwise dysregulated. Because of the multiplicity of LGMD subtypes (greater than 35 subtypes identified to date – each with a number and unique letter to identify it), is it likely that not all subtypes will be targeted simultaneously for drug development in the short term. For example, in contrast to Duchenne muscular dystrophy (DMD), where a dysregulation of the protein dystrophin is deemed to be the mechanism of action, LGMD presents as a complex of multiple subtypes of disease with sarcoglycan and dysferlin as the primary dysregulated proteins.
Several sponsors are developing potential gene therapies for LGMD patients. Therefore, LGMD patients have a choice of clinical trials, which can raise new challenges for recruitment. Additionally, trial sites are selected based on a myriad of factors, which may include sponsor choice (for an individual company’s business purposes or rapid enrolment considerations), the physical location of the clinical sites (based on feasibility and capability), the site’s principal investigator, regional marketing considerations, and ease of patient access. These complexities must be balanced against the risk of conducting trials in countries/regions that may lack the ability, expertise, or capacity to participate.
This article describes the results of a proactive, global LGMD feasibility study conducted by IQVIA in 2019, including insights from investigators currently providing LGMD patient care. The feasibility study asked 32 questions about physician interest in and motivation for LGMD clinical trial participation and solicited data regarding the investigator-estimated numbers of LGMD patients available for clinical trials by age group and for subtypes of LGMD identified by Nigro and Savarese. Over 200 investigators were polled in 48 countries with a response from 116 investigators across 36 countries.
Overview of the Muscular Dystrophies and US Impact

Muscular dystrophies (MD) comprise a group of multi-systemic diseases caused by defects in genes for the production of muscle proteins. These diseases manifest clinically as progressive muscle weakness, with associated loss of mobility, agility and body movements. Ongoing elucidation of the proteins and structures involved in certain disease processes has boosted the number of potential pharmaceutical targets, with significantly heightened interest in investment, partnership and collaboration as a result. The research community could benefit from tools to identify where patients are eligible and willing to participate in clinical research and
where trial sites have adequate experience and resources to conduct the complex protocols that will be forthcoming.
Although the muscular dystrophies are rare in terms of individual disorders, when combined with other neuromuscular disorders, they have a significant influence on the global economy. For example, the IQVIA Institute estimates that, collectively, the neuromuscular disorders impact 250,000 patients and their caregivers in the US alone. Analysis of healthcare charges using IQVIA real-world data indicates that total annual charges across all neuromuscular patients in the US exceed $46 billion.1
Overview of the Limb Girdle Muscular Dystrophies
The LGMD group contains heterogeneous autosomal muscular dystrophies under the phenotype of progressive proximal weakness at the hip and shoulder girdles. There are multiple forms of LGMD due to mutations in different genes (such as CAPN3, DYSF, SGCA, SGCB, SGCG, SGCD, TTN and ANO5).2
Determinants of specific diagnoses include the distribution of weakness; ethnicity; family history; age at onset; rate of disease progression; presence of contracture, rigidity, rippling muscle, muscle hypertrophy or atrophy; and systemic involvement including cardiac, pulmonary, and skin complications.3,4 By definition, the term “limb girdle muscular dystrophy” usually excludes other defined types of muscular dystrophies such as Duchenne and Becker MD, myotonic dystrophies, and facioscapulohumeral muscular dystrophy.5 Ethnicity and family history may reveal recessive vs. dominant variants endemic to certain regions, e.g., LGMD 2A in southern Europe. In some families, the inheritance pattern and the exact gene mutation cannot be determined. Complicating the diagnostic process are substantial overlaps, as mutations in different proteins that share similar cellular functions can result in almost identical clinical phenotypes.
There is no standard of care for patients with LGMD,6 though as with Duchenne MD, physicians often prescribe corticosteroids. In addition, supportive therapy is often prescribed for those patients with cardiopulmonary issues and cognitive issues. Many LGMD patients take various vitamin supplements, although little is known or proven about vita min supplementation in patients with LGMD.7
Increasing Number of Clinical Trials in LGMD
In addition to the heterogeneity of LGMD, the ultra-rare characteristics of each subtype add to clinical trial recruitment challenges. This article addresses the global LGMD landscape for a better understanding of patient distribution, aimed at overcoming these challenges.
30 Journal for Clinical Studies Volume 15 Issue 1 Therapeutics
LGMD clinical trial success relies upon site selection, which may include sponsor choice (for business purposes or rapid enrolment considerations, based on feasibility and capability), advice from patient advocacy groups specific for the subtype (e.g., Jain Foundation, Coalition to Cure Calpain 3, Family Group of Beta-sarcogylcanopathy, etc.), locations of natural history studies (e.g., Defining Clinical Endpoints in Limb Girdle Muscular Dystrophy or GRASP study),6 the physical location of the clinical site, the site’s principal investigator’s speciality, regulatory requirements, marketing considerations and ease of access. These complexities must be considered when considering conducting trials in countries/regions that may lack the ability, expertise, capacity or willingness to implement LGMD studies.

It is worth highlighting that some sponsors of LGMD treatments hesitate to work in certain markets and geographies because of access and reimbursement challenges. Also, some sponsors are hesitant to partner with trial sites outside of traditional and known facilities, which could be due to long start-up timelines (e.g., in Eastern Europe and Latin America), lack of knowledge of the local environment and lack of previous rare disease clinical trial experience, especially when conducting first-in-human (FIH) clinical trials.
According to clinicaltrials.gov, a database of privately and publicly funded clinical studies conducted around the world, a total of 33 current trials mention the term “limb girdle muscular dystrophy” as of December 3, 2019 – although some are specified by the exact subtype (e.g., 2E, 2C, 2 and 2B). This compares with 266 DMD trials (search term “Duchenne muscular dystrophy”). Some of the postings on clinicaltrials. gov reflect the fact that LGMD clinical trial development is at an early stage, such as those seeking to identify clinical trial endpoints as well as those seeking to understand the natural history of a specific LGMD subtype. The number of LGMD clinical trials is expected to increase with cell and gene therapy development on the rise.
This paper will describe the results of a proactive LGMD feasibility study which was conducted by IQVIA (a leading provider of advanced analytics, technology solutions and contract research services to the life sciences industry) in the second half of 2019 with the objective of obtaining unique and current insights from global investigators currently providing LGMD patient care.
Methods
Our 32-question survey for LGMD clinical trial “readiness” –defined here as physician interest/motivation in LGMD clinical trial participation – was sent to more than 200 physicians treating LGMD patients across 48 countries.
The objective was to evaluate investigator access to LGMD patients, research experience, and challenges in recruiting and
treating the LGMD patient population, and to gain an awareness of access to equipment and diagnostic capabilities in different geographies.
The broad categories of questions included: investigator interest, patient population (including age [3–6 years old, 7–17 years old, 18 years and older], subtype, and how patients are identified), diagnosis and treatment, and site experience and logistics.
The main physician specialities this survey interviewed were neurologists, paediatric neurologists, neuromuscular specialists, and paediatric neuromuscular specialists (see Figure 1).

The survey was launched in two phases: the first survey was distributed to treating physicians in 12 countries (the US, France, Italy, Spain, UK, Germany, Denmark, Finland, Malaysia, Korea, Japan and Brazil); while the second phase included physicians in 36 additional countries (Argentina, Belgium, Bulgaria, Czech Republic, Hungary, India, Mexico, New Zealand, Poland, Romania, Turkey, South Africa, Australia, Bosnia, Canada, Chile, China, Colombia, Costa Rica, Croatia, Greece, Israel, Latvia, Lithuania, Netherlands, Norway, Peru, Philippines, Portugal, Russia, Serbia, Slovakia, Slovenia, Sweden, Taiwan and Ukraine). Responses were summarised descriptively and by region.
Results
IQVIA received completed surveys from 166 investigators across 36 countries (see Figure 2) over a three-month period. There was no difference in the response rate between the two phases of the survey.
In terms of general interest in participating in LGMD trials, a majority (70%) of physicians responded positively (see Figure 3). More than 50% of the physicians, identified largely as neurologists or paediatric neurologists, who had expressed their interest to participate in a LGMD trial, were found to be associated with an academic hospital (see Figure 4).
Sites in the United States, Canada, Brazil, Russia, Portugal, Ukraine, China and Spain were among those having the largest numbers of interested physicians. It is important to note that physician interest may not necessarily imply that there is the ability to conduct a clinical trial or the capacity to enroll patients.
Journal for Clinical Studies 31 www.journalforclinicalstudies.com Therapeutics
Figure 1 – An Overview of the Physician Specialities Responding



32 Journal for Clinical Studies Volume 15 Issue 1 Therapeutics
Figure 2 – Total Number of Survey Responses by Country
Figure 3 – Number of Interested Physicians by Country
Figure 4 – Interested Investigators’ Practice Setting
Investigator Descriptions of Patients
Investigators who responded to the survey believe patients and caregivers treated in the last six months across all three age groups (3–6 years old, 7–17 years old, and ≥ 18 years of age) would be interested in being enrolled in LGMD clinical trials, with the largest number of patients coming from the adult (≥ 18 years) age group (see Figure 5 for country-by-country tallies).
In the 3–6-years age group, investigators from Russia, India, Ukraine and US reported the highest patient volume who may be interested in participating in a trial for LGMD. In the age group of 7-17 years, investigators from Russia, Ukraine, Germany and India reported a higher volume.
In the adult population, investigators from countries like Spain, Brazil, Russia and US have estimated ≥100 LGMD patients may be interested in participating in an upcoming LGMD trial (see Figure 5).
Figure 6 illustrates that investigators predict that a large number of LGMD patients would be willing to participate in an LGMD trial
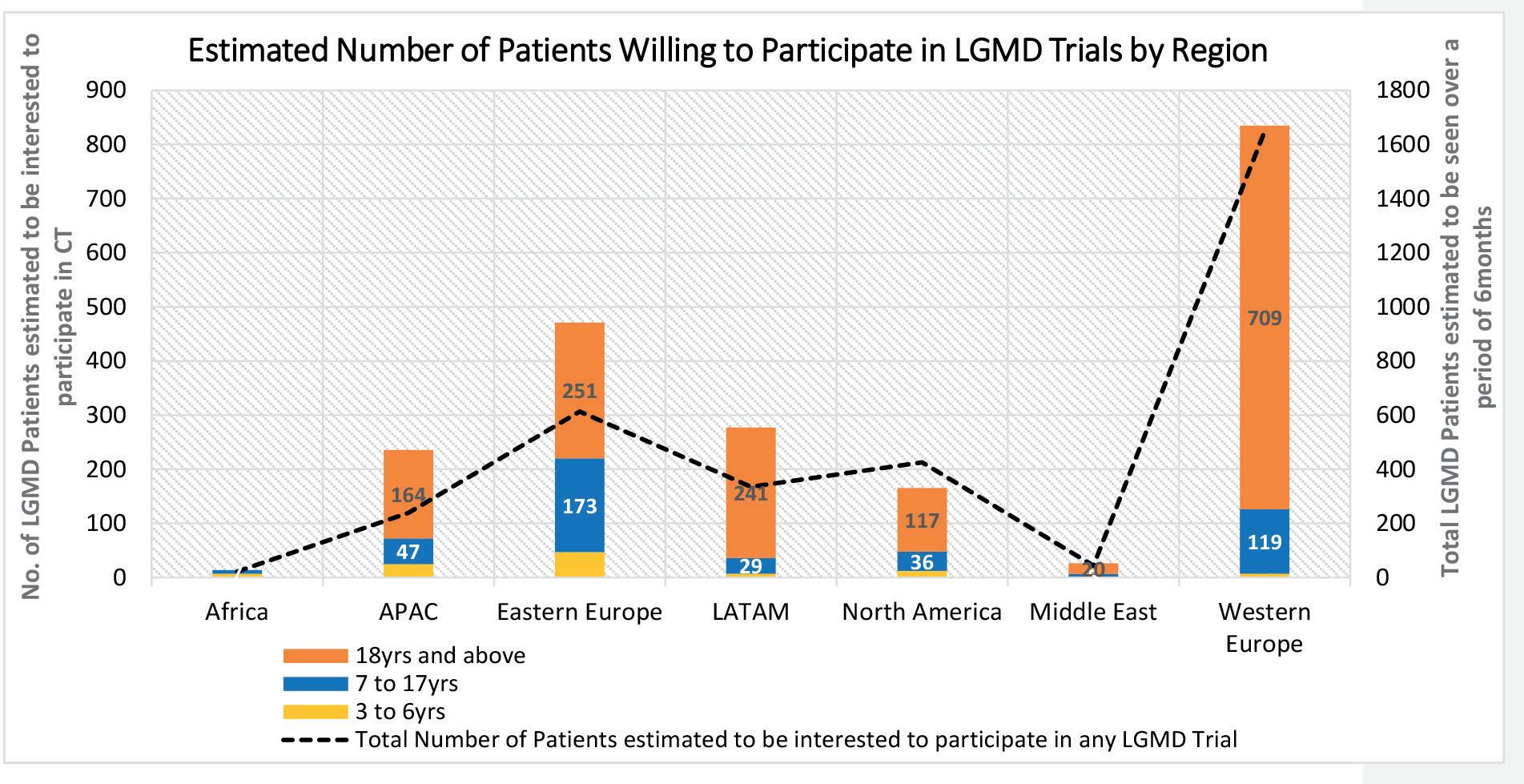

from Western Europe, followed by Eastern Europe, Latin America, Asia Pacific and North America. Regions/countries seeing a relatively smaller number of total / interested LGMD patients is mainly due to lack of access to appropriate genetic testing to confirm diagnosis and a lack of trial-ready infrastructure.
Across all regions, investigators reported that most of the patients in their clinical trials were identified through self-referral (34%), followed by patient registries / databases (23%) (see Figure 7). “Registries” referred to investigators’ own patient databases, local registries or national/international neuromuscular disease registries.
It was also noted across regions that there was a variance in the distance travelled by the LGMD patients to reach their treating physician. In some regions, LGMD patients travelled long distances, some up to 75 miles (or more) to reach their treating physician (see Figure 8).
Only one LGMD treatment guideline exists within the ICH (International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use) countries. Dated 2014,5 this
Journal for Clinical Studies 33 www.journalforclinicalstudies.com Therapeutics
Figure 5 – Physician Estimates of Numbers of Patients Willing to Participate in LGMD Trials Based on Age Group – By Country
Figure 6 - Number of Patients Estimated by Physicians to be Willing to Participate in LGMD Trials Based on Age Group – By Region
Some regions are currently conducting clinical trials either for LGMD or other types of muscular dystrophies as seen in Figures 11 and 12.


Antisense oligonucleotides (ASOs), such as nusinersen, inotersen and drisapersen) were the most commonly used class of drugs for trials in treatment of LGMD globally (24%), followed by small molecules such as edasalonexent and risidiplam (20%), others (which

34 Journal for Clinical Studies Volume 15 Issue 1 Therapeutics
Figure 7 – Regional Distribution of Patients by Source of Identification
Figure 8 – Distance Travelled by Patients to Reach the Site
Figure 9 – Classes of Medications Used as Supportive Care for LGMD states that any experimental treatment offered to patients with LGMD (e.g., myostatin, gene therapy, etc.) should be administered only within the clinical trial setting.
Figure 9 shows supportive care for LGMD under treatment by the responding investigators. Figure 10 summarises the types of treatments used as “standards of care” by country.
Most investigators started LGMD treatment before the motor function plateaued, with some preferring to start the treatment when the motor function begins to decline.
Conclusions
In summary, responses to the global proactive feasibility study from 116 investigators treating LGMD patients in 36 countries yielded the following findings:

• In terms of general interest in participating in LGMD trials, a majority (70%) of physicians responded positively.


Journal for Clinical Studies 35 www.journalforclinicalstudies.com
Figure 10 – LGMD Standard of Care by Country
Figure 11 – Site Experience with Investigational Products for LGMD and Other Muscular Dystrophy Clinical Trials
Figure 12 – Site Experience in Terms of Treatment Modalities: Regional Distribution include enzyme replacement and adrenocorticotropic hormone or ACTH therapy) (19%), gene therapies (14%) and monoclonal antibodies (14%).
Therapeutics
• The typical speciality of principal investigators for LGMD clinical trials is neurology.
• More than half of the physicians – identified largely as neurologists or paediatric neurologist – who had expressed their interest in participating in a LGMD trial, were found to be associated with an academic hospital.
• Sites in the United States, Brazil, Russia, Portugal, Ukraine, China and Spain were among those having the largest numbers of interested physicians.
• There is an opportunity to enroll more LGMD patients in global clinical trials, beyond North America and Western Europe.
• Investigators rely on self-referral, LGMD patient registries and site databases to identify patients for clinical trials.
• Travel distance for LGMD patients may be an important barrier for recruitment.
LGMD is one of the most severe forms of muscular dystrophy with currently no available cure and no approved disease-modifying treatments. Since LGMD can be life-threatening and severely debilitating due to progressive muscle weakness, new diseasemodifying therapies aiming to slow or halt disease progression – as well as curative treatments – are desperately needed.
LGMD in all of its forms represents a formidable challenge for drug developers. Due to the disparity of subtypes, some forms might not be amenable to all types of currently proposed therapies (e.g., gene therapies using AAV vector technology). Although advanced / precision therapy use is currently a small part of the LGMD treatment regimen, this is expected to increase considerably in importance if disease-modifying therapies are approved.
As the pathology of each of the subtypes becomes more elucidated, additional targets will be available for study. As the number of clinical trials rises, so will the need for patients with certain subtypes. Given the small size of the clinical trials, as shown in the clinicaltrials.gov postings, concierge services (including reimbursement for travel costs, and vouchers for meals and accommodations) and language support services (e.g., a translator) should be considered to ease patient and caregiver burden after enrolling in a clinical trial and to ensure compliance.
The speciality of principal investigators for LGMD clinical trials is typically neurology; although other specialities are either already part of the LGMD medical care team (respiratory pathologist, cardiologist, physical therapist, speech pathologist, etc.) or may be added to the patient’s management team as the disease progresses.
Based on all global investigator feedback received, the authors believe that while LGMD patient enrolment will be challenging in certain geographies (such as the US and Europe), other regions are not yet fully explored (including Eastern Europe, Latin America, China and India).
As clinical research is still at an early stage in LGMD, some expert sites have never participated in industry-sponsored clinical trials, especially in FIH studies. LGMD clinical trial expertise and knowledge could be provided to investigative trial sites by the pharmaceutical companies and/or designee, directly. Once a trial has been identified and initiated, LGMD training should be provided from the early stages and throughout the study.
It is hoped that with further LGMD feasibility analyses, we will gain a better grasp on investigator interest and experience with the potential to allow all patients with LGMD – regardless of subtype or geography – to enroll in a clinical trial to explore products that could provide desperately needed disease-modifying treatments or a cure.
Diagnostic Approach to LGMD
The diagnosis of limb girdle muscular dystrophy is often challenging because of significant disease heterogeneity. Historically, the muscular dystrophies were classified as either type 1 (dominant) or type 2 (recessive) depending on the mode of inheritance. According to Nigro and Savarese (2014), there are currently eight subtypes of autosomal dominant (type 1) limb girdle muscular dystrophy.
Calpainopathies or LGMD type 2A, are the most common form of LGMD, and are caused by mutations in CAPN3. LGMD type 2B, also known as dysferlinopathies, are caused by mutations in the DYSF gene. While sarcoglycanopathies, also known as LGMD types 2D, 2E, 2C, and 2F are caused by mutations of the following genes: SGCA, SGCB, SGCG, and SGCD genes. Mutations of the ANO5 gene cause type 2L LGMD. Several other gene mutations cause LGMD forms called dystroglycanopathies, including types 2I, 2K, 2M, and 2N. The disorders are labelled alphabetically according to when the individual genes were identified. The main classes of proteins involved in these conditions include extracellular matrix and external membrane proteins, enzymes or proteins with putative enzymatic function, sarcolemma-associated proteins, nuclear membrane proteins, and sarcomeric proteins.
The differential diagnosis of limb girdle muscular dystrophy is broad which, at least in the past, has led to misdiagnoses. For example, the authors have met several patients with LGMD who had previously been diagnosed with DMD or another dystrophy before their LGMD and specific subtype diagnosis. The differential diagnosis spectrum includes other muscular dystrophies such as congenital muscular dystrophies, myotonic dystrophy, facioscapulohumeral muscular dystrophy, and Emery-Dreifuss muscular dystrophy, as well as congenital myopathies, myofibrillar myopathies, distal myopathies, metabolic myopathy (such as Pompe or lipid storage disease), channelopathies, inflammatory myopathies, neurogenic disorders, and neuromuscular junction transmission disorders.
Similar to other muscular dystrophies, the approach to LGMD requires a detailed history, a thorough physical examination, and measurement of a serum creatine kinase level. Other genetic and acquired causes of proximal muscular weakness should be excluded. The diagnosis may be confirmed by molecular genetic testing, muscle biopsy, or a combination of both. Muscle biopsy will typically reveal the characteristic dystrophic features; further immunostaining may demonstrate the presence or absence of specific muscle proteins such as dystrophin, dysferlin, sarcoglycans, emerin, collagen VI, merosin, and glycosylated alpha-dystroglycan. The future of molecular testing may shift away from targeted genetic analysis toward whole genome or exome sequencing that will allow rapid and cost-efficient confirmation of the diagnosis. General treatment principles include offering genetic counselling for affected individuals and families; connecting them with patient organisations and disease registries; providing rehabilitation through multidisciplinary clinics to maximise function; supporting education, career, social, and financial needs; screening and treating the associated complications; and evaluating new treatment options for specific diseases when available.
Acknowledgment
The authors wish to thank the entire IQVIA Feasibility Team, which
36 Journal for Clinical Studies Volume 15 Issue 1
Therapeutics
was supported by Marie Trad, MD, Vice President and Therapeutic Area Head, CNS, at IQVIA Biotech, for the design of the survey.
REFERENCES
1. Understanding Neuromuscular Disease Care: Current State and Future Prospects; Institute Report, October 30, 2018; https://www.iqvia.com/ insights/the-iqvia-institute/reports/understanding-neuromuscular-diseasecare, accessed November 12, 2019.
2. Nigro V, Savarese M. Genetic Basis of Limb-Girdle Muscular Dystrophies: the 2014 Update. Acta Myol. 2014 May; 33(10):1-2; https://www.ncbi.nlm.nih.gov/ pubmed/24843229, accessed November 27, 2019.
3. Mercuri E, Muntoni F. Muscular Dystrophies. Lancet. 2013; 381:845-60.
4. Mah JK. Chapter 5: An Overview of the Other Muscular Dystrophies: Underlying Genetic and Molecular Mechanisms in Muscular Dystrophy: A Concise Guide (Huml RA, Editor), Springer Publishing (ISBN 978-3-319-17361-0), Copyright 2015.
5. Narayanaswami P, Weiss M, Selcen D, David W, Raynor E, Carter G, et al. Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2014;83:1453-63.
6. Evidence Based Guideline: Diagnosis and Treatment of Limb-Girdle and Distal Dystrophies; American Association of the Neuromuscular & Electrodiagnostic Medicine (AANEM) group: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine at https://www.aan.com/Guidelines/home/ GetGuidelineContent/672, accessed November 20, 2019.
7. Life Extension: The Source of a Healthier Life at https://www.lifeextension.com/ protocols/neurological/muscular-dystrophy#, accessed November 27, 2019.
8. Defining Clinical Endpoints in Limb Girdle Muscular Dystrophy (LGMD) (GRASP); https://clinicaltrials.gov/ct2/show/NCT03981289, accessed November 20, 2019.
Raymond A. Huml

Raymond A. Huml, MS, DVM, RAC is Vice President and Head of IQVIA’s Global Biosimilars Center of Excellence, has a personal interest in MD and works with the IQVIA/Muscular Dystrophy Association (MDA) team on their national patient registry. Dr Huml is a member of the FSH Society, co-founder and member of the North Carolina Chapter of the FSH Society, and a member of MD STARnet’s NC Advisory Committee. He edited and co-wrote eight chapters for the Springer book, “Muscular Dystrophy: A Concise Guide (2015).”
Email: raymond.huml@iqvia.com
Olja Tanjga
Olja Tanjga, MD is Director of IQVIA’s CNS Biotech solution. Dr Tanjga is a neurologist with 17 years’ combined industry and clinical research experience. Dr Tanjga is a boardcertified neurologist with a special interest in MS, neuromuscular diseases, extrapyramidal disorders, stroke and pain with experience in many other neurology indications. She has served as both medical and scientific advisor on over 40 clinical trials including the following indications: multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, ALS, epilepsy, stroke, acute / chronic pain (headache, low lumbar pain, neurogenic, herpetic pain, diabetic neuropathy, trigeminal neuralgia), and myasthenia gravis.
Email: olja.tanjga@quintiles.com
Therapeutics
Tiffany Chow
Tiffany Chow, MD is Director and Medical Strategy Lead for IQVIA’s CNS Center of Excellence. A neurologist by training, she is an experienced industry physician with over 20 years of clinical research in both academic neuroscience and research on elucidating the neurobiology of neurodegenerative disorders, applying PET imaging and functional neuroimaging methods, neuropsychiatric assessments and genomic testing in order to develop clinically meaningful therapies.
Email: tiffany.chow@quintiles.com
Siddharth
Aras
Siddharth Aras, MS is a Clinical Planning Strategist and the Global Feasibility Manager at IQVIA. Some of his key responsibilities include overseeing and analysing business requests for designing, developing and delivering analytical solutions to support and enhance IQVIA R&D and RWI solutions opportunities and trial programmes. Siddharth has 12 years of industry experience in the areas of regulatory affairs and global feasibility.

Email: siddharth.aras@quintiles.com
Marie Trad
Marie Trad, MD is Vice President and Head of IQVIA’s CNS Biotech division. Dr Trad has over 30 years of CNS experience, including 11 years as a clinical Neurologist / Neuroradiologist and 19 years of pharmaceutical industry experience as a Clinical Research Specialist / Consultant. She also has 19 years of CNS drug development and clinical trial management experience, providing medical, clinical and global strategic support to neurology and psychiatry trials in: epilepsy (both monotherapy and add-on therapy), idiopathic Parkinson’s disease (early and advanced), Alzheimer’s disease, amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), chronic and acute pain, including diabetic neuropathic pain and post herpetic neuralgia, migraine, major depressive disorder, schizophrenia, bipolar disorders, generalised anxiety disorder, traumatic brain injury and stroke. She has significant experience in neuroradiology, including magnetic resonance imaging, magnetic resonance angiography, and computed tomography (e.g., scans of head, neck and spine).
Email: marie.trad@novellaclinical.com
Jill Dawson
Jill Dawson, PhD, is a medical writing consultant to IQVIA, and a co-author of multiple papers on muscular dystrophy, biosimilars, celiac disease and other topics. She has a BSc and a PhD in Life Sciences from Imperial College London.



Email: jilldawson100mac.com

Journal for Clinical Studies 37 www.journalforclinicalstudies.com
Shipping Clinical Trials, Robustly and Reliably
According to recent forecasts by Fortune Business Insights, the global clinical trials market is projected to reach nearly $100 billion in 2030. That represents huge growth – yet achieving it simply won’t be possible without sensible investment across the supply chain.
When it comes to the transportation of clinical trial samples, the industry faces the same problems as the wider pharmaceutical market, but further intensified and complicated by the very nature of clinical trials.
It’s important to understand both the similarities with the pharmaceutical cold chain, and the very different dynamics involved in transporting clinical trial samples, in order to evolve a robust and reliable system for shipping samples around the world.
Product Efficacy
First and foremost, clinical trial samples need to work at the point of delivery. This is the core rationale for all cold chain businesses; product efficacy is sacrosanct. The billions invested in biologic specimens, new vaccines or investigational drugs are worthless if they have failed by the time they reach patients.
In the best-case scenario, they simply won’t work, skewing data and delaying regulatory validation. In the worst-case scenario, poorly protected samples may harm the patient.
Whatever else you require from a clinical trials cold chain solution, product efficacy always comes first.
The Increasing Complexity of Clinical Trials
The rapid growth of the clinical trials market is due in part to the huge advances being made by researchers. Consider biologics, for example. Global Data reported in 2022 that sales of biologics will significantly overtake innovative small molecule sales by 2027 – a testament to the exciting work being done in this field.
Of course, biologics are much more complex to transport. They are sensitive to heat and susceptible to microbial contamination, so maintaining a consistent ambient and secure environment during transit is even more important for biologics than other medicines. As biologics grow in influence and importance, cold increasingly becomes the new normal.
The Regulatory Framework
Accordingly, the safety-first mantra of any medicine is more strictly regulated when it comes to clinical trial samples. Regulatory authorities, such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), have specific guidelines
and regulations in place to ensure the quality and safety of samples. Typically, these regulations require strict temperature monitoring and documentation during transportation to demonstrate compliance.
At Once Global, and Local
Clinical trials are increasingly conducted on a global scale, with samples transported across different countries and continents. This globalisation poses challenges in terms of maintaining temperature control during long-distance transportation and varying climates.
At the same time, the smaller sample sizes of clinical trials compared to mass-market pharmaceuticals, requires a more agile, sub-pallet solution.
These issues come together especially when you consider remote locations. How do you deliver clinical trials safely? You need the same robustness of cold chain solution to maintain internal temperatures and prevent damage in-transit, but portable enough to ensure delivery wherever in the world it is needed – which might be in a region where, for example, access to electricity is fallible or non-existent. In short, a container which strikes the optimum balance between high performance, durability and optimised size and weight, is key.
Effective Monitoring
With so much at stake for the clinical trials industry – financially and reputationally – it’s imperative that sample deliveries can be effectively monitored and tracked. The main source of reassurance at the end of a sample’s journey is its temperature data logger, which provides ongoing and real-time insight into whether there has been any temperature excursion.
Handily, as more journeys are made and their data is recorded, providers of pharmaceutical cold chain solutions can advise potential customers of their typical excursion rates – the lower, the better. Shipment data confirms that, with modern pharmaceutical cold chain solutions, you can expect fewer than 0.1% temperature excursions per 25,000 shipments.

This means you can overlay a statistically modelled macro level of reassurance, onto the micro level from each individual transit.
Reducing Risk
Clearly, there is a lot for clinical trials manufacturers to think about. All of the points above must be considered, but they aren’t always in alignment. Sometimes, a decision must be made that comprises on one factor in favour another, more important, one.
Bearing in mind the primary consideration – patient safety, through product efficacy – it is crucial to weigh up all options, and conduct a risk assessment of all available solutions. What takes precedence? The length of time that the temperature can be maintained? The functionality of the datalogger? The size and weight
38 Journal for Clinical Studies Volume 15 Issue 1 Logistics & Supply Chain
of the container? The choice of materials used to maintain internal temperatures, especially whether electricity is required?
Towards the Optimum Solution
The good news is that pharmaceutical cold chain solutions providers are always working to close the gap, so that no one element needs to be sacrificed. It’s worth understanding the latest innovations in product design and material choice, to help you move towards the optimum solution for clinical trial samples.
Passive vs Active
The first thing to decide when choosing a temperature-controlled pharmaceutical container is whether you prefer an active, or a passive packaging system.
An active container uses mechanical and electric systems powered by an energy source, combined with thermostatic control in order to maintain proper product temperatures.
In comparison, passive packaging systems consist of materials intended to keep the internal contents of the package within a specific temperature range for a defined period of transport without any means of mechanical assist or manual intervention during transit.
Passive packaging systems generally comprise two main components, which are insulation (for example, lightweight vacuum panels) and coolants, such as phase-change technology.
From the clinical trial company’s viewpoint, the critical issue is reliability, and some understandably take comfort in having a powered solution. Yet, as advances in passive technology have progressed, containers can comfortably maintain internal temperatures for in excess of the industry-standard 120 hours.
For clinical trials, the use of a passive system offers an obvious benefit, as it can used around the world, regardless of local environmental conditions. The samples, therefore, will stay safe even in the event of a power cut.
Robust Design
Temperature of course not the only risk factor during transit. It’s also vital to ensure that a container is completely sealed, so that it is fully weatherproof and washable, to minimise the risk of microbial and bacterial product contamination.
At the same time, this robustness shouldn’t come at the expense of manoeuvrability. Heavily protected need not mean heavy. Innovative product design has, for example, enabled the use of recyclable polyethylene foam, strategically placed in critical areas to protect against potential damage through impact and shock sustained during transportation. Such design permits a robust but lightweight, manually handleable solution with the same reliable, thermal protection but at a size easily carried by fewer cargo agents.
Portable Containers
Huge strides have been made in recent years to ensure that the same quality of product protection can be achieved, within a smaller container size ideal for clinical trials or ‘last mile’ delivery. As such, it becomes possible to find a robust lightweight handleable solution with the same reliable thermal protection and durability of palletsized containers.
From a design perspective, the container needs to strike the optimum balance between high performance, durability, and optimised weight. For example, using a lightweight webbing system
that runs around the external body of the container can help to minimise fittings and fixtures, while adding greater strength and stability. Such webbing can also be designed with integral handles to simplify the manual handling process.
Reusable vs Single-use
While this article has focussed on how clinical trial cold chain solutions can protect and maintain product efficacy, it’s worth noting the increasing importance of sustainability in supply chain decisionmaking.

You should also therefore aim to prioritise a reusable cold chain system, which is a compelling proposition for any manufacturer seeking to reduce its carbon footprint. The benefit is simple, namely that reusable containers stay in market circulation for a long time – and thus minimise the impact of carbon generated during manufacture.
Inevitably, reusable solutions must also be robust to withstand years of freighting – as documented in the consideration of design above – but there’s an obvious advance to having a system that significantly decreases packaging waste, in comparison to single-use solutions which are discarded after one journey.
As a final aside, research demonstrates that reusable passive shippers afford the lowest cost solution when compared to alternative systems. In the White Paper ‘The Total Cost of Shipping,’ reusable passive shippers were compared to single-use passive shippers and active shippers, on a range of cost factors. It was, respectively, 9% and 34% less expensive than the alternatives.
Box Ticking in the Best Sense
Whatever your specific requirements in terms of shipping clinical trial samples, the innovation within the pharmaceutical cold chain sector means it’s feasible to choose a solution that offers the best possible mix of robust product protection and reliable performance – with long-term reusability as an added advantage.
The ability to select smaller box sizes ideal for clinical trials –which are capable of maintaining temperatures for 120+ hours without electricity and can be manually handled for a truly global reach – further enhances the capabilities now available.
In short, by looking carefully at the options on the market, you can find the container that meets all of your needs: a case of ‘box ticking’ in the best sense.
Nick Gilmore
Nick Gilmore has 24 years sales and marketing experience in international public and private equity owned companies. He is an energetic business leader working cross-functionally and with boards to deliver winning commercial strategies delivering long term sustainable growth from multi-site manufacturing facilities supplying global customers.

Journal for Clinical Studies 39 www.journalforclinicalstudies.com Logistics & Supply Chain
Arena Healthcare Events IFC Catalyst Clinical Research
IBC CPHI Worldwide
5 Ramus Medical Ltd Page 3 Tower Cold Chain
OBC Trilogy Writing & Consulting GmbH
Journal for Clinical Studies

I hope this journal guides you progressively, through the maze of activities and changes taking place in the pharmaceutical industry JCS is also now active on social media. Follow us on:
www.twitter.com/jforcsjournal www.facebook.com/Journal-for-Clinical-Studies www.plus.google.com/+Jforcsjournal www.journalforclinicalstudies.com

40 Journal for Clinical Studies Volume 15 Issue 1 Ad Index Page 29
Page
25 ELRIG Page
Subscribe
today at Email: info@senglobalcoms.com www.journalforclinicalstudies.com
24-26 October 2023

Barcelona, Spain

Exclusive Entry Into The Global Pharma Market
At the heart of Pharma
Book Your Stand Now
Pharma’s largest event is making an exciting return to Barcelona on 24-26 October 2023. SCAN ME



A podcast to discuss topics important for the profession of medical writing Available on Apple Podcasts, Google Podcasts, Spotify, YouTube Podcasts, and TrilogyWriting.com/TriloTalk
